
1 Phänomenologische Therm., Quantenmechanik, Statistische ...pciii/PC32.pdf · - 45 - Statistische...
81
- 45 - Statistische Thermodynamik 1 Phänomenologische Therm., Quantenmechanik, Statistische Thermodynamik In diesem einführenden Kapitel soll etwas zu den Möglichkeiten, Vorzügen und Nachteilen dieser Theorien gesagt werden. Phänomenologische Thermodynamik Wir haben uns bisher in der PC I und im ersten Teil der PC III mit der Phänomenologischen Thermo- dynamik beschäftigt. Diese Theorie gestattet es, ausgehend von einigen Hauptsätzen ein umfangreiches theoretisches Gebäude zu entwickeln, das eine sehr effektive Beschreibung vieler Phänomene in der Natur erlaubt. Wir wollen noch einmal einige Gleichungen herausgreifen und sie kritisch durchleuchten. Mit Hilfe der Clausius-Clapeyronschen Gleichung gelang es uns, den Dampfdruck von Flüssigkeiten zu beschreiben. So konnten wir den Dampfdruck für beliebige Temperaturen vorhersagen, falls ein p,T-Paar und die Verdampfungsenthalpie bekannt waren. Eine ab initio-Berechnung des Dampfdrucks einer vor- gegebenen Flüssigkeit war dagegen nicht möglich. Ganz ähnlich verhält es sich mit dem MWG. Die thermodynamische ab initio-Berechnung der Gleichgewichtskonstante eines Systems ist nicht möglich. Dagegen können wir mit der van't Hoffschen Reaktionsisobaren die Gleichgewichtskonstante bei beliebigen Temperaturen vorhersagen, falls ein K,T-Wertepaar und die Reaktionsenthalpie bekannt sind. Die Unmöglichkeit der Durchführung von ab initio-Berechnungen ist sicher ein erheblicher Nachteil der Phänomenologischen Thermodynamik. Auf der anderen Seite ist es ein gewaltiger Vorteil der Phänomenologischen Thermodynamik, dass ihre Anwendung unabhängig von der Kenntnis des Aufbaus der untersuchten Materie funktioniert. So könnte die Materie auch aus anderen Teilchen mit anderen Wechselwirkungen als in unserem Universum aufgebaut sein, ohne dass sich die Gleichungen der Phänomenologischen Thermodynamik ändern würden. Was sich ändern würde, wären nur die Werte der Variablen in den Gleichungen. Quantenmechanik Um die Quantenmechanik anwenden zu können, ist die Kenntnis der das System aufbauenden Teilchen samt ihren Wechselwirkungen notwendig. Dann ist es möglich, die Schrödinger-Gleichung des Systems (1.1) zu formulieren, in deren Hamiltonoperator H die Operatoren für die kinetischen Energien und die Wechselwirkungen der Teilchen stehen. Für ein makroskopisches System führt dieses zu einer Gleichung mit .10 kinetischen Energietermen und o10 Wechselwirkungstermen. Die Lösung einer 23 23 derartigen Gleichung ist i. a. nicht möglich. In einigen Fällen - z. B. für ein ideales Gas - kann die Komplexität des Problems drastisch reduziert werden. Dazu wird vorausgesetzt, dass die aus den elementaren Bausteinen der Materie gebildeten Atome bzw. Moleküle nicht mehr untereinander wechselwirken, d. h. (1.2) wobei i über die Atome/Moleküle läuft. Wir wollen weiterhin annehmen, dass (1.3) d. h. die Schrödinger-Gleichung des Atoms/Moleküls, lösbar oder in guter Näherung lösbar sei. Dann ergibt sich die Gesamtenergie mit (1.4)
Transcript of 1 Phänomenologische Therm., Quantenmechanik, Statistische ...pciii/PC32.pdf · - 45 - Statistische...

- 45 -
Statistische Thermodynamik
1 Phänomenologische Therm., Quantenmechanik, Statistische ThermodynamikIn diesem einführenden Kapitel soll etwas zu den Möglichkeiten, Vorzügen und Nachteilen dieserTheorien gesagt werden.
Phänomenologische ThermodynamikWir haben uns bisher in der PC I und im ersten Teil der PC III mit der Phänomenologischen Thermo-dynamik beschäftigt. Diese Theorie gestattet es, ausgehend von einigen Hauptsätzen ein umfangreichestheoretisches Gebäude zu entwickeln, das eine sehr effektive Beschreibung vieler Phänomene in derNatur erlaubt.Wir wollen noch einmal einige Gleichungen herausgreifen und sie kritisch durchleuchten. Mit Hilfe derClausius-Clapeyronschen Gleichung gelang es uns, den Dampfdruck von Flüssigkeiten zu beschreiben.So konnten wir den Dampfdruck für beliebige Temperaturen vorhersagen, falls ein p,T-Paar und dieVerdampfungsenthalpie bekannt waren. Eine ab initio-Berechnung des Dampfdrucks einer vor-gegebenen Flüssigkeit war dagegen nicht möglich. Ganz ähnlich verhält es sich mit dem MWG. Diethermodynamische ab initio-Berechnung der Gleichgewichtskonstante eines Systems ist nicht möglich.Dagegen können wir mit der van't Hoffschen Reaktionsisobaren die Gleichgewichtskonstante beibeliebigen Temperaturen vorhersagen, falls ein K,T-Wertepaar und die Reaktionsenthalpie bekanntsind.Die Unmöglichkeit der Durchführung von ab initio-Berechnungen ist sicher ein erheblicher Nachteilder Phänomenologischen Thermodynamik. Auf der anderen Seite ist es ein gewaltiger Vorteil derPhänomenologischen Thermodynamik, dass ihre Anwendung unabhängig von der Kenntnis desAufbaus der untersuchten Materie funktioniert. So könnte die Materie auch aus anderen Teilchen mitanderen Wechselwirkungen als in unserem Universum aufgebaut sein, ohne dass sich die Gleichungender Phänomenologischen Thermodynamik ändern würden. Was sich ändern würde, wären nur dieWerte der Variablen in den Gleichungen.
QuantenmechanikUm die Quantenmechanik anwenden zu können, ist die Kenntnis der das System aufbauenden Teilchensamt ihren Wechselwirkungen notwendig. Dann ist es möglich, die Schrödinger-Gleichung desSystems
(1.1)
zu formulieren, in deren Hamiltonoperator H die Operatoren für die kinetischen Energien und dieWechselwirkungen der Teilchen stehen. Für ein makroskopisches System führt dieses zu einerGleichung mit .10 kinetischen Energietermen und o10 Wechselwirkungstermen. Die Lösung einer23 23
derartigen Gleichung ist i. a. nicht möglich.In einigen Fällen - z. B. für ein ideales Gas - kann die Komplexität des Problems drastisch reduziertwerden. Dazu wird vorausgesetzt, dass die aus den elementaren Bausteinen der Materie gebildetenAtome bzw. Moleküle nicht mehr untereinander wechselwirken, d. h.
(1.2)
wobei i über die Atome/Moleküle läuft. Wir wollen weiterhin annehmen, dass
(1.3)
d. h. die Schrödinger-Gleichung des Atoms/Moleküls, lösbar oder in guter Näherung lösbar sei. Dannergibt sich die Gesamtenergie mit
(1.4)

- 46 -
iund die Gesamtwellenfunktion ist eine Funktion der R .Ist die Berechnung der Eigenschaften eines idealen Gases damit gelöst? Zur Diskussion dieser Fragewollen wir ein Gedankenexperiment mit 100 nicht unterscheidbaren und nicht wechselwirkendenOszillatoren durchführen. Für die Energieniveaus jedes (harmonischen) Oszillators möge
(1.5)
gelten.Fall 1: Die im System der Oszillatoren befindliche Energie möge gerade dem Zustand entsprechen,
in dem sich alle Oszillatoren im Grundzustand befinden. Das Problem ist damit gelöst, da dieGesamtwellenfunktion bekannt ist und alle Observablen mit Hilfe der quantenmechanischenMethoden daraus berechenbar sind.
Fall 2: Die im System befindliche Energie möge gerade 1 h< über dem Grundzustand betragen. Mankönnte vielleicht noch die Frage stellen, welcher Oszillator gerade dieses h< aufweist. Da dieOszillatoren aber ununterscheidbar sind und die Gesamtwellenfunktion so konstruiert werdenmuss, dass die Oszillatoren in ihr ununterscheidbar sind, ist diese Frage ohne Belang. Auchdieses Problem ist damit gelöst.
Fall 3: Die Systemenergie möge 100 h< betragen, wobei die Energieskala von jetzt ab aus Gründeneiner übersichtlicheren Beschreibung vom Grundzustand aus gerechnet wird. In diesemBeispiel können wir quantenmechanisch und experimentell mehrere Fälle unterscheiden. a) 100 Oszillatoren mit 1 h< b) 99 Oszillatoren mit 0 h<, 1 Oszillator mit 100 h< c) 50 Oszillatoren mit 0 h<, 50 Oszillatoren mit 2 h< d) ..........
Die Gesamtwellenfunktionen dieser Systeme unterscheiden sich. In der Quantenmechanikwird das Auftreten mehrerer Wellenfunktionen zu einem Energieniveau als Entartungbezeichnet. Große Systeme mit großen Inneren Energien weisen riesige Entartungszahlen auf.Die Quantenmechanik kann keine Aussage zu der Frage machen, welche Wellenfunktion ineinem entarteten System auftritt oder mit welcher Wahrscheinlichkeit sie auftritt. Die obenaufgeführten, unterschiedlichen quantenmechanischen Zustände des Systems führen zuunterschiedlichen Eigenschaften. So würden sich z. B. die mittleren Abstände in den Oszilla-toren für den Fall eines anharmonischen Potenzials ändern. Die Quantenmechanik istoffensichtlich mit der Berechnung von Systemeigenschaften in Systemen mit großen Ent-artungszahlen überfordert.
Eine zweite Schwierigkeit ergibt sich daraus, dass i. a. gar nicht die Energie eines Systems festgelegtwird wie in den oben diskutierten Fällen, sondern die Temperatur. Die Quantenmechanik kennt dieTemperatur als Variable nicht und kann daher mit dieser Vorgabe nichts anfangen.
Statistische ThermodynamikDie Statistische Thermodynamik schafft nun einen Ausweg aus diesem Dilemma. Die StatistischeThermodynamik setzt voraus, dass die quantenmechanische Lösung des Problems auf mikroskopischerEbene bekannt ist - z. B. Teilchen im Kasten, Rotatoren, Oszillatoren. Dann wird eine Annahme überdie Häufigkeit des Auftretens der einzelnen Wellenfunktionen gemacht und schließlich mit statisti-schen Methoden die daraus resultierenden Systemeigenschaften berechnet.Gegenüber der Phänomenologischen Thermodynamik hat dieses Vorgehen einen großen Vorteil. Istdie quantenmechanische Lösung des mikroskopischen Problems bekannt und besteht das System auseiner genügend großen Anzahl von Teilchen, so liefert die Statistische Thermodynamik exakteAbsolutberechnungen der Systemeigenschaften. Der Zusatz "exakt" ist weitgehend korrekt, obwohldie Quantenmechanik gewisse Variable nur innerhalb der Unschärferelation anzugeben gestattet. Fürmakroskopische Systeme ist jedoch die so hervorgerufene Unschärfe vernachlässigbar klein. Weiter-

- 47 -
hin unterliegen statistische Verfahren Schwankungen. Legt man es darauf an, so kann man in kleinenSystemen Schwankungen beobachten. Beispielsweise schwankt die Teilchenzahl in einem offenenSystem mit festgelegtem p, V, T. Der Begriff exakt ist daher im Zusammenhang mit einer genügendgroßen Teilchenzahl zu sehen; genauer gesagt: Die Statistische Thermodynamik liefert exakteErgebnisse für den Fall Teilchenzahl 6 4.Auf der anderen Seite weist die Statistische Thermodynamik eine Reihe gravierender Nachteilegegenüber der Phänomenologischen Thermodynamik auf. Ist die quantenmechanische Lösung desbetreffenden Problems auf mikroskopischer Ebene nicht bekannt, so lässt sich mit der StatistischenThermodynamik wenig anfangen. Dieses ist z. B. der Fall für fast alle Berechnungen in flüssigenSystemen. Die quantenmechanische Lösung des Gesamtsystems ist aus den oben diskutierten Gründennicht zugänglich. Der bei den Gasen beschreitbare Weg über die Zerlegung des Systems in nichtwechselwirkende Teilchen ist nicht möglich, da die Wechselwirkung der Teilchen untereinander beiweitem zu groß sind. Zur Thermodynamik flüssiger Systeme kann daher die Statistische Thermodyna-mik vergleichsweise wenig aussagen.Bei Festkörpern ist die Lage für die Statistische Thermodynamik günstiger wegen der hohen Symme-trie der Festkörper. Im Gegensatz zu den Flüssigkeiten lässt sich hier wegen der hohen Translations-symmetrie des Problems die Gesamtwellenfunktion des Systems für viele Zwecke mit ausreichenderGenauigkeit berechnen. Die Statistische Thermodynamik erlaubt dann die Berechnung der inter-essierenden thermodynamischen Größen.Zum Schluß dieses Kapitels soll noch etwas zur Abgrenzung des Vorlesungsstoffs bemerkt werden.Bei der Phänomenologischen Thermodynamik gibt es zwei große Gebiete: die bereits behandeltePhänomenologische Thermodynamik der Gleichgewichtszustände und die PhänomenologischeThermodynamik der irreversiblen Prozesse. Entsprechende Analoga gibt es auch im Bereich derStatistischen Thermodynamik. Während die Statistische Thermodynamik der Gleichgewichtszuständeheute eine gut entwickelte Theorie darstelle, ist die Statistische Thermodynamik der irreversiblenProzesse heute noch Gegenstand der Forschung (Prigogine u. a.). Aus Zeitgründen und wegen derKomplexität der Theorie der irreversiblen Prozesse wird auf eine Darstellung dieses Teils vollständigverzichtet.

- 48 -
Abb. 26 Mikrokanonisches Ensemble
2 GesamtheitenFür die folgende Diskussion wollen wir folgende Begriffe verwenden.
TeilchenUnter einem Teilchen wollen wir ein Atom, ein Molekül, einen Spin, einen Oszillator oder ähnlichesverstehen. Der innere Aufbau der Teilchen interessiert die Statistische Thermodynamik nicht. Bekanntsein müssen dagegen die Energieniveaus (z. B. Rotationsniveaus), die ein derartiges Teilchen aufweist.
SystemDer Systembegriff stimmt mit dem bereits in der PC I verwendeten überein. Ein System enthält i. a.viele Teilchen. Wir werden jedoch später bei der Berechnung der Zustandssummen auch durchausSysteme, die nur aus einem Teilchen bestehen, zulassen. Die Systeme werden durch ihre thermody-namischen Variablen charakterisiert, z. B. - Teilchenzahl N - Energie E - Volumen VDas stimmt mit dem Beschreibungsverfahren in der PC I weitgehend überein, da die Teilchenzahl unddie Stoffmenge über einen konstanten Faktor zusammenhängen und sonst nur S gegen E ausgetauschtist. Obwohl das System nun thermodynamisch festgelegt ist, ist es die Gesamtwellenfunktion desSystems nicht. So sagt die Angabe - N = 100 - E = 500 h< - V = 1 dm3
für ein System aus Oszillatoren nichts über die vorliegende Wellenfunktion aus.
GesamtheitUnter einer Gesamtheit oder einem Ensemble versteht man eine große Anzahl von Systemen, diemiteinander in einer noch zu beschreibenden Weise gekoppelt werden. Die auf den ersten Blickvielleicht etwas merkwürdig anmutende Konstruktion, die zu einer Erhöhung der Komplexität führt,wobei das einzelne System schon genügend komplex ist, stammt vom Großmeister der Thermodyna-mik, J. W. Gibbs, und bringt folgenden Vorteil mit sich. Eine Gesamtheit kann man in einemGedankenexperiment beliebig vergrößern und damitdie Grundforderung der Statistik nach Teilchenzahl 64 erfüllen. Diese Möglichkeit wird uns weiterhin spä-ter bei einer Vereinfachung gute Dienste leisten (sieheKap. 4, Verwendung der Stirlingschen Näherungs-formel).Gibbs hat drei verschiedene Typen von Ensembleseingeführt, von denen wir zwei benutzen werden.
Mikrokanonische GesamtheitWir stellen M Systeme zu einer Gesamtheit zusam-men. Die Wände, welche die einzelnen Systeme von-einander trennen sind sowohl energie- als auch mate-rieundurchlässig. In der Sprache der Phänomenologi-schen Thermodynamik könnte man daher auch sagen:Ein mikrokanonisches Ensemble ist eine Ansammlungvon isolierten Systemen. Die einzelnen Systeme sinddurch N, E und V definiert, die für alle Systeme über-einstimmen sollen. Es gilt für die Gesamtheit:

- 49 -
Abb. 27 Kanonisches Ensemble
(2.1)
Kanonische GesamtheitDer einzige Unterschied zum mikrokanonischen Ensem-ble besteht darin, dass die einzelnen Systeme durch N, Tund V definiert werden. Die Wände sollen wärmedurch-lässig und materieundurchlässig sein (geschlossene Sys-teme). Die Herstellung des Ensembles geschieht wiefolgt. Die einzelnen Systeme oder auch alle zusammenwerden in Kontakt mit einem genügend großen Wärme-reservoir der Temperatur T gebracht und der Tempera-turausgleich abgewartet. Dann werden die M Systemezum Ensemble zusammengestellt. Dabei bilden für jedesSystem die restlichen M - 1 Systeme das Wärmereser-voir. Dann wird das ganze Systeme durch eine wärmeun-durchlässige (adiabatische) Hülle umschlossen. Es gilt:
(2.2)
iIm Gegensatz zum mikrokanonischen Ensemble kann jetzt die Energie E der einzelnen Systemeschwanken. Die Energie E des Gesamtsystems ist dagegen durch die adiabatische Hülle, dasG
festgelegte Volumen und die Materieundurchlässigkeit der Wände der Systeme fixiert.
Große kanonische GesamtheitDieser Ensembletyp enthält wärme- und materiedurchlässige Wände um die einzelnen Systeme. Fürdie uns interessierenden Probleme ist dieser Typ von geringerem Interesse.

- 50 -
Abb. 28 Mikrokanonisches Ensemble mit quan-tenmechanischen Zuständen
3 PostulateDie bisher per definitionem eingeführten Begriffe unterliegen keinem Zweifel, ob sie richtig oderfalsch sind; sie könnten allenfalls unpraktisch sein.Wie alle anderen Theorien (Phänomenologische Thermodynamik, Quantenmechanik, Elektrodynamik(Maxwell-Gleichungen), Mechanik (Newtonsche Axiome), Mathematik) kommt die StatistischeThermodynamik nicht ohne unbewiesene Postulate aus. Unter einem Postulat versteht man nichtsanderes als unter einem Axiom oder einem Hauptsatz. Diese Begriffe haben die gleiche Bedeutung;es haben sich einfach innerhalb der verschiedenen Zweige der Wissenschaft unterschiedliche Bezeich-nungen entwickelt.Die Statistische Thermodynamik benötigt zwei Postulate, zu deren Verständnis einige Vorbemerkun-gen notwendig sind.
Zuerst konstruieren wir ein mikrokanonischesEnsemble, indem wir jeden mit der BedingungE = const. verträglichen quantenmechanischen
iZustand R berechnen und diese Zustände alsmögliche Zustände für unseren thermodynamischdefinierten Zustand geordnet nach einer willkür-lich festgelegten Indizierung i in die Systemzellenhineinschreiben. Wir wollen jeden möglichenZustand nur einmal aufführen. Wie wird sich einthermodynamisch definiertes, reales System alsFunktion der Zeit verhalten? Es wird sich z. B.
3 17eine gewisse Zeit in R aufhalten, dann nach Rspringen usw. Sieht man dieses Ensemble alsmöglichen Zustandsraum für ein reales Systeman, so bewegt sich das System auf einer Bahndurch diesen Raum. Im Experiment wird man nunden Verlauf dieser Bahn sicherlich nicht zeitlichauflösen können. So weiß man z. B. bei einemGas, dass sich bei fast jedem Stoß Drehimpuls(Rotationsenergie) und translatorischer Impuls(kinetische Energie) ändern. Die Gesamtstoßfre-
quenz in einem Gas liegt nun so hoch, dass an eine zeitaufgelöste Messung der Einzelimpulse nichtzu denken ist. Messtechnisch sind nur zeitliche Mittelwerte zugänglich. So kann z. B. in einemMolekularstrahl die Geschwindigkeitsverteilung gemessen werden. Die Berechnung dieser Mittelwertemit der Theorie wäre möglich, wenn man wüßte, wie lange sich das reale System in den einzelnenquantenmechanischen Zuständen aufhält, oder wenn man wüßte, wie der zeitliche Mittelwert derinteressierenden Variablen mit dem Ensemblemittelwert zusammenhängt.Diese Lücke schließen nun die beiden Postulate der Statistischen Thermodynamik.
Postulat IDas zeitliche Mittel einer Variablen eines thermodynamischen Systems stimmt mit dem Ensemble-mittelwert für diese Variable überein, wenn die das System definierenden thermodynamischen Größenmit denen der Systeme des Ensembles übereinstimmen und die Systemzahl gegen unendlich geht.Oder in Kurzfassung: Zeitmittel = GesamtheitsmittelFür den Begriff Gesamtheitsmittel wird auch manchmal "Scharmittel" verwendet.
Postulat IIIn einem mikrokanonischen Ensemble treten die mit N, E und V verträglichen quantenmechanischenZustände mit gleicher Häufigkeit auf.

- 51 -
Das kleinste, vollständige mikrokanonische Ensemble enthält also jeden quantenmechanischenZustand gerade einmal. Es ist jedoch erlaubt, alle Zustände mehrfach aufzuführen und damit derForderung nach M 6 4 Genüge zu tun.Postulat II ist auch als "Prinzip der gleichen a priori-Wahrscheinlichkeit" bekannt. "a priori" ist einBegriff aus der Erkenntnis-Theorie und charakterisiert eine Behauptung, die nicht durch ein Exper-iment belegt werden kann, sondern von vornherein als wahr angenommen wird. Dagegen ist eineBehauptung "a posteriori" nach Durchführung eines Experiments.Aus den Postulaten I und II folgt: Ein thermodynamisch definiertes System verbringt über lange Zeitengemittelt gleiche Zeiten in den einzelnen quantenmechanischen Zuständen. Dieser Satz ist auch unterdem Namen Ergodenhypothese bekannt. Eine Ergode ist die Bahn eines Systems im Phasenraum.Ob die beiden Postulate der Statistischen Thermodynamik wahr sind, ist nicht nachgewiesen und wirdsich wohl auch nicht nachweisen lassen. Z. Z. ist jedoch kein Experiment bekannt, das diesenPostulaten widerspricht.

- 52 -
Abb. 29 Ensemble mit einem Supersystem
4 BoltzmannverteilungDie Boltzmannverteilung beschreibt die Energieverteilung in einem System mit (quantenmechanisch)vorgegebenen Energieniveaus bei vorgegebener Temperatur. Zur Berechnung der Energieverteilungwird wie folgt vorgegangen. Statt einzelne Teilchen in einem System werden wir zuerst Systeme ineinem Ensemble betrachten und später nur noch ein Teilchen pro System zulassen. Mit einemmikrokanonischen Ensemble werden wir die Berechnung nicht durchführen können, da dann die
iSystemenergien E festliegen würden. Das richtige Ensemble für diese Berechnung ist sicher das
ikanonische. In diesem Ensemble dürfen wir die Temperatur festlegen, und die Energie E der einzelnenSysteme schwankt. Wir werden dann später nur noch ein Teilchen pro System der Berechnungzugrunde legen und damit den zeitlichen Mittelwert für die Energieverteilung in einem System mit NTeilchen finden. Bei dieser Berechnung geht leider die Temperatur verloren, da nur noch die EnergieE der Gesamtheit festliegt. Die Temperatur werden wir daher anderweitig bestimmen müssen.G
Eine Schwierigkeit besteht darin, dass die Aussage des Postulats II nur für ein mikrokanonischesEnsemble gilt. Für das zu verwendende kanonische Ensemble steht uns leider keine Aussage über dieHäufigkeit des Auftretens der quantenmechanischen Zustände zur Verfügung. Diese Schwierigkeitwird wie folgt überwunden. Im kanonischen Ensemble ist wegen der Konstruktion der Wände undwegen der adiabatischen Hülle die Energie konstant. Wir dürfen daher das ganze Ensemble als Systemeines größeren mikrokanonischen Ensembles betrachten. Um Verwechslungen bei diesem schwierigenSchritt zu vermeiden, wollen wir das jetzt als System betrachtete kanonische Ensemble als Supersys-tem bezeichnen.
Abb. 29 zeigt ein Supersystem mit seiner Wel-lenfunktion R . Die verschiedenen SupersystemeSS
entstehen durch Vertauschung der Wellenfunktio-
1 2nen R , R , . . . aus der jetzt ins Auge gefasstenVerteilung der Wellenfunktionen. Jede dieserVertauschungen ergibt eine unterschiedliche Wel-lenfunktion R und damit ein neues Supersystem.SS
Jedes dieser durch Vertauschungen entstandenenSupersysteme tritt im mikrokanonischen Ensem-ble der Supersysteme mit gleicher Häufigkeit auf.Die Anzahl dieser möglichen Wellenfunktionenwollen wir zählen und das Ergebnis, die thermo-dynamische Wahrscheinlichkeit, mit W bezeich-nen.
Dazu soll folgendes Beispiel diskutiert werden. Die Energieverteilung auf
4 4 1*R mit 1*E
3 3 2*R mit 2*E
2 2 3*R mit 3*E
1 1 4*R mit 4*E
4 3 2 1sei mit der Energie E = 1*E + 2*E + 3*E + 4*E des Supersystems mit M = 10 Systemen ver-G
träglich. Zwei der vielen möglichen Wellenfunktionen des Supersystems sind
4 3 3 2 2 3 4 3 2 2R R R R R R R R R R
2 1 1 1 1 2 1 1 1 1R R R R R R R R R R
Insgesamt gibt es 12 600 verschiedene Wellenfunktionen des Supersystems, wie in einem der folgen-den Beispiele gezeigt werden wird.

- 53 -
i i i iM E /h< M * E /h<
1 4 4
2 3 6
3 2 6
4 1 4
E = 10 E = 20
iSind alle System-R verschieden, so gibt es M! unterscheidbare Anordnungsmöglichkeiten (Zahl der
1 1Permutationen von M Elementen ohne Wiederholungen). Falls M Systeme mit gleichem R vorhan-
1den sind, muss obiges Ergebnis durch die Zahl der Anordnungsmöglichkeiten von M Systemen , d.
1 i ih. durch M !, geteilt werden. Im nächsten Schritt lassen wir für alle i M Systeme mit gleichem R zu.Dies führt zu folgendem Ergebnis
(4.1)
Neben dieser Gleichung gibt es zwei weitere Bedingungen, die erfüllt sein müssen:
(4.2)
Beachten Sie bitte, dass dieses die drei ersten ordentlichen Gleichungen dieses Teils des Skripts sind.Die Gleichungsdichte nimmt von jetzt an jedoch exponentiell zu.Die Gl. (4.1) und (4.2) bilden die Basis der Ableitung der Boltzmannverteilung. Wenn Sie dieseGleichungen mit denen in einfacheren Lehrbüchern vergleichen, werden Sie feststellen, dass die
iUnterschiede gering sind. Hier sind die M Systemzahlen, die im Gedankenexperiment beliebig großgemacht werden können, dort sind es Besetzungszahlen, die oft sehr klein (n1) sind.
i iDas korrekte weitere Vorgehen zur Berechnung von M = f(E ) wäre das folgende. Man müsste für allemit Gl. (4.2) verträglichen Verteilungen W berechnen und über alle Verteilungen mit W gewichtet
imitteln und damit die mittleren M ausrechnen. Diese Verfahren wurde von Darwin und Fowler unteranderem mit Hilfe der Theorie komplexer Funktionen durchgeführt. Wir wollen ein sehr viel ein-facheres Verfahren anwenden, das für M 6 4 zum gleichen Ergebnis führt.Von den oben aufgeführten Verteilungen wollen wir nur die Verteilung berücksichtigen, welche diegrößte thermodynamische Wahrscheinlichkeit aufweist. Alle anderen Verteilungen werden ver-nachlässigt. Dass dieses Verfahren mit seiner anscheinend groben Näherung zum richtigen Ergebnisführt, hat folgenden Grund. Alle Verteilungen, die wesentlich von der wahrscheinlichsten abweichen,weisen eine im Verhältnis zur häufigsten äußerst geringe thermodynamische Wahrscheinlichkeit auf.Alle Verteilungen, die große thermodynamische Wahrscheinlichkeiten aufweisen, sind der wahr-scheinlichsten sehr ähnlich.Dieses soll an zwei Beispielen erläutert werden.
1) Das betrachtete System möge einen Oszillator pro System aufweisen. Die Gesamtsystemzahl M soll10 und die Gesamtenergie E soll 20 h< über dem Grundzustand betragen. Wir untersuchen jetztG
einige mit dieser thermodynamischen Festlegung verträgliche quantenmechanische Zustände.
Verteilung 1: Jedes der 10 Systeme weise die
2Energie E = 2 h< auf. Nach (4.1) ergibt sichW = 10!/10! = 1. Die 10! im Nenner entsteht da-durch, dass nur ein Niveau (i = 2) mit 10 Syste-men besetzt ist.Verteilung 2: Ein System weise 20 h< auf. Fürdiesen Fall ist W = 10!/(1! 9!) = 10. Die 9! entstehtdadurch, dass sich 9 Systeme im Grundzustandbefinden.Verteilung 3: Als letzte Verteilung wollen wir dienebenstehende Verteilung untersuchen, die derendgültigen nahekommt.
Die thermodynamische Wahrscheinlichkeit be-trägt:

- 54 -
Abb. 30 Drei verschiedene Verteilungen
Die drei untersuchten Verteilungen sind in Abb. 30noch einmal dargestellt. Die thermodynamischeWahrscheinlichkeit von Verteilung 3 ist somit erheb-lich größer als die der anderen. Dieses hängt damitzusammen, dass in dieser Verteilung viele unter-schiedliche Niveaus besetzt sind und deswegen die
iM ! im Nenner klein sind. Die endgültige Verteilungwird sich daher über eine möglichst große Zahl vonNiveaus erstrecken. Da die Gesamtenergie festliegt,werden die energetisch tiefer liegenden Niveaushöhere Besetzungszahlen aufweisen.
1 2 12) Im zweiten Beispiel soll ein einfaches System mit zwei Zuständen R und R gleicher Energie Euntersucht und W für die wahrscheinlichste sowie ähnliche Verteilungen bei großen Systemzahlen
1berechnet werden. Die Systemzahl soll 2M und die Gesamtenergie E gerade 2ME betragen. Also z.G
B.
1 - M = 100: 200 Systeme mit 200 E
1 - M = 1 000: 2000 Systeme mit 2000 E
1 - M = 10 000: 20000 Systeme mit 20000 EDie wahrscheinlichste Verteilung ist in allen Fällen
1 2 M Systeme in R und M Systeme in RWir wollen nun das Verhältnis V der thermodynamischen Wahrscheinlichkeit für eine Verteilung, die
1 2) mehr Systeme in R und dementsprechend ) weniger in R aufweist, zur wahrscheinlichstenausrechnen.
(4.3)
) soll immer 1 % von M betragen. Für den Fall 2M = 200 ergibt sich daher
Für große M gelingt die Berechnung nach zwei Verfahren:1) Durch Kürzen der Fakultäten entsteht
(4.4)
2) Die Fakultäten werden mit der Stirlingschen Näherungsformel (siehe Anhang 14.2) berechnet.
(4.5)
Die Berechnung von V ergibt

- 55 -
M V
100 0,99
1 000 0,90
10 000 0,37
100 000 0,000 05
1 000 000 4@10-44
Einige Werte für V = f(M) sind in der nebenste-henden Tabelle aufgeführt. Aus dieser Berech-nung folgt, dass die alleinige Berücksichtigungder wahrscheinlichsten Verteilung für alle makro-skopischen Systeme ausreichend ist. Weiterhinkonnte gezeigt werden, dass die Darwin-Fowler-Methode für große Systeme zum gleichen Ergeb-nis führt.
iWir werden daher die M so bestimmen, dass unter Beachtung der Nebenbedingungen (4.2) diethermodynamische Wahrscheinlichkeit nach (4.1) maximal wird. Das Verfahren der Wahl zur Lösungdieses Variationsproblems ist die bereits in der PC I benutzte Lagrangesche Multiplikatormethode.Anstelle von W werden wir ln W maximal machen; das ist zulässig und macht die Berechnung beizwei Schritten (Differenziation von W und Anwendung der Stirlingschen Näherungsformel) einfacher.Zuerst wird das totale Differenzial von ln W gebildet.
(4.6)
Die Abhängigkeit von M ist dabei nicht berücksichtigt, da wegen der Nebenbedingung dM = 0 gilt. Fürdie wahrscheinlichste Verteilung muss nun gelten
(4.7)
oder
(4.8)
i iWären alle M völlig unabhängig voneinander variierbar, so würde daraus MlnW/MM = 0 folgen. DieseUnabhängigkeit liegt nun wegen der Nebenbedingungen (4.2) nicht vor. Im Lagrangeschen Multiplika-torverfahren werden die Nebenbedingungen als Differenziale geschrieben und mit den LagrangeschenMultiplikatoren multipliziert zur Hauptbedingung addiert.
(4.9)
Die Bedingungsgleichung für ein Extremum von W ist daher:
(4.10)
wobei " und $ die unbekannten Lagrangeschen Multiplikatoren sind. Einige Umstellungen ergeben:
(4.11)
(4.12)
Bei der Differenziation wird die Stirlingsche Näherungsformel verwendet
(4.13)

- 56 -
(4.14)
(Verwendet man in (4.13) die erste Nebenbedingung von Gl. (4.2), so bleibt in (4.14) eine 1 stehen,
idie gegen ln M vernachlässigt werden kann). Aus Gl. (4.12) wird daher
(4.15)
(4.16)
Der nächste Schritt ist die Bestimmung von " aus der ersten Nebenbedingung (4.2).
(4.17)
(4.18)
Schwieriger ist die Bestimmung von $. $ muss die Dimension einer reziproken Energie aufweisen.Weiterhin muss in $ die bei unserer Berechnung verlorengegangene Temperatur enthalten sein. Da
i ihohe Temperaturen die M bei hohen E begünstigen sollen, erwarten wir
(4.19)
Um diese Gleichung dimensionsmäßig richtig zu machen, nehmen wir
(4.20)
an, wobei k die Boltzmannkonstante mit dem Wert 1,3806@10 J/K ist. Später werden wir diese-23
Annahme genauer durch einen Vergleich eines aus der Statistischen Thermodynamik gewonnenErgebnisses mit experimentellen Daten begründen können. Diese Schwierigkeit wird immer nochdurch das alte Problem hervorgerufen: Die Quantenmechanik kennt die Temperatur nicht, und diebisher durchgeführten Berechnungen mit der Statistischen Mechanik können daran auch nichts ändern.Um bereits jetzt zu den endgültigen Formeln zu gelangen, wollen wir die Annahme (4.20) benutzen
iund erhalten damit die Verteilung der Energien E in einem kanonischen Ensemble
(4.21)
Wegen der zentralen Bedeutung dieser Gleichung sollen die eingehenden Größen noch einmalbeschrieben werden: - M: Systemzahl
i i - M : Zahl der Systeme mit der Wellenfunktion R .Die Wellenfunktion wird auch als Zustand i bezeichnet.
iZum Zustand i gehört die Energie E .
i - E : Energie des Zustands i. - E: Die Summe läuft über die Zustände i, nicht über die Energien.
jEine andere Möglichkeit der Darstellung besteht darin, die Summe über die Energieniveaus E laufen
j j jzu lassen. Ist das Energieniveau E g -fach entartet, d. h. gibt es zu diesem Niveau g linear un-
i jabhängige R , so muss bei einer Summenbildung über die Energien g als Faktor in der Verteilungberücksichtigt werden.
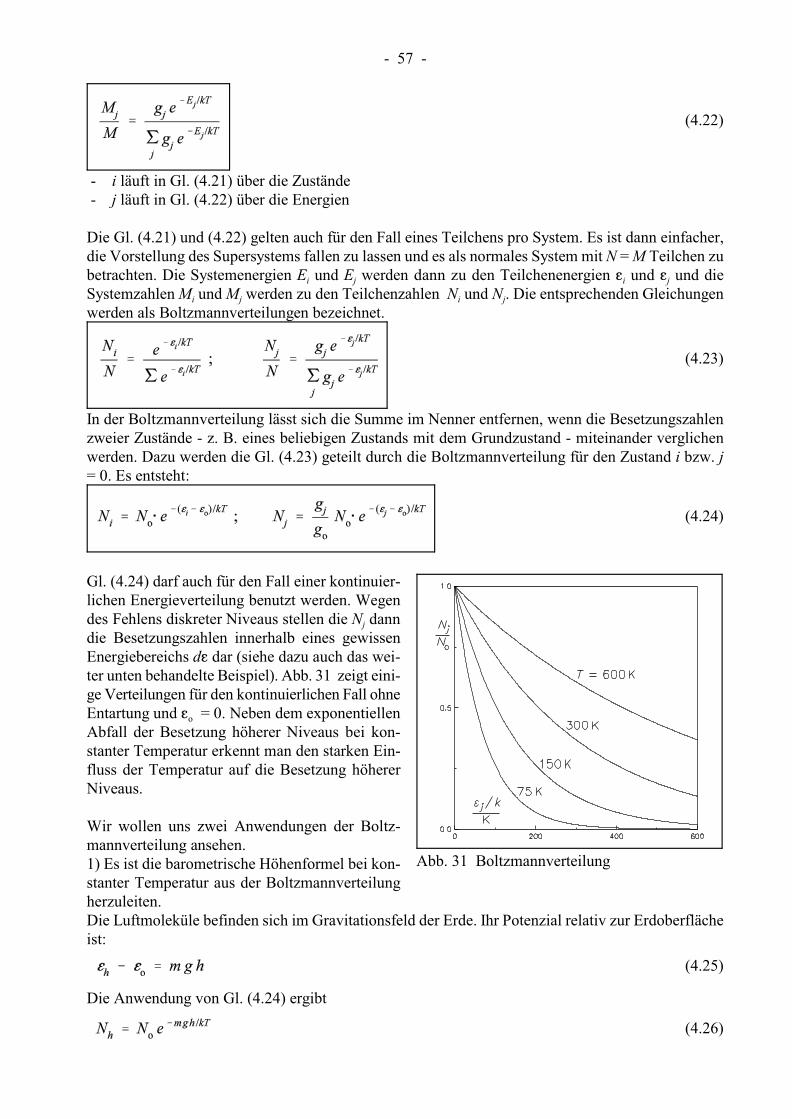
- 57 -
Abb. 31 Boltzmannverteilung
(4.22)
- i läuft in Gl. (4.21) über die Zustände - j läuft in Gl. (4.22) über die Energien
Die Gl. (4.21) und (4.22) gelten auch für den Fall eines Teilchens pro System. Es ist dann einfacher,die Vorstellung des Supersystems fallen zu lassen und es als normales System mit N = M Teilchen zu
i j i jbetrachten. Die Systemenergien E und E werden dann zu den Teilchenenergien g und g und die
i j i jSystemzahlen M und M werden zu den Teilchenzahlen N und N . Die entsprechenden Gleichungenwerden als Boltzmannverteilungen bezeichnet.
(4.23)
In der Boltzmannverteilung lässt sich die Summe im Nenner entfernen, wenn die Besetzungszahlenzweier Zustände - z. B. eines beliebigen Zustands mit dem Grundzustand - miteinander verglichenwerden. Dazu werden die Gl. (4.23) geteilt durch die Boltzmannverteilung für den Zustand i bzw. j= 0. Es entsteht:
(4.24)
Gl. (4.24) darf auch für den Fall einer kontinuier-lichen Energieverteilung benutzt werden. Wegen
jdes Fehlens diskreter Niveaus stellen die N danndie Besetzungszahlen innerhalb eines gewissenEnergiebereichs dg dar (siehe dazu auch das wei-ter unten behandelte Beispiel). Abb. 31 zeigt eini-ge Verteilungen für den kontinuierlichen Fall ohne
oEntartung und g = 0. Neben dem exponentiellenAbfall der Besetzung höherer Niveaus bei kon-stanter Temperatur erkennt man den starken Ein-fluss der Temperatur auf die Besetzung höhererNiveaus.
Wir wollen uns zwei Anwendungen der Boltz-mannverteilung ansehen.1) Es ist die barometrische Höhenformel bei kon-stanter Temperatur aus der Boltzmannverteilungherzuleiten.Die Luftmoleküle befinden sich im Gravitationsfeld der Erde. Ihr Potenzial relativ zur Erdoberflächeist:
(4.25)
Die Anwendung von Gl. (4.24) ergibt
(4.26)

- 58 -
hN ist die Teilchenzahl innerhalb einer gewissen Höhendifferenz dh. Bezieht man die Teilchenzahlenzusätzlich auf die Fläche dx@dy, so gehen sie in die Teilchendichten N über. Da die Teilchendichten1
bei konstanter Temperatur den Drücken proportional sind, gilt schließlich
(4.27)
Das ist die barometrische Höhenformel.
2) Es ist der Besetzungsunterschied der beiden Kernspinniveaus eines Protons im Magnetfeld eines100 MHz-Kernresonanzspektrometers zu berechnen. Dieser Besetzungsunterschied ist für dieFunktion eines Kernresonanzspektrometers von zentraler Wichtigkeit, da bei einer Gleichbesetzungder Niveaus bei Einstrahlung der elektromagnetischen Welle genausoviel Absorptions- wie Emis-sionsprozesse stattfinden würden, d. h. in diesem Fall wäre keine Detektion der Resonanz möglich.Die Empfindlichkeit eines Kernresonanzspektrometers ist dem Besetzungsunterschied direkt propor-tional.
1 0 1Der Energieunterschied der beiden Niveaus ist h< mit < = 100 MHz, d. h. g - g = 6,626@10 J. g!26
0und g sollen 1 sein. Gl. (4.24) ergibt daher bei 300 K
Der relative Besetzungsunterschied beträgt daher nur etwa 10 . Er ist proportional zur Messfreqenz-5
und damit zur Magnetfeldstärke. Ein 600 MHz-Spektrometer ergibt daher bei sonst gleichen Bedin-gungen die 6-fache Empfindlichkeit.

- 59 -
5 Thermodynamische Zustandsgrößen, Zustandssummen
5.1 Innere Energie und ZustandssummeDie Energieverteilung (4.21) erlaubt nun die Berechnung der mittleren Energie eines Systems ineinfacher Weise. Für die berechnete Energie wollen wir U als Symbol benutzen, da wir uns nur mitden in der Inneren Energie enthaltenen Termen beschäftigen werden. Für die mittlere Energie einesSystems finden wir
(5.1.1)
Im Prinzip könnte man die Gleichung so stehen lassen. Diese Form ist jedoch wegen der zweiSummen unpraktisch. So wird z. B. die Berechnung der Molwärmen durch eine Differenziation nachT sehr aufwändig. Zur Vereinfachung wird die Zustandssumme Z eingeführt:
i läuft über die Zustände (5.1.2)
j läuft über die Energieniveaus (5.1.3)
Diese Zustandssummen haben keine direkte physikalische Bedeutung; sie sind ein Hilfsmittel, um diefolgenden Gleichungen einfacher schreiben zu können. Später werden wir jedoch sehen, dass einrelativ enger Zusammenhang zwischen Z und der Freien Energie besteht (siehe Kap. 5.4). ZurDarstellung der Inneren Energie mit der Zustandssumme berechnen wir
(5.1.4)
Mit (5.1.1) folgt daraus
(5.1.5)
Der Vorteil der Verwendung der Zustandssumme besteht darin, dass die Gl. für die Innere Energie nur
Lnoch eine Summe enthält. Die dafür eingekaufte Differenziation ist unproblematisch. c lässt sicheinfach durch eine weitere Differenziation berechnen.
5.2 Systemzustandssummen, TeilchenzustandssummenDie oben eingeführte Zustandssumme "Groß-Z" stellt die Zustandssumme für ein System dar (System-
izustandssumme), da die E Systemenergien sind. Bekannt sind aus der Quantenmechanik nur dieTeilchenzustandssummen "Klein-z"
(5.2.1)
iwobei die g die Teilchenenergien darstellen und i über die Teilchenzustände und j über die Teil-chenniveaus läuft. Im folgenden wird der Zusammenhang zwischen Z und z hergeleitet.
a) Unterscheidbare TeilchenUnterscheidbare Teilchen findet man z. B. bei Atomen oder Molekülen in einem Kristall, da dieseanhand ihrer Position unterscheidbar sind. Wir nehmen nun an, dass die Teilchen entweder nichtmiteinander wechselwirken sollen, was in einem Kristall kaum möglich ist, oder dass die Wechsel-

- 60 -
Systemeigenschaften
R E
11 21 11 21R @ R g + g
11 22 11 22R @ R g + g
12 21 12 21R @ R g + g
12 22 12 22R @ R g + g
Teilcheneigenschaften
Teilchen 1 Teilchen 2 g
11 21 1R R g
12 22 2R R g
wirkung der Teilchen mit ihrer Umgebung durch ein gleiches, mittleres Potenzial, das von derUmgebung herrührt, beschrieben werden kann. Für diese Fälle gelten die Gl. (1.1) bis (1.4), wobei dieUmgebung im Hamiltonoperator des Teilchens als Potenzial auftritt. Für die weitere Diskussionwollen wir zwei identische, aber unterscheidbare Teilchen 1 und 2 betrachten, die jeweils zweiZustände 1 und 2 aufweisen sollen. Insgesamt entstehen die unten aufgeführten, vier Systemzustände.Der erste Index bezieht sich auf die Teilchen, der zweite auf die Zustände.
Die Systemzustandssumme ist:
(5.2.2)
Der letzte Gleichungsteil resultiert aus der Gleichartigkeit der Teilchen. Für N unterscheidbareTeilchen gilt analog
(5.2.3)
Es könnten Zweifel an der Formel bezüglich der Teilchenzahlabhängigkeit mit der Potenz N entstehen.Es gilt U % N, da aber U = f(ln Z) ist, ergibt Gl. (5.2.3) die richtige Abhängigkeit von der Teilchenzahl.
b) Ununterscheidbare TeilchenDieser Fall liegt z. B. bei Atomen in der Gasphase vor und ist leider schwieriger zu durchschauen. Wir
11 12 21 22betrachten dazu N identische Teilchen 1, 2, 3, . . . mit den Energieniveaus g , g , . . ., g , g , . . . . undnehmen zuerst noch einmal an, dass die Teilchen unterscheidbar sind. Dann gilt:
(5.2.4)
wobei k, l, m, . . . über alle Zustände laufen. Gl. (5.2.4) entspricht Gl. (5.2.2) als Summe geschrieben.Die Ununterscheidbarkeit bewirkt nun folgendes. Die Quantenmechanik verlangt, dass die Wellen-funktion des Systems (in einer Determinante) so geschrieben werden muss, dass sie bei einer Teilchen-vertauschung bis auf einen möglichen Vorzeichenwechsel erhalten bleibt (siehe dazu auch Kap. 13.1mit genauerer Information). Daher müssen alle Terme in der Vielfachsumme der ersten Zeile von(5.2.4) gestrichen werden, die sich nur durch Teilchenvertauschungen unterscheiden, d. h. z. B.
1,k 2,l 2,k 1,l g ;g ; . . . und g ;g ; . . . . .Diese stellen im Sinne der Quantenmechanik keine unterschiedlichen Wellenfunktionen dar. Wenn

- 61 -
alle k, l, m, . . . verschieden sind, sind das insgesamt N! Vertauschungen, d. h. z ist durch N! zu teilen.N
Dieses Verfahren bewirkt jedoch einen Fehler, da auch Terme von der Form
11 21 31 g ;g ;g ; . . . .und ähnliche auftreten, die es (in diesem Fall) nur einmal gibt und an denen daher nichts zu streichenist. Nun beschreibt dieser Zustand eine Mehrfachbesetzung, d. h. alle Teilchen (bzw. mehrereTeilchen) befinden sich im gleichen Zustand. Für die uns interessierenden Teilchen in der Gasphaseist die Wahrscheinlichkeit des Auftretens solcher Zustände vernachlässigbar klein, da die Zahl der
ierreichbaren Translationsniveaus (g . kT) immer riesig im Verhältnis zur Teilchenzahl ist (siehe Kap.6). Wir machen daher keinen großen Fehler, wenn auch diese Terme durch N! geteilt werden. Wirwerden im Kap. 13 sehen, dass die Boltzmannverteilung sowieso nur für den Fall geringer Besetzungs-dichten gilt, die aber i. a. vorliegen.Für den Fall der ununterscheidbaren Teilchen gilt daher unter den genannten Bedingungen
(5.2.5)
5.3 EntropieIm folgenden soll der Zusammenhang zwischen der Zustandssumme und den weiteren Zustandsgrößenhergeleitet werden. Der einfachste Weg besteht darin, zuerst den Zusammenhang mit der Entropieherzuleiten und dann aus U und S die Freie Energie zu berechnen, deren Ableitung nach dem Volumenden Druck ergibt. Die restlichen Zustandsgrößen sind dann einfach zugänglich.
iDer Zusammenhang zwischen U und S ist durch die GFF gegeben. Bei Konstanthaltung von V, n , . .gilt
(5.3.1)
Zur Berechnung von dU/T wird wie folgt vorgegangen
(5.3.2)
oder
(5.3.3)
Da alle Variablen außer T voraussetzungsgemäß konstant gehalten werden, darf der letzte Term wiefolgt umgeformt werden:
(5.3.4)
Daraus folgt
(5.3.5)
(5.3.6)
wobei die Integrationskonstante Null gesetzt wurde. Dieses ist eine willkürliche Setzung, da eineDifferenzialgleichung für die Entropie diese immer nur bis auf ein konstantes Glied ergeben kann!

- 62 -
Bei der Behandlung der Entropie in der Phänomenologischen Thermodynamik hatten wir die Entropiefür einen idealen Kristall bei 0 K Null gesetzt. Dieses muss aus Konsistenzgründen mit dem nach Gl.(5.3.6) berechneten Wert übereinstimmen. Dazu ist der Grenzwert von (5.3.6) für T 6 0 zu bilden. In
oz entfallen alle Terme mit höheren j außer g , da der Exponent bei höheren j nach -4 geht. Es verbleibt:
(5.3.7)
und
(5.3.8)
und daher
(5.3.9)
Gibt es keine Entartung des tiefsten Niveaus, so ergibt sich auch hier S = 0.Das oben vorgestellte Verfahren zur Berechnung der Entropie benutzte weitgehend die Phänomeno-logische Thermodynamik. Umgekehrt ist es möglich, weitgehend in der Statistischen Thermodynamikzu bleiben und erst zum Schluß die Phänomenologische Thermodynamik zu verwenden. Obwohl dieDurchführung der Rechnung etwas schwieriger ist, soll sie durchgeführt werden, da dabei zwei
iwichtige Formeln anfallen. Bei der Herleitung spielt das Verhältnis P der Zahl der Systeme in einemZustand i zur Gesamtzahl eine wichtige Rolle.
(5.3.10)
Durch Logarithmieren folgt:
(5.3.11)
Gl. (5.1.1) wird jetzt in der Form
(5.3.12)
geschrieben. Das Differenzial von U wird damit
(5.3.13)
Mit (5.3.11) folgt daraus
(5.3.14)
iDer letzte Term der ersten Summe verschwindet wegen GdP = 0. Weiterhin wird der letzte Term
iwegen der Unabhängigkeit von E von T als alleinige Funktion von V geschrieben.
(5.3.15)
Durch Vergleich mit der GFF finden wir:
(5.3.16)
wobei man die letzten beiden Gleichungen am einfachsten von hinten her ableitet. Durch Integrationfolgt schließlich:
(5.3.17)
wobei die Integrationskonstante Null gesetzt worden ist, da nach unserer Konvention bei einem nicht

- 63 -
1entarteten, tiefsten Niveau bei T = 0 mit P = 1 S = 0 entstehen muss. Der nächste Schritt ist dienochmalige Verwendung von (5.3.11)
(5.3.18)
(5.3.19)
Es entsteht die bereits bekannte Gleichung.Gl. (5.3.17) bildet die Ausgangsgleichung für die Herleitung der schon aus der PC I bekanntenGleichung
(5.3.20)
Zur Herleitung dieser Gleichung gehen wir etwas unorthodox von ihr aus (jeder Schritt ist auchrückwärts ausführbar).
(5.3.21)
iSchreibt man in (5.3.17) die P aus
(5.3.22)
und berücksichtigt, dass (5.3.21) für ein Supersystem und (5.3.22) für ein System gilt, so ist (5.3.20)nachgewiesen.Gl. (5.3.20) wird meistens anders gesehen. Im kanonischen Ensemble ist die Gesamtenergie E eineG
Konstante. Die unterschiedlichen quantenmechanischen Zustände dieses Supersystems sind nichtsanderes als die Entartung dieses Systems auf dem Energieniveau E . W kann daher auch als EntartungG
eines Systems mit gegebener Energie gesehen werden. Gl. (5.3.20) geht auf Boltzmann zurück; explizit wurde sie jedoch von ihm nicht angegeben. Sie wurdeihm auf seinen Grabstein auf dem Zentralfriedhof in Wien gemeißelt, wo er unweit von Beethoven,Brahms, Mozart, Schubert und J. Strauss 1906 beerdigt wurde.Nachdem jetzt die Entropie phänomenologisch und statistisch eingehend behandelt wurde, soll nocheinmal das Problem der Nullpunktsentropie kurz diskutiert werden. Alle Gleichungen, mit denen dieEntropie absolut berechnet werden kann, sind die Lösungen von Differenzialgleichungen (die GFF,
revdQ /T, (5.3.1) - (5.3.5), (5.3.16) - (5.3.17) und damit auch (5.3.20)). Die Lösungen dieser Diffe-renzialgleichungen enthalten eine frei wählbare Konstante - die Nullpunktsentropie. Die in der PC Ibeschriebenen Messungen und die Form von (5.3.20) legen es nahe, die Entropie eines idealenKristalls bei 0 K Null zu setzen, ein "Muss" ist dieses jedoch nicht.
5.4 Andere ZustandsgrößenAus der Inneren Energie und der Entropie ist direkt die Freie Energie zugänglich
(5.4.1)
(5.4.2)
Die Zustandssumme ist also ein Maß für F/kT.

- 64 -
Wegen
(5.4.3)
folgt mit (5.4.2)
(5.4.4)
Für die Größen H und G wird das Produkt pV benötigt.
(5.4.5)
Damit wird G:
(5.4.6)
und H
(5.4.7)
(5.4.8)
Als letzte Größe wollen wir das chemische Potenzial berechnen. Wir hatten bei der Berechnung vonZ aus z bisher angenommen, dass die Systeme aus einer Teilchensorte bestehen. Die Gleichungen zurBerechnung der Zustandsgrößen aus Z bleiben auch für Mischungen richtig; der Zusammenhangzwischen Z und z muss jedoch für Mischungen modifiziert werden.Das chemische Potenzial für eine reine Substanz stimmt mit der Freien Enthalpie für 1 mol überein.Für diesen Fall ist also das chemische Potenzial ohne Schwierigkeiten zu berechnen. Als nächsteswollen wir uns den Zusammenhang zwischen Z und z für eine Mischung idealer Gase, d. h. nichtwechselwirkender Teilchen, ansehen. Das Analogon zu Gl. (5.2.4) enthält dann im Exponenten beideTeilchensorten a und b
(5.4.9)
Für den Fall der Ununterscheidbarkeit der Teilchen innerhalb jeder Sorte ist noch durch die Fakultätder entsprechenden Teilchenzahl zu teilen:
(5.4.10)
Zur Berechnung des chemischen Potenzials dieser Mischung geht man am besten von der FreienEnthalpie aus, da in diesem Fall bei der Ableitung die Definitionsgrößen des thermodynamischenSystems V und T konstant gehalten werden. Zusätzlich weist die Freie Energie den einfachstenZusammenhang mit Z auf.

- 65 -
(5.4.11)
Im Gegensatz zur Phänomenologischen Thermodynamik ist es in der Statistischen Thermodynamiküblich, das chemische Potenzial auf ein Teilchen und nicht auf 1 mol zu beziehen. Beide Größenhängen über die Avogadrosche Konstante als Faktor zusammen.
(5.4.12)
(5.4.13)
Diese Gleichung wird bei der Berechnung von Gleichgewichtskonstanten für das MWG benutztwerden.

- 66 -
6 Translationszustandssumme für ideale GaseDie Berechnung der Translationszustandssumme gibt uns die Möglichkeit, die thermodynamischenDaten eines einatomigen, idealen Gases, d. h. z. B. für die Edelgase, zu berechnen. Aus der Quanten-mechanik sind die Energieniveaus eines Teilchens der Masse m in einem rechtwinkligen Kasten mitunendlich hohen Potenzialwänden bekannt.
(6.1)
x y zwobei a, b und c die Kastenlängen in x-, y- bzw. z-Richtung sind. Die Quantenzahlen n , n und ndürfen die Werte 1, 2, 3, . . . annehmen. Die Zustandssumme z ist daher
(6.2)
Die Summen dürfen durch Integrale ersetzt werden, falls die Änderungen der Summanden zwischen
xzwei n -Werten klein sind. Dazu untersuchen wir folgendes Beispiel: 1 mol Argon bei 273 K in einemKubus von 1 m Kantenlänge. Gl. (6.1) geht dann über in
(6.3)
Aus der Phänomenologischen Thermodynamik wissen wir, dass die mittlere Teilchenenergie bei 3/2kT liegt, d. h. das mittlere n beträgt nach (6.3)2
(6.4)
(6.5)
Das mittlere n beträgt daher 4,8@10 . Das Verhältnis Q zweier aufeinander folgender Summanden10
wächst mit n und beträgt beim mittleren n
(6.6)
Wir dürfen daher statt der Summation eine Integration durchführen. Als untere Integralgrenze darf 0
xbenutzt werden. Der Integrand ist dort 1; die nächsten n ergeben jedoch auch eine 1, so dass diezusätzliche 1 einen vernachlässigbaren Fehler bewirkt (implizit ist dieses auch bereits in der Abschät-zung mit Gl. (6.6) enthalten).
(6.7)
Das erste Integral wird durch die Substitution

- 67 -
(6.8)
gelöst
(6.9)
Insgesamt entsteht daher
(6.10)
Die System-Translationszustandssumme für 1 mol ununterscheidbarer Teilchen beträgt daher
(6.11)
Für die Berechnung der Zustandsgrößen benötigen wir ln Z
(6.12)
wobei die Stirlingsche Näherungsformel verwendet wurde. Eine Umrechnung ergibt
(6.13)
Für die Ableitung nach T darf dieses umgeschrieben werden in
(6.14)
(6.15)
(6.16)
(6.17)
Dieses Ergebnis stimmt nach PC I, Kap. 9.1.1 mit den experimentellen Beobachtungen überein. Daherdürfen wir jetzt mit Einschränkungen feststellen, dass die Annahme $ = 1/kT korrekt war. Auf etwassicheren Beinen als Nachweis für diese Annahme steht folgende Argumentation. Das Produkt pV
Awurde bereits in Gl. (5.4.5) als Funktion von Z berechnet. Mit Z gemäß Gl. (6.13) und N = N erhältman
(6.18)
und schließlich

- 68 -
(6.19)
d. h. die ideale Gasgleichung. Wir rechtfertigen daher die Annahme $ = 1/kT durch Reproduktion derDefinitionsgleichung für die gasthermometrische Temperatur.
Als nächstes wollen wir die Entropie berechnen.
(6.20)
(6.21)
Das ist die Gleichung von Sackur und Tetrode. Sie gestattet die Berechnung der Entropie vonEdelgasen mit hoher Genauigkeit. Dazu soll folgendes Beispiel gerechnet werden: 1 mol Argon bei 1atm und 25 C. Argon ist weitgehend ein Reinelement (99,6 % Ar), so dass keine Probleme durch die� 40
unterschiedlichen Isotope zu befürchten sind. Für m wird die mittlere Atommasse verwandt.
Dieses ist bis zur letzten Stelle der Wert, den wir im Tabellenanhang von PC I für Argon finden. Dasist kein Zufall; mit großer Wahrscheinlichkeit wurde der Tabellenwert für Argon auch gerechnet.Offensichtlich ist es aber möglich, mit Hilfe der Sackur-Tetrode-Gleichung die Entropie eines idealen,einatomigen Gases mit der Genauigkeit der eingehenden Naturkonstanten zu berechnen.Um die Naturkonstanten nicht immer einsetzen zu müssen, soll die Sackur-Tetrode-Gleichungentsprechend umgeformt werden. Dazu wird in Gl. (6.21) V mit Hilfe der idealen Gasgleichungentfernt
(6.22)
(6.23)
M ist die molare Masse in g/molT ist die absolute Temperatur in Kp ist der Druck in atm
Das Argonbeispiel ergibt
Als letzter Punkt der Entropiediskussion soll die Abhängigkeit der Entropie von der Stoffmengeuntersucht werden. Wird Gl. (6.20), 1. Zeile nicht für 1 mol in V sondern für n mol in nV hergeleitet,
Aso ist das Glied 3R/2 mit n zu multiplizieren und der Vorfaktor vor dem Logarithmus kN ist auch mitn zu multiplizieren; innerhalb des Logarithmus hebt sich die Stoffmengenabhängigkeit heraus. Die

- 69 -
Entropie ist daher korrekt proportional zu Stoffmenge.
AWäre dagegen die Division durch N! in Gl. (5.2.5) nicht eingeführt worden, so würde der Nenner Nim Logarithmus fehlen. Dieses würde dazu führen, dass bei einer Systemverdopplung, d. h. 2n in 2V,ein zusätzlicher Faktor 2 im Logarithmus entstünde. Genau diese Schwierigkeit hat zu der Argumenta-tion mit der Ununterscheidbarkeit der Teilchen geführt.Mischt man dagegen zwei unterschiedliche ideale Gase 1 und 2, so gilt nach Gl. (5.4.10) für dieMischung
(6.24)
1,m 1wobei sich z auf die Mischung bezieht; z dagegen auf das ungemischte System. Mit (5.3.5) und
1 2U = U + U folgt daraus:
(6.25)
Die Mischungsentropie ist der nichtadditive Anteil der Entropie
(6.26)
Für 1 mol der Mischung finden wir mit
(6.27)
das bereits aus der Phänomenologischen Thermodynamik bekannte Ergebnis (I 12.2.4). Der Übergangauf eine beliebig große Anzahl von Mischungskomponenten ist hier leicht zu überblicken.Schließlich wollen wir noch die im Kap. 5.2 benutzte Annahme untersuchen, dass bei einem Gas unterden üblichen Bedingungen immer sehr viel mehr "erreichbare" Niveaus vorhanden sind als Teilchen.Wir schreiben Gl. (6.3) für die Translationsniveaus in einem Kubus etwas um:
(6.28)
x y z x y zn , n und n werden jetzt in einem Oktanten eines n , n , n -Koordinatensystems als Punkte mit
x y zganzzahligen n , n und n aufgetragen (siehe auch die entsprechende Abb. in Kap. 10.2). Alle Punkte
x y zin diesem Oktanten mit Ausnahme der Seitenflächen wegen n , n , n � 0 stellen daher Energieniveausdar. Die Punkte bilden ein Gitter. Jeder Gitterwürfel weist ein Volumen von 1 auf. Für eine vor-
maxgegebene Energiegrenze g ergibt daher das Volumen des Kugeloktanten mit dem Radius R
(6.29)
max gdie Zahl aller Energieniveaus mit g # g . Die Zahl N dieser Niveaus ist daher
(6.30)
maxSetzt man jetzt g . 3kT/2, so folgt

- 70 -
(6.31)
Für 1 mol Argon bei 273 K in 1 m finden wir3
Diese Zahl ist groß im Vergleich zur Avogadroschen Konstante. Selbst bei höheren Drücken undtiefen Temperaturen ist dieses immer noch der Fall. Bei tiefen Temperaturen stellt dann auch derDampfdruck eine Obergrenze für den Druck dar. Schließlich gelten die hergeleiteten Gleichungensowieso nur für ideale Gase.

- 71 -
7 Rotationszustandssumme für ideale Gase
7.1 AllgemeinesWir betrachten die mit der Quantenmechanik berechenbaren Energieniveaus eines vielatomigenMoleküls in einer idealen Gasphase, d. h. ohne zwischenmolekulare Wechselwirkungen. An und fürsich hätten wir die Schrödinger-Gleichung für dieses Molekül zu formulieren und zu lösen. Auchdieses Problem ist - obwohl einfacher als ein System mit vielen Atomen bzw. Molekülen - sehrkomplex. Die Quantenmechanik und experimentelle Ergebnisse zeigen, dass das Molekül eine Reihevon Energieniveaus aufweist, die unterschiedlichen Bewegungsformen des Gesamtmoleküls, derKerne und der Elektronen zugeordnet werden können. Die Translationsniveaus liegen so dicht, dass
transdie Translationsübergänge )g unter den üblichen experimentellen Bedingungen nicht aufgelöst
rotwerden können. Die Rotationsübergänge )g lassen sich mit elektromagnetischer Strahlung im
vibMikrowellenbereich (8 . 0,2 mm) anregen, Schwingungsübergänge )g mit 8 . 5 :m und dieelektronische Anregung liegt bei etwa 0,1 - 500 nm. Schließlich gibt es noch die mit der Quanten-mechanik nicht beschreibbaren Kernübergänge bei 8 . 0,1 pm.Die Struktur der Schrödinger Gleichung erlaubt nun i. a. eine Separation dieser Bewegungsformen. Z.B. wird der Translationsanteil separiert, indem die Schrödinger-Gleichung in Schwerpunktskoordina-ten geschrieben wird und die Bewegung des Molekülschwerpunkts als Bewegung eines Massepunktsin einem Potenzialkasten betrachtet wird. Diese Separation ist immer möglich, wenn der Potenzialkas-ten wesentlich größer als die Moleküldimension ist. Dieses führt zu der bereits beschriebenenBehandlung der Translationszustandssumme. Im nächsten Schritt schreibt man die Schrödingerglei-chung in einem mit dem Molekül rotierenden Koordinatensystem auf. Die Molekülrotation führt zuden Rotationsniveaus und wird quantenmechanisch als Rotation eines starren Körpers behandelt.Diese Separation kann zu Fehlern führen, wenn bei "weichen" Molekülen die Rotation zu einerVergrößerung des Trägheitsmoments führt (Zentrifugaldistorsion). Der letzte Schritt ist die Separationder Kern- und Elektronenbewegungen (Born-Oppenheimer-Näherung). Es wird angenommen, dasssich die Elektronen aufgrund ihrer geringeren Masse erheblich schneller bewegen als die Kerne. Manlöst daher die Schrödinger-Gleichung für fixierte Kerne. Bei den Molekülschwingungen treten die soberechneten Energien als Potenziale auf, welche die rücktreibenden Kräfte bei einer Molekül-schwingung bewirken. Diese Separation kann bei Molekülen mit sehr niedrig liegenden elektronischenAnregungsniveaus Schwierigkeiten machen. Hoch angeregte Molekülschwingungen können auchVeränderungen der Molekülgeometrie ergeben, wodurch die Separation der Rotationsanteile dannverhindert wird.In der Sprache der Quantenmechanik bedeutet die Separation eine Auftrennung des Hamiltonoperatorsentsprechend
(7.1.1)
und damit
(7.1.2)
Diese Additivität der Energieniveaus führt zu den charakteristischen Gasphasenspektren der Moleküle,in denen auf jeder elektronischen Anregungsbande - falls nicht durch ein Verbot unterdrückt -zusätzlich die Schwingungsniveaus aufgesetzt sind ("Schwingungsprogression"), auf die noch - beigenügend kleinen Molekülen beobachtbar - die Rotationsniveaus aufgesetzt sind.Die Additivität der Energieniveaus nach Gl. (7.1.2) führt zu einer drastischen Vereinfachung derZustandssumme komplexer Moleküle
(7.1.3)

- 72 -
(7.1.4)
d. h. wir müssen nicht die sehr komplexe Zustandssumme über die Gesamtenergie des Molekülsberechnen, sondern dürfen diese Berechnung in separierter Form durchführen.Für die Systemzustandssumme gilt:
(7.1.5)
Der N!-Term wird üblicherweise zur Translationszustandssumme geschlagen und ist auch dort bereitsim vorherigen Kapitel verrechnet worden.
7.2 Lineare MoleküleFür linear aufgebaute Moleküle sind die Rotationsniveaus quantenmechanisch relativ einfach zuberechnen.
(7.2.1)
I ist das Trägheitsmoment für die Rotation um die Normale zur Moleküllängsachse und J die Rota-tionsquantenzahl mit den erlaubten Werten 0, 1, 2, . . . Jeder Rotationszustand ist (2J + 1)-fachentartet, da es zu jedem J 2J+1 (m = -J, -(J-1), . . . . ,(J-1), J) linear unabhängige Wellenfunktionengibt, die man als unterschiedliche Richtungen der Rotationsachse im Raum ansehen kann. Daher giltfür die Rotationszustandssumme:
(7.2.2)
Zur Abkürzung führen wir die charakteristische Temperatur 1 ein:
(7.2.3)
(7.2.4)
J läuft über die ganzen Zahlen von 0 bis 4, falls dem nicht Verbote entgegenstehen.Um die Berechnungen nicht unnötig zu komplizieren, wollen wir uns zuerst auf zweiatomigeMoleküle beschränken. 7.2.1 Heteronukleare zweiatomige MoleküleFür heteronukleare zweiatomige Moleküle AB gibt es keine Einschränkung für die Rotations-quantenzahlen. Da die Summe in (7.2.4) nicht geschlossen angegeben werden kann, müssen wir alsnächstes die Frage diskutieren, ob wie bei der Translation die Summe durch ein Integral ersetzt werdendarf. Dieses hängt vom Verhältnis der Temperatur zur charakteristischen Temperatur ab, die für einige
rotMoleküle in der untenstehenden Tabelle aufgeführt ist. Wir benötigen z bei einer bestimmtenTemperatur (nicht etwa ein Integral von 0 bis 4) und müssen daher wie bei der Translation feststellen,wie groß die Änderung der einzelnen Summanden bei dieser Temperatur ist. Wir untersuchenfolgenden Fall: 1 = 2,73 K und T = 273 K. Die Summanden sind:

- 73 -
J f(J)
0 1
1 2,94
2 4,71
3 6,21
4 7,37
5 8,15
6 8,54
7 8,56
8 8,27
9 7,72
10 6,99
Molekül 1/K
HD 65,6
OH 27
HF 30
HBr 12,1
NO 2,4
CO 2,8
Die nebenstehende Tabelle zeigt die Summanden in Abhängigkeit von J. Die Verhältnisse liegen hierlängst nicht mehr so günstig wie bei der Translation. Die großen Summanden bleiben etwa konstant.Bei den Summanden für kleine J ist die Änderung zwar groß, sie tragen aber wenig zur Summe bei.Weiterhin hebt sich durch die unterschiedlichen Krümmungen ein Teil der Fehler heraus. Wir ersetzendaher bei genügend hohen T/1 die Summe durch das Integral
(7.2.1.1)
Das Integral wird mit der Substitution
(7.2.1.2)
gelöst.
(7.2.1.3)
Für 1 mol finden wir für die Innere Energie
(7.2.1.4)
(7.2.1.5)
und für die Molwärme
(7.2.1.6)
Bei niedrigeren T/1-Werten kann eine Reihenentwicklung für die Zustandssumme anstelle von Gl.
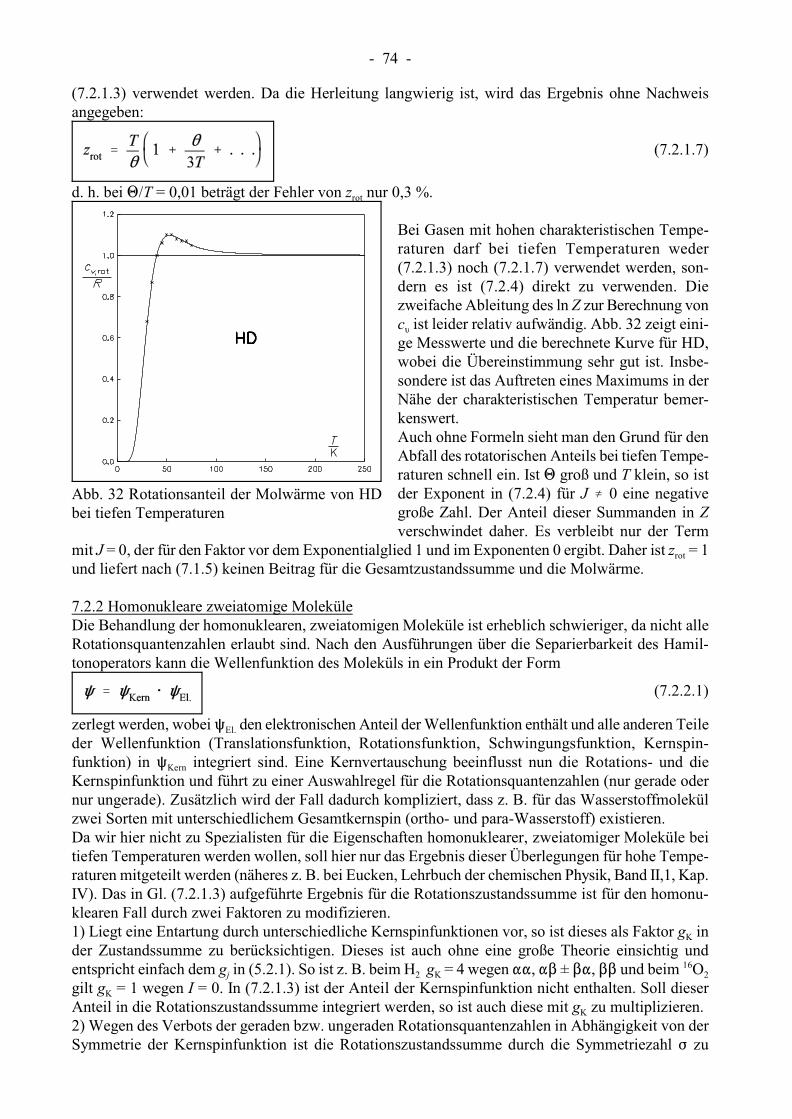
- 74 -
Abb. 32 Rotationsanteil der Molwärme von HDbei tiefen Temperaturen
(7.2.1.3) verwendet werden. Da die Herleitung langwierig ist, wird das Ergebnis ohne Nachweisangegeben:
(7.2.1.7)
rotd. h. bei 1/T = 0,01 beträgt der Fehler von z nur 0,3 %.
Bei Gasen mit hohen charakteristischen Tempe-raturen darf bei tiefen Temperaturen weder(7.2.1.3) noch (7.2.1.7) verwendet werden, son-dern es ist (7.2.4) direkt zu verwenden. Diezweifache Ableitung des ln Z zur Berechnung von
Lc ist leider relativ aufwändig. Abb. 32 zeigt eini-ge Messwerte und die berechnete Kurve für HD,wobei die Übereinstimmung sehr gut ist. Insbe-sondere ist das Auftreten eines Maximums in derNähe der charakteristischen Temperatur bemer-kenswert.Auch ohne Formeln sieht man den Grund für denAbfall des rotatorischen Anteils bei tiefen Tempe-raturen schnell ein. Ist 1 groß und T klein, so istder Exponent in (7.2.4) für J � 0 eine negativegroße Zahl. Der Anteil dieser Summanden in Zverschwindet daher. Es verbleibt nur der Term
rotmit J = 0, der für den Faktor vor dem Exponentialglied 1 und im Exponenten 0 ergibt. Daher ist z = 1und liefert nach (7.1.5) keinen Beitrag für die Gesamtzustandssumme und die Molwärme.
7.2.2 Homonukleare zweiatomige MoleküleDie Behandlung der homonuklearen, zweiatomigen Moleküle ist erheblich schwieriger, da nicht alleRotationsquantenzahlen erlaubt sind. Nach den Ausführungen über die Separierbarkeit des Hamil-tonoperators kann die Wellenfunktion des Moleküls in ein Produkt der Form
(7.2.2.1)
El.zerlegt werden, wobei R den elektronischen Anteil der Wellenfunktion enthält und alle anderen Teileder Wellenfunktion (Translationsfunktion, Rotationsfunktion, Schwingungsfunktion, Kernspin-
Kernfunktion) in R integriert sind. Eine Kernvertauschung beeinflusst nun die Rotations- und dieKernspinfunktion und führt zu einer Auswahlregel für die Rotationsquantenzahlen (nur gerade odernur ungerade). Zusätzlich wird der Fall dadurch kompliziert, dass z. B. für das Wasserstoffmolekülzwei Sorten mit unterschiedlichem Gesamtkernspin (ortho- und para-Wasserstoff) existieren.Da wir hier nicht zu Spezialisten für die Eigenschaften homonuklearer, zweiatomiger Moleküle beitiefen Temperaturen werden wollen, soll hier nur das Ergebnis dieser Überlegungen für hohe Tempe-raturen mitgeteilt werden (näheres z. B. bei Eucken, Lehrbuch der chemischen Physik, Band II,1, Kap.IV). Das in Gl. (7.2.1.3) aufgeführte Ergebnis für die Rotationszustandssumme ist für den homonu-klearen Fall durch zwei Faktoren zu modifizieren.
K1) Liegt eine Entartung durch unterschiedliche Kernspinfunktionen vor, so ist dieses als Faktor g inder Zustandssumme zu berücksichtigen. Dieses ist auch ohne eine große Theorie einsichtig und
j 2 K 2entspricht einfach dem g in (5.2.1). So ist z. B. beim H g = 4 wegen "", "$ ± $", $$ und beim O16
Kgilt g = 1 wegen I = 0. In (7.2.1.3) ist der Anteil der Kernspinfunktion nicht enthalten. Soll dieser
KAnteil in die Rotationszustandssumme integriert werden, so ist auch diese mit g zu multiplizieren.2) Wegen des Verbots der geraden bzw. ungeraden Rotationsquantenzahlen in Abhängigkeit von derSymmetrie der Kernspinfunktion ist die Rotationszustandssumme durch die Symmetriezahl F zu

- 75 -
teilen.Für hohe Temperaturen gilt daher
(7.2.2.2)
F gibt die Anzahl der Kernvertauschungsmöglichkeiten durch Molekülrotationen an. Da nun beiheteronuklearen Systemen F = 1 und bei homonuklearen F = 2 gilt, darf (7.2.2.2) anstelle von (7.2.1.3)für beide Fälle benutzt werden.
LBei der Berechnung der thermodynamischen Größen u und c gehen nur die Ableitungen des Logarith-
Kmus von Z ein. g und F beeinflussen daher diese beiden Größen nicht. Die Entropie wird jedoch
Kwegen des Gliedes ln Z geändert. Für 1 mol gasförmiger Rotatoren ist R ln g /F zu addieren. Im
KKristall bei 0 K bleibt nun die Entartung durch g erhalten. Üblicherweise wird jedoch die Entropieder kristallinen Substanzen bei 0 K Null gesetzt, d. h. die experimentellen Werte enthalten das Glied
KR ln g nicht. Wir werden daher auch in der Theorie dieses Glied fallenlassen. Nur bei Kernumwand-lungen werden dadurch Probleme entstehen.
LDie allgemeinen Gleichungen für den rotatorischen Anteil für u, c und s eines zweiatomigen Mole-küls sind daher bei genügend hohen Temperaturen
(7.2.2.3)
(7.2.2.4)
(7.2.2.5)
(7.2.2.6)
Diese Gleichungen lassen sich auch für den allgemeinen Fall der linearen Moleküle aus folgendenGründen verwenden. Ist das Molekül unsymmetrisch aufgebaut (HCN), so gibt es keine Auswahlregelfür J. Daher gelten (7.2.2.3) - (7.2.2.6) mit F = 1. Im Falle symmetrisch aufgebauter linearer Moleküle(HCCH) gelten die gleichen Auswahlregeln für die Rotationsquantenzahlen wie für die zweiatomigenMoleküle. Daher gelten auch hier (7.2.2.3) - (7.2.2.6) mit F = 2.Als Beispiel soll die Entropie von Stickstoff bei 25 C und 1 atm berechnet werden. Stickstoff eignet�
sich für eine derartige Berechnung besonders gut, da wegen der hochfrequenten Streckschwingung des
2N der vibratorische Anteil zur Entropie besonders klein ist. Stickstoff ist weitgehend ein Reinelement(99,63 % N), so dass weder beim translatorischen noch beim rotatorischen Anteil bei Verwendung14
mittlerer Massen Schwierigkeiten zu befürchten sind. Der translatorische Anteil ist nach (6.23)
und der rotatorischen Anteil wird mit

- 76 -
Molekül F
HCl 1
2N 2
2H O 2
3NH 3
6 6C H 12
4CH 12
Die Entropie des Stickstoffs sollte daher insgesamt s = 191,44 J K mol betragen. Aus dem Tabellen--1 -1
anhang zur PC I entnimmt man s = 191,50 J K mol , d. h. auch in diesem Fall ist die Überein--1 -1
stimmung sehr gut. 7.3 Nichtlineare MoleküleHier sollen die Zustandssummen nichtlinearer Moleküle mit drei unterschiedlichen Trägheits-momenten untersucht werden. Leider ist die Schrödinger-Gleichung für die Rotation entsprechenderstarrer Körper nicht lösbar. Dagegen ist die Rotationsenergie eines starren Körpers mit drei ver-schiedenen Trägheitsmomenten im Rahmen der klassischen Mechanik einfach darstellbar:
(7.3.1)
A B C A B Cwobei I , I und I die drei Hauptträgheitsmomente des Körpers und T , T und T die Winkelge-schwindigkeiten um die Achsen der Hauptträgheitsrichtungen sind.Auch für die Zustandssumme gibt es ein Analogon in der klassischen Mechanik, das Zustandsintegralder klassischen Mechanik. Da eine Diskussion dieses Zustandsintegrals die Kenntnis der Hamilton-
Kfunktion voraussetzt, soll hier nur das Endergebnis für g = 1 angegeben werden.
(7.3.2)
Vergleicht man diesen Ausdruck mit dem für zwei gleiche Träg-heitsmomente gültigen Ausdruck (7.2.2.2) unter Verwendungvon (7.2.3), so stellt (7.3.2) nur eine Erweiterung auf drei unter-schiedliche Trägheitsmomente mit einem neu dazugekommenenFaktor B dar. F hat die bereits bekannte Bedeutung: Es stellt½
die Zahl der durch Molekülrotationen möglichen Vertauschun-
3gen dar (siehe nebenstehende Tabelle). Beim NH sind es die
4Rotationen um die 3-zählige Achse. Beim CH ist die Berech-nung schwieriger. Die Zahl aller möglichen Vertauschungen derH wäre 4!. Dabei werden dann aber auch Vertauschungen ge-zählt, die sich nicht durch Rotationen allein erreichen lassen, dadas Molekül spiegelverkehrt entsteht. Es sind daher alle Vertau-schungen zu streichen, die durch eine Spiegelung entstehen. Daskorrekte Ergebnis ist daher 4!/2. Beim Benzol entsteht die 12durch die 6- und eine der 2-zähligen Achsen.Der Übergang von der quantenmechanischen Zustandssummezum klassischen Zustandsintegral ist nur dann erlaubt, wenn T/1genügend groß ist, wobei 1 das für den nichtlinearen Fall gültige Analogon zu (7.2.3) darstellt.
(7.3.3)
Die obige Bedingung T o 1 ist üblicherweise erfüllt. Die Moleküle müssen aus mindestens dreiAtomen bestehen, die nicht nur Wasserstoffatome sein können. Die hoch liegenden Siedepunktewerden eine Messung bei sehr tiefen Temperaturen verhindern. Wegen der großen Trägheitsmomentewird 1 klein sein. Gl. (7.3.2) darf daher bei allen, real existierenden Gasen angewendet werden.Der rotatorische Anteil für die Innere Energie beträgt daher

- 77 -
2Abb. 33 Geometrie des SO -Mo-leküls
(7.3.4)
(7.3.5)
(7.3.6)
Dieses stimmt mit dem bereits aus der PC I bekannten Rezept überein: Jeder molekulare Freiheitsgrad
Lergibt für c einen Anteil von R/2.Der rotatorische Anteil zur Entropie ergibt sich nach (5.3.5) zu
(7.3.7)
(7.3.8)
2Als Beispiel soll die Entropie von SO unter Standardbedingungen (1 atm, 25 C) berechnet werden.�
2Bezüglich der Isotopenreinheit liegen die Verhältnisse beim SO ungünstiger als bei den vorstehendenVerbindungen (95,00 % S, 4,22 % S und 0,78 % S). Da erfahrungsgemäß die Verwendung32 34 33
mittlerer Massen zu guten Ergebnissen führt und sonst die Rechnung sehr aufwändig werden würde,soll hier mit mittleren Massen gerechnet werden. Der Translationsbeitrag ist nach (6.23)
Für die Berechnung des Rotationsbeitrages werden die Haupt-trägheitsmomente benötigt. In Abb. 33 sind die Geometrie des
2SO -Moleküls und die Hauptträgheitsachsen angegeben. Die Lageder Hauptträgheitsachse B fällt mit der 2-zähligen Achse desMoleküls zusammen. Die beiden anderen Achsen müssen in denSpiegelebenen des Moleküls liegen und durch den Schwerpunkt
2gehen. Da die molaren Massen von S und O fast übereinstim-men, liegt der Schwerpunkt fast genau auf der Mitte der Höhe derO-O-Verbindungslinie nach S. Die genaue Berechnung der Träg-
i iheitsmomente Gm r ergibt mit der mittleren Molmasse:2
A B CI = 1,386@10 ; I = 8,143@10 ; I = 9,529@10 kg m-46 -46 -46 2
Damit wird der Rotationsbeitrag:

- 78 -
Die Gesamtentropie sollte daher
2betragen. Im Tabellenanhang zur PC I findet man jedoch 248,10 J K mol für SO . Diese Differenz-1 -1
ist weitgehend auf den noch nicht berücksichtigten Anteil der Molekülschwingungen zurückzuführen(siehe nächstes Kapitel).

- 79 -
8 Schwingungszustandssumme für ideale Gase
8.1 Zweiatomige MoleküleIn einem zweiatomigen Molekül gibt es nur eine mögliche Schwingung. Die quantenmechanischeBehandlung der harmonischen Schwingung führt zu folgenden Energieniveaus:
(8.1.1)
n ist die Schwingungsquantenzahl, < ist die klassische Schwingungsfrequenz (und gleichzeitig dieFrequenz des absorbierten Lichts bei einem Übergang mit )n = 1). Bei Schwingungen mit kleinenSchwingungsquantenzahlen kann i. a. davon ausgegangen werden, dass die Schwingung harmonischist, d. h. dass die rücktreibende Kraft proportional zur Auslenkung aus der Gleichgewichtslage ist. Beihöher angeregten Schwingungen ist dieses i. a. nicht mehr der Fall. Anstelle von (8.1.1) müssen dannAusdrücke mit höheren Potenzen von n verwendet werden. Der ' dient nur dazu, die so eingeführteng'-Werte von später eingeführten g-Werten mit einem verschobenen Energieskalennullpunkt zuunterscheiden. Die Zustandssumme ist daher
(8.1.2)
Unser bisher angewendetes Verfahren, die Summe durch ein Integral zu ersetzen, ist hier nicht mehrerlaubt. Für eine Schwingung mit einer Absorptionswellenlänge von 8 = 5 :m wird
Zwei aufeinander folgende Glieder der Summe verhalten sich wie 1 : e . Die Verwendung der-9,6
Integration ist in diesem Fall auch unnötig, da die Reihensumme dieser unendlichen geometrischenReihe mit dem Quotienten q = exp(-h</kT) leicht berechnet werden kann.
(8.1.3)
An dieser Stelle ist eine Diskussion der Energieskala, auf der g gemessen wird, notwendig. WelcheAussage macht eigentlich Gl. (6.16)? Damit ist offensichtlich nicht die gesamte Innere Energie deseinatomigen, idealen Gases gemeint, sondern - wie der Index zeigt - nur der translatorische Anteil. Dieentsprechende Entropiegleichung (6.21) ist aber offensichtlich vollständig, wie durch den Vergleichmit dem Experiment festgestellt worden war und wie die Diskussion zu Gl. (8.1.18) zeigt.Die Energieniveaus g sind auf einer für alle Atome bzw. Moleküle gemeinsamen Energieskala zumessen, über deren Nullpunkt jetzt befunden werden soll. Eine Wahl wäre: Dem Vakuum wird dieEnergie g = 0 zugeordnet. 1) Es werden die Elementarteilchen erzeugt. 2) Die Atomkerne werdengebildet. 3) Die isolierten Atome bzw. Moleküle bei 0 K werden gebildet. 4) Den Atomen bzw.Molekülen werden die translatorischen Energien usw. zugefügt. Dieses Verfahren hat den schwer-wiegenden Nachteil, dass der Schritt 2) heute mit keiner Theorie mit ausreichender Genauigkeitzugänglich ist.Erheblich günstiger ist es, den Energieskalennullpunkt g = 0 wie folgt zu definieren: Die Atomkerneund die Elektronen befinden sich isoliert bei 0 K. Daraus werden isolierte Atome bzw. Moleküle bei0 K gebildet, welche die Nullpunktsenergie für die Schwingung oder die Schwingungen aufweisen.
oDie bei diesem Schritt auftretenden Energieänderungen g sind prinzipiell mit quantenmechanischenMethoden berechenbar. Es ist nun zu untersuchen, welche Auswirkung diese Definition des Skalen-nullpunkts auf die Gleichungen der Statistischen Thermodynamik hat. Anstelle von (7.1.2) gilt daher
(8.1.4)

- 80 -
Weiter verfahren wird entsprechend (7.1.3ff)
(8.1.5)
(8.1.6)
trans x y z rotBei z ist der Skalennullpunkt das nicht erlaubte Niveau mit n = n = n = 0, bei z ist es das mit
vibJ = 0. Bei z ist die Lage anders, da wir die Schwingungsnullpunktsenergie - verabredungsgemäß und
onur deswegen - bereits in g berücksichtigt haben. Wir definieren daher ohne den Strich
(8.1.7)
(8.1.8)
wobei das heute übliche Symbol für die neu eingeführte Größe
(8.1.9)
leider schlecht gewählt wurde.Die Innere Energie wird daher ohne den elektronischen Term
(8.1.10)
oDen ersten Term werden wir zukünftig mit u bezeichnen. Er stellt die quantenmechanisch bere-chenbare Energie eines Mols isolierter Moleküle mit der Schwingungsnullpunktsenergie bei 0 Kbezogen auf die isolierten Atomkerne und Elektronen bei 0 K dar. Wenn man beachtet, dass x von Tabhängt, ergibt der letzte Term :
(8.1.11)
(8.1.12)
Für die Innere Energie finden wir daher
(8.1.13)

- 81 -
(8.1.14)
An dieser Gleichung kann man sehr schön das "Auftauen" eines Schwingungsfreiheitsgrades sehen.Ist x >> 1, d. h. h< >> kT, so wird der Exponentialterm sehr groß und der Beitrag der Schwingung zurInneren Energie wird sehr klein. Ist umgekehrt x << 1, d. h. h< << kT, so darf der Exponentialterm ineine Reihe entwickelt werden, die nach dem ersten Glied abgebrochen wird.
(8.1.15)
d. h. der Schwingungsfreiheitsgrad ergibt den vollen Beitrag von RT zur Inneren Energie.Als nächstes soll die Molwärme berechnet werden.
(8.1.16)
(8.1.17)
LFür x 6 0 findet man wie erwartet c = 7/[email protected] der Entropie spielt die Verschiebung des Skalennullpunkts der g keine Rolle, da in
(8.1.18)
o A o oim 1. Term ein u /T dazukommt, im hinteren Term jedoch wegen kN g /kT = u /T wieder abgezogenwird. Wir müssen also nur den durch die Schwingung hervorgerufenen Anteil zu den bereits bekann-ten Gleichungen addieren.
(8.1.19)
(8.1.20)
Beispiele sollen zu den zweiatomigen Molekülen nicht gerechnet werden. Entweder ist der Beitrag
2 2 2 2sehr klein, wie bei H , N oder CO; bei anderen Molekülen wie I oder Cl machen sich Nicht-idealitäten störend bemerkbar.
8.2 Mehratomige MoleküleIn der Quantenmechanik wird die Schwingung eines Moleküls in Normalschwingungen aufgespalten,d. h. der Hamiltonoperator wird in Operatoren für die einzelnen Normalschwingungen zerlegt.Dementsprechend ist die Schwingungsenergie die Summe der Normalschwingungsenergien.Auch die klassische Mechanik kennt den Begriff der Normalschwingung. Jede Schwingung einesMoleküls muss als Überlagerung der Normalschwingungen eines Moleküls darstellbar sein. Keine
NNormalschwingung darf als Überlagerung anderer Normalschwingungen darstellbar sein. Die Zahl nder Normalschwingungen ist mit einem einfachen Verfahren bestimmbar. Die Summe der trans-latorischen und rotatorischen Freiheitsgrade muss zusammen mit der Zahl der Normalschwingungen
A Agerade die Zahl der Freiheitsgrade der das Molekül aufbauenden Atome ergeben, d. h. 3 n , wobei ndie Zahl der Atome im Molekül ist. Daher gilt für lineare Moleküle
(8.2.1)

- 82 -
2Abb. 34 Normalschwingungen des SO -Moleküls
und für nichtlineare Moleküle
(8.2.2)
NFür ein zweiatomiges Molekül gilt daher n = 1, d. h. es gibt eine Normalschwingung - die Streck-schwingung - des Moleküls. Bei nichtlinearen dreiatomigen Molekülen gibt es drei Normalschwingun-gen: eine symmetrische und eine antisymmetrische Streckschwingung und eine Biegeschwingung
2(siehe Berechnungen am SO weiter unten). Bei linearen dreiatomigen Molekülen muss es vierNormalschwingungen geben. Auf den ersten Blick sieht man die drei Normalschwingungen desvorigen Beispiels. Zu berücksichtigen ist jedoch, dass die Biegeschwingung aus folgendem Grunddoppelt zu zählen ist. Legt man das Molekül in die x-Achse eines Koordinatensystems und bezeichnetneben den Streckschwingungen diejenige Biegeschwingung, bei der das Molekül in y-Richtungdeformiert wird, als Normalschwingung, so lässt sich die entsprechende Biegeschwingung in z-Richtung mit den angeführten Normalschwingungen nicht darstellen. Es gibt daher zwei entarteteBiegeschwingungen mit senkrecht aufeinander stehenden Schwingungsrichtungen und natürlichgleicher Schwingungsfrequenz. Die Berechnung der statistischen und thermodynamischen Größenerfolgt nach der bereits mehrfach durchgeführten Prozedur.
(8.2.3)
(8.2.4)
(8.2.5)
(8.2.6)
Zwei Beispiele sollen vorgerechnet werden.
21. SO
2Beim SO -Molekül gibt es folgende Normalschwingungen:
Die folgende Tabelle enthält die Berechnung des Schwingungsanteils. Die Frequenz < und die Wel-lenzahl < = 1/8 hängen über c< = < zusammen. Die Summe der Schwingungsentropien beträgt daher
- -
S = 2,866 J/K mol. Für die gesamte Entropie finden wir daher

- 83 -
vib</cm h</kT s /JK mol-1 -1 -1-
1151,4 5,5626 0,210
517,7 2,5011 2,5682
1361,8 6,5791 0,088
Das Experiment ergibt 248,11 J/K mol. DieÜbereinstimmung ist hervorragend, obwohl
2SO sicher nicht vollständig ideales Verhaltenzeigt und Fehler bei der Vernachlässigung derIsotopenzusammensetzung begangen wordensind.Zum letzten Punkt ist folgendes zu bemerken.Die unterschiedlichen Isotope ergeben unter-schiedliche Beiträge zur Entropie. I. a. stellt jedoch die Verwendung von mittleren molaren Massenbeim Translations- und Rotationsbeitrag und die Verwendung mittlerer Frequenzen beimSchwingungsbeitrag eine gute Näherung dar.
22. CO
2Als zweites Beispiel soll die Entropie und die Molwärme von CO als linearem, dreitatomigenMolekül bei 25 C und 1 atm berechnet werden. Hier soll auch das Vorliegen unterschiedlicher Isotope�
korrekt berücksichtigt werden. Schließlich tritt bei der Berechnung des Schwingungsbeitrages eineBesonderheit auf.
2Natürliches CO ist eine komplexe Mischung von Molekülisotopen. Beim Sauerstoff überwiegt jedochdas Isotop O so stark (99,7 %), dass die durch die anderen Isotope bewirkten Abweichungen16
vernachlässigbar sind. Beim Kohlenstoff beträgt der Gehalt an C 1,11 %. Dieses soll bei der13
Berechnung berücksichtigt werden. Dabei ist die Frage zu beantworten, wie sich eine Isotopenmi-schung auf die Entropie bzw. die Molwärme auswirkt. Die Mischungsentropie wird nicht berück-sichtigt, da das Material, an dem die experimentelle Bestimmung durchgeführt wird, sowohl bei 0 Kals auch bei der Standardtemperatur gemischt vorliegt und die Entropie bei 0 K Null gesetzt wird. Wir
2 2haben daher die Entropien von CO und CO zu berechnen und entsprechend dem Gehalt der12 13
natürlichen Mischung zu mitteln.Die Berechnung der Translationsanteile erfolgt nach Gl. (6.23). Zur Unterscheidung der Isotope wirddie Massenzahl des Kohlenstoffs als Index angefügt.
Translatorischer Anteil:
Rotatorischer Anteil:
Da das Kohlenstoffatom bei einer Drehung um die Querachse ruht, ist das Trägheitsmoment un-abhängig von der Kohlenstoffmasse. Der C-O-Abstand beträgt 115,98 pm.
Schwingungsanteil:
2Das CO -Molekül weist drei Normalschwingungen auf, von denen die Biegeschwingung entartet ist.

- 84 -
2Abb. 35 Normalschwingungen des CO -Moleküls
2Für das CO -Molekül findet man folgende Wellenzahlen:12
Die symmetrische Streckschwingung ist im Ramanspektrum bei etwa 1340 cm zu erwarten und fällt-1
damit relativ genau mit der 1. Oberschwingung der Biegeschwingung (2< = 1334,6 cm ) zusammen.-1-
In solchen Fällen entstehen durch Wechselwirkungen - die Separierbarkeit in die Normalschwingun-gen ist dann aufgehoben - zwei weiter auseinander liegende Schwingungsniveaus. Durch diese sog.
2Fermiresonanz entstehen beim CO zwei Linien bei 1388,3 und 1285,5 cm (siehe Herzberg). An und-1
für sich müssten für derartige Fälle die Zustandssummen für den Schwingungsanteil modifiziertwerden. Dieses führt jedoch zu großen Schwierigkeiten, da dann die Bildung der Reihensumme nichtmehr in einfacher Weise möglich ist. Sehr viel einfacher durchführbar ist folgendes Verfahren. DieSchwingungen bei 1340 cm werden sowieso nicht viel zur Entropie beitragen (siehe weiter unten).-1
Daher ist es erlaubt, Gl. (8.2.6) mit fiktiven, aber einigermaßen korrekten Werten für die symmetrischeStreckschwingung und die Biegeschwingung zu verwenden. Bei der Biegeschwingung ist es die 1.Oberschwingung mit 2<, die in die geometrische Reihe passt. Bei der Streckschwingung soll der
-
Mittelwert der beiden Fermiresonanzen, d. h. 1337 cm verwendet werden.-1
2 2Die Schwingungsfrequenzen des CO -Moleküls lassen sich aus denen des CO -Moleküls, für die13 12
sie zur Verfügung stehen (siehe oben), relativ einfach berechnen. Die symmetrische Streckschwingungwird durch die Masse des Kohlenstoffatoms nicht beeinflusst. Bei den beiden anderen Schwingungenwird wie folgt verfahren. In erster Näherung ändert sich bei beiden Schwingungen der O-O-Abstand
2nicht. Die beiden Sauerstoffatome dürfen daher zu einem Pseudo-O -Atom in der Mitte zwischenihnen zusammengefasst werden. Die Änderung der Schwingungsfrequenzen dieses pseudo-zwei-atomigen Moleküls lässt sich über die Veränderung der reduzierten Masse
(8.2.7)
berechnen. Bei gleicher Kraftkonstante gilt
(8.2.8)
12 13wobei : = 8,7273 und : = 9,2461 g/mol (in molaren Einheiten) beträgt.

- 85 -
2 2Tab. 8.2.1 Schwingungszustandssumme für CO (oben) und CO (unten)12 13
</cm x S/R Faktor x exp x /(1 - exp x)-1 2 2-
1337,0 6,4548 0,0117 1 0,0657
2349,3 11,3421 0,0001 1 0,0015
667,3 3,2216 0,1746 2 0,4491
G = 0,3610 E = 0,9654
1337,0 6,4548 0,0117 1 0,0657
2282,4 11,0191 0,0002 1 0,0020
648,3 3,1299 0,1878 2 0,4683
E = 0,3875 E = 1,0043
Die Gesamtentropien für die Isotope betragen daher:
Die mittlere Entropie beträgt daher:
Der experimentelle Wert von 213,69 J/K mol aus dem Anhang zur PC I befindet sich damit in besterÜbereinstimmung.
L 2Weiterhin soll die Molwärme c für CO bei der Standardtemperatur berechnet werden. Nach Gl.(8.1.17) gilt für den translatorischen und rotatorischen Anteil unabhängig von der Masse des Kohlens-toffatoms
(8.2.9)
Der Schwingungsanteil muss nach
(8.2.10)
für die Isotope getrennt berechnet werden (siehe Tab. 8.2.1). Der gesamte Schwingungsanteil beträgtdaher
Die gesamte Molwärme sollte daher
betragen. Der experimentelle Wert von 28,82 J/K mol stimmt damit sehr gut überein. Das Fazit der
2 2Berechnungen am SO und CO muss wohl das folgende sein: Ist die Gasphase von einfach aufgebau-ten Molekülen weitgehend ideal, so führt die Berechnung der Entropie, der Molwärme und - an-
ozunehmenderweise - der Funktionen u - u usw. zu Ergebnissen, deren Genauigkeit dem Experimentnicht nachsteht.
- 86 -
Abb. 36 Energieniveaus für die Rotation einer Methylgruppe
9. Statistische Thermodynamik größerer MoleküleDie bisherige Diskussion hat sich mit der Untersuchung der einfachsten Fälle beschäftigt. Zwei häufigauftretende Probleme sollen hier wenigstens erwähnt werden.
Viele Moleküle weisen Schwingungenbzw. Rotationen um eine Einfachbin-dung auf (z. B. organische Molekülemit einer Methylgruppe). Bei tiefenTemperaturen liegt eine (anharmoni-sche) Schwingung in einem flachenperiodischen Potenzial vor (schemati-sche Abb. 36). Bei mittleren Tempe-raturen erfolgt eine gehinderte Rota-tion und bei hohen Temperaturen eineweitgehend freie Rotation. Von derTheorie her einfach zugänglich sinddie Fälle bei tiefen Temperaturen(harmonische Schwingung) und beihohen Temperaturen. Im letzteren Fallwird eine Rotation um eine Achse an-gesetzt. Ohne detaillierte Theorie siehtman, dass dieses einem molekularenFreiheitsgrad entspricht und daher bei
Lu und c zu einem Term ½RT bzw. ½R führt.Anregungszustände des Elektronensystems brauchen i. a. im Bereich normaler Temperaturen nichtberücksichtigt zu werden. So ergibt ein Anregungszustand, der mit Licht von 500 nm erreicht werdenkann, entsprechend der Rechnung am Anfang von Kap. 8.1 bei Raumtemperatur eine Besetzungswahr-scheinlichkeit von 1:e , d. h. die Zustandssumme besteht nur aus dem ersten Glied. Es gibt jedoch-96
Moleküle, die aufgrund ihrer Elektronenstruktur sehr niedrig liegende Anregungszustände aufweisen.
1/2So ist z. B. beim NO durch seine ungerade Elektronenzahl der tiefste Zustand A (2: Multiplizität2
mit S = ½, A: Bahndrehimpuls um die Molekülachse mit 7 = 1, ½: Gesamtdrehimpuls J = ½) Teil
3/2eines Dubletts. Der obere Dublettzustand A liegt in der Energie nur so wenig darüber, dass seine2
Anregung bereits bei 50 K deutlich bemerkbar ist. Die ausführliche Rechnung zeigt, dass ein Maxi-
Lmum der Molwärme c bei ca 70 K entstehen sollte. Leider ist dieser Temperaturbereich wegen derKondensation des NO nicht zugänglich. Die bei höheren Temperaturen gemessenen Werte zeigenjedoch deutlich den von der Theorie geforderten Anstieg mit abnehmender Temperatur.Schließlich sollen noch die Grenzen der Statistischen Thermodynamik bei der Berechnung derthermodynamischen Größen aufgezeigt werden. Für viele der bereits diskutierten Probleme, wieAnharmonizität, Zentrifugaldistorsion, Nichtidealität, gibt es Möglichkeiten, dies durch entsprechendeErweiterungen der Theorie angenähert zu berücksichtigen. Unlösbar wird jedoch das Problem, wenndie Moleküle zu kompliziert aufgebaut sind. Als Beispiel soll das noch vergleichsweise einfachaufgebaute n-Hexan-Molekül diskutiert werden. Dieses Molekül weist 3@20 - 6 = 54 Normalschwin-gungen auf! Diese liegen teilweise im langwelligen Bereich, so dass es im Bereich zwischen 3 und 10:m zu einer unentwirrbaren Anhäufung von Grundschwingungen, Oberschwingungen, Kombinations-schwingungen und Fermiresonanzen im IR- und Ramanspektrum kommt. Es sind daher weder dieFrequenzen der Normalschwingungen bestimmbar, noch darf man die oben angegebenen Gleichungenwegen der Fermiresonanzen verwenden. Die teilweise langwelligen Schwingungen ergeben großeBeiträge zur Zustandssumme, so dass grobe Näherungen zwecks Vereinfachung nicht erlaubt sind.

- 87 -
10 Kristalline FestkörperNach der ausführlichen Besprechung der Gase sollen jetzt die kristallinen Festkörper behandeltwerden. Wir wollen uns auf die Untersuchung der aus Elementen aufgebauten kristallinen Festkörperbeschränken; eine Ausweitung auf andere Festkörper ist in einfacher Weise möglich.Festkörper unterscheiden sich von den Gasen dadurch, dass 1) keine Translationsfreiheitsgrade und 2) keine Rotationsfreiheitsgrade im mikroskopischen Bereich vorhanden sind sowie 3) die Atome aufgrund ihrer Ortskoordinaten unterscheidbar sind.Von den 3N Translationsfreiheitsgraden der isolierten Atome verbleiben 3 Translations- und 3Rotationsfreiheitsgrade des gesamten Kristalls. Bei Vernachlässigung dieser 6 Freiheitsgrade muss derKristall daher 3N Normalschwingungen aufweisen. Die Aufgabe der Statistischen Thermodynamik istes nun, die Frequenzen dieser 3N Normalschwingungen herauszufinden und daraus die Zustands-summe sowie die thermodynamischen Funktionen zu berechnen.An dieser Stelle taucht wieder die Frage auf, wie der Nullpunkt der Energieskala, auf der die Schwin-gungsenergien angegeben werden sollen, festzulegen ist. Entsprechend dem im Kap. 8 angewandtenVerfahren bezeichnen wir den Zustand mit g = 0, bei dem die Atomkerne und Elektronen sich isoliertbei 0 K befinden. Aufgabe der Quantenmechanik ist es, aus diesen Elementarteilchen die Atome (bzw.Moleküle) zu bilden und sie bei 0 K zu einem Kristall zusammenzusetzen. Diese Energie wollen wir
o omit g für ein Teilchen bzw. mit u für 1 mol des Kristalls bezeichnen. Die Nullpunktsenergien für die
oNormalschwingungen wollen wir in diesem Fall nicht in g integrieren, da die Feststellung derNormalschwingungen und ihrer Frequenzen quantenmechanisch nicht ohne weiteres möglich ist undein wesentlicher Teil des statistischen Problems ist.
10.1 Einsteinsche TheorieEin einfaches Modell zur Berechnung der Zustandssumme wurde von Einstein vorgeschlagen. Diewesentlichen Annahmen des Modells sind:1) Die Schwingungen sind harmonisch.2) Die Normalschwingungen erfolgen alle mit der gleichen Frequenz. Dies könnte man sich sovorstellen, dass jedes Atom in drei aufeinander senkrechten stehenden Richtungen gegen den restli-chen, ruhenden Kristall schwingen kann. Ist der Kristall isotrop, so sollten die Schwingungen in denunterschiedlichen Richtungen mit gleicher Frequenz erfolgen.Man sieht ein, dass insbesondere die Annahme 2) mit starken Mängeln behaftet ist. Die Kritik solljedoch bis zur Diskussion des Debyeschen Modells verschoben werden.Mit obigen Annahmen und Gl. (8.1.2) gilt für eine Schwingung:
(10.1.1)
Für die drei Normalschwingungen eines Atoms gilt
(10.1.2)
Aund für die 3N unabhängigen Normalschwingungen eines Mols Atome gilt
(10.1.3)
Wegen der Unterscheidbarkeit der Atome entfällt hier im Vergleich zu den Gasen die Division durch
AN !. Die Summation ergibt wie in Kap. 8

- 88 -
(10.1.4)
Ähnlich wie bei der Rotation und nicht wie bei der Schwingung von Molekülen in der Gasphase mitx = h</kT - der einzige Grund dafür ist, dass es alle so machen - wird eine charakteristische Tempera-tur 1 eingeführt.
(10.1.5)
(10.1.6)
LZum Vergleich mit dem Experiment eignet sich besonders die Molwärme c mit ihrem komplexenVerlauf in Abhängigkeit von der Temperatur (Kap. I 9.1.2).
(10.1.7)
(10.1.8)
owobei - wie oben bereits definiert - die Energie des Kristalls bei 0 K ohne Schwingungsenergie mit ubezeichnet wurde. Die Molwärme wird durch eine weitere Differenziation erhalten.
(10.1.9)
(10.1.10)
Zuerst soll die Vorhersage bei hohen Temperaturen überprüft werden. Mit
(10.1.11)
entsprechend der Definition bei den Schwingungen der Moleküle in der Gasphase folgt
(10.1.12)
Es ist jetzt der Grenzwert dieses für x 6 0 unbestimmten Ausdrucks zu untersuchen. Ohne dasVerfahren von Bernoulli/L'Hospital bemühen zu müssen, wird die Exponentialfunktion im Nenner ineine Reihe entwickelt und nach dem zweiten Glied abgebrochen. Für den Quotienten folgt damit 1 undinsgesamt
(10.1.13)
d. h. die Einsteinsche Theorie sagt korrekt die Dulong-Petitsche Regel (I 9.1.2.1) voraus.Bei tiefen Temperaturen, d. h. für x 6 4, ist der Ausdruck von der Form 4/4. Man darf dann die 1 imNenner streichen und erhält

- 89 -
Abb. 37 Schwingungen eines langen Quaders
(10.1.14)
Das stimmt nicht mit den Experiment überein, da bei tiefen Temperaturen die Molwärme mit T gehen3
sollte (Debyesches T -Gesetz, Kap. I 9.1.2). Offensichtlich treffen die Annahmen, die zu dieser3
Theorie führten, nicht zu.
10.2 Debyesche TheorieDer wesentliche Fehler der Einsteinschen Theorie ist die Annahme einer einzigen Schwingungs-frequenz. Das führt zu einer einfachen Theorie, die deswegen auch hier vorgeführt wurde. IR-spektroskopische und andere Untersuchungen zeigen jedoch, dass es ein ganzes Spektrum solcherSchwingungen gibt. Im langwelligen Teil gibt es Schwingungen des gesamten Kristalls. Diese liefernden wesentlichen Beitrag bei tiefen Temperaturen, da nur sie bei tiefen Temperaturen angeregt werden
können. Abb. 37 zeigt die drei langsamsten Mo-den. Diese Schwingungen muss man sich auf dreiDimensionen ausgedehnt vorstellen. Die Fre-quenz wächst mit der Zahl der Knoten. Eine obereGrenze für diese Frequenz ist dadurch gegeben,dass in einer Dimension die Knotenzahl derAtomzahl entspricht, d. h. die Atome schwingengerade gegenphasig. Später werden wir jedoch dieobere Grenzfrequenz als anpaßbaren Parametereinführen.
Sind die Schwingungen des Kristalls unabhängigvoneinander, so darf man
(10.2.1)
lansetzen, wobei der Index l über die Schwingungen läuft und n die Schwingungsquantenzahlen der
oNiveaus der Schwingung l darstellen. , ist die gesamte Energie des Kristalls mit ruhenden Atomen.Für diejenigen, die Schwierigkeiten mit dieser Formel haben, sei noch folgendes angemerkt. Manstelle sich den Kristall als schwingungsfähiges, großes Molekül vor; die verschiedenen l entsprechen
ldann den einzelnen Normalschwingungen und die n entsprechen den normalen Schwingungs-quantenzahlen und sind damit ein Maß für die Amplituden der Schwingungen. Die ZustandssummeZ wird
(10.2.2)
lwobei in den ersten beiden Zeilen die Summe über die n eine Kurzschreibweise für die Vielfachsum-
1 2 lme über alle n , n , . . , n darstellt. Der letzte Teil der Gleichung ist im Prinzip die Quintessenz derbereits öfter gefundenen Regel: Können die Energieterme separiert werden, so dürfen die Zustands-
o osummen als Produkte dieser separierten Teile geschrieben werden. Weiterhin wird g durch u ersetzt.
(10.2.3)
Analog zu Gl. (8.1.3) folgt
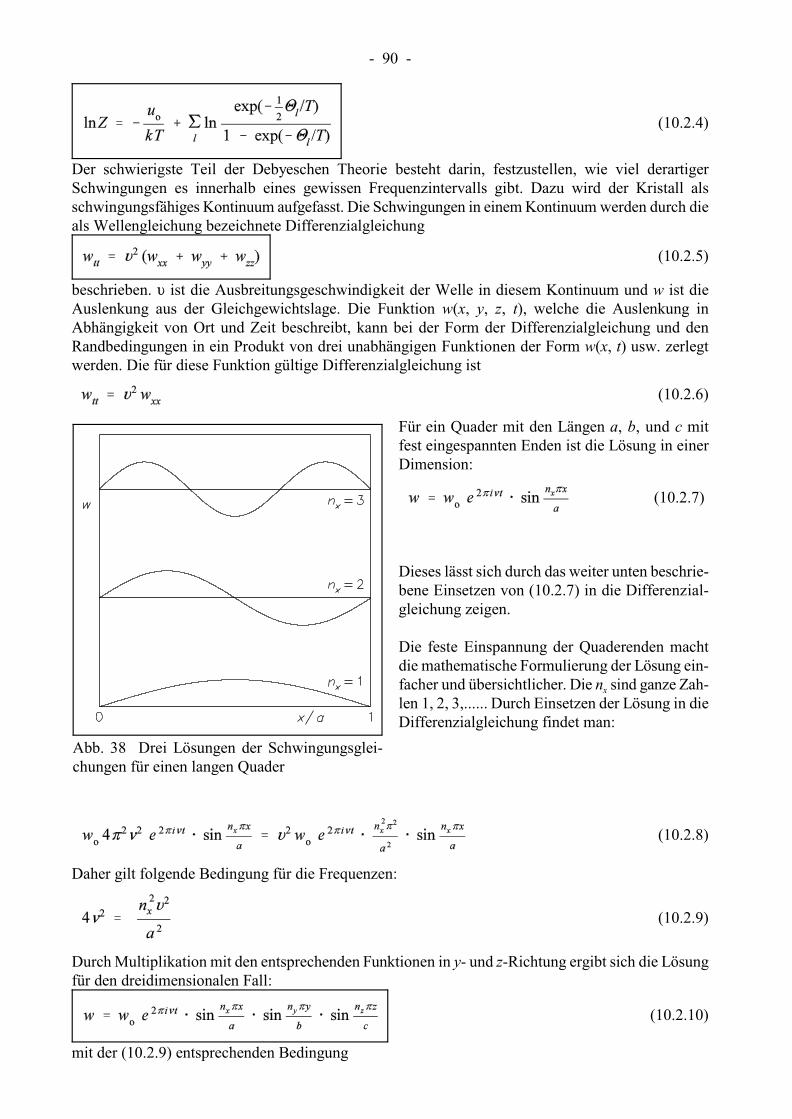
- 90 -
Abb. 38 Drei Lösungen der Schwingungsglei-chungen für einen langen Quader
(10.2.4)
Der schwierigste Teil der Debyeschen Theorie besteht darin, festzustellen, wie viel derartigerSchwingungen es innerhalb eines gewissen Frequenzintervalls gibt. Dazu wird der Kristall alsschwingungsfähiges Kontinuum aufgefasst. Die Schwingungen in einem Kontinuum werden durch dieals Wellengleichung bezeichnete Differenzialgleichung
(10.2.5)
beschrieben. L ist die Ausbreitungsgeschwindigkeit der Welle in diesem Kontinuum und w ist dieAuslenkung aus der Gleichgewichtslage. Die Funktion w(x, y, z, t), welche die Auslenkung inAbhängigkeit von Ort und Zeit beschreibt, kann bei der Form der Differenzialgleichung und denRandbedingungen in ein Produkt von drei unabhängigen Funktionen der Form w(x, t) usw. zerlegtwerden. Die für diese Funktion gültige Differenzialgleichung ist
(10.2.6)
Für ein Quader mit den Längen a, b, und c mitfest eingespannten Enden ist die Lösung in einerDimension:
(10.2.7)
Dieses lässt sich durch das weiter unten beschrie-bene Einsetzen von (10.2.7) in die Differenzial-gleichung zeigen.
Die feste Einspannung der Quaderenden machtdie mathematische Formulierung der Lösung ein-
xfacher und übersichtlicher. Die n sind ganze Zah-len 1, 2, 3,...... Durch Einsetzen der Lösung in dieDifferenzialgleichung findet man:
(10.2.8)
Daher gilt folgende Bedingung für die Frequenzen:
(10.2.9)
Durch Multiplikation mit den entsprechenden Funktionen in y- und z-Richtung ergibt sich die Lösungfür den dreidimensionalen Fall:
(10.2.10)
mit der (10.2.9) entsprechenden Bedingung

- 91 -
Abb. 39 Zur Berechnung der Zahl der Transla-tionsniveaus
(10.2.11)
Für einen würfelförmigen Kristall mit der Kantenlänge a gilt:
(10.2.12)
Wie bei der Berechnung der Zahl der Transla-
x ytionsniveaus in einem Gas werden die n , n und
zn in einem entsprechenden Koordinatensystemdargestellt (siehe nebenstehende Abb. für zweiDimensionen) und es wird eine Kugel mit demRadius R eingezeichnet.
(10.2.13)
Die Zahl der zulässigen Schwingungen innerhalbeiner Kugelschale mit dem Radius R und der Di-cke dR entspricht dem Volumen dieser Kugel-schale in einem Oktanten und beträgt daher:
(10.2.14)
Zur Umrechnung des Radius auf die Frequenzwird die Wurzel aus (10.2.13) gezogen unddifferenziert
(10.2.15)
Aus (10.2.14) wird daher
(10.2.16)
Die Zahl der zulässigen Schwingungen wird nun durch die Dichtefunktion g(<) beschrieben.
(10.2.17)
Die vielleicht einschränkend erscheinende Berechnung in einem Würfel kann mit viel Mathematik aufandere Formen erweitert werden. Die entsprechende Berechnung in einem Quader ist z. B. in ähnlicherWeise möglich, indem das Analogon zu (10.2.13) anstelle von (10.2.12) aus (10.2.11) hergeleitet wird.Anstelle eines Kugeloktanten entsteht dann ein Ellipsoidoktant. Die Durchrechnung führt auch indiesem Fall zu (10.2.17). Weiterhin sieht man, dass g(<) proportional zum Volumen ist. Man darfdaher ein gegebenes, unregelmäßig begrenztes Volumen in kleinere Würfel oder Quader aufteilen undkommt dann so zu unregelmäßig begrenzten Körpern.
In einem Festkörper können sich sowohl Longitudinal- als auch Transversalwellen ausbreiten, letzteresogar mit zwei aufeinander senkrecht stehenden Schwingungsrichtungen. Dies entspricht völlig denmöglichen Normalschwingungen eines mehratomigen, linearen Moleküls, wo die Longitudinal-

- 92 -
schwingungen den Streckschwingungen und die doppelt zu zählenden Transversalschwingungen denentarteten Biegeschwingungen entsprechen. Die schematische Darstellung der niederfrequentenModen am Anfang dieses Kapitels gibt nur eine dieser drei Moden wieder. Wie sehen die anderen aus?Die Frequenzdichte (10.2.17) ist daher noch mit einem Faktor 3 zu multiplizieren.
(10.2.18)
Aus dieser Gleichung soll noch die Fortpflanzungsgeschwindigkeit L der Welle eliminiert und durch
maxdie maximale Frequenz < ersetzt werden. Dies gelingt durch Integration der vorstehenden Glei-chung
(10.2.19)
Daraus folgt für 1 mol
(10.2.20)
so dass die endgültige Gleichung
(10.2.21)
lautet.Die Summe in Gl. (10.2.4) wird jetzt durch ein Integral ersetzt. Dieses ist erlaubt, da der Summendsich beim Übergang von einer Schwingungsmode zur nächsten kaum ändert, wie die folgendeRechnung zeigt.
(10.2.22)
wobei die Wurzel aus Gl. (10.2.9) verwendet wurde. Für die Änderung folgt daher
(10.2.23)
mit L = 5000 m/s und a = 0,01 m. Da nun nicht mehr über Quantenzahlen, sondern über Frequenzenintegriert wird, muss der Integrand noch mit der Zustandsdichte der erlaubten Schwingungen ent-sprechend Gl. (10.2.21) multipliziert werden. Das ergibt:
(10.2.24)
Auch für die Debyesche Theorie sollen die Molwärmen berechnet werden. Dieses ist leider mit einerReihe langwieriger mathematischer Operationen verbunden, die daher in den Anhang 14.3 verbanntworden sind. Das Ergebnis ist:
(10.2.25)

- 93 -
VAbb. 40 Verlauf der Molwärme c nach derDebyeschen Theorie
mit
(10.2.26)
Wie bei der Einsteinschen Theorie soll der Grenzwert für hohe und niedrige Temperaturen untersuchtwerden.
maxDa bei hohen Temperaturen 1 /T klein ist, darf der Nenner des letzten Terms in eine Reihe entwi-ckelt und nach dem 2. Glied abgebrochen werden. Der letzte Term ergibt daher eine 1. Der vordereTerm wird wie folgt berechnet. Wegen der kleinen Werte der oberen Integralgrenze liegt die In-tegrationsvariable immer bei kleinen Werten. Daher darf auch hier die Exponentialfunktion entwickeltund nach dem 2. Glied abgebrochen werden, so dass der Integrand nur noch x enthält und das Integral2
max(1 /T) /3 ergibt. Insgesamt entsteht daher der korrekte Wert3
(10.2.27)
Bei niedrigen Temperaturen verschwindet der letzte Term wegen des Exponentialausdrucks im
maxNenner. Beim Integral darf die obere Grenze durch 4 ersetzt werden, da 1 /T sehr große Werteannimmt und der Integrand bei großen Werten der Variablen klein wird, d. h. die Beiträge des
maxIntegrals zwischen 1 /T und 4 können vernachlässigt werden. Es verbleibt daher:
(10.2.28)
Das Integral ergibt B /15, so dass insgesamt4
(10.2.29)
entsteht. Das ist das Debyesche T -Gesetz. Es3
stimmt gut mit den experimentellen Beobachtun-gen überein. Im Bereich mittlerer Temperaturenstimmt das Ergebnis der Debyeschen Theorie (sie-he Abb. 40) trotz des - wie auch bei tiefen Tempe-raturen - für jedes Element frei anpaßbaren Para-
maxmeters 1 nur befriedigend bis mangelhaft mitden experimentellen Beobachtungen überein. DieDebyesche Theorie stimmt deswegen im Bereichtiefer Temperaturen mit dem Experiment so gutüberein, weil der Ansatz für die Schwingungs-frequenzdichte im Bereich niedriger Frequenzenweitgehend korrekt ist. Die niedrigen Frequenzensind eben die wichtigen Frequenzen für die Mol-wärmen bei tiefen Temperaturen. Der Grenzwertbei hohen Temperaturen stimmt unabhängig vomAnsatz für die Frequenzdichte, da sowieso alleSchwingungen angeregt sind. Schwierig ist derBereich der mittleren Temperaturen. Aus der Ab-
maxbildung ist das nicht zu erkennen, da 1 dort angepasst wurde. Passt man dagegen so an, dass dieWerte bei tiefen Temperaturen stimmen, so entstehen große Fehler im Bereich mittlerer Temperaturen.Das Experiment zeigt, dass der Ansatz für die hohen Frequenzen nicht korrekt ist. So beobachtet manin den IR- und Ramanspektren scharfe Frequenzen im Hochfrequenzbereich. Von der Theorie her istdie Behandlung des Kristalls als Kontinuum bei hohen Frequenzen nicht korrekt. Exaktere Theorien

- 94 -
behandeln den Kristall als ein Gitter von Massenpunkten, die durch ein harmonisches Potenzialgekoppelt sind. Das System gekoppelter Differenzialgleichungen für die Bewegung der Massenpunktewird genähert gelöst. Das Ergebnis ist eine etwas höhere Frequenzdichte bei hohen Frequenzen imVergleich zum Debyeschen Ansatz.

- 95 -
11 Chemisches GleichgewichtDie folgende Behandlung beschränkt sich aus naheliegenden Gründen auf die Gasphase.Da das chemische Potenzial bereits im Kap. 5.4 berechnet wurde, gelingt der schnellste Zugang zumMWG über die Gleichgewichtsbedingung
(11.1)
aus. Einsetzen von (5.4.13) ergibt
(11.2)
Daraus folgt:
(11.3)
(11.4)
(11.5)
Das ist das MWG aus statistisch thermodynamischer Sicht. Zur weiteren Behandlung wollen wir, wie
i oam Anfang von Kap. 8 beschrieben, bei den z den g -Wert herausziehen, d. h. analog zu Gl. (8.1.6)
(11.6)
ansetzen, wobei der elektronische Term fallengelassen wurde. Eingesetzt in (11.5) ergibt sich:
(11.7)
(11.8)
owobei )g die "Reaktionsenergie" für einen molekularen (nicht molaren!) Formelumsatz bei 0 K unterEinschluß der Nullpunktsschwingungen darstellt. Diese Größe kann die Statistische Thermodynamiknicht zur Verfügung stellen; sie muss quantenmechanisch berechnet werden oder aus anderen Quellenstammen. Gl. (11.8) stellt den einfachsten, noch allgemein gültigen Ausdruck für das MWG dar. Dieweitere Behandlung ist systemspezifisch.
2 2Als Beispiel soll die Isotopenaustauschreaktion H + D W 2HD behandelt werden. Die Gleichge-wichtskonstante soll bei 383 K berechnet werden, da für diese Temperatur die Gleichgewichtskon-stante experimentell bestimmt wurde.
0Für diese Reaktion ist )g = 0, wenn die Energieniveaus für alle Komponenten vom Minimum desPotenzialtopfs, d. h. im Gegensatz zur obigen Beschreibung, mit ½h< gerechnet werden. Es gilt dann:

- 96 -
(11.9)
Da bei der Berechnung wegen der Ähnlichkeit der Moleküle viele Größen herausfallen, werden ameinfachsten nicht die z'-Werte vollständig berechnet, sondern es wird gleich der Quotient entsprechenddem letzten Teil von (11.9) für die einzelnen Energieformen berechnet. Für den translatorischen
transQuotienten Q gilt
(11.10)
Beim rotatorischen Quotienten verbleibt von (7.2.2.2) nur der Nenner F1, d. h.
(11.11)
Die Abstände in I = :r fallen mit sehr großer Genauigkeit heraus2
(11.12)
Für den Schwingungsanteil muss (8.1.3) benutzt werden, da vom Potenzialminimum aus gerechnetwerden muss.
(11.13)
Für HD gilt < = 3770 cm . Der Nenner wird daher-1-
Zusätzlich heben sich die Nenner für die einzelnen Reaktanden noch weitgehend gegenseitig weg. Siewerden daher vernachlässigt und es folgt:
(11.14)
Weiterhin gilt
(11.15)
Jetzt kommt das große Finale

- 97 -
(11.16)
D HMit ausreichender Genauigkeit darf man m = 2m setzen.
(11.17)
Einsetzen der Konstanten ergibt
(11.18)
Experimentell findet man 3,50. Wahrscheinlich ist in diesem Fall dem theoretischen Ergebnis sogarder Vorzug zu geben.Nur wenige Berechnungen von Gleichgewichtskonstanten laufen so glatt wie dieses Beispiel. Vielfach
olassen sich nur schwer Werte für )g finden. In einigen Fällen, z. B. bei der Dimerisierung vonMetalldämpfen in der Gasphase, ist die Berechnung aus spektroskopischen Daten möglich.
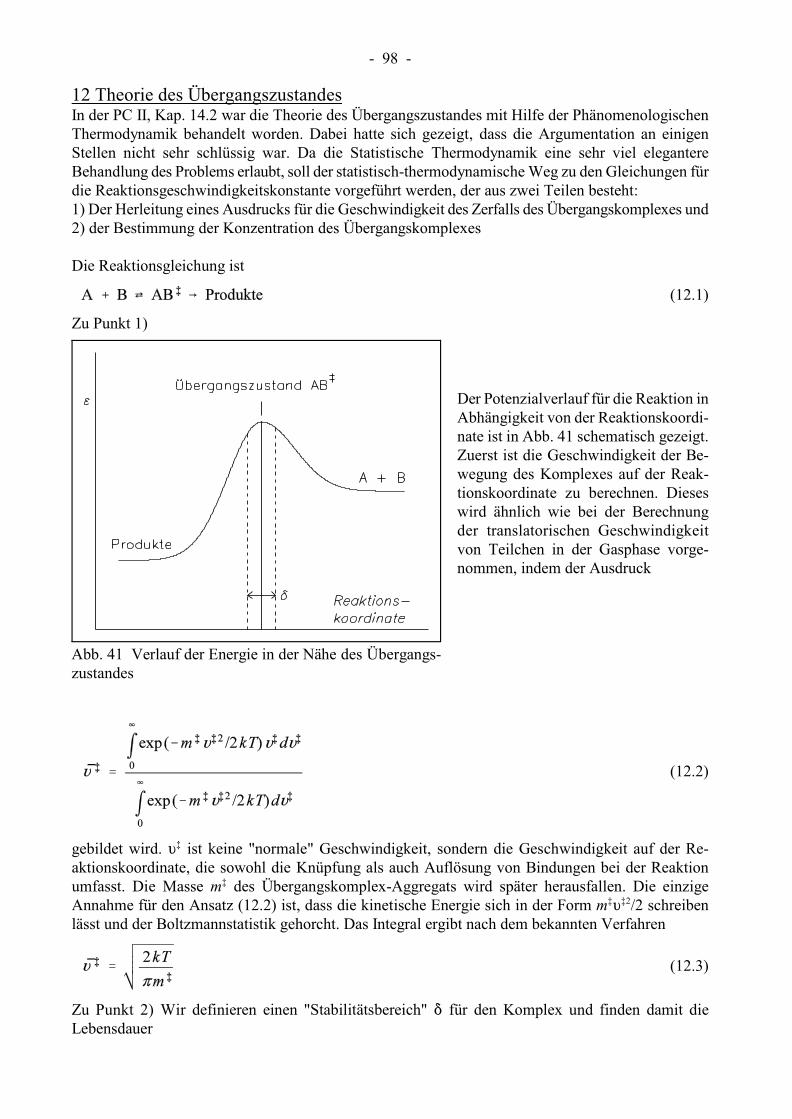
- 98 -
Abb. 41 Verlauf der Energie in der Nähe des Übergangs-zustandes
12 Theorie des ÜbergangszustandesIn der PC II, Kap. 14.2 war die Theorie des Übergangszustandes mit Hilfe der PhänomenologischenThermodynamik behandelt worden. Dabei hatte sich gezeigt, dass die Argumentation an einigenStellen nicht sehr schlüssig war. Da die Statistische Thermodynamik eine sehr viel elegantereBehandlung des Problems erlaubt, soll der statistisch-thermodynamische Weg zu den Gleichungen fürdie Reaktionsgeschwindigkeitskonstante vorgeführt werden, der aus zwei Teilen besteht:1) Der Herleitung eines Ausdrucks für die Geschwindigkeit des Zerfalls des Übergangskomplexes und2) der Bestimmung der Konzentration des Übergangskomplexes
Die Reaktionsgleichung ist
(12.1)
Zu Punkt 1)
Der Potenzialverlauf für die Reaktion inAbhängigkeit von der Reaktionskoordi-nate ist in Abb. 41 schematisch gezeigt.Zuerst ist die Geschwindigkeit der Be-wegung des Komplexes auf der Reak-tionskoordinate zu berechnen. Dieseswird ähnlich wie bei der Berechnungder translatorischen Geschwindigkeitvon Teilchen in der Gasphase vorge-nommen, indem der Ausdruck
(12.2)
gebildet wird. L ist keine "normale" Geschwindigkeit, sondern die Geschwindigkeit auf der Re-‡
aktionskoordinate, die sowohl die Knüpfung als auch Auflösung von Bindungen bei der Reaktionumfasst. Die Masse m des Übergangskomplex-Aggregats wird später herausfallen. Die einzige‡
Annahme für den Ansatz (12.2) ist, dass die kinetische Energie sich in der Form m L /2 schreiben‡ ‡2
lässt und der Boltzmannstatistik gehorcht. Das Integral ergibt nach dem bekannten Verfahren
(12.3)
Zu Punkt 2) Wir definieren einen "Stabilitätsbereich" * für den Komplex und finden damit dieLebensdauer

- 99 -
(12.4)
Auch * wird später herausfallen. Als Reaktionsgeschwindigkeit r wollen wir die pro Volumen undZeit zum Produkt umgesetzten Stoffmenge definieren. Das ergibt
(12.5)
Der Faktor ½ resultiert aus folgender Überlegung. Der Übergangskomplex auf dem höchsten Punktdes Potenzials sieht das nach beiden Seiten in gleicher Weise abfallende, in erster Näherung parabo-lische Potenzial. Die Wahrscheinlichkeit des Zerfalls in die Produkte und Edukte sollte daher gleich
AB‡sein, so dass für jede Zerfallsrichtung gerade der in Gl. (12.5) angegebene Wert entsteht. c wird alsKonzentration des Übergangskomplexes im Gleichgewicht A + B W AB berechnet, d. h. es wird die‡
Annahme verwandt, dass die Einstellungsgeschwindigkeit des Gleichgewichts genügend groß ist.
(12.6)
und daher
(12.7)
cAls nächstes ist die (einheitenbehaftete) Gleichgewichtskonstante K ' zu berechnen. Nach (11.8) gilt
(12.8)
und nach (I 16.1.14)
(12.9)
so dass
(12.10)
folgt. Dieser Ausdruck wird jetzt mit dem reaktionskinetischen Ansatz
(12.11)
verglichen
(12.12)
ABDie Zustandssumme z' für den Komplex im Übergangszustand spalten wir nun in zwei Teile auf: In
R ABeinen Teil für die Bewegung in der Reaktionskoordinatenrichtung z' und den Rest z ' . Der erste Teil‡
entspricht der Schwingung in Reaktionsrichtung. Da diese Schwingung zum Zerfall des Komplexesführt, wird sie durch eine Translation in Richtung der Reaktionskoordinate ersetzt. Die restlichen

- 100 -
ABFreiheitsgrade werden in z ' zusammengefasst.‡
(12.13)
R z' ist die Zustandssumme für eine durch die Länge * eingeschränkte Translation. Verwandt wirdhierfür in grober Näherung die Zustandssumme für die eindimensionale Translation eines Teilchensin einem Kastenpotenzial, d. h. mit unendlich hohen Wänden an den Kastenenden und nicht mit einemstark abfallenden Potenzial. Nach Gl. (6.9) beträgt die Zustandssumme
(12.14)
Insgesamt entsteht daher
(12.15)
(12.16)
Im Vergleich zur phänomenologischen Herleitung in der PC II stellen wir fest, dass der dort mit einernur sehr undurchsichtigen Argumentation eingeführte Faktor kT/h sich hier zwanglos ergibt. DasProdukt der Zustandssummen und des Exponentialausdrucks entspricht im wesentlichen der Gleich-
ABgewichtskonstante, wobei aber in z ' der Schwingungsfreiheitsgrad in Zerfallsrichtung des Kom-‡
plexes fehlt.Der Vorteil der hier vorgestellten Gleichung besteht darin, dass die rechte Seite im Prinzip explizit
oberechenbar ist, wobei für )g natürlich die Quantenmechanik zu Hilfe genommen werden muss. In
2einfachen Fällen (z. B. H + D W HD + H) ist der Rechenaufwand noch vergleichsweise gering, daviele Terme herausfallen und im angeführten Beispiel sogar der Exponentialterm.Ein sehr einfaches Beispiel soll durchgerechnet werden: Die Reaktion von zwei "strukturlosen"Partikeln A und B. Darunter sollen Partikel verstanden werden, die keine Trägheitsmomente undSchwingungen aufweisen, d. h. z. B. Atome. Die Zustandssummen der Partikel sind:
(12.17)
Rotations- und Schwingungszustandssummen brauchen für die strukturlosen Partikeln nicht berück-sichtigt zu werden. Im Übergangskomplex muss dagegen die Rotationszustandssumme berücksichtigtwerden; die Schwingungszustandssumme entfällt entsprechend der obigen Festsetzung. Man findetgemäß Gl. (7.2.1.3)
(12.18)
(12.19)
AB ABwobei d der Abstand der Molekülschwerpunkte A, B im Übergangskomplex und F der Stoßquer-schnitt sind. Insgesamt ergibt sich daher

- 101 -
(12.20)
(12.21)
oder in der Schreibweise der PC II
(12.22)
Es ergibt sich also die Gleichung der einfachen Stoßtheorie, d. h. die Theorie des Übergangszustandesliegt nicht vollständig daneben.Rechnungen an nur wenig komplizierteren Reaktionen werden leider sehr viel aufwändiger und lassensich nur noch mit Abschätzungen durchführen. Ein Beispiel ist die bereits angesprochene, sehr
2einfache Reaktion H + D W H@@@@@H@@@@@D W H + HD. Für die Translations- und Rotationszustands-
2, osummen von H D und HHD lassen sich exakte Ausdrücke oder gute Näherungen finden. )g
2verschwindet. Auch die Schwingungszustandssumme von H ist einfach berechenbar. Schwierig wirdaber die Berechnung der Schwingungszustandssumme von HHD. Die asymmetrische Streckschwin-gung entfällt zwar, da sie auf der Reaktionskoordinate verläuft. Es verbleiben aber noch die symmetri-sche Streckschwingung und die beiden entarteten Biegeschwingungen, für die quantenmechanischeRechnungen ausgeführt oder Näherungen verwendet werden müssen. Die Frequenzen dieser Schwin-gungen gehen direkt in die Aktivierungsenergie ein und beeinflussen das Ergebnis stark, so dass dieVerwendung grober Näherungen nicht zulässig ist.
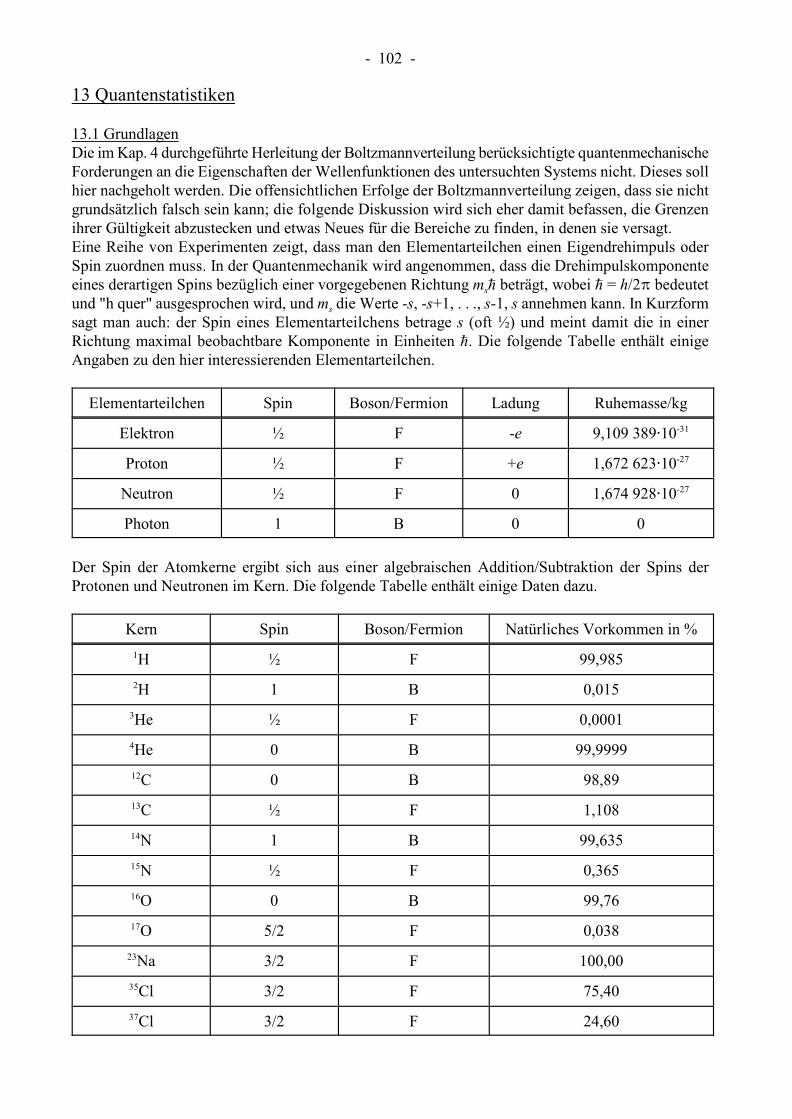
- 102 -
13 Quantenstatistiken
13.1 GrundlagenDie im Kap. 4 durchgeführte Herleitung der Boltzmannverteilung berücksichtigte quantenmechanischeForderungen an die Eigenschaften der Wellenfunktionen des untersuchten Systems nicht. Dieses sollhier nachgeholt werden. Die offensichtlichen Erfolge der Boltzmannverteilung zeigen, dass sie nichtgrundsätzlich falsch sein kann; die folgende Diskussion wird sich eher damit befassen, die Grenzenihrer Gültigkeit abzustecken und etwas Neues für die Bereiche zu finden, in denen sie versagt.Eine Reihe von Experimenten zeigt, dass man den Elementarteilchen einen Eigendrehimpuls oderSpin zuordnen muss. In der Quantenmechanik wird angenommen, dass die Drehimpulskomponente
seines derartigen Spins bezüglich einer vorgegebenen Richtung m hG beträgt, wobei hG = h/2B bedeutet
sund "h quer" ausgesprochen wird, und m die Werte -s, -s+1, . . ., s-1, s annehmen kann. In Kurzformsagt man auch: der Spin eines Elementarteilchens betrage s (oft ½) und meint damit die in einerRichtung maximal beobachtbare Komponente in Einheiten hG. Die folgende Tabelle enthält einigeAngaben zu den hier interessierenden Elementarteilchen.
Elementarteilchen Spin Boson/Fermion Ladung Ruhemasse/kg
Elektron ½ F -e 9,109 389@10-31
Proton ½ F +e 1,672 623@10-27
Neutron ½ F 0 1,674 928@10-27
Photon 1 B 0 0
Der Spin der Atomkerne ergibt sich aus einer algebraischen Addition/Subtraktion der Spins derProtonen und Neutronen im Kern. Die folgende Tabelle enthält einige Daten dazu.
Kern Spin Boson/Fermion Natürliches Vorkommen in %
H ½ F 99,9851
H 1 B 0,0152
He ½ F 0,00013
He 0 B 99,99994
C 0 B 98,8912
C ½ F 1,10813
N 1 B 99,63514
N ½ F 0,36515
O 0 B 99,7616
O 5/2 F 0,03817
Na 3/2 F 100,0023
Cl 3/2 F 75,4035
Cl 3/2 F 24,6037

- 103 -
Die Teilchen mit einem halbzahligen Spin werden als Fermionen und die mit einem ganzzahligen alsBosonen bezeichnet. Daraus folgt, dass Kerne mit gerader Massenzahl grundsätzlich Bosonen undsolche mit ungerader Massenzahl Fermionen sein müssen.Schließlich werden die Kerne und Elektronen zu Atomen bzw. Molekülen zusammengesetzt. Bei denleichteren Elementen erhält man durch algebraische Addition/Subtraktion der Elektronenspins denGesamtelektronenspin, der mit dem Kernspin zu einem Gesamtspin zusammengefasst wird. Wegender algebraischen Addition/Subtraktion gilt auch hier: Atome und Moleküle mit gerader Elementarteil-
2chenzahl sind Bosonen; solche mit ungerader Zahl Fermionen. Beispielsweise ist H in allen Zu-1
2ständen ein Boson. Dies gilt sowohl für die verschiedenen Kernspinfunktionen (ortho-H : I = 1, para-
2H : I = 0), als auch für die verschiedenen Elektronenspinfunktionen (Singuletts mit S = 0, Tripletts mitS = 1).Die Unterscheidung in Bosonen und Fermionen ist wichtig, da die Quantenmechanik eine unter-schiedliche Konstruktion der Gesamtwellenfunktion für ein System aus mehreren Teilchen vor-schreibt. Der Hamilton-Operator des Systems der N Teilchen möge in die Operatoren der einzelnenTeilchen separabel sein, zu denen die Wellenfunktionen
1 2 3 R (1), R (2), R (3), . . . .gehören mögen, wobei der Index verschiedene Wellenfunktionen anzeigt und die Zahl in der Klammerdie Teilchen unterschiedet.Die Vorschrift für die Konstruktion der Wellenfunktion eines Vielteilchensystems aus Fermionen lässtsich nicht der Quantenmechanik entnehmen. Pauli hat gezeigt, dass diese Vorschrift eine Folge derQuantenfeldtheorie ist. Das Ergebnis ist:
(13.1.1)
wobei P der Permutationsoperator ist, der zwei Teilchen vertauscht. P bedeutet eine <-fache Permuta-<
tion, wobei Rückpermutationen nicht erlaubt sind. Die Summation ist über alle möglichen Permutatio-nen zu erstrecken und jeder Term ist mit einem Vorzeichen entsprechend (-1) zu versehen. Gl.<
(13.1.1) ist nichts anderes als eine kompakte Schreibweise für die vielleicht eher bekannte De-terminante
(13.1.2)
Eine Determinante - wie auch sofort an Gl. (13.1.1) erkennbar - hat folgende Eigenschaft: Stimmen
1 2zwei der Wellenfunktionen überein (z. B. R und R ), so verschwindet die Determinante wegen derÜbereinstimmung von zwei Spalten, d. h. diese Wellenfunktion stellt keine Lösung des Problems dar.Dieses ist nichts anderes als die Aussage des Pauliprinzips.Für Bosonen gilt nun:
(13.1.3)
d. h. es fehlt nur der Vorzeichenwechsel im Vergleich zur Wellenfunktion für die Fermionen. Mansagt daher auch: Wellenfunktionen für Bosonen müssen symmetrisch bezüglich einer Teilchenver-tauschung sein, während sie für die Fermionen antisymmetrisch sein müssen. Im Unterschied zu denFermionen ist es für Bosonen durchaus erlaubt, ein Niveau mehrfach zu besetzen.Wie wirkt sich nun die Unterschiedlichkeit der Wellenfunktionen auf die Statistische Thermodynamikaus? Bei der Berechnung der Systemzustandssummen in der Gasphase in Kap. 5.2 hatte es Schwierig-

- 104 -
Funktion 1 2 3 4
Realisierung 1 x x
Realisierung 2 x x
Realisierung 3 x x
Realisierung 4 x x
Realisierung 5 x x
Realisierung 6 x x
keiten bei der Division durch N! gegeben. Es musste angenommen werden, dass sehr viel mehrbesetzbare Niveaus als Teilchen vorhanden sind, so dass Mehrfachbesetzungen eines Niveaus eineuntergeordnete Rolle spielen. Bei Mehrfachbesetzungen führt daher die Anwendung der Boltzmann-verteilung zu falschen Ergebnissen. Gerade unter diesen Bedingungen unterscheiden sich auch dieWellenfunktionen für Fermionen und Bosonen, wobei bei den ersteren die Mehrfachbesetzung ebennicht erlaubt ist. Dieses führt z. B. dazu, dass die Sackur-Tetrode-Gleichung in der Form (6.21), d. h.noch ohne Verwendung der idealen Gasgleichung, für T 6 0 zu dem unsinnigen Ergebnis S = -4 führt!An diesem Fehler führt auch die Argumentation nicht vorbei, dass die meisten Substanzen dortkristallin sind. Helium ist bei 0 K durchaus gasförmig.Vor der eigentlichen Herleitung der Quantenstatistiken für Fermionen bzw. Bosonen sollten wir nocheinmal die Herleitung der Energieverteilung (4.21) bzw. (4.22) in einem kanonischen Ensemble unddie der Boltzmannverteilung (4.23) rekapitulieren. Zuerst wurden die Verteilungen (4.21) und (4.22)für ein kanonisches Ensemble hergeleitet. Dabei wurde ein Ensemble makroskopischer Systeme mitder Möglichkeit des Wärmeaustauschs vorausgesetzt. Die einzelnen Systeme wurden als unterscheid-bar angenommen. Dieses ist für makroskopische Systeme durchaus sinnvoll. Falsch ist nun derÜbergang zur Boltzmannverteilung durch Annahme eines Teilchens pro System, da die Teilchen nicht
imehr unterscheidbar sind! Die Boltzmannverteilung ergibt dann N Teilchen auf einem (nicht ent-
iarteten) Energieniveau mit der Energie g im Widerspruch zu den Aussagen des Pauliprinzips fürFermionen.Der Fehler bei der Herleitung der Boltzmannverteilung besteht in dem grundsätzlich falschenAbzählverfahren: Alle Teilchen einer Sorte sind ununterscheidbar. Es kommt also nicht darauf an,welche Teilchen sich in den einzelnen Zuständen befinden, sondern nur darauf an, wie viel Teilchensich in den einzelnen Zuständen befinden. Die unterschiedlichen Verteilungen für Fermionen bzw.Bosonen werden dann durch die nicht erlaubten bzw. erlaubten Mehrfachbesetzungen bewirkt. In derBoltzmannstatistik wurde die Ununterscheidbarkeit der Teilchen zwar später wegen offensichtlicherFehler der sonst entstehenden Gleichungen eingeführt (Kap. 5.2, Fall b); diese Einführung erfolgt abererheblich zu spät.Der schnellste und mathematisch unkomplizierteste Weg zu den Analoga zur Boltzmannverteilung fürFermionen und Bosonen besteht in einem grundsätzlich anderen Vorgehen als bei der Herleitung derBoltzmannverteilung. Leider muss dabei eine Schwierigkeit mit etwas mageren Argumenten umgan-gen werden.
13.2 Fermi-Dirac-StatistikDie Fermi-Dirac-Statistik (FD-Statistik) ist die nach ihren Entdeckern benannte Statistik für Fermio-nen.Im Gegensatz zum Vorgehen bei der Boltzmannstatistik betrachten wir hier nur ein System aus N
j jTeilchen. Das System möge die Energieniveaus g mit der Entartung g aufweisen. Es soll die thermo-dynamische Wahrscheinlichkeit W fürdie Verteilung der Fermionen auf dieNiveaus berechnet werden, d. h. dieZahl der unterscheidbaren Realisie-rungsmöglichkeiten bei gegebenerTeilchenzahl und Gesamtenergie.Als Beispiel betrachten wir das Ener-
1gieniveau g , das vierfach entartet sein
1 1soll (g = 4) und verteilen N = 2 Fer-mionen darüber. Die nicht unterscheidbaren Teilchen & daher wurden auchkeine Nummern sondern Kreuze ver-wendet & können auf 6 erkennbarunterschiedliche Arten auf die 4 Funk-tionen des entarteten Niveaus verteilt

- 105 -
jwerden. Für die allgemeine Berechnung wird die Gleichung für die Zahl der Kombinationen von g
jElementen zur N -ten Klasse herangezogen.
(13.2.1)
jwobei der vordere Teil dieser Gleichung die Definition der linken Seite der Gleichung darstellt, die "g
jüber N " gelesen wird. Aus dem letzten Teil der Gleichung, die aus dem mittleren Teil durch Er-
j jweiterung mit (g - N )! entsteht, kann man auch die Gültigkeit für dieses Problem erkennen: Es ist die
j j j jZahl der möglichen Permutationen von g zu berechnen (g !). Dabei sind die Permutationen N ! der N
j jbesetzten Niveaus und die der freien Niveaus ((g - N )!) zu streichen, da zwischen diesen nicht mehrunterschieden werden kann. Für alle Niveaus gilt daher:
(13.2.2)
Der weitere Gang der Berechnung erfolgt wie bei der Herleitung der Gleichung für das kanonischeEnsemble. Die oben angekündigte Schwierigkeit besteht darin, dass bei der Berechnung die Stirling-
j j jsche Näherungsformel verwendet werden muss. Da die g oft klein sind und N nur zwischen 0 und gschwanken kann, ist die Verwendung der nur für sehr große Zahlen gültigen Stirlingschen Näherungs-formel an und für sich unzulässig. Das Argument für die doch erfolgende Verwendung ist dasfolgende: Bei allen Gasen (Translationsniveaus), Elektronen in Metallen und Photonen (die beidenletzteren Systeme sollen später genauer diskutiert werden) ist die Niveaudichte so extrem hoch, dass
jviele Niveaus zu einem fiktiven Niveau mit einem g -Wert zusammengefasst werden dürfen, für das
j jdann g und N entsprechend größer sind. Nach erfolgter Berechnung wird diese Zusammenfassungwieder rückgängig gemacht.Es folgt die eigentliche Durchrechnung, wobei auf die Kommentare zur Rechnung in Kap. 4 verwiesenwird.
(13.2.3)
(13.2.4)
Die Nebenbedingungen
(13.2.5)
(13.2.6)
werden dazuaddiert und das Extremum gesucht
(13.2.7)
(13.2.8)
(13.2.9)

- 106 -
(13.2.10)
(13.2.11)
(13.2.12)
(13.2.13)
Wie in Kap. 4 darf $ durch 1/kT ersetzt werden
(13.2.14)
Der zweite Lagrangesche Multiplikator kann hier nun nicht mehr so einfach wie in Kap. 4 mit derGesamtteilchenzahl in Verbindung gebracht werden. Es ist einfacher " durch das chemische Potenzialauszudrücken. Dazu wird von der allgemein gültigen Gl. (5.3.20) in Differenzialschreibweiseausgegangen
(13.2.15)
dlnW wird aus der Extremalbedingung und den etwas anders formulierten Nebenbedingungen (13.2.5und 13.2.6)
; (13.2.16, 13.2.17)
gewonnen
(13.2.18)
oder
(13.2.19)
Das chemische Potenzial ist nun
(13.2.20)
Damit folgt
(13.2.21)
Das chemische Potenzial der Fermionen ist keine Konstante für ein bestimmtes System, sondern hängtvon den äußeren Bedingungen ab (siehe z. B. Gl. (13.2.1.9)). Die endgültige Form der Fermi-Dirac-Verteilung lautet daher
(13.2.22)
Wären wir dagegen von unterscheidbaren Teilchen ausgegangen, die keiner Einschränkung bei derNiveaubesetzung unterliegen, so lautete die Ausgangsgleichung

- 107 -
(13.2.23)
Diese Gleichung kommt wie folgt zustande: N! stellt die Zahl der Anordnungsmöglichkeiten von N
j j junterscheidbaren Teilchen dar. g gibt die Zahl der Variationen von g Elementen zur N -ten KlasseNj
j jmit Wiederholungen an. Dieses soll an einem Beispiel verdeutlicht werden: g sei 3 und N sei 2. DieNummern in der Tabelle stellen die unterscheidbaren Teilchen dar.
Funktion 1 2 3
Realisierung 1 1,2
Realisierung 2 1 2
Realisierung 3 1 2
Realisierung 4 2 1
Realisierung 5 1,2
Realisierung 6 1 2
Realisierung 7 2 1
Realisierung 8 2 1
Realisierung 9 1,2
Es gibt daher 3 = 9 Realisierungsmöglichkeiten. Die Realisierungen mit den Besetzungen 1,22
jenthalten die erlaubten Mehrfachbesetzungen; sie entsprechen den Wiederholungen der g Elemente.
jMan sieht nun relativ einfach, dass der Term g in Ordnung ist, da jedes dazukommende TeilchenNj
j jauf die g Funktionen gesetzt werden kann und dadurch eine Multiplikation mit g bewirkt. Schließlich
j jist noch durch die Permutationen der N zu teilen, da diese in N! und g enthalten sind. Rechnet manNj
diese Statistik bis zum Ende durch (siehe Anhang 14.4), so folgt
(13.2.24)
Das entspricht der Boltzmannverteilung (4.23), wenn hier noch " durch die Gesamtteilchenzahlausgedrückt wird.Der in Kap. 4 benutzte Ansatz für das kanonische Ensemble entspricht bei Betrachtung eines Systemsohne Entartung
(13.2.25)
Die Entartung wurde dann später in Gl. (4.22) bzw. (4.23) eingeführt. Das in Kap. 4 verwendeteVerfahren ist einfacher; es wurde hier nicht angewendet, da bei der entsprechenden Herleitung derFermi-Dirac- und Bose-Einstein-Statistik die Entartung von Anfang an berücksichtigt werden muss.Der Unterschied zwischen den Gl. (13.2.14) und (13.2.24) besteht im Fehlen der 1 im Nenner. Die
jbeiden Gleichungen gehen ineinander über für genügend große Werte von exp((g - :)/kT), d. h. beikleinen Besetzungsdichten. Dieses ist der Fall bei kleinen Dichten und hohen Temperaturen. DieBoltzmannstatistik stellt daher den Grenzfall der FD-Statistik für geringe Besetzungsdichten dar.
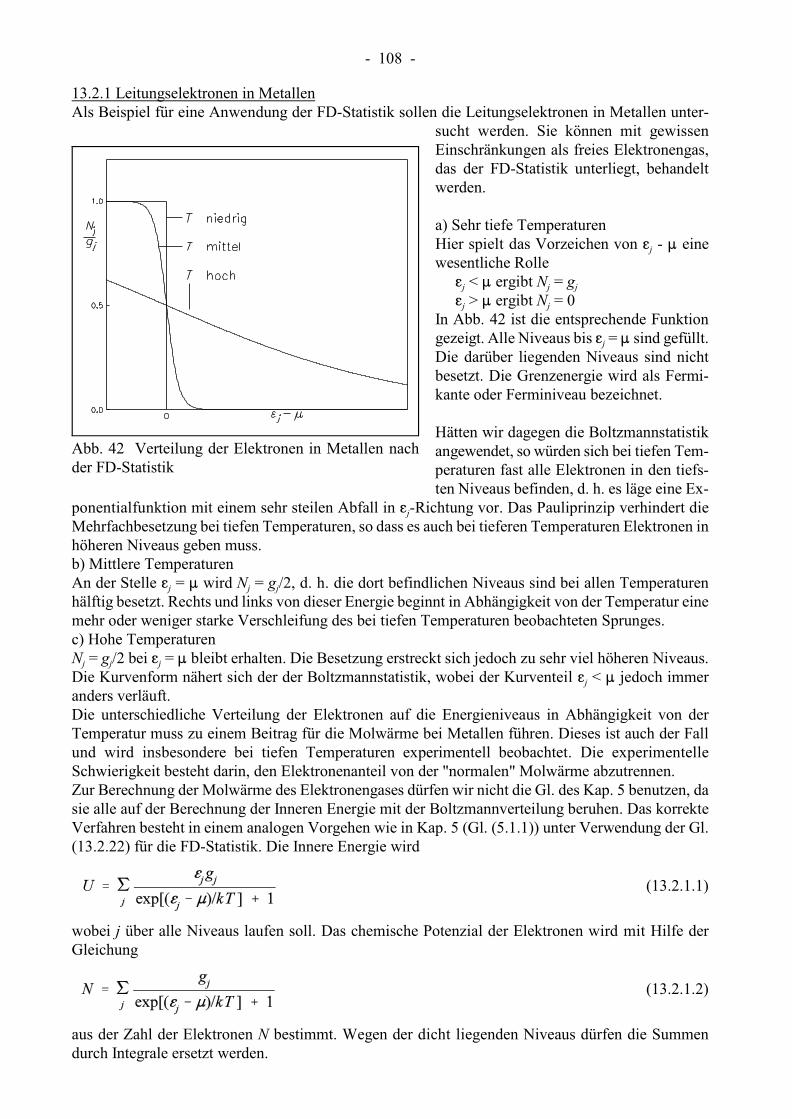
- 108 -
Abb. 42 Verteilung der Elektronen in Metallen nachder FD-Statistik
13.2.1 Leitungselektronen in MetallenAls Beispiel für eine Anwendung der FD-Statistik sollen die Leitungselektronen in Metallen unter-
sucht werden. Sie können mit gewissenEinschränkungen als freies Elektronengas,das der FD-Statistik unterliegt, behandeltwerden.
a) Sehr tiefe Temperaturen
jHier spielt das Vorzeichen von g - : einewesentliche Rolle
j j j g < : ergibt N = g
j j g > : ergibt N = 0In Abb. 42 ist die entsprechende Funktion
jgezeigt. Alle Niveaus bis g = : sind gefüllt.Die darüber liegenden Niveaus sind nichtbesetzt. Die Grenzenergie wird als Fermi-kante oder Ferminiveau bezeichnet.
Hätten wir dagegen die Boltzmannstatistikangewendet, so würden sich bei tiefen Tem-peraturen fast alle Elektronen in den tiefs-ten Niveaus befinden, d. h. es läge eine Ex-
jponentialfunktion mit einem sehr steilen Abfall in g -Richtung vor. Das Pauliprinzip verhindert dieMehrfachbesetzung bei tiefen Temperaturen, so dass es auch bei tieferen Temperaturen Elektronen inhöheren Niveaus geben muss.b) Mittlere Temperaturen
j j jAn der Stelle g = : wird N = g /2, d. h. die dort befindlichen Niveaus sind bei allen Temperaturenhälftig besetzt. Rechts und links von dieser Energie beginnt in Abhängigkeit von der Temperatur einemehr oder weniger starke Verschleifung des bei tiefen Temperaturen beobachteten Sprunges.c) Hohe Temperaturen
j j jN = g /2 bei g = : bleibt erhalten. Die Besetzung erstreckt sich jedoch zu sehr viel höheren Niveaus.
jDie Kurvenform nähert sich der der Boltzmannstatistik, wobei der Kurventeil g < : jedoch immeranders verläuft.Die unterschiedliche Verteilung der Elektronen auf die Energieniveaus in Abhängigkeit von derTemperatur muss zu einem Beitrag für die Molwärme bei Metallen führen. Dieses ist auch der Fallund wird insbesondere bei tiefen Temperaturen experimentell beobachtet. Die experimentelleSchwierigkeit besteht darin, den Elektronenanteil von der "normalen" Molwärme abzutrennen.Zur Berechnung der Molwärme des Elektronengases dürfen wir nicht die Gl. des Kap. 5 benutzen, dasie alle auf der Berechnung der Inneren Energie mit der Boltzmannverteilung beruhen. Das korrekteVerfahren besteht in einem analogen Vorgehen wie in Kap. 5 (Gl. (5.1.1)) unter Verwendung der Gl.(13.2.22) für die FD-Statistik. Die Innere Energie wird
(13.2.1.1)
wobei j über alle Niveaus laufen soll. Das chemische Potenzial der Elektronen wird mit Hilfe derGleichung
(13.2.1.2)
aus der Zahl der Elektronen N bestimmt. Wegen der dicht liegenden Niveaus dürfen die Summendurch Integrale ersetzt werden.

- 109 -
(13.2.1.3)
(13.2.1.4)
wobei D(g) die Zustandsdichte der Niveaus beim Niveau g beschreibt und der Faktor 2 vor denIntegralen durch die Doppelbesetzung der Niveaus mit s = ±½ bewirkt wird.Die Berechnung des Integrals (13.2.1.3) ist leider sehr aufwändig (siehe z. B. Reif, Statistische Physikund Theorie der Wärme, Kap. Leitungselektronen in Metallen). Die Berechnung unter Verwendungvon (13.2.1.4) ergibt
(13.2.1.5)
owobei der Index o sich auf T = 0 K bezieht, d. h. U ist die Innere Energie der Elektronen bei 0 K und
oD(: ) ist die Zustandsdichte der Elektronen beim Ferminiveau bei 0 K. Die Abhängigkeit der Inneren
o oEnergie von der Zahl der Elektronen steckt in U und D(: ).Zur Berechnung der Energie des Ferminiveaus bei 0 K betrachten wir das Elektronengas als einSystem von freien Teilchen in einem Kasten. Die Zahl der Niveaus bis zu einer Energie , war bereitsin Kap. 6 in Gl. (6.30) berechnet worden. Diese Gleichung wird mit einem Faktor 2 für die beidenSpineinstellungen übernommen.
(13.2.1.6)
Bei T = 0 K sind die Niveaus bis zum Ferminiveau mit den N Elektronen besetzt
(13.2.1.7)
(13.2.1.8)
Die Energie des Ferminiveaus ergibt sich daher zu
(13.2.1.9)
und hängt von der Elektronendichte N/V ab. Für Kupfer findet man bei 0 K mit D � 9000 kg/m3
o FRechnet man das mit : = kT in eine Temperatur, die Fermi-Temperatur T , um, so findet man
FT = 82000 K!! Selbst bei 0 K sind daher wegen des Pauli-Prinzips die Energieniveaus bis zu einer82000 K entsprechenden Höhe vollständig besetzt. Die zusätzlichen 300 K bei Raumtemperaturführen dementsprechend nur zu einer geringfügigen Änderung in der Verteilung. Die Zustandsdichte D(g) - nicht zu verwechseln mit der Energiedichte D bei der Planckschen Formel -wird aus Gleichung (13.2.1.6) durch Bildung des Differenzials berechnet, wobei der Faktor 2 für dieSpins entfällt.

- 110 -
(13.2.1.10)
Die Zahl dN der Niveaus zwischen g und g + dg beträgt:
(13.2.1.11)
oder
(13.2.1.12)
oDie Zustandsdichte bei : ist daher
(13.2.1.13)
Schließlich wird noch das Volumen mit (13.2.1.9) eliminiert
(13.2.1.14)
(13.2.1.15)
(13.2.1.16)
Damit wird die Innere Energie
(13.2.1.17)
(13.2.1.18)
und die Molwärme ergibt sich zu
(13.2.1.19)
Die Boltzmannstatistik hätte dagegen den erheblich größeren Wert von 3R/2 vorhergesagt, der imGegensatz zu Gl. (13.2.1.19) nicht von der Temperatur abhängt.
Die gesamte Molwärme von Metallen unter Einschluß des Debyeschen T -Gesetzes beträgt daher bei3
tiefen Temperaturen
(13.2.1.20)
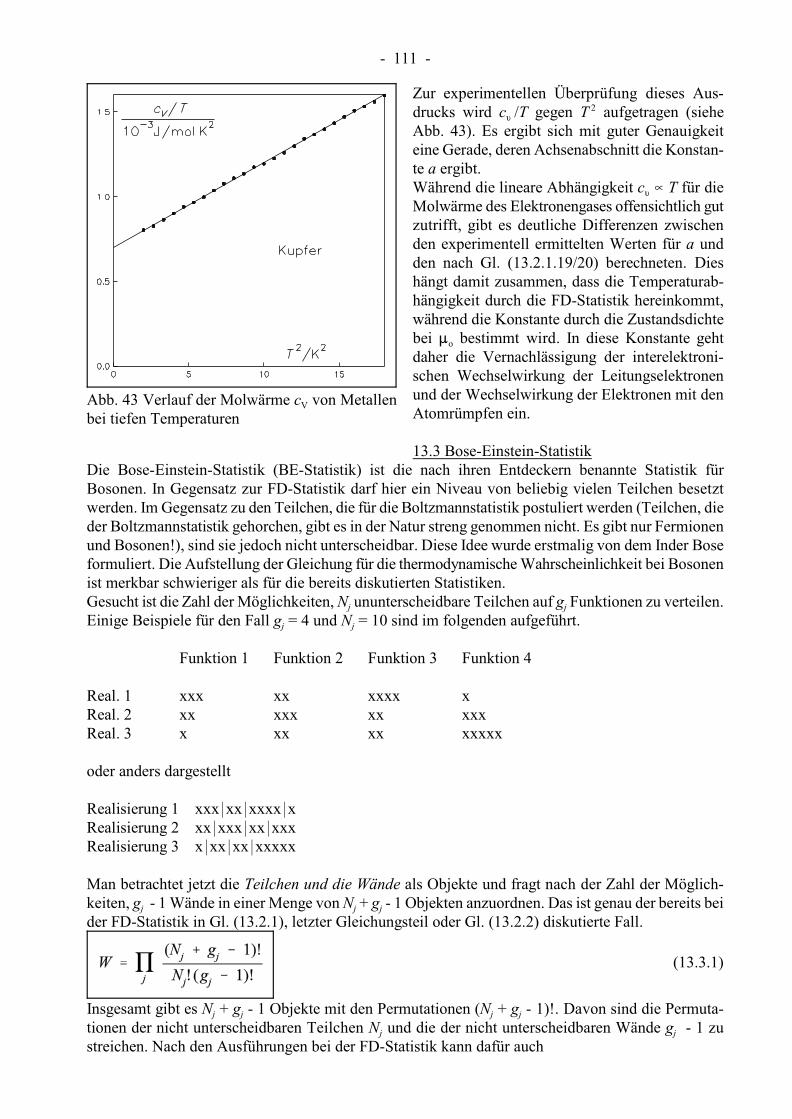
- 111 -
VAbb. 43 Verlauf der Molwärme c von Metallenbei tiefen Temperaturen
Zur experimentellen Überprüfung dieses Aus-
Ldrucks wird c /T gegen T aufgetragen (siehe2
Abb. 43). Es ergibt sich mit guter Genauigkeiteine Gerade, deren Achsenabschnitt die Konstan-te a ergibt.
LWährend die lineare Abhängigkeit c % T für dieMolwärme des Elektronengases offensichtlich gutzutrifft, gibt es deutliche Differenzen zwischenden experimentell ermittelten Werten für a undden nach Gl. (13.2.1.19/20) berechneten. Dieshängt damit zusammen, dass die Temperaturab-hängigkeit durch die FD-Statistik hereinkommt,während die Konstante durch die Zustandsdichte
obei : bestimmt wird. In diese Konstante gehtdaher die Vernachlässigung der interelektroni-schen Wechselwirkung der Leitungselektronenund der Wechselwirkung der Elektronen mit denAtomrümpfen ein.
13.3 Bose-Einstein-StatistikDie Bose-Einstein-Statistik (BE-Statistik) ist die nach ihren Entdeckern benannte Statistik fürBosonen. In Gegensatz zur FD-Statistik darf hier ein Niveau von beliebig vielen Teilchen besetztwerden. Im Gegensatz zu den Teilchen, die für die Boltzmannstatistik postuliert werden (Teilchen, dieder Boltzmannstatistik gehorchen, gibt es in der Natur streng genommen nicht. Es gibt nur Fermionenund Bosonen!), sind sie jedoch nicht unterscheidbar. Diese Idee wurde erstmalig von dem Inder Boseformuliert. Die Aufstellung der Gleichung für die thermodynamische Wahrscheinlichkeit bei Bosonenist merkbar schwieriger als für die bereits diskutierten Statistiken.
j jGesucht ist die Zahl der Möglichkeiten, N ununterscheidbare Teilchen auf g Funktionen zu verteilen.
j jEinige Beispiele für den Fall g = 4 und N = 10 sind im folgenden aufgeführt.
Funktion 1 Funktion 2 Funktion 3 Funktion 4
Real. 1 xxx xx xxxx xReal. 2 xx xxx xx xxxReal. 3 x xx xx xxxxx
oder anders dargestellt
Realisierung 1 xxx*xx*xxxx*xRealisierung 2 xx*xxx*xx*xxxRealisierung 3 x*xx*xx*xxxxx
Man betrachtet jetzt die Teilchen und die Wände als Objekte und fragt nach der Zahl der Möglich-
j j jkeiten, g - 1 Wände in einer Menge von N + g - 1 Objekten anzuordnen. Das ist genau der bereits beider FD-Statistik in Gl. (13.2.1), letzter Gleichungsteil oder Gl. (13.2.2) diskutierte Fall.
(13.3.1)
j j j jInsgesamt gibt es N + g - 1 Objekte mit den Permutationen (N + g - 1)!. Davon sind die Permuta-
j jtionen der nicht unterscheidbaren Teilchen N und die der nicht unterscheidbaren Wände g - 1 zustreichen. Nach den Ausführungen bei der FD-Statistik kann dafür auch
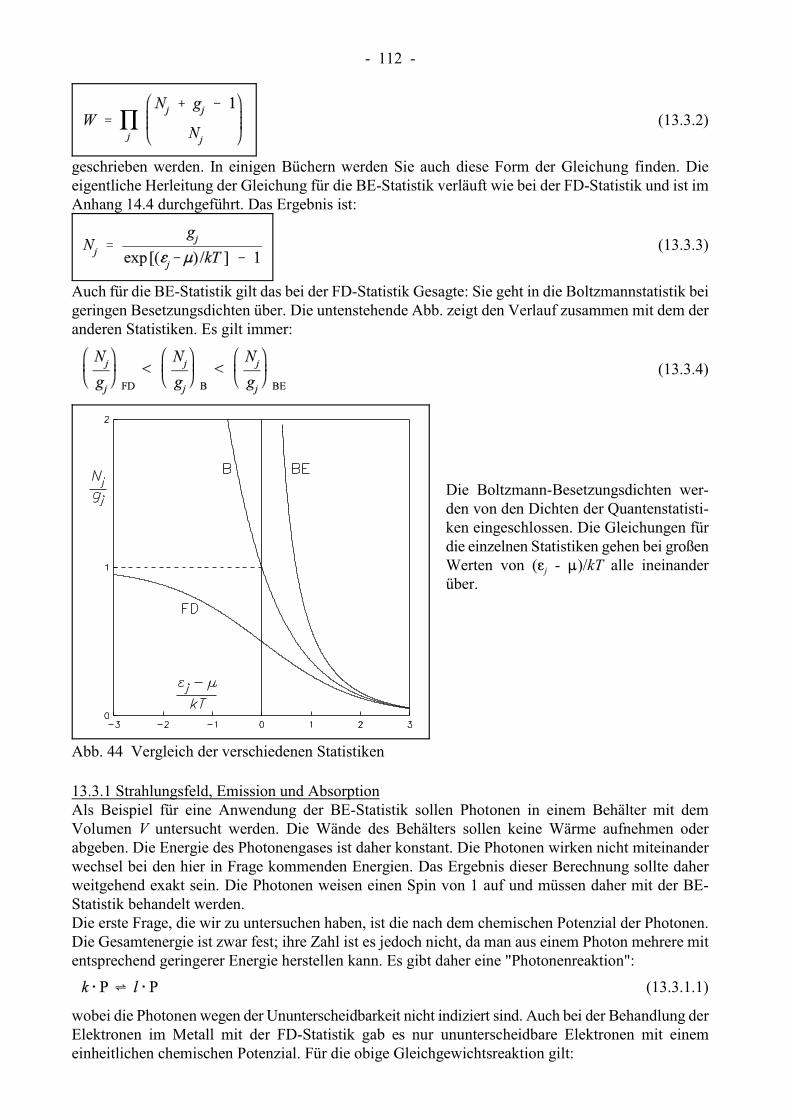
- 112 -
Abb. 44 Vergleich der verschiedenen Statistiken
(13.3.2)
geschrieben werden. In einigen Büchern werden Sie auch diese Form der Gleichung finden. Dieeigentliche Herleitung der Gleichung für die BE-Statistik verläuft wie bei der FD-Statistik und ist imAnhang 14.4 durchgeführt. Das Ergebnis ist:
(13.3.3)
Auch für die BE-Statistik gilt das bei der FD-Statistik Gesagte: Sie geht in die Boltzmannstatistik beigeringen Besetzungsdichten über. Die untenstehende Abb. zeigt den Verlauf zusammen mit dem deranderen Statistiken. Es gilt immer:
(13.3.4)
Die Boltzmann-Besetzungsdichten wer-den von den Dichten der Quantenstatisti-ken eingeschlossen. Die Gleichungen fürdie einzelnen Statistiken gehen bei großen
jWerten von (g - :)/kT alle ineinanderüber.
13.3.1 Strahlungsfeld, Emission und AbsorptionAls Beispiel für eine Anwendung der BE-Statistik sollen Photonen in einem Behälter mit demVolumen V untersucht werden. Die Wände des Behälters sollen keine Wärme aufnehmen oderabgeben. Die Energie des Photonengases ist daher konstant. Die Photonen wirken nicht miteinanderwechsel bei den hier in Frage kommenden Energien. Das Ergebnis dieser Berechnung sollte daherweitgehend exakt sein. Die Photonen weisen einen Spin von 1 auf und müssen daher mit der BE-Statistik behandelt werden.Die erste Frage, die wir zu untersuchen haben, ist die nach dem chemischen Potenzial der Photonen.Die Gesamtenergie ist zwar fest; ihre Zahl ist es jedoch nicht, da man aus einem Photon mehrere mitentsprechend geringerer Energie herstellen kann. Es gibt daher eine "Photonenreaktion":
(13.3.1.1)
wobei die Photonen wegen der Ununterscheidbarkeit nicht indiziert sind. Auch bei der Behandlung derElektronen im Metall mit der FD-Statistik gab es nur ununterscheidbare Elektronen mit einemeinheitlichen chemischen Potenzial. Für die obige Gleichgewichtsreaktion gilt:

- 113 -
(13.3.1.2)
d. h. das chemische Potenzial der Elektronen beträgt Null. Eine andere, etwas elegantere Sicht ist diefolgende. Da die Photonenzahl nicht konstant ist, gibt es die Nebenbedingung dN = 0 bei der Herlei-tung der BE-Statistik für die Photonen nicht. Dementsprechend wird der Lagrangesche Multiplikator" nicht benötigt, so dass das chemische Potenzial in der Endgleichung überhaupt nicht auftritt. Die BE-Gleichung für den vorliegenden Fall wird daher:
(13.3.1.3)
jDie Energien , entsprechen den Photonenenergien h<. Die erlaubten Werte für die Frequenzen < kannman mit dem gleichen Formalismus berechnen wie die Schwingungen in einem Kristall in Kap. 10.2.Die Photonen sind eine kurzwellige elektromagnetische Strahlung. Es bilden sich daher stehendeelektromagnetische Wellen in dem Behälter aus, die bezüglich der Berechnung der Frequenzdichte mitdem gleichen Formalismus wie die mechanischen Schwingungen in einem Kristall behandelt werdendürfen. Ähnlich wie bei den Schwingungen in einem Kristall ist von Gl. (10.2.17) ausgehend eineMultiplikation mit dem Faktor 2 durchzuführen, um die beiden unabhängigen Polarisationsrichtungenzu berücksichtigen. Weiterhin wird die Geschwindigkeit L durch die Lichtgeschwindigkeit c ersetzt.
(13.3.1.4)
Man beachte: Zur Berechnung der Frequenzdichte haben wir das Photonengas auf die Translations-niveaus des Behältervolumens gesetzt. Zur Berechnung der Photonenenergie muss jedoch die Energieh< des Photons verwandt werden!Als nächstes wird diese Dichtefunktion in die BE-Statistik eingesetzt.
(13.3.1.5)
Weiterhin soll die Energiedichte berechnet werden.
(13.3.1.6)
(13.3.1.7)
Das ist die berühmte Plancksche Strahlungsformel für die Energiedichte in einem Hohlraum! Sie gibtin der hier dargestellten Form und in Abb. 45 die Energiedichte pro Frequenzintervall an. AndereDarstellungen ergeben die Energiedichte pro Wellenlängenintervall oder die Photonendichte (siehe(13.3.1.5)). Dies führt zu Verschiebungen des Maximums. Hohe Temperaturen ergeben eine Be-vorzugung der hohen Frequenzen bzw. kurzen Wellenlängen. Dies führt zu einer Verschiebung desMaximums zu hohen Frequenzen (Wiensches Verschiebungsgesetz) und zu einer stark erhöhtenGesamtenergiedichte (Stefan-Boltzmannsches Gesetz) mit steigender Temperatur. Letzteres sollhergeleitet werden.
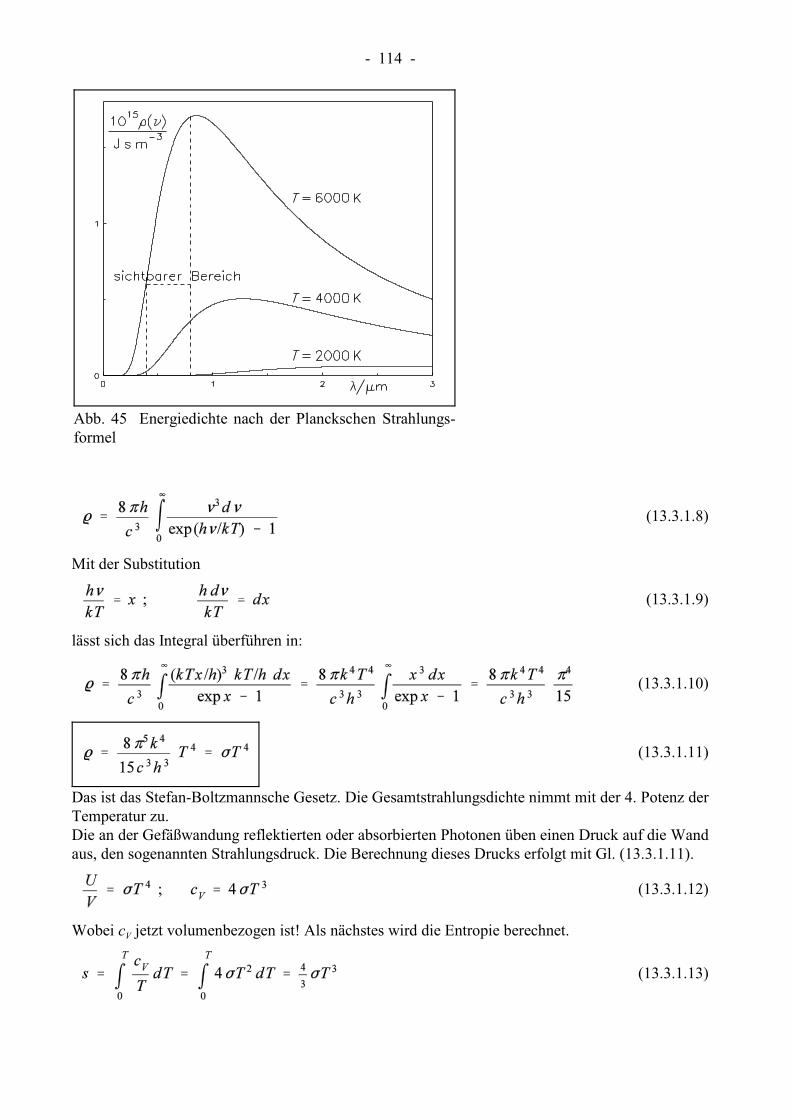
- 114 -
Abb. 45 Energiedichte nach der Planckschen Strahlungs-formel
(13.3.1.8)
Mit der Substitution
(13.3.1.9)
lässt sich das Integral überführen in:
(13.3.1.10)
(13.3.1.11)
Das ist das Stefan-Boltzmannsche Gesetz. Die Gesamtstrahlungsdichte nimmt mit der 4. Potenz derTemperatur zu.Die an der Gefäßwandung reflektierten oder absorbierten Photonen üben einen Druck auf die Wandaus, den sogenannten Strahlungsdruck. Die Berechnung dieses Drucks erfolgt mit Gl. (13.3.1.11).
(13.3.1.12)
VWobei c jetzt volumenbezogen ist! Als nächstes wird die Entropie berechnet.
(13.3.1.13)

- 115 -
(13.3.1.14)
(13.3.1.15)
Nun gilt für eine reine Komponente
(13.3.1.16)
d. h. mit Hilfe der beiden letzten Gleichungen kann der Druck berechnet werden.
(13.3.1.17)
oder
(13.3.1.18)
Vergleicht man das mit dem Druck eines idealen, einatomigen Gases, so findet man
(13.3.1.19)
(13.3.1.20)
Die Drücke des Photonengases und eines idealen, einatomigen Gases sind daher bei gleicher Energie-dichte vergleichbar. Wegen der unterschiedlichen Abhängigkeit der Energiedichte von der Temperaturnimmt der Strahlungsdruck mit steigender Temperatur erheblich stärker zu als der Druck des idealenGases. Für 10 K ergibt sich für den Strahlungsdruck folgender Wert:6
(13.3.1.21)
(13.3.1.22)
Beobachtbar ist der Strahlungsdruck bei Kometenschweifen, die aus vom Kometen abdampfendenGasen bestehen. Sie werden durch den Druck der von der Sonne ausgehenden Strahlung in diesonnenabgewandte Richtung getrieben. Bei Sternen, wie z. B. unserer Sonne mit Temperaturen voneinigen 10 K im Inneren, wird ein Teil des Drucks im Sterninneren, der ein Kollabieren des Sterns7
verhindert, durch den Strahlungsdruck aufgebracht.Um die aus einer kleinen Öffnung im Hohlraum oder von einer schwarzen Fläche emittierte Strahlungzu berechnen, wird wie folgt vorgegangen. Nach den Überlegungen bei der Maxwellschen Ge-schwindigkeitsverteilung ist die Stoßzahl auf die Einheitsfläche
(13.3.1.23)
Diese Gleichung wenden wir jetzt auf die Photonen an. Die Photonendichte N wird aus der Energie-1
Wdichte durch Division durch h< erhalten oder direkt aus (13.3.1.5). Da wir aber später aus Z dieemittierte Strahlung berechnen wollen, ist anschließend wieder mit h< zu multiplizieren. Es verbleibtdaher nur der Faktor L6/4 = c/4, d. h. für die in den Halbraum emittierte Strahlung(Energie/(Fläche@Zeit)) gilt:

- 116 -
(13.3.1.24)
und das Integral über die gesamte Strahlung beträgt
(13.3.1.25)
Die Thermodynamik reguliert noch eine Reihe weiterer Dinge bei der Strahlung. Nicht alle Körpersind schwarz, d. h. absorbieren die Strahlung vollständig. Es wird daher der Absorptionsgrad A undder Reflexionsgrad R definiert
(13.3.1.26)
(13.3.1.27)
o A Rwobei I die einfallende, I die absorbierte und I die reflektierte Strahlungsintensität darstellen. Stelltman nun zwei gleich große Flächen mit unterschiedlichen Temperaturen gegenüber, wobei freieÖffnungen zwischen den Flächen durch ideale Spiegel verschlossen werden und die gesamte Appara-tur von der Umgebung isoliert wird, so muss nach dem 2. Hauptsatz ein Ausgleich der Temperaturenerfolgen. Im Gleichgewichtszustand ist die Energiebilanz der Flächen
Fläche Emittierte Str. Absorbierte Str. Reflektierte Str. Gesamte abgegebene Str.
1 2 1 2 1 1 2 11 Q Q A Q (1 - A ) Q + Q (1 - A )
2 1 2 1 2 2 1 22 Q Q A Q (1 - A ) Q + Q (1 - A )
Im Gleichgewicht muss daher
(13.3.1.28)
gelten. Oder
(13.3.1.29)
d. h. für alle Körper ist bei gegebener Temperatur das Verhältnis von emittierter zu absorbierter
2 Strahlung eine Konstante. Ist die Fläche 2 eine schwarze Fläche, so ist A = 1, und es gilt: DasVerhältnis von emittierter zu absorbierter Strahlung entspricht der bei gleicher Temperatur vomschwarzen Körper emittierten Strahlung. Das ist das Kirchhoffsche Gesetz.
Ein weiteres Problem ist die Emission und Absorption von Strahlung in Systemen mit diskreten
1 2Energieniveaus (Einstein, 1917). Wie nehmen ein System mit zwei Energieniveaus g und g an undfragen nach der Zahl der Übergänge zwischen den Niveaus in Gegenwart eines Strahlungsfeldes,dessen Photonen den Übergang zwischen 1 und 2 energetisch gerade ermöglichen. Die Zahl derAbsorptionsprozesse von 1 nach 2 ist proportional zur Energiedichte D(<) ("Lichtintensität") und zur
1Teilchenzahl N in 1.

- 117 -
Abb. 46 Zwei Energieniveaus mit Beset-zungen
(13.3.1.30)
12wobei der Proportionalitätskoeffizient B als Einstein-Koeffizient bezeichnet wird. Ähnlich gilt für den Über-gang von 2 nach 1
(13.3.1.31)
Diese Vorgänge werden als induzierte Emissionen be-zeichnet. Bei der induzierten Emission erfolgt die Emis-sion von Strahlung kohärent, d. h. phasengleich, zumvorhandenen Strahlungsfeld. Dieses Verhalten lässt sich
in einfacher Weise als Emission von Strahlung eines im Strahlungsfeld phasengleich schwingendenEmitters verstehen. Für die Funktion eines Lasers ist diese Eigenschaft von zentraler Wichtigkeit.Neben der induzierten Emission gibt es noch die spontane Emission, die unabhängig vom Strahlungs-feld erfolgt und daher auch nicht kohärent ist (kohärent wozu bei D = 0?). Der entsprechende Koeffi-
21zient der spontanen Emission wird mit A bezeichnet.
(13.3.1.32)
2Zusammengefasst ergibt sich auf N bezogen
(13.3.1.33)
Im Gleichgewicht muss daher gelten:
(13.3.1.34)
2 1Das Verhältnis N /N lässt sich mit der Boltzmannstatistik berechnen (die Anwendung der Quanten-statistik ist unnötig).
(13.3.1.35)
wobei < die für den Übergang notwendige Frequenz der Strahlung darstellt. Daraus folgt:
(13.3.1.36)
Die Gleichung wird nach D aufgelöst und mit der Planckschen Strahlungsformel verglichen.
(13.3.1.37)
(13.3.1.38)
Der Vergleich mit (13.3.1.7) ergibt
(13.3.1.39)
Wir stellen daher fest: Die Einstein-Koeffizienten für induzierte Emission und Absorption sind gleich.Der erste Teil von (13.3.1.39) gestattet, des Verhältnis der Koeffizienten der spontanen zur induzier-ten Emission zu berechnen. Die Absolutwerte der Koeffizienten können mit Hilfe der Statistischen

- 118 -
Abb. 47 Molwärme von Helium bei tiefen Tempe-raturen
Thermodynamik nicht berechnet werden.; hierzu muss die Quantenmechanik bemüht werden.
13.3.2 Superfluidität von HeliumNach der langen Ausführung über das Strah-lungsfeld soll noch kurz über ein weiteres Phä-nomen berichtet werden, das bei Bosonen be-obachtet wird. Kühlt man He ab, so geht es4
bei 4,2 K in den flüssigen Zustand über. EineÜberführung in den festen Zustand ist bis 0 Kbei 1 bar nicht möglich und gelingt nur beihöheren Drücken. Helium bleibt daher unterNormaldruck bis 0 K flüssig. Bei 2,2 K wirdjedoch eine weitere Umwandlung beobachtet,wobei die Umwandlung selbst als auch dieentstehende Tieftemperaturphase außerge-wöhnliche Eigenschaften aufweisen.
Untersucht man Helium in diesem Tempera-turbereich kalorimetrisch (siehe Abb. 47), sostellt man einen starken Anstieg der Molwärme
Lc in der Nähe dieser Umwandlung fest, dienach der Form der Kurve auch 8-Umwandlunggenannt wird. Die kalorimetrische Untersu-chung mit sehr viel höherer Temperaturauflö-sung in den :K-Bereich hinein zeigt, dass die-se Umwandlung keine Umwandlungsenthalpie
aufweist. Die Umwandlung erfolgt sozusagen kontinuierlich im Verlauf einiger K; daher auch dieBezeichnung "kontinuierliche Umwandlung".Wodurch unterscheiden sich nun die Hochtemperaturphase He-I und die Tieftemperaturphase He!II?Die He-I-Phase ist ein sich normal verhaltendes flüssiges Helium. Die Untersuchung von He-II ergabdagegen eine Reihe außergewöhnlicher Eigenschaften, an deren erster Stelle die Superfluidität zunennen ist. Versucht man die Viskosität von He-II in einem Kapillarviskosimeter zu messen, so findetman den Wert Null. Sorgfältige Messungen ergaben, dass die Viskosität des flüssigen He-II mindes-tens 10 -fach kleiner als die des gasförmigen Heliums oberhalb des Siedepunkts ist. Wird He-II in11
einem torusförmigen Gefäß zum Strömen gebracht, so klingt diese Strömung selbst nach Tagen nichtin messbarer Weise ab. Dieses Phänomen stellt daher ein mechanisches Analogon zum Stromfluss ineiner supraleitenden Spule dar.Die Erklärung für dieses Phänomen ist in der sog. Bose-Kondensation des Heliums unterhalb 2,2 Kzu suchen. He-Atome sind Bosonen und unterliegen daher der Bose-Einstein-Statistik. Bei genügend4
tiefen Temperaturen ist es daher möglich, eine makroskopische He-Menge in einem quantenmecha-nischen Niveau unterzubringen. Das ist die Bose-Kondensation. Die Berechnung ergibt, dass die Bose-Kondensation aus dem Gas heraus erst bei sehr viel tieferen Temperaturen erfolgen sollte; die durchdie Kondensation bei 4,2 K erfolgende Dichtevergrößerung bringt die Bose-Kondensations-Tempera-tur dann auf 2,2 K. Fließendes superfluides Helium ist daher Helium in einem quantenmechanischenTranslationszustand.Dass die Dinge jedoch nicht ganz so einfach sind, zeigt ein Experiment zur Untersuchung derViskosität in einem Rotationsviskosimeter. Hier findet man normale Viskositätswerte. Zur Erklärungdieser Eigenschaft stellte Tisza das Zweiflüssigkeitsmodell auf. He-II stellt eine Mischung "normalen"Heliums und Bose-kondensierten Heliums dar. Das "normale" Helium kann man auch als Anregungs-zustände des Bose-kondensierten Heliums ansehen. Der Anteil des "normalen" Heliums nimmt mitabnehmender Temperatur bis 0 K auf den Wert 0 ab. Das Strömungsexperiment im Kapillarviskosi-meter bzw. im Torus sieht nur das Bose-kondensierte Helium, unbeeinflusst und ohne Impulsübertrag

- 119 -
vom normalen Helium.Neben vielen anderen außergewöhnlichen Eigenschaften zeigt auch die Wärmeleitfähigkeit einbemerkenswertes Verhalten. Sie wird bei He-II sehr groß; nicht unendlich groß, aber immerhin 100-fach größer als die von Kupfer! Dieses führt zu einer besonderen Form des Siedens von He-II.Normale Flüssigkeiten sieden unter Blasenbildung. Bei He-II ist die Wärmeleitfähigkeit so groß, dassdie Wärme von der Wärmeeintragstelle so schnell abgeführt wird, dass die Temperatur überallhomogen bleibt. Unter diesen Bedingungen erfolgt der Übergang in die Gasphase von der Oberflächeaus, da dort der Druck wegen des verschwindenden hydrostatischen Drucks am geringsten ist. He-IIsiedet daher ohne jegliche Blasenbildung von der Oberfläche aus.
- 120 -
14 Anhänge
14.1 Werte einiger Naturkonstanten
Bezeichnung Symbol Wert
AAvogadrosche Konstante N 6,022 137@10 mol23 -1
Boltzmann-Konstante k 1,380 658@10 J/K-23
Elementarladung e 1,602 177@10 C-19
Faraday-Konstante F 9,648 531@10 C/mol4
eRuhemasse des Elektrons m 9,109 389@10 kg -31
Gaskonstante R 8,314 510 J/K mol
oDielektrizitätskonstante des Vakuums g 8,854 188@10 As/Vm-12
Plancksche Wirkungskonstante h 6,626 075@10 J s -34
Vakuumlichtgeschwindigkeit c 299 792 458 m/s
Nullpunkt der Celsiusskala 273,15 K
Standardtemperatur 25 C; 298,15 K�
Standarddruck 1 atm; 1,013 25@10 Pa5

- 121 -
14.2 Stirlingsche NäherungsformelDie Fakultäten großer Zahlen lassen sich mit der Stirlingschen Näherungsformel berechnen, die sichwie folgt herleiten lässt. Für große Werte von N kann man den Differenzialquotienten von ln N! dementsprechenden Differenzenquotienten mit )N = 1 gleichsetzen.
(14.2.1)
(14.2.2)
Eine bestimmte Integration ergibt:
(14.2.3)
Wird die Integration links ausgeführt und rechts die 1 gestrichen, so resultiert die StirlingscheNäherungsformel
(14.2.4)
Die Stirlingsche Näherungsformel konvergiert schlecht - die nächsten Glieder in der Reihenentwick-lung sind ½lnN und ½ln(2B) - und gilt daher nur für sehr große N. Da sie in der Statistischen Thermo-dynamik öfter verwendet wird, sollen in der folgenden Tabelle einige Zahlenwerte zum Vergleich
relgegenübergestellt werden. ) stellt die Differenz zwischen dem korrekten Wert von ln N! und demnach der Stirlingschen Formeln berechneten bezogen auf den korrekten Wert dar ((Spalte 2 - Spalte3)/Spalte 2). Die Tabelle zeigt die schlechte Annäherung mit wachsendem N.
N ln N! N ln N - N N ln N - N + ½ln NN ln N - N +
½ln N + ½ln (2B) rel)
10 0 -1 -1 -0.1 -0
10 15,1 13,0 14,2 15,1 0,141
10 363,7 360,5 362,8 363,7 0,0092
10 5 912,1 5 907,7 5 911,2 5 912,1 0,000 73
10 82 108,9 82 103,4 82 108,0 82 108,9 0,000 074
Weiterhin könnten noch Zweifel entstehen, ob der Vergleich der Logarithmen der Funktion korrekt ist.Für den Vergleich der Logarithmen spricht, dass die thermodynamischen Größen logarithmisch mitder thermodynamischen Wahrscheinlichkeit zusammenhängen, z. B. S = k lnW (siehe Kap. 5.3). Dadie Zahl der Systeme eines Ensembles in einem Gedankenexperiment beliebig groß gemacht werdenkann, ist die Verwendung der besseren Näherungen unnötig.

- 122 -
L14.3 Berechnung der Molwärme c aus der Zustandssumme entsprechend der Debyeschen TheorieDie Zustandssumme ist nach Gl. (10.2.24)
(14.3.1)
Daraus soll jetzt die Innere Energie berechnet werden.
(14.3.2)
Die Differenziation nach T darf in das Integral hineingezogen werden.
(14.3.3)
Daher gilt:
(14.3.4)
Wie bereits bei anderen Gelegenheiten wird für die Integration mit
(14.3.5)
substituiert. An der oberen Integralgrenze gilt:
(14.3.6)
gesetzt wurde. Weiterhin gilt:
(14.3.7)
so dass das folgende Integral entsteht:

- 123 -
(14.3.8)
(14.3.9)
LDurch Differenziation nach T wird c gewonnen.
(14.3.10)
wobei die Regeln für die Differenziation eines Integrals nach der oberen Integralgrenze verwendetwurden. Es entsteht dabei der Integrand an der oberen Integralgrenze. Falls die obere Integralgrenzenicht direkt die Variable ist, nach der abgeleitet werden soll, ist noch mit der Ableitung der oberenIntegralgrenze nach dieser Variablen zu multiplizieren. Mit einigen Umstellungen findet man für dieMolwärme:
(14.3.11)

- 124 -
14.4 Herleitung der Boltzmann- und Bose-Einstein-Statistik
BoltzmannstatistikDie thermodynamische Wahrscheinlichkeit für ein System, das der Boltzmannstatistik gehorcht, ist inKap. 13.2 hergeleitet worden.
(14.4.1)
Die Herleitung der Boltzmannstatistik erfolgt ähnlich wie die in Kap. 12.2 angegebene Herleitung derFD-Statistik.
(14.4.2)
(14.4.3)
Nebenbedingungen:
(14.4.4)
(14.4.5)
(14.4.6)
(14.4.7)
(14.4.8)
Bose-Einstein-StatistikDie thermodynamische Wahrscheinlichkeit für ein System, das der Boltzmannstatistik gehorcht, ist inKap. 13.2 hergeleitet worden.
(14.4.9)
Wegen der Ähnlichkeit zur vorstehenden Ableitung erfolgt die weitere Herleitung ohne Kommentar.
(14.4.10)
(14.4.11)
(14.4.12)
(14.4.13)

- 125 -
(14.4.14)
(14.4.15)
j jDie 1 im Zähler wird jetzt gegen N + g vernachlässigt (siehe dazu auch die Diskussion zu Gl.(13.2.3).
(14.4.16)
(14.4.17)
(14.4.18)
(14.4.19)
(14.4.20)
(14.4.21)


















