137 1 8 6 9 ? 81 +$8 I= A#5J487 $8 I= A#7J48 7 7$8 I= A#...
4
This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution 4.0 International License. Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung 4.0 Lizenz. Studies on Heterocyclic Enamines: New Synthesis of Pyrano[2,3-b]pyndine, Pyrano[2,3-d]pyrimidine and Pyrano[2,3-c]pyrazole Derivatives Nosrat Mustafa Abed*, Nadia Sobhy Ibrahim, and Mohamed Helmy Elnagdi Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt Z. Naturforsch. 41b, 925-928 (1986); received January 21, 1986 Heterocyclic Enaminonitriles, Trichloroacetonitrile, Pyrano[2,3-c]pyrazole, Ring-Chain Tautomerism in Pyrans A variety of novel pyrano[2,3-d]pyrimidines could be obtained via reaction of ethyl 2-amino- 3-cyano-6-methylpyran-4-carboxylate with a variety of reagents. Evidence for the existance of this pyran derivative as a ring chain tautomer is presented. Although 2-amino-3-cyanopyrans became recently readily obtainable via Soto’s pyran synthesis [1], util ity of these enaminonitriles in heterocyclic synthesis has received only limited interest compared with enaminofurans [2]. In conjunction of our effort di rected toward exploring the synthetic potential of enaminonitriles [3 — 5], we report here a synthesis of a new enaminopyran derivative and the chemistry of this enaminonitrile. Benzylidenemalononitrile 1 reacted with ethyl acetoacetate in refluxing ethanolic triethylamine to yield a 1:1 adduct. This could be proved to be the pyran derivative 2 and not the possible intermediate Michael adduct 3 based on 'H NMR of the reaction product. Thus, *H NMR revealed the absence of sig nals for protons in the region 3—6, other than the methylene quartet and pyran H-4 singlet at (3 4.8 ppm. If the reaction product was the acyclic 3, a multiplet should have appeared at 6 3—6 ppm. In contrast to the observed ready reaction of 2- amino-3-cyano-4,5-dihydrofurans with ethyl cyano- acetate [6], compound 2 was recovered unreacted when treated with ethyl cyanoacetate under the same reaction conditions. However, when 2 was heated at 160 °C (bath T) a product of molecular formula C 19 H 17 N304 was obtained. Two isomeric structures were considered (cf. structures 4 and 5). Structure 4 was eliminated based on IR spectrum which revealed only one CN signal and a highly chelated NH and OH bands extending from 3500 to 2700 cm-1. Moreover, the reaction product failed to couple with * Reprint requests to Dr. N. M. Abed. Verlag der Zeitschrift für Naturforschung, D-7400 Tübingen 0340 - 5087/86/0700 - 0925/$ 01.00/0 benzenediazonium chloride under conditions re ported [7] to effect ready coupling of aryldiazonium salts with cyanoacetamide derivatives. In contrast to ethyl cyanoacetate, malononitrile reacted with 2 in refluxing ethanolic triethylamine to yield an adduct which was formulated as 6 and not 7, 8 or 9 based on IR spectrum which revealed two CN signals. Al though 'H NMR of the reaction product points to structure 6 it can be also interpreted for 7 or 8 . Thus, other evidence to support form 6 was needed. Since the reaction product failed to couple with ben zenediazonium chloride, structure 6 was considered most likely, as 7 or 8 is expected to couple readily with aryldiazonium salts [7], Compound 6 could be converted into 9 on long reflux in dioxane. Com pound 9 was also obtained from fusion of 2 with malononitrile at 160 °C for 2 h. Compound 2 reacted with trichloroacetonitrile to yield the pyranopyrimidine derivative 10. The forma tion of 10 from reaction of 2 with trichloroacetonitrile is a further example of the usefullness of the new ly reported synthesis of pyrimidine from enamino nitriles [8 , 9]. However, the limitations of this reac tion recently reported by Gewald and Hain should be considered [ 10], Compound 2 also reacted with ethyl 3-amino- 2-cyano-4-trichlorocrotonate to yield pyrano[2,3-d]- pyrimidine derivative 11. Compound 2 reacted with benzoylisothiocyanate to yield the pyranopyrimidine derivative 12 , formed most likely via intermediacy of the thiourea deriva tive 13. In contrast, ethoxycarbonylisothiocyanate reacted with 2 to yield the ethoxycarbonylaminopy- ran 14. Ethoxycarbonylation of aminoheterocycles by this reagent has been previously reported [8 , 11 ]
Transcript of 137 1 8 6 9 ? 81 +$8 I= A#5J487 $8 I= A#7J48 7 7$8 I= A#...

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Studies on Heterocyclic Enamines: New Synthesis of Pyrano[2,3-b]pyndine, Pyrano[2,3-d]pyrimidine and Pyrano[2,3-c]pyrazole DerivativesNosrat Mustafa Abed*, Nadia Sobhy Ibrahim, and Mohamed Helmy ElnagdiD epartm ent of Chemistry, Faculty of Science, Cairo University, Giza, Egypt
Z. Naturforsch. 41b, 925-928 (1986); received January 21, 1986
Heterocyclic Enaminonitriles, Trichloroacetonitrile, Pyrano[2,3-c]pyrazole,Ring-Chain Tautomerism in Pyrans
A variety of novel pyrano[2,3-d]pyrimidines could be obtained via reaction of ethyl 2-amino-3-cyano-6-methylpyran-4-carboxylate with a variety of reagents. Evidence for the existance of this pyran derivative as a ring chain tautom er is presented.
Although 2-amino-3-cyanopyrans became recently readily obtainable via Soto’s pyran synthesis [1], utility of these enaminonitriles in heterocyclic synthesis has received only limited interest compared with enaminofurans [2]. In conjunction of our effort directed toward exploring the synthetic potential of enaminonitriles [3 — 5], we report here a synthesis of a new enaminopyran derivative and the chemistry of this enaminonitrile.
Benzylidenemalononitrile 1 reacted with ethyl acetoacetate in refluxing ethanolic triethylamine to yield a 1:1 adduct. This could be proved to be the pyran derivative 2 and not the possible intermediate Michael adduct 3 based on 'H NMR of the reaction product. Thus, *H NMR revealed the absence of signals for protons in the region 3—6, other than the methylene quartet and pyran H-4 singlet at (34.8 ppm. If the reaction product was the acyclic 3, a multiplet should have appeared at 6 3—6 ppm.
In contrast to the observed ready reaction of 2- amino-3-cyano-4,5-dihydrofurans with ethyl cyano- acetate [6], compound 2 was recovered unreacted when treated with ethyl cyanoacetate under the same reaction conditions. However, when 2 was heated at 160 °C (bath T) a product of molecular formula C19H 17N304 was obtained. Two isomeric structures were considered (cf. structures 4 and 5). Structure 4 was eliminated based on IR spectrum which revealed only one CN signal and a highly chelated NH and OH bands extending from 3500 to 2700 cm-1. M oreover, the reaction product failed to couple with
* Reprint requests to Dr. N. M. Abed.
V erlag de r Z eitschrift für N aturforschung, D-7400 Tübingen0340 - 5087/86/0700 - 0925/$ 01.00/0
benzenediazonium chloride under conditions reported [7] to effect ready coupling of aryldiazonium salts with cyanoacetamide derivatives. In contrast to ethyl cyanoacetate, malononitrile reacted with 2 in refluxing ethanolic triethylamine to yield an adduct which was formulated as 6 and not 7, 8 or 9 based on IR spectrum which revealed two CN signals. Although 'H NMR of the reaction product points to structure 6 it can be also interpreted for 7 or 8 . Thus, other evidence to support form 6 was needed. Since the reaction product failed to couple with benzenediazonium chloride, structure 6 was considered most likely, as 7 or 8 is expected to couple readily with aryldiazonium salts [7], Compound 6 could be converted into 9 on long reflux in dioxane. Compound 9 was also obtained from fusion of 2 with malononitrile at 160 °C for 2 h.
Compound 2 reacted with trichloroacetonitrile to yield the pyranopyrimidine derivative 10. The formation of 10 from reaction of 2 with trichloroacetonitrile is a further example of the usefullness of the newly reported synthesis of pyrimidine from enaminonitriles [8, 9]. However, the limitations of this reaction recently reported by Gewald and Hain should be considered [10],
Compound 2 also reacted with ethyl 3-amino- 2-cyano-4-trichlorocrotonate to yield pyrano[2,3-d]- pyrimidine derivative 11.
Compound 2 reacted with benzoylisothiocyanate to yield the pyranopyrimidine derivative 12 , formed most likely via intermediacy of the thiourea derivative 13. In contrast, ethoxycarbonylisothiocyanate reacted with 2 to yield the ethoxycarbonylaminopy- ran 14. Ethoxycarbonylation of aminoheterocycles by this reagent has been previously reported [8, 11 ]
926 N. M. A bed et al. • Studies on H eterocyclic E nam ines
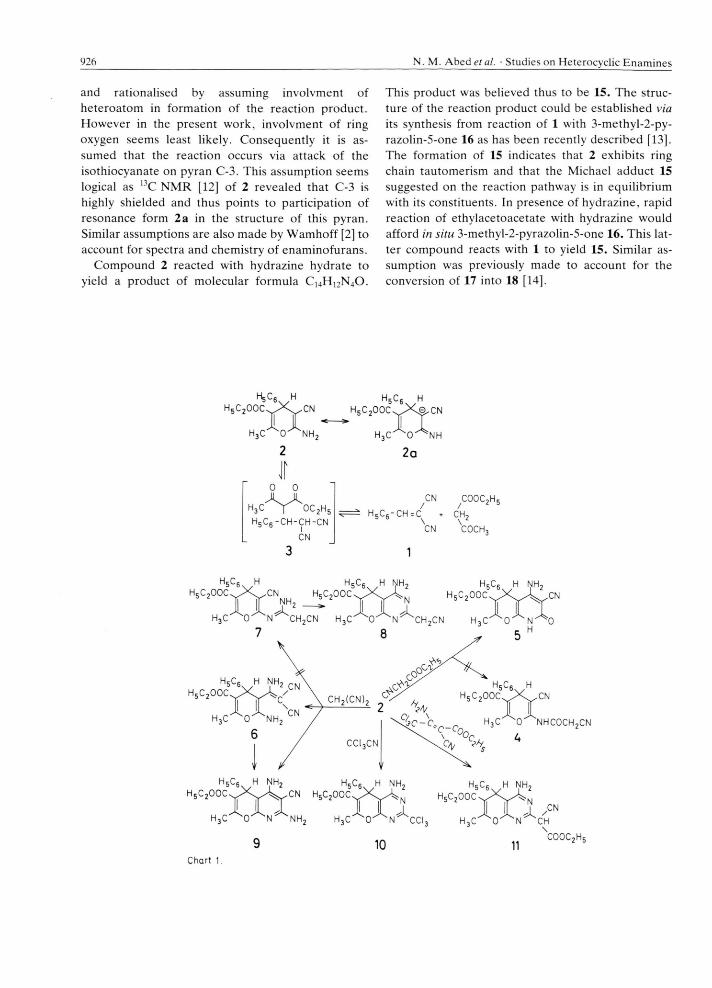
and rationalised by assuming involvment of heteroatom in formation of the reaction product. However in the present work, involvment of ring oxygen seems least likely. Consequently it is assumed that the reaction occurs via attack of the isothiocyanate on pyran C-3. This assumption seems logical as °C NMR [12] of 2 revealed that C-3 is highly shielded and thus points to participation of resonance form 2a in the structure of this pyran. Similar assumptions are also made by Wamhoff [2] to account for spectra and chemistry of enaminofurans.
Compound 2 reacted with hydrazine hydrate to yield a product of molecular formula C 14H 12N40 .
This product was believed thus to be 15. The structure of the reaction product could be established via its synthesis from reaction of 1 with 3-methyl-2-py- razolin-5-one 16 as has been recently described [13]. The formation of 15 indicates that 2 exhibits ring chain tautomerism and that the Michael adduct 15 suggested on the reaction pathway is in equilibrium with its constituents. In presence of hydrazine, rapid reaction of ethylacetoacetate with hydrazine would afford in situ 3-methyl-2-pyrazolin-5-one 16. This latter compound reacts with 1 to yield 15. Similar assumption was previously made to account for the conversion of 17 into 18 [14].
^ c 6 H h5c2oocy X . cn
H3C ^O "% H 2
2
if0 0
H,C O C ,H c31“ I ^ 2 n5 H5C6 -C H -C H -C N
CN
H5C6 Hh5c2ooc^ X © . cn
XXH3C ' " O '
2aNH
CN
h5c6- c h=c
CN
2 5COOC,H
CH?\COCH
2CN
H5C6 \ y H y H z H 5 C 6 w H NH2 H5C6 H NH2H5C2O O C y Y V c N H5C200Cy < A N H ^ O O C ^ O ^
h 3c ^ cA n ^ n h , H ,C 0 N CCIi H , C ^ O ^ N ^
9 10C hart 1.
x c o o c 2h 5

N. M. A bed et al. ■ S tudies on H eterocyclic E nam ines 927
H5C6 h H5C2OOC._Xv .CN
XX.H,C 0 N,COOC2H5
H5C6 H NH
H5c2o o c ^ X ^ A NCOCe.H;.
H,C O ^ N ^ S 3 H
12
t
14j-H N C S
h^ h cn ^h 5c 2ooc
NHCOOC2H5
H,C 0 NH
h5c6whHcCoOOC. X . X N
H,C 0 N S
13
H,C N
NHCOCcHcA . 6 5
H5C2OOCNCS
n h 2n h 2- h 2o
NC NH,
Chart 2
Ethyl-5-amino-6-cyano-7,8-dihydro-2-methyl-7-oxo- 4 H -pyrano[2,3-b]pyridine-3-carboxylate (5)
Compound 2 (0.01 mol) was fused with ethyl cyanoacetate (0.01 mol) in presence of few drops of piperidine at 160 °C (bath T) for 2 h. The solid product, so formed, was crystallized from acetic acid as brown powder 5, yield 65%, m.p. 210 °C. IR: 3400, 3200 (NH2); 2220 (CN); 1720, 1680 (CO).
h 5c 6 h h 5c 6 h
h 5c 2o o c ^ X ^ c n Ch 2 (CN)2 N c y V GN
h 2n o c 6h 5 h 2n o c 6h 5
17 18
In support of this view, 2 reacted with 16 to yield also 15.
ExperimentalAll melting points are uncorrected. IR spectra
were recorded on a Beckman spectrophotometer. 'H NM R on a Varian EM-390-90 MHz spectrometer using TMS as internal indicator and chemical shifts are expressed as ppm. The microanalysis were performed by the microanalytical unit at Cairo University.
CI9H I7N30 4 (351)Calcd C 64.9 Found C 64.8
H 4.8 N 11.9, H 4.6 N 11.8.
Reaction o f 2 with malononitrile
(a) Compound 2 (0.01 mol) was refluxed with malononitrile (0.01 mol) in ethanol (20 ml) with catalytic amount of triethylamine for 4 h. The solid product, so formed on cooling, was collected by filtration and crystallized from ethanol as yellow crystals 6, yield (75%), m.p. 235 °C. IR: 3410, 3310, 3200 (N H 0; 2220, 2190, (CN); 1700 (CO). 'H NMR:1.2 (t, 3H , CH3); 2.4 (s, 3H , CH3); 3.4 (s, 2H , N H 2);4.2 (q, 2H , CH2); 4.8 (s, 1H, H4); 5.9 (s, 2H , N H 2); 7.2—7.6 (m, 5H , aromatic protons).
(b) Compound 2 (0.01 mol) was heated with malononitrile (0.01 mol) in presence of few drops of piperidine at 160 °C (bath T) for 2 h. The solid product, so formed, was crystallized from acetic acid as brown powder 9, yield (70%), m.p. 290 °C. IR: 3400, 3260 (NH2); 2210 (CN); 1720 (CO).
(c) Compound 6 (0.01 mol) was refluxed in dioxane (30 ml) for 10 h. The solid product, so formed on standing, was collected by filtration, crystallized from acetic acid and identified as compound 9 (m.p. and mixed m.p.).
C19H 18N40 3 (350)Calcd C 65.1 H 5.1 N 16.0,Found C 65.3 H 5.2 N 16.3.
Ethyl-4-amino-7-methyl-5 -phenyl-2-trichloromethyl- 4 H -pyrano[2,3-d]pyrimidine-6-carboxylate (10)
Equimolecular amounts of compound 2 (0.01 mol) and trichloroacetonitrile (0.01 mol) were refluxed in dry toluene (30 ml) with catalytic amount of piperidine for 6 h. The product, so formed, was collected by filtration and crystallized from toluene as brown crystals 10 yield (70%), m.p. 235°C. IR: 3400, 3200 (NH2), 1710 (CO). 'H NMR: 1.3 (t, 3H , CH 3);2.2 (s, 3H , CH3); 4.2 (q, 2H , CH2); 4.6 (s, 1H, H-4); 6.8 (s, 2H , NH2); 7 .2 - 7 .8 (m, 5H , aromatic protons).C18H 16N30 3C13 (428.5)
Calcd C 50.4 H 3.7 Cl 24.8,Found C 50.6 H 3.5 Cl 24.6.

928 N. M. A bed et al. ■ S tudies on H eterocyclic E nam ines
Reaction o f com pound 2 with ethyl-3-amino-2-cyano-4-trichlorocrotonate
Equimolecular amounts of compound 2 (0.01 mol) and ethyl-3-amino-2-cyano-4-trichlorocrotonate (0.01 mol) were refluxed in 20 ml pyridine for 3 h. The reaction mixture was cooled and poured onto water. The oil, so formed, was collected by extraction with ether and crystallized from acetic acid as brown crystals 11; yield (80%), m.p. >300 °C. IR: 3380 (NH2); 2220 (CN); 1720 (CO).
C22H21N 40 5 (421)Calcd C 62.7 H 4.9 N 13.3,Found C 62.9 H 4.9 N 13.2.
Reaction o f com pound 2 with isothiocyanates
Equimolecular amounts (0.01 mol) of 2 and the appropriate isothiocyanate were refluxed in dry di- oxane (50 ml) for 4 h. The reaction mixture was cooled and poured onto water. The oil, so formed, was extracted with ether and crystallized from ethanol.
Compound 12, orange crystals; yield 80%; m.p. 110 °C. IR: 3200 (NH); 1720, 1690 (CO). !H NMR:1.3 (t, 3H , CH3); 2.4 (s, 3H , CH3); 3.4 (s, 1H, NH);4.2 (q, 2H , CH2) ; 5.0 (s, 1H, H-4); 7 .2 -1 .1 (m, 10H, aromatic protons); 8.2 (s, br, 1H, NH).
C24H2IN30 4S (447)Calcd C 64.4 H 4.6 N 9.3 S 7.1,Found C 64.2 H 4.5 N 9.3 S 7.2.
Compound 14, yellow crystals; yield 75%; m.p. 175 °C. IR: 3250, 3100 (NH); 2200 (CN); 1720, 1700 (CO).
CI9H20N2O5 (356)Calcd C 64.0 H 5.6 N 7.8,Found C 64.1 H 5.4 N 7.8.
7-Amino-6-cyano-3-methyl-4-phenyl- 4 H -pyrano[2,3-c]pyrazole (15)
(a) Compound 2 (0.01 mol) was refluxed with hydrazine hydrate (0.01 mol) in 20 ml ethanol for 1/2 h. The solid product was collected by filtration and crystallized from DM F/H20 as colourless crystals 15; yield (70%), and the reaction product was identified (m.p. and mixed m .p.) as 15 [13].
(b) Compound 15 was also prepared via reaction of 2 (0.01 mol) with 16 (0.01 mol) in refluxed pyridine (20 ml) for 1 h. The solid product, so formed, was collected by filtration, crystallized from DMF/ H20 and identified as compound 15 (m.p. and mixed imp. 250 °C). IR: 3400, 3320, 3200 (NH2); 2200 (CN). !H NMR: 1.9 (s, 3H , CH3); 4.5 (s, 1H, H-4);6.8 (s, 2H , NH2); 7 .2 - 7 .6 (m, 5H , aromatic protons); 10.1 (s, br, 1H, NH).
C14H 12N40 (252)Calcd C 66.6 H 4.7 N 22.2,Found C 66.6 H 4.6 N 22.1.
[1] M. Q uinteiro, C. Seoune, and J. L. Sotto, T etrahedron Lett. 1977, 1835.
[2] H. W amhoff, Lectures in Heterocyclic Chemistry 6 , 11 (1982); M. H. Elnagdi and H. Wamhoff, J. Heterocycl. Chem. 18, 1287 (1981).
[3] M. H. Elnagdi, H. A. El-Fahham, and G. E. H. El- gomeie, Heterocycles 20, 519 (1983).
[4] Z. E. Kandeel, F. M. Abdelrazek, M. E. M. Salah Eldin, and M. H. Elnagdi, J. Chem. Soc., Perkin Trans. I 1985, 1499.
[5] N. M. Abed, N. S. Ibrahim, S. M. Fahmy, and M. H. Elnagdi, Organic Preparations and Procedures Int. 17, 107 (1985).
[6 ] Z. Huang and H. Wamhoff, Chem. Ber. 117, 1856 (1984).
[7] M. R. H. Elmoghayar, E. A. Chali, M. M. M. Bamiz, and M. H. Elnagdi, Liebigs Ann. Chem. 1985, 1962.
[8 ] M. H. Elnagdi, S. M. Fahmy, E. A. A. Hafez, M. R. H. Elmoghayar, and S. A. R. Am er, J. Heterocycl. Chem. 16, 1109 (1979).
[9] M. H. Elnagdi, H. A. Elfahham, S. A. Ghozlan, andG. E. Elgemeie, J. Chem. Soc., Perkin Trans. I 1982, 2667.
[10] K. Gewald, U. H ain, and M. G runer, Chem. Ber. 1985, 2198.
[11] S. M. Fahmy, E. M. Kandeel, E. R. Elsayed, and M. H. Elnagdi, J. Heterocycl. Chem. 15, 1391 (1978).
[12] N. S. Ibrahim. Heterocycles 24, 935 (1986).[13] S. Abdou, S. M. Fahmy, K. U. Sadek, and M. H.
Elnagdi, Heterocycles 16, 2177 (1981).[14] M. R. H. Elmoghayar, M. A. E. Khalifa, M. K. A.
Ibrahim, and M. H. Elnagdi, Monatsh. Chem. 113, 53(1982).
















![Flur 26 Hst Freiligrathplatz - Düsseldorf€¦ · 15 I 13 I 18 16 17 II I I I 14 12 II 10 II I I 4a II I 6 II 8 II 8 II 10 II I I-I-I-I I I I I I I I I I I /LOLHQWKDOVWUD H B8 'DQ]LJHU](https://static.fdokument.com/doc/165x107/6061ca6ec0f4fb4c5004df74/flur-26-hst-freiligrathplatz-dsseldorf-15-i-13-i-18-16-17-ii-i-i-i-14-12-ii.jpg)

