
5. Elektrophoretischen Trennverfahren
16
Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren 60 5. Elektrophoretischen Trennverfahren 5.1 Einleitung Elektrophorese ist die Bewegung (Wanderung/Transport) von geladenen Teilchen in einem flüssigen Medium unter Einfluss eines elektrischen Feldes. Bei einer elektrophoretischen Trennung werden geladene Teilchen aufgrund ihrer unterschiedlichen Wanderungsgeschwindigkeiten in einem elektrischen Feld getrennt. Die Wanderungsgeschwindigkeit hängt von der Ladung, der Form und der Umgebung des Teilchens, sowie von der Grösse des elektrischen Feldes ab. Es findet also im Prinzip keine Verteilung zwischen zwei Phasen statt womit sich diese Methoden grundlegend von den chromatographischen Trennungen unterscheiden. Abb. 5.1 Elektrophoretische Trennung Dem schwedischen Forscher Arne Tiselius (1902-1971) wird die Urheberschaft der Elektrophorese zugeordnet. Er hat die Technik als erster analytisch-chemisch genutzt. Für seine Arbeiten wurde er 1948 mit dem Nobelpreis ausgezeichnet. Im Laufe der Zeit wurden verschiedene Variationen der Elektrophorese entwickelt. In ihrer Variationsbreite wurden diese elektrophoretischen Methoden die wichtigsten Werkzeuge in der biochemischen Analytik, da sie sehr gut geeignet sind für die Trennung von Peptiden, Proteinen, Polynucleotiden und anderen biologisch interessanten Makromolekülen. In ersten Anwendungen elektrophoretischer Trennverfahren wurden Glasplatten verwendet, die mit Gelen beschichtet sind. Der apparative Aufbau ist ähnlich wie bei Dünnschichtchromatographie: Abb. 5.2 Aufbau von Glasplatten-Elektrophorese Die Probe wird auf die mit Puffer getränkte Gelplatte aufgetragen. Nach Anlegen einer Spannung mit Hilfe von zwei Elektroden, die in die beiden Reservoirs getaucht sind, wandern die Analytionen entsprechend ihrer Ladung und Grösse unterschiedlich schnell zu einer der beiden Elektroden und trennen sich dabei. Nach Ende der Entwicklungszeit wird die Lage der Analyten off-line detektiert. ! EOF ! EP
Transcript of 5. Elektrophoretischen Trennverfahren

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
60
5. Elektrophoretischen Trennverfahren
5.1 Einleitung Elektrophorese ist die Bewegung (Wanderung/Transport) von geladenen Teilchen in einem flüssigen Medium unter Einfluss eines elektrischen Feldes. Bei einer elektrophoretischen Trennung werden geladene Teilchen aufgrund ihrer unterschiedlichen Wanderungsgeschwindigkeiten in einem elektrischen Feld getrennt. Die Wanderungsgeschwindigkeit hängt von der Ladung, der Form und der Umgebung des Teilchens, sowie von der Grösse des elektrischen Feldes ab. Es findet also im Prinzip keine Verteilung zwischen zwei Phasen statt womit sich diese Methoden grundlegend von den chromatographischen Trennungen unterscheiden.
Abb. 5.1 Elektrophoretische Trennung
Dem schwedischen Forscher Arne Tiselius (1902-1971) wird die Urheberschaft der Elektrophorese zugeordnet. Er hat die Technik als erster analytisch-chemisch genutzt. Für seine Arbeiten wurde er 1948 mit dem Nobelpreis ausgezeichnet. Im Laufe der Zeit wurden verschiedene Variationen der Elektrophorese entwickelt. In ihrer Variationsbreite wurden diese elektrophoretischen Methoden die wichtigsten Werkzeuge in der biochemischen Analytik, da sie sehr gut geeignet sind für die Trennung von Peptiden, Proteinen, Polynucleotiden und anderen biologisch interessanten Makromolekülen. In ersten Anwendungen elektrophoretischer Trennverfahren wurden Glasplatten verwendet, die mit Gelen beschichtet sind. Der apparative Aufbau ist ähnlich wie bei Dünnschichtchromatographie:
Abb. 5.2 Aufbau von Glasplatten-Elektrophorese
Die Probe wird auf die mit Puffer getränkte Gelplatte aufgetragen. Nach Anlegen einer Spannung mit Hilfe von zwei Elektroden, die in die beiden Reservoirs getaucht sind, wandern die Analytionen entsprechend ihrer Ladung und Grösse unterschiedlich schnell zu einer der beiden Elektroden und trennen sich dabei. Nach Ende der Entwicklungszeit wird die Lage der Analyten off-line detektiert.
!EOF
!EP

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
61
Entscheidende Nachteile der Plattengelelektrophorese sind die relativ schwierigere produzierbare Herstellung der Platten und die umständliche off-line Detektion. Dies führte in den 60er Jahren zur Entwicklung von elektrophoretische Trennverfahren in Kapillaren – Kapillarelektrophorese (capillary electrophoreses, CE).
5.2 Grundlagen
5.2.1 Elektrophoretische Beweglichkeit Die elektrophoretische Beweglichkeit von Ionen, das Trennprinzip der Elektrophorese, ist proportional zur elektrischen Kraft, welche die Ionen erfahren und indirekt proportional zum Reibungswiderstand durch das Puffermedium. Auf ein geladenes Teilchen der Ladung q wirkt in
einem elektrischen Feld der Feldstärke E (V/cm) folgende Kraft Fe:
€
Fe = q ⋅ E = z ⋅ e ⋅ E 5.1
wobei z Ladungszahl der Komponente ist und e Elementarladung [C] ist, E die elektrische Feldstärke [V/cm] beschreibt. Nach Stokeschem Gesetz erfährt das Teilchen (unter der Voraussetzung einer kugelförmigen Gestalt) mit dem Radius r, wenn es sich bei dieser Einwirkung in einem Medium mit der Viskosität η befindet und bei einer (elektrophoretischen) Geschwindigkeit v eine Gegenkraft FR:
€
FR = k ⋅η ⋅ v = 6π ⋅ r ⋅η ⋅ν 5.2
wobei k Konstante [cm] (k = 6πr für sphärische Partikel), η Viskosität der Lösung [Pa⋅s] und v die Wanderungsgeschwindigkeit der Komonente [cm/s]. Diese Überlegungen gelten für idealisierte Teilchen in einem trägerfreien, unendlich verdünnten, praktisch salzfreien Medium und haben so nur theoretische Bedeutung! Im stationären Zustand ist die Summe aller Kräfte gleich Null. Damit ergibt sich für die elektrophoretische Geschwindigkeit der Ausdruck:
€
v =z ⋅ e
6π ⋅η ⋅ r⋅ E 5.3
und für die elektrophoretische Mobilität µEP (in der Elektrophoreseliteratur findet man auch den Begriff Beweglichkeit):
€
µEP =vE
=z ⋅ e
6π ⋅η ⋅ r=
q6π ⋅η ⋅ r 5.4
wobei q die Ladung des Ions ist, η die Viskosität des Puffermediums und r der Radius des Ions.
5.2.2 Elektroosmotischer Fluss Zusätzlich zur elektrophoretischen Beweglichkeit der Analytionen tritt im elektrischen Feld auch ein elektroosmotischer Fluss (electroosmotic flow, EOF) in einer CE-Kapillare auf. Die Ursache dieses EOF liegt in der Beschaffenheit der Kapillarwände. Bei geeigneten Pufferbedingungen sind die Silanolgruppen (Si–OH, pKa≈5.3) deprotoniert und demnach negativ geladen. Dies bewirkt, dass Kationen der Pufferlösung sich an die Kapillarwand anlagern und eine elektrische Doppelschicht bilden. Durch das an die Kapillare angelegte

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
62
elektrische Feld, wandern die (hydratisierten) Kationen zur negativen Elektrode und bewirken dadurch eine Bewegung der gesamten Lösung in der Kapillare hin zur Kathode.
Abb. 5.3 Schema eines elektroosmotischen Flusses (EOFs)
Der EOF, µEOF, kann beschrieben werden als
€
µEOF =ζ ⋅ ε4π ⋅η 5.5
wobei ε die Dielektrizitätskonstante und η die Viskosität der Pufferlösung ist, E die elektrische Feldstärke und ζ das Zeta-Potential (in Volt), das die Abnahme der Ladungsdichte von der Kapillarwand zum Innern der Kapillare beschreibt. Der EOF hat ein einzigartiges, beinahe flaches Flussprofil, wohin gegen ein hydrostatisch verursachtes Flussprofil (wie in der HPLC üblich) parabolförmig ausgestaltet ist, das aufgrund von Reibungskräften an der Wand zustande kommt.
Abb. 5.4a EOF Flussprofil (elektrisches Potential: stempelförmig)
Abb. 5.4b Flussprofil in der HPLC (hydrostatischer Druck: parabelförmig)
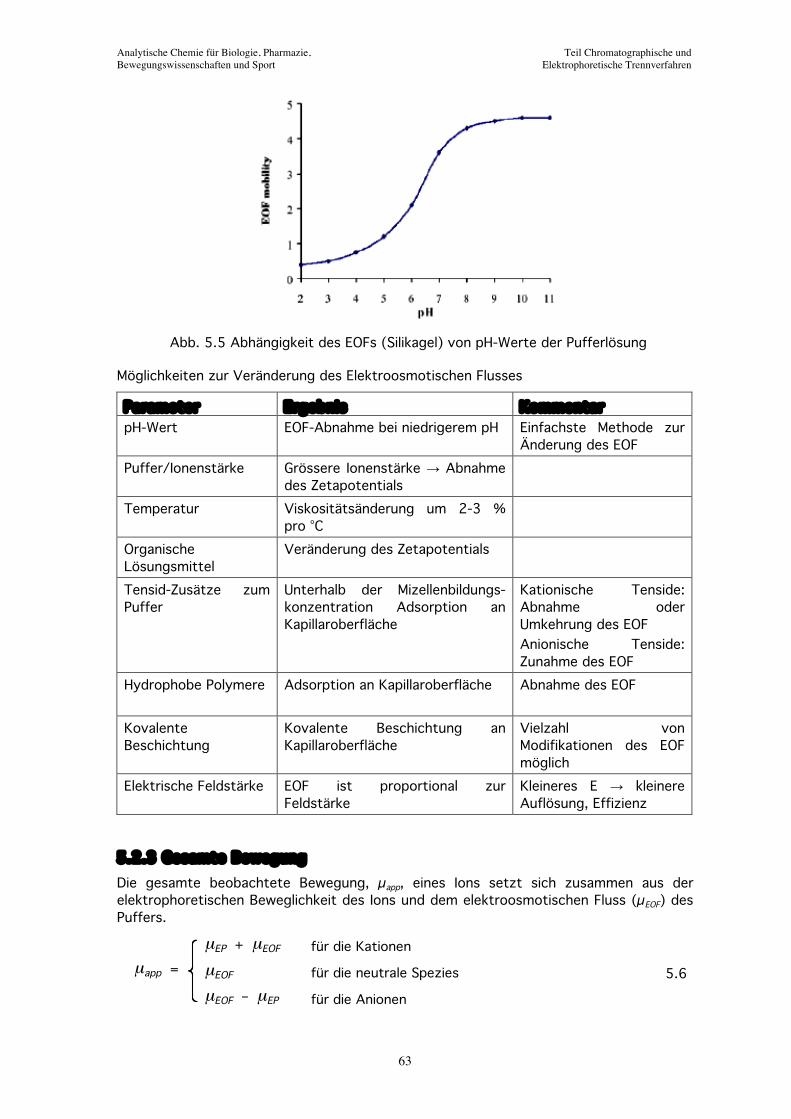
Die Intensität des EOF hängt vor allem von Eigenschaften der Pufferlösung ab. Je höher die pH-Werte der Pufferlösung, desto grosser die Intensität des EOF.
! Zetapotenzial
resultierender EOF!Anode! Anode!
Kapillarwand!
Kathode
parabelförmig
stempelförmig
Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
63
Abb. 5.5 Abhängigkeit des EOFs (Silikagel) von pH-Werte der Pufferlösung
Möglichkeiten zur Veränderung des Elektroosmotischen Flusses
Parameter Ergebnis Kommentar pH-Wert EOF-Abnahme bei niedrigerem pH Einfachste Methode zur
Änderung des EOF Puffer/Ionenstärke Grössere Ionenstärke → Abnahme
des Zetapotentials
Temperatur Viskositätsänderung um 2-3 % pro °C
Organische Lösungsmittel
Veränderung des Zetapotentials
Tensid-Zusätze zum Puffer
Unterhalb der Mizellenbildungs-konzentration Adsorption an Kapillaroberfläche
Kationische Tenside: Abnahme oder Umkehrung des EOF Anionische Tenside: Zunahme des EOF
Hydrophobe Polymere Adsorption an Kapillaroberfläche Abnahme des EOF
Kovalente Beschichtung
Kovalente Beschichtung an Kapillaroberfläche
Vielzahl von Modifikationen des EOF möglich
Elektrische Feldstärke EOF ist proportional zur Feldstärke
Kleineres E → kleinere Auflösung, Effizienz
5.2.3 Gesamte Bewegung Die gesamte beobachtete Bewegung, µapp, eines Ions setzt sich zusammen aus der elektrophoretischen Beweglichkeit des Ions und dem elektroosmotischen Fluss (µEOF) des Puffers.
5.6
µapp =
µEP + µEOF
µEOF
µEOF – µEP
für die neutrale Spezies
für die Kationen
für die Anionen

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
64
Für die meisten Analyten gilt µEP < µEOF. Unter dieser Vorraussetzung können sowohl Kationen als auch Anionen mit einem einzigen Detektor, platziert am Katoden-Ende der Kapillare, detektiert werden. Die Kationen wandern hierbei mit dem EOF und haben kurze Migrationszeiten, wogegen die Anionen gegen den EOF wandern und dementsprechend längere Migrationszeiten haben. Neutrale Analyten (zwei oder mehr) können wegen gleichmässiges EOF in der klassischen Kapillarelektrophorese nicht getrennt werden.
Abb. 5.6 Gesamte Bewegungsreihefolge von Kationen, Neutrale und Anionen
Sehr bewegliche Anionen (meist kleine anorganische Ionen wie Chlorid oder Bromid) deren µEP-Wert grösser ist als der EOF, bewegen sich in die entgegengesetzte Richtung und können in der konventionellen Anordnung nicht detektiert werden.
5.2.4 Migrationszeit, Böden und Auflösung Diese beobachtete Bewegung kann berechnet werden aus der Länge der Kapillare, Ld, der Migrationszeit vom Startpunkt zum Detektor, tM, und der Stärke des elektrischen Feldes, E.
€
tM =Ld
µapp ⋅ E 5.7a
€
µapp=Ld
tM ⋅ E 5.7b
Der grösste Faktor, der in der CE Bandenverbreiterung verursacht, ist die Longitudinal-Diffusion (siehe Diskussion der van-Deemter-Gleichung). Die Anzahl theoretische Böden, N, wird hauptsächlich beeinflusst von der Länge der Kapillare, Ld, der Wanderungszeit, tM, und des Diffusionskoeffizienten, D, des Analytions. Typischerweise werden Böden von 100'000 – 1'000'000 erreicht.
€
Nmax =Ld2
2DtM 5.8
Die Auflösung (R) von zwei Analyten kann analog zu chromatographischen Methoden beschrieben werden, wobei µ die elektrophoretischen Mobilitätsunterschiede der Ionen und die durchschnittliche beobachtete Bewegung der Ionen bezeichnet.

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
65
€
R =N4
Δµµapp
5.9
5.3 Kapil larelektrophorese – Apparatur Die wichtigsten Bestandteile einer Kapillarelektrophorese sind die Kapillare, das Puffersystem, die Hochspannungsversorgung und der Detektor. Die Spannungsversorgung HV liefert Gleichspannungen in der Grösse von 0 – 30 kV. Zusätzlich können Timerfunktionen zur Steuerung der Injektion und der Analyse vorhanden sein.
Abb. 5.7 Schematischer Aufbau einer CE-Anlage.
Typische Bedingungen zur EP Trennung Kapillaren: Länge: 10-100 cm, Innendurchmesser: 10-100 µm, Material: fused
silica Jedes Ende der Kapillare ist in ein Reservoir getaucht, die über Elektroden mit
einer HV-Versorgung verbunden sind. Reservoirs und Kapillare sind mit Elektrolyt (Pufferlösung) gefüllt. Auftragen der Probe: das Inlet-reservoir wird mit einem Probenreservoir
vertauscht. Durch einen kurzen Druck- oder Spannungspuls wird typischerweise 1-10nl Probe in die Kapillare injiziert.
Nach Anlegen der Spannung wandern die Ionen Richtung Outlet-Elektrode Oft werden optische Methoden zur Detektion verwendet (UV, Fluoreszenz), wobei
der Detektor meist katodenseitig platziert ist.
Puffersystem Die gängigsten verwendeten Puffersysteme sind Phosphat- (pH-Wert um den Neutralpunkt) und Boratpuffer (pH um 9). Zu hohe Pufferkonzentrationen sollten vermieden werden, da es sonst zu einem erhöhten Stromfluss kommt, in dessen Folge die Wärmeentwicklung zunimmt. Auf der anderen Seite darf die Pufferkonzentration nicht kleiner sein als die Analytkonzentration, da dieses zu einem Effekt führt, der der Überladung einer HPLC-Säule entspricht (erschöpfte Pufferkapazität).
•! L: 10-100 cm
•! ID 10-100 !m
Probenaufgabe (1-10 nl) durch •! Hydrostatische Injektion (Siphoneffekt)
•! Druck/Vakuum Injektion (Druckdifferenz)
•! Elektrokinetische Injektion (!EOF, !EP)
•! UV – 10–6 mol/L
•! Fluoreszenz – Attomol
•! Kopplung an MS
On-column (durchsichtlich)
Fused silica mit
Polyimidbeschichtung
bis 30 kV

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
66
Weiterhin sind in der Pufferlösung alle für den entsprechenden Elektrophoresemodus benötigten Zusätze wie Methanol (oder ein anderer organischer Modifier), SDS, Cyclodextrine (oder andere Komplexbildner) und zusätzliche Salze (Elektrolyte), die die Ionenstärke beeinflussen, enthalten.
Kapil lare Als Kapillaren kommen vor allem die aus der GC bekannten fused-silica Kapillaren mit Polyimidbeschichtung zum Einsatz. Gängige Längen sind zwischen 10 und 100 cm, typische Durchmesser 25 bis 100 µm (Vergleich GC: 5 - 50 m, I.D. 220 - 500 µm). Eine Kapillare von 50 cm Länge und einem Durchmesser von 50 µm besitzt ein Volumen von etwa einem Mikroliter. Dies bedeutet, dass sich die Injektionsmengen in einem Bereich von 1 -10 nL bewegen müssen. Für Kapillare sind kleine Durchmesser günstig für eine gute Wärmeabstrahlung und damit für hohe mögliche Felder. Aus diesem Grund wurde bereits mit Kapillaren experimentiert, die nur einen Durchmesser von neun bzw. sogar nur von zwei Mikrometern hatten. Solche Kapillaren stellen aber extremste Anforderungen an Injektions- und Detektionstechniken (Picoliter Injektions und Detektionsvolumina!). Trotz der guten Wärmeabstrahlung einer Kapillare sollte in Anbetracht der Erhöhung der Reproduzierbarkeit eine zusätzliche Thermostatisierung der Kapillare erfolgen. Dies dient nicht nur dazu, zu verhindern, dass sich ein parabolisches Strömungsprofil aufgrund der Joule’schen Wärme ausbildet, sondern auch zum Konstanthalten der Wanderungsgeschwindigkeiten, da diese sich pro °C um etwa 2% ändern. Das Optimum für eine Trennung ergibt sich nach Jorgenson und Lukacs bei der höchstmöglichen Spannung und der kürzestmöglichen Kapillare.
Injektion Eine Injektion wie in der GC oder HPLC ist in der Kapillarelektrophorese (CE) nicht möglich, da sich die Injektionsvolumina etwa im Bereich von 1/100 des Kapillarenvolumens bewegen. Dies entspricht bei gängigen Kapillaren etwa 1-10 nL. Die Injektionstechniken in der CE funktionieren deshalb nach dem Prinzip, eine Flüssigkeitsmenge (mindestens 1 µL) vorzulegen, und Aliquote davon in die Kapillare zu transportieren. Die drei gängigsten Verfahren sind
Elektrokinetische Injektion Siphonmethode Anlegen von Druck und/oder Vakuum
Elektrokinetische Injektion: Bei dieser Injektionsmethode wird die Probe durch Anlegen einer Spannung für eine bestimmte Zeit in die Kapillare hinein migriert. Die Reproduzierbarkeit mit dieser Methode ist relativ hoch, allerdings tritt eine Diskriminierung der Probe auf. Falls kein elektroosmotischer Fluss vorhanden ist, werden nur geladene Probenbestandteile mit der richtigen Polarität in die Säule hinein bewegt. Selbst bei Vorhandensein eines elektroosmotischen Flusses werden die verschiedenen Probenbestandteile aufgrund ihrer unterschiedlichen elektrophoretischen Mobilitäten unterschiedlich gut auf die Kapillare aufgegeben (Moleküle mit grosser Mobilität besser als Moleküle mit kleiner Mobilität). Siphoninjektion: Es lässt sich auch Probe auf die Kapillare aufgeben, indem man das Probengefäss um etwa 5–20 cm anhebt bzw. das Puffergefäss am anderen Ende absenkt und so durch den Niveau-Unterschied einen hydrostatischen Fluss erzeugt. Die aufgegebene Menge hängt hierbei vom Höhenunterschied und der Zeit ab. Es erfolgt hier keine Diskriminierung, jedoch ist die Reproduzierbarkeit kleiner als im ersten Fall.

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
67
Vakuum- oder Druckinjektion: Durch Anlegen von Druck am Einlass oder Vakuum am Auslass der Kapillare lässt sich ebenfalls Probe auf die Säule aufgeben. Die injizierte Probenmenge wird durch die Grösse von Druck oder Vakuum und der Zeit bestimmt. Diese Methode der Injektion ist technisch etwa aufwendiger als die beiden obengenannten, bietet aber sehr gute Werte für Reproduzierbarkeit und Diskriminierung. Weitere Methoden, die sich aber noch im Experimentierstadium bewegen, sind Techniken mit einem elektrischen Splitter oder einem Probenschleifenanalogon.
Detektion Die Detektion stellt ein Problem in der CE dar, da die Detektoren auf die winzigen Säulendimensionen und die sehr scharfen Banden angepasst sein sollten. Bei durchaus möglichen 600’000 theoretischen Böden hat in einer 100 µm Kapillare eine typische Bande eine Breite von 0.2 mm, was einem Elutionsvolumen von 1.5 nL entspricht. Die verbreitetste Detektionsart in der CE ist die optische Detektion, also UV-Detektion oder Fluoreszenz-Detektion. Da Standard-Detektorzellen wie z.B. aus der HPLC zu grosse Volumina hätten, erfolgt die Detektion in der Regel "on-column". Hierzu wird in den Polyimidfilm ein Fenster gebrannt, geätzt oder gekratzt, durch das detektiert wird. Zusätzliche Vorrichtungen unterstützen ggf. die Fokussierung des Lichtstrahls auf die Kapillarmitte bzw. unterdrücken den Einfluss von Streulicht. Aufgrund des kleinen Lichtwegs sind die erreichbaren Empfindlichkeiten nicht grundsätzlich besser als in der HPLC (d.h. eine bestimmte Konzentration ist erforderlich, auch wenn kleinere Absolutmengen detektiert werden können). Bei gut absorbierenden Verbindungen liegen die Nachweisgrenzen mit UV-Detektion bei etwa 10–6 mol/L. Wesentlich empfindlicher (und selektiver) ist ein Fluoreszenzdetektor. Besonders mit laserinduzierter Fluoreszenz sind Aminosäuren bis in den Attomolbereich detektierbar. Eine Alternative zur direkten Detektion sind die indirekten Methoden, bei denen die Pufferlösung ein Grundsignal liefert (z.B. ein fluoreszierender Bestandteil im Puffer), das dann vom Analyten gestört wird. Diese Methode ist natürlich im allgemeinen weniger empfindlich, dafür aber sehr universell, z.B. für Verbindungen, die keine Fluoreszenz zeigen. Weitere Detektionsmöglichkeiten sind elektrochemische Detektoren (vor allem Leitfähigkeitsdetektoren), Radioaktivitätsdetektoren, thermooptische Detektoren, RI- und CD Detektoren. Besonders interessant ist auch die Kopplung von CE und MS, da die niedrigen Flussraten der CE diese Kopplung im Gegensatz zur konventionellen LC-MS-Kopplung besonders begünstigen.
5.4 Varianten der Elektrophorese Der schwerwiegendste Nachteil der Elektrophorese in freier Lösung ist, dass die bereits getrennte Banden durch den Abtransport der Joule’schen-Wärme verzerrt werden. Der Durchbruch der Elektrophorese als analytische Trennmethode erfolgte erst als es gelang, diese Störungen durch Verwendung von stabilisierenden Gelen, die auf Glasplatten gegossen wurden, oder durch puffergetränkte Papierstreifen einzuschränken bzw. ganz auszuschalten. Die Gelelektrophorese entwickelte sich zu einem verbreitetsten analytischen Werkzeug für die Trennung von Biopolymers. Aber die klassische Flachbetteelektrophorese hat einige entscheidende Nachteile wie die schwierige reproduzierbare Herstellung und Handhabung der Gelplatten, Austrocknen der Gele und direkte Detektion. Der endgültige Durchbruch gelang mit der Verwendung von Quarzkapillaren mit Innendurchmessern im Bereich von 50 – 100 µm. In diesen Kapillaren kann wegen des grossen Verhältnisses von Oberfläche zu Volumen die Joule’sche Wärme effizient abgeführt werden. Dadurch wird der störende Einfluss der thermischen

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
68
Konvektion drastisch reduziert. Dies ermöglicht die Durchführung äusserst effizienter Trennungen. Gleichzeitig lässt die gute Transparenz der Quarzkapillaren auch die Detektion der getrennten Analyten im unteren UV-Bereich zu. Unter dem Oberbegriff „Kapillarelektrophorese“ werden verschiedene Trenntechniken zusammengefasst.
5.4.1 Kapil larzonenelektrophorese (CZE) Es gibt verschieden Varianten der Kapillarelektrophorese, wobei Kapillarzonen-elektrophorese (CZE) die klassische und am weitesten verbreitete Methode ist. Mit CZE können kleine und mittelgrosse Ionen getrennt werden (z.B. Aminosäuren, Peptiden). Es werden meist unmodifizierte Quarzkapillaren verwendet.
Abb. 5.8 Trennprinzip der Kapillarzonenelektrophorese (CZE)
Die Kapillarzonenelektrophorese ist eine Methode, die den elektroosmotischen Fluss ausnutzt. Durch diesen Effekt ist es nämlich möglich, in einem analytischen Run sowohl Kationen wie auch Anionen und Neutralteilchen zu untersuchen. Die Gesamtmobilität ergibt sich für die einzelnen Spezies als Summe der elektrophoretischen Mobilität und der elektroosmotischen Mobilität. Da sich die beiden Mobilitäten bei den Kationen addieren, wandern diese insgesamt am schnellsten. Innerhalb der Kationen kommt es zu einer Trennung aufgrund der verschieden elektrophoretischen Mobilitäten. Nach den Kationen erreichen die Neutralteilchen den Detektor. Sie wandern mit der Geschwindigkeit des elektroosmotischen Flusses (gleichmässig µEOF), eine Auftrennung in einzelne Komponenten erfolgt in der Regel nicht. Als dritte Gruppe eluieren schliesslich die Anionen, da bei ihnen die elektrophoretische und die elektroosmotische Bewegung entgegengesetzt sind. Innerhalb der Anionen eluieren die Komponenten mit der kleinsten elektrophoretischen Mobilität zuerst.
5.4.2 Kapil lar-Gelelektrophorese (Capil lary Gel Electrophoreses, CGE) Die Trennung von grossen geladenen Molekülen, z.B. Proteine, ist mit CZE kaum möglich, da der Ladung/Masse Unterschied oft zu gering ist (Gl. 5.4). Die meisten mit Gelgefüllten Kapillaren (oft Acrylamid-Gel) bewirken eine Trennung der Analyten aufgrund ihrer Grösse (ähnlich GPC in der Flüssigkeitschromatographie). Zudem verringern die Gele die Diffusion der Analyten, was sehr effiziente Trennungen erlaubt. Mit CGE werden hauptsächlich Proteine und DNA-Moleküle getrennt.
!
µEOF
!
µEP
!
µEP

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
69
Abb. 5.9 Trennprinzip der Kapillargelelektrophorese (CGE)
5.4.3 Isoelektrische Fokussierung (IEF) Ein völlig anderes Trennprinzip liegt den elektrophoretischen Fokussierungsmethoden zugrunde. Bei der isoelektrischen Fokussierung erfolgt die Elektrophorese in einem pH-Gradienten, der durch ein komplexes Puffergemisch (Trägerampholyte, meist Aminocarbonsäuremischungen mit unterschiedlichen Verhältnissen an Amino- und Säuregruppen) stabilisiert wird. Dabei nimmt der pH-Wert zwischen Anode und Kathode stetig und im Idealfall linear zu. Ampholyte der aufgetragenen Probe (i. a. Proteine) werden somit an jenen Stellen im pH-Gradienten angereichert, die dem isoelektrischen Punkt der betreffenden Komponenten entsprechen (maximale Häufigkeit von elektrisch neutralen Zwitterionen H3N+-X-COO–). Die Methode wird hauptsächlich zur Trennung von Proteinen eingesetzt; die maximale Auflösung beträgt ca. 0.01 pH-Einheiten (Unterschied in den isoelektrischen Punkten). Die Detektion der getrennten Ampholyte erfolgt meist photometrisch (nach Anfärben) oder UV-spektrometrisch.
Abb. 5.10 Probenaufgabe in den pH-Gradienten.
Die Probe wird irgendwo in das Gemisch mit aufgegeben und das Feld angelegt. Die amphoteren Probenbestandteile beginnen nun zu wandern. Komponenten mit einer negativen Nettoladung wandern zur Anode, kommen dabei in Zonen mit immer kleinerem pH-Wert und werden dabei immer stärker protoniert. Schliesslich wird ein Punkt erreicht, an dem die Nettoladung Null ist (der isoelektrische Punkt) und die Komponente hört auf zu wandern. Auf die gleiche Weise wandert jede Komponente bis zu ihrem isoelektrischen Punkt, dann endet die Wanderung.

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
70
Abbildung 5.11: Fokussierung der Probenbestandteile am jeweiligen isoelektrischen Punkt.
Diese Technik hat eine fokussierende Eigenschaft. Diffundiert nämlich eine Komponente in einen Bereich mit einem anderen pH-Wert, wird sie sofort protoniert oder deprotoniert und bekommt damit wieder eine von Null verschiedene Nettoladung. Sie beginnt dann wieder zu ihrem isoelektrischen Punkt zurück zu migrieren. Das Ende der Trennung ist am abnehmenden Stromfluss zu erkennen. Das Feld wird dann abgeschaltet und der Kapillareninhalt durch Druck aus der Kapillare befördert, wobei die Komponenten nach ihren pl-Werten geordnet am Detektor vorbei kommen. Sowohl bei der IEF als auch bei der unten beschriebenen Isotachophorese (ITP) muss der elektroosmotische Fluss aus naheliegenden Gründen unterdrückt bzw. verhindert werden.
5.4.4 Isotachophorese (ITP) Der Name rührt daher, dass sich am Ende alle Zonen mit der gleichen Geschwindigkeit bewegen. Die Probe befindet sich in der Kapillare zwischen zwei Elektrolyten, dem ,leading electrolyte‘ (Leitelektrolyten) und dem ,terminating electrolyte‘ (Endelektrolyten), wobei die Leitionen eine möglichst hohe und die Endionen eine möglichst niedrige elektrische Beweglichkeit aufweisen sollten. Unter diesen Voraussetzungen stellt sich ein stationärer Zustand ein, d. h. es bildet sich ein selbst stabiles Trennmuster, wobei alle Elektrolytzonen (einzelne Komponenten, in der Reihenfolge abnehmender Beweglichkeit angeordnet) mit der gleichen Geschwindigkeit wandern. In einer Analyse lassen sich so nur entweder Anionen oder Kationen bestimmen.
Abb. 5.12 Probenaufgabe zwischen den leading und den terminating electrolyte
Kation mit einer höheren Mobilität als
alle Probenkationen (H+, H3O+)
Kation mit niedrigerer Mobilität als
alle Probenkationen
Kathodenseite Anodenseite

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
71
Bei einer Kationenanalyse ist der ,leading electrolyte‘ ein Kation mit einer höheren Mobilität als alle Probenkationen (in der Regel das Proton oder entsprechend solvatisierte Spezies), und der ,terminating electrolyte‘ ein Kation mit niedrigerer Mobilität als alle Probenkationen. Der ,leading electrolyte‘ befindet sich auf der Kathodenseite, der ,terminating electrolyte‘ auf der Anodenseite: Wird ein elektrisches Feld angelegt, kommt es zur Ausbildung eines Potenzialgradienten, der letztendlich dafür sorgt, dass sich alle Ionen mit der gleichen Geschwindigkeit bewegen. In Bereichen mit Kationen geringer Mobilität ist die Feldstärke grösser als in Bereichen mit Kationen höherer Mobilität. Die Kationen werden praktisch nach ihrer individuellen Leitfähigkeit geordnet.
Abb. 5.13 Gleichgewichtszustand – alle Zonen wandern mit der gleichen Geschwindigkeit.
Im Verlauf der Durchführung bildet sich ein stationärer Zustand, in dem alle Kationen gleicher Mobilität als ein reines Band, eingeschlossen von einem Band mit Kationen der nächst höheren Mobilität und einem Band mit Kationen der nächst niedrigeren Mobilität, wandern. Da die Konzentration an Anionen im ganzen System konstant ist, muss gleiches auch für die Kationen gelten. Das heisst, alle Banden haben die gleiche Konzentration, die Länge einer Bande ist ein Mass für die Absolutmenge an Substanz. Eine spezielle Charakteristik dieser Technik ist die Fokussierung, die zu scharfen Bandengrenzen führt. Falls ein Ion durch Diffusion in eine Zone vor oder hinter seiner Zone gelangt, kommt es in einen Bereich niedrigerer bzw. höherer Feldstärke und wird dadurch gebremst bzw. beschleunigt, bis es wieder in seiner Zone ist. Der Potenzialgradient bleibt bestehen und hat am Ende eine Treppenform. Innerhalb einer Zone sind Elektrolytkonzentration und elektrische Feldstärke konstant. Die Konzentrationen der getrennten Komponenten sind im Wesentlichen durch die Konzentration des Leitelektrolyten bestimmt und nehmen zwischen Leit- und Endelektrolyt ab. Da sich in der Isotachophorese die Analyten in direkt hintereinander liegenden Zonen anordnen (und nicht in getrennten Peaks) ergeben sich Detektionsprobleme, so dass diese Methode nur selten angewendet wird.
5.4.5 Mizellar elektrokinetische Kapil larchromatographie (MEKC) Einer der grössten Nachteile der klassischen Elektrophorese wie CZE ist, dass neutrale Analyten nicht getrennt werden können. Mit der Entwicklung der MEKC wurde jedoch
Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
72
auch dies möglich. Sie wurde zum ersten Mal 1984 von Terabe et al. erwähnt. Der Grundgedanke ist die Trennung von ungeladenen Substanzen und von relativ unpolaren Verbindungen mit der Kapillarelektrophorese. Wie oben bei der Zonenelektrophorese gezeigt, können ungeladene Verbindungen zwar unter Ausnutzung des elektroosmotischen Flusses von den Anionen und Kationen getrennt werden, jedoch ist eine weitere Auftrennung der einzelnen Neutralverbindungen dort nicht möglich. Ebenso bereiten sehr unpolare Substanzen Schwierigkeiten, da sie sich in den üblichen Puffergemischen schlecht lösen, und die Puffergemische selber nicht mehr als etwa 50% Methanol enthalten sollten, da sonst die Leitfähigkeit zu stark herabgesetzt wäre.
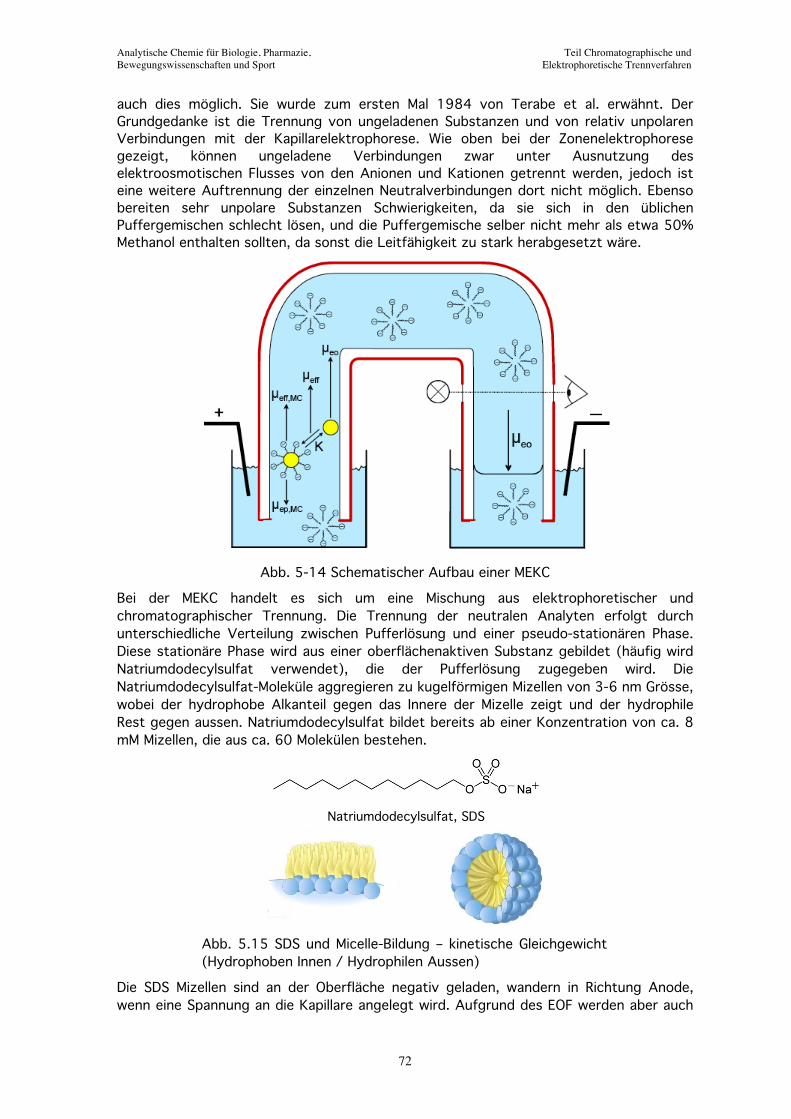
Abb. 5-14 Schematischer Aufbau einer MEKC
Bei der MEKC handelt es sich um eine Mischung aus elektrophoretischer und chromatographischer Trennung. Die Trennung der neutralen Analyten erfolgt durch unterschiedliche Verteilung zwischen Pufferlösung und einer pseudo-stationären Phase. Diese stationäre Phase wird aus einer oberflächenaktiven Substanz gebildet (häufig wird Natriumdodecylsulfat verwendet), die der Pufferlösung zugegeben wird. Die Natriumdodecylsulfat-Moleküle aggregieren zu kugelförmigen Mizellen von 3-6 nm Grösse, wobei der hydrophobe Alkanteil gegen das Innere der Mizelle zeigt und der hydrophile Rest gegen aussen. Natriumdodecylsulfat bildet bereits ab einer Konzentration von ca. 8 mM Mizellen, die aus ca. 60 Molekülen bestehen.
Abb. 5.15 SDS und Micelle-Bildung – kinetische Gleichgewicht (Hydrophoben Innen / Hydrophilen Aussen)
Die SDS Mizellen sind an der Oberfläche negativ geladen, wandern in Richtung Anode, wenn eine Spannung an die Kapillare angelegt wird. Aufgrund des EOF werden aber auch
Natriumdodecylsulfat, SDS !

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
73
die Mizellen langsam Richtung Kathode geführt. Als Nettowirkung bewegen sich SDS Mizellen sehr langsam oder sogar still bleiben wie pseudo-stationäre Phase. Eine sehr hydrophile neutrale Analyt-Substanz, die überhaupt nicht in Wechselwirkung mit den Mizellen tritt, wird mit dem EOF wandern und zur Zeit t0 an Detektor ankommen. Im anderen Extremfall wird ein sehr hydrophober Analyt sich fast vollständig in den Mizellen aufhalten und dementsprechend zur gleichen Zeit, tmc, detektiert werden wie die Mizellen selbst. Alle andern Analyten, die sich zwischenden Mizellen und der Pufferlösung verteilen, werden Migrationszeiten haben die zwischen t0 und tmc liegen. Dieses Zeitfenster (Retentionsfenster) definiert in der MEKC die Kapazität der Methode. Der Grad der Verteilung der Analyten in die Mizellen kann wesentlich durch die Wahl der oberflächenaktiven Substanz beeinflusst werden, sowie durch deren Konzentration. Bei Natriumdodecylsulfat werden typischerweise Konzentrationen von 20-100 mM eingesetzt. Ein grosser Vorteil der MEKC liegt in der einfachen Austauschbarkeit der pseudo-stationären Phase; wesentlich einfacher und billiger ist als bei HPLC-Säulen.
Abb. 5.16 µEP der negativ geladenen Mizellen entgegensetzt µEOF der Puffenlösung => Micelle bewegt sich fast nicht oder gar leicht umzukehren, wie stationäre Phase bei HPLC und unpolar Innenraum wie Flüssigfilm bei HPLC.
Abb. 5.17 Trennung der neutralen Verbindungen durch MEKC
5.4.7 Kapil larelektrochromatographie (CEC)
Ein weiteres Hybrid zwischen Chromatographie und Elektrophorese ist die Elektrochromatographie. Dabei werden Kapillaren mit stationären Phasen aus der HPLC beladen (in der Regel RP-Kieselgele), als mobile Phasen kommen Methanol-Puffer- oder Acetonitril-Puffer-Gemische zum Einsatz. Im Unterschied zur ,klassischen‘ HPLC wird nun

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
74
nicht eine Pumpe zum Vorantreiben der mobilen Phase benutzt, sondern der elektroosmotische Effekt ausgenutzt.
Die Idee, eine flüssigchromatographische Trennung von neutralen Analyten mit Strom zu betreiben, äusserte Pretorius bereits 1974. Diese Idee konnte jedoch erst 10 Jahre später umgesetzt werden, nachdem die Anfangsprobleme der CE, der Miniaturisierung der Detektoren und anderer dafür notwendiger Bestandteile gelöst waren. Durch die Verwendung des elektroosmotischen Flusses bekommt man die Möglichkeit, dessen Vorteile (vor allem das fast flache Strömungsprofil) mit den Selektivitäten der HPLC zu verknüpfen. Weiterhin sind im Gegensatz zur klassischen HPLC Materialien mit Partikelgrösse < 3 µm möglich, was zu viel höheren Trennleistungen führt (in der klassichen HPLC gäbe es bei so kleinen Teilchen Probleme mit dem Druck). Ausserdem lassen sich ganz einfach polare und unpolare sowie geladene und ungeladene Verbindungen nebeneinander analysieren.
Die Schwierigkeiten der CEC liegen hier auf der apparativen Seite: Das Packungsmaterial muss mit geeigneten Fritten in der Kapillare gehalten werden, die Packungsprozedur ist schwierig, der pH Bereich der mobilen Phase ist eingeschränkt, eine Gasblasenbildung muss unterdrückt werden und die Reproduzierbarkeit des Flusses ist nicht immer einfach.
Das Trennprinzip der CEC ist in nachfolgender Abbildung gezeigt:
4.3.6 Plattenelektrophoretische Methode – 2-D PAGE Eine viel versprechende Methode zur Analyse von Polypeptidkomponenten in komplexen Proteingemischen oder DNA-Gemischen wurde in den 1970er Jahren eingeführt. Durch Erweiterung klassischer Elektrophoresetechniken auf zwei Dimensionen ergab sich eine enorme Verbesserung des Auflösungsvermögens. Das Probengemisch wird in der einen Richtung durch isoelektrische Fokussierung und anschliessend senkrecht dazu mittels SDS-PAGE getrennt (SDS: sodium dodecylsulfate, PAGE: polyacrylamide gel electrophoresis). Da sich ohne weiteres Hunderte von Polypeptidkomponenten entsprechend ihrem isoelektrischen Punkt und ihrer Molmasse trennen lassen, ist die 2-D-Elektrophorese für die Biochemie oder die klinische Chemie von ausserordentlicher Relevanz.

Analytische Chemie für Biologie, Pharmazie, Teil Chromatographische und Bewegungswissenschaften und Sport Elektrophoretische Trennverfahren
75
5.5 Zusammenfassung
Vor- und Nachteile der Kapil larelektrophorese (CE/HPCE) Vorteile und Charakteristika der HPCE (alle oben beschriebenen Methoden):
sehr gute Abstrahlung der überschüssigen Hitze und damit keine Bandenverbreiterung durch Konvektion
flaches Strömungsprofil bei Ausnutzung des elektroosmotischen Flusses keine feste stationäre Phase kleine Dimension der Mizellen in MEKC → günstiger Massentransfer prädestiniert für Makromoleküle mit kleinem Diffusionskoeffizienten hohe Trennleistung (wegen geringer Bandenverbreiterung) extrem wenig Lösungsmittelverbrauch (Kosten, Abfall) nur kleine Probenmengen nötig schonende Technik (wichtig für Biopolymere) in der Regel schnelles Verfahren (10 - 20 Minuten pro Analyse) automatisierbar hohe Bodenzahl, kleine reduzierte Bodenhöhe
Nachteile und Einschränkungen: Thermische Effekte sind begrenzend geringe Selektivität (verglichen mit HPLC) geringe Konzentrationsempfindlichkeit kleiner dynamischer Bereich Säulen wenig belastbar ungünstig für sehr unpolare Verbindungen Reproduzierbarkeit z.T. schwierig
Tabelle: Übersicht über die CE-Modi und ihre Trennprinzipien
Kapil larelektrophorese-Modus Trennprinzip
Kapillarzonenelektrophorese (Kationen und Anionen)
µEOF und µEP, EOF ausgenutzt
Gel Elektrophorese Grösse, Ladung
Isoelektrische Fokussierung (Proteine) pI-Werte + pH-Gradient
Isotachophorese (Kationen oder Anion) Potenzial-Gradient
MEKC (Kationen, Anionen und Neutrale) µEP und µEOF, Mizelle als RP bei HPLC
Kapillarelektrochromatographie (alle) wie HPLC, aber µEOF als Antriebskraft
2D-PAGE wie DC, IEF und Gelelektrophorese
Neuerungen für die HPCE, die erprobt werden:
wide bore Kapillaren (I.D. > 100 µm) und hohe Felder → hyperbolisches Strömungsprofil → Abhilfe durch genau gesteuerten laminaren Gegenstrom
Kapillarbündel → Parallelanalysen bzw. mikropräparative Technik dünne rechteckige Kapillare → bessere Hitzeabstrahlung, mehr Lichtweg für
die Detektion verschiedenste Gradienten (pH, Modifier, Temperatur, elektrisches Feld) zweidimensionale Techniken


















