
Ab-initio Beschreibung des Ladungstransports …...Ab-initio Beschreibung des Ladungstransports...
45
Ab-initio Beschreibung des Ladungstransports durch Kohlenstoffnanoröhren Bachelorarbeit von Mario Behrens An der Fakultät für Physik Institut für Theoretische Festkörperphysik (TFP) Referent: Prof. Dr. Gerd Schön Betreuer: Dr. Fabian Pauly Bearbeitungszeit: 10. September 2011 – 7. Dezember 2011 KIT – Universität des Landes Baden-Württemberg und nationales Forschungszentrum der Helmholtz-Gesellschaft www.kit.edu
Transcript of Ab-initio Beschreibung des Ladungstransports …...Ab-initio Beschreibung des Ladungstransports...

Ab-initio Beschreibung desLadungstransports durchKohlenstoffnanoröhren
Bachelorarbeitvon
Mario Behrens
An der Fakultät für PhysikInstitut für Theoretische Festkörperphysik (TFP)
Referent: Prof. Dr. Gerd SchönBetreuer: Dr. Fabian Pauly
Bearbeitungszeit: 10. September 2011 – 7. Dezember 2011
KIT – Universität des Landes Baden-Württemberg und nationales Forschungszentrum der Helmholtz-Gesellschaft www.kit.edu


Inhaltsverzeichnis
1 Einleitung 11.1 Gliederung der Arbeit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2 Theoretischer Hintergrund 52.1 Kohlenstoffnanorohren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.1 Klassifikation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52.1.2 Symmetrie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72.1.3 Elektronische Struktur . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.1.3.1 Bandstruktur des Graphens . . . . . . . . . . . . . . . . . . 72.1.3.2 Bandstruktur und Zustandsdichte der Kohlenstoffnanorohren 9
2.1.4 Ladungstransport der Kohlenstoffnanorohren . . . . . . . . . . . . . 122.2 Dichtefunktionaltheorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.2.1 Hohenberg-Kohn-Theoreme . . . . . . . . . . . . . . . . . . . . . . . 132.2.1.1 1. Theorem . . . . . . . . . . . . . . . . . . . . . . . . . . . 132.2.1.2 2. Theorem . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.2.2 Kohn-Sham Ansatz . . . . . . . . . . . . . . . . . . . . . . . . . . . . 142.2.3 LCAO Ansatz der Kohn-Sham-Orbitale . . . . . . . . . . . . . . . . 15
2.3 Ladungstransport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162.3.1 Greenscher Formalismus . . . . . . . . . . . . . . . . . . . . . . . . . 162.3.2 Konstruktion der Greenschen Funktion der Elektroden . . . . . . . . 17
3 Methoden 193.1 Elektronische Struktur der Kohlenstoffnanorohren . . . . . . . . . . . . . . 193.2 Einteilung der Rohre in Schichten . . . . . . . . . . . . . . . . . . . . . . . . 21
3.2.1 Kopplung entlang der Nanorohre . . . . . . . . . . . . . . . . . . . . 213.2.2 Wahl der Schichtgroße . . . . . . . . . . . . . . . . . . . . . . . . . . 223.2.3 Konstruktion der Schicht . . . . . . . . . . . . . . . . . . . . . . . . 233.2.4 Kopplung zweier Schichten . . . . . . . . . . . . . . . . . . . . . . . 24
3.3 Test des Dezimationsverfahrens mit einem Tight-Binding-Hamiltonian . . . 26
4 Ergebnisse 294.1 Zustandsdichte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294.2 Transmission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
5 Zusammenfassung 35
iii


1. Einleitung
Robert F. Curl, Richard E. Smalley und Harold W. Kroto erhielten 1996 den Nobelpreisin Chemie fur den experimentellen Nachweis der Existenz von Fullerene im Jahr 1985.Die Struktur dieser spharischen Kohlenstoffmolekule war jedoch schon viele Jahre zuvorbekannt.
Abbildung 1.1: Zeichnung von Leonardo da Vinci aus dem Werk “De divina proportione”des italienischen Mathematikers Luca Pacioli aus dem Jahr 1509. [1]
Der Ikosaeder aus Abbildung 1.1 entsprang der Feder von Leonardo da Vinci. Er besitzt12 funf- und 20 sechseckige Seitenflachen und entspricht der Form des bekannten C60
“Buckminster-fullerene”, bei dem die Eckpunkte der Seitenflachen die Positionen der Koh-lenstoffatome sind [2]. Der Name des besagten Molekuls beruht auf der Ahnlichkeit zur
1

2 1. Einleitung
geodatischen Kuppel des Architekten R. Buckminster Fuller. Die Entdeckung dieser fuß-ballahnlichen Molekule mit hoher Symmetrie im Jahre 1985 machte viele Wissenschaftlerauf diese Kohlenstoffsysteme aufmerksam.Sechs Jahre spater entdeckte Sumio Iijima die Kohlenstoffnanorohren.
Abbildung 1.2: Blick in eine Kohlenstoffnanorohre hinein.[3]
Diese zylinderformigen Gebilde versprechen aufgrund ihrer hohen Strombelastbarkeit, Warm-leitfahigkeit und Zugfestigkeit vielseitige Anwendungen. Das Besondere dieser eindimen-sionalen Kohlenstoffsysteme ist, dass allein ihre Geometrie die Leitungseigenschaften be-stimmt. So legt der Durchmesser und die Chiralitat fest, ob die Nanorohre metallischenoder halbleitenden Charakter besitzt.Diese Bachelorarbeit soll als Vorarbeit fur spinpolarisierte Transportbetrachtungen durchGraphensheets dienen und liefert die notwendigen Informationen uber die Elektroden, wel-che metallische Kohlenstoffnanorohren sind. Zudem wird als eigenstandiges Ergebnis dieserAusarbeitung die Zustandsdichte und die Transmission drei verschiedener Nanorohre be-rechnet.
1.1 Gliederung der Arbeit
Es wird eine Methode realisiert, welche eine ab-initio Beschreibung des Ladungstransportsdurch Kohlenstoffnanorohren ermoglicht. Dazu wird im Rahmen der Dichtefunktionaltheo-rie (DFT) die elektronische Struktur ermittelt. Die damit gewonnene Information liefert imLandauer-Buttiker-Formalismus in Verbindung mit den Nichtgleichgewichts-GreenschenFunktionen die Transmission der Kohlenstoffnanorohren. Um diese Ergebnisse mit einemanalytisch losbaren Modell zu vergleichen, wird zudem eine Tight-Binding-Berechnungdurchgefuhrt.
Um ferner den Einsatz der in dieser Arbeit untersuchten Nanorohren als Elektroden im Be-reich der molekularen Elektronik fur z.B. spinpolarisierte Transportuntersuchungen durchGraphen zu ermoglichen, beschrankt sich die Betrachtung auf metallische Kohlenstoffnano-rohren. Bei der Wahl des Durchmessers wird berucksichtigt, dass in Kohlenstoffnanorohrenmit zu kleinen Radien die π und σ Zustande aufgrund der starken Krummung hybridisieren[4]. Dadruch andert sich die Bandstruktur und folglich der metallische Charakter und dieLeitungseigenschaft. Fur große Radien hingegen wird die Berechnung der elektronischenStruktur aufgrund der großen Atomanzahl zu umfangreich. Untersucht werden somit Na-norohren mit der Klassifikation (n,m) = (6, 6), (7, 7) und (8, 8).
2

1.1. Gliederung der Arbeit 3
Als Ausgangspunkt dieser Ausarbeitung werden die Atomkoordinaten der Nanorohrenbestimmt. Dabei werden die Rohren so konstruiert, dass ihre Achsen entlang der z-Achseverlaufen und ihre Endungen mit Wasserstoffatomen terminiert sind. Die Ausrichtung derRohrenachse legt die Transportrichtung fest. Das Quantenchemiepaket TURBOMOLEV.6.3 [5] berechnet anschließend anhand dieser Geometriedaten die elektronische Struk-tur. Dies geschieht im Rahmen der DFT mit dem Perdew-Burke-Ernzerhof [6] (PBE)Austausch-Korrelations-Funktional. Zudem wird eine Vergleichsrechnung mit dem Hyprid-funktional PBE0 [7] durchgefuhrt, welches zusatzlichen Hartree-Fock-Austausch beinhal-tet. Zur Beschreibung der Kohlenstofforbitale werden die gaußschen atomaren Basisfunk-tionen def-SV(P) [8] und def-TZVP [9] verwendet.
Im verwendeten Transportformalismus wird das System in drei Bereiche eingeteilt: dieElektroden (L und R) und das Zentrum (C). Im Allgemeinen unterscheiden sich die Elek-troden von dem Zentrum. Hier wird jedoch das gesamte System aus einer Kohlenstoff-nanorohre aufgebaut. Die elektronische Struktur wird diesen drei Bereichen und derenKopplungen untereinander zugewiesen. Das Zentrum muss groß genug sein, damit dieKopplung zwischen L und R verschwindet. Wichtig fur die Transportbeschreibung sindunter anderem die Greenschen Funktionen der Elektroden, welche als Greensche Funk-tionen der Oberflache konstruiert werden. Diese Vereinfachung ist gerechtfertigt, da dieKopplung des Zentrums an die Elektroden eine endliche Reichweite hat.Die Greenschen Funktionen der Oberflache werden mit Hilfe des iterativen Dezimations-verfahrens von Guinea et al. [10] ermittelt. Bei diesem Verfahren muss die Kohlenstoffna-norohre entlang ihrer Achse in Schichten eingeteilt werden, wobei die Lange der Schichtso gewahlt wird, dass die Kopplung zwischen zwei ubernachsten Schichten verschwindet.Dazu werden in Translationsrichtung die Kopplungen zwischen Einheitszellen mit stei-gender Entfernung zueinander verglichen. Der Abfall dieser Kopplungen gibt schließlichAufschluss uber die Große der Schicht.
3


2. Theoretischer Hintergrund
2.1 Kohlenstoffnanorohren
2.1.1 Klassifikation
Eine Kohlenstoffnanorohre besitzt eine zylindrische Form und kann als ein zusammenge-rolltes Graphensheet beschrieben werden. Die geometrische Struktur der Rohre wird durchderen Chiralitatsvektor ~Ch definiert [11, 12].
Abbildung 2.1: Konstruktion der Kohlenstoffnanorohre aus einem zweidimensionalen Gra-phensheet mit Gittervektoren ~a1 und ~a2. [12]
Als Grundlage zum Verstandnis der Rohre dient ein Graphensheet. Die Einheitszelle des
Graphens wird durch zwei Gittervektoren ~a1 =(√
32 a,
12a)
und ~a2 =(√
32 a,−
12a)
auf-
gespannt. Die Große | ~a1,2 |= a ist die Gitterkonstante des Graphens und folgt aus derBeziehung a =
√3 ·acc, wobei acc = 1, 42 ·10−10 m die C-C-Bindungslange ist. Der Chirali-
tatsvektor der Kohlenstoffnanorohre setzt sich aus diesen beiden Gittervektoren zusammen
~Ch = n~a1 +m~a2 ≡ (n,m) . (2.1)
5

6 2. Theoretischer Hintergrund
Die ganzzahligen Koeffizienten n und m legen den Typ der Rohre fest. Man unterscheidethier die drei Typen Armchair, Zig-zag und chiral, welche in Tabelle 2.1 angegeben sind.
~Ch Typ
(n, n) Armchair
(n, 0) Zig-zag
(n,m) chiral
Tabelle 2.1: Klassifikation der Kohlenstoffnanorohre.
Um nun vom Graphen aus Abbildung 2.1 zur Rohre zu gelangen, wird das Sheet entlangdes Chiralitatsvektors aufgerollt. Demnach bestimmt ~Ch die Struktur des Rands der Rohreund | ~Ch | deren Umfang. Die Bezeichnungen “Armchair” und “Zig-zag” beschreiben dabeianschaulich diesen Rand (Abbildung 2.2).
Abbildung 2.2: Armchair- und Zig-zag-Kohlenstoffnanorohre [13].
Den Rohrendurchmesser dt = Chπ erhalt man aus der Lange des Chiralitatsvektors
dt =a
π
√n2 +m2 + nm . (2.2)
Der Vektor ~Ch bildet mit dem Gittervektor ~a1 den Chiralitatswinkel θ. Im Falle einerZig-zag-Kohlenstoffnanorohre ist θ = 0° und fur Armchair-Nanorohren 30°. Bei chiralenNanorohren liegt θ zwischen diesen beiden Grenzfallen.
Das Rechteck OAB’B aus Abbildung 2.1 bildet die Einheitszelle der Kohlenstoffnanorohre.Dabei gibt der Translationsvektor ~T die kurzeste Strecke entlang der Rohrenachse an, nachwelcher sich die Anordnung der Kohlenstoffatome wiederholt. Er steht senkrecht auf demChiralitatsvektor und kann auch aus den Gittervektoren des Graphens aufgebaut werden
~T = t1~a1 + t2~a2 ≡ (t1, t2) . (2.3)
Wie man Abbildung 2.1 leicht entnehmen kann, gilt fur eine Armchair- bzw. Zig-zag-Kohlenstoffnanorohre ~T = (1,−1) bzw. ~T = (1,−2). Fur chirale Rohren erhalt man denTranslationsvektor unter Verwendung der Bedingung ~Ch · ~T = 0
t1 =2m+ n
dR, t2 = −2n+m
dR(2.4)
6

2.1. Kohlenstoffnanorohren 7
dR ist dabei der großte gemeinsame Teiler von (2m+ n) und (2n+m).Die Flache |~a1 × ~a2|, welche von den Gittervektoren ~a1 und ~a2 des Graphens aufgespanntwird, umfasst zwei Kohlenstoffatome. In der Einheitszelle der Nanorohre (|~Ch × ~T |) sindsomit 2N C-Atome, wobei
N =|~Ch × ~T ||~a1 × ~a2|
=2(m2 + n2 +mn)
dR(2.5)
ist.
2.1.2 Symmetrie
Die Symmetrie [14] einer Einheitszelle der armchair (n, n) oder zigzag (n, 0) Kohlenstoff-nanorohre erhalt man aus dem direkten Produkt der Punktsymmetrien Dn und Ci
Dn ⊗ Ci =
Dnh n gerade
Dnd n ungerade. (2.6)
Fur die Kohlenstoffnanorohre beschreibt die Punktgruppe Dn eine n-zahlige Rotationsach-se (Cn), welche mit der Rohrenachse zusammenfallt, und n-mal 2-zahlige Rotationsachsensenkrecht zur Rohrenachse (nC2).Die Punktgruppe Ci beinhaltet eine Inversion i deren Inversionszentrum auf dem Mittel-punkt der Rohrenachse liegt.Entscheidend fur die vollstandige Symmetriegruppe der Rohre ist nun der Wert von n. Istn gerade, so bleibt bei einer Rotation um die Rohrenachse um π die Struktur der Rohreerhalten. D.h. es gibt eine Cm2m = C2 Rotation um die Rohrenachse. Findet daraufhin eineInversion statt, so entspricht dies einer Spiegelung an einer Ebene senkrecht zur Rohren-achse (Cm2m ⊗ i = σh).Ist n jedoch ungerade, so gibt es keine Cm2m Symmetrie und somit keine σh Operation. Diesfuhrt zur Aufteilung in Dnh und Dnd.
2.1.3 Elektronische Struktur
Die elektronische Struktur einer Kohlenstoffnanorohre lasst sich aus der Struktur des zuGrunde liegenden Graphens ableiten. Dazu wird die Bandstruktur des Graphens im Tight-Binding-Modell bestimmt. Der Ubergang zur Rohre wird schließlich mit Hilfe von periodi-schen Randbedingungen an die Wellenfunktion entlang des Rohrenumfangs realisiert. Esgibt somit entlang der ~Ch-Richtung nur eine bestimmte Anzahl an erlaubten Wellenvekto-ren. In der Translationsrichtung ~T der Tube, bleibt ~k jedoch kontinuierlich. Somit erhaltman fur die Kohlenstoffnanorohre eine eindimensionale Energiedispersion.Ein Lehrbuch, welches dieses Thema gut diskutiert, ist [11]. Die folgende Herleitung derelektronischen Struktur orientiert sich an dieses Buch.
2.1.3.1 Bandstruktur des Graphens
Die pz-Orbitale der Kohlenstoffatome des Graphens, welche nicht fur die Bindung benotigtwerden, bilden delokalisierte Elektronenzustande. Die fur den Transport relevanten Elek-tronen sind somit die π Valenzelektronen, welche diese Zustande einnehmen. Im Folgendenwird die Bandstruktur dieser Elektronen mit dem Tight-Binding-Modell bestimmt [15, 16].Ausgangspunkt ist die Schrodingergleichung
Hφ = Eφ (2.7)
7

8 2. Theoretischer Hintergrund
mit dem Hamiltonian H und den Energieeigenwerten E. Um die Wellenfunktion φ zubeschreiben, werden Bloch-Orbitale verwendet. Dabei muss berucksichtigt werden, dassGraphen eine zweiatomige Einheitszelle besitzt (Atom A und Atom B). Daher folgt dieEinteilung in φA und φB
φα(~r) =1√N
∑~Rα
ei~k ~Rαϕα(~r − ~Rα), (α = A,B) . (2.8)
Die Hamiltonmatrix hat somit die Eintrage Hαβ (α, β = A,B)
HAA =1
N
∑~RA
∑~RA′
eik( ~RA′−~RA) < ϕA(~r − ~RA)|H|ϕA(~r − ~RA′) >= HBB (2.9)
HAB =1
N
∑~RA
∑~RB
ei~k(~RB−~RA) < ϕA(~r − ~RA)|H|ϕB(~r − ~RB) >= H∗BA . (2.10)
Dabei ist N die Anzahl der Einheitszellen, ~Rx die Position des Atoms X und ϕX das zu-gehorige Atomorbital. Betrachtet man lediglich Wechselwirkung zwischen nachsten Nach-barn, so vereinfachen sich HAA und HAB. Das Matrixelement HAA reduziert sich auf dieBeitrage fur ~RA = ~RA′ :
HAA =1
N
∑~RA
< ϕA(~r − ~RA)|H|ϕA(~r − ~RA) >= ε
Ein Atom A ist von drei B Atomen umgeben. ~R1,2,3 seien die Vektoren zwischen demAtom A und seinen drei nachsten Nachbarn. Wechselwirkung zwischen weiter voneinanderentfernten Atomen wird vernachlassigt. HAB wird dadurch zu:
HAB =1
N
∑~RA
(ei~k ~R1 < ϕA(~r − ~RA)|H|ϕB(~r − ~RA − ~R1) > (2.11)
+ei~k ~R2 < ϕA(~r − ~RA)|H|ϕB(~r − ~RA − ~R2) (2.12)
+ei~k ~R3 < ϕA(~r − ~RA)|H|ϕB(~r − ~RA − ~R3) >) (2.13)
= t(ei~k ~R1 + ei
~k ~R2 + ei~k ~R3) (2.14)
= tf(~k) . (2.15)
Die Wellenfunktionen ϕ seien normiert. Demnach ist der Uberlapp SAA = SBB = 1. DieElemente SAB = S∗BA = sf(~k) mit s =< ϕA(~r − ~RA)|ϕB(~r − ~RA − ~R1,2,3) > erhalt mananalog zu Gl. (2.11-2.15).Die Schrodingergleichung (Gleichung 2.7) ist losbar, falls
det [H − ES] = 0 . (2.16)
Diese Beziehung liefert die Eigenwerte
EGraphen(~k) =ε± tω(~k)
1± sω(~k)(2.17)
mit
ω(~k) =
√|f(~k)|2 =
√√√√1 + 4 cos
(√3kxa
2
)cos
(kya
2
)+ 4 cos2
(kya
2
)(2.18)
8

2.1. Kohlenstoffnanorohren 9
wobei in f(~k) die Vektoren ~R1,2,3 durch die Gittervektoren ~a1,2 ersetzt wurden. Die zwei Lo-sungen der Energiedispersion stehen fur das Band der π-Bindung und der π∗-Antibindung.
Abbildung 2.3: Energiedispersion von Graphen [17].
Bei der zweidimensionalen Energiedispersion des Graphens beruhren sich die Bander inden K-Punkten. Die K-Punkte sind die Ecken der wabenformigen Brillouin-Zone.
2.1.3.2 Bandstruktur und Zustandsdichte der Kohlenstoffnanorohren
Die reziproken Gittervektoren der Kohlenstoffnanorohre haben die Gestalt
~K1 =1
N(−t2~b1 + t1~b2), ~K2 =
1
N(m~b1 − n~b2) (2.19)
~K1 zeigt entlang der ~Ch-Richtung und liefert diskrete k-Werte. ~K2 zeigt entlang der Roh-renachse und ist parallel zur ersten Brillouin-Zone der Nanorohre. N folgt aus Gl. (2.5).~b1 =
(2π√3a, 2πa
)und ~b2 =
(2π√3a,−2π
a
)sind die reziproken Gittervektoren des Graphens.
Der Vektor N ~K1 = −t2~b1 + t1~b2 ist als Linearkombination von ~b1 und ~b2 selbst reziprokerGittervektor des Graphens. Demnach sind zwei Wellenvektoren, die sich um N ~K1 unter-scheiden aquivalent. Fur die Vektoren µ ~K1 (µ = 0, ..., N − 1) trifft dies jedoch nichtzu. Diese N Vektoren liefern N diskrete k-Vektoren beim Ubergang vom Graphen zurNanorohre. Hieraus ergeben sich N eindimensionale Energiebander der Gestalt
Eµ(k) = EGraphen
(k~K2
| ~K2|+ µ ~K1
)(µ = 0, .., N − 1, und − π
T< k <
π
T) . (2.20)
Abbildung 2.4: Links: VerbindungWW ′ entspricht der ersten Brillouin Zone mit der Lange2πT und kontinuierlichen k-Werten. Rechts: Die Vektoren µ ~K1 bewirken Nparallele Verschiebungen der Brillouin Zone [11].
9

10 2. Theoretischer Hintergrund
Mit Hilfe der Abbildung 2.4 lasst sich die Bedingung fur metallische Nanorohren herleiten.Liegt eine Verbindung WW ′ auf dem K-Punkt der Brillouin Zone des Graphens, wo dieπ und π∗ Bander entartet sind, so beruhren sich die eindimensionalen Energiebander unddie Nanorohre zeigt metallischen Charakter. Diese Bedingung ist erfullt, wenn die Langedes Vektors ~Y K Vielfaches der Lange des Vektors ~K1 ist
~Y K =2n+m
3~K1 . (2.21)
Es ergeben sich metallische Kohlenstoffnanorohren, falls (n−m) ein Vielfaches von 3 ist. Indem vereinfachten Tight-Binding-Modell, welches keine Krummungseffekte berucksichtigt,sind Armchair-Nanorohren immer metallisch.
In diesem Abschnitt wird die Energiedispersion der Armchair-Nanorohren genauer be-trachtet. Wie bereits erwahnt gilt als Ausgangspunkt EGraphen(k) Gl. (2.17). Zur besserenHandhabung wird das Uberlappintegral s = 0 gesetzt
EGraphen(kx, ky) = ±t
√√√√1 + 4cos
(√3kxa
2
)cos
(kya
2
)+ 4cos2
(kya
2
)(ε = 0, s = 0) .
(2.22)
Fur | ~K1| sind nur diskrete Werte erlaubt:
| ~K1,q| =2π
|~Ch|· q , (q = 1, ..., 2n) . (2.23)
Mit der Lange des Chiralitatsvektors |~Ch| = π · dt (Gl. (2.2)) erhalt man
K1,q =2πq√3na
= kx,q . (2.24)
Setzt man dies in EGraphen(kx, ky) ein, so ergeben sich 2n eindimensionale Energiedisper-sionen fur die Armchair-Nanorohre (gekennzeichnet durch den Index a).
Eq,±a (k) = ±t
√1± 4 cos
(qπn
)cos
(ka
2
)+ 4 cos
(ka
2
), (−π < ka < π) , (q = 1, ..., 2n) .
(2.25)
10

2.1. Kohlenstoffnanorohren 11
Abbildung 2.5: Bandstruktur und Zustandsdichte fur (5,5) Nanorohre [18].
Auf der linken Seite der Abbildung 2.5 ist die Bandstruktur fur eine (5,5) Nanorohre dar-gestellt. Man erkennt sechs Leitungs- und sechs Valenzbander. Vier der Leitungs- bzw.Valenzbander sind zweifach entartet, sodass insgesamt zehn (2n = 10) bindende (+ Lo-sung) und zehn antibindende (− Losung) Zustande moglich sind. Die Energiedispersio-nen sind entlang der WW’ Richtung aus Abbildung 2.4 aufgetragen. Fur k = 0 befindetman sich beim Γ Punkt der Brillouin Zone. Die X Punkte sind bei k = ±π
a . Hier wirdEqa(k = ±π
a ) = ±γ0 fur alle q. γ0 = t ≈ 2, 9 eV ist die Tight-Binding-Kopplung nachsterNachbarn. Bei kF = ±2π
3a beruhren sich das Valenz- und das Leitungsband bei der Fermi-energie EF = 0. D.h. der ursprungliche K-Punkt des Graphens wird durch Gl. (2.20) aufzweidrittel der Strecke ΓX gefaltet.Die rechte Seite der Abbildung 2.5 zeigt die Zustandsdichte. Nanorohren sind eindimensio-nale Leiter und haben eine 1√
Evan Hove Singularitat an der Bandkante. Extrema in der
Dispersionsrelation sind demnach Singularitaten in der Zustandsdichte. Der metallischeCharakter der Armchair-Nanorohre spiegelt sich darin wider, dass die Zustandsdichte beider Fermienergie nicht verschwindet.Der Zusammenhang zwischen Energiedispersion und Zustandsdichte ist unten kurz an-gedeutet. Die Zustandsdichte gibt die Anzahl erlaubter Zustande je Energieintervall an[16, 18]. Fur sie gilt
ρ(E) =2
Ω
∑q
∑s=±
ˆdkδ(E − Eq,sa (k)) (2.26)
mit Ω = 4π| ~Ch|√3a2
.
ρ(E) =2
Ω
∑Ω
∑s=±
ˆdkδ(k − kq,s)|
∂Eq,sa (k)
∂k|−1 (2.27)
∂Eq,s=±a (k)∂k ist die Gruppengeschwindigkeit und lasst sich aus Gl. (2.25) berechnen. Extrema
in der Dispersionsrelation (∂Eq,s=±a (k)∂k = 0) sind demnach Singularitaten in der Zustands-
dichte.Daruber hinaus haben Mintmire und White gezeigt dass die Zustandsdichte fur eine Ein-heitslange entlang der Rohrenachse bei der Fermienergie eine Konstante ist und sich rezi-prok zum Umfang der Rohre verhalt [19].
11

12 2. Theoretischer Hintergrund
2.1.4 Ladungstransport der Kohlenstoffnanorohren
Der Ladungstransport der Kohlenstoffnanorohren wird mit Hilfe des Leitwerts
G = G0
∑n
τn(EF ) (2.28)
beschrieben (siehe Abschnitt 2.3). G0 = 2e2
h ist das Leitwertsquant. Der darin enthalte-ne Faktor 2 folgt aus der Spinentartung. τn ist die Transmission fur den Kanal n undhat fur idealen ballistischen Transport den Wert eins. Wie man Abbildung 2.5 entneh-men kann, kreuzen bei armchair Nanorohren zwei Energiebander die Fermienergie. JedesBand bildet einen Kanal aus mit einer Transmission von τn = 1 und stellt zwei Elektronenmit entgegengesetztem Spin zur Verfugung. An der Fermienergie ist der Leitwert demnach
G = G0
2∑n=1
τn(EF ) = 2 ·G0. Betrachtet man den Leitwert fur Energien an einer Bandkante,
so erkennt man einen Sprung in G(E), da ein neues Energieband einen weiteren Transport-kanal ausbildet. Zwischen den Bandkanten ist der Leitwert konstant. Er zeigt somit einengequantelten Verlauf. Abbildung 2.6 illustriert den Zusammenhang zwischen Bandstruktur(links), Zustandsdichte (Mitte) und Leitwert (rechts) fur eine (5,5) Armchair-Nanorohre.
Abbildung 2.6: Bandstruktur, Zustandsdichte und Leitwert einer (5,5) Kohlenstoffnano-rohre [18].
Die Summe∑nτn(E) = T (E) uber die Transmissionen der einzelnen Kanale ergibt die
gesamte Transmission der Nanorohre.
12

2.2. Dichtefunktionaltheorie 13
2.2 Dichtefunktionaltheorie
Die Dichtefunktionaltheorie ermoglicht die stationare Schrodingergleichung
HΨ(~x1, ..., ~xN , ~R1, ..., ~RM
)= EΨ
(~x1, ..., ~xN , ~R1, ..., ~RM
)(2.29)
fur ein System im Zustand Ψ, welches aus N Elektronen und M Atomkernen besteht,
naherungsweise zu losen [20, 21]. Die Koordinaten~Ri
umfassen hierbei 3M Ortsfrei-
heitsgrade der Atomkerne. Die Koordinaten ~xi umfassen 3N Ortsfreiheitsgrade undzusatzlich N Spinfreiheitsgrade der Elektronen. Der Hamiltonian eines solchen Systemshat folgende Gestalt
H = − 2
2me
N∑µ=1
∇2µ −
2
2
M∑A=1
1
MA∇2A −
N∑µ=1
M∑A=1
ZAe2
4πε0rµA(2.30)
+N∑µ=1
N∑ν>µ
e2
4πε0rµν+
M∑A=1
M∑B>A
ZAZBe2
4πε0RAB. (2.31)
Hier ist zum einen ein kinetischer Anteil der Elektronen und der Kerne und zum anderenein potentieller Anteil aufgrund der Coulomb-Wechselwirkung der Teilchen untereinanderzu erkennen.Mit Hilfe der Born-Oppenheimer-Naherung wird der Hamiltonian in einen rein elektroni-schen Hamiltonian ubergefuhrt. Im Rahmen dieser Naherung vereinfacht man das Systemzu sich bewegenden Elektronen im Feld der fixen Kerne. Diese Naherung ist aufgrundder im Vergleich zur Elektronenmasse sehr viel hoheren Kernmasse gerechtfertigt. Damitreduziert sich der Hamilton-Operator auf:
Hel = − 2
2me
N∑µ=1
∇2µ −
N∑µ=1
M∑A=1
ZAe2
4πε0rµA+
N∑µ=1
N∑ν>µ
e2
4πε0rµν= T + VNe + Vee (2.32)
(Zu beachten ist, dass damit die ausHel resultierende Energie nichtmehr der Gesamtenergiedes Systems entspricht.)Die entscheidende Große in der DFT ist die Elektronendichte des Systems. Sie lasst sichaus der Wellenfunktion fur die Elektronen gewinnen
ρ(~r) = N
ˆ...
ˆ| ψ(~x1, ..., ~xn) |2 ds1d
4x2...d4xN (2.33)
und gibt die Wahrscheinlichkeit an, eines der N Elektronen im Volumenelement d3r mitwillkurlichem Spin zu finden. Die besondere Rolle von ρ folgt aus den Hohenberg-KohnTheoremen.
2.2.1 Hohenberg-Kohn-Theoreme
2.2.1.1 1. Theorem
Das 1. Hohenberg-Kohn-Theorem enthalt die wichtige Aussage, dass die Grundzustand-selektronendichte ein System aus Elektronen mit externen Potential Vext(~r) (in unseremFall ware Vext = VNe) vollstandig beschreibt, da sie den Hamiltonian eindeutig festlegt[22, 23],
ρ0 ⇒N,ZA, ~RA
⇒ Hel ⇒ ψ0 ⇒ E0 . (2.34)
13

14 2. Theoretischer Hintergrund
Der Grundzustand eines Systems ist Funktional der Grundzustandsdichte (ψ0 = ψ [ρ0]).Diese eindeutige Abbildung zwischen Grundzustand und Grundzustandsdichte hat alsKonsequenz, dass der Grundzustandserwartungswert einer Observablen auch Funktionalder Grundzustandsdichte ist
< ψ [ρ0] |O|ψ [ρ0] >= O [ρ0] . (2.35)
Hieraus folgt der in (2.34) angedeutete Zusammenhang zwischen der Grundzustandsenergieund der Grundzustandsdichte. Das Energiefunktional lasst sich nun wie folgt schreiben
E0 [ρ0] = T [ρ0] + Eee [ρ0] + ENe [ρ0] . (2.36)
Dadurch vereinfacht sich die Bestimmung der Energie, da statt der 3N Freiheitsgradeder Wellenfunktion lediglich 3 Freiheitsgrade der Elektronendichte berucksichtigt werdenmussen.
2.2.1.2 2. Theorem
Das 2. Theorem besagt, dass nur mit der Grundzustandsdichte das Funktional E [ρ] dieGrundzustandsenergie liefert [22, 23].Das Theorem lasst sich wie folgt vereinfacht darstellen
E0 = E [ρ0] ≤ E [ρ] . (2.37)
2.2.2 Kohn-Sham Ansatz
Bei der Anwendung der Hohenberg-Kohn-Theoreme stoßt man auf das Problem, dassdie exakte kinetische Energie und der nicht-klassische Beitrag zur Elektron-Elektron-Wechselwirkung unbekannt sind. Kohn und Sham entwickelten eine Methode wie die un-bekannten Funktionale approximiert werden konnen. Dazu definiert man ein wechselwir-kungsfreies Referenzsystem mit dem Hamiltonian Hs, welcher ein effektives Potential Vs(~r)beinhaltet
Hs = − 2
2me
N∑µ=1
∇2i +
N∑µ=1
Vs(~r) , (2.38)
Hsφ = Esφ . (2.39)
Die Wellenfunktion φ dieses Systems ist als Slater Determinante aus Einelektron-Wellenfunktionen(Kohn-Sham-Orbitalen) ϕi konstruiert. Sie erfullen die Gleichung
fKSϕµ = εµϕµ (2.40)
mit dem Kohn-Sham Operator
fKS = − 2
2me∇2 + Vs(~r) (2.41)
Der Zusammenhang zum eigentlichen System mit Wechselwirkung wird nun hergestellt,indem das effektive Potential Vs so gewahlt wird, dass die Dichte des Referenzsystems derGrundzustandsdichte des Systems mit Wechselwirkung entspricht
ρs(~r) =N∑µ=1
1/2∑si=−1/2
|ϕµ(~r, s)|2 = ρ0(~r) . (2.42)
14

2.2. Dichtefunktionaltheorie 15
Die kinetische Energie dieses Referenzsystems
Ts = − 2
2me
N∑µ=1
< ϕµ|∇2|ϕµ > (2.43)
entspricht aufgrund fehlender Wechselwirkung nicht der kinetischen Energie des eigentli-chen Systems (T 6= Ts).Kohn und Sham fuhren an dieser Stelle ein sogenanntes Austausch-Korrelations-FunktionalEXC [ρ] ein, welches alle unbekannten Beitrage erfassen soll. Somit ist in EXC der unbe-kannte Teil der kinetischen Energie Tc [ρ] = T [ρ]−Ts [ρ], sowie der nichtklassische BeitragEncl [ρ] = Eee [ρ] − J [ρ] der Elektron-Elektron-Wechselwirkung enthalten. J [ρ] steht furdie klassische Coulomb-Wechselwirkung der Elektronen untereinander
J [ρ] =e2
8πε0
ˆ ˆρ(~r1)ρ(~r2)
r12d3r1d
3r2 . (2.44)
Das Energie- und das Austausch-Korrelations-Funktional haben nun die Gesalt
E [ρ] = Ts [ρ] + J [ρ] + ENe [ρ] + EXC [ρ] (2.45)
EXC [ρ] = (T [ρ]− Ts [ρ]) + (Eee [ρ]− J [ρ]) . (2.46)
Vergleicht man nun die Variation von E [ρ] mit der von Es [ρ]
0 =δE [ρ]
δρ=δTs [ρ]
δρ+δJ [ρ]
δρ+δENe [ρ]
δρ+δEXC [ρ]
δρ(2.47)
=δTs [ρ]
δρ+ VNe + VJ + VXC (2.48)
0 =δEs [ρ]
δρ=δTs [ρ]
δρ+ Vs (2.49)
so erhalt man die Form von Vs
Vs(~r1) =e2
4πε0
ˆρ(~r2)
r12d3r2︸ ︷︷ ︸
VJ
+ VXC(~r1)− e2
4πε0
M∑A=1
ZAr1A︸ ︷︷ ︸
VNe
. (2.50)
2.2.3 LCAO Ansatz der Kohn-Sham-Orbitale
Ziel ist es nun die Kohn-Sham Gleichungen zu losen. Eine effiziente Strategie ist dabei derLCAO Ansatz. Hierbei schreibt man die Kohn-Sham Orbitale als Linearkombination vonatomaren Orbitalen
ϕµ =
L∑i=1
ciµφi . (2.51)
Der endliche Satz an Basisfunktionen φi wird so gewahlt, dass die Kohn-Sham Orbitalebestmoglichst approximiert werden. Setzt man diesen Ansatz in Gleichung (2.38) ein, soergibt sich die Matrix-Gleichung
FKS(C)C = SCε (2.52)
15

16 2. Theoretischer Hintergrund
mit der Fock-Matrix
FKSi,j =
ˆφi(~r1)fKSφj(~r1)d3r1 , (2.53)
der Uberlapp-Matrix
Si,j =
ˆφi(~r1)φj(~r1)d3r1 , (2.54)
der Matrix der Entwicklungskoeffizienten
Ciµ = ciµ (2.55)
und der Matrix der Eigenenergien
εµν = δµνεµ (2.56)
Damit reduziert sich die Auswertung auf ein lineares Eigenwertproblem mit den Variablenciµ, welches iterativ gelost werden kann.
2.3 Ladungstransport
2.3.1 Greenscher Formalismus
Zur Beschreibung des Transports durch Kontakte von atomarer Großenordnung wird derLandauer-Buttiker-Formalismus in Verbindung mit der Greenschen Funktion verwendet[24, 25, 26, 27]. Der experimentelle Aufbau lasst sich in drei Bereiche einteilen - links(L), Zentrum (C), rechts (R). L und R sind halbunendlich ausgedehnte Elektroden unddienen als unendlich große Elektronenreservoirs mit den chemischen Potentialen µL undµR. Dazwischen ist die Region C durch welche der Transport untersucht werden soll.Der Stromfluß durch das Zentrum ist
I =2e
h
ˆdE(f(E − µL)− f(E − µR))T (E) . (2.57)
Dabei beschreibt f(E−µL,R) die Fermifunktion von L bzw. R und T (E) die Transmission
T (E) = Tr [ΓL(E)GrCC(E)ΓR(E)GaCC(E)] = Tr[t†(E)t(E)
](2.58)
mit der Green’schen Funktion des Zentrums
GrCC(E) = [ESCC −HCC − ΣrL(E)− Σr
R(E)]−1 , (2.59)
GaCC = [GrCC ]† ,
den Selbstenergien
Σrx(E) = (HCX − ESCX)grxx(E)(HXC − ESXC) (2.60)
und Linienverbreiterungsmatrizen
Γx(E) = −2Im [Σrx(E)] , (2.61)
16

2.3. Ladungstransport 17
wobei grXX = (ESXX −HXX)−1 die Green’sche Funktion der Elektrode X = L,R ist.An C liegt die Spannung V = µL−µR
e an. Der Leitwert ist dann
G =dI
dV|V=0 =
2e2
h
ˆdE
[− ∂
∂Ef(E, T )
]T (E) . (2.62)
Fur tiefe Temperaturen wird die Fermifunktion zur Stufenfunktion und− ∂∂E f = δ(E−EF ).
Damit ist der Leitwert fur T ≈ 0 K das Produkt aus dem Leitwertquant G0 und derTransmission fur E = EF
G =2e2
hT (EF ) = G0T (EF ) . (2.63)
Aufgrund der endlichen Reichweite der Kopplungselemente HCX − ESCX reicht es aus,grXX fur die ersten Atomschichten zu konstruieren. Die Green’sche Funktion der Elektrodewird deshalb mit Hilfe des Dezimationsverfahrens von Guinea et al. [10] als Green’scheFunktion der Oberflache konstruiert.
2.3.2 Konstruktion der Greenschen Funktion der Elektroden
Ziel des Dezimationsverfahrens [10] ist es den Oberflachenanteil der Greenschen Funkti-on der halbunendlichen Elektroden zu extrahieren. Dazu wird die Elektrode in Schichteneingeteilt. Die Schichtgroße wird so gewahlt, dass nur noch Kopplung zwischen nachstenNachbarschichten vorhanden ist.Ausgangspunkt fur die Konstruktion der Greenschen Funktion der Oberflache ist die De-finition
[(E + iη)S −H]Gr = 1 . (2.64)
Die Matrizen S,H,Gr werden gemaß der Schichtwahl in Blocke eingeteilt
∞∑n=0
[(E + iη)Sm,n −Hm,n]Grn,l = 1m,l . (2.65)
Die Indizes parametrisieren jeweils eine Schicht. Ein Element (m,n) beschreibt demnachdie Kopplung zwischen der Schicht m und der Schicht n. Ziel ist es nun dieses Gleichungs-system (Gl. (2.65)) fur Gr0,0 zu losen. Dabei wird ausgenutzt, dass Sm,m′ = Sm−m′,0 bzw.Hm,m′ = Hm−m′,0 und Sm,0 = Hm,0 = 0 fur m ≥ 2 gilt.Zunachst erhalt man aus Gl. (2.65) die Gleichungen
WGr0,0 + τ1Gr1,0 = 1 , (2.66)
τ2Grm−1,0 +WGrm,0 + τ1G
rm+1,0 = 0 fur m ≥ 1 (2.67)
mit den Abkurzungen
W = (E + iη)S0,0 −H0,0 , (2.68)
τ1 = (E + iη)S0,1 −H0,1 , (2.69)
τ2 = (E + iη)S1,0 −H1,0 . (2.70)
17

18 2. Theoretischer Hintergrund
Das Verfahren [10] fuhrt mit den Gln. (2.66) und (2.67) auf die iterativen Gleichungen
W(n)sR = W
(n−1)sR − τ (n−1)
1
(W
(n−1)b
)−1τ
(n−1)2 , (2.71)
W(n)sL = W
(n−1)sL − τ (n−1)
2
(W
(n−1)b
)−1τ
(n−1)1 , (2.72)
W(n)b = W
(n−1)b − τ (n−1)
1
(W
(n−1)b
)−1τ
(n−1)2 − τ (n−1)
2
(W
(n−1)b
)−1τ
(n−1)1 , (2.73)
τ(n)1 = −τ (n−1)
1
(W
(n−1)b
)−1τ
(n−1)1 , (2.74)
τ(n)2 = −τ (n−1)
2
(W
(n−1)b
)−1τ
(n−1)2 (2.75)
mit den Anfangswerten W(0)b = W
(0)s = W , τ
(0)1 = τ1, τ
(0)2 = τ2. Sind τ
(n)1 und τ
(n)2
nach n = ν Schritten vernachlassigbar klein, so erhalt man die Greensche Funktion derOberflache der linken (Index L) bzw. der rechten (Index R) Elektrode aus den Gleichungen
Gr0,0;L =(W
(ν)sL
)−1, (2.76)
Gr0,0;R =(W
(ν)sR
)−1(2.77)
und die Greensche Funktion des Festkorpers
Grn,n =(W
(ν)b
)−1. (2.78)
18

3. Methoden
In dieser Bachelorarbeit wird die Transmission und die Zustandsdichte fur eine (6,6), (7,7)und (8,8) Kohlenstoffnanorohre berechnet. Die Nanorohren werden gemaß Abschnitt 2.1.1konstruiert und die offenen Bindungen an den Enden der Rohren werden mit Wasserstof-fatomen terminiert. Das Quantenchemie-Programm TURBOMOLE V.6.3 [5] liefert dieelektronische Struktur innerhalb der DFT, welche in Verbindung mit dem Dezimations-verfahren (Abschnitt 2.3.2) die Greensche Funktion des Festkorpers sowie die GreenscheFunktion der Oberflache liefert. Die Greensche Funktion des Festkorpers gibt Aufschlussuber die Zustandsdichte der Kohlenstoffnanorohre. Die Greensche Funktion der Oberflachehingegen kann verwendet werden zur Berechnung der Transmission.
3.1 Elektronische Struktur der Kohlenstoffnanorohren
Fur die drei erwahnten Nanorohren wird die elektronische Struktur bestimmt. D.h. eswerden die Matrixelemente des Hamiltonians Hkiα,ljβ =< k, i, α|H|l, j, β > ermittelt. DieIndizes k und l stehen fur die Einheitszellen. i und j legen die Positionen der Atome inden jeweiligen Einheitszellen fest. α und β sind atomare Basisfunktionen (die φi aus Gl.(2.49)).Bei der Berechnung mit TURBOMOLE werden folgende Einstellungen gewahlt. Fur dieKohlenstoff- und Wasserstoffatome der Nanorohre wird die def-SV(P) Basis [8] verwendet.Der Wasserstoff wird durch insgesamt 2 Basisfunktionen (2 s-Orbitale) beschrieben. DerKohlenstoff erhalt 3 s-Orbitale, 2 p-Orbitale und 1 d-Orbital (insgesamt 14 Basisfunktio-nen). Eine zusatzliche Berechnung der (6,6) Rohre mit der erweiterten Basis def-TZVP [9]soll zum Vergleich dienen. Als Symmetrie der Rohre wird Dnh oder Dnd gewahlt (sieheAbschnitt 2.1.2). Die DFT-Rechnungen werden fur alle drei Rohren mit dem Austausch-Korrelations-Funktional PBE [6] (GGA) durchgefuhrt. Zudem wird fur die (6,6) Rohreeine Vergleichsrechnung mit dem Funktional PBE0 [7] (hybrid) durchgefuhrt. Um die kor-rekte Besetzung der Molekulorbitale zu ermitteln, wird die Temperatur des elektronischenSystems von 900 K auf 30 K abgekuhlt (“fermi smearing of occupation numbers”) [28].Das Konvergenzkriterium der totalen Energie wird auf 10−7 H gesetzt.
Bei der Verwendung des Programms TURBOMOLE sind wir auf Systeme mit endlicherGroße beschrankt. Die Kohlenstoffnanorohren konnen deshalb nicht beliebig lang gewahltwerden. Die (6, 6) Nanorohre wird aus 19 Einheitszellen aufgebaut. Die (7, 7) Nanorohrebesitzt 17 Einheitszellen und die (8, 8) Nanorohre umfasst 15 Einheitszellen.
19
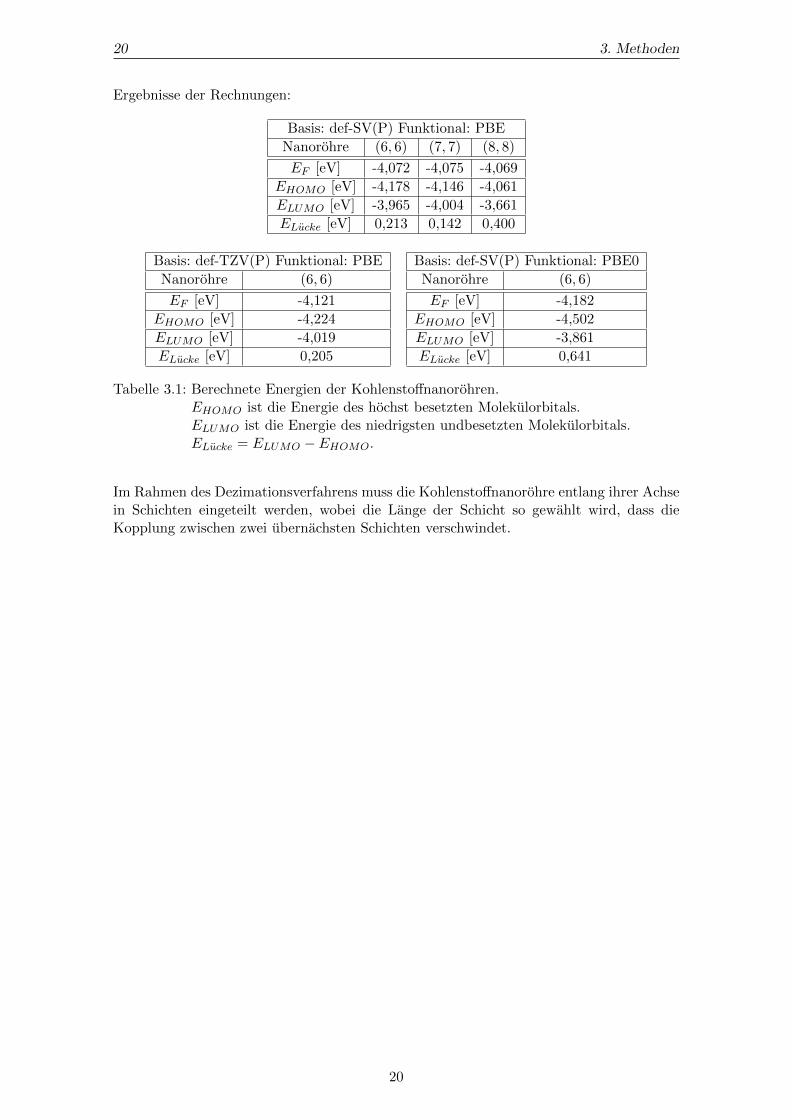
20 3. Methoden
Ergebnisse der Rechnungen:
Basis: def-SV(P) Funktional: PBE
Nanorohre (6, 6) (7, 7) (8, 8)
EF [eV] -4,072 -4,075 -4,069
EHOMO [eV] -4,178 -4,146 -4,061
ELUMO [eV] -3,965 -4,004 -3,661
ELucke [eV] 0,213 0,142 0,400
Basis: def-TZV(P) Funktional: PBE
Nanorohre (6, 6)
EF [eV] -4,121
EHOMO [eV] -4,224
ELUMO [eV] -4,019
ELucke [eV] 0,205
Basis: def-SV(P) Funktional: PBE0
Nanorohre (6, 6)
EF [eV] -4,182
EHOMO [eV] -4,502
ELUMO [eV] -3,861
ELucke [eV] 0,641
Tabelle 3.1: Berechnete Energien der Kohlenstoffnanorohren.EHOMO ist die Energie des hochst besetzten Molekulorbitals.ELUMO ist die Energie des niedrigsten undbesetzten Molekulorbitals.ELucke = ELUMO − EHOMO.
Im Rahmen des Dezimationsverfahrens muss die Kohlenstoffnanorohre entlang ihrer Achsein Schichten eingeteilt werden, wobei die Lange der Schicht so gewahlt wird, dass dieKopplung zwischen zwei ubernachsten Schichten verschwindet.
20

3.2. Einteilung der Rohre in Schichten 21
3.2 Einteilung der Rohre in Schichten
3.2.1 Kopplung entlang der Nanorohre
Um nun herauszufinden, wieviele Atome eine Schicht umfasst, wird die Kopplung zwischenverschiedenen Atomen betrachtet, welche auf einer Strecke parallel zur Rohrenachse liegen(siehe Abbildung 3.1).
Abbildung 3.1: Aufgerollte Kohlenstoffnanorohre. Der rote Pfeil gibt die Ausrichtung derRohrenachse an. Die betrachteten Kopplungen sind durch die blauen Pfeiledargestellt.
Extrahiert werden die Eintrage
H0iα,ljβ =< k = 0, i, α|H|l, i, β > (3.1)
Der Index k = 0 steht fur die Einheitszelle, welche das in Abbildung 3.1 mit 0 markierteAtom beinhaltet. Das Atom mit der Markierung 1 befindet sich in der benachbarten Ein-heitszelle. Der Index l aus Gleichung (3.1) lauft somit von 1-5 und reprasentiert die auf-einanderfolgenden Einheitszellen in Translationsrichtung. Die Kopplungen zwischen denEinheitszellen wird fur diejenigen Atome extrahiert, welche auf eine Verbindungstreckeparallel zur Rohrenachse liegen. Daher gilt i = j. Zur Beschreibung der Kohlenstoffatomewerden 14 Basisfunktionen gewahlt. Demnach gibt es 142 verschiedene H0i,li. Wichtig istjeweils der großte Beitrag, welcher in Abbildung 3.2 zu sehen ist.
21
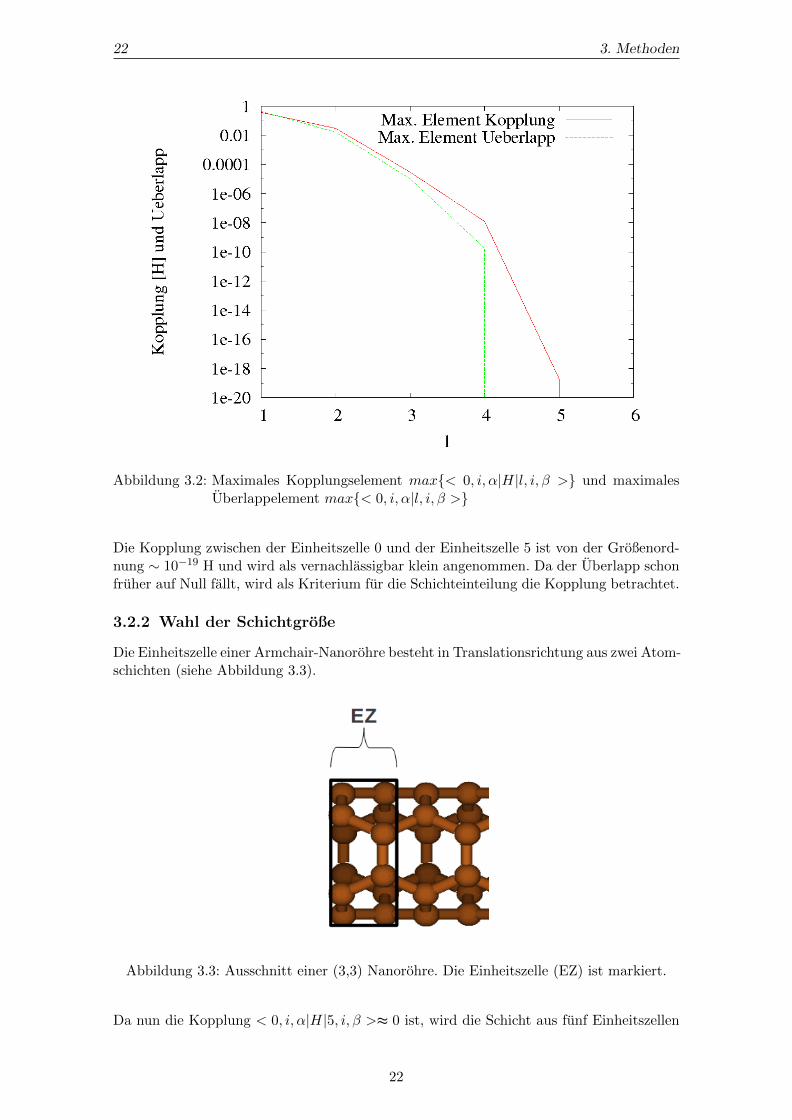
22 3. Methoden
Abbildung 3.2: Maximales Kopplungselement max< 0, i, α|H|l, i, β > und maximalesUberlappelement max< 0, i, α|l, i, β >
Die Kopplung zwischen der Einheitszelle 0 und der Einheitszelle 5 ist von der Großenord-nung ∼ 10−19 H und wird als vernachlassigbar klein angenommen. Da der Uberlapp schonfruher auf Null fallt, wird als Kriterium fur die Schichteinteilung die Kopplung betrachtet.
3.2.2 Wahl der Schichtgroße
Die Einheitszelle einer Armchair-Nanorohre besteht in Translationsrichtung aus zwei Atom-schichten (siehe Abbildung 3.3).
Abbildung 3.3: Ausschnitt einer (3,3) Nanorohre. Die Einheitszelle (EZ) ist markiert.
Da nun die Kopplung < 0, i, α|H|5, i, β >≈ 0 ist, wird die Schicht aus funf Einheitszellen
22

3.2. Einteilung der Rohre in Schichten 23
aufgebaut. Fortan wird eine vereinfachte graphische Darstellung der Einheitszelle und derSchicht verwendet:
Abbildung 3.4: Vereinfachte Darstellung Einheitszellen (EZ).
Abbildung 3.5: Vereinfachte Darstellung Schicht (aus 5 EZ).
Die Matrixelemente Hkiα,ljβ =< k, i, α|H|l, j, β > fur alle i, α der Einheitszelle k und allej, β der Einheitszelle l werden nun zusammengefasst zur Matrix HEZ
k,l .
Der Hamiltonian der Schicht hat die Gestalt
HSchicht =
HEZ
0,0 HEZ0,1 HEZ
0,2 HEZ0,3 HEZ
0,4
HEZ1,0 HEZ
1,1 HEZ1,2 HEZ
1,3 HEZ1,4
HEZ2,0 HEZ
2,1 HEZ2,2 HEZ
2,3 HEZ2,4
HEZ3,0 HEZ
3,1 HEZ3,2 HEZ
3,3 HEZ3,4
HEZ4,0 HEZ
4,1 HEZ4,2 HEZ
4,3 HEZ4,4
. (3.2)
Sieht man die Schicht in mitten einer idealen, unendlich langen Rohre, so sind aufgrundder Translationsinvarianz die Matrixblocke HEZ
k,l mit gleichem k−l identisch. Im Folgenden
wird die Bezeichnung EZ bei HEZi,j nicht weiter mitgefuhrt. Der Hamiltonian der Schicht
vereinfacht sich damit zu
HSchicht =
H0,0 H0,1 H0,2 H0,3 H0,4
H1,0 H0,0 H0,1 H0,2 H0,3
H2,0 H1,0 H0,0 H0,1 H0,2
H3,0 H2,0 H1,0 H0,0 H0,1
H4,0 H3,0 H2,0 H1,0 H0,0
. (3.3)
Zudem gilt
Hk,l = (Hl,k)T . (3.4)
Bei der Berechnung der Eintrage vonHSchicht (Gl. (3.3)) sind wir wegen TURBOMOLE aufeine endliche Rohre beschranken. Der Einfluss der Rander stort die Symmetrie in HSchicht.Die folgende Aufgabe besteht nun darin, bestmoglichste Werte fur Hk,l zu ermitteln.
3.2.3 Konstruktion der Schicht
Um die Auswirkung der Rander gering zu halten, wird eine moglichst lange Rohre verwen-det. Des weiteren wird die Rohre aus einer ungeraden Anzahl an Einheitszellen aufgebaut,damit es eine mittig gelegene Einheitszelle gibt.
23

24 3. Methoden
Abbildung 3.6: Ausschnitt der Nanorohre.
Die Abbildung 3.6 zeigt einen Ausschnitt der Nanorohre. Der Block 0 ist die Einheitszellein der Mitte. Fur sie wurde H0,0 extrahiert und in HSchicht verwendet. H0,0 hat damit dengeringsten Randeinfluss.Um die Translationssymmetrie der Kopplungen zwischen zwei Einheitszellen entlang derRohrenachse zu gewahrleisten, werden die Kopplungen symmetrisiert [26]
Hsym0,l =
1
2
(H0,l + (H0,−l)
T)
mit l = 1, 2, 3, 4 . (3.5)
In Abbildung 3.6 sind als Beispiel die Kopplungen H0,3 und H0,−3 dargestellt. Der Einflussder Symmetrisierung ist in Tabelle 3.2 einzusehen. Hier wird der maximale Unterschiedzwischen einem Hsym
0,j Element und dem zugehorigen H0,j Element berechnet (Werte fur(6,6) Nanorohre).
l max|Hsym
0,l −H0,l|
[H]
1 5, 67 · 10−5
2 3, 54 · 10−6
3 3, 28 · 10−9
4 5, 01 · 10−11
Tabelle 3.2: Einfluss Symmetrisierung bei (6,6) Nanorohre.
Nun sind alle Voraussetzungen gegeben, um HSchicht gemaß Gl. (4.3) zu konstruieren.Fur das Dezimationsverfahren (siehe Abschnitt 2.3.2) wird zudem die Kopplung zwischenzwei benachbarten Schichten benotigt (Gl. (2.69) und (2.70)). Fur diese Kopplung sind -wie der nachste Abschnitt zeigt - schon alle notigen Informationen gegeben.
3.2.4 Kopplung zweier Schichten
Abbildung 3.7: Kopplung zweier Schichten.
24

3.2. Einteilung der Rohre in Schichten 25
Der Hamiltonian des Systems aus Abbildung 3.7 hat die Gestalt
H =
(HSchicht 1 HSchicht 12
HSchicht 21 HSchicht 2
)(3.6)
HSchicht 12 beschreibt die Kopplung zwischen Schicht 1 und Schicht 2
HSchicht 12 =
H0,0′ H0,1′ H0,2′ H0,3′ H0,4′
H1,0′ H1,1′ H1,2′ H1,3′ H1,4′
H2,0′ H2,1′ H2,2′ H2,3′ H2,4′
H3,0′ H3,1′ H3,2′ H3,3′ H3,4′
H4,0′ H4,1′ H4,2′ H4,3′ H4,4′
. (3.7)
Nach Abschnitt 3.2.1 verschwinden Kopplungen Hk,l mit |l − k| > 4. Dies trifft fur dieDiagonaleintrage vonHSchicht 12 und die Eintrage oberhalb der Diagonalen zu. Die Eintrageunterhalb der Diagonalen wurden bereits in Abschnitt 3.3 bestimmt. HSchicht 12 vereinfachtsich zu
HSchicht 12 =
0 0 0 0 0
H0,4 0 0 0 0H0,3 H0,4 0 0 0H0,2 H0,3 H0,4 0 0H0,1 H0,2 H0,3 H0,4 0
. (3.8)
Die Kopplung zwischen Schicht 2 und Schicht 1 HSchicht 21 entspricht (HSchicht 12)T .
Auf analoge Weise wird SSchicht und SSchicht 12 bzw. SSchicht 21 aus den Eintragen Skiα,ljβ =<k, i, α|l, j, β > der Uberlappmatrix konstruiert.
Die Motivation fur die Schichteinteilung folgt aus dem Dezimationsverfahren aus Abschnitt2.3.2. Hier wird der Hamiltonian (in diesem Fall der Hamiltonian der Kohlenstoffnanoroh-re) in Blocke Hm,n eingeteilt. Die Indizes m und n parametrisieren jeweils die Schicht. Desweiteren gilt Hm,0 = Sm,0 = 0 fur m ≥ 2. Diese Bedingung, dass die Kopplung zwischenubernachsten Schichten verschwindet, wurde bei der Konstruktion der Schicht der Nano-rohre verwendet.H0,0, H0,1 und H1,0 sowie S0,0, S0,1 und S1,0 aus den Gleichungen (2.68)-(2.70) entsprechensomit HSchicht, HSchicht 12 und HSchicht 21 sowie SSchicht, SSchicht 12 und SSchicht 21.
25
26 3. Methoden
3.3 Test des Dezimationsverfahrens mit einem Tight-Binding-Hamiltonian
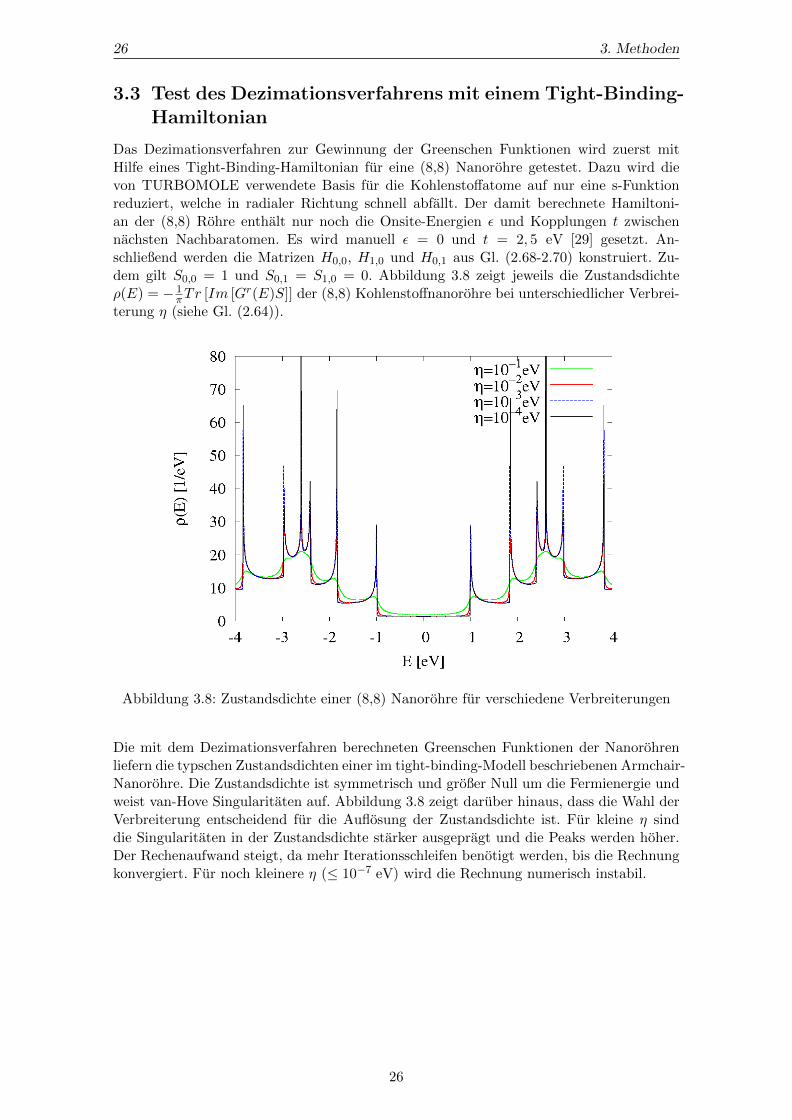
Das Dezimationsverfahren zur Gewinnung der Greenschen Funktionen wird zuerst mitHilfe eines Tight-Binding-Hamiltonian fur eine (8,8) Nanorohre getestet. Dazu wird dievon TURBOMOLE verwendete Basis fur die Kohlenstoffatome auf nur eine s-Funktionreduziert, welche in radialer Richtung schnell abfallt. Der damit berechnete Hamiltoni-an der (8,8) Rohre enthalt nur noch die Onsite-Energien ε und Kopplungen t zwischennachsten Nachbaratomen. Es wird manuell ε = 0 und t = 2, 5 eV [29] gesetzt. An-schließend werden die Matrizen H0,0, H1,0 und H0,1 aus Gl. (2.68-2.70) konstruiert. Zu-dem gilt S0,0 = 1 und S0,1 = S1,0 = 0. Abbildung 3.8 zeigt jeweils die Zustandsdichteρ(E) = − 1
πTr [Im [Gr(E)S]] der (8,8) Kohlenstoffnanorohre bei unterschiedlicher Verbrei-terung η (siehe Gl. (2.64)).
Abbildung 3.8: Zustandsdichte einer (8,8) Nanorohre fur verschiedene Verbreiterungen
Die mit dem Dezimationsverfahren berechneten Greenschen Funktionen der Nanorohrenliefern die typschen Zustandsdichten einer im tight-binding-Modell beschriebenen Armchair-Nanorohre. Die Zustandsdichte ist symmetrisch und großer Null um die Fermienergie undweist van-Hove Singularitaten auf. Abbildung 3.8 zeigt daruber hinaus, dass die Wahl derVerbreiterung entscheidend fur die Auflosung der Zustandsdichte ist. Fur kleine η sinddie Singularitaten in der Zustandsdichte starker ausgepragt und die Peaks werden hoher.Der Rechenaufwand steigt, da mehr Iterationsschleifen benotigt werden, bis die Rechnungkonvergiert. Fur noch kleinere η (≤ 10−7 eV) wird die Rechnung numerisch instabil.
26

3.3. Test des Dezimationsverfahrens mit einem Tight-Binding-Hamiltonian 27
Mit Hilfe der Greenschen Funktion der Oberflache lasst sich die Transmission der Nano-rohre berechnen, welche in Abbildung 3.9 fur eine Verbreiterungen von η = 10−4 eV undη = 10−1 eV aufgetragen ist.
Abbildung 3.9: Transmission einer (8,8) Nanorohre fur η = 10−1 eV und η = 10−4 eV
In der Abbildung 3.9 ist die Transmission fur η = 10−1 eV an der Fermienergie T (EF =0) < 2. Hier ist η relativ groß gewahlt. In Abbildung 3.8 ist die zugehorige Zustandsdich-te dargestellt. Dort erkennt man, dass die Zustande nicht genau aufgelost werden. Diesspiegelt sich in der Transmission wider. Der Sprung an den Bandkanten ist nicht eindeutigund zwischen zwei Bandkanten ist die Transmission nicht konstant. Fur eine Verbreiterungvon η = 10−4 eV hingegen ist wie erwartet T (EF = 0) = 2. Die Transmission zeigt dascharakteristische, quantisierte Verhalten fur Armchair-Kohlenstoffnanorohren.
27
28 3. Methoden
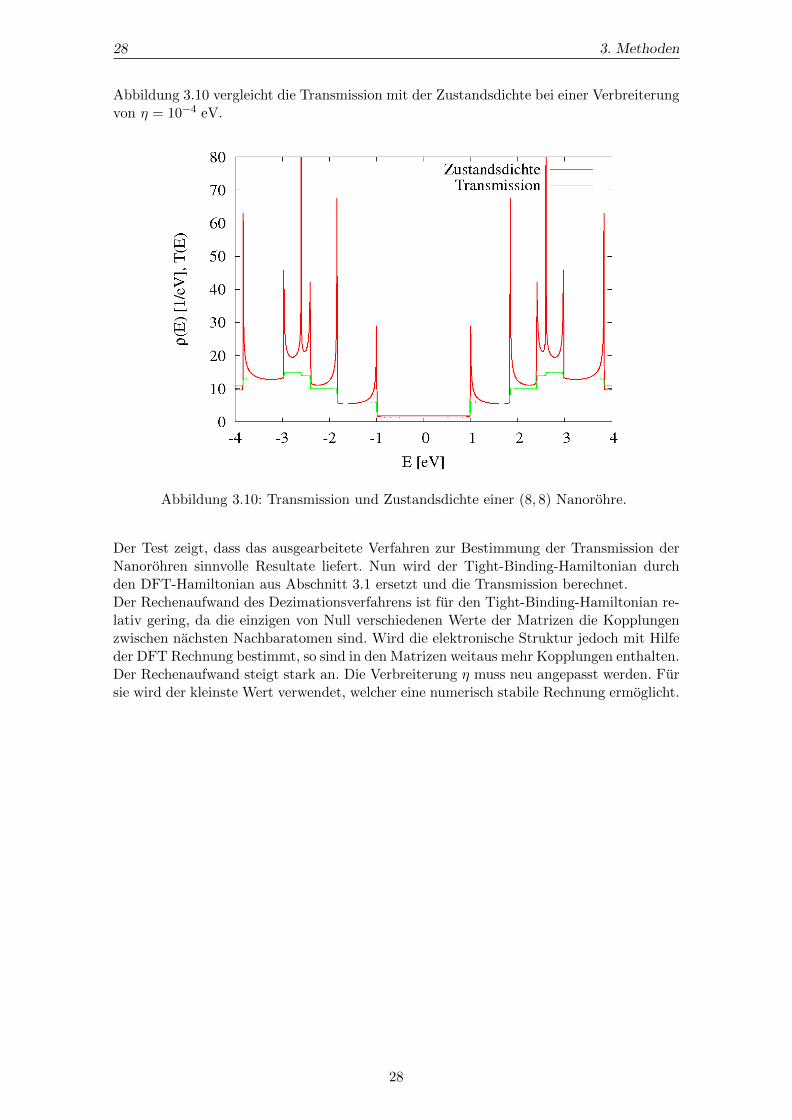
Abbildung 3.10 vergleicht die Transmission mit der Zustandsdichte bei einer Verbreiterungvon η = 10−4 eV.
Abbildung 3.10: Transmission und Zustandsdichte einer (8, 8) Nanorohre.
Der Test zeigt, dass das ausgearbeitete Verfahren zur Bestimmung der Transmission derNanorohren sinnvolle Resultate liefert. Nun wird der Tight-Binding-Hamiltonian durchden DFT-Hamiltonian aus Abschnitt 3.1 ersetzt und die Transmission berechnet.Der Rechenaufwand des Dezimationsverfahrens ist fur den Tight-Binding-Hamiltonian re-lativ gering, da die einzigen von Null verschiedenen Werte der Matrizen die Kopplungenzwischen nachsten Nachbaratomen sind. Wird die elektronische Struktur jedoch mit Hilfeder DFT Rechnung bestimmt, so sind in den Matrizen weitaus mehr Kopplungen enthalten.Der Rechenaufwand steigt stark an. Die Verbreiterung η muss neu angepasst werden. Fursie wird der kleinste Wert verwendet, welcher eine numerisch stabile Rechnung ermoglicht.
28

4. Ergebnisse
Die elektronische Struktur der Kohlenstoffnanorohren wird durch approximatives Losender Schrodingergleichung gewonnen. Hierzu wird das DFT-Programm aus dem Quanten-chemiepaket TURBOMOLE eingesetzt (siehe Abschnitt 3.1). Tabelle 4.1 fasst zusammen,welche Basen und Funktionale fur die Rechnungen verwendet wurden. Bei der Berechnungder Greenschen Funktion wurde eine Verbreiterung von η = 5 · 10−5 H gewahlt.
Nanorohre Basis Funktionaldef-SV(P) def-TZVP PBE PBE0
(6, 6) x x
(6, 6) x x
(6, 6) x x
(7, 7) x x
(8, 8) x x
Tabelle 4.1: DFT Rechnungen
4.1 Zustandsdichte
In der Abbildung 4.1 sind die Zustandsdichten D(E) = − 1πTr [Im [Gr(E)S]] der drei Na-
norohren aufgetragen. Zum besseren Vergleich wurde die Fermienergie jeweils auf Nullgesetzt.
29
30 4. Ergebnisse
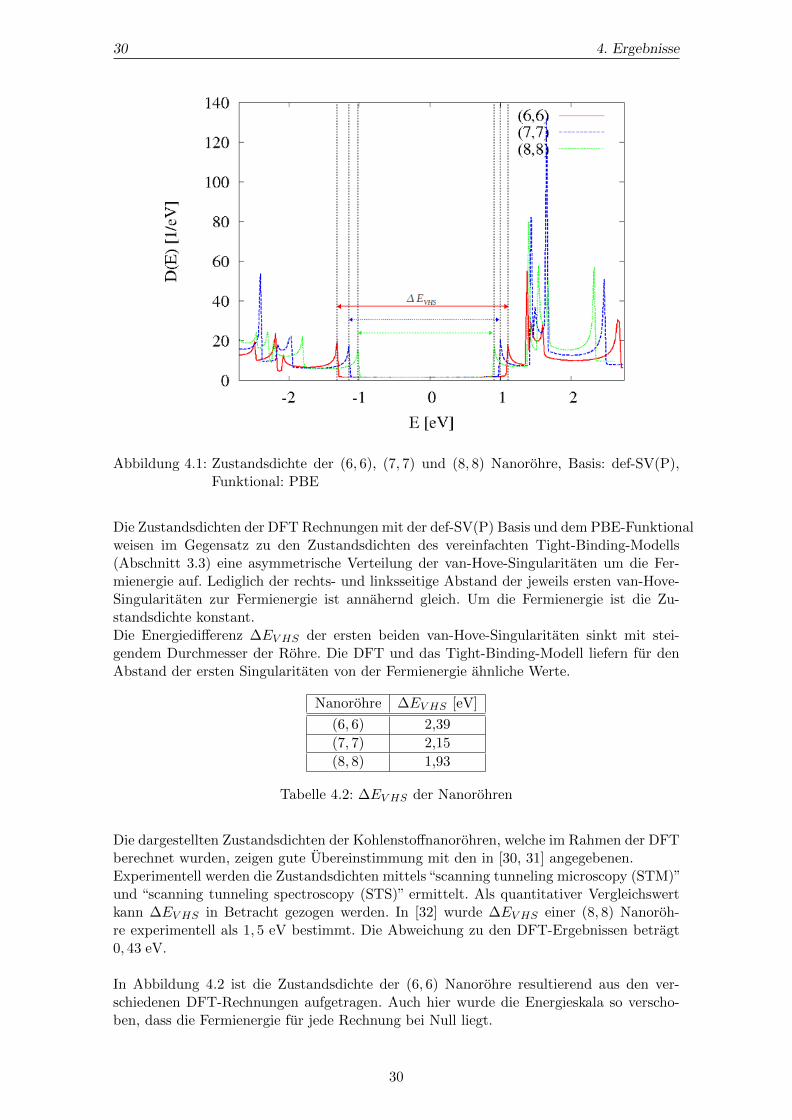
Abbildung 4.1: Zustandsdichte der (6, 6), (7, 7) und (8, 8) Nanorohre, Basis: def-SV(P),Funktional: PBE
Die Zustandsdichten der DFT Rechnungen mit der def-SV(P) Basis und dem PBE-Funktionalweisen im Gegensatz zu den Zustandsdichten des vereinfachten Tight-Binding-Modells(Abschnitt 3.3) eine asymmetrische Verteilung der van-Hove-Singularitaten um die Fer-mienergie auf. Lediglich der rechts- und linksseitige Abstand der jeweils ersten van-Hove-Singularitaten zur Fermienergie ist annahernd gleich. Um die Fermienergie ist die Zu-standsdichte konstant.Die Energiedifferenz ∆EV HS der ersten beiden van-Hove-Singularitaten sinkt mit stei-gendem Durchmesser der Rohre. Die DFT und das Tight-Binding-Modell liefern fur denAbstand der ersten Singularitaten von der Fermienergie ahnliche Werte.
Nanorohre ∆EV HS [eV]
(6, 6) 2,39
(7, 7) 2,15
(8, 8) 1,93
Tabelle 4.2: ∆EV HS der Nanorohren
Die dargestellten Zustandsdichten der Kohlenstoffnanorohren, welche im Rahmen der DFTberechnet wurden, zeigen gute Ubereinstimmung mit den in [30, 31] angegebenen.Experimentell werden die Zustandsdichten mittels“scanning tunneling microscopy (STM)”und “scanning tunneling spectroscopy (STS)” ermittelt. Als quantitativer Vergleichswertkann ∆EV HS in Betracht gezogen werden. In [32] wurde ∆EV HS einer (8, 8) Nanoroh-re experimentell als 1, 5 eV bestimmt. Die Abweichung zu den DFT-Ergebnissen betragt0, 43 eV.
In Abbildung 4.2 ist die Zustandsdichte der (6, 6) Nanorohre resultierend aus den ver-schiedenen DFT-Rechnungen aufgetragen. Auch hier wurde die Energieskala so verscho-ben, dass die Fermienergie fur jede Rechnung bei Null liegt.
30
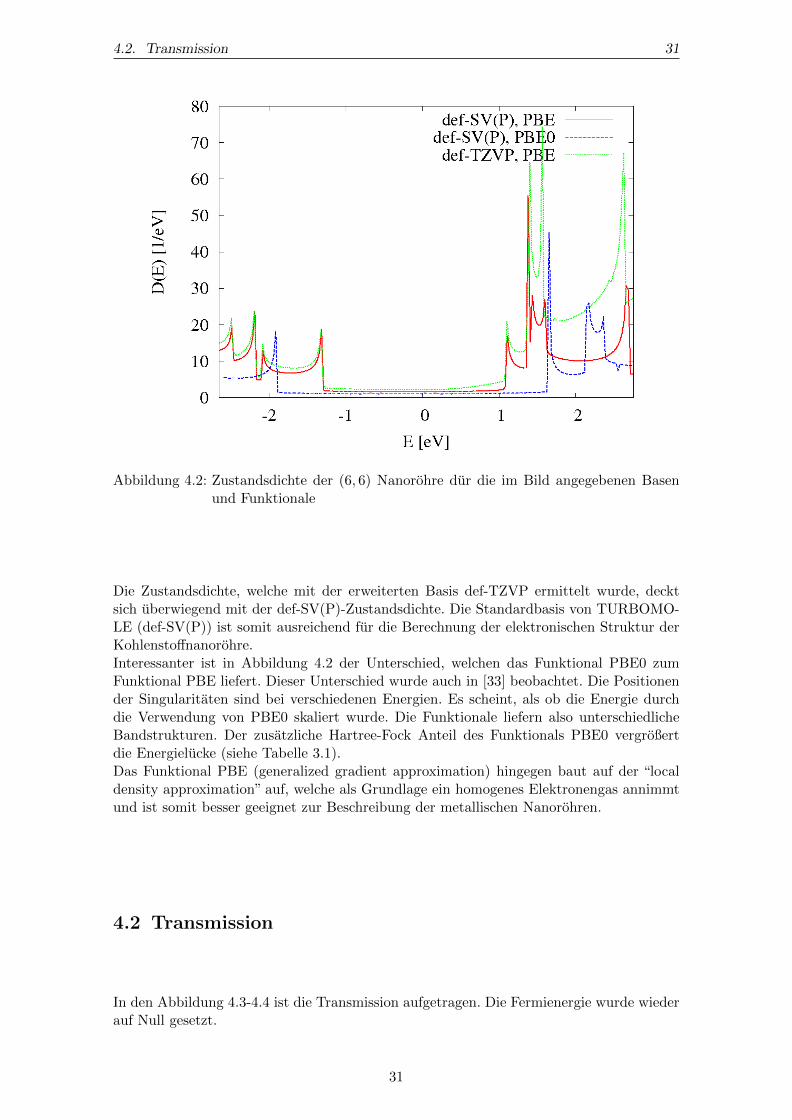
4.2. Transmission 31
Abbildung 4.2: Zustandsdichte der (6, 6) Nanorohre dur die im Bild angegebenen Basenund Funktionale
Die Zustandsdichte, welche mit der erweiterten Basis def-TZVP ermittelt wurde, decktsich uberwiegend mit der def-SV(P)-Zustandsdichte. Die Standardbasis von TURBOMO-LE (def-SV(P)) ist somit ausreichend fur die Berechnung der elektronischen Struktur derKohlenstoffnanorohre.Interessanter ist in Abbildung 4.2 der Unterschied, welchen das Funktional PBE0 zumFunktional PBE liefert. Dieser Unterschied wurde auch in [33] beobachtet. Die Positionender Singularitaten sind bei verschiedenen Energien. Es scheint, als ob die Energie durchdie Verwendung von PBE0 skaliert wurde. Die Funktionale liefern also unterschiedlicheBandstrukturen. Der zusatzliche Hartree-Fock Anteil des Funktionals PBE0 vergroßertdie Energielucke (siehe Tabelle 3.1).Das Funktional PBE (generalized gradient approximation) hingegen baut auf der “localdensity approximation” auf, welche als Grundlage ein homogenes Elektronengas annimmtund ist somit besser geeignet zur Beschreibung der metallischen Nanorohren.
4.2 Transmission
In den Abbildung 4.3-4.4 ist die Transmission aufgetragen. Die Fermienergie wurde wiederauf Null gesetzt.
31
32 4. Ergebnisse
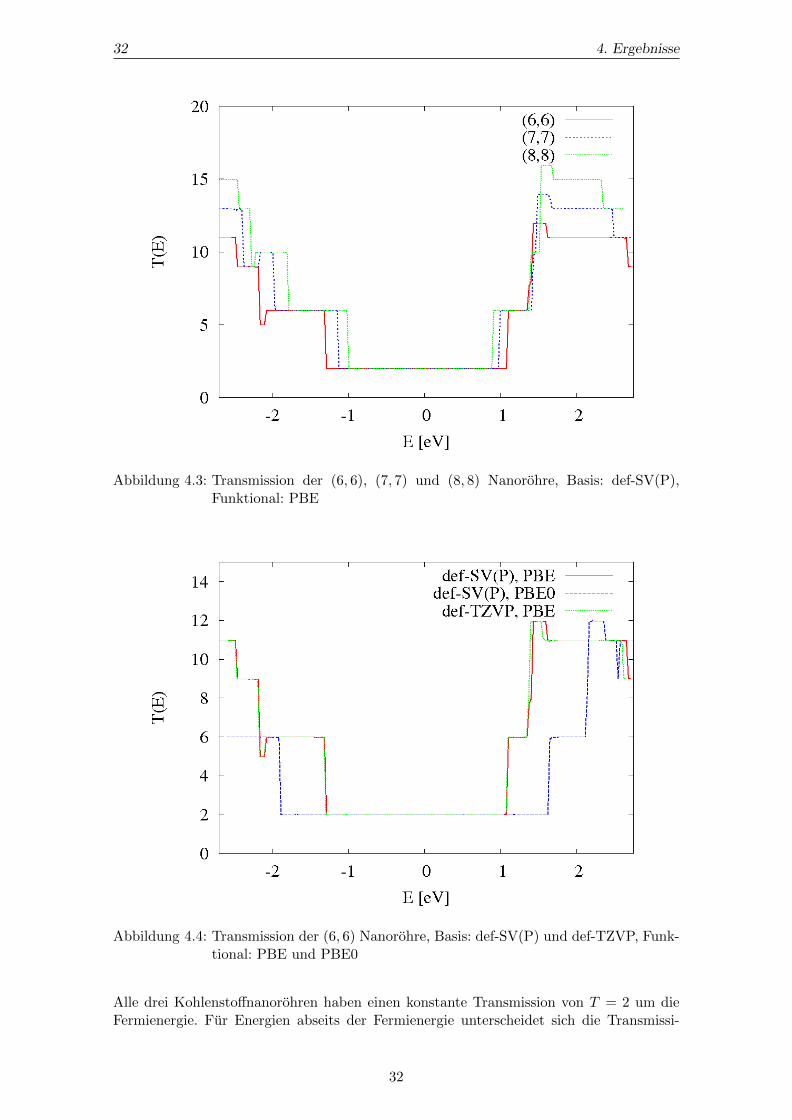
Abbildung 4.3: Transmission der (6, 6), (7, 7) und (8, 8) Nanorohre, Basis: def-SV(P),Funktional: PBE
Abbildung 4.4: Transmission der (6, 6) Nanorohre, Basis: def-SV(P) und def-TZVP, Funk-tional: PBE und PBE0
Alle drei Kohlenstoffnanorohren haben einen konstante Transmission von T = 2 um dieFermienergie. Fur Energien abseits der Fermienergie unterscheidet sich die Transmissi-
32

4.2. Transmission 33
on der Nanorohren den jeweiligen Zustandsdichten entsprechend. Vergleicht man die Zu-standsdichte mit der dazugehorigen Transmission, so erkennt man, dass bei Energien,an denen Bander geschnitten werden, die Transmission Sprunge aufweist. Zwischen zweiBandkanten ist die Transmission konstant. Dieses Verhalten deckt sich mit der in Ab-schnitt 2.1.4 beschriebenen Theorie.Wie man Abbildung 4.4 entnehmen kann, stehen die Unterschiede bei der Transmissionder (6, 6) Nanorohre fur die drei verschiedenen DFT-Rechnungen im Einklang mit den Un-terschieden der zugehorigen Zustandsdichten. Die Tranmission ermittelt mit der Basis def-TZVP deckt sich uberwiegend mit der Transmission der def-SV(P) Basis. Das FunktionalPBE0 liefert großere Bandlucken, daher ist der Leitwert fur einen großeren Energiebereichum EF = 0 konstant.
33


5. Zusammenfassung
In dieser Arbeit wurde der Transport durch drei verschiedene Kohlenstoffnanorohren imLandauer-Buttiker-Formalimus unter Verwendung der Nichtgleichgewichts-Greenschen Funk-tionen untersucht. Neben dem Transport konnten als zusatzliche Informationen die Zu-standsdichten der Kohlenstoffnanorohren ermittelt werden.
Ein wichtiges Ergebnis ist, dass die Zustandsdichte bei der Fermienergie fur (6, 6), (7, 7)und (8, 8) Armchair-Kohlenstoffnanorohren großer Null ist. Dies zeigt ihren Charakter.Des weiteren sind die typischen van-Hove Singularitaten eines eindimensionalen Leiters inden Zustandsdichten zu erkennen. Diese Singularitaten resultieren aus den Extrema dereindimensionalen Energiedispersion der Rohre und reprasentieren die Bandkanten.
Die Analyse des Ladungstransports im Landauer-Buttiker-Formalismus zeigt, dass dieTransmission fur einen Kanal τn ≈ 1 ist, da der Transport durch die Kohlenstoffnano-rohren durch eine Quantelung des Leitwerts gepragt ist. Jedes Energieband bildet einenKanal aus. Die Transmission fur eine gewisse Energie wird somit durch die Anzahl der beidieser Energie zum Transport beitragenden Energiebander bestimmt. Sie ist zwischen denBandkanten konstant und steigt oder sinkt an den Bandkanten sprunghaft. Fur alle dreiberechneten Nanorohren ist die Transmission bei der Fermienergie T ≈ 2. Dies beruht aufder Gemeinsamkeit der armchair Nanorohren, dass zwei Energiebander die Fermienergiekreuzen. Daher bezeichnet man diese Nanorohren als “metallisch”.
Die elektronische Struktur der Nanorohren wurde ab initio bestimmt. Dabei traten großereAbweichungen zwischen dem PBE und dem PBE0 Funktional auf. Bei den Berechnungenmit dem Funktional PBE0 erhalt man um die Fermienergie großere Bereiche konstanterZustandsdichte und Transmission. ∆EV HS fallt großer aus als bei der PBE-Rechnung. Dieuntersuchten Kohlenstoffnanorohren sind metallisch und sollten daher keine Energieluckezwischen der HOMO- und LUMO-Energie haben. In Tabelle 3.1 sind die von den beidenFunktionalen berechneten HOMO-LUMO-Lucken angegeben. Das Funktional PBE lieferthier kleinere Werte verglichen mit dem Funktional PBE0 und ist demnach die bessereWahl fur metallische Kohlenstoffnanorohren.
Ferner wurden die theoretisch bestimmten Zustandsdichten mit experimentell ermittel-ten Zustandsdichten verglichen. Als Vergleichwert galt dabei die Energiedifferenz ∆EV HSzwischen den ersten beiden van-Hove-Singularitaten rechts und links der Fermienergie.Fur die (8, 8) Nanorohre wurde ∆EV HS experimentell als 1, 5 eV bestimmt. Die in die-
35

36 5. Zusammenfassung
ser Arbeit theoretisch berechneten Werte fur ∆EV HS betragen im Tight-Binding-Modell(Abschnitt 3.3) 2 eV und im Rahmen der DFT (Abschnitt 4) 1, 93 eV.
36

Literaturverzeichnis
[1] http://iffwww.iff.kfa-juelich.de/ ekoch/theses/habil.html.
[2] M.S. Dresselhaus, G. Dresselhaus, and P.C. Eklund. Science of fullerenes and carbonnanotubes. Academic Press, 1996.
[3] http://www.planet-wissen.de/natur technik/forschungszweige/nanotechnologie /in-terview vengels.jsp.
[4] X. Blase, L. X. Benedict, E. L. Shirley, and S. G. Louie. Hybridization effects andmetallicity in small radius carbon nanotubes. Physical Review Letters, 72:1878–1881,1994.
[5] R. Ahlrichs, M. Bar, M. Haser, H. Horn, and C. Kolmel. Electronic structure calcu-lations on workstation computers: The program system turbomole. Chemical PhysicsLetters, 162(3):165 – 169, 1989.
[6] J. P. Perdew, K. Burke, and M. Ernzerhof. Generalized gradient approximation madesimple. Physical Review Letters, 77(18):3865–3868, 1996.
[7] C. Adamo and V. Barone. Toward reliable density functional methods without adjust-able parameters: The PBE0 model. The Journal of Chemical Physics, 110(13):6158–6170, 1999.
[8] A. Schafer, H. Horn, and R. Ahlrichs. Fully optimized contracted Gaussian basis setsfor atoms Li to Kr. The Journal of Chemical Physics, 97(4):2571–2577, 1992.
[9] A. Schaefer, C. Huber, and R. Ahlrichs. Fully optimized contracted gaussian basissets of triple zeta valence quality for atoms li to kr. The Journal of Chemical Physics,100(8):5829, 1994.
[10] F. Guinea, C. Tejedor, F. Flores, and E. Louis. Effective two-dimensional hamiltonianat surfaces. Physcal Review B, 28(8):4397–4402, 1983.
[11] R. Saito, G. Dresselhaus, and M. S. Dresselhaus. Physical Properties of Carbon Na-notubes. World Scientific Publishing Company, 1998.
[12] M.S. Dresselhaus, G. Dresselhaus, and A. Jorio. Unusual properties and structure ofcarbon nanotubes. Annual Review of Materials Research, 34(1):247–278, 2004.
[13] http://phycomp.technion.ac.il/ talimu/structure.html.
[14] D.M. Bishop. Group theory and chemistry. Dover books on physics and chemistry.Dover, 1993.
[15] S. Reich, J. Maultzsch, C. Thomsen, and P. Ordejon. Tight-binding description ofgraphene. Physical Review B, 66(3):1–5, 2002.
[16] G. Czycholl. Theoretische Festkoerperphysik: Von Den Klassischen Modellen Zu Mo-dernen Forschungsthemen. Springer-Verlag.
37

38 Literaturverzeichnis
[17] A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov, and A. K. Geim. Theelectronic properties of graphene. Reviews of Modern Physics, 81:109–162, 2009.
[18] J.-C. Charlier, X. Blase, and S. Roche. Electronic and transport properties of nano-tubes. Reviews of Modern Physics, 79(2):677–732, 2007.
[19] J. W. Mintmire and C. T. White. Universal density of states for carbon nanotubes.Physical Review Letters, 81:2506–2509, 1998.
[20] W. Koch and M. C. Holthausen. A Chemist’s Guide to Density Functional Theory.WILEY-VCH Verlag, 2002.
[21] F. Pauly. Phase-coherent electron transport through metallic atomic-sized contactsand organic molecules. 2007.
[22] P. Hohenberg and W. Kohn. Inhomogeneous electron gas. Physcal Review,136(3B):B864–B871, 1964.
[23] K. Capelle. A bird ’ s-eye view of density-functional theory. Brazilian Journal ofPhysics, 36(4a):1318, 2006.
[24] S. Datta. Electronic transport in mesoscopic systems. Cambridge studies in semicon-ductor physics and microelectronic engineering. Cambridge University Press, 1997.
[25] M. Buttiker, Y. Imry, R. Landauer, and S. Pinhas. Generalized many-channel con-ductance formula with application to small rings. Physical Review B, 31:6207–6215,May 1985.
[26] F. Pauly, J. K. Viljas, U. Huniar, M. Hafner, S. Wohlthat, M. Burkle, J. C. Cuevas, andG. Schon. Cluster-based density-functional approach to quantum transport throughmolecular and atomic contacts. New Journal of Physics, 10(12):125019, 2008.
[27] J. K. Viljas. Molecular electronics - a brief introduction. 2011.
[28] Turbomole 6.3 user’s manual. March 2011.
[29] J.W. Mintmire and C.T. White. Electronic structural properties of carbon nanotubes.Carbon, 33(7):893 – 902, 1995.
[30] Y. Akai and S. Saito. Physica E, 29:555, 2005.
[31] http://www.stat.phys.titech.ac.jp/saito/ldados.html.
[32] M. Ouyang, J.-L. Huang, C. L. Cheung, and C. M. Lieber. Energy gaps in ”metallic”single-walled carbon nanotubes. Science, 292(5517):702–705, 2001.
[33] P. V. Avramov, K. N. Kudin, and G. E. Scuseria. Single wall carbon nanotubesdensity of states: comparison of experiment and theory. Chemical Physics Letters,370(5-6):597 – 601, 2003.
38

Danksagung
An dieser Stelle mochte ich mich in erster Linie bei Herrn Prof. Dr. Gerd Schon dafurbedanken, dass er mir ermoglicht hat meine Bachelorarbeit im Institut fur theoretischeFestkorperphysik anzufertigen.
Daruber hinaus mochte ich diese Gelegenheit nutzen, um mich bei all denen zu bedanken,die mich unterstutzt haben. Ein ganz besonderer Dank gilt Dr. Fabian Pauly, Dr. MariusBurkle und Thomas Hellmuth fur die hervorragende Betreuung. Außerdem bedanke ichmich herzlich bei meinen Eltern ohne die mein Studium niemals moglich gewesen ware.


Erklarung
Hiermit erklare ich, die vorliegende Arbeit selbststandig angefertigt und dabei nur dieangegebenen Quellen und Hilfsmittel verwendet zu haben.
Mario Behrens
Karlsruhe im Dezember 2011


















