
Albumin als Carrier zur laserinduzierten Fluoreszenz ... · 3 Inhaltsverzeichnis Kapitel 1...
112
Abteilung für Neurochirurgie der Universitätsklinik Heidelberg (Ärztlicher Direktor: Prof. Dr. Stefan Kunze) Albumin als Carrier zur laserinduzierten Fluoreszenz- diagnostik und Chemotherapie maligner Tumoren Habilitationsschrift zur Erlangung der Venia Legendi für das Fach Neurochirurgie der Medizinischen Fakultät Heidelberg der Ruprecht-Karls-Universität vorgelegt von Dr. Paul Kremer aus Bad Säckingen 2002
Transcript of Albumin als Carrier zur laserinduzierten Fluoreszenz ... · 3 Inhaltsverzeichnis Kapitel 1...

Abteilung für Neurochirurgieder Universitätsklinik Heidelberg
(Ärztlicher Direktor: Prof. Dr. Stefan Kunze)
Albumin als Carrier zur laserinduzierten Fluoreszenz-diagnostik und Chemotherapie maligner Tumoren
Habilitationsschriftzur Erlangung der Venia Legendi
für das Fach Neurochirurgieder Medizinischen Fakultät Heidelberg
der Ruprecht-Karls-Universität
vorgelegt vonDr. Paul Kremer
ausBad Säckingen
2002

2
Euntes eunt et plorantsemen spargendum portantes:Venientes venient cum exsultatione,portantes manipulos suos.Psalm 126

3
InhaltsverzeichnisKapitel 1 Einleitung und Fragestellung1.1 Das Konzept der gezielten Tumortherapie mit makromolekularen Trägerstoffen1.2 Die Eigenschaften von Serum-Albumin1.3 Der Albuminmetabolismus in malignen Tumoren1.4 Die Entwicklung von Albuminkonjugaten1.5 Methotrexat-Albumin1.6 Aufgabenstellung und Zielsetzung der ArbeitKapitel 2Intraoperative Fluoreszenzdiagnostik maligner Gliome mit 5-Amino-fluorescein - HSA (AFLc-HSA)2.1 Einführung2.2 Physikalische Eigenschaften von 5-Aminofluorescein (AFL)2.3 Die kovalente Bindung von 5-Aminofluorescein an Albumin (AFLc-HSA)2.4 Die Makierung von AFLc-HSA mit radioaktivem 111Indium2.5 Das C6-Gliom Modell2.6 Plasmaclearance von AFLc-HSA2.7 Die Aufnahme von 111In-DTPA-AFLc-HSA in das C6-Gliom2.8 Fuoreszenzdiagnostik von AFLc-HSA und AFL am C6-Gliom der Ratte2.9 Fluoreszenzdiagnostik von AFLc-HSA in Zellkultur2.10 Konfokale Mikroskopie mit AFLc-HSA und an C6-Gliomzellen2.11 Klinische Studien mit 111In-DTPA-HSA2.12 Klinische Studien mit AFLc-HSA2.13 DiskussionKapitel 3 3.1 Albuminvermittelte Chemotherapie mit Aminopterin-HSA(AMPT-HSA) in vivo
3.1.1 Einführung3.1.2 Die Koppelung von Aminopterin (AMPT) an Albumin (HSA)3.1.3 Die Markierung von Aminopterin-Albumin (AMPT-HSA) mit 111Indium3.1.4 Tumormodell (Walker-256-Karzinom) und Versuchstiere3.1.5 Die Plasmaclearance von AMPT-HSA3.1.6 Die Bioverteilung und Tumoraufnahme von AMPT-HSA3.1.7 Vergleichende Toxizität von AMPT-HSA versus AMPT an Walker-256 Karzi
nom tragenden Ratten3.1.8 Wirksamkeit von AMPT-HSA versus AMPT an Walker-256-Karcinom tragen-
den Ratten

4
3.2 Untersuchungen von AMPT-HSA in Zellkultur3.2.1 Einführung3.2.2 Optimierung der Kulturbedingungen3.2.3 Das Wachstumsverhalten von C6-Gliomzellen unter dem Einfluß von AMPT-
HSA3.2.4 Der Einfluß von lysosomalen Proteaseinhibitoren auf die Wirkung von
AMPT-HSA3.2.5 Der Einfluß von AMPT-HSA auf die Walker-256-Karzinomzelllinie3.2.6 DiskussionKapitel 4 Klinische Phase-I und II-Studien mit Methotrexat-Albumin (MTX-HSA) bei Patienten mit rezidivierenden malignen Gehirntumoren3.3 Einleitung3.4 Klinische Phase-I-Studie mit zweiwöchentlichen Applikationen von MTX-HSA
bei Patienten mit rezidivierenden malignen Gehirntumoren4.3 Vergleichende Phase-II-Studie von MTX-HSA und Carmustin/Lomustin bei
Patienten mit primären und sekundären Glioblastomrezidiven4.4 DiskussionKapitel 5Albuminvermittelte Lymphdiagnostik mit TCPC- oder TCPP-HSAund Gadolinium-HSA5.1 Einleitung5.2 Die Diagnostik von Lymphwegen mit TCPC – HSA5.3 Diagnostik von Lymphwegen und reaktiv veränderten Lymphknoten des
Kaninchens mit TCPC – HSA5.4 Die Darstellung von Lymphbahnen und Lymphknoten mit Gd-HSA5.5 DiskussionKapitel 6Perspektiven 6.1 Tumordiagnostik mit Gadolinium-HSA6.2 Die Behandlung der rheumatoiden Arthitis mit Methotrexat-Albumin6.3 Die Konjugierung von Hydrocortison, Prednison und Dexamethason an HSA

5
Kapitel 7Zusammenfassung7.1.1 Laserfluoreszenzdiagnostik mit 5-Aminofluorescein - Albumin (AFLc-HSA)7.1.2 Albuminvermittelte Chemotherapie7.1.3 Albuminvermittelte Sentinel-Lymph-Node-Diagnostik7.2 Schlußfolgerung
Kapitel 8Literatur
Kapitel 9Danksagung

6
Kapitel 1Einleitung und Fragestellung1.1 Das Konzept der gezielten Tumortherapie mit makromolekularen Träger-stoffen
Paul Ehrlich (1854 – 1915) prägte als erster den Begriff der Chemotherapie (1). Chemi-sche Substanzen sollten ohne Mithilfe körpereigener Abwehrvorgänge im Stande sein,Mikroorganismen zu töten, ohne den Makroorganismus zu schädigen ( = Therapiamagna sterilisans). Gewarnt durch Nebenwirkungen erster Präparate und angeregt durchdie Entdeckung von Antitoxinen im Serum, entwickelte Ehrlich schon damals Vorstel-lungen einer gezielten medikamentösen Therapie, welche über den Einsatz von Träger-stoffen, der „Haptophore“, den Wirkstoff, die „Toxophore“, in den Tumor einschließensoll. Dieses Prinzip wurde von ihm mit dem einer Zauberkugel ( = magic bullet) vergli-chen.
Heutzutage wird dieses Prinzip des gezielten Einschließens von Wirksubstanzen imTumorgewebe „drug targeting“ genannt. Jedoch ist bislang aber nur wenig realisiert.Die meisten der zur Tumortherapie verwendeten Substanzen sind niedermolekular(Molmasse < 1000 g/Mol) und passieren Tumorgewebe nur über einen kurzen Zeit-raum. Zudem bewirkt das geringe Molekulargewicht eine weite Verbreitung im gesun-dem Organismus mit einer dadurch bedingten Schädigung physiologisch proliferieren-den Gewebes. Auch werden die meisten Wirksubstanzen rasch eleminiert, was zur Fol-ge hat, daß die Plasmazeiten der meisten Zytostatika unter einer Stunde liegen (2-5).Dies wiederum hat zur Folge, daß das Zeitdauer, in dem niedermolekulare Substanzenin das Tumorgewebe eindringen und dort ihre Wirkung entfalten sollten, sehr kurz ist(6). Eigentlich müßte in Kenntnis der pharmakokinetischen Eigenschaften niedermole-kularer Substanzen die Applikation wiederholt und in kurzzeitigen Abständen erfolgen,doch gerade bei den meisten Chemotherapeutika verbietet die systemische Toxizität einsolches Behandlungsschema.

7
Daher wurde schon in den 60er Jahren versucht, die Wirkung von Zytostatika durchKoppelung an makromolekulare Trägersysteme zu verbessern (7-9). Doch bislang bliebein durchschlagender Erfolg in der Tumortherapie aus.
Eine wesentliche Eigenschaft maligner Tumoren ist die Neoangiogenese. Induziert wirdsie durch das Tumorwachstum selbst, denn ohne eine Einsprossung neuer Gefäße würdedie Tumorversorgung bald zum Erliegen kommen. Bereits ab einer Tumorgröße von 3mm3 ist die Ernährung des Tumores durch Diffusion nicht mehr gewährleistet (10).Durch eine Vielzahl von Mediatoren, wie z. B. dem „basic fibroblast growth factor“(BFGF) oder dem „vascular endothelial growth factor „(VEGF) wird die Ge-fäßeinsprossung in das Tumorgewebe induziert (11-14). Aber nicht alle Tumorarealewerden gleichmäßig durchblutet. Gerade in den zentralen Tumoranteilen bilden sichNekrosen. Denn hier übersteigt der deutlich erhöhte interstitielle Druck im Tumor vonbis zu 50 mm Hg den intravasalen Druck (15-17), was wiederum zu einer Verminde-rung des Blutflusses und zu einem Gefäßkollaps führt (18). Begünstigt wird dieses Phä-nomen durch das in der Regel weitgehende Fehlen funktionierender Lymphdrainagen,welches einen Rückstau makromolekularer Substanzen im Tumor bewirkt.
Ganz anders ist die Situation in der Tumorperipherie, den Arealen höchster Zellprolife-ration. Hier übersteigt der intravasale Druck den interstitiellen. Eine Vielzahl von fe-nestrierten Kapillaren fördert hohe kapilläre Filtrationsraten, welche 10 - 1000fach hö-her sein können als im normalen Gewebe (19). So werden bei verschiedenen Rattentu-moren 6 – 14 % des Plasmas in den extravasalen Raum der Tumorperipherie abgegeben(20-21).
Mit dieser hohen Filtrationsrate geht die Besonderheit einher, daß fenestrierte Tumor-kapillare Makromoleküle nicht zurückhalten. Und weil bei malignen Tumoren in derRegel ein funktionstüchtiges lymphatisches System fehlt (22), kommt es zu einer Ak-kulumation von Makromolekülen im Tumorgewebe, im Englischen „enhanced permea-bility and retention“ genannt (23).

8
Während für den Eintritt der Makromoleküle in das Interstitium des Tumors der Blut-fluß (Konvektion) verantwortlich ist, bestimmt nun die Diffusion die weitere Vertei-lung. Diese ist in erster Linie abhängig von der Molekülgröße (24-25). Makromoleküle,wie z. B. Liposomen (400 – 1.000 kDa) oder Immunglobuline (ca. 150 kDa) diffun-dieren nur sehr langsam. Ein IgG benötigt für die Überwindung einer Strecke von 1 mmca. 2 Tage, ein kleineres Makromolekül wie Albumin (66439 Da) diffundiert ca. 10fachschneller (26-28). Niedermolekulare Substanzen diffundieren jedoch weitaus schneller.Substanzen mit einem MG von bis zu 1.000 diffundieren binnen weniger Minuten bisStunden aus dem Bereich der Tumorproliferationszone entweder zurück in die Blutzir-kulation oder werden aus dem umgebenden Interstitium abgeschwemmt. Zusammenfas-send gilt jedoch, daß gerade in den proliferationsaktiven Arealen solider Tumoren Mak-romoleküle akkumulieren.
Das Konzept der makromolekularen Trägersysteme wurde an zahlreichen Verbindungenerprobt, von denen allerdings nur wenige Eingang in die klinische Prüfung erfuhren.Grundsätzlich lassen sich zwei verschiedene Trägersysteme unterscheiden.
Partikuläre TrägerssystemeEine klinische Bedeutung für partikuläre Trägersysteme haben Liposomen erlangt, wel-che mit ihrer Doppelschichtmembran die eigentliche Wirksubstanz umschließen (29-32). Je nach Herstellungsverfahren haben diese Partikel einen Durchmesser von 50 nmbis 1 µm. Liposomen können mit der Zellmembran verschmelzen und so die in der inne-ren wässrigen Phase enthaltene Wirksubstanzen in die Tumorzellen einschleusen. Durcheine selektive Instillation von Liposomen soll dadurch eine gezielte Abgabe von Sub-stanzen in den Tumor erreicht werden.
Polymere TrägersystemePolymere Trägersysteme sind entweder biologische Makromoleküle, wie z. B. mono-klonale Antikörper, oder synthetische Makromoleküle, wie z. B. Polysaccaride oderPolyäthylenglycol. Monoklonale Antikörper, 1975 von Köhler und Milstein erstmalshergestellt (33), sollen im Rahmen einer passiven Immuntherapie im Patienten spezi-fisch Tumorzellen auffinden und zerstören. Ihre Aktivität entfalten Antikörper nach

9
Bindung an der Tumorzelle über die Blockierung von Signaltransduktionswegen, wie z.B. Inhibierung von Proliferationsreizen, lokale Initiierung der Komplementkaskade oderdurch Rekrutierung von Effektorzellen (34-36). Trotz zahlreicher Therapiestudien blie-ben die Erfolge jedoch aus. Probleme bereiteten vor allem die nach wiederholter An-wendung regelmäßig auftretenden blockierenden humanen Anti-Maus-Antikörper. Erstdie Entwicklung rekombinanter humanisierter Antikörper Mitte der 90er Jahre ermög-lichte wiederholte Therapiezyklen. Zwischenzeitlich sind von der amerikanischen Foodand Drug Administration (FDA) neun Antikörper mit unterschiedlicher Indikation zu-gelassen. Auch in Europa wurde der humanisierte Antikörper Tratuzumab (Herceptin)zugelassen, welcher HER2-überexprimierende Mammakarzinome erkennt. Währendhingegenen dieser Antikörper als Monotherapeutikum in ca. 15% ein Ansprechen zeigt(37), kann dieser Effekt durch gleichzeitige Verwendung verschiedener Chemothera-peutika auf ca. 60% gesteigert werden (38). Ähnliches gilt für einen weiteren Antikör-per C225, der den von vielen epithelialen Tumoren überexprimierenden „epidermalgrowth factor receptor“ (EGF-R) erkennt (39). Auch hier kann die alleinige Wirkungdes Antikörpers durch den gleichzeitigen Einsatz chemotherapeutischer Substanzendeutlich gesteigert werden (40). Ein generelles Problem der Antikörpertherapie soliderTumoren liegt aber zum einen in der unzureichenden Zugänglichkeit des Zielantigensund zum anderen dem fehlenden Vorhandensein eines tumorassoziierten Antigens über-haupt. Ebenfalls ist die Verwendung von Antikörperkonjugaten, bei denen der Antikör-per als Vehikel für Chemotherapeutika oder Radionukleide dient, Gegenstand klinischerStudien an verschiedenen Tumorentitäten (41-42).Bei den synthetischen Makromolekülen konnte ein Polyethylenglycolkonjugat, diePEG-L-Asparaginase, bis zur klinischen Zulassung entwickelt werden. Hierbei wirdAsparaginase kovalent an Polyethylenglykol (PEG) gebunden, wodurch eine verlän-gerte Halbwertszeit und ein verbessertes Ansprechen auf Lymphosarkome und andereMäusetumore erreicht werden konnte (43). Inzwischen ist L-Asparaginase-PEG (On-caspar) bei akuten lymphoblastischen Leukämien im Kindesalter als therapeutischeOption etabliert (44-46), wobei auch hier über eine Wirkungsverstärkung in Kombinati-on mit Methotrexat, Vincristin und Prednison berichtet wird (47). Auch eine kovalenteBindung von PEG an Interferon α2a (Pegasys) oder PEG-Interferon α2b (PegIntron)

1 0
bewirkt eine Wirkungsverstärkung in der Behandlung der chronischen Hepatitis C ge-genüber der Kombinationstherapie ohne PEG (48-49).Weitere synthetische Makromoleküle stellen die supramagnetischen monocristallinenEisenoxide (MION) oder ultrakleine supramagnetische Eisenoxidpartikel (USPIO) dar.MIONs bestehen aus einem kristallinen Eisenoxidkern (Durchmesser ca. 6,5 nm) undeiner Carboxydextranhülle. Der hydrodynamische Durchmesser der Partikel beträgt ca.20 – 40 nm. MIONs werden von der Tumorzelle phagozytiert, wobei das dadurch ein-geschleuste Eisenoxid kernspintomographisch nachgewiesen werden kann. Allerdingssind die Zirkulationszeiten kurz, da die Polysaccharide effektiv vom retikuloendotheli-schen System (RES) oder der Leber abgefangen werden (50-51).
Auf Grund der bisherigen Erfahrungen mit makromolekularen Trägersystemen sollteein Konjugat, welches als „drug carrier“ fungieren soll, folgende Eigenschaften besitzen(52):• Keine Immunogenität oder Toxizität des Trägermoleküles• Gewährleistung einer stabilen chemischen Koppelung zwischen Trägermolekül und
Wirksubstanz• Lange Verweildauer des Trägermoleküles im Kreislauf• Hohe Aufnahmerate des Trägermoleküles im Tumor• Die Wirksubstanz soll erst im Tumorgewebe freigesetzt werden.
1.2 Die Eigenschaften von Serum-Albumin
Humanes Serumalbumin besteht aus 585 Aminosäuren. Sein Molekulargewicht beträgt66.439 Dalton (D). Albumin hat die Form eines rotationselipsoiden Körpers ähnlichdem einer Zigarre. Die Längsachse mißt 14 nm, die Querachse 4 nm. Im Inneren befin-det sich ein hydrophober Kanal. Während an den Öffnungen positive Ladungen vorherr-schen, so befinden sich an der Gesamtoberfläche negative Ladungen (53).
Albumine sind gut wasserlösliche Proteine mit einem isoelektrischen Punkt bei pH 4,8.Im Blutplasma zirkulieren ca. 100 weitere Proteine, jedoch stellt Albumin mit ca. 60 %

1 1
den größten Anteil dar. Beim Menschen schwankt der Serumalbuminspiegel zwischen3,5 – 5,5 g/dl.Bereits kurze Zeit nach intravenöser Injektion sind ca. 50 % der applizierten Albumin-menge ins Interstitium diffundiert. Von dort gelangt Albumin über das Lymphbahnsy-stem in das Blut zurück. Im Schnitt zirkuliert jedes Albuminmolekül 15.000 mal durchden Kreislauf, macht dabei 15 Passagen durch das Gefäßendothel um über das lympha-tische Gewebe innerhalb weniger Tage wieder in die systemische Zirkulation zu gelan-gen (54-56). Die Halbwertszeit von humanem Serumalbumin beträgt ca. 19 Tage. Täg-lich müssen ca. 2,5 % Albumin durch Neusynthese ersetzt werden. Der Albuminverlustist vor allem durch Verstoffwechselung bedingt, die renale Filtration ist ohne Belang(57). Der Hauptteil des physiologischen Albuminabbaues findet in den Lysosomen vonFibroblasten der Haut und der Muskulatur statt (58-59). Hauptsyntheseort von Albuminsind die Hepatozyten der Leber. Täglich produziert eine gesunde Person ca. 11 ±3 gAlbumin (60-64).
Bereits 1839 entdeckte H. Ancell, daß Albumin bei der Verhinderung von Ödemen eineRolle spielt und in die Transportprozesse im Blut involviert sei (65). In der Tat tragendie Plasmaproteine und davon vor allem Albumin mit 60 % wesentlich zur Aufrechter-haltung des kolloidosmotischen Druckes bei. Ein Mangel an Albumin reduziert somitden onkotischen Druck im Plasma, was zu einer generellen Ödemneigung führt.Schließlich stellen Plasmaproteine, und damit vor allem Albumine, als schwache Säureneine Pufferkapazität dar, welche stärkere pH-Schwankungen des Blutes zu verhindernhelfen (66). Eine weitere wichtige Eigenschaft von Albumin ist der Transport von ver-schiedenen Stoffen im Blut. Die zu transportierenden Substanzen werden dabei abernicht kovalent, sondern adhäsiv gebunden. Eine Vielzahl von Liganden, wie z. B. Fett-säuren, Bilirubin, Gallensäure, Hämatin, Histamin, Thyroxin oder Thryptophan, sindbeschrieben. Auch Kalzium oder Chlorid werden durch Albumin gebunden. Ebenfallswerden über 90 % der niedermokelularen Medikamente, wie z. B. Penizillin, Sulfon-amide, Diazepan, Digitoxin, Methotrexat, Indozyanin, temporär durch Albumin imPlasma transportiert (67). Die Liganden werden vorwiegend in der Leber oder denGlomeruli der Nieren von Albumin abgelöst. Gerade in der Leber wird dann ein Groß-teil der so freigesetzten Medikamente metabolisiert und ausgeschieden (68).

1 2
Albumin stellt mit 150 – 200 g im Plasma eine wichtige Eiweißreserve dar. Ein MolekülAlbumin beinhält dabei 779 Stickstoffatome. Diese Menge stellt eine bei Bedarf sehrschnell verfügbare Eiweiß- und Stickstoffreserve im Organismus dar (54). Die Ami-nosäuren des dominierenden Plasmaproteins Albumin enthalten insgesamt ca. 30 malmehr Energie als das Glucoseangebot des Blutes. Etwa 200 mal mehr Aminosäuren sindalbumingebunden verfügbar als frei zirkulierende Aminosäuren (57).
1.3 Der Albuminmetabolismus in malignen Tumoren
Die Vorstellung, daß Eiweiße, wie z. B. Albumin, eine wichtige Rolle in der Versor-gung maligner Tumoren darstellen, wurde bereits 1948 von Mider (69) beschrieben.Ihnen war aufgefallen, daß sich maligne Tumoren wie Stickstofffallen verhalten, d. h.mehr stickstoffhaltige Substrate, wie z. B. Aminosäuren, aufnehmen als abgeben. We-nige Jahre später zeigte Babson und Winnick (70), daß maligne Tumoren eher Plasma-proteine als Aminosäuren verstoffwechseln, und folgerten daraus, daß die Tumorkache-xie erkrankter Patienten durch den erhöhten Eiweißstoffwechsel maligner Tumoren be-dingt ist. Erstmals wurde von diesen Autoren auf die Möglichkeit, Medikamente an Al-bumin zu binden und in maligne Tumoren einzuschleusen, hingewiesen (71-72). Aller-dings wurde dem Plasmaeiweißkatabolismus maligner Tumoren im weiteren nicht vielBeachtung geschenkt. In Übersichtsarbeiten über Tumorkachexie finden sich diesbe-züglich nur wenig Hinweise (73-77). Möglicherweise war dies methodisch bedingt,denn nach Markierung von Plasmaeiweißen mit Radiojod, 3H oder 14 C entstehen nachAbbau des Proteins sehr rasch eine Vielzahl niedermolekularer Katabolite, die eine ex-akte Zuordnung und Verteilung des Proteins im Körper nicht möglich machen (78-81).Diese radioaktiv markierten Aminosäuren oder Proteinspaltprodukte unterliegen dannnormalen niedermolekularen Verteilungsvorgängen und werden entweder ausgeschie-den oder in die Neusynthese anderer Proteine eingeschleust (82-83).
Um diesen Schwierigkeiten zu begegnen, wurden in den letzten Jahren metabolisch sta-bile sog. „kumulative“ Markierungstechniken entwickelt (84-87). Wesentliches Merk-mal dieser stabilen Markierung ist, daß der radioaktive Marker am Ort des Abbaus, also

1 3
bei Proteinen am Lysosom, verbleibt. Diese metabolisch stabilen Marker bestehen meistaus einem Kohlenhydrat, welches mit dem radioaktiven Marker verknüpft ist. Mit Hilfedieser metabolischen Marker war es möglich, den Nachweis des Plasmaabaues von Ge-sunden zu untersuchen (88-89). Hierbei wurden Fibroblasten als wesentlicher Ort desAlbuminabbaues erkannt (59).
1.4 Die Entwicklung von Albuminkonjugaten
Ende der 80er Jahre übernahmen Hansjörg Sinn und seine Mitarbeiter das Prinzip dermetabolisch stabilen bwz. kumulativen Markierungstechniken, um der Frage nachzuge-hen, ob und in welcher Weise Albumin als Trägersubstanz genutzt werden kann. Zudemsollte untersucht werden, ob Albumin durch die Koppelungstechnik in seiner pharma-kokinetischen und biologischen Funktion beeinflußt wird und wie hoch Albumin alsTrägersubstanz beladen werden darf. Ausgehend von den Untersuchungen von Pitman(84), welcher Tyraminzellobiose (TCB) zur Markierung von Low-Density-Lipopro-teinen in der Arterioskleroseforschung genutzt hatte, wurde ein kumulativer 131Jod-Label mit Tyramindesoxysorbitol (TDS) entwickelt, welcher kovalent an Albumin ge-bunden werden konnte (87). Nach intravenöser Injektion dieses kumulativ radioaktivmarkierten 131I-TDS-Albumins zeigte sich bei O-342 (tierexperimenteller Ovarialtumor)tragenden Ratten auch 72 h nach i. v. Applikation eine deutlich erhöhte Aufnahme desmarkierten Albumins im Tumor. In weiteren Untersuchungen mit 131I-TDS-Albuminfand sich gegenüber nativem Albumin keine Veränderung in der Plasmahalbwertszeit.Auch war als Ausdruck der stabilen kovalenten Bindung und alleinigen intrazellulärenMetabolisierung von 131I-TDS-Albumin gegenüber konventionell markiertem 131I-Albumin keine Aufnahme von 131 I-Albumin-Spaltprodukten in der Schilddrüse derVersuchstiere zu finden (90). Diese Ergebnisse wurden auch mit einem weiteren kumu-lativen Marker, dem 111Indium-Diethylentriamin-Pentaessigsäure (111 In-DTPA) bestä-tigt. So zeigte sich bei Walker-256-Karzinom tragenden Ratten 48 h nach intravenöserApplikation ca. 20 % der initial verabreichten Substanz im Tumor (91). Sowohl 131Jod-TDS wie auch 111Indium-DTPA markiertes Albumin war in der Lage, den Albumin-stoffwechsel im tumortragenden Tier darzustellen. Die Pharmakokinetik entsprach da-bei natürlichem Albumin, wobei sich bei der Ratte keine Unterschiede in den Zirkula-
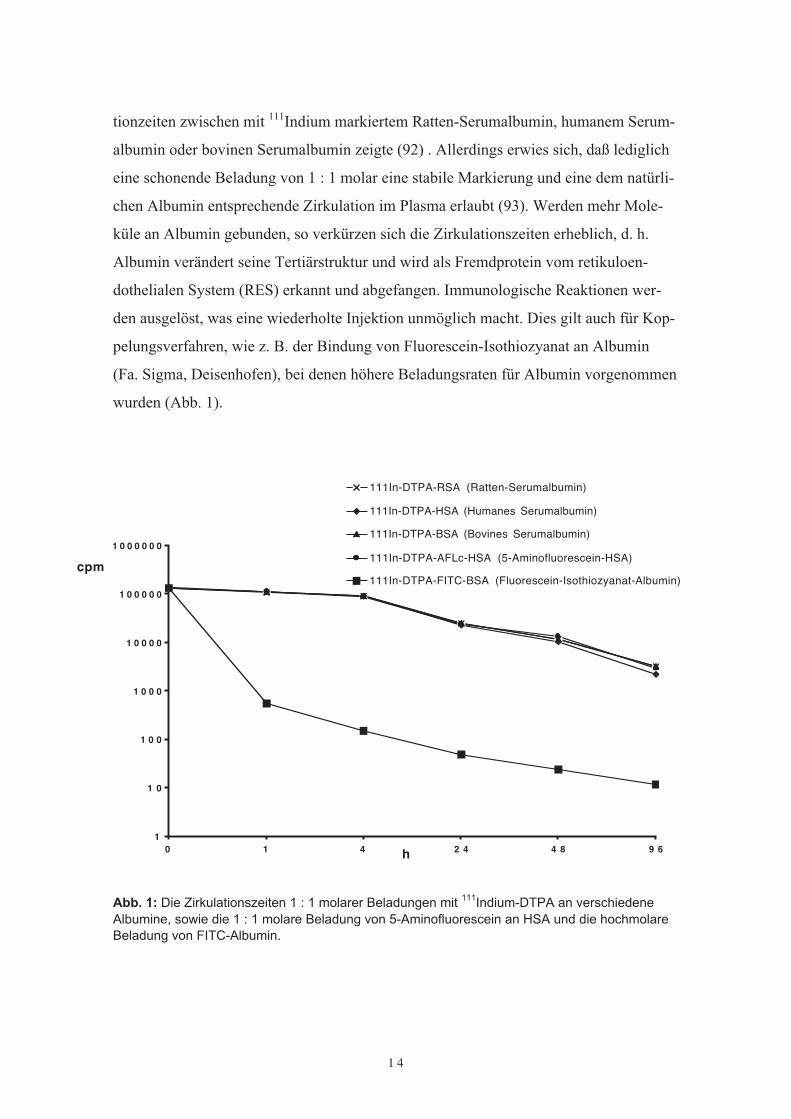
1 4
tionzeiten zwischen mit 111Indium markiertem Ratten-Serumalbumin, humanem Serum-albumin oder bovinen Serumalbumin zeigte (92) . Allerdings erwies sich, daß lediglicheine schonende Beladung von 1 : 1 molar eine stabile Markierung und eine dem natürli-chen Albumin entsprechende Zirkulation im Plasma erlaubt (93). Werden mehr Mole-küle an Albumin gebunden, so verkürzen sich die Zirkulationszeiten erheblich, d. h.Albumin verändert seine Tertiärstruktur und wird als Fremdprotein vom retikuloen-dothelialen System (RES) erkannt und abgefangen. Immunologische Reaktionen wer-den ausgelöst, was eine wiederholte Injektion unmöglich macht. Dies gilt auch für Kop-pelungsverfahren, wie z. B. der Bindung von Fluorescein-Isothiozyanat an Albumin(Fa. Sigma, Deisenhofen), bei denen höhere Beladungsraten für Albumin vorgenommenwurden (Abb. 1).
Abb. 1: Die Zirkulationszeiten 1 : 1 molarer Beladungen mit 111Indium-DTPA an verschiedeneAlbumine, sowie die 1 : 1 molare Beladung von 5-Aminofluorescein an HSA und die hochmolareBeladung von FITC-Albumin.
1
1 0
1 0 0
1 0 0 0
1 0 0 0 0
1 0 0 0 0 0
1 0 0 0 0 0 0
0 1 4 2 4 4 8 9 6
111In-DTPA-RSA (Ratten-Serumalbumin)111In-DTPA-HSA (Humanes Serumalbumin)111In-DTPA-BSA (Bovines Serumalbumin)111In-DTPA-AFLc-HSA (5-Aminofluorescein-HSA)111In-DTPA-FITC-BSA (Fluorescein-Isothiozyanat-Albumin)
cpm
h
1 5
1.5 Methotrexat-Albumin
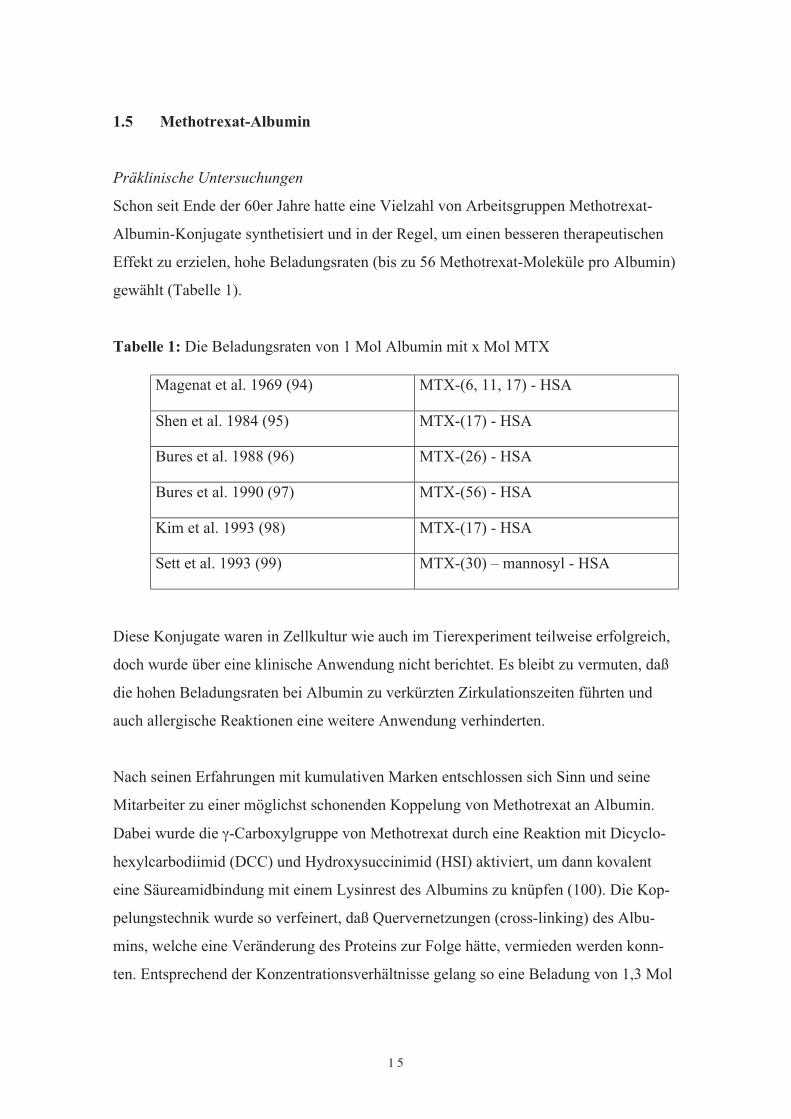
Präklinische UntersuchungenSchon seit Ende der 60er Jahre hatte eine Vielzahl von Arbeitsgruppen Methotrexat-Albumin-Konjugate synthetisiert und in der Regel, um einen besseren therapeutischenEffekt zu erzielen, hohe Beladungsraten (bis zu 56 Methotrexat-Moleküle pro Albumin)gewählt (Tabelle 1).
Tabelle 1: Die Beladungsraten von 1 Mol Albumin mit x Mol MTXMagenat et al. 1969 (94) MTX-(6, 11, 17) - HSAShen et al. 1984 (95) MTX-(17) - HSABures et al. 1988 (96) MTX-(26) - HSABures et al. 1990 (97) MTX-(56) - HSAKim et al. 1993 (98) MTX-(17) - HSASett et al. 1993 (99) MTX-(30) – mannosyl - HSA
Diese Konjugate waren in Zellkultur wie auch im Tierexperiment teilweise erfolgreich,doch wurde über eine klinische Anwendung nicht berichtet. Es bleibt zu vermuten, daßdie hohen Beladungsraten bei Albumin zu verkürzten Zirkulationszeiten führten undauch allergische Reaktionen eine weitere Anwendung verhinderten.
Nach seinen Erfahrungen mit kumulativen Marken entschlossen sich Sinn und seineMitarbeiter zu einer möglichst schonenden Koppelung von Methotrexat an Albumin.Dabei wurde die γ-Carboxylgruppe von Methotrexat durch eine Reaktion mit Dicyclo-hexylcarbodiimid (DCC) und Hydroxysuccinimid (HSI) aktiviert, um dann kovalenteine Säureamidbindung mit einem Lysinrest des Albumins zu knüpfen (100). Die Kop-pelungstechnik wurde so verfeinert, daß Quervernetzungen (cross-linking) des Albu-mins, welche eine Veränderung des Proteins zur Folge hätte, vermieden werden konn-ten. Entsprechend der Konzentrationsverhältnisse gelang so eine Beladung von 1,3 Mol

1 6
Methotrexat auf 1 Mol Albumin. In pharmakokinetischen Studien an Ratten zeigte sich,daß die Verweildauer von MTX-HSA natürlichem HSA entsprach. Auch konnte 1 - 2Tage nach intravenöser Applikation des Albuminkonjugates an verschiedenen tierexpe-rimentellen Tumoren eine bis zu 20%ige Aufnahme im Tumor nachgewiesen werden.Diese Konjugateigenschaften konnten jedoch durch eine höhere Beladungsrate des Al-bumin deutlich beeinflußt werden. Waren nach Applikation von MTX-RSA 1 : 1 molar15 % der Substanz nach 24 h in Zirkulation, so wurde dies bei einer 10 : 1 molaren Be-ladung auf 3 % verkürzt (93). Entsprechend zeigte die Leber eine 7fach erhöhte Auf-nahme des durch die höhere Beladung denaturierten Proteins, wie Analysen in derhochauflösenden Flüssigkeitschromatographie (High Performance Liquid Chromato-graphie, HPLC) bestätigten. Denn je höher Albumin beladen wird, desto breiter undatypischer ist das Albuminsignal in der HPLC verglichen mit nativem Albumin. Auchdie Aufnahme des Konjugates im Tumor wurde durch die hohe Beladung beeinflußt.Aufgrund dieser Ergebnisse wurden für weitere präklinische Untersuchungen das MTX-Albumin (MTX-SA) Konjugat mit der nahezu 1 : 1 molaren Beladungsrate gewählt, dahier lange Zirkulationszeiten, hohe Tumoraufnahmeraten und fehlende allergische Re-aktionen zu vermuten waren.
Die in-vivo Prüfung des Konjugates erfolgte am Walker-256-Karzinom der Ratte, wo-bei die Wirkung des Konjugates mit der von freiem MTX verglichen wurde. Als Dosislimitierende Toxizität (DLT) wurde für beide Gruppen eine 4fach wiederholte i. v. In-jektion an den Tagen 1, 3, 8 und 10 festgestellt. Danach traten typische MTX-Neben-wirkungen, wie Durchfall, Mukositis und struppiges Fell auf. Somit wurde die Effekti-vität in Höhe der maximal tolerablen Dosis (MTD) für beide Gruppen als repetitive In-jektionen von 3 x 2mg MTX/kg Körpergewicht (KG) oder 3 x 2mg MTX-SA/kg KGuntersucht. MTX erreichte dabei eine Heilrate von 55 %, während hingegen sie fürMTX-SA bei 60 % lag. Durch eine Mischung von freiem MTX mit MTX-SA wurde beigleichem Applikationsschema und gleicher Dosis eine Heilrate von 70 % erreicht (101).
Die Effektivität von MTX-Konjugaten gegenüber freiem MTX bestätigte sich ebenfallsbei Dunning R 3327 Prostataadenokarzinomen an Copenhagen-Ratten, wo in einer Do-sierung 4 x 2 mg/MTX-SA/kg KG eine deutlich verzögerte Tumorprogression gegen-

1 7
über freiem MTX erreicht wurde (102). Auch an verschiedenen Xenograft-Tumoren,wie z. B. Nierenzell-, Blasen-, Kolon-, Bronchial-, Pankreas-, Mamma-Karzinomen wieauch bei Mesotheliomen und malignen Melanomen, konnte eine verbesserte Wirksam-keit von MTX-HSA gegenüber freiem MTX nachgewiesen werden. Zwischenzeitlichfand sich auch eine verbesserte Wirksamkeit von MTX-HSA an 2 von 4 malignenGliomxenografts (103-104).
Klinische Studien mit MTX-HSA
Ermutigt durch zahlreiche präklinische Untersuchungen mit MTX-HSA erfolgte 1997eine erste klinische Phase-I-Studie an 17 Tumorpatienten mit MTX-HSA. Es erfolgteeine vorsichtige Dosiseskalation mit wöchentlichen Injektionen ab 20 mg/m2. Ab einerDosis von 40 mg/m2 pro Woche wurden erste Toxizitätssymptome (Common-Toxic-Criteria: CTC Grade) beobachtet. Insgesamt erschien eine wöchentliche Applikationvon 50 – 60 mg/m2 als gut verträglich.Unerwünschte Arzneimittelreaktionen, wie z. B. leichte Übelkeit, Diarrhoe, Obstipation,Epistaxis, Mukositis oder Müdigkeit, bildeten sich durch eine Therapieunterbrechunginnerhalb von 1 – 2 Wochen komplett zurück. In keinem Fall war die Gabe von Folin-säure (Leucovorin) notwendig. An hämatologischen oder laborchemischen Verände-rungen fanden sich dreimal eine Thrombozytopenie (2 x Grad 2, 1 x Grad 4) sowieviermal ein Anstieg der Transaminasen auf Grad 3. Auch hier konnte eine Rückbildungder Veränderungen durch eine Spreizung des Applikationsschemas erreicht werden.Bei den 10 Patienten, welche mit einer wöchentlichen Applikation von 50 bzw. 60mg/m2 behandelt wurden, kam es in drei Fällen (1 x Pleuramesotheliom, 2 x Hyperne-phrom) zu einer langanhaltenden Remission (105).Die Ergebnisse der präklinischen und klinischen Erfahrungen mit MTX-HSA bildetendie Grundlage für eine Lizenzvergabe von MTX-HSA an Klinge-Pharma, München,jetzt Fujisawa-Deutschland, um MTX-HSA im Rahmen eines Zulassungsverfahrensweiter zu untersuchen.

1 8
1.6 Fragestellung und Zielsetzung der Arbeit
Aufgabe dieser Arbeit war es, die Möglichkeit von Albumin als vielfach verwendbarerTrägerstoff in der Diagnostik und Therapie maligner Tumoren aufzuzeigen. Albuminwurde hierzu bei verschiedenen Aufgabestellungen eingesetzt:
1.6.1 Die laserinduzierte Fluoreszenzdiagnostik mit 5-Aminofluorescein-HSA beimalignen Gliomen
Es sollte die Frage geklärt werden, ob durch eine albuminvermittelte Fluoreszenzanfär-bung bei malignen Gliomen eine intraoperative Tumordarstellung möglich sei, wobeifolgende Eigenschaften an den albumingebundenen Farbstoff gestellt wurden:
• Fehlende Toxizität• Hohe, selektive und langanhaltende Anreicherung des Farbstoffkonjugates im
Tumorgewebe• Keine oder nur unwesentliche Ausbleichung des Farbstoffes• Hohe Kontrastierung und Detektion des Farbstoffes ohne Zuhilfenahme optischer
Verstärkersysteme.
1.6.2 Albuminvermittelte Chemotherapie mit Aminopterin-HSA
Gerade besonders toxische Chemotherapeutika erscheinen für eine Albuminbeladunginteressant, da durch die kovalente Bindung des Chemotherapeutikums an den Träger-stoff Albumin ein „pro-drug“ ensteht, welcher dadurch bedingt eine reduzierte systemi-sche Toxizität aufweist und die Wirksubstanz erst im Lysosom der Tumorzelle freisetzt.Ziel war es, das bereits Mitte der 50er Jahre verwendete hochwirksame aber systemischzu toxische Antifolat Aminopterin an Albunim zu binden und es im Tierexperiment aufseine pharmakokinetischen Eigenschaften, seine Verteilung, seine Verträglichkeit undWirksamkeit zu untersuchen.

1 9
1.6.3 Klinische Phase-I und II-Studien mit Methotrexat-Albumin (MTX-HSA)bei Patienten mit rezidivierenden malignen Gehirntumoren
Nach Auslizensierung der von Sinn entwickelten Patente (MTX-HSA; AFlc-HSA etc.)an Firma Klinge-Pharma, jetzt Fujisawa-Deutschland, wurde entsprechend dem Arz-neimittelgesetz eine zweite Phase-I-Studie mit aufsteigenden Dosierungen von 50mg/m2; 70 mg/m2; 80 mg/m2 und 90 mg/m2 MTX-HSA in zweiwöchentlichen Abstän-den bei insgesamt 23 Karzinompatienten initiiert. Wir selbst nahmen an dieser Studiemit vier Patienten teil. Danach wurde unter der Leitung der Neurochirurgischen Univer-sitätsklinik Heidelberg eine vergleichenden Phase-II-Studie bei Patienten mit primärenund sekundären Glioblastomrezidiven veranlaßt.
1.6.4 Albuminvermittelte Lymphdiagnostik mit Tetra(4-Carboxy-Phenyl-Chlorin- (TCPC) oder Tetra(4-Carboxy-Phenyl-Porphyrin (TCPP) HSAund Gadolinium-HSA
Durch Einführung des „sentinel lymph note“ (SLN) Konzeptes hat die Diagnostik undTherapie von tumordrainierende Lymphbahnen und deren Lymphknoten eine Renais-sance erfahren. Mit 99mTc-HSA ist präoperativ eine Lymphabstromszintigraphie mög-lich, intraoperativ kann dann der SLN mit speziellen Gammasonden und auch unterZuhilfenahme von intraoperativ appliziertem Patentblau aufgesucht werden. Da bislanghauptsächlich kolloidales 99mTc-HSA mittels der Lymphabstromszintigraphie und in-traoperativ Gammasonden zum Einsatz kommen, sollte versucht werden, ob nicht auchdurch eine Fluoreszenzbeladung von HSA eine direkte Darstellung von Lymphwegenund von im Abstromgebiet liegenden Lymphknoten (SLN) möglich sei. Dies solltetierexperimentell durch eine Beladung von Tetra(4-Carboxy-Phenyl-Chlorin (TCPC)oder Tetra(4-Carboxy-Phenyl-Porphyrin (TCPP) an HSA und auch kernspintomogra-phisch mit Gadolinium-HSA untersucht werden.

2 0
Kapitel 2Intraoperative Fluoreszenzdiagnostik maligner Gliome mit5-Aminofluorescein-HSA (AFLc-HSA)2.1 Einführung
Als Anfang der 90er Jahre in unserer Klinik unter Leitung von Albert (1-2) das frühepostoperative Kernspintomogramm vermehrt zur Beurteilung der Radikalität nach mi-krochirurgischer Resektion von Glioblastomen herangezogen wurde, stellten er undseine Mitarbeiter fest, daß in vielen Fällen solide kontrastmittelaufnehmende Tumoran-teile verblieben waren, die der intraoperativen Wahrnehmung des Neurochirurgen trotzVerwendung moderner Operationsmikroskope entgangen waren. Diese soliden und imfrühen postoperativen Kernspintomogramm entdeckten Tumoranteile stellten dann imweiteren Verlauf den Ursprung des klinisch manifesten Tumorrezidives dar und warenstatistisch gesehen prognostisch von entscheidender Bedeutung.
Aus diesen Beobachtungen heraus ergab sich unmittelbar die Konsequenz einer verbes-serten intraoperativen Darstellung von solidem Tumorgewebe. Auch wenn andere Ver-fahren, wie z. B. Neuronavigation, intraoperatives CT oder Kernspintomogramm undUltraschall (3-9), ebenfalls Gegenstand intensiver wissenschaftlicher Betrachtungenbezüglich der verbesserten intraoperativen Tumordarstellung sind, so galten unsereÜberlegungen der Verwendung fluoreszenzaktiver Substanzen, wie z. B. dem Fluore-scein, welches bereits 1948 seinen Einsatz zur intraoperativen Darstellung von Gehirn-tumoren fand (10). Ähnlich den sonst zur Tumordiagnostik applizierten Kontrastmittelpassiert das Fluroescein-Natriumsalz die bei malignen Gliomen gestörte Blut-Hirn-Schranke und lagert sich dann interstitiell im Tumorgewebe ab. Auf Grund der aberschon im Jahre 1948 beschriebenen Penetration von Fluorescein ins umliegende Tumo-rödem ist eine lang anhaltende und selektive Darstellung von Tumorgewebe jedoch nurbedingt möglich.
2 1
2.2 Physikalische Eigenschaften von 5-Aminofluorescein (AFL)
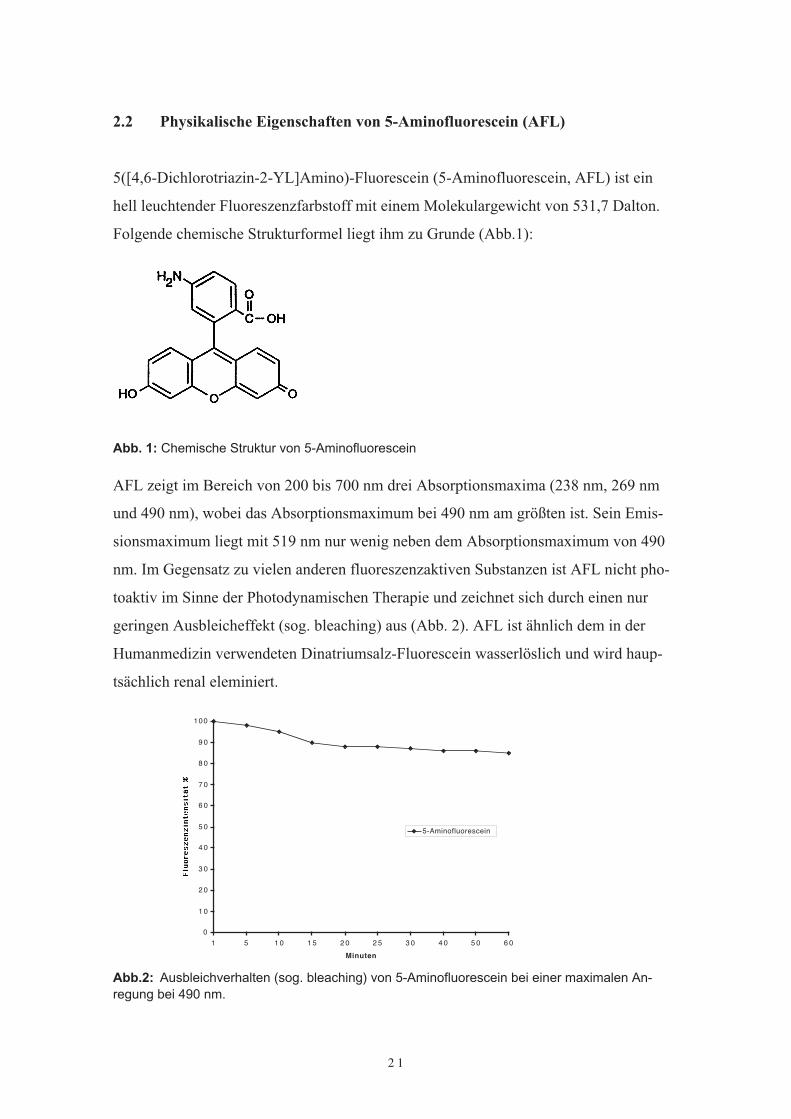
5([4,6-Dichlorotriazin-2-YL]Amino)-Fluorescein (5-Aminofluorescein, AFL) ist einhell leuchtender Fluoreszenzfarbstoff mit einem Molekulargewicht von 531,7 Dalton.Folgende chemische Strukturformel liegt ihm zu Grunde (Abb.1):
Abb. 1: Chemische Struktur von 5-Aminofluorescein
AFL zeigt im Bereich von 200 bis 700 nm drei Absorptionsmaxima (238 nm, 269 nmund 490 nm), wobei das Absorptionsmaximum bei 490 nm am größten ist. Sein Emis-sionsmaximum liegt mit 519 nm nur wenig neben dem Absorptionsmaximum von 490nm. Im Gegensatz zu vielen anderen fluoreszenzaktiven Substanzen ist AFL nicht pho-toaktiv im Sinne der Photodynamischen Therapie und zeichnet sich durch einen nurgeringen Ausbleicheffekt (sog. bleaching) aus (Abb. 2). AFL ist ähnlich dem in derHumanmedizin verwendeten Dinatriumsalz-Fluorescein wasserlöslich und wird haup-tsächlich renal eleminiert.
Abb.2: Ausbleichverhalten (sog. bleaching) von 5-Aminofluorescein bei einer maximalen An-regung bei 490 nm.
0
1 0
2 0
3 0
4 0
5 0
6 0
7 0
8 0
9 0
100
1 5 1 0 1 5 2 0 2 5 3 0 4 0 5 0 6 0Minuten
5-Aminofluorescein

2 2
2.3 Die kovalente Bindung von 5-Aminofluorescein an Albumin (AFLc-HSA)
Ein Volumen von 65 mg 5-([4,6-Dichlorotriazin-2-yl]-Amino)-Fluorescein (AFL, Sig-ma, Deisenhofen) wird in 3 ml Dimethylsulfoxid (DMSO) gelöst und wird unter konti-nuierlichem langsamen Rühren zu 4 g humanem Serumalbumin 20 % (Pharma DessauGmbH, Dessau), in 20 ml Albuminlösung und 60 ml 0,34 Mol NaHCO3 gelöst, hinzuge-fügt. Während des Hinzufügens von AFL zur Proteinlösung färbt diese sich grün-gelb,bleibt aber absolut klar. Ungefähr 45 Minuten später wird die Proteinlösung mit 350 mlAmpuwa verdünnt. Verunreinigungen, wie z. B. ungebundenes Fluorescein oderDMSO, werden dann vom gebundenen Protein AFLc-HSA durch Ultrafiltration (YM30 Millipore) abgetrennt. Die Reinheit des hergestellten Proteinkonjugates wurde mit-tels der hochauflösenden Flüssigkeitschromatographie (HPLC) überprüft:
HPLC Bedingungen:Vorsäule: Phenomenex GFC - 4000, 4 mm L x 3 mm IDSäule: Zorbax GF 2501. Laufmittel: 0,25 Mol Li-acetat, 0,05 Mol Li-citrate pH 7,4 (Ampuwa)2. Laufmittel: 95% Methanol, 5% Wasser (HPLCgrad/Am-puwa)Fluß: 0 -15 min Laufmittel 1 (100%); 1,0 ml/minGradient: 15 - 30 min Laufmittel 2 (von 0 auf 100%); 1,3 ml/min
30 - 45 min Laufmittel 2 (100%)45 - 50 min Laufmittel 2 (von 100 auf 0%)50 - 60 min Laufmittel 1 (100%); 1,0 ml/min
Druck: ungefähr 65 Bar
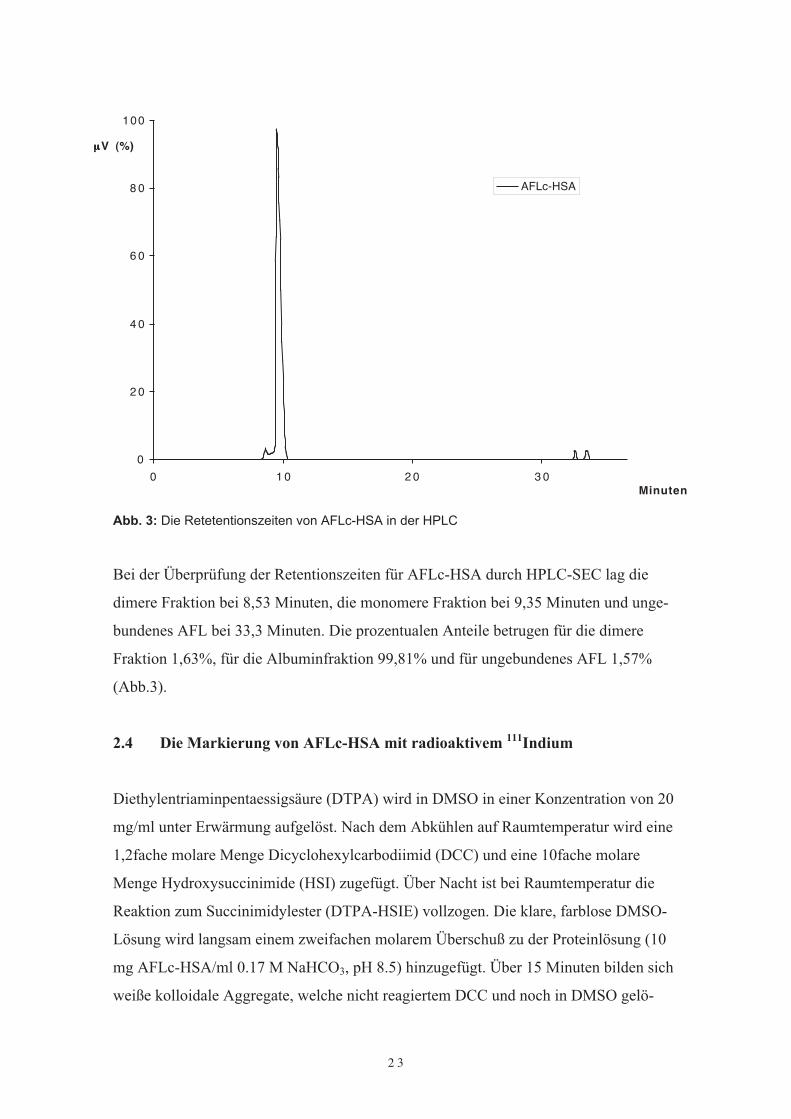
Retentionszeiten fürAFLc-HSA: dimere Albuminfraktion 8,25 min
monomere Albuminfraktion 9,16 minungebundene Albuminfraktion 31,34 min
2 3
Abb. 3: Die Retetentionszeiten von AFLc-HSA in der HPLC
Bei der Überprüfung der Retentionszeiten für AFLc-HSA durch HPLC-SEC lag diedimere Fraktion bei 8,53 Minuten, die monomere Fraktion bei 9,35 Minuten und unge-bundenes AFL bei 33,3 Minuten. Die prozentualen Anteile betrugen für die dimereFraktion 1,63%, für die Albuminfraktion 99,81% und für ungebundenes AFL 1,57%(Abb.3).
2.4 Die Markierung von AFLc-HSA mit radioaktivem 111Indium
Diethylentriaminpentaessigsäure (DTPA) wird in DMSO in einer Konzentration von 20mg/ml unter Erwärmung aufgelöst. Nach dem Abkühlen auf Raumtemperatur wird eine1,2fache molare Menge Dicyclohexylcarbodiimid (DCC) und eine 10fache molareMenge Hydroxysuccinimide (HSI) zugefügt. Über Nacht ist bei Raumtemperatur dieReaktion zum Succinimidylester (DTPA-HSIE) vollzogen. Die klare, farblose DMSO-Lösung wird langsam einem zweifachen molarem Überschuß zu der Proteinlösung (10mg AFLc-HSA/ml 0.17 M NaHCO3, pH 8.5) hinzugefügt. Über 15 Minuten bilden sichweiße kolloidale Aggregate, welche nicht reagiertem DCC und noch in DMSO gelö-
0
2 0
4 0
6 0
8 0
100
0 1 0 2 0 3 0Minuten
µµµµV (%)
AFLc-HSA

2 4
stem di-Cyclohexyl-Harnstoff (DCH) entsprechen. Diese werden mit Hilfe eines Filtersentfernt (0,22 µm Millipore), während hingegen DMSO und freies DTPA durch Ultra-filtration (C30 Millipore) abgetrennt werden. DTPA-aktiviertes AFLc-HSA wird dannmit 111InCl3 durch Hinzufügen einer entprechenden Menge Radioaktivität als ein 111In-Citrat-Komplex an10 mg Protein gebunden. Direkt nach Hinzufügens des 111In-Citrat-Komplexes werden ungebundenes 111In durch Ultrafiltration mit einer C30 MilliporeEinheit abgetrennt. Mit Hilfe dieser Technik können 98 % des verwendeten 111In kova-lent an DTPA-AFLc-HSA gebunden werden. Um die Plasmazirkulationszeiten zu ver-gleichen, wurde AFLc-HSA auch in einer 3:1 molaren Beladung mit 111In-DTPA mar-kiert. Ebenfalls wurde das käuflich zu erwerbende Fluorescein-Isothiozyanat (FITC-SA) mit 111In-DTPA beladen.
2.5 Das C6-Gliom Modell
Weibliche, pathogenfreie (specific pathogen free, SPF) Sprague-Dawley-Ratten (n = 25)mit einem Körpergewicht von 180 - 200 g wurden für die Fluoreszenzuntersuchungenmit AFLc-HSA verwendet. Die Tiere wurden unter standardisierten SPF Bedingungenin einem Mikroisolator gehalten.Die C6-Gliom Zelllinie (11) wurde in Gewebekultur in 90 % DMEM Medium (PAALaboratories, Östereich) und 10 % fetalen Kälberserum (Gibco, Eggenstein) gehalten.Die Analgosedierung der Versuchstiere erfolgte durch intraperitoneale Injektionen vonKetanest (Park-Davis & Company, Berlin) und Rompun (Bayer, Leverkusen), danachkonnte der Kopf in einem stereotaktischen Ringsystem (David Kopf Instruments, USA)fixiert werden. Nach Bohrlochtrepanation wurde in einer Geschwindigkeit von 2mm/Minute eine 10 µl Hamilton Spritze, versehen mit einer 26-Gauge Nadel, in daslinke frontale Marklager der Ratte in einer Tiefe von 6 mm, 4 mm lateral des Bregmaseingebracht. Nach Zurückziehen der Nadel um einen Millimeter erfolgte die intrazere-brale Injektion in einer Dosierung von 1 x 105 C6-Gliomzellen in 5 µl serumfreiemDMEM Medium über einen Zeitrahmen von 5 Minuten (1 µl/Minute). Nach langsamemZurückziehen der Nadel (2 mm/Minute) wurde das Bohrloch mit Knochenwachs ver-schlossen und die Haut vernäht. In mehr als 90 % der Tiere entwickelte sich ein soliderTumor im linken frontalen Marklager. Typischerweise war eine Gewichtsstagnation ein
2 5
Frühsymptom bei ausreichender Tumorgröße von 4 - 5 mm im Durchmesser. DieTumorgewichte lagen zwischen 80 - 120 mg.
2.6 Die Plasmaclearance von AFLc-HSA
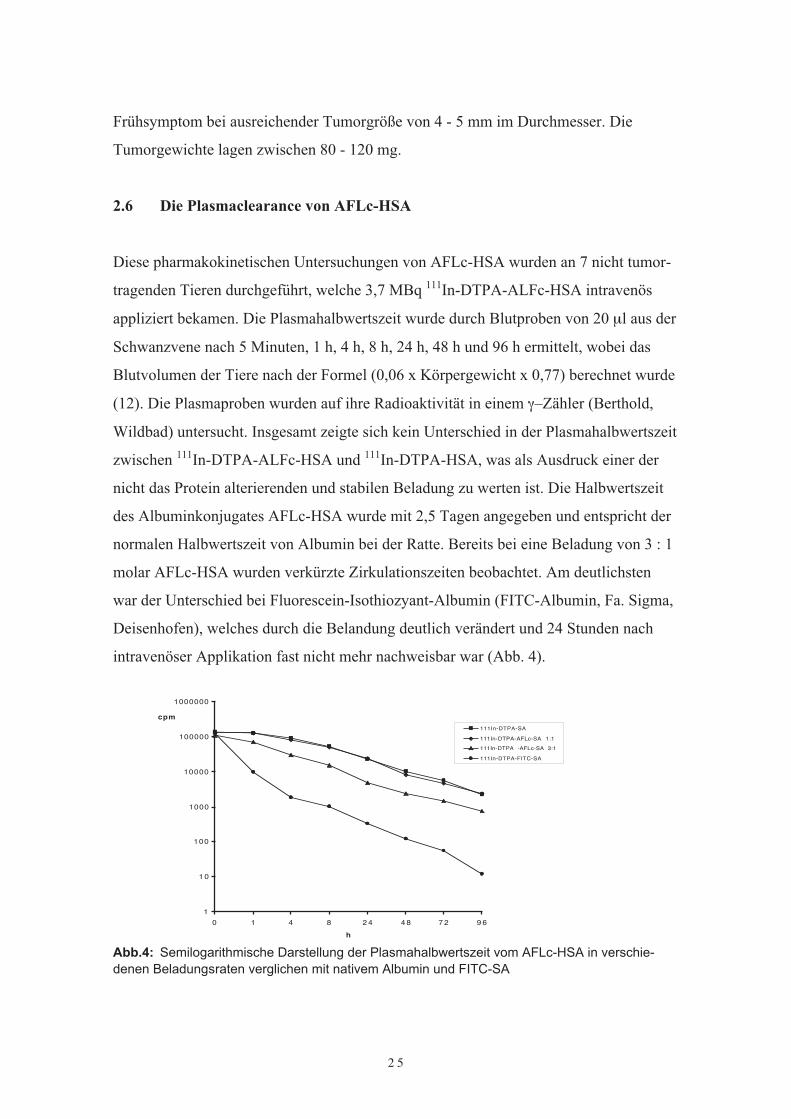
Diese pharmakokinetischen Untersuchungen von AFLc-HSA wurden an 7 nicht tumor-tragenden Tieren durchgeführt, welche 3,7 MBq 111In-DTPA-ALFc-HSA intravenösappliziert bekamen. Die Plasmahalbwertszeit wurde durch Blutproben von 20 µl aus derSchwanzvene nach 5 Minuten, 1 h, 4 h, 8 h, 24 h, 48 h und 96 h ermittelt, wobei dasBlutvolumen der Tiere nach der Formel (0,06 x Körpergewicht x 0,77) berechnet wurde(12). Die Plasmaproben wurden auf ihre Radioaktivität in einem γ–Zähler (Berthold,Wildbad) untersucht. Insgesamt zeigte sich kein Unterschied in der Plasmahalbwertszeitzwischen 111In-DTPA-ALFc-HSA und 111In-DTPA-HSA, was als Ausdruck einer dernicht das Protein alterierenden und stabilen Beladung zu werten ist. Die Halbwertszeitdes Albuminkonjugates AFLc-HSA wurde mit 2,5 Tagen angegeben und entspricht dernormalen Halbwertszeit von Albumin bei der Ratte. Bereits bei eine Beladung von 3 : 1molar AFLc-HSA wurden verkürzte Zirkulationszeiten beobachtet. Am deutlichstenwar der Unterschied bei Fluorescein-Isothiozyant-Albumin (FITC-Albumin, Fa. Sigma,Deisenhofen), welches durch die Belandung deutlich verändert und 24 Stunden nachintravenöser Applikation fast nicht mehr nachweisbar war (Abb. 4).
Abb.4: Semilogarithmische Darstellung der Plasmahalbwertszeit vom AFLc-HSA in verschie-denen Beladungsraten verglichen mit nativem Albumin und FITC-SA
1
1 0
100
1000
10000
100000
1000000
0 1 4 8 2 4 4 8 7 2 9 6h
cpm111In-DTPA-SA111In-DTPA-AFLc-SA 1:1111In-DTPA -AFLc-SA 3:1111In-DTPA-FITC-SA

2 6
2.7 Die Aufnahme von 111In-DTPA-AFLc-HSA in das C6-Gliom
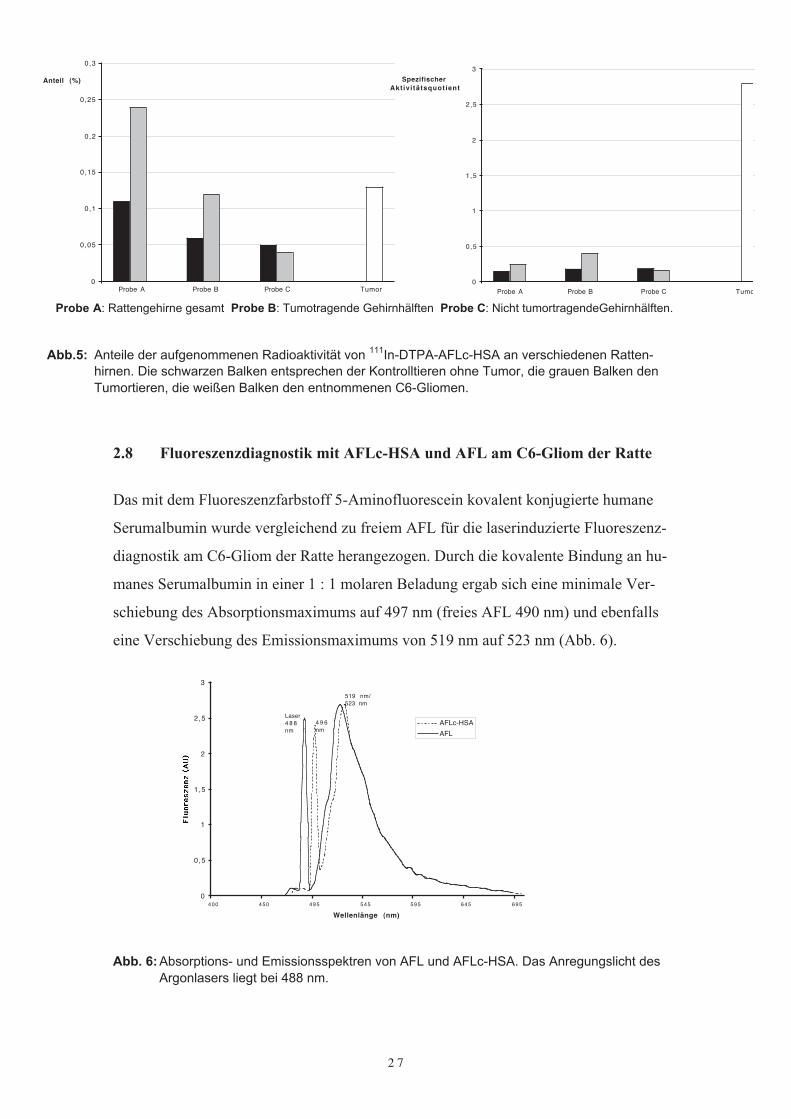
Szintigraphische Untersuchungen sollten die Aufnahme des Proteinkonjugates im Tu-mor sowie seine Bioverteilung aufzeigen. Dazu wurden die Versuchstiere (n=5) 5 Mi-nuten, 1 h, 4 h, 8 h und 24 h nach Applikation von 3,7 MBq 111In-DTPA-ALFc-HSAszintigraphisch untersucht. Zudem wurden 24 h nach Applikation des radioaktiv mar-kierten Proteinkonjugates die Rattengehirne entnommen, um die Radioaktivität im Ge-hirn selbst im γ-Zähler untersuchen zu können. Die Aufnahme der Radioaktivität wurdesowohl im gesamten Gehirn wie auch der rechten nicht tumortragenden Hemisphäre,der linken tumortragenden Hemissphäre und nach Entnahme des Tumors in ihm selbstgemessen.Szintigraphisch fand sich kein Unterschied zwischen tumortragenden und nicht tumor-tragenden Tieren, so daß die Bestimmung der Aufnahme der Radioaktivität im C6-Gliom der Rattengehirne nur im γ-Zähler möglich war. Insgesamt war der Anteil anRadioaktivität mit 0,04 – 0,34 % an der Gesamtverteilung im Körper gering. Dennochfanden sich deutliche Unterschiede bei den einzelnen Gewebeproben zwischen den Tie-ren mit Tumor und denen ohne Tumor. Die Messung der gesamten Rattengehirne ohneTumor ergab lediglich eine Aufnahme 0,1 % an Radioaktivität. Bei den tumortragendenRattengehirnen dagegen steigerte sich die Aufnahme der applizierten Radioaktivität auf0,23 % (Probe A). Die Messung der linken Gehirnhemisphären (Probe B) ergab dannaber eine Steigerung der Aufnahme an Radioaktivität bei den tumortragenden Tierenum das Dreifache. Kein Unterschied ergab sich bei der Messung der rechten Gehirn-hälften (Probe C).Der spezifische Aktivitätsquotient berechnet den Anteil der applizierten Radioaktivitätin Relation zum Tumorgewicht und dem Gewicht der Versuchstiere. Bei einer homoge-nen Verteilung berechnet sich somit ein spezifischer Aktivitätsquotient von 1, währendhingegen ein Quotient von > 1 eine spezifische Aufnahme beweist.Die spezifischen Aktivitätsquotienten bei den nicht tumortragenden Rattengehirnen lagbei 0,12, bei den tumortragenen Rattengehirnen bei 0,26 und bei den linken Tumortragenden Hemissphären bei 0,4. Die Berechnung des spezifischen Aktivitätsquotientenim Tumor selbst wies mit 2,8 eine 23fach höhere Aufnahme gegenüber dem normalenGehirn nach (Abb.5).
2 7
Probe A: Rattengehirne gesamt Probe B: Tumotragende Gehirnhälften Probe C: Nicht tumortragendeGehirnhälften.
Abb.5: Anteile der aufgenommenen Radioaktivität von 111In-DTPA-AFLc-HSA an verschiedenen Ratten-hirnen. Die schwarzen Balken entsprechen der Kontrolltieren ohne Tumor, die grauen Balken denTumortieren, die weißen Balken den entnommenen C6-Gliomen.
2.8 Fluoreszenzdiagnostik mit AFLc-HSA und AFL am C6-Gliom der Ratte
Das mit dem Fluoreszenzfarbstoff 5-Aminofluorescein kovalent konjugierte humaneSerumalbumin wurde vergleichend zu freiem AFL für die laserinduzierte Fluoreszenz-diagnostik am C6-Gliom der Ratte herangezogen. Durch die kovalente Bindung an hu-manes Serumalbumin in einer 1 : 1 molaren Beladung ergab sich eine minimale Ver-schiebung des Absorptionsmaximums auf 497 nm (freies AFL 490 nm) und ebenfallseine Verschiebung des Emissionsmaximums von 519 nm auf 523 nm (Abb. 6).
Abb. 6:Absorptions- und Emissionsspektren von AFL und AFLc-HSA. Das Anregungslicht desArgonlasers liegt bei 488 nm.
0
0,05
0,1
0,15
0,2
0,25
0,3
1
Anteil (%)
Probe A Probe B Probe C Tumor0
0,5
1
1,5
2
2,5
3
1
Spezifischer Akt iv i tätsquot ient
Probe A Probe B Probe C Tumo
0
0,5
1
1,5
2
2,5
3
400 450 495 545 595 645 695Wellenlänge (nm)
AFLc-HSAAFL
Laser4 8 8nm
4 9 6nm
519 nm/523 nm

2 8
Das Farbstoffkonjugat wurde den tumortragenden Tieren in Konzentrationen von 0,5mg/kg Körpergewicht sowie 1,0 und 2,0 mg/kg Körpergewicht (5 Tiere pro Dosie-rungsgruppe) intravenös appliziert und die Rattenhirne 24 Stunden später in toto ent-nommen und bei - 70 ° C tiefgefroren. Vergleichend zur Fluoreszenzdarstellung mitAFLc-HSA wurde ungebundenes AFL bei 5 Tieren in einer Konzentration von 2,0mg/kg Körpergewicht ebenfalls intravenös appliziert und ebenfalls die Rattengehirne 24h danach entnommen.Als Anregungsquelle diente ein 10 Watt Argonlaser (Spectra Physics, USA). AFLc-HSA oder AFL wurden bei 488 nm angeregt (Ausgang Argonlaser 1,2 Watt, Beschei-nung bei 150 – 200 mWatt/cm2). Zur Fluoreszenzbetrachtung wurde das Anregungslichtdes Lasers durch einen Langwellengelbfilter bei 515 nm abgeschnitten.
Makroskopische Fluoreszenzbetrachtung
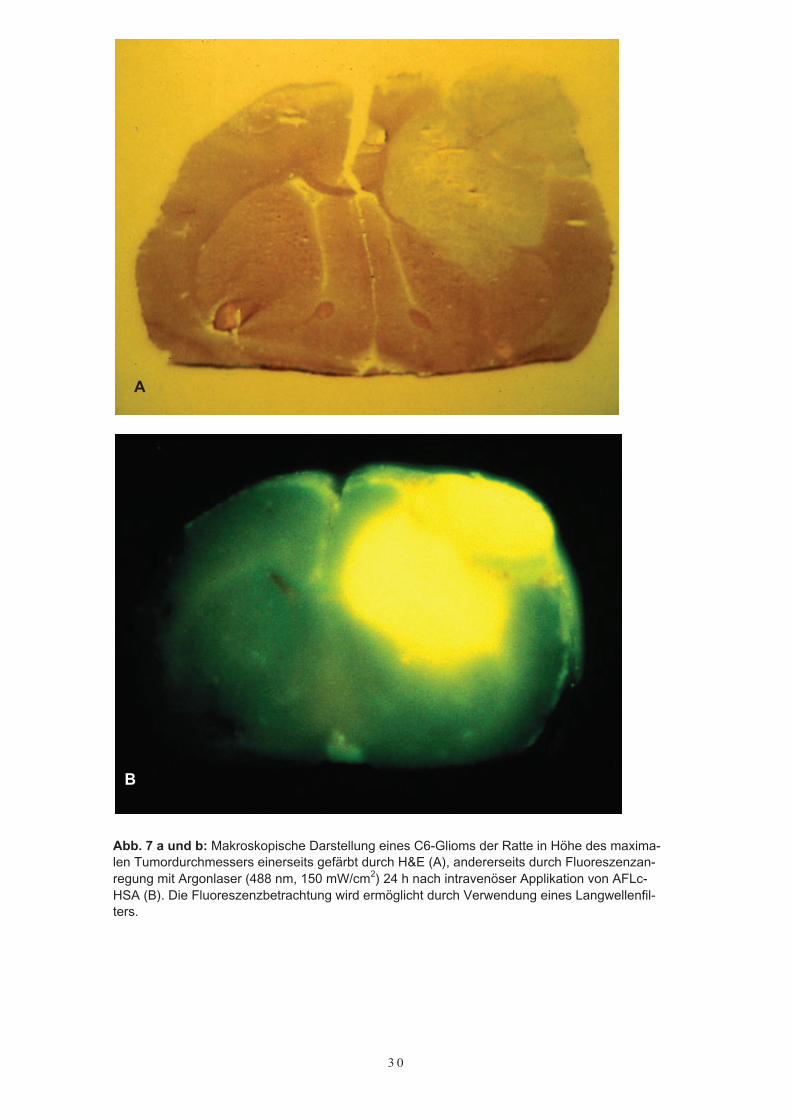
An koronaren Schnitten der 24 Stunden nach intravenöser Farbstoffapplikation ent-nommenen Gehirne wurden die tiefgefrorenen Proben in Höhe des maximalen Tumor-durchmessers untersucht. Bei allen C6-Gliomen zeigte sich unter der laserinduziertenFluoreszenzanregung eine gute Darstellung des durch AFLc-HSA markierten Tumors,wobei die Betrachtung der Fluoreszenzemission bereits mit bloßem Auge möglich war.Subjektiv war kein Unterschied in der Fluoreszenz zwischen den verschiedenen Kon-zentrationen bemerkbar. Im Gegensatz zu der sehr deutlichen Tumordarstellung mitAFLc-HSA fehlte diese 24 Stunden nach intravenöser Applikation von freiem AFLgänzlich (Abb. 7 a und b). Eine Autofluoreszenz des Gehirns wurde bei einer Anregungvon 488 nm nicht beobachtet.
Mikroskopische Fluoreszenzbetrachtung
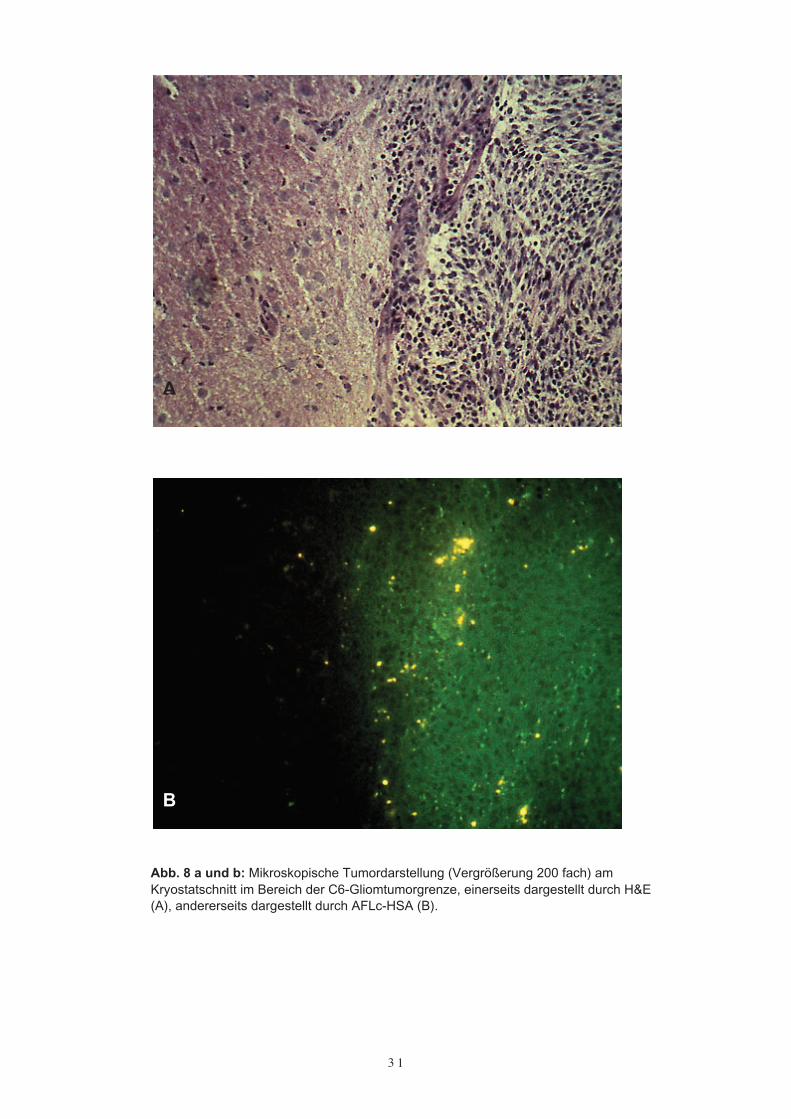
Um die Fluoreszenzdarstellung mit AFLc-HSA der histopathologischen Tumorgrenzezuordnen zu können, wurden koronare Kryostatschnitte in einer Dicke von 10 µm ange-fertigt. Die so gewonnenen Kryostatschnitte wurden im Fluoreszenzmikroskop be-trachtet und mit einem direkt benachbarten H & E gefärbten Kryostatschnitt verglichen.

2 9
Dabei zeigte sich ein gutes Übereinstimmen der fluoreszenzoptischen Tumordarstellungmit der durch H & E markierten histopathologischen Tumorgrenze (Abb. 8 a und b).
2.9 Fluoreszenzlokalisation von AFLc-HSA in der C6-Gliom Zellkultur
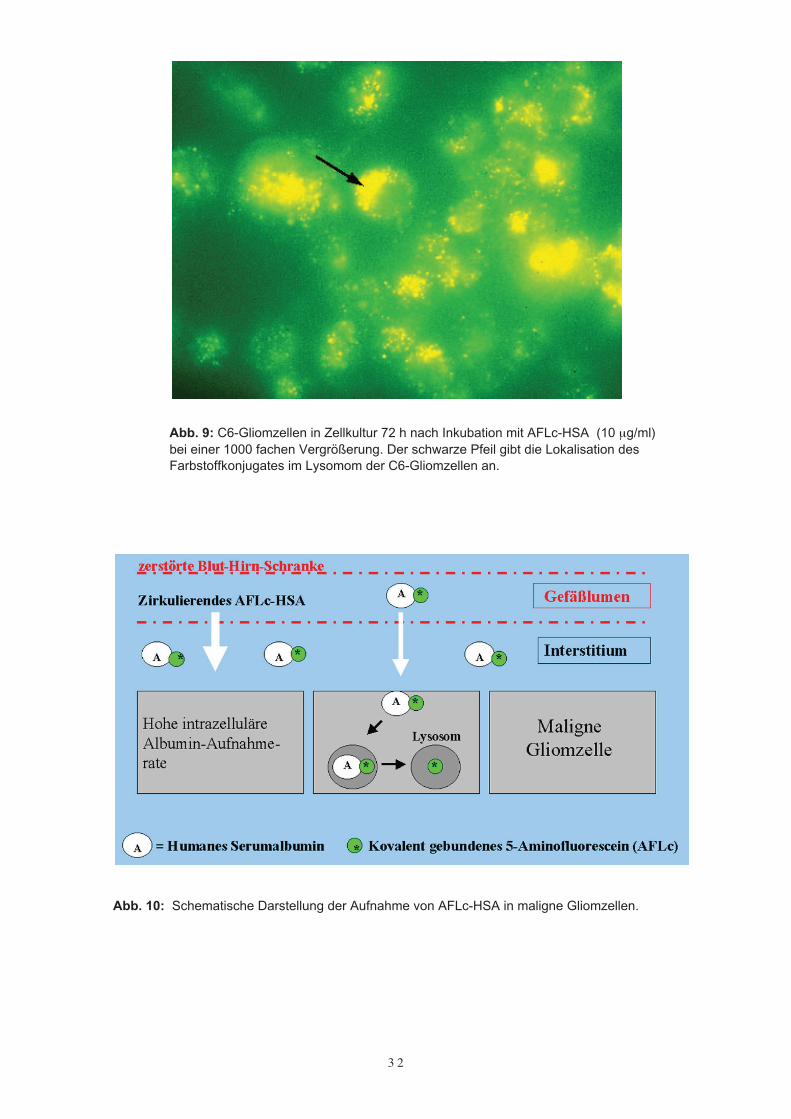
Um die Lokalisation des Farbstoffkonjugates in der Tumorzelle selbst genauer definie-ren zu können, wurden C6-Gliom Zellkulturen über 72 Stunden mit AFLc-HSA in einerKonzentration von 10 µg/ml inkubiert. Der nicht zellulär verbliebene Farbstoff wurdedann durch zweimaliges Waschen der Zellkultur mit Phosphatpuffer entfernt. Danachkonnten die C6-Gliomzellen im Fluoreszenzmikroskop bei einer 1.000fachen Vergröße-rung betrachtet werden. Dabei zeigte sich eine punktuelle, den Lysosomen entsprechen-de Akkumulation des Farbstoffkonjugates in den C6-Gliomzellen (Abb. 9). Somit kannvermutet werden, daß das Albuminkonjugat via Endozytose intrazellulär aufgenommenwird und sich dann im Lysosom der Zelle anreichert (Abb. 10). Trotz des sauren pH-Wertes im Lysosom (die Fluoreszenzemission ist pH abhängig) fand sich eine guteFluoreszenz von AFLc-HSA in der Tumorzelle. Somit war die aktive Aufnahme desFarbstoffkonjugates in die Tumorzelle bewiesen, welche im Gegensatz zu den meistengängigen Kontrastmitteln AFLc-HSA als intrazelluläres Kontrastmittel aufweist.
3 0
Abb. 7 a und b: Makroskopische Darstellung eines C6-Glioms der Ratte in Höhe des maxima-len Tumordurchmessers einerseits gefärbt durch H&E (A), andererseits durch Fluoreszenzan-regung mit Argonlaser (488 nm, 150 mW/cm2) 24 h nach intravenöser Applikation von AFLc-HSA (B). Die Fluoreszenzbetrachtung wird ermöglicht durch Verwendung eines Langwellenfil-ters.
A
B
3 1
Abb. 8 a und b: Mikroskopische Tumordarstellung (Vergrößerung 200 fach) amKryostatschnitt im Bereich der C6-Gliomtumorgrenze, einerseits dargestellt durch H&E(A), andererseits dargestellt durch AFLc-HSA (B).
A
B
3 2
Abb. 9: C6-Gliomzellen in Zellkultur 72 h nach Inkubation mit AFLc-HSA (10 µg/ml)bei einer 1000 fachen Vergrößerung. Der schwarze Pfeil gibt die Lokalisation desFarbstoffkonjugates im Lysomom der C6-Gliomzellen an.
Abb. 10: Schematische Darstellung der Aufnahme von AFLc-HSA in maligne Gliomzellen.

3 3
2.10 Konfokale Mikroskopie mit AFLc-HSA und lysosomaler Markierung anC6-Gliomzellen
Die Kultivierung der C6-Gliomzellen erfolgte in RMPI 1640 mit 10 % FCS und 1 %Glutamin. Für die konfokale Laserscanningmikroskopie (cLSM, Fa. Zeiss, Oberkochen)wurden die Zellen abtrypsiniert, in einer Neubauer Kammer gezählt und auf 5 x 104
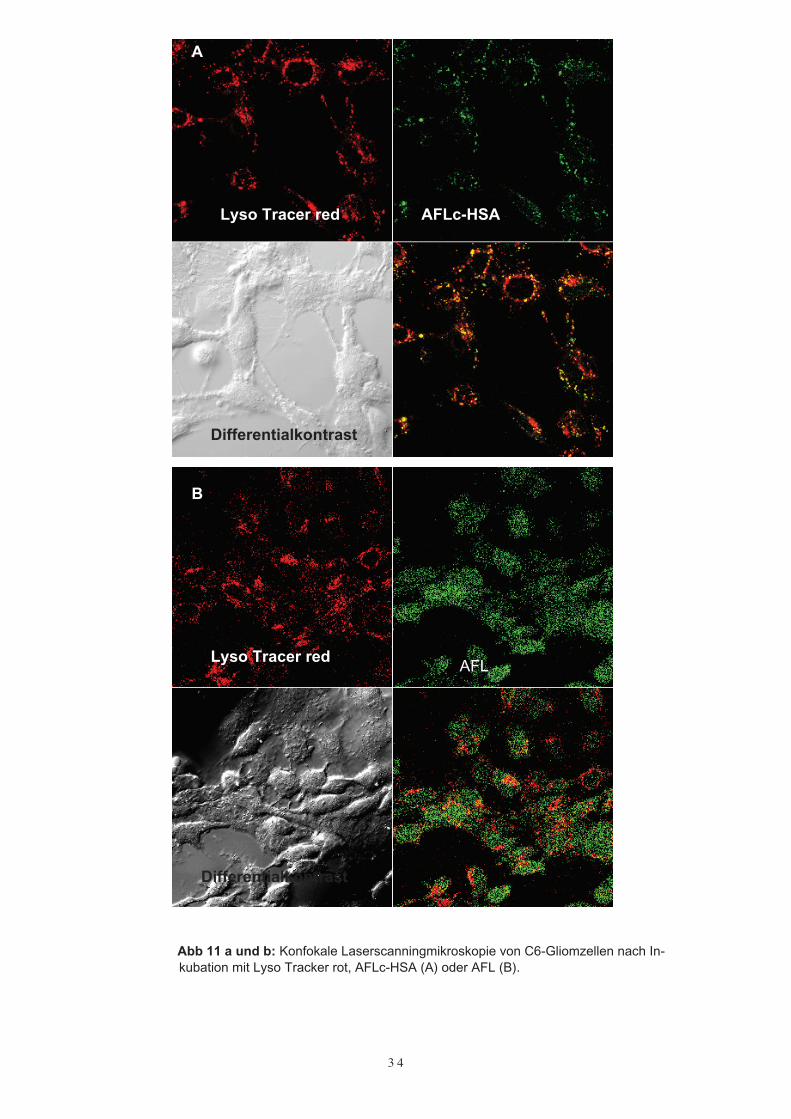
Zellen/ml eingestellt. Davon wurden 50 – 100 µl auf Deckgläschen ausgesät (Durch-messer 12 mm) und für 24 Stunden bei 37°C mit 5 % CO2 inkubiert. Auf 1 % reduzier-tem FCS Medium wurden die Zellen mit 50 µg/ml AFLc-HSA bzw. 50 µg/ml AFLüberflutet und für weitere 24h kultiviert.Für die Lysosomenmarkierung wurden die C6-Gliomzellen dreimal mit RMPI ohneZusätze gewaschen. Danach wurde Lyso Tracker Red (Absorption: 577 nm; Emission:590 nm; Molecular Probes, USA) in einer Konzentration von 50 nM über 40 Minutenhinzugefügt. Wiederum wurden die Zellen dreimal mit RMPI ohne Zusätze gewaschenund anschließend am konfokalen Laserscanningmikroskop untersucht.Einerseits wurden die Zellen im Differentialkontrast inspiziert, andererseits wurde derLyso Tracer bei 577 nm (rote Fluoreszenz) und AFL oder AFLc-HSA bei 488 nm (grü-ne Fluoreszenz) angeregt. Werden beide Fluoreszenzen übereinandergelegt, zeigen Or-te, an denen beide Farstoffe lokalisiert sind, eine gelbe Fluoreszenz (Abb. 11 a). Somitwar die Aufnahme von AFLc-HSA im Lysosom der C6-Gliomzellen bewiesen. Dage-gen war nach Inkubation der Zellen mit freiem AFL keine punktförmige, den Lysoso-men entsprechende Aufnahme zu erkennen. Hier konnte man eine unspezifische Ver-teilung von AFL im Zytoplasma beobachten (Abb. 11b).
3 4
Abb 11 a und b: Konfokale Laserscanningmikroskopie von C6-Gliomzellen nach In-kubation mit Lyso Tracker rot, AFLc-HSA (A) oder AFL (B).
Lyso-tracer rot AFLc-HSA
Lyso-tracer + AFLc-HSA
Lyso-tracer rot AFL
Lyso-tracer + AFL
LsoTracker red AFLc-HSA
Differentialkontrast
AFL
A
B
A
Lyso Tracer red AFL
A
Lyso Tracer red AFLc-HSA
Differentialkontrast
Lyso Tracer red AFL
Differentialkontrast
B

3 5
2.11 Präklinische Studien mit 111In-DTPA-HSA bei Patienten mit Glioblastomen
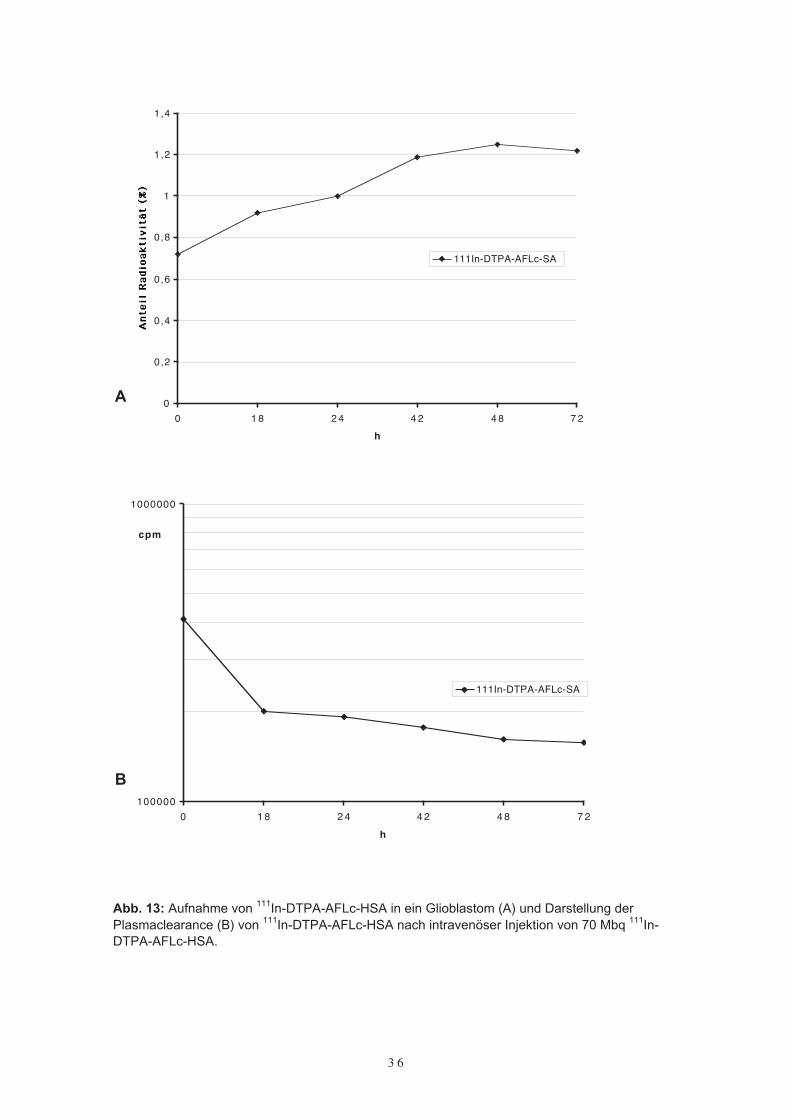
Zunächst wurde die Aufnahme des mit 111In-DTPA-Farbstoffkonjugates bei Patientenmit Glioblastomen im SPECT (Single-Photon-Emmissions-Tomogramm) untersucht.Bei drei Patienten, welche nach MR-Kriterien an einem Glioblastom erkrankt waren,wurden 70 MBq 111In-DTPA-ALFc-HSA intravenös appliziert. Über einen Beobach-tungszeitraum von drei Tagen (die Halbwertszeit von 111In beträgt 2,7 Tage) wurdenwiederholt Untersuchungen im SPECT wie auch Blutproben zur Bestimmung der Plas-mahalbwertszeit vorgenommen (Abb. 12).Dabei zeigte sich ein Maximum an Aufnahme von 111In-DTPA-ALFc-HSA nach 48 h.Ebenfalls waren zu diesem Zeitpunkt die Tumoren im SPECT sehr deutlich zu erken-nen. Die Bestimmung der Plasmahalbwertszeit erfolgte durch Proben aus 5 ml Vollblut.Hier stellte sich ein langsamer und kontinuierlicher Abfall der Konzentration an 111In-DTPA-ALFc-HSA dar. Die Halbwertszeit des Proteinkonjugates wurde bei den dreiPatienten im Mittel mit 4,7 Tagen angegeben. Somit bestätigte sich die hohe und lang-anhaltende Aufnahme des Farbstoffkonjugates AFLc-HSA bei Patienten mit Gliobla-stomen. Allerdings waren die Untersuchungen wegen der kurzen Halbwertszeit von111In in ihrer Dauer und Aussage nur eingeschränkt verwertbar.
Abb. 12: Darstellung eines links temporalen Glioblastoms 48 h nach intravenöser Injektion von70 MBq 111In-DTPA-AFLc-HSA im SPECT
3 6
Abb. 13: Aufnahme von 111In-DTPA-AFLc-HSA in ein Glioblastom (A) und Darstellung derPlasmaclearance (B) von 111In-DTPA-AFLc-HSA nach intravenöser Injektion von 70 Mbq 111In-DTPA-AFLc-HSA.
0
0,2
0,4
0,6
0,8
1
1,2
1,4
0 1 8 2 4 4 2 4 8 7 2h
111In-DTPA-AFLc-SA
100000
1000000
0 1 8 2 4 4 2 4 8 7 2h
cpm
111In-DTPA-AFLc-SA
A
B

3 7
2.12 Präklinische Studien zur laserinduzierten Fluoreszenzdiagnostik mit AFLc-HSA
Drei weitere Patienten, welche gemäß MRT-Untersuchungen an einem Glioblastomerkrankt waren, wurden nach Einwillligung für die laserinduzierte Fluoreszenzdiagno-stik mit AFLc-HSA zur intraoperativen Tumordarstellung vorgesehen. Die intravenöseApplikation des Farbstoffkonjugates erfolgte in einer Dosierung von 0,5 mg/kg Körper-gewicht in einem Zeitraum von 3 - 5 Tagen vor der geplanten Operation. Alle drei Pati-enten wurden unter Neuronavigation und einer Radikalitätskontrolle durch ein intraope-ratives MRT und frühes postoperatives MRT operiert. Die Aktivierung des Farbstoff-konjugates erfolgte durch einem dem OP benachbarten Argon-Laser bei einer Anre-gungswelle von 488 nm. Die Ausleuchtung des OP-Gebietes wurde mit Hilfe eines ste-rilisierbaren FD1-Applikators bei einer Leistung von 150 – 200 mWatt/cm2 gewährlei-stet. Die Fluoreszenzbetrachtung erfolgte dann über einen im Operationsmikroskop ein-gebrachten Schwenkfilter, welcher das Anregungslicht des Lasers bei einer Wellenlängevon 515 nm abschnitt und nur Fluoreszenzlicht über 515 nm passieren ließ (Abb. 14).Die Bestimmung der Plasmahalbwertszeit erfolgte aus Proben von 5 ml Vollblut übereinen Zeitraum von bis zu 20 Tagen nach der Operation.
Abb. 14: Integrierbarer Langwellen-Schwenkfilter für das Operationsmikroskop MKM, Zeiss.
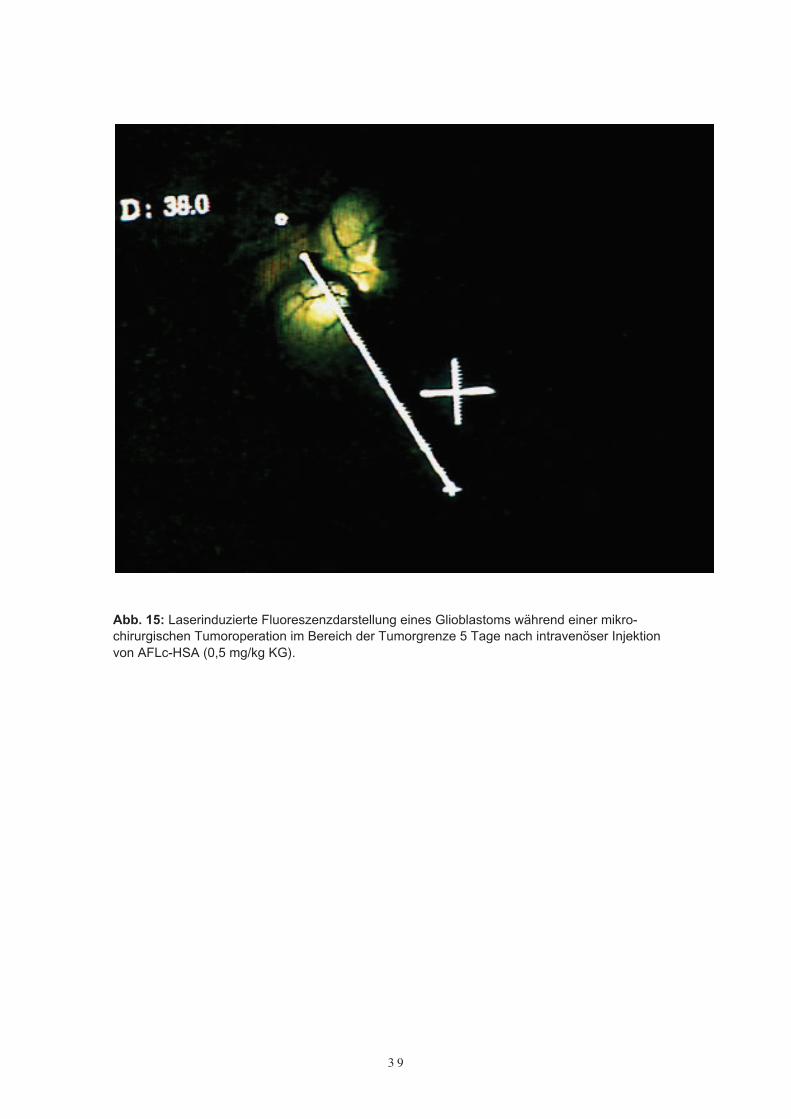
Bei allen drei Patienten gelang eine intraoperative Fluoreszenzdarstellung der Tumorenmit AFLc-HSA über die gesamte Dauer der Operation. Vor allem im Bereich derTumorgrenze, d. h. der proliferationsaktiven Zone, waren die Tumoren fluoreszenzop-tisch gut erkennbar. Gegenüber normalem Gehirngewebe zeigte sich eine scharfe Ab-

3 8
grenzung in der Fluoreszenzdarstellung. Dagegen war in den zentralen nekrotischenTumoranteilen keine Fluoreszenzdarstellung erkennbar. Ein Übertritt von AFLc-HSAaus eröffneten Gefäßen führte zu einer Kontamination der Resektionshöhle. Allerdingskonnte dies durch ein sorgfältiges Absaugen vor der Fluoreszenzbetrachtung verhindertwerden. Üblicherweise erfolgte die Fluoreszenzbetrachtung wiederholt während derTumorpräparation und abschließend nach Entfernung des Tumors. Nur in einem Fallmußte wegen einer beginnenden Infiltration des Tumors in das Wernicke-Sprachzen-trum ein kleiner solider durch AFLc-HSA wie auch durch das intraoperative MRT zuerkennender Tumorrest belassen werden. Die histopathologischen Untersuchungen wie-sen in allen Fällen fluoreszenzpositiver Proben ein Glioblastom oder seine Infiltrations-zone nach. Entsprechend den Kriterien der intraoperativen Radikalitätskontrolle durchein intraoperatives und frühes postoperatives Kernspintomogram konnte bei zwei Pati-enten eine radikale Tumorresektion unter Zuhilfenahme der intraoperativen Laserfluo-reszenz erreicht werden. Trotz mehrfacher Bescheinung und langanhaltender Tumoro-peration war ein Ausbleichen des Farbstoffes oder eine Penetration des Konjugates inumgebendes Gehirngewebe nicht beobachtet worden (Abb.15).Ein Patient entwickelte eine Woche nach der Applikation von AFLc-HSA ein generali-siertes allergisches Hautexanthem, welches mit Antihistaminika behandelt werdenmußte. Nach Abklingen des Hautexanthems konnte die allergische Reaktion im Intra-kutantest durch AFLc-HSA provoziert werden. Weitere Nebenwirkungen oder Labor-veränderungen wurden nicht beobachtet.Ebenfalls wurden in der Abteilung für Mund- Zahn- und Kieferchirurgie der UniversitätHeidelberg drei Patienten mit Plattenepithelkarzinomen der Mundhöhle mit AFLc-HSAbehandelt. Auch hier gelang durch AFLc-HSA eine Darstellung der Mundhöhlenkarzi-nome, wobei die Fluoreszenzdarstellung der Tumoren nach Abklingen der Hintergrund-fluoreszenz in der Schleimhaut ca. 1 – 2 Wochen nach der i. v. Applikation von AFLc-HSA deutlich verbessert werden konnte.
3 9
Abb. 15: Laserinduzierte Fluoreszenzdarstellung eines Glioblastoms während einer mikro-chirurgischen Tumoroperation im Bereich der Tumorgrenze 5 Tage nach intravenöser Injektionvon AFLc-HSA (0,5 mg/kg KG).

4 0
2.13 Diskussion
Die fluoreszenzoptische Darstellung von Gehirntumorgewebe findet sich erstmals 1948bei Moore (10). Zu dieser Zeit war die Lokalisation eines Gehirntumors nur auf Grundvon neurologischen, einige Jahre später durch ventrikulographische Untersuchungenvermutbar. In der Regel erfolgte eine ausgedehnte Trepanation, bei der Veränderungenam Cortex aufgesucht wurden, oder es erfolgten vorsichtige Punktionen in das Gehirn,sogenannte Dandy-Nadel, welche eine Lokalisation vor allem subkortikaler Läsionenvermuten ließ. Verständlich, daß man nach Wegen suchte, Gehirngeschwülste selbstsichtbar zu machen.
Grundlage des von Moore verwendeten Dinatriumfluoresceins ist die Tatsache, daßmaligne Gehirntumoren eine Störung der Blut-Hirn-Schranke aufweisen und somit Sub-stanzen, wie das Fluorescein, in den Tumor penetrieren lassen. Eine Fenestration vonBlutgefäßen im Tumor (sog. Blut-Tumor-Schranke) findet sich aber auch bei extraaxi-alen Gehirntumoren. Somit konnte Moore bei einer Vielzahl unterschiedlicher Gehirn-tumoren (Glioblastome, Astroblastome, Meningiome, Akustikusneurinome, Ependy-mome und Hypophysenadenome) eine ausreichende Fluoreszenzdarstellung währendder Operation beobachten. Verwendet wurde das Dinatriumfluorescein 20 % in 5 mlLösung, was einer Gesamtapplikation von 1 g entspricht. Der Farbstoff wurde unmittel-bar präoperativ intravenös injiziert und während der Operation mit einer Quecksilber-dampflampe angeregt. Die Fluoreszenzbetrachtung erfolgte mit einem entsprechendenLangwellenfilter (Wood‘s-Filter). Das Maximum an Fluoreszenz wurde 2 Stunden nachintravenöser Applikation erreicht, jedoch war die Fluoreszenzdarstellung nach 5 Stun-den deutlich vermindert. Wenn auch Moore eine sehr gute Fluoreszenzdarstellung vonTumorgewebe beobachtete, so wies er in seiner Publikation von 1948 bereits auf dasProblem der Penetration des Farbstoffes ins umgebende ödematös veränderte Gehirn-gewebe hin.
Auch andere fluoreszenzaktive Substanzen, wie z. B. Atebrin oder Trypaflavin (13),wurden zu dieser Zeit zur intraoperativen Fluoreszenzdiagnostik bei Gehirntumorenherangezogen. Auch mit diesen Substanzen gelang eine intraoperative Fluoreszenzdar-

4 1
stellung, aber auch hier wiesen die Autoren wiederum auf eine unscharfe Begrenzungder Fluoreszenz, vor allem bei den infiltrativ wachsenden maligen Gliomen, hin.Warum durch fluoreszierende Substanzen eine optische Anfärbung von Gehirntumorenmöglich war, war den Autoren Anfang der 50er Jahre nicht ganz klar. Zwar wurde eineFenestrierung von Tumorgefäßen vermutet, doch erst die ausgedehnten Untersuchungenvon Klatzo Anfang der 60er Jahre zeigten den Übertritt von nieder- und hochmolekula-rem Fluorescein als Ausdruck einer Störung der Blut-Hirn-Schranke (14). Dieser Über-tritt nieder- und hochmolekularer Substanzen in Gehirntumoren wurden im weiterenauch zur Isotopendarstellung von Gehirntumoren verwendet (15-16).
Erst 34 Jahre nach Moores Publikation wurde die intraoperative Fluoreszenzdarstellungmit Na-Fluorescein von Murray (17) wieder aufgegriffen, welcher sich in dieser Metho-de eine Verbesserung der operativen Radikalität bei malignen Gliomen erhoffte. Weiterverbessert wurde die Methode der intraoperativen Fluoreszenzdarstellung mit Na-Fluorescein Ende der 90er Jahre durch zwei japanische Arbeitgruppen (18-20), welcheeine Xenonlampe als Lichtquelle zur Anregung des Farbstoffes in ein Operationsmikro-skop integrierten.
Zwischenzeitlich sind auch andere Farbstoffe zur optischen Darstellung von Gehirntu-moren untersucht worden. Zu erwähnen ist der nicht fluoreszierende Farbstoff Indozya-nin, welcher ebenfalls eine gestörte Blut-Hirn-Schranke passiert und sich im Tumorge-webe anreichert. Nach tierexperimentellen Untersuchungen am C6-Gliom der Ratte (21)wurde der Farbstoff auch am Menschen untersucht. Allerdings mußte die ursprünglicham Tiermodell verwendete Dosis von 60 – 120 mg/kg wegen zu erwartender Nebenwir-kungen deutlich auf 2 mg/kg reduziert werden, weswegen zur intraoperativen Farbstof-ferkennung eine Restlichtverstärkerkamera verwendet werden mußte (22-23). Ein wei-terer Farbstoff ist das Phthalozyanin. Auch Phthalozyanin erlaubt eine Darstellung beimalignen Gehirntumoren. Seine Verwendung wurde aber bislang nur tierexperimentellam C6-Gliom der Ratte untersucht (24).
Von größerer Bedeutung in der optischen Darstellung von Gehirntumoren ist die Ver-wendung photoaktiver Substanzen, welche den Porphyrinen zuzuordnen sind (25-27).

4 2
Diese besitzen neben fluoreszierenden vor allem photoaktive Eigenschaften, wie sie beider Photodynamischen Therapie (PDT) erwünscht sind. Hier bilden sich nach Anregungdes Farbstoffes toxische Metabolite (Phase-I-Reaktion) oder Sauerstoffradikale (Phase-II-Reaktion), die das umgebende Tumorgewebe zerstören. Die am häufigsten bei malig-nen Gehirntumoren verwendete Substanz ist das lipophile Porphyringemisch Hämato-porphyrinderivat (HPD, Photofrin), welches sich im Tumorgewebe anreichert und dannnach Anregung durch eine Lichtquelle aktiviert werden kann. Da die photoaktiven Ei-genschaften die fluoreszierenden Eigenschaften überwiegen, ist seine Anwendung imwesentlichen auf die photodynamische Therapie konzentriert. Derzeit wird diese Sub-stanz in einer prospektiven, randomisierten Phase-III-Studie bei Patienten mit Gliobla-stomrezidiven untersucht, wobei die Bestrahlung nach Abschluß der Resektion nochvorhandenes Resttumorgewebe zerstören soll (28).
Eine weitere zur Zeit vielfach untersuchte Substanz ist die 5-Aminolävulinsäure (5-ALA). Diese Substanz wird intrazellulär im Tumorgewebe zur eigentlichen Wirksub-stanz Protoporphyrin IX metabolisiert, welche fluoreszenzaktiv ist. Ursprünglich wurde5-ALA zur Diagnostik von Blasenkarzinomen eingesetzt (29), jedoch findet diese Sub-stanz zwischenzeitlich bei oberflächlich liegenden Tumoren eine immer breitere An-wendung. Es ist der Münchener Arbeitsgruppe zu verdanken, daß 5-ALA zur intraope-rativen Fluoreszenzdiagnostik bei malignen Gliomen eingesetzt wurde. Auch hier wirdzur Anregung des Fluoreszenzfarbstoffes eine Xenonlampe verwendet, welche im OP-Mikroskop integriert ist. Somit ist eine direkte Betrachtung der Fluoreszenzemissiondurch das Operationsmikroskop möglich. Nach tierexperimenteller Testung befindetsich 5-ALA zwischenzeitlich in einer multizentrischen Phase-III-Studie. Die bisherigenErgebnisse bestätigen eine Verbesserung der durch 5-ALA bedingten Operationsradika-lität mit Verbesserung der Überlebenszeit bei Patienten mit Glioblastomen (30-34). Wiealle Porphyrinderivate besitzt Protoporphyrin IX die Eigenschaft des raschen Ausblei-chens, weswegen im OP-Mikroskoplicht gezielt eine Wellenlänge ausgefiltert werdenmuß. Seine phototoxischen Eigenschaften sind gegenüber anderen Porphyrinderivatendeutlich geringer ausgeprägt.

4 3
Bei der Frage der selektiven Darstellung von Gehirntumoren während einer mikrochir-urgischen Tumoroperation sollten unseren Überlegungen nach folgende Eigenschaftenvon dem zu verwendeten Farbstoff erfüllt werden:
• Gute Kontrastierung des Tumorgewebes• Selektive und langanhaltende Aufnahme des Fluoreszenzfarbstoffes im Tumor• Fehlende Toxizität• Stabile Fluoreszenzintensität• Geringe Penetration ins umgebende Gewebe.
Aus den genannten Anforderungen entschlossen wir uns zu dem bekannten Fluores-zenzfarbstoff Fluorescein. Dieser Fluoreszenzfarbstoff zeichnet sich durch eine hoheLichtquantenausbeute aus und ist auch nach langanhaltender Anregung weitgehendfarbstoffstabil. Seine fehlenden photoaktiven Eigenschaften bedingen eine geringe lo-kale und systemische Toxizität. Durch die kovalente Koppelung von Fluorescein anhumanes Serumalbumin (AFLc-HSA) konnte die sonst durch die niedermolekularenEigenschaften bedingte Pharmakokinetik des Farbstoffes gänzlich im Sinne eines intra-zellulären und lange im Tumorgewebe verbleibenden Kontrastmittels verändert werden.In unseren tierexperimentellen Untersuchungen, den Untersuchungen in Zellkultur undder konfokalen Mikroskopie, wie auch in den individuellen Heilversuchen bei Glio-blatompatienten zeigte AFLc-HSA eine selektive und langanhaltende Fluoreszenzanfär-bung im malignen Tumorgewebe (35). Mit hoher Wahrscheinlichkeit verbleibt dieserFarbstoff auch nach lysosomaler Abspaltung vom Albumin sehr lange in der Tumor-zelle, so daß ein Auswaschen der Fluoreszenz verhindert wird. Sein klinischer Einsatzin einer dafür notwendigen Phase-I/II-Studie ist, nachdem die Voraussetzungen bewäl-tigt werden konnten, inzwischen von der hiesigen Ethikkommission genehmigt worden.Wie gut die Tumordarstellung mit AFLc-HSA ist und welche Information sie für denoperativ tätigen Neurochirurgen bringt, soll im direkten Vergleich der Tumordarstellungdurch Systeme der Neuronavigation, der Tumordarstellung im intra- und frühen post-operativen MR und dem intraoperativen Ultraschall untersucht werden.

4 4
Kapitel 3
3.1 Albuminvermittelte Chemotherapie mit Aminopterin-HSA(AMPT-HSA) in vivo
3.1.1 Einführung
Bereits 1940 fand das Antifolat Aminopterin (AMPT) seine erste klinische Anwendungbei Kindern, welche an Leukämie erkrankt waren (1-2). Es zeigte einen guten zytostati-schen Effekt und leitete die Entwicklung der modernen Chemotherapie ein. WenigeJahre später wurde AMPT durch ein ähnliches Antifolat, Methotrexat (MTX), ersetzt,nachdem tierexperimentelle Untersuchungen eine ähnliche Wirksamkeit, aber bessereVerträglichkeit gegenüber AMPT bestätigten (3). MTX, meist in Kombination mit an-deren zytostatischen Substanzen, findet heutzutage eine breite klinische Anwendung inder Behandlung akuter Leukämien im Kindesalter, bei Osteosarkomen, Choriokar-zinomen, Lymphomen, Kopf-Hals-Tumoren und anderen soliden Tumoren. Aber auchchronisch entzündliche Prozesse, wie z. B. die der rheumatoiden Arthritis oder der Pso-riasis, stellen eine Indikation für die Verwendung von MTX dar. Vergleicht man aberdie Wirksamkeit von AMPT gegenüber MTX, so findet sich für AMPT eine 20fachhöherer Vmax/Km Quotient für das Enzym Folylpolyglutamatsynthetase (4) und einestärkere Aufnahme und Polyglutamierung in Lymphoblasten und Myeloblasten (5). Soist es nicht verwunderlich, daß AMPT wieder Einzug in eine klinische Studie fand. Rat-liff (6) untersuchte AMPT bei 20 therapieresistenten Malignompatienten und konnteeine komplette Remission bei einem Patienten mit Endometriumkarzinom und eine Sta-bilisierung des Krankheitsverlaufes bei 7 weiteren Patienten beobachten.
Nachdem die präklinischen und klinischen Ergebnisse mit dem Albuminkonjugat MTX-HSA eine gegenüber MTX zumindest gleichwertige, wenn nicht sogar leicht bessere,Wirkung zeigten, galt unser Interesse der Albuminkonjugierung eines anderen Antifo-lates, dem AMPT (Abb. 1). Es sollte, ähnlich wie MTX-HSA, in seiner Verteilung, sei-ner Plasmahalbwertszeit, seiner Aufnahme in den Tumor, wie auch in seiner Verträg-lichkeit und Effektivität untersucht werden.
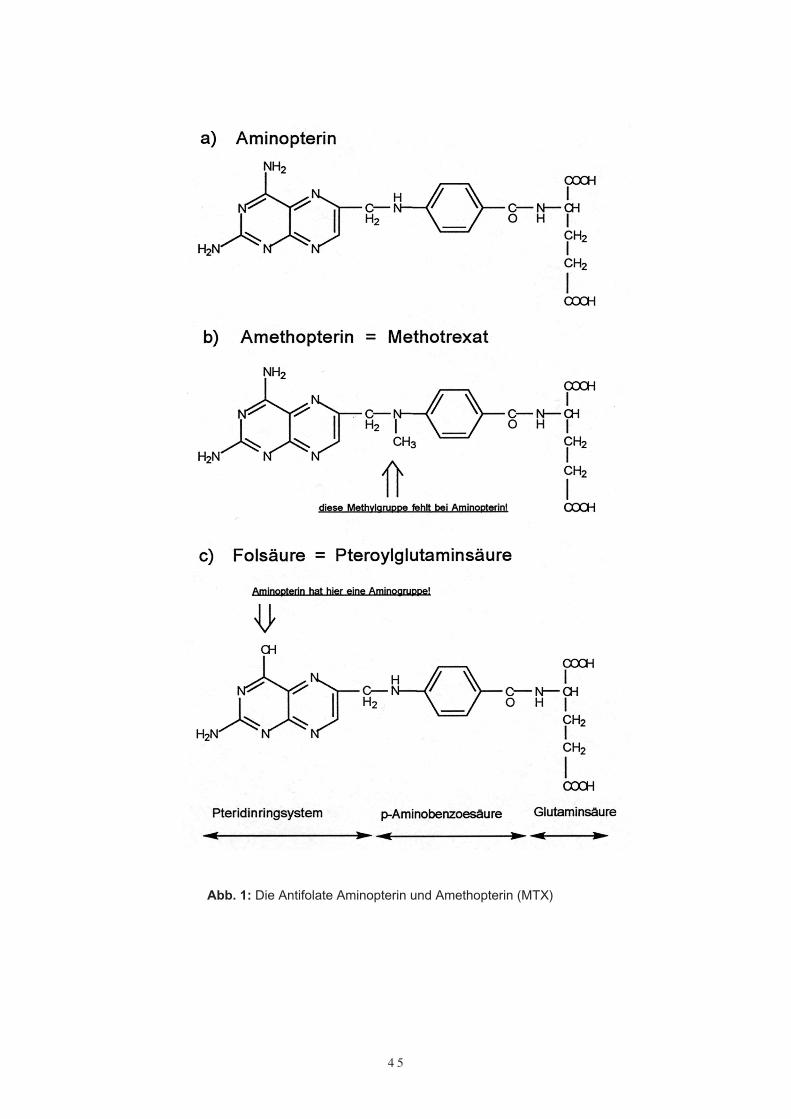
4 5
Abb. 1: Die Antifolate Aminopterin und Amethopterin (MTX)

4 6
3.2.1 Die Koppelung von Aminopterin (AMPT) an Albumin (HSA)
Aminopterin Aktivierung
Aminopterin-Hydrat (AMPT; Molekulargewicht 440.42 Dalton; Aldrich, Steinheim)wird in einer Konzentration von 10 mg/ml in Dimethylsulfoxid (DMSO) gelöst. Zu derklaren gelben Lösung wird eine 1,5fach molare Menge von Dicyclohexyl-carbodiimid(DCC) und eine 10fach molare Menge an Hydroxysuccinimid (HSI) hinzugefügt. Nach14 – 15 Stunden ist die Reaktion bei Raumtemperatur zum Succinimidylester (AMPT-HSIE) weitgehend abgeschlossen, was durch die auskristallierte Menge an Dicyclohe-xyl-Harnstoff (DCHU) zu erkennen ist.
Kovalente Koppelung des aktivierten Aminopterin an humanes Serumalbumin
Zu einer Lösung von humanem Serumalbumin (HSA, 66.439 Da, Pharma DessauGmbH, Dessau, 50 mg HSA/ml 0.17 M NaHCO3, pH 8.5) wird die klare gelbe AMPT-HSI Lösung in DMSO wird unter langsamem Rühren in einem 1,5 : 1 molaren Verhält-nis hinzugefügt. Nicht reagiertes DCC und noch in DMSO gelöstes DCHU trübt 15Minuten später die ursprünglich klare Lösung durch Bildung kolloidaler Aggregate.Nach weiteren 30 Minuten Reaktionszeit werden die kolloidalen Aggregate mit einemsterilen Filter (0.22 µm; Millipore, Molsheim) abgetrennt, während hingegen DMSOand nicht kovalent gebundenes AMPT durch Ultrafiltration über eine Druckrührzellemit passendem Membranfilter (YM 30, Millipore) abgetrennt werden. Die Reinheits-kontrolle erfolgt dann durch die hochauflösende Flüssigkeitschromatographie (HPLC):
HPLC Bedingungen:Vorsäule: Phenomenex GFC - 4000, 4 mm L x 3 mm IDSäule: Zorbax GF 250,1. Laufmittel: 0.25 M Li-acetat, 0,05M Li-citrat pH 7,4 (Ampuwa)2. Laufmittel: 95% Methanol (HPLC-grade), 5% Wasser (Ampuwa)Fluß: 0 -15 min Laufmittel 1 (100%); 1.0 ml/minGradient: 15 - 30 min Laufmittel 2 (von 0 auf 100%); 1.3 ml/min
4 7
30 - 45 min Laufmittel 2 (100%)45 - 50 min Laufmittel 2 (von 100 auf 0%)50 - 60 min Laufmittel 1 (100%); 1.0 ml/min
Detection: 370 nmDruck: ungefähr 65 Bar
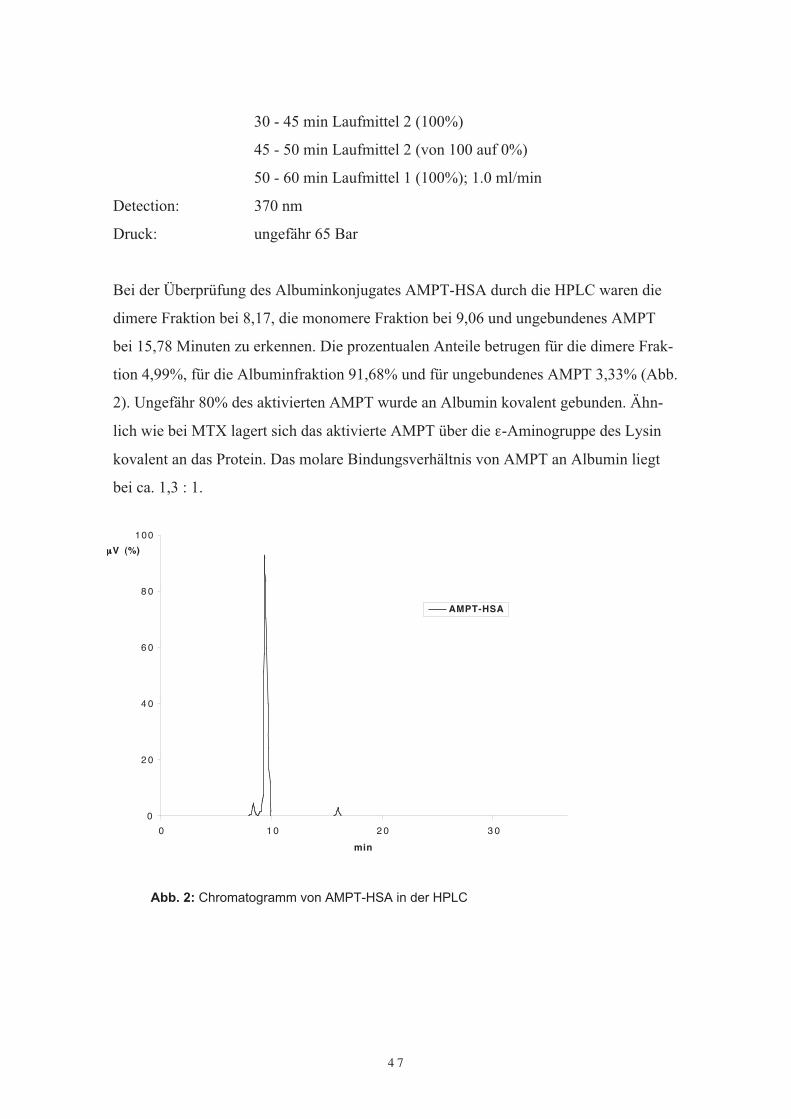
Bei der Überprüfung des Albuminkonjugates AMPT-HSA durch die HPLC waren diedimere Fraktion bei 8,17, die monomere Fraktion bei 9,06 und ungebundenes AMPTbei 15,78 Minuten zu erkennen. Die prozentualen Anteile betrugen für die dimere Frak-tion 4,99%, für die Albuminfraktion 91,68% und für ungebundenes AMPT 3,33% (Abb.2). Ungefähr 80% des aktivierten AMPT wurde an Albumin kovalent gebunden. Ähn-lich wie bei MTX lagert sich das aktivierte AMPT über die ε-Aminogruppe des Lysinkovalent an das Protein. Das molare Bindungsverhältnis von AMPT an Albumin liegtbei ca. 1,3 : 1.
Abb. 2: Chromatogramm von AMPT-HSA in der HPLC
0
2 0
4 0
6 0
8 0
100
0 1 0 2 0 3 0min
µµµµV (%)
AMPT-HSA

4 8
3.1.3 Die Markierung von Aminopterin-Albumin (AMPT-HSA) mit 111Indium
Man löst Diethylentriaminpentaessigsäure (DTPA) mit einer Konzentration von 20mg/ml unter Erhitzung in DMSO. Nach dem Abkühlen dieser Lösung auf Zimmertem-peratur fügt man die 1,2fache molare Menge an Dicyclohexylcarbodiimid (DCC) unddie 10fache molare Menge an Hydroxysuccinimid (HSI) hinzu. Nach einer Reaktions-zeit von etwa 24 Stunden ist die Aktivierung zum Succinimidylester weitgehend abge-schlossen und an der auskristallisierten Menge von Dicyclohexylharnstoff (DCH) er-kennbar. Die klare DMSO-Lösung wird nun im 3fach molaren Überschuß zum Proteinunter ständigem Rühren sehr langsam (10 mg AMPT-HSA/ml in 0,17 M NaCO3, pH7,9 - 8,0) zugegeben, wobei nach einiger Zeit eine weiße Trübung von nicht umgesetz-tem DCC und noch in DMSO gelöstem DCH auftritt. Nach einer Reaktionszeit von et-wa 30 Minuten wird die Reaktionslösung mit 0,17 M NaHCO3 verdünnt, die weißeTrübung über einen Sterilfilter (0,22 µm, Millipore) und das DMSO und nicht gebunde-nes DTPA durch Ultrafiltration über eine C 30 Mikrokonzentratoreinheit (Millipore)abgetrennt.Die Markierung des mit DTPA geladenem AMPT-HSA mit 111In erfolgt durch Zugabevon der gewünschten Radioaktivitätsmenge in Form des 111In-Citratkomplexes (herge-stellt aus 111InCl3 und 10 µl einer 0,2 M Na-Citratlösung, pH 7,5) zu 10 mg Protein.Schon unmittelbar nach Zugabe des 111In-Citrats kann das nicht an AMPT-HSA gebun-dene 111In durch Ultrafiltration über eine C30 Einheit abgetrennt werden. Die Markie-rungsausbeute beträgt über 98 % und entpricht einem „residualizing label“ (7, 8, 9).
3.1.4 Tumormodell und Versuchstiere
Walker-256 Karzinom (W-256)
Wegen seiner Eigenschaft als rasch proliferierender und antifolat-sensitiver Tumorwurde für unsere Versuche das Walker-256 Karzinom (W-256) ausgewählt (10) . DieTumorzellen wurden aus der Tumorbank des Deutschen Krebsforschungszentrums Hei-delberg gewonnen, wo sie bei -196 °C aufbewahrt wurden. Nach Auftauen der W-256-Karzinomzellen wurden diese mit RPMI Medium, welches mit 10 % Hitze inaktivier-

4 9
tem fätalen Kälberserum (FCS) und 1 % L-Glutamin angereichert war, kultiviert. DasKulturmedium wurde alle 48 Stunden ausgetauscht. Die Endkonzentration der Zellkul-tur betrug 106 Zellen/ml in PBS. Zur intramuskulären Tumorzellimplantation wurden 5x 106 W-256 Zellen in ein Volumen von 200 µl RPMI appliziert.
Versuchstiere und Datenerhebung
Insgesamt wurden 139 weibliche, apathogene (SPF) Sprague-Dawley (SD) Ratten fürdie tierexperimentellen Untersuchungen herangezogen. Die Tiere wurden standardisiertin Mikroisolatoren gehalten. Nach intramuskulärer Tumorimplantation im rechten Hin-terbein wurden die Tumorradii täglich mit Meßschablonen bestimmt. Das Tumorvolu-men wurde aus der Formel (Länge x Breite2)/2 errechnet, wobei die Länge (a) die grö-ßere Seite und die Breite (b) die kürzere Seite darstellt (11). Die Datenerhebung erfolgtedurch eine Software, in der die Tumorvolumina gegenüber der Zeit aufgelistet wurden.Die Wachstumskurven der Tumoren bezüglich der maximalen Tumorinhibierung wur-den folgendermaßen analysiert: Behandlungsgruppe/Kontrollgruppe (treated/control,T/C), errechnet als medianes Tumorgewicht in der behandelten Rate/medianes Tumor-gewicht in der unbehandelten Ratte x 100. Die Tumorverdoppelungszeit nach intramus-kulärer Implantation lag bei 48 Stunden. Sie entspricht im wesentlichen den Angabender Literatur (12). Statistische Berechnungen bezüglich der Tumorinhibition, Tumor-heilung oder Tumorrezidivrate erfolgten im Fisher‘s Exakttest.
3.1.5 Plasmaclearence von 111In-DTPA-HSA und 111In-DTPA-AMPT-HSA
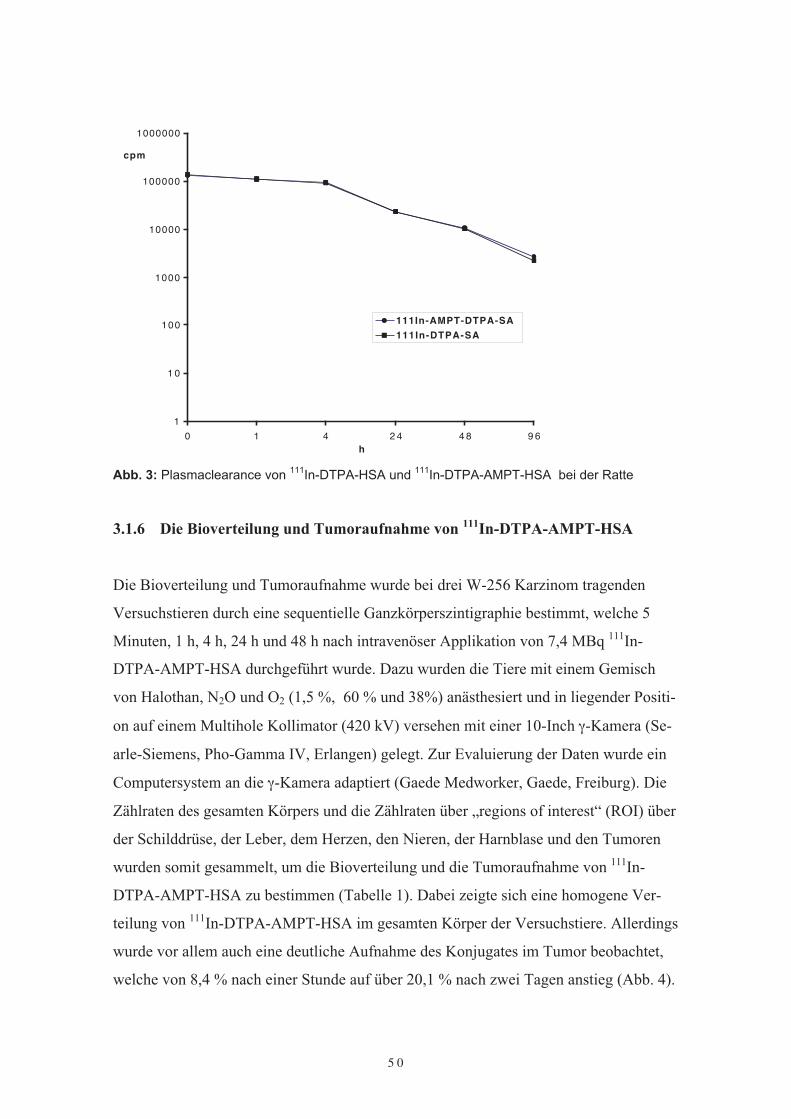
Die Plasmaclearance der Proteinkonjugates erfolgte nach Markierung mit radioaktivem111In-DTPA. Zwei Tieren wurde 111In-DTPA-HSA (3,7 MBq) intravenös appliziert,zwei weitere Tiere erhielten die gleiche Menge an 111In-DTPA-AMPT-HSA. Die Plas-mahalbwertszeit wurde aus Blutproben von 20 µl nach 5 Minuten, 1 Stunde, 4, 8, 24,48, 72 und 96 Stunden bestimmt. Die so aus der Schwanzvene der Versuchstiere ge-wonnenen Blutproben wurden im γ-Zähler (Berthold, Wildbad) untersucht. Dabei zeigtesich kein Unterschied in der Plasmahalbwertszeit von 111In-DTPA-AMPT-HSA und111In-DTPA-HSA (Abb. 3). Die errechnete Plasmahalbwertszeit lag bei 2,5 Tagen undentspricht der Halbwertszeit von nativem Albumin bei der Ratte.
5 0
Abb. 3: Plasmaclearance von 111In-DTPA-HSA und 111In-DTPA-AMPT-HSA bei der Ratte
3.1.6 Die Bioverteilung und Tumoraufnahme von 111In-DTPA-AMPT-HSA
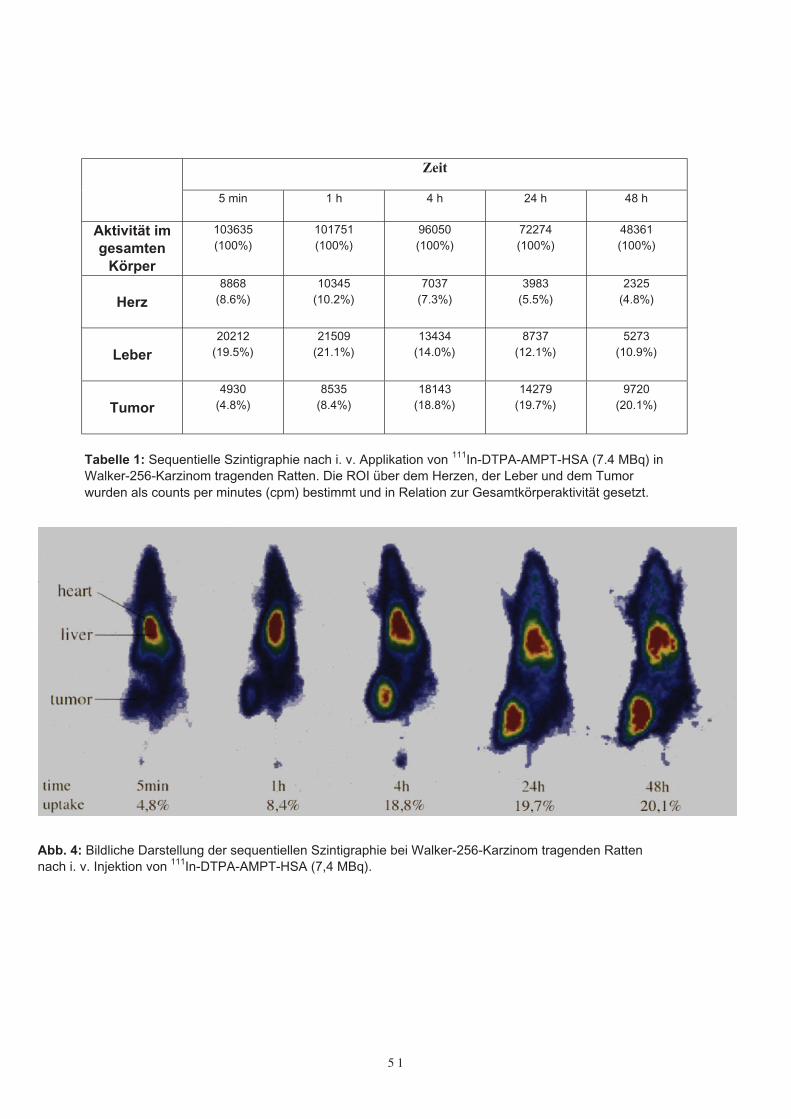
Die Bioverteilung und Tumoraufnahme wurde bei drei W-256 Karzinom tragendenVersuchstieren durch eine sequentielle Ganzkörperszintigraphie bestimmt, welche 5Minuten, 1 h, 4 h, 24 h und 48 h nach intravenöser Applikation von 7,4 MBq 111In-DTPA-AMPT-HSA durchgeführt wurde. Dazu wurden die Tiere mit einem Gemischvon Halothan, N2O und O2 (1,5 %, 60 % und 38%) anästhesiert und in liegender Positi-on auf einem Multihole Kollimator (420 kV) versehen mit einer 10-Inch γ-Kamera (Se-arle-Siemens, Pho-Gamma IV, Erlangen) gelegt. Zur Evaluierung der Daten wurde einComputersystem an die γ-Kamera adaptiert (Gaede Medworker, Gaede, Freiburg). DieZählraten des gesamten Körpers und die Zählraten über „regions of interest“ (ROI) überder Schilddrüse, der Leber, dem Herzen, den Nieren, der Harnblase und den Tumorenwurden somit gesammelt, um die Bioverteilung und die Tumoraufnahme von 111In-DTPA-AMPT-HSA zu bestimmen (Tabelle 1). Dabei zeigte sich eine homogene Ver-teilung von 111In-DTPA-AMPT-HSA im gesamten Körper der Versuchstiere. Allerdingswurde vor allem auch eine deutliche Aufnahme des Konjugates im Tumor beobachtet,welche von 8,4 % nach einer Stunde auf über 20,1 % nach zwei Tagen anstieg (Abb. 4).
1
1 0
100
1000
10000
100000
1000000
0 1 4 2 4 4 8 9 6h
cpm
111In-AMPT-DTPA-SA111In-DTPA-SA
5 1
Zeit5 min 1 h 4 h 24 h 48 h
Aktivität imgesamtenKörper
103635(100%)
101751(100%)
96050(100%)
72274(100%)
48361(100%)
Herz8868
(8.6%)10345
(10.2%)7037
(7.3%)3983
(5.5%)2325
(4.8%)
Leber20212
(19.5%)21509
(21.1%)13434
(14.0%)8737
(12.1%)5273
(10.9%)
Tumor4930
(4.8%)8535
(8.4%)18143
(18.8%)14279
(19.7%)9720
(20.1%)
Tabelle 1: Sequentielle Szintigraphie nach i. v. Applikation von 111In-DTPA-AMPT-HSA (7.4 MBq) inWalker-256-Karzinom tragenden Ratten. Die ROI über dem Herzen, der Leber und dem Tumorwurden als counts per minutes (cpm) bestimmt und in Relation zur Gesamtkörperaktivität gesetzt.
Abb. 4: Bildliche Darstellung der sequentiellen Szintigraphie bei Walker-256-Karzinom tragenden Rattennach i. v. Injektion von 111In-DTPA-AMPT-HSA (7,4 MBq).

5 2
3.1.7 Vergleichende Toxizität von AMPT-HSA versus AMPT
Daten über die maximal tolerable Dosis für wiederholte Injektionen von AMPT oderAMPT-HSA in SD-Ratten lagen nicht vor. Aus diesem Grunde entschlossen wir uns, inAnlehnung an vorbestehende tierexperimentelle Untersuchungen mit MTX-HSA beiSD-Ratten auf ein ähnliches Applikationsschema von drei bis vier intravenösen Appli-kationen zurückzugreifen. Aufgrund der bekannten erhöhten Toxizität von AMPT ver-glichen mit aequimolaren Dosen von MTX wurde zunächst eine reduzierte Dosis von0,5 mg AMPT/kg oder 0,5 mg/AMPT-HSA/kg gewählt. Die intravenösen Injektionenerfolgten an den Tagen 1, 3, 7 und dann, wenn möglich, drei Tage später am Tag 10, bistypische durch das Antifolat bedingte Nebenwirkungen, wie z. B. Gewichtsverlust, Mu-kositis, Diarrhoe oder struppiges Fell, beobachtet wurden. Während der Behandlungwurden das Körpergewicht, das Verhalten und die Gesundheit der Versuchstiere alsAusdruck einer möglichen Toxizität wie auch die Tumorvolumina täglich festgehalten.Die MTD für AMPT und AMPT-HSA wurde bei tumortragenden Tieren in einer Grup-pengröße von 10 Tieren/Dosis bestimmt. Eine Dosiseskalation auf 1,0 mg/kg Körper-gewicht entsprechend des oben genannten Applikationsschemas wurde nur dann in Er-wägung gezogen, wenn keine schwerwiegenden Nebenwirkungen beobachtet wurden.Die MTD bei tumortragenden Ratten, welche wiederholte intravenöse Applikationender Prüfsubstanzen erhielten, wurde definiert als die Dosis, bei welcher 20 % der Ver-suchstiere starben (LD 20) und/oder ein Gewichtsverlust von 20 % auftrat.Insgesamt wurde die Verträglichkeit von AMPT-HSA und ungebundenem AMPT bei60 Versuchstieren (10 Tiere/Substanzgruppe und Dosis) untersucht. Im ersten Ver-suchsansatz erhielten die Tiere wiederholte intravenöse Applikationen in einer Dosie-rung von 0,5 mg AMPT oder 0,5 mg AMPT-HSA. Bereits nach der dritten intravenösenInjektion von 0,5 mg AMPT zeigten 2/3 der Tiere typische durch das Antifolat indu-zierte Nebenwirkungen, wie Mukositis, Diarroe und struppiges Fell, während hingegendie Tiere, die drei wiederholte intravenöse Applikationen von 0,5 mg AMPT-HSA er-hielten, keinerlei Nebenwirkungen aufwiesen. Die Fortführung der Behandlung mit ei-ner vierten intravenösen Injektion von 0,5 mg AMPT ging mit einer deutlichen Steige-rung der Toxizität einher. Alle Tiere erkrankten und zeigten einen deutlichen Gewichts-verlust, so daß drei von ihnen starben. Im Gegensatz dazu wiesen alle Tiere, die vier

5 3
intravenöse Injektionen von 0,5 mg AMPT-HSA erhielten, keine schwerwiegenden Ne-benwirkungen auf. Dieser Unterschied in der Verträglichkeit ist statistisch hochsignifi-kant (p = 0,0006, Fischer’s Exact). Über einen Zeitraum von vier Tagen erholten sichdie überlebenden Tiere vollständig. Aufgrund der gewonnenen Ergebnisse bezüglich derVerträglichkeit von AMPT konnte hierfür die MTD für drei wiederholte intravenöseInjektionen in einer Dosierung von 0,5 mg AMPT/kg Körpergewicht appliziert an denTagen 1, 3 und 7 festgelegt werden. Wegen der fehlenden Toxizität in der AMPT-HSAGruppe wurde eine Dosiseskalation zu wiederholten intravenösen Injektionen auf 1,0mg AMPT-HSA/kg Körpergewicht vorgenommen. Ähnlich den Injektionen bei 0,5 mgAMPT fand sich nun nach dreifacher Applikation von 1,0 mg AMPT-HSA auch eineNebenwirkungsrate bei 70 % der Versuchstiere. Eine Steigerung der Toxizität mit einerLetalität bei vier Versuchstieren wurde nach der vierten intravenösen Applikation beob-achtet, so daß die MTD für AMPT-HSA bei drei wiederholten intravenösen Applikatio-nen in einer Dosierung von 1,0 mg/kg Körpergewicht, gegeben an den Tagen 1, 3 und 7,festgelegt wurde. Dies entspricht einer Verdoppelung der Verträglichkeit gegenüberungebundenem AMPT (Tabelle 2).
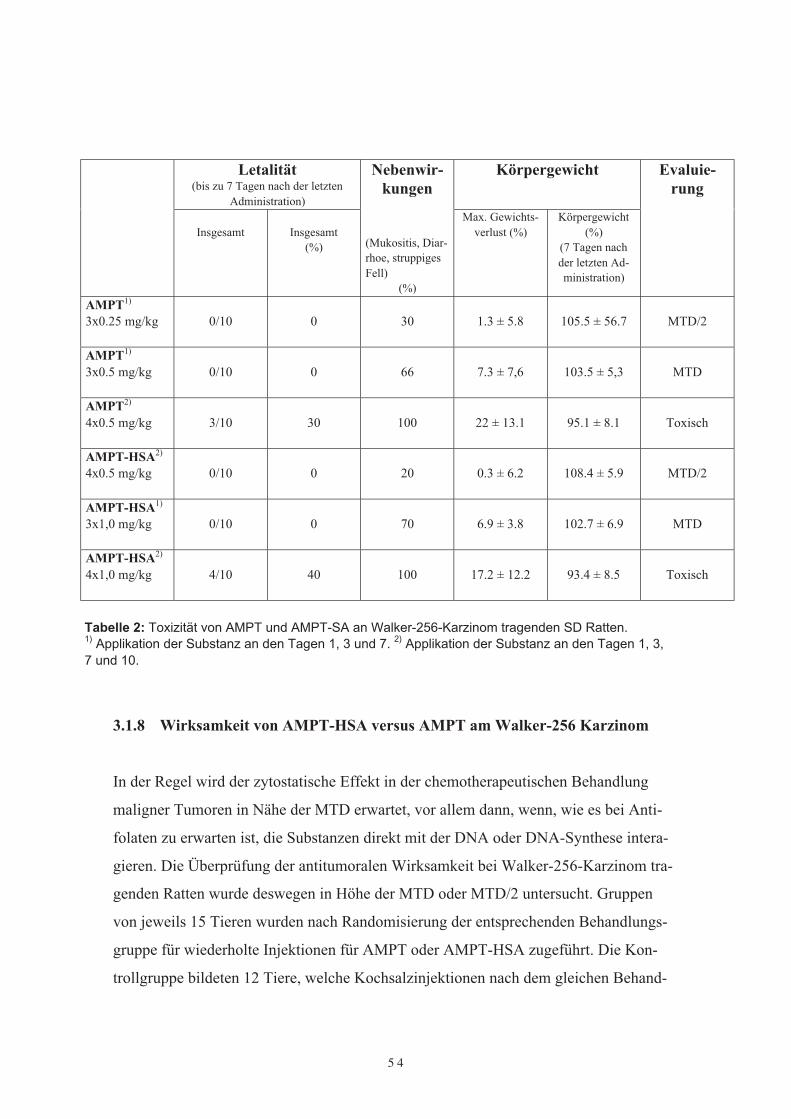
5 4
Letalität(bis zu 7 Tagen nach der letzten
Administration)Körpergewicht
Insgesamt Insgesamt(%)
Nebenwir-kungen
(Mukositis, Diar-rhoe, struppigesFell)
(%)
Max. Gewichts-verlust (%)
Körpergewicht(%)
(7 Tagen nachder letzten Ad-ministration)
Evaluie-rung
AMPT1)3x0.25 mg/kg 0/10 0 30 1.3 ± 5.8 105.5 ± 56.7 MTD/2AMPT1)3x0.5 mg/kg 0/10 0 66 7.3 ± 7,6 103.5 ± 5,3 MTDAMPT2)4x0.5 mg/kg 3/10 30 100 22 ± 13.1 95.1 ± 8.1 ToxischAMPT-HSA2)4x0.5 mg/kg 0/10 0 20 0.3 ± 6.2 108.4 ± 5.9 MTD/2AMPT-HSA1)3x1,0 mg/kg 0/10 0 70 6.9 ± 3.8 102.7 ± 6.9 MTDAMPT-HSA2)4x1,0 mg/kg 4/10 40 100 17.2 ± 12.2 93.4 ± 8.5 Toxisch
Tabelle 2: Toxizität von AMPT und AMPT-SA an Walker-256-Karzinom tragenden SD Ratten.1) Applikation der Substanz an den Tagen 1, 3 und 7. 2) Applikation der Substanz an den Tagen 1, 3,7 und 10.
3.1.8 Wirksamkeit von AMPT-HSA versus AMPT am Walker-256 Karzinom
In der Regel wird der zytostatische Effekt in der chemotherapeutischen Behandlungmaligner Tumoren in Nähe der MTD erwartet, vor allem dann, wenn, wie es bei Anti-folaten zu erwarten ist, die Substanzen direkt mit der DNA oder DNA-Synthese intera-gieren. Die Überprüfung der antitumoralen Wirksamkeit bei Walker-256-Karzinom tra-genden Ratten wurde deswegen in Höhe der MTD oder MTD/2 untersucht. Gruppenvon jeweils 15 Tieren wurden nach Randomisierung der entsprechenden Behandlungs-gruppe für wiederholte Injektionen für AMPT oder AMPT-HSA zugeführt. Die Kon-trollgruppe bildeten 12 Tiere, welche Kochsalzinjektionen nach dem gleichen Behand-

5 5
lungsschema erhielten. Die Behandlung wurde initiiert, nachdem die Tumoren ein Vo-lumen von ≥ 1.500 mm3 erreichten, was normalerweise 3 - 4 Tage nach der muskulärenTumorimplantation geschah. Die Körpergewichte, Nebenwirkungen und Tumorvolu-mina wurden täglich kontrolliert. Die Studien wurden dann beendet, wenn die Tumoren:a) Eine kritische Größe von mehr als 3 cm im Durchmesser erreichtenb) Eine lang anhaltende Tumorremission bis zum Wiederauftreten des Tumors beob-achtet wurdec) Bis zu vier Wochen nach der letzten Applikation.Wiederholte intravenöse Injektionen an den Tagen 1, 3 und 7 wurden bei insgesamt 60Walker-256-Karzinom tragenden Ratten in der Höhe der MTD oder MTD/2 durchge-führt. Unter diesem Behandlungsschema wurden keine schwerwiegenden Nebenwir-kungen beobachtet. Allerdings entwickelten die Tiere in der Kontrollgruppe innerhalbvon 10 Tagen nach der Tumorimplantation sehr rasch eine deutliche Tumorprogression,so daß 11 von 12 Kontrolltieren entsprechend der Empfehlung der Tierschutzkommissi-on getötet werden mußten.In Höhe der MTD und MTD/2 zeigte das Antifolatkonjugat AMPT-HSA eine sehr hoheWirksamkeit, welche mit langanhaltenden Tumorremissionen und einer sehr guten Ver-träglichkeit einherging (Tabelle 3). Dagegen zeigte ungebundenes AMPT lediglich inder MTD-Gruppe eine hohe antitumorale Wirksamkeit, während hingegen AMPT inHöhe der MTD/2 eine signifikant niedrigere Tumorremissionsrate (53.3 %) verglichenmit der MTD/2 bei AMPT-HSA aufwies. Dieser Unterschied in der Wirksamkeit inHöhe der MTD/2 beider Prüfsubstanzen zeigte sich als statistisch signifikant (p =0,0032). Ebenfalls war das Albuminkonjugat in der Tumorrezidivrate dem niedermole-kularen AMPT überlegen. So wurden für AMPT-HSA im aequimolaren Vergleich bei0,5 mg/kg KG nur 2 Tumorrezidive an den Tagen 10 und 24 gegenüber 4 Tumorrezidi-ven am Tag 14 für AMPT beobachtet. Vergleicht man die Tumorrezidivrate in Höhe derMTD/2 so findet sich wiederum ein statistisch signifikanter Vorteil für AMPT-HSA (p= 0,05). In Höhe der MTD zeigte AMPT-HSA wiederum eine 100%ige Heilrate 7 Tagenach der letzten Applikation. Hier wurde nur bei einem Tier ein Tumorrezidiv 22 Tagenach der letzten Applikation beobachtet (Abb. 5).
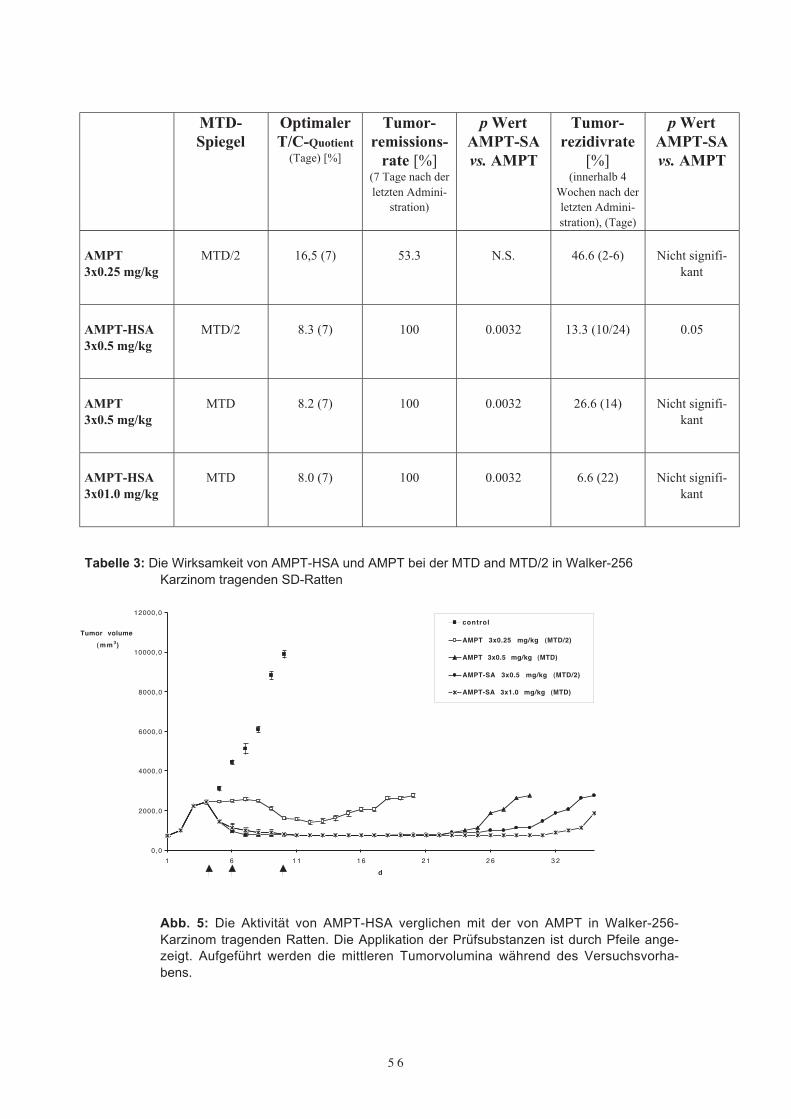
5 6
MTD-Spiegel
OptimalerT/C-Quotient
(Tage) [%]
Tumor-remissions-rate [%]
(7 Tage nach derletzten Admini-
stration)
p WertAMPT-SAvs. AMPT
Tumor-rezidivrate
[%](innerhalb 4
Wochen nach derletzten Admini-stration), (Tage)
p WertAMPT-SAvs. AMPT
AMPT3x0.25 mg/kg
MTD/2 16,5 (7) 53.3 N.S. 46.6 (2-6) Nicht signifi-kant
AMPT-HSA3x0.5 mg/kg
MTD/2 8.3 (7) 100 0.0032 13.3 (10/24) 0.05
AMPT3x0.5 mg/kg
MTD 8.2 (7) 100 0.0032 26.6 (14) Nicht signifi-kant
AMPT-HSA3x01.0 mg/kg
MTD 8.0 (7) 100 0.0032 6.6 (22) Nicht signifi-kant
Tabelle 3: Die Wirksamkeit von AMPT-HSA und AMPT bei der MTD and MTD/2 in Walker-256Karzinom tragenden SD-Ratten
Abb. 5: Die Aktivität von AMPT-HSA verglichen mit der von AMPT in Walker-256-Karzinom tragenden Ratten. Die Applikation der Prüfsubstanzen ist durch Pfeile ange-zeigt. Aufgeführt werden die mittleren Tumorvolumina während des Versuchsvorha-bens.
0,0
2000,0
4000,0
6000,0
8000,0
10000,0
12000,0
1 6 1 1 1 6 2 1 2 6 3 2d
Tumor volumecontrol
AMPT 3x0.25 mg/kg (MTD/2)
AMPT 3x0.5 mg/kg (MTD)
AMPT-SA 3x0.5 mg/kg (MTD/2)
AMPT-SA 3x1.0 mg/kg (MTD)
(mm 3)

5 7
3.2 Untersuchungen von AMPT-HSA in Zellkultur
3.2.1 Einführung
Neben den Anwendungen am Walker-256 Karzinom der Ratte sollte der Effekt desProteinkonjugates AMPT-HSA aber auch in Zellkultur untersucht werden. Erstmalshatte Stehle (13) einen Effekt von MTX-HSA in Zellkultur nachweisen können, dabeifiel allerdings ein höherer Konzentrationsbedarf gegenüber ungebundenem MTX auf.Diese Ergebnisse wurden auch durch andere Arbeitsgruppen bestätigt (14, 15). Gründehierfür liegen wohl an den Bedingungen in der Zellkultur selbst, in der in der Regel eingesättigtes Nährmedium vorliegt, so daß eine Albuminaufnahme in die Tumorzelle zuderen Energieabdeckung gar nicht notwendig ist.
3.2.2 Optimierung der Kulturbedingungen
Die Zusammensetzung herkömmlicher Zellkulturmedien ist so gewählt, daß Tumorzel-len darin optimal mit Nährsubstraten versorgt werden und somit bestmögliche Wachs-tumsbedingungen vorfinden. In der Regel setzen sich die Zellkulturmedien aus soge-nannten Basismedien (z. B. RPMI 1640) und einem Anteil von 5 - 10 % fetalem Käl-berserum (FCS) oder anderen Seren, wie z. B. Pferdeserum, zusammen, welche die fürdie Zellproliferation notwendigen Wachstumsfaktoren enthalten. Da es sich bei Zell-kulturen um ein geschlossenes System ohne Austausch, d. h. ohne Blutzirkulation, han-delt, enthalten die Basismedien im Vergleich zum menschlichen Blut einen bis zu20fachen Überschuß an freien Aminosäuren, Glucose, Hormonen und Vitaminen. Al-lerdings hat dies zur Folge, daß die kultivierten Tumorzellen zunächst auf einfach zuverstoffwechselnde, d. h. auf die niedermolekularen Substrate zugreifen, und wenigermakromolekulare Substanzen, wie z. B. Proteine, verstoffwechseln, die ja erst intrazel-lulär zu Aminosäuren degradiert werden müssten. Die Arbeitsgruppe von Rannels (16-17) beschrieb bereits in den 80er Jahren, daß durch Verminderung überhöhter Ami-nosäurenkonzentrationen in den Kulturmedien verschiedene Zelllinien gezwungen wer-den, vermehrt auf die im Medium vorhandenen Eiweiße als Nährstoffquelle zuzugrei-fen.
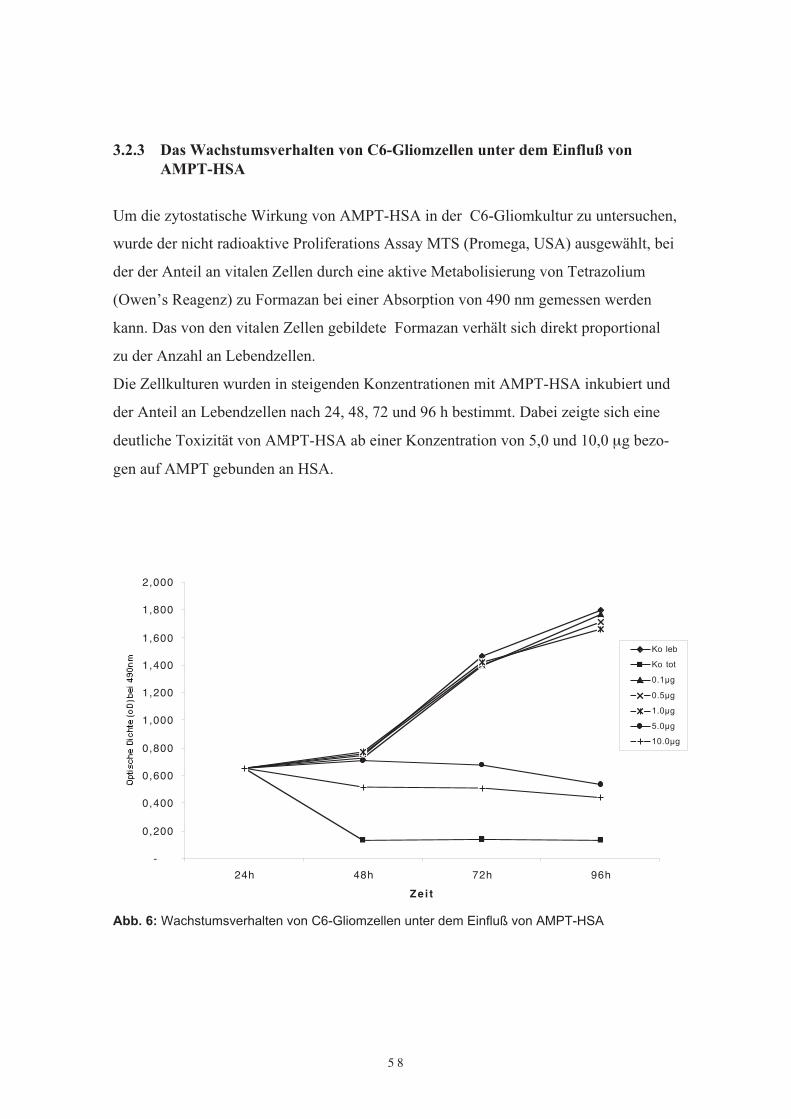
5 8
3.2.3 Das Wachstumsverhalten von C6-Gliomzellen unter dem Einfluß vonAMPT-HSA
Um die zytostatische Wirkung von AMPT-HSA in der C6-Gliomkultur zu untersuchen,wurde der nicht radioaktive Proliferations Assay MTS (Promega, USA) ausgewählt, beider der Anteil an vitalen Zellen durch eine aktive Metabolisierung von Tetrazolium(Owen’s Reagenz) zu Formazan bei einer Absorption von 490 nm gemessen werdenkann. Das von den vitalen Zellen gebildete Formazan verhält sich direkt proportionalzu der Anzahl an Lebendzellen.Die Zellkulturen wurden in steigenden Konzentrationen mit AMPT-HSA inkubiert undder Anteil an Lebendzellen nach 24, 48, 72 und 96 h bestimmt. Dabei zeigte sich einedeutliche Toxizität von AMPT-HSA ab einer Konzentration von 5,0 und 10,0 µg bezo-gen auf AMPT gebunden an HSA.
Abb. 6: Wachstumsverhalten von C6-Gliomzellen unter dem Einfluß von AMPT-HSA
-
0,200
0,400
0,600
0,800
1,000
1,200
1,400
1,600
1,800
2,000
24h 48h 72h 96hZei t
Ko lebKo tot0.1µg0.5µg1.0µg5.0µg10.0µg
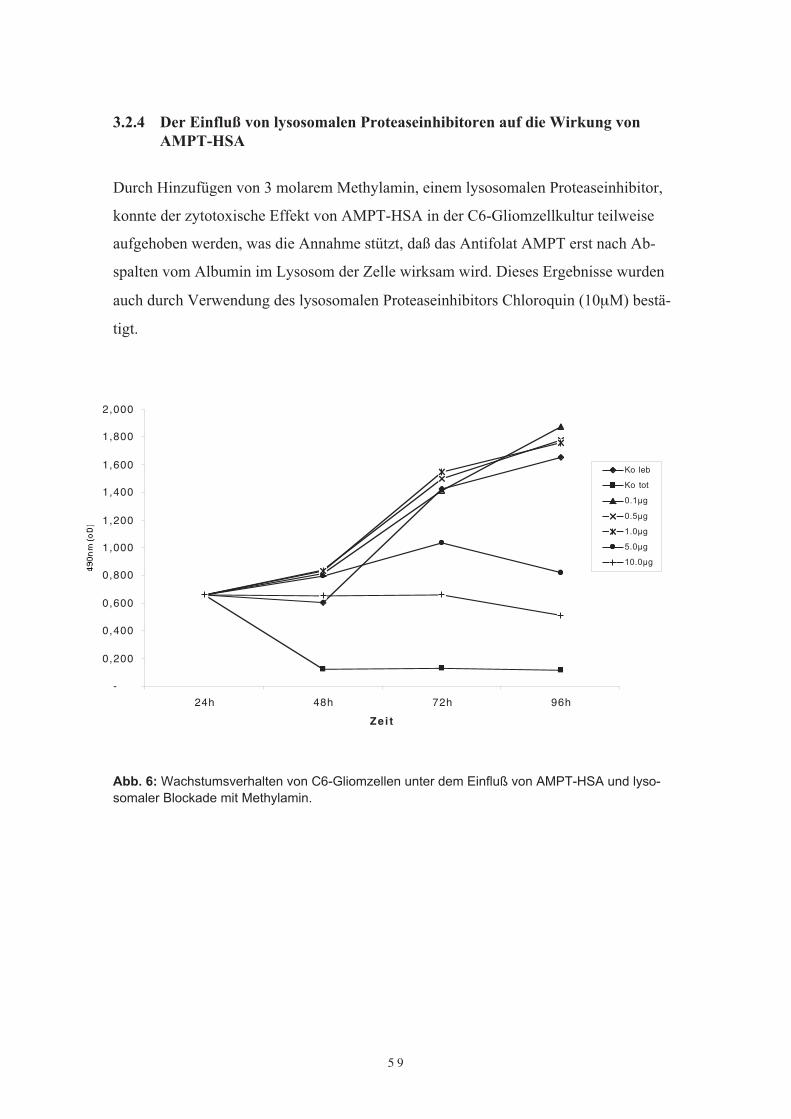
5 9
3.2.4 Der Einfluß von lysosomalen Proteaseinhibitoren auf die Wirkung vonAMPT-HSA
Durch Hinzufügen von 3 molarem Methylamin, einem lysosomalen Proteaseinhibitor,konnte der zytotoxische Effekt von AMPT-HSA in der C6-Gliomzellkultur teilweiseaufgehoben werden, was die Annahme stützt, daß das Antifolat AMPT erst nach Ab-spalten vom Albumin im Lysosom der Zelle wirksam wird. Dieses Ergebnisse wurdenauch durch Verwendung des lysosomalen Proteaseinhibitors Chloroquin (10µM) bestä-tigt.
Abb. 6: Wachstumsverhalten von C6-Gliomzellen unter dem Einfluß von AMPT-HSA und lyso-somaler Blockade mit Methylamin.
-
0,200
0,400
0,600
0,800
1,000
1,200
1,400
1,600
1,800
2,000
24h 48h 72h 96hZei t
Ko lebKo tot0.1µg0.5µg1.0µg5.0µg10.0µg
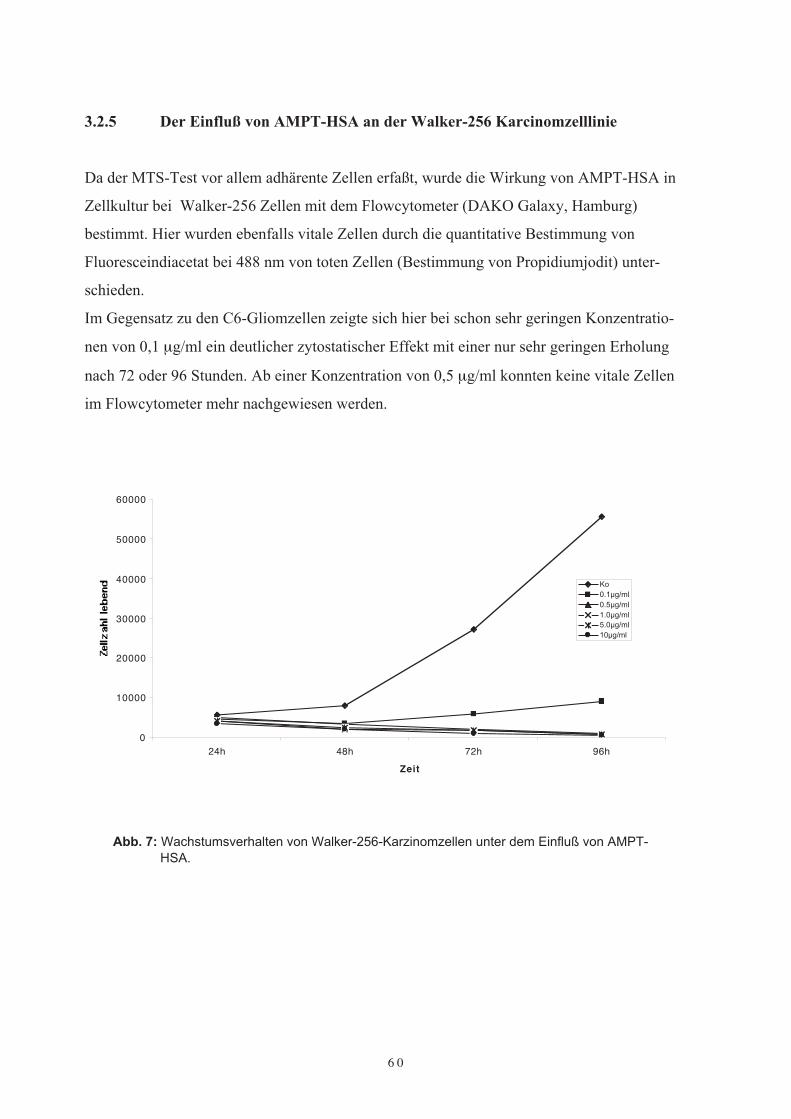
6 0
3.2.5 Der Einfluß von AMPT-HSA an der Walker-256 Karcinomzelllinie
Da der MTS-Test vor allem adhärente Zellen erfaßt, wurde die Wirkung von AMPT-HSA inZellkultur bei Walker-256 Zellen mit dem Flowcytometer (DAKO Galaxy, Hamburg)bestimmt. Hier wurden ebenfalls vitale Zellen durch die quantitative Bestimmung vonFluoresceindiacetat bei 488 nm von toten Zellen (Bestimmung von Propidiumjodit) unter-schieden.Im Gegensatz zu den C6-Gliomzellen zeigte sich hier bei schon sehr geringen Konzentratio-nen von 0,1 µg/ml ein deutlicher zytostatischer Effekt mit einer nur sehr geringen Erholungnach 72 oder 96 Stunden. Ab einer Konzentration von 0,5 µg/ml konnten keine vitale Zellenim Flowcytometer mehr nachgewiesen werden.
Abb. 7: Wachstumsverhalten von Walker-256-Karzinomzellen unter dem Einfluß von AMPT-HSA.
0
10000
20000
30000
40000
50000
60000
24h 48h 72h 96hZeit
Ko 0.1µg/ml0.5µg/ml1.0µg/ml5.0µg/ml10µg/ml

6 1
3.2.6 Diskussion
Mehrfach war in den letzten Jahren versucht worden, Albumin als Carrier für zytosta-tisch aktive Substanzen zu verwenden. Die Aktivitäten, zytostatische Albuminkonjugatezu entwickeln, konzentrierten sich im Wesentlichen auf Methotrexat (MTX), wobeiversucht wurde, den zu erhoffenden therapeutischen Effekt durch höhere Beladungsra-ten von bis zu 56 Mol MTX/ 1 Mol Albumin zu steigern. Dies hatte jedoch zur Folge,daß die räumliche Tertiärstruktur von Albumin und somit seine biologische Integritätverändert wurde, weswegen allergische Reaktionen gegenüber nativem Albumin, ver-kürzte Zirkulationszeiten und ein Abfangen des hoch beladenen Albumins im retikulo-endothelialen System (RES) beobachtet wurden (18 - 22). Kaum eine dieser Albumin-MTX-Konjugate erreichte das Stadium einer klinischen Studie. Wie zuvor in Kapitel 1berichtet, sollte Albumin aber nur in einem Verhältnis von nahezu 1 : 1 beladen werden,um seine physiologischen Eigenschaften nicht zu verändern. Dann aber bietet Albumindie Vorteile eines biologisch verfügbaren Carriersystemes:
• lange Zirkulationszeiten• niedrige Toxizität• hohe Aufnahme im Tumor.
Mittlerweile wurde MTX-HSA in einer Phase-I/II-Studie am Städtischen KlinikumMannheim untersucht, wobei neben der guten Verträglichkeit bei 3/17 Patinten langanhaltende Remissionen beobachtet wurden (23). MTX-HSA befindet sich zwischen-zeitlich in einer multizentrischen Phase-II-Studie (Fujisawa-Deutschland) bei Hyperne-phromen, Pleuramesotheliomen und Glioblastomen. Die viel versprechenden Ergebnis-se mit MTX-HSA wie auch die erfolgreiche und lang anhaltende Aufnahme von AFLc-HSA in malignen Gehirntumoren (24) ermutigte uns, die Albuminkoppelungstechnikfür ein weiteres Antifolat, wie dem des Aminopterins, einzusetzen. In den 40er Jahrenwar Aminopterin das erste Antifolat, welches in der Behandlung akuter Leukämien beiKindern eingesetzt wurde. Seine Remissionsrate war in der damaligen Zeit für die be-handelnden Ärzte sehr eindrucksvoll und läutete die Entwicklung der modernen Che-motherapie ein. Jedoch wurde Aminopterin in den 50er Jahren wegen seiner doch deut-

6 2
lichen systemischen Toxizität durch ein ähnliches Antifolat, dem MTX, ersetzt. Im Ge-gensatz zu MTX weist AMPT jedoch eine erhöhte therapeutische Wirksamkeit auf, dadurch AMPT ein 20fach höherer Vmax/Km Quotient für das Enzym FolylpolyglutamatSynthetase erreicht wird (4). Zudem zeigt sich für Lymphoblasten und Myeloblasteneine größere Aufnahmerate und Polyglutamatbildung (5). In jüngster Zeit wurde AMPTerneut in einer Phase-I-Studie bei 20 Patienten mit therapieresistenten Karzinomerkran-kungen eingesetzt und erreichte bei einer Patientin mit einem Endometriumkarzinomeine komplette Remission. Bei 7 Patienten wurde zwischen zwei und neun Monaten einStillstand der initial vorhandenen Tumorprogression beobachtet (6). Die initial appli-zierte Dosis von 2,5 mg/m2 alle 12 Stunden (2 Applikationen pro Woche) mußte vorallem wegen einer beobachteten Mukositis auf 2 mg/m2 reduziert werden. Zudem wareine Gabe von Leukoverin in einer Dosierung von 5 mg/ m2 notwendig.
In unserer Studie wurde AMPT in einem 1 : 1 molaren Verhältnis an Albumin gebun-den und wurde bezüglich seiner Plasmahalbwertszeit, Bioverteilung, Verträglichkeitund Wirksamkeit am Walker-256 Karzinom der Ratte untersucht (25). Wie erwartet,zeigte sich im Vergleich zu nativem Albumin eine unveränderte Plasmahalbwertszeitvon ungefähr 2,5 Tagen bei der Ratte. Auch war in der Ganzkörperszintigraphie einenormale Verteilung des Proteinkonjugates erkennbar. Jedoch wurde über dem Walker-256 Karcinom eine hohe und lang anhaltende Aufnahme von bis zu 20,1 % der nach 2Tagen im ganzen Körper verbliebenen Dosis beobachtet.
Bezüglich der Verträglichkeit fanden sich nach wiederholten Applikationen von 0,5 mgAMPT/kg KG typische durch das Antifolat induzierte Nebenwirkungen. Nach viermali-gen Applikationen von 0,5 mg AMPT/kg KG wurde eine schwere Toxizität (maximalerGewichtsverlust 17,2 %) und einer Letalität von 30 % beobachtet. Dagegen waren vierApplikationen von 0,5 mg AMPT-HSA/kg KG vollkommen nebenwirkungsfrei, so daßdas Konjugat höher dosiert werden konnte. Erst nach vier Applikationen von 1,0 mgAMPT-HSA/kg Körpergewicht erreichte das Konjugat eine Toxizität. Die MTD für dasniedermolekulare AMPT lag bei 3 x 0,5 mg/kg, für das Konjugat aber bei 3 x 1,0mg/kg KG. Somit wurde durch die Konjugierung des Antifolates eine Verdoppelung inder MTD erreicht. Diese Untersuchungen bestätigen eine Reduzierung der systemischen

6 3
Toxizität von AMPT durch die kovalente Koppelung an Albumin, obwohl das Konjugatverglichen mit dem ungebundenen, niedermolekularen AMPT weitaus länger in Zirku-lation bleibt. Mit hoher Wahrscheinlichkeit läßt sich vermuten, daß das KonjugatAMPT-HSA normal proliferierendes Gewebe nicht in der gleichen Art und Weise er-reicht oder belastet wie niedermolekular zirkulierendes AMPT.
Sowohl in der MTD und MTD/2 Dosierung war AMPT-HSA sehr wirksam und er-reichte lang anhaltende Tumorremissionen. Im Gegensatz dazu war AMPT in derMTD/2 Dosierung deutlich weniger wirksam und wies frühere Tumorrezidive auf. Die-ser Unterschied in den T/C-Werten (p = 0,0032) und Tumorrezidiven (p = 0,05) erwiessich als statistisch signifikant. Der Vergleich beider Substanzen in einer aequimolarenDosierung von 0,5 mg/kg Körpergewicht für AMPT (MTD) und AMPT-HSA (MTD/2)zeigte für beide Substanzen eine komplette Remissionsrate über 7 Tage nach der letztenApplikation. Allerdings wurde für das Konjugat eine niedrigere und spätere Tumorrezi-divrate (AMPT: 4 Rezidive am Tag 14 nach der letzten Applikation versus 2 Rezidivefür AMPT-HSA an den Tagen 10 und 24 nach der letzten Applikation) beobachtet. ImHinblick auf die hohe Wirksamkeit und die deutlich reduzierte systemische Toxizitätdes Konjugates AMPT-HSA gegenüber ungebundenem AMPT läßt sich vermutetn, daßdie eigentliche Wirksubstanz erst nach intrazellulärer Abspaltung von Albumin durchlysosomale Proteasen in der Tumorzelle entfaltet wird, um dann, wie bereits beschrie-ben, mit hoher Affinität die Folylpolyglutamatsynthetase zu inhibieren. Insgesamt im-poniert AMPT-HSA verglichen mit dem Albuminkonjugat MTX-HSA als die aktivereSubstanz, denn lang anhaltende Remissionen für MTX-HSA in einer Höhe von 60 %beim Walker-256 Karzinom der Ratte wurden erst in einer Dosierung von 3 x 2,0 mg/kgKG beobachtet. Gerade aber bei den Karzinomen, welche eine erhöhte Tumorpermea-bilität aufweisen (27 – 29), verspricht AMPT-HSA aufgrund seiner pharmakokineti-schen Eigenschaften eine mögliche Wirksamkeit. Neben MTX-HSA und dem vonPommerenke untersuchten Doxorubicin-HSA an Doxorubicin-resistenten L1210 Tumo-ren bei Mäusen (30) liegt nun mit AMPT-HSA ein weiteres Albuminkonjugat vor, wel-ches für weitere präklinische und klinische Studien auf Grund der durch die Albumin-konjugierung deutlich erniedrigten systemischen Toxizität als geeignet erscheint.

6 4
Kapitel 4
Klinische Phase-I und II-Studien mit Methotrexat-Albumin (MTX-HSA) bei Patienten mit rezidivierenden malignen Gehirntumoren
4.1 Einleitung
Auf die tierexperimentellen Untersuchungen mit MTX-RSA und die erste klinischePhase-I-Studie mit MTX-HSA am Onkologischen Zentrum des Städtischen KlinikumsMannheim wurde bereits in Kapitel 1 verwiesen. Sowohl die tierexperimentellen Ar-beiten wie auch die klinische Phase-I-Studie waren die Grundlage für eine Auslizensie-rung der von Sinn entwickelten Patente (MTX-HSA; AFlc-HSA etc.) an Firma Klinge-Pharma, jetzt Fujisawa-Deutschland. Nach Übernahme der Patente erfolgte eine zweitevon Klinge-Pharma initiierte, dem Arzneimittelgesetz entsprechende Phase-I-Studie mitaufsteigenden Dosierungen von 50 mg/m2, 70 mg/ m2, 80 mg/ m2 und 90 mg/ m2 MTX-HSA in zweiwöchentlichen Abständen bei insgesamt 23 Karzinompatienten. Die Studi-endauer verlief über vier Administrationen, d. h. die Behandlungsdauer betrug insge-samt zwei Monate. Bei Studienende war bei 10 von 23 Patienten keine weitere Tumor-progression mehr zu beobachten. Die akute Toxizität der ambulant behandelten Patien-ten war gering. Erst ab zweiwöchentlichen Dosierungen von 80 mg MTX-HSA/m2 tra-ten typische MTX-induzierte Nebenwirkungen (CTC-Grade gemäß der common-toxic-critera) auf:
• Thrombozytopenie: 2 x Grad 4 bei 80 mg/m2; 1 x Grad 3 und 1 x Grad 4 bei90 mg/m2.
• Stomatitis: 1 x Grad 3 und 1 x Grad 4 bei 80 mg/ m2; 1 x Grad 3 bei90 mg/ m2
• Bilirubinämie: 1 x Grad 3 bei 80 mg/m2;• Transaminasenerhöhung: 1 x Grad 3 bei 90 mg/m2.
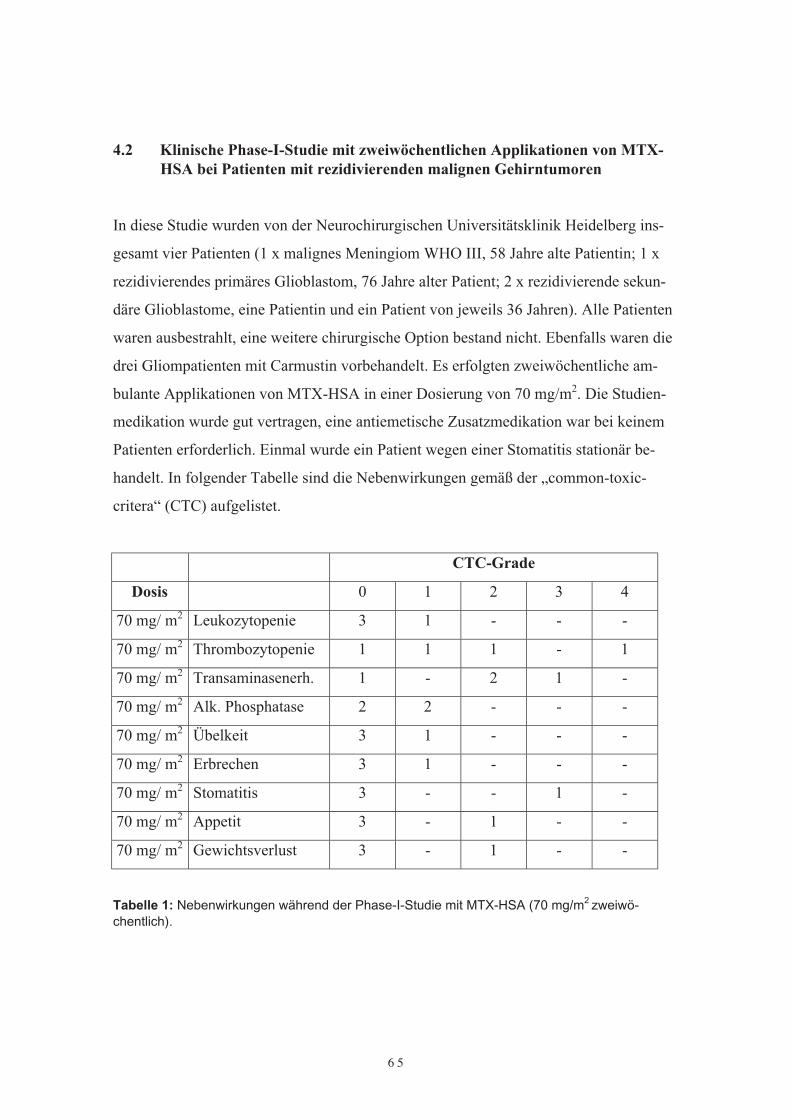
6 5
4.2 Klinische Phase-I-Studie mit zweiwöchentlichen Applikationen von MTX-HSA bei Patienten mit rezidivierenden malignen Gehirntumoren
In diese Studie wurden von der Neurochirurgischen Universitätsklinik Heidelberg ins-gesamt vier Patienten (1 x malignes Meningiom WHO III, 58 Jahre alte Patientin; 1 xrezidivierendes primäres Glioblastom, 76 Jahre alter Patient; 2 x rezidivierende sekun-däre Glioblastome, eine Patientin und ein Patient von jeweils 36 Jahren). Alle Patientenwaren ausbestrahlt, eine weitere chirurgische Option bestand nicht. Ebenfalls waren diedrei Gliompatienten mit Carmustin vorbehandelt. Es erfolgten zweiwöchentliche am-bulante Applikationen von MTX-HSA in einer Dosierung von 70 mg/m2. Die Studien-medikation wurde gut vertragen, eine antiemetische Zusatzmedikation war bei keinemPatienten erforderlich. Einmal wurde ein Patient wegen einer Stomatitis stationär be-handelt. In folgender Tabelle sind die Nebenwirkungen gemäß der „common-toxic-critera“ (CTC) aufgelistet.
CTC-GradeDosis 0 1 2 3 4
70 mg/ m2 Leukozytopenie 3 1 - - -70 mg/ m2 Thrombozytopenie 1 1 1 - 170 mg/ m2 Transaminasenerh. 1 - 2 1 -70 mg/ m2 Alk. Phosphatase 2 2 - - -70 mg/ m2 Übelkeit 3 1 - - -70 mg/ m2 Erbrechen 3 1 - - -70 mg/ m2 Stomatitis 3 - - 1 -70 mg/ m2 Appetit 3 - 1 - -70 mg/ m2 Gewichtsverlust 3 - 1 - -
Tabelle 1: Nebenwirkungen während der Phase-I-Studie mit MTX-HSA (70 mg/m2 zweiwö-chentlich).

6 6
Bei Studienende zeigten beide sekundäre Glioblastompatienten über 2,5 bzw. drei Mo-nate einen Stillstand der zuvor kernspintomographisch nachgewiesenen Tumorprogres-sion. Bei der Patientin mit dem multifokalen malignen Meningiom wie auch bei demPatienten mit dem primären Glioblastomrezidiv war eine weitere Tumorprogressionnach Abschluß der Studiendauer jedoch nicht aufhaltbar. Erfreulicherweise konnte dannaber in der Extension-Studie bei dem Patienten mit dem progredienten primären Glio-blastomrezidiv ein weiteres Tumorwachstum unter sechs weiteren MTX-HSA Applika-tionen über vier Monate verhindert werden.
PatientenTumorprogressionwährend der Studien-dauer (Monate)
Extension-Studie
01sekund. Glioblastom NC über 2,5 Monate PD nach 1,5 Monaten02malignes Meningeom PD03sekund. Glioblastom NC über 3 Monate PD nach 1 Monat04primäres Glioblastom PD
NC über 4 Monate, dannPD
Tabelle 2: Kernspintomographisch nachgewiesene Tumorprogression unter der Phase-I-Studie mit MTX-HSA (NC = no change; PD = progressive disease)
Im Gegensatz zur ersten Phase-I-Studie in Mannheim, in der die Halbwertszeit vonMTX-HSA mit 21± 12 Tagen angegeben wurde (1), lag diese nun nach pharmakokine-tischen Bestimmungen durch Klinge-Pharma bei 9,96 ± 5.65 Tagen. Insgesamt ermu-tigten uns die Ergebnisse in der Phase-I-Studie, MTX-HSA in einer vergleichendenPhase-II-Studie bei Patienten mit primären und sekundären Glioblastomrezidiven zuuntersuchen. Zudem wurden von Klinge-Pharma weitere klinische Phase-II-Studien,sowohl für das malignen Hypernephrom (2) wie auch für das maligne Mesotheliom (3),veranlaßt.

6 7
4.3 Vergleichende Phase-II-Studie von MTX-HSA und Carmustin/Lomustinbei Patienten mit primären und sekundären Glioblastomrezidiven
Um die Wirkung von MTX-HSA bei primären und sekundären Glioblastompatientennachzuweisen, wurde eine randomisierte Vergleichsstudie mit den NitrosoharnstoffenCarmustin (BCNU; Carmubris) bzw. Lomustin (CCNU; Cecenu) als weitere Prüfsub-stanzen konzipiert. Dabei galten folgende Einschlußkriterien:
• Kernspintomographisch nachgewiesenes Tumorrezidiv eines primären Glioblastoms• Solider Tumorrest nach mikrochirurgischer Resektion oder bioptischer Sicherung
eines primären Glioblastoms• Kernspintomographisch nachgewiesene, eindeutige maligne Transformation eines
ursprünglich niedergradigen Glioms zum sekundären Glioblastom.
Als Ausschlußkriterien wurden formuliert:
• Vorbehandlung mit Nitrosoharnstoffen• Operation vor < 2 Wochen, Radiotherapie < 4 Wochen• Hormontherapie < 4 Wochen oder Immuntherapie < 2 Wochen• Zweitkarzinomerkrankung oder akute/chronische Infektionen• Schwangerschaft• Allergische Reaktionen gegenüber Nitrosoharnstoffen oder MTX
MTX-HSA wurde wöchentlich in einer Dosierung von 50 mg/ m2 ambulant, BCNU(Carmustin) bzw. CCNU (Lomustin) sechswöchentlich in einer Dosierung von 200mg/m2 kurzstationär verabreicht. Ab dem 01.05.01 wurde das MTX-HSA Dosierungs-schema wie folgt geändert: Initialdosis (loading-dose) von 110 mg/m2, gefolgt von wö-chentlichen Applikationen mit 40 mg/m2 oder bei fehlender Toxizität von wöchentli-chen Applikationen mit 50 mg/m2.
Mit dem Stand vom 30.08.02 sind nun 17 Patienten in diese Vergleichstudie einge-schleust (10 x primäre Glioblastome; 4 x sekundäre Glioblastome, 3 x Gliosarkome).

6 8
Alle Patienten hatten einen Karnofsky-Index von > 70 %, das mittlere Alter lag bei 47Jahren. Wiederum wurde der Effekt der Therapie kernspintomographisch in zwei- bisdreimonatigen Abständen kontrolliert. Mit dem Stand vom 31.08.02 ergibt sich folgen-de Nebenwirkungsrate:
CTC-Grade
1 2 3 4BCNU/CCNU
Leukozytopenie 1 1 - -Transaminasen - 3 1 -Anämie 1 - - -Thrombozytopenie - - - -MTX-HSA
Leukozytopenie - - - -Transaminasen - 1 2 -Anämie - - - -Thrombozytopenie 2 1 1 -Stomatitis 2 1 - -Nausea 1 - - -
Tabelle 3: Nebenwirkungsrate von Carmustin und Lomustin im Vergleich zu MTX-HSA
Gegenwärtig ergibt sich mit dem Stand vom 31.08.02 folgende Wirksamkeit für diebeiden Substanzgruppen, welche bei nachgewiesener Tumorprogression auch gegenein-ander ausgetauscht werden konnten (cross-over):
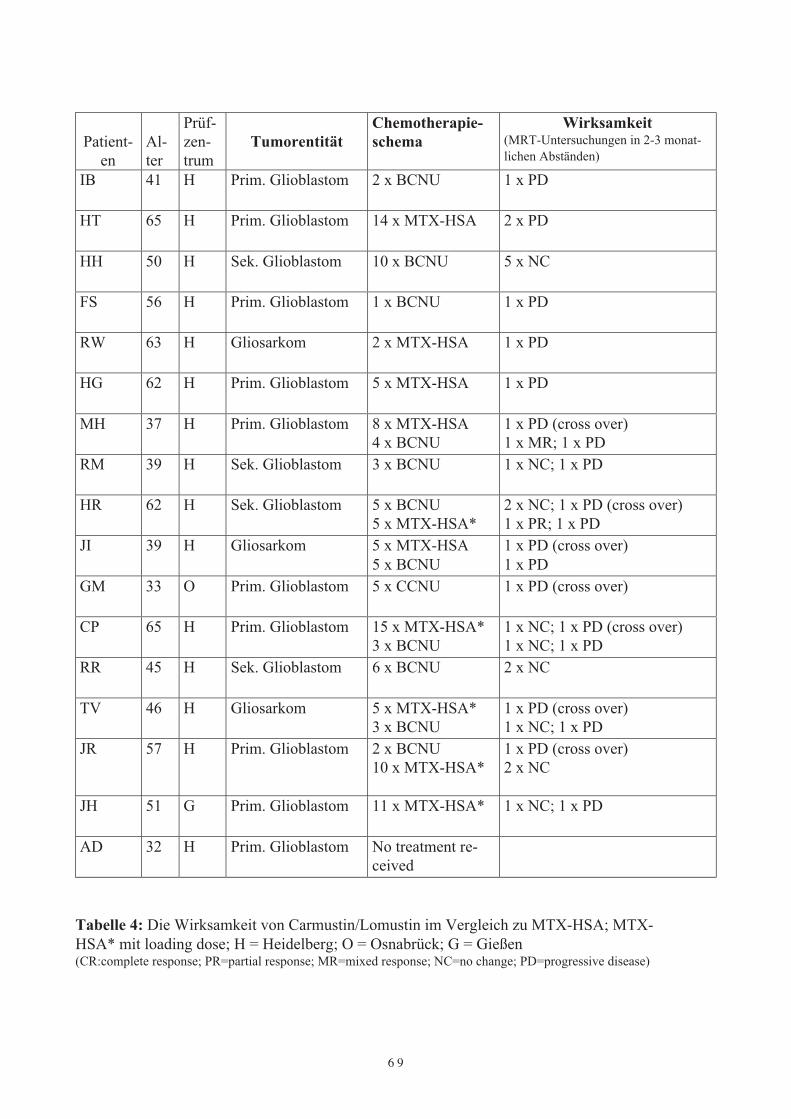
6 9
Patient-en
Al-ter
Prüf-zen-trum
TumorentitätChemotherapie-schema
Wirksamkeit(MRT-Untersuchungen in 2-3 monat-lichen Abständen)
IB 41 H Prim. Glioblastom 2 x BCNU 1 x PDHT 65 H Prim. Glioblastom 14 x MTX-HSA 2 x PDHH 50 H Sek. Glioblastom 10 x BCNU 5 x NCFS 56 H Prim. Glioblastom 1 x BCNU 1 x PDRW 63 H Gliosarkom 2 x MTX-HSA 1 x PDHG 62 H Prim. Glioblastom 5 x MTX-HSA 1 x PDMH 37 H Prim. Glioblastom 8 x MTX-HSA
4 x BCNU1 x PD (cross over)1 x MR; 1 x PD
RM 39 H Sek. Glioblastom 3 x BCNU 1 x NC; 1 x PDHR 62 H Sek. Glioblastom 5 x BCNU
5 x MTX-HSA*2 x NC; 1 x PD (cross over)1 x PR; 1 x PD
JI 39 H Gliosarkom 5 x MTX-HSA5 x BCNU
1 x PD (cross over)1 x PD
GM 33 O Prim. Glioblastom 5 x CCNU 1 x PD (cross over)CP 65 H Prim. Glioblastom 15 x MTX-HSA*
3 x BCNU1 x NC; 1 x PD (cross over)1 x NC; 1 x PD
RR 45 H Sek. Glioblastom 6 x BCNU 2 x NCTV 46 H Gliosarkom 5 x MTX-HSA*
3 x BCNU1 x PD (cross over)1 x NC; 1 x PD
JR 57 H Prim. Glioblastom 2 x BCNU10 x MTX-HSA*
1 x PD (cross over)2 x NC
JH 51 G Prim. Glioblastom 11 x MTX-HSA* 1 x NC; 1 x PDAD 32 H Prim. Glioblastom No treatment re-
ceived
Tabelle 4: Die Wirksamkeit von Carmustin/Lomustin im Vergleich zu MTX-HSA; MTX-HSA* mit loading dose; H = Heidelberg; O = Osnabrück; G = Gießen(CR:complete response; PR=partial response; MR=mixed response; NC=no change; PD=progressive disease)

7 0
Als Prüfzentren dienten neben der Neurochirurgischen Universitätsklinik Heidelberg(15 Patienten) die Neurochirurgische Abteilung der Paracelsus-Klinik Osnabrück (1Patient), die Neuroonkologische Abteilung des AKH Wien (kein Patient), die Neuro-chirurgische Universitäsklinik Giessen (1 Patient) und erst seit kurzem die Onkologi-sche Abteilung der Universitätsklinik Rostock (bislang noch kein Patient). Alle vierPatienten mit sekundären Glioblastomen zeigten initial ein Ansprechen auf die chemo-therapeutische Behandlung mit BCNU mit einem unveränderten Tumorstatus über 3, 5,6 und 10 Behandlungszyklen. Somit erscheinen gerade sekundäre Glioblastome im Ge-gensatz zu den primären Glioblastom- oder Gliosarkomrezidiven als besonders sensitivgegenüber einer Chemotherapie mit Nitrosoharnstoffen (p = 0,0082; Fischer’s ExactTest). Bei den vier Patienten mit primären Glioblastomrezidiven, welche mit BCNUbehandelt wurden, fand sich bei keinem ein Stillstand des Tumorwachstums. Dagegenzeigten zwei von vier Patienten mit primären Glioblastomrezidiven, welche mit MTX-HSA behandelt wurden, ein „no change“ über jeweils 3 Monate. Allerdings wies dieserUnterschied in der Prüfmedikation zwischen MTX-HSA und den Nitrosoharnstoffenkeine statistische Signifikanz auf (p = 0,42; Fischer’s Exact Test). Überraschend wardas Ansprechen auf die chemotherapeutische Behandlung unter einem Wechsel der Stu-dienmedikation, sog. „cross over“, welches bei 7 Patienten angeboten, aber nur bei 6Patienten durchgeführt wurde. Hierbei fand sich bei 5 der 6 Patienten ein Ansprechenauf die chemotherapeutische Behandlung: 1 Patient mit MR (mixed response) vonMTX-HSA zu BCNU (prim. Glioblastom); 1 Patient mit PR (partial response) vonBCNU auf MTX-HSA (sek. Glioblastom); 2 Patienten mit NC (no change) von MTX-HSA auf BCNU (prim. Glioblastome); 1 Patient mit NC von BCNU auf MTX-HSA(prim. Glioblastom). Auch hier zeigte sich eine statistische Signifikanz (p = 0,0014;Fischer’s Exact Test) gegenüber den Patienten, bei denen kein „cross over“ stattfand.

7 1
4.4 Diskussion
Immer noch wird die chemotherapeutische Behandlung von Glioblastomen kontroversdiskutiert. Manche Autoren geben eine Ansprechrate von 20 – 25 % an (4, 5). Insgesamtaber wird eine durch die Chemotherapie bedingte Verlängerung der Überlebenszeit füreinen Zeitraum von nur wenigen Wochen erreicht (6, 7). Erschwerend kommt hinzu,daß in vielen klinischen Studien nicht zwischen primären und sekundären Glioblasto-men unterschieden wird. Auch findet sich unter der Bezeichnung „maligner Gliome“eine Vermischung von anaplastischen Astrozytomen WHO III, anaplastischen Oligo-dendrogliomen WHO III und Glioblastomen, WHO Grad IV (5, 8). Zudem kann ver-mutet werden, daß ein gewisser Anteil der Ansprechrate unter der Chemotherapie mitNitrosoharnstoffen auf das Vorhandensein von nicht diagnostizierten, aber chemosensi-tiven anaplastischen Oligodendrogliomen zurückzuführen ist (9). Vielen Studien fehlteine Vergleichsgruppe nach Randomisierung, so daß eigentlich in den meisten Fällennur gesammelte Fallberichte vorliegen. Auch findet die Radikalität der Operation undder dadurch bedingte Einfluß auf die Prognose bei Glioblastomen nur wenig Beachtung(10, 11).
Die am häufigsten verwendeten Substanzen sind die seit den 80er Jahren eingesetztenNitrosoharnstoffe (BCNU, Carmustin; CCNU, Lomustin und ACNU, Fotemustin), wel-che als Anti-Alkylantien direkt in die DNA- Synthese eingreifen (12). Trotz ihrer gutenVerträglichkeit hat die, wenn auch immer wieder Einzelerfolge beobachtet werden, ge-ringe Ansprechrate nur wenige Zentren veranlaßt, diese Substanzen, zumindest bei Gli-oblastompatienten, regelmäßig einzusetzen. Mit Temozolamid, einem ebenfalls alkylie-rendem Wirkstoff, hat die chemotherapeutische Behandlung von Glioblastomen einegewisse Renaissance erfahren, obwohl in einer vergleichenden Phase-II-Studie von Te-mozolamid versus Procarbazin nur ein leichter Überlebensvorteil bei einem Zeitpunktvon sechs Monaten nachgewiesen werden konnte (13).
Methotrexat ist ein Folsäureantagonist, welcher in die S-Phase eingreift und die Pyrimi-dinsynthese blockiert. MTX findet Verwendung bei den akuten lymphoblastischen Leu-kämien im Kindesalter, bei Osteosarkomen, Chorionkarzinomen, Lymphomen, Kopf -

7 2
Hals - Tumoren und anderen soliden Tumorerkrankungen. MTX wird ebenfalls beichronisch entzündlichen Erkrankungen, wie z. B. der rheumatoiden Arthritis oder beiPsoriasis, klinisch angewendet.
Die Dosierungen reichen von 30 mg/m2 wöchentlich oder 50 - 100 mg/m2 drei- bisvierwöchentlich bis zu Dosierungen von 100 - 1500 oder 8000 - 12000 mg/m2 (Hochdo-sistherapie). Hier ist ein Einsatz von Folsäureersatz unabdingbar (14). Nach intravenö-ser Gabe lagert sich MTX zu 35 - 50 % unspezifisch an Plasmaeiweiße. Seine Halb-wertszeit, welche vor allem durch die glomeruläre Filtration bedingt ist, wird mit 0,5 - 3Stunden angegeben (15). Eine intakte Blut - Hirn - Schranke passiert MTX nicht. MTXinduzierte Nerbenwirkungen bestehen vor allen von Seiten des Blutbildes (Thrombo-zytopenie, Leukozytopenie), der Schleimhäute (Mucositis, Stomatitis), des Urogenital-traktes (Nierenfunktionsstörungen, Zystitis), der Leber (Hepatopathien, Lebernekrosen)und der Haut (Exantheme, Erytheme, Alopezie).
Die Verwendung von Methotrexat in der Behandlung von zerebralen Malignomen kon-zentriert sich vor allem auf die intrathekale Applikation bei zerebralen Lymphomen (16)und der leptomeningialen Aussaat von systemischen Tumorerkrankungen (17). Jedochwurde Methotrexat in den 70er Jahren auch bei Glioblastomen getestet, wobei teilweisesehr hohe Dosierungen in Kombination mit Folsäureersatz verwendet wurden (18).Intraarterielle (19) oder intratumorale Injektionen (20, 21, 22) von Methotrexat oderlokale Applikationen von MTX, welche teilweise an synthetische Träger, wie z. B. Po-lylactit (23) oder Polymethylmethacrylat (24), dienten der Verbesserung der Pharmako-kinetik mit höheren lokalen Wirkstoffkonzentationen. Weiterführende klinische Studienhierüber existieren jedoch nicht. Insgesamt muß man davon ausgehen, daß Methotrexatbei Glioblastomen eher schwach wirksam ist. Neben den bekannten systemischen Ne-benwirkungen kann bei intrathekaler Applikation eine schwere Neurotoxizität beob-achtet werden, vor allem dann, wenn MTX intrathekal in Kombination mit einer Ganz-hirnbestrahlung appliziert wird (25, 26).
In Kenntnis der doch deutlichen systemischen Nebenwirkungsrate bei Dosierungen von50 mg MTX/m2 wöchentlich erscheinen die in den Phase-I und II-Studien erreichten

7 3
Toxizitäten von 50 mg MTX-HSA/m2 trotz der deutlich längeren Zirkulationszeiten desKonjugates im Vergleich zu freiem MTX als gering (2 x Transaminasenerhöhung Grad3 und 1 x Trombozytopenie Grad 3 bei 50 mg MTX-HSA /m2 wöchentlich; 1 x Throm-bozytopenie Grad 4 bei 70 mg MTX-HSA /m2 zweiwöchentlich). In allen Fällen konntedie Toxizität durch eine Spreizung der Studienmedikation beherrscht werden. Eine Zu-nahme an Toxizität nach Einführung der „loading dose“ von 110 mg MTX-HSA/m2
wurde nicht beobachtet. Insgesamt bestätigte sich, daß mit MTX-HSA ein sog. „pro-drug“ vorliegt, welches trotz der langen Zirkulationszeit physiologisch proliferierendesGewebe kaum belastet.
Bezüglich der Effektivität konnte in der Phase-I-Studie unter Behandlung mit MTX-HSA bei zwei Patienten mit sekundären Glioblastomen, welche unter BCNU eine Tu-morprogression aufwiesen, ein Stillstand des Tumorwachstums über 2,5 bzw. drei Mo-nate erreicht werden. In der Phase-II-Studie wurden nach Randomisierung alle sekundä-ren Glioblastome primär mit BCNU behandelt. Hierbei zeigte sich eine Ansprechratemit einem „no change“ über drei, fünf, sechs und zehn Behandlungszyklen (p = 0,0082;sekundäre versus primäre Glioblastome). Deutlich niedriger war die Ansprechrate beiden primären Glioblastomen. Hier hatte die primäre Nitrosoharnstoffbehandlung beikeinem Patienten zu einem Stillstand des Tumorwachstums geführt. Allerdings zeigtenzwei Patienten mit primärem Glioblastomrezidiven, welche initial mit MTX-HSA be-handelt wurden, einen Stillstand der zuvor beobachteten Tumorprogression auf. Amhäufigsten aber, nämlich bei fünf von sechs Patienten, finden sich Effekte der Chemo-therapie bei einem Wechsel der Studienmedikation, sog. „cross over“, so daß ein syner-gistischer Effekt des Folsäureantagonisten und des Anti-Alkylans vermutet werden kann(p = 0,0014; cross over versus kein cross over).
Insgesamt aber sind die Effekte vor allem bei den primären Glioblastomen oder Glio-sarkomen von nur kurzer Dauer und erreichen einen Stillstand in der Tumorprogressionvon ungefähr vier bis fünf Monaten. Auch wenn dies insgesamt gesehen ein nur kurzerZeitraum ist, so erscheint dies bei der sehr guten Verträglichkeit beider Substanzgrup-pen für den Einzelfall durchaus als Teilerfolg. Auch wenn eine abschließende Beurtei-lung der Studie noch nicht vorliegt, so stellt sich die Frage, ob nicht gerade das Ausnut-

7 4
zen eines vermuteten synergistischen Effektes zwischen Antifolat und Anti-Alkylans(27) eine Fortführung der Studie im Sinne einer Kombinationstherapie rechtfertigt.

7 5
Kapitel 5Albuminvermittelte Lymphdiagnostik mit TCPC- oder TCPP-HSAund Gadolinium-HSA
5.1. Einleitung
Eine Hauptaufgabe des Lymphgefäßsystems besteht in der Bewältigung der extravas-kulären Zirkulation von Plasmaproteinen. Bis auf eine geringe Menge, welche überBlutkapillaren wieder in den Kreislauf zurückgelangen, muß der überwiegende Teil derdas Kapillarbett verlassenden Plasmaproteine über das Lymphbahnsystem in das venöseSystem über den Ductus thoracicus zurückgeführt werden. Für diese Eigenschaft sindLymphgefäße mit Klappen ausgestattet, können aber auch eine gewisse Eigenkontrakta-bilität entwickeln. Äußere Einflüsse, wie die Muskelpumpe, Atemexkursion und ateri-elle Pulsation unterstützen den Lymphfluß (1).
Neben dem Transport von Plasmaproteinen stellen Lymphwege aber auch ein wichtigesAusbreitungsnetz bei Karzinomerkrankungen dar. Eine Vielzahl maligner Tumoren,wie z. B. das Mammakarzinom, maligne Melanom und Prostatakarzinom, metastasierenüber Lymphbahnen, weswegen der Diagnostik von tumordrainierenden Lymphwegenund deren Lymphknoten in der letzten Zeit vermehrt Beobachtung geschenkt wird. Sowurde das Konzept des Wächterlymphknotens, d. h. des ersten im Lymphabstromgebietdes malignen Tumors befallenen Lymphknoten, entwickelt. Dieses Konzept des „senti-nel lymph node“ (SLN) wurde 1977 von Canabas (2) anhand lymphangiographischerUntersuchungen bei Peniskarzinomen entwickelt und 1992 von Morton (3) auf das ma-ligne Melanom übertragen. Während beide Autoren für die intraoperative Darstellungvon Lymphwegen noch Patentblau verwendeten, wurde wenige Zeit später radioaktivmit 99mTechnetium markiertes kolloidales humanes Serumalbumin (99mTc-HSA) zurIdentifizierung des SLN eingesetzt ( 4, 5, 6, 7, 8).
Durch Einführung des Sentinel-Lymph-Node-Konzeptes (SLN) hat die Diagnostik undTherapie von tumordrainierenden Lymphbahnen und deren Lymphknoten eine Renais-
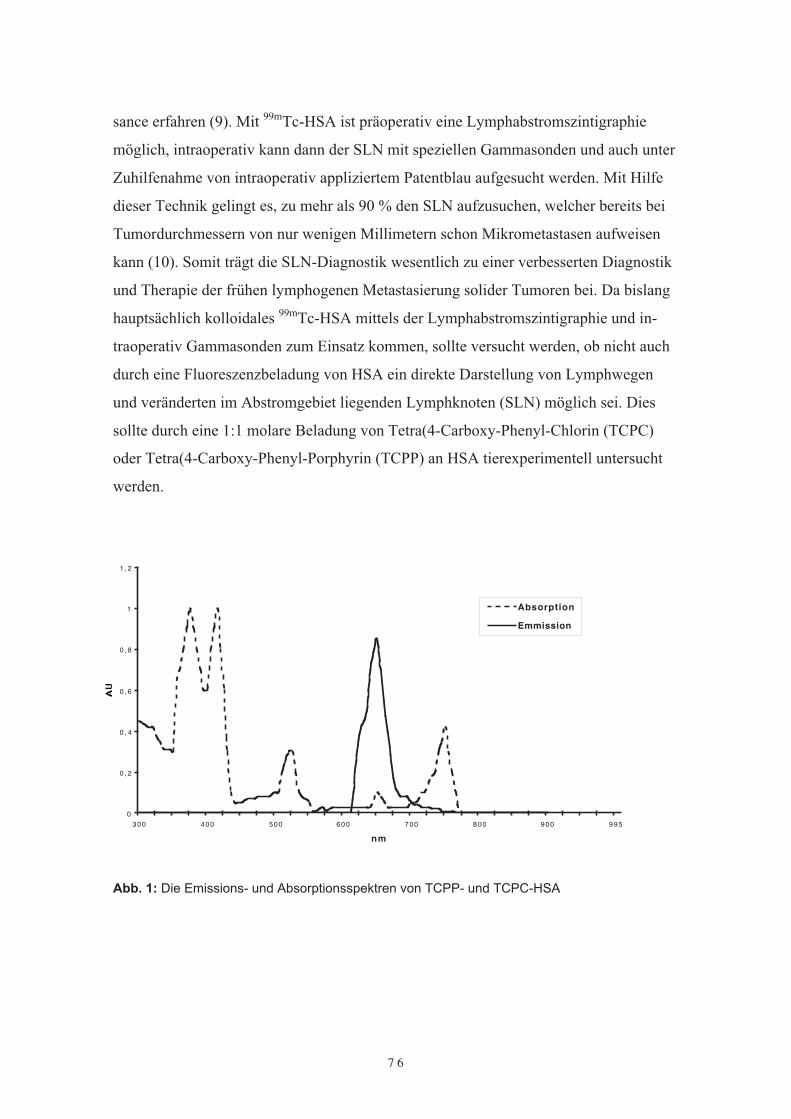
7 6
sance erfahren (9). Mit 99mTc-HSA ist präoperativ eine Lymphabstromszintigraphiemöglich, intraoperativ kann dann der SLN mit speziellen Gammasonden und auch unterZuhilfenahme von intraoperativ appliziertem Patentblau aufgesucht werden. Mit Hilfedieser Technik gelingt es, zu mehr als 90 % den SLN aufzusuchen, welcher bereits beiTumordurchmessern von nur wenigen Millimetern schon Mikrometastasen aufweisenkann (10). Somit trägt die SLN-Diagnostik wesentlich zu einer verbesserten Diagnostikund Therapie der frühen lymphogenen Metastasierung solider Tumoren bei. Da bislanghauptsächlich kolloidales 99mTc-HSA mittels der Lymphabstromszintigraphie und in-traoperativ Gammasonden zum Einsatz kommen, sollte versucht werden, ob nicht auchdurch eine Fluoreszenzbeladung von HSA ein direkte Darstellung von Lymphwegenund veränderten im Abstromgebiet liegenden Lymphknoten (SLN) möglich sei. Diessollte durch eine 1:1 molare Beladung von Tetra(4-Carboxy-Phenyl-Chlorin (TCPC)oder Tetra(4-Carboxy-Phenyl-Porphyrin (TCPP) an HSA tierexperimentell untersuchtwerden.
Abb. 1: Die Emissions- und Absorptionsspektren von TCPP- und TCPC-HSA
0
0 , 2
0 , 4
0 , 6
0 , 8
1
1 , 2
300 400 500 600 700 800 900 995nm
AbsorptionEmmission
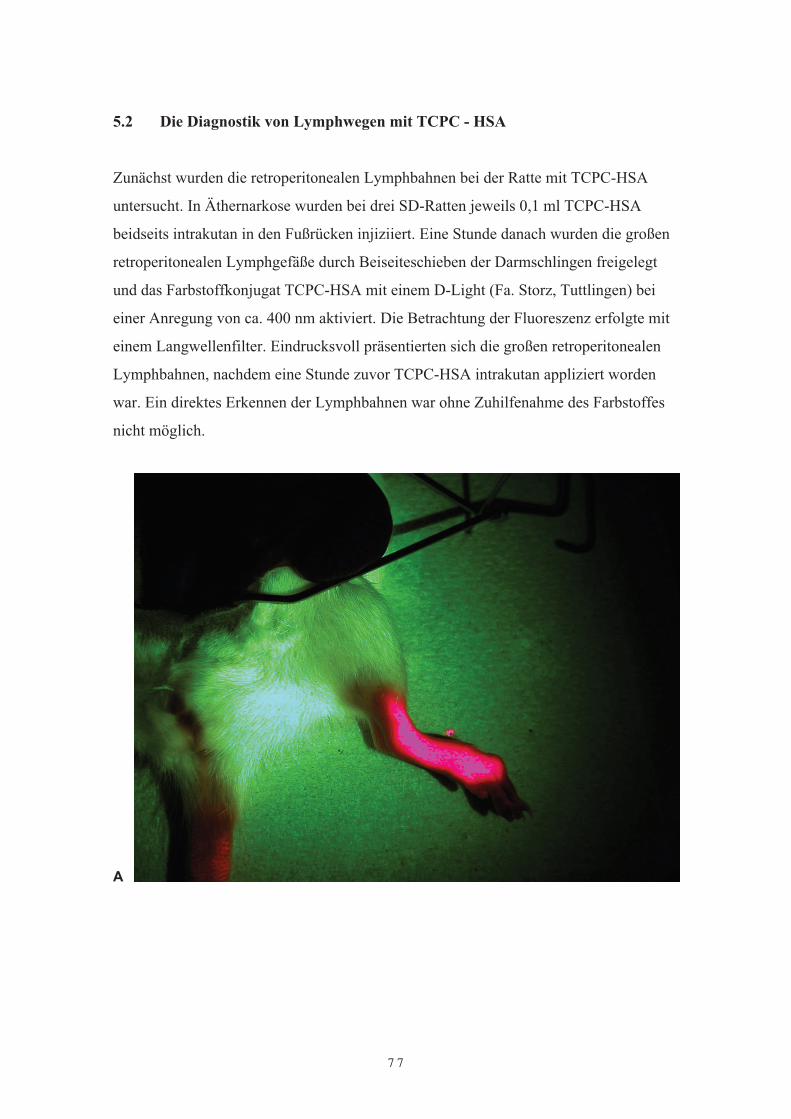
7 7
5.2 Die Diagnostik von Lymphwegen mit TCPC - HSA
Zunächst wurden die retroperitonealen Lymphbahnen bei der Ratte mit TCPC-HSAuntersucht. In Äthernarkose wurden bei drei SD-Ratten jeweils 0,1 ml TCPC-HSAbeidseits intrakutan in den Fußrücken injiziiert. Eine Stunde danach wurden die großenretroperitonealen Lymphgefäße durch Beiseiteschieben der Darmschlingen freigelegtund das Farbstoffkonjugat TCPC-HSA mit einem D-Light (Fa. Storz, Tuttlingen) beieiner Anregung von ca. 400 nm aktiviert. Die Betrachtung der Fluoreszenz erfolgte miteinem Langwellenfilter. Eindrucksvoll präsentierten sich die großen retroperitonealenLymphbahnen, nachdem eine Stunde zuvor TCPC-HSA intrakutan appliziert wordenwar. Ein direktes Erkennen der Lymphbahnen war ohne Zuhilfenahme des Farbstoffesnicht möglich.
A
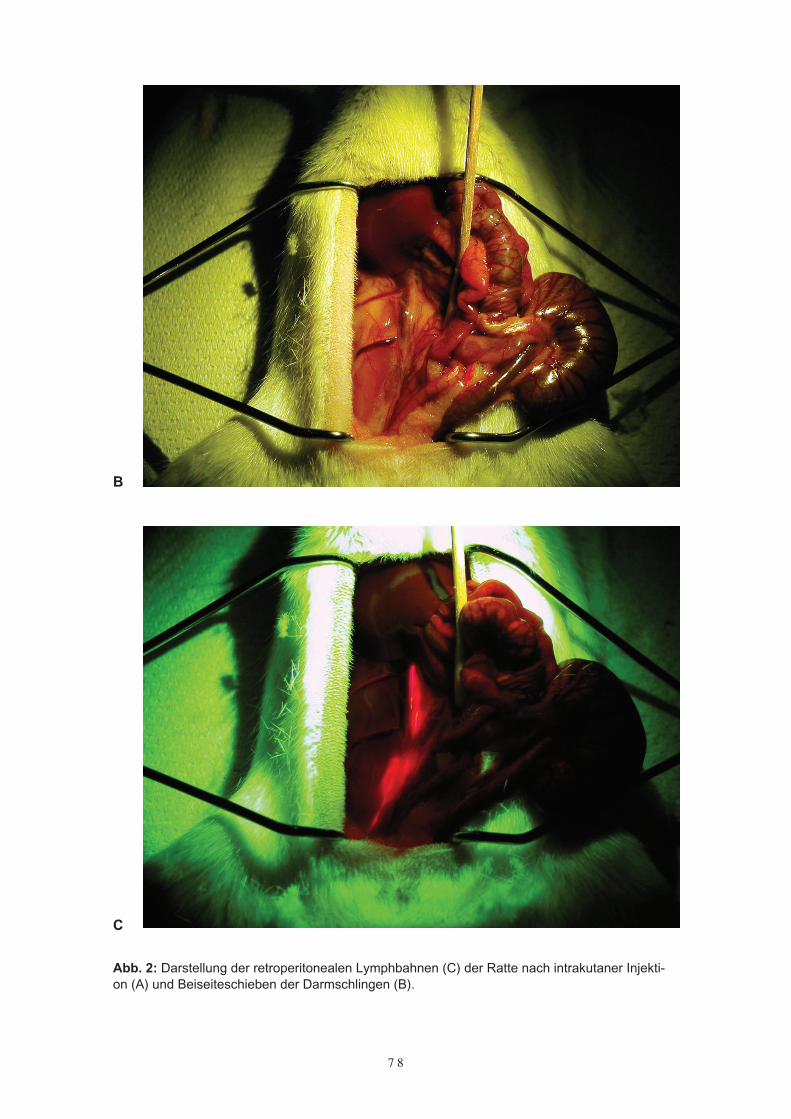
7 8
B
C
Abb. 2: Darstellung der retroperitonealen Lymphbahnen (C) der Ratte nach intrakutaner Injekti-on (A) und Beiseiteschieben der Darmschlingen (B).
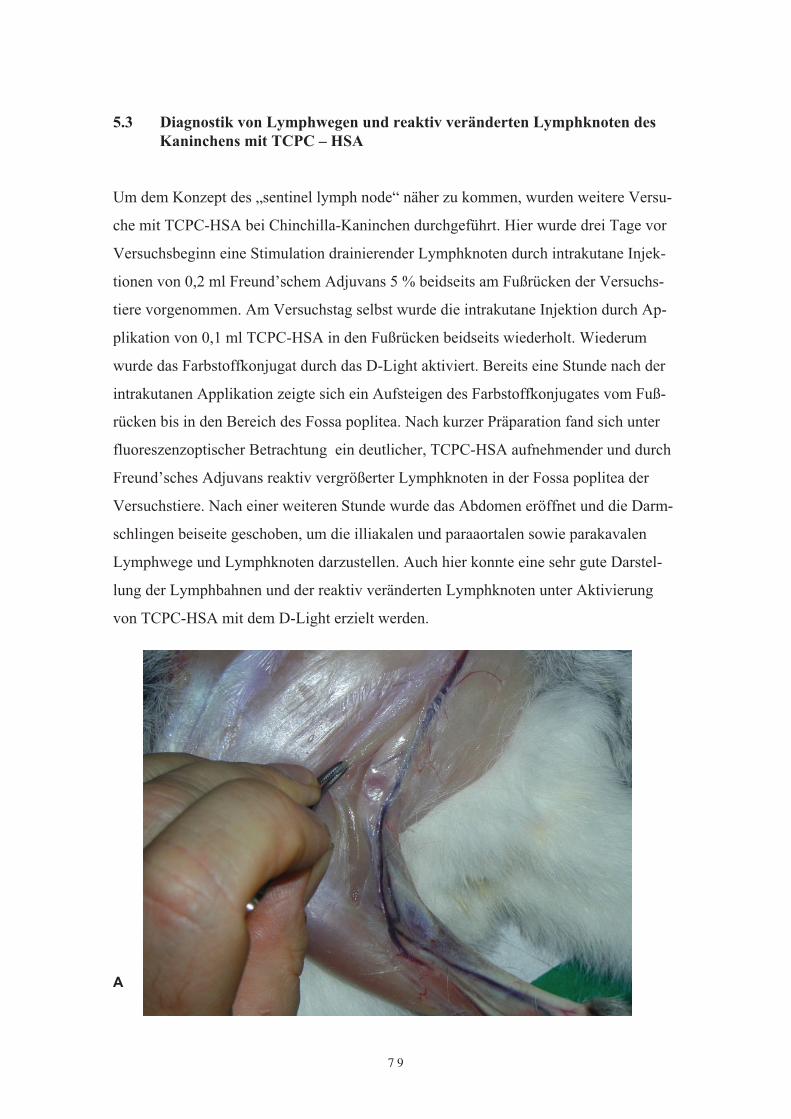
7 9
5.3 Diagnostik von Lymphwegen und reaktiv veränderten Lymphknoten desKaninchens mit TCPC – HSA
Um dem Konzept des „sentinel lymph node“ näher zu kommen, wurden weitere Versu-che mit TCPC-HSA bei Chinchilla-Kaninchen durchgeführt. Hier wurde drei Tage vorVersuchsbeginn eine Stimulation drainierender Lymphknoten durch intrakutane Injek-tionen von 0,2 ml Freund’schem Adjuvans 5 % beidseits am Fußrücken der Versuchs-tiere vorgenommen. Am Versuchstag selbst wurde die intrakutane Injektion durch Ap-plikation von 0,1 ml TCPC-HSA in den Fußrücken beidseits wiederholt. Wiederumwurde das Farbstoffkonjugat durch das D-Light aktiviert. Bereits eine Stunde nach derintrakutanen Applikation zeigte sich ein Aufsteigen des Farbstoffkonjugates vom Fuß-rücken bis in den Bereich des Fossa poplitea. Nach kurzer Präparation fand sich unterfluoreszenzoptischer Betrachtung ein deutlicher, TCPC-HSA aufnehmender und durchFreund’sches Adjuvans reaktiv vergrößerter Lymphknoten in der Fossa poplitea derVersuchstiere. Nach einer weiteren Stunde wurde das Abdomen eröffnet und die Darm-schlingen beiseite geschoben, um die illiakalen und paraaortalen sowie parakavalenLymphwege und Lymphknoten darzustellen. Auch hier konnte eine sehr gute Darstel-lung der Lymphbahnen und der reaktiv veränderten Lymphknoten unter Aktivierungvon TCPC-HSA mit dem D-Light erzielt werden.
A
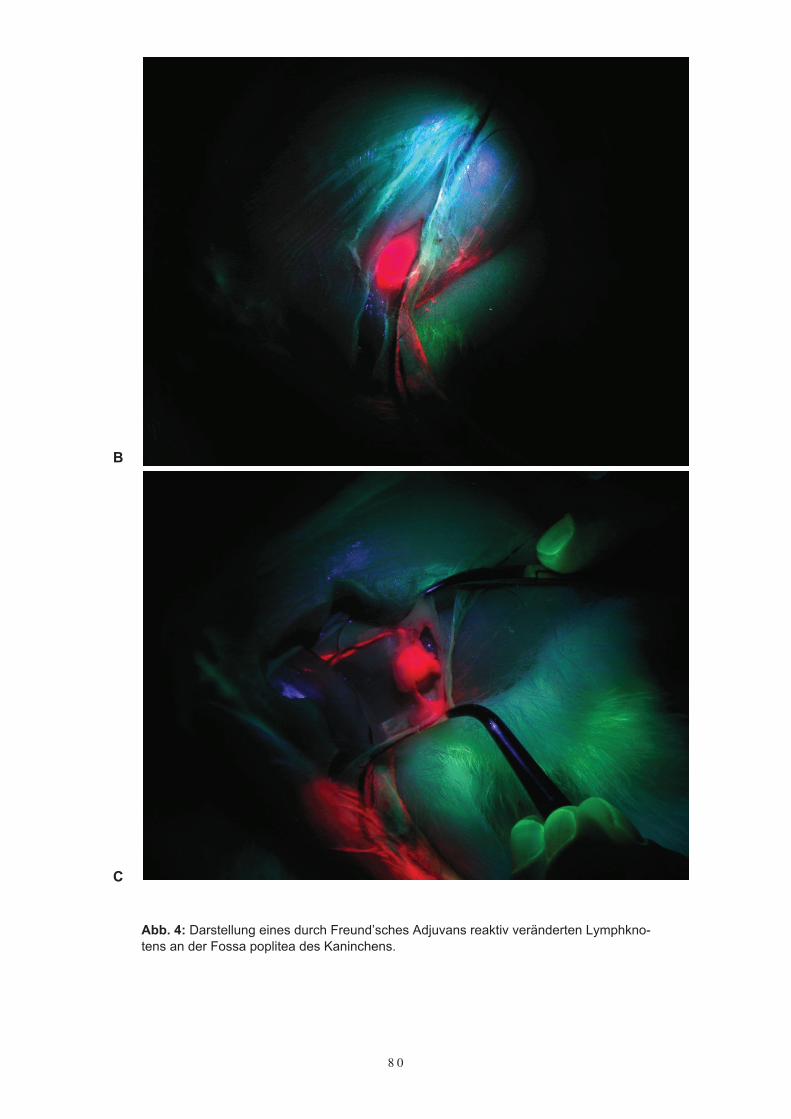
8 0
B
C
Abb. 4: Darstellung eines durch Freund’sches Adjuvans reaktiv veränderten Lymphkno-tens an der Fossa poplitea des Kaninchens.
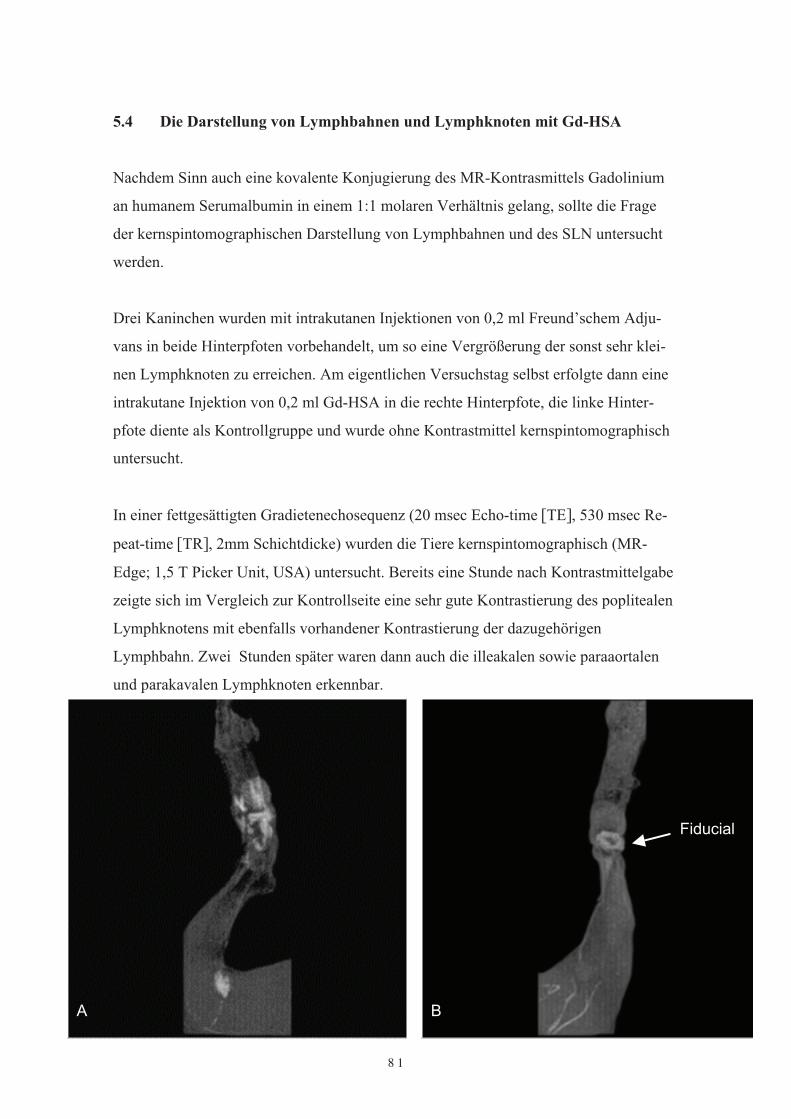
8 1
5.4 Die Darstellung von Lymphbahnen und Lymphknoten mit Gd-HSA
Nachdem Sinn auch eine kovalente Konjugierung des MR-Kontrasmittels Gadoliniuman humanem Serumalbumin in einem 1:1 molaren Verhältnis gelang, sollte die Frageder kernspintomographischen Darstellung von Lymphbahnen und des SLN untersuchtwerden.
Drei Kaninchen wurden mit intrakutanen Injektionen von 0,2 ml Freund’schem Adju-vans in beide Hinterpfoten vorbehandelt, um so eine Vergrößerung der sonst sehr klei-nen Lymphknoten zu erreichen. Am eigentlichen Versuchstag selbst erfolgte dann eineintrakutane Injektion von 0,2 ml Gd-HSA in die rechte Hinterpfote, die linke Hinter-pfote diente als Kontrollgruppe und wurde ohne Kontrastmittel kernspintomographischuntersucht.
In einer fettgesättigten Gradietenechosequenz (20 msec Echo-time [TE], 530 msec Re-peat-time [TR], 2mm Schichtdicke) wurden die Tiere kernspintomographisch (MR-Edge; 1,5 T Picker Unit, USA) untersucht. Bereits eine Stunde nach Kontrastmittelgabezeigte sich im Vergleich zur Kontrollseite eine sehr gute Kontrastierung des poplitealenLymphknotens mit ebenfalls vorhandener Kontrastierung der dazugehörigenLymphbahn. Zwei Stunden später waren dann auch die illeakalen sowie paraaortalenund parakavalen Lymphknoten erkennbar.
A B
Fiducial

8 2
Abb. 5: Darstellung eines reaktiv veränderten Lymphknotens (A) sowie drainierenderLymphwege (C) beim Kaninchen mit Gd-HSA; (B) entspricht der Kontrollseite ohneGd-HSA
LK der Fossa popliteaLK der Fossa poplitea
C

8 3
5.5 Diskussion
Noch Mitte der 50er Jahre erforderte eine Lymphangiographie eine operative Freilegungeines oberflächlichen Lymphgefäßes, welches zuvor nach subkutaner Injektion von Pa-tentblau, z. B. in der ersten Interdigitalfalte, dargestellt wurde (1). In dieses operativfreigelegte Lymphgefäß wurde dann ein öliges, jodhaltiges Kontrastmittel langsam ein-gespritzt. Dargestellt werden konnten so die im Abstromgebiet liegenden Lymphbahnenund Lymphknoten, wie z. B. die inguinalen Stationen, die Gruppen der Illiaca commu-nis und Illiaca externa, die paraaortalen und paracavalen Lymphstationen und der Duc-tus thoracicus.
Durch die Entwicklung des Sentinel-Lymph-Node-Konzeptes (SLN) hat das Untersu-chungsverfahren der Lymphwegediagnostik eine Renaissance erhalten (9). Hierbei wirdvor allem mit 99Tc markiertes kolloidales Serumalbumin intradermal oder peritumoralinjiziert, so daß wenige Minuten bis zu einigen Stunden danach Lymphabflußwege undvor allem Wächterlymphknoten (SLN) dargestellt werden können. Die intraoperativeDetektion eines Lymphknotens wird dann unter Zuhilfenahme einer speziellen Gamma-sonde teilweise aber auch durch den intraoperativen Einsatz von Patentblau ermöglicht.Mit Hilfe dieser Methode lassen sich über 95 % der SNL (10) erkennen. Da sich auchschon bei kleinen Tumorvolumina immer wieder Mikrometastasen in den drainierendenLymphknoten finden, erscheint dieses Verfahren nicht nur von diagnostischer Bedeu-tung sondern auch von chirurgischem Interesse.
In dem von uns vorgestellten Konzept der Darstellung lymphatischen Gewebes wurdezunächst eine fluoreszenzoptische Darstellung der Lymphbahnen und Lymphknoten mitTCPC/TCPP-HSA durchgeführt. TCPC und TCPP sind stark leuchtende hellrote Fluo-reszenzfarbstoffe, welche im Rotlichtbereich erkannt werden. In den Tierversuchenkonnten die drainierenden Lymphbahnen und die veränderten Lymphknoten im Sinnedes SLN mit TCPC/TCPP-HSA sehr gut dargestellt werden. Albumin, welches intra-kutan appliziert wurde, blieb eindeutig auf das lymphatische System beschränkt. Auchwenn Angaben über die Abflußgeschwindigkeiten von TCPC-HSA aus den bisherigenDaten noch nicht erhoben werden können, so imponiert der eigentliche Vorteil der fluo-

8 4
reszenzoptischen Darstellung der Lymphabstrombahnen darin, daß seine Wege erstdurch Aktivierung des Farbstoffes sichtbar werden, ohne daß eine Kontamination desOP-Gebietes durch einen immer sichtbaren Farbstoff, wie z. B. dem Patentblau, vor-liegt. Wie selektiv, wie spezifisch und wie gut das „lymphatic mapping“ mit TCPC/TCPP-HSA wirklich ist, kann erst in klinischen Studien überprüft werden. Allerdingserfordert dies eine dem Arzneimittelgesetz entsprechende Phase-I/II-Zulassungsstudie.
Im Gegensatz zu der direkten Darstellung des lymphatischen Gewebes mit TCPC- oderTCPP-HSA erlaubt die Lymphdiagnostik mit Gd-HSA eine präoperative Lokalisationvon Lymphbahnen und Lymphknoten im Kernspintomogramm. Erstaunlich gut konntendiese Strukturen in den Tierversuchen in hochauflösenden und Fettgewebe supprimie-renden MR-Sequenzen dargestellt werden. Da uns ein Tumormodell mit lymphogenerMetastasierung bislang nicht bekannt ist, wurde eine Aktivierung von im Abstromgebietliegenden Lymphknoten mit Freund’schem Adjuvans vorgenommen und dadurch dasKonzept des SLN imitiert. Somit stünde für die Lymphdiagnostik ein MR-Kontrast-mittel zur Verfügung, welches in den bisherigen tierexperimentellen Untersuchungeneine hohe Auflösung der entsprechenden anatomischen Strukturen aufzeigte. Die Ver-wendung von 3D-Datensätzen und der Einsatz von Navigationsverfahren erscheinen alsgeeignete Methoden mit Hilfe des MR-Kontrastmittels Gd-HSA Lymphbahnen undSLN intraoperativ gezielt aufzusuchen. Allerdings bedarf es auch für Gd-HSA einer Zu-lassungsstudie entsprechend dem Arzneimittelgesetz. Insgesamt aber kann vermutetwerden, daß besonders tumorbefallene drainierende Lymphknoten durch die endozytoti-sche Aufnahme der Albuminkonjugate TCPC/TCPP-HSA und/oder GD-HSA und derenlangen Verweildauer zur Darstellung gebracht werden (11).

8 5
Kapitel 6Perspektiven 6.1 Tumordiagnostik mit Gadolinium-HSAKooperation mit: Dr. Th. Egelhof, Neuroradiologische Abteilung, Universitätsklinikum Essen, Dr.F. Kiessling, Abteilung für Tumordiagnostik und Therapie, Deutsches KrebsforschungszentrumHeidelberg und Frau PD Dr. S. Heiland, Neuroradiologische Abteilung, UniversitätsklinikumHeidelberg
Berichte über die Konjugierung des MR-Kontrastmittels Gadolinium an humanesSerumalbumin (Gd-HSA) liegen bereits sei Ende der 80er Jahre vor (1,2). Ähnlich wiebei den Albuminbeladungen mit Methotrexat wurde, wohl um eine Erhöhung der Kon-trastdarstellung zu erreichen 18, 19, 24 oder 30 Moleküle Gadolinium pro Albuminmo-lekül geladen ( 1, 2, 3, 4, 5). Dies aber führt unweigerlich zu einer uns schon bekanntenAlteration von Albumin, so daß allergische Reaktionen und deutlich verkürzte Zirkula-tionszeiten eine Anwendung am Menschen nicht als möglich erscheinen lassen (6).Bislang wird Gadolinium-HSA vor allem zur Darstellung der Mikrozirkulation oder zurPerfusionsmessung verwendet (7, 8, 9).
Durch die von Sinn, DKFZ, entwickelte niedrige Beladungsrate von ca. 1 : 1 oder 3 : 1molarem Gd-HSA war nun aber eine Anwendung auch zur Tumordiagnostik möglich.Sowohl Egelhoff wie auch Kiesling gelangen lang anhaltende Tumordarstellungen mitdem niedermolar beladenen Gd-HSA bei malignen Rattengliomen wie auch bei Xe-notransplantaten an Nacktmäusen (10, 11). Allergische Reaktionen oder ein Abfangenvon Gd-HSA im retikuloendothelialen System oder der Leber wurden nicht beobachtet.
Nicht immer ist eine diagnostische Differenzierung zwischen benignen und malignenTumoren trotz Verwendung moderner bildgebender Verfahren möglich. Aber geradeder Hinweis, daß durch makromolekulare Kontrastmittel, welche von Tumorzellen viaEndozytose aufgenommen werden, eine Unterscheidung von benignen und malignenTumoren, wie z. B. dem Fibroadenom der Brust versus maligner Mammatumoren, läßtan eine unmittelbare klinische Anwendung denken (6, 12). Monokristalline Eisenparti-kel (MIONS) werden ebenfalls von Tumorzellen phagozytiert (13, 14) und können da-
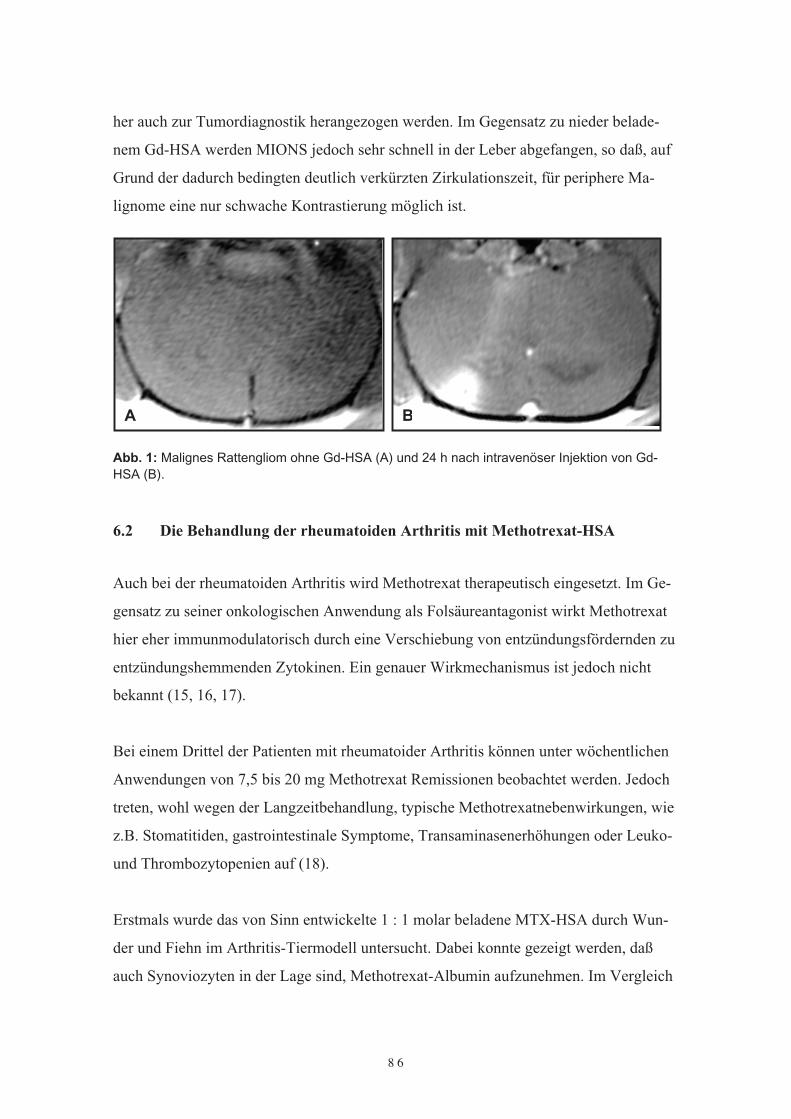
8 6
her auch zur Tumordiagnostik herangezogen werden. Im Gegensatz zu nieder belade-nem Gd-HSA werden MIONS jedoch sehr schnell in der Leber abgefangen, so daß, aufGrund der dadurch bedingten deutlich verkürzten Zirkulationszeit, für periphere Ma-lignome eine nur schwache Kontrastierung möglich ist.
Abb. 1: Malignes Rattengliom ohne Gd-HSA (A) und 24 h nach intravenöser Injektion von Gd-HSA (B).
6.2 Die Behandlung der rheumatoiden Arthritis mit Methotrexat-HSA
Auch bei der rheumatoiden Arthritis wird Methotrexat therapeutisch eingesetzt. Im Ge-gensatz zu seiner onkologischen Anwendung als Folsäureantagonist wirkt Methotrexathier eher immunmodulatorisch durch eine Verschiebung von entzündungsfördernden zuentzündungshemmenden Zytokinen. Ein genauer Wirkmechanismus ist jedoch nichtbekannt (15, 16, 17).
Bei einem Drittel der Patienten mit rheumatoider Arthritis können unter wöchentlichenAnwendungen von 7,5 bis 20 mg Methotrexat Remissionen beobachtet werden. Jedochtreten, wohl wegen der Langzeitbehandlung, typische Methotrexatnebenwirkungen, wiez.B. Stomatitiden, gastrointestinale Symptome, Transaminasenerhöhungen oder Leuko-und Thrombozytopenien auf (18).
Erstmals wurde das von Sinn entwickelte 1 : 1 molar beladene MTX-HSA durch Wun-der und Fiehn im Arthritis-Tiermodell untersucht. Dabei konnte gezeigt werden, daßauch Synoviozyten in der Lage sind, Methotrexat-Albumin aufzunehmen. Im Vergleich
A B
BA

8 7
zu freiem Methrotrexat war bei dem Konjugat nur ein Fünftel der Dosis notwendig, denEntzündungsprozess im Tiermodell günstig zu beeinflussen (19).
In Kenntnis der deutlich reduzierten systemischen Toxizität von MTX-HSA erscheinteine klinische Prüfung bei Patienten mit rheumatoider Arthritis vor allem in Hinblickauf die durch die Konjugierung deutlich verminderte systemische Toxizität als sehrvielversprechend.

8 8
Kapitel 77.1 Zusammenfassung
In der vorliegenden Arbeit wurde die Eigenschaft von Albumin als Carrier für verschie-dene diagnostische und therapeutische Vorhaben aufgezeigt.
7.1.1 Laserfluoreszenzdiagnostik mit 5-Aminofluorescein - Albumin (AFLc-HSA)
In tierexperimentellen und präklinischen Untersuchungen wurde der Fluoreszenzfarb-stoff AFLc-HSA vorgestellt, welcher in der intraoperativen Tumordiagnostik bei malig-nen Gehirntumoren im Rahmen einer Phase I/II-Studie untersucht werden soll. Die hoheLichtstärke von Aminofluorescein, sein geringes Ausbleichen und seine durch dieKonjugierung an Albumin bedingte lange intrazelluläre Verweildauer im Tumor bietenneue Ansätze in der intraoperativen Tumordiagnostik durch Laserfluoreszenz. Eine fürmaligne Gehirntumoren vorgesehene Phase-I/II-Studie wurde im April d. J. von derhiesigen Ethikkommission genehmigt und kann nun nach Erteilung der Prüfnummerdurch das Bundesministerium für Arzneimittel begonnen werden. Alle technischenVoraussetzungen hierfür sind erfolgt. Der Vergleich der laserinduzierten Fluoreszenzdurch AFLc-HSA mit Systemen der Neuronavigation, dem intraoperativen MR-Openund dem intraoperativen Ultraschall stellt für die Neurochirurgische UniversitätsklinikHeidelberg einen weiteren Schwerpunkt in der Bedeutung der intraoperativen Bildge-bung und Radikalitätskontrolle dar.
7.1.2 Albuminvermittelte Chemotherapie
Aminopterin-Albumin (AMPT-HSA)Tierexperimentell wurde das Antifolat Aminopterin gegenüber dem AlbuminkonjugatAMPT-HSA in seiner Pharmakokinetik wie auch Verträglichkeit und Wirksamkeit amWalker-256-Karzinom der Ratte untersucht. Unzweifelhaft bewirkt die Konjugierungvon Aminopterin an Albumin eine deutliche Reduzierung der systemischen Toxizitätdes Antifolates trotz der Tatsache, daß AMPT-HSA vielfach länger in Zirkulation

8 9
bleibt. Dies ist dadurch zu verstehen, daß Albumin oder Albuminkonjugate physiolo-gisch proliferierendes Gewebe nicht in der gleichen Quantität wie freies und niedermo-lekulares AMPT erreichen und die eigentliche Wirksubstanz erst nach der lysosomalenAbspaltung in der Tumorzelle freigesetzt wird. Neben Methotrexat-Albumin (MTX-HSA) steht nun mit AMPT-HSA ein weiteres hoch wirksames Antifolatkonjugat zurVerfügung, welches in Xenographtmodellen aber auch in klinischen Studien weiter un-tersucht werden kann.
Methotrexat-Albumin (MTX-HSA)Seit Februar 2000 befindet sich MTX-HSA in einer vergleichenden Phase-II-Studie beiPatienten mit primären und sekundären Glioblastomrezidiven. MTX-HSA wird ambu-lant wöchentlich bis zweiwöchentlich in einer Dosierung von 50 mg/m2 intravenös alsKurzinfusion appliziert und zeigt eine durch die Albuminkonjugierung bedingte deut-lich reduzierte systemische Toxizität im Vergleich zu freiem MTX. Bislang ist jedochkeine Überlegenheit in der Wirksamkeit gegenüber den Nitrosoharnstoffen BCNU undCCNU zu erkennen, auch wenn positive Effekte unter der Behandlung von MTX-HSAgesehen wurden. Überraschend ist jedoch die hohe Ansprechrate der Glioblastomrezidi-ve bei einem Wechsel der Studienmedikation bei insgesamt 5/6 Patienten, so daß über-legt werden muß, ob die klinische Prüfung mit MTX-HSA als Kombinationsbehandlungmit z. B. Nitrosoharnstoffen oder Temozolomid fortgeführt werden soll.
7.1.3 Albuminvermittelte Sentinel-Lymph-Node-Diagnostik
Entsprechend der Sentinel-Lymph-Node-(SNL)-Diagnostik, bei der vor allem kolloida-les 99mTechnetium-Albumin verwendet wird, zeigen die Albuminkonjugate Tetra(4-Carboxy-Phenyl-Chlorin- (TCPC) oder Tetra(4-Carboxy-Phenyl-Porphyrin TCPP-HSAeindrucksvoll die drainierenden Lymphwege und deren Lymphknoten unter Fluoreszen-zaktivierung. Eine unter Fluoreszenzbedingungen durchgeführte Lymphwegedissektionmit TCPP- und TCPC-HSA erlaubt eine exakte Zuordnung und anatomische Differen-zierung der Lymphstrukturen, ohne daß das Operationsgebiet, wie z. B. durch Patent-blau, permanent kontaminiert wird. Eine SLN-Diagnostik unter Zuhilfenahme dieserFluoreszenzkonjugate bei Mammakarzinomen, malignen Melanomen oder Kopf - Hals -

9 0
Tumoren, erscheint vielversprechend. Auch insofern, als daß durch eine ebenfalls nie-dermolare Konjugierung von Gadolinium an Albumin eine präoperative Lymphdiagno-stik kernspintomographisch als möglich erscheint.
7.2 Schlußfolgerung
Mit Albumin existiert ein vielfach verwendbarer und biologisch verfügbarer Carrier, derunterschiedliche Wirksubstanzen gezielt via Endozytose in malignes oder chronischentzündlich verändertes Gewebe einschleust. Eine grundsätzliche Voraussetzung für diekovalente Albuminkonjugierung ist jedoch eine schonende, d.h. 1 : 1 molare Beladung,so daß das Protein weder in seiner Tertiärstruktur noch in seiner biologischen Funktionoder Integrität verändert wird. Dies hätte nämlich deutlich verringerte Zirkulationszei-ten, ein Abfangen im retikuloendothelialen System oder der Leber und, bei wiederhol-ten Applikationen, allergische Reaktionen zur Folge. Zwischenzeitlich befinden sichbereits zwei Albuminkonjugate, nämlich MTX-HSA und AFLc-HSA, in klinischer Prü-fung. Und trotz vielfacher und oft wiederholter Anwendung ist für MTX-HSA in denmultizentrischen Phase - I/II - Studien bei über 100 Patienten bislang keine einzige all-ergische Reaktion beobachtet worden.
Gerade die niedermolar beladenen Albuminkonjugate erreichen in malignen oder chro-nisch entzündlich veränderten Geweben hohe und lang anhaltende Wirkstoffkonzentra-tionen. Zudem wird durch die kovalente Konjugierung an Albumin die sonst vor allembei niedermolekularen Zytostatika häufig beobachtete systemische Toxizität deutlichreduziert. Insgesamt erscheint das Konzept der niedermolaren kovalenten Albumin-konjugierung als vielversprechend für ganz unterschiedliche diagnostoische und thera-peutische Fragestellungen.

9 1
Literaturverzeichnis
Kapitel 1
1. Schwyzer R. Schöpfung und Entwicklung einer gezielten Chemotherapie: Paul Ehr-lich zum Gedächtnis. Angewandte Chemie 1954; 66:345-428.
2. Juliano RL. Targeted drug delivery. Berlin, Heidelberg: Springer Verlag, 1991.3. Poznansky MJ, Juliano RL. Biological approaches to the controlled delivery of
drugs: a critical review. Pharmacol Rev 1984; 36:277-336.4. Bertino JR. Antineoplastic drugs. In: Melmon KL, Morelli HF, Hoffman BB, Nie-
renberg DW, editors. Clinical pharmacology: basic principles in therapeutics. NewYork: Mc Graw Hill, 1992: 600-641.
5. Evans WE, Crom WR, Yalowich JC. Methotrexate. In: Evans WE, Schentag JJ,Jusko WJ, Harrison H, editors. Applied pharmacokinetics: principles of therapeuticdrug monitoring. Spokane, WA: Applied Therapeutics, Inc, 1986: 1009-1056.
6. Jain RK. Therapeutic implications of tumor physiology. Curr Opin Oncol 1991;3:1105-1108.
7. Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res1987; 47:3039-3051.
8. Takakura Y, Hashida M. Macromolecular drug carrier systems in cancer chemothe-rapy: macromolecular prodrugs. Crit Rev Oncol Hematol 1995; 18:207-231.
9. Duncan R. Drug-polymer conjugates: potential for improved chemotherapy. Anti-Cancer Drugs 1992; 3:175-210.
10. Folkman J. Tumor angiogenesis. Adv Cancer Res 1985; 43: 175-203.11. Aguayo A, Estey E, Kantarijan H et al. Cellular vascular endothelial growth factor
ia a predictior of outcome in patients with acute myeloid leukemia. Blood 1999; 94:3717-3721.
12. Chen CA, Cheng WF, Lee CN et al. Serum vascular growth factor in epithelial ova-rian neoplasmas: correlation with patient survival. Gynecol Oncol 1999; 74: 235-240.
13. Fiedler W, Graeven U, Ergün S et al. Vascular endothelial growth factor, a possibleparacrine growth factor in human acute myeloid leukemia. Blood 1997; 6: 1870 –1875.

9 2
14. Imoto H, Osaki T, Taga S et al. Vascular endothelial growth factor expression innon-small-cell lung cancer: prognostic significance in squamous cell Karzinoma. JThorac Cardiovasc Surg 1998; 115: 1007-1014.
15. Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated andsubcutaneous tumors: implications for therapy. Cancer Res 1990; 50:4478-4484.
16. Boucher Y, Jain RK. Microvascular pressure is the principal driving force for inter-stitial hypertension in solid tumors: implications for vascular collapse. Cancer Res1992; 52:5110-5114.
17. Gutmann R, Leunig M, Feyh J, Goetz AE, Messmer K, Kastenbauer E, Jain RK.Interstitial hypertension in head and neck tumors in patients: correlation with tumorsize. Cancer Res 1992; 52:1993-1995.
18. Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role ofinterstitial pressure and convection. Microvasc Res 1989; 37:77-104.
19. Jain RK. Determinants of tumor blood flow: a review. Cancer Res 1988; 48:2641-2658.
20. Sevick EM, Jain RK. Measurement of capillary filtration coefficient in a solid tu-mor. Cancer Res 1991; 51:1352-1355.
21. Sevick EM, Jain RK. Blood flow and venous pH of tissue-isolated Walker 256 Kar-zinoma during hyperglycemia. Cancer Res 1988; 48:1201-1207.
22. Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. II. Role ofheterogeneous perfusion and lymphatics. Microvasc Res 1990; 40:246-263.
23. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancerchemotherapy: mechanism of tumoritropic accumulation of proteins and the antitu-mor agent smancs. Cancer Res 1986; 46:6387-6392.
24. Jain RK. Haemodynamic and transport barriers to the treatment of solid tumours. IntJ Radiat Biol 1991; 60:85-100.
25. Jain RK. Physiological barriers to delivery of monoclonal antibodies and othermacromolecules in tumors. Cancer Res 1990; 50:814-819.
26. Jain RK. Delivery of novel therapeutic agents in tumors: physiological barriers andstrategies. J Natl Cancer Inst 1989; 81:570-576.
27. Jain RK. Vascular and interstitial barriers to delivery therapeutic agents in tumors.Cancer Metastasis Rev 1990; 9:253-266.
28. Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal anti-bodies and other macromolecules in tumors: significance of elevated interstitialpressure. Cancer Res 1988; 48:7022-7032.

9 3
29. Hwang KJ, Beaumier PL. Disposition of liposomes in vivo. In: Gregoriadis G, edi-tor. Liposomes as drug carriers: recent trends and progress. Chichester: John Wiley& Sons, 1988: 19-35.
30. Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta1992; 1113:171-199.
31. Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes:effect of dose. Biochim Biophys Acta 1991; 1068:133-141.
32. Kim S. Liposomes as carriers of cancer chemotherapy. Current status and futureprospects. Drugs 1993; 46:618-638.
33. Köhler G, Milstein C. Contineous cultures of fused cells secreting antibody of pre-defined specifity. Nature 1975; 256: 495-497.
34. Clynes RA, Towers TL, Presta LG, Ravetch JV: Inhibitory Fc receptors modulate invivo cytotoxicity against tumor targets. Nat Med 2000; 6: 443-446.
35. Flieger D, Renoth S, Beier I, Sauerbruch T, Schmidt-Wolf I: Mechanism of cytoto-xicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 inCD20-expressing lymphoma cell lines. Cell Immunol 2000; 204: 55-63.
36. Shin DM, Donato NJ, Perez-Soler R et al.: Epidermal growth factor receptor-targeted therapy with C225 and cisplatin in patients with head and neck cancer. ClinCancer Res 2001; 7: 1204-1213.
37. Cobleigh MA, Vogel CL, Tripathy D et al.: Efficacy and safety of HerceptinTM(humanized anti-Her2 antibody) as a single agent in 222 women with Her2 overex-pression who relapsed following chemotherapy for metastatic breast cancer. ProcAmer Soc Clin Oncol 1998; 17: 97a.
38. Slamon DJ, Leyland-Jones B, Shak S et al.: Use of chemotherapy plus a monoclonalantibody against HER2 for metastatic breast cancer that overexpresses HER2. NEngl J Med 2001; 344: 783-792.
39. Mendelsohn J, Baselga J: The EGF receptor family as targets for cancer therapy.Oncogene 2000; 19: 6550-6565.
40. Baselga J, Pfister D, Cooper MR et al.: Phase I studies of anti-epidermal growthfactor receptor chimeric antibody C225 alone and in combination with cisplatin. JClin Oncol 2000; 18: 904-914.
41. Goldenberg M: The role of radiolabeled antibodies in the treatment of non-Hodgkin's lymphoma:the coming of age of radioimmunotherapy. Crit Rev OncolHematol 2001; 39: 195-201.

9 4
42. Tolcher AW, Sugarman S, Gelmon KA et al.: Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol 1999;17: 478-484.
43. Abuchowski A, Kazo GM, Verhoest CR Jr, Van Es T, Kafkewitz D, Nucci ML,Viau AT, Davis FF. Cancer therapy with chemically modified enzymes. I. Antitu-mor properties of polyethylene glycol-asparaginase conjugates. Cancer BiochemBiophys 1984;7: 175-186.
44. Ettinger LJ, Kurtzberg J, Voute PA, Jurgens H, Halpern SL. An open-label, mul-ticenter study of polyethylene glycol-L-asparaginase for the treatment of acute lym-phoblastic leukemia. Cancer 1995; 75:1176-1181.
45. Graham ML, Asselin BL, Herndon JE 2nd, Casey JR, Chaffee S, Ciocci GH,Daeschner CW, Davis AR, Gold S, Halperin EC, Laughlin MJ, Martin PL, OlsonJF, Kurtzberg J. Toxicity, pharmacology and feasibility of administration of PEG-L-asparaginase as consolidation therapy in patients undergoing bone marrow trans-plantation for acute lymphoblastic leukemia. Bone Marrow Transplant 1998;21:879-885.
46. Vieira Pinheiro JP, Lanvers C, Boos J. Use of PEG-asparaginase in the treatment ofpatients with solid tumors. Cancer Chemother Pharmacol 2001 Nov;48(5):421-422.
47. Aguayo A, Cortes J, Thomas D, Pierce S, Keating M, Kantarjian H. Combinationtherapy with methotrexate, vincristine, polyethylene-glycol conjugated-asparaginase, and prednisone in the treatment of patients with refractory or recurrentacute lymphoblastic leukemia. Cancer 1999;86:1203-1209.
48. Fried MW, Shiffman ML, Reddy RK et al.: Pegylated (40 kDa) interferon alfa-2a(Pegasys) in combination with ribavirin: efficiacy and safety results from a phaseIII, randomized, actively-controlled, multicenter study. Gastroenterology 2001; 5Suppl. 1: 55.
49. Manns MP, McHutchison JG, Gordon SC et al.: Peginterferon alfa-2b plus ribavirincompared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepa-titis C: a randomised trial. Lancet 2001; 358: 958-965.
50. Egelhof T, Delbeck N, Hartmann M, Roth SU, Elste V, Heiland S, Sartor K. Cansuperparamagnetic contrast media improve MRI-tomographic images of experi-mental gliomas? Radiologe 1998;38: 943-947.
51. Zimmer C, Weissleder R, Poss K, Bogdanova A, Wright SCJ, Enochs WS: MRimaging of phagocytosis in experimental gliomas. Radiology 1995; 197: 533-538, .
52. Jain RK, Janda KD, Saltzman WM. Drug discovery and delivery. Mol Med Today1995; 1:4-4.
53. Carter DC and Ho JX. Structure of serum albumin. Adv Protein Chem 1994, 45:153-203.

9 5
54. Peters T. All about albumin: biochemestry, genetics and medical applications. SanDiego, New York: Academic Press, 1996.
55. Rothschild MA, Oratz M, schreiber SS. Extravasal albumin. N Eng J Med 1979;301: 497-498.
56. Rothschild MA, Oratz M, Schreiber SS. Albumin metabolism. Prog Liver Dis 1972;4:19-29.
57. Ballmer PE, McNurlan MA, Milne E, Heys SD, Buchan V, Calder AG, Garlic PJ.Measurement of albumin synthesis in humans: a new approach employing stableisoptopes. Am J Physiol 1990; 259: 797-803.
58. Yedgar S, Carew TE, Pittman RC, Belz WF, Steinberg D. Tissue sites of catabolismof albumin in rabbits. Am J Physiol 1983; 244: 101-107.
59. Strobel JL, Cady SG, Borg TK, Terracio L, Baynes JW, Thorpe SR. Identificationof fibroblasts as a major site of albumin catabolism in peripheral tissues. J BiolChem 1986; 261: 7989-7994.
60. Doweiko JP, Nompleggi DJ. Role of albumin in human physiology and pathophy-siology. JPEN J Parenter Enteral Nutr 1991; 15:207-211.
61. Feldmann G, Penaud Laurencin J, Crassous J, Benhamou JP. Albumin synthesis byhuman liver cells: its morphological demonstration. Gastroenterology 1972; 63:1036-1048.
62. Franz CP, Croze EM, Morre JD, Schreiber G. Albumin secreted by rat liver bapas-ses golgi apparatus cisternae. Acta Biochem Biophys 1981; 678: 395-402.
63. Le Rumeur E, Beaumont C, Guillouzo C, Rissel M, Bourel M, Guillouzo A. Allnormal rat hepatozytes produce albumin as a rate related to their degree of ploidy.Biochem Biophys Res Commun 1981; 101: 1038-1046.
64. Tstsumi T, Nakao K, Mitsuoka S, Hamasaki K, Tsuruta S, Shima M, Nakata K, Ta-maoki T, Nagataki S. Regulation of albumin and alpha-fetoprotein gene expressionby colloid osmotic pressure in human hepatoma cells. Gastroenterology 1993; 104:256-262.
65. Ancell H. Course of lectures on the physiology and pathology of the blood and theother animal fluids. Lancet 1839; I:222-231.
66. Weil MA, Hemming RJ, Puri VK. Colloid osmotic pressure: clinical significance.Crit Care Med 1979; 7: 113-116.
67. Kragh-Hansen U. Structure and ligand binding properties of human serum albumin.Dan Med Bull 1990; 37:57-84.

9 6
68. Reed RG, Burrington CM, The albumin receptor effect may be due to a surface-induced conformational change in albumin. J Biol Chem 1989; 264: 9867-9872.
69. Mider GB, Tesluk H, Morton JJ. Effects of Walker Karzinoma 256 on food intake,body weight and nitrogen metabolism of growing rats. Acta Union internat contracancrum 1948; 6:409-420.
70. Babson AL, Winnick T. Protein transfer in tumor-bearing rats. Cancer Res 1954;14:606-611.
71. Busch H, Fuchiwara E, Firsze DC. Studies on the metabolism of radioactive albu-min in tumor bearing rats. Cancer Res 1961; 21:371-377.
72. Busch H, Green HSN. Studies on the metabolism of plasma proteins in tumor bea-ring rats. The Yale Journal of Biology and Medicine 1954; 27:339 - 349.
73. Theologides A. Cancer cachexia. Cancer 1979; 43:2004-2012.74. DeWys WD. Pathophysiology of cancer cachexia: current understanding and areas
for future research. Cancer Res 1982; 42:721-726.75. Morrison SD. Cancer cachexia. In: Liotta LA, editor. Influence of tumor develop-
ment on the host. Dordrecht: Kluwer Academic Publishers, 1989: 176 - 213.76. Beck SA, Mulligan HD, Tisdale MJ. Lipolytic factors associated with murine and
human cancer cachexia. J Natl Cancer Inst 1990; 82:1922-1926.77. DeWys WD. Anorexia as a general effect of cancer. Cancer 1979; 43:2013-2019.78. Steinfeld JL. 131-I-albumin degradation in patients with neoplastic diseases. Cancer
1960; 13:974.79. Marcante ML. Uptake of 3H ascites albumin by Yoshida ascites tumor cells. Z
Krebsforsch Klin Onkol 1969; 72:305-311.80. Karlberg I, Edstrom S, Ekman L, Schersten T, Lundholm K. Reutilization of amino-
acid-carbons in relation to albumin turnover in non growing sarcoma bearing mice.Cancer Res 1982; 42:2284-2288.
81. Kremer P. Die Resorption von humanem Serumalbumin aus dem Dickdarm desKaninchens. Dissertation, Klinik für Allgemeine Chirurgie und Thoraxchirurgie derChristian-Albrechts-Universität Kiel, 1992.
82. Ehrenreich BA, Cohn ZA. The uptake and digestion of iodinated serum albumin bymacrophages in vitro. J Exp Med 1967; 126:941-949.
83. De Nardo GL, De Nardo SJ, Miyao NP, Mills SL, Peng JS, O'Grady LF, EpsteinAL, Young WC. Non-dehalogenation mechanisms for excretion of radioiodine afteradministration of labeled antibodies. Int J Biol Markers 1988; 3:1-9.

9 7
84. Pittman RC, Carew TE, Glass CK, Green SR, Taylor CAJ, Attie AD. A radioiodi-nated, intracellularly trapped ligand for determining the sites of plasma protein de-gradation in vivo. Biochem J 1983; 212:791-800.
85. Thorpe SR, Baynes JW, Chroneos ZC. The design and application of residualizinglabels for the studies of protein catabolism. FASEB J 1993; 7:399-405.
86. Sinn H, Schrenk HH, Friedrich EA, Via DP, Dresel HA. Radioiodination of proteinsand lipoproteins using N-bromosuccinimide as oxidizing agent. Anal Biochem1988; 170:186-192.
87. Sinn H, Schrenk HH, Friedrich EA, Schilling U, Maier Borst W. Design of com-pounds having an enhanced tumour uptake, using serum albumin as a carrier. Part I.Int J Rad Appl Instrum B 1990; 17:819-827.
88. Henderson LA, Baynes JW, Thorpe SR. Identification of the sites of IgG catabolismin the rat. Arch Biochem Biophys 1982; 215:1-11.
89. Chroneos ZC, Baynes JW, Thorpe SR. Identification of liver endothelial cells as theprimary site of IgM catabolism in the rat. Arch Biochem Biophys 1995; 319:63-73.
90. Schilling U, Friedrich EA, Sinn H, Schrenk HH, Clorius JH, Maier Borst W. Designof compounds having enhanced tumour uptake, using serum albumin as a carrier--Part II. In vivo studies. Int J Rad Appl Instrum B 1992; 19:685-695.
91. Wunder A, Stehle G, Sinn H, Schrenk HH, Hoff-Biederbeck D, Bader F, FriedrichEA, Peschke P, Maier-Borst W, Heene DL. Enhanced albumin uptake by rat tumors.Int J Oncol 1997; 11:497-507.
92. Stehle G, Wunder A, Schrenk HH, Hartung G, Heene DL, Sinn H. Distribution pat-tern of radiolabeled human, bovine, and rat albumin in tumor bearing rats. Anti-Cancer Drugs 1999; 10:405-411.
93. Stehle G, Sinn H, Wunder A, Schrenk HH, Schütt S, Heene DL, Maier-Borst W.The loading rate determines tumor targeting of methotrexate-albumin conjugates inrats. Anti-Cancer Drugs 1997; 8:677-685.
94. Magnenat G, Schindler R, Isliker H. Transport d'agents cytostatiques par lesprotéines plasmatiques: III. Activité antitumorale in vitro de conjugués cytostatique-azoprotéines. Europ J Cancer 1969; 5:33 - 40.
95. Shen WC, Ryser HJ. Selective killing of Fc-receptor-bearing tumor cells throughendocytosis of a drug-carrying immune complex. Proc Natl Acad Sci U S A 1984;81:1445-1447.
96. Bures L, Bostik J, Motycka K, Spundova M, Rehak L. The use of protein as a carri-er of methotrexate for experimental cancer chemotherapy. III. Human serum albu-min-methotrexate derivative, its preparation and basic testing. Neoplasma 1988;35:329-342.

9 8
97. Bures L, Lichy A, Bostik J, Spundova M. The use of protein as a carrier of me-thotrexate for experimental cancer chemotherapy. V. Alternative method for prepa-ration of serum albumin-methotrexate derivative. Neoplasma 1990; 37:225-231.
98. Kim CK, Hwang SJ, Lee MG. The organ targetability of small and large albuminmicrospheres containing free and HSA conjugated methotrexate. Int J Pharm 1993;89:91-102.
99. Sett R, Sarkar HS, Das PK. Pharmacokinetics and biodistribution of methotrexateconjugated to mannosyl human serum albumin. J Antimicrob Chemother 1993;31:151-159.
100. Sinn H, Schrenk HH, Maier-Borst W, Friedrich E, Graschew G, Elias P. Konju-gate aus tumoraktiver Verbindung und Protein sowie Verfahren zu deren Herstel-lung. DE 4122210; 1991.
101. Wunder A, Stehle G, Schrenk HH, et al. Antitumor activity of methotrexate-albumin conjugates in rats bearing a Walker-256 Karzinoma. Int J Cancer 1998; 76:884-890.
102. Stehle G, Wunder A, Schrenk HH, Hartung G, Heene DL, Sinn H. Methotrexate-albumin conjugate causes tumor growth delay in Dunning R3327 HI prostate can-cer-bearing rats. Anticancer Drugs 1999; 10: 405-411.
103. Rattel B, Breiter N, Biedermann E, Löser R, Wosikowski K. In vivo anti-tumoractivities of methotrexate conjugated to human serum albumin in human tumor xe-nografts. Clin Cancer Res 2000; 6(Suppl):4490.
104. Burger AM, Hartung G, Stehle G, Sinn H, Fiebig HH. Pre-clinical evaluation of amethotrexate-albumin conjugate (MTX-HSA) in human tumor xenografts in vivo.Int J Cancer 2001; 92: 718-724.
105. Hartung G, Stehle G, Sinn H, et al. Phase I trial of a methotrexate-albumin (MTX-HSA) conjugate in a weekly intraveneous bolus regimen in cancer patients. ClinCancer Res 1999; 5: 753-759.

9 9
Kapitel 2
1. Albert FK, Forsting M, Sartor K, Adams HP, Kunze S: Early postoperative magne-tic resonance imaging after resection of malignant glioma: objective evaluation ofresidual tumor and its influence on regrowth and prognosis. Neurosurgery 1994; 34:45- 61.
2. Forsting M, Albert FK, Kunze S, Adams HP, Zenner D, Sartor K: Radiologic fol-low-up after extirpation of glioblastoma: Residual tumor and regrowth pattern.AJNR 14: 77-87: 1993. Anderson C, Iresjo BM, Lundholm K: Identification of tis-sue sites for an increased albumin degradation in sarcoma-bearing mice. J Surg Res1991; 50: 156-162.
3. Becker G, Perez J, Krone A, Demuth K, Lindner A, Hofmann E, Winkler J,Bogdahn U: Transcranial color-coded real-time sonography in the evaluation of in-tracranial neoplasms and arteriovenous malformations. Neurosurgery 1992; 31: 420-427.
4. Koivukangas J, Louhisalmi Y, Alakuijala J, Oikarienen J: Ultrasound controlledneuronavigator-guided brain surgery. J Neurosurg 1993; 79: 36-42.
5. Okudera H, Kyoshina K, Kobayashi S, Sugita K: Intraoperative CT-scan findingsduring resection of glial tumors. Neurol Res 1994; 6: 265-267.
6. Tronnier VM, Wirtz CR, Knauth M, Lenz G, Pastyr O, Bonsannto MM, Albert FK,Kuth R, Staubert A, Schlegel W, Sartor K, Kunze S. Intraoperative diagnostic andinterventional MRI in neurosurgery. Neurosurgery 1997; 40: 891-902.
7. Wirtz CR, Bonsanto MM, Knauth M, Tronnier VM, Albert FK, Staubert A, KunzeS: Intraoperative magnetic resonance imaging to update interactive navigation inneurosurgery: Method and preliminary experience. Comput Aided Surg 1997; 2:172-179.
8. Wirtz CR, Knauth M, Staubert A, Bonsanto MM, Sartor K, Kunze S, Tronnier VM.Clinical evaluation and follow-up results for intraoperative magnetic resonanceimaging in neurosurgery. Neurosurgery 2000; 46:1112-1120.
9. Bonsanto MM, Staubert A, Wirtz CR, Tronnier V, Kunze S. Initial experience withan ultrasound-integrated single-RACK neuronavigation system. Acta Neurochir(Wien) 2001;143:1127-1132.
10. Moore GE, Peyton WT, French LA, Walker WW: The clinical use of fluorescein inneurosurgery. The localization of brain tumors. J Neurosurg 1948; 5: 392-398.

1 0 0
11. Benda P, Lightbody J, Sato G, Levine L, Sweet W: Differentiated rat glial cell strainin tissue culture. Science 1968; 16: 370-371.
12. Lee HB, Blaufox MD: Blood volume in the rat. J Nucl Med 1985; 26: 72-76.13. Brilmayer C, Cramer H, Schürmann K. Die Bedeutung bestimmter Farbstoffe in der
Diagnostik der Hirntumoren. Zentralbl Neuroch 1952; 4: 210-218.14. Klatzo I, Miquel J, Otenasek R: The application of fluorescein labeled serum pro-
teins (FLSP) to the study of vascular permeability in the brain. Acta Neruopath1962; 2: 144-160.
15. Chou SN, Aust JB,Moore GE, Peyton WT: Radioactive iodinated human serumalbumin a tracer agent for diagnosing and localizing intracranial lesions. Proc SocExp Biol Med 1951; 77: 193-195.
16. Wilcke O: Isotopendiagnostik in der Neurochirurgie. Acta Neurochirurgica (Suppl)XV 1966; 1-160.
17. Murray KJ: Improved surgical resection of human brain tumors: Part 1. A prelimi-nary study. Surg Neurol 1982; 17: 316-319.
18. Kabuto M, Kubota T, Kobayashi H, Nakagawa T, Ishii H, Takeuchi H, Kitai R, Ko-dera T. Experimental and clinical study of detection of glioma at surgery using fluo-rescent imaging by a surgical microscope after fluorescein administration. NeurolRes 1997; 19: 9-16.
19. Kuriowa T, Kajiamoto Y, Ohta T. Comparison between operative findings on ma-lignant glioma by fluorescein surgical microscopy and histological findings. NeurolRes1999, 21: 130-134.
20. Kuriowa T, Kajiamoto Y, Ohta T. Developement of a fluorescein operative micros-cope for the use during malignant glioma surgery. A technical note and preliminaryreport. Surg Neurol 1998; 50: 41-49.
21. Hansen DA, Spence AM, Carski T, Berger MS: Indocanine green (ICG) stainingand demarcation of tumor margins in a rat glioma model. Surg Neurol 1993; 40:451-456.
22. Haglund MM, Berger MS, Hochman DW: Enhanced optical imaging of humangliomas and tumor margins. Neurosurgery 1996; 38: 308-317.
23. Haglund MM, Hochman DW, Spence AM, Berger MS: Enhanced optical imagingof rat gliomas and tumor margins. Neurosurgery 1994; 35: 930-941.

1 0 1
24. Poon WS, Schomacker KT, Deutsch TF, Martuza RL: Laser-induced fluorescence:experimental intraoperative delineation of tumor resection margins. J Neurosurg1992; 76: 679-686.
25. Kaye AH, Morstyn G, Ashcroft R: Uptake and retention of hematoporphyrin deri-vative in an in vivo/in vitro model of cerebral glioma. Neurosurgery 1985; 17: 883-890.
26. Kostron H, Bellnier DA, Lin C-W, Swartz MR, Martuza RL: Distribution, retention,and phototoxicity of hematoporphyrin derivative in a rat glioma. J Neurosurg 1986;64: 768-774.
27. Perria C, Carai M, Falzoi A, Orunesu G, Rocca A, Massarelli G, Francaviglia N,Jori G: Photodynamic therapy of malignant brain tumors: Clinical results of diffi-culties with questions about and future prospects for the neurosurgical applications.Neurosurgery 1988; 23: 557-563.
28. Muller PJ and wilson BC. Photodynamic therappy for recurrent supratentorial glio-mas. Sem Surg Oncol 1995; 11: 347-354.
29. Stepp H, Baumgartner R, Beyer W et al. In: Optical Biopsies (Eds: Cubeddu R,Mordon SR, Svanberg K) Proc SPIE 1995; 2627: 13-24.
30. Stummer W, Stocker S, Novotny A et al. In vitro and in vivo porphyrin accummu-lation by C6 glioma cells after exposure to 5-aminolevulinic acid. J PhotochemPhotobiol B 1998; 45: 160-169.
31. Stummer W, Stepp H, Möller G et al. Technical principles for protoporphyrin-IX-fluorescence guided microsurgical resection of malignant glioma tissue. Acta Neu-rochir 1998; 140: 995-1000.
32. Stummer W, Stocker S, Wagner S, Stepp H, Fritsch C, Goetz C, Kiefmann R, Reu-len HJ: Intraoperative detection of malgnant gliomas by 5-aminolavulinic acid-induced porhyrin fluorescence. Neurosurgery 1998; 42: 518-526.
33. Stummer W, Novotny A, Stepp H, Goetz C, Bise K, Reulen HJ. Fluorescence-guided resection of glioblastoma multiforme by using 5-aminolevulinic acid-induced porphyrins: a prospective study in 52 consecutive patients. J Neurosurg2000; 93:1003-1013.
34. Hebeda KM, Saarnak AE, Olivo M, Sterenborg HJCM, Wolbers JG. 5-aminolavu-linic acid induced endogenous porphyrin fluorescence in 9L and C6 brain tumorsand in the normal rat brain. Acta Neurochirurg (Wien) 1998; 140: 503-513.
35. Kremer P, Wunder A, Sinn H, Haase T, Rheinwald M, Zillmann U, Albert FK,Kunze S. Laser-induced fluorescence detection of malignant gliomas using fluore-

1 0 2
scein-labeled serum albumin: Experimental and preliminary clinical results, NeurolRes 2000; 22: 481-489.

1 0 3
Kapitel 3
1. Farber S., Diamond LK, Mercer RD, Sylvester RF, Wolff JA. Temporary remissionsin acute leukemia in children produced by folic acid antagonist 4-aminopteroyl-glutamic acid (aminopterin). N Engl J Med 1948; 238: 787-793.
2. Bertino JR. Ode to methotrexate. J Clin Oncol 1993; 11: 5-14.3. Goldin A, Venditti JM, Humphreys SR, Don D, Mantel N, Greenhouse SW. A
quantitative comparison of the antileukemic effectiveness of two folic acid antago-nists in mice. J Natl Cancer Inst. 1955; 15: 1657-1664.
4. Garrow TA, Shane B. Purification and general properties of human folylpolyglut-amate synthetase. In: Ayling J.E.; Nair M.G. Baugh C.M. eds. Chemistry and Bio-logy of Pteridines and Folates. New York, NY, Plenum; 1993: 659-662.
5. Smith A, Hum M, Winick N, et al. A case for the use of aminopterin in treatment ofpatients with leukemia based upon metabolic studies of blasts in vitro. Clin CancerRes 1996; 2: 1-5.
6. Ratliff AF, Wilson JW, Hum M, et al. Phase I and pharmacokinetic trial of aminop-terin in patients with refractory malignancies. J Clin. Oncol 1998; 16: 1458-1464.
7. Jain RK. Delivery of novel therapeutic agents in tumors: physiological barriers andstrategies. J Natl Cancer Inst 1989; 81: 570-576.
8. Sinn H, Schrenk HH, Friedrich EA, Schilling U, Maier-Borst W. Design of com-pounds having an enhanced tumour uptake, using serum albumin as a carrier. Part I.Nucl Med Biol 1990; 17: 819 – 827.
9. Schilling U, Friedrich EA, Sinn H, Schrenk HH, Clorius JH, Maier-Borst W. Designof compounds having enhanced tumour uptake, using serum albumin as a carrier.Part II. In vivo studies. Nucl Med Biol 1992; 19: 685-695.
10. Smith GM. A comparison of the effects of cytotoxic agents on the Walker 256 tu-mour growing in the rat and at the hamster cheek pouch. Brit J Cancer 1969; 23: 88-94.
11. Geran RI, Greenberg NH, MacDonald MM, et al. Protocols for screening chemicalagents and natural products against tumor and other biological systems. CancerChemother. Rep. 1972; 3: 1-103.
12. Fogt F, Wan J, O’Hara C, et al. Flow cytometricmeasurement of cell cycle kineticsin rat Walker-256 Karzinoma following in vivo pulse labelling with bromodeoxyu-ridine. Cytometry 1991; 12: 33-41.

1 0 4
13. Stehle G, Sinn H, Wunder A, et al. Plasma protein (albumin) catabolism by the tu-mor itself - implications for tumor metabolism and the genesis of cachexia. Crit RevOncol Hematol 1997; 26: 77 - 100.Babson AL and Winnick T. Protein transfer intumor-bearing rats. Cancer Res 1954; 14: 606-611.
14. Biedermann E, Fujisawa-Deutschland; Investigator Meeting, Amsterdam, 2001(personal communication).
15. Weigand M, Hartung G, Roboz J, Sieger S, Wolf M, Sinn H, Schrenk HH, WiesslerM, Frei E. Mode of action of methotrexate-albumin in a human T-cell leukemia lineand activity against an MTX-resistant clone. Anticancer Drug Des 2001;16: 227-237.
16. Hammer JA, Rannels DE. Protein turnover in pulmonary macrophages. Utilizationof amino acids derived from protein degradation. Biochem J 1981; 198:53-65.
17. Besterman JM, Airhart JA, Low RB, Rannels DE. Pinocytosis and intracellular de-gradation of exogenous protein: modulation by amino acids. J Cell Biol 1983;96:1586-1591.
18. Magnenat G, Schindler R, and Isliker H. Transport d'agents cytostatiques par lesprotéines plasmatiques: III. Activité antitumorale in vitro de conjugués cytostatique-azoprotéines. Europ J Cancer 1969; 5: 33-40.
19. Jacobs SA, D‘Urso-Scott M, Bertino JR. Some biochemical and pharmacologic pro-perties of amethopterin-albumin. Ann N Y Acad Sci 1971; 186: 284-286.
20. Chu BC, Fan CC, Howell SB. Activity of free and carrier-bound methotrexateagainst transport-deficient and high dihydrofolate dehydrogenase-containing me-thotrexate-resistant L1210 cells. J Nat Cancer Inst 1981; 66: 121 - 124.
21. Halbert GW, Florence AT, Stuart JF. Characterization of in-vitro drug release andbiological activity of methotrexate-bovine serum albumin conjugates. J PharmPharmacol 1987; 39: 871-876.
22. Bures L, Bostik J, Motycka K, Spundova M, Rehak L. The use of protein as a carri-er of methotrexate for experimental cancer chemotherapy. III. Human serum albu-min-methotrexate derivative, its preparation and basic testing. Neoplasma 1988; 35:329-342.
23. Hartung G, Stehle G, Sinn H, et al. Phase I trial of a methotrexate-albumin (MTX-HSA) conjugate in a weekly intraveneous bolus regimen in cancer patients. ClinCancer Res 1999; 5: 753-759.
24. Kremer P, Wunder A, Sinn H, et al. Laser-induced fluorescence detection of ma-lignant gliomas using fluorescein-labeled serum albumin: Experimental and preli-minary clinical results. Neurol Res 2000; 22: 481-489.

1 0 5
25. Kremer P, Hartung G, Bauder-Wüst U, Schrenk HH, Wunder A, Heckl S, ZillmannU, Sinn H: Efficacy and tolerability of an aminopterin-albumin conjugate in tumor-bearing rats. Anti-Cancer-Drugs 2002; 13: 615-623.
26. Loadman PM, Bibby M, Double JA, et al. Pharmacokinetics of PK1 and doxorubi-cin in experimental colon tumor models with differing responses to PK1. Clin. Can-cer Res 1999; 5: 3682-3688.
27. Muggia FM. Doxorubicin-polymer conjugates: further demonstration of the conceptof enhanced permeability and retention. Clin Cancer Res 1999; 5: 7-8.
28. Vasey PA, Kaye SB, Morrsion R, et al. Phase I clinical and pharmacokinetic studyof PK1 (HPMA co-polymer doxorubicin): first member of a new class of chemothe-rapeutic agents – drug polymer conjugates, Clin. Cancer Res. 1999; 5; 83-94.
29. Pommerenke EW, Sinn H, Volm M. Cirumvention of Doxorubicin-resistance intumours by albumin-conjugated Doxorubicin. Eur J Cancer 1995; 31: 283-284.

1 0 6
Kapitel 4
1. Hartung G, Stehle G, Sinn H, et al. Phase I trial of a methotrexate-albumin (MTX-HSA) conjugate in a weekly intraveneous bolus regimen in cancer patients. ClinCancer Res 1999; 5: 753-759.
2. Vis N, van-der-Gast A, van-Rhijn-G, Catsburg K, Schimdt C, Mickisch J. A phaseII trial of methotrexate-human serum albumin (MTX-HSA) in patients with meta-static renal cell Karzinoma who progressed under immunotherapy. Cancer Chemo-ther Pharmacol 2002; 49: 342-345.
3. Scheulen ME, Gatzemeier U, v. Pawel J, Hossfeld DK, Kaukel E, Mross K, HartungG, Gastl G, Burk K, Schmidt C, Queisser W. Clinical phase II study of methotrexatebound to human serum albumin (MTX-HSA) in first line chemotherapy of patientswith advanced malignant mesothelioma (a study with participation of CESAR-EWIV). J Cancer Res Clinical Oncol (Suppl) 2002; 142.
4. Kornblith PL, Walker M. Chemotherapy for malignant gliomas. J Neurosurg 1988;68: 1-17.
5. Fine HA. The basis for current treatment recommondations for malignant gliomas. JNeuro Oncol 1994; 20: 111-120.
6. Fine HA, Dear KB, Loeffler JS, Black PM, Canellos GP. Meta-Analysis of radiationtherapy with and without adjuvant chemotherapy for malignant gliomas in adults.Cancer 1993;71: 2585-2597.
7. Thomas DGT, Bleehen NM, Roberts JT, Abram P, Brada M, Lantos PL, Moss TH,Ironside JW, Whaley JB and Stenning SP for the MRC Brain Tumor Working Party,Vcambidge UK. MRC randomized trial of adjuvant chemotherapy in high gradglioma. J Neuro Oncol 1998; 39: 102.
8. Mahaley MS. Neuro-oncology index and review (adult primary brain tumors). JNeuro Oncol 1991; 11: 85-147.
9. Fortin D, Cairncross GJ, Hammond RR. Oligodendroglioma: An appraisal of recentdata pertaining to diagnosis and treatment. Neurosurgery 1999; 45: 1279-1291.
10. Albert FK, Forsting M, Sartor K, Adams HP, Kunze S: Early postoperative magne-tic resonance imaging after resection of malignant glioma: objective evaluation ofresidual tumor and its influence on regrowth and prognosis. Neurosurgery 1994; 34:45-61.
11. Wirtz CR, Knauth M, Staubert A, Bonsanto MM, Sartor K, Kunze S, Tronnier VM.Clinical evaluation and follow-up results for intraoperative magnetic resonanceimaging in neurosurgery.Neurosurgery. 2000; 46: 1112-20.

1 0 7
1 2 . Levin VA, Wilson CB:. Nitrosoureas: Clinical and experimental considerations inthe treatment of brain tumors. In: Hellmann K, Conners TA (ed.): Chemotherapy,Vol 7 New York, Plenum press, 1977, 277-283.
13. Yung A, Levin VA, Albright R, Olson MH, Fredericks R, Fink K, Prados M, BradaM, Spence A, Brunner J, Yue N, Dugan MH, Zaknoen S and Temodal Brain TumorGroup. Randomized trial of temodal(TEM) vs. Procarbazine (PCB) in glioblastomamultiforme (GBM) at first relaps. Proc Am Soc Clin Oncol 1999; 18: 139.
14. Chabner BA, Clenndennin N, Curt GA, Jolivet J. Biochemistry of methotrexate. In:Kimura K, Wang YM, editors. Methotrexate in cancer therapy. New York: RavenPress, 1983: 1-38.
15. Huffman DH, Wan SH, Azarnoff DL, Hogstraten B. Pharmacokinetics of me-thotrexate. Clin Pharmacol Ther 1973; 14:572-589.
16. DeAngelis LM, Yahalom J, Heinemann MH, Cirricione C, Thaler HT, Krol G: Pri-mary CNS lymphoma: Combined treatment with chemotherapy and radiotherapy.Neurology 1990; 40: 80-86.
17. Wilson CB and Norreil HA. Brain tumor chemotherapy with intrathecal me-thotrexate. Cancer 1969; 23: 1038 – 1044.
18. Djerassi I, Kim JS. High-dose methotrexate-citrovorum factor rescue in the mana-gement of brain gliomas. Cancer Treat Rep 1977; 61: 691-694.
19. Rosen G, Ghavimi F, Vanucci R. Poutrin glioma: High-dose methotrexate and leu-covorin rescue. JAMA 1974; 230: 1149-1152.
20. Garfield J, Dayan AD. Postoperative intracavitary chemotherapy of malignant glio-mas. A preliminary study using methotrexate. J Neurosurg 1973; 39: 315-322.
21. Tator CH, Wassenaar W. Intraneoplastic injection of methotrexate for experimentalbrain-tumor chemotherapy. J Neurosurg 1977; 46: 165-174.
22. Nierenberg D, Harbaugh R, Maurer LH, Reeder T, Scott G, Fratkin J, Newman E.Continuous intratumoral infusion of methotrexate for recurrent glioblastoma: a pilotstudy. Neurosurgery. 1991; 28: 752-761.
23. Wowra B, Zeller WJ, Remmele T, Sturm V, Stricker H. Local methotrexate che-motherapy with polylactide implants for experimental rat glioma. Reg Cancer Treat1989; 2: 26-31.
24. Rama B, Mandel T, Jansen J, Dingeldein E, Mennel HD. The intraneoplastic che-motherapy in a rat brain tumour model utilizing methotrexate-polymethyl-methacrylate-pellets. Acta Neurochir (Wien). 1987; 87: 70-75.

1 0 8
25. DeAngelis LM, Yahalom J, Thaler HAT, Kher U: Combined modality therapy forprimary CNS lymphoma. J Clin Oncol 1992; 10: 635-645.
26. Thiel E, Korfel A. Successful treatment of non-Hodgin’s lymphoma of the CNSwith the BMPD chemotherapy protocol followed by radiotherapy. Exp Hematol1997; 25: 846.
27. Wang AM, Elion GB, Friedman HS, Bodell WJ, Bigner DD, Schold SC Jr. Positivetherapeutic interaction between thiopurines and alkylating drugs in human gliomaxenografts. Cancer Chemother Pharmacol 1991; 27: 278-284.

1 0 9
Kapitel 5
1. Kaindl, F, Mannheimer E, Pfleger-Schwarz L, Thurnher B: Lymphangiographie undLymphadenographie der Extremitäten. Thieme, Stuttgart 1960.
2. Cabanas RM: An approach for the treatment of penile Karzinoma: Cancer 1997; 39:456 -466.
3. Morton DL, Wen DR, Wong JH, et al.: Technical details of intraoperative lymphaticmapping for early stage melanoma: Arch Surg 1992; 127: 392-399.
4. Alex JC, Krag DN. Gamma-probe-guided localization of lymph nodes. Surg Oncol1993; 2: 137-144.
5. Bachter D, Balda BR, Vogt H, Büchels H: Die „sentinel“-Lymphonodektomie mit-tels Szintillationsdetektor. Hautarzt 1996; 47: 754-758.
6. Krag DN, Meijer SJ, Weaver DL. Minimal-access surgery for staging of malignantmelanoma: Arch Surg 1995; 130: 654-658.
7. Pijpers R, Borgstein PJ, Meijer S, Hoekstra OS, Van Hattum LH, Teule GJJ. Senti-nel node biopsy in melanoma patients: Dynamic lymphoscintigraphy followed byintraoperative gamma probe and vital dye guydance. World J Surg 1997; 21: 788-793.
8. Reintgen D, Curse CW, Wells K et al. The orderly progression of melanoma nodalmetastases. Ann Surg 1994; 220: 759-767.
9. Heidenreich P, Vogt H, Bachter D, Büchels H., Steinfeld D, Wawroschek F, Wen-genmair H., Wagner T. Das Konzept des Wächterlymphknotens. DÄ 2001; 9: 534 –540.
10. Faries MB, Bedrosian I, Reynolds C, Nguyen HQ, Alavi A, Czerniecki BJ. Activemacromolecule uptake by lymph node antigen-presenting cells: A novel mechanismin determining sentinel lymph node status. Ann of Surg Oncol 2000; 7: 98-105.
11. Bedrosian I, Scheff AM, Mick R, Callans LS, Bucky LP, Spitz FR, Helsabeck C,Elder DE, Alavi A, Fraker DF, Czerniecki BJ: 99mTC-human serum albumin: An ef-fective radiotracer for identifying sentinel lymph nodes in melanoma. J Nucl Med1999; 40: 1143-1150.

1 1 0
Kapitel 61. Schmiedl U, Ogan M, Paajanen H, Marotti M, Crooks LE, Brito AC, Brasch RC.
Albumin labeled with Gd-DTPA as an intravascular, blood pool-enhancing agent forMR imaging: biodistribution and imagingn studies. Radiology 1987; 162: 205-210.
2. Schmiedl U, Moseley ME, Ogan MD, Chew WM, Brasch RC. Comparison of initi-al biodistribution patterns of Gd-DTPA and albumin-(Gd-DTPA) using rapid spinecho MR imaging. J Comput Assist Tomogr 1987; 11: 306-313.
3. Roberts HC, Saeed M, Roberts TP, Muhler A, Shames DM, Mann JSA, Stiskal M,Demsar F, Brasch RC. Comparison of albumin-(Gd-DTPA) 30 and GD-DTPA-24-cascade-polymer for measurements of normal and abnormal microvascular permea-bility. J Magn Reson Imaging 1997; 7: 331-338.
4. Daldrup H, Shames DM, Wendland M, Okuhate Y, Link TM, Rosenau W, Lu Y,Brasch RC. Correlation of dynamic contrast-enhanced magnetic resonance imagingwith histologic tumor grade: comparison of macromolecular and small-molecularcontrast media. Pediatr Radiol 1998; 28: 67-78.
5. Ogan MD, Schmiedl U, Moseley ME, Grodd W, Paajanen H, Brasch RC. Albuminlabeled with Gd-DTPA. An intravascular contrast-enhancing agent for magnetic re-sonance blood pool imaging: Preparation and characterization. Invest Radiol 1987;22: 665-671.
6. Daldrup, HE, Roberts TPL, Mühler A, Gossmann A, Roberts HC, Wendland M,Rosenau W, Brasch RC. Makromolekulare Kontrastmittel für die MR-Mammographie. Ein neuer Ansatz für die Charakterisierung von Mammatumoren.Radiologe 1997; 37: 733 - 740.
7. Mellin AF, Cofer CP, Smith BP, et al. Three dimensional magnetic resonancemicroangiography of rat neurovasculare. Magn Reson Med 1994; 32: 199-205.
8. Schwickert HC, Stiskal M, van Dijke CF, et al. Tumor angiography using high-resolution, three-dimensional magnetic resonance imaging: comparison of gado-pentetate dimeglumine and a macromolecular blood-pool contrast agent. Acad Ra-diol 1995; 2: 851-858.
9. Schmiedl U, Brasch RC, Ogan MD, et al. Albumin labeled with Gd-DTPA. An in-travascular contrast-enhancing agent for magnetic resonance blood pool and perfu-sing imaging. Acta Radiol 1990; Suppl 274: 99-102.
10. Kiessling F, Fink C, Hansen M, Bock M, Sinn H, Schrenk HH, Krix M, Egelhof T,Fusenig NE, Delorme S. Magnetic resonance imaging of nud mice with heterotrans-planted high-grade squamous cell Karzinomas. Use of a low-loaded, covalentlybound Gd-Hsa conjugate as contrast agent with high tumor affinity. Invest Radiol2002; 37: 193-198.

1 1 1
11. Egelhof T (unpublished data)12. Su MY, Wang Z, Carpenter PM, Lao X, Muhler A, Nalciouglu O. Characterization
of N-ethyl-N-nitrosourea-induced malignant and benign breast tumors in rats byusing three MR contrast agents. J Magn Reson Imaging 1999; 9: 177-186.
13. Zimmer C, Weissleder R, Poss K, Bogdanova A, Wright SCJ, Enochs WS: MRimaging of phagocytosis in experimental gliomas. Radiology 1995;197: 533-538.
14. Egelhof T, Delbeck N, Hartmann M, Roth SU, Elste V, Heiland S, Sartor K. Cansuperparamagnetic contrast media improve MRI-tomographic images of experi-mental gliomas? Radiologe 1998; 38: 943-947.
15. Bannwarth B, Labat L, Moride Y, Schaeverbeke T. Methotrexate in rheumatoidarthirtis. An update. Drugs 1994; 47: 25-50.
16. Kremer JM. The mechanism of action of methotrexate in rheumatoid arthirtis: thesearch continues. J Rheumatol 1994; 21: 1-5.
17. Cronstein BN. The mechanism of action of methotrexate. Rheum Dis Clin NorthAm 1997; 23: 739-755.
18. Schnabel A, Gross WL. Low-dose methotrexate in rheumatic dieseases-efficacy,side effects, and risk factors for side effects. Semin Arthritis Rheum 1994; 23: 310-327.
19. Wunder A. Habilitationsschrift der Ruprechts-Karls-Universität Heidelberg, Medi-zinische Fakultät Klinikum Mannheim, 2000.

1 1 2
Danksagung
Zum Schluß dieser Arbeit möchte ich all denen danken, die mir mit ihren Ideen oder mitRat und Tat zur Seite standen.Allen voran danke ich Herrn Professor Dr. Kunze, meinem akademischen Lehrer, der esmir ermöglicht hat, diese Arbeit zu verwirklichen.Die Idee, Resttumorgewebe intraoperativ anzufärben, stammte von Herrn Professor Dr.Albert, jetzt Paracelsus-Klinik Osnabrück. Durch ihn kam der Kontakt zu Herrn Dr.Sinn, DKFZ, zu Stande, der das Konzept der niedermolaren Albuminkonjugierung ent-wickelte. In zahlreichen Stunden wurden verschiedene Projekte besprochen und durch-geführt. Nun darf Herr Dr. Sinn sich stolz Vater zweier vom DKFZ initiierter Phase-I/II-Studien nennen. Ebenfalls danke ich seinen Mitarbeitern, Herrn Schrenk, Frau Bau-der-Wüst wie auch den Herren Dr. Haase und Dr. Wunder, für ihre wertvolle Zusam-menarbeit.Ohne die großzügige Unterstützung von Herrn Dr. Zillmann, Leiter des Zentralen Tier-labores des DKFZ, und seinen Mitarbeitern, den Herren Dähmel, Eskersky und Lehr,wären die zahlreichen und zeitaufwendigen tierexperimentellen Untersuchungen nichtmöglich gewesen.Auch danke ich Herrn PD Dr. Hartung, Onkologische Abteilung der UniversitätsklinikRostock, sowie Frau Dr. Frei, DKFZ, für ihre wertvollen Anregungen im Hinblick aufdie albuminvermittelte Chemotherapie.Danken möchte ich auch allen meinen Kollegen aus der Neurochirurgischen Universi-tätsklinik, die mir vor allem bei der Durchführung der klinischen Studien zur Seite stan-den.Der Dank gilt besonders meiner Familie, meiner Frau für das Schreiben und meinemSchwiegervater, Herrn Dr. Inhoffen, für die aufmerksame Durchsicht des Manuskriptes.Ganz im Sinne von „euntes eunt et plorant...“ gilt mein aufrichtiger Dank den Patientenund ihren Angehörigen. Denn auch sie tragen in Geduld und Hoffnung zu dem medizi-nischen Fortschritt, sei es bei individuellen Heilversuchen oder klinischen Phase-I/II-Studien, einen großen Anteil bei.


![3. Fluoreszenz- und UV/VIS-spektroskopische ......49 3. Fluoreszenz- und UV/VIS-spektroskopische Untersuchungen der dargestellten Imidazo[4,5-c]carbazole, Carbazole und Phenazine 3.1.](https://static.fdokument.com/doc/165x107/60b7520dfe96d2539945bd31/3-fluoreszenz-und-uvvis-spektroskopische-49-3-fluoreszenz-und-uvvis-spektroskopische.jpg)















