
Allgemeine Chemie In.ethz.ch/~nielssi/download/1. Semester/Allgemeine Chemie I (OC... · benutzt...
148
HS 2011 Allgemeine Chemie I Organische Chemie Einführung in die organische Chemie Klassische Strukturlehre, Stereochemie, die chemische Bindung, Symmetrielehre, organische Thermochemie, Konformationsanalyse. Prof. Dr. Peter Chen Dr. Andreas Bach
Transcript of Allgemeine Chemie In.ethz.ch/~nielssi/download/1. Semester/Allgemeine Chemie I (OC... · benutzt...

HS 2011
Allgemeine Chemie I Organische Chemie
Einführung in die organische Chemie Klassische Strukturlehre, Stereochemie, die chemische Bindung, Symmetrielehre, organische Thermochemie,
Konformationsanalyse.
Prof. Dr. Peter Chen Dr. Andreas Bach

2 Skript AC-OC I Vorlesung: Freitag, 7.45 – 9.30 HCI G3
Übungsstunden: Montag, 8.45 – 9.30 HCI D2
HCI J3 HIT F31.2 10.45 – 11.30 HIT F12 HIT F31.2 HCI J8 Freitag, 14.45 – 15.30 HCI J6
Übungsassistenten/innen: Lukas Fritsche [email protected]
David Ringger HCI G220 Tel.: 044 632 4359 [email protected]
Nicolas Bennett [email protected]
Felicitas Flohr [email protected]
Florian Krausbeck [email protected]
Raffael Huber [email protected]
Michael Lerch [email protected]

KAPITEL 1:
EINFÜHRUNG In diesem Kapitel werden folgende Themen behandelt: 1.1. Organische Chemie 1.2. Zielsetzung der Vorlesung
Entdeckung des Elements Phosphor durch Hennig Brand 1669

4 Skript AC-OC I
1.1. Organische Chemie Die organische Chemie beschäftigt sich mit den Beziehungen zwischen Struktur, Funktion und Reaktivität von kohlenstoffhaltigen Verbindungen. Diese Verbindungen bestehen aus einem Grundgerüst aus C- und H-Atomen (Ketten, Ringe oder Netzwerke) an dem weitere Elemente wie z. B. O, N, S, P, F, Cl, Br, I, usw. gebunden sein können. Die praktisch unendliche Zahl von möglichen Kombinationen dieser Elemente führt zu einer Vielzahl von organischen Verbindungen. Dies beinhaltet z. B. alle Plastike, natürliche und synthetische Fibern (Wolle, Baumwolle, Nylon, ...), die meisten Farbstoffe, Heilmitteln, Pestizide, Aromen, Parfums sowie alle petrochemische Produkte (Benzin, Diesel, ...). Weiter bestehen Nahrungsmittel meistens aus organischen Komponenten wie Kohlenhydraten, Fette, Proteine und Vitamine. Diese Beispiele erläutern, wie verbreitet organische Moleküle in unserem alltäglichen Leben sind. Der Name "Organische Chemie" bezieht sich auf lebende Organismen. In der Tat waren Pflanzen und Tiere während vieler Jahrzehnte die einzige Quelle von organischen Verbindungen. Heutzutage werden sie meistens ausgehend von Erdöl oder anorganischen Verbindungen synthetisiert. Im März 2003 waren 8.6 Millionen organische Verbindungen bekannt. Die erste Synthese eines organischen Molekül wurde 1828 durch Friedrich Wöhler publiziert. Er stellte Harnstoff aus Ammoniak und Knallsäure her:
NH3 + NCOH H2NCONH2 1845 synthetisierte Hermann Kolbe Essigsäure durch Elektrolyse aus C, FeS2 und Cl2:
CFeS2 CS2
Cl2 CCl4
H3CCOOH
C2Cl4
Cl3CCO2H
heisses
Rohr
Licht, H2O
e–, H2O
Seitdem wurde eine Vielzahl von Methoden entdeckt, die organische Verbindungen ineinander umwandeln. Die Kenntnis dieser Verfahren ist das Ziel der späteren organisch-chemischen Vorlesungen. Die Struktur einer organischen Verbindung ist essentiell für das Verständnis seiner Eigenschaften, wie z. B. seine Farbe, sein Geschmack, sein Geruch, seine biologische Aktivität, seine Reaktivität usw. Um die Wichtigkeit, die Struktur der Verbindungen zu kennen, können die Perkin-Geschichte und die Herstellung von Vanillin sehr nützlich sein.
1.2. Zielsetzung der Vorlesung Im Rahmen dieser Vorlesung werden wir uns mit der Struktur von organischen Verbindungen beschäftigen. Wir werden verschiedene Modelle diskutieren, die während der Zeit vorgeschlagen wurden, um die experimentellen Beobachtungen zu erklären. Während dieser Vorlesung werden, nach der Einführung der klassischen Konzepte der Strukturlehre, die Grundlagen des aktuellsten Molekularmodells besprochen. In der Folge werden

Skript AC-OC I 5 die energetischen Aspekte von Molekülen erklärt. Der physikalische Gesichtspunkt von Umwandlungen zwischen chemischen Verbindungen wird am Ende diskutiert. In diesem Skript wird die Energieeinheit Kalorie (cal) verwendet, da sie in der organischen Chemie weit verbreitet ist, obwohl sie vom Internationalen System nicht empfohlen wird. 1 cal = 4.184 J = 2.61·1019 eV. Dieses Skript ist als Hilfsmittel zur Erleichterung des Verständnisses der Vorlesung gedacht. Die wichtigen Konzepte, die bekannt sein müssen, sind hier enthalten. Zusatzmaterial und Beispiele sind im Anhang „Beispiele, Bemerkungen und Vertiefungen zum Skript AC-OC I“ enthalten und können durch Links abgerufen werden. Weiter können aufgrund der limitierten Zeit einige Beispiele und Teilkapitel nicht besprochen werden. Einige Themen, wie z. B. die Nomenklatur und die Symmetrielehre werden ausschliesslich in den Übungen besprochen. Es ist daher empfohlen, immer den Vorlesungs- und Übungsstunden zu folgen und sich nicht nur auf dieses Skript zu stützen. Als Prüfungsstoff dienen alle Themen, die in der Vorlesung und den Übungen behandelt werden.

6 Skript AC-OC I
KAPITEL 2:
NOMENKLATUR In diesem Kapitel werden folgende Themen behandelt: 2.1. Terminologie 2.2. Kohlenwasserstoffe 2.2.1. Gesättigte offenkettige Kohlenwasserstoffe 2.2.2. Ungesättigte offenkettige Kohlenwasserstoffe 2.2.3. Monozyklische Kohlenwasserstoffe 2.2.4. Verbrückte polyzyklische Kohlenwasserstoffe 2.2.5. Kondensierte polyzyklische Kohlenwasserstoffringe 2.2.6. Spirokohlenwasserstoffe 2.2.7. Einfach- oder doppelbindungsverknüpfte Kohlenwasserstoffringe 2.3. Heterocyclen 2.3.1. Trivialnamen 2.3.2. Austauschnomenklatur 2.3.3. Hantzsch-Widman-System 2.4. Funktionelle Derivate der Kohlenwasserstoffe 2.4.1. Substitutive Nomenklatur 2.4.2. Funktionsklassennamen 2.4.3. Bestimmung der Hauptkette funktionalisierter Verbindungen 2.5. Exemplifizierung der Nomenklatur funktionalisierter Verbindungen 2.5.1. Kationen 2.5.2. Carbon- und Sulfonsäuren 2.5.3. Carbonsäurederivate 2.5.4. Sulfonsäurederivate 2.5.5. Aldehyde und Thioaldehyde 2.5.6. Ketone und Derivate 2.5.7. Alkohole, Phenole, Thiole und Derivate 2.5.8. Amine, Hydroxylamine und Imine 2.5.9. Ether, Epoxide, Sulfide, Sulfoxide und Sulfone 2.5.10. Halogenide 2.5.11. Azide, Isocyanide, Nitroso- und Nitroverbindungen

Skript AC-OC I 7 Ziel dieses Kapitels ist die kompakte Darstellung der grundlegenden Prinzipien der organisch-chemischen Nomenklatur. Dabei wird bewusst auf allzu komplizierte und spitzfindige Beispiele sowie die ausführliche Behandlung der entsprechenden Regeln verzichtet. Wichtig ist im Rahmen dieser Vorlesung vielmehr die Schulung des Vermögens, Strukturelemente zu erkennen, zu benennen und zu einem Ganzen zusammenzufügen. Ist dieses Ziel erreicht, wird man in komplizierten praktischen Fällen schnell anhand der erforderlichen Regeln den korrekten Namen für eine Verbindung konstruieren, bzw. aus diesem die Struktur eines Moleküls ableiten können. Als praktische Orientierungshilfe für die im Rahmen dieser Vorlesung erforderlichen Kenntnisse dienen in erster Linie die Übungsaufgaben. Von Bedeutung sind vor allem die rationellen Verbindungsnamen. Die Trivialnamen, die sehr wichtig sind und bekannt sein müssen, sind explizit im Text enthalten. Die vollständigen Listen dienen nur als Hilfsmittel für eventuelle zukünftige Nomenklaturprobleme und müssen nicht gelernt werden. Die Welt der Nomenklatur ist ständig im Wandel und man muss immer berücksichtigen, dass in einigen Fällen, wie z. B. bei Chemical Abstract, die benutzte Nomenklatur leicht verschieden sein kann (siehe z. B. Handbuch für die systematische Nomenklatur der organischen Chemie, metallorganischen Chemie und Koordinationschemie). In diesem Teil des Skriptes werden nur die wichtigsten Nomenklaturregeln gemäss der IUPAC Empfehlungen 1993 aufgelistet.
2.1. Terminologie Zum bessern Verständnis der folgenden Ausführungen werden an dieser Stelle einige Definitionen von häufig benutzten Begriffen aufgelistet. Trivialnamen und systematische (oder rationelle) Namen Trivialnamen sind individuelle Namen einzelner Verbindungen, die keine systematische Beziehung zu deren Struktur haben. Diesem Nachteil steht die Einfachheit gegenüber, die vor allem beim Benennen von grossen, komplexen Molekülen geschätzt wird. Systematische Namen setzen sich dagegen aus speziellen Silben für Stammnamen und Affixe, aus den Namen der Substituenten sowie aus Positionsangaben zusammen. Aus diesen Informationen kann man die chemische Struktur der Verbindungen abzuleiten. Ein Nachteil systematischer Namen ist, dass bei grossen Molekülen rasch sehr kompliziert werden können. Beispiele. Stammverbindungen Sehr oft kann man sich vorstellen, dass einige Verbindungen aus wenigen Stammverbindungen durch Ersetzen von H-Atomen durch andere Atome oder Atomgruppen abgeleitet werden können. Der Name der Stammverbindung ist deshalb die Basis (Stammname), auf die der Name der tatsächlichen Verbindung aufgebaut wird. Z. B. darf CH3F als Derivat der Stammverbindung CH4 (Methan) betrachtet werden und wird deshalb Fluormethan genannt. Substituenten Als Substituenten bezeichnet man Atome oder Atomgruppen, die H-Atome der Stammverbindung ersetzen. In CH3F ist das F-Atom der Substituent. In Abhängigkeit von der Zahl der Bindungen, die den Substituenten an die Stammverbindung binden, kann er einwertig, zweiwertig, usw. sein. Reste Reste sind über ein Kohlenstoffatom gebundene Substituenten. Sie können selbst wiederum substituiert sein und werden oft mit R abgekürzt.

8 Skript AC-OC I Funktionelle Gruppe (oder charakteristische Gruppe) Atomgruppen, die Heteroatome (Atome, die nicht C oder H sind) enthalten und häufig in organischen Verbindungen vorkommen, werden als funktionelle Gruppe bezeichnet. Sie verleihen einer Verbindung ihre charakteristischen Eigenschaften, darunter insbesondere ihre chemische Reaktivität. Beispiele funktioneller Gruppen sind –OH für Alkohole oder –Br für Bromide. Lokanten Zur Lokalisierung von Substituenten, Bindungen und Verknüpfungsstellen innerhalb einer Struktur benutzt man Zahlen, Buchstaben oder Präfixe, die man als Lokanten bezeichnet. Affixe, Präfixe und Suffixe Affixe sind definierte Silben, die zur Beschreibung von charakteristischen Strukturmerkmalen benutzt werden. Man unterschiedet zwischen Präfixen (Vorsilben) und Suffixen (Nachsilben). Z. B. bezeichnet das Suffix –al Aldehyde, während das Präfix cyclo- typisch für cyclische Verbindungen ist.
2.2. Kohlenwasserstoffe 2.2.1. Gesättigte offenkettige Kohlenwasserstoffe Lineare Alkane Die gesättigten Kohlenwasserstoffe gehören zur Klasse der Alkane (oder Paraffine). Die ersten vier unverzweigten Alkane besitzen Halbtrivialnamen (Trivialteil + Suffix -an), während die höheren durch systematische Namen (griechisches oder lateinisches Zahlwort + Suffix -an) benannt werden. CH4 Methan H3C-(CH2)4-CH3 Hexan H3C-CH3 Ethan H3C-(CH2)5-CH3 Heptan H3C-CH2-CH3 Propan H3C-(CH2)6-CH3 Octan H3C-CH2-CH2-CH3 Butan H3C-(CH2)7-CH3 Nonan H3C-(CH2)3-CH3 Pentan H3C-(CH2)8-CH3 Decan Zur besonderen Hervorhebung der Tatsache, dass ein Alkan unverzweigt ist, wird es oft als n-Alkan (z. B. n-Octan oder n-C8H18) bezeichnet. Der Name von n-Alkanen mit mehr als zehn C-Atomen wird aus zwei Teilen gebildet: ein Präfix stellt den Einer dar, während der Zehner durch den zweiten Teil wiedergegeben ist (vgl. auch die deutsche Schreibweise von Nummern).
Einer Zehner 1 hen- 10 Decan 2 do- 20 Cosan3 tri- 30 Triacontan 4 tetra- 40 Tetracontan5 penta- 50 Pentacontan6 hexa- 60 Hexacontan 7 hepta- 70 Heptacontan8 octa- 80 Octacontan 9 nona- 90 Nonacontan 100 Hectan
Ausnahmen dieser Regel sind Undecan (n-C11H24), Icosan (n-C20H42) und Henicosan (n-C21H44).

Skript AC-OC I 9 Die Namen der abgeleiteten einwertigen Reste werden durch Änderung des Suffixes -an durch -yl gebildet. Beispiele. Die Reste Methyl-, Ethyl-, Propyl- und Butyl- werden sehr oft mittels Me, Et, Pr bzw. Bu abgekürzt (z. B. CH3OH = MeOH). Verzweigte Alkane Um verzweigte Alkane zu benennen, werden sie als Derivate einer Hauptkette betrachtet. Ausgehend von der Zahl Reste, die an einem C-Atom gebunden sind, werden die Kohlenstoffatome in primären (H3CR), sekundären (H2CRR'), tertiären (HCRR'R'') und quaternären Zentren (CRR'R''R''') geteilt. Für die Festlegung der Hauptkette von Kohlenwasserstoffen gelten die folgenden nach Priorität geordneten Bedingungen. Die Hauptkette ist diejenige mit:
1. den meisten Mehrfachbindungen; 2. den meisten Atomen (längste Kette); 3. den meisten Doppelbindungen; 4. den meisten Seitenketten; 5. dem niedrigsten Lokantensatz für Substituenten; 6. den meisten C-Atomen in kleineren Seitenketten; 7. den am wenigsten verzweigten Seitenketten.
Die letzten zwei Bedingungen müssen nur in sehr seltenen Fällen erfüllt werden. Die Namen der Seitenketten werden in Form der einwertigen Reste dem Namen der Hauptkette vorangestellt. Die Hauptkette muss dann nummeriert werden. Die Nummerierung erfolgt in der Weise, dass man den niedrigsten Lokantensatz erhält. Zur Ermittlung desselben vergleicht man die verschiedene Sätze miteinander. Sobald ein Unterschied gefunden wird, gilt derjenige Satz als der niedrigste, der den kleinsten Lokanten an dieser Stelle aufweist. Z. B. der Satz 1,2,6,7,7,9,11 ist gegenüber 1,3,3,4,8,9 bevorzugt. Wenn zwei oder mehrere Seitenketten in äquivalenten Positionen sind, wird der niedrigere Lokant für die alphabetisch niedrigere Kette gewählt. Der Name der Hauptkette dient als Stammname. Die Namen der Seitenketten werden in alphabetischer Reihenfolge, jeweils mit den entsprechenden Lokanten versehen, als Präfixe vorgestellt. Die Lokanten müssen nur eingeführt werden, wenn die Position der Substituenten nicht eindeutig ist. Die erste Buchstabe des Gesamtnamens wird im Deutschen gross geschrieben, alle weiteren Teile müssen klein geschrieben werden. Zahlen und Buchstaben werden durch Bindestriche voneinander getrennt, was i. A. nicht auf verschiedene Namenteile zutrifft. Wenn dieselbe Seitekette mehrmals anwesend ist, werden die multiplikativen Präfixe di-, tri-, tetra-, penta-, hexa-, usw. vorangestellt. Jede Seitekette erhält ihren eigenen Lokanten. Die multiplikativen Präfixe müssen nicht für die alphabetische Aufzählung der Substituenten berücksichtig werden. Z. B. Dimethyl- besitzt eine niedrigere Priorität als Ethyl-. Beispiele. Wenn die Seitenketten weitere Verzweigungen besitzen, wird analog obiger Hierarchie verfahren. Innerhalb der Seitenkette wird eine "Hauptkette" bestimmt, usw. Man muss beachten, dass die Nummerierung immer an der Verknüpfungsstelle mit der Hauptkette beginnt. Um zu zeigen, dass es sich bei der verzweigten Seitenkette um eine zusammengehörende Substruktur handelt, werden ihre Namenbestandteile in eine runde Klammer gesetzt. Die Klammern werden benutzt, wenn Untereinheiten wiederum aus Einzelbestandteilen aufgebaut sind. Wenn komplexe Hierarchien weitere Klammern erforderlich machen, muss man die Untereinheiten mit eckigen und weiter geschweiften Klammer umhüllen. Wenn noch weitere Klammern nötig sind, beginnt man wiederum mit runden Klammer, usw.

10 Skript AC-OC I Verzweigte Untereinheiten besitzen eigene multiplikative Präfixe: Es wird bis-, tris-, tetrakis-, pentakis-, hexakis-, usw. mitverwendet. Sie werden der entsprechenden Klammer ohne Bindestrich vorangestellt. Obwohl die multiplikativen Präfixe bei der alphabetischen Ordnung in der Regel nicht mitgezählt werden, ist bei zusammengesetzten Substituenten der erste Buchstabe massgebend, auch wenn er ein Zahlwort ist. Beispiele. Einige verzweigte Alkane und Reste behalten Trivialnamen bei. Daraus werden einige Reste durch spezielle Präfixe abgeleitet. Die Beispiele, die bekannt sein müssen, sind hier gegeben:
Isopropyl- Isobutyl-
~ ~
sec-Butyl-
~
tert-Butyl-
~
Im Gegensatz zu den Präfixen iso- und neo-, müssen sec- und tert- nicht bei der alphabetischen Aufstellung berücksichtig werden, sie sind aber immer kursiv geschrieben. Isopropyl-, Isobutyl- und tert-Butyl- werden oft mittels iPr, iBu und tBu abgekürzt. Beispiele. 2.2.2. Ungesättigte offenkettige Kohlenwasserstoffe Die ungesättigte Kohlenwasserstoffe werden nach Alkenen (eine oder mehrere Doppelbindungen) und Alkinen (eine oder mehrere Dreifachbindungen) unterschieden. Die Endung -an der gesättigten Ketten wird durch das Suffix -en bzw. -in ersetzt. Wenn eine Verbindung mehrere Doppel- oder Dreifachbindungen besitzt, werden die Endungen -adien, -atrien, ... bzw. -adiin, -atriin, ... verwendet. Die Doppelbindungen besitzen eine höhere Priorität bezüglich der Dreifachbindungen und werden immer zuerst geschrieben. Die Lage der ungesättigten Bindungen wird in Form eines Lokanten des ersten an der Mehrfachbindung beteiligten C-Atoms angegeben und dem entsprechenden Suffix vorangestellt. Für die Bezifferung einer ungesättigten Hauptketten gelten die Regeln des Kapitels 2.2.1. Bei der Zusammensetzung des Gesamtnamens geht man wie bei den Alkanen vor. Die Seitenketten werden durch Anhängen der Endsilbe -yl gebildet und ausgehend von der Verknüpfungsstelle mit der Hauptkette beziffert. Beispiele. Einige Alkene und Alkine sowie einige Reste behalten Trivialnamen bei (vollständige Liste):
C
Allyl-
Kohlenwasserstoffe:
Allen (Propadien)
Acetylen (Ethin)
Reste:
Vinyl- (Ethenyl-)
(Prop-2-enyl-)~
~
Die Benützung von Ethylen statt Ethen ist nicht mehr erlaubt. Neben einwertigen Resten existieren auch zwei-, drei- und mehrwertige Substituenten.

Skript AC-OC I 11 Eine erste Klasse mehrwertiger Reste besteht aus Substituenten, die mehrmals durch Einfachbindungen an der Hauptkette gebunden sind. Um den Namen zu bilden, werden Stammname, Lokanten, und multiplikative Silben vor das Suffix -yl geschrieben. Eine Ausnahme ist –CH2–, das den Trivialnamen Methylen- besitzt. Ethylen- (–CH2–CH2–, Ethan-1,2-diyl-) kann auch benützt werden. Beispiele. Zweiwertige unverzweigte Resten werden noch oft durch deren alte Nomenklatur benannt, obwohl sie nach IUPAC nicht mehr erlaubt ist. Die Namen zweiwertiger Reste, die durch eine Doppelbindung an der Hauptkette gebunden sind, werden durch Änderung der Endung -yl der relativen einwertigen Substituenten mit -yliden gekennzeichnet. Methyliden- (H2C=) darf auch durch den Trivialnamen Methylen- gekennzeichnet werden. Dies wird aber nicht empfohlen, um Methyliden- von –CH2– zu unterscheiden. Einige häufige zweiwertige Reste besitzen eigene Trivialnamen. Name endständiger dreiwertiger Reste sind durch Austausch von -yl durch -ylidin gebildet. Beispiele mehrwertiger Reste. 2.2.3. Monozyklische Kohlenwasserstoffe Monocyclische Kohlenwasserstoffe und deren Reste werden durch den Präfix Cyclo- gekennzeichnet. Ansonst erfolgt die Namensbildung in der üblichen Weise. Beispiele. Zur "Cyclohexatrienderivaten" und deren Resten werden Trivial- oder Halbtrivialnamen zugeordnet. Fundamentale Beispiele sind Benzol (C6H6) und Phenyl- (–C6H5 = –Ph). Um disubstituierte Verbindungen zu bezeichnen wird sehr oft eine spezielle Konvention angewendet: der Buchstabe o (ortho) wird für 1,2-Substitution verwendet, m (meta) für 1,3 und p (para) für 1,4:
1,2-Diethylbenzolo-Diethylbenzol
1,3-Diethylbenzolm-Diethylbenzol
1,4-Diethylbenzolp-Diethylbenzol
Zudem werden einigen substituierten Benzolringen Trivialnamen zugeordnet. In der folgenden Abbildung sind die wichtigste Beispiele von monozyklischen Kohlenwasserstoffen und relative Reste eingetragen (vollständige Liste):

12 Skript AC-OC I
HC CH C6H5C6H5
Toluol*Benzol*
m-Xylol**(auch o- und p-Xylol)
Mesitylen** Cumol**
Styrol* Stilben*
Fulven**
~
Mesityl-**
~
Trityl-*
Benzyl-* o-Tolyl-**(auch m- und p-Tolyl-)
~
~Phenyl-*
~
*) Darf auch für am Ring substituierte Derivate angewendet werden. **) Darf nur für unsubstituierte Verbindungen angewendet werden.
Zweiwertige Substituenten werden gemäss der üblichen Nomenklatur identifiziert, ausser zweiwertige Benzolreste, die o-, m- oder p-Phenylen- genannt werden: ~ ~ ~
~
~ ~
o-Phenylen- m-Phenylen- p-Phenylen- Zyklische Kohlenwasserstoffe mit azyklischen Ketten können grundsätzlich sowohl als kettensubstituierte Ringsysteme als auch als ringsubstituierte Ketten behandelt werden. Im allgemein verfährt man so, dass die Grundstruktur möglichst viele Substituenten trägt oder die

Skript AC-OC I 13 kleinere Einheit als Substituent der grösseren betrachtet wird. Oft wird in solchen Fällen auch der einfachste Name gewählt, oder derjenige, der den chemischen Absichten am besten entspricht. Beispiele. 2.2.4. Verbrückte polyzyklische Kohlenwasserstoffe Gesättigte cyclische Kohlenwasserstoffe mit zwei oder mehr Ringen, in denen mindestens zwei Ringe wenigstens zwei gemeinsame C-Atome aufweisen, werden als bi-, tri-, tetra-, usw. -cycloalkane bezeichnet. Die Anzahl der Ringe ergibt sich aus der Zahl hypothetischer C–C-Spaltungen, die notwendig sind, um eine offenkettige Verbindung zu erhalten. Die mehreren Ringen gemeinsamen Atome werden als Brückenköpfe bezeichnet. Zur Festlegung des Verbindungsnamens geht man wie folgt vor:
1. Im dreidimensionalen Formelbild oder einer geeigneten planaren Projektion wird derjenige Ring als Hauptring definiert, der die meisten C-Atome enthält.
2. Die längstmögliche C-Kette, die zwei C-Atome des aus den zwei Zweigen bestehenden Hauptrings zusätzlich miteinander verbindet, wird als Hauptbrücke festgelegt, die entsprechenden Verknüpfungsgellen heissen Hauptbrückenköpfe.
3. Wenn mehrere Brücken dieselbe Länge besitzen, ist die Hauptbrücke diejenige, die den Hauptring so symmetrisch wie möglich teilt.
4. Alle anderen Brücken werden als Sekundärbrücken bezeichnet. Ihre Verknüpfungsstellen heissen Nebenbrückenköpfe. Unabhängige Sekundärbrücken binden Brückenkopfe, die zum Hauptring oder zur Hauptbrücke gehören. Die anderen heissen abhängige Sekundärbrücken.
5. Die Bezifferung der C-Atome beginnt an einem Hauptbrückenkopf und läuft innerhalb des Hauptrings auf dem längsten Weg über den zweiten Hauptbrückenkopf dahin zurück. Dann folgt die Hauptbrücke. Die Sekundärbrücken werden fortlaufend – unabhängig von ihrer Länge – der Reihe sinkender Brückenkopf-Lokanten nach weiter nummeriert. Die Bezifferung beginnt jeweils beim höher nummerierten Brückenkopf. Abhängige Sekundärbrücken werden zuletzt nummeriert.
6. Die Lokanten der Nebenbrückenköpfe sollen so niedrig wie möglich sein. Der Name wird gemäss folgender Ordnung zusammengesetzt: erstens kommt das multiplikative Präfix gefolgt durch -cyclo-, dann in eckigen Klammern die Anzahl C-Atome der verschiedenen Zweigen und letztlich der Name des Stammalkans. In den eckigen Klammern wird erstens die Anzahl der C-Atome des längsten Zweiges des Hauptringes geschrieben, gefolgt durch diejenige des kürzeren Zweigs, der Hauptbrücke, der unabhängigen und schliesslich der abhängigen Sekundärbrücke. Alle diese Zahlen sind durch Punkte getrennt. Die Lokanten der Brückenköpfe der Sekundärbrücken müssen als Superskripte (durch Komma getrennt) angegeben sein. Unabhängige Sekundärbrücken werden in der Reihenfolge absteigender Länge und zunehmender Lokanten angegeben, während die abhängigen Sekundärbrücken unabhängig von ihrer Länge nach abnehmenden Lokanten geordnet werden müssen. Beispiele. Wenn diese Regeln nicht ausreichen, um eine Verbindung eindeutig zu benennen, können weitere Regeln und Beispiele in den IUPAC Empfehlungen 1999 (G. P. Moss, Pure Appl. Chem. 1999, 71, 513) gefunden werden. Bei Derivaten verbrückter polyzyklischer Kohlenwasserstoffe (Systeme mit Mehrfachbindungen oder Substituenten) wird bei der Zusammenstellung des Gesamtnamens analog verfahren wie bei den offenkettigen Verbindungen. Bei der Bezifferung des Grundgerüsts ist darauf zu achten, dass – sofern es nach Anwendung der soeben beschriebenen Regeln noch mehrere Möglichkeiten gibt –

14 Skript AC-OC I für entsprechende Strukturelemente niedrigste Lokanten zu wählen sind. Für den Fall dass ein verbrücktes polyzyklisches Kohlenwasserstoffgerüst als Rest auftritt, erhält die Verknüpfungsstelle den niedrigsten Lokanten, der mit der Nummerierung des Grundgerüsts vereinbar ist. Wenn eine Doppelbindung zwei Atome verknüpft, die nicht aufeinander folgend nummeriert sind, muss man beide Lokanten der Atome schreiben. Beispiele. Einige verbrückte Polycyclen behalten Trivialnamen bei. Polycyclen, die Doppelbindungen enthalten, besitzen oft eine spezielle Nomenklatur (vgl. Kapitel 2.2.5). 2.2.5. Kondensierte polyzyklische Kohlenwasserstoffe Die rationelle Nomenklatur polyzyklischer kondensierter (oder anellierter) Kohlenwasserstoffe ist kompliziert und für die Ziele dieser Vorlesung nicht wichtig (G. P. Moss, Pure Appl. Chem. 1998, 70, 143). Nur die Trivialnamen einiger wichtiger Grundgerüste müssen gelernt werden. Die Gerüste besitzen normalerweise eine festgesetzte Bezifferung, die unabhängig von eventuellen Substituenten ist. Dies gilt auch für den Fall, dass sie als Reste auftreten. Im Folgenden sind die Trivialnamen und die Nummerierungen drei wichtiger anellierter polyzyklischer Kohlenwasserstoffe wiedergegeben (vollständige Liste):
1
2
3
45
6
7
8
Naphthalin
1
2
3
4
5
6
7
8
9
10
PhenanthrenAnthracen
1
2
3
45
6
7
8 9
10
8a
4a
8a
10a
9a
4a
8a
4b4a
10a
Die davon abgeleiteten Reste heissen Naphthyl-, Anthryl- und bzw. Phenanthryl-. Trägt ein Gerüst in Position 1 einen Rest, so kann man auch von -Substitution sprechen, im Fall der Position 2 von -Substitution, usw. Die Positionen 1 und 8 sowie 4 und 5 von Naphthalin können als peri-Positionen bezeichnet werden. Kondensationsstellen werden nicht fortlaufend nummeriert, sondern sie bekommen die Nummer des vorherigen Atoms und einen Buchstaben (a, b, usw.). Ausser in einem Fall, ist die Benützung von Trivialnamen für gesättigte und partiell ungesättigte Derivate kondensierter Polycyclen nicht mehr erlaubt. Sie werden als Produkte der Hydrierung der vollständig ungesättigten Stammverbindungen betrachtet. Die Nummerierung wird beibehalten und die Ringatome, die nicht mehr an Doppelbindungen beteiligt sind, werden durch das Präfix hydro- gekennzeichnet. Beispiele. 2.2.6. Spirokohlenwasserstoffe Eine polyspirozyklische Verbindung liegt vor, wenn Ringe über ein einziges Zentrum miteinander verbunden sind. Das Atom, das die zwei Ringe verknüpft, wird als Spiroatom definiert. Im Rahmen dieser Vorlesung werden nur Monospirocyclen behandelt. Für eine ausführlichere Betrachtung vgl. die IUPAC Empfehlungen 1999 (G. P. Moss, Pure Appl. Chem. 1999, 71, 531). Monospirocyclen enthalten nur zwei miteinander gebundene Ringe. Ihr Name wird durch wie folgt zusammengesetzt: das Präfix spiro-, die Gliederzahlen der das Spiroatom überbrückenden C-Ketten in eckigen Klammern und schliesslich der Name des Stammalkans. Anders als bei den verbrückten polyzyklischen Kohlenwasserstoffen beginnt die Nummerierung von Spirosystemen an einer dem

Skript AC-OC I 15 Spiroatom benachbarten Position und verläuft dann über den kleineren Zweig und das Spiroatom in den grösseren Zweig. Bei Derivaten von Spirocyclen sind in Übereinstimmung mit der genannten Bezifferungsregel die kleinsten Lokanten für entsprechende Strukturelemente zu wählen. Beispiele. 2.2.7. Einfach- oder doppelbindungsverknüpfte Kohlenwasserstoffringe Im Fall von über Einfach- oder Doppelbindungen verknüpften Kohlenwasserstoffringen muss man generell einen Ring als Grundkomponente und die restlichen als Substituenten betrachten. Wenn aber die Komponenten ähnlich sind, tritt eine spezielle Nomenklatur in Kraft. Sind zwei identische monozyklische Kohlenwasserstoffe oder zwei Benzolderivate über eine Einfachbindung miteinander verknüpft, spricht man von bi-...-yl-Derivaten. In den anderen Fällen darf man auch bi- vor den Kohlenwasserstoffnamen setzen. Die Lokanten der Verknüpfungspositionen müssen vorangestellt werden, ausser wenn sie schon zur Benennung der Substituenten explizit angegeben waren. Die Nummerierung ist für die zwei Nomenklaturmethoden unterschiedlich. Im ersten Fall muss der Verknüpfungspunkt den tiefstmöglichen Lokanten bekommen. Im zweiten Fall behalten die Teilstrukturen ausnahmslos ihre Nummerierung, die ohne Berücksichtigung der Ring-Ring-Verknüpfungsstelle festgelegt sein muss. Um die Ringe zu unterscheiden, wird die Nummerierung des einen Systems mit Apostrophen versehen. In der Regel wird derjenige mit der höher nummerierten Verknüpfungsstelle apostrophiert. Beispiele. Wenn die Ringe über Doppelbindungen verbunden sind, muss man normalerweise einen Ring als Stammsystem und die anderen als Substituenten betrachten. Im Fall von zwei identischen Ringen darf man aber auch das Suffix –yliden benützen. Beispiele. Bei drei oder mehr identischen Cyclen setzt man die Präfixe ter-, quarter-, quinque-, sexi-, septi-, usw. vor den Stammnamen. Ketten aus über Einfachbindungen verknüpften Benzolringen werden durch die Trivialnamen Bi-, Tri-, Tetra-, usw. -phenyle bezeichnet. Einem der endständigen Ringe werden Ziffern ohne Apostroph zugeordnet, während die folgenden der Reihe nach mit einfachen, zweifachen, usw. Apostrophen versehen werden. Die Verknüpfungen sollen so niedrig wie möglich sein. Ein Doppelpunkt dient im Namen als Separator zwischen nicht direkt miteinander verknüpften Positionen. Beispiele. Sind nicht identische Ringsysteme über Einfach- und Doppelbindungen miteinander verbunden, so wird ein Zyklus als Stammsystem und die anderen Ringe als Substituenten betrachtet. Die Wahl des Stammsystems erhält man in abnehmender Priorität:
1. nach der Anzahl der Ringe; 2. nach dem grössten vorhandenen Ring; 3. nach dem höchstens Grad der Unsättigung; 4. nach der Liste der beibehaltenen Trivialnamen (vgl. Literatur).
Beispiele.
2.3. Heterocyclen Eine zyklische Struktur, die Heteroatome enthält, nennt man einen Heterocyclus. Die Nomenklatur ist nicht einfach. IUPAC schlägt zwei alternative Methoden vor, die zudem sehr viele Trivialnamen erlauben. Die Nomenklatur von kondensierten Heterocyclen ist noch komplexer und wird hier nicht besprochen. Heterocyclen haben gegenüber den entsprechenden Kohlenwasserstoffen höhere Priorität.

16 Skript AC-OC I 2.3.1. Trivialnamen Häufig vorkommende Heterocyclen besitzen eigene Trivialnamen. Ihre Bezifferung ist definiert, so dass die Heteroatome die kleinstmöglichen Lokanten erhalten. Heteroatome, die sich an einer Kondensationsstelle befinden, werden im Gegensatz zu den polyzyklischen Kohlenwasserstoffe fortlaufend durchnummeriert. Die folgenden wichtigen Verbindungen besitzen Trivialnamen (eine vollständigere Liste ist in jedem Nomenklaturbuch zu finden):
OHN SO
N
HN
HN
HN
O N N
N N
N N
HN
Purin(7H-Purin)
Piperidin Morpholin Pyridin Pyrimidin
Furan Tetrahydrofuran(THF)
Pyrrol Imidazol Thiophen
1
3
1
3
1
3
1
3
1
3
1
31
4
1
3
1
3 1
2
39
5
4
67
8
Reste, die ausgehend von diesen Verbindungen abgeleitet werden, sind durch Anhängen der üblichen Endsilben -yl, -diyl, -yliden, usw. gekennzeichnet. Die Verknüpfungsstelle muss unter Erhaltung der definierten Nummerierung den tiefstmöglichsten Lokant bekommen. Der Lokant muss vor dem Suffix -yl angegeben werden. Ausnahmen dieser Regel sind die folgenden wichtigen Substituenten (vollständige Liste). Wenn diese Trivialnamen verwendet werden, muss der Lokant der Verknüpfungsstelle vor dem Substituentnamen stehen.
Furan Furyl- Piperidin Piperidyl- (1-Piperidyl- darf auch Piperidino- geschrieben werden) Pyridin Pyridyl- Chinolin Chinolyl- Isochinolin Isochinolyl- Thiophen Thienyl- (systematische Nomenklatur nicht erlaubt für dieser Substituenten)
2.3.2. Austauschnomenklatur Die Austauschnomenklatur (oder a-Nomenklatur) ist die einfachste Methode, Heterocyclen zu benennen. Sie wird vor allem für grosse und siliziumhaltige Heterocyclen verwendet. Sie kann aber im Prinzip auf alle Heterocyclen und sogar auf offenkettige Verbindungen angewendet werden. Nur kleine Heteromonocyclen werden gemäss dem Hantzsch-Widman-System beschrieben. Gemäss der Austauschnomenklatur wird der Name des zu Grunde liegenden Kohlenwasserstoffes durch vorangestellte a-Terme für die Heteroatome ergänzt. Im Folgenden sind einige a-Terme mit abnehmender Priorität geordnet. (vollständige Liste im Handbuch für die systematische Nomenklatur der organischen Chemie, metallorganischen Chemie und Koordinationschemie):

Skript AC-OC I 17
Atom a-Term Ion a-Term
O ~~ oxa- O ~
~
~ Oxonia-
S ~~ thia- S ~
~
~ Thionia-
N ~
~
~
aza- N ~~
~
~ Azonia-
P ~
~
~
phospha- P ~~
~
~ phosphonia-
Si ~~
~
~
sila-
Sn ~~
~
~
stanna-
B ~
~
~
bora- B ~~
~
~ Borata-
Jedes Heteroatom wird im Verbindungsnamen durch sein Präfix dargestellt. Wenn mehrere Heteroatome anwesend sind, werden die Präfixe mit abnehmender Priorität angeordnet. Ionen werden nach den entsprechenden Atomen aufgelistet. Vor jedem Term müssen die Lokanten stehen. Beispiele. 2.3.3. Hantzsch-Widman-System Das Hantzsch-Widman-System wird benützt, um kleine Monoheterocyclen systematisch zu benennen. Seine Regeln müssen nicht im Rahmen dieser Vorlesung bekannt sein. Man muss nur einige wichtige Verbindungsnamen kennen, die gemäss dieser Methode gebildet werden:
OHN O NH
O
O
1,4-DioxanOxiran Aziridin Oxetan Azetidin
1
2
1
2
1
3
1
3 1
4
2.4. Funktionelle Derivate der Kohlenwasserstoffe Organische Moleküle enthalten meinst neben C- und H-Atome eine Vielzahl von Heteroatomen. Diese Substituenten bestimmen die chemischen und physikalischen Eigenschaften des Moleküls und werden daher charakteristische oder funktionelle Gruppen genannt. Für funktionelle Derivate gibt es verschiedene Nomenklaturtypen. Hier werden die zwei häufigsten Methoden besprochen: die substitutive Nomenklatur und die Funktionsklassennomenklatur (alt

18 Skript AC-OC I radikofunktionelle Nomenklatur). Weitere Nomenklaturtypen können sich in bestimmten Fällen als praktisch erweisen, aber spielen im Rahmen dieser Vorlesung nur eine untergeordnete Rolle. 2.4.1. Substitutive Nomenklatur Dieses Abschnitt bildet die Grundlage der Nomenklatur funktioneller Verbindungen. Der folgende Teil des Skripts enthält nur Erläuterungen dieser Regeln. Bei diesem vielseitigsten Nomenklaturtyp, der bevorzugt angewandt werden sollte, werden die Substituenten dem Stammnamen in Form von Präfixen oder Suffixen voran- bzw. nachgestellt. In diesem Zusammenhag werden die funktionellen Gruppe hierarchisch angeordnet. Die ranghöchste funktionelle Gruppe wird grundsätzlich als Suffix genommen und bestimmt damit die Verbindungsklasse. Sie erhält in der Regel den niedrigstmöglichen Lokanten und wenn er 1 ist, muss er wie üblich nicht spezifiziert werden. Die weiteren Reste und funktionellen Gruppen werden als Präfixe vor den Namen des Grundgerüsts gestellt. Zur Ermittlung des Suffixes wird eine hierarchische Gliederung verwendet. Im Folgenden sind die wichtigsten funktionellen Gruppe nach absteigender Priorität angeordnet (vollständigere Liste; R sind Reste, M ist ein beliebiges Metall, X ist ein Halogenid; C-Atome, die zwischen eckigen Klammern stehen, sind im Stammnamen einbezogen):
Verbindungsklasse Charakteristische Gruppe
Präfix Suffix
Kationen –(NR3)+, ... z. B. -io- z. B. -ium
Carbonsäuren –C(=O)OH –[C](=O)OH
Carboxy- -carbonsäure -säure
Sulfonsäuren –S(=O)2OH Sulfo- -sulfonsäure
Carbonsäuresalze –C(=O)OM –[C](=O)OM
M-carboxylato- M -carboxylat M -oat
Sulfonsäuresalze –S(=O)2OM M-sulfonato- M -sulfonat
Carbonsäureanhydride –C(=O)OC(=O)– --- -säure…säureanhydrid -säureanhydrid
Carbonsäureester –C(=O)OR –[C](=O)OR
…yloxycarbonyl- ---
-yl…carboxylat -yl…oat
Sulfonsäureester –S(=O)2OR -yloxysulfonyl- -yl…sulfonat
Lactone*
O
OR
--- -carbolacton -olacton
Carbonsäurehalogenide –C(=O)X –[C](=O)X
Halogencarbonyl- -carbonylhalogenid -oylhalogenid

Skript AC-OC I 19
Sulfonsäurehalogenide –S(=O)2X Halogensulfonyl- -sulfonylhalogenid
Carbonsäureamide –C(=O)NH2 –[C](=O)NH2
Carbamoyl- ---
-carboxamid -amid
Sulfonsäureamide –S(=O)2NH2 Sulfamoyl- -sulfonamid
Lactame*
O
NHR
--- -lactam
Carbonsäureimide* O
HN O
R
--- -dicarboximid -imid
Amidine –C(=NH)NH2
–[C](=NH)NH2 Carbamimidoyl- ---
-carboximidamid -imidamid
Nitrile –CN –[C]N
Cyan- -carbonitril -nitril
Aldehyde –C(=O)H –[C](=O)H
Formyl- Oxo-
-carbaldehyd -al
Thioaldehyde –C(=S)H –[C](=S)H
Thioformyl- Thioxo-
-carbothioaldehyd -thial
Ketone >C=O Oxo- -on
Thioketone >C=S Thioxo- -thion
Acetale >C(OR)(OR') …yloxy…yloxy- -al…yl…ylacetal -on…yl…ylacetal
Oxime >C=N-OH Hydroxyimino- -aloxim -onoxim
Alkohole, Phenole –OH Hydroxy- -ol
Thiole –SH Sulfanyl- -thiol
Alkoholate, Phenolate –OM M-oxido- M -olat
Amine –NH2 Amino- -amin
Hydroxylamine –NH-OH Hydroxylamino- -N-…hydroxylamin
Imine >C=NH Imino- -imin
*) Diese Verbindungen werden immer häufiger mit der Austauschnomenklatur benannt.

20 Skript AC-OC I Einige funktionelle Gruppen werden nur als Präfixe verwendet. Ihre Priorität in der substitutiven Nomenklatur ist deshalb die niedrigste (vollständigere Liste; C-Atome, die zwischen eckigen Klammern sind, sind im Stammnamen einbezogen):
Charakteristische Gruppe
Präfix
Fluoride –F Fluor- Chloride –Cl Chlor- Bromide –Br Brom- Iodide –I Iod- Azide –N3 Azido- Isocyanide –NC Isocyan- Nitrosoverbindungen –NO Nitroso- Nitroverbindungen –NO2 Nitro- Ether –OR …yloxy- Sulfide –SR …ylsulfanyl- Sulfoxide –S(=O)R …ylsulfinyl- Sulfone –S(=O)2R …ylsulfonyl-
Epoxide [C] [C]
O ~~~~
Epoxy-
2.4.2. Funktionsklassennomenklatur Kleine, einfache Moleküle, die nur eine funktionelle Gruppe besitzen, werden sehr oft gemäss der Funktionsklassennomenklatur benannt. Es werden keine Suffixe verwendet, sondern die Stammnamen werden in Form ihrer Radikale benannt (d. h. mit der -yl-Endung), denen die meist anionisierten Namen der entsprechenden Verbindungsklasse nachgestellt sind. In der folgenden Tabelle sind die Endungen der häufigsten funktionellen Gruppe wiedergegeben (vollständigere Liste; C-Atome, die zwischen eckigen Klammern sind, sind im Stammnamen einbezogen):
Charakteristische Gruppe Radikofunktioneller Verbindungsname Ester -ester Säurehalogenide (z. B. R[C](=O)X, RS(=O)2X)
-fluorid, -chlorid, -bromid, -iodid
Nitrile; Isocyanide -cyanid; -isocyanid Ketone -keton Alkohole -alkohol Ether; Sulfide -ether; -sulfidSulfoxide; Sulfone -sulfoxid; -sulfon Halogenide -fluorid, -chlorid, -bromid, -iodid Azide -azid Amine -amin
Beispiele. 2.4.3. Bestimmung der Hauptkette von funktionalisierten Verbindungen

Skript AC-OC I 21 Um die Hauptkette einer Verbindung, die funktionelle Gruppen trägt, zu identifizieren, sind modifizierte Regeln anzuwenden. Die Hauptkette ist diejenige mit:
1. den meisten Hauptgruppen (d. h. der meisten Zahl von Substituenten, die als Suffix geschrieben werden können, siehe zudem Kapitel 2.4.1.). Beispiel:
Cl
OH
OH
3-(4-Chlorbutyl)pentan-1,4-diol
2. den meisten Mehrfachbindungen; 3. der meisten Atomen (längste Kette); 4. den meisten Doppelbindungen; 5. den niedrigsten Lokanten für das Suffix. Beispiel:
Cl
HO
Cl
HO
OH
8-Chlor-5-(1-chlor-3-hydroxypropyl)octan-1,7-diol
6. den niedrigsten Lokanten für die Mehrfachbindungen; 7. den niedrigsten Lokanten für die Doppelbindungen; 8. den meisten durch Präfixe benannten Substituenten. Beispiel:
OH
OH
Cl
OH
3-Chlor-5-(3-hydroxybutyl)-4,6-dimethylnonan-2,8-diol
9. dem Satz niedrigster Lokanten für alle durch Präfixe benannten Substituenten; 10. den Substituenten, dessen Name im Alphabet zuerst erscheint; 11. den niedrigsten Lokanten für den Substituenten, dessen Name im Alphabet zuerst erschient.

22 Skript AC-OC I
2.5. Beispiele für die Nomenklatur funktionalisierter Verbindungen Im Folgenden werden die substitutive und, wenn möglich, die radikofunktionelle Nomenklatur der wichtigsten Klassen funktioneller Verbindungen mit Hilfe von Beispielen erläutert. IUPAC erlaubt sehr viele Trivialnamen, meistens für einfache oder biologisch wichtige Moleküle. Diese Trivialnamen sind nicht im Rahmen dieser Vorlesung auswendig zu lernen, aber sie werden sehr häufig in der chemischen Literatur verwendet. Für eine ausführlichere Betrachtung und für die Nomenklatur seltener funktioneller Verbindungen siehe z. B. Handbuch für die systematische Nomenklatur der organischen Chemie, metallorganischen Chemie und Koordinationschemie. 2.5.1. Kationen Im Rahmen dieser Vorlesung wird nur das Kation Ammonium (R4N
+) betrachtet:
NN
Br
Tetramethylammonium tert-Butyldimethyl-pentylammoniumbromid
2.5.2. Carbon und Sulfonsäuren Carbonsäuren Je nachdem, ob das C-Atom der Carbonylgruppe (>C=O) im Stammnamen enthalten ist, oder nicht, benutzt man die Bezeichnung -säure oder -carbonsäure, die dem Namen der Stammverbindung folgt.
O
OHCOOH HOOC
COOH
COOHCOOH
3-Methylbutansäure Cyclopropancarbonsäure Butandisäure
Naphthalin-2-carbonsäure 2-Benzylpentansäure Sehr viele Carbonsäuren behalten Trivialnamen bei. Obwohl sie noch zugelassen sind, ist es empfohlen, nur systematische Namen zu benutzten. Nur drei Säuren sollten mittels ihren Trivialnamen bezeichnet werden:

Skript AC-OC I 23
H
O
OH H3C
O
OH
COOH
Ameisensäure Essigsäure Benzoesäure Die den Carbonsäuren entsprechenden Reste werden als Acylreste bezeichnet. Deren Namen werden durch Anhängen des Suffixes -oyl an den Stammnamen gebildet. Wenn das Carbonyl-C-Atom nicht im Stammnamen enthalten ist, wird die Endung -carbonyl verwendet. Acetyl-, Formyl- und Benzoyl- sind Ausnahmen:
O
R
O
O
O
O
H
O
O
Benzoyl-
~
~ ~
~
~
Acylrest Cyclopentancarbonyl- Butandioyl-
Acetyl-
~
~
Formyl- Sulfonsäure Für die Benennung von Sulfonsäuren wird die Vorsilbe -carbon- durch -sulfon- ersetzt:
S
O
O
OH
S
O
O
HO
SOH
O
O
N
SO3H
Propan-1,2-disulfonsäure 4-Methylbenzolsulfonsäure(p-Toluolsulfonsäure)
Pyrrol-1-sulfonsäure
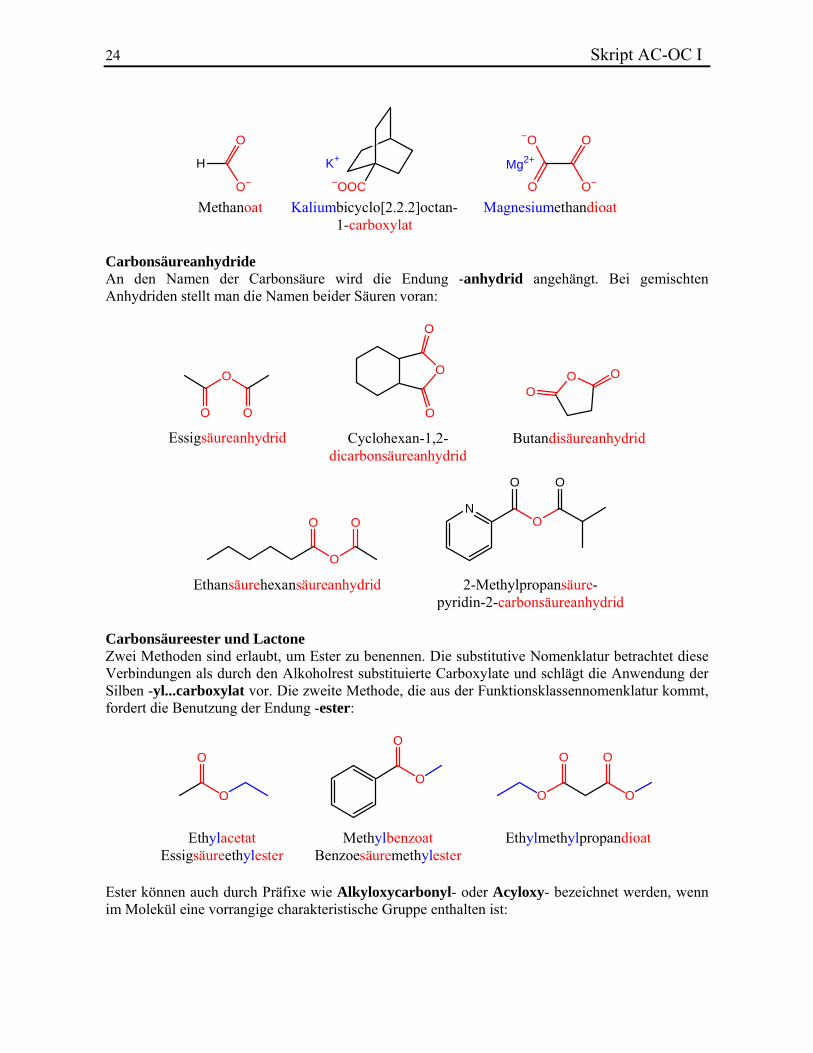
2.5.3. Carbonsäurederivate Carbonsäuresalze Die den Carbonsäuren entsprechenden Anionen (Carboxylate) werden durch Anhängen des Suffixes -oat an den Stammnamen gebildet. Ist das Carbonyl-C-Atom nicht in diesem enthalten, so lautet die Endung -carboxylat. Für die Benennung der entsprechenden Salze wird der Name des Kations vorangestellt:
24 Skript AC-OC I
H
O
O–
–O
O
O
O–
Mg2+
–OOC
K+
Methanoat Kaliumbicyclo[2.2.2]octan-1-carboxylat
Magnesiumethandioat
Carbonsäureanhydride An den Namen der Carbonsäure wird die Endung -anhydrid angehängt. Bei gemischten Anhydriden stellt man die Namen beider Säuren voran:
O
O
O
O
O
O
O
O
O
OO
O
Essigsäureanhydrid Cyclohexan-1,2-dicarbonsäureanhydrid
Butandisäureanhydrid
Ethansäurehexansäureanhydrid
NO
O O
2-Methylpropansäure-pyridin-2-carbonsäureanhydrid
Carbonsäureester und Lactone Zwei Methoden sind erlaubt, um Ester zu benennen. Die substitutive Nomenklatur betrachtet diese Verbindungen als durch den Alkoholrest substituierte Carboxylate und schlägt die Anwendung der Silben -yl...carboxylat vor. Die zweite Methode, die aus der Funktionsklassennomenklatur kommt, fordert die Benutzung der Endung -ester:
O
O
O
OO
O O
O
EthylacetatEssigsäureethylester
MethylbenzoatBenzoesäuremethylester
Ethylmethylpropandioat
Ester können auch durch Präfixe wie Alkyloxycarbonyl- oder Acyloxy- bezeichnet werden, wenn im Molekül eine vorrangige charakteristische Gruppe enthalten ist:
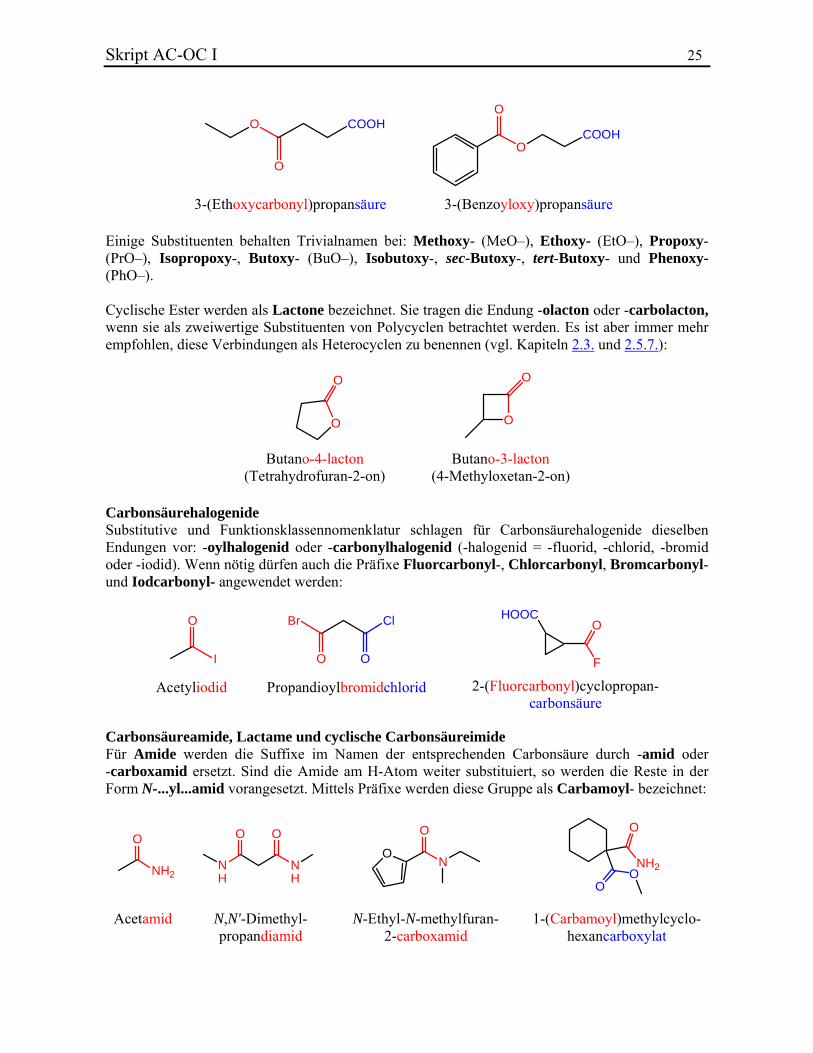
Skript AC-OC I 25
O
O
COOHO
O
COOH
3-(Benzoyloxy)propansäure3-(Ethoxycarbonyl)propansäure Einige Substituenten behalten Trivialnamen bei: Methoxy- (MeO–), Ethoxy- (EtO–), Propoxy- (PrO–), Isopropoxy-, Butoxy- (BuO–), Isobutoxy-, sec-Butoxy-, tert-Butoxy- und Phenoxy- (PhO–). Cyclische Ester werden als Lactone bezeichnet. Sie tragen die Endung -olacton oder -carbolacton, wenn sie als zweiwertige Substituenten von Polycyclen betrachtet werden. Es ist aber immer mehr empfohlen, diese Verbindungen als Heterocyclen zu benennen (vgl. Kapiteln 2.3. und 2.5.7.):
O
O
Butano-4-lacton(Tetrahydrofuran-2-on)
O
O
Butano-3-lacton(4-Methyloxetan-2-on)
Carbonsäurehalogenide Substitutive und Funktionsklassennomenklatur schlagen für Carbonsäurehalogenide dieselben Endungen vor: -oylhalogenid oder -carbonylhalogenid (-halogenid = -fluorid, -chlorid, -bromid oder -iodid). Wenn nötig dürfen auch die Präfixe Fluorcarbonyl-, Chlorcarbonyl, Bromcarbonyl- und Iodcarbonyl- angewendet werden:
O
I
O
F
HOOCBr
O O
Cl
Acetyliodid 2-(Fluorcarbonyl)cyclopropan-carbonsäure
Propandioylbromidchlorid
Carbonsäureamide, Lactame und cyclische Carbonsäureimide Für Amide werden die Suffixe im Namen der entsprechenden Carbonsäure durch -amid oder -carboxamid ersetzt. Sind die Amide am H-Atom weiter substituiert, so werden die Reste in der Form N-...yl...amid vorangesetzt. Mittels Präfixe werden diese Gruppe als Carbamoyl- bezeichnet:
O
NH2
O
O
N
OO
O
NH2NH
O O
NH
Acetamid N,N'-Dimethyl-propandiamid
N-Ethyl-N-methylfuran-2-carboxamid
1-(Carbamoyl)methylcyclo-hexancarboxylat

26 Skript AC-OC I Die Amide von Ameisensäure, Essigsäure und Benzoesäure behalten die Trivialnamen Formamid, Acetamid und Benzamid bei. Intracyclische Amide werden als Lactame bezeichnet. Sie werden als Heterocyclen oder unter Verwendung des Suffixes -lactam benannt:
HN
OButano-4-lactam
(Tetrahydropyrrol-2-on) Carbonsäureimide sind die Stickstoffanaloga der cyclischen Anhydride. Sie werden durch Ersetzung der Endungen -disäure oder -dicarbonsäure des systematischen oder triviales Namen der entsprechenden Säure durch -imid bzw. -dicarboximid benannt. Sie können auch gemäss der Heterocyclennomenklatur bezeichnet werden:
HNO O
Butanimid(Trivialname: Succinimid)
Amidine Amidine werden durch Verwendung der Endungen -imidamid und -caroximidamid oder des Präfixes Carbamimidoyl- bezeichnet:
NH
NH2
ONH
NH2COOH
NH
H2N
Pentanimidamid Furan-2-carboximidamid 3-Carbamimidoyl-propansäure
Nitrile In Analogie zu den Säuren werden die entsprechenden Nitrile (oder Cyanide) mit -nitril (C-Atom zählt zur Kette), -carbonitril oder Cyan- bezeichnet. Alternativ darf die Funktionsklassennomenklatur angewendet werden, die die Endung -cyanid vorschlägt. H3C–CN besitzt den Trivialnamen Acetonitril; H–CN wird Blausäure genannt.
CN
CNNC CNC N
AcetonitrilMethylcyanid
PentannitrilPentylcyanid
CyclobutancarbonitrilCyclobutylcyanid
Propandinitril
Viele einfache Nitrile werden oft mit Trivialnamen bezeichnet, obwohl die systematische Nomenklatur zu bevorzugen ist (vgl. Kapitel 2.5.2.).
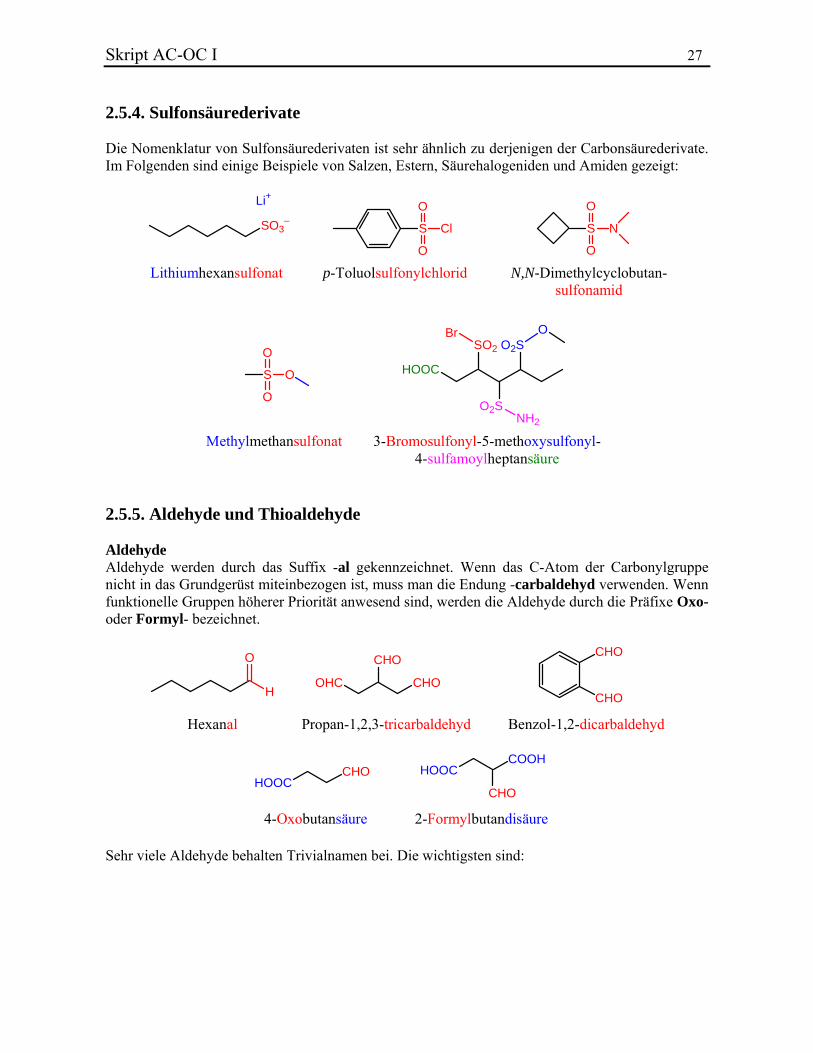
Skript AC-OC I 27 2.5.4. Sulfonsäurederivate Die Nomenklatur von Sulfonsäurederivaten ist sehr ähnlich zu derjenigen der Carbonsäurederivate. Im Folgenden sind einige Beispiele von Salzen, Estern, Säurehalogeniden und Amiden gezeigt:
SO3–
Li+
S
O
O
Cl S
O
O
N
S
O
O
O HOOC
SO2
BrO2S
O2SNH2
O
Lithiumhexansulfonat p-Toluolsulfonylchlorid
Methylmethansulfonat 3-Bromosulfonyl-5-methoxysulfonyl-4-sulfamoylheptansäure
N,N-Dimethylcyclobutan-sulfonamid
2.5.5. Aldehyde und Thioaldehyde Aldehyde Aldehyde werden durch das Suffix -al gekennzeichnet. Wenn das C-Atom der Carbonylgruppe nicht in das Grundgerüst miteinbezogen ist, muss man die Endung -carbaldehyd verwenden. Wenn funktionelle Gruppen höherer Priorität anwesend sind, werden die Aldehyde durch die Präfixe Oxo- oder Formyl- bezeichnet.
HOOCCOOH
CHOHOOC
CHO
OHC CHO
CHO
H
O CHO
CHO
Hexanal Propan-1,2,3-tricarbaldehyd
4-Oxobutansäure
Benzol-1,2-dicarbaldehyd
2-Formylbutandisäure Sehr viele Aldehyde behalten Trivialnamen bei. Die wichtigsten sind:
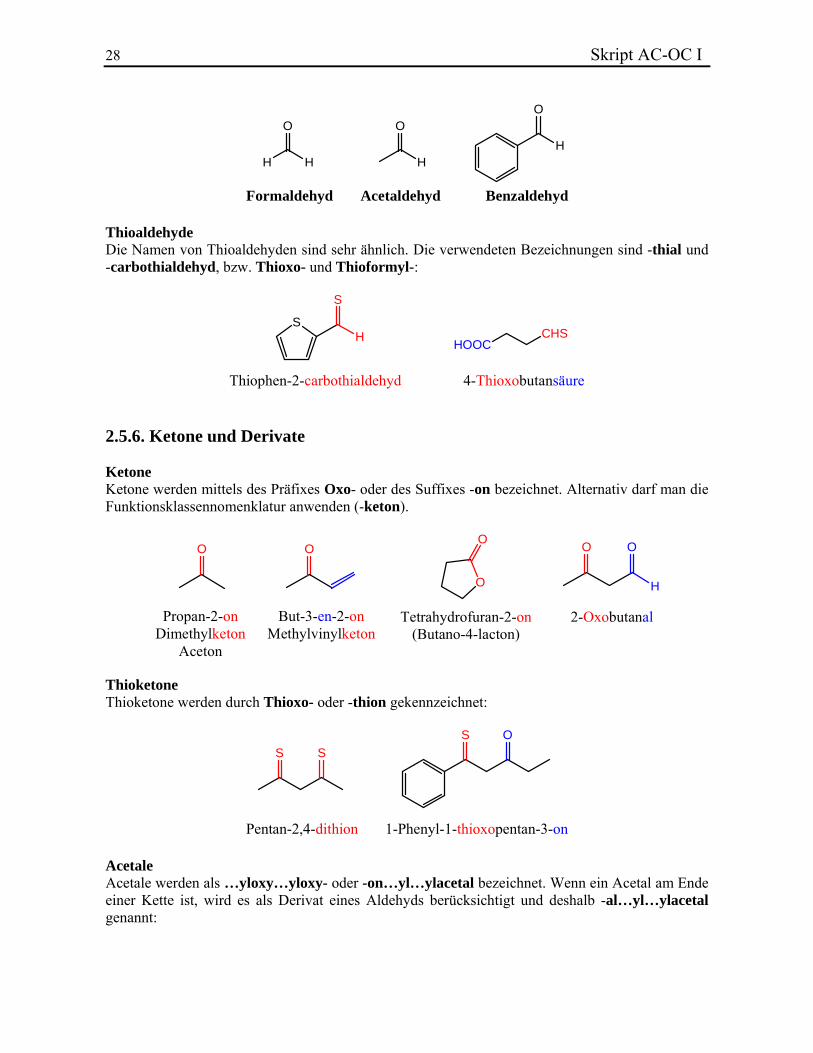
28 Skript AC-OC I
O
H H H
OO
H
Formaldehyd Acetaldehyd Benzaldehyd Thioaldehyde Die Namen von Thioaldehyden sind sehr ähnlich. Die verwendeten Bezeichnungen sind -thial und -carbothialdehyd, bzw. Thioxo- und Thioformyl-:
HOOCCHS
S
S
H
4-ThioxobutansäureThiophen-2-carbothialdehyd 2.5.6. Ketone und Derivate Ketone Ketone werden mittels des Präfixes Oxo- oder des Suffixes -on bezeichnet. Alternativ darf man die Funktionsklassennomenklatur anwenden (-keton).
O O
O
OO O
H
Propan-2-onDimethylketon
Aceton
But-3-en-2-onMethylvinylketon
Tetrahydrofuran-2-on(Butano-4-lacton)
2-Oxobutanal
Thioketone Thioketone werden durch Thioxo- oder -thion gekennzeichnet:
S S
S O
Pentan-2,4-dithion 1-Phenyl-1-thioxopentan-3-on Acetale Acetale werden als …yloxy…yloxy- oder -on…yl…ylacetal bezeichnet. Wenn ein Acetal am Ende einer Kette ist, wird es als Derivat eines Aldehyds berücksichtigt und deshalb -al…yl…ylacetal genannt:
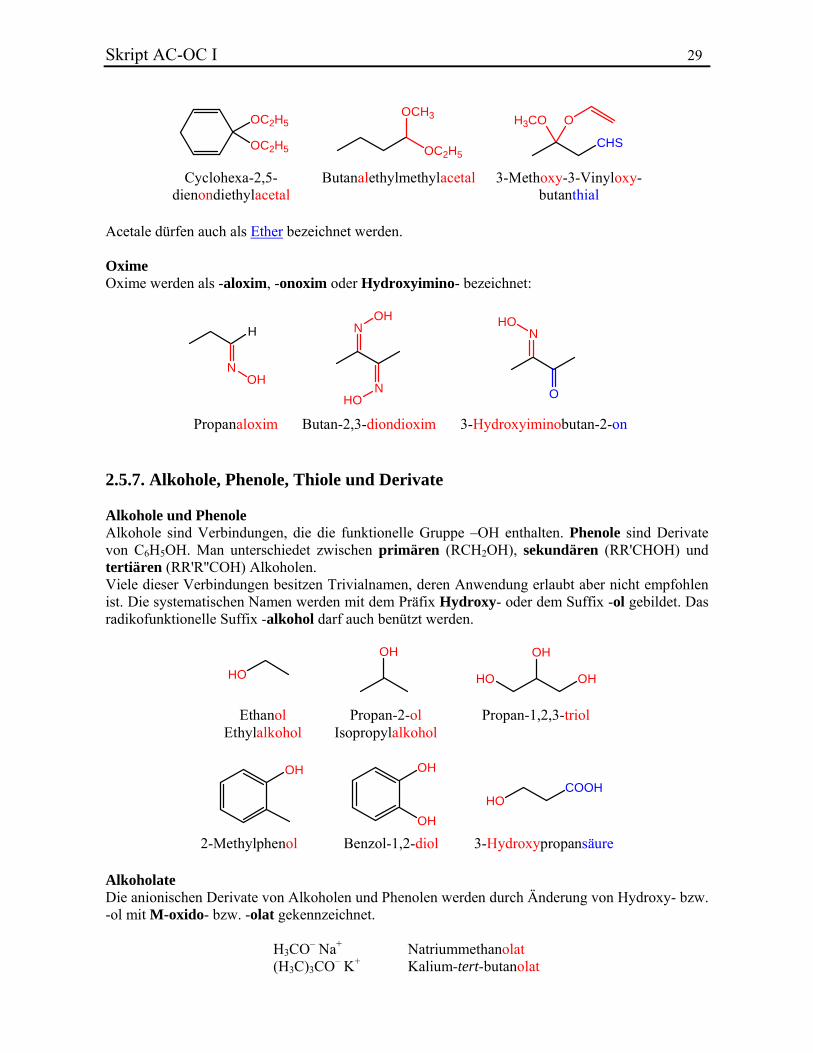
Skript AC-OC I 29
OC2H5
OC2H5
OCH3
OC2H5CHS
H3CO O
Cyclohexa-2,5-dienondiethylacetal
Butanalethylmethylacetal 3-Methoxy-3-Vinyloxy-butanthial
Acetale dürfen auch als Ether bezeichnet werden. Oxime Oxime werden als -aloxim, -onoxim oder Hydroxyimino- bezeichnet:
NOH
H NOH
NHO
NHO
O
Propanaloxim Butan-2,3-diondioxim 3-Hydroxyiminobutan-2-on 2.5.7. Alkohole, Phenole, Thiole und Derivate Alkohole und Phenole Alkohole sind Verbindungen, die die funktionelle Gruppe –OH enthalten. Phenole sind Derivate von C6H5OH. Man unterschiedet zwischen primären (RCH2OH), sekundären (RR'CHOH) und tertiären (RR'R''COH) Alkoholen. Viele dieser Verbindungen besitzen Trivialnamen, deren Anwendung erlaubt aber nicht empfohlen ist. Die systematischen Namen werden mit dem Präfix Hydroxy- oder dem Suffix -ol gebildet. Das radikofunktionelle Suffix -alkohol darf auch benützt werden.
HO
OH
HO
OH
OH
OH OH
OH
COOHHO
EthanolEthylalkohol
Propan-2-olIsopropylalkohol
Propan-1,2,3-triol
2-Methylphenol Benzol-1,2-diol 3-Hydroxypropansäure Alkoholate Die anionischen Derivate von Alkoholen und Phenolen werden durch Änderung von Hydroxy- bzw. -ol mit M-oxido- bzw. -olat gekennzeichnet.
H3CO– Na+ Natriummethanolat (H3C)3CO– K+ Kalium-tert-butanolat
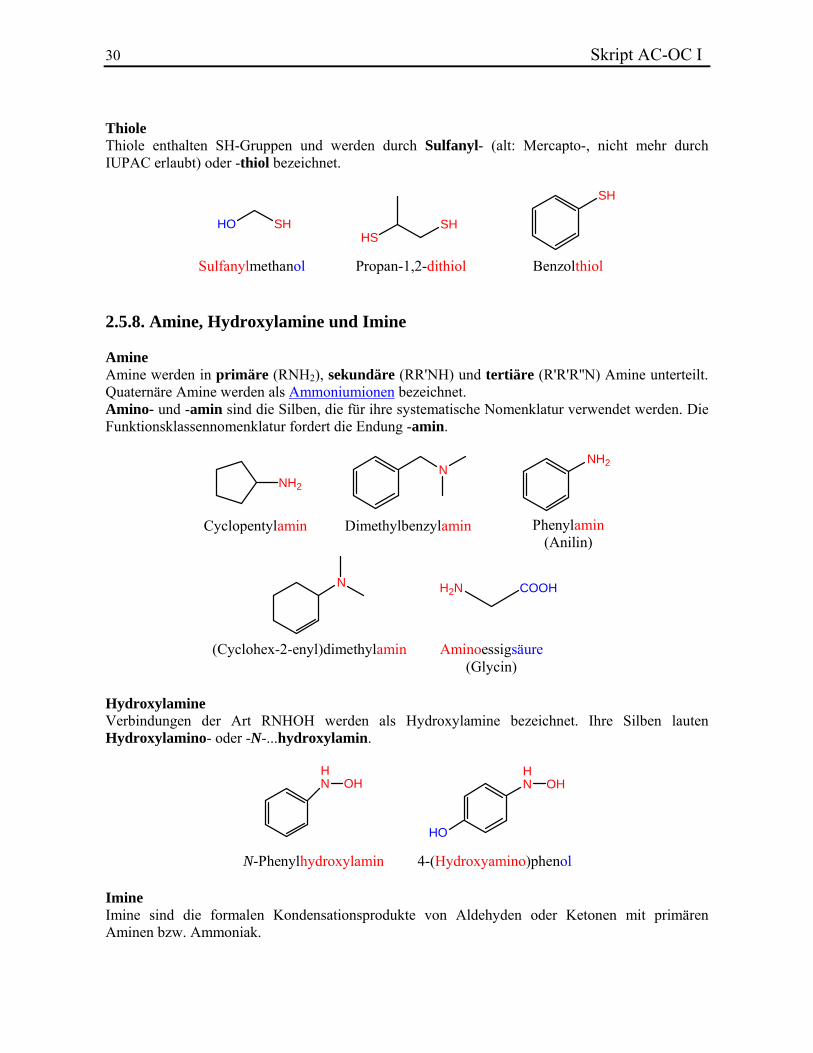
30 Skript AC-OC I Thiole Thiole enthalten SH-Gruppen und werden durch Sulfanyl- (alt: Mercapto-, nicht mehr durch IUPAC erlaubt) oder -thiol bezeichnet.
HO SHHS
SH
SH
Sulfanylmethanol Propan-1,2-dithiol Benzolthiol 2.5.8. Amine, Hydroxylamine und Imine Amine Amine werden in primäre (RNH2), sekundäre (RR'NH) und tertiäre (R'R'R''N) Amine unterteilt. Quaternäre Amine werden als Ammoniumionen bezeichnet. Amino- und -amin sind die Silben, die für ihre systematische Nomenklatur verwendet werden. Die Funktionsklassennomenklatur fordert die Endung -amin.
NH2
NNH2
N H2N COOH
Cyclopentylamin Dimethylbenzylamin Phenylamin(Anilin)
(Cyclohex-2-enyl)dimethylamin Aminoessigsäure(Glycin)
Hydroxylamine Verbindungen der Art RNHOH werden als Hydroxylamine bezeichnet. Ihre Silben lauten Hydroxylamino- oder -N-...hydroxylamin.
HN OH
HO
HN OH
N-Phenylhydroxylamin 4-(Hydroxyamino)phenol Imine Imine sind die formalen Kondensationsprodukte von Aldehyden oder Ketonen mit primären Aminen bzw. Ammoniak.
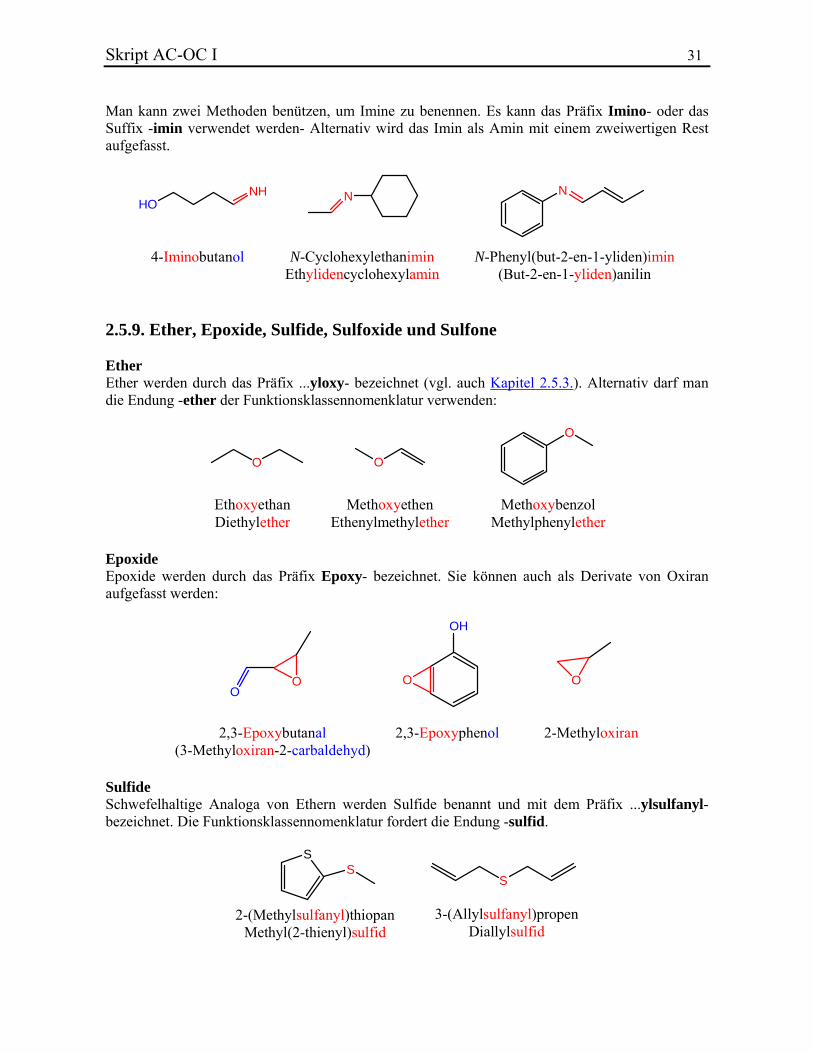
Skript AC-OC I 31 Man kann zwei Methoden benützen, um Imine zu benennen. Es kann das Präfix Imino- oder das Suffix -imin verwendet werden- Alternativ wird das Imin als Amin mit einem zweiwertigen Rest aufgefasst.
HONH N N
4-Iminobutanol N-CyclohexylethaniminEthylidencyclohexylamin
N-Phenyl(but-2-en-1-yliden)imin(But-2-en-1-yliden)anilin
2.5.9. Ether, Epoxide, Sulfide, Sulfoxide und Sulfone Ether Ether werden durch das Präfix ...yloxy- bezeichnet (vgl. auch Kapitel 2.5.3.). Alternativ darf man die Endung -ether der Funktionsklassennomenklatur verwenden:
O
O
O
EthoxyethanDiethylether
MethoxyethenEthenylmethylether
MethoxybenzolMethylphenylether
Epoxide Epoxide werden durch das Präfix Epoxy- bezeichnet. Sie können auch als Derivate von Oxiran aufgefasst werden:
OH
OOO
O
2,3-Epoxybutanal(3-Methyloxiran-2-carbaldehyd)
2,3-Epoxyphenol 2-Methyloxiran
Sulfide Schwefelhaltige Analoga von Ethern werden Sulfide benannt und mit dem Präfix ...ylsulfanyl- bezeichnet. Die Funktionsklassennomenklatur fordert die Endung -sulfid.
SS
S
2-(Methylsulfanyl)thiopanMethyl(2-thienyl)sulfid
3-(Allylsulfanyl)propenDiallylsulfid
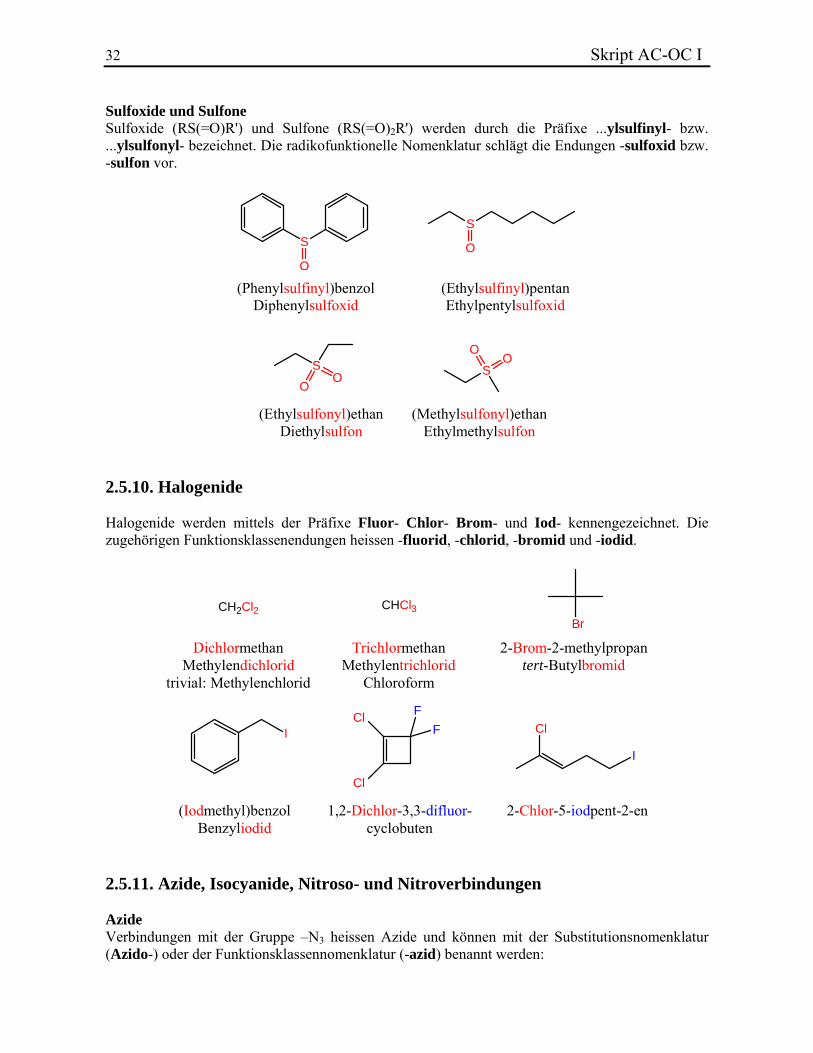
32 Skript AC-OC I Sulfoxide und Sulfone Sulfoxide (RS(=O)R') und Sulfone (RS(=O)2R') werden durch die Präfixe ...ylsulfinyl- bzw. ...ylsulfonyl- bezeichnet. Die radikofunktionelle Nomenklatur schlägt die Endungen -sulfoxid bzw. -sulfon vor.
S
O
S
O
S SO
O
OO
(Phenylsulfinyl)benzolDiphenylsulfoxid
(Ethylsulfinyl)pentanEthylpentylsulfoxid
(Ethylsulfonyl)ethanDiethylsulfon
(Methylsulfonyl)ethanEthylmethylsulfon
2.5.10. Halogenide Halogenide werden mittels der Präfixe Fluor- Chlor- Brom- und Iod- kennengezeichnet. Die zugehörigen Funktionsklassenendungen heissen -fluorid, -chlorid, -bromid und -iodid.
CH2Cl2 CHCl3Br
I
Cl
F
FCl
Cl
I
DichlormethanMethylendichlorid
trivial: Methylenchlorid
TrichlormethanMethylentrichlorid
Chloroform
2-Brom-2-methylpropantert-Butylbromid
(Iodmethyl)benzolBenzyliodid
1,2-Dichlor-3,3-difluor-cyclobuten
2-Chlor-5-iodpent-2-en
2.5.11. Azide, Isocyanide, Nitroso- und Nitroverbindungen Azide Verbindungen mit der Gruppe –N3 heissen Azide und können mit der Substitutionsnomenklatur (Azido-) oder der Funktionsklassennomenklatur (-azid) benannt werden:
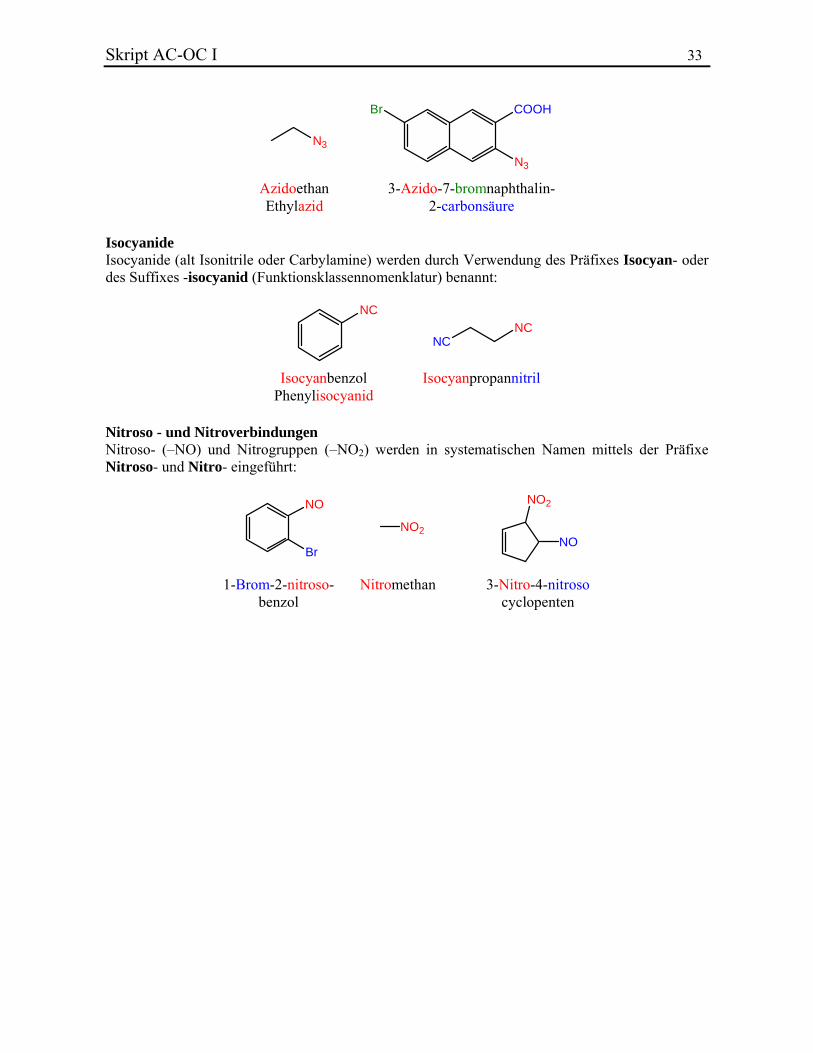
Skript AC-OC I 33
N3
N3
Br COOH
AzidoethanEthylazid
3-Azido-7-bromnaphthalin-2-carbonsäure
Isocyanide Isocyanide (alt Isonitrile oder Carbylamine) werden durch Verwendung des Präfixes Isocyan- oder des Suffixes -isocyanid (Funktionsklassennomenklatur) benannt:
NC
NCNC
IsocyanbenzolPhenylisocyanid
Isocyanpropannitril
Nitroso - und Nitroverbindungen Nitroso- (–NO) und Nitrogruppen (–NO2) werden in systematischen Namen mittels der Präfixe Nitroso- und Nitro- eingeführt:
NO2
NO
Br
NO2
NO
1-Brom-2-nitroso-benzol
Nitromethan 3-Nitro-4-nitrosocyclopenten

34 Skript AC-OC I
KAPITEL 3:
KLASSISCHE STRUKTURLEHRE In diesem Kapitel werden folgende Themen behandelt: 3.1. Valenz und Konstitutionsisomere 3.2. Chemische Bindung 3.2.1. Lewis-Bindungsmodell 3.2.2. Formalladungen 3.2.3. Elektronegativität und Bindungsdipol 3.2.4. Homolyse und Heterolyse 3.2.5. Resonanztheorie 3.3. Stereochemie 3.3.1. Das Tetraedermodell 3.3.2. Optische Aktivität 3.3.3. Stereoisomere, Enantiomere und Diastereoisomere 3.3.4. Beispiel: Zuckerstrukturaufklärung nach Fischer 3.3.5. Absolute Konfiguration und CIP-Regeln 3.3.6. Stereoisomerie bei Doppelbindungen 3.3.7. Stereoisomere ohne Stereozentrum 3.4. Symmetrielehre 3.4.1. Punktgruppen 3.4.2. Molekülsymmetrie und Chiralität 3.4.3. Topizität

Skript AC-OC I 35
3.1. Valenz und Isomere Die chemischen und physikalischen Eigenschaften der organischen Verbindungen sind nicht nur durch ihre elementare Zusammensetzung bestimmt, sondern durch ihre Konstitution, das heisst die Art wie die Atome in einem Molekül miteinander verbunden sind. Das heutzutage verwendete Modell wurde durch Kekulé und Couper 1858 vorgeschlagen. Die Zahl möglicher Konstitutionen wird durch die Valenz (oder Wertigkeit) der Atome bestimmt. Jedes Atom besitzt eine maximale Zahl von Bindungen, die es mit benachbarten Atomen bilden darf. Die folgende Tabelle zeigt, die Valenz von Atomen, die in der organischen Chemie wichtig sind:
C 4-wertig N 3-wertig O 2-wertig H 1-wertig
Mehrwertiger Atome (meistens Kohlenstoff) können Ketten, Ringe oder Mehrfachbindungen bilden, was zu einer grossen Zahl von möglichen Konstitutionen führt. Diese werden eindeutig durch ein graphisches Schema dargestellt (Konstitutionsformel) oder durch den Namen der Verbindung kennengezeichnet. Verbindungen gleicher Summenformel (oder Molekularformel, d. h. gleicher Zahl und Typ von Atomen) aber mit verschiedener Konstitution werden als Konstitutionsisomere bezeichnet. Sie unterscheiden sich in ihren physikalischen und chemischen Eigenschaften. Z. B. sind Essigsäure, Ameisensäuremethylester und Hydroxyethanal die stabilsten Konstitutionsisomere mit Summenformel C2H4O2, aber sie verhalten sich drastisch unterschiedlich:
Konstitutionsformel
O
OH
H
O
O
HO
O
H
Schmelzpunkt 17 °C –99 °C 97 °C Siedepunkt 118 °C 31 °C Zersetzung
Die Zahl der Konstitutionsisomere nimmt mit ansteigender Zahl der Atome rasch zu:
Konstitutionsisomere CH4 1 C2H6 1 C3H8 1 C4H10 2 C5H12 3 C6H14 5 C10H22 75 C20H42 366319 C40H82 ~ 6·1013
Die Anzahl Doppelbindungsäquivalente (Ringe, Doppel- oder Dreifachbindungen, vgl. Kapitel 3.2.1.) kann aus der Summenformel bestimmt werden. Man betrachte das Molekül AaBbCcDd, in dem A die 4-wertigen (z. B. C, Si), B die 1-werigen (z. B. H, F, Cl, Br, I), C die 2-wertigen (z. B. O, S) und D die 3-wertigen Atome (z. B. N, P) sind. Die Zahl Doppelbindungsäquivalente x ist dann

36 Skript AC-OC I
2
22 bdax
Eine Dreifachbindung zählt als zwei Doppelbindungen. Z. B. besitzt eine Verbindung mit Summenformen C20H25NO 9 Doppelbindungsäquivalente. Im folgenden Isomer sind sie in 3 Doppelbindungen, 1 Dreifachbindung und 5 Ringe verteilt:
HO
CH3NH2
Weitere Arten von Isomerie werden im Kapitel 3.3. beschreiben.
3.2. Chemische Bindung 3.2.1. Lewis-Bindungsmodell In den vorherigen Kapiteln wurde die Struktur von vielen Verbindungen gezeigt, in denen die Atome durch Striche verbunden werden. Man sprach dabei von Bindungen diskutiert, ohne eine Definition anzugeben. Dies nachzuholen ist das Ziel dieses Kapitels. Nach der Entdeckung der Elektronen wurde versucht, die periodischen Eigenschaften der Elemente damit zu interpretieren. Zwei der wichtigsten Atomeigenschaften sind das Ionisationspotential und die Elektronenaffinität. Das Ionisationspotential IP ist die Energie, die für die Freisetzung eines Elektrons entsprechend folgender Gleichung benötigt wird: A A+ + e–. Die Elektronenaffinität EA ist die Energie, die beim Aufnehmen eines Elektrons laut folgender Gleichung freigesetzt wird: A + e– A–. Lewis schlug vor, dass die Atome versuchen, die leeren Stellen eines Kubus mit Elektronen zu füllen. Atome, in denen schon alle acht Ecken besetzt sind, werden Edelgase genannt und zeigen grosse Stabilität und Unreaktivität: sie sind chemisch inert. Dieser hypothetische Kubus wurde Valenzschale genannt und der Zustand, wobei die Valenzschale vollständig gefüllt ist, heisst Edelgaskonfiguration (vgl. auch Kapitel 4.1.).

Skript AC-OC I 37
Atome mit ungefüllter Valenzschale versuchen, die Konfiguration des nächsten Edelgases durch Bindungen zu anderen Atomen zu erreichen. Für Atome der tieferen Periode der Periodensystem bedeutet dies, dass in der Valenzschale normalerweise acht Elektronen anwesend sein müssen. Diese Beobachtung wird als Oktettregel bezeichnet. Ausnahme dazu sind H und He, die nur zwei Elektronen beherbergen können. In Abhängigkeit der Atome, die miteinander wechselwirken können, werden drei verschiedene Arten von Bindungen unterschieden: metallische, ionische und kovalente Bindung.
Metallische Bindung: Atome wie z. B. Na oder Li bilden metallische Aggregate der Form Nax bzw. Lix. Die Atome geben die Elektronen der entfernsten Schale ab und erreichen damit die Konfiguration des vorhergehenden Edelgases. Die abgegebenen Elektronen sind im ganzen Gitter delokalisiert.
Ionische Bindung: In Salzen geben die Atome eines Elements mit niedrigem IP die äussersten Elektronen einer Komponenten mit höherer Elektroaffinität ab. Beide erreichen die Konfiguration des vorhergehenden bzw. nachfolgenden Edelgases. Dies ist z. B. der Fall in LiF oder MgCl2.
Kovalente Bindung: In einer kovalenten Bindung gehören die Bindungselektronen beider aneinander gebundener Atome. Der Aufenthalt der Elektronen zwischen den Kernen verringert die Abstossung der Atomkerne. Solche Bindungen sind die wichtigste Bindungsart der organischen Chemie. Sie werden vor allem für Elemente beobachtet, die ein grosses IP besitzen, wo die Abgabe mehrerer Elektronen zu viel Energie brauchen würde. Diese Elemente bilden Oktettschalen aus, indem sie Elektronenpaare unter Ausbildung kovalenter Bindungen teilen.
Das Kubusmodell wurde eingeführt, um die Oktettregel physikalisch zu begründen. Das Molekül Cl2 kann z. B. durch die Überlappung von zwei unvollständigen Kuben erklärt werden:

38 Skript AC-OC I
Cl Cl
+
Cl Cl In den meisten Fällen teilen zwei an einer kovalenten Bindung beteiligte Atome nur zwei Elektronen. Es kommt aber vor, dass sie vier oder sogar sechs Elektronen gemeinsam haben. Diese Bindungen werden Doppel- bzw. Dreifachbindungen genannt. Doppelbindungen sind länger als Dreifachbindungen aber kürzer als Einfachbindungen (C–C 1.54 Å; C=C 1.34 Å; CC 1.21 Å). Weiter sind Mehrfachbindungen nicht um die Bindungsachse frei drehbar. Die Doppelbindung, wie diejenige in O2, kann gemäss dem Kubusmodell erklärt werden:
O O
+
O O Das Modell kann aber nicht die Entstehung von Dreifachbindungen wie in N2 begründen. Anstatt einen Würfel zu verwenden, schlug Lewis daher vor, die Valenzschale eines Atoms als vier Paare von Elektronenstellen in den Ecken eines Tetraeders zu betrachten:
Cl Cl
Eine solche tetraedrische Geometrie wird tatsächlich für die meisten organische Atome gefunden, die keine Mehrfachbindung bilden. Die Ecken können entweder durch Bindungselektronenpaare oder durch freie, nichtbindende Elektronenpaare besetzt werden. Die Unterteilung der Elektronen in Paaren führte zur Idee, dass eine Bindungen durch zwei Elektronen erzeugt werden. Die Entstehung von Bindungen wird durch die Überlappung solcher Tetraeder erklärt:
Cl2 O2 N2
Dieses Modell besagt die freie Rotation um Einfachbindungen und die eingeschränkte Drehbarkeit von Doppel- und Dreifachbindungen, wobei die Atome durch mehreren Ecken gebunden sind. Weiter kann auch den Abstand zwischen den zwei Kerne durch einfachen, geometrischen Rechnungen berechnet werden. Dessen berechnete Abnahme von Einfach- bis Dreifachbindungen wird tatsächlich beobachtet. Obwohl dieses Modell viele Eigenschaften der Moleküle gut erklärt, wurde es vollständig durch die quantenchemische Betrachtung der Atome ersetzt (vgl. Kapitel 4.). Wasserstoff und Helium sind die einzigen Elemente, die nur zwei Elektronen benötigen, um die Edelgaskonfiguration zu erreichen. Zwei H-Atome werden daher ihre Elektronen billigen und eine kovalente Bindung erzeugen, unter Bildung des stabilen H2-Molekül:

Skript AC-OC I 39
H· + ·H H:H H–H In Methan (CH4) besitzt das C-Atom vier leere Valenzstellen, die durch die vier Elektronen der H-Atome besetzt werden. Durch die Bildung von vier kovalenten Bindungen erhalten alle Atome eine Edelgaskonfiguration.
C::
: :HH
HH
Methan Ammoniak
N::
: :HH
H B:: :FF
F
BF3 In Ammoniak (NH3) sind drei kovalente Bindungen und ein freies Elektronenpaar anwesend. Verbindungen mit nichtbindenden Elektronenpaaren werden oft Lewis-Basen, Nukleophile oder Elektronenpaar-Donoren genannt. Einige Atome wie B und Al bilden stabile Moleküle auch wenn die Oktettregel nicht erfüllt wird. Z. B. sind in BF3 nur drei kovalenten Bindungen anwesend. Die F-Atome besitzen zwar acht Elektronen in der Valenzschale, aber das B-Atom hat noch zwei Valenzstellen frei. Dieses Molekül wird daher versuchen die Elektronenlücke durch Bildung weiterer Bindungen zu füllen. Solche Moleküle werden Lewis-Säuren, Elektrophile oder Elektronenpaar-Akzeptoren genannt. Im Folgenden werden Elektronenpaare durch Striche statt Punkte dargestellt. 3.2.2. Formalladungen Wie im vorhergehenden Abschnitt besprochen wurde, haben Lewis-Säuren eine grosse Elektronenaffinität, während Lewis-Basen nichtbindende Elektronenpaare besitzen. Analog zu den normalen Säure/Base Reaktionen finden auch bei diesen Verbindungen Neutralisationsreaktionen statt:
B
F
F
F
N
H
H
H
B
F
F
F
N
H
H
H Durch Bildung einer neuen Bildung mit dem freien Elektronenpaar des Ammoniak-Moleküls, wird jetzt die Oktettregel auch beim B-Atom erfüllt. Diese Art von Bindungen wird dative kovalente Bindung genannt. Das neue Molekül darf aber auch als das Produkt von zwei geladenen Teilchen (BF3
·– und NH3·+)
betrachtet werden, da die Elektronen nicht mehr vollständig zum N-Atom gehören. Die Ladung dieser fiktiven Teilchen wird als Formalladung bezeichnet. Sie wird durch folgende Gleichung bestimmt:
Formalladung eines Atoms in einer Lewis-Formel
= Anzahl der Valenzelektronen des freien Atoms
–
Anzahl der Elektronen in einsamen Elektronenpaaren
– 1/2 Anzahl der Bindungselektronen

40 Skript AC-OC I Beispiele:
CO2
NH4
+
N
H
H
H HN: 5 – 0 – 4 = +1H: 1 – 0 – 1 = 0
CO3
2-
––– –
––
3.2.3. Elektronegativität und Bindungsdipol Die Elemente weisen unterschiedliches Verhalten bezüglich ihre Elektronen in den entferntesten Schalen auf. Z. B. gibt K sein Elektron leicht ab, während F eine sehr grosse Elektronenaffinität besitzt. Die Elektronegativität EN besagt, wie ausgeprägt diese Anziehung ist. In der folgenden Abbildung ist die Pauling-Elektronegativität der Elemente dargestellt:
Wenn zwei Atome verschiedener Elemente eine kovalente Bindung bilden, werden sie die zwei gemeinsamen Elektronen mit unterschiedlicher Kraft anziehen. Z. B. werden die Elektronen in HCl mehr durch das Cl-Atom (EN = 3.16) als durch das H-Atom (EN = 2.20) angezogen. Die Bindung wird daher polarisiert, wobei sich die Bindungselektronen räumlich und zeitlich bevorzugt bei dem

Skript AC-OC I 41 Atom mit grösserer Elektronegativität aufhalten. Im Fall einer C–Li-Bindung zieht C (EN = 2.55) die Elektronen stärker an sich als Li (EN = 0.98). Dagegen sind in einer C–F-Bindung die Elektronen näher beim F-Atom (EN = 3.98) lokalisiert. Kovalente Bindungen, in denen die Elektronen sich nicht gleichmässig um die zwei Kerne verteilen, werden polare Bindungen genannt. Da sich die Elektronen zeitlich länger beim elektronegativeren Atom aufhalten, bildet sich ein gerichtetes Dipolmoment. Die Summe aller Dipolmomentvektoren eines Moleküls ergibt das Moleküldipol.
H Cl
Dipolmoment
H3C Cl
H
H
H
Cl ClCl
Cl
Cl ClCl
ClCl
Kein Moleküldipol
Moleküldipol Im Fall von Tetrachlormethan führt die Summe der Dipolmomentvektoren zur Aufhebung der Moleküldipol, obwohl die vier Bindungen polar sind. Die Moleküldipole sind sehr wichtig, um z. B. das Verhalten zwischen den Molekülen zu verstehen. 3.2.4. Homolyse und Heterolyse Kovalente Bindungen können auf zwei Arten gespalten werden. Bei der Homolyse entstehen nach der Zerstörung der Bindung zwei Atome mit leeren Valenzstellen. Dagegen findet bei der Heterolyse eine asymmetrische Trennung der Bindungen statt, wodurch zwei ionische Teilchen gebildet werden.
Homolyse: CH3–H CH3· + ·H Heterolyse: CH3–H CH3
– + H+
Die Energie, die für die Heterolyse notwendig ist, ist normalerweise sehr viel grösser als diejenige der Homolyse. Im Fall von Methan braucht z. B. die Homolyse 104 kcal/mol und die Heterolyse 400 kcal/mol. 3.2.5. Resonanztheorie In einigen Fällen ist es nicht möglich, die Elektronenstruktur mit Hilfe der Lewis-Formel zu beschreiben. Benzol dient als Paradebeispiel. Es wird normalerweise als ein Cyclohexanderivat mit drei Doppelbindungen dargestellt. Man würde daher erwarten, dass die drei C–C-Einfachbindungen länger als die C=C-Doppelbindungen sind. In Realität ist die Länge aller sechs Bindungen gleich. Sie liegt mit 1.40 Å zwischen derjenigen einer Einfachbindung und derjenigen einer Doppelbindung. In den Fällen, wo mehr als eine sinnvolle Lewis-Struktur für ein Molekül geschrieben werden kann, spricht man von Resonanz. Dieses Konzept wurde eingeführt, um die Beschränkungen des üblichen Modells überzuwinden. Die elektronischen Eigenschaften werden dann durch eine Überlagerung

42 Skript AC-OC I der verschiedenen möglichen Resonanzstrukturen mit unterschiedlicher Bewichtung der einzelnen Strukturen beschrieben. Diese unterscheiden sie sich nur durch die Verteilung der Elektronen in der Bindungen. Die räumliche Lage der Atomkerne im Molekül in den verschiedenen Strukturen ist identisch. Resonanzstrukturen werden durch Doppelpfeile () verbunden. Die Überlappung der einzelner Strukturen wird oft auch eine spezielle Schreibweise gekennzeichnet, die die Resonanzstrukturen in gekrümmten Linien zusammenfasst.
Br
Cl
Br
Cloder
Br
Cl
Ein Molekül, das durch mehrere Resonanzstrukturen beschrieben wird, ist stabiler als eine isolierte Lewis-Struktur. Dieser Effekt wird Resonanzstabilisierung genannt. Neben der Anzahl von Resonanzstrukturen ist es auch wichtig, deren relative Stabilitäten zu berücksichtigen. Eine Resonanzstruktur die ziemlich unstabil ist, wird kaum Einfluss auf die Stabilisierung des Moleküls haben. Um das Gewicht einer einzelnen Resonanzstruktur abzuschätzen, müssen die folgenden Punkte berücksichtigt werden:
Oktettstrukturen sind weitaus am stabilsten. In Resonanzstrukturen mit Ladungstrennung ist die negative Ladung bevorzugt auf dem
Atom mit der höchsten Elektronegativität lokalisiert. Die Ladungstrennung kostet Energie. Resonanzstrukturen mit Ladungstrennung tragen
weniger zur aktuellen Elektronenverteilung im Molekül bei als ungeladene Resonanzstrukturen.
Selbst wenn ein Molekül durch viele Resonanzstrukturen beschrieben werden kann, existiert in Wirklichkeit nur eine einzige Geometrie und eine einzige Elektronenverteilung! Die Resonanzstrukturen sind nur ein Hilfsmittel, um die Lewis-Formeln zu verwenden, auch wenn sie die Realität nicht richtig darstellen können. Beispiele.
3.3. Stereochemie Die Stereochemie beschäftigt sich mit der dreidimensionalen Struktur von Molekülen. 3.3.1. Das Tetraedermodell und seine Ausnahmen Die tetraedrische Anordnung der Valenzelektronenpaare eines Atoms führt zu einer tetraedrischen Anordnung der Substituenten. Dies gilt für die meisten Atome, die in der organischen Chemie wichtig sind und an Mehrfachbindungen beteiligt sind.
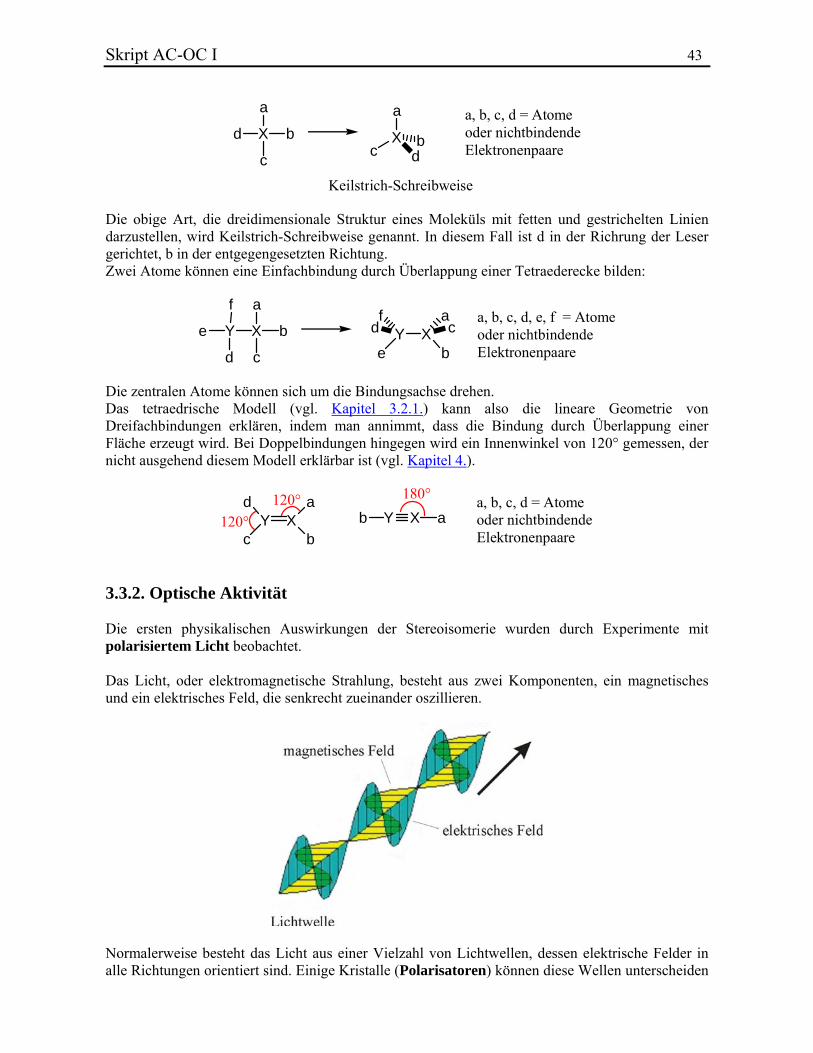
Skript AC-OC I 43
a
Xd
c
b
a
Xc d
b
a, b, c, d = Atomeoder nichtbindendeElektronenpaare
Keilstrich-Schreibweise Die obige Art, die dreidimensionale Struktur eines Moleküls mit fetten und gestrichelten Linien darzustellen, wird Keilstrich-Schreibweise genannt. In diesem Fall ist d in der Richrung der Leser gerichtet, b in der entgegengesetzten Richtung. Zwei Atome können eine Einfachbindung durch Überlappung einer Tetraederecke bilden:
a
XY
c
ba, b, c, d, e, f = Atomeoder nichtbindendeElektronenpaare
f
e
d
Y Xe b
f ad c
Die zentralen Atome können sich um die Bindungsachse drehen. Das tetraedrische Modell (vgl. Kapitel 3.2.1.) kann also die lineare Geometrie von Dreifachbindungen erklären, indem man annimmt, dass die Bindung durch Überlappung einer Fläche erzeugt wird. Bei Doppelbindungen hingegen wird ein Innenwinkel von 120° gemessen, der nicht ausgehend diesem Modell erklärbar ist (vgl. Kapitel 4.).
a, b, c, d = Atomeoder nichtbindendeElektronenpaare
Y Xad
c b
Y X ab120°
120°
180°
3.3.2. Optische Aktivität Die ersten physikalischen Auswirkungen der Stereoisomerie wurden durch Experimente mit polarisiertem Licht beobachtet. Das Licht, oder elektromagnetische Strahlung, besteht aus zwei Komponenten, ein magnetisches und ein elektrisches Feld, die senkrecht zueinander oszillieren.
Normalerweise besteht das Licht aus einer Vielzahl von Lichtwellen, dessen elektrische Felder in alle Richtungen orientiert sind. Einige Kristalle (Polarisatoren) können diese Wellen unterscheiden

44 Skript AC-OC I und nur diejenige mit einer bestimmten Orientierung des elektrischen Feldes durchlassen. Das Licht, dessen Wellen alle dieselbe Richtung des elektrischen Feldes besitzen, ist linear polarisiert. Jean-Baptiste Biot beobachtete im Jahr 1815, dass einige organischen Flüssigkeiten die Polarisationsebene von Licht drehen können. Darunter befinden sich z. B. Lösungen von Rohzucker und in einigen Fällen Weinsäure, je nach Herkunft. Um den Drehwinkel zu bestimmen, wird die folgende Apparatur verwendet:
Eine Lichtquelle erzeugt eine Reihe von Lichtwellen, deren Polarisationsebene zufällig angeordnet sind. Ein Polarisator (Nikolsches Prisma) ist nur für Wellen mit einer wohldefinierten Orientierung durchlässig. Das linear polarisierte Licht geht durch die optisch aktive Probe und seine Polarisationsebene wird um den Winkel gedreht. Ein Beobachter kann dann das polarisierte Licht nur dann sehen, wenn er einen zweiten Polarisator ebenfalls um den Winkel dreht. Der Betrag von ist eine stoffspezifische Grösse, die auch von der Wellenlänge des Lichtes, dem Lösungsmittel, der Temperatur, der Konzentration der optisch aktiven Verbindung und der Länge l der Probe abhängt. Sehr oft werden die spezifische Drehung [] oder die molare Drehung [] in der chemischen Literatur angetroffen:
100
,,LM
,,LM
Mcl
clcl
TT
T
ist der Drehwinkel in °, T die Temperatur, die Wellenlänge in nm, LM das Lösungsmittel, l die Länge der Probe in dm, c die Konzentration in g/mL und M die Molekularmasse. Drehungen im Uhrzeigersinn werden durch einen positiven Winkel bezeichnet, diejenigen im Gegenuhrzeigersinn durch einen negativen. Louis Pasteur (1822-1895) bewies im Jahr 1847, dass es in Natur zwei Formen von Weinsäure gab: eine, die die Polarisationsebene rotiert und eine zweite die darauf keinen Effekt hat. Weiter beobachtete er, dass die zweite Form nach Kristallisation zwei Arten von Kristallen gibt, die die Polarisation in entgegengesetzten Richtungen drehen. Im Laufe der Zeit wurden viele natürliche Verbindungen isoliert, die polarisiertes Licht drehen; sie wurden als optisch aktive Verbindungen bezeichnet. Optisch aktive Moleküle, die die Polarisationsebene im Uhrzeigersinn drehen, werden mit (+) vor dem Namen gekennzeichnet (z. B. (+)-Weinsäure), die andere durch (–). Es ist das Verdienst von Pasteur, die physikalische messbare Drehung von polarisiertem Licht auf die räumliche Struktur von Molekülen zurückzuführen.

Skript AC-OC I 45 3.3.3. Stereoisomere, Enantiomere und Diastereoisomere Kohlenstoff ist ein 4-wertiges Element und kann daher vier Substituenten binden. Wenn sie alle unterschiedlich sind, wird eine neue Isomerieart erzeugt. Stereoisomere sind Verbindungen, die dieselbe Konnektivität (Atom-Verbundenheit) besitzen und sich nicht durch Drehungen um Einfachbindungen ineinander überführen lassen (Konformationsisomerie, vgl. Kapitel 6.). Die unterstehenden Verbindungen zeigen ein C-Zentralatom, das ein F-, ein Cl-, ein Br- und ein I-Atom bindet. Obwohl ihre Konstitution gleich ist, sind sie räumlich unterschiedlich angeordnet. Es ist unmöglich, das eine Stereoisomer durch Drehungen in das andere überzuführen. Sie besitzen eine unterschiedliche Konfiguration
I
F
ClBr I
F
BrCl
Solche asymmetrische Verbindungen, die sich wie Bild und Spiegelbild zueinander verhalten, d. h. nicht zur Deckung gebracht werden können, sind chiral und werden Enantiomere oder optische Isomere genannt. Die Chiralität ist die notwendige und hinreichende Beziehung für das Auftreten von Enantiomeren. Ein Zentrum, das chiral ist, wird stereogenes Zentrum oder Stereozentrum genannt. Ethanol ist z. B. nicht chiral, da Bild und Spiegelbild gleich sind. Dagegen existieren zwei Enantiomere von 2-Iodbutan:
CH3
H OHH
CH3
HHOH
CH2CH3
H3C IH
CH2CH3
CH3IH
Spiegel
Spiegel
Ethanolkeine Stereoisomereachirales Molekülkein Drehwinkel
(+)-2-Iodbutan[] = + 15.9°rechtsdrehend
24589
(–)-2-Iodbutan[] = – 15.9°linksdrehend
24589
Die Komponenten eines Enantiomerenpaars haben immer dieselben chemischen und physikalischen Eigenschaften. Sie können aber durch Messung ihrer optischen Aktivität unterschieden werden. Der Betrag der spezifischen Drehung eines Enantiomerenpaars ist gleich, nur die Vorzeichen sind umgekehrt. Mischungen, die beide Enantiomere einer Verbindung in gleicher Menge enthalten, werden Racemate genannt. Die gegenseitigen Effekte der zwei Enantiomere heben sich auf; es wird daher keine optische Aktivität gemessen. Enantiomere können auch stark unterschiedliche biologische Aktivitäten zeigen. Z. B. riechen (–)-Carvon (aus Krauseminze) und (+)-Carvon (aus Kümmel) sehr verschieden.
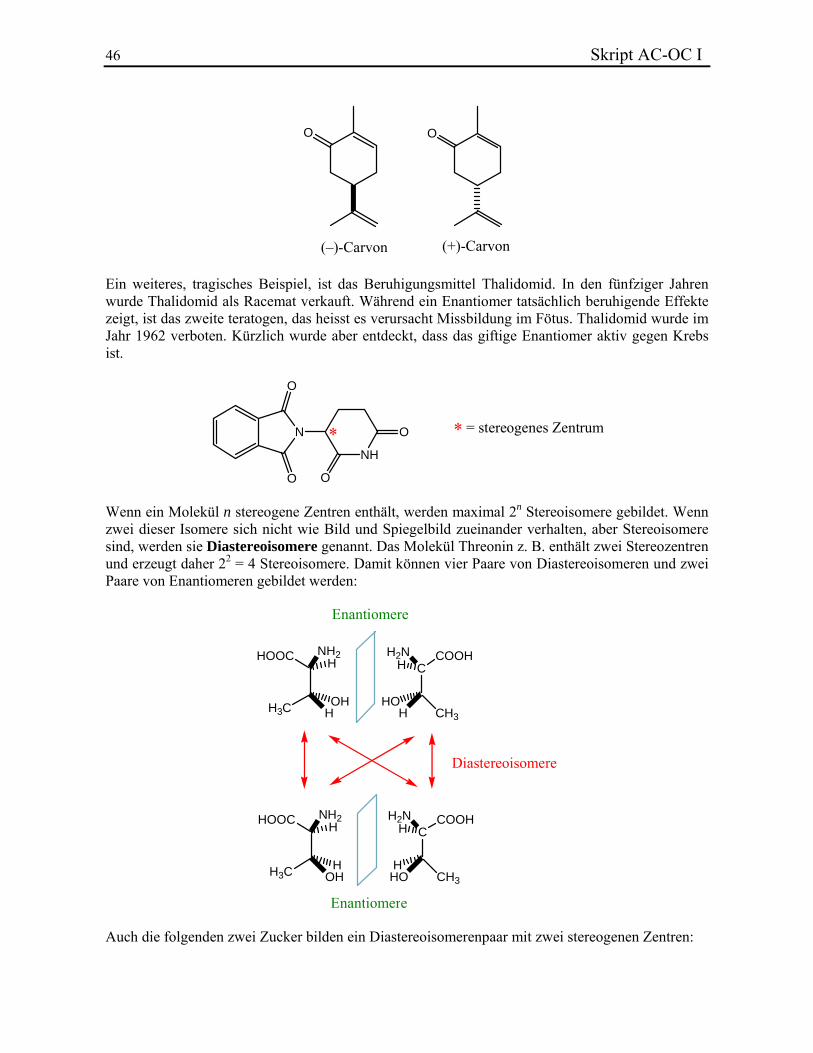
46 Skript AC-OC I
O O
(–)-Carvon (+)-Carvon Ein weiteres, tragisches Beispiel, ist das Beruhigungsmittel Thalidomid. In den fünfziger Jahren wurde Thalidomid als Racemat verkauft. Während ein Enantiomer tatsächlich beruhigende Effekte zeigt, ist das zweite teratogen, das heisst es verursacht Missbildung im Fötus. Thalidomid wurde im Jahr 1962 verboten. Kürzlich wurde aber entdeckt, dass das giftige Enantiomer aktiv gegen Krebs ist.
N
O
O
NH
O
O* = stereogenes Zentrum*
Wenn ein Molekül n stereogene Zentren enthält, werden maximal 2n Stereoisomere gebildet. Wenn zwei dieser Isomere sich nicht wie Bild und Spiegelbild zueinander verhalten, aber Stereoisomere sind, werden sie Diastereoisomere genannt. Das Molekül Threonin z. B. enthält zwei Stereozentren und erzeugt daher 22 = 4 Stereoisomere. Damit können vier Paare von Diastereoisomeren und zwei Paare von Enantiomeren gebildet werden:
H3C HOH
HOOC NH2H C
CH3HHO
COOHH2NH
H3C OHH
HOOC NH2H C
CH3HOH
COOHH2NH
Diastereoisomere
Enantiomere
Enantiomere Auch die folgenden zwei Zucker bilden ein Diastereoisomerenpaar mit zwei stereogenen Zentren:

Skript AC-OC I 47
HOH
O
H
HO
H OH
H
H
HOH
O
H
HO
HO H
H
H
(–)-Erythrose (–)-Threose In Gegensatz zu Enantiomeren zeigen Diastereoisomere unterschiedliche chemische und physikalische Eigenschaften. Das folgende Schema fasst die Beziehungen zwischen den verschiedenen Isomeriearten zusammen:
Isomere (gleiche Summenformel)
Ist die Konstitution gleich?Nein
Ja
Stereoisomere
Verhalten sie sich wieBild und Spiegelbild?
Konstitutionsisomere
NeinDiastereoisomere
Ja
Enantiomere 3.3.4. Beispiel: Zuckerstrukturaufklärung nach Fischer Dieses Beispiel wird hilfreich sein, um das Verständnis der obigen abstrakten Konzepte zu erleichtern. Dazwischen werden auch einige weitere Begriffe eingeführt. Die Zuckerstrukturaufklärung nach Fischer ist eines der schönsten Beispiele für logische Rückschlüsse in der Chemie. Emil Fischer publizierte im Jahr 1891 die exakte chemische Struktur von Glucose und anderen natürlich vorkommenden Zuckern. Zu dieser Zeit stand fast keine analytische Werkzeuge und fast keine unnatürliche Verbindung zu Verfügung. Er verfügte lediglich über folgende Informationen:
1. Die Molekularformel (durch Verbrennung bestimmbar) und die Molekularmasse. 2. Physikalische Eigenschaften wie Schmelzpunkt, Siedepunkt, Brechungsindex, optische
Aktivität, Farbe, Geruch und Geschmack. 3. Säuren und Basen.
Mit diesen primitiven Hilfsmittel dauerte die Arbeit an der Strukturaufklärung fast dreissig Jahre. Fischer gewann dafür den Nobelpreis im Jahr 1902.

48 Skript AC-OC I Fischer verwendete für die Strukturbestimmung zwei wichtigen Reaktionsklassen: die Oxidation und die Reduktion. Im Folgenden wird Oxidation ([O]) der Einfachheit wegen als Reaktion, die den Sauerstoffgehalt eines Moleküls erhöht, definiert. Dagegen erhöht eine Reduktion ([R]) die Anzahl der H-Atome. Z. B. können Alkohole einfach zu Aldehyden und danach zu Carbonsäuren oxidiert werden.
R OH
H H [O]
[R] R
O
H [R]
[O]
R
O
OH
mehr Hweniger O
weniger Hmehr O
Salpetersäure ist z. B. ein Reagenz, das Alkohole und Aldehyde direkt zu Carbonsäuren oxidieren kann. Fischer kannte nur vier natürlich vorkommende Zucker mit Summenformel (CH2O)6 (Hexose): Glucose, Mannose (aus Nüssen), Galactose (aus Feigen) und Fructose. Anhand der Summenformel lässt sich berechnen, dass das Molekül über ein Doppelbindungsäquivalent verfügt. Fischer beobachtete dann, dass Glucose, Mannose und Galactose in Gegensatz zu Fructose andere Verbindungen reduzieren können, was eine charakteristische Eigenschaft von Aldehyden ist. Er nahm daher an, dass Glucose, Mannose und Galactose Aldehyde sind (Aldose), während Fructose zu den Ketonen gehört (Ketose). Fischer wusste auch, dass jedes C-Atom maximal ein O-Atom trägt. Die allgemeine Struktur einer Aldose mit n C-Atomen kann daher wie folgt dargestellt werden:
C CH
O
H
OH
CH2
OH
n-2 (+)-Glyzerinaldehyd ist die einfachste Aldose, die eine optische Aktivität zeigt und daher ein stereogenes Zentrum besitzt:
HO
O
H
H
H
H
OH
[O]
[R]
HO
O
OH
H OH
O
HO OH
H
H
H
OH
H H*
Hydroxymalonsäure(nicht chiral)
(+)-Glyzerinaldehyd(Fischer kannte noch nichtdie exakte Konfiguration)
Glyzerin(nicht chiral)
Die Produkte der Oxidation und der Reduktion zeigen keine optische Aktivität. Zucker bilden sehr häufig Sirupe. Fischer suchte damals übrig einen Weg, Derivate dieser Verbindungen zu kristallisieren, um sie zu analysieren. Es war bekannt, dass Phenylhydrazin, unter Verlust von Wasser, mit Aldehyden eine neue Verbindungsklasse bildet (Phenylhydrazone). Fischer

Skript AC-OC I 49 entdeckte im Jahr 1884, dass Aldose mit drei Äquivalenten Phenylhydrazin ganz neue Verbindungen (Osazone) bildet:
RH
O
OH
HNNH2
RN
H
NNH
NH
C6H5
C6H5
* + 3
+ C6H5NH2 + NH3 + H2O Die Osazonbildung zerstört das stereogene Zentrum in Position 2, aber die Produkte sind einfach zu kristallisieren. Fischer wollte auch aus einfachen Zuckern die komplexere synthetisieren. Er entdeckte dafür, dass die Zugabe von Blausäure zu einer Zucker ergibt, nach säure Aufarbeitung, das erwünschte Produkt (Kiliani-Fischer-Reaktion). Damit konnte er ausgehend von (+)-Glyzerinaldehyd die zwei (CH2O)4-Aldose herstellen:
HO
O
H
H OH
HCN
HO CN
H OH
HO H
HO CN
H OH
H OH
HO
H OH
HO H
O
H
HO
H OH
H OH
O
H
*
(+)-Glyzerinaldehyd
*
* *
*
+
* *
* *
(–)-Threose
(–)-Erythrose
1) H3O+
2)
3) NaBH4
1) H3O+
2)
3) NaBH4
Die Verwendung der üblichen Keilstrich-Schreibweise kann für Zucker ziemlich mühsam werden. Fischer führte daher eine neue Konvention ein, die Fischer-Projektion genannt wird. Die längste Kette im Molekül wird durch eine vertikale Linie dargestellt. Die Bindungen des Moleküls werden so gedreht, dass alle Substituenten über dieser Kette zu liegen kommen. Schliesslich wird das Molekül auf die Ebene projiziert:
c
d
b
ac a
b d
d
b
c a
Fischer-Projektion Einer Drehung der Fischer-Projektion um 90° führt ein Enantiomer in das andere über:
50 Skript AC-OC I
H
CHO
CH2OH
OH
HOH2C CHO
H OH
H
CHOHOH2C
OH
CHO
CH2OH
H OH
HOH2C CHO
H OH
HOH2C CHO
HO H
H
CHO
CH2OH
OH
H
CHO
CH2OH
HO
HOH2C CHO
H OH
(+)-Glyzerinaldehyd
90° 90°
(+)-Glyzerinaldehyd (–)-Glyzerinaldehyd (+)-Glyzerinaldehyd Die bisher verwendete (+),(–)-Nomenklatur stützt sich auf der optische Aktivität der chiralen Verbindungen, deren Messung nicht immer möglich ist und nicht einfach mit der dreidimensionalen Struktur eines Moleküls in Beziehung gebracht werden kann. Ausgehend von der Fischer-Projektion wurde ein neues Nomenklatursystem vorgeschlagen (D,L-System), das noch in der Zuckerchemie verwendet wird. Im Fall von Zuckern, muss man die Fischer-Projektion so drehen, dass das höchst oxidierte Kettenende oben liegt. Wenn die am weitesten entfernte Hydroxygruppe eines chiralen Zentrums nach rechts scheint, wird die Konfiguration als D bezeichnet, sonst als L.
CHO
H OH
CH2OH
H OH
CHO
HO H
CH2OH
H OH
CHO
H OH
CH2OH
D-Glyzerinaldehyd D-Threose D-Erythrose Oft wird noch die Richtung der spezifischen Drehung angegeben (z. B. D-(–)-Threose, L-(+)-Threose). Wir sind bisher davon ausgegangen, dass wir die richtige, absolute Konfiguration der Zucker kennen, dies traf auf Fischer natürlich nicht zu. Er begann daher seine Arbeit unter der Annahme, dass die absolute Konfiguration von (+)-Glyzerinaldehyd D war. Die absolute Konfiguration wurde erst im Jahr 1954 eindeutig bestimmt. Fischer war in der Lage, die relativen Konfigurationen der Zucker mit Hilfe der Oxidation mit HNO3 zu bestimmen. Im Folgenden wird das Beispiel von Threose und Erythrose besprochen:

Skript AC-OC I 51
CHO
HO H
CH2OH
H OH
HNO3
CHO
H OH
CH2OH
HO H
HNO3
COOH
H OH
COOH
HO H
COOH
HO H
COOH
H OHD-Threose
L-Threose
D-Weinsäure
L-Weinsäure
CHO
H OH
CH2OH
H OH
HNO3
CHO
HO H
CH2OH
HO H
HNO3
COOH
HO H
COOH
HO H
COOH
H OH
COOH
H OHD-Erythrose
L-Erythrose
selbe Verbindungoptisch inaktiv
meso-Weinsäure
Die Oxidation von L- und D-Threose führt zu zwei verschiedenen Enantiomeren von Weinsäure, die natürlich eine unterschiedliche optische Aktivität zeigen. Die Oxidation von D- und L-Erythrose führt dagegen zu derselben Verbindung, die optisch inaktiv ist. Moleküle, die zwar über stereogene Zentren verfügen, aber dennoch optisch inaktiv sind, nennt man meso-Verbindungen. Die Ursache liegt in einer internen Spiegelebene, die die optische Aktivität gegenseitig aufhebt. Im vorliegenden Fall ist dies nur möglich, wenn die zwei Hydroxygruppen auf derselben Seite der Fischer-Projektion zu liegen kommen. Ob es sich dabei um einen D- oder L-Zucker handelt, spielt keine Rolle. Es folgt daher, dass in der Threose die Hydroxygruppen aneinander gegenüber liegen. Die D-Zucker wurden so genannt, weil sie nur ausgehend von D-Glyzerinaldehyd hergestellt werden konnten. Fischer war aber nicht sicher, ob die entfernste chirale Hydroxygruppe tatsächlich auf der rechten Seite der Fischer-Projektion war. Analog nahm Fischer für die Strukturaufklärung der Hexose an, dass die natürliche Glucose die Konfiguration D besitzt. Er hatte nur eine 50%-ige Wahrscheinlichkeit, aber 1954 wurde bewiesen, dass er richtig lag. Da nun ein Stereozentrum eine fixierte Konfiguration hat, bleiben drei stereogene Zentren, die die folgenden acht Strukturen erzeugen:

52 Skript AC-OC I
CHO
OHH
OHH
OHH
OHH
CH2OH
CHO
HHO
OHH
OHH
OHH
CH2OH
CHO
OHH
HHO
OHH
OHH
CH2OH
CHO
HHO
HHO
OHH
OHH
CH2OH
CHO
OHH
OHH
HHO
OHH
CH2OH
CHO
HHO
OHH
HHO
OHH
CH2OH
CHO
OHH
HHO
HHO
OHH
CH2OH
CHO
HHO
HHO
HHO
OHH
CH2OH
1 2 3 4
5 6 7 8 Das weiter Vorgehen war wie folgt:
1. D-Glucose und Mannose bilden dasselbe Osazon. Da während der Synthese nur das stereogene Zentrum in Position 2 zerstört wird, müssen die Stereozentren an den Stellen 3, 4 und 5 beider Zucker gleich sein. Daher müssen Glucose und Mannose einem der Paare (1;2), (3;4), (5;6) und (7;8) entsprechen.
2. D-Glucose und D-Mannose werden durch Salpetersäure zu optisch aktiven Carbonsäuren
oxidiert. 1 und 7 würden aber meso-Verbindungen erzeugen, die keine optische Aktivität besitzen. Daher können 1 mit 2 und 7 mit 8 ausgeschlossen werden. Folglich verbleiben nur noch die Paare (3;4) und (5;6).
3. Eine Kiliani-Fischer-Reaktion bildet ausgehend von Arabinose D-Glucose und D-Mannose.
Es folgt, dass Arabinose dieselbe Konfiguration von D-Glukose in Position 2, 3 und 4 besitzt. Wenn D-Arabinose die Konfiguration 9 hat, werden 3 und 4 gebildet, hingegen führt 10 zu 5 und 6:
CHO
HO H
H OH
CH2OH
H OH
CHO
H OH
HO H
CH2OH
H OH
CHO
OHH
HHO
OHH
OHH
CH2OH
CHO
OHH
OHH
HHO
OHH
CH2OH
CHO
HHO
HHO
OHH
OHH
CH2OH
CHO
HHO
OHH
HHO
OHH
CH2OH
3 4
5 6
9
10

Skript AC-OC I 53
4. D-Arabinose gibt nach Oxidation eine optisch aktive Carbonsäure. Die Oxidation von 10 wurde aber zu einer optisch inaktiven Verbindung führen. D-Arabinose besitzt daher die Konfiguration dargestellt in 9 und D-Glucose und D-Mannose entsprechen dem Paar (3;4).
CHO
H OH
HO H
CH2OH
H OH
CHO
HO H
H OH
CH2OH
H OH
COOH
H OH
HO H
COOH
H OH
COOH
HO H
H OH
COOH
H OH
9
10
optisch aktiv
optisch inaktivmeso-Verbindung
5. Fischer entdeckte eine neue Methode, um die zwei Enden eines Zuckers auszutauschen. Wenn die –CHO und die –CH2OH Gruppe ausgetauscht werden, stellt man aus 3 ein neues Produkt her. Dagegen würde 4 nur das Ausgangsmaterial liefern.
CHO
OHH
HHO
OHH
OHH
CH2OH
CHO
HHO
HHO
OHH
OHH
CH2OH
CH2OH
HHO
HHO
OHH
OHH
CHO
CH2OH
OHH
HHO
OHH
OHH
CHO
CHO
HHO
HHO
OHH
OHH
CH2OH
CH2OH
HO H
H OH
HO H
HO H
CHO
3
4
180°
4
180°
D-Glucose erzeugt eine neue Verbindung, daher ist D-Glucose 3 und D-Mannose 4:
CHO
OHH
HHO
OHH
OHH
CH2OH
CHO
HHO
HHO
OHH
OHH
CH2OH
D-Glucose D-Mannose

54 Skript AC-OC I
6. Fructose hat keine reduzierende Wirkung, folglich muss er sich um eine Ketose handeln. Sie liefert aber dasselbe Osazon wie D-Glucose. Die drei entfernsten Hydroxygruppen müssen daher dieselbe Konfiguration, nämlich derjenige der D-Glucose besitzen. Daher hat D-Fructose die folgende Struktur:
CH2OH
O
HO H
H OH
H OH
CH2OH
D-Fructose 3.3.5. Absolute Konfiguration und CIP-Regeln Bis zu den fünfziger Jahren konnte nur die relative Konfiguration von chiralen Verbindungen bestimmt werden. Nach der Einführung einer neuen analytischen Methode (Röntgenstrukturanalyse) wurde es aber möglich, die absolute Konfiguration, d. h. eine Konfiguration ohne relative Bezüge zu benachbarten Substanzen, jedem Stereozentrum zuzuordnen. Um die verschiedenen Stereoisomeren zu benennen, wurde ursprünglich das D,L-System verwendet. Heutzutage wird aber das systematischere und allgemeingültige R,S-System benützt. Dieses Nomenklatursystem geht direkt von der dreidimensionalen Struktur des Moleküls aus, um die absolute Konfiguration von Stereoisomeren zu spezifizieren. Die vier verschiedene Substituenten eines chiralen Zentrums werden dabei in einer Sequenz angeordnet, aus der sich die Konfiguration (R oder S) abgeleiten lässt. Man folgt der folgenden Prozedur:
1. Die vier Substituenten eines stereogenen Zentrums werden identifiziert. Jedem der vier Substituenten wird unter der Benutzung bestimmter Sequenzregeln die Prioritäten a, b, c, d zugeordnet, so dass gilt: a > b > c > d.
2. Das Molekül wird räumlich so angeordnet, dass man entlang der Bindung vom
Stereozentrum zum Substituenten mit der niedrigsten Priorität d schaut:
a
d cb
3. Man betrachtet nun das Molekül in dieser Orientierung und verbinde die Substituenten in der Reihenfolge a über b zu c. Bewegt man sich dabei im Uhrzeigersinn, so wird das Stereozentrum R (rectus) genannt, verläuft die Bewegung entgegen dem Uhrzeigersinn, dann wird das Stereozentrum S (sinister) genannt.

Skript AC-OC I 55
a
c b b
a
c
d d
R S Für die Zuordnung der Prioritäten werden die Regeln von Cahn, Ingold und Prelog (CIP-Regeln) angewendet. Für die Zuordnung gelten die folgenden Punkte in der abnehmender Priorität:
1. Höchste Ordnungszahl des ersten Atoms am Stereozentrum: z. B. 53I > 35Br > 17Cl > 16S > 9F > 8O > 7N > 6C > 1H > freies Elektronenpaar.
2. Höchste Massenzahl des ersten Atoms am Stereozentrum: z. B. 2D > 1H. 3. Wenn mehrere Substituenten dieselbe Ordnungszahl und dieselbe Massenzahl besitzen, so
wird der Substitutionsgrad berücksichtigt. Dasjenige Atom, welches mit anderen Atomen höherer Ordnungszahl verbunden ist, geht voran. Sind die beide Atome auch in dieser Richtung gleichwertig, so hat dasjenige Atom die höhere Priorität, welches mit mehr Atomen höherer Ordnungszahl verbunden ist. Erlaubt das zweitinnerste Atom keine Entscheidung, so geht man zum dritten über, usw. Z. B. CCl3 > COCl > COOR > COOH > CONH2 > COR > CHO.
4. Mehrfach gebundene Atome werden durch Verdoppelung (bei Doppelbindungen) bzw. Verdreifachung (bei Dreifachbindungen) durch anbringen von Phantomatomen dargestellt:
O
R
R
R
R
R
R
R R
(O)
CR
R
O
(C)
R C C R
(C)(C)
(C) (C)
(C)
CR
R
C
(C)
R
R R N R C N
(C)(N)
(N) (C) Weitere Unterregeln können in der Literatur gefunden werden. Beispiele. Dieselbe Regeln gelten auch für chirale Zentren, die nicht C-Atome sind:
N
O
N Si P As
Auch 3-wertige Elemente bilden Stereozentren, wenn das zentrale Atom nicht in der Ebene der drei Substituenten liegt. Das freie Elektronenpaar dient als vierter Substituent:
S SO
P
Bei Aminen und Oxoniumsalzen ist das Chiralitätszentrum nicht stabil, da die Konfiguration sehr schnell invertiert:

56 Skript AC-OC I
X
b
a c
X
b
ac
X = N, O
S R 3.3.6. Stereoisomerie bei Doppelbindungen Doppelbindungen können sich bei Raumtemperatur nicht um die Bindungsachse drehen. Unterschiedliche Substituenten können daher zu Diastereoisomere führen:
H3C
CH3
H
H
H3C
H H
CH3
Eine alte Nomenklatur, die aber noch häufig verwendet wird, gibt die relative Anordnung der Substituenten durch die Silben cis (auf derselben Seite) und trans (auf verschiedenen Seiten) an. Diese Nomenklatur wird auch für die Angabe der relativen Konfigurationen von Substituenten verwendet und muss nicht zwingend auf den CIP-Regeln basieren. Die Verwendung von cis und trans sei daher mit Vorsicht zu geniessen.
H3C
CH3
H
H
H3C
H H
CH3
cis-2-Butentrans-2-Buten Da diese Nomenklatur nicht eindeutig ist, wurde das E,Z-System eingeführt, das den CIP-Regeln für die Bestimmung der Priorität der Substituenten folgt. Befinden sich die Gruppen höchster Priorität jedes Atoms auf derselben Seite der Doppelbindung, ist die Verbindung ein Z-Isomer (zusammen). Umgekehrt spricht man von E-Isomer (entgegen). Beispiele. 3.3.7. Stereoisomere ohne Stereozentrum Bisher haben wir nur über Stereoisomerie in Verbindung mit Stereozentren oder Doppelbindungen gesprochen. Ein Chiralitätszentrum muss aber nicht immer Sitz eines Atoms sein. Z. B. liegt im folgenden Molekül das stereogene Zentrum im Molekülzentrum:
d
a c
b Weiter können Achsen, Ebenen oder Helizes Chiralitätselemente sein.
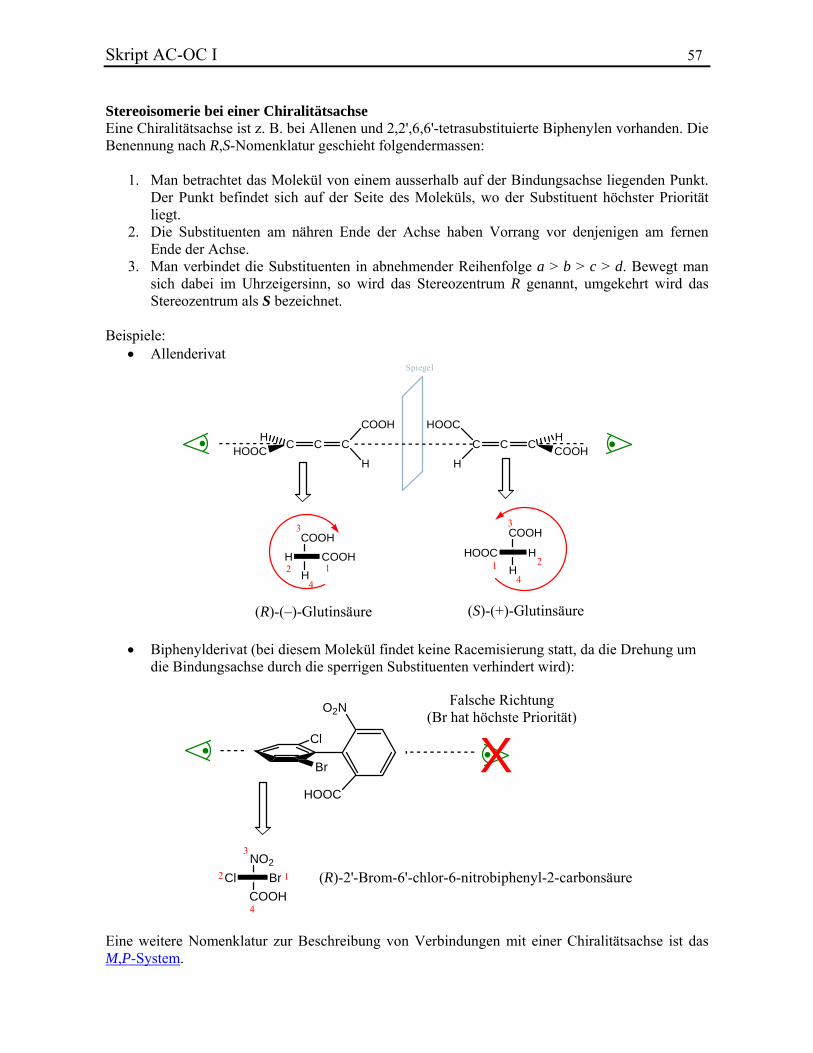
Skript AC-OC I 57 Stereoisomerie bei einer Chiralitätsachse Eine Chiralitätsachse ist z. B. bei Allenen und 2,2',6,6'-tetrasubstituierte Biphenylen vorhanden. Die Benennung nach R,S-Nomenklatur geschieht folgendermassen:
1. Man betrachtet das Molekül von einem ausserhalb auf der Bindungsachse liegenden Punkt. Der Punkt befindet sich auf der Seite des Moleküls, wo der Substituent höchster Priorität liegt.
2. Die Substituenten am nähren Ende der Achse haben Vorrang vor denjenigen am fernen Ende der Achse.
3. Man verbindet die Substituenten in abnehmender Reihenfolge a > b > c > d. Bewegt man sich dabei im Uhrzeigersinn, so wird das Stereozentrum R genannt, umgekehrt wird das Stereozentrum als S bezeichnet.
Beispiele:
Allenderivat
C C C
COOH
HHOOC
HCCC
HOOC
HCOOHH
H
COOH
HOOC H
H
COOH
H COOH
Spiegel
12
3
4
1 2
3
4
(R)-(–)-Glutinsäure (S)-(+)-Glutinsäure
Biphenylderivat (bei diesem Molekül findet keine Racemisierung statt, da die Drehung um die Bindungsachse durch die sperrigen Substituenten verhindert wird):
Br
Cl
O2N
HOOC
X
COOH
NO2
Cl Br
Falsche Richtung(Br hat höchste Priorität)
12
3
4
(R)-2'-Brom-6'-chlor-6-nitrobiphenyl-2-carbonsäure
Eine weitere Nomenklatur zur Beschreibung von Verbindungen mit einer Chiralitätsachse ist das M,P-System.
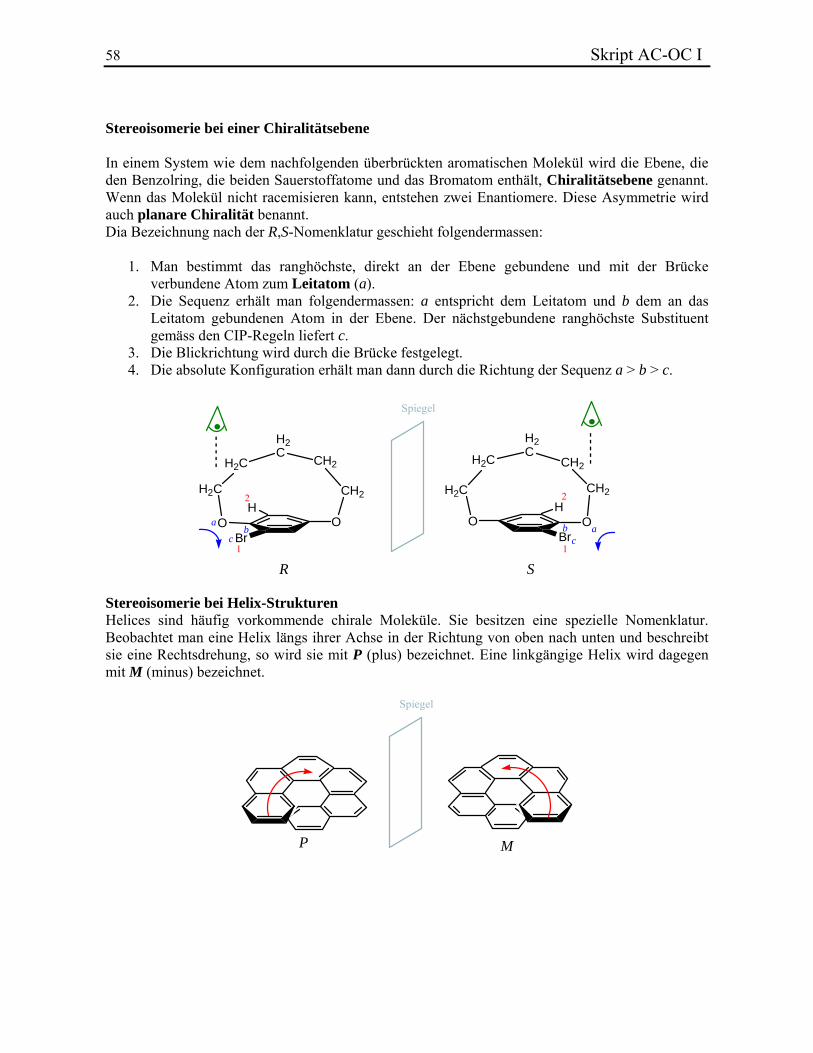
58 Skript AC-OC I Stereoisomerie bei einer Chiralitätsebene In einem System wie dem nachfolgenden überbrückten aromatischen Molekül wird die Ebene, die den Benzolring, die beiden Sauerstoffatome und das Bromatom enthält, Chiralitätsebene genannt. Wenn das Molekül nicht racemisieren kann, entstehen zwei Enantiomere. Diese Asymmetrie wird auch planare Chiralität benannt. Dia Bezeichnung nach der R,S-Nomenklatur geschieht folgendermassen:
1. Man bestimmt das ranghöchste, direkt an der Ebene gebundene und mit der Brücke verbundene Atom zum Leitatom (a).
2. Die Sequenz erhält man folgendermassen: a entspricht dem Leitatom und b dem an das Leitatom gebundenen Atom in der Ebene. Der nächstgebundene ranghöchste Substituent gemäss den CIP-Regeln liefert c.
3. Die Blickrichtung wird durch die Brücke festgelegt. 4. Die absolute Konfiguration erhält man dann durch die Richtung der Sequenz a > b > c.
BrO O
H2C
H2C
H2C
CH2
CH2
Spiegel
BrOO
CH2
CH2
H2C
H2C
H2C
ab
c1
2H H
2
1c
b a
R S Stereoisomerie bei Helix-Strukturen Helices sind häufig vorkommende chirale Moleküle. Sie besitzen eine spezielle Nomenklatur. Beobachtet man eine Helix längs ihrer Achse in der Richtung von oben nach unten und beschreibt sie eine Rechtsdrehung, so wird sie mit P (plus) bezeichnet. Eine linkgängige Helix wird dagegen mit M (minus) bezeichnet.
Spiegel
MP

Skript AC-OC I 59
3.4. Symmetrielehre 3.4.1. Punktgruppen Um die Geometrie eines Moleküls zu beschreiben, werden Symmetrieelemente wie z. B. Punkte, Gerade und Ebene verwendet. Eine Symmetrieoperation ist eine Bewegung eines dreidimensionalen Gebildes in einem festgelegten Koordinatensystem. Eine Spiegelung z. B. entspricht einer Drehung um 180° so dass der Anfangs- und Endzustand ununterscheidbar sind. Es werden vier Arten von Symmetrieelementen definiert:
-Ebenen sind Spiegelebenen, die eine geometrische Figur so in zwei Teile unterteilt, dass diejenige Hälfte auf der einen Seite der Ebene, genau dem Spiegelbild der Hälfte auf der anderen Seite entspricht.
H
HBrBr
Cn-Achsen sind Geraden, die so durch ein Molekül verlaufen, dass man bei einer Drehung des Moleküls um 360°/n (n = 1,2,...) um diese Gerade eine dreidimensionale Anordnung erhält, die von der ursprünglichen Figur nicht zu unterscheiden ist.
I
I
C2
Drehspiegelachsen Sn sind eine Kombination von - und Cn-Symmetrieelementen. Diese Symmetrie wird beobachtet, wenn ein Molekül mit einer Spiegelung an einer -Ebene und einer Drehung um 360°/n um eine Gerade, die senkrecht zur Spiegelebene liegt, in sich selbst übergeführt wird.
H Cl
Cl H
C2 S2
1) Spiegelung2) Drehung um 180°
Inversionszentrum i: Ein Molekül ist inversionssymmetrisch, wenn durch die Spiegelung aller Substituenten am Inversionszentrum das Molekül unverändert bleibt.

60 Skript AC-OC I
CH3
H3C
i
Die Moleküle können in Abhängigkeit ihrer Symmetrieelemente in verschiedene Gruppen unterteilt werden. Diese Gruppe werden als Punktgruppen bezeichnet. Im Folgenden sind die Abkürzungen der verschiedenen Punkgruppen aufgelistet: Nicht axiale Punktgruppen C1 oder I Identität, keine Symmetrieelement. Cs Nur eine Symmetrieebene . Ci Nur ein Inversionszentrum i. Axiale Punktgruppen Punktgruppe Cn
Hauptachse
C2' Nebenachsen senkrecht zur Hauptachse
v vertikale Spiegelebenen enthaltend die Hauptachse
h horizontale Spiegelebenen senkrecht zur Hauptachse
Sonstige
Cn 1 Dn 1 n Cnv 1 n Cnh 1 1 Sn; i bei geradem n Dnh 1 n n 1 Sn; i bei geradem n Dnd 1 n n S2n; i bei
ungeradem n Sn Cn/2 Sn, i bei n/2
ungerade Kubische Punktgruppen Td Tetraedrische Symmetrie: 4 C3, 3 C2, 6 , 3 S4 (z. B. CH4). Oh Oktaedrische Symmetrie: 3 C4, 4 C3, 6 C2, 9 , 3 S4, 4 S6, i (z. B. SF6). ... Spezielle Punktgruppen Cv C, (z. B. Cl–CC–H). Dh C, v, i, S, C2', h (z. B. H–CC–H). Ih Insgesamt 120 Symmetrieelemente (z. B. C60). Zur Bestimmung der Punktgruppe eines Moleküls ist es oft sehr hilfreich, ein Flussdiagramm zu verwenden. Beispiele. 3.4.2. Molekülsymmetrie und Chiralität Die Punktgruppen finden viele Anwendungen in allen Bereichen der Chemie. Im Rahmen dieser Vorlesung werden wir nur die Beziehung zwischen Chiralität und Symmetrie betrachten.

Skript AC-OC I 61 Moleküle sind chiral, wenn Bild und Spiegelbild nicht deckungsgleich sind. Ausgehend aus der Symmetrielehre kann man beweisen, dass ein Molekül chiral ist, wenn es zu den Punktgruppen C1, Cn oder Dn gehört.
CH3
CH3
H
H
H
CH3
CH3
H
H3C CH3
H H
H3C HH CH3
C2
C2
cis-IsomerCs Punktgruppeachiral
trans-IsomerC2 Punktgruppechiral
Weitere Beispiele. 3.4.3. Topizität Die Topizität beschreibt die Äquivalenz oder Nichtäquivalenz von Atomen oder Gruppen in einem Molekül. Zwei Gruppen sind homotop (oder äquivalent), wenn sie durch eine Drehung des Moleküls um eine Drehachse ineinander überführt werden können. Moleküle mit homotopen Gruppen müssen also eine Cn-Achse (n > 1) aufweisen. Solche Gruppen befinden sich in gleicher Umgebung innerhalb des Moleküls und sind nicht voneinander unterscheidbar. Die Substitution der einen oder anderen homotopen Gruppe durch irgendeine andere Gruppe liefert identische Moleküle. Man nennt Gruppen enantiotop, wenn sie durch eine Drehspiegelung des Moleküls ineinander überführt werden können. Moleküle mit enantiotopen Gruppen müssen also eine Sn-Achse (n 1) aufweisen. Solche Gruppen befinden sich in zueinander enantiomeren Umgebungen innerhalb des Moleküls und sind in achiralen Medien nicht unterscheidbar, können aber in chiralen Medien oder mit chiralen Reagenzien unterschieden werden. Die Substitution der einen oder der anderen enantiotopen Gruppe durch irgendeine andere achirale Gruppe liefert zueinander enantiomere Moleküle, durch irgendeine andere chirale Gruppe zueinander diastereomere Moleküle. Man nennt Gruppen diastereotop, wenn sie die gleiche Konstitution besitzen, aber durch keinerlei Symmetrieoperationen ineinander überführt werden können. Solche Gruppen befinden sich in zueinander diastereomeren Umgebungen innerhalb des Moleküls und sind deshalb in achiralen und chiralen Medien unterscheidbar. Die Substitution der einen oder der anderen diastereotopen Gruppe durch irgendeine andere Gruppe liefert zueinander diastereomere Moleküle. In der folgenden Abbildung ist ein Flussdiagramm zur Klassifizierung der Topizität von Gruppen gezeigt:

62 Skript AC-OC I
Haben die Gruppen die gleichechemische Verbundenheit?
Nein
Ja
Sind die Gruppen mittels einerDrehung ineinander überführbar?
Sind die Gruppen durchDrehspiegelungen (Sn)
ineinander überführbar?
verschieden
Nein
enantiotopJa
diastereotop
homotopJa
Nein
Die Topizität spielt eine wichtige Rolle in der Kernresonanzspektroskopie (vgl. Vorlesungen Analytische Chemie). Zueinander homotope und enantiotope Atomkerne ergeben in achiralen Lösungsmitteln Signale an der gleichen Stelle. In chiralen Lösungsmitteln sind enantiotope Kerne unterscheidbar. Diastereotope und verschiedene Kerne geben immer verschiedene Signale. Beispiele.

KAPITEL 4:
EINFÜHRUNG IN DIE QUANTENCHEMIE In diesem Kapitel werden folgende Themen behandelt: 4.1. Atomorbitale 4.2. Chemische Bindungen 4.3. Geometrie eines Moleküls und Hybridisierung 4.4. Konjugierte Systeme
Werner Heisenberg

64 Skript AC-OC I
4.1. Atomorbitale Um die Bewegung eines Elektrons um einen Kern zu beschreiben, muss man seine Position und seine Energie kennen. Um diese Grössen zu beschreiben, wurden viele Atommodelle vorgeschlagen. Das aktuellste wurde nach der Einführung der Quantenphysik vorgelegt. Gemäss diesem Modell verhalten sich die Elektronen nicht wie Planeten zu ihrer Sonne, um die alle denkbaren Bahnen besetzt sein können. Für Elektronen um einen Kern sind dagegen nur ganz bestimmte Werte für Energie und Abstand erlaubt. Dazu wurde es auch bewiesen, dass das klassische Konzept von Teilchen und Welle nicht auf Elementarteilchen angewendet werden darf (De Broglie). Jedes Elektron wird in der Tat durch eine Wellenfunktion charakterisiert. Obwohl keine klassische physikalische Bedeutung hat, gibt die Grösse 2 die Wahrscheinlichkeitsverteilung des Elektrons an, d. h. die Wahrscheinlichkeit dass sich das Elektron innerhalb eines bestimmten Raums befindet. Ausgehend von den Wellenfunktionen können die erlaubten Energiezustände der Elektronen berechnet werden. Generell finden die chemischen Reaktionen statt, nur um die Gesamtenergie eines Moleküls zu erniedrigen. Es ist deshalb sehr wichtig, die relativen Energien der Elektronen zu kennen. Die Energiezustände sind durch vier ganze Zahlen charakterisiert:
Die Hauptquantenzahl n unterscheidet die Hauptenergieniveaus oder Schalen. Sie hängt vom mittleren Abstand zwischen Elektron und Kern ab und seine mögliche Werte gehen von 1 bis .
Die Orbitalquantenzahl (oder Nebenquantenzahl) l bestimmt den Drehimpuls des Elektrons. l kann nur Werte zwischen 0 und n-1 annehmen (0 l < n), d. h. dass jede Schale n Unterschalen besitzt. Normalerweise werden die Unterschalen nicht durch Nummern bezeichnet, sondern durch Buchstaben. Zustände mit l = 0 werden als s definiert, mit l = 1 als p, mit l = 2 als d, usw.
Die magnetische Quantenzahl m bezeichnet die möglichen Orientierungen des Elektrons in einem magnetischen Feld. -m l m: jede Unterschale besitzt deshalb 2l + 1 Atomorbitale.
Die Spinquantenzahl s gibt die Orientierung des Elektronenspins an, der als eine Art von Drehimpuls angesehen werden kann. Sie darf nur die Werte 1/2 oder –1/2 nehmen.
Gemäss den obigen Definitionen wird ein Atomorbital durch eine Kombination von n, l und m eindeutig beschrieben. In der folgenden Tabelle sind die energetisch niedrigsten Atomorbitale gezeigt (1s ist das energieärmste Orbital):
n L m Symbol vierte Schale 4 3 -3, -2, -1, 0, 1, 2, 3 4f 2 -2, -1, 0, 1, 2 4d 1 -1, 0, 1 4p 0 0 4s dritte Schale 3 2 -2, -1, 0, 1, 2 3d 1 -1, 0, 1 3p 0 0 3s zweite Schale 2 1 -1, 0, 1 2p 0 0 2s erste Schale 1 0 0 1s

Skript AC-OC I 65 Da das Wichtigste in der Chemie die Minimierung der Gesamtenergie eines Systems ist, ist es sehr wichtig die relative Energie der Orbitale zu kennen. Einige Regeln sind dafür nützlich:
Je grösser n ist, desto grösser ist die mittlere Energie der Schale. Innerhalb derselben Schale, besitzen Orbitale mit grösseren l die grössere Energie. In Abwesenheit eines äusseren magnetischen Felds besitzen die Orbitale mit gleichen n und
l dieselbe Energie. Orbitale, die dieselbe Energie haben, sind entartet.
In der folgenden Abbildung ist ein qualitatives Energieniveaudiagramm der ersten Atomorbitale des Wasserstoffatoms gezeigt:
Die Gesamtenergie eines Systems ist durch die Summe der Energien aller Elektronen gegeben. Die Gestalt (die Form) und die Symmetrie eines Orbitals sind wichtig für qualitative Aussagen über der Reaktivität von Atomen. In der folgenden Abbildung sind die Wellenfunktionen der Orbitale s, pz, py und px gezeigt. Orbitalen mit l > 0 besitzen verschiedene Teile, deren Vorzeichen unterschiedlich sind. Sie werden durch hellere und dunklere Farben unterschieden.
x
y
z
x
y
z
s(l = 0)
x
y
z
pz
(l = 1)
x
y
z
py
(l = 1)px
(l = 1)

66 Skript AC-OC I Um die Elektronen in den Orbitalen anzuordnen, müssen drei Regeln berücksichtigt werden (Aufbauprinzip):
Je weniger Energie ein System besitzt, desto stabiler ist es. Jedes Orbital darf mit maximal zwei Elektronen besetzt sein (Pauliprinzip). Wenn es mehrere Orbitale gleicher Energie gibt, besetzen die Elektronen unterschiedliche
Orbitale (Hund’sche Regel). Die Elektronenkonfiguration beschreibt die Verteilung der Elektronen innerhalb der Atomorbitale. Das Wasserstoff Atom besitzt nur ein Elektron, das sich im tiefliegenden 1s-Orbital befindet. Eine solche Konfiguration wird durch 1s1 abgekürzt. Im Helium ist auch das zweite Elektron im 1s-Orbital: die Konfiguration ist deswegen 1s2. Das dritte Elektron in Lithium muss das nächste Orbital besetzen, das 2s-Orbital: die Konfiguration ist daher 1s22s1. Mit derselben Methode können die Elektronenkonfigurationen aller Elemente geschrieben werden:
H 1s1 He 1s2 1. Schale vollständig besetzt Li 1s22s1
Be 1s22s2 B 1s22s22p1 C 1s22s22p2 N 1s22s22p3 O 1s22s22p4 F 1s22s22p5 Ne 1s22s22p6 2. Schale vollständig besetzt … … Kr 1s22s22p63s23p63d104s24p6 3. Schale vollständig besetzt … …
Die äusserste Schale wird Valenzschale genannt. Die Elektronen dieser Schale sind die am schwächsten gebundenen Elektronen und nehmen an der Bildung chemischer Bindungen teil. Die tiefer liegenden Elektronen sind nicht wichtig für das chemische Verhalten der Elemente. Eine Lücke in der Valenzschale bedeutet, dass das Atom ein Elektron relativ leicht akzeptieren kann (vgl. z. B. Fluor), während ein isoliertes Elektron aus einem Valenzorbital relativ leicht abgegeben wird (vgl. z. B. Lithium oder Natrium). Da die Valenzschale der Edelgase vollständig ist, sind diese Atomen besonders stabil und unreaktiv (Oktettregel, vgl. Kapitel 3.2.1.). Wenn die Elektronen eines Atoms in der stabilsten Konfiguration sind, ist das Atom in seinem Grundzustand. Alle andere Konfigurationen der Elektronen entsprechen angeregte Zustände.
4.2. Chemische Bindung Atomorbitale können miteinander wechselwirken. Das führt zur Bildung chemischer Bindungen. Um die Stärke einer Bindung zu quantifizieren, wird die Dissoziationsenergie benützt, d. h. die Energie die notwendig ist, um die chemische Bindung zu spalten. Je stabiler die Bindung ist, desto mehr wird Energie benötigt (vgl. Kapitel 5.1.). Quantenchemisch werden Bindungen als Wechselwirkungen zwischen Atomorbitalen betrachtet (Molekülorbitalmodell). Folgende Eigenschaften sind für ihre Verständnis wichtig:

Skript AC-OC I 67
Wenn sich zwei Atome annähern, entsteht zwischen den Atomorbitalen eine Wechselwirkung, die neue Molekülorbitale erzeugt. Nur Atomorbitale gleicher Symmetrie bezüglich der Kern-Kern-Achse wirken aufeinander.
Eine chemische Bindung wird gebildet, wenn diese Wechselwirkung die Gesamtenergie des Systems sinkt. D. h., dass die besetzten Orbitale energetisch abgesenkt werden müssen. Die Energie der leeren Orbitalen hat keinen Einfluss auf die Gesamtenergie des Moleküls.
Die Wechselwirkung ist grösser, wenn die Orbitale gute Überlappung haben und ähnlich in der Energie sind.
Die Wechselwirkung zwischen zwei Orbitalen führt zu einer Aufspaltung der Energieniveaus.
Die Summe der Atomorbitale und diejenige der Molekülorbitale muss stets gleich sein. Um die Entstehung von chemischen Bindungen zu analysieren, ist es immer nützlich die Situation grafisch in Energieniveaudiagrammen darzustellen:
Die Atomorbitale A und B wechselwirken miteinander und erzeugen die zwei Molekülorbitale C und D, beide mit einer Energie die anders als die Energien der Ausgangsorbitale ist. Mit einem Energieniveaudiagramm kann man dann sehen, ob eine Bindung gebildet wird. Wenn z. B. zwei H-Atome wechselwirken, können beiden Elektronen in das tiefliegende Orbital C gehen und das System wird deshalb stabiler. In der Tat existiert die Molekül H2 (Bindungsstärke 104 kcal/mol). Im Fall von zwei He-Atome, muss man vier Elektronen verteilen. Da in einem Orbital nur maximal zwei Elektronen sein können, werden beide Molekülorbitale vollständig besetzt und keine Systemstabilisierung findet statt. Da es sich keine Bindung bilden kann, existiert He2 nicht. Wenn die Energie eines Molekülorbitales niedriger als diejenige der Atomorbitale ist, wird es als bindend bezeichnet. Wenn seine Energie höher ist, ist es antibindend und wenn praktisch keine Energieänderung stattfindet, ist das Molekülorbital nichtbindend. Die Wellenfunktionen der Molekülorbitale können gemäss dem LCAO-Modell (linear combination of atomic orbitals) berechnet werden. Die Grundidee ist, dass die Molekülorbitale praktisch nur eine lineare Kombination von Atomorbitalen sind. In den folgenden Abbildungen sind die Gestalten einiger Molekülorbitale ausgehend von s und p Atomorbitalen gezeigt:
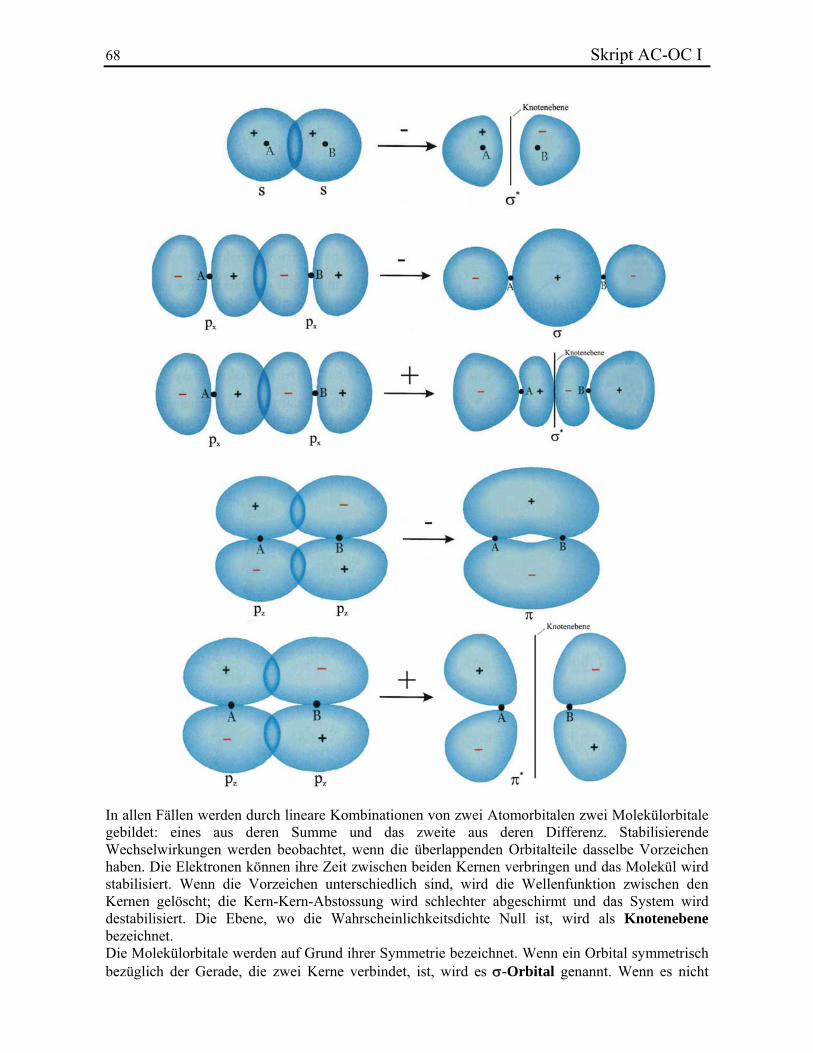
68 Skript AC-OC I
In allen Fällen werden durch lineare Kombinationen von zwei Atomorbitalen zwei Molekülorbitale gebildet: eines aus deren Summe und das zweite aus deren Differenz. Stabilisierende Wechselwirkungen werden beobachtet, wenn die überlappenden Orbitalteile dasselbe Vorzeichen haben. Die Elektronen können ihre Zeit zwischen beiden Kernen verbringen und das Molekül wird stabilisiert. Wenn die Vorzeichen unterschiedlich sind, wird die Wellenfunktion zwischen den Kernen gelöscht; die Kern-Kern-Abstossung wird schlechter abgeschirmt und das System wird destabilisiert. Die Ebene, wo die Wahrscheinlichkeitsdichte Null ist, wird als Knotenebene bezeichnet. Die Molekülorbitale werden auf Grund ihrer Symmetrie bezeichnet. Wenn ein Orbital symmetrisch bezüglich der Gerade, die zwei Kerne verbindet, ist, wird es -Orbital genannt. Wenn es nicht

Skript AC-OC I 69 symmetrisch ist, heisst es -Orbital. Antibindende Orbitale werden durch einen Stern (*) kennengezeichnet. Bindungen, die durch - bzw. -Molekülorbitale charakterisiert sind, werden - bzw. -Bindungen bezeichnet. Da die -Orbitale nicht symmetrisch bezüglich Drehungen sind, kann die Überlappung nur stattfinden, wenn die Atomorbitale parallel sind. Die Energie, die benötigt wird, um diese Überlappung aufzubrechen ist so hoch, dass bei Raumtemperatur keine Drehung beobachtet wird. Bei -Bindungen dagegen ist kaum Energie notwendig und sie sind frei drehbar (vgl. Kapitel 6.). -Orbitale sind weniger stabilisiert (bzw. destabilisiert) als -Orbitale, weil im ersten Fall die Überlappung zwischen den Atomorbitalen kleiner ist. In der Tat beträgt die Dissoziationsenergie einer C-C--Bindung 90 kcal/mol, während für eine C-C--Bindung nur 70 kcal/mol genügen. Die Stabilität der Bindungen wird, neben ihren Typ, auch durch die Energien der Atomorbitale bestimmt: je ähnlicher sie sind, desto grösser werden Wechselwirkung und Stabilisierung (bzw. Destabilisierung).
4.3. Geometrie eines Moleküls und Hybridisierung Das Energieniveaudiagramm eines H2-Moleküls wird wie folgt dargestellt:
Die 1s-Orbitale der zwei H-Atome erzeugen zwei Molekülorbitale. Beide Elektronen können im bindenden Orbital bleiben und eine stabile Bindung wird gebildet. Zweiatomige Moleküle müssen unbedingt linear sein, während für dreiatomige Verbindungen auch eine eventuelle gewinkelte Geometrie berücksichtigt werden muss. Mit der Anwendung der Molekülorbitale kann man in der Tat begründen, warum Wasser diese zweite Möglichkeit bevorzugt.

70 Skript AC-OC I
HO
HH O H
nichtbindend
bindend
antibindend
Nur die sechs Atomorbitale der Valenzschalen (vier aus dem O-Atom und ein aus jedem H-Atom) werden in der Bildung von Wasser involviert, d. h. man erwartet sechs Molekülorbitale. Im Fall des theoretischen linearen Wassers können zwei p-Orbitale des O-Atoms weder bindende noch antibindende Wechselwirkungen mit den H-Atomen bilden, weil sich aus geometrischen Gründen alle Wechselwirkungen gegenseitig aufheben. Sie werden dann zwei nichtbindende Molekülorbitale erzeugen. Beim linearen Wasser bekommt man daher vier bindende und vier nichtbindende Elektronen. Wenn das Molekül gewinkelt ist, findet eine Änderung der Überlappungen statt, was die Energie der Molekülorbitale ändert. Ein signifikanter Fall ist dasjenige des pz-O-Orbitals, das jetzt ein bindendes Molekülorbital erzeugen kann. Das gewinkelte Wasser besitzt sechs bindende und nur zwei nichtbindende Elektronen und ist deshalb stabiler. Die Schwierigkeiten der Anwendung der Molekülorbitalmodell nehmen leider mit der Anzahl Atomen rasch zu, weil die Zahl der möglichen Atomorbitalkombinationen immer grösser wird. Wechselwirkungen zwischen mehr als zwei Orbitalen sind schwierig qualitativ zu schätzen. Deshalb wurde das Konzept von Hybridisierung vorgestellt, in der die Wechselwirkungen zwischen den Atomen stufenweise aufgebaut werden. Die Idee der Hybridisierung ist, dass die Atomorbitale vor der Bildung der Molekülorbitale zusammenwechselwirken und neue Atomorbitale erzeugen. Diese Mischung wird einfach als eine lineare Addition der Atomorbitale abgeschätzt, was oft missverstanden ist. Die Hybridisierung bleibt trotzdem sehr hilfreich zur Beschreibung organisch-chemischer Moleküle.
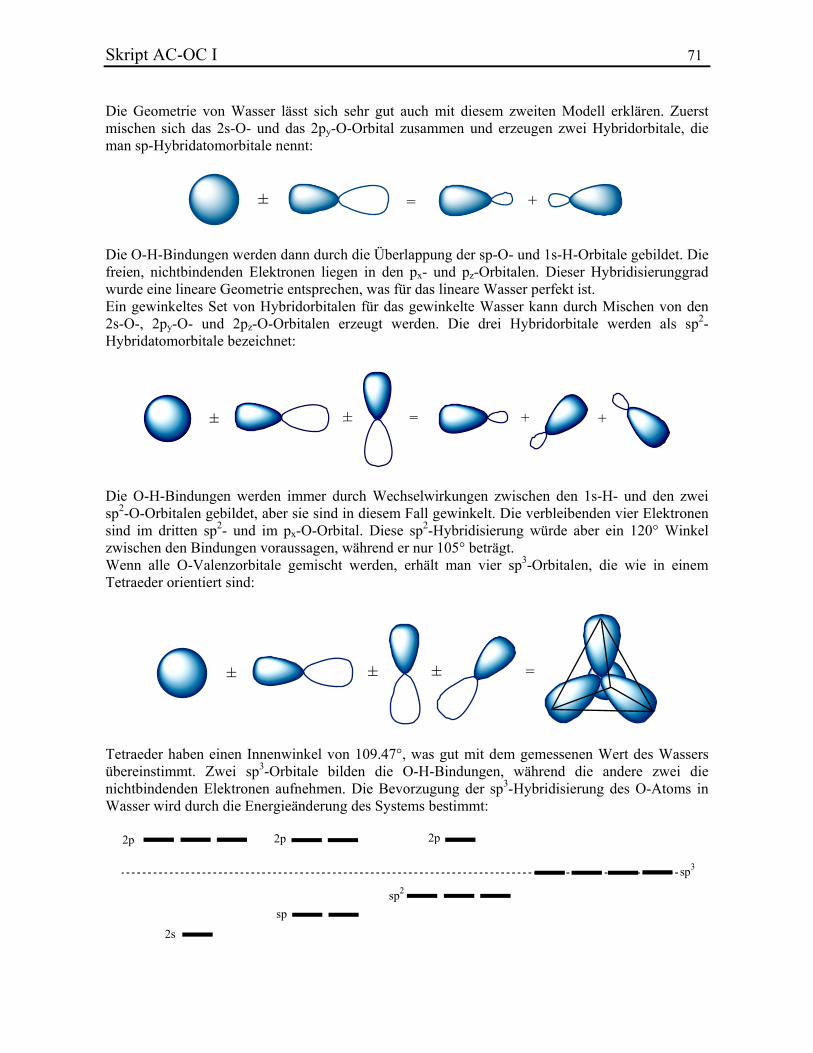
Skript AC-OC I 71 Die Geometrie von Wasser lässt sich sehr gut auch mit diesem zweiten Modell erklären. Zuerst mischen sich das 2s-O- und das 2py-O-Orbital zusammen und erzeugen zwei Hybridorbitale, die man sp-Hybridatomorbitale nennt:
± = +
Die O-H-Bindungen werden dann durch die Überlappung der sp-O- und 1s-H-Orbitale gebildet. Die freien, nichtbindenden Elektronen liegen in den px- und pz-Orbitalen. Dieser Hybridisierunggrad wurde eine lineare Geometrie entsprechen, was für das lineare Wasser perfekt ist. Ein gewinkeltes Set von Hybridorbitalen für das gewinkelte Wasser kann durch Mischen von den 2s-O-, 2py-O- und 2pz-O-Orbitalen erzeugt werden. Die drei Hybridorbitale werden als sp2-Hybridatomorbitale bezeichnet:
± = +± +
Die O-H-Bindungen werden immer durch Wechselwirkungen zwischen den 1s-H- und den zwei sp2-O-Orbitalen gebildet, aber sie sind in diesem Fall gewinkelt. Die verbleibenden vier Elektronen sind im dritten sp2- und im px-O-Orbital. Diese sp2-Hybridisierung würde aber ein 120° Winkel zwischen den Bindungen voraussagen, während er nur 105° beträgt. Wenn alle O-Valenzorbitale gemischt werden, erhält man vier sp3-Orbitalen, die wie in einem Tetraeder orientiert sind:
± ± ± =
Tetraeder haben einen Innenwinkel von 109.47°, was gut mit dem gemessenen Wert des Wassers übereinstimmt. Zwei sp3-Orbitale bilden die O-H-Bindungen, während die andere zwei die nichtbindenden Elektronen aufnehmen. Die Bevorzugung der sp3-Hybridisierung des O-Atoms in Wasser wird durch die Energieänderung des Systems bestimmt:
2s
2p 2p
sp
sp2
2p
sp3
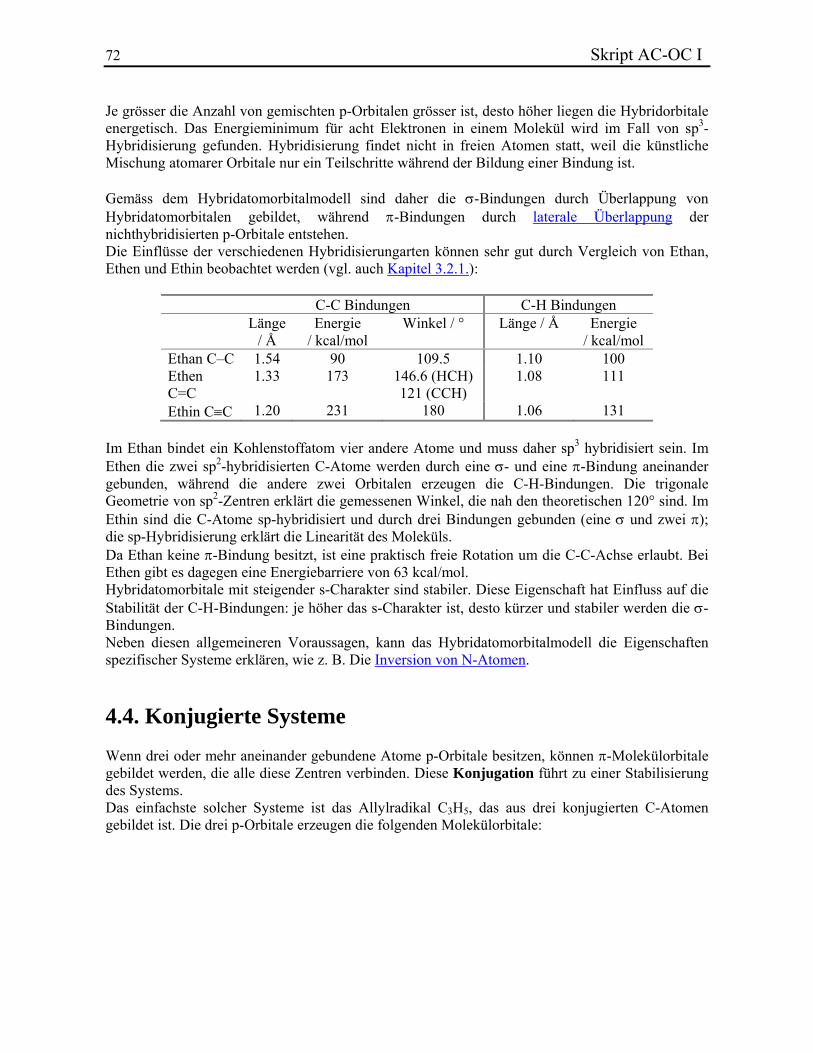
72 Skript AC-OC I Je grösser die Anzahl von gemischten p-Orbitalen grösser ist, desto höher liegen die Hybridorbitale energetisch. Das Energieminimum für acht Elektronen in einem Molekül wird im Fall von sp3-Hybridisierung gefunden. Hybridisierung findet nicht in freien Atomen statt, weil die künstliche Mischung atomarer Orbitale nur ein Teilschritte während der Bildung einer Bindung ist. Gemäss dem Hybridatomorbitalmodell sind daher die -Bindungen durch Überlappung von Hybridatomorbitalen gebildet, während -Bindungen durch laterale Überlappung der nichthybridisierten p-Orbitale entstehen. Die Einflüsse der verschiedenen Hybridisierungarten können sehr gut durch Vergleich von Ethan, Ethen und Ethin beobachtet werden (vgl. auch Kapitel 3.2.1.):
C-C Bindungen C-H Bindungen Länge
/ Å Energie
/ kcal/mol Winkel / ° Länge / Å Energie
/ kcal/mol Ethan C–C 1.54 90 109.5 1.10 100 Ethen C=C
1.33 173 146.6 (HCH) 121 (CCH)
1.08 111
Ethin CC 1.20 231 180 1.06 131 Im Ethan bindet ein Kohlenstoffatom vier andere Atome und muss daher sp3 hybridisiert sein. Im Ethen die zwei sp2-hybridisierten C-Atome werden durch eine - und eine -Bindung aneinander gebunden, während die andere zwei Orbitalen erzeugen die C-H-Bindungen. Die trigonale Geometrie von sp2-Zentren erklärt die gemessenen Winkel, die nah den theoretischen 120° sind. Im Ethin sind die C-Atome sp-hybridisiert und durch drei Bindungen gebunden (eine und zwei ); die sp-Hybridisierung erklärt die Linearität des Moleküls. Da Ethan keine -Bindung besitzt, ist eine praktisch freie Rotation um die C-C-Achse erlaubt. Bei Ethen gibt es dagegen eine Energiebarriere von 63 kcal/mol. Hybridatomorbitale mit steigender s-Charakter sind stabiler. Diese Eigenschaft hat Einfluss auf die Stabilität der C-H-Bindungen: je höher das s-Charakter ist, desto kürzer und stabiler werden die -Bindungen. Neben diesen allgemeineren Voraussagen, kann das Hybridatomorbitalmodell die Eigenschaften spezifischer Systeme erklären, wie z. B. Die Inversion von N-Atomen.
4.4. Konjugierte Systeme Wenn drei oder mehr aneinander gebundene Atome p-Orbitale besitzen, können -Molekülorbitale gebildet werden, die alle diese Zentren verbinden. Diese Konjugation führt zu einer Stabilisierung des Systems. Das einfachste solcher Systeme ist das Allylradikal C3H5, das aus drei konjugierten C-Atomen gebildet ist. Die drei p-Orbitale erzeugen die folgenden Molekülorbitale:

Skript AC-OC I 73
E
2p
Um die relativen Energien der Orbitale zu beschreiben, werden sie zur Energie eines isolierten 2p-Orbitales in Beziehung gebracht. Diese Energie wird mit bezeichnet. Die Aufspaltung zwischen und dem Orbital wird durch die Energieeinheit ausgedrückt. Da < 0 ist, hat ein Orbital bei + eine tiefere Energie als . Die Energie der Orbitale hängt von der Zahl der Knotenebenen ab: je mehr Knotenebene es gibt, desto energiereicher ist das Orbital. Allylkationen, Allylradikale und Allylanionen sind alle relativ stabile Moleküle, weil alle Elektronen bindende und nichtbindende Molekülorbitale besetzen. Die Bindungslänge des Allylkations ist 1.4 Å und die Rotationsbarriere beträgt 15.7 kcal/mol für das Allylradikal (H.-G. Korth, H. Trill, R. Sustmann, J. Am. Chem. Soc. 1981, 103, 4483). Diese Werte bestätigen, dass die Atome weder durch Einfach- noch durch Doppelbindungen gebunden sind. Analog können die Molekülorbitale grösserer konjugierter Systeme gebildet werden. Die folgende Abbildung zeigt die Orbitale von Butadien:
HOMO
1.46 Å
1.36 Å
LUMO
E
Auch in diesem Fall entsprechen die Bindungslängen nicht den normalen Werten für Einfach- und Doppelbindungen. Die Energieaufspaltung zwischen dem LUMO (lowest unoccupied molecular orbital) und dem HOMO (highest occupied molecular orbital) hängt von der Anzahl konjugierter Doppelbindungen ab. Je grösser das konjugierte System ist, desto kleiner wird die Aufspaltung. Die Energiedifferenz zwischen den Orbitalen kann aus der Wellenlänge des Lichts berechnet werden, das benötigt ist, um ein Elektron aus dem HOMO in das LUMO anzuregen. Das

74 Skript AC-OC I Absorptionsmaximum von Ethen liegt bei ~165 nm (173 kcal/mol) und bei ~215 nm (133 kcal/mol) für Butadien. Sehr lange konjugierte Systeme besitzen Absorptionsmaxima im sichtbaren Bereich. Z. B. hat -Carotin, ein wichtiges Pigment für das Sehen, ein Absorptionsmaximum im blauen Bereich, bei 452 nm (63 kcal/mol). Konjugierte Systeme können auch eine cyclische Struktur besitzen. Das bekannteste Beispiel ist Benzol, dessen -Molekülorbitale in der folgenden Abbildung dargestellt sind:
E
Einige cyclische konjugierte Systeme besitzen eine aussergewöhnliche Stabilität relativ zu ihrer offenkettigen Analoga und werden als aromatisch bezeichnet. Z. B. ist die Gesamtenergie von Benzol gegenüber derjenigen von drei Ethenmolekülen um 2 stabiler, während die Energie von Benzol ist um 1 niedriger als diejenige von Hexatrien. Ein aromatisches Molekül muss planar und cyclisch sein und 2, 6, 10, 14, ... (4n + 2) -Elektronen besitzen (Hückel Regel). Wenn eine dieser Bedingungen nicht erfüllt wird, ist die Molekül nichtaromatisch. Wenn das System planar und cyclisch ist aber 4n -Elektron besitzt, ist es antiaromatisch. Aromatische Moleküle sind z. B. Naphthalin (10 -e-), Anthracen (14 -e-) oder [18]-Annulen (18 -e-). Cyclooctatetraen ist dagegen nicht aromatisch, weil es 4n -Elektronen besitzt aber auch nicht planar ist. Cyclobutadien ist ein antiaromatisches Molekül, dessen Gesamtenergie 4 + 2 beträgt. Zwei freie Ethenmoleküle besitzen eine Energie von nur 4 + 4. Die darausfolgende relative Unstabilität von Cyclobutadien hat mit seiner Antiaromatizität zu tun.

KAPITEL 5:
ORGANISCHE THERMOCHEMIE In diesem Kapitel werden folgende Themen behandelt: 5.1. Dissoziationsenergie, Enthalpie und freie Enthalpie 5.2. Standardbildungsenthalpie 5.3. Homodesmische Reaktionen
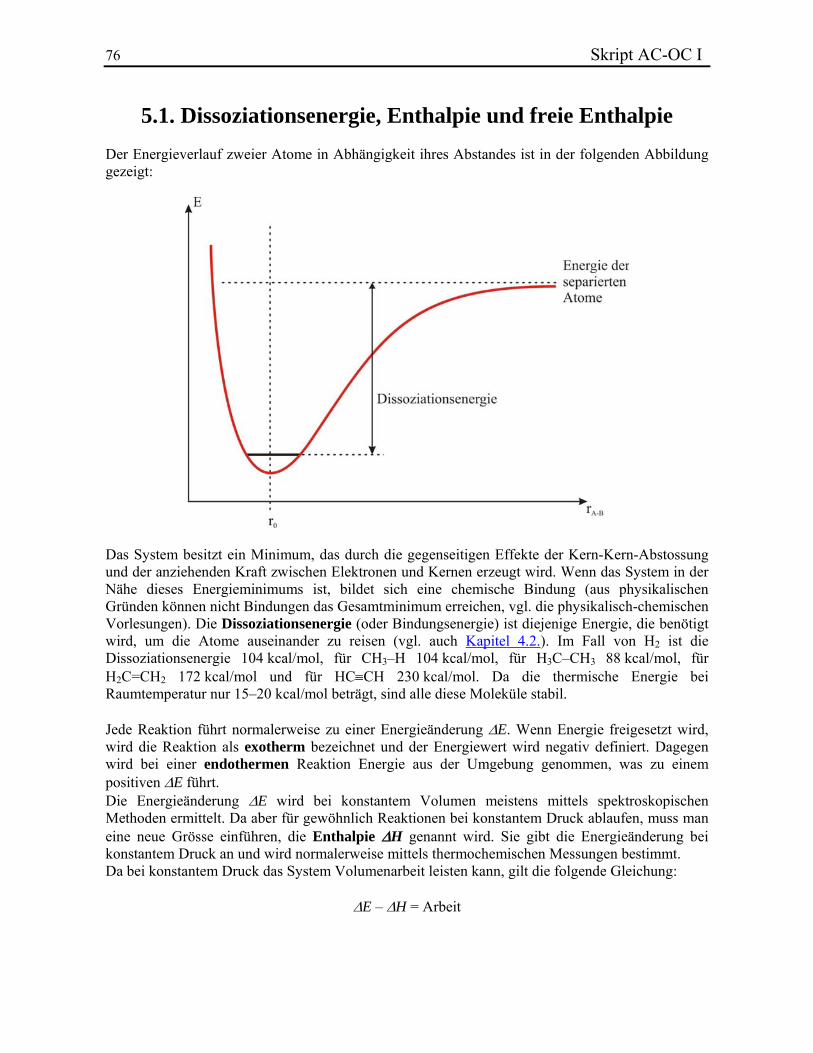
76 Skript AC-OC I
5.1. Dissoziationsenergie, Enthalpie und freie Enthalpie Der Energieverlauf zweier Atome in Abhängigkeit ihres Abstandes ist in der folgenden Abbildung gezeigt:
Das System besitzt ein Minimum, das durch die gegenseitigen Effekte der Kern-Kern-Abstossung und der anziehenden Kraft zwischen Elektronen und Kernen erzeugt wird. Wenn das System in der Nähe dieses Energieminimums ist, bildet sich eine chemische Bindung (aus physikalischen Gründen können nicht Bindungen das Gesamtminimum erreichen, vgl. die physikalisch-chemischen Vorlesungen). Die Dissoziationsenergie (oder Bindungsenergie) ist diejenige Energie, die benötigt wird, um die Atome auseinander zu reisen (vgl. auch Kapitel 4.2.). Im Fall von H2 ist die Dissoziationsenergie 104 kcal/mol, für CH3–H 104 kcal/mol, für H3C–CH3 88 kcal/mol, für H2C=CH2 172 kcal/mol und für HCCH 230 kcal/mol. Da die thermische Energie bei Raumtemperatur nur 15–20 kcal/mol beträgt, sind alle diese Moleküle stabil. Jede Reaktion führt normalerweise zu einer Energieänderung E. Wenn Energie freigesetzt wird, wird die Reaktion als exotherm bezeichnet und der Energiewert wird negativ definiert. Dagegen wird bei einer endothermen Reaktion Energie aus der Umgebung genommen, was zu einem positiven E führt. Die Energieänderung E wird bei konstantem Volumen meistens mittels spektroskopischen Methoden ermittelt. Da aber für gewöhnlich Reaktionen bei konstantem Druck ablaufen, muss man eine neue Grösse einführen, die Enthalpie H genannt wird. Sie gibt die Energieänderung bei konstantem Druck an und wird normalerweise mittels thermochemischen Messungen bestimmt. Da bei konstantem Druck das System Volumenarbeit leisten kann, gilt die folgende Gleichung:
E – H = Arbeit

Skript AC-OC I 77 Zum Beispiel wird ein Teil der Energie, die in einem Ottomotor durch Verbrennung von Benzin freigesetzt wird, in benutzbare Arbeit umgewandelt, während der Rest als Wärme in die Umwelt zerstreut wird. Die freie Enthalpie G, dass heisst die in Arbeit überführbare Energie, ist eine weitere wichtige Grösse der Thermochemie. Sie beinhaltet sowohl einen enthalpischen Term (H) als auch einen Entropieterm (S) (T ist die absolute Temperatur):
G = H – T·S Die Entropieänderung kann als eine Änderung der Bewegungsfreiheitsgrade (z. B. Rotation, Vibration, Translation) des Systems betrechtet werden. Z. B. nimmt die Entropie ab, wenn die Bewegung eines Moleküls durch die Umsetzung eingeschränkt wird oder wenn aus zwei Molekülen eines entstehet. Sie nimmt dagegen zu, wenn aus einer Ausgangsverbindung zwei oder mehrere Produkte gebildet werden. Ein negativer Wert für G bedeutet, dass Energie freigesetzt wird und dass die Reaktion spontan abläuft, obwohl keine Aussage über deren Geschwindigkeit gemacht werden kann (vgl. Kapitel 7.). Wenn die thermochemischen Werte beim Normalzustand (298 K, 1 atm) gemessen werden, sind sie durch ein ° ergänzt (z. B. G°). Eine tiefere Beschreibung dieser Grösse wird im Rahmen der physikalisch chemischen Vorlesung ausgeführt.
5.2. Standardbildungsenthalpie Die Standardbildungsenthalpie ist die Energieänderung bei konstantem Druck und Normalzustand, die bei der Bildung einer Verbindung aus den Elementen resultiert. Sie ist eine der am häufigsten verwendeten Grössen der organischen Chemie und wird als Hf° bezeichnet. Definitionsgemäss ist für die stabilste Modifikation eines Elements Hf° = 0. Beispiele:
Butan: 4 C (Graphit) + 5 H2 (g) n-C4H10 (g) H° = Hf° = –30.36 kcal/mol
2-Methylpropan:
4 C (Graphit) + 5 H2 (g) (H3C)3CH (g) H° = Hf° = –32.41 kcal/mol Butan und 2-Methylpropan sind daher stabiler als vier C-Atome und 5 H2-Moleküle im Normalzustand. Die Bildungsenthalpie von 2-Methylpropan ist um 2.05 kcal/mol niedriger als diejenige des Butans. Im folgenden hypothetischen Gleichgewicht wird daher 2-Methylpropan dominieren (vgl. also Kapitel 7.2.):
n-C4H10 (g) (H3C)3CH (g) H° = –2.05 kcal/mol

78 Skript AC-OC I Folgende Grafik erleichtert das Verständnis:
H
Normalzustand(Graphit + H2)
Butan
2-Methylpropan
Hf° =
–32.41
kcal/mol
Hf° =
–30.36
kcal/mol
H° = –2.05kcal/mol
~~ ~~
Gemessene Hf° für eine Vielzahl von Verbindungen können in fast jedem allgemeinem Chemiebuch gefunden werden. Mit Hilfe der Bildungsenthalpien können die Reaktionsenthalpien H° abgeschätzt werden:
C2H4 + H2 C2H6
H° = Hf° (Produkte) – Hf° (Edukte) =
= Hf° (C2H6) – [Hf° (C2H4) + Hf° (H2)] = (–20) – (13 + 0) = –33 kcal/mol Analog kann die freie Reaktionsenthalpie G° definiert werden.

KAPITEL 6:
KONFORMATIONS-ANALYSE In diesem Kapitel werden folgende Themen behandelt: 6.1. Konformationen offenkettiger Verbindungen 6.1.1. Ethan 6.1.2. Butan 6.2. Konformationsanalyse ringförmiger Moleküle 6.2.1. Cyclopropan 6.2.2. Cyclobutan 6.2.3. Cyclopentan 6.2.4. Cyclohexan 6.2.5. Monosubstituierte Cyclohexane 6.2.6. Disubstituierte Cyclohexane 6.2.7. Weitere Cyclohexanderivate 6.3. Zusammenfassung wichtiger Konzepte der Konformationsanalyse

80 Skript AC-OC I
6.1. Konformationen offenkettiger Verbindungen Wie im Kapitel 4.2. erklärt wurde, sind die cis- und trans-Isomere eines Olefins bei Raumtemperatur konformationsstabil, d. h. es findet keine Rotation um die C=C-Bindung statt. Die zwei Diastereoisomere stehen im dynamischen Gleichgewicht nur oberhalb 400°C. Bei gesättigten offenkettigen Verbindungen ist hingegen das Verhalten anders. Da die Rotationsbarriere um Einfachbindungen relativ klein ist, findet bei Raumtemperatur praktisch freie Rotation statt:
Cl
H
H
Cl
Cl
H
Cl
H
H
HH
H
Cl
Cl HH
Cl
H
HCl
Die Energie, die für die Rotation gebraucht wird, ist so klein, dass unter normalen Bedingungen keine Isomerie beobachtet werden kann. Bei Raumtemperatur sind im Allgemein zwei Isomere trennbar, wenn die Energiebarriere wenigstens 25 kcal/mol beträgt. Die Analyse der möglichen Isomere solcher einfacher Systeme ist aber Hilfreich, um das chemische Verhalten komplexerer Moleküle zu verstehen. 6.1.1. Ethan Um die Rotation um Einfachbindungen zu analysieren, ist es vorteilhaft, einige Konventionen einzuführen. Es gibt mehrere Methoden, um die dreidimensionale Struktur eines Moleküls darzustellen. Neben der üblichen Keilstrich-Schreibweise und der Fischer-Projektion, gibt es die Newman-Projektionen, die für Konformationsanalysen sehr hilfreich sind. Dazu betrachtet man das Molekül entlang der Achse, um die die Rotation erfolgt:
H
HH
H
H
H
H
H HH
HH
Newman-Projektion
Keilstrich-Schreibweise
Bei der Newman-Projektion wird ein Diederwinkel definiert, der den Winkel der beiden Ebenen, welche durch die C-C-H-Bindungen verlaufen, angibt. Wenn = 0° ist, sind die Substituenten verdeckt (oder ekliptisch, engl. eclipsed), wenn am grössten ist, sind sie gestaffelt (engl. staggered):

Skript AC-OC I 81
H
H HH
HHH
H HH
H
H
gestaffelt verdecktekliptisch
Obwohl die H-Atome sehr klein sind, sind sie genügend gross, um die gegenseitige Abstossung ihrer Elektronen zu spüren. Je näher sie sind, desto unstabiler ist das System. In einem Diagramm lässt sich die Energie des System in Abhängigkeit des Winkels darzustellen:
Die möglichen relativen Anordnungen werden als Konformationen bezeichnet. Die Energieminima eines solchen Diagramms heissen Konformere, die Maxima Übergangszustände. Die Spannung, die durch die sterische Abstossung verdeckter H-Atome erzeugt wird, heisst Pitzer-Spannung und beträgt 1 kcal/mol. Die Energiedifferenz zwischen der gestaffelten und der verdeckten Anordnung beträgt daher für Ethan nur 3 x 1 = 3 kcal/mol. Bei Raumtemperatur ist genügend Energie vorhanden, um die Konformere ineinander überzuführen. Während einer Sekunde geht Ethan ca. 3·1011 mal von einem Minimum in ein anderes über. 6.1.2. Butan Während es bei Ethan nur ein Konformer gibt, ist bei Butan die Situation komplizierter. Die Anwesenheit von Methylgruppen führt zu drei möglichen Konformeren, wenn man das Molekül entlang der C(2)-C(3)-Bindung betrachtet:
H
H CH3
H3C
H
H
= 0°
verdeckt
synperiplanar
C2v
H
H CH3
H3C
HH
= 60°
gestaffelt
gauche, synclinal
C2
H
H CH3
H
H
H3C
= 120°
verdeckt
C2
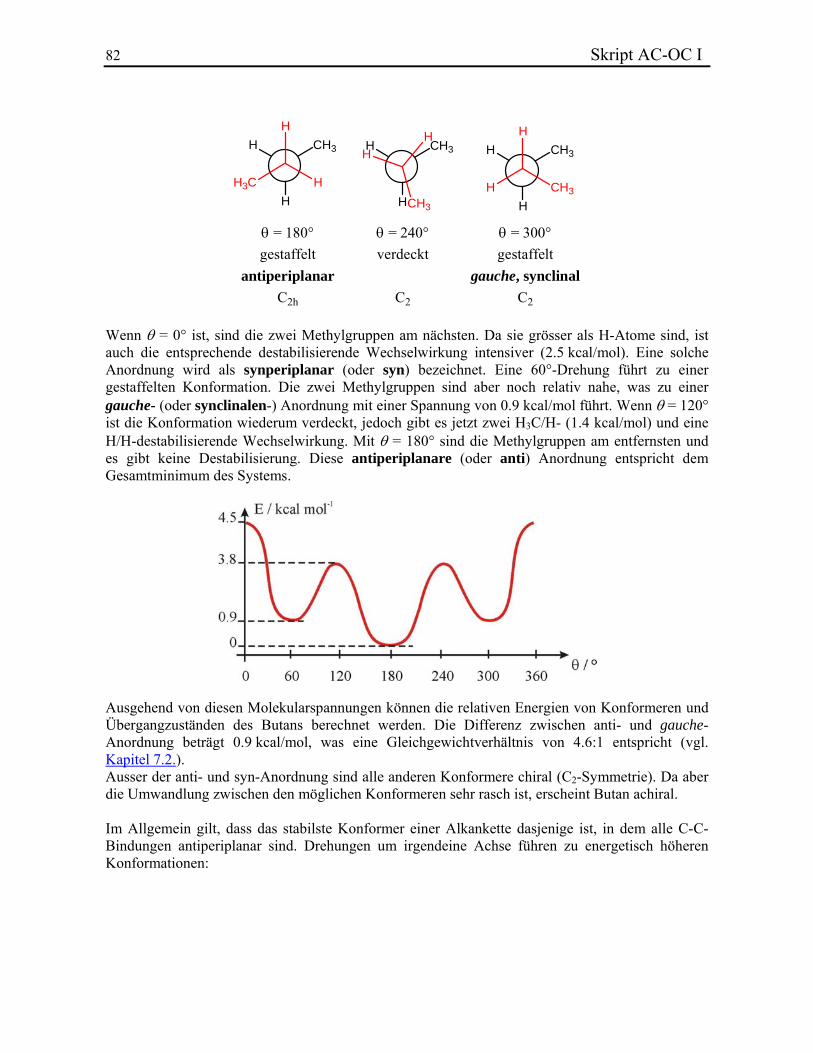
82 Skript AC-OC I
H
H CH3
H
HH3C
= 180°
gestaffelt
antiperiplanar
C2h
H
H CH3H
CH3
H
= 240°
verdeckt
C2
H
H CH3
H
CH3H
= 300°
gestaffelt
gauche, synclinal
C2
Wenn = 0° ist, sind die zwei Methylgruppen am nächsten. Da sie grösser als H-Atome sind, ist auch die entsprechende destabilisierende Wechselwirkung intensiver (2.5 kcal/mol). Eine solche Anordnung wird als synperiplanar (oder syn) bezeichnet. Eine 60°-Drehung führt zu einer gestaffelten Konformation. Die zwei Methylgruppen sind aber noch relativ nahe, was zu einer gauche- (oder synclinalen-) Anordnung mit einer Spannung von 0.9 kcal/mol führt. Wenn = 120° ist die Konformation wiederum verdeckt, jedoch gibt es jetzt zwei H3C/H- (1.4 kcal/mol) und eine H/H-destabilisierende Wechselwirkung. Mit = 180° sind die Methylgruppen am entfernsten und es gibt keine Destabilisierung. Diese antiperiplanare (oder anti) Anordnung entspricht dem Gesamtminimum des Systems.
Ausgehend von diesen Molekularspannungen können die relativen Energien von Konformeren und Übergangzuständen des Butans berechnet werden. Die Differenz zwischen anti- und gauche-Anordnung beträgt 0.9 kcal/mol, was eine Gleichgewichtverhältnis von 4.6:1 entspricht (vgl. Kapitel 7.2.). Ausser der anti- und syn-Anordnung sind alle anderen Konformere chiral (C2-Symmetrie). Da aber die Umwandlung zwischen den möglichen Konformeren sehr rasch ist, erscheint Butan achiral. Im Allgemein gilt, dass das stabilste Konformer einer Alkankette dasjenige ist, in dem alle C-C-Bindungen antiperiplanar sind. Drehungen um irgendeine Achse führen zu energetisch höheren Konformationen:

Skript AC-OC I 83
H3CCH3
H3C CH3
H
H
H
H
H H H H
HHH
H
H H H
HHH
Butan Nonan
6.2. Konformationsanalyse ringförmiger Moleküle Obwohl die Konformationsanalyse offenkettiger Verbindungen oft keine praktische Konsequenz mit sich bringt, erlaubt ihre Anwendung auf ringförmige Systeme in einigen Fälle ein besseres Verständnis für ihre Reaktivität. Neben den üblichen destabilisierenden Wechselwirkungen zwischen Substituenten, gibt es einen weiteren Effekt, der für die Analyse zyklischer Verbindungen berücksichtigt werden muss: die Ringspannung. Sie ist die Energie, die benötigt wird, um die normale Geometrie eines sp3-hybridisierten Atoms (109.5°) zu ändern, so dass die Bildung des Zyklus möglich wird. Z. B. ist die Ringspannung für Cyclopropan sehr gross, da der Innenwinkel im Dreiring 60° beträgt. Diese Spannung kann mit Hilfe von homodesmischen Reaktionen abgeschätzt werden (vgl. das Beispiel "Ringspannung" im Kapitel 5.). Im Folgenden wird die Konformationsanalyse der wichtigsten und häufigsten cyclischen Kohlenwasserstoffe durchgeführt. 6.2.1. Cyclopropan Cyclopropan ist der kleinste zyklische Kohlenwasserstoff, bestehend aus nur 3 C-Atomen. Aus geometrischen Gründen müssen die C-C-C-Winkel 60° statt der normalen 109.47° für sp3-hybridisierte Atome betragen. Diese ungünstige Anordnung führt zu einer Destabilisierung, die Baeyer-Spannung genannt wird. Diese Wechselwirkung, zusammen mit sechs Pitzer-Spannungen (alle H-Atome sind ekliptisch angeordnet), führen zu einer relativ grossen Ringspannung von 28 kcal/mol.
H
H
H
H
H
H
H
H
H
H
H
H
60°
Um die C-C-C-Bindungswinkeln im Cyclopropan mit dem Hybridatomorbitalmodell zu erklären, muss man annehmen, dass die C-C--Bindungen gebogen sind, so dass der Winkel zwischen den Orbitalen grösser werden:

84 Skript AC-OC I
H
H
H
HH
H 6.2.2. Cyclobutan Aus geometrischen Gründen muss Cyclobutan einen Innenwinkel von 90° besitzen. Diese Tatsache und acht Pitzer-Spannungen würden zu einer sehr hohen Ringspannung führen. Um den Betrag der Pitzer-Spannungen des planaren Cyclobutans zu reduzieren, besitzt dieses Molekül ein nichtplanares Konformer. Wenn Cyclobutan eine planare Konformation annehmen würde, wäre es noch energiereicher (ca. 1.3 kcal/mol). Die beiden gefalteten (engl. puckered) Konformere stehen miteinander in raschem Gleichgewicht über den planaren Übergangzustand (‡):
H
H H
H
H
H
H
HHH
H H
H
H
H
HH H
HHH
HH
H
Bei den gefalteten Konformeren sind die H-Atome nicht mehr ekliptisch angeordnet, was das Molekül stabilisiert. Die Konformere sind um 25° bezüglich der planaren Konformation gewinkelt. Um die dreidimensionale Anordnung eines Moleküls zu zeichnen, ist es oft nützlich die folgende Konvention anzuwenden:
HH
H H
H
H
H
H+
+
––
Die Atome, die unter einer fiktiver Ringebene liegen, werden durch ein – gekennzeichnet; diejenigen, die darüber liegen, tragen ein +. 6.2.3. Cyclopentan Planares Cyclopentan würde einen Innenwinkel von 108° besitzen. Ein Wert, der sehr nah an die 109.47° eines Tetraeders kommt. Die geringe Baeyer-Spannung würde aber von 10 ungünstigen H-C-C-H Pitzer-Spannungen (10 x 1 kcal/mol) begleitet sein. Experimentell findet man, dass in der Tat die Ringspannung nur ca. 6 kcal/mol beträgt und das Molekül nicht planar ist:

Skript AC-OC I 85
HH H
H
HH
H
H
H H
H
H
H
H
H H
H
H H
H
108°
Diese bevorzugte Anordnung heisst envelope-Konformation (Briefumschlag). Eine Pseudorotation (d. h. eine Bewegung einiger Atome, die als eine Rotation des ganzen Moleküls erscheint) führt die verschiedenen envelope-Konformere ineinander über. Diese Pseudorotation benötigt sehr wenig Energie und verläuft daher sehr schnell.
...
...
+ +
––
+
+
+
+
+
+
+–
– –
–
6.2.4. Cyclohexan Eine planare Konformation im Cyclohexan würde zu einem Innenwinkel von 120° führen, was wiederum eine beträchtlicher Baeyer-Spannung erzeugen würde. Experimentell weist aber dieses Molekül einen Winkel von 109.5° und keine Ringspannung auf. Das Molekül bevorzugt eine sesselförmige Konformation, in der alle Bindungen gestaffelt vorliegen:
H
H
H
HH
HH
H
HH
H H
H H
H
H
H
H
H
H
H
H
H
H
Die H-Atome lassen sich in zwei Gruppen einteilen. Atome, die senkrecht zur Ringebene stehen, werden als axiale (engl. axial) Wasserstoffe bezeichnet, die anderen sechs heissen äquatoriale (engl. equatorial) Wasserstoffe. Einer der wichtigsten Eigenschaften der Sesselkonformation, ist dass sie umklappen kann. Dieser Vorgang wird als Ringinversion bezeichnet und seine Energiebarriere beträgt 10.8 kcal/mol. Durch Inversion werden die axialen Wasserstoffe in die äquatorialen Positionen übergeführt und umgekehrt.

86 Skript AC-OC I
HH
H
H
H
H
H
H
H
H
H
H
H H
H
H
H
H
H
H
H
H
H
H
a = axial; e = äquatorial
e
e
e
e
e
e e
e
e
e
ee
a a
a
a
aa
a
a a
a
a
a
Sesselkonformationen kommen sehr oft in verschiedene Bereiche der Chemie vor, wie z. B. die Zuckerchemie. Neben Grundzustandskonformationen besitzen häufig auch Übergangzustände eine Sesselkonformation. Es ist daher sehr wichtig, dieser Sessel akkurat und gleichbleibend zu zeichnen. Die sechs Verknüpfungen des Ringes bilden drei Paare von gegenüberliegenden, parallelen Bindungen. Weiter sind die Bindungen zu den äquatorialen Substituenten parallel zu den Ringbindungen. Für einige Problemstellungen ist es auch nützlich, die doppelte Newmann-Projektion anzuwenden:
HH
H
H
H
H
H
H
H
H
H
H
H
HH
HH
HH
H
H
H
H H
+
–
+ +
–
–
Die Ringinversion kann experimentell mit Hilfe der magnetischen Kernresonanz (NMR) nachgewiesen werden. Mit dieser analytischen Methode, lassen sich 1H-Kerne in Abhängigkeit ihrer chemischen Umgebung unterscheiden. Axiale und äquatoriale Wasserstoffkerne haben eine unterschiedliche Umgebung. Im Fall von [D11]-Cyclohexan, wo nur ein 1H-Kern vorhanden ist, würde man zwei Signale erwarten. Da aber die Ringinversion bei Raumtemperatur schneller als die Zeitauflösung von NMR ist, ist nur ein Signal zu beobachten, da das Proton sich zeitlich gemittelt sowohl in der axialen als auch in der äquatorialen Position befindet. Man könnte sagen, dass es nur ein gemitteltes Molekül vorliegt. Wenn man aber die Temperatur erniedrigt, wird die Ringinversion langsamer und bei –90°C wird das Proton sowohl in der axialen als auch in der äquatorialen Lage gemessen, was zu zwei verschiedenen Signalen führt:
Signal von [D11]-Cyclohexan bei Raumtemperatur Nur ein gemitteltes Signal ist messbar.
Signale von [D11]-Cyclohexan bei –90°C. Die zwei Positionen im Sessel werden unterschieden.

Skript AC-OC I 87
DH
D
D
D
D
D
D
D
D
D
D
D D
D
D
H
D
D
D
D
D
D
D
Die Ringinversion erfolgt nicht über einen planaren Übergangzustand, sondern sie verläuft über eine Serie von Konformeren:
Die sesselförmige Konformation (Punktgruppe D3d) geht über einen Halbsessel-Übergangzustand (Cs-Symmetrie) in ein twist-Konformer (Punktgruppe D2) über. Die Energiedifferenz von 5.5 kcal/mol entspricht im Gleichgewicht einem Sessel/Twist Verhältnis von 8000:1. Die Wanne (oder Boot, C2v) ist wesentlich unstabiler (7.1 kcal/mol). Diese hohe Spannung wird durch zwei Arten destabilisierender Wechselwirkungen erzeugt:
H H
HH
flagpole H/H-Wechselwirkungen
H
H
H
H
H
HH
H
4 Pitzer-Spannungen 6.2.5. Monosubstituierte Cyclohexane Obwohl die zwei Sesselkonformere im Cyclohexan nicht unterscheidbar sind, ist die Situation für Cyclohexanderivate drastisch anders (vgl. auch [D11]-Cyclohexan).

88 Skript AC-OC I Im Gleichgewicht liegen die zwei Sesselkonformere von Methylcyclohexan im Verhältnis 95:5, zugunsten des Konformers mit äquatorialer Methylgruppe, vor.
H
H
H
H HH
HH
HH
H
95% 5% Wenn die Methylgruppe in der äquatorialen Position liegt, sind keine starken sterischen Wechselwirkungen mit den Ringprotonen vorhanden. Durch Ringinversion kommt die äquatoriale Methylgruppe axial zu liegen, was zu zwei ungünstigen gauche-Wechselwirkungen führt. Solche destabilisierende Abstossungen in Cyclohexanderivaten werden 1,3-diaxiale Wechselwirkungen genannt. Die Spannung beträgt 1.7 kcal/mol, was das beobachtete Gleichgewicht erklärt. Ein ähnlicher Fall wurde schon für die gauche-Konformere von Butan beobachtet, wo eine Destabilisierung um 0.9 kcal/mol gemessen wird. Die Ähnlichkeit zwischen 1,3-diaxialen Wechselwirkungen und gauche-Abstossungen kann in der folgenden Abbildung beobachtet werden:
H
HH
HH
HCH3
H
H
H
HCH3
HH
H
H
H
1 x gauche-WW0.9 kcal/mol
2 x 1,3-diaxiale WW1.7 kcal/mol
H HH
H
H
Dieses Verhalten wird für alle Substituenten beobachtet. Sie liegen in Allgemein bevorzugt in der äquatorialen Position. Die Energiedifferenz zwischen dem stabilen äquatorialen Konformer und dem axialen wird A-Parameter genannt. In der folgende Tabelle sind die Werte einiger wichtigen Substituenten eingetragen:

Skript AC-OC I 89
Substituent A / kcal mol-1 Substituent A / kcal mol-1
F 0.2 Me 1.7 Cl 0.4 Et 1.8 Br 0.5 iPr 2.1 I 0.4 n-Pr 2.1 OH 0.3 n-Bu 2.1 OMe 0.7 tBu 5.5 OEt 0.9 Neopentyl 2.0 OAc 0.7 Cyclohexyl 2.2 OTs 0.7 Ph 3.1 OBn 1.0 COOH 1.2 SH 0.9 COO– 2.3 SPh 0.9 COOMe 1.1 NH2 1.2 COOEt 1.1 NH3
+ 1.9 C≡ N 0.2 NMe2 2.1 C≡ CH 0.2 NHMe2
+ 2.4 HgBr 0 NMe3
+ 4.1 6.2.6. Disubstituierte Cyclohexane Es existieren verschiede Isomere von Dimethylcyclohexan, die ganz verschiedene Standardbildungsenthalpien besitzen. Im Folgenden werden diese Energieunterschiede dank der Konformationsanalyse erklärt. 1,1-Dimethylcyclohexan Im 1,1-Dimethylcyclohexan (achiral) besitzen beide Sesselkonformere eine axiale und eine äquatoriale Methylgruppe, was zu keiner Energiedifferenz zwischen den Konformeren führt:
CH3
CH3
CH3
CH3
1,2-Dimethylcyclohexan Es existieren zwei Isomere dieses Moleküls: cis und trans. Das chirale trans-Isomer kann zwei verschiedene Konformationen mit unterschiedlicher Energie einnehmen:
CH3
CH3
CH3 CH3
CH3
CH3

90 Skript AC-OC I Das diäquatoriale Konformer hat nur eine ungünstige Me/Me-gauche-Wechselwirkung (0.9 kcal/mol), während im diaxialen Fall vier 1,3-diaxialen Abstossungen vorhanden sind, die insgesamt 3.4 kcal/mol betragen. Eine Energiedifferenz von 2.5 kcal/mol entspricht einen Verhältnis von ca. 99:1. Für das achirale cis-Isomer ist dagegen keine Energiedifferenz zwischen den beiden Sesselkonformeren zu erwarten, weil in beiden Fällen nur eine 1,3-diaxiale und eine gauche-Wechselwirkung anwesend sind:
CH3
CH3CH3H3C
CH3
CH3
R
S
S R CH3
CH3
SR
CH3
CH3
Die zwei Konformere sind chiral. Da sie aber in einem raschen Gleichgewicht miteinander stehen, scheint diese Verbindung von einem makroskopischen Gesichtspunkt achiral zu sein. Der Unterschied in den Standardbildungsenthalpien der zwei Isomere (das trans Isomer ist um 1.6 kcal/mol stabiler als das cis) wird deswegen durch die Anwesenheit von zwei gauche-Wechselwirkungen bei dem trans Isomer erklärt:
CH3
CH3H
cis
Hf° = – 50.6
trans
Hf° = – 52.2
CH3CH3
H
1,3-Dimethylcyclohexan Das cis-Isomer ist achiral und seine Sesselkonformere weisen eine grosse Energiedifferenz auf. Der diaxiale Sessel besitzt eine Me/Me- (3.7 kcal/mol) und zwei Me/H- (1.7 kcal/mol) 1,3-diaxiale Wechselwirkungen, was zu einem Verhältnis von 10000:1 führt.
CH3
CH3 HH3C
H3C
CH3
CH3
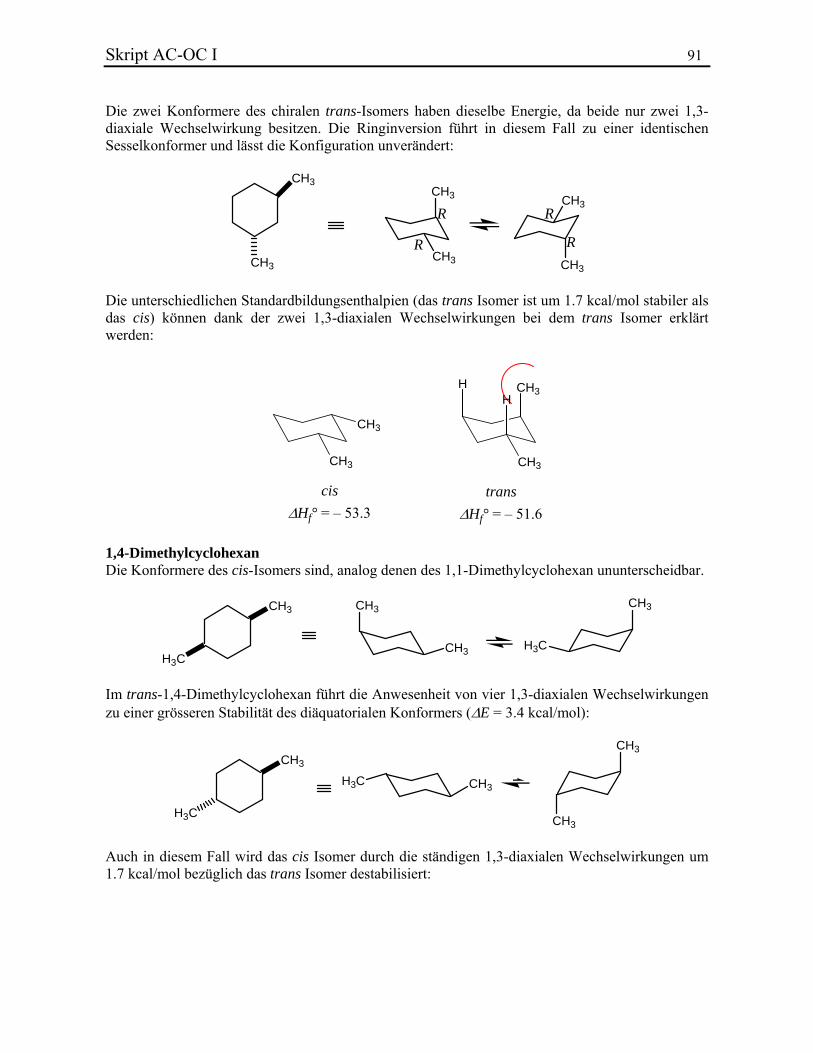
Skript AC-OC I 91 Die zwei Konformere des chiralen trans-Isomers haben dieselbe Energie, da beide nur zwei 1,3-diaxiale Wechselwirkung besitzen. Die Ringinversion führt in diesem Fall zu einer identischen Sesselkonformer und lässt die Konfiguration unverändert:
CH3
CH3CH3
CH3 CH3
CH3
R
R
R
R
Die unterschiedlichen Standardbildungsenthalpien (das trans Isomer ist um 1.7 kcal/mol stabiler als das cis) können dank der zwei 1,3-diaxialen Wechselwirkungen bei dem trans Isomer erklärt werden:
CH3
CH3
CH3
CH3H
cis
Hf° = – 53.3
trans
Hf° = – 51.6
H
1,4-Dimethylcyclohexan Die Konformere des cis-Isomers sind, analog denen des 1,1-Dimethylcyclohexan ununterscheidbar.
CH3
CH3 H3C
CH3CH3
H3C Im trans-1,4-Dimethylcyclohexan führt die Anwesenheit von vier 1,3-diaxialen Wechselwirkungen zu einer grösseren Stabilität des diäquatorialen Konformers (E = 3.4 kcal/mol):
H3C CH3
CH3
CH3CH3
H3C
Auch in diesem Fall wird das cis Isomer durch die ständigen 1,3-diaxialen Wechselwirkungen um 1.7 kcal/mol bezüglich das trans Isomer destabilisiert:

92 Skript AC-OC I
HH3C
cis
Hf° = – 51.5
trans
Hf° = – 53.2
CH3CH3
H3CH
6.2.7. Weitere Cyclohexanderivate Wenn die Energiedifferenz zwischen zwei Sesselkonformeren gross ist, kann der Ring bei Raumtemperatur praktisch nicht mehr invertieren. Dies ist z. B. der Fall bei tert-Butylcyclohexan. Die sehr starken 1,3-diaxialen Wechselwirkungen schieben das Gleichgewicht zu 99.99% auf die Seite der äquatorialen Konformation (E = 5.5 kcal/mol).
In trans-Decalin (IUPAC: Decahydronaphthalin) würde die Ringinversion eines Ringes zu grossen Spannungen im anderen Teil des Bicyclus führen. trans-Decalin ist daher konformationsstabil, was sich leicht mit Hilfe von Molekülmodellen überprüfen lässt:
H
H
cis-Decalin kann hingegen rasch invertieren, da es keine Energiedifferenz zwischen den beiden Konformeren gibt:
H
H H
H Die starre Struktur polycyclischer Verbindungen ist wichtig für natürlich vorkommende Moleküle wie z. B. Steroide, deren dreidimensionale Anordnung die biologische Aktivität mitbestimmt. Das Cholestangerüst besitzt 7 Stereozentren, was theoretisch 64 diastereoisomeren Paaren entspricht. Jedoch existiert in der Natur nur dasjenige Diastereoisomer, beim die ungünstigen Wechselwirkungen minimiert werden:
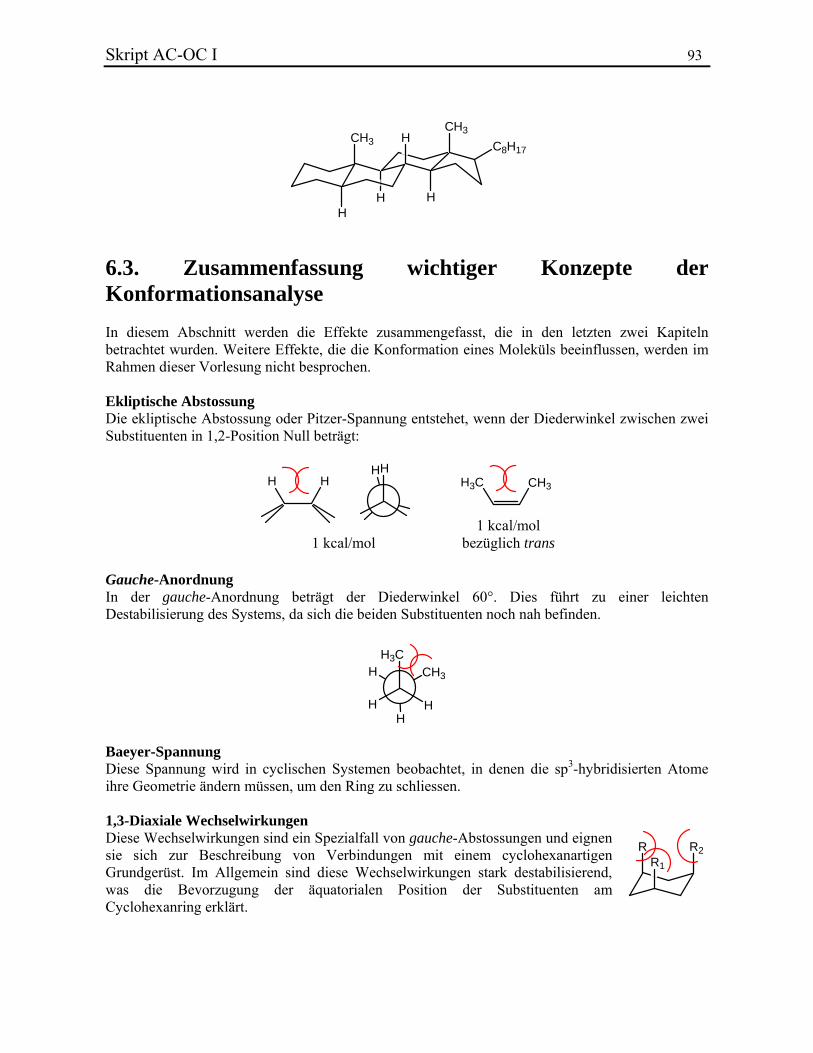
Skript AC-OC I 93
C8H17
CH3H
HH
CH3
H
6.3. Zusammenfassung wichtiger Konzepte der Konformationsanalyse In diesem Abschnitt werden die Effekte zusammengefasst, die in den letzten zwei Kapiteln betrachtet wurden. Weitere Effekte, die die Konformation eines Moleküls beeinflussen, werden im Rahmen dieser Vorlesung nicht besprochen. Ekliptische Abstossung Die ekliptische Abstossung oder Pitzer-Spannung entstehet, wenn der Diederwinkel zwischen zwei Substituenten in 1,2-Position Null beträgt:
H HHH
1 kcal/mol
CH3H3C
1 kcal/molbezüglich trans
Gauche-Anordnung In der gauche-Anordnung beträgt der Diederwinkel 60°. Dies führt zu einer leichten Destabilisierung des Systems, da sich die beiden Substituenten noch nah befinden.
H
H CH3
H3C
HH
Baeyer-Spannung Diese Spannung wird in cyclischen Systemen beobachtet, in denen die sp3-hybridisierten Atome ihre Geometrie ändern müssen, um den Ring zu schliessen. 1,3-Diaxiale Wechselwirkungen Diese Wechselwirkungen sind ein Spezialfall von gauche-Abstossungen und eignen sie sich zur Beschreibung von Verbindungen mit einem cyclohexanartigen Grundgerüst. Im Allgemein sind diese Wechselwirkungen stark destabilisierend, was die Bevorzugung der äquatorialen Position der Substituenten am Cyclohexanring erklärt.
RR1
R2

94 Skript AC-OC I
KAPITEL 7:
CHEMISCHE REAKTIONSLEHRE In diesem Kapitel werden folgende Themen behandelt: 7.1. Einführung 7.2. Thermodynamik und Reaktionsgleichgewicht 7.3. Reaktionskinetik 7.3.1. Reaktionsgeschwindigkeit 7.3.2. Aktivierungsenergie und Arrhenius-Gleichung

Skript AC-OC I 95
7.1. Einführung Im Kapitel 5.1. wurde erklärt, dass eine Reaktion prinzipiell spontan abläuft wenn G = H – T·S < 0. Ein gutes Beispiel dafür ist die Verbrennung von Glukose:
C6H12O6 + 6 O2 6 CO2 + 6 H2O Die Reaktionsenthalpie H° beträgt – 608 kcal/mol und die Entropieänderung S ist stark positiv, da aus sieben Teilchen zwölf Teilchen entstehen. Die freie Reaktionsenthalpie G hat mit -676 kcal/mol daher einen sehr hohen Wert. Trotz der günstigen Energieänderung entzünden sich Glukose und deren Polymerisationsprodukte (darunter Zucker, Mehl, Baumwolle, Cellulose, Holz, Papier,...) glücklicherweise nicht spontan. Dies wird durch die Tatsache erklärt, dass die Reaktion extrem langsam abläuft. Der Verlauf chemischer Reaktionen wird in der Tat durch zwei völlig verschiedene Konzepte charakterisiert. Die Thermodynamik beschreibt die Energieänderung einer chemischen Reaktion, die Kinetik hingegen beschäftigt sich mit der Reaktionsgeschwindigkeit. Die Thermodynamik macht also nur eine Aussage über den Anfangs- und Endzustand einer Reaktion und die Kinetik beschreibt den Weg, der dabei eingeschlagen wird. Diese beiden Konzepte dürfen absolut nicht verwechselt werden!
7.2. Thermodynamik und Reaktionsgleichgewicht In den vorherigen Kapiteln wurde manchmal der Begriff "Gleichgewicht" benützt, ohne dass er eindeutig definiert war. Dies ist das Ziel dieses Abschnittes. Normalerweise sind Reaktionen in der organischen Chemie reversibel, dass heisst dass neben der Umwandlung von Edukten in Produkte auch die Rückreaktion (Produkte zu Edukten) stattfinden kann. Als Beispiel hierfür betrachte man die Reaktion zwischen Chlormethan und Hydroxidanionen:
HO– + CH3Cl CH3OH + Cl– G° = – 22 kcal/mol Die Reaktion endet nicht, wann alle die Chlormethan-Moleküle in Methanol umgewandelt sind, sondern sie dauert fort, bis das Energieminimum erreicht wird. Die Verringerung der Energie des Gesamtsystems ist die Triebkraft der Reaktion. Im Minimum sind noch alle vier Komponenten der Reaktion vorhanden. Dieser Zustand wird thermodynamisches Gleichgewicht genannt. Die Lage dieses Gleichgewichtes wird durch die Gleichgewichtskonstante K beschrieben, die das Verhältnis von Produkten zu Edukten definiert:
...EE
...PP21
21
21
21
Reaktanden
Produkte
i
i
i
i
c
cK
wobei i die stöchiometrischen Koeffizienten der verschiedenen Verbindungen sind. Im Fall der obigen Reaktion ist K = 1016, was die praktisch vollständige Umsetzung der Edukte entspricht. Das Vorhandensein eines Gleichgewichts wird durch doppelte Reaktionspfeile ( ) angegeben.

96 Skript AC-OC I Wenn die Gleichgewichtskonstante sehr gross ist, sagt man "die Reaktion läuft vollständig ab", oder "das Gleichgewicht liegt vollständig auf der Seite der Produkte". Das ist dann der Fall, wenn im Gleichgewicht weniger als 0.1% der Edukte vorhanden sind. Die Reaktion wird dann mit einem einfachen Pfeil () geschrieben. Beispiel. Die Gleichgewichtskonstante kann auch mit Hilfe der freien Reaktionsenthalpie G° berechnet werden,
G° = – R T ln K wobei T die absolute Temperatur (in K) und R die universelle Gaskonstante (1.987 cal mol-1 K-1) ist. Bei exergonischen Reaktionen (G° < 0) ist daher K > 1 und das Gleichgewicht liegt auf der Seite der Produkte. Dagegen liegen bei endergonischen Reaktionen (G° > 0, K < 1) die Edukte im Überschuss vor. Für G° – 4.1 kcal/mol läuft die Reaktion vollständig ab. Beispiel. Mit folgender nützlicher Beziehung, können Gleichgewichte einfach abgeschätzt werden:
log K = G° / 1.4 (bei 25 °C) Somit wird für jede Änderung von G° um 1.4 kcal/mol die Gleichgewichtskonstante um den Faktor 10 verändert:
G° 0 – 1.4 – 2.8 – 4.2 – 5.6 – 7.0 K 1 10 100 1000 10000 100000
Um die Gleichgewichtskonstante für nichtstandardisierte Zustände zu berechnen, muss die Temperaturabhängigkeit von G berücksichtigt werden (vgl. Kapitel 5.1.):
G = f(T) = H – T · S Da H und S in ersten Näherung temperaturunabhängig sind, darf man die Standardwerte H° und S° auch bei anderen Temperaturen verwenden.
7.3. Reaktionskinetik Obwohl die Reaktion zwischen Chlormethan und Hydroxydionen eine hohe Triebkraft (grosses negatives G) besitzt, läuft sie recht langsam ab. Nach zwei Tagen bei Raumtemperatur wird z. B. in einer 0.05 M Lösung von CH3Cl in 0.1 M wässriger NaOH nur 10% Umsatz gefunden. Analog, obwohl die Verbrennung von Cellulose energetisch sehr günstig ist, ist die Reaktion extrem langsam. Diese Beobachtungen werden durch ein fundamentales Prinzip der Chemie erklärt: Die Energieänderung eines Systems während einer Reaktion erlaubt keine Aussagen über deren Geschwindigkeit.

Skript AC-OC I 97 7.3.1. Reaktionsgeschwindigkeit Die Reaktionsgeschwindigkeit eines chemischen Systems ist proportional zur Konzentration der Moleküle im System. Die Proportionalitätskonstante wird Geschwindigkeitskonstante k genannt. Man unterscheidet grundsätzlich zwischen der Zahl an einem Schritt beteiligten Moleküle. Kinetik erster Ordnung Die einfachste Reaktion ist die Umwandlung eines Moleküls A in ein oder mehrere Produkte, wie z. B. bei Konformationsänderungen oder bei dem radioaktiven Zerfall. Solche Reaktionen werden als Reaktionen erster Ordnung oder unimolekulare Reaktionen bezeichnet. Die Geschwindigkeit wird durch die folgende Gleichung, das Geschwindigkeitsgesetz, beschrieben ([A] ist die Konzentration von A):
Ad
A dk
t
Nach Integration dieser Differentialgleichung erhält man den Konzentrationsverlauf in Abhängigkeit der Zeit ([A]0 ist die Anfangskonzentration):
kte
tk
0AA
KonstanteAln
In diesem Fall besitzt k die Einheit s-1 und man kann daraus eine Lebensdauer berechnen,
= 1/k die die Zeit der Abnahme von [A] bis 1/e seines Anfangswert beschreibt. Reaktionen zweiter Ordnung Reaktionen der Art
A + B Produkte werden Reaktionen zweiter Ordnung oder bimolekulare Reaktionen genannt und ihr Geschwindigkeitsgesetz lautet
BAd
B d
d
A d k
tt
k besitzt nun die Einheit M-1 s-1. Wenn die Konzentration einer Komponente sehr grösser als diejenige der zweiten ist ([B] » [A]), kann man eine Reaktion zweiter Ordnung als quasi erster Ordnung betrachten. Da [B] praktisch konstant bleibt, wird eine neue Geschwindigkeitskonstante k' = k[B] definiert und die Reaktionsgeschwindigkeit hängt dann effektiv nur von der Konzentration von A ab. Wenn A = B, wird das Geschwindigkeitsgesetz

98 Skript AC-OC I
2Ad
A d k
t
und nach Integration bekommt man
tk 0A
1
A
1
Komplexere Kinetik Die meisten Reaktionen folgen nicht den oben beschriebenen, einfachen Geschwindigkeitsgesetzen. Auch im Fall von zwei aufeinanderfolgenden irreversiblen Reaktionen, wie z. B.
A 1k B
B + C 2k D
benötigt man für die Beschreibung des Geschwindigkeitsgesetzes die Lösung des Differentialgleichungssystems
CBAd
Bd
CBd
Dd
21
2
kkt
kt
Wenn k2 » k1 wird die Konzentration von B nicht gross werden ([B] 0). Diese Tatsache erlaubt die steady-state-Näherung, die hilfreich für die Lösung des Systems ist:
0CBAd
Bdist dann 0 [B]Wenn 21 kk
t
Es folgt daraus
Ad
Dd
C
AB
1
2
1
kt
k
k
Das Geschwindigkeitsgesetz ist daher gleich demjenigen der hypothetischen Reaktion erster Ordnung
A 1k D. Im obigen Beispiel hat die Teilreaktion B + C D keinen Einfluss auf die Reaktionsgeschwindigkeit. Sie wird durch die langsame Transformation A B bestimmt. Die Beobachtung, dass verschiedene Reaktionsschritte die Geschwindigkeit einer Reaktion unterschiedlich beeinflussen, führt zum Konzept des geschwindigkeitsbestimmenden Schritts.

Skript AC-OC I 99 Damit ist derjenige Reaktionsschritt gemeint, der die Geschwindigkeit der ganzen Reaktion bestimmt. Man betrachtet jetzt ein System, in dem eine Verbindung A in Gleichgewicht mit B ist und B mit C zu D reagiert:
k1
k-1A B
B + C Dk2
Die Hin- und Rückreaktion besitzen unterschiedliche Reaktionsgeschwindigkeiten, die durch k1 bzw. k-1 bestimmt sind. Die Produktbildungsgeschwindigkeit ist gegeben durch
CBd
Dd2 k
t
Wenn B als steady-state-Intermediat betrachtet wird, gilt:
0CBBAd
Bd211 kkk
t
Nach Umformen bekommt man:
AC
C
C
CA
d
Dd
C
AB
21
21
21
21
21
1
kk
kk
kk
kk
t
kk
k
Wenn die Konzentration von C gross ist, wird die Produktbildungsgeschwindigkeit scheinbar erster Ordnung in [A]. Mit der gemessenen Geschwindigkeitskonstante kgem ist:
121
1
21
21
1
C
11
C
C
Ad
Dd
kkk
k
k
kk
kkk
kt
gem
gem
gem
Eine lineare Regression von 1/kgem gegen 1/[C] erlaubt uns daher k1 (Achsenabschnitt) und das Verhältnis k-1/(k1·k2) (Steigung) zu berechnen. Wenn aber k-1 » k2 ist, ist die Rückreaktion des Gleichgewichts schneller als die Reaktion zu D, d. h. A und B liegen in einem sogenannten vorgelagerten Gleichgewicht vor. B ist nicht mehr ein steady-state-Intermediat, da sich B anreichert und damit [B] 0. Im Gleichgewicht gilt:
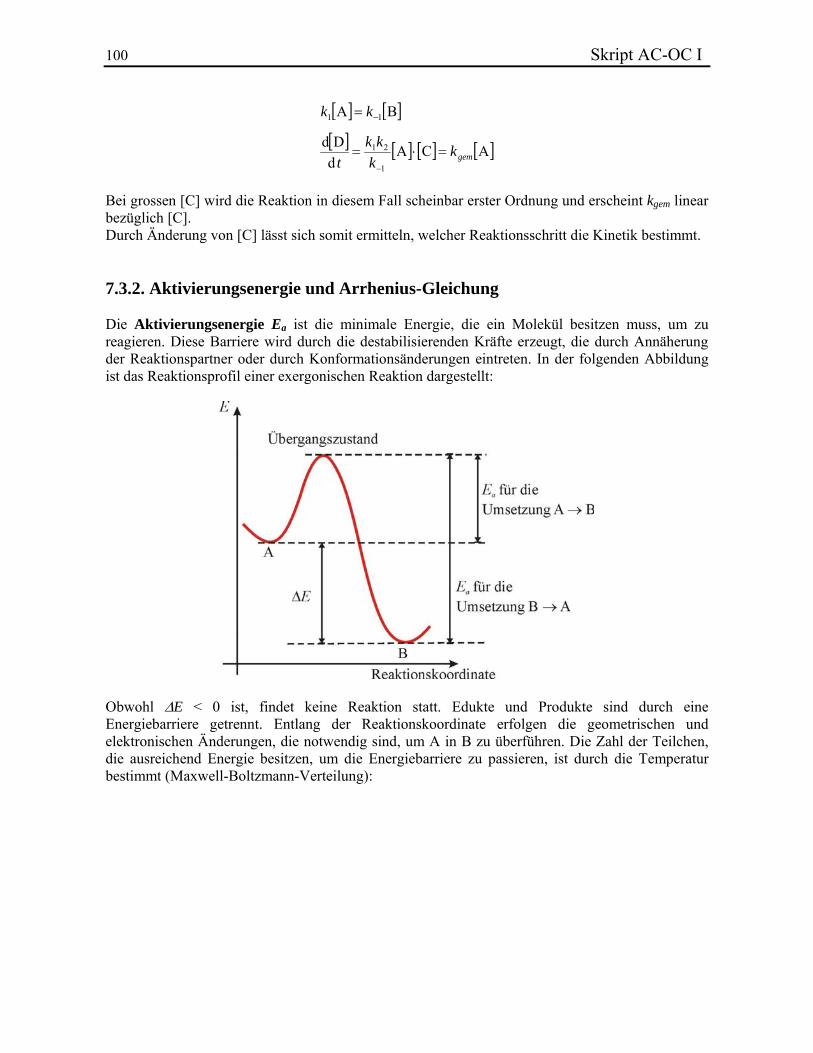
100 Skript AC-OC I
ACAd
Dd
BA
1
21
11
gemkk
kk
t
kk
Bei grossen [C] wird die Reaktion in diesem Fall scheinbar erster Ordnung und erscheint kgem linear bezüglich [C]. Durch Änderung von [C] lässt sich somit ermitteln, welcher Reaktionsschritt die Kinetik bestimmt. 7.3.2. Aktivierungsenergie und Arrhenius-Gleichung Die Aktivierungsenergie Ea ist die minimale Energie, die ein Molekül besitzen muss, um zu reagieren. Diese Barriere wird durch die destabilisierenden Kräfte erzeugt, die durch Annäherung der Reaktionspartner oder durch Konformationsänderungen eintreten. In der folgenden Abbildung ist das Reaktionsprofil einer exergonischen Reaktion dargestellt:
Obwohl E < 0 ist, findet keine Reaktion statt. Edukte und Produkte sind durch eine Energiebarriere getrennt. Entlang der Reaktionskoordinate erfolgen die geometrischen und elektronischen Änderungen, die notwendig sind, um A in B zu überführen. Die Zahl der Teilchen, die ausreichend Energie besitzen, um die Energiebarriere zu passieren, ist durch die Temperatur bestimmt (Maxwell-Boltzmann-Verteilung):

Skript AC-OC I 101
Je höher die Temperatur ist, umso mehr Teilchen besitzen die benötigte Energie. Die Fläche unter der Kurve bleibt stets konstant, da sie alle Teilchen im Reaktionsgefäss umfasst. Eine Faustregel besagt, dass jede Erhöhung der Reaktionstemperatur um 10 K eine zwei bis dreifache Erhöhung der Reaktionsgeschwindigkeit bewirkt. Bei Raumtemperatur besitzen Moleküle eine durchschnittliche Energie von 0.6 kcal/mol. Für das Beispiel
HO– + CH3Cl CH3OH + Cl– Ea beträgt 25 kcal/mol, was die niedrige Reaktionsgeschwindigkeit erklärt. Die Temperaturabhängigkeit der Reaktionsgeschwindigkeit wird durch die Konstante k beschrieben. k ist mit Hilfe der Arrhenius-Gleichung berechenbar:
RT
Ea
eAk
wobei R die universelle Gaskonstante (1.987 cal mol-1 K-1) und T die absolute Temperatur ist. Die Berechnung des Faktors A ist kompliziert und ist in späteren Vorlesungen ausführlich betrachtet. Der Faktor A ist eine temperaturabhängige Konstante, die weiter die Entropie des Systems berücksichtigt. Für eine unimolekulare Reaktion besitzt A die Einheit s-1. Sie darf die Streckschwingungsfrequenz der zerbrechenden Bindung nicht überschreiten. Dies entspricht für gewöhnlich eine Frequenz von A 1014 s-1. Für bimolekulare Reaktionen muss A kleiner als die Diffusionsgeschwindigkeit der Edukte in der Lösung sein. Für übliche organische Lösungsmittel gilt A 1010 M-1 s-1. Die Arrhenius-Aktivierungsenergie für typische Reaktionen der organischen Chemie liegt gewöhnlich zwischen 0 und 100 kcal/mol. In einigen Fällen erhält man mit der Arrhenius-Gleichung negative Ea, was jedoch physikalisch nicht sinnvoll ist. Folglich beschreibt diese Gleichung die physikalische Wirklichkeit in diesen Fällen nicht ausreichend.

102 Skript AC-OC I Mit Hilfe der Arrhenius-Gleichung und anderen Parametern, die nicht näher erklärt werden sollen, kann die Geschwindigkeitskonstante einer Reaktion erster Ordnung bei verschiedenen Temperaturen und Energiebarrieren berechnet werden:
Ea / kcal mol-1 k / s-1 (298 K) t1/2 / s (†) k / s-1 (100 K) 0 6.2·1012 1.2·10-13 2.1·1012 5 1.9·109 3.6·10-10 28 10 4.7·105 1.4·10-6 3.9·10-10 15 1.2·102 5.7·10-3 5·10-21 20 3.7·10-2 18 25 9.5·10-6 7.2·104 30 2.2·10-9 2.8·108 35 6.0·10-13 1.2·1012
(†) Halbwertszeit, das heisst die Zeit bis die Konzentration der Edukte auf die Hälfte der Anfangskonzentration abgesunken ist (t1/2 = ln 2 / k). Zum Vergleich: 1 Jahr 3·107 s; Alter der Erde 1017 s.
Die Arrheniusparameter Ea und A können mit Hilfe einer linearen Regression von ln k gegen 1/T ermittelt werden:
TR
EAk a 1
lnln

LITERATUR
Allgemeine Bücher
- CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton. - L. Eberson, A. Senning, Organische Chemie 1, VCH Verlag, Weinheim, 1983 (Geht nicht
wesentlich weiter als die Vorlesung.) - H.-R. Christen, F. Vögtle, Grundlagen der organischen Chemie, O. Salle Verlag, Frankfurt,
1989. (Deckt den Stoff der ersten beiden Jahre gut ab.) - A. Streitwieser, C. H. Heathcock, E. M. Kosower, Organische Chemie, VCH Verlag,
Weinheim, 1994. (Das wohl beste Lehrbuch der organischen Chemie, kann zu grossen Teilen der Ausbildung in organischer Chemie herangezogen werden.)
- D. W. Oxtoby, H. P. Gillis, N. H. Nachtrieb, Principles of Modern Chemistry, Saunders, Fort Worth, 1999. (Enthalt eine ausführliche Beschreibung der physikalischen Teile dieser Vorlesung.)
Nomenklatur
- U. Bünzli-Trepp, Handbuch für die systematische Nomenklatur der organischen Chemie, metallorganischen Chemie und Koordinationschemie, Logos Verlag, Berlin, 2001. (Ganz aktuelles und vollständiges Buch, das die Nomenklaturregeln von IUPAC und Chemical Abstracts beschreibt.)
- Gesellschaft Deutscher Chemiker, IUPAC Nomenklatur der organischen Chemie, VCH Verlag, Weinheim, 1997.
- R. Panico, W. H. Powell, J.-C. Richter, A Guide to IUPAC Nomenclature of Organic Com-pounds, Blackwell Scientific Publications, Oxford, 1993.
- IUPAC, Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F and H, Pergamon Press, Oxford, 1979.
Klassische Strukturlehre
- E. L. Eliel, S. H. Wilen, M. P. Doyle, Basic Organic Stereochemistry, Wiley, New York, 2001.
- E. L. Eliel, S. H. Wilen, Organische Stereochemie, Wiley-VCH, Weinheim, 1998.

104 Skript AC-OC I
Quantenchemie
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 1998. (Sehr gutes Buch für die physikalisch-chemischen Vorlesungen).
Organische Thermochemie
- J. F. Liebman, A. Greenberg, Strained Organic Molecules, Academic Press, New York, 1978.
Konformationsanalyse
- E. Juaristi, Conformational Behaviour Six-Membered Rings, VCH, New York, 1995. - H. Dodziuk, Modern Conformational Analysis, VCH, New York, 1995.

APPENDIX:
BEISPIELE, BEMERKUNGEN, VERTIEFUNGEN

106 Skript AC-OC I
1. Einführung
1.1. Vertiefung: Die Perkin-Geschichte William Henry Perkin (1838-1907), ein englischer Student, wollte 1856 Chinin herstellen, das gegen Malaria aktiv ist.
N
O
NH
HOH
Chinin, C20H24N2O2 Im Jahr 1856 war sehr wenig über organische Verbindungen bekannt. Die Strukturtheorie wurde erst im Jahr 1858, zwei Jahre später, publiziert. Bekannt waren lediglich die Summenformeln, die durch Verbrennung bestimmbar sind. Perkin versuchte Chinin ausgehend von Anilin, ein damals neues Derivat aus Kohle, zu synthetisieren. Die Summenformel von Anilin würde in C7H7N bestimmt. Perkin schlug daher die folgende Synthese vor:
C7H7N
2 C10H13N
C3H5I
O
H
C20H24N2O2
C10H13N
Anilin Allyliodid
?
?
+ +
Chinin Perkin verwendete für den zweiten Schritt K2Cr2O7. Es war bekannt, dass diese Verbindung den Gehalt von Sauerstoff in einem Molekül erhöht und denjenigen von Wasserstoff erniedrigt. Heutzutage weißt man, dass eine solche Synthese sinnlos ist, da sie die Struktur der verschiedenen Moleküle nicht berücksichtigt. Weiter war das damals verfügbare Anilin nicht rein, was zu einer falschen Summenformel führte. Es ist daher für uns nicht überraschend, dass Perkin aus diesem verunreinigten Anilin, Allyliodid und K2Cr2O7 ein ganz unerwartetes Endprodukt bekam:

Skript AC-OC I 107
NH2
CH3
NH2 NH2
CH3
N
N
NH
H2N
H3C CH3
N
N
NH
H2N
HSO4HSO4
Verbindungenin der Anilinmischungvon Perkin
Anilin
K2Cr2O7 / H2SO4
+ +
+
Pseudomauvein Mauvein Nach Reinigung der Mischung isolierte er einen violetten Farbstoff, den er Mauvein nannte. Perkins Mauvein war der erste künstliche Anilinfarbstoff, der beim Färben im Gegensatz zu den natürlichen Farbstoffen leuchtend und dauerhaft gefärbte Stoffe ergab, die sich sehr bald grosser Beliebtheit erfreuten. Ein Jahr später gründete er eine Fabrik in London. 1862 zog die Königin Viktoria eine seidene Bekleidung an, die mit diesem synthetischen Farbstoff gefärbt wurde, wodurch die Farbstoffindustrie erfolgreich wurde. Diese Firma ist jetzt ein Teil der AstraZeneca. Die Forschung auf dem Gebiet der Farbstoffe führte später zu derjenigen der synthetischen Parfums und Aromen, Spreng- und Kunststoffe, und letztlich zu Arzneimittel und unzähligen anderen Stoffen. Das erste künstliche Antibiotikum (Sulfanilamid) wurde in den dreissiger Jahren in der Farbstoffindustrie entdeckt:
H2N S
OO
NH2
H2N
O
OH
Sulfanilamid p-Aminobenzoesäure Diese Verbindung ähnelt der Struktur der natürlich vorkommenden p-Aminobenzoesäure. Sie stört daher der Stoffwechsel der letzteren, was zur Hemmung des Wachstums von unerwünschten Organismen führt. Man kann sagen, dass die aktuelle chemische Industrie ihrem Ursprung in der Farbstoffchemie hat. Weiter führte Perkin im Jahr 1904 das Konzept der Synthese als Strukturaufklärungsmethode ein. Er wollte die Struktur von -Terpinol bestimmen, ein Naturstoff, der aus Keifernnadelöl extrahiert und als Desinfektionsmittel verwendet wurde. Er entwickelte dazu eine gezielte Synthese, die zur vorgeschlagenen Struktur führte. Am Ende erhielt er eine Verbindung, die dieselben Eigenschaften
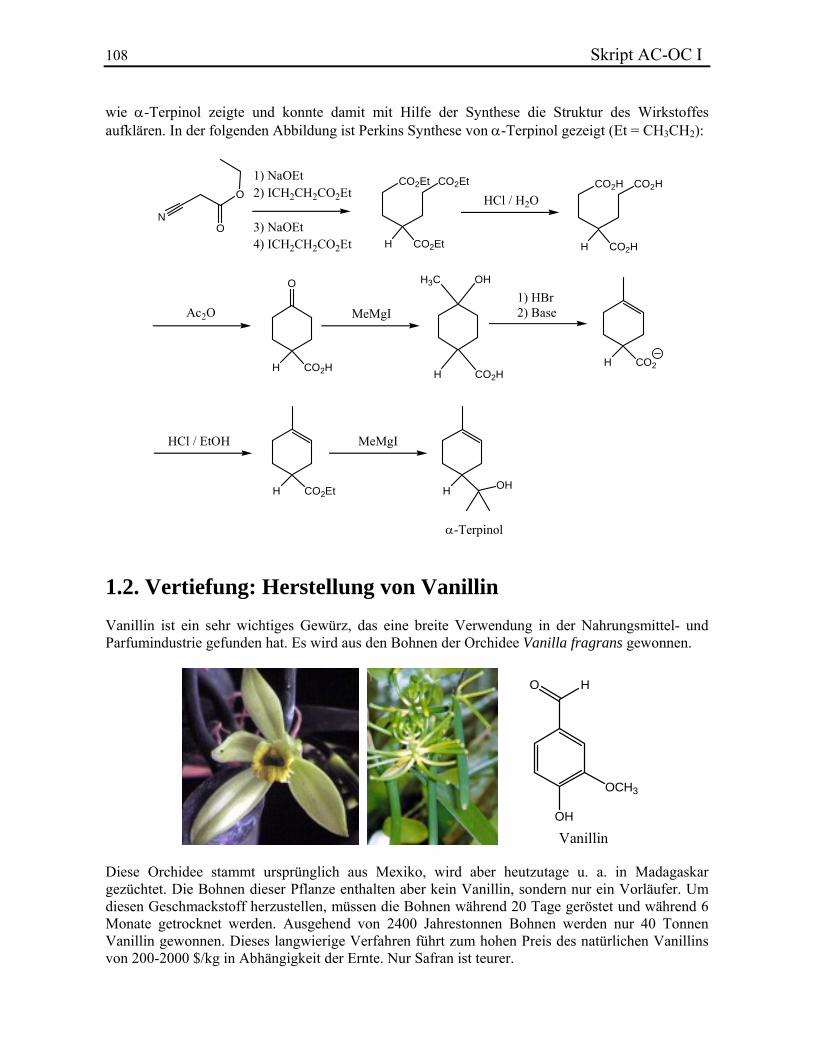
108 Skript AC-OC I wie -Terpinol zeigte und konnte damit mit Hilfe der Synthese die Struktur des Wirkstoffes aufklären. In der folgenden Abbildung ist Perkins Synthese von -Terpinol gezeigt (Et = CH3CH2):
NO
O
O
CO2HH
CO2EtH
Ac2O MeMgI
MeMgI
CO2Et CO2Et
H CO2Et
CO2HH
H3C OH
H OH
HCl / H2OCO2H CO2H
H CO2H
CO2H
-Terpinol
1) NaOEt2) ICH2CH2CO2Et
3) NaOEt4) ICH2CH2CO2Et
1) HBr2) Base
HCl / EtOH
1.2. Vertiefung: Herstellung von Vanillin Vanillin ist ein sehr wichtiges Gewürz, das eine breite Verwendung in der Nahrungsmittel- und Parfumindustrie gefunden hat. Es wird aus den Bohnen der Orchidee Vanilla fragrans gewonnen.
O H
OH
OCH3
Vanillin Diese Orchidee stammt ursprünglich aus Mexiko, wird aber heutzutage u. a. in Madagaskar gezüchtet. Die Bohnen dieser Pflanze enthalten aber kein Vanillin, sondern nur ein Vorläufer. Um diesen Geschmackstoff herzustellen, müssen die Bohnen während 20 Tage geröstet und während 6 Monate getrocknet werden. Ausgehend von 2400 Jahrestonnen Bohnen werden nur 40 Tonnen Vanillin gewonnen. Dieses langwierige Verfahren führt zum hohen Preis des natürlichen Vanillins von 200-2000 $/kg in Abhängigkeit der Ernte. Nur Safran ist teurer.

Skript AC-OC I 109 Da Vanillin so wichtig ist, wurden zwei neue Methoden entwickelt, um es aus billigen Ausgangsmaterialien zu synthetisieren. Es kann aus Lignin, eine komplexe Mischung aus Holz, oder aus Eugenol (aus Nelkeöl) durch Oxidation hergestellt werden. Das auf diese Weise hergestellte Vanillin besitzt selbstverständlich dieselben chemischen, physikalischen und organoleptischen Eigenschaften wie dasjenige aus den Orchideen. Der einzige wichtige Unterschied ist der Preis, der ca. 15$/kg beträgt. Heutzutage stammt nur 5% des Vanillins aus den Bohnen. Obwohl mit diesen drei Methoden dasselbe Molekül hergestellt wird (das Rösten der Bohnen produziert Vanillin durch Oxidation anderer Verbindungen) ist natürliches und künstliches Vanillin in gesetzlicher Hinsicht nicht zu vergleichen. In einigen Staaten darf nur das Vanillin aus den Bohnen als natürliches Vanillin verkauft werden. Die künstliche Herkunft des Produkts muss immer angegeben werden. Dies ist ein Nachteil für die Industrie, weil die Verbrauchern die Produkte mit dem natürlichen Vanillin bevorzugen und die Gewinnmarge kleiner wird. Um die Quelle der Herkunft eindeutig zu identifizieren, verwendet der Gesetzgeber geringe Unterschiede, die in den Isotopenverhältnissen von natürlichem und künstlichem Vanillin gibt. In der Tat hängt das Isotopenverhältnis einer Pflanze von klimatischen Faktoren wie z. B. der Temperatur ab. Eine Pflanze aus Madagaskar wird daher ein Isotopenverhältnis haben, der sich vom demjenigen einer Pflanze aus Skandinavien unterscheidet. Das Vanillin wird nur dann als natürlich bezeichnet, wenn sein Isotopenverhältnis in einem bestimmten Bereich liegt. Da aber die Preisunterschied von Vanillin so hoch ist, lohnt es sich heutzutage das Isotopenverhältnis während der Synthese aus Lignin und Eugenol zu kontrollieren, so dass es im gesetzlichen Bereich liegt. Das so hergestellte Vanillin kann daher als natürliches Vanillin bezeichnet werden, obwohl sie nicht aus den Bohnen stammt.
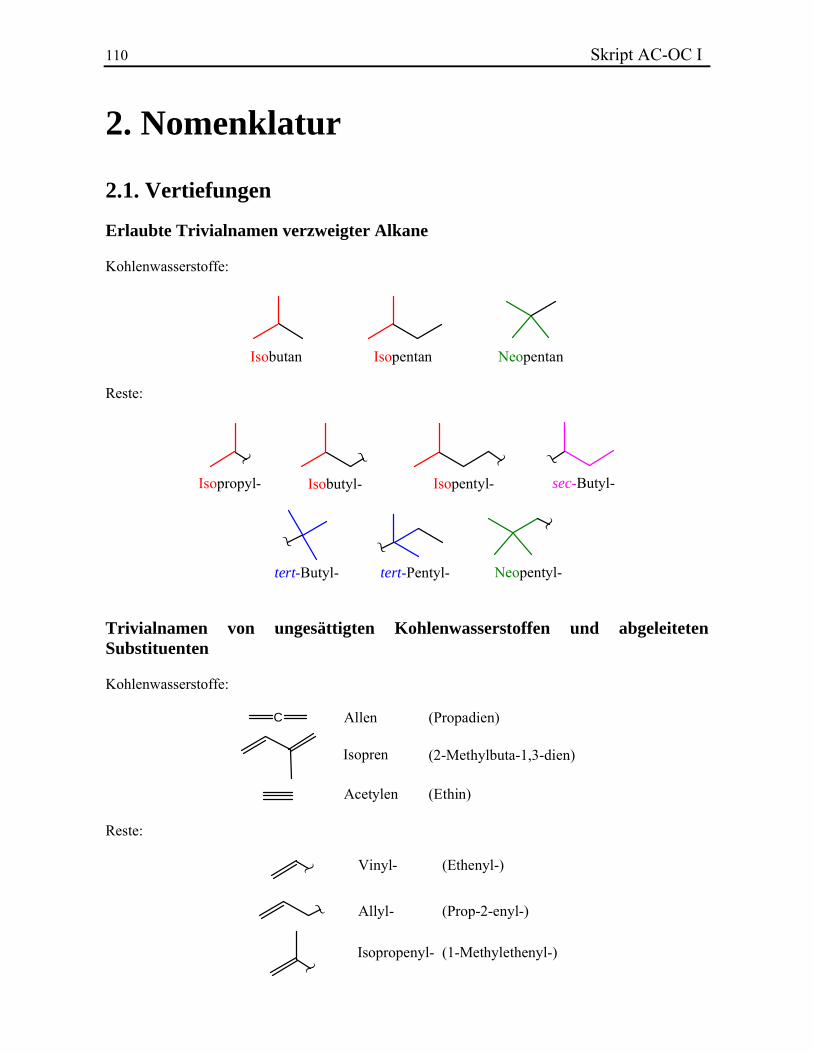
110 Skript AC-OC I
2. Nomenklatur
2.1. Vertiefungen Erlaubte Trivialnamen verzweigter Alkane Kohlenwasserstoffe:
Isopentan NeopentanIsobutan
Reste:
Isopropyl-
tert-Butyl- tert-Pentyl-
sec-Butyl-Isobutyl- Isopentyl-
Neopentyl-
~ ~ ~ ~
~ ~
~
Trivialnamen von ungesättigten Kohlenwasserstoffen und abgeleiteten Substituenten Kohlenwasserstoffe:
C Allen (Propadien)
Isopren (2-Methylbuta-1,3-dien)
Acetylen (Ethin)
Reste:
Allyl-
Isopropenyl- (1-Methylethenyl-)
Vinyl- (Ethenyl-)
(Prop-2-enyl-)~
~
~

Skript AC-OC I 111 Alte Nomenklatur für zweiwertige Substituenten Zweiwertige Reste, die aus zwei endständigen Bindungsstellen bestehen und als einwertige Substituenten dienen können, wurden durch das Suffix -ylen gekennzeichnet. Ausser dem Fall von –CH2–CH2– (Ethylen-), wurden die anderen Substituenten als eine Kette von Methyleneinheiten aufgefasst und durch ein multiplikatives Präfix + -methylen gekennzeichnet. Beispiele:
~~ Methylen-
Ethylen-
Trimethylen-
Tetramethylen-
~
~
~
~~~
Trivialnamen zweiwertiger Substituenten
C
C C
Isopropyliden- (1-Methylethyliden-)
Vinyliden- (Ethenyliden-)
Allenyliden- (Propadienyliden-)~~
~
Trivialnamen der monocyclischen Benzolderivate und abgeleiteten Reste Kohlenwasserstoffe:
HC CH C6H5C6H5
Toluol*Benzol*
m-Xylol**(auch o- und p-Xylol)
Mesitylen** Cumol**
Styrol* Stilben* Fulven**
o-Cymol**(auch m- und p-Cymol)

112 Skript AC-OC I Reste:
Phenyl-* Benzyl-* o-Tolyl-**(auch m- und p-Tolyl-)
Styryl-*Benzyliden-*
HC CH
~ ~
~
~
~
Phenethyl-* Cinnamyl-*
~
Benzhydryl-*
~
~~
Trityl-* Mesityl-**
~
*) Darf auch für am Ring substituierte Derivate angewendet werden. **) Darf nur für unsubstituierte Verbindungen angewendet werden. Trivialnamen verbrückter polycyclischer Alkane
Adamantan
12
3
456
7
8
9
10
Cuban Prisman Indan
1
1
1
2
2
2
3
3
3
4
4 4
5
5
5
6
6
6
7 77a
3a
8
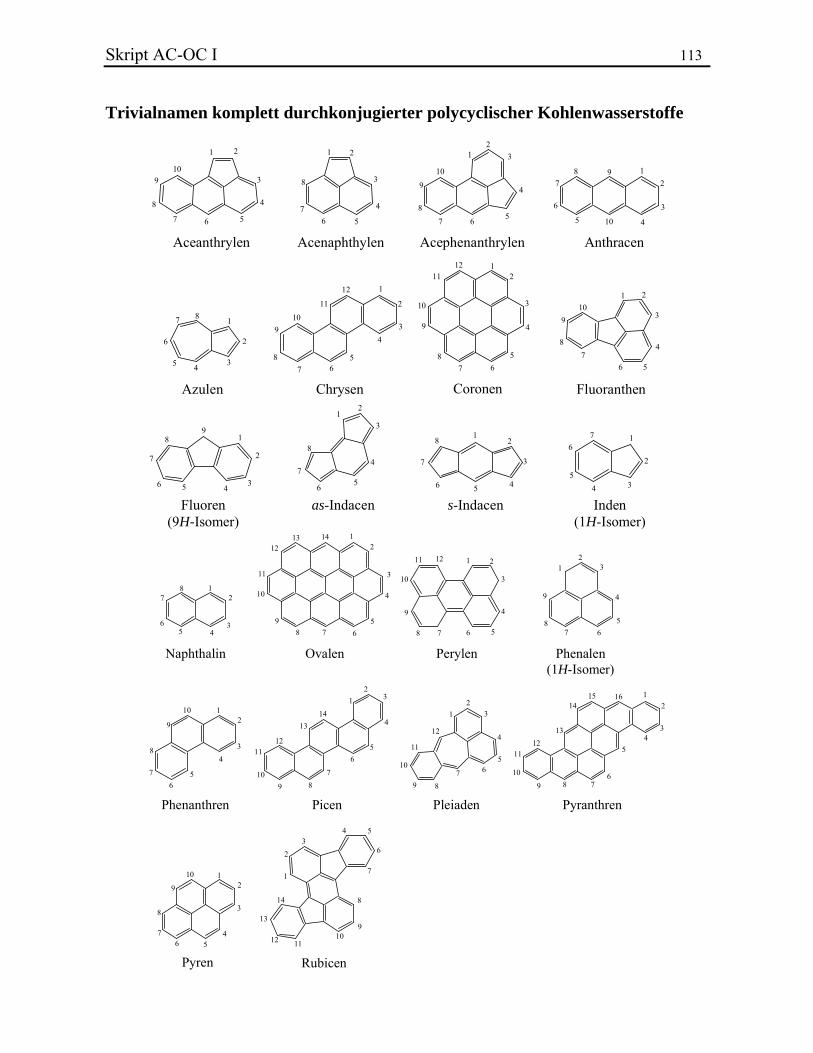
Skript AC-OC I 113 Trivialnamen komplett durchkonjugierter polycyclischer Kohlenwasserstoffe
Aceanthrylen Acenaphthylen Acephenanthrylen Anthracen
Azulen Chrysen Coronen Fluoranthen
Fluoren(9H-Isomer)
as-Indacen s-Indacen Inden(1H-Isomer)
1 2
3
4
567
8
910
1 2
3
4
56
7
8
12
3
4
567
8
9
10 1
2
3
45
6
7
8 9
10
1
2
34
5
6
7 8
1
2
3
4
567
8
9
10
11
12
12
3
4
5
67
8
9
10
1112
1 2
3
4
56
7
8
9
10
1
2
3456
7
89
12
3
4
56
7
8
12
3
456
7
8 1
2
34
5
6
7
12
345
6
78
12
3
4
5
678
9
10
11
13 14
12
1 2
3
4
5678
9
10
11 121
23
4
5
678
9
Naphthalin Ovalen Perylen Phenalen(1H-Isomer)
12
3
4
5
6
7
8
9
10
1
2
34
5
6789
10
11
13
14
12
15 16
1
2
3
4
567
89
10
11
12
1
23
4
5
6
7
89
10
11
13
14
12
Phenanthren Picen Pleiaden Pyranthren
1
2
34 5
6
7
8
910
11
13
14
12
12
3
456
7
8
9
10
Pyren Rubicen

114 Skript AC-OC I Geschlossene Ketten von o-verknüpften Benzolringen werden als Bi-, Tri-, Tetra-, Penta-, usw. -phenylene bezeichnet. Jeder Polycyclus besitzt eine eigene Priorität bezüglich der anderen. Vgl. IUPAC Nomenklatur der Organischen Chemie, S. 199. Trivialnamen heterocyclischer Substituenten
Furan Furyl- Piperidin Piperidyl- (1-Piperidyl- darf auch Piperidino- geschrieben werden) Morpholin Morpholino- (nur statt Morpholin-4-yl-) Pyridin Pyridyl- Chinolin Chinolyl- Isochinolin Isochinolyl- Thiophen Thienyl- (systematische Nomenklatur nicht erlaubt für dieses
Substituent)
Die Trivialnamen Thenyl- ((2-Thienyl)methyl-) und Furfuryl- ((2-Furyl)methyl-) dürfen nicht mehr benützt werden (H. A. Favre, K.-H. Hellwich, G.P. Moss, W. H. Powell, J. G. Traynham, Pure Appl. Chem. 1999, 71,1327). Hantzsch-Widman-System Das Hantzsch-Widman-System (W. H. Powell, Pure Appl. Chem. 1983, 55, 409) wird benützt, um Verbindungen mit drei- bis zehngliedrigen Heteromonocyclen mit einem oder mehreren Heteroatomen systematisch zu benennen. Die Heteroatome werden durch dieselben Präfixe der Austauschnomenklatur bezeichnet. Die Grösse und der Sättigungsgrad der Ringe werden durch spezielle Endbezeichnungen angegeben:
Ringglieder Maximal ungesättigt Gesättigt 3 -iren, -irin* -iran, -iridin* 4 -et -etan, -etidin* 5 -ol -olan, -olidin* 6 (N, ...) -in -inan 6 (O, S, ...) -in -an 6 (P, B, ...) -inin -inan 7 -epin -epan 8 -ocin -ocan 9 -onin -onan 10 -ecin -ecan
*) Diese Suffixe werden nur im Fall von stickstoffhaltigen Heterocyclen aus historischen Gründen bevorzugt verwendet. Bei Heterocyclen mit mehreren Heteroatomen erfolgt die Bezifferung in der Weise, dass das ranghöchste Heteroatom den Lokanten 1 erhält. Innerhalb des Periodensystems haben dabei die Elemente der höchsten Gruppe und der niedrigsten Periode den Vorrang. Die Heteroatome werden in der Reihenfolge abnehmender Priorität angeordnet. Wenn bei der Kombination zwei Vokale

Skript AC-OC I 115 aufeinander treffen, entfällt das endständige -a des Präfixes. Die Endsilben hängen von dem Heteroatom ab, das die niedrigste Priorität besitzt. Die Lokanten müssen natürlich der Ordnung der Heteroatome entsprechen und werden zusammen dem Namen vorangestellt. Partiell ungesättigte Heterocyclen werden durch Verwendung des hydro-Präfixes benannt. Beispiele:
NB
O
N
NHO
HB
P
P
S
NO
Borinan
1,2-Oxazinan
Phosphinin
1,3,5,2-Oxadiazaborol
2,3-Dihydrophosphet
5,6-Dihydro-1,4,2-oxathiazin
1
1
2
1
1
23
4
5
1
23
4
1
2
3
4
5
6
Weitere Nomenklaturtypen Neben der substitutiven und der radikofunktionellen Nomenklatur, die gelernt werden müssen, gibt es noch weitere Methoden, die in speziellen Fällen sehr hilfreich sind. Additive Nomenklatur Bei diesem Nomenklaturtyp wird eine Addition von Atomen oder Atomgruppen an die Stammverbindung vorgenommen. Die wichtigste Anwendung ist die Benennung hydrierter cyclischer Verbindungen, die in enger Beziehung zu einem völlig ungesättigten System stehen. Ein weiterer Fall sind Aminoxide und Nitrone. Beispiele:
N
O N
O
1,4,5,8-Tetrahydronaphthalin Trimethylamin-N-oxid (N-Benyliden)methylamin-N-oxid
Subtraktive Nomenklatur Man verwendet subtraktive Präfixe oder Suffixe, um die Entfernung von Atomen oder Atomgruppen aus einem trivialen oder systematischen Stammbegriff anzuzeigen. Die wichtigste Anwendung bezieht sich auf die Entfernung von Wasserstoff (Präfix Dehydro-) was zu einem stärker ungesättigten System führt. Bei komplexen trivial benannten Naturstoffen werden manchmal Derivate mit dem Präfix des- gekennzeichnet:
R2N-CH3 R2NH des-N-methyl- RCH2-OH RCH3 desoxy-

116 Skript AC-OC I Konjunktive Nomenklatur Diese Nomenklatur wird selten benützt. Insbesondere acyclische Carbonsäuren, Aldehyde, Alkohole und Amine, die terminal einen cyclischen Substituenten tragen, werden dadurch benannt, indem man den unveränderten Stammnamen des Cyclus mit dem unveränderten Namen der acyclischen Komponente kombiniert:
OH
O
OH
Cyclohexanmethanol(substitutiv: Cyclohexylmethanol)
2-Naphthalinessigsäure(substitutiv: 2-Naphthylessigsäure)
Relative Priorität wichtiger funktioneller Gruppe Im Folgenden sind einige wichtigen funktionellen Gruppen nach absteigender Priorität angeordnet (R sind Reste, M ist ein beliebiges Metall, X ist ein Halogenid; C-Atome, die zwischen eckigen Klammern stehen, sind im Stammnamen einbezogen):
Verbindungsklasse Charakteristische Gruppe
Präfix Suffix
Freie Radikale R·, RO·, … --- z. B. -yl
Anionen* R–, R2N–, … --- -id
Kationen –(NR3)+, R+, ... z. B. -io- z. B. -ium
Carbonsäuren –C(=O)OH –[C](=O)OH
Carboxy- -carbonsäure -säure
Sulfonsäuren –S(=O)2OH Sulfo- -sulfonsäure
Sulfinsäuren –S(=O)OH Sulfino- -sulfinsäure
Sulfensäuren –SOH Sulfeno- -sulfensäure
Boronsäuren –B(OH)2 Borono- -boronsäure
Carbonsäuresalze –C(=O)OM –[C](=O)OM
M-carboxylato- M -carboxylat M -oat
Sulfonsäuresalze –S(=O)2OM M-sulfonato- M -sulfonat
Sulfinsäuresalze –S(=O)OM M-sulfinato- M -sulfinat
Sulfensäuresalze –SOM M-sulfenato- M -sulfenat
Carbonsäureanhydride –C(=O)OC(=O)– --- -säure…säureanhydrid -säureanhydrid

Skript AC-OC I 117
Carbonsäureester –C(=O)OR –[C](=O)OR
…yloxycarbonyl- ---
-yl…carboxylat -yl…oat
Sulfonsäureester –S(=O)2OR -yloxysulfonyl- -yl…sulfonat
Sulfinsäureester –S(=O)OR -yloxysulfinyl- -yl…sulfinat
Sulfensäureester –SOR -yloxysulfenyl- -yl…sulfenat
Lactone**
O
OR
--- -olacton -carbolacton
Sultone** S
O
OR
O--- -sulton
Carbonsäurehalogenide –C(=O)X –[C](=O)X
Halogencarbonyl--carbonylhalogenid -oylhalogenid
Sulfonsäurehalogenide –S(=O)2X Halogensulfonyl- -sulfonylhalogenid
Sulfinsäurehalogenide –S(=O)X Halogensulfinyl- -sulfinylhalogenid
Sulfensäurehalogenide –SOX Halogensulfenyl- -sulfenylhalogenid
Carbonsäureamide –C(=O)NH2 –[C](=O)NH2
Carbamoyl- ---
-carboxamid -amid
Sulfonsäureamide –S(=O)2NH2 Sulfamoyl- -sulfonamid
Sulfinsäureamide –S(=O)NH2 Sulfinamoyl- -sulfinamid
Sulfensäureamide –SONH2 Sulfenamoyl- -sulfenamid
Lactame**
O
NHR
--- -lactam
Lactime**
OH
NHR
--- -lactim
Carbonsäureimide** O
HN O
R
--- -dicarboximid -imid
Carbonsäurehydrazide –C(=O)-NH-NH2 –[C](=O)-NH-NH2
--- -carbohydrazid -hydrazid

118 Skript AC-OC I
Hydroxamsäure –C(=O)-NH-OH –[C](=O)-NH-OH
--- -carbohydroxamsäure -hydroxamsäure
Amidine –C(=NH)NH2
–[C](=NH)NH2 Carbamimidoyl- ---
-carboximidamid -imidamid
Nitrile –CN –[C]N
Cyan- -carbonitril -nitril
Aldehyde –C(=O)H –[C](=O)H
Formyl- Oxo-
-carbaldehyd -al
Thioaldehyde –C(=S)H –[C](=S)H
Thioformyl- Thioxo-
-carbothioaldehyd -thial
Ketone >C=O Oxo- -on
Thioketone >C=S Thioxo- -thion
Acetale >C(OR)(OR') …yloxy…yloxy- -al…yl…ylacetal -on…yl…ylacetal
Oxime >C=N-OH Hydroxyimino- -aloxim -onoxim
Hydrazone >C=N-NH2 Hydrazono- -alhydrazon -onhydrazon
Azine >C=N-N=C< Azinodi- -ylidenhydrazon
Alkohole, Phenole –OH Hydroxy- -ol
Thiole –SH Sulfanyl- -thiol
Alkoholate, Phenolate –OM M-oxido- M -olat
Amine –NH2 Amino- -amin
Hydroxylamine –NH-OH Hydroxylamino- -N-…hydroxylamin
Imine >C=NH Imino- -imin
*) Ausser Anionen von Säuren, Alkoholen, Thiole, Selenole und Tellorole. **) Diese Verbindungen werden immer häufiger mit der Austauschnomenklatur benannt. Wichtige charakteristische Gruppe, die nur als Präfix auftreten C-Atome, die zwischen eckigen Klammern stehen, sind im Stammnamen einbezogen.

Skript AC-OC I 119
Charakteristische Gruppe
Präfix Charakteristische Gruppe
Präfix
–F Fluor- –NH-NH2 Hydrazino- oder Diazanyl-
–Cl Chlor- –N3 Azido- –Br Brom- =N2 Diazo- –I Iod- –NC Isocyan- –ClO Chlorosyl- –NO Nitroso- –ClO2 Chloryl- –NO2 Nitro- –ClO3 Perchloryl- –OCN Cyanat- –IO Iodosyl- –NCO Isocyanat- –IO2 Iodyl- –SCN Thiocyanat- –SR …ylsulfanyl- –NCS Isothiocyanat- –S(=O)R …ylsulfinyl- –OR …yloxy- –S(=O)2R …ylsulfonyl- –OOH Hydroperoxy- >C(SR)2 Di-…ylthio- –OOR …ylperoxy-
[C] [C]
S ~~~~
Epithio- [C] [C]
O ~~~~
Epoxy-
Radikofunktionelle Verbindungsnamen C-Atome, die zwischen eckigen Klammern stehen, sind im Stammnamen einbezogen.
Charakteristische Gruppe Radikofunktioneller Verbindungsname –C(=O)OR -ester Halogenide in Säurederivaten (z. B. R[C](=O)X, RS(=O)2X)
-fluorid, -chlorid, -bromid, -iodid
–CN;–NC -cyanid; -isocyanid –OCN; –NCO -cyanat; -isocyanat –SCN; –NCS -thiocyanat; -isothiocyanat >C=O; >C=S -keton; -thioketon >C=C=O -keten –OH; –SH -alkohol; -hydrosulfid –OOH; –SSxSH -hydroperoxid; -hydropolysulfid (oder -
hydropolysulfan) –O–; –O-O– -ether (oder –oxid); -peroxid –S–; –S-S– -sulfid; -disulfid –S(=O)–; –S(=O)2– -sulfoxid; -sulfon Halogenide; –N3 -fluorid, -chlorid, -bromid, -iodid; -azid –NH2, –NH–, >N– -amin –NH-NH2 -hydrazin –NH-OH -hydroxylamin –N=C=N– -carbodiimid
Wichtige zugelassene Trivialnamen für Carbonsäuren

120 Skript AC-OC I Neben Ameisensäure, Essigsäure und Benzoesäure sind weitere Trivialnamen für Carbonsäuren erlaubt. Darunter sind z. B. die Aminosäuren, die eine zentrale Rolle in der Biochemie spielen. Weitere häufig verwendete Trivialnamen sind im Folgenden wiedergegeben. Obwohl sie noch zugelassen sind, ist es nicht empfohlen, sie zu benützen.
Verbindung Säure Rest Anion n-C2H5-COOH Propionsäure Propionyl- Propionat n-C3H7-COOH Buttersäure Butyryl- Butyrat n-C4H9-COOH Valeriansäure Valeryl- Vallerat tert-C4H9-COOH Pivalinsäure Pivaloyl- Pivalat n-C5H11-COOH Capronsäure Caproyl- Caproat n-C11H23-COOH Laurinsäure Lauroyl- Laurat n-C15H31-COOH Palmitinsäure Palmitoyl- Palmitat n-C17H35-COOH Stearinsäure Stearoyl- Stearat H2C=CH-COOH Acrylsäure Acryloyl- Acrylat
COOH Crotonsäure Crotonoyl- Crotonat
COOH
Zimtsäure Cinnamoyl- Cinnamat
HCC-COOH Propiolsäure Propioyl- Propiolat HOOC-COOH Oxalsäure Oxalyl- Oxalat HOOC-CH2-COOH Malonsäure Maloyl- Malonat HOOC-(CH2)2-COOH Bernsteinsäure Succinyl- Succinat HOOC-(CH2)3-COOH Glutarsäure Glutaryl- Glutarat
COOHHOOC Maleinsäure Maleoyl- Maleat
HOOCCOOH
Fumarsäure Fumaroyl- Fumarat
Viele Carbonsäurederivate besitzen eigene Trivialnamen. Für weitere Beispiele siehe IUPAC Nomenklatur der organischen Chemie.
2.2. Beispiele Triviale und systematische Namen Die systematischen Namen können rasch kompliziert werden und sehr oft werden noch die Trivialnamen benützt. Im Folgenden sind die trivialen und systematischen Namen zwei gewöhnlicher Verbindungen geschrieben (die Bedeutung aller Symbole und Silben wird am Ende dieser Vorlesung klar sein):

Skript AC-OC I 121
OH
(-)-Menthol(1R,2S,5R)-2-Isopropyl-5-methylcyclohexanol
N
NN
N
NH2
O
HOH
HH
HH
HO
Adenosin2-(6-Aminopurin-9-yl)-5-
hydroxymethyltetrahydrofuran-3,4-diol Beispiele unverzweigter gesättigter Ketten und daraus abgeleiteter Reste n-C45H92 Pentatetracontan n-C81H164 Henoctacontan CH3– Methyl- CH3CH2– Ethyl- CH3(CH2)10CH2– Dodecyl- Beispiel systematischer Namen eines verzweigten Alkans Als Beispiel der Anwendung der Nomenklaturregeln für verzweigte Alkane betrachte man die folgende Verbindung:
Regel (2) bestimmt die Hauptkette eindeutig. Da sie zehn C-Atome enthält, ist die Verbindung ein Derivat des Decans. Zwei Lokantensätze sind möglich: Nummerierung von links oder von rechts. Im ersten Fall hätten die Substituenten die Lokanten 3,5,5,8, im zweiten Fall 3,6,6,8. Der richtige Lokantensatz ist daher von links nach rechts. Es sind zwei Arten von Substituenten vorhanden: Dreimal Ethyl und einmal Methyl. Der systematische Name der Verbindung ist also 3,5,5-Triethyl-8-methyldecan. Weiteres Beispiel:
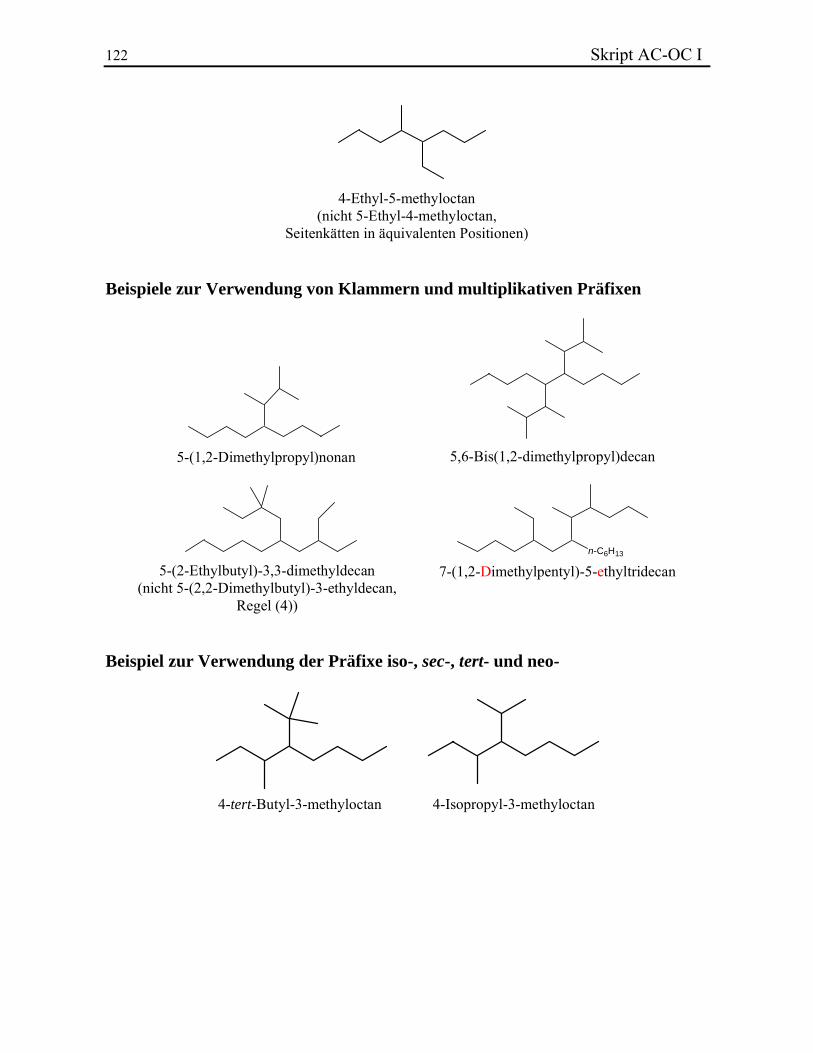
122 Skript AC-OC I
4-Ethyl-5-methyloctan(nicht 5-Ethyl-4-methyloctan,
Seitenkätten in äquivalenten Positionen) Beispiele zur Verwendung von Klammern und multiplikativen Präfixen
5-(1,2-Dimethylpropyl)nonan 5,6-Bis(1,2-dimethylpropyl)decan
5-(2-Ethylbutyl)-3,3-dimethyldecan(nicht 5-(2,2-Dimethylbutyl)-3-ethyldecan,
Regel (4))
n-C6H13
7-(1,2-Dimethylpentyl)-5-ethyltridecan
Beispiel zur Verwendung der Präfixe iso-, sec-, tert- und neo-
4-tert-Butyl-3-methyloctan 4-Isopropyl-3-methyloctan
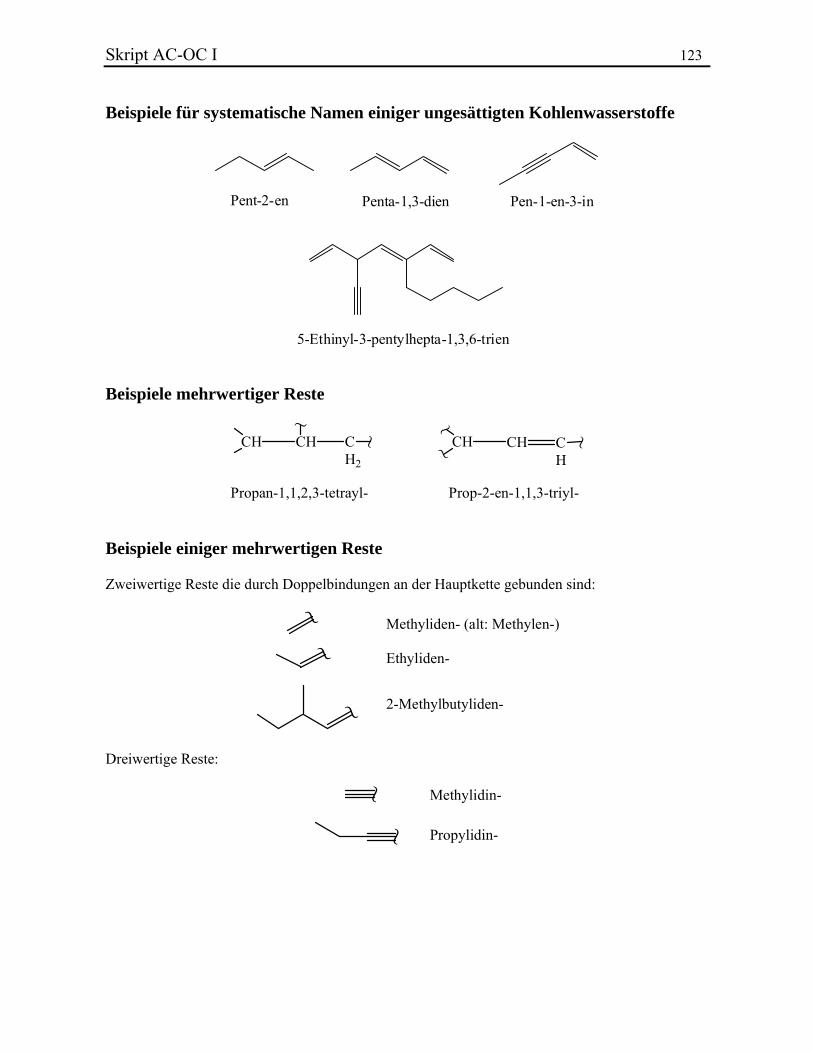
Skript AC-OC I 123 Beispiele für systematische Namen einiger ungesättigten Kohlenwasserstoffe
Pent-2-en Penta-1,3-dien Pen-1-en-3-in
5-Ethinyl-3-pentylhepta-1,3,6-trien Beispiele mehrwertiger Reste
CH CH CH
~
~ ~CH CH CH2
~
~
Propan-1,1,2,3-tetrayl- Prop-2-en-1,1,3-triyl- Beispiele einiger mehrwertigen Reste Zweiwertige Reste die durch Doppelbindungen an der Hauptkette gebunden sind:
Methyliden- (alt: Methylen-)
Ethyliden-
2-Methylbutyliden-
~~
~
Dreiwertige Reste:
Methylidin-
Propylidin-~~
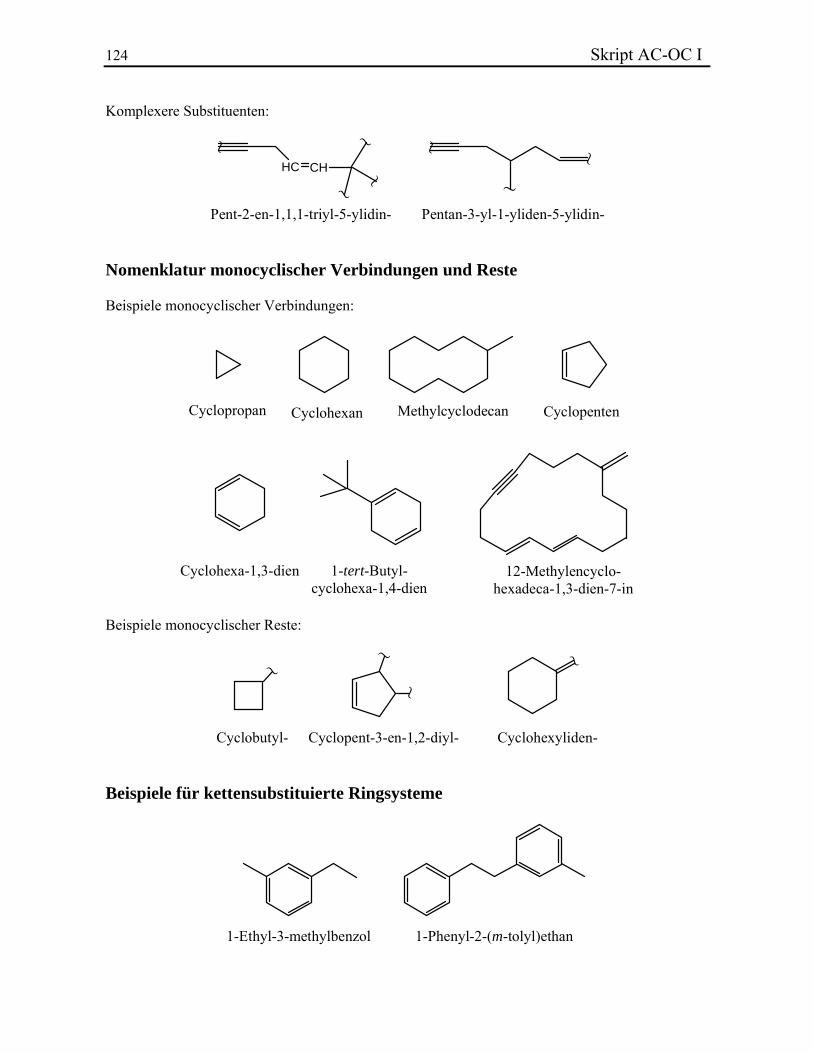
124 Skript AC-OC I Komplexere Substituenten:
~
HC CH
~
~~
~ ~
~
Pent-2-en-1,1,1-triyl-5-ylidin- Pentan-3-yl-1-yliden-5-ylidin- Nomenklatur monocyclischer Verbindungen und Reste Beispiele monocyclischer Verbindungen:
Cyclopropan Cyclohexan Methylcyclodecan
Cyclohexa-1,3-dien
Cyclopenten
1-tert-Butyl-cyclohexa-1,4-dien
12-Methylencyclo-hexadeca-1,3-dien-7-in
Beispiele monocyclischer Reste:
Cyclobutyl- Cyclopent-3-en-1,2-diyl- Cyclohexyliden-
~~
~
~
Beispiele für kettensubstituierte Ringsysteme
1-Ethyl-3-methylbenzol 1-Phenyl-2-(m-tolyl)ethan

Skript AC-OC I 125 Beispiele polyzyklischer verbrückter Verbindungen
Bicyclo[3.2.1]octan
Tetracyclo[4.4.1.13,9.02,4]dodecan(nicht Tetracyclo[5.3.1.14,9.02,10]dodecan, weil derHauptring weniger symmetrisch geteilt ist)
Tricyclo[3.2.1.02,4]octan (Brücke mit einem C-Atomsind besser als Brücke mit keinem Atom)
Hexacyclo[15.3.2.23,7.12,12.013,21.011,25]pentacosan(013,21 und 011,25sind abhängige Sekundärbrücke,d. h. abnehmende Anordnung der Lokanten)
1 2 3
7
111213
17
18
20
2122
2324
25
Beispiele für Derivate verbrückter polycyclischer Verbindungen
Bicyclo[2.2.1]hept-2-en Bicyclo[6.4.1]tridec-1(13)-en
21
8
12
13
Nomenklatur nicht vollständig ungesättigter Polycyclen
1-Methyl-1,2,3,4-tetrahydronaphthalin
(alt: Tetralin)
Decahydronaphthalin(alt: Decalin)
1-Methyliden-1,2,4b,8a-tetrahydrophenanthrene

126 Skript AC-OC I Das multiplikative Präfix von hydro- muss für die alphabetische Aufzählung berücksichtigt werden. Die Hydro-Präfixe werden im Namen beibehalten, auch wenn eines oder mehrere der beigeführten H-Atome nachträglich durch andere Substituenten ersetzt werden. Beispiele monospirocyclischer Verbindungen
Spiro[3.3]heptan
Spiro[2.4]heptan
Spiro[3.5]nona-1,5,7-trien
Beispiele über Einfachbindungen gebundener Ringe
1,1'-Bicyclobutyl Bi(cyclobut-2-en-1-yl) 1,1'-Biphenyl
3,4'-Dimethyl-1,1'-biphenyl 1,2'-Binaphtyl(oder 1,2'-Binaphthalin)
Bi(bicyclo[2.2.1]hept-5-en-2-yl)(oder 5,5'-Bibicyclo[2.2.1]hept-2-en)Verschiedene Nummerierungsregeln!
Beispiele für Nomenklatur durch Doppelbindungen verbundener Verbindungen
1,1'-Bicyclohexyliden(Cyclohexylidencyclohexan)
1,1'-Bi(cyclopenta-2,4-dien-1-yliden)(5-(Cyclopenta-2,4-dien-1-yliden)cyclopenta-1,3-dien)
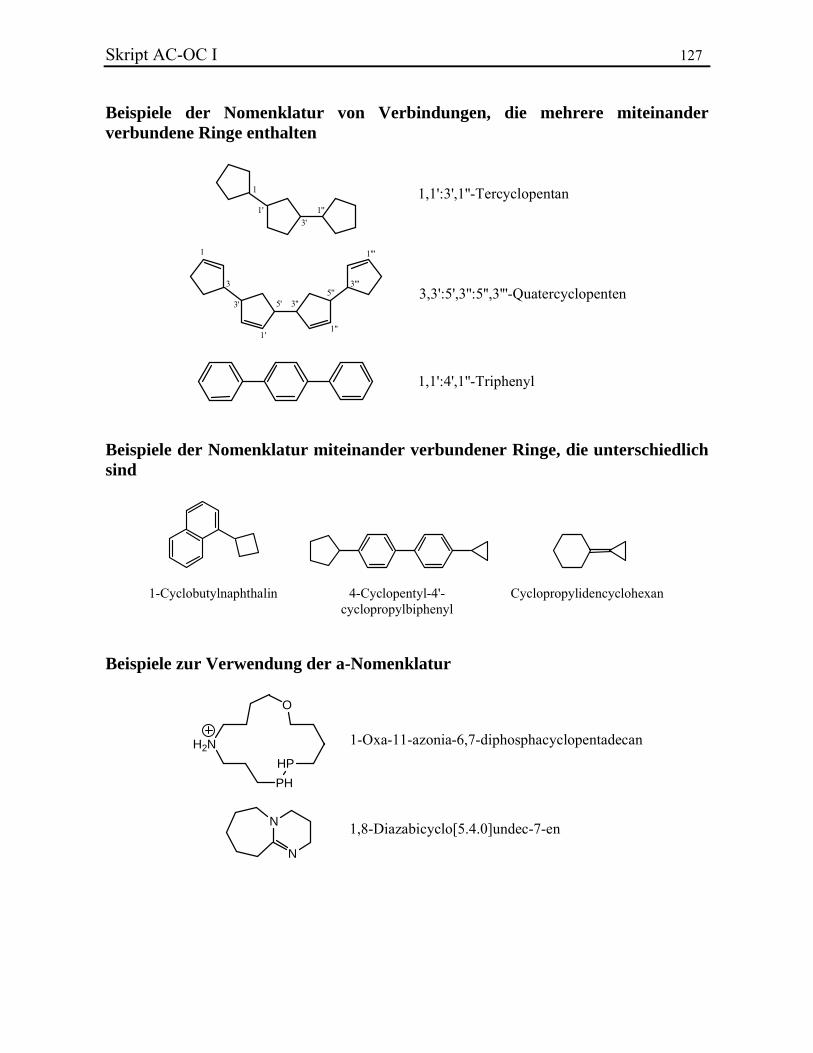
Skript AC-OC I 127 Beispiele der Nomenklatur von Verbindungen, die mehrere miteinander verbundene Ringe enthalten
1,1':3',1''-Tercyclopentan
3,3':5',3'':5'',3'''-Quatercyclopenten
1,1':4',1''-Triphenyl
1
1'
3'
1''
1
3
1'
3' 5' 3''
1''
5''
1'''
3'''
Beispiele der Nomenklatur miteinander verbundener Ringe, die unterschiedlich sind
1-Cyclobutylnaphthalin 4-Cyclopentyl-4'-cyclopropylbiphenyl
Cyclopropylidencyclohexan
Beispiele zur Verwendung der a-Nomenklatur
N
N
1,8-Diazabicyclo[5.4.0]undec-7-en
HP
O
H2N
PH
1-Oxa-11-azonia-6,7-diphosphacyclopentadecan

128 Skript AC-OC I
O
S
N
S
Si
N
HB
9-Borabicyclo[3.3.1]nonan
6-Oxa-1,9-dithia-3-azaspiro[4.4]non-2-en
9-Aza-4a-silaphenanthren
Beispiele radikofunktioneller Nomenklatur n-C5H11-COF Hexanoylfluorid H3C-CN Methylcyanid H3C-CO-C2H5 EthylmethylketonH3C-O-CH3 Dimethylether (H3C)2SO Dimethylsulfoxid

Skript AC-OC I 129
3. Klassische Strukturlehre
3.1. Vertiefungen Natürlich vorkommende Zucker Geschichtlicher Überblick Der Zucker ist mit der Kulturgeschichte der Menschheit eng verbunden. Die ältesten süssen, zuckerhaltigen Nahrungsmittel, die den Menschen zur Verfügung standen, waren der Bienenhonig und süsse Pflanzensäfte. Der Honig ist vermutlich bereits in vorgeschichtlicher Zeit ein beliebtes Genussmittel gewesen. Später wurde er durch den zuckerhaltigen Zellsaft des Zuckerrohrs (wird tropischen Ländern angebaut) zurückgedrängt. Erst sehr viel später kam die Zuckergewinnung aus der Zuckerrübe (wird in gemässigten Klimazonen angebaut) hinzu. Die Araber brachten das Zuckerrohr nach Europa, und um 750 n. Chr. wurde es auf Sizilien und in Spanien kultiviert. Der Anbau des Rohrs erreichte hier bald eine grosse Blüte. Um 1000 n. Chr. wurden bereits einige 10000 t Rohr verarbeitet. Er diente allerdings mehr als Luxusartikel oder Arzneimittel. Die Zuckerrübe hat dagegen erst verhältnismässig spät als Rohstoffquelle für die Zuckergewinnung Bedeutung erlangt. Im Jahre 1801 errichtete der Berliner Chemiker und Akademiedirektor A. S. Margraf mit Unterstützung von König Friedrich Wilhelm III von Preussen die erste Rübenzuckerfabrik der Welt in Cunern in Schlesien. Starken Auftrieb erfuhr kurze Zeit später die junge Zuckerindustrie durch die 1806 von Napoleon verhängte Kontinentalsperre, wodurch der Import von Rohrrohzucker (Kolonialzucker) zu den Raffinerien der mitteleuropäischen Hafenstädte verhindert wurde. Die Fabrikationsprozesse für die Herstellung von Zucker gehören zu den am längsten bekannten und am sorgfältigsten studierten Vorgängen der chemischen Industrie. Die Weltjahresproduktion liegt zur Zeit bei über 108 t! 100 g Saccharose enthalten 410 Kalorien. Zucker zeichnet sich über seinen Nähr- und Genusswert hinaus durch leichte Verdaulichkeit und schnelle Resorbierbarkeit im menschlichen Organismus aus. Der Prokopfverbrauch pro Jahr beläuft sich in der Schweiz auf etwa 30 kg. Glucose D-Glucose (Dextrose, Traubenzucker) ist in fast allen süssen Früchten (meist mit D-Fructose zur Saccharose verbunden) verbreitet, besonders in Weintrauben, woher auch der Name Traubenzucker stammt. D-Glucose ist auch am Aufbau von Polysacchariden beteiligt (Cellulose Pergamentpapier-Poster, Stärke Enzym-Poster) und kann aus diesen durch Hydrolyse mit Hilfe von Säuren oder Enzymen gewonnen werden. Die Spaltung von Stärke in ihre Glucosebausteine durch im Speichel vorhandene Enzyme ist dafür verantwortlich, dass Stärke nach etwa 1-2 min süss schmeckt. Durch Mikroorganismen kann Glucose zu Ethanol, Essig-, Milch- oder Buttersäure vergoren werden. Fructose D-Fructose (Laevulose, Fruchtzucker) ist die wichtigste Ketohexose unter den natürlich vorkommenden Zuckerarten. In freier Form findet sie sich neben der D-Glucose in vielen süssen Früchten und im Honig. Als Bestandteil von Di- und Oligosacchariden finden wir die Fructose u. a. in Saccharose und Raffinose. Ausserdem enthalten viele stärkeähnliche Polysaccharide (Polyfructosane) des Pflanzenreichs wie Inulin als Baustein fast ausschliesslich D-Fructose.

130 Skript AC-OC I Mannose D-Mannose kommt in der Natur z. T. frei, häufig jedoch gebunden in Form der Mannane, z. B. in der Steinnuss und in Johannisbrotsamen, vor. Es wird ein süss-bitterer Geschmack berichtet. Für Bienen ist Mannose giftig. Saccharose Saccharose (Haushaltszucker, Rübenzucker, Rohrzucker, Sucrose) ist ein nichtreduzierendes Disaccharid, das durch Säuren oder Enzyme (Invertase) leicht in die Bausteine D-Glucose und D-Fructose gespalten wird. Das Gemisch wird Invertzucker genannt. Saccharose ist in zahlreichen Pflanzen in kleinen Mengen anzutreffen, z. B. im Süssmais oder der Zuckerhirse, in Früchten, Samen und Baumsäften. Grössere Zuckergehalte finden sich fast nur im Zuckerrohr und in der Zuckerrübe. Relative Süsswerte bei 25°C:
Saccharose 100 D-Glucose 69 D-Fructose 114 D-Mannose 59
M,P-Nomenklatur für Moleküle mit Chiralitätsachse Die Spezifikation der Chiralität von Verbindungen mit einer Chiralitätsachse kann auch mit Hilfe des Torsionswinkels zwischen den Bezugsgruppen erfolgen. Die Bezugsgruppen sind die am nächsten beieinanderliegenden, direkt an Atome in der Achse gebundenen Atome. Man legt zunächst die Prioritäten der Bezugsgruppen wie im Falle des gestreckten Tetraeders fest. Als nächstes betrachtet man den Diederwinkel wxyz, wobei die Positionen w und z jeweils den Bezugsatomen höchster Priorität am nähren bzw. am entfernsten Ende der Chiralitätsachse entsprechen. Ist dieser Diederwinkel positiv, so erhält die Struktur den Deskriptor P (plus); ist er negativ, so erhält sie den Deskriptor M (minus).
Br
Cl
O2N
HOOC
(M)-2'-Brom-6'-chlor-6-nitrobiphenyl-2-carbonsäure
z
y
x ww
xy
z
M
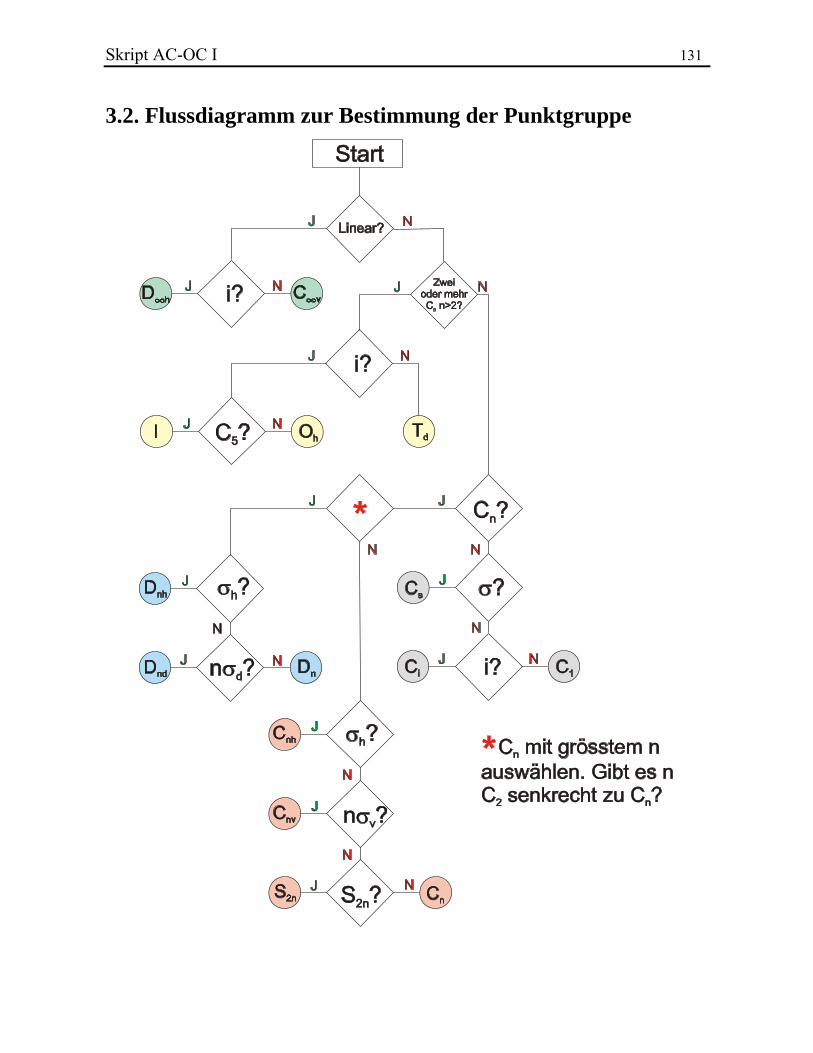
Skript AC-OC I 131
3.2. Flussdiagramm zur Bestimmung der Punktgruppe
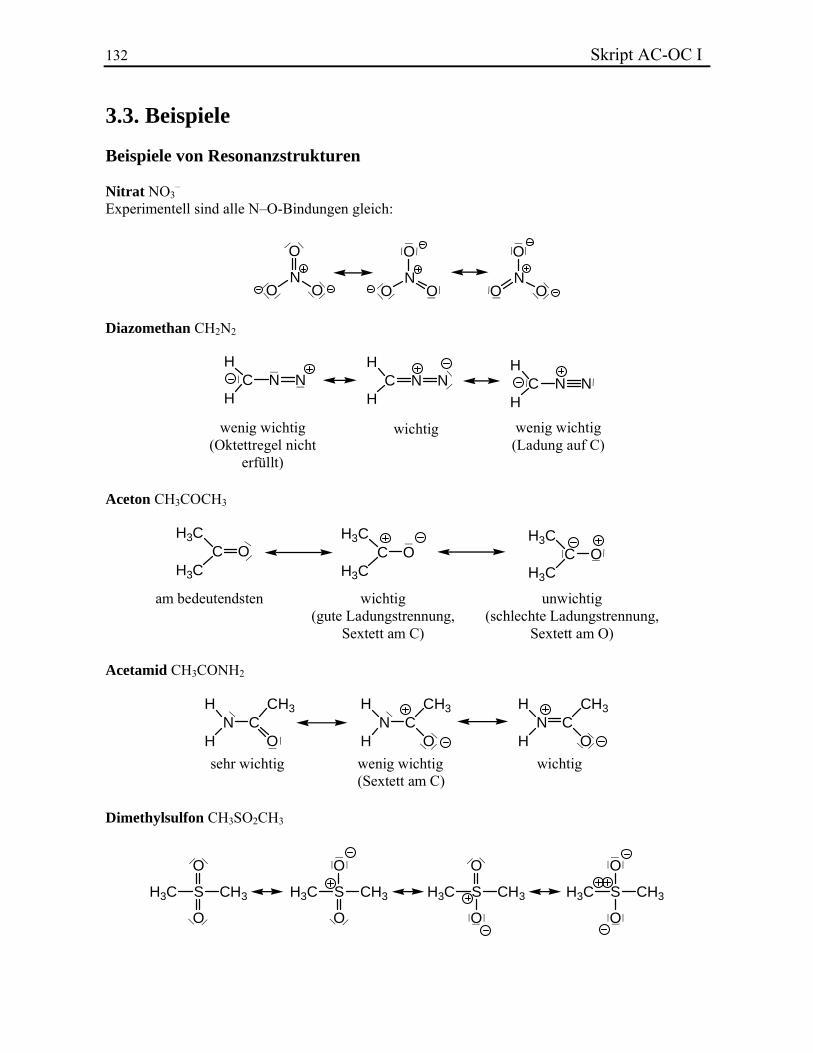
132 Skript AC-OC I
3.3. Beispiele Beispiele von Resonanzstrukturen Nitrat NO3
–
Experimentell sind alle N–O-Bindungen gleich:
N
O
O ON
O
O ON
O
O O–––
––– ––
–
––
––
––
–
–
– –
– –
– ––
Diazomethan CH2N2
–
–– –C N NH
HC N N
H
HC N N
H
H
–
–
–
wenig wichtig(Oktettregel nicht
erfüllt)
wenig wichtig(Ladung auf C)
wichtig
Aceton CH3COCH3
H3CC
H3CO
H3CC
H3CO
H3CC
H3CO
–– ––
––
am bedeutendsten wichtig(gute Ladungstrennung,
Sextett am C)
unwichtig(schlechte Ladungstrennung,
Sextett am O) Acetamid CH3CONH2
N CCH3
OH
HN C
CH3
OH
HN C
CH3
OH
H
––
––
––
–
– – –
sehr wichtig wenig wichtig(Sextett am C)
wichtig
Dimethylsulfon CH3SO2CH3
H3C S
O
O
CH3 H3C S
O
O
CH3 H3C S
O
O
CH3 H3C S
O
O
CH3
–
– –
––
– –
––
– –
–– –
– –
– –– –
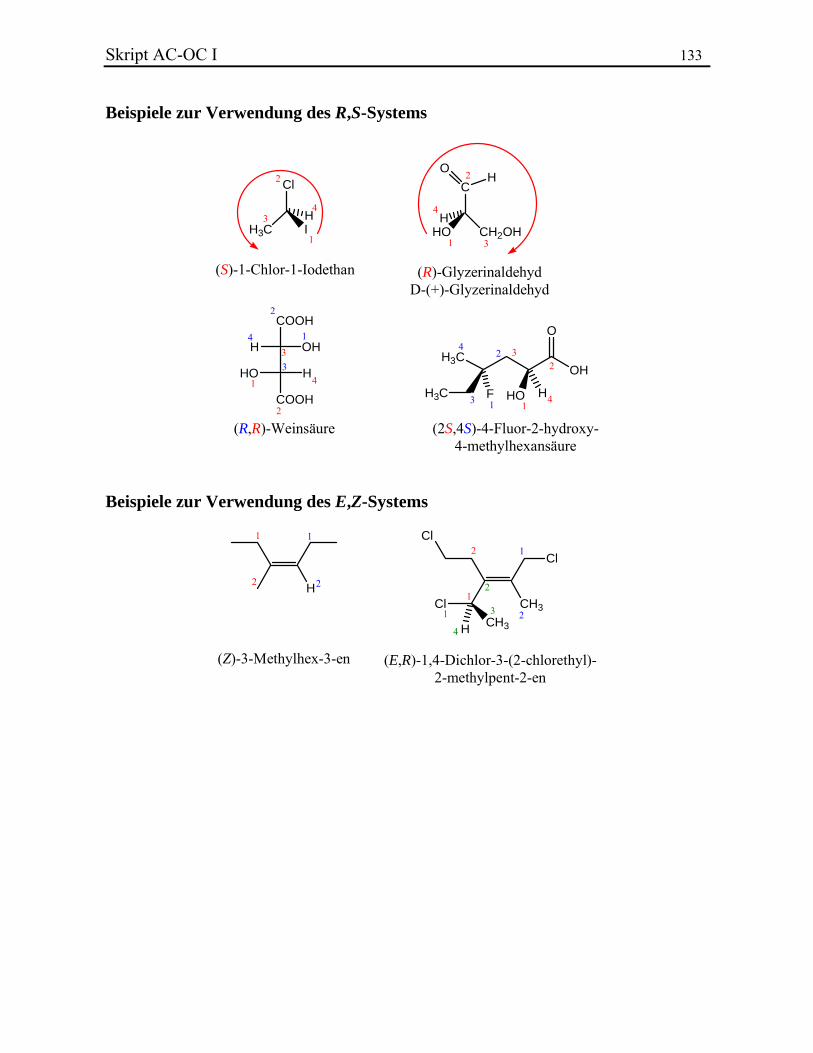
Skript AC-OC I 133 Beispiele zur Verwendung des R,S-Systems
Cl
H3C IH
C
CH2OHHOH
OH
H3C
O
OH
HOF HH3C
COOH
COOH
HO H
H OH
1
2
34
(S)-1-Chlor-1-Iodethan
1
2
3
4
(R)-GlyzerinaldehydD-(+)-Glyzerinaldehyd
1
23
4
4
3
2
1
(2S,4S)-4-Fluor-2-hydroxy-4-methylhexansäure
1
2
3
4
1
2
3
4
(R,R)-Weinsäure
Beispiele zur Verwendung des E,Z-Systems
H
1
2
1
2
(Z)-3-Methylhex-3-en
CH3
Cl
Cl
Cl
CH3H
1
2
2
1
1
2
3
4
(E,R)-1,4-Dichlor-3-(2-chlorethyl)-2-methylpent-2-en

134 Skript AC-OC I Beispiele zur Bestimmung der Punktgruppe Olefine:
C2'
C2'
C2C CHH
H Hi
v
v
h
Hauptachse
Nebenachsen
1 C2, 2 C2', 1 h, 2 v, i D2h
H
H
H
Cl
H
H
Cl
Cl C2
H
Cl
H
Cl
H
Cl
Cl
HC2
C2
1 Cs
1 C2, 2 v
C2v
1 C2, 1 h
C2h
1 C2, 2 v
C2v
Tetraedrische Moleküle:
H
H HH
Cl
Br HH
Cl
H ClCl
H
H ClCl
I
H BrCl
C3
C2
1 Cs
Kein Symmetrieelement (C1)chirales Zentrum
Td
1 C3, 3 v
C3v
1 C2, 2 v
C2v
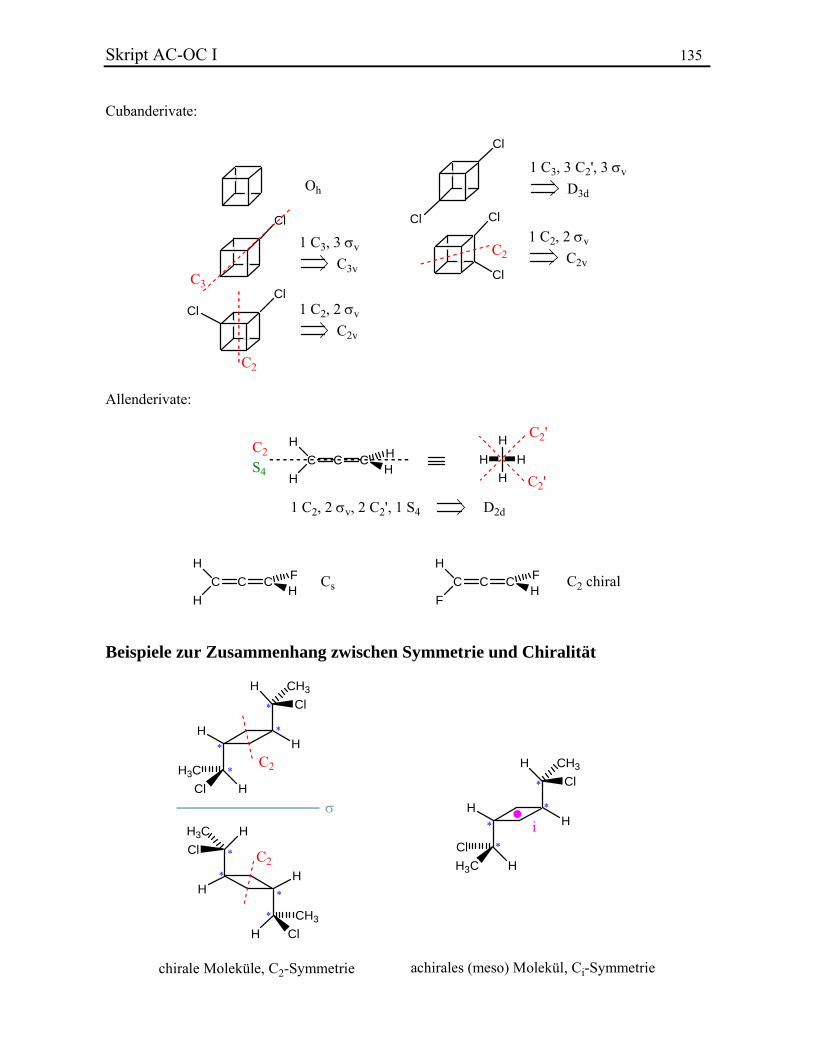
Skript AC-OC I 135 Cubanderivate:
Cl
ClCl
C3
C2
Cl
Cl
Cl
Cl
C21 C3, 3 v
C3v
1 C2, 2 v
C2v
Oh
1 C3, 3 C2', 3 v
D3d
1 C2, 2 v
C2v
Allenderivate:
C C C
H
HHH H H
H
HC2'
C2'
1 C2, 2 v, 2 C2', 1 S4 D2d
C C C
H
HHF C C C
H
FHFCs C2 chiral
C2
S4
Beispiele zur Zusammenhang zwischen Symmetrie und Chiralität
H
HH
HCl
Cl
CH3
H3C
H
HH
H Cl
Cl
H3C
CH3
H
HH
HH3C
Cl
CH3
Cl
C2
C2
*
*
*
*
*
*
*
*
*
*
*
*
i
chirale Moleküle, C2-Symmetrie achirales (meso) Molekül, Ci-Symmetrie
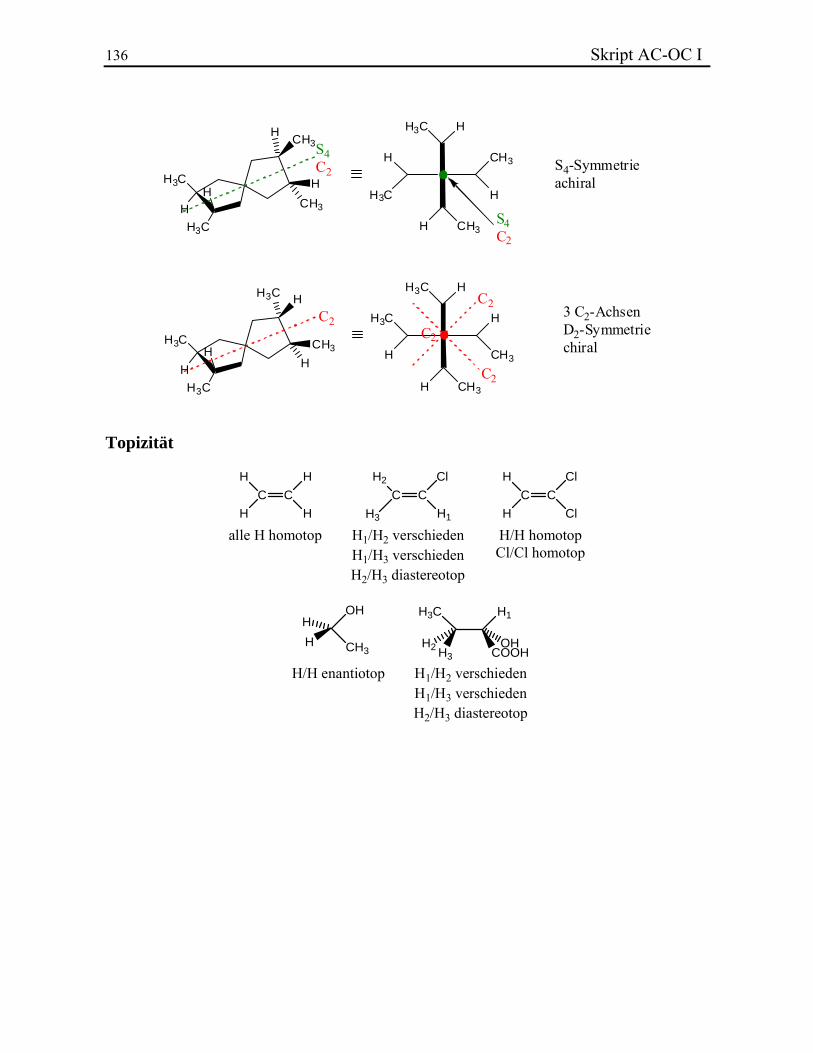
136 Skript AC-OC I
C2
H
CH3
H3C
H
H3CH
CH3
H
H
H3C
H3C H
H CH3
CH3
H
S4C2
S4C2
S4-Symmetrieachiral
H3C H
H CH3
H3C
H
H
CH3
C2
H3C
H
H3C
H
H3CH
H
CH3
C2
C2
3 C2-AchsenD2-Symmetriechiral
Topizität
C C
H H
H H
C C
H2 Cl
H3 H1
C C
H Cl
H Cl
OH
CH3H
HH1H3C
H3 COOHOHH2
alle H homotop H1/H2 verschiedenH1/H3 verschiedenH2/H3 diastereotop
H/H homotopCl/Cl homotop
H/H enantiotop H1/H2 verschiedenH1/H3 verschiedenH2/H3 diastereotop

Skript AC-OC I 137
4. Quantenchemie
4.1. Vertiefungen Quantenphysik Am Ende des 19. Jahrhunderts wurden einige physikalischen Beobachtungen gemacht, die nicht im Rahmen der klassischen Physik erklärbar waren. Sie führten zur Entwicklung einer neuen Physik, die als Quantenphysik bezeichnet wurde. Eines der revolutionierenden Konzepte war, dass einige Grössen gequantelt sind: z. B. ein Teilchen darf nicht beliebige Werte für Position und Geschwindigkeit annehmen. Es sind nur bestimmte Werte erlaubt. Weiter wurde auch bewiesen, dass einige Grössen nicht gleichzeitig mit beliebiger Genauigkeit bestimmt werden können: z. B. kann man nicht gleichzeitig die exakte Position und Geschwindigkeit eines Teilchens kennen. Für eine ausführlichere Betrachtung dieses Thema siehe z. B. P. W. Atkins, Physical Chemistry, Kap. 11-12. De Broglie Gleichung Am Anfang des 20. Jahrhunderts gab es Zweifel über die wirkliche Natur der Elementarteilchen. In der Tat treten Elektronen, Protonen, Photonen, u. a. als klassische Teilchen oder Wellen je nach Experiment. Es wurde deshalb das Konzept von Welle-Teilchen-Dualismus eingeführt. 1924 zeigte De Broglie dass jedem Teilchen auch eine Welle mit der Wellenlänge
vm
h
zugeordnet werden kann (h ist die Planck Konstante, m die Masse und v die Geschwindigkeit). Da diese Wellenlänge für makroskopische Körper praktisch Null ist, dieses Dualismus ist nur für mikroskopische Teilchen wie Elektronen und Atomkerne relevant. Für eine ausführlichere Betrachtung dieses Thema siehe z. B. P. W. Atkins, Physical Chemistry, Kap. 11. Nomenklatur der Orbitalquantenzahlen Aus historischen Gründen wird die Orbitalquantenzahl normalerweise nicht durch Zahlen sondern durch Buchstaben beschrieben:
l 0 1 2 3 4 5 ... Symbol s p d f g h ...
Die ersten vier Abkürzungen stammen aus der Sonnespektroskopie und bedeuten sharp (s), principal (p), diffuse (d) und fine (f).

138 Skript AC-OC I Energie der Schalen Die relative Energie zwischen den Schalen wird durch die folgende Beziehung beschrieben:
2
11
nIPE
wo IP die Ionisierungspotential eines Elektrons des 1s-Orbitals ist. Pauliprinzip Das Pauliprinzip besagt, dass zwei Elektronen im selben Atom nicht durch dieselben vier Quantenzahlen beschrieben werden können. Da es im Fall von Elektronen nur zwei Werte für s gibt, kann daher ein Orbital nur von zwei Elektronen mit antiparallelen Spin besetzt werden. Elektronenkonfigurationen In der folgenden Abbildung sind die Elektronenkonfigurationen der Elementen der ersten und zweiten Periode angegeben:
2p
2s
1s
2p
2s
1s
2p
2s
1s
2p
2s
1s
H: 1s1 He: 1s2 Li: 1s22s1 Be: 1s22s2
2p
2s
1s
B: 1s22s22p1
2p
2s
1s
2p
2s
1s
2p
2s
1s
2p
2s
1s
C: 1s22s22p2 N: 1s22s22p3 O: 1s22s22p4 F: 1s22s22p5
2p
2s
1s
Ne: 1s22s22p6
Nichtbindende Orbitale In einigen Fällen können die Wechselwirkungen zwischen Atomorbitalen aus geometrischen Gründen nicht zu stabilisierenden oder destabilisierenden Wechselwirkungen führen. Z. B. im Fall des linearen Wassers ist die bindende Wechselwirkung zwischen einem H-1s-Orbital und dem Teil

Skript AC-OC I 139 des p-Orbitals gleicher Vorzeichen vollständig durch die antibindende Wechselwirkung mit dem anderen Lappen kompensiert:
Es ist daher, wie wenn es keine Wechselwirkung gäbe. Hybridatomorbitalmodell für Ethen und Ethin Ethen besitzt zwei sp2-hybridisierte C-Atome. Die sechs Hybridatomorbitale werden benützt, um die C-H- und C-C--Bindungen zu erzeugen. Jedes C-Atom besitzt daneben noch ein p-Orbital, das zur Bildung der C-C--Bindung führt:
H
H
H
H
Auf ähnlichen Weise können die zwei senkrechten p-Orbitale eines sp hybridisierten Atoms Dreifachbindungen erzeugen:
HH
Inversion des N-Atoms Obwohl ein N-Atom mit drei verschiedenen Substituenten theoretisch chiral sein sollte, ist es praktisch unmöglich die reinen Stereoisomere zu erhalten. Das sp3-hybridisierte Atom ist nicht imstande, eine bestimmte Konfiguration zu erhalten. In der Tat kann das N-Atom kontinuierlich zwischen den beiden Isomeren durch ein sp2-Zentrum oszillieren:
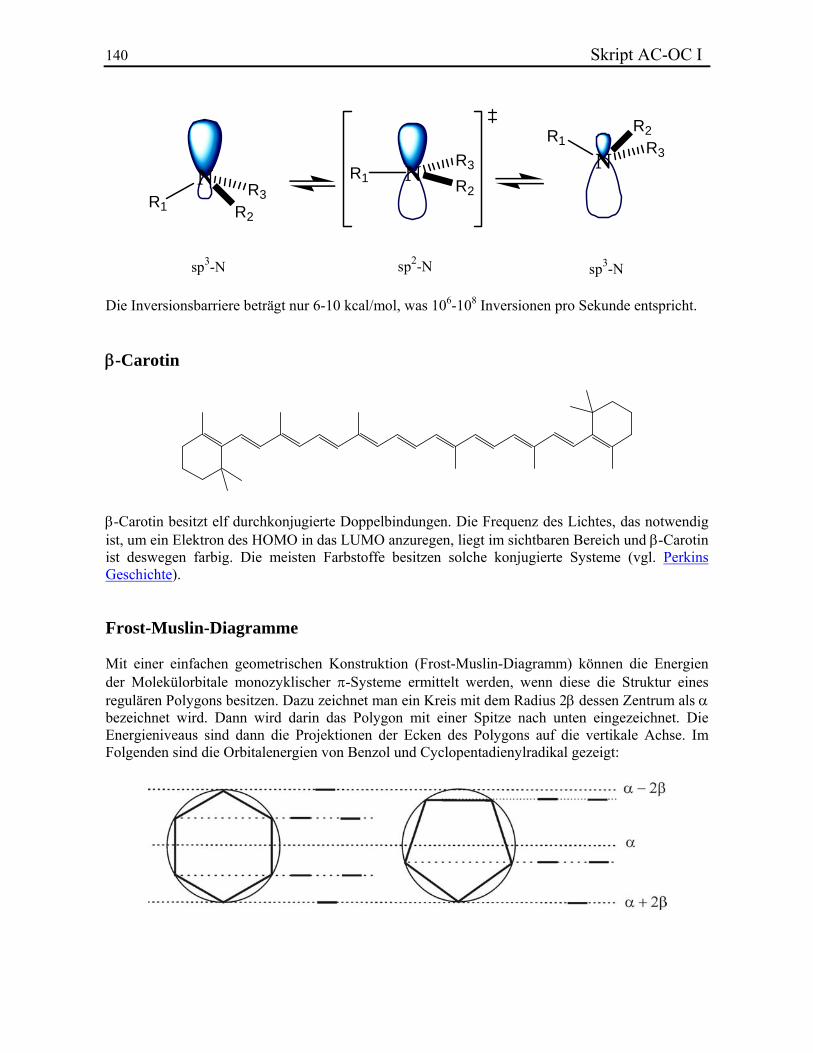
140 Skript AC-OC I
NR1
R3
R2
NR1 R3
R2
NR1R2
R3
sp3-N sp3-Nsp2-N Die Inversionsbarriere beträgt nur 6-10 kcal/mol, was 106-108 Inversionen pro Sekunde entspricht.
-Carotin
-Carotin besitzt elf durchkonjugierte Doppelbindungen. Die Frequenz des Lichtes, das notwendig ist, um ein Elektron des HOMO in das LUMO anzuregen, liegt im sichtbaren Bereich und -Carotin ist deswegen farbig. Die meisten Farbstoffe besitzen solche konjugierte Systeme (vgl. Perkins Geschichte). Frost-Muslin-Diagramme Mit einer einfachen geometrischen Konstruktion (Frost-Muslin-Diagramm) können die Energien der Molekülorbitale monozyklischer -Systeme ermittelt werden, wenn diese die Struktur eines regulären Polygons besitzen. Dazu zeichnet man ein Kreis mit dem Radius 2 dessen Zentrum als bezeichnet wird. Dann wird darin das Polygon mit einer Spitze nach unten eingezeichnet. Die Energieniveaus sind dann die Projektionen der Ecken des Polygons auf die vertikale Achse. Im Folgenden sind die Orbitalenergien von Benzol und Cyclopentadienylradikal gezeigt:

Skript AC-OC I 141 Beispiele aromatischer Moleküle
Naphthalin Anthracen
[18]Annulen
[n]Annulene sind n-gliedrige, zyklisch konjugierte Monocyclen. Cyclooctatetraen

142 Skript AC-OC I
5. Thermochemie
5.1. Vertiefung: Thermodynamischer Effekt der Aromatizität Die homodesmischen Reaktionen wurden eingeführt, um die Stabilität cyclischer Moleküle zu quantifizieren. Sie sind aber auch nützlich, um die Anwesenheit von neuen thermochemischen Eigenschaften zu beweisen. In diesem Beispiel wird die Homoaromatizität besprochen, ein Thema das während einiger Jahre hitzig diskutiert wurde (vgl. folgende Literaturzitate). Quinacene sind starre, schalförmige Moleküle:
Hf =
Triquinacen
53.5 ± 1.0 30.5 ± 1.0 3.0 ± 1.0 -24.5 ± 0.9 kcal/mol In Triquinacen sind die drei Doppelbindungen in einer bestimmten Anordnung festgelegt. Die C=C Bindungslänge und der Abstand zwischen nichtgebundenen benachbarten C-Atomen betragen 1.33 bzw. 2.55 Å. Obwohl die Doppelbindungen ziemlich weit auseinander liegen, wurde eine Überlappung zwischen den -Orbitalen vorgeschlagen. Dieses spezielle System mit sechs -Elektronen könnte daher dem Molekül einen aromatischen Charakter verleihen (Homoaromatizität). Spektroskopische Untersuchungen ergaben keine eindeutigen Beweise. Da aber die Aromatizität eine thermochemische Eigenschaft ist, kann man dafür eine homodesmische Reaktion betrachten (J. F. Liebman, L. A. Paquette, J. R. Peterson, D. W. Rogers, J. Am. Chem. Soc. 1986, 108, 8267). Die Bildungsenthalpien (siehe oben) wurden mit Hilfe der Verbrennungswärme bestimmt, ausser für Triquinacen, bei dem die Hydrierwärme benützt wurde. Man kann daher die folgende homodesmische Reaktion schreiben:
+ 2 3
(53.5 ± 1.0) 2·(–24.5 ± 0.9) 3·(3.0 ± 1.0)
4.5 ± 1.6 kcal/mol 9.0 ± 1.4 kcal/mol
Wenn es keine Homoaromatizität gäbe, sollte die Reaktionsenthalpie Null betragen. Da aber die Reaktion um 4.5 kcal/mol endotherm ist, sollte man annehmen, dass Triquinacen bezüglich den Referenzmolekülen stabilisiert ist. Eine mögliche Kritik dieser Methode ist, dass nicht alle möglichen Effekte (z. B. 1,3-diaxiale Wechselwirkung zwischen H-Atomen) berücksichtigt werden. Wenn man aber eine ähnliche homodesmische Reaktion für Dihydrotriquinacen schreibt, findet man dass die Reaktionsenthalpie Null ist. Dies schliesst den Beitrag durch diaxiale Wechselwirkungen aus.

Skript AC-OC I 143
+ 2
(30.5 ± 1.0) (–24.5 ± 0.9) 2·(3.0 ± 1.0)
6.0 ± 1.4 kcal/mol 6.0 ± 1.4 kcal/mol Eine geeignete Summe der zwei Gleichungen kann weiter die sp3-sp3, sp3-sp2 und die sp2-sp2 1,3-diaxialen Wechselwirkungen beiseitigen. Dies sprengt jedoch den Rahmen dieser Vorlesung. Der Vollständigkeit halber muss man berichten, dass 1998 die Bildungsenthalpie von Triquinacen mittels Verbrennung bestimmt wurde, was einen zuverlässigen Wert lieferte (S. P. Verevkin, H.-D. Beckhaus, C. Rüchardt, R. Haag, S. I. Kozhushkov, T. Zywietz, A. de Meijere, H. Jiao, P. von Ragué Schleyer, J. Am. Chem. Soc. 1998, 120, 11130). Mit einer Bildungsenthalpie von 57.01 ± 0.70 kcal/mol findet man dass die Reaktionsenthalpie praktisch Null ist. Dass heisst, dass der Einfluss der Homoaromatizität sehr klein wäre, wenn er überhaupt wäre.
5.2. Vertiefung: Berechnung der Ringspannung In Ringen weichen die Winkel oft beträchtlich von den Idealwinkeln der entsprechenden Hydridorbitale ab. Das augenfälligste Beispiel ist Cyclopropan, dessen sp3-Atome einen Winkel von 60° statt die normalen 109.5° besitzen müssen. Diese Destabilisierung wird Baeyer-Spannung genannt (vgl. Kapitel 6.2.). Im Folgenden werden die Ringspannungen der ersten sechs monocyclischen ungesättigten Kohlenwasserstoffe mit Hilfe von homodesmischen Reaktionen berechnet (Hf° (Ethan) = – 20 kcal/mol; Hf° (Propan) = – 25 kcal/mol; alle Werte sind in kcal/mol angegeben):
Cyclus Hf exp.
Homodesmische Reaktion
Hf ber. Spannung
Cyclopropan 12.7 H2C
H2CCH2
H2C+ 33 – 15 28
Cyclobutan 6.8 + 44 – 20 27
Cyclopentan – 18.7 + 55 – 25 6
Cyclohexan – 29.5 + 66 – 30 ~ 0
Cycloheptan – 28.3 + 77 – 35 7
Cyclooctan – 29.7 + 88 – 40 10
Nach dem stark gespannten Cyclopropan und Cyclobutan, sinkt die Spannung in Cyclohexan auf Null. Mit steigender Zahl der Ringglieder nimmt die Spannung wieder zu.

144 Skript AC-OC I
5.3. Vertiefung: Berechnung der Ringspannung Im Kapitel 4.4. wurde gezeigt, wie mehrere direkt gebundene Atome mit parallel angeordneten p-Orbitalen durch Konjugation stabilisiert werden können. Mit Hilfe von homodesmischen Reaktionen kann diese Stabilisierung quantifiziert werden. Als erster Fall sei Penta-1,4-dien (Hf° = 25 kcal/mol) betrachtet. Das sp3-hybridisierte C-Atom in Position 3 sollte die Konjugation unterbrechen:
+ 2 2 +
(25) 2·(–20) 2·(5) (–25)
–15 kcal/mol –15 kcal/mol Tatsächlich zeigt die vorgeschlagene homodesmische Reaktion keinen Unterschied zwischen Reaktanden und Produkte. Im Fall von Penta-1,3-dien ist dagegen eine Wechselwirkung zwischen den zwei Doppelbindungen möglich:
+ +
(18) (–20) (5) (–3)
–2 kcal/mol +2 kcal/mol Penta-1,3-dien ist daher bezüglich zwei separierten Doppelbindungen um 4 kcal/mol stabilisiert. Homodesmische Reaktionen hängen stark von den gewählten Referenzmolekülen ab. Eine mögliche alternative Reaktion für Penta-1,3-dien ist:
+ +
(18) (5) (26) (–3)
23 kcal/mol 23 kcal/mol Die Reaktion zeigt keine Enthalpieänderung. Dies bedeutet, dass Penta-1,3-dien gegenüber Butadien nicht stabiler ist. Dies macht Sinn, da Butadien selbst schon durch Konjugation stabilisiert wird.
5.4. Thermodynamischen Daten Atomisierungsenthalpien Die Atomisierungsenthalpie ist die Energie, die notwendig ist, um gasförmige Atome aus den Elementen herzustellen. Z. B. gilt
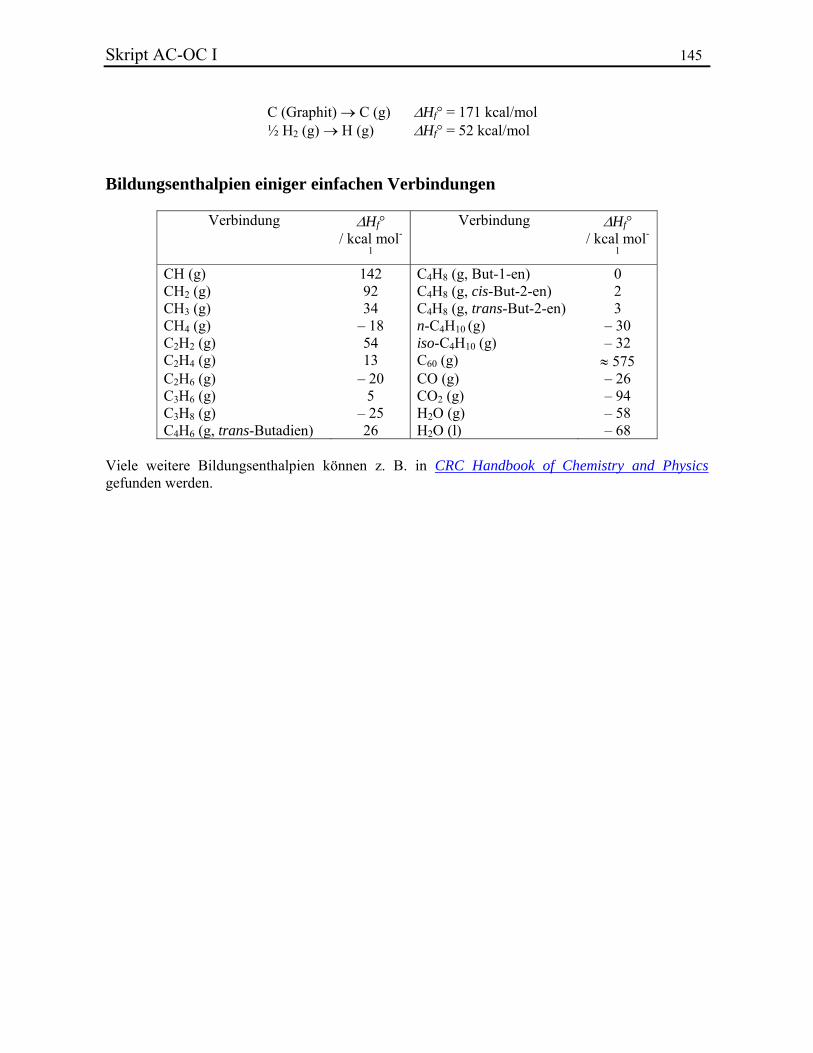
Skript AC-OC I 145
C (Graphit) C (g) Hf° = 171 kcal/mol ½ H2 (g) H (g) Hf° = 52 kcal/mol
Bildungsenthalpien einiger einfachen Verbindungen
Verbindung Hf° / kcal mol-
1
Verbindung Hf°
/ kcal mol-
1
CH (g) 142 C4H8 (g, But-1-en) 0 CH2 (g) 92 C4H8 (g, cis-But-2-en) 2 CH3 (g) 34 C4H8 (g, trans-But-2-en) 3 CH4 (g) – 18 n-C4H10 (g) – 30 C2H2 (g) 54 iso-C4H10 (g) – 32 C2H4 (g) 13 C60 (g) 575 C2H6 (g) – 20 CO (g) – 26 C3H6 (g) 5 CO2 (g) – 94 C3H8 (g) – 25 H2O (g) – 58 C4H6 (g, trans-Butadien) 26 H2O (l) – 68
Viele weitere Bildungsenthalpien können z. B. in CRC Handbook of Chemistry and Physics gefunden werden.

146 Skript AC-OC I
6. Konformationsanalyse
6.1. Wie man ein Cyclohexanderivat zeichnet Um Cyclohexanderivate akkurat zu zeichnen, ist die folgende Prozedur sehr nützlich.
1. Man zeichnet ein breites V:
2. Darunter wird ein breites, umgekehrtes und leicht verschobenes V hinzufügt:
3. Die zwei V werden verknüpft:
4. Die axialen Substituenten werden durch vertikalen Segmente zum Cyclohexanring
verknüpft:
5. Die äquatorialen Substituenten sind durch Segmente dargestellt, die parallel zu den
Ringbindungen sein müssen:

Skript AC-OC I 147
6.2. Thermodynamischen Daten Standardbildungsenthalpien der verschieden Isomere von Dimethylcyclohexan
Verbindung Anzahl gauche-WW
*
Destabilisierung/ kcal mol-1 **
Hf° / kcal mol-1
1,1-Dimethylcyclohexan 2 1.8 – 52.3 cis-1,2-Dimethylcyclohexan 2 1.8 – 50.6 trans-1,2-Dimethylcyclohexan 0 0 – 52.2 cis-1,3-Dimethylcyclohexan 0 0 – 53.3 trans-1,3-Dimethylcyclohexan 2 1.8 – 51.6 cis-1,4-Dimethylcyclohexan 2 1.8 – 51.5 trans-1,4-Dimethylcyclohexan 0 0 – 53.2
*) Anzahl der gauche-Wechselwirkungen (1,3-diaxialen Wechselwirkungen) im stabilsten Konformer. **) Theoretische Destabilisierung des Moleküls durch die Anwesenheit von gauche-Wechselwirkungen (jede beträgt ca. 0.9 kcal/mol).

148 Skript AC-OC I
7. Chemische Reaktionslehre
7.1. Numerische Beispiele Beispiel zur Verwendung der Gleichgewichtskonstante Man betrachte die folgende Reaktion in Wasser bei 25°C:
HO– + CH3Cl CH3OH + Cl–
Wenn die Anfangskonzentrationen [CH3Cl]0 = 0.1 M und [NaOH]0 = 0.2 M sind, ist ihre Konzentration im Gleichgewicht [CH3Cl] = (0.1 – x) M und [NaOH] = (0.2 – x) M, wobei x die Konzentration der Moleküle ist, die die Reaktion vollzogen haben. Analog gilt im Gleichgewicht [CH3OH] = [Cl–] = x M. Da die stöchiometrischen Koeffizienten aller Verbindungen 1 betragen, ist die folgende Gleichung gültig:
16–
3
–3 10
)2.0)(1.0(
)()(
HOClCH
ClOHCH
xx
xxK
Durch Lösen dieser Gleichung bekommt man für x die Werte 0.1 und 0.2. Da maximal 0.1 M von CH3Cl reagiert haben können, hat man in Gleichgewicht die folgende Konzentrationen: [CH3Cl] 0.0 M; [NaOH] = 0.1 M; [CH3OH] = 0.1 M und [Cl–] = 0.1 M. Beispiel zur Berechnung der Gleichgewichtskonstante Man betrachte die folgende Reaktion in Wasser bei 25°C:
HO– (aq) + CH3Cl (aq) CH3OH (aq) + Cl– (aq) Gf°: – 37.58 – 13.72 – 41.90 – 31.36 kcal/mol
Die freie Reaktionsenthalpie ist daher
Gf°(CH3OH) + Gf°(Cl–) – Gf°(HO–) – Gf°(CH3Cl) = = – 41.90 – 31.36 – (– 37.58) – (– 13.72) = – 21.96 kcal/mol
was einer Gleichgewichtskonstante
1610
RT
G
eK entspricht.


















