
Aus dem Institut für experimentelle und klinische ... · kontrollierten, randomisierten,...
99
Aus dem Institut für experimentelle und klinische Pharmakologie und Toxikologie der Universität Erlangen-Nürnberg Geschäftsführender Direktor: Prof. Dr. med. M. F. Fromm Pharmakokinetik und Pharmakodynamik von zwei freiverkäuflichen Analgetika in einem experimentellen Schmerzmodell – Teil 1– Paracetamol mit Koffein Inaugural-Dissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg vorgelegt von Gregor Muth
Transcript of Aus dem Institut für experimentelle und klinische ... · kontrollierten, randomisierten,...

Aus dem Institut für experimentelle und klinische Pharmakologie
und Toxikologie der Universität Erlangen-Nürnberg
Geschäftsführender Direktor: Prof. Dr. med. M. F. Fromm
Pharmakokinetik und Pharmakodynamik von zwei freiverkäuflichen Analgetika
in einem experimentellen Schmerzmodell – Teil 1– Paracetamol mit Koffein
Inaugural-Dissertation
zur Erlangung der Doktorwürde
der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
vorgelegt
von
Gregor Muth

Gedruckt mit Erlaubnis der
Medizinischen Fakultät der Friedrich-Alexander-Universität
Erlangen-Nürnberg
Dekan: Prof. Dr. Schüttler
Referent: Prof. Dr. med Dr. h.c. Kay Brune
Korreferent: Prof. Dr. med Martin F. Fromm
Tag der mündlichen Prüfung: 27.10.2010

Für Enzo Frech

1. Synopsis ........................................................................................................................ 1
1.1 Hintergrund und Ziele ............................................................................................. 1
1.2 Methoden ................................................................................................................ 1
1.3 Ergebnisse ............................................................................................................... 2
1.4 Praktische Schlussfolgerung ................................................................................... 2
1.5 Abstract ................................................................................................................... 2
1.5.1 Background and objectives .............................................................................. 2
1.5.2 Methodology .................................................................................................... 3
1.5.3 Results .............................................................................................................. 3
1.5.4 Conclusion ....................................................................................................... 3
2. Einleitung ..................................................................................................................... 5
2.1 Paracetamol ............................................................................................................. 6
2.1.1 Pharmakokinetik .............................................................................................. 6
2.1.2 Pharmakodynamik ........................................................................................... 7
2.1.3 Unerwünschte Arzneimittelwirkungen und Toxizität ...................................... 9
2.2 Koffein .................................................................................................................. 10
2.2.1 Pharmakokinetik ............................................................................................ 12
2.2.2 Pharmakodynamik ......................................................................................... 12
2.2.3 Koffein und Antinozizeption ......................................................................... 15
2.3 Ziele der Studie ..................................................................................................... 17
3. Material und Methoden ............................................................................................ 18
3.1 Studiendesign ........................................................................................................ 18
3.2 Studienteilnehmer ................................................................................................. 18
3.3 Schmerzmodell ...................................................................................................... 20
3.3.1 Phasische Schmerzreizung ............................................................................. 20
3.3.2 Tonische Schmerzreizung .............................................................................. 25
3.4 Prüfmedikation ...................................................................................................... 26
3.5 Trainingssitzung .................................................................................................... 26
3.6 Ablauf der Studientage .......................................................................................... 27
3.6.1 Ablauf der analgesimetrischen Untersuchung ............................................... 29
3.7 Erfassung der analgetischen Effekte ..................................................................... 32
3.7.1 Messung der chemo-somatosensorisch evozierten Potentiale (CSSEP) ........ 33
3.7.2 Subjektive Intensitätsschätzung der phasischen Schmerzreize ...................... 34
3.7.3 Intensitätsschätzung der tonischen Schmerzreize .......................................... 35
3.8 Erfassung unspezifischer Effekte .......................................................................... 36

3.8.1 Powerspektren des Hintergrund-Elektroencephalogramms ........................... 36
3.8.2 Tracking Performance .................................................................................... 36
3.8.3 Unerwünschte Arzneimittelwirkungen .......................................................... 37
3.9 Erfassung der pharmakokinetischen Effekte ......................................................... 38
3.9.1 Analytik des Probenmateriales ....................................................................... 38
3.10 Good Clinical Practice und Standard Operating Procedure ................................ 39
3.11 Zielparameter ...................................................................................................... 40
3.11.1 Primäre Endpunkte ....................................................................................... 40
3.11.2 Sekundäre Endpunkte .................................................................................. 41
3.12 Statistische Methoden ......................................................................................... 41
4. Ergebnisse .................................................................................................................. 42
4.1 Unerwünschte Ereignisse und Beurteilung der Sicherheit der Prüfmedikation .... 42
4.2 Pharmakokinetik der Prüfmedikation ................................................................... 43
4.3 Subjektive, psychophysische Daten – Schmerzintensitätsschätzungen (VAS) und
Vigilazprüfung (TP) .............................................................................................. 44
4.4 Objektive Daten – chemo-somatosensorisch evozierter Potentiale (CSSEP) ....... 47
5. Diskussion .................................................................................................................. 49
5.1 Pharmakokinetische Ergebnisse ............................................................................ 49
5.2 Pharmakodynamische Effekte ............................................................................... 51
5.2.1 Subjektive Schmerzintensitätsschätzung (VAS) ............................................ 51
5.2.2 Tracking Performance (TP) – Einflüsse auf die Vigilanz .............................. 52
5.2.3 Schmerzkorrelierte evozierte Potentiale (CSSEP) ......................................... 53
5.3 Schlussfolgerung ................................................................................................... 55
6. Literaturverzeichnis .................................................................................................. 56
7. Abkürzungsverzeichnis............................................................................................. 68
8. Anhang ....................................................................................................................... 71
8.1 Pharmakokinetische Daten von Paracetamol ........................................................ 71
8.2 Pharmakokinetischen Daten von Koffein ............................................................. 72
8.3 Demographische Daten der Studienteilnehmer ..................................................... 73
8.4 Im Rahmen der Studie erhobene Blutdruckwerte der Probanden ......................... 74
8.5 Im Rahmen der Studie erhobene Laborparameter der Probanden ........................ 76
8.6 Entblindung der Studienmedikation ...................................................................... 89
8.7 Standardisierte Probandenanleitung für die phasische und tonische Schmerz-
reizung nach Standard Operating Procedure (SOP) .............................................. 89
9. Danksagung ............................................................................................................... 92

10. Curiculum vitae ....................................................................................................... 93

1
1. Synopsis
1.1 Hintergrund und Ziele
Bereits in früheren Studien mit dem hier verwendeten Schmerzmodell konnten die
analgetische Wirksamkeit von Paracetamol und Propyphenazon sowie die beschleunigte
Absorption und verlängerten analgetischen Effekte von Paracetamol durch dessen
Kombination mit Koffein gezeigt werden. Bei der hier vorliegenden Arbeit sollte nun
erstmals die Wirksamkeit von Paracetamol plus Koffein gegenüber Placebo innerhalb
eines Arzneimittelprüfungsverfahren der Phase I bestätigt werden. Gleichzeitig wurden
die aktuellen GCP-Methoden (Good Clinical Practice) in den Prüfplan aufgenommen,
um die Pharmakodynamik und Pharmakokinetik der Kombinationsmedikation
Paracetamol plus Koffein nach den derzeitigen Gesetzesvorgaben zu erfassen.
Ziele dieser unter klinischen Prüfungsbedingungen durchgeführten Studie waren der
direkte Vergleich einer Einmaldosis Panadol Extra® (1000mg Paracetamol + 130mg
Koffein) mit Placebo (Panadol Placebo®, GSK), die Analyse und Beurteilung der
pharmakokinetischen Daten sowie die Reproduktion früher erhobener Studiendaten.
1.2 Methoden
Diese Studie wurde anhand eines monozentrisch, doppelt blinden, Placebo
kontrollierten, randomisierten, cross-over Studiendesigns an 26 gesunden, freiwilligen
Versuchsteilnehmern durchgeführt. Die Medikation wurde im Gegensatz zur
Voruntersuchung mittels Verkapselung zusätzlich verblindet. Alle in die Studie
eingeschlossenen Probanden wurden zu drei, mindestens je sieben Tage voneinander
getrennt liegenden Versuchstagen einbestellt. An jedem dieser Versuchstage wurden
die analgetischen Effekte an Hand eines bereits mehrfach etablierten experimentellen
Schmerzmodells beurteilt. Dieses Schmerzmodell basiert zum einen auf der Erhebung
und Analyse objektiver, chemo-somatosensorisch evozierter Potentiale sowie
subjektiver Schmerzintensitätsschätzungen, als Reaktion auf eine periodische Reizung
der nasalen Schleimhaut bzw. deren Nozizeptoren mit gasförmigem CO2. Zum anderen
basiert es auf der Intensitätsschätzung eines tonischen, durch Einleitung trockener,
gleichmäßig temperierter Luft in die Nasenhöhle, hervorgerufenen Schmerzreizes.
Zeitgleich wurden die Vigilanz der Probanden zu jedem Versuchsabschnitt sowie die
pharmakokinetischen Daten der Prüfmedikation erfasst.

2
1.3 Ergebnisse
Beim tonisch generierten Schmerzzustand konnte 72 Minuten nach Gabe von
Paracetamol plus Koffein eine signifikante Schmerzreduktion beobachtet werden. Für
die phasisch induzierten Schmerzreize konnten unter Einfluss der Prüfmedikation
sowohl für Amplitude P2, als auch für die Amplitude N1 an vereinzelten Messpunkten
signifikante Reduktionen im Vergleich zu Placebo beobachtet werden.
Die Probanden zeigten unter der Koffein enthaltenden Medikation eine signifikant
höhere Vigilanz im Vergleich zu Placebo.
Die gewonnenen pharmakokinetischen Daten zu Paracetamol plus Koffein sind im
Wesentlichen mit denen der Vorstudie vergleichbar. Die Anflutungsphase der beiden
Substanzen war jedoch in der aktuellen Untersuchung verzögert.
1.4 Praktische Schlussfolgerung
Die vorliegende Arbeit konnte belegen, dass die Kombination von Paracetamol und
Koffein antinoziceptiv und analgetisch aktiv ist. Dies ließ sich unter anderem an den
Veränderungen der schmerzkorrelierten evozierten Potentiale (CSSEP) sowie an den
dazu positiv korrelierenden subjektiven Schmerzintensitätsschätzungen (VAS)
aufzeigen. Zudem konnte eine deutliche Erhöhung der Vigilanz der Probanden unter
Paracetamol und Koffein im Vergleich zu Placebo beobachtet werden. Die verzögerte
systemische Verfügbarkeit der Studienmedikation kann auf die geänderte Galenik der
Studienmedikation (Verblindung) zurückgeführt werden.
Die Ergebnisse der vorliegenden Arbeit konnten frühere Daten vorangegangener
Studien dieses Hauses bestätigen und somit deren Validität untermauern.
Des Weiteren konnte gezeigt werden, dass das der Studie zugrunde liegende
Schmerzmodell unter GCP-Bedingungen durchführbar ist und reproduzierbare Daten
liefern kann.
1.5 Abstract
1.5.1 Background and objectives
The pain model used in this study has been applied previously to demonstrate the
activity of a variety of analgesic drugs like propyphenazone, paracetamol and its
combination with caffeine.
In the actual study, the previous observation should be confirmed for the first time in a
clinical Phase I trial in order to show a superior activity of paracetamol plus caffeine

3
compared to placebo. Additionally, the actual GCP-Guidelines (Good Clinical Practice)
were included in the study protocol to access the pharmacodynamics and
pharmacokinetics of paracetamol plus caffeine in accordance with current law. Within
this study, the aim was to compare the effects of a single dose of Panadol Extra®
(1000mg paracetamol + 130mg caffeine) against placebo (Panadol Placebo®, GSK), to
summarise descriptively the pharmacokinetics of a single dose of Panadol Extra® and
furthermore to repilcate data of previous conducted studies.
1.5.2 Methodology
This study was a single centre, double-blind, placebo-controlled, randomized, cross-
over study, conducted in 26 healthy volunteers. In contrast to the previous study, the
actual medication was encapsulated in a double blind capsule. All subjects attended
three treatment visits, each separated by a minimum of seven days. At each treatment
visit the analgesic effects have been assessed by means of a well established
experimental pain model based on chemo-somatosensory pain-related evoked potentials
and pain ratings after phasic painful stimulation of the nasal mucosa with gaseous
carbon dioxide, and on pain ratings after tonic pain stimulation of the nasal mucosa with
dry air of controlled flow and temperature. Pharmacokinetics parameters were
investigated and the subjects’ vigilance recorded simultaneously.
1.5.3 Results
Paracetamol plus caffeine resulted in significantly less tonic pain at 72 minutes after
drug administration. The treatments produced, compared to placebo, significant
reductions in phasic evoked potentials P2 and at seperate time points also for N1.
As expected the subject’s vigilance, in terms of an increased TP was significantly higher
after administration of the caffeine containing medication compared to placebo.
The pharmacokinetic data of paracetamol plus caffeine were comparable with previous
results except the delay during the initial absorption period in the actual study.
1.5.4 Conclusion
This work confirms that the combination of paracetamol plus caffeine can have an
antinociceptiv effect. We were able to demonstrate this by observing changes in pain-
related evoked potentials (CSSEP) that correlate positively with the subjective pain
ratings (VAS).

4
The vigilance tracking performance also suggested an improvement on paracetamol plus
caffeine compared to placebo.
The results of this survey were consistent with former studies from our department and
therefore endorse their validity. The delayed bioavailability of the study medication may
be caused by the encapsulated formulation (double blind capsule).
Furthermore, we could show that the experimental pain model, on which this study is
based, is useful for objective examinations (according to GCP) and is applicable in
clinical investigations comparing analgesic drugs.

5
2. Einleitung
Schmerz (lat. dolor oder gr. algos) ist, auf den physischen oder psychosomatischen
Bereich bezogen, nach einer Definition der internationalen Gesellschaft zum Studium
des Schmerzes von 1979 „ein unangenehmes Sinnes- oder Gefühlserlebnis, das mit
aktueller oder potentieller Gewebeschädigung verknüpft ist oder mit Begriffen einer
solchen Schädigung beschrieben wird“ (Bonica 1979).
Schmerzen und Fieber gelten als die wesentlichen Charakteristika bzw. Symptome von
Leid und Krankheit. Es ist daher nicht verwunderlich, dass sich die ersten Hinweise
über die Verwendung natürlich vorkommender Materialien zur Schmerzlinderung
bereits 3.000 Jahre vor Christus finden (Haas 1983). Als einer der wesentlichen
Meilensteine auf der Suche nach Schmerzmitteln kann sicherlich die Entdeckung des
Opiums ca. 2.000 Jahre vor Christus angesehen werden. Auch bei Hippokrates und den
griechischen und römischen Wundärzten findet sich bereits die Empfehlung,
salicylathaltige Extrakte und Abkochungen aus Weide, Pappel und Immergrün zur
äußerlichen und innerlichen Behandlung des Wundschmerzes und bei Verletzungen zu
verwenden (Brune und Egger 2002). Die entscheidenden Entwicklungen und
Fortschritte in der Verwendung und Isolierung analgetischer Substanzen wurden
allerdings erst in den letzten 200 Jahren erzielt. Diese begannen 1803/1804 mit der
Isolierung des „Morphiums“ und damit der ersten Reindarstellung eines Analgetikums.
Die Strukturaufklärung und Synthese der Salicylsäure gelang 1859, die Entdeckung des
Antipyrins in Erlangen 1884 und schließlich des 1886 Acetanilids sowie 1887 des
Phenacetins (Brune und Egger 2002, Brune und Niederweis 2007). Aus den beiden
letzteren konnte nur wenige Jahre später als wesentlicher aktiver Metabolit Paracetamol,
eine der heute am weitesten verbreitete analgetisch und antipyretisch wirksame
Substanz, synthetisiert werden (Brodie und Axelrod 1948).
Heutzutage steht neben der Neu- und Weiterentwicklung analgetischer Substanzen auch
die Kombinierung synergistischer bzw. sich additiv ergänzender Wirkstoffe im
Mittelpunkt pharmakologischer und medizinischer Interessen. Als gutes Beispiel hierfür
gilt die Kombination von Paracetamol mit Koffein: Durch den Zusatz von Koffein zu
Paracetamol wird nicht nur dessen Absorption beschleunigt sondern auch dessen
analgetische Wirksamkeit verstärkt (Renner et al. 2007).

6
2.1 Paracetamol
Paracetamol, im angloamerikanischen Raum besser bekannt als Acetaminophen, ist, als
wesentlicher aktiver Metabolit des Phenacetin und Acetanilid, ein Anilin-Derivat,
welches zu der großen Gruppe der nicht sauren antipyretischen Analgetika zählt.
Abbildung 1: Strukturformel von N-Acetyl-p-Aminophenol bzw. Paracetamol (modif. nach Aktories et al. 2005).
2.1.1 Pharmakokinetik
Acetaminophen wird nach oraler Applikation rasch und nahezu vollständig aus dem
Intestinum und hier zum überwiegenden Anteil aus dem Duodenum resorbiert (Forrest
et al. 1982; Clements et al 1978).
Die orale Bioverfügbarkeit unterliegt dosisabhängig, bei Einzeldosen von 500mg bis 2g,
aufgrund eines ausgeprägten First-pass-Metabolismus in der Leber, einer großen Breite
von 65% bis 90% (Rawlins et al. 1977, Eandi et al. 1984). Wobei die maximalen
Serumkonzentrationen, wiederum nach oraler Gabe, nach 20 Minuten bis zu zwei
Stunden zu verzeichnen sind und dabei proportional zur verabreichten Dosis liegen (Mc
Gliveray and Mattok 1972, Eandi et al. 1984). Paracetamol verteilt sich hierbei
vergleichsweise einheitlich in allen Körpergeweben und erreicht innerhalb einer Stunde
einen Plasma-Gewebe-Quotienten von nahezu 1:1. Die Eliminationshalbwertszeit
beträgt im Durchschnitt etwa 1,5 bis 2,5 Stunden (Forrest et al. 1982, Eandi et al. 1984).
Die Bindung an Plasmaproteine ist variabel und abhängig von der Dosierung und ist im
Rahmen therapeutischer Applikationen mit einem Anteil von 10% relativ gering
(Forrest et al. 1982).
Ein Großteil des aufgenommen Paracetamols wird innerhalb der ersten 24 Stunden v. a.
in der Leber durch Glucuronidierung (ca. 60%), Sulfatierung (ca. 35%) und
Konjugation mit Cystein und Mercaptursäure metabolisiert und anschließend renal
eliminiert. Lediglich ca. 2,5% werden in unveränderter Form über die Niere
ausgeschieden. Daneben wird Paracetamol aber auch in geringem Umfang über

7
Cytochrom-P450-Oxygenasen in das hepatotoxische N-Acetyl-p-benzochinonimin
verstoffwechselt, welches jedoch innerhalb therapeutischer Dosierung schnell durch
reduziertes Glutathion inaktiviert und als Mercaptursäurekonjugat über den Harn
ausgeschieden werden kann (Prescott 1992).
Tabelle 1: Zusammenfassende Übersicht der pharmakokinetischen Daten von Paracetamol und Koffein.
2.1.2 Pharmakodynamik
Paracetamol zeichnet sich durch eine deutlich antipyretische und analgetische Wirkung
aus. Seine antiphlogistische Wirkung ist, im Gegensatz zu den klassischen Nicht-
steroidalen-Antiphlogistika, sehr gering bzw. als nicht nennenswert einzustufen
(Glissold 1986). Der molekulare Wirkmechanismus Paracetamols ist seit Jahrzehnten
Mittelpunkt intensivster Forschung und zahlreicher Publikationen, bleibt jedoch, trotz
alledem, auch bis zum heutigen Tage noch nicht vollständig geklärt. Diskutiert wurde
vor allem eine zentrale, zerebrale Inhibition der Prostaglandin-Synthese, welche mit
einer schwachen peripheren COX-1 und COX-2 Hemmung und damit verminderten
peripheren Prostaglandinkonzentration einhergeht (Flower und Vane 1972; Vane und
Botting 1995). Dies galt auch für die Begründung des nur schwachen antiphlogistischen
Effekts von Paracetamol. Nachfolgende Studien widersprachen jedoch einer
gewebespezifischen Wirkung (Lanz et al. 1986; Swierkosz et al. 2002; Warner et al.
2004).
In wie weit eine u. a. im zentralen Nervensystem exprimierte, Intron-1 beibehaltende
Spleiß-Variante der COX-1, die sog. COX-3, mit am Wirkungsmechanismus von
Paracetamol Koffein Eliminationshalbwerteszeit (h) 1.5 - 2.5 3.5 - 6.0 Verteilungsvolumen (l/kg) 0.86 0.6 Plasmaclearance (ml/(min*kg) 5.7 0.9 - 2.0 Bioverfügbarkeit (%) 65 - 90 100 Plasmaproteinbindung (%) 10 10 - 35

8
Paracetamol beteiligt ist, ist Gegenstand kontroverser Diskussionen und bedarf weiterer
klärender Untersuchungen (Chandrasekharan et al. 2002; Schwab et al. 2003; Kis et al.
2005; Simmons et al. 2005; Qin et al. 2005).
Das Wirkungsprofil Paracetamols weist einige Ähnlichkeiten mit dem selektiver
Cyclooxygenase-2-Inihibitoren, sog. Coxibe, auf. Beide zeigen einen lediglich sehr
geringen toxischen Effekt auf den Gastrointestinaltrakt (Garcia und Hernandez-Diaz
2001), haben keinen Einfluss auf die Thrombozytenfunktion (Mielke 1981; Catella-
Lawson 2001) und führen kaum zu Konstriktionen der Bronchien bei Aspirin-sensitiven
Asthmatikern (Jenkins et al. 2004). Vor diesem Hintergrund konnten Hinz et al. in einer
erst kürzlich publizierten Studie eine nahezu vollständige in vitro und in vivo Hemmung
der COX-2-Aktivität durch Paracetamol nachweisen, wohingegen eine lediglich geringe
Hemmung der COX-1 beobachtet werden konnte (Hinz et al. 2008). Die Inhibition der
Cyclooxygenase 2 zeigte sich hierbei sogar deutlicher als bei Einmalgabe von 200mg
Celecoxib bzw. 25mg Rofecoxib (Hinz et al 2008, 2006).
Die antiphlogistische Wirkung von Acetaminophen wurde aufgrund mehrerer bereits
länger zurückliegender Studien für lange Zeit als eher gering bzw. nicht vorhanden
eingestuft (Boardman et al. 1967; Ring et al. 1974; Glissold 1986), gleichwohl häufen
sich zunehmend Hinweise auf eine durchaus vorhandene antiinflammatorische
Wirkungskomponente (Skjelbred und Lokken 1979; Honore et al. 1995; Bjornsson et al.
2003), die auch wiederum in Zusammenhang mit der durch Paracetamol induzierten
COX-2 Inhibition gebracht wird (Hinz et al. 2008).
In der Literatur finden sich im Tiermodell aber auch zahlreiche Hinweise auf
Wirkmechanismen, unabhängig von der Prostaglandin-Synthesehemmung. Im
Mittelpunkt stehen dabei Serotonin-Rezeptoren bzw. das serotinerge System.
Paracetamol führt dabei zu verschiedenen Veränderungen wie einer Abnahme von 5-
HT2-Rezeptoren mit nachfolgender Analgesie (Pini et al. 1996, Courade et al. 2001).
In wie weit es daneben auch zur direkten Involvierung von Serotoninrezeptoren (u. a. 5-
HT3 und 5-HT1A/B-Rezeptoren) oder bisher noch nicht klassifizierten Rezeptoren
kommt, ist Gegenstand aktueller Forschung (Pelissier et al. 1995; Alloui et al. 1996,
2002; Roca-Vinardell et al. 2003; Sandrini et al 2004; Libert et al. 2004).
Ebenfalls für einen zentralen Wirkmechanismus sprechen die Ergebnisse von Pickering
et al. (2006), die eine Wirkungsabschwächung von oral appliziertem Paracetamol bei
gleichzeitiger Gabe von Tropisetron oder Granisetron (5-HT3-Anatagonisten) zeigen.
Dies weist erstmals auch beim Menschen auf einen zentralen, serotinergen
Wirkmechanismus hin.

9
Die pharmakologische Wirkung von Paracetamol tritt etwa 30 Minuten nach Gabe von
1g auf, bei einer durchschnittlichen Wirkdauer von vier bis sechs Stunden. Maximale
Wirkungen werden nach 1 (Bentley und Head 1987) bzw. 2 Stunden (Gretzbein et. al
1986; Arendt-Nielson et al. 1991) erreicht, wobei in der Regel eine deutliche
Verzögerung zwischen dem Auftreten des Wirkmaximums und der maximalen
Blutplasmaspiegel besteht (Arendt-Nielson et al. 1991; Seymour und Rawlins 1981).
2.1.3 Unerwünschte Arzneimittelwirkungen und Toxizität
In empfohlen therapeutischen Dosierungen ist Paracetamol ein wirksames und in der
Regel gut verträgliches Medikament (Rumack 2004). In seltenen Fällen können
Hautausschläge und Überempfindlichkeitsreaktionen wie Quincke-Ödem, Atemnot,
Schweißausbruch, Übelkeit, Hypotension bis hin zum Schock sowie bei prädisponierten
Patienten ein Bronchospasmus beobachtet werden (Prescott 1992). Allergische Reaktionen
mit Exanthembildung und Störungen der Hämatopoese (Thrombozytopenie,
Leukozytopenie, Agranulozytose, Panzytopenie) werden nur äußerst selten gesehen
(Gürsoy et al. 1996), siehe hierzu auch Tabelle 2.
Tabelle 2: Nebenwirkungsprofil von Paracetamol (modif. nach Roter Liste® 2008)
Wie bereits oben erwähnt spiegelt sich vermutlich die lediglich geringe Inhibition der
COX-1 durch Acetaminophen in den geringen gastrointestinalen Nebenwirkungen und
der fehlenden Beeinflussung der Thrombozytenfunktion wider. Dies trifft, wie mehrere
Studien zeigen konnten jedoch lediglich auf die Applikation geringer Dosen zu. Bei
höher applizierten Mengen konnten sowohl eine Inhibition der Thrombozytenfunktion
Nebenwirkung
Häufigkeit
Haut
Hautrötung bis hin zu Urtikaria sehr selten (<0,01%)
Leber
Anstieg der Lebertransaminasen Seltenst akutes Leberversagen
Selten (≥0,01%-<0,1%)
Atemwege
Bronchospasmus, bei prädisponierten Patienten (Analgetika-Asthma)
sehr selten (<0,01%)
Blut
Veränderungen im Blutbild wie Thrombozytopenie, Agranulozytose
sehr selten (<0,01%)
Immunsystem
Überempfindlichkeitsreaktionen von einfacher Hautrötung bis hin zu Urtikaria und anaphyl.aktischem Schock
sehr selten (<0,01%)

10
als auch eine Zunahme gastrointestinaler Nebenwirkungen wie Verdauungsstörungen
beobachtet werden (Nieme et al. 2000; Munsterhjelm et al. 2005).
Bezüglich der Einflüsse Paracetamols auf den Blutdruck konnten Forman et al. (2005)
zeigen, dass die regelmäßige Einnahme von Acetaminophen, im Vergleich zur Karenz
mit einem signifikant höheren Risiko der Entwicklung eines Bluthochdrucks einhergeht.
Damit einhergehend sind die Ergebnisse von Chan et al. (2006), die zeigen, dass die
Einnahme von mehr als 15 Tabletten Paracetamol pro Woche das nahezu gleiche Risiko
kardiovaskulärer Nebenwirkungen aufweist wie das gewöhnlicher nichtsteroidaler
Antiphlogistika.
Eine der gefürchtetsten Nebenwirkungen Paracetamols ist die bei Überdosierung
auftretende, u. U. tödlich verlaufenden akuten Leberzellnekrose (Jaeschke und Bajt,
2006). Die leberzellschädigende Wirkung von Acetaminophen beruht auf einer durch
Cytochrom-P-450 Enzyme katalysierten Metabolisierung zu N-Acetyl-p-
benzochinonimin. Dieser aktive Metabolit wird unter üblichen Dosierungen jedoch
schnell durch Bildung ungiftiger Konjugate mittels Glutathion abgefangen. Sind die
intrazellulären Glutathion-Speicher auf Grund einer Leberinsuffizienz oder einer zu
hohen Paracetamol-Dosis jedoch erschöpft, interagieren die Chinonimin-Metabolite mit
zellulären, einschließlich mitochondrialen Proteinen der Hepatozyten. In der Folge
treten zytotoxische Reaktionen, im Sinne einer Leberzellnekrose, auf (Prescott 1992;
Jaeschke and Bajt 2006).
Diese Nebenwirkungen können beim Erwachsenen ab Plasmakonzentrationen von
Paracetamol > 200mg/l bzw. Einmaldosen > 4g/Tag auftreten (Rainsford et al. 1997),
wobei zu beachten ist, dass eine Paracetamol bedingte Hepatotoxizität bei
vorgeschädigter Leber bereits bei Konzentrationen bzw. Dosen unter diesen Werten
auftreten kann (Rampal et al. 2002).
Eine Überdosierung mit Paracetamol ist aufgrund der einfachen, rezeptfreien
Zugänglichkeit und weiten Verbreitung in den USA und Großbritannien die häufigste
Ursache für eine medikamenteninduzierte Leberschädigung (Lee 2004).
2.2 Koffein
Koffein oder 1,3,7-Trimethylxanthin, mit lateinischem Namen auch als Coffeinum
anhydricum (purum) bezeichnet, ist ein zu den Purinen (Methylxanthinen) zählendes,
natürlich vorkommendes Pflanzen-Alkaloid. Es zählt zu einem der ältesten von
Menschen genutzten und weltweit am weitesten verbreiteten Stimulanzien.

11
Abbildung 2: Strukturformel von Koffein (C8H10N4O2) (modif. nach Aktories et al.
2005).
Bei Koffein handelt es sich unter Normalbedingungen um ein weißes, geruchsloses,
kristallines Pulver mit bitterem Geschmack. Es findet sich neben den Bohnen des
Kaffeestrauches auch zu unterschiedlich hohen Anteilen in den Beeren, Samen und
Blättern des Teestrauches, in der afrikanischen Kolanuss, dem Kakao und Guarana
sowie dem südamerikanischen Mate. Schätzungen über den Koffeingehalt
verschiedener Nahrungsmittel sind in Tabelle 3 dargestellt.
Tabelle 3: Koffeingehalt in verschiedenen Genussmitteln (modif. nach Mandel 2002).
Allgemeingültige Angaben über den Koffeingehalt einer Tasse Kaffe sind nur schwer
zu erheben, da die Koffeinkonzentration zum einen abhängig ist von der Art der
verwendeten Kaffeebohne, deren Anbau und Röstung sowie zum anderen von den
zahlreichen verschiedenen Zubereitungsverfahren (Mandel 2002).
Produkt Koffeingehalt
Kaffee: `- Filterkaffee 84 - 112mg/150ml `- Pulverkaffee 60 - 71mg/150ml `- entkoffeiniert 1 - 4mg/150ml Tee (Schwarztee) 27 - 40mg/Beutel Milchschokolade 6mg/28g Erfrischungsgetränke: `- Coca Cola 46mg/340ml `- Pepsi Cola 38mg/340ml Energy-Drink (Red Bull) 80mg/250ml

12
2.2.1 Pharmakokinetik
Koffein wird nach oraler Applikation schnell (ca. 90% des aufgenommenen Koffeins in
20 Minuten) und nahezu vollständig enteral resorbiert. Die Bioverfügbarkeit liegt bei
annähernd 100% (Axelrod und Reichenthal 1953; Chvasta und Cook 1971).
Aufgrund seines lipophilen Charakters überwindet Koffein nahezu ungehindert sowohl
die Blut-Hirn- als auch die Blut-Placenta-Schranke und verteilt sich in allen
Körpergeweben entsprechend ihres Wassergehaltes (Axelrod und Reichenthal 1953;
Sawynok und Yaksh 1993). Die höchsten Plasmakonzentrationen werden 30 – 60
Minuten nach oraler Applikation beobachtet, wobei die Zeitspanne zwischen 15 bis 120
Minuten, abhängig von der Geschwindigkeit der enteralen Resorption, liegen kann
(Grab und Reinstein 1968; Bonati et al. 1982).
Die Bindung an Plasmaproteine ist variabel und liegt zwischen 10% und 35%, das
Verteilungsvolumen schwankt zwischen 0,5 und 0,7 l/kg (Axelrod und Reichenthal
1953; Arnaud 1987).
In der Leber wird Koffein zu 84% mittels Demethylierung durch Cytochrom-P-450-IA2
zu Paraxanthin verstoffwechselt, während lediglich etwa 12% zu 3,7-Dimethylxanthin
(Theobromin) und etwa 4% zu 1,3-Dimethylxanthin (Theophyllin) metabolisiert
werden (Arnaud 1984; Butler et al. 1989; Lelo et al. 1986a).
Bei wiederholtem Koffeinkonsum während des Tages, wie beim durchschnittlichen
Kaffeekonsumenten zu finden, steigen die Plasmaspiegel von Paraxanthin auf etwa 2/3
des Koffeinspiegels an (Lelo et al 1986b). Dies ist insofern interessant, da Benowitz et
al. (1995) zeigen konnten, dass Paraxanthin, als Hauptmetabolit des Koffeins, ähnliche
pharmakologische Aktivitäten zeigt, und somit mitbeteiligt ist an dessen Wirkung auf
den menschlichen Organismus.
Die beim Menschen ermittelte Plasmahalbwertszeit für Koffein liegt zwischen 3,5 und
6,0 Stunden, verlängert sich jedoch entsprechend bei vermehrter Zufuhr oder
eingeschränkter Leberfunktion (Kaplan et al 1997).
Koffein, davon lediglich 1-5% in unveränderter Form, und seine Metabolite werden
überwiegend renal eliminiert (Axelrod und Reichenthal 1953).
Tabelle 1 gibt einen Überblick über die pharmakokinetischen Eigenschaften Koffeins.
2.2.2 Pharmakodynamik
Koffein übt eine Vielzahl verschiedener Wirkungen auf zentraler und peripherer Ebene
aus. Es führt zu einer Relaxation glatter Muskulatur, zeigt im Sinne einer

13
Bronchodilatation antiasthmatische Wirkungen, hat einen stimulierenden Effekt auf das
Herz und das zentrale Nervensystem und zeigt zudem diuretische Eigenschaften.
Als wesentlicher molekularer Wirkmechanismus konnte ein kompetitiver, nicht
selektiver Antagonismus an Adenosin A1 und A2 Rezeptoren ausgemacht werden
(Fredholm 1980; Phillis und Wu 1981; Daly 1982; Rall 1982), welche sich sowohl
zentral, im ZNS, als auch peripher u. a. im Blutgefäßsystem, in Nieren, Herz, Magen-
Darm- und Respirationstrakt befinden.
Andere Wirkungen, wie die Inhibition des Enzyms Phosphodiesterase und die Ca2+-
Mobilisierung in Zellen, allen voran Muskel- und Nervenzellen, scheinen, in Anbetracht
der dafür notwendigen hohen Plasmakonzentrationen, weniger wahrscheinlich an der
Wirkung von Koffein im menschlichen Organismus beteiligt zu sein (Sawynok und
Yaksh, 1993). Nichts desto trotz darf diese Option, angesichts der, je nach
Applikationsweg schwankenden lokalen Konzentrationen sowie der toxischen
Eigenschaften Koffeins, nicht vollkommen außer Acht gelassen werden (Sawynok und
Yaksh, 1993).
Nachfolgend seien die Wirkungen sowie die unerwünschten Arzneimittelwirkungen
Koffeins kurz näher erläutert.
Koffein bewirkt am Herzen eine positive Inotropie, Dromotropie und Bathmotrophie,
sowie einen Anstieg der Herzfrequenz, die bei massivem Konsum und sensitiven
Personen bis hin zu Tachykardien und Tachyarrhythmien reichen kann (Dobmeyer et al.
1983). Eine Blutdrucksteigerung tritt nur bei Individuen mit geringem bzw. nicht
regelmäßigem Koffeinkonsum auf, wohingegen es bei chronischer Zufuhr relativ
schnell zur Entwicklung einer Toleranz kommt (Ammon et al. 1983). Nach einer
Abstinenzphase von etwa einer Woche kann diese Toleranz jedoch unterbrochen und
durch Koffeinapplikation eine erneute Blutdrucksteigerung beobachtet werden (Shi et
al. 1993).
An der glatten Muskelzelle der Gefäße und insbesondere des Bronchialbaumes bewirkt
Koffein, über die oben erwähnte Blockade der Adenosinrezeptoren, eine Relaxation und
führt somit, im direkten Vergleich zu Theophyllin jedoch geringer, zu einer
Bronchodilatation (Arnaud 1987; Gong et al. 1986; Rall 1990).
Die Wirkung auf das Gefäßsystem ist gegensätzlich: Während Koffein bei zentralen
Gefäßen zu einer Zunahme des Widerstandes, damit zu einer reduzierten cerebralen
Perfusion, führt (Mathew und Wilson, 1985), nimmt dieser in der Peripherie hingegen
ab (Sechzer 1979).

14
Neben der bereits oben erwähnten bronchodilatatorischen Wirkung des Koffeins besteht
die Hauptwirkung / Haupteffekt auf den Respirationstrakt in erster Linie jedoch in einer
Steigerung der Atemfrequenz, vermutlich durch Sensibilisierung des medullären
Atemzentrums gegenüber Kohlendioxid (Murat et al. 1981). Dies erklärt die
Anwendung Koffeins bei Neugeborenen mit rezidivierenden apnoeischen Phasen.
An der Niere führt die Applikation von Koffein zu einer kurzfristigen Zunahme der
Diurese sowie zu einer vermehrten Ausscheidung von Elektrolyten wie Natrium,
Kalium, Chlorid, Kalzium und Magnesium (Dorfman und Jarvik 1970; Massey und
Wise 1984). Der hierfür zugrunde liegende Wirkmechanismus scheint ebenfalls eine
Blockade von Adenosin A1 Rezeptoren zu sein (Rieg et al. 2005).
Neben einer Konzentrationssteigerung von Renin im Plasma kommt es weiterhin zu
einer Erhöhung der Konzentrationen von Adrenalin und Noradrenalin unter
Koffeineinfluss (Robertson et al. 1978; Greenberg et al. 2006).
Koffein führt zu einer verstärkten Sekretion von Magensäure und Pepsin, was erstmals
von Krasnow und Grossman nachgewiesen werden konnte (Krasnow und Grossman
1945) und in mehreren nachfolgenden Studien bestätigt wurde. Auch diese Wirkung ist
wahrscheinlich auf die Antagonisierung der inibitorischen Wirkung Adenosins, auf die
Magensäure bildenden Parietalzellen, bedingt (Gerber et al. 1985). Zudem bewirkt
Koffein eine Abnahme des Tonus des unteren Ösophagussphinkters.
Die Einflüsse Koffeins auf den Stoffwechsel zeigen sich in einer gesteigerten Lipolyse
mit Anstieg der freien Fettsäuren im Serum, einer Erhöhung der Blutglukosewerte und
Abnahme der Insulinsensitivität, vermutlich bedingt durch eine vermehrte
Katecholaminsekretion, sowie in einer Aktivierungen der Thermogenese (Robertson et
al. 1978, Greenberg et al. 2006).
Einige Ergebnisse epidemiologischer Untersuchungen sehen zudem einen
Zusammenhang zwischen chronischem Koffeinkonsum und erhöhten Serum-
Cholesterol-Werten (Williams et al. 1985).
Auf das zentrale Nervensystem wirkt Koffein als potentes Stimulans. Niedrigere
Dosierungen (32 – 256mg) führen zu einer deutlich verbesserten Vigilanz und
Konzentrationsfähigkeit, zu einer verbesserten visuellen Reaktionszeit sowie zu einer
Abnahme der Müdigkeit (Lieberman et al. 1987). Konsum höherer Dosen (>500mg)
hingegen bewirkt zunehmende Nervosität und Angstgefühle, Unruhe, Schlaflosigkeit

15
und gesteigerte Erregbarkeit (Victor et al. 1981; Smith 2002). Einnahme toxischer
Mengen (mehrere Gramm-Einheiten) induzieren generalisierte Krampfanfälle.
Es finden sich zudem zahlreiche Quellen in der Literatur die auf eine, durch chronischen
Koffeinkonsum induzierte körperliche Abhängigkeit hinweisen (Griffiths und Woodson
1988; Griffiths et al. 1990). Kopfschmerzen und Müdigkeit sind hierbei die mit Abstand
am häufigsten beobachteten Symptome, gefolgt von einer großen Bandbreite seltenerer
Anzeichen wie z. B. Angstgefühl, verschlechterte psychomotorische Leistungen,
Suchtgefühl und Übelkeit und Erbrechen. Die Entzugserscheinungen treten in der Regel
nach 12 bis 24-stündiger Abstinenz auf, erreichen ihren Höhepunkt nach 20 bis 48
Stunden und halten etwa eine Woche an.
2.2.3 Koffein und Antinozizeption
Koffein übt, wie bereits oben dargelegt eine Vielzahl verschiedener Wirkungen aus, die
es wahrscheinlich machen, dass unterschiedliche Mechanismen an ihrer Genese beteiligt
sind. In wieweit Koffein per se, direkt oder indirekt antinozizeptive Wirkungen besitzt,
ist seit vielen Jahren, bis zum aktuellen Zeitpunkt, Gegenstand kontrovers geführter
Diskussionen. Nachfolgend sollen nun die wesentlichen, möglicherweise an der
Verarbeitung von Schmerzen bzw. Antinozizeption beteiligten Prozesse (Mechanismen)
erwähnt werden.
Koffein bewirkt über eine verstärkte enterale Resorption eines gleichzeitig applizierten
Analgetikums, wie z. B. Acetylsalicylsäure, eine Erhöhung dessen Bioverfügbarkeit
(Yoovathaworn et al. 1986). Darüber hinaus reduziert Koffein die hepatische Perfusion
(Onrot et al. 1986), was zu einer verminderten Clearance primär über die Leber
metabolisierter Substanzen, wie z. B. Acetaminophen und somit zu einer Verlängerung
deren Halbwertszeit führt. Zugleich bewirkt eine verstärkte Durchblutung des
Gastrointestinaltraktes eine erhöhte intestinale Absorbtionsrate (Beubler and Lembeck
1976, Iqbal et al. 1995).
Die unter hohen Dosen auftretende, wenn auch nur schwach potente Inhibition der
Phosphodiesterase (PDE) ist eher unwahrscheinlich an einer Antinozizeption beteiligt,
da Taiwo und Levine 1990 zeigen konnten, dass der durch eine PDE-Inhibition
induzierte Anstieg der cAMP-Konzentration an primär nozizeptiven Afferenzen eine
Hyperalgesie hervorruft (Sawynok und Yaksh, 1993; Ukena et al. 1993; Taiwo und
Levine, 1990).

16
In wieweit eine durch Koffein induzierte, über Aktivierung des cGMP-Signalweges,
gesteigerte NO-Produktion zur Antinozizeption beiträgt, ist auch hier wiederum auf
Grund der dafür notwendigen hohen Koffein Dosen, sowie der teilweise
widersprüchlichen Ergebnisse bezüglich der nozizeptiven bzw. antinozizeptiven
Eigenschaften von NO, nicht zufrieden stellend geklärt (Lopez-Munoz et al. 1996;
Aguirre-Banuelos et al. 1999; Wu et al. 2001; Machelska et al. 1998).
In mehreren Tierversuchen konnte gezeigt werden, dass Koffein die Ausschüttung von
Neurotransmittern wie Serotonin und Noradrenalin erhöht, wobei auch hier hohe Dosen
(>50mg/kg) verabreicht werden müssen (Sawynok 1995, Sawynok and Reid 1996).
Möglicherweise trägt darüber hinaus eine durch Koffein initiierte Aktivierung des
cholinergen Systems zur antinozizeptiven Aktivität bei (Ghelardini 1997).
Ein weiterer Ansatzpunkt könnte die, von Fiebich und Kollegen gezeigte, durch Koffein
hervorgerufene Inhibition der COX-2 Induktion in Mikrogliazellen von Ratten und
damit eine verminderte Synthese von, an Entzündungsprozessen und
Schmerzweiterleitung beteiligten, Prostaglandinen sein. Dieser inhibitorische Effekt ist
vermutlich bedingt durch eine antagonistische Aktivität des Koffeins an Adenosin A2-
Rezeptoren (Fiebich et al. 1996; Fiebich et al. 2000; Fredholm et al. 1994).
Der wesentliche molekulare Wirkmechanismus von Koffein besteht, wie bereits weiter
oben erwähnt, in einem kompetitiven, nicht selektiven Antagonismus an Adenosin A1
und A2 Rezeptoren. Die Wirkung von Adenosin ist dabei auf den verschiedenen Ebenen
des Organismuses nicht identisch. So hemmt Adenosin Schmerzimpulse auf spinaler
und supraspinaler Ebene, wohingegen es in der Peripherie an der Entstehung bzw. der
Verstärkung von Schmerzinformationen beteiligt ist (Sylven 1993, Sawynok and Reid
1996). Auf diese Weise könnte Koffein zumindest auf peripherer Ebene ebenfalls einen
Einfluss auf die Schmerzverarbeitung nehmen, wobei auch hier weitere klärende
Studien nötig sind.
Ungeachtet der dargestellten, vielfältigen Wirkmechanismen besitzt Koffein bei
alleiniger Gabe lediglich eine moderate analgetisch-antinozizeptive Potenz. Zahlreiche
Studien sowohl am Menschen als auch am Tier zeigen jedoch, dass Koffein als
Adjuvans in Kombinationsanalgetika einen wirkungsverstärkenden Effekt besitzt
(Sawynok und Yaksh 1993, Iqbal et al. 1995). Auch eine erst kürzlich publizierte Studie

17
aus unserem Haus zeigt, dass Koffein die Absorption und die analgetische Wirksamkeit
von Acetaminophen verstärkt kann (Renner et al. 2007).
2.3 Ziele der Studie
Für die hier vorliegende Studie lassen sich folgende Fragestellungen und Ziele
formulieren:
1. Bestehen signifikante Unterschiede bezüglich des analgetischen Effektes zwischen
Paracetamol plus Koffein und Placebo?
2. In wie weit lassen sich hiermit an einem neuen Probandenkollektiv die Daten und
Ergebnisse vorangegangener Studien (Renner et al. 2007) reproduzieren und somit
in ihrer Aussagekraft untermauern?
3. Ist das zu Grunde liegende experimentelle Schmerzmodell anwendbar bzw. aus
technisch-organisatorischer Sicht realisierbar für die Durchführung einer
Arzneimittelgesetz-Studie (AMG 12. Novelle, 2004) nach GCP-Methoden?
4. Die Erhebung der Pharmakokinetik von einer Einmalgabe Paracetamol plus Koffein.
5. Die Beurteilung der Sicherheit (anhand klinischer Laborparameter) und die
Erfassung von unerwünschten Ereignissen (’adverse events’) und unerwünschten
Arzneimittelwirkungen einer Einmalapplikation von Paracetamol plus Koffein und
Placebo.
6. Lediglich erwähnt werden soll hier der in einer separaten Arbeit abgehandelte
Vergleich einer Einmaldosis Nurfen® (400mg Ibuprofen) mit Placebo

18
3. Material und Methoden
3.1 Studiendesign
Bei der vorliegenden Arbeit handelt es sich um eine monozentrische, doppelt blinde,
Placebo kontrollierte, randomisierte, drei-fach cross over Studie, durchgeführt nach
GCP-Methoden an erwachsenen, freiwilligen Probanden.
Sie wurde von der Ethikkommission der Friedrich-Alexander-Universität Erlangen-
Nürnberg begutachtet und vom Bundesinstitut für Arzneimittel und Medizinprodukte
(BfArM) genehmigt. Die Durchführung erfolgte nach den Richtlinien der Deklaration
von Helsinki, Summerset West für biomedizinische Forschung am Menschen.
Jedem der an der Studie teilnehmenden Probanden wurde pro begonnenem Versuchstag
eine Aufwandsentschädigung von 150€ gewährt.
3.2 Studienteilnehmer
An den im Rahmen dieser Studie durchgeführten Experimenten nahmen 26 gesunde
Probanden (nach Prüfplan vorgesehen waren max. 28 Probanden, damit mind. 24
Probanden die Studie abschlossen), im Alter von 23-30 Jahren (s. h. Anhang 8.3) und
mit einem BMI von 19-27 kg/m2, auf freiwilliger Basis teil. 12 Probanden waren
männlichen, 14 weiblichen Geschlechts.
Die Rekrutierung der Probanden erfolgte nach ausführlicher Aufklärung sowie
medizinischer Untersuchung (Prescreening/Screening) durch den Versuchsleiter der
Studie. Im Rahmen dieser Untersuchung wurden alle den Anforderungen der Studie
entsprechenden relevanten medizinischen Daten erhoben (s. h. Anhang 8.3, 8.4 und
8.5). Des Weiteren war die Aufnahme in die Studie abhängig von zuvor festgelegten
Ein- und Ausschlusskriterien, siehe Tabelle 4 und 5.

19
Einschlusskriterien Unterschrift der Einverständniserklärung körperliche und geistige Gesundheit Alter zwischen 18-45 Jahre normales Körpergewicht bei einem BMI von 19-27 regelmäßiger täglicher Koffeinkonsum in Form von Kaffee, Tee, Cola usw. Kooperationsbereitschaft und Verständnis des Ablaufs positiv abgeschlossene Trainingssitzung (oder Zusatztraining) mit Beherrschung der Atemtechnik und Toleranz der Schmerzen absolute Alkoholkarenz für mindestens 24 Stunden vor Beginn der Messung absolute Nahrungskarenz von mindestens 6 Stunden vor Beginn der Messung keine Einnahme von Medikamenten (im speziellen die Gruppe der COX-
Hemmer), die die Leber und Nierenfunktion belasten, über mindestens 7 Tage vor Beginn der Messung (Einnahme oraler Kontrazeptiva war gestattet)
negativer Urin-Schwangerschaftstest und sichere Kontrazeption, negativer Urin- Drogentest (auf Barbiturate, Benzodiazepine, Amphetamine, Cocain, Opiate, Cannabis), negativer Atemalkoholtest
Tabelle 4: Einschlusskriterien vor Aufnahme in die Studie
Ausschlusskriterien Verdacht oder Hinweise auf klinisch relevante Abnormitäten aufgrund der
aktuellen Anamnese, der medizinischen Vorgeschichte oder der erhobenen Befunde, insbesondere Leber-, Gastrointestinal-, Atemwegs- oder Nierenerkrankungen
Schwangerschaft, Stillzeit, Frauen ohne sichere Kontrazeption Raucher mit einem Konsum von mehr als 15 Zigaretten pro Tag Positive Serologie für Hepatitis B/C oder HIV Allergien oder Intoleranzen Drogenkonsum (s. h. oben) regelmäßiger Koffeinkonsum von >5 Tassen am Tag Blutspende von >1500ml in den letzten 12 Monaten akute oder chronische Entzündungen Teilnahme an anderen Studien in den letzten 30 Tagen Angestellte des Institutes oder des Labors
Tabelle 5: Ausschlusskriterien vor Aufnahme in die Studie

20
3.3 Schmerzmodell
Dieser Studie lag ein objektives Schmerzmodell zu Grunde, welches auf dem Prinzip
der Ableitung kortikaler chemo-somatosensorisch evozierter Potentiale (CSSEP)
basiert.
Diese Potentiale werden als elektrophysiologisches Korrelat der zentralnervösen
Verarbeitungsreaktion schmerzhafter Stimulation der in diesem Fall durch den Nervus
Trigeminus sensibel innervierten, nasalen Schleimhaut, angesehen (Huttunen et al.
1986). Zur zeitgleichen Erfassung der subjektiven Schmerzerlebnisse bediente man sich
psychophysikalischer Messmethoden, im Sinne einer visuellen Analogskala.
Etabliert hat sich dieser Versuchsaufbau bereits in mehreren, in unserem Hause
vorangegangen Studien zur Objektivierung nozizeptiver Effekte verschiedener opioider
(Hummel et al. 1994b; Hummel et al. 1995c; Kobal et al. 1990a; Lötsch et al. 1997b;
Thürauf et al. 1996) und nicht-opioider Analgetika (Hummel et al. 1995a; Kobal et al.
1990b; Kraetsch et al. 1996; Lötsch et al. 1995b; Lötsch et al. 1995a).
Die Stimulation nasaler Mukosa erfolgte zum einen periodisch mittels zweier definierter
Gemische aus Luft und Kohlendioxid (sog. phasische Stimulation zur Simulation eines
200 msec. dauernden, stechenden, nicht entzündlichen Akutschmerzes, in der linken
Nasenöffnung), zum anderen tonisch durch das Einleiten von trockener, geruchsloser
Luft (zur Simulation eines entzündlichen Schmerzzustandes, in der rechten
Nasenöffnung).
Während der Durchführung der Experimente saßen die Versuchsteilnehmer auf einem
bequemen Stuhl, innerhalb eines klimatisierten Messraumes. Um eventuell auftretende
Nebengeräusche aus der Umgebung sowie Schaltgeräusche des Versuchsaufbaus zu
maskieren, wurden die Probanden mittels eines Akustikstimulator-generierten so
genannten weißen Rauschens (Zeisberg, Ingolstadt Deutschland) über einen, während
der Messung getragenen Kopfhörer, mit 50 dB Schalldruck beschallt.
Eine Videoüberwachung des Messplatzes diente, neben der Überwachung der
Kompliance, auch der Sicherheit des Probanden, um zeitnah auf eventuell auftretende
unerwünschte Arzneimittelwirkungen oder andere Alterationen von Gesundheit oder
Befindlichkeit reagieren zu können.
3.3.1 Phasische Schmerzreizung
Die phasische Irritation der Nasenschleimhaut erfolgte in diesem Versuch mittels eines
Gemisches aus Luft und CO2-Gas in zwei verschiedenen Mischverhältnissen mit 60%-

21
(Klasse 1) oder 70%igem (Klasse 2) CO2-Anteil. Die Verwendung zweier
unterschiedlicher Reizstärkeklassen war dabei notwendig, um die Verzerrung des
Ergebnisses durch sog. „Response Bias“ (’Antwort-Voreingenommenheit’) von Seiten
des Probanden möglichst gering zu halten, indem diesem die Möglichkeit gegeben
wurde, zwischen zwei Schmerzintensitäten zu unterscheiden. Dabei lag die CO2
Konzentration zu jedem Zeitpunkt sicher über der erforderlichen
Schwellenkonzentration von ungefähren 30% CO2-Gas-Anteil in Luft (Kobal 1985), ab
welcher stechend-scharfe Schmerzempfindungen von akutem Charakter ohne
zusätzliche olfaktorische oder gustatorische Sensation reproduzierbar sind.
Die Reize sind, abhängig von der CO2-Konzentration der Stimuli und deren Dauer,
durchaus schmerzhaft, führen aber zu keinerlei Schädigung der nasalen Mukosa (Kobal
et al. 1986).
Um die subjektiv empfundene Schmerzintensität der jeweiligen Probanden zu erheben,
wurden sog. visuelle Analogskalen (VAS) zur Quantifizierung eingesetzt. Abbildung 3
liefert einen schematisch dargestellten, orientierenden Überblick über den
Versuchsaufbau.
Abbildung 3: Schema des verwendeten Schmerzmodells. Darstellung der phasischen (schwarz) und tonischen (grau) Schmerzreizung unter Registrierung der CSSEP und des Hintergrund EEGs (FFT) im Fünfkanal-EEG (links im Bild). Darstellung des subjektiven Schmerzempfindens (VAS) durch psychophysikalische Meßmethoden (Subjektivskalen, rechts im Bild) (Abb. modif. nach Neuwald 2007).

22
Die Kohlendioxid-Stimuli wurden mit einer Dauer von 200 ms in das linke Nasenloch
appliziert. Zwischen den einzelnen Stimuli lag ein reizfreies Interstimulus-Intervall von
25-30s Dauer. Dadurch können Adaptations- und Habituationsphänomene weitgehend
ausgeschaltet werden (Hummel und Kobal 1999; Kobal 1981; Kobal et al. 1986;
Miyazaki et al. 1994). Der Trägergasstrom bestand im Interstimulus-Intervall aus
Reinluft mit einer Temperatur von 36,5 °C sowie einer Luftfeuchtigkeit von 80%.
Während der Reizapplikation kam es lediglich zu einer Änderung der molekularen
Zusammensetzung des Trägergasstorms, d.h. zu einem Wechsel zwischen reiner Luft
und dem reizklassenabhängigem CO2-Gas/Luftgemisch. Daneben durfte es jedoch zu
keinerlei weiteren Veränderungen, seien sie z.B. thermischer oder mechanischer Natur
des auf die Schleimhaut treffenden Gasstrahles im Vergleich zum Interstimulus-Strom
kommen, da dies eine zeitgleiche Erregung von Rezeptoren anderer Sinnesmodalitäten
mit sich bringen würde (Kobal 1981; Kobal 1985; Thürauf et al. 1991; Thürauf et al.
1993).
Um neben diesen Anforderungen gleichzeitig sehr kurze Stimulusanschlagszeiten mit
Zeiten unter 20 ms zu erreichen, die für die Erzeugung evozierter Potentiale durch eine
möglichst zeitsynchrone Aktivierung zerebraler Neuronen in ausreichenden Anzahl von
Bedeutung sind (Kobal 1981, Kobal 1985, Kobal et al. 1988), wurde die phasische
Schmerzreizung mit Hilfe des in Abbildung 4 dargestellten Olfaktometers durchgeführt.
Dieses wurde computergesteuert (Labview™, National Instruments, Austin, TX, USA)
mit dem Programm Bioresponse Olfactometry Program, BOMP.O2b (Kobal) betrieben.
Abbildung 4: Darstellung des verwendeten Olfaktometers (Modell OM4, Firma Burghart Instruments, Wedel, Deutschland)

23
Ein Weiteres wesentliches Prinzip dieser von G. Kobal 1981 entwickelten Konstruktion
(Kobal et al. 1981) ist, neben der Verwirklichung einer genau zeitlich, qualitativ und
quantitativ kontrollierbaren, monomodalen Rechteck-Reizung, die absolute Passivität
des Versuchsteilnehmers, welche die reizsynchrone Erregung anderer, nicht mit der
Schmerzverarbeitung assoziierter, kortikaler Neurone weitestgehend unterbindet.
Eine schnelle Spülung der Nasenhöhlen von verbliebenen CO2-Gas-Molekülen wird
durch die hohe Flussrate des Trägerstroms von insgesamt 8 l*min-1 erreicht. Dies
vermeidet zudem eventuelle, durch Diffusion bedingte, Konzentrationsänderungen. Die
Dauer des phasischen Reizabschnittes beträgt insgesamt 20 Minuten.
Das technische Grundprinzip des von uns für diesen Versuch verwendeten
Olfaktometers sei nun im Genaueren erläutert. Abbildung 5 zeigt hierzu den Schaltplan.
Abbildung 5: technischer Schaltplan des Olfaktometers: P = Druckluftquelle, G = Gastank, O = Reizleitung, Gas = Gasleitung, D = Verdünnungsleitung, C = Reinluft-Leitung, ME = Hauptabsaugung, CCo = Absaugung der Luftschranke, CCi = Zuteilung Luftschranke, J = Einfüllstutzen der Fritte, M = Y-Magnetventil, E1= Absaugung des Reizgas enthaltenden Luftstroms, E2 = Absaugung des reizgasfreien Luftstroms, N = Ausgang des Olfaktometers zur Probandennase (Abb. modif. nach Neuwald 2007).

24
Ein Kompressor P liefert durch Aktivkohle geleitete und gereinigte Druckluft für die
Leitungen Reinluft C, Verdünnungsluft D sowie Zuluft für den Querstrom CCi.
Die Leitung Gas G wird in diesem Versuchsaufbau von einem mit CO2-Gas gefüllten
Flaschentank (Air Liquide®, Deutschland GmbH, Kohlendioxid 4.5) gespeist. Neben
diesen Leitungen besitzt das Gerät zudem eine Anzahl verschiedener Duftleitungen O,
die ebenfalls vom Kompressor mit Reinluft versorgt werden und je nach Fragestellung
mit reinen Duftstoffen oder trigeminal und olfaktorisch aktiven Substanzen (wie z.B.
Methanol) versetzt werden können.
An allen Leitungen befinden sich Durchflussmessgeräte (Tylan® Flow-Controller,
Mykrolis, Eching, Deutschland), mit deren Hilfe die jeweiligen Stromstärken gemessen
und entsprechend der gewünschten Reizklassenstärke justiert werden können.
Die so regulierten Trägerströme i0, iD und iC werden durch speziell konstruierte
Gaswaschflaschen geleitet, in denen sie entweder mit Duftstoff (O) oder mit
Wasserdampf (C und D) bei 46,5°C angereichert werden.
Nach Verlassen der Waschflaschen werden die Gasströme durch Teflonschläuche zu
einem ventillosen, aus Glas angefertigten Schaltstück geleitet, welches sich, um die
Bildung von Pendelvolumina in Toträumen weitestmöglichst auszuschalten, direkt vor
der Nase des Probanden befindet. Der Funktionsweise dieses ventillosen, unter Einsatz
von Sperrströmen, als Luftschranke arbeitenden Schaltwerkes kommt dabei eine
besondere Rolle zu, weshalb diese mit Hilfe von Abbildung 6 kurz dargestellt sei:
Die rechte Bildhälfte zeigt die Gasströme im Interstimulusintervall (d.h. kein CO2-
Gas/Luftgemisch erreicht die Nasenschleimhaut des Probanden). Über die Leitung C
erreicht die nasale Mukosa lediglich CO2-freie Reinluft.
Durch ein zwischen den beiden Hauptabsaugungen E1, E2 und deren Hauptausgang ME
geschaltetes Y-Magnetschaltventil (Kobal und Plattig 1978), welches neben dem
ventillosen Schaltstück wesentlich am schnellen und strömungskonstanten
Umschaltvorgang beteiligt ist, kann nun zwischen diesen beiden Saugschenkeln
umgeschaltet werden. Dies ist möglich, ohne dass es währenddessen zu wesentlichen
Veränderungen des Gesamtquerschnittes kommt, welche zu Verwirbelungen und somit
zu einer Erregung weiterer Sinnesmodalitäten führen würden.
Zur Reizapplikation müssen nun lediglich, wie in der rechten Bildhälfte dargestellt, mit
Hilfe des Magnetschaltventils die Unterdruckverhältnisse durch Öffnen von E2 und
Schließen von E1 spiegelbildlich umgekehrt werden. Auf diese Weise lässt sich
zwischen den beiden Zuständen Stimulus und Interstimulus-Intervall umschalten.

25
Abbildung 6: Schaltstück eines Olfaktometers; linker Teil: Schaltzustand bei Stimulusgabe, rechter Teil: Schaltzustand im Interstimulus-Intervall; N = Probandennase, E2 = Absaugung der Reinluft, C = Reinluft, E1 = Absaugung des Reizstoff-Luft-Gemisches, O1 = möglicher Duftstoff oder Gas – in dieser Studie CO2, D = Verdünnungsluft, CCo1 = Absaugungen der Luftschranken, CCi1 = Zuleitungen Luftschranken (Abb. modif. nach Kobal 1981).
Die durch die Querströme (CCi1/CCo1) realisierte Luftschranke verhindert zudem
jegliche Kontamination des CO2-Luftgemisches mit anderen Reizstoffen.
Die Luft- bzw. Gas-führenden Leitungen in diesem Versuchsaufbau bestehen
ausschließlich aus biochemischen größtenteils inerten Materialien wie Teflon oder Glas
die bis kurz vor erreichen der Probandennase mittels einer Wasserummantelung
thermostabilisiert sind.
Mit Hilfe dieses hier dargestellten Versuchsaufbaus konnte die geforderte monomodale,
nozizeptive Rechteck-Stimulation zur Evozierung möglichst rein schmerz-korrelierter
Potentiale umgesetzt werden.
3.3.2 Tonische Schmerzreizung
Ein weiterer Teil des Experimentes bestand in der tonischen, einen dumpfen
Dauerschmerz induzierenden (Kobal et al. 1990b), die Verhältnisse eines entzündlichen
Schmerzustandes nachbildenden Reizung (Mohammadian et al. 1997) der nasalen
Mukosa, die im Anschluss an die phasischen Reizblöcke durchgeführt wurde.

26
Hierbei wurde ein kontinuierlicher Strom von auf 32°C gleichmäßig temperierter,
getrockneter Luft mit einer relativen Luftfeuchtigkeit von 20% sowie einem konstanten
Fluss von 8 l*min-1 über einen Zeitraum von 16 min in das rechte Nasenloch appliziert
(Lötsch et al. 1998; Mohammadian et al. 1997).
Der durch einen in einem separaten Raum stehenden Kompressor generierte Druckluft-
Fluss wurde mittels eines Dosierventils (2 bar) und einer Feineinstellung durch ein
Rotameter (Rotameter, Rota YOKOGAWA® GmbH + Co KG) adjustiert.
Durch diese Form der Reizapplikation kam es zu einer einseitigen, leichten Schwellung
der Nasenschleimhaut, sowie zu Schmerzen, welche in der Regel innerhalb der ersten
Stunden reversibel sind (Lötsch et al. 1995b; Lötsch et al. 1998).
3.4 Prüfmedikation
Glaxo-Smith-Kline (GSK) Consumer Health Care lieferte die Medikation an ein die
‘Good Manufacturing Practice’ (GMP) Kriterien erfüllendes Unternehmen, in unserem
Fall die Apotheke am Bohlenplatz in 91054 Erlangen, aus. Diese stellte die in Swedish
Orange double blind Gelatine Kapseln verblindete Prüfmedikation, gemäß eines von
GSK vorgegebenen Randomisierungsplanes, her.
Die Prüfmedikationen, sowie das Placebo, wurden dem Probanden zusammen mit
150ml „stillem“ Wasser gereicht. Auf kohlensäurehaltiges Wasser wurde aufgrund
möglicher Interferenzen mit dem Schmerzmodell sowie einer Ablenkung des Probanden
oder Störung der EEG-Aufzeichnungen, im Sinne von Muskelartefakten durch
eventuelles Aufstoßen verzichtet.
Die verblindeten Probanden erhielten an jedem Versuchstag, der Randomisierung
entsprechend, je eine der drei Prüfmedikationen:
Entweder 1000mg Paracetamol + 130mg Koffein zu je zwei Tabletten a 500mg
Paracetamol + 65mg Koffein, oder 400mg Ibuprofen zu je zwei Tabletten a 200mg
Ibuprofen oder, als dritte Möglichkeit je zwei Tabletten Placebo.
Die Probanden mussten mindestens sechs Stunden vor Erhalt der Versuchsmedikation
auf feste Nahrungsmittel sowie eine Stunde vor Erhalt der Versuchsmedikation auf
flüssige Nahrungsmittel verzichten.
3.5 Trainingssitzung
Alle an der Studie teilnehmenden Probanden nahmen innerhalb von zwei Wochen vor
Beginn der Studientage an mindestens einer Trainingssitzung (Prescreening/Screening)

27
teil, die ihnen dazu diente, sich mit allen relevanten praktischen Elementen und
Abläufen des Versuchsaufbaus vertraut zu machen.
Im Besonderen wurde hierbei Wert gelegt auf das Erlernen einer speziellen
Atemtechnik mit velo-pharyngealem Verschluss (Kobal und Hummel 1992).
Bei dieser Technik presst der Proband sein Gaumensegel durch willkürliche
Kontraktion der Mm. tensor und levator veli palatini gegen die Pharynx-Hinterwand,
sodass die oberen Luftwege völlig von den unteren abgetrennt werden. Dies führte zu
einer ausschließlichen Mundatmung ohne jeglichen Luftfluss durch die Nase, sodass es
zu keinen atmungsbedingten Luftbewegungen während der Reizintervalle und dadurch
möglicherweise hervorgerufenen artifiziellen Konzentrationsveränderungen des CO2-
Gas-Anteils in der Nase des Probanden kommt.
Zum Erlernen dieses Verfahrens, so wie zur Objektivierung und Kontrolle des
Lernerfolges aus Sicht der Versuchsleiter, wurde eine Biofeedbackvorrichtung zu Hilfe
genommen, welche mittels eines Thermistors (Respirant X®, Siemens, München,
Deutschland) nasale Restluftbewegungen bei nicht korrekter Durchführung als
oszilloskopische Ausschläge (HM203-6®, Hameg, Mainhausen, Deutschland) darstellt.
Die Probanden wurden zudem zur Einnahme einer möglichst entspannten Sitzposition
veranlasst sowie darin geschult, mögliche, durch zu häufiges Augenzwinkern
entstehende EEG Artefakte zu vermeiden.
3.6 Ablauf der Studientage
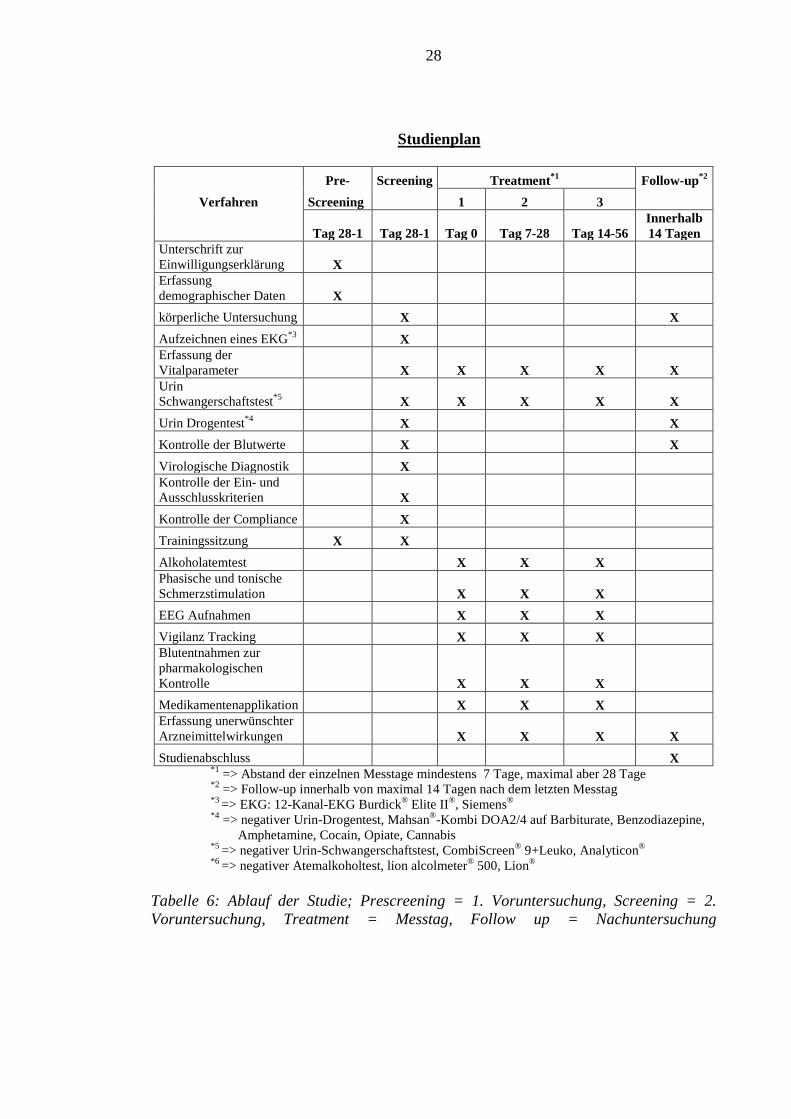
Einen Überblick über den Studienablauf bietet Tabelle 6.
Die Probanden fanden sich an den drei, mindestens sieben, maximal 28 Tage
voneinander getrennt liegenden Versuchstagen ca. eine Stunde vor Applikation der
Versuchsmedikation in unserem Labor ein.
Hierzu wurde die an der Studie teilnehmende Population in zwei Gruppen eingeteilt.
Gruppe eins, bestehend aus sieben Männern und sieben Frauen, erhielt ihre
Prüfmedikation um 8:00 Uhr. Gruppe zwei, bestehend aus fünf Männern und 7 Frauen,
erhielt ihre Prüfmedikation um 14:00 Uhr. Die Probanden blieben über die gesamte
Dauer des Experiments in der Ihnen zugewiesenen Gruppe. Die Abläufe der drei
Versuchstage waren identisch und wegen der zu erwartenden langen Halbwertszeit der
28
Studienplan
Pre- Screening Treatment*1 Follow-up*2 Verfahren Screening 1 2 3
Tag 28-1 Tag 28-1 Tag 0 Tag 7-28 Tag 14-56 Innerhalb 14 Tagen
Unterschrift zur Einwilligungserklärung X Erfassung demographischer Daten X körperliche Untersuchung X X Aufzeichnen eines EKG*3 X Erfassung der Vitalparameter X X X X X Urin Schwangerschaftstest*5 X X X X X Urin Drogentest*4 X X Kontrolle der Blutwerte X X Virologische Diagnostik X Kontrolle der Ein- und Ausschlusskriterien X Kontrolle der Compliance X Trainingssitzung X X Alkoholatemtest X X X Phasische und tonische Schmerzstimulation X X X EEG Aufnahmen X X X Vigilanz Tracking X X X Blutentnahmen zur pharmakologischen Kontrolle X X X Medikamentenapplikation X X X Erfassung unerwünschter Arzneimittelwirkungen X X X X Studienabschluss X
*1 => Abstand der einzelnen Messtage mindestens 7 Tage, maximal aber 28 Tage *2 => Follow-up innerhalb von maximal 14 Tagen nach dem letzten Messtag *3 => EKG: 12-Kanal-EKG Burdick® Elite II®, Siemens®
*4 => negativer Urin-Drogentest, Mahsan®-Kombi DOA2/4 auf Barbiturate, Benzodiazepine, Amphetamine, Cocain, Opiate, Cannabis *5 => negativer Urin-Schwangerschaftstest, CombiScreen® 9+Leuko, Analyticon®
*6 => negativer Atemalkoholtest, lion alcolmeter® 500, Lion®
Tabelle 6: Ablauf der Studie; Prescreening = 1. Voruntersuchung, Screening = 2. Voruntersuchung, Treatment = Messtag, Follow up = Nachuntersuchung

29
Medikation, zum Ausschluss möglicher Interferenzen zwischen den Substanzen, durch
eine mindestens siebentätige Pause unterbrochen.
Um an den Experimenten des jeweiligen Messtages teilnehmen zu können, mussten die
in Tabelle 7 aufgelisteten, zu Beginn des Versuchstages kontrollierten Kriterien erfüllt
werden.
subjektiv unbehinderte Nasenatmung ohne Rhinorrhoe subjektives Gefühl völliger Gesundheit Nahrungskarenz fester Nahrung von mindestens sechs Stunden, Nahrungskarenz
flüssiger Nahrung von mindestens einer Stunde vor Beginn des Studientages Alkoholkarenz von mindestens 24 Stunden, kontrolliert mittels eines Alkohol-
Atemtest Nikotin- und Koffeinkarenz von mindestens 12 Stunden vor Beginn des
Studientages Bei Frauen negativer Schwangerschaftstest negative Medikamentenanamnese Blutdruck, sitzend: systolisch zw. 90 - 150 mmHg, diastolisch zw. 60 - 90 mmHg Herzfrequenz im Sitzen: zwischen 50 – 100 Schläge pro Minute
Tabelle 7: in dem Prüfprotokoll (CRF) des entsprechenden Versuchtages protokollierte Kriterien
Vor Beginn des Messtages wurden über den gesamten Studienzeitraum hinweg der
Versuchsaufbau, die Flussraten der verschiedenen Gase und Gasgemische
(Bubblemeter, GilibratorTM, Gilian Instrument, NJ, USA), die Temperatur sowie die
relative Feuchte (Tri-SenseTM, Cole Parmer, IL, USA) von den Untersuchern
routinemäßig überprüft und protokolliert.
3.6.1 Ablauf der analgesimetrischen Untersuchung
Die analgesimetrischen Untersuchungen wurden in insgesamt vier Messblöcke
unterteilt, mit einer Gesamtdauer von 216 Minuten. Einen schematischen Überblick
hierzu zeigt Abbildung 7.
Der erste Messblock (sog. ‚Baseline’) mit einer Dauer von 36 Minuten wurde direkt vor
Applikation der Prüfmedikation durchgeführt, um Referenzdaten, sog. Baseline-Daten,
für den jeden Messtag und damit den Status der Prüfparameter vor Medikamentengabe
zu erhalten. Innerhalb dieses Messblocks bekam der Proband für 20 Minuten insgesamt
40 phasische CO2-Stimuli, davon 20 mit einer CO2-Konzentration von 70%, sowie
weitere 20 mit einer Konzentration von 60% CO2, in randomisierter Abfolge über die
linke Nasenseite appliziert.
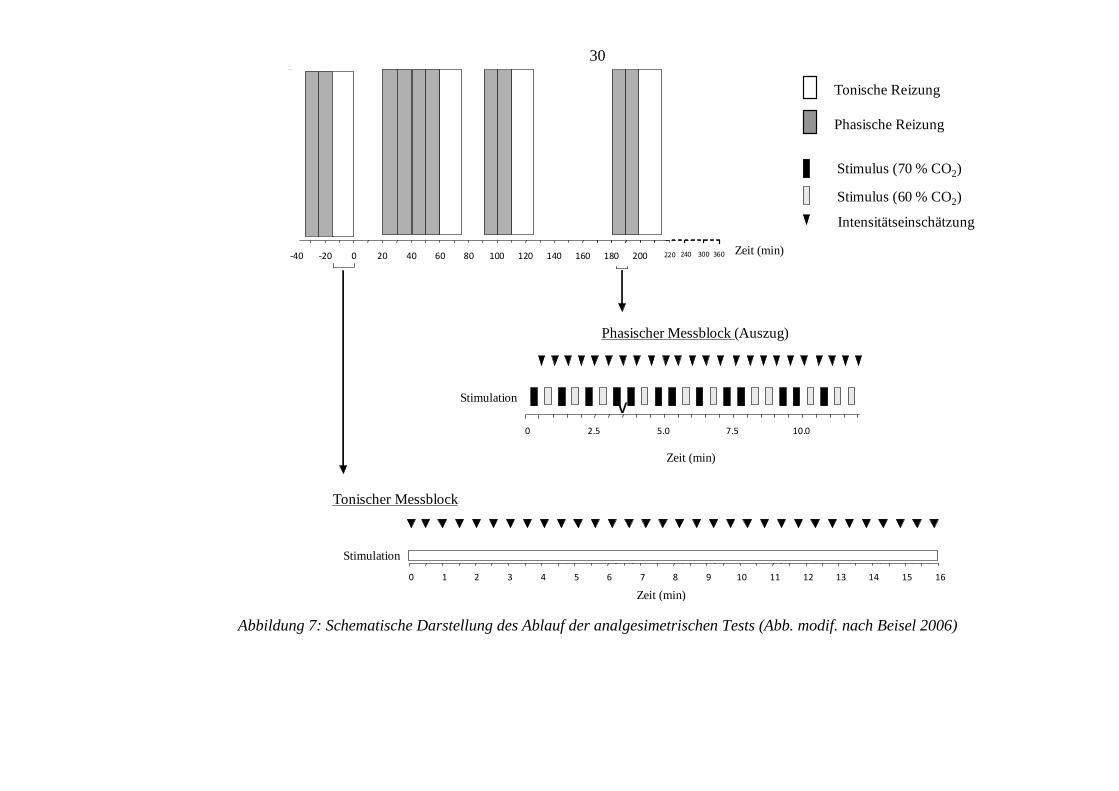
30
Abbildung 7: Schematische Darstellung des Ablauf der analgesimetrischen Tests (Abb. modif. nach Beisel 2006)
Tonischer Messblock
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Stimulation
Zeit (min)
Stimulus (70 % CO2)
Stimulus (60 % CO2)
Intensitätseinschätzung
Tonische Reizung
Phasische Reizung
Zeit (min)-40 -20 0 20 40 60 80 100 120 140 160 180 200 220 240 300 360
Phasischer Messblock (Auszug)
Zeit (min)
0 2.5 5.0 7.5
Stimulation
10.0
v

31
Darauf folgte nach kurzem Umbau der Versuchsapparatur in direktem Anschluss eine
16 minütige tonische Schmerzreizung über die rechte Nasenseite.
Unmittelbar anschließend an diesen ersten Messblock erfolgte die Verabreichung der
Prüfmedikation zusammen mit 150ml „stillem“ Wasser.
Messblock zwei (sog. ‚Session one’) mit einer Dauer von 56 Minuten folgte nach einer
20-minütigen Pause im Anschluss an die Medikation. Dieser Abschnitt bestand
seinerseits aus einer 40 Minuten dauernden (2 x 20 Minuten, durch eine 5-minütige
Pause voneinander getrennten) phasischen CO2 Applikation, wobei 40 Stimuli mit einer
Konzentration von 70% CO2-Gasanteil und 40 Stimuli mit 60% CO2-Konzentration
verabreicht wurden. Hieran schloss sich ein tonischer Reizblock von 16-minütiger
Dauer an.
Messblock drei (sog. ‚Session two’) und vier (sog. ‚Session three’), 90 bzw. 180
Minuten nach Medikation, dauerten jeweils 36 Minuten und waren ihrerseits aufgebaut
wie Messblock eins (sog. ‚Baseline’).
Zwischen zweitem und drittem Messblock war es den Probanden gestattet, koffeinfreie
und kohlensäurefreie Flüssigkeit zu sich zu nehmen. Im Anschluss an den dritten
Messblock durften feste Nahrungsmittel in Form kleinerer, fettreduzierter Snacks
konsumiert werden. Abbildung 8 zeigt den zeitlichen Ablauf eines Versuchstages.
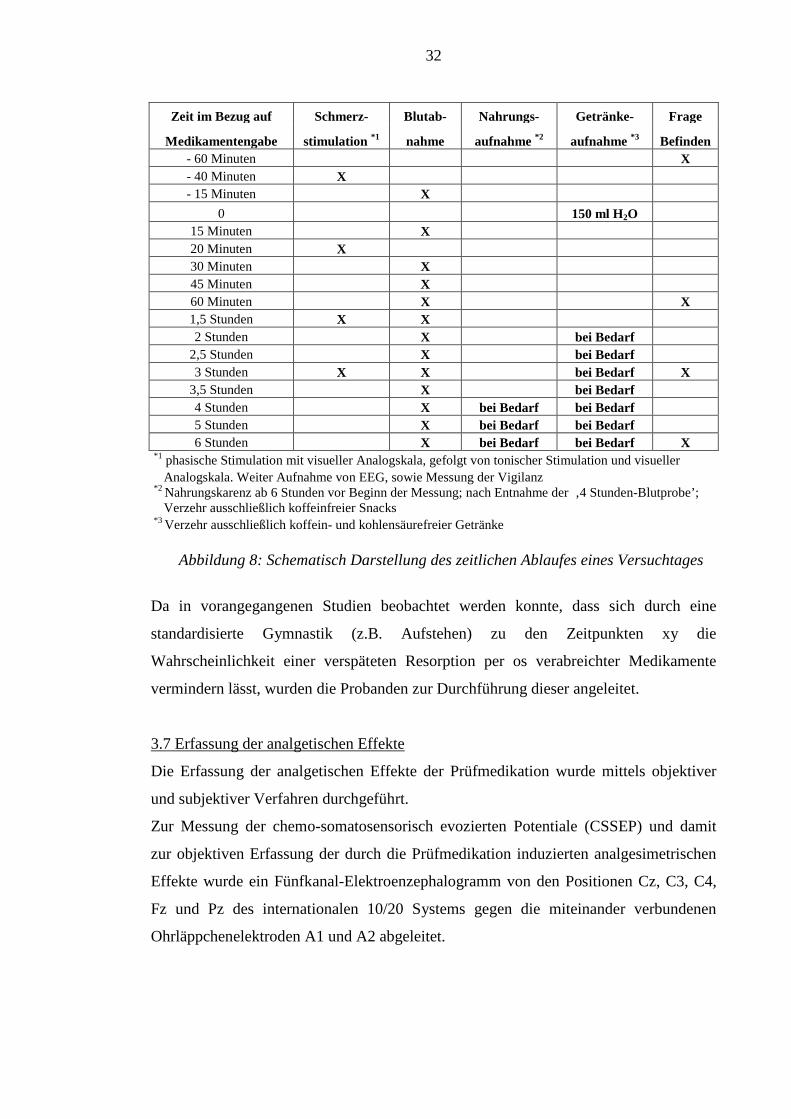
32
Zeit im Bezug auf Schmerz- Blutab- Nahrungs- Getränke- Frage
Medikamentengabe stimulation *1 nahme aufnahme *2 aufnahme *3 Befinden - 60 Minuten X - 40 Minuten X - 15 Minuten X
0 150 ml H2O 15 Minuten X 20 Minuten X 30 Minuten X 45 Minuten X 60 Minuten X X 1,5 Stunden X X 2 Stunden X bei Bedarf
2,5 Stunden X bei Bedarf 3 Stunden X X bei Bedarf X
3,5 Stunden X bei Bedarf 4 Stunden X bei Bedarf bei Bedarf 5 Stunden X bei Bedarf bei Bedarf 6 Stunden X bei Bedarf bei Bedarf X
*1 phasische Stimulation mit visueller Analogskala, gefolgt von tonischer Stimulation und visueller Analogskala. Weiter Aufnahme von EEG, sowie Messung der Vigilanz *2 Nahrungskarenz ab 6 Stunden vor Beginn der Messung; nach Entnahme der ‚4 Stunden-Blutprobe’; Verzehr ausschließlich koffeinfreier Snacks *3 Verzehr ausschließlich koffein- und kohlensäurefreier Getränke Abbildung 8: Schematisch Darstellung des zeitlichen Ablaufes eines Versuchtages
Da in vorangegangenen Studien beobachtet werden konnte, dass sich durch eine
standardisierte Gymnastik (z.B. Aufstehen) zu den Zeitpunkten xy die
Wahrscheinlichkeit einer verspäteten Resorption per os verabreichter Medikamente
vermindern lässt, wurden die Probanden zur Durchführung dieser angeleitet.
3.7 Erfassung der analgetischen Effekte
Die Erfassung der analgetischen Effekte der Prüfmedikation wurde mittels objektiver
und subjektiver Verfahren durchgeführt.
Zur Messung der chemo-somatosensorisch evozierten Potentiale (CSSEP) und damit
zur objektiven Erfassung der durch die Prüfmedikation induzierten analgesimetrischen
Effekte wurde ein Fünfkanal-Elektroenzephalogramm von den Positionen Cz, C3, C4,
Fz und Pz des internationalen 10/20 Systems gegen die miteinander verbundenen
Ohrläppchenelektroden A1 und A2 abgeleitet.

33
Abbildung 9: Schematische Darstellung der Elektrodenplatzierung eines 5-Kanal EEG
Zur Kontrolle von durch eventuelles Augenzwinkern oder Muskelbewegungen der
Probanden entstandenen Artefakten diente eine zusätzliche frontopolare Ableitung Fp2
gegen A1 und A2.
Das EEG wurde mit Hilfe von 5mm Silber/Silberchlorid-Napfelektroden an, in
Abhängigkeit der Kopfgröße definierten Arealen der Kopfhaut mittels EC2™-
Elektrodenpaste (Grass, West Warwick, RI, USA) angebracht. Zur Optimierung der
Leitungseigenschaften wurde die Kopfhaut mit NuPrep™-Gel (Weaver&Co. Aurora,
CO, USA) vorbehandelt.
Zur Erfassung der, neben diesen objektiven Methoden, subjektiv empfundenen
Schmerzintensität dienten visuelle Analogskalen (VAS).
3.7.1 Messung der chemo-somatosensorisch evozierten Potentiale (CSSEP)
Während der phasischen und tonischen Reizblöcke wurden, mit einer Abtastfrequenz
von 250Hz (band pass 0,2-30 Hz), 540ms vor, bis 1508ms nach Stimulusapplikation
insgesamt je 2048ms lang andauernde EEG-Abschnitte verstärkt (SIR, Röttenbach,
Deutschland), von analoger in digitale Form konvertiert und auf mehreren, voneinander
getrennten Computersystemen zur Weiterverarbeitung archiviert.
Diese EEG-Aufzeichnungen wurden anschließend für beide CO2-Konzentrationen und
für jede der entsprechenden Ableitungspositionen getrennt gemittelt, wobei durch
Artefakte verfälschte Einzelaufnahmen der Mittelung ausgeschlossen und verworfen
wurden (Kobal 1981; Kobal und Hummel 1988).
Die zentralnervösen, durch die zwei verschiedenen CO2-Stimuli induzierten
Reizantworten zeigten sich in der entsprechenden Veränderung der Form der
schmerzkorrelierten chemo-somatosensorisch evozierten Potentiale (CSSEP).
Abbildung 10 zeigt die charakteristische Form sowie die Einzelkomponenten eines
CSSEP.
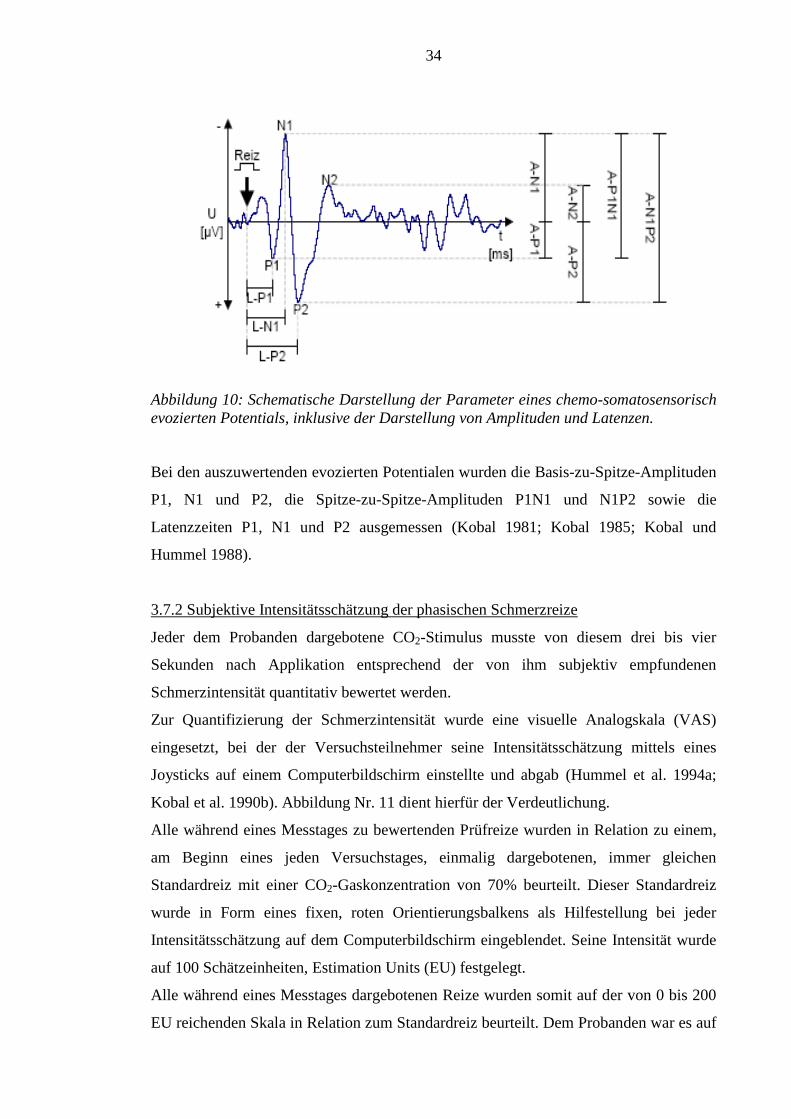
34
Abbildung 10: Schematische Darstellung der Parameter eines chemo-somatosensorisch evozierten Potentials, inklusive der Darstellung von Amplituden und Latenzen.
Bei den auszuwertenden evozierten Potentialen wurden die Basis-zu-Spitze-Amplituden
P1, N1 und P2, die Spitze-zu-Spitze-Amplituden P1N1 und N1P2 sowie die
Latenzzeiten P1, N1 und P2 ausgemessen (Kobal 1981; Kobal 1985; Kobal und
Hummel 1988).
3.7.2 Subjektive Intensitätsschätzung der phasischen Schmerzreize
Jeder dem Probanden dargebotene CO2-Stimulus musste von diesem drei bis vier
Sekunden nach Applikation entsprechend der von ihm subjektiv empfundenen
Schmerzintensität quantitativ bewertet werden.
Zur Quantifizierung der Schmerzintensität wurde eine visuelle Analogskala (VAS)
eingesetzt, bei der der Versuchsteilnehmer seine Intensitätsschätzung mittels eines
Joysticks auf einem Computerbildschirm einstellte und abgab (Hummel et al. 1994a;
Kobal et al. 1990b). Abbildung Nr. 11 dient hierfür der Verdeutlichung.
Alle während eines Messtages zu bewertenden Prüfreize wurden in Relation zu einem,
am Beginn eines jeden Versuchstages, einmalig dargebotenen, immer gleichen
Standardreiz mit einer CO2-Gaskonzentration von 70% beurteilt. Dieser Standardreiz
wurde in Form eines fixen, roten Orientierungsbalkens als Hilfestellung bei jeder
Intensitätsschätzung auf dem Computerbildschirm eingeblendet. Seine Intensität wurde
auf 100 Schätzeinheiten, Estimation Units (EU) festgelegt.
Alle während eines Messtages dargebotenen Reize wurden somit auf der von 0 bis 200
EU reichenden Skala in Relation zum Standardreiz beurteilt. Dem Probanden war es auf

35
diese Weise möglich, vergleichsweise schwach, gleichstark bis hin zu eben noch
tolerierbar empfundene Reizstärken zu wählen.
Die von der Versuchsperson vorgenommenen Bewertungen der Prüfreize wurden ihrer
Reizklasse entsprechend getrennt gespeichert.
Abbildung 11: Darstellung der verwendeten visuellen Analogskala (VAS) unter phasischer Reizung. Der untere, rote Balken entspricht dem Standardreiz, auf den sich alle folgenden Reize beziehen. Lediglich der obere grüne Balken ist durch den Probanden regulierbar und gibt eine Skala, entsprechend der empfundenen Reizintensität von 0 EU (keine Schmerzempfindung, links) bis max. 200 EU (eben noch tolerierbare Schmerzempfindung, rechts), an (s.h. auch Anhang 8.7).
3.7.3 Intensitätsschätzung der tonischen Schmerzreize
Die Beurteilung der Schmerzintensität der tonischen Schmerzreize wurde ebenfalls mit
einer, in ihrer Methodik leicht veränderten, visuellen Analogskala vorgenommen (s. h.
Abbildung Nr. 12). Im Vergleich zur phasischen Schmerzreizung bekommt der Proband
hier keinen Standardreiz dargeboten, so dass die visuelle Analogskala diesbezüglich
entsprechend modifiziert werden musste.
Geschätzt wurde hier mittels einer Zeigereinstellung auf einem Balken, dessen linkes
Ende mit 0 EU, das heißt keinerlei Schmerzempfindung des Probanden, und dessen
rechtes Ende mit 100 EU, das heißt eben noch tolerierbarer Schmerzempfindung,
definiert wurde. Auf diese Weise gaben die Versuchspersonen alle 30 Sekunden eine
Bewertung ihrer zu diesem Zeitpunkt empfundenen Schmerzintensität ab.
Da, wie sich in vorangegangenen Studien zeigte, die Schmerzwahrnehmung nach acht
Minuten ein Plateau erreichte, wurde die Gesamtstimulationszeit von 16 Minuten in
zwei Hälften zu je acht Minuten getrennt analysiert und zur statistischen Auswertung
gespeichert (Lötsch et al. 1998).

36
Abbildung 12: Darstellung der verwendeten visuellen Analogskala (VAS) unter tonischer Reizung. Mittels des weißen Balkens konnte die entsprechend empfundene Schmerzintensität auf einer Skala von 0 EU (links) bis 100 EU (rechts) angegeben werden (s. h. auch Anhang 8.7).
3.8 Erfassung unspezifischer Effekte
Um eventuelle weitere Effekte und Begleitwirkungen der zu prüfenden Substanzen
erfassen und beurteilen zu können, wurden ein sog. Hintergrund-EEG, motorische
Koordination und Vigilanz sowie Veränderungen der Befindlichkeit der Probanden
nach Quantität und Qualität protokolliert.
3.8.1 Powerspektren des Hintergrund-Elektroencephalogramms
Während des Interstimulus-Intervalls wurde vor Applikation eines jeden CO2-Reizes
das aktuelle Spontan-EEG für 4096ms mit einer Abtastrate von 125Hz aufgezeichnet.
Die gewonnenen Daten wurden einer Frequenzanalyse mittels Fast Fourier
Transformation (FFT) unterzogen.
Die daraus resultierenden Powerspektren wurden in sechs Frequenzbändern gemittelt
(Delta 1-3,5 Hz, Theta 3,5-7 Hz, Alpha1 7-10 Hz, Alpha2 10-13 Hz, Beta1 13-18 Hz
und Beta2 18-21 Hz), sowie die Spectral-Edge- und Median-Frequenzen für jeden
Einzelkanal bestimmt.
3.8.2 Tracking Performance
Um während der phasischen und tonischen Schmerzstimulation die Vigilanz des
Probanden aufrecht zu erhalten und zu erfassen, wurde dieser mit einem einfachen, in
Abbildung Nr. 13 dargestellten Computerspiel beschäftigt.

37
Abbildung 13: Computerspiel zur Erfassung der Vigilanz des Probanden. Das kleine rote Rechteck musste mittels eines Joysticks im größeren, sich zufällig über den Bildschirm bewegenden, grünen Rechteck gehalten werden.
Aufgabe dieses Spieles war es, ein kleines, mit einem Joystick frei zu bewegendes
Rechteck innerhalb eines sich zufällig über den gesamten Computerbildschirm
bewegenden großen Rechtecks zu halten. Dabei wurde registriert, wie häufig und wie
lange sich das kleine Rechteck außerhalb des großen Rechtecks befand, um daraus die
mögliche Erfolgsrate, die so genannte Tracking Performance, welche zwischen 0 bis
100% liegen konnte, zu ermitteln.
Diese Daten wurden getrennt für jede einzelne Sitzung der Versuchstage gemittelt und
statistisch ausgewertet (Kobal et al. 1990b).
Mit Hilfe dieser Aufgabe gelang es, neben der Erfassung eventueller Änderungen der
Vigilanz und motorischen Koordination, die Aufmerksamkeit auf den Bildschirm zu
richten, um somit eine Verbesserung der Qualität der evozierten Potentiale im EEG
(Haider et al. 1964) und eine Verringerung von Artefakten in den Aufnahmen durch
Muskelpotentiale oder willkürliche Augenbewegungen zu erreichen.
Lediglich zur Abfrage der einzelnen Schmerzbewertungen wurde das Computerspiel
kurz unterbrochen, um im Anschluss an gleicher Stelle fortgeführt zu werden.
3.8.3 Unerwünschte Arzneimittelwirkungen
Zu Beginn eines jeden Versuchstages wurde eine ausführliche Anamnese über den
zurückliegenden Zeitraum erhoben. Diese hatte zum Ziel, neben potenziell
unerwünschten Arzneimittelwirkungen der verabreichten Studienmedikation auch
weitere, während des messfreien Intervalls aufgetretene, eventuell die Studie
betreffende Ereignisse, zu erfassen.

38
Darüber hinaus wurden die Probanden zur Selbstbeobachtung angehalten, um im Falle
des Auftretens möglicher unerwünschter Arzneimittelwirkungen diese dem die Studie
durchführenden Personal mit zu teilen.
Am Ende des letzten Messtages wurden die Probanden dazu aufgefordert, jedem der
drei Studientage bezüglich der Wahrnehmung einer Medikamentenwirkung je eine der
Beschreibungen - kein Effekt, schwacher Effekt, starker Effekt - zuzuordnen. Diese
Daten wurden protokolliert und sind in Tabelle 19 im Anhang wiedergegeben.
Innerhalb von 14 Tagen nach Absolvierung des dritten Messtages wurden die
Probanden zu einer letzten Abschlussuntersuchung (‚Follow up’), an der die in Tabelle
6 angegebenen Punkte durchgeführt und notiert wurden, einbestellt und anschließend
offiziell aus der Studie entlassen.
3.9 Erfassung der pharmakokinetischen Effekte
Zu Beginn eines jeden Versuchstages wurde im Bereich des linken Armes des
Probanden ein venöser Zugang mittels einer Verweilkanüle geschaffen. Über diesen
Zugang konnten, entsprechend eines vorgegeben fixen Zeitregimes kurz vor, 15 min, 30
min, 45 min, 60 min sowie 1 h, 1,5 h, 2 h, 2,5 h, 3 h, 3,5 h, 4 h, 5 h und 6 h nach Gabe
der Medikation venöse Blutproben in einem EDTA Röhrchen mit einem Volumen von
9ml entnommen werden. Die jeweiligen Abnahmezeiten wurden in einem Protokoll
(Case Report Form, CRF) dokumentiert, wobei die Abnahmezeiten nicht mehr als +/- 5
min von dem zuvor fixierten Zeitschema abweichen durften.
Die gewonnenen Blutproben wurden umgehend mit 3500 Umdrehungen pro Minute bei
4°C für 15 Minuten zentrifugiert, von diesen ca. 4ml Plasma abpipettiert und innerhalb
der ersten Stunde nach Blutentnahme bei -20°C tiefgefroren. Am Ende des jeweiligen
Versuchstages wurden alle Proben bei -75°C tiefgefroren und für die spätere Analytik
gelagert.
Die Summe aller Blutentnahmen bei jedem einzelnen Probanden während der gesamten
Studie betrug ca. 420 ml, dabei an den 3 Messtagen jeweils 13*9 ml, sowie für
Screening und Follow Up zusammen ca. 60 ml.
3.9.1 Analytik des Probenmateriales
Die Analyse der pharmakokinetischen Daten zu Paracetamol plus Koffein und
Ibuprofen wurde nach GLP-Richtlinien (Good Laboratory Practice) im Labor
MEDEVAL LTD, Skelton House, Manchester Science Park, Manchester im Auftrag
von GlaxoSmithKline Consumer Healthcare, Weybridge, Surrey, England durchgeführt.

39
Die laborchemischen Blutuntersuchungen wurden durch das Zentrallabor des
Universitätsklinikums Erlangen (Leiter Dr. med. H. Parsch) durchgeführt.
3.10 Good Clinical Practice und Standard Operating Procedure
Bei der Durchführung der klinischen Prüfung eines Arzneimittels am Menschen müssen
gemäß § 40 des deutschen Arzneimittelgesetztes (AMG) die Anforderungen der guten
klinischen Praxis (Good Clinical Practice) nach Maßgabe des Artikels 1 Abs. 3 der
Richtlinie des Europäischen Parlamentes und des Rates 2001/20/EG eingehalten werden
(AMG §40).
Bei diesen Anforderungen handelt es sich um vom Bundesinstitut für Arzneimittel und
Medizinprodukte (BfArM) sowie der europäischen Arzneimittelagentur (European
Medicines Agency, EMEA) international anerkannte, nach ethischen und
wissenschaftlichen Gesichtspunkten aufgestellte Regeln für die Durchführung von
klinischen Studien. Im Mittelpunkt der GCP-Leit- und Richtlinien stehen neben dem
Schutz der Studienteilnehmer, welcher nochmals gesondert durch die Deklaration von
Helsinki (World Medical Association Declaration of Helsinki) sichergestellt wird, die
Qualität und Glaubwürdigkeit der Studienergebnisse (ICH Topic 6 Guideline for Good
Clinical Practice). So wurden z. B. alle die zu einem Probanden während dieser Studie
erhobenen Daten in einem Prüfbogen, sog. Case Report Form (CRF) notiert. Dies
beinhaltete neben denen in Tabelle 6 und 7 erhobenen Informationen u. a. auch die
Dokumentation der Start- und Endzeiten der tonischen und phasischen Reizblöcke
sowie die exakten Zeitpunkte der Blutproben-Gewinnung.
Im Rahmen dieser sog. Standard Operating Procedure (SOP, d.
Standardvorgehensweise) wurden jedoch nicht nur die Erhebung und Dokumentation
der Studiendaten geregelt, sondern zudem auch zahlreiche standardisierte
Arbeitsanweisungen festgelegt. Diese beinhalteten neben der Bedienung, regelmäßigen
Wartung und Eichung des in diesem Versuchsaufbau benutzten Olfaktometers auch den
direkten Umgang mit den Probanden. So wurden z. B. zu Beginn eines neuen
Messblockes die Probanden mit Hilfe standardisierter Ansagen (s. h. Anhang 8.7) über
den folgenden Versuchsabschnitt erneut informiert und so u. a. an die Notwendigkeit
des Einhaltens der erlernten Atemtechnik erinnert.
Die Studie wurde über den gesamten Studienzeitraum von einem unabhängigen
klinischen Monitor (Clinical Research Associate od. Site Manager) überwacht sowie im
Rahmen der Qualitätssicherung eines stichprobenartigen Audits (lat. Anhörung)
unterzogen.
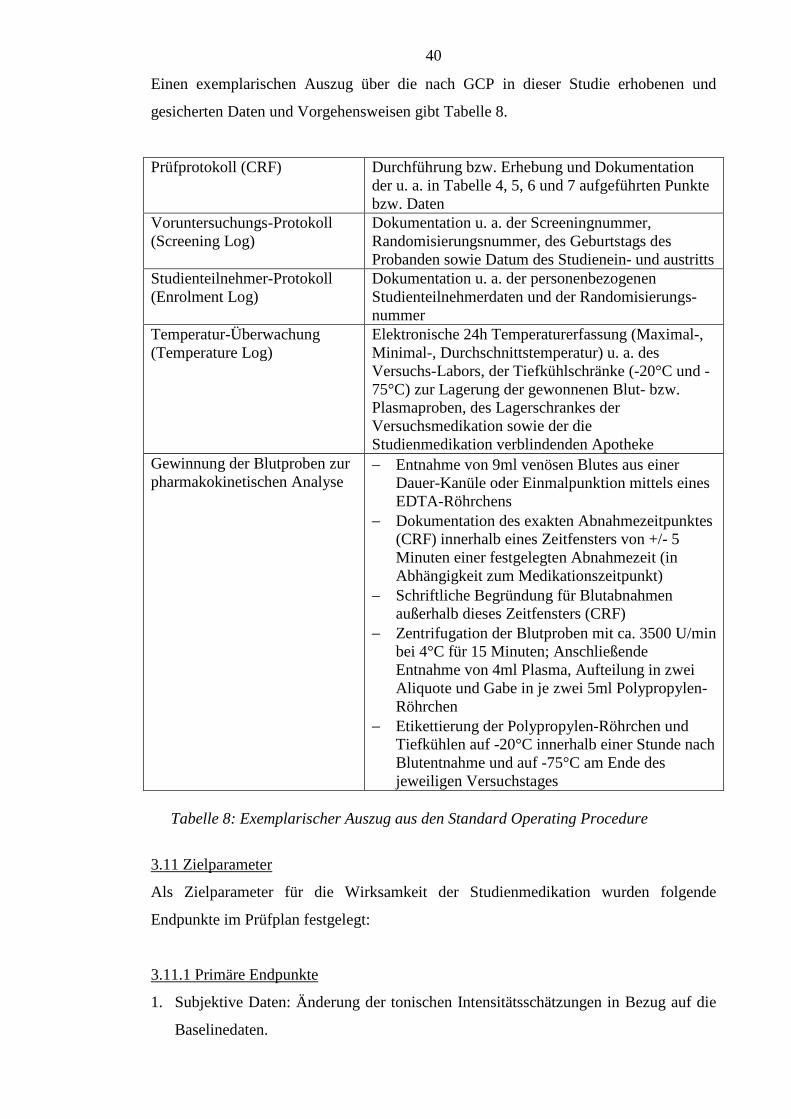
40
Einen exemplarischen Auszug über die nach GCP in dieser Studie erhobenen und
gesicherten Daten und Vorgehensweisen gibt Tabelle 8.
Prüfprotokoll (CRF) Durchführung bzw. Erhebung und Dokumentation der u. a. in Tabelle 4, 5, 6 und 7 aufgeführten Punkte bzw. Daten
Voruntersuchungs-Protokoll (Screening Log)
Dokumentation u. a. der Screeningnummer, Randomisierungsnummer, des Geburtstags des Probanden sowie Datum des Studienein- und austritts
Studienteilnehmer-Protokoll (Enrolment Log)
Dokumentation u. a. der personenbezogenen Studienteilnehmerdaten und der Randomisierungs-nummer
Temperatur-Überwachung (Temperature Log)
Elektronische 24h Temperaturerfassung (Maximal-, Minimal-, Durchschnittstemperatur) u. a. des Versuchs-Labors, der Tiefkühlschränke (-20°C und -75°C) zur Lagerung der gewonnenen Blut- bzw. Plasmaproben, des Lagerschrankes der Versuchsmedikation sowie der die Studienmedikation verblindenden Apotheke
Gewinnung der Blutproben zur pharmakokinetischen Analyse
− Entnahme von 9ml venösen Blutes aus einer Dauer-Kanüle oder Einmalpunktion mittels eines EDTA-Röhrchens
− Dokumentation des exakten Abnahmezeitpunktes (CRF) innerhalb eines Zeitfensters von +/- 5 Minuten einer festgelegten Abnahmezeit (in Abhängigkeit zum Medikationszeitpunkt)
− Schriftliche Begründung für Blutabnahmen außerhalb dieses Zeitfensters (CRF)
− Zentrifugation der Blutproben mit ca. 3500 U/min bei 4°C für 15 Minuten; Anschließende Entnahme von 4ml Plasma, Aufteilung in zwei Aliquote und Gabe in je zwei 5ml Polypropylen-Röhrchen
− Etikettierung der Polypropylen-Röhrchen und Tiefkühlen auf -20°C innerhalb einer Stunde nach Blutentnahme und auf -75°C am Ende des jeweiligen Versuchstages
Tabelle 8: Exemplarischer Auszug aus den Standard Operating Procedure
3.11 Zielparameter
Als Zielparameter für die Wirksamkeit der Studienmedikation wurden folgende
Endpunkte im Prüfplan festgelegt:
3.11.1 Primäre Endpunkte
1. Subjektive Daten: Änderung der tonischen Intensitätsschätzungen in Bezug auf die
Baselinedaten.

41
2. Objektive Daten: Änderung der Amplituden P2 und N1 getrennt für beide
Reizstärken.
3.11.2 Sekundäre Endpunkte
1. Änderung der phasischen Intensitätsschätzungen in Bezug auf die Baselinedaten
2. Änderung der phasischen Tracking performance (TP in %) in Bezug auf die
Baselinedaten, getrennt für beide Reizstärken.
3. Änderung der tonischen Tracking performance (TP in %) in Bezug auf die
Baselinedaten.
4. Die Beurteilung der Sicherheit und Nebenwirkungen der Prüfmedikation erfolgte
anhand einer ausführlichen Anamnese der Probanden sowie mit Hilfe von
Laboruntersuchungen nach Applikation der entsprechenden Prüfmedikation.
3.12 Statistische Methoden
Zu den Zielparametern der psychophysischen Daten zählten die Tracking Performance
(TP) und die Intensitätsschätzung des tonischen bzw. phasischen Schmerzes (VAS).
Die Mittelwerte der tonischen Schmerz-Intensitätsschätzung wurden aus den
Einzelwerten der zweiten Hälfte (ab der achten Minute nach Reizbeginn) für jeden
Messblock berechnet. Für die TP unter tonischer Stimulation erfolgte die Berechnung in
gleicher Weise. Demgegenüber wurden die Mittelwerte für die phasische Stimulation
getrennt für die beiden zugehörigen Reizkonzentrationen (60 % bzw. 70% CO2) und für
jeden Messblock berechnet. Alle Mittelwerte der psychophysischen Daten sowie die
Einzelwerte aus den evozierten Potentialen wurden der statistischen Auswertung
zugeführt.
Als statistischer Test diente eine Kovarianzanalyse für wiederholte Messungen in Form
eines linearen gemischten Modells. Die zeitlichen Änderungen der Zielparameter im
Bezug zur Baseline (erster Messblock) wurden als Differenz berechnet. In diesem
Modell wurden als feste Effekte die Medikation, der Studientag, die Zeit sowie deren
Interaktion (Zeit versus Medikation) festgelegt. Die entsprechenden Baselinewerte (aus
Messblock 1) dienten hier als Kovariaten. Die Probanden wurden als zufälliger Faktor
in die Analyse aufgenommen. Das Signifikanzniveau wurde auf 5% festgelegt.
Korrekturen für Mehrfachvergleiche wurden nicht vorgenommen.

42
4. Ergebnisse
Insgesamt 41 Probanden willigten zur Teilnahme an der Studie ein, von denen 27
randomisiert werden konnten und 24 die Studie komplett abschlossen. Ein Proband
schied aufgrund einer Laryngitis, ein weiterer aufgrund einer Erkältung aus. Ein dritter
Proband wurde auf falsche Weise randomisiert (jedoch ohne eine Medikation
eingenommen zu haben) und somit als ’screening failure’ vorzeitig aus der Studie
ausgeschlossen. Die Daten von 26 Probanden gingen in die Auswertung dieser Studie
ein (ITT = 26). 12 Probanden waren männlichen, 14 weiblichen Geschlechts, mit einem
Durchschnittsalter von 25,9 Jahren. Bezüglich der ethnischen Rasse waren 25
Probanden Indoeuropäer und ein Proband asiatischer Abstammung. Die
demographischen Daten der Studienpopulation sind im Anhang unter 8.3 tabellarisch
aufgeführt.
4.1 Unerwünschte Ereignisse (’adverse events’) und Beurteilung der Sicherheit der
Prüfmedikation
Während der Studie wurden insgesamt neun unerwünschte Ereignisse (’adverse events’,
AE) an acht Probanden von geringer bis mäßiger Intensität beobachtet. Zwei Probanden
schieden aufgrund einer Erkältung (unter Placebo-Medikation) sowie wegen einer
Laryngitis (unter Paracetamol plus Koffein) aus der Studie aus. Es konnte keinerlei
Bezug zwischen Prüfmedikation und AE hergestellt werden. Die häufigste Anzahl an
unerwünschten Ereignissen ereignete sich nach Placebo-Applikation (fünf
unerwünschte Ereignisse an vier Probanden). Tabelle 9 fasst die Art und Häufigkeiten
von unerwünschten Ereignissen zusammen.
Probanden Anzahl der betroffenen Anzahl der AE AE im Speziellen (n=26) Probanden (%) (adverse Events) Gesamt 6 (23.1) 7
Paracetamol + 2 (7.7) 2 milde Harnwegsinfektion
Koffein
Placebo 4 (15.4) 5 Parästhesie am linken Unterarm milde orthostatische Hypotension Harnwegsinfektion milde Laryngitis milde Erkältung
Tabelle 9: Art und Häufigkeit der unerwünschten Ereignisse unter der Prüfmedikation
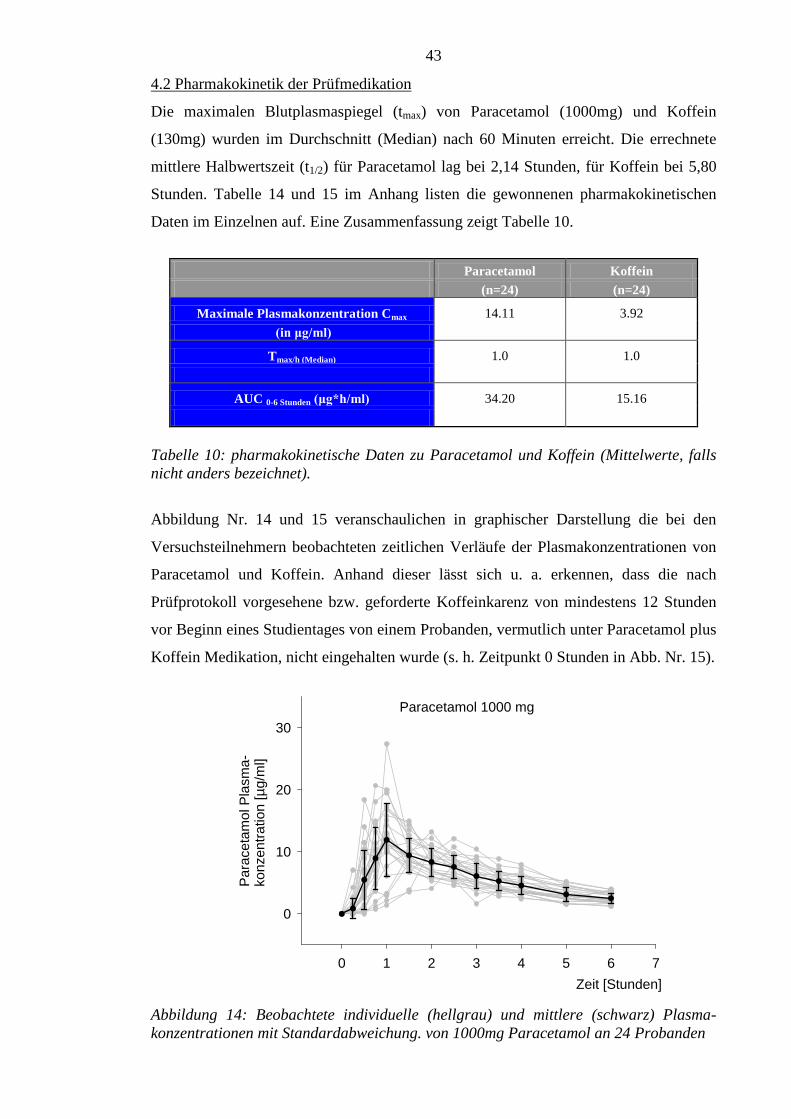
43
4.2 Pharmakokinetik der Prüfmedikation
Die maximalen Blutplasmaspiegel (tmax) von Paracetamol (1000mg) und Koffein
(130mg) wurden im Durchschnitt (Median) nach 60 Minuten erreicht. Die errechnete
mittlere Halbwertszeit (t1/2) für Paracetamol lag bei 2,14 Stunden, für Koffein bei 5,80
Stunden. Tabelle 14 und 15 im Anhang listen die gewonnenen pharmakokinetischen
Daten im Einzelnen auf. Eine Zusammenfassung zeigt Tabelle 10.
Paracetamol Koffein (n=24) (n=24)
Maximale Plasmakonzentration Cmax 14.11 3.92 (in μg/ml)
Tmax/h (Median) 1.0 1.0
AUC 0-6 Stunden (μg*h/ml) 34.20 15.16
Tabelle 10: pharmakokinetische Daten zu Paracetamol und Koffein (Mittelwerte, falls nicht anders bezeichnet).
Abbildung Nr. 14 und 15 veranschaulichen in graphischer Darstellung die bei den
Versuchsteilnehmern beobachteten zeitlichen Verläufe der Plasmakonzentrationen von
Paracetamol und Koffein. Anhand dieser lässt sich u. a. erkennen, dass die nach
Prüfprotokoll vorgesehene bzw. geforderte Koffeinkarenz von mindestens 12 Stunden
vor Beginn eines Studientages von einem Probanden, vermutlich unter Paracetamol plus
Koffein Medikation, nicht eingehalten wurde (s. h. Zeitpunkt 0 Stunden in Abb. Nr. 15).
Abbildung 14: Beobachtete individuelle (hellgrau) und mittlere (schwarz) Plasma-konzentrationen mit Standardabweichung. von 1000mg Paracetamol an 24 Probanden
Paracetamol 1000 mg
Zeit [Stunden]0 1 2 3 4 5 6 7
Para
ceta
mol
Pla
sma-
konz
entra
tion
[µg/
ml]
0
10
20
30
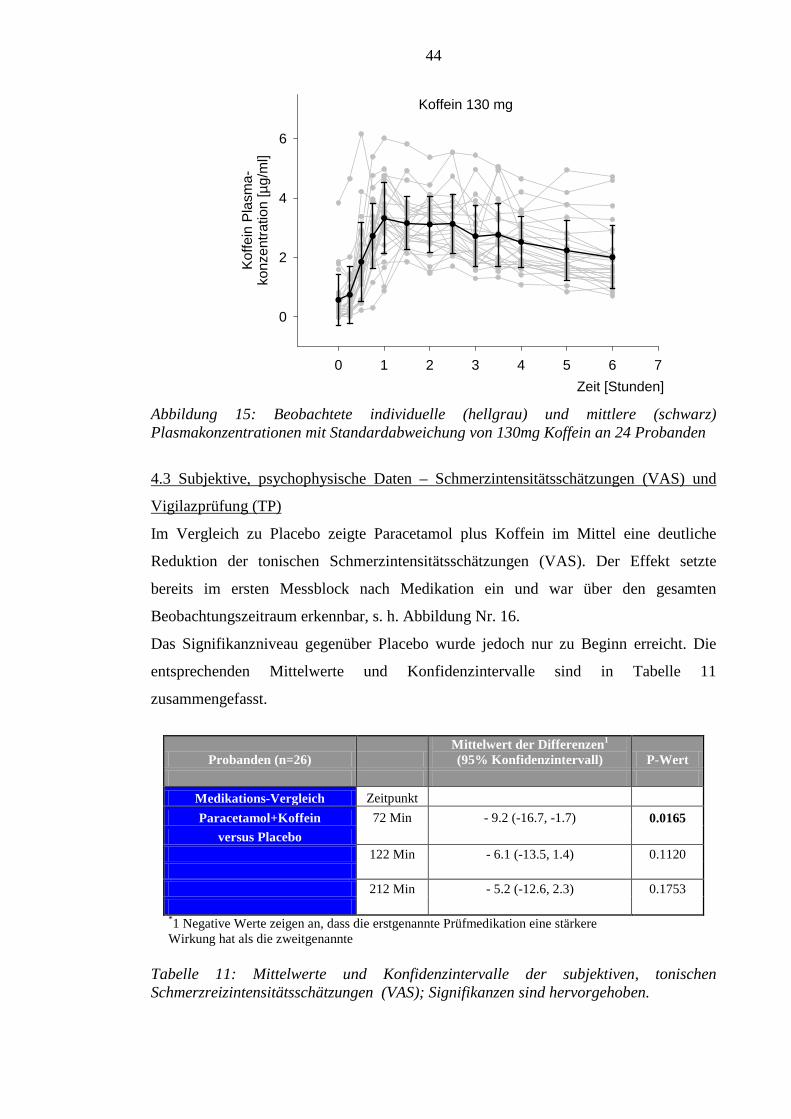
44
Abbildung 15: Beobachtete individuelle (hellgrau) und mittlere (schwarz) Plasmakonzentrationen mit Standardabweichung von 130mg Koffein an 24 Probanden
4.3 Subjektive, psychophysische Daten – Schmerzintensitätsschätzungen (VAS) und
Vigilazprüfung (TP)
Im Vergleich zu Placebo zeigte Paracetamol plus Koffein im Mittel eine deutliche
Reduktion der tonischen Schmerzintensitätsschätzungen (VAS). Der Effekt setzte
bereits im ersten Messblock nach Medikation ein und war über den gesamten
Beobachtungszeitraum erkennbar, s. h. Abbildung Nr. 16.
Das Signifikanzniveau gegenüber Placebo wurde jedoch nur zu Beginn erreicht. Die
entsprechenden Mittelwerte und Konfidenzintervalle sind in Tabelle 11
zusammengefasst.
Probanden (n=26) Mittelwert der Differenzen1
(95% Konfidenzintervall) P-Wert
Medikations-Vergleich Zeitpunkt Paracetamol+Koffein 72 Min - 9.2 (-16.7, -1.7) 0.0165
versus Placebo 122 Min - 6.1 (-13.5, 1.4) 0.1120 212 Min - 5.2 (-12.6, 2.3) 0.1753
*1 Negative Werte zeigen an, dass die erstgenannte Prüfmedikation eine stärkere Wirkung hat als die zweitgenannte
Tabelle 11: Mittelwerte und Konfidenzintervalle der subjektiven, tonischen Schmerzreizintensitätsschätzungen (VAS); Signifikanzen sind hervorgehoben.
Koffein 130 mg
Zeit [Stunden]0 1 2 3 4 5 6 7
Koffe
in P
lasm
a-ko
nzen
tratio
n [µ
g/m
l]
0
2
4
6
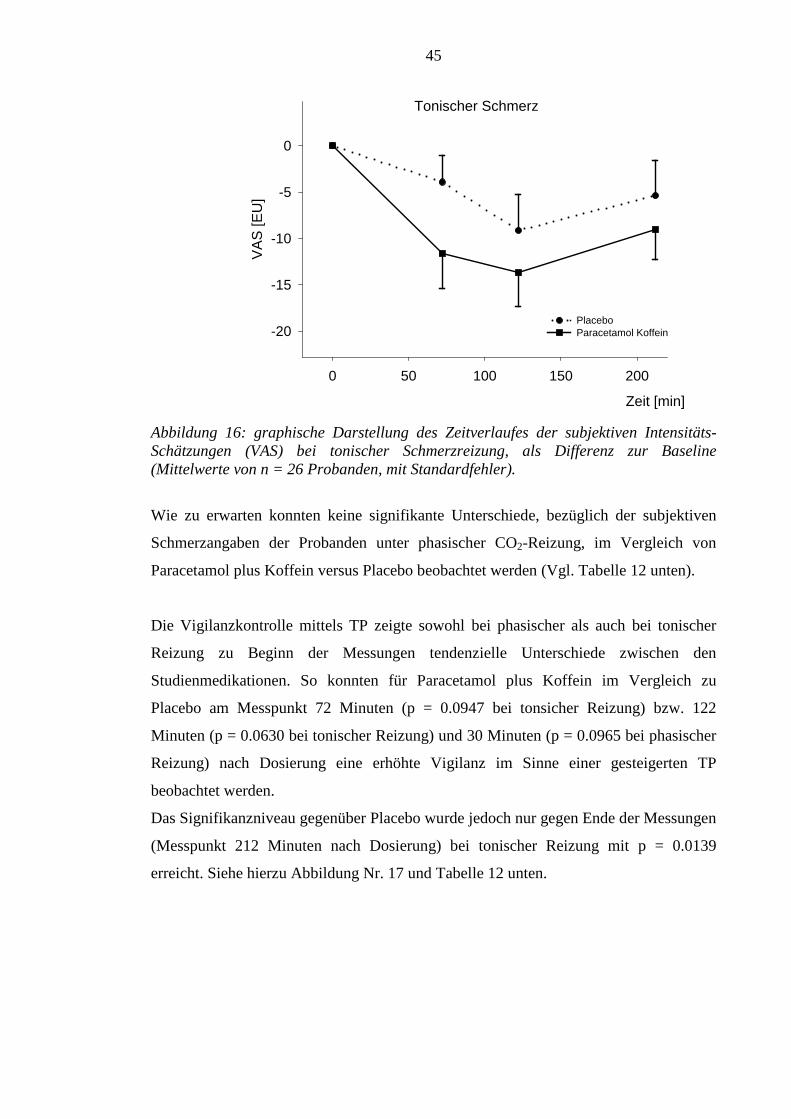
45
Abbildung 16: graphische Darstellung des Zeitverlaufes der subjektiven Intensitäts-Schätzungen (VAS) bei tonischer Schmerzreizung, als Differenz zur Baseline (Mittelwerte von n = 26 Probanden, mit Standardfehler).
Wie zu erwarten konnten keine signifikante Unterschiede, bezüglich der subjektiven
Schmerzangaben der Probanden unter phasischer CO2-Reizung, im Vergleich von
Paracetamol plus Koffein versus Placebo beobachtet werden (Vgl. Tabelle 12 unten).
Die Vigilanzkontrolle mittels TP zeigte sowohl bei phasischer als auch bei tonischer
Reizung zu Beginn der Messungen tendenzielle Unterschiede zwischen den
Studienmedikationen. So konnten für Paracetamol plus Koffein im Vergleich zu
Placebo am Messpunkt 72 Minuten (p = 0.0947 bei tonsicher Reizung) bzw. 122
Minuten (p = 0.0630 bei tonischer Reizung) und 30 Minuten (p = 0.0965 bei phasischer
Reizung) nach Dosierung eine erhöhte Vigilanz im Sinne einer gesteigerten TP
beobachtet werden.
Das Signifikanzniveau gegenüber Placebo wurde jedoch nur gegen Ende der Messungen
(Messpunkt 212 Minuten nach Dosierung) bei tonischer Reizung mit p = 0.0139
erreicht. Siehe hierzu Abbildung Nr. 17 und Tabelle 12 unten.
Tonischer Schmerz
Zeit [min]
0 50 100 150 200
VA
S [E
U]
-20
-15
-10
-5
0
PlaceboParacetamol Koffein
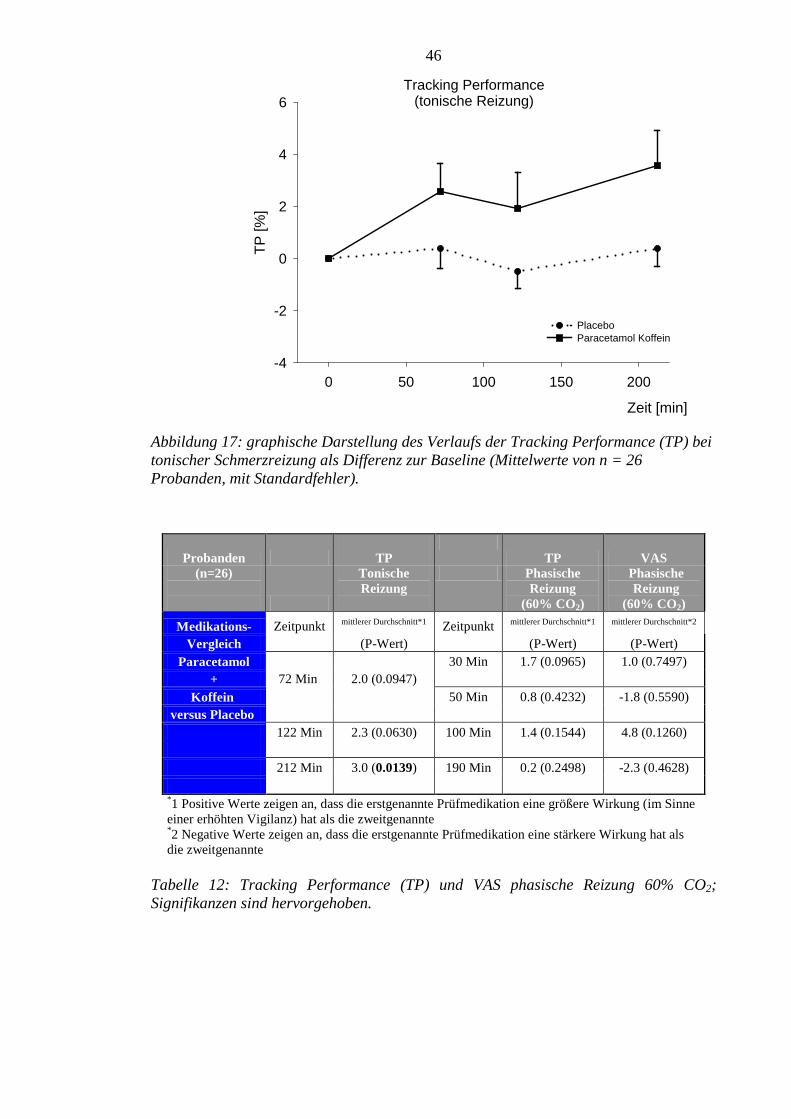
46
Abbildung 17: graphische Darstellung des Verlaufs der Tracking Performance (TP) bei tonischer Schmerzreizung als Differenz zur Baseline (Mittelwerte von n = 26 Probanden, mit Standardfehler).
Tabelle 12: Tracking Performance (TP) und VAS phasische Reizung 60% CO2; Signifikanzen sind hervorgehoben.
Probanden
TP
TP VAS (n=26)
Tonische Reizung
Phasische Reizung
(60% CO2)
Phasische Reizung
(60% CO2)
Medikations- Zeitpunkt mittlerer Durchschnitt*1 Zeitpunkt mittlerer Durchschnitt*1 mittlerer Durchschnitt*2 Vergleich (P-Wert) (P-Wert) (P-Wert)
Paracetamol 30 Min 1.7 (0.0965) 1.0 (0.7497) + 72 Min 2.0 (0.0947)
Koffein 50 Min 0.8 (0.4232) -1.8 (0.5590) versus Placebo
122 Min 2.3 (0.0630) 100 Min 1.4 (0.1544) 4.8 (0.1260) 212 Min 3.0 (0.0139) 190 Min 0.2 (0.2498) -2.3 (0.4628)
*1 Positive Werte zeigen an, dass die erstgenannte Prüfmedikation eine größere Wirkung (im Sinne einer erhöhten Vigilanz) hat als die zweitgenannte *2 Negative Werte zeigen an, dass die erstgenannte Prüfmedikation eine stärkere Wirkung hat als die zweitgenannte
Tracking Performance(tonische Reizung)
Zeit [min]
0 50 100 150 200
TP [%
]
-4
-2
0
2
4
6
PlaceboParacetamol Koffein
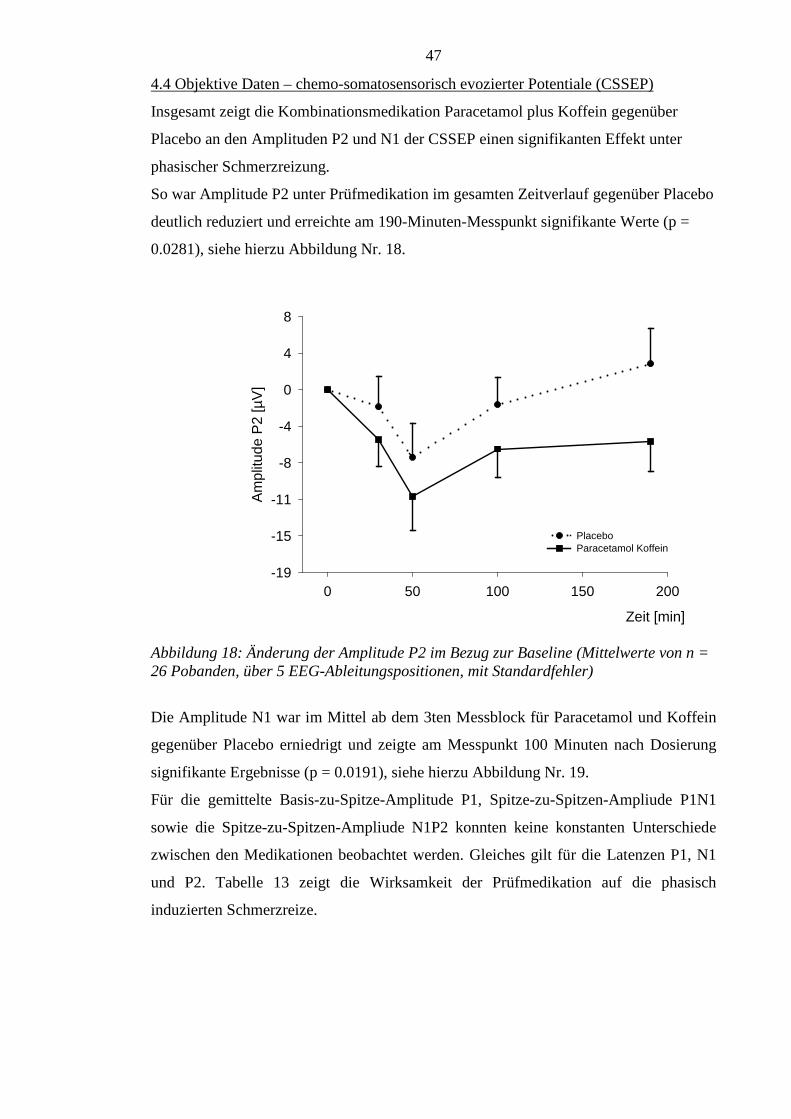
47
4.4 Objektive Daten – chemo-somatosensorisch evozierter Potentiale (CSSEP)
Insgesamt zeigt die Kombinationsmedikation Paracetamol plus Koffein gegenüber
Placebo an den Amplituden P2 und N1 der CSSEP einen signifikanten Effekt unter
phasischer Schmerzreizung.
So war Amplitude P2 unter Prüfmedikation im gesamten Zeitverlauf gegenüber Placebo
deutlich reduziert und erreichte am 190-Minuten-Messpunkt signifikante Werte (p =
0.0281), siehe hierzu Abbildung Nr. 18.
Abbildung 18: Änderung der Amplitude P2 im Bezug zur Baseline (Mittelwerte von n = 26 Pobanden, über 5 EEG-Ableitungspositionen, mit Standardfehler)
Die Amplitude N1 war im Mittel ab dem 3ten Messblock für Paracetamol und Koffein
gegenüber Placebo erniedrigt und zeigte am Messpunkt 100 Minuten nach Dosierung
signifikante Ergebnisse (p = 0.0191), siehe hierzu Abbildung Nr. 19.
Für die gemittelte Basis-zu-Spitze-Amplitude P1, Spitze-zu-Spitzen-Ampliude P1N1
sowie die Spitze-zu-Spitzen-Ampliude N1P2 konnten keine konstanten Unterschiede
zwischen den Medikationen beobachtet werden. Gleiches gilt für die Latenzen P1, N1
und P2. Tabelle 13 zeigt die Wirksamkeit der Prüfmedikation auf die phasisch
induzierten Schmerzreize.
Zeit [min]
0 50 100 150 200
Am
plitu
de P
2 [µ
V]
-19
-15
-11
-8
-4
0
4
8
PlaceboParacetamol Koffein
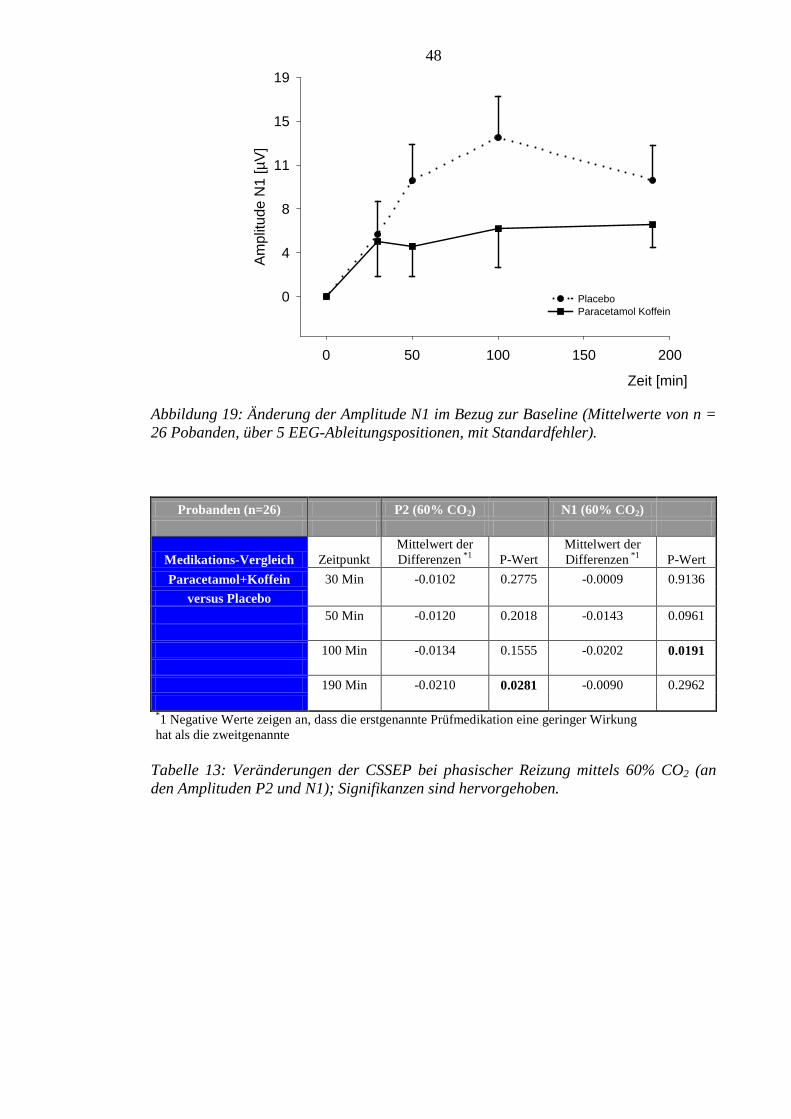
48
Abbildung 19: Änderung der Amplitude N1 im Bezug zur Baseline (Mittelwerte von n = 26 Pobanden, über 5 EEG-Ableitungspositionen, mit Standardfehler).
Probanden (n=26) P2 (60% CO2) N1 (60% CO2)
Medikations-Vergleich Zeitpunkt Mittelwert der Differenzen *1 P-Wert
Mittelwert der Differenzen *1 P-Wert
Paracetamol+Koffein 30 Min -0.0102 0.2775 -0.0009 0.9136 versus Placebo
50 Min -0.0120 0.2018 -0.0143 0.0961 100 Min -0.0134 0.1555 -0.0202 0.0191 190 Min -0.0210 0.0281 -0.0090 0.2962
*1 Negative Werte zeigen an, dass die erstgenannte Prüfmedikation eine geringer Wirkung hat als die zweitgenannte
Tabelle 13: Veränderungen der CSSEP bei phasischer Reizung mittels 60% CO2 (an den Amplituden P2 und N1); Signifikanzen sind hervorgehoben.
Zeit [min]
0 50 100 150 200
Am
plitu
de N
1 [µ
V]
0
4
8
11
15
19
PlaceboParacetamol Koffein
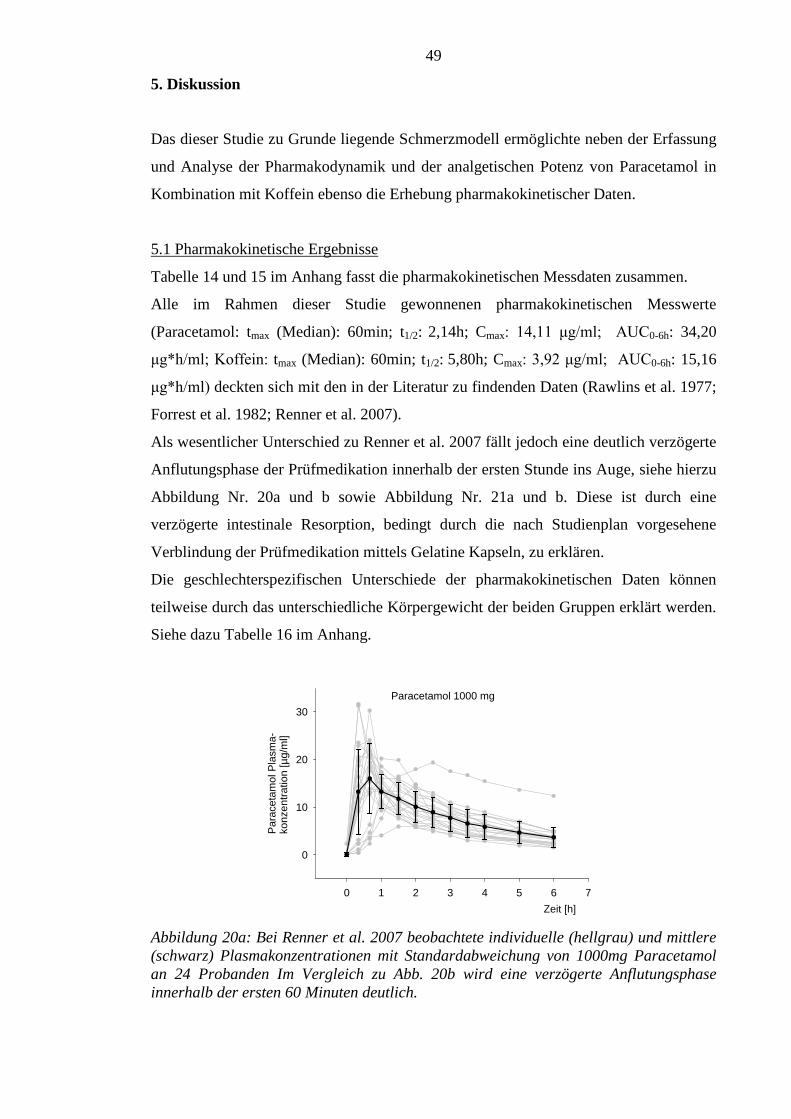
49
5. Diskussion
Das dieser Studie zu Grunde liegende Schmerzmodell ermöglichte neben der Erfassung
und Analyse der Pharmakodynamik und der analgetischen Potenz von Paracetamol in
Kombination mit Koffein ebenso die Erhebung pharmakokinetischer Daten.
5.1 Pharmakokinetische Ergebnisse
Tabelle 14 und 15 im Anhang fasst die pharmakokinetischen Messdaten zusammen.
Alle im Rahmen dieser Studie gewonnenen pharmakokinetischen Messwerte
(Paracetamol: tmax (Median): 60min; t1/2: 2,14h; Cmax: 14,11 μg/ml; AUC0-6h: 34,20
μg*h/ml; Koffein: tmax (Median): 60min; t1/2: 5,80h; Cmax: 3,92 μg/ml; AUC0-6h: 15,16
μg*h/ml) deckten sich mit den in der Literatur zu findenden Daten (Rawlins et al. 1977;
Forrest et al. 1982; Renner et al. 2007).
Als wesentlicher Unterschied zu Renner et al. 2007 fällt jedoch eine deutlich verzögerte
Anflutungsphase der Prüfmedikation innerhalb der ersten Stunde ins Auge, siehe hierzu
Abbildung Nr. 20a und b sowie Abbildung Nr. 21a und b. Diese ist durch eine
verzögerte intestinale Resorption, bedingt durch die nach Studienplan vorgesehene
Verblindung der Prüfmedikation mittels Gelatine Kapseln, zu erklären.
Die geschlechterspezifischen Unterschiede der pharmakokinetischen Daten können
teilweise durch das unterschiedliche Körpergewicht der beiden Gruppen erklärt werden.
Siehe dazu Tabelle 16 im Anhang.
Abbildung 20a: Bei Renner et al. 2007 beobachtete individuelle (hellgrau) und mittlere (schwarz) Plasmakonzentrationen mit Standardabweichung von 1000mg Paracetamol an 24 Probanden Im Vergleich zu Abb. 20b wird eine verzögerte Anflutungsphase innerhalb der ersten 60 Minuten deutlich.
Paracetamol 1000 mg
Zeit [h]0 1 2 3 4 5 6 7
Para
ceta
mol
Pla
sma-
konz
entra
tion
[µg/
ml]
0
10
20
30
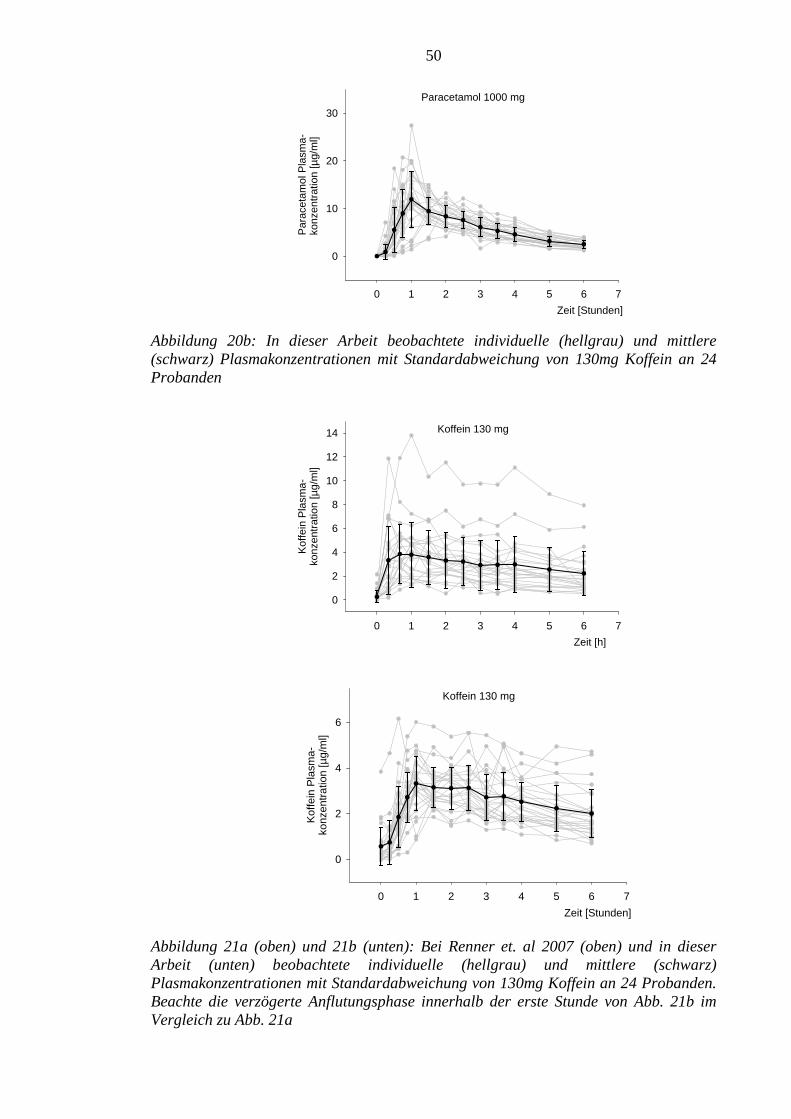
50
Abbildung 20b: In dieser Arbeit beobachtete individuelle (hellgrau) und mittlere (schwarz) Plasmakonzentrationen mit Standardabweichung von 130mg Koffein an 24 Probanden
Abbildung 21a (oben) und 21b (unten): Bei Renner et. al 2007 (oben) und in dieser Arbeit (unten) beobachtete individuelle (hellgrau) und mittlere (schwarz) Plasmakonzentrationen mit Standardabweichung von 130mg Koffein an 24 Probanden. Beachte die verzögerte Anflutungsphase innerhalb der erste Stunde von Abb. 21b im Vergleich zu Abb. 21a
Paracetamol 1000 mg
Zeit [Stunden]0 1 2 3 4 5 6 7
Para
ceta
mol
Pla
sma-
konz
entra
tion
[µg/
ml]
0
10
20
30
Koffein 130 mg
Zeit [h]0 1 2 3 4 5 6 7
Koffe
in P
lasm
a-ko
nzen
tratio
n [µ
g/m
l]
0
2
4
6
8
10
12
14
Koffein 130 mg
Zeit [Stunden]0 1 2 3 4 5 6 7
Koffe
in P
lasm
a-ko
nzen
tratio
n [µ
g/m
l]
0
2
4
6

51
5.2 Pharmakodynamische Effekte
Durch die Ergebnisse der vorliegenden Studie konnte dargelegt werden, dass die
Kombination von Paracetamol und Koffein in dem von uns verwendeten
Schmerzmodell analgetisch aktiv ist. Dies ließ sich unter anderem an den
Veränderungen der schmerzkorrelierten evozierten Potentiale (CSSEP) sowie an den
dazu positiv korrelierenden subjektiven Schmerzintensitätsschätzungen aufzeigen
(Kobal und Hummel 1988; Chen 1993).
Die Ausprägungen beider Parameter zeigten somit in der vorliegenden Studie eine
gleichartige Sensibilität gegenüber der analgetisch und antinozizeptiv wirksamen
Prüfmedikation. Dies ließ sich bereits in früheren Studien zeigen (Chen 1993; Kobal et
al. 1990b; Rohdewald et al. 1982).
5.2.1 Subjektive Schmerzintensitätsschätzung (VAS)
Wie erwartet und die Resultate mehrerer vorangegangener Studien bestätigend zeigte
sich kein signifikanter Effekt der Prüfmedikation auf die subjektive
Schmerzeinschätzung des phasischen Schmerzreizes. So konnte für mehrere
nichtsteroidale Analgetika wie Acetylsalicylsäure (Kobal et al. 1985), Propyphenazon
(Kraetsch et al. 1996), Ibuprofen (Hummel et al. 1997) und ebenso für Paracetamol
(Renner at al. 2003a, Renner et al. 2003b), welche ebenfalls mittels des hier
verwendeten Schmerzmodells untersucht wurden, eine signifikante Reduktion der
CSSEP-Amplituden, nicht jedoch der subjektiven Intensitätsschätzung, gezeigt werden.
Die dabei beobachtete deutlich größere Varianzbreite der subjektiven
Schmerzintensitätsschätzungen, verglichen mit den objektiven Messdaten, liegt offenbar
in ihrer Anfälligkeit auf unbeeinflussbare Faktoren, wie der aktuellen Gemütsverfassung
der Probanden, deren Voreingenommenheit sowie deren Bereitschaft auf schmerzhafte
Stimuli zu antworten, begründet (Kraetsch et al. 1996). Um dennoch die analgetische
Wirkung von Paracetamol mit Hilfe subjektiver Schmerzintensitätsschätzung zu
ermitteln, wäre demzufolge eine größere Anzahl an Probanden notwendig.
Im Gegensatz zur phasischen Schmerzintensitätsschätzung spiegelten sich dagegen die
in den CSSEP-Amplituden beobachteten analgetischen Effekte der Prüfmedikation in
den subjektiven Einschätzungen des tonischen Schmerzes wider. So konnte in unserer
Studie gezeigt werden, dass die Kombination von Paracetamol und Koffein, wie bereits
in einer aktuellen Studie beschrieben (Renner et al. 2007), zu einer signifikanten
Reduktion des tonisch induzierten Schmerzzustandes führte. Das beobachtete Ausmaß
des analgetischen Effektes war jedoch sowohl in Bezug auf die subjektiven, tonischen

52
Schmerzintensitätsschätzungen als auch, wie bereist weiter oben erwähnt, an den beiden
Ableitepositionen N1 und P2 der CSSEP ähnlich, nicht aber identisch. Dies könnte auf
mehrere Gründe zurückzuführen sein: Zum einen lag beiden Studien ein anderes
Probandenkollektiv zu Grunde, zum anderen gab es deutliche Unterschiede innerhalb
des Aufbaus und des Ablaufs der phasischen Schmerzreizung. So waren nicht nur die
Reizanzahl an CO2-Stimuli insgesamt sowie deren Interstimuluszeiten verschieden,
sondern auch Reizabfolge und insbesondere Anzahl an 60% und 70% CO2-Reizen
waren andere. Renner et al. verwendeten innerhalb der einzelnen Messblöcke deutlich
mehr 70% CO2-Stimuli (z.B. Messblock 2 mit insgesamt 64 x 70% CO2 Stimuli gegen
40 x 70% CO2 in der vorliegenden Studie). Es ist somit nicht sicher auszuschließen,
dass die phasische Reizung mit CO2 auch die tonische Schmerzreizung bzw. deren
subjektive Schätzungen beeinflusst.
Die unterschiedlichen Effekte der Prüfmedikation Paracetamol plus Koffein auf die
Intensitätsschätzungen des tonischen und phasischen Schmerzreizes sind offensichtlich
auf die zugrunde liegenden unterschiedlichen Schmerz verarbeitenden Systemen zurück
zu führen. Es wird vermutet, dass der tonische, lang anhaltende, einen entzündlichen
Zustand imitierende Schmerz überwiegend über neuronale C-Fasern verarbeitet bzw.
weitergeleitet wird, wohingegen der phasische, kurz anhaltende und nicht entzündliche
Schmerzstimulus v. a. über Adelta -Fasern vermittelt zu werden scheint (Hummel et al.
2003; Lucier und Egizii 1989). Dies wird durch eine kürzlich von Hinz und Mitarbeitern
(Hinz et al. 2007) publizierte Studie unterstützt. Hinz et al. konnten zeigen, dass
Paracetamol neben seiner analgetischen Aktivität im Gegensatz zu bisherigen
Erkenntnissen zudem durch Hemmung der Cyclooxygenase-2 (COX-2) eine
antiinflammatorische Wirkung zeigt. Auf diese Weise könnte Paracetamol einen Effekt
auf die nachweislich durch Entzündungsmediatoren beeinflussbare Sensibilität von C-
Fasern (Millan 1999) ausüben.
5.2.2 Tracking Performance (TP) – Einflüsse auf die Vigilanz
Sowohl für die phasische als auch die tonische Schmerzreizung konnte eine Erhöhung
der Vigilanz, im Sinne einer gesteigerten TP, der Probanden unter Paracetamol und
Koffein im Vergleich zu Placebo beobachtet werden. Deutlich signifikante Unterschiede
zeigten sich jedoch nur während der tonischen Stimulation. Dieses Ergebnis war
erwartet und reiht sich in die Ergebnisse vorangegangener Studien ein (Renner et al.
2003a; Renner et al. 2007).

53
Positive Einflüsse auf diesen Vigilanzparameter zeigten sich neben Kombination von
Paracetamol mit Koffein auch bei Verbindungen von Propyphenazon mit Koffein
(Kraetsch et al. 1996). Als ursächlich für die Steigerung der Vigilanz ist der weiter oben
erläuterte Wirkmechanismus des Koffeins anzusehen (Lieberman et al. 1987).
5.2.3 Schmerzkorrelierte evozierte Potentiale (CSSEP)
Eine Abnahme der Amplituden der schmerzkorrelierten evozierten Potentiale nach
Gabe von Analgetika (Chen und Chapman 1980) wird als abhängig angesehen zum
einen von der verabreichten Dosis (Rohdewald et al. 1982), zum anderen von der
analgetischen Potenz der applizierten Substanz (Bromm 1985). So konnten u. a. Renner
et al. (Renner at al. 2003b) zeigen, dass die Applikation von Paracetamol 1000mg zu
einer signifikanten Reduktion der Amplitude der CSSEP führte, wohingegen keine
signifikanten Effekte bei Dosierungen von 500mg Paracetamol beobachtet werden
konnten.
Die Applikation von 1000mg Paracetamol plus 130mg Koffein bewirkte im Vergleich
zu Placebo in unserer Studie eine signifikante Erniedrigung der auf eine schmerzhafte
Reizung der Nasenschleimhaut induzierten Basis-zu-Spitzen-Amplitude P2 und N1 der
schmerzkorrelierten evozierten Potentiale, wohingegen keinerlei signifikante
Veränderungen auf die Amplitude P1N1 beobachtet werden konnten. Dieses Resultat
bestätigt das vorangegangener Studien, bei denen ebenfalls die Auswirkung von
Paracetamol plus Koffein vor allem auf die späte Komponente P2 beobachtet werden
konnte (Renner et al. 2007).
In einer anderen Studie konnten Renner et al. (Renner et al. 2003b) durch die alleinige
Applikation von 1000mg Paracetamol eine signifikante Abnahme der Amplitude P1N1
zeigen sowie eine jedoch nicht signifikante Abnahme bei Gabe von 500mg Paracetamol.
Dies mag zum einen auf die den Studien zugrunde liegenden jeweils unterschiedlichen
Probandenkollektive zurück zu führen sein, zum anderen aber auch auf den Aufbau
bzw. die verschiedenartigen Komponenten der chemo-somatosensibel-evozierten
Potentiale (CSSEP). Das CSSEP zeigt wie bereits oben erläutert (Abschnitt 3.7.1) mit
seinen verschiedenen Einzelkomponenten eine charakteristische Form, wobei die
Amplitude P1N1 als so genannte „frühe“ Komponente und die Amplitude P2 als
„spätere“ Komponente anerkannt sind. P1N1 wird dabei als direkte Antwort auf den
Schmerzreiz, d. h. als reizkorreliert betrachtet. Demgegenüber werden die späteren
Anteile, wie z. B. P2, als Ausdruck der assoziativen bzw. endogenen Verarbeitung des
Schmerzreizes angesehen, die Bezug zu den subjektiven Intensitätseinschätzungen

54
zeigen (Chen et al. 1979, Picton und Hillyard 1988; Kobal und Hummel 1991). Diese
Begebenheit lässt sich auch in anderen experimentellen Schmerzmodellen nachweisen
(Miyazaki et al. 1994).
Da Koffein, wie Ward et al. (Ward et al. 1991) zeigen konnten, in die Stimmungs- und
Gefühlswelt eingreift, ließe sich vermuten, dass es somit auch einen Einfluss auf die
kognitive bzw. endogene Verarbeitung von Schmerzreizen ausübt und auf diese Weise
zu einer Veränderung der Amplitude P2 beiträgt. Die Ergebnisse von Renner (Renner et
al. 2007) zeigen jedoch, dass Koffein alleine nur zu Beginn (Anflutungsphase) sonst
jedoch zu keiner signifikanten Reduktion der Amplitude P2 führt, wohingegen die
Kombination von Paracetamol mit Koffein P2 in stärkerem Maße beeinflusst als
Paracetamol alleine. Dieser Summationseffekt konnte von Renner nicht nur bei den
CSSEP, sondern auch bei den subjektiven Schmerzintensitätsschätzungen (VAS)
gezeigt werden (s. u.).
Als Generationszentren der durch Stimulation mittels CO2-Gas induzierten chemo-
somatosensibel-evozierte Potentiale (CSSEP) wird nach derzeitigem Stand des Wissens
die Area II des somatosensorischen Kortex (Huttunen et al. 1986), die als primäres
Projektionsfeld für nozizeptive Efferenzen gilt (Chudler et al. 1985), angesehen.
In diesem Experiment wurde CO2-Gas in zwei verschiedenen Reizstärkeklassen von
60% und 70% CO2-Gas-Anteil in Luft zur schmerzhaften Stimulation der
Nasenschleimhaut verwandt. Dies war, wie bereits erwähnt, zum einen notwendig, um
durch Verwendung zweier unterschiedlicher Reizstärken die Verzerrung des
Ergebnisses durch sog. „Response Bias“ (’Antwort-Voreingenommenheit’) von Seiten
des Probanden möglichst gering zu halten. Zum anderen bot dies zudem die
Möglichkeit, eventuelle Rückschlüsse hinsichtlich der Veränderung der CSSEP zu
ziehen. In der vorliegenden Arbeit konnte ein signifikanter, antinozizeptiver Effekt, d. h.
eine Verminderung der Basis-zu-Spitzen-Amplitude P2, lediglich bei den schwächeren,
60%igen CO2-Reizen beobachtet werden. Dieses Resultat ist jedoch gut in
Übereinstimmung zu bringen mit in der Literatur zu findenden Ergebnissen
vorangegangener Studien. So konnte bei Untersuchungen von Acetylsalicylsäure (Kobal
et al. 1990b) und Azapropazon (Lötsch et al. 1995b) mittels des gleichen
Schmerzmodells ebenfalls eine signifikante Reduktion der Amplitude der CSSEP auf
den schwächeren Schmerzreiz gesehen werden.

55
5.3 Schlussfolgerung
Die Ergebnisse der vorliegenden, im Rahmen eines Arzneimittelprüfungverfahrens nach
dem neuen Arzneimittelgesetz (AMG 12. Novelle, 2004) und nach GCP-Kriterien
(Good Clinical Practice) durchgeführten Studie, lassen mehrere Schlussfolgerungen zu:
• Es konnte gezeigt werden, dass das experimentelle Schmerzmodell, welches auf der
Erhebung und Analyse objektiver, chemo-somatosensorisch evozierter Potentiale
sowie subjektiver Schmerzintensitätsschätzungen basiert, geeignet ist für die
analgesimetrische Untersuchung der Schmerzmittelkombination Paracetamol +
Koffein und unter den aktuell gültigen Kriterien einer klinischen Prüfung technisch
realisierbar ist.
• Die Veränderungen der EEG Parameter P2 und N1 zeigen, dass Paracetamol +
Koffein im Vergleich zu Placebo eine analgetische, früh- und spät-kognitive und
antinociceptive Wirkung aufweist. Unsere Ergebnisse bestätigen die Resultate
früherer Studien mit diesem Schmerzmodell.
• Die Resultate der Intensitätsschätzungen während der tonischen Applikation
trockener Luft in die Nasenhöhle zeigten eine Überlegenheit der Kombination
Paracetamol + Koffein gegenüber Placebo.
• Die Kombination von Paracetamol mit Koffein erhöhte im Vergleich zu Placebo die
Vigilanz der Probanden, was sich in den Veränderungen der Tracking Performance
Daten widerspiegelt. Auch hier befinden sich unsere Daten, jedoch nicht mit
derselben Deutlichkeit, in Einklang mit dem aktuellen Stand der Literatur.
• Die in unserer Arbeit erhobenen pharmakokinetischen Daten der Prüfmedikation
Paracetamol + Koffein sind mit denen vorangegangener Studien vergleichbar (mit
Ausnahme der Anflutungsphase).

56
6. Literaturverzeichnis
1. Aguirre-Banuelos P, Castaneda-Hernandez G, Lopez-Munoz FJ, Granados-Soto V
(1999): Effect of coadministration of caffeine and either adenosine agonists or cyclic nucleotides on ketorolac analgesia. Eur J Pharmacol 377: 175-182
2. Aktories K, Förstermann U, Hofmann F, Starke K; Allgemeine und spezielle
Pharmakologie und Toxikologie, 9. Auflage, Urban und Fischer Verlag, München, 2005: 188, 240
3. Alloui A, Chassaing C, Schmidt J, Ardid D, Dubray C, Cloarec A, Eschalier A
(2002): Paracetamol exerts a spinal, tropisetron-reversible, antinociceptive effect in an inflammatory pain model in rats. Eur J Pharmacol 443(1-3): 71-7
4. Alloui A, Pelissier T, Dubray C, Lavarenne J, Eschalier A (1996): Tropisetron
inhibits the antinociceptive effect of intrathecally administered paracetamol and serotonin. Fundam Clin Pharmacol 10(4): 406-7.
5. AMG §40: http://bundesrecht.juris.de/amg_1976/BJNR024480976.html 6. Ammon HPT, Bieck PR, Mandalaz D, Verspohl EJ (1983): Apation of blood
pressure to continuous heavy coffee drinking in young volunteers. A double-blind crossover study. Br J Clin Pharmacol 15: 701-706
7. Arendt-Nielsen L, Nielsen JC, Bjerring P (1991): Double-blind, placebo controlled
comparison of paracetamol and paracetamol plus codeine - a quantitative evaluation by laser induced pain. Eur J Clin Pharmacol 40: 241-247
8. Arnaud MJ: Products of metabolism of caffeine. In Caffeine: Perspectives
from Recent Research Springer-Verlag, New York, 1983, 3-32 9. Arnaud MJ (1987): The pharmacology of caffeine. Prog Drug Res 31: 273-313 10. Axelrod J, Reichenthal J (1982): The fate of caffeine in man and a method for its
estimaion in biological material. J Pharmac Exp Therap 107: 519-523 11. Beisel A, Untersuchung der analgetischen Wirksamkeit von Paracetamol in
Kombination mit Coffein; Med. Diss., Universität Erlangen/Nürnberg, 2006: 32 12. Benowitz NL, Jakob PIII, Mayan H and Denaro C (1995): Sympothomimetic effects
of Paraxanthine and caffeine in humans. Clin Pharmacol Ther 58: 684-69 13. Bentley KC, Head TW (1987): The additive analgesic efficacy of acetaminophen,
1000 mg, and codeine, 60 mg, in dental pain. Clin Pharmacol Ther 42(6): 634-640 14. Beubler E, Lembeck P (1976): Methylxanthines and intestinal drug absorption.
Naunym Schmiedebergs Arch Pharmacol 292: 73-77 15. Bevan S, Yeats J (1991): Protons activate a cation conductance in a sub-population
of rat dorsal root ganglion neurones. J Physiol 433: 145-161.

57
16. Bjornsson GA, Haanaes HR, Skoglund LA (2003): A randomized, double-blind crossover trial of paracetamol 1000 mg four times daily vs ibuprofen 600 mg: effect on swelling and other postoperative events after third molar surgery. Br J Clin Pharmacol 55: 405–412
17. Boardman PL, Hart FD (1967): Clinical measurement of the anti-inflammatory
effects of salicylates in rheumatoid arthritis. BMJ 4: 264–268 18. Bonati M, Latini R, Galletti F, Young JF, Tognoni G, Garattini S (1982): Caffeine
disposition after oral doses. Clin Pharmacol Ther 32(1): 98-106 19. Bonica JJ (1979): The need of a taxonomy. Pain 6(3): 247-248 20. Bromm B (1985): Modern techniques to measure pain in healthy man. Methods
Find Exp Clin Pharmacol 7(3): 161-169 21. Brodie BB, Axelrod J (1948): The fate of acetanilide in man. J Pharmacol Exp Ther.
94(1): 29-38. 22. Brune K, Egger T (2002): Weltweit umsatzstärkste Arzneimittelgruppe. Die
Entwicklung der antipyretischen Analgetika. Pharm Unserer Zeit 31(2): 133-9 23. Brune K, Niederweis U (2007): Von der Weidenrinde zu den Coxiben. Entwicklung
der antiphlogistischen Analgetika. Schmerz 21(4): 318-330 24. Butler MA, Iwasaki M, Guengerich FP, Kadlubar FF (1989): Human cytochrome P-
450PA (P-450IA2), the phenacetin-o-deethylase, is primarily responsible for the hepatic 3-methylation of caffeine and N-oxidation of cardinogenic arylamines. Proc Nat Acad Sci USA 86: 7696-7790
25. Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B,
Vyas SN, Fitz Gerald GA (2001): Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med 345: 1809–1817
26. Chan AT, Manson JE, Albert CM, Chae CU, Rexrode KM, Curhan GC, Rimm, EB,
Willett WC, Fuchs CS (2006): Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk of cardiovascular events. Circulation 113: 1578–1587
27. Chandrasekharan NV, Dai H, Roos KL (2002): COX-3, a cyclooxygenase-1 variant
inhibited by acetaminophen and other analgesic / antipyretic drugs: cloning, structure and expression. Proc Natl Acad Sci 99: 13926-13931
28. Chen AC (1993): Human brain measures of clinical pain: a review. I. Topographic
mappings. Pain 54(2): 115-132 29. Chen AC, Chapman CR (1980): Aspirin analgesia evaluated by event-related
potentials in man: possible central action in brain. Exp Brain Res 39(4): 359-364 30. Chen AC, Chapman CR, Harkins SW (1979): Brain evoked potentials are functional
correlates of induced pain in man. Pain 6(3): 365-374

58
31. Chudler EH, Dong WK, Kawakami Y. (1985): Tooth pulp-evoked potentials in the monkey: cortical surface and intracortical distribution. Pain 22(3): 221-233.
32. Chvasta TE; Cook AR (1971): Emptying and absorption of caffeine from the
human stomach. Gastroenterology 61: 838-843 33. Clements JA, Heading RC, Nimmo WS, Prescott LF (1978): Kinetics of
acetaminophen absorption and gastric emptying in man. Clin Pharmacol Ther 24(4): 420-431
34. Courade JP, Caussade F, Martin K, Besse D, Delchambre C, Hanoun N, Hamon M,
Eschalier A, Cloarec A (2001): Effects of acetaminophen on monoaminergic systems in the rat central nervous system. Naunyn Schmiedebergs Arch Pharmacol 364(6): 534-537
35. Daly JW (1982): Adenosine receptors: target sites for drugs. J Med Chem 25: 197-
207 36. Dobmeyer DJ, Stine RA, Leier CV, Greenberg R, Schaal SF (1983): The
arrhythmogenic effects of caffeine in human beings. N Engl J Med 308: 814-816 37. Dorfman LJ, Jarvik ME (1970): Comparative stimulant and diuretic actions of
caffeine and theobromine in man. Clin Pharmacol Ther 11(6): 869-872 38. Eandi M, Viano I, Gamalero SR (1984):Absolute bioavailability of paracetamol
after oral or rectal administration in healthy volunteers. Arzneimittelforschung 34: 903-907
39. Forman JP, Stampfer MJ, Curhan GC (2005): Non-narcotic analgesic dose and risk
of incident hypertension in US women. Hypertension 46: 500–507 40. Forrest JAH, Clements JA, Prescott LF (1982): Clinical Pharmacokinetics of
Paracetamol Clin Pharmacokinet 7: 93-107 41. Flower RJ, Vane JR (1972): Inhibition of prostaglandin synthetase in brain explains
the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature 240: 410-411 42. Fredholm BB (1980): Are methylxanthine effects due to antagonism of endogenous
adenosine? Trends Pharmacol Sci 1: 129-132 43. Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden KT, Jacobson KA,
Leff P, Williams M (1994): VI. Nomenclature and classification of purinoceptors. Pharmacol Re. 46: 143–156
44. Garcia Rodriguez, LA, Hernandez-Diaz S (2001): Relative risk of upper
gastrointestinal complications among users of acetaminophen and nonsteroidal anti-inflammatory drugs. Epidemiology 12: 570–576
45. Gerber JG, Nies AS, Payne NA (1985): Adenosine receptors on canine parietal cells
modulate gastric acid secretion to histamine. J Pharmacol Exp Ther 233: 623-627

59
46. Gertzbein SD, Tile M, McMurty RY, Kellam JF, Hunter GA, Keith RG, Harsanyi Z, Luffman J (1986): Analysis of the analgesic efficacy of acetaminophen 1000 mg, codeine phosphate 60 mg, and the combination of acetaminophen 1000 mg and codeine phosphate 60 mg in the relief of postoperative pain. Pharmacotherapy 6(3): 104-107
47. Glissold SP (1986): Paracetamol and Phenacetin. Drugs 32(4): 46-59 48. Gong H, Simmons MS, Tashkin DP, Hui KK, Lee EY (1986): Bronchodilator
effects of caffeine in coffee. A dose-response study of asthmatic subjects. Chest 89: 335-342
49. Grab FL, Reinstein JA (1968): Determination of caffeine in plasma by gas chromatography. J Pharm Sci 57: 1703-1706
50. Greenberg JA, Boozer CN, Geliebter A (2006): Coffee, diabetes, and weight
control. Am J Clin Nutr 84(4): 682-93 51. Griffiths RR, Evans SM, Heishman SJ (1990): Low-dose caffeine physical
dependence in humans. J Pharm Exp Ther 255: 1123-1132 52. Griffiths RR, Woodson PP (1988): Caffeine physical dependence: a review of
human and laboratory animal studies. Psychopharmacology 94: 437-451 53. Gursoy M, Haznedaroglu IC, Celik I, Sayinalp N, Ozcebe OI, Dundar SV (1996):
Granulocytosis, and thrombocytosis followed by a leukemoid reaction due to acute acetaminophen toxicity. Ann Pharmacother 30: 762-765
54. Haas H (1983): History of Antipyretic Analgesic Therapy. Am. J. Med. 75(5A): 1-3 55. Hinz B, Cheremina O, Brune K (2008): Acetaminophen (paracetamol) is a selective
cyclooxygenase-2 inhibitor in man. FASEB J 22(2): 383-90 56. Hinz B, Dormann H, Brune K (2006): More pronounced inhibition of
cyclooxygenase 2, increase in blood pressure, and reduction of heart rate by treatment with diclofenac compared with celecoxib and rofecoxib. Arthritis Rheum 54: 282–291
57. Honore P, Buritova J, Besson JM (1995): Aspirin and acetaminophen reduced both
Fos expression in rat lumbar spinal cord and inflammatory signs produced by carrageen in inflammation. Pain 63: 365–375
58. Hummel T, Cramer O, Mohammadian P, Geisslinger G, Pauli E, Kobal G (1997):
Comparison of the antinociception produced by two oral formulations of ibuprofen: ibuprofen effervescent versus ibuprofen tablets. Eur J Clin Pharmacol 52(2): 107-114
59. Hummel T, Gruber M, Pauli E, Kobal G. (1994a): Chemo-somatosensory event-
related potentials in response to repetitive painful chemical stimulation of the nasal mucosa. Electroencephalogr Clin Neurophysiol 92(5): 426-432.

60
60. Hummel T, Huber H, Kobal G (1995a): Comparison of analgesic effects of ibuprofen-lysinate and ibuprofen-acid. Pharmacology Communications 5(2): 101-108.
61. Hummel T, Huber H, Menzel S, Kobal G. (1995b): Tonic versus phasic pain: dose-
related effects of ketoprofen. Eur J Clin Pharmacol. 49(1-2):7-14. 62. Hummel T, Hummel C, Friedel I, Pauli E, Kobal G. (1994b): A comparison of the
antinociceptive effects of imipramine, tramadol and anpirtoline. Br J Clin Pharmacol 37(4): 325-333.
63. Hummel T, Kobal G (1999): Chemosensory event-related potentials to trigeminal
stimuli change in relation to the interval between repetitive stimulation of the nasal mucosa. Eur Arch Otorhinolaryngol 256(1): 16-21.
64. Hummel T, Kraetsch HG, Lötsch J, Hepper M, Liefhold J, Kobal G. (1995c):
Analgesic effects of dihydrocodeine and tramadol when administered either in the morning or evening. Chronobiol Int 12(1): 62-72.
65. Hummel T, Mohammadian P, Marchl R, Kobal G, Lotsch J (2003): Pain in the
trigeminal system: irritation of the nasal mucosa using short and long-lasting stimuli. Int J Psychophysiol 47: 147-158.
66. Huttunen J, Kobal G, Kaukoranta E, Hari R (1986): Cortical responses to painful
CO2 stimulation of nasal mucosa; a magnetoencephalographic study in man. Electroencephalogr Clin Neurophysiol 64(4): 347-349.
67. ICH Topic 6 Guideline for Good Clinical Practice CPMP/ICH/135/95 17th July 1996.
68. Iqbal N, Ahmad B, Janbaz KH, Gilani AU, Niazi SK (1995): The effect of caffeine on the pharmacokinetics of acetaminophen in man. Biopharm Drug Dispos 16: 481-487
69. Jaeschke H, Bajt ML (2006): Intracellular signaling mechanisms of acetaminophen
induced liver cell death. Toxicol Sci 89: 31–41 70. Jenkins C, Costello J; Hodge L (2004): Systematic review of prevalence of aspirin
induced asthma and its implications for clinical practice. BMJ 328: 434 71. Kaplan GB, Greenblatt DJ, Ehrenberg BL, Goddard JE, Cotreau MM, Harmatz JS,
Shader RI (1997): Dose-dependent pharmacokinetics and psychomotor effects of caffeine in humans. Journal of Clinical Pharmacology 37: 693–703
72. Kis B, Snipes JA, Busija DW (2005): Acetaminophen and the cyclooxygenase-3
puzzle: sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther 31: 1–7 73. Kobal G: Elektrophysiologische Untersuchungen des menschlichen Geruchssinns.
Thieme, Stuttgart, 1981 74. Kobal G (1985): Pain-related electrical potentials of the human nasal mucosa
elicited by chemical stimulation. Pain 22(2): 151-163.

61
75. Kobal G, Hummel C (1988): Cerebral chemosensory evoked potentials elicited by chemical stimulation of the human olfactory and respiratory nasal mucosa. Electroencephalogr Clin Neurophysiol 71(4): 241-250.
76. Kobal G, Hummel C, Hoesl M (1990a): Pain related electrical evoked potentials by
chemical stimuli effects of fentanyl. Advances in Pain Research and Theapy 13: 95-98.
77. Kobal G, Hummel C, Gruber M, Geisslinger G, Hummel T. (1994): Dose-related
effects of ibuprofen on pain-related potentials. Br J Clin Pharmacol. 37(5):445-52. 78. Kobal G, Hummel C, Nuernberg B, Brune K (1990b): Effects of pentazocine and
acetylsalicylic acid on pain-rating, pain-related evoked potentials and vigilance in relationship to pharmacokinetic parameters. Agents Actions 29(3-4): 342-359.
79. Kobal G, Hummel T (1991): Olfactory evoked potentials in humans. In: Smell and
Taste in Health and Disease. Raven Press, New York, 255-275 80. Kobal G, Hummel T (1992): Human Electro-olfactograms and Brain Responses to
Olfactory Stimulation. In: The human sense of smell, Springer, Berlin 135-151. 81. Kobal G, Kamp HD, Brunner M (1986): Analgesimetrie mit Hilfe
schmerzkorrelierter evozierter Potentiale während der Narkose. Anaesthesiologie und Intensivmedizin, Anaestesiology and Intensive Care 194: 40-45.
82. Kobal G, Plattig KH (1978): Methodische Anmekungen zur Gewinnung
olfaktorischer EEG-Antworten des wachen Menschen (objektive Olfaktometrie). Zeitschrift für Elektroenzephalographie, Elektromyographie und verwandte Gebiete 9(3): 135-145.
83. Komai M, Bryant BP (1993): Acetazolamide specifically inhibits lingual trigeminal
nerve responses to carbon dioxide. Brain Res 612(1-2): 122-129. 84. Kraetsch HG, Hummel T, Lötsch J, Kussat R, Kobal G (1996): Analgesic effects of
propyphenazone in comparison to its combination with caffeine. Eur J Clin Pharmacol 49(5): 377-382.
85. Krasnow S, Grossman MI (1949): Stimulation of gastric secretion in man by
theophylline ethylenediamine. Proc Soc Exp Biol Med 71: 335-336 86. Lanz R, Polster P, Brune K (1986): Antipyretic analgesics inhibit prostaglandin
release from astrocytes and macrophages similarly. Eur J Pharmacol 130: 105–109 87. Lee WM (2004): Acetaminophen and the U.S Acute Liver Failure Study Group:
lowering the risks of hepatic failure. Hepatology 40: 6–9 88. Lelo A, Miners JO, Robson R, Birkett DJ (1986a): Quantitative assessment of
caffeine partial clearences in man. Br J Clin Parmacol 22: 183-186 89. Lelo A, Miners JO, Robson R, Birkett DJ (1986b): Assessment of caffeine
exposure: Caffeine content of beverages, caffeine intake and plasma concentration of methylxanthines. Clin Pharmacol Ther 39: 54-59

62
90. Libert F, Bonnefont J, Bourinet E, Doucet E, Alloui A, Hamon M, Nargeot J, Eschalier A (2004): Acetaminophen: a central analgesic drug that involves a spinal tropisetron-sensitive, non-5-HT(3) receptor-mediated effect. Mol Pharmacol 66(3): 728-34
91. Lieberman HR,Wurtman RJ, Emde GG, Roberts C, Coviella ILG (1987): The
effects of low doses of caffeine on human perforromance and mood. Psychopharmacology 92: 308-312
92. Lötsch J, Ahne G, Kunder J, Kobal G, Hummel T (1998): Factors affecting pain
intensity in a pain model based upon tonic intranasal stimulation in humans. Inflamm Res 47(11): 446-450.
93. Lötsch J, Ditterich W, Hummel T, Kobal G (1997a): Antinociceptive effects of the
kappa-opioid receptor agonist RP 60180 compared with pentazocine in an experimental human pain model. Clin Neuropharmacol 20(3): 224-233.
94. Lötsch J, Geisslinger G, Mohammadian P, Brune K, Kobal G (1995a): Effects of
flurbiprofen enantiomers on pain-related chemo-somatosensory evoked potentials in human subjects. Br J Clin Pharmacol 40(4): 339-346.
95. Lötsch J, Mohammadian P, Hummel T, Florin S, Brune K, Geisslinger G et al.
(1995b): Effects of azapropazone on pain-related brain activity in human subjects. Br J Clin Pharmacol 40(6): 545-552.
96. Lopez-Munoz FJ, Castaneda-Hernandez G, Flores-Murrieta FJ, Granados-Soto V
(1996): Effect of caffeine coadministration and of nitric oxide synthesis inhibition on the antinociceptive action of ketorolac. Eur J Pharmacol 308: 275-277
97. Lucier GE, Egizii R (1989): Characterization of cat nasal afferents and brain stem
neurones receiving ethmoidal input. Exp Neurol 103: 83-89 98. Machelska H, Przewlocki R, Radomski MW, Przewlocka B (1998): Differential
effects of intrathecally and intracerebroventricularly administered nitric oxide donors on noxious mechanical and thermal stimulation. Pol J Pharmacol 50: 407-415
99. Mandel HG (2002): Update on caffeine consumption, disposition and action. Food
Chem Toxicol 40: 1231-1234 100. Massey LK, Wise KJ (1984): The effect of dietary caffeine on urinary excretion
of calcium, magnesium, sodium and potassium in healthy young females. Nutr Res 4: 43-50
101. Mathew RJ, Wilson WI (1985): Caffeine induced changes in cerebral
circulation. Stroke 16: 814-17 102. Mc Gilveray IJ and Mattok GL (1972): Some factors affecting the absorption of
paracetamol, J Pharm Pharmacol 24(8): 615-619 103. Mielke CH Jr (1981): Comparative effects of aspirin and acetaminophen on
hemostasis. Arch. Intern. Med. 141: 305–310

63
104. Millan MJ (1999): The induction of pain: an integrative review. Prog Neurobiol,
57: 1-164. 105. Miyazaki M, Shibasaki H, Kanda M, Xu X, Shindo K, Honda M et al.
(1994): Generator mechanism of pain-related evoked potentials following CO2 laser stimulation of the hand: scalp topography and effect of predictive warning signal. J Clin Neurophysiol 11(2): 242-254.
106. Mohammadian P, Hummel T, Loetsch J, Kobal G (1997): Bilateral hyperalgesia
to chemical stimulation of the nasal mucosa following unilateral inflammation. Pain 73(3): 407-412.
107. Munsterhjelm E, Munsterhjelm NM, Niemi TT, Ylikorkala O, Neuvonen PJ,
Rosenberg PH (2005): Dose-dependent inhibition of platelet function by acetaminophen in healthy volunteers. Anesthesiology 103: 712–717
108. Murat I, Moriette G, Blin MC, Couchard M, Flouvat B (1981): The efficacy of
caffeine in the treatment of recurrent idiopathic apnea in premature infants. J Pediatr 99: 984-989
109. Neuwald S R P, Pharmakokinetische und pharmakodynamische Effekte von
Etoricoxib und Valdecoxib im Vergleich zu Placebo im experimentellen Schmerzmodell; Med. Diss., Universität Erlangen/Nürnberg, 2007: 43, 46
110. Nieme TT, Backman JT, Syrja¨la¨ MT, Vinikka LU, Rosenberg PH (2000):
Platelet dysfunction after intravenous ketorolac or propacetamol. Acta Anaesthesiol Scand 44: 69–74
111. Onrot J, Shaheen O, Biaggione I, Goldberg MR, Feely J, Wilkinson GR,
Hollister S, Robertson D (1986): Reduction of liver plasma flow by caffeine and theophylline. Clin Pharmacol Ther 40: 506-510
112. Pelissier T, Alloui A, Paeile C, Eschalier A (1995): Evidence of a central
antinociceptive effect of paracetamol involving spinal 5HT3 receptors. Neuro Report 6: 1546-1548
113. Phillis JW, Wu PH (1981): The role of adenosine and its nucleotides in central synaptic transmission. Frog Neurobiol 16: 187-239
114. Pini LA, Sandrini M, Vitale G (1996): The antinociceptive action of paracetamol
is associated with changes in the serotonergic system in the rat brain. Eur J Pharmacol 308: 31-40
115. Pickering G, Loriot MA, Libert F, Eschalier A, Beaune P, Dubray C (2006):
Analgesic effect of acetaminophen in humans: first evidence of a central serotonergic mechanism. Clin Pharmacol Ther 79(4): 371-8
116. Picton TW, Hillyard SA: Endogenous event-related potentials. In: Picton T.W.
EEG-Handbook Revised series, Volume 3 Elsevier, Amsterdam, 1988, 361-426

64
117. Prescott LF (1992): The hepatotoxicity of non-steroidal anti-inflammatory drugs. In: Side-effects of Anti-inflammatory Drugs 3: Inflammation and Drug Therapy, edited by Rainsford KD, Velo GP Lancaster (UK): Kluwer Academic Publisher 176-187
118. Qin N, Zhang, SP, Reitz, TL, Mei JM, Flores CM (2005): Cloning, expression,
and functional characterization of human cyclooxygenase-1 splicing variants: evidence for intron 1retention. J Pharmacol Exp Ther 315: 1298–1305
119. Rainsford KD, Robertson SC, Brown S (1997): Ibuprofen and paracetamol:
relative safety in non-prescription dosages. J Pharm Pharmacol 49: 345-376 120. Rall TW (1982): Evolution of the mechanism of action of methylxanthines: from
calcium mobilizers to antagonists of adenosine receptors. Pharmacologist 24: 277-287
121. Rall TW: Drugs used in the treatment of asthma. The methylxanthines, cromolyn
sodium, and other agents. In: Goodman and Gilman’s, The Pharmalogical Basis Therapeutics, Pergamon Press, New York, 1990, 307-312
122. Rampal P, Moore N, Van Ganse E, Le Parc JM, Wall R, Schneid H, Verriere F
(2002): Gastrointestinal tolerability of ibuprofen compared with paracetamol and aspirin at over-the-counter doses. J Int Med Res 30: 301-308
123. Rawlins MD, Henderson DB, Hijab AR (1977): Pharmacokinetics of
paracetamol (acetaminophen) after intravenous and oral administration. Eur J Clin Pharmacol 11: 283-286
124. Renner B, Clarke G, Grattan T, Beisel A, Mueller C, Werner U, Kobal G, Brune
K (2007): Caffeine accelerates absorption and enhances the analgesic effect of acetaminophen. J Clin Pharmacol 47(6): 715-26
125. Renner B, Clarke G, Grattan T, Beisel A, Wille C, Mueller C (2003a): Analgesic
effects of paracetamol in comparison to its combination with caffeine. J Clin Pharmacol 43: 1038; Ref Type: Abstract
126. Renner B, Clarke G, Grattan T, Lang B, Ahne G, Brune K (2003b): Assessment
of dose dependent analgesic effects of paracetamol tablet compared to placebo. J Clin Pharmacol 43: 1038; Ref Type: Abstract
127. Rieg T, Steigele H, Schnermann J, Richter K, Osswald H, Vallon V (2005):
Requirement of intact adenosine A1 receptors for the diuretic and natriuretic action of the methylxanthines theophylline and caffeine. J Pharmacol Exp Ther 313(1): 403-409
128. Ring EF, Collins AJ, Bacon PA, Cosh JA (1974): Quantitation of thermography
in arthritis using multi-isothermal analysis. II. Effect of nonsteroidal anti-inflammatory therapy on the thermographic index. Ann Rheum Dis 33: 353–356
129. Robertson D, Fröhlich JC, Carr RK, Watson JT, Hollifield JW, Shand DG, Oates
JA (1978): Effects of caffeine on plasma renin activity, catecholamines and blood pressure. N Engl J Med 298: 181-186

65
130. Roca-Vinardell A, Ortega-Alvaro A, Gibert-Rahola J, Micó JA (2003): The role of 5-HT1A/B autoreceptors in the antinociceptive effect of systemic administration of acetaminophen. Anesthesiology 98(3): 741-7.
131. Rohdewald P, Derendorf H, Drehsen G, Elger CE, Knoll O (1982): Changes in
cortical evoked potentials as correlates of the efficacy of weak analgesics. Pain 12(4): 329-341
132. Rote Liste®, Fachinformation Paracetamol, Rote Liste® Service GmbH, Berlin,
Stand 2008: 1 133. Rumack BH (2004): Acetaminophen misconceptions. Hepatology 40: 10–15 134. Sandrini M, Pini LA, Vitale G (2003): Differential involvement of central 5-
HT1B and 5-HT3 receptor subtypes in the antinociceptive effect of paracetamol. Inflamm Res 52(8): 347-52
135. Sawynok J, Reid AR (1996): Neurotoxin-induced lesions to central serotonergic,
noradrenergic and dopaminergic systems modify caffeine-induced antinociception in the formalin test and locomotor stimulation in rats. J Pharmacol Exp Ther 277: 646-653
136. Sawynok J, Reid AR, Doak GJ (1995): Caffeine antinociception in the rat hot-
plate and formalin tests and locomotor stimulation: involvement of noradrenergic mechanisms. Pain 61: 203-213
137. Sawynok J, Yaksh TL (1993): Caffeine as an analgetic adjuvant: A review of
pharmacology and mechanism of action. Pharmacol Rev 45: 43-85 138. Schwab JM, Beiter T, Linder JU, Laufer S, Schulz JE, Meyermann R,
Schluesener HJ (2003): COX-3 – a virtual pain target in humans? FASEB J 17: 2174 – 2175
139. Seymour RA, Rawlins MD (1981): Pharmacokinetics of parenteral paracetamol and its analgesic effects in post-operative dental pain. Eur J Clin Pharmacol 20(3): 215-218
140. Sechzer PH (1978): Postspinal anaesthesia headache treated with caffeine (II).
Intracranial distention, a key factor. Curr Ther Res 26: 307-312 141. Shi J, Benowitz NL, Denaro CP, Sheiner LB (1993): Pharmacokinetic
pharmacodynamic modeling of caffeine: tolerance to pressor effects. Clin Pharmacol Ther 53:6-14
142. Simmons DL, Chandrasekharan NV, Hu D, Roos KL, Tomsik J (2005):
Comments on acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther 315(1): 1-7
143. Skjelbred P, Lokken P (1979): Paracetamol versus placebo: effects on post
operative course. Eur J Clin Pharmacol 15: 27–33

66
144. Smith A (2002): Effects of caffeine on human behavior. Food Chem Toxicol 40: 1243–1255
145. Steen KH, Reeh PW, Anton F, Handwerker HO (1992): Protons selectively
induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci 12(1):86-95.
146. Steen KH, Wegner H, Reeh PW (1999): The pH response of rat cutaneous
nociceptors correlates with extracellular [Na+] and is increased under amiloride. Eur J Neurosci 11(8): 2783-2792.
147. Swierkosz TA, Jordan L, McBride M, McGough K, Devlin J, Botting RM
(2002): Actions of paracetamol on cyclooxygenases in tissue and cell homogenates of mouse and rabbit. Med Sci Monit 8: BR496–BR503
148. Sylven C (1993): Mechanisms of pain in angina pectoris: a critical view of the
adenosine hypothesis. Cardiovasc Drugs Ther 7: 745-759 149. Thürauf N, Fleischer WK, Liefhold J, Schmid O, Kobal G (1996): Dose
dependent time course of the analgesic effect of a sustained-release preparation of tramadol on experimental phasic and tonic pain. Br J Clin Pharmacol 41(2): 115-123.
150. Thürauf N, Friedel I, Hummel C, Kobal G (1991): The mucosal potential elicited
by noxious chemical stimuli with CO2 in rats: is it a peripheral nociceptive event? Neurosci Lett 128(2): 297-300.
151. Thürauf N, Hummel T, Kettenmann B, Kobal G (1993): Nociceptive and
reflexive responses recorded from the human nasal mucosa. Brain Res 629(2): 293-299.
152. Ukena D, Schudt C, Sybrecht GW (1993): Adenosine receptor-blocking
xanthines as inhibitors of phosphodiesterase isozymes. Biochem Pharmacol 45: 847-851
153. Vane JR, Botting RM (1995): New insights into the mode of action of anti-
inflammatory drugs. Inflamm Res 44: 1-10 154. Victor BS, Lubetsky M, Greden JF (1981): Somatic manifestations of
caffeinism. J Clin Psychiatry 42: 185-188
155. Ward N, Whitney C, Avery D, Dunner D (1991): The analgesic effects of caffeine in headache. Pain 44: 151-155
156. Warner TD, Vojnovic I, Giuliano F, Jimenez R, Bishop-Bailey D, Mitchell JA
(2004): Cyclooxygenases 1, 2, and 3 and the production of prostaglandin I2: investigating the activities of acetaminophen and cyclooxygenase-2-selective inhibitors in rat tissues. J Pharmacol Exp Ther 310: 642–647
157. Williams PT, Wood PD, Vranizan KM, Albers JJ, Garay SC (1985): Coffee
intake and elevated cholesterol and apolipoprotein B levels in men. J Am Med Assoc 253: 1407-11

67
158. World Medical Association Declaration of Helsinki, 48th World Medical Assembly, Somerset West, Republic of South Africa, October 1996.
159. Wu J, Fang L, Lin Q, Willis WD (2001): Nitric oxide synthase in spinal cord
central sensitization following intradermal injection of capsaicin. Pain 94: 47-58 160. Yoovathaworn KC, Sriwatanakul K, Thithapandha A (1986): Influence of
caffeine on aspirin pharmacocinetics. Eur J Drug Metab Pharmacokinet 11: 71-76

68
7. Abkürzungsverzeichnis
Abs. Absatz
AE Adverse Event
al. andere
ALT Alanin-Aminotransferase
AMG Arzneimittelgesetz
AST Aspertat-Aminotransferase
AUC Area under the curve
BfArM Bundesinstitut für Arzneimittel und Medizinprodukte
BMI Body Mass Index
cAMP cyclisches Adenosin3‘-5‘-monophosphat
Ca2+ Kalzium
cGMP cyclisches Guanosinmonophosphat
CL Clearance
Cmax maximal erreichte Plasmaspiegel
CO2 Kohlendioxid
COX Cyclooxygenase
CRF Case Report Form, Prüfprotokoll
CSSEP Chemo-somatosensorisch evoziertes Potential
dB Dezibel
diast. diastolisch
dl Deziliter
EDTA Ethylendiamintetraacetat
EEG Elektroencephalogramm
EG Europäische Gemeinschaft
EKG Elektrokardiogramm
EMEA European Medicines Agency
eng. englisch
EP evozierte Potentiale
EU Estimation Units
€ Euro
FFT Fast Fourier Transformation
fl Femtoliter
g Gramm

69
GCP Good clinical Practice
GLP Good Laboratory Practice
GMP Good Manufacturing Practice
GOT Glutamat-Oxalaxetat-Transaminase
GPT Glutamat-Pyruvat-Transaminase
GSK Glaxo-Smith-Kline
h Stunde
Hb Hämoglobin
HBs Hepatitis B surface Antigen
HCV Hepatitis CVirus
HIV Humanes-Insuffizienz-Virus
Hz Hertz
5-HT 5-Hydroxytryptamin (= Serotonin)
HWZ Halbwärtszeit
ICH International Conference on Harmonisation of Technical Requirements
for Registration of Pharmaceuticals for Human Use
IL Illinois
ITT intention-to-treat
kg Kilogramm
KI Konfidenzintervall
l Liter
lat. lateinisch
m männlich
m2 Quadratmeter
max. maximal
MCH mittleres korpuskuläres Hämoglobin
MCHC mittlere korpuskuläre Hämoglobinkonzentration
MCV mittleres korpuskuläres Volumen
mg Milligramm
μg Mikrogramm
Min Minute
mind. mindestens
mmHg Millimeter-Quecksilbersäule
mmol Milli-Mol
modif. modifiziert

70
MPV Mean Platelet Volume
MW Mittelwert
NJ New Jersey
NMP negative Mucosa-Potentiale
NO Stickstoffmonoxid
Nr. Nummer
NSAID Nonsteroidal Antiinflammatory Drug
PDE Phosphodiesterase
pg Pikogramm
PGE Prostaglandin E
RDW Red Blood Cell Distribution Width
s. h. siehe
SOP Standard Operating Procedure
SPL Sound pressure level
Std. Stunde
syst. systolisch
tmax Zeit bis zum Erreichen maximaler Plasmaspiegel
t1/2 Halbwärtszeit
TP Tracking Performance
U Units
u. a. unter anderem
u. U. unter Umständen
USA United States of America
VAS Visuelle Analogskala
Vgl. Vergleiche
vs. versus
VZ Verteilungsvolumen
w weiblich

71
8. Anhang
8.1 Pharmakokinetische Daten von Paracetamol
Tabelle 14: Pharmakokinetik der 26 Probanden zu Paracetamol; MW = Mittelwert, SD = Standardabweichung
Proband / t max c max t 1/2 AUC 0-6 h AUC 0-t∞ CL-tot VZ Geschlecht [h] [μg*ml-1] [h] [μg*h*ml-1] [μg*h*ml-1] [ml*min-1] [ml]
1 M 0,98 9,99 1,73 24,24 27,19 613,07 91946,60 2 M 1,00 27,38 2,57 38,78 49,60 336,03 74692,67 3 M 0,98 12,69 1,83 31,49 35,86 464,74 73432,35 4 M 0,93 13,03 2,26 28,90 35,77 465,93 91019,45 5 M 0,98 11,33 1,92 28,19 33,07 504,00 83778,44 6 M 0,47 18,39 2,31 31,25 37,59 443,36 88757,64 7 M 0,77 20,69 2,65 46,14 59,61 279,60 64214,00 8 M 0,95 9,90 2,21 27,70 33,96 490,74 94058,11 9 M 1,98 13,21 3,14 36,72 54,73 304,53 82660,67
10 W 0,98 19,51 1,85 43,38 49,77 334,88 53702,64 11 W 1,03 17,19 2,53 40,14 50,85 327,75 71879,47 12 W 1,52 11,47 2,48 41,48 51,86 321,37 69030,07 13 W 1,00 11,50 1,68 28,38 31,69 525,90 76258,76 14 W 1,97 11,19 2,16 37,72 47,95 347,60 65027,56 15 W 1,00 17,35 2,36 40,56 47,82 348,54 71282,61 16 W 1,00 15,11 2,19 42,17 54,51 305,78 58025,65 17 M 2,00 10,72 2,42 23,60 31,18 534,60 112009,33 18 W 1,00 14,29 2,18 48,25 59,35 280,83 53105,01 19 M 0,98 11,50 1,67 27,13 31,04 536,91 77750,89 20 W 2,50 7,73 2,44 23,36 31,06 536,62 113131,90 21 W 0,50 10,43 1,83 24,92 29,36 567,70 89892,69 22 W 1,00 16,74 1,38 41,72 48,89 340,92 40729,19 23 W 2,50 10,78 1,10 30,94 37,05 449,85 42697,72 24 W 1,00 15,79 1,99 39,92 46,77 356,33 61303,94 26 W 0,48 14,04 1,79 32,85 37,81 440,84 68231,31 27 W 0,75 14,96 3,02 29,31 38,77 429,91 112403,65
Parameter t max c max t 1/2 AUC 0-6 h AUC 0-t∞ CL-tot VZ [h] [μg*ml-1] [h] [μg*h*ml-1] [μg*h*ml-1] [ml*min-1] [ml] MW gesamt 1,16 14,11 2,14 34,20 42,04 418,78 76193,17
MW w 1,24 13,82 2,01 36,67 44,45 393,58 67366,11 MW m 1,08 14,41 2,28 31,74 39,63 443,98 85020,22
SD 0,56 4,25 0,47 7,49 10,00 99,49 19594,58 Median 1,00 13,12 2,19 32,17 38,29 435,38 74062,51
Median m 0,98 12,86 2,28 29,11 35,82 465,34 86268,04 Median w 1,00 14,17 2,07 40,03 47,88 348,07 66629,44
Min. 0,47 7,73 1,10 23,36 27,19 279,60 40729,19 Max. 2,50 27,38 3,14 48,25 59,61 613,07 113131,90
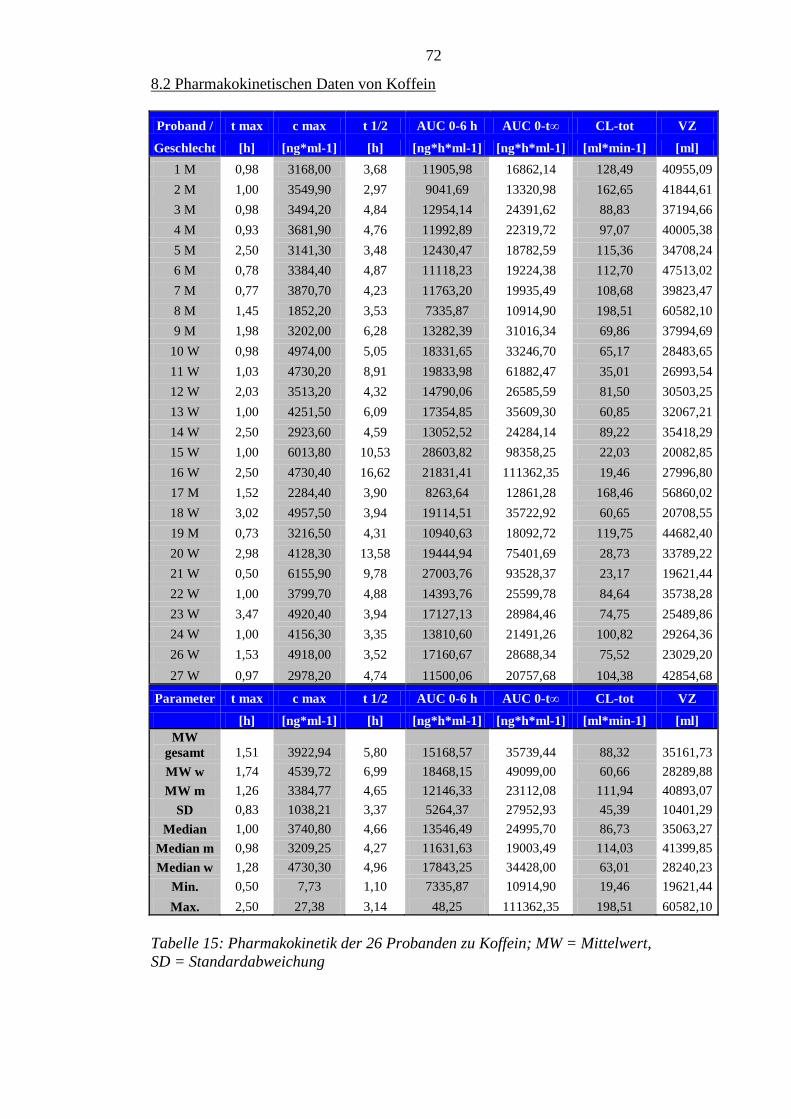
72
8.2 Pharmakokinetischen Daten von Koffein
Tabelle 15: Pharmakokinetik der 26 Probanden zu Koffein; MW = Mittelwert, SD = Standardabweichung
Proband / t max c max t 1/2 AUC 0-6 h AUC 0-t∞ CL-tot VZ
Geschlecht [h] [ng*ml-1] [h] [ng*h*ml-1] [ng*h*ml-1] [ml*min-1] [ml] 1 M 0,98 3168,00 3,68 11905,98 16862,14 128,49 40955,09 2 M 1,00 3549,90 2,97 9041,69 13320,98 162,65 41844,61 3 M 0,98 3494,20 4,84 12954,14 24391,62 88,83 37194,66 4 M 0,93 3681,90 4,76 11992,89 22319,72 97,07 40005,38 5 M 2,50 3141,30 3,48 12430,47 18782,59 115,36 34708,24 6 M 0,78 3384,40 4,87 11118,23 19224,38 112,70 47513,02 7 M 0,77 3870,70 4,23 11763,20 19935,49 108,68 39823,47 8 M 1,45 1852,20 3,53 7335,87 10914,90 198,51 60582,10 9 M 1,98 3202,00 6,28 13282,39 31016,34 69,86 37994,69
10 W 0,98 4974,00 5,05 18331,65 33246,70 65,17 28483,65 11 W 1,03 4730,20 8,91 19833,98 61882,47 35,01 26993,54 12 W 2,03 3513,20 4,32 14790,06 26585,59 81,50 30503,25 13 W 1,00 4251,50 6,09 17354,85 35609,30 60,85 32067,21 14 W 2,50 2923,60 4,59 13052,52 24284,14 89,22 35418,29 15 W 1,00 6013,80 10,53 28603,82 98358,25 22,03 20082,85 16 W 2,50 4730,40 16,62 21831,41 111362,35 19,46 27996,80 17 M 1,52 2284,40 3,90 8263,64 12861,28 168,46 56860,02 18 W 3,02 4957,50 3,94 19114,51 35722,92 60,65 20708,55 19 M 0,73 3216,50 4,31 10940,63 18092,72 119,75 44682,40 20 W 2,98 4128,30 13,58 19444,94 75401,69 28,73 33789,22 21 W 0,50 6155,90 9,78 27003,76 93528,37 23,17 19621,44 22 W 1,00 3799,70 4,88 14393,76 25599,78 84,64 35738,28 23 W 3,47 4920,40 3,94 17127,13 28984,46 74,75 25489,86 24 W 1,00 4156,30 3,35 13810,60 21491,26 100,82 29264,36 26 W 1,53 4918,00 3,52 17160,67 28688,34 75,52 23029,20 27 W 0,97 2978,20 4,74 11500,06 20757,68 104,38 42854,68
Parameter t max c max t 1/2 AUC 0-6 h AUC 0-t∞ CL-tot VZ [h] [ng*ml-1] [h] [ng*h*ml-1] [ng*h*ml-1] [ml*min-1] [ml]
MW gesamt 1,51 3922,94 5,80 15168,57 35739,44 88,32 35161,73 MW w 1,74 4539,72 6,99 18468,15 49099,00 60,66 28289,88 MW m 1,26 3384,77 4,65 12146,33 23112,08 111,94 40893,07
SD 0,83 1038,21 3,37 5264,37 27952,93 45,39 10401,29 Median 1,00 3740,80 4,66 13546,49 24995,70 86,73 35063,27
Median m 0,98 3209,25 4,27 11631,63 19003,49 114,03 41399,85 Median w 1,28 4730,30 4,96 17843,25 34428,00 63,01 28240,23
Min. 0,50 7,73 1,10 7335,87 10914,90 19,46 19621,44 Max. 2,50 27,38 3,14 48,25 111362,35 198,51 60582,10
73
8.3 Demographische Daten der Studienteilnehmer
Proband Geschlecht Alter Größe Gewicht BMI Raucher
[Jahre] [m] [kg] [kg/m2] 1 M 26 1,94 90 23,91 Nein 2 M 30 1,84 71 20,97 Ja 3 M 24 1,75 67 21,88 Nein 4 M 25 1,74 60 19,82 Nein 5 M 26 1,74 62 20,48 Ja 6 M 27 1,82 80 24,15 Ja 7 M 26 1,83 73 21,80 Nein 8 M 27 1,68 55 19,49 Nein 9 M 26 1,77 66 21,07 Ja
10 W 23 1,54 53 22,35 Ja 11 W 24 1,63 58 21,83 Nein 12 W 25 1,72 58 19,61 Ja 13 W 25 1,68 68 24,09 Ja 14 W 24 1,68 65 23,03 Nein 15 W 25 1,54 53 22,35 Nein 16 W 26 1,76 61 19,69 Nein 17 M 24 1,9 82 22,71 Nein 18 W 24 1,66 52 18,87 Ja 19 M 25 1,79 83 25,90 Nein 20 W 25 1,8 67 20,68 Nein 21 W 24 1,68 75 26,57 Nein 22 W 24 1,67 56,5 20,26 Ja 23 W 24 1,67 55 19,72 Nein 24 W 24 1,64 52 19,33 Nein 26 W 30 1,68 60 21,26 Nein 27 M 25 1,85 75 21,91 Nein Mittelwert 25,31 1,73 65,29 21,68 Minimum 23 1,54 52 18,87 Maximum 30 1,94 90 26,57 Mittelwert ♀ 24,79 1,67 59,54 21,40 Mittelwert ♂ 25,92 1,80 72,00 21,68
Tabelle 16: Demographische Daten der Probanden
74
8.4 Im Rahmen der Studie erhobene Blutdruckwerte der Probanden
Proband 1 Proband 2 Proband 3 Proband 4 Proband 5
Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Screening 129 70 120 64 121 73 134 78 130 79 1. Visit 119 75 114 75 131 70 138 94 113 67 2. Visit 132 66 106 60 134 80 145 82 121 71 3. Visit 114 72 118 73 139 82 135 85 121 63 Follow up 126 75 124 71 136 78 140 89 132 82 Mittelwerte 124 71,6 116,4 68,6 132,2 76,6 138,4 85,6 123,4 72,4 Proband 6 Proband 7 Proband 8 Proband 09 Proband 10
Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Screening 142 72 115 69 124 76 128 72 95 60 1. Visit 123 70 109 68 127 77 118 63 104 66 2. Visit 126 75 110 74 132 85 120 70 98 57 3. Visit 123 74 103 73 141 77 121 81 91 69 Follow up 131 82 117 67 135 74 131 71 105 60 Mittelwerte 129 74,6 110,8 70,2 131,8 77,8 123,6 71,4 98,6 62,4 Proband 11 Proband 12 Proband 13 Proband 14 Proband 15
Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Screening 120 80 125 83 134 87 142 83 136 90 1. Visit 103 62 115 82 132 86 144 78 123 86 2. Visit 117 88 111 75 132 89 133 87 129 77 3. Visit 105 68 128 87 131 88 139 82 115 74 Follow up 118 76 115 76 132 85 135 80 118 80 Mittelwerte 112,6 74,8 118,8 80,6 132,2 87 138,6 82 124,2 86 Proband 16 Proband 17 Proband 18 Proband 19 Proband 20
Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Screening 119 75 118 69 105 68 143 77 121 81 1. Visit 112 69 109 70 117 70 138 72 113 85 2. Visit 115 72 111 66 112 75 130 74 126 75 3. Visit 118 74 117 65 121 75 133 77 124 72 Follow up 116 77 123 73 107 77 123 51 113 69 Mittelwerte 116 73,4 115,6 68,6 112,4 73 133,4 70,2 119,4 76,4
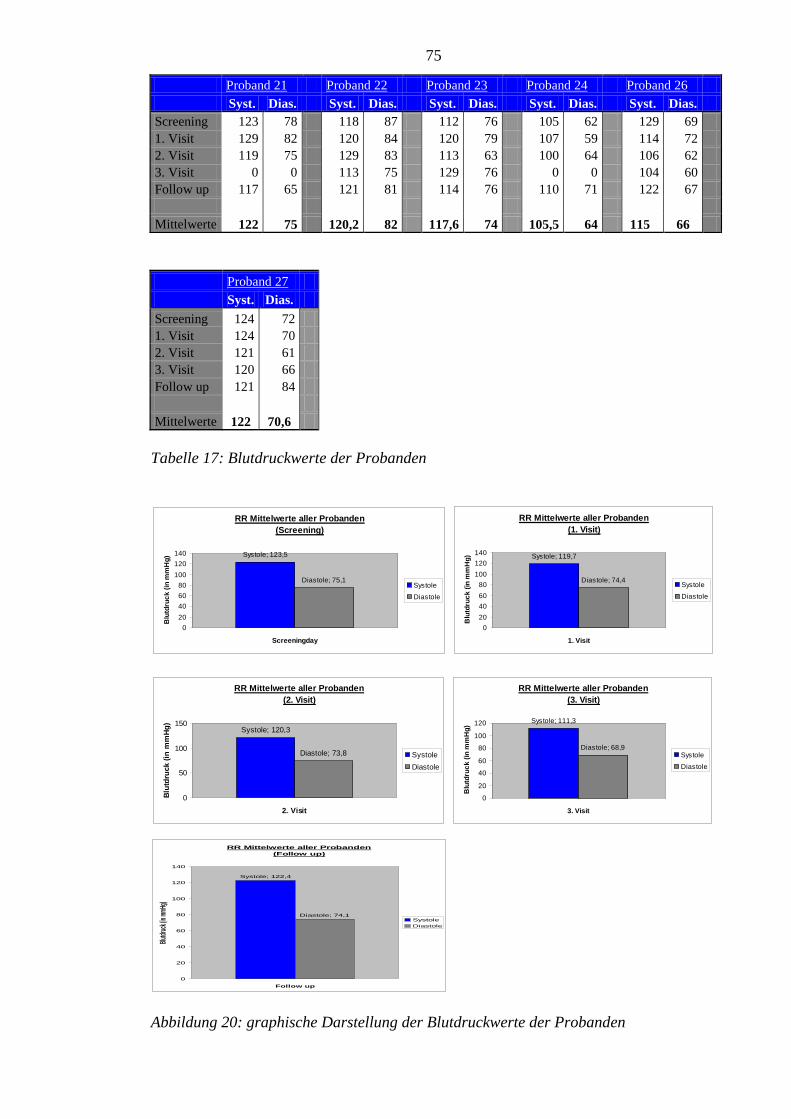
75
Proband 21 Proband 22 Proband 23 Proband 24 Proband 26 Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias. Syst. Dias.
Screening 123 78 118 87 112 76 105 62 129 69 1. Visit 129 82 120 84 120 79 107 59 114 72 2. Visit 119 75 129 83 113 63 100 64 106 62 3. Visit 0 0 113 75 129 76 0 0 104 60 Follow up 117 65 121 81 114 76 110 71 122 67 Mittelwerte 122 75 120,2 82 117,6 74 105,5 64 115 66 Proband 27
Syst. Dias. Screening 124 72 1. Visit 124 70 2. Visit 121 61 3. Visit 120 66 Follow up 121 84 Mittelwerte 122 70,6 Tabelle 17: Blutdruckwerte der Probanden
Abbildung 20: graphische Darstellung der Blutdruckwerte der Probanden
RR Mittelwerte aller Probanden(Screening)
Systole; 123,5
Diastole; 75,1
020406080
100120140
Screeningday
Blu
tdru
ck (i
n m
mH
g)
Systole
Diastole
RR Mittelwerte aller Probanden(1. Visit)
Systole; 119,7
Diastole; 74,4
020406080
100120140
1. Visit
Blu
tdru
ck (i
n m
mH
g)
Systole
Diastole
RR Mittelwerte aller Probanden(2. Visit)
Systole; 120,3
Diastole; 73,8
0
50
100
150
2. Visit
Blut
druc
k (in
mm
Hg)
SystoleDiastole
RR Mittelwerte aller Probanden(3. Visit)
Systole; 111,3
Diastole; 68,9
0
20
40
60
80
100
120
3. Visit
Blu
tdru
ck (i
n m
mH
g)
Systole
Diastole
RR Mittelwerte aller Probanden(Follow up)
Systole; 122,4
Diastole; 74,1
0
20
40
60
80
100
120
140
Follow up
Blutdr
uck (in
mmHg
)
SystoleDiastole
76
8.5 Im Rahmen der Studie erhobene Laborparameter der Probanden
Laborparameter Proband 1 Proband 2 Proband 3
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 3,90 4,30 4,50 4,80 4,20 4,50 Sodium [mmol/L] 140,00 139,00 140,00 147,00 138,00 139,00 Chloride [mmol/L] 106,00 104,00 1,03 106,00 103,00 101,00 Glucose [mg/dl] 89,00 91,00 93,00 100,00 94,00 86,00 Calcium [mmol/L] 2,20 2,30 2,50 2,50 2,20 2,30 anorganic Phosphates [mg/dl] 4,30 3,50 3,10 4,00 3,20 4,30 Uric acis [mg/dl] 5,20 4,70 6,10 6,00 3,60 4,60 Urea [mg/dl] 16,00 20,00 31,00 24,00 33,00 40,00 Creatinine [mg/dl] 1,06 1,05 1,19 1,37 0,88 1,00 Bilirubin in total [mg/dl] 0,40 0,70 0,90 0,60 0,80 0,60 AST/GOT [U/l] 18,00 20,00 22,00 19,00 23,00 21,00 ALT/GPT [U/l] 29,00 28,00 18,00 15,00 24,00 31,00 Gamma-GT [U/l] 19,00 21,00 16,00 13,00 17,00 23,00 alk. Phosphatase [U/l] 92,00 112,00 55,00 49,00 45,00 51,00 Protein in total [g/l] 68,50 70,40 72,80 69,60 74,60 81,70 Albumins [g/l] 46,10 45,60 49,70 48,90 48,10 50,70 Cholesterol [mg/dl] 243,00 234,00 136,00 159,00 164,00 223,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Laborparameter Proband 4 Proband 5 Proband 6
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 4,50 4,30 3,90 3,70 4,20 4,50 Sodium [mmol/L] 139,00 142,00 138,00 138,00 138,00 139,00 Chloride [mmol/L] 103,00 105,00 103,00 102,00 103,00 105,00 Glucose [mg/dl] 105,00 74,00 106,00 94,00 104,00 94,00 Calcium [mmol/L] 2,50 2,40 2,40 2,50 2,50 2,50 anorganic Phosphates [mg/dl] 3,10 3,40 3,40 3,80 2,90 3,50 Uric acis [mg/dl] 6,00 6,30 5,50 4,20 5,50 6,70 Urea [mg/dl] 31,00 45,00 24,00 20,00 34,00 29,00 Creatinine [mg/dl] 0,87 0,92 1,02 0,86 1,02 1,02 Bilirubin in total [mg/dl] 0,90 1,10 1,20 1,00 0,60 0,50 AST/GOT [U/l] 30,00 24,00 24,00 19,00 25,00 33,00 ALT/GPT [U/l] 27,00 20,00 14,00 13,00 33,00 45,00 Gamma-GT [U/l] 23,00 35,00 14,00 16,00 35,00 32,00 alk. Phosphatase [U/l] 97,00 91,00 94,00 88,00 77,00 84,00 Protein in total [g/l] 76,60 76,00 78,30 79,20 71,60 77,80 Albumins [g/l] 50,40 49,30 48,70 50,50 48,30 51,40 Cholesterol [mg/dl] 247,00 203,00 195,00 205,00 235,00 151,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Tabelle 18a: Plasmaparameter und Infektiologie der Probanden 1 bis 6
77
Laborparameter Proband 7 Proband 8 Proband 9
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 4,40 4,20 4,10 4,40 4,30 4,40 Sodium [mmol/L] 140,00 139,00 139,00 142,00 139,00 138,00 Chloride [mmol/L] 102,00 103,00 104,00 106,00 104,00 103,00 Glucose [mg/dl] 88,00 99,00 100,00 79,00 82,00 103,00 Calcium [mmol/L] 2,40 2,50 2,40 2,30 2,40 2,40 anorganic Phosphates [mg/dl] 4,70 4,30 3,00 2,20 4,10 4,50 Uric acis [mg/dl] 6,00 5,80 4,40 4,80 5,50 5,30 Urea [mg/dl] 33,00 26,00 22,00 24,00 35,00 27,00 Creatinine [mg/dl] 1,02 1,07 1,00 1,03 1,16 1,17 Bilirubin in total [mg/dl] 0,50 0,80 1,20 0,70 0,50 0,50 AST/GOT [U/l] 33,00 28,00 29,00 30,00 25,00 24,00 ALT/GPT [U/l] 49,00 38,00 27,00 28,00 18,00 19,00 Gamma-GT [U/l] 40,00 30,00 20,00 19,00 20,00 12,00 alk. Phosphatase [U/l] 109,00 97,00 46,00 46,00 45,00 43,00 Protein in total [g/l] 76,70 73,60 70,60 71,20 74,30 70,50 Albumins [g/l] 49,30 49,30 48,50 46,10 47,00 45,20 Cholesterol [mg/dl] 219,00 228,00 229,00 195,00 231,00 216,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Laborparameter Proband 10 Proband 11 Proband 12
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 4,20 4,10 4,50 4,30 4,10 4,20 Sodium [mmol/L] 135,00 139,00 140,00 139,00 138,00 141,00 Chloride [mmol/L] 100,00 102,00 104,00 102,00 103,00 104,00 Glucose [mg/dl] 87,00 89,00 106,00 76,00 99,00 71,00 Calcium [mmol/L] 2,40 2,50 2,50 2,50 2,40 2,50 anorganic Phosphates [mg/dl] 4,20 4,30 3,40 3,70 3,10 3,40 Uric acis [mg/dl] 4,20 4,00 5,20 4,40 2,90 3,70 Urea [mg/dl] 26,00 20,00 27,00 24,00 24,00 27,00 Creatinine [mg/dl] 0,71 0,78 1,02 0,89 0,94 0,97 Bilirubin in total [mg/dl] 0,40 3,00 0,30 0,40 0,50 0,40 AST/GOT [U/l] 19,00 19,00 23,00 18,00 23,00 24,00 ALT/GPT [U/l] 18,00 17,00 15,00 13,00 17,00 18,00 Gamma-GT [U/l] 15,00 13,00 14,00 10,00 28,00 29,00 alk. Phosphatase [U/l] 54,00 54,00 61,00 62,00 51,00 62,00 Protein in total [g/l] 72,70 76,70 77,50 76,70 66,60 74,00 Albumins [g/l] 46,80 47,60 45,10 43,30 39,80 45,80 Cholesterol [mg/dl] 175,00 184,00 224,00 232,00 183,00 221,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Tabelle 18b: Plasmaparameter und Infektiologie der Probanden 7 bis 12
78
Laborparameter Proband 13 Proband 14 Proband 15
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 3,80 4,30 4,20 4,20 4,40 5,40 Sodium [mmol/L] 139,00 135,00 139,00 139,00 130,00 137,00 Chloride [mmol/L] 103,00 102,00 102,00 102,00 94,00 102,00 Glucose [mg/dl] 105,00 99,00 106,00 90,00 76,00 86,00 Calcium [mmol/L] 2,40 2,50 2,40 2,50 2,40 2,50 anorganic Phosphates [mg/dl] 2,90 3,40 3,50 3,90 4,00 4,30 Uric acis [mg/dl] 3,60 3,40 3,70 3,10 3,30 2,60 Urea [mg/dl] 23,00 19,00 17,00 18,00 20,00 32,00 Creatinine [mg/dl] 0,90 1,02 0,76 0,81 0,90 0,91 Bilirubin in total [mg/dl] 0,40 0,60 0,70 0,40 0,60 0,30 AST/GOT [U/l] 23,00 21,00 31,00 27,00 23,00 16,00 ALT/GPT [U/l] 16,00 13,00 26,00 23,00 14,00 10,00 Gamma-GT [U/l] 15,00 13,00 25,00 29,00 25,00 23,00 alk. Phosphatase [U/l] 57,00 59,00 65,00 76,00 42,00 52,00 Protein in total [g/l] 71,40 75,30 76,00 82,20 75,20 71,30 Albumins [g/l] 47,20 47,30 51,50 55,40 43,50 41,10 Cholesterol [mg/dl] 188,00 194,00 188,00 201,00 221,00 253,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Laborparameter Proband 16 Proband 17 Proband 18
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 3,70 4,30 4,20 4,80 4,20 4,00 Sodium [mmol/L] 134,00 139,00 137,00 138,00 139,00 135,00 Chloride [mmol/L] 99,00 103,00 102,00 103,00 105,00 103,00 Glucose [mg/dl] 99,00 87,00 82,00 96,00 82,00 76,00 Calcium [mmol/L] 2,40 2,40 2,40 2,60 2,50 2,40 anorganic Phosphates [mg/dl] 3,50 4,40 3,00 3,70 2,70 3,20 Uric acis [mg/dl] 4,20 3,30 5,90 5,70 4,50 4,10 Urea [mg/dl] 26,00 25,00 26,00 29,00 20,00 21,00 Creatinine [mg/dl] 0,99 0,94 1,07 1,07 0,94 1,16 Bilirubin in total [mg/dl] 0,30 0,30 0,60 0,90 0,70 0,30 AST/GOT [U/l] 25,00 28,00 23,00 23,00 32,00 32,00 ALT/GPT [U/l] 32,00 29,00 22,00 20,00 20,00 18,00 Gamma-GT [U/l] 18,00 17,00 18,00 22,00 13,00 14,00 alk. Phosphatase [U/l] 49,00 53,00 44,00 44,00 69,00 60,00 Protein in total [g/l] 72,20 74,10 71,50 75,30 74,60 72,10 Albumins [g/l] 45,70 44,80 46,90 50,00 45,70 42,70 Cholesterol [mg/dl] 228,00 260,00 241,00 240,00 168,00 175,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Tabelle 18c: Plasmaparameter und Infektiologie der Probanden 13 bis 18
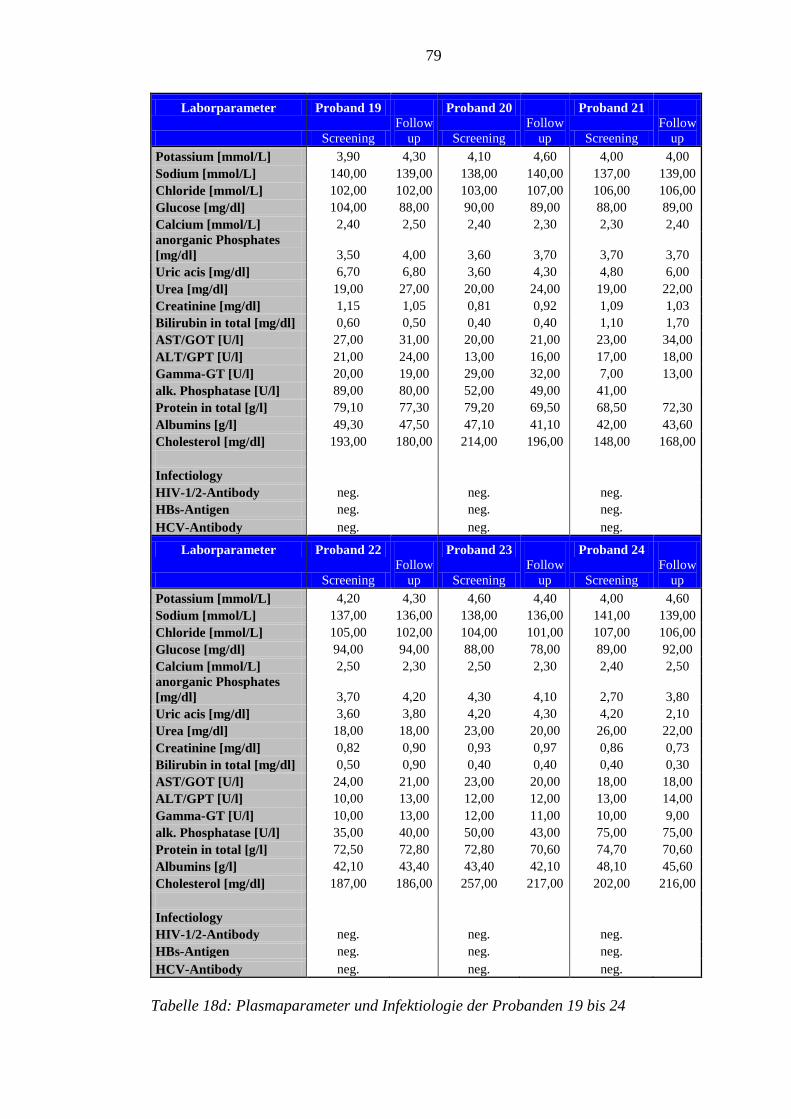
79
Laborparameter Proband 19 Proband 20 Proband 21
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 3,90 4,30 4,10 4,60 4,00 4,00 Sodium [mmol/L] 140,00 139,00 138,00 140,00 137,00 139,00 Chloride [mmol/L] 102,00 102,00 103,00 107,00 106,00 106,00 Glucose [mg/dl] 104,00 88,00 90,00 89,00 88,00 89,00 Calcium [mmol/L] 2,40 2,50 2,40 2,30 2,30 2,40 anorganic Phosphates [mg/dl] 3,50 4,00 3,60 3,70 3,70 3,70 Uric acis [mg/dl] 6,70 6,80 3,60 4,30 4,80 6,00 Urea [mg/dl] 19,00 27,00 20,00 24,00 19,00 22,00 Creatinine [mg/dl] 1,15 1,05 0,81 0,92 1,09 1,03 Bilirubin in total [mg/dl] 0,60 0,50 0,40 0,40 1,10 1,70 AST/GOT [U/l] 27,00 31,00 20,00 21,00 23,00 34,00 ALT/GPT [U/l] 21,00 24,00 13,00 16,00 17,00 18,00 Gamma-GT [U/l] 20,00 19,00 29,00 32,00 7,00 13,00 alk. Phosphatase [U/l] 89,00 80,00 52,00 49,00 41,00 Protein in total [g/l] 79,10 77,30 79,20 69,50 68,50 72,30 Albumins [g/l] 49,30 47,50 47,10 41,10 42,00 43,60 Cholesterol [mg/dl] 193,00 180,00 214,00 196,00 148,00 168,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Laborparameter Proband 22 Proband 23 Proband 24
Screening Follow
up Screening Follow
up Screening Follow
up Potassium [mmol/L] 4,20 4,30 4,60 4,40 4,00 4,60 Sodium [mmol/L] 137,00 136,00 138,00 136,00 141,00 139,00 Chloride [mmol/L] 105,00 102,00 104,00 101,00 107,00 106,00 Glucose [mg/dl] 94,00 94,00 88,00 78,00 89,00 92,00 Calcium [mmol/L] 2,50 2,30 2,50 2,30 2,40 2,50 anorganic Phosphates [mg/dl] 3,70 4,20 4,30 4,10 2,70 3,80 Uric acis [mg/dl] 3,60 3,80 4,20 4,30 4,20 2,10 Urea [mg/dl] 18,00 18,00 23,00 20,00 26,00 22,00 Creatinine [mg/dl] 0,82 0,90 0,93 0,97 0,86 0,73 Bilirubin in total [mg/dl] 0,50 0,90 0,40 0,40 0,40 0,30 AST/GOT [U/l] 24,00 21,00 23,00 20,00 18,00 18,00 ALT/GPT [U/l] 10,00 13,00 12,00 12,00 13,00 14,00 Gamma-GT [U/l] 10,00 13,00 12,00 11,00 10,00 9,00 alk. Phosphatase [U/l] 35,00 40,00 50,00 43,00 75,00 75,00 Protein in total [g/l] 72,50 72,80 72,80 70,60 74,70 70,60 Albumins [g/l] 42,10 43,40 43,40 42,10 48,10 45,60 Cholesterol [mg/dl] 187,00 186,00 257,00 217,00 202,00 216,00 Infectiology HIV-1/2-Antibody neg. neg. neg. HBs-Antigen neg. neg. neg. HCV-Antibody neg. neg. neg.
Tabelle 18d: Plasmaparameter und Infektiologie der Probanden 19 bis 24
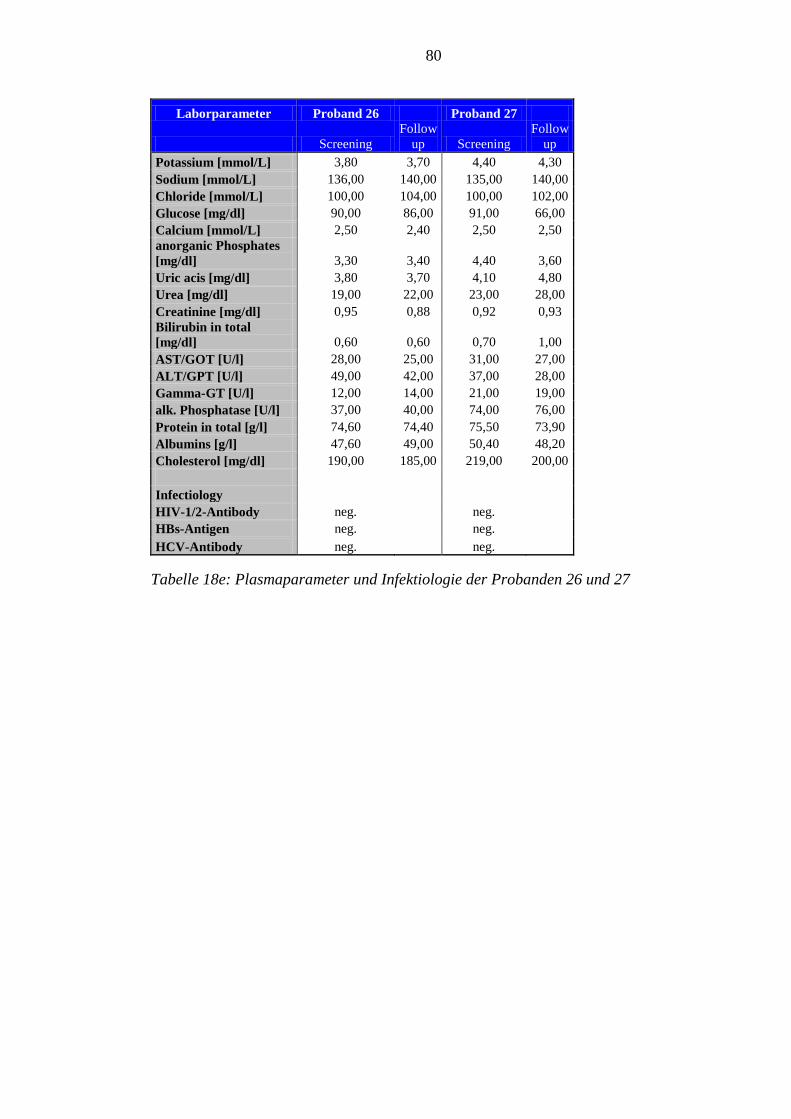
80
Tabelle 18e: Plasmaparameter und Infektiologie der Probanden 26 und 27
Laborparameter Proband 26 Proband 27
Screening Follow
up Screening Follow
up Potassium [mmol/L] 3,80 3,70 4,40 4,30 Sodium [mmol/L] 136,00 140,00 135,00 140,00 Chloride [mmol/L] 100,00 104,00 100,00 102,00 Glucose [mg/dl] 90,00 86,00 91,00 66,00 Calcium [mmol/L] 2,50 2,40 2,50 2,50 anorganic Phosphates [mg/dl] 3,30 3,40 4,40 3,60 Uric acis [mg/dl] 3,80 3,70 4,10 4,80 Urea [mg/dl] 19,00 22,00 23,00 28,00 Creatinine [mg/dl] 0,95 0,88 0,92 0,93 Bilirubin in total [mg/dl] 0,60 0,60 0,70 1,00 AST/GOT [U/l] 28,00 25,00 31,00 27,00 ALT/GPT [U/l] 49,00 42,00 37,00 28,00 Gamma-GT [U/l] 12,00 14,00 21,00 19,00 alk. Phosphatase [U/l] 37,00 40,00 74,00 76,00 Protein in total [g/l] 74,60 74,40 75,50 73,90 Albumins [g/l] 47,60 49,00 50,40 48,20 Cholesterol [mg/dl] 190,00 185,00 219,00 200,00 Infectiology HIV-1/2-Antibody neg. neg. HBs-Antigen neg. neg. HCV-Antibody neg. neg.
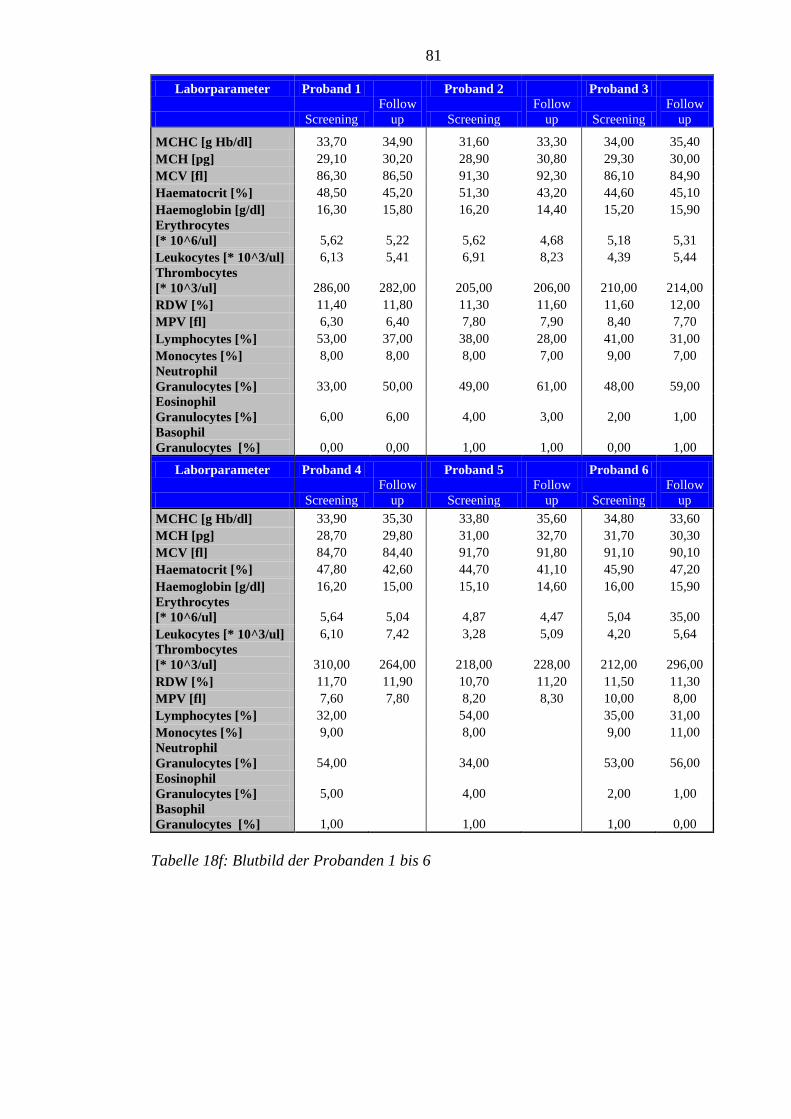
81
Laborparameter Proband 1 Proband 2 Proband 3
Screening Follow
up Screening Follow
up Screening Follow
up
MCHC [g Hb/dl] 33,70 34,90 31,60 33,30 34,00 35,40 MCH [pg] 29,10 30,20 28,90 30,80 29,30 30,00 MCV [fl] 86,30 86,50 91,30 92,30 86,10 84,90 Haematocrit [%] 48,50 45,20 51,30 43,20 44,60 45,10 Haemoglobin [g/dl] 16,30 15,80 16,20 14,40 15,20 15,90 Erythrocytes [* 10^6/ul] 5,62 5,22 5,62 4,68 5,18 5,31 Leukocytes [* 10^3/ul] 6,13 5,41 6,91 8,23 4,39 5,44 Thrombocytes [* 10^3/ul] 286,00 282,00 205,00 206,00 210,00 214,00 RDW [%] 11,40 11,80 11,30 11,60 11,60 12,00 MPV [fl] 6,30 6,40 7,80 7,90 8,40 7,70 Lymphocytes [%] 53,00 37,00 38,00 28,00 41,00 31,00 Monocytes [%] 8,00 8,00 8,00 7,00 9,00 7,00 Neutrophil Granulocytes [%] 33,00 50,00 49,00 61,00 48,00 59,00 Eosinophil Granulocytes [%] 6,00 6,00 4,00 3,00 2,00 1,00 Basophil Granulocytes [%] 0,00 0,00 1,00 1,00 0,00 1,00
Laborparameter Proband 4 Proband 5 Proband 6
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 33,90 35,30 33,80 35,60 34,80 33,60 MCH [pg] 28,70 29,80 31,00 32,70 31,70 30,30 MCV [fl] 84,70 84,40 91,70 91,80 91,10 90,10 Haematocrit [%] 47,80 42,60 44,70 41,10 45,90 47,20 Haemoglobin [g/dl] 16,20 15,00 15,10 14,60 16,00 15,90 Erythrocytes [* 10^6/ul] 5,64 5,04 4,87 4,47 5,04 35,00 Leukocytes [* 10^3/ul] 6,10 7,42 3,28 5,09 4,20 5,64 Thrombocytes [* 10^3/ul] 310,00 264,00 218,00 228,00 212,00 296,00 RDW [%] 11,70 11,90 10,70 11,20 11,50 11,30 MPV [fl] 7,60 7,80 8,20 8,30 10,00 8,00 Lymphocytes [%] 32,00 54,00 35,00 31,00 Monocytes [%] 9,00 8,00 9,00 11,00 Neutrophil Granulocytes [%] 54,00 34,00 53,00 56,00 Eosinophil Granulocytes [%] 5,00 4,00 2,00 1,00 Basophil Granulocytes [%] 1,00 1,00 1,00 0,00
Tabelle 18f: Blutbild der Probanden 1 bis 6
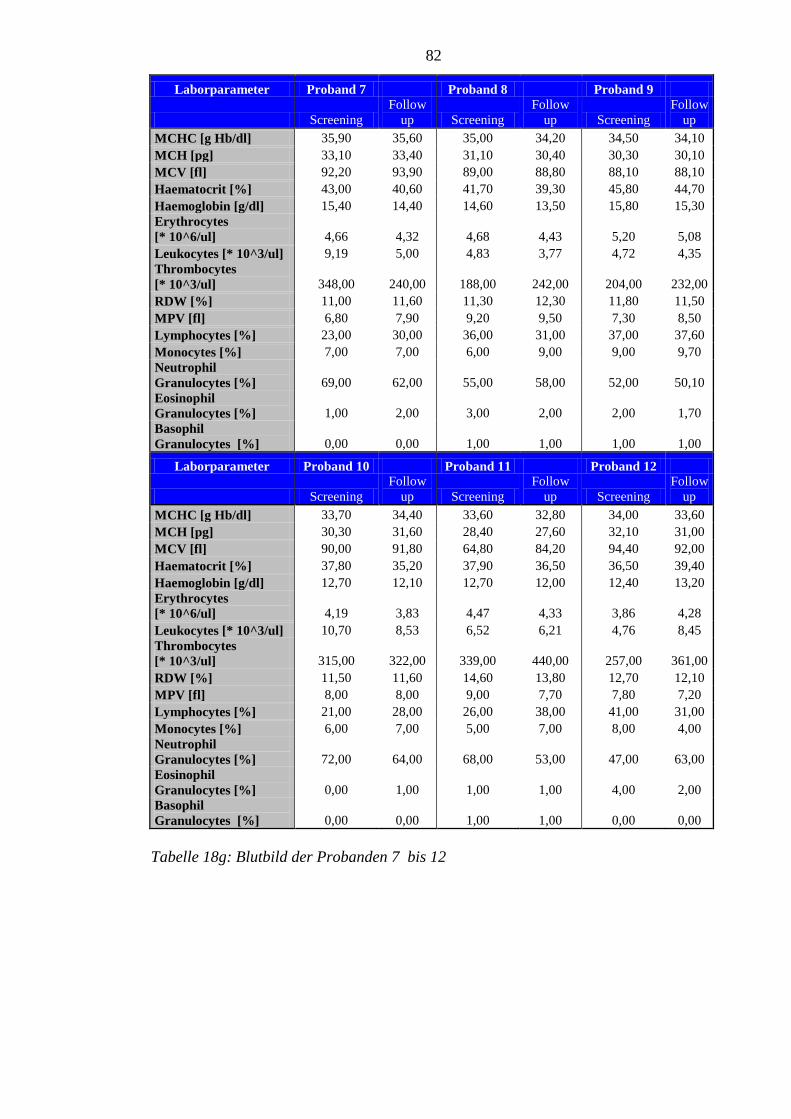
82
Laborparameter Proband 7 Proband 8 Proband 9
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 35,90 35,60 35,00 34,20 34,50 34,10 MCH [pg] 33,10 33,40 31,10 30,40 30,30 30,10 MCV [fl] 92,20 93,90 89,00 88,80 88,10 88,10 Haematocrit [%] 43,00 40,60 41,70 39,30 45,80 44,70 Haemoglobin [g/dl] 15,40 14,40 14,60 13,50 15,80 15,30 Erythrocytes [* 10^6/ul] 4,66 4,32 4,68 4,43 5,20 5,08 Leukocytes [* 10^3/ul] 9,19 5,00 4,83 3,77 4,72 4,35 Thrombocytes [* 10^3/ul] 348,00 240,00 188,00 242,00 204,00 232,00 RDW [%] 11,00 11,60 11,30 12,30 11,80 11,50 MPV [fl] 6,80 7,90 9,20 9,50 7,30 8,50 Lymphocytes [%] 23,00 30,00 36,00 31,00 37,00 37,60 Monocytes [%] 7,00 7,00 6,00 9,00 9,00 9,70 Neutrophil Granulocytes [%] 69,00 62,00 55,00 58,00 52,00 50,10 Eosinophil Granulocytes [%] 1,00 2,00 3,00 2,00 2,00 1,70 Basophil Granulocytes [%] 0,00 0,00 1,00 1,00 1,00 1,00
Laborparameter Proband 10 Proband 11 Proband 12
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 33,70 34,40 33,60 32,80 34,00 33,60 MCH [pg] 30,30 31,60 28,40 27,60 32,10 31,00 MCV [fl] 90,00 91,80 64,80 84,20 94,40 92,00 Haematocrit [%] 37,80 35,20 37,90 36,50 36,50 39,40 Haemoglobin [g/dl] 12,70 12,10 12,70 12,00 12,40 13,20 Erythrocytes [* 10^6/ul] 4,19 3,83 4,47 4,33 3,86 4,28 Leukocytes [* 10^3/ul] 10,70 8,53 6,52 6,21 4,76 8,45 Thrombocytes [* 10^3/ul] 315,00 322,00 339,00 440,00 257,00 361,00 RDW [%] 11,50 11,60 14,60 13,80 12,70 12,10 MPV [fl] 8,00 8,00 9,00 7,70 7,80 7,20 Lymphocytes [%] 21,00 28,00 26,00 38,00 41,00 31,00 Monocytes [%] 6,00 7,00 5,00 7,00 8,00 4,00 Neutrophil Granulocytes [%] 72,00 64,00 68,00 53,00 47,00 63,00 Eosinophil Granulocytes [%] 0,00 1,00 1,00 1,00 4,00 2,00 Basophil Granulocytes [%] 0,00 0,00 1,00 1,00 0,00 0,00 Tabelle 18g: Blutbild der Probanden 7 bis 12
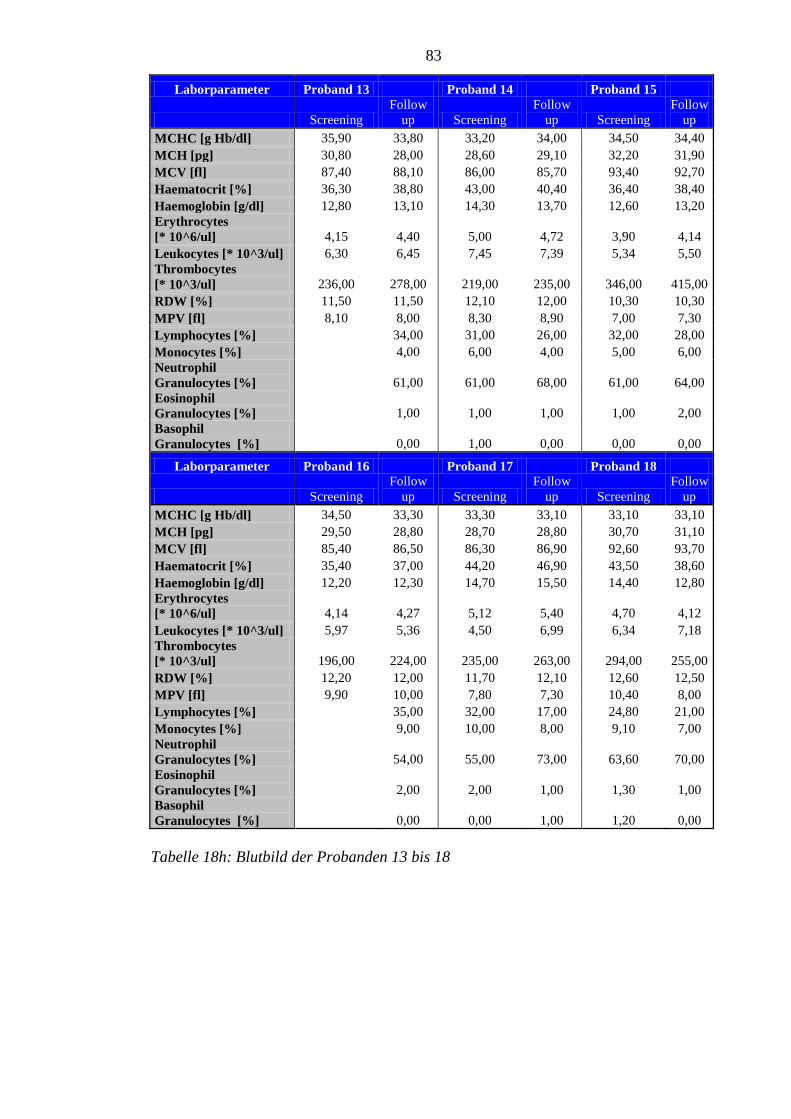
83
Laborparameter Proband 13 Proband 14 Proband 15
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 35,90 33,80 33,20 34,00 34,50 34,40 MCH [pg] 30,80 28,00 28,60 29,10 32,20 31,90 MCV [fl] 87,40 88,10 86,00 85,70 93,40 92,70 Haematocrit [%] 36,30 38,80 43,00 40,40 36,40 38,40 Haemoglobin [g/dl] 12,80 13,10 14,30 13,70 12,60 13,20 Erythrocytes [* 10^6/ul] 4,15 4,40 5,00 4,72 3,90 4,14 Leukocytes [* 10^3/ul] 6,30 6,45 7,45 7,39 5,34 5,50 Thrombocytes [* 10^3/ul] 236,00 278,00 219,00 235,00 346,00 415,00 RDW [%] 11,50 11,50 12,10 12,00 10,30 10,30 MPV [fl] 8,10 8,00 8,30 8,90 7,00 7,30 Lymphocytes [%] 34,00 31,00 26,00 32,00 28,00 Monocytes [%] 4,00 6,00 4,00 5,00 6,00 Neutrophil Granulocytes [%] 61,00 61,00 68,00 61,00 64,00 Eosinophil Granulocytes [%] 1,00 1,00 1,00 1,00 2,00 Basophil Granulocytes [%] 0,00 1,00 0,00 0,00 0,00
Laborparameter Proband 16 Proband 17 Proband 18
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 34,50 33,30 33,30 33,10 33,10 33,10 MCH [pg] 29,50 28,80 28,70 28,80 30,70 31,10 MCV [fl] 85,40 86,50 86,30 86,90 92,60 93,70 Haematocrit [%] 35,40 37,00 44,20 46,90 43,50 38,60 Haemoglobin [g/dl] 12,20 12,30 14,70 15,50 14,40 12,80 Erythrocytes [* 10^6/ul] 4,14 4,27 5,12 5,40 4,70 4,12 Leukocytes [* 10^3/ul] 5,97 5,36 4,50 6,99 6,34 7,18 Thrombocytes [* 10^3/ul] 196,00 224,00 235,00 263,00 294,00 255,00 RDW [%] 12,20 12,00 11,70 12,10 12,60 12,50 MPV [fl] 9,90 10,00 7,80 7,30 10,40 8,00 Lymphocytes [%] 35,00 32,00 17,00 24,80 21,00 Monocytes [%] 9,00 10,00 8,00 9,10 7,00 Neutrophil Granulocytes [%] 54,00 55,00 73,00 63,60 70,00 Eosinophil Granulocytes [%] 2,00 2,00 1,00 1,30 1,00 Basophil Granulocytes [%] 0,00 0,00 1,00 1,20 0,00 Tabelle 18h: Blutbild der Probanden 13 bis 18

84
Laborparameter Proband 19 Proband 20 Proband 21
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 34,50 34,80 34,20 33,80 34,80 34,40 MCH [pg] 31,80 32,00 32,40 32,40 29,80 29,20 MCV [fl] 92,30 91,90 94,60 95,70 85,70 85,00 Haematocrit [%] 44,90 42,50 43,90 36,40 41,00 39,90 Haemoglobin [g/dl] 15,50 14,80 15,00 12,30 14,30 13,70 Erythrocytes [* 10^6/ul] 4,87 4,63 4,64 3,80 4,78 4,70 Leukocytes [* 10^3/ul] 8,73 5,77 6,64 4,79 6,11 7,31 Thrombocytes [* 10^3/ul] 326,00 326,00 254,00 228,00 288,00 267,00 RDW [%] 10,80 10,90 11,40 11,70 11,20 11,40 MPV [fl] 7,50 7,00 9,70 9,80 8,80 8,20 Lymphocytes [%] 32,00 35,00 25,00 28,00 46,00 28,00 Monocytes [%] 5,00 8,00 5,00 12,00 9,00 8,00 Neutrophil Granulocytes [%] 58,00 51,00 68,00 58,00 36,00 58,00 Eosinophil Granulocytes [%] 4,00 6,00 2,00 1,00 8,00 4,00 Basophil Granulocytes [%] 0,00 1,00 0,00 1,00 0,00 2,00
Laborparameter Proband 22 Proband 23 Proband 24
Screening Follow
up Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 34,30 33,90 34,00 32,80 33,10 33,80 MCH [pg] 30,70 30,30 27,80 26,30 30,90 31,10 MCV [fl] 89,60 89,20 81,70 80,10 93,20 92,00 Haematocrit [%] 39,00 37,00 37,10 32,70 43,00 38,40 Haemoglobin [g/dl] 13,40 12,50 12,60 10,70 14,20 13,00 Erythrocytes [* 10^6/ul] 4,35 4,14 4,55 4,08 4,61 4,17 Leukocytes [* 10^3/ul] 5,41 4,49 4,45 5,62 5,87 6,05 Thrombocytes [* 10^3/ul] 302,00 323,00 355,00 338,00 274,00 288,00 RDW [%] 11,10 11,20 11,90 12,40 11,20 11,10 MPV [fl] 6,90 7,00 8,30 8,80 6,80 6,40 Lymphocytes [%] 30,00 51,00 51,00 43,00 44,00 42,00 Monocytes [%] 5,00 8,00 8,00 6,00 4,00 4,00 Neutrophil Granulocytes [%] 64,00 39,00 37,00 48,00 48,00 50,00 Eosinophil Granulocytes [%] 1,00 2,00 3,00 3,00 4,00 3,00 Basophil Granulocytes [%] 0,00 0,00 0,00 0,00 1,00 1,00 Tabelle 18i: Blutbild der Probanden 19 und 24
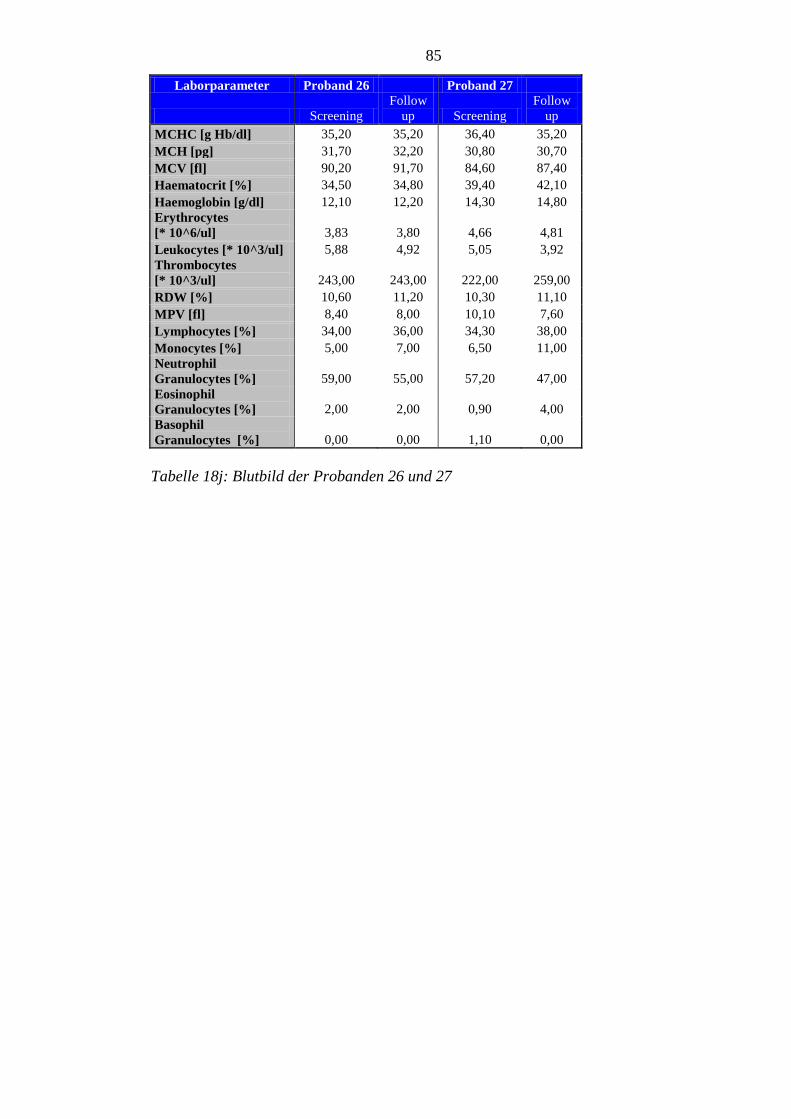
85
Laborparameter Proband 26 Proband 27
Screening Follow
up Screening Follow
up MCHC [g Hb/dl] 35,20 35,20 36,40 35,20 MCH [pg] 31,70 32,20 30,80 30,70 MCV [fl] 90,20 91,70 84,60 87,40 Haematocrit [%] 34,50 34,80 39,40 42,10 Haemoglobin [g/dl] 12,10 12,20 14,30 14,80 Erythrocytes [* 10^6/ul] 3,83 3,80 4,66 4,81 Leukocytes [* 10^3/ul] 5,88 4,92 5,05 3,92 Thrombocytes [* 10^3/ul] 243,00 243,00 222,00 259,00 RDW [%] 10,60 11,20 10,30 11,10 MPV [fl] 8,40 8,00 10,10 7,60 Lymphocytes [%] 34,00 36,00 34,30 38,00 Monocytes [%] 5,00 7,00 6,50 11,00 Neutrophil Granulocytes [%] 59,00 55,00 57,20 47,00 Eosinophil Granulocytes [%] 2,00 2,00 0,90 4,00 Basophil Granulocytes [%] 0,00 0,00 1,10 0,00 Tabelle 18j: Blutbild der Probanden 26 und 27

86
Urinuntersuchung Nitrite pH Proteins Glucose Ascorbinsäure Leukozyten Ketonkörper Urobilinogen Bilirubin Blut Gravidität Drogen auf [mg/dl] [mg/dl]
Proband 1 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg.
Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 2 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 3 Screening neg. 7 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 4 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 5 Screening neg. 7 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 6 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Poband 7 Screening neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 8 Screening neg. 7 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 9 Screening neg. 6 neg. neg. pos. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 10 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. pos. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg.
Tabelle 18k: Urinuntersuchung der Probanden 1 bis 10
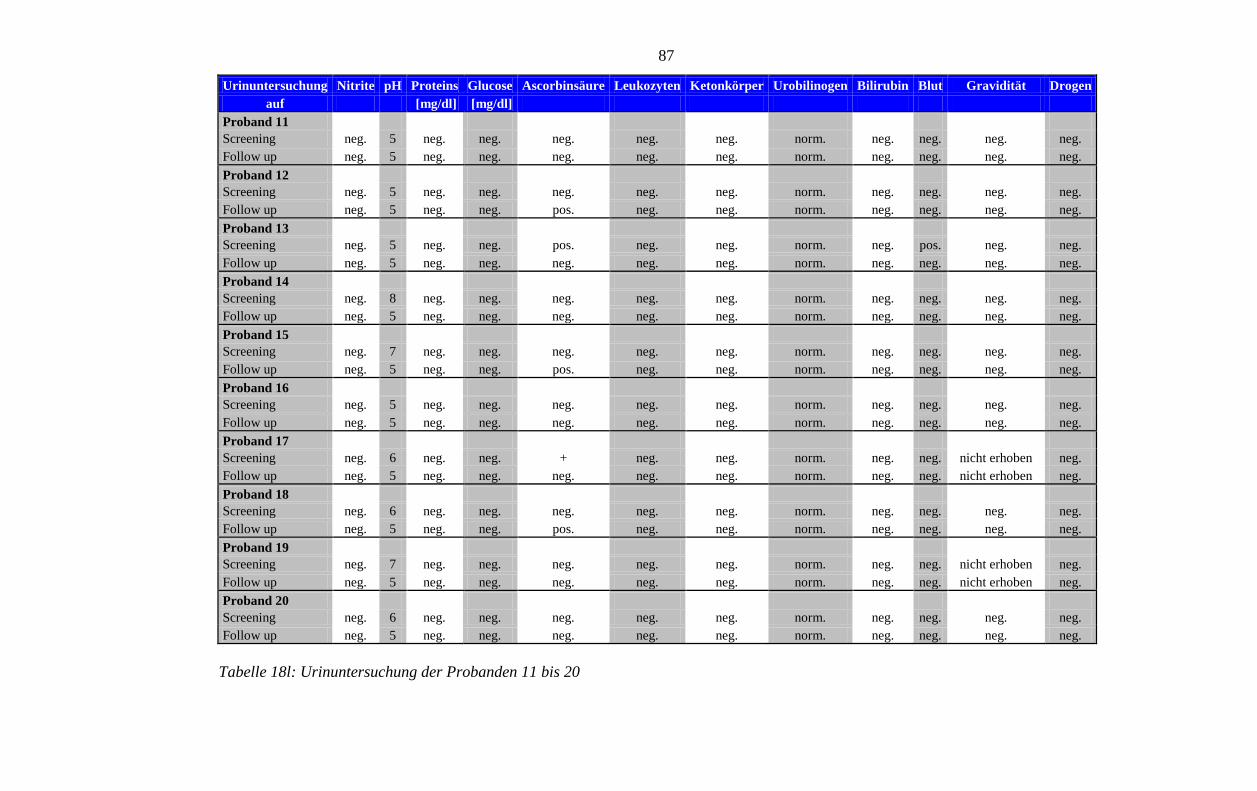
87
Urinuntersuchung Nitrite pH Proteins Glucose Ascorbinsäure Leukozyten Ketonkörper Urobilinogen Bilirubin Blut Gravidität Drogen auf [mg/dl] [mg/dl]
Proband 11 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 12 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Proband 13 Screening neg. 5 neg. neg. pos. neg. neg. norm. neg. pos. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 14 Screening neg. 8 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 15 Screening neg. 7 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Proband 16 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 17 Screening neg. 6 neg. neg. + neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 18 Screening neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Proband 19 Screening neg. 7 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. nicht erhoben neg. Proband 20 Screening neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg.
Tabelle 18l: Urinuntersuchung der Probanden 11 bis 20
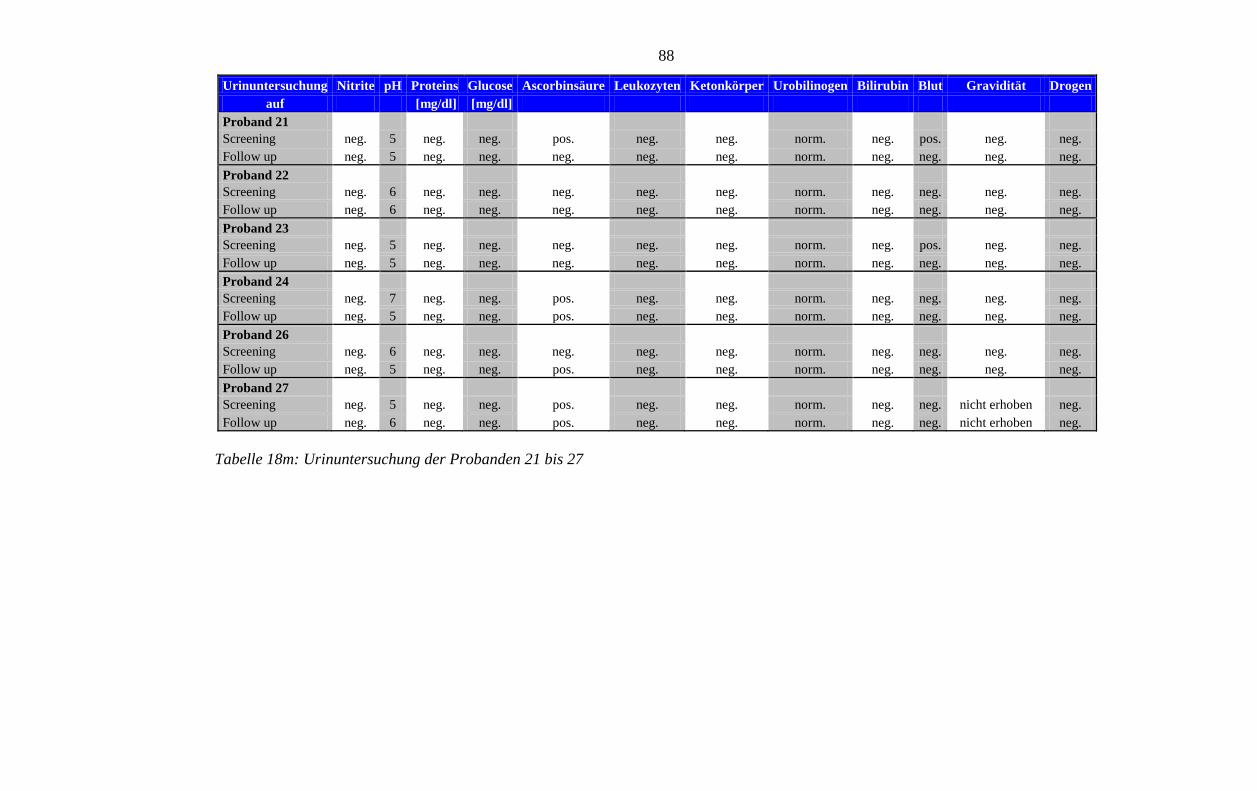
88
Urinuntersuchung Nitrite pH Proteins Glucose Ascorbinsäure Leukozyten Ketonkörper Urobilinogen Bilirubin Blut Gravidität Drogen auf [mg/dl] [mg/dl]
Proband 21 Screening neg. 5 neg. neg. pos. neg. neg. norm. neg. pos. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 22 Screening neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 23 Screening neg. 5 neg. neg. neg. neg. neg. norm. neg. pos. neg. neg. Follow up neg. 5 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Proband 24 Screening neg. 7 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Proband 26 Screening neg. 6 neg. neg. neg. neg. neg. norm. neg. neg. neg. neg. Follow up neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. neg. neg. Proband 27 Screening neg. 5 neg. neg. pos. neg. neg. norm. neg. neg. nicht erhoben neg. Follow up neg. 6 neg. neg. pos. neg. neg. norm. neg. neg. nicht erhoben neg.
Tabelle 18m: Urinuntersuchung der Probanden 21 bis 27
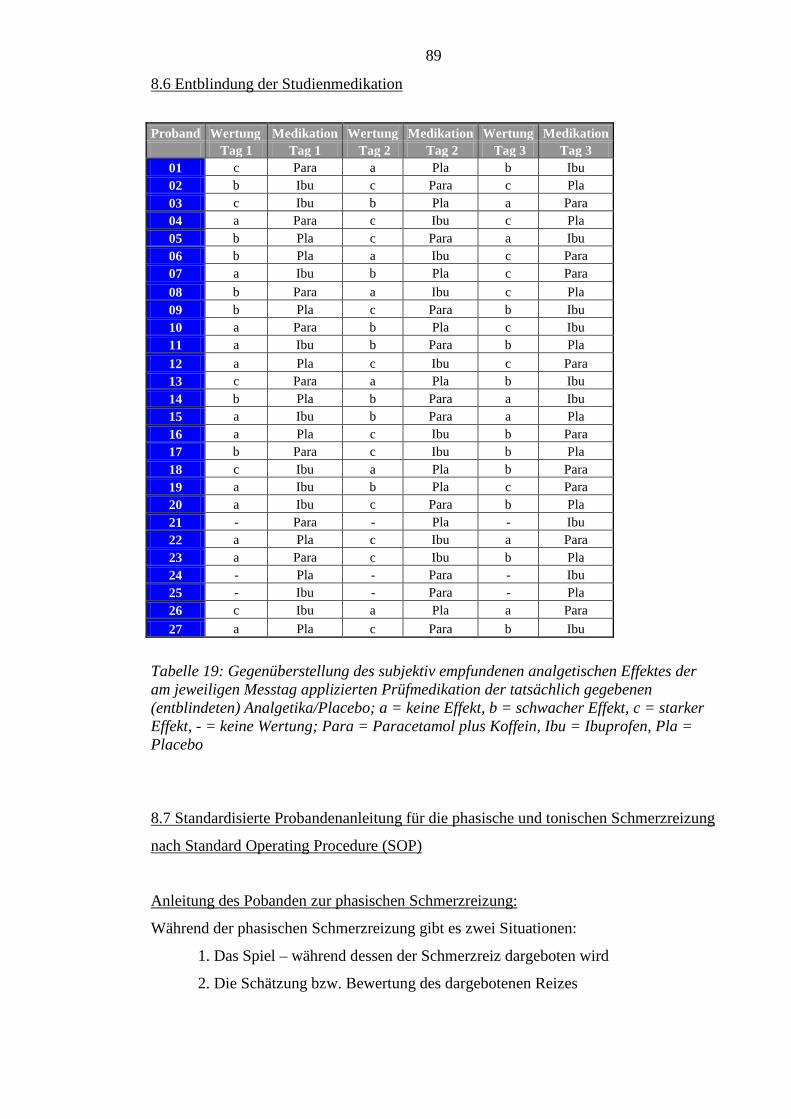
89
8.6 Entblindung der Studienmedikation
Tabelle 19: Gegenüberstellung des subjektiv empfundenen analgetischen Effektes der am jeweiligen Messtag applizierten Prüfmedikation der tatsächlich gegebenen (entblindeten) Analgetika/Placebo; a = keine Effekt, b = schwacher Effekt, c = starker Effekt, - = keine Wertung; Para = Paracetamol plus Koffein, Ibu = Ibuprofen, Pla = Placebo
8.7 Standardisierte Probandenanleitung für die phasische und tonischen Schmerzreizung
nach Standard Operating Procedure (SOP)
Anleitung des Pobanden zur phasischen Schmerzreizung:
Während der phasischen Schmerzreizung gibt es zwei Situationen:
1. Das Spiel – während dessen der Schmerzreiz dargeboten wird
2. Die Schätzung bzw. Bewertung des dargebotenen Reizes
Proband Wertung Medikation Wertung Medikation Wertung Medikation Tag 1 Tag 1 Tag 2 Tag 2 Tag 3 Tag 3
01 c Para a Pla b Ibu 02 b Ibu c Para c Pla 03 c Ibu b Pla a Para 04 a Para c Ibu c Pla 05 b Pla c Para a Ibu 06 b Pla a Ibu c Para 07 a Ibu b Pla c Para 08 b Para a Ibu c Pla 09 b Pla c Para b Ibu 10 a Para b Pla c Ibu 11 a Ibu b Para b Pla 12 a Pla c Ibu c Para 13 c Para a Pla b Ibu 14 b Pla b Para a Ibu 15 a Ibu b Para a Pla 16 a Pla c Ibu b Para 17 b Para c Ibu b Pla 18 c Ibu a Pla b Para 19 a Ibu b Pla c Para 20 a Ibu c Para b Pla 21 ´- Para ´- Pla ´- Ibu 22 a Pla c Ibu a Para 23 a Para c Ibu b Pla 24 ´- Pla ´- Para ´- Ibu 25 ´- Ibu ´- Para ´- Pla 26 c Ibu a Pla a Para 27 a Pla c Para b Ibu

90
(1.) Während des Spieles:
- Bitte nicht zwinkern
- Schlucken vermeiden
- Auf entspannte Kopfhaltung und entspannten Schulter- und Nackenbereich achten
- Atemtechnik beachten
(2.) Während der Schätzung:
- Bitte kurz Zwinkern, Schlucken und den Mund anfeuchten
Erläuterungen zur Baseline-Messung:
- Es folgen zwei Adaptationsreize und ein Standard-Reiz, der auf 100 (grauer Balken
parallel zum roten Balken) eingeschätzt werden muss.
- Der Standardreiz wird nur einmal am Messtag gegeben. Diesen bitte für den
gesamten Messtag merken und alle Reize in Bezug auf den Standardreiz
einschätzen.
- Die empfundene Reizstärke kann, in Relation zum Standardreiz, von ’kein
Schmerzempfinden’ (= grüner Balken ganz am linken Bildschirmrand = 0) bis ’eben
noch tolerierbares Schmerzempfinden’ (= grüner Balken ganz am rechten
Bildschirmrand = 200) gewählt werden.
- Bei Ausbleiben einer Schätzung bitte sofort melden!
- Es folgen jetzt zwei Adaptationsreize von den der Erste (Adaptation 1) angekündigt
und der Zweite (Adaptation 2) unangekündigt sind
- Adaptationsreiz 1 folgt jetzt – wurde dieser erhalten?
- Adaptationsreiz 2 unangekündigt – wurde dieser erhalten?
- Das Spiel wird jetzt, nach Anschalten des Kopfhörer gestartet. Es folgt der
Standard-Reiz, der auf 100 (grauer Balken parallel zum roten Balken) eingeschätzt
werden muss. Diesen bitte für den gesamten Messtag merken und alle Reize in
Bezug auf den Standardreiz einschätzen
- Bitte an die Atemtechnik denken und nicht zwinkern!
- Ich schalte den Kopfhörer jetzt an und dann beginnt die Messung
Erläuterungen zum Messblock 1 – 4:
- Es folgen jetzt zwei Adaptationsreize, von den der Erste (Adaptation 1) angekündigt
und der Zweite (Adaptation 2) unangekündigt sind. Es folgt kein Standard-Reiz
mehr!

91
- Adaptationsreiz 1 folgt jetzt – wurde dieser erhalten?
- Adaptationsreiz 2 unangekündigt – wurde dieser erhalten?
- Alle jetzt folgenden Reize bitte in Bezug auf den Standardreiz einschätzen
- Bitte an die Atemtechnik denken und nicht zwinkern!
- Ich schalte den Kopfhörer jetzt an und dann beginnt die Messung
Anleitung des Pobanden für die tonische Schmerzreizung
Während der tonischen Schmerzreizung gibt es zwei Situationen:
1. Das Spiel
2. Die Schätzung bzw. Bewertung des aktuellen Schmerzempfindens
(1.) Während des Spieles:
- bitte nicht zwinkern
- Schlucken vermeiden
- auf entspannte Kopfhaltung und entspannten Schulter und Nackenbereich acht
(2.) Während der Schätzung:
- bitte kurz Zwinkern, Schlucken und den Mund anfeuchten
- Die empfundene Reizstärke kann von ’kein Schmerzempfinden’ (= weißer Balken
ganz am linken Bildschirmrand = 0) bis ’eben noch tolerierbares
Schmerzempfinden’ (= weißer Balken ganz am rechten Bildschirmrand = 100)
gewählt werden.
An die Atemtechnik muss hier (während der tonischen Reizung) nicht gedacht werden!
Ich schalte den Kopfhörer jetzt an und dann beginnt die Messung.

92
9. Danksagung
Herrn Prof. Dr. med. Dr. h.c. K. Brune danke ich für die freundliche Überlassung dieses
Themas sowie für die Möglichkeit, die Studie am Institut für experimentelle und
klinische Pharmakologie und Toxikologie der Friedrich-Alexander-Universität
Erlangen-Nürnberg durch zu führen.
Bei Herrn Dr. med. Bertold Renner möchte ich mich von ganzem Herzen für seine
stetige Einsatzbereitschaft sowie seine beständige und geduldige Unterstützung bei allen
kleinen und großen Problemen bedanken.
Meinem Kommilitonen und Arbeitskollegen Daniel Busse, den Mitarbeitern der
Arbeitsgruppe Physiologische Pharmakologie sowie all denen im Rahmen der Studie
beteiligten Probanden, die so manche gewollten und ungewollten Schmerzen über sich
ergehen lassen mussten sei hier ausdrücklich gedankt.
Mein abschließender Dank geht an meine Eltern und meine Großmutter für ihre
immerwährende Unterstützung in allen Bereichen, ihr Vertrauen und ihren tiefen
Glauben an mich.

93
10. Curriculum vitae
Persönliche Daten:
Name: Gregor Johannes Muth
Geburtsdatum: 06.05.1980
Geburtsort: Achern, Baden
Familienstand: Ledig
Konfession: Römisch-katholisch
Eltern: Carl Muth, Sicherheitsingenieur
Christiane Muth, dipl. Bibliothekarin
Geschwister: Claudius Muth, Opernsänger
Bildungsgang:
1986 – 1990 Grundschule Lichtenau, Scherzheim
1990 – 1999 Gymnasium Heimschule Lender, Sasbach; Abschluss: Abitur
1999 – 2000 Zivildienst: DRK Kreisverband Bühl / Baden, Rettungshelfer
2002 – 2004 Rettungssanitäter, DRK Bühl/Baden; Malteser Hilfsdienst, Köln
2002 – 2008 Studium der Humanmedizin, Friedrich-Alexander-Universität Erlangen-
Nürnberg, Abschluss: Staatsexamen
Seit 10/2008 Assistenzarzt Innere Medizin, Bethesda AK Bergedorf, Lehrkrankenhaus
der Universität Hamburg


















