
Blut und Blutgerinnung Teil 2 - boku.ac.at · 2 (I) Bestandteile und Funktionen des Blutes (II)...
124
1 Blut und Blutgerinnung Teil 2
Transcript of Blut und Blutgerinnung Teil 2 - boku.ac.at · 2 (I) Bestandteile und Funktionen des Blutes (II)...

1
Blut und Blutgerinnung
Teil 2

2
(I) Bestandteile und Funktionen des Blutes
(II) Blutgerinnung und Fibrinolyse
(III) Regulation der Blutgerinnung
(IV) Wechselwirkung von Gerinnung und Entzündung
(V) Antikoagulation und Methoden der Blutreinigung
INHALT

3
Inhalt
• Blutgruppen
• zelluläre und plasmatische Gerinnung
• Regulation der Gerinnung
• Fibrinolyse

Die Blutgruppen
4

5
Blutgruppen
verschiedene Blutgruppensysteme
am wichtigsten: AB0-System
Rhesus-Faktor
Karl Landsteiner 1901
beschrieb A, B, 0
Castello&Sturli 1902 AB
Nobelpreis für Medizin 1930

6
Karl Landsteiner1868 - 1943
2655

7
Blutgruppenantigene
Glykolipide und Glykoproteine auf der
Oberfläche von Erythrozyten, Leukozyten, Thrombozyten, Endothelzellen
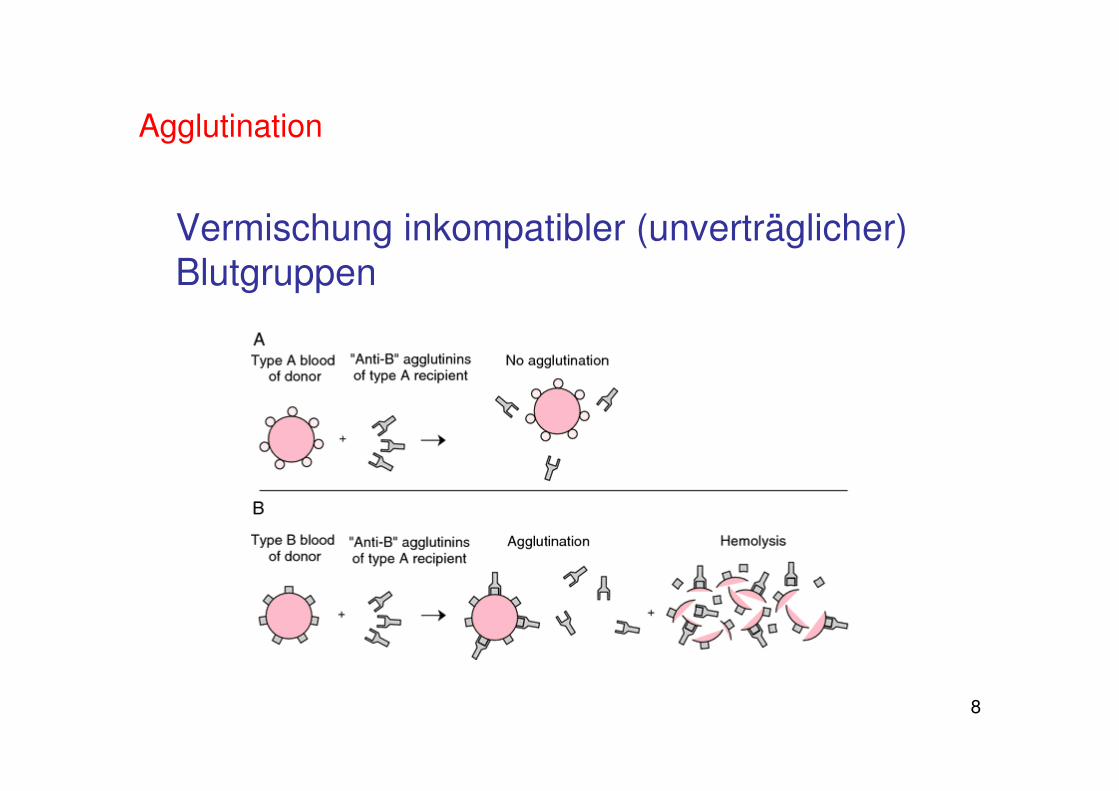
gegen die Blutgruppenantigene der jeweils anderen BG gibt es Antikörper im Blut
Mischung von Blut verschiedener Blutgruppen führt zu Verklumpung: Agglutination
8
Agglutination
Vermischung inkompatibler (unverträglicher) Blutgruppen

9
AB0-System
• 4 Hauptgruppen: A, B, AB, 0
• angehängtes + oder – bezeichnet den Rhesusfaktor
Erythrozyt
Blutgruppenantigen
YAntikörper

10
11
AB0-System
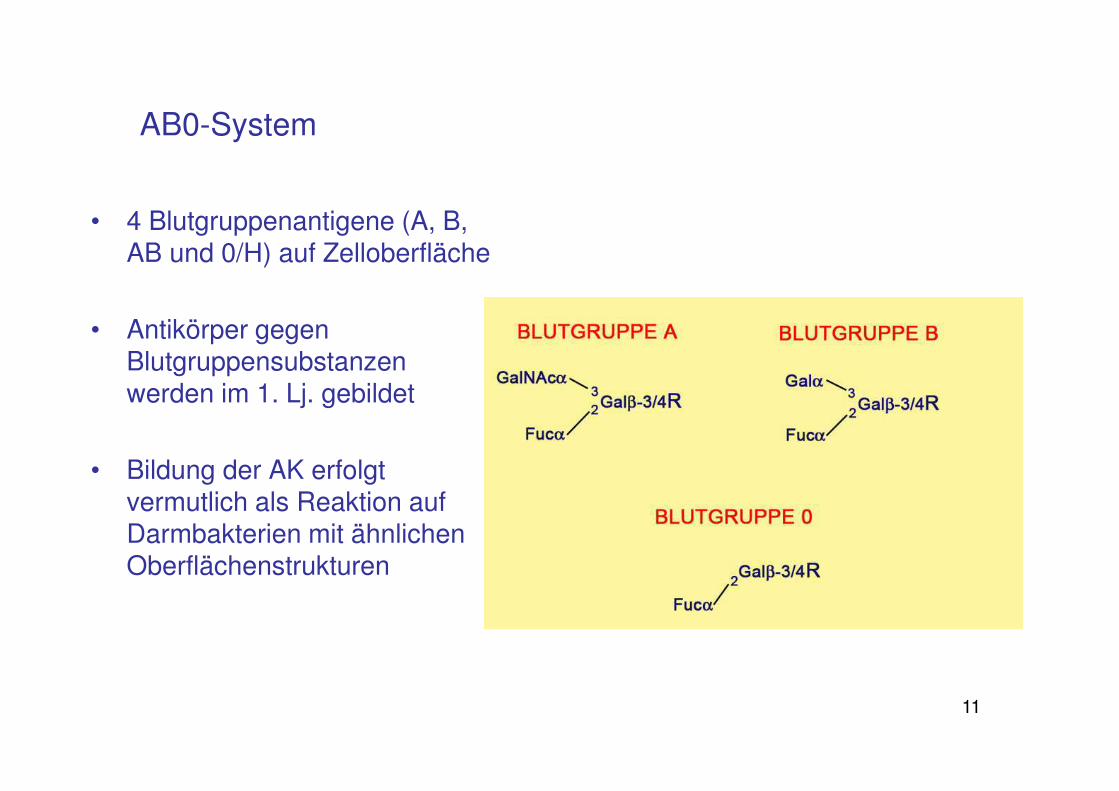
• 4 Blutgruppenantigene (A, B, AB und 0/H) auf Zelloberfläche
• Antikörper gegen Blutgruppensubstanzen werden im 1. Lj. gebildet
• Bildung der AK erfolgt vermutlich als Reaktion auf Darmbakterien mit ähnlichen Oberflächenstrukturen

12
Häufigkeit der Blutgruppen
Häufigkeitsverteilung der Blutgruppen in Österreich Quelle: ÖRK

13
Bedeutung
• Bluttransfusion
• AK sind beim AB0-System bereits vorhanden, sofortige Reaktion
• bei anderen Blutgruppensystemen werden AK erst gebildet und sind daher erst bei Nachfolgetransfusionen von Bedeutung
• inkompatible Blutübertragung führt zur Hämolyse

14
Rhesusfaktor
• Gruppe von Proteinen auf Erythrozytenoberfläche
• wichtigster: Faktor D
• Entdeckt von Karl Landsteiner im Blut von Rhesusaffen (1930)
• ca.15% der Europäer sind Rh-negativ

15
15% Rh- dd homozygot 50% Rh+ Dd heterozygot35% Rh+ DD homozygot
Mutter Rh- ddVater Rh+ DD Kind dD und somit Rh+
Mutter Rh- ddVater Rh+ dD Kind entweder dd (Rh-) oder dD (Rh+)
Rhesusfaktor

16
Rhesus-Inkompatibilität
• Rh- Mutter und Rh+ Kind
Morbus haemolyticus neonatorum:
• Geburt: Vermischung von Blut von Mutter und Kind • Mutter wird sensibilisiert und bildet Antikörper gegen das Rhesus-
Antigen • In einer nachfolgenden Schwangerschaft können diese AK über die
Placenta in den Kreislauf des Kindes übertreten und dort an Erythrozyten binden
• daraus resultiert eine hämolytische Anämie • vorbeugende Gabe von anti-D-Antikörper an die Rh- Mutter in der
ersten Schwangerschaft (inaktiviert Rh+ Blutkörperchen des Kindes beim Übertritt in den Kreislauf der Mutter, verhindert Antikörperbildung bei der Mutter)

17
Geschichte der Bluttransfusion lat. transfundere: hinübergießen
1492 Erster bekannter Versuch einer BlutübertragungPapst Innocens VIII (1432 - 1492 †) trinkt das Blut von drei Buben
1628 William Harvey entdeckt Blutkreislauf
1649 Bluttransfusion zwischen zwei Hennendurch Francis Potter
18
Bluttransfusion Schaf-Mensch (1667)

19
• 1818 James Blundell
• Transfusionen an Frauen mit großem Blutverlust während der Geburt
• 4 von 12 überlebten
Geschichte der Bluttransfusion

20
Am 18. Dezember 1891 hielt der Vorstand des chemischen Laboratoriums an der k. k. Krankenanstalt „Rudolf-Stiftung" Dr. Ernst Freund (1863·1946) vor der Gesellschaft der Ärzte in Wien einen Vortrag„Über die Ursachen der Blutgerinnung", in welchem er ausführte:
„Wenn das Blut, das ja bekanntlich aus der Blutflüssigkeit (Plasma) und den Blutkörperchen besteht, die Ader verlässt, dann verliert es unter gewöhnlichen Verhältnissen den Charakter einer Flüssigkeit. Verbleibt das Blut darauf in Ruhe, dann gerinnt dasselbe zu einer gallertartigen Masse, welche sich allmälig in zwei Theile scheidet, in eine klare, gelbgefärbte Flüssigkeit, die man Blutserum nennt, und in eine feste, rothe Masse, die man Blutkuchen nennt . . ."
•Blutgruppen •Blutgerinnung

21
1914Luis Agote
erste erfolgreiche Bluttransfusion (Buenos Aires)
Antikoagulation mit Natriumcitrat
Spanischer Bürgerkrieg
erster großer Einsatz von Bluttransfusionen (20.000 Konserven)

22
Blutgerinnung und Fibrinolyse

23
Begriffe
Hämostase griech αιµααιµααιµααιµα, Blut; στασισστασισστασισστασισ, Stillstand
Primäre (zelluläre) Hämostase
Blutstillung
Sekundäre (plasmatische) Hämostase
Blutgerinnung

24
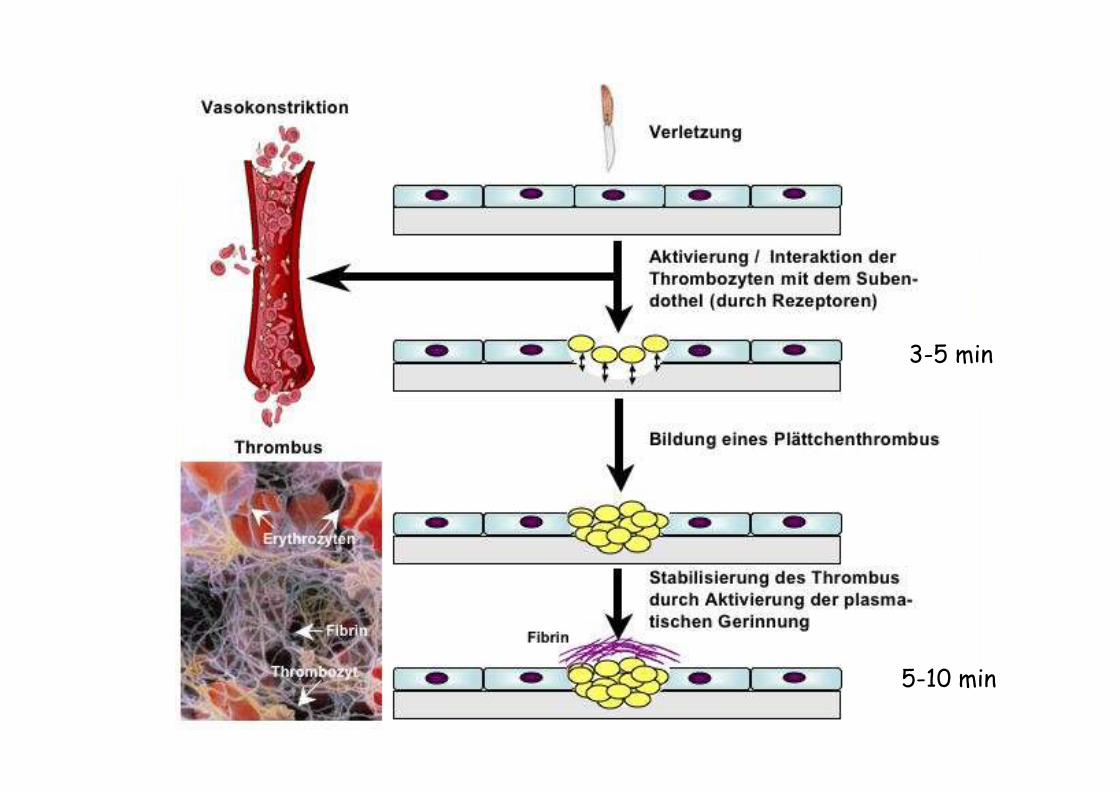
3-5 min
5-10 min

25
Koagulation
Antikoagulation
Fibrinolyse

26
Spronk et al 2003, BioEssays 25:1220-1228

27
Gerinnungssystem besteht aus
Zellen Thrombozyten
Membranmikrovesikeln von verschiedenen Zellen
ProteinenGerinnungsfaktoren antikoagulatorische Faktoren
Cofaktoren Calcium

Release of MVs and exosomes.
Raposo G , and Stoorvogel W J Cell Biol 2013;200:373-383
© 2013 Raposo and Stoorvogel

Microvesicle shedding.
Muralidharan-Chari V et al. J Cell Sci 2010;123:1603-1611
©2010 by The Company of Biologists Ltd
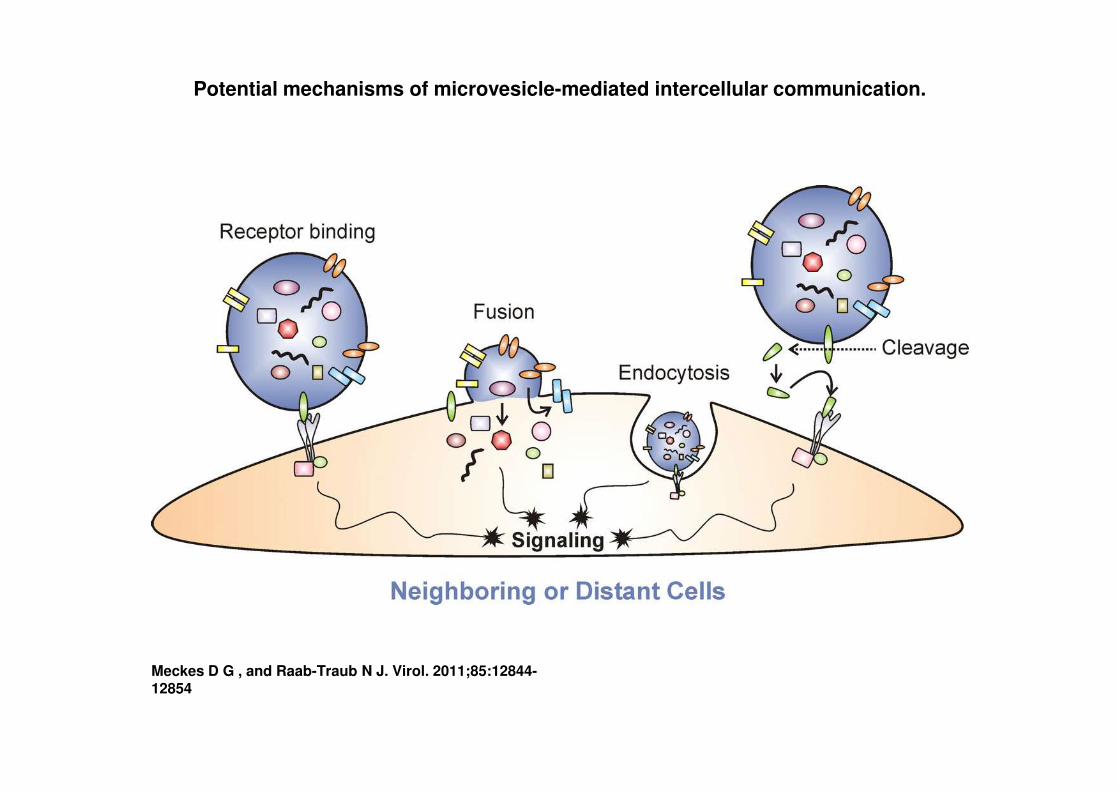
Potential mechanisms of microvesicle-mediated intercellular communication.
Meckes D G , and Raab-Traub N J. Virol. 2011;85:12844-12854

31
Interaktion dieser Faktoren ....
...führt zu
Bildung von Fibrin aus Fibrinogen

32
Hippokrates (460-377 v. Chr.)Über die Natur des Menschen: tierisches Blut gerinnt „beim Abkühlen“
Steven Blankaart (1650-1702) Opera Medica, Theoretica, Practica et Chirurgica:
„Viele Krankheiten entstehen durch ein Übermaß der Säuren im Blut. Wenn sich Partikel verhaken und nicht mehr recht bewegen können, oder auch durch Abkühlung des Blutes,entstehen gefährliche Gefäßverstopfungen".
Geschichte

33
Geschichte
Buchanan 1836frisch geronnenes Blut beschleunigt die Gerinnung (Thrombin!)
Hammersten 1879 Reinigung von Fibrinogen
Bizzozero 1882Beschreibung der Blutplättchen
Arthus 1890Rolle von Calcium bei der Blutgerinnung
Morawitz 1904
Thrombokinase, CaProthrombin > Thrombin
ThrombinFibrinogen > Fibrin

34
Geschichte
1900-1950
• Entdeckung und Charakterisierung der weiteren Gerinnungsfaktoren und Co-Faktoren
• Synthese der Gerinnungsfaktoren erfolgt in der Leber
• Faktor II, VII, IX, und X sind Vitamin-K-abhängig und enthalten γ-Carboxy-Glutamat

35
Geschichte
1964 Beschreibung des Gerinnungsablaufs als Kaskaden- oder Wasserfallmodell
Macfarlane R
An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier.
Nature 1964, 202:498-499
Davie E, Ratnoff O
Waterfall sequence for intrinsic blood clotting.
Science 1964, 145: 1310-1312
36

37
Phase I
Primäre Hämostase
Blutstillung
38
3-5 min
5-10 min

39
Blutung nach einer Verletzung sistiert nach 2-5 min
- Gefäßverengung (Vasokonstriktion)
- Thrombozytenadhäsion
40
Plättchen im Blut Adhäsion & Aktivierung Aggregation

41

42
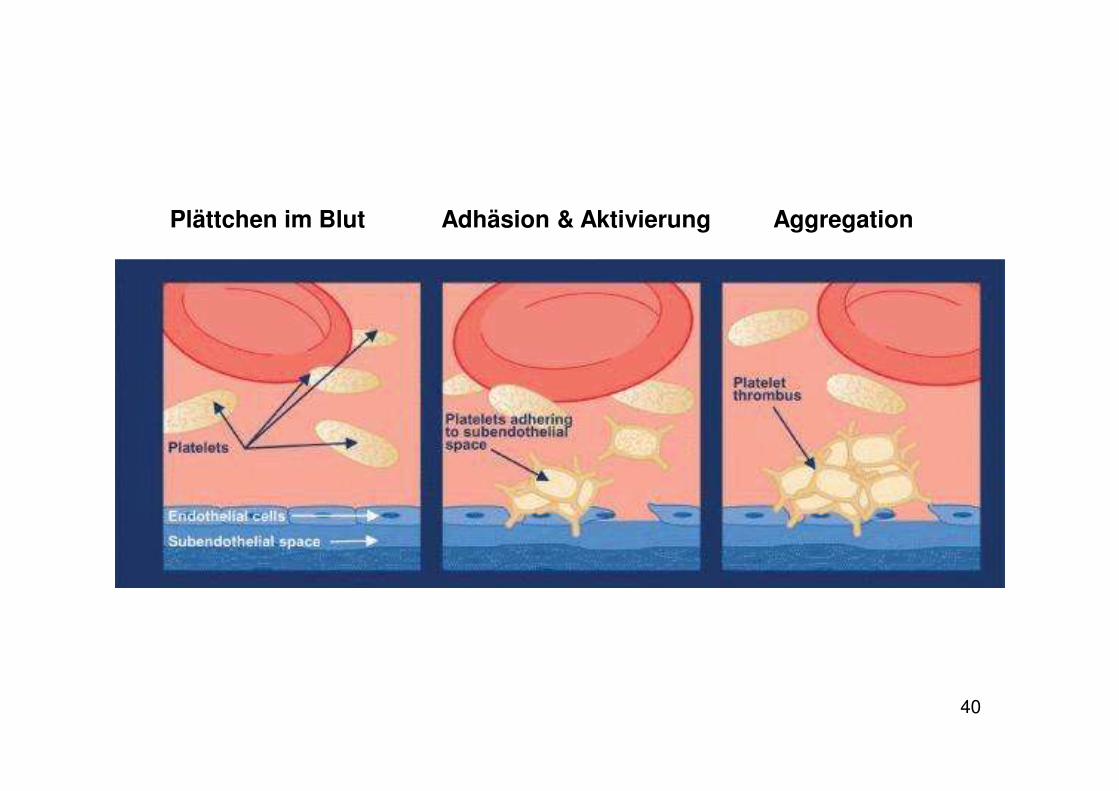
Keine Adhäsion auf gesundem Endothel
Kontakt mit subendothelialem Gewebe:� Adhäsion und Aktivierung
Plättchen

43
Aktivierte Plättchen: • Geänderte Morphologie • Anheftung an das Subendothel • Freisetzung von Mediatoren (z. B. Thromboxan A2, ADP,
Serotonin, Ca ++) • Aus den α-Granula werden außerdem Gerinnungsfaktoren und
Wachstumsfaktoren und freigesetzt• Aggregation der Plättchen • Aktivierung der plasmatischen Gerinnung und
Fibrinbildung
Plättchen

44
Plättchenadhäsion: Anheftung der Plättchen an das Subendothel
Interaktion der Plättchen mit der freiliegenden extrazellulären Matrix bei Verletzungen des Endothels
Die extrazelluläre Matrix enthält adhäsive Makromoleküle:
Laminin
Fibronectin
Kollagen
Interaktion mit von Willebrand Faktor (vWF)

45
Von Willebrand-Faktor (vWF)
• multimeres Glykoprotein
• Bindungsstellen für Kollagen GPIb (von Willebrand-Rezeptor auf Plättchen) Integrin αIIβIII (GPIIb/IIIa)
findet sich in Endothelzellen, in Granula der Plättchen, sowie im Plasma (10 µg/mL)
unter normalen Bedingungen interagiert löslicher vWF nicht mit Rezeptoren auf Plättchen
bei Endothelverletzung bindet vWF an Kollagen in der ECM> stark adhäsives Substrat

46
GPIb-vWF Komplex
• Wechselwirkung zwischen GPIb (auf der Plättchenoberfläche) und vWF (an Kollagen gebunden ) stellt den
ersten Schritt der Plättchenadhäsion dar
• keine feste Verankerung
• bringt Plättchen an den Ort der Verletzung
• verlangsamt die Fließschwindigkeit der Plättchen am Ort der Verletzung („Rollen“ der Plättchen über die Oberfläche)

47
Plättchenadhäsion
• Scherkräfte !
• Blut fließt mit größerer Geschwindigkeit in der Mitte der Gefäße (parabolisches Profil)
• Geschwindigkeit nimmt zur Gefäßwand hin ab
• Scherkräfte sind an der Gefäßwand und in kleinen Gefäßen am größten
48
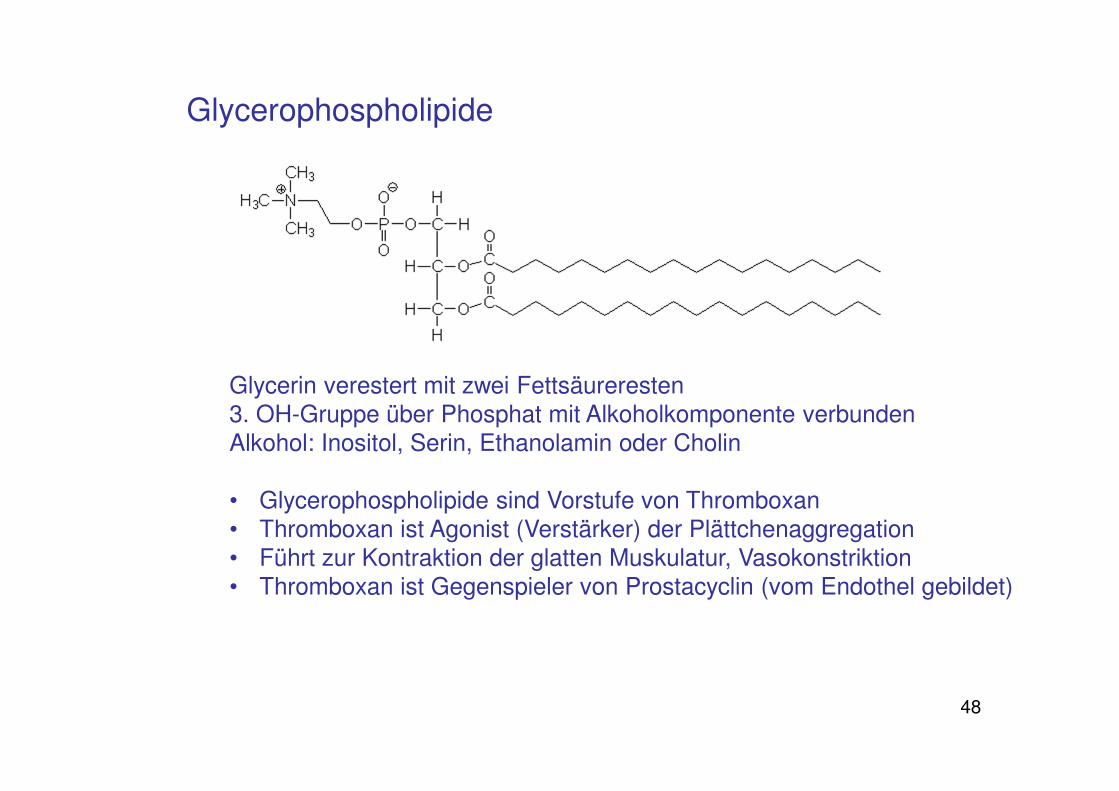
Glycerophospholipide
Glycerin verestert mit zwei Fettsäureresten 3. OH-Gruppe über Phosphat mit Alkoholkomponente verbundenAlkohol: Inositol, Serin, Ethanolamin oder Cholin
• Glycerophospholipide sind Vorstufe von Thromboxan• Thromboxan ist Agonist (Verstärker) der Plättchenaggregation• Führt zur Kontraktion der glatten Muskulatur, Vasokonstriktion • Thromboxan ist Gegenspieler von Prostacyclin (vom Endothel gebildet)

49
Integrine
Transmembranproteine
große extrazelluläre N-terminale Domäne, Transmembrandomäne, kleine zytoplasmatische Domäne
werden auf der Oberfläche nicht aktivierter Plättchen exprimiert (niedrige Affinität; „OFF“)
Konformationsänderung in Zustand hoher Affinität („ON“) im Fall der Plättchenaktivierung

50
Integrine
• verschiedene Integrine:
Kollagen-Rezeptor
Fibronectin-Rezeptor
Laminin-Rezeptor
2 wichtigste: GPIa/IIa
GPIIb/IIIa

51
GPIIb/IIIa
60.000 - 80.000 Kopien/Plättchen
bindet Fibrinogen
Fibronectin
vWF
GPIa/IIa
erster beschriebener
Kollagenrezeptor
2000-4000 Kopien/Plättchen
bindet Kollagen

52
GPIIb/IIIa
Liganden enthalten die Sequenz
Arginin-Glycin-Asparaginsäure (RGD)
im „OFF-Zustand“ stark gebogene Konformation - -> RGD-Bindungsstelle ist versteckt
vgl. Klappmesser
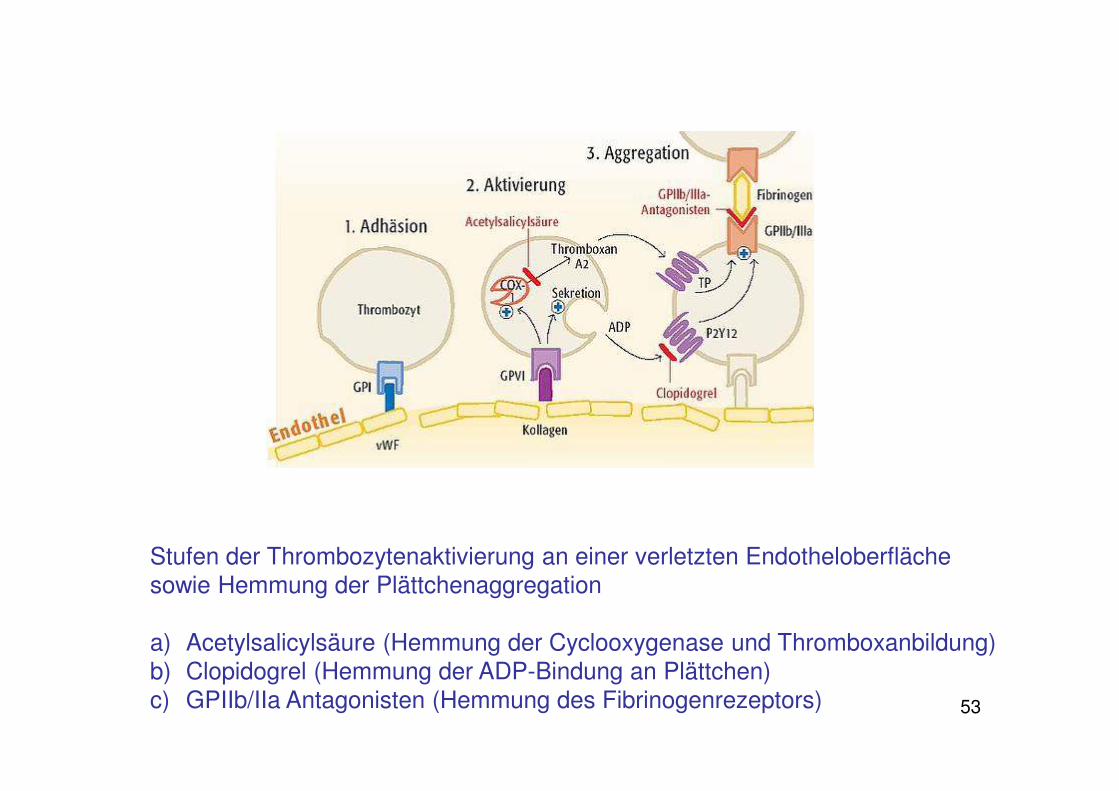
53
Stufen der Thrombozytenaktivierung an einer verletzten Endotheloberflächesowie Hemmung der Plättchenaggregation
a) Acetylsalicylsäure (Hemmung der Cyclooxygenase und Thromboxanbildung)b) Clopidogrel (Hemmung der ADP-Bindung an Plättchen) c) GPIIb/IIa Antagonisten (Hemmung des Fibrinogenrezeptors)

54
Phase II
Sekundäre Hämostase
Blutgerinnung
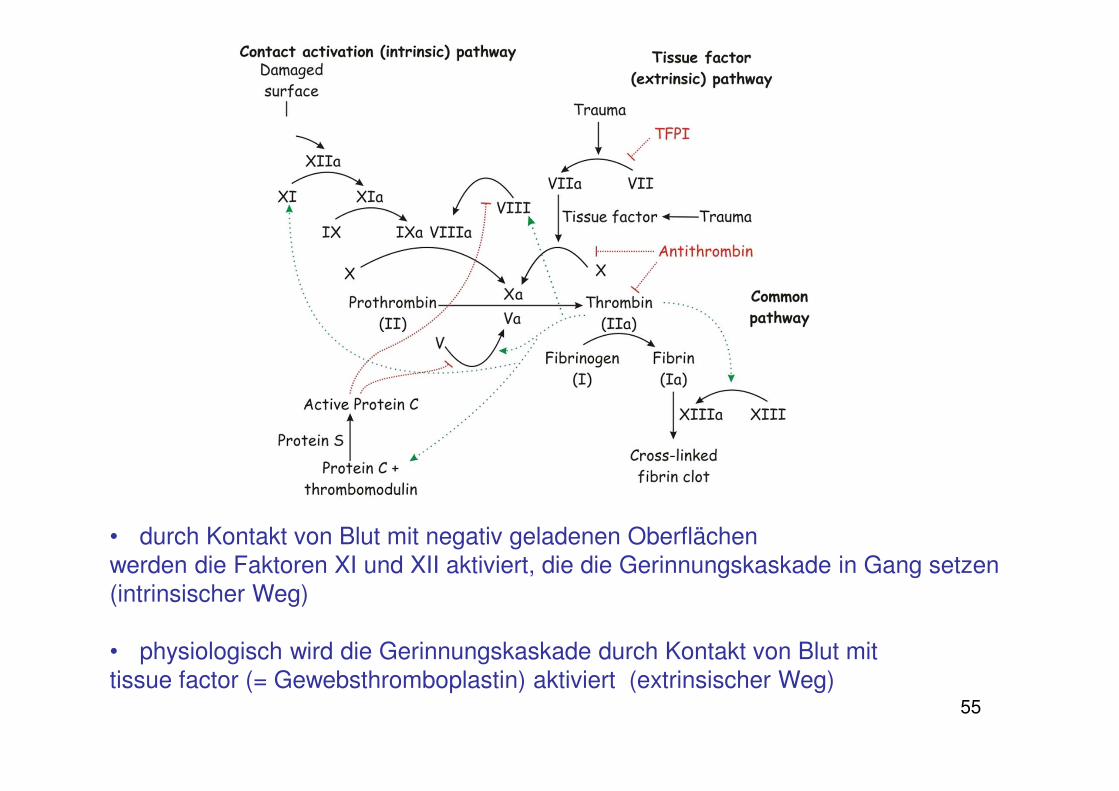
55
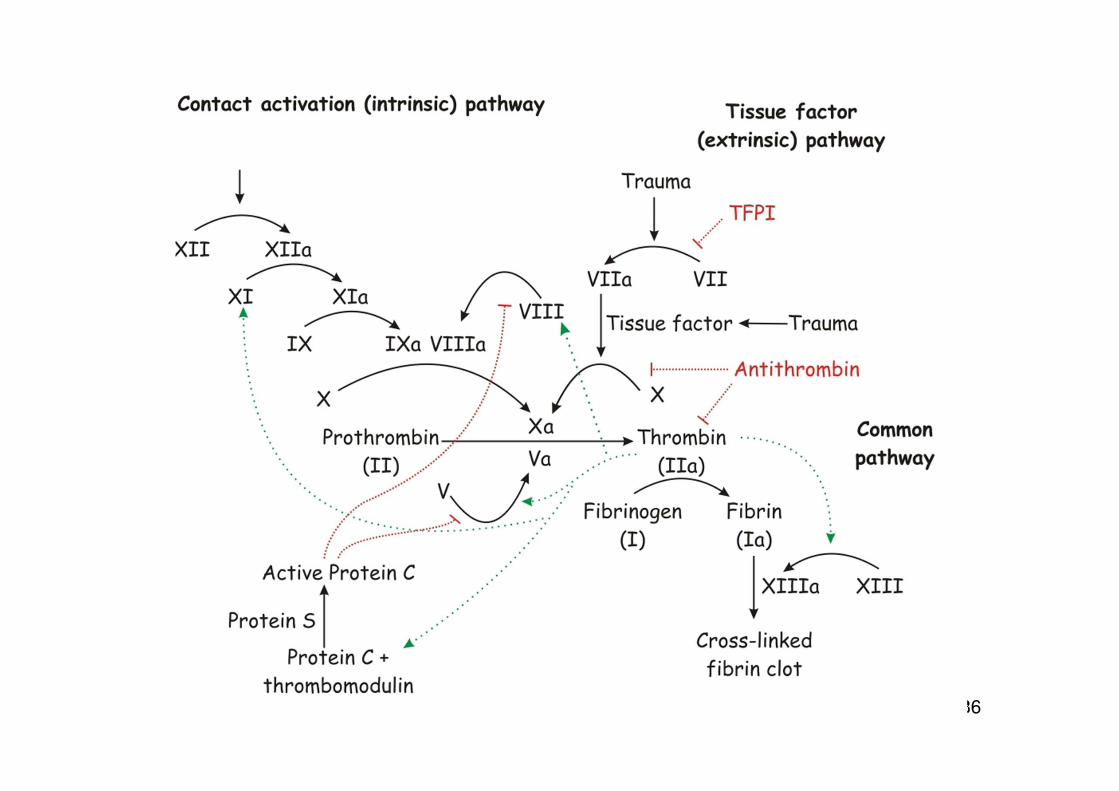
• durch Kontakt von Blut mit negativ geladenen Oberflächen werden die Faktoren XI und XII aktiviert, die die Gerinnungskaskade in Gang setzen (intrinsischer Weg)
• physiologisch wird die Gerinnungskaskade durch Kontakt von Blut mit tissue factor (= Gewebsthromboplastin) aktiviert (extrinsischer Weg)

56
Nomenklatur der Gerinnungsfaktoren
Proenzyme (inaktiv) römische Ziffer X, FX
Enzyme (aktiviert) römische Ziffer+a Xa, FXa
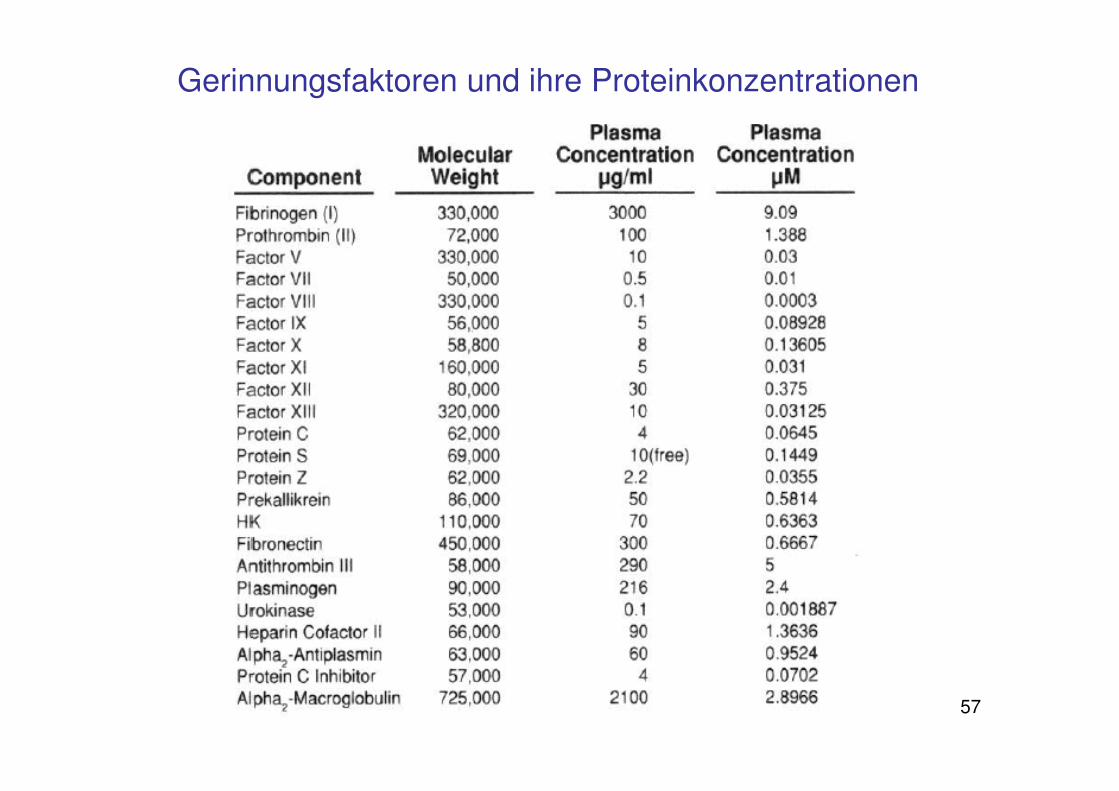
57
Gerinnungsfaktoren und ihre Proteinkonzentrationen

58
FIII Thromboplastin, tissue factor
(subendotheliales Gewebe)
FIV Ca2+
wird von vielen Faktoren zur Bindung an negativ geladene Membranphospholipide benötigt
FVI = FVa
Weitere Faktoren
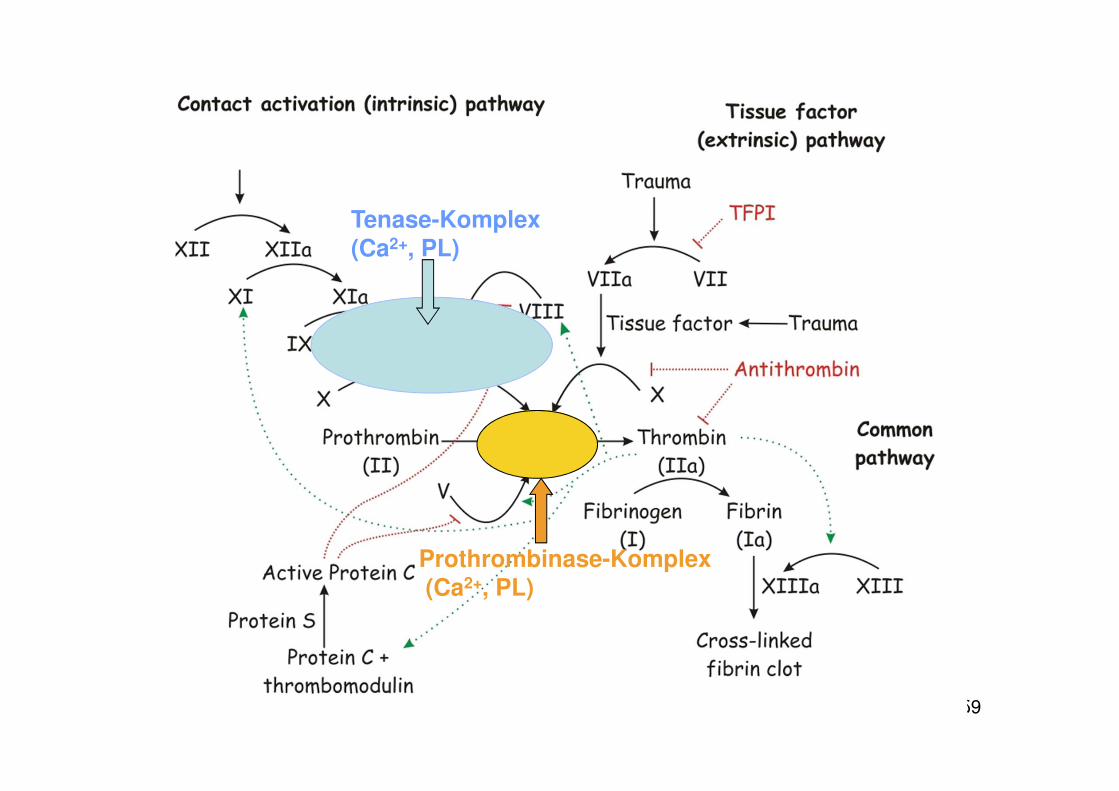
59
Tenase-Komplex(Ca2+, PL)
Prothrombinase-Komplex(Ca2+, PL)

60
Kaskadenmodell der Gerinnung
• ...gültig, wird aber in der jüngeren Literatur teilweise modifiziert
• „revised model of coagulation“
z.B. Hoffman & Monroe 2001; A cell-based model of hemostasisThromb. Haemost. 85: 958-965

61
Warum ?
• Kaskadenmodell stellt den Gerinnungsablauf als eine Abfolge proteolytischer Reaktionen dar
• Rolle der Zellen steht sehr im Hintergrund (als Lieferant für eine Phospholipidoberfläche)
• Rolle der Zellen wird in der neuen Sicht stärker betont

62
Revised Model of Coagulation
• Zentrale Rolle von tissue factor
• Rolle der Zellen: geht über die Bereitstellung einer Phospholipid-Oberfläche hinaus
-- Zelluläre Komponenten regulieren wesentlich den Ablauf der Gerinnung

63
Revised Model of Coagulation
Dreiphasenmodell der Gerinnung:
Initiation: auf Zellen, die tissue factor exprimieren
Amplifikation: Verlagerung des Geschehens auf die Plättchenoberfläche, PlättchenadhäsionPlättchenaktivierung
Propagation: Thrombinbildung und Fibrinpolymerisation

64
Tissue factor
• primärer physiologischer Auslösefaktor für Gerinnung• integrales Membranprotein • exprimiert auf extravaskulären Zellen (im Gewebe) • bei Entzündung auch auf Monozyten und Endothelzellen,
Abschnürung von TF-haltigen Vesikeln (Mikrovesikel) • Schaden in der Gefäßwand führt zu Kontakt von Plasma mit tissue-
factor exprimierenden Zellen, dadurch wird die Gerinnung aktiviert• Wichtig ist auch die Verbindung von Entzündung und Gerinnung

65
Rolle der Phospholipid-Oberflächen bei der Gerinnung
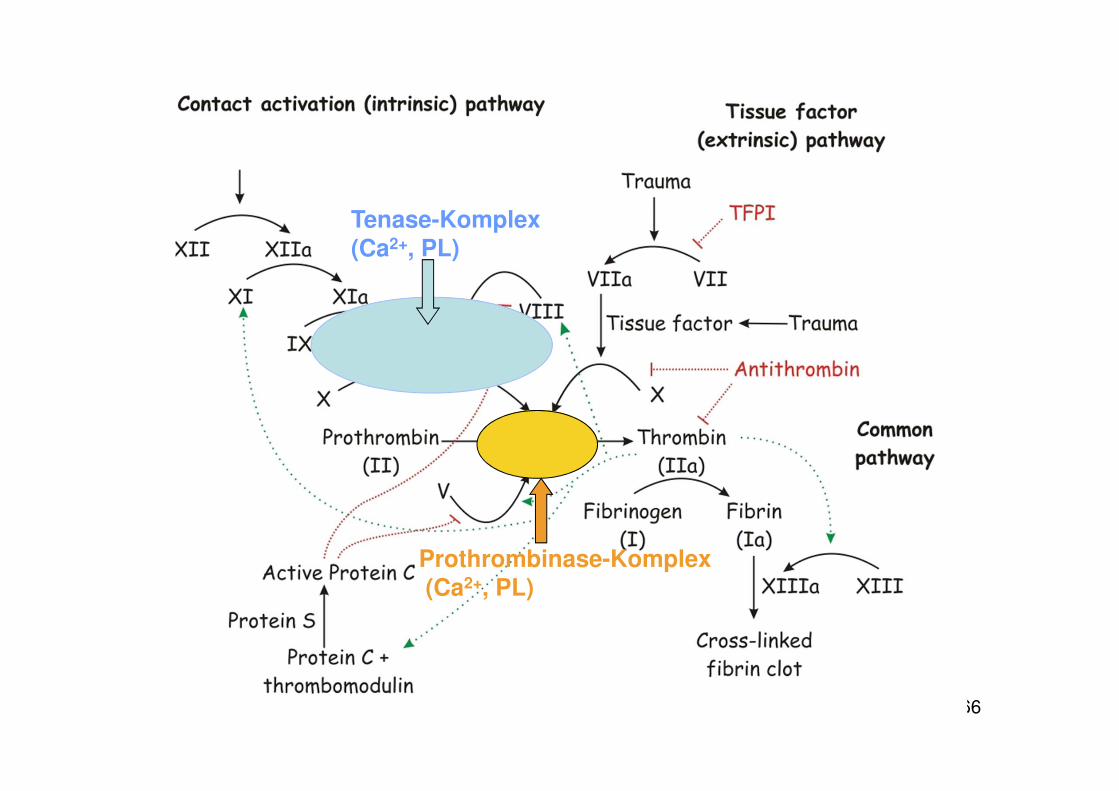
66
Tenase-Komplex(Ca2+, PL)
Prothrombinase-Komplex(Ca2+, PL)

67
Spronk et al 2003, BioEssays 25:1220-1228
Umwandlung von Prothrombin in ThrombinFXa, Va
Grau: Phosphatidylserin auf der Membran Schwarz: Ca 2+
Prothrombin Xa, Va Thrombin

68
Phospholipid-Oberflächen
• alle wichtigen Reaktionen der Gerinnungskaskade laufen auf Phospholipid-Oberflächen ab
• Phosphatidylserin (PS; negativ geladen) wandert bei Plättchenaktivierung aus der inneren in die äußere Schicht der Membran (flip-flop)
• Ca 2+ als Brücke zwischen PS und Gerinnungsfaktoren
• Co-Lokalisation von Komponenten auf einer Oberfläche erleichtert enzymatische Reaktion

69Spronk et al 2003, BioEssays 25:1220-1228
Bedeutung von Vitamin K
Ca²+ als Vermittler der Bindung zwischen Phosphatidylserin auf der Membranoberfläche und den Gerinnungsfaktoren
Faktoren II, VII, IX, Xenthalten γ-Carboxyglutaminsäure-Reste„Gla-Domänen“

70
Bedeutung von Vitamin K
Für die Bildung der Gla-Domänen (γ-Carboxylierung)wird Vitamin K benötigt
Vitamin K ist Co-Faktor der γ-Glutamylcarboxylase
Fehlen oder Hemmung von Vitamin K bewirkt fehlende Carboxylierung unddamit fehlende Interaktion der Gerinnungsfaktorenmit der Membran, Gerinnungsstörungen
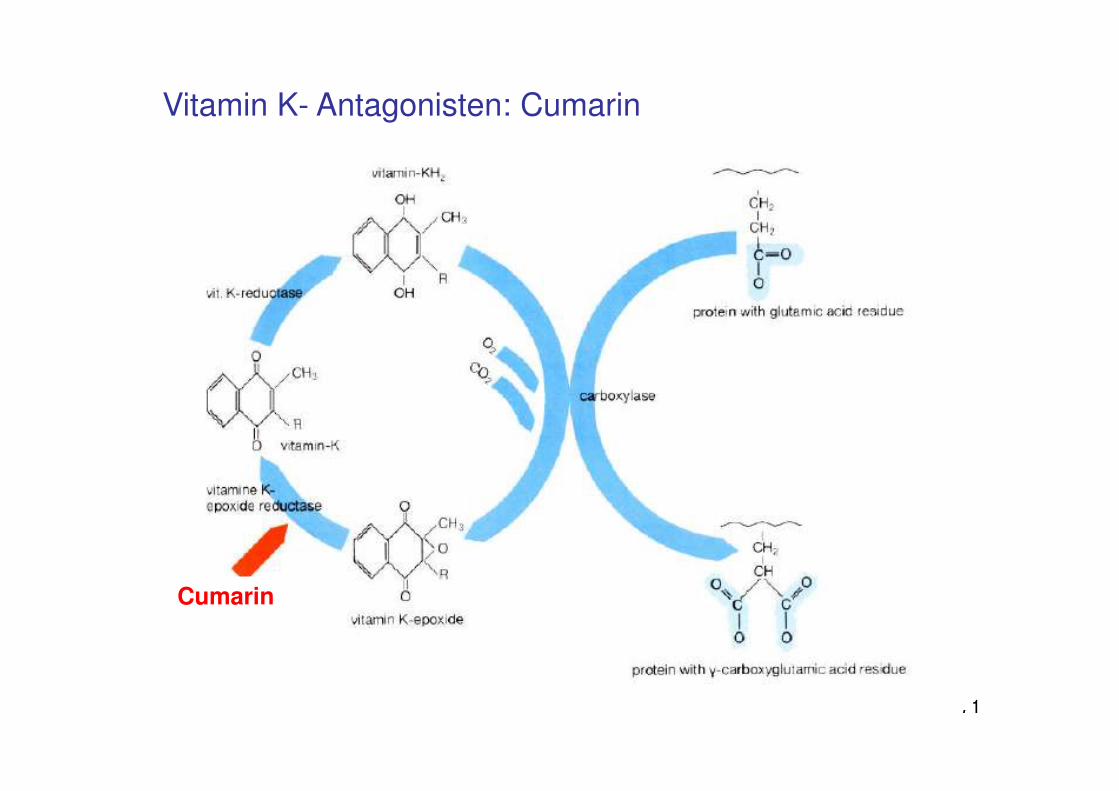
71
Cumarin
Vitamin K- Antagonisten: Cumarin
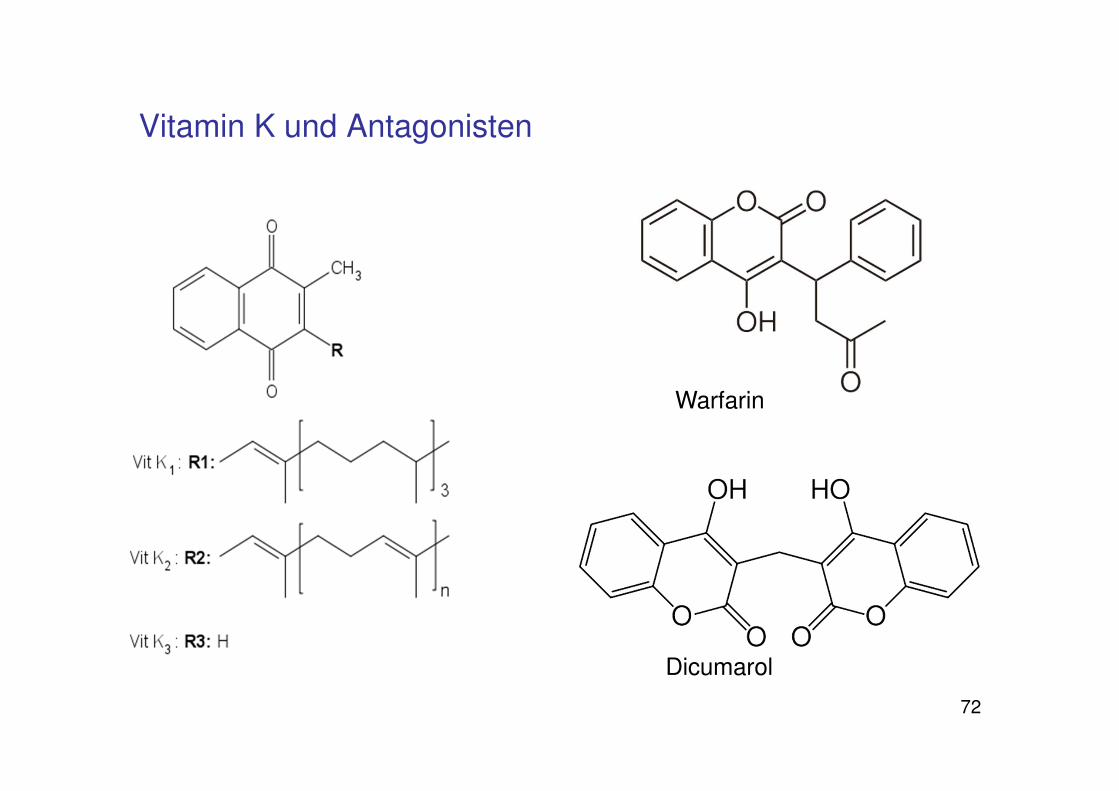
72
Vitamin K und Antagonisten
Warfarin
Dicumarol

73
Warfarin:
Rattengift
Cumarinderivat
schleichendes Verbluten
Wirkt auf Protein S,C,FII,VII,IX,X
(alle Faktoren mit Gla-Domänen)
Karl Paul Link 1948 patentiert als RattengiftWisconsin Alumni Research Foundation„-arin“ von Cumarin
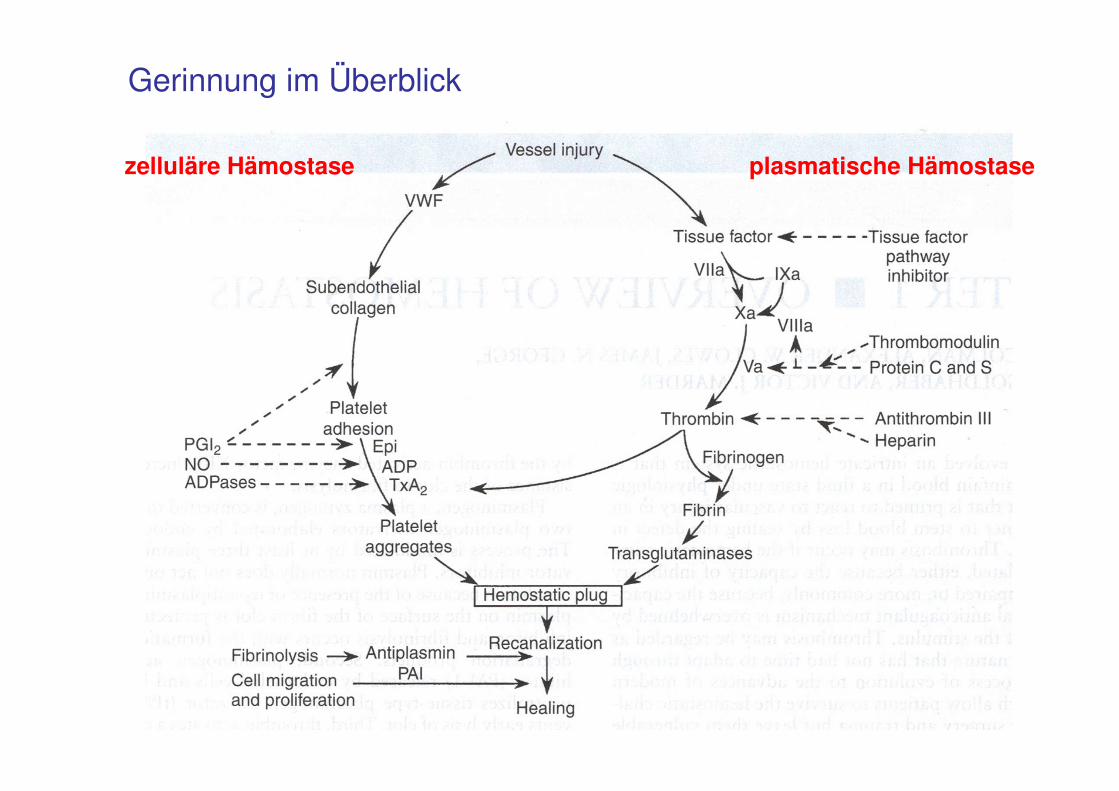
74
Gerinnung im Überblick
zelluläre Hämostase plasmatische Hämostase

75
Das Endothel
Im Normalzustand: gerinnungshemmende Oberfläche
- Bildung von Inhibitoren der Gerinnung (Thrombomodulin, Heparansulfat)und derPlättchenaggregation (Prostacyclin, NO)
- Modulation der Gefäßspannung Endotheline (Vasokonstriktion)Prostacycline, NO (Vasodilatation)
- Trennung von Blut und reaktiven subendothelialen Strukturen
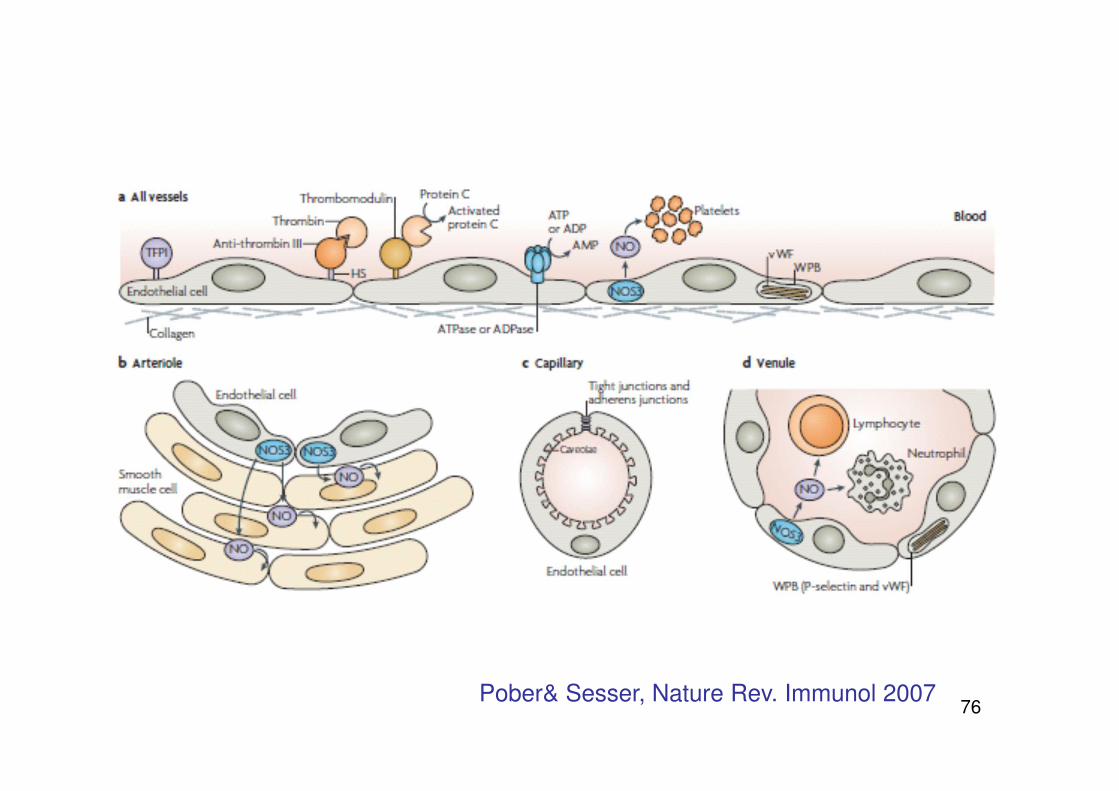
76Pober& Sesser, Nature Rev. Immunol 2007

77
Das Endothel
...zentrale Rolle für die Hämostase !
Verlust der gerinnungshemmenden Eigenschaften kann induziert werden durch z. B.
- Thrombin
- Entzündungsprozesse (z.B. Schädigung des Endothels)
- Endotoxin & Zyktokine (Vermittlung von Entzündung)

78
Fibrinolyse

79

80
Letzte Schritte der Gerinnung
Thrombin
Fibrinogen Fibrin
Thrombin
XIII XIIIa
kovalente Bindungen zwischen
Fibrinmolekülen
Stabilisierung des Gerinnsels
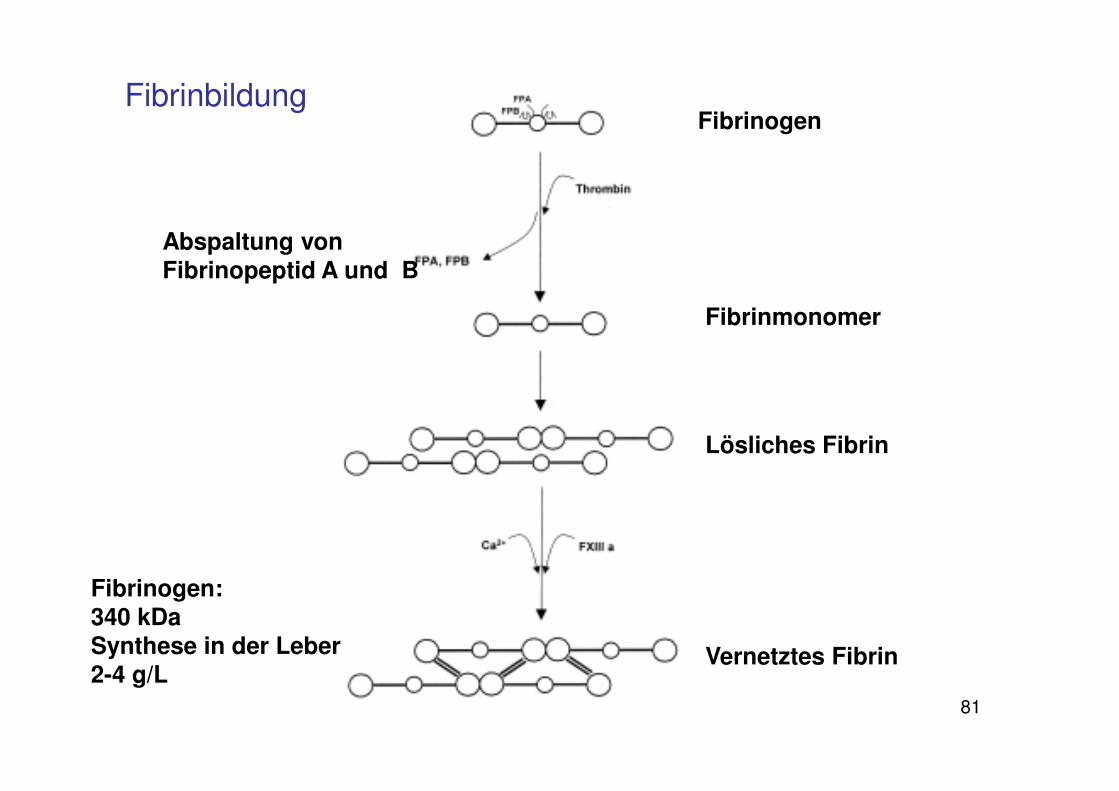
81
Fibrinbildung
Abspaltung von Fibrinopeptid A und B
Fibrinogen
Fibrinmonomer
Lösliches Fibrin
Vernetztes Fibrin
Fibrinogen: 340 kDaSynthese in der Leber2-4 g/L

82
Fibrin sorgt für Wundverschluss,
ist aber keine Dauerlösung !
Fibrinabbau

83
• Umwandlung des Plasmaproteins Plasminogen in Plasmin
• Plasmin bindet an Fibrin und spaltet es in lösliche Abbauprodukte
• Dadurch zerfällt der Thrombus
Fibrinolyse
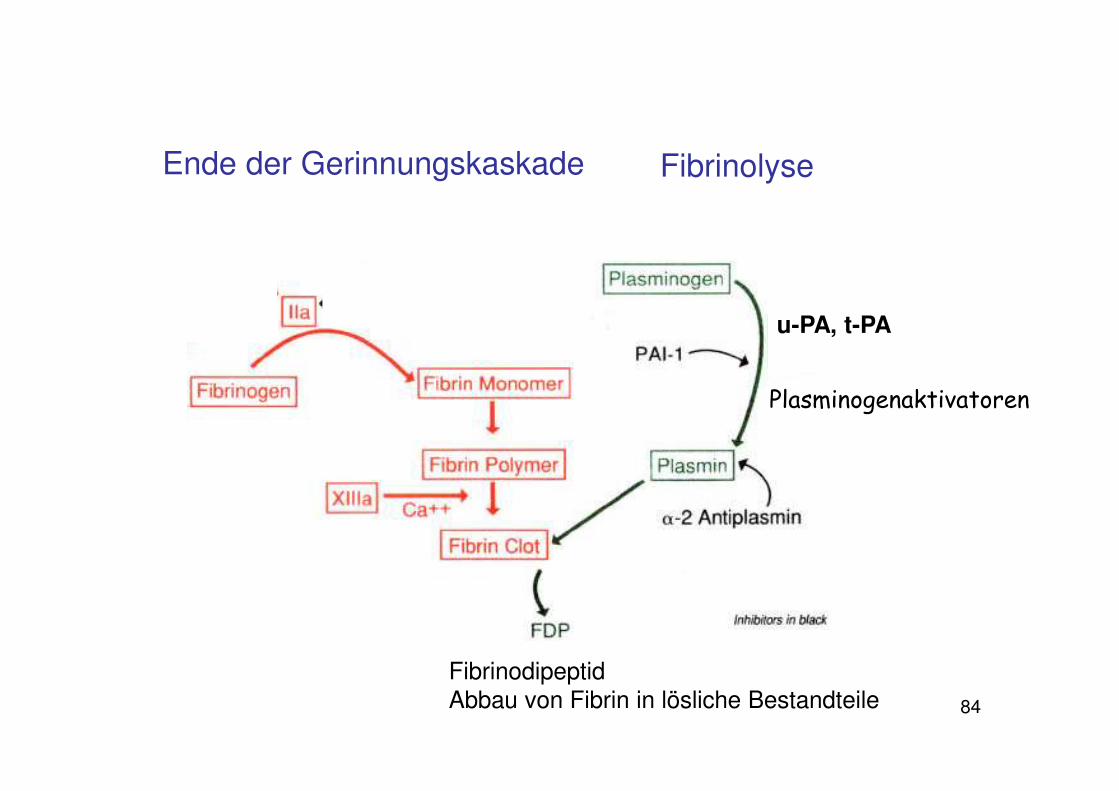
84
Fibrinolyse
u-PA, t-PA
Plasminogenaktivatoren
FibrinodipeptidAbbau von Fibrin in lösliche Bestandteile
Ende der Gerinnungskaskade

85
Regulation: PAI Plasminogenaktivator-Inhibitor PAI-1 hemmt sowohl t-PA als auch u-PA
Großteil (90%) von Thrombozyten gespeichert Thrombozyten setzen bei Aktivierung PAI frei
-> hemmt die Fibrinolyse, erhöht die Stabilität des Thrombus
Fibrinolyse

86
Regulation der Blutgerinnung
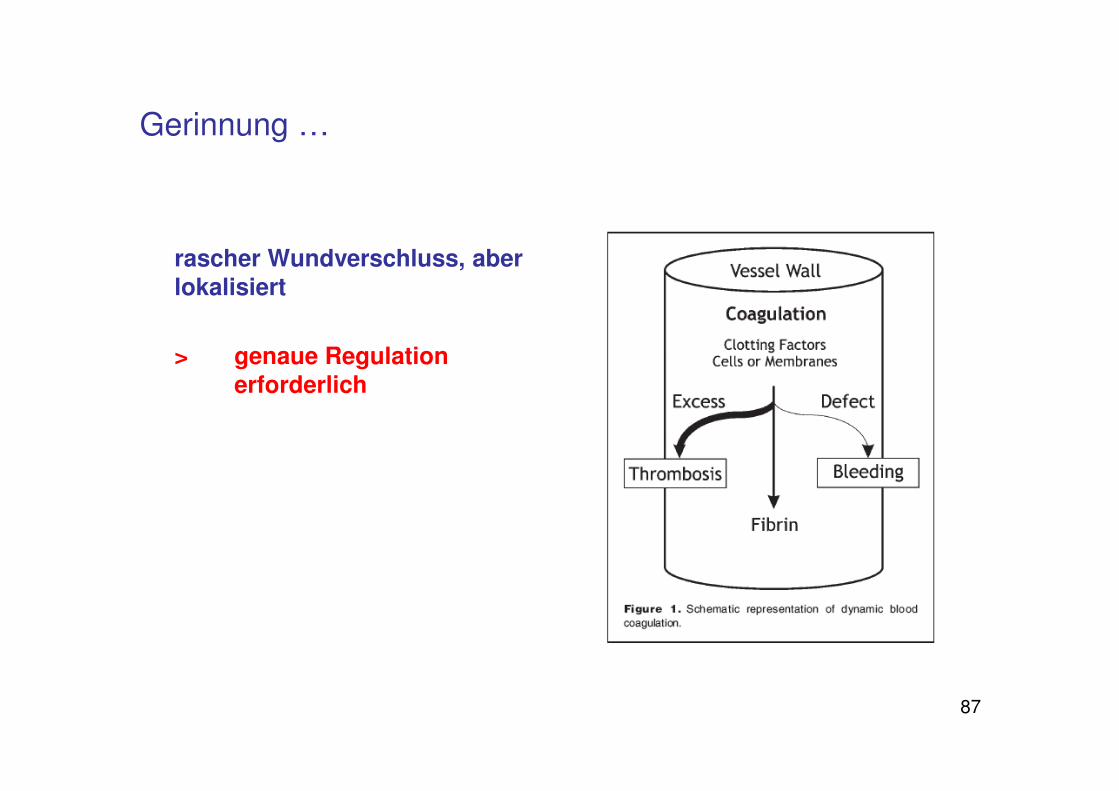
87
Gerinnung …
rascher Wundverschluss, aber lokalisiert
> genaue Regulation erforderlich

88
Gerinnungsstörungen
• Thrombophilien (erhöhte Thrombosebereitschaft)
arterielle Thrombose
venöse Thrombose
disseminierte intravasale Koagulation (DIC)
• Hämorrhagische Diathesen (erhöhte Blutungsbereitschaft)
Mangel an Gerinnungsfaktoren
Vitamin K Mangel
Leberinsuffizienz
Gestörte Plättchenbildung
Skorbut

89
Arterielle Thrombose
Auslöser: • Ruptur oder Erosion eines arteriosklerotischen Plaques
• Plättchen spielen wichtige Rolle
Lipoprotein-induced hypothesis
Joseph Leonard Goldstein (Nobelpreis 1985 für Arbeiten zum (Cholesterinstoffwechsel)

Arteriosklerose
• Systemische Erkrankung der Arterien
• Ablagerung von Blutfetten, Thromben, Bindegewebe in den Gefäßwänden
• Verhärtung und Verdickung der Gefäßwände
• Verengung, abnehmende Elastizität
• Häufigste Todesursache in westl. Industrienationen
90

ArterioskleroseEntstehung und Ursachen
• Response to injury hypothesisVerletzung der inneren Arterienwandschicht (z.B. durch Bluthochdruck, bakterielle Toxine, Endothelschädigung allgemein)
• Lipoprotein-induced hypothesisJoseph Leonard Goldstein: Aufnahme von oxidiertem LDL durch Makrophagen, Umwandlung der Makrophagen in Schaumzellen
91
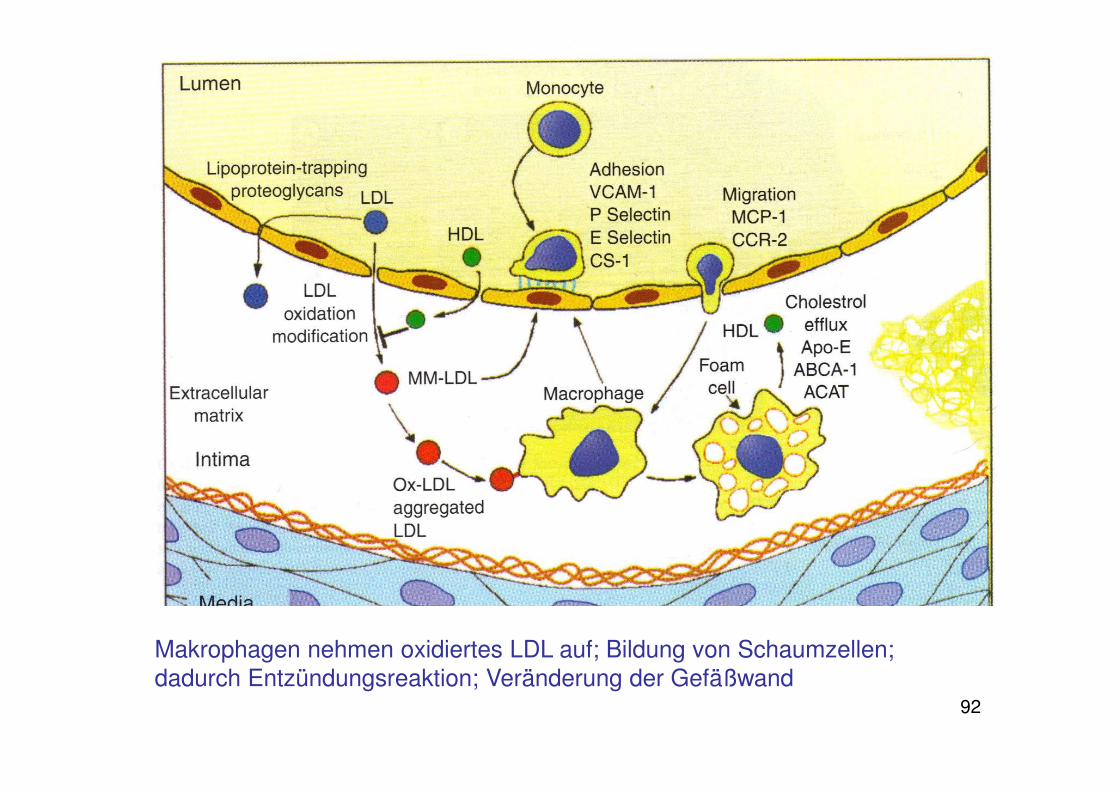
92
Makrophagen nehmen oxidiertes LDL auf; Bildung von Schaumzellen; dadurch Entzündungsreaktion; Veränderung der Gefäßwand

93
Muskelzellen proliferieren, bilden eine Kappe über den absterbendenSchaumzellen.

94
RupturMonozyten produzieren Metalloproteinasen
> Schwächung der Gefäßwand > Defekt der Endotheloberfläche> Freisetzung von tissue factor und Thrombusbildung

95
Folgen
• Ischämie (Blutleere; griech. ισχηαιµια)
= Unterversorgung eines Gewebes oder Organs mit Sauerstoff
• Herzinfarkt
• Schlaganfall
> anti-platelet drugs (greifen auf verschiedenen Ebenen ein, unterbinden Plättchenaktivierung und Plättchenaggregation
96
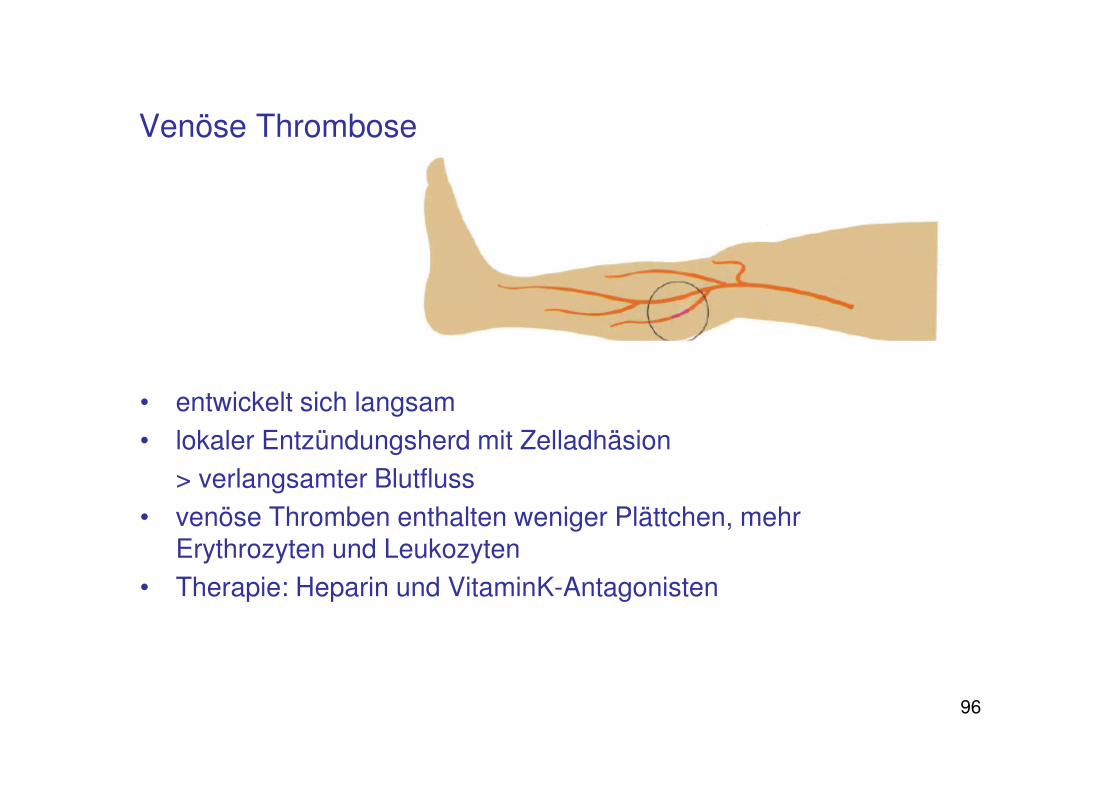
• entwickelt sich langsam
• lokaler Entzündungsherd mit Zelladhäsion
> verlangsamter Blutfluss
• venöse Thromben enthalten weniger Plättchen, mehr Erythrozyten und Leukozyten
• Therapie: Heparin und VitaminK-Antagonisten
Venöse Thrombose

97
Virchow´sche TriasRudolf Virchow, 1821-1902
3 Risikofaktoren für Gerinnung:
WANDFAKTOR Veränderung an den Gefäßwänden (Endothelschäden -> Thrombozytenadhäsion) z.B. durch Entzündung, Sklerose, Unfall, Verletzungen, Hypoxie (Herz/Lungenerkrankungen)
BLUTFAKTOR erhöhte Gerinnungsneigung
z.B. Störung der Blutgerinnung/Thrombolyse; erhöhte Viskosität, etwadurch Exsikkose
KREISLAUFFAKTOR langsamere Strömungsgeschwindigkeit
z.B. Fehlen der Muskel-Venen-Pumpe bei Bettlägrigkeit oder Ruhigstellung durch Schienen, Herzinsuffizienz, Krampfadern, Venenklappeninsuffizienz

98
Therapie
Initial: Heparin + Vitamin K-Antagonisten
Vitamin K-Antagonisten für 3-6 Monate

99
Thromben
Nach dem Erscheinungsbild
- weißer Thrombus: plättchenreich, Peripherie der Gefäße
= Abscheidungsthrombus
- roter Thrombus: bei verlangsamtem Blutfluss, Gefäßverschluss, reich an Erythrozyten
= Gerinnungsthrombus

100
Embolie
= Verschleppung von partikulärem Material in den Blutstrom
Thromboembolie (am häufigsten)
Entstehung vorwiegend in den Venen, Wirbelbildung hinter den Venenklappen > Gerinnungsaktivierung & Thrombinbildung
Mobilisierung von Thromben > Verschluss von Pulmonalarterien („Lungenembolie“)

101
Disseminierte intravasale Gerinnung (DIC)
Diffuse Aktivierung der Gerinnung im Gefäßsystem
Auslöser: z.B. Infektionen, bakterielle Toxine, Schlangengift,…
Bildung winziger Abscheidungsthromben
> Stagnation des Blutflusses, Hypoxie, Gewebsschädigung
> weitere Thrombusbildung
> Verbrauch an Komponenten der Gerinnungskaskade und Plättchenmangel (Thrombozytopenie)
„Verbrauchskoagulopathie“
102
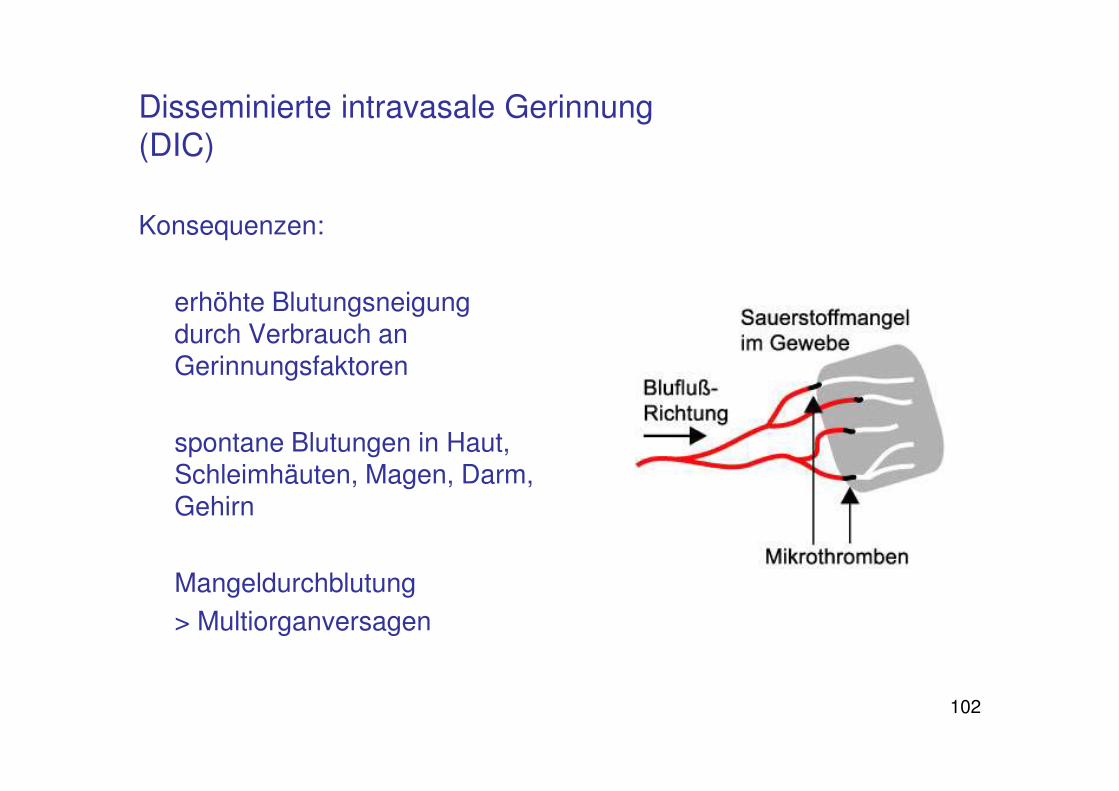
Disseminierte intravasale Gerinnung (DIC)
Konsequenzen:
erhöhte Blutungsneigung durch Verbrauch an Gerinnungsfaktoren
spontane Blutungen in Haut, Schleimhäuten, Magen, Darm, Gehirn
Mangeldurchblutung
> Multiorganversagen

Blutungsneigungen
103

104
Hämorrhagische DiatheseUrsachen:
1) Mangel an Gerinnungsfaktoren
erblich, Hämophilie, Bluterkrankheit
2) Vitamin K-Mangel
mangelnde γ-Carboxylierung der Gerinnungsfaktoren, daher keine Interaktion mit Ca2+ und mit Phospholipidmembranen
3) Leberinsuffizienz
da Gerinnungsfaktoren in der Leber gebildet werden
4) gestörte Plättchenbildung
5) Bindegewebserkrankungen (Skorbut)

105
Mangel an Gerinnungsfaktoren Hämophilie, Bluterkrankheit
griech. haima, Blut; philos, Freund
Erbkrankheit, X-chromosomal rezessiv vererbt
daher vorwiegend Männer betroffen
Hämophilie A: Faktor VIII-Mangel
Hämophilie B: Faktor IX-Mangel
von Willebrand Disease: Mangel an funktionsfähigem vWF

106
Hämophilie
Symptome:
� verlangsamte Blutgerinnung
� Spontanblutungen
� Gelenksblutungen
Schnittverletzungen o.ä. führen nicht zu stärkerem Bluten als beim Gesunden, da Thrombozyten intakt sind;
Verletzungen brechen aber immer wieder auf

107
Hämophilie
Therapie:
- früher Bluttransfusion
- heute Ersatz des fehlenden Faktors (prophylaktisch oder bei Bedarf; intravenös)
- Faktoren früher aus menschlichem Plasma gewonnen > Gefahr der Infektion mit HIV, Hepatitis C, Hepatitis B,...
- heute rekombinant hergestellt

108
Faktor VIII-Präparate
Komplikationen bei der Verabreichung:
• Bildung von Antikörpern gegen FVIII, dadurch Inaktivierung des zugeführten Faktors (Hemmkörperhämophilie)bei ca. 30% der Patienten
Gegenmaßnahmen:
> Immunadsorption Entfernung der Antikörper durch Bindung an Adsorber mit Protein A
> Desensibilisierung hohe Dosen von FVIII

109
Von Willebrand Disease
• Häufigste erbliche Bluterkrankheit (0.1 – 1%)• Klinisch manifest nur in schweren Fällen (ca. 250 Fälle in D)• beschrieben von Prof. Erik von Willebrand, Helsinki 1926
Ursache: Mangel an funktionellem vWF
verschiedene Typen;
Mutationen in vWF beeinträchtigen - Bildung des multimeren Proteins - Stabilität/Halbwertszeit- Interaktion mit Liganden

110
Klinisches Leitsymptom:
Verlängerte Schleimhautblutungen
� Zahnextraktion
� Nasenbluten
� Oberflächliche Hämatome
� Tonsillektomie
� Magen-/Darmblutungen
� Blutungen nach Geburten
Von Willebrand Disease

111
Von Willebrand-Faktor
multimeres Glykoprotein, 2050 Aminosäuren
Bindungsstellen für Kollagen GPIb GPIIb/IIIa
findet sich in Endothelzellen, in α-Granula der Plättchen, sowie im Plasma (10 µg/mL)
unter normalen Bedingungen interagiert löslicher vWF nicht mit Rezeptoren auf Plättchen
Bei Endothelverletzung bindet vWF an Kollagen in der ECM>stark adhäsives Substrat
112
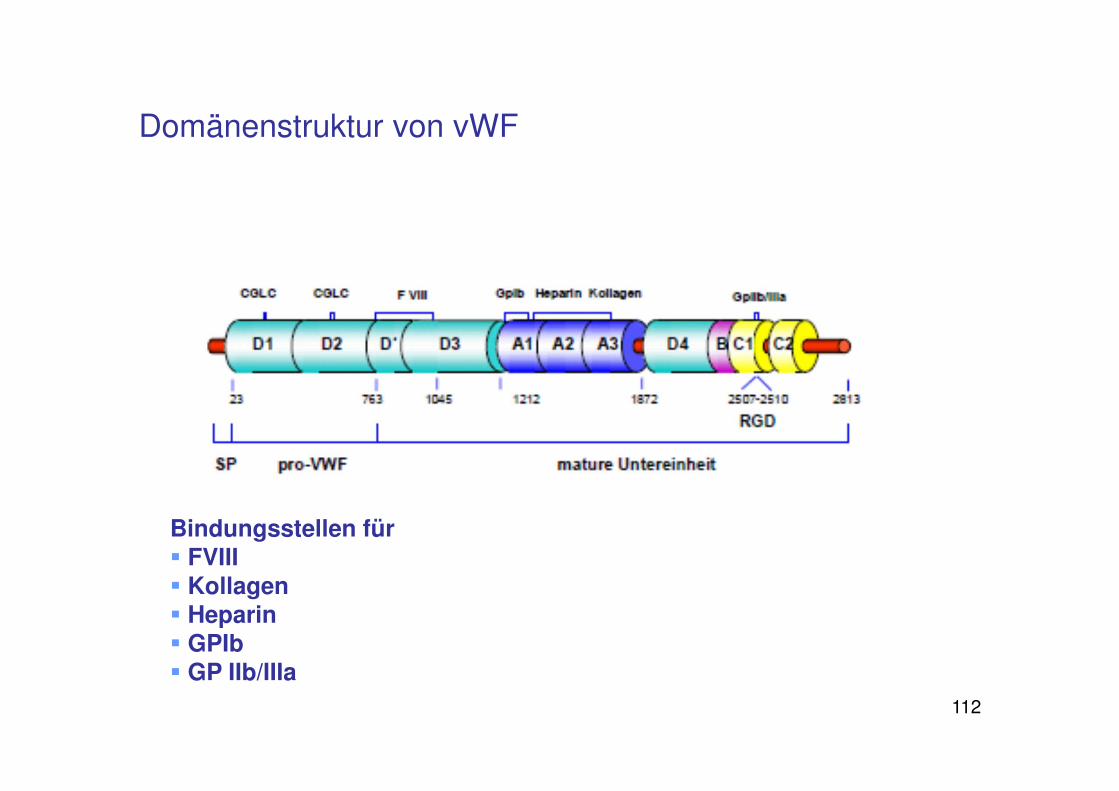
Domänenstruktur von vWF
Bindungsstellen für � FVIII� Kollagen� Heparin � GPIb� GP IIb/IIIa
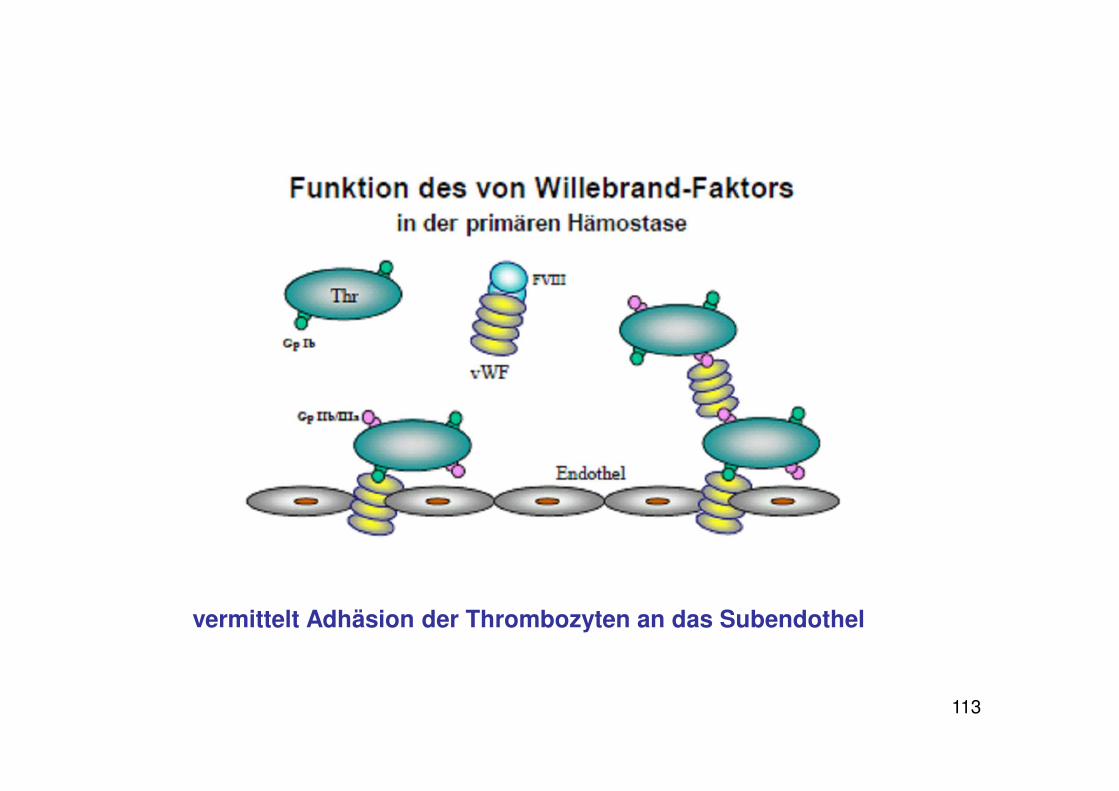
113
vermittelt Adhäsion der Thrombozyten an das Subendothel
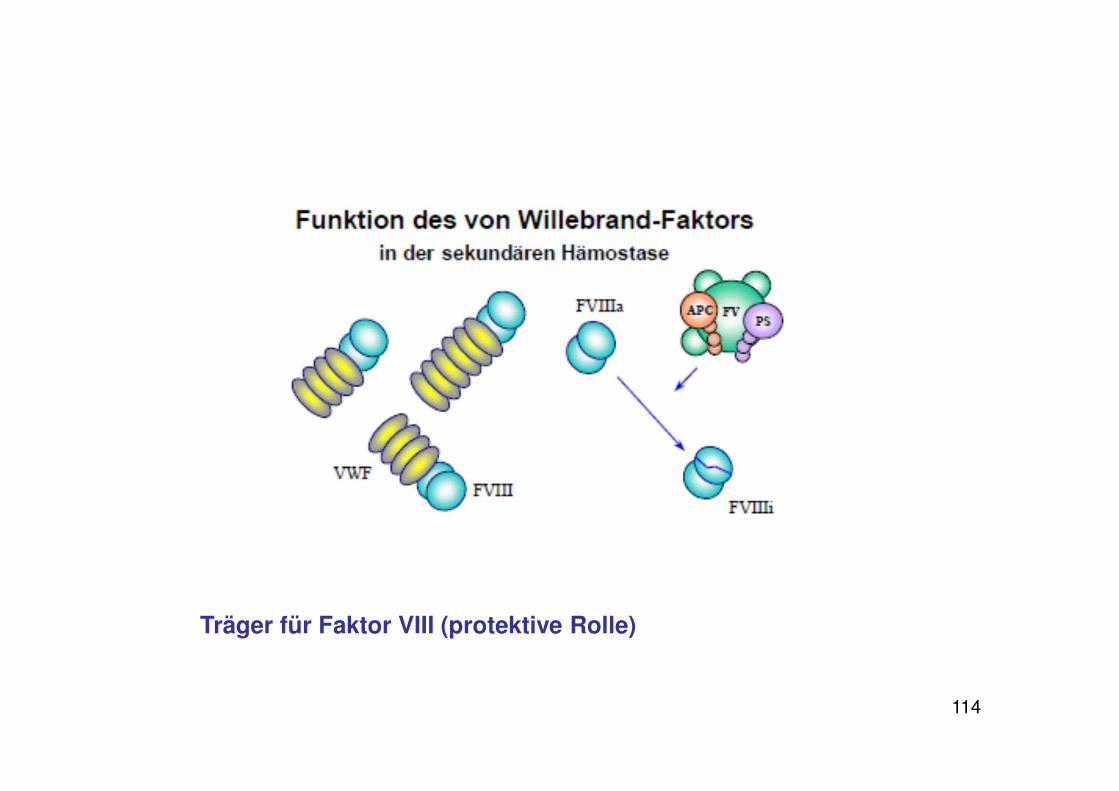
114
Träger für Faktor VIII (protektive Rolle)

115
Von Willebrand-Faktor
Zwei Funktionen:
1) Vermittlung der Plättchenadhäsion bei Endothelverletzungen
vWF bindet an Kollagen in der extrazellulären Matrix, dadurch bildet sich eine stark adhäsive Oberfläche, Plättchen heften sich an
2) Bindung und Stabilisierung von Faktor VIII

116
Hämorrhagische DiatheseUrsachen:
1) Mangel an Gerinnungsfaktoren
erblich, Hämophilie, Bluterkrankheit
2) Vitamin K-Mangel
mangelnde γ-Carboxylierung der Gerinnungsfaktoren, daher keine Interaktion mit Ca2+ und mit Phospholipidmembranen
3) Leberinsuffizienz
da Gerinnungsfaktoren in der Leber gebildet werden
4) gestörte Thrombopoese
5) Bindegewebserkrankungen (Skorbut)
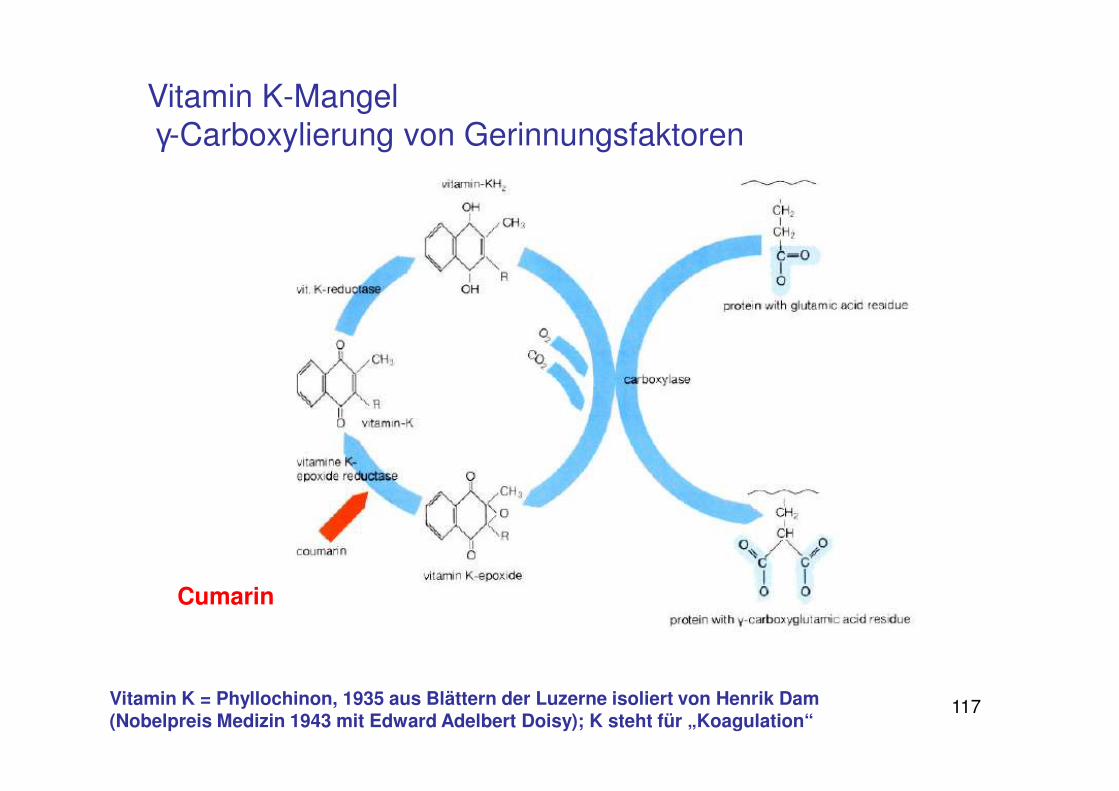
117
Cumarin
Vitamin K-Mangel γ-Carboxylierung von Gerinnungsfaktoren
Vitamin K = Phyllochinon, 1935 aus Blättern der Luzerne isoliert von Henrik Dam (Nobelpreis Medizin 1943 mit Edward Adelbert Doisy); K steht für „Koagulation“
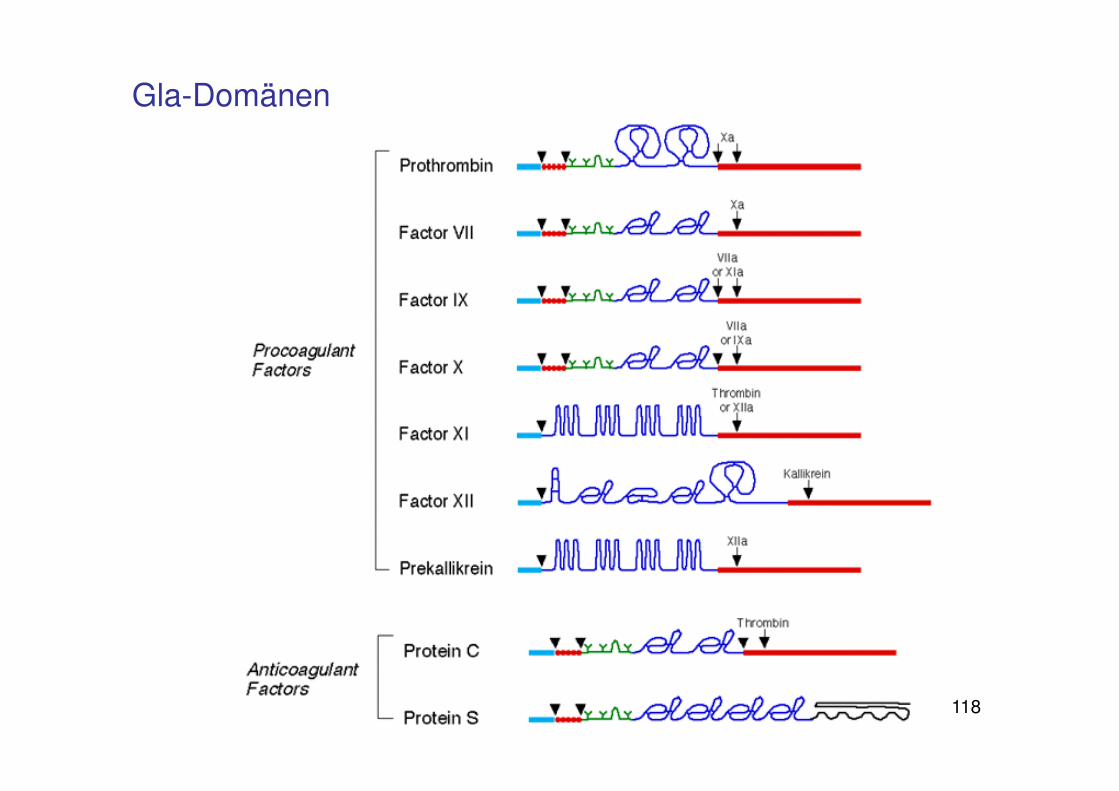
118
Gla-Domänen

119
Blutungsneigung - Ursachen:
1) Mangel an Gerinnungsfaktoren
erblich, Hämophilie, Bluterkrankheit
2) Vitamin K-Mangel
mangelnde γ-Carboxylierung der Gerinnungsfaktoren, daher keine Interaktion mit Ca2+ und mit Phospholipidmembranen
3) Leberinsuffizienz
da Gerinnungsfaktoren in der Leber gebildet werden
4) gestörte Thrombopoese
5) Bindegewebserkrankungen (Skorbut)

120
Skorbut
Vitamin C-Mangel (< 20 mg/Tag)
> mangelnde Kollagen-Biosynthese, Brüchigkeit der Blutgefäße
untersucht von James Lind 1747 > Zitronensaft als „Gegenmittel“
(Zitrone: 53 mg Vitamin C/100g; Paprika 120 mg/100g; Hagebutte: 1250 mg/100g!)
Tagesbedarf ca. 100 mg, Einzeldosen bis 5000 mg werden gut toleriert
Ausscheidung über die Niere; bei langfristig hohen Dosen Risiko der Bildung von Nierensteinen (Oxalat)

121
Skorbut
Kollagen: Strukturprotein des Bindegebes und der extrazellulären Matrix
Typ I fibrillär: Tripelhelix
Aminosäuresequenz: G-x-y
x: oft Prolin
y: oft 4-Hydroxyprolin
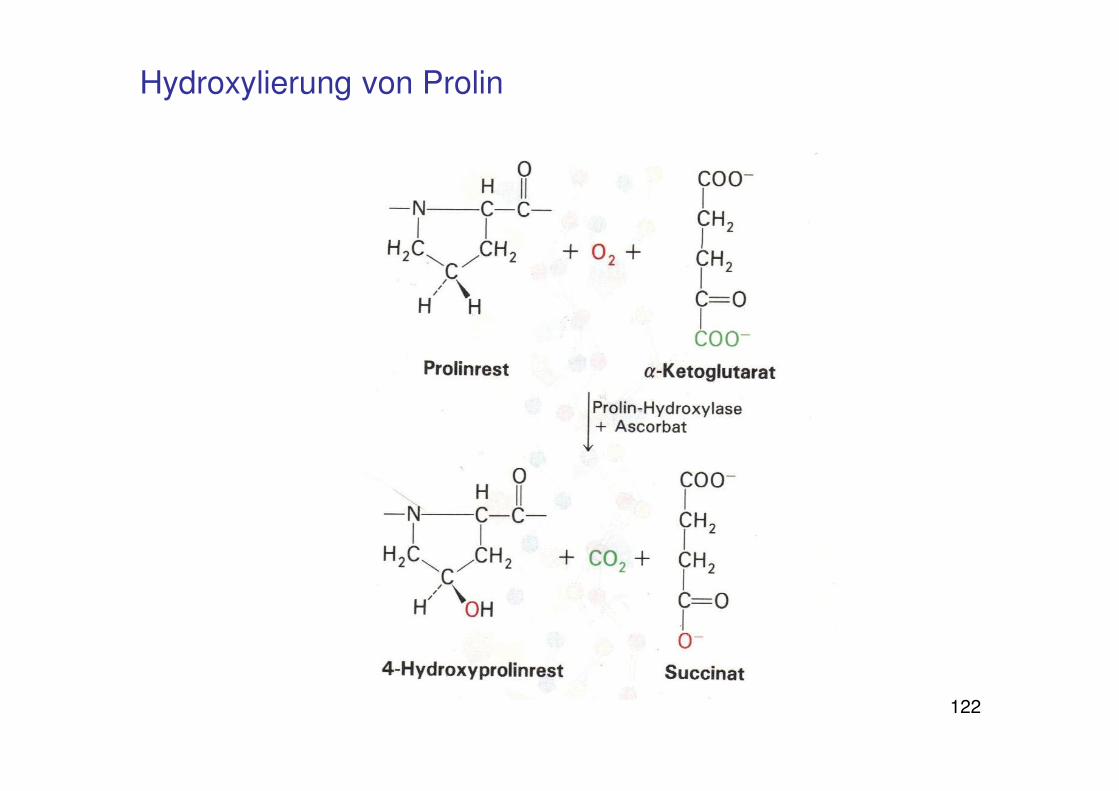
122
Hydroxylierung von Prolin
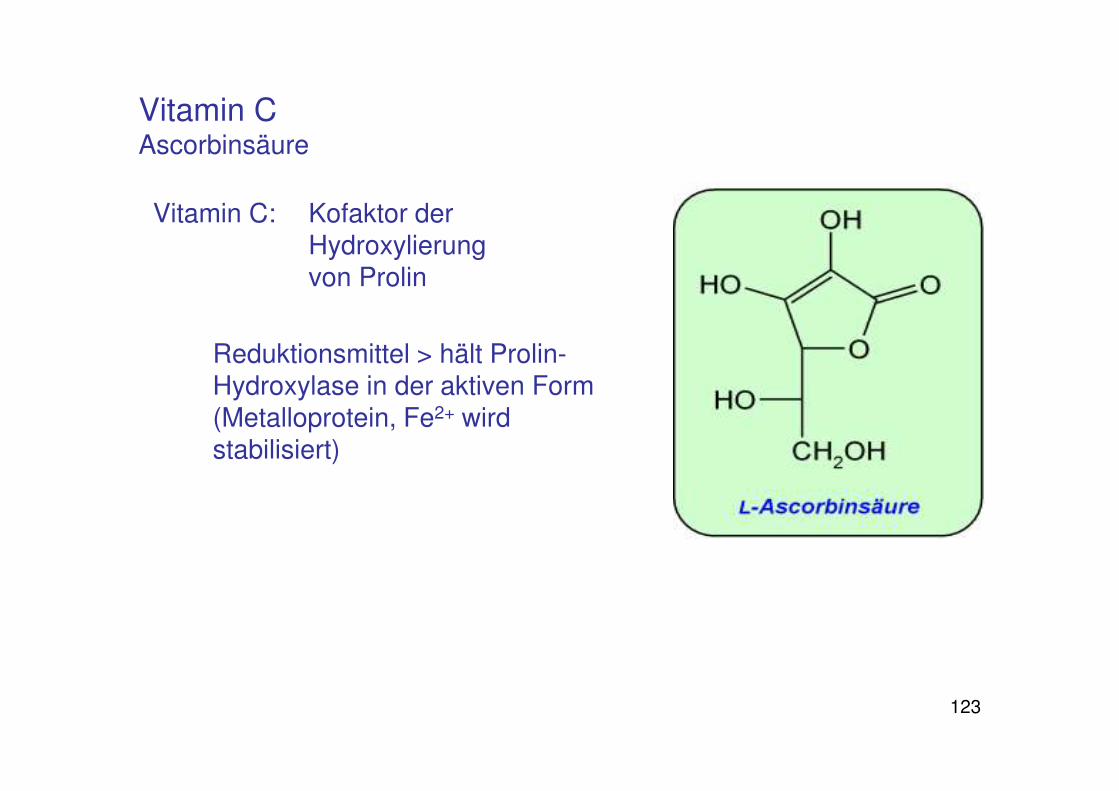
123
Vitamin C Ascorbinsäure
Vitamin C: Kofaktor der Hydroxylierung von Prolin
Reduktionsmittel > hält Prolin-Hydroxylase in der aktiven Form (Metalloprotein, Fe2+ wird stabilisiert)

124
Vasco da Gama, Seeweg nach Indien (1496 – 1498) 100/160 Besatzungsmitgliedern sterben an Skorbut


















