
Consensus-StatementderÖsterreichischenGesellschaften ...
10
consensus report Wien Klin Wochenschr (2021) 133 (Suppl 2):S23–S32 https://doi.org/10.1007/s00508-021-01874-3 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur Definition, Evaluation und Therapie von progredient fibrosierenden interstitiellen Lungenerkrankungen (pfILD) David Lang · Florentine Moazedi-Fürst · Judith Sautner · Helmut Prosch · Sabin Handzhiev · Klaus Hackner · Ivan Tancevski · Holger Flick · Hubert Koller · Hans Peter Kiener · Christian Prior · Bernd Lamprecht Online publiziert: 22. April 2021 © Der/die Autor(en) 2021 Zusammenfassung Interstitielle Lungenerkrankun- gen (ILD) sind eine heterogene Gruppe von Patho- logien, die zunehmend als relevanter Faktor pul- monaler Morbidität und Mortalität erkannt werden. Verschiedene ILD wie die idiopathische Lungenfi- brose (IPF), aber auch durch Autoimmunprozesse oder durch exogene Noxen bedingte ILD können zu progredienter, meist irreversibler Lungenfibrose füh- ren. Die antifibrotischen Substanzen Nintedanib und Pirfenidon können den Krankheitsverlauf bei IPF- Patienten günstig beeinflussen. Dagegen werden ILD, die auf entzündlichen Prozessen wie z. B. rheumatolo- gischen Grunderkrankungen oder exogen-allergischer Alveolitis beruhen, bis dato klassischerweise immun- suppressiv therapiert. Immer wieder kommt es aber trotz einer solchen Behandlung zu progredienter Fi- brosierung. Eine positive Wirkung antifibrotischer Dr. D. Lang () · B. Lamprecht Klinik für Lungenheilkunde, Kepler Universitätsklinikum Linz, Krankenhausstraße 9, Linz, Österreich [email protected] F. Moazedi-Fürst Klinische Abteilung für Rheumatologie und Immunologie, Landeskrankenhaus Universitätsklinikum Graz, Graz, Österreich J. Sautner 2. Medizinische Abteilung mit Rheumatologie, Landesklinikum Stockerau, Niederösterreichisches Zentrum für Rheumatologie, Stockerau, Österreich H. Prosch Universitätsklinik für Radiologie und Nuklearmedizin, Klinische Abteilung für Allgemeine Radiologie und Kinderradiologie, Medizinische Universität Wien am Allgemeinen Krankenhaus der Stadt Wien, Wien, Österreich S. Handzhiev · K. Hackner Klinische Abteilung für Pneumologie, Universitätsklinikum Krems, Krems, Österreich Medikation auf progredient fibrosierende (pf)ILD ab- seits der IPF konnte in rezenten Studien demonstriert werden, auch wenn der Stellenwert der Antifibrotika in solchen Situationen noch nicht vollständig geklärt ist. Dieses Consensus-Statement beruht auf einem virtuellen, multidisziplinären Expertenmeeting von Rheumatologen, Pneumologen und Radiologen und wurde durch die jeweiligen ILD-Arbeitskreise der Ös- terreichischen Gesellschaften für Pneumologie sowie Rheumatologie (ÖGP und ÖGR) akkordiert. Ziel war es, den aktuellen Stand von klinischer Praxis und wis- senschaftlicher Datenlage zu Definition, Evaluation und Therapie von pfILD darzustellen. Zusammenfas- send sollen ILD-Patienten einer standardisierten Ab- klärung unterzogen, in einem multidisziplinären ILD- Board diskutiert und dementsprechend therapiert werden. Kern dieser Empfehlungen ist, auch Non- I. Tancevski Universitätsklinik für Innere Medizin II, Infektiologie, Rheumatologie und Pneumologie, Medizinische Universität Innsbruck, Innsbruck, Österreich H. Flick Universitätsklinik für Innere Medizin, Klinische Abteilung für Pulmonologie, Landeskrankenhaus Universitätsklinikum Graz, Graz, Österreich H. Koller Abteilung für Atemwegs- und Lungenkrankheiten, Wiener Gesundheitsverbund – Klinik Penzing, Wien, Österreich H. P. Kiener Universitätsklinik für Innere Medizin III, Klinische Abteilung für Rheumatologie, Medizinische Universität Wien am Allgemeinen Krankenhaus der Stadt Wien, Wien, Österreich C. Prior Facharztordination, Heiliggeiststr. 1, Innsbruck, Österreich K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S23
Transcript of Consensus-StatementderÖsterreichischenGesellschaften ...

consensus report
Wien Klin Wochenschr (2021) 133 (Suppl 2):S23–S32https://doi.org/10.1007/s00508-021-01874-3
Consensus-Statement der Österreichischen Gesellschaftenfür Pneumologie und Rheumatologie zur Definition,Evaluation und Therapie von progredient fibrosierendeninterstitiellen Lungenerkrankungen (pfILD)
David Lang · Florentine Moazedi-Fürst · Judith Sautner · Helmut Prosch · Sabin Handzhiev · Klaus Hackner ·Ivan Tancevski · Holger Flick · Hubert Koller · Hans Peter Kiener · Christian Prior · Bernd Lamprecht
Online publiziert: 22. April 2021© Der/die Autor(en) 2021
Zusammenfassung Interstitielle Lungenerkrankun-gen (ILD) sind eine heterogene Gruppe von Patho-logien, die zunehmend als relevanter Faktor pul-monaler Morbidität und Mortalität erkannt werden.Verschiedene ILD wie die idiopathische Lungenfi-brose (IPF), aber auch durch Autoimmunprozesseoder durch exogene Noxen bedingte ILD können zuprogredienter, meist irreversibler Lungenfibrose füh-ren. Die antifibrotischen Substanzen Nintedanib undPirfenidon können den Krankheitsverlauf bei IPF-Patienten günstig beeinflussen. Dagegen werden ILD,die auf entzündlichen Prozessen wie z.B. rheumatolo-gischen Grunderkrankungen oder exogen-allergischerAlveolitis beruhen, bis dato klassischerweise immun-suppressiv therapiert. Immer wieder kommt es abertrotz einer solchen Behandlung zu progredienter Fi-brosierung. Eine positive Wirkung antifibrotischer
Dr. D. Lang (�) · B. LamprechtKlinik für Lungenheilkunde, Kepler UniversitätsklinikumLinz, Krankenhausstraße 9, Linz, Ö[email protected]
F. Moazedi-FürstKlinische Abteilung für Rheumatologie und Immunologie,Landeskrankenhaus Universitätsklinikum Graz, Graz,Österreich
J. Sautner2. Medizinische Abteilung mit Rheumatologie,Landesklinikum Stockerau, Niederösterreichisches Zentrumfür Rheumatologie, Stockerau, Österreich
H. ProschUniversitätsklinik für Radiologie und Nuklearmedizin,Klinische Abteilung für Allgemeine Radiologie undKinderradiologie, Medizinische Universität Wien amAllgemeinen Krankenhaus der Stadt Wien, Wien, Österreich
S. Handzhiev · K. HacknerKlinische Abteilung für Pneumologie, UniversitätsklinikumKrems, Krems, Österreich
Medikation auf progredient fibrosierende (pf)ILD ab-seits der IPF konnte in rezenten Studien demonstriertwerden, auch wenn der Stellenwert der Antifibrotikain solchen Situationen noch nicht vollständig geklärtist. Dieses Consensus-Statement beruht auf einemvirtuellen, multidisziplinären Expertenmeeting vonRheumatologen, Pneumologen und Radiologen undwurde durch die jeweiligen ILD-Arbeitskreise der Ös-terreichischen Gesellschaften für Pneumologie sowieRheumatologie (ÖGP und ÖGR) akkordiert. Ziel wares, den aktuellen Stand von klinischer Praxis und wis-senschaftlicher Datenlage zu Definition, Evaluationund Therapie von pfILD darzustellen. Zusammenfas-send sollen ILD-Patienten einer standardisierten Ab-klärung unterzogen, in einem multidisziplinären ILD-Board diskutiert und dementsprechend therapiertwerden. Kern dieser Empfehlungen ist, auch Non-
I. TancevskiUniversitätsklinik für Innere Medizin II, Infektiologie,Rheumatologie und Pneumologie, Medizinische UniversitätInnsbruck, Innsbruck, Österreich
H. FlickUniversitätsklinik für Innere Medizin, Klinische Abteilungfür Pulmonologie, Landeskrankenhaus UniversitätsklinikumGraz, Graz, Österreich
H. KollerAbteilung für Atemwegs- und Lungenkrankheiten, WienerGesundheitsverbund – Klinik Penzing, Wien, Österreich
H. P. KienerUniversitätsklinik für Innere Medizin III, Klinische Abteilungfür Rheumatologie, Medizinische Universität Wien amAllgemeinen Krankenhaus der Stadt Wien, Wien, Österreich
C. PriorFacharztordination, Heiliggeiststr. 1, Innsbruck, Österreich
K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S23

consensus report
IPF-Patienten mit dokumentiert progredient fibro-sierendem ILD-Verlauf antifibrotisch zu behandeln,insbesondere wenn Honigwabenzysten oder eine be-reits ausgedehnte Erkrankung vorliegen. Patientenmit fibrotischer ILD, die auf Basis der ILD-Board-Empfehlung primär keiner oder ausschließlich einerimmunsuppressiven Therapie unterzogen werden,sollten engmaschig hinsichtlich eines progredientenVerlaufes überwacht werden.
Schlüsselwörter Idiopathische Lungenfibrose ·Exogen-allergische Alveolitis · AntifibrotischeTherapie · Sarkoidose · Computertomographie ·Immunsuppression
Einleitung
Interstitielle Lungenerkrankungen (ILD) sind eine he-terogene Gruppe an Pathologien, bei denen es durchInflammation und/oder Fibrose des pulmonalen In-terstitiums durch unterschiedliche Ursachen zu einerEinschränkung der physiologischen Lungenfunktionkommt [1, 2].
Idiopathische Lungenfibrose (IPF) stellt die klassi-sche Form einer idiopathischen progredient fibrosie-renden (pf)ILD dar. Die IPF betrifft v. a. Männer imhöheren Lebensalter (>60 Jahre) mit Raucheranamne-se und führt bei fast allen Betroffenen zu rasch progre-dienter Lungenfunktionseinschränkung mit ungüns-tiger Prognose [3, 4]. In den letzten Jahren kamenmit Pirfenidon und Nintedanib 2 antifibrotische Wirk-stoffe zur Zulassung, die den Verlauf dieser Erkran-kung bremsen können [5–8]. Da beide Medikamen-te den kontinuierlichen Lungenfunktionsverlust abernicht gänzlich aufhalten können, ist eine frühe Dia-gnose der IPF mit rascher Therapieeinleitung essen-ziell.
Schon lange ist bekannt, dass auch andere fibrosie-rende ILD, allen voran die chronische exogen-allergi-sche Alveolitis (EAA), die Sarkoidose und ILD im Rah-men von verschiedenen Autoimmunerkrankungentrotz adäquater Diagnose und – zumeist immunsup-pressiver – Therapie einen progredient fibrosierendenVerlauf mit einer ebenso unvorteilhaften Prognoseaufweisen können [1, 9–12].
Klassisch hierfür ist die systemische Sklerose (SSc),die sich in 70–80% in der Lunge manifestiert, waszu einer substanziellen Prognoseeinschränkung führt[12–14]. Für die SSc-ILD wurde in den Scleroder-ma Lung Studies I und II die Wirksamkeit von Cy-clophosphamid und Mycophenolat-Mofetil (MMF)nachgewiesen, wobei sich MMF als nebenwirkungs-ärmer herausstellte [15, 16]. Andere Therapieoptionenin therapierefraktären Fällen umfassen auch Tocilizu-mab, Rituximab, autologe Stammzelltransplantationund Lungentransplantation [12, 13, 17–20]. Im Jahr2019 konnte in der SENSCIS-Studie an 576 SSc-ILD-PatientInnen mit Nintedanib analog zur IPF eineReduktion des Verlustes an forcierter Vitalkapazität
(FVC) gezeigt werden, was zur Zulassung dieser Sub-stanz durch die Europäische Arzneimittel-Agentur(EMA) bei SSc-ILD führte. Patienten, die Nintedanibund zusätzlich eine Begleitmedikation mit MMF er-hielten, wiesen den geringsten FVC-Abfall auf [21].Dies legt nahe, dass zumindest bei einem Teil derSSc-ILD-Patienten eine Kombination aus antifibro-tischer und immunsuppressiver Therapie vorteilhaftsein könnte.
Bei pfILD verschiedener Genese abseits der IPFwurden zuletzt in der INBUILD-Studie zu Nintedanibähnliche Ergebnisse gezeigt: Auch hier konnte derAbfall der FVC signifikant reduziert werden, unab-hängig davon, ob der bekannt ungünstige Progno-sefaktor eines „usual-interstitial pneumonia“(UIP)-artigen Musters in der hochauflösenden Computerto-mographie (HRCT) vorlag [22]. In dieser Studie waren663 PatientInnen mit verschiedensten progredient fi-brosierenden ILD eingeschlossen, davon jeweils ca.ein Viertel mit EAA und autoimmun assoziierten ILD,zum Teil mit bestehender immunsupprimierenderBasistherapie. Über alle Subgruppen hinweg konntenerneut konsistente Effekte bezüglich der FVC gezeigtwerden [23]. Dies führte 2020 zur EMA-Zulassung vonNintedanib für die Indikation pfILD.
Auch für die Substanz Pirfenidon wurden in Stu-dien an Patienten mit unklassifizierbarer oder mitGrunderkrankungen assoziierter pfILD ähnliche Er-gebnisse berichtet [24, 25]. Aufgrund des Studiende-signs wurden hier aber die primären Endpunkte nichterreicht, weswegen bei der EMA bis dato auch keineZulassung in diesen Indikationen beantragt wurde.
Innerhalb weniger Jahre hat sich also die Therapie-landschaft bei fibrosierenden ILD unterschiedlicherGenese deutlich erweitert. Speziell die antifibrotischeTherapie mit Nintedanib – bis vor Kurzem IPF-Pati-enten vorbehalten – hat deutlich an Gewicht gewon-nen. Wichtig sind daher nun einerseits die Definitionsolcher Erkrankungen und andererseits die standardi-sierte Abklärung und Diagnosestellung sowie in Folgedie richtige Auswahl der Therapieoptionen.
Dieses Consensus-Statement soll dazu dienen, Ab-klärung und Therapie von pfILD-Patienten auf einegemeinsame, aktuelle und evidenzbasierte Basis zustellen.
Methoden
Dieses Dokument beruht auf den Diskussionen imRahmen eines virtuellen Expertenmeetings am 05.10.2020, an dem alle Autoren aktiv teilgenommen haben.Auf Basis der Aufzeichnung wurde das Manuskript vonDL und BL verfasst und in 2 Review-Runden von al-len anderen Autoren bearbeitet und schlussendlichakzeptiert.
Das Expertenmeeting wurde von Boehringer Ingel-heim RCV GmbH & Co KG (BI) in Form eines Adviso-ry Board Meetings unterstützt. BI hatte weder Einflussauf den Prozess der Recherche, des Verfassens desMa-
S24 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . K

consensus report
nuskriptes und der Überarbeitung durch die Autorennoch auf den Inhalt dieses Consensus-Statements. BIwurde die Gelegenheit gegeben, das Manuskript aufmedizinische und wissenschaftliche Richtigkeit in Be-zug auf die erwähnte BI-Substanz zu überprüfen.
Dieses Dokument erhebt nicht den Anspruch aufVollständigkeit wie etwa eine Leitlinie. Vielmehr sollhier eine gemeinsame Diskussion über ein vielschich-tiges Thema, welches multidisziplinär angegangenwerden muss, möglichst komplett abgebildet werden.
Wir möchten betonen, dass die Abklärung und The-rapie einzelner Erkrankungen, wie z.B. der IPF, EAA,der SSc-ILD oder RA-ILD, prinzipiell nach Maßgabeder entsprechenden Leitlinien in Zusammenarbeit derjeweiligen Fachdisziplinen erfolgen sollen.
Definition von pfILD
Die häufigsten Ursachen von fibrosierenden ILD sindIPF, chronische EAA, mit Autoimmunerkrankungenassoziierte ILD, unklassifizierbare ILD und Sarkoido-se, die Prävalenzwird zusammen bei ca. 50–70/100.000angesetzt [1]. Abseits der in den meisten Fällen pro-gredienten IPF wird davon ausgegangen, dass es beianderen fibrosierenden ILD in bis zu 30% zu einemprogredienten Verlauf kommt [1, 9, 11].
In mehreren Übersichtsarbeiten und internationa-len Positionspapieren wurde zuletzt klar empfohlen,pfILD abseits der IPF als „progredient trotz optima-ler aktueller Abklärung und Therapie“ zu definieren[1, 11]. Dies bedeutet, dass der Terminus pfILD keineDiagnose darstellen soll, sondern lediglich einen Phä-notyp beschreibt, der bei verschiedenen ILD auftretenkann. Jedenfalls sind weiterhin eine genaue Abklärungzur exakten Diagnosestellung und eine genaue Ursa-chenforschung insbesondere im Hinblick auf die EAAund auf Autoimmunerkrankungen durchzuführen.
Wenn sich jedoch trotz dieser optimalen Aufarbei-tung und ggf. trotz korrekt durchgeführter Therapiedas Bild einer pfILD zeigt, besteht derzeit auf Basisder vorliegenden Studiendaten die Empfehlung zurEinleitung einer antifibrotischen Therapie. Eine EMA-Zulassung in dieser Indikation liegt auf Basis derINBUILD-Studie derzeit nur für Nintedanib vor [22](Abb. 1).
Standardisierte ILD-Abklärung
Anamnese, Status
Die allermeisten pfILD-Patienten präsentieren sichmit progredienter Belastungsdyspnoe und Reizhus-ten. Symptome, die auf autoimmun-assoziierteGrunderkrankungen hinweisen können, wie Gelenk-beschwerden, Raynaud-Phänomen, Muskelschwächeoder Hauterscheinungen sollten gezielt abgefragt wer-den [1, 3]. Im Status ist die Auskultation essenziell, dadie klassische „Sklerosiphonie“, also das spät-inspi-
Fibrosierende ILD
Standardisierte Evaluation, ILD-Board
Primär progredient fibrosierend, IPF
Antifibrotische Therapie*
Entzündlich mediiert; EAA, V.a. autoimmun-assoziiert
Antigenkarenz bei EAA,Therapie der
Grunderkrankung bzw. antiinflammatorische
Therapie für 3-6 Monate
Add-on/switch antifibrotische Therapie bei weiter progredienter
Fibrose*
Abb. 1 Schematischer Ablauf zum Management von thera-piebedürftiger progredient fibrosierender ILD (interstitielle Lun-generkrankung). IPF idiopathische Lungenfibrose, EAA exo-gen-allergische Alveolitis; Stern derzeit ist durch die EMA nurdie Substanz Nintedanib auf Basis der INBUILD-Studie in derIndikation progredient fibrosierende ILD abseits der IPF zuge-lassen
ratorische feine Knisterrasseln, sehr prädiktiv für dasVorliegen von Fibrose ist [1, 26].
In der Anamnese ist es wichtig, den Beschwerdebe-ginn zu erheben; so kann die Dynamik der Erkrankungabgeschätzt werden. Aktuelle und frühere Medikati-on sollte abgefragt werden (cave: insbesondere Amio-daron und Tumortherapien), ebenso Nikotinkonsumund sämtliche Expositionen im beruflichen und pri-vaten Bereich, die auf eine EAA hinweisen können (or-ganische Stäube z.B. im Stall, Vogelhaltung, Schimmelim Wohnbereich).
K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S25

consensus report
a
b
c
d
Abb. 2 Progredient fibrosierender Verlauf einer ILD (inters-titielle Lungenerkrankung), die initial als idiopathische NSIP(nichtspezifische interstitielle Pneumonie) klassifiziert, spä-ter aufgrund histologischer Untersuchungsergebnisse als IPF(idiopathische Lungenfibrose) behandelt wurde. Im zeitlichenVerlauf von der Diagnosestellung (a), nach 2 (b), 4 (c) und6 Jahren (d) zeigen sich zunehmende peripher betonte Reti-kulation und Ausbildung von Traktionsbronchiektasen (c, d).(Bildquelle: Kepler Universitätsklinikum Linz)
Ebenso soll eine Familienanamnese für pulmonalewie auch rheumatologische Erkrankungen erfolgen.
Computertomographie
Die Diagnose einer ILD kann nur anhand einer hoch-auflösenden Computertomographie (HRCT) des Tho-rax gestellt werden [27, 28]. Die anatomische Vertei-lung der Veränderungen, die vorherrschenden Musteroder – wenn verfügbar – der zeitliche Verlauf könnenbereits wichtige Hinweise auf die zugrunde liegendeKrankheit liefern [29].
Das klassische Thoraxröntgen eignet sich nicht zurspezifischen Diagnose einer ILD, kann aber helfen,den Verlauf der Erkrankung anhand von Vorbildern
abzuschätzen oder Differenzialdiagnosen (z.B. kardia-le Dekompensation) auszuschließen [27].
Hinweise auf eine fibrosierende Lungenerkrankungin der CT-Bildgebung sind netzartige (retikuläre) Ver-änderungen und/oder Milchglasverdichtungen. Beimzusätzlichen Nachweis von begleitenden periphe-ren Traktionsbronchiektasien/-bronchiolektasien isteine Fibrose sehr wahrscheinlich. Als beweisendesZeichen der Fibrose gelten Honigwabenzysten (Ho-neycombing) [27], die aber bei Weitem nicht beijedem Patienten vorhanden sind, sondern je nachEntität nur in 30–40% [30]. Die HRCT ermöglichtdie Einteilung von ILD in verschiedene radiologischeMuster, die ihrerseits für die Klassifizierung und Dia-gnose der ILD wegweisend sind. Häufige Muster sinddie UIP, die durch peripher, subpleural und basalbetonte Retikulation mit Honeycombing und Trak-tionsbronchiektasen gekennzeichnet sind und dienichtspezifische interstitielle Pneumonie (NSIP), dieklassischerweise Milchglasverdichtungen und/oderretikuläre Verdichtungen in basaler und subpleuralerVerteilung mit begleitenden Bronchiektasien zeigt [27,28].
Wichtig ist aber, dass sich das radiologische Er-scheinungsbild in der HRCT bei pfILD im Krankheits-verlauf verändern kann (Abb. 2) und dass auch schonbei Diagnosestellung Mischbilder verschiedener Mus-ter vorliegen können. Die genaue Diagnose einer ILDbenötigt also die Zusammenschau von HRCT mit kli-nischen Daten, wie z.B. einer genauen Anamnese hin-sichtlich Antigenexposition, und anderen diagnosti-schen Tests wie Laboruntersuchungen und Lungen-funktionstestung.
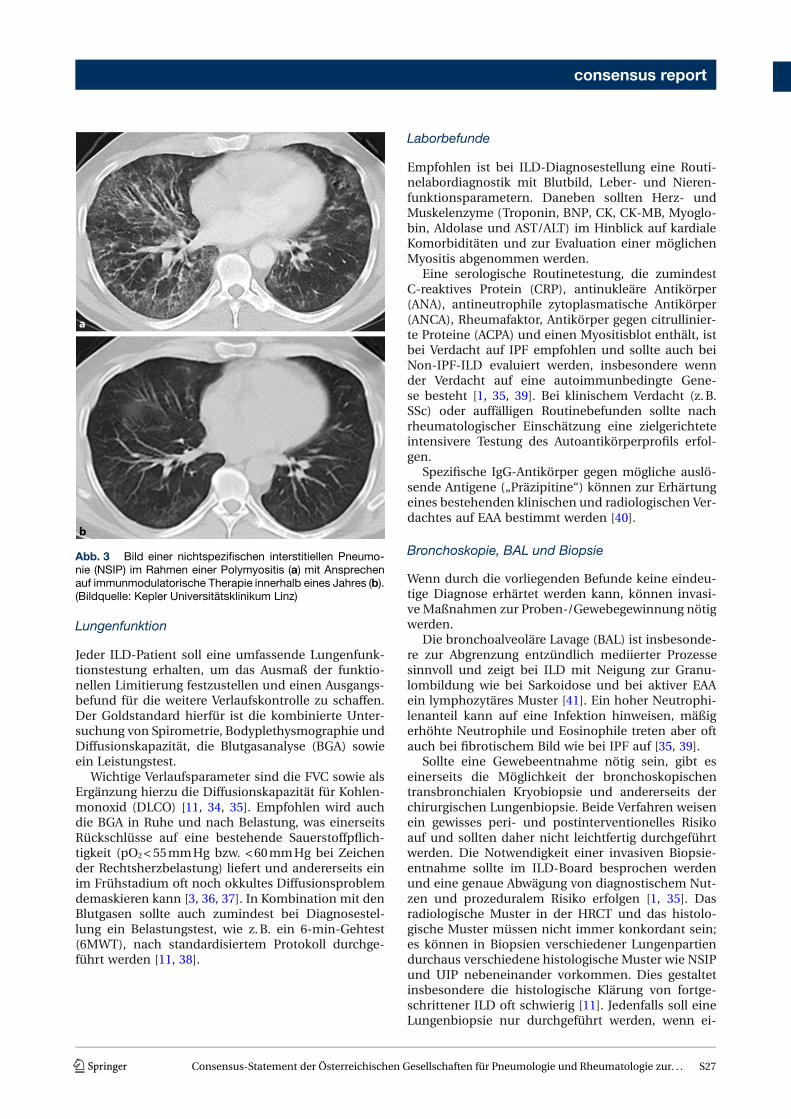
Neben der Unterscheidung verschiedener Musterermöglicht die HRCT in gewissem Maße auch eineAbschätzung der Prognose [27]. Das klassische UIP-Muster mit Honeycombing und Traktionsbronchiek-tasen hat – ungeachtet seiner Ursache – die ungüns-tigste Prognose [1, 3, 27, 30]. Muster mit Milchglasver-dichtungen oder retikulären Verdichtungen, wie z.B.das der NSIP, sind prognostisch günstiger und oftmalseiner immunsuppressiven Therapie eher zugänglich(Abb. 3; [31, 32]) Dennoch können auch primär über-wiegend inflammatorische Veränderungen später zuirreversibler Fibrose führen bzw. liegen oft von Be-ginn an Mischbilder aus fibrotischen und inflamma-torischen Veränderungen vor.
Die HRCT kann zur Verlaufsbeurteilung bei pfILDherangezogen werden [27], allerdings sollten immerauch andere Verlaufsparameter, z.B. die Lungen-funktion einschließlich der Diffusionskapazität unddie Symptomlast, berücksichtigt werden, da die Ver-gleichbarkeit von HRCT-Schnitten auch durch tech-nische oder patientenseitige Faktoren eingeschränktsein kann. Zukünftig könnten hier computerbasier-te quantitative Auswertungsalgorithmen zum Einsatzkommen, die derzeit in der klinischen Routine abernoch nicht verfügbar sind [27, 33].
S26 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . K
consensus report
a
b
Abb. 3 Bild einer nichtspezifischen interstitiellen Pneumo-nie (NSIP) im Rahmen einer Polymyositis (a) mit Ansprechenauf immunmodulatorische Therapie innerhalb eines Jahres (b).(Bildquelle: Kepler Universitätsklinikum Linz)
Lungenfunktion
Jeder ILD-Patient soll eine umfassende Lungenfunk-tionstestung erhalten, um das Ausmaß der funktio-nellen Limitierung festzustellen und einen Ausgangs-befund für die weitere Verlaufskontrolle zu schaffen.Der Goldstandard hierfür ist die kombinierte Unter-suchung von Spirometrie, Bodyplethysmographie undDiffusionskapazität, die Blutgasanalyse (BGA) sowieein Leistungstest.
Wichtige Verlaufsparameter sind die FVC sowie alsErgänzung hierzu die Diffusionskapazität für Kohlen-monoxid (DLCO) [11, 34, 35]. Empfohlen wird auchdie BGA in Ruhe und nach Belastung, was einerseitsRückschlüsse auf eine bestehende Sauerstoffpflich-tigkeit (pO2< 55mmHg bzw. <60mmHg bei Zeichender Rechtsherzbelastung) liefert und andererseits einim Frühstadium oft noch okkultes Diffusionsproblemdemaskieren kann [3, 36, 37]. In Kombination mit denBlutgasen sollte auch zumindest bei Diagnosestel-lung ein Belastungstest, wie z.B. ein 6-min-Gehtest(6MWT), nach standardisiertem Protokoll durchge-führt werden [11, 38].
Laborbefunde
Empfohlen ist bei ILD-Diagnosestellung eine Routi-nelabordiagnostik mit Blutbild, Leber- und Nieren-funktionsparametern. Daneben sollten Herz- undMuskelenzyme (Troponin, BNP, CK, CK-MB, Myoglo-bin, Aldolase und AST/ALT) im Hinblick auf kardialeKomorbiditäten und zur Evaluation einer möglichenMyositis abgenommen werden.
Eine serologische Routinetestung, die zumindestC-reaktives Protein (CRP), antinukleäre Antikörper(ANA), antineutrophile zytoplasmatische Antikörper(ANCA), Rheumafaktor, Antikörper gegen citrullinier-te Proteine (ACPA) und einen Myositisblot enthält, istbei Verdacht auf IPF empfohlen und sollte auch beiNon-IPF-ILD evaluiert werden, insbesondere wennder Verdacht auf eine autoimmunbedingte Gene-se besteht [1, 35, 39]. Bei klinischem Verdacht (z.B.SSc) oder auffälligen Routinebefunden sollte nachrheumatologischer Einschätzung eine zielgerichteteintensivere Testung des Autoantikörperprofils erfol-gen.
Spezifische IgG-Antikörper gegen mögliche auslö-sende Antigene („Präzipitine“) können zur Erhärtungeines bestehenden klinischen und radiologischen Ver-dachtes auf EAA bestimmt werden [40].
Bronchoskopie, BAL und Biopsie
Wenn durch die vorliegenden Befunde keine eindeu-tige Diagnose erhärtet werden kann, können invasi-ve Maßnahmen zur Proben-/Gewebegewinnung nötigwerden.
Die bronchoalveoläre Lavage (BAL) ist insbesonde-re zur Abgrenzung entzündlich mediierter Prozessesinnvoll und zeigt bei ILD mit Neigung zur Granu-lombildung wie bei Sarkoidose und bei aktiver EAAein lymphozytäres Muster [41]. Ein hoher Neutrophi-lenanteil kann auf eine Infektion hinweisen, mäßigerhöhte Neutrophile und Eosinophile treten aber oftauch bei fibrotischem Bild wie bei IPF auf [35, 39].
Sollte eine Gewebeentnahme nötig sein, gibt eseinerseits die Möglichkeit der bronchoskopischentransbronchialen Kryobiopsie und andererseits derchirurgischen Lungenbiopsie. Beide Verfahren weisenein gewisses peri- und postinterventionelles Risikoauf und sollten daher nicht leichtfertig durchgeführtwerden. Die Notwendigkeit einer invasiven Biopsie-entnahme sollte im ILD-Board besprochen werdenund eine genaue Abwägung von diagnostischem Nut-zen und prozeduralem Risiko erfolgen [1, 35]. Dasradiologische Muster in der HRCT und das histolo-gische Muster müssen nicht immer konkordant sein;es können in Biopsien verschiedener Lungenpartiendurchaus verschiedene histologische Muster wie NSIPund UIP nebeneinander vorkommen. Dies gestaltetinsbesondere die histologische Klärung von fortge-schrittener ILD oft schwierig [11]. Jedenfalls soll eineLungenbiopsie nur durchgeführt werden, wenn ei-
K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S27

consensus report
Tab. 1 Verschiedene vorgeschlagene Definitionssysteme für progredient fibrosierende ILDIn den letzten 24 Monaten trotz Standardbehandlung:INBUILD-
Studie (Nin-tedanib) [22]
pfILD außer IPF mit≥10% CT-Beteiligung Relat. FVC-Abfall
≥10%Relat. FVC-Abfall ≥5% und:Verschlechterung der Symptome oderProgress in CT
Verschlechterung Symptome undProgression in CT
In den letzten 6 Monaten trotz Therapie:Maher et al.(Pirfenidon)[24]
Unklassifizierbare pfILD
≥5% FVC-Verlustoder:
Symptomverschlechterung ohne andere Ursache (kardial etc.)
Trotz adäquater Therapie:RELIEF-Studie (Pirfe-nidon) [25]
pfILD inklusive CVD-assoz., fibrotischer NSIP,EAA, Asbest-induz. ILD
≥5% FVC-Verlust/Jahr über zumindest 6–24 Monate in zumindest 3 Messungen
In den letzten 24 Monaten:
FVC-Abfall ≥5% relativ+
George et al.(„positionpaper“) [11]
Definition pfILD
FVC-Abfall ≥10%relativ DLCO-Abfall ≥15% Zunehmende Fibrose in CT oder
zunehmende Symptome
Progrediente Symptome mitzunehmender Fibrose in CT
pfILD progredient fibrosierende ILD, IPF idiopathische Lungenfibrose, ILD interstitielle Lungenerkrankung, CT Computertomographie, FVC forcierte Vitalkapazität,CVD „collagen vascular disease“, NSIP nichtspezifische interstitielle Pneumonie, EAA exogen-allergische Alveolitis, DLCO Diffusionskapazität für Kohlenmonoxid
ne unmittelbare therapeutische Konsequenz besteht.In der klinischen Praxis ist dies v. a. in frühen ILD-Stadien gegeben, wo z.B. bei inflammatorisch me-diierter ILD mit einer immunsuppressiven Therapienoch eine Verbesserung erzielt werden kann, oderbei EAA, wo dann die Antigenkarenz rasch etabliertwerden könnte [11]. Die histologische Aufarbeitungsollte durch Pathologen mit großer ILD-Erfahrung aneinem Referenzzentrum durchgeführt werden.
ILD-Board
Abklärung und Diagnose von ILD sind nicht auf einFachgebiet zu beschränken, vielmehr bedarf dies ei-ner multidisziplinären Zusammenarbeit [42]. ILD-Boards, zumeist bestehend aus Pneumologen, Rheu-matologen, Radiologen und Pathologen, die der erfor-derlichen Multidisziplinarität Rechnung tragen sindan vielen Kliniken bereits Standard. Ihnen obliegt diediagnostische und therapeutische Entscheidung inden oftmals komplexen ILD-Fragestellungen [1, 11,35, 39, 43]. Im Bereich der IPF konnte gezeigt werden,dass die Fallbesprechung im ILD-Board die diagnos-tische Sicherheit deutlich erhöht und auch prognosti-sche Relevanz besitzt [35, 44, 45]. In den letzten Jahrensind auch ILD bei rheumatischer Grunderkrankungdurch die zunehmenden therapeutischen Möglich-keiten und die steigende Awareness verstärkt in denFokus gerückt. Umso wichtiger ist daher auch derDiskurs zwischen Pneumologie und Rheumatologieim ILD-Board geworden [1, 11, 13].
Erfassung der Progredienz
Wichtig ist die Einholung von Vorbefunden, wie z.B.älteren Röntgenaufnahmen, CT-Bildern (z.B. auchAbdomen-CT mit Lungenanschnitten!) und Lungen-funktionsbefunden; so kann häufig ein bereits länge-rer, subklinischer Verlauf einer pfILD nachgewiesenwerden.
Einige Risikofaktoren hinsichtlich Gefahr der Pro-gression einer ILD können aus den Routinebefundenabgeleitet werden: So ist das UIP-Muster mit Honey-combing in der HRCT mit einer ungünstigeren Prog-nose verbunden [30], insbesondere bei Patienten mitRA-ILD oder EAA [46, 47]. Auch haben Patienten mithöherem Lebensalter generell eine schlechtere Pro-gnose, ebenso wie Patienten mit bereits bei Beginnausgedehnten fibrotischen CT-Veränderungen, einge-schränkter Lungenfunktion oder rascher Krankheits-progression [11, 34, 48].
Es existiert noch keine klare Empfehlung, an wel-chen Biomarkern die Progression bei pfILD gemessenwerden soll. Verschiedene, in klinischen Studien ver-wendete und vorgeschlagene Kriterien für pfILD sindin Tab. 1 aufgeführt.
Für IPF wird die FVC derzeit als bester longitudi-naler Parameter angesehen, da er mit dem Überle-ben korreliert und daher auch als primärer Endpunktin den Studien bezüglich der Antifibrotika verwendetwurde [5, 6, 34]. Auch die DLCO korreliertmit derMor-talität bei IPF [34] und wird in der klinischen Routinewie auch in Studien herangezogen. Sie gilt als anfälli-ger für Schwankungen und Störungen, kann aber dieFVC ergänzen [11]. Da alle gemessenen Lungenfunk-tionsparameter immer auch eine inhärente Schwan-kungsbreite besitzen, empfehlen wir, mehrere Funk-tionsparameter unterschiedlicher Messmethoden zuerheben, insbesondere auch Belastungstests wie den6MWT [1, 11, 38]. Ebenso sollen die Beschwerdesym-ptomatik des Patienten und die subjektive Symptom-wahrnehmung in die Entscheidungsfindung einflie-ßen.
Unter erheblicher Diskussion steht derzeit das Zeit-intervall, das abgewartet werden soll, bevor eine pfILDdiagnostiziert werden kann: Es ist evident, dass pfILD-Patienten, die in die oben genannten Studien einge-schlossen wurden, in den Placebogruppen einen ähn-lichen Lungenfunktionsverlust erlitten, wie IPF-Stu-dienpatienten unter Placebo, obwohl sie im Durch-schnitt jünger waren. Es erscheint daher sinnvoll, das
S28 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . K

consensus report
Zeitintervall der Verlaufsbeobachtung vor Einleitungeiner antifibrotischen Therapie möglichst kurz zu hal-ten und bei Zeichen der Progression eher großzügigund rasch eine solche Therapie einzuleiten [9]. Eben-so unter Kritik stehen die relativ strengen Kriterienfür Progression bei pfILD, welche in den Placeboar-men im gegebenen Zeitintervall oft gar nicht erreichtwurden [9]. Zu hinterfragen ist auch die starke Kon-zentration auf den Biomarker der FVC, insbesonderebei der beträchtlichen Fraktion an Patienten mit be-gleitendem Lungenemphysem, da dies die FVC gegen-läufig verfälschen kann [9, 49, 50].
Es wurde daher vorgeschlagen, das Risiko derProgression nicht ausschließlich am vorherigen Ver-lauf festzumachen, sondern Hochrisikopopulationenschon anhand von initial vorhandenen Biomarkernzu identifizieren [9]. Deutliche Assoziationen mit derMortalität bestehen hier schon bekannterweise fürdas Vorhandensein von Honeycombing [30] sowie einFibroseausmaß in der HRCT von >20% [9, 48, 51]. BeiPatienten mit diesen Befunden könnte die sofortigeEinleitung einer antifibrotischen Therapie analog zurIPF bzw. ggf. auch eine Kombination mit immunsup-pressiver Therapie bei autoimmun-assoziierter ILDerwogen werden, auch wenn (noch) keine Progres-sion dokumentiert wurde [9]. Prospektive Daten zueinem solchen Vorgehen liegen jedoch derzeit nochnicht vor.
Therapie und Management von pfILD
Allgemeine Therapieziele
Therapieziel bei pfILD ist naturgemäß die Besserungvon Symptomen, Lebensqualität und funktionellerEinschränkung, was aber nicht immer gelingen kann.Oftmals ist die Stabilisierung oder die Verlangsamungder Verschlechterung ein realistischeres Ziel. Da pfILDwie die IPF nicht heilbar sind und zumeist chronischprogredient verlaufen, ist immer – unabhängig vonder medikamentösen Therapie und deren Erfolgsaus-sichten – auch schon parallel ein palliativer Thera-pieansatz zu verfolgen: Die individuell belastendenSymptome sollen priorisiert behandelt bzw. gelindertwerden. Hierzu können verschiedenste Interventio-nen beitragen, von Rehabilitation zu psychologisch/psychotherapeutischer Betreuung bis hin zur Linde-rung von Atemnot mit Opiaten. Essenziell sind dieenge Kommunikation mit Patienten und Angehöri-gen und die gemeinsame Erarbeitung der Ziele undErwartungen. Bei allen medikamentösen Therapienmuss der Nutzen auf den Krankheitsverlauf genausowie die möglichen Nebenwirkungen und deren Aus-wirkungen auf die Lebensqualität besprochen werden[12].
Medikamentöse Therapie
Die zielgerichtete Therapie von pfILD-Patienten solltejedenfalls in einem ILD-Board besprochen werden.Hier sollten multidisziplinär sämtliche erhobenen Be-funde aus der Abklärung durchgesehen werden undauch eine Einschätzung getroffen werden, ob sichdie vorliegende ILD rein „fibrotisch“ im Sinne einerIPF verhält oder ob Zeichen einer aktiv inflammatori-schen Erkrankung vorliegen.
Bezüglich der einzelnen Therapien in verschiede-nen klinischen Situationen, z.B. ILD bei rheumatoiderArthritis, SSc oder EAA, sei auf die aktuellen Emp-fehlungen der relevanten Leitlinien verwiesen. JedeTherapie soll nach individueller Nutzen-Risiko-Abwä-gung, nach genauer Aufklärung und Diskussion mitPatienten und ggf. Angehörigen und in komplexenFällen auch nach ILD-Board-Besprechung erfolgen.
Generell empfehlen wir, folgende Punkte in derEntscheidungsfindung bezüglich medikamentöserTherapie bei pfILD zu bedenken:
1. Nicht alle pfILD, abgesehen von IPF, benötigen ei-ne sofortige Therapie. Bei langjährigem Verlauf,langsamer Progression und geringer Symptomlast,insbesondere bei älteren, multimorbiden Patienten,kann ein beobachtendes Procedere sinnvoller seinals ein nebenwirkungsreicher Therapieversuch.
2. Die diagnostischen Kriterien für die IPF und diedaraus abgeleiteten Therapieindikationen bleibenaufrecht [35, 39, 52]. ImHinblick auf die aktuelleDa-tenlage und die rezente Indikationserweiterung vonNintedanib auf pfILD [22] empfehlen wir, bei UIP-artigen Mustern, die nicht als IPF zu diagnostizie-ren sind, sehr engmaschig zu kontrollieren und beiProgredienz rasch eine antifibrotische Therapie ein-zuleiten. Dies betrifft insbesondere ILD-Patientenmit hohem Fibroseausmaß bei Diagnosestellung(>20% des Lungenvolumens) oder Vorhandenseinvon Honeycombing in der HRCT [9].
3. Bei Hinweisen auf eine zugrunde liegende Auto-immunerkrankung, soll jedenfalls ein/e Rheuma-tologe/in hinzugezogen werden und primär dieGrunderkrankung bestmöglich therapiert werden.Zeigt sich im Verlauf dann dennoch ein progredientfibrosierendes Geschehen, ist eine antifibrotischeTherapie als „add-on“ oder anstatt der spezifischenimmunsuppressiven Therapie (je nach extrapulmo-naler Krankheitsaktivität undNebenwirkungsprofil)anzuraten.
4. Ähnliches gilt für Fälle von fibrosierender EAA: Hierist das primäre Ziel die Identifikation der auslösen-den Noxe und deren konsequente Vermeidung. Jemehr Hinweise auf aktuell inflammatorische Pro-zesse vorliegen (z.B. lymphozytäre BAL, Milchglas/Noduli in der CT), desto eher sollte auch eine im-munsuppressive Therapie gegeben werden. Beitrotzdem anhaltend progredient fibrosierendem
K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S29

consensus report
Verlauf sollte eine antifibrotische Therapie zusätz-lich oder stattdessen angedacht werden.
5. Für alle anderen ILD, z.B. idiopathische NSIP oderunklassifizierbare ILD, gilt der obige Ansatz: Stan-dardisierte Abklärung und Besprechung sowie The-rapieentscheidung in einem ILD-Board; hier solltedann nach oben genannten Gesichtspunkten abge-wogen werden, ob:a.eine medikamentöse Therapie sinnvoll ist,b.primär ein Versuch mit immunsuppressiver The-rapie erfolgen soll,
c. eine antifibrotische Therapie bei progredient fi-brosierendem Phänotyp allein oder in Kombina-tion mit (b) erfolgen soll.
6. Der Erfolg und die Verträglichkeit jeder eingeleite-ten Therapie sollten spätestens nach 3 bis 6 Mo-naten reevaluiert werden. Gegebenenfalls kanndann eine Therapieadaptation, ein Wechsel bzw.ein „add-on“ erfolgen. Die Betreuung bzw. regel-mäßige Vorstellung von pfILD-Patienten an einerspezialisierten ILD-Ambulanz ist vorteilhaft, da hierdie üblicherweise langjährige Behandlungserfah-rung vorhanden ist und ggf. Zugang zu klinischenStudien besteht.
7. Zu bedenken ist, dass die wissenschaftliche Evidenzfür die Wirksamkeit immunsuppressiver Substan-zen bei den meisten pfILD gering ist und solcheTherapien ein oft deutliches Nebenwirkungsrisiko(z.B. Infektneigung, Verschlechterung von Komor-biditäten) mit sich bringen. Antifibrotische Thera-pien hingegen sind bei IPF (Pirfenidon, Nintedanib)[7, 8], SSc-ILD (Nintedanib) [21] und pfILD (Ninte-danib) [22] mittlerweile in großen Studien erprobtund zugelassen. Die Rate und Intensität vonNeben-wirkungen sind akzeptabel.
8. Kombinationen aus immunsuppressiver Therapie(insbesondere MMF, Methotrexat) und antifibro-tischer Therapie (Nintedanib) erscheinen in derderzeitigen Datenlage als sicher [21, 22, 24]. Gewis-se Subgruppen profitierenwahrscheinlich von einersolchen Kombination. Um mögliche Nebenwirkun-gen zu differenzieren, sollten diese Therapien abernicht gleichzeitig, sondern sequenziell eingeleitetwerden.
Nichtmedikamentöse Therapien
Keinesfalls sollten neben diesen pharmakologischenInterventionen nichtmedikamentöse Maßnahmenvergessen werden. Diese umfassen insbesonderedie Vermeidung des auslösenden Antigens bei EAA[1, 53, 54], Nikotinkarenz, regelmäßige ambulanteoder stationäre Lungenrehabilitation [55] und Sau-erstofftherapie, wenn indiziert. Eine Erstvorstellungan einem Lungentransplantationszentrum bei infra-ge kommenden Patienten muss zeitgerecht erfolgen.Ähnliches gilt für Patienten mit SSc, für die eine au-tologe Stammzelltransplantation in Betracht kommt.Sämtliche Impfungen laut Impfplan sollten aktuell
durchgeführt bzw. ggf. aufgefrischt werden, insbe-sondere sind alle Patienten mit chronischen Lun-generkrankungen gegen Pneumokokken, Influenza,Pertussis und SARS-CoV-2 zu impfen [56, 57].
Funding Open access funding provided by Kepler Universi-tätsklinikum Linz.
Interessenkonflikt D. Lang: Vortragshonorare und Bera-tertätigkeit von/für BI, Bristol-Myers Squibb (BMS); Reise-/Fortbildungsfinanzierung durch BI, Roche, Merck, Sharp &Dohme (MSD); F. Moazedi-Fürst: Vortragshonorare von BI,Pfizer, Roche, BMS, MSD, Lilly; J. Sautner: Beratertätigkeitvon/fürBI;H.Prosch:VortragshonorarevonBI,Roche,Novar-tis, MSD, BMS, AstraZeneca (AZ); Beratertätigkeit für BI, Ro-che, MSD, BMS, AZ; Reise-/Fortbildungsfinanzierung von BI,Bayer; Forschungsfinanzierung durch BI; S. Handzhiev: Be-ratertätigkeit von/für BI und Roche; K.Hackner: Vortragsho-norare vonNovartis, AZ; Beratertätigkeit für Novartis; Reise-/Fortbildungsfinanzierung durch Roche, BI; I. Tancevski: Vor-tragshonorare von BI, AZ, Novartis, Sanofi; Beratertätigkeitfür BI, AZ, Novartis, Roche; Reise-/Fortbildungsfinanzierungvon BI, AZ, Novartis; H. Flick: Vortragshonorare von BI,Novartis, MSD, Roche, GlaxoSmithKline; Beratertätigkeit fürBI, Reise-/Fortbildungsfinanzierung von BI, Astropharma;H. Koller: Vortragshonorare von und Beratertätigkeit für BI;H.P. Kiener: Vortragshonorare und Beratertätigkeit von/fürBIK: Vortragshonorare von BMS, Novartis, MedMedia Verlag,MediaserviceGmbH,MSD,BI;Beratertätigkeit fürBI;C. Prior:Vortragshonorare von und Reise-/Fortbildungsfinanzierungdurch BI; B. Lamprecht: Vortragshonorare, Beratertätigkeit,Reise-/Fortbildungsfinanzierung von/für BI und Roche.
OpenAccess DieserArtikelwirdunter derCreativeCommonsNamensnennung 4.0 International Lizenz veröffentlicht,wel-che die Nutzung, Vervielfältigung, Bearbeitung, Verbreitungund Wiedergabe in jeglichem Medium und Format erlaubt,sofern Sie den/die ursprünglichen Autor(en) und die Quelleordnungsgemäß nennen, einen Link zur Creative CommonsLizenzbeifügenundangeben, obÄnderungenvorgenommenwurden.
Die in diesem Artikel enthaltenen Bilder und sonstiges Dritt-material unterliegen ebenfalls der genannten Creative Com-mons Lizenz, sofern sich aus der Abbildungslegende nichtsanderes ergibt. Sofern das betreffende Material nicht unterder genannten Creative Commons Lizenz steht und die be-treffende Handlung nicht nach gesetzlichen Vorschriften er-laubt ist, ist für die oben aufgeführten Weiterverwendungendes Materials die Einwilligung des jeweiligen Rechteinhaberseinzuholen.
Weitere Details zur Lizenz entnehmen Sie bitte der Lizenz-information auf http://creativecommons.org/licenses/by/4.0/deed.de.
Literatur
1. WijsenbeekM,Cottin V. Spectrumoffibrotic lungdiseases.N Engl J Med. 2020;383:958–68. https://doi.org/10.1056/NEJMra2005230.
2. European Respiratory Society. Interstitial lung dis-eases. In: European Respiratory Society, Hrsg. EurLung White B. Sheffield: European Respiratory Society;2013. S. 256–69. https://www.erswhitebook.org/chapters/interstitial-lung-diseases/.
S30 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . K

consensus report
3. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis.N Engl J Med. 2018;378:1811–23. https://doi.org/10.1056/NEJMra1705751.
4. Raghu G, Chen S-Y, YehW-S,Maroni B, Li Q, Lee Y-C, et al.Idiopathicpulmonaryfibrosis inUSMedicarebeneficiariesaged 65 years and older: incidence, prevalence, and sur-vival, 2001–11. Lancet Respir Med. 2014;2:566–72. https://linkinghub.elsevier.com/retrieve.
5. Noble PW, Albera C, BradfordWZ, Costabel U, du Bois RM,Fagan EA, et al. Pirfenidone for idiopathic pulmonaryfibrosis: analysis of pooled data from three multinationalphase3 trials.EurRespir J. 2016;47:243–53.https://doi.org/10.1183/13993003.00026-2015.
6. Richeldi L, Cottin V, du Bois RM, SelmanM, Kimura T, Bai-les Z, et al. Nintedanib in patients with idiopathic pulmo-nary fibrosis: Combined evidence from the TOMORROWandINPULSIS®trials.RespirMed.2016;113:74–9.
7. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA,Glaspole I, Glassberg MK, et al. A phase 3 trial of pir-fenidone in patients with idiopathic pulmonary fibrosis.N Engl J Med. 2014;370:2083–92. https://doi.org/10.1056/NEJMoa1402582.
8. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK,Costabel U, et al. Efficacy and safety of Nintedanib in idio-pathicpulmonaryfibrosis.NEngl JMed.2014;370:2071–82.https://doi.org/10.1056/NEJMoa1402584.
9. De Sadeleer LJ, Goos T, Yserbyt J, Wuyts WA. Towards theessence of progressiveness:bringingprogressiveFibrosinginterstitial lung disease (PF-ILD) to the next stage. J ClinMed.2020;9:1722.
10. Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ.What’s inaname?Thatwhichwecall IPF,byanyothernamewould act the same. Eur Respir J. 2018;51:1800692.https://doi.org/10.1183/13993003.00692-2018.
11. George PM, Spagnolo P, KreuterM, Altinisik G, BonifaziM,MartinezFJ, et al. Progressivefibrosing interstitial lungdis-ease: clinical uncertainties, consensus recommendations,andresearchpriorities.LancetRespirMed.2020;8:925–34.
12. Hoffmann-Vold A-M, Allanore Y, Bendstrup E, Bruni C,DistlerO,Maher TM, et al. Theneed for a holistic approachforSSc-ILD—achievementsandambiguity inadevastatingdisease. Respir Res. 2020;21:197. https://doi.org/10.1186/s12931-020-01459-0.
13. Hoffmann-VoldA-M,MaherTM,PhilpotEE,AshrafzadehA,Barake R, Barsotti S, et al. The identification and ma-nagement of interstitial lung disease in systemic sclerosis:evidence-based European consensus statements. LancetRheumatol.2020;2:e71–83.
14. Cottin V, Brown KK. Interstitial lung disease associatedwith systemic sclerosis (SSc-ILD). Respir Res. 2019;20:13.https://doi.org/10.1186/s12931-019-0980-7.
15. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D,Kleerup EC, et al. Mycophenolate mofetil versus oral cy-clophosphamide in scleroderma-related interstitial lungdisease (SLS II): a randomised controlled, double-blind,parallelgrouptrial.LancetRespirMed.2016;4:708–19.
16. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD,Furst DE, et al. Cyclophosphamide versus placebo in Scle-roderma lung disease. N Engl J Med. 2006;354:2655–66.https://doi.org/10.1056/NEJMoa055120.
17. Jordan S, Distler JHW, Maurer B, Huscher D, van Laar JM,Allanore Y, et al. Effects and safety of rituximab in systemicsclerosis: an analysis from the European SclerodermaTrial and Research (EUSTAR) group. Ann Rheum Dis.2015;74:1188–94. https://doi.org/10.1136/annrheumdis-2013-204522.
18. KhannaD, LinCJF, FurstDE,Goldin J, KimG,KuwanaM, etal.Tocilizumabinsystemicsclerosis:arandomised,double-blind,placebo-controlled,phase3 trial.LancetRespirMed.2020;8:963–74.
19. ChanEY,Goodarzi A, SinhaN,NguyenDT, Youssef JG, Sua-rezEE, et al. Long-termsurvival inbilateral lung transplan-tation for Scleroderma-related lung disease. Ann ThoracSurg.2018;105:893–900.
20. Sottile PD, Iturbe D, Katsumoto TR, Connolly MK, Col-lard HR, Leard LA, et al. Outcomes in systemic sclero-sis–related lung disease after lung transplantation. Trans-plantJ.2013;95:975–80.
21. Distler O, Highland KB, Gahlemann M, Azuma A, Fi-scher A, Mayes MD, et al. Nintedanib for SystemicSclerosis–Associated Interstitial Lung Disease. N Engl JMed. 2019;380:2518–28. https://doi.org/10.1056/NEJMoa1903076.
22. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, In-oue Y, et al. Nintedanib in progressiveFibrosing interstitiallungdiseases.NEngl JMed. 2019;381:1718–27. https://doi.org/10.1056/NEJMoa1908681.
23. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A,Richeldi L, et al. Nintedanib in patients with progressivefibrosing interstitial lung diseases—subgroup analyses byinterstitial lung disease diagnosis in the INBUILD trial:a randomised, double-blind, placebo-controlled, parallel-grouptrial.LancetRespirMed.2020;8:453–60.
24. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ,Molina-Molina M, et al. Pirfenidone in patients with un-classifiable progressive fibrosing interstitial lung disease:a double-blind, randomised, placebo-controlled, phase 2trial.LancetRespirMed.2020;8:147–57.
25. Guenther A, Prasse A, Kreuter M, Neuser P, Rabe K, Bo-nella F, et al. Late breaking abstract—exploring efficacyand safety of oral pirfenidone for progressive, non-IPFlung fibrosis (RELIEF). Idiopathic interstitial pneumonias.Eur Respir Soc. 2019; https://doi.org/10.1183/13993003.congress-2019.RCT1879.
26. Sgalla G, Walsh SLF, Sverzellati N, Fletcher S, Cerri S,Dimitrov B, et al. “Velcro-type” crackles predict specificradiologic features of fibrotic interstitial lungdisease. BMCPulm Med. 2018;18:103. https://doi.org/10.1186/s12890-018-0670-0.
27. Walsh SLF, Devaraj A, Enghelmayer JI, Kishi K, Silva RS,Patel N, et al. Role of imaging in progressive-fibrosinginterstitial lung diseases. Eur Respir Rev. 2018;27:180073.https://doi.org/10.1183/16000617.0073-2018.
28. Jeny F, Brillet P-Y, Kim Y-W, Freynet O, Nunes H, Valeyre D.Theplace of high-resolution computed tomography imag-ing in the investigation of interstitial lung disease. ExpertRev Respir Med. 2019;13:79–94. https://doi.org/10.1080/17476348.2019.1556639.
29. Gruden JF, Naidich DP, Machnicki SC, Cohen SL, Girvin F,Raoof S. An algorithmic approach to the interpretationof diffuse lung disease on chest CT imaging. Chest. 2019.https://doi.org/10.1016/j.chest.2019.10.017
30. Adegunsoye A, Oldham JM, Bellam SK, Montner S,Churpek MM, Noth I, et al. Computed tomographyhoneycombing identifies a progressive fibrotic phenotypewith increased mortality across diverse interstitial lungdiseases. Ann Am Thorac Soc. 2019;16:580–8. https://doi.org/10.1513/AnnalsATS.201807-443OC.
31. Ito Y, Arita M, Kumagai S, Takei R, Noyama M, Tokioka F,et al. Serological and morphological prognostic factorsin patients with interstitial pneumonia with autoimmunefeatures. BMC PulmMed. 2017;17:111. https://doi.org/10.1186/s12890-017-0453-z.
K Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . S31

consensus report
32. ParkIN,JegalY,KimDS,DoK-H,YooB,ShimTS,etal.Clinicalcourse and lung function change of idiopathic nonspecificinterstitialpneumonia.EurRespirJ.2009;33:68–76.https://doi.org/10.1183/09031936.00158507.
33. Sverzellati N, Brillet P-Y. When Deep Blue first defeatedKasparov: is a machine stronger than a radiologist at pre-dictingprognosis in idiopathicpulmonaryfibrosis?EurRe-spir J.2017;49:1602144.https://doi.org/10.1183/13993003.02144-2016.
34. PaternitiMO,BiY,RekicD,WangY,Karimi-ShahBA,Chowd-hury BA. Acute exacerbation anddecline in forced vital ca-pacityareassociatedwith increasedmortality in idiopathicpulmonaryfibrosis.AnnAmThoracSoc.2017;14:1395–402.https://doi.org/10.1513/AnnalsATS.201606-458OC.
35. Behr J, Günther A, Bonella F, Dinkel J, Fink L, Geiser T, etal. S2K-Leitlinie zur Diagnostik der idiopathischen Lun-genfibrose. Pneumologie. 2020;74:e1–e2. https://doi.org/10.1055/a-1179-2905.
36. RaghuG,CollardHR,EganJJ,MartinezFJ,BehrJ,BrownKK,et al. An official ATS/ERS/JRS/ALAT statement: idiopa-thic pulmonary fibrosis: evidence-based guidelines fordiagnosis and management. Am J Respir Crit Care Med.2011;183:788–824. https://doi.org/10.1164/rccm.2009-040GL.
37. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A,Behr J, et al. Anofficial ATS/ERS/JRS/ALATclinical practiceguideline: treatment of idiopathic pulmonary fibrosis. Anupdate of the 2011 clinical practice guideline. Am J RespirCrit Care Med. 2015;192:e3–e19. https://doi.org/10.1164/rccm.201506-1063ST.
38. Holland AE, Spruit MA, Troosters T, Puhan MA, Pepin V,Saey D, et al. An official European Respiratory Socie-ty/American Thoracic Society technical standard: fieldwalking tests in chronic respiratory disease. Eur Re-spir J. 2014;44:1428–46.https://doi.org/10.1183/09031936.00150314.
39. Raghu G, Remy-JardinM,Myers JL, Richeldi L, RyersonCJ,LedererDJ,etal.Diagnosisofidiopathicpulmonaryfibrosis.An official ATS/ERS/JRS/ALAT clinical practice guideline.Am J Respir Crit Care Med. 2018;198:e44–e68. https://doi.org/10.1164/rccm.201807-1255ST.
40. RaghuG, Remy-JardinM, RyersonCJ, Myers JL, KreuterM,Vasakova M, et al. Diagnosis of hypersensitivity pneumo-nitis in adults. An official ATS/JRS/ALAT clinical practiceguideline. Am J Respir Crit Care Med. 2020;202:e36–e69.https://doi.org/10.1164/rccm.202005-2032ST.
41. Meyer KC. The clinical utility of bronchoalveolar lavage ininterstitial lung disease—is it really useful? Expert Rev Re-spirMed.2014;8:133–5.https://doi.org/10.1586/17476348.2014.879827.
42. American Thoracic Society/European Respiratory Socie-ty. International multidisciplinary consensus classifica-tion of the idiopathic interstitial pneumonias. Am J RespirCritCareMed. 2002;165:277–304. https://doi.org/10.1164/ajrccm.165.2.ats01.
43. Travis WD, Costabel U, Hansell DM, King TE, Lynch DA,NicholsonAG, et al. An official AmericanThoracic Society/European Respiratory Society statement: update of theinternational multidisciplinary classification of the idio-pathic interstitial Pneumonias. Am J Respir Crit CareMed.2013;188:733–48. https://doi.org/10.1164/rccm.201308-1483ST.
44. DeSadeleer LJ,MeertC, Yserbyt J, SlabbynckH,Verschake-len JA, Verbeken EK, et al. Diagnostic ability of a dynamic
multidisciplinary discussion in interstitial lung diseases.Chest.2018;153:1416–23.
45. Thomeer M, Demedts M, Behr J, Buhl R, Costabel U,Flower CDR, et al. Multidisciplinary interobserver agree-ment in the diagnosis of idiopathic pulmonary fibro-sis. Eur Respir J. 2008;31:585–91. https://doi.org/10.1183/09031936.00063706.
46. Walsh SLF, Sverzellati N, Devaraj A, Wells AU, Han-sell DM. Chronic hypersensitivity pneumonitis: high re-solution computed tomography patterns and pulmonaryfunction indices as prognostic determinants. Eur Radi-ol. 2012;22:1672–9. https://doi.org/10.1007/s00330-012-2427-0.
47. Kim EJ, Elicker BM, Maldonado F, Webb WR, Ryu JH, VanUden JH, et al. Usual interstitial pneumonia in rheuma-toid arthritis-associated interstitial lung disease. Eur Re-spir J. 2010;35:1322–8. https://doi.org/10.1183/09031936.00092309.
48. GohNSL,DesaiSR,VeeraraghavanS,HansellDM,CopleySJ,MaherTM,etal. Interstitial lungdisease in systemicsclero-sis. Am J Respir Crit Care Med. 2008;177:1248–54. https://doi.org/10.1164/rccm.200706-877OC.
49. Cottin V. The impact of emphysema in pulmonary fibrosis.Eur Respir Rev. 2013;22:153–7. https://doi.org/10.1183/09059180.00000813.
50. Cottin V, Hansell DM, Sverzellati N, Weycker D, Anto-niou KM, Atwood M, et al. Effect of emphysema extent onserial lung function in patients with idiopathic pulmonaryfibrosis.AmJRespirCritCareMed.2017;196:1162–71.
51. Ryerson CJ, Urbania TH, Richeldi L, Mooney JJ, Lee JS,Jones KD, et al. Prevalence and prognosis of unclassifiableinterstitiallungdisease.EurRespirJ.2013;42:750–7.https://doi.org/10.1183/09031936.00131912.
52. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV,GalvinJR,etal.Diagnosticcriteriaforidiopathicpulmonaryfibrosis: a Fleischner Society White Paper. Lancet RespirMed.2018;6:138–53.
53. Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Marti-nez FJ, Flaherty KR. Diagnosis and treatment of Fibrotichypersensitivity pneumonia. Where we stand and wherewe need to go. Am J Respir Crit Care Med. 2017;196:690–9.https://doi.org/10.1164/rccm.201608-1675PP.
54. Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hy-persensitivity pneumonitis: perspectives in diagnosis andmanagement.AmJRespirCritCareMed.2017;196:680–9.
55. Cheng L, Tan B, Yin Y, Wang S, Jia L, Warner G, et al.Short- and long-term effects of pulmonary rehabilitationfor idiopathic pulmonary fibrosis: a systematic review andmeta-analysis.ClinRehabil.2018;32:1299–307.https://doi.org/10.1177/0269215518779122.
56. Bundesministerium für Arbeit, Soziales G undK. ImpfplanÖsterreich 2020. 2020. https://www.sozialministerium.at/Themen/Gesundheit/Impfen/Impfplan-Österreich.html.Zugegriffen: Internet.
57. Bundesministerium Soziales, Gesundheit P und K.COVID-19-Impfungen: Empfehlungen des Nationa-len Impfgremiums zur Priorisierung.. https://www.sozialministerium.at/dam/jcr:12f12b2b-375e-483f-8a80-d6c58b0c848c/COVID-19_Empfehlung_des_Nationalen_Impfgremiums_zur_Priorisierung_Version_2.1-26.12.2020.pdf.Zugegriffen:05.01.2021
Hinweis des Verlags Der Verlag bleibt in Hinblick auf geo-grafische Zuordnungen und Gebietsbezeichnungen in veröf-fentlichten Karten und Institutsadressen neutral.
S32 Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur. . . K


















