
Deckblatt Untersuchungen zu Struktur und Funktion eines...
101
Deckblatt Untersuchungen zu Struktur und Funktion eines Porenproteins der äußeren Chloroplastenmembran von Diplom-Biochemiker Dirk Linke aus Leonberg von der Fakultät II - Mathematik und Naturwissenschaften der Technischen Universität Berlin zur Erlangung des akademischen Grades Doktor der Naturwissenschaften -Dr. rer.nat.- genehmigte Dissertation Promotionsausschuß: Vorsitzender: Prof. Dr. R. Schomäcker, TU Berlin Berichter: Priv. Doz. Dr. Petra Fromme, TU Berlin Berichter: Prof. Dr. G. H. Findenegg, TU Berlin Tag der wissenschaftlichen Aussprache: 10. Juli 2002 Berlin 2002 D 83
-
Upload
trinhnguyet -
Category
Documents
-
view
220 -
download
0
Transcript of Deckblatt Untersuchungen zu Struktur und Funktion eines...

Deckblatt Untersuchungen zu Struktur und Funktion
eines Porenproteins der äußeren Chloroplastenmembran
von Diplom-Biochemiker
Dirk Linke
aus Leonberg
von der Fakultät II - Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
-Dr. rer.nat.-
genehmigte Dissertation
Promotionsausschuß:
Vorsitzender: Prof. Dr. R. Schomäcker, TU Berlin
Berichter: Priv. Doz. Dr. Petra Fromme, TU Berlin
Berichter: Prof. Dr. G. H. Findenegg, TU Berlin
Tag der wissenschaftlichen Aussprache: 10. Juli 2002
Berlin 2002 D 83

1
INHALT
DECKBLATT.............................................................................................................. 0
INHALT....................................................................................................................... 1
ZUSAMMENFASSUNG.............................................................................................. 4
ABKÜRZUNGSVERZEICHNIS .................................................................................. 5
EINLEITUNG .............................................................................................................. 6
Die Erbse (Pisum Sativum)......................................................................................................6
Die Pflanzenzelle .......................................................................................................................7
Der Chloroplast.........................................................................................................................8 Aufbau.....................................................................................................................................8 Funktion des Chloroplasten in der pflanzlichen Zelle ............................................................9 Endosymbiontentheorie...........................................................................................................9
Die äußere Chloroplastenmembran......................................................................................10 Lipide der äußeren Chloroplastenmembran ..........................................................................10 Proteine der äußeren Chloroplastenmembran und ihre Funktion..........................................12 Proteinimport über die äußere und innere Chloroplastenmembran ......................................13 Proteinimport in die äußere Chloroplastenmembran ............................................................14
OEP16 ......................................................................................................................................14 Sequenzen..............................................................................................................................15 Aufbau...................................................................................................................................16 Funktion und Regulation.......................................................................................................16
Faltung von Membranproteinen ...........................................................................................19 Allgemeine strukturelle Eigenschaften von Membranproteinen...........................................19 Mechanismen der Insertion in die Membran in vivo .............................................................19 Thermodynamische Stabilität von Membranproteinen .........................................................20 Der Faltungsprozeß ...............................................................................................................21
Zielsetzung...............................................................................................................................23
MATERIAL UND METHODEN ................................................................................. 25
Überexpression von OEP16 in E.coli ....................................................................................25
Reinigung von OEP16 ............................................................................................................25
SDS-Gelelektrophorese ..........................................................................................................26

2
Faltung von OEP16 ................................................................................................................27 Liposomen.............................................................................................................................27 Detergenz-basierte Methoden für die Rekonstitution ...........................................................28
Spektroskopische Methoden..................................................................................................29 Proteinbestimmung nach Bradford........................................................................................29 Fluoreszenz-Spektroskopie ...................................................................................................30 CD-Spektroskopie .................................................................................................................30 IR-Spektroskopie...................................................................................................................31 Zeitaufgelöste Spektroskopie ................................................................................................31
Physikalisch-chemische Methoden........................................................................................32 DSC .......................................................................................................................................32
Elektronenmikroskopie..........................................................................................................33
Kristallisation..........................................................................................................................33 Probenvorbereitung ...............................................................................................................33 Kristallisationsansätze...........................................................................................................34
Messung der Röntgenbeugung ..............................................................................................35
Theoretische Strukturvorhersage .........................................................................................35 "Multiple Sequence Alignment" ...........................................................................................35 Vorhersage transmembraner Bereiche ..................................................................................36 Hydrophobe Cluster-Analyse................................................................................................37
ERGEBNISSE .......................................................................................................... 38
Von der Proteinreinigung zum gefalteten Protein (eine Einleitung) .................................38
Proteinreinigung .....................................................................................................................40
Faltungsmethoden ..................................................................................................................42 Dialyse...................................................................................................................................42 Gelfiltration ...........................................................................................................................44 Verdünnung...........................................................................................................................44 Fluoreszenzspektroskopie .....................................................................................................45 CD-Spektroskopie .................................................................................................................55 IR-Spektroskopie...................................................................................................................59
Biophysikalische Daten des gefalteten Proteins...................................................................62 DSC .......................................................................................................................................62 Zeitaufgelöste Spektroskopie ................................................................................................65 Zeitaufgelöste Fluoreszenzspektroskopie .............................................................................65 Zeitaufgelöste CD-Spektroskopie .........................................................................................67 Zeitkonstanten und Aktivierungsenergien ............................................................................68
Das Protein wird sichtbar ......................................................................................................73 Elektronenmikroskopie .........................................................................................................73 Kristallisation ........................................................................................................................75

3
Theoretische Ansätze zur Strukturaufklärung....................................................................81 Multiple Sequence Alignment...............................................................................................82 Hydropathie und Vorhersage transmembraner Segmente.....................................................83 Hydrophobe Cluster-Analyse................................................................................................85 Topologie ..............................................................................................................................87
ZUSAMMENFASSUNG DER ERGEBNISSE........................................................... 90
LITERATUR ............................................................................................................. 92
DANKSAGUNG...................................................................................................... 100

4
Zusammenfassung
In der vorliegenden Arbeit wurde die biophysikalischen Eigenschaften eines kleinen Kanalproteins aus
der äußeren Chloroplastenmembran der Erbse, genannt OEP16 („outer envelope Protein 16 kDa“),
untersucht und ein neues Strukturmodell entwickelt, auf dessen Basis die Funktion des Proteins mit
molekularbiologischen Methoden nun genauer erforscht werden kann.
OEP16 ist ein Membranprotein mit ungewöhnlichen Eigenschaften. Als Protein der äußeren
Chloroplastenmembran insertiert es selbständig in geeignete Lipiddoppellschichten und in
Detergenzmizellen. Bei vielen eukaryontischen Membranproteinen kommt eine bakterielle
Überexpression zur Gewinnung großer Proteinmengen nicht in Frage, da zumeist ein korrekter Einbau
in eine Membran mangels der komplexen eukaryontischen Faltungsmaschinerie in diesen Systemen
nicht gegeben ist. OEP16 hingegen erlaubt es, aufgrund seiner selbsttätigen Renaturierung das Protein
ohne Rücksicht auf die native Struktur in E.coli überzuexprimieren und erst anschließend
rückzufalten.
Es konnte gezeigt werden, daß man große Mengen korrekt gefalteten Proteins erhalten kann, indem
man in chaotropen Puffersystemen aufgereinigtes Protein durch einen Verdünnungsschritt in einen
detergenzhaltigen Puffer überführt. Der Nachweis, daß sich OEP16 in Detergenzmizellen in eine
native Form faltet, ist schwierig, da die Funktion des Proteins, seine Kanalaktivität, nur in einem
Membransystem meßbar ist. Durch den Vergleich von Fluoreszenz- und CD-Spektren von
detergenzhaltigen Proteinlösungen und dem in Liposomen eingebautem Protein (mit nachgewiesener
Funktionalität) konnte gezeigt werden, daß die Struktur von OEP16 in den auf unterschiedliche Weise
rekonstituierten Proben übereinstimmte. Zeitaufgelöste Messungen mit denselben spektroskopischen
Methoden lieferten Daten über den Verlauf und die Geschwindigkeit der Proteinfaltung bei dieser
Renaturierungsmethode, einschließlich der Aktivierungsenergien für den Prozeß. Kalorimetrische
Messungen (DSC) erlaubten eine Abschätzung der Faltungsenthalpie. Durch hochauflösende CD-
Messungen und Infrarotspektroskopie wurde schließlich am in vitro gefalteten Protein gezeigt, daß es
sich bei OPE16 entgegen bisheriger Voraussagen um ein rein α-helikales Membranprotein handelt.
Dieser Befund konnte durch theoretische Methoden der Strukturbestimmung bestätigt werden. Ein
neues Strukturmodell wurde erstellt. Erste vielversprechende Ansätze der Proteinkristallisation und
der Elektronenmikroskopie zeigen, daß eine detaillierte Struktur von OEP16 in absehbarer Zeit
erhältlich sein könnte. Dies wäre weltweit die erste hochaufgelöste Struktur eines im Reagenzglas
gefalteten Membranproteins. In einer Zeit, in der Proteomics, also das Überexprimieren und
Analysieren von Proteinen, die Arbeit der größtenteils abgeschlossenen Sequenzierungprojekte
fortführen sollen, konnte mit OEP16 ein Modellsystem für die detaillierte Struktur- und
Funktionsanalyse von Membranproteinen etabliert werden.

5
Abkürzungsverzeichnis
ATP Adenosintriphosphat C8E4 Tetraethylenglykol-Monooctylether C10E6 Hexaethylenglykol-Monodecylether C12E6 Hexaethylenglykol-Monododecylether C12E8 Octaethylenglykol-Monododecylether Capso 3-Cyclohexylamino-2-hydroxy-1-propansulfonsäure CD Zirkulardichroismus (Circular Dichroism) CMC Kritische Mizellare Konzentration (critical micellaer concentration) DGDG Digalaktosyldiacylglycerin β-DM β-D-Dodecylmaltosid DNA Desoxyribonukleinsäure DTT Dithiothreitol EDTA Ethylendiamintetraessigsäure E.coli Escherichia coli ER Endoplasmatisches Retikulum FTIR Fourier-Transform-Infrarotspektroskopie HCA Hydrophobe Cluster-Analyse Hepes N-2-Hydroxyethylpiperazin-N'-2-Ethansulfonsäure in vitro „im Reagenzglas“ in vivo „im lebenden Organismus“ IPTG Isopropyl–β–D–thiogalactopyranoside kDa Kilodalton MDGD Monogalaktosyldiacylglyzerin MPD 2-Methyl-2,4-Pentandiol mRNA Boten-Ribonukleinsäure (m für engl. „messenger“) NMR Kernspinresonanz (nuclear magnetic resonance) OEP „Outer Envelope Protein“ β-OG β-D-Octylglukosid PC Phosphatidylcholin PCR Polymerasekettenreaktion (polymerase chain reaction) PE Phosphatidylethanolamin PEG Polyethylenglykol PG Phosphatidylserin PI Phospahtidyinositol PS Phosphatidylserin R Reynolds-Konstante, R = 8,314 J/mol*K RNA Ribonukleinsäure SB12 n-Dodecyl-N,N-Dimethyl-3-ammoniopropansulfonat SDS Natriumdodecylsulfat SQDG Sulfoquinovosyldiacylglyzerin

6
Einleitung
Die Erbse (Pisum Sativum)
Die Erbse ist schon seit den berühmten Experimenten zur Vererbungslehre von Mendel ein
wichtiger Modellorganismus der biologischen Forschung. Für die Untersuchung von
Chloroplasten ist die Erbse besonders geeignet, weil sich diese Plastiden aus den schnell
wachsenden Erbsenkeimlingen vergleichsweise einfach und vor allem intakt isolieren lassen.
Es ist daher nur konsequent, für Experimente zum Stofftransport über die
Chloroplastenmembranen, aber auch für weiterführende molekularbiologische und
biochemische Forschung gerade die Erbsenpflanze als Modell zu wählen.
Abbildung 1:
Foto eines Erbsenkeimlings, wenige Tage alt.

7
Die Pflanzenzelle
Abbildung 2:
Schematische Darstellung einer Pflanzenzelle.
Eukaryontische Zellen unterscheiden sich von den einfacher aufgebauten bakteriellen Zellen
durch die Existenz eines Zellkerns, der das Erbgut vom Cytosol der Zelle abtrennt. Die
Pflanzenzelle stellt eine besondere Form der eukaryontischen Zelle dar. Zusätzlich zur
Zellmembran, einem Zellkern, den Mitochondrien und Lysosomen, dem endoplasmatischen
Reitculum (ER) und dem Golgiapparat, die praktisch allen eukaryontischen Zellen gemeinsam
sind, besitzt eine pflanzliche Zelle eine Zellwand, Plastiden und eine oder mehrere Vakuolen.
Die Zellmembran umhüllt die gesamte Zelle und grenzt sie gegenüber der Außenwelt ab.
Stofftransport durch diese Membran ist nur durch Transportproteine möglich. Die Zellwand
der Pflanzen ist eine harte, poröse Struktur aus komplexen Kohlenhydraten, die die Zelle
mechanisch schützt. Die Flüssigkeit innerhalb der Zelle wird als Cytosol bezeichnet. Es
umgibt alle Organellen der Zelle und ist Ort der Biosynthese vieler für die Zelle
lebensnotwendiger Substanzen. Im Cytosol befindet sich auch der Apparat zur Zellteilung.
Der Zellkern enthält die Chromosomen der Pflanzenzelle. Er ist von einer Doppelmembran,
der Kernmembran, umgeben, die Verbindung zum endoplasmatischen Retikulum hat. Am
rauhen ER bzw. den daran angelagerten Ribosomen findet die Proteinbiosynthese statt. Die
Proteine werden dann durch ein Erkennungs- und Transportsystem, den Golgi-Apparat, mit
letzten Modifikationen versehen und mit Hilfe von Vesikeln an ihren Bestimmungsort
geschleust. Am glatten ER werden hauptsächlich chemische Signale von der Zellmembran
verarbeitet und weitergegeben (z.B. in den Zellkern zur Veränderung der Genexpression). In
den Lysosomen werden überflüssige Zellbestandteile zerlegt und giftige Substanzen abgebaut.
Die Vakuole dient als Speicher für Wasser und Nährstoffe und kann Giftstoffe wie z.B.
Schwermetalle einlagern und sie so für die Zelle unschädlich machen. Die Mitochondrien sind
die Kraftwerke der Zelle. Hier wird der Großteil der Energie gewonnen, der zum
Aufrechterhalten der Zellfunktionen notwendig ist. Bei der „Atmung“ werden Kohlenhydrate

8
zu Wasser und CO2 oxidiert und die frei werdende Energie in Form von ATP und anderen
biochemischen Energieträgern gespeichert. Die Mitochondrien verfügen über ein
Doppelmembransystem und ein eigenes Genom, das für einen kleinen Teil der in der
Organelle vorhandenen Proteine codiert. Auch die Plastiden, deren prominenteste Vertreter
die Chloroplasten sind, verfügen über eigenes Erbgut. Ihr Aufbau und ihre Funktion werden
im folgenden detaillierter erläutert.
Der Chloroplast
Plastiden sind die Zellorganellen, durch die sich Pflanzenzellen von den Zellen aller anderen
Eukaryonten unterscheiden. Die grünen Chloroplasten sind die wichtigsten und bekanntesten
Plastiden. Erwähnenswert sind aber auch die Chromoplasten, die Pflanzenblüten und Früchten
vor allem durch ihren hohen Carotinoid-Gehalt ihre Farbe verleihen und die Amyloplasten,
die den Stärkespeicher in Pflanzensamen, Wurzeln und Knollen bilden. Auch andere
Plastidentypen in hoch spezialisierten Pflanzengeweben sind bekannt, so sind zum Beispiel
die Plastiden in Wurzeln hauptsächlich mit der Assimilierung von anorganischem Stickstoff
befaßt (Joyard, 1998) (Lam, 1996).
Aufbau
Chloroplasten bestehen aus mehreren Membranschichten von sehr unterschiedlichem Aufbau
und Funktion. Vom Cytosol ist der Chloroplast durch die äußere (1) und die innere
Chloroplastenmembran (2) getrennt. Der Kern eines Chloroplasten wird von gestapelten
Ausstülpungen der Thylakoidmembran, den Grana (3,4) gebildet. Diese Membran umschließt
das Lumen und trennt es vom Stroma (5), dem Raum zwischen den Thylakoiden und der
inneren Chloroplastenmembran, ab. Das Lumen ist ein durchgehender Raum, die Grana sind
durch Membrankanäle, die sogenannten Stromathylakoide, verbunden. Der Chloroplast
besitzt ein eigenes, im Stroma lokalisiertes Genom, das jedoch nur für einen Teil der
Chloroplastenproteine codiert, ebenso eigene Ribosomen für die Translation von RNA in
Proteine. Chloroplasten sind in der Lage, sich unabhängig von der Pflanzenzelle zu teilen und
besitzen den gesamten hierfür notwendigen biochemischen Apparat (Pyke, 1999).

9
Abbildung 3:
Schematischer Aufbau eines Chloroplasten.
1: Äußere Chloroplastenmembran
2: Innere Chloroplastenmembran
3: Thylakoide (Querschnitt, darin das Lumen)
4: Thylakoide (Außenansicht)
5: Stroma
Funktion des Chloroplasten in der pflanzlichen Zelle
Die Hauptaufgabe der Chloroplasten ist die Erzeugung von chemischer Energie aus Licht,
Kohlendioxid und Wasser, die Photosynthese. Dieser Prozeß läuft an den
Thylakoidmembranen („Lichtreaktion“, das Aufbauen eines elektrochemischen Gradienten)
und im Stroma („Dunkelreaktion“, die CO2-Fixierung) ab. Als Produkte entstehen
Kohlenhydrate, die die Grundlage des Stoffwechsels einer Pflanzenzelle darstellen.
Gewissermaßen als Nebenprodukt entsteht Sauerstoff, der in die Atmosphäre abgegeben wird.
Darüber hinaus enthalten Chloroplasten Enzyme für eine ganze Reihe grundlegender
Stoffwechselwege, insbesondere für die Lipidbiosynthese und den Aminosäuremetabolismus
(Lam, 1996). Auch eine Reihe von Pflanzenhormonen wird in den Chloroplasten aus
Lipidvorstufen synthetisiert (Joyard, 1998). Ein Austausch von Stoffwechselprodukten und
deren Vorstufen, aber auch anorganischen Ionen über die innere und äußere
Chloroplastenmembran wird durch eine Vielzahl hoch spezialisierter Transportsysteme
ermöglicht (Flügge, 1998).
Endosymbiontentheorie
Chloroplasten sind nicht in der Lage, außerhalb der pflanzlichen Zelle zu leben. Dennoch geht
man davon aus, daß Chloroplasten von Vorläufern der Cyanobakterien abstammen, die eine
symbiotische Verbindung mit frühen eukaryontischen Zellen eingegangen sind (Cavalier-
Smith, 2000; Margulis, 1996). Dafür spricht zum Beispiel, daß diese Organellen ihr eigenes

10
Genom besitzen, das allerdings im Lauf der Evolution stark reduziert wurde und inzwischen
von Genen ergänzt wird, die im Zellkern der Pflanze liegen (Leon, 1998). Die Ribosomen im
Stroma der Chloroplasten unterscheiden sich signifikant von den Ribosomen im Cytosol der
Zelle. Sie sind bakteriellen, nicht eukaryontischen Typs(Harris, 1994). Die Untereinheiten des
Proteinimportkomplexes, der im Cytosol translatierte Proteine ins Innere der Chloroplasten
transportiert, haben große Sequenzähnlichkeiten zu cyanobakteriellen Membranproteinen
(McFadden, 1999; Reumann, 1999). Auch die große Ähnlichkeit des in den Thylakoiden
lokalisierten Photosyntheseapparates zu dem der Cyanobakterien zeigt das enge
verwandtschaftliche Verhältnis der Plastiden zu ihren Vorfahren(Blankenship, 1998). Die
Lipidzusammensetzung der Chloroplastenmembranen ist sehr ähnlich zu der
photosynthetischer Bakterien und unterscheidet sich grundlegend von allen anderen
Membranen der pflanzlichen Zelle. So kommt das anionische Lipid
Sulfoquinovosyldoacylglcerin fast ausschließlich in photosynthetischen Bakterien und
Chloroplasten vor(Benning, 1998).
Die äußere Chloroplastenmembran
Gemeinsam bilden die innere und äußere Chloroplastenmembran 5-10% der gesamten
Membranfläche der Chloroplasten. Dennoch enthalten sie nur 1-2% des Gesamtproteins der
Organelle. Die äußere Membran besteht aus 2,5 bis 3 mg Lipid pro mg Protein, dem höchsten
Lipidanteil aller pflanzlichen Membransysteme, und hat damit auch eine sehr geringe Dichte
von 1,08 g/cm3. Dies kann man sich für die Abtrennung von der inneren Plastidenmembran zu
Nutze machen(Joyard, 1991). Wegen ihrer scheinbaren Durchlässigkeit für Substanzen mit
einem Molekulargewicht von unter 10000 g/mol wurde bisher angenommen, daß die äußere
Chloroplastenmembran wie die entsprechende mitochondriale Membran in vivo keinerlei
Membranpotential besitzt. Sollte der Transport durch dieses Membransystem jedoch einer
Regulation unterliegen, wofür es Hinweise gibt(Flügge, 2000; Soll, 2000), ist unter
bestimmten physiologischen Bedingungen ein durch unterschiedliche
Metabolitenkonzentrationen dies- und jenseits der Membran gebildetes Potential
vorstellbar(Lemeshko, 2000).
Lipide der äußeren Chloroplastenmembran
Biologische Membranen bestehen aus einer Lipid-Doppelschicht. Ihre Hauptbestandteile bei
Pflanzen sind anionische Phospholipide (hauptsächlich Phosphatidylcholin (PC),
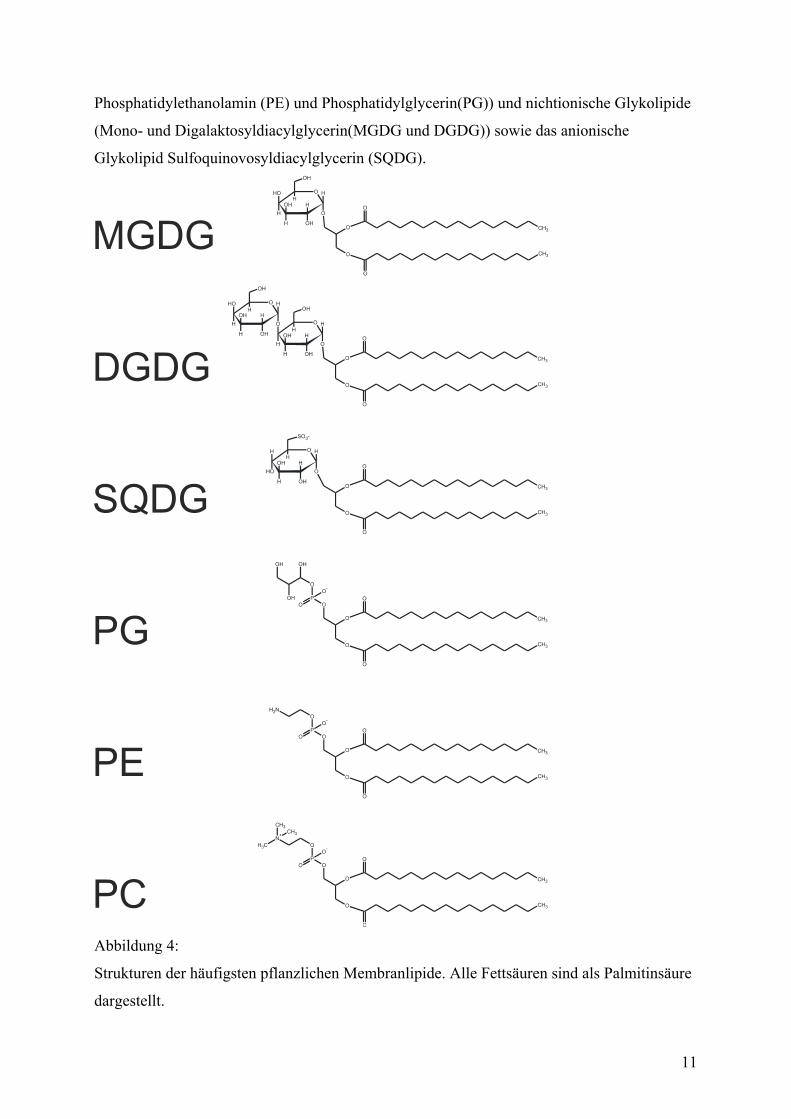
11
Phosphatidylethanolamin (PE) und Phosphatidylglycerin(PG)) und nichtionische Glykolipide
(Mono- und Digalaktosyldiacylglycerin(MGDG und DGDG)) sowie das anionische
Glykolipid Sulfoquinovosyldiacylglycerin (SQDG).
Abbildung 4:
Strukturen der häufigsten pflanzlichen Membranlipide. Alle Fettsäuren sind als Palmitinsäure
dargestellt.

12
In kleineren Mengen kommen auch pflanzliche Sterole und Phosphatidylinositol (PI),
Phosphatidyserin (PS) und Cardiolipin (Diphosphatidydiacylglycerin) vor. Interessant ist, daß
sich diese Lipide sehr ungleichmäßig auf die verschiedenen pflanzlichen Membransysteme
verteilen. Insbesondere die Chloroplastenmembranen weichen deutlich von der
Zusammensetzung anderer eukaryontischer Membranen ab. So kommen die beiden
Galaktolipide und das Sulfolipid ausschließlich in Chloroplasten vor. Im Gegensatz dazu
findet man in den Chloroplastenmembranen kein Phosphatidylethanolamin(Block, 1983).
Auch unter den verschiedenen Membranen innerhalb der Plastiden gibt es gravierende
Unterschiede. Phosphatidylcholin findet sich zum Beispiel kaum in den Thylakoiden, in
großen Mengen jedoch in den beiden umhüllenden Membranen der Organelle. Die äußere
Chloroplastenmembran besteht bei den gut untersuchten Spinatchloroplasten aus etwa 20%
MGDG, 30% DGDG, 5% SQDG, 30% PC, 10% PG und 5% PI (Block, 1983). Auch kleine
Mengen Sterole wurden gefunden(Poincelot, 1973).
Proteine der äußeren Chloroplastenmembran und ihre Funktion
In vielen Textbüchern zur Physiologie und Biochemie der Pflanzen werden die äußere
Chloroplastenmembran und ihre Bestandteile nur am Rande erwähnt. Dies liegt hauptsächlich
daran, daß bis vor wenigen Jahren nur eine Handvoll Proteinkomponenten dieser Membran
identifiziert worden waren. Bekannt war der Proteinimportapparat der Chloroplasten, der sich
über die äußere und innere Membran der Organelle erstreckt, und bekannt war auch, daß es
Porenproteine geben mußte, die dafür sorgten, daß Stoffwechselprodukte die
Lipiddoppelschicht passieren können. Man ging davon aus, daß es sich hierbei um Proteine
des Porin-Typs handeln mußte, und tatsächlich konnten in Wurzelplastiden der Erbse solche
Porine identifiziert werden. Allerdings wurden diese den mitochondrialen Porinen ähnlichen
Proteine in vitro nur in nicht-grüne Plastiden importiert. In grünen Chloroplasten scheinen sie
nicht vorzukommen (Fischer, 1994; Popp, 1997). Hinweise auf unterschiedliche spezifische
Kanalpoteine lieferten elektrophysiologische Messungen an intakten äußeren
Chloroplastenmembranen(Heiber, 1995). Später wurden die entsprechenden Gene mit
molekularbiologischen Methoden identifiziert(Bölter, 1999; Pohlmeyer, 1997a; Pohlmeyer,
1998) und die Proteine genauer charakterisiert(Linke, 2000; Röhl, 1999; Steinkamp, 2000).
Auch viele für den Stoffwechsel der Pflanze wichtige Enzyme sind an oder in der äußeren
Membran der Plastiden lokalisiert. Dies konnte unter anderem für das Enzym DGDG-
Synthase gezeigt werden, welches das Membranlipid Monogalaktosyldiacylglycerin in

13
Digalaktosyldiacylglycerin umwandelt(Froehlich, 2001) und für die Acyl-Coenzym-A-
Synthase, ein zentrales Enzym des Fettsäurestoffwechsels(Block, 1983). Auch die pflanzliche
Hexokinase, die Glucose und auch Fructose unter ATP-Verbrauch in energiereiche
Zuckerphosphate umwandelt, ist in der Hüllmembran verankert und verhindert dort durch die
Umsetzung möglicherweise einen Rücktransport von Glucose ins Innere der Organelle durch
spezifische Glucose-Translokatoren in der inneren Chloroplastenmembran(Wiese, 1999).
Proteinimport über die äußere und innere Chloroplastenmembran
Ein großer Teil der Proteine in den Chloroplasten wird von Genen im Zellkern codiert. Diese
Proteine, die von den Ribosomen im Cytosol der Pflanzenzelle synthetisiert werden, müssen
mittels spezifischer Transportprozesse in die Chloroplasten gelangen. Dazu ist nicht nur ein
Importsignal innerhalb der Aminosäuresequenz des Proteins nötig, sondern auch ein spezieller
Import-Apparat, der das Peptid über die äußere und innere Chloroplastenmembran
transportiert.
Im allgemeinen enthalten für den Chloroplasten bestimmte Proteine ein sogenanntes
Signalpeptid an ihrem N-Terminus, das während des Importvorgangs in die Organelle
abgespalten wird. Signalpeptide für den Transport ins Stroma und in die innere Membran der
Chloroplasten enthalten oft viele basische und hydroxylierte Aminosäuren und werden von
einer cytosolischen Proteinkinase an einem Serin- oder Threoninrest phosphoryliert. Die
Dephosphorylierung erfolgt vor dem eigentlichen Importprozess vermutlich an der äußeren
Chloroplastenmembran(Heins, 1998). Das Signalpeptid wird dann von einer im Stroma
befindlichen spezifischen Protease beim Durchtritt durch den Importapparat entfernt(Bruce,
2000). Im Gegensatz zu mitochondrialen Signalsequenzen bilden die Signalpeptide der
Chloroplasten selbst keinerlei Sekundärstruktur aus(von Heijne, 1991). Eine solche Struktur
entsteht erst durch Wechselwirkungen mit den Lipiden der äußeren Chloroplastenmembran
und ermöglicht damit den Import des Proteins(Pinnaduwage, 1996; Van't Hof, 1993). Dieser
Vorgang ist vermutlich deshalb selektiv für die äußere Chloroplastenmembran, weil sie als
einzige zum Cytosol exponierte Membran die Glykolipide Mono- und
Digalaktosyldiacylglycerin enthält und die Signalpeptide nachweislich mit diesen Lipiden
wechselwirken können(Pinnaduwage, 1996). Auch hat dieses Membransystem ein hohes
Lipid/Protein-Verhältnis von 3.0(Keegstra, 1989), was eine starke Exponierung von Lipid-
Kopfgruppen zum Cytosol zur Folge hat. Veränderungen in der Lipidzusammensetzung dieser
Membran haben eine Veränderung des Import-Verhaltens zur Folge(Kerber, 1992).

14
Die Signalsequenzen für die innere Chloroplastenmembran unterscheiden sich nicht von den
für das Stroma codierenden Signalen. Es wird vermutet, daß die Information für das
Inserieren in die Membran während des Importvorganges in hydrophoben Bereichen des
Proteins außerhalb der abspaltbaren Signalsequenz liegt(Heins, 1998). Für den Import in das
Lumen der Thylakoide oder in die Thylakoidmembran muß das Signalpeptid hingegen aus
zwei Teilen bestehen, einem abspaltbaren Peptid für den Transport ins Stroma und einer
weiteren Sequenz, die nach dem Durchtritt durch die Thylakoidmembran von einer im Lumen
befindlichen spezifischen Protease abgeschnitten wird(Keegstra, 1999). Die
Importmechanismen in die Thylakoide haben Ähnlichkeit mit Transportvorgängen in
Bakterien und im endoplasmatischen Reticulum der Eukaryonten(Cline, 1996; Settles, 1998).
Proteinimport in die äußere Chloroplastenmembran
Der Importweg von Proteinen der äußeren Chloroplastenmembran ist nur in Einzelfällen
komplett aufgeklärt.
OEP75 („outer envelope protein 75 kDa“), selbst Bestandteil der Proteinimport-Maschinerie
der äußeren Chloroplastenmembran, enthält ein Signalpeptid für den Transport ins Stroma.
Eine weitere, ebenfalls abspaltbare Signalsequenz sorgt jedoch dafür, daß OEP75 nicht
komplett ins Stroma überführt wird, sondern im Importkanal stecken bleibt und in der äußeren
Membran verbleibt(Tranel, 1996). Andere, kleinere Proteine wie OEP34 werden jedoch direkt
und ohne Signalpeptid in die äußere Chloroplastenmembran insertiert(Keegstra, 1999).
Hierbei spielt besonders eine hydrophobe Sequenz am C-Terminus des Proteins eine
entscheidende Rolle(Li, 1991). Für OEP14 konnte eine nicht abspaltbare Signalsequenz im N-
Terminus des Proteins identifiziert werden, die für den spezifischen Import verantwortlich
ist(Li, 1996). Die kleinen Kanalproteine der Hüllmembran der Chloroplasten integrieren
spontan durch hydrophobe Wechselwirkungen in die Lipid-Doppelschicht(Cline, 1996; Heins,
1998). Man kann davon ausgehen, daß die besondere Lipidzusammensetzung auch hier eine
entscheidende Rolle für die Selektivität des Imports in die Chloroplastenmembran
spielt(Joyard, 1991; Keegstra, 1989).
OEP16
OEP16 („Outer Envelope Protein 16 kDa“) ist ein in der äußeren Chloroplastenmembran
lokalisiertes Kanalprotein. Entdeckt wurde es, als man versuchte, durch systematische
Analyse von Proteinbestandteilen der äußeren Chloroplastenmembran neue Untereinheiten

15
des Proteinimportkomplexes der Chloroplasten und andere Transportproteine zu finden. Das
Protein wurde ansequenziert und seine Gensequenz mittels PCR mit degenerierten Primern
aus einer Genbank ermittelt(Pohlmeyer, 1997a). Schnell stellte sich heraus, daß es sich um
einen eigenständigen Kanal handelte, ohne direkten Bezug zur Import-Maschinerie. Neben
OEP16 wurden weitere Kanalproteine in der äußeren Chloroplastenmembran entdeckt,
namentlich OEP21(Bölter, 1999) und OEP24(Pohlmeyer, 1998). OEP16 konnte in
Chloroplasten aus Blättern und Stengeln, aber auch in dunkel adaptierten Chloroplasten und
in Wurzelplastiden nachgewiesen werden(Pohlmeyer, 1997a). OEP16 inseriert ebenso wie
OEP24 in die äußere Chloroplastenmembran, ohne daß ein Signalpeptid abgespalten wird.
Der Import ist unabhängig vom Proteinimportkomplex der Chloroplasten und von ATP, und
auch Deletionsmutanten, denen der N-Terminus (20 Aminosäuren) oder der C-Terminus (53
Aminosäuren) fehlt, werden korrekt in die äußere Membran integriert(Pohlmeyer, 1997b).
Sequenzen
1 MPRSSFSGSL SSPKLDVVID MGNPFLNLTV DGFLKIGAVA ATRSVAEDTF HIIRKGSISS
61 NDFEKSLKKM CKEGAYWGAI AGVYVGMEYG VERIRGTRDW KNAMFGGAVT GALVSAASNN
121 KKDKIAVDAI TGAAIATAAE FINYLT Abbildung 5: Aminosäuresequenz von OEP16 aus Erbse (Pisum sativum)
Inzwischen kennt man die Gensequenz von OEP16 nicht nur aus der Erbse(Pohlmeyer,
1997a), sondern auch aus Gerste (Baldi, 1999) und der Ackerschmalwand (Arabidopsis
thaliana), deren komplettes Genom inzwischen sequenziert wurde(Arabidopsis Genome
Initiative, 2000; Lin, 1999; Mayer, 1999; Salanoubat, 2000; Tabata, 2000; Theologis, 2000).
OEP16 aus Erbse wurde auch auf Ähnlichkeiten zu anderen, bekannten Proteinen untersucht.
Das Kanalprotein hat in Teilbereichen starke Homologien zu Tim17, Tim22 und Tim23,
Untereinheiten verschiedener Präproteinimportkanäle in der inneren Mitochondrienmembran,
und zu LivH, einem Aminosäuretransporter aus E.coli(Rassow, 1999). Ein auffälliger
Sequenzbereich (Aminosäuren 72-92) mit fünf Glycylresten in einem Abstand von je drei
Aminosäuren hat erstaunlich hohe Homologie zu einem Abschnitt aus den Sequenzen von
Na+-Glucose-Kotransportern aus Mensch, Huhn, Schaf und Ratte. Dasselbe gilt für Teile
eines Na+-Nukleosid-Kotransporters aus Kaninchen, für Permeasen für verzweigte
Aminosäuren aus Salmonella und Pseudomonas, und für einen Na+-Kotransporter für neutrale
Aminosäuren aus dem Schwein(Pohlmeyer, 1997b).

16
Aufbau
Der N-Terminus von OEP16 kann in Chloroplasten und Vesikeln der äußeren
Chloroplastenmembran, die mit der Außenseite außen ("right side out") präpariert
wurden(Waegemann, 1992), von der Protease Thermolysin abgespalten werden und ist somit
in vivo zu Cytosol hin exponiert(Pohlmeyer, 1997b). Der Sequenzbereich, der für die Bildung
der eigentlichen Pore zuständig ist, konnte auf die Aminosäuren 21-93 eingeengt
werden(Steinkamp, 2000).
Erste Untersuchungen an nativen Membranen ergaben, daß OEP16 ein Homodimer sein
könnte. Dafür sprechen Crosslink-Experimente(Pohlmeyer, 1997a) und die Tatsache, daß sich
OEP16 durch Zugabe von Kupfer(II)-Chlorid oxidativ dimerisieren läßt(Seedorf, 1995), und
zwar über das in der Sequenz nur ein einziges Mal vorkommende Cystein in Position
Cys71(Pohlmeyer, 1997a). Dieser Vorgang ist reversibel.
Funktion und Regulation
Elektrophysiologische Eigenschaften
Die Existenz von OEP16 als Kanalprotein konnte indirekt durch Messung seiner
elektrophysiologischen Eigenschaften in nativen äußeren Chloroplastenmembranen
nachgewiesen werden(Heiber, 1995), zusammen mit anderen spezifischen Kanalproteinen
dieses Membransystems. Genaue Kenntnis über Leitfähigkeit und Permeabilität für
verschiedene Substrate brachten jedoch Untersuchungen an rekombinantem Protein, das in
Liposomen rekonstituiert und zur Messung in planare Lipid-Doppelschichten eingebaut
wurde(Pohlmeyer, 1997a). Die Lipiddoppelschicht überspannt hierbei ein Loch in einer
Teflon-Membran, welche zwei Elektrodenkammern voneinander trennt. Durch Zugabe
verschiedener Puffer oder Substrate in die beiden Kammern können damit die spezifischen
physiologischen Eigenschaften von OEP16 bestimmt werden. Gemessen in 250 mM KCl auf
beiden Seiten der Membran hat OEP16 die höchste Offen-Wahrscheinlichkeit (etwa 80%
offene Kanäle) bei einer äußeren Spannung von 0 mV. Dies dürfte dem physiologischen
Zustand in vivo nahekommen, auch wenn geringe Membranpotentiale für ähnliche
Membransysteme postuliert wurden(Lemeshko, 2000). Bei einer angelegten Spannung von ±
150 mV waren praktisch alle Kanäle geschlossen.

17
Substrate
Es zeigte sich, daß der Kanal über eine gewisse Selektivität für Kationen verfügt, auch wenn
kleine Anionen wie Cl- praktisch ungehindert passieren können. Etwas größere, organische
Moleküle können aber nur noch dann durch die Pore diffundieren, wenn sie Aminogruppen
und/oder positive Ladungen tragen.
Selektivität des rekonstituierten OEP16-Kanals
Substrat Permeabilität Glycin Ja*
Valin Ja
Arginin Ja*
Lysin Ja
Glutamat Ja
Glutamin Ja*
Kadaverin Ja
Sucrose Nein*
Glucose Nein
Fructose Nein*
1-Phosphoglycerat Nein*
Dihydroxyaceton Nein*
Tetraethylamin Nein*
Tris-(hydroxymethyl)-aminomethan Ja*
Sorbitol Nein
Mannitol Nein
Abbildung 6:
Die in der Tabelle gezeigten Substrate wurden mittels Leitfähigkeitsmessungen auf ihre
Fähigkeit, durch die OEP16-Pore zu diffundieren, getestet. Mit * markierte Ergebnisse
wurden durch osmotisch induzierte Fusion von OEP16-Liposomen mit planaren
Lipiddoppelschichten bestätigt. (Pohlmeyer, 1997a)
Eine ganze Reihe Stoffwechselprodukte müssen die äußere Chloroplastenmembran passieren,
um die Funktion der Pflanzenzelle aufrecht erhalten zu können. OEP16 ist jedoch lediglich
für einen kleinen Ausschnitt dieser organischen Moleküle selektiv. OEP16 ist für alle
Aminosäuren durchlässig, ebenso wie für biologisch relevante Amine wie Cadaverin. Auch
das unphysiologische Tris-Kation konnte die Pore passieren. Praktisch undurchlässig ist

18
OEP16 hingegen für Kohlenhydratprodukte der Glycolyse und Zuckerphosphate, tertiäre
Amine und von Kohlenhydraten abgeleitete Substanzen wie Sorbitol und
Mannitol(Pohlmeyer, 1997a).
Oxidative Zerstörung der Kanalfunktion
Kupfer(II)-Chlorid ist ein effektiver Katalysator zur Bildung von Disulfidbrücken(Seedorf,
1995). Diese kovalente Verknüpfung zweier benachbarter Cysteinreste kann auch zwischen
Cysteinen verschiedener Peptidketten entstehen und spielt in vielen Proteinen und
Proteinkomplexen eine wichtige strukturelle Rolle. OEP16 wird durch Zugabe von CuCl2
dimerisiert(Pohlmeyer, 1997a). Dabei geht die Kanalleitfähigkeit durch die Membran
verloren, der Vorgang ist jedoch umkehrbar. Ob dieser Vorgang auch in vivo reversibel ist,
durch welche biologisch relevanten Oxidationsmittel er ausgelöst werden kann und ob er der
Regulation der Kanalaktivität dient, ist bisher ungeklärt.
Phosphorylierung
Natives OEP16 in der äußeren Chloroplastenmembran ist vermutlich phosphoryliert. Bei in
vitro Phosphorylierungsexperimenten wurde OEP16 durch eine in der äußeren
Chloroplastenmembran befindliche Kinase mit radioaktivem Phosphat verestert. An
Deletionsmutanten konnte gezeigt werden, daß die Phosphorylierungsstelle im Bereich des N-
Terminus, genauer innerhalb der ersten 20 Aminosäuren in der Sequenz, liegen
muß(Pohlmeyer, 1997b). In diesem Bereich befinden sich sechs Serinreste, die als
Phosphatakzeptoren in Frage kommen.
Genexpression
In Gerste ist bekannt, daß die Expression von OEP16 bei Kälte stark zunimmt(Baldi, 1999).
Dies konnte sowohl auf RNA- als auch auf Proteinebene gezeigt werden. Welche Funktion
OEP16 bei der Kälteregulation zukommt, ist bisher ungeklärt. In Spinat macht OEP16 einen
beträchtlichen Teil der Gesamtproteinmenge in der äußeren Chloroplastenmembran
aus(Koike, 1998).

19
Faltung von Membranproteinen
Allgemeine strukturelle Eigenschaften von Membranproteinen
Anders als lösliche, globuläre Proteine müssen Membranproteine bestimmten strukturellen
Vorgaben gehorchen, die aus dem Kontakt mit dem hydrophoben Inneren der biologischen
Membranen resultieren. Entsprechend zeigen die bekannten Strukturen von
Membranproteinen weit weniger verschiedene Strukturmotive, als ihre vielfältigen
Funktionen vermuten lassen(Cowan, 1994). Typische Strukturelemente von
Membranproteinen sind amphiphile Helices, die parallel zur Lipiddoppelschicht angeordnet
sind und mit ihrem hydrophoben Teil in sie hineinragen, und membranspannende Strukturen,
die entweder aus sogenannten „β-Barrels“, antiparallelen β-Faltblattstrukturen, oder Bündeln
von α-Helices bestehen(White, 1999). Große Domänen von Membranproteinen, die nicht in
direktem Kontakt mit den Lipiden stehen, verhalten sich hingegen wie lösliche Proteine(von
Heijne, 1996). Während die genannten amphiphilen Helices häufig bei Proteinen vorkommen,
die lediglich teilweise in die Membran hineinragen, bilden transmembrane Proteine entweder
eine β-Barrel-Struktur oder Helixbündel aus. Gemischte Strukturen sind nicht bekannt.
Typische Vertreter der β-Barrel-Struktur sind die bakteriellen Porine, Diffusionskanäle mit
einem großen Innendurchmesser(Buchanan, 1999; Hirsch, 1997; Klebba, 1998; Kreusch,
1994). Die am besten untersuchten Vertreter der Helixbündel-Proteine sind die bakteriellen
Reaktionszentren(Allen, 1987; Deisenhofer, 1985) und das Bakteriorhodopsin(Griegorieff,
1996). Auch bakterielle(McDermott, 1995) und eukaryontische(Kühlbrandt, 1994)
Lichtsammelkomplexe, die Photosysteme I und II(Jordan, 2001; Zouni, 2001), die ATP-
Synthase(Stock, 1999), die Calcium-ATPase(Toyoshima, 2000) und die Cytochrom C
Oxidase(Iwata, 1995; Tsuhikara, 1996) bestehen in ihrem transmembranen Bereich
ausschließlich aus Helixstrukturen.
Mechanismen der Insertion in die Membran in vivo
In eukaryontischen Zellen werden die meisten Membranproteine direkt bei der Translation
durch Ribosomen in die Membran eingebaut. Die Ribosomen können mRNA-Moleküle, die
für Membranproteine codieren, von denen für lösliche Proteine unterscheiden. Sie haften sich
dann an das sogenannte Translocon, einen großen Proteinkomplex in der Membran des
endoplasmatischen Reticulums an. Das entstehende Polypeptid wird vom Translocon in die

20
Membran eingefädelt. Hier geschehen auch die ersten posttranslationalen Modifikationen.
Später werden die neuen Membranproteine je nach ihren Signalsequenzen über
endoplasmatisches Reticulum und Golgi-Apparat an ihren Bestimmungsort in der Zelle
transportiert. Eine ganze Reihe von Membranproteinen können aber auch spontan in
Membranen eingebaut werden, ohne Mithilfe von anderen Proteinen. Die bestuntersuchten
Vertreter dieser Proteine sind bakterielle Toxine, zum Beispiel das Diphterie-Toxin(White,
1999). Generell werden ungefaltete Proteine in Zellen meist von Chaperonen in Lösung
gehalten. Diese Faltungshelfer verhindern die Aggregation von Peptiden und helfen ihnen,
ihren Bestimmungsort zu erreichen. Auch in Organellen wie den Mitochondrien und Plastiden
gibt es eigene Chaperone(Miernyk, 1999).
Für lösliche Proteine ist aufgrund einer Vielzahl von thermodynamischen, strukturellen und
funktionellen Daten bekannt, daß sich ihre Strukturen unter normalen physiologischen
Bedingungen in einem lokalen Minimum der freien Enthalpie der Proteinfaltung befinden. Es
gibt viele Belege dafür, daß dies auch für Membranproteine gilt(White, 1999). So konnte
gezeigt werden, daß sich auch in Stücke geschnittene Membranproteine in der
Lipiddoppelschicht zu funktionellen Komplexen zusammenlagern können. Dies gilt sowohl
für α-helikale Proteine(Ridge, 1995) als auch für β-Barrel-Proteine(Koebnick, 1996). Auch
Proteine, die wie das Diphterie-Toxin eine stabile lösliche Struktur haben, können in der
Lipidumgebung thermodynamisch stabile Strukturen annehmen(Zhan, 1995).
Thermodynamische Stabilität von Membranproteinen
Drei unterschiedliche Typen von Wechselwirkungen bestimmen Struktur und Stabilität von
Membranproteinen: die Wechselwirkungen von Proteinketten untereinander, die
Wechselwirkungen mit dem wäßrigen Medium und mit der Lipidmembran. Meßbar werden
diese Kräfte durch Untersuchungen des Faltungsprozesses oder des umgekehrten Vorganges,
der Denaturierung, mittels Temperaturänderungen oder durch Zugabe chaotroper Reagenzien,
welche die Proteinstruktur destabilisieren(White, 1999). Das Hauptproblem hierbei ist, daß
Membranproteine häufig nicht komplett denaturieren, weil die in die Lipidumgebung
eingebetteten Bereiche besonders stabil sind. Erfaßt werden dann nur die Dissoziation von
Untereinheiten und die Entfaltung von Domänen außerhalb der Membran. Zudem sind diese
Prozesse oft irreversibel(Haltia, 1995).

21
Die freie Enthalpie ∆G für das Eindringen von hydrophoben Peptiden in eine
Lipiddoppelschicht läßt sich in zwei Faktoren unterteilen. Hydrophobe Aminosäure-
Seitenketten partitionieren freiwillig in die Lipid-Doppelschicht. Die Peptidbindungen des
Protein-Rückrads hingegen können aufgrund ihrer polaren Struktur in der Wasserphase
energetisch günstigere Wasserstoffbrückenbindungen ausbilden. Nur bei der Ausbildung von
Sekundärstrukturelementen, in denen sämtliche beteiligten Peptidbindungen untereinander H-
Brücken bilden, also bei α-Helices und β-Faltblattstrukturen, ist ein Verbleiben in der
Membran bei direktem Kontakt mit den Lipiden energetisch möglich. In diesem Fall
kompensieren die neu gebildeten hydrophoben Wechselwirkungen der Seitenketten mit der
Membran den Verlust an freier Enthalpie, der durch den Übergang des hydrophilen
Peptidrückrads in das lipophile Medium entsteht(White, 1999). Alternativ dazu können
hydrophile Bereiche in der Membran durch benachbarte Proteinstrukturen bedeckt werden, so
daß sie praktisch nicht mit den Lipiden in Berührung kommen.
Der Faltungsprozeß
Es gibt eine Reihe von Hypothesen und Modellrechnungen über den genauen Ablauf des
Faltungsprozesses, deren komplette Darstellung den Rahmen dieser Arbeit sprengen würde.
Im Folgenden wird die gängigste Hypothese zur Membranproteinfaltung kurz erläutert.
Danach läßt sich der Prozeß der Membranproteinfaltung in vier separate Teilschritte zerlegen.
Zunächst partitioniert ein hydrophobes Peptid aus der Wasserphase in die Grenzfläche
zwischen Wasserphase und Lipidmembran. Im nächsten Schritt bilden sich dort
Sekundärstrukturelemente aus, die das Passieren der Grenzfläche erlauben, im allgemeinen
sind das α-Helices(White, 1999). Diese Sekundärstrukturelemente dringen im dritten Schritt
in die Membran ein und lagern sich zuletzt zu einem funktionell intakten Membranprotein
zusammen(Bechinger, 2000).
Die Anlagerung an die Grenzfläche der Membran ist in erster Linie auf den hydrophoben
Effekt zurückführbar, der lipophile Aminosäureseitenketten aus der Wasserphase verdrängt.
Bei der Assoziation von Membranproteinen mit der Lipiddoppelschicht spielen aber auch
elektrostatische Wechselwirkungen positiv geladener Seitenketten mit den anionischen
Phospholipiden eine entscheidende Rolle. In nativen Membranproteinen liegt oft ein Großteil
der positiv geladenen Seitenketten in dem wäßrigen Medium zugänglichen Bereichen auf

22
einer Seite der Membran. Vermutlich spielen sie eine entscheidende Rolle bei der Frage, in
welcher Orientierung ein Membranprotein in die Membran eingebaut wird.
Das Ausbilden von Sekundärstrukturelementen ist notwendig, um Peptiden das Eindringen in
die Membran zu ermöglichen. Sind alle Peptidbindungen zum Beispiel zu einer α-Helices
verbrückt, können diese im dritten Schritt der Faltung als helikale Haarnadelstrukturen in die
Membran eindringen. Für den letzten Schritt, die Anordnung der Sekundärstrukturelemente
zum aktiven Membranprotein, ist eine Vielzahl unterschiedlicher Faktoren verantwortlich.
Transmembrane Strukturelemente werden oft durch aromatische Aminosäurereste in der
Grenzfläche der Membran verankert. Untereinander können sie durch
Wasserstoffbrückenbindungen, Disulfidbrücken, hydrophobe Wechselwirkungen und ionische
Wechselwirkungen zusammengelagert sein. Auch die Bindung von Kofaktoren und das
Zusammenwirken von Domänen außerhalb der Membran kann für die korrekte Anlagerung
von transmembranen Elementen aneinander eine Rolle spielen(Booth, 1999).

23
Zielsetzung
Proteine sind wichtigster Bestandteil aller Lebensvorgänge. Neben vielfältigen strukturellen
Aufgaben in der Zelle katalysieren sie vor allem die Stoffwechselreaktionen und sorgen für
den gezielten Transport lebenswichtiger Substanzen an ihren Bestimmungsort. Die
funktionelle Vielseitigkeit der Proteine liegt in der Vielzahl der möglichen Sequenzen
begründet, die aus den 20 Grundbausteinen, den Aminosäuren, gebildet werden können, und
in den komplexen Strukturen, zu denen sich solch ein Proteinstrang zusammenlagern kann.
Um detaillierte Informationen über Struktur und Funktion eines Proteins zu gewinnen, bedarf
es großer Mengen des hochreinen Proteins. In vielen Fällen kann dieser Bedarf nicht durch
Aufreinigung des Zielproteins aus dem ursprünglichen Organismus gedeckt werden. Dies gilt
besonders für Membranproteine, die schwierig zu isolieren sind und oft nur in geringsten
Mengen in der Zelle vorkommen. So gibt es zum Beispiel Rezeptoren in medizinisch
relevanten Signaltransduktionsprozessen, die nur in wenigen oder gar einer Kopie pro Zelle
vorliegen. Diese geringe Verfügbarkeit ist zusammen mit den Schwierigkeiten bei der
Kristallisation der Grund dafür, daß bisher weniger als 30 verschiedene
Membranproteinstrukturen aufgeklärt wurden, gegenüber mehr als 5000 Strukturen löslicher
Proteine.
Die Überexpression – das Produzieren eines Proteins in einem Wirtsorganismus mit
gentechnischen Methoden – ist ein häufig genutztes Mittel zur Herstellung großer
Proteinmengen. Als Wirtsorganismen eignen sich besonders Bakterien, nicht nur weil sie
einfach und kostengünstig zu handhaben sind, sondern auch weil die Proteinausbeuten weit
über denen eukaryontischer Expressionssysteme liegen. Bei der Überexpression von
eukaryontischen Proteinen in Bakterien (meist E.coli) können jedoch verschiedene Probleme
auftreten. Insbesondere Membranproteine werden von Bakterien zumeist nicht in ihrer
korrekten Konformation hergestellt. Dies trifft auch auf das in dieser Arbeit untersuchte
Protein OEP16 zu.
OEP16 ist ein Protein aus der Erbse, das den spezifischen Transport von Aminosäuren und
Aminen durch die äußere Chloroplastenmembran ermöglicht. Es wird von E.coli in Form von
„Inclusion bodies“, großen Proteinaggregaten, produziert. Da OEP16 in der Erbsenpflanze nur
in relativ kleinen Mengen vorkommt und bisher keine Reinigungsprozedur für das native
Protein besteht, sollten in der vorliegenden Arbeit Wege gefunden werden, das fehlgefaltete

24
Protein aus E.coli für strukturelle und funktionelle Untersuchungen zugänglich zu machen. Im
Erfolgsfall würde dies OEP16 darüber hinaus zu einem Modell für die Strukturbildung von
Membranproteinen in vitro machen – ein Vorgang, über dessen Mechanismen bisher nur sehr
wenig bekannt ist. Der erste Teil dieser Arbeit beschäftigt sich daher mit der Faltung des
Membranproteins in vitro.
Bisher existierende Strukturmodelle von OEP16, die sich hauptsächlich auf Crosslink-
Experimente, CD-Spektren und Primärsequenzanalysen stützten, sprachen für ein Homodimer
mit einer gemischten Struktur aus transmembranen α-Helices und transmembranen β-
Faltblattstrukturen. Bisher kennt man Membranproteine dieser Größe nur als rein α-helikal
(z.B. Bakteriorhodopsin) oder als reine β-Faltblattstrukturen (z.B. Porine). Bekannte
Transportproteine der äußeren Membranen von Chloroplasten, Mitochondrien und auch gram-
negativen Bakterien sind ausschließlich aus β-Faltblättern aufgebaut. OEP16 ist nach den
bisherigen Untersuchungen demnach ein Vertreter einer neuen Familie von
Membranproteinen. Dies macht weitergehende Untersuchungen zur Struktur des Proteins
besonders interessant.
Im zweiten Teil der Arbeit sollten physikochemische und spektroskopische Daten erfaßt
werden, die Schlüsse auf die Struktur von OEP16 zulassen. Die gewonnenen Daten und
theoretischen Betrachtungen zur Aminosäuresequenz von OEP16 sollten genutzt werden, um
ein neues, detailliertes Modell für die Struktur dieses ungewöhnlichen Transportproteins zu
erstellen. Darüber hinaus sollten mit Methoden der Proteinkristallographie und
Elektronenmikroskopie Vorarbeiten geleistet werden, auf denen aufbauend in Zukunft ein
Strukturmodell mit nahezu atomarer Auflösung geschaffen werden kann.

25
Material und Methoden
Überexpression von OEP16 in E.coli
Die Überexpression von OEP16 erfolgte in der Arbeitsgruppe von Prof. Soll im Botanischen
Institut der Universität Kiel. Verwendet wurde ein pET-Vektor der Firma Novagen. Der
E.coli-Expressionsstamm BL21(DE3) wuchs in 2YT-Medium (1,6% Caseinhydrolysat, 1%
Hefeextrakt und 0,5% NaCl) mit dem Selektionsantibiotikum Ampicillin (135 mg/l) in 500
ml-Schüttelkulturen. OEP16 lag nach Induktion der Überexpression mit IPTG aggregiert in
Form von so genannten Inclusionbodies vor. Diese Proteinpartikel wurden nach Lyse der
E.coli-Zellen bei 1200 psi in einer „French Press“ in einem Lysepuffer (20 mM Tris/HCl pH
8, 1 mM EDTA, 25% Saccharose) und anschließender Ultraschallbehandlung zur Zerstörung
von DNA mehrfach durch Zentrifugieren (10 min bei 10000 g in einer Sorvall Zentrifuge mit
GSA-Rotor) und Resuspendieren in detergenzhaltigem Puffer gewaschen. Der erste
Waschschritt erfolgte in einem Detergenzpuffer (20 mM Tris/HCl pH 7,5, 200 mM NaCl, 1%
Desoxycholat, 1% Nonidet P40, 1 mM EDTA, 10 mM β-Mercaptoethanol). Anschließend
wurde zweimal mit einem ähnlichen Puffer (mit 0,5% MEGA9 als Detergenz und ohne
Kochsalz) resuspendiert und erneut abzentrifugiert
und anschließend in Puffer (Tris/HCl pH 7,5, 1 mM EDTA, 10 mM DTT) bei –80°C gelagert
bzw. auf Trockeneis versandt.
Reinigung von OEP16
Die OEP16-Inclusionbodies (bis zu 500 µl Pelletvolumen) wurden in 10 ml harnstoffhaltigem
Solubilisierungspuffer (Puffer C) gelöst. Dies geschah entweder durch Schütteln im Kühlraum
über Nacht oder durch Beschallen mit der Mikrospitze eines Branson Sonifiers (30% Puls,
maximale Energie) für etwa 5 Minuten auf Eis. Die Lösung wurde anschließend 10 min bei
5000 g zentrifugiert und anschließend durch einen 0,2 µm Spritzenfilter filtriert.
Abzentrifugieren und Filtrieren der Probe wurde notwendig, um feine Verunreinigungen zu
entfernen, die regelmäßig die Pumpenköpfe der verwendeten Chromatographieanlage
verstopften.
Die chromatographische Reinigung erfolgte bei Raumtemperatur in einem Abzug. Verwendet
wurde eine Knauer-HPLC-Anlage mit zwei Pumpen und einer Programm-Einheit, die das
Mischen des Salzgradienten und die Flußraten steuert. Die Detektion des Proteins erfolgte mit

26
einem Filter-UV-Detektor bei 280 nm Wellenlänge. Chromatogramme (UV-Signal) und
Gradient (Leitfähigkeit) wurden mit einem Zweikanalschreiber aufgezeichnet. Die
Präpaprationspuffer (Puffer A und B) für die Säulenchromatographie entsprechen dem Puffer
zum Lösen der Inclusionbodies, enthielten aber eine geringere Harnstoffkonzentration (6M).
Der Hochsalzpuffer B enthielt zusätzlich 1 M NaCl. Der erste Schritt zur
chromatographischen Reinigung des Proteins erfolgte mit einer Source30Q
Anionenaustauschsäule (Pharmacia) mit einem Säulenvolumen von 30 ml und einem
Querschnitt von 25 mm. Die Flußrate betrug 3 ml/min. Das Protein befindet sich aufgrund
seines hohen isoelektrischen Punktes von 9 im Druchbruch der Säule, während ein Großteil
der Verunreinigungen, vor allem Proteine mit negativer Nettoladung und Nukleinsäuren,
zurückgehalten werden. Details zum Gradienten und das Elutionsprofil werden im
Ergebnisteil dieser Arbeit gezeigt und diskutiert. Der aufgefangene Druchbruch hat ein
Volumen von 15-25 ml und eine geringe Ionenstärke (50-70 µS).
In einem zweiten Schritt wird das so vorgereinigte OEP16 direkt auf eine MonoS
Kationenaustauschsäule mit 1 ml Säulenvolumen (Pharmacia) aufgetragen. Bei einer
Fließgeschwindigkeit von 1 ml/min bindet das Protein an die Kationenaustauschmatrix und
eluiert nach starten des Salzgradienten bei einer Salzkonzentration von 120 mM NaCl.
Typischerweise erhält man so aus 300 µl Inclusionbodies 3-4 ml einer OEP16-Lösung mit
einer Konzentration von 1 mg/ml.
Die für die Präparation verwendeten Puffer sind:
Puffer A (Präpapration) Puffer B (Präparation) Puffer C (Solubilisierung)
6 M Harnstoff 6 M Harnstoff 8 M Harnstoff
20 mM Hepes/KOH pH 7,5 20 mM Hepes/KOH pH 7,5 20 mM Hepes/KOH pH 7,5
10 mM ß-Mercaptoethanol 10 mM ß-Mercaptoethanol 10 mM ß-Mercaptoethanol
1 mM EDTA 1 mM EDTA 1 mM EDTA
1 M NaCl
SDS-Gelelektrophorese
10 µl des gereinigten Proteins in Harnstoff wurden unverdünnt mit 10 µl Probenpuffer (20
mM Tris/HCl pH 7,5, 2 mM EDTA, 0,2% SDS und 0,2% DTT) versetzt und 5 min auf 95°C

27
erhitzt, um Cysteinreste zu reduzieren und das Protein vollständig zu denaturieren. jeweis 1 µl
der entstandenen Proteinlösung wurden dann mit einem Probenkamm (Pharmacia) auf ein
SDS-HD-Gel (ebenfalls Pharmacia) aufgetragen. Die elektrophoretische Trennung erfolgte
mit einem PhastSystem (Pharmacia) mit einem Standardprogramm (500 V, 10 mA, 158 Vh
bei 15°C). Die Gele wurden einer Silberfärbung nach Vorschrift des Herstellers unterzogen.
Faltung von OEP16
Liposomen
In vivo insertiert OEP16 vermutlich spontan in die äußere Chloroplastenmembran(Cline,
1996; Heins, 1998). Dieser Vorgang läßt sich durch Rekonstitution in Liposomen imitieren.
Für elektrophysiologische Messungen an OEP16 werden Liposomen aus Phosphatidylcholin
verwendet (siehe unten), die mit dem Detergenz MEGA-9 aufgelöst und mit OEP16 in
Harnstoff versetzt wurden. Nach anschließender Dialyse zur Entfernung von Harnstoff und
Detergenz erhält man Liposomen mit eingebautem OEP16, das dieselbe Kanalleitfähigkeit
wie das native Protein zeigt(Pohlmeyer, 1997a). Diese Liposomen lassen sich auch für
spektroskopische Untersuchungen von OEP16 in seiner Lipidumgebung verwenden.
Als Lipid wurde entweder eine Mischung aus hochreinem Phosphatidylcholin mit
Phosphatidsäure (Firma Lipoid) im Verhältnis 20:1 verwendet, oder selbst nachgereinigtes
Phosphatidylcholin (Typ S-IV, Sigma). Zum Reinigen des S-IV-Lipids wurde die
Lipidsuspension (100 mg/ml in 10 mM Tricin/KOH pH 8,2) mit sechs Volumenteilen
Methanol/Chloroform versetzt (Methanol/Chloroform 2:1) und nach Zusatz von zwei
Volumenteilen 1M KCl, 0.2M H3PO4 im Scheidetrichter ausgeschüttelt. Danach wird die
Lösung stehengelassen und gewartet, bis sich die Phasen getrennt haben. Die (untere)
Lipidphase wird abgelassen und der pH der Wasserphase mit KOH auf ca. 8 eingestellt. Dann
wird die Wasserphase erneut mit Lösungsmittel ausgeschüttelt. Anschließend werden die
organischen Phasen vereinigt und das Lipid im Rotationsverdampfer getrocknet. Dieses Lipid
enthält neben Phosphatidylcholin Spuren vieler anderer pflanzlicher Membranlipide.

28
Die Liposomen werden wie folgt hergestellt:
500 µl OEP16-Lösung (1 mg/ml) im Präparationspuffer (Puffer A mit ca. 150 mM NaCl)
werden mit 500 µl einer Lipidlösung (50 mg/ml gelöst in Puffer D mit 160 mM MEGA-9)
versetzt und durch Schütteln und Beschallen im Ultraschallbad bei Raumtemperatur gemischt,
bis das Lipid vollständig gelöst ist. Die klare Lösung wird in einen Dialyseschlauch mit 3500
Da maximaler Porengröße (Spectrum) gefüllt. Die Dialyse zur Bildung der Liposomen erfolgt
gegen 5 l Puffer D zunächst bei Raumtemperatur, da das Detergenz MEGA-9 bei diesen
hohen Konzentrationen in der Kälte ausfällt. Nach etwa zwei Stunden wird die Dialyse über
Nacht im Kühlraum fortgesetzt.
Detergenz-basierte Methoden für die Rekonstitution
Detergenzien können Proteine aus Lipidmembranen herauslösen, indem sie die Bestandteile
dieser Membranen imitieren und ersetzen. Ähnlich wie Membranlipide besitzen Detergenzien
langkettige, hydrophobe Bereiche, die an einem hydrophilen Kopf sitzen. Sie bilden in
wäßrigen Medien Mizellen mit einem hydrophoben Kernbereich. Da OEP16 spontan in
Lipidmembranen eingebaut und korrekt gefaltet werden kann, sollte dieser Vorgang auch in
Detergenzmizellen zu imitieren sein, wenn man das geeignete Detergenz wählt.
Gelfiltration
Getestet wurden verschiedene Sephadex-Materialien (G10, G25 und G50) sowie analytische
Gelfiltrationssäulen mit Superdex-Matrix (Superdex75 und Superdex200).
Sephadex-Säulen mit einem Durchmesser von 0,9 cm und einer Bettlänge von 8 cm wurden
selbst nach Vorschriften des Herstellers (Pharmacia) gegossen. Als Laufpuffer diente der für
die Dialysemethode verwendete Puffer (siehe dort) unter Zusatz einer Detergenzkonzentration
von 0,03% C12E8. Es wurde dieselbe Knauer HPLC-Anlage wie für die Proteinreinigung
verwendet. Die Flußraten lagen bei 0,5 ml/min für selbst gegossene Säulen und bei 1 ml/min
für die fertigen Superdex-Säulen.
Von der Matrix gebundenes Protein wurde mit 0,5 M NaOH eluiert und die Säule
anschließend mit 30% Isopropanol gewaschen. Wiederfindungsraten wurden aus dem UV-
Signal des HPLC-Detektors abgeschätzt.

29
Dialyse
Zur Rekonstitution des Proteins mittels Dialyse wurden der Proteinlösung in Harnstoffpuffer
die notwendige Menge hochkonzentrierter Detergenz-Stammlösung zugesetzt, um die für
weitergehende Untersuchungen notwendige Endkonzentration an Detergenz zu erhalten ohne
das Protein dabei unnötig zu verdünnen. Die Lösung wurde anschließend in Dialyseschläuche
oder in Dialysebuttons (Hampton Research) gefüllt und diese in ein großes Reservoir
Dialysepuffer versenkt. Das Endvolumen der Dialyseansätze betrug (je nach Bedarf des
anschließenden Experiments) 0,2 bi 2 ml. Die Dialyse erfolgte im allgemeinen über Nacht.
Verwendet wurden ausschließlich Dialysemembranen der Forma Spectrum mit einer
Porenweite von 2500 Da. Der Dialysepuffer enthielt 20 mM Hepes/KOH pH 7,5 und
1 mM EDTA.
Verdünnung
OEP16-Proben, die im weiteren Verlauf dieses Kapitels erwähnt werden, wurden (wenn nicht
anders angegeben) durch Verdünnen hergestellt. Hierzu werden 100 µl der aus der
chromatographischen Reinigung erhaltenen OEP16-Lösung in Puffer A ohne weitere
Behandlung mit 900 µl detergenzhaltigem Puffer in einem Reaktionsgefäß (1,5 ml,
Plastibrand) durch vortexen gemischt. Der Verdünnungspuffer enthielt neben der
gewünschten Detergenzkonzentration 20 mM Hepes/KOH (pH 7,6) und 1 mM EDTA. Die
Arbeitskonzentrationen der Detergenzien lagen üblicherweise oberhalb ihrer CMC, im
Bereich 1 x CMC bis 10 x CMC. Die Faltungsreaktion erfolgt bei Raumtemperatur.
Spektroskopische Methoden
Proteinbestimmung nach Bradford
Da die meisten Proteinbestimmungmethoden auf das Reduktionsmittel β-Mecaptoethanol
emfindlich reagieren, das in allen OEP16-Proben enthalten ist, konnte die Konzentration des
Proteins nur mittels der Bradford-Methode bestimmt werden. Da diese Methode durch die
Gegenwart von Detergenzien erheblich gestört wird, beziehen sich alle
Proteinkonzentrationen auf die Messung der Ausgangskonzentration von OEP16 in der

30
Harnstofflösung (Präparationspuffer A mit 150 mM NaCl). Ist von Endkonzentrationen die
Rede, sind diese durch Berücksichtigung des Verdünnungsfaktors aus der
Ausgangskonzentration berechnet.
Fluoreszenz-Spektroskopie
Die in dieser Arbeit gezeigten Spektren wurden mit einem FluoroMax-2 Spektrometer der
Firma ISA in 1 ml Halbmikro-Quartzküvetten mit einer Schichtdicke von 0,5 cm in der
Anregungsrichtung und 1 cm in der Meßrichtung aufgenommen. Die Bandbreiten für die
Anregungs- und Meßwellenlängen betrugen üblicherweise 1 bis 2 nm. Zur Aufnahme der
Spektren wurden in den meisten Fällen 100 µl einer OEP16-Lösung in Puffer A mit 900 µl
einer geeigneten Detergenzlösung (Detergenzkonzentration wie beim jeweiligen Experiment
angegeben, in 20 mM Hepes/KOH pH 7,6, 1 mM EDTA) in einem 1,5 ml Reaktionsgefäß
gemischt und anschließend in die Küvette gefüllt. Das Mischen in der Küvette ist aufgrund
der unterschiedlichen Viskositäten ond Dichten der Lösungen schwierig. Die
Endkonzentration von OEP16 in diesen Ansätzen betrug je nach Experiment 0,03 bis 0,1
mg/ml. Die Messungen erfolgten bei Raumtemperatur.
Zum Messen von Differenzspektren (OEP16-Liposomen/leere Liposomen) wurden die wie im
Abschnitt „Liposomen“ beschrieben hergestellten Liposomen mit den oben genannten
Parametern ohne weitere Verdünnung gemessen. Die Spektren wurden ohne weitere
Bearbeitung voneinander abgezogen, um das störende Fluoreszenzsignal der
Liposomenlösung zu eliminieren.
CD-Spektroskopie
Alle CD-Spektren wurden mit einem Jasco J500 CD-Spektropolarimeter bei Raumtemperatur
aufgenommen. Die verwendeten Küvetten hatten eine Schichtdicke von 0,2 mm, die
Probenkammer wurde ständig mit Stickstoff gespült, um Absorptionseffekte durch Sauerstoff
sowie oxidative Beschädigung der empfindlichen Spiegelsysteme des Geräts zu vermeiden.
Die Spektren wurden mit einer Auflösung von 0,1 nm und einer Aufnahmezeit von 10s/nm
aufgenommen. Es wurden jeweils fünf Spektren gemittelt. Die Proteinkonzentrationen lagen
je nach Messung zwischen 0,1 und 0,5 mg/ml. Die Proben wurden wie im Abschnitt
„Detergenz-basierte Methoden für die Rekonstitution“ erläutert hergestellt.

31
Für hochauflösende Spektren im gesamten Wellenlängenbereich wurden Proben mit 0,5
mg/ml OEP16, dialysiert gegen 10 mM Natrimcacodylatpuffer (pH 7,0, mit 50 mM
Natriumsulfat), in Tonnenküvetten mit einer Schichtdicke von 0,05 mm gemessen. Es wurden
wiederum fünf Spektren gemittelt. Alle CD-Messungen erfolgten bei Raumtemperatur.
IR-Spektroskopie
FT-IR-Spektren (Fuorier-Transform-Infrarotspektren) wurden mit einem IFS28/B
Spektrometer der Firma Bruker Optik GmbH, Ettlingen aufgenommen. Die Probenkammer
wurde mit getrockneter Luft gespült. Die Signale wurden mit einem DTGS-Detektor erfaßt
und mittels Fourier-Transformation („Happ-Genzel apodization“) in Infrarotspektren
umgewandelt. Die Geräteauflösung betrug 4 Wellenzahlen. Es wurden jeweils 32 Messungen
gemittelt, um das Rauschen zu reduzieren. Die Spektren sind in ihrer zweiten Ableitung
dargestellt.
Da Protein-FTIR-Spektren wegen der störenden anregbaren Schwingungen des Wassers
(insbesondere der symmetrischen Deformationsschwingung δ bei 1595 cm-1) im
interessierenden Wellenlängenbereich (Wellenzahlen zwischen 2000 und 1400 cm-1) nur in
D2O meßbar sind, mußten die Proben vorher in D2O überführt werden.
Den Liposomen wurden hierzu über nacht in einer Gefriertrocknungsanlage das Wasser
entzogen; sie wurden erst kurz vor der Messung in reinem D2O wieder aufgenommen.
Gleichzeitig mit dem Wechsel des Lösungsmittels kann so auch eine höhere
Probenkonzentration erreicht werden. Liposomensuspensionen mit einem ursprünglichen
Volumen von 1 ml wurden in 100 µl aufgenommen. Die OEP16-Endkonzentration in diesen
Proben betrug für die Messung etwa 5 mg/ml. Die Suspension wird in eine IR-Küvette mit
CaCl2-Fenstern gefüllt, die mit Dichtungsringen zu einer Schichtdicke von 50 µm
zusammengepreßt werden. Gemessen wurde bei Raumtemperatur; Die Probenkammer wurde
vor der Messung ausreichend (mindestens 20 Minuten) mit getrockneter Luft gespült, um den
störenden Einfluß von Wasserdampf auf die Spektren zu minimieren.
Zeitaufgelöste Spektroskopie
Zeitaufgelöste Intensitätsunterschiede der Tryptophanfluoreszenz von OEP16 während des
Faltungsprozesses wurden im Zeitfenster von 1 bis 100 ms mit einem SX-18MV stopped flow

32
Apparat der Firma Applied Photophysics (Leatherhead, Großbrittanien) gemessen. UV-
Anregung erfolgte durch eine Xenondrucklampe bei einer Wellenlänge von 280 ± 20 nm. Die
Detektion wurde mit einem Interferenzfilter auf 334 ± 5 nm Wellenlänge eingeschränkt. Das
Mischungsverhältnis von Proteinprobe zu Verdünnungspuffer betrug 1:10, was einer
Enkonzentration von 0,07 mg/ml OEP16 in 600 mM Harnstoff, 20 mM Hepes/KOH (pH 7,6),
1 mM EDTA und 1 mM ß-Mercaptoethanol mit unterschiedlicher Detergenzkonzentration (je
nach Experiment) entsprach. Die erhaltenen Signale wurden mittels nichtlinearer Regression
in einem Bereich von 3-100 ms mit der Methode der kleinten Quadrate als doppelt
exponentielle Kurven angenähert. Einzelne Meßkurven wurden mit einem π∗-180 stopped
flow Apparat desselben Herstellers aufgenommen, der ein besseres Signal-Rausch-Verhältnis
hat, was den relativen Fehler für die mathematische Bestimmung der Zeitkonstanten von
<10% auf <1% reduziert. Bei niedrigen Detergenzkonzentrationen konnten hier 3-fach
exponentielle Kurven angenähert werden. Die Standardmessungen erfolgten bei
Raumteperatur (25°C). Die temperaturabhängigen Messungen wurden im Temperaturbereich
von 10°C bis 35°C durchgeführt.
Messungen mit CD-Detektion erfolgten ebenfalls mit einem π∗-180 stopped flow Apparat
unter denselben Bedingungen (25°C). Die Änderung der Elliptizität wurde bei einer
Wellenlänge von 228 nm im Bereich von 2-200 ms aufgenommen.
Physikalisch-chemische Methoden
DSC
Denaturierungskurven wurden in einem Microcal MC-2 Scanning-Kalorimeter aufgenommen.
Die Proteinkonzentration für die Messungen betrug 0,55 mg/ml in einem Meßvolumen von
1,225 ml. Der Puffer enthielt 20 mM Hepes/KOH (pH 7,6), 1 mM EDTA und variable
Mengen Detergenz. Die Proben wurden wie im Abschnitt „Detergenz-basierte Methoden für
die Rekonstitution“ erläutert hergestellt. Die Referenzprobe bestand aus demselben Puffer
ohne Protein. Die Heizrate betrug 30°C/h, gemessen wurde im Bereich zwischen 20° und
95°C. Meßgröße war die Wärmekapazität als eine Funktion der Temperatur, die unter
Berücksichtigung von Volumen und Konzentration der Probe auf die molare Wärmekapazität
umgerechnet wurde. Denaturierungstemperatur (Kurvenmaximum) und –enthalpie (Fläche
unter der Kurve) wurden aus der gemessenen Kurve nach Abziehen des Untergrundsignals
bestimmt.

33
Elektronenmikroskopie
Für die Transmissions-Elektronenmikroskopie an OEP16 wurden 5 µl der durch Verdünnung
hergestellten Proteinlösung mit einer Endkonzentration von 0,01 mg/ml auf einem
hydrophilisierten kohlenstoffbeschichteten Gitter (hergestellt durch 60 s Plasmabehandlung
bei 8 W Leistung mit einem Baltec MED 020) aufgetragen, überschüssige Flüssigkeit
abgesaugt und die Probe luftgetrocknet. Ein Tropfen Phospho-Wolframsäure (2% w/v, pH
7,0) wurde für 45 s zugegeben und anschließend wieder abgesaugt. Diese Vorgänge erfolgten
bei Raumtemperatur. Die erneut getrocknete Probe wurde dann in einem Philips CM12
Transmissions-Elektronenmikroskop bei einer 58300-fachen primären Vergrößerung
betrachtet. Hierzu wurde ein „Nieder-Dosis-Protokoll“ des Herstellers verwendet, um
unnötige Strahlungsschäden an der Probe zu vermeiden. Die Bildaufzeichnung erfolgte
fotografisch.
Kristallisation
Probenvorbereitung
Erste Kristallisationsexperimente wurden mit OEP16, rekonstituiert in C12E8, durchgeführt.
Die Proben wurden wie im Abschnitt „Detergenz-basierte Methoden für die Rekonstitution“
erläutert hergestellt. Die Detergenzkonzentration betrug 0,03%. Um eine möglichst hohe
Proteinkonzentration zu erreichen, wurde das gefaltete Protein in Puffer mit gesättigter
Ammoniumsulfatlösung im Verhältnis 1:1 versetzt und dabei präzipitiert. Das Protein wurde
in Wasser wiederaufgenommen, so daß das Endvolumen einem zehntel des
Ausgangsvolumens entsprach, was einer Proteinkonzentration von etwa 10 mg/ml entspricht.
Da auch das verwendete Detergenz mit Ammoniumsulfat aus der Lösung ausfällt, erhöht sich
auch die Detergenzkonzentration entsprechend. Die erhaltene Lösung wurde mit maximaler
Geschwindigkeit (13.000 upm in einer Heraeus Mikrizentrifuge) abzentrifugiert, um reichlich
vorhandene Aggregate abzutrennen und mit den Pufferlösungen der Kristallisationsansätze
versetzt.

34
Kristallisationsansätze
Grundsätzlich wurde wegen der einfachen Handhabbarkeit die Methode der hängenden
Tropfen genutzt. Hierbei wird ein kleiner Tropfen der Proteinlösung mit Puffer hängend an
einem Deckglas angebracht, der mittels Dampfdiffusion mit einem Pufferreservoir in Kontakt
steht. Die Salzkonzentration im Reservoir ist höher als im Tropfen, so daß Salz- und
Proteinkonzentration während des Diffusionsprozesses im Tropfen zunehmen. Hierbei wird
idealerweise die Phasengrenze für die Löslichkeit des Proteins überschritten – eine
notwendige Bedingung für die Kristallisation. Das Verhältnis von Proteinlösung zu
Präzipitationslösung betrug 4:1. Auf diese Weise wurde eine weitere Aufkonzentrierung der
Proteinlösung durch den Diffusionsvorgang erreicht. Das Startvolumen der hängenden
Tropfen zu Beginn des Diffusionsvorganges betrug 5 µl, die Tropfen wurden dann auf
unbehandelte Deckgläser aufgebracht. Deckgläser und Pufferreservoire wurden durch
Silikonpaste gasdicht miteinander verbunden und derart abgedichtet, um ein Austrocknen der
Ansätze zu vermeiden.
Die Löslichkeit von OEP16 wurde durch Variation der Parameter Fällungsmittel, pH,
Kristallisationsansätze und Temperatur verringert. Fällungsmittel, pH-Werte und Zusätze
wurden zunächst durch die fertigen Lösungen zur Proteinkristallisation der Firma Hampton
Research bestimmt. Die Zusammensetzung dieser Lösungen beruht auf empirischen
Erfahrungen bei der Kristallisation von Membranproteinen. Die Ansätze wurden bei 18°C in
einem Temperierschrank und bei 4°C im Kühlraum inkubiert und täglich mit einem
Mikroskop überprüft, ob sich Präzipitat oder Kristalle in den Tropfen gebildet hatten. In
einigen Ansätzen trat auch eine Phasentrennung in zwei nicht mischbare, wäßrige Phasen auf.
Die besten Kristalle wurden mit Natriumzitrat als Fällungsmittel erhalten. Die auftretende
Phasentrennung entsteht wohl in erster Linie durch die geringe Löslichkeit von
Polyethylenglykolen (und damit auch von PEG-haltigen Detergenzien) in Zitrat-
Lösungen(Marcos, 1999). Die Proteinkristalle bilden sich in der entstandenen
detergenzreichen Phase erst nach der Phasentrennung aus. Unter dem Mikroskop läßt sich die
Veränderung von runden, öligen Tropfen (detergenzreiche Phase) zu kantigeren,
doppelbrechenden Kristallen im Lauf von 1-3 Tagen gut beobachten.

35
Messung der Röntgenbeugung
Die einzelnen OEP16-Kristalle wurden vorsichtig mit einer kleinen Kunststofföse (0,5 mm
Durchmesser) zusammen mit einem Tropfen Kristallsiationslösung aus den Ansätzen
„gefischt“. Die Öse („Loop“) wurde in eine dünne Quarzkapillare mit 2 mm Durchmesser
gesteckt und mit dieser so mit Wachs verbunden, daß der Kristall die Quarzoberfläche nicht
berühren konnte (bei Berührung lösten sich die Kristalle auf) und das Röhrchen gasdicht
verschlossen war. Die Kapillare wurde dann auf einen Gonometerkopf aufgebracht und in den
Röntgenstrahl gebracht.
Die Röntgenbeugung der OEP16-Kristalle wurde mit einer rotierenden Kupferanode bei einer
Wellenlänge von 15,4 nm gemessen. Der Generator wurde mit 40 kV und 65 mA betrieben,
die Blenden für die Strahlbegranzung waren auf 0,3 mm eingestellt. Reflexe wurden mit einer
Imageplate (MAR Research) mit 18 cm Durchmesser erfaßt. Die Messung und die
anschließende Auswertung der Beugungsbilder mit dem Programm DENSO in Verbindung
mit dem Grafikprgramm XDISPLAYF erfolgte bei der AG Sänger, FU Berlin.
Theoretische Strukturvorhersage
Sämtliche im Folgenden beschriebenen Methoden sind im Internet abrufbar. Besonders
hilfreich für das Finden solcher Analysewerkzeuge sind die Server des Europäischen
Molekularbiologie-Netzwerks (http://www.embnet.org), des Proteinanalyse-Systems Expasy
(http://www.expasy.ch) oder des Institut Pasteur (http://bioweb.pasteur.fr). Im Folgenden
werden die für diese Arbeit verwendeten Methoden kurz beschrieben und genaue
Internetadressen angegeben.
"Multiple Sequence Alignment"
Um Sequenzen eines Proteins aus verschiedenen verwandten Organismen überhaupt
miteinander vergleichen zu können, muß man sie in geeigneter Weise nebeneinander
darstellen. Was sich zunächst wie ein triviales Problem anhört, wird um so schwieriger, je
geringer der Verwandtschaftsgrad der untersuchten Proteine ist. Das so genannte "Multiple

36
Sequence Alignment" berücksichtigt auch Sequenzbereiche, die nicht in allen Proteinen einer
untersuchten Gruppe vorkommen und markiert diese entsprechend.
Hier wurde der Algorithmus Dialign (Version 2.1) verwendet(Morgenstern, 1999), der unter
der Adresse http://bibiserv.techfak.uni-bielefeld.de/dialign/ genutzt werden kann. Dialign gibt
nicht nur die entsprechend ihrer Homologie angeordneten Sequenzen aus, sondern berechnet
für jede Position in der Sequenz einen Homologie-Index mit einem Wert zwischen 0 und 5,
wobei 0 komplette Abweichung und 5 eine komplette Übereinstimmung im untersuchten
Sequenzbereich bedeutet.
Vorhersage transmembraner Bereiche
Die Vorhersage von transmembranen Bereichen wird im allgemeinen über sogenannte
Hydrophobizitäts-Plots(Kyte, 1982) vorgenommen. Diese Methode errechnet die
durchschnittliche Hydrophobizität eines kurzen Sequenzbereichs. Anhand solcher
Hydrophobizitätswerte aus Proteinen, deren Struktur bekannt ist, kann man einen Grenzwert
der Hydrophobizität bestimmen, über dem ein Sequenzbereich mit hoher Wahrscheinlichkeit
innerhalb einer Membran liegen muß. Leider versagt diese Methode bei transmembranen β-
Faltblattstrukturen ebenso wie bei amphiphilen α-Helices, da diese relativ viele hydrophile
Aminosäuren enthalten können, wenn sie zum Beispiel einen Kanal durch die Membran
bilden.
Hilfreich ist hier die Verwendung von "Multiple Sequence Alignments". Das Programm
„Pepwindowall“ (http://bioweb.pasteur.fr/seqanal/interfaces/pepwindowall.html) erlaubt das
Erstellen von gemittelten Hydrophobizitätsplots aus mehreren verwandten Sequenzen. Im Fall
von OEP16 treten hierdurch die transmembranen Segmente deutlicher zu Tage.
Auf ganz andere Weise funktioniert derAlgorithmus TMPred zur Vorhersage transmembraner
Helices(Hofmann, 1993), verfügbar unter der Adresse
http://www.ch.embnet.org/software/TMPRED_form.html. Hier werden Sequenzbereiche mit
einer Sequenzdatenbank bekannter transmembraner Helices direkt verglichen und so ein
Topologiemodell erstellt.

37
Hydrophobe Cluster-Analyse
Die hydrophobe Cluster-Analyse HCA(Callebaut, 1997) erlaubt ebenfalls Vorhersagen über
transmembrane Bereiche und erkennt auch amphiphile Bereiche in vielen Fällen. Die
zweidimensionale Darstellung einer Sequenz zeigt jede Aminosäure umgeben von den sechs
Aminosäuren in ihrer näheren Umgebung (Position -3 bis -1 und 1 bis drei in der Sequenz um
die untersuchte Aminosäure). So lassen sich Sequenzbereiche mit hoher Hydrophobizität
einfach bildlich darstellen. Amphiphile Bereiche und transmembrane β-Faltblattstrukturen
ergeben auffällige Muster. HCA-Plots können unter der Adresse
http://smi.snv.jussieu.fr/hca/hca-form.html erstellt werden.

38
Ergebnisse
Von der Proteinreinigung zum gefalteten Protein (eine Einleitung)
OEP16 ist ein kleines Membranprotein aus der äußeren Chloroplastenmembran der Erbse. Bis
heute gibt es kein Reinigungsprotokoll, das die Isolierung großer Mengen des nativen Proteins
aus Erbsenpflanzen erlaubt. Zum einen ist der Anteil von OEP16 am Gesamtprotein einer
Pflanzenzelle sehr gering, zum anderen ist selbst bei einer Isolierung äußerer
Chloroplastenmembranen und der damit verbundenen Anreicherung von OEP16 das
Abtrennen anderer Membrankomponenten, vor allem der anderen OEPs mit vergleichbaren
isoelektrischen Punkten, schwierig.
Bei Überexpression des Gens in E.coli hingegen kann man große Mengen des Proteins
gewinnen. Dieses Protein ist jedoch fehlgefaltet und läßt sich nur in Form sogenannter
„Inclusion bodies“ isolieren, großen stabilen Proteinaggregaten mit eingeschlossenen
Verunreinigungen. Diese bestehen meist aus RNA und anderen, bakteriellen Proteinen(Brill,
2000). Als Voraussetzung für alle Experimente zur Struktur- und Funktionsaufklärung
müssen diese Proteinaggregate gelöst und das darin enthaltene OEP16 gereinigt werden.
Anschließend gilt es, Methoden zu finden, die eine Faltung in die native Struktur in vitro
erlauben. Abbildung 7 zeigt die möglichen Strategien, um zu korrekt gefaltetem, für
Funktionsuntersuchungen und die Kristallisation geeignetem Protein zu gelangen.
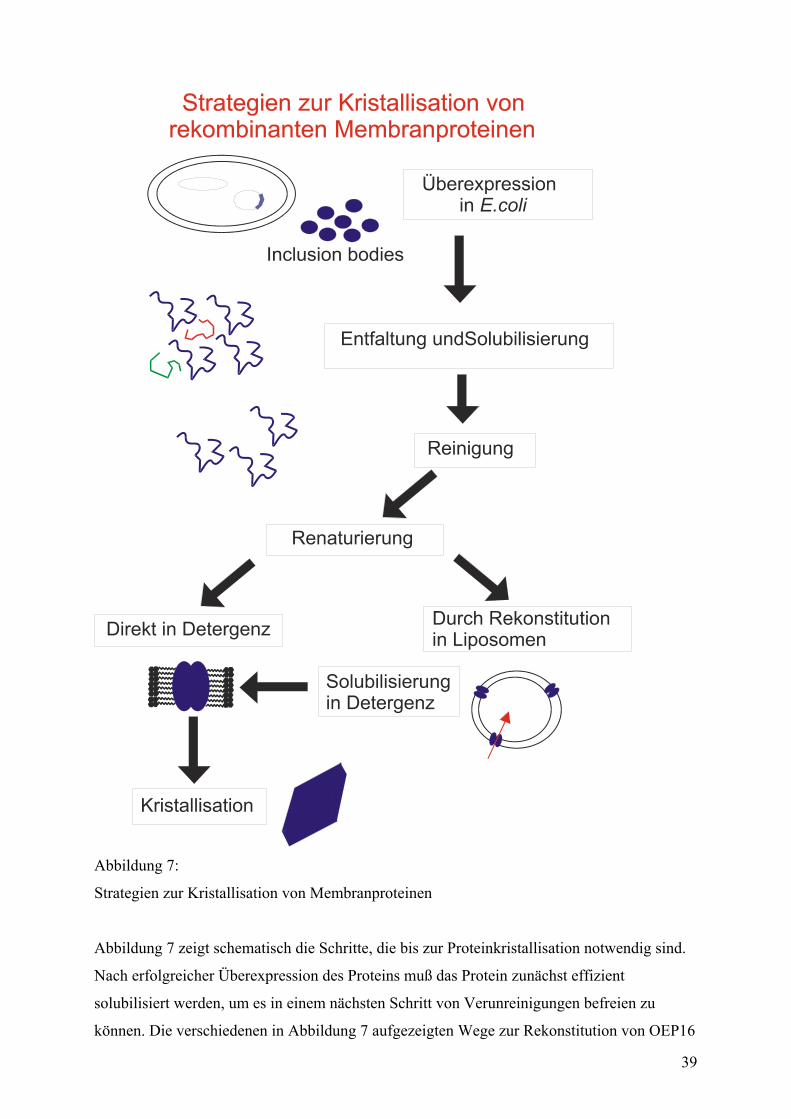
39
Abbildung 7:
Strategien zur Kristallisation von Membranproteinen
Abbildung 7 zeigt schematisch die Schritte, die bis zur Proteinkristallisation notwendig sind.
Nach erfolgreicher Überexpression des Proteins muß das Protein zunächst effizient
solubilisiert werden, um es in einem nächsten Schritt von Verunreinigungen befreien zu
können. Die verschiedenen in Abbildung 7 aufgezeigten Wege zur Rekonstitution von OEP16

40
unterscheiden sich in ihrer Handhabbarkeit und ihrer Effizienz. Der Einbau in eine künstliche
Lipid-Doppelschicht ermöglicht es, bei Wahl einer geeigneten Lipidzusammensetzung die
natürliche Membranumgebung des Proteins möglichst genau nachzuahmen. Dies sollte zu
hohen Rekonstitutionsraten führen, hat aber den Nachteil, daß das Protein in einer Membran
für viele biophysikalische Methoden und vor allem für die Proteinkristallisation nicht
verwendbar ist und erst wieder solubilisiert, d.h. mit Detergenzien aus der Membran
herausgelöst werden muß.
Ein System, das es ermöglicht, das Protein direkt in Detergenz-Mizellen einzubringen und
dabei in seine native Form zu falten, könnte ohne Umwege große Mengen Protein liefern.
OEP16 ist aber ein Porenprotein, dessen Aktivität sich nur in Membranen messen läßt. Die
Schwierigkeit eines reinen Detergenzsystems ist weniger seine Handhabung, als die Frage,
wie man die korrekte Strukturbildung eines Kanalproteins in Lösung bzw. in Mizellen zeigen
kann.
Im Folgenden soll gezeigt werden, daß mit spektroskopischen Methoden in Detergenz
solubilisiertes Protein mit in Membranen eingebautem Protein verglichen werden kann. Der
Beweis der Faltung in Detergenzmizellen ist hierbei indirekter Natur. Die Aktivität als
wichtigster Beleg für eine korrekte Struktur kann nur in Liposomen gezeigt werden. Hier läßt
sich ein Membranpotential aufbauen und die Diffusion von Substraten durch den Kanal wird
meßbar. Die in Verlauf dieser Arbeit erläuterten Methoden der Fluoreszenz-, CD- und
Infrarotspektroskopie ermöglichen dann vergleichende Untersuchungen der unterschiedlich
präparierten Proben. Nur wenn alle verwendeten vergleichenden Methoden eine
Übereinstimmung der Proben in Liposomen und Mizellen zeigen, kann man von einer
korrekten Faltung von OEP16 in Detergenzmizellen ausgehen und Strukturdaten gewinnen.
Zunächst wird der erste Arbeitsschritt, die Reinigung des Proteins im ungefalteten Zustand,
genauer betrachtet.
Proteinreinigung
Die im Kapitel „Material und Methoden“ beschriebene Proteinreinigung basiert auf der in der
Literatur(Pohlmeyer, 1997a; Pohlmeyer, 1997b) beschriebenen Reinigungsmethode für
denaturiertes OEP16 und wurde weiter optimiert.
Zur Solubilisierung wurde ein Beschallungsschritt eingeführt, um das langwierige Lösen des
Proteins im Solubilisierungspuffer zu beschleunigen (siehe Material und Methoden). Durch
Abzentrifugieren und Filtrieren der Probe konnten feine Verunreinigungen entfernt werden,
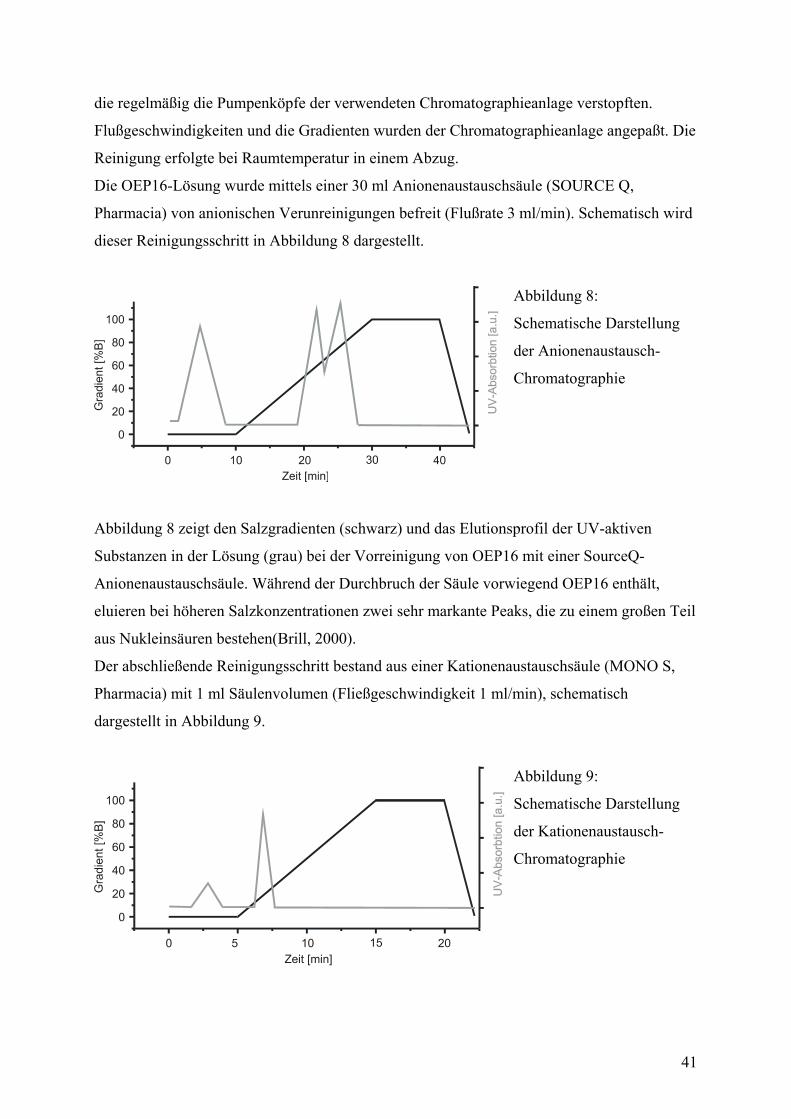
41
die regelmäßig die Pumpenköpfe der verwendeten Chromatographieanlage verstopften.
Flußgeschwindigkeiten und die Gradienten wurden der Chromatographieanlage angepaßt. Die
Reinigung erfolgte bei Raumtemperatur in einem Abzug.
Die OEP16-Lösung wurde mittels einer 30 ml Anionenaustauschsäule (SOURCE Q,
Pharmacia) von anionischen Verunreinigungen befreit (Flußrate 3 ml/min). Schematisch wird
dieser Reinigungsschritt in Abbildung 8 dargestellt.
Abbildung 8:
Schematische Darstellung
der Anionenaustausch-
Chromatographie
Abbildung 8 zeigt den Salzgradienten (schwarz) und das Elutionsprofil der UV-aktiven
Substanzen in der Lösung (grau) bei der Vorreinigung von OEP16 mit einer SourceQ-
Anionenaustauschsäule. Während der Durchbruch der Säule vorwiegend OEP16 enthält,
eluieren bei höheren Salzkonzentrationen zwei sehr markante Peaks, die zu einem großen Teil
aus Nukleinsäuren bestehen(Brill, 2000).
Der abschließende Reinigungsschritt bestand aus einer Kationenaustauschsäule (MONO S,
Pharmacia) mit 1 ml Säulenvolumen (Fließgeschwindigkeit 1 ml/min), schematisch
dargestellt in Abbildung 9.
Abbildung 9:
Schematische Darstellung
der Kationenaustausch-
Chromatographie

42
Abbildung 9 zeigt schematisch den zweiten Reinigungsschritt von OEP16 (schwarz
dargestellt ist der Salzgradient, grau das Elutionsprofil). OEP16 eluiert von der
Kationenaustauschsäule mit 120 mM NaCl. Im Durchbruch der Säule befindet sich eine
Substanz, die zwar UV-Licht absorbiert, jedoch laut SDS-Gelelektrophorese keine
nennenswerten Mengen Nukleinsäuren oder Protein enthält. Möglicherweise handelt es sich
hier um kleine Bruchstücke von Proteinen und RNA/DNA.
Das Eluat enthält gereinigtes OEP16 in einer Konzentration von 0,5-1 mg/ml. Es dient als
Ausgangslösung für die im Folgenden beschriebenen Experimente zur Faltung des Proteins in
seine native Struktur.
Faltungsmethoden
Zunächst soll die Wechselwirkung von OEP16 mit verschiedenen Detergenzien betrachtet
werden. Das Protein wird in präparativem Maßstab in einem Puffersystem mit 6 M Harnstoff
gereinigt. Der Harnstoff, ein sogenanntes chaotropes Reagenz, verhindert die
Sekundärstrukturbildung. Dies geschieht sowohl durch das Aufbrechen von
Wasserstoffbrückenbindungen des Proteinrückrates, die für α-Helix- und β-Faltblatt-
Strukturen hauptsächlich verantwortlich sind, als auch durch das Stören von
Wechselwirkungen zwischen benachbarten Aminosäureseitenketten. Um diese Wirkung
aufzuheben und gleichzeitig einen Einbau des Proteins in Detergenzmizellen zu erreichen,
wurden verschiedene Methoden getestet.
Dialyse
Ähnlich zu den Verfahren zur Liposomenherstellung kann man die Protein-Harnstofflösung
mit Detergenz versetzen und den Harnstoff anschließend durch Dialyse entfernen. Dabei gilt
es zu beachten, daß Detergenzmizellen im Gleichgewicht mit Detergenzmonomeren in der
Lösung stehen. Detergenzien mit kurzem hydrophobem Alkylrest haben im allgemeinen eine
hohe CMC (kritische mizellare Konzentration), solche mit einem langen hydrophoben Rest
eine niedrige. Oberhalb dieser Konzentration bilden sich Mizellen, eine konstante
Konzentration an monomerem Detergenz in der Lösung bleibt aber erhalten. Benutzt man
Dialysemembranen mit einem Ausschlußvolumen kleiner als die Mizellengröße, können nur

43
Detergenzmonomere bei der Dialyse entfernt werden. In der Probe lösen sich Mizellen auf,
um eine konstante Monomerkonzentration entsprechend der CMC aufrechtzuerhalten. Die
Dialysegeschwindigkeit hängt generell vom Konzentrationsgefälle über die Dialysemembran
ab. Im Falle von Detergenzien ist das Gefälle und damit die Geschwindigkeit der
Detergenzentfernung um so höher, je höher die CMC und damit die Konzentration an
monomerem Detergenz in der Probe ist.
Membranproteine können durch Detergenzmizellen solubilisiert und in Lösung gehalten
werden. Eine Membranproteinlösung muß also unbedingt eine Detergenzkonzentration
oberhalb der CMC enthalten. Unterhalb dieser Konzentration neigen die hydrophoben
Proteine dazu, zu denaturieren und zu aggregieren. In der Dialyse muß also dem
Dialysepuffer Detergenz zugesetzt werden, um die Konzentration konstant zu halten.
Da hochreine Detergenzien sehr teuer sind, kann das Zusetzen von Detergenz zum
Dialysepuffer ein Nachteil der Dialysemethode sein, insbesondere bei Detergenzien mit hoher
CMC. Andere Detergenzien (solche mit langem hydrophobem Rest) sind aufgrund der
geringen Monomerkonzentration und der kinetischen Stabilität der Mizellen durch Dialyse
nur sehr langsam entfernbar und somit für diese Methode besser geeignet. Generell ist die
Dialyse ein langsamer Prozeß, bei dem die Harnstoffkonzentration in einem Zeitraum von
Stunden auf ein Minimum herabgesetzt wird.
Alle Experimente zur Rekonstitution durch Dialyse wurden mit Membranen durchgeführt, die
3500 Da Porenweite besitzen. Die enge Porenweite ist notwendig, um sicherzustellen, daß
denaturierte Proteinstränge nicht durch die Membran dringen können. Für das korrekt
gefaltete Protein sollten Membranen mit 10 bis 12 kDa Ausschlußgrenze ausreichend sein.
Der Proteinlösung in Harnstoffpuffer wurde die notwendige Menge hochkonzentrierter
Detergenz-Stammlösung zugesetzt, um die für das Folgeexperiment benötigte
Endkonzentration an Detergenz zu erhalten ohne das Protein unnötig zu verdünnen. Die
Lösung wurde anschließend in Dialyseschläuche oder in Dialysebuttons (Hampton Research)
gefüllt und diese gegen 5 l Dialysepuffer dialysiert.
Die Dialysemethode führte generell zu denselben Ergebnissen wie das Verdünnen des
Proteins (siehe dort). Besonders für die Probenvorbereitung zur CD-Spektroskopie erwies sie
sich als hilfreich, weil störende Pufferkomponenten wie z.B. Harnstoff komplett entfernt
werden können. Wegen des verhältnismäßig hohen Zeitaufwandes wurde jedoch bei einem
Großteil der Experimente die Verdünnungsmethode bevorzugt.

44
Gelfiltration
Gelfiltrationsmaterialien sind grobkörnige Matrizes, die von Poren definierten Durchmessers
durchzogen werden. Partikeln, die in diese Poren eindringen können, steht ein größeres
Aufenthaltsvolumen zur Verfügung, kleine Partikel wandern also langsamer durch eine
Gelfiltrationssäule als große. Um Protein aus einer Harnstofflösung mittels Gelfiltration in
eine Detergenzlösung zu überführen, muß die Säule mit dem detergenzhaltigen Puffer
äquilibriert werden. Das Protein wandert schneller als der Harnstoff durch das Säulenbett und
wird dadurch in das Laufpuffersystem überführt. Ein Nachteil dieser Methode ist, daß die oft
recht hydrophoben Matrizes der Gelfiltrationsmaterialien stark mit Detergenzien
wechselwirken. Detergenz im Laufpuffer bindet an die Säulenmatrix, der eluierende Puffer
hat eine unbekannte Detergenzkonzentration. Auch wechselwirken hydrophobe Proteine
häufig mit dem Gelfiltrationsmaterial.
Die Methode ist schneller als die Dialysemethode, aber in vielen Fällen konnte die Adsorption
von Protein an das Säulenmaterial beobachtet werden, mit Substanzverlusten bis zu 100% je
nach Gelfiltrationsmaterial und verwendetem Detergenz. Für OEP16 konnte kein
Gelfiltrationsmaterial gefunden werden, das akzeptable Wiederfindungsraten des Proteins
erbrachte. Getestet wurden verschiedene Sephadex-Materialien (G10, G25 und G50) sowie
analytische Gelfiltrationssäulen mit Superdex-Matrix (Superdex75 und Superdex200). Von
den Superdexsäulen konnte das Protein erst mit 0,5M NaOH wieder entfernt werden. Bei den
Sephadexmaterialien fand sich nach dem Lauf nur etwa 10-20% des eingesetzten Proteins
wieder, der Rest eluierte erst mit 0,5 M NaOH oder 30% Isopropanol.
Verdünnung
Die einfachste und schnellste Methode zur Verringerung hoher Harnstoffkonzentrationen ist
das Verdünnen mit detergenzhaltigem Puffer. Dem Protein wird schlagartig ein Großteil des
denaturierenden Harnstoffs entzogen (siehe auch Abbildung 11 weiter unten), gleichzeitig
kommt es mit Detergenzmizellen in Kontakt. Nachteil ist, daß auch das Protein entsprechend
verdünnt wird und oft für spektroskopische Messungen anschließend wieder ankonzentriert
werden muß. Große Vorteile bieten die Geschwindigkeit und einfache Handhabbarkeit der
Methode.

45
Während den Versuchen zeigte sich, daß – abgesehen von Substanzverlusten bei der
Gelfiltration – die angesprochenen Methoden alle bei der Wahl eines geeigneten Detergenz zu
erfolgreicher Rekonstitution von OEP16 führen können. Leider ist es nur mittels IR-
Spektroskopie möglich, genaue Aussagen über die Rekonstitutionseffizienz, also das
Verhältnis von ungefaltetem zu gefaltetem Protein in einer Probe zu machen. Da die IR-
Spektroskopie (siehe dort) aber hohe Probenkonzentrationen benötigt, ist eine Festlegung auf
die effizienteste Methode der Rekonstitution nicht möglich. Im Folgenden wurde daher meist
die einfachste und schnellste Methode, die Verdünnung, verwendet, um mittels verschiedener
spektroskopischer Methoden die korrekte Faltung des Membranproteins OEP16
nachzuweisen.
Fluoreszenzspektroskopie
Kann eine Substanz Lichtquanten bestimmter Wellenlänge absorbieren, so ist der
resultierende angeregte Zustand nur von kurzer Lebensdauer. Die Rückführung der
elektronischen Struktur in den energetisch günstigeren Grundzustand kann unter Aussendung
von Licht erfolgen. Der Grundzustand ist im allgemeinen durch ein Elektronenpaar
gekennzeichnet, das nach dem Pauli-Prinzip antiparallele Spins trägt. Kommt es bei der
Anregung durch Lichtabsorption zur Spinumkehr bei einem der Elektronen, bildet sich durch
diesen quantenmechanisch eigentlich verbotenen Übergang ein Triplett-Zustand. Der
Übergang dieses Triplett-Zustandes in den Grundzustand unter Emission von Licht wird als
Phosphoreszenz bezeichnet. Verläuft die Anregung ohne Spinumkehr, entsteht ein Singulett-
Zustand, dessen Rückführung in den Grundzustand Fluoreszenz genannt wird. Wegen der
erforderlichen erneuten, „verbotenen“ Spinumkehr ist die Lebensdauer der Phosphoreszenz
deutlich größer als die der Fluoreszenz.
Fluoreszenz wird zumeist bei aromatischen und heterozyklischen Molekülen mit zwei oder
mehr kondensierten Ringen beobachtet. Da die angeregten Singulett-Zustände solcher
Moleküle eine höhere Polarität als ihr Grundzustand besitzen, zeigen sie eine stärkere Dipol-
Wechselwirkung mit polaren Lösungsmitteln. Die Folge davon ist, daß die Fluoreszenz-
Emission gegenüber dem Absorptionsspektrum des Grundzustandes zu größeren
Wellenlängen verschoben wird als in unpolaren Lösungsmitteln. Die Dipol-
Wechselwirkungen stabilisieren den angeregten Zustand, die Energie des emittierten Lichts ist

46
geringer. Somit kann man aus dem Fluoreszenz-Emissionsspektrum eines Moleküls auf die
Polarität seiner Umgebung schließen.
In Proteinen ist Tryptophan die Aminosäure, die aufgrund hoher Fluoreszenzausbeute, einem
Absorptionsmaximum von 280 nm (ε280 = 5600 cm-1M-1) und ihrer relativen Seltenheit
innerhalb einer Proteinsequenz als intrinsische Fluoreszenzprobe am häufigsten untersucht
wird(Chen, 1998). Potentiell fluoreszieren auch die aromatischen Aminosäuren Phenylalanin
(ε280 < 100 cm-1M-1) und Tyrosin (ε280 < 1000 cm-1M-1) , wegen ihrer geringeren
Extinktionskoeffizienten im Wellenlängenbereich um 280 nm können sie aber für die im
folgenden gezeigten Fluoreszenzspektren vernachlässigt werden. Da Tryptophan im Protein in
eine lange Kette von Aminosäuren eingebaut ist, bestimmt sich die Hydrophobizität seiner
Umgebung selten durch das Lösungsmittel, sondern im allgemeinen aus der
Aminosäurezusammensetzung seiner Umgebung im gefalteten Protein. Bei
Membranproteinen ist zusätzlich zu beachten, daß Tryptophanreste, die aus der
Proteinstruktur herausragen und so Kontakt mit dem Lösungsmittel haben, potentiell auch im
transmembranen Bereich des Proteins angesiedelt sein können und somit mit der
Lipiddoppelschicht einer Membran oder – nach Solubilisierung – mit der hydrophoben
Detergenzmizelle wechselwirken können. Auch die Fluoreszenzintensität der einzelnen
Tryptophanreste in einem Protein kann sich deutlich unterscheiden. So ist bekannt, daß die
Aminosäuren Lysin und Tyrosin sowie Cystein und, schwächer, Glutamin, Asparagin,
Glutaminsäure, Asparaginsäure und (protoniertes) Histidin die Fluoreszenz von Tryptophan
quenchen können(Chen, 1998). Auch die Halbwertsbreite des Fluoreszenzpeaks von
Tryptophanresten kann Informationen über deren Umgebung liefern. Grundsätzlich ist der
Emissionspeak um so schmaler, je kleiner die Wellenlänge des Emissionsmaximums ist.
Diese Abhängigkeit ist linear und kann genutzt werden, um vergleichsweise breite
Fluoreszenzspektren aus mehreren Tryptophan-Resten eines Proteins in ihre Bestandteile zu
zerlegen und somit auf die Umgebung des einzelnen Tryptophans
zurückzuschließen(Burstein, 1973). Abbildung 10 zeigt diesen Effekt in einem sehr einfachen
Experiment.
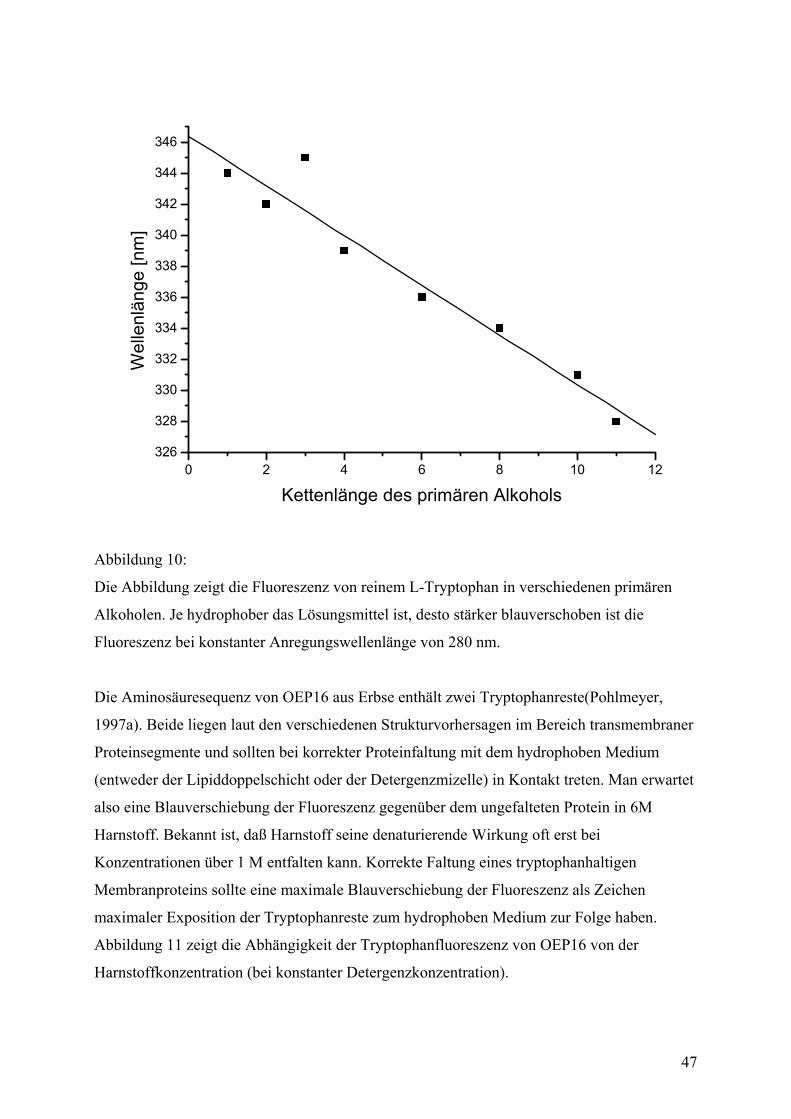
47
0 2 4 6 8 10 12326
328
330
332
334
336
338
340
342
344
346W
elle
nlän
ge [n
m]
Kettenlänge des primären Alkohols
Abbildung 10:
Die Abbildung zeigt die Fluoreszenz von reinem L-Tryptophan in verschiedenen primären
Alkoholen. Je hydrophober das Lösungsmittel ist, desto stärker blauverschoben ist die
Fluoreszenz bei konstanter Anregungswellenlänge von 280 nm.
Die Aminosäuresequenz von OEP16 aus Erbse enthält zwei Tryptophanreste(Pohlmeyer,
1997a). Beide liegen laut den verschiedenen Strukturvorhersagen im Bereich transmembraner
Proteinsegmente und sollten bei korrekter Proteinfaltung mit dem hydrophoben Medium
(entweder der Lipiddoppelschicht oder der Detergenzmizelle) in Kontakt treten. Man erwartet
also eine Blauverschiebung der Fluoreszenz gegenüber dem ungefalteten Protein in 6M
Harnstoff. Bekannt ist, daß Harnstoff seine denaturierende Wirkung oft erst bei
Konzentrationen über 1 M entfalten kann. Korrekte Faltung eines tryptophanhaltigen
Membranproteins sollte eine maximale Blauverschiebung der Fluoreszenz als Zeichen
maximaler Exposition der Tryptophanreste zum hydrophoben Medium zur Folge haben.
Abbildung 11 zeigt die Abhängigkeit der Tryptophanfluoreszenz von OEP16 von der
Harnstoffkonzentration (bei konstanter Detergenzkonzentration).
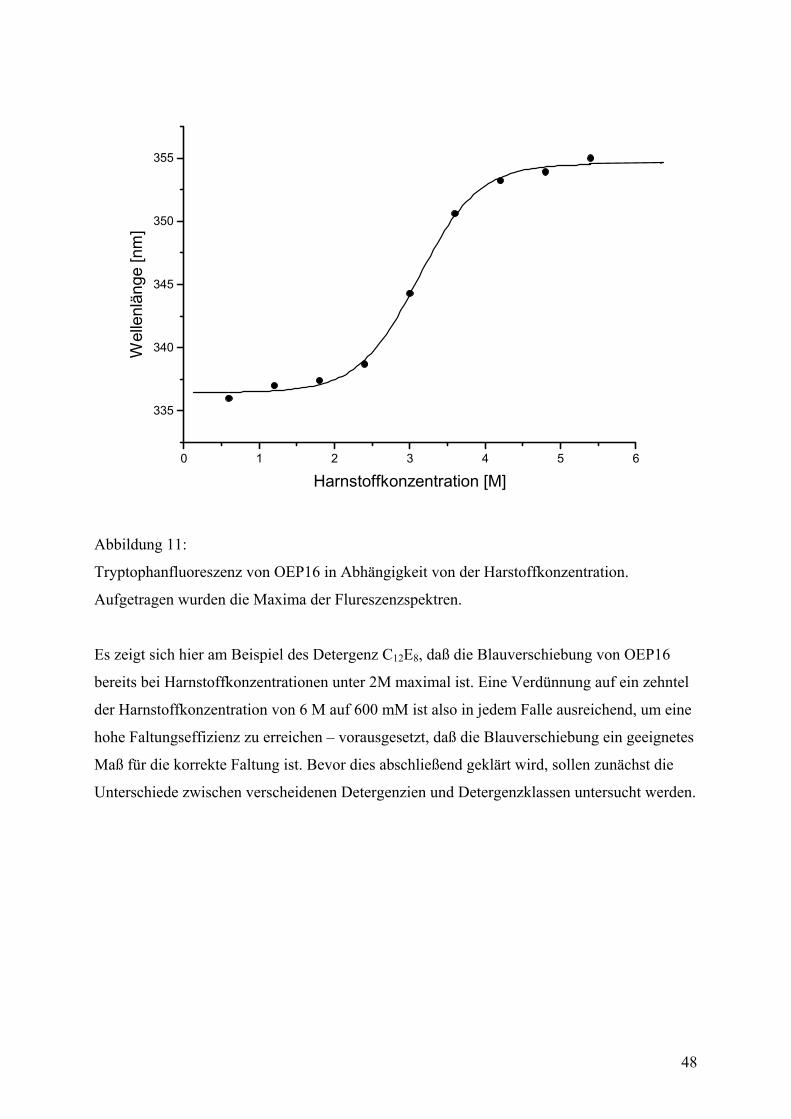
48
0 1 2 3 4 5 6
335
340
345
350
355W
elle
nlän
ge [n
m]
Harnstoffkonzentration [M]
Abbildung 11:
Tryptophanfluoreszenz von OEP16 in Abhängigkeit von der Harstoffkonzentration.
Aufgetragen wurden die Maxima der Flureszenzspektren.
Es zeigt sich hier am Beispiel des Detergenz C12E8, daß die Blauverschiebung von OEP16
bereits bei Harnstoffkonzentrationen unter 2M maximal ist. Eine Verdünnung auf ein zehntel
der Harnstoffkonzentration von 6 M auf 600 mM ist also in jedem Falle ausreichend, um eine
hohe Faltungseffizienz zu erreichen – vorausgesetzt, daß die Blauverschiebung ein geeignetes
Maß für die korrekte Faltung ist. Bevor dies abschließend geklärt wird, sollen zunächst die
Unterschiede zwischen verscheidenen Detergenzien und Detergenzklassen untersucht werden.
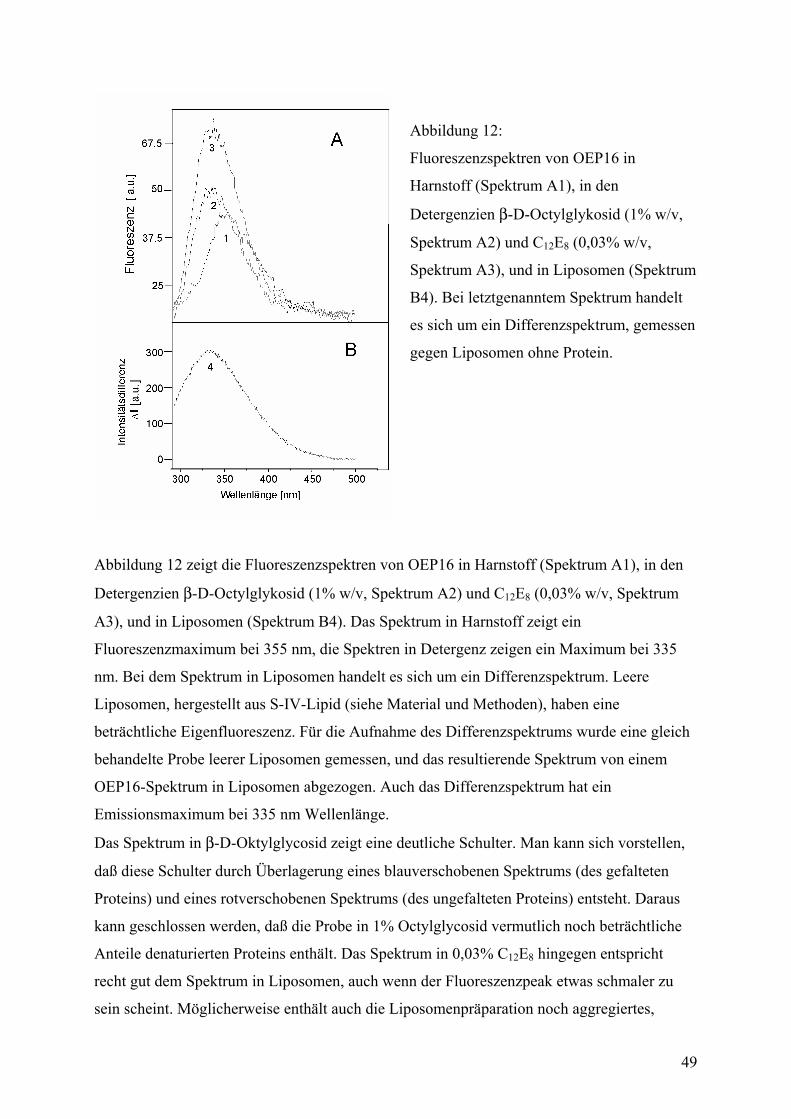
49
Abbildung 12:
Fluoreszenzspektren von OEP16 in
Harnstoff (Spektrum A1), in den
Detergenzien β-D-Octylglykosid (1% w/v,
Spektrum A2) und C12E8 (0,03% w/v,
Spektrum A3), und in Liposomen (Spektrum
B4). Bei letztgenanntem Spektrum handelt
es sich um ein Differenzspektrum, gemessen
gegen Liposomen ohne Protein.
Abbildung 12 zeigt die Fluoreszenzspektren von OEP16 in Harnstoff (Spektrum A1), in den
Detergenzien β-D-Octylglykosid (1% w/v, Spektrum A2) und C12E8 (0,03% w/v, Spektrum
A3), und in Liposomen (Spektrum B4). Das Spektrum in Harnstoff zeigt ein
Fluoreszenzmaximum bei 355 nm, die Spektren in Detergenz zeigen ein Maximum bei 335
nm. Bei dem Spektrum in Liposomen handelt es sich um ein Differenzspektrum. Leere
Liposomen, hergestellt aus S-IV-Lipid (siehe Material und Methoden), haben eine
beträchtliche Eigenfluoreszenz. Für die Aufnahme des Differenzspektrums wurde eine gleich
behandelte Probe leerer Liposomen gemessen, und das resultierende Spektrum von einem
OEP16-Spektrum in Liposomen abgezogen. Auch das Differenzspektrum hat ein
Emissionsmaximum bei 335 nm Wellenlänge.
Das Spektrum in β-D-Oktylglycosid zeigt eine deutliche Schulter. Man kann sich vorstellen,
daß diese Schulter durch Überlagerung eines blauverschobenen Spektrums (des gefalteten
Proteins) und eines rotverschobenen Spektrums (des ungefalteten Proteins) entsteht. Daraus
kann geschlossen werden, daß die Probe in 1% Octylglycosid vermutlich noch beträchtliche
Anteile denaturierten Proteins enthält. Das Spektrum in 0,03% C12E8 hingegen entspricht
recht gut dem Spektrum in Liposomen, auch wenn der Fluoreszenzpeak etwas schmaler zu
sein scheint. Möglicherweise enthält auch die Liposomenpräparation noch aggregiertes,
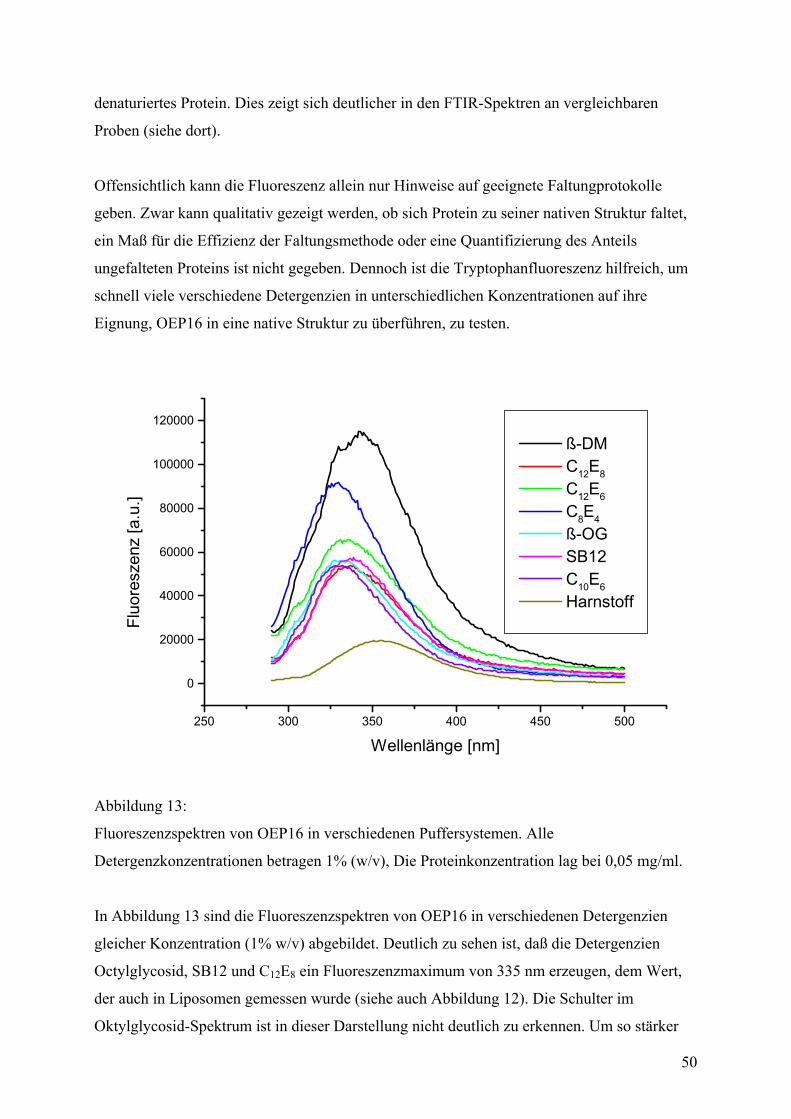
50
denaturiertes Protein. Dies zeigt sich deutlicher in den FTIR-Spektren an vergleichbaren
Proben (siehe dort).
Offensichtlich kann die Fluoreszenz allein nur Hinweise auf geeignete Faltungprotokolle
geben. Zwar kann qualitativ gezeigt werden, ob sich Protein zu seiner nativen Struktur faltet,
ein Maß für die Effizienz der Faltungsmethode oder eine Quantifizierung des Anteils
ungefalteten Proteins ist nicht gegeben. Dennoch ist die Tryptophanfluoreszenz hilfreich, um
schnell viele verschiedene Detergenzien in unterschiedlichen Konzentrationen auf ihre
Eignung, OEP16 in eine native Struktur zu überführen, zu testen.
250 300 350 400 450 500
0
20000
40000
60000
80000
100000
120000
ß-DM C12E8
C12E6
C8E4
ß-OG SB12 C10E6
Harnstoff
Fluo
resz
enz
[a.u
.]
Wellenlänge [nm]
Abbildung 13:
Fluoreszenzspektren von OEP16 in verschiedenen Puffersystemen. Alle
Detergenzkonzentrationen betragen 1% (w/v), Die Proteinkonzentration lag bei 0,05 mg/ml.
In Abbildung 13 sind die Fluoreszenzspektren von OEP16 in verschiedenen Detergenzien
gleicher Konzentration (1% w/v) abgebildet. Deutlich zu sehen ist, daß die Detergenzien
Octylglycosid, SB12 und C12E8 ein Fluoreszenzmaximum von 335 nm erzeugen, dem Wert,
der auch in Liposomen gemessen wurde (siehe auch Abbildung 12). Die Schulter im
Oktylglycosid-Spektrum ist in dieser Darstellung nicht deutlich zu erkennen. Um so stärker
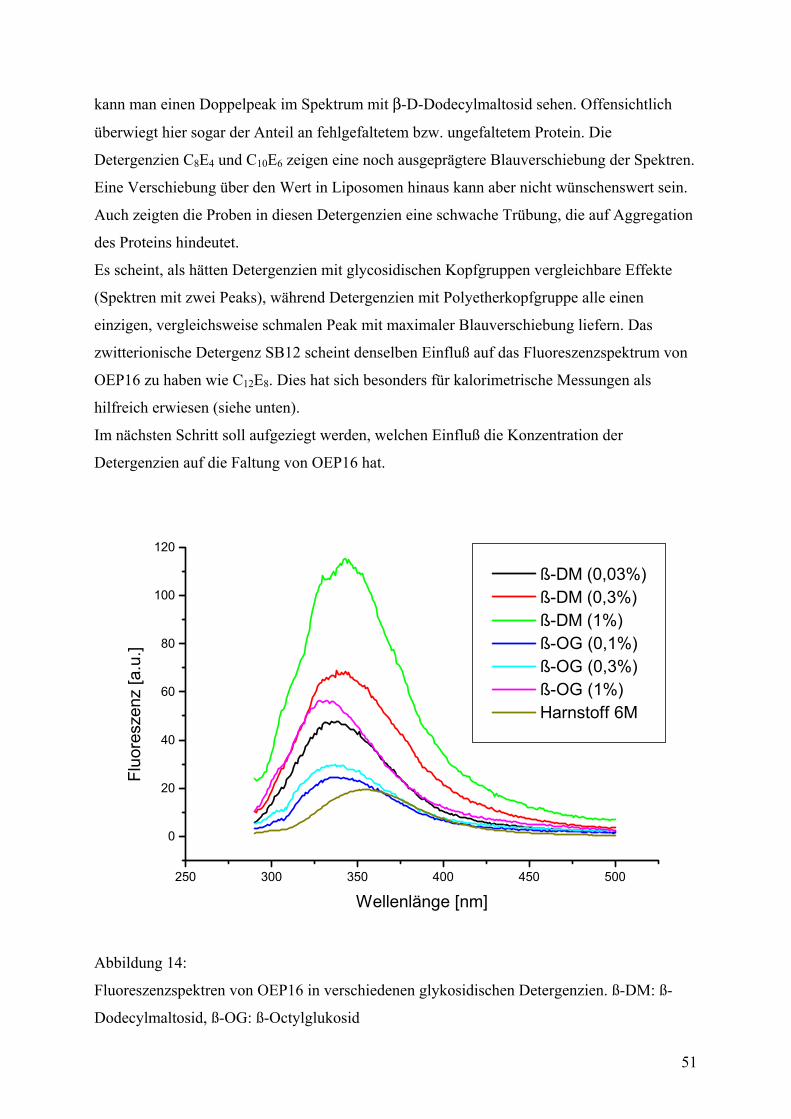
51
kann man einen Doppelpeak im Spektrum mit β-D-Dodecylmaltosid sehen. Offensichtlich
überwiegt hier sogar der Anteil an fehlgefaltetem bzw. ungefaltetem Protein. Die
Detergenzien C8E4 und C10E6 zeigen eine noch ausgeprägtere Blauverschiebung der Spektren.
Eine Verschiebung über den Wert in Liposomen hinaus kann aber nicht wünschenswert sein.
Auch zeigten die Proben in diesen Detergenzien eine schwache Trübung, die auf Aggregation
des Proteins hindeutet.
Es scheint, als hätten Detergenzien mit glycosidischen Kopfgruppen vergleichbare Effekte
(Spektren mit zwei Peaks), während Detergenzien mit Polyetherkopfgruppe alle einen
einzigen, vergleichsweise schmalen Peak mit maximaler Blauverschiebung liefern. Das
zwitterionische Detergenz SB12 scheint denselben Einfluß auf das Fluoreszenzspektrum von
OEP16 zu haben wie C12E8. Dies hat sich besonders für kalorimetrische Messungen als
hilfreich erwiesen (siehe unten).
Im nächsten Schritt soll aufgeziegt werden, welchen Einfluß die Konzentration der
Detergenzien auf die Faltung von OEP16 hat.
250 300 350 400 450 500
0
20
40
60
80
100
120
ß-DM (0,03%) ß-DM (0,3%) ß-DM (1%) ß-OG (0,1%) ß-OG (0,3%) ß-OG (1%) Harnstoff 6M
Fluo
resz
enz
[a.u
.]
Wellenlänge [nm]
Abbildung 14:
Fluoreszenzspektren von OEP16 in verschiedenen glykosidischen Detergenzien. ß-DM: ß-
Dodecylmaltosid, ß-OG: ß-Octylglukosid

52
250 300 350 400 450 500
0
20
40
60 C10E6 (0,3%) C12E8 (0,3%) C12E6 (0,3%) C8E4 (0,3%) Harnstoff 6M
Fluo
resz
enz
[a.u
.]
Wellenlänge [nm]
Abbildung 15:
Fluoreszenzspektren von OEP16 in verschiedenen Detergenzien mit Ether-Kopfgruppen.
Beispielhaft gezeigt sind die Spektren für eine Detergenzkonzentration von 0,3%. Eine
Variation der Detergenzkonzentration veränderte die Spektren nicht.
Die glykosidischen Detergenzien zeigen eine Konzentrationsabhängigkeit der
Fluoreszenzverschiebung. β-OG verschiebt die Fluoreszenz von OEP16 unterhalb seiner
CMC von 0,7% w/v weniger stark als oberhalb seiner CMC (Abbildung 14). In vivo baut sich
ungefaltetes OEP16 selbsttätig in die äußere Chloroplastenmembran ein. Geht man davon aus,
daß Detergenzmizellen die Membranoberfläche und -struktur simulieren können, kann eine
Proteinfaltung nur oberhalb der CMC – also in Anwesenheit von Mizellen – erfolgen.
Unterhalb der CMC läßt sich eine geringe Blauverschiebung der Fluoreszenz durch
Anlagerung von Detergenzmonomeren an die hydrophoben Bereiche des Proteins erklären.
Möglicherweise entstehen dabei bereits Teilstrukturen, die sich aber nicht zur nativen Struktur
zusammenlagern können. Der bereits gezeigte breite Peak oberhalb der CMC von β-OG
deutet an, daß auch hier mehrere Faltungszustände existieren könnten. Offensichtlich ist die
Faltung unvollständig.

53
β-DM hingegen zeigt bei hohen Konzentrationen einen weiteren Effekt. Es scheint, als
könnten auch hohe Detergenzkonzentrationen Fehlfaltung verursachen. Deutlich zu sehen ist
ein Doppelpeak im Spektrum bei einer Detergenzkonzentration von 1%. Offensichtlich liegt
hier zu etwa gleichen Teilen gefaltetes und ungefaltetes Protein vor. Interessant ist auch die
Zunahme der Fluoreszenz bei steigender Detergenzkonzentration. Die Ursache hierfür könnte
eine Abnahme der Proteinaggregation bei steigender Detergenzmenge sein. In diesem Fall
würde die Fluoreszenz weniger gequencht.
Die Detergenzien mit Etherkopfgruppe zeigen ein ganz anderes Bild. Hier sind die
Fluoreszenzintensitäten praktisch unabhängig von der Detergenzkonzentration (Abbildung 15
zeigt deshalb nur je eine beispielhaft ausgewählte Konzentration), und auch die Unterschiede
zwischen den Detergenzien unterschiedlicher Alkylkettenlängen und Kopfgruppengrößen sind
geringer. Die beiden Detergenzien C10E6 und C8E4 zeigen eine noch stärkere
Blauverschiebung als die beiden langkettigen Tenside und auch stärker als die Verschiebung
in Liposomen. Leider trübten diese Lösungen bei jedem Versuch, höhere
Proteinkonzentrationen zu erreichen, stark ein, was eine Eignung zur korrekten Faltung
unwahrscheinlich macht (Aggregatbildung). Die hohe Verschiebung der Fluoreszenz kommt
vermutlich durch Anlagerung der kleinen Detergenzmoleküle im Bereich der Tryptophanreste
zustande.
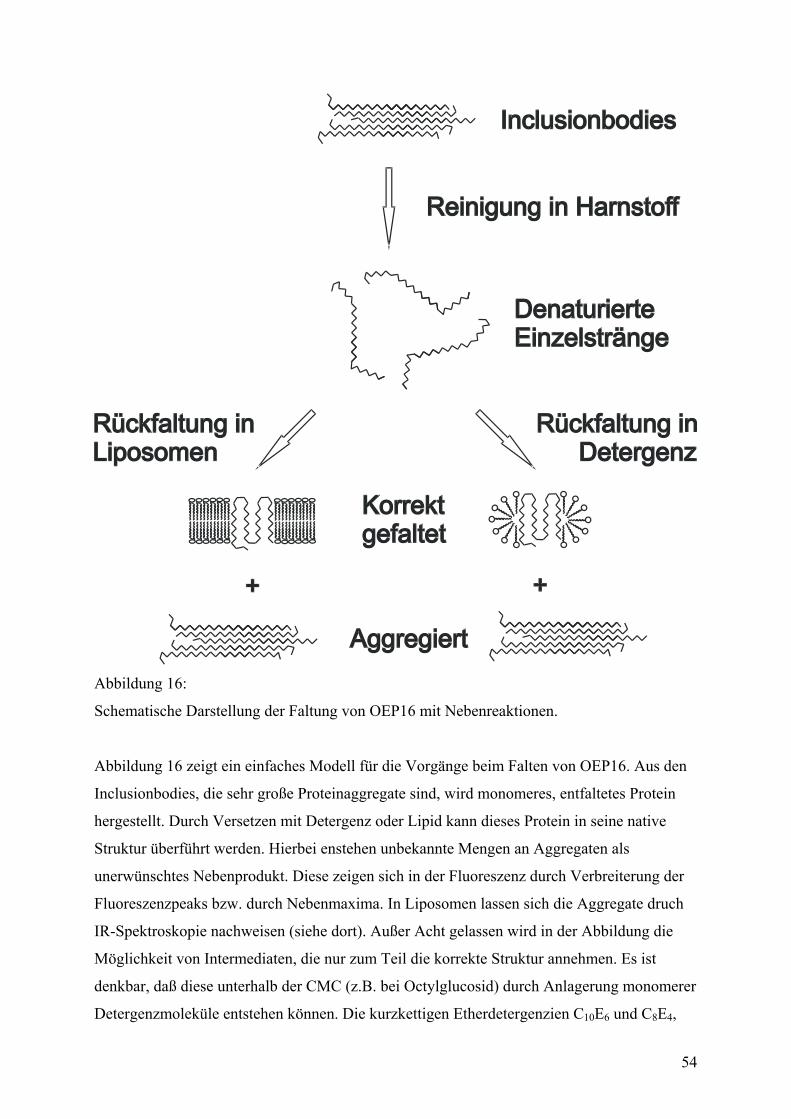
54
Abbildung 16:
Schematische Darstellung der Faltung von OEP16 mit Nebenreaktionen.
Abbildung 16 zeigt ein einfaches Modell für die Vorgänge beim Falten von OEP16. Aus den
Inclusionbodies, die sehr große Proteinaggregate sind, wird monomeres, entfaltetes Protein
hergestellt. Durch Versetzen mit Detergenz oder Lipid kann dieses Protein in seine native
Struktur überführt werden. Hierbei enstehen unbekannte Mengen an Aggregaten als
unerwünschtes Nebenprodukt. Diese zeigen sich in der Fluoreszenz durch Verbreiterung der
Fluoreszenzpeaks bzw. durch Nebenmaxima. In Liposomen lassen sich die Aggregate druch
IR-Spektroskopie nachweisen (siehe dort). Außer Acht gelassen wird in der Abbildung die
Möglichkeit von Intermediaten, die nur zum Teil die korrekte Struktur annehmen. Es ist
denkbar, daß diese unterhalb der CMC (z.B. bei Octylglucosid) durch Anlagerung monomerer
Detergenzmoleküle entstehen können. Die kurzkettigen Etherdetergenzien C10E6 und C8E4,

55
die die Fluoreszenz trotz offensichtlicher Aggregation des Proteins oberhalb ihrer CMC stark
verschieben, lagern sich möglicherweise als Monomere in die Aggregate ein.
Geht man davon aus, daß für eine vergleichsweise gute Rekonstitutionsrate der
Fluoreszenzpeak von OEP16 in Detergenz möglichst schmal sein soll (also keine
Nebenmaxima haben darf, die auf Fehlfaltung hindeuten), dieselbe Verschiebung wie das
Spektrum in Liposomen haben soll (wo die Proteinaktivität nachgewiesen wurde) und eine
möglichst hohe Intensität haben sollte (weil auch Fluoreszenzquenching auf Aggregation des
Proteins hinweist), scheinen C12E8 und C12E6 besonders geeignet. Dies gilt es nun mittels
anderer Methoden zu überprüfen.
CD-Spektroskopie
Bei der CD-Spektroskopie nutzt man die Tatsache, daß optisch aktive Substanzen nicht nur
mit linear, sondern auch mit zirkular polarisiertem Licht wechselwirken können. Die
Extinktionskoeffizienten εL und εR für links und rechts zirkular polarisiertes Licht
unterscheiden sich, die eigentliche Meßgröße ist die Differenz der Extinktionskoeffizienten,
die positive und negative Werte annehmen kann. Praktisch gibt man die sogenannte
Elliptizität Θλ an, die wie folgt definiert ist:
LR
LR
IIII
+−=Θλtan
Hierbei sind I die Intensitäten des rechts und links zirkular polarisierten Lichts nach der
Absorption durch die Probe. Die Elliptizität ist wellenlängenabhängig (daher der Index λ) und
erlaubt somit das Aufnehmen von Spektren. Unter Berücksichtigung von Schichtdicke und
Probenkonzentration kann man auch eine molare Elliptizität ΘM in Abhängigkeit von der
Wellenlänge angeben.
CD-aktive Substanzen zeichnen sich durch große, übergeordnete Strukturen aus. Insbesondere
bei Proteinen kann die CD-Spektroskopie wertvolle Informationen über den
Sekundärstrukturgehalt liefern. Während ein denaturiertes, ungeordnetes Protein nur wenig
Auslenkung des CD-Signals zeigt, haben α-Helices und β-Faltblätter deutlich voneinander
unterscheidbare Maxima und Minima im CD-Spektrum. Der Meßbereich liegt hierbei etwa
zwischen 250 und 180 nm. Die Untergrenze des Meßbereichs wird hierbei durch
Geräteparameter festgelegt. Wird die UV-Absorption der Probe zu hoch, verschlechtert sich
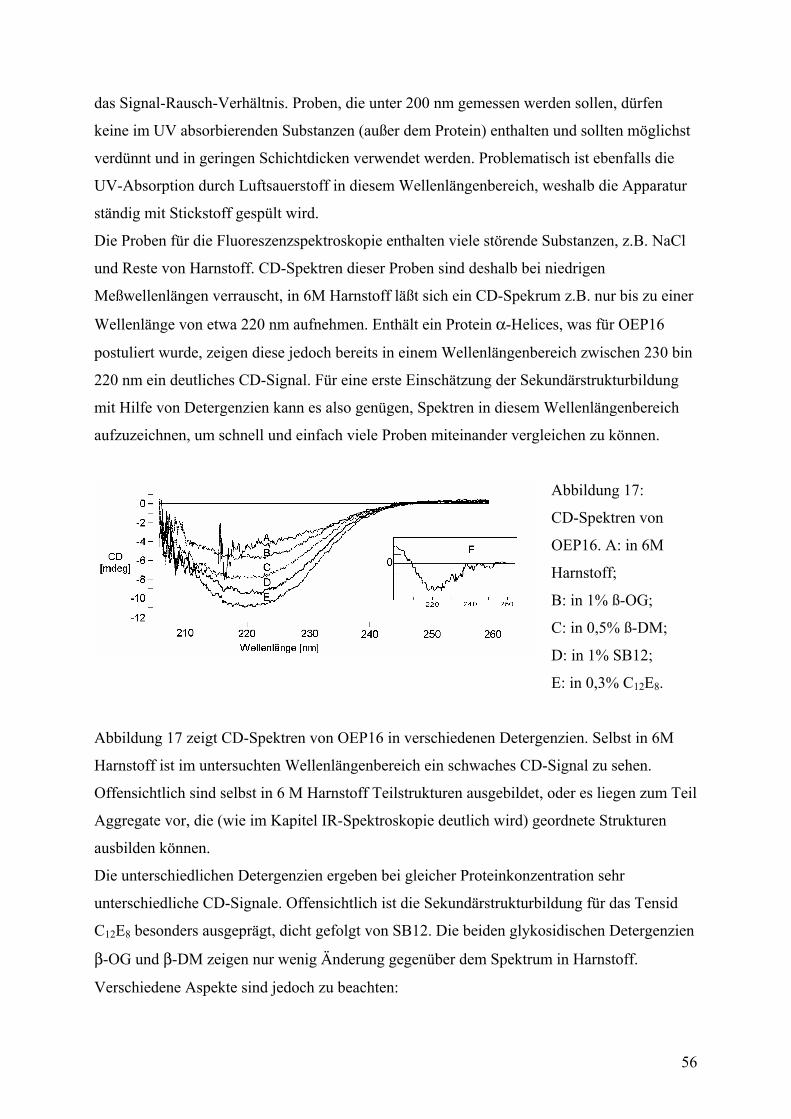
56
das Signal-Rausch-Verhältnis. Proben, die unter 200 nm gemessen werden sollen, dürfen
keine im UV absorbierenden Substanzen (außer dem Protein) enthalten und sollten möglichst
verdünnt und in geringen Schichtdicken verwendet werden. Problematisch ist ebenfalls die
UV-Absorption durch Luftsauerstoff in diesem Wellenlängenbereich, weshalb die Apparatur
ständig mit Stickstoff gespült wird.
Die Proben für die Fluoreszenzspektroskopie enthalten viele störende Substanzen, z.B. NaCl
und Reste von Harnstoff. CD-Spektren dieser Proben sind deshalb bei niedrigen
Meßwellenlängen verrauscht, in 6M Harnstoff läßt sich ein CD-Spekrum z.B. nur bis zu einer
Wellenlänge von etwa 220 nm aufnehmen. Enthält ein Protein α-Helices, was für OEP16
postuliert wurde, zeigen diese jedoch bereits in einem Wellenlängenbereich zwischen 230 bin
220 nm ein deutliches CD-Signal. Für eine erste Einschätzung der Sekundärstrukturbildung
mit Hilfe von Detergenzien kann es also genügen, Spektren in diesem Wellenlängenbereich
aufzuzeichnen, um schnell und einfach viele Proben miteinander vergleichen zu können.
Abbildung 17:
CD-Spektren von
OEP16. A: in 6M
Harnstoff;
B: in 1% ß-OG;
C: in 0,5% ß-DM;
D: in 1% SB12;
E: in 0,3% C12E8.
Abbildung 17 zeigt CD-Spektren von OEP16 in verschiedenen Detergenzien. Selbst in 6M
Harnstoff ist im untersuchten Wellenlängenbereich ein schwaches CD-Signal zu sehen.
Offensichtlich sind selbst in 6 M Harnstoff Teilstrukturen ausgebildet, oder es liegen zum Teil
Aggregate vor, die (wie im Kapitel IR-Spektroskopie deutlich wird) geordnete Strukturen
ausbilden können.
Die unterschiedlichen Detergenzien ergeben bei gleicher Proteinkonzentration sehr
unterschiedliche CD-Signale. Offensichtlich ist die Sekundärstrukturbildung für das Tensid
C12E8 besonders ausgeprägt, dicht gefolgt von SB12. Die beiden glykosidischen Detergenzien
β-OG und β-DM zeigen nur wenig Änderung gegenüber dem Spektrum in Harnstoff.
Verschiedene Aspekte sind jedoch zu beachten:

57
Die Proben wurden zwar gleich behandelt, wurden aber nach der Rekonstitution mit
Konzentratoren, also Ultra-Filtrationseinheiten, ankonzentriert. Substanzverluste an den
Filtern oder den Kunststoffgehäusen der Zentrifugationseinheiten könnten also zumindest
teilweise mitverantwortlich sein für die unterschiedlichen Auslenkungen der CD-Spektren. Da
eine Proteinbestimmung in dem verwendeten Puffersystem praktisch nicht durchführbar ist
(Detergenzien, Harnstoff und Reduktionsmittel wie β-Mercaproethanol stören praktisch alle
Methoden der kolorimetrischen Proteinbestimmung), kann dies jedoch nicht quantitativ
nachvollzogen werden.
Die Proben könnten Aggregate enthalten, die keine korrekte Struktur ausgebildet haben, aber
dennoch einen Beitrag zum CD-Spektrum liefern. Tatsächlich war in vielen der Proben eine
schwach Trübung erkennbar, die auf Aggregation hinweist.
Um ein CD-Spektrum zu erhalten, das auf den Sekundärstrukturgehalt von OEP16
quantitative Schlüsse zuläßt, musste die Probe anders vorbereitet werden. Es galt, sämtliche
UV-aktiven Substanzen zu entfernen. Dies geschah mit Hilfe von Dialysezellen und einer
Dialysemembran von 3.500 kDa Porengröße, die weder für OEP16 noch für Mizellen des
Detergenz (hier C12E8) durchlässig ist. Harnstoff, Reduktionsmittel, Salze und
Puffersubstanzen werden durch einen Puffer aus 10 mM Natrium-Cacodylat und 50 mM
NaSO4 ersetzt. Die Probe wird zur Entfernung potentiell noch enthaltener Aggregate
zusätzlich filtriert. Das resultierende CD-Spektrum, aufgenommen in einer Tonnenküvette mit
0,1 mm Schichtdicke, ist in Abbildung 18 gezeigt.
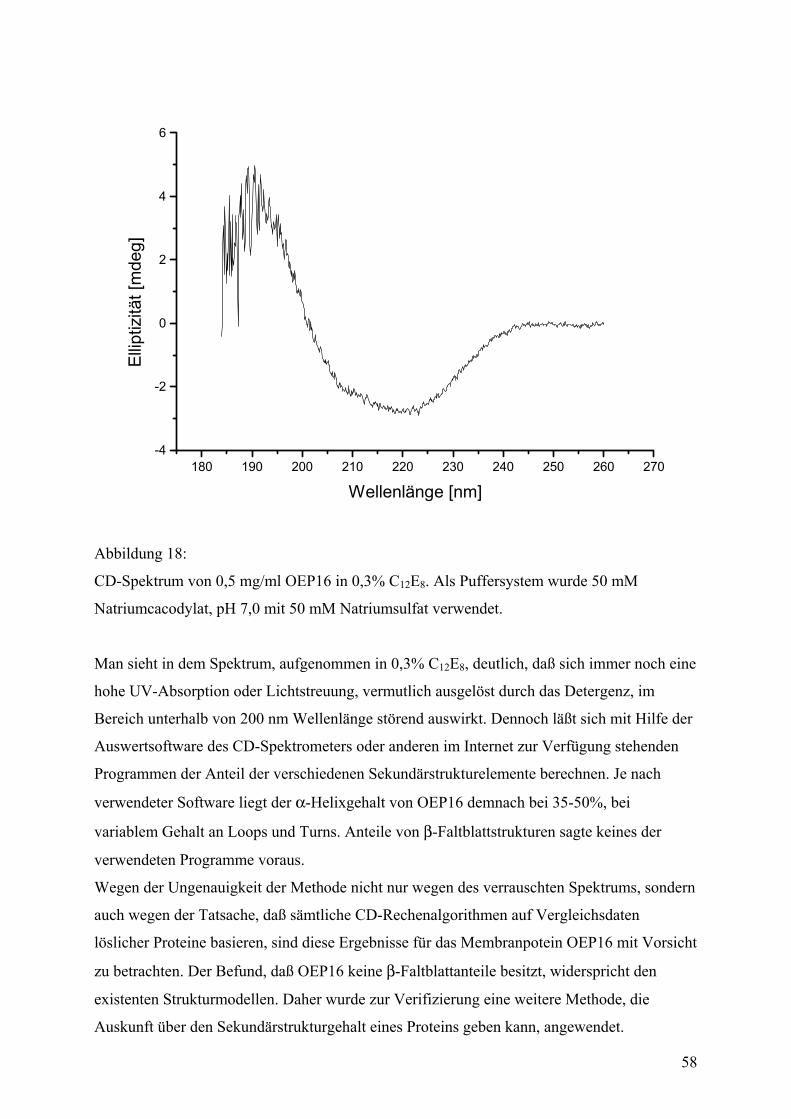
58
180 190 200 210 220 230 240 250 260 270-4
-2
0
2
4
6El
liptiz
ität [
mde
g]
Wellenlänge [nm]
Abbildung 18:
CD-Spektrum von 0,5 mg/ml OEP16 in 0,3% C12E8. Als Puffersystem wurde 50 mM
Natriumcacodylat, pH 7,0 mit 50 mM Natriumsulfat verwendet.
Man sieht in dem Spektrum, aufgenommen in 0,3% C12E8, deutlich, daß sich immer noch eine
hohe UV-Absorption oder Lichtstreuung, vermutlich ausgelöst durch das Detergenz, im
Bereich unterhalb von 200 nm Wellenlänge störend auswirkt. Dennoch läßt sich mit Hilfe der
Auswertsoftware des CD-Spektrometers oder anderen im Internet zur Verfügung stehenden
Programmen der Anteil der verschiedenen Sekundärstrukturelemente berechnen. Je nach
verwendeter Software liegt der α-Helixgehalt von OEP16 demnach bei 35-50%, bei
variablem Gehalt an Loops und Turns. Anteile von β-Faltblattstrukturen sagte keines der
verwendeten Programme voraus.
Wegen der Ungenauigkeit der Methode nicht nur wegen des verrauschten Spektrums, sondern
auch wegen der Tatsache, daß sämtliche CD-Rechenalgorithmen auf Vergleichsdaten
löslicher Proteine basieren, sind diese Ergebnisse für das Membranpotein OEP16 mit Vorsicht
zu betrachten. Der Befund, daß OEP16 keine β-Faltblattanteile besitzt, widerspricht den
existenten Strukturmodellen. Daher wurde zur Verifizierung eine weitere Methode, die
Auskunft über den Sekundärstrukturgehalt eines Proteins geben kann, angewendet.

59
IR-Spektroskopie
Bei der Fourier-Transform-Infrarotspektroskopie (FTIR) wird anders als in gewöhnlichen IR-
Spektrometern nicht der Wellenlängenbereich abgetastet, sondern simultan ein großer
Wellenlängenbereich bestrahlt. Mit einem Interferometer werden die Änderungen von
Interferenzen erfaßt, die durch Absorption einzelner IR-Banden entstehen. Dieses
Interferogramm kann mittels Fouriertransformation in ein komplettes IR-Spektrum
umgerechnet werden. Hauptvorteil dieser Methode ist die Geschwindigkeit, mit der ein
Spektrum aufgezeichnet werden kann, sowie die hohe Strahlintensität und damit ein
geringeres Rauschen.
Proteine haben charakteristische IR-Absorptionsbanden. Insbesondere die Amid-I-Bande
(gemessen in D2O im Wellenzahlenbereich von 1600-1700 cm-1), die hauptsächlich von den
C=O-Streckschwingungen der Carbonylgruppe im Polypeptidrückrad herrührt, wird
maßgeblich von der Sekundärstruktur des Proteins beeinflußt. Dies rührt daher, daß die
Carbonylgruppe je nach Sekundärstruktur unterschiedliche Wasserstoffbrückenbindungen
eingehen kann. α-Helices zeigen charakteristische Amid-I-Banden im Bereich von 1650-1658
cm-1, während typische β-Faltblattstrukturen im Bereich um 1620-1640 cm-1 absorbieren.
Loopstrukturen (1670-1680 cm-1) und ungeordnete Bereiche (1640-1650 cm-1) haben
ebenfalls charakteristische Absorptionsbanden.
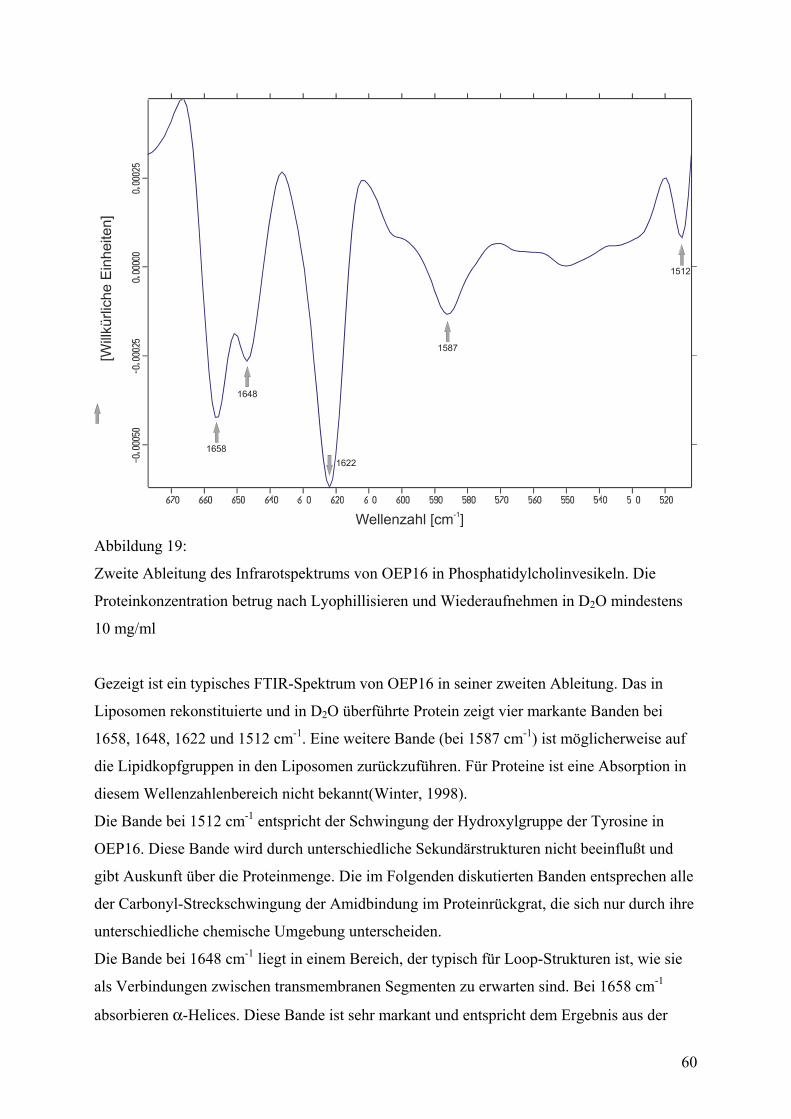
60
Abbildung 19:
Zweite Ableitung des Infrarotspektrums von OEP16 in Phosphatidylcholinvesikeln. Die
Proteinkonzentration betrug nach Lyophillisieren und Wiederaufnehmen in D2O mindestens
10 mg/ml
Gezeigt ist ein typisches FTIR-Spektrum von OEP16 in seiner zweiten Ableitung. Das in
Liposomen rekonstituierte und in D2O überführte Protein zeigt vier markante Banden bei
1658, 1648, 1622 und 1512 cm-1. Eine weitere Bande (bei 1587 cm-1) ist möglicherweise auf
die Lipidkopfgruppen in den Liposomen zurückzuführen. Für Proteine ist eine Absorption in
diesem Wellenzahlenbereich nicht bekannt(Winter, 1998).
Die Bande bei 1512 cm-1 entspricht der Schwingung der Hydroxylgruppe der Tyrosine in
OEP16. Diese Bande wird durch unterschiedliche Sekundärstrukturen nicht beeinflußt und
gibt Auskunft über die Proteinmenge. Die im Folgenden diskutierten Banden entsprechen alle
der Carbonyl-Streckschwingung der Amidbindung im Proteinrückgrat, die sich nur durch ihre
unterschiedliche chemische Umgebung unterscheiden.
Die Bande bei 1648 cm-1 liegt in einem Bereich, der typisch für Loop-Strukturen ist, wie sie
als Verbindungen zwischen transmembranen Segmenten zu erwarten sind. Bei 1658 cm-1
absorbieren α-Helices. Diese Bande ist sehr markant und entspricht dem Ergebnis aus der

61
CD-Spektroskopie, daß OEP16 praktisch ausschließlich aus helikalen Strukturen besteht. Die
letzte Absorptionsbande bei 1622 cm-1 liegt in einem Wellenzahlenbereich, der üblicherweise
β-Faltblattstrukturen vorbehalten ist. Dieses Ergebnis ist zunächst unerwartet. Betrachtet man
jedoch mehrere Spektren unterschiedlich präparierter Liposomenproben, stellt man fest, daß
das Verhältnis der beiden Banden bei 1648 cm-1 und 1658 cm-1 zu der β-Faltblattbande
(normiert auf die Tyrosinbande) variabel ist. Das bedeutet: der Sekundärstrukturgehalt der
Probe in Liposomen variiert mit der Probenpräparation. FTIR-Messungen in Detergenzlösung
zum direkten Vergleich sind nicht möglich, weil in Detergenz die notwendigen
Probenkonzentrationen nicht erreicht werden können. Mißt man eine OEP16-Probe in
Detergenz, die bis zur Aggregation ankonzentriert wurde (also stark getrübt ist), sieht man
neben der Tyrosinbande praktisch nur die Bande bei 1622 cm-1.
Wie im Kapitel über die Fluoreszenz bereits andiskutiert, enthalten insbesondere die
Liposomen-Proben also größere Mengen aggregiertes und fehlgefaltetes Protein. Vermutlich
sind diese Aggregate nicht in die Lipiddoppelschichten eingebaut, sondern nur lose daran
assoziiert. Diese Aggregation zeigt sich im IR als Bande bei 1622 cm-1, eine Wellenzahl, bei
der typischerweise antiparallele β-Faltblattstrukturen absorbieren, z.B. solche aus
Aggregationsfibrillen. Solche Fibrillen entstehen aus fehlgefalteten Membranproteinen unter
anderem bei Prionenkrankheiten (Scrapie(Callahan, 2001), BSE und die Creutzfeld-Jakub-
Krankheit, aber auch Alzheimer und Parkinson(Conway, 2000)). Transmembrane
Faltblattstrukturen, wie sie hauptsächlich in Porinen vorkommen, absorbieren bei höheren
Wellenzahlen (ab 1630 cm-1)(Abrecht, 2000).
Während für OEP16 in Detergenzlösung mittels CD-Spektroskopie eine rein α-helikale
Struktur nachgewiesen werden konnte, sind in Liposomen-Proben noch beträchtliche Anteile
an ungefaltetem bzw. fehlgefaltetem Protein enthalten. Diese zeigen sich in der
Fluoreszenzspektroskopie als eine Nebenbande bei höherer Wellenlänge, die das
Gesamtspektrum scheinbar verbreitert. In der IR-Spektroskopie sind die Aggregate als
antiparallele β-Faltblattstrukturen mit einem Absorptionsmaximum von 1622 cm-1
nachweisbar. Offensichtlich handelt es sich bei den Aggregaten um Strukturen, wie sie auch
von vielen anderen aggregierten Membranproteinen bekannt sind.
Es wurde gezeigt, daß sich OEP16 in Lösung wie in Liposomen korrekt falten läßt.
Spektroskopische Methoden zeigten auch, daß OEP16 hauptsächlich aus α-Helices besteht. In

62
den folgenden Abschnitten werden physikalisch-chemische Experimente zur Proteinstabilität
und Faltung vorgestellt.
Biophysikalische Daten des gefalteten Proteins
DSC
Die Differenzkalorimetrie beruht auf der Messung der Wärmekapazität einer Probe gegenüber
der bekannten Wärmekapazität einer Referenz. Probe und Referenz werden während der
Messung kontinuierlich aufgeheizt, wobei die Heizquellen so eingestellt sind, daß die
Temperatur in der Probenzelle etwas langsamer ansteigt als in der Referenzzelle. Ein
Thermoelement kontrolliert die resultierende Temperaturdifferenz zwischen den
Meßkammern und regelt eine Hilfs-Heizquelle. Die Heizleistung P (Energie pro Zeit), die
benötigt wird, um die Temperaturdifferenz zwischen Referenz und Probe auszugleichen, wird
genutzt, um die Wärmekapazität bei konstantem Druck cP zu bestimmen. In die Berechnung
geht die Geschwindigkeit der Temperaturerhöhung χ=∆T/t wie folgt ein:
1−= χPcP
Man erhält eine Kurve, die die Änderung der Wärmekapazität cP in Abhängigkeit von der
Temperatur darstellt. Eine solche Änderung erwartet man zum Beispiel bei einem
Phasenübergang. Da cP als partielle Ableitung der Enthalpie ∆H nach der Temperatur bei
konstantem Druck definiert ist, kann man durch Integration der Fläche unter der gemessenen
Kurve die Enthalpie eines Phasenüberganges berechnen.
Spektroskopische Methoden haben gezeigt, daß OEP16 eine dem nativen Zustand
entsprechende Konformation annehmen kann, wenn es in detergenzhaltigen Puffer
eingebracht wird. Da dieser Faltungsprozeß freiwillig abläuft, muß er mit einem Gewinn an
freier Energie verbunden sein. Diese freie Energie setzt sich aus einem Enthalpie- und einem
Entropieanteil zusammen, es gilt:
( ) )()( FaltungFaltungFaltung STHG ∆−∆=∆

63
Die Entropieänderung für den betrachteten Prozeß dürfte in erster Linie aus dem hydrophoben
Effekt entstehen. Das hydrophobe Protein begibt sich in die hydrophobe Umgebung der
Mizelle, während es zuvor dem Wasser eine geordnete Struktur um seine hydrophoben
Bereiche aufgezwungen hat.
Die Enthalpieänderung setzt sich aus verschiedenen Anteilen zusammen.
1. Das Protein faltet sich in seine Sekundärstrukturelemente. Hierbei werden viele
Wasserstoffbrückenbindungen gebildet. Unabhängig davon, ob es sich um α-Helices oder β-
Faltblattstrukturen handelt, dürfte dies den größten Anteil der Enthalpieänderung ausmachen.
2. Das Protein "löst" sich im Detergenz, die neuen Wechselwirkungen mit der
Proteinumgebung werden sich in der Enthalpieänderung niederschlagen.
3. Eventuell lagern sich mehrere Proteineinheiten unter Ausbildung unterschiedlicher
Wechselwirkungen zusammen.
Erhitzt man das gefaltete Protein in Detergenzlösung in einem geeigneten Kalorimeter, kann
man die Denaturierungswärme des Proteins bestimmen. Diese entspricht dem Umkehrprozeß
der genannten Effekte, mit dem Unterschied, daß das hitze-denaturierte Protein
möglicherweise in der Detergenzmizelle verbleibt - oder aggregiert und aus der Lösung
ausfällt.
In einem Diferenziellen "Scanning"-Kalorimeter (DSC) wurden 0,55 mg/ml OEP16 in
verschiedenen Detergenzlösungen graduell bis auf 95°C erwärmt. Gemessen wurde die
Änderung der Wärmekapazität der Lösung gegenüber einer Referenzlösung, die nur die
detergenzhaltige Pufferlösung ohne Protein enthielt. Die Enthalpieänderung entspricht bei
dieser Messung der Fläche unter einem eventuell auftretenden Denaturierungspeak. Vor allem
Proteinkomplexe mit mehreren Untereinheiten können schrittweise danaturieren, haben also
mehrere Denaturierungspeaks.
Das Detergenz C12E8 lieferte ein starkes, aber extrem verrauschtes Denaturierungssignal bei
ca. 85°C. Da das Detergenz in diesem Temperaturbereich bei der gegebenen Konzentration
von 0,3% selbst eine Phasenumwandlung durchmacht(Mitchell, 1983), ist die
Enthalpieänderung in diesem Fall praktisch nicht auswertbar.
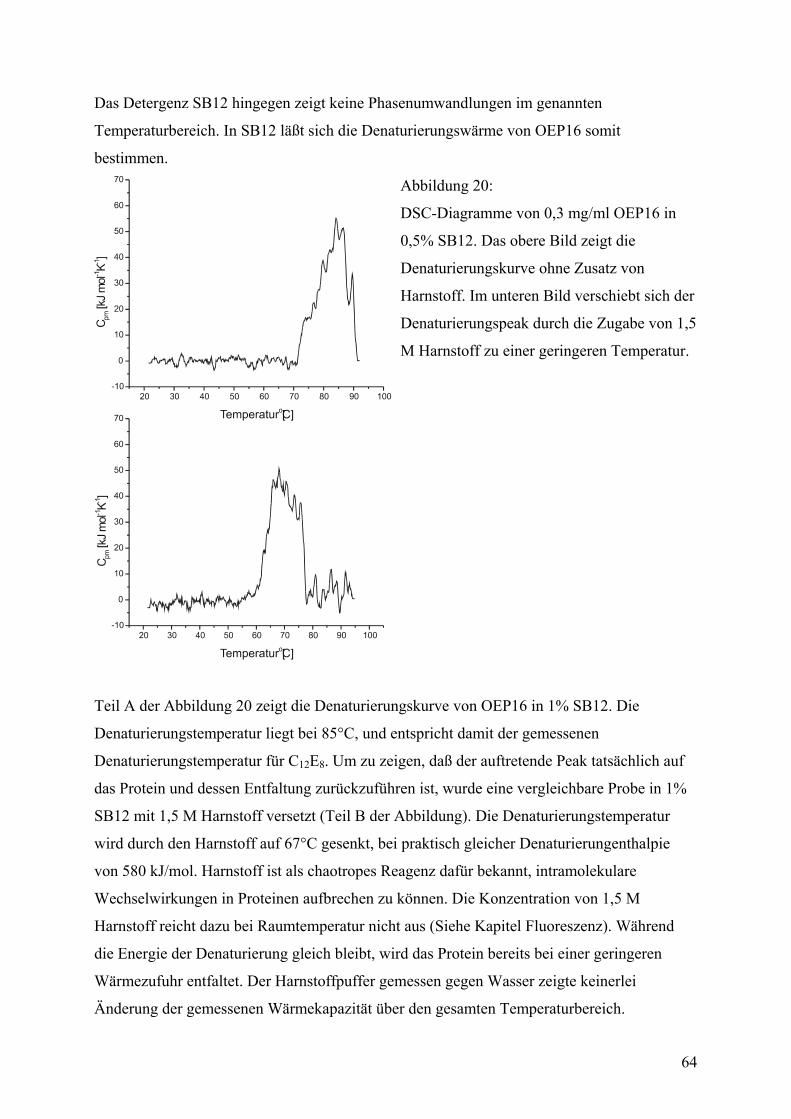
64
Das Detergenz SB12 hingegen zeigt keine Phasenumwandlungen im genannten
Temperaturbereich. In SB12 läßt sich die Denaturierungswärme von OEP16 somit
bestimmen.
Abbildung 20:
DSC-Diagramme von 0,3 mg/ml OEP16 in
0,5% SB12. Das obere Bild zeigt die
Denaturierungskurve ohne Zusatz von
Harnstoff. Im unteren Bild verschiebt sich der
Denaturierungspeak durch die Zugabe von 1,5
M Harnstoff zu einer geringeren Temperatur.
Teil A der Abbildung 20 zeigt die Denaturierungskurve von OEP16 in 1% SB12. Die
Denaturierungstemperatur liegt bei 85°C, und entspricht damit der gemessenen
Denaturierungstemperatur für C12E8. Um zu zeigen, daß der auftretende Peak tatsächlich auf
das Protein und dessen Entfaltung zurückzuführen ist, wurde eine vergleichbare Probe in 1%
SB12 mit 1,5 M Harnstoff versetzt (Teil B der Abbildung). Die Denaturierungstemperatur
wird durch den Harnstoff auf 67°C gesenkt, bei praktisch gleicher Denaturierungenthalpie
von 580 kJ/mol. Harnstoff ist als chaotropes Reagenz dafür bekannt, intramolekulare
Wechselwirkungen in Proteinen aufbrechen zu können. Die Konzentration von 1,5 M
Harnstoff reicht dazu bei Raumtemperatur nicht aus (Siehe Kapitel Fluoreszenz). Während
die Energie der Denaturierung gleich bleibt, wird das Protein bereits bei einer geringeren
Wärmezufuhr entfaltet. Der Harnstoffpuffer gemessen gegen Wasser zeigte keinerlei
Änderung der gemessenen Wärmekapazität über den gesamten Temperaturbereich.

65
Die Faltungsenthalpie von OEP16 liegt bei ca. 600 kJ/mol. Die Hitzedenaturierung erfolgt in
einem einzigen Schritt und ist irreversibel, weil das Protein anschließend aggregiert und
ausfällt (zu sehen an der Trübung der Probe). Der umgekehrte Prozeß, die Faltung des
Proteins, läßt sich demnach kalorimetrisch nicht verfolgen. Messen läßt sich allerdings bei
geeignetem Versuchsaufbau die Geschwindigkeit der Proteinfaltung. Dadurch sind
Rückschlüsse auf die Aktivierungsenergie des Faltungsprozesses und auf seine
Reaktionsordnung möglich.
Zeitaufgelöste Spektroskopie
Um Prozesse wie die hier untersuchte Proteinfaltung besser verstehen zu können, ist es
hilfreich, nicht nur statische Spektren des gefalteten und entfalteten Zustandes zu messen,
sondern auch die Veränderung während dem Prozeß zu verfolgen. Im Prinzip sind bei
geeignetem Versuchsaufbau solche zeitaufgelösten Messungen mit jeder spektroskopischen
Methode möglich. Oft macht aber eine zu geringe Empfindlichkeit der notwendigen
Detektoren das Messen schneller Prozesse sehr schwierig.
Im Verlauf dieser Arbeit wurden zeitaufgelöste Messungen mit Fluoreszenz- und CD-
Detektion durchgeführt. Die Meßprinzipien dieser beiden spektroskopischen Methoden
wurden bereits erläutert. Der Versuchsaufbau ist abgesehen vom unterschiedlichen Detektor
für beide Methoden identisch. Zur Vereinfachung der Messung wird nur eine feste
Wellenlänge detektiert, anstatt zu jedem Zeitpunkt ein komplettes Spektrum aufzunehmen.
Sinnvollerweise wählt man diese Wellenlänge in einem spektralen Bereich, in dem die
größten Änderungen zwischen Anfangs- und Endzustand zu erwarten sind. Der Übergang von
einem Zustand zum anderen wird durch schnelles Vermischen zweier Lösungen in einer
druckbetriebenen Mischkammer, einer sogenannten "stopped-flow" Apparatur erreicht. Diese
Mischkammer dient gleichzeitig als Meßküvette, in der nach einer gerätebedingten Totzeit die
Änderung des Meßsignals erfaßt werden kann.
Zeitaufgelöste Fluoreszenzspektroskopie
Es konnte bereits gezeigt werden, daß sich die Fluoreszenz der Tryptophanreste in OEP16
signifikant ändert, wenn sich die native Struktur des Proteins bildet. Der Faltungsprozeß ist
jedoch so schnell, daß sich mit einem herkömmlichen Spektrometer lediglich der Anfangs-
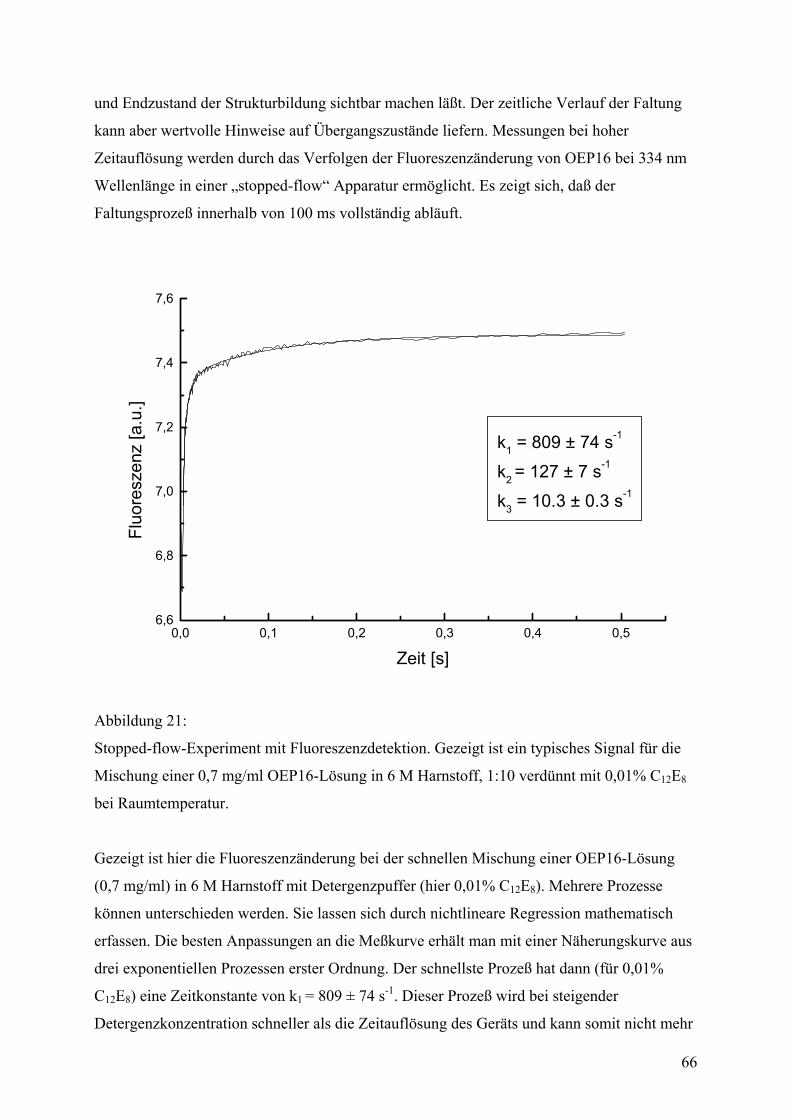
66
und Endzustand der Strukturbildung sichtbar machen läßt. Der zeitliche Verlauf der Faltung
kann aber wertvolle Hinweise auf Übergangszustände liefern. Messungen bei hoher
Zeitauflösung werden durch das Verfolgen der Fluoreszenzänderung von OEP16 bei 334 nm
Wellenlänge in einer „stopped-flow“ Apparatur ermöglicht. Es zeigt sich, daß der
Faltungsprozeß innerhalb von 100 ms vollständig abläuft.
0,0 0,1 0,2 0,3 0,4 0,56,6
6,8
7,0
7,2
7,4
7,6
k1 = 809 ± 74 s-1
k2 = 127 ± 7 s-1
k3 = 10.3 ± 0.3 s-1
Fluo
resz
enz
[a.u
.]
Zeit [s]
Abbildung 21:
Stopped-flow-Experiment mit Fluoreszenzdetektion. Gezeigt ist ein typisches Signal für die
Mischung einer 0,7 mg/ml OEP16-Lösung in 6 M Harnstoff, 1:10 verdünnt mit 0,01% C12E8
bei Raumtemperatur.
Gezeigt ist hier die Fluoreszenzänderung bei der schnellen Mischung einer OEP16-Lösung
(0,7 mg/ml) in 6 M Harnstoff mit Detergenzpuffer (hier 0,01% C12E8). Mehrere Prozesse
können unterschieden werden. Sie lassen sich durch nichtlineare Regression mathematisch
erfassen. Die besten Anpassungen an die Meßkurve erhält man mit einer Näherungskurve aus
drei exponentiellen Prozessen erster Ordnung. Der schnellste Prozeß hat dann (für 0,01%
C12E8) eine Zeitkonstante von k1 = 809 ± 74 s-1. Dieser Prozeß wird bei steigender
Detergenzkonzentration schneller als die Zeitauflösung des Geräts und kann somit nicht mehr
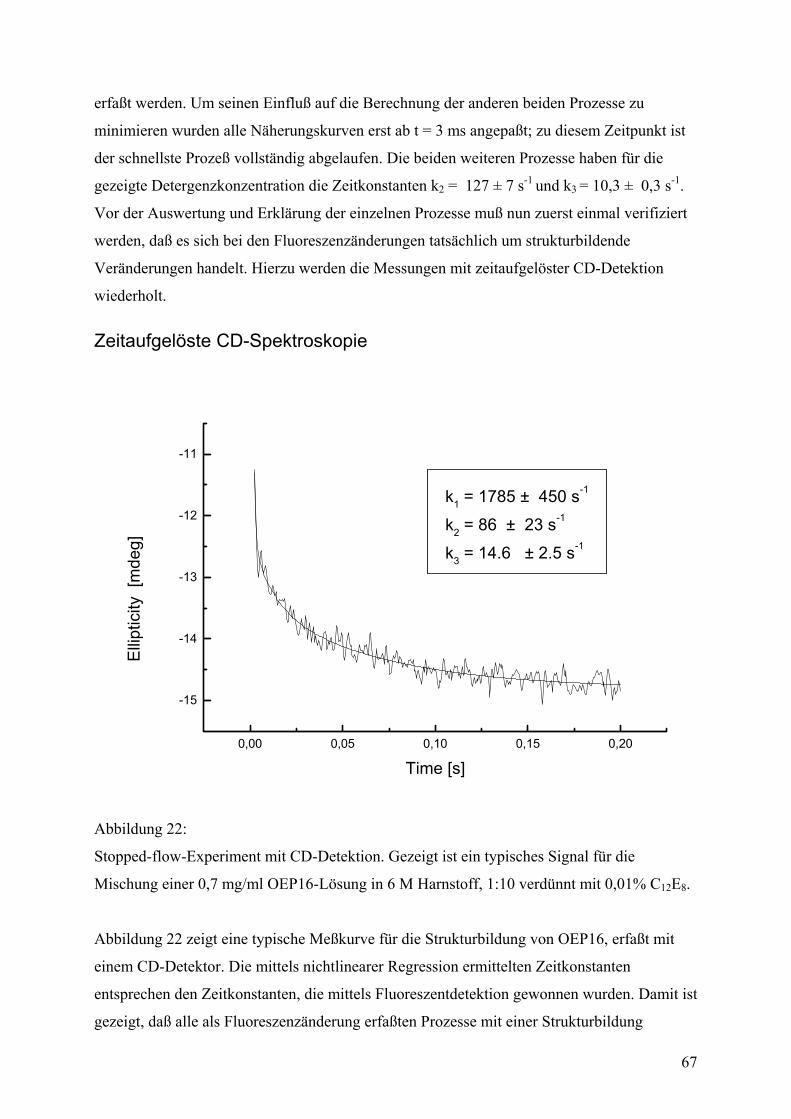
67
erfaßt werden. Um seinen Einfluß auf die Berechnung der anderen beiden Prozesse zu
minimieren wurden alle Näherungskurven erst ab t = 3 ms angepaßt; zu diesem Zeitpunkt ist
der schnellste Prozeß vollständig abgelaufen. Die beiden weiteren Prozesse haben für die
gezeigte Detergenzkonzentration die Zeitkonstanten k2 = 127 ± 7 s-1 und k3 = 10,3 ± 0,3 s-1.
Vor der Auswertung und Erklärung der einzelnen Prozesse muß nun zuerst einmal verifiziert
werden, daß es sich bei den Fluoreszenzänderungen tatsächlich um strukturbildende
Veränderungen handelt. Hierzu werden die Messungen mit zeitaufgelöster CD-Detektion
wiederholt.
Zeitaufgelöste CD-Spektroskopie
0,00 0,05 0,10 0,15 0,20
-15
-14
-13
-12
-11
k1 = 1785 ± 450 s-1
k2 = 86 ± 23 s-1
k3 = 14.6 ± 2.5 s-1
Ellip
ticity
[m
deg]
Time [s]
Abbildung 22:
Stopped-flow-Experiment mit CD-Detektion. Gezeigt ist ein typisches Signal für die
Mischung einer 0,7 mg/ml OEP16-Lösung in 6 M Harnstoff, 1:10 verdünnt mit 0,01% C12E8.
Abbildung 22 zeigt eine typische Meßkurve für die Strukturbildung von OEP16, erfaßt mit
einem CD-Detektor. Die mittels nichtlinearer Regression ermittelten Zeitkonstanten
entsprechen den Zeitkonstanten, die mittels Fluoreszentdetektion gewonnen wurden. Damit ist
gezeigt, daß alle als Fluoreszenzänderung erfaßten Prozesse mit einer Strukturbildung

68
einhergehen und nicht etwa allein durch das Mischen mit detergenzhaltigem und damit
hydrophobem Puffer zustandekommen.
Zeitkonstanten und Aktivierungsenergien
Es konnte gezeigt werden, daß sich Faltungs-Teilprozesse einzeln erfassen lassen. Dies
funktioniert gut für das Detergenz C12E8 bis zu Konzentrationen um 0,03% w/v. Bei höheren
Detergenzkonzentrationen werden die Prozesse so schnell, daß eine präzise Berechnung von
Zeitkonstanten nicht mehr möglich ist (jedenfalls nicht mit den gegebenen Meßgeräten).
Ähnliches gilt auch für andere Detergenzien, der Faltungsprozeß läßt sich im Prinzip auch bei
β-Dodecylmaltosid und SB12 verfolgen, ebenfalls nur bei niedrigen Konzentrationen. Da die
Faltungsausbeute bei diesen Detergenzien etwas niedriger ist, sind die Signalintensitäten
entsprechend geringer und die Berechnungen ungenauer. Den schnellsten Prozeß (und
eventuell noch schnellere, gar nicht erfaßte Prozesse) kann man bei diesen Detergenzien wie
auch bei relativ hohen C12E8-Konzentrationen gar nicht beobachten.
Trotz dieser Einschränkungen lassen sich eine Reihe von Ergebnissen aus den zeitaufgelösten
Messungen gewinnen. Diese werden im Folgenden ausschließlich für das Detergenz C12E8
näher ausgeführt, da es sich als das für die Faltung von OEP16 geeignetste Detergenz
erwiesen hat. Zunächst wird die Änderung der Zeitkonstanten mit steigender
Detergenzkonzentration betrachtet:
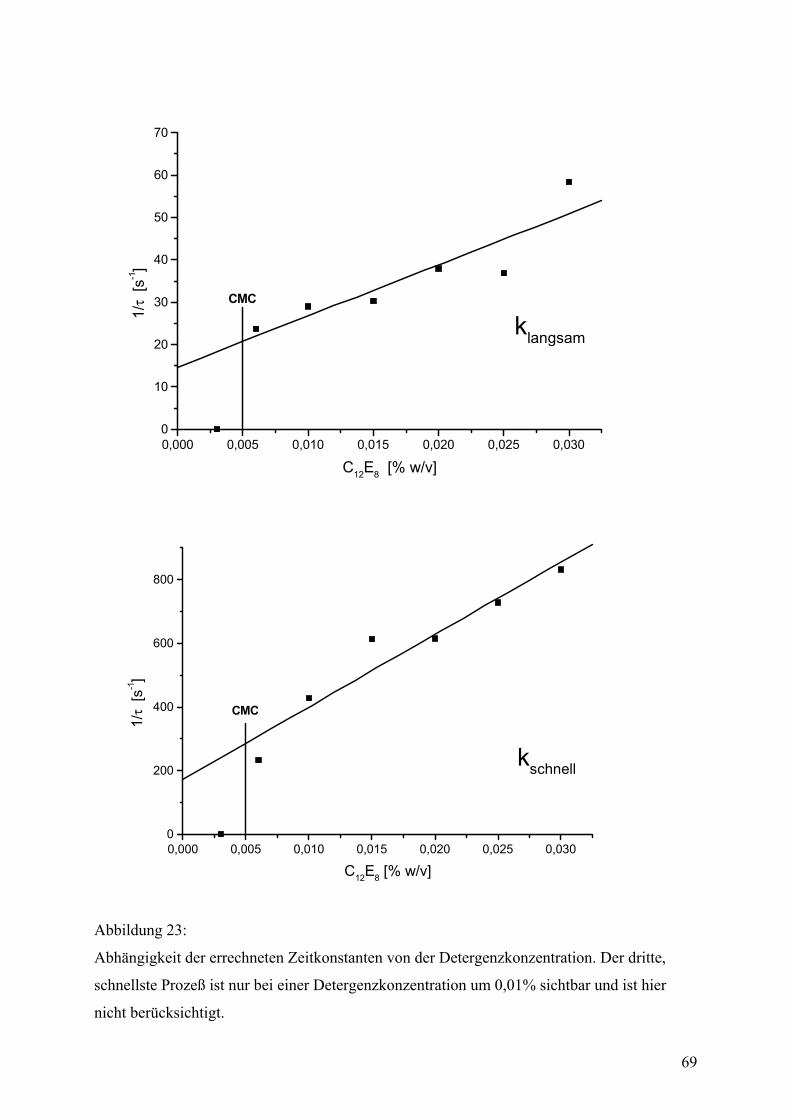
69
0,000 0,005 0,010 0,015 0,020 0,025 0,0300
10
20
30
40
50
60
70
CMC
klangsam
1/τ
[s-1]
C12E8 [% w/v]
0,000 0,005 0,010 0,015 0,020 0,025 0,0300
200
400
600
800
CMC
kschnell
1/τ
[s-1]
C12E8 [% w/v]
Abbildung 23:
Abhängigkeit der errechneten Zeitkonstanten von der Detergenzkonzentration. Der dritte,
schnellste Prozeß ist nur bei einer Detergenzkonzentration um 0,01% sichtbar und ist hier
nicht berücksichtigt.
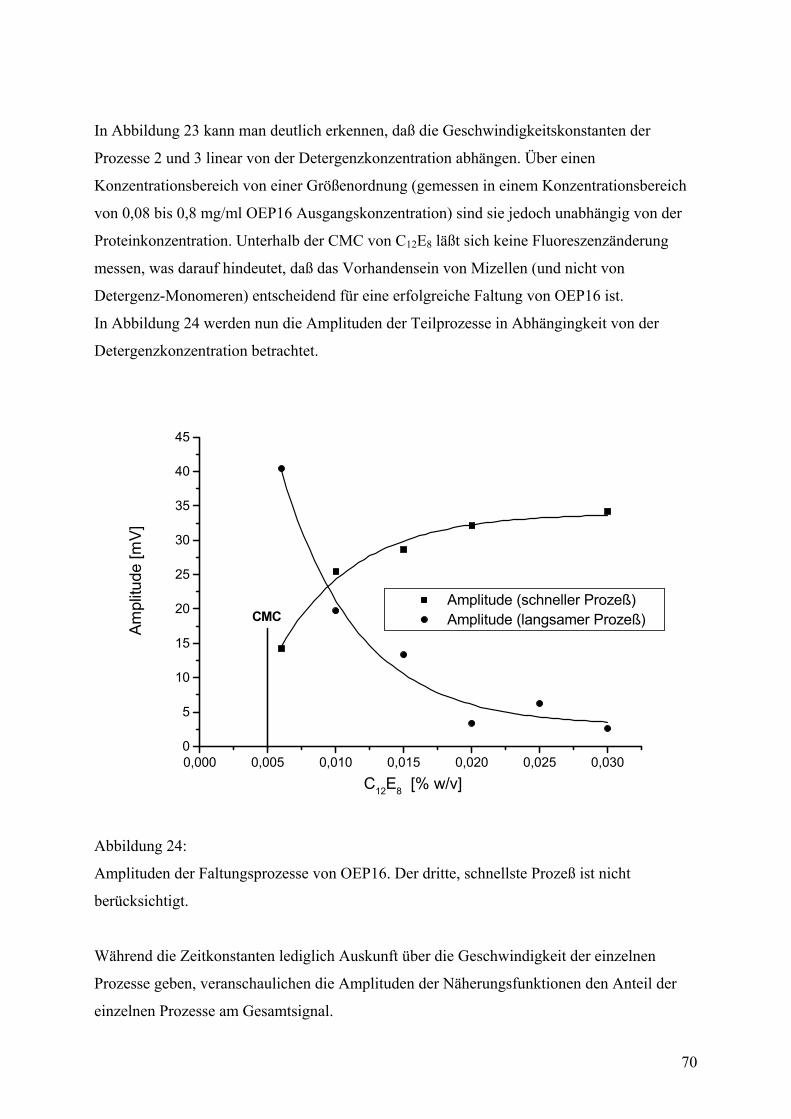
70
In Abbildung 23 kann man deutlich erkennen, daß die Geschwindigkeitskonstanten der
Prozesse 2 und 3 linear von der Detergenzkonzentration abhängen. Über einen
Konzentrationsbereich von einer Größenordnung (gemessen in einem Konzentrationsbereich
von 0,08 bis 0,8 mg/ml OEP16 Ausgangskonzentration) sind sie jedoch unabhängig von der
Proteinkonzentration. Unterhalb der CMC von C12E8 läßt sich keine Fluoreszenzänderung
messen, was darauf hindeutet, daß das Vorhandensein von Mizellen (und nicht von
Detergenz-Monomeren) entscheidend für eine erfolgreiche Faltung von OEP16 ist.
In Abbildung 24 werden nun die Amplituden der Teilprozesse in Abhängingkeit von der
Detergenzkonzentration betrachtet.
0,000 0,005 0,010 0,015 0,020 0,025 0,0300
5
10
15
20
25
30
35
40
45
CMC
C12E8 [% w/v]
Ampl
itude
[mV]
Amplitude (schneller Prozeß) Amplitude (langsamer Prozeß)
Abbildung 24:
Amplituden der Faltungsprozesse von OEP16. Der dritte, schnellste Prozeß ist nicht
berücksichtigt.
Während die Zeitkonstanten lediglich Auskunft über die Geschwindigkeit der einzelnen
Prozesse geben, veranschaulichen die Amplituden der Näherungsfunktionen den Anteil der
einzelnen Prozesse am Gesamtsignal.

71
Es wird deutlich, daß Prozeß 2 erst bei höheren Detergenzkonzentrationen dominiert, während
der langsamste Prozeß 3 dort praktisch nicht mehr meßbar ist. Beide Amplitudenänderungen
lassen sich als exponentieller Zusammenhang zwischen Meßgröße und
Detergenzkonzentration darstellen. In beiden Fällen nähern sich die exponentiellen Kurven
nicht der Abszisse, sondern der kritischen mizellaren Konzentration (CMC) asymptotisch an.
Unterhalb dieser Konzentration – also ohne die Gegenwart von Detergenzmizellen – kann der
Faltungsprozeß nicht stattfinden, die Amplituden und die Zeitkonstanten selbst betragen 0.
Oberhalb dieser Konzentration werden die Prozesse um so schneller, je mehr Detergenz im
Puffer zur Verfügung steht. Bei noch höheren Detergenzkonzentrationen stirbt der langsamere
der beiden Prozesse aus. Möglicherweise handelt es sich hierbei um einen Übergangszustand,
der nur unter relativem Detergenz-Mangel bzw. bei einem Mangel an verfügbaren
Detergenzmizellen auftritt.
Zuletzt soll die Temperaturabhängigkeit der Geschwindigkeitskonstanten gezeigt werden. Aus
der Abhängigkeit der Reaktionsgeschwindigkeit von der Temperatur läßt sich die
Aktivierungsenergie eines Prozesses nach Arrhenius berechnen:
21
21
11lnln
TT
kkREeAk ART
EA
−−∗−=⇔∗= −
Hierbei ist EA die Aktivierungsenergie des Teilprozesses, A der sogenannte Stoßfaktor oder
Arrhenius-Faktor, eine Größe aus der statistischen Themodynamik, und R die allgemeine
Gaskonstante. Durch Auftragung der logarithmierten Zeitkonstanten gegen den Kehrwert der
Temperatur läßt sich die Aktivierungsenergie als Steigung der resultierenden Geraden ablesen
(Abbildung 25).
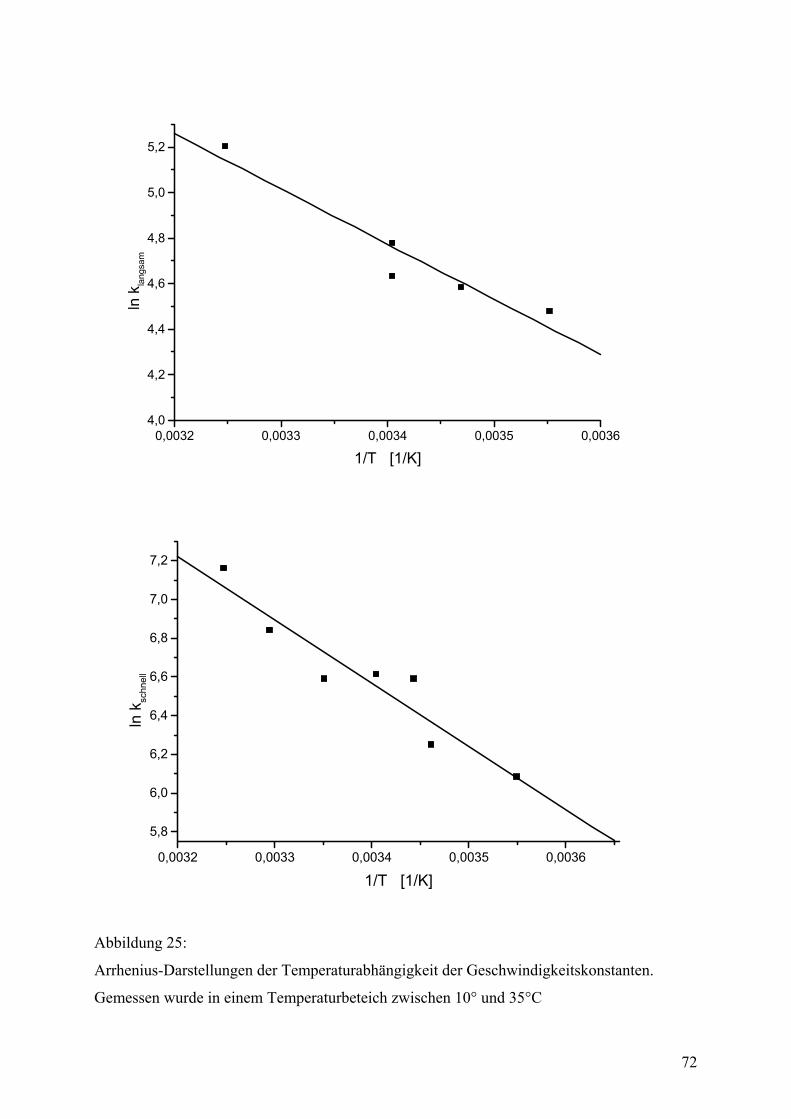
72
0,0032 0,0033 0,0034 0,0035 0,00364,0
4,2
4,4
4,6
4,8
5,0
5,2ln
kla
ngsa
m
1/T [1/K]
0,0032 0,0033 0,0034 0,0035 0,0036
5,8
6,0
6,2
6,4
6,6
6,8
7,0
7,2
ln k
schn
ell
1/T [1/K]
Abbildung 25:
Arrhenius-Darstellungen der Temperaturabhängigkeit der Geschwindigkeitskonstanten.
Gemessen wurde in einem Temperaturbeteich zwischen 10° und 35°C

73
Die Aktivierungsenergien betragen, abgelesen aus Abbildung 25 bzw. gewonnen aus den
Daten der liearen Regression der Datenpunkte, EA = 27 ± 4 kJ mol-1 für den schnelleren
Prozeß und 20 ± 3 kJ mol-1 für den langsameren Prozeß.
Das Protein wird sichtbar
Bisher wurde gezeigt, daß OEP16 in E.coli überexprimiert und in denaturiertem Zustand
gereinigt werden kann, um es anschließend in seine native Form zurückzufalten. Die
zugehörigen strukturbildenden Prozesse konnten detailliert untersucht werden. All dies sind
notwendige Voraussetzungen, um die Struktur des Proteins im Detail aufklären zu können.
Die folgenden Untersuchungen widmen sich direkt der Strukturbestimmung.
Elektronenmikroskopie
Bei der Elektronenmikroskopie macht man sich zu nutze, daß sich Elektronen nach De
Broglie auch wie Wellen verhalten können. Die Wellenlänge eines Elektrons ist umgekehrt
proportional zu seinem Impuls p = mev :
vmhe
=λ
wobei h das Plancksche Wirkungsquantum und me die Masse des Elektrons sind. Die
Geschwindigkeiten der Elektronen bei dieser Methode sind so regelbar, daß man
Wellenlängen um 10-3 nm erreicht. Bei einer Apertur von etwa 10-3 für Elektronenmikroskope
ergibt sich eine um etwa einen Faktor von 1000 bessere Auflösung gegenüber einem
Lichtmikroskop.
Anders als in der Lichtmikroskopie muß der optische Weg des Elektronenmikroskops
evakuiert sein, da Elektronen durch Gase sehr stark gestreut werden. Ansonsten ist der
Aufbau eines Elektronenmikroskops mit dem eines Lichtmikroskops durchaus vergleichbar.
Anstelle einer Lichtquelle wird eine Glühkathode als Elektronenquelle eingesetzt. Kondensor,
Objektiv und Okular werden durch elektromagnetische Linsen ersetzt, und sichtbar gemacht
wird das Abbild der Probe mittels eines durch Elektronen anregbaren Leuchtschirms. Kontrast
entsteht in der Transmissions-Elektronenmikroskopie durch die Streuung und Absorption von
Elektronen an dem Untersuchungsobjekt in Abhängigkeit von dessen Masse und Dicke. Der

74
Kontrast kann durch Zugabe von geeigneten Kontrastmitteln verstärkt werden. Im Falle von
Protein-Proben benutzt man häufig Schwermetalle, die bei der Probenvorbereitung an die
Moleküle binden und sowohl Streuung als auch Absorption den Elektronenstrahls verstärken.
Man spricht in diesem Fall von „negative stain“, da man ein Negativabbild des Proteins
erhält. Transmissions-Elektronenmikroskopie kann, bei geeigneter Wahl eines
Kontrastmittels, einzelne Proteinpartikel sichtbar machen und – wenn auch bei
vergleichsweise niedriger Auflösung – Strukturinformationen liefern. Besonders geeignet ist
diese Methode, wenn es gelingt, aus dem elektronenmikoskopischen Abbild des Proteins auf
dessen Orientierung im Raum zu schließen. In diesem Fall kann man mit Hilfe moderner
Computermethoden hunderte von Aufnahmen mitteln und so ein dreidimensionales Bild des
Proteins generieren.
OEP16, rekonstituiert in C12E8, wurde mit Phospho-Wolframsäure als Kontrastmittel in einem
Kryo-Elektronenmikroskop bei maximaler Vergrößerung (siehe Material und Methoden)
abgebildet. Das Ergebnis ist in Abbildung 26 gezeigt.
Abbildung 26:
Elektronenmikroskopische Aufnahme von
OEP16-Partikeln. Der Balken entspricht 30
nm.
Man sieht deutlich die einzelnen Proteinpartikel, der Maßstab in der unteren rechten Bildecke
entspricht 30 nm. Die Partikel haben also einen Durchmesser von etwa 5 nm. Dieser Wert ist
für ein Protein von 16-48 kDa Größe (je nach dem ob es sich um ein Monomer, Dimer oder
Trimer handelt), sinnvoll. Bemerkenswerterweise taucht derselbe Wert als
röntgenkristallographische Größe im Folgenden nocheinmal auf (siehe Abschnitt
Kristallisation). Bedauerlicherweise sind die Partikel damit zu klein für detailliertere
Strukturaussagen oder 3D-Rekonstitutionsmethoden. Die Methode der 3D-Rekonstitution aus

75
Einzelbildern wird ständig verbessert, höher auflösende Elektronenmikroskope und
automatisierte Bildbearbeitung werden möglicherweise schon in naher Zukunft bessere
Resultate auch mit kleinen Proteinen ermöglichen.
Aus dem Experiment lassen sich aber dennoch Strukturinformationen gewinnen. Man kann
bei einzelnen Partikeln eine Kleeblatt-ähnliche Form, also eine dreizählige Symmetrieachse
ausmachen. Bisherige Strukturmodelle gehen von einem Dimer (mit gemischter α-Helix/β-
Faltblattstruktur(Pohlmeyer, 1997a)) oder von einem Tetramer (reiner β-Faltblattkanal mit
assoziierten Helices(Steinkamp, 2000)) aus. Beides macht eine dreizählige Symmetrieachse
unmöglich. Der Partikeldurchmesser von 5 nm würde zu einem Trimer passen, betrachtet man
das durchschnittliche Volumen von Proteinen entsprechender Größe (etwa 50 kDa).
Die bisher diskutierten spektroskopischen Messungen erlauben keine Aussage über die
Oligomerisierung von OEP16. Es ist denkbar, daß einer der kinetischen Prozesse aus den
zeitaufgelösten Messungen einer Oligomerisierung von OEP16-Monomeren entspricht. Auch
die Kalorimetrie zeigte keinen separaten Monomerisierungs-Prozeß, durch den sich auf die
Enthalpie der Oligomer-Bildung hätte schließen lassen. Möglicherweise geschehen
Denaturierung und Aggregation in der Hitze ohne vorherigen Zerfall in Monomere. Um das
Problem des Oligomerisierungszustandes von OEP16 zu lösen, wurde versucht, das Protein zu
kristallisieren und seine Struktur mittels Röntgenstrukturanalyse zu bestimmen.
Kristallisation
Die Kristallisation und anschließende Röntgenstrukturanalyse ist nach wie vor die effektivste
Methode, um zu hochauflösenden Proteinstrukturen zu gelangen. Inzwischen gibt es
allerdings eine ganze Reihe von Strukturen, die mittels Elektronenmikroskopie und –
kristallographie sowie mit Kernspinresonanzmethoden (NMR) gelöst wurden. Hauptnachteil
der klassischen Kristallisationsmethoden ist der enorme Zeit- und Materialaufwand.
Die Kristallisation von Membranproteinen folgt im Prinzip denselben Regeln wie die löslicher
Proteine. Die Kristallisation erfolgt aus einer übersättigten Lösung, wenn die freie Enthalpie
∆G für diesen Vorgang einen negativen Wert annimmt. Die Übersättigung kann sowohl durch
Erhöhung der Proteinkonzentration als auch durch andere Faktoren, die die Löslichkeit des
Proteins beeinflussen, erreicht werden.

76
Detergenzien
Bei der Kristallisation von Membranproteinen wird deren Löslichkeit natürlich maßgeblich
von den verwendeten Detergenzien beeinflußt. Dies geschieht auch durch Wechselwirkung
des Detergenz mit den verschiedenen Salzen und Kristallisationszusätzen. Oft kristallisieren
Membranproteine dadurch, daß die Löslichkeitsgrenze des Detergenz (und nur indirekt die
des darin gelösten Proteins) unterschritten wird. Auf diesem Prinzip beruht auch die
Kristallisation von OEP16.
Erste Kristallisationsexperimente wurden mit OEP16, rekonstituiert in C12E8, durchgeführt.
Um eine möglichst hohe Proteinkonzentration zu erreichen, wurde das gefaltete Protein in
Puffer mit gesättigter Ammoniumsulfatlösung im Verhältnis 1:1 versetzt und dabei
präzipitiert. Das Protein wurde in Wasser wiederaufgenommen, so daß das Endvolumen
einem zehntel des Ausgangsvolumens entsprach. Die erhaltene Lösung wurde abzentrifugiert,
um Aggregate abzutrennen und mit den Pufferlösungen der Kristallisationsansätze versetzt.
Es wurde die Methode der hängenden Tropfen genutzt. Hierbei wird ein kleiner Tropfen der
Proteinlösung mit Puffer hängend an einem Deckglas angebracht, der mittels Dampfdiffusion
mit einem Pufferreservoir in Kontakt steht. Die Salzkonzentration im Reservoir ist höher als
im Tropfen, so daß Salz- und Proteinkonzentration während des Diffusionsprozesses im
Tropfen zunehmen. Hierbei wird idealerweise eine Übersättigung der Proteinlösung erreicht
und so die Bildung von Kristallen ermöglicht. Variiert werden die Parameter pH, Salztyp und
Salzkonzentration, die Konzentration von Präzipitationsadditiven wie Polyethylenglykol oder
hydrophoben Alkoholen und – bei Membranproteinen – potentiell auch das Detergenz und
dessen Konzentration.
Für OEP16 in C12E8 zeigten sich folgende Änderungen der Löslichkeit in Abhängigkeit von
physikochemischen Parametern:
pH
Der pH-Wert beeinflußt direkt die Oberflächenladung des Proteins. Saure und basische
Aminosäurereste können protoniert oder deprotoniert werden. Häufig ist die Löslichkeit eines
Proteins am geringsten im pH-Bereich um seinen isoelektrischen Punkt. Hier hat das Protein

77
nach außen keine Nettoladung mehr und wandert entsprechend nicht mehr im elektrischen
Feld.
OEP16 hat einen berechneten isoelektrischen Punkt von 9, seine Löslichkeit sollte mangels
eigener Nettoladung bei einem pH um etwa 9 am geringsten sein. Bei den
Kristallisationsexperimenten konnte Präzipitation und die Bildung von Mikrokristallen bei
allen untersuchten pH-Werten im Bereich von 4,5 bis 9,5 beobachtet werden. Dabei zeigte
sich jedoch wie erwartet, daß die Präzipitation bei höheren pH-Werten bei niedrigeren
Fällungsmittelkonzentrationen erfolgt.
Temperatur
Wie praktisch bei allen Lösungen hängt die maximale Löslichkeit von Proteinen auch von der
Temperatur ab. Es läßt sich nicht sicher vorhersagen, ob ein Protein bei höheren
Temperaturen besser oder schlechter löslich ist als bei niedrigen. Da auch die meisten
Kristallisationszusätze abhängig von der Temperatur das Phasendiagramm des Proteins
unterschiedlich beeinflussen, werden in der Praxis meist Kristallisationsansätze parallel bei
verschiedenen Temperaturen gemacht, um empirische Daten über das Temperaturverhalten zu
bekommen.
OEP16 wurde bei verschiedenen Temperaturen kristallisiert. Zur Verfügung standen ein
Temperierschrank bei 18°C und ein Kühlraum mit 4°C Innentemperatur. Im allgemeinen
waren bei 4°C Phasentrennung und Proteinpräzipitation deutlich ausgeprägter. Die
Zitratansätze (siehe später), die doppelbrechende OEP16-Kristalle lieferten, wurden ebenfalls
bei den verscheidenen Temperaturen angesetzt. Die deutlich besseren Kristalle wurden bei
18°C erhalten. Möglicherweise ging die Phasentrennung bei 4°C so schnell, daß eine
geordnete Kristallisation des Proteins dort verhindert wurde.
Präzipitationszusätze
So genannte Kristallisationszusätze sind zumeist organische Moleküle oder Lösungsmittel,
die Wasser binden oder ordnen (hydrophober Effekt). Dem Protein steht weniger Wasser zur
Verfügung, seine Löslichkeit wird herabgesetzt. Typische Kristallisationszusätze dieser Art
sind mehrwertige Alkohole wie Glyzerin oder MPD (2-Methyl-2,4-Pentandiol) oder
Polyethylenglykole.

78
In Gegenwart von Polyethylenglykolen kurzer Kettenlänge (z.B. PEG 400) bildeten sich in
den OEP16-Ansätzen zwei nicht mischbare flüssige Phasen aus, während Zusatz von sehr
langkettige PEGs (ab etwa PEG 6000) meist zu amorpher Präzipitation des Proteins führten.
PEGs mittlerer Kettenlänge verursachten im allgemeinen Beides, d.h. Phasentrennung bei
gleichzeitigem amorphem Niederschlag. Die Löslichkeit des Proteins nahm mit steigendem
pH auch hier ab. Dennoch konnte in keinem Fall mit PEG Kristallwachstum induziert werden.
Zusatz von Alkoholen wie MPD (2-Methyl-2,4-Paentandiol) verursachten eine Trübung der
Proteinlösung bei 4°C, bei 18°C einen amorphen Niederschlag. Es ist anzunehmen, daß die
Trübung von einer Denaturierung und anschließenden Aggregation der Proteinmoleküle
herrührt.
Ionenstärke
Die Ionenstärke einer Lösung beeinflußt die Löslichkeit von Proteinen maßgeblich. Ionen
wechselwirken mit geladenen und polaren Gruppen auf der Oberfläche des Proteins.
Typischerweise haben Proteine ihre höchste Löslichkeit bei mittleren Ionenstärken. Bei hohen
Ionenstärken konkurrieren die Ionen mit dem Protein um das verfügbare Wasser zur
Hydratation. Hierbei spielt nicht nur die absolute Ionenstärke, sondern auch der Typ des Ions
eine große Rolle, auch weil entstehende Kristallkontakte durch entsprechende Ionen erst
vermittelt werden können. Bei niedrigen Ionenstärken hingegen werden Gegenionen der
geladenen Aminosäurereste im Protein abgelöst, die durch sie vermittelte Abschirmung
entfällt und das Protein kann durch direkte Kontakte aggregieren bzw. kristallisieren.
Komplettes Entfernen aller ionischen Pufferkomponenten durch Dialyse gegen Wasser führte
nicht zu einer ausreichenden Verringerung der Löslichkeit von OEP16, eingesetzt in einer
Konzentration von ca. 1 mg/ml. Als Kristallisiationsmethode kommt diese Methode also nur
bei noch höheren (bei OEP16 z.Z. nicht erreichbaren) Proteinkonzentrationen in Betracht.
Starkes Anheben der Ionenstärke hatte im Gegensatz dazu dramatische Effekte auf die
Löslichkeit von OEP16. Die Löslichkeit ist unter den beobachteten Bedingungen generell bei
4°C niedriger als bei 18°C. Mikrokristalle bildeten sich in Gegenwart verschiedener Salze,
wobei die Art des Salzes ebenso wichtig ist wie seine Konzentration. Es konnte gezeigt
werden, daß der Einfluß verschiedener Anionen auf die Löslichkeit des Proteins deutlich
ausgeprägter als der der verwendeten Kationen ist. Am schwächsten ist der Einfluß von
Chlorid, stärker der von Phosphat und Sulfat-Ionen. Am deutlichsten senkte Citrat die

79
Löslichkeit von OEP16. Kleine Kristalle von OEP16 konnten mit Citrat in einem pH-Bereich
von 6,5 bis 9,5 beobachtet werden, wobei die Größe der Kristalle mit steigendem pH zunahm.
Betrachtete man die Kristallisationsansätze regelmäßig, so konnte man feststellen, daß nach
etwa 48 Stunden eine Phasentrennung eintritt, bei der sich zähe, ölige Tropfen in der
wäßrigen Lösung ausbildeten, die vermutlich in erster Linie aus dem Detergenz C12E8
bestehen. Manche dieser Tropfen wandelten sich dann im Laufe von Tagen langsam in
kantige Kristalle um. Citratkonzentrationen unter 1M führten nicht zu Phasentrennung bei
4°C. Bei 18°C genügte zur Phasentrennung eine Konzentration von 800 mM Citrat.
Die erhaltenen Kristalle wurden fotografiert, zum Teil röntgenkristallographisch untersucht
und mittels SDS-Gelelektrophorese analysiert.
Abbildung 27:
Gelelektrophoretische Analyse der OEP16-Kristalle.
Rechts sind die Banden der Markerproteine angedeutet.
Spur A enthielt den Überstand der Kristallisationsansätze.
Spur B enthält die plättchenförmigen Kristalle,
aufgenommen in SDS-Probenpuffer. Spur C enthält die
Tröpfchen (siehe auch Abbildung 25), soweit sich diese
vom Überstand trennen ließen.
Im Gel sieht man neben den Markerproteinen eine Lösung aus den Kristallen, dem Überstand
des Ansatzes und der kleinen, nicht polarisierenden öligen Tropfen, jeweils vor dem
Auftragen in SDS-Probenpuffer erhitzt. Nur die Kristalle enthalten nennenswerte Mengen an
OEP16. Die öligen Tropfen wurden beim Erhitzen so zäh, daß sie sich kaum auf das Gel
auftragen ließen. Es ist anzunehmen, daß diese Tropfen aus einer detergenzreichen Phase
bestehen. Der Überstand enthält praktisch kein Protein mehr.

80
Abbildung 28:
Kristallisationsansatz von OEP16 in 1 M
Natriumzitrat.
Bild A zeigt den Ansatz in normalem Licht,
Bild B unter polarisiertem Licht.
Abbildung 28 zeigt OEP16-Kristalle. Sie wurden mit 1 M Natriumcitrat und 100 mM
CAPSO-Puffer bei pH 9,5 in C12E8 gezüchtet. Oben wurden die Kristalle unter normalem
Licht fotografiert, unten unter polarisiertem Licht. Deutlich zu sehen ist, daß die kantigen

81
Kristalle die Ebene des polarisierten Lichts drehen können und somit leuchten, während die
kleinen, „öligen“ Tropfen das nicht tun. Die Kristalle sind sehr flach und haben eine
Abmessung von 100-200 µm Länge, 50-100 µm Breite und nur etwa 10 µm Dicke.
Die gezeigten Kristalle ergaben nur ein extrem schwaches Beugungsbild. Röntgendiffraktion
konnte bis zu einer Auflösung von 2 nm beobachtet werden. Schon ein zweites Beugungsbild
nach Drehen des Kristalls um 1° zeigte praktisch keine Diffraktion mehr, der Kristall wurde
im Röntgenstrahl also zerstört. Dennoch läßt sich aus den ersten Beugungsbildern die
Gitterdimension der Elementarzelle in einer der Raumrichtungen auf 5,7 nm abschätzen. Dies
paßt sehr gut zu dem elektronenmikroskopisch gefundenen Partikeldurchmesser. Um bessere
Beugungsbilder zu erhalten müssen Bedingungen gefunden werden, unter denen die Kristalle
größer und vor allem stabiler werden. Auch eine geeignete Kryolösung, in der sich die
Kristalle ohne Eisbildung einfrieren lassen, konnte bisher nicht gefunden werden. Getestet
wurden Lösungen mit 10-30% Glyzerin, 1-2 M Sucrose oder Glukose sowie Trehalose. In
jeder dieser Lösungen waren die Kristalle nicht stabil.
Bei weiterführenden Kristallisationsversuchen sollte der Augenmerk vor allem auf die
Detergenzkonzentration gelegt werden. Die durch das Detergenz mit Citrat verursachte
Phasentrennung ermöglicht zwar erst die Bildung der gezeigten Kristalle. Das in den
Kristallen in großer Menge vorhandene Detergenz und möglicherweise auch das
überschüssige Detergenz in Lösung dürften aber auch verantwortlich für die weiche
Konsistenz und die geringe Stabilität der Kristalle gegen typische Kryoreagenzien sein. Eine
Verringerung der Detergenzkonzentration auf Werte, die eine Kristallisation gerade noch
erlauben, dürfte stabilere und möglicherweise auch größere Kristalle liefern.
Theoretische Ansätze zur Strukturaufklärung
In Verlauf dieser Arbeit konnte mit spektroskopischen Methoden gezeigt werden, daß OEP16
eine rein α-helikale Struktur aufweist. Dies widerspricht allen bisherigen Vorstellungen vom
Aufbau dieses integralen Membranproteins(Pohlmeyer, 1997a; Steinkamp, 2000). Auch wenn
elektronenmikroskopische Untersuchungen und Kristallisationsversuche noch nicht zu einer
Auflösung führen, die eine detaillierte Aussage über die Struktur von OEP16 erlaubt, kann
aufgrund theoretischer Überlegungen ein neues Strukturmodell erstellt werden.

82
Auch wenn die genaue Vorhersage einer Proteinstruktur allein aus der Primärsequenz zum
jetzigen Zeitpunkt unmöglich ist, lassen sich aus den bereits verfügbaren Modellen und
Algorithmen viele nützliche Informationen gewinnen. Hilfreich ist es hierbei, mehrere
Sequenzen eines Proteins aus unterschiedlichen Organismen zur Verfügung zu haben.
Multiple Sequence Alignment
Grundvoraussetzung jeder theoretischen Strukturvorhersage ist eine Aminosäuresequenz.
Diese ist für OEP16 aus den Organismen Erbse, Gerste und der Ackerschmalwand vollständig
bekannt. Da theoretische Ansätze an der Sequenz aus Erbse bisher offensichtlich zu falschen
Annahmen führten, sollen im folgenden die Sequenzen aus allen drei Organismen für die
Erstellung eines neuen Modells herangezogen werden. Grundvoraussetzung hierfür ist das
„Multiple Sequence Alignment“, bei dem die Sequenzen verglichen und homologe
Aminosäuren untereinander gestellt werden.
Erbse 1 MPRSSFSGSL SSPKLDVVID MGNPFLNLTV DGFLKIGAVA ATRSVAEDTFGerste 1 MPTAGLAAG- -SNKVDVAID LGNPLLNRTV DGFLKIGAVG ACRVVAEDAFArabidopsis 1 MPSSTFSGTV STPKLSVAVD MGNPFLNLTV DAFLKIGAVG VTKSLAEDTY
Homologie 2222222222 2334444444 4444444444 4444444443 2333444444
Erbse 51 HIIRKGSISS NDFEKSLKKM CKEGAYWGAI AGVYVGMEYG VERIRGTRDWGerste 49 DCLHRGDISK RQLEETLKKM CKEGAYWGAV AGVYVGMEYG VERVRGDRDWArabidopsis 51 KAIDKGSLSK STLEHALKKL CKEGVYWGAA GGVYIGTEYG IERIRGSRDW
Homologie 3333333333 3333333555 5555555553 3555555555 5555555555
Erbse 101 KNAMFGGAVT GALVSAASNN KKDKIAVDAI TGAAIATAAE FINYLT--Gerste 99 KNALIGGIAT GALVSAASNN KGNKIAQDAI TGGAIATAVE FINYLT--Arabidopsis 101 KNAMLAGAAT GAVLSAVGKK GKDTIVIDAI LGGALATASQ FVNNHYFY
Homologie 5555555335 5555552222 2222222333 3333333333 333222--
Abbildung 29:
Die Abbildung zeigt die Sequenzen von OEP16 der verschiedenen Pflanzen. Die Zeile
„Homologie“ gibt die Übereinstimmung im betreffenden Bereich an. Augenscheinlich ist die
Sequenzhomologie über den gesamten Bereich hoch. Welche Funktion den am besten
konservierten Bereichen zukommt (Homologie-Werte von 4-5) und warum, soll im Folgenden
geklärt werden.

83
Hydropathie und Vorhersage transmembraner Segmente
Hydrophatieplots der OEP16-Sequenz aus Erbse ließen den Schluß zu, daß OEP16 aus drei
transmembranen Helices und zwei β-Faltblattstrukturen besteht. Ermittelt man die mittleren
Hydrophobizitäten der Aminosäuren aus den bekannten Sequenzen, kann man aus den
Mittelwerten einen Hydrophobizitätsplot erstellen, der ein besseres Bild von potentiellen
transmembranen Segmenten liefert. In diesem Plot überschreitet der Sequenzbereich etwa um
die Aminosäuren 25-43 deutlich den gesetzten Schwellenwert für ein transmembranes
Segment, ein Befund, der sich allein aus der Erbsensequenz, aus der die vermeintlichen
Faltblattstrukturen abgeleitet wurden, nicht ergibt(Steinkamp, 2000). Die drei
transmembranen Segmente in den Sequenzbereichen 70-91, 102-118 und 125-144 bestätigen
sich, zusätzlich tritt ein weiterer, hydrophober Bereich am n-Terminus der Sequenz zu Tage.
Abbildung 30:
Gezeigt ist die mittlere Hydropathie der Aminosäurereste des „multiple sequence alignments“
aus Abbildung 29

84
Was bedeutet das für das Strukturmodell? Da transmembrane (hydrophobe) Sequenzbereiche
nach bisherigen Erkenntnissen ausschließlich aus Helices und in Sonderfällen aus antiparallel
angeordneten β-Faltblättern bestehen können, OEP16 aber ausschließlich helikalen Charakter
hat, sollten die transmembranen Segmente jeweils membranspannende Helix-Strukturen
ausbilden. Um dies zu verifizieren, wurde der Algorithmus TMPred angewandt, der erlaubt,
Sequenzen mit einer Datenbank bekannter Helix-Strukturen zu vergleichen. Die Ergebnisse
dieser Datenbankabfrage sind in Abbildung 29 durch Unterstreichung der entsprechenden
Sequenzbereiche dargestellt. Demnach besteht OEP16 aus mindestens vier transmembranen
Helices in den Bereichen 25-43, 70-91, 102-118 und 125-144, von hier an Helix I-IV genannt.
Der hydrophobe Bereich im n-Terminus ergibt nur für die Arabidopsis-Sequenz eine positive
Vorhersage (Aminosäuren 1-19). Vermutlich handelt es sich hier um den Signalbereich, der in
Kontakt mit der äußeren Chloroplastenmembran beim Proteinimport eine Helix ausbildet und
damit das Einfädeln in die Lipiddoppelschicht initiiert(Pinnaduwage, 1996). Gegen ein echtes
transmembranes Segment in diesem Bereich spricht vor allem der experimentelle Befund, daß
sich von OEP16 in nativen Erbsenmembranen durch Proteasebehandlung ein etwa 1 kDa
großes Stück (etwa 10 Aminosäuren) abspalten läßt(Pohlmeyer, 1997b). Dies kann nur am n-
Terminus geschehen, da der c-Terminus nach allen bisherigen Ergebnissen in die Membran
eingebettet sein muß bzw. kaum aus ihr herausragt.
Die bereits postulierten drei transmembranen Helices in den Sequenzbereichen 70-91, 102-
118 und 125-144 bestätigen sich. Helices III und IV können mittels Mutagenese entfernt
werden, ohne die Kanalaktivität zu beeinflussen, ebenso wie der Signalbereich (Reste 1-20).
Helix II hingegen wurde lange als entscheidend für die Kanalaktivität betrachtet. Sie hat
große Ähnlichkeiten mit Helices aus vielen Aminosäuretransportproteinen aus Bakterien und
Säugetieren. Mit diesen gemeinsam hat sie vor allem einen Bereich von fünf jeweils durch
drei Aminosäuren getrennten Gylcinresten(Pohlmeyer, 1997b). Auch enthält sie das einzige
Cystein der Sequenz (Cys71), das unter oxidativen Bedingungen den Kanal zu schließen
vermag – möglicherweise über eine Dimerisierung von OEP16-Untereinheiten. Mutagenese
an hydrophilen Resten dieser Helix änderten jedoch nichts an der Kanalaktivität, lediglich ein
Austausch des Cysteins gegen einen Serinrest verhinderte die oxidative Zerstörung der
Kanalfunktion. Es stellte sich sogar heraus, daß die Helix komplett mittels Mutagenese durch
die ebenfalls sehr lange Helix 4 ersetzt werden kann, ohne die Kanalaktivität zu verändern.
Damit dürfte ihr lediglich eine strukturelle Bedeutung für den Kanal zukommen, die
Selektivität muß von einem anderen Sequenzbereich gesteuert werden.

85
Für den Sequenzbereich 25-43 ist das Ergebnis von TMPred nicht eindeutig. Für die
Gerstesequenz wurde hier kein transmembraner Bereich vorhergesagt. Dies liegt vermutlich
an der Häufung geladener und polarer Aminosäuren in diesem Bereich, der in der
Gerstensequenz noch stärker ausgeprägt ist als in den anderen beiden Sequenzen. Diese
polaren Reste dürften auch für die niedrige Hydropathie der Erbsensequenz verantwortlich
sein, die dort auf transmembrane β-Faltblätter hindeutete. Tatsächlich dürfte es sich um eine
amphiphile Helix handeln, die nur zum Teil in Kontakt mit der hydrophoben Lipidmembran
steht, während die hydrophilen Reste das Innere (oder Teile des Inneren) der Kanalstruktur
bilden, die den Aminosäuretransport von OEP16 ermöglichen. Bei 3,6 Aminosäuren pro
Helixwindung kann auf diese Weise jeder dritte bis vierte Aminosäurerest polar sein – und
damit die Helix den klassischen Analysemethoden entgehen.
Hydrophobe Cluster-Analyse
Die Hydrophobe Cluster-Analyse (HCA) erlaubt, sich ein zweidimensionales Bild der
dreidimensionalen Hydropathie-Verhältnisse in einem Protein zu machen. Eine Aminosäure
in einer Helix, zum Beispiel, ist nicht nur von ihren „Sequenznachbarn“ umgeben, sondern
auch von Aminosäuren, die etwa 3-4 Positionen vor oder hinter ihr in der Sequenz angeordnet
sind. Durch geeignete zweidimensionale Darstellung der Sequenz können solche
Nachbarschaftsverhältnisse aufgezeigt werden. Hydrophobe Bereiche werden markiert.
Ergeben sich großflächige hydrophobe Bereiche, handelt es sich mit hoher
Wahrscheinlichkeit um transmembrane Helices. Auch transmembrane β-Faltblätter können
mit dieser Methode sichtbar gemacht werden. Sie zeichnen sich durch ein Alternieren von
hydrophilen und hydrophoben Aminosäureresten aus und werden durch Zickzack-Linien im
HCA-Plot dargestellt.

86
Abbildung 31:
Hydrophobe Cluster-Analyse von OEP16. In der Gerstesequenz wurden zur besseren
Veranschaulichung bzw. Vergleichbarkeit die beiden Prolin-Reste 10 und 11 eingeführt (Rot
markiert). Die Gerstesequenz ist in diesem Bereich zwei Reste kürzer, siehe auch Abbildung
29).
Gezeigt sind die HCA-Plots für die Sequenzen aus Ackerschmalwand, Gerste und Erbse.
Grau unterlegt wurden die Bereiche, die laut TMPred transmembrane Helices darstellen.
Hydrophobe Bereiche sind umrahmt. Prolinreste sind durch einen Stern, Glycinreste durch
eine Raute und Serin- und Threoninreste durch ein Quadrat (mit bzw. ohne Punkt darin)
gekennzeichnet. Diese besondere Markierung hebt ebenso wie die Hervorhebung der
Cysteinreste die besonderen Struktureigenschaften dieser Aminosäuren hervor. Während
Prolin oft am Beginn oder Ende einer Helix steht, ermöglicht das kleine Glycin Knicke in
Helix- und Faltblattstrukturen. Serin- und Threoninreste können durch
Wasserstoffbrückenbindungen ihrer Hydroxylreste mit dem Proteinrückrat unter Umständen
ihren hydrophilen Charakter innerhalb einer Membran verbergen – wiederum ist ein Knick in
einer α-Helix die Folge.

87
Klar zu sehen ist eine Häufung hydrophober Cluster in den vorhergesagten transmembranen
Segmenten. Putative Faltblattstrukturen sind kurz und nicht in Kernbereichen angeordnet, die
als transmembrane Helix in Frage kommen. Nur ein kurzer Faltblattbereich am Ende von
Helix zwei wird in allen drei Sequenzen sichtbar – in einem Bereich von fünf Aminosäuren,
zu kurz für ein transmembranes Strukturelement. Schön zu sehen in dieser Darstellung ist der
Bereich von fünf nebeneinander liegenden Gylcinresten in Helix II, dem vermutlich eine
wichtige Rolle bei der Kanalbildung zukommt (möglicherweise bei der Oligomerisierung).
Auffällig sind auch die praktisch identischen Hydrophobizitätsmuster in Helix I, was für ihre
besondere Rolle für die Kanalselektivität spricht, und die auffällige Übereinstimmung der
Muster im Loopbereich zwischen den ersten beiden Helices. Für viele Kanal- und
Transportproteine wurde beschrieben, daß Substraterkennung und –selektivität durch Loops
vermittelt wird(Slotboom et al., 2001), die zum Teil in die Membran hineinragen. Es ist
denkbar, daß dies auch bei OEP16 der Fall ist.
Topologie
Zusammengenommen ergeben die genannten theoretischen Untersuchungen folgendes Bild:

88
AN
GG F
M
AT
V
VL A
G
SA
AS
LF
VT L
N
DF
G
GI K
L
AA
VA
DL
V
CM
AG E
K
YG
W
GA I
A
VV
Y
YE M
G
G
AI
IA D
V
TA
G
TA I
A
AE
A
YN I
F
L
G
IT
K
R
DG
I
E
K
D
FE
K
V
R
NK
R
NK
W
I
RT
S
R
ND F
IA
H
S
V L
S
VG I
SS
D
S
P
R
ND
LF GS
K
P
M
E
T
P
S S
S
S
K
KK
II III IVI
C
M
N
T
Abbildung 32:
Topologiemodell von OEP16
Nach Proteolyseexperimenten ist der n-Terminus von OEP16 im Chloroplsten dem Cytosol
zugewandt(Pohlmeyer, 1997b). Das OEP16-Monomer bildet vier transmembrane Helices aus,
wobei Helix I vermutlich die Reste enthält, die die Selektivität des Aminosäurekanals
bestimmen. Die Helix beginnt nach einem Prolinrest. Sie enthält (gezeigt für die Erbsen-
Sequenz) die hydrophilen Aminosäuren Asparagin 28, Aspartat 32 und Lysin 36. Besonders
die beiden geladenen Reste Asp32 und Lys36 dürften für zukünftige Mutageneseexperimente
ein interessantes Ziel darstellen, da sie aus thermodynamischen Gründen unbedingt in das
Innere des Kanals ragen müssen. Helix II scheint ein gemeinsames Strukturmotiv vieler
Aminosäuretransporter darzustellen und hat offensichtlich für den Kanal vor allem eine
strukturgebende Bedeutung. Ob das erwähnte Cystein Cys71 in vivo regulatorisch für die
Kanalaktivität wirkt, bleibt eine interessante Fragestellung für weitere Untersuchungen. Der
„Loop“ zwischen den Helices I und II ist in Gerste möglicherweise durch eine Disulfidbrücke
(Cys40-Cys50) stabilisiert. Helices III und IV haben keine Bedeutung für die Bildung des
Kanals. Möglicherweise sind sie an der Oligomerisierung von OEP16-Monomeren beteiligt.

89
OEP16 muß als intakter Kanal ein Oligomer bilden, da zwei α-Helices für sich genommen
noch keinen Kanal bilden können.

90
Zusammenfassung der Ergebnisse
In der vorliegenden Arbeit wurde die biophysikalischen Eigenschaften eines kleinen
Kanalproteins aus der äußeren Chloroplastenmembran der Erbse, genannt OEP16 („outer
envelope Protein 16 kDa“), untersucht und ein neues Strukturmodell entwickelt, auf dessen
Basis die Funktion des Proteins mit molekularbiologischen Methoden nun genauer erforscht
werden kann.
OEP16 ist ein Membranprotein mit ungewöhnlichen Eigenschaften. Als Protein der äußeren
Chloroplastenmembran insertiert es selbständig in geeignete Lipiddoppellschichten und in
Detergenzmizellen. Bei vielen eukaryontischen Membranproteinen kommt eine bakterielle
Überexpression zur Gewinnung großer Proteinmengen nicht in Frage, da zumeist ein
korrekter Einbau in eine Membran mangels der komplexen eukaryontischen
Faltungsmaschinerie in diesen Systemen nicht gegeben ist. OEP16 hingegen erlaubt es,
aufgrund seiner selbsttätigen Renaturierung das Protein ohne Rücksicht auf die native
Struktur in E.coli überzuexprimieren und erst anschließend rückzufalten.
Es konnte gezeigt werden, daß man große Mengen korrekt gefalteten Proteins erhalten kann,
indem man in chaotropen Puffersystemen aufgereinigtes Protein durch einen
Verdünnungsschritt in einen detergenzhaltigen Puffer überführt. Der Nachweis, daß sich
OEP16 in Detergenzmizellen in eine native Form faltet, ist schwierig, da die Funktion des
Proteins, seine Kanalaktivität, nur in einem Membransystem meßbar ist. Durch den Vergleich
von Fluoreszenz- und CD-Spektren von detergenzhaltigen Proteinlösungen und dem in
Liposomen eingebautem Protein (mit nachgewiesener Funktionalität) konnte gezeigt werden,
daß die Struktur von OEP16 in den auf unterschiedliche Weise rekonstituierten Proben
übereinstimmte. Zeitaufgelöste Messungen mit denselben spektroskopischen Methoden
lieferten Daten über den Verlauf und die Geschwindigkeit der Proteinfaltung bei dieser
Renaturierungsmethode, einschließlich der Aktivierungsenergien für den Prozeß.
Kalorimetrische Messungen (DSC) erlaubten eine Abschätzung der Faltungsenthalpie. Durch
hochauflösende CD-Messungen und Infrarotspektroskopie wurde schließlich am in vitro
gefalteten Protein gezeigt, daß es sich bei OPE16 entgegen bisheriger Voraussagen um ein
rein α-helikales Membranprotein handelt. Dieser Befund konnte durch theoretische Methoden
der Strukturbestimmung bestätigt werden. Ein neues Strukturmodell wurde erstellt. Erste
vielversprechende Ansätze der Proteinkristallisation und der Elektronenmikroskopie zeigen,
daß eine detaillierte Struktur von OEP16 in absehbarer Zeit erhältlich sein könnte. Dies wäre

91
weltweit die erste hochaufgelöste Struktur eines im Reagenzglas gefalteten Membranproteins.
In einer Zeit, in der Proteomics, also das Überexprimieren und Analysieren von Proteinen, die
Arbeit der größtenteils abgeschlossenen Sequenzierungprojekte fortführen sollen, konnte mit
OEP16 ein Modellsystem für die detaillierte Struktur- und Funktionsanalyse von
Membranproteinen etabliert werden.

92
Literatur
Abrecht, H., Goormaghtigh, E., Ruysschaert, J.-M., and Homble, F. (2000) Structure and
Orientation of Two Voltage-dependent Anion-selective Channel Isoforms: An
Attenuated Total Reflection Fourier-Trnasform Infrared Spectroscopy Study. Journal
of Biological Chemistry, 275, 40992-40999.
Allen, J., Feher, G., Yeates, T., Komiya, H., and Rees, D. (1987) Structure of the reaction
center from Rhodobacter sphaeroides R-26: The protein subunits. Proceedings of the
National Academy of Science, USA, 84, 6162-6166.
Arabidopsis Genome Initiative. (2000) Analysis of the genome sequence of the flowering
plant Arabidopsis thaliana. Nature, 408, 796-815.
Baldi, P., Grossi, M., Pecchioni, N., Vale G., and Cattivelli, L. (1999) High expression level
of a gene coding for a chloroplastic amino acid selective channel protein is correlated
to cold acclimation in cereals. Plant Molecular Biology, 41, 233-243.
Bechinger, B. (2000) Understanding peptide interactions with the lipid bilayer: a guide to
membrane protein engineering. Current Opinion in Chemical Biology, 4, 639-644.
Benning, C. (1998) Biosynthesis and Funktion of the Sulfolipid
Sulfoquinovosyldiacylglycerol. Annual Reviews of Plant Physiology and Plant
Molecular Biology, 49, 53-75.
Blankenship, R.E., and Hartman, H. (1998) The origin and evolution of oxygenic
photosynthesis. Trends in Biochemical Science, 23, 94-97.
Block, M.A., Dorne, A.-J., Joyard, J., and Douce, R. (1983) Preparation and Characterization
of Membrane Fractions Enriched in Outer and Inner Envelope Membranes from
Spinach Chloroplasts. Journal of Bilogical Cemistry, 258, 13281-13286.
Bölter, B., Soll, J., Hill, K., Hemmler, R., Wagner, R. (1999) A rectifying ATP-regulated
solute channel in the chloroplastic outer envelope from pea. EMBO Journal, 18, 5505-
5516.
Booth, P.J., Curran, A.R. (1999) Membrane protein folding. Current Opinion in Structural
Biology, 9, 115-121.
Brill, L.M., Nunn, R.S., Kahn, T.W., Yeager, M., and Beachy, R.N. (2000) Recombinant
tobacco mosaic virus movement protein is an RNA-binding, a-helical membrane
protein. Proceedings of the National Academy of Science, USA, 97, 7112-7117.
Bruce, B.D. (2000) Chloroplast transit peptides: structure, function and evolution. Trends in
Cell Biology, 10, 440-447.

93
Buchanan, S.K. (1999) ß-Barrel proteins from bacterial outer membranes: structure, function
and refolding. Current Opinion in Structural Biology, 9, 455-461.
Burstein, E.A., Vedenkina, N.S., Ivkova, M.N. (1973) Fluorescence and the location of
tryptophan residues in protein molecules. Photochemistry and Photobiology, 18, 263-
279.
Callahan, M.A., Xiong, L.-W., and Caughey, B. (2001) Reversibility of Scrapie-associated
Prion Protein Aggregation. Journal of Biological Chemistry, 276, 28022–28028.
Callebaut, I., Labesse, G., Durand, P., Poupon, A., Canard, L., Chomilier, J., Henrissat B., and
Mornon, J.P. (1997) Deciphering protein sequence information through hydrophobic
cluster analysis (HCA): current status and perspectives. Cellular and Molecular Life
Science, 53, 621–645.
Cavalier-Smith, T. (2000) Membrane heredity and early chloroplast evolution. Trends in
Plant Science, 5, 174-182.
Chen, Y., BArkley, M.D. (1998) Toward Understanding Tryptophan Fluorescence in Proteins.
Biochemistry, 37, 9976-9982.
Cline, K., and Henry, R. (1996) Import and Routing of Nucleus-encoded Chloroplast Proteins.
Annual Reviews of Cell and Developmental Biology, 12, 1-26.
Conway, K.A., Harper, J.D., and Lansbury, P.T.Jr. (2000) Fibrils Formed in Vitro from R-
Synuclein and Two Mutant Forms Linked to Parkinson’s Disease are Typical
Amyloid. Biochemistry, 39, 2552-2563.
Cowan, S.W., Rosenbusch, J.P. (1994) Folding Pattern Diversity of Integral Membrane
Proteins. Science, 264, 914-916.
Deisenhofer, J., Epp, O., Miki, K., Huber, R., and Michel, H. (1985) Structure of the protein
subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3 A
resolution. Nature, 318, 618-624.
Fischer, K., Weber, A., Brink, S., Arbinger, B., Schünemann, D., Borchert, S., Heldt, H.W.,
Popp, B., Benz, R., Link, T.A., Eckerskorn, C., and Flügge, U.-I. (1994) Porins from
Plants. Journal of Biological Chemistry, 269, 25754-25760.
Flügge, U.I. (1998) Metabolite transporters in plastids. Current Opinion in Plant Biology, 1,
201-206.
Flügge, U.I. (2000) Transport in and out of plastids: does the outer envelope membrane
control the flow? Trends in Plant Science, 5, 135-137.
Froehlich, J.E., Benning, C., and Dörmann, P. (2001) The Digalactosyldiacylglycerol
(DGDG) Synthase DGD1 Is Inserted into the Outer Envelope Membrane of

94
Chloroplasts in a Manner Independent of the General Import Pathway and Does Not
Depend on Direct Interaction with Monogalactosyldiacylglycerol Synthase for DGDG
Biosynthesis. Journal of Biological Chemistry, 276, 31806–31812.
Griegorieff, N., Ceska, T.A., Downing, K.H., Baldwin, J.M., and Henderson, R. (1996)
Electron-crystallographic refinement of the structure of bacteriorhodopsin. Journal of
Molecular Biology, 259, 393-421.
Haltia, T., and Freire, E. (1995) Forces and factors that contribute to the structural stability of
membrane proteins. Biochimica et Biophysica Acta, 1228, 1-27.
Harris, E.H., Boynton, J.E., and Gillham, N.W. (1994) Chloroplast Ribosomes and Protein
Synthesis. Microbiological Reviews, 58, 700-754.
Heiber, T., Steinkamp, T., Hinnah, S., Schwarz, M., Flügge, U.I., Weber, A., Wagner, R.
(1995) Ion Channels in the Chloroplast Envelope Membrane. Biochemistry, 34,
15906-15917.
Heins, L., Collinson, I., Soll, J. (1998) The protein translocation apparatus of chloroplast
envelopes. Trends in Plant Science, 3, 56-61.
Hirsch, A., Breed, J., Saxena, K., Richter, O.M.H., Ludwig, B., Diedrichs, K., Welte, W.
(1997) The structure of porin from Paracoccus denitrificans at 3.1 A resolution. FEBS
letters, 404, 208-210.
Hofmann, K., and Stoffel,W. (1993) TMbase - A database of membrane spanning proteins
segments. Biological Chemistry Hoppe-Seyler, 374, 166.
Iwata, S., Ostermeier, C., Ludwig, B., and Michel, H. (1995) Structure at 2.8 A resolution of
cytochrome c oxidase from paracoccus denitrificans. Nature, 376, 660-669.
Jordan, P., Fromme, P., Witt, H.T., Klukas, O., Saenger, W., and Krauss, N. (2001) Three-
dimensional structure of cyanobacterial photosystem I at 2.5 angstrom resolution.
Nature, 411, 909-917.
Joyard, J., Block, M.A., and Douce, R. (1991) Molecular aspects of plastid envelope
biochemistry. European Journal of Biochemistry, 199, 489-509.
Joyard, J., Teyssier, E., Miege, C., Berny-Seigneurin, D., Marechal, E., Block, M.A., Dorne,
A.-J., Rolland, N., Ajlani, G., and Douce, R. (1998) The Biochemical Machinery of
Plastid Envelope Membranes. Plant Physiology, 118, 715-723.
Keegstra, K. (1989) In Briggs, W. (ed.) Photosynthesis. Alan R. Liss, New York, pp. 347-357.
Keegstra, K., Cline, K. (1999) Protein Import and Routing Systems of Chloroplasts. Plant
Cell, 11, 557-570.

95
Kerber, B., and Soll, J. (1992) Transfer of a chloroplast-bound precursor protein into the
translocation apparatus is impaired after phospholipase C treatment. FEBS Letters,
306, 71-74.
Klebba, P.E., Newton, S.M.C. (1998) Mechanisms of solute transport through outer
membrane porins: burning down the house. Current Opinion in Microbiology, 1, 238-
248.
Koebnick, R. (1996) In vivo membrane assembly of split variants of the E.coli outer
membrane protein OmpA. EMBO Journal, 15, 3529-3537.
Koike, H., Yoshio, M., Kashino, Y., and Satoh, K. (1998) Polypeptide composition of
envelopes of spinach chloroplasts: Two major proteins occupy 90% of outer envelope
membranes. Plant and Cell Physiology, 39, 526-532.
Kreusch, A., Schulz, G.E. (1994) Refined Structure of the Porin from Rheudopseudomonas
blastica. Journal of Molecular Biology, 243, 891-905.
Kühlbrandt, W., Wang, D.N., and Fujioshi, Y. (1994) Atomic model of plant kght harvesting
complex by electron crystallography. Nature, 367, 614-621.
Kyte, J., and Doolittle, R.F. (1982) A simple method for displaying the hydropatic character
of a protein. Journal of Molecular Biology, 157, 105-132.
Lam, H.M., Coschingano, K.T., Oliveira, I.C., Melo-Oliveira, R., Coruzzi, G.M. (1996) The
Molecular Genetics of Nitrogen Assimilation into Amino Acids in Higher Plants.
Annual Reviews of Plant Physiology and Plant Molecular Biology, 47, 569-593.
Lemeshko, S.V., and Lemeshko, V.V. (2000) Metabolically Derived Potential on the Outer
Membrane of Mitochondria: A Computational Model. Biophysical Journal, 79, 2785-
2800.
Leon, P., Arroyo, A. (1998) Nuclear Control of Plastid and Mitochondrial Development in
Higher Plants. Annual Reviews of Plant Physiology and Plant Molecular Biology, 49,
453-480.
Li, H.M., Moore, T., Keegstra, K. (1991) Targeting of Proteins to the Outer Envelope
Membrane Uses a Different Pathway tha Transport into Chloroplasts. Plant Cell, 3,
709-717.
Li, H.M., Chen, L.J. (1996) Protein Targeting and Integration Signal for the Chloroplastic
Outer Envelope Membrane. Plant Cell, 8, 2117-2126.
Lin, X., Samir Kaul, Steve Rounsley, Terrance P. Shea, Maria-Ines Benito, Christopher D.
Town, Claire Y. Fujii, Tan et al. (1999) Sequence and analysis of chromosome 2 of
the plant Arabidopsis thaliana. Nature, 402, 761 - 768.

96
Linke, D., Frank, J., Holzwarth, J.F., Soll, J., Boettcher, C., and Fromme, P. (2000) In Vitro
Reconstitution and Biophysical Characterization of OEP16, an Outer Envelope Pore
Protein of Pea Chloroplasts. Biochemistry, 39, 11050-11056.
Marcos, J.C., Fonseca, L.P., Ramalho, M.T., Cabral, J.M.S. (1999) Partial purification of
pennicilin acylase from Escherichia coli in poly(ethylene glycol)-sodium citrate
aqueous two-phase systems. Journal of Chromatography B, 734, 15-22.
Margulis, L. (1996) Archeal-eubacterial mergers in the origin of Ekarya: Phylogenic
classification of life. Proceedings of the National Academy of Science, USA, 93, 1071-
1076.
Mayer, K., C. Schuller, R. Wambutt, G. Murphy, G. Volckaert, T. Pohl, A. Dusterhoft, W.
Stiekema, K.-D. Entian, N. Terryn et al. (1999) Sequence and analysis of chromosome
4 of the plant Arabidopsis thaliana. Nature, 402, 769 - 777.
McDermott, G., Price, S.M., Freer, A.A., Hawthornthwaite-Lawless, A.M., Papiz, M.Z.,
Cogdell, R.J., and Isaacs, N.W. (1995) Crystal structure of an integral membrane light-
harvesting complex from photosynthetic bacteria. Nature, 374, 517-521.
McFadden, G.I. (1999) Endosymbiosis and evolution of the plant cell. Current Opinion in
Plant Biology, 2, 513-519.
Miernyk, J.A. (1999) Protein Folding in the Plant Cell. Plant Physiology, 121, 695-703.
Mitchell, D.J., Tiddy, J.T., Waring, L., Bostok, T., and McDonald, M.P. (1983) Journal of the
Chemical Society, Faraday Transactions, 79, 975-1000.
Morgenstern, B. (1999) DIALIGN 2: improvement of the segment-to-segment approach to
multiple sequence alignment. Bioinformatics, 15, 211-218.
Pinnaduwage, P., and Bruce, P.D. (1996) In vitro interaction between a chloroplast transit
peptide and chloroplast outer envelope lipids is sequence-specific and lipid class-
dependent. Journal of Biological Chemistry, 271, 32907-32915.
Pohlmeyer, K., Soll, J., Steinkamp, T., Hinnah, S., Wagner, R. (1997a) Isolation and
characterization of an amino acid-selective channel protein present in the chloroplastic
outer envelope membrane. Proceedings of the National Academy of Science, USA, 94,
9504-9509.
Pohlmeyer, K. (1997b) Porenbildende Proteine in der äußeren Hüllmembran von
Chloroplasten aus Erbse (Pisum sativum L.). Mathematisch-Natrurwissenschaftliche
Fakultät. Christian-Albrechts-Universität, Kiel, p. 88.
Pohlmeyer, K., Soll, J., Grimm, R., Hill, K., Wagner, R. (1998) A High-Conductance Solute
Channel in the Chloroplastic Outer Envelope from Pea. The Plant Cell, 10, 1207-1216.

97
Poincelot, R.P. (1973) Isolation and Lipid Composition of Spinach Chloroplast Envelope
Membranes. Archives of Biochemistry and Biophysics, 159, 134-142.
Popp, B., Gbauer, S., Fischer, K., Flügge, U.I., Benz, R. (1997) Study of Structure and
Function of Recombinant Pea Root Plastid Porin by Biophysical Methods.
Biochemistry, 36, 2844-2852.
Pyke, K.A. (1999) Plastid Division and Development. Plant Cell, 11, 549-556.
Rassow, J., Dekker, P.J.T., van Wilpe, S., Meijer, M., Soll, J. (1999) Ther Preprotein
Translocase of the Mitochondrial Inner Membrane: Function and Evolution. Journal
of Molecular Biology, 286, 105-120.
Reumann, S., Keegstra, K. (1999) The endosymbiotic origin of the protein import machinery
of chloroplastic envelope membranes. Trends in Plant Science, 4, 302-307.
Ridge, K.D., Lee, S.S.J., and Yao, L.L. (1995) In vivo assembly of rhodopsin from expressed
polypeptide fragments. Proceedings of the National Academy of Science, USA, 92.
Röhl, T., Motzkus, M., Soll, J. (1999) The outer envelope protein OEP24 from pea
chloroplasts can functionally replace the mitochondrial VDAC in yeast. FEBS letters,
460, 491-494.
Salanoubat, M., Lemcke K., Rieger M., Ansorge W., Unseld M., Fartmann B., Valle G.,
Blocker H., Perez-Alonso M., Obermaier B., Delseny M., Boutry M., Grivell L.A.,
Mache R., Puigdomenech P., De Simone V., Choisne N., Artiguenave F, Robert C,
Brottier P, Wincker P, et.al. (2000) Sequence and analysis of chromosome 3 of the
plant Arabidopsis thaliana. Nature, 408, 820-822.
Seedorf, M., Soll, J. (1995) Copper chloride, an inhibitor of protein import into chloroplasts.
FEBS letters, 367, 19-22.
Settles, A.M., Martienssen, R. (1998) Old and new pathways of protein export in chloroplasts
and bacteria. Trends in Cell Biology, 8, 494-501.
Slotboom, D.J., Konings, W.N. and Lolkema, J.S. (2001) Glutamate transporters combine
transporter- and channel-like features. Trends in Biochemical Science, 26, 534-9.
Soll, J., Bölter, B., Wagner, R., Hinnah, S.C. (2000) response: The chloroplast outer envelope:
a molecular sieve? Trends in Plant Science, 5, 137-138.
Steinkamp, T., Hill, K., Hinnah, S.C., Wagner, R., Röhl, T., Pohlmeyer, K., and Soll, J.
(2000) Identification of the Pore-forming Region of the Outer Chloroplast Envelope
Protein OEP16. Journal of Biological Chemistry, 275, 11758–11764.
Stock, D., Leslie, A.G.W., and Walker, J.E. (1999) Molecular architecture of the rotary motor
in ATP synthase. Science, 286, 1700-1705.

98
Tabata, S., Kaneko T, Nakamura Y, Kotani H, Kato T, Asamizu E, Miyajima N, Sasamoto S,
Kimura T, Hosouchi T, Kawashima K, Kohara M, Matsumoto M, Matsuno A, Muraki
A, Nakayama S, Nakazaki N, Naruo K, Okumura S, Shinpo S, Takeuchi C, Wada T,
Watanabe A, Yamada M, et.al. (2000) Sequence and analysis of chromosome 5 of the
plant Arabidopsis thaliana. Nature, 408, 823-826.
Theologis, A., Joseph R. Ecker, Curtis J. Palm, Nancy A. Federspiel, Samir Kaul, Owen
White, Jose Alonso, et al. (2000) Sequence and analysis of chromosome 1 of the plant
Arabidopsis thaliana. Nature, 408, 816 - 820.
Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000) Crystal structure of the
calcium pump of sarcoplasmatic reticulum at 2.6 A resolution. Nature, 405, 647-655.
Tranel, P.J., and Keegstra, K. (1996) A novel, bipartite transit peptide targets OEP75 to the
outer membrane of the chloroplast envelope. Plant Cell, 8, 2093-2104.
Tsuhikara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K.,
Nakashima, R., Yaono, R., and Yoshikawa, S. (1996) The whole structure of the 13-
subunit oxidized bovine heart cytochrome c oxidase at 2.8 A resolution. Science, 272,
1136-1144.
Van't Hof, R., van Klompenburg, W., Pilon, M., Kozubek, A., de Korte-Kool, G., Demel,
R.A., Weisbeek, P.J., and de Kruijff, B. (1993) The transit sequence mediates the
specific interaction of the precursor ferredoxin with chloroplast envelope membrane
lipids. Journal of Biological Chemistry, 268, 4037-4042.
von Heijne, G., and Nishikawa, K. (1991) Chloroplast transit peptides. The perfect random
coil? FEBS Letters, 278, 1-3.
von Heijne, G. (1996) Principles of membrane protein assembly and structure. Progress in
Biophysics and Molecular Biology, 66, 113-139.
Waegemann, K., Eichacker, S., and Soll, J. (1992) Outer envelope membranes from
chloroplasts are isolated as right-side-out vesicles. Planta, 187, 89-94.
White, S.H. (1999) Membrane Protein Folding and Stability: Physical Principles. Annual
Reviews of Biophysics and Biomolecular Structure, 28, 319-365.
Wiese, A., Gröner, F. , Sonnewald, U., Deppner, H., Lerchl, J., Hebbeker, U., Flügge, U.-I.,
and Weber, A. (1999) Spinach hexokinase I is located in the outer envelope membrane
of plastids. FEBS Letters, 461, 13-18.
Winter, R., and Noll, F. (1998) Methoden der Biophysikalischen Chemie.

99
Zhan, H., Oh, K.J., Shin, Y.-K., Hubbel, W.L., and Collier, R.J. (1995) Interaction of the
isolated transmembrane domain of diphteria toxin with membranes. Biochemistry, 34,
4856-4863.
Zouni, A., Witt, H.T., Kern, J., Fromme, P., Krauss, N., Saenger, W., Orth, P. (2001) Crystal
structure of photosystem II from Synechococcus elongatus at 3.8 angstrom resolution.
Nature, 409, 739-743.

100
Danksagung
Ich danke Frau Prof. Dr. Petra Fromme nicht nur für die interessante Aufgabenstellung und
die dafür bereitgestellten finanziellen Mittel aus ihren Projekten, sondern auch insbesondere
für die große Freiheit, die ich beim Berarbeiten des Themas genossen habe. Die Arbeit wurde
hauptsächlich aus Mitteln der DFG (Fördernummer Fr 320/3-1 und 3-2) finanziert.
Herrn Prof. Soll (Kiel, inzwischen München) danke ich für die fruchtbare Zusammenarbeit
bei mehreren Publikationen und ihm und seiner gesamten Arbeitsgruppe für das Produzieren
großer Mengen „inclusion bodies“, ohne die diese Arbeit nicht möglich gewesen wäre. Herrn
Prof. Wagner (Osnabrück) und seinen Mitarbeitern sind die Leitfähigkeitsmessungen an
OEP16-Liposomen zu verdanken.
Herrn Prof. Findenegg (TU Berlin) danke ich für die Bereitschaft, sehr kurzfristig die
Begutachtung meiner Arbeit zu übernehmen.
Uwe Fink, Dr. Joachim Frank, Jan Kern, Dr. Athina Zouni und Dr. Priv.Doz. Petra Fromme
haben mich ständig durch Fachwissen, neue Ideen und hilfreiche Diskussionen unterstützt.
Ein solch produktives und angenehmes Arbeitsumfeld kann nicht genug gewürdigt werden.
Meinen Eltern, Herrn Timm Krause-Plonka sowie Erwin und Anneliese Schäuble danke ich
für moralische und finanzielle Unterstützung während des gesamten Studiums.
Meiner Frau Daniela gilt besonderer Dank für viel Geduld.
Diese Arbeit ist meinem Sohn Lukas gewidmet, der mit knapp fünf Jahren mehr über
Dinosaurier, Autos und Fische weiß als ich.


















