Differentialkalorimetrie (DSC) und ... · Differentialkalorimetrie (DSC) und...
179
Differentialkalorimetrie (DSC) und Differentialthermoanalyse (DTA) bei hohen Drücken Untersuchungen zum Phasenverhalten ausgewählter Triacylglycerine, Flüssigkristalle und Anthrachinonfarbstoffe bis 200 MPa Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften der Fakultät für Chemie der Ruhr-Universität Bochum vorgelegt von Stefan Masberg aus Bottrop Referent: Prof. Dr. G. M. Schneider Korreferent: Prof. Dr. A. Würflinger Bochum, Dezember 1999
Transcript of Differentialkalorimetrie (DSC) und ... · Differentialkalorimetrie (DSC) und...

Differentialkalorimetrie (DSC) und Differentialthermoanalyse (DTA)
bei hohen Drücken
Untersuchungen zum Phasenverhalten ausgewählter
Triacylglycerine, Flüssigkristalle und Anthrachinonfarbstoffe
bis 200 MPa
Dissertationzur Erlangung des Doktorgrades
der Naturwissenschaften
der Fakultät für Chemie
der Ruhr-Universität Bochum
vorgelegt von
Stefan Masberg
aus Bottrop
Referent: Prof. Dr. G. M. Schneider
Korreferent: Prof. Dr. A. Würflinger
Bochum, Dezember 1999

Referent: Prof. Dr. G. M. Schneider, Lehrstuhl für Physikalische Chemie II,
Ruhr-Universität Bochum
Korreferent: Prof. Dr. A. Würflinger, Lehrstuhl für Physikalische Chemie II,
Ruhr-Universität Bochum
Drittprüfer: Prof. Dr. H.-J. Götze, Lehrstuhl für Analytische Chemie,
Ruhr-Universität Bochum
Disputation: 8. Dezember 1999

Meinen Eltern

Inhaltsverzeichnis
I
Inhaltsverzeichnis I
Zusammenfassung V
1 Einleitung 1
1.1 Mesomorphe Übergänge kristalliner Festkörper 1
1.2 Plastische Kristalle 3
1.3 Flüssigkristalle 4
1.3.1 Klassifizierung der Flüssigkristalle 5
1.3.2 Thermotrope calamitische Flüssigkristalle 8
1.3.2.1 Die nematische Phase 9
1.3.2.2 Die cholesterische Phase 12
1.3.2.3 Die smektischen Phasen 13
1.3.3 Polymorphismus der thermotropen Flüssigkristalle 15
1.3.4 Hochdruckuntersuchungen an Flüssigkristallen 16
1.3.5 Identifizierung der flüssigkristallinen Phasen 17
2 Theoretische Grundlagen 19
2.1 Thermodynamische Grundlagen der Kalorimetrie 19
2.2 Klassifikation der Phasenumwandlungen 26
3 Differential Scanning Calorimetry(DSC) 32
3.1 DSC-Meßprinzip 32
3.1.1 Normaldruck-DSC 34
3.1.2 Hochdruck-DSC 36
3.2 Apparativer Aufbau 37
3.2.1 Übersicht über die Meßanordnung 37

Inhaltsverzeichnis
II
3.2.2 Hochdruckautoklav 40
3.2.3 Meßkopf 41
3.2.4 Elektrische Durchführung 45
3.2.5 Meßzellen 48
3.2.6 Temperiermantel 50
3.2.7 Druckerzeugung und -messung 50
3.2.8 Meßwerterfassung 52
3.3 Auswertung von DSC-Peaks 53
3.3.1 Einfluß der Versuchsbedingungen auf die Peakform 54
3.3.2 Festlegung des Umwandlungspunktes 54
3.3.3 Peakfläche und Umwandlungsenthalpie 56
3.3.4 Umwandlungsentropie und –volumen 58
3.4 Meßtechnik 59
3.4.1 Meßmethode 59
3.4.2 Kalibrierung des DSC-Kalorimeters 59
3.4.2.1 Temperaturkalibrierung 60
3.4.2.2 Enthalpiekalibrierung 65
3.4.3 Meßgenauigkeit 68
3.4.3.1 Fehlerabschätzung bei der Temperatur- und Druckmessung 68
3.4.3.2 Fehlerabschätzung bei der Enthalpiebestimmung 69
4 Differentialthermoanalyse (DTA) 71
4.1 Definition und Grundlagen der Differentialthermoanalyse 71
4.2 DTA-Apparatur 76
4.2.1 Aufbau des Autoklavs 76
4.2.2 Druckversorgung 79
4.2.3 Temperaturregelung 79
4.2.4 Meßwerterfassung 80
4.2.5 Meßzellen 80
4.3 Kalibrierung der Thermoelemente 83

Inhaltsverzeichnis
III
5 Untersuchungen zum Phasenverhalten des ausgewählten Triacylglycerins
1,2,3-Tris-eicosanoyloxypropan (Triarachidin, AAA) 86
5.1 Einführung 86
5.2 Chemischer Aufbau und Nomenklatur der Fette 87
5.3 Modifikationen der Triacylglycerine 89
5.4 Umwandlungen zwischen den polymorphen Modifikationen 93
5.5 Meßergebnisse und Diskussion 95
5.5.1 Phasendiagramm 97
5.5.2 Phasenumwandlungsenthalpie, -entropie und -volumen 99
6 Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle 105
6.1 Flüssigkristalle mit lateralen aromatischen Verzweigungen 105
6.1.1 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylester 107
6.1.2 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-cyan-benzylester 109
6.1.3 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-2-cyan-benzylester 110
6.1.4 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester 111
6.2 Phasenverhalten der 1-[4-Alkylbiphenyl]-2-[4-isothiocyanatophenyl]ethane
6-TPEB und 10-TPEB 115
6.2.1 1-[4-Hexylbiphenyl]-2-[4-isothiocyanatophenyl]ethan (6-TPEB) 116
6.2.2 1-[4-Decylbiphenyl]-2-[4-isothiocyanatophenyl]ethan (10-TPEB) 117
7 Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe 126
7.1 Überblick über die Meßsubstanzen 126
7.2 DSC-Messungen bei Normaldruck 128
7.3 DSC- und DTA-Hochdruckmessungen 133
7.3.1 Phasenverhalten von 1,4-Bis-(n-propylamino)-9,10-anthrachinon
(AC03) 133
7.3.1.1 Meßergebnisse und Diskussion 134

Inhaltsverzeichnis
IV
7.3.2 Phasenverhalten von 1,4-Bis-(n-butylamino)-9,10-anthrachinon
(AC04) 139
7.3.2.1 Meßergebnisse und Diskussion 139
7.3.2.2 Abschätzung des gelösten Gasanteils 143
8 Wärmekapazitätsbestimmung 147
8.1 Grundlagen der Wärmestromkalibrierung 147
8.2 Meßergebnisse und Diskussion 149
9 Ausblick 154
10 Anhang 155
A1 Verzeichnis der Abbildungen 155
A2 Verzeichnis der Tabellen 159
A3 Verzeichnis der Lieferfirmen 161
A4 Literaturverzeichnis 162

Zusammenfassung
V
Zusammenfassung
In der vorliegenden Arbeit wurde das Phasenverhalten ausgewählter Triacylglycerine,
Flüssigkristalle und Anthrachinonfarbstoffe mit thermischen Meßverfahren, der Differential-
kalorimetrie (DSC) und der Differentialthermoanalyse (DTA), unter hohen Drücken
untersucht.
Das verwendete Hochtemperatur-DSC-Hochdruckkalorimeter, das nach dem Prinzip der
leistungskompensierenden DSC arbeitet, kann in einem Druckbereich von Normaldruck bis
etwa 200 MPa und in einem Temperaturbereich von 293 K bis etwa 500 K eingesetzt werden.
Die DTA-Hochdruckapparatur ist für Drücke bis 300 MPa ausgelegt, der nutzbare
Temperaturbereich liegt bei 273 K bis 465 K und ist durch das Zellenmaterial (Indium bzw.
Blei) begrenzt.
Als Vertreter der Triacylglycerine wurde das 1,2,3-Tris-eicosanoyloxypropan (Triarachidin,
AAA) gewählt. DTA-Hochdruckuntersuchungen wurden bereits von Ernst [65], Becker [93]
und Wagner [110] vorgenommen. Die in dieser Arbeit mittels DSC gewonnenen
Meßergebnisse wurden mit denen von Ernst verglichen. Aus den erhaltenen Umwandlungs-
temperaturen und -enthalpien wurden Entropie- und Volumenänderungen der Phasen-
übergänge in Abhängigkeit vom Druck entlang der jeweiligen Koexistenzlinie berechnet.
Unter den gewählten experimentellen Bedingungen waren für Triarachidin zwei
Phasenübergänge zu beobachten, wobei die Phasen folgendermaßen durchlaufen wurden:
α→β→l. Für den Übergang α→β ist aus den Thermogrammen deutlich ersichtlich, wie der
zugehörige Peak von endotherm nach exotherm wechselt. Ollivon und Perron [106] nehmen
an, daß es sich bei der Umwandlung nicht um eine reine fest-fest-Umwandlung unter
Reorganisation der Kristallstruktur handelt, sondern daß intermediär eine Schmelze
durchlaufen wird. Demnach vollzieht sich die Umwandlung in zwei Schritten: Schmelzen der
α-Modifikation zur instabilen α-Schmelze, die exotherm zur stabilen β-Modifikation
kristallisiert.
Die verzweigten flüssigkristallinen Substanzen 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-4-nitro-benzylester, -4-cyan-benzylester, -3-cyan-benzylester und -2-
cyan-benzylester wurden von Prof. Dr. W. Weißflog, Universität Halle, zur Verfügung

Zusammenfassung
VI
gestellt. An allen vier Substanzen wurden DSC-Messungen unter Normaldruck
vorgenommen, der -3-cyan-benzylester wurde zusätzlich unter Druck mittels DTA untersucht.
Es zeigt sich, daß die strukturell sehr ähnlichen Substanzen deutliche Unterschiede im
Phasenverhalten besitzen.
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylester zeigt beim Erwärmen die
Phasenabfolge kristallin → smektisch A → isotrop flüssig. Beim ersten Aufheizen konnte
eine weitere, metastabile kristalline Phase beobachtet werden.
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-cyan-benzylester durchläuft ebenfalls die
Phasenabfolge kristallin → smektisch A → isotrop flüssig. Der Austausch der Nitrogruppe
durch die Cyangruppe bewirkt keine grundsätzliche Änderung im Phasenverhalten unter
Normaldruck. Die Phasenumwandlungen treten jedoch bei niedrigeren Temperaturen auf.
Beim 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester tritt die
Phasenabfolge kristallin → nematisch → isotrop flüssig auf. Je nach thermischer
Vorbehandlung weisen die Thermogramme außerdem eine fest-fest-Umwandlung auf, wobei
die metastabile kristalline Phase nach längerem Tempern nicht mehr zu beobachten ist. Das
Phasenverhalten dieser Substanz wurde bis zu einem Druck von 257.5 MPa untersucht.
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-2-cyan-benzylester zeigt unter Normaldruck
beim Aufheizen einen fest-fest-Übergang und das Schmelzen der kristallinen Phase. Beim
Abkühlen aus der isotrop flüssigen Phase wird monotrop eine nematische Phase durchlaufen.
Weiteres Abkühlen führt zur Bildung einer metastabilen kristallinen Phase, welche sich dann
beim darauf folgenden Aufheizen exotherm in eine stabile kristalline Phase umwandelt.
Das Hochdruckphasenverhalten der Flüssigkristalle 6-TPEB und 10-TPEB, zweier Homologe
der 1-[4-Alkylbiphenyl]-2-[4-iso-thiocyanatophenyl]ethane (n-TPEBs), wurde von Ernst [65]
mittels DTA untersucht. Nach Angaben von Jadzyn [129] sollten Homologe mit n ≥ 4 die
Phasenabfolge kristallin → smektisch B → nematisch → isotrop flüssig zeigen.
DSC-Normaldruckmessungen an 6-TPEB zeigen die erwarteten Umwandlungen.
Thermogramme des 10-TPEBs weisen darüberhinaus eine fest-fest-Umwandlung auf. Das
Phasenverhalten des 10-TPEBs wurde bei 140 MPa an einer Substanz hoher Reinheit
(> 99.6%) untersucht. Das Auftreten einer zusätzlichen smektischen Phase (siehe Ernst [65])
konnte nicht bestätigt werden.

Zusammenfassung
VII
Bei den Dispersionsfarbstoffen, die bezüglich ihres Phasenverhaltens untersucht wurden,
handelt es sich um acht Homologe der Reihe der 1,4-Bis-(alkylamino)-9,10-anthrachinone
(mit Alkyl = Methyl (AC01), Ethyl (AC02), Propyl (AC03), Isopropyl (iso-AC03), Butyl
(AC04), Pentyl (AC05), Octyl (AC08) und Octadecyl (AC18), sowie um die Substanzen C. I.
Disperse Red 60 (DRed60) und C. I. Disperse Blue 60 (DBlue60). Von Wagner [132] mit
einer Strömungsmethode durchgeführte Untersuchungen zur Löslichkeit von Anthrachinon-
farbstoffen in nah- und überkritischem CO2 ergaben, daß sich die Löslichkeit beim Übergang
von AC02 zu den Homologen mit zusätzlichen CH2-Gruppen beträchtlich erhöht. Wagner
stellte fest, daß offensichtlich ein starker Einfluß der Kristallstruktur der untersuchten
Dispersionsfarbstoffe auf die Löslichkeit vorliegt. Im Einklang damit stehen die im Rahmen
dieser Arbeit gewonnenen Ergebnisse der DSC-Normaldruckmessungen, nach denen bei den
gut löslichen Derivaten AC03, iso-AC03, AC04 und AC05 polymorphes Verhalten zu
beobachten ist, bei den schlecht löslichen Farbstoffen AC01 und AC02 dagegen nicht.
Hochdruckuntersuchungen wurden an AC03 und AC04 vorgenommen. Bei den an AC03
durchgeführten DSC-Messungen in offenen Meßzellen unter Verwendung von Argon und
CO2 als Druckgas konnte über den gesamten Druckbereich nur der Schmelzpeak beobachtet
werden. Unter Argoneinfluß ist beim Höchstdruck von 136 MPa eine Temperaturerhöhung
von 14.1 K gegenüber dem Normaldruckwert festzustellen. Besonders interessant ist die
Tatsache, daß das berechnete Umwandlungsvolumen beim Höchstdruck um den Faktor drei
größer ist, als der unter Normaldruck ermittelte Wert. In Gegenwart von CO2 tritt für die feste
Gleichgewichtsphase zunächst eine Schmelzpunktserniedrigung ein, wobei im Bereich bis 8.5
MPa eine Abnahme der Schmelztemperatur um 8.2 K zu verzeichnen ist.
An AC04 wurden DTA-Messungen in geschlossenen und offenen Meßzellen unter
Argondruck durchgeführt. Im Gegensatz zu AC03 zeigt AC04 in beiden Fällen auch unter
Druck polymorphes Verhalten. Im Druckbereich bis 225 MPa ist die Phasenabfolge kristallin
(s2) → kristallin (s1) → isotrop flüssig zu beobachten.

Einleitung
1
1 Einleitung
1.1 Mesomorphe Übergänge kristalliner Festkörper
Ein kristalliner Festkörper zeichnet sich normalerweise durch zwei Ordnungsprinzipien aus:
die regelmäßige Anordnung der Molekülschwerpunkte im Kristallgitter, auch positionelle
Fernordnung genannt, und die Orientierungsfernordnung, welche die Ausrichtung der
Molekülachsen der auf den Gitterplätzen sitzenden Moleküle zueinander beschreibt. Beim
Aufheizen eines solchen Kristalls gehen bei einer bestimmten Temperatur die positionelle
Fernordnung und die Orientierungsfernordnung gleichzeitig verloren, der Kristall schmilzt.
Begleitet wird dieser Vorgang von drastischen Änderungen der zugehörigen
thermodynamischen Größen Enthalpie, Entropie und Volumen.
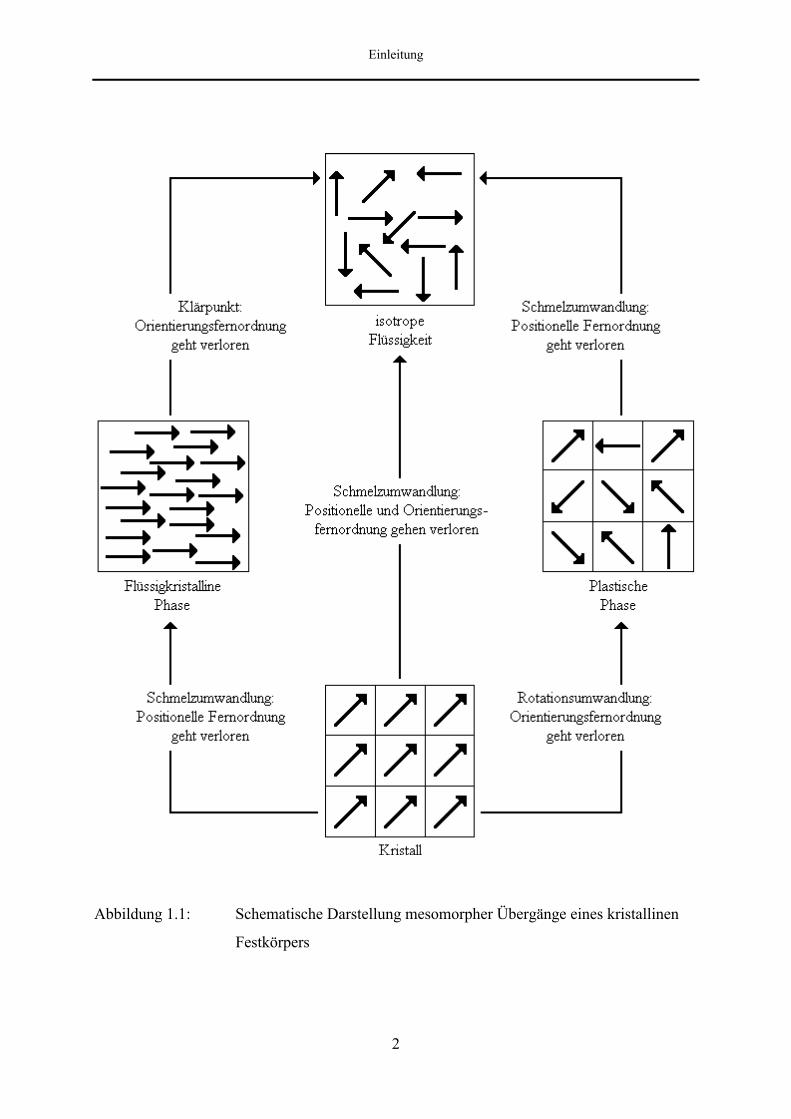
Es gibt eine Vielzahl von hauptsächlich organischen Verbindungen, die beim Erwärmen nicht
direkt vom kristallinen in den isotrop-flüssigen Zustand übergehen, sondern innerhalb
begrenzter Temperaturbereiche eine oder auch mehrere zusätzliche Phasenumwandlungen
durchlaufen, bei denen die Bewegungsfreiheit der einzelnen Moleküle jeweils sprunghaft
zunimmt [1, 2]. Diese mesogenen Phasen besitzen Eigenschaften, die zwischen denen des
kristallinen Festkörpers und der isotropen Flüssigkeit liegen.
Man unterscheidet zwischen flüssigkristallinen (positionell fehlgeordneten) und plastisch-
kristallinen (orientierungsfehlgeordneten) Phasen, die beide eine geringere Ordnung als der
Kristall, aber eine höhere Ordnung als die isotrope Schmelze aufweisen. Flüssigkristalline
Mesophasen bilden sich, wenn beim Schmelzvorgang zunächst die positionelle Fernordnung
verlorengeht, aber zumindest noch ein Teil der Orientierungsfernordnung erhalten bleibt. Die
sogenannte plastisch-kristalline Mesophase entsteht, wenn, ausgehend vom kristallinen
Festkörper, zunächst der Verlust der Orientierungsfernordnung unter Beibehaltung der
positionellen Ordnung erfolgt. Bei höheren Temperaturen gehen beide mesogenen Phasen in
eine isotrope flüssige Phase über.
Einleitung
2
Abbildung 1.1: Schematische Darstellung mesomorpher Übergänge eines kristallinen
Festkörpers

Einleitung
3
Die vorliegende Separierung des Schmelzvorgangs dieser Substanzen in einzelne Teilschritte
ermöglicht eine getrennte Untersuchung der sich sonst überlagernden Effekte. Neben
Verfahren wie Kernresonanzspektroskopie, Röntgenstrukturanalyse, Infrarotspektroskopie,
Polarisationsmikroskopie, dielektrische Messungen oder Neutronenstreuung dient dazu auch
die Differentialkalorimetrie, insbesondere, wenn als zusätzlicher Parameter der Druck variiert
werden kann [6]. Einen umfassenden Überblick über die verschiedenen Meßmethoden an
Flüssigkristallen und an plastischen Kristallen geben Sherwood [2], Schmid [3], Gray [4],
Parsonage und Staveley [1] sowie Aston [5]. Ferner berichtet Randzio [149] sehr ausführlich
über Entwicklung und Fortschritte kalorischer Meßmethoden in neuerer Zeit.
1.2 Plastische Kristalle
Bereits im Jahre 1935 lieferte Timmermans [7, 8] eine erste Zusammenstellung über
”plastische Kristalle”, denen er aufgrund ihrer wachsartigen Konsistenz diesen Namen gab.
Ähnlich dem Verhalten der Flüssigkristalle, repräsentieren plastische Kristalle einen Zustand,
der zwischen dem Idealkristall und der isotropen Flüssigkeit liegt. In der plastischen Phase
liegen die Moleküle mit ihren Schwerpunkten auf definierten Gitterplätzen. Aufgrund einer
gewissen ”Globularität” der Moleküle, das heißt, bedingt durch ihre meist sphärische Gestalt
(z. B. tetraedrische Moleküle wie t-Butylchlorid [10, 11, 12]), können sie jedoch im
Gegensatz zu Molekülen im Idealkristall mehr oder weniger freie Rotationsbewegungen
ausführen [15]. In der Literatur werden plastische Phasen aufgrund dieser
Rotationsbewegungen auch häufig als ”Rotatorphasen” bezeichnet. Nach Timmermans
können Moleküle auch dann plastische Phasen ausbilden, wenn durch Rotation um eine ihrer
Hauptträgheitsachsen ein sphärischer Rotationskörper gebildet wird. Dies trifft beispielsweise
auf scheibenförmige Moleküle wie Cyclohexan [13] oder Cyclohexen [10, 12] zu. Eine
weitere Gruppe globularer Moleküle sind Käfigmoleküle, wie Adamantan oder Diamantan
[14, 12]. Da beim Übergang vom geordneten Kristall in den plastisch-kristallinen Zustand die
Orientierungsfernordnung verlorengeht, während die positionelle Fernordnung erhalten bleibt,
werden plastische Kristalle heute zunehmend als ”Orientationally Disordered Crystals
(ODIC)” bezeichnet.

Einleitung
4
Im folgenden werden einige charakteristische Eigenschaften plastischer Kristalle aufgeführt:
- Als wesentliches Merkmal für das Vorliegen einer plastisch-kristallinen Phase ist die geringe
molare Schmelzentropie anzusehen. Sie liegt im Bereich von 0.4 R S 2.5 Rsl
m⋅ < < ⋅∆ , also
zwischen 3.3 und 21 J⋅mol-1⋅K-1, während bei Molekülkristallen im allgemeinen Schmelz-
entropien größer als 42 J⋅mol-1⋅K-1 zu beobachten sind [7].
- Dampfdruck, Schmelztemperatur und Tripelpunktsdaten liegen im Vergleich zu den nicht
plastisch-kristallinen Isomeren relativ hoch [9].
- Es tritt mindestens eine Fest-Fest-Umwandlung mit erheblicher Enthalpie- und
Entropieänderung auf. Die molaren Umwandlungsenthalpien beim Übergang von der
kristallinen zur plastischen Phase sind dabei in der Regel um das Fünffache größer als die
molaren Schmelzenthalpien [15].
- Die plastische oder Hochtemperaturphase besitzt ein hohes Maß an
Orientierungsfehlordnung und Selbstdiffusion [3]. Die Aktivierungsenergie für die
Reorientierung beträgt ≈ 10 kJ⋅mol-1, für die Selbstdiffusion liegt sie dagegen bei ≈ 50 - 150
kJ⋅mol-1.
- Im allgemeinen ist die Symmetrie der Kristallgitter in der plastischen Phase weit höher, als
die Symmetrie der Moleküle es erwarten läßt [16]. Die Rotatorphase weist fast immer
kubische [16, 17], seltener hexagonale [18] oder orthorhombische Gitter [19] auf.
1.3 Flüssigkristalle
Das Phänomen des flüssigkristallinen Zustandes wurde vor über einem Jahrhundert zum
ersten Mal beobachtet. Der Botaniker Friedrich Reinitzer [20] entdeckte im Jahre 1888 bei
zwei von ihm synthetisierten Estern des Cholesterins, dem Benzoat und dem Acetat, ein ”
anomales ” Schmelzverhalten. In einem Brief an den Physiker Otto Lehmann [21] berichtete
Reinitzer, er habe bei den Estern ” zwei Schmelzpunkte ” gefunden. Die Substanzen
schmelzen ” zu einer trüben, jedoch völlig flüssigen Flüssigkeit ”, die bei höheren
Temperaturen plötzlich völlig klar wird.
Erst zu einem späteren Zeitpunkt ließ sich diese Beobachtung damit erklären, daß sich die
Moleküle in einer zwischen dem Zustandsgebiet des kristallinen Festkörpers und der isotropen

Einleitung
5
Flüssigkeit gelegenen ” Mesophase ” befinden können, in der noch ein gewisses Maß an
Orientierungsfernordnung erhalten ist. Reinitzer hat also das thermische Verhalten einer
thermotropen, mesogenen Substanz beschrieben, ohne daß er damals jedoch das seiner
Beobachtung zugrundeliegende physikalische Phänomen sofort erkannt hat. Die von Reinitzer
bemerkte Trübung beruht auf der Bildung kleiner orientierter Bereiche, an deren Grenzflächen
das Licht gestreut wird. Die Schmelze wird beim Übergang von der flüssigkristallinen in die
isotrop flüssige Phase klar. Deshalb wird diese Zustandsänderung als Klärpunkt bezeichnet.
Die Erforschung der theoretischen Grundlagen über den Zusammenhang zwischen der
Struktur und dem Auftreten flüssigkristalliner Phasen seit ihrer Entdeckung 1888 bis etwa
1930 wurde neben F. Reinitzer und O. Lehmann noch maßgeblich von G. Friedel geprägt.
Nach 1930 kam die Erforschung der Flüssigkristalle zunächst zum Stillstand, da man ihre
technischen Anwendungsmöglichkeiten nicht zu nutzen wußte. Dies änderte sich erst Mitte
der sechziger Jahre. Flüssigkristalle werden heute vor allem in der Displaytechnik sowie der
Thermographie und der Gaschromatographie als stationäre Phase verwendet.
Nähere Informationen über die historische Entwicklung auf dem Gebiet der
Flüssigkristallforschung bietet sehr ausführlich der Übersichtsartikel ”History of Liquid
Crystals” von Kelker [22]. Ein detaillierter Überblick über theoretische Überlegungen,
experimentelle Methoden und Anwendungen der Flüssigkristalle in den verschiedenen
Forschungsbereichen wird von den Autoren Kelker und Hatz [23] in ihrem ”Handbook of
Liquid Crystals” und von Stegemeyer [41] in einer Zusammenstellung der Arbeiten
verschiedener Autoren vermittelt.
1.3.1 Klassifizierung der Flüssigkristalle
Flüssigkristalle sind kondensierte Aggregate, die in Ein- oder Mehrstoffsystemen als Phasen
in bestimmten Temperatur-, Druck- und Konzentrationsbereichen existieren [24]. Molekulare
Einheiten mit einer ausgeprägten Formanisotropie sind für die Bildung dieser
Phasenstrukturen verantwortlich.
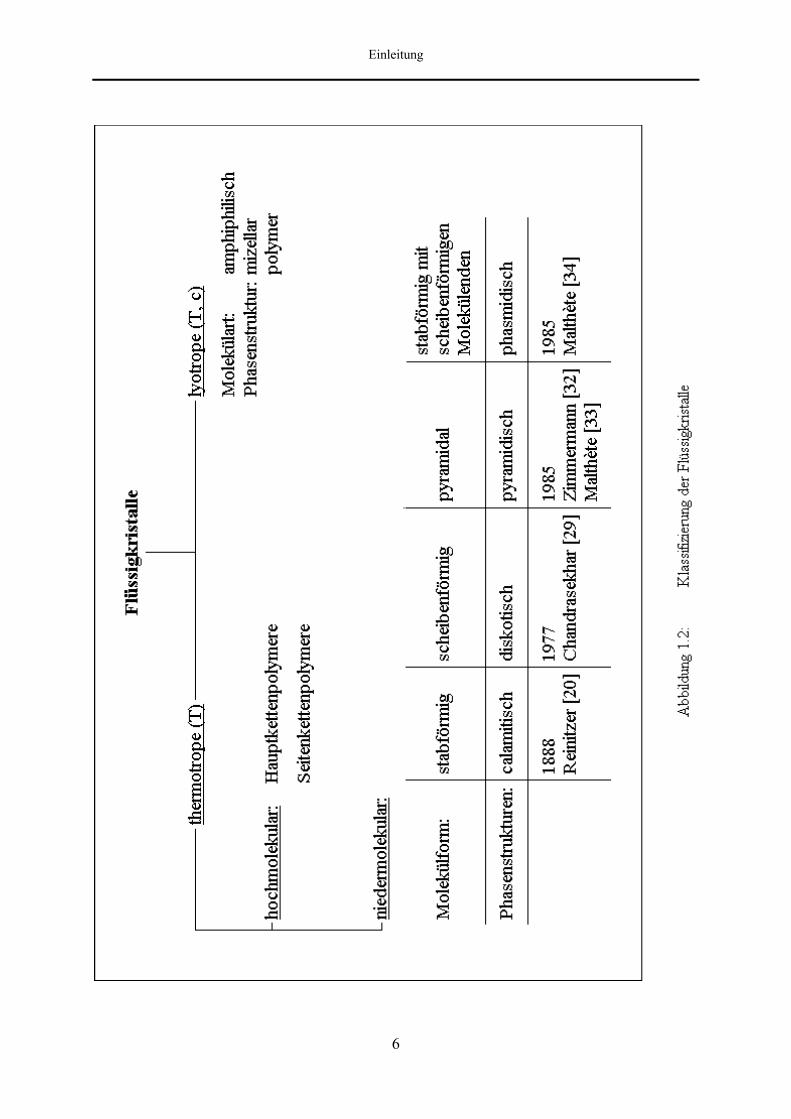
Grundsätzlich lassen sich Flüssigkristalle vom thermodynamischen Standpunkt in zwei
Klassen unterteilen: die thermotropen und die lyotropen Flüssigkristalle (Abbildung 1.2).
Einleitung
6

Einleitung
7
Das Auftreten und Verschwinden flüssigkristalliner Mesophasen thermotroper Flüssigkristalle
wird allein durch die Temperatur bestimmt. Dem gegenüber stehen die lyotropen
Flüssigkristalle, welche Mischungssysteme amphiphiler Substanzen mit Lösungsmitteln
darstellen. Neben der Temperatur ist hierbei vor allem die Konzentration des Lösungsmittels
für die Bildung des mesomorphen Bereichs entscheidend. Abhängig von der Beschaffenheit
der amphiphilischen Einheiten unterscheidet man mizellare und polymere Systeme. Die dabei
möglichen Phasenstrukturen sowie Eigenschaften dieser Systeme sind in der Literatur
beschrieben [4, 25, 26].
Flüssigkristalle, die in der Lage sind, sowohl thermotrope als auch lyotrope Phasen
auszubilden, werden als amphotrop bezeichnet.
Die Unterteilung der thermotropen Flüssigkristalle beruht auf der Unterscheidung zwischen
hochmolekularen oder polymeren Flüssigkristallen und niedermolekularen Flüssigkristallen.
Die erstgenannte Gruppe läßt sich weiterhin in Haupt- und Seitenkettenpolymere einteilen, je
nachdem, ob die flüssigkristallinen Monomereinheiten linear zu einem Makromolekül
verknüpft sind oder als Seitenkette einer nicht mesomorphen Hauptkette fungieren [27, 28].
Die Aufteilung innerhalb der Gruppe der niedermolekularen thermotropen Flüssigkristalle
erfolgt nach der Geometrie der Moleküle, weil durch diese spezifische Phasenstrukturen
induziert werden. Die erste der im folgenden aufgeführten vier Untergruppen bilden die
calamitischen Flüssigkristalle, für deren Bildung stabförmige Moleküle verantwortlich sind.
Zu dieser Gruppe zählen auch die von Reinitzer untersuchten Cholesterylester, weshalb die
Phasentypen und Strukturen dieser Mesophase die wohl am besten untersuchte und gegenüber
den anderen Gruppen am genauesten charakterisierte Gruppe ist. Die stabförmige
Molekülform (rod-like), meist aus einem starren, aromatischen System bestehend, welches
mit flexiblen terminalen Gruppen substituiert ist, induziert Phasenstrukturen der Typen
nematisch (n), cholesterisch (ch) und smektisch (sm) (siehe Kapitel 1.3.2).
Die zweite Gruppe niedermolekularer thermotroper Flüssigkristalle bilden die
scheibenförmigen Moleküle (disk-like). Bis zum Zeitpunkt der Synthese diskotischer
Mesogene durch Chandrasekhar [29] galt die starre, stabförmige Molekülform als
Voraussetzung für die Entstehung flüssigkristalliner Phasen. Im einfachsten Fall bestehen die
Moleküle der diskotischen Mesophasen aus hexasubstituierten Benzolringen, welche durch

Einleitung
8
Stapelung Säulenaggregate ausbilden können. Verschiedene Anordnungen dieser Formationen
führen zu der für diskotische Flüssigkristalle typischen Polymorphie [30, 31].
Eine Weiterentwicklung der diskotischen Flüssigkristalle führt von den planaren Molekülen
zu den pyramidischen, schalenförmigen (bowl-like) Anordnungen [32, 33]. Die
Phasenstrukturen dieser der dritten Gruppe niedermolekularer thermotroper Flüssigkristalle
zuzuordnenden pyramidischen Mesogenen unterscheiden sich wesentlich von den
diskotischen Modifikationen.
Die vierte Gruppe niedermolekularer thermotroper Flüssigkristalle bilden die sogenannten
Phasmide. Malthète et al. [34] synthetisierten erstmalig flüssigkristalline Verbindungen mit
starrer, stabähnlicher Geometrie und scheibenförmigen Molekülenden. Dem entsprechend
weisen Moleküle dieser Substanzklasse mesomorphe Eigenschaften auf, die zwischen denen
der calamitischen und diskotischen Flüssigkristalle liegen.
Darüberhinaus sind in neuerer Zeit noch andere Arten von Molekülen untersucht worden.
Dazu zählen z. B. die sogenannten Schwalbenschwanz- bzw. Doppelschwalbenschwanz-
moleküle oder auch gebogene, bananenförmige Verbindungen [148].
Es zeigt sich, daß es zunehmend schwieriger wird, eine Einteilung in die einzelnen Gruppen -
calamitisch, diskotisch, pyramidisch und phasmidisch - vorzunehmen, da die
Phasenstrukturen nicht mehr getrennt beziehungsweise unabhängig voneinander betrachtet
werden können.
1.3.2 Thermotrope calamitische Flüssigkristalle
Die im Rahmen dieser Arbeit untersuchten flüssigkristallinen Verbindungen gehören zur
Substanzklasse der thermotropen calamitischen Flüssigkristalle. Im folgenden werden deshalb
die Phasenstrukturen dieser Gruppe etwas ausführlicher behandelt.
Die heute üblichen Bezeichnungen für die flüssigkristallinen Phasen beruhen auf historischen
Beobachtungen und wurden von G. Friedel [35] geprägt. Folgt man der Nomenklatur von
Friedel, so wird eine Klassifizierung der thermotropen Flüssigkristalle in drei Kategorien
vorgenommen: nematisch (n), cholesterisch (ch) und smektisch (sm). Bei diesen
Bezeichnungen handelt es sich um aus dem Griechischen entnommene Ausdrücke für die
Beschreibung makroskopischer Eigenschaften der verschiedenen flüssigkristallinen Phasen.

Einleitung
9
So wurden smektische Phasen (griech.: Seife) zuerst bei Seifen beobachtet, während
nematische Phasen (griech.: Faden) bei polarisationsmikroskopischer Vergrößerung
charakteristische Fadentexturen zeigen.
1.3.2.1 Die nematische Phase
In Abbildung 1.3 ist die Anordnung langgestreckter Moleküle in einer nematischen Phase
schematisch dargestellt. Im Gegensatz zu der völlig ungeordneten, isotropen Flüssigkeit gibt
es hier eine Fernordnung der Orientierung. Eine Translationsfernordnung der Molekülschwer-
punkte ist in der nematischen Phase, ebenso wie in der isotropen Flüssigkeit, nicht gegeben.
Abbildung 1.3: Schematische Darstellung der unterschiedlichen Strukturmerkmale
verschiedener flüssigkristalliner Phasen

Einleitung
10
Die nematische Phase zeigt aufgrund von Vielfachstreunung eine starke Trübung. Die
Temperatur, bei der die nematische und auch alle anderen flüssigkristallinen Phasen in die
isotrop flüssige Phase übergehen, wird als Klärpunkt bezeichnet. In der Regel wird dieser
Übergang als Umwandlung erster Ordnung interpretiert [37]. Die zugehörigen Enthalpie-
änderungen liegen im Bereich von 0.4 bis 4.2 kJ⋅mol-1. Besitzt eine Substanz mehrere
flüssigkristalline Phasen, so ist die nematische Mesophase die Hochtemperaturphase.
Wichtigstes Strukturmerkmal nematischer Phasen ist die bevorzugte Parallelorientierung der
Moleküllängsachsen. Diese Vorzugsrichtung wird als Direktor n bezeichnet. Abbildung 1.4
zeigt die schematische Anordnung der Moleküle in der nematischen Mesophase.
Abbildung 1.4: Anordnung der Moleküle in einem nematischen Flüssigkristall [36]
Eine weitgehend vollständige Parallelorientierung ist aufgrund thermischer Fluktuationen der
Moleküle jedoch nicht gegeben. Vielmehr ordnen sich die Moleküle mit ihrer Längsachse
unter einem Winkel ϑ in einer statistischen Verteilung um den Direktor n an (siehe
Abbildung 1.5). Ein Maß für die Parallelstellung der Moleküle stellt der Ordnungsparameter S
dar:
S = −12
3 12cos ϑi (1.1)

Einleitung
11
Der Ordnungsparameter ist von der Temperatur abhängig. Er kann theoretisch Werte
zwischen S = 0 in der isotropen Phase und S = 1 im Falle eines idealen Flüssigkristalls
annehmen. Eine ideale Parallelorientierung aller Moleküle, also S = 1, läßt sich in der Praxis
z. B. durch eine Ausrichtung in einem elektrischen oder in einem magnetischen Feld erreichen
[38]. Im Normalfall finden sich in der Nähe des Klärpunktes Werte um S = 0.4, die mit
abnehmender Temperatur auf S = 0.8 ansteigen [39]. Maier und Saupe [40] beschreiben dieses
Verhalten in ihrer molekularstatistischen Theorie.
Abbildung 1.5: Schematische Darstellung der Orientierung der Molekülachse eines
Einzelmoleküls zum Direktor n
Da den Molekülen in der nematischen Phase die laterale Kohäsion fehlt, kann es im
Gegensatz zu den smektischen Phasen (vgl. Abbildung 1.3 und Kapitel 1.3.2.3) nicht zu
einem schichtförmigen Aufbau kommen. Die Moleküle können vielmehr frei aneinander
vorbeigleiten. Aus diesem Grunde sind nematische Phasen viel dünnflüssiger als die
smektischen Phasen.

Einleitung
12
1.3.2.2 Die cholesterische Phase
Die Struktur der cholesterischen Mesophase steht in enger Verwandtschaft mit der
nematischen Phase. Beiden flüssigkristallinen Phasen ist eine Parallelorientierung der
Moleküllängsachsen gemeinsam. Bei der cholesterischen Phase ist jedoch der Direktor in
benachbarten Schichten um einen konstanten Winkel gleichsinnig verdreht und beschreibt
eine Schraube mit konstanter Ganghöhe um die zu ihm senkrechte Helixachse (Abbildung
1.6). Die Helix kann unterschiedlichen Drehsinn besitzen, den man durch das Vorzeichen der
Ganghöhe unterscheidet.
Die cholesterische Phase tritt bei Molekülen mit einer chiralen Molekülstruktur auf, oder sie
entsteht durch Zugabe einer chiralen Verbindung zu einer nematischen Phase. Prinzipiell
lassen sich cholesterische Phasen als spontan verdrillte nematische Phasen auffassen, die oft
auch als chiral-nematische Phasen bezeichnet werden.
Abbildung 1.6: Schematische Darstellung der Helixstruktur der cholesterischen Phase

Einleitung
13
1.3.2.3 Die smektischen Phasen
Ebenso wie in der nematischen Phase richten sich die Moleküle in den smektischen Phasen
parallel zu einer Vorzugsrichtung aus. Zusätzlich tritt jedoch eine Positionsfernordnung der
Molekülschwerpunkte auf, wodurch sich als besonderes Merkmal der verschiedenen
smektischen Phasen die Anordnung der Moleküle in gegeneinander verschiebbaren Schichten
ergibt. Daraus resultiert die große Viskosität und Oberflächenspannung dieser Phasen im
Gegensatz zu der niederviskosen nematischen Phase. Aufgrund des höheren
Ordnungszustandes treten die smektischen Phasen in der Regel bei tieferen Temperaturen als
die nematische und cholesterische Phase auf.
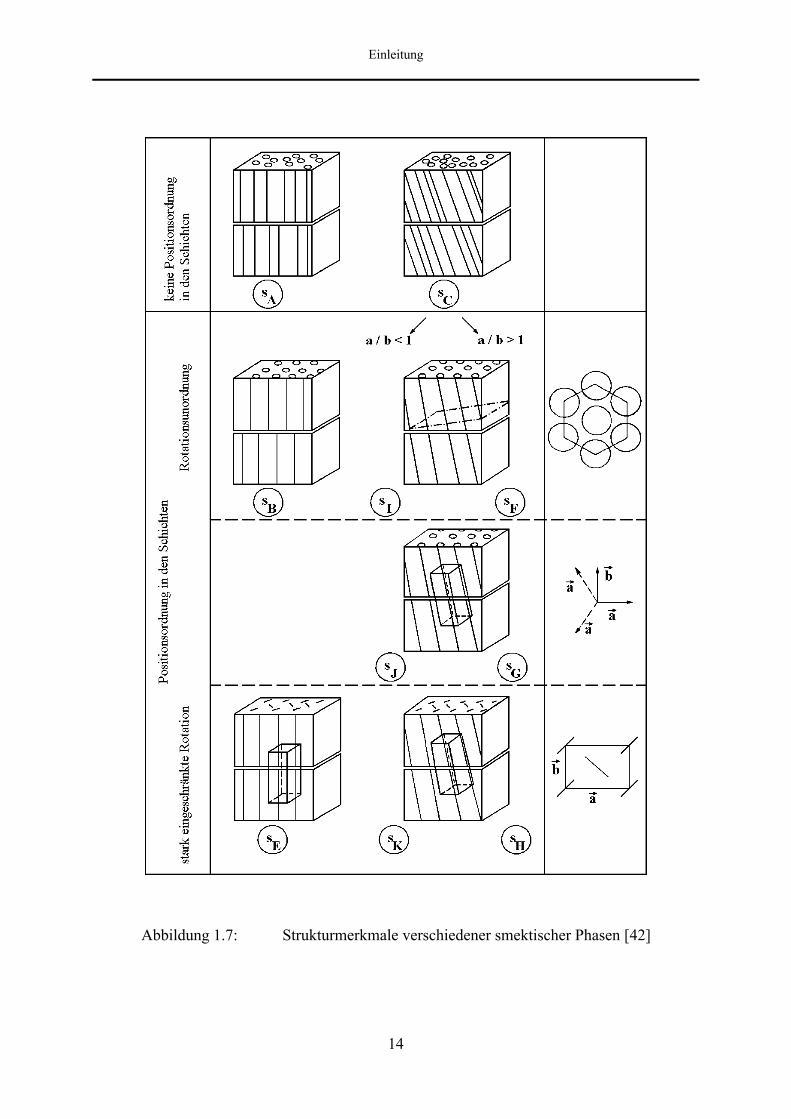
Es existieren ungefähr 20 verschiedene Typen von smektischen Phasen, von denen einige in
Abbildung 1.7 dargestellt sind. Diese Vielfalt ist zurückzuführen auf die unterschiedlichen
Orientierungen des Direktors n zu den Schichtebenen sowie auf Unterschiede in der
Ausprägung der Schichtstruktur und der Positionsordnung innerhalb der Schichten. Nach
Diele und Sackmann [42] lassen sich die verschiedenen smektischen Phasen auf der Basis von
umfangreichen Mischbarkeitsuntersuchungen klassifizieren. Sie werden dabei chronologisch
nach der Reihenfolge ihrer Entdeckung mit smektisch A (smA), smektisch B (smB),
smektisch C (smC), ... bezeichnet.
Prinzipiell kann eine Unterteilung der smektischen Phasen in zwei Gruppen vorgenommen
werden, je nachdem, ob die Molekülschwerpunkte innerhalb der Schichten statistisch oder
regelmäßig verteilt sind.
Die erstgenannte Gruppe wird z. B. durch die Phasen smektisch A (smA) und smektisch C
(smC) vertreten. In diesen smektischen Phasen besteht keinerlei Positionsordnung der
Moleküle; jede Schicht stellt quasi eine zweidimensionale Flüssigkeit dar. Besondere
Eigenschaften zeigen smektisch A-Phasen von stark polaren Molekülen, die ein Dipolmoment
in Richtung der Moleküllängsachse aufweisen. Eine Zuordnung der unterschiedlichen
Modifikationen smektisch A1 (smA1), smektisch A2 (smA2), smektisch Ad (smAd), etc. erfolgt
hier aufgrund des Quotienten aus Schichtdicke und Moleküllänge [43].
Der zweiten Gruppe gehören die sogenannten hexatisch smektischen und die kristallin
smektischen Phasen an. In den Schichten der Phasen smektisch Bhex (smBhex), smektisch I
(smI) und smektisch F (smF) besteht eine hexagonale Nahordnung der Molekülposition und
eine Orientierungsfernordnung der hexagonalen Einheitszelle.
Einleitung
14
Abbildung 1.7: Strukturmerkmale verschiedener smektischer Phasen [42]

Einleitung
15
Demgegenüber liegt bei den Phasen smektisch Bcryst (smBcryst), smektisch J (smJ), smektisch
G (smG), smektisch E (smE), smektisch K (smK) und smektisch H (smH) eine
dreidimensionale Fernordnung der Molekülpositionen vor.
Die smektischen Phasen in den einzelnen Gruppen unterscheiden sich jeweils in dem Winkel
des Direktors n zur Schichtebene. Bei den orthogonalen Phasen steht der Direktor senkrecht
auf den Schichtebenen, während er bei den getilteten Phasen gegenüber der Schichtnormalen
geneigt ist.
1.3.3 Polymorphismus der thermotropen Flüssigkristalle
Flüssigkristalle weisen häufig eine ausgeprägte Polymorphie auf. Bei vielen thermotropen
Flüssigkristallen können sich zwischen der festen Phase und der isotropen Flüssigkeit mehrere
flüssigkristalline Phasen ausbilden, welche durch thermodynamisch definierte Umwandlungs-
punkte voneinander getrennt sind. Als ”enantiotrop” werden dabei anisotrope Phasen
bezeichnet, die oberhalb des Schmelzpunktes liegen und deren Umwandlungen in benachbarte
Phasen reversibel verlaufen. Man kann sie sowohl durch Erwärmung aus der fest-kristallinen
Phase als auch durch Abkühlung der isotropen Flüssigkeit erhalten. Im Gegensatz dazu stehen
die sogenannten ”monotropen” Phasen. Diese sind stabil gegenüber benachbarten
flüssigkristallinen Phasen aber metastabil bezüglich nicht benachbarten festen Phasen. Sie
treten beim Abkühlen unterhalb des Schmelzpunktes auf, da sich der Schmelzpunkt in vielen
Fällen weit unterkühlen läßt.
Aufgrund der Tatsache, daß die smektischen Phasen einen höheren Ordnungszustand
aufweisen als die nematische oder cholesterische Phase, sind die smektischen Phasen bei allen
polymorphen Flüssigkristallen immer die Tieftemperaturmodifikationen der flüssigkristallinen
Phasen. Bezüglich der in den einzelnen Phasen vorliegenden Ordnungszuständen ergibt sich
beim Abkühlen eine thermische Abfolge flüssigkristalliner Phasen [41], wobei natürlich nicht
alle Phasen von einer Verbindung durchlaufen werden müssen:
isotrop → nematisch (cholesterisch) → smA → smC → smBhex → smI → smF →
smBcryst → smJ → smG → smE → smK → smH → kristallin [44].

Einleitung
16
Einige Flüssigkristalle weisen eine Abweichung von dieser Abfolge auf. Bei diesen
Flüssigkristallen tritt beim Abkühlen eine Phase, die schon bei höheren Temperaturen zu
beobachten ist, bei tieferen Temperaturen noch einmal auf. Die wiederholt auftretende Phase
wird als ”Reentrantphase” bezeichnet. Cladis [45] entdeckte dieses Phänomen zuerst in
binären Mischungen, dann auch bei Hochdruckuntersuchungen an 4-n-Octyloxy-4’-
cyanobiphenyl (8 OCB) [46]. Später konnte das Reentrantverhalten von Hardouin et al.
[47] auch bei einer reinen Substanz unter Normaldruck nachgewiesen werden.
Untersuchungen von Shashidhar [48] ergaben, daß das Reentrantverhalten einer
flüssigkristallinen Verbindung an das Vorhandensein von zwei oder drei Phenylringen mit
stark polaren, endständigen Gruppen wie -CN oder -NO2 geknüpft ist. Eine
Zusammenfassung auf dem Gebiet der Reentrantphänomene gibt Cladis [49]. Grundsätzlich
tritt dieses Verhalten jedoch nur selten auf und sollte als Ausnahmeerscheinung angesehen
werden [41].
1.3.4 Hochdruckuntersuchungen an Flüssigkristallen
Neben der starken Temperaturabhängigkeit des mesomorphen Zustandes zeigen
Flüssigkristalle ebenfalls eine Abhängigkeit von den herrschenden Druckverhältnissen.
Aufgrund vieler an flüssigkristallinen Verbindungen durchgeführten Hochdruckmessungen
können folgende Charakteristika und Phänomene der Flüssigkristalle zusammengestellt
werden:
- Die Umwandlungstemperaturen zeigen eine starke Druckabhängigkeit.
- Es können sowohl druckinduzierte Phasen auftreten als auch bestimmte flüssigkristalline
Zustände durch Druckeinfluß verschwinden.
- Die Existenz trikritischer Punkte auf einer T(p)-Kurve weist auf einen Wechsel der Ordnung
einer Phasenumwandlung von erster zu höherer Ordnung hin.
- Bei einigen flüssigkristallinen Verbindungen kann beim Abkühlen die ungewöhnliche
Phasensequenz i → n → smA → nre → cr beobachtet werden, wobei eine reentrant
nematische Phase auftritt. Das Reentrantphänomen ist nicht nur auf die nematische Phase
beschränkt, sondern läßt sich auch bei der smektisch A- und smektisch C-Phase beobachten.

Einleitung
17
- Der Ordnungsgrad S zeigt in der nematischen Phase eine deutliche Druck- und
Temperaturabhängigkeit.
- Bei Hochdruckuntersuchungen ist eine Änderung der Ganghöhe der Helix einer
cholesterischen Phase festzustellen.
Einen Überblick über Hochdruckuntersuchungen an Flüssigkristallen gibt der Review-Artikel
von Chandrasekhar und Shashidhar [50].
1.3.5 Identifizierung der flüssigkristallinen Phasen
Mit Hilfe von thermischen Untersuchungsmethoden, wie die in dieser Arbeit benutzte
Differential Scanning Calorimetry (DSC) und Differentialthermoanalyse (DTA), lassen sich
Phasenumwandlungen detektieren und Umwandlungstemperaturen bestimmen. Über die Art
der Umwandlung und Zuordnung von Phasen lassen sich allerdings nur begrenzt Aussagen
machen. Schmelzumwandlungen sind meist mit großen Enthalpieänderungen, d. h. großen
Peakflächen, verbunden, während Klärpeaks dagegen typischerweise extrem scharf sind und
sich über ein sehr kleines Temperaturintervall erstrecken. Um beispielsweise verschiedene
auftretende smektische Phasen im T(p)-Diagramm zuordnen zu können, benötigt man jedoch
zusätzliche Informationen.
Eine eindeutige Identifizierung eines flüssigkristallinen Phasentyps ist nur durch
Röntgenstrukturanalyse möglich. Diese Untersuchungsmethode liefert Informationen über die
Moleküllängen, Schichtdicken und Schichtabstände, wodurch Rückschlüsse auf die
Phasenstrukturen gezogen werden können. Da in der Vergangenheit eine Vielzahl
flüssigkristalliner Phasenstrukturen derart bestimmt worden sind, ist heute oft nur eine
Zuordnung zu bereits bekannten Strukturen erforderlich.
Eine weitverbreitete Methode der Zuordnung, welche auf der optischen Anisotropie der
Flüssigkristalle basiert, ist die polarisationsmikroskopische Untersuchung. Nach Friedel wird
das mikroskopische Bild einer flüssigkristallinen Phase im polarisierten Licht als Textur
bezeichnet. Diese Texturen sind, abhängig vom Phasentyp, von unterschiedlichen
Erscheinungsformen, wie z. B. Fantextur, Mosaikstruktur, Schlierentextur, usw. [23, 41]. Sie
können optisch verfolgt und zur Charakterisierung der flüssigkristallinen Phasentypen

Einleitung
18
herangezogen werden. Die bei Normaldruck erhaltenen Phasensequenzen (vergleiche Kapitel
1.3.3) basieren auf dieser Untersuchungsmethode.
Eine geeignete Meßmethode zur Identifizierung flüssigkristalliner Phasen unter Druck bietet
die von Hermann [51] konzipierte Diamanthochdruckzange (DAC). Mit der DAC ist es
möglich, polarisationsmikroskopische Untersuchungen unter sehr hohen Drücken durch-
zuführen.

Theoretische Grundlagen
19
2 Theoretische Grundlagen
2.1 Thermodynamische Grundlagen der Kalorimetrie
Ein Kalorimeter ist eine Meßvorrichtung, mit der die Änderung der inneren Energie
∆U = U2 - U1 indirekt bestimmt werden kann, nachdem eine Zustandsänderung in einem
System stattgefunden hat [52]. Für eine infinitesimale Zustandsänderung gilt nach dem ersten
Hauptsatz der Thermodynamik:
dU = dQ + dW = dQ + dWrev + dW’ (2.1)
Q ist die mit der Umgebung ausgetauschte Wärme, Wrev stellt die am Gesamtsystem geleistete
reversible Volumen-, Deformations-, Elektrisierungs-, Magnetisierungsarbeit usw. dar und in
W’ sind alle übrigen Arbeitsanteile (z. B. Reibungsarbeit, elektrische Arbeit von äußeren
Stromquellen) zusammengefaßt [53]. Falls an Arbeitsanteilen nur reversible Volumenarbeit
zu berücksichtigen ist, gilt:
dU = dQ - pdV (2.2)
Ist das System thermisch isoliert, d. h. adiabatisch (dQ = 0), und wird gleichzeitig das
Volumen konstant gehalten (dV = 0), so ist in einem geschlossenen System (dn = 0) dU
= 0 bzw. U = const (Satz von der Erhaltung der Energie).
Da die Innere Energie U eine Zustandsfunktion ist, während Q und W im allgemeinen keine
Zustandsfunktionen sind, folgt, daß zwar dU, aber nicht dQ und dW totale Differentiale sind.
Die Erfahrung zeigt, daß für einen reversiblen Vorgang in einem geschlossenen System die
Größe
dSdQ
Trev≡ (2.3)
ein totales Differential und die so definierte Entropie S eine Zustandsfunktion ist. Für alle
irreversiblen Vorgänge in einem geschlossenen System ist

Theoretische Grundlagen
20
dSdQT
> . (2.4)
Die Beziehungen (2.3) und (2.4) sind Formulierungen des zweiten Hauptsatzes der
Thermodynamik. Beschränkt man sich auf reversible Vorgänge und berücksichtigt an
Arbeitsanteilen nur reversible Volumenarbeit, so ergibt eine Kombination des ersten
Hauptsatzes (Gleichung 2.2) und des zweiten Hauptsatzes (Gleichung 2.3) für ein
geschlossenes System:
dU = TdS - pdV (2.5)
Die Berücksichtigung einer Stoffmengenänderung in Gleichung (2.5), z. B. bei chemischen
Reaktionen oder Materieaustausch mit der Umgebung (offenes System, dn ≠ 0), führt zu:
dU TdS pdV dni ii
k= − +
=∑µ
1, (2.6)
wo ∂∂
µUni S V n
ij j i
≡
≠, , ( )
das chemische Potential der Komponente i ist.
Für viele Anwendungen ist die Benutzung der Enthalpie H anstelle der Inneren Energie U
günstiger:
H ≡ U + pV
(2.7)
dH = dU + Vdp + pdV
Einsetzen von (2.5) in (2.7) liefert
dH = TdS + Vdp (2.8)

Theoretische Grundlagen
21
Weil in der Definitionsgleichung von H nur Zustandsgrößen auftreten, ist die Enthalpie H
selbst eine Zustandsfunktion mit den Variablen Temperatur, Druck und Stoffmengen der
beteiligten Komponenten k: H = f (T, p, n1, n2, ..., nk); dH ist ein totales Differential. Eine
Enthalpieänderung während eines Phasenübergangs oder einer chemischen Reaktion innerhalb
eines geschlossenen Systems wird demnach beschrieben durch:
dHHT
dTHp
dp Hn
dnp n T n i T p n
ii
k
i i j j i
= +
+
= ≠
∑∂∂
∂∂
∂∂, , , , ( )1
(2.9)
Weil gilt:
∂∂
∂∂
Sp
VTT p
= −
,
∂∂HT
Cp
p
≡ und
∂∂
Hn
Hi T p n
ij j i
≡
≠, , ( )
läßt sich Beziehung (2.9) umformen zu:
dH C dT V TVT
dp H dnpp n
i ii
k
i
= + −
+
=∑∂
∂ , 1, (2.10)
wobei Hi die partielle molare Enthalpie der Komponente i und Cp die Wärmekapazität für
p=const ist.
Bei chemischen Reaktionen gilt ferner:
H dn H di ii
k
r i ii
k
= =∑ ∑= ⋅
1 1∆ ξ , (2.11)
mit d dnii
iξν
≡1
,
wo ξi die sogenannte Reaktionslaufzahl bezüglich der Komponente i ist.

Theoretische Grundlagen
22
Einsetzen der Beziehung (2.11) in Gleichung (2.10) liefert die “Grundgleichung der
Kalorimetrie”:
dH C dT V TVT
dp H d dQ Vdp dWpp n
r ii
k
i eli
= + −
+ ⋅ = + +
=∑∂
∂ξ
,.∆
1(2.12)
Für die Koexistenz zweier Phasen ‘‘ und ‘ bei einer streng reversiblen Phasenumwandlung
erster Ordnung in einem Einstoffsystem (Gesamtstoffmenge 1 mol) müssen folgende zwei
Gleichgewichtskriterien erfüllt sein:
1. ∆ ∆ ∆trs m trs m trs trs mG H T S= − = 0 (2.13)
mit: ∆ trs m m mG G G= −'' ' Änderung der molaren Gibbs-Energie
∆ trs m m mH H H= −' ' ' Änderung der molaren Enthalpie
∆ trs m m mS S S= −' ' ' Änderung der molaren Entropie
2. ( ) ( ) ( )d G
Gp
dpG
TdTtrs m
trs m
T
trs m
p
∆∆ ∆
=
+
=
∂∂
∂∂
0
bzw. (2.14)
( )d G V dp S dTtrs m trs m trs m∆ ∆ ∆= − = 0
Die für die Beziehungen (2.13) und (2.14) notwendigen Gleichgewichtsbedingungen lauten:
T T' ''= ,
p p' ''= ,
G Gmi mi*' *''= bzw. allgemein µ µi i
' ''=
und beschreiben das thermische, mechanische und stoffliche Gleichgewicht.

Theoretische Grundlagen
23
Aus Gleichung (2.13) folgt für die Entropieänderung ∆trsSm bei der Umwandlungstemperatur
Ttrs unmittelbar:
∆∆
trs mtrs m
trsS
HT
= (2.15)
Gleichung (2.14) liefert die sogenannte verallgemeinerte Clausius-Clapeyron-Gleichung für
die Steigung der Phasenkoexistenzlinie im p-T-Diagramm:
dpdT
SVkoex
trs m
trs m
=
∆∆
(2.16)
Bei bekannter p-T-Abhängigkeit und bekannten ∆trsHm-Werten, die direkt aus den DSC-
Messungen erhalten werden, sind somit aus den Gleichungen (2.15) und (2.16) die Daten für
die Umwandlungsentropien und die Umwandlungsvolumina zugänglich.
Die Druckabhängigkeit der Enthalpieänderung und der Temperatur einer Phasenumwandlung
der Phase ‘ in die Phase ‘‘ entlang der Koexistenzlinie in einem geschlossenen System (keine
chemische Reaktion, dni = 0) ist aus dem totalen Differential der molaren
Umwandlungsenthalpie erhältlich. Die Gleichungen (2.9) und (2.10) lauten dann:
d HH
TdT
Hp
dptrs mtrs m
p
trs m
T∆ =
+
∂∆∂
∂∆∂
(2.17)
d Hdp
CdTdp
V TV
Ttrs m
koextrs p m
koextrs m
trs m
p
∆∆ ∆
=
+ −
,
∂∆∂
(2.18)
Planck-Gleichung
Darüberhinaus können auch Ausdrücke für die Druckabhängigkeit der Umwandlungsentropie
und des Umwandlungsvolumens entlang der Koexistenzlinie angegeben werden:

Theoretische Grundlagen
24
d SS
TdT
Sp
dptrs mtrs m
p
trs m
T∆ =
+
∂∆∂
∂∆∂
(2.19)
d Sdp
CT
dTdp
VT
trs m
koex
trs p m
koex
trs m
p
∆ ∆
=
−
, ∂∆∂
(2.20)
d VV
TdT
Vp
dptrs mtrs m
p
trs m
T∆ =
+
∂∆∂
∂∆∂
(2.21)
d Vdp
VT
dTdp
Vp
trs m
koex
trs m
p koex
trs m
T
∆
=
+
∂∆∂
∂∆∂
(2.22)
Die Einführung des isobaren thermischen Ausdehnungskoeffizienten αp (Gleichung 2.23)
bietet die Möglichkeit, Gleichung (2.18) umzuformulieren:
α∂∂p
m
m
pVVT
≡
1(2.23)
[ ]( )d Hdp
CdTdp
V T V Vtrs m
koextrs p m
koextrs m m p m p
∆∆ ∆
=
+ − −,
' ' ' ' ' 'α α (2.24)
wobei [ ]∂∆∂
∂∂
∂∂
α αtrs m
p
m
p
m
pm p m p
VT
VT
VT
V V
=
−
= −
' ' '' ' ' ' ' '
d Hdptrs m
koex
∆
und
dTdp koex
sind aus den DSC-Messungen direkt ableitbar, ∆ trs mV wird
aus diesen Größen berechnet, und mit Meß- oder Literaturdaten für die Dichten der

Theoretische Grundlagen
25
koexistierenden Phasen ‘ und ‘‘ als Funktion der Temperatur läßt sich ∂∂
∂∂
VT
VT
m
p
m
p
' ' '
−
und damit auch ∆ trs p mC , bestimmen.
Außerdem kann der Ausdruck ∂∂
∂∂
VT
VT
m
p
m
p
' ' '
−
auch über isotherme
Kompressibilitätsdaten κT errechnet werden. Verwendung und Umformung von Gleichung
(2.21) liefert dann:
d VdT
VT
Vp
dpdT
trs m
koex
trs m
p
trs m
T koex
∆
=
+
∂∆∂
∂∆∂
bzw. (2.25)
∂∂
∂∂
VT
VT
m
p
m
p
' ' '
−
= d V
dTtrs m
koex
∆
−−−−
∂∂
∂∂
Vp
Vp
m
T
m
T
' ' '
−
dpdT koex
und mit: κ∂∂T
m
m
TVVp
≡ −
1(2.26)
∂∂
∂∂
VT
VT
m
p
m
p
' ' '
−
= d V
dTtrs m
koex
∆
−−−− [ ]V Vm T m T
' ' '' ''κ κ−dpdT koex
(2.27)
Die Terme, die sich auf die Koexistenz beziehen, sind aus den Messungen erhältlich. Die
Kompressibilitätsdaten können für viele Substanzen der Literatur entnommen werden.

Theoretische Grundlagen
26
2.2 Klassifikation der Phasenumwandlungen
Betrachtet man einfache Phasenübergänge wie z. B. das Schmelzen, so sind im allgemeinen
starke Änderungen von Enthalpie, Entropie und Volumen am Umwandlungspunkt
charakteristisch. Die chemischen Potentiale zweier im Gleichgewicht nebeneinander
vorliegenden Phasen sind zwar gleich, aber die Werte der molaren Enthalpien und Entropien,
sowie der Molvolumina sind meistens sehr verschieden. Außerdem stellt man fest, daß sich
bei gewöhnlichen Phasenübergängen die Werte der Wärmekapazität Cp und der
Kompressibilität κ nicht sehr schnell ändern, wenn man sich aus der Phase ‘‘ bzw. ‘ kommend
dem Umwandlungspunkt nähert [54]. Es sind jedoch auch Phasenübergänge zu beobachten,
bei denen weder eine Volumenänderung, noch eine Enthalpie- oder Entropieänderung auftritt.
Die Klassifizierung von Phasenumwandlungen im Hinblick auf deren Ordnung erfolgt nach
der Theorie von Ehrenfest [55]. Demnach weist eine Phasenumwandlung n-ter Ordnung für
einen reinen Stoff an der Phasengrenze eine Unstetigkeit der n-ten Ableitung der molaren
Gibbsenergie Gm nach der Temperatur und dem Druck auf:
∂
∂
∂
∂
nm
np
nm
np
GT
GT
' ' '
≠
bzw.
∂∂
∂∂
nm
nT
nm
nT
Gp
Gp
' ''
≠
(2.28)
Für eine Phasenumwandlung erster Ordnung zwischen den Phasen ‘ und ‘‘ gelten folgende
Beziehungen:
∆ trs m m mG G G= − =' ' ' 0 (2.29)
∂∆∂
∂∂
∂∂
trs m m
p
m
pm m trs m
GT
GT
GT
S S S
=
−
= − + = − ≠
' ' '' ' ' ∆ 0 (2.30)
T S Htrs m trs m⋅ = ≠∆ ∆ 0 (2.31)
∂∆∂
∂∂
∂∂
trs m m
T
m
Tm m trs m
Gp
Gp
Gp
V V V
=
−
= − = ≠
' ' '' ' ' ∆ 0 (2.32)

Theoretische Grundlagen
27
Umwandlung erster Ordnung Umwandlung zweiter Ordnung
Abbildung 2.1: Klassifizierung von Phasenübergängen nach Ehrenfest (nach [1]). Dargestellt ist
u. a. der Verlauf der molaren Gibbsenergie als Funktion der Temperatur für eine Umwandlung
zweiter Ordnung (rechts oben). Die Abbildung macht deutlich, daß die Ehrenfest-
Klassifizierung bei Umwandlungen zweiter Ordnung nicht anwendbar ist, da “1” immer die
stabilere Phase ist und somit keine Umwandlung eintritt. Eine genauere Betrachtung dieser
Problematik findet sich bei Parsonage und Staveley [1].

Theoretische Grundlagen
28
Die sprunghaften Änderungen der molaren Größen Enthalpie, Entropie und Volumen sind
also charakteristisch für Phasenumwandlungen erster Ordnung. Die erste Ableitung der
molaren Gibbsenergie zeichnet sich demnach durch eine Diskontinuität aus. In Abbildung
(2.1) sind die Änderungen einiger thermodynamischer Größen am Phasenübergang
schematisch dargestellt.
Liegt eine Umwandlung zweiter Ordnung vor, so zeigt sich die Diskontinuität erst in der
zweiten Ableitung der molaren Gibbsenergie nach der Temperatur und dem Druck. Es gelten
dann folgende Zusammenhänge:
∆ trs mG = 0 (2.33)
∂∆∂trs m
ptrs m
GT
S
= − =∆ 0 (2.34)
∂
∂
2
2 0∆ ∆trs m
p
trs p mGT
CT
= − ≠
,(2.35)
beziehungsweise:
∂∆∂trs m
Ttrs m
Gp
V
= =∆ 0 (2.36)
∂
∂κ
2
2 0∆
∆trs m
Tm trs
Gp
V
= − ⋅ ≠ (2.37)
∂∂ ∂
α2
0∆
∆trs mm trs p
Gp T
V
= ⋅ ≠ (2.38)
Diese Umwandlungen sind also mit einer kontinuierlichen Änderung der molaren Größen
Enthalpie, Entropie und Volumen verbunden. Eine Diskontinuität tritt jedoch in der molaren

Theoretische Grundlagen
29
Wärmekapazität Cp, im isobaren thermischen Ausdehnungskoeffizienten αp und in der
isothermen Kompressibilität κT auf.
Für Phasenumwandlungen erster Ordnung kann die verallgemeinerte Clausius-Clapeyron-
Gleichung
dpdT
SV
HT Vkoex
trs m
trs m
trs m
trs m
= =
⋅∆∆
∆∆
(2.39)
zur Beschreibung der Gleichgewichtskurve der koexistierenden Phasen benutzt werden.
Bezogen auf Umwandlungen zweiter Ordnung ergeben sich entsprechende Beziehungen:
d SS
TdT
Sp
dptrs mtrs m
p
trs m
T∆ =
+
=
∂∆∂
∂∆∂
0 (2.40)
d VV
TdT
Vp
dptrs mtrs m
p
trs m
T∆ =
+
=
∂∆∂
∂∆∂
0 (2.41)
Unter Verwendung von α∂∂p
m
m
pVVT
=
1 und κ
∂∂T
m
m
TVVp
= −
1 erhält man durch
Umformen Ausdrücke, welche die Druckabhängigkeit der Umwandlungstemperatur für die
Koexistenzkurve analog zu Gleichung (2.39) beschreiben:
dpdT
CT Vkoex
trs p m
m trs p
=
⋅ ⋅∆
∆,
α(2.42)
dpdT koex
trs p
trs T
=
∆
∆
α
κ(2.43)
Phasenumwandlungen höherer Ordnung sind gewöhnlich nur selten zu beobachten. In den
meisten Fällen ist vor der Phasenumwandlung in einem weiten Bereich eine kontinuierliche
Theoretische Grundlagen
30
Änderung der Wärmekapazität zu verzeichnen. Bei Annäherung an den Umwandlungspunkt
nimmt die Wärmekapazität von niedrigen Temperaturen herkommend immer stärker zu. Am
Umwandlungspunkt selbst ist, wie bei Umwandlungen erster Ordnung, eine Unendlichkeits-
stelle vorhanden (siehe dazu A. B. Pippard in [1]). Wegen der Ähnlichkeit des Kurvenverlaufs
mit dem griechischen Buchstaben Lambda (λ), werden solche Phasenumwandlungen auch als
λ-Übergänge bezeichnet.
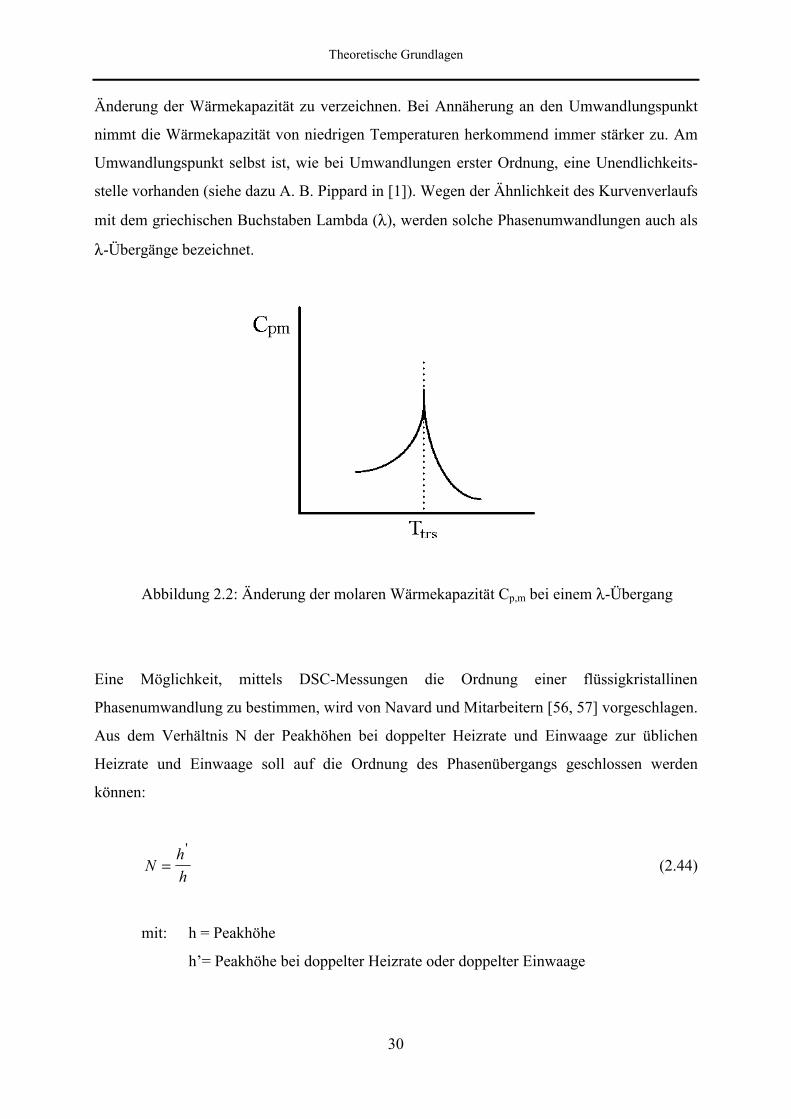
Abbildung 2.2: Änderung der molaren Wärmekapazität Cp,m bei einem λ-Übergang
Eine Möglichkeit, mittels DSC-Messungen die Ordnung einer flüssigkristallinen
Phasenumwandlung zu bestimmen, wird von Navard und Mitarbeitern [56, 57] vorgeschlagen.
Aus dem Verhältnis N der Peakhöhen bei doppelter Heizrate und Einwaage zur üblichen
Heizrate und Einwaage soll auf die Ordnung des Phasenübergangs geschlossen werden
können:
Nhh
='
(2.44)
mit: h = Peakhöhe
h’= Peakhöhe bei doppelter Heizrate oder doppelter Einwaage

Theoretische Grundlagen
31
Ergibt sich N = 2, handelt es sich um eine Umwandlung zweiter Ordnung. Liegt N in den
Grenzen 1 < N < 2 , so ist dies eine Umwandlung erster Ordnung. Experimentelle
Ergebnisse zeigen, daß sich nur bei sehr reinen Substanzen Aussagen über die Ordnung der
Phasenumwandlung machen lassen, da verunreinigte Proben ebenfalls den Wert N = 2 liefern
können, obwohl es sich um einen Übergang erster Ordnung handelt. Außerdem können
zusätzlich Schwierigkeiten bei der Bestimmung der Peakhöhe auftreten, wenn die
Umwandlung keinen scharfen Peak zeigt oder der Peak sehr klein ist.

Differential Scanning Calorimetry (DSC)
32
3 Differential Scanning Calorimetry (DSC)
Bei der Untersuchung temperaturabhängiger Stoffeigenschaften besitzen die
Differentialthermoanalyse (DTA) und die Differential Scanning Calorimetry (DSC) ein weites
Anwendungsfeld bei wissenschaftlichen Forschungsaufgaben und in der Industrie.
Die DSC ist wie die DTA eine weitverbreitete Methode der thermischen Analyse und findet
Anwendung bei der Bestimmung thermodynamischer Daten von chemischen Substanzen. Es
lassen sich Wärmeumsätze bei physikalischen Umwandlungen (z. B. Modifikations-
umwandlung, Verdampfung, Schmelze) und bei chemischen Reaktionen (z. B. Zersetzung,
Wasserabspaltung) bestimmen, sowie Reinheitsuntersuchungen durchführen [58]. Des
weiteren eröffnet sich die Möglichkeit, Wärmekapazitäten zu messen, reaktionskinetische
Untersuchungen durchzuführen, sowie, anhand gemessener Daten, Phasendiagramme zu
erstellen.
3.1 DSC-Meßprinzip [58, 59, 60, 61]
Im allgemeinen wird bei beiden Verfahren eine Probe (sample) neben einer
Vergleichssubstanz (reference) einem streng linearen Temperaturprogramm unterworfen.
Über Temperatursensoren, die sich an jeder der beiden Meßstellen befinden, werden jeweils
die Proben- und die Referenztemperatur, sowie die Systemtemperatur in Abhängigkeit von der
Zeit registriert. Befindet sich das System im thermischen Gleichgewicht, so ist im Idealfall die
Temperaturdifferenz zwischen beiden Meßstellen gleich Null oder nimmt im realen Fall
zumindest einen konstanten Wert an. Eine Änderung der Temperaturdifferenz zwischen Probe
und Referenz zeigt eine Änderung im thermischen Verhalten einer der beiden Substanzen an.
Dieser thermische Effekt ist solange beobachtbar (Peak), bis er durch Wärmefluß aus der
Umgebung, d.h. aus dem gesamten System, zur Seite mit der niedrigeren Temperatur
kompensiert ist.
Als Vergleichssubstanz wählt man meist Stoffe, die im untersuchten Temperaturbereich dem
thermischen Verhalten der Probe ähnlich sind, jedoch keine Peaks zeigen (Inertsubstanz).

Differential Scanning Calorimetry (DSC)
33
Im Unterschied zur DTA sind bei der DSC Probe und Referenz mit getrennten elektrischen
Heizelementen, die durch eine Regelelektronik gesteuert werden, versehen, was einen
Unterschied hinsichtlich der Art der Meßsignale und der daraus folgenden Auswertung
bewirkt.
Abbildung 3.1: Meßanordnung der DTA (a) und der DSC (b) [61]
Die DTA liefert lediglich die Differenztemperatur als Meßgröße. Um Aussagen über
Enthalpieänderungen machen zu können, müssen die Wärmekapazitäten von Probe und
Referenz, sowie deren Wärmeübergangskoeffizienten zur Umgebung und zur Meßstelle bei
der jeweiligen Meßtemperatur bekannt sein.
Bei der DSC können Probe und Referenz stets auf gleicher Temperatur gehalten werden. Ein
thermischer Effekt, sei er endotherm oder exotherm, wird dadurch kompensiert, daß die
entsprechend kältere Seite während einer entsprechenden Zeit mit einer zusätzlichen
Heizleistung versorgt wird. Hier ist also die Differenzheizleistung zwischen beiden Seiten das
Meßsignal, wodurch die DSC auch als ”Dynamische Leistungs-Differenz-Kalorimetrie”
bezeichnet wird.

Differential Scanning Calorimetry (DSC)
34
3.1.1 Normaldruck-DSC
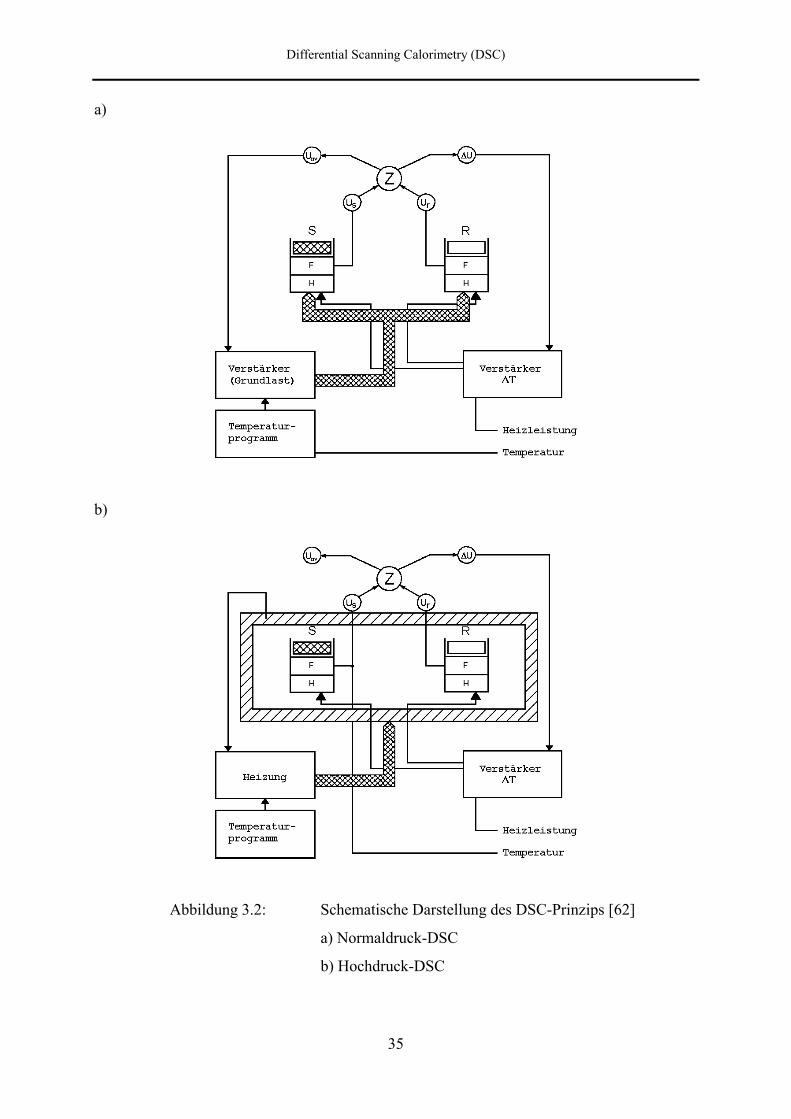
In Abbildung 3.2a) ist die prinzipielle Funktionsweise der DSC, so wie sie in den DSC-
Geräten der Firma Perkin-Elmer verwirklicht wird, schematisch dargestellt. Die Proben- (S)
und die Referenzzelle (R) befinden sich jeweils in einem zylindrisch geformten
Meßpfännchen mit eingebauten Platinwiderständen, welche als Widerstandsthermometer (TS,
TR) und als Heizwiderstände (HS, HR) fungieren. Die Drähte sind durch eine
Aluminiumoxidschicht voneinander isoliert. Mit den getrennten Heizelementen, deren
elektronische Steuerung über zwei getrennte Regelkreise erfolgt, werden die Meßpfännchen
streng linear aufgeheizt. Der erste Regelkreis dient dabei zur Einstellung der
Durchschnittstemperatur, während der zweite Regelkreis aus dem Differenztemperaturregler
besteht. Ein massiver Aluminiumblock, der die beiden DSC-2-Halter umschließt, sorgt für
eine konstante Umgebungstemperatur.
Die an den Platinwiderstandsthermometern abgegriffenen Spannungen US und UR werden
einem Zerhacker (Z) zugeführt. Dieser besteht aus zwei elektronischen Einheiten, der
Eingangsstufe und der Zerhackerstufe. Zunächst bildet die Eingangsstufe die
Differenzgleichspannung und die Mittelwertgleichspannung. Die Zerhackerstufe erzeugt dann
aus diesen Gleichspannungen auf elektromechanischem Weg mit einer Frequenz von 60 Hz
zwei Wechselausgangsspannungen ∆U und Uav.
Uav dient als Stellgröße für den ersten Regelkreis. Die Durchschnittstemperatur der
Meßstellen wird mit der Programmtemperatur verglichen. Ein Programmverstärker bewirkt
dann die Einstellung eines Durschnittsheizstromes, um ein Temperaturgleichgewicht
herzustellen. Ist die Programmtemperatur höher als die der Spannung proportionalen
Durchschnittspfännchen-temperatur, wird die Heizleistung erhöht, ist sie geringer als die
Durchschnittstemperatur, wird die Heizleistung gesenkt.
Das Auftreten eines thermischen Effektes bewirkt eine Änderung der Temperaturdifferenz
zwischen Probe und Referenz und damit einhergehend eine Änderung des Widerstandes des
Probenfühlers. Die zur Widerstandsänderung proportionale Spannungsdifferenz ∆U dient als
Stellgröße für den zweiten Regelkreis, der die elektrischen Heizer ansteuert. Die
Temperaturdifferenz wird dann mit Hilfe eines Verstärkers, der einen differentiellen
Heizstrom mit positivem oder negativem Vorzeichen liefert, sofort wieder auf Null geregelt.
Differential Scanning Calorimetry (DSC)
35
a)
b)
Abbildung 3.2: Schematische Darstellung des DSC-Prinzips [62]
a) Normaldruck-DSC
b) Hochdruck-DSC

Differential Scanning Calorimetry (DSC)
36
Dieses Prinzip wird auch ”Null-Balance” Prinzip genannt. Die benötigte Zusatzheizleistung
wird in Abhängigkeit von der Zeit aufgetragen. Die Fläche unter dem Peak ergibt die
elektrische Energie, die zur Kompensation des thermischen Effektes in der Probe aufgebracht
werden muß. Diese Energie entspricht direkt der Umwandlungsenthalpie.
3.1.2 Hochdruck-DSC
Das Prinzip der Hochdruck-DSC veranschaulicht Abbildung 3.2b). Aufgrund der großen
Autoklavenmassen ist es nicht mehr möglich, die Temperaturführung allein durch die
Platinwiderstandsdrähte in den Meßpfännchen zu bewerkstelligen. Der Regelkreis zur
Einstellung der Durchschnittstemperatur wird daher notwendigerweise durch einen externen
Regelkreis, d. h. durch eine von außen auf die Autoklaven wirkende Heizung mit einem
eigenen Programmgeber, ersetzt. Ein Pt-100 Widerstandsthermometer im Temperiermantel, in
welchem die Autoklaven symmetrisch angeordnet sind, liefert die Ist-Temperatur und dient
als Regelgröße für den externen Regelkreis. Der Temperaturprogrammgeber liefert den
Sollwert und ermöglicht die Einstellung unterschiedlicher Temperaturprogramme.
Im Gegensatz zur Normaldruck-DSC erfolgt die Temperaturbestimmung an den
Meßpfännchen bei der Hochdruck-DSC nicht mehr über die mittlere Spannung Uav, sondern
über den direkt an dem Probenmeßpfännchen abgegriffenen Spannungsabfall. Auf diese
Weise wird also nicht die mittlere Temperatur von Proben- und Referenzstelle, sondern
unmittelbar die Probentemperatur gemessen.
Der Regelkreis für den differentiellen Heizstrom bleibt wie bei dem Normaldruck-Prinzip
bestehen, so daß die Leistungskompensation weiterhin stattfindet. Da die Temperatur des
Temperiermantels beim Aufheizen allerdings immer um ein ∆T größer ist als die eigentliche
Meßpfännchentemperatur, kann es bei starken thermischen Effekten neben der Kompensation
durch die eingebauten Heizdrähte zu einer teilweisen Kompensation durch den sogenannten
Umgebungsanteil [14] kommen. Dies muß bei der Kalibrierung des Kalorimeters
berücksichtigt werden.

Differential Scanning Calorimetry (DSC)
37
3.2 Apparativer Aufbau
Die in dieser Arbeit für die Messungen von Phasenübergängen bei hohen Drücken und hohen
Temperaturen verwendete DSC-Apparatur ermöglicht Messungen in einem Arbeitsbereich
von 293 K bis 480 K bei Drücken von Normaldruck bis zu 200 MPa.
Die von Schmidt [63] aufgebaute Autoklaveneinheit entstand in Anlehnung an eine bereits
vorhandene Tieftemperatur-Hochdruck-DSC-Anlage, welche von Arntz [64] konstruiert und
von Wenzel [10] und Ellert [59] weiterentwickelt wurde. Die Druckversorgung, ein Großteil
der Regel- und Steuerelektronik, sowie die Datenerfassung wurden von der existierenden
Apparatur übernommen und stehen wahlweise beiden Autoklaveneinheiten zur Verfügung.
Bei der in den folgenden Kapiteln beschriebenen Apparatur handelt es sich ausschließlich um
die Hochtemperatur-DSC-Anlage.
Ergänzend zur Hochdruck-DSC steht ein Gerät des Typs DSC-7 der Firma Perkin-Elmer für
Normaldruckmessungen mit einem Arbeitsbereich von 273 K bis 773 K zur Verfügung. Über
ein Interface können die Meßdaten einem IBM-kompatiblen Rechner zugänglich gemacht
werden. Die Auswertung und Bearbeitung der Meßdaten erfolgt über die mitgelieferte Pyris-
Software, welche in einer aktuelleren Version neben Temperatur- und
Enthalpiebestimmungen auch die Bestimmung von Wärmekapazitäten ermöglicht.
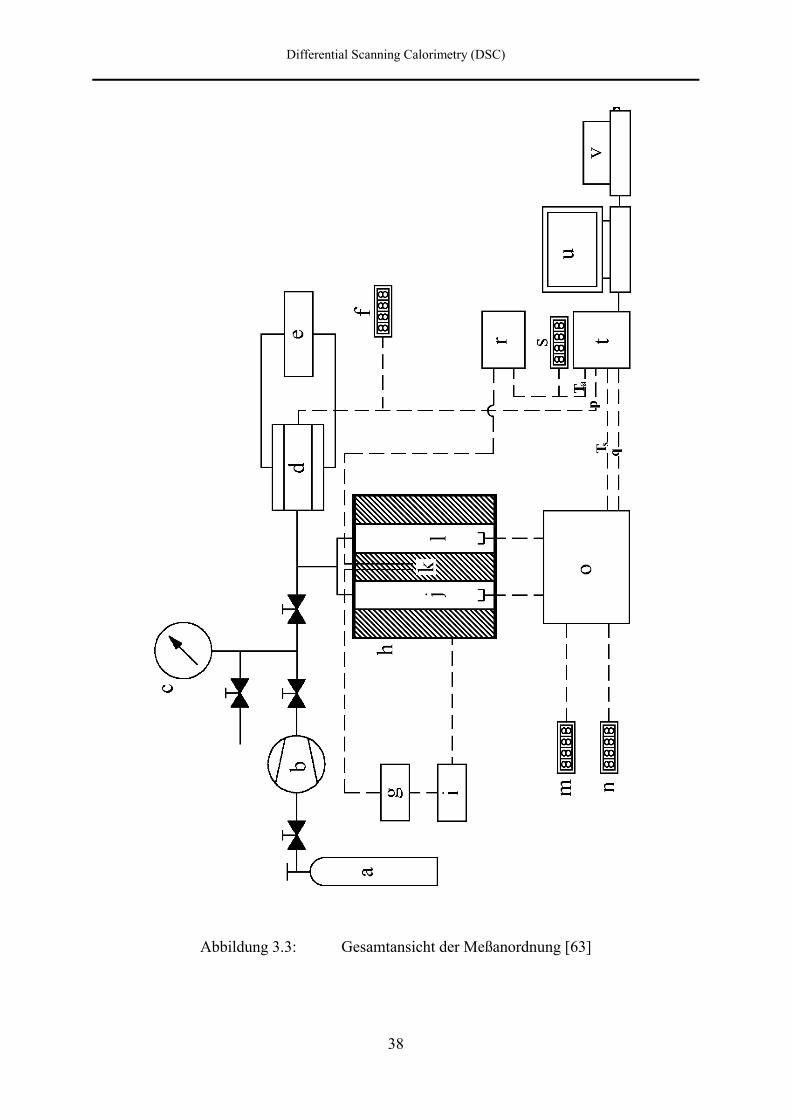
3.2.1 Übersicht über die Meßanordnung
Abbildung 3.3 liefert eine schematische Übersicht über die gesamte Meßanordnung. Die
beiden identisch angefertigten Hochdruckautoklaven (j und l) bilden das Herzstück dieser
Anlage. Sie enthalten die Meßköpfe mit dem Probenmeßpfännchen und dem
Referenzmeßpfännchen der Firma Perkin-Elmer [L 1].
Die Autoklaven sind von einem Temperiermantel (h) aus Aluminium umschlossen, welcher
über insgesamt dreizehn Heizpatronen beheizt und über Bohrungen mit Preßluft gekühlt
werden kann. Die Heizpatronen sind dabei so angeordnet, daß jeder Autoklav gleichmäßig
von acht Heizpatronen umgeben ist. Die Temperaturmessung des Mantels erfolgt über zwei
Differential Scanning Calorimetry (DSC)
38
Abbildung 3.3: Gesamtansicht der Meßanordnung [63]

Differential Scanning Calorimetry (DSC)
39
a Druckgasvorrat b Kompressor
c Manometer d Druckmeßdose
e Thermostat f Digitalvoltmeter
g Temperaturprogrammgeber h Temperiermantel
i Heizungsregler j, l Autoklaven
k Pt-100 Widerstandsthermometer m, n Digitalvoltmeter
o DSC-2 Gerät r Meßverstärker
s Digitalvoltmeter t Interface
u Computer v Drucker
Ta Autoklavtemperatur
Ts Pfännchentemperatur der Probe
p Druck
q Differenzheizleistung

Differential Scanning Calorimetry (DSC)
40
Pt-100 Platinwiderstandsthermometer (k), wobei eines zur Meßwertaufnahme dient, während
das andere Pt-100 zusammen mit dem Temperaturprogrammgeber (g) und dem
Heizungsregler (i) die im Temperiermantel befindlichen Heizpatronen ansteuert.
Die Druckversorgung erfolgt mittels eines durch Preßluft angetriebenen zweistufigen
Kompressors (b) [L 2], mit dem das Druckgas, wahlweise Helium [L 3] oder Argon [L 3], aus
einer Vorratsflasche (a) durch ein Kapillarsystem in die Autoklaven gedrückt wird.
Zur Druckkontrolle stehen ein Manometer (c) [L 4] mit einem Druckbereich von 0,1 MPa bis
400 MPa, sowie eine über einen Thermostaten (e) [L 5] temperierte Dehnungsmeßstreifen-
Druckdose (d) [L 6] mit angeschlossenem Digitalvoltmeter (f) [L 7] zur Verfügung.
Die Meßköpfe sind über druckdichte elektrische Zuleitungen mit einem DSC-2 Gerät (o) [L 1]
verbunden, von dem aus die Meßgrößen für die Meßpfännchentemperatur (Ts) und die
Differenzheizleistung (q) über Digitalvoltmeter (m und n) angezeigt werden und einem
Interface (t) [L 8] für die Rechnerauswertung zugeführt werden. Außerdem ist das Interface
mit der Druckmeßdose und über einen Meßverstärker (r) mit einem der beiden
Platinwiderstandsthermometer des Temperiermantels verbunden.
Die Meßdaten werden innerhalb des Interface über einen 12 bit Analog/Digital-Wandler
digitalisiert und einem Computer (u) [L 9] mit angeschlossenem Drucker (v) [L 10] zugeführt.
Mit Hilfe eines von Ellert [59] erstellten Programmes werden die Meßdaten ausgewertet und
die Meßergebnisse grafisch dargestellt.
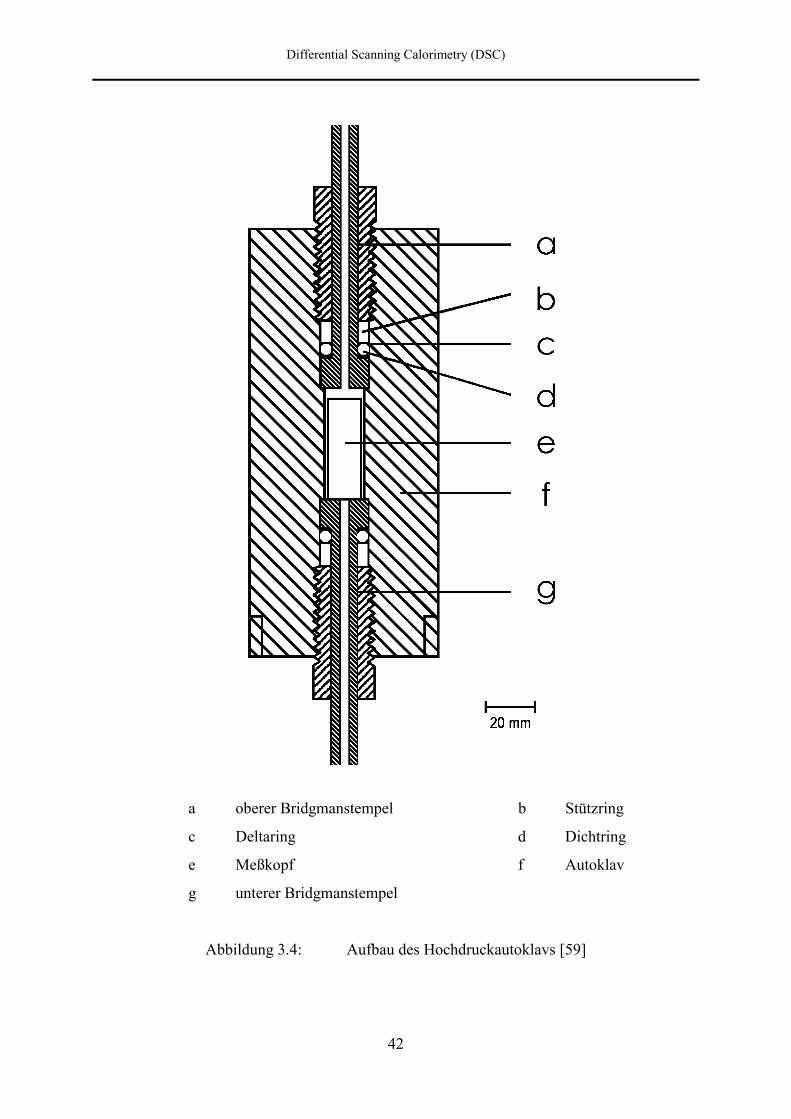
3.2.2 Hochdruckautoklav
Abbildung 3.4 zeigt schematisch den Aufbau eines der beiden identischen
Hochdruckautoklaven (f). Er ist aus Nimonic 90 gefertigt, einem Werkstoff, der gute
Hochdruckeigenschaften, z. B. eine hohe Fließgrenze und eine große Zugfestigkeit, besitzt.
Der obere und untere Teil des Autoklavkörpers wird jeweils durch einen axial durchbohrten,
aus einer Chrom-Nickel-Legierung (Inconel 716) gefertigten Stempel (a und g) verschlossen.
Die Stempel arbeiten nach dem Bridgman-Prinzip, wobei die eigentliche Abdichtung durch

Differential Scanning Calorimetry (DSC)
41
Dichtringe (d) aus Polyethylen (RCH 100) erfolgt, welche, um ein Wegfließen des Materials
unter Druck zu verhindern, in Deltaringe aus Messing (c) eingebettet sind. Oberhalb des
Deltaringes befindet sich ein Stützring (b) aus 17-4 PH.
Der obere Bridgmanstempel muß zum Einsetzen der Meßzellen herausgeschraubt werden.
Durch ihn wird das Druckgas in den Autoklaven geleitet. Durch den unteren
Bridgmanstempel werden die elektrischen Zuleitungen des Meßkopfes (e) über ein
Kupplungsstück nach außen geführt.
3.2.3 Meßkopf
Die Autoklaven enthalten jeweils einen der beiden identisch ausgelegten
Hochdruckmeßköpfe. Abbildung 3.5 stellt schematisch den Aufbau eines Meßkopfes (c bis l)
und seine Anordnung im Autoklav dar, während Abbildung 3.6 eine perspektivische Ansicht
des Meßkopfes liefert.
Wiedergegeben sind zunächst die Autoklavwand (b), die beiden den Autoklaven
verschließenden Bridgmanstempel (a und n) und der Meßkopf selbst (c bis l), der über ein
Schraubgewinde fest mit dem unteren Stempel verbunden ist.
Das aus einer Platin-Iridium-Legierung bestehende Meßpfännchen (e), das die Meßzelle (d)
aufnimmt, ist ein Standardmeßpfännchen aus dem für Normaldruckmessungen ausgelegten
Meßkopf eines DSC-2-Gerätes der Firma Perkin-Elmer [L 1], kann aber ohne Veränderungen
für Hochdruckmessungen verwendet werden. Das zylindrische Meßpfännchen ist auf einer
Kupferscheibe befestigt und enthält in seinem Boden zwei identische, um 90° versetzt
übereinander angeordnete Platindraht-Widerstände, von denen einer als Temperaturfühler und
der zweite als Heizwiderstand arbeitet. In die Kupferscheibe sind außerdem vier elektrisch
voneinander isolierte Metalldrähte montiert, an die, oberhalb der Kupferscheibe, dünne
Platinbändchen zur elektrischen Verbindung an die Platinwiderstände des Meßpfännchens
angelötet sind. Unterhalb der Kupferscheibe sind an diesen Metalldrähten die
Kupferdrahtzuleitungen (m) angelötet.
Differential Scanning Calorimetry (DSC)
42
a oberer Bridgmanstempel b Stützring
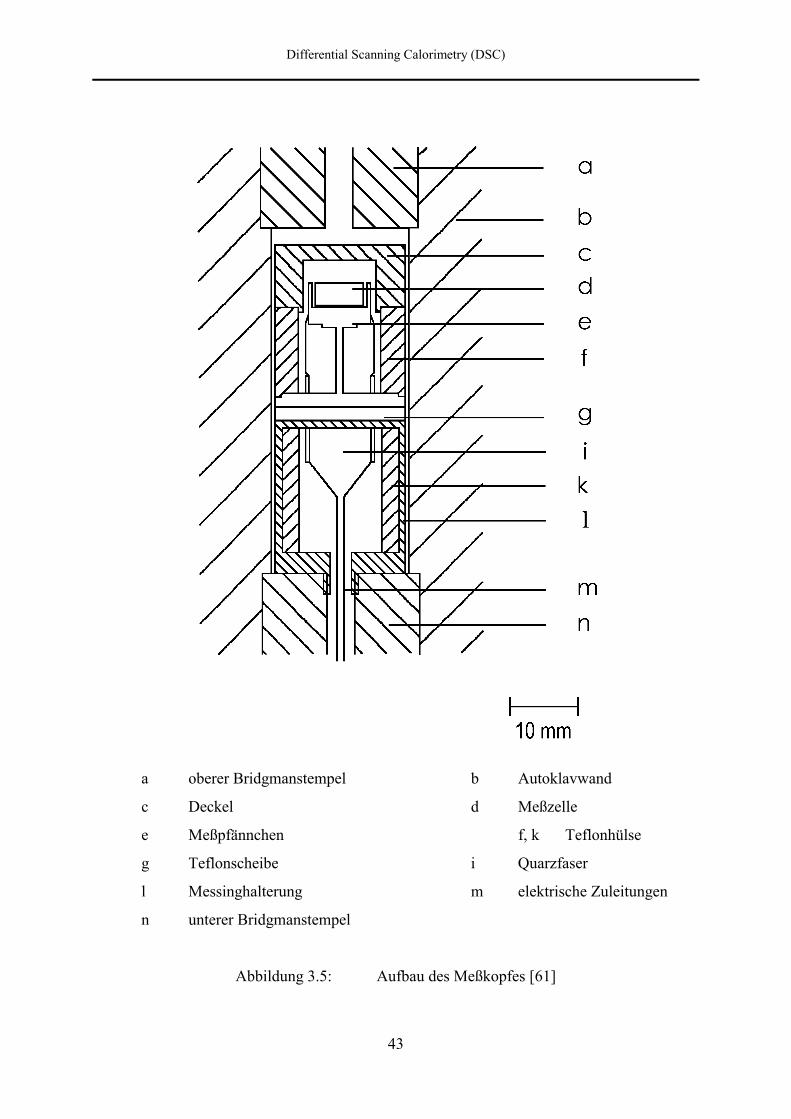
c Deltaring d Dichtring
e Meßkopf f Autoklav
g unterer Bridgmanstempel
Abbildung 3.4: Aufbau des Hochdruckautoklavs [59]
Differential Scanning Calorimetry (DSC)
43
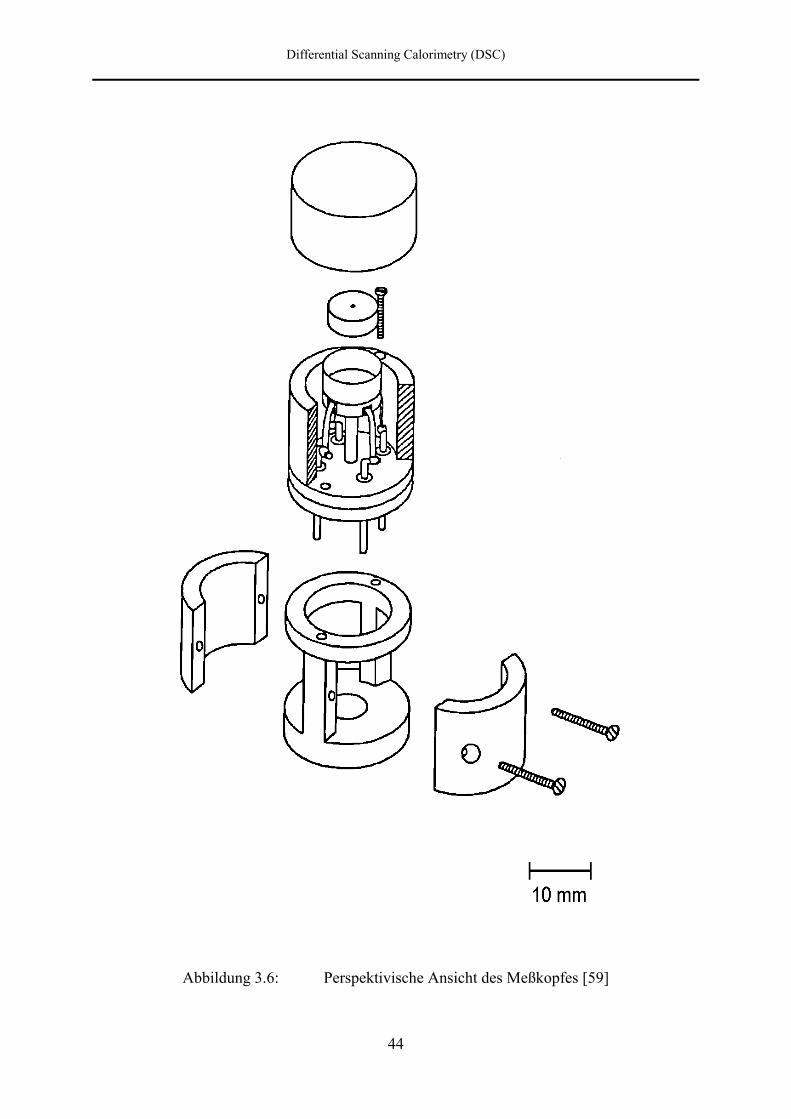
a oberer Bridgmanstempel b Autoklavwand
c Deckel d Meßzelle
e Meßpfännchen f, k Teflonhülse
g Teflonscheibe i Quarzfaser
l Messinghalterung m elektrische Zuleitungen
n unterer Bridgmanstempel
Abbildung 3.5: Aufbau des Meßkopfes [61]
Differential Scanning Calorimetry (DSC)
44
Abbildung 3.6: Perspektivische Ansicht des Meßkopfes [59]

Differential Scanning Calorimetry (DSC)
45
Die Kupferscheibe mit dem Meßpfännchen ist mit zwei Schrauben über eine Teflonscheibe
(g) auf einer Messinghalterung (l) befestigt, die an der Unterseite mit einem Gewinde in den
unteren Bridgmanstempel eingeschraubt ist. Aus konstruktionstechnischen Gründen mußte die
Kupferscheibe dazu von 22 mm Durchmesser auf 19 mm abgedreht werden.
Da Temperaturunterschiede zwischen der Autoklavwand und dem Meßpfännchen Gaswirbel
verursachen können, ist das Meßpfännchen von einer Teflonhülse (f) umgeben. Über der
Hülse befindet sich ein Deckel (c) aus Polyethylen. Zwei weitere Teflonhalbschalen (k)
umgeben die Messinghalterung. Die Zwischenräume (i) sind mit Quarzfaser ausgestopft.
3.2.4 Elektrische Durchführung
Bei den erwähnten elektrischen Zuleitungen handelt es sich pro Autoklav um vier mit Capton-
Folie isolierte Kupferdrähte mit einem Durchmesser von 0.8 mm. Um die elektrischen
Zuleitungen gegen den Außendruck abzudichten, werden sie durch den unteren
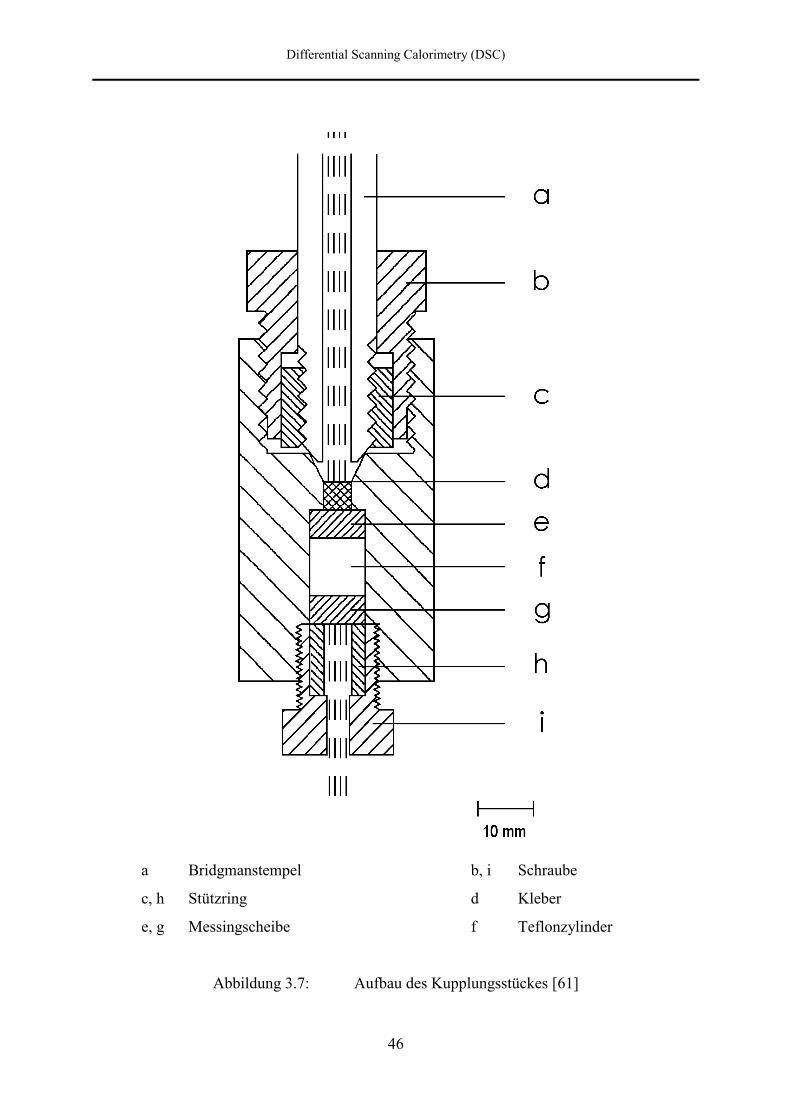
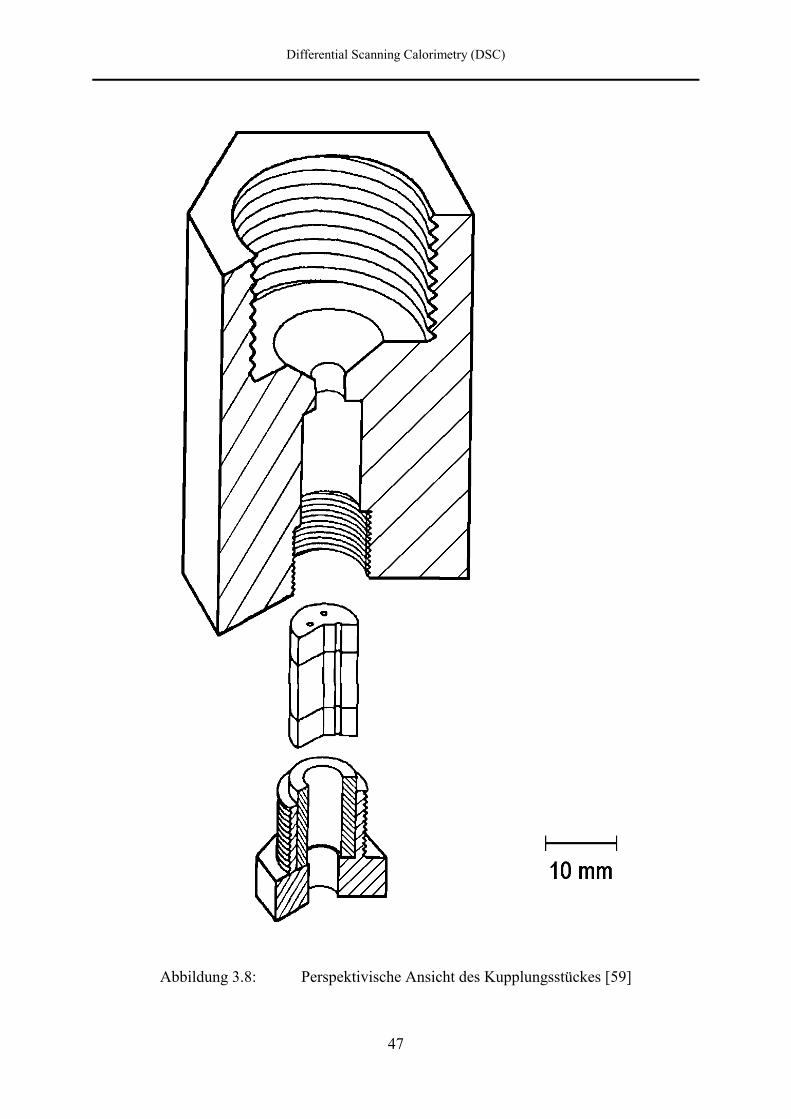
Bridgmanstempel einem Kupplungsstück zugeführt, das in Abbildung 3.7 schematisch und in
Abbildung 3.8 perspektivisch mit einem 120° Schnitt dargestellt ist.
Der Bridgmanstempel (a) wird mit einer Schraube (b) und einem Stützring (c) über eine
Konusdichtung mit dem Kupplungsstück verbunden. Die Dichtung der Kupferdrähte erfolgt
durch einen 10 mm langen Teflonzylinder (f), der zwischen zwei 5 mm dicken
Messingscheiben (e und g) mittels einer Schraube (i) und eines Stützringes aus gehärtetem
Stahl (h) zusammengepreßt wird. Der Teflonzylinder und die Messingscheiben enthalten vier
Bohrungen mit einem Durchmesser von 1 mm, durch die die Kupferdrähte geführt sind.
Durch das Zusammenpressen des Zylinders beginnt die Teflonmasse zu fließen und quetscht
die Drähte ein. Zusätzlich sind die Bohrungen (d) mit dem Klebstoff UHU Plus [L 11] gefüllt.
Außerdem soll ein kleiner Edelstahlkeil in der Nut des Kupplungsstückes und der unteren
Messingscheibe verhindern, daß sich beim Anziehen der Schraube Teflonzylinder und
Messingscheibe gegeneinander verschieben und somit die Isolierung der Kupferdrähte
beschädigt wird.
Differential Scanning Calorimetry (DSC)
46
a Bridgmanstempel b, i Schraube
c, h Stützring d Kleber
e, g Messingscheibe f Teflonzylinder
Abbildung 3.7: Aufbau des Kupplungsstückes [61]
Differential Scanning Calorimetry (DSC)
47
Abbildung 3.8: Perspektivische Ansicht des Kupplungsstückes [59]

Differential Scanning Calorimetry (DSC)
48
3.2.5 Meßzellen
Für DSC-Messungen unter hohem Druck ist es erforderlich, daß die Meßzellen mehrere
Bedingungen gleichzeitig erfüllen. Zunächst muß eine hysteresefreie Druckübertragung auf
die Substanz gewährleistet sein. Die verwendeten Meßzellen dürfen sich unter Druck nicht
verformen, da ein schlechter Kontakt zum Meßpfännchen eine Änderung des
Wärmeübergangskoeffizienten bewirkt. Außerdem darf das druckübertragende Medium nicht
mit der Substanz in Berührung kommen, da dies eine Verunreinigung zur Folge hätte, die sich
auf die Umwandlungsgrößen auswirkt.
Bei den in dieser Arbeit verwendeten Meßzellen handelt es sich um Hochdruckzellen der
Firma Perkin-Elmer in etwas abgewandelter Form (Abbildung 3.9a). Die Meßzellen sind aus
rostfreiem, korrosionsgeschütztem Stahl gefertigt. Sie bestehen aus einem Unterteil (e) und
einem aufschraubbaren Deckel (a), wiegen etwa 0.62 g, haben ein nutzbares Volumen von
25 µl und können bis zu maximal 400° C benutzt werden. Diese Zellen sind normalerweise
für Messungen an flüchtigen Substanzen vorgesehen und halten mit entsprechenden Kupfer-
Dichtungsringen einem Innendruck von 15 MPa stand. Um einen hohen äußeren Druck auf
das Zelleninnere übertragen zu können, wurde in den Deckel der Zelle ein Loch (Durchmesser
1 mm) gebohrt. Eine Membran aus Aluminium (b) schützt die Substanz (d) nach dem
Einfüllen in das Unterteil der Zelle vor dem direkten Kontakt mit dem Druckgas. Die
Membran, eine kreisförmige Aluminiumscheibe mit einem Durchmesser von 6.5 mm und
einer Dicke von 0.1 mm, wird mit UHU Plus (c) [L 11] auf das Unterteil geklebt.
Anschließend wird der Deckel auf das Zellenunterteil geschraubt.
Membran und Klebestelle stellen die Schwachpunkte dieser Konstruktion dar. Insbesondere
organische Flüssigkeiten neigen dazu, über den Rand der Zelle zu kriechen und ein sicheres
Verkleben der Membran zu verhindern. Auch das Aufschrauben des Deckels muß mit
Vorsicht geschehen, um die Membran nicht zu beschädigen, da diese sonst bei hohen Drücken
reißt.
Für Normaldruckmessungen oder für die Messungen an offenen Zellen wurden die schon in
früheren Arbeiten [59, 63] benutzten ”large volume” Zellen von Perkin-Elmer [L 1]
verwendet (Abbildung 3.9b). Das Unterteil, welches ein Fassungsvermögen von ungefähr 60
µl besitzt, und der dazugehörige Deckel sind aus korrosionsgeschütztem, rostfreiem Stahl
gefertigt und werden durch einen O-Ring abgedichtet.
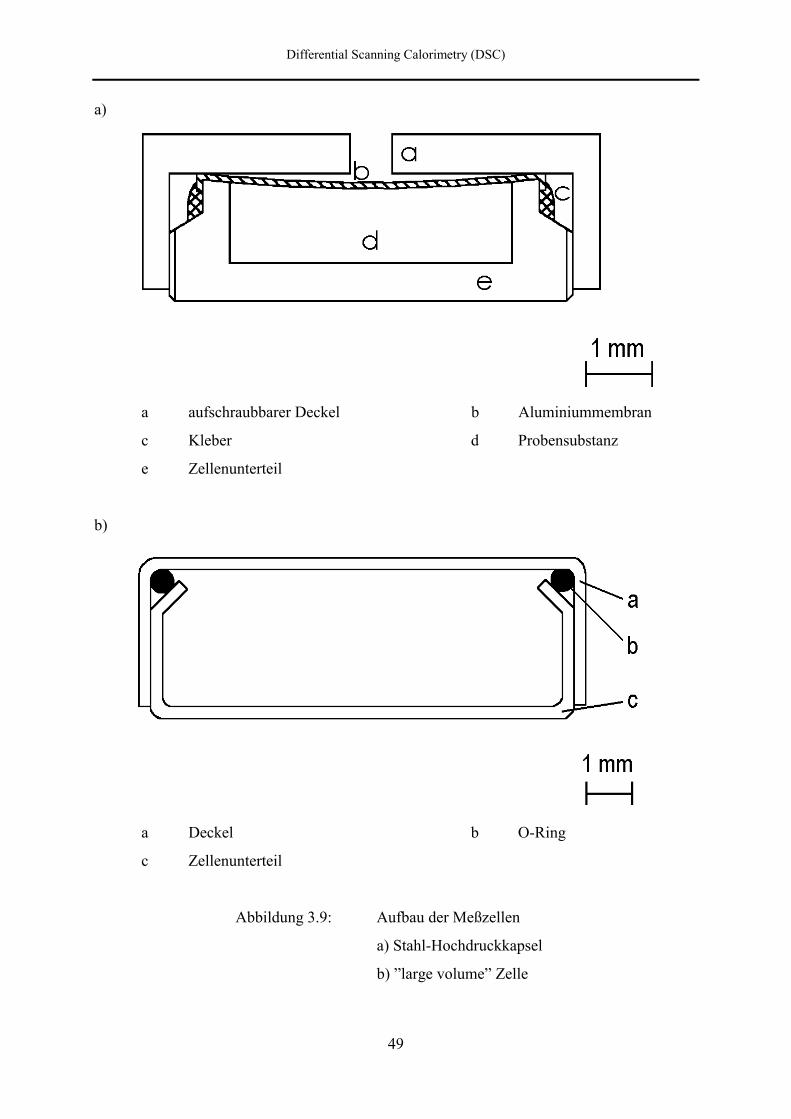
Differential Scanning Calorimetry (DSC)
49
a)
a aufschraubbarer Deckel b Aluminiummembran
c Kleber d Probensubstanz
e Zellenunterteil
b)
a Deckel b O-Ring
c Zellenunterteil
Abbildung 3.9: Aufbau der Meßzellen
a) Stahl-Hochdruckkapsel
b) ”large volume” Zelle

Differential Scanning Calorimetry (DSC)
50
Der Vorteil dieser Zelle gegenüber der zuvor beschriebenen Hochdruckkapsel liegt darin, daß
sie bei gleicher Substanzmenge noch nicht vollständig gefüllt ist und somit ein Auslaufen der
Substanz aus der Zelle vermieden wird.
3.2.6 Temperiermantel
Der in Abbildung 3.10 dargestellte Temperiermantel ist aus Aluminium gefertigt und so
konstruiert, daß die beiden identischen Hochdruckautoklaven symmetrisch von insgesamt
dreizehn Heizpatronen umgeben sind. Die Heizpatronen, die eine Länge von 160 mm und
einen Durchmesser von 8 mm aufweisen, erbringen eine Heizleistung von je 200 Watt [63].
Sie werden über einen Programmgeber und einen Regler angesteuert, welcher für den
Temperaturbereich von 273 K bis 773 K ausgelegt ist und zusammen mit dem
Temperaturprogrammgeber Heizraten von 0.1 K ⋅ min -1 bis 10 K ⋅ min -1 erlaubt. Die Ist-
Werte für diese Regelung liefert ein über eine Bohrung in den Temperiermantel eingelassenes
Pt-100 Widerstandsthermometer. Durch die symmetrische Anordnung der Heizelemente soll
eine lineare Aufheizrate beider Autoklaven und der darin befindlichen Meßköpfe
gewährleistet werden. Das Auftreten zu unterschiedlicher Temperaturen des Proben- und
Referenzmeßkopfes soll auf diese Weise verhindert und eine Grundliniendrift minimiert
werden. Darüberhinaus reduziert ein den Temperiermantel umgebender, mit einem Asbest-
Ersatzstoff gefüllter Blechmantel die Wärmeableitung an die Umgebung während des
Aufheizens.
Nach einer erfolgten Messung kann über weitere, insgesamt vierzehn Bohrungen Preßluft
durch den Temperiermantel geleitet werden, um die Autoklaven wieder abzukühlen. Die
Abkühlgeschwindigkeit ist dabei allerdings nicht regelbar.
3.2.7 Druckerzeugung und -messung
Zur Druckerzeugung wird ein mit Preßluft angetriebener zweistufiger Kompressor [L 2] ver-
wendet. Als druckübertragendes Medium stehen wahlweise Helium [L 3] oder Argon [L 3]
zur Verfügung. Die Druckkontrolle erfolgt über ein Präzisionsmanometer [L 4] mit einem
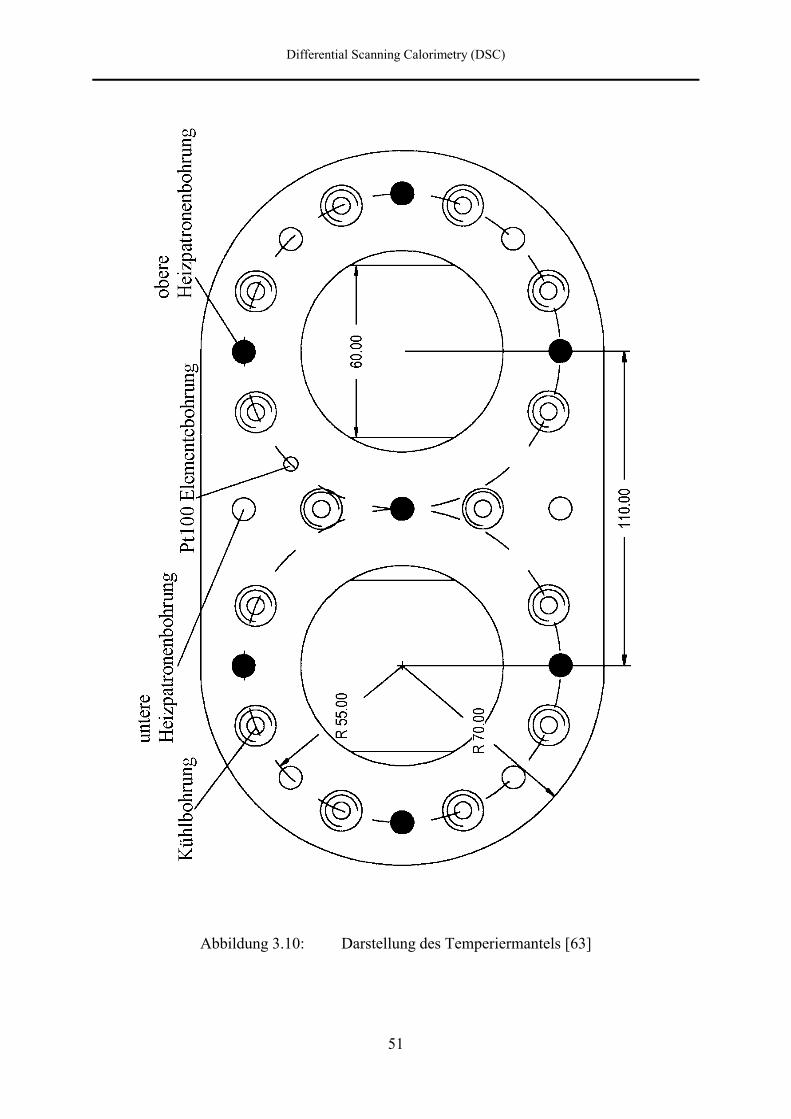
Differential Scanning Calorimetry (DSC)
51
Abbildung 3.10: Darstellung des Temperiermantels [63]

Differential Scanning Calorimetry (DSC)
52
Meßbereich von 0.1 MPa bis 400 MPa und über eine Dehnungsmeßstreifen-Druckdose [L 6],
die von einem Temperiermantel aus Kupfer umgeben ist und über einen Thermostaten [L 5]
auf 313 K temperiert wird, um von der veränderlichen Raumtemperatur unabhängig zu sein.
Ein an die Druckdose angeschlossener Rechenverstärker ermöglicht es, daß der aktuelle
Druck während der Messung durch ein Digitalvoltmeter [L 7] angezeigt und über einen A/D-
Wandler kontinuierlich von einem Rechner [L 9] erfaßt werden kann. Eine kontinuierliche
Druckmessung ist hier notwendig, da der Druck während des Aufheizens der Autoklaven
ständig steigt.
3.2.8 Meßwerterfassung
Zur Meßwerterfassung dient ein von Ellert [59] angeschlossenes Rechnersystem und die dazu
erstellte Software. Die Meßsignale der bei der Hochdruck-DSC benötigten vier Meßgrößen:
Differenzheizleistung, Zellentemperatur, Autoklavtemperatur und Druck werden einem am
Lehrstuhl gebauten Interface zugeführt. Über vier unabhängige Eingangskanäle werden die
analogen Ausgangsspannungen der jeweiligen Meßstellen einem 12-bit Analog/Digital-
Wandler zugewiesen, wobei softwaremäßig zwischen den einzelnen Eingangskanälen
umgeschaltet werden kann. Die digitalisierten Signale werden über einen IEEE-Bus [L 8]
einem Computer [L 9] zur Aufnahme sowie weiteren Verarbeitung und Speicherung
zugeführt.
Der erste Eingangskanal besitzt einen Eingangsspannungsbereich von -500 bis +500 mV und
wird für die der Differenzheizleistung proportionale Spannung verwendet.
Dem zweiten Kanal wird die am Probenpfännchen abgegriffene Spannung zugeführt. Der
Eingangsbereich von 0 bis 500 mV ist softwaremäßig auf einen Bereich von 0 bis 2500 mV
erweiterbar. Da ein Spannungswert von 500 mV ungefähr Raumtemperatur zuzuordnen ist,
wird für die Hochtemperatur-DSC-Anlage der größere Bereich benutzt. Dabei ist jedoch zu
berücksichtigen, daß die maximale Auflösung der Meßsignale vom
Eingangsspannungsbereich abhängt. Die maximale Empfindlichkeit liegt auf Grund des 12-bit
A/D-Wandlers bei 1/4096 des zu digitalisierenden analogen Spannungsbereiches, so daß bei
vergrößertem Meßbereich ein Genauigkeitsverlust in Kauf genommen werden muß.
Über den dritten Kanal kann wahlweise die der Temperaturanzeige des DSC-Gerätes
proportionale Spannung eingelesen oder die vom Pt-100 gelieferte und über einen

Differential Scanning Calorimetry (DSC)
53
Meßverstärker verstärkte, zur Temperatur im Temperiermantel proportionale Spannung
aufgenommen werden. Der ursprüngliche Eingangsspannungsbereich von 0 bis 5000 mV
reichte für die hohen Temperaturen des Heizmantels nicht mehr aus und wurde von Schmidt
[63] mit Hilfe eines Spannungsteilers verändert, so daß jetzt drei extern einstellbare Bereiche
zur Verfügung stehen: 0 bis 5000 mV, 0 bis 8300 mV und 0 bis 10000 mV. In dieser Arbeit
wurde für alle Messungen mit dem zweiten Eingangsbereich gearbeitet.
Der vierte Eingangskanal ist für einen Spannungsbereich von 0 bis 5000 mV ausgelegt und
wird zur Aufnahme der von der Druckmeßdose über einen Verstärker gelieferten
Spannungswerte benutzt.
3.3 Auswertung von DSC-Peaks
Die Peakauswertung erfolgt durch eine von Ellert [59] speziell entwickelte Software voll
rechnergestützt und wird in den folgenden Abschnitten etwas näher erläutert.
Die Signalspannungen der vier Meßgrößen: Differenzheizleistung, Zellentemperatur,
Autoklavtemperatur und Druck werden in Abhängigkeit von der Zeit registriert und
aufgezeichnet. Eine Phasenumwandlung der Probensubstanz zeigt sich in einer Abweichung
des Differenzheizleistungssignals von der Grundlinie und ergibt in der Aufzeichnung den für
DSC-Messungen typischen Peakverlauf.
Die digitalisierten DSC-Kurven enthalten oftmals Datenpunkte, die als “Ausreißer”
bezeichnet werden können, da sie im allgemeinen deutlich vom restlichen Datenverlauf
abweichen.
Diese Ausreißer, welche zum Beispiel durch fehlerhafte Digitalisierung, durch
Spannungsspitzen der Signalquellen, durch sogenanntes “Prellen” der Umschaltrelais oder
allgemein durch Störungen in den Datenleitungen zustande kommen können, führen bei der
Auswertung des Peaks zu Fehlern. Sie werden entfernt, indem der Kurvenverlauf aus der
Steigung des Signals vor und nach dem Ausreißer auf den ursprünglichen Kurvenverlauf
zurückgerechnet wird.

Differential Scanning Calorimetry (DSC)
54
3.3.1 Einfluß der Versuchsbedingungen auf die Peakform
Das Aussehen eines Thermogramms wird durch eine Reihe apparativer Faktoren und
Eigenschaften der Proben- und Referenzsubstanz beeinflußt. Dazu zählen u.a. [65, 66]:
- die Ofenatmosphäre
- das Material und die Anordnung der Meßzellen
- die Geometrie der Thermoelemente
- die Wärmeleitung zwischen Wärmequelle und Meßstelle
- die Anfangstemperaturen der Substanzen
- Wärmeleitfähigkeit und Wärmekapazität der Substanzen
- Teilchengröße und Packungsdichte
- die Aufheizgeschwindigkeit
- die Probenmenge, ...
Schulze [67] und Wendlandt [68] geben eine ausführliche Beschreibung der oben
aufgeführten Parameter. Im folgenden wird nur kurz der Einfluß der Aufheizgeschwindigkeit
etwas näher betrachtet. Die Größe der Peakfläche und die Lage des Peakmaximums sind stark
von der Aufheizrate abhängig. Eine Erhöhung der Aufheizgeschwindigkeit bewirkt eine
Verschiebung des Peakmaximums zu höheren Temperaturen. Da die pro Zeiteinheit
umgewandelte Stoffmenge größer wird, werden die Peaks höher und breiter, so daß sich auch
die Peakfläche vergrößert. Bei hohen Heizraten besteht die Gefahr, daß benachbarte Peaks
nicht mehr getrennt aufgelöst werden.
3.3.2 Festlegung des Umwandlungspunktes
In der Literatur werden unterschiedliche Methoden zur Bestimmung der Umwandlungs-
temperatur aus DSC- bzw. DTA-Meßkurven beschrieben [69].
Ein sehr einfaches Verfahren ist die Bestimmung der Umwandlungstemperatur am
Peakmaximum. Da die Peakform, insbesondere die Lage des Peakmaximums, von der
Wärmekapazität und Wärmeleitfähigkeit der Probe, sowie von der Probenmenge und der

Differential Scanning Calorimetry (DSC)
55
Aufheizrate abhängig ist, was eine gute Reproduzierbarkeit erschwert, wird diese Methode
häufig nur bei der routinemäßigen Untersuchung gleicher Proben angewendet.
Einige Autoren [69, 70, 71] verweisen auf die Möglichkeit, die Bestimmung der
Umwandlungstemperatur mit Hilfe der maximalen Steigung der Peakanstiegsflanke einer sehr
reinen Referenzsubstanz im interessierenden Temperaturbereich durchzuführen. Dabei wird
eine Gerade mit dieser Steigung vom Peakmaximum zur verlängerten Grundlinie des
auszuwertenden Peaks gezogen, wobei der Schnittpunkt mit der Grundlinie den
Umwandlungspunkt mit der entsprechenden Temperatur liefert. Diese Methode eignet sich
allerdings ebenfalls nur für Substanzen, die eine ähnliche Peakform aufweisen. Wird als
Referenzsubstanz beispielsweise ein Metall mit einem scharfen Peak verwendet, so ist es
nicht sinnvoll, die Steigung dieses Peaks bei der Auswertung einer organischen Substanz mit
einem relativ breiten Peak zu benutzen.
Weitere Methoden zur Bestimmung des Umwandlungspunktes werden in den Arbeiten von
Hamann [66] und Ernst [65] diskutiert. Hamann benutzt die halbe Peakhöhe zur Festlegung
des Umwandlungspunktes, während Ernst den Wendepunkt der Peakanstiegsflanke des
auszuwertenden Peaks als Umwandlungspunkt definiert.
Bei dem in dieser Arbeit verwendeten Verfahren [145] wird der Schnittpunkt aus der am
Wendepunkt der Peakanstiegsflanke angelegten Tangente mit der Verlängerung der
Grundlinie vor dem Peak ermittelt und als Umwandlungspunkt festgelegt. Die zu diesem
Zeitpunkt registrierten Werte für die Probentemperatur und den Druck werden als
Umwandlungstemperatur und Umwandlungsdruck definiert. Wie Untersuchungen von Reuter
[73] zeigen, ist zu berücksichtigen, daß die nach dem oben beschriebenen Verfahren
ermittelten Umwandlungstemperaturen bei unterschiedlichen Heizraten nicht genau
reproduzierbar sind. Auf die in der Literatur beschriebene Extrapolation der
Umwandlungstemperatur auf eine Heizrate von 0 K ⋅ min -1 wurde jedoch verzichtet, da die
Abhängigkeit der Umwandlungstemperatur von der Heizrate erst bei höheren Heizraten (> 5
K ⋅ min -1) signifikant wird [145]. Die in dieser Arbeit bei allen Messungen verwendete
Aufheizrate lag bei 1 K ⋅ min -1 beziehungsweise 2 K ⋅ min -1 und entsprach immer der
Heizrate, mit der die Kalibrierung durchgeführt wurde.
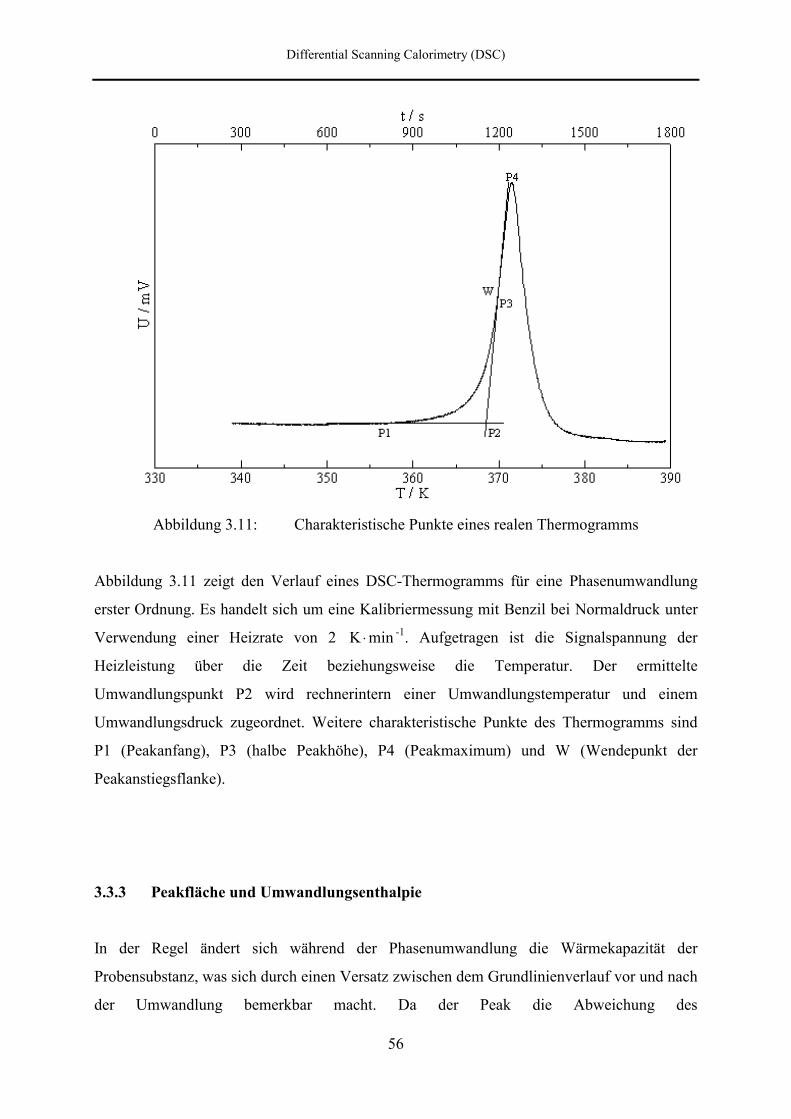
Differential Scanning Calorimetry (DSC)
56
Abbildung 3.11: Charakteristische Punkte eines realen Thermogramms
Abbildung 3.11 zeigt den Verlauf eines DSC-Thermogramms für eine Phasenumwandlung
erster Ordnung. Es handelt sich um eine Kalibriermessung mit Benzil bei Normaldruck unter
Verwendung einer Heizrate von 2 K ⋅ min -1. Aufgetragen ist die Signalspannung der
Heizleistung über die Zeit beziehungsweise die Temperatur. Der ermittelte
Umwandlungspunkt P2 wird rechnerintern einer Umwandlungstemperatur und einem
Umwandlungsdruck zugeordnet. Weitere charakteristische Punkte des Thermogramms sind
P1 (Peakanfang), P3 (halbe Peakhöhe), P4 (Peakmaximum) und W (Wendepunkt der
Peakanstiegsflanke).
3.3.3 Peakfläche und Umwandlungsenthalpie
In der Regel ändert sich während der Phasenumwandlung die Wärmekapazität der
Probensubstanz, was sich durch einen Versatz zwischen dem Grundlinienverlauf vor und nach
der Umwandlung bemerkbar macht. Da der Peak die Abweichung des
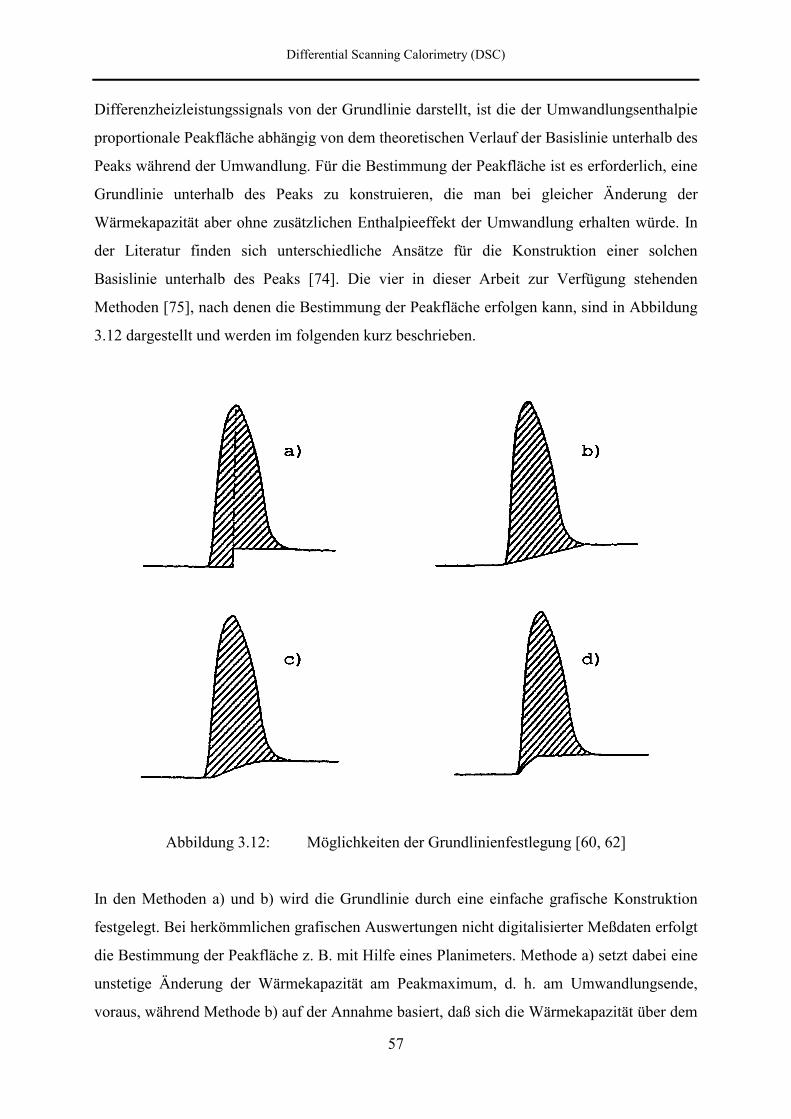
Differential Scanning Calorimetry (DSC)
57
Differenzheizleistungssignals von der Grundlinie darstellt, ist die der Umwandlungsenthalpie
proportionale Peakfläche abhängig von dem theoretischen Verlauf der Basislinie unterhalb des
Peaks während der Umwandlung. Für die Bestimmung der Peakfläche ist es erforderlich, eine
Grundlinie unterhalb des Peaks zu konstruieren, die man bei gleicher Änderung der
Wärmekapazität aber ohne zusätzlichen Enthalpieeffekt der Umwandlung erhalten würde. In
der Literatur finden sich unterschiedliche Ansätze für die Konstruktion einer solchen
Basislinie unterhalb des Peaks [74]. Die vier in dieser Arbeit zur Verfügung stehenden
Methoden [75], nach denen die Bestimmung der Peakfläche erfolgen kann, sind in Abbildung
3.12 dargestellt und werden im folgenden kurz beschrieben.
Abbildung 3.12: Möglichkeiten der Grundlinienfestlegung [60, 62]
In den Methoden a) und b) wird die Grundlinie durch eine einfache grafische Konstruktion
festgelegt. Bei herkömmlichen grafischen Auswertungen nicht digitalisierter Meßdaten erfolgt
die Bestimmung der Peakfläche z. B. mit Hilfe eines Planimeters. Methode a) setzt dabei eine
unstetige Änderung der Wärmekapazität am Peakmaximum, d. h. am Umwandlungsende,
voraus, während Methode b) auf der Annahme basiert, daß sich die Wärmekapazität über dem

Differential Scanning Calorimetry (DSC)
58
gesamten Peak (d.h. auch nach dem Maximum) linear ändert. Beide Methoden liefern bei
symmetrischen Peaks identische Flächenwerte.
Die in dieser Arbeit angewendete Methode c) stellt ein von Brennan, Miller und Whitwell
[76] vorgeschlagenes Verfahren zur rechnergestützten Auswertung dar. Es besteht darin, den
Grundlinienverlauf unter dem Peak der Peakform anzupassen und wird in ähnlicher Form
auch in den Arbeiten von Xiang und Yunliang [77] und von Bandara [78] beschrieben. Die
Auswertung nach dieser Methode beruht auf einem Iterationsverfahren, bei dem der Verlauf
der Grundlinie unter dem Peak solange iterativ wiederholt berechnet wird, bis sich die
Grundlinie nicht mehr signifikant ändert. Schmidt [63] und Wilmers [62] geben einen guten
und detaillierten Überblick über die mathematischen Grundlagen dieser Auswertemethode.
Methode d) beruht ebenfalls auf dem zuvor beschriebenen Verfahren, jedoch erklärt man hier
die Umwandlung bereits am Peakmaximum für beendet und nimmt über dem restlichen Peak
den Grundlinienverlauf der Nachperiode an.
3.3.4 Umwandlungsentropie und -volumen
Die Umwandlungsentropie und das Umwandlungsvolumen sind thermodynamische Größen,
die sich für Umwandlungen erster Ordnung aus der jeweiligen gemessenen
Umwandlungstemperatur und der zugehörigen Umwandlungsenthalpie, bzw. aus der
zusätzlich zu bestimmenden Druckabhängigkeit der Umwandlungstemperatur, mit den in
Kapitel 2.1 hergeleiteten Gleichungen (2.15) und (2.16)
∆∆
trs mtrs m
trsS
HT
= unddpdT
SVkoex
trs m
trs m
=
∆∆
rechnerisch ermitteln lassen.

Differential Scanning Calorimetry (DSC)
59
3.4 Meßtechnik
3.4.1 Meßmethode
Zwischen 20 mg und 30 mg Substanz wurden in die Meßzellen gefüllt und aufgeschmolzen.
Anschließend wurden die Zellen in der in Kapitel 3.2.5 beschriebenen Weise verschlossen.
Als Referenz wurde eine leere Zelle verwendet. Nach dem Einsetzen der Meßzellen in die
Autoklaven und nach dem Anschließen an die Druckversorgung wurde der
Aluminiumtemperiermantel mit konstanter Heizrate von entweder 1 K⋅min-1 oder 2 K⋅min-1
aufgeheizt. Es erwies sich als günstig, die Anfangstemperatur etwa 30 K unter der zu
erwartenden Umwandlungstemperatur anzusetzen. Auf den Temperaturregler des DSC-2
Gerätes wurde verzichtet, lediglich der Regelkreis der Zusatzheizleistung, der das Meßsignal
liefert, blieb in Betrieb. Er arbeitete unabhängig davon, wie die Meßpfännchen aufgeheizt
wurden. Die Pfännchentemperatur lag somit immer etwa 5 K unter der Manteltemperatur.
Nach Beendigung des Temperaturprogramms wurden der Druck abgelassen und die
Autoklaven über die in den Aluminiummantel eingelassenen Bohrungen mit Preßluft gekühlt.
Zuvor wurde die Preßluft durch eine Kupferspirale geleitet, welche sich in einem
Eiswasserbad befand. Auf diese Weise konnte eine Abkühlrate von 1.2 K⋅min-1 bis 1.3 K⋅min-
1 erreicht werden.
3.4.2 Kalibrierung des DSC-Kalorimeters
Bevor die Umwandlungsgrößen einer zu untersuchenden Probensubstanz bestimmt werden
können, muß das Kalorimeter zunächst mit genau bekannten Referenzsubstanzen kalibriert
werden. Die Kalibrierung des DSC-Kalorimeters wird dabei in zwei Bereiche aufgeteilt: die
Temperaturkalibrierung und die Enthalpiekalibrierung.
Zur Bestimmung der Probentemperatur wird der Spannungsabfall am Platinwiderstands-
thermometer des Probenmeßpfännchens herangezogen. Dazu ist es erforderlich, die
registrierte Spannung einer Temperaturskala zuzuordnen.
Zur Kalibrierung der Umwandlungsenthalpie muß der Bezug der gemessenen Peakfläche zu
einer bekannten Energieänderung hergestellt werden.

Differential Scanning Calorimetry (DSC)
60
An die Kalibriersubstanzen werden folgende Anforderungen gestellt: Neben einer hohen
Reinheit sollen sie genau bekannte Umwandlungstemperaturen und Umwandlungsenthalpien
besitzen. Außerdem sollen sie thermisch stabil, nicht hygroskopisch, nicht lichtempfindlich
und gegenüber dem Zellenmaterial inert sein.
Zur Kalibrierung besonders gut geeignet erscheinen Metalle. In der Regel erfüllen sie die oben
genannten Anforderungen und liefern darüberhinaus scharfe und reproduzierbare
Umwandlungspeaks. Es sollten aber auch organische Kalibriersubstanzen verwendet werden
[69], da organische Substanzen eine geringere Wärmeleitfähigkeit als Metalle besitzen und
sich dies auf die Bestimmung der Umwandlungstemperatur auswirkt.
Für Messungen bei hohen Drücken muß zusätzlich eine Druckkalibrierung vorgenommen
werden, da einerseits die Platinwiderstände druckabhängig sind und sich andererseits die
Wärmeleitfähigkeit des Druckmediums in Abhängigkeit vom Druck verändert und damit die
Temperatur- und Enthalpiebestimmung beeinflußt werden. Die Druckkorrektur geschieht mit
Hilfe der Schmelzumwandlung von hochreinem Indium, da sowohl die Druckabhängigkeit der
Umwandlungstemperatur als auch der Umwandlungsenthalpie genau bekannt sind [72, 79,
80].
3.4.2.1 Temperaturkalibrierung
Hinsichtlich des zu kalibrierenden Temperaturbereichs von 300 K bis 450 K werden eine
Anzahl genügend reiner Kalibriersubstanzen, deren Umwandlungsdaten hinreichend genau
bekannt sind, vermessen. Die Auftragung der in der Literatur angegebenen
Umwandlungstemperaturen gegen die experimentell erhaltenen Meßwerte, d. h. die am
Probenmeßpfännchen abgegriffenen und registrierten Spannungswerte am
Umwandlungspunkt, und anschließende Berechnung der Ausgleichsfunktion ergibt die
entsprechende Kalibrierfunktion für die Temperaturbestimmung.
In Tabelle 3.1 und Abbildung 3.13 sind Werte für eine Kalibrierung bei einer Heizrate von
1 K⋅min-1 dargestellt. Bei den aufgeführten Umwandlungstemperaturen Ttrs/K handelt es sich
um Normaldruckwerte. Die Reinheit der zur Kalibrierung herangezogenen Substanzen ist im
allgemeinen größer als 99%.
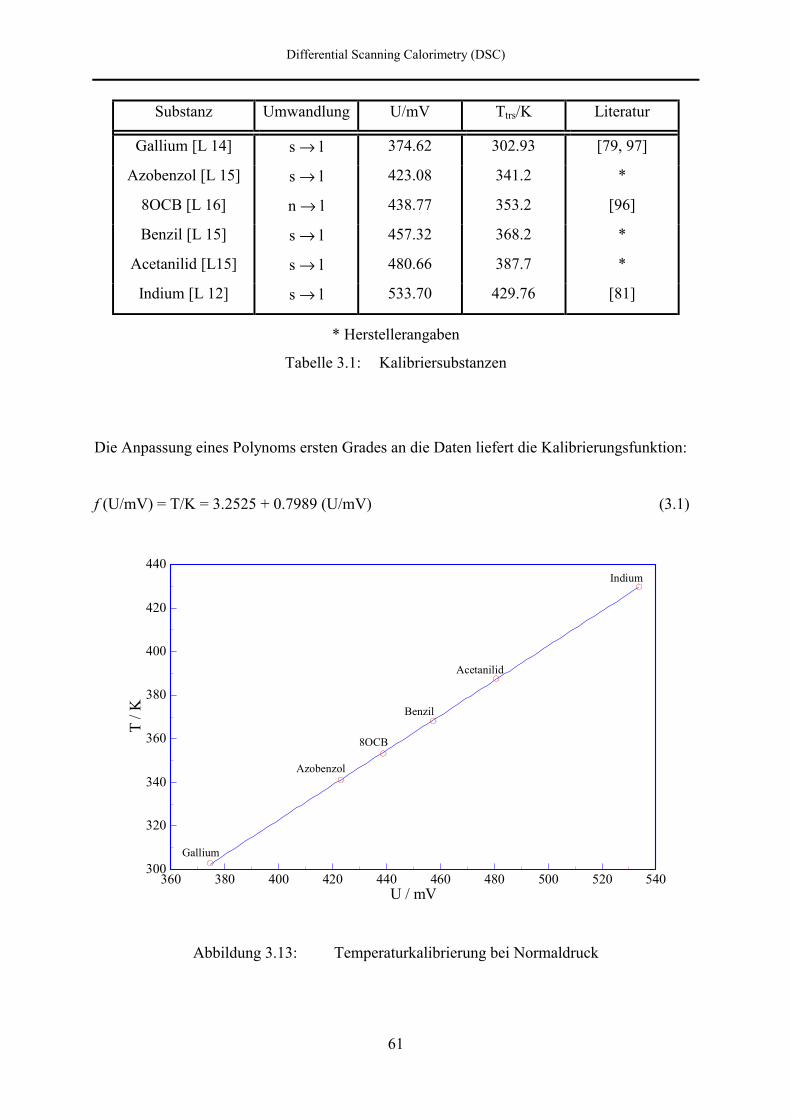
Differential Scanning Calorimetry (DSC)
61
Substanz Umwandlung U/mV Ttrs/K Literatur
Gallium [L 14] s → l 374.62 302.93 [79, 97]
Azobenzol [L 15] s → l 423.08 341.2 *
8OCB [L 16] n → l 438.77 353.2 [96]
Benzil [L 15] s → l 457.32 368.2 *
Acetanilid [L15] s → l 480.66 387.7 *
Indium [L 12] s → l 533.70 429.76 [81]
* Herstellerangaben
Tabelle 3.1: Kalibriersubstanzen
Die Anpassung eines Polynoms ersten Grades an die Daten liefert die Kalibrierungsfunktion:
f (U/mV) = T/K = 3.2525 + 0.7989 (U/mV) (3.1)
360 380 400 420 440 460 480 500 520 540U / mV
300
320
340
360
380
400
420
440
T / K
Gallium
Azobenzol
8OCB
Benzil
Acetanilid
Indium
Abbildung 3.13: Temperaturkalibrierung bei Normaldruck
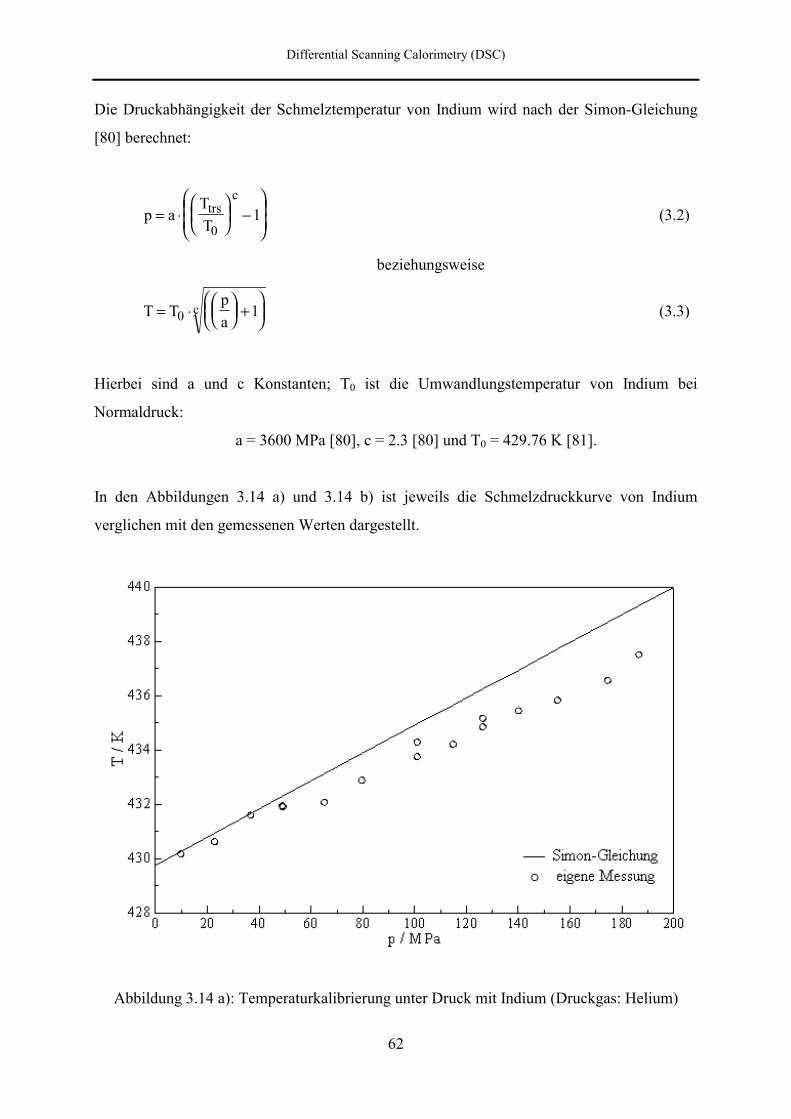
Differential Scanning Calorimetry (DSC)
62
Die Druckabhängigkeit der Schmelztemperatur von Indium wird nach der Simon-Gleichung
[80] berechnet:
p aTTtrs
c= ⋅
−
0
1 (3.2)
beziehungsweise
T Tpa
c= ⋅
+
0 1 (3.3)
Hierbei sind a und c Konstanten; T0 ist die Umwandlungstemperatur von Indium bei
Normaldruck:
a = 3600 MPa [80], c = 2.3 [80] und T0 = 429.76 K [81].
In den Abbildungen 3.14 a) und 3.14 b) ist jeweils die Schmelzdruckkurve von Indium
verglichen mit den gemessenen Werten dargestellt.
Abbildung 3.14 a): Temperaturkalibrierung unter Druck mit Indium (Druckgas: Helium)
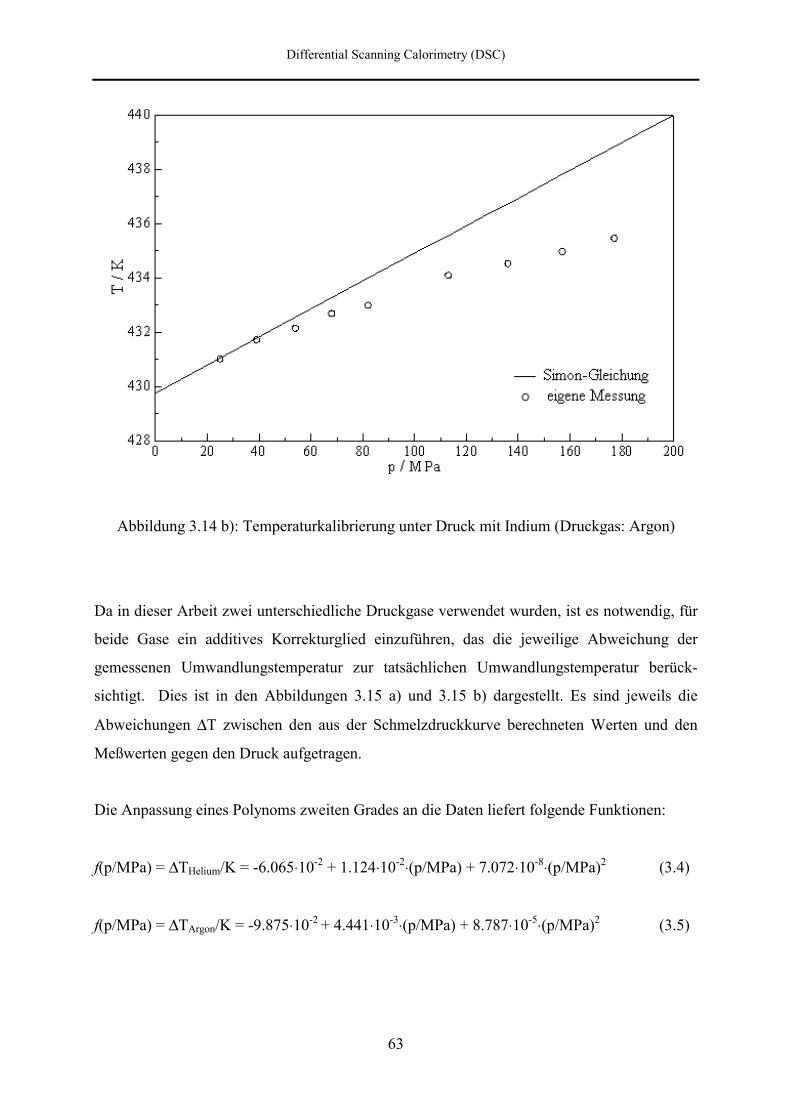
Differential Scanning Calorimetry (DSC)
63
Abbildung 3.14 b): Temperaturkalibrierung unter Druck mit Indium (Druckgas: Argon)
Da in dieser Arbeit zwei unterschiedliche Druckgase verwendet wurden, ist es notwendig, für
beide Gase ein additives Korrekturglied einzuführen, das die jeweilige Abweichung der
gemessenen Umwandlungstemperatur zur tatsächlichen Umwandlungstemperatur berück-
sichtigt. Dies ist in den Abbildungen 3.15 a) und 3.15 b) dargestellt. Es sind jeweils die
Abweichungen ∆T zwischen den aus der Schmelzdruckkurve berechneten Werten und den
Meßwerten gegen den Druck aufgetragen.
Die Anpassung eines Polynoms zweiten Grades an die Daten liefert folgende Funktionen:
f(p/MPa) = ∆THelium/K = -6.065⋅10-2 + 1.124⋅10-2⋅(p/MPa) + 7.072⋅10-8⋅(p/MPa)2 (3.4)
f(p/MPa) = ∆TArgon/K = -9.875⋅10-2 + 4.441⋅10-3⋅(p/MPa) + 8.787⋅10-5⋅(p/MPa)2 (3.5)
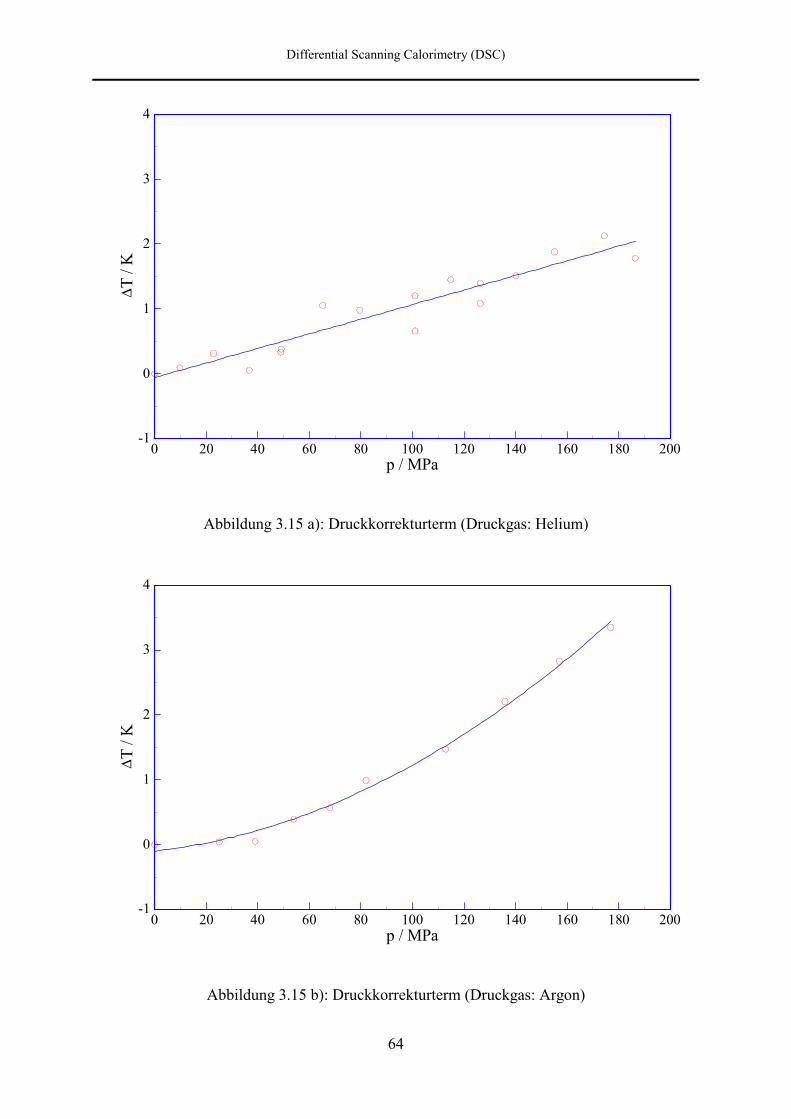
Differential Scanning Calorimetry (DSC)
64
0 20 40 60 80 100 120 140 160 180 200p / MPa
-1
0
1
2
3
4∆ T
/ K
Abbildung 3.15 a): Druckkorrekturterm (Druckgas: Helium)
0 20 40 60 80 100 120 140 160 180 200p / MPa
-1
0
1
2
3
4
∆ T /
K
Abbildung 3.15 b): Druckkorrekturterm (Druckgas: Argon)

Differential Scanning Calorimetry (DSC)
65
Für die Temperaturbestimmung ergibt sich somit folgende druckabhängige Kalibrierfunktion:
f(U/mV, p/MPa) = T/K - A + B⋅(p/MPa) + C⋅(p/MPa)2 (3.6)
mit: AArgon = 9.875⋅10-2, BArgon = 4.441⋅10-3 und CArgon = 8.787⋅10-5
AHelium = 6.065⋅10-2, BHelium = 1.124⋅10-2 und CHelium = 7.072⋅10-8
3.4.2.2 Enthalpiekalibrierung
Die Enthalpiekalibrierung geschieht ebenfalls unter Verwendung von hochreinem Indium
[L 12], wobei auch hier die Druckabhängigkeit berücksichtigt werden muß. Es wird daher ein
druckabhängiger Kalibrierfaktor bestimmt, der sich aus dem Verhältnis der bekannten
Umwandlungsenthalpie der Kalibriersubstanz und der gemessenen Peakfläche bei bekannter
Einwaage und Molmasse der Substanz nach Gleichung 3.7 berechnet:
km HA Mslm( )
( )( )
T,pT,p
T,p=
⋅⋅
∆(3.7)
mit: m / g = Einwaage der Kalibriersubstanz
∆slmH / J⋅mol-1 = molare Schmelzenthalpie der Kalibriersubstanz
A / mV⋅s = gemessene Peakfläche
M / g⋅mol-1 = Molmasse der Kalibriersubstanz
Der Kalibrierfaktor k besitzt die Dimension J⋅mV-1⋅s-1, da die Peakfläche durch die Integration
der Signalspannung der Heizleistung über die Zeit erhalten wird.
Wie schon bei Ellert [59] und Schmidt [63] beschrieben, hat es sich herausgestellt, daß der
Kalibrierfaktor bei Messungen unter Normaldruck im Rahmen der Meßgenauigkeit
unwesentlich von der jeweiligen Umwandlungstemperatur abhängig ist und über den
gesamten Temperaturbereich den Wert k = (3.90 ± 0.01)⋅10-4 J⋅mV-1⋅s-1 besitzt. Aus diesem

Differential Scanning Calorimetry (DSC)
66
Grund wurde darauf verzichtet, eine Temperaturabhängigkeit der Peakfläche in die
Kalibrierungsfunktion einzubeziehen.
Da die Wärmeleitfähigkeit des verwendeten Druckgases einen Einfluß auf die Peakgröße hat,
ist es auch bei der Enthalpiekalibrierung wiederum nötig, für beide Druckgase eine eigene
Kalibrierung durchzuführen.
Zunächst muß jedoch die Druckabhängigkeit der Schmelzenthalpie von Indium etwas näher
betrachtet werden.
Für die Umwandlung einer Phase a zur Phase b entlang der Koexistenzlinie gilt die bereits in
Kapitel 2.1 hergeleitete Planck-Gleichung:
d Hdp
CdTdp
V TVT
abm
koexabp m
koexabm
abm
p
∆∆ ∆
=
+ −
,
∂∆∂
(3.8)
Im folgenden sind die für Indium zur Berechnung benötigten Daten aufgeführt:
∆s
l Cpm / J⋅K-1⋅mol-1 = 1.22 [82]
(dT/dp)koex / K⋅MPa-1 = 4.9⋅10-4 [83]
∆s
l Vm / cm3⋅mol-1 = 0.398 [84]
∆s
l Hm / J⋅mol-1 = 3290 [85]
(∂(∆s
l Vm)/∂T)p / cm3⋅K-1⋅mol-1 = 0.20⋅10-3 [84]
T(s→l) / K = 429.76 [81]
Durch Einsetzen der Werte in Gleichung (3.8) ergibt sich für die Druckabhängigkeit der
Schmelzenthalpie von Indium:
(d(∆s
l Hm)/dp)koex = (3.7 ± 0.1)⋅10-1 J⋅MPa-1⋅mol-1
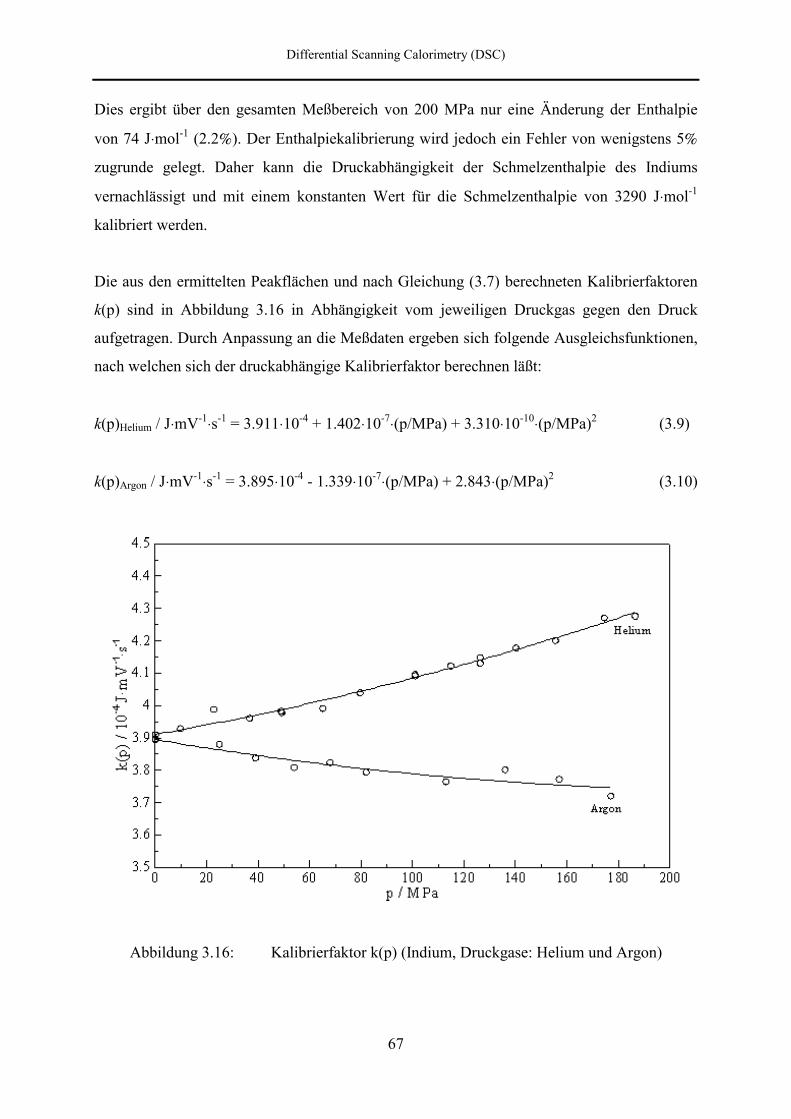
Differential Scanning Calorimetry (DSC)
67
Dies ergibt über den gesamten Meßbereich von 200 MPa nur eine Änderung der Enthalpie
von 74 J⋅mol-1 (2.2%). Der Enthalpiekalibrierung wird jedoch ein Fehler von wenigstens 5%
zugrunde gelegt. Daher kann die Druckabhängigkeit der Schmelzenthalpie des Indiums
vernachlässigt und mit einem konstanten Wert für die Schmelzenthalpie von 3290 J⋅mol-1
kalibriert werden.
Die aus den ermittelten Peakflächen und nach Gleichung (3.7) berechneten Kalibrierfaktoren
k(p) sind in Abbildung 3.16 in Abhängigkeit vom jeweiligen Druckgas gegen den Druck
aufgetragen. Durch Anpassung an die Meßdaten ergeben sich folgende Ausgleichsfunktionen,
nach welchen sich der druckabhängige Kalibrierfaktor berechnen läßt:
k(p)Helium / J⋅mV-1⋅s-1 = 3.911⋅10-4 + 1.402⋅10-7⋅(p/MPa) + 3.310⋅10-10⋅(p/MPa)2 (3.9)
k(p)Argon / J⋅mV-1⋅s-1 = 3.895⋅10-4 - 1.339⋅10-7⋅(p/MPa) + 2.843⋅(p/MPa)2 (3.10)
Abbildung 3.16: Kalibrierfaktor k(p) (Indium, Druckgase: Helium und Argon)

Differential Scanning Calorimetry (DSC)
68
3.4.3 Meßgenauigkeit
Zur Angabe der Meßgenauigkeit der Apparatur werden zunächst die Meßfehler in der
Temperatur-, der Druck- und der Enthalpiebestimmung betrachtet, da die weiteren thermo-
dynamischen Umwandlungsgrößen, wie Umwandlungsentropie und Umwandlungsvolumen,
aus diesen berechnet werden.
3.4.3.1 Fehlerabschätzung bei der Temperatur- und Druckmessung
Es muß zunächst berücksichtigt werden, daß die Auflösung des Temperatursignals im
Meßbereich von 300 K bis 450 K durch Verwendung eines 12-bit Analog/Digital-Wandlers
bei 1/4096 des zu digitalisierenden Meßbereichs liegt und deshalb nicht besser als 0.07 K bis
0.11 K sein kann.
Zur Bestimmung des Fehlers für die Umwandlungstemperatur wurden die bei den
Kalibriermessungen erhaltenen Peaks mehrfach in der in Kapitel 3.3.2 beschriebenen Weise
ausgewertet. DSC-Messungen mit Indium, unter gleichen Bedingungen und mit derselben
Indiumprobe durchgeführt, ergaben dabei eine Reproduzierbarkeit der Umwandlungs-
temperatur von ± 0.2 K. Die bei der Temperaturkalibrierung verwendeten organischen
Substanzen lieferten jedoch Peaks, bei denen aufgrund eines erhöhten Grundliniendrifts diese
Reproduzierbarkeit nicht mehr gegeben war. In diesen Fällen traten Abweichungen von
± 0.5 K auf. Es erscheint daher sinnvoll, den maximalen Fehler bei der
Temperaturbestimmung nicht kleiner als mit ± 0.5 K anzugeben.
Die Unsicherheit bei der Bestimmung des Umwandlungsdruckes ergibt sich aus der
Ablesegenauigkeit des Manometers und durch die Digitalisierung der Analogspannung der
Druckmeßdose. In beiden Fällen bewegt sich der Fehler in der Druckbestimmung im Rahmen
von ± 0.5 MPa.

Differential Scanning Calorimetry (DSC)
69
3.4.3.2 Fehlerabschätzung bei der Enthalpiebestimmung
Die Genauigkeit bei der Enthalpiebestimmung hängt von einer Reihe von Parametern ab.
Die apparative Genauigkeit ergibt sich aus der Ablesegenauigkeit der digitalisierten
Heizleistung und der Zeit. Es läßt sich hier ein Maximalfehler von einem Prozent angeben.
Problematisch ist die Erfassung der Fehlergrenzen des verwendeten DSC-Gerätes im Hinblick
auf die Langzeitkonstanz der eingestellten Geräteparameter. Um mit einem DSC-Gerät unter
Normaldruck Enthalpien mit einer Unsicherheit von unter 3% zu bestimmen, wird in der
Literatur empfohlen, mindestens einmal pro Tag eine Justierung und Kalibrierung des Gerätes
durchzuführen [85, 86]. Diese Maßnahme ist bei einer Hochdruckapparatur nicht
durchführbar.
Eine wichtige Rolle bei der Fehlerbetrachtung spielt die Wahl der Methode der
Grundlinienfestlegung. Bei Peaks mit einer zur Zeitachse nahezu parallelen Grundlinie vor
und nach der Umwandlung liefern alle vier in Kapitel 3.3.3 vorgestellten
Auswertungsverfahren nahezu dasselbe Ergebnis. Bei den Kalibriermessungen mit Indium
ergaben die vier Methoden im Meßergebnis lediglich einen Unterschied von einem Prozent.
Aus diesem Grund wurde hier jeder Peak nach allen vier Verfahren ausgewertet und
anschließend ein Mittelwert gebildet. Die absolute Genauigkeit bei der Enthalpiebestimmung
von Indium ergab sich dabei zu ± 30 J⋅mol-1. Anders verhielt es sich bei den zur
Kalibrierung benutzten organischen Substanzen. Die gegenüber Metallen geringere
Wärmeleitfähigkeit organischer Substanzen wirkt sich auf die Form der Peaks und damit auf
das Meßergebnis aus [69]. Stark driftende Grundlinien führen zu größeren Unterschieden in
den vier Auswertungsverfahren. In diesem Fall hängt der Flächenwert besonders von den
vorgegebenen Grenzwerten der Integration und damit vom ”Geschick” des Experimentators
ab. Methode c), nach der der Grundlinienverlauf unter dem Peak der Peakform angepaßt wird,
lieferte hierbei gegenüber den anderen drei Verfahren wesentlich reproduzierbarere
Ergebnisse.
Als weitere Fehlerquelle ist aufzuführen, daß die zur Enthalpiekalibrierung benutzten Metalle
oft nicht in dem Temperaturbereich der Umwandlung der vermessenen Substanz liegen. Da
der Kalibrierfaktor auch eine Funktion der Temperatur ist, könnte er bei höheren oder tieferen

Differential Scanning Calorimetry (DSC)
70
Temperaturen in Abhängigkeit vom Druck anders verlaufen. Außerdem müssen die bei der
Kalibrierung vorgenommenen Vereinfachungen hinsichtlich der Druckabhängigkeit der
Umwandlungsenthalpie und der Temperaturabhängigkeit der Peakfläche berücksichtigt
werden.
Alles in allem erscheint es deshalb nicht sinnvoll, den Größtfehler für die
Enthalpiebestimmung unter Druck kleiner als 5% anzugeben.
Es sei an dieser Stelle darauf hingewiesen, daß die in den Kapiteln 5 bis 7 aufgeführten
Meßdaten für die Umwandlungsenthalpien, -entropien und -volumina in Abhängigkeit vom
Druck größtenteils mit einer zu hohen Genauigkeit angegeben sind. Dies geschah absichtlich,
um Rundungsfehler bei Folgeberechnungen zu vermeiden. In allen Fällen wurden an die
angegebenen druckabhängigen Umwandlungsdaten Ausgleichsfunktionen erster bzw. zweiter
Ordnung angepaßt. Wird bei gegebenem Druck ein Einzelwert betrachtet, so gilt die in diesem
Abschnitt diskutierte Fehlertoleranz, und der Meßwert ist entsprechend zu modifizieren
(Streichung der Nachkommastellen bzw. Aufrunden).

Differentialthermoanalyse (DTA)
71
4 Differentialthermoanalyse (DTA)
4.1 Definition und Grundlagen der Differentialthermoanalyse
Gemäß der ICTA-Definition (ICTA: International Confederation for Thermal Analysis) [87]
stellt die Differentialthermoanalyse eine Meßtechnik dar, bei der die Temperaturdifferenz
zwischen einer Probensubstanz und einem Referenzmaterial als Funktion der Temperatur
gemessen wird, während Probe und Referenz einem geregelten Temperaturprogramm
unterworfen werden. Die Aufzeichnung ∆T = f (T) wird als Thermogramm bezeichnet und
erfolgt in der Regel über Thermoelemente, die sich entweder in der Proben- und
Referenzsubstanz befinden (klassische DTA) oder in metallische Tiegel (Boersma-DTA), die
die Substanzen enthalten, eingeführt sind. Die beiden Thermoelemente sind dabei so
gegeneinander geschaltet, daß eine gleichzeitige Registrierung der Differenztemperatur
∆T = TRef - TP und der Absoluttemperatur, die in dieser Arbeit stets in der Probensubstanz
bestimmt wurde, möglich ist. Eine Änderung der Differenztemperatur, hervorgerufen durch
einen wärmeerzeugenden oder -verbrauchenden Prozeß in der Probensubstanz, wird im
Thermogramm durch einen Peak sichtbar. Am Vorzeichen der Differenztemperatur kann
abgelesen werden, ob der stattfindende Prozeß exotherm oder endotherm verläuft. Bei den in
dieser Arbeit dargestellten Thermogrammen zeigt das Meßsignal bei endothermen Prozessen
nach oben.
Entsprechend obiger Definition sollen die Proben- und Referenzsubstanz einem geregelten
Temperatur-Zeit-Programm unterworfen werden. Zur exakten Auswertung der DTA-
Thermogramme ist daher ein reproduzierbarer, linearer Temperaturanstieg nötig, wobei die
optimale Aufheizgeschwindigkeit der jeweils untersuchten Substanz angepaßt werden muß.
Wünschenswert ist eine möglichst rasche und schwingungsfreie Temperatureinstellung.
Ausgehend von der Annahme, daß der Temperiermantel des Autoklavs mit einer konstanten
Heizrate β aufgeheizt wird, gilt für die Manteltemperatur TM zum Zeitpunkt t:
TM(t) = TM(t=0) + β t (4.1)

Differentialthermoanalyse (DTA)
72
mit: TM(t=0): Anfangstemperatur des Temperiermantels
Die Temperatur der Probe TP ist niedriger als die Manteltemperatur. Setzt man eine
einheitliche Probentemperatur voraus, kann für die Probe folgende Wärmebilanz aufgestellt
werden:
CP⋅dTP - L⋅A⋅(TM - TP)⋅dt = 0 (4.2)
mit: CP: Wärmekapazität der Probe
L: Wärmeübergangskoeffizient zwischen Autoklavwand und Probe
A: wärmeaustauschende Fläche der Probe
Einsetzen von Gleichung (4.2) in (4.1) und Einführung der Zeitkonstanten τ =⋅
CL A
P liefert:
τ βdTdt
T t t TPM P= = + −( )0 (4.3)
Die Lösung der Differentialgleichung ergibt einen Ausdruck für das gesuchte „Nachhinken“
der Probentemperatur δT (t) = (TM - TP) (t) hinter der Manteltemperatur:
( ) ( )( ) ( ) ( )( )δτ
βτ βττ
T t T T t T t T tt t
M P M P= − = = − = ⋅−
+ − ⋅
−
0 0 exp exp (4.4)
mit: TP(t=0): Anfangstemperatur der Probe
Für t ≤ τ wird die Größe des „Nachhinkens“ (TM - TP) (t) maßgeblich durch die Differenz
(TM(t=0) - TP(t=0)) bestimmt. Für t > τ, also nach dem Abklingen des Anfangsverhaltens,
erhält man als bleibende konstante Temperaturabweichung:
(TM - TP) (t) = β⋅τ (4.5)
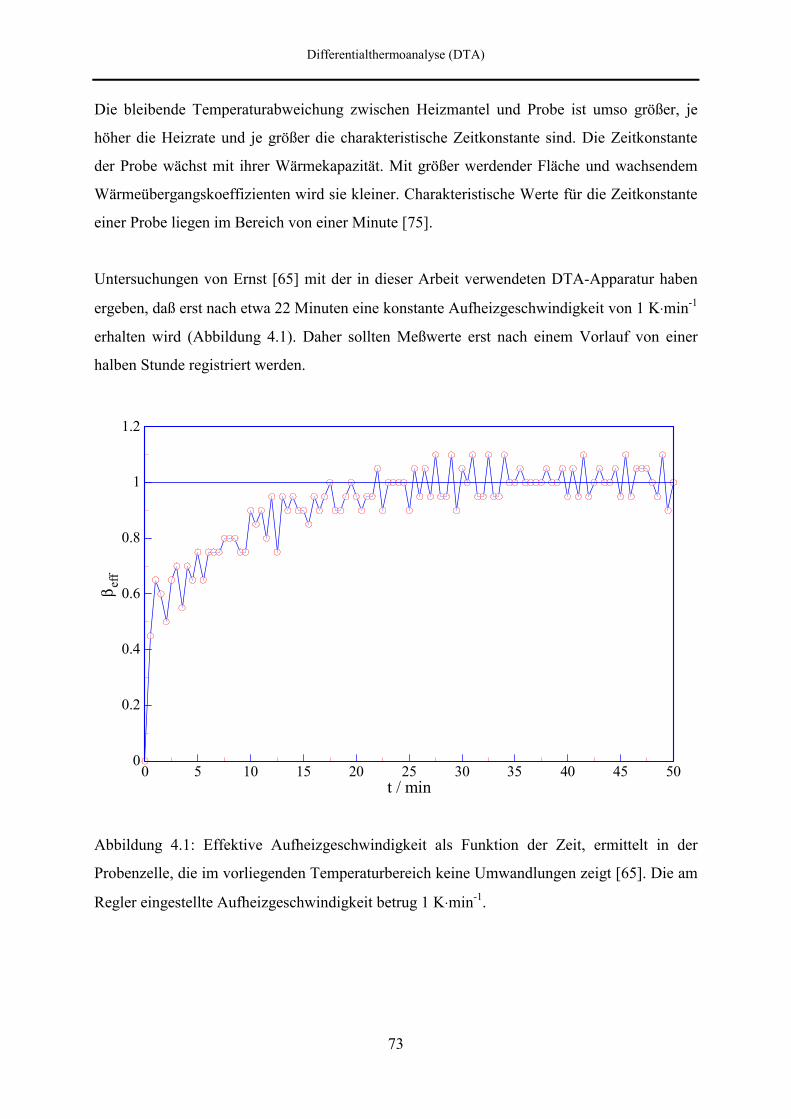
Differentialthermoanalyse (DTA)
73
Die bleibende Temperaturabweichung zwischen Heizmantel und Probe ist umso größer, je
höher die Heizrate und je größer die charakteristische Zeitkonstante sind. Die Zeitkonstante
der Probe wächst mit ihrer Wärmekapazität. Mit größer werdender Fläche und wachsendem
Wärmeübergangskoeffizienten wird sie kleiner. Charakteristische Werte für die Zeitkonstante
einer Probe liegen im Bereich von einer Minute [75].
Untersuchungen von Ernst [65] mit der in dieser Arbeit verwendeten DTA-Apparatur haben
ergeben, daß erst nach etwa 22 Minuten eine konstante Aufheizgeschwindigkeit von 1 K⋅min-1
erhalten wird (Abbildung 4.1). Daher sollten Meßwerte erst nach einem Vorlauf von einer
halben Stunde registriert werden.
0 5 10 15 20 25 30 35 40 45 50t / min
0
0.2
0.4
0.6
0.8
1
1.2
β eff
Abbildung 4.1: Effektive Aufheizgeschwindigkeit als Funktion der Zeit, ermittelt in der
Probenzelle, die im vorliegenden Temperaturbereich keine Umwandlungen zeigt [65]. Die am
Regler eingestellte Aufheizgeschwindigkeit betrug 1 K⋅min-1.

Differentialthermoanalyse (DTA)
74
Beabsichtigt man, mit DTA-Messungen Reaktionsenthalpien oder Wärmekapazitäten zu
bestimmen, so muß der funktionale Zusammenhang zwischen der gesuchten Größe und dem
Meßsignal bekannt sein. In der folgenden Herleitung [88] wird der Zusammenhang zwischen
der Peakfläche und der Umwandlungsenthalpie dargestellt. Da die der Herleitung zugrunde
liegenden Voraussetzungen in der Praxis meist nur unzureichend erfüllt sind, ist der
Bestimmung der Umwandlungsenthalpien mittels DTA jedoch eher ein abschätzender
Charakter zuzuordnen. Steht, wie in dieser Arbeit, auch eine DSC-Apparatur zur Verfügung,
so sollte diese zur Bestimmung von Umwandlungsenthalpien eingesetzt werden.
Für die Wärmebilanzen von Proben- und Referenzzelle gelten folgende Beziehungen:
( )K T T dt C dT H dnP u P P P⋅ − ⋅ = ⋅ + ⋅∆ (4.6)
( )K T T dt C dTR u R R R⋅ − ⋅ = ⋅ (4.7)
mit: KP, KR = Wärmedurchgangszahlen (multipliziert mit der Austauschfläche)
Tu = Umgebungstemperatur im Autoklav
TP, TR = Temperatur der Proben- bzw. Referenzzelle
∆H = Umwandlungsenthalpie
dn = die sich im Zeitintervall dt umwandelnde Stoffmenge
CP, CR = Wärmekapazitäten der Proben- bzw. Referenzzelle
Unter Berücksichtigung der Definitionen für die Differenztemperatur ∆T und die Heizrate Φ
∆T T TR P= − (4.8)
( )Φ = = ⋅ −dTdt
KC
T TR R
Ru R (4.9)
ergibt sich für die zeitliche Änderung der Differenztemperatur:
( ) ( )d Tdt
KC
T TKC
T TH
Cdndt
R
Ru R
P
Pu P
P
∆ ∆= ⋅ − − ⋅ − + ⋅ (4.10)

Differentialthermoanalyse (DTA)
75
( )= ⋅ − + ⋅KC
T TH
Cdndt
p
P P∆ ∆
∆0 (4.11)
mit: ( ) ( )∆ ∆TCK
KC
T T T T TP
P
R
Ru R u P0 ≡ ⋅ ⋅ − − − + (4.12)
( )= − ⋅ ⋅ −
T T
KC
CK
CKu R
R
R
P
P
R
R(4.13)
= ⋅ −
Φ
CK
CK
P
P
R
R(4.14)
Aus Beziehung (4.14) läßt sich ableiten, daß ∆T0 = 0 für den Fall gilt, daß CK
CK
P
P
R
R= ist.
Bleiben sowohl die Wärmekapazitäten als auch die Wärmedurchgangszahlen während des
Umwandlungsprozesses konstant, so resultiert ein konstantes ∆T0. In der Praxis ist dies
aufgrund der Temperaturabhängigkeit der Wärmekapazitäten jedoch nicht realisierbar. Eine
konstante Heizrate Φ wird durch einen linearen Aufheizprozeß gewährleistet.
Integration von Gleichung (4.11) über das Zeitintervall t1 bis t2 während des Umwandlungs-
prozesses ergibt:
( )d TKC
T T dtH
Cdn
T t
T tP
P t
t
P∆ ∆ ∆
∆
∆
∆
( )
( )
1
2
1
2
00
1
∫ ∫ ∫= ⋅ − ⋅ + ⋅ (4.15)
Für die Enthalpieänderung folgt unter der Annahme ∆ ∆ ∆T t T t T( ) ( )1 2 0= = :
∆H K AP= ⋅ (4.16)
mit: ( )A T T dtt
t
= − ⋅∫ ∆ ∆ 02
1

Differentialthermoanalyse (DTA)
76
4.2 DTA-Apparatur
Die in dieser Arbeit durchgeführten DTA-Messungen wurden an einer bereits vorhandenen
Hochdruck-DTA-Anlage, welche für Drücke von 0.1 MPa bis 300 MPa ausgelegt ist,
vorgenommen (siehe Ernst [65] und Becker [93]). Da Reparaturarbeiten am Heiz- und
Kühlmantel des Autoklavs zu Beschädigungen der Thermoelemente führten, mußten diese
ausgetauscht und neu kalibriert werden. Der maximal nutzbare Temperaturbereich liegt bei
273 K bis 465 K und ist durch das Zellenmaterial begrenzt (465 K = Schmelztemperatur des
Blei-Zinn-Eutektikums bei 0.1 MPa [81]). Den schematischen Aufbau der verwendeten
Apparatur zeigt Abbildung 4.2.
4.2.1 Aufbau des Autoklavs
Der in Abbildung 4.3 dargestellte Hochdruckautoklav ist aus Nimonic 90 gefertigt. Die
Temperaturmessung erfolgt über zwei von unten in den Autoklav eingeführte Chromel-
Alumel-Stahlmantelthermoelemente (i), welche in Dichtungskonen (k) hart eingelötet sind
und mittels Überwurfmuttern mit dem Autoklav druckdicht verschraubt werden. Die
Vergleichs-lötstellen der Thermoelemente befinden sich in einem mit Decan gefüllten
Glasgefäß, welches in einen mit Eiswasser gefüllten Dewar eintaucht. Um einen thermischen
Kontakt mit dem Autoklav zu vermeiden, sind die Thermoelemente von Korundhülsen (j)
umgeben. Probenmeßzelle und Referenzzelle werden auf die Spitzen der Thermoelemente
aufgesetzt und sind von einem Kalorimeterblock aus Kupfer (h) umschlossen. Zur
Reduzierung des Druckgasvolumens im Autoklav wird der verbleibende freie Raum mit
einem Block aus Nimonic 90 (g) ausgefüllt. Der Autoklav wird mit einem Bridgmanstempel
(a) verschlossen, der gleichzeitig auch durch eine Bohrung die Druckübertragung
gewährleistet. Als Dichtungsmaterial dienen Teflon- und weichgeglühte Kupferringe (e). Die
Druckerzeugung erfolgt mit Hilfe eines Gasmembrankompressors. Helium und Argon dienen
dabei als druckübertragendes Medium.Von außen ist der Autoklav mit einem Heiz- und
Kühlmantel (c) umgeben, wobei die Kühlung mit Preßluft oder Eiswasser erfolgen kann. Um
eine möglichst gute thermische Isolierung zu erreichen, wird der gesamte Autoklav
einschließlich Heiz- und Kühlmantel mit einem speziellen Isoliermaterial (Keramikvlies)
umgeben.
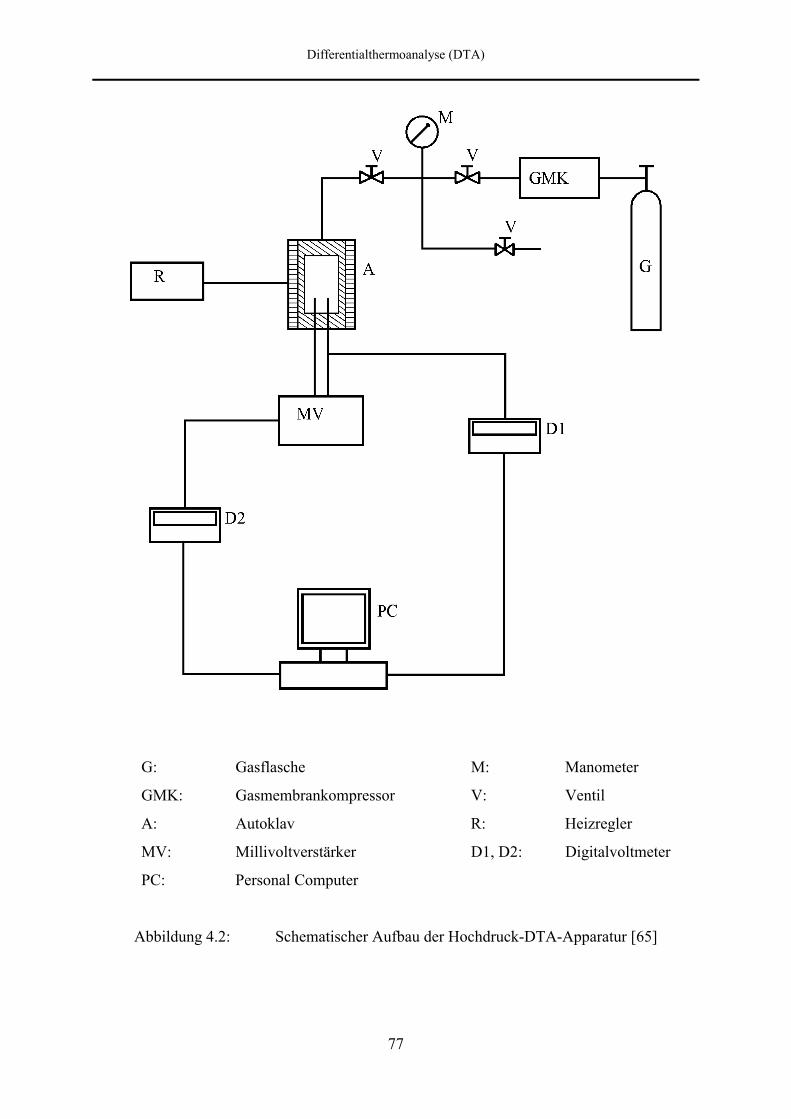
Differentialthermoanalyse (DTA)
77
G: Gasflasche M: Manometer
GMK: Gasmembrankompressor V: Ventil
A: Autoklav R: Heizregler
MV: Millivoltverstärker D1, D2: Digitalvoltmeter
PC: Personal Computer
Abbildung 4.2: Schematischer Aufbau der Hochdruck-DTA-Apparatur [65]
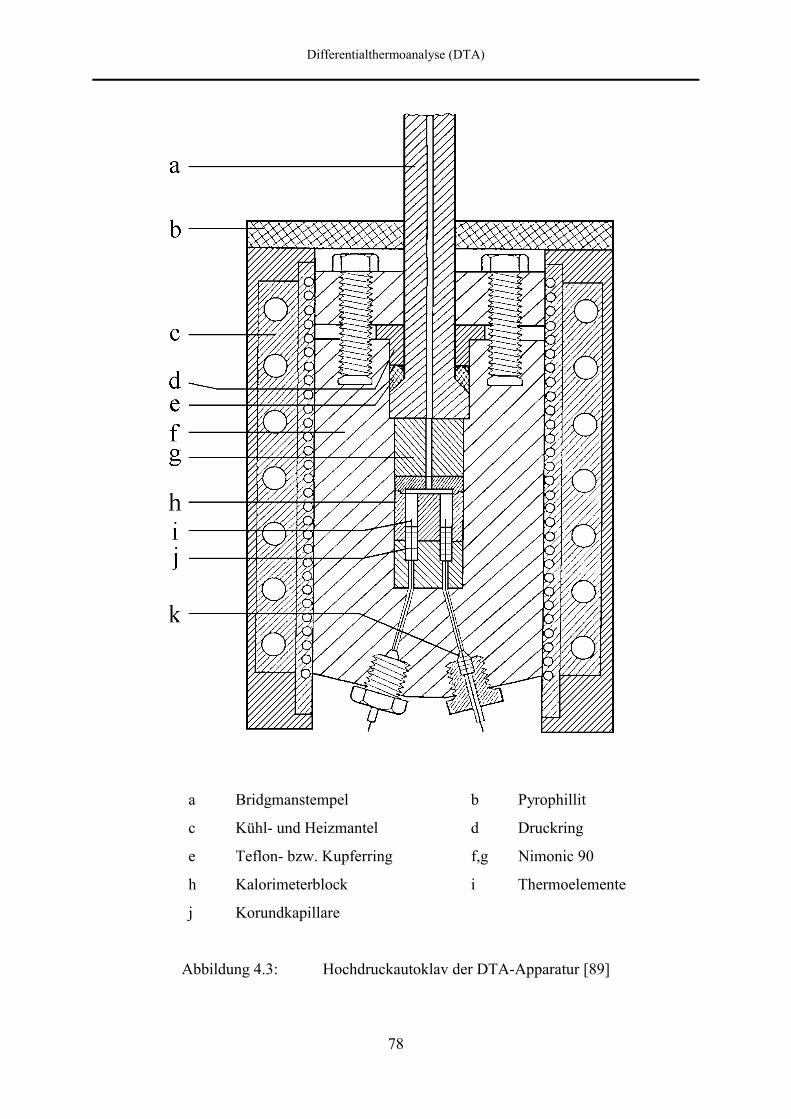
Differentialthermoanalyse (DTA)
78
a Bridgmanstempel b Pyrophillit
c Kühl- und Heizmantel d Druckring
e Teflon- bzw. Kupferring f,g Nimonic 90
h Kalorimeterblock i Thermoelemente
j Korundkapillare
Abbildung 4.3: Hochdruckautoklav der DTA-Apparatur [89]

Differentialthermoanalyse (DTA)
79
4.2.2 Druckversorgung
Die Druckversorgung erfolgt über einen Gasmembrankompressor [L 13], mit dem das Druck-
gas, wahlweise Argon oder Helium [L 3], aus einer Vorratsflasche durch ein Kapillarsystem
in den Autoklav gedrückt wird. Mit einem Bourdon-Präzisionsmanometer [L 4], das für einen
Druckbereich von bis zu 300 MPa ausgelegt ist und auf 0.1 MPa genau abgelesen werden
kann, wird der Druck am Umwandlungspunkt bestimmt. Die Genauigkeit des Manometers
beträgt nach Aussagen der Lieferfirma 0.1% des Endausschlages.
4.2.3 Temperaturregelung
Zur Temperaturregelung steht ein hochpräziser, quarzgesteuerter Regler mit einer Stromstärke
von 28 A (bei 60 V) und 48 A (bei 35 V) zur Verfügung, welcher einen linearen Anstieg der
Temperatur von Probenmeßzelle und Referenzzelle in Abhängigkeit von der Zeit
gewährleistet. Mit einer Heizwicklung aus Cronix-Heizdraht (0.448 Ω / m) wird die
elektrische Leistung auf den Autoklav übertragen. Der Heizdraht ist so dimensioniert, daß die
elektrische Energie optimal ausgenutzt wird und eine ausreichend homogene
Temperaturverteilung im Autoklav ermöglicht wird. Um Wärmeverluste der Heizung an die
äußere Umgebung gering zu halten, ist der Heizdraht von mehreren Lagen Keramikvlies
umgeben. Zur Kühlung kann Preßluft oder Eiswasser durch die Bohrungen im
Temperiermantel geleitet werden. Mit Hilfe eines Pt-100-Widerstandsthermometers, das im
Heiz- und Kühlblock des Autoklavs gut wärmeleitend angebracht ist, wird durch ständiges
Messen die Ofentemperatur bestimmt. Auf diese Weise ist dem Regler zu jedem Zeitpunkt der
Istwert der Temperatur bekannt. Regelintern wird der durch die vorgewählte Heizrate bedingte
Sollwert der Temperatur gebildet. Besteht zwischen Sollwert und Istwert kein Unterschied, so
befindet sich der Regler auf der Regelkurve. Dieser Idealzustand wird bei dem hier
verwendeten Proportionalregler jedoch nicht erreicht, da gerade die Abweichung von Ist- und
Solltemperatur als Regelgröße benutzt wird. Die übertragene Heizleistung ist dabei der
Abweichung proportional und kann über einen einstellbaren Verstärkungsfaktor an die
Autoklavgröße angepaßt werden. Wird der Verstärkungsfaktor zu groß gewählt, resultiert ein
Regelschwingen mit großer Amplitude und niedriger Frequenz um die Regelkurve. Ist der
Verstärkungsfaktor zu klein eingestellt, wird die Regelkurve erst nach sehr langer Zeit oder

Differentialthermoanalyse (DTA)
80
aber überhaupt nicht erreicht. Zusätzlich zum Verstärkungsfaktor kann ein empirisch zu
ermittelnder Differenzanteil zwischen Soll- und Istwert der Temperatur eingestellt werden.
Dadurch ist die Geschwindigkeit, mit der sich der Regler auf die Regelkurve zubewegt,
bekannt. Sind Verstärkungsfaktor und Differenzanteil ideal gewählt, werden bereits sehr
kleine Abweichungen von der Regelkurve erkannt. Ein hochfrequentes Schwingen mit kleiner
Amplitude um die Regelkurve ist die Folge. Im Autoklavinnern erhält man so eine fast lineare
Aufheizgeschwindigkeit.
4.2.4 Meßwerterfassung
Da die Thermospannungen für die Temperaturwerte im Bereich einiger Millivolt liegen,
während die Spannungen der Differenztemperaturen nur einige Mikrovolt betragen, werden
letztere vor der Weiterverarbeitung mit einem Meßverstärker der Firma Knick um den Faktor
500 verstärkt. Die Genauigkeit der Verstärkung liegt bei 1%. Über einen Analog-Digital-
Wandler werden jede Sekunde die Thermospannungen der Absoluttemperatur bzw. die
verstärkten Spannungen der Differenztemperatur direkt in einen IBM-kompatiblen Rechner
eingelesen und als Wertepaare von Absoluttemperatur und Differenztemperatur gespeichert.
Während der Messung können die differenzierten DTA-Signale auf einem Monitor verfolgt
werden. Die Auswertung der Thermogramme erfolgt nach den Messungen mittels eines von
Ernst [65] geschriebenen Computerprogramms, das eine genaue Bestimmung der
Umwandlungstemperaturen erlaubt. Darüberhinaus ist es möglich, Bereiche des
Thermogramms vergrößert darzustellen und Peakflächen zu bestimmen.
4.2.5 Meßzellen
Bei dem in dieser Arbeit für Hochdruckuntersuchungen verwendeten Zellentyp erfolgt die
Druckübertragung auf die Meßsubstanz über die Wände der Meßzellen. Um eine
hysteresefreie Druckübertrageung zu gewährleisten, müssen die Zellen aus Werkstoffen
gefertigt werden, die leicht verformbar und gasdicht verschließbar sind. Zusätzlich sollte die
Zelle samt Dichtung soweit belastbar sein, daß sie den bei Phasenumwandlungen auftretenden
Volumensprüngen folgen kann. Als Zellenmaterial bieten sich Indium, Blei und Kupfer an.

Differentialthermoanalyse (DTA)
81
Dabei ist zu berücksichtigen, daß der nutzbare Temperaturbereich durch den Schmelzpunkt
des jeweiligen Zellenmaterials eingeschränkt wird.
Die in dieser Arbeit verwendeten Blei- und Indiumzellen wurden eigens in einer speziell dafür
angefertigten Gießform (Abbildung 4.4) in einem Stück gegossen.
Abbildung 4.4: Gießform
Anschließend wurde in den Zapfen, der in die Mitte der Zelle hineinragt, eine 2.8 mm tiefe
Bohrung mit einem Durchmesser von 0.55 mm eingebracht (Abbildung 4.5a)). Diese dient
zum Aufstecken der Zelle auf das Thermoelement.
Eine wichtige Voraussetzung bei Hochdruckmessungen ist die Vermeidung eines Kontakts
des Druckgases mit der Meßsubstanz. Untersuchungen von Würflinger [90] an n-Alkanen,
Krombach [91, 92] und Schmidt [63] an flüssigkristallinen Substanzen sowie Becker [93] und
Ernst [65] an Triacylglycerinen in offenen Zellen zeigen einen deutlichen Einfluß des
druckübertragenden Gases auf das Verhalten der Phasenumwandlungstemperaturen, was auf
die mit steigendem Druck zunehmende Löslichkeit des Druckgases in der Probe
zurückzuführen ist. Da dieser Effekt die Meßergebnisse erheblich verfälschen kann, ist es
unbedingt notwendig, die Zellen gasdicht zu verschließen. Dazu wird die Meßzelle mit der
flüssigen Probensubstanz bis zum Rand gefüllt und danach im oberen Drittel mit einer
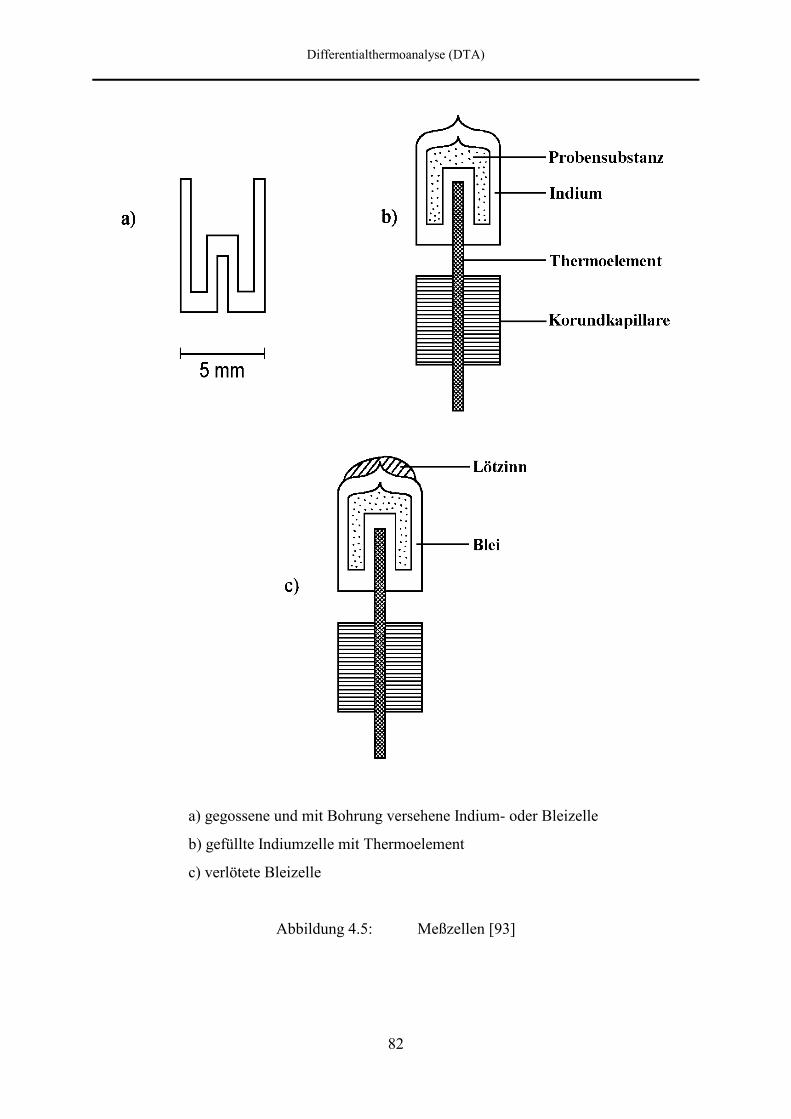
Differentialthermoanalyse (DTA)
82
a) gegossene und mit Bohrung versehene Indium- oder Bleizelle
b) gefüllte Indiumzelle mit Thermoelement
c) verlötete Bleizelle
Abbildung 4.5: Meßzellen [93]

Differentialthermoanalyse (DTA)
83
Rundzange abgequetscht. Eine Probenzelle enthält so ca. 30 mg bis 40 mg Substanz. Die auf
diese Weise erhaltenen Indiumzellen (Abbildung 4.5b)) haben sich als ausreichend belastbar
erwiesen, um den bei den Phasenumwandlungen auftretenden Volumensprüngen zu folgen.
Durch den niedrigen Indium-Schmelzpunkt von 430 K bei Normaldruck wird der nutzbare
Temperaturbereich jedoch stark eingeschränkt. Dieser sollte durch die Verwendung von Blei
(Schmelztemperatur: 600 K bei Normaldruck [94]) vergrößert werden. Im Gegensatz zu den
weitgehend problemlos verlaufenden Messungen mit Indiumzellen, traten beim Einsatz von
Bleizellen immer wieder Undichtigkeiten auf, wobei sich zeigte, daß besonders die
Quetschnaht den Hochdruckbelastungen nicht standhalten konnte. Ein von Bartelt [95]
entwickeltes Verfahren, bei dem die Quetschnaht mit handelsüblichem Radiolot verlötet wird,
beseitigte diesbezügliche Probleme (Abbildung 4.5c)). Damit das Lötzinn auf der Zelle haften
bleibt, wird die Oberfläche der Zelle mit einem Flußmittel gesäubert und aufgerauht. Der
maximal verfügbare Temperaturbereich wird hier allerdings durch den Schmelzpunkt des
entstehenden Blei-Zinn-Eutektikums auf 465 K bei Normaldruck [81] begrenzt.
Als Referenzzelle wird eine leere Platinzelle bzw. eine leere Probenzelle benutzt. Es ist
möglich, auf eine Referenzsubstanz zu verzichten, wenn deren Masse und Wärmekapazität im
Vergleich zu denen der Meßzelle klein ist [75].
4.3 Kalibrierung der Thermoelemente
Die Kalibrierung der Thermoelemente erfolgte mit Substanzen, die einen scharfen,
reproduzierbaren Umwandlungspeak und eine genau bekannte Umwandlungstemperatur
aufweisen. Als geeignete Kalibriersubstanzen erwiesen sich die Metalle Gallium, Indium und
Blei, deren Reinheit von den Lieferfirmen mit > 99.9% angegeben wurde. Da zur Bestimmung
der Umwandlungstemperaturen nach dem in Kapitel 3.3.2 beschriebenen Verfahren auch
solche Substanzen eingesetzt werden sollten, die den später untersuchten Proben ähnlich sind,
wurde zur Kalibrierung außerdem die Umwandlung nematisch → isotrop der
flüssigkristallinen Verbindung 4-Octyloxy-4’-cyanobiphenyl (8 OCB) herangezogen.
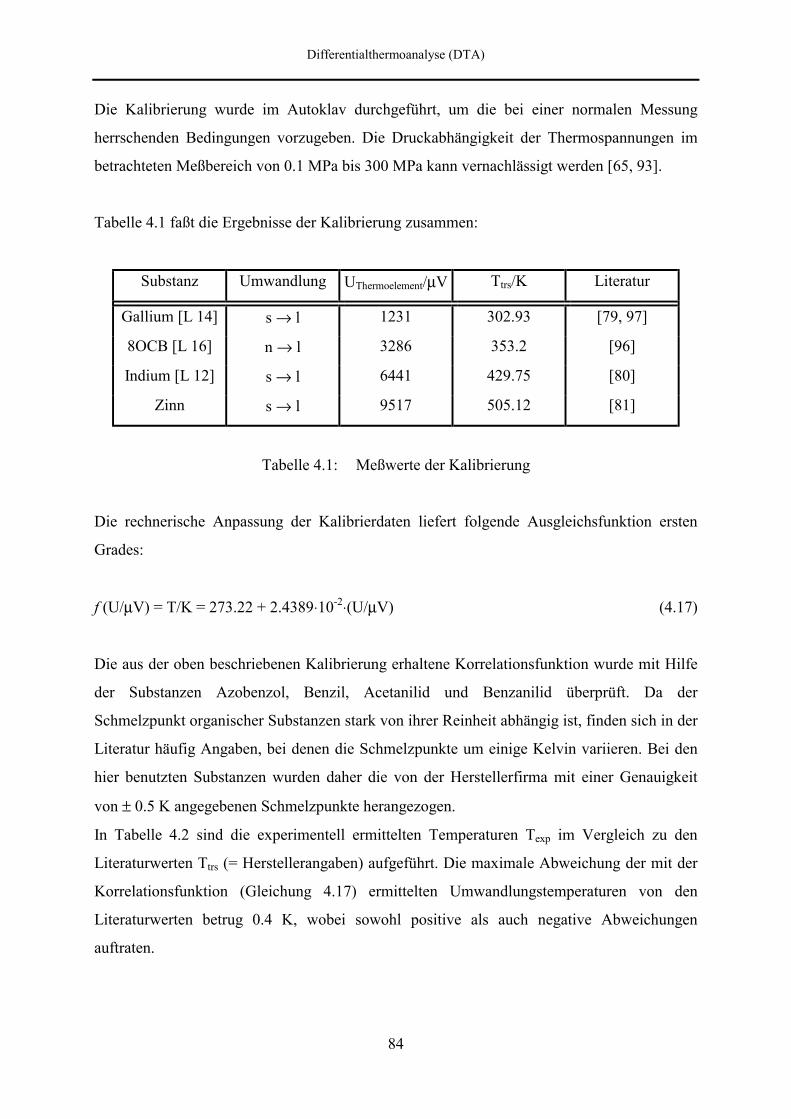
Differentialthermoanalyse (DTA)
84
Die Kalibrierung wurde im Autoklav durchgeführt, um die bei einer normalen Messung
herrschenden Bedingungen vorzugeben. Die Druckabhängigkeit der Thermospannungen im
betrachteten Meßbereich von 0.1 MPa bis 300 MPa kann vernachlässigt werden [65, 93].
Tabelle 4.1 faßt die Ergebnisse der Kalibrierung zusammen:
Substanz Umwandlung UThermoelement/µV Ttrs/K Literatur
Gallium [L 14] s → l 1231 302.93 [79, 97]
8OCB [L 16] n → l 3286 353.2 [96]
Indium [L 12] s → l 6441 429.75 [80]
Zinn s → l 9517 505.12 [81]
Tabelle 4.1: Meßwerte der Kalibrierung
Die rechnerische Anpassung der Kalibrierdaten liefert folgende Ausgleichsfunktion ersten
Grades:
f (U/µV) = T/K = 273.22 + 2.4389⋅10-2⋅(U/µV) (4.17)
Die aus der oben beschriebenen Kalibrierung erhaltene Korrelationsfunktion wurde mit Hilfe
der Substanzen Azobenzol, Benzil, Acetanilid und Benzanilid überprüft. Da der
Schmelzpunkt organischer Substanzen stark von ihrer Reinheit abhängig ist, finden sich in der
Literatur häufig Angaben, bei denen die Schmelzpunkte um einige Kelvin variieren. Bei den
hier benutzten Substanzen wurden daher die von der Herstellerfirma mit einer Genauigkeit
von ± 0.5 K angegebenen Schmelzpunkte herangezogen.
In Tabelle 4.2 sind die experimentell ermittelten Temperaturen Texp im Vergleich zu den
Literaturwerten Ttrs (= Herstellerangaben) aufgeführt. Die maximale Abweichung der mit der
Korrelationsfunktion (Gleichung 4.17) ermittelten Umwandlungstemperaturen von den
Literaturwerten betrug 0.4 K, wobei sowohl positive als auch negative Abweichungen
auftraten.
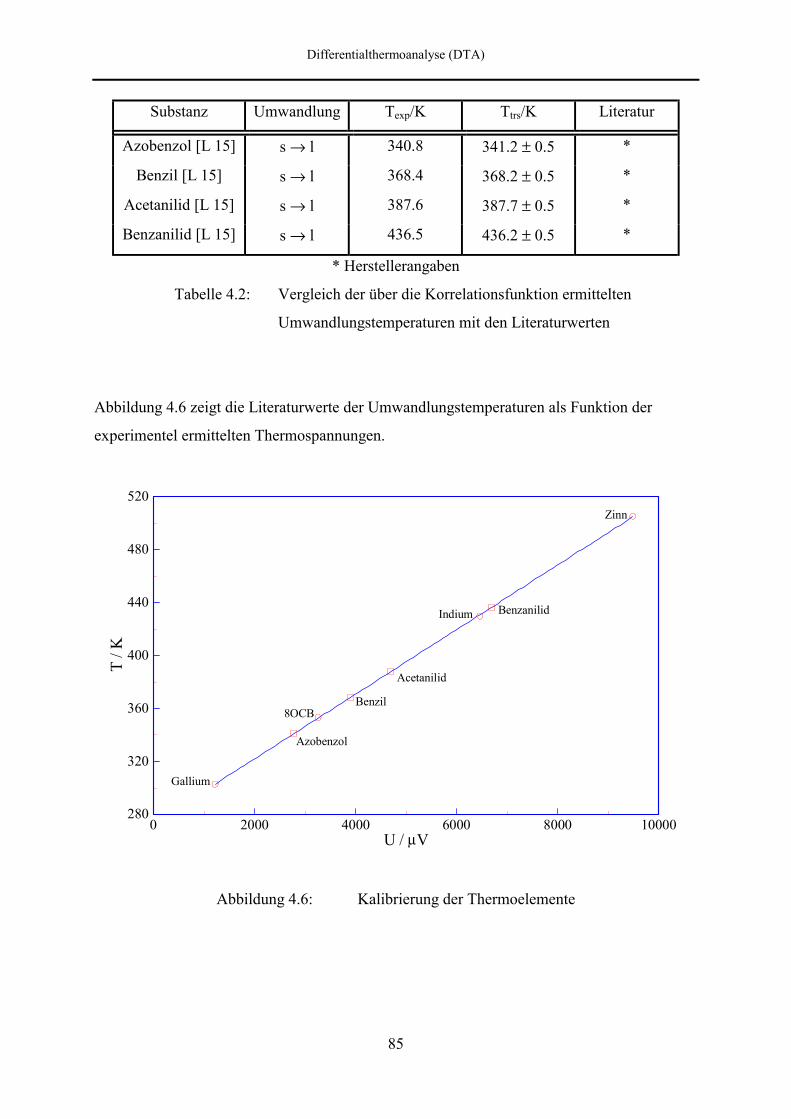
Differentialthermoanalyse (DTA)
85
Substanz Umwandlung Texp/K Ttrs/K Literatur
Azobenzol [L 15] s → l 340.8 341.2 ± 0.5 *
Benzil [L 15] s → l 368.4 368.2 ± 0.5 *
Acetanilid [L 15] s → l 387.6 387.7 ± 0.5 *
Benzanilid [L 15] s → l 436.5 436.2 ± 0.5 *
* Herstellerangaben
Tabelle 4.2: Vergleich der über die Korrelationsfunktion ermittelten
Umwandlungstemperaturen mit den Literaturwerten
Abbildung 4.6 zeigt die Literaturwerte der Umwandlungstemperaturen als Funktion der
experimentel ermittelten Thermospannungen.
0 2000 4000 6000 8000 10000U / µV
280
320
360
400
440
480
520
T / K
Gallium
Indium
Zinn
8OCB
Azobenzol
Benzil
Acetanilid
Benzanilid
Abbildung 4.6: Kalibrierung der Thermoelemente

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
86
5 Untersuchungen zum Phasenverhalten des ausgewählten Triacyl-
glycerins 1,2,3-Tris-eicosanoyloxypropan (Triarachidin, AAA)
5.1 Einführung
Das Schmelzverhalten der Triacylglycerine wird aufgrund der kommerziellen Bedeutung der
eßbaren Fette bereits seit mehr als 150 Jahren untersucht. Uneinheitliche und widersprüchlich
erscheinende Ergebnisse sorgten dabei lange Zeit für Verwirrung und führten zu kontroversen
Diskussionen. Im Jahre 1844 beobachtete Heintz [98] zwei Schmelzpunkte für Tristearin.
Bemerkenswerterweise gelang es Duffy [99] nur wenige Jahre später, 1853, das Schmelz-
verhalten des Tristearins korrekt zu beschreiben, als er von drei Schmelzpunkten berichtete.
Zu jener Zeit war man sich jedoch der Ursache für dieses Schmelzverhalten nicht bewußt. Es
galt die allgemeine Meinung, das Schmelzverhalten sei auf eine Form der Isomerie
zurückzuführen. 1934 erbrachten Malkin und Clarkson [100] mit Hilfe von
Röntgenstrukturuntersuchungen den Beweis dafür, daß die Beobachtung verschiedener
Schmelzpunkte für dasselbe Triacylglycerin tatsächlich auf Polymorphie zurückzuführen ist.
Es gelang ihnen, die unterschiedlichen Schmelzpunkte eines Fettes verschiedenen durch
Röntgenbeugung unterscheidbaren Kristallstrukturen zuzuordnen. Trotzdem dauerte es noch
bis in die Anfänge der sechziger Jahre, ehe Übereinstimmung in Bezug auf Anzahl, Struktur
und Nomenklatur der verschiedenen polymorphen Formen der Triacylglycerine erzielt werden
konnte [101, 102]. Danach scheint jedoch gesichert, daß Fette in drei verschiedenen
polymorphen Hauptmodifikationen vorliegen können, welche nach steigendem Schmelzpunkt
mit α, β´ und β bezeichnet werden. Sie unterscheiden sich durch ihre Stabilität und sind
jeweils durch die Art der Packung der Kohlenstoffketten im Kristallgitter charakterisiert. Die
Anwendung verfeinerter experimenteller Techniken in Kombination mit einer größeren
Probenreinheit hat in jüngster Zeit zu einer neuen Diskussion über die Existenz, Anzahl und
Nomenklatur von Submodifikationen jeder der drei polymorphen Hauptmodifikationen
geführt [103, 104, 105]. Darüberhinaus sind Phasenübergänge zwischen den Modifikationen
zumeist monotrop. In der Literatur finden sich noch heute widersprüchliche Schemata, welche
die möglichen Umwandlungen zwischen den drei Hauptmodifikationen und der Schmelze
beschreiben [106, 107, 108].

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
87
5.2 Chemischer Aufbau und Nomenklatur der Fette
Fette sind chemisch gesehen Carbonsäureester des Glycerins, eines dreiwertigen Alkohols:
Abbildung 5.1: Strukturformel des Glycerins (1,2,3-Propantriol)
Als Säuren enthalten sie unverzweigte Monocarbonsäuren, die Fettsäuren, unter denen
Palmitinsäure (C15H31COOH), Stearinsäure (C17H35COOH) und Ölsäure (C17H33COOH) am
häufigsten vertreten sind. Das Glycerin kann als dreiwertiger Alkohol Mono-, Di- und Triester
bilden, welche nach der heute gültigen Nomenklatur als Monoacyl-, Diacyl- und
Triacylglycerine bezeichnet werden. Die Triacylglycerine bilden dabei mit anteilig 96% den
Hauptbestandteil aller tierischen und pflanzlichen Fette, während Mono- und Diacylglycerine
im allgemeinen nur zu 3% bzw. 1% auftreten [109].
Die chemischen, physikalischen und biologischen Eigenschaften der Triacylglycerine werden
durch die Art der Fettsäuren und durch ihre Position im Molekül beeinflußt. Das in dieser
Arbeit untersuchte Triarachidin ist ein Vertreter der einsäurigen, gesättigten Triacylglycerine,
die in der Nahrungsmittelindustrie und in der pharmazeutischen Industrie eingesetzt werden
und dort u. a. als Gleitmittel in Tabletten und als Grundlage für fetthaltige Puder dienen [65].
In der Literatur findet sich für die verschiedenen Triacylglycerine eine abkürzende
Nomenklatur [108], welche üblicherweise aus einem Code von drei Buchstaben besteht. Jeder
Buchstabe kennzeichnet dabei eine der drei Fettsäuren, mit denen das Glycerin verestert ist,
wobei der mittlere Buchstabe der Sequenz stets die an der 2-Position des Glycerins veresterte
Fettsäure anzeigt.

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
88
Im folgenden sind die am Lehrstuhl für Physikalische Chemie II der Ruhr-Universität Bochum
im Rahmen dieser und früherer Arbeiten [110, 111, 93, 65] untersuchten Triacylglycerine mit
ihren Trivialnamen und den entsprechenden Abkürzungen angegeben:
Palmitinsäure : P ⇒ Tripalmitin : PPP
(Hexadecansäure) (1,2,3-Tris-hexadecanoyloxypropan)
Stearinsäure : S ⇒ Tristearin : SSS
(Octadecansäure) (1,2,3-Tris-octadecanoyloxypropan)
Arachidinsäure : A ⇒ Triarachidin : AAA
(Eicosansäure) (1,2,3-Tris-eicosanoyloxypropan)
Behininsäure : B ⇒ Tribehinin : BBB
(Docosansäure) (1,2,3-Tris-docosanoyloxypropan)
Abbildung 5.2 gibt die Strukturformel der oben aufgeführten Triacylglycerine wieder.
mit n = 13:PPP
n = 15:SSS
n = 17:AAA
n = 19:BBB
Abbildung 5.2: Strukturformel einsäuriger, gesättigter Triacylglycerine

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
89
5.3 Modifikationen der Triacylglycerine
Triacylglycerine können in drei verschiedenen polymorphen Hauptmodifikationen α, β´ und β
auftreten [108]. In allen drei Modifikationen liegen die Triacylglycerine in bestimmter
Konformation im Kristallgitter vor. Darüberhinaus bilden sie alle Schichtstrukturen aus.
Die Triacylglycerine liegen im Kristall in der sogenannten Stimmgabelkonformation (tuning-
fork conformation) vor (Abbildung 5.3). Diese bereits 1950 von Lutton [112] vorgeschlagene
Konformation wurde 1963 in unabhängig voneinander durchgeführten Röntgenstruktur-
analysen von Larsson [113] sowie von Jensen und Mabis [114] bestätigt. Die Kohlen-
wasserstoffketten in der 1- und 3- Position des Glycerins weisen in die entgegengesetzte
Richtung wie die in der 2- Position.
ausgefüllte Kreise: C-Atome
hohle Kreise: O-Atome
Abbildung 5.3: Stimmgabel-Konformation eines Triacylglycerins [nach 102]
In der β´-Phase und mehr noch in der β-Phase liegen die Kohlenwasserstoffketten der
Moleküle bevorzugt in der all-trans-Konformation vor. Die Schichtstruktur ist dabei derart
aufgebaut, daß sich die mehr oder weniger langen Kohlenwasserstoffketten innerhalb der
Schicht parallel zueinander anordnen, wobei sich für jede Modifikation zwei Arten der

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
90
Schichtbildung unterscheiden lassen (Abbildung 5.4). Die erste Form setzt sich aus zwei
(Bezeichnung z. B. β-2), die zweite Form aus drei (Bezeichnung z. B. β-3) Fettsäure-
segmenten zusammen. Die Anzahl der Segmente wird als Kettenlängenmultiplizität
bezeichnet. In der Regel zeigen gesättigte einsäurige Triacylglycerine zweifache Multiplizität.
Eine Anordnung der Schichten mit dreifacher Kettenlängenmultiplizität erfolgt zumeist dann,
wenn die erste Form der Anordnung aus sterischen Gründen unvorteilhaft wäre. Dies trifft
beispielsweise für Triacylglycerine mit cis-ungesättigten Fettsäuren oder für solche
Triacylglycerine zu, in denen sich die Anzahl der Kohlenstoffatome der Fettsäuren um mehr
als sechs unterscheiden.
Abbildung 5.4: β-2- und β-3-Anordnungen von Triacylglycerinen [aus 93]
Die drei bei den Triacylglycerinen vorkommenden Hauptmodifikationen α, β´ und β
unterscheiden sich durch die laterale Anordnung der Fettsäurereste innerhalb der Schichten
sowie durch den Winkel, den die Kohlenwasserstoffketten mit den gebildeten Schichtebenen
einschließen. Zur Beschreibung der lateralen Anordnung der Kohlenwasserstoffketten
innerhalb der Schichten werden sogenannte Subzellen definiert, welche die Lage der C2H4-
Einheiten angeben. Zur vollständigen Beschreibung der Anordnung der Kohlenwasserstoff-
ketten im Gitter muß zusätzlich die relative Orientierung der Ebenen, die durch die Zick-
Zack-Anordnung der Ketten gebildet werden, angegeben werden [115, 116].
Die thermodynamisch instabilste Modifikation ist die α-Modifikation, welche durch schnelles
Abkühlen der Schmelze erhalten werden kann. Sie ist durch eine hexagonale Anordnung der
Kohlenwasserstoffketten innerhalb der Schichten charakterisiert. Die Ketten weisen einen

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
91
Neigungswinkel null bezüglich der Schichtebenen auf, jedoch sind die Ebenen, die durch die
Zick-Zack-Anordnung der Ketten gebildet werden, willkürlich zueinander ausgerichtet. Man
schreibt den Ketten Rotationsfreiheitsgrade um ihre Längsachsen zu. Hagemann und Rothfus
[117, 118] stellten hierzu Modellrechnungen an, nach denen Torsions-Oszillationen
wahrscheinlicher als eine freie Rotation der Ketten sind.
Abbildung 5.5 : Hexagonale Subzelle der α-Modifikation. Die Doppelkreise deuten an,
daß die Ketten Rotationsfreiheitsgrade um ihre Längsachsen besitzen.
Die β´-Modifikation, welche gegenüber der β-Modifikation metastabil ist, ist durch eine
orthorhombische Anordnung der Kohlenwasserstoffketten innerhalb der Schichten
gekennzeichnet. Die von der Zick-Zack-Anordnung der Ketten gebildeten Ebenen stehen
senkrecht zueinander. Außerdem weisen die Ketten einen Neigungswinkel zwischen 50° und
70° bezüglich der Schichtebenen auf [108, 119].
Abbildung 5.6: Orthorhombische Subzelle der β´-Modifikation

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
92
In der β-Modifikation, welche die am dichtesten gepackte Modifikation darstellt, sind die
Fettsäureketten innerhalb der Schichten triklin angeordnet [102]. Die von der Zick-Zack-
Konformation der Kohlenwasserstoffketten gebildeten Ebenen sind parallel zueinander
ausgerichtet [119]. Wie in der β´-Modifikation schließen die Ketten mit der Schichtebene
einen Winkel zwischen 50° und 70° ein. Die β-Form stellt die thermodynamisch stabilste
Modifikation der Triacylglycerine dar. Sie kann durch langsames Abkühlen der Schmelze
oder durch Auskristallisieren aus einer Lösung erhalten werden.
Abbildung 5.7: Trikline Subzelle der β-Modifikation
Neben diesen drei Hauptmodifikationen wird die Existenz von weiteren Submodifikationen
diskutiert, wobei jedoch zwischen gesättigten und ungesättigten Triacylglycerinen
unterschieden werden muß. Nähere Informationen zur Existenz von Submodifikationen
ungesättigter Triglyceride finden sich z. B. bei Sato [104]. Für gesättigte Triglyceride gilt die
Existenz einer sub-α-Phase als gesichert. Hierbei handelt es sich um eine orthorhombische
Variante der α-Phase [106, 107]. 1982 leiteten Simpson und Hageman [120] aus ihren
Röntgenstrukturuntersuchungen und DSC-Messungen an Tristearin die Existenz von zwei
verschiedenen β´-Phasen ab. Hernquist und Larsson [105] fanden unabhängig davon mit Hilfe
von Röntgenmessungen ebenfalls zwei β´-Phasen bei Triundecanoin (1,2,3-Tris-
undecanoyloxypropan). In weiterführenden DSC-Messungen an ”symmetrischen”
Triglyceriden gelang Hageman und Rothfus [103] etwas später sogar in Abhängigkeit von der
Kettenlänge, sowie von einer geraden oder ungeraden Kohlenstoffatomanzahl der
Fettsäurekette, die Unterscheidung von zwei α-, drei β´- und zwei β-Submodifikationen.

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
93
Aufgrund der schnellen Umwandlung in die jeweils stabilste Modifikation, konnten diese
Submodifikationen bisher jedoch nicht isoliert werden. Andere Autoren, wie z. B. Kellens
[121], bezweifeln daher, ob es sich tatsächlich um polymorphe Modifikationen im
thermodynamischen Sinne handelt. Zeitaufgelöste Röntgenuntersuchungen führen Kellens zu
dem Schluß, daß es sich bei den aus DSC-Messungen abgeleiteten Submodifikationen
lediglich um Übergangszustände handelt, die bei der Umwandlung zwischen den
Hauptmodifikationen auftreten.
5.4 Umwandlungen zwischen den polymorphen Modifikationen
Triacylglycerine zeigen in der Regel monotrope Polymorphie, d. h. nur eine Modifikation ist
thermodynamisch stabil. Lediglich die Umwandlung einer auftretenden sub-α-Phase in die
α-Phase ist enantiotrop [101, 106].
Die einzelnen metastabilen Modifikationen können aus der Schmelze durch Variation des
Grades der Unterkühlung erhalten werden, jedoch niemals aus der stabilen Modifikation. Eine
metastabile Modifikation wandelt sich stets in die stabile Form um, die Phasenübergänge sind
irreversibel. Bei den meisten Triacylglycerinen stellt die β-Modifikation die thermodynamisch
stabile Phase dar.
Für die möglichen Umwandlungen zwischen den Modifikationen sowie der Schmelze werden
von Wesdorp [108] einerseits sowie von Ollivon und Perron [106, 107] andererseits
unterschiedliche Schemata angegeben (Abbildungen 5.8 und 5.9).
Abbildung 5.8: Phasenumwandlungsschema nach Wesdorp [108]

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
94
Abbildung 5.9: Phasenumwandlungsschema nach Ollivon und Perron [106, 107]
Die Unterschiede zwischen den Schemata bestehen im wesentlichen darin, daß nach Wesdorp
bei der Umwandlung von der α- in die β-Modifikation immer die β´-Modifikation
durchlaufen wird, während sich dagegen nach Ollivon und Perron die α-Modifikation direkt
in die stabile β-Modifikation umwandeln kann. Allerdings handelt es sich dabei nicht um
einen reinen fest-fest-Übergang. Es entsteht vielmehr durch Schmelzen der α-Phase
intermediär eine instabile Flüssigkeit, aus der dann die stabile β-Modifikation auskristallisiert.
Gemäß Ollivon und Perron kann die Schmelze lediglich über die stabile β-Phase erhalten
werden. Dagegen läßt sich nach Wesdorp die Flüssigkeit auch direkt durch Schmelzen der α-
sowie der β´-Modifikation erhalten.
Aus weiterführenden Untersuchungen mittels Röntgenstrukturanalyse und Raman-
spektroskopie konnten Erkenntnisse über die Struktur der Triacylglycerinschmelze gewonnen
werden. Larsson [122] und Hernquist [123] fanden heraus, daß die ”tuning-fork”-
Konformation über den Schmelzpunkt der β-Phase hinaus beibehalten wird und daß eine
lamellare Anordnung der Triacylglycerinschichten vorliegt, die einer thermotropen oder
lyotropen flüssigkristallinen Phase ähnelt.
Für detailliertere Informationen zu Untersuchungen mittels Ramanspektroskopie sei auf die
Literatur verwiesen [122, 123]. Eine ausführliche Zusammenfassung der dort gewonnenen
Ergebnisse findet sich in der Arbeit von Becker [93].

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
95
5.5 Meßergebnisse und Diskussion
Bei dem in der vorliegenden Arbeit untersuchten 1,2,3-Tris-eicosanoyloxypropan, das im
folgenden nur noch mit seinem Trivialnamen oder der entsprechenden Abkürzung
(Triarachidin, AAA) bezeichnet wird (vgl. Abschnitt 5.2), handelt es sich um ein
Triacylglycerin, das an allen drei OH-Gruppen des Glycerins mit der gleichen gesättigten
Fettsäure mit gerader Kohlenstoffatomanzahl verestert ist (SEMT, ”saturated even monoacid
triglyceride”).
Die Reinheit der Substanz wird von der Lieferfirma Sigma [L 17] mit annähernd 99%
angegeben.
Triarachidin wurde unter Normaldruck bereits von verschiedenen Autoren mittels DTA und
DSC untersucht. DTA-Hochdruckuntersuchungen wurden erst von Becker [93], Wagner [110,
111] und Ernst [65] durchgeführt.
In dieser Arbeit wurde das Phasenverhalten des Triarachidins mittels Hochdruck-DSC im
Temperaturbereich zwischen 330 K und 380 K und bei Drücken bis zu 175 MPa untersucht.
Die Messungen wurden mit einer Aufheizrate von 1 K⋅min-1 unter Verwendung von Helium
als Druckgas durchgeführt. Nach den Druckmessungen wurde jeweils unter Normaldruck
abgekühlt.
In Abbildung 5.10 sind drei DSC-Thermogramme, die den Einfluß des Druckes auf die Lage
und Gestalt der einzelnen Phasenübergangspeaks veranschaulichen, dargestellt.
Eine Abkühlgeschwindigkeit von etwa 1,5 K⋅min-1 reichte aus, um die α-Modifikation zu
erhalten. Beim nachfolgenden Aufheizen konnte die Phasenabfolge α→α-Schmelze→β→l
beobachtet werden. Für die Umwandlung α→α-Schmelze→β zeigt sich dabei deutlich ein
Wechsel zwischen endothermem und exothermem Peak, wodurch der Mechanismus der
Umwandlung, nämlich ein endothermes Schmelzen der α-Modifikation zur instabilen
α-Schmelze, die exotherm zur β-Modifikation auskristallisiert, bestätigt wird. Die Phasen-
abfolge α→β´→β wurde nicht gefunden. Die beiden Umwandlungen α→β und β→l konnten
in der vorliegenden Arbeit über den gesamten Druckbereich verfolgt werden. Im Gegensatz
dazu stehen die Meßergebnisse von Ernst [65], wonach bei Drücken oberhalb von 76.5 MPa
nur noch das Schmelzen einer festen Phase (vermutlich der α-Phase) zu erkennen ist.
Hagemann und Rothfus [103] konnten das Ausbleiben des Auskristallisierens der β-Phase,
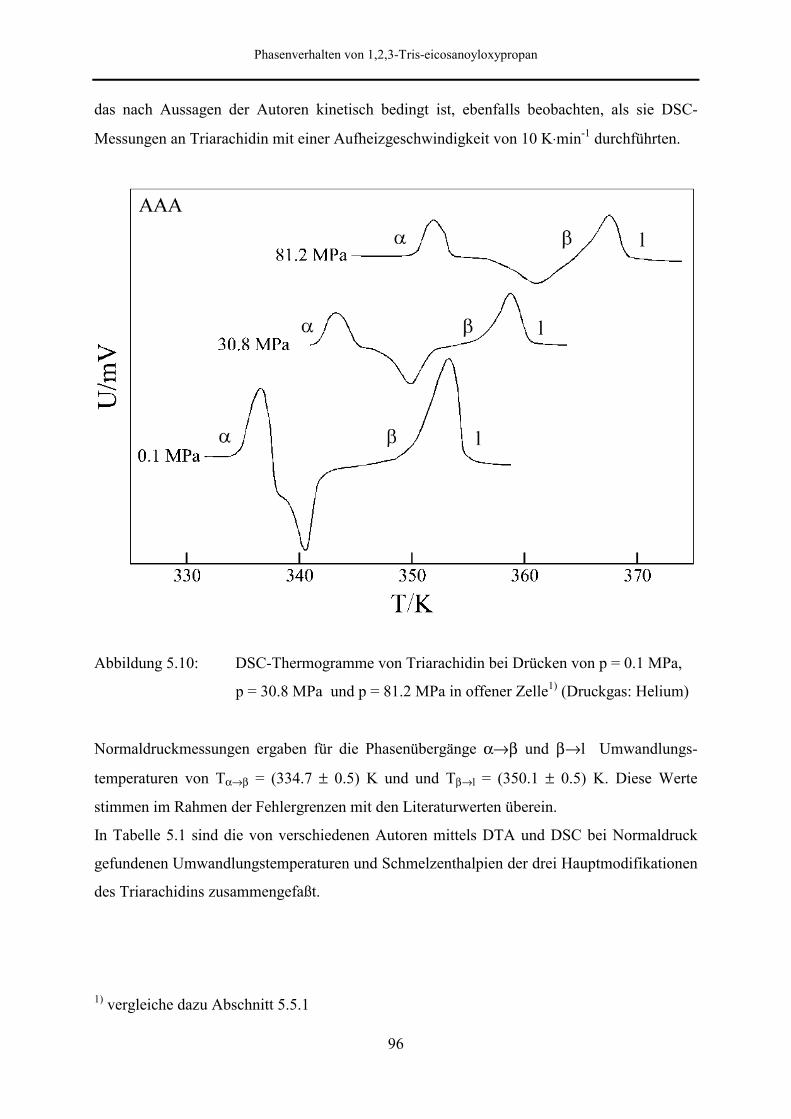
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
96
das nach Aussagen der Autoren kinetisch bedingt ist, ebenfalls beobachten, als sie DSC-
Messungen an Triarachidin mit einer Aufheizgeschwindigkeit von 10 K⋅min-1 durchführten.
Abbildung 5.10: DSC-Thermogramme von Triarachidin bei Drücken von p = 0.1 MPa,
p = 30.8 MPa und p = 81.2 MPa in offener Zelle1) (Druckgas: Helium)
Normaldruckmessungen ergaben für die Phasenübergänge α→β und β→l Umwandlungs-
temperaturen von Tα→β = (334.7 ± 0.5) K und und Tβ→l = (350.1 ± 0.5) K. Diese Werte
stimmen im Rahmen der Fehlergrenzen mit den Literaturwerten überein.
In Tabelle 5.1 sind die von verschiedenen Autoren mittels DTA und DSC bei Normaldruck
gefundenen Umwandlungstemperaturen und Schmelzenthalpien der drei Hauptmodifikationen
des Triarachidins zusammengefaßt.
1) vergleiche dazu Abschnitt 5.5.1
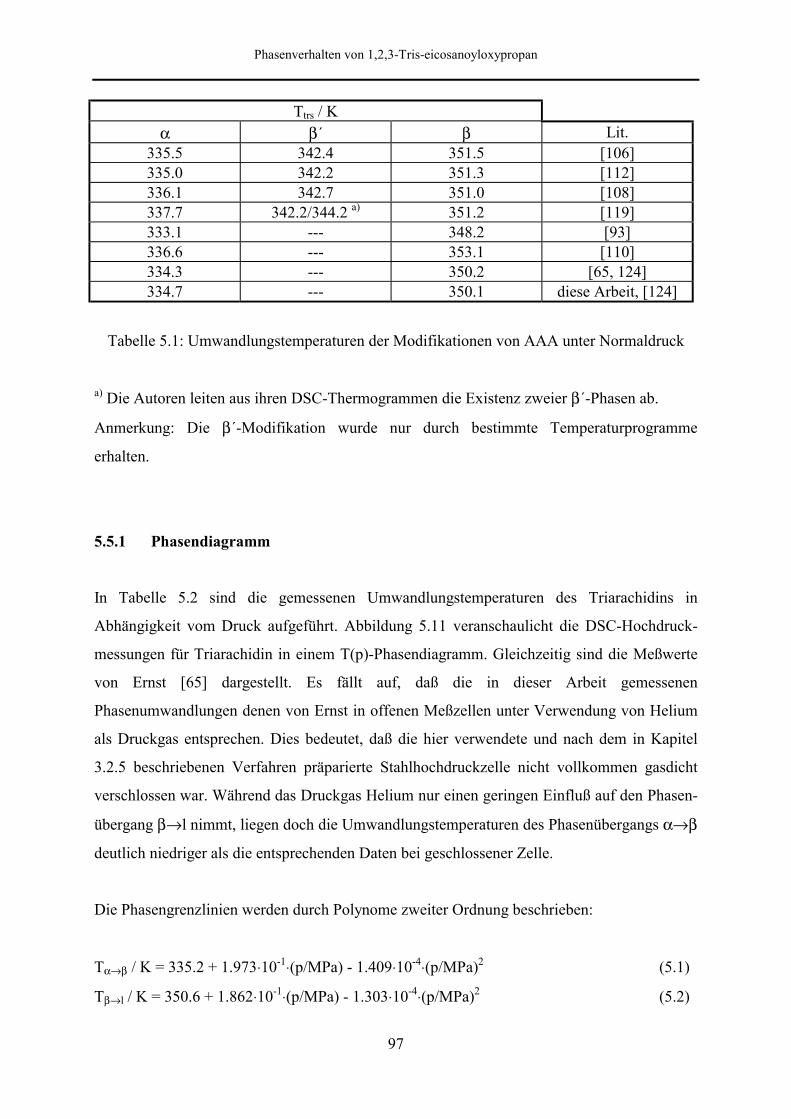
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
97
Ttrs / Kα β´ β Lit.
335.5 342.4 351.5 [106]335.0 342.2 351.3 [112]336.1 342.7 351.0 [108]337.7 342.2/344.2 a) 351.2 [119]333.1 --- 348.2 [93]336.6 --- 353.1 [110]334.3 --- 350.2 [65, 124]334.7 --- 350.1 diese Arbeit, [124]
Tabelle 5.1: Umwandlungstemperaturen der Modifikationen von AAA unter Normaldruck
a) Die Autoren leiten aus ihren DSC-Thermogrammen die Existenz zweier β´-Phasen ab.
Anmerkung: Die β´-Modifikation wurde nur durch bestimmte Temperaturprogramme
erhalten.
5.5.1 Phasendiagramm
In Tabelle 5.2 sind die gemessenen Umwandlungstemperaturen des Triarachidins in
Abhängigkeit vom Druck aufgeführt. Abbildung 5.11 veranschaulicht die DSC-Hochdruck-
messungen für Triarachidin in einem T(p)-Phasendiagramm. Gleichzeitig sind die Meßwerte
von Ernst [65] dargestellt. Es fällt auf, daß die in dieser Arbeit gemessenen
Phasenumwandlungen denen von Ernst in offenen Meßzellen unter Verwendung von Helium
als Druckgas entsprechen. Dies bedeutet, daß die hier verwendete und nach dem in Kapitel
3.2.5 beschriebenen Verfahren präparierte Stahlhochdruckzelle nicht vollkommen gasdicht
verschlossen war. Während das Druckgas Helium nur einen geringen Einfluß auf den Phasen-
übergang β→l nimmt, liegen doch die Umwandlungstemperaturen des Phasenübergangs α→β
deutlich niedriger als die entsprechenden Daten bei geschlossener Zelle.
Die Phasengrenzlinien werden durch Polynome zweiter Ordnung beschrieben:
Tα→β / K = 335.2 + 1.973⋅10-1⋅(p/MPa) - 1.409⋅10-4⋅(p/MPa)2 (5.1)
Tβ→l / K = 350.6 + 1.862⋅10-1⋅(p/MPa) - 1.303⋅10-4⋅(p/MPa)2 (5.2)
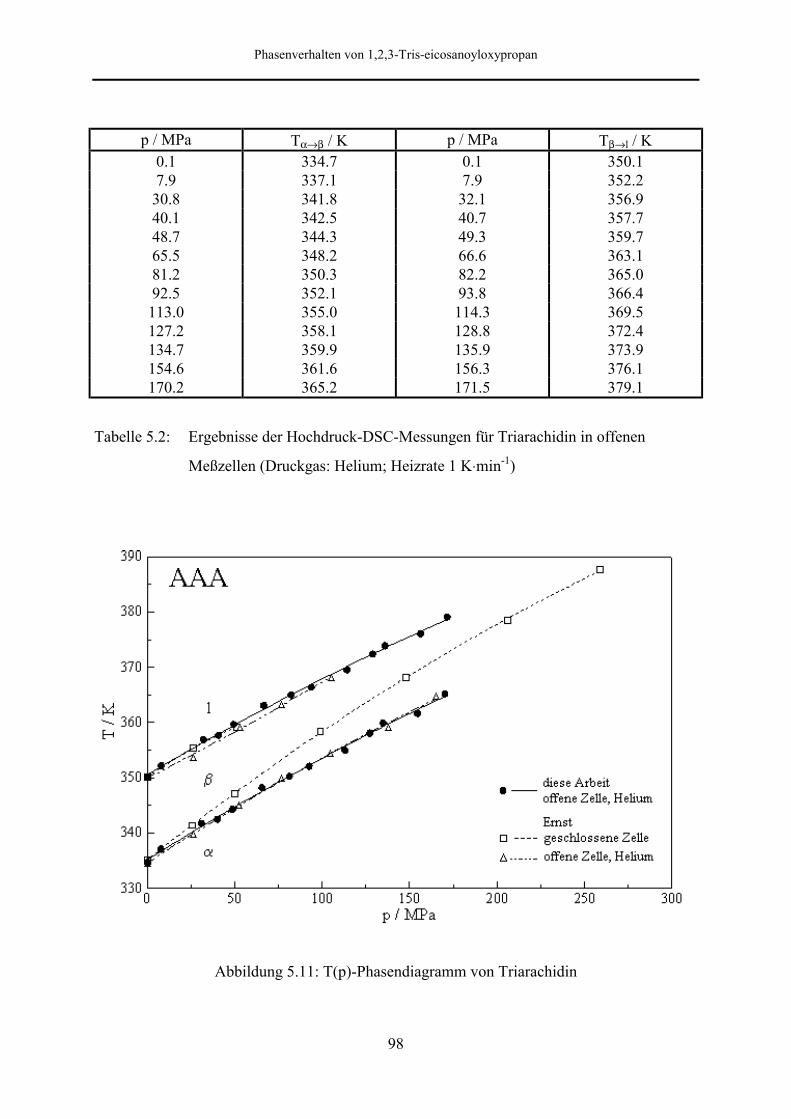
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
98
p / MPa Tα→β / K p / MPa Tβ→l / K0.1 334.7 0.1 350.17.9 337.1 7.9 352.230.8 341.8 32.1 356.940.1 342.5 40.7 357.748.7 344.3 49.3 359.765.5 348.2 66.6 363.181.2 350.3 82.2 365.092.5 352.1 93.8 366.4113.0 355.0 114.3 369.5127.2 358.1 128.8 372.4134.7 359.9 135.9 373.9154.6 361.6 156.3 376.1170.2 365.2 171.5 379.1
Tabelle 5.2: Ergebnisse der Hochdruck-DSC-Messungen für Triarachidin in offenen
Meßzellen (Druckgas: Helium; Heizrate 1 K⋅min-1)
Abbildung 5.11: T(p)-Phasendiagramm von Triarachidin

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
99
5.5.2 Phasenumwandlungsenthalpie, -entropie und -volumen
In Abbildung 5.12 sind die Umwandlungsenthalpien der Phasenübergänge α→β und β→l von
Triarachidin gegen den Druck aufgetragen. Die Umwandlungsenthalpie des β→l -Übergangs
nimmt mit steigendem Druck leicht ab. Für den Übergang α→β findet sich dagegen ein
nahezu druckunabhängiger Verlauf der Umwandlungsenthalpie.
Die folgenden Ausgleichsfunktionen beschreiben den Verlauf der Umwandlungsenthalpien
für die Phasenübergänge α→β und β→l mit dem Druck:
∆α→βHm / kJ⋅mol-1 = 124.66 - 3.279⋅10-2⋅(p/MPa) + 2.364⋅10-4⋅(p/MPa)2 (5.3)
∆β→lHm / kJ⋅mol-1 = 202.07 - 4.013⋅10-1⋅(p/MPa) + 1.762⋅10-3⋅(p/MPa)2 (5.4)
Die Ableitungen dieser Funktionen nach dem Druck liefern Ausdrücke für die Plancksche
Gleichung (2.18) für diese Phasenübergänge. Die bei Normaldruck ermittelten Werte der
Umwandlungsenthalpien
∆α→βHm = (125 ± 14) kJ⋅mol-1
∆β→lHm = (200 ± 16) kJ⋅mol-1
stimmen im Rahmen der Fehlergrenzen mit den in Tabelle 5.3 aufgeführten Literaturwerten
überein.
∆trsHm / kJ⋅mol-1
α β´ β Lit.124.8 160.3 207.7 [106]122.3 --- 220.7 [103]125 --- 200 diese Arbeit
Tabelle 5.3: Umwandlungsenthalpien der Modifikationen von Triarachidin bei
Normaldruck (mit DSC ermittelt)
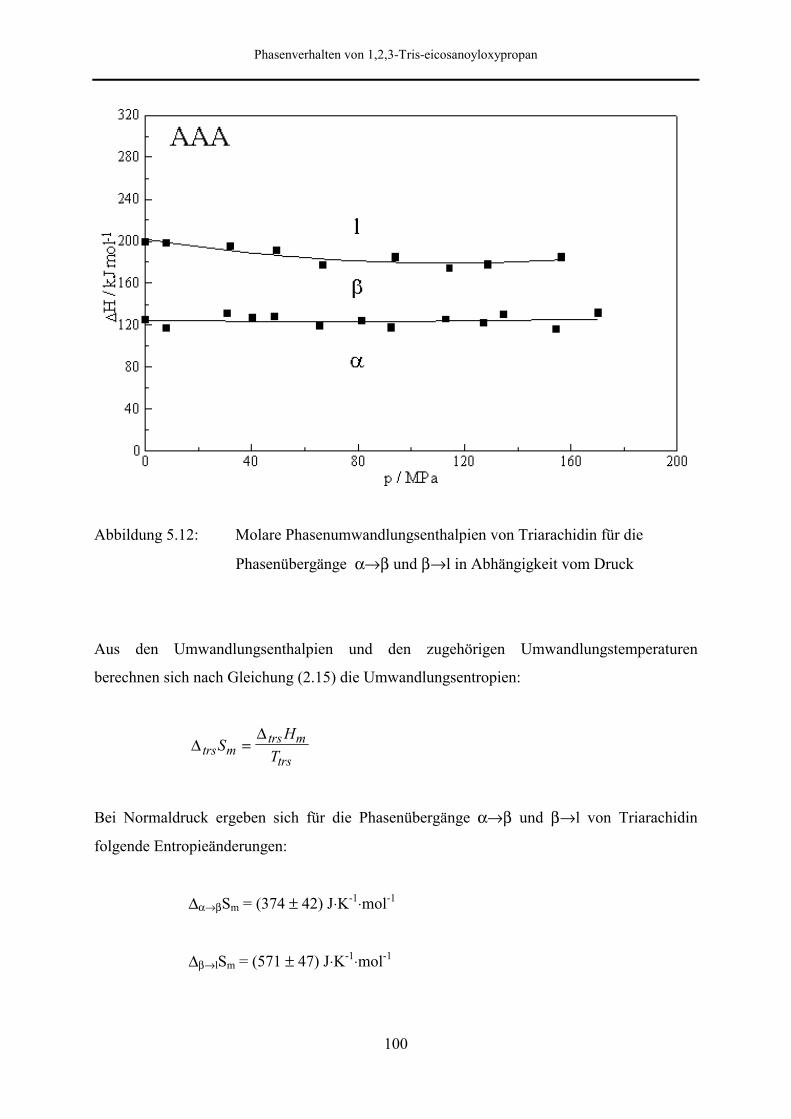
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
100
Abbildung 5.12: Molare Phasenumwandlungsenthalpien von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck
Aus den Umwandlungsenthalpien und den zugehörigen Umwandlungstemperaturen
berechnen sich nach Gleichung (2.15) die Umwandlungsentropien:
∆∆
trs mtrs m
trsS
HT
=
Bei Normaldruck ergeben sich für die Phasenübergänge α→β und β→l von Triarachidin
folgende Entropieänderungen:
∆α→βSm = (374 ± 42) J⋅K-1⋅mol-1
∆β→lSm = (571 ± 47) J⋅K-1⋅mol-1
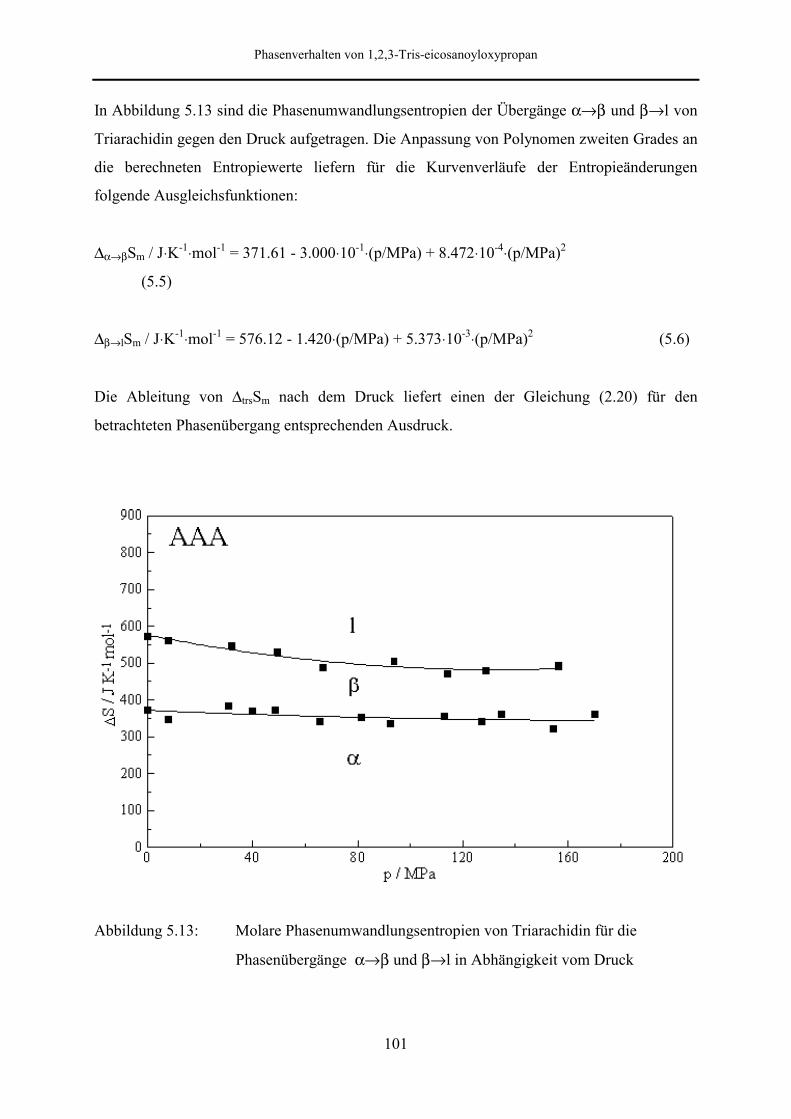
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
101
In Abbildung 5.13 sind die Phasenumwandlungsentropien der Übergänge α→β und β→l von
Triarachidin gegen den Druck aufgetragen. Die Anpassung von Polynomen zweiten Grades an
die berechneten Entropiewerte liefern für die Kurvenverläufe der Entropieänderungen
folgende Ausgleichsfunktionen:
∆α→βSm / J⋅K-1⋅mol-1 = 371.61 - 3.000⋅10-1⋅(p/MPa) + 8.472⋅10-4⋅(p/MPa)2
(5.5)
∆β→lSm / J⋅K-1⋅mol-1 = 576.12 - 1.420⋅(p/MPa) + 5.373⋅10-3⋅(p/MPa)2 (5.6)
Die Ableitung von ∆trsSm nach dem Druck liefert einen der Gleichung (2.20) für den
betrachteten Phasenübergang entsprechenden Ausdruck.
Abbildung 5.13: Molare Phasenumwandlungsentropien von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck

Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
102
Die Umwandlungsvolumina berechnen sich nach der Clausius-Clapeyron-Gleichung (2.16):
dpdT
SVkoex
trs m
trs m
=
∆∆
⇒ ∆ ∆trs m trs mkoex
V SdTdp
= ⋅
Hierbei ist dTdp koex
die Steigung der Koexistenzlinie des jeweiligen Phasenübergangs im
T(p)-Diagramm. Es ergeben sich für die Steigungen folgende Gleichungen:
dTdp
→α β= 0.1973 - 2.818⋅10-4⋅(p/MPa) (5.7)
dTdp
→β l = 0.1862 - 2.606⋅10-4⋅(p/MPa) (5.8)
Die Anpassung der berechneten Umwandlungsvolumina für die entsprechenden Phasen-
übergänge liefert folgende Korrelationsfunktionen:
∆α→βVm / 10-6⋅m3⋅mol-1 = 73.21 - 1.276⋅10-1⋅(p/MPa) + 1.653⋅10-4⋅(p/MPa)2 (5.9)
∆β→lVm / 10-6⋅m3⋅mol-1 = 107.12 - 3.634⋅10-1⋅(p/MPa) + 1.022⋅10-3⋅(p/MPa)2 (5.10)
Leitet man die Gleichungen (5.9) und (5.10) nach dem Druck ab, so erhält man für die
betrachteten Phasenübergänge Ausdrücke, die der Gleichung (2.22) entsprechen.
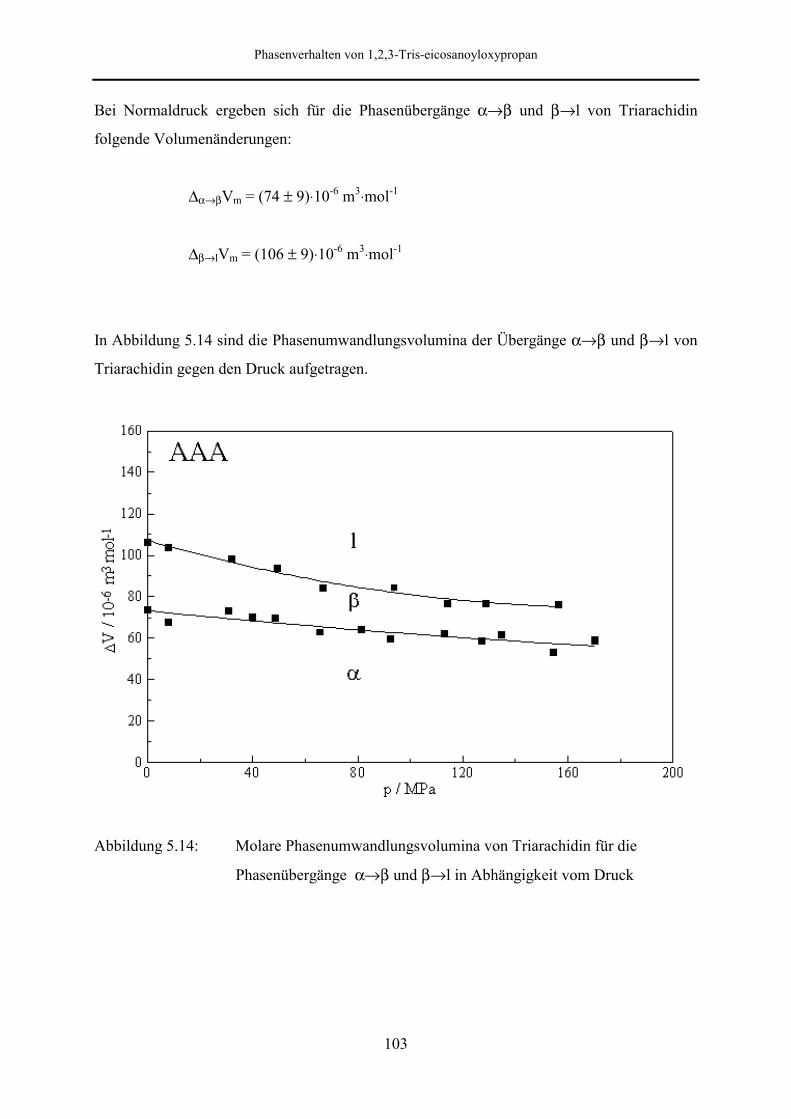
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
103
Bei Normaldruck ergeben sich für die Phasenübergänge α→β und β→l von Triarachidin
folgende Volumenänderungen:
∆α→βVm = (74 ± 9)⋅10-6 m3⋅mol-1
∆β→lVm = (106 ± 9)⋅10-6 m3⋅mol-1
In Abbildung 5.14 sind die Phasenumwandlungsvolumina der Übergänge α→β und β→l von
Triarachidin gegen den Druck aufgetragen.
Abbildung 5.14: Molare Phasenumwandlungsvolumina von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck
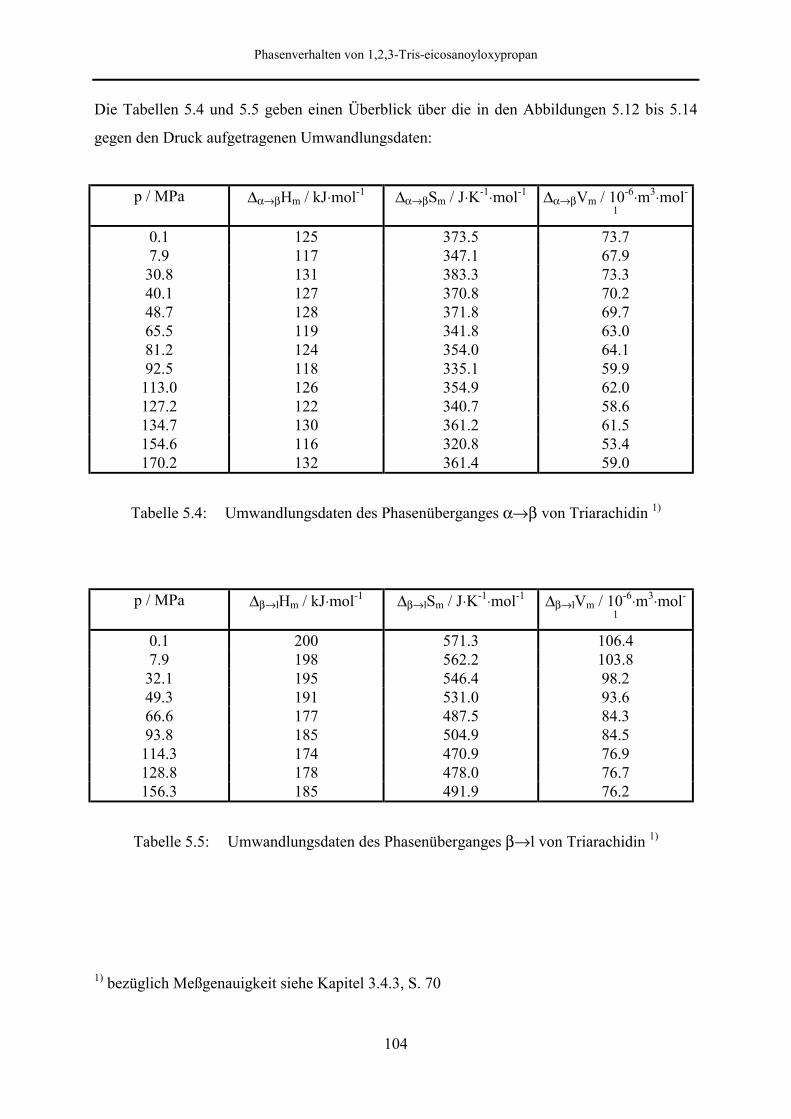
Phasenverhalten von 1,2,3-Tris-eicosanoyloxypropan
104
Die Tabellen 5.4 und 5.5 geben einen Überblick über die in den Abbildungen 5.12 bis 5.14
gegen den Druck aufgetragenen Umwandlungsdaten:
p / MPa ∆α→βHm / kJ⋅mol-1 ∆α→βSm / J⋅K-1⋅mol-1 ∆α→βVm / 10-6⋅m3⋅mol-
1
0.1 125 373.5 73.77.9 117 347.1 67.930.8 131 383.3 73.340.1 127 370.8 70.248.7 128 371.8 69.765.5 119 341.8 63.081.2 124 354.0 64.192.5 118 335.1 59.9113.0 126 354.9 62.0127.2 122 340.7 58.6134.7 130 361.2 61.5154.6 116 320.8 53.4170.2 132 361.4 59.0
Tabelle 5.4: Umwandlungsdaten des Phasenüberganges α→β von Triarachidin 1)
p / MPa ∆β→lHm / kJ⋅mol-1 ∆β→lSm / J⋅K-1⋅mol-1 ∆β→lVm / 10-6⋅m3⋅mol-
1
0.1 200 571.3 106.47.9 198 562.2 103.832.1 195 546.4 98.249.3 191 531.0 93.666.6 177 487.5 84.393.8 185 504.9 84.5114.3 174 470.9 76.9128.8 178 478.0 76.7156.3 185 491.9 76.2
Tabelle 5.5: Umwandlungsdaten des Phasenüberganges β→l von Triarachidin 1)
1) bezüglich Meßgenauigkeit siehe Kapitel 3.4.3, S. 70

Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
105
6 Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
6.1 Flüssigkristalle mit lateralen aromatischen Verzweigungen
Die im folgenden aufgelisteten Flüssigkristalle wurden von der Max-Planck-Arbeitsgruppe
”Flüssigkristalline Systeme” an der Martin-Luther-Universität Halle-Wittenberg unter Leitung
von Prof. Dr. Weißflog synthetisiert und gereinigt. Aufgeführt sind außerdem die
Strukturformeln und die in der Arbeit von Ernst [65] mittels DTA gefundenen
Phasenumwandlungstemperaturen:
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylester
kristallin 368 2. K → smektisch A 435 6. K → isotrop flüssig
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-cyan-benzylester
kristallin 363 9. K → smektisch A 423 3. K → isotrop flüssig
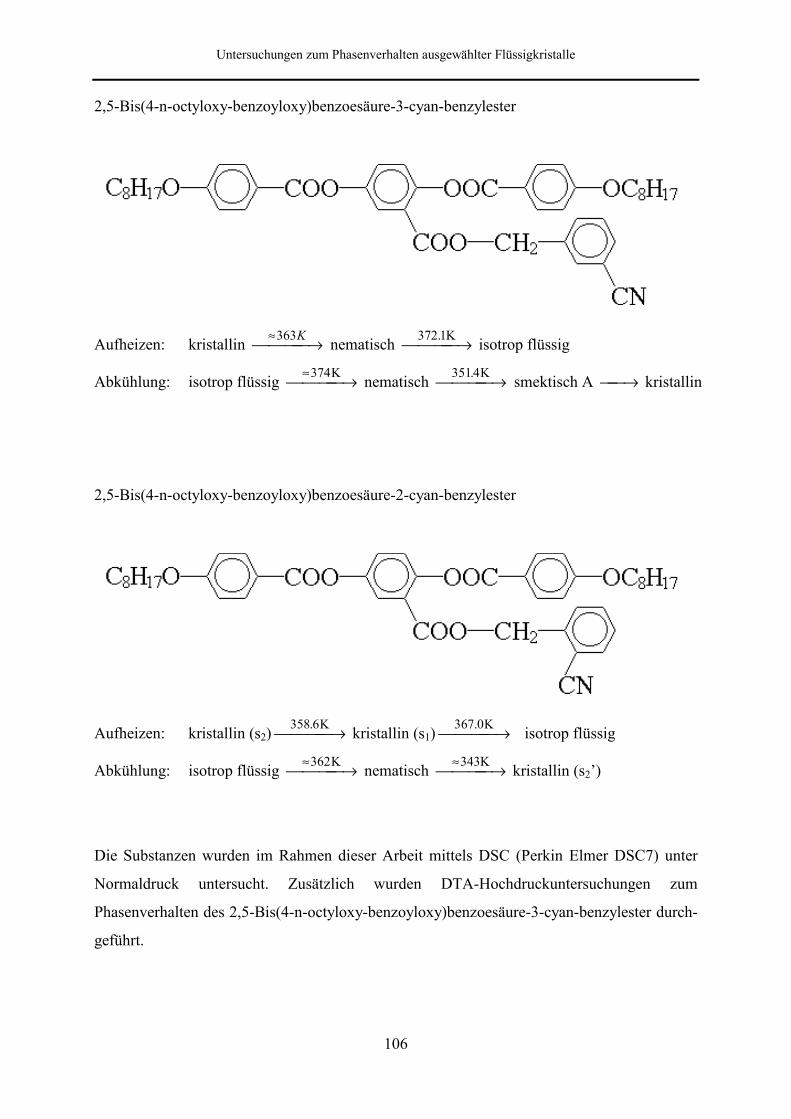
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
106
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester
Aufheizen: kristallin ≈ →363K nematisch 372.1K → isotrop flüssig
Abkühlung: isotrop flüssig ≈ →374K nematisch 3514. K → smektisch A → kristallin
2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-2-cyan-benzylester
Aufheizen: kristallin (s2)358 6. K → kristallin (s1)
367 0. K → isotrop flüssig
Abkühlung: isotrop flüssig ≈ →362K nematisch ≈ →343K kristallin (s2’)
Die Substanzen wurden im Rahmen dieser Arbeit mittels DSC (Perkin Elmer DSC7) unter
Normaldruck untersucht. Zusätzlich wurden DTA-Hochdruckuntersuchungen zum
Phasenverhalten des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester durch-
geführt.
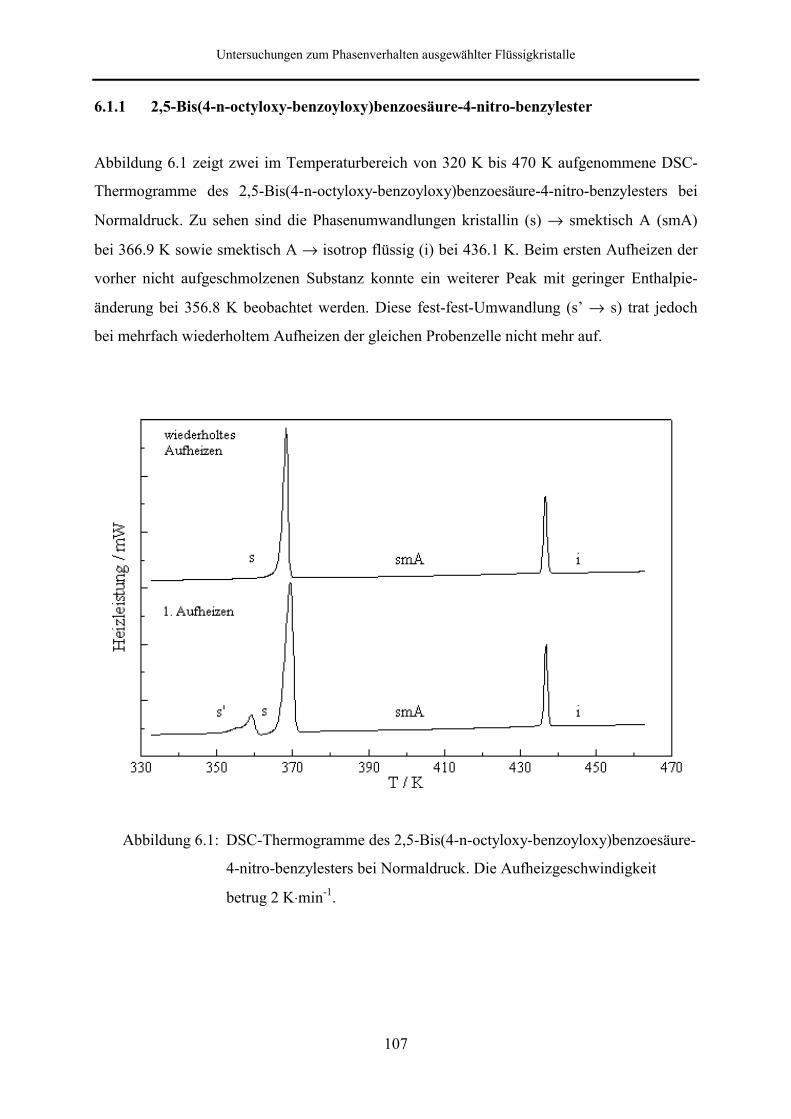
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
107
6.1.1 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylester
Abbildung 6.1 zeigt zwei im Temperaturbereich von 320 K bis 470 K aufgenommene DSC-
Thermogramme des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylesters bei
Normaldruck. Zu sehen sind die Phasenumwandlungen kristallin (s) → smektisch A (smA)
bei 366.9 K sowie smektisch A → isotrop flüssig (i) bei 436.1 K. Beim ersten Aufheizen der
vorher nicht aufgeschmolzenen Substanz konnte ein weiterer Peak mit geringer Enthalpie-
änderung bei 356.8 K beobachtet werden. Diese fest-fest-Umwandlung (s’ → s) trat jedoch
bei mehrfach wiederholtem Aufheizen der gleichen Probenzelle nicht mehr auf.
Abbildung 6.1: DSC-Thermogramme des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-
4-nitro-benzylesters bei Normaldruck. Die Aufheizgeschwindigkeit
betrug 2 K⋅min-1.
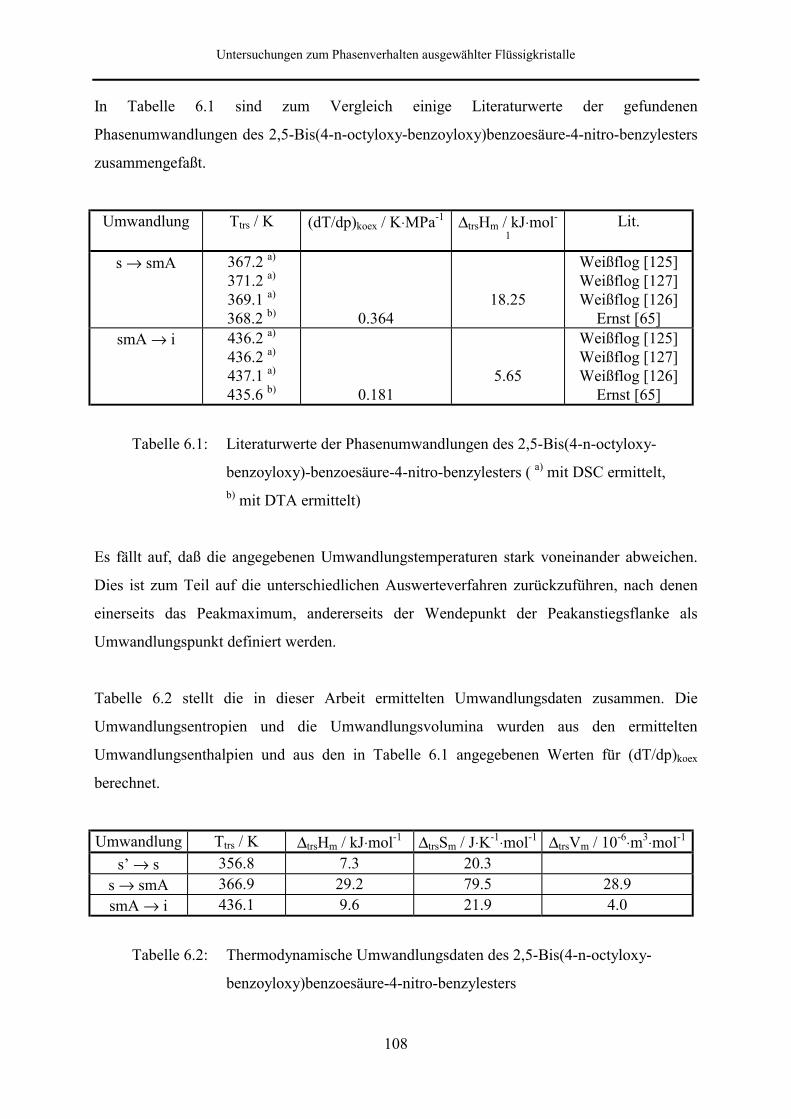
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
108
In Tabelle 6.1 sind zum Vergleich einige Literaturwerte der gefundenen
Phasenumwandlungen des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-nitro-benzylesters
zusammengefaßt.
Umwandlung Ttrs / K (dT/dp)koex / K⋅MPa-1 ∆trsHm / kJ⋅mol-
1Lit.
s → smA 367.2 a)
371.2 a)
369.1 a)
368.2 b) 0.36418.25
Weißflog [125]Weißflog [127]Weißflog [126]
Ernst [65]smA → i 436.2 a)
436.2 a)
437.1 a)
435.6 b) 0.1815.65
Weißflog [125]Weißflog [127]Weißflog [126]
Ernst [65]
Tabelle 6.1: Literaturwerte der Phasenumwandlungen des 2,5-Bis(4-n-octyloxy-
benzoyloxy)-benzoesäure-4-nitro-benzylesters ( a) mit DSC ermittelt,b) mit DTA ermittelt)
Es fällt auf, daß die angegebenen Umwandlungstemperaturen stark voneinander abweichen.
Dies ist zum Teil auf die unterschiedlichen Auswerteverfahren zurückzuführen, nach denen
einerseits das Peakmaximum, andererseits der Wendepunkt der Peakanstiegsflanke als
Umwandlungspunkt definiert werden.
Tabelle 6.2 stellt die in dieser Arbeit ermittelten Umwandlungsdaten zusammen. Die
Umwandlungsentropien und die Umwandlungsvolumina wurden aus den ermittelten
Umwandlungsenthalpien und aus den in Tabelle 6.1 angegebenen Werten für (dT/dp)koex
berechnet.
Umwandlung Ttrs / K ∆trsHm / kJ⋅mol-1 ∆trsSm / J⋅K-1⋅mol-1 ∆trsVm / 10-6⋅m3⋅mol-1
s’ → s 356.8 7.3 20.3s → smA 366.9 29.2 79.5 28.9smA → i 436.1 9.6 21.9 4.0
Tabelle 6.2: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-4-nitro-benzylesters
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
109
6.1.2 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-cyan-benzylester
Das Phasenverhalten des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-4-cyan-benzylesters
ist in Abbildung 6.2 im Temperaturbereich von 320 K bis 450 K bei Normaldruck dargestellt.
Abbildung 6.2: DSC-Normaldruckthermogramm des 2,5-Bis(4-n-octyloxybenzoyloxy)-
benzoesäure-4-cyan-benzylesters (Heizrate: 2 K⋅min-1)
Die Phasenumwandlung kristallin (s) → smektisch A (smA) erfolgt bei 360.0 K und liegt
damit deutlich unter der von Ernst angegebenen Umwandlungstemperatur (363.9 K). Der
Klärpeak ist im Thermogramm bei 423.0 K aufzufinden. Das Aufheizprogramm wurde
mehrfach wiederholt; auch nach mehrtägigem Tempern der Probe bei 293 K ergab sich stets
der gleiche Kurvenverlauf.
Die Meßergebnisse der DSC-Untersuchungen sind Tabelle 6.3 zu entnehmen. Aufgeführt sind
auch die von Ernst ermittelten Steigungen der Phasenkoexistenzlinien (dT/dp)koex, mit denen
die Volumenänderungen der entsprechenden Umwandlungen berechnet wurden.
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
110
Umwandlung Ttrs / K (dT/dp)koex /K⋅MPa-1
∆trsHm /kJ⋅mol-1
∆trsSm /J⋅K-1⋅mol-1
∆trsVm /10-6⋅m3⋅mol-1
s → smA 360.0 0.304 [65] 25.6 71.2 21.6smA → i 423.0 0.241 [65] 8.1 19.1 4.6
Tabelle 6.3: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-4-cyan-benzylesters
6.1.3 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-2-cyan-benzylester
Das DSC-Thermogramm in Abbildung 6.3 zeigt eine Aufheiz- und die sich daran
anschließende Abkühlkurve des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-2-cyan-
benzylesters bei Normaldruck.
Abbildung 6.3: Aufheiz- und Abkühlthermogramm des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-2-cyan-benzylesters bei Normaldruck
(Heiz-/Kühlrate: 2 K⋅min-1)
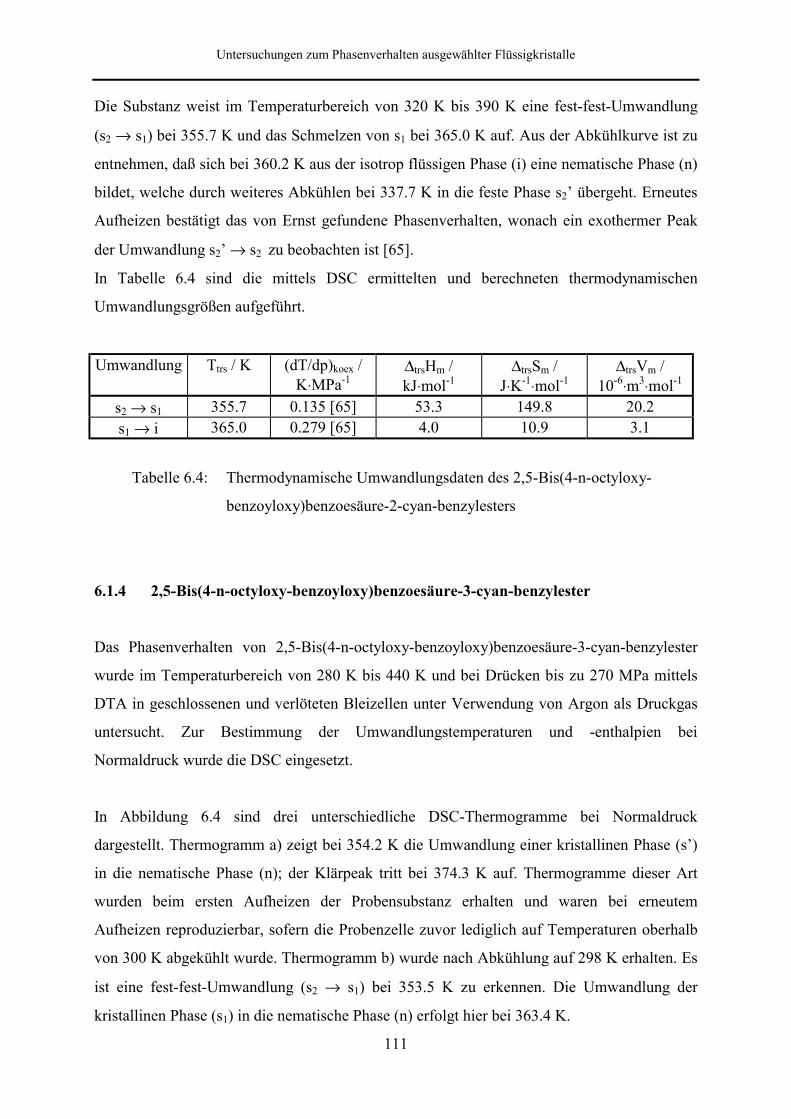
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
111
Die Substanz weist im Temperaturbereich von 320 K bis 390 K eine fest-fest-Umwandlung
(s2 → s1) bei 355.7 K und das Schmelzen von s1 bei 365.0 K auf. Aus der Abkühlkurve ist zu
entnehmen, daß sich bei 360.2 K aus der isotrop flüssigen Phase (i) eine nematische Phase (n)
bildet, welche durch weiteres Abkühlen bei 337.7 K in die feste Phase s2’ übergeht. Erneutes
Aufheizen bestätigt das von Ernst gefundene Phasenverhalten, wonach ein exothermer Peak
der Umwandlung s2’ → s2 zu beobachten ist [65].
In Tabelle 6.4 sind die mittels DSC ermittelten und berechneten thermodynamischen
Umwandlungsgrößen aufgeführt.
Umwandlung Ttrs / K (dT/dp)koex /K⋅MPa-1
∆trsHm /kJ⋅mol-1
∆trsSm /J⋅K-1⋅mol-1
∆trsVm /10-6⋅m3⋅mol-1
s2 → s1 355.7 0.135 [65] 53.3 149.8 20.2s1 → i 365.0 0.279 [65] 4.0 10.9 3.1
Tabelle 6.4: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-2-cyan-benzylesters
6.1.4 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester
Das Phasenverhalten von 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylester
wurde im Temperaturbereich von 280 K bis 440 K und bei Drücken bis zu 270 MPa mittels
DTA in geschlossenen und verlöteten Bleizellen unter Verwendung von Argon als Druckgas
untersucht. Zur Bestimmung der Umwandlungstemperaturen und -enthalpien bei
Normaldruck wurde die DSC eingesetzt.
In Abbildung 6.4 sind drei unterschiedliche DSC-Thermogramme bei Normaldruck
dargestellt. Thermogramm a) zeigt bei 354.2 K die Umwandlung einer kristallinen Phase (s’)
in die nematische Phase (n); der Klärpeak tritt bei 374.3 K auf. Thermogramme dieser Art
wurden beim ersten Aufheizen der Probensubstanz erhalten und waren bei erneutem
Aufheizen reproduzierbar, sofern die Probenzelle zuvor lediglich auf Temperaturen oberhalb
von 300 K abgekühlt wurde. Thermogramm b) wurde nach Abkühlung auf 298 K erhalten. Es
ist eine fest-fest-Umwandlung (s2 → s1) bei 353.5 K zu erkennen. Die Umwandlung der
kristallinen Phase (s1) in die nematische Phase (n) erfolgt hier bei 363.4 K.
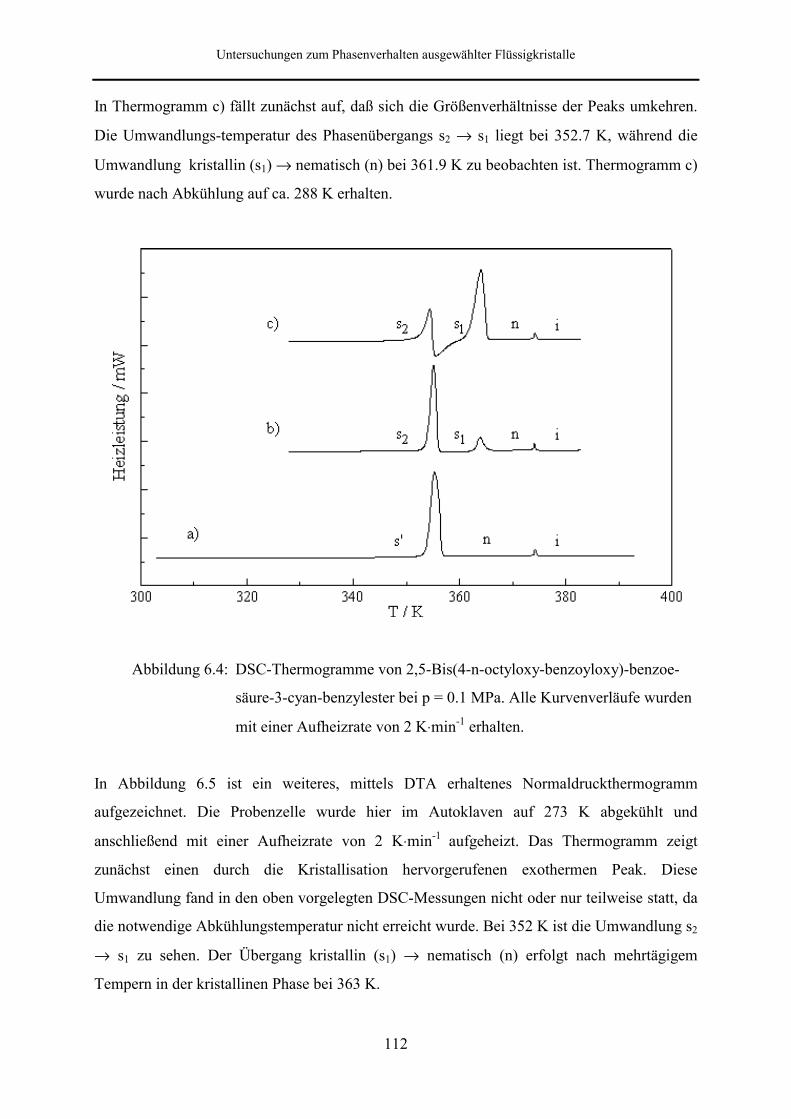
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
112
In Thermogramm c) fällt zunächst auf, daß sich die Größenverhältnisse der Peaks umkehren.
Die Umwandlungs-temperatur des Phasenübergangs s2 → s1 liegt bei 352.7 K, während die
Umwandlung kristallin (s1) → nematisch (n) bei 361.9 K zu beobachten ist. Thermogramm c)
wurde nach Abkühlung auf ca. 288 K erhalten.
Abbildung 6.4: DSC-Thermogramme von 2,5-Bis(4-n-octyloxy-benzoyloxy)-benzoe-
säure-3-cyan-benzylester bei p = 0.1 MPa. Alle Kurvenverläufe wurden
mit einer Aufheizrate von 2 K⋅min-1 erhalten.
In Abbildung 6.5 ist ein weiteres, mittels DTA erhaltenes Normaldruckthermogramm
aufgezeichnet. Die Probenzelle wurde hier im Autoklaven auf 273 K abgekühlt und
anschließend mit einer Aufheizrate von 2 K⋅min-1 aufgeheizt. Das Thermogramm zeigt
zunächst einen durch die Kristallisation hervorgerufenen exothermen Peak. Diese
Umwandlung fand in den oben vorgelegten DSC-Messungen nicht oder nur teilweise statt, da
die notwendige Abkühlungstemperatur nicht erreicht wurde. Bei 352 K ist die Umwandlung s2
→ s1 zu sehen. Der Übergang kristallin (s1) → nematisch (n) erfolgt nach mehrtägigem
Tempern in der kristallinen Phase bei 363 K.
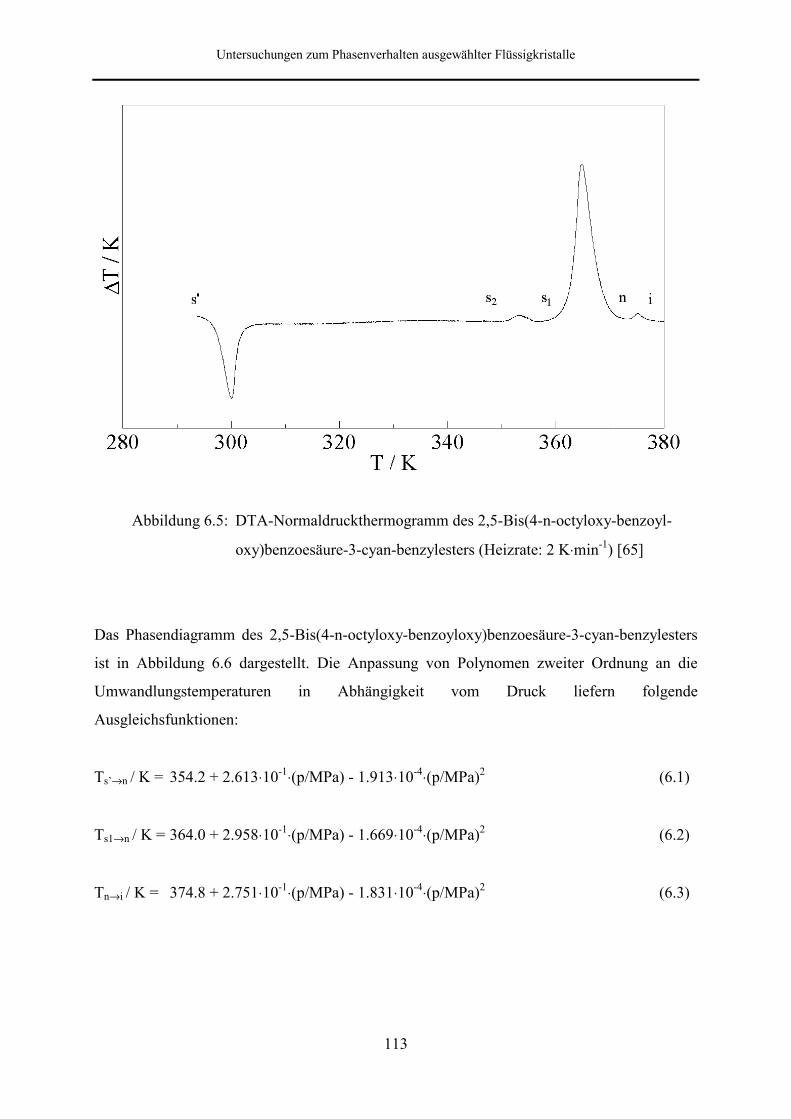
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
113
Abbildung 6.5: DTA-Normaldruckthermogramm des 2,5-Bis(4-n-octyloxy-benzoyl-
oxy)benzoesäure-3-cyan-benzylesters (Heizrate: 2 K⋅min-1) [65]
Das Phasendiagramm des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoesäure-3-cyan-benzylesters
ist in Abbildung 6.6 dargestellt. Die Anpassung von Polynomen zweiter Ordnung an die
Umwandlungstemperaturen in Abhängigkeit vom Druck liefern folgende
Ausgleichsfunktionen:
Ts’→n / K = 354.2 + 2.613⋅10-1⋅(p/MPa) - 1.913⋅10-4⋅(p/MPa)2 (6.1)
Ts1→n / K = 364.0 + 2.958⋅10-1⋅(p/MPa) - 1.669⋅10-4⋅(p/MPa)2 (6.2)
Tn→i / K = 374.8 + 2.751⋅10-1⋅(p/MPa) - 1.831⋅10-4⋅(p/MPa)2 (6.3)
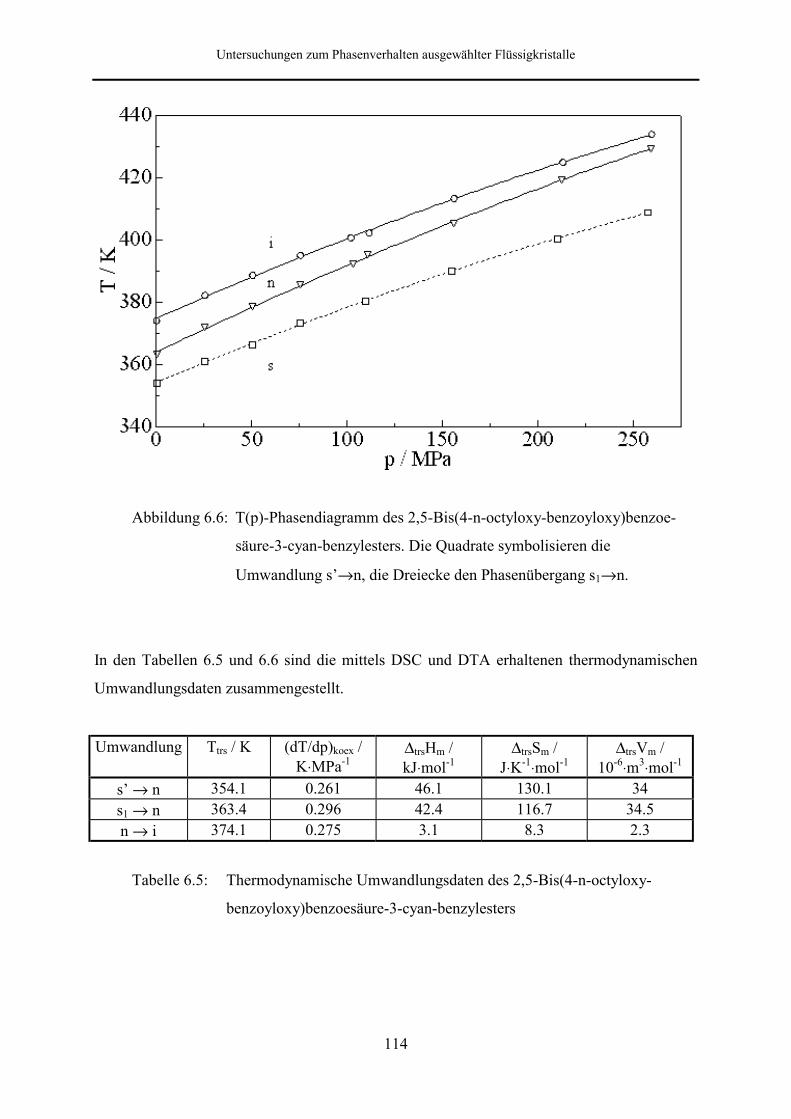
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
114
Abbildung 6.6: T(p)-Phasendiagramm des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoe-
säure-3-cyan-benzylesters. Die Quadrate symbolisieren die
Umwandlung s’→n, die Dreiecke den Phasenübergang s1→n.
In den Tabellen 6.5 und 6.6 sind die mittels DSC und DTA erhaltenen thermodynamischen
Umwandlungsdaten zusammengestellt.
Umwandlung Ttrs / K (dT/dp)koex /K⋅MPa-1
∆trsHm /kJ⋅mol-1
∆trsSm /J⋅K-1⋅mol-1
∆trsVm /10-6⋅m3⋅mol-1
s’ → n 354.1 0.261 46.1 130.1 34s1 → n 363.4 0.296 42.4 116.7 34.5n → i 374.1 0.275 3.1 8.3 2.3
Tabelle 6.5: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-3-cyan-benzylesters
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
115
p / MPa Ts’→n / K p / MPa Ts1→n / K p / MPa Tn→i / K0.1 354.1 0.1 363.4 0.1 374.125.5 360.9 25.5 372.1 25.5 382.350.5 366.5 50.5 378.6 50.5 388.775.5 373.4 75.5 385.6 75.5 395.1109.5 380.3 103.0 392.3 102.0 400.8154.5 390.0 110.5 395.2 111.5 402.3210.5 400.5 156.0 405.3 156.0 413.4257.5 408.9 212.5 419.5 213.0 425.0
259.0 429.5 259.5 434.0
Tabelle 6.6: Ergebnisse der Hochdruck-DTA-Messungen des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-3-cyan-benzylesters (Druckgas: Argon;
Heizrate 2 K⋅min-1)
6.2 Phasenverhalten der 1-[4-Alkylbiphenyl]-2-[4-isothiocyanatophenyl]ethane
6-TPEB und 10-TPEB
Die Flüssigkristalle 6-TPEB und 10-TPEB wurden am Institut für Chemie der
Militärakademie für Technologie in Warschau synthetisiert und gereinigt [128]. Es handelt
sich um zwei Homologe der Reihe der 1-[4-Alkylbiphenyl]-2-[4-isothiocyanatophenyl]ethane,
welche in der Literatur häufig in der abkürzenden Schreibweise n-TPEB zu finden sind (das n
bezeichnet dabei die Anzahl der Kohlenstoffatome in der Alkylkette). Die Strukturformel der
n-TPEBs ist in Abbildung 6.7 dargestellt. Nach Angaben von Jadzyn [129] sollten Homologe
mit n ≥ 4 die Phasenabfolge kristallin → smektisch B → nematisch → isotrop flüssig zeigen.
Abbildung 6.7: Strukturformel der n-TPEBs
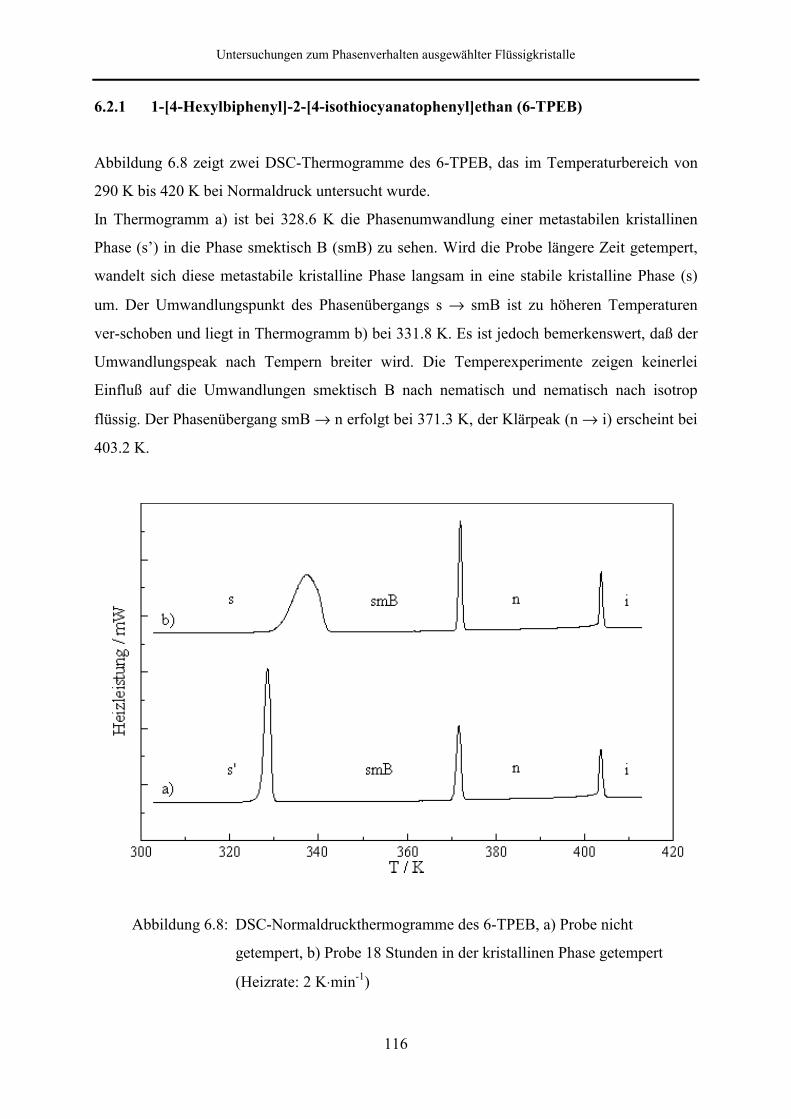
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
116
6.2.1 1-[4-Hexylbiphenyl]-2-[4-isothiocyanatophenyl]ethan (6-TPEB)
Abbildung 6.8 zeigt zwei DSC-Thermogramme des 6-TPEB, das im Temperaturbereich von
290 K bis 420 K bei Normaldruck untersucht wurde.
In Thermogramm a) ist bei 328.6 K die Phasenumwandlung einer metastabilen kristallinen
Phase (s’) in die Phase smektisch B (smB) zu sehen. Wird die Probe längere Zeit getempert,
wandelt sich diese metastabile kristalline Phase langsam in eine stabile kristalline Phase (s)
um. Der Umwandlungspunkt des Phasenübergangs s → smB ist zu höheren Temperaturen
ver-schoben und liegt in Thermogramm b) bei 331.8 K. Es ist jedoch bemerkenswert, daß der
Umwandlungspeak nach Tempern breiter wird. Die Temperexperimente zeigen keinerlei
Einfluß auf die Umwandlungen smektisch B nach nematisch und nematisch nach isotrop
flüssig. Der Phasenübergang smB → n erfolgt bei 371.3 K, der Klärpeak (n → i) erscheint bei
403.2 K.
Abbildung 6.8: DSC-Normaldruckthermogramme des 6-TPEB, a) Probe nicht
getempert, b) Probe 18 Stunden in der kristallinen Phase getempert
(Heizrate: 2 K⋅min-1)
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
117
In Tabelle 6.7 sind einige Literaturwerte der Phasenumwandlungen des 6-TPEB aufgeführt.
Die in dieser Arbeit bestimmten Enthalpien der Phasenübergänge bei Normaldruck stehen in
gutem Einklang mit den in Warschau ermittelten Umwandlungsenthalpien. Tabelle 6.8 faßt
die Ergebnisse der thermodynamischen Berechnungen zusammen.
Umwandlung Ttrs / K[131] a)
(dT/dp)koex / K⋅MPa-1
[131] a)∆trsHm / kJ⋅mol-1
[aus 65] b)
s’ → smB 329.1 0.286s → smB 333.5 0.319 21smB → n 372.6 0.442 5.8
n → i 403.8 0.557 3.2
Tabelle 6.7: Literaturwerte der Phasenumwandlungen des 6-TPEB (a) DTA, b) DSC)
Umwandlung Ttrs / K ∆trsHm / kJ⋅mol-1 ∆trsSm / J⋅K-1⋅mol-1 ∆trsVm / 10-6⋅m3⋅mol-1
s’ → smB 328.6 13.7 41.7 11.9s → smB 331.8 21.3 64.2 20.5smB → n 371.3 5.4 14.5 6.4
n → i 403.2 2.95 7.3 4.1
Tabelle 6.8: Thermodynamische Umwandlungsdaten des 6-TPEB
6.2.2 1-[4-Decylbiphenyl]-2-[4-isothiocyanatophenyl]ethan (10-TPEB)
Untersuchungen zum Phasenverhalten des 10-TPEB wurden bereits von Ernst [65] mittels
DTA bei Drücken bis etwa 300 MPa an einer Probensubstanz mit einer Reinheit von
annähernd 98% vorgenommen. Die angegebene Phasenabfolge s → sm? → smBcryst → n → i
weist eine unbekannte smektische Phase auf, deren Zuordnung mit Hilfe der
Polarisationsmikroskopie nicht möglich war. Außerdem konnte Ernst eine metastabile
kristalline Phase finden, die sich beim Tempern langsam in eine stabile kristalline Phase
umwandelte.
Im Rahmen dieser Arbeit sollten die von Ernst gefundenen Meßergebnisse mit einer
Probensubstanz höherer Reinheit (> 99.6% [130]) überprüft und ergänzt werden. DSC-
Hochdruckmessungen im Temperaturbereich von 280 K bis 440 K bei Drücken bis zu

Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
118
160 MPa ergaben dabei die Phasenabfolge s2 → s1 → smBcryst → n → i. In Warschau
durchgeführte Röntgenstrukturuntersuchungen bestätigten, daß es sich bei der gefundenen
smektischen Phase um die Phase smektisch Bcryst (smBcryst) handelt, welche in der
Nomenklatur der smektischen Phasen auch als smektisch L (smL) bezeichnet wird (smBcryst =
smL).
Abbildung 6.9 zeigt zwei DSC-Normaldruckthermogramme. In Thermogramm a) ist zunächst
ein durch die Kristallisation hervorgerufener exothermer Peak zu erkennen. Längeres
Tempern in der kristallinen Phase führt dazu, daß sich die wahrscheinlich metastabile
kristalline Phase s2 langsam in die stabile kristalline Phase s1 umwandelt. Die weiteren
Umwandlungen werden durch das Tempern nicht beeinflußt. Bei allen durchgeführten
Temperexperimenten konnte jedoch stets eine fest-fest-Umwandlung (s2 → s1) mit kleiner
Enthalpieänderung beobachtet werden. Thermogramm b) gibt als Beispiel den Kurvenverlauf
nach einer Temperzeit von mehr als 48 Stunden wieder. Abbildung 6.10 veranschaulicht,
daß sich das beschriebene Phasen-verhalten auch bei höheren Drücken beobachten läßt.
In Tabelle 6.9 sind die bei Normaldruck mit DTA und DSC ermittelten Umwandlungs-
temperaturen aufgeführt. Die Phasenübergangstemperaturen der verunreinigten Proben-
substanz liegen deutlich niedriger als die des reinen 10-TPEB.
Umwandlung Ttrs / K(DTA, [65])
Umwandlung Ttrs / K(DSC, diese Arbeit)
s’ → sm? 296.4 s2 → s1 298.4s → sm? 313.3 s1 → smBcryst 311.7
sm? → smBcryst 355.4 smBcryst → n 376.4smBcryst → n 367.3 n → i 398.2
n → i 393.4
Tabelle 6.9: Umwandlungstemperaturen des 10-TPEB bei Normaldruck
Abbildung 6.11 zeigt das Phasendiagramm des 10-TPEB. Die (metastabile) Phasengrenzlinie
der Umwandlung s2 → s1 ist gestrichelt dargestellt.
Die Ergebnisse der DSC-Hochdruckmessungen sind in Tabelle 6.10 zusammengestellt.
Die Koeffizienten der Polynome zweiter Ordnung, die an die Umwandlungstemperaturen in
Abhängigkeit vom Druck angepaßt wurden, können Tabelle 6.11 entnommen werden.
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
119
Abbildung 6.9: DSC-Thermogramme von 10-TPEB bei p = 0.1 MPa. In a) ist der durch
die Kristallisation hervorgerufene exotherme Peak zu sehen; b) wurde nach
längerem Tempern in der kristallinen Phase (> 48 h) erhalten.
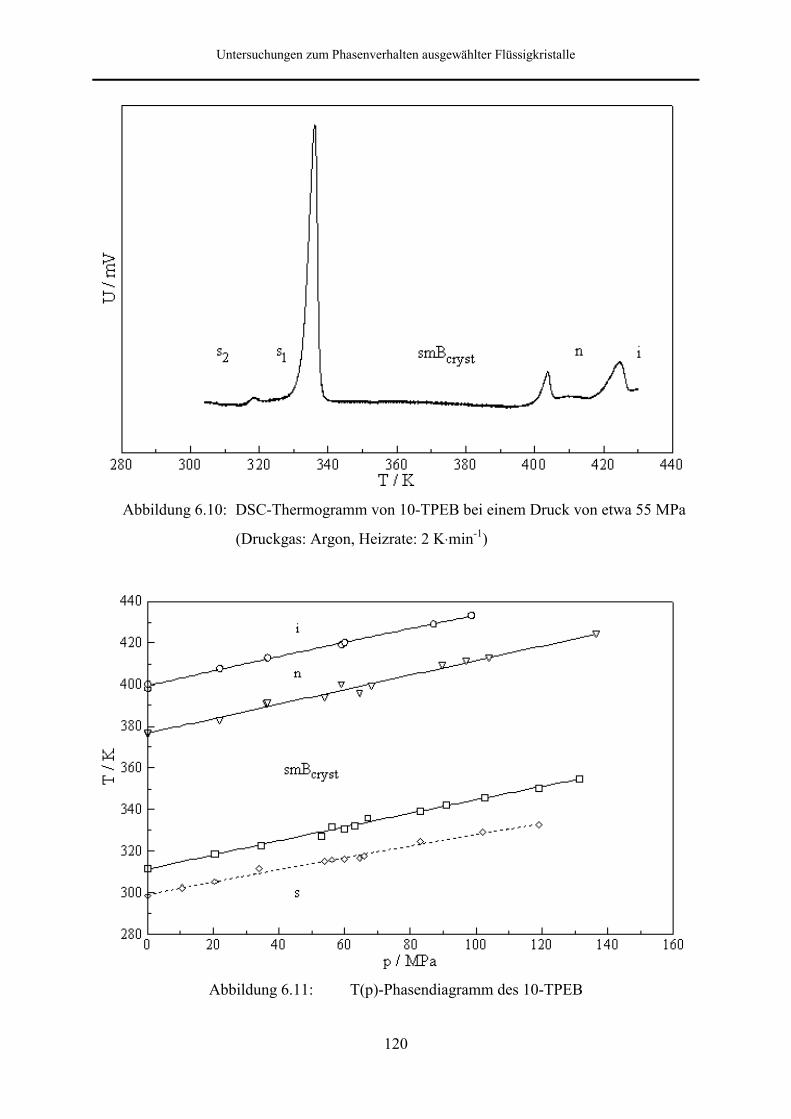
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
120
Abbildung 6.10: DSC-Thermogramm von 10-TPEB bei einem Druck von etwa 55 MPa
(Druckgas: Argon, Heizrate: 2 K⋅min-1)
Abbildung 6.11: T(p)-Phasendiagramm des 10-TPEB
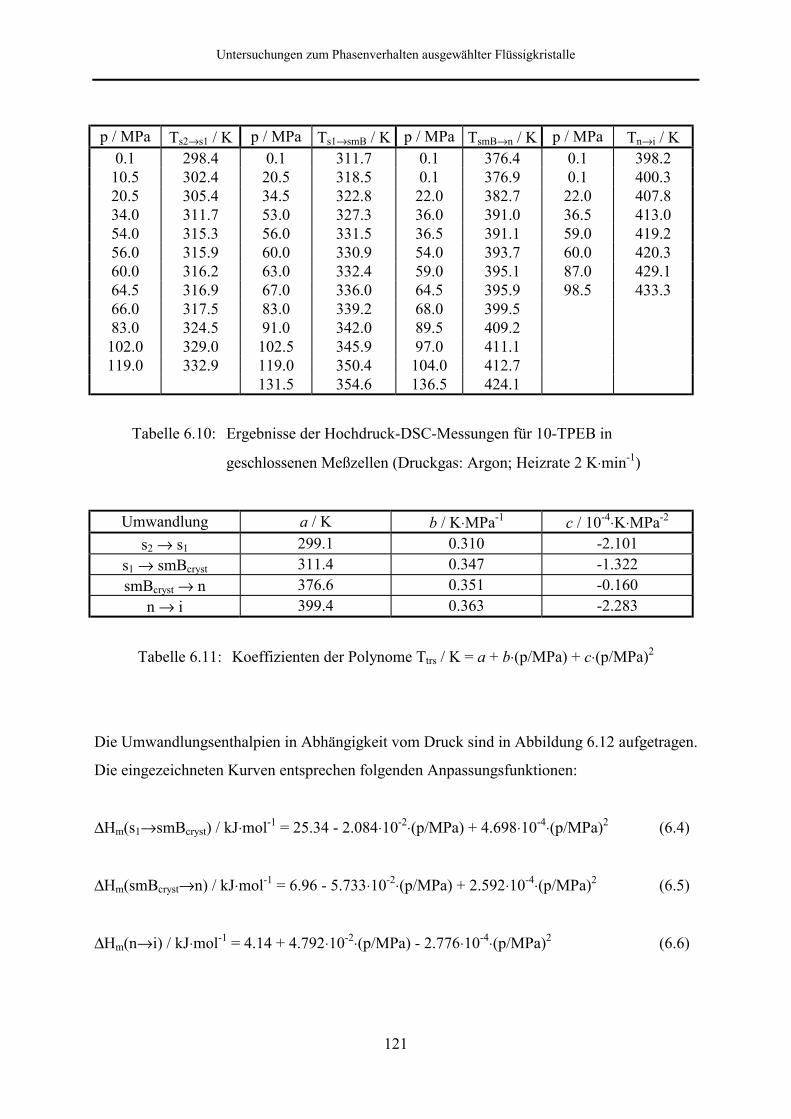
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
121
p / MPa Ts2→s1 / K p / MPa Ts1→smB / K p / MPa TsmB→n / K p / MPa Tn→i / K0.1 298.4 0.1 311.7 0.1 376.4 0.1 398.210.5 302.4 20.5 318.5 0.1 376.9 0.1 400.320.5 305.4 34.5 322.8 22.0 382.7 22.0 407.834.0 311.7 53.0 327.3 36.0 391.0 36.5 413.054.0 315.3 56.0 331.5 36.5 391.1 59.0 419.256.0 315.9 60.0 330.9 54.0 393.7 60.0 420.360.0 316.2 63.0 332.4 59.0 395.1 87.0 429.164.5 316.9 67.0 336.0 64.5 395.9 98.5 433.366.0 317.5 83.0 339.2 68.0 399.583.0 324.5 91.0 342.0 89.5 409.2102.0 329.0 102.5 345.9 97.0 411.1119.0 332.9 119.0 350.4 104.0 412.7
131.5 354.6 136.5 424.1
Tabelle 6.10: Ergebnisse der Hochdruck-DSC-Messungen für 10-TPEB in
geschlossenen Meßzellen (Druckgas: Argon; Heizrate 2 K⋅min-1)
Umwandlung a / K b / K⋅MPa-1 c / 10-4⋅K⋅MPa-2
s2 → s1 299.1 0.310 -2.101s1 → smBcryst 311.4 0.347 -1.322smBcryst → n 376.6 0.351 -0.160
n → i 399.4 0.363 -2.283
Tabelle 6.11: Koeffizienten der Polynome Ttrs / K = a + b⋅(p/MPa) + c⋅(p/MPa)2
Die Umwandlungsenthalpien in Abhängigkeit vom Druck sind in Abbildung 6.12 aufgetragen.
Die eingezeichneten Kurven entsprechen folgenden Anpassungsfunktionen:
∆Hm(s1→smBcryst) / kJ⋅mol-1 = 25.34 - 2.084⋅10-2⋅(p/MPa) + 4.698⋅10-4⋅(p/MPa)2 (6.4)
∆Hm(smBcryst→n) / kJ⋅mol-1 = 6.96 - 5.733⋅10-2⋅(p/MPa) + 2.592⋅10-4⋅(p/MPa)2 (6.5)
∆Hm(n→i) / kJ⋅mol-1 = 4.14 + 4.792⋅10-2⋅(p/MPa) - 2.776⋅10-4⋅(p/MPa)2 (6.6)
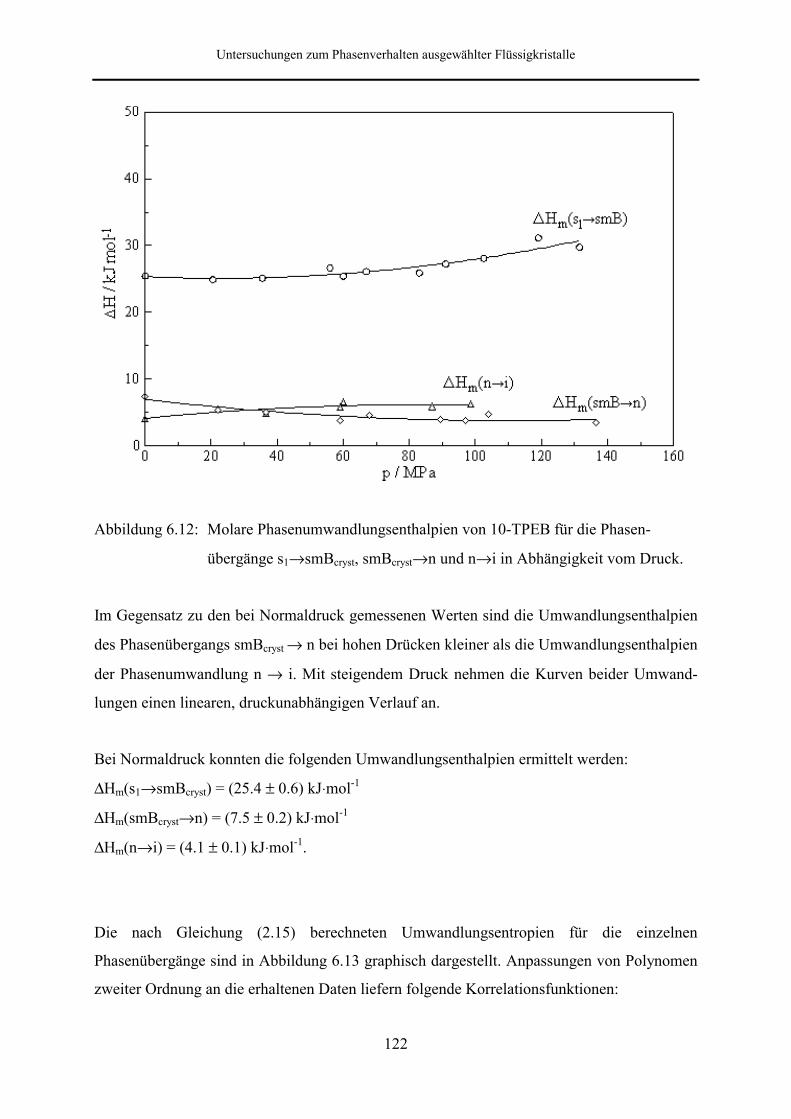
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
122
Abbildung 6.12: Molare Phasenumwandlungsenthalpien von 10-TPEB für die Phasen-
übergänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck.
Im Gegensatz zu den bei Normaldruck gemessenen Werten sind die Umwandlungsenthalpien
des Phasenübergangs smBcryst → n bei hohen Drücken kleiner als die Umwandlungsenthalpien
der Phasenumwandlung n → i. Mit steigendem Druck nehmen die Kurven beider Umwand-
lungen einen linearen, druckunabhängigen Verlauf an.
Bei Normaldruck konnten die folgenden Umwandlungsenthalpien ermittelt werden:
∆Hm(s1→smBcryst) = (25.4 ± 0.6) kJ⋅mol-1
∆Hm(smBcryst→n) = (7.5 ± 0.2) kJ⋅mol-1
∆Hm(n→i) = (4.1 ± 0.1) kJ⋅mol-1.
Die nach Gleichung (2.15) berechneten Umwandlungsentropien für die einzelnen
Phasenübergänge sind in Abbildung 6.13 graphisch dargestellt. Anpassungen von Polynomen
zweiter Ordnung an die erhaltenen Daten liefern folgende Korrelationsfunktionen:
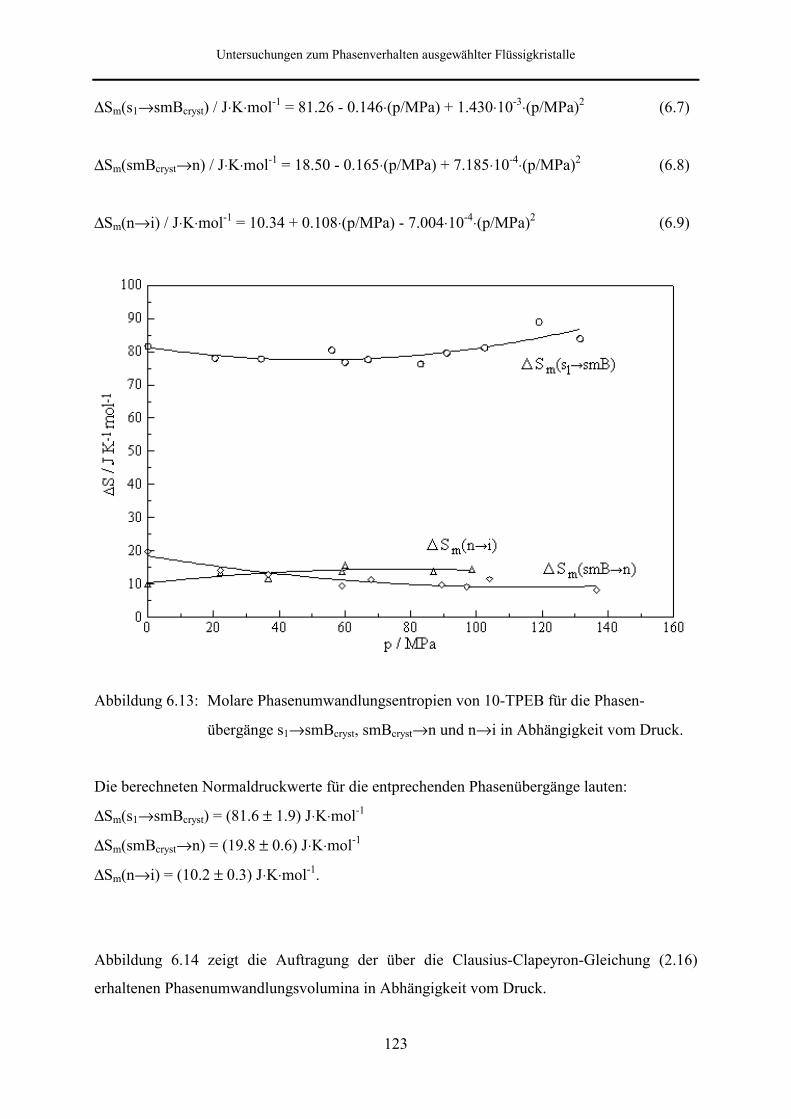
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
123
∆Sm(s1→smBcryst) / J⋅K⋅mol-1 = 81.26 - 0.146⋅(p/MPa) + 1.430⋅10-3⋅(p/MPa)2 (6.7)
∆Sm(smBcryst→n) / J⋅K⋅mol-1 = 18.50 - 0.165⋅(p/MPa) + 7.185⋅10-4⋅(p/MPa)2 (6.8)
∆Sm(n→i) / J⋅K⋅mol-1 = 10.34 + 0.108⋅(p/MPa) - 7.004⋅10-4⋅(p/MPa)2 (6.9)
Abbildung 6.13: Molare Phasenumwandlungsentropien von 10-TPEB für die Phasen-
übergänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck.
Die berechneten Normaldruckwerte für die entprechenden Phasenübergänge lauten:
∆Sm(s1→smBcryst) = (81.6 ± 1.9) J⋅K⋅mol-1
∆Sm(smBcryst→n) = (19.8 ± 0.6) J⋅K⋅mol-1
∆Sm(n→i) = (10.2 ± 0.3) J⋅K⋅mol-1.
Abbildung 6.14 zeigt die Auftragung der über die Clausius-Clapeyron-Gleichung (2.16)
erhaltenen Phasenumwandlungsvolumina in Abhängigkeit vom Druck.
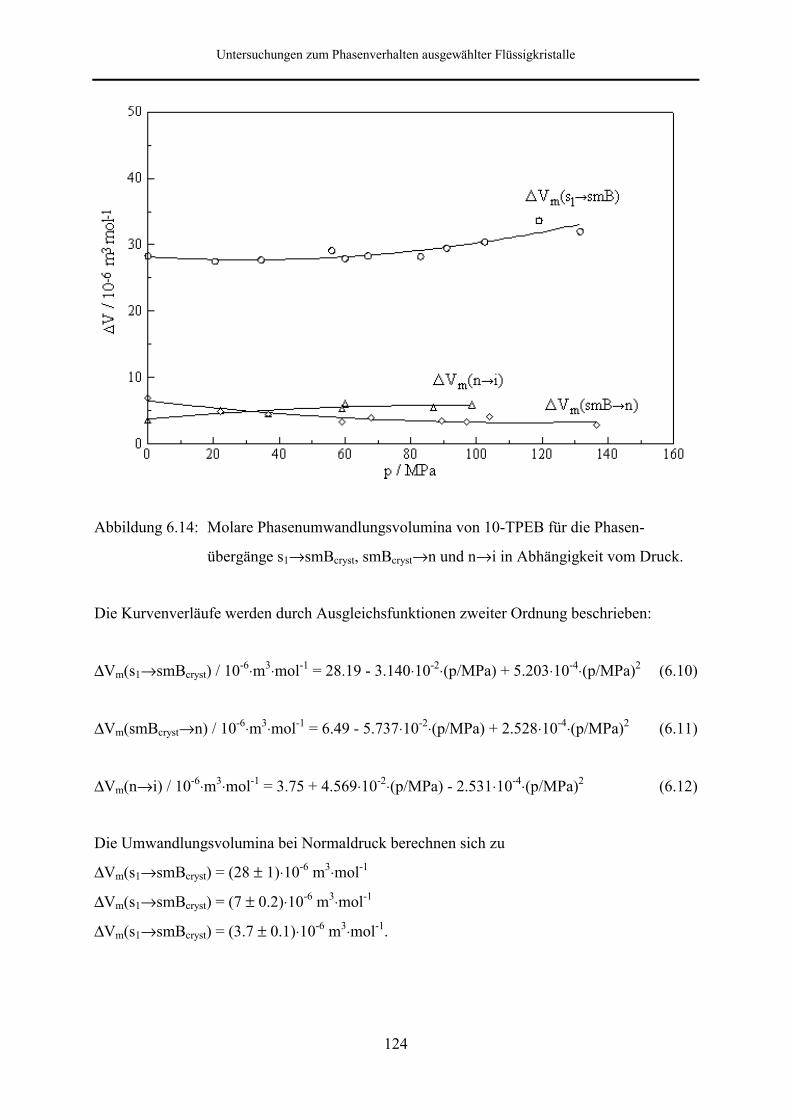
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
124
Abbildung 6.14: Molare Phasenumwandlungsvolumina von 10-TPEB für die Phasen-
übergänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck.
Die Kurvenverläufe werden durch Ausgleichsfunktionen zweiter Ordnung beschrieben:
∆Vm(s1→smBcryst) / 10-6⋅m3⋅mol-1 = 28.19 - 3.140⋅10-2⋅(p/MPa) + 5.203⋅10-4⋅(p/MPa)2 (6.10)
∆Vm(smBcryst→n) / 10-6⋅m3⋅mol-1 = 6.49 - 5.737⋅10-2⋅(p/MPa) + 2.528⋅10-4⋅(p/MPa)2 (6.11)
∆Vm(n→i) / 10-6⋅m3⋅mol-1 = 3.75 + 4.569⋅10-2⋅(p/MPa) - 2.531⋅10-4⋅(p/MPa)2 (6.12)
Die Umwandlungsvolumina bei Normaldruck berechnen sich zu
∆Vm(s1→smBcryst) = (28 ± 1)⋅10-6 m3⋅mol-1
∆Vm(s1→smBcryst) = (7 ± 0.2)⋅10-6 m3⋅mol-1
∆Vm(s1→smBcryst) = (3.7 ± 0.1)⋅10-6 m3⋅mol-1.
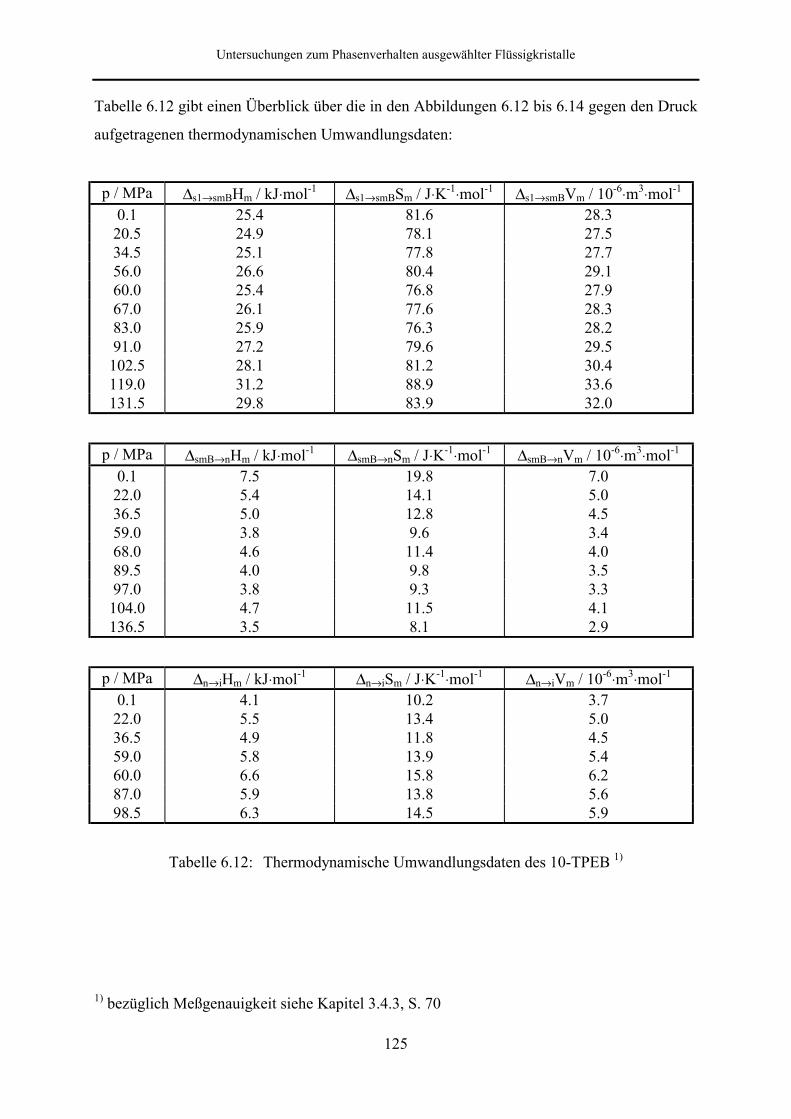
Untersuchungen zum Phasenverhalten ausgewählter Flüssigkristalle
125
Tabelle 6.12 gibt einen Überblick über die in den Abbildungen 6.12 bis 6.14 gegen den Druck
aufgetragenen thermodynamischen Umwandlungsdaten:
p / MPa ∆s1→smBHm / kJ⋅mol-1 ∆s1→smBSm / J⋅K-1⋅mol-1 ∆s1→smBVm / 10-6⋅m3⋅mol-1
0.1 25.4 81.6 28.320.5 24.9 78.1 27.534.5 25.1 77.8 27.756.0 26.6 80.4 29.160.0 25.4 76.8 27.967.0 26.1 77.6 28.383.0 25.9 76.3 28.291.0 27.2 79.6 29.5102.5 28.1 81.2 30.4119.0 31.2 88.9 33.6131.5 29.8 83.9 32.0
p / MPa ∆smB→nHm / kJ⋅mol-1 ∆smB→nSm / J⋅K-1⋅mol-1 ∆smB→nVm / 10-6⋅m3⋅mol-1
0.1 7.5 19.8 7.022.0 5.4 14.1 5.036.5 5.0 12.8 4.559.0 3.8 9.6 3.468.0 4.6 11.4 4.089.5 4.0 9.8 3.597.0 3.8 9.3 3.3104.0 4.7 11.5 4.1136.5 3.5 8.1 2.9
p / MPa ∆n→iHm / kJ⋅mol-1 ∆n→iSm / J⋅K-1⋅mol-1 ∆n→iVm / 10-6⋅m3⋅mol-1
0.1 4.1 10.2 3.722.0 5.5 13.4 5.036.5 4.9 11.8 4.559.0 5.8 13.9 5.460.0 6.6 15.8 6.287.0 5.9 13.8 5.698.5 6.3 14.5 5.9
Tabelle 6.12: Thermodynamische Umwandlungsdaten des 10-TPEB 1)
1) bezüglich Meßgenauigkeit siehe Kapitel 3.4.3, S. 70

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
126
7 Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
7.1 Überblick über die Meßsubstanzen
In der vorliegenden Arbeit wurden Untersuchungen zum Phasenverhalten der folgenden
Anthrachinonfarbstoffe vorgenommen:
1) 1,4-Bis-(methylamino)-9,10-anthrachinon ⇒ AC01
2) 1,4-Bis-(ethylamino)-9,10-anthrachinon ⇒ AC02
3) 1,4-Bis-(n-propylamino)-9,10-anthrachinon ⇒ AC03
4) 1,4-Bis-(iso-propylamino)-9,10-anthrachinon ⇒ iso-AC03
5) 1,4-Bis-(n-butylamino)-9,10-anthrachinon ⇒ AC04
6) 1,4-Bis-(n-pentylamino)-9,10-anthrachinon ⇒ AC05
7) 1,4-Bis-(n-octylamino)-9,10-anthrachinon ⇒ AC08
8) 1,4-Bis-(n-octadecylamino)-9,10-anthrachinon ⇒ AC18
9) C. I. Disperse Red 60 ⇒ DRed60
10) C. I. Disperse Blue 60 ⇒ DBlue60
Bei den Substanzen 1) bis 8) handelt es sich um Homologe der Reihe der 1,4-Bis-
(alkylamino)-9,10-anthrachinone. Diese allesamt blauen Dispersionsfarbstoffe weisen die in
Abbildung 7.1 dargestellte allgemeine Strukturformel auf. Die Stickstoffatome in 1- und 4-
Position sind dabei jeweils mit demselben Alkylrest R substituiert.
Abbildung 7.1: Allgemeine Strukturformel der untersuchten Aminoanthrachinonfarbstoffe
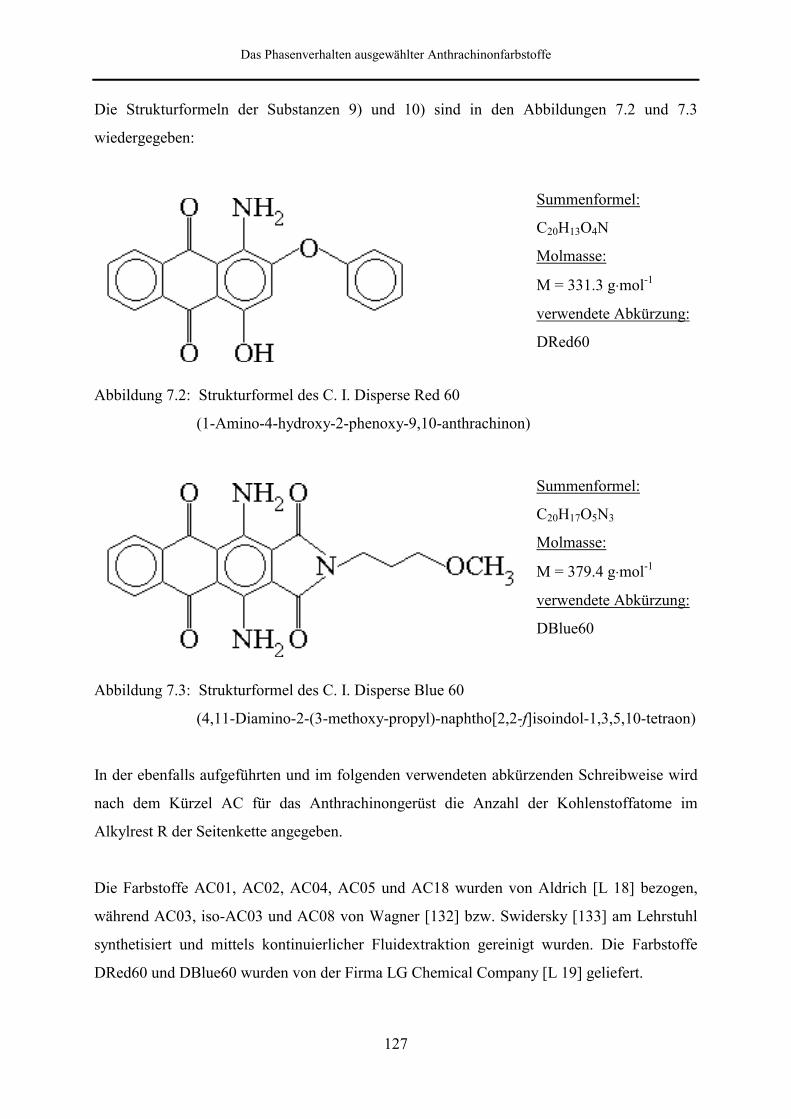
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
127
Die Strukturformeln der Substanzen 9) und 10) sind in den Abbildungen 7.2 und 7.3
wiedergegeben:
Abbildung 7.2: Strukturformel des C. I. Disperse Red 60
(1-Amino-4-hydroxy-2-phenoxy-9,10-anthrachinon)
Abbildung 7.3: Strukturformel des C. I. Disperse Blue 60
(4,11-Diamino-2-(3-methoxy-propyl)-naphtho[2,2-f]isoindol-1,3,5,10-tetraon)
In der ebenfalls aufgeführten und im folgenden verwendeten abkürzenden Schreibweise wird
nach dem Kürzel AC für das Anthrachinongerüst die Anzahl der Kohlenstoffatome im
Alkylrest R der Seitenkette angegeben.
Die Farbstoffe AC01, AC02, AC04, AC05 und AC18 wurden von Aldrich [L 18] bezogen,
während AC03, iso-AC03 und AC08 von Wagner [132] bzw. Swidersky [133] am Lehrstuhl
synthetisiert und mittels kontinuierlicher Fluidextraktion gereinigt wurden. Die Farbstoffe
DRed60 und DBlue60 wurden von der Firma LG Chemical Company [L 19] geliefert.
Summenformel:
C20H13O4N
Molmasse:
M = 331.3 g⋅mol-1
verwendete Abkürzung:
DRed60
Summenformel:
C20H17O5N3
Molmasse:
M = 379.4 g⋅mol-1
verwendete Abkürzung:
DBlue60

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
128
Die Reinheit der Substanzen ist in Tabelle 7.1 zusammengefaßt.
Substanz Herkunft ReinheitAC01 Aldrich [L 18] ~ 97 %AC02 Aldrich [L 18] ~ 98 %AC03 eigene Synthese (Wagner [132]) ~ 99 % (Kapillar-SFC)
iso-AC03 eigene Synthese (Wagner [132]) ~ 96 % (Kapillar-SFC)AC04 Aldrich [L 18] ~ 98 %AC05 Aldrich [L 18] ~ 97 %AC08 eigene Synthese (Swidersky [133]) > 99% (Kapillar-SFC) [142]AC18 Aldrich [L 18] ~ 98 %
DRed60 LG Chemical Company [L 19] > 99% (Kapillar-SFC) [141]DBlue60 LG Chemical Company [L 19] ~ 97 % (Kapillar-SFC)
Tabelle 7.1: Reinheit der verwendeten Farbstoffe
Die kommerziell erworbenen Substanzen wurden vor der Durchführung der DSC-Messungen
in heißem Methanol umkristallisiert.
7.2 DSC-Messungen bei Normaldruck
Die Schmelzpunkte und die Umwandlungsenthalpien der im vorherigen Abschnitt
aufgeführten Anthrachinonfarbstoffe wurden bei Normaldruck mittels DSC (Perkin-Elmer
DSC7) bestimmt. Alle Messungen wurden mit einer Heizrate von 2 K⋅min-1 durchgeführt.
Die Meßergebnisse sind in Tabelle 7.2 zusammengefaßt. Außerdem sind die berechneten
Umwandlungsentropien angegeben. Die Umwandlungstemperaturen sind auf ± 0.3 K genau,
bei den Werten für die Umwandlungsenthalpien und -entropien ist ein mittlerer relativer
Fehler von ± 3% zu berücksichtigen.
Die jeweils erste Messung wurde an den gereinigten Farbstoffen durchgeführt. Alle weiteren
Messungen wurden dann nach Abkühlen der Meßzelle mit der zuvor aufgeschmolzenen
Substanz vorgenommen.
Bei den untersuchten Farbstoffen AC01 bis AC08 ist mit wachsender Länge der Alkylseiten-
kette eine Abnahme der Schmelztemperatur festzustellen, wobei die Schmelztemperatur von
iso-AC03 deutlich oberhalb der von AC03 liegt. Die an AC18 durchgeführten Messungen be-
stätigen den von Swidersky [133] bei den längerkettigen Homologen AC08, AC12, AC16 und
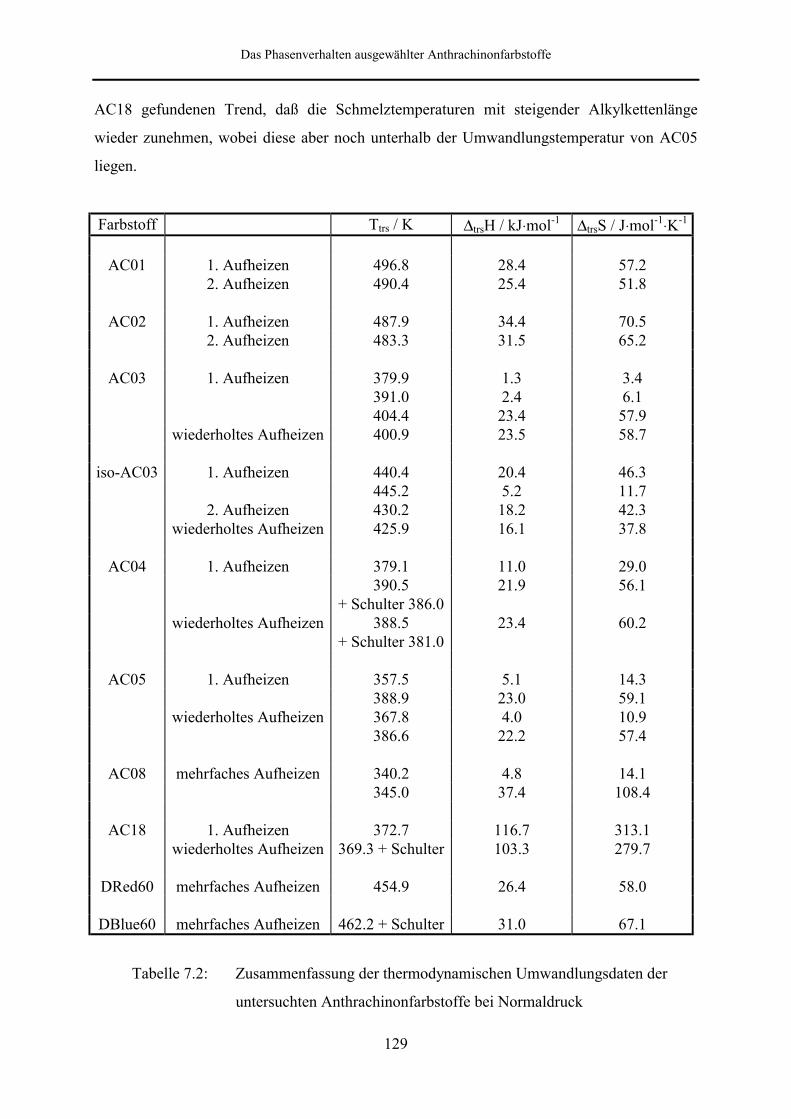
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
129
AC18 gefundenen Trend, daß die Schmelztemperaturen mit steigender Alkylkettenlänge
wieder zunehmen, wobei diese aber noch unterhalb der Umwandlungstemperatur von AC05
liegen.
Farbstoff Ttrs / K ∆trsH / kJ⋅mol-1 ∆trsS / J⋅mol-1⋅K-1
AC01 1. Aufheizen 496.8 28.4 57.22. Aufheizen 490.4 25.4 51.8
AC02 1. Aufheizen 487.9 34.4 70.52. Aufheizen 483.3 31.5 65.2
AC03 1. Aufheizen 379.9 1.3 3.4391.0 2.4 6.1404.4 23.4 57.9
wiederholtes Aufheizen 400.9 23.5 58.7
iso-AC03 1. Aufheizen 440.4 20.4 46.3445.2 5.2 11.7
2. Aufheizen 430.2 18.2 42.3wiederholtes Aufheizen 425.9 16.1 37.8
AC04 1. Aufheizen 379.1 11.0 29.0390.5
+ Schulter 386.021.9 56.1
wiederholtes Aufheizen 388.5+ Schulter 381.0
23.4 60.2
AC05 1. Aufheizen 357.5 5.1 14.3388.9 23.0 59.1
wiederholtes Aufheizen 367.8 4.0 10.9386.6 22.2 57.4
AC08 mehrfaches Aufheizen 340.2 4.8 14.1345.0 37.4 108.4
AC18 1. Aufheizen 372.7 116.7 313.1wiederholtes Aufheizen 369.3 + Schulter 103.3 279.7
DRed60 mehrfaches Aufheizen 454.9 26.4 58.0
DBlue60 mehrfaches Aufheizen 462.2 + Schulter 31.0 67.1
Tabelle 7.2: Zusammenfassung der thermodynamischen Umwandlungsdaten der
untersuchten Anthrachinonfarbstoffe bei Normaldruck
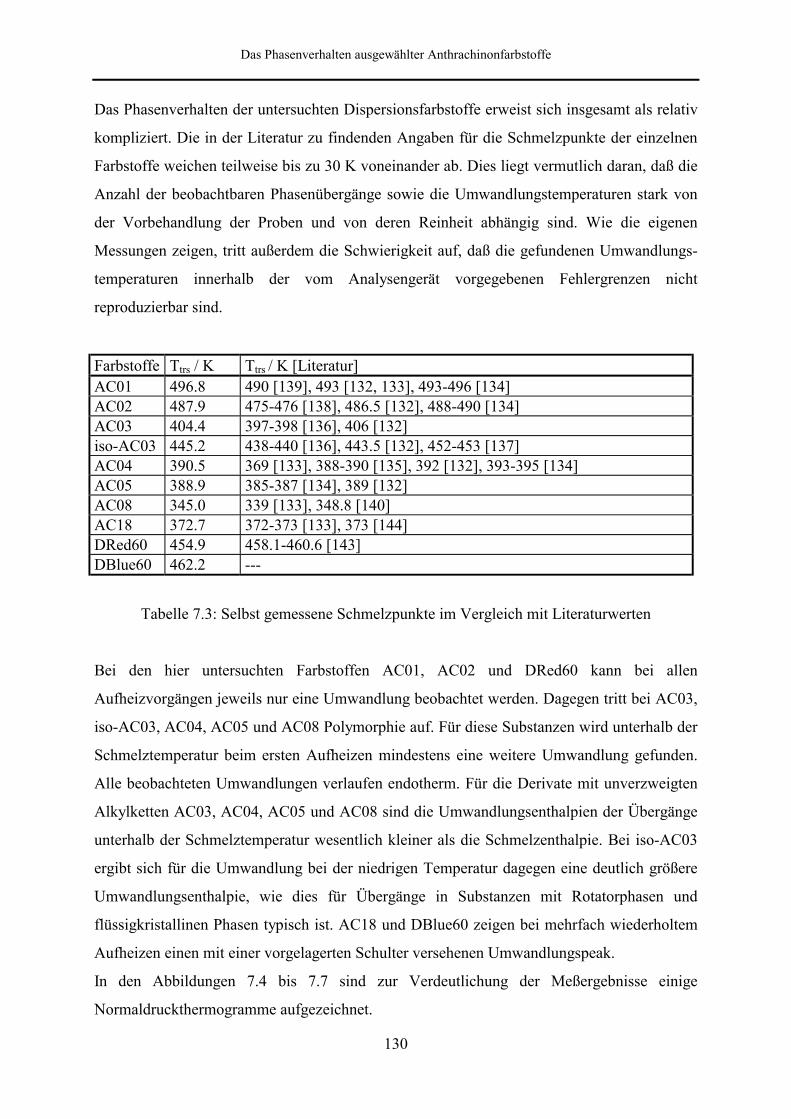
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
130
Das Phasenverhalten der untersuchten Dispersionsfarbstoffe erweist sich insgesamt als relativ
kompliziert. Die in der Literatur zu findenden Angaben für die Schmelzpunkte der einzelnen
Farbstoffe weichen teilweise bis zu 30 K voneinander ab. Dies liegt vermutlich daran, daß die
Anzahl der beobachtbaren Phasenübergänge sowie die Umwandlungstemperaturen stark von
der Vorbehandlung der Proben und von deren Reinheit abhängig sind. Wie die eigenen
Messungen zeigen, tritt außerdem die Schwierigkeit auf, daß die gefundenen Umwandlungs-
temperaturen innerhalb der vom Analysengerät vorgegebenen Fehlergrenzen nicht
reproduzierbar sind.
Farbstoffe Ttrs / K Ttrs / K [Literatur]AC01 496.8 490 [139], 493 [132, 133], 493-496 [134]AC02 487.9 475-476 [138], 486.5 [132], 488-490 [134]AC03 404.4 397-398 [136], 406 [132]iso-AC03 445.2 438-440 [136], 443.5 [132], 452-453 [137]AC04 390.5 369 [133], 388-390 [135], 392 [132], 393-395 [134]AC05 388.9 385-387 [134], 389 [132]AC08 345.0 339 [133], 348.8 [140]AC18 372.7 372-373 [133], 373 [144]DRed60 454.9 458.1-460.6 [143]DBlue60 462.2 ---
Tabelle 7.3: Selbst gemessene Schmelzpunkte im Vergleich mit Literaturwerten
Bei den hier untersuchten Farbstoffen AC01, AC02 und DRed60 kann bei allen
Aufheizvorgängen jeweils nur eine Umwandlung beobachtet werden. Dagegen tritt bei AC03,
iso-AC03, AC04, AC05 und AC08 Polymorphie auf. Für diese Substanzen wird unterhalb der
Schmelztemperatur beim ersten Aufheizen mindestens eine weitere Umwandlung gefunden.
Alle beobachteten Umwandlungen verlaufen endotherm. Für die Derivate mit unverzweigten
Alkylketten AC03, AC04, AC05 und AC08 sind die Umwandlungsenthalpien der Übergänge
unterhalb der Schmelztemperatur wesentlich kleiner als die Schmelzenthalpie. Bei iso-AC03
ergibt sich für die Umwandlung bei der niedrigen Temperatur dagegen eine deutlich größere
Umwandlungsenthalpie, wie dies für Übergänge in Substanzen mit Rotatorphasen und
flüssigkristallinen Phasen typisch ist. AC18 und DBlue60 zeigen bei mehrfach wiederholtem
Aufheizen einen mit einer vorgelagerten Schulter versehenen Umwandlungspeak.
In den Abbildungen 7.4 bis 7.7 sind zur Verdeutlichung der Meßergebnisse einige
Normaldruckthermogramme aufgezeichnet.
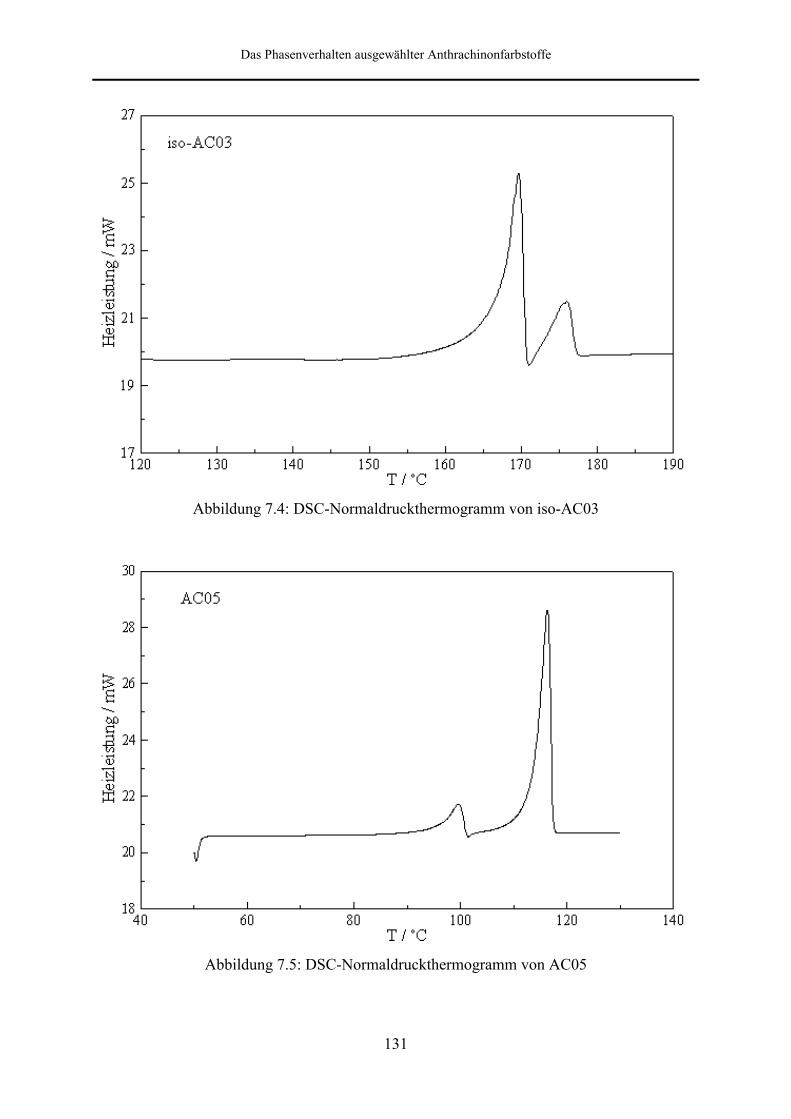
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
131
Abbildung 7.4: DSC-Normaldruckthermogramm von iso-AC03
Abbildung 7.5: DSC-Normaldruckthermogramm von AC05
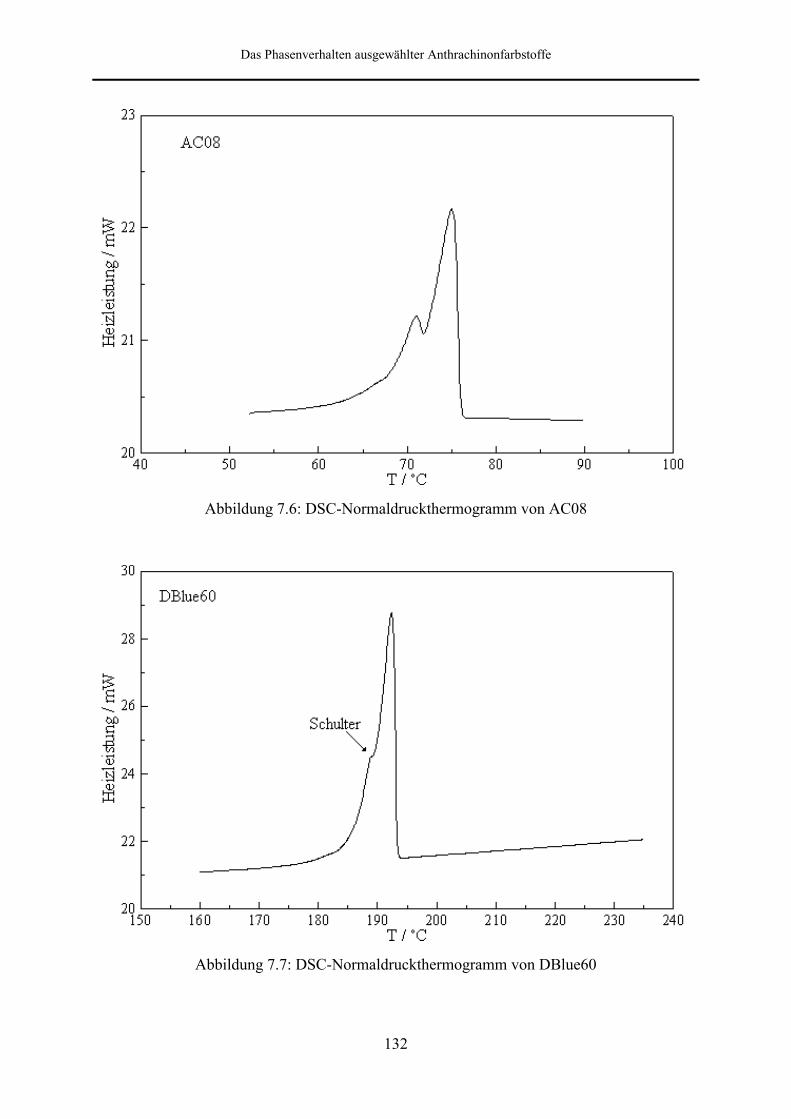
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
132
Abbildung 7.6: DSC-Normaldruckthermogramm von AC08
Abbildung 7.7: DSC-Normaldruckthermogramm von DBlue60

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
133
7.3 DSC-und DTA-Hochdruckmessungen
Die von Wagner [132] am Lehrstuhl für Physikalische Chemie II der Ruhr-Universität
Bochum vorgenommenen Untersuchungen zur Löslichkeit von Anthrachinonfarbstoffen in
nah- und überkritischem Kohlendioxid waren Ausgangspunkt für die hier durchgeführten
DSC- und DTA-Hochdruckmessungen an AC03 und AC04 (Kapitel 7.3.1 und 7.3.2).
Wagner fand heraus, daß sich die Löslichkeit beim Übergang von AC02 zu den Homologen
mit einer zusätzlichen CH2-Gruppe beträchtlich erhöht, und er stellte fest, daß offensichtlich
ein starker Einfluß der Kristallstruktur der untersuchten Dispersionsfarbstoffe auf die
Löslichkeit vorliegt. Im Einklang damit stehen die Ergebnisse der DSC-Normaldruck-
messungen, nach denen bei den gut löslichen Derivaten AC03, iso-AC03, AC04 und AC05
polymorphes Verhalten zu beobachten ist, bei den schlecht löslichen Farbstoffen AC01 und
AC02 dagegen nicht.
Da die von Wagner durchgeführten Löslichkeitsuntersuchungen im CO2-Strom bei Drücken
bis 20 MPa erfolgten, erschien es sinnvoll, das Phasenverhalten einiger Farbstoffe unter Druck
zu verfolgen. Neben den herkömmlichen Druckgasen Helium und Argon sollte dabei auch
CO2 als Druckgas verwendet werden. Als Meßsubstanzen für die Hochdruckuntersuchungen
wurden AC03 und AC04 ausgesucht, da die zu erwartenden Umwandlungstemperaturen in
den für die Meßapparaturen ausgelegten Temperaturbereichen liegen.
7.3.1 Phasenverhalten von 1,4-Bis-(n-propylamino)-9,10-anthrachinon (AC03)
Das Phasenverhalten von AC03 wurde im Temperaturbereich von 320 K bis 430 K bei
Drücken bis 140 MPa (Argon) bzw. 10 MPa (CO2) mittels DSC untersucht. Die eingestellte
Heizrate betrug bei allen Messungen 2 K⋅min-1. Die Argon-Messungen sollten in
geschlossenen, die CO2-Messungen in offenen Meßzellen durchgeführt werden. Die
Handhabung der Meßsubstanz und die Durchführung der Messungen gestaltete sich jedoch als
ausgesprochen schwierig. So gelang es auch nach mehreren Versuchen nicht, wie in Kapitel
3.2.5 beschrieben, druckdicht verschlossene Meßzellen zu präparieren, da entweder die
Klebestelle den Anforderungen nicht genügte oder die verwendete Aluminiummembran dem
Druck nicht standhielt. Darüberhinaus trat bei den CO2-Messungen die Schwierigkeit auf, daß
die Substanz dermaßen gut in Kohlendioxid löslich war, daß das Druckgas die Meßzelle
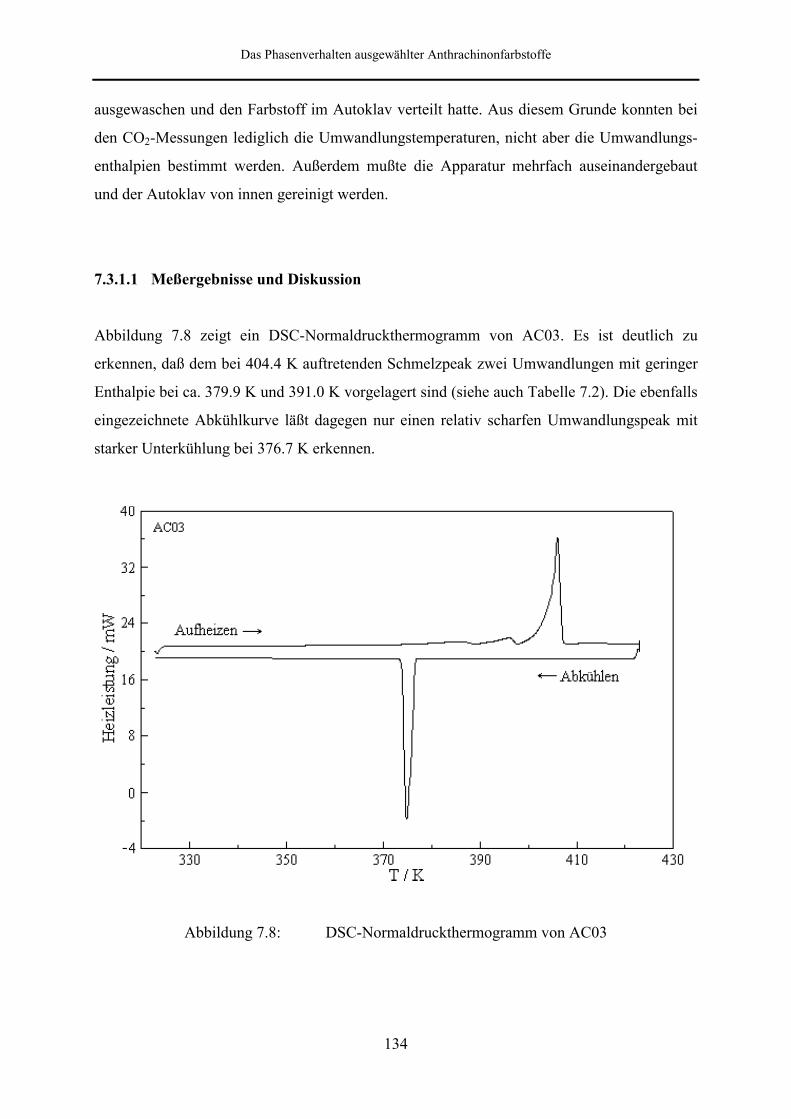
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
134
ausgewaschen und den Farbstoff im Autoklav verteilt hatte. Aus diesem Grunde konnten bei
den CO2-Messungen lediglich die Umwandlungstemperaturen, nicht aber die Umwandlungs-
enthalpien bestimmt werden. Außerdem mußte die Apparatur mehrfach auseinandergebaut
und der Autoklav von innen gereinigt werden.
7.3.1.1 Meßergebnisse und Diskussion
Abbildung 7.8 zeigt ein DSC-Normaldruckthermogramm von AC03. Es ist deutlich zu
erkennen, daß dem bei 404.4 K auftretenden Schmelzpeak zwei Umwandlungen mit geringer
Enthalpie bei ca. 379.9 K und 391.0 K vorgelagert sind (siehe auch Tabelle 7.2). Die ebenfalls
eingezeichnete Abkühlkurve läßt dagegen nur einen relativ scharfen Umwandlungspeak mit
starker Unterkühlung bei 376.7 K erkennen.
Abbildung 7.8: DSC-Normaldruckthermogramm von AC03
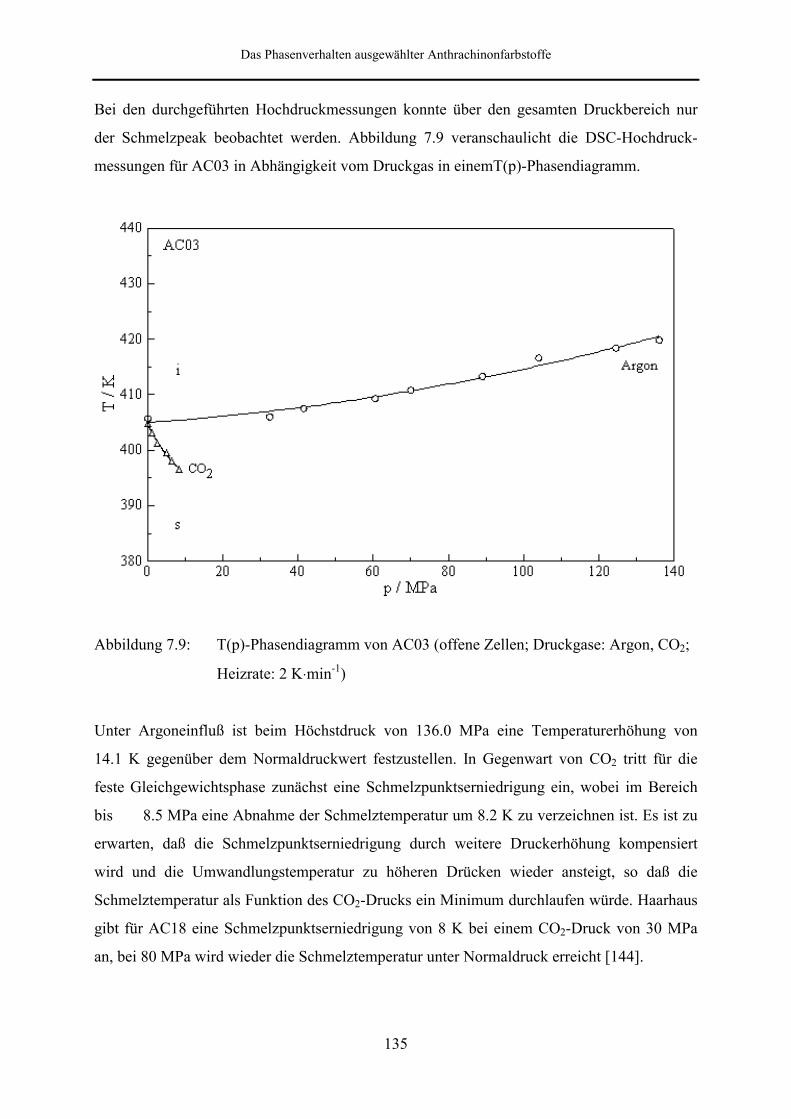
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
135
Bei den durchgeführten Hochdruckmessungen konnte über den gesamten Druckbereich nur
der Schmelzpeak beobachtet werden. Abbildung 7.9 veranschaulicht die DSC-Hochdruck-
messungen für AC03 in Abhängigkeit vom Druckgas in einemT(p)-Phasendiagramm.
Abbildung 7.9: T(p)-Phasendiagramm von AC03 (offene Zellen; Druckgase: Argon, CO2;
Heizrate: 2 K⋅min-1)
Unter Argoneinfluß ist beim Höchstdruck von 136.0 MPa eine Temperaturerhöhung von
14.1 K gegenüber dem Normaldruckwert festzustellen. In Gegenwart von CO2 tritt für die
feste Gleichgewichtsphase zunächst eine Schmelzpunktserniedrigung ein, wobei im Bereich
bis 8.5 MPa eine Abnahme der Schmelztemperatur um 8.2 K zu verzeichnen ist. Es ist zu
erwarten, daß die Schmelzpunktserniedrigung durch weitere Druckerhöhung kompensiert
wird und die Umwandlungstemperatur zu höheren Drücken wieder ansteigt, so daß die
Schmelztemperatur als Funktion des CO2-Drucks ein Minimum durchlaufen würde. Haarhaus
gibt für AC18 eine Schmelzpunktserniedrigung von 8 K bei einem CO2-Druck von 30 MPa
an, bei 80 MPa wird wieder die Schmelztemperatur unter Normaldruck erreicht [144].
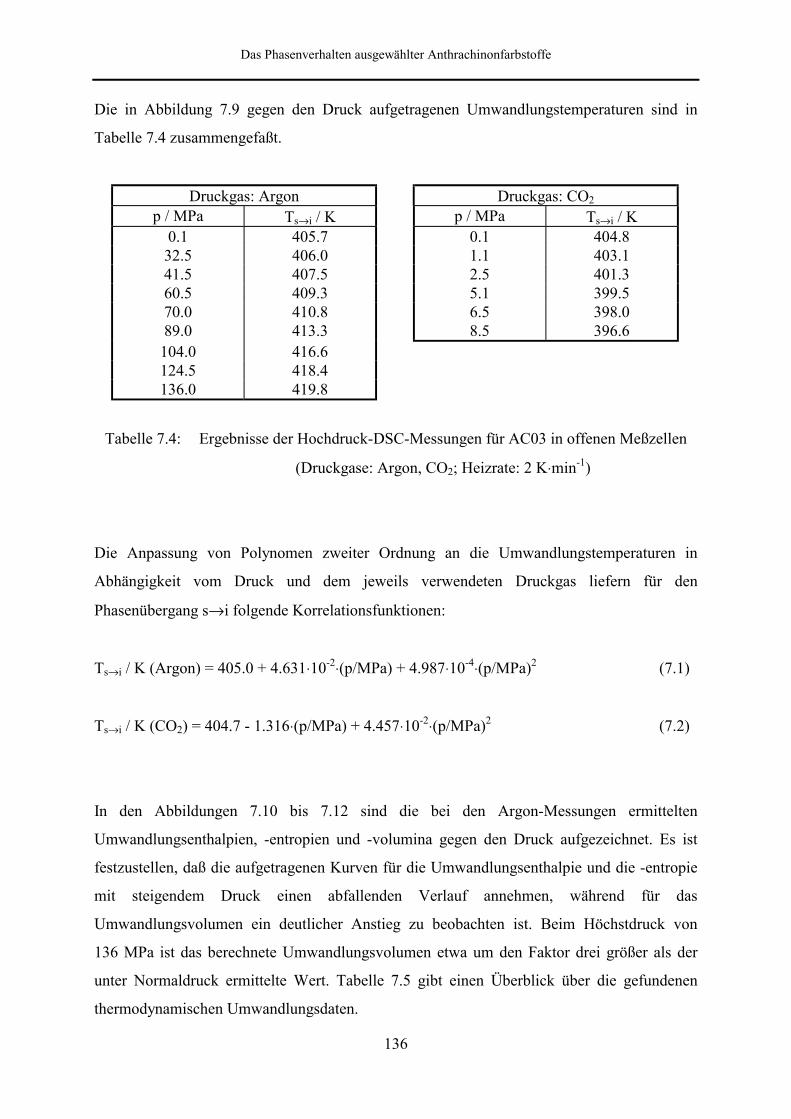
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
136
Die in Abbildung 7.9 gegen den Druck aufgetragenen Umwandlungstemperaturen sind in
Tabelle 7.4 zusammengefaßt.
Druckgas: Argon Druckgas: CO2
p / MPa Ts→i / K p / MPa Ts→i / K0.1 405.7 0.1 404.832.5 406.0 1.1 403.141.5 407.5 2.5 401.360.5 409.3 5.1 399.570.0 410.8 6.5 398.089.0 413.3 8.5 396.6104.0 416.6124.5 418.4136.0 419.8
Tabelle 7.4: Ergebnisse der Hochdruck-DSC-Messungen für AC03 in offenen Meßzellen
(Druckgase: Argon, CO2; Heizrate: 2 K⋅min-1)
Die Anpassung von Polynomen zweiter Ordnung an die Umwandlungstemperaturen in
Abhängigkeit vom Druck und dem jeweils verwendeten Druckgas liefern für den
Phasenübergang s→i folgende Korrelationsfunktionen:
Ts→i / K (Argon) = 405.0 + 4.631⋅10-2⋅(p/MPa) + 4.987⋅10-4⋅(p/MPa)2 (7.1)
Ts→i / K (CO2) = 404.7 - 1.316⋅(p/MPa) + 4.457⋅10-2⋅(p/MPa)2 (7.2)
In den Abbildungen 7.10 bis 7.12 sind die bei den Argon-Messungen ermittelten
Umwandlungsenthalpien, -entropien und -volumina gegen den Druck aufgezeichnet. Es ist
festzustellen, daß die aufgetragenen Kurven für die Umwandlungsenthalpie und die -entropie
mit steigendem Druck einen abfallenden Verlauf annehmen, während für das
Umwandlungsvolumen ein deutlicher Anstieg zu beobachten ist. Beim Höchstdruck von
136 MPa ist das berechnete Umwandlungsvolumen etwa um den Faktor drei größer als der
unter Normaldruck ermittelte Wert. Tabelle 7.5 gibt einen Überblick über die gefundenen
thermodynamischen Umwandlungsdaten.
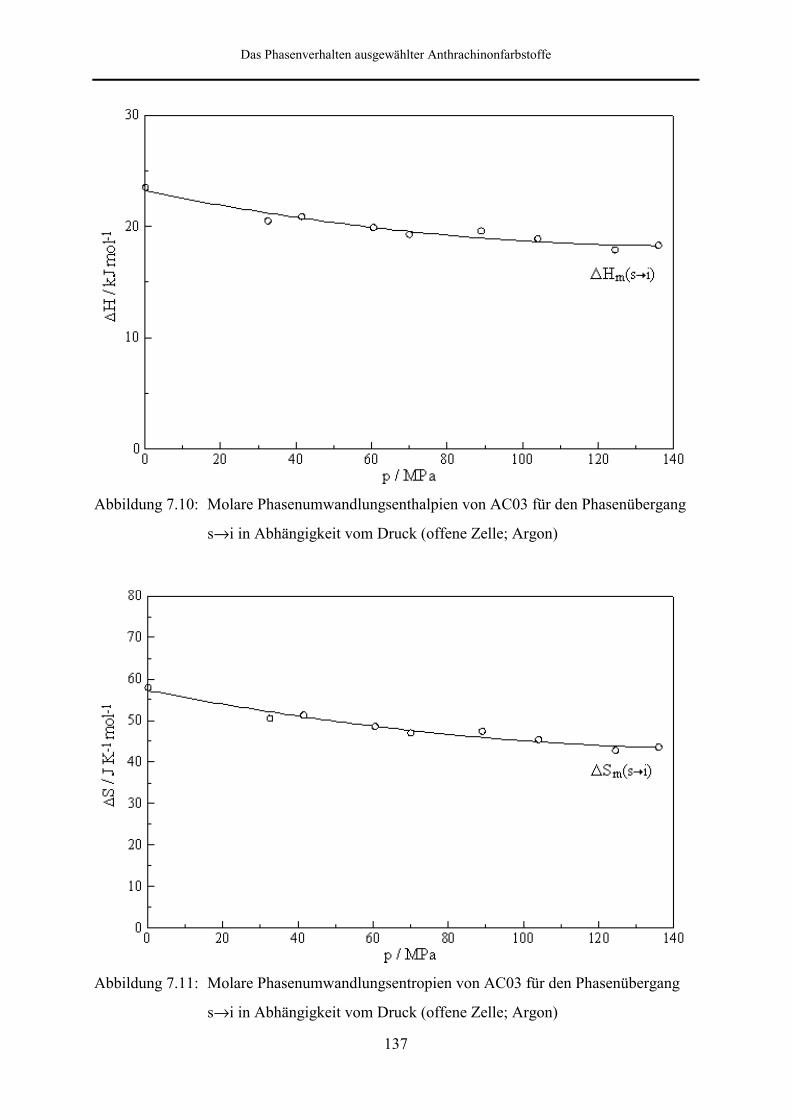
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
137
Abbildung 7.10: Molare Phasenumwandlungsenthalpien von AC03 für den Phasenübergang
s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
Abbildung 7.11: Molare Phasenumwandlungsentropien von AC03 für den Phasenübergang
s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
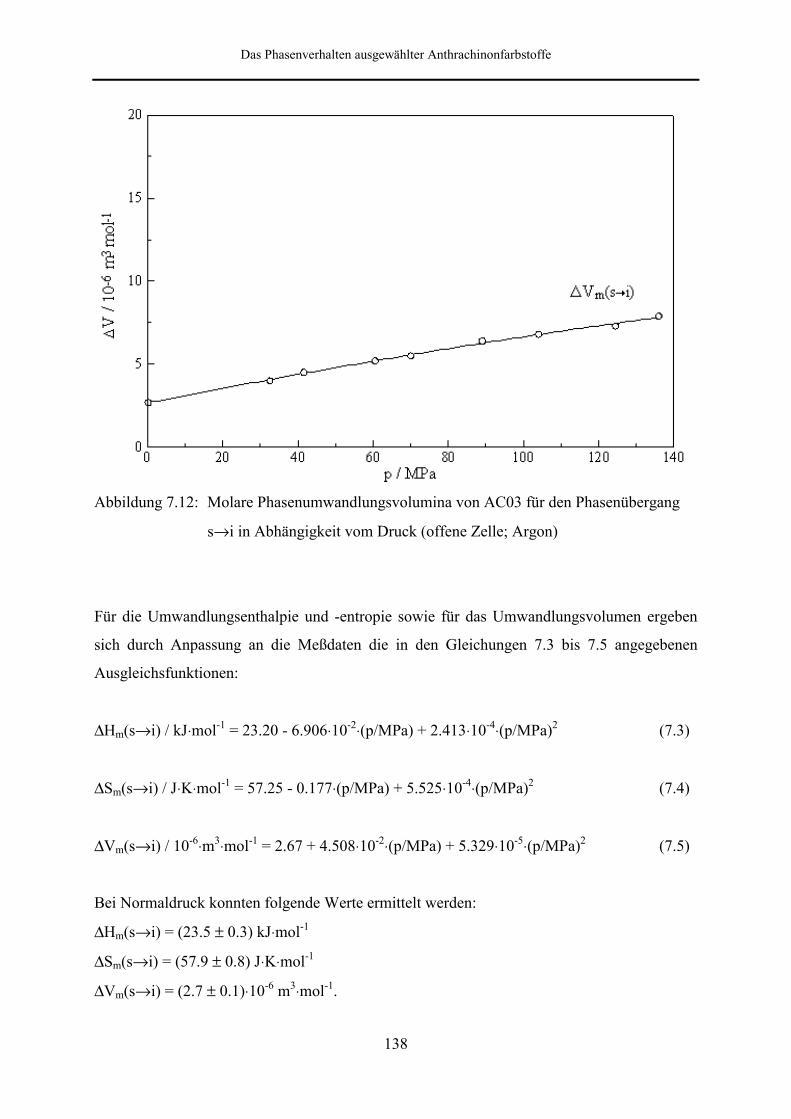
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
138
Abbildung 7.12: Molare Phasenumwandlungsvolumina von AC03 für den Phasenübergang
s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
Für die Umwandlungsenthalpie und -entropie sowie für das Umwandlungsvolumen ergeben
sich durch Anpassung an die Meßdaten die in den Gleichungen 7.3 bis 7.5 angegebenen
Ausgleichsfunktionen:
∆Hm(s→i) / kJ⋅mol-1 = 23.20 - 6.906⋅10-2⋅(p/MPa) + 2.413⋅10-4⋅(p/MPa)2 (7.3)
∆Sm(s→i) / J⋅K⋅mol-1 = 57.25 - 0.177⋅(p/MPa) + 5.525⋅10-4⋅(p/MPa)2 (7.4)
∆Vm(s→i) / 10-6⋅m3⋅mol-1 = 2.67 + 4.508⋅10-2⋅(p/MPa) + 5.329⋅10-5⋅(p/MPa)2 (7.5)
Bei Normaldruck konnten folgende Werte ermittelt werden:
∆Hm(s→i) = (23.5 ± 0.3) kJ⋅mol-1
∆Sm(s→i) = (57.9 ± 0.8) J⋅K⋅mol-1
∆Vm(s→i) = (2.7 ± 0.1)⋅10-6 m3⋅mol-1.

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
139
p / MPa ∆s→iHm / kJ⋅mol-1 ∆s→iSm / J⋅K-1⋅mol-1 ∆s→iVm / 10-6⋅m3⋅mol-
1
0.1 23.5 57.9 2.732.5 20.5 50.5 4.041.5 20.9 51.3 4.560.5 19.9 48.6 5.270.0 19.3 47.0 5.589.0 19.6 47.4 6.4104.0 18.9 45.4 6.8124.5 17.9 42.8 7.3136.0 18.3 43.6 7.9
Tabelle 7.5: Thermodynamische Umwandlungsdaten für den Phasenübergang s→i von
AC03, ermittelt in offener Zelle (Druckgas: Argon)
7.3.2 Phasenverhalten von 1,4-Bis-(n-butylamino)-9,10-anthrachinon (AC04)
Aufgrund der aufgetretenen und in Abschnitt 7.3.1 dargelegten Probleme bei der
Durchführung der DSC-Hochdruckmessungen an AC03 wurde das Phasenverhalten von
AC04 mittels DTA untersucht; auf CO2-Messungen wurde verzichtet. Die im
Temperaturbereich von 290 K bis 480 K und bei Drücken bis 240 MPa vorgenommenen
Messungen erfolgten in geschlossenen und verlöteten Bleizellen sowie in offenen Bleizellen
unter Verwendung von Argon als Druckgas. Alle Messungen wurden bei einer Aufheizrate
von 2 K⋅min-1 durchgeführt.
7.3.2.1 Meßergebnisse und Diskussion
In den Abbildungen 7.13 und 7.14 sind jeweils mehrere DTA-Thermogramme, die den
Einfluß des Druckes auf die Lage und Gestalt der einzelnen Phasenübergangspeaks
veranschaulichen, dargestellt. Im Gegensatz zu AC03 ist bei AC04 auch bei hohen Drücken
sowohl mit als auch ohne Gaseinfluß polymorphes Verhalten zu beobachten. Im
Normaldruckthermogramm in Abbildung 7.13 erfolgt die Schmelzumwandlung bei 388.5 K.
Eine fest-fest-Umwandlung wird durch die am Schmelzpeak vorhandene, vorgelagerte
Schulter angedeutet. Eine Erhöhung des Druckes bewirkt eine zunehmende Trennung der
beiden Umwandlungspeaks.
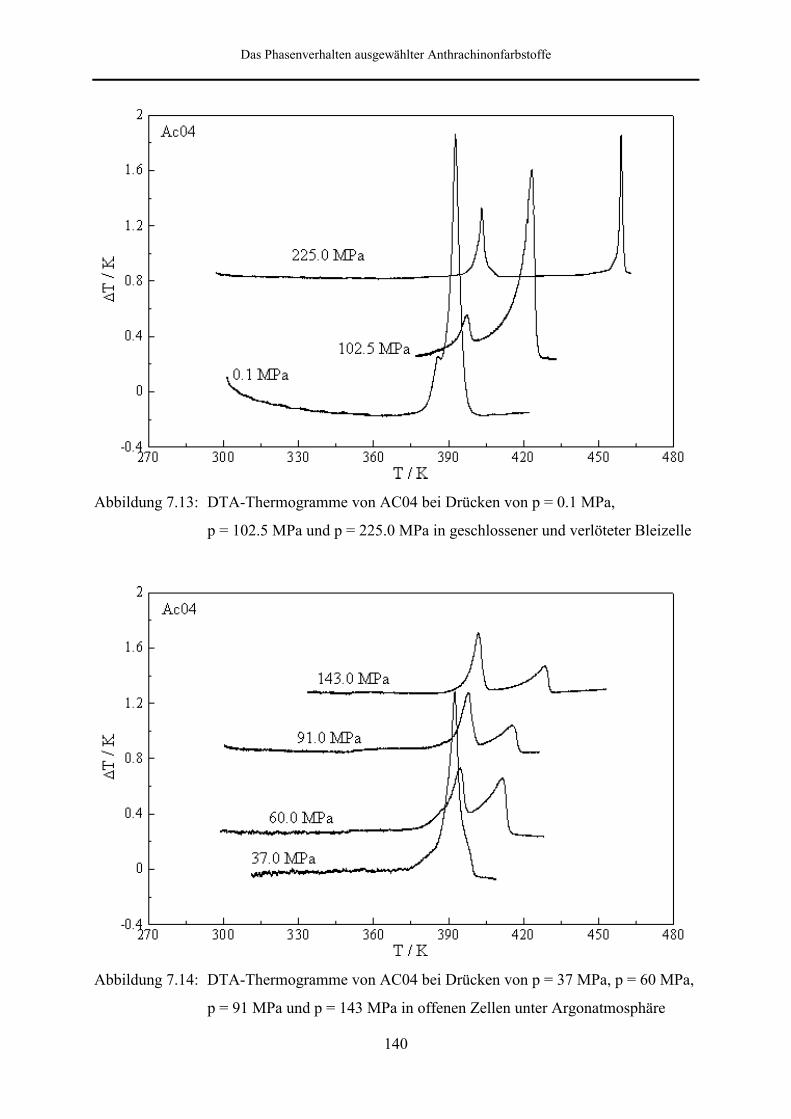
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
140
Abbildung 7.13: DTA-Thermogramme von AC04 bei Drücken von p = 0.1 MPa,
p = 102.5 MPa und p = 225.0 MPa in geschlossener und verlöteter Bleizelle
Abbildung 7.14: DTA-Thermogramme von AC04 bei Drücken von p = 37 MPa, p = 60 MPa,
p = 91 MPa und p = 143 MPa in offenen Zellen unter Argonatmosphäre
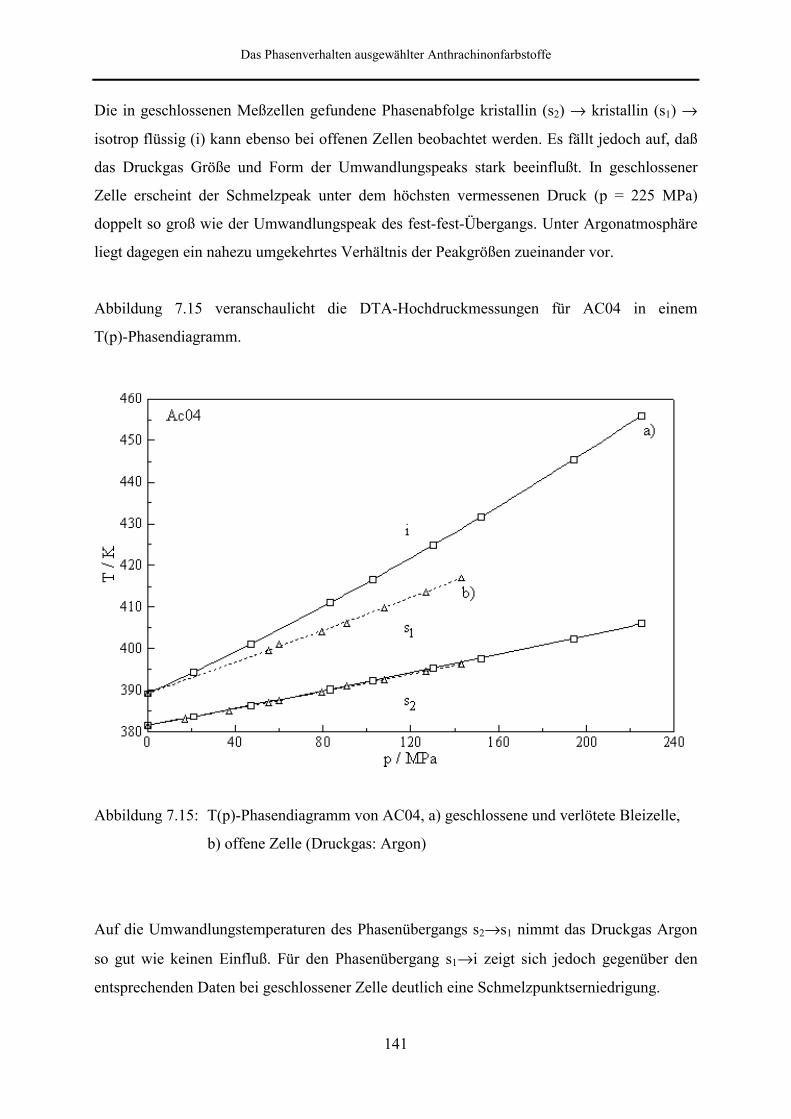
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
141
Die in geschlossenen Meßzellen gefundene Phasenabfolge kristallin (s2) → kristallin (s1) →
isotrop flüssig (i) kann ebenso bei offenen Zellen beobachtet werden. Es fällt jedoch auf, daß
das Druckgas Größe und Form der Umwandlungspeaks stark beeinflußt. In geschlossener
Zelle erscheint der Schmelzpeak unter dem höchsten vermessenen Druck (p = 225 MPa)
doppelt so groß wie der Umwandlungspeak des fest-fest-Übergangs. Unter Argonatmosphäre
liegt dagegen ein nahezu umgekehrtes Verhältnis der Peakgrößen zueinander vor.
Abbildung 7.15 veranschaulicht die DTA-Hochdruckmessungen für AC04 in einem
T(p)-Phasendiagramm.
Abbildung 7.15: T(p)-Phasendiagramm von AC04, a) geschlossene und verlötete Bleizelle,
b) offene Zelle (Druckgas: Argon)
Auf die Umwandlungstemperaturen des Phasenübergangs s2→s1 nimmt das Druckgas Argon
so gut wie keinen Einfluß. Für den Phasenübergang s1→i zeigt sich jedoch gegenüber den
entsprechenden Daten bei geschlossener Zelle deutlich eine Schmelzpunktserniedrigung.

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
142
Die Phasengrenzlinien werden durch Polynome zweiter Ordnung beschrieben:
Ohne Gaseinfluß (geschlossene Zelle):
Ts2→s1 / K = 381.59 + 1.011⋅10-1⋅(p/MPa) + 3.266⋅10-5⋅(p/MPa)2 (7.6)
Ts1→i / K = 389.18 + 2.429⋅10-1⋅(p/MPa) + 2.403⋅10-4⋅(p/MPa)2 (7.7)
Mit Gaseinfluß (offene Zelle):
Ts2→s1 / K (Argon) = 381.54 + 9.955⋅10-2⋅(p/MPa) + 2.785⋅10-5⋅(p/MPa)2 (7.8)
Ts1→i / K (Argon) = 389.27 + 1.846⋅10-1⋅(p/MPa) + 5.984⋅10-5⋅(p/MPa)2 (7.9)
In Tabelle 7.6 sind die gemessenen Umwandlungstemperaturen der Phasenübergänge s2→s1
und s1→i von AC04 in Abhängigkeit vom Druck aufgeführt.
geschlossene Bleizelle offene Zelle (Argon)p / MPa Ts2→s1 / K Ts1→i / K p / MPa Ts2→s1 / K Ts1→i / K
0.1 381.6 389.2 0.1 381.6 389.221.0 383.7 394.4 17.0 383.247.0 386.4 401.1 37.0 385.283.0 390.2 411.0 55.0 387.1 399.6102.5 392.3 416.6 60.0 387.5 401.0130.0 395.3 424.8 79.0 389.6 404.2152.0 397.7 431.7 91.0 391.0 406.1194.0 402.4 445.4 108.0 392.6 409.9225.0 406.0 456.0 127.0 394.6 413.7
143.0 396.3 417.0
Tabelle 7.6: Ergebnisse der Hochdruck-DTA-Messungen für AC04 in geschlossenen und
offenen Meßzellen (Druckgas: Argon; Heizrate 2 K⋅min-1)

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
143
7.3.2.2 Abschätzung des gelösten Gasanteils
Wie im vorherigen Kapitel gezeigt, bewirkt der direkte Kontakt des Druckgases mit der
Probensubstanz im vermessenen Druckbereich eine Absenkung der Schmelztemperatur. Im
folgenden wird versucht, den Molenbruch des im Dispersionsfarbstoff AC04 gelösten Gases
(Argon) in Abhängigkeit vom Druck abzuschätzen.
Wird das Schmelzgleichgewicht einer binären Mischung betrachtet, so sind im
Gleichgewichts-zustand das chemische Potential der Komponente i in der flüssigen Phase
µ il
ilT p x( , , ) und das chemische Potential der Komponente i in der festen Phase
µ is
isT p x( , , ) gleich groß:
µ µil
il
is
isT p x T p x( , , ) ( , , )= (7.10)
Mit Hilfe des chemischen Standardpotentials µ i* und der Raoult’schen Aktivitätskoeffizienten
fi lassen sich die chemischen Potentiale der Komponente i der festen und der flüssigen Phase
folgendermaßen ausdrücken:
µ µis
is
is
isisT p x T p RT x f( , , ) ( , ) ln*= + ⋅ (7.11)
µ µil
il
il
ililT p x T p RT x f( , , ) ( , ) ln*= + ⋅ (7.12)
Durch Kombination der obigen Gleichungen ergibt sich für den Molenbruch xil der
Komponente i in der der flüssigen Phase:
( )x
RTx f
fil
is
il
isis
il=
−
exp* *µ µ
(7.13)

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
144
Da die Differenz der chemischen Standardpotentiale µ µil
is* *− der Änderung der molaren
Gibbsenergie ∆ slm iG ,* entspricht, wobei diese wiederum über die Enthalpie- und Entropie-
änderungen beschrieben werden kann (Beziehung 7.14), läßt sich für den Molenbruch xil der
in Gleichung (7.15) angegebene Ausdruck formulieren:
∆ ∆ ∆slm i i
lis
slm i s
lm iG H T S,
* * *,
*,
*= − = −µ µ (7.14)
x
GRT
x f
fil
slm
isis
il=
−
exp*∆
bzw.
( )x
H T S
RTx f
fil
slm s
lm
isis
il=
−−
exp
* *∆ ∆
(7.15)
Wird die molare Entropieänderung durch die bereits in Gleichung (2.15) angegebene
Beziehung ∆∆
slm i
slm iSHT,
* ,*
= ersetzt, so ergibt sich für den Molenbruch xil :
x
HR T T
x f
fil
slm
sl i
sis
il=
−
exp*
*∆ 1 1
(7.16)
Betrachtet man nunmehr das gelöste Gas (Komponente 2) als Verunreinigung der isotrop
flüssigen Phase des AC04 (Komponente 1), so kann unter der Voraussetzung, daß der reine
Feststoff auskristallisiert, folgende Vereinfachung vorgenommen werden:
a x fs s s1 1 1 1= ⋅ = (7.17)
Die weitergehende Annahme, daß für die (gasgesättigte) isotrop flüssige Phase das
Raoult’sche Grenzgesetz gilt (d.h. f l1 1= ), führt dann zu dem in Gleichung (7.18)
angegebenen Ausdruck für den Molenbruch des gelösten Gases x l2 [111]:

Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
145
xHR
TT T
l slm
sl2
11= −⋅
exp ,*
*∆ ∆
mit ∆T T Tsl= − * (7.18)
Aufgrund der durchgeführten DTA-Messungen sind die Druckabhängigkeiten der
Umwandlungstemperatur Tsl* des reinen AC04 und der Temperaturerniedrigung ∆T durch
das gelöste Gas bekannt. Für die molare Enthalpieänderung ∆ slmH ,*
1 wird ein
druckunabhängiges Verhalten angenommen. Bei der Berechnung des Molenbruchs in
Abhängigkeit vom Druck wird hierfür der mittels DSC gefundene Normaldruckwert
eingesetzt.
x pHR
T pT p T p
l slm
sl2
11( ) exp( )
( ) ( ),
*
*= −⋅
∆ ∆(7.18a)
mit T p p psl* ( ) / . . ( / ) . ( / )K MPa MPa= + ⋅ ⋅ + ⋅ ⋅− −389 18 2 429 10 2 403 101 4 2 (7.7)
T p p psl ( ) / . . ( / ) . ( / )K MPa MPa= + ⋅ ⋅ + ⋅ ⋅− −389 27 1846 10 5984 101 5 2 (7.9)
∆T p T p T psl
sl( ) ( ) ( )*= − (7.19)
∆ slmH ,* / .1
1 234kJ mol⋅ =− (Tabelle 7.2)
In Abbildung 7.16 sind die Verläufe des Molenbruchs x l2 und des Massenbruchs w l2 für das
in AC04 gelöste Gas Argon in Abhängigkeit vom Druck dargestellt. Die Aussagekraft dieser
Auftragung wird durch die gemachten Annahmen stark eingeschränkt. Aufgrund der an AC03
vorgenommenen DSC-Messungen ist zu vermuten, daß auch AC04 eine Druckabhängigkeit
der Umwandlungsenthalpie aufweist. Hinweise darauf zeigen sich auch in den in Abbildung
7.14 dargestellten DTA-Thermogrammen, wonach der Umwandlungspeak des Schmelzüber-
gangs bei Zunahme des Druckes kleiner wird. Außerdem muß berücksichtigt werden, daß die
Annahme der Gültigkeit des Raoult’schen Grenzgesetzes problematisch ist.
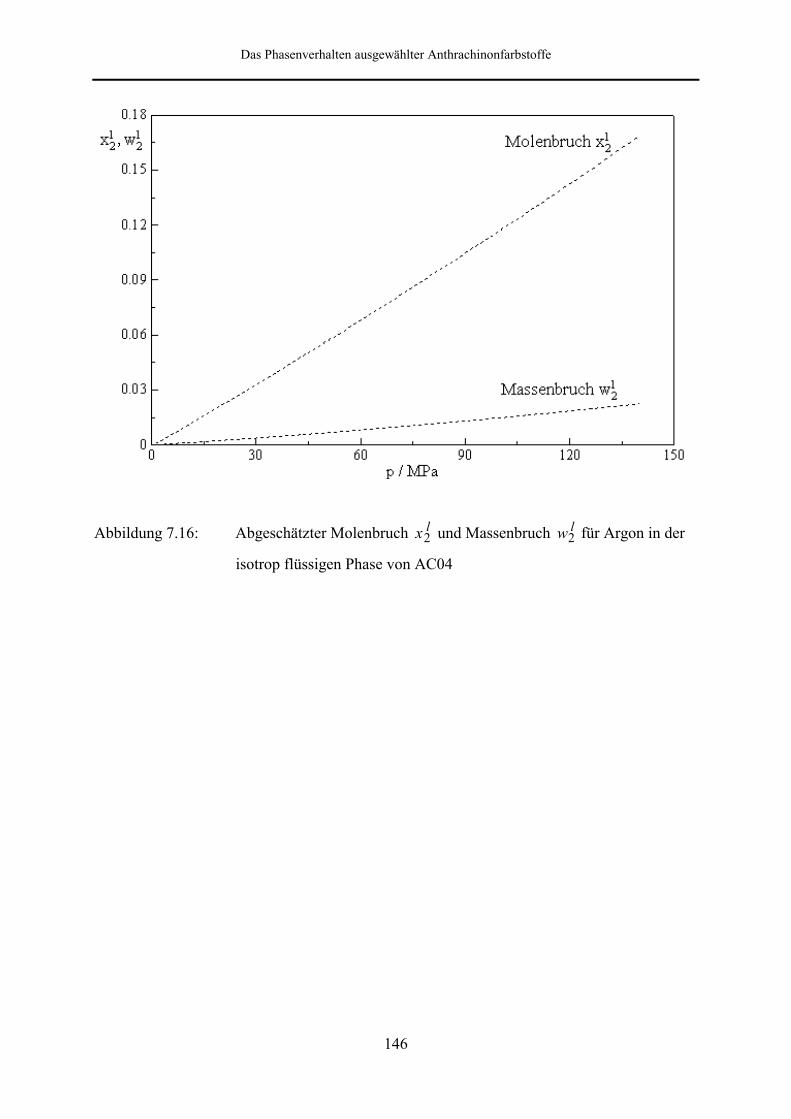
Das Phasenverhalten ausgewählter Anthrachinonfarbstoffe
146
Abbildung 7.16: Abgeschätzter Molenbruch x l2 und Massenbruch w l2 für Argon in der
isotrop flüssigen Phase von AC04

Bestimmung von Wärmekapazitäten
147
8 Wärmekapazitätsbestimmung
Die im Rahmen der vorliegenden Arbeit durchgeführte Bestimmung der Wärmekapazität
basiert auf dem Verfahren der Wärmestromkalibrierung Dynamischer Differenz Kalorimeter
(DSC), das weitgehend gerätetyp-unabhängig ist und das den Einfluß wesentlicher
Versuchsparameter (Temperatur, Heizrate, Probenmasse) und Probenparameter (thermischer
Effekt, Temperaturleitfähigkeit) berücksichtigt. Im folgenden Abschnitt sollen kurz einige
Grundlagen vermittelt werden. Es werden die notwendigen Begriffe definiert sowie das
entsprechende Verfahren und die Auswertemethode dargelegt [145, 146, 147]. Darüberhinaus
sei auf Arbeiten von B. Wunderlich [151] und I. Czarnota [152] verwiesen.
8.1 Grundlagen der Wärmestromkalibrierung
Die Wärmestromkalibrierung soll eine eindeutige Zuordnung des vom Gerät angezeigten
Wärmestroms Φm zum wahren, von der Probe aufgrund ihrer Wärmekapazität Cp auf-
genommenen oder abgegebenen Wärmestrom Φw gewährleisten.
Es gilt:
Φ ΦΦw m pK T C= ⋅ = ⋅( ) β (8.1)
wo ( )K TΦ der temperaturabhängige Kalibrierfaktor bezüglich des wahren Wärmestroms und
β die im Temperaturprogramm eingestellte Heizrate ist.
Die Wärmestromkalibrierung wird in der folgenden Weise durchgeführt:
Zuerst ist die Kalibriersubstanz entsprechend dem interessierenden Temperaturbereich
auszuwählen. In diesem Fall wurde als Kalibriersubstanz Korund (α-Al2O3), d.h. ein
synthetischer Saphir (Masse: 8.723 mg), verwendet.

Bestimmung von Wärmekapazitäten
148
Bei der Auswahl der Tiegel ist darauf zu achten, daß diese bezüglich Material, Form,
Verschlußart, Emissionsgrad und Masse möglichst ähnlich oder identisch sind mit den
Tiegeln für die Leermessungen und Meßprobenmessungen. Die von Perkin-Elmer [L 1]
gelieferten Aluminiumtiegel erfüllen diese Bedingungen.
Das Temperaturprogramm unterteilt sich in drei Abschnitte (siehe dazu Abbildung 8.1):
isothermer Abschnitt zur Bestimmung der isothermen Anfangslinien (Φiso,st,S und Φiso,st,0),
dynamischer Abschnitt (Heizrate: 10 K⋅min-1, Dauer: 10 - 30 min) (ΦS und Φ0), sowie
isothermer Abschnitt zur Bestimmung der isothermen Endlinien (Φiso, end,S und Φiso, end,0).
Jeder Abschnitt muß mindestens so lang sein, daß sich quasi-stationäre Verhältnisse einstellen
können (im allgemeinen nach 2 - 10 min).
Jede Kalibrierprobenmessung ist im Wechsel mit einer Leermessung durchzuführen, wobei
Kalibrierprobenmessung und Leermessung am gleichen Tag mit dem gleichen Temperatur-
programm auszuführen sind.
Zusammengehörige Paare von Leer- und Kalibrierprobenmeßwerten sind im quasi-stationären
Bereich des dynamischen Abschnitts nach Gleichung 8.2 auszuwerten (für die Bedeutung der
benutzten Symbole siehe Abbildung 8.1):
K T)C T)
t tt t
t tt t
p
S iso st Siso end S iso st S
end stst iso st
iso end iso st
end stst
Φ
Φ ΦΦ Φ
Φ ΦΦ Φ
((
( ) ( ), ,, , , ,
, ,, , , ,
=⋅
− +−−
−
− − +
−−
−
β
0 00 0
Gleichung (8.2)
Die berechneten Mittelwerte von KΦ(T) für die entsprechende Kalibrierprobe werden als
Funktion der Temperatur Tw dargestellt. Ist KΦ(T) bekannt, so werden die oben beschriebenen
Messungen im gleichen Temperaturbereich an der Probensubstanz durchgeführt. Gemäß
Gleichung 8.2 ist es dann möglich, die Wärmekapazität der Probe Cp(T) zu bestimmen.
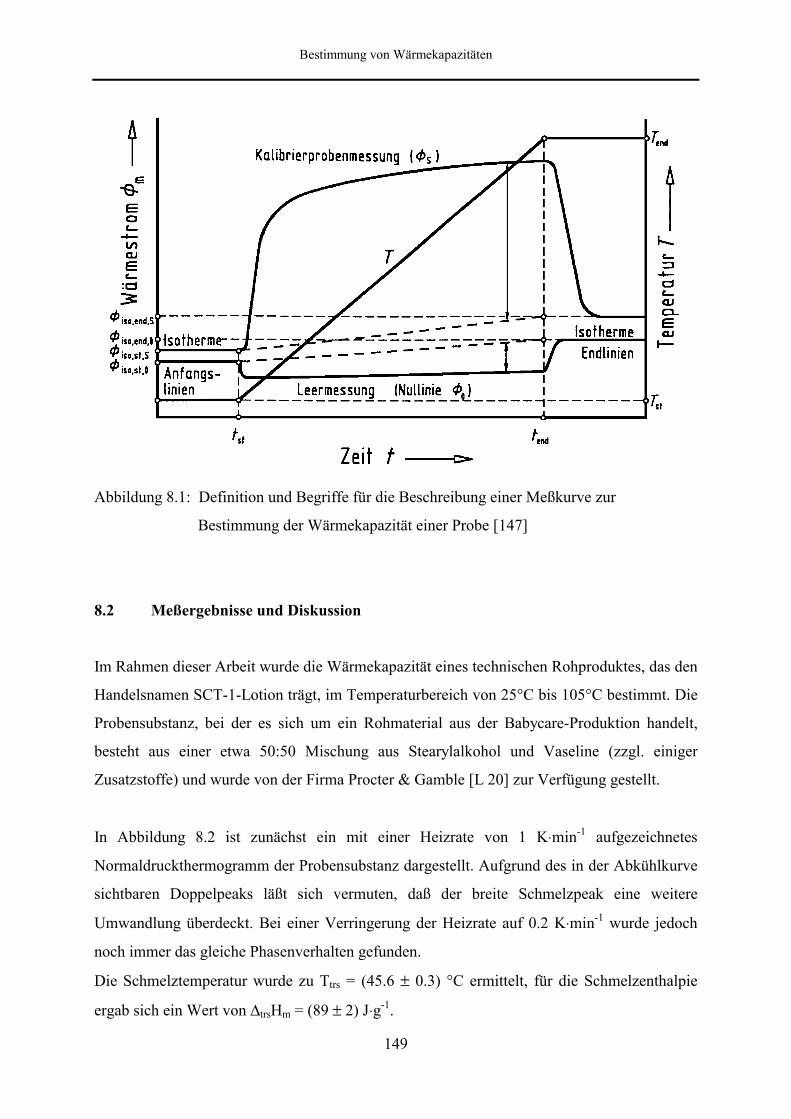
Bestimmung von Wärmekapazitäten
149
Abbildung 8.1: Definition und Begriffe für die Beschreibung einer Meßkurve zur
Bestimmung der Wärmekapazität einer Probe [147]
8.2 Meßergebnisse und Diskussion
Im Rahmen dieser Arbeit wurde die Wärmekapazität eines technischen Rohproduktes, das den
Handelsnamen SCT-1-Lotion trägt, im Temperaturbereich von 25°C bis 105°C bestimmt. Die
Probensubstanz, bei der es sich um ein Rohmaterial aus der Babycare-Produktion handelt,
besteht aus einer etwa 50:50 Mischung aus Stearylalkohol und Vaseline (zzgl. einiger
Zusatzstoffe) und wurde von der Firma Procter & Gamble [L 20] zur Verfügung gestellt.
In Abbildung 8.2 ist zunächst ein mit einer Heizrate von 1 K⋅min-1 aufgezeichnetes
Normaldruckthermogramm der Probensubstanz dargestellt. Aufgrund des in der Abkühlkurve
sichtbaren Doppelpeaks läßt sich vermuten, daß der breite Schmelzpeak eine weitere
Umwandlung überdeckt. Bei einer Verringerung der Heizrate auf 0.2 K⋅min-1 wurde jedoch
noch immer das gleiche Phasenverhalten gefunden.
Die Schmelztemperatur wurde zu Ttrs = (45.6 ± 0.3) °C ermittelt, für die Schmelzenthalpie
ergab sich ein Wert von ∆trsHm = (89 ± 2) J⋅g-1.
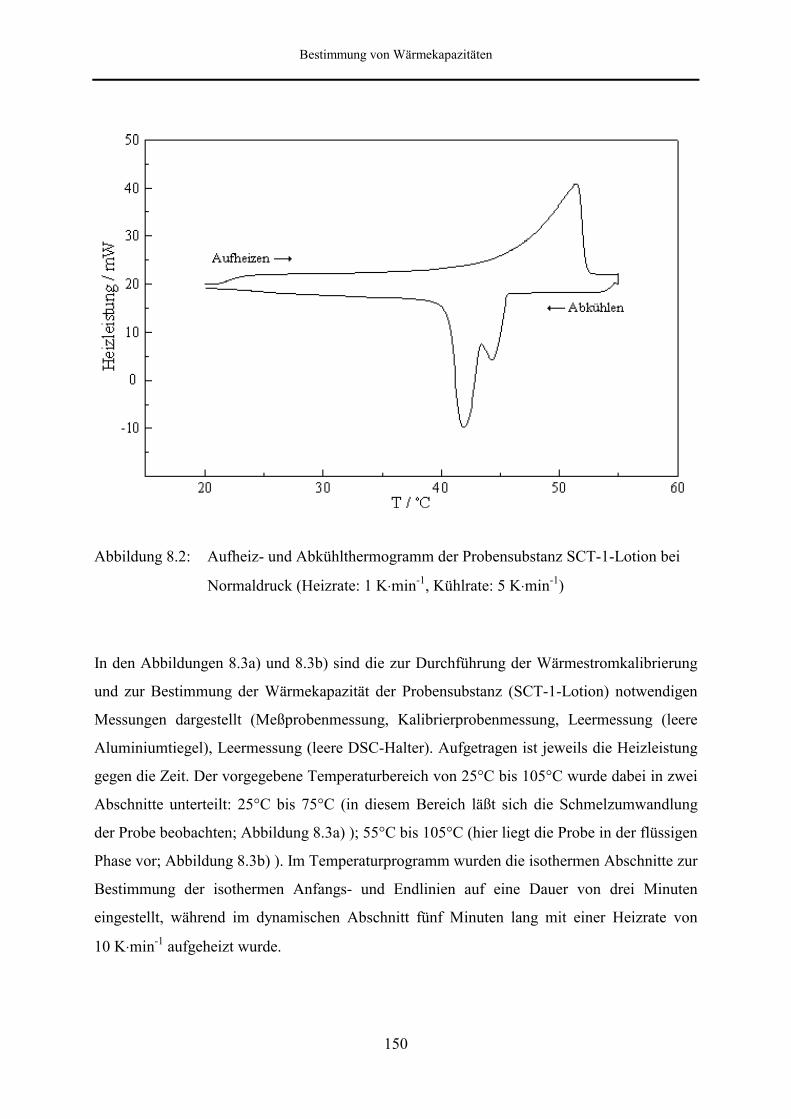
Bestimmung von Wärmekapazitäten
150
Abbildung 8.2: Aufheiz- und Abkühlthermogramm der Probensubstanz SCT-1-Lotion bei
Normaldruck (Heizrate: 1 K⋅min-1, Kühlrate: 5 K⋅min-1)
In den Abbildungen 8.3a) und 8.3b) sind die zur Durchführung der Wärmestromkalibrierung
und zur Bestimmung der Wärmekapazität der Probensubstanz (SCT-1-Lotion) notwendigen
Messungen dargestellt (Meßprobenmessung, Kalibrierprobenmessung, Leermessung (leere
Aluminiumtiegel), Leermessung (leere DSC-Halter). Aufgetragen ist jeweils die Heizleistung
gegen die Zeit. Der vorgegebene Temperaturbereich von 25°C bis 105°C wurde dabei in zwei
Abschnitte unterteilt: 25°C bis 75°C (in diesem Bereich läßt sich die Schmelzumwandlung
der Probe beobachten; Abbildung 8.3a) ); 55°C bis 105°C (hier liegt die Probe in der flüssigen
Phase vor; Abbildung 8.3b) ). Im Temperaturprogramm wurden die isothermen Abschnitte zur
Bestimmung der isothermen Anfangs- und Endlinien auf eine Dauer von drei Minuten
eingestellt, während im dynamischen Abschnitt fünf Minuten lang mit einer Heizrate von
10 K⋅min-1 aufgeheizt wurde.
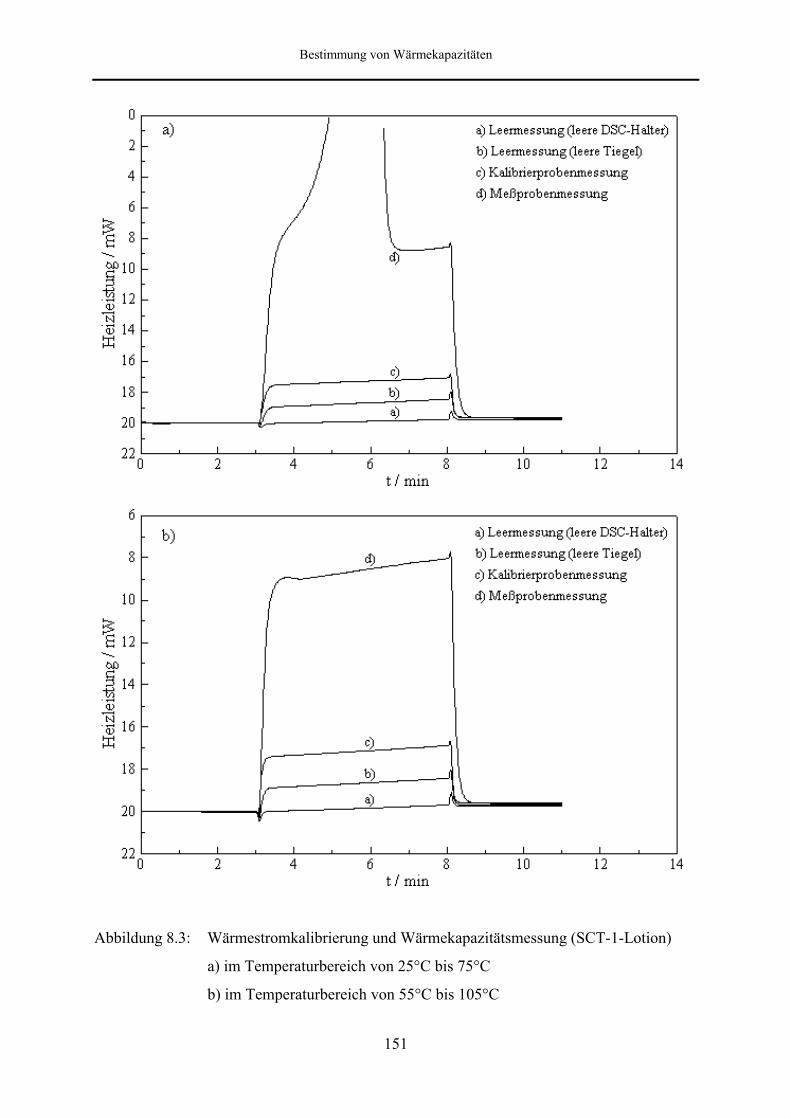
Bestimmung von Wärmekapazitäten
151
Abbildung 8.3: Wärmestromkalibrierung und Wärmekapazitätsmessung (SCT-1-Lotion)
a) im Temperaturbereich von 25°C bis 75°C
b) im Temperaturbereich von 55°C bis 105°C

Bestimmung von Wärmekapazitäten
152
Die in den Abbildungen 8.3a) und 8.3b) dargestellten Thermogramme werden wie
beschrieben gemäß Abbildung 8.1 und nach Gleichung 8.2 rechnerintern ausgewertet. Für die
hier vermessene Probensubstanz SCT-1-Lotion ergibt sich über den Temperaturbereich von
25°C bis 105°C der in Abbildung 8.4 gegen die Temperatur aufgetragene Verlauf der
Wärmekapazität Cp(T).
Abbildung 8.4: Wärmekapazität Cp(T) von SCT-1-Lotion im Temperaturbereich von 25°C
bis 105°C
Für den Temperaturbereich von 60°C bis 105°C, in dem die Probe keine Umwandlung zeigt,
läßt sich die Wärmekapazität als Funktion der Temperatur durch folgende Ausgleichsfunktion
beschreiben:
Cp(T) / J⋅g-1⋅grd-1 = 1.1054 + 5.5628⋅10-3⋅(T / °C) (8.3)
(im Temperaturbereich: T = 60°C - 105°C)
Bestimmung von Wärmekapazitäten
153
Für den Temperaturbereich unterhalb der Schmelztemperatur können hier nur abschätzende
Aussagen gemacht werden. Aufgrund einer fehlenden Kühlvorrichtung war es nicht möglich,
bereits ab 0°C zu messen. Aus Abbildung 8.4 läßt sich für die Meßsubstanz bei 35°C ein Wert
für die Wärmekapazität abschätzen: Cp(35°C) ≈ 1.83 J⋅g-1⋅K-1.
In Tabelle 8.1 sind einige vergleichende Literaturwerte der Wärmekapazitäten ähnlicher
Substanzen angegeben.
Substanz Cp / J⋅mol-1⋅K-1 Cp / J⋅g-1⋅K-1 Literatur
1-Hexadecanol (C16H34O) 422.0 (25°C) 1.74 (25°C) [81]Stearinsäre (C18H36O) 501.5 (25°C) 1.76 (25°C) [81]Octadecan (C18H38) 485.6 (25°C) 1.91 (25°C) [81]
SCT-1-Lotion(50:50 Stearylalkohol:Vaseline)
--- 1.83 (35°C) eigene Messung
Tabelle 8.1: Literaturwerte

Ausblick
154
9 Ausblick
Zur Verbesserung der im Rahmen der vorliegenden Arbeit benutzten DSC- und DTA-
Hochdruckapparatur gibt es einige Möglichkeiten.
Zur Optimierung beider Apparaturen wären vor allem geregelte Abkühlvorrichtungen
sinnvoll, da insbesondere den Abkühlthermogrammen bei der Untersuchung zum
Phasenverhalten große Bedeutung zukommt. Schmelzsicherungen könnten die Gefahr des
Überheizens der Autoklaven während mehrtägiger Temperexperimente verringern. Des
weiteren sollten beide Apparaturen mit moderneren Rechnersystemen ausgestattet werden, so
daß die Arbeitsleistung erhöht und der zeitliche Aufwand bei den Auswertungen reduziert
wird.
Bei der DTA wird der Temperaturbereich durch das verwendete Lötzinn stark eingeschränkt.
Daher sollte nach anderen Möglichkeiten, wie z. B. höher schmelzende Lote, gesucht werden,
um die Bleizellen zu verschließen.
Bei der Durchführung von DSC-Hochdruckmessungen ist besonders die Wahl der Meßzellen
zu überdenken. Membran und Klebestelle stellen die Schwachpunkte dar. Organische
Flüssigkeiten neigen generell dazu, über den Rand der Zelle zu kriechen und ein sicheres
Verkleben der Membran zu verhindern. Auch das Aufschrauben des Deckels muß mit
Vorsicht geschehen, um die Membran nicht zu beschädigen, da diese sonst bei hohen Drücken
reißt. Es ist möglicherweise sinnvoll, auf den aufschraubbaren Deckel der Meßzelle gänzlich
zu verzichten. Die Membran könnte an die Zelle angelötet werden.
Da sich viele der verwendeten Flüssigkristalle stark unterkühlen ließen, wären
Untersuchungen an einer für tiefe Temperaturen ausgelegten DTA- bzw. DSC-Apparatur
nützlich. Von den in dieser Arbeit untersuchten flüssigkristallinen Verbindungen zeigen die in
Kapitel 6.1 besprochenen Benzylester ein recht komplexes Phasenverhalten. So konnte
Weissflog [150] in neueren DSC-Normaldruckmessungen an 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-3-cyan-benzylester beim Abkühlen das Auftreten zweier smektischer
Phasen (smA und smC) beobachten. DSC-Messungen unter Hochdruck wurden bislang nicht
durchgeführt.
Die Untersuchungen zum Phasenverhalten der Anthrachinonfarbstoffe können ebenfalls als
noch nicht abgeschlossen angesehen werden. Bei den Experimenten dieser Arbeit traten hier
Schwierigkeiten bei der Präparation der Meßzellen auf (siehe oben).

Anhang
155
10 Anhang
A1 Verzeichnis der Abbildungen
Abbildung 1.1: Schematische Darstellung mesomorpher Übergänge eines kristallinen
Festkörpers
Abbildung 1.2: Klassifizierung der Flüssigkristalle
Abbildung 1.3: Schematische Darstellung der unterschiedlichen Strukturmerkmale
verschiedener flüssigkristalliner Phasen
Abbildung 1.4: Anordnung der Moleküle in einem nematischen Flüssigkristall
Abbildung 1.5: Schematische Darstellung der Orientierung der Molekülachse eines
Einzelmoleküls zum Direktor n
Abbildung 1.6: Schematische Darstellung der Helixstruktur der cholesterischen Phase
Abbildung 1.7: Strukturmerkmale verschiedener smektischer Phasen
Abbildung 2.1: Klassifizierung von Phasenübergängen nach Ehrenfest
Abbildung 2.2: Änderung der molaren Wärmekapazität Cp,m bei einem λ-Übergang
Abbildung 3.1: Meßanordnung der DTA (a) und der DSC (b)
Abbildung 3.2: Schematische Darstellung des DSC-Prinzips
a) Normaldruck-DSC, b) Hochdruck-DSC
Abbildung 3.3: Gesamtansicht der Meßanordnung
Abbildung 3.4: Aufbau des Hochdruckautoklavs
Abbildung 3.5: Aufbau des Meßkopfes
Abbildung 3.6: Perspektivische Ansicht des Meßkopfes
Abbildung 3.7: Aufbau des Kupplungsstückes
Abbildung 3.8: Perspektivische Ansicht des Kupplungsstückes
Abbildung 3.9: Aufbau der Meßzellen
a) Stahl-Hochdruckkapsel
b) “large volume“ Zelle
Abbildung 3.10: Darstellung des Temperiermantels
Abbildung 3.11: Charakteristische Punkte eines realen Thermogramms
Abbildung 3.12: Möglichkeiten der Grundlinienfestlegung
Abbildung 3.13: Temperaturkalibrierung bei Normaldruck

Anhang
156
Abbildung 3.14: a) Temperaturkalibrierung unter Druck mit Indium (Druckgas: Helium)
b) Temperaturkalibrierung unter Druck mit Indium (Druckgas: Argon)
Abbildung 3.15: a) Druckkorrekturterm (Druckgas: Helium)
b) Druckkorrekturterm (Druckgas: Argon)
Abbildung 3.16: Kalibrierfaktor k(p) (Indium, Druckgase: Helium und Argon)
Abbildung 4.1: Effektive Aufheizgeschwindigkeit als Funktion der Zeit, ermittelt in der
Probenzelle, die im vorliegenden Temperaturbereich keine Umwandlungen
zeigt
Abbildung 4.2: Schematischer Aufbau der Hochdruck-DTA-Apparatur
Abbildung 4.3: Hochdruckautoklav der DTA-Apparatur
Abbildung 4.4: Gießform
Abbildung 4.5: Meßzellen
a) gegossene und mit Bohrung versehene Indium- oder Bleizelle
b) gefüllte Indiumzelle mit Thermoelement
c) verlötete Bleizelle
Abbildung 4.6: Kalibrierung der Thermoelemente
Abbildung 5.1: Strukturformel des Glycerins (1,2,3-Propantriol)
Abbildung 5.2: Strukturformel einsäuriger, gesättigter Triacylglycerine
Abbildung 5.3: Stimmgabel-Konformation eines Triacylglycerins
Abbildung 5.4: β-2- und β-3-Anordnungen von Triacylglycerinen
Abbildung 5.5: Hexagonale Subzelle der α-Modifikation
Abbildung 5.6: Orthorhombische Subzelle der β´-Modifikation
Abbildung 5.7: Trikline Subzelle der β-Modifikation
Abbildung 5.8: Phasenumwandlungsschema nach Wesdorp
Abbildung 5.9: Phasenumwandlungsschema nach Ollivon und Perron
Abbildung 5.10: DSC-Thermogramme von Triarachidin bei Drücken von p = 0.1 MPa,
p = 30.8 MPa und p = 81.2 MPa in offener Zelle1) (Druckgas: Helium)
Abbildung 5.11: T(p)-Phasendiagramm von Triarachidin
Abbildung 5.12: Molare Phasenumwandlungsenthalpien von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck
Abbildung 5.13: Molare Phasenumwandlungsentropien von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck

Anhang
157
Abbildung 5.14: Molare Phasenumwandlungsvolumina von Triarachidin für die
Phasenübergänge α→β und β→l in Abhängigkeit vom Druck
Abbildung 6.1: DSC-Thermogramme des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoe-
säure-4-nitro-benzylesters bei Normaldruck
Abbildung 6.2: DSC-Normaldruckthermogramm des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-4-cyan-benzylesters (Heizrate: 2 K⋅min-1)
Abbildung 6.3: Aufheiz- und Abkühlthermogramm des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-2-cyan-benzylesters bei Normaldruck (Heizrate: 2 K⋅min-1)
Abbildung 6.4: DSC-Thermogramme von 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoe-
säure-3-cyan-benzylester bei p = 0.1 MPa
Abbildung 6.5: DTA-Normaldruckthermogramm des 2,5-Bis(4-n-octyloxy-benzoyl-
oxy)benzoesäure-3-cyan-benzylesters (Heizrate: 2 K⋅min-1)
Abbildung 6.6: T(p)-Phasendiagramm des 2,5-Bis(4-n-octyloxy-benzoyloxy)benzoe-
säure-3-cyan-benzylesters
Abbildung 6.7: Strukturformel der n-TPEBs
Abbildung 6.8: DSC-Normaldruckthermogramme des 6-TPEB, a) Probe nicht
getempert, b) Probe 18 Stunden in der kristallinen Phase getempert
(Heizrate: 2 K⋅min-1)
Abbildung 6.9: DSC-Thermogramme von 10-TPEB bei p = 0.1 MPa
Abbildung 6.10: DSC-Thermogramm von 10-TPEB bei einem Druck von etwa 55 MPa
(Druckgas: Argon, Heizrate: 2 K⋅min-1)
Abbildung 6.11: T(p)-Phasendiagramm des 10-TPEB
Abbildung 6.12: Molare Phasenumwandlungsenthalpien von 10-TPEB für die Phasenüber-
gänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck
Abbildung 6.13: Molare Phasenumwandlungsentropien von 10-TPEB für die Phasenüber-
gänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck
Abbildung 6.14: Molare Phasenumwandlungsvolumina von 10-TPEB für die Phasenüber-
gänge s1→smBcryst, smBcryst→n und n→i in Abhängigkeit vom Druck
Abbildung 7.1: Allgemeine Strukturformel der untersuchten Aminoanthrachinonfarbstoffe
Abbildung 7.2: Strukturformel des C. I. Disperse Red 60
(1-Amino-4-hydroxy-2-phenoxy-9,10-anthrachinon)

Anhang
158
Abbildung 7.3: Strukturformel des C. I. Disperse Blue 60
Abbildung 7.4: DSC-Normaldruckthermogramm von iso-AC03
Abbildung 7.5: DSC-Normaldruckthermogramm von AC05
Abbildung 7.6: DSC-Normaldruckthermogramm von AC08
Abbildung 7.7: DSC-Normaldruckthermogramm von DBlue60
Abbildung 7.8: DSC-Normaldruckthermogramm von AC03
Abbildung 7.9: T(p)-Phasendiagramm von AC03 (offene Zellen; Druckgase: Argon, CO2;
Heizrate: 2 K⋅min-1)
Abbildung 7.10: Molare Phasenumwandlungsenthalpien von AC03 für den
Phasenübergang s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
Abbildung 7.11: Molare Phasenumwandlungsentropien von AC03 für den
Phasenübergang s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
Abbildung 7.12: Molare Phasenumwandlungsvolumina von AC03 für den
Phasenübergang s→i in Abhängigkeit vom Druck (offene Zelle; Argon)
Abbildung 7.13: DTA-Thermogramme von AC04 bei Drücken von p = 0.1 MPa,
p = 102.5 MPa und p = 225 MPa in geschlossener und verlöteter Bleizelle
Abbildung 7.14: DTA-Thermogramme von AC04 bei Drücken von p = 37 MPa, p = 60
MPa, p = 91 MPa und p = 143 MPa in offenen Zellen unter
Argonatmosphäre
Abbildung 7.15: T(p)-Phasendiagramm von AC04, a) geschlossene und verlötete Bleizelle,
b) offene Zelle (Druckgas: Argon)
Abbildung 7.16: Abgeschätzter Molenbruch x l2 und Massenbruch w l2 für Argon in der
isotrop flüssigen Phase von AC04
Abbildung 8.1: Definition und Begriffe für die Beschreibung einer Meßkurve zur
Bestimmung der Wärmekapazität einer Probe
Abbildung 8.2: Aufheiz- und Abkühlthermogramm der Probensubstanz SCT-1-Lotion bei
Normaldruck (Heizrate: 1 K⋅min-1, Kühlrate: 5 K⋅min-1)
Abbildung 8.3: Wärmestromkalibrierung und Wärmekapazitätsmessung (SCT-1-Lotion)
a) im Temperaturbereich von 25°C bis 75°C
b) im Temperaturbereich von 55°C bis 105°C
Abbildung 8.4: Wärmekapazität Cp(T) von SCT-1-Lotion im Temperaturbereich von
25°C bis 105°C

Anhang
159
A2 Verzeichnis der Tabellen
Tabelle 3.1: Kalibriersubstanzen
Tabelle 4.1: Meßwerte der Kalibrierung
Tabelle 4.2: Vergleich der über die Korrelationsfunktion ermittelten Umwandlungs-
temperaturen mit den Literaturwerten
Tabelle 5.1: Umwandlungstemperaturen der Modifikationen von AAA unter Normaldruck
Tabelle 5.2: Ergebnisse der Hochdruck-DSC-Messungen für Triarachidin in offenen
Meßzellen (Druckgas: Helium; Heizrate 1 K⋅min-1)
Tabelle 5.3: Umwandlungsenthalpien der Modifikationen von Triarachidin bei Normaldruck
(mit DSC ermittelt)
Tabelle 5.4: Umwandlungsdaten des Phasenüberganges α→β von Triarachidin
Tabelle 5.5: Umwandlungsdaten des Phasenüberganges β→l von Triarachidin
Tabelle 6.1: Literaturwerte der Phasenumwandlungen des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-4-nitro-benzylesters ( a) mit DSC ermittelt, b) mit DTA ermittelt)
Tabelle 6.2: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-4-nitro-benzylesters
Tabelle 6.3: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-4-cyan-benzylesters
Tabelle 6.4: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-2-cyan-benzylesters
Tabelle 6.5: Thermodynamische Umwandlungsdaten des 2,5-Bis(4-n-octyloxy-benzoyloxy)-
benzoesäure-3-cyan-benzylesters
Tabelle 6.6: Ergebnisse der Hochdruck-DTA-Messungen des 2,5-Bis(4-n-octyloxy-
benzoyloxy)benzoesäure-3-cyan-benzylesters (Druckgas: Argon; Heizrate
2 K⋅min-1)
Tabelle 6.7: Literaturwerte der Phasenumwandlungen des 6-TPEB (a) DTA, b) DSC)
Tabelle 6.8: Thermodynamische Umwandlungsdaten des 6-TPEB
Tabelle 6.9: Umwandlungstemperaturen des 10-TPEB bei Normaldruck
Tabelle 6.10: Ergebnisse der Hochdruck-DSC-Messungen für 10-TPEB in geschlossenen
Meßzellen (Druckgas: Argon; Heizrate 2 K⋅min-1)
Tabelle 6.11: Koeffizienten der Polynome Ttrs / K = a + b⋅(p/MPa) + c⋅(p/MPa)2

Anhang
160
Tabelle 6.12: Thermodynamische Umwandlungsdaten des 10-TPEB
Tabelle 7.1: Reinheit der verwendeten Farbstoffe
Tabelle 7.2: Zusammenfassung der thermodynamischen Umwandlungsdaten der
untersuchten Anthrachinonfarbstoffe bei Normaldruck
Tabelle 7.3: Selbst gemessene Schmelzpunkte im Vergleich mit Literaturwerten
Tabelle 7.4: Ergebnisse der Hochdruck-DSC-Messungen für AC03 in offenen Meßzellen
(Druckgase: Argon, CO2; Heizrate: 2 K⋅min-1)
Tabelle 7.5: Thermodynamische Umwandlungsdaten für den Phasenübergang s→i von AC03,
ermittelt in offener Zelle (Druckgas: Argon)
Tabelle 7.6: Ergebnisse der Hochdruck-DTA-Messungen für AC04 in geschlossenen und
offenen Meßzellen (Druckgas: Argon; Heizrate 2 K⋅min-1)
Tabelle 8.1: Literaturwerte

Anhang
161
A3 Verzeichnis der Lieferfirmen
[L 1] Bodenseewerk Perkin-Elmer & Co. GmbH, Überlingen
[L 2] American Instruments Company, Silver Spring, Maryland, USA
[L 3] Messer Griesheim, Düsseldorf
[L 4] Heise Bourdon Tube Co., Newtown, Connecticut, USA
[L 5] Haake Thermostate, Karlsruhe
[L 6] Burster Präzisionsmeßtechnik, Gernsbach
[L 7] Keithley Instruments GmbH, München
[L 8] Rhothron GmbH, Homburg/Saar
[L 9] Atari Corp. (Deutschland) GmbH, Raunheim
[L 10] Star Micronics, Deutschland GmbH, Frankfurt
[L 11] Uhu Vertrieb GmbH, München
[L 12] Th. Goldschmidt AG, Essen
[L 13] Nova Werke AG, Effretikon, Schweiz
[L 14] Preussag Metall, Goslar
[L 15] Wagner + Munz, München
[L 16] Merck AG, Darmstadt
[L 17] Sigma Chemical Co., St. Louis, USA
[L 18] Sigma-Aldrich Chemie GmbH, Steinheim
[L 19] LG Chemical Company, Seoul, Korea
[L 20] Procter & Gamble GmbH, Schwalbach

Anhang
162
A4 Literaturverzeichnis
[1] N. G. Parsonage, L. A. K. Staveley; “Disorder in Crystals“, Clarendon Press, Oxford
(1978)
[2] J. N. Sherwood (Ed.); “The Plastically Crystalline State“, Wiley & Sons, New York
(1979)
[3] D. Schmid; Chemiker-Zeitung, 99, 12 (1975)
[4] G. W. Gray, P. A. Winsor; “Liquid Crystals and Plastic Crystals“, Vol. I und II, Ellis
Horwood Limited, Chichester (1974)
[5] J. G. Aston in D. Fox, M. Labes, A. Weissgerber; Physics and Chemistry of the
Organic Solid State, Vol. I, Interscience Publishers, 77 (1963)
[6] G. M. Schneider; Thermochim. Acta, 88, 159 (1985)
[7] J. Timmermans; Bull. Soc. Chim. Belges., 44, 17 (1935)
[8] J. Timmermans; J. Chim. Phys., 35, 33 (1938)
[9] J. Timmermans; J. Phys. Chem. Solids, 18, 1 (1961)
[10] U. Wenzel; Dissertation, Ruhr-Universität Bochum, (1986)
[11] U. Wenzel, G. M. Schneider; Mol. Cryst. Liq. Cryst. (Lett.), 72, 25 (1982)
[12] R. Sandrock, U. Wenzel, H. Arntz, G. M. Schneider; Thermochim. Acta, 49, 23 (1981)
[13] H. Arntz, G. M. Schneider; Faraday Diss., 69, 139 (1980)
[14] R. Sandrock; Dissertation, Ruhr-Universität Bochum, (1982)
[15] D. Schmid, U. Wannagat; Chemiker-Zeitung, 98, 575 (1974)
[16] W. J. Dunning; J. Phys. Chem. Solids, 18, 21 (1961)
[17] I. Nitta; Z. Krist., 112, 234 (1959)
[18] B. Post, R. S. Schwarz, I. Fankuchen; J. Am. Chem. Soc., 73, 5113 (1951)
[19] K. J. Tauer, W. N. Lipscomb; Acta Cryst., 5, 606 (1952)
[20] F. Reinitzer; Monatsh. Chem. 9, 421 (1888)
[21] O. Lehmann; Z. Physik. Chem. 4, 462 (1889)
[22] H. Kelker; Mol. Cryst. Liq. Cryst., 21, 1 (1973)
[23] H. Kelker, R. Hatz; Handbook of Liquid Crystals, Verlag Chemie, Weinheim (1980)
[24] A. Bartelt; Dissertation, Bochum, (1988)
[25] P. S. Pershan; Phys. Today, 35, 34 (1982)

Anhang
163
[26] W. Helfrich, G. Heppke; “Liquid Crystals of One- and Two-Dimensional Order“,
Springer Verlag, Berlin, Heidelberg, New York (1980)
[27] A. Cifferi, W. R. Krigbaum, R. B. Meyer; Polymer Liquid Crystals, Academic Press,
London (1982)
[28] E. T. Samulski; Phys. Today, 35, 40 (1982)
[29] S. Chandrasekhar, B. K. Sadashiva, B. K. Suresh; Pramana 9, 471 (1977)
[30] S. Chandrasekhar in G. H. Brown: Advances in Liquid Crystals, Vol. 5, Academic
Press, London, (1982)
[31] C. Destrade, P. Foucher, H. Gasparoux, Nguyen Huu Tinh, A. M. Levelut,
J. Malthete; Mol. Cryst. Liq. Cryst., 106, 121 (1984)
[32] H. Zimmermann, R. Poupko, Z. Luz, J. Billard; Z. Naturforsch., A40, 149 (1985)
[33] J. Malthète, A. Collet; Nouv. J. Chim., 9, 151 (1985)
[34] J. Malthète, A. M. Levelut, Nguyen Huu Tinh; J. Phys. Lett., 46, 875 (1985)
[35] G. Friedel; Ann. Phys. (Paris); 18, 273 (1922)
[36] W. H. de Jeu: “Introduction to Thermotropic Liquid Crystals“, in S. Martelucci, A. N.
Chester; “Phase Transitions in Liquid Crystals“, Plenum Press, New York, London,
(1992)
[37] J. Thoen; Int. J. Mod. Phys., 9, 2157 (1995)
[38] R. G. Horn, T. E. Faber; Proc. Roy. Soc. London, A 368, 199 (1994)
[39] R. Demus; Z. Chem., 26, 6 (1986)
[40] W. Maier, A. Saupe; Z. Naturforsch., A 14, 882 (1959)
[41] H. Stegemeyer, Guest Ed.: Liquid Crystals, Steinkopf: Darmstadt, Springer:
New York, (1994)
[42] S. Diele, H. Sackmann; Ber. Bunsenges. Phys. Chem., 93, 467-477 (1989)
[43] D. Demus, S. Diele, S. Grande, S. Sackmann in G. H. Brown (ed.): Advances in
Liquid Crystals, Vol. 6, Academic Press, London (1983)
[44] A. Adamczyk; Liquid and Solid State Crystals, 2, 1845 (1992)
[45] P.E. Cladis; Phys. Rev. Lett., 35, 48 (1975)
[46] P.E. Cladis, R. K. Borgadus, W. B. Daniels, G. N. Taylor; Phys. Rev. Lett., 39, 720
(1977)
[47] F. Hardouin, G. Sigaud, M. F. Achard, H. Gasparoux; Phys. Lett., A 71, 347 (1979)
[48] R. Shashidhar, S. Somasekhar, B. R. Ratna, Mol. Cryst. Liq. Cryst., 133, 19 (1986)

Anhang
164
[49] P.E. Cladis; Mol. Cryst. Liq. Cryst., 165, 85 (1988)
[50] S. Chandrasekhar, R. Shashidhar in G. H. Brown (ed.): Advances in Liquid Crystals,
Vol. 4, Academic Press, London, (1979)
[51] J. Hermann; Dissertation, Bochum, (1982)
[52] M. L. McGlashan; Chemical Thermodynamics, Academic Press, London 1979
[53] G. M. Schneider; Grundlagen der chemischen Thermodynamik,
aus Ullmanns Encyklopädie der technischen Chemie, 4. Aufl., Bd. 1, 2, Verlag Chemie,
Weinheim (1972)
[54] M. K. Rittmeier-Kettner; Dissertation, Ruhr-Universität Bochum, (1996)
[55] P. Ehrenfest; Proc. Acad. Sci. Amst. 36, 153 (1933)
[56] P. Navard, J. M. Haudin; J. Thermal Anal., 29, 405 (1984) ibid. 29, 415 (1984)
[57] P. Navard, R. Cox; Mol. Cryst. Liq. Cryst. Lett. 102, 261 (1984) ibid. 102, 265 (1984)
[58] Skript zum anorganisch-chemischen F-Praktikum, Ruhr-Universität Bochum, (1993)
[59] J. Ellert; Dissertation, Ruhr-Universität Bochum, (1991)
[60] C. Schmidt; Diplomarbeit, Ruhr-Universität Bochum, (1991)
[61] C. Schmidt, M. Rittmeier-Kettner, H. Becker, J. Ellert, R. Krombach and
G. M. Schneider; Thermochim. Acta, 238, 321-336 (1994)
[62] J. Wilmers; Dissertation, Ruhr-Universität Bochum, (1990)
[63] C. Schmidt; Dissertation, Ruhr-Universität Bochum, (1997)
[64] H. Arntz; Dissertation, Ruhr-Universität Bochum, (1980)
[65] C. R. Ernst; Dissertation, Ruhr-Universität Bochum, (1997)
[66] F. Hamann; Dissertation, Ruhr-Universität Bochum, (1998)
[67] Schulze; “Differentialthermoanalyse“, 2. Auflage, Verlag Chemie, Weinheim (1972)
[68] W. W. Wendlandt; “Thermal Methods of Analysis“, John Wiley & Sons, New York
(1964)
[69] E. Pella, M. Nebuloni; J. Thermal Anal., 3, 292 (1971)
[70] J. L. McNaughton, C. T. Mortimer; “Differential Scanning Calorimetry“ in H. A.
Skinner; “Physical Chemistry“, Ser. 2, Vol. 10, Butterworths London (1975)
[71] Perkin-Elmer Corp.; Thermal Analysis News Letter No. 5, Norwalk Conn. (1970)
[72] G. W. H. Höhne, W. Dollkopf, P. U. Mayr; Thermochim. Acta, 273, 17-24 (1996)
[73] J. Reuter; Dissertation, Ruhr-Universität Bochum, (1996)
[74] W. F. Hemminger, S. M. Sarge; J. Thermal Anal., 37, 1455 (1991)

Anhang
165
[75] W. F. Hemminger, H. K. Cammenga; Methoden der Thermischen Analyse, Springer
Verlag: Berlin, Heidelberg (1989)
[76] W. P. Brennan, B. Miller, J. C. Whitwell; Ind. Eng. Chem. Fund., 8, 314 (1969)
[77] G. Xiang, L. Yunliang; Thermochim. Acta, 150, 53 (1989)
[78] U. Bandara; J. Thermal Anal., 31, 1063 (1986)
[79] P. W. Bridgman; Phys. Rev., 48, 893 (1935)
[80] J. D. Dudley, H. T. Hall; Phys. Rev., 118, 1211 (1960)
[81] Handbook of Chemistry and Physics, 74th Edition, CRC-Press, (1993/94)
[82] A. Roth, J. Meyer, H. Zenner; Z. anorg. allg. Chem., 214, 309 (1933)
[83] V. E. Fadeev; Russ. J. Phys. Chem., 45, 1051 (1971)
[84] Landolt-Börnstein, Neue Serie, Gruppe III, Bd. 6, S. 13
[85] Perkin-Elmer, DSC-2 Handbuch, (1974)
[86] J. E. Callanan, S. A. Sullivan; Rev. Sci. Instrum., 57, 2584 (1986)
[87] G. Lombardi; For Better Thermal Analysis, 2nd ed.; published by the International
Confederation for Thermal Analysis (1980)
[88] Versuchsanleitung: Praktikumsversuch F2, Phys. Chem. Praktikum, Ruhr-Universität
Bochum
[89] A. Hamm, Dissertation, Ruhr-Universität Bochum, (1989)
[90] A. Würflinger; Dissertation, Ruhr-Universität Bochum, (1972)
[91] R. Krombach; Dissertation, Ruhr-Universität Bochum, (1991)
[92] R. Krombach, G. M. Schneider; Thermochim. Acta; 231, 169 (1994)
[93] H. Becker; Dissertation, Ruhr-Universität Bochum, (1996)
[94] M. L. McDaniel, S. E. Bablo jr., G. J. Scott; J. Chem. Phys., 37, 822 (1962)
[95] A. Bartelt; Dissertation, Ruhr-Universität Bochum, (1988)
[96] P. E. Cladis, R. K. Borgadus, W. B. Daniels, G. N. Taylor; Phys. Rev. Lett., 39, 720
(1977)
[97] A. Jayaraman, W. Klement jr., R. C. Newton, G. C. Kennedy; J. Phys. Chem. Solids,
24, 7 (1963)
[98] W. Heintz; Jahresber., 2, 342 (1849)
[99] P. Duffy; J. Chem. Soc., 5, 197 (1853)
[100] T. Malkin, C. E. Clarkson; J. Chem. Soc., 666 (1934)
[101] D. Chapman; Chem. Rev., 62, 433 (1962)

Anhang
166
[102] K. Larsson; Arkiv Kemi, 23, 35 (1964)
[103] J. W. Hagemann, J. A. Rothfus; J. Am. Oil Chem. Soc., 60, 1123 (1983)
[104] K. Sato et al.; J. Am. Oil Chem. Soc., 66, 664 (1989)
[105] L. Hernquist, K. Larrson; Fette-Seifen-Anstrichmittel, 84, 349 (1982)
[106] M. Ollivon, R. Perron; Thermochim. Acta, 53, 183 (1982)
[107] M. Ollivon, R. Perron; Dev. Food Sci., 11, 107 (1985)
[108] L. H. Wesdorp; “Liquid-Multiple Solid Phase Equilibria in Fats“, Dissertation,
Technische Universität Delft, (1990)
[109] A. Thomas; “Fette und Öle“, Ullmanns Encyklop., 4. Aufl., 11, 455 (1976)
[110] B. Wagner; Diplomarbeit, Ruhr-Universität Bochum, (1994)
[111] B. Wagner, G. M. Schneider; Thermochim. Acta, 274, 179 (1996)
[112] E. S. Lutton; J. Am. Oil Chem. Soc., 27, 276 (1950)
[113] K. Larsson; Proc. Chem. Soc., 87 (1963)
[114] L. H. Jensen, A. J. Mabis; Nature 197, 681 (1963)
[115] G. Strobl, B. Ewen, E. W. Fischer, W. Piesczek; J. Chem. Phys., 61, 5257 (1974)
[116] W. Piesczek, G. Strobl, K. Malzahn; Acta Crys., 30, 1278 (1974)
[117] J. W. Hagemann, J. A. Rothfus; J. Am. Oil Chem. Soc., 60, 1308 (1983)
[118] J. W. Hagemann, J. A. Rothfus; J. Am. Oil Chem. Soc., 65, 638 (1988)
[119] J. W. Hagemann, W. H. Tallent; J. Am. Oil Chem. Soc., 49, 118 (1972)
[120] T. D. Simpson, J. W. Hagemann; J. Am. Oil Chem. Soc., 59, 118 (1982)
[121] M. Kellens, W. Meussen, C. Riekel, H. Reynaers; Chem. Phys. Lipids, 52, 79 (1990)
[122] K. Larsson; Fette-Seifen-Anstrichmittel, 74, 136 (1972)
[123] L. Hernquist; Fette-Seifen-Anstrichmittel, 86, 297 (1984)
[124] C. Schmidt, C. Ernst, S. Masberg, C. Meyer-Inci, H. Becker, G. M. Schneider; Polish
J. Chem., 71, 1742 (1997)
[125] W. Weißflog; Liq. Cryst., 3(2), 275 (1988)
[126] S. Diele, W. Weißflog, G. Pelzl, H. Manke, D. Demus; Liq. Cryst., 1, 101 (1986)
[127] S. Haddawi, S. Diele, H. Kresse, W. Weißflog; Cryst. Res. Tech., 29(5), 745 (1994)
[128] J. Jadzyn, C. Legrand, N. Isaert, A. Cartier, P. Bonnet, G. Czechowski, B. Zywucki;
J. Mol. Liq., 62, 53 (1994)
[129] J. Jadzyn, G. Czechowski, B. Zywucki, C. Legrand, P. Bonnet, R. Dabrowski;
Z. Naturforsch., 48 a, 871 (1993)

Anhang
167
[130] S. Masberg, C. Ernst, G. M. Schneider, A. Würflinger, R. Dabrowski;
Z. Naturforsch., 54 a, 287 (1999)
[131] C. Ernst, G. M. Schneider, A. Würflinger, J. Jadzyn, R. Dabrowski; Z. Naturforsch.,
52 a, 490 (1997)
[132] B. Wagner; Dissertation, Ruhr-Universität Bochum, (1998)
[133] P. Swidersky; Dissertation, Ruhr-Universität Bochum, (1994)
[134] Aldrich, Katalog: Handbuch Feinchemikalien (1994/95)
[135] K. Yoshida, M. Matsuoka, T. Ueyama, Y. Yamashita, T. Kitao; Chem. Lett., 765
(1978)
[136] H. Falk, N. Müller, M. Oberreiter; Monatsh. Chem., 125, 313 (1994)
[137] M. S. Simon; J. Am. Chem. Soc., 85, 1974 (1963)
[138] K. C. Murdock, R. G. Child, P. F. Fabio, R. B. Angier, R. E. Wallace, F. E. Durr,
R. V. Citarella; J. Med. Chem., 22, 1024 (1979)
[139] Y. Suda, F. Nakajima, H. Ujigawa; Kogyo Kagaku Zasshi, 72, 898 (1969);
zitiert in „Beilstein Datenbank“: Chem. Abstr., 71, 71896 (1969)
[140] C. Kautz; Dissertation, Ruhr-Universität Bochum, (1996)
[141] D. Tuma, G. M. Schneider; Wissenschaftliche Berichte FZKA, 6271, 155-158 (1999)
[142] D. Tuma, G. M. Schneider; Fluid Phase Equilibria, 158-160, 743-757 (1999)
[143] J. Cognard, T. Hieu Phan; Mol. Cryst. Liq. Cryst., 70, 1-19 (1981)
[144] U. Haarhaus; Dissertation, Ruhr-Universität Bochum, (1992)
[145] G. W. H. Höhne, H. K. Cammenga, W. Eysel, E. Gmelin, W. Hemminger;
Thermochim. Acta, 160, 1 (1990)
[146] H. K. Cammenga et.al.; Thermochim. Acta, 219, 333-342, (1993)
[147] S. M. Sarge et.al.; Thermochim. Acta, 247, 129-168 (1994)
[148] G. Pelzl, S. Diele, W. Weissflog; Adv. Mater., 11(9), 707 (1999)
[149] S. L. Randzio; Annual Reports, Physical Chemistry, Vol. 94 C, 433 (1998)
[150] W. Weissflog; mündliche Mitteilung, (Oktober 1999)
[151] B. Wunderlich, A. Boller, I. Okazaki, K. Ishikiriyama; Thermochim. Acta,
304/305, 125-136 (1997)
[152] I. Czarnota; Meas. Sci. Technol., 5, 1345-1349 (1994)

Danksagung
Mein besonders herzlicher Dank gilt Herrn Prof. Dr. G. M. Schneider für die Möglichkeit zur
Durchführung dieser Arbeit, die interessante Aufgabenstellung, die jederzeit gewährte
Unterstützung und Betreuung sowie für viele wertvolle Anregungen und Diskussionen.
Herrn Prof. Dr. A. Würflinger danke ich herzlich für die Übernahme des Korreferats und für
sein Interesse an dieser Arbeit.
Herrn Prof. Dr. H.-J. Götze danke ich für seine Bereitschaft, in der Disputation als Drittprüfer
zu fungieren.
Frau Prof. Dr. M. Havenith-Newen, Herrn Prof. Dr. H. Weingärtner und Herrn Prof. Dr. C.
Wöll gilt mein Dank für die großzügige finanzielle Unterstützung.
Für das gute Arbeitsklima und die freundschaftliche Zusammenarbeit danke ich Herrn Dr. U.
Klask, Herrn Dr. D. Klante und Herrn Dr. C. R. Ernst. Weiterhin gilt mein Dank Herrn Dr. B.
Wagner, Herrn Dr. D. Tuma und anderen hier nicht namentlich genannten Kollegen für ihre
stete Bereitschaft zum Gespräch und ihre großzügige Hilfestellung.
Für die stets gewährte Unterstützung bei der Behebung apparativer Probleme danke ich Herrn
R. Renzewitz und Herrn D. Rösener.
Frau I. Mildt, Herrn H. Fahnenschmidt und Herrn J. Masuch danke ich für ihre vielfältige
Unterstützung während des Entstehens dieser Arbeit.
Allen Mitarbeiterinnen und Mitarbeitern des Lehrstuhls für Physikalische Chemie II danke ich
für die freundliche Atmosphäre. Des weiteren danke ich allen, die zum Gelingen dieser Arbeit
beigetragen haben.

Lebenslauf
Persönliche Daten
Name: Stefan Masberg
Geburtsdatum: 09.10.1969
Geburtsort: Bottrop
Familienstand: ledig
Ausbildung
8/1976 - 7/1980 Grundschule in Bottrop
8/1980 - 5/1989 Josef-Albers-Gymnasium in Bottrop
12.05.1989 Abitur
6/1989 - 8/1990 Grundwehrdienst
WS 1990/91 Beginn des Chemiestudiums an der Ruhr-Universität Bochum
30.03.1993 Vordiplom
30.05.1996 Diplom
seit 7/1996 Promotion am Lehrstuhl für Physikalische Chemie II der
Ruhr-Universität Bochum
Arbeitsverhältnisse
11/1995 - 6/1996 studentische Hilfskraft
7/1996 - 9/1996 wissenschaftliche Hilfskraft am Lehrstuhl für
10/1996 - 8/1998 wissenschaftlicher Mitarbeiter Physikalische Chemie II
9/1998 - 3/1999 wissenschaftliche Hilfskraft der Ruhr-Universität Bochum
seit 4/1999 wissenschaftlicher Mitarbeiter