
Empfehlungen zu Diagnostik, Therapie und ... · 2 Basaliom 2.1 Epidemiologie 2.2 Klinik und...
62
Hauttumore Empfehlungen zu Diagnostik, Therapie und Nachsorgeuntersuchungen in Tirol
Transcript of Empfehlungen zu Diagnostik, Therapie und ... · 2 Basaliom 2.1 Epidemiologie 2.2 Klinik und...

HauttumoreEmpfehlungen zu Diagnostik, Therapie und
Nachsorgeuntersuchungen in Tirol


„Hautkrebs“, Melanome, Non-melanoma-skin cancer der Haut und Schleim-häute zusammengefasst, repräsentiert die häufigste maligne Neoplasie des Menschen und die Tendenz dieser Entitäten ist steigend.
Während Basaliome und Plattenepithelkarzinome besonders häufig in der Bevölkerung sind, besteht beim fortgeschrittenen Melanom eine frühe Tendenz zur Metastasierung und damit eine ungünstige Prognose. Macht die Anzahl der Melanome zwar nur etwa 5-10% aller Hauttumore aus, so ist diese Tumorerkrankung doch für mehr als 90% aller Todesfälle an Hautkrebs verant wortlich.
In den frühen Stadien ist Hautkrebs gut behandelbar. Früherkennungs- und Präventionprogramme sind überaus erfolgreich („Sonne ohne Reue“). Waren bis vor ein paar Jahren die Therapieoptionen für fortgeschrittene Hautkrebs-erkrankungen äußerst beschränkt und die Wirksamkeit bescheiden, so geben uns die letzten Jahre Hoffnung, diese Erkrankungen besser behandeln zu können. Dies ist vor allem den sogenannten „Targeted Therapies“ und Immuntherapien mit Checkpointinhibitoren zu verdan ken, die es uns erlauben, unsere Patienten individueller und effektiver zu behandeln.
Die Arbeitsgruppe „Melanom und dermatologische Onkologie“ der Öster-reich ischen Gesellschaft für Dermatologie und Venerologie (ÖGDV) orientiert sich in ihren Empfehlungen an den deutschsprachigen und internationalen Richtlinien. Internationale Leitlinien haben immer den Nachteil, dass sie einen Kompromiss, einen „kleinsten gemeinsamen Nenner“, darstellen, da sie auf lokale Gegebenheiten, Ressourcen und sozialversicherungstechnische, spezifische Gesetze Rücksicht nehmen müssen. Aber auch nationale oder lokale Leitlinien stellen lediglich Empfehlungen dar, die keinen Gesetzes-charakter haben können. Sie sind für Ärzte unverbindlich und haben weder haftungs begründende noch haftungsbefreiende Wirkung. Die letztendliche Entscheidung und Verantwortung, wie der onkologische Patient zu behandeln ist, verbleibt somit beim betreuenden Arzt.
Es wurde unsererseits versucht, die Kapitel knapp und möglichst übersichtlich zu strukturieren, sodass der dermato-onkologisch tätige Arzt rasch die geeigneten Informationen für Diagnostik und Therapie erhalten kann. Wir hoffen, dass unsere an die Tiroler Gegebenheiten regional angepasste Synthese geltender Richtlinien den PatientInnen zugutekommt.
Prof. Georg Weinlich Prof. Van Anh Nguyen
Hauttumore Vorwort 3
Vorwort

Obmann1. Obmann-Stv.2. Obmann-Stv.
Schriftführer1. Schriftführer-Stv.2. Schriftführer-Stv.
KassierÄrztekammervertreter
kooptiert
Koordinator
4 Hauttumore TAKO Vorstand und Mitwirkende
TAKOVorstand und Mitwirkende
TAKO VorstandUniv.-Prof. DI Dr. Peter Lukas Strahlentherapie-Radioonkologie, InnsbruckN.N. Univ.-Prof. Dr. Günther Gastl Hämatologie & Onkologie, InnsbruckPrim. ao. Univ-Prof. Dr. Ewald Wöll Innere Medizin, Zamsao. Univ.-Prof. Dr. Wolfgang Eisterer Hämatologie & Onkologie, Klagenfurtao. Univ.-Prof. Dr. Reinhard Stauder Hämatologie & Onkologie, InnsbruckUniv.-Doz. Dr. Eberhard Gunsilius Hämatologie & Onkologie, InnsbruckDr. Stefan Kastner Ärztekammer bzw. Chirurgie, Zams
Prim. Univ.-Prof. Dr. med. Klaus Gattringer
Mitwirkende in der Arbeitsgruppe HauttumoreAo. Univ.-Prof. Dr. Georg Weinlich Dermatologie & Venerologie, Innsbruck Ao. Univ.-Prof. Dr. Van Anh Nguyen Dermatologie & Venerologie, Innsbruck Dr. Arpad Sztankay Strahlentherapie-Radioonkologie, Innsbruck Univ.-Prof. Dr. Gerhard Pierer Plastische-, Rekonstruktive- und Ästhetische Chirurgie InnsbruckUniv.-Prof. Dr. Dr. Michael Rasse Zahn-, Mund- & Kieferheilkunde, InnsbruckUniv.-Prof. Dr. Herbert Riechelmann HNO, Innsbruck Univ.-Prof. Dr. Johann Pratschke Chirurgie, Innsbruck Univ.-Doz. Dr. Eberhard Gunsilius Hämatologie, InnsbruckAo. Univ.-Prof. Dr. Reto Bale Radiologie, InnsbruckDr. Renate Frank Radiologie, Innsbruck
Satz, Gestaltung und VersionenDr. Eugen Preuß pdl, InnsbruckVersion 1.0 2012Version 2.0 2016Copyright: pdl 2016

Hauttumore Inhaltsverzeichnis 5
7
1011
1314
151617181920
2425
29
30
3132
33
35
37
4142
1 Melanom1.1 Epidemiologie1.2 Klinische und histopathologische Subtypen 1.3 Diagnostik1.3.1 Prognose und Staging1.3.2 Histopathologische Diagnostik1.3.3 Molekularbiologische Diagnostik1.3.4 Ausbreitungsdiagnostik1.4 Chirurgische Therapie1.4.1 Wächterlymphknotenbiopsie / Sentinel-lymphnode biopsy (SLNB)1.4.2 Operative Therapie in metastasierten Stadien1.5 Bestrahlung1.6 Adjuvante Therapie1.7 Systemische Therapie des metastasierenden Melanoms1.7.1 Zielgerichtete, molekulare Therapien1.7.2 Chemotherapie und Chemoimmuntherapie1.8 Nachsorge
2 Basaliom2.1 Epidemiologie2.2 Klinik und Histopathologie 2.3 Diagnostik2.4 Therapie 2.4.1 Chirurgie2.4.2 Nicht-chirurgische Therapien2.5 Nachsorge2.6 Therapie von Rezidiven2.7 Systemtherapien
3 Plattenepithelkarzinom3.1 Epidemiologie3.2 Subtypen und Histologie3.2.1 In situ Plattenepithelkarzinom3.2.2 Invasives Plattenepithelkarzinom3.3 Stadieneinteilung und Prognose3.4 Diagnostik3.5 Therapie3.6 Nachsorge3.7 Rehabilitation3.8 Psychosoziale Versorgung
Inhaltsverzeichnis

6 Hauttumore Inhaltsverzeichnis
43
444547
48
49
51
52
53
55
4 Merkelzellkarzinom4.1 Epidemiologie4.2 Klinik und Histopathologie4.3 Diagnostik4.4 Prognose und Staging4.5 Therapie4.5.1 Operative Therapie4.5.2 Strahlentherapie4.5.3 Chemotherapie4.5.4 Experimentelle Therapien4.6 Nachsorge
5 Dermatofibrosarkomaprotuberans5.1 Epidemiologie und Stadieneinteilung5.2 Klinik und Histopathologie5.3 Diagnostik5.4 Therapie 5.4.1 Operative Therapie5.4.2 Andere Therapieverfahren5.5 Nachsorge
6 Literatur
Hauttumore Melanom 7
Entstehung
Inzidenzsteigtstetig
MeistpolygeneVererbung
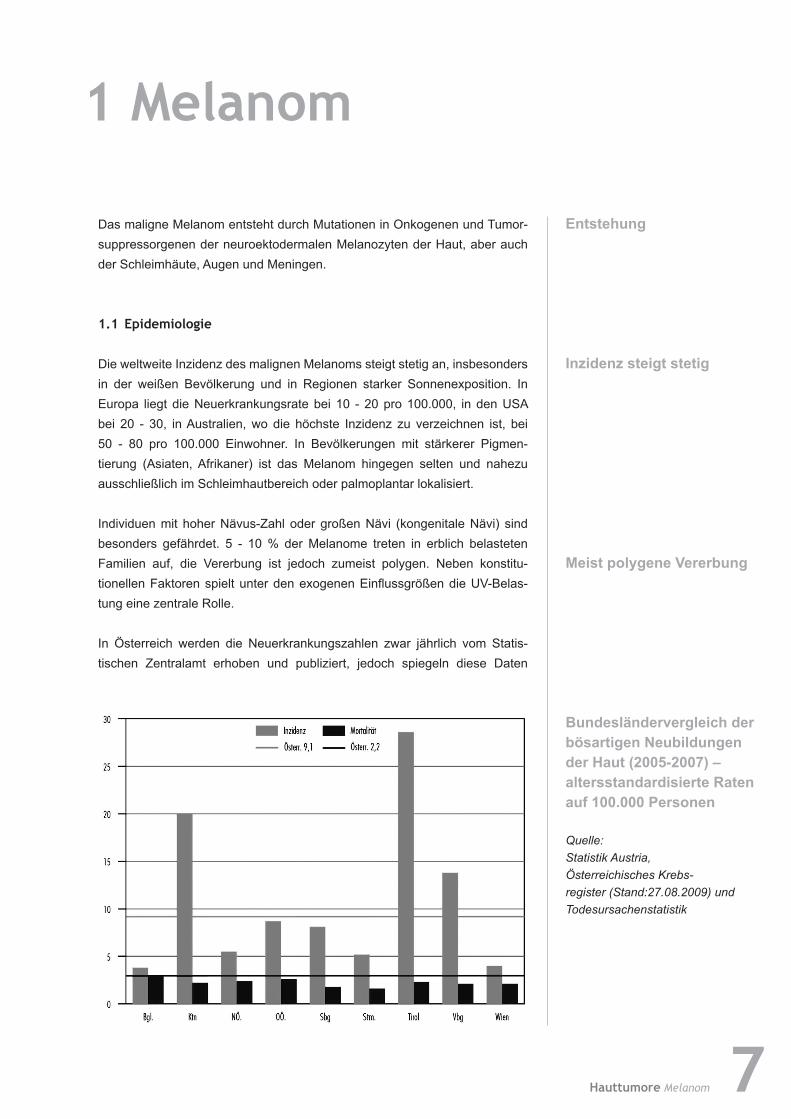
BundesländervergleichderbösartigenNeubildungenderHaut(2005-2007)–altersstandardisierte Raten auf100.000Personen
Quelle: Statistik Austria, Österreichisches Krebsregister (Stand:27.08.2009) und Todesursachenstatistik
1 Melanom
Das maligne Melanom entsteht durch Mutationen in Onkogenen und Tumor-suppressorgenen der neuroektodermalen Melanozyten der Haut, aber auch der Schleimhäute, Augen und Meningen.
1.1 Epidemiologie
Die weltweite Inzidenz des malignen Melanoms steigt stetig an, insbesonders in der weißen Bevölkerung und in Regionen starker Sonnenexposition. In Europa liegt die Neuerkrankungsrate bei 10 - 20 pro 100.000, in den USA bei 20 - 30, in Australien, wo die höchste Inzidenz zu verzeichnen ist, bei 50 - 80 pro 100.000 Einwohner. In Bevölkerungen mit stärkerer Pigmen-tierung (Asiaten, Afrikaner) ist das Melanom hingegen selten und nahezu aus schließ lich im Schleimhautbereich oder palmoplantar lokalisiert.
Individuen mit hoher Nävus-Zahl oder großen Nävi (kongenitale Nävi) sind besonders gefährdet. 5 - 10 % der Melanome treten in erblich belasteten Familien auf, die Vererbung ist jedoch zumeist polygen. Neben konstitu-tionellen Faktoren spielt unter den exogenen Einflussgrößen die UV-Belas-tung eine zentrale Rolle.
In Österreich werden die Neuerkrankungszahlen zwar jährlich vom Statis-tischen Zentralamt erhoben und publiziert, jedoch spiegeln diese Daten

8Hauttumore Melanom
Tiroler Daten
nach Meinung der Österreichischen Gesellschaft für Dermatologie und Venerologie (ÖGDV) nicht die wahre Situation wider, da das Meldungs-verhalten sehr unterschiedlich ist. So ist ein deutliches West-/Ostgefälle der Inzidenz zu verzeichnen, hingegen ist die Mortalitätsrate in allen Bundes-ländern etwa gleich; eine mögliche Erklärung ist die mangelnde Meldung von Neuerkrankungsfällen.
In Tirol erhebt das Tumorregister des Institutes für klinische Epidemiologie nicht nur passiv, sondern auch aktiv Tumordaten, sodass die Dunkelziffer an nicht bekannten Neuerkrankungsfällen relativ gering ist. Die Inzidenz des Melanoms steht bei Frauen in Tirol bereits an der 3. Stelle aller Tumor-erkran kungen, bei den Männern an der 5. Stelle, dies entspricht dem inter-nationalen Vergleich.
Im Jahre 2009 wurde bei rund 300 Personen in Tirol ein invasives Melanom diagnostiziert, dazu kommen weitere 250 nicht invasive in-situ-Melanome.
DiehäufigstenTumorlokalisationeninTirol2005-2009
Quelle: IET - Institut für klinische Epidemiologie der TILAK GmbH, „Inzidenz und Mortalität bösartiger Neubildungen in Tirol Diagnosejahr 2009‟
Frauen Männer
Hauttumore Melanom 9
Durchschnittsalterbei 53 Jahren
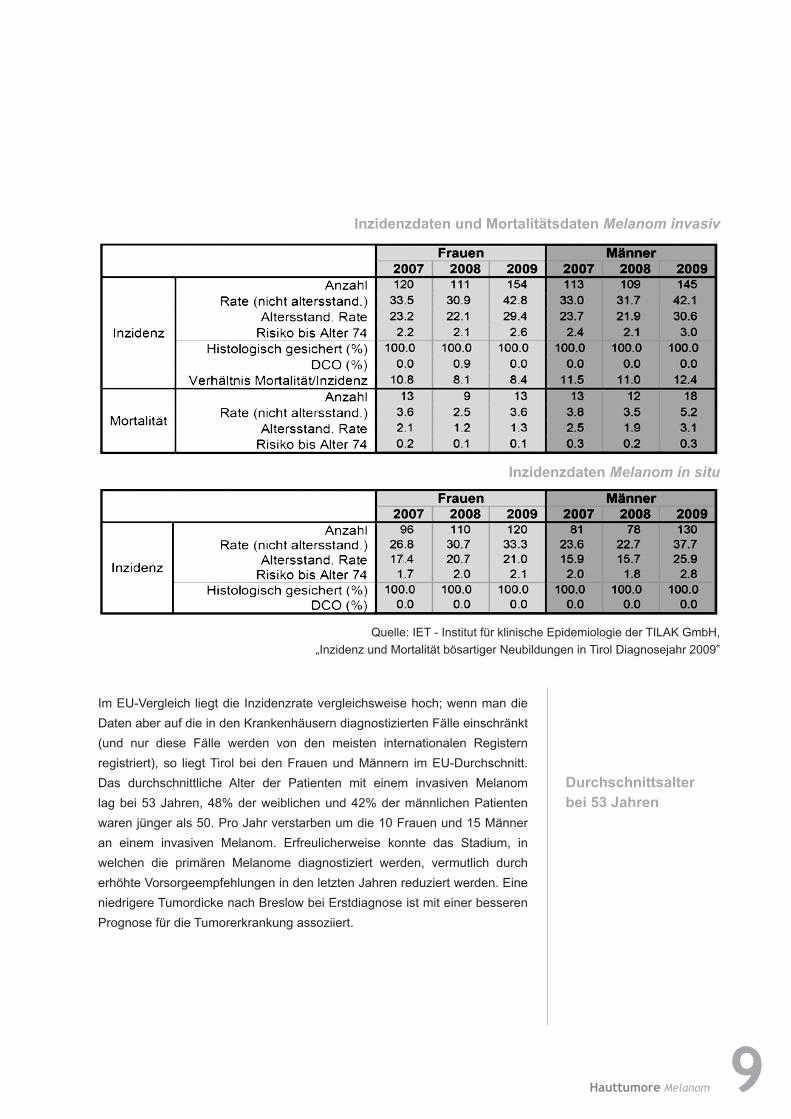
Im EU-Vergleich liegt die Inzidenzrate vergleichsweise hoch; wenn man die Daten aber auf die in den Krankenhäusern diagnostizierten Fälle einschränkt (und nur diese Fälle werden von den meisten internationalen Registern registriert), so liegt Tirol bei den Frauen und Männern im EU-Durchschnitt. Das durchschnittliche Alter der Patienten mit einem invasiven Melanom lag bei 53 Jahren, 48% der weiblichen und 42% der männlichen Patienten waren jünger als 50. Pro Jahr verstarben um die 10 Frauen und 15 Männer an einem invasiven Melanom. Erfreulicherweise konnte das Stadium, in welchen die primären Melanome diagnostiziert werden, vermutlich durch erhöhte Vorsorgeempfehlungen in den letzten Jahren reduziert werden. Eine niedrigere Tumordicke nach Breslow bei Erstdiagnose ist mit einer besseren Prognose für die Tumorerkrankung assoziiert.
InzidenzdatenundMortalitätsdatenMelanom invasiv
Inzidenzdaten Melanom in situ
Quelle: IET - Institut für klinische Epidemiologie der TILAK GmbH, „Inzidenz und Mortalität bösartiger Neubildungen in Tirol Diagnosejahr 2009‟
10Hauttumore Melanom
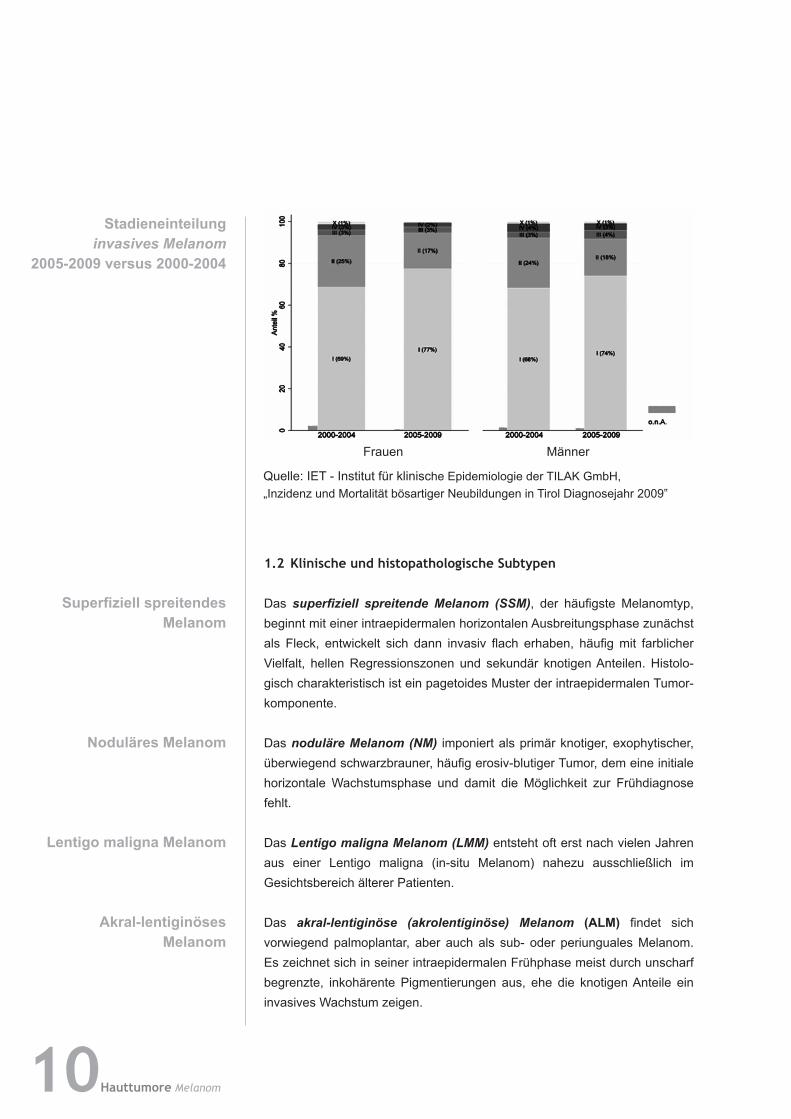
Stadieneinteilunginvasives Melanom
2005-2009versus2000-2004
Superfiziellspreitendes Melanom
NoduläresMelanom
LentigomalignaMelanom
Akral-lentiginöses Melanom
1.2 Klinische und histopathologische Subtypen
Das superfiziell spreitende Melanom (SSM), der häufigste Melanomtyp, beginnt mit einer intraepidermalen horizontalen Ausbreitungsphase zunächst als Fleck, entwickelt sich dann invasiv flach erhaben, häufig mit farb licher Viel falt, hellen Regressionszonen und sekundär knotigen Anteilen. Histo lo-gisch charakteristisch ist ein pagetoides Muster der intraepidermalen Tumor -komponente.
Das noduläre Melanom (NM) imponiert als primär knotiger, exophytischer, überwiegend schwarzbrauner, häufig erosiv-blutiger Tumor, dem eine initiale horizontale Wachstumsphase und damit die Möglichkeit zur Frühdiagnose fehlt.
Das Lentigo maligna Melanom (LMM) entsteht oft erst nach vielen Jahren aus einer Lentigo maligna (in-situ Melanom) nahezu ausschließlich im Gesichts bereich älterer Patienten.
Das akral-lentiginöse (akrolentiginöse) Melanom (ALM) findet sich vorwiegend palmoplantar, aber auch als sub- oder periunguales Melanom. Es zeichnet sich in seiner intraepidermalen Frühphase meist durch unscharf begrenzte, inkohärente Pigmentierungen aus, ehe die knotigen Anteile ein invasives Wachstum zeigen.
Quelle: IET - Institut für klinische Epidemiologie der TILAK GmbH, „Inzidenz und Mortalität bösartiger Neubildungen in Tirol Diagnosejahr 2009‟
Frauen Männer

Hauttumore Melanom 11
Mischformen
A-B-C-DRegel
Tumorspezifisches10-Jahres Überleben bei 75 - 80%
Einige Melanome sind jedoch nach diesem Schema nicht klassifizierbar oder repräsentieren Mischformen. Klinische Sonderformen sind z.B. amelano-tische Melanome, sowie Schleimhaut- oder andere extrakutane Melanome, die etwa 5 % aller Melanome ausmachen. Eine zunehmend wichtige Rolle übernimmt die molekularpathologische Klassifizierung der Melanome (siehe unten).
1.3 Diagnostik
Pigmentläsionen werden nach der sogenannten A-B-C-D Regel beurteilt:
A = Asymmetrie des AufbausB = Begrenzung unregelmäßigC = Colorit inhomogenD = Durchmesser > 5 mm
Zur differentialdiagnostischen Abklärung sollte in der Regel die Auflicht-mikroskopie (Dermatoskopie) herangezogen werden, welche die diagnos-tischen Treffsicherheit und damit Sensitivität und Spezifität des trainierten Befunders erhöhen.Mehrere dermatoskopische Scores zur Melanomdiagnose wurden entwickelt. Dazu zählen die modifizierte Musteranalyse, die ABCD-Regel der Dermato-skopie, die Bewertungsmethode nach Menzies oder die 7-Punkte-Checkliste. Eine Verlaufsbeobachtung ist mit der computergestützten Dermatoskopie möglich. Änderungen der Pigmentmale können so leichter erkannt und bewertet, stärker vergrößert und mit digitalen Bildanalyseprogrammen unter-sucht werden.
1.3.1 Prognose und StagingCa. 90 % aller malignen Melanome kommen derzeit als Primärtumor ohne erkenn bare Metastasierung zur ersten Diagnose. Die tumorspezifische 10-Jahres -Überlebensrate im Gesamtkollektiv beträgt ca. 75 - 80 %. Die wichtig sten prognostischen Faktoren beim primären malignen Melanom ohne Metastasen sind: • Die vertikale Tumordicke nach Breslow: TD < 1,0 mm: ca. 88 - 95 %
tumorspezifische 10-Jahres-Überlebensrate (10-JÜR); 1,01 - 2,0 mm: ca. 79 - 84 % 10-JÜR; 2,01 - 4,0 mm ca. 64 - 73 % 10-JÜR; > 4,0 mm: ca. 52 - 54% 10-JÜR.

12Hauttumore Melanom
Ulzeration
Mitoserate
Metastasierung
• Obige Werte gelten für Tumoren ohne Ulzeration, welche die Prognose verschlechtert. Das Vorhandensein einer histologisch erkennbaren Ulzeration führt daher in der aktuellen AJCC-Klassifikation zur Gruppierung in ein höheres Stadium.
• Eine Mitoserate ≥ 1/mm2 im histologischen Präparat• Der Nachweis von Mikrometastasierung in den regionären Lymphknoten
durch Wächterlymphknotenbiopsie (SLNB)
Weitere nicht signifikante Prognosefaktoren sind der Invasionslevel nach Clark, die Tumorlokalisation oder das Geschlecht.
Das maligne Melanom kann sowohl primär lymphogen als auch primär hämatogen metastasieren. Etwa 2/3 aller Erstmetastasierungen sind zunächst auf das regionäre Lymphabflußgebiet beschränkt.
Eine regionäre Metastasierung kann mit SatellitenMetastasen (bis 2 cm um den Primärtumor), IntransitMetastasen (in der Haut bis zur ersten Lymphknoten-Station) und regionären Lymphknotenmetastasen manifest werden. Die 10-Jahres-Überlebenswahrscheinlichkeit beträgt bei Patienten mit Satelliten- und In-transit-Metastasen ca. 30 - 50 % und bei Patienten mit klinisch manifesten regionären LK-Metastasen ca. 20 - 40 %.
Bei Fernmetastasierung ist die Prognose zumeist infaust, die mediane Überlebenszeit ohne Behandlung beträgt nur ca. 6 - 9 Monate, wobei je nach Organbefall eine erhebliche Variationsbreite vorliegt.
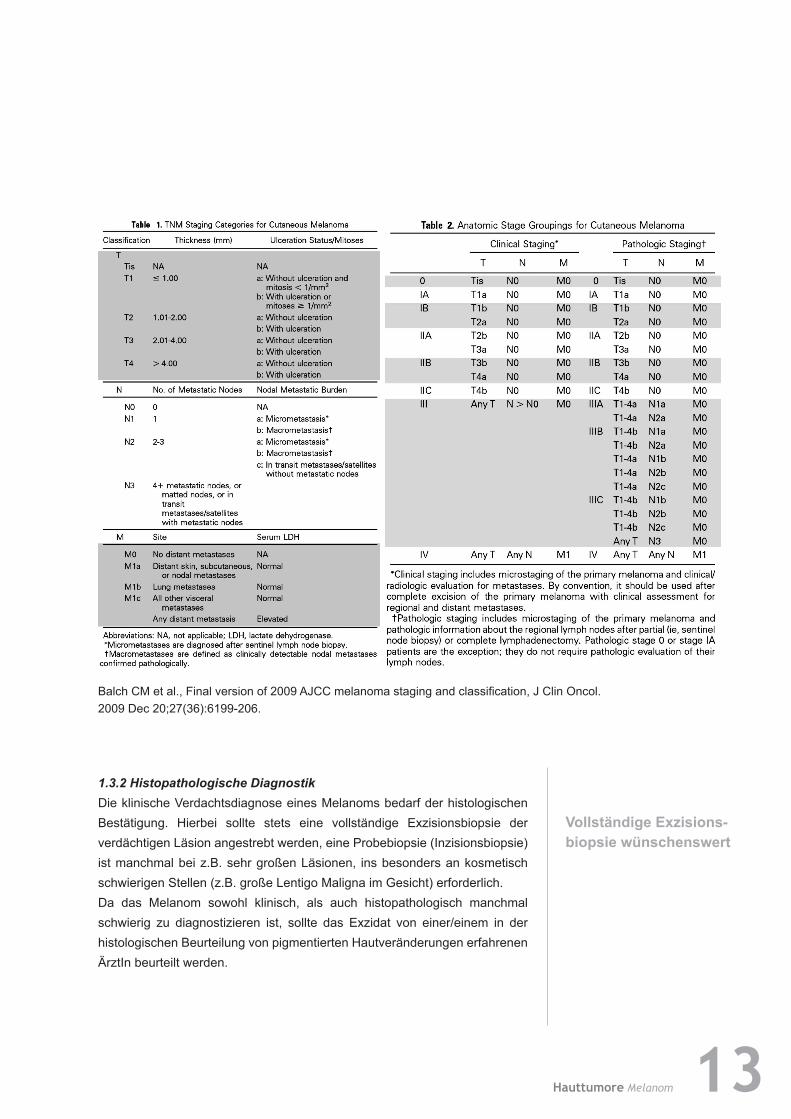
Für das maligne Melanom wurde vom AJCC (American Joint Committee on Cancer) 2009 eine neue TNM-Klassifikation und Stadieneinteilung publiziert, welche den prognostischen Parametern Rechnung trägt und international akzeptiert wird.
Hauttumore Melanom 13
VollständigeExzisions-biopsie wünschenswert
1.3.2 Histopathologische DiagnostikDie klinische Verdachtsdiagnose eines Melanoms bedarf der histologischen Bestätigung. Hierbei sollte stets eine vollständige Exzisionsbiopsie der verdächtigen Läsion angestrebt werden, eine Probebiopsie (Inzisionsbiopsie) ist manchmal bei z.B. sehr großen Läsionen, ins besonders an kosmetisch schwierigen Stellen (z.B. große Lentigo Maligna im Gesicht) erforderlich.Da das Melanom sowohl klinisch, als auch histopathologisch manchmal schwierig zu diagnostizieren ist, sollte das Exzidat von einer/einem in der histologischen Beurteilung von pigmentierten Hautveränderungen erfahrenen ÄrztIn beurteilt werden.
Balch CM et al., Final version of 2009 AJCC melanoma staging and classification, J Clin Oncol. 2009 Dec 20;27(36):6199-206.

14Hauttumore Melanom
HistologischeMerkmale
Zusatzinformationen
Immunphänotypus
MolekularbiologischeDiagnostik
Ausbreitungsdiagnostik
Der histologische Befund soll folgende Merkmale enthalten:• den Melanomtyp• die Tumordicke in mm nach Breslow• Zeichen der Ulzeration• Mitoserate ≥ 1/mm2
• Angabe der Resektionsränder nach lateral und in die Tiefe
Weiter mögliche histologische Zusatzinformationen können beinhalten:• die Eindringtiefe nach Clark• Regressionszeichen• Einbruch in Lymph-oder Blutgefäße• Vorhandensein von Tumor infiltrierenden Lymphozyten (TILs)
Bei histologisch unklaren Tumoren, amelanotischen Tumoren oder auch Melanommetastasen kann eine weiterführende immunophänotypische Charak terisierung (z.B. S-100 Protein, HMB-45 Antigen, Melan A, als Proliferations marker MIB-1) erforderlich sein.
1.3.3 Molekularbiologische Diagnostik In den letzten Jahren gewinnen weiterführende molekularbiologische Analysen zunehmend an Bedeutung. Gerade bei PatientInnen mit Fern-metas tasen, die einer Systemtherapie zugeführt werden sollen (ab Stadium IIIb), kann eine Mutationsanalyse einer Metastase, aber auch des ursprünglichen primären Melanoms die Therapiewahl beeinflussen. Durch die Entwicklung neuer, spezifischer Medikamente ist die Detektion von bestimmten Mutationen z.B. BRAF-V600, NRAS, c-KIT (bei akralen oder mukosalen Melanomen) für die Therapieentscheidung zum Standard geworden.
1.3.4 AusbreitungsdiagnostikDer Wert einer Ausbreitungsdiagnostik im Primärtumorstadium ist inter-national umstritten und wird unterschiedlich gehandhabt. Früher war es üblich, bei allen invasiven Melanomen eine initiale Staginguntersuchung durch zuführen. Die Ausbreitungsdiagnostik umfasste dabei eine Lymphknotensonographie des regionären Abflußgebietes, die Sonographie des Abdomens einschließ-lich Becken und Retroperitoneum und eine Röntgen-Thoraxaufnahme (2 Ebenen). Diese erhobenen Untersuchungsbefunde sind auch bei dünnen Tumoren geeignet, auffällige, aber harmlose Befunde wie z.B. Häm angiome der Leber, Nierenzysten bereits initial zu erkennen und bei späterer Feststellung aufwendige Untersuchungen z.B. zum Ausschluss

Hauttumore Melanom 15
UnterschiedNiedrig-undHochrisikopatienten
MöglichstvollständigeExcision
PrimäroperativeTherapie
von Metastasen zu vermeiden. Bei Patienten mit Melanomen mit einer Tumordicke > 1 mm, bei denen eine Wächterlymphknotenbiopsie geplant ist, wurde präoperativ eine zusätzliche Ausbreitungsdiagnostik durchgeführt, um eine Fernmetastasierung auszuschließen (z.B. Computertomographie, MRT-Schädel).
Heute wird zwischen Niedrig- und Hochrisikopatienten unterschieden. Bei Niedrig-Risikopatienten wird auf Bildgebungen verzichtet, weil die Wahr-schein lichkeit Tumor-relevanter Befunde niedrig ist, die Strahlen belastung vermeidbar und die mit falsch-positiven Befunden verbunden Unsicher heit vermeidbar ist. Nach den aktuellen deutschsprachigen S3-Empfehlungen werden ab dem Stadium IB eine Lymphknoten-Sonographie und Messung des Tumormarkers S-100 empfohlen, erst ab dem Stadium IIC/III (Tumor-dicke > 4mm, LK-Befall) neben der Lymphknotensonographie ein CT bzw. MRT, sowie die Bestimmung von S-100 und LDH.
1.4 Chirurgische Therapie
Bei malignen Melanomen, die noch nicht histologisch gesichert sind, ist primär eine Exzisionsbiopsie der verdächtigen Läsion anzustreben. Es sollte immer eine möglichst vollständige Exzision der pigmentierten Läsion ange strebt werden, um eine sichere Beurteilung des dermatohistologischen Präparates, aber auch der Resektionsränder zu ermöglichen. Die Exzision sollte innerhalb von 4 - 6 Wochen ab Diagnosestellung erfolgen.
Probebiopsien (Inzisionsbiopsie) aus Arealen eines pigmentierten Haut-tumors werden im Allgemeinen nicht durchgeführt, wenn Exzisionsbiopsien technisch möglich sind. Sie können bei bestimmten Indikationen (z.B. bei Verdacht auf eine ausgedehnte Lentigo maligna im Gesichtsbereich, bei akrolentiginösem Melanom oder auch bei Schleimhautveränderungen) notwendig sein.
Bei gesicherter Melanomdiagnose ist die Therapie im klinischen Stadium I primär operativ. Die Wahl des Sicherheitsabstandes der Exzision richtet sich nach der Tumordicke und dem Metastasierungsrisiko. Bei Patienten mit dünnen Melanomen sind ausgedehnte Eingriffe nicht notwendig, und bei Patienten mit dicken Primärtumoren bleibt ein radikaleres operatives Vorgehen ebenfalls ohne Einfluss auf das Risiko der Fernmetastasierung. Allerdings ist ein zu kleiner Abstand möglicherweise mit dem Risiko von vermehrten Lokalrezidiven verbunden.

16Hauttumore Melanom
Sicherheitsabstand
Sentinel-Lymphknoten
Wächterlymphknoten-biopsie
Diesem Umstand trägt eine abgestufte Exzisionsstrategie Rechnung. In der Regel sind die Eingriffe in Lokalanästhesie mit Defektversorgung möglich. Bei eindeutigem Verdacht auf das Vorliegen eines invasiven Melanom sollte ein primärer Sicherheitsabstand von 10 mm gewählt werden. Größere Sicherheitsabstände sind bei primärer Exzision wegen einer gegebenenfalls später noch durchzuführenden Schild wächterlymphknotenbiopsie mit Lymph-abstromszintigrafie aus dem Tumorgebiet zu vermeiden. Nach den derzeit international gültigen Empfeh lungen werden folgende Sicherheits abstände empfohlen:
Tumordicke nach Breslow – Sicherheitsabstandin situ – 0,5 cmTD bis 2 mm – 1 cmTD > 2 mm – 2 cm
1.4.1 Wächterlymphknotenbiopsie / Sentinel-lymphnode biopsy (SLNB)Die Wächterlymphknotenbiopsie wurde entwickelt, um selektiv den ersten drainierenden Lymphknoten des regionären Lymphabstromgebietes darzu-stellen. Das Verfahren ist für klinisch Lymphknoten-negative Patienten geeignet, die unter Verwendung aller üblichen diagnostischen Verfahren (Lymph knoten palpation, Lymphknotensonographie) keine Hinweise für manif este Lymphknotenmetastasen aufweisen. Nur in wenigen Fällen(< 5 %) wird der Wächterlymphknoten übersprungen.
Die Wächterlymphknotenbiopsie ist eine Staging-Untersuchung („patho lo-gisches Staging“) und keine therapeutische Maßnahme mit nachgewiesenem Wert im Hinblick auf eine Verlängerung der Gesamt über lebenszeit.
Eine Wächterlymphknotenbiopsie ist laut internationalen Leitlinien ab einer Tumordicke von 1,0 mm nach Breslow angezeigt. Nur bei Vorliegen weiterer ungünstiger Prognoseparameter (Mitoserate > 1, Ulzeration des Primär tumors, Clark-Level IV/V) kann auch bei geringeren Tumor dicken eine Wächterlymphknotenbiopsie erwogen werden. Die Wächterlymph-knoten biopsie sollte vorrangig in mit dieser Operationstechnik und nuklear-medizinischen Darstellungstechniken vertrauten Zentren vorgenommen werden.
Bei fehlendem Nachweis einer Mikrometastasierung im Wächterlymph knoten sind keine weiteren operativen Maßnahmen an der regionalen Lymph knoten-station indiziert.

Hauttumore Melanom 17
Operative Therapie von Metastasen
Bei Nachweis einer Mikrometastasierung > 1 mm im Wächterlymphknoten sollte eine Ausräumung der entsprechenden Lymphknotenstation (radikale Lymphadenektomie) angeboten werden, wenngleich der prognos tische Über lebens vorteil nicht durch Studien gesichert ist.
Ein positives Ergebnis der histologischen Aufarbeitung des Wächterlymph-knoten hat Einfluss auf die Stadieneinteilung (upgrading) und damit auch auf den Einsatz einer möglichen adjuvanten Therapie.
1.4.2 Operative Therapie in metastasierten Stadien Bei Satelliten- und/oder In-transit-Metastasen erfolgt möglichst die operative Entfernung aller Metastasen im Gesunden. Bei multiplen Satelliten- und/oder In-transit-Metastasen im Extremitätenbereich ohne weitere Metastasierung, ist die hypertherme Zytostatikaperfusion in therapeutischer Indikation zu erwägen. Alternativen sind unter anderem intraläsionales Interleukin-2 und Elektrochemotherapie oder T-VEC (Talimogene laherparepvec, einem onko-lytischem Herpesvirus).
Bei klinisch oder sonographisch diagnostizierten regionären Lymphknoten-metastasen ist eine radikale Lymphadenektomie (bzw. modifiziert radikale Neck-Dissektion, ggf. auch selektive Neck-Dissektion) mit kurativer Indikation indiziert. Im Anschluss daran kann eine lokale Irradiatio sinnvoll sein (siehe unten).
Im Stadium IV sollte beim Vorliegen operabler Metastasen (z.B. Lunge oder Hirn) die operative Exstirpation der Metastase(n) (R0-Resektion) als Therapie der ersten Wahl angestrebt werden, wenn Tumorfreiheit erzielt werden kann. Ein Vorteil lediglicher Tumormassenreduktion ist nicht gesichert und sollte interdisziplinär abgewogen werden, wenn funktionelle Einschränkungen damit behebbar sind.

18Hauttumore Melanom
Strahlentherapienichtprimärindiziert
AusgeprägteTendenzzuzerebralerMetastasierung
1.5 Bestrahlung
Die alleinige Strahlentherapie ist als Primärbehandlung bei malignem Melanom nur in den Einzelfällen indiziert, bei denen ein operativer Eingriff unmöglich oder nicht sinnvoll ist (z.B. große Lentigo maligna Melanome bei älteren Personen). Es werden schnelle Elektronen oder konventionelle Röntgenstrahlen in Weichstrahltechnik in konventioneller Fraktionierung mit Einzeldosen zwischen 2,0 und 3,0 Gy und einer Zielvolumendosis zwischen 50 und 60 Gy eingesetzt. Die lokale Tumorkontrolle mittels Bestrahlung ist der chirurgischen Therapie unterlegen. Die Strahlentherapie ist indiziert, wenn eine Operation regionärer Lymph-knotenstationen non in sano (R1-Resektion) verläuft bzw. bei Inoperabilität, obwohl die Literatur hierzu widersprüchlich ist.
Die Strahlentherapie von Knochenmetastasen kann eine effektive Palliation erzielen. Die Ansprechrate liegt zwischen 67% und 85 % und wird häufig bereits unter Therapie beobachtet. Generell stellt sich die Indikation zur palliativen Bestrahlung bei Schmerzen und/oder Statikgefährdung bzw. Kompression des Spinalkanales mit oder ohne neurologische Symptomatik.
Das maligne Melanom zeichnet sich gegenüber anderen Malignomen durch eine ausgeprägte Tendenz zur zerebralen Metastasierung aus, die mit einer Prognose von 3-5 Monaten einhergeht. Neben einer Hirndruckprophylaxe mit Dexamethason können solitäre Hirn-metastasen mittels stereotaktischer Einzeitbestrahlung (oder operativer Resektion) behandelt werden. Der Vorteil der stereotaktischen Einzeit-behandlung liegt in der geringen Toxizität. Die Behandlung von Hirn metas-tasen (stereotaktisch oder operativ) kombiniert mit Bestrahlung des Ganz-hirns kann die mediane Überlebenszeit verlängern.

Hauttumore Melanom 19
Interferon-alpha
1.6 Adjuvante Therapie
Bei Melanompatienten mit einem erhöhten Metastasierungsrisiko ab Stadium IIA (= 2,0 mm Tumordicke, mit oder ohne Ulzeration) wird eine adjuvante Immun therapie angeboten. Da ihre Nebenwirkungen die Lebensqualität allerdings einschränken kann, sind die Indikation, und die Risiken (z.B. Fieber, Laborveränderungen, Depression) und Alternativen sorgfältig abzu-wägen.
Interferon-alpha ist die erste Substanz in der adjuvanten Therapie des malignen Melanoms, die in prospektiv randomisierten Studien zu einem signifikanten Vorteil bezüglich Rezidivfreiheit für die Behandelten geführt hat. Nur in wenigen Studien zeigte sich ein Trend zum verbesserten Überleben. Eine adjuvante Therapie mit Interferon-alpha sollte jedenfalls allen Patienten mit erhöhtem Metastasierungsrisiko angeboten werden, soweit keine Kontra-indikationen bestehen.
Für das Stadium der Lymphknotenmetastasierung wurden international verschiedene randomisierte Therapiestudien mit unterschiedlichen Inter-feron-Dosierungen durchgeführt. Die klarsten Therapieergebnisse liegen momentan zur Hochdosis-Interferon-Therapie vor. Zwei prospektiv-randomi-sierte Studien zeigten übereinstimmend einen Vorteil in Bezug auf die rezidiv freie Überlebenszeit zu unbehandelten Kontrollpatienten.
Vergleichsstudien zwischen IFN-alpha 3 x 3 Mill E / Woche und Hochdosis-schemen haben keinen klaren Vorteil der Hochdosis herausarbeiten können.
Die Akzeptanz dieser Ergebnisse als Therapiestandard im Stadium der Lymphknotenmetastasierung ist in den USA und Kanada vorhanden, während in verschiedenen europäischen Ländern aufgrund der relativ hohen Toxizität des Hochdosis-Interferons und des Therapievorteils nur für eine begrenzte Subgruppe von Patienten (z.B. Ulzeration des Primärtumors, Makro-Metastasen des LK) höhere Interferondosierungen selektiv eingesetzt werden.

20Hauttumore Melanom
InoperableTumoreoderMetastasen als Indikation
TargetedTherapy
1.7 Systemische Therapie des metastasierenden Melanoms
Prinzipielle Indikationen zur systemischen Therapie sind inoperable Rezidiv-tumoren, inoperable regionäre Metastasen und Fernmetastasen, wobei in den letzten Jahren die zielgerichteten Therapieoptionen die klassischen Chemotherapien als Erstlinientherapie abgelöst haben: „Bei BRAF-V600-Mutation soll eine Therapie mit einem BRAF-Inhibitor in Kombination mit einem MEK-Inhibitor oder Checkpoint-Inhibitor-therapie (PD1-Mono therapie oder PD1 + CTLA4 Antikörpertherapie) durchgeführt werden.“ (Konsultations-fassung der S3-Leitlinie April 2016)
1.7.1 Zielgerichtete, molekulare TherapienIn den letzten Jahren konnten mehrere neue Medikamente entwickelt werden, welche zielgerichtet, auf eine kurative Behandlung abzielen.
Targeted Therapy im Stadium IV (BRAF, MEK, c-Kit Inhibitoren)
Bei 40-60% der kutanen Melanome können Mutationen in BRAF detektiert werden. 90% dieser Mutationen führen zu einem Aminosäurenaustausch von Valin (V) durch Glutamat (E) - (BRAFV600E). Seltener sind andere BRAF-Inhibitor sensitive Mutationen wie z.B. BRAFV600K. Dies führt zu einer konstitutiven Aktivierung des sogenannten RAF-MEK-ERK Signaltrans-duktions weges, der für die Tumorentwicklung und -progression des Melanoms relevant ist.
Das objektive Ansprechen in den Zulassungsstudien betrug für den BRAF-Inhibitor Vemurafenib 57%. Vemurafenib führte zu einer signifikanten Verbesserung der Überlebensraten nach 6 Monaten von 84% im Vemura-fenib-Arm im Vergleich zu 56% mit der konventionellen Chemo therapie. Bemerkens wert ist, dass Melanompatienten mit hoher Tumorlast gleicher-maßen von der Behandlung mit Vemurafenib profitierten.Die empfohlene Dosis beträgt 960 mg per os alle 12 Stunden. Eine Dosis-reduktion um mehr als 50% wird nicht empfohlen. Die häufigsten Neben-wirkungen sind Arthralgien, Exantheme, Alopezie, Abgeschlagenheit, photo-toxische Reaktionen, Nausea, Pruritus, das Auftreten von Papillomen und Plattenepithelkarzinomen (häufig vom Keratoakanthom-Typ). Ein transientes Ansprechen mit konsekutiver Tumorresistenz ist häufig.
Die Daten, welche schlussendlich zur Zulassung eines weiteren BRAF-Inhibitors Dabrafenib führten (BREAK III), zeigte gegenüber Darcabazin einen Anstieg des medianen PFS unter Dabrafenib auf 6,9 Monate

Hauttumore Melanom 21
Kombinationstherapie
c-KITMutationen
gegenüber weiterhin 2,7 Monaten unter Dacarbazin. Das mittlere Gesamt-überleben in der Dabrafenib Gruppe, in welcher allerdings eine Behandlung über die Progression (bei fehlender Alternative) möglich war und bei 20% der Patienten erfolgte, lag bei doch beachtlichen 18.2 Monaten.Die empfohlene Dosis beträgt 150 mg per os alle 12 Stunden, das Neben-wirkungs profil ist jenem von Vemurafenib ähnlich, wenngleich es häufiger zur Pyrexie kommt, die Phototoxizität ist allerdings deutlich geringer.
Da es bei doch einem relativ hohen Prozentsatz der Patienten schon inner-halb von 12 Monaten zu einer erneuten Tumorprogression durch Resistenz-entwicklung kommt, wurden Kombinationstherapie aus BRAF und MEK-Inhibitoren getestet.
Eine aktuelle Metaanalyse von 16 randomisierten Studien mit BRAF- oder/und MEK-Inhibitoren (u.a. Phase III-Studien Combi-d, Combi-v, co-BRIM-Studie) bestätigt die Überlegenheit der Kombinationstherapie mit einem BRAF-Inhibitor und einem MEK-Inhibitor gegenüber einer Monotherapie mit einem BRAF-Inhibitor oder einem MEK-Inhibitor. Die Kombination BRAF-Inhibitor plus MEK-Inhibitor verlängert das Gesamtüberleben im Vergleich zu BRAF-Inhibitoren (HR: 0,67, 95%CI: 0,56-0,81, P< 0,0001) oder MEK-Inhibitoren (HR: 0,29, 95%CI: 0,22-0,37, P< 0,0001) sowie das progressionsfreie Überleben im Vergleich zu BRAF-Inhibitoren (HR: 0,58, 95%CI: 0,51-0,67, P< 0,0001) oder MEK-Inhibitoren (HR: 0,48, 95%CI: 0,36-0,65, P < 0,0001).
Ende 2015 wurden daher beide Kombinationstherapien (Vemurafenib + Cobimetinib, Dabrafenib + Trametinib) als Erstlinientherapien bei BRAF-mutierten Melanomen durch die Europäische Behörde zugelassen.Interessant ist, dass das Nebenwirkungsprofil sich durch die Kombination nicht wesentlich verändert hat, es kommt sicher häufiger zum Auftreten von Fieber, allerdings sinkt das Risiko, andere Hautkrebsarten zu entwickeln, signifikant.
c-KIT Mutationen finden sich seltener, am ehesten in akral-lentiginösen und Schleimhautmelanomen. Bisherige Beobachtungen aus Phase II Studien sprechen dafür, dass Patienten mit c-KIT Aberration auf eine Behandlung mit einem c-KIT Kinase-Inhibitor ansprechen können. Patienten mit einer c-KIT Mutation in Exon 11 bzw. in Exon 13 sprachen am besten auf den c-KIT Kinase-Inhibitor Imatinib ( 400 mg/d ) an. Die häufigsten Nebenwirkungen dieses Medikaments sind Ödeme, Abgeschlagenheit, Diarrhoe, Appetitlosigkeit, Nausea, Neutropenie und Leberenzymanstieg.

22Hauttumore Melanom
StadiumIVIpilimumab
NivolumabundPembrolizumab
Immuntherapie im Stadium IV (PD1-Antikörper, CTLA-4 Antikörper)
Ipilimumab ist ein humaner IgG1 monoklonaler Antikörper, der das zytotoxische T- Lymphozyten assoziierte Antigen (CTLA-4) auf der T-Zelle blockiert. Hierdurch wird die Anti-Tumorimmunität durch die Proliferation und Aktivierung von T-Zellen, aber auch die Autoimmunität, gesteigert.
In Patienten im Stadium IV ihrer Tumorerkrankung können vier Zyklen Ipilimumab 3 mg/kg KG i.v. über 90 min alle 3 Wochen bei ca. 15 - 20% der Behandelten einen Überlebensvorteil bewirken. Das Ansprechen auf Ipilimumab ist bis zu 12 Wochen verzögert und kann erst Monate nach Therapiebeginn eintreten. Entsprechend wird die Beurteilung des Tumor-ansprechens auf Ipilimumab erst nach Abschluss der vier Zyklen empfohlen. Die Radiodiagnostik dieser Patienten muss nach eigenen Kriterien erfolgen, welche die hervorgerufenen Entzündungsreaktionen berück sichtigen.
Da Ipilimumab, ebenfalls um Wochen verzögert, schwere immun-vermittelte Nebenwirkungen induzieren kann, sind Verlaufskontrollen wichtig. Es treten insbesondere kutane (Exantheme), gastrointestinale (Colitis), hepatische (Hepatitis), endokrine (Hypophysitis) und neurologische Nebenwirkungen auf, die mittels konsequentem Nebenwirkungsmanagement kontrolliert werden können.
Neben Ipilimumab stehen nun zwei weitere Immuntherapeutika, Nivolumab und Pembrolizumab, zur Behandlung des nicht-resektablen oder metastasierten Melanoms zur Verfügung. Die gemeinsame Zielstruktur dieser monoklonalen Antikörper ist der Rezeptor Programmed Death 1 (PD-1) ist. PD-1 reguliert T-Zellen bei Interaktion mit seinen Liganden, insbesondere PD-L1, negativ, so dass die Blockade von PD-1 die Antitumor-Immunität enthemmen und Autoimmunität verursachen kann.
In therapienaiven Patienten mit metastasiertem Melanom mit einer wild-typ-Sequenz in BRAF konnte in einer prospektiven, randomisierten Phase III Studie gezeigt werden, dass Nivolumab (3 mg/kg, i.v., q14) der Vergleichs substanz Dacarbazine (1000mg/m2, i.v., q21) in Bezug auf das Gesamtüberleben (HR für Tod 0,42; 99,79% CI, 0,25 - 0,73; p<0.001, medianes Gesamtüberleben noch nicht erreicht) und Ansprechrate (40,0% (95% CI, 33,3 - 47,0) für Nivolumab vs. 13,9% (95% CI, 9,5 -,19,4) für DTIC, p<0.001) signifikant überlegen ist.

Hauttumore Melanom 23
Pembrolizumab
CheckMate067-Studie
Die Wirksamkeit des PD-1 Antikörpers Pembrolizumab wurde in Therapie-naiven, bei Nachweis einer BRAF-Mutation auch in Patienten mit einer Vorbehandlung mit einer zielgerichteten Therapie in einer pro-spektiven, randomisierten Phase III Studie untersucht [507]. In dieser drei-armigen Studie wurden Pembrolizumab 10mg/kg i.v. alle 2 Wochen und Pembrolizumab 10 mg/kg i.v. alle 3 Wochen mit Ipilimumab 3 mg/m2 i.v. alle 3 Wochen (4 Zyklen) verglichen. Es konnte gezeigt werden, dass das PFS für beide Pembrolizumab-Arme signifikant länger war als für Ipilimumab (HR für Krankheitsprogress, 0,58; p<0.001 für beide Pembrolizumab-Arme vs Ipilimumab; 95% CI, 0,46 - 0,72 und 0,47 - 0,72).
Die bei Ipilimumab bereits bekannten, teilweise schweren, immunvermittelten Nebenwirkungen können bei den PD1-Antikörpern zwar auftreten, sind aber insgesamt seltener und selten Grad 3/4. Neben kutanen (Exantheme), gastrointestinalen (Colitis), hepatischen (Hepatitis), endokrinen (Hypo-physitis) oder neurologische Nebenwirkungen treten vor allem auch immun-vermittelte Lungenenzündungen auf, die aber mit den bekannten konse-quenten Nebenwirkungsmanagement meist gut behandelt werden können.
Aufgrund der hervorragenden Studienergebnisse der CheckMate 067-Studie wurde Anfang 2016 auch die Kombinationstherapie von Ipilimumab und Nivolumab als Erstlinientherapie zugelassen. Das mittlere progressionsfrei Überleben betrug 11,5 Monate (95% confidence interval [CI], 8,9 - 16,7) bei der Kombination, aber nur 2,9 Monate (95% CI, 2,8 - 3,4) bei Ipilimumab (hazard ratio für Tod oder Progression, 0,42; 99,5% CI, 0,31 - 0,57; p<0.001), und 6,9 Monate (95% CI, 4,3 - 9,5) mit Nivolumab (hazard ratio im Vergleich zu Ipilimumab, 0,57; 99,5% CI, 0,43 to 0,76; p<0.001). Fast 60% aller Patienten sprachen auf diese Kombinationstherapie an, allerdings beobachtete man in mehr als 80% der Patienten immunvermittelte Nebenwirkungen, davon in gut 50% Grad 3 oder 4. Allerdings dürften auch jene Patienten, die auf die Therapie prinzipiell ansprachen, die aber auf Grund der Toxizitäten diese absetzen mussten, längerfristig von dieser Therapie profitieren.

24Hauttumore Melanom
Objektive Remission mit Dacarbazinnurselten
KombinationstherapieZytostatika mit Zytokinen
1.7.2 Chemotherapie und ChemoimmuntherapieFür die systemische Chemotherapie des fortgeschrittenen Melanoms stehen seit vielen Jahren verschiedene Substanzen zur Verfügung, deren klinische Wirksamkeit vergleichbar ist. Die älteste und am besten unter-suchte Substanz ist Dacarbazin (DTIC). Objektive Remissionen, d.h. eine Rückbildung der Tumormassen um mehr als 50% werden aber nur in etwa 10% der Patienten nach einer Monochemotherapie mit Dacarbazin beobachtet. In einem noch geringeren Anteil werden komplette Remissionen erzielt, die meistens nur von kurzer Dauer (3 - 6 Monate) sind. Das Ein-Tages-Regime (850 mg/m2 i.v. alle 3 - 4 Wochen) führt zu vergleichbaren Ergebnissen wie das Fünf-Tages-Regime (250 mg/m2 i.v. Tag 1 - 5 alle 3 - 4 Wochen), das keine höhere Toxizität besitzt.
Alternativen inkludieren den Nitrosoharnstoff Fotemustin, der aufgrund seiner Liquorgängigkeit auch bei Hirnmetastasen eingesetzt werden kann, und Temozolomid, ein Alkylans, das im Gegensatz zu den vorgenannten Chemo therapeutika oral verabreicht wird. Aufgrund der Liquorgängigkeit ist Temozolomid im Gegensatz zu Dacarbazin, wie Fotemustine auch bei Hirnmetastasen wirksam. Temozolomid wird in einer Dosierung von 150 mg/m2 Tag 1 - 5 alle 4 Wochen eingesetzt, alternativ täglich in metro-nomischer Dosierung von 20 - 40 mg tgl. p.o.
Durch eine Kombination von Zytostatika mit Zytokinen kann eine Steigerung der objektiven Ansprechraten erreicht werden, allerdings ergab sich in allen bisher durchgeführten Studien hierdurch keine signifikante Verlängerung der Gesamtüberlebenszeit. Die subjektive und objektive Verträglichkeit einer Monochemotherapie wird durch die Zugabe von IFN bzw. IL-2 deutlich verschlechtert.
In gleicher Weise bewirkt die Kombination verschiedener Chemotherapeutika (Polychemotherapie) bzw. Chemotherapeutika mit Zytokinen (Polychemo-immuntherapie) im Vergleich zur Monotherapie z.T. höhere Remissionsraten, ohne dass hierdurch aber eine Verlängerung der Gesamtüberlebensraten erreicht wird. Dafür ist aber die Toxizität kombinierter Chemotherapien im Vergleich zur Monotherapie signifikant erhöht.
Hauttumore Melanom 25
Hazard-RatenfürdasAuf-treten des ersten Rezidivs beimmalignenMelanom.Eine Analyse des Zentralre-gistersMalignesMelanomderDGGan33.384Fällen
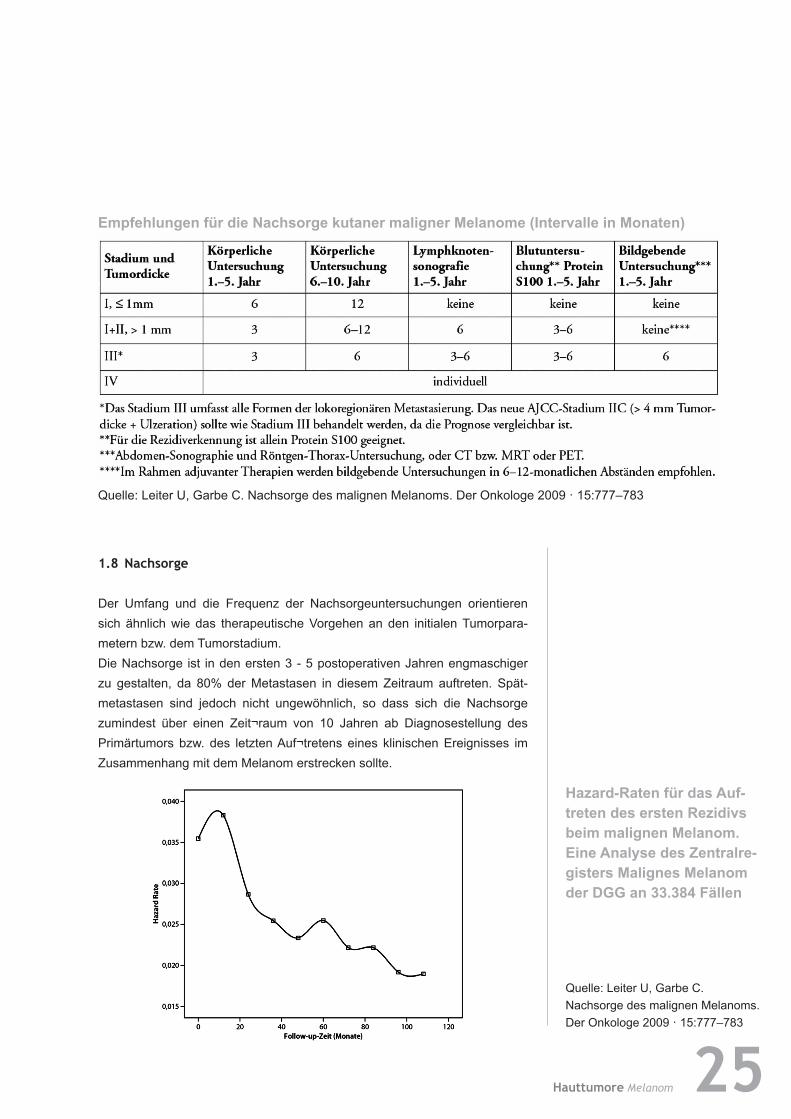
1.8 Nachsorge
Der Umfang und die Frequenz der Nachsorgeuntersuchungen orientieren sich ähnlich wie das therapeutische Vorgehen an den initialen Tumor para-metern bzw. dem Tumorstadium. Die Nachsorge ist in den ersten 3 - 5 postoperativen Jahren engmaschiger zu gestalten, da 80% der Metastasen in diesem Zeitraum auftreten. Spät-metas tasen sind jedoch nicht ungewöhnlich, so dass sich die Nachsorge zumindest über einen Zeit¬raum von 10 Jahren ab Diagnosestellung des Primärtumors bzw. des letzten Auf¬tretens eines klinischen Ereignisses im Zusammenhang mit dem Melanom erstrecken sollte.
EmpfehlungenfürdieNachsorgekutanermalignerMelanome(IntervalleinMonaten)
Quelle: Leiter U, Garbe C. Nachsorge des malignen Melanoms. Der Onkologe 2009 · 15:777–783
Quelle: Leiter U, Garbe C. Nachsorge des malignen Melanoms. Der Onkologe 2009 · 15:777–783

26Hauttumore Melanom
Ziele der Nachsorgeuntersuchungen
Folgende Ziele werden mit den Nachsorgeuntersuchungen verfolgt:1. Feststellung der Tumorfreiheit bzw. Früherkennung einer Progression2. Überwachung zur Früherkennung von Rezidiven und Zweitmalignomen (melanozytär und nicht-melanozytär)3. Psychosoziale Betreuung4. Dokumentation der Krankheitsverläufe5. Durchführung und Überwachung einer adjuvanten Therapie
Insgesamt gibt es zwischen verschiedenen Ländern und auch zwischen Experten eine große Variationsbreite, wie die Nachsorge bei Melanom-patienten zu erfolgen hat. Bislang wurde kein internationaler Konsensus erreicht, die Datenlage ist unzureichend. In Frankreich und in den Niederlanden erfolgen nur klinische Unter-suchungen. In Deutschland, der Schweiz und Österreich wurden bislang regelmäßige engmaschige Untersuchungen unter Einschluss bildgebender Verfahren (Sonographie, CT) empfohlen.
Von der Deutschen Dermatologischen Gesellschaft (DDG) wurde 1994 ein umfangreiches Schema empfohlen, das für alle Stadien zusätzlich zu den klinischen Untersuchungen bildgebende Verfahren in regelmäßigen Abständen empfahl. Heutzutage stellt sich jedoch die Frage, ob die Vielzahl der Untersuchungen wirklich sinnvoll und für die Patienten von prognostischer Relevanz sind. So wurde bezweifelt, dass die Untersuchungen und Maßnahmen der Nachs orge das Leben metastasierender Patienten tatsächlich verlängern. Kritisch beurteilt wurde insbesondere der Einsatz bildgebender Verfahren, da Rezidive überwiegend durch Selbstuntersuchung des Patienten oder aufgrund der körperlichen Untersuchung durch den Arzt entdeckt werden.
Dies führte in den letzten Jahren zu einer Modifizierung des empfohlenen Nachsorgeschemas nach den klinischen Stadien. Ein reduziertes Nachsorge-programm in den Stadien I und II, und eine intensivierte Nach sorge im Stadium III scheint ein kosteneffektives und sinnvolles Verfahren zu sein. Das Stadium III umfasst alle Formen der lokoregionären Metastasierung. Das neue AJCC-Stadium IIC (>4 mm Tumordicke + Ulzeration) sollte wie Stadium III behandelt werden, da die Prognose vergleichbar ist.
Abdomensonographie und Röntgenthoraxuntersuchung oder CT/MRT oder PET sollten nicht routinemäßig zum Einsatz kommen, solange keine kurative Therapie daraus folgt. Eine aktualisierte deutschsprachige S3-Leitlinie, die diesen Prinzipien folgt, ist im Juli 2016 erschienen.

Hauttumore Melanom 27
Die Arbeitsgruppe Dermatologische Onkologie der Österreichischen Gesell-schaft für Dermatologie und Venerologie (ÖGDV) hat in Anpassung an diese S3-Leitlininien 12/2013 österreichweit geltende Nachsorge empfehlungen herausgegeben.
NachsorgeempfehlungenfürdasMaligneMelanomderArbeitsgruppeDermatologischeOnkologie
Quelle: Empfehlungen der AG Dermatologie der ÖGDV, Stand 12/2013


Hauttumore Basaliom 29
MaligneNeoplasieder Keratinozyten
HäufigsteNeoplasiedes Menschen
SuperfiziellerTyp
2 Basaliom
Das Basaliom ist eine maligne Neoplasie, die von Keratinozyten mit Haarkeim-Differenzierung ausgeht und sich durch langsam, lokal-invasives Wachstum auszeichnet. Metastasen sind eine Rarität. Es gibt verschiedene Wachstumsmuster, die sich lichtmikroskopisch in der Histologie wider-spiegeln und das therapeutische Vorgehen bestimmen. UV-Licht spielt eine wichtige pathogenetische Rolle. Das Basaliom findet sich etwa 80% im Kopf-Hals-Bereich.
2.1 Epidemiologie
Basaliome sind die häufigsten Neoplasien des Menschen. Die genaue Inzidenz in Österreich oder Tirol liegt nicht vor, sie ist jedoch ca. 10x höher, als jene des malignen Melanoms. International wurden 105/128 Frauen/Männer pro 100.000 Einwohner in England, aber auch Deutschland und 1195/2058 Frauen/Männer pro 100.000 Einwohner in Australien berichtet, d.h. Männer sind häufiger betroffen als Frauen. Basaliome reflektieren die UV-Exposition der vergangenen Jahre, insbesondere der Kindheit. UV-Schutz reduziert die Inzidenz. Bei älteren PatientInnen ist die Lokalisation im Kopfbereich häufiger, bei jüngeren PatientInnen am Stamm. Immunsuppression und genetische Faktoren, z.B. multiple Basaliome beim Basalzellnävusssyndrom (Gorlin-Goltz), sind prädisponierend.
2.2 Klinik und Histopathologie
Basalzellkarzinome beginnen zumeist als flach erhabene, umschriebene, gelblich-rötliche Papeln mit einem perlschnurartigen Randsaum. Daneben existieren andere Varianten wie die als rote Flecken erscheinenden Rumpf-hautbasaliome (superfizieller Typ) oder die an Vernarbungen erinnern den sklerodermiformen Basaliome. Fortgeschrittenere Basalzell karzinome gehen in Erosionen und Ulzerationen über. Das Wachstum kann auch knorpelige und knöcherne Strukturen zerstören.
Histogenetisch stammen Basalzellkarzinome von den Zellen der Basalzell-schicht und/oder der äußeren Wurzelscheide der Haarfollikel ab. Zum Teil zeigen sie eine Differenzierung, die an Merkmale von Adnexorganen, (Follikel, Talgdrüsen, ekkrine/apokrine Schweißdrüsen) erinnern. Die histo lo-gische Subtypisierung der Basalzellkarzinome basiert auf unter schied lichen Differenzierungsmustern, die auch in der aktuellen histolo gi schen Klassi-fizierung der WHO zum Ausdruck kommen.

30 Hauttumore Basaliom
ChirurgischeExzision
2.3 Diagnostik
Die Diagnose wird in der Regel klinisch gestellt. Die Biopsie und Histologie erlaubt die eindeutige Diagnose und Ausdehnungsdiagnostik und ist der Bild gebung (CT, MRT) überlegen, weshalb diese nur in den seltenen Fällen mangelnder Biopsierbarkeit notwendig werden. Der histologische Subtyp bestimmt das weitere therapeutische Vorgehen. Es werden unter anderem noduläre, solid-zystische, oberflächliche, sklerodermiforme, fibroepitheliale Subtypen unterschieden. Molekularpathologisch sind vielfach Mutationen der Sonic-Hedgehog-Genkaskade, insbesondere Mutationen des Smoothened und Patched I Gen nachweisbar.
2.4 Therapie
2.4.1 Chirurgie Das übergeordnete Ziel jeder therapeutischen Handlung beim Basaliom ist die vollständige Tumorentfernung mittels chirurgischer Exzision. Jedoch können hohes Alter und Komorbiditäten eine palliative statt einer kurativen Vorgehensweise rechtfertigen.
Unbehandeltes Basaliom: Die chirurgische Exzision mit histologischer Kon-trolle der seitlichen und tiefen Resektionsränder ist der effektivste Therapie-modus.
Solange kein sklerodermiformes Wachstumsmuster vorliegt erzielt die klinische Resektion mit 3 mm Abstand in 85% der Fälle und mit 5 mm in 95% der Fälle Tumorfreiheit der Ränder. Größere Sicherheitsabstände sind notwendig für sklerodermiforme Basaliome (13 - 15 mm für >95% Tumor freiheit) und solche mit einem Durchmesser > 20 mm. Tumorreste im Resektionsrand führen in einem Viertel der Fälle zu einem Rezidiv, im Gesichtsbereich führen sie sogar in der Hälfte der Fälle zum Rezidiv. Zur Randkontrolle stehen verschiedene Varianten der histographischen/mikro-graphischen Chirurgie zur Verfügung, z.B. „Tübinger Torte“ oder „Münchner Methode“.
Inkomplett exzidiertes Basaliom: Eine Nachexzision mit Randkontrolle ist indiziert. Wenn es sich um ein Niedrig-Risiko Basaliom (nicht sklero-dermiform, kein Rezidiv, < 1 cm im Gesicht, < 2 cm am Stamm und Extremi-täten, keine Immunsuppression) handelt und sich die Tumorreste im seit-lichen Resektionsrand und nicht im basalen Resektionsrand befinden,

Hauttumore Basaliom 31
Physikalische Therapiemodalitäten
Bestrahlung
Medikamentöse Therapie
sowie keine Lappenplastik vorliegt, kann in Ausnahmefällen auch eine engmaschige Verlaufsbeobachtung gerechtfertigt sein.
2.4.2 Nicht-chirurgische Therapien 2.4.2.1 Physikalische Therapiemodalitäten Oberflächliche, klar abgrenzbare Läsionen in begünstigter anatomischer Lokalisation (Stamm) sind mittels Kürretage und Kauterung mit Heilungsraten von 95% behandelbar. Diese Methode ist jedoch nicht für Hochrisiko- und große Basaliome geeignet. Ähnliche Heilungsraten wie mit Kürretage und Kauterung wurden mittels Kryochirurgie (flüssiger Stickstoff) erzielt. Die Erfolgs rate hängt jeweils stark von der Technik ab (Spray-Verfahren, Kontakt-technik, Doppel und Dreifach Vereisung). Kohlendioxid-Laser Behandlungen sind ähnlich zu bewerten, die Heilungsraten dieser Methode sind weniger gut in der Literatur dokumentiert.
2.4.2.2 Bestrahlung Oberflächliche Basaliome können mittels Licht im Rahmen einer photo-dyna mischen Therapie, nach Applikation eines Photosensitizers (z. B. delta-Aminolävulinsäure) bestrahlt werden. Nebenwirkungen sind Brennen, Schmerzen, Erytheme, Erosionen. Bei inkomplett exzidierten Basaliomen ist eine Strahlentherapie möglich, die in 91% Rezidivfreiheit herstellt (gegen-über 61% bei reiner Observation). In nicht-resektierbaren Basaliomen (alte PatientInnen, anatomisch-chirurgisch nicht behandelbar) kann ebenfalls eine Strahlentherapie Heilungsraten von 90% erzielen. Allerdings sind die Ergebnisse der Strahlentherapie für große Basaliome (> 40 mm), für solche im Gesichtsbereich und in Regionen erhöhter mechanischer Belastung weniger gut. Aufgrund des Risikos für vermehrte Tumorbildung wird die Strahlen therapie bei PatientInnen mit Basalzellnävussyndrom nicht durch-geführt. Nebenwirkungen der Strahlentherapie sind Teleangiektasien, Atrophie und Fibrose.
2.4.2.3 Medikamentöse Therapie Oberflächliche Basaliome (<2 mm, nicht nodulär) können mittels topischem 5-Fluoruracil (5-FU) oder Imiquimod 5% Creme behandelt werden. Letzteres stimuliert das Immunsystem (Zytokinsekretion, TLR7-Agonist) und induziert die Apoptose. Heilungsraten von ca. 80% werden berichtet (tägliche Gabe für 6 Wochen). Seit kurzem steht für Patienten mit lokal fortgeschrittenen oder metasta-sie renden Basaliomen, die nicht durch eine Operation oder Bestrahlung behandelt werden können, eine medikamentöse Therapie mit Vismodegib (150 mg täglich) zur Verfügung. Vismodegib hemmt selektiv die über-

32 Hauttumore Basaliom
6-monatliche Verlaufskontrolle
aktive Signalgebung im Sonic-Hedgehog-Signalweg und führte in einer Studie zu einer Verkleinerung der Tumore bei 43% der PatientInnen mit fort geschrittenem Basaliom und bei 30% der PatientInnen mit metastasie-rendem Basaliom. Die häufigsten Nebenwirkungen sind Muskel krämpfe, Haarausfall, veränderte Geschmacksempfindung, Müdigkeit, Übel keit, Durch fall, Appetitlosigkeit und Gelenkschmerzen.
2.5 Nachsorge
Rezidive treten gehäuft in den ersten 5 Jahren nach Diagnosestellung auf (< 0% im 1. Jahr, 50% innerhalb der ersten 2 Jahre, 66% innerhalb 3 Jahre). Verlaufskontrollen sollten deshalb 6-monatlich stattfinden und einen vollständigen dermatologischen Status beinhalten, Ziele sind: • Feststellung der Tumorfreiheit• Früherkennung von Rezidiven oder Zweitmalignomen • Dokumentation der Krankheitsverläufe • Beratung über UV-Schutz
Patienten mit lokal rezidivierenden oder nicht in sano exzidierten Basaliomen bzw. solche mit erhöhtem Risiko für weitere neue Tumore (Immun-suppression, genetische Disposition) sollten individuell engmaschiger nach-kontrolliert werden.
2.6 Therapie von Rezidiven
Die Behandlungsergebnisse sind generell schlechter als die von Primär-tumoren empfohlene. Das Vorgehen ist chirurgisch, größere Sicherheits-abstände sind erforderlich. 5 - 10 mm Sicherheitsabstand werden empfohlen.
2.7 Systemtherapien
Eine zielgerichtete, molekulare Therapie steht mit Vismedogib für Patienten mit multiplen Basaliomen zur Verfügung. Chemotherapien sind nur in den seltenen Ausnahmefällen einer Metastasierung angezeigt und beinhalten Cisplatin-hältige Regimes (Remissionen 28 - 40%). Aufgrund dieser begrenz ten Wirksamkeit ist auch ein palliativ-symptomatisches Vorgehen im Einzelfall zu diskutieren (alternativ Tumorlastreduktion durch Bestrahlung).

Hauttumore Plattenepithelkarzinom 33
Entstehenausin-situLäsionen
AuslösendeundbegünstigendeFaktoren
Aktinische Keratose
3 Plattenepithelkarzinom
Plattenepithelkarzinome der Haut und Schleimhäute sind maligne Neo-plasien, die von Keratinozyten ausgehen und lokal destruierend wachsen, nur selten metastasieren. Sie entstehen aus in situ Plattenepithel karzinomen, am häufigsten darunter aus aktinischen Keratosen (AK), jedoch auch aus anderen in situ Vorläuferläsionen.
3.1 Epidemiologie
Für invasive Plattenepithelkarzinome der Haut wurde die Inzidenz in Mittel-europa in den 1990er Jahren mit ca. 20 - 30 Neuerkrankungen pro 100.000 Einwohnern ermittelt. Männer sind häufiger betroffen als Frauen. Die Präva-lenz von in situ Plattenepithelkarzinomen (aktinischen Keratosen - AK) wird in der der kaukasischen Bevölkerung Europas mit 15% bei Männern, 6% bei Frauen, im Alter > 70 Jahren 34% bei Männern, 18% bei Frauen mit steigender Tendenz angegeben. Exakte Daten aus Österreich oder Tirol liegen nicht vor. Neben Geschlecht und Alter sind auch UV-Licht und ionisierende Strahlen, insbesondere in der Kindheit, auslösende Faktoren. UV-Schutz reduziert die Inzidenz. Auch genetische Prädisposition (Pigmen -tierungsgrad, Xeroderma pigmentosum), Röntgenbestrahlungen, Arsen-exposition und Immunsupression begünstigen die Karzinogenese. Darüber hinaus werden Papillomviren, Verbrennungen, chronische Wunden und Entzündungen wie Ulcera crurum, Narben, bullöse Dermatosen, Lichen ruber als ätiologische Faktoren gesehen.
3.2 Subtypen und Histologie
3.2.1 In situ PlattenepithelkarzinomAktinische Keratosen (AK) imponieren als keratotische Papeln und Plaques auf erythematösem Grund. Die Prädilektionsstellen von AK inklu dieren den Gesichts-Kopfbereich, insbesondere die besonders UV-exponierten Stirn und Nasenrücken, Ohren, Nacken, außerdem Dekolleté, Streckseiten der Extremitäten und (Unter)Lippen. AK können als Einzel-läsionen, nicht selten jedoch auch multipel an Stirn und Handrücken als Field Cancerisation auftreten. Die Wahrscheinlichkeit der Progression in ein invasives Plattenepithelkarzinom variiert zwischen 0,025-16%, als Durch-schnitt werden 10% angegeben, bei Immunsupprimierten 40%.
Histopathologisch sind AK auf die Epidermis beschränkt, zeigen Para-keratose und eine gestörte epidermale Zytoarchitektur bestehend aus

34 Hauttumore Plattenepithelkarzinom
MorbusBowen
ErythroplasieQueyrat
atypischen proliferierenden Keratinozyten. Frühe AK zeigen licht mikro sko-pisch eine alternierende Ortho- und Parakeratose, fokal atypische Keratino-zyten mit pleomorphen, hyperchromatischen Kernen die vielfach eng zusammengelagert sind (Crowding) und eine solare Elastose des dermalen Bindegewebes. Mit längerer Bestandsdauer entsteht eine epi dermale Hyperplasie, in der atypische Keratinozyten mit Mitosen in allen Schichten zu finden sind und einhergehen mit dermalen Entzündungs zellinfiltraten.
Histologische Subtypen inkludieren lichenoide AK mit bandförmigen Infiltraten, akantholytische AK mit dyskeratotischen Keratinozyten, hyper-trophe, bowenoide und pigmentierte AK. Die Auflichtmikroskopie und neue bild gebende Verfahren wie optische Kohärenztomorgraphie, Hochfrequenz-ultraschall und konfokale Laserscanning-Mikroskopie (mit einer Sensitivität und Spezifität von nahezu 98%) können in der Differential diagnose zu Pigmentläsionen und Basaliomen hilfreich sein.
Der Morbus Bowen imponiert klinisch als scharf begrenzte erythematöse Plaques mit Schuppen oder Krusten, die Ekzemen ähneln können. Histopathologisch zeigen sich Parakeratose, Akanthose und fehlende Differenzierung von atypischen Keratinozyten mit großen Kernen.
Die Erythroplasie Queyrat besteht aus scharf begrenzten, rötlichen Plaques an der Genitalschleimhaut, am häufigsten bei Männern im Alter >40 Jahre. Histopathologisch zeigen sich Veränderungen wie bei Morbus Bowen.
Die Grenze zwischen in situ und invasiven Plattenepithelkarzinomen ist klinisch schwer zu ziehen. Klinische Kriterien werden unter dem Akronym IDBREU zusammengefasst: I (Induration/Inflammation), D (Diameter > 1 cm), B (Blutung), R (Rapide Größenzunahme), E (Erythem) und U (Ulzeration)
Minor-Kriterien sind Schmerzen, Juckreiz und Pigmentierung.
In der Histopathologie besteht die Grenze in der Überschreitung der Basal-membran zone, aber auch diese ist lichtmikroskopisch nicht immer eindeutig zu bestimmen. Der Übergang ist ein Kontinuum. In der Epidermis am Rand von invasiven Plattenepithelkarzinomen finden sich häufig in situ Anteile.

Hauttumore Plattenepithelkarzinom 35
Subtypen
Keratoakanthome
HistologischmessbareTumordicke
3.2.2 Invasives PlattenepithelkarzinomBesteht in der Histopathologie eine Überschreitung der Basalmembranzone, handelt es sich um ein invasives Plattenepithelkarzinom. Histopathologisch imponieren atypische Keratinozyten, die in die Dermis hinein reichen. Die Zellen zeigen eine Verhornung, mitunter mit Ausbildung von Hornperlen. Für die Abgrenzung von Basaliomen können immun histolo-gische Marker hilfreich sein (z.B. EMA). Es gibt mehrere Subtypen:
• Plattenepithelkarzinom mit Hornbildung• Spindelzelliges Plattenepithelkarzinom der Haut (aggressives Verhalten)• Akantholytisches Plattenepithelkarzinom der Haut• Verruköses Plattenepithelkarzinom der Haut• Lymphoepitheliomartiges Plattenepithelkarzinom der Haut• Desmoplastisches Plattenepithelkarzinom der Haut
Keratoakanthome zeichnen sich durch ihr rasches Entstehen (z.B. Größen-zunahme von 1 mm zu 25 mm in 3 - 8 Wochen) aus. Es handelt sich um hyper keratotische Papeln mit scharfer Begrenzung und zentralem Hornpfropf. Histopathologisch findet sich ein zentraler Krater aus eosinophilem Keratin. Im Randbereich ist die Epidermis invaginiert und weist eine Akanthose auf. In der Umgebung sind gemischtzellige Entzündungsinfiltrate nachweisbar.
3.3 Stadieneinteilung und Prognose
Die Ausdehnung der Plattenepithelkarzinome wird nach dem derzeit gültigen TNM-System der AJCC (American Joint Committee on Cancer) angegeben (Tabelle 1 und 2). Die rein klinische Klassifizierung wird durch histopathologische Parameter wie das Grading (Bestimmung des Differen-zierungsgrades) angegeben, wobei sich die vier Abstufungen nach Broders nicht bewährt haben und in der internationalen Literatur die besser vierstufige Einteilung (gut, intermediär, schlecht und undifferenziert) propa-giert wird. Wichtiger für die Klassifizierung scheint die histologisch messbare Tumor dicke und den histologischen Tumortyp (z.B. desmo plastisches Platten epithelkarzinom) zu sein. Durch sie ist eine bessere Schätzung des Metastasierungsrisikos möglich. Eine Verschlechterung der Prognose wird bei immunsupprimierten Patienten nach Organtransplantation oder nach hochdosierter Chemotherapie beobachtet. Auch Lokalrezidive werden als prognostisch schlechtes Zeichen eingestuft. Dabei bleibt offen, ob das Lokal-rezidiv selbst zu dieser Verschlechterung beiträgt oder ein Zeichen des aggressiven Wachstumsverhaltens des Tumors darstellt.

36 Hauttumore Plattenepithelkarzinom
Tabelle 1: TNM-Klassifikationvon Plattenepithel-
karzinomenderHaut(AJCC2010)
TNMKlassifikation
Tumormerkmale
TX Primärtumor* kann nicht beurteilt werden
T0 Kein Anhalt für Primärtumor
Tis Carcinoma in situ**T1 Tumor 2 cm oder weniger in größter horizontaler Ausdehnung
mit 0-1 „high-risk-feature“***T2 Tumor 2 cm oder weniger in größter horizontaler Ausdehnung
mit 2-5 „high-risk-feature“*** oder Tumor mehr als 2 cm in größter horizontaler Ausdehnung
T3 Infiltration der Gesichts-und Schädelknochen
T4 Infiltration der Skelettknochen oder Schädelbasis
NX Regionäre Lymphknoten können nicht beurteilt werden
N0 keine regionären Lymphknotenmetastasen N1 solitäre ipsilaterale Lymphknotenmetastase, Durchmesser
3 cm oder weniger N2a solitäre ipsilaterale Lymphknotenmetastase, Durchmesser
mehr als 3 cm bis max. 6 cmN2b multiple ipsilaterale Lymphknotenmetastasen, alle nicht mehr
als 6 cm in DurchmesserN2c multiple bilaterale und kontralaterale
Lymphknotenmetastasen, alle nicht mehr als 6 cm in Durchmesser
N3 Lymphknotenmetastase, Durchmesser 6 cm oder mehrMX Das Vorliegen von Fernmetastasen kann nicht beurteilt
werden M0 Keine Fernmetastasen
M1 Fernmetastasen
* Im Falle multipler simultaner Tumoren wird der Tumor mit der höchsten T-Kategorie klassifiziert und die Anzahl abgrenzbarer Tumoren in Klammern angegeben, z.B. T2.
** Tis (Carcinoma in situ): Eine Metastasierung des in situ Karzinoms ist ausgeschlossen. *** Hochrisikomerkmale: Tumordicke (>2 mm, Clark Level IV/V), perineurale Invasion,
Lokalisation (Ohr oder Haar tragende Lippe); und Differenzierungsgrad (schlecht oder undifferenziert).
Quelle: Farasat S et al. A new American Joint Committee on Cancer staging system for cutaneous squamous cell carcinoma: creation and rationale for inclusion of tumor (T) characteristics. J Am Acad Dermatol. 2011;64:1051-1059.

Hauttumore Plattenepithelkarzinom 37
Tabelle 2: Stadieneinteilungvon Plattenepithel-karzinomen (AJCC2010)
Biopsie
Therapieentscheidung
Stadium Primärtumor Lymphknoten Fernmetastasen
Stadium 0 Tis N0 M0
Stadium I T1 N0 M0
Stadium II T2 N0 M0
Stadium III T3 N0 oder N1 M0
T1 oder T2 N1 M0
Stadium IV
T1, 2 oder 3 N2 M0
jedes T N3 M0
T4 jedes N M0
jedes T jedes N M1Quelle: Farasat S et al. A new American Joint Committee on Cancer staging
system for cutaneous squamous cell carcinoma: creation and rationale for inclusion of tumor (T) characteristics. J Am Acad Dermatol. 2011;64:1051-1059.
3.4 Diagnostik
Die Verdachtsdiagnose wird klinisch gestellt. Notwendig ist die histopatho-logische Sicherung durch Biopsie, die in manchen Situationen auch die komplette Entfernung des Tumors umfassen kann. Prätherapeutisch empfiehlt sich die Fotodokumentation des Tumorbefundes.
Die klinische Untersuchung des Lymphabstromgebietes durch Palpation ist in jedem Fall erforderlich. Bei Plattenepithelkarzinomen ab einer Tumor dicke von > 2 mm sollte eine Ausbreitungsdiagnostik mit bildgebenden Verfahren zur Abklärung einer Lymphknotenmetastasierung erfolgen. Bei klinischem Verdacht auf eine Fernmetastasierung werden organspezifische Unter-suchungen zur weiterführenden Diagnostik durchgeführt.
3.5 Therapie
Weil der Übergang von der in situ Plattenepithelkarzinome zum invasiven Plattenepithelkarzinom nicht immer eindeutig bestimmt oder vorausgesagt werden kann, ist das in situ Plattenepithelkarzinom zu therapieren. Die Therapieentscheidung erfordert eine genaue Anamnese, dazu gehören die Erhebung früherer Hautkrebserkrankungen, Bestandsdauer, Verlauf, Lokalisation, Ausmaß/Ausdehnung der Läsion(en), Alter, Komorbititäten und Abschätzung der Adhärenz.

38 Hauttumore Plattenepithelkarzinom
Ablative Therapien
Topische Therapien
Kryochirurgie
5-Fluoruracil(5-FU)
Imiquimod
Ablative Therapien (Exzision, Laserablation, Kürettage, Kryochirurgie) sind bei in situ Plattenepithelkarzinomen zumeist auf Einzelläsionen ausgerichtet. Zu beachten ist, dass einer Behandlung mit diesen Modalitäten, wenn sie nicht in toto erfolgen, von raschen Lokalrezidiven gefolgt sind. Eine Exzision ist bei der Diagnose in situ Plattenepithelkarzinom nur selten notwendig, wenngleich auch eine Biopsie zur Diagnosesicherung angezeigt sein kann.
Topische Therapien (photodynamische Therapie, 5% Fluoruracil, 3% Diclofenac und 5% Imiquimod) können sowohl auf Einzelläsionen als auch auf multiple Läsionen (Field Cancerisation) abzielen, welche besonders bei Immunsupprimierten ein Problem darstellen können.
Mit der Kryochirurgie werden Heilungsraten von ca. 67 - 99% erreicht, abhängig vom BehandlerIn, der Länge der Einfrier-Auftau-Zyklen und Lokalisation (AK am Handrücken sprechen schlechter an). Dennoch ist die Rezidiv rate relativ hoch. Die Nebenwirkungen bestehen vorwiegend aus Schmerzen, Pigmentverschiebungen und Narbenbildungen.
5-Fluoruracil (5-FU) entspricht einer topischen Chemotherapie. In den proliferierenden Zellen von AK führt dieser Wirkmechanismus zum Sis tieren der Keratinozytenproliferation und Apoptoseinduktion. 5-FU wird üblicher-weise 2x täglich für 1 - 2 Wochen appliziert, wonach an der Stelle vormaliger AK Erosionen entstehen. Bei verzögertem Ansprechen kann okklusiv und/oder prolongiert (bis zu 4 Wochen) behandelt werden. Die Heilungsrate liegt bei 50%, die Rezidivrate bei 50%. Nebenwirkungen von topischem 5-FU beinhalten ausgeprägte irritative Ekzemreaktionen, Erosione, Ulzerationen, Wund infektionen, Narbenbildung, Schmerzen und Pruritus. Ein kommerziell erhält liches Produkt (Actikerall®) kombiniert 5 mg / 1 g Fluoruracil mit 100 mg / 1 g Salicylsäure und wird täglich bis zu 12 Wochen angewendet.
Imiquimod bewirkt eine Zytokinsekretion am Ort der Applikation. Der Wirkmechanismus läuft über den Toll-like Rezeptor 7. Die sezernierten Zytokine aktivieren das zelluläre Immunsystem. Entsprechend kommt es zu Behandlungsbeginn zur vermehrten Rötung der behandelten Läsionen, subklinische Areale (Field Cancerisation) können klinisch sichtbar werden, ein Vorteil dieser Modalität. Diese Reaktion korreliert mit dem Ansprechen. Die Heilungsrate von Imiquimod liegt nach derzeitigem Stand bei 50-70%. Die Rezidivrate beträgt 10% pro Jahr. In immunsupprimierten Trans -plantations patientInnen hat sich Imiquimod als sicher erwiesen. Imiqui-mod wurde anfangs bei dem Präparat Aldara® in einer Konzentration von 5% 3x pro Woche für 4 - 16 Wochen verschrieben. Ein akzeleriertes

Hauttumore Plattenepithelkarzinom 39
Ingenolmebutat
Diclofenac
Photodynamische Therapie
Behandlungsschema über 6 Wochen (2 Wochen 2x täglich, 2 Wochen Pause, 2 Wochen 1x täglich) mit dem Präparat Zyclara® ist ebenfalls wirksam. Nebenwirkungen von topischem Imiquimod beinhalten ausge prägte Erytheme und irritative Ekzemreaktionen (10 - 30%), Erosionen, Ulzerationen und Krustenbildungen.
Ingenolmebutat ist ein makrozyklischer Diterpenester aus der Gartenwolfs-milch Euphorbia peplus. Der Wirkmechanismus ist noch nicht vollständig geklärt: In-vivo- und In-vitro-Modelle lassen auf einen dualen Mechanismus schließen – eine direkte zytotoxische Wirkung und die Förderung einer Entzündungs reaktion. Das Ansprechen des Patienten auf die Therapie ist ungefähr acht Wochen nach der Therapie beurteilbar. Im Unterschied dazu wird Ingenolmebutat (150 µg oder 500 µg, Picato®) im Gesicht in der niedrigeren Konzentration für 3 Tage, 1x täglich appliziert, an den übrigen Körperregionen in der höheren Dosierung allerdings nur an 2 aufeinander-folgenden Tagen.
3% Diclofenac in 2,5% Hyaluronsäure besitzt den Wirkmechanismus der Cyclooxygenase-2-Hemmung und Induktion der Arachidonsäure-Kaskade und Prostaglandinproduktion. Neben der anti-inflammatorischen Aktivität wirkt Diclofenac auch direkt anti-proliferativ, Apoptose-induzierend (extrin-sischer Pathway) und anti-angiogenetisch.Diclofenac in Hyaluronsäure wird üblicherweise 2x täglich für 2 - 3 Monate appliziert. Die Heilungsrate liegt bei 50%, Zahlen zur Rezidivrate liegen nicht vor. Nebenwirkungen von topischem Diclofenac sind mild und beinhalten Erytheme, Schuppung, Juckreiz, Hypo- und Parästhesien. Photoallergische Reaktionen und allergische Kontaktezeme sind als seltene Nebenwirkungen eben falls beschrieben.
Die photodynamische Therapie (PDT) wirkt durch die Destruktion atypischer, proliferender Keratinozyten durch die Kombination eines Photo-sensitizers mit Lichtexposition (oftmals Schmalspektrum) und daraus resul-tierender Sauerstoffradikalbildung. Neoplastische Zellen reichern den Photo-sensitizer in höherem Ausmaß an als unbetroffene Keratinozyten. Dieser selektive Effekt kann durch verschiedene Photosensitizer erzielt werden, prinzipiell handelt es sich jedoch um Vorläufer des Protoporphyrin IX. Am häufigsten werden derzeit 5-Aminolaevulin Säure (ALA) und Deriva tive verwendet. Der Vorteil der photodynamischen Therapie liegt in der Behandelbarkeit größerer Hautareale. Wegen der limitierten Eindringtiefe des Lichtes werden vor der Behandlung Hyperkeratosen und Krusten mechanisch entfernt (z.B.

40 Hauttumore Plattenepithelkarzinom
Systemische Retinoide
Strahlentherapie beiInoperabilität
Kürettage) und der Photosensitizer wird 3 - 5 Stunden vor der Lichtexposition okklusiv appliziert. Die Heilungsrate wird mit 80 - 90% angegeben, Rezidiv-raten mit 19%. In immunsupprimierten TransplantationspatientInnen ist die Heilungsrate niedriger. Nebenwirkungen der PDT betreffen vor allem Schmerzen, die üblicherweise auf die Zeit der Lichtexposition beschränkt sind. Systemische Retinoide, z.B. niedrig dosiert Acitretin 20 mg, sind für die sekundäre Chemoprävention von Plattenepithelkarzinomen bei Xero derma pigmentosum, Basalzellnaevussyndrom und immunsupprimierte Trans-plantations patientInnen verwendet worden, werden aber in den aktuellen Leitlinien nicht mehr erwähnt, da sie in Phase III-Studien im Vergleich zu Inter feron keinen Hinweis auf einen protektiven Effekt hinsichtlich des Auftretens von weiteren Plep-Ca erbrachte. Häufige Nebenwirkungen sind Photosensitivität, Hautatrophie, Erosionen und Pruritus.
Für invasive Plattenepithelkarzinome ist die vollständige chirurgische Exzision mit histopathologischer Kontrolle der Schnittränder die Therapie der ersten Wahl. Beim desmoplastischen Typ bedarf es, über die festge-stellten tumorfreien Schnittränder hinaus, noch einen zusätzlichen Sicher-heits nachresektion von ca. 5 mm. Bei weiteren Risikofaktoren (z.B. Tumor-dicke > 2 cm ) werden noch größere Sicherheitsabstände empfohlen.Bei Inoperabilität oder NoninsanoResektion besteht die Indikation einer Strahlenbehandlung. Die Ergebnisse der Bestrahlung invasiver Platten-epithelkarzinome der Haut sind mit denen einer Exzision vergleichbar, wobei die Tumorkontrollraten zwischen 70 und 100 % angegeben werden.
Quelle: Diagnosis and Treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur J Cancer. 2015 Sep;51(14):1989-2007 (2015)

Hauttumore Plattenepithelkarzinom 41
Regionäre Lymphknotenmetastasen
Fernmetastasen
Nachsorgemehr als 5 Jahre
Abzuwägen bleibt die primäre Indikationsstellung zur Strahlentherapie, wenn chirurgisch ein ungünstiges ästhetisches Resultat zu erwarten ist. Insbesondere im Gesichts -/Hals- und Handbereich ist jedoch die Operation vorzuziehen, wenn die vollständige Resektion des Tumors erwartet werden kann. Eine Sentinel-Lymphknoten Biopsie kann zur Ausbreitungsdiagnostik bei Stamm- oder Extremitäten-Lokalisation eines Primärtumors mit Hochrisiko-profil angewandt werden, ist allerdings nicht obligat.
Beim Vorliegen von regionären Lymphknotenmetastasen ist eine thera-peutische Lymphadenektomie angezeigt. Die regionären Lymph ab strom-gebiete ipsilateral sollten einer Strahlentherapie unterzogen werden, falls nach Lymphknotendissektion histologisch Befall verifiziert werden konnte, sowie bei inoperablen Lymphknotenmetastasen oder bei Rezidivmetastasen. Die elektive Bestrahlung der regionären Lymphabstrom gebiete führt zu keiner Prognoseverbesserung.
Im Stadium der Fernmetastasierung ist die Prognose zumeist infaust. Die Remissionsraten sind bei der Verwendung von Polychemotherapieschemata (Cisplatin mit 5-FU oder Doxirubicin) höher (60%-80%), als bei einer Mono-therapie mit Cisplatin, die man Patienten mit eingeschränktem Allgemein-zustand anbieten sollte. Hinsichtlich der Überlebenszeit scheint die Anwen-dung der kombinierten Schemata gegenüber der Monotherapie, oder mit Metho threxat (20 - 40% Remissionsrate) aber keine Vorteile zu bieten. Bei Nichtansprechen kann auch eine Therapie mit Cetuximab angeboten werden. Dieses hat ein günstiges Nebenwirkungsprofil mit einer Aktivität, die vergleich bar mit jener von Methotrexat ist.
3.6 Nachsorge
Obwohl die Rezidive und die primär lymphogen lokoregionären Metas tasen in der Mehrzahl innerhalb der ersten 2 Jahre auftreten, wird empfohlen, die Nachsorge über 5 Jahre hinweg durchzuführen. Die Nach sorge unter such-ungen sind vorwiegend klinische Untersuchungen zur Beur teilung des loko-regionären Befundes. Eine Sonographie der regionären Lymphknoten wird bei unklarem oder schwierig zu erhebendem Palpations befund durchgeführt. Bei Hoch-Risiko-Patienten werden klinische Nach sorge kontrollen und Sono-graphien der regionären Lymph knoten stationen alle 3 Monate über die ersten Jahre nach Diagnosestellung empfohlen. Prävention durch UV-Schutz, Selbstunter suchung und Früh erkennung sollte stets erfolgen.

42 Hauttumore Plattenepithelkarzinom
3.7 Rehabilitation
Narbenbildungen nach Operationen mit Funktionseinschränkungen, Strahlen therapie und potentiell toxische Systemtherapien machen Behand-lungs massnahmen zur Rehabilitation notwendig. Bei PatientInnen mit Axilla-, Leisten- und Neck-Dissektion z.B. Versorgung mit Informations material, Arm-/Beinkompressionsstrumpf, Lymphdrainage. Allen PatientInnen sollten Physio therapie, Bewegungstherapie und Anleitung zum Selbstüben zu Hause angeboten werden.
3.8 Psychosoziale Versorgung
Allen PatientInnen sollte eine psycho-onkologische Betreuung angeboten werden, die mittels validierter Messinstrumente den Bedarf erhebt und in die das Gesamtkonzept der dermato-onkologischen Behandlung integriert ist.

Hauttumore Merkelzellkarzinom 43
SehrseltenerHauttumor
Häufigkeit
4.1 Epidemiologie
Das sehr aggressive Merkelzellkarzinom (MCC) oder kutanes neuro-endokrines Karzinom ist ein sehr seltener bösartiger Hauttumor, der von den Merkel-Zellen der Oberhaut ausgeht. Die Tumorinzidenz ist stark zunehmend, derzeit ca. 0,4/100.000/Jahr und gleich häufig bei beiden Geschlechtern. Gute Epidemiologische Daten gibt es international aber bisher leider nicht. Der Tumor manifestiert sich innerhalb von Wochen bis wenigen Monaten typischerweise in chronisch lichtgeschädigten Hautarealen.
Das MCC wurde erstmalig von Toker 1972 als trabekuläres Karzinom der Haut beschrieben. Einer 2008 im Journal „Science“ publizierten Studie von Feng et al. zufolge liegt der Tumorentstehung eine virale Onkogenese zugrunde. Dabei geht die Integration des Merkelzellpolyomavirus in das Wirtsgenom der Expansion der Tumorzellen voran. Polyomaviren verursachen oft latente Infektionen, bei bestehender Immunsuppression ist jedoch die Entwicklung einer manifesten Tumorerkrankung möglich.
Merkelzellkarzinome kommen z. B. bei organtransplantierten oder HIV-Patien ten viel häufiger (12/100.000/Jahr) und in deutlich jüngerem Alter (ca. 50 % < 50 Jahre) vor. Übereinstimmend damit besteht eine hohe Assoziation des Merkelzellkarzinoms mit Spinaliomen, Basaliomen, Morbus Bowen und Malig nomen innerer Organe sowie Leukämien. Auch die UV-Karzinogenese spielt ebenso eine wichtige Rolle in der Tumor-entstehung.
4.2 Klinik und Histopathologie
Das MCC ist ein Karzinom des höheren Lebensalters (Median ca. 70 Jahre) mit bevorzugter Lokalisation im Kopf/Halsbereich und Extremitäten (je ca. 40%) und seltener am Körperstamm (10%).
Die Klinik des Merkelzellkarzinoms (MCC) ist nicht sehr charakteristisch. Das MCC imponiert meist als derb-elastischer, rötlicher bis livider Tumor mit glatter, glänzender Oberfläche. Neben den häufigen halbkugeligen oder knotigen Formen kommen aber auch plaqueartige Varianten vor. Letztere insbsonders am Stamm. Ulzerationen sind sehr selten und werden erst im Spät stadium beobachtet. Die Diagnose wird daher in den meisten Fällen erst anhand des bioptischen Befundes gestellt.
4 Merkelzellkarzinom

44 Hauttumore Plattenepithelkarzinom
IntermediärerTyp
Diagnoseimmerimmunhistologisch
Inzisions- oder Exzisionsbiopsie
Ausbreitungsdiagnostik
Histologisch breitet sich der Tumor in der retikulären Dermis und Subkutis aus, die papilläre Dermis, Epidermis und Adnexen bleiben ausgespart. Bei der Routine HE-Färbung sind die Zellen ausgesprochen monomorph. Es existieren 3 histologische Sub-Typen:Der intermediäre Typ ist am häufigsten (ca. 80 %). Die Tumorzellen sind mittelgroß, zeigen große, gelappte Zellkerne und wenig schwach gefärbtes Zytoplasma. Die Mitoserate ist meist hoch, Nekrosen sind selten. Der klassische, ursprünglich trabekuläre Typ ist selten (ca. 10 %). Die großen mono morphen Zellen wachsen trabekulär in der Dermis. Der kleinzellige Typ mit kleinen Zellen und stark hyperchromatischen Zellkernen, gelegentlich Nekrosen, macht ebenfalls ca. 10 % aus. Misch- und Übergangsformen zwischen den 3 Typen sind sehr häufig. Die Diagnose muss immer immunhistologisch bestätigt werden. Wie auch normale, nicht maligne veränderte Merkelzellen exprimieren Merkel zell-karzinome sowohl epitheliale als auch neuroendokrine Antigene. Die Expression von Zytokeratin 20 und Neurofilamenten dient der eindeutigen Identi fizierung von Merkelzellkarzinomen. An neuroendokrinen Markern ist Chromo granin A ein typischer und spezifischer Marker, der jedoch stark variiert. Negativ ist der Transkriptionsfaktor (TTF-1), der in kleinzelligen Lungen karzinomen positiv ist; ebenso sind HMB 45, S 100, LCA, Vimentin, saures Gliafaser- Protein und Desmin negativ.
4.3 Diagnostik
Je nach Größe des Tumors wird die primäre Diagnose mittels Inzisions-biopsie oder Exzisionsbiopsie erfolgen. Da charakteristische klinische Merk-male, die eine sichere Diagnose erlauben, fehlen, werden die Tumoren oft verkannt und unter der Annahme eines häufigeren Hauttumors wie z. B. eines Plattenepithelkarzinoms, Basalzell karzinoms oder eines kutanen malignen Lymphoms exzidiert.Nach histologischen Diagnosesicherung sollte eine Ausbreitungsdiagnostik mittels Lymphknotensonographie der drainierenden Lymphknotenstation erfolgen. Auch eine Abdomen-Sonographie ist ebenso wie eine Röntgen-Thorax-Untersuchung indiziert. Bei klinischem Verdacht auf eine Fern metas-tasierung sind die entsprechenden bildgebenden Verfahren verschiedener Organe durchzuführen (z.B. Magnetresonanztomographie des Schädels, Computertomographie des Thorax / Abdomens oder [68Ga]-DOTATOC-PET-Untersuchung).

Hauttumore Merkelzellkarzinom 45
UngünstigePrognose
UngünstigeprognostischeFaktoren
HistologischeTypisierung
4.4 Prognose und Staging
Aufgrund seiner aggressiven Biologie ist das MCC nach wie vor mit einer ungünstigen Prognose behaftet: Die 5-Jahres-Überlebensrate bei Vorliegen eines Primärtumors beträgt 64%, nach Eintreten einer Fernmetastasierung sinkt dieser Prozentsatz auf 18%Die meisten Rezidive treten während der ersten 2 Jahre auf. Retrospektive Studien an mehr als 400 in der Literatur publizierten Patienten zeigten folgende ungünstige prognostische Faktoren auf: • Befall des Wächterlymphknotens (ca. 30%)• Histologisch kleine Zellen, hohe Mitoserate, Gefäßreichtum• Fortgeschrittenes Tumorstadium (lokoregionäre Metastasen oder Fern-
metas tasen)• Männliches Geschlecht• Lokalisation des Primärtumors in der Kopf-Hals-Region oder am Rumpf• Immunsuppression
Eine prognostische Bedeutung wird auch dem histologischen Typ zuge-messen: Der trabekuläre Typ ist der bestdifferenzierte, während der klein-zellige Typ am wenigsten differenziert ist; auch die Bestimmung der Tumor-dicke scheint eine prognostische Abgrenzung zu ermöglichen. Kleine Zellen, hohe Mitoserate und Gefäßreichtum in der Histologie ist eher mit einer schlechten Prognose vergesellschaftet, ein entzündliches Infiltrat oder eine solare Elastose mit einer Besseren.
Eine allgemein eingeführte Stadieneinteilung für Merkelzell-Karzinome existierte jahrelang nicht. Meist wurden die Einteilungen nach Yiegpruk-sawan et al. (1991) und Boyle et al. (1995) verwendet. 2010 wurde eine neue Stadieneinteilung des MCC von der AJCC (American Joint Committee on Cancer) publiziert.

46 Hauttumore Merkelzellkarzinom
Quelle: Lemos BD et al.;Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. 2010 Nov;63(5):75161.
TNM-KriterienundStadieneinteilung

Hauttumore Merkelzellkarzinom 47
Operatoíve Therapie
Strahlentherapie
4.5 Therapie
4.5.1 Operative Therapie Bei Primärtumoren ohne Hinweise auf das Vorliegen von Organmetastasen ist die vollständige chirurgische Exzision als Basistherapie anzusehen. Wegen der hohen Rate von Lokalrezidiven, die in der Regel auf sub-klinische Satellitenmetastasen zurückzuführen sind, sollte möglichst ein Sicherheitsabstand von 2-3 cm angestrebt werden. In besonderer Loka-li sation, wo in Abwägung der Gesamtsituation nur ein geringerer Sicher-heitsabstand möglich ist, sollte diesem Umstand durch eine entspre ch ende lückenlose histologische Darstellung der Exzidatschnitt ränder Rechnung getragen werden.
Wegen des hohen Frequenz einer lymphogenen Metastasierung erfolgt in der Regel die Durchführung einer Wächterlymphknotenbiopsie (SLNB), da die Präsenz von Mikrometastasen im Wächterlymphknoten prog-nostisch ungünstig ist. Beim Nachweis einer Mikrometastasierung oder Makrometastasierung sollte im Anschluss eine komplette Lymph adenek tomie erfolgen. Bei Lokalrezidiven oder spontanem Auftreten von Lymphknotenmetastasen ist die chirurgische Sanierung nach wie vor die Therapie der Wahl. Diese sollte mit kurativer Intention vorgenommen werden.
4.5.2 Strahlentherapie Merkelzellkarzinome sind in aller Regel radiosensitiv. Retrospektive Analysen zeigen, dass die lokale Rezidivrate nach alleiniger R0-Operation des Primärtumors durch eine kombinierte lokoregionäre adjuvante Strahlen-behandlung (3 cm Umgebung der Exzisionsnarbe + regionäre Lymph knoten-station) deutlich gesenkt werden kann. Ebenso ergaben sich Hinweise, dass diese Vorgehen zu einer relevanten Verlängerung des rezidivfreien und Gesamt überlebenszeit führt.
Für Primärtumoren und lokoregionäre Rezidive, einschließlich Lymph-knoten metastasen ist daher die postoperative, adjuvante Strahlen therapie der Tumorregion und der regionären Lymphknotenstationen indiziert. Als erfor der liche Gesamtdosis werden in der adjuvanten Situation 50 Gray mit einer Einzeldosis von je 2 Gy fünf Mal wöchentlich empfohlen. Bei metastasierendem Merkelzellkarzinom wird die Bestrahlung häufig im Rahmen multimodaler Therapiekonzepte neben chirurgischen Exzisionen und/oder einer systemischen Chemotherapie durchgeführt

48 Hauttumore Merkelzellkarzinom
Chemotherapie
ExperimentelleTherapien
4.5.3 Chemotherapie Das Merkelzellkarzinom ist prinzipiell ein chemosensitiver Tumor. Es gibt allerdings keine Standard-Chemotherapie-Schemata. Wegen der morpho-logischen Ähnlichkeiten wurden oft Regime gewählt, die bei den klein-zelligen Lungenkarzinomen etabliert sind (u.a. Anthrazykline, Anti meta-bo lite, Bleomycin, Cyclophosphamid, Etoposid, Platinderivate allein oder in Kombination). Es finden sich zwar hohe Ansprechraten (first line bis zu 57%, second line bis zu 45%, third line bis zu 20%), aber keine Korrela-tion zwischen Ansprechen und Überleben, ebenso wie keine Korrelation zwischen Dosisintensität und Ansprechen.Dafür aber eine hohe Rate von Therapie-assoziierten Todesfällen. Heilungen scheinen in diesem Tumorstadium nicht aufzutreten. Daher ist eine systemische Chemotherapie als Palliativmaßnahme bei Vorliegen von Fernmetastasen zwar indiziert, sollte aber insbesondere wegen der hohen Toxizität der meisten Chemotherapeutika für alte Patienten (eingeschränkte Leber- und Nierenfunktion sowie Hämatopoese) auf den individuellen Fall angepasst werden. Als gut verträgliche Monotherapeutika stehen Etoposid oder Anthrazykline, z. B. liposomal verkapseltes Doxorubicin, zur Verfügung.
4.5.4 Experimentelle Therapien Der genaue Stellenwert der Immuntherapeutika, Signaltransduktions-inhibi toren oder epigentischen Modulatoren kann aufgrund der geringen Fallzahlen derzeit noch nicht beurteilt werden. Neue therapeutische Strate-gien (Ipilimumab, Pembrolizumab, Avelumab, αCD56-Toxin-Konjugate, Kinase-Inhibitoren, Survivin-Vakzine) müssen erst in randomisierten Studien getestet werden.Ein ebenso interessanter Therapieansatz ist der therapeutische Einsatz eines DOTATOC-Radiopeptides nach erfolgter Detektion von Somato statin-rezeptoren des Tumors mittels [68Ga]-DOTATOC-PET-Scan.

Hauttumore Merkelzellkarzinom 49
HalbjährlicheKontrollen
4.6 Nachsorge
Bis heute existieren keine wissenschaftlich gesicherten Studien zur Nach-sorge des Merkelzell-Karzinoms. Wegen der bekannten Gefahr von Lokal-rezidiven oder regionären Lymphknotenmetastasen innerhalb der ersten Jahre nach Entfernung des Primärtumors wird eine engmaschige Nachsorge zunächst in vierteljährlichen Abständen und später in halbjährlichen Abständen empfohlen.Im Rahmen dieser Nachsorgeuntersuchung erfolgt neben der klinischen Untersuchung mit Lymphknotenpalpation, eine Lymphknotensonographie vor allem der regionären Lymphknotenstationen. Einmal jährlich sollten eine Oberbauchsonographie und eine Röntgen-Thorax-Untersuchung vorge-nommen werden.


Hauttumore Dermatofibrosarkoma protuberans 51
Selten,aberhäufigstesSarkomderHaut
SelteneMetastasierung
MolekulareUntersuchung
5.1 Epidemiologie und Stadieneinteilung
Das Dermatofibrosarcoma protuberans (DFSP) ist ein, fibrohistiozytärer, vorwiegend am Stamm sowie an den proximalen Extremitätenabschnitten vorkommender Tumor von intermediärer Malignität. Obwohl es ein seltener Tumor ist (unter 1 pro 100.000 Einwohner und Jahr), stellt es das häufigste Sarkom der Haut dar.
Das Durchschnittsalter der Patienten liegt bei 40 Jahren. Männer und Frauen sind gleich häufig betroffen. Die Mortalität ist als gering einzuschätzen, da das DSFP langsam lokal infiltrierend wächst, sowohl seitlich als auch in die Tiefe, nur selten metastasiert (< 5%) und zu lokalen Rezidiven neigt (10 - 30%). Das fibrosarkomatöse DFSP als Progressionsform des DSFP besitzt eine erhöhte Metastasierungsrate und wird als ein G2-Tumor eingeordnet.
Eine verbindliche Stadieneinteilung des DSFP existiert nicht. In der Regel wird das Primärstadium als Stadium I, die Lymphknotenmetastasierung als Stadium II und die Fernmetastasierung als Stadium III angegeben.
Neuere molekulare Untersuchungen zeigten ein gehäuftes Vorliegen chro-mo somaler Translokationen in DFSP-Zellen. Hierbei handelt es sich in über 90% der Fälle um die Translokation 17q22; 22q13, die mit einer Fusion der Gene COL1A1 und PDGFß einhergeht. Das Genprodukt, ein COL1A1-PDGFß-Fusionsprotein, wirkt über eine Bindung an den PDGF-Rezeptor als autokriner Wachstums-Stimulus für die DFSP-Zellen. Diese Kenntnisse ermöglichten erstmals eine neue ätiopathogenetisch fundierte Therapie des DFSP.
5.2 Klinik und Histopathologie
Das DSFP ist ein meist derber, hautfarbener bis bräunlich-rötlicher, höckriger Tumor, der aus einem knotigen und einem darunterliegenden platten-artigen Anteil („Eisbergphänomen“) besteht und eine teilweise jahre lange Bestandsdauer aufweist.
Histologisch findet sich eine diffuse Infiltration der Haut und des subkutanen Fett gewebes durch dicht gelagerte, wenig pleomorphe, spindelförmige, CD34-positive Tumorzellen, die in storiformen (bastmattenartigen) Forma-tionen angeordnet sind. Charakteristisch sind die Ausbreitung der Tumor-
5Dermatofibrosarkomaprotuberans

52 Hauttumore Merkelzellkarzinom
Keine sichere klinische Diagnostik
MikrographischeChirurgie
zellen entlang der Septen des subkutanen Fettgewebes sowie eine diffuse Durch setzung desselben. Das fibrosarkomatöse DFSP zeichnet sich histo-logisch durch einen abrupten bzw. graduellen Übergang in zellreiche Spindel zellfaszikel mit erhöhten Zellatypien und –mitosen aus.
5.3 Diagnostik
Eine sichere klinische Diagnose ist nicht möglich. Charakteristisch ist die derbe Konsistenz der Läsion. Die Diagnose wird in der Regel durch eine Inzisionsbiopsie, seltener durch Exzisionsbiopsie gestellt. Die Ausbreitung des Tumors erfolgt meist intra- und subkutan. Ultraschall-untersuchungen und MRT-Aufnahmen lassen nur bedingt Aussagen über die wirkliche Infiltration zu, im Einzelfall können diese Untersuchungen präoperativ nützlich sein. Bei Rezidiven sind zur Ausbreitungsdiagnostik eine Lymphknoten sono-graphie, ein Röntgen-Thorax sowie eine Abdomen-Sonographie notwendig.
5.4 Therapie
5.4.1 Operative Therapie Die Therapie des Primärtumors besteht in der chirurgischen Exzision. Der hierbei einzuhaltende Sicherheitsabstand wird in der Literatur sehr variabel zwischen 1 und 5 cm angegeben. Wird der Tumor unter anschlie ßender lückenloser histologischer Darstellung der Schnittränder exzidiert (mikro-graphische Chirurgie), kann ein Sicherheitsabstand von 1 cm als ausreichend angesehen werden, da die lokalen Heilungsraten selbst bei Lokalrezidiven 98% betragen. Bei anderen histopathologischen Auf arbeitungs verfahren ist jedoch aufgrund des erhöhten Rezidivrisikos ein höherer Sicherheitsabstand von 2 bis 3 cm angezeigt.
5.4.2 Andere Therapieverfahren Neben der operativen Therapie steht für nicht-operable primäre, lokal rezidivierende, oder metastasierte DFSP eine molekular ausgerichtete Therapie mit dem Ziel einer Unterbrechung des autokrinen PDGF-gesteu-erten Wachstums-Stimulus zur Verfügung. Studien mit dem PDGF-Rezeptor -selektiven oralen Tyrosinkinase-Inhibitor Imatinib zeigten ein thera peu tisches Ansprechen von ca. 70%. Imatinib kann auch bei ausge dehnten Tumoren zur präoperativen Reduktion der Tumormasse eingesetzt werden.

Hauttumore Melanom 53
R1- oder R2-Resektion
HalbjährlicheKontrollenzurErfassungvon Lokalrezidiven oder Metastasen
Häufige Nebenwirkungen sind gastrointestinale Beschwerden, periphere Ödeme, Myalgien, Abgeschlagenheit und Exantheme.
Bei einem Therapieansprechen wird eine operative Entfernung des resi-dualen Tumors nach Abschluss der Behandlung zur histologischen Siche-rung des Therapieerfolgs sowie zur Vermeidung lokaler Rezidive empfohlen. Sowohl primäre als auch sekundäre Resistenzen gegen Imatinib sind beschrieben. Bei Therapieresistenz kann eine Therapie mit anderen PDGFR-Inhibitoren wie Suntinitib oder Nilotinib versucht werden.
Als weitere Behandlungsmöglichkeit bei primärer Inoperabilität, R1- oder R2-Resektion, sowie bei Z. n. mehrfachen Rezidiven kann die Strahlentherapie angesehen werden. Das Bestrahlungsareal umfasst den Primärtumor, post-operative Narben und einen Sicherheitsabstand von 3 - 5 cm. Die Einzel-dosis beträgt dabei 2 Gy, 5 × pro Woche, mit einer Gesamtdosis von 60 - 70 Gy bei kurativer Zielsetzung.
Eine wirksame Chemotherapie in der Behandlung des DFSP ist nicht bekannt.
5.5 Nachsorge
Daten zur Nachsorge des DSFP liegen nicht vor. Die Nachsorge richtet sich vor allem auf die frühzeitige Erfassung von Lokalrezidiven oder Lymph knoten metastasierungen. Hierzu sind klinische Untersuchungen in halbjährlichen Abständen für mindestens fünf Jahre empfehlenswert. Technische Untersuchungen mittels Sonographie oder MRI sind nicht routine mäßig, sondern nur bei Bedarf, erforderlich.

54Hauttumore Melanom

Hauttumore Melanom 55
Kapitel 1
6 Literatur
IET - Institut für klinische Epidemiologie der TILAK GmbH, „Inzidenz und Mortalität bösartiger Neubildungen in Tirol Diagnosejahr 2009“
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011 Jun 30;364(26):2507-16. doi: 10.1056/NEJMoa1103782
S3-Leitlinie „Diagnostik, Therapie und Nachsorge des Melanoms“, Kurz-version 2.0, Juli 2016, Konsultationsfassung AWMF (Arbeits gemein-schaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V.) der Deutschen Krebsgesellschaft e.V. und deutschen Krebshilfe e.V., Registrie rungs nummer: 032-024OL
Garbe C, Hauschild A, Volkenandt M, Schadendorf D, Stolz W, Kortmann RD, Kettelhack C, Frerich B, Keilholz U, Dummer R, Sebastian G, Tilgen W, Schuler G, Mackensen A, Kaufmann R. „Deutsche Leitlinie: Malignes Melanom“. In: Garbe C (Hrsg.) Interdisziplinäre Leitlinien zur Diagnostik und Behandlung von Hauttumoren. Stuttgart, New York: Georg Thieme Verlag, S. 23-55, 2005.
Garbe C, Schadendorf D, Stolz W, Volkenandt M, Reinhold U, Kortmann RD, Kettelhack Ch, Frerich B, Keilholz U, Dummer R, Sebastian G, Tilgen W, Schuler G, Mackensen A, Kaufmann R, Hauschild A. „Kurzleitlinie – Malignes Melanom der Haut“. JDDG, Supplement 1, 2008 (Band 6), DOI: 10.1111/j.1610-0387.2008.06711.x
Garbe C, Peris K, Hauschild A, Saiag P, Middleton M, Spatz A, Grob JJ, Malvehy J, Newton-Bishop J, Stratigos A, Pehamberger H, Eggermont AM. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline - Update 2012. Eur J Cancer. 2012 Oct;48(15):2375-90. doi: 10.1016/j.ejca.2012.06.013
Hersh EM, O'Day SJ, Powderly J, Khan KD, Pavlick AC, Cranmer LD, Samlowski WE, Nichol GM, Yellin MJ, Weber JS. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naïve patients with advanced melanoma. Invest New Drugs. 2011 Jun;29(3):489-98. doi: 10.1007/s10637-009-9376
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved

56Hauttumore Melanom
Kapitel 2
survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010 Aug 19;363(8):711-23. doi: 10.1056/NEJMoa1003466
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schaden dorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD, Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med, 2015. 373(1): p. 23-34.
Leiter U, Garbe C. „Nachsorge des malignen Melanoms“ Zentrum für Dermato-Onkologie, Südwestdeutsches Tumorzentrum, Universitäts-Hautklinik, Eberhard-Karls-Universität, Tübingen, Der Onkologe 2009 · 15:777–783, DOI 10.1007/s00761-009-1634-z
Mai R, Zhou S, Zhong W, Rong S, Cong Z, Li Y, Xie Q, Chen H, Li X, Liu S, Cheng Y, Huang Y, Zhou Y, Zhang G, Therapeutic efficacy of combined BRAF and MEK inhibition in metastatic melanoma: a comprehensive network meta-analysis of randomized controlled trials. Oncotarget, 2015, Sep 29;6(29):28502-12. doi: 10.18632/oncotarget.4375.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med, 2015. 372(4): p. 320-30.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A; KEYNOTE-006 investigators, Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med, 2015. 372(26): p. 2521-32.
Diepgen TL, Mahler VM. The epidemiology of skin cancer. Br J Dermatol 2002;146 (suppl 61):1-6.
Berlin j, Katz KH, Helm KF, Maloney ME. The significance of tumor persistence after incomplete excision of basal cell carcinoma. J Am Acad Dermatol. 2002;46 (4):639.
Geisse JK, Rich P, Pandya A, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: a double-blind, randomized, vehicle-controlled study. J Am Acad Dermatol 2002; 47: 390-8.

Hauttumore Melanom 57
Kapitel 3
Morton CA, Brown SB, Collins S et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatolgy Group. Br J Dermatol 2002; 146:552-567.
Marghoob A, Kopf AW, Bart R. Set al Risk of another basal cell carcinoma developing after treatment of a basal cell carcinoma. J Am Acad Dermatol 1993; 28: 22-8.
Hauschild A, Breuninger H, Kaufmann R, Kortmann RD, Schwipper V, Werner J, Reifenberger J, Dirschka T, Garbe C; „Kurzleitlinie - Basalzellkarzinom der Haut“ 2008, basierend auf: Garbe C „Interdisziplinäre Leitlinien zur Diagnostik und Behandlung von Hauttumoren. Georg Thieme Verlag, Stuttgart, New York S. 1-11.
Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A. N Engl J Med. 2012;366:2171-9.
Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006 Jul;155(1):9-22.
Alomar A, Bichel J, McRae S. Vehicle-controlled, randomized, double-blind study to assess safety and efficacy of Imiquimod 5% cream applied once daily 3 days per week in one or two courses of treatment of actinic keratoses on the head. Br J Dermatol. 2007; 157:133-141.
Askew DA, Mickan SM, Soyewr HP, Wilkinson D. Effectiveness of 5-fluorouracil treatment for actinic keratosis--a systematic review of randomized controlled trials. Int J Dermatol. 2009;48:453-463.
Braathen LR, Szeimies RM, Basset-Seguin N, Bissonnette R, Foley P, Pariser D, Roelandts R, Wennberg AM, Morton CA. International Society for Photodynamic Therapy in Dermatology. Guidelines on the use of photodynamic therapy for nonmelanoma skin cancer: an international consensus. International Society for Photodynamic Therapy in Dermatology, 2005. J Am Acad Dermatol. 2007;56:125-143.
Breuninger H, Bootz F, Hauschild A Kortmann RD, Wolff K, Stockfleth E, Szeimies M, Rompel R, Garbe C. „Onkologische Leitlinien: Platten-epithelkarzinom der Haut. J Dtsch Dermatol Ges. 2008 .6 Suppl 1:5-8.
Falagas ME, Angelousi AG, Peppas G. Imiquimod for the treatment of actinic keratosis: Ameta-analysis of randomized controlled trials. J Am Acad Dermatol. 2006;55:537-538.
Farasat S, Yu SS, Neel VA, Nehal KS, Lardaro T, Mihm MC, Byrd DR, Balch CM, Califano JA, Chuang AY, Sharfman WH, Shah JP, Nghiem P, Otley CC, Tufaro AP, Johnson TM, Sober AJ, Liégeois NJ. A new American Joint Committee on Cancer staging system for cutaneous squamous cell

58Hauttumore Melanom
Kapitel 4
carcinoma: creation and rationale for inclusion of tumor (T) characteristics. J Am Acad Dermatol. 2011;64:1051-1059.
Motley R, Kersey P, Lawrence C; British Association of Dermatologists; British Association of Plastic Surgeons; Royal College of Radiologists, Faculty of Clinical Oncology. Multiprofessional guidelines for the management of the patient with primary cutaneous squamous cell carcinoma. Br J Dermatol. 2002;14:18-25.
Pirard, D, Vereecken, P, Mélot, C, Heenen, M. Three percent diclo fe-nac in 2.5% hyaluronan gel in the treatment of actinic keratoses: a meta-analysis of the recent studies. Arch Dermatol Res. 2005;297:185-189.
Stratigos A, Garbe C, Lebbe C, Malvehy J, del Marmol V, Pehamberger H, Peris K, Becker JC, Zalaudek I, Saiag P, Middleton MR, Bastholt L, Testori A, Grob JJ; European Dermatology Forum (EDF); European Asso ci ation of Dermato-Oncology (EADO); European Organization for Research and Treatment of Cancer (EORTC), Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur J Cancer. 2015 Sep;51(14):1989-2007.
Trakatelli M, Ulrich C, del Marmol V, Euvrard S, Stockfleth E, Abeni D. Epidemiology of nonmelanoma skin cancer (NMSC) in Europe: accurate and comparable data are needed for effective public health monitoring and interventions. Br J Dermatol. 2007;156 Suppl 3:1-7.
Zouboulis ChC. Principles of cutaneous cryosurgery: An update. Dermatology. 1999; 198:11-117.
Becker J, Mauch C, Kortmann RD, Keilholz U, Bootz F, Garbe C, Hauschild A, Moll I; „Kutanes neuroendokrines Karzinom (Merkelzellkarzinom)“, 2008, basierend auf den „Interdisziplinären Leitlinien zur Diagnostik und Behandlung von Hauttumoren” (C.Garbe, ed., 2005, Georg Thieme Verlag, Stuttgart, New York S.),
Bichakjian CK, Lowe L, Lao CD, Sandler HM, Bradford CR, Johnson TM, Wong SL. Merkel cell carcinoma: critical review with guidelines for multidisciplinary management. Cancer, 2007 1;110:1-12.
Feng H, Shuda M, Chang Y, Moore PS; Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008 Feb 22;319(5866):1096-100.
Heute D, Kostron H, von Guggenberg E, Ingorokva S, Gabriel M, Dobrozemsky G, Stockhammer G, Virgolini IJ.; Response of recurrent high-grade glioma to treatment with (90)Y-DOTATOC. J Nucl Med. 2010 Mar;51(3):397-400.

Hauttumore Melanom 59
Kapitel 5
Lemos BD, Storer BE, Iyer JG, Phillips JL, Bichakjian CK, Fang LC, Johnson TM, Liegeois-Kwon NJ, Otley CC, Paulson KG, Ross MI, Yu SS, Zeitouni NC, Byrd DR, Sondak VK, Gershenwald JE, Sober AJ, Nghiem P; Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. 2010 Nov;63(5):751-61.
Meier G, Waldherr C, Herrmann R, Maecke H, Mueller-Brand J, Pless M.; Successful targeted radiotherapy with 90Y-DOTATOC in a patient with Merkel cell carcinoma. A Case Report. Oncology. 2004;66(2):160-3.
Breuninger H, Sebastian G, Garbe C (2005): Deutsche Leitlinie Dermato-fibro sarcoma protuberans. In: Garbe C (Hrsg.) Interdisziplinäre Leit-linien zur Diagnostik und Behandlung von Hauttumoren. Georg Thieme Verlag, Stuttgart, New York pp62–68.
Fletcher CD, Evans BJ, MacArtney JC, Smith N, Wilson Jones E, McKee PH. Dermatofibrosarcoma protuberans: a clinicopathological and immunohistochemical study with a review of the literature. Histopathology. 1985;9:921-938.
Hafner HM, Moehrle M, Eder S, Trilling B, Rocken M, Breuninger H. 3D-Histological evaluation of surgery in dermatofibrosarcoma pro-tuberans and malignant fibrous histiocy-toma: Differences in growth patterns and outcome. Eur J Surg Oncol. 2008;34:680-686.
Kransdorf MJ, Meis Kindblom JM. Dermatofibrosarcoma protuberans: radiologic appearance. AJR Am J Roentgenol. 1994;163: 391-394.
Marks LB, Suit HD, Rosenberg AE, Wood WC. Dermatofibrosarcoma protuberans treated with radiation therapy. Int J Radiat Oncol Biol Phys. 1989;17: 379-384.
McArthur GA, Demetri GD, van Oosterom A, Heinrich MC, Debiec-Tychter M, Corless CL, Nikolova, Dimitijevic S. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
Parker TL, Zitelli JA. Surgical margins for excision of dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1995;32:233-236.
Smola MG, Soyer HP, Scharnagl E (1991) Surgical treatment of dermatofibrosarcoma protuberans. A retrospective study of 20 cases with review of literature. Eur J Surg Oncol 17: 447-453.
Ugurel S, Kortmann RD, Mohr P, Mentzel T, Garbe C, Breuninger H. „Onkologische Leitlinien: Dermatofibrosarcoma protuberans. J Dtsch Dermatol Ges. 2008 May;6 Suppl 1:19-20.





















