
ATLAS Der Kalorimetertrigger Experimentelle Teilchen- und Astroteilchen- Physik Das Trigger-System.

Entwicklung säurelabiler Trigger-Gruppen auf Basis von Schiffschen Basen des
p-Aminobenzylalkohols
Inaugural-Dissertation
zur Erlangung der Doktorwürde
der Fakultät für Chemie, Pharmazie und Geowissenschaften
der Albert-Ludwigs-Universität
Freiburg im Breisgau
vorgelegt von
Ivonne Anita Müller geboren in Fulda
2010

Die vorliegende Arbeit entstand im Zeitraum von Oktober 2006 bis
September 2010 in der Klinik für Tumorbiologie, Freiburg im Breisgau unter
Anleitung von Herrn Dr. A. Warnecke und Herrn Dr. F. Kratz in Kooperation mit
Herrn Prof. Dr. M. Jung am Institut für Pharmazeutische Chemie der Albert-
Ludwigs-Universität, Freiburg im Breisgau.
Vorsitzender des Promotionsausschusses: Prof. Dr. R. Schubert
Referent: Prof. Dr. M. Jung
Koreferent: Prof. Dr. C. Unger
Verkündigung des Prüfungsergebnisses: 04.11.10

Posterbeiträge
I. A. Müller, F. Kratz, A. Warnecke; Self-immolative linkers for the design of acid-
sensitive polymer conjugates of Camptothecin, 7th International Symposium on
Polymer Therapeutics: From Laboratory to Clinical Practice, Valencia, 2008,
Abstract book p. 141.
I. A. Müller, F. Kratz, M. Jung, A. Warnecke; Imines based on p-aminobenzyl
alcohol as versatile trigger groups for the design of pH-sensitive anticancer
prodrugs, 10th Tetrahedron Symposium, Paris, 2009, Abstract book B038.
A. Warnecke, I. A. Müller, F. Kratz; Linear self-eliminating systems – oligomers for
a controlled release of effector molecules, 10th Tetrahedron Symposium, Paris,
2009, Abstract book B019.
Publikationen
F. Kratz, I. A. Müller, C. Ryppa, A. Warnecke; Prodrug strategies in anticancer
chemotherapy, ChemMedChem, 2008, 3 (1), 20–53 (Review).
I. A. Müller, F. Kratz, M. Jung, A. Warnecke; Schiff bases derived from
p-aminobenzyl alcohol as trigger groups for pH-dependent prodrug activation,
Tetrahedron Lett., 2010, 51 (33), 4371–4374.
Patent
I. A. Müller, A. Warnecke; Acid-labile trigger units, Europäische Patentanmeldung,
EP 09008150.6.

Danksagung
Mein besonderer Dank gilt meinem Doktorvater Prof. Dr. Manfred Jung für seine
hilfreichen Ratschläge, aufbauenden Worte und für sein großes Engagement bei
der Beschaffung finanzieller Mittel.
Weiterhin möchte ich Herrn Prof. Dr. Clemens Unger für die Annahme des
Koreferats danken.
Für die große Unterstützung bei der Überwindung zahlreicher Hindernisse und das
allseits offene Ohr danke ich Herrn Dr. André Warnecke.
Bei Herrn Dr. Felix Kratz möchte ich mich für die freundliche Aufnahme in seine
Arbeitsgruppe und die finanzielle Unterstützung bedanken.
An dieser Stelle gilt mein Dank Volker Brecht für die Aufnahme der NMR-
Spektren. Dr. Jürgen Wörth und Christoph Warth bin ich für das Erstellen der
Massenspektren zu Dank verpflichtet. Florian Tönnies danke ich für die
Durchführung der Elementaranalysen.
Vielen Dank sage ich auch unserer fleißigen Laborantin Tamara Huck und Florian
Vonberg, der im Rahmen seiner Ausbildung einen wesentlichen Beitrag zu dieser
Arbeit geleistet hat.
Lisa, Katrin und Steffi möchte ich für die Zerstreuung an so manch
feuchtfröhlichen und witzigen Abenden danken.
Abschließend möchte ich noch ein herzliches Dankeschön an alle aktuellen und
ehemaligen Kollegen der Arbeitsgruppen Kratz und Jung aussprechen, die durch
eine angenehme Arbeitsatmosphäre zum Gelingen dieser Arbeit beigetragen
haben.

Abkürzungsverzeichnis
Å Längeneinheit (1 Å = 10-10 m) LC liquid chromatography
abs. absolut (engl.)
Ac Acetyl LFER linear free energy
MeCN Acetonitril relationship (engl.)
AMC 7-Amino-4-methylcumarin Me Methyl
Boc tert.-Butoxycarbonyl MeOH Methanol
CI Chemische Ionisation mPEGxk Monomethoxypoly-
CuAAC Kupfer-katalysierte Alkin-Azid- (ethylenglykol) (x kDa)
Cycloaddition Ms Methansulfonyl
∆ Erwärmen MS Massenspektrometrie
DBTL Dibutylzinndilaurat NEt3 Triethylamin
DC Dünnschichtchromatographie NMR nuclear magnetic
DCM Dichlormethan resonance (engl.)
DIEA N-Ethyldiisopropylamin Np p-Nitrophenyl
DMAP N,N-Dimethyl-4-aminopyridin PABA p-Aminobenzylalkohol
DMF N,N-Dimethylformamid PABC p-Aminobenzyloxy-
DMSO Dimethylsulfoxid carbonyl
EI Elektronenstoß-Ionisation PE Poly(ethylen)
em. Emission PEG Poly(ethylenglykol)
ESI Elektrospray-Ionisation PMDETA N,N,N’,N’,N’’-Penta-
Et2O Diethylether methyldiethylentriamin
EtOH Ethanol PPTS Pyridinium-p-
EPR enhanced permeability and toluolsulfonat
retention (engl.) RP reversed phase (engl.)
eq equivalent (engl.) RT Raumtemperatur
ex. Excitation Smp. Schmelzpunkt
ges. gesättigt t1/2 Halbwertszeit
Glch. Gleichung TBS tert.-Butyldimethylsilyl
HSA Humanserumalbumin TFA Trifluoressigsäure
I Fluoreszenzintensität THF Tetrahydrofuran
konz. konzentriert TMS Tetramethylsilan

IUPAC-Nomenklatur ausgewählter Verbindungen
Für die aufgeführten Verbindungen werden im Folgenden ausschließlich
Trivialnamen verwendet.
Doxorubicin (7S,9S)-7-[(2R,4S,5S,6S)-4-Amino-5-hydroxy-6-methyloxan-2-
yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-
dihydro-7H-tetracen-5,12-dion
OHOH
O
OO OH
OH O
OO
NH2
HO
Paclitaxel 4α,10β-Bis(acetyloxy)-13α-[(2R,3S)-3-(benzoylamino)-2-
hydroxy-3-phenylpropanoyloxy]-1,7β-dihydroxy-9-oxo-5β,20-
epoxytax-11-en-2α-yl-benzoesäureester
O
O
OO
OH
OOO
HO
NH
OH
OO
O
O

Inhaltsverzeichnis 1. Einleitung ........................................................................................................1 1.1 Prodrugstrategien in der antitumoralen Chemotherapie ...................................1
1.1.1 Aktives Tumor-Targeting ...................................................................3
1.1.2 Passives Tumor-Targeting unter Ausnutzung des EPR-Effekts ........3
1.2 Strategien für eine tumorspezifische Aktivierung von Prodrugs – Konzept der
säurelabilen Freisetzung...................................................................................5
1.3 Strategie der doppelten Prodrugs...................................................................12
1.4 Linker-Systeme für eine gezielte Freisetzung von Wirkstoffen .......................15
1.4.1 Selbsteliminierende Linker ..............................................................15
1.4.2 Chemische Adapter-Systeme..........................................................18
2. Zielsetzung....................................................................................................20 3. Ergebnisse und Diskussion.........................................................................22
3.1 Entwicklung niedermolekularer AMC-Modellverbindungen mit säurelabil
getriggertem PABC-Linker..............................................................................22
3.1.1 Synthese niedermolekularer, säurelabiler Modellverbindungen .......22
3.1.2 Synthese einer Modellsubstanz zur Untersuchung der
1,6-Eliminierungsreaktion.................................................................27
3.1.3 Untersuchung der pH-abhängigen Spaltungseigenschaften der
niedermolekularen AMC-Modellverbindungen 1b, 4b–14b und 17 ..28
3.1.4 Untersuchung der Substituenteneffekte hinsichtlich der Linearen
Freien Enthalpie-Beziehung (LFER).................................................34
3.1.5 Untersuchung der Hydrolyseeigenschaften von 13b und 14b im
physiologisch relevanten pH-Bereich ...............................................36
3.1.6 Untersuchung der Plasmastabilität ausgewählter Modell-
verbindungen ...................................................................................39
3.2 Entwicklung makromolekularer, säurelabiler Verbindungen unter Verwendung
eines neuen trifunktionellen Linker-Systems auf Basis von 3-Amido-4-
hydroxymethylanilin ........................................................................................42
3.2.1 Syntheseversuche makromolekularer PEG-geträgerter
Verbindungen...................................................................................45
3.2.1.1 Synthesestrategien ......................................................................45

3.2.1.2 Synthese niedermolekularer Vorstufen zur Darstellung
makromolekularer AMC-Modellverbindungen bzw. Prodrugs ......46
3.2.1.3 Untersuchungen der pH-abhängigen Spaltungseigenschaften der
niedermolekularen Modellverbindungen und Prodrugs 28, 29 und
38–41 ...........................................................................................50
3.2.1.4 Versuche zur Darstellung makromolekularer AMC-Modellverbin-
dungen und Prodrugs mittels CuAAC ..........................................53
3.2.2 Alternative Synthesestrategien zur Darstellung makromolekularer
AMC-Modellverbindungen................................................................58
3.2.3 Abschließende Untersuchungen zur Eignung von 3-Amido-4-
hydroxymethylanilin als Adapter-Baustein........................................62
3.3 Säurelabile PEG-Konjugate auf Basis des 2,4-Bis(hydroxymethyl)anilin-
Linkers ............................................................................................................64
3.3.1 Synthese und Charakterisierung der makromolekularen AMC-
Modellverbindungen.........................................................................65
3.3.2 Synthese einer pH-unempfindlichen Vergleichsubstanz ..................68
3.3.3 Untersuchung der Stabilität der Vergleichsubstanzen (16 und 55)
sowie der Spaltungseigenschaften der PEG-Konjugate 53 und 54 ..70
3.3.4 Untersuchung der Plasmastabilität von 53 und 54 ..........................75
3.4 Untersuchung der nachgelagerten 1,4-Benzyleliminierung anhand
niedermolekularer AMC-Modellverbindungen.................................................76
4. Fazit und Ausblick ........................................................................................81
5. Zusammenfassung .......................................................................................82 6. Experimenteller Teil .....................................................................................87 6.1 Materialien und Methoden ..............................................................................87
6.1.1 Verwendete Reagenzien .................................................................87
6.1.2 Verwendete Geräte und Materialien................................................87
6.1.3 Chromatographie.............................................................................89
6.1.4 Fluorometrische Bestimmung der Halbwertszeiten der AMC-Modell-
verbindungen in Puffer-, Plasma- und Albuminlösungen..................89
6.2 Chemische Synthesen....................................................................................91
6.2.1 Synthese der niedermolekularen AMC-Modellverbindungen 1b und
4b–14b.............................................................................................91

6.2.2 Synthese der Modellsubstanz 17 ..................................................111
6.2.3 Synthese niedermolekularer Vorstufen für die Darstellung von PEG-
geträgerten AMC-Modellverbindungen mittels CuAAC ..................113
6.2.4 Synthese niedermolekularer Vorstufen für die Darstellung von PEG-
geträgerten Prodrugs mittels CuAAC .............................................125
6.2.5 PEG-Synthesen am Beispiel von PEG2k ........................................136
6.2.6 Synthese niedermolekularer Vorstufen zur Darstellung von AMC-
Modellverbindungen über zur CuAAC alternative Reaktionen .......138
6.2.7 Synthese der makromolekularen Modellverbindungen 53–55.......140
6.2.8 Synthese der niedermolekularen Modellverbindungen 56 und 57 .145
7. Literatur .......................................................................................................152
Anhang ...............................................................................................................158

Einleitung _________________________________________________________________
- 1 -
1. Einleitung
1.1 Prodrugstrategien in der antitumoralen Chemotherapie
Nach Herz- und Kreislauferkrankungen ist Krebs noch immer die zweithäufigste
Todesursache in Deutschland. Die Anzahl der an Krebs neuerkrankten Patienten
wächst vor allem aufgrund der gestiegenen Lebenserwartung kontinuierlich
(Tab. 1.1). Tab. 1.1: Zahlen zu registrierten Krebsneuerkrankungen und -sterbefällen in Deutschland aus dem
Jahr 2006 und eine Schätzung für 2010. Mit Ausnahme der Erkrankungen am malignen Melanom
wurden Hautkrebserkrankungen nicht berücksichtigt.1
Männer Frauen
Erkrankungsfälle 2006 229.200 197.600
Projektion für 2010 246.200 204.000
Sterbefälle 2006 112.438 98.492
Häufigste Krebserkrankung; Prostatakrebs Brustkrebs
daran in 2006 neu Erkrankte 60.100 58.000
Auch wenn die Überlebensraten in den letzten Jahren für einzelne Krebsarten wie
z. B. Prostatakrebs durch moderne Therapien deutlich gesteigert werden konnten,
gibt es noch immer viele Krebserkrankungen, bei denen konventionelle
Behandlungsmethoden versagen. Aus diesem Grund wurden auf der Suche nach
neuen Therapieoptionen in den letzten Jahrzehnten intensiv die Entstehung und
Pathophysiologie von Krebserkrankungen erforscht.2-4 Insbesondere die neuen
Erkenntnisse zu den auf biochemischer und molekularbiologischer Ebene
ablaufenden Vorgängen führten zu zahlreichen neuen Therapieansätzen. Dazu
gehören beispielsweise die Behandlung mit monoklonalen Antikörpern5-8 sowie mit
Inhibitoren von Tyrosinkinasen,9-11 DNA-Methyltransferasen bzw. Histondes-
acetylasen,12-15 der Angiogenese16,17 und des Proteasoms.18-20 Weitere neue
Entwicklungen sind Impfungen21 und die somatische Gentherapie.22

Einleitung _________________________________________________________________
- 2 -
Da sie zumeist nur gegen bestimmte Tumorentitäten gerichtet sind, ließen sich
diese neuen, vielversprechenden Methoden bisher nur in begrenztem Maße
erfolgreich etablieren. Die klassische und vielseitig einsetzbare Chemotherapie
von Tumoren mit zytostatischen, d. h. zellteilungshemmenden Wirkstoffen bildet
daher weiterhin eine der tragenden Säulen bei der Behandlung von
Krebserkrankungen. Gravierende systemische Nebenwirkungen wie Haarausfall,
Blutbildstörungen, Übelkeit und Diarrhöen, die sich dosislimitierend auswirken,
verhindern jedoch in vielen Fällen, dass sich das therapeutische Potenzial dieser
Arzneistoffe vollständig ausschöpfen lässt. Das grundlegende Problem hierfür liegt
in der mangelnden Tumorselektivität der klinisch eingesetzten Zytostatika. Zur
Verbesserung der Behandlungsmöglichkeiten von Krebspatienten sind deshalb
therapeutische Konzepte erforderlich, die das pharmakologische Potenzial eines
tumorhemmenden Wirkstoffs durch ein Anreichern (Tumor-Targeting, Abb. 1.1)
und gezieltes Freisetzen im Tumor erhöhen und gleichzeitig die toxischen
Wirkungen auf den gesunden Teil des Körpers verringern. In diesem
Zusammenhang wurden in der Vergangenheit sowohl aktive als auch passive
Targeting-Ansätze verfolgt, von denen sich insbesondere die Verwendung von
Makromolekülen als Transportvehikel bewährt hat.23-26
Träger-Wirkstoff-Konjugat
WirkstoffTargeting-Einheit
Aktives Targeting (Kap. 1.1.1)
monoklonale Antikörper
Rezeptor-affine Liganden
Passives Targeting (Kap. 1.1.2)
Polymere oder Proteine miteiner molaren Masse >30 kDa
Abb. 1.1: Schematische Darstellung der Tumor-Targeting-Strategie, bei der Wirkstoffe an einen
Träger gebunden werden.

Einleitung _________________________________________________________________
- 3 -
1.1.1 Aktives Tumor-Targeting
Das aktive Targeting basiert auf Antigenen und Rezeptoren, die in gesunden und
malignen Geweben an der Zelloberfläche unterschiedlich stark exprimiert werden.
Beispielsweise können Zellmarker wie die „Unterscheidungsgruppen“ (cluster of
differentiation (CD)),27,28 Integrine29-31 und der Folatrezeptor32,33 als Angriffsziele
für Wirkstoffkonjugate dienen, in die zum Erkennen eines spezifischen
Oberflächenmerkmals monoklonale Antikörper oder Liganden mit hoher
Rezeptoraffinität integriert wurden. Nach Bindung an der Zelloberfläche werden
die Konjugate schließlich entweder extrazellulär oder im Anschluss an die
endozytotische Aufnahme intrazellulär gespalten.
Nachteilig für dieses vielversprechende Konzept ist die oftmals nicht sonderlich
stark ausgeprägte Tumor-Spezifität der Liganden und Antikörper.34,35 Außerdem
wird der Therapieeffekt von der Anzahl der an der Zelloberfläche vorhandenen
Marker sowie deren Heterogenität begrenzt, und nur geringe Mengen des
Konjugats gelangen in die häufig schlecht vaskularisierten Regionen des
Tumors.36 Dennoch wurden mit der aktiven Targeting-Strategie Erfolge erzielt.37,38
Beispielsweise wurde das Antikörper-Wirkstoff-Konjugat Gemtuzumab
Ozogamicin (Handelsname: Mylotarg®) (siehe Kap. 1.2) zur Behandlung der
akuten myeloischen Leukämie zugelassen.39
Gegenüber der aktiven Targeting-Strategie sind passive Ansätze deutlich
preisgünstiger zu realisieren. Daher lohnt es sich, diese trotz weniger stark
ausgeprägtem Targeting-Effekt zu verfolgen.
1.1.2 Passives Tumor-Targeting unter Ausnutzung des EPR-Effekts
Die Strategie des passiven Targetings basiert auf dem Phänomen, dass sich
synthetische Polymere, aber auch Serumproteine mit einem Molekulargewicht
>30 kDa im Tumorgewebe auf eine im Vergleich zu gesundem Gewebe um den
Faktor 5-10040 erhöhte Konzentration anreichern. Dieser passive Effekt, der auch
als EPR-Effekt (enhanced permeability and retention) bezeichnet wird (Abb. 1.2,
S. 4), beruht auf einer für Makromoleküle erhöhten kapillaren Durchlässigkeit des

Einleitung _________________________________________________________________
- 4 -
Tumorgewebes, verbunden mit einem defekten abführenden Lymphsystem.
Zudem verfügen Makromoleküle über eine gegenüber niedermolekularen
Verbindungen veränderte Pharmakokinetik, die aus einer herabgesetzten renalen
Filtration resultiert und zu höheren Plasmahalbwertszeiten führt.41,42 In späteren
Arbeiten konnte zudem gezeigt werden, dass sich der EPR-Effekt, z. B. durch
Gabe von blutflusserhöhenden Mitteln wie Glycerintrinitrat40 bzw. blutdrucker-
höhenden Mitteln wie Angiotensin II,43 zusätzlich verstärken lässt.
Die Anreicherung im Tumorgewebe wurde bereits für eine ganze Reihe
synthetischer Polymere wie Styrol-Maleinsäure-Copolymere (z. B. SMANCS),44 N-
(2-Hydroxypropyl)methacrylamid- (HPMA)-Copolymere,45-47 Poly(ethylenglyko-
le),48,49 Poly(L-glutaminsäure),50 Poly(vinylalkohol),51,52 aber auch für Biomoleküle
wie Serumproteine53,54 nachgewiesen. Diese Ergebnisse deuten darauf hin, dass
der EPR-Effekt eine universelle Erscheinung ist, die weitgehend unabhängig von
der chemischen Natur des Makromoleküls auftritt.
Lymphatic System
Normal tissue
pH ~7
Blood streamMacromolecules
Tumor tissue
pH ~6
Small Molecules
Lymphatic system
Small molecules
Macromolecules
Lymphatisches System
Makromoleküle
BlutstromKleine
Moleküle
Normales Gewebe
Tumorgewebe
Abb. 1.2: Schematische Darstellung eines an gesundes und tumorales Gewebe grenzenden
Blutgefäßes.23
Neben der Größe müssen bei der Auswahl eines geeigneten Trägers für
Wirkstoffe weitere Faktoren berücksichtigt werden.55 Einerseits sollte das
Makromolekül einen gezielten Transport von eingeschlossenen oder gebundenen
Arzneistoffen ins Zielgewebe gewährleisten, andererseits ist ein effizientes und
möglichst spezifisches Freisetzen des Wirkstoffs am Zielort für eine erfolgreiche
Anwendung zwingend erforderlich. Weitere Anforderungen an den Träger sind

Einleitung _________________________________________________________________
- 5 -
Bioverträglichkeit, gute Wasserlöslichkeit sowie synthetisch-chemische Diversität.
All diese gewünschten Eigenschaften besitzt das synthetische Polymer
Poly(ethylenglykol) (PEG), weshalb es sich besonders für die Entwicklung
therapeutischer Polymerkonjugate eignet. So konnte durch "Pegylierung" von
Proteinen56-58 deren Immunogenität und Pharmakokinetik verbessert werden.
Zudem schützt PEG Wirkstoffe59-61 vor Angriffen durch Enzyme oder Antikörper.
Neben der Wahl des Trägermoleküls ist der Einbau einer geeigneten
Sollbruchstelle zwischen Wirkstoff und Makromolekül von entscheidender
Bedeutung für die Wirksamkeit von Konjugaten.
1.2 Strategien für eine tumorspezifische Aktivierung von Prodrugs – Konzept der säurelabilen Freisetzung
Zur Realisierung einer tumorspezifischen Freisetzung wurden zahlreiche
Methoden62 entwickelt, die biochemische oder pathophysiologische
Besonderheiten des Zielgewebes bzw. der Zielzellen zur Freisetzung des
Wirkstoffs ausnutzen. So wurden in der Vergangenheit erfolgreich
tumorassoziierte, d. h. im Tumor (über-)exprimierte, Proteasen für die Aktivierung
von Prodrugs (siehe Kap. 1.3) ausgenutzt. Zu diesen Enzymen gehören
beispielsweise Cathepsine,63,64 Matrixmetalloproteinasen,65 das Prostata-
spezifische Antigen66 und zahlreiche weitere Biokatalysatoren.67-69
Darüber hinaus können hypoxische Tumorzellen, die häufig nicht auf eine
klassische Behandlung durch Strahlentherapie oder Zytostatika ansprechen,
gezielt durch reduktiv-spaltbare Prodrugs abgetötet werden.70-75
Eine in der Regel vielseitigere Methode als beispielsweise die enzymatische
Aktivierung, die eine (Über-)Expression der Zielenzyme bei den zu behandelnden
Tumorentitäten voraussetzt, ist die Verwendung von säurelabilen Sollbruch-
stellen. Diese Freisetzungsstrategie beruht auf einem universellen, in allen
Tumorzellen beobachtbaren Phänomen. Durch nicht-invasive Techniken konnte
gezeigt werden, dass der interstitielle pH-Wert in Tumorgewebe häufig um 0.5–1.0
pH-Einheiten niedriger liegt als in gesundem Gewebe,73 während sich der

Einleitung _________________________________________________________________
- 6 -
zytosolische pH-Wert von Tumorzellen nicht von dem anderer Zellen
unterscheidet.
Einen deutlich größeren pH-Abfall erfährt ein makromolekulares Prodrug während
seiner zellulären Aufnahme. Makromoleküle werden – anders als niedermole-
kulare Verbindungen – nicht mittels Diffusion durch die Zellmembran, sondern
über Endozytose in eine Zelle aufgenommen. Abhängig von der chemischen Natur
des Makromoleküls kann dieser Vorgang unspezifisch und/oder rezeptorvermittelt
ablaufen (siehe Abb. 1.3).
Endosom
Primäre oder sekundäre Lysosome
pH 4
pH 6.5-5Recycling
Diffusion des Wirkstoffsin das Zytoplasma
Flüssig-Phasen-EndozytoseAdsorptive Endozytose
Rezeptor-vermittelteEndozytoseMakromolekulares
Prodrug
ExtrazellulärerRaumpH 7.2-7.4
Abb. 1.3: Zelluläre Mechanismen zur Aufnahme von makromolekularen Prodrugs.23
Im Verlauf der Endozytose werden durch Einstülpungen der Zellmembran kleine
Vesikel, die Endosomen, gebildet. In deren Inneren sinkt – verursacht durch eine
Adenosintriphosphat- (ATP-) abhängige Protonenpumpe – der pH-Wert von
ursprünglich 7.2–7.4 im extrazellulären Raum auf 6.5–5.0. Bei weiterer
Umwandlung in primäre oder sekundäre Lysosomen kann der pH-Wert bis auf
pH 4 abfallen. Eindrucksvolle Aufnahmen von Endosomen in HeLa-Zellen wurden
kürzlich von SERRESI et al.76 veröffentlicht (Abb. 1.4, S. 7). Durch Fusion zweier
Proteine (des ratiometrischen pH-Indikators E1GFP mit HIV-Tat, das eine
endozytotische Aufnahme des fusionierten Proteins gewährleistet) und

Einleitung _________________________________________________________________
- 7 -
anschließender Inkubation der Zellen ließen sich Echtzeitaufnahmen des
vollständigen endozytotischen Prozesses gewinnen. Abbildung 1.4 zeigt eine
Momentaufnahme des untersuchten Vorgangs 6 h nach Inkubation der Zellen mit
Tat-E1GFP. Zu diesem Zeitpunkt lag der pH-Wert im Inneren der Endosomen bei
pH 5.83.
Abb. 1.4: Messungen der pH-Änderungen in Vesikeln 6 h nach Inkubation mit Tat-E1GFP.
a) Konfokale Aufnahme von HeLa-Zellen, b) Ratiometrische Übersicht der pH-Werte in mit
Tat-E1GFP-angereicherten Vesikeln, c) Vergrößerter Ausschnitt aus Abbildung a, d) Vergrößerter
Ausschnitt aus Abbildung b (Diese Abbildung wurde entnommen aus SERRESI et al.76).
Insbesondere der deutliche pH-Sprung von 2–3 Einheiten nach zellulärer
Aufnahme lässt sich für das Design von Prodrugs mit einer säureempfindlichen
Bindungsstelle zwischen Wirkstoff und Träger ausnutzen. Chemisch lassen sich
pH-abhängige Sollbruchstellen beispielsweise durch Acetal-77,78 bzw. Orthoester-
Bindungen,79,80 cis-Aconityl-51,81,82 sowie Trityl-Spacer,83 N-Alkoxyalkylimidazole,84
Acylhydrazone85-89 und Imine26,90,91 realisieren (Abb. 1.5, S. 8). Bei der Wahl einer
für die Entwicklung von säurelabilen Prodrugs geeigneten Sollbruchstelle sind

Einleitung _________________________________________________________________
- 8 -
verschiedene Faktoren zu berücksichtigen. Die Bindung sollte bei neutralem
pH-Wert und im Blut stabil sein und im leicht Sauren rasch hydrolysieren.
Außerdem sollte sie vielseitig einsetzbar sein sowie einfach und effektiv zwischen
Wirkstoff und Träger aufgebaut werden können. Die Möglichkeit,
Spaltungseigenschaften durch geringfügige Strukturvariationen maßzuschneidern,
wäre ein weiterer Vorteil. Während Acylhydrazone zumeist für eine medizinische
Anwendung geeignete Spaltungseigenschaften besitzen, weisen Acetale und
Orthoester häufig eine zu hohe Stabilität bei pH 4–5 auf.77,79 Aliphatische Imine
sind in der Regel äußerst säureempfindlich, jedoch besitzen aromatische Imine
günstige Hydrolyseeigenschaften. Als Sollbruchstelle eignet sich ebenfalls die
cis-Aconityl-Bindung. Allerdings entsteht beim Aufbau dieser Bindung ein oftmals
deutlich stabileres trans-Isomer, welches aufwändig aus dem Gemisch entfernt
werden muss.51 Unter Berücksichtigung der säurelabilen Eigenschaften und des
Syntheseaufwands eignen sich Acylhydrazon- bzw. aromatische Imin-Bindungen
besonders für eine Anwendung als Sollbruchstelle in pH-empfindlichen Prodrugs
zu empfehlen.
R
NNH
R
NNH
O
R
N
NH
NH
O OHO O
NH
O O
R R
O O
R H
Imin-Bindung Hydrazon-Bindung Acylhydrazon-Bindung
Acetal-BindungKetal-Bindung
cis-Aconityl-Bindung Trityl-Bindung
O O
H
O O
Roder oder
Abb. 1.5: Die wichtigsten pH-sensitiven funktionellen Gruppen für die Entwicklung säurelabiler
Prodrugs.23
Ein säurelabiles Antikörper-Wirkstoff-Konjugat mit integrierter Acylhydrazon-
Bindung, das bereits zur Behandlung der akuten myeloischen Leukämie
zugelassen wurde, ist Gemtuzumab Ozogamicin (Handelsname: Mylotarg®).

Einleitung _________________________________________________________________
- 9 -
Dieses Prodrug besteht aus dem Antikörper P67.6 (für selektives Targeting von
CD33) und einem Acylhydrazid-Derivat des hochpotenten Endiin-Antibiotikums
Calicheamicin.39 Verbunden sind die beiden Einheiten über einen
4-(4’-Acetylphenoxy)butansäure-Linker. Nach endozytotischer Aufnahme des
Antikörper-Wirkstoff-Konjugats wird zunächst die säurelabile Acylhydrazon-
Bindung zwischen Linker und Wirkstoff gespalten und anschließend durch
Reduktion der Disulfid-Bindung das Endiin aktiviert (Abb. 1.6).
OO
OHMeOHO
I
OMe
OMe
O
OS
OH
O O
HO ONH
O
O
MeON
H
O
S
NHCO2MeHO
S
NHN
O
OO
NH
Et
O2-3
1. Hydrolyse
2. Reduktion
Abb. 1.6: Struktur des säurelabilen Prodrugs Mylotarg, das sich aus dem Antikörper P67.6, einem
4-(4’-Acetylphenoxy)butansäure-Linker und einem Derivat des Wirkstoffs Calicheamicin
zusammensetzt.23
Nicht immer lassen sich ein säurelabiler Ansatz und eine aktive Targeting-
Strategie so erfolgreich verbinden wie im Fall des Mylotarg. YANG et al.92 zeigten,
dass Folat-Wirkstoff-Konjugate mit integrierter Acylhydrazon-Bindung in Zellen nur
sehr langsam gespalten werden. Durch Fluoreszenzmessungen ermittelten sie
den pH-Wert in Endosomen, die durch Folatrezeptor-vermittelte Endozytose
entstanden. Dieser lag mit pH ~ 6.5 deutlich zu hoch für eine rasche Hydrolyse der
Acylhydrazon-Bindung. Darüber hinaus scheinen bei der Folatrezeptor-
vermittelten Endozytose keine Lysosomen gebildet zu werden, weshalb die
Konjugate keinen weiteren pH-Abfall erfahren können. Stattdessen wird der
Rezeptor durch Zurückkehren an die Zelloberfläche „recycelt“. Der Erfolg einer
säurelabilen Strategie ist folglich stark von der Art des endozytotischen
Aufnahmewegs abhängig.

Einleitung _________________________________________________________________
- 10 -
Beim passiven Targeting erfolgt die zelluläre Aufnahme über Flüssig-Phasen-
Endozytose oder adsorptive Endozytose.93 Neben Polymer-Wirkstoff-Konjugaten,
bei denen Wirkstoffe kovalent über säurelabile Bindungen mit ihrem Träger
verbunden werden, können ebenso Aggregate aus säurelabilen Polymeren wie
Micellen oder Hydrogele eine passive Anreicherung erfahren. Bei dieser neueren
Entwicklung werden Effektoren durch Polymere verkapselt, die in Folge des
pH-Abfalls während der endozytotischen Aufnahme gespalten werden und dabei
die eingeschlossenen Wirk- oder Farbstoffe freisetzen. Der Vorteil dieser Systeme
liegt in ihrer biologischen Abbaubarkeit. Zusätzlich zur Verhinderung einer
Vakuolbildung können solche Polymere nach ihrem Zerfall in eine große Anzahl
niedermolekularer Fragmente durch Destabilisierung der endosomalen Membran
den Austritt eines Effektors aus dem Endosom erleichtern.94 Als säurelabile
Sollbruchstellen wurden bislang hauptsächlich Imin-,26,95 Ketal-96,97 und Acetal-
Bindungen98-100 verwendet. Erfolgreich umgesetzt wurde die Strategie
beispielsweise mit dem Kinaseinhibitor Imatinib, der durch das aliphatische
Poly(ketal)-Copolymer (PK3) verkapselt wurde. In in-vivo-Experimenten konnte
eine verbesserte Wirksamkeit dieser Formulierung gegenüber freiem Inhibitor bei
Concanavalin A-induzierten Leberschäden in einem Mausmodell gezeigt
werden.97 Obgleich dieses Beispiel vielversprechend klingt, ist für eine breite
medizinische Anwendung der biologisch abbaubaren Polymere eine Optimierung
der Spaltungseigenschaften erforderlich. Bislang weisen die meisten Polymere mit
pH-empfindlichen Sollbruchstellen entweder eine zu hohe Stabilität im leicht
sauren oder eine zu geringe Stabilität im neutralen Milieu auf.26,99
Mehr Relevanz erlangten die säurelabilen Polymer-Wirkstoff-Konjugate, die in
Abbildung 1.7 (S. 11) dargestellt sind. Ein fortgeschrittenes klinisches Prüfstadium
erreichte beispielsweise das säurelabile, albuminbindende Doxorubicin-Derivat
INNO-206.85,101 In diesem Prodrug wurde der Wirkstoff über eine Acylhydrazon-
Bindung mit dem Linker 6-Maleinimidocapronsäurehydrazid verknüpft, der nach
i.v.-Applikation in situ an das Humanserumalbumin bindet. In Tiermodellen konnte
bereits gezeigt werden, dass aufgrund der passiven Anreicherung und der
gezielten säurelabilen Freisetzung in den Tumorzellen INNO-206 bei gleicher
Dosis viel wirksamer und dabei verträglicher ist als freies Doxorubicin.85 In Kürze

Einleitung _________________________________________________________________
- 11 -
wird die Wirksamkeit von INNO-206 in drei klinischen Phase-II-Studien gegen
Pankreas- und Magenkrebs sowie Weichteilsarkome untersucht werden.
Eine neuere Entwicklung stellen Nanopartikel dar, die aus PEG3.5k-PLA-Cisplatin-
Konjugaten (PLA = Poly(L-Lactid)) gebildet wurden.86 Ein aktiv wirksames
Cisplatin-Analogon wurde dabei kovalent über Acylhydrazon-Bindungen mit
jeweils zwei Copolymer-Ketten verknüpft. Die Halbwertszeiten für die
Wirkstofffreisetzung aus den Nanopartikeln betrugen in Pufferlösungen t1/2 = 4 h
(pH 5.0) und t1/2 = 22 h (pH 7.4). In-vitro-Untersuchungen ergaben, dass die
Nanopartikel in Tumorzellen zytotoxischer wirken als freies Cisplatin.
Über Imin-Bindungen wurde in einem weiteren Beispiel das Antimykotikum
Amphotericin B (AMB) an ein Copolymer aus PEG und Poly(L-Lysin) gebunden.102
In Pufferlösung bei pH 5.0 betrug die Halbwertszeit für die Wirkstofffreisetzung
t1/2 = 2 min. Hingegen wurden bei pH 7.4 innerhalb von 24 h weniger als 5 % des
AMB abgespalten.
NHN
O
OOCH3 OH
OH
OO
NH2
HO
H3C
OH
NO
O
O
OH
* HCl
S Cys34 HSA
INNO-206
PtCl
Cl NH3
NH3
O
O
O
O
N
NHN
NH
O
O
O
O
O
O
O
OHOOC-PEG-O
O
HOOC-PEG-O
O
90
90
Bi(PEG-PLA)-Pt (IV)-Konjugat
HN
O
NH
O
NAMB
NAMB
NH
OO
NH
O HN
HN
O
O
NAMB
NAMB
5 5228
Poly(ethylenglykol)-[b-poly(L-lysin)5]2-(AMB)12
O
OCOOH
H
OOH
OHOH
OH
OHOHO
OH
HO
O
OH
OHNH2
Amphotericin B (AMB)
A
B
Abb. 1.7: Beispiele für säurelabile Polymer-Wirkstoff-Konjugate mit Acylhydrazon-Bindung (A)
bzw. Imin-Bindung (B).
Die in den vorgestellten Beispielen verwendeten Wirkstoffe haben den Vorteil,
dass sie über funktionelle Gruppen verfügen, mit denen sie durch Aufbau
säurelabiler Acylhydrazon- oder Imin-Bindungen an einen makromolekularen
Träger gebunden werden können. Für potente Zytostatika, deren Strukturen keine

Einleitung _________________________________________________________________
- 12 -
reaktiven Carbonyl- bzw. Aminogruppen aufweisen, ist eine säurelabile
Formulierung durch Einbau tumorspezifisch spaltbarer Acylhydrazon- oder Imin-
Bindungen schwieriger zu realisieren. So müssten hydroxy-funktionalisierte
Wirkstoffe wie Paclitaxel und Camptothecin zunächst so derivatisiert werden, dass
eine Carbonyl- oder Aminofunktion in das Molekül integriert wird (Abb. 1.8).
N
N O
O
OOHO
O
OO
OH
OO
O
HO
NH
OH
OO
O
O
OHOH
O
OO OH
OH O
OO
NH2
HO
Paclitaxel Camptothecin Doxorubicin
Ester, Carbonat
Ester, CarbonatAmid, Carbamat
Hydrazon, Imin
Ester, Carbonat
Abb. 1.8: Möglichkeiten der Derivatisierung für Doxorubicin und die hydroxy-funktionalisierten
Wirkstoffe Camptothecin und Paclitaxel.
Dies kann beispielsweise durch Acylierung mit einem bifunktionalen Linker
geschehen.103 Allerdings ist bei dieser Strategie zwingend erforderlich, dass die
Wirksamkeit des Arzneistoffs auch nach der Derivatisierung erhalten bleibt, d. h.
die modifizierten Wirkstoffe eine der Stammverbindung vergleichbare
in-vivo-Wirksamkeit aufweisen. Da dies nicht immer gewährleistet ist, empfiehlt
sich die Verwendung einer doppelten Prodrug-Strategie, wie im Folgenden
erläutert wird.
1.3 Strategie der doppelten Prodrugs
Prodrugs im Allgemeinen sind inaktive Derivate von Arzneistoffen, die in vivo zur
aktiven Form metabolisiert werden.104,105 Insgesamt lassen sich ca. 5–7 %106 der
weltweit zugelassenen Wirkstoffe als Prodrugs klassifizieren. Diese können
abhängig von ihrem Anwendungsgebiet und ihrer Darreichungsform gegenüber

Einleitung _________________________________________________________________
- 13 -
klassischen Wirkstoffen verschiedene Vorteile bieten, z. B. eine geringere
Toxizität, erhöhte Wasserlöslichkeit oder Lipophilie, höhere chemische Stabilität,
verbesserte Passage der Blut-Hirn-Schranke, verbesserte orale Absorption oder
gesteigerte Selektivität für malignes Gewebe durch Drug Targeting*.
Die beiden häufigsten Methoden104 zur Derivatisierung von Wirkstoffen sind die
Veresterung von Carboxyl-, Hydroxyl- oder Thiol-Funktionalitäten und die
Synthese von Phosphatestern aus Hydroxyl- oder Amino-Gruppen. Darüber
hinaus können amino-funktionalisierte Wirkstoffe unter anderem in Prodrugs mit
Amid-, Carbamat- oder Imin-Funktion überführt werden. Beispiele für
niedermolekulare, bereits zugelassene Prodrugs sind in Abbildung 1.9 dargestellt.
NH
OOH H
N
OO OH
HNH
OHOH H
N
OO OH
H
Esterasen
Enalapril Enalaprilat
NHN
O
O
OPOH
HO
O
NHHN
O
O
Phosphatasen
Fosphenytoin Phenytoin
OHHO
NH2
O OH
OHHO
NH2
DOPA-Decarboxylase
L(evo)-DOPA Dopamin
ermöglichtorale Applikation
Vorteil des Prodrugs
erhöhteWasserlöslichkeit
verbesserte Passage derBlut-Hirn-Schranke
Anwendung
ACE-Hemmer
Epilepsie
Parkinson-Krankheit
Prodrug Aktiver Wirkstoff
Abb. 1.9: Beispiele für niedermolekulare Prodrugs.
Eine Weiterentwicklung des Prodrug-Konzepts stellt die doppelte Prodrug-
Strategie dar.107 In Abbildung 1.10 (S. 14) ist der schematische Aufbau und das
Prinzip der Wirkstofffreisetzung eines doppelten Prodrugs dargestellt.
* Der Begriff Drug Targeting lässt sich mit „zielgerichteter Wirkstofftransport“ nur schlecht ins Deutsche übersetzen und umfasst die in Kapitel 1.1.1 und 1.1.2 vorgestellten Targeting-Ansätze.

Einleitung _________________________________________________________________
- 14 -
Linker Wirkstoff
Wirkstoff
enzymatische oderchemische Aktivierung
spontaner Zerfall
WirkstoffLinker
Trigger
Abb. 1.10: Prinzip der Wirkstofffreisetzung bei doppelten Prodrugs.
Der Wirkstoff wird in diesem Fall über ein Linkermolekül an einen Trigger
(Auslöser, Sollbruchstelle) gebunden (bei makromolekularen Prodrugs kann der
Trigger mit der Bindungsstelle des Trägers gleichgesetzt werden). Am Zielort
erfolgt in zwei Schritten eine „kontrollierte“ Freisetzung des Wirkstoffs (engl.
controlled release). Zunächst wird der Trigger durch eine tumorspezifische
chemische oder enzymatische Reaktion gespalten, wodurch ein Wirkstoff-Linker-
Derivat freigesetzt wird. Dieses ist unter den vorherrschenden Bedingungen
instabil und zerfällt spontan unter Freisetzung des aktiven Wirkstoffs.
Beispielsweise entsteht durch enzymatische Spaltung des Phosphatesters in
Fosphenytoin (Abb.1.9, S. 13) ein instabiles Intermediat, das neben dem aktiven
Wirkstoff Phenytoin auch Formaldehyd freisetzt.
Trotz oftmals aufwändigerer Synthese bieten doppelte Prodrugs entscheidende
Vorteile. So kann beispielsweise die Spaltbarkeit von Peptid-Sollbruchstellen bei
einer direkten Kopplung des Peptids an einen amino-funktionalisierten Wirkstoff
herabgesetzt sein. Der Einbau von selbsteliminierenden Linkern* (siehe Kap. 1.4)
wie Aminobenzylcarbamoyl-Linkern108,109 zwischen Arzneistoff und Peptidsequenz
kann in vielen Fällen durch eine Reduktion der sterischen Hinderung am
Reaktionszentrum eine verbesserte Spaltbarkeit durch Proteasen bewirken.
* Im angloamerikanischen Sprachraum wurde für solche Linker-Systeme der Begriff self-immolative geprägt, der sich nur schlecht mit „selbstaufopfernd“ ins Deutsche übersetzen lässt. Daher werden im Folgenden alle Linker unabhängig vom ihnen zugrunde liegenden Reaktionstyp (Eliminierung, Zyklisierung) als „selbsteliminierend“ bezeichnet.

Einleitung _________________________________________________________________
- 15 -
Ebenso ist der Einbau selbsteliminierender Linker in Konjugate erforderlich, wenn
für die Realisierung der angestrebten, tumorspezifischen Spaltung funktionelle
Gruppen eingeführt werden müssen, die im Wirkstoff nicht vorhanden sind (z. B.
Carbonyl- oder Hydrazidgruppen für säurelabile Acylhydrazon-Bindungen).
1.4 Linker-Systeme für eine gezielte Freisetzung von Wirkstoffen
1.4.1 Selbsteliminierende Linker
In den letzten Jahren wurde eine beachtliche Anzahl selbsteliminierender Linker
für das Design von doppelten Prodrugs entwickelt, die sich infolge Eliminierungs-
oder Zyklisierungsreaktionen rasch vom Wirkstoff abspalten.110 In Tabelle 1.2
werden einige Vertreter beider Klassen vorgestellt.
Tab. 1.2: Beispiele für selbsteliminierende Linker und deren zugrunde liegender
Spaltungsmechanismus.
Linker Reaktions-typ Spaltungsmechanismus Ref.
R
Z
O X
O
Wirkstoff
Trigger
X, Z = O, NH
1,6-Benzyl-eliminierung
R
Z
O X
O
X, Z = O, NH
Wirkstof f
111-114
N
O
O X
O
Trigger
X = O, NH
Wirkstoff
1,6-Eliminierung
N
HO
O X
O
X = O, NH
Wirkstoff
115
R
ZTrigger
X, Z = O, NH
O X
O
Wirkstoff
1,4-Benzyl-eliminierung
R
Z
X, Z = O, NH
O X
O
Wirkstof f
112,114
NH
Trigger
O
O
O Wirkstoff
1,8-Eliminierung
H2N
O
O
O Wirkstof f
116
R
Trigger
X = O, NH
XOO
H O
Wirkstoff
β-Eliminierung
R
X = O, NH
XO
H
HO
O
Wirkstof f
117

Einleitung _________________________________________________________________
- 16 -
X
OO
R
Trigger
X = O, NH
Wirkstoff
Zyklisierung X
OOH
R
X = O, NH
Wirkstof f
118
NN O
O
TriggerWirkstoff
Zyklisierung
HNN O
O
Wirkstoff
119
NH
O O
O
TriggerWirkstoff
Zyklisierung
H2NO O
O
Wirkstoff
120
NN O
O
Trigger
O
OS
R
N Wirkstoff Zyklisierung NN O
O
O
OS
R
HN Wirkstof f
121
Verwendung fanden solche Linker bislang in chemisch-aktivierten, d. h. unter
nicht-physiologischen Bedingungen aktivierten, Modellverbindungen und in
enzymatisch- bzw. (bio-)reduktiv-aktivierten Prodrugs.113,118-123 Jedoch existieren
keine Beispiele für selbsteliminierende Linker-Systeme, die sich für die
Realisierung säurelabiler Prodrugs von Wirkstoffen mit Amino-, Hydroxy- oder
Thiol-Funktionalitäten eignen.
Auf Eliminierungsreaktionen beruhende selbsteliminierende Linker zeigen in den
meisten Fällen eine schnellere Freisetzung und werden seit einigen Jahren
bevorzugt eingesetzt.124-126 1981 beschrieben KATZENELLENBOGEN et al.111
erstmals die Verwendung eines p-Aminobenzyloxycarbonyl- (PABC-) Linkers, der
zwischen das Trypsin-Substrat Boc-Lys und den Farbstoff p-Nitroanilin integriert
wurde. Eine enzymatische Spaltung der Bindung zwischen Aminosäure und Linker
führte zu einer schnellen Freisetzung des Farbstoffs, was auf eine rasch
verlaufende 1,6-Benzyl-Eliminierung des PABC-Linkers (Tab 1.2, Zeile 1;
Abb. 1.11) zurückgeführt wurde.112 Heute ist das PABC-System der bekannteste
und meistverwendete selbsteliminierende Linker und wurde für die Entwicklung
verschiedener Krebsmedikamente114,116,124-129 wie auch für komplexere
selbsteliminierende Strukturen wie Oligomere,130 Polymere,131,132 und
Dendrimere133 ausgenutzt.
Eine gezielte Eliminierung des PABC-Linkers wird durch Freisetzen der
aromatischen Aminogruppe aus einer maskierten Vorstufe ausgelöst. In früheren
Arbeiten wurde gezeigt, dass das Anilin des PABC-Systems beispielsweise als
Azid, als Nitrogruppe oder durch Acylierung effektiv maskiert, d. h. so deaktiviert

Einleitung _________________________________________________________________
- 17 -
werden kann, dass eine Eliminierung unter physiologischen Bedingungen nicht
stattfindet. Eingeleitet wurde die Benzyleliminierung dieser Systeme schließlich
durch Reduktion,130,133 enzymatische Spaltung109,134-136 oder mittels Staudinger-
Reaktion.137
X
O Z
O
stabil
im wässrigen Milieuspontane 1,6-Benzyl-
eliminierung
X = maskierte Aminogruppe
z.B. N3, NO2, R-CO-NH
Demaskierungvon X H2N
O Z
OEE
E = Effektor (Wirk- oderFarbstoff)
Z = NH, O, S
H2N
OHCO2 HZ E+ +
A B
C D
Abb. 1.11: Aktivierung und Eliminierung des PABC-Linkers.
Abbildung 1.11 zeigt den Mechanismus der Eliminierung des PABC-Linkers einer
Verbindung (A), bestehend aus Trigger, PABC-Linker und einem amino-, thiol-
oder hydroxy-funktionalisierten Effektor. Nach Aktivierung des Triggers (Spaltung
der Bindung zwischen Trigger und Linker) entsteht ein vinyloges Halbaminal (B),
das nach 1,6-Eliminierung des PABC-Derivats intermediär ein Carbamin- bzw.
Kohlensäurederivat des Effektors sowie ein chinoides Zwischenprodukt
(p-Iminochinomethan) bildet. Beide Intermediate sind instabil. Unter
CO2-Abspaltung wird einerseits der Effektor (D) freigesetzt, während das
p-Iminochinomethan unter Addition von Wasser zu p-Aminobenzylalkohol (C)
reagiert.
Alle bisher vorgestellten doppelten Prodrugs besaßen einen linearen Aufbau
(Abb. 1.12, S. 18). Neben Spaltung einer Sollbruchstelle zwischen Wirkstoff und
Träger gibt es eine elegantere Methode, makromolekulare Prodrugs (bzw.
allgemein: Träger-Wirkstoff-Konjugate) zu aktivieren. Diese beruht auf der
Verwendung sogenannter Adapter-Bausteine (siehe Kap. 1.4.2).

Einleitung _________________________________________________________________
- 18 -
A
B
WirkstoffLinkerTräger
Trigger
Trigger
Träger WirkstoffLinker
Abb. 1.12: Schematische Darstellung spaltbarer Träger-Wirkstoff-Konjugate mit linearem
Linker (A) und mit chemischem Adapter-System (B).
1.4.2 Chemische Adapter-Systeme
Als Adapter bezeichnet man in diesem Zusammenhang einen trifunktionellen,
selbsteliminierenden Linker (orange), der makromolekularen Träger (grün),
Wirkstoff (blau) und die zur Aktivierung notwendige Trigger-Gruppe (rot)
miteinander verbindet (Abb. 1.12). Im Vergleich zur linearen Anordnung bietet der
Aufbau mit Hilfe eines chemischen Adapters mehrere Vorteile. Dazu gehört die
exponierte Stellung der Trigger-Gruppe, die zur Beschleunigung verschiedener
Aktivierungsreaktionen, z. B. enzymatische Spaltungen, beitragen kann. Zudem
erlaubt der Einsatz von Adapter-Bausteinen eine modulare Entwicklung
makromolekularer Prodrugs, bei der mit geringerem synthetischen Aufwand
Träger, Trigger-Gruppen und Effektoren variiert werden können.
In einer Veröffentlichung von SHABAT et al.138 wurden verschiedene trifunktionelle
Linker-Systeme vorgestellt, die nach Aktivierung entweder über Zyklisierungs-
oder über Eliminierungsreaktionen Effektoren freisetzen können. Abbildung 1.13
(S. 19) zeigt ein Beispiel für ein makromolekulares Prodrug des Wirkstoffs
Etoposid,138 das als Adapter-Einheit ein Phenylessigsäure-Derivat verwendet. Die
Aktivierung erfolgt über eine vom Antikörper 38C2 katalysierte Retro-Aldol-/Retro-
Michael-Tandemreaktion und der dadurch ausgelösten Kaskade von

Einleitung _________________________________________________________________
- 19 -
Zyklisierungs- und Eliminierungsreaktionen. Anschließend wird der Wirkstoff
freigesetzt, während die Adapter-Einheit am Träger verbleibt.
OO
N CH3
NH3CO
O
CH3OH
H3CO
O N
ONCH3
O
N
O
H3CH3C
H3CO
H3CO
OO
OO
O
O
OO
CH3
H
OHH
OHH
O
NH
H3COH
NH
HN
O
O
On m
CH3 CH3
NO CH3
Abb. 1.13: Beispiel für ein chemisches Adapter-System als makromolekulares Prodrug des
Wirkstoffs Etoposid.
Zahlreiche weitere Strukturen für chemische Adapter-Systeme sind denkbar.
Generell sollten sich für eine Verwendung als Adapter-Gruppe alle trifunktionellen
Linker eignen, mit denen einerseits eine stabile Bindung zu einem
(makromolekularen) Träger aufgebaut werden kann und die andererseits die
Weiterleitung eines Bindungsbruchs (infolge der Aktivierung des Triggers) durch
Zyklisierungs- oder Eliminierungsreaktionen ermöglichen.
Bislang ist kein Adapter bekannt, der auf dem häufig verwendeten PABC-Linker
basiert. Die Verwendung dieses effektiv-spaltenden Systems als Grundlage für
einen Adapter-Baustein könnte zu einer modularen und rationalen Konzeption
makromolekularer Prodrugs beitragen.

Zielsetzung _________________________________________________________________
- 20 -
2. Zielsetzung
Im Rahmen dieser Arbeit sollte eine rationale und modulare Strategie zur
säurelabilen Aktivierung makromolekularer Prodrugs von hydroxy- oder amino-
funktionalisierten Wirkstoffen entwickelt werden. Dazu wurde das bereits gut
untersuchte und effektiv spaltende PABC-System als selbsteliminierender Linker
für die Entwicklung von doppelten Prodrugs ausgewählt. Obgleich für dieses
Linker-System bereits zahlreiche Aktivierungsmethoden bekannt sind, wurde die
Aminogruppe des Linkers bislang nicht über eine säurelabile Spaltung demaskiert.
Daher sollte zunächst anhand niedermolekularer Modellverbindungen mit dem
Fluorophor 7-Amino-4-methylcumarin (AMC)* untersucht werden, ob eine effektive
Maskierung, d.h. Stabilität der Sollbruchstelle bei pH 7.4 und rasche Spaltung bei
pH 5.0, durch Ausbildung einer säureempfindlichen Imin-Bindung realisiert werden
kann (Abb. 2.1).
R
O
N
H NH
O
O O
Abb. 2.1: Allgemeine Struktur der geplanten niedermolekularen AMC-Modellverbindungen.
Im Fall einer erfolgreichen Maskierung sollten durch Strukturvariation der für die
Synthese der Schiffschen Base verwendeten aromatischen Aldehyde die
Substituenteneinflüsse untersucht und anschließend die Hydrolyseeigenschaften
der Imin-Bindung in Pufferlösungen bei pH 5.0 und 7.4 sowie im Plasma optimiert
werden. Dabei sollte der Farbstoff AMC als Effektor und Modell für einen amino-
funktionalisierten Wirkstoff dienen, was eine einfachere und schnellere
Untersuchung von Freisetzungskinetiken einer Vielzahl verschiedener
Modellverbindungen ermöglicht.
Trigger-Gruppen mit besonders interessanten Spaltungseigenschaften sollten im
nächsten Schritt für die Synthese makromolekularer AMC-Modellverbindungen
* Dieser Farbstoff verfügt über eine stark blaue Fluoreszenz (ex. 390 nm; em. 460 nm), die selbst noch bei Konzentrationen im nanomolaren Bereich gemessen werden kann. Durch Acylierung der Aminogruppe lässt sich die Intensität der Fluoreszenz bei 460 nm stark herabsetzen. Aufgrund dieser besonderen Eigenschaft eignet sich AMC für die Bestimmung von Kinetiken solcher Reaktionen, in deren Verlauf der Farbstoff aus seiner acylierten Form freigesetzt wird.

Zielsetzung _________________________________________________________________
- 21 -
bzw. Prodrugs mit einem amino- bzw. hydroxy-funktionalisierten Wirkstoff (z. B.
Doxorubicin und Paclitaxel) ausgewählt werden. Als makromolekulare Träger
sollten sowohl das synthetische Polymer PEG als auch HSA verwendet und
bezüglich ihrer Einflüsse auf die pharmakokinetischen Eigenschaften der
jeweiligen Modellverbindung miteinander verglichen werden. Makromolekulare
Prodrugs sollten hierbei auf Basis eines zentralen Adapter-Bausteins entwickelt
werden. Dazu sollte ein um eine Funktionalität erweiterter PABC-Linker zum
Einsatz kommen, der über einen geeigneten Anknüpfungspunkt für den Träger
verfügt (Abb. 2.2).
NH
HN
O
O
O
O
Trigger
Effektor
Ankergruppe zur Bindungan einen polymeren
Träger über Kupfer-katalysierteAzid-Alkin-Cycloaddition
Abb. 2.2: Allgemeine Struktur des geplanten Adapter-Bausteins.
Nach erfolgreicher Synthese von makromolekularen Zielstrukturen sollten durch
kinetische Untersuchungen analog zu den niedermolekularen Messungen in
Pufferlösung bei pH 5.0 und 7.4 sowie im Plasma Antworten auf folgende Fragen
gefunden werden:
Lassen sich die Ergebnisse aus den kinetischen Untersuchungen der
niedermolekularen Modellverbindungen auf die Spaltungseigenschaften der
makromolekularen Verbindungen übertragen?
Welchen Einfluss übt die Einführung eines chemischen Adapters und eines
polymeren Trägers auf die Spaltungseigenschaften des Triggers aus?
Welches Polymer eignet sich besser als Träger für diese
Modellverbindungen?
Verfügen die neu entwickelten Trigger-Gruppen über geeignete
Hydrolyseeigenschaften zur Aktivierung von makromolekularen Prodrugs in
einer medizinischen Anwendung?
Welches Potenzial steckt im modularen Ansatz?

Ergebnisse und Diskussion _________________________________________________________________
- 22 -
3. Ergebnisse und Diskussion
3.1 Entwicklung niedermolekularer AMC-Modellverbindungen mit säurelabil getriggertem PABC-Linker
Um eine auf dem PABC-Linker basierende, säurelabile Trigger-Gruppe zu
entwickeln, müssen folgende Voraussetzungen erfüllt sein:
1. effektive Maskierung der PABC-Aminogruppe, um eine vorzeitige
Eliminierung zu verhindern;
2. rasche Demaskierung bei pH 5, welche die für die Prodrug-Aktivierung
nötige Eliminierungsreaktion initiiert.
Neben Acylhydrazon-85-89 und Acetal-Bindung77,78 ist die Imin-Bindung26,90,91
bekannt für ihre pH-abhängigen Hydrolyseeigenschaften. Imine werden durch
Reaktion von Aldehyden oder Ketonen mit Ammoniak oder primären Aminen
erhalten. Am Stickstoffatom substituierte Imine werden oftmals nach ihrem
Entdecker (H. SCHIFF, 1834–1915) auch als Schiffsche Basen bezeichnet.139
Im Folgenden sollte zunächst durch Synthese einer Modellverbindung gezeigt
werden, dass der PABC-Linker in Form einer Schiffschen Base maskiert werden
kann. Anschließend sollten durch Strukturvariationen Substituenteneinflüsse
untersucht und mit Hilfe der erhaltenen Ergebnisse die Hydrolyseeigenschaften
der Imin-Bindung optimiert werden. Der Fluoreszenzfarbstoff AMC sollte als
Modell für einen amino-funktionalisierten Wirkstoff dienen und dabei eine
einfachere und schnellere Bestimmung der Freisetzungskinetiken ermöglichen.140
3.1.1 Synthese niedermolekularer, säurelabiler Modellverbindungen
Die Synthese von Schiffschen Basen basierend auf p-Aminobenzylalkohol (PABA)
wurde zunächst in verschiedenen Varianten durchgeführt. Tabelle 3.1 (S. 23) gibt

Ergebnisse und Diskussion _________________________________________________________________
- 23 -
eine Übersicht der Reaktionsbedingungen der einzelnen Syntheseversuche. In
jeder Variante wurde Benzaldehyd im Überschuss (3–5 eq) zugesetzt.
Tab. 3.1: Übersicht der durchgeführten Synthesevarianten zur Darstellung einer Schiffschen Base
basierend auf p-Aminobenzylalkohol.
H
O
H2N
OHOH
N
H
Variante Lösungsmittel Katalysatorsystem Methodea
1 MeOH - I
2 MeOH PPTS, NEt3 I
3 MeOH AcOH I
4 MeOH BF3·OEt2 I
5 MeOH Sc(O3SCF3)3 I
6 THF TMSCl, NEt3 I
7 Toluol PPTS II
8 Benzol PPTS, NEt3 II
9 Benzol AcOH II
a) Dem Reaktionssystem wurde das während der Umsetzung freigesetzte Wasser durch den
Zusatz von Molekularsieb (I) oder Erhitzen am Wasserabscheider (II) entzogen.
Die Verfolgung des Reaktionsverlaufs mittels DC erwies sich als problematisch, da
die entstehenden Schiffschen Basen sowohl auf Kieselgel- als auch auf
Diol-beschichteten* Platten instabil waren, wodurch eine unvollständige
Umsetzung suggeriert wurde. Auch unter Verwendung von MeCN/H2O-Gemischen
und RP-Beschichtung konnte der Endpunkt der jeweiligen Reaktion nicht ermittelt
werden. Dies gelang erst durch Abbruch der Umsetzung nach unterschiedlichen
Zeitintervallen, Aufarbeitung und anschließender Reinheitskontrolle durch 1H-NMR. Die optimale Reaktionszeit für die Reaktion mit höchster Effizienz
(Variante 9), die im Folgenden als Standardmethode verwendet wurde, betrug 3 h.
* mit –(CH2)3OCH2CH(OH)CH2OH-Gruppen modifizierte Kieselgelplatten.

Ergebnisse und Diskussion _________________________________________________________________
- 24 -
Zur Darstellung von AMC-Modellverbindungen war es zunächst erforderlich, die
Aminogruppe des AMC für eine Reaktion zu aktivieren. Erste Versuche mit
Triphosgen misslangen, was sich auf die nur schwach ausgeprägte Reaktivität des
AMC zurückführen lässt.141 Ebenso führte eine direkte Umsetzung mit
Chlorameisensäurephenylester nicht zum Erfolg. Allerdings gelang die Synthese
von Phenyl-N-4-methylcumarin-7-yl-carbamat (2), einem geschützten Isocyanat,142
nach einer Vorbehandlung des AMC mit Chlortrimethylsilan (Abb. 3.1).
O OH2N O ONH
O
O
272 %
1. ChlortrimethylsilanDIEADCM (abs.), Δ
O Cl
O
2. DCM (abs.)0 °C RT
AMC
Abb. 3.1: Synthese von Phenyl-N-4-methylcumarin-7-yl-carbamat (2).
Für die Darstellung der ersten AMC-Modellverbindung (1b) wurde 2 mit dem aus
der Reaktion von PABA mit Benzaldehyd (rot) erhaltenen Imin 1a umgesetzt
(Abb. 3.2). Dabei reagiert 2 beim Erhitzen unter Eliminierung von Phenol zum
7-Isocyanato-4-methylcumarin (AMC-NCO, 3) und mit 1a direkt weiter zum
Carbamat 1b.
OH
N
H
O
N
H NH
O
O OO ON
HO
O
1b52 %
1aDBTL, Dioxan (abs.), 80 °C
2
Abb. 3.2: Synthese der AMC-Modellverbindung 1b.
Die Ausbeute der Modellverbindung konnte erhöht werden (siehe Abb. 3.4, S. 25),
indem 3 nicht erst in situ sondern vorab generiert und isoliert wurde. Die
Herstellung erfolgte durch Erhitzen von 2 im Vakuum einer Sublimationsapparatur
(Abb. 3.3, S. 25). Dieses neu entwickelte Verfahren stellt im Vergleich zur
herkömmlichen Methode, der Umsetzung von AMC mit Phosgen,143 eine
gefahrlose Alternative dar.

Ergebnisse und Diskussion _________________________________________________________________
- 25 -
O ONH
O
O
O ONC
O
2 3
Sublimations-apparatur
150 °C, Vakuum
Abb. 3.3: Synthese von AMC-NCO (3).
Durch Reaktion von 3 mit 1a wurde das Carbamat 1b schließlich in verbesserter
Ausbeute erhalten und mittels NMR charakterisiert (Abb. 3.4).
O ONC
O
DBTL, THF (abs.)
3OH
N
H
O
N
H NH
O
O O
1b66 %
1a
Abb. 3.4: Optimierte Synthese der AMC-Modellverbindung 1b.
Nach erfolgreicher Synthese von 1b wurden die Halbwertszeiten für die AMC-
Freisetzung fluorometrisch in Pufferlösungen bei zwei verschiedenen pH-Werten
bestimmt (siehe Kapitel 3.1.3). Bei pH 5.0 wurde AMC sehr schnell (t1/2 = 34 min)
freigesetzt. Bei pH 7.4 hingegen erwies sich 1b mit einer Halbwertszeit von 8 h als
deutlich stabiler. Diese Ergebnisse zeigten, dass der PABC-Linker als Schiffsche
Base effektiv maskiert werden kann. Allerdings ist eine Optimierung der
Hydrolyseeigenschaften von 1b notwendig, da für eine medizinische Anwendung
neben rascher Spaltung im leicht sauren Milieu eine Langzeitstabilität des Triggers
im Neutralen von t1/2 > 48 h wünschenswert ist. Für die weitere Entwicklung der
Trigger-Gruppe sollten daher die Faktoren, welche die Hydrolyseeigenschaften
der Modellverbindung beeinflussen, näher untersucht werden. Diese
Informationen sollten anschließend die Darstellung eines maßgeschneiderten
Triggers mit erhöhter Stabilität im Neutralen ermöglichen.
Dazu wurden zunächst analog zur Synthese von 1a eine kleine Bibliothek von
Schiffschen Basen ausgehend von PABA und verschiedenen Benzaldehyd-
derivaten mit elektronenziehenden oder elektronenschiebenden Substituenten
dargestellt. Nach zu 1b analoger Umsetzung mit 3 wurden die
Modellverbindungen (4b–14b) erhalten. Die Ausbeuten dieser Reaktionen sind der
Tabelle 3.2 zu entnehmen.

Ergebnisse und Diskussion _________________________________________________________________
- 26 -
Tab. 3.2: Ausbeuten für die Synthesen der Modellverbindungen 1b und 4b–14b.
OH
N
H
O
N
H NH
O
O O
R
Rxa
xb
O ONC
O
DBTL, THF (abs.)
3
O
H
R
OH
H2N
AcOHBenzol (abs.), Δ
Nr. R = Ausbeute
xa [%]
Ausbeute xb [%]
4a/b NMe2 77 87
5a/b OMe 81 95
1a/b H
77 66
6a/b F
73 84
7a/b Cl
71 76
8a/b O
34 37
9a/b CF3 90 85
10a/b NO2 67 74
11a/b
ClCl
Cl
36 63
12a/b F
F 66 63
13a/b
F F
F F
75 82
14a/b F
F F
F F
84 87

Ergebnisse und Diskussion _________________________________________________________________
- 27 -
3.1.2 Synthese einer Modellsubstanz zur Untersuchung der 1,6-Eliminierungsreaktion
In der Vergangenheit wurde die Kinetik der 1,6-Benzyleliminierung bereits von
SHABAT et al.144 und WARNECKE et al.130 näher untersucht. Die Arbeitsgruppe um
SHABAT verwendete für ihre Studien einen PABC-Linker, der nach Demaskierung
durch das Enzym Penicillin-G-Amidase den Farbstoff p-Nitroanilin freisetzt. Da die
schnelle Eliminierungsreaktion von der langsameren enzymatischen Reaktion
überlagert wurde, konnte keine Quantifizierung der Eliminierungsgeschwindigkeit
erfolgen. Durch Reduktion einer Nitrogruppe mit Zink und Essigsäure
demaskierten WARNECKE et al. den PABC-Linker, der durch anschließende
Eliminierung den Effektor Tryptamin freisetzte. Die mittels HPLC-Messungen
bestimmte Halbwertszeit dieser 1,6-Benzyleliminierung betrug 4 min. Die
Spaltungsversuche wurden bei pH 3 durchgeführt, da im neutralen pH-Bereich
Löslichkeitsprobleme auftraten. Halbwertszeiten für die im physiologischen
pH-Bereich verlaufende 1,6-Benzyleliminierung waren bislang nicht bekannt.
Da die Freisetzungskinetiken der Modellverbindungen 1b und 4b–14b bei den
physiologisch relevanten pH-Werten pH 5.0 und 7.4 untersucht werden sollten,
sollte zunächst die Geschwindigkeit der 1,6-Benzyleliminierung bei diesen
pH-Werten ermittelt werden. Sollte die Eliminierung mit einer sehr viel höheren
Geschwindigkeit ablaufen als die Hydrolyse der Imin-Bindung, so ist sie bei der
Betrachtung der Kinetiken von 1b und 4b–14b vernachlässigbar.
Für die Untersuchung der 1,6-Benzyleliminierung wurde eine weitere
Modellverbindung ohne Trigger-Gruppe synthetisiert (Abb. 3.5, S. 28). Im ersten
Schritt wurde die Aminogruppe von PABA durch Reaktion mit (Boc)2O geschützt
und das erhaltene Produkt (15) anschließend analog zur Synthese von 1b mit
AMC-NCO (3) zum Carbamat (16) umgesetzt. Nach Abspaltung der Boc-
Schutzgruppe mit einem TFA/DCM-Gemisch konnte schließlich die Zielverbindung
17 isoliert werden. 17 liegt als Trifluoracetat vor und ist als Feststoff über längere
Zeit stabil.

Ergebnisse und Diskussion _________________________________________________________________
- 28 -
(Boc)2ODCM (abs.)
RHN
OH
BocHN
O NH
O
O ODBTLTHF (abs.)
+H3N
O NH
O
O O
CF3COO-
TFADCM (abs.)
N O OC
O
1663 %
1798 %
R = H15: R = Boc (71 %)
3
Abb. 3.5: Synthese von Modellverbindung 17.
Die erhaltenen Modellverbindungen 1b, 4b–14b und 17 wurden anschließend im
Rahmen einer kinetischen Studie hinsichtlich ihrer pH-abhängigen Spaltungs-
eigenschaften untersucht, deren Durchführung und Ergebnisse im folgenden
Kapitel ausführlich diskutiert werden.
3.1.3 Untersuchung der pH-abhängigen Spaltungseigenschaften der niedermolekularen AMC-Modellverbindungen 1b, 4b–14b und 17
Der postulierte Mechanismus für die pH-abhängige Spaltung der Modellverbin-
dungen 1b, 4b–14b und 17 ist in Abbildung 3.6 dargestellt. Im ersten Schritt wird
die Aminogruppe des Linkers durch eine säurekatalysierte Hydrolyse der Imin-
Bindung demaskiert. Anschließend findet spontan eine irreversible 1,6-Benzyl-
eliminierung des PABC-Linkers statt, wobei AMC freigesetzt und infolgedessen die
Fluoreszenzintensität der Lösung bei 460 nm stark erhöht wird.
N
OH
O
NH
O O
O
H
H2N
OH
H2N O O
CO2+ +
AMCfluoreszierend
R
R
Imin-Hydrolysebei pH 5.0
stabil bei pH 7.4 H2N
O
O
NH
O O
spontane1,6-Benzyleliminierung
k1
k2
H2O
Abb. 3.6: Postulierter Mechanismus für die säurekatalysierte Freisetzung von AMC.
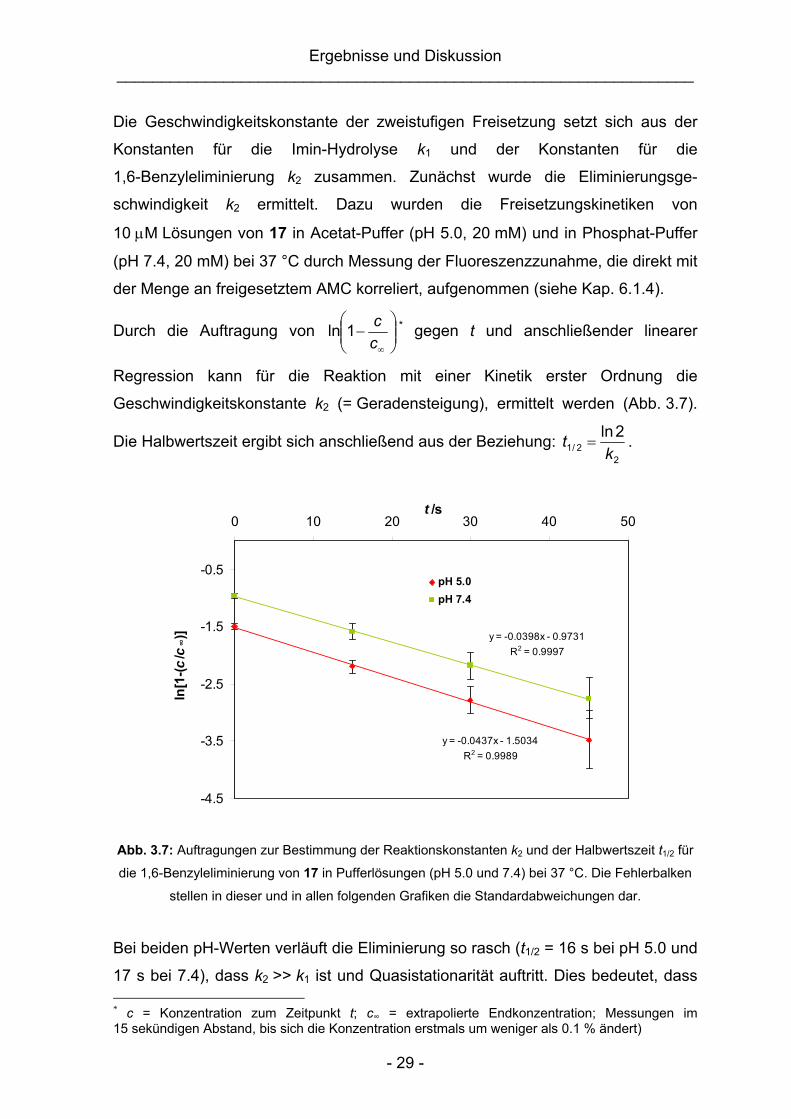
Ergebnisse und Diskussion _________________________________________________________________
- 29 -
Die Geschwindigkeitskonstante der zweistufigen Freisetzung setzt sich aus der
Konstanten für die Imin-Hydrolyse k1 und der Konstanten für die
1,6-Benzyleliminierung k2 zusammen. Zunächst wurde die Eliminierungsge-
schwindigkeit k2 ermittelt. Dazu wurden die Freisetzungskinetiken von
10 μM Lösungen von 17 in Acetat-Puffer (pH 5.0, 20 mM) und in Phosphat-Puffer
(pH 7.4, 20 mM) bei 37 °C durch Messung der Fluoreszenzzunahme, die direkt mit
der Menge an freigesetztem AMC korreliert, aufgenommen (siehe Kap. 6.1.4).
Durch die Auftragung von ⎟⎟⎠
⎞⎜⎜⎝
⎛−
∞cc1ln * gegen t und anschließender linearer
Regression kann für die Reaktion mit einer Kinetik erster Ordnung die
Geschwindigkeitskonstante k2 (= Geradensteigung), ermittelt werden (Abb. 3.7).
Die Halbwertszeit ergibt sich anschließend aus der Beziehung: 2
2/12ln
kt = .
y = -0.0437x - 1.5034R2 = 0.9989
y = -0.0398x - 0.9731R2 = 0.9997
-4.5
-3.5
-2.5
-1.5
-0.5
0 10 20 30 40 50t /s
ln[1
-(c/c
∞)]
pH 5.0pH 7.4
Abb. 3.7: Auftragungen zur Bestimmung der Reaktionskonstanten k2 und der Halbwertszeit t1/2 für
die 1,6-Benzyleliminierung von 17 in Pufferlösungen (pH 5.0 und 7.4) bei 37 °C. Die Fehlerbalken
stellen in dieser und in allen folgenden Grafiken die Standardabweichungen dar.
Bei beiden pH-Werten verläuft die Eliminierung so rasch (t1/2 = 16 s bei pH 5.0 und
17 s bei 7.4), dass k2 >> k1 ist und Quasistationarität auftritt. Dies bedeutet, dass * c = Konzentration zum Zeitpunkt t; c∞ = extrapolierte Endkonzentration; Messungen im 15 sekündigen Abstand, bis sich die Konzentration erstmals um weniger als 0.1 % ändert)
Ergebnisse und Diskussion _________________________________________________________________
- 30 -
der Eliminierungsschritt bei den Kinetikmessungen der Modellverbindungen
vernachlässigbar ist. Die Hydrolyse der Schiffschen Base ist daher der alleinige
geschwindigkeitsbestimmende Schritt der AMC-Freisetzung. Mit Hilfe der
Bodenstein´schen Näherung vereinfacht sich das Geschwindigkeitsgesetz
infolgedessen zu dem einer Kinetik pseudo-erster Ordnung (siehe Glch. 1).
(1)
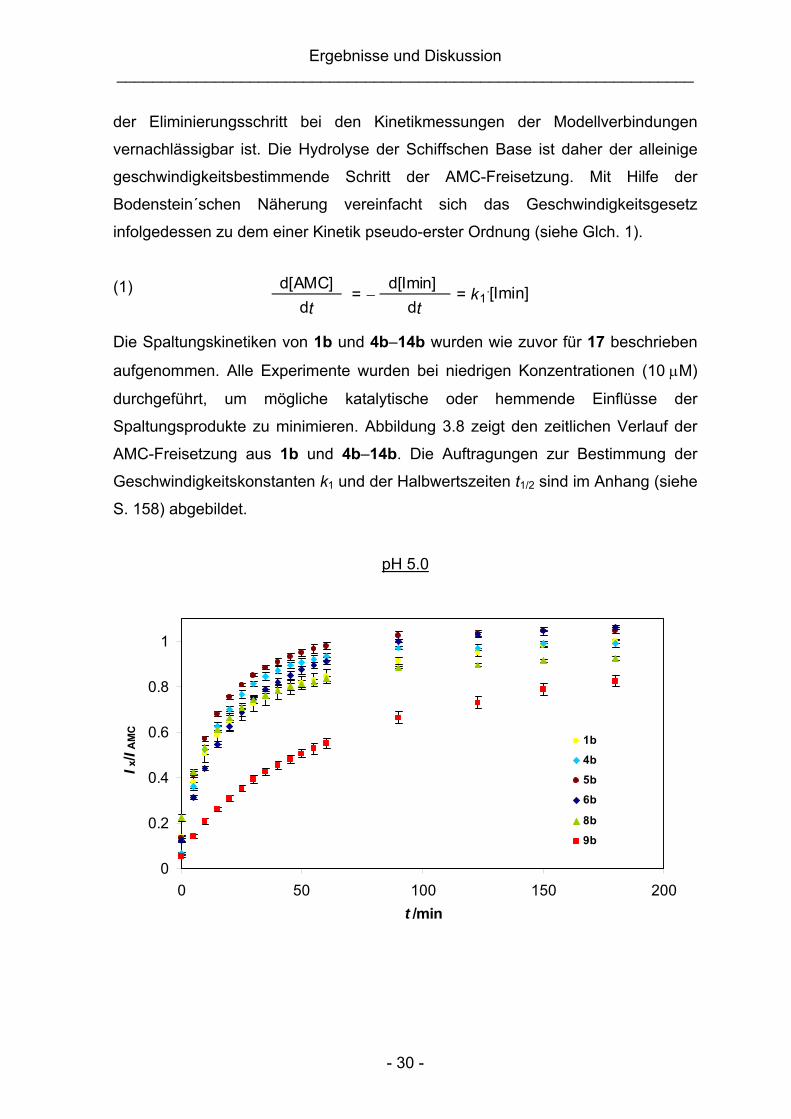
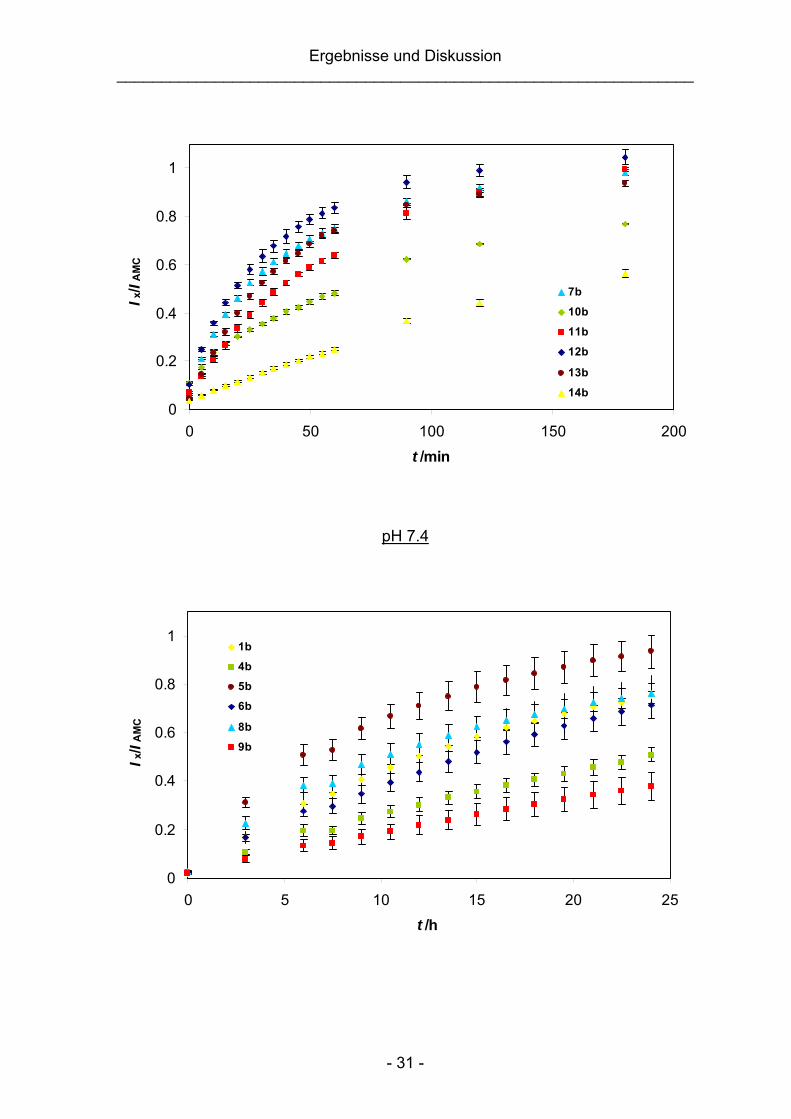
Die Spaltungskinetiken von 1b und 4b–14b wurden wie zuvor für 17 beschrieben
aufgenommen. Alle Experimente wurden bei niedrigen Konzentrationen (10 μM)
durchgeführt, um mögliche katalytische oder hemmende Einflüsse der
Spaltungsprodukte zu minimieren. Abbildung 3.8 zeigt den zeitlichen Verlauf der
AMC-Freisetzung aus 1b und 4b–14b. Die Auftragungen zur Bestimmung der
Geschwindigkeitskonstanten k1 und der Halbwertszeiten t1/2 sind im Anhang (siehe
S. 158) abgebildet.
pH 5.0
0
0.2
0.4
0.6
0.8
1
0 50 100 150 200t /min
I x/I
AM
C
1b
4b
5b
6b
8b
9b
d[Imin]dt
= k1.[Imin]d[AMC]
dt= −
Ergebnisse und Diskussion _________________________________________________________________
- 31 -
0
0.2
0.4
0.6
0.8
1
0 50 100 150 200t /min
I x/I
AM
C
7b
10b
11b
12b
13b
14b
pH 7.4
0
0.2
0.4
0.6
0.8
1
0 5 10 15 20 25t /h
I x/I
AM
C
1b
4b
5b
6b
8b
9b

Ergebnisse und Diskussion _________________________________________________________________
- 32 -
0
0.2
0.4
0.6
0.8
1
0 5 10 15 20 25t /h
I x/I
AM
C
7b
10b
11b
12b
13b
14b
Abb. 3.8: Zeitlicher Verlauf der AMC-Freisetzung aus 1b und 4b–14b bei pH 5.0 und 7.4 und
37 °C (Ix: Fluoreszenzintensität der Verbindung zum Zeitpunkt t; IAMC: Fluoreszenzintensität einer
10 μM AMC-Kontrolllösung zum Zeitpunkt t).
Aus den in Tabelle 3.3 (S. 33) aufgeführten Halbwertszeiten ergibt sich, dass die
Imin-Bindung generell rasch bei pH 5.0 gespalten wird (t1/2 = 17–173 min). Bei pH
7.4 hingegen zeigten die Modellverbindungen eine erheblich größere Stabilität
(t1/2 = 8−365 h). Ein deutlicher Einfluss der Substituenten am aromatischen Ring
des Benzaldehyds auf die Geschwindigkeitskonstante ist bei beiden pH-Werten zu
beobachten, wobei elektronenziehende Substituenten im Allgemeinen einen
stabilisierenden Effekt ausüben. Für die para-substituierten Benzaldehydderivate
1b und 4b–10b wurden die Substituenteneffekte unter Berücksichtigung von
Linearen Freien Enthalpie-Beziehungen näher untersucht (siehe Kapitel 3.1.4).

Ergebnisse und Diskussion _________________________________________________________________
- 33 -
Tab. 3.3: Halbwertszeiten von 1b und 4b–14b für die AMC-Freisetzung in Pufferlösungen (pH 5.0
und 7.4) bei 37 °C.
R
O
N
H NH
O
O O
a) Acetat-Puffer, 20 mM; b) Phosphat-Puffer, 20 mM.
Nr. R = t1/2
pH 5.0a
[min]
t1/2 pH 7.4b
[h]
4b NMe2 17 26
5b OMe 20 13
1b H
34 8
6b F
27 14
7b Cl 37 36
8b O
28 9
9b CF3 61 36
10b NO2 81 65
11b
ClCl
Cl
50 50
12b F
F 33 39
13b
F F
F F
34 89
14b F
F F
F F
173 365

Ergebnisse und Diskussion _________________________________________________________________
- 34 -
3.1.4 Untersuchung der Substituenteneffekte hinsichtlich der Linearen Freien Enthalpie-Beziehung (LFER)
Im Rahmen einer empirischen Korrelationsanalyse fand L. P. HAMMETT in der Mitte
der dreißiger Jahre145,146 eine Lineare Freie Enthalpie-Beziehung (LFER) zwischen
den logarithmierten Geschwindigkeitskonstanten der basenkatalysierten Hydrolyse
substituierter Benzoesäureethylester und den ebenfalls logarithmierten
Gleichgewichtskonstanten für die Ionisierung entsprechend substituierter
Benzoesäuren. In der Hammett-Gleichung (Glch. 2) fasste er später die bis dahin
gesammelten Daten über die Seitenkettenreaktivität von meta- und para-
substituierten Benzol-Derivaten zusammen:
(2) k0: Geschwindigkeitskonstante für die unsubstituierte Bezugsverbindung
ρ: Reaktionskonstante, Maß für die Empfindlichkeit der Reaktion gegenüber Substituenten-
einflüssen
σ: Substituentenkonstante, Maß für den polaren Effekt (Summe der induktiven und mesomeren
Effekte sowie der Feldeffekte) des Substituenten gegenüber Wasserstoff
Daraufhin wurden in den folgenden Jahren hunderte σp-Werte (Konstanten für
Substituenten in para-Stellung) bestimmt und gezeigt, dass die Hammett-
Gleichung auch auf zahlreiche, andere Reaktionssysteme erfolgreich angewendet
werden kann. Allerdings wurden ebenso Reaktionen gefunden, welche die
Hammett-Gleichung nicht erfüllen.147,148 Die Anwendbarkeit der Gleichung
ermöglicht Rückschlüsse auf den vorliegenden Reaktionsmechanismus. Eine
Gerade weist beispielsweise daraufhin, dass sowohl der Mechanismus als auch
der geschwindigkeitsbestimmende Schritt der Reaktion von der Art der
Substituenten unabhängig ist. Abweichungen von der Linearität sind hingegen ein
Indikator dafür, dass die Art der Substituenten einen Einfluss auf Mechanismus
und geschwindigkeitsbestimmenden Schritt ausübt. Um zu prüfen, ob die
Hammett-Gleichung auf die para-substituierten Modellverbindungen 1b und 4b–
14b anwendbar ist, wurde für beide pH-Werte 0logkk gegen σp
146 aufgetragen
(Abb. 3.9, S. 35).
ρσ+= 0loglog kk

Ergebnisse und Diskussion _________________________________________________________________
- 35 -
-0,4
-0,2
0
0,2
0,4
-0,8 -0,6 -0,4 -0,2 0 0,2 0,4 0,6 0,8
σ p
log
(k/k
0)
p-NMe2 p-OMe
p-H
p-F
p-Cl
p-CF3p-NO2
p-Ac
-1
-0,8
-0,6
-0,4
-0,2
0
0,2
-0,8 -0,6 -0,4 -0,2 0 0,2 0,4 0,6 0,8
σ p
log
(k/k
0)
p-NMe2
p-OMep-F
p-H
p-Cl
p-Ac
p-CF3
p-NO2
A
B
Abb. 3.9: Hammett-Auftragung für die Spaltung von 1b und 4b–14b bei pH 5.0 (A) und pH 7.4 (B).
Ein linearer Zusammenhang ist in guter Näherung für die Auftragung bei pH 5.0 zu
beobachten. Allerdings weichen die Datenpunkte für die Substituenten p-Ac und
p-NMe2 signifikant von der erhaltenen Gerade ab. Eine mögliche Erklärung für die
Abweichung des Dimethylamino-Substituenten liegt darin, dass die partielle
Protonierung bei pH 5.0 nicht berücksichtigt wird, da sich der entsprechende
σp-Wert nur auf die freie Base bezieht. Für die gravierende Abweichung des
Acetyl-Substituenten wurde bisher keine plausible Erklärung gefunden.
Bei der weiteren Auswertung der Daten ergab sich eine Reaktionskonstante
(ρ-Wert) von –0.57, was auf einen moderaten Einfluss der Substituenten bei
pH 5.0 hinweist, wobei eine Abnahme der Reaktionsgeschwindigkeit durch
Einführen von elektronenziehenden Substituenten in para-Stellung erreicht wird.
Im Gegensatz dazu wurde bei pH 7.4 keine Lineare Freie Enthalpie-Beziehung
gefunden. Der Einfluss der Substituenten auf die Stabilität war jedoch im
Allgemeinen stärker ausgeprägt als bei pH 5.0.

Ergebnisse und Diskussion _________________________________________________________________
- 36 -
Der generelle Trend der pH-Abhängigkeit innerhalb des Spektrums der
untersuchten Modellverbindungen ist offensichtlich. Dennoch ist zu bedenken,
dass der Imin-Hydrolyse ein relativ komplexer Reaktionsmechanismus zugrunde
liegt149,150 und bei der Auswertung der kinetischen Daten nicht alle Effekte
berücksichtigt wurden, die durch allgemeine Säure-Base-Katalyse hervorgerufen
werden können.151
Durch das Einführen zusätzlicher elektronenziehender Substituenten gelang es
erwartungsgemäß, die Stabilität der mono-halogenierten Schiffschen Basen bei
pH 7.4 deutlich zu erhöhen (vgl. Tab. 3.3, S. 33, 11b–14b).
3.1.5 Untersuchung der Hydrolyseeigenschaften von 13b und 14b im physiologisch relevanten pH-Bereich
Besonders das 2,3,5,6-Tetrafluor- und das Pentafluorbenzaldehyd-Derivat (13b,
14b) verfügen über eine hinreichende Stabilität im neutralen Milieu (siehe Kapitel
3.1.3), was sie zu interessanten Kandidaten für eine medizinische Anwendung
macht. Deswegen wurden beide Verbindungen für eine ausführlichere
Betrachtung ihrer Hydrolyseeigenschaften im physiologisch relevanten pH-Bereich
(pH 4.5–7.4, Abb. 3.10) ausgewählt.
y = -0.8891x + 2.8414
y = -0.8748x + 1.9739
-5
-4
-3
-2
-1
04.0 5.0 6.0 7.0 8.0
pH
log
k obs
13b14b
Abb. 3.10: pH-Abhängigkeit der Geschwindigkeitskonstanten k1 von 13b und 14b (Acetat-Puffer:
4.5–6; Phosphat-Puffer: 6.5–7.4, 40 mM, 37 °C).

Ergebnisse und Diskussion _________________________________________________________________
- 37 -
Beide Derivate zeigen im gesamten untersuchten pH-Bereich eine konstante
Abnahme der Hydrolysegeschwindigkeit. Zwischen pH und log kobs wurde in guter
Näherung eine lineare Beziehung gefunden. Erklären lässt sich diese
Beobachtung bei näherer Betrachtung des zweistufigen Spaltungsmechanismus
für die säurekatalysierte Imin-Hydrolyse (siehe Abb. 3.11):152
R H
NR' +H+
-H+R H
NR' H
R H
NR' H + H2O
- H2O R O
NR' H
HH
H
- H++ H+
R OH
NR' H
H
R OH
NR' H
H
H
- H+
+ H+
R H
OR'NH2
- H+
+ H+
Abb. 3.11: Mechanismus der säurekatalysierten Imin-Hydrolyse.152
Durch Protonierung entsteht ein Iminiumion, welches den Angriff eines
Wassermoleküls einleitet. Das daraus resultierende tetraedrische Additionsprodukt
zerfällt anschließend unter Austritt des Amins. Die Reaktionsgeschwindigkeit der
Hydrolyse stellt dabei eine Funktion des pH-Wertes und der Basizität des Amins
dar. Da nur die protonierte Spezies (Iminiumion) des vorgelagerten Säure-Base-
Gleichgewichts in den nächsten (geschwindigkeitsbestimmenden) Schritten weiter
zum Produkt reagiert, zählt die Imin-Hydrolyse zu den Reaktionen mit spezifischer
Säurekatalyse (Glch. 3).151
(3)
Das Produkt AMC wird folglich mit folgender Geschwindigkeit gebildet (Glch. 4):
(4)
Imin + H+ Imin.H+KS kAMC + Aldehyd + Amin
d[AMC]dt
= k .[Imin.H+]

Ergebnisse und Diskussion _________________________________________________________________
- 38 -
Für die Gleichgewichtskonstante des vorgelagerten Säure-Base-Gleichgewichts
gilt (Glch. 5):
(5)
Einsetzen von (5) in das Geschwindigkeit-Zeit-Gesetz (4) liefert Ausdruck (6):
(6)
Bei konstantem pH liegt ein Geschwindigkeitsgesetz erster Ordnung bezüglich des
Imins vor. Die Säure selbst tritt – wie für eine Reaktion mit spezifischer
Säurekatalyse zu erwarten – nicht im Geschwindigkeitsgesetz auf.
Für die gemessene Geschwindigkeitskonstante erster Ordnung (kobs) gilt im
vorliegenden Fall (Glch. 7):
(7)
kcat: Geschwindigkeitskonstante der katalysierten Reaktion.
Nach Logarithmieren von (7) erhält man Gleichung (8), die die beobachtete lineare
Beziehung zwischen log kobs und pH wiedergibt:
(8)
Gleichung 8 ist zu entnehmen, dass die Steigung der Geraden im Idealfall der
spezifischen Säurekatalyse theoretisch den Wert 1 annimmt. Für 13b und 14b
wurde jeweils eine Steigung von ~0.9 berechnet, was nur eine leichte Abweichung
vom Idealfall darstellt. Darüber hinaus liefert das lineare Verhalten einen Hinweis
darauf, dass sich der geschwindigkeitsbestimmende Schritt innerhalb des
untersuchten pH-Bereichs nicht ändert. Da diese beiden Modellverbindungen im
Vergleich zu den anderen Verbindungen am stärksten auf eine pH-Änderung
[Imin.H+][H+][Imin]
= KS [Imin.H+] = KS [H+][Imin]
d[AMC]dt
= k.KS[H+][Imin]
kobs
kobs = k.KS[H+]
kcat
log kobs = log kcat − pH

Ergebnisse und Diskussion _________________________________________________________________
- 39 -
ansprechen, d. h. ihre Halbwertszeiten bei pH 5.0 und 7.4 annähernd den
größtmöglichen Abstand zueinander aufweisen und zudem für medizinische
Anwendungen günstige Werte annehmen, bieten sich 13b und 14b für die
Verwendung als Trigger in säurelabilen Prodrugs an.
3.1.6 Untersuchung der Plasmastabilität ausgewählter Modellverbindungen
Bei der Entwicklung pharmakologisch aktiver Substanzen ist die Berücksichtigung
der pharmakokinetischen Eigenschaften erforderlich. Hohe Bioverfügbarkeit,
gezielte Freisetzung des Wirkstoffs am Zielort und hinreichende Langzeitstabilität
in der Blutbahn sind unter anderem Grundvoraussetzungen für eine medizinische
Anwendung. Daher wurde eine Auswahl der Modellverbindungen (1b, 10b, 13b,
14b) hinsichtlich ihrer Plasmastabilität untersucht. Analog zu den Messungen in
Pufferlösungen (Kap. 3.1.3) wurden 10 μM Lösungen in Plasma hergestellt und
die Fluoreszenzzunahme, die aus der Freisetzung von AMC resultiert, nach 0 h,
2 h, 4 h, und 6 h bzw. 24 h für 13b und 14b gemessen (Abb. 3.12).
0
0.2
0.4
0.6
0.8
1
0 1 2 3 4 5 6 7t /h
I x/I
AM
C
1b10b13b14b
Abb. 3.12: Zeitlicher Verlauf der AMC-Freisetzung von ausgewählten Verbindungen in humanem
Plasma.

Ergebnisse und Diskussion _________________________________________________________________
- 40 -
Da die Auftragungen von ⎟⎟⎠
⎞⎜⎜⎝
⎛−
∞cc1ln gegen t keine linearen Beziehungen ergaben,
wurden die Halbwertszeiten aus der graphischen Darstellung des zeitlichen
Verlaufs der Fluoreszenzzunahme näherungsweise bestimmt. Die Halbwertszeit
entsprach dabei dem Zeitpunkt, an dem die Hälfte der aus der Verbindung
insgesamt freigesetzten Menge an AMC erreicht wurde. Die untersuchten
Verbindungen wiesen überaschenderweise nur eine geringe Stabilität im Plasma
auf. Entgegen der Erwartung entsprachen ihre Halbwertszeiten für die Freisetzung
von AMC im Plasma eher den Halbwertszeiten der Spaltung bei pH 5.0 statt bei
pH 7.4. (Tab. 3.4). Um die Ursache für die geringe Plasmastabilität zu ermitteln,
wurden die Messungen in Plasma wiederholt, aus dem zuvor durch
Ultrazentrifugalfiltration alle Moleküle der Größen > 10 kDa entfernt worden waren
(Tab. 3.4, Abb. 3.13, S. 41).
Tab. 3.4: Vergleich der Halbwertszeiten von 1b, 10b, 13b und 14b für die AMC-Freisetzung in
Pufferlösungen (5.0, 7.4) und im Plasma (pur bzw. nach Ultrazentrifugalfiltration).
t1/2 [h]
Nr. R = pH 5.0a pH 7.4b Plasmac Plasmad
1b H
0.57 8 < 0.33 ~ 7
10b NO2
1.35 65 < 0.33 > 72
13b F F
F F
0.57 ~ 89 1.30 > 72
14b F
F F
F F
2.88 ~ 365 2.33 > 72
a) Acetat-Puffer, 20 mM; b) Phosphat-Puffer, 20 mM; c) humanes Plasma; d) humanes Plasma, aus
dem alle Bestandteile >10 kDa durch Ultrazentrifugalfiltration abgetrennt wurden (siehe
Kap. 6.1.4).
In diesem Medium war die Stabilität der Verbindungen im Vergleich zum reinen
Plasma deutlich erhöht. Die Halbwertszeiten für die AMC-Freisetzung lagen dabei
in der Größenordnung der Halbwertszeiten, die in Phosphatpuffer (pH 7.4)
gemessen wurden. Anhand dieser Ergebnisse lässt sich vermuten, dass die

Ergebnisse und Diskussion _________________________________________________________________
- 41 -
Instabilität durch im Plasma vorliegende Proteine mit Molmassen >10 kDa
verursacht wurde.
0
0.2
0.4
0.6
0.8
1
0 10 20 30 40 50t /h
I x/I
AM
C
1b10b13b14b
Abb. 3.13: Stabilität von 1b, 10b, 13b und 14b in Plasma, dem zuvor durch Ultrazentrifugalfiltration
Moleküle >10 kDa entfernt wurden.
Das Protein mit der höchsten Konzentration im Blutplasma (~ 750 μM) ist das
Humanserumalbumin (HSA) mit einer Molmasse von 66.5 kDa. Als
Transporterprotein verfügt HSA über ausgezeichnete und vielfältige
Bindungseigenschaften. Neben körpereigenen Liganden wie z. B. Fettsäuren,
Hormonen oder Bilirubin können auch zahlreiche Wirkstoffe oder Fluoreszenz-
Marker von HSA gebunden werden.153-155 Ebenso sind Beispiele für katalytische
Aktivitäten von Albuminen dokumentiert.131 Es war daher denkbar, dass HSA für
die Plasmainstabilität der Modellverbindungen verantwortlich ist. Um diese
Vermutung zu überprüfen, wurde die Stabilität von 14b analog zu den vorherigen
Messungen in HSA-Lösungen (750 μM, 500 μM, 250 μM) untersucht. Daraus
ergab sich für 14b in HSA (750 μM) eine Halbwertszeit von t1/2 = 272 min
(Abb. 3.14, S. 42). Der Verdacht, dass Albumin die AMC-Freisetzung katalysiert,
wurde mit diesem Ergebnis bestätigt. Die Abweichung zur Halbwertszeit im
Plasma (t1/2 = 140 min) lässt sich auf die zusätzlichen Bestandteile in diesem
Medium zurückführen.
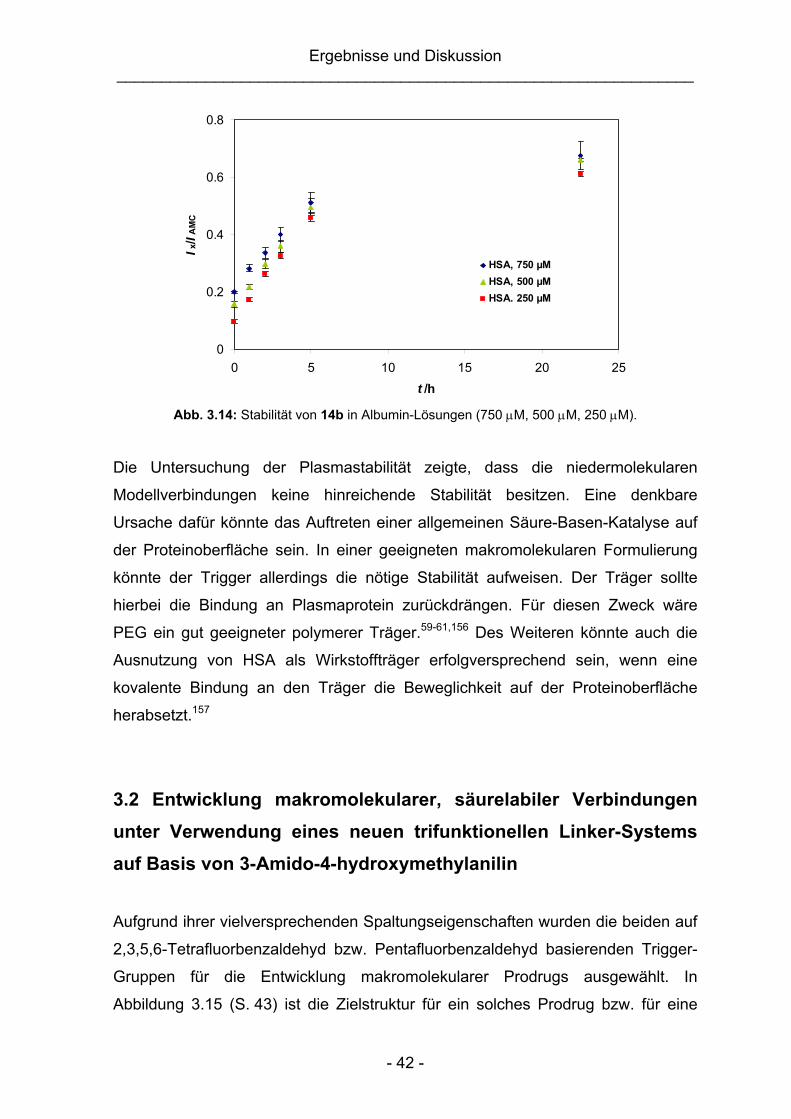
Ergebnisse und Diskussion _________________________________________________________________
- 42 -
0
0.2
0.4
0.6
0.8
0 5 10 15 20 25t /h
I x/I
AM
C
HSA, 750 µM HSA, 500 µMHSA. 250 µM
Abb. 3.14: Stabilität von 14b in Albumin-Lösungen (750 μM, 500 μM, 250 μM).
Die Untersuchung der Plasmastabilität zeigte, dass die niedermolekularen
Modellverbindungen keine hinreichende Stabilität besitzen. Eine denkbare
Ursache dafür könnte das Auftreten einer allgemeinen Säure-Basen-Katalyse auf
der Proteinoberfläche sein. In einer geeigneten makromolekularen Formulierung
könnte der Trigger allerdings die nötige Stabilität aufweisen. Der Träger sollte
hierbei die Bindung an Plasmaprotein zurückdrängen. Für diesen Zweck wäre
PEG ein gut geeigneter polymerer Träger.59-61,156 Des Weiteren könnte auch die
Ausnutzung von HSA als Wirkstoffträger erfolgversprechend sein, wenn eine
kovalente Bindung an den Träger die Beweglichkeit auf der Proteinoberfläche
herabsetzt.157
3.2 Entwicklung makromolekularer, säurelabiler Verbindungen unter Verwendung eines neuen trifunktionellen Linker-Systems auf Basis von 3-Amido-4-hydroxymethylanilin
Aufgrund ihrer vielversprechenden Spaltungseigenschaften wurden die beiden auf
2,3,5,6-Tetrafluorbenzaldehyd bzw. Pentafluorbenzaldehyd basierenden Trigger-
Gruppen für die Entwicklung makromolekularer Prodrugs ausgewählt. In
Abbildung 3.15 (S. 43) ist die Zielstruktur für ein solches Prodrug bzw. für eine

Ergebnisse und Diskussion _________________________________________________________________
- 43 -
entsprechende AMC-Modellverbindung dargestellt. Die Trigger-Gruppen sollten
über ein PABC-analoges Adapter-System (orange) auf Basis von 3-Amido-4-
hydroxymethylanilin mit dem jeweiligen Effektor verbunden werden. Mögliche
Effektoren stellen amino- oder hydroxy-funktionalisierte Wirk- bzw. Farbstoffe dar,
die über eine Carbamat- oder Carbonat-Bindung mit dem Adapter verknüpft
werden sollten. Das Adapter-System sollte außerdem über eine Alkin-
Ankergruppe zur Kupplung an makromolekulare Träger mittels Kupfer-katalysierter
Alkin-Azid-Cycloaddition (CuAAC) verfügen.
NH
HN
O
O
O
O
O
X
N
H
R
X = O, NH
Effektor
Ankergruppe zur Kupplungan makromolekulare Träger
über CuAACTrigger-Gruppe
Abb. 3.15: Beispielhafte Zielstruktur makromolekularer säurelabiler Verbindungen mit chemischem
Adapter (orange).
Kupfer-katalysierte Azid-Alkin-Cycloaddition (CuAAC) 2001 stellte die Gruppe von SHARPLESS ein neues Konzept unter dem Namen
„Click“-Chemie vor, das verschiedene Reaktionstypen umfasst.158 „Click“-
Reaktionen bieten gegenüber vielen klassischen Methoden in der Regel
zahlreiche Vorteile wie beispielsweise kurze Reaktionszeiten, hohe Ausbeuten
und große Selektivität. Zudem können sie in Anwesenheit einer Vielzahl
unterschiedlicher Lösungsmittel und funktioneller Gruppen durchgeführt werden.
Gerade die Reaktionsfähigkeit im wässrigen Milieu prädestiniert diese
Reaktionsklasse für eine breite Anwendung in der pharmazeutischen
Chemie.159,160 Die populärste und am häufigsten angewandte „Click“-Reaktion ist
die Kupfer-katalysierte Azid-Alkin-Cycloaddition, bei der regioselektiv ein
gegenüber vielen Reaktionsbedingungen inertes Triazol gebildet wird.
N N NR1
H R2 Cu(I)
NN
N
R2
R1

Ergebnisse und Diskussion _________________________________________________________________
- 44 -
Klassisch wird eine solche Cycloaddition im wässrigen Milieu mit Kupfer(II)sulfat,
welches durch Natriumascorbat in situ zu Cu(I) reduziert wird, durchgeführt. Doch
ebenso effektiv ist eine Umsetzung in organischen Lösungsmitteln mit Cu(I)-
Salzen als Katalysatoren.
Als potenzielle Träger für die säurelabilen Verbindungen wurden das synthetische
Polymer PEG sowie das körpereigene Protein HSA ausgewählt, da beide
kostengünstig in größeren Mengen zu erwerben sind und über folgende
vorteilhafte Eigenschaften verfügen:
passives Tumor-Targeting;
verbesserte Pharmakokinetik, d. h. erhöhte Plasmahalbwertszeiten durch
Zurückdrängen der raschen Eliminierung;
Aufnahme über Endozytose (Grundvoraussetzung für säurelabile
Spaltung);
bessere Applizierbarkeit durch Erhöhung der Wasserlöslichkeit;
schwächere Bindung an Plasmaproteine und damit höhere Stabilität im
Plasma.
Welchen Einfluss die Einführung eines polymeren Trägers sowie eines
chemischen Adapter-Systems auf die Spaltungseigenschaften der Trigger-Gruppe
ausübt, sollte anhand von AMC-Modellverbindungen untersucht werden.

Ergebnisse und Diskussion _________________________________________________________________
- 45 -
3.2.1 Syntheseversuche makromolekularer PEG-geträgerter Verbindungen
3.2.1.1 Synthesestrategien
Im Rahmen eines anderen Projekts der Arbeitsgruppe wurde von BAKHEET
ELSADEK ein PABC-analoger Baustein synthetisiert (Abb. 3.16).
NH
N3
O
O
O
O
O
NH
O O
"geschütztes"Isocyanat
gegenüber Nucleophilenaktivierte Funktionalität
Abb. 3.16: In der Arbeitsgruppe vorhandener Adapter-Baustein.
Die Aminogruppe des PABC-Systems war in ein Phenylcarbamat, ein „maskiertes“
Isocyanat,142 überführt worden, welches nach thermischer Aktivierung mit
Alkoholen Carbamate bilden kann. In para-Stellung wurde AMC über eine
Carbamat-Bindung in Benzylstellung eingeführt. Darüber hinaus verfügte der
Baustein in meta-Position zur Aminogruppe über eine Acylazid-Funktion, die eine
Reaktivität gegenüber Nucleophilen besitzt und damit den Einbau einer
Ankergruppe wie beispielsweise einer Alkingruppe ermöglicht. Über diese
Ankergruppe sollte durch CuAAC eine Bindung an einen polymeren Träger
erfolgen. Da dieser Baustein über die benötigten Funktionalitäten für den Aufbau
makromolekularer, säurelabiler AMC-Modellverbindungen verfügte und dessen
Synthese in ihren Grundzügen bekannt war, wurde er als Adapter-System für die
Darstellung säurelabiler AMC-Modellverbindungen ausgewählt.
Ein Nachteil der bekannten Syntheseroute zu diesem Baustein bestand darin,
dass sie sich nicht für den Aufbau makromolekularer Prodrugs eignet. Manche
Wirkstoffe wie beispielsweise Paclitaxel neigen bei niedrigen pH-Werten zur
Zersetzung.161 In der erprobten Syntheseroute ist die Verwendung starker Säuren
nach Einführen des Effektors allerdings unumgänglich (siehe Abb. 3.18, S. 47).
Daher wurde eine alternative Synthesestrategie entwickelt, die in Abbildung 3.17
(S. 46) retrosynthetisch dargestellt ist.

Ergebnisse und Diskussion _________________________________________________________________
- 46 -
NH
HN
O
O
O
O
O
N
HFF
RF
F
Wirkstoff
R = H, F
NH
HN
O
O
O
O
O
N
HFF
RF
FR = H, F
NH
HN
O
O
O
O
O
O
O
NO2
NO2
H2NHN
O
OH
O2NHN
O
OH
O2NO
O
6-Nitrophthalidkäuflich
Abb. 3.17: Retrosynthese des Adapter-Bausteins für die Darstellung makromolekularer Prodrugs.
Im letzten Syntheseschritt sollte der Wirkstoff durch Reaktion mit der als
p-Nitrophenylcarbonat aktivierten Benzylalkohol-Funktion des Linkers eingeführt
werden. Die Integration der Trigger-Gruppe sollte in Analogie zur Darstellung der
AMC-Modellverbindungen über ein „maskiertes“ Isocyanat erfolgen. Die erforderte
Carbonat- bzw. Carbamat-Bindung sollte aus einem verzweigten PABC-Derivat
aufgebaut werden. Als käufliche Ausgangsverbindung bot sich 6-Nitrophthalid an,
dessen Lactonring durch nucleophilen Angriff der späteren Ankergruppe geöffnet
werden sollte.
Eine ausführliche Diskussion der einzelnen Syntheseschritte zur Darstellung
niedermolekularer AMC-Modellverbindungen bzw. Prodrugs, die über eine
Ankergruppe durch CuAAC an makromolekulare Träger gekuppelt werden
können, erfolgt im nächsten Kapitel.
3.2.1.2 Synthese niedermolekularer Vorstufen zur Darstellung makromolekularer AMC-Modellverbindungen bzw. Prodrugs
Zwei niedermolekulare AMC-Modellverbindungen wurden ausgehend vom
käuflichen 6-Nitrophthalid in einer 11-stufigen Synthese (Abb. 3.18, S. 47)
dargestellt, wobei die ersten Syntheseschritte in Anlehnung an die Arbeiten von
BAKHEET ELSADEK162 durchgeführt und dabei optimiert wurden.

Ergebnisse und Diskussion _________________________________________________________________
- 47 -
OO2N
O
N2H4 H2OMeOH (abs.)
O2NHN
ONH2
OH(Boc)2ODioxan (abs.)
t-Butanol O2NHN
ONH
OH
Boc
TBSClImidazolDMF (abs.) O2N
HN
ONH
OTBS
Boc
Ni(NO3)2 / SiO2NaBH4, -18 °CMeOH (abs.)
H2NHN
ONH
OTBS
BocO Cl
O
DIEA, 0 °CDCM (abs.)N
H
HN
ONH
OTBS
BocO
OMeOH/1 M HCl
50:1
NH
HN
ONH
OH
BocO
O
NH
HN
ONH
O
BocO
O
O
AMC
DBTLTHF (abs.)
N O OCO
TFA/DCM 1:1
NH
HN
ONH3
+
O
O
O
O
AMC
NH
N3
O
O
O
O
O
AMC1 M HClNaNO2, 0 °C
H2N
NH
HN
O
O
O
O
O
AMC
DBTL in ToluolDioxan (abs.), Δ
NH
HN
O
O
O
AMC
O
O
DMF (abs.)
OHN
HFF
F F
R
2357 %
2270 %
2172 %
2495 %
3
2596 %
2691 %
2774 %
R = H: 13aR = F: 14a
1894 %
1997 %
2093 %
R = H: 28 (64 %)R = F: 29 (66 %)
CF3COO-
N
HFF
RF
F
Abb. 3.18: Darstellung der säurelabilen AMC-Modellverbindungen 28 und 29.
Im ersten Schritt wurde der Lactonring des käuflichen 6-Nitrophthalids mit
Hydrazin geöffnet. Die Hydrazidfunktion des entstandenen Produkts 18 wurde im
Folgenden Boc-geschützt. Ebenso musste der erhaltene Benzylalkohol 19 in den
TBS-Ether 20 überführt werden, da die anschließende Reduktion der Nitrogruppe
zum Amin im Fall einer freien Alkoholfunktion zu zahlreichen Nebenprodukten
führte. Als Reduktionsmittel lieferte Natriumborhydrid in Kombination mit einem
durch Kieselgel aktivierten Nickelnitrat-Katalysator163 21 in einer deutlich höheren
Ausbeute als ein Zink/Essigsäure-Gemisch. Die nun freie Aminogruppe wurde
schließlich mit Chlorameisensäurephenylester zum Phenylcarbamat 22, einem
maskierten Isocyanat,142 umgesetzt. Nach Spaltung des TBS-Ethers mit einem
MeOH/HCl-Gemisch (50:1) wurde durch Reaktion des Benzylalkohols 23 mit
AMC-NCO (3) der Farbstoff AMC über eine Carbamat-Bindung eingeführt (24).
Abspaltung der Boc-Gruppe mit einem TFA/DCM-Gemisch und Behandlung der

Ergebnisse und Diskussion _________________________________________________________________
- 48 -
Hydrazidfunktion von 25 mit HCl und Natriumnitrit lieferte das Acylazid 26. Durch
nucleophilen Angriff von Propargylamin auf das Azid wurde das Amid 27 gebildet.
Im letzten Syntheseschritt wurde das Phenylcarbamat durch Erhitzen in ein
Isocyanat überführt, welches in situ mit den Benzylalkoholen 13a bzw. 14a zu den
niedermolekularen Modellverbindungen 28 bzw. 29 reagierte.
Parallel zu den Arbeiten an den AMC-Modellverbindungen wurde mit der Synthese
niedermolekularer Vorstufen für makromolekulare Prodrugs begonnen.
Doxorubicin und Paclitaxel wurden hierbei als Vertreter für amino- bzw. hydroxy-
funktionalisierte Wirkstoffe ausgewählt. Im ersten Syntheseschritt wurde der
Lactonring durch Propargylamin geöffnet (Abb. 3.19, S. 49). Der entstandene
Benzylalkohol 30 wurde zunächst als TBS-Ether (31) geschützt und die
Nitrogruppe anschließend unter Standardbedingungen mit Zink-Pulver und
Essigsäure zum Amin 32 reduziert. Nach Umsetzung mit Chlorameisensäure-
phenylester zum geschützten Isocyanat 33 wurde der TBS-Ether mit einem
MeOH/Wasser-Gemisch (50:1) gespalten. Das Produkt 34 mit freier
Benzylalkoholgruppe wurde durch Reaktion mit Chlorameisensäure-p-
nitrophenylester in die aktivierte Carbonatform 35 überführt. In einer zur Synthese
von 28 bzw. 29 analog durchgeführten Reaktion gelang es, die jeweilige Trigger-
Gruppe über eine Isocyanat-Zwischenstufe einzuführen.
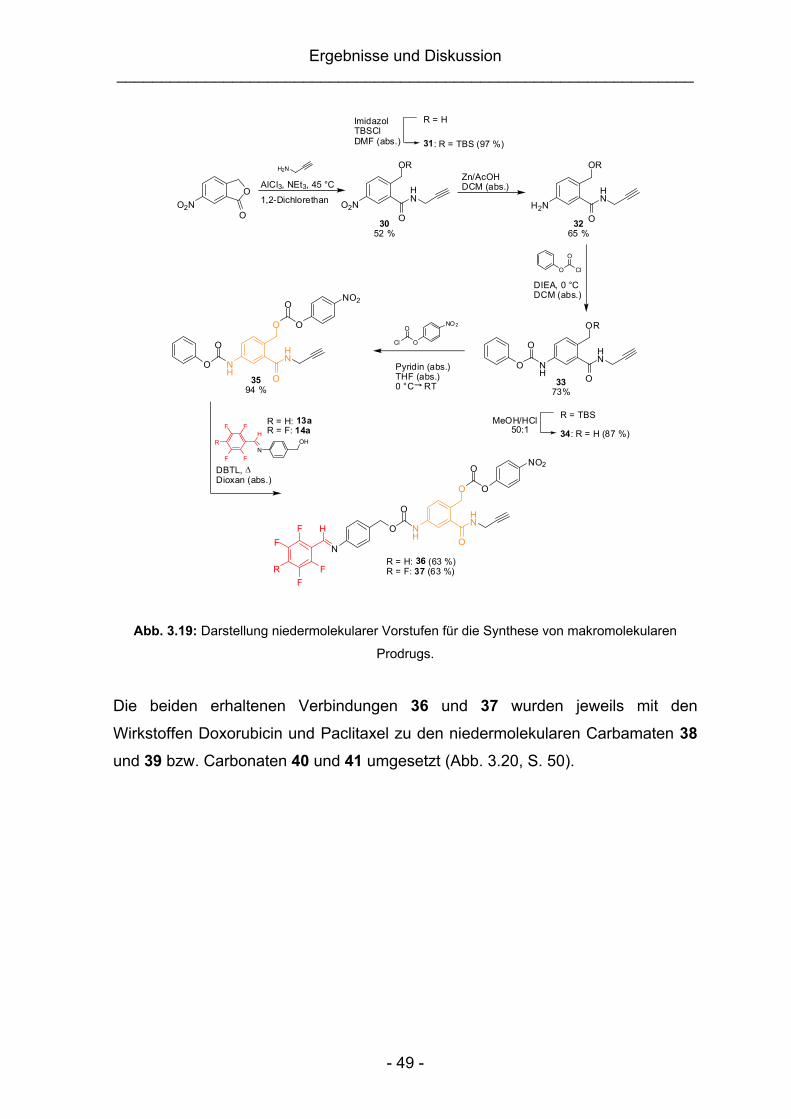
Ergebnisse und Diskussion _________________________________________________________________
- 49 -
OO2N
O
H2N
AlCl3, NEt3, 45 °C
1,2-Dichlorethan O2NHN
OH2N
HN
O
Zn/AcOHDCM (abs.)
NH
HN
OO
O
OR OR
OR
31: R = TBS (97 %)
ImidazolTBSClDMF (abs.)
34: R = H (87 %)MeOH/HCl
50:1
DIEA, 0 °CDCM (abs.)
OCl
ONO2
NH
HN
O
O
O
O
O
O
NO2
DBTL, ΔDioxan (abs.)
NH
HN
O
O
O
O
O
O
OHN
HFF
F F
R
NO2
N
HFF
RF
F
O Cl
O
Pyridin (abs.)THF (abs.)0 °C RT
3052 %
3265 %
3594 %
R = H: 36 (63 %)R = F: 37 (63 %)
R = H
R = TBS
3373%
R = H: 13aR = F: 14a
Abb. 3.19: Darstellung niedermolekularer Vorstufen für die Synthese von makromolekularen
Prodrugs.
Die beiden erhaltenen Verbindungen 36 und 37 wurden jeweils mit den
Wirkstoffen Doxorubicin und Paclitaxel zu den niedermolekularen Carbamaten 38
und 39 bzw. Carbonaten 40 und 41 umgesetzt (Abb. 3.20, S. 50).

Ergebnisse und Diskussion _________________________________________________________________
- 50 -
NH
HN
O
O
O
O
O
O
NO2
N
HFF
RF
FR = H: 36R = F: 37
Doxurubicin HClDIEADMF (abs.)
PaclitaxelDIEADMAPDCM (abs.)
NH
HN
O
O
O
O
O
N
HFF
RF
F
O
O
O
H3CO
HO
OH
O
O
NH
HOCH3
OHOH
NH
HN
O
O
O
N
HFF
FFR
R = H: 38 (72 %)R = F: 39 (61 %)
R = H: 40 (75 %)R = F: 41 (40 %)
O
O
O
OO
O
OH
OO
O
HO
HN
OO
O
O
O
Abb. 3.20: Synthese niedermolekularer Prodrugs mit einer Alkin-Ankergruppe zur Bindung an
PEG-Diazid via CuAAC.
Im Unterschied zu den AMC-Modellverbindungen, die im Kapitel 3.1 vorgestellt
wurden, verfügen die erhaltenen niedermolekularen Vorstufen 28, 29 und 38–41
über ein zwischen PABC-Linker und Effektor integriertes zusätzliches PABC-
analoges System. Welchen Einfluss die Einführung eines solchen chemischen
Adapters auf die Freisetzungsgeschwindigkeit von AMC ausübt, sollte im
Folgenden anhand der niedermolekularen Vorstufen 28 und 29 untersucht
werden.
3.2.1.3 Untersuchungen der pH-abhängigen Spaltungseigenschaften der niedermolekularen Modellverbindungen und Prodrugs 28, 29 und 38–41
Die Halbwertszeiten der AMC-Freisetzung in Pufferlösungen (pH 5.0 und 7.4) und
im Plasma wurden für 28 und 29 analog zu den Kinetikmessungen der
Modellverbindungen 1b und 4b–14b durch Messung der Fluoreszenzzunahme
bestimmt (siehe Kapitel 6.1.4, S. 89). Nach säurekatalysierter Hydrolyse der

Ergebnisse und Diskussion _________________________________________________________________
- 51 -
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 100 200 300 400t /min
I x/I
AM
C
2829
0
0.2
0.4
0.6
0.8
1
0 40 80 120 160t /h
I x/I
AM
C
2829
Schiffschen Base sollten die beiden Linker spontan in zwei aufeinander folgenden
1,6-Benzyleliminierungen unter Abspaltung von AMC reagieren (Abb. 3.21).
NH
HN
O
O
O
AMC
O
O
R = H: 28R = F: 29
N
HFF
RF
F
H2NHN
O
OH
AMCOH
H2N
HFF
RF
F
O
2 x CO2
1. Benzyleliminierungen2. "Recycling" der PABC-Systeme
Abb. 3.21: Spaltung der Verbindungen 28 und 29 unter AMC-Freisetzung nach saurer Hydrolyse.
Im Folgenden sind die Ergebnisse der durchgeführten Spaltungsstudien
abgebildet (Abb. 3.22).
A
B

Ergebnisse und Diskussion _________________________________________________________________
- 52 -
0
0.2
0.4
0.6
0.8
1
0 5 10 15 20 25t /h
I x/I
AM
C
2829
C
Abb. 3.22: Zeitlicher Verlauf der AMC-Freisetzung von 28 und 29 in Pufferlösungen und im Plasma
(pH 5.0 (A), pH 7.4 (B), Plasma (C)).
Die Auftragung von ⎟⎟⎠
⎞⎜⎜⎝
⎛−
∞cc1ln gegen t zeigte keinen linearen Zusammenhang.
Daher wurden die Halbwertszeiten aus den graphischen Darstellungen des
zeitlichen Verlaufs der Fluoreszenzzunahme näherungsweise bestimmt. Die
Halbwertszeit entsprach dabei dem Zeitpunkt, an dem die Hälfte der aus dieser
Verbindung insgesamt freigesetzten Menge an AMC erreicht wurde.
Da sich der Kurvenverlauf nicht durch eine Exponentialfunktion beschreiben lässt,
scheinen sich mehrere, voneinander unabhängige Reaktionsschritte zu
überlagern. Ein Vergleich der Halbwertszeiten von 13b und 14b bzw. 28 und 29
lieferte ein überraschendes Ergebnis (Tab. 3.5, S. 53). Entgegen der Erwartungen
verlief die AMC-Freisetzung aus den komplexeren Verbindungen 28 und 29 in den
Pufferlösungen tendenziell rascher als aus den entsprechenden Analoga. Im
Plasma lagen die Halbwertszeiten hingegen in einer ähnlichen Größenordnung.

Ergebnisse und Diskussion _________________________________________________________________
- 53 -
Tab. 3.5: Vergleich der Halbwertszeiten von 13b und 14b bzw. 28 und 29 in Pufferlösungen bei
pH 5.0, pH 7.4 und im Plasma.
t1/2 [h] bei pH 5.0a t1/2 [h] bei pH 7.4b t1/2 [h] im Plasma
R = 13b/14b 28/29 13b/14b 28/29 13b/14b 28/29
F F
F F
0.57 0.28 96 ~ 72 ~ 1.30 ~ 1.25
F
F F
F F
2.88 1.00 360 ~ 72 ~ 2.33 ~ 1.25
a) Acetat-Puffer, 20 mM; b) Phosphat-Puffer, 20 mM.
Analog zu den niedermolekularen AMC-Modellverbindungen 28 und 29 sollten die
Spaltungseigenschaften der niedermolekularen Prodrugs (38–41) untersucht und
deren Halbwertszeiten miteinander verglichen werden. Da der Effektor als
Abgangsgruppe bei der Freisetzung kaum einen Einfluss auf die
Spaltungsgeschwindigkeit haben sollte, wurde angenommen, dass sich die
Halbwertszeiten von 28 und 29 bzw. 38, 39 und 40, 41 in einer ähnlichen
Größenordnung bewegen. In ersten Voruntersuchungen konnte mittels DC gezeigt
werden, dass alle vier Verbindungen (38–41) in Pufferlösungen den Wirkstoff
freisetzen. Die Spaltung verlief bei pH 5.0 deutlich schneller als bei pH 7.4. Auf
eine Quantifizierung mittels High Performance Liquid Chromatography (HPLC)
wurde an dieser Stelle jedoch aufgrund der sich andeutenden Löslichkeits-
probleme verzichtet.
3.2.1.4 Versuche zur Darstellung makromolekularer AMC-Modellverbin-dungen und Prodrugs mittels CuAAC
Nach der Untersuchung ihrer Hydrolyseeigenschaften sollten die
niedermolekularen AMC-Modellverbindungen 28 und 29 sowie die
niedermolekularen Prodrugs 38–41 durch CuAAC an PEG-Diazide
unterschiedlicher Kettenlänge (2 kDa, 4 kDa, 35 kDa) gekuppelt werden. Für eine
gezielte Anreicherung im Tumor unter Ausnutzung des EPR-Effekts ist ein
Prodrug mit hoher Molmasse (>30 kDa) erforderlich. Allerdings lassen sich

Ergebnisse und Diskussion _________________________________________________________________
- 54 -
Moleküle in dieser Größenordnung nur schwer charakterisieren. Daher sollten
zusätzlich durch Reaktionen unter identischen Bedingungen analoge
Modellverbindungen mit PEG2k bzw. PEG4k dargestellt und anstelle des PEG35k
charakterisiert werden. Außerdem sollte durch Variation der Kettenlänge des
Polymers der Einfluss des Trägers auf die Stabilität der Verbindungen in
Pufferlösungen und im Plasma näher untersucht werden.
Zunächst wurden die PEG-Diazide in einer zweistufigen Synthese (Abb. 3.23)
hergestellt. Durch Reaktion von PEG mit Mesylchlorid und Triethylamin wurde 42
erhalten, welches nach nucleophilem Angriff von aktiviertem Natriumazid164 zum
PEG-Diazid (43) reagierte.
OOHHO n
OOMsMsO n
ON3N3 n
DCM (abs.)0 °C
NEt3MsCl
NaN3DMF (abs.)70 °C
4292 %
4377 %
Abb. 3.23: Darstellung von PEG-Diaziden.
Mittels 1H- und 13C-NMR-Spektroskopie wurde schließlich die Struktur des PEG2k-
Diazids bestätigt. Durch Integration konnte das Verhältnis der Protonen c und d zu
den vier Protonen a in Nachbarstellung des Azidsubstituenten bestimmt werden.
Daraus ergab sich für 43 eine Beladung von ungefähr 94 % (Abb. 3.24, S. 55).

Ergebnisse und Diskussion _________________________________________________________________
- 55 -
1H-NMR
c, d
b a DMSO
13C-NMR
c, d
DMSO
b a
Abb. 3.24: 1H- und 13C-NMR-Spektren von PEG2k-Diazid (43) (n ~ 45).
PPM 3.70 3.60 3.50 3.40 3.30 3.20 3.10 3.00 2.90 2.80 2.70 2.60 2.50 2.40
0.
993
47
.83
3.
6106
3.
5985
3.
5965
3.
5859
3.
5453
3.
5391
3.
5347
3.
5318
3.
5070
3.
3944
3.
3814
3.
3696
3.
3186
N3O
OnN3
a
ab
b
c
d
N3O
OnN3
a
ab
b
c
d
PPM 68.0 66.0 64.0 62.0 60.0 58.0 56.0 54.0 52.0 50.0 48.0 46.0 44.0 42.0 40.0
69.
6331
69.
0929
49.
8591

Ergebnisse und Diskussion _________________________________________________________________
- 56 -
Vor der Umsetzung der erhaltenen PEG-Diazide durch CuAAC mit den
niedermolekularen Vorstufen wurden in Testansätzen mit 29 und käuflichem
Benzylazid geeignete Reaktionsbedingungen ermittelt. Der erste Syntheseversuch
erfolgte in einem Wasser/DMSO-Gemisch mit dem klassischen Katalysatorsystem
bestehend aus Kupfersulfat und Natriumascorbat. Es konnte mittels DC keine
Reaktion detektiert werden, was vermutlich auf die äußerst geringe Löslichkeit von
29 in Wasser und Wassergemischen zurückzuführen ist. Die Bedingungen der
anschließenden Probeansätze, bei denen sowohl DMSO als auch DMF als
Lösungsmittel (LM) dienten, sind in Tabelle 3.6 zusammengestellt.
Tab. 3.6: Übersicht der verschiedenen getesteten Bedingungen zur Darstellung des CuAAC-
Produkts aus 29 und Benzylazid.
CuINatriumascorbatAminLM (abs.)
NH
HN
O
O
O
O
O
NH
O O
NH
HN
O
O
O
O
O
NH
O O
N
HFF
FF
NNN
F29
N3
N
HFF
FF
F
Variante
Reagenz 1 2 3 4 5
29 1 eq 1 eq 1 eq 1 eq 1 eq
Benzylazid 1 eq 1 eq 1 eq 1 eq 1 eq
Cu(I)I 10 mol%
10 mol%
20 mol%
20 mol%
10 mol%
DIEA 2 eq - 2 eq - 1 eq
PMDETA - 1.6 eq - 1.6 eq -
Neben den beiden Lösungsmitteln wurden die Katalysatormenge sowie die amino-
funktionalisierten Liganden variiert. In DMF wurde keine Reaktion beobachtet, was
vermutlich auf die hohe Verdünnung aufgrund geringer Löslichkeit zurückzuführen
ist. In DMSO hingegen konnte eine Umsetzung der Reaktanden festgestellt
werden. Nach Auswertung der DC zeigte Variante 1 in DMSO über Nacht den
größten Umsatz. Die ersten Reaktionsversuche mit 29 und PEG-Diazid wurden

Ergebnisse und Diskussion _________________________________________________________________
- 57 -
daher in Anlehnung an diese Synthesevariante durchgeführt. Allerdings wurden
die Bedingungen an das veränderte Edukt angepasst und optimiert (Abb. 3.25). Im
Vergleich zu Reaktionen zwischen zwei niedermolekularen Reaktionspartnern sind
bei Reaktionen mit makromolekularer Komponente aufgrund des erhöhten
Lösungsmittelbedarfs längere Reaktionszeiten zu erwarten. Um zu gewährleisten,
dass dauerhaft das aktive Cu(I) statt des unreaktiven Cu(II) für die Reaktion zur
Verfügung stand, wurde Natriumascorbat zum Recyceln des stark sauerstoff-
empfindlichen Katalysators zugesetzt. Ebenso wurde das Verhältnis der einzelnen
Reagenzien zueinander angepasst: PEG-Diazid (1 eq), 29 (3 eq), Cu(I)I (2 eq),
Natriumascorbat (2 eq), DIEA (4 eq). Als Lösungsmittel dienten entweder DMSO,
THF oder DMF; in Dioxan und MeOH konnte aufgrund der geringen Löslichkeit der
Edukte nicht konzentriert genug (30 – 40 mM) gearbeitet werden.
CuINatriumascorbatDIEALM (abs.)
NH
HN
O
O
O
NH
O O
PEG-Diazid (43)
NH
HN
O
O
O
O
O
NH
O O
N
HFF
FF
NNN
PEG
2
RR = H 28R = F 29
O
O
N
HFF
RF
F
instabil
Abb. 3.25: Versuchte Synthese zur Darstellung makromolekularer AMC-Modellverbindungen
mittels CuAAC.
Mittels DC konnte die Umsetzung zu einem Produkt verfolgt werden, welches, wie
für die Zielverbindung erwartet, das Lauf- und Anfärbeverhalten eines PEG
aufwies und darüber hinaus UV- bzw. Fluoreszenzaktivität besaß. Allerdings
ließen sich auch Nebenprodukte beobachten. Es wurden zahlreiche Versuche
unternommen, um das vermeintliche Produkt zu isolieren. Fällungen mit unpolaren
Lösungsmitteln wie Et2O oder Hexan und Extraktionen inklusive Waschschritten
(z. B.: DCM/Phosphat-Puffer pH 7.4) lieferten ebenso wenig die Zielverbindung
wie Säulenchromatographien unter Verwendung verschiedener stationärer
Phasen (Kieselgel, Diol, RP). Stattdessen wurde ausschließlich AMC isoliert, das
sich während der Aufreinigung aus dem mutmaßlichen Produkt bildete.

Ergebnisse und Diskussion _________________________________________________________________
- 58 -
In Anlehnung an die AMC-Modellverbindungen wurden Syntheseversuche zur
Darstellung makromolekularer Prodrugs durch Umsetzung der niedermolekularen
Vorstufen 38–41 mit PEG-Diaziden in einer CuAAC unternommen (Abb. 3.26).
NH
HN
O
O
O
O
O
N
HFF
RF
F
Wirkstoff
R = H 38, 40R = F 39, 41
CuINatriumascorbatDIEALM (abs.)
PEG-Diazid (43)
NH
HN
O
O
O
O
O
Wirkstoff
N
HFF
FF
NNN
PEG
2
R
instabil
Abb. 3.26: Versuchte Darstellung von makromolekularen Prodrugs durch Umsetzung der
niedermolekularen Vorstufen 38–41 mit PEG-Diaziden in einer CuAAC.
Analog zu den Beobachtungen, die bei den Syntheseversuchen der AMC-
Modellverbindungen gemacht wurden, konnte mittels DC zunächst die Bildung
eines Produkts mit zu der Zielverbindung passenden Eigenschaften verfolgt
werden. Spätestens während der zahlreichen Isolierungsversuche durch
beispielsweise Fällungen, Chromatographien unter Verwendung verschiedener
Trägermaterialien oder Extraktionen entstand allerdings ein nicht zu
charakterisierender, geleeartiger Niederschlag, welcher auf Aggregatbildung
hindeuten könnte.
3.2.2 Alternative Synthesestrategien zur Darstellung makromolekularer AMC-Modellverbindungen
Da bislang alle Versuche scheiterten, den polymeren Träger durch eine CuAAC
mit einer der niedermolekularen Vorstufen zu verknüpfen, wurden schließlich
alternative Synthesestrategien verfolgt. Zur Evaluierung geeigneter
Reaktionsbedingungen wurden die Synthesen ausschließlich mit
niedermolekularen Vorstufen der AMC-Modellverbindungen durchgeführt.
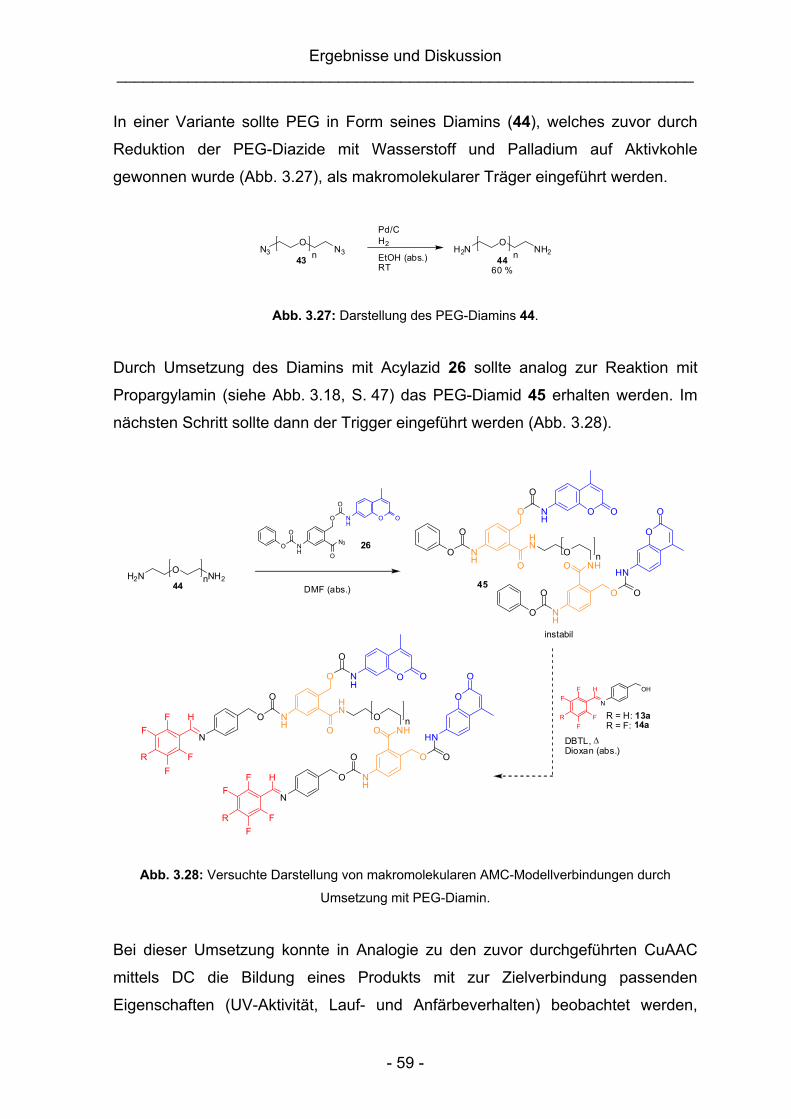
Ergebnisse und Diskussion _________________________________________________________________
- 59 -
In einer Variante sollte PEG in Form seines Diamins (44), welches zuvor durch
Reduktion der PEG-Diazide mit Wasserstoff und Palladium auf Aktivkohle
gewonnen wurde (Abb. 3.27), als makromolekularer Träger eingeführt werden.
ON3 N3n
OH2N NH2n
Pd/CH2
EtOH (abs.)RT
43 4460 %
Abb. 3.27: Darstellung des PEG-Diamins 44.
Durch Umsetzung des Diamins mit Acylazid 26 sollte analog zur Reaktion mit
Propargylamin (siehe Abb. 3.18, S. 47) das PEG-Diamid 45 erhalten werden. Im
nächsten Schritt sollte dann der Trigger eingeführt werden (Abb. 3.28).
H2NO
NH2DMF (abs.)
NH
NHO
O O
HN
O
O
ONH
HN
O
O
O
O
O
NH
O O
DBTL, ΔDioxan (abs.)
NH
NHO
O O
HN
O
O
ONH
HN
O
O
O
O
O
NH
O O
N
HFF
RF
F
26
44 45
R = H: 13aR = F: 14a
NH
N3
O
O
O
O
O
NH
O O
n
n
n
OH
N
HFF
RF
F
O
O
O
O
N
HFF
RF
F
instabil
Abb. 3.28: Versuchte Darstellung von makromolekularen AMC-Modellverbindungen durch
Umsetzung mit PEG-Diamin.
Bei dieser Umsetzung konnte in Analogie zu den zuvor durchgeführten CuAAC
mittels DC die Bildung eines Produkts mit zur Zielverbindung passenden
Eigenschaften (UV-Aktivität, Lauf- und Anfärbeverhalten) beobachtet werden,

Ergebnisse und Diskussion _________________________________________________________________
- 60 -
dessen Isolierung abermals scheiterte. Statt des erwarteten Produkts wurden
AMC und ein nicht zu charakterisierendes, geleeartiges Nebenprodukt, welches
auf Aggregatbildung hindeuten könnte, erhalten.
Da alle Versuche scheiterten, das Polymer PEG mittels CuAAC oder durch Aufbau
einer Amid-Bindung mit dem Adapter zu verknüpfen, sollten in einer weiteren
Variante AMC-Modellverbindungen hergestellt werden, die an HSA als
Trägermolekül binden können. Dazu sollte eine Maleinimido-Ankergruppe anstelle
der Propargylgruppe in den bereits bekannten Linker basierend auf 3-Amido-4-
hydroxymethylanilin integriert werden. Diese Maleinimidofunktion bindet selektiv
und kovalent an die Cystein-34-Position von HSA.165
Für die Synthese der Zielverbindungen (Abb. 3.29) wurde zunächst
6-Maleinimidocapronsäurehydrazid durch Reaktion mit Natriumnitrit in HCl (1 M) in
das Acylazid 46 überführt. Diese labile Verbindung wurde mit DCM extrahiert und
direkt nach Entfernen des Lösungsmittels in Dioxan aufgenommen. Durch äußerst
langsames Zutropfen der organischen Phase zu siedendem, mit einem Äquivalent
konz. HCl versetztem Wasser und anschließendem Lyophilisieren konnte
1-Maleinimidopentan-5-amin (47) erhalten werden. Dieses Amin sollte im
nächsten Schritt mit dem Acylazid 26 zum Amid reagieren. Durch zur Synthese
von 28 bzw. 29 analoge Umsetzung mit den Benzylalkoholen 13a und 14a
(Abb. 3.18, S. 47) sollten schließlich die beiden Zielverbindungen erhalten werden.
+H3NHN
ON
O
O
CF3COO-1 M HClNaNO2, 0° C N3
ON
O
O
Dioxan/H2Okonz. HCl, Δ
+H3N N
O
O
26NH
N3
O
O
O
O
O
NH
O O DIEADMF (abs.)
NH
HN
O
O
O
O
O
NH
O O
N
O
O
NH
HN
O
O
O
O
O
NH
O O
N
HFF
FF
DBTL, ΔDioxan (abs.)
OH
N
HFF
FFR
R
N
O
O
Cl-
4790 %
46
R = H: 13aR = F: 14a
Abb. 3.29: Versuchte Darstellung von AMC-Modellverbindungen mit Maleinimido-Ankergruppe zur
Bindung an HSA.

Ergebnisse und Diskussion _________________________________________________________________
- 61 -
Obwohl in einer analogen Reaktion von 26 mit Propargylamin das Amid 27
erhalten werden konnte (Abb. 3.18, S. 47), war eine Umsetzung mit dem Amin 47
nicht erfolgreich. Langsames Zutropfen der mit Lösungsmittel verdünnten Base
DIEA zum Reaktionsgemisch mit dem basenlabilen Maleinimid führte zur Bildung
zahlreicher Produkte. Die säulenchromatographisch isolierten, zum Teil instabilen
Verbindungen wurden mittels NMR-Spektroskopie untersucht und entsprachen
nicht der Zielverbindung. Da sich 47 mit Acetanhydrid acetylieren ließ, kann eine
Empfindlichkeit von 47 als alleinige Ursache für das Scheitern der Synthese
ausgeschlossen werden.
In einem weiteren Versuch sollte eine abgeänderte Variante des Linkers
synthetisiert werden (Abb. 3.30), bei der die Ankergruppe nicht als Amid sondern
über eine Hydrazid-Bindung mit dem Linker verknüpft wird. Dazu wurde Hydrazid
25 zunächst mit 6-Maleinimidohexanoyl-N-hydroxysuccinimidester zu 48
umgesetzt. Bei den Versuchen eine Trigger-Gruppe analog zu der Reaktion von
27 mit den Benzylalkoholen 13a bzw. 14a (Abb. 3.18, S. 47) einzuführen, wurde
jedoch wiederum ausschließlich das Zersetzungsprodukt AMC erhalten. Ob diese
Beobachtung auf ein Stabilitätsproblem des Produkts oder auf die Inkompatibilität
der Maleinimidofunktion mit den Reaktionsbedingungen zurückzuführen war,
konnte nicht geklärt werden.
NH
HN
ONH3
+
O
O
O
O
NH
O O
NH
HN
ONH
O
O
O
O
NH
O O
ON
O
ODIEADMF (abs.)
DBTL in ToluolDioxan (abs.), Δ
OHN
HFF
F F
R
NH
HN
ONH
O
O
NH
O O
ON
O
O
N
HF
FF
F
R
25
N
O
O
O
O
N OO
R = H: 13aR = F: 14a
4845 %
O
O
CF3COO-
Abb. 3.30: Versuchte Darstellung von AMC-Modellverbindungen mit Maleinimido-Ankergruppe zur
Bindung an HSA.

Ergebnisse und Diskussion _________________________________________________________________
- 62 -
3.2.3 Abschließende Untersuchungen zur Eignung von 3-Amido-4-hydroxymethylanilin als Adapter-Baustein
Bei Betrachtung der Ergebnisse aus den Kapiteln 3.2.1.4 und 3.2.2 zeigt sich,
dass alle bisherigen Versuche misslangen, makromolekulare Systeme mit dem
integrierten Adapter-Baustein 3-Amido-4-hydroxymethylanilin darzustellen. Das
Scheitern der Synthesen ist auf die Bildung von Produkten zurückzuführen, die
aus unbekannten Gründen nicht stabil waren und sich daher nicht isolieren ließen.
Die gewonnenen Erkenntnisse werfen die Frage auf, ob sich der Linker auf Basis
von 3-Amido-4-hydroxymethylanilin überhaupt als Adapter-Baustein eignet. Zur
Verifizierung, dass nicht die Methoden selbst einer erfolgreichen Synthese im
Wege standen, sollten zu den gescheiterten Synthesen aus den vorherigen
Kapiteln analoge Reaktionen mit niedermolekularen Reaktionspartnern
durchgeführt werden. Folgende Vorteile wurden sich davon versprochen: kürzere
Reaktionszeiten, erleichterte Verfolgung des Reaktionsverlaufs mittels DC,
geringerer Lösungsmittelbedarf und Verfügbarkeit zahlreicher Charakterisierungs-
methoden. Zudem wurden für die Reaktionen Vorstufen ohne säurelabile Imin-
Bindung ausgewählt, um mögliche Einflüsse einer vorzeitigen Imin-Hydrolyse
ausschließen zu können.
Zunächst wurde eine CuAAC von 27 mit dem niedermolekularen Reaktionspartner
Benzylazid durchgeführt (Abb. 3.31, S. 63). Dabei wurden zu den Umsetzungen
mit den PEG-Diaziden analoge Bedingungen gewählt. In einem weiteren Versuch
sollte 26 mit Methoxyethylamin anstelle des PEG-Diamins in einer nucleophilen
Substitution zum Amid umgesetzt werden. Interessanterweise wurde statt der
Zielverbindungen in beiden Fällen nahezu ausschließlich das Zersetzungsprodukt
AMC erhalten.

Ergebnisse und Diskussion _________________________________________________________________
- 63 -
NH
HN
O
O
O
O
O
NH
O ON3
NH
HN
O
O
O
O
O
NH
O O
NNNCuI
NatriumascorbatDIEATHF (abs.)
NH
N3
O
O
O
O
O
NH
O OH2N
DMF (abs.)
NH
HN
O
O
O
O
O
NH
O OO
O
27
26
instabil
instabil
Abb. 3.31: Zu den Syntheseversuchen der makromolekularen Verbindungen aus den Kapiteln
3.2.1.4 und 3.2.2 analoge Reaktionen mit niedermolekularem Reaktionspartner.
In Analogie zu den Versuchen, makromolekulare AMC-Modellverbindungen und
Prodrugs über CuAAC, Amid-Bindungen oder Einführung eines HSA-bindenden
Linkers zu synthetisieren, wurden bei den analytisch gut zu verfolgenden
Umsetzungen mit niedermolekularen Reaktionspartnern keine stabilen Produkte
erhalten. Da das Versagen unabhängig von der jeweiligen Darstellungsmethode
und der Struktur des Effektors ist, scheint eine Unbeständigkeit der
Zielverbindungen vorzuliegen. Diese wird vermutlich durch die im Vergleich zu den
stabilen Modellverbindungen 1b und 4b–14b zusätzlich integrierte Amid-Bindung
zur Kupplung an den polymeren Träger hervorgerufen. Der verwendete Linker
eignet sich daher nicht als Adapter-Baustein. In Abbildung 3.32 werden denkbare
Zyklisierungsreaktionen vorgestellt, die zur vorzeitigen Abspaltung des Effektors
führen könnten. Ebenso könnten katalytische Effekte der Amid-Funktion sowie die
Ausbildung von Wasserstoffbrückenbindungen zwischen dem Proton am
Stickstoffatom und dem Sauerstoffatom der Carbonyl-Funktion die Freisetzung
des Effektors beschleunigen.
NH
HN
O
O
O
R''
O
OR'
XEffektor
X = NH, O
Abb. 3.32: Mögliche Zyklisierungsreaktionen, die zur Abspaltung des Effektors führen.

Ergebnisse und Diskussion _________________________________________________________________
- 64 -
Die Erkenntnis, dass der Linker sich nicht für den Aufbau der makromolekularen
Verbindungen eignet, wurde leider erst nach zahlreichen Bemühungen gewonnen.
Dies lag vorwiegend daran, dass Verbindung 27 und ihre Folgestufen
bemerkenswerte Ausnahmen darstellten. So gelang ihre Isolierung und
Charakterisierung, was vermutlich auf die durch die Ethinylgruppe hervorgerufene
starrere Struktur der Verbindungen zurückzuführen ist. Allerdings zeigten auch
diese Verbindungen Anzeichen von Instabilität (vgl. Freisetzungsstudien, Kapitel
3.2.1.3, S. 50). Entgegen der Erwartung setzten sie AMC schneller frei als die
analogen Modellsubstanzen 13b und 14b ohne chemischen Adapter.
Für die weitere Entwicklung ist die Verwendung eines anderen Adapters
erforderlich. Da sich sowohl die Trigger-Gruppen als auch die benzylischen
Carbamat-Bindungen den Modellsubstanzen 1b und 4b–14b als stabil erwiesen,
sollte in einem neuen System das Polymer z. B. über eine benzylische Carbamat-
Bindung gekuppelt werden.
3.3 Säurelabile PEG-Konjugate auf Basis des 2,4-Bis(hydroxymethyl)anilin-Linkers
Da alle Versuche zur Synthese von makromolekularen Modellverbindungen und
Prodrugs mit dem auf 3-Amido-4-hydroxymethylanilin basierenden chemischen
Adapter-System nicht zum Ziel führten, wurde als Alternative der verzweigte
Linker 2,4-Bis(hydroxymethyl)anilin130 eingesetzt. Zur Evaluierung des neuen
Systems sollten zunächst ausschließlich AMC-Modellverbindungen dargestellt
werden. Die verfolgte Synthesestrategie wird retrosynthetisch in Abbildung 3.33
dargelegt. Als Ausgangsverbindung für die Synthese diente ein Baustein, der von
A. WARNECKE zur Verfügung gestellt wurde.166

Ergebnisse und Diskussion _________________________________________________________________
- 65 -
NHNO
OO
O CO
NH
O
O
O
HN
ONH
O
O
O O
NH
O
O
O
N3
O
HNO
OO
O
O O
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
R
NH
O
n
nNH
OHNO
OO
O
NH
O
n
n
O
NH
O O
NH
O
O
OH
HN
ONH
O
O
O O
vorhandener Baustein
R = H, F
Abb. 3.33: Retrosynthese von makromolekularen AMC-Modellverbindungen auf Basis eines
2,4-Bis(hydroxymethyl)anilin-Adapters.
Im letzten Schritt der Darstellung sollte der Trigger durch Umsetzung des
jeweiligen säurelabilen Imins mit einem Isocyanat, welches durch eine Curtius-
Umlagerung aus einem Acylazid intermediär gebildet wird, eingeführt werden. Das
Acylazid kann durch Behandlung mit HCl und Natriumnitrit aus einer Hydrazid-
Vorstufe dargestellt werden. Diese lässt sich wiederum durch Reaktion eines
PEG-Isocyanats mit dem vorliegenden Baustein erhalten. Im folgenden Kapitel
werden die einzelnen Syntheseschritte zur Darstellung der PEG-Konjugate
ausführlich erläutert.
3.3.1 Synthese und Charakterisierung der makromolekularen AMC-Modellverbindungen
Die makromolekularen AMC-Modellverbindungen wurden in mehrstufigen
Synthesen dargestellt. Im ersten Schritt wurde mPEG5k dabei durch Umsetzung
mit einem großen Überschuss an 1,6-Hexyldiisocyanat in PEG-Isocyanat 49
überführt (Abb. 3.34, S. 66). Anschließend wurde durch Reaktion mit dem von
A. WARNECKE zur Verfügung gestellten Benzylalkohol 50166 der trifunktionelle
Linker durch den Aufbau einer Carbamat-Bindung eingeführt. Nach Abspaltung

Ergebnisse und Diskussion _________________________________________________________________
- 66 -
der Boc-Schutzgruppe in 5 M HCl wurde durch Zugabe von Natriumnitrit aus dem
Hydrazid 51 das Acylazid 52 erhalten. Dieses reagierte beim Erhitzen in einer
Curtius-Umlagerung unter Stickstoffabspaltung zum Isocyanat, welches in situ mit
den Schiffschen Basen 13a bzw. 14a umgesetzt wurde. Die Produkte 53 und 54
wurden schließlich nach Fällung mit Et2O erhalten.
NHNO
OO
O CO
NH
O
O
O
HN
ONH
O
O
DBTL, 80 °CDioxan (abs.)
OH
N
HFF
RF
F
1. 5 M HCl2. NaNO2, H2O
NH
O
O
OH
HN
ONH
O
O
O O
O O
NH
O
O
O
N3
O
HNO
OO
O
O O
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
R53: R = H (21 %)54: R = F (35 %)
NH
O
n
13a: R = H14a: R = F
n
4998 %
OHO
On
NH
OHNO
OO
O
50
NH
O
n
n
5168 %
DBTL, 80 °CDioxan (abs.)
DBTLBenzol (abs.)
NN
CO
CO
5292 %
O
NH
O O
Abb. 3.34: Synthese der makromolekularen AMC-Modellverbindungen 53 und 54 (n ~ 114).
Zur Charakterisierung und zur Bestimmung der Beladung am Polymer wurden von
den beiden Zielverbindungen 53 und 54 1H-NMR-Spektren in DMSO-d6
aufgenommen (Abb. 3.35, S. 67). Im Bereich von 1.21–1.23 ppm und 1.33–
1.40 ppm treten die Signale von vier CH2-Gruppen des Hexyllinkers auf. Der Peak
bei 2.36 ppm ist charakteristisch für die Methylgruppe des AMC. Die zum Teil sehr
breiten Peaks bei 2.90–2.97 ppm, 3.24 ppm, 3.44–3.66 ppm und 4.02–4.04 ppm
lassen sich den zahlreichen Protonen am Polymer (CH2 und OCH3) und zwei
weiteren CH2-Gruppen des Hexyllinkers zuordnen. Die Signale der drei

Ergebnisse und Diskussion _________________________________________________________________
- 67 -
benzylischen CH2-Gruppen erscheinen bei 4.98 ppm und 5.19–5.21 ppm. Das
Proton am Lactonring des AMC erzeugt ein charakteristisches Signal bei
6.19 ppm. Im Bereich von 7.16–7.69 ppm treten die Signale der aromatischen
Protonen sowie die der beiden Protonen an den Stickstoffatomen des Hexyllinkers
auf. Dem aldehydischen Proton lässt sich der Peak bei 8.61 ppm und den beiden
übrigen Carbamat-Protonen die Peaks bei 9.34 und 10.30 ppm zuordnen.
Zusätzlich zu den bereits aufgelisteten Peaks zeigt das Spektrum von 53 bei 8.05–
8.14 ppm ein Signal für das einzelne Proton am fluorierten Benzolring.
Durch Integration konnte das Verhältnis der Protonen am PEG zu den zwölf
Protonen im Bereich von 7.16–7.69 ppm (zehn aromatische Protonen und zwei
NH) bestimmt werden. Daraus ergab sich für 53 eine Beladung von 61 % und für
54 eine Beladung von 69 %.
A PEG, c DMSO i h g Aromaten, f e d b a
B PEG, c DMSO b g Aromaten, f e d a h
Abb. 3.35: 1H-NMR-Spektren von 53 (A) und 54 (B).
PPM 9.6 9.2 8.8 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2
12
.00
742
.65
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
H
NH
O
n
O
NH
O O
53
a
a
a
ab
c
c
dd
d
e
f
f
g
h
i
i
PPM 9.6 9.2 8.8 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2
659
.68
12
.00
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
F
NH
O
n
O
NH
O O
54
a
a
a
ab
c
c
dd
d
e
f
f
g
h
h

Ergebnisse und Diskussion _________________________________________________________________
- 68 -
Die Stabilität der beiden PEG-Konjugate 53 und 54 sowie zweier
Vergleichsubstanzen (16 und 55; siehe Kapitel 3.3.2) ohne Trigger-Gruppe sollte
in Analogie zu den zuvor durchgeführten Freisetzungsstudien (vgl. Kapitel 3.1.3) in
Pufferlösungen (pH 5.0 und 7.4; siehe Kapitel 3.3.3, S. 70) und im Plasma (siehe
Kapitel 3.3.4, S. 75) untersucht werden.
3.3.2 Synthese einer pH-unempfindlichen Vergleichsubstanz
Die Strukturen der AMC-Modellverbindungen 1b, 4b–14b, 53 und 54 verfügen
über zahlreiche Carbamat-Bindungen, die unspezifisch gespalten werden könnten.
Um die Stabilität dieser Bindungen untersuchen zu können, wurde eine
Vergleichsubstanz ohne säurelabile Sollbruchstelle synthetisiert. Dazu wurde das
zu 53 und 54 analoge Methylcarbamat 55 durch Erhitzen des Azids 52 in MeOH
synthetisiert und – wie für die PEG-Konjugate in Kapitel 3.3.1 beschrieben –
aufgearbeitet (Abb. 3.36).
NH
O
O
O
N3
O
HNO
OO
O
O O
NH
O
n
52
MeOH (abs.), Δ
NH
O
O
O
NH
HNO
OO
O
O O
NH
O
n
5537 %
O
O
Abb. 3.36: Synthese der Vergleichsubstanz 55 ohne säurelabile Trigger-Gruppe.
Die Charakterisierung und Bestimmung der Beladung von 55 erfolgte mittels 1H-NMR-Spektroskopie in DMSO-d6 (Abb. 3.37, S. 69). Im Bereich von 1.21–
1.22 ppm und 1.35–1.36 ppm treten die Signale von vier CH2-Gruppen des
Hexyllinkers auf. Der charakteristische Peak bei 2.39 ppm lässt sich der
Methylgruppe des AMC zuschreiben. Die zum Teil sehr breiten Peaks bei 2.93–
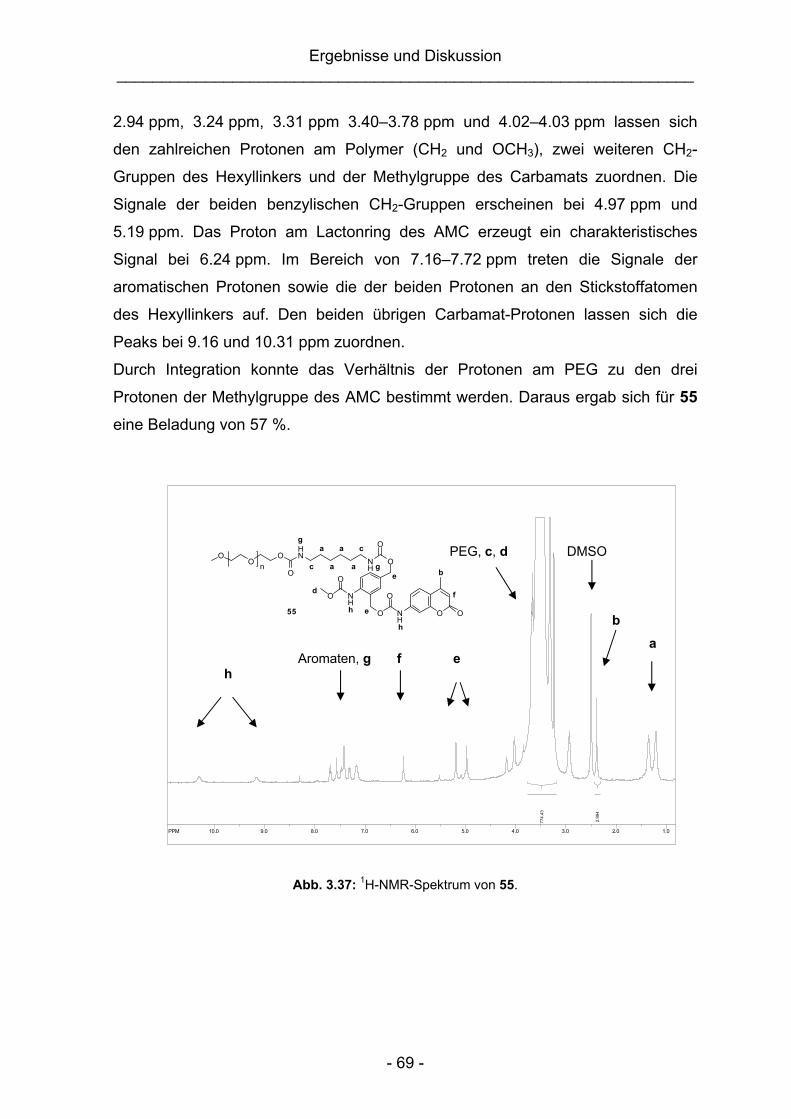
Ergebnisse und Diskussion _________________________________________________________________
- 69 -
2.94 ppm, 3.24 ppm, 3.31 ppm 3.40–3.78 ppm und 4.02–4.03 ppm lassen sich
den zahlreichen Protonen am Polymer (CH2 und OCH3), zwei weiteren CH2-
Gruppen des Hexyllinkers und der Methylgruppe des Carbamats zuordnen. Die
Signale der beiden benzylischen CH2-Gruppen erscheinen bei 4.97 ppm und
5.19 ppm. Das Proton am Lactonring des AMC erzeugt ein charakteristisches
Signal bei 6.24 ppm. Im Bereich von 7.16–7.72 ppm treten die Signale der
aromatischen Protonen sowie die der beiden Protonen an den Stickstoffatomen
des Hexyllinkers auf. Den beiden übrigen Carbamat-Protonen lassen sich die
Peaks bei 9.16 und 10.31 ppm zuordnen.
Durch Integration konnte das Verhältnis der Protonen am PEG zu den drei
Protonen der Methylgruppe des AMC bestimmt werden. Daraus ergab sich für 55
eine Beladung von 57 %.
PEG, c, d DMSO
b a Aromaten, g f e h
Abb. 3.37: 1H-NMR-Spektrum von 55.
NH
O
O
O
O
NH
OHNO
OO
On
55
O
NH
O O
d
a
a
a
ab
c
c
e
e
f
g
g
h
h
PPM 10.0 9.0 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0
2.
994
774
.43

Ergebnisse und Diskussion _________________________________________________________________
- 70 -
3.3.3 Untersuchung der Stabilität der Vergleichsubstanzen (16 und 55) sowie der Spaltungseigenschaften der PEG-Konjugate 53 und 54
Zur Bestimmung der Stabilität der Carbamat-Bindungen gegenüber unspezifischer
Spaltung wurden die beiden Vergleichsubstanzen 16 und 55 (Abb. 3.38) ohne
säurelabile Trigger-Gruppe in Analogie zu den bisher durchgeführten
Spaltungsstudien (siehe Kapitel 6.1.4) in Puffer- (pH 5.0 und 7.4) bzw.
Plasmalösungen untersucht.
NH
O NH
O
O O
O
O NH
O
O
O
O
NH
OHNO
OO
On
16 55
O
NH
O O
Abb. 3.38: Vergleichsubstanzen 16 und 55 ohne säurelabile Trigger-Gruppe.
Beide Verbindungen setzten in einem Zeitraum von drei Tagen in diesen Medien
nur äußerst geringfügige Mengen an AMC frei. Diese Ergebnisse zeigten, dass
unter den Messbedingungen keine unspezifische Spaltung der Carbamat-
Bindungen auftritt und die bei den Modellverbindungen 1b, 4b–14b beobachtete
AMC-Freisetzung allein auf der Hydrolyse der Imin-Bindung und den
anschließenden Eliminierungsreaktionen beruht. Eine unspezifische Spaltung der
beiden PEG-Konjugate 53 und 54 sollte aufgrund der Stabilität von 55 in den
folgenden Spaltungsstudien nicht auftreten.
In kinetischen Untersuchungen wurde gezeigt, dass sowohl die 1,6- als auch die
1,4-Benzyleliminierung sogar im sauren Milieu rasch verlaufen, wobei die 1,6-
Benzyleliminierung schneller als die 1,4-Eliminierung ist.130,144 Daher kann
folgender Mechanismus für die spezifische AMC-Freisetzung aus 53 und 54
angenommen werden (Abb. 3.39, S. 71). Im ersten Schritt wird die Imin-Bindung
säurekatalysiert gespalten. Dabei wird die Aminogruppe des PABC-Linkers
freigesetzt, was in spontan ablaufenden 1,6-Benzyleliminierungen der beiden
Linker-Systeme resultiert. Nach Regeneration des PABC-Linkers durch den Angriff
von Wasser auf die p-Iminochinomethan-Zwischenstufe wird AMC durch eine
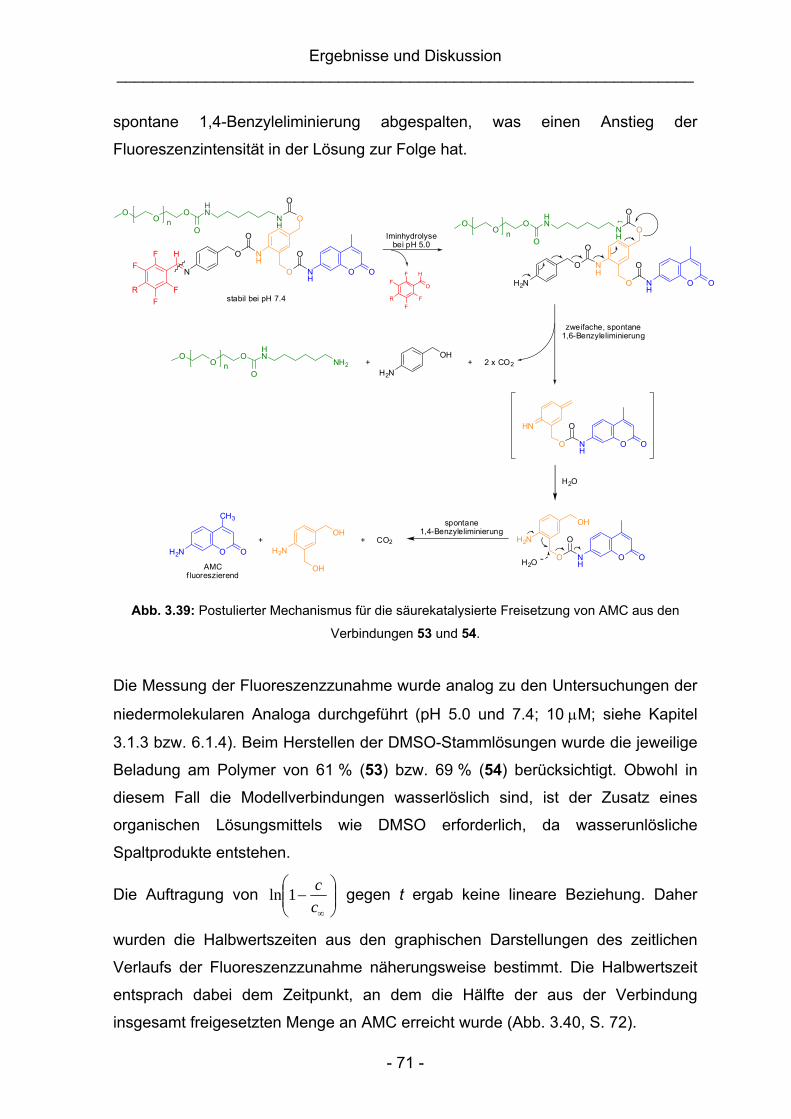
Ergebnisse und Diskussion _________________________________________________________________
- 71 -
spontane 1,4-Benzyleliminierung abgespalten, was einen Anstieg der
Fluoreszenzintensität in der Lösung zur Folge hat.
H2N
OH
H2N O O
CH3
2 x CO2++
AMCfluoreszierend
Iminhydrolysebei pH 5.0
stabil bei pH 7.4
zweifache, spontane1,6-Benzyleliminierung
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
R
NH
O
n
O
NH
O O H
O
FF
FF
R
NH
O
O
O
H2N O
HNO
OO
ONH
O
n
O
NH
O O
HN
O
O
NH
O O
HNO
OO
ONH2n
H2N
OHCO2++
OH
H2O
H2N
O
O
NH
O O
OHspontane1,4-Benzyleliminierung
H2O
Abb. 3.39: Postulierter Mechanismus für die säurekatalysierte Freisetzung von AMC aus den
Verbindungen 53 und 54.
Die Messung der Fluoreszenzzunahme wurde analog zu den Untersuchungen der
niedermolekularen Analoga durchgeführt (pH 5.0 und 7.4; 10 μM; siehe Kapitel
3.1.3 bzw. 6.1.4). Beim Herstellen der DMSO-Stammlösungen wurde die jeweilige
Beladung am Polymer von 61 % (53) bzw. 69 % (54) berücksichtigt. Obwohl in
diesem Fall die Modellverbindungen wasserlöslich sind, ist der Zusatz eines
organischen Lösungsmittels wie DMSO erforderlich, da wasserunlösliche
Spaltprodukte entstehen.
Die Auftragung von ⎟⎟⎠
⎞⎜⎜⎝
⎛−
∞cc1ln gegen t ergab keine lineare Beziehung. Daher
wurden die Halbwertszeiten aus den graphischen Darstellungen des zeitlichen
Verlaufs der Fluoreszenzzunahme näherungsweise bestimmt. Die Halbwertszeit
entsprach dabei dem Zeitpunkt, an dem die Hälfte der aus der Verbindung
insgesamt freigesetzten Menge an AMC erreicht wurde (Abb. 3.40, S. 72).

Ergebnisse und Diskussion _________________________________________________________________
- 72 -
0
0.2
0.4
0.6
0.8
1
0 50 100 150 200 250 300t /min
I x/I
AM
C
13b14b5354
0
0.2
0.4
0.6
0.8
1
0 5 10 15 20 25t /h
I x/I
AM
C
13b14b5354
A
B
Abb. 3.40: Ergebnisse der Spaltungsstudien an 53 und 54 im Vergleich zu ihren
niedermolekularen Analoga 13b bzw. 14b in Pufferlösungen bei pH 5.0 (A) und pH 7.4 (B).
Die Halbwertszeiten der Spaltung lagen in Acetat-Puffer (pH 5.0) bei t1/2 = 15 min
(53) bzw. t1/2 = 17 min (54) und in Phosphat-Puffer (pH 7.4) bei t1/2 = 65 min (53)
bzw. t1/2 = 104 min (54). Beide makromolekularen Verbindungen waren in den
Pufferlösungen demzufolge deutlich labiler als die vergleichbaren
niedermolekularen Verbindungen (siehe Tab. 3.7, S. 73). Auch der zuvor stark
ausgeprägte Effekt des Substituenten in para-Stellung der Benzaldehyd-
komponente verlor drastisch an Einfluss. Eine mögliche Erklärung für die raschere
Freisetzung liegt im Auftreten von übergeordneten Strukturen innerhalb der

Ergebnisse und Diskussion _________________________________________________________________
- 73 -
Lösung. So sind beispielsweise Block-Copolymere aus einem PEG-Block und
einem unpolaren Block dafür bekannt, in wässrigen Lösungen Micellen
auszubilden. 167,168
Tab. 3.7: Vergleich der Halbwertszeiten für die AMC-Freisetzung von 13b und 14b
(niedermolekular) bzw. 53 und 54 (makromolekular) in Pufferlösungen bei pH 5.0 und 7.4.
O
NR
H NH
O
O O NH
O
O
O
NR
H
O
NH
O O O
O
NH
HNO
OO
On
53 bzw. 5413b bzw. 14b
R = Nr. t1/2 [h] bei pH 5.0
t1/2 [h] bei pH 7.4
13b 0.57 89
F F
F F
53 0.25 1.10
14b 2.88 365 F
F F
F F
54 0.28 1.70
Micellen sind sphärische Aggregate aus amphiphilen
Substanzen, bei denen sich die unpolaren Gruppen in
wässrigen Systemen spontan zusammenlagern,
während die hydrophilen Teile nach außen ragen und
dort eine hydratisierte Hülle bilden (Abb. 3.41). Bedingt
durch die wasserfreie Umgebung und die räumliche
Nähe der hydrophoben Reste könnte die Hydrolyse der
Imin-Bindung möglicherweise aufgrund autokata-
lytischer Effekte begünstigt sein. Abb. 3.41: Aufbau einer Micelle.169
Zudem ist der pH-Wert nur in wässrigen Systemen eindeutig definiert, so dass
dieser als Maß für die Protonierung innerhalb des hydrophoben Kerns einer
Micelle nicht verwendet werden kann.

Ergebnisse und Diskussion _________________________________________________________________
- 74 -
Ferner wurde bei der Messung der Freisetzungskinetiken von 53 und 54
beobachtet, dass nur ca. 40 % (pH 5.0) bzw. 55 % (pH 7.4) des Farbstoffs
freigesetzt wurden. DE GROOT et al. lieferten in einem Patent aus dem Jahr 2004
den Hinweis,170 dass bei Verwendung von 2,4,6-Tri(hydroxymethyl)anilin als
Mehrfach-Eliminierungslinker im Fall eines als Carbamat gebundenen amino-
funktionalisierten Effektors nur eine unvollständige Freisetzung zu beobachten
war. Im Gegensatz dazu konnten mit demselben Linker-System über eine
Carbonat-Bindung gekuppelte hydroxy-funktionalisierte Effektoren vollständig
freigesetzt werden (Abb. 3.42).
O2N
O
OO NH
O
NH
O
Effektor
Ef fektor
NH
OEffektor
O2N
O
OO O
O
O
O
Effektor
Effektor
O
OEffektor
Reduktion
Reduktion
H2N
OH
OH
HO
3 CO2 3 H2N-Effektor
H2N
OH
OH
HO
3 CO2 3 HO-Effektor
Abb. 3.42: In Untersuchungen von DE GROOT et al. setzte der Mehrfach-Eliminierungslinker
2,4,6-Tri(hydroxymethyl)anilin nur hydroxy-funktionalisierte Effektoren vollständig frei.170
In späteren Arbeiten von SHABAT et al.144 und WARNECKE et al.130 wurde
2,4-Bis(hydroxymethyl)anilin als Mehrfach-Eliminierungslinker für amino-
funktionalisierte Effektoren eingesetzt. Eine Quantifizierung des freigesetzten
Effektors erfolgte in diesen Untersuchungen allerdings nicht. Bei WARNECKE et al.
lag dies vor allem an aufgetretenen Löslichkeitsproblemen.130
Im Unterschied zu den Modellverbindungen 1b und 4b–14b setzten 53 und 54 den
Farbstoff nicht direkt, sondern erst nach einer vorgelagerten 1,6-Benzyl-
eliminierung und anschließender Regeneration des PABC-Systems durch Angriff
von Wasser auf das intermediär gebildete Iminochinomethan frei. Es ist denkbar,
dass unerwünschte Nebenreaktionen auftreten, die durch Bildung stabiler
Produkte bzw. von Verbindungen mit Quencheigenschaften eine vollständige
Freisetzung verhindern. Zwar zeigten die nach dem postulierten Mechanismus
entstehenden Produkte (PABA- oder Benzaldehydderivate) in einer Untersuchung

Ergebnisse und Diskussion _________________________________________________________________
- 75 -
keine Quencheffekte, aber es ist nicht ausgeschlossen, dass weitere, bisher nicht
identifizierte Produkte mit einer solchen Eigenschaft entstanden sind. Eine
umfangreiche Literaturrecherche ergab, dass die Regenerierung von PABA durch
Addition von Wasser an das intermediär entstehende Iminochinomethan zwar
stets postuliert, aber nur unzureichend untersucht wurde.
3.3.4 Untersuchung der Plasmastabilität von 53 und 54
Bei der Untersuchung der Plasmastabilität von 53 und 54 wurde analog zu den
Messungen in den Pufferlösungen verfahren. Der zeitliche Verlauf der AMC-
Freisetzung ist in Abbildung 3.43 dargestellt.
0
0.2
0.4
0.6
0.8
1
1.2
0 100 200 300 400t /min
I x/I
AM
C
13b14b5354
Abb. 3.43: AMC-Freisetzung aus den makromolekularen Modellverbindungen 53 und 54 und ihren
niedermolekularen Analoga 13b und 14b im Plasma.
Die graphische Auswertung ergab, dass beide Verbindungen AMC mit einer
Halbwertszeit von t1/2 = 374 min freisetzten. Damit erwiesen sie sich als deutlich
stabiler als ihre niedermolekularen Analoga (t1/2 = 78 min (13b) bzw. t1/2 = 140 min
(14b)). Die Ergebnisse der Hydrolysestudien zeigten, dass es prinzipiell möglich
ist, die Freisetzung von Effektoren aus makromolekularen Verbindungen durch
säurelabile Trigger-Gruppen zu initiieren. Wie erhofft konnte die Plasmastabilität

Ergebnisse und Diskussion _________________________________________________________________
- 76 -
der Imin-Bindungen durch die Kupplung an einen polymeren Träger etwas erhöht
werden. Die durch HSA katalysierte Hydrolyse der Imin-Bindung scheint in der
makromolekularen Formulierung zurückgedrängt zu werden. Der Vergleich der
Halbwertszeiten von 53 und 54 in Plasma und in Pufferlösung bei pH 7.4 zeigte,
dass die Stabilität im Plasma um ungefähr Faktor sechs höher ist als in der
Pufferlösung. Die Ausprägung des stabilisierenden Effekts, der durch das
Einführen eines Fluoratoms in para-Stellung der Benzaldehydkomponente bewirkt
wurde, war im Plasma und in den Pufferlösungen für die PEG-geträgerten
Verbindungen gegenüber 13b und 14b stark vermindert. Da im Plasma – anders
als im Puffer – bemerkenswerterweise ein Endwert von 100 % Freisetzung
gemessen wurde, scheint der Freisetzung in Plasma ein anderer Mechanismus
zugrunde zu liegen.
3.4 Untersuchung der nachgelagerten 1,4-Benzyleliminierung anhand niedermolekularer AMC-Modellverbindungen
Bei der Untersuchung der Spaltungseigenschaften von 53 und 54 fiel auf, dass der
Farbstoff AMC in Pufferlösungen nur zu maximal 55 % freigesetzt wird. Um zu
klären, ob die unvollständige Spaltung auf die Verwendung von PEG als Träger
zurückzuführen ist oder ob diese generell bei Mehrfacheliminierungen aus
2,4-Bis(hydroxymethyl)anilin auftritt, sollten niedermolekulare AMC-Modellverbin-
dungen mit sowohl einfachem (56) als auch mit verzweigtem (57) PABC-analogen
System (Abb. 3.44, S. 77) synthetisiert und hinsichtlich ihrer Spaltungseigen-
schaften miteinander verglichen werden. Während anhand von Verbindung 56 die
Kinetik der direkten 1,4-Benzyleliminierung bestimmt werden sollte, ermöglicht 57
eine Untersuchung der Spaltungseigenschaften einer 1,4-Benzyleliminierung, die
einer 1,6-Benzyleliminierung sowie der Regeneration des PABC-Systems
nachgelagert ist.

Ergebnisse und Diskussion _________________________________________________________________
- 77 -
56
+H3N
O NH
O
O O
CF3COO-
57
+H3N
O
O
NH
O NH
O
O O
CF3COO-
Abb. 3.44: Zielstrukturen niedermolekularer AMC-Modellverbindungen zur Untersuchung der
1,4-Benzyleliminierung.
Die Synthese von 56 (Abb. 3.45) erfolgte ausgehend von o-Aminobenzylalkohol
analog zur Darstellung von 17 (siehe Kapitel 3.1.2, S. 27).
H2N
DBTLTHF (abs.)
N O OCO
TFA/DCM 1:1
35891 %
NH
Boc
5987 %
NH
Boc
OH O NH
O
O O
5668 %
+H3N
O NH
O
O O
CF3COO-
OH
(Boc)2ODCM (abs.)
Abb. 3.45: Synthese der Modellverbindung 56.
Für die Synthese der Modellverbindung 57 wurde zunächst 2-((tert.-
Butyldimethylsilyloxy)methyl)-4-(hydroxymethyl)anilin*166 durch Umsetzung mit
(Boc)2O geschützt (60) (Abb. 3.46, S. 78). Im nächsten Schritt wurde mit
Propylisocyanat zum Propylcarbamat 61 umgesetzt. Nach Spaltung des TBS-
Ethers mit einem MeOH/HCl-Gemisch (50:1) wurde durch Reaktion des
Benzylalkohols 62 mit AMC-NCO (3) der Farbstoff AMC über eine Carbamat-
Bindung eingeführt. Abspaltung der Boc-Schutzgruppe von 63 mit einem
TFA/DCM-Gemisch lieferte schließlich die Zielverbindung 57.
* Diese Verbindung wurde von A. WARNECKE zur Verfügung gestellt.

Ergebnisse und Diskussion _________________________________________________________________
- 78 -
H2N
OH
MeOH/1 M HCl50:1
DBTLTHF (abs.)
N O OCO
TFA/DCM 1:1
3
6099 %
NH
OH
Boc
DBTLTHF (abs.)
NC
O
6198 %
NH
O
Boc
O
NH
6279 %
NH
O
Boc
O
NH
6394 %
NH
O
Boc
O
NH
OTBSOTBS
OHO NH
O
O O
5764 %
+H3N
O
O
NH
O NH
O
O O
CF3COO-
OTBS
(Boc)2ODCM (abs.)
Abb. 3.46: Synthese der Modellverbindung 57.
Beide Verbindungen lagen als TFA-Salze vor und waren als Feststoffe stabil. Die
Bestimmung ihrer Kinetiken zur Freisetzung von AMC erfolgte analog zu den
bisherigen Messungen (vgl. Kapitel 6.1.4). Da die Eliminierungsreaktionen im
Allgemeinen mit hoher Geschwindigkeit ablaufen und geringfügige
pH-Änderungen keinen entscheidenden Einfluss auf die Reaktionsgeschwindigkeit
ausüben, wurden die Messungen ausschließlich im Phosphat-Puffer (pH 7.4)
durchgeführt. In Abbildung 3.47 (S. 79) ist der zeitliche Verlauf für die AMC-
Freisetzung durch 1,4-Benzyleliminierung von 56 und 57 dargestellt.
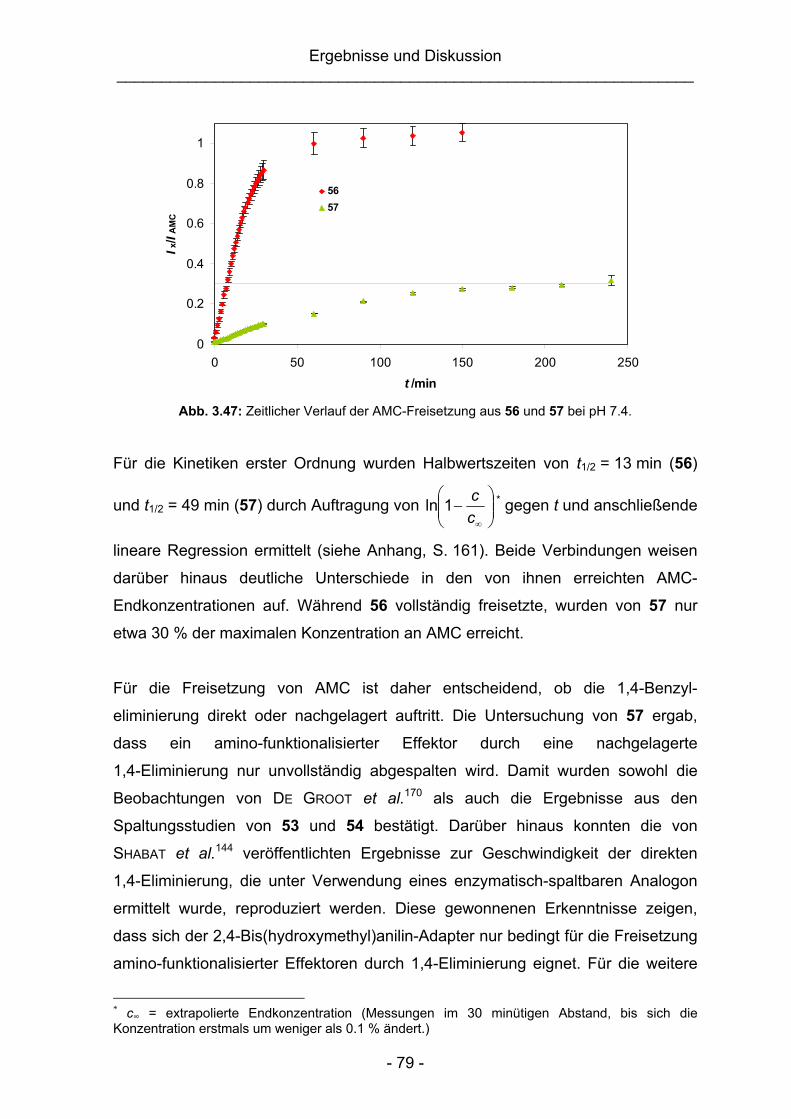
Ergebnisse und Diskussion _________________________________________________________________
- 79 -
0
0.2
0.4
0.6
0.8
1
0 50 100 150 200 250t /min
I x/I
AM
C
5657
Abb. 3.47: Zeitlicher Verlauf der AMC-Freisetzung aus 56 und 57 bei pH 7.4.
Für die Kinetiken erster Ordnung wurden Halbwertszeiten von t1/2 = 13 min (56)
und t1/2 = 49 min (57) durch Auftragung von ⎟⎟⎠
⎞⎜⎜⎝
⎛−
∞cc1ln * gegen t und anschließende
lineare Regression ermittelt (siehe Anhang, S. 161). Beide Verbindungen weisen
darüber hinaus deutliche Unterschiede in den von ihnen erreichten AMC-
Endkonzentrationen auf. Während 56 vollständig freisetzte, wurden von 57 nur
etwa 30 % der maximalen Konzentration an AMC erreicht.
Für die Freisetzung von AMC ist daher entscheidend, ob die 1,4-Benzyl-
eliminierung direkt oder nachgelagert auftritt. Die Untersuchung von 57 ergab,
dass ein amino-funktionalisierter Effektor durch eine nachgelagerte
1,4-Eliminierung nur unvollständig abgespalten wird. Damit wurden sowohl die
Beobachtungen von DE GROOT et al.170 als auch die Ergebnisse aus den
Spaltungsstudien von 53 und 54 bestätigt. Darüber hinaus konnten die von
SHABAT et al.144 veröffentlichten Ergebnisse zur Geschwindigkeit der direkten
1,4-Eliminierung, die unter Verwendung eines enzymatisch-spaltbaren Analogon
ermittelt wurde, reproduziert werden. Diese gewonnenen Erkenntnisse zeigen,
dass sich der 2,4-Bis(hydroxymethyl)anilin-Adapter nur bedingt für die Freisetzung
amino-funktionalisierter Effektoren durch 1,4-Eliminierung eignet. Für die weitere
* c∞ = extrapolierte Endkonzentration (Messungen im 30 minütigen Abstand, bis sich die Konzentration erstmals um weniger als 0.1 % ändert.)

Ergebnisse und Diskussion _________________________________________________________________
- 80 -
Entwicklung von säurelabilen makromolekularen Prodrugs ist die Verwendung
eines anderen Linkers zu empfehlen, der eine vollständige Freisetzung des
Effektors gewährleistet.

Fazit und Ausblick _________________________________________________________________
- 81 -
4. Fazit und Ausblick
Im Rahmen dieser Arbeit wurden säurelabile Trigger-Gruppen auf Basis von
Schiffschen Basen des PABC-Linkers entwickelt und gezeigt, dass deren
Hydrolyseeigenschaften durch die Verwendung verschiedener Substituenten am
aromatischen Ring des Benzaldehyds maßgeschneidert werden können. Die
dargestellten pH-empfindlichen Imine können vielseitig eingesetzt werden. So
wurde anhand eines PEG-geträgerten Modell-Prodrugs basierend auf einem
2,4-Bis(hydroxybenzyl)anilin-Adapter gezeigt, dass durch saure Hydrolyse der
Trigger-Gruppe ein Effektor freigesetzt werden kann. Weitere denkbare
Anwendungen wären ein Einsatz als pH-Sensoren oder in der Entwicklung von
pH-empfindlichen Materialien, z. B. basierend auf selbsteliminierenden Polymeren,
die säurelabil getriggert werden.26,95
Ein Nachteil des verwendeten chemischen Adapters auf Basis von
2,4-Bis(hydroxybenzyl)anilin ist, dass über die nachgelagerte 1,4-Benzylelimi-
nierung ein amino-funktionalisierter Effektor nur unvollständig freigesetzt wird.
Daher ist dieser Baustein nur bedingt für die Entwicklung makromolekularer
Prodrugs geeignet.
Zukünftige Strategien sollten folgende im Rahmen dieser Arbeit gewonnenen
Erkenntnisse berücksichtigen:
Für eine effektive und rasche Abspaltung des Effektors sollte ein
alternativer Adapter-Baustein verwendet werden, der den Farb- oder
Wirkstoff über eine 1,6-Benzyleliminierung freisetzt.
Zur Erhöhung der Wasserlöslichkeit könnte auf den Einbau eines
zusätzlichen Linkers verzichtet werden, indem der Adapter-Baustein direkt
durch Aufbau einer Imin-Bindung getriggert wird.
Um die Spaltungseigenschaften besser vorhersagen zu können, sollten
Aggregatbildungen möglichst verhindert werden. Dies könnte z. B. durch
Einsatz wasserlöslicher Linker geschehen,115 welche die Amphiphilie des
Prodrugs und damit die Ausbildung supramolekularer Strukturen in
wässrigen Systemen herabsetzen können.

Zusammenfassung _________________________________________________________________
- 82 -
5. Zusammenfassung
Im Zuge einer klassischen Chemotherapie zur Behandlung von Krebser-
krankungen treten oftmals gravierende systemische Nebenwirkungen auf, die sich
dosislimitierend auswirken. Das grundlegende Problem hierfür liegt in der
mangelnden Tumorselektivität der klinisch eingesetzten Zytostatika. Das Konzept
des Drug Targeting, eines gezielten Wirkstofftransports, bei dem die Wirkstoffe mit
Trageting-Einheiten wie beispielsweise Antikörper (aktives Targeting) oder
Polymere mit einer Molmasse >30 kDa (passives Targeting) verknüpft werden,
ermöglicht eine Anreicherung von Wirkstoffen in Tumorgewebe. Die pH-abhängige
Freisetzung des Wirkstoffs nach zellulärer Aufnahme ist eine der universellsten
und effektivsten Methoden zur Aktivierung solch makromolekularer Prodrugs. Bei
dieser Strategie wird der nach endosomaler Aufnahme auftretende pH-Sprung von
mehr als zwei Einheiten für die Spaltung von säurelabilen Sollbruchstellen
ausgenutzt. Da dieser Ansatz bislang nahezu ausschließlich für Wirkstoffe mit
einer reaktiven Carbonylgruppe verwirklicht wurde, sollte im Rahmen dieser Arbeit
eine rationale und modulare Strategie zur säurelabilen Aktivierung
makromolekularer Prodrugs von hydroxy- und amino-funktionalisierten Wirkstoffen
entwickelt werden. Dazu wurde das bereits gut untersuchte und effektiv spaltende
PABC-System als selbsteliminierender Linker für die Entwicklung der Prodrugs
ausgewählt. Anhand von strukturverwandten, niedermolekularen AMC-
Modellverbindungen (1b, 4b–14b) konnte gezeigt werden, dass die
Hydrolyseeigenschaften des Triggers durch Einführung verschiedener
Substituenten am aromatischen Ring des Benzaldehyds maßgeschneidert werden
können. Während im neutralen Milieu die 1,6-Benzyleliminierung des Linkers
blockiert wurde, erfolgte im schwach sauren pH-Bereich durch rasche Hydrolyse
der Imin-Bindung und anschließender Eliminierung eine Freisetzung des
Farbstoffs.

Zusammenfassung _________________________________________________________________
- 83 -
O
NR
H NH
O
O O
Me2N
4b MeO
5b H
1b F
6b Cl
7b O
8b
F3C
9b
O2N
10b
ClCl
Cl
11b
FF
12b
F
FF
F
13b
F
FF
F
F
14b
Für die para-substituierten Benzaldehydderivate 1b und 4b–10b wurden durch
Auftragung von 0logkk gegen σp die Substituenteneffekte unter Berücksichtigung
von Linearen Freien Enthalpie-Beziehungen näher untersucht. Für pH 5.0 wurde
die Hammett-Gleichung in guter Näherung erfüllt und ein moderater Einfluss der
Substituenten festgestellt. Bei pH 7.4 war der Substituenteneinfluss zwar
ausgeprägter, eine deutliche LFER-Beziehung jedoch nicht erkennbar.
Erwartungsgemäß ließ sich die Stabilität durch das Einfügen zusätzlicher
elektronenziehender Substituenten erhöhen. Besonders gute Eigenschaften bei
pH 5.0 und 7.4 wiesen die auf Tetrafluor- (13b) bzw. Pentafluorbenzaldehyd (14b)
basierenden Imine auf, deren Freisetzungskinetiken zusätzlich zwischen pH 4.5
und 7.4 genauer untersucht wurden. Dabei ergab sich, dass für beide
Verbindungen im physiologisch relevanten pH-Bereich ein linearer
Zusammenhang zwischen pH und log kobs bestand. Diese lineare Beziehung ist für
eine Reaktion mit spezifischer Säurekatalyse zu erwarten.
Im Plasma wurden die Imin-Bindungen der Modellverbindungen 1b, 10b, 13b und
14b überraschend schnell gespalten (t1/2 < 140 min). In weiteren Untersuchungen
konnte gezeigt werden, dass diese Instabilität hauptsächlich durch eine
katalytische Aktivität von HSA
hervorgerufen wird.
Im Folgenden sollten auf Basis der
meistversprechenden Trigger-Gruppen
makromolekulare Modellverbindungen und
NH
HN
O
O
O
O
O
X
Träger
N
H
R
Effektor
X = O, NH

Zusammenfassung _________________________________________________________________
- 84 -
Prodrugs synthetisiert werden. Dazu wurden zunächst mit einem chemischen
Adapter-Baustein auf Basis von 3-Amido-4-hydroxymethylanilin ausgehend von
käuflichem 6-Nitrophthalid niedermolekulare AMC-Modellverbindungen (28, 29) in
einer elfstufigen Synthese in Anlehnung an die von BAKHEET ELSADEK162
entwickelte Route dargestellt. Außerdem konnten über eine alternative achtstufige
Syntheseroute analoge Prodrugs der Zytostatika Doxorubicin und Paclitaxel (38–
41) erhalten werden.
Im letzten Syntheseschritt sollten die niedermolekularen Vorstufen schließlich über
eine Alkin-Ankergruppe mittels „Click-Chemie“ (CuAAC) an PEG gebunden
werden, was an der Instabilität der gebildeten Produkte scheiterte.
NH
HN
O
O
O
O
OTrigger
Effektor
NH
N3
O
O
O
O
O
Effektor
NH
HN
ONH
O
O
O
O
Effektor
ON
O
O
OH
N
HFF
R'F
F R' = H, FΔ
NH
HN
O
O
O
O
O
Effektor
Trigger NNN
PEG
2instabil
H2NO
NH2
NH
NHO
O O
Effektor
ONH
HN
O
O
O
O
O
Effektor
n
n
O
O
instabil
NH
HN
ONH
O
O
OTrigger
O
Effektor
ON
O
O
N3O
N3n
Da es ebenso nicht möglich war, stabile Produkte durch Umsetzung einer
niedermolekularen Vorstufe mit PEG-Diamin sowie durch Reaktion des Triggers
mit einer HSA-bindenden Vorstufe zu erhalten, scheint die Struktur des Adapter-
Bausteins ausschlaggebend für die Unbeständigkeit der Verbindungen zu sein.
Als alternativen Adapter-Baustein wurde ein 2,4-Bis(hydroxymethyl)anilin-Linker
verwendet, an den das Polymer über
eine benzylische Carbamat-Bindung
gekuppelt werden kann. Unter
Verwendung eines bereits vorhanden-
den Bausteins166 gelang es in einer
jeweils vierstufigen Synthese, die
säurelabilen PEG-Konjugate (53, 54) darzustellen. Mittels NMR-Spektroskopie
NH
O
O
O
N
HFF
FF
O
NH
O O OR
O
NH
HNO
OO
On
R = H: 53R = F: 54

Zusammenfassung _________________________________________________________________
- 85 -
wurden die Zielverbindungen eindeutig charakterisiert und deren Beladung
ermittelt. Die sich anschließenden Spaltungs- und Stabilitätsstudien zeigten
allerdings, dass die makromolekularen Verbindungen in Pufferlösungen AMC
deutlich schneller freisetzen als ihre niedermolekularen Analoga. Im Gegensatz
dazu konnte die Plasmastabilität durch die makromolekulare Formulierung erhöht
werden. Überraschenderweise verlief die Freisetzung des Farbstoffs aus 53 und
54 in den Pufferlösungen unvollständig. Um zu klären, ob die unvollständige
Spaltung auf die PEG-Trägerung zurückzuführen ist oder ob diese generell bei
Mehrfacheliminierungen aus 2,4-Bis(hydroxymethyl)anilin auftritt, wurden
niedermolekulare AMC-Modellverbindungen mit sowohl einfachem (56) als auch
mit verzweigtem (57) PABC-analogen System synthetisiert und hinsichtlich ihrer
Spaltungseigenschaften miteinander verglichen. Während anhand von Verbindung
56 die Kinetik der einfachen 1,4-Benzyleliminierung bestimmt wurde, ermöglichte
57 eine Untersuchung der Spaltungseigenschaften einer 1,4-Benzyleliminierung,
die einer 1,6-Benzyleliminierung sowie der Regeneration des PABC-Systems
nachgelagert ist.
56
+H3N
O NH
O
O O
CF3COO-
57
+H3N
O
O
NH
O NH
O
O O
CF3COO-
Spaltungsstudien ergaben, dass der verzweigte PABC-analoge Linker 57 in
Pufferlösung (pH 7.4) AMC ebenfalls nur unvollständig freisetzt, wogegen 56 den
Farbstoff vollständig eliminiert. Adapter-Systeme auf Basis von
2,4-Bis(hydroxymethyl)anilin sind daher für eine Prodrug-Entwicklung nur bedingt
geeignet.
Im Rahmen dieser Arbeit wurde ein neuartiges Konzept vorgestellt, das Imine des
bekannten selbsteliminierenden PABC-Linkers als säurelabile Trigger-Gruppen
nutzt, die in der Lage sind, Kaskadenreaktionen zu initiieren und sich daher
modular und vielseitig verwenden lassen. Durch Strukturvariationen der Aldehyd-
Komponente ließen sich die säurelabilen Eigenschaften gezielt beeinflussen. Die

Zusammenfassung _________________________________________________________________
- 86 -
Versuche, makromolekulare Prodrugs auf Basis der entwickelten Trigger-Gruppen
aufzubauen, waren hingegen durch vielfältige Probleme geprägt, die aus der
Verwendung eines zentralen Adapter-Bausteins als Bindeglied von Trigger,
makromolekularem Träger und Wirkstoff resultierten. Aus den Untersuchungen
zweier verschiedener Adapter-Systeme konnten schließlich wertvolle Erkenntnisse
gewonnen werden, die zum Gelingen zukünftiger Prodrug-Strategien beitragen
können.

Experimenteller Teil _________________________________________________________________
- 87 -
6. Experimenteller Teil
6.1 Materialien und Methoden
6.1.1 Verwendete Reagenzien
Die für die Synthesen verwendeten Reagenzien und Lösungsmittel wurden von
den Firmen Merck (Darmstadt), Sigma-Aldrich und Fluka (Seelze) sowie Roth
(Karlsruhe) in handelsüblicher Qualität bezogen. AMC wurde von Echinachem
(China) und Doxorubicin-Hydrochlorid sowie Paclitaxel von Yick-Vic (China)
geliefert. 2,3,5,6-Tetrafluorbenzaldehyd und Chlortrimethylsilan wurden vor ihrer
Verwendung frisch destilliert; alle weiteren Reagenzien wurden ohne zusätzliche
Aufreinigung eingesetzt. Für die Herstellung der verschiedenen Lösungen und
Puffer wurde destilliertes Wasser verwendet. Als Trockenmittel für die organischen
Phasen diente wasserfreies Natriumsulfat von Roth (Karlsruhe).
Das Anfärben von DC-Platten erfolgte durch Eintauchen in eine
Anisaldehydlösung (2.14 mL Anisaldehyd, 100 mL EtOH, 1 mL konz.
Schwefelsäure) und anschließendem Entwickeln im heißen Luftstrom einer
Heißluftpistole.
6.1.2 Verwendete Geräte und Materialien
Schmelzpunktbestimmung Die Schmelzpunkte wurden mit dem Digital Melting Point Apparatus (Barnstead
international/Electrothermal, IA9000 series) bestimmt und sind unkorrigiert.
pH-Messungen Das Einstellen des pH-Wertes von Pufferlösungen erfolgte unter Verwendung
eines Microprocessor pH/ION Meter pMX 3000 von den Wissenschaftlich-
Technischen Werkstätten.

Experimenteller Teil _________________________________________________________________
- 88 -
Elementaranalysen Die Elementaranalysen wurden mit einem VarioEL der Firma Elementaranalysen-
systeme GmbH am Institut für Organische Chemie und Biochemie der Universität
Freiburg aufgenommen. Bei fluorierten und nichtkristallinen Verbindungen wurde
auf die Durchführung einer Elementaranalyse verzichtet und die Reinheit mittels
NMR-Spektroskopie ermittelt.
Massenspektrometrie (MS) Die EI- und CI-Massenspektren wurden mit einem TSQ700 MS instrument
(Thermoelectron) und die ESI-Massenspektren mit einem LCQ-Advantage MS
instrument (Thermoelectron) am Institut für Organische Chemie und Biochemie
der Universität Freiburg aufgenommen.
Am Institut für Organische Chemie und Biochemie der Freien Universität Berlin
diente ein Agilent 6210 ESI-TOF (Agilent Technologies) zur Aufnahme der HRMS-
Massenspektren.
Kernresonanzspektrometrie (NMR) Für die Aufnahme der 1H- und 13C-NMR-Spektren wurden ein 400 MHz Avance
DRX Spectrometer (Bruker) bzw. ein 300 MHz Varian Unity 300 Spectrometer
verwendet. Die Messungen erfolgten bei Raumtemperatur in den deuterierten
Lösungsmitteln DMSO-d6, THF-d8 und D2O. Die chemischen Verschiebungen (δ)
der Signale bzw. derer Schwerpunkte sind in ppm zum internen Standard
Tetramethylsilan (TMS, δ = 0 ppm) und die Kopplungskonstanten (J) in Hz
angegeben. Für die Multiziplitäten werden folgende Abkürzungen verwendet:
(breites) Singulett ((br) s), Dublett (d), Triplett (t), Quartett (q), Multiplett (m). Die 13C-NMR-Spektren wurden protonenbreitbandentkoppelt aufgenommen.
Durchgeführt wurden die Messungen am Institut für Pharmazie der Universität
Freiburg.
Fluoreszenzmessungen Zur Messung von Fluoreszenzintensitäten wurde ein POLARstar OPTIMA der
BMG LABTECH GmbH mit der Software FLUOstar OPTIMA verwendet. Die 96-
Well-Platten Fluotrac 200 (schwarz) wurden von Greiner Microlon geliefert. Die

Experimenteller Teil _________________________________________________________________
- 89 -
Messungen wurden im Arbeitskreis von Prof. Dr. M. Jung am Institut für
Pharmazeutische Wissenschaften der Universität Freiburg durchgeführt.
6.1.3 Chromatographie
Dünnschichtchromatographie (DC) Die Reaktionskontrollen erfolgten mittels Dünnschichtchromatographie auf
Fertigplatten von Merck (Darmstadt) mit Kieselgel 60 F254 auf Aluminiumfolie bzw.
auf HPTLC-Platten DIOL F254s. Für die Detektion der einzelnen Substanzen auf
den Platten wurden eine UV-Lampe (254 nm, 366 nm) von Benda (Wiesloch) bzw.
Anisaldehyd-Entwicklerlösung verwendet.
Säulenchromatographie Zur säulenchromatographischen Trennung wurde, wenn in der entsprechenden
Vorschrift nicht anders vermerkt, als Säulenmaterial Kieselgel 60 (Korngröße:
0.040–0.063 mm) der Firma Roth (Karlsruhe) eingesetzt. Die Chromatographie
erfolgte unter Verwendung von Druckluft. Die als Eluens dienenden
Lösungsmittelgemische sind den Synthesevorschriften der einzelnen
Verbindungen zu entnehmen.
6.1.4 Fluorometrische Bestimmung der Halbwertszeiten der AMC-Modellverbindungen in Puffer-, Plasma- und Albuminlösungen Messbedingungen:
200 μL Lösung pro Well
Wellenlängen: λex = 390 nm, λem = 460 nm
Messtemperatur: 37 °C
Medien: Acetat-Puffer (40 mM, pH 4.0, 4.5, 5.0, 5.5):
1.64 g CH3COONa, 1.14 mL konz. CH3COOH, 1 L H2O

Experimenteller Teil _________________________________________________________________
- 90 -
Phosphat-Puffer (40 mM, pH 6.0, 6.5, 7.0, 7.4):
3.12 g NaH2PO4 · 2 H2O, 3.56 g Na2HPO4 · 2 H2O, 1 L H2O
Acetat-Puffer (20 mM, pH 5.0):
820 mg CH3COONa, 572 μL konz. CH3COOH, 1 L H2O
Phosphat-Puffer (20 mM, pH 7.4):
1.56 g NaH2PO4 · 2 H2O, 1.78 g Na2HPO4 · 2 H2O, 1 L H2O
Die Einstellung der pH-Werte erfolgte durch Zugabe von 1 M HCl bzw. 1 M
Natronlauge.
Plasma:
Das durch Blutentnahme und anschließender Zentrifugation gewonnene Plasma
wurde in der Regel ohne weitere Vorbehandlung eingesetzt. Lediglich die
Verbindungen 1b, 10b, 13b und 14b wurden zusätzlich in Plasmalösungen
untersucht, denen zuvor durch Ultrazentrifugalfiltration alle Bestandteile > 10 kDa
entfernt wurden (s. Kap.3.1.4). Zur Bereitung dieser Lösungen wurden Centriprep®
Zentrifugen-Filtereinheiten (Ultragel YM-10) der Millipore corporation (USA)
verwendet.
Albumin:
Die verwendete Albuminlösung (HSA, ~750 μM) enthielt 5 % Octalbin und wurde
von OCTAPHARMA GmbH (Langenfeld) bezogen.
Probenbereitung:
Endkonzentration: 10 μM in Puffer-, Plasma- oder Albumin-Lösungen mit 5 %
DMSO
Stammlösungen: 1 mM in DMSO; vor den Messungen frisch hergestellt
Verdünnung auf 200 μM: 200 μL der 1 mM Stammlösung mit 800 μL DMSO
versetzen

Experimenteller Teil _________________________________________________________________
- 91 -
Auftragung auf die Platte: in jedes Well 10 μL der 200 μM Lösung einfüllen und
direkt vor der Messung 190 μL Puffer zugeben (jede Verbindung wurde sechsmal
gemessen)
Vergleichslösungen: Auf jeder Platte wurden folgende Vergleichslösungen vierfach mitgemessen:
10 μM AMC-Lösung (Puffer-, Plasma- oder Albumin-Lösungen mit 5 % DMSO)
15 μM AMC-Lösung (Puffer-, Plasma- oder Albumin-Lösungen mit 5 % DMSO)
Stammlösung: 1 mM in DMSO
Verdünnungen:
100 μM: 100 μL der 1 mM Stammlösung mit 900 μL des für die Messung
relevanten Mediums;
10 μM: 100 μL der 100 μM Lösung mit 40 μL DMSO und 860 μL Medium;
15 μM: 150 μL der 100 μM Lösung mit 35 μL DMSO und 815 μL Medium;
Auftragung auf die Platte: 200 μL pro Well.
6.2 Chemische Synthesen
Alle Synthesen in organischen Medien wurden in wasserfreien Lösungsmitteln
unter einer trockenen Stickstoffatmosphäre durchgeführt. Die anschließende
Aufarbeitung erfolgte ohne Schutzatmosphäre. Die Lösungsmittel wurden
üblicherweise in einem Wasserbad bei 40 °C am Rotationsverdampfer entfernt.
6.2.1 Synthese der niedermolekularen AMC-Modellverbindungen 1b und 4b–14b
Allgemeine Synthesevorschrift zur Darstellung der 4-(Benzylidenamino)benzylalkohole 1a und 4a–14a Eine Suspension von 200 mg (1.62 mmol) 4-Aminobenzylalkohol in 20 mL Benzol
wird unter Rühren mit einem Benzaldehydderivat (4.86 mmol) versetzt. Zum
Reaktionsgemisch werden 5 μL konz. Essigsäure als Katalysator zugegeben. Der

Experimenteller Teil _________________________________________________________________
- 92 -
Ansatz wird 3 h am Wasserabscheider erhitzt. Nach Entfernen des Lösungsmittels
unter vermindertem Druck wird das Produkt bei – 20 °C aus einem Gemisch aus
Ethylacetat und n-Hexan kristallisiert. Die Charakterisierung der Produkte wird für
jede Verbindung im Folgenden beschrieben:
4-(Benzylidenamino)benzylalkohol (1a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 1a in einer
Ausbeute von 252 mg (77 % d. Theorie) erhalten.
OHN
H
Smp.: 62–63 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.51 (d, J = 5.6 Hz, 2H, CH2), 5.20 (t,
J = 5.6 Hz, 1H, OH), 7.23–7.25 (m, 2H, Ar-H), 7.35–7.37 (m, 2H, Ar-H), 7.52–7.53
(m, 3H, Ar-H), 7.93–7.94 (m, 2H, Ar-H), 8.63 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.5 (CH2), 120.7, 127.2, 128.5, 128.7,
131.3, 136.0, 140.4, 149.8, 160.0 (Ar-C, C(N)H).
APCI-MS (5 μA, MeOH): m/z (%) = 212 (100) [M + H]+.
Elementaranalyse: C14H13NO [211.26]
berechnet: C: 79.59 % H: 6.20 % N: 6.63 %
gefunden: C: 79.35 % H: 6.29 % N: 6.55 %
4-(4-Dimethylaminobenzylidenamino)benzylalkohol (4a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 4a in einer
Ausbeute von 319 mg (77 % d. Theorie) erhalten.
OHN
HN

Experimenteller Teil _________________________________________________________________
- 93 -
Smp.: 144–146 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.00 (s, 6H, CH3), 4.49 (d, J = 5.6 Hz, 2H,
CH2), 5.16 (t, J = 5.6 Hz, 1H, OH), 6.76–6.79 (m, 2H, Ar-H), 7.14–7.16 (m, 2H, Ar-
H), 7.30–7.32 (m, 2H, Ar-H), 7.72–7.75 (m, 2H, Ar-H), 8.40 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.6 (CH2), 111.4, 120.5, 123.8, 127.3,
130.1, 139.2, 150.8, 152.2, 159.4 (Ar-C, C(N)H).
Das 13C-Signal der Methylgruppen am Stickstoffatom (N(CH3)2) wird vom
Lösungsmittelsignal (DMSO-d6: δ = 39.4 ppm) überlagert.
ESI-MS (5 kV, MeCN): m/z (%) = 255 (100) [M + H]+.
Elementaranalyse: C16H18N2O [254.33]
berechnet: C: 75.56 % H: 7.13 % N: 11.01 %
gefunden: C: 75.29 % H: 7.46 % N: 10.96 %
4-(4-Methoxybenzylidenamino)benzylalkohol (5a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 5a in einer
Ausbeute von 318 mg (81 % d. Theorie) erhalten.
OHN
HO
Smp.: 114–115 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.83 (s, 3H, CH3), 4.50 (d, J = 5.7 Hz, 2H,
CH2), 5.18 (t, J = 5.7 Hz, 1H, OH), 7.05–7.08 (m, 2H, Ar-H), 7.18–7.21 (m, 2H, Ar-
H), 7.32–7.35 (m, 2H, Ar-H), 7.86–7.90 (m, 2H, Ar-H), 8.54 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 55.3 (CH3), 62.5 (CH2), 114.2, 120.6, 127.3,
129.0, 130.3, 139.9, 150.2, 159.2, 161.7 (Ar-C, C(N)H).

Experimenteller Teil _________________________________________________________________
- 94 -
APCI-MS (5 μA, MeOH): m/z (%) = 242 (100) [M + H]+.
Elementaranalyse: C15H15NO2 [241.29]
berechnet: C: 74.67 % H: 6.27 % N: 5.80 %
gefunden: C: 74.43 % H: 6.33 % N: 5.68 %
4-(4-Fluorbenzylidenamino)benzylalkohol (6a) Nach dem Trocknen des hellbeigen Feststoffs im Hochvakuum wird 6a in einer
Ausbeute von 270 mg (73 % d. Theorie) erhalten.
OHN
HF
C14H12FNO
Smp.: 88–89 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.51 (d, J = 5.7 Hz, 2H, CH2), 5.20 (t,
J = 5.7 Hz, 1H, OH), 7.23–7.25 (m, 2H, Ar-H), 7.33–7.38 (m, 4H, Ar-H), 7.98–8.02
(m, 2H, Ar-H), 8.63 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.5 (CH2), 115.8 (d, J = 21.8 Hz, FCCH),
120.7, 127.2 (Ar-C), 130.8 (d, J = 8.8 Hz, FCCHCH), 132.7 (d, J = 2.9 Hz,
FCCHCHC), 140.4, 149.7, 158.7 (Ar-C, C(N)H), 163.8 (d, J = 249.2 Hz, FC).
ESI-MS (5 kV, MeCN): m/z (%) = 230 (100) [M + H]+.
4-(4-Chlorbenzylidenamino)benzylalkohol (7a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 7a in einer
Ausbeute von 281 mg (71 % d. Theorie) erhalten.
OHN
HCl

Experimenteller Teil _________________________________________________________________
- 95 -
Smp.: 142–143 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.51 (d, J = 5.7 Hz, 2H, CH2), 5.23 (t,
J = 5.7 Hz, 1H, OH), 7.24–7.26 (m, 2H, Ar-H), 7.35–7.37 (m, 2H, Ar-H), 7.57–7.60
(m, 2H, Ar-H), 7.94–7.96 (m, 2H, Ar-H), 8.65 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.5 (CH2), 120.8, 127.3, 128.9, 130.1,
134.9, 135.9, 140.7, 149.5, 158.8 (Ar-C, C(N)H).
ESI-MS (5 kV, MeCN): m/z (%) = 246 (100) [M + H]+.*
Elementaranalyse: C14H12ClNO [245.70]
berechnet: C: 68.44 % H: 4.92 % N: 5.70 %
gefunden: C: 68.19 % H: 4.94 % N: 5.74 %
4-(4-Acetylbenzylidenamino)benzylalkohol (8a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 8a in einer
Ausbeute von 138 mg (34 % d. Theorie) erhalten.
OHN
H
O
Smp.: 142–144 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.63 (s, 3H, CH3), 4.53 (d, J = 5.6 Hz, 2H,
CH2), 5.19 (t, J = 5.6 Hz, 1H, OH), 7.29–7.39 (m, 4H, Ar-H), 8.06–8.08 (m, 4H, Ar-
H), 8.74 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 27.3 (CH3), 63.0 (CH2), 121.4, 127.8, 129.0,
129.1, 138.9, 140.3, 141.5, 149.9, 159.5 (Ar-C, C(N)H), 198.1 (C(O)CH3).
ESI-MS (5 kV, MeOH): m/z (%) = 254 (100) [M + H]+. * Neben dem angegebenen Basispeak wiesen die Spektren der chlorhaltigen Verbindungen 7a, 7b, 11a und 11b charakteristische Isotopensignale auf.

Experimenteller Teil _________________________________________________________________
- 96 -
Elementaranalyse: C16H15NO2 [253.30]
berechnet: C: 75.87 % H: 5.97 % N: 5.53 %
gefunden: C: 74.28 % H: 5.86 % N: 5.23 %
4-(4-Trifluormethylbenzylidenamino)benzylalkohol (9a) Nach dem Trocknen des hellgelben Feststoffs im Hochvakuum wird 9a in einer
Ausbeute von 405 mg (90 % d. Theorie) erhalten.
OHN
HF3C
C15H12F3NO
Smp.: 124–126 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.53 (d, J = 5.5 Hz, 2H, CH2), 5.23 (t,
J = 5.5 Hz, 1H, OH), 7.29–7.32 (m, 2H, Ar-H), 7.37–7.40 (m, 2H, Ar-H), 7.87–7.89
(m, 2H, Ar-H), 8.13–8.15 (m, 2H, Ar-H), 8.77 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.5 (CH2), 120.9 (Ar-C), 124.0 (q,
J = 272.3 Hz, CF3), 125.6 (q, J = 3.7 Hz, CHCCF3), 127.3, 129.1 (Ar-C), 130.8 (q,
J = 31.7 Hz, CCF3), 139.6, 141.1, 149.2, 158.7 (Ar-C, C(N)H).
ESI-MS (5 kV, MeOH): m/z (%) = 280 (100) [M + H]+.
4-(4-Nitrobenzylidenamino)benzylalkohol (10a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 10a in einer
Ausbeute von 280 mg (67 % d. Theorie) erhalten.
OHN
HO2N
Smp.: 154–155 °C

Experimenteller Teil _________________________________________________________________
- 97 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.53 (d, J = 5.7 Hz, 2H, CH2), 5.25 (t,
J = 5.7 Hz, 1H, OH), 7.32–7.40 (m, 4H, Ar-H), 8.17–8.19 (m, 2H, Ar-H), 8.35–8.37
(m, 2H, Ar-H), 8.82 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.5 (CH2), 121.1, 124.0, 127.3, 129.5,
141.5, 141.6, 148.7, 149.0, 158.1 (Ar-C, C(N)H).
ESI-MS (5 kV, MeCN): m/z (%) = 257 (100) [M + H]+.
Elementaranalyse: C14H12N2O3 [256.26]
berechnet: C: 65.62 % H: 4.72 % N: 10.93 %
gefunden: C: 65.25 % H: 4.77 % N: 10.89 %
4-(2,3,6-Trichlorbenzylidenamino)benzylalkohol (11a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 11a in einer
Ausbeute von 182 mg (36 % d. Theorie) erhalten.
OHN
HClCl
Cl
Smp.: 84–87 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.53 (d, J = 5.7 Hz, 2H, CH2), 5.24 (t,
J = 5.7 Hz, 1H, OH), 7.25–7.27 (m, 2H, Ar-H), 7.39–7.41 (m, 2H, Ar-H), 7.63–7.65
(m, 1H, Ar-H), 7.77–7.80 (m, 1H, Ar-H), 8.71 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.4 (CH2), 120.5, 127.3, 129.8, 131.4,
131.7, 131.9, 132.2, 134.5, 141.5, 148.9, 156.0 (Ar-C, C(N)H).
CI-MS (130 eV, 300 μA, NH3): m/z (%) = 314 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 98 -
Elementaranalyse: C14H10Cl3NO [314.59]
berechnet: C: 53.45 % H: 3.20 % N: 4.45 %
gefunden: C: 53.35 % H: 3.22 % N: 4.43 %
4-(3,4-Difluorbenzylidenamino)benzylalkohol (12a) Nach dem Trocknen des gelben Feststoffs im Hochvakuum wird 12a in einer
Ausbeute von 264 mg (66 % d. Theorie) erhalten.
OHN
HF
F
C14H11F2NO
Smp.: 86–87 °C
1H-NMR (299.9 MHz, DMSO-d6): δ = 4.51 (d, J = 5.5 Hz, 2H, CH2), 5.21 (t,
J = 5.5 Hz, 1H, OH), 7.24–7.26 (m, 2H, Ar-H), 7.35–7.38 (m, 2H, Ar-H), 7.58–7.64
(m, 1H, Ar-H), 7.77–7.80 (m, 1H, Ar-H), 7.92–7.99 (m, 1H, Ar-H), 8.63 (s, 1H,
C(N)H).
13C-NMR (75.4 MHz, DMSO-d6): δ = 62.5 (CH2), 116.4 (d, J = 17.7 Hz, FCCH),
118.0 (d, J = 16.9 Hz, FCCH), 120.8 (Ar-C), 126.0 (m, Ar-C), 127.3 (Ar-C), 133.8
(m, Ar-C), 140.8 (Ar-C), 148.9 (dd, J = 111.5 Hz, J = 13.1 Hz, FC), 149.2 (Ar-C),
152.1 (dd, J = 115.7 Hz, J = 13.1, FC), 157.8 (m, C(N)H).
APCI-MS (5 μA, MeOH): m/z (%) = 248 (100) [M + H]+.
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzylalkohol (13a) Nach dem Trocknen des hellbeigen Feststoffs im Hochvakuum wird 13a in einer
Ausbeute von 345 mg (75 % d. Theorie) erhalten.
OHN
HF F
F F

Experimenteller Teil _________________________________________________________________
- 99 -
C14H9F4NO
Smp.: 164–165 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.53 (d, J = 5.7 Hz, 2H, CH2), 5.25 (t,
J = 5.7 Hz, 1H, OH), 7.28–7.31 (m, 2H, Ar-H), 7.38–7.41 (m, 2H, Ar-H), 8.06–8.11
(m, 1H, Ar-H), 8.69 (s, 1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.4 (CH2), 108.8 (dd, J1 = J2 = 19.6 Hz, Ar-
C), 115.8 (dd, J1 = J2 = 11.1 Hz, Ar-C), 120.8, 127.4, 141.9 (Ar-C), 144.0 (m, CF),
146.4 (m, CF), 149.3, 149.4 (Ar-C, C(N)H).
ESI-MS (5 kV, MeCN): m/z (%) = 284 (100) [M + H]+.
4-(Pentafluorbenzylidenamino)benzylalkohol (14a) Nach dem Trocknen des farblosen Feststoffs im Hochvakuum wird 14a in einer
Ausbeute von 408 mg (84 % d. Theorie) erhalten.
OHN
HF F
F F
F
C14H8F5NO
Smp.: 135–136 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.53 (d, J = 5.7 Hz, 2H, CH2), 5.25 (t,
J = 5.7 Hz, 1H, OH), 7.28–7.30 (m, 2H, Ar-H), 7.38–7.40 (m, 2H, Ar-H), 8.65 (s,
1H, C(N)H).
13C-NMR (100.6 MHz, DMSO-d6): δ = 62.4 (CH2), 111.1 (ddd, J1 = 11.3 Hz,
J2 = 7.1 Hz, J3 = 3.9 Hz, Ar-C), 120.8, 127.3 (Ar-C), 137.3 (m, CF), 141.7 (m, CF),
141.8 (Ar-C), 145.5 (m, CF), 148.5, 149.2 (Ar-C, C(N)H).
ESI-MS (5 kV, MeCN): m/z (%) = 302 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 100 -
Phenyl-N-4-methylcumarin-7-yl-carbamat (2) Zu einer Suspension von 1.50 g (8.56 mmol) AMC in 18 mL DCM werden 3.22 mL
(18.8 mmol) DIEA und 2.40 mL (18.8 mmol) Chlortrimethylsilan gegeben. Das
Reaktionsgemisch wird für 3 h unter Reflux erhitzt und direkt im Anschluss im
Eisbad auf 0 °C abgekühlt. Innerhalb von 5 min werden 2.15 mL (17.1 mmol)
Chlorameisensäurephenylester zugetropft und die Mischung weitere 16 h bei RT
gerührt. Nach beendeter Reaktion wird die Reaktionslösung mit 600 mL DCM
versetzt und zweimal mit 400 mL Wasser und einmal mit 400 mL ges.
Natriumchloridlösung gewaschen. Die vereinigten wässrigen Phasen werden mit
900 mL DCM extrahiert und die organische Phase anschließend erneut einmal mit
900 mL ges. Natriumchloridlösung gewaschen und über Natriumsulfat getrocknet.
Nach Entfernen des Lösungsmittels unter vermindertem Druck wird das Produkt
aus 1 L Ethylacetat kristallisiert. Nach dem Trocknen des farblosen Feststoffs im
Hochvakuum wird 2 in einer Ausbeute von 1.83 g (72 % d. Theorie) erhalten.
O ONH
O
O
Smp: 139 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.39 (s, 3H, CH3), 6.26 (s, 1H, CH3CCH),
7.26–7.31 (m, 3H, Ar-H), 7.43–7.48 (m, 3H, Ar-H), 7.55–7.56 (m, 1H, Ar-H), 7.72–
7.74 (m, 1H, Ar-H), 10.73 (s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 104.7, 112.1, 114.4, 114.7
(CH3CCH, Ar-C), 121.9, 125.6, 126.1, 129.4, 142.1, 150.2, 151.5, 153.0, 153.7,
159.9 (C(O)NH, CH3C, Ar-C, C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 296 (100) [M + H]+.
Elementaranalyse: C17H13NO4 [295.29]
berechnet: C: 69.15 % H: 4.44 % N: 4.74 %
gefunden: C: 68.97 % H: 4.40 % N: 4.64 %

Experimenteller Teil _________________________________________________________________
- 101 -
7-Isocyanato-4-methylcumarin (3) 1 g (3.39 mmol) 2 werden in einer Sublime im Vakuum (2 × 10-3 bis 5 × 10-2 mbar)
für vier Tage auf 150 °C erhitzt. Das Produkt wird als farbloser Feststoff in einer
Ausbeute von 382 mg (56 % d. Theorie) erhalten und direkt mit den Schiffschen
Basen 1a und 4a–14a umgesetzt.
O ONC
O
Allgemeine Synthesevorschrift zur Darstellung der 4-(Benzyliden-amino)benzyl-N-4-methylcumarin-7-yl-carbamate 1b und 4b–14b
Zu einer Lösung von 100 mg (497 μmol) 3 in 8 mL THF werden 20 mg
Molekularsieb 4 Å und 4-(Benzylidenamino)benzylalkohol (497 μmol) gegeben.
Nach Hinzufügen von 24.9 μL (2.49 μmol) einer 100 mM Lösung von DBTL in
Toluol wird das Reaktionsgemisch 0.5 h bei RT gerührt. Der dabei entstandene
Feststoff wird vorsichtig ohne Molekularsieb abfiltriert und mit 15 mL n-Hexan
gewaschen. Die weitere Aufreinigung und Charakterisierung der Produkte wird für
jede Verbindung im Folgenden beschrieben:
4-(Benzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (1b) Der Feststoff wird in ca. 40 mL DCM gelöst. Die resultierende Lösung wird unter
vermindertem Druck auf ein Volumen von ca. 4 mL eingeengt und auf – 20 °C
abgekühlt, wobei das Produkt erneut als farbloser Feststoff ausfällt. Nach der
Isolierung durch Filtration und anschließender Trocknung im Hochvakuum wird 1b
in einer Ausbeute von 135 mg (66 % d. Theorie) erhalten.
N
OH NH
O
O O
C25H20N2O4
Smp.: 231 °C (unter Zersetzung)

Experimenteller Teil _________________________________________________________________
- 102 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.39 (s, 3H, CH3), 5.22 (s, 2H, CH2), 6.22 (s,
1H, CH3CCH), 7.28–7.30 (m, 2H, Ar-H), 7.43–7.56 (m, 7H, Ar-H), 7.68–7.70 (m,
1H, Ar-H), 7.93–7.95 (m, 2H, Ar-H), 8.63 (s, 1H, C(N)H), 10.18 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.6 (CH3), 65.7 (CH2), 104.4, 111.7, 114.1,
114.2 (CH3CCH, Ar-C), 120.7, 125.6, 128.4, 128.5, 129.0, 131.2, 133.6, 135.7,
142.4, 151.1 (Ar-C), 152.7, 152.9, 153.6, 159.6, 160.6 (C(O)NH, CH3C, Ar-C,
C(N)H, C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 413 (100) [M + H]+.
4-(4-Dimethylaminobenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (4b) Nach Trocknung des gelben Feststoffs im Hochvakuum wird 4b in einer Ausbeute
von 197 mg (87 % d. Theorie) erhalten.
N
OH NH
O
O O
N
C27H25N3O4
Smp.: 235 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.39 (s, 3H, CH3C), 3.01 (s, 6H, N(CH3)2),
5.19 (s, 2H, CH2), 6.24 (s, 1H, CH3CCH), 6.77–6.79 (m, 2H, Ar-H), 7.20–7.22 (m,
2H, Ar-H), 7.41–7.46 (m, 3H, Ar-H), 7.56 (s, 1H, Ar-H), 7.69–7.75 (m, 3H, Ar-H),
8.41 (s, 1H, C(N)H), 10.29 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3C), 66.1 (CH2), 104.3, 111.4,
111.8, 114.2, 114.3 (CH3CCH, Ar-C), 120.9, 123.6, 126.0, 129.4, 130.3, 132.5,
142.7 (Ar-C), 152.27, 152.34, 153.1, 153.2, 153.8, 160.0, 160.2 (Ar-C, C(O)NH,
CH3C, C(N)H, C(O)CH).

Experimenteller Teil _________________________________________________________________
- 103 -
Das 13C-Signal der Methylgruppen am Stickstoffatom (N(CH3)2) wird vom
Lösungsmittelsignal (DMSO-d6: δ = 39.4 ppm) überlagert.
APCI-MS (5 μA, MeOH): m/z (%) = 456 (100) [M + H]+.
4-(4-Methoxybenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (5b) Nach Trocknung des farblosen Feststoffs im Hochvakuum wird 5b in einer
Ausbeute von 208 mg (95 % d. Theorie) erhalten.
N
OH NH
O
O O
O
C26H22N2O5
Smp.: 221 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3C), 3.83 (s, 3H, CH3O), 5.20
(s, 2H, CH2), 6.23 (s, 1H, CH3CCH), 7.06–7.08 (m, 2H, Ar-H), 7.25–7.27 (m, 2H,
Ar-H), 7.41–7.49 (m, 3H, Ar-H), 7.56 (s, 1H, Ar-H), 7.68–7.70 (m, 1H, Ar-H), 7.87–
7.89 (m, 2H, Ar-H), 8.53 (s, 1H, C(N)H), 10.29 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3C), 55.3 (CH3O), 66.0 (CH2),
104.3, 111.8, 114.1, 114.2, 114.3 (CH3CCH, Ar-C), 120.9, 126.0, 128.8, 129.4,
130.4, 133.2, 142.6, 151.7, 153.10, 153.12, 153.8, 160.0, 160.1, 161.9 (C(O)NH,
CH3C, Ar-C, C(N)H, C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 443 (100) [M + H]+.
4-(4-Fluorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (6b) Nach Trocknung des farblosen Feststoffs im Hochvakuum wird 6b in einer
Ausbeute von 179 mg (84 % d. Theorie) erhalten.

Experimenteller Teil _________________________________________________________________
- 104 -
N
OH NH
O
O O
F
C25H19FN2O4
Smp.: 221 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.21 (s, 2H, CH2), 6.23 (s,
1H, CH3CCH), 7.28–7.30 (m, 2H, Ar-H), 7.34–7.43 (m, 3H, Ar-H), 7.49–7.51 (m,
2H, Ar-H), 7.56–7.57 (m, 1H, Ar-H), 7.68–7.70 (m, 1H, Ar-H), 7.99–8.02 (m, 2H,
Ar-H), 8.63 (s, 1H, C(N)H), 10.30 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.9 (CH2), 104.3, 111.8, 114.1,
114.3 (CH3CCH, Ar-C), 115.8 (m, Ar-C), 121.0, 126.0, 129.4 (Ar-C), 131.0 (m, Ar-
C), 132.5 (m, Ar-C), 133.8, 142.6, 151.1, 153.0, 153.1, 153.7, 159.7, 159.9 (Ar-C,
C(O)NH, CH3C, C(N)H, C(O)CH), 164.0 (m, CF).
APCI-MS (5 μA, MeCN): m/z (%) = 431 (100) [M + H]+.
4-(4-Chlorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (7b) Nach Trocknung des hellgelben Feststoffs im Hochvakuum wird 7b in einer
Ausbeute von 169 mg (76 % d. Theorie) erhalten.
N
OH NH
O
O O
Cl
C25H19ClN2O4
Smp.: 240 °C (unter Zersetzung)

Experimenteller Teil _________________________________________________________________
- 105 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.21 (s, 2H, CH2), 6.23 (s,
1H, CH3CCH), 7.30–7.32 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.50–7.51 (m,
2H, Ar-H), 7.56–7.60 (m, 3H, Ar-H), 7.68–7.70 (m, 1H, Ar-H), 7.94–7.97 (m, 2H,
Ar-H), 8.65 (s, 1H, C(N)H), 10.30 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.9 (CH2), 104.3, 111.8, 114.1,
114.3 (CH3CCH, Ar-C), 121.1, 126.0, 128.9, 129.4, 130.2, 134.0, 134.7, 136.0,
142.6, 150.9, 153.1, 153.2, 153.7, 159.7, 159.9 (Ar-C, C(O)NH, CH3C, C(N)H,
C(O)CH).
APCI-MS (5 μA, MeCN): m/z (%) = 447 (100) [M + H]+.
4-(4-Acetylbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (8b) Nach Lösen des Feststoffs in 12 mL DCM werden 15 mL n-Hexan zugegeben und
das Gemisch auf – 20 °C abgekühlt. Das Produkt wird im Anschluss erneut
abfiltriert und im Hochvakuum getrocknet. 8b wird als gelber Feststoff in einer
Ausbeute von 101 mg (37 % d. Theorie) erhalten.
N
OH NH
O
O O
O
C27H22N2O5
Smp.: 196 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3CCH), 2.63 (s, 3H,
C(O)CH3), 5.22 (s, 2H, CH2), 6.23 (s, 1H, CH3CCH), 7.34–7.43 (m, 3H, Ar-H),
7.51–7.56 (m, 3H, Ar-H), 7.68–7.70 (m, 1H, Ar-H), 8.07–8.08 (m, 4H, Ar-H), 8.74
(s, 1H, C(N)H), 10.31 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3CCH), 26.9 (C(O)CH3), 65.9
(CH2), 104.3, 111.8, 114.2, 114.3 (CH3CCH, Ar-C), 121.2, 126.0, 128.6, 128.8,

Experimenteller Teil _________________________________________________________________
- 106 -
129.4, 134.3, 138.5, 139.6, 142.6, 150.8, 153.1, 153.2, 153.8, 159.9, 160.1 (Ar-C,
CH3C, C(O)NH, C(N)H, C(O)CH), 197.6 (C(O)CH3).
APCI-MS (5 μA, MeOH): m/z (%) = 455 (100) [M + H]+.
4-(4-Trifluormethylbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (9b) Die Reaktionslösung wird unter vermindertem Druck auf ein Volumen von ca.
3 mL eingeengt. Der dabei entstandene, gelbe Feststoff wird anschließend
abfiltriert und im Hochvakuum getrocknet. 9b wird in einer Ausbeute von 203 mg
(85 % d. Theorie) erhalten.
N
OH NH
O
O O
F3C
C26H19F3N2O4
Smp.: 236 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.22 (s, 2H, CH2), 6.22 (s,
1H, CH3CCH), 7.35–7.37 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.51–7.56 (m,
3H, Ar-H), 7.67–7.69 (m, 1H, Ar-H), 7.87–7.89 (m, 2H, Ar-H), 8.13–8.15 (m, 2H,
Ar-H), 8.76 (s, 1H, C(N)H), 10.30 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.9 (CH2), 104.3, 111.8, 114.2,
114.3 (CH3CCH, Ar-C), 121.2 (Ar-C), 123.9 (q, J = 272.5 Hz, CF3), 125.7 (q,
J = 3.7 Hz, CHCHCF3), 125.9, 129.2, 129.4, (Ar-C), 131.0 (q, J = 31.7 Hz,
CHCF3), 134.4, 139.4, 142.6, 150.6, 153.08, 153.09, 153.8, 159.7, 159.9 (Ar-C,
C(O)NH, CH3C, C(O)CH, C(N)H).
ESI-MS (5 kV, DCM): m/z (%) = 481 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 107 -
4-(4-Nitrobenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (10b) Nach Trocknung des gelben Feststoffs im Hochvakuum wird 10b in einer
Ausbeute von 167 mg (74 % d. Theorie) erhalten.
N
OH NH
O
O O
O2N
C25H19N3O6
Smp.: 240 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.24 (s,
1H, CH3CCH), 7.38–7.43 (m, 3H, Ar-H), 7.53–7.56 (m, 3H, Ar-H), 7.68–7.71 (m,
1H, Ar-H), 8.18–8.21 (m, 2H, Ar-H), 8.36–8.38 (m, 2H, Ar-H), 8.83 (s, 1H, C(N)H),
10.31 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.8 (CH2), 104.3, 111.8, 114.2,
114.3 (CH3CCH, Ar-C), 121.3, 124.0, 126.0, 129.4, 129.6, 134.7, 141.3, 142.6,
148.8, 150.4, 153.1, 153.2, 153.7, 159.2, 159.9 (Ar-C, C(O)NH, CH3C, C(N)H,
C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 458 (100) [M + H]+.
4-(2,3,6-Trichlorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (11b) Nach beendeter Reaktion wird das Molekularsieb abfiltriert und das Lösungsmittel
unter vermindertem Druck entfernt. Zum Rückstand werden 15 mL Et2O gegeben
und die Lösung auf – 20 °C abgekühlt. Der dabei entstandene hellgelbe Feststoff
wird abfiltriert und anschließend mit 5 mL Et2O gewaschen. Nach Trocknung im
Hochvakuum wird 11b in einer Ausbeute von 163 mg (63 % d. Theorie) erhalten.

Experimenteller Teil _________________________________________________________________
- 108 -
N
OH NH
O
O OClCl
Cl
C25H17Cl3N2O4
Smp.: 138 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.23 (s,
1H, CH3CCH), 7.30–7.32 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.53–7.56 (m,
3H, Ar-H), 7.63–7.70 (m, 2H, Ar-H), 7.78–7.80 (m, 1H, Ar-H), 8.71 (s, 1H, C(N)H),
10.31 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.7 (CH2), 104.3, 111.8, 114.1,
114.3 (CH3CCH, Ar-C), 120.8, 125.9, 129.3, 129.8, 131.4, 131.85, 131.89, 132.2,
134.3, 134.9, 142.6, 150.2, 153.04, 153.05, 153.7, 157.0, 159.9 (C(O)NH, CH3C,
Ar-C, C(N)H, C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 515 (100) [M + H]+.
4-(3,4-Difluorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (12b) Nach Trocknung des farblosen Feststoffs im Hochvakuum wird 12b in einer
Ausbeute von 141 mg (63 % d. Theorie) erhalten.
N
OH NH
O
O OF
F
C25H18F2N2O4
Smp.: 226 °C (unter Zersetzung)

Experimenteller Teil _________________________________________________________________
- 109 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.21 (s, 2H, CH2), 6.23 (s,
1H, CH3CCH), 7.30–7.32 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.50–7.62 (m,
4H, Ar-H), 7.68–7.70 (m, 1H, Ar-H), 7.79–7.81 (m, 1H, Ar-H), 7.96–7.98 (m, 1H,
Ar-H), 8.63 (s, 1H, C(N)H), 10.30 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.9 (CH2), 104.3, 111.8, 114.1,
114.3 (CH3CCH, Ar-C), 116.5 (d, J = 17.7 Hz, FCCH), 118.0 (d, J = 17.7 Hz,
FCCH), 121.1, 126.0 (Ar-C), 126.2 (m, Ar-C), 129.4 (Ar-C), 133.6 (m, Ar-C), 134.1,
142.6 (Ar-C), 149.3 (m, CF), 150.6 (Ar-C), 151.7 (m, CF), 153.0, 153.1, 153.7,
158.8, 159.9 (C(O)NH, CH3C, Ar-C, C(O)CH, C(N)H).
APCI-MS (5 μA, MeOH): m/z (%) = 449 (100) [M + H]+.
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (13b) Nach Trocknung des farblosen Feststoffs im Hochvakuum wird 13b in einer
Ausbeute von 197 mg (82 % d. Theorie) erhalten.
N
OH NH
O
O OF
F
FF
C25H16F4N2O4
Smp.: 255 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.24 (s,
1H, CH3CCH), 7.33–7.37 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.53–7.56 (m,
3H, Ar-H), 7.68–7.71 (m, 1H, Ar-H), 8.08–8.13 (m, 1H, CFCH), 8.69 (s, 1H,
C(N)H), 10.32 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.8 (CH2), 104.4 (Ar-C), 109.0
(dd, J1 = J2 = 23.1 Hz, CFCCF), 111.9, 114.2, 114.4 (CH3CCH, Ar-C), 115.6 (dd,
J1 = J2 = 10.8 Hz, CFCCF), 121.1, 126.0, 129.4, 135.1, 142.6 (Ar-C), 144.0 (m,

Experimenteller Teil _________________________________________________________________
- 110 -
CF), 146.4 (m, CF), 150.4, 150.7, 153.10, 153.13, 153.8, 160.0 (Ar-C, C(O)NH,
CH3C, C(N)H, C(O)CH).
ESI-MS (5 kV, MeCN): m/z (%) = 485 (100) [M + H]+.
HRMS (ESI, 4 kV): C25H16F4N2O4
berechnet: 507.0944 [M + Na]+ 485.1124 [M + H]+
gefunden: 507.0942 [M + Na]+ 485.1124 [M + H]+
4-(Pentafluorbenzylidenamino)benzyl-N-4-methylcumarin-7-yl-carbamat (14b) Nach Trocknung des gelben Feststoffs im Hochvakuum wird 14b in einer
Ausbeute von 218 mg (87 % d. Theorie) erhalten.
N
OH NH
O
O OF
F
FFF
C25H15F5N2O4
Smp.: 257 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.39 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.24 (s,
1H, CH3CCH), 7.34–7.36 (m, 2H, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.53–7.56 (m,
3H, Ar-H), 7.69–7.71 (m, 1H, Ar-H), 8.66 (s, 1H, C(N)H), 10.32 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 65.7 (CH2), 104.3 (Ar-C), 110.9
(m, CFCCF), 111.8, 114.1, 114.3 (CH3CCH, Ar-C), 121.1, 126.0, 129.3, 135.1 (Ar-
C), 137.1 (m, CF), 139.3 (m, CF), 142.6 (Ar-C), 146.1 (m, CF), 149.6, 150.5,
153.06, 153.10, 153.7, 159.9 (Ar-C, C(O)NH, CH3C, C(N)H, C(O)CH).
APCI-MS (5 μA, MeOH): m/z (%) = 503 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 111 -
HRMS (ESI, 4 kV): C25H15F5N2O4
berechnet: 525.0850 [M + Na]+ 503.1030 [M + H]+
gefunden: 525.0841 [M + Na]+ 503.1021 [M + H]+
6.2.2 Synthese der Modellsubstanz 17
tert.-Butyl-N-4-(hydroxymethyl)phenylcarbamat (15) Zu einer Lösung von 1 g (8.12 mmol) 4-Aminobenzylalkohol in 40 mL DCM
werden 2.66 g (12.2 mmol) (Boc)2O gegeben. Nach drei Tagen Rühren bei RT
wird das Lösungsmittel unter vermindertem Druck entfernt und das Produkt aus
einem Gemisch aus n-Hexan und Ethylacetat bei – 20 °C kristallisiert. Nach dem
Trocknen des farblosen Feststoffs im Hochvakuum wird 15 in einer Ausbeute von
1.28 g (71 % d. Theorie) erhalten.
NH
OH
O
O
Smp.: 82–83 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.47 (s, 9H, CH3), 4.39 (d, J = 5.6 Hz, 2H,
CH2), 5.04 (t, J = 5.6 Hz, 1H, OH), 7.16–7.19 (m, 2H, Ar-H), 7.38–7.40 (m, 2H, Ar-
H), 9.27 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.0 (CH3), 62.5 (CH2), 78.7 (C(CH3)3),
117.7, 126.8, 136.0, 138.0 (Ar-C), 152.7 (C(O)NH).
APCI-MS (5 μA, MeOH): m/z (%) = 222 (100) [M – H]–.
Elementaranalyse: C12H17NO3 [223.27]
berechnet: C: 64.55 % H: 7.67 % N: 6.27 %
gefunden: C: 64.47 % H: 7.87 % N: 6.24 %

Experimenteller Teil _________________________________________________________________
- 112 -
4-(tert.-Butoxycarbonylamino)benzyl-N-4-methylcumarin-7-yl-carbamat (16)
Zu einer Lösung von 180 mg (896 μmol) 3 in 15 mL THF werden Molekularsieb
4 Å (20 mg), 200 mg (896 μmol) 15 und 45 μL (4.5 μmol) einer 100 mM Lösung
von DBTL in Toluol gegeben. Nach 18 h Rühren bei RT wird das Molekularsieb
abfiltriert und das Lösungsmittel unter vermindertem Druck entfernt. Kristallisation
aus einem Gemisch von n-Hexan und THF bei – 20 °C und anschließendes
Trocknen des farblosen Feststoffs im Hochvakuum liefert 16 in einer Ausbeute von
239 mg (63 % d. Theorie).
NH
O NH
O
O O
O
O
Smp.: 200 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.47 (s, 9H, OC(CH3)3), 2.37 (s, 3H,
CH3CCH), 5.09 (s, 2H, CH2), 6.23 (s, 1H, CH3CCH), 7.32–7.34 (m, 2H, Ar-H),
7.39–7.41 (m, 1H, Ar-H), 7.46–7.48 (m, 2H, Ar-H), 7.54 (s, 1H, Ar-H), 7.67–7.69
(m, 1H, Ar-H), 9.42 (br s, 1H, NH), 10.24 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3CCH), 28.0 (OC(CH3)3), 66.1
(CH2), 79.0 (OC(CH3)3), 104.3, 111.8, 114.1, 114.2 (CH3CCH, Ar-C), 117.8, 125.9,
129.1, 129.3, 139.5, 142.6, 152.6, 153.07, 153.1, 153.7, 159.9 (C(O)NH, CH3CCH,
C(O)CH, Ar-C).
APCI-MS (5 μA, MeCN): m/z (%) = 423 (100) [M – H]–.
Elementaranalyse: C23H24N2O6 [424.45]
berechnet: C: 65.08 % H: 5.70 % N: 6.60 %
gefunden: C: 65.18 % H: 5.73 % N: 6.32 %

Experimenteller Teil _________________________________________________________________
- 113 -
4-Aminobenzyl-N-4-methylcumarin-7-yl-carbamat-Trifluoracetat (17)
Zu einer Lösung von 50 mg (118 μmol) 16 in 2 mL DCM werden 2 mL TFA
gegeben. Die Reaktionslösung wird 35 min bei RT gerührt und das Produkt
anschließend mit 30 mL Et2O (abs.) gefällt. 17 wird als gelber Feststoff in einer
Ausbeute von 50.8 mg (98 % d. Theorie) erhalten und weist laut DC keine
Verunreinigungen auf.
+H3N
O NH
O
O O
CF3COO-
C20H17F3N2O6
APCI-MS (5 μA, MeCN): m/z (%) = 323 (11) [M – H]–, 113 (100) [CF3COO]–.
6.2.3 Synthese niedermolekularer Vorstufen für die Darstellung von PEG-geträgerten AMC-Modellverbindungen mittels CuAAC
2-(Hydroxymethyl)-5-nitrobenzohydrazid (18) Zu einer Suspension von 50 g (279 mmol) 6-Nitrophthalid in 1.7 L MeOH werden
21.0 g (20.3 mL, 419 mmol) Hydrazin-Hydrat gegeben. Nach 23 h Rühren bei RT
wird der farblose Feststoff abfiltriert. Das Filtrat wird auf ein Volumen von ca.
100 mL eingeengt und auf 4 °C abgekühlt, wobei sich ein farbloser, schaumartiger
Feststoff bildet. Nach erneuter Filtration werden die Feststoffe vereint und im
Hochvakuum getrocknet. 18 wird in einer Ausbeute von 55.5 g (94 % d. Theorie)
erhalten.
O2NHN
ONH2
OH
C8H7N3O4

Experimenteller Teil _________________________________________________________________
- 114 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 4.60 (br s, 2H, NH2), 4.72 (s, 2H, CH2), 5.57
(br s, 1H, OH), 7.85–7.87 (m, 1H, Ar-H), 8.15–8.16 (m, 1H, Ar-H), 8.31–8.33 (m,
1H, Ar-H), 9.78 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 60.2 (CH2), 122.0, 124.4, 128.0, 133.9,
145.6, 148.9 (Ar-C), 165.6 (C(O)NH).
APCI-MS (5 μA, MeOH): m/z (%) = 210 (100) [M – H]–.
1-tert.-Butylcarbonyl-2-(2-(hydroxymethyl)-5-nitrobenzoyl)hydrazin (19) Zu einer Suspension von 55.0 g (260 mmol) 18 in 900 mL Dioxan und 1.8 L tert.-
Butanol werden 113 g (111 mL, 520 mmol) (Boc)2O gegeben. Nach 88 h Rühren
bei RT werden die Lösungsmittel unter vermindertem Druck entfernt. Das Produkt
wird bei 4 °C aus einem Gemisch aus Ethylacetat und n-Hexan kristallisiert,
abfiltriert und im Hochvakuum getrocknet. 19 wird als farbloser Feststoff in einer
Ausbeute von 78.7 g (97 % d. Theorie) erhalten.
O2NHN
ONH
OH
Boc
Smp.: 123–125 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.43 (s, 9H, CH3), 4.73 (d, J = 5.3 Hz, 2H,
CH2), 5.59 (t, J = 5.3 Hz, 1H, OH), 7.89–7.91 (m, 1H, Ar-H), 8.15–8.16 (m, 1H, Ar-
H), 8.35–8.38 (m, 1H, Ar-H), 9.06 (br s, 1H, NH), 10.25 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.0 (CH3), 59.9 (CH2), 79.5 (C(CH3)3),
121.8, 124.9, 127.9, 133.0, 145.6, 149.1 (Ar-C), 155.1, 166.1 (C(O)NH).
ESI-MS (5 kV, MeCN): m/z (%) = 310 (100) [M – H]–.

Experimenteller Teil _________________________________________________________________
- 115 -
Elementaranalyse: C13H17N3O6 [311.29]
berechnet: C: 50.16 % H: 5.50 % N: 13.50 %
gefunden: C: 49.95 % H: 5.87 % N: 13.35 %
1-tert.-Butylcarbonyl-2-[2-((tert.-butyldimethylsilyloxy)methyl)-5-nitrobenzoyl]hydrazin (20) Zu einer Lösung von 41.3 g (274 mmol) tert.-Butyldimethylchlorsilan und 25.5 g
(374 mmol) Imidazol in 300 mL DMF werden 77.4 g (249 mmol) 19, gelöst in
450 mL DMF, innerhalb von 20 min zugetropft. Nach 2.5 h Rühren bei RT wird die
Reaktionslösung auf 700 mL Wasser gegeben und das Produkt anschließend
dreimal mit 800 mL DCM extrahiert. Die vereinigten organischen Phasen werden
einmal mit 800 mL Wasser gewaschen und über Natriumsulfat getrocknet. Nach
Entfernen des Trockenmittels durch Filtration wird das Lösungsmittel unter
vermindertem Druck entfernt. Das Produkt wird bei 4 °C aus einem Gemisch aus
MeOH und Wasser kristallisiert. Filtration und Trocknung des farblosen Feststoffs
im Hochvakuum liefert 20 in einer Ausbeute von 98.6 g (93 % d. Theorie).
O2NHN
ONH
OTBS
Boc
Smp.: 176–178 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.10 (s, 6H, SiCH3), 0.92 (s, 9H, SiC(CH3)3),
1.44 (s, 9H, OC(CH3)3), 4.96 (s, 2H, CH2), 7.87–7.89 (m, 1H, Ar-H), 8.19–8.20 (m,
1H, Ar-H), 8.40–8.43 (m, 1H, Ar-H), 9.07 (br s, 1H, NH), 10.29 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -5.6, 17.9, 25.7, 27.9 (SiCH3, SiC(CH3)3,
SiC(CH3)3, OC(CH3)3), 61.5 (CH2), 79.5 (OC(CH3)3), 121.7, 125.1, 127.5, 132.7,
145.7, 147.7 (Ar-C), 155.2, 166.0 (C(O)NH).
APCI-MS (5 μA, MeCN): m/z (%) = 426 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 116 -
Elementaranalyse: C19H31N3O6Si [425.55]
berechnet: C: 53.63 % H: 7.34 % N: 9.87 %
gefunden: C: 53.67 % H: 7.38 % N: 9.82 %
Aktivierung des Nickelkatalysators für die Synthese von 21 Zu einer Lösung von 50.0 g Nickelnitrat-Hexahydrat in 400 mL Wasser werden
100 g Kieselgel 60 (0.040–0.063 mm) gegeben. Das Reaktionsgemisch wird 1 h
bei RT gerührt und anschließend für 15 h stehengelassen. Das Lösungsmittel wird
unter vermindertem Druck entfernt und der Rückstand im Hochvakuum getrocknet.
1-tert.-Butylcarbonyl-2-[5-amino-2-((tert.-butyldimethylsilyloxy)methyl)-benzoyl]hydrazin (21) Zu einer Lösung von 40.0 g (94.0 mmol) 20 in 1 L MeOH werden 18.8 g des zuvor
aktivierten Ni(NO3)2/SiO2-Katalysators gegeben. Das Reaktionsgemisch wird mit
einem Eis/NaCl-Bad auf – 18 °C abgekühlt. Innerhalb von 40 min werden 8.80 g
(233 mmol) Natriumborhydrid portionsweise bis zur intensiven Schwarzfärbung
des Gemisches zugegeben. Nach beendeter Zugabe werden weitere 15 min im
Kältebad gerührt. Die Reaktionslösung wird mit 240 mL Wasser versetzt und das
Produkt anschließend dreimal mit 1 L Ethylacetat extrahiert. Die vereinigten
organischen Phasen werden über Natriumsulfat getrocknet. Nach einer Filtration
wird das Lösungsmittel unter vermindertem Druck entfernt und das Rohprodukt
mittels Flash-Säulenchromatographie gereinigt (Kieselgel, n-Hexan/Ethylacetat 1:1
(v/v)). Einengen der Lösung und Trocknung des braunen, schaumartigen
Feststoffs im Hochvakuum liefert 21 in einer Ausbeute von 53.8 g (72 % d.
Theorie).
H2NHN
ONH
OTBS
Boc
C19H33N3O4Si

Experimenteller Teil _________________________________________________________________
- 117 -
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.05 (s, 6H, SiCH3), 0.88 (s, 9H, SiC(CH3)3),
1.42 (s, 9H, OC(CH3)3), 4.67 (s, 2H, CH2), 5.21 (br s, 2H, NH2), 6.63–6.66 (m, 2H,
Ar-H), 7.14–7.16 (m, 1H, Ar-H), 8.85 (br s, 1H, NH), 9.71 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -5.4, 17.9, 25.7, 28.0 (SiCH3, SiC(CH3)3,
SiC(CH3)3, OC(CH3)3), 61.8 (CH2), 79.0 (OC(CH3)3), 112.6, 115.2, 125.6, 128.1,
133.4, 147.3 (Ar-C), 155.1, 168.4 (C(O)NH).
APCI-MS (5 μA, MeCN): m/z (%) = 394 (100) [M – H]–.
1-tert.-Butylcarbonyl-2-[2-((tert.-butyldimethylsilyloxy)methyl)-5-(phenoxy-carbonylamino)benzoyl]hydrazin (22) Zu einer Lösung von 53.1 g (134 mmol) 21 in 750 mL DCM werden bei 0 °C
17.8 mL (22.1 g, 141 mmol) Chlorameisensäurephenylester und 24.1 mL (18.2 g,
141 mmol) DIEA gegeben. Die Reaktionslösung wird für 1 h im Eisbad gerührt und
anschließend dreimal mit 600 mL ges. Natriumhydrogencarbonatlösung sowie
einmal mit 600 mL ges. Natriumchloridlösung gewaschen. Die vereinigten
organischen Phasen werden über Natriumsulfat getrocknet. Nach einer Filtration
wird die Lösung bis zur Trockene eingeengt und das Rohprodukt mittels Flash-
Säulenchromatographie gereinigt (Kieselgel, n-Hexan/Ethylacetat 4:1 (v/v)).
Entfernen der Lösungsmittel unter vermindertem Druck und Trocknung des
farblosen, schaumartigen Feststoffs im Hochvakuum liefert 22 in einer Ausbeute
von 48.7 g (70 % d. Theorie).
NH
HN
ONH
OTBS
BocO
O
C26H37N3O6Si
1H-NMR (400.1 MHz, THF-d8): δ = 0.12 (s, 6H, SiCH3), 0.95 (s, 9H, SiC(CH3)3),
1.46 (s, 9H, OC(CH3)3), 4.92 (s, 2H, CH2), 7.15–7.19 (m, 3H, Ar-H), 7.33–7.36 (m,

Experimenteller Teil _________________________________________________________________
- 118 -
2H, Ar-H), 7.52–7.54 (m, 1H, Ar-H), 7.62–7.63 (m, 1H, Ar-H), 7.70–7.72 (m, 1H,
Ar-H), 8.23 (br s, 1H, NH), 9.32–9.36 (m, 2H, NH).
13C-NMR (100.6 MHz, THF-d8): δ = -5.2, 19.0, 26.4, 28.5 (SiCH3, SiC(CH3)3,
SiC(CH3)3, OC(CH3)3), 63.3 (CH2), 80.1 (OC(CH3)3), 118.4, 120.5, 122.4, 125.8,
129.0, 129.9, 134.6, 135.2, 138.8, 152.2, 152.3, 156.7, 168.6 (Ar-C, C(O)NH).
ESI-MS (5 kV, MeCN): m/z (%) = 538 (100) [M + Na]+.
1-tert.-Butylcarbonyl-2-[2-(hydroxymethyl)-5-(phenoxycarbonylamino)-benzoyl]hydrazin (23) Eine Lösung von 48.1 g (93.3 mmol) 22 in 1 L eines 50:1-Gemisches aus MeOH
und HCl (1M) wird bei RT 2 h gerührt und anschließend mit 1 L Ethylacetat und
500 mL Wasser versetzt. Das Produkt wird dreimal mit 700 mL Ethylacetat
extrahiert. Die vereinigten organischen Phasen werden zweimal mit 500 mL ges.
Natriumhydrogencarbonatlösung sowie einmal mit 500 mL ges. Natriumchlorid-
lösung gewaschen und über Natriumsulfat getrocknet. Nach Abtrennen des
Trockenmittels wird das Lösungsmittel unter vermindertem Druck entfernt. Das
Rohprodukt wird aus einem Gemisch aus Ethylacetat und n-Hexan bei – 20 °C
kristallisiert. Filtration und Trocknung des farblosen Feststoffs im Hochvakuum
liefert 23 in einer Ausbeute von 21.5 g (57 % d. Theorie).
NH
HN
ONH
OH
BocO
O
Smp.: 156–158 °C
1H-NMR (400.1 MHz, THF-d8): δ = 1.46 (s, 9H, CH3), 4.53 (t, J = 5.9 Hz, 1H, OH),
4.61 (d, J = 5.9 Hz, 2H, CH2), 7.17–7.19 (m, 3H, Ar-H), 7.32–7.39 (m, 3H, Ar-H),
7.64–7.71 (m, 2H, Ar-H), 8.30 (br s, 1H, NH), 9.38 (br s, 1H, NH), 9.62 (br s, 1H,
NH).

Experimenteller Teil _________________________________________________________________
- 119 -
13C-NMR (100.6 MHz, THF-d8): δ = 28.5 (CH3), 63.2 (CH2), 80.2 (C(CH3)3), 119.0,
120.6, 122.4, 125.8, 129.9, 130.7, 135.7, 136.2, 139.1, 152.2, 152.3, 156.7, 169.2
(Ar-C, C(O)NH).
ESI-MS (5 kV, MeCN): m/z (%) = 424 (100) [M + Na]+.
Elementaranalyse: C20H23N3O6 [401.41]
berechnet: C: 59.84 % H: 5.78 % N: 10.47 %
gefunden: C: 59.95 % H: 5.64 % N: 10.32 %
1-tert.-Butylcarbonyl-2-[2-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-5-(phenoxycarbonylamino)benzoyl]hydrazin (24) Zu einer Lösung von 2.99 g (7.46 mmol) 23 in 150 mL THF werden 20 mg
zerstoßenes Molekularsieb 4 Å, 1.50 g (7.46 mmol) 3 und 221 μL (22.1 μmol)
einer 100 mM Lösung von DBTL in Toluol gegeben. Die Reaktionslösung wird
30 min bei RT gerührt und das Molsieb anschließend abfiltriert. Nach Entfernen
des Lösungsmittels unter vermindertem Druck wird das Rohprodukt aus einem
Gemisch aus Ethylacetat und n-Hexan bei – 20 °C kristallisiert. Filtration und
Trocknung des farblosen Feststoffs im Hochvakuum liefert 24 in einer Ausbeute
von 4.27 g (95 % d. Theorie).
NH
HN
ONH
O
BocO
O
O
NH
O O
Smp.: 137 °C (unter Zersetzung)
1H-NMR (400.1 MHz, THF-d8): δ = 1.55 (s, 9H, OC(CH3)3), 2.39 (s, 3H, CH3CCH),
5.39 (s, 2H, CH2), 6.08 (s, 1H, CH3CCH), 7.20–7.23 (m, 3H, Ar-H), 7.37–7.41 (m,
2H, Ar-H), 7.43–7.49 (m, 2H, Ar-H), 7.57–7.59 (m, 2H, Ar-H), 7.65–7.67 (m, 1H,
Ar-H), 7.73–7.74 (m, 1H, Ar-H), 8.54 (br s, 1H, NH), 9.47–9.50 (m, 3H, NH).

Experimenteller Teil _________________________________________________________________
- 120 -
13C-NMR (100.6 MHz, THF-d8): δ = 18.3 (CH3CCH), 28.5 (OC(CH3)3), 64.6 (CH2),
80.7 (OC(CH3)3), 105.6, 113.3, 114.5, 115.5 (CH3CCH, Ar-C), 118.4, 120.0, 122.4,
125.9, 126.0, 129.9, 130.8, 131.0, 136.5, 139.8, 144.0, 152.1, 152.3, 152.7, 153.9,
155.7, 157.3, 160.3, 168.9 (C(O)NH, CH3CCH, C(O)CH, Ar-C).
ESI-MS (5 kV, MeOH): m/z (%) = 625 (100) [M + Na]+.
Elementaranalyse: C31H30N4O9 [602.59]
berechnet: C: 61.79 % H: 5.02 % N: 9.30 %
gefunden: C: 60.55 % H: 5.31 % N: 8.77 %
[2-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-5-(phenoxycarbonyl-amino)benzoyl]hydrazin-Trifluoracetat (25) Eine Lösung von 4.05 g (6.72 mmol) 24 in 40 mL eines 1:1-Gemisches aus DCM
und TFA wird 45 min bei RT gerührt. Nach beendeter Reaktion wird das Produkt
mit 2 L Et2O gefällt und zur Vervollständigung der Fällung für 1 h auf 4 °C
abgekühlt. Filtration und Trocknung des farblosen, schaumartigen Feststoffs im
Hochvakuum liefert 25 in einer Ausbeute von 3.25 g (96 % d. Theorie).
NH
HN
ONH3
+
O
O
O
O
NH
O O
CF3COO-
C28H23F3N4O9
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 5.29 (s, 2H, CH2), 6.23 (s,
1H, CH3CCH), 7.23–7.30 (m, 3H, Ar-H), 7.39–7.46 (m, 3H, Ar-H), 7.58–7.60 (m,
2H, Ar-H), 7.68–7.70 (m, 2H, Ar-H), 7.76–7.78 (m, 1H, Ar-H), 8.00–10.18 (br s,
3H, NH3), 10.30 (s, 1H, NH), 10.58 (s, 1H, NH), 11.24 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3), 63.3 (CH2), 104.4, 111.9, 114.2,
114.3 (CH3CCH, Ar-C), 116.5 (q, J = 296.4 Hz, CF3), 117.5, 120.6, 121.8, 125.6,

Experimenteller Teil _________________________________________________________________
- 121 -
125.9, 129.1, 129.4, 130.3, 132.6, 138.5, 142.6, 150.2, 151.7, 153.0, 153.1, 153.8
(C(O)NH, CH3CCH, C(O)CH, Ar-C), 158.3 (q, J = 33.3 Hz, CCF3), 160.0, 167.0
(C(O)NH).
ESI-MS (5 kV, MeOH): m/z (%) = 548 (100) [M + 2 Na]+.
[2-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-5-(phenoxycarbonyl-amino)benzoyl]azid (26) Zu einer Suspension von 3.17 g (6.31 mmol) 25 in 100 mL HCl (1M) wird im
Eisbad eine Lösung von 653 mg (9.47 mmol) Natriumnitrit in 2 mL Wasser
gegeben. Nach 3 h Rühren bei 0 °C wird der gelbe Feststoff abfiltriert, mit ca.
15 mL Wasser gewaschen und im Hochvakuum getrocknet. 26 wird in einer
Ausbeute von 2.95 g (91 % d. Theorie) erhalten und ohne weitere Aufreinigung
eingesetzt.
NH
N3
O
O
O
O
O
NH
O O
C26H19N5O7
Phenyl-N-[4-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-3-(prop-2-ynylcarbamoyl)phenyl]carbamat (27)
Zu einer Lösung von 179 mg (349 μmol) 26 in 3 mL DMF werden 23.9 μL (192 mg,
349 μmol) Propargylamin gegeben. Nach 4 h Rühren bei RT wird die Lösung bis
zur Trockene eingeengt und das Rohprodukt mittels Flash-
Säulenchromatographie gereinigt (Kieselgel, Chloroform/MeOH 50:1 (v/v)). Nach
Entfernen der Lösungs-mittel unter vermindertem Druck und Trocknung des
farblosen Feststoffs im Hochvakuum wird 27 in einer Ausbeute von 136 mg (74 %
d. Theorie) erhalten.

Experimenteller Teil _________________________________________________________________
- 122 -
NH
HN
O
O
O
O
O
NH
O O
C29H23N3O7
Smp.: 168 °C (unter Zersetzung)
1H-NMR (400.1 MHz, THF-d8): δ = 2.36 (s, 3H, CH3), 2.61 (t, J = 2.6 Hz, 1H,
C≡CH), 4.15 (dd, J1 = 5.4 Hz, J2 = 2.6 Hz, 2H, NHCH2), 5.36 (s, 2H, OCH2), 6.07
(s, 1H, CH3CCH), 7.16–7.20 (m, 3H, Ar-H), 7.33–7.37 (m, 2H, Ar-H), 7.39–7.42
(m, 1H, Ar-H), 7.46–7.50 (m, 2H, Ar-H), 7.55–7.57 (m, 1H, Ar-H), 7.61–7.63 (m,
1H, Ar-H), 7.68–7.69 (m, 1H, Ar-H), 8.02 (t, J = 5.4 Hz, 1H, NHCH2), 9.31 (s, 1H,
NH), 9.42 (s, 1H, NH).
13C-NMR (100.6 MHz, THF-d8): δ = 18.2 (CH3CCH), 29.4 (NCH2), 64.7 (OCH2),
71.9 (C≡CH), 81.2 (C≡CH), 105.8, 113.4, 114.6, 115.6 (CH3CCH, Ar-C), 118.1,
120.1, 122.4, 125.9, 126.1, 129.9, 130.6, 130.8, 137.7, 139.7, 144.0, 152.1, 152.3,
152.6, 154.0, 155.7, 160.3, 168.4 (C(O)NH, CH3CCH, C(O)CH, Ar-C).
APCI-MS (5 μA, MeOH): m/z (%) = 526 (67) [M + H]+, 432 (100) [M – PhOH + H]+.
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzyl-N-4-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (28)
Zu einer Lösung von 400 mg (761 μmol) 27 und 216 mg (761 μmol) 13a in 25 mL
Dioxan werden 381 μL (38.1 μmol) einer 100 mM Lösung von DBTL in Toluol und
30 mg Molekularsieb 4 Å gegeben. Nach 3 h Rühren unter Reflux wird das
Molekularsieb durch Filtration entfernt und das Lösungsmittel unter vermindertem
Druck auf ein Volumen von ca. 7 mL eingeengt, wobei das Produkt als hellgelber
Feststoff ausfällt. Dieses wird anschließend abfiltriert, mit ca. 4 mL iso-Hexan
gewaschen und im Hochvakuum getrocknet. 28 wird in einer Ausbeute von
348 mg (64 % d. Theorie) erhalten.

Experimenteller Teil _________________________________________________________________
- 123 -
NH
HN
O
O
O
O
O
NH
O O
N
HFF
FF
C37H26F4N4O7
Smp.: 186 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 3.13 (t, J = 2.5 Hz, 1H,
C≡CH), 4.02 (dd, J1 = 5.6 Hz, J2 = 2.5 Hz, 2H, NHCH2), 5.21 (s, 2H, OCH2), 5.24
(s, 2H, OCH2), 6.23 (s, 1H, CH3CCH), 7.34–7.36 (m, 2H, Ar-H), 7.40–7.47 (m, 2H,
Ar-H), 7.51–7.53 (m, 3H, Ar-H), 7.56–7.58 (m, 1H, Ar-H), 7.63–7.64 (m, 1H, Ar-H),
7.67–7.70 (m, 1H, Ar-H), 8.08–8.13 (m, 1H, Ar-H), 8.69 (s, 1H, C(N)H), 8.86 (t,
J = 5.6 Hz, 1H, NHCH2), 10.03 (s, 1H, NH), 10.24 (s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 18.4 (CH3), 28.8 (NCH2), 64.0, 65.9 (OCH2),
73.4 (C≡CH), 81.4 (C≡CH), 104.8 (Ar-C), 109.5 (dd, J1 = J2 = 23.6 Hz, CFCCF),
112.3, 114.7, 114.8 (CH3CCH, Ar-C), 116.1 (dd, J1 = J2 = 10.9 Hz, CFCCF), 117.4,
119.6, 121.6, 126.4, 128.3, 129.7, 130.3, 135.9, 136.7, 139.3, 143.2 (Ar-C), 145.3
(m, CF), 145.9 (m, CF), 150.8 (dd, J1 = J2 = 3.0 Hz, C(N)H), 151.1, 153.5, 153.6,
153.7, 154.2, 160.5, 168.1 (C(O)NH, CH3CCH, C(O)CH, Ar-C).
ESI-MS (5 kV, MeCN): m/z (%) = 715 (100) [M + H]+.
4-(Pentafluorbenzylidenamino)benzyl-N-4-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (29)
Zu einer Lösung von 400 mg (761 μmol) 27 und 229 mg (761 μmol) 14a in 25 mL
Dioxan werden 381 μL (38.1 μmol) einer 100 mM Lösung von DBTL in Toluol und
30 mg Molekularsieb 4 Å gegeben. Nach 2.5 h Rühren unter Reflux wird das
Molekularsieb durch Filtration entfernt und das Lösungsmittel unter vermindertem
Druck auf ein Volumen von ca. 7 mL eingeengt, wobei das Produkt als hellgelber

Experimenteller Teil _________________________________________________________________
- 124 -
Feststoff ausfällt. Dieses wird anschließend abfiltriert, mit ca. 4 mL iso-Hexan
gewaschen und im Hochvakuum getrocknet. 29 wird in einer Ausbeute von
366 mg (66 % d. Theorie) erhalten.
NH
HN
O
O
O
O
O
NH
O O
N
HFF
FF
F
C37H25F5N4O7
Smp.: 205 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 2.38 (s, 3H, CH3), 3.13 (t, J = 2.5 Hz, 1H,
C≡CH), 4.04 (dd, J1 = 5.5 Hz, J2 = 2.5 Hz, 2H, NHCH2), 5.21 (s, 2H, OCH2), 5.24
(s, 2H, OCH2), 6.23 (s, 1H, CH3CCH), 7.34–7.36 (m, 2H, Ar-H), 7.40–7.48 (m, 2H,
Ar-H), 7.51–7.59 (m, 4H, Ar-H), 7.64–7.70 (m, 2H, Ar-H), 8.66 (s, 1H, C(N)H), 8.86
(t, J = 5.5 Hz, 1H, NHCH2), 10.03 (s, 1H, NH), 10.24 (s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 18.4 (CH3), 28.8 (NCH2), 64.0, 65.9 (OCH2),
73.4 (C≡CH), 81.4 (C≡CH), 104.8 (Ar-C), 111.4 (m, Ar-C), 112.3, 114.7, 114.8
(CH3CCH, Ar-C), 117.4, 119.6, 121.6, 126.4, 128.3, 129.7, 130.3, 136.0, 136.7
(Ar-C), 137.8 (m, CF), 139.3 (Ar-C), 142.3 (m, CF), 143.2 (Ar-C), 146.0 (m, CF),
150.1, 150.9, 153.5, 153.6, 153.7, 154.2, 160.5, 168.1 (C(O)NH, C(N)H, CH3CCH,
C(O)CH, Ar-C).
ESI-MS (5 kV, MeCN): m/z (%) = 733 (100) [M + H]+.

Experimenteller Teil _________________________________________________________________
- 125 -
6.2.4 Synthese niedermolekularer Vorstufen für die Darstellung von PEG-geträgerten Prodrugs mittels CuAAC
2-(Hydroxymethyl)-5-nitro-N-(prop-2-ynyl)benzamid (30) Zu einer Suspension von 14.9 g (112 mmol) Aluminiumchlorid in 75 mL
Dichlorethan werden im Eisbad innerhalb von 45 min 27.0 mL (19.7 g, 195 mmol)
Triethylamin in 35 mL Dichlorethan zugetropft. Anschließend werden zu der
Mischung 10.0 g (55.8 mmol) 6-Nitrophthalid und 13.3 mL (10.7 g, 195 mmol)
Propargylamin in 240 mL Dichlorethan innerhalb von 45 min zugetropft. Nach
Entfernen des Kühlbades wird das Reaktionsgemisch für 22 h auf 45 °C erwärmt.
Das Lösungsmittel wird unter vermindertem Druck entfernt (Wasserbad: 30 °C)
und der Rückstand in ges. Natriumchloridlösung aufgenommen. Das Produkt wird
mit 15 L Ethylacetat extrahiert. Die vereinigten organischen Phasen werden im
Anschluss nochmals mit ges. Natriumchloridlösung gewaschen, über Natrium-
sulfat getrocknet und unter vermindertem Druck auf ein Volumen von ca. 2 L
eingeengt. Nach Kristallisation bei – 20°C und Trocknung im Hochvakuum wird 30,
ein beigefarbener Feststoff, in einer Ausbeute von 6.85 g (52 % d. Theorie)
erhalten.
O2NHN
O
OH
Smp.: 142 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.20 (t, J =2.5 Hz, 1H, C≡CH), 4.06 (dd,
J1 = 5.4 Hz, J2 = 2.5 Hz, 2H, NHCH2), 4.72 (d, J = 5.6 Hz, 2H, OCH2), 5.59 (t,
J = 5.6 Hz, 1H, OH), 7.87–7.90 (m, 1H, Ar-H), 8.20–8.21 (m, 1H, Ar-H), 8.33–8.36
(m, 1H, Ar-H), 9.13 (t, J = 5.4 Hz, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.9 (NHCH2), 60.6 (CH2OH), 73.7 (C≡CH),
81.1 (C≡CH), 122.4, 125.1, 128.5, 134.9, 146.2, 149.3 (Ar-C), 166.5 (C(O)NH).

Experimenteller Teil _________________________________________________________________
- 126 -
ESI-MS (5 kV, MeCN): m/z (%) = 217 (100) [M – H2O + H]+.
Elementaranalyse: C11H10N2O4 [234.21]
berechnet: C: 56.41 % H: 4.30 % N: 11.96 %
gefunden: C: 56.34 % H: 4.54 % N: 11.86 %
2-((tert.-Butyldimethylsilyloxy)methyl)-5-nitro-N-(prop-2-ynyl)benzamid (31) Zu einer Lösung von 3.20 g (47.0 mmol) Imidazol und 3.90 g (25.9 mmol) tert.-
Butyldimethylsilylchlorid in 130 mL DMF werden nach 30 min Rühren bei RT
5.50 g (23.5 mmol) 30 zugegeben. Nach weiteren 2.5 h Rühren werden 150 mL
DCM zugegeben. Die organische Phase wird dreimal mit je 300 mL Natrium-
hydrogencarbonatlösung gewaschen und über Natriumsulfat getrocknet. Nach
Entfernen des Lösungsmittels unter vermindertem Druck wird der Rückstand
mittels Flash-Säulenchromatographie (Kieselgel, n-Hexan/Ethylacetat 10:1 (v/v))
gereinigt. Trocknen des farblosen Öls im Hochvakuum liefert 31 in einer Ausbeute
von 7.94 g (97 % d. Theorie).
O2NHN
O
OTBS
Smp.: 62–64 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.10 (s, 6H, SiCH3), 0.92 (s, 9H, SiC(CH3)3),
3.20 (t, J = 2.5 Hz, 1H, C≡CH), 4.07 (dd, J1 = 5.4 Hz, J2 = 2.5 Hz, 2H, NHCH2),
4.95 (s, 2H, OCH2), 7.84–7.86 (m, 1H, Ar-H), 8.23–8.24 (m, 1H, Ar-H), 8.36–8.39
(m, 1H, Ar-H), 9.14 (t, J = 5.4 Hz, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -5.0, 18.4, 26.2, 28.9 (SiCH3, SiC(CH3)3,
SiC(CH3)3, NHCH2), 62.2 (CH2O), 73.7 (C≡CH), 81.0 (C≡CH), 122.5, 125.3, 128.2,
134.6, 146.4, 147.9 (Ar-C), 166.2 (C(O)NH).
APCI-MS (5 μA, MeCN): m/z (%) = 349 (100) [M + H]+, 217 (50) [M – OTBS]+.

Experimenteller Teil _________________________________________________________________
- 127 -
Elementaranalyse: C17H24N2O4Si [348.47]
berechnet: C: 58.59 % H: 6.94 % N: 8.04 %
gefunden: C: 58.94 % H: 7.12 % N: 7.78 %
5-Amino-2-((tert.-butyldimethylsilyloxy)methyl)-N-(prop-2-ynyl)benzamid (32) Zu einer Suspension von 1.55 g (4.45 mmol) 31 und 1.03 g (15.8 mmol) Zink in
80 mL DCM werden 1.52 mL (1.60 g, 26.7 mmol) konz. Essigsäure gegeben.
Nach vier Tagen Rühren bei RT wird das Zink abfiltriert und mit 30 mL MeOH
ausgewaschen. Die organischen Phasen werden vereint und die Lösungsmittel
unter vermindertem Druck entfernt. Der Rückstand wird mittels Flash-Säulen-
chromatographie (Kieselgel, n-Hexan/Ethylacetat 5:1 (v/v)) gereinigt. Trocknen
des gelben Feststoffs im Hochvakuum liefert 32 in einer Ausbeute von 928 mg
(65 % d. Theorie).
H2NHN
O
OTBS
C17H26N2O2Si
Smp.: 75–77 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.03 (s, 6H, SiCH3), 0.87 (s, 9H, SiC(CH3)3),
3.09 (t, J = 2.5 Hz, 1H, C≡CH), 3.97 (dd, J1 = 5.6 Hz, J2 = 2.5 Hz, 2H, NHCH2),
4.62 (s, 2H, OCH2), 5.20 (s, 2H, NH2), 6.58–6.63 (m, 2H, Ar-H), 7.08–7.10 (m, 1H,
Ar-H), 8.56 (t, J = 5.6 Hz, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -4.9, 18.4, 26.2, 28.5 (SiCH3, SiC(CH3)3,
SiC(CH3)3, NHCH2), 62.6 (CH2O), 73.1 (C≡CH), 81.5 (C≡CH), 113.2, 115.2, 125.8,
129.2, 135.7, 148.0 (Ar-C), 168.8 (C(O)NH).
APCI-MS (5 μA, MeOH): m/z (%) = 317 (100) [M – H]–.

Experimenteller Teil _________________________________________________________________
- 128 -
Phenyl-N-4-((tert.-butyldimethylsilyloxy)methyl)-3-(prop-2-ynylcarbamoyl)-phenylcarbamat (33) Zu einer Lösung von 1.93 g (6.06 mmol) 32 in 35 mL DCM werden im Eisbad
801 μL (996 mg, 6.36 mmol) Chlorameisensäurephenylester und 1.09 mL
(822 mg, 6.36 mmol) DIEA gegeben. Nach 4.5 h Rühren bei 0 °C wird die
Reaktionslösung dreimal mit je 50 mL ges. Natriumhydrogencarbonatlösung und
zweimal mit je 50 mL ges. Natriumchloridlösung gewaschen und über
Natriumsulfat getrocknet. Nach Entfernen des Lösungsmittels unter vermindertem
Druck wird der Rückstand mittels Flash-Säulenchromatographie (Kieselgel,
n-Hexan/Ethylacetat 4:1 (v/v)) gereinigt. Trocknen des farblosen Feststoffs im
Hochvakuum liefert 33 in einer Ausbeute von 1.93 g (73 % d. Theorie).
NH
HN
O
OTBS
O
O
Smp.: 146–148 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.07 (s, 6H, SiCH3), 0.89 (s, 9H, SiC(CH3)3),
3.13 (t, J = 2.5 Hz, 1H, C≡CH), 4.00 (dd, J1 = 5.5 Hz, J2 = 2.5 Hz, 2H, NHCH2),
4.76 (s, 2H, OCH2), 7.22–7.29 (m, 3H, Ar-H), 7.42–7.47 (m, 3H, Ar-H), 7.56–7.58
(m, 2H, Ar-H), 8.77 (t, J = 5.5 Hz, 1H, NHCH2), 10.36 (s, 1H, OC(O)NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -4.9, 18.4, 26.2, 28.7 (SiCH3, SiC(CH3)3,
SiC(CH3)3, NHCH2), 62.2 (CH2O), 73.3 (C≡CH), 81.4 (C≡CH), 117.7, 119.9, 122.3,
125.9, 128.2, 129.9, 133.7, 135.3, 137.7, 150.8, 152.2, 168.2 (Ar-C, C(O)NH).
ESI-MS (5 kV, MeCN): m/z (%) = 439 (87) [M + H]+, 307 (100) [M – OTBS]+.
Elementaranalyse: C24H30N2O4Si [438.59]
berechnet: C: 65.72 % H: 6.89 % N: 6.39 %
gefunden: C: 65.61 % H: 7.00 % N: 6.37 %

Experimenteller Teil _________________________________________________________________
- 129 -
Phenyl-N-4-(hydroxymethyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (34) Eine Lösung von 1.92 g (4.38 mmol) 33 in 30 mL eines 50:1-Gemisches aus
MeOH und HCl (1M) wird für 15 min bei RT gerührt. Die Reaktionslösung wird mit
50 mL Wasser und 150 mL Ethylacetat versetzt und die Phasen getrennt. Nach
der Extraktion des Produkts aus der wässrigen Phase mit Ethylacetat (2 × 75 mL)
werden die vereinigten organischen Phasen zweimal mit je 200 mL ges.
Natriumhydrogencarbonatlösung und einmal mit 200 mL ges. Natriumchlorid-
lösung gewaschen und über Natriumsulfat getrocknet. Das Produkt wird in der
organischen Phase unter vermindertem Druck aufkonzentriert und anschließend
unter Zugabe von n-Hexan bei – 20 °C kristallisiert. Trocknen des beigen
Feststoffs im Hochvakuum liefert 34 in einer Ausbeute von 1.24 g (87 % d.
Theorie).
NH
HN
O
OH
O
O
Smp.: 135–137 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.14 (t, J = 2.5 Hz, 1H, C≡CH), 4.03 (dd,
J1 = 5.5 Hz, J2 = 2.5 Hz, 2H, NHCH2), 4.53 (d, J = 5.6 Hz, 2H, OCH2), 5.27 (t,
J = 5.6 Hz, 1H, OH), 7.22–7.29 (m, 3H, Ar-H), 7.42–7.48 (m, 3H, Ar-H), 7.56–7.60
(m, 2H, Ar-H), 8.84 (t, J = 5.5 Hz, 1H, NHCH2), 10.34 (s, 1H, OC(O)NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.8 (NHCH2), 60.9 (CH2OH), 73.4 (C≡CH),
81.4 (C≡CH), 117.8, 120.0, 122.3, 125.9, 128.9, 129.9, 134.9, 135.6, 137.6, 150.9,
152.2, 168.4 (Ar-C, C(O)NH).
ESI-MS (5 kV, MeOH): m/z (%) = 347 (65) [M + Na]+, 307 (100) [M – H2O + H]+.
Elementaranalyse: C18H16N2O4 [324.33]
berechnet: C: 66.66 % H: 4.97 % N: 8.64 %
gefunden: C: 66.14 % H: 5.21 % N: 8.56 %

Experimenteller Teil _________________________________________________________________
- 130 -
Phenyl-N-4-[((4-nitrophenoxy)carbonyloxy)methyl]-3-(prop-2-ynylcarbamoyl)-phenylcarbamat (35) Zu einer Lösung von 1.54 g (7.64 mmol) Chlorameisensäure-4-nitrophenylester
und 614 μL (604 mg, 7.64 mmol) Pyridin in 15 mL THF werden nach 20 min
Rühren im Eisbad innerhalb von 13 min 1.65 g (5.09 mmol) 34 in 35 mL THF
zugetropft. Das Kältebad wird entfernt und die Reaktionsmischung für 2.5 h bei RT
gerührt. Nach Filtration und Waschen des Filterkuchens mit THF wird das
Lösungsmittel unter vermindertem Druck entfernt und der Rückstand mittels Flash-
Säulenchromatographie (Kieselgel, n-Hexan/Ethylacetat 3:1 (v/v)) gereinigt.
Trocknen des beigen Feststoffs im Hochvakuum liefert 35 in einer Ausbeute von
2.35 g (94 % d. Theorie).
NH
HN
O
O
O
O
O
O
NO2
Smp.: 139 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.36 (t, J = 2.5 Hz, 1H, C≡CH), 4.04 (dd,
J1 = 5.5 Hz, J2 = 2.5 Hz, 2H, NHCH2), 5.40 (s, 2H, OCH2), 7.24–7.28 (m, 3H, Ar-
H), 7.43–7.46 (m, 2H, Ar-H), 7.51–7.57 (m, 3H, Ar-H), 7.63–7.65 (m, 1H, Ar-H),
7.69–7.70 (m, 1H, Ar-H), 8.31–8.34 (m, 2H, Ar-H), 8.96 (t, J = 5.5 Hz, 1H,
NHCH2), 10.51 (s, 1H, OC(O)NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.8 (NHCH2), 68.2 (CH2O), 73.4 (C≡CH),
81.2 (C≡CH), 117.9, 119.9, 122.3, 123.0, 125.8, 126.0, 127.3, 129.9, 130.8, 136.9,
139.3, 145.5, 150.7, 152.1, 152.2, 155.7, 168.0 (Ar-C, C(O)NH, OC(O)O).
ESI-MS (5 kV, MeOH): m/z (%) = 512 (100) [M + Na]+, 307 (91) [M – OC(O)ONp]+.

Experimenteller Teil _________________________________________________________________
- 131 -
Elementaranalyse: C25H19N3O8 [489.43]
berechnet: C: 61.35 % H: 3.91 % N: 8.59 %
gefunden: C: 61.27 % H: 4.20 % N: 8.35 %
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzyl-N-4-[((4-nitrophenoxy)carbonyl-oxy)methyl]-3-(prop-2-ynylcarbamoyl)phenylcarbamat (36)
Zu einer Lösung von 250 mg (511 μmol) 35 und 145 mg (511 μmol) 13a in 10 mL
Dioxan werden 256 μL (25.6 μmol) einer 100 mM Lösung von DBTL in Toluol und
20 mg Molekularsieb 4 Å gegeben. Nach 3 h Rühren unter Reflux wird das
Molekularsieb abfiltriert und das Lösungsmittel unter vermindertem Druck entfernt.
Kristallisation aus einem Gemisch von n-Hexan und Ethylacetat bei – 20 °C und
anschließendes Trocknen des gelben Feststoffs im Hochvakuum liefert 36 in einer
Ausbeute von 218 mg (63 % d. Theorie).
NH
HN
O
O
O
O
O
O
NO2
N
HFF
FF
C33H22F4N4O8
Smp.: 130 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.14 (m, 1H, C≡CH), 4.03 (m, 2H, NHCH2),
5.22 (s, 2H, OCH2), 5.37 (s, 2H, OCH2), 7.35–7.37 (m, 2H, Ar-H), 7.47–7.61 (m,
6H, Ar-H), 7.66 (m, 1H, Ar-H), 8.10–8.11 (m, 1H, Ar-H), 8.31–8.33 (m, 2H, Ar-H),
8.70 (s, 1H, C(N)H), 8.92–8.93 (m, 1H, NHCH2), 10.08 (s, 1H, OC(O)NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.8 (NHCH2), 65.9, 68.3 (CH2O), 73.4
(C≡CH), 81.3 (C≡CH), 109.5 (dd, J1 = J2 = 23.6 Hz, CFCCF), 116.1 (dd,
J1 = J2 = 12.0 Hz, CFCCF), 117.6, 119.6, 121.6, 123.0, 125.8, 126.7, 129.7, 130.8,
135.9, 136.9, 139.8 (Ar-C), 145.4 (m, CF), 145.5 (Ar-C), 146.0 (m, CF), 150.8,
151.1, 152.2, 153.7, 155.7, 168.1 (Ar-C, C(O)NH, OC(O)O, C(N)H).

Experimenteller Teil _________________________________________________________________
- 132 -
ESI-MS (5 kV, MeCN): m/z (%) = 679 (100) [M + H]+, 496 (56) [M – OC(O)ONp]+.
4-(Pentafluorbenzylidenamino)benzyl-N-4-[((4-nitrophenoxy)carbonyloxy)-methyl]-3-(prop-2-ynylcarbamoyl)phenylcarbamat (37)
Zu einer Lösung von 250 mg (511 μmol) 35 und 154 mg (511 μmol) 14a in 10 mL
Dioxan werden 256 μL (25.6 μmol) einer 100 mM Lösung von DBTL in Toluol und
20 mg Molekularsieb 4 Å gegeben. Nach 2.5 h Rühren unter Reflux wird das
Molekularsieb abfiltriert und das Lösungsmittel unter vermindertem Druck entfernt.
Kristallisation aus einem Gemisch von n-Hexan und Ethylacetat bei – 20 °C und
anschließendes Trocknen des hellgelben Feststoffs im Hochvakuum liefert 37 in
einer Ausbeute von 223 mg (63 % d. Theorie).
NH
HN
O
O
O
O
O
O
NO2
N
HFF
FFF
C33H21F5N4O8
Smp.: 109 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 3.14 (m, 1H, C≡CH), 4.02 (m, 2H, NHCH2),
5.22 (s, 2H, OCH2), 5.37 (s, 2H, OCH2), 7.35–7.66 (m, 9H, Ar-H), 8.31–8.33 (m,
2H, Ar-H), 8.67 (s, 1H, C(N)H), 8.93–8.94 (m, 1H, NHCH2), 10.08 (s, 1H,
OC(O)NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.8 (NHCH2), 65.9, 68.3 (CH2O), 73.4
(C≡CH), 81.3 (C≡CH), 111.5 (m, Ar-C), 117.6, 119.6, 121.6, 123.0, 125.8, 126.7,
129.7, 130.8, 135.9, 136.9 (Ar-C), 137.8 (m, CF), 139.8 (Ar-C), 142.3 (m, CF),
145.5 (Ar-C), 146.0 (m, CF), 150.1 (m, C(N)H), 151.0, 152.2, 153.7, 155.7, 168.1
(Ar-C, C(O)NH, OC(O)O).
ESI-MS (5 kV, MeCN): m/z (%) = 697 (46) [M + H]+, 514 (100) [M – OC(O)ONp]+.

Experimenteller Teil _________________________________________________________________
- 133 -
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzyl-N-4-((doxorubicin-4’-yl-carbamato)methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (38)
Zu einer Lösung von 80.0 mg (147 μmol) Doxorubicin-Hydrochlorid und 27.7 μL
(20.9 mg, 162 μmol) DIEA in 7 mL DMF werden nach 5 min Rühren bei RT
100 mg (147 μmol) 36 in 3 mL DMF gegeben. Nach 26 h Rühren bei RT wird das
Lösungsmittel unter vermindertem Druck entfernt. Der Rückstand wird in 50 mL
Ethylacetat aufgenommen und die unlöslichen Bestandteile abfiltriert. Nach
Einengen der Lösung unter vermindertem Druck auf ca. 30 mL wird das Produkt
durch Zugabe von 50 mL iso-Hexan bei – 20 °C kristallisiert. Wiederholte
Kristallisation und anschließendes Trocknen des roten Feststoffs im Hochvakuum
liefert 38 in einer Ausbeute von 114 mg (72 % d. Theorie).
NH
HN
O
O
O
O
O
N
HFF
FF
O
O
O
H3CO
HO
OH
O
O
NH
HOCH3
OHOH
C54H46F4N4O16
Smp.: 130 °C (unter Zersetzung)
ESI-MS (5 kV, MeCN): m/z (%) = 1081 (100) [M – H]–.
4-(Pentafluorbenzylidenamino)benzyl-N-4-((doxorubicin-4’-yl-carbamato)-methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (39)
Zu einer Lösung von 19.5 mg (35.9 μmol) Doxorubicin-Hydrochlorid und 12.3 μL
(9.28 mg, 71.8 μmol) DIEA in 4 mL DMF werden nach 5 min Rühren bei RT 25 mg
(35.9 μmol) 37 in 2 mL DMF gegeben. Nach 4.5 h Rühren bei RT wird das
Lösungsmittel unter vermindertem Druck entfernt. Der Rückstand wird in 25 mL

Experimenteller Teil _________________________________________________________________
- 134 -
Ethylacetat aufgenommen und die unlöslichen Bestandteile abfiltriert. Nach
Einengen der Lösung unter vermindertem Druck auf ca. 10 mL wird das Produkt
durch Zugabe von 4 mL iso-Hexan bei – 20 °C kristallisiert. Trocknen des roten
Feststoffs im Hochvakuum liefert 39 in einer Ausbeute von 24 mg (61 % d.
Theorie).
NH
HN
O
O
O
O
O
N
HFF
FF
F
O
O
O
H3CO
HO
OH
O
O
NH
HOCH3
OHOH
C54H45F5N4O16
Smp.: 119 °C (unter Zersetzung)
ESI-MS (5 kV, MeCN): m/z (%) = 1123 (100) [M + Na]+.
4-(2,3,5,6-Tetrafluorbenzylidenamino)benzyl-N-4-((paclitaxel-2’-yl-carbonato)-methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (40)
Zu einer Lösung von 41.9 mg (49.1 μmol) Paclitaxel und 8.41 μL (6.35 mg,
49.1 μmol) DIEA in 3 mL DCM werden nach 5 min Rühren bei RT 50 mg
(73.7 μmol) 36 und 9.00 mg (73.7 μmol) DMAP in 2 mL DCM gegeben. Nach 4.5 h
Rühren bei RT wird das Lösungsmittel unter vermindertem Druck entfernt. Der
Rückstand wird in 5 mL Ethylacetat aufgenommen. Durch Zugabe von 10 mL
iso-Hexan bei – 20 °C wird das Produkt kristallisiert. Wiederholte Kristallisation
und anschließendes Trocknen des gelben Feststoffs im Hochvakuum liefert 40 in
einer Ausbeute von 51 mg (75 % d. Theorie).

Experimenteller Teil _________________________________________________________________
- 135 -
NH
HN
O
O
O
O
O
N
HFF
FF
O
OO
O
OH
OO
O
HO
HN
OO
O
O
O
C74H68F4N4O19
Smp.: 131 °C (unter Zersetzung)
ESI-MS (5 kV, MeCN): m/z (%) = 1393 (100) [M + H]+.
4-(Pentafluorbenzylidenamino)benzyl-N-4-((paclitaxel-2’-yl-carbonato)-methyl)-3-(prop-2-ynylcarbamoyl)phenylcarbamat (41)
Zu einer Lösung von 40.9 mg (47.9 μmol) Paclitaxel und 8.20 μL (6.19 mg,
47.9 μmol) DIEA in 3 mL DCM werden nach 5 min Rühren bei RT 50 mg
(71.8 μmol) 37 und 8.77 mg (71.8 μmol) DMAP in 2 mL DCM gegeben. Nach 4 h
Rühren bei RT wird das Lösungsmittel unter vermindertem Druck entfernt. Der
Rückstand wird in 7 mL Ethylacetat aufgenommen und die unlöslichen
Bestandteile abfiltriert. Durch Zugabe von 20 mL iso-Hexan bei – 20 °C wird das
Produkt kristallisiert. Wiederholte Kristallisation und anschließendes Trocknen des
hellgelben Feststoffs im Hochvakuum liefert 41 in einer Ausbeute von 27 mg
(40 % d. Theorie).
NH
HN
O
O
O
O
O
N
HFF
FF
F
O
OO
O
OH
OO
O
HO
HN
OO
O
O
O

Experimenteller Teil _________________________________________________________________
- 136 -
C74H67F5N4O19
Smp.: 129 °C (unter Zersetzung)
ESI-MS (5 kV, MeCN): m/z (%) = 1411 (100) [M + H]+.
6.2.5 PEG-Synthesen am Beispiel von PEG2k
PEG2k-Dimesylat (42) Zu einer Lösung von 5.00 g (2.50 mmol) PEG2k in 40 mL DCM werden im Eisbad
innerhalb von 1 min 693 μL (506 mg, 5.00 mmol) Triethylamin zugetropft. Nach
10 min Rühren bei 0 °C werden 580 μL (859 mg, 7.50 mmol) Mesylchlorid in
10 mL DCM innerhalb von 5 min zugegeben. Das Reaktionsgemisch wird für
weitere 10 h gerührt, wobei es langsam auf RT auftaut. Nach beendeter Reaktion
wird das Produkt mit 500 mL Et2O gefällt, abfiltriert, in 15 mL MeOH gelöst und
erneut mit 500 mL Et2O gefällt. Der Feststoff wird abfiltriert und mit n-Pentan
gewaschen. Nach Trocknung des farblosen Feststoffs im Hochvakuum wird 42 in
einer Ausbeute von 4.97 g (92 % d. Theorie) erhalten.
MsOO
OMsn
Aktivierung von Natriumazid für die Synthese von 43164 Zu einer Lösung von 15.9 g (245 mmol) Natriumazid in 50 mL Wasser werden
2.37 mL (2.45 g, 49.0 mmol) Hydrazin-Hydrat gegeben. Nach 15 min Rühren bei
RT wird die Lösung filtriert und das Salz mit 1 L Aceton gefällt. Nach Filtration und
Trocknung des farblosen Feststoffs im Hochvakuum wird das aktivierte
Natriumazid direkt bei der Synthese von 43 eingesetzt.
PEG2k-Diazid (43) Zu einer Lösung von 4.97 g (2.31 mmol) 42 in 80 mL DMF werden 1.50 g
(23.1 mmol) aktiviertes Natriumazid gegeben. Die dabei entstandene Suspension
wird unter Rühren für 18.5 h im Ölbad auf 70 °C erhitzt. Nach beendeter Reaktion
wird die Reaktionsmischung heiß filtriert und das Lösungsmittel unter

Experimenteller Teil _________________________________________________________________
- 137 -
vermindertem Druck entfernt. Das Produkt wird mit 250 mL 2-Propanol in der
Siedehitze umkristallisiert, abfiltriert und anschließend mit 15 mL 2-Propanol und
20 mL n-Pentan gewaschen. Nach Trocknung des farblosen Feststoffs im
Hochvakuum wird 43 in einer Ausbeute von 3.71 g (77 % d. Theorie) erhalten.
N3O
OnN3
a
ab
b
c
d
siehe Abb. 3.24, S. 55 1H-NMR (400.1 MHz, DMSO-d6): δ = 3.39 (t, J = 5.0 Hz, 4H, CH2N), 3.51 (br s,
176H, OCH2CH2O), 3.61 (t, J = 5.0 Hz, 4H, CH2CH2N).
13C-NMR (100.6 MHz, DMSO-d6): δ = 50.0 (CH2N), 69.2 (CH2CH2N), 69.6
(CH2OCH2CH2N), 69.7 (OCH2CH2O).
Durch Integration konnte das Verhältnis der Protonen c und d zu den vier
Protonen a in Nachbarstellung des Azidsubstituenten bestimmt werden. Daraus
ergab sich für 43 eine Beladung von ungefähr 94 %.
PEG2k-Diamin (44)
Zu einer Lösung von 653 mg (314 μmol) 43 in 75 mL EtOH werden 900 mg
Palladium auf Aktivkohle (10 % Pd) gegeben. Unter einer Wasserstoffatmosphäre
wird für fünf Tage bei RT gerührt. Nach beendeter Reaktion wird der Katalysator
über einen Glasfasermikrofilter abfiltriert. Das Lösungsmittel wird unter
vermindertem Druck entfernt und der Rückstand anschließend in 20 mL MeOH
aufgenommen. Nach erneuter Filtration wird die organische Phase unter
vermindertem Druck auf ein Volumen von ca. 5 mL eingeengt und das Produkt mit
75 mL Et2O gefällt. Nach Filtration, Waschen mit 10 mL n-Pentan und Trocknung
des farblosen Feststoffs im Hochvakuum wird 44 in einer Ausbeute von 358 mg
(60 % d. Theorie) erhalten.
H2NO
NH2n

Experimenteller Teil _________________________________________________________________
- 138 -
6.2.6 Synthese niedermolekularer Vorstufen zur Darstellung von AMC-Modellverbindungen über zur CuAAC alternative Reaktionen
1-Maleinimidopentan-5-amin-Hydrochlorid (47) Zu einer Lösung von 15 g (44.2 mmol) 6-Maleinimidocapronsäurehydrazid-
Trifluoracetat in 100 mL HCl (1M) werden im Eisbad 4.57 g (66.3 mmol)
Natriumnitrit in 20 mL Wasser gegeben. Nach 1.5 h Rühren bei 0 °C wird das
entstandene Azid dreimal mit 200 mL DCM extrahiert und die vereinigten
organischen Phasen anschließend über Natriumsulfat getrocknet. Nach Entfernen
des Lösungsmittels unter vermindertem Druck wird der Rückstand in 10 mL
Dioxan aufgenommen und unter starkem Rühren innerhalb von 5 min zu 300 mL
siedendem Wasser mit 3.68 mL (44.2 mmol) konz. HCl getropft. Nach beendeter
Zugabe wird das Reaktionsgemisch für weitere 2 min zum Sieden erhitzt, filtriert
und das Wasser durch lyophylisieren entfernt. 47 wird als beigefarbener Feststoff
in einer Ausbeute von 8.75 g (90 % d. Theorie) erhalten.
+H3N N
O
O
Cl-
Smp.: 134–136 °C
1H-NMR (400.1 MHz, D2O): δ = 1.34–1.37 (m, 2H, CH2), 1.57–1.69 (m, 4H, CH2),
2.97 (t, J = 7.6 Hz, 2H, CH2), 3.51 (t, J = 7.0 Hz, 2H, CH2), 6.82 (s, 2H, CH).
13C-NMR (100.6 MHz, D2O): δ = 22.7, 26.1, 27.0, 37.1, 39.2 (CH2), 134.2 (CH),
173.3 (C(O)).
CI-MS (130 eV, 300 μA, NH3): m/z (%) = 183 (100) [M + H]+.
Elementaranalyse: C9H15ClN2O2 [218.68]
berechnet: C: 49.43 % H: 6.91 % N: 12.81 %
gefunden: C: 49.13 % H: 6.95 % N: 12.52 %

Experimenteller Teil _________________________________________________________________
- 139 -
Phenyl-N-4-((4-methylcumarin-7-yl-carbamoyloxy)methyl)-3-(2-(6-malein-imidohexanoyl)hydrazincarbonyl)phenylcarbamat (48) Zu einer Suspension von 300 mg (597 μmol) 25 in 20 mL DMF werden 368 mg
(1.19 mmol) 6-Maleinimidohexanoyl-N-hydroxysuccinimidester und 102 μL
(597 μmol) DIEA gegeben. Nach 24 h Rühren bei RT wird die Lösung zur
Trockene eingeengt und der Rückstand anschließend mittels Flash-
Säulenchromatographie gereinigt (Kieselgel, Chloroform/MeOH 40:1 (v/v)). Nach
Entfernen der Lösungsmittel unter vermindertem Druck und Trocknung des
farblosen Feststoffs im Hochvakuum wird 48 in einer Ausbeute von 186 mg (45 %
d. Theorie) erhalten.
NH
HN
ONH
O
O
O
O
NH
O O
ON
O
O
C36H33N5O10
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.21–1.29 (m, 2H, CH2), 1.48–1.54 (m, 4H,
CH2), 2.12 (t, J = 7.80 Hz, 2H, CH2), 2.38 (s, 3H, CH3), 3.38 (t, J = 7.21 Hz, 2H,
CH2), 5.28 (s, 2H, OCH2), 6.23 (s, 1H, CH3CCH), 6.99 (s, 2H, NC(O)CH), 7.22–
7.31 (m, 3H, Ar-H), 7.40–7.46 (m, 3H, Ar-H), 7.51–7.56 (m, 2H, Ar-H), 7.63–7.71
(m, 3H, Ar-H ), 9.96 (s, 1H, NH), 10.18 (s, 1H, NH), 10.24 (s, 1H, NH), 10.49 (s,
1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 18.4 (CH3), 24.9, 26.1, 28.1, 33.4, 37.3
(CH2), 64.0 (OCH2), 104.8, 112.3, 114.6, 114.8, 115.6 (CH3CCH, Ar-C), 122.3,
126.0, 126.4, 129.0, 129.8, 129.9, 130.2, 134.8, 135.4, 138.8, 143.1, 150.8, 152.1,
153.4, 153.6, 154.2, 160.4, 167.5, 171.5, 172.0 (Ar-C, C(O)NH, CH3CCH,
C(O)CH, NC(O)CH, NC(O)CH).

Experimenteller Teil _________________________________________________________________
- 140 -
6.2.7 Synthese der makromolekularen Modellverbindungen 53–55 Da für 13C-NMR-Spektren von PEG`s mit hoher Molmasse große
Substanzmengen und lange Messzeiten erforderlich sind, wurde bei den
folgenden Verbindungen auf die Aufnahme solcher Spektren verzichtet.
Neutralisierung von mPEG5k Zu einer Lösung von 25 g (5 mmol) mPEG5k in 100 mL DCM werden 1 g
Amberlyst 15 gegeben. Nach 18 h Rühren bei RT wird filtriert und das PEG
anschließend in 2 L Et2O gefällt. Zur Vervollständigung der Fällung wird die
Suspension für 30 min bei 5 °C gelagert. Der farblose Feststoff wird im Anschluss
abfiltriert, mit Et2O und n-Pentan gewaschen und im Hochvakuum getrocknet. Es
werden 24.3 g (97 % d. Theorie) neutralisiertes mPEG5k erhalten.
6-Hexylisocyanato-1-mPEG5k-carbamat (49) Eine Lösung von 10 g (2 mmol) des zuvor aktivierten mPEG5k in 100 mL Benzol
wird für eine Stunde am Wasserabscheider erhitzt. Nach Abkühlen auf RT werden
zur Lösung 3.20 mL (20 mmol) 1,6-Hexyldiisocyanat und 50 μL DBTL in 10 mL
Benzol innerhalb von 1 h tropfenweise unter starkem Rühren zugesetzt. Nach 3 h
Rühren bei RT wird das PEG in 1 L abs. Et2O gefällt und unter Schutzgas
abfiltriert. Zur weiteren Aufreinigung wird der Feststoff in 20 mL DCM gelöst und
die Fällung wiederholt. Nach Waschen mit Et2O und n-Pentan sowie Trocknen des
farblosen Feststoffs im Hochvakuum wird 49 in einer Ausbeute von 9.80 g (98 %
d. Theorie) erhalten.
OO
O NH
NO
COn
tert.-Butyl-2-[4-(hydroxymethyl)-2-((4-methylcumarin-7-yl-carbamoyloxy)methyl)benzoyl]hydrazincarboxylat (50) Diese Verbindung war innerhalb der Arbeitsgruppe vorrätig und wurde von einer
Mitarbeiterin in einer 8-stufigen Synthese ausgehend von 2,4-Dimethylbenzoe-
säure dargestellt.166

Experimenteller Teil _________________________________________________________________
- 141 -
O ONH
O
O
OHHN
ONH
O
O
mPEG5k-OC(O)N-Hexyl-[4-(2-(tert.-butoxycarbonyl)hydrazincarbonyl)-3-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)]benzylcarbamat (51)
Zu einer Lösung von 2.54 g (489 μmol) 49 und 364 mg (734 μmol) 50 in 30mL
Dioxan werden 250 mg zerstoßenes Molekularsieb 4 Å gegeben. Nach 30 min
Rühren bei RT werden 245 μL (24.5 μmol) einer 100 mM Lösung von DBTL in
Dioxan zugetropft. Das Reaktionsgemisch wird für 3 h auf 80 °C erhitzt und
anschließend über Celite filtriert, wobei zweimal mit heißem Dioxan gewaschen
wird. Nach Lyophilisieren wird der Rückstand in 38 mL Toluol aufgenommen und
die erhaltene Lösung erneut über Celite filtriert. Das Produkt wird mit 280 mL Et2O
gefällt und die Suspension für 1 h bei RT gerührt. Zur weiteren Aufreinigung wird
der Feststoff in 5 mL DCM gelöst und die Fällung mit 200 mL Et2O wiederholt.
Nach Waschen mit Et2O, n-Pentan und PE 30-50 sowie Trocknen des farblosen
Feststoffs im Hochvakuum wird 51 in einer Ausbeute von 1.91 g (68 % d. Theorie)
erhalten.
NH
O
O
O
HN
ONH
O
O
O O
NH
OHNO
OO
On
mPEG5k-OC(O)N-Hexyl-[4-(azidocarbonyl)-3-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)]benzylcarbamat (52)
Eine Lösung von 1.80 g (316 μmol) 51 in 11 mL HCl (5M) wird für 30 min bei RT
gerührt. Anschließend wird der Ansatz mit 44 mL Wasser verdünnt und auf 0 °C
abgekühlt. Nach Zugabe einer Lösung von 87.0 mg (1.26 mmol) Natriumnitrit in
1 mL Wasser wird das Reaktionsgemisch für weitere 30 min im Eisbad gerührt.
Schließlich werden 50 mL einer gesättigten Natriumchloridlösung zugegeben und
das Produkt mit fünfmal je 20 mL DCM extrahiert. Nach Trocknen der organischen
Phase über Natriumsulfat wird das Lösungsmittel unter vermindertem Druck

Experimenteller Teil _________________________________________________________________
- 142 -
entfernt und der Rückstand mit 180 mL Et2O gefällt. Zur Vervollständigung der
Fällung wird die Suspension für 10 min bei 5 °C gelagert. Der farblose Feststoff
wird im Anschluss abfiltriert, mit Et2O und n-Pentan gewaschen und im
Hochvakuum getrocknet. 52 wird in einer Ausbeute von 1.63 g (92 % d. Theorie)
erhalten.
NH
O
O
O
N3
O
HNO
OO
O
O O
NH
O
n
mPEG5k-OC(O)N-Hexyl-[4-(4-(2,3,5,6-tetrafluorbenzylidenamino)benzyl-carbamat)-3-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)]benzylcarbamat (53)
Zu einer Lösung von 285 mg (50.9 μmol) 52 und 28.8 mg (102 μmol) 13a in
2.5 mL Dioxan werden 100 mg zerstoßenes Molekularsieb 4 Å und 25 μL
(2.5 μmol) einer 100 mM Lösung von DBTL in Dioxan gegeben. Nach 30 min
Rühren bei RT wird das Reaktionsgemisch für 3 h auf 80 °C erhitzt und
anschließend über Celite filtriert. Das Lösungsmittel wird unter vermindertem
Druck entfernt und der Rückstand in 4 mL Toluol aufgenommen. Nach Filtration
über Celite und Waschen mit 1 mL Toluol wird das Produkt in 28 mL Et2O gefällt.
Zur Vervollständigung der Fällung wird die Suspension für 30 min bei 5 °C
gelagert. Der farblose Feststoff wird im Anschluss abfiltriert, mit Et2O und n-
Pentan gewaschen und im Hochvakuum getrocknet. 53 wird in einer Ausbeute von
63 mg (21 % d. Theorie) erhalten.
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
H
NH
O
n
O
NH
O O

Experimenteller Teil _________________________________________________________________
- 143 -
siehe Abb. 3.35, S. 67: 1H-NMR (400.1 MHz, DMSO-d6): δ = 1.21–1.23 (br m, CH2), 1.33–1.40 (br m,
CH2), 2.36 (s, CH3), 2.90–2.97 (m, CH2), 3.24 (s, OCH3), 3.44–3.66 (br m, CH2),
4.02–4.04 (m, CH2), 4.98 (s, CH2), 5.19–5.21 (m, CH2), 6.19 (s, CH3CCH), 7.16–
7.69 (m, Ar-H, NH), 8.05–8.14 (m, Ar-H), 8.61 (s, C(N)H), 9.34 (br s, NH), 10.30
(br s, NH).
Durch Integration konnte das Verhältnis der Protonen am PEG zu den zwölf
Protonen im Bereich von 7.16–7.69 ppm (zehn aromatische Protonen und zwei
NH) bestimmt werden. Daraus ergab sich für 53 eine Beladung von 61 %.
mPEG5k-OC(O)N-Hexyl-[4-(4-(pentafluorbenzylidenamino)benzylcarbamat)-3-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)]benzylcarbamat (54)
Zu einer Lösung von 283 mg (50.5 μmol) 52 und 30.4 mg (101 μmol) 14a in
2.5 mL Dioxan werden 100 mg zerstoßenes Molekularsieb 4 Å und 25 μL
(2.5 μmol) einer 100 mM Lösung von DBTL in Dioxan gegeben. Nach 30 min
Rühren bei RT wird das Reaktionsgemisch für 3 h auf 80 °C erhitzt und
anschließend über Celite filtriert. Das Lösungsmittel wird unter vermindertem
Druck entfernt und der Rückstand in 4 mL Toluol aufgenommen. Nach Filtration
über Celite und Waschen mit 1 mL Toluol wird das Produkt in 28 mL Et2O gefällt.
Zur Vervollständigung der Fällung wird die Suspension für 30 min bei 5 °C
gelagert. Der farblose Feststoff wird im Anschluss abfiltriert, mit Et2O und n-
Pentan gewaschen und im Hochvakuum getrocknet. 54 wird in einer Ausbeute von
103 mg (35 % d. Theorie) erhalten.
NH
O
O
O
N
HFF
FF
O
HNO
OO
O
F
NH
O
n
O
NH
O O

Experimenteller Teil _________________________________________________________________
- 144 -
siehe Abb. 3.35, S. 67: 1H-NMR (400.1 MHz, DMSO-d6): δ = 1.21–1.23 (br m, CH2), 1.32–1.39 (br m,
CH2), 2.37 (s, CH3), 2.90–2.95 (m, CH2), 3.24 (s, OCH3), 3.41–3.67 (br m, CH2),
4.00–4.05 (m, CH2), 4.97 (s, CH2), 5.19–5.21 (m, CH2), 6.21 (s, CH3CCH), 7.16–
7.69 (m, Ar-H, NH), 8.59 (m, C(N)H), 9.34 (br s, NH), 10.30 (br s, NH).
Durch Integration konnte das Verhältnis der Protonen am PEG zu den zwölf
Protonen im Bereich von 7.16–7.69 ppm (zehn aromatische Protonen und zwei
NH) bestimmt werden. Daraus ergab sich für 54 eine Beladung von 69 %.
mPEG5k-OC(O)N-Hexyl-[4-(methylcarbamat)-3-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)]benzylcarbamat (55)
Zu einer Lösung von 152 mg (27.1 μmol) 52 in 3 mL MeOH werden 50 mg
zerstoßenes Molekularsieb 3 Å und 14 μL (1.4 μmol) einer 100 mM Lösung von
DBTL in Dioxan gegeben. Nach 10 min Rühren bei RT wird das Reaktionsgemisch
für 3 h unter Reflux erhitzt und anschließend über Celite filtriert. Das Produkt wird
in 15 mL Et2O gefällt. Zur Vervollständigung der Fällung wird die Suspension für
30 min bei – 20 °C gelagert. Der farblose Feststoff wird im Anschluss abfiltriert, mit
Et2O und n-Pentan gewaschen und im Hochvakuum getrocknet. 55 wird in einer
Ausbeute von 111 mg (73 % d. Theorie) erhalten.
NH
O
O
O
O
NH
OHNO
OO
On
O
NH
O O
siehe Abb. 3.37, S. 69: 1H-NMR (400.1 MHz, DMSO-d6): δ = 1.21–1.23 (br m, CH2), 1.33–1.40 (br m,
CH2), 2.36 (s, CH3), 2.90–2.97 (m, CH2), 3.24 (s, CH2OCH3), 3.44–3.66 (br m,
CH2, NC(O)OCH3), 4.02–4.04 (m, CH2), 4.96 (s, CH2), 5.19 (s, CH2), 6.19 (s,
CH3CCH), 7.13–7.68 (m, Ar-H, NH), 9.18 (br s, NH), 10.25 (br s, NH).

Experimenteller Teil _________________________________________________________________
- 145 -
Durch Integration konnte das Verhältnis der Protonen am PEG zu den drei
Protonen im Bereich von 7.16–7.69 ppm bestimmt werden. Daraus ergab sich für
55 eine Beladung von 57 %.
6.2.8 Synthese der niedermolekularen Modellverbindungen 56 und 57
tert.-Butyl-N-2-(hydroxymethyl)phenylcarbamat (58) Zu einer Lösung von 500 mg (4.06 mmol) 2-Aminobenzylalkohol in 10 mL DCM
werden 2.19 mL (10.2 mmol) (Boc)2O gegeben. Nach 24 h Rühren bei RT wird
das Lösungsmittel unter vermindertem Druck entfernt und das Rohprodukt mittels
Flash-Säulenchromatographie gereinigt (Kieselgel, iso-Hexan/Ethylacetat 4:1
(v/v)). Nach Trocknen des farblosen Öls im Hochvakuum wird 58 in einer
Ausbeute von 824 mg (91 % d. Theorie) erhalten.
NH
O
O
OH
C12H17NO3
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.46 (s, 9H, CH3), 4.50 (d, J = 5.2 Hz, 2H,
CH2), 5.44 (t, J = 5.2 Hz, 1H, OH), 7.03–7.07 (m, 1H, Ar-H), 7.19–7.23 (m, 1H, Ar-
H), 7.29–7.31 (m, 1H, Ar-H), 7.56–7.58 (m, 1H, Ar-H), 8.52 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 28.0 (CH3), 60.7 (CH2), 79.0 (C(CH3)3),
121.8, 123.3, 127.2, 127.4, 132.7, 136.3 (Ar-C), 152.9 (C(O)NH).
APCI-MS (5 μA, MeOH): m/z (%) = 222 (100) [M – H]–.
2-(tert.-Butoxycarbonylamino)benzyl-N-4-methylcumarin-7-yl-carbamat (59)
Zu einer Lösung von 102 mg (507 μmol) AMC-NCO (3) in 12 mL THF werden
zerstoßenes Molekularsieb 4 Å (20 mg), 103 mg (461 μmol) 58 und 231 μL

Experimenteller Teil _________________________________________________________________
- 146 -
(23.1 μmol) einer 100 mM Lösung von DBTL in Dioxan gegeben. Nach 4.5 h
Rühren bei RT wird das Molekularsieb über Celite abfiltriert und das Lösungsmittel
im Wasserbad bei 30 °C unter vermindertem Druck entfernt. Das Rohprodukt wird
mittels Flash-Säulenchromatographie gereinigt (Kieselgel, Chloroform/Ethylacetat
8:1 (v/v)). Nach Trocknen des farblosen Feststoffs im Hochvakuum wird 59 in
einer Ausbeute von 169 mg (87 % d. Theorie) erhalten.
NH
O
O
O
O
NH
O O
Smp.: 189–191 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 1.45 (s, 9H, OC(CH3)3), 2.38 (s, 3H,
CH3CCH), 5.20 (s, 2H, CH2), 6.23 (s, 1H, CH3CCH), 7.16–7.20 (m, 1H, Ar-H),
7.29–7.34 (m, 1H, Ar-H), 7.39–7.44 (m, 3H, Ar-H), 7.56 (s, 1H, Ar-H), 7.67–7.70
(m, 1H, Ar-H), 8.82 (br s, 1H, NH), 10.29 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 17.9 (CH3CCH), 28.0 (OC(CH3)3), 62.8
(CH2), 79.0 (OC(CH3)3), 104.3, 111.8, 114.1, 114.2 (CH3CCH, Ar-C), 124.5, 124.7,
125.9, 128.4, 129.0, 129.3, 136.3, 142.6, 153.05, 153.11, 153.5, 153.7, 159.9
(C(O)NH, CH3CCH, C(O)CH, Ar-C).
Elementaranalyse: C23H24N2O6 [424.45]
berechnet: C: 65.08 % H: 5.70 % N: 6.60 %
gefunden: C: 65.43 % H: 6.02 % N: 6.19 %
2-(Amino)benzyl-N-4-methylcumarin-7-yl-carbamat-Trifluoracetat (56)
Zu einer Lösung von 30 mg (70.7 μmol) 59 in 1.5 mL DCM werden 1.5 mL TFA
gegeben. Die Reaktionslösung wird 5 min bei RT gerührt und das Produkt
anschließend mit 10 mL Et2O (abs.) gefällt. Nach Waschen des farblosen
Feststoffs mit n-Pentan (abs.) und Trocknen im Hochvakuum wird 56 in einer
Ausbeute von 21 mg (68 % d. Theorie) erhalten.

Experimenteller Teil _________________________________________________________________
- 147 -
+H3N
O
O
NH
O OCF3COO-
C20H17F3N2O6
APCI-MS (5 μA, MeCN): m/z (%) = 323 (100) [M – H]–.
tert.-Butyl-N-[2-((tert.-butyldimethylsilyloxy)methyl)-4-(hydroxymethyl)]-phenylcarbamat (60) Zu einer Lösung von 2.50 g (9.35 mmol) 2-((tert.-Butyldimethylsilyloxy)methyl)-4-
(hydroxymethyl)anilin*166 in 25 mL DCM werden 5 mL (23.4 mmol) (Boc)2O
gegeben. Nach 40 h Rühren bei RT wird das Lösungsmittel unter vermindertem
Druck entfernt und das Rohprodukt mittels Flash-Säulenchromatographie gereinigt
(Kieselgel, iso-Hexan/Ethylacetat 3:1 (v/v)). Nach Trocknen des farblosen
Feststoffs im Hochvakuum wird 60 in einer Ausbeute von 3.42 g (99 % d. Theorie)
erhalten.
NH
O
O
OTBS
OH
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.08 (s, 6H, SiCH3), 0.90 (s, 9H, SiC(CH3)3),
1.44 (s, 9H, OC(CH3)3), 4.43 (d, J = 5.6 Hz, 2H, CH2), 4.69 (s, 2H, CH2), 5.14 (t,
J = 5.6 Hz, 1H, OH), 7.15–7.17 (m, 1H, Ar-H), 7.26–7.27 (m, 1H, Ar-H), 7.46–7.48
(m, 1H, Ar-H), 8.41 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -5.5, 17.8, 25.7, 28.0 (SiCH3, SiC(CH3)3,
SiC(CH3)3, OC(CH3)3), 62.4, 62.6 (CH2), 79.0 (OC(CH3)3), 121.9, 125.4, 125.6,
131.5, 134.6, 137.7 (Ar-C), 152.9 (C(O)NH).
* Diese Verbindung war innerhalb der Arbeitsgruppe vorrätig und wurde von einer Mitarbeiterin in einer 6-stufigen Synthese ausgehend von 3-Methyl-4-nitrobenzoesäureethylester dargestellt.

Experimenteller Teil _________________________________________________________________
- 148 -
Elementaranalyse: C19H33NO4Si [367.56]
berechnet: C: 62.09 % H: 9.05 % N: 3.81 %
gefunden: C: 62.16 % H: 9.11 % N: 3.47 %
tert.-Butyl-N-[2-((tert.-butyldimethylsilyloxy)methyl)-4-(hydroxymethyl-N-propylcarbamat)]phenylcarbamat (61) Zu einer Suspension von 400 mg (1.09 mmol) 60 in 5 mL THF werden
zerstoßenes Molekularsieb 4 Å (20 mg) und 544 μL (54.4 μmol) einer 100 mM
Lösung von DBTL in Dioxan gegeben. Nach 30 min Rühren bei RT werden 164 μL
(1.74 mmol) Propylisocyanat zugetropft. Die nach 26 h Rühren bei RT
entstandene Lösung wird über Celite filtriert, und das Lösungsmittel wird
anschließend unter vermindertem Druck entfernt. Das Rohprodukt wird mittels
Flash-Säulenchromatographie gereinigt (Kieselgel, iso-Hexan/Ethylacetat 7:1
(v/v)). Nach Trocknen des farblosen Feststoffs im Hochvakuum wird 61 in einer
Ausbeute von 483 mg (98 % d. Theorie) erhalten.
NH
O
O
OTBS
O
O
NH
C23H40N2O5Si
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.08 (s, 6H, SiCH3), 0.81–0.86 (m, 3H,
CH2CH2CH3), 0.91 (s, 9H, SiC(CH3)3), 1.37–1.43 (m, 2H, CH2CH2CH3), 1.45 (s,
9H, OC(CH3)3), 2.91–2.96 (m, 2H, CH2CH2CH3), 4.71 (s, 2H, OCH2), 4.95 (s, 2H,
OCH2), 7.18–7.22 (m, 2H, Ar-H, NH), 7.30–7.31 (m, 1H, Ar-H), 7.51–7.53 (m, 1H,
Ar-H), 8.48 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = -5.6, 11.1, 17.7, 22.5, 25.6, 27.9 (SiCH3,
SiC(CH3)3, SiC(CH3)3, CH2CH2CH3, CH2CH2CH3, OC(CH3)3), 41.9, 62.1, 64.7
(OCH2, CH2CH2CH3), 79.0 (OC(CH3)3), 121.8, 126.6, 127.0, 131.6, 132.4, 135.5
(Ar-C), 152.7, 156.0 (C(O)NH).

Experimenteller Teil _________________________________________________________________
- 149 -
tert.-Butyl-N-[2-(hydroxymethyl)-4-((hydroxymethyl)-N-propylcarbamat)]-phenylcarbamat (62) Eine Lösung von 2.46 g (5.44 mmol) 61 in 90 mL eines 50:1-Gemisches aus
MeOH und HCl (1M) wird 22 h bei RT gerührt. Es werden 250 mL Wasser
zugegeben und das Produkt dreimal mit je 100 mL DCM extrahiert. Die
organischen Phasen werden anschließend dreimal mit je 100 mL einer gesättigten
Natriumchloridlösung gewaschen und über Natriumsulfat getrocknet. Kristallisation
aus einem Gemisch von Ethylacetat und iso-Hexan bei – 20 °C, anschließendes
Waschen mit n-Pentan und Trocknen des farblosen Feststoffs im Hochvakuum
liefert 62 in einer Ausbeute von 1.45 g (79 % d. Theorie).
NH
O
O
OH
O
O
NH
Smp.: 68–70 °C
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.80–0.84 (m, 3H, CH2CH2CH3), 1.37–1.42
(m, 2H, CH2CH2CH3), 1.45 (s, 9H, OC(CH3)3), 2.91–2.96 (m, 2H, CH2CH2CH3),
4.49 (d, J = 5.3 Hz, 2H, OCH2), 4.94 (s, 2H, OCH2), 5.47 (t, J = 5.3 Hz, 1H, OH),
7.17–7.20 (m, 2H, Ar-H, NH), 7.29–7.30 (m, 1H, Ar-H), 7.53–7.55 (m, 1H, Ar-H),
8.53 (br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 11.2, 22.6, 28.0 (CH2CH2CH3, CH2CH2CH3,
OC(CH3)3), 42.0, 60.6, 64.9 (OCH2, CH2CH2CH3), 79.2 (OC(CH3)3), 121.7, 126.9,
127.2, 132.1, 132.7, 135.9 (Ar-C), 152.9, 156.1 (C(O)NH).
Elementaranalyse: C17H26N2O5 [338.40]
berechnet: C: 60.34 % H: 7.74 % N: 8.28 %
gefunden: C: 60.09 % H: 8.01 % N: 8.07 %

Experimenteller Teil _________________________________________________________________
- 150 -
tert.-Butyl-N-[2-((hydroxymethyl)-N-4-methylcumarin-7-yl-carbamat)-4-((hydroxymethyl)-N-propylcarbamat)]phenylcarbamat (63) Zu einer Lösung von 400 mg (1.18 mmol) 62 in 5 mL THF werden zerstoßenes
Molekularsieb 4 Å (20 mg) und 590 μL (59.0 μmol) einer 100 mM Lösung von
DBTL in Dioxan gegeben. Nach 10 min Rühren bei RT werden 238 mg
(1.18 mmol) 3 zugesetzt. Zur entstandenen Suspension werden nach 19 h Rühren
bei RT 10 mL THF gegeben, und das Gemisch wird anschließend bis zur Lösung
leicht erwärmt. Das Molekularsieb wird über Celite abfiltriert und das
Lösungsmittel unter vermindertem Druck entfernt. Kristallisation aus einem
Gemisch von Ethylacetat und iso-Hexan bei – 20 °C, anschließendes Waschen
mit n-Pentan und Trocknen des farblosen Feststoffs im Hochvakuum liefert 63 in
einer Ausbeute von 598 mg (94 % d. Theorie).
NH
O
O
O
O
O
NH
O
NH
O
O
Smp.: 161 °C (unter Zersetzung)
1H-NMR (400.1 MHz, DMSO-d6): δ = 0.78–0.82 (m, 3H, CH2CH2CH3), 1.35–1.41
(m, 2H, CH2CH2CH3), 1.45 (s, 9H, OC(CH3)3), 2.39 (s, 3H, CH3CCH), 2.89–2.94
(m, 2H, CH2CH2CH3), 4.98 (s, 2H, OCH2), 5.19 (s, 2H, OCH2), 6.24 (s, 1H,
CH3CCH), 7.21–7.23 (m, 1H, NH), 7.28–7.30 (m, 1H, Ar-H), 7.41–7.42 (m, 3H, Ar-
H), 7.56–7.57 (m, 1H, Ar-H), 7.69–7.71 (m, 1H, Ar-H), 8.84 (br s, 1H, NH), 10.31
(br s, 1H, NH).
13C-NMR (100.6 MHz, DMSO-d6): δ = 11.2, 18.0, 22.6, 28.0 (CH3CCH,
CH2CH2CH3, CH2CH2CH3, OC(CH3)3), 42.0, 62.7, 64.7 (OCH2, CH2CH2CH3), 79.2
(OC(CH3)3), 104.4, 111.9, 114.2, 114.3 (CH3CCH, Ar-C), 124.7, 126.0, 128.1,

Experimenteller Teil _________________________________________________________________
- 151 -
128.7, 129.2, 133.5, 135.9 (Ar-C), 142.7, 153.17, 153.21, 153.6, 153.8, 156.0,
160.0 (C(O)NH, CH3CCH, C(O)CH, Ar-C).
ESI-MS (5 kV, MeCN): m/z (%) = 1101 (15) [2M + Na]+, 562 (100) [M + Na]+.
Elementaranalyse: C28H33N3O8 [539.58]
berechnet: C: 62.33 % H: 6.16 % N: 7.79 %
gefunden: C: 61.90 % H: 6.25 % N: 7.63 %
[2-Amino-5-((hydroxymethyl)-N-propylcarbamat)]benzyl-N-4-methylcumarin-7-yl-carbamat-Trifluoracetat (57)
Zu einer Lösung von 50 mg (92.7 μmol) 63 in 500 μL DCM werden 500 μL TFA
gegeben. Die Reaktionslösung wird 45 min bei RT gerührt und das Produkt
anschließend mit 5 mL Et2O (abs.) gefällt. Nach Waschen des gelben Feststoffs
mit n-Pentan (abs.) und Trocknen im Hochvakuum wird 57 in einer Ausbeute von
33 mg (64 % d. Theorie) erhalten.
+H3N
O
O
O
NH
O
NH
O
O
CF3COO-
C25H26F3N3O8
APCI-MS (5 μA, MeCN): m/z (%) = 396 (100) [X1 + H]–, 337 (30) [X2 + H]–.
H2N
O NH
O
O O
NH
X1
HN
O NH
O
O O
X2

Literatur _________________________________________________________________
- 152 -
7. Literatur 1. Robert Koch-Institut (Hrsg.) und die Gesellschaft der epidemiologischen
Krebsregister in Deutschland e. V. (Hrsg.): Krebs in Deutschland 2005/2006. Häufigkeiten und Trends. 7. Ausgabe. Berlin, 2010.
2. Heng, H. H.; Stevens, J. B.; Bremer, S. W.; Ye, K. J.; Liu, G.; Ye, C. J., J. Cell. Biochem. 2010, 109 (6), 1072-1084.
3. Brooks, S. A.; Lomax-Browne, H. J.; Carter, T. M.; Kinch, C. E.; Hall, D. M., Acta Histochem. 2010, 112 (1), 3-25.
4. Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G., Curr. Pharm. Des. 2010, 16 (1), 3-10.
5. Reichert, J. M., MAbs 2010, 2 (1), 84-100. 6. Shapira, S.; Lisiansky, V.; Arber, N.; Kraus, S., Expert Opin. Invest. Drugs
2010, 19 Suppl 1, S67-77. 7. Cuesta, A. M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Alvarez-Vallina, L.,
Trends Biotechnol. 2010, 28 (7), 355-362. 8. Riaz, W.; Hernandez-Ilizaliturri, F. J.; Czuczman, M. S., Immunol. Res.
2010, 46 (1-3), 192-205. 9. Agrawal, M.; Garg, R. J.; Cortes, J.; Quintas-Cardama, A., Curr. Hematol.
Malig. Rep. 2010, 5 (2), 70-80. 10. Stegmeier, F.; Warmuth, M.; Sellers, W. R.; Dorsch, M., Clin. Pharmacol.
Ther. 2010, 87 (5), 543-552. 11. Klyuchnikov, E.; Kroger, N.; Brummendorf, T. H.; Wiedemann, B.; Zander,
A. R.; Bacher, U., Biol. Blood Marrow Transplant. 2010, 16 (3), 301-310. 12. Jovanovic, J.; Ronneberg, J. A.; Tost, J.; Kristensen, V., Mol. Oncol. 2010,
4 (3), 242-254. 13. Albert, M.; Helin, K., Semin. Cell Dev. Biol. 2010, 21 (2), 209-220. 14. Spannhoff, A.; Sippl, W.; Jung, M., Int. J. Biochem. Cell Biol. 2009, 41 (1),
4-11. 15. Müller, S.; Kramer, O. H., Curr. Cancer Drug Targets 2010, 10 (2), 210-228. 16. Ghayad, S. E.; Cohen, P. A., Recent Pat. Anti-Cancer Drug Discovery
2010, 5 (1), 29-57. 17. Kessler, T.; Bayer, M.; Schwoppe, C.; Liersch, R.; Mesters, R. M.; Berdel,
W. E., Recent Results Cancer Res. 2010, 180, 137-163. 18. Allende-Vega, N.; Saville, M. K., Semin. Cancer Biol. 2010, 20 (1), 29-39. 19. Genin, E.; Reboud-Ravaux, M.; Vidal, J., Curr. Top. Med. Chem. 2010, 10
(3), 232-256. 20. Colland, F., Biochem. Soc. Trans. 2010, 38 (Pt 1), 137-143. 21. Fry, T. J.; Lankester, A. C., Hematol. Oncol. Clin. North Am. 2010, 24 (1),
109-127. 22. Bell, D. W., J. Pathol. 2010, 220 (2), 231-243. 23. Kratz, F.; Müller, I. A.; Ryppa, C.; Warnecke, A., ChemMedChem 2008, 3
(1), 20-53. 24. Thanou, M.; Duncan, R., Curr. Opin. Invest. Drugs 2003, 4 (6), 701-709. 25. Ouchi, T.; Ohya, Y., Prog. Polym. Sci. 1995, 20, 211-257. 26. Xu, S.; Kramer, M.; Haag, R., J. Drug Targeting 2006, 14 (6), 367-374. 27. Wahl, R. L., J. Nucl. Med. 2005, 46 Suppl 1, 128S-140S. 28. Luczynski, W.; Krawczuk-Rybak, M., Postepy Hig. Med. Dosw. 2002, 56 (6),
789-801.

Literatur _________________________________________________________________
- 153 -
29. Mousa, S. A., Curr. Opin. Chem. Biol. 2002, 6 (4), 534-541. 30. Meyer, A.; Auernheimer, J.; Modlinger, A.; Kessler, H., Curr. Pharm. Des.
2006, 12 (22), 2723-2747. 31. Desgrosellier, J. S.; Cheresh, D. A., Nat. Rev. Cancer 2010, 10 (1), 9-22. 32. Low, P. S.; Kularatne, S. A., Curr. Opin. Chem. Biol. 2009, 13 (3), 256-262. 33. Ross, J. F.; Chaudhuri, P. K.; Ratnam, M., Cancer 1994, 73 (9), 2432-2443. 34. Richards, F. F.; Konigsberg, W. H.; Rosenstein, R. W.; Varga, J. M.,
Science 1975, 187 (4172), 130-137. 35. Cameron, D. J.; Erlanger, B. F., Nature 1977, 268 (5622), 763-765. 36. Jain, R. K., Annu. Rev. Biomed. Eng. 1999, 1, 241-263. 37. Alley, S. C.; Okeley, N. M.; Senter, P. D., Curr. Opin. Chem. Biol. 2010, 14
(4), 529-537. 38. Markman, B.; Tabernero, J., Curr. Clin. Pharmacol. 2010, 5 (3), 160-165. 39. Stasi, R., Expert Opin. Biol. Ther. 2008, 8 (4), 527-540. 40. Seki, T.; Fang, J.; Maeda, H., Cancer Sci. 2009, 100 (12), 2426-2430. 41. Fox, M. E.; Szoka, F. C.; Frechet, J. M., Acc. Chem. Res. 2009, 42 (8),
1141-1151. 42. Torchilin, V., Adv. Drug Delivery Rev. 2010, in press. 43. Maeda, H., Bioconjugate Chem. 2010, 21 (5), 797-802. 44. Matsumura, Y.; Maeda, H., Cancer Res. 1986, 46 (12 Pt 1), 6387-6392. 45. Kopecek, J.; Kopeckova, P., Adv. Drug Delivery Rev. 2010, 62 (2), 122-149. 46. Duncan, R., Pharm. Sci. Technol. Today 1999, 2 (11), 441-449. 47. Kissel, M.; Peschke, P.; Subr, V.; Ulbrich, K.; Schuhmacher, J.; Debus, J.;
Friedrich, E., PDA J. Pharm. Sci. Technol. 2001, 55 (3), 191-201. 48. Yamaoka, T.; Tabata, Y.; Ikada, Y., J. Pharm. Sci. 1994, 83 (4), 601-606. 49. Murakami, Y.; Tabata, Y.; Ikada, Y., Drug Delivery 1997, 4, 23-31. 50. Singer, J. W.; Bhatt, R.; Tulinsky, J.; Buhler, K. R.; Heasley, E.; Klein, P.; de
Vries, P., J. Control. Release 2001, 74, 243-247. 51. Kakinoki, A.; Kaneo, Y.; Ikeda, Y.; Tanaka, T.; Fujita, K., Biol. Pharm. Bull.
2008, 31 (1), 103-110. 52. Kakinoki, A.; Kaneo, Y.; Tanaka, T.; Hosokawa, Y., Biol. Pharm. Bull. 2008,
31 (5), 963-969. 53. Wunder, A.; Stehle, G.; Sinn, H.; Schrenk, H. H.; Hoff-Biederbeck, D.;
Bader, F.; Friedrich, E. A.; Peschke, P.; Maier-Borst, W.; Heene, D. L., Int. J. Oncol. 1997, 11, 497-507.
54. Qu, X.; Yang, C.; Zhang, J.; Ding, N.; Lu, Y.; Huang, L.; Xiang, G., J. Drug Targeting 2010, 18 (5), 351-361.
55. Ghosh, S., J. Chem. Res. 2004, 241-246. 56. Leonard, M.; Dellacherie, E., Biochim. Biophys. Acta 1984, 791 (2), 219-
225. 57. Katre, N. V.; Knauf, M. J.; Laird, W. J., Proc. Natl. Acad. Sci. U. S. A. 1987,
84 (6), 1487-1491. 58. Okada, M.; Matsushima, A.; Katsuhata, A.; Aoyama, T.; Ando, T.; Inada, Y.,
Int. Arch. Allergy Appl. Immunol. 1985, 76 (1), 79-81. 59. Pasut, G.; Canal, F.; Dalla Via, L.; Arpicco, S.; Veronese, F. M.; Schiavon,
O., J. Control. Release 2008, 127 (3), 239-248. 60. Pasut, G.; Veronese, F. M., Adv. Drug Delivery Rev. 2009, 61 (13), 1177-
1188. 61. Zhu, S.; Hong, M.; Tang, G.; Qian, L.; Lin, J.; Jiang, Y.; Pei, Y., Biomaterials
2010, 31 (6), 1360-1371.

Literatur _________________________________________________________________
- 154 -
62. D'Souza, A. J.; Topp, E. M., J. Pharm. Sci. 2004, 93 (8), 1962-1979. 63. Guiotto, A.; Canevari, M.; Orsolini, P.; Lavanchy, O.; Deuschel, C.; Kaneda,
N.; Kurita, A.; Matsuzaki, T.; Yaegashi, T.; Sawada, S.; Veronese, F. M., J. Med. Chem. 2004, 47 (5), 1280-1289.
64. Dubowchik, G. M.; Mosure, K.; Knipe, J. O.; Firestone, R. A., Bioorg. Med. Chem. Lett. 1998, 8 (23), 3347-3352.
65. Hofmann, U. B.; Westphal, J. R.; Waas, E. T.; Zendman, A. J.; Cornelissen, I. M.; Ruiter, D. J.; van Muijen, G. N., Br. J. Cancer 1999, 81 (5), 774-782.
66. Jones, G. B.; Crasto, C. F.; Mathews, J. E.; Xie, L.; Mitchell, M. O.; El-Shafey, A.; D'Amico, A. V.; Bubley, G. J., Bioorg. Med. Chem. 2006, 14 (2), 418-425.
67. Gullbo, J.; Wickström, M.; Tullberg, M.; Ehrsson, H.; Lewensohn, R.; Nygren, P.; Luthman, K.; Larsson, R., J. Drug Targeting 2003, 11 (6), 355-363.
68. Shamis, M.; Lode, H. N.; Shabat, D., J. Am. Chem. Soc. 2004, 126 (6), 1726-1731.
69. Duimstra, J. A.; Femia, F. J.; Meade, T. J., J. Am. Chem. Soc. 2005, 127 (37), 12847-12855.
70. Denny, W. A., Future Oncol. 2010, 6 (3), 419-428. 71. Granchi, C.; Funaioli, T.; Erler, J. T.; Giaccia, A. J.; Macchia, M.; Minutolo,
F., ChemMedChem 2009, 4 (10), 1590-1594. 72. Huang, B.; Tang, S.; Desai, A.; Cheng, X. M.; Kotlyar, A.; Van Der Spek, A.;
Thomas, T. P.; Baker, J. R., Jr., Bioorg. Med. Chem. Lett. 2009, 19 (17), 5016-5020.
73. Tannock, I. F.; Rotin, D., Cancer Res. 1989, 49 (16), 4373-4384. 74. Hay, M. P.; Sykes, B. M.; Denny, W. A.; O`Connor, C. J., J. Chem. Soc.,
Perkin Trans. 1 1999, 2759-2770. 75. Senter, P. D.; Pearce, W. E.; Greenfield, R. S., J. Org. Chem. 1989, 55 (9),
2975-2978. 76. Serresi, M.; Bizzarri, R.; Cardarelli, F.; Beltram, F., Anal. Bioanal. Chem.
2009, 393 (4), 1123-1133. 77. Gillies, E. R.; Goodwin, A. P.; Frechet, J. M., Bioconjugate Chem. 2004, 15
(6), 1254-1263. 78. Svarovsky, S. A.; Taraban, M. B.; Barchi Jr., J. J., Org. Biomol. Chem.
2004, 2 (21), 3155-3161. 79. Bruyère, H.; Westwell, A. D.; Jones, A. T., Bioorg. Med. Chem. Lett. 2010,
20, 2200-2203. 80. Huang, X.; Du, F.; Cheng, J.; Dong, Y.; Liang, D.; Ji, S.; Lin, S.-S.; Li, Z.,
Macromol. 2009, 42, 783-790. 81. Remenyi, J.; Csik, G.; Kovacs, P.; Reig, F.; Hudecz, F., Biochim. Biophys.
Acta 2006, 1758 (3), 280-289. 82. Lavignac, N.; Nicholls, J. L.; Ferruti, P.; Duncan, R., Macromol. Biosci.
2009, 9 (5), 480-487. 83. Kratz, F.; Beyer, U.; Schütte, M. T., Crit. Rev. Ther. Drug Carrier Sys. 1999,
16 (3), 245-288. 84. Yang, J.; Kong, S. D.; Luong, A.; Howell, S., Acid-sensitive linkers for drug
delivery. WO 2007/114946 A2, 2006. 85. Kratz, F., Expert Opin. Invest. Drugs 2007, 16 (6), 855-866. 86. Aryal, S.; Hu, C. M.; Zhang, L., ACS Nano 2010, 4 (1), 251-258. 87. Kale, A. A.; Torchilin, V. P., Bioconjugate Chem. 2007, 18 (2), 363-370.

Literatur _________________________________________________________________
- 155 -
88. Rodrigues, P. C. A.; Roth, T.; Fiebig, H. H.; Unger, C.; Mülhaupt, R.; Kratz, F., Bioorg. Med. Chem. 2006, 14 (12), 4110-4117.
89. Bae, Y.; Fukushima, S.; Harada, A.; Kataoka, K., Angew. Chem. Int. Ed. Engl. 2003, 42 (38), 4640-4643.
90. Yao, K.; Peng, T.; Xu, M.; Yuan, C., Polym. Int. 1994, 34, 213-219. 91. Sedlak, M.; Pravda, M.; Staud, F.; Kubicova, L.; Tycova, K.; Ventura, K.,
Bioorg. Med. Chem. 2007, 15 (12), 4069-4076. 92. Yang, J.; Chen, H.; Vlahov, I. R.; Cheng, J. X.; Low, P. S., J. Pharmacol.
Exp. Ther. 2007, 321 (2), 462-468. 93. Mukherjee, S.; Ghosh, R. N.; Maxfield, F. R., Physiol. Rev. 1997, 77 (3),
759-803. 94. Jain, R.; Standley, S. M.; Fréchet, J. M. J., Macromol. 2007, 40, 452-457. 95. Ding, C.; Gu, J.; Qu, X.; Yang, Z., Bioconjug. Chem. 2009, 20 (6), 1163-
1170. 96. Knorr, V.; Ogris, M.; Wagner, E., Pharm. Res. 2008, 25 (12), 2937-2945. 97. Yang, S. C.; Bhide, M.; Crispe, I. N.; Pierce, R. H.; Murthy, N., Bioconjug.
Chem. 2008, 19 (6), 1164-1169. 98. Murthy, N.; Xu, M.; Schuck, S.; Kunisawa, J.; Shastri, N.; Frechet, J. M.,
Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (9), 4995-5000. 99. Chan, Y.; Wong, T.; Byrne, F.; Kavallaris, M.; Bulmus, V.,
Biomacromolecules 2008, 9 (7), 1826-1836. 100. Chen, W.; Meng, F.; Li, F.; Ji, S. J.; Zhong, Z., Biomacromolecules 2009,
10, 1727-1735. 101. Kratz, F.; Warnecke, A.; Schmid, B.; Chung, D.-E.; Gitzel, M., Curr. Med.
Chem. 2006, 13, 763-771. 102. Sedlak, M.; Pravda, M.; Kubicova, L.; Mikulcikova, P.; Ventura, K., Bioorg.
Med. Chem. Lett. 2007, 17 (9), 2554-2557. 103. Etrych, T.; Sirova, M.; Starovoytova, L.; Rihova, B.; Ulbrich, K., Mol. Pharm.
2010, 7 (4), 1015-1026. 104. Campo, V. L.; Carvalho, I., Curr. Methods Med. Chem. Biol. Phys. 2008, 2,
187-214. 105. Ettmayer, P.; Amidon, G. L.; Clement, B.; Testa, B., J. Med. Chem. 2004,
47 (10), 2393-2404. 106. Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.;
Savolainen, J., Nat. Rev. Drug Discov. 2008, 7 (3), 255-270. 107. Bundgaard, H., Adv. Drug Delivery Rev. 1989, 3, 39-65. 108. Elsadek, B.; Gräser, R.; Warnecke, A.; Unger, C.; Saleem, T.; El-Melegy,
N.; Madkor, H.; Kratz, F., ACS Med. Chem. Lett. 2010, 1 (5), 234-238. 109. Dubowchik, G. M.; Firestone, R. A., Bioorg. Med. Chem. Lett. 1998, 8 (23),
3341-3346. 110. Tranoy-Opalinski, I.; Fernandes, A.; Thomas, M.; Gesson, J. P.; Papot, S.,
Anti-Cancer Agents Med. Chem. 2008, 8 (6), 618-637. 111. Carl, P. L.; Chakravarty, P. K.; Katzenellenbogen, J. A., J. Med. Chem.
1981, 24 (5), 479-480. 112. Wakselman, M., Nouv. J. Chem. 1983, 7, 439-447. 113. de Groot, F. M.; de Bart, A. C.; Verheijen, J. H.; Scheeren, H. W., J. Med.
Chem. 1999, 42 (25), 5277-5283. 114. Greenwald, R. B.; Pendri, A.; Conover, C. D.; Zhao, H.; Choe, Y. H.;
Martinez, A.; Shum, K.; Guan, S., J. Med. Chem. 1999, 42, 3657-3667.

Literatur _________________________________________________________________
- 156 -
115. Perry-Feigenbaum, R.; Baran, P. S.; Shabat, D., Org. Biomol. Chem. 2009, 7 (23), 4825-4828.
116. Damen, E. W. P.; Nevalainen, T. J.; van den Bergh, T. J. M.; de Groot, F. M. H.; Scheeren, H. W., Bioorg. Med. Chem. 2002, 10, 71-77.
117. Rivault, F.; Tranoy-Opalinski, I.; Gesson, J. P., Bioorg. Med. Chem. 2004, 12, 675-682.
118. Greenwald, R. B.; Choe, Y. H.; Conover, C. D.; Shum, K.; Wu, D.; Royzen, M., J. Med. Chem. 2000, 43 (3), 475-487.
119. de Groot, F. M.; van Berkom, L. W.; Scheeren, H. W., J. Med. Chem. 2000, 43 (16), 3093-3102.
120. Ge, Y.; Wu, X.; Zhang, D.; Hu, L., Bioorg. Med. Chem. Lett. 2009, 19 (3), 941-944.
121. Meyer, Y.; Richard, J. A.; Delest, B.; Noack, P.; Renard, P. Y.; Romieu, A., Org. Biomol. Chem. 2010, 8, 1777-1780.
122. Wang, B.; Zhang, H.; Wang, W., Bioorg. Med. Chem. Lett. 1996, 6, 945. 123. Wang, B.; Zhang, H.; Zheng, A.; Wang, W., Bioorg. Med. Chem. 1998, 6
(4), 417-426. 124. Abu Ajaj, K.; Biniossek, M. L.; Kratz, F., Bioconjugate Chem. 2009, 20 (2),
390-396. 125. Burke, P. J.; Senter, P. D.; Meyer, D. W.; Miyamoto, J. B.; Anderson, M.;
Toki, B. E.; Manikumar, G.; Wani, M. C.; Kroll, D. J.; Jeffrey, S. C., Bioconjugate Chem. 2009, 20 (6), 1242-1250.
126. Miller, K.; Erez, R.; Segal, E.; Shabat, D.; Satchi-Fainaro, R., Angew. Chem. Int. Ed. Engl. 2009, 48 (16), 2949-2954.
127. Niculescu-Duvaz, D.; Niculescu-Duvaz, I.; Friedlos, F.; Martin, J.; Spooner, R.; Davies, L.; Marais, R.; Springer, C. J., J. Med. Chem. 1998, 41, 5297-5309.
128. Barthel, B. L.; Zhang, Z.; Rudnicki, D. L.; Coldren, C. D.; Polinkovsky, M.; Sun, H.; Koch, G. G.; Chan, D. C.; Koch, T. H., J. Med. Chem. 2009, 52 (23), 7678-7688.
129. Gao, S. Q.; Lu, Z. R.; Petri, B.; Kopeckova, P.; Kopecek, J., J. Control. Release 2006, 110 (2), 323-331.
130. Warnecke, A.; Kratz, F., J. Org. Chem. 2008, 73 (4), 1546-1552. 131. Sagi, A.; Weinstain, R.; Karton, N.; Shabat, D., J. Am. Chem. Soc. 2008,
130 (16), 5434-5435. 132. Weinstain, R.; Baran, P. S.; Shabat, D., Bioconjugate Chem. 2009, 20 (9),
1783-1791. 133. de Groot, F. M.; Albrecht, C.; Koekkoek, R.; Beusker, P. H.; Scheeren, H.
W., Angew. Chem. Int. Ed. 2003, 42 (37), 4490-4494. 134. Niculescu-Duvaz, I.; Niculescu-Duvaz, D.; Friedlos, F.; Spooner, R.; Martin,
J.; Marais, R.; Springer, C. J., J. Med. Chem. 1999, 42 (13), 2485-2489. 135. de Groot, F. M. H., Bioorg. Med. Chem. Lett. 2002, 12, 2371-2376. 136. Jeffrey, S. C.; Torgov, M. Y.; Andreyka, J. B.; Boddington, L.; Cerveny, C.
G.; Denny, W. A.; Gordon, K. A.; Gustin, D.; Haugen, J.; Kline, T.; Nguyen, M. T.; Senter, P. D., J. Med. Chem. 2005, 48, 1344-1358.
137. van Brakel, R.; Vulders, R. C.; Bokdam, R. J.; Grull, H.; Robillard, M. S., Bioconjugate Chem. 2008, 19 (3), 714-718.
138. Shabat, D.; Amir, R. J.; Gopin, A.; Pessah, N.; Shamis, M., Chemistry 2004, 10 (11), 2626-2634.
139. Tidwell, T. T., Angew. Chem. 2008, 120 (6), 1032-1036.

Literatur _________________________________________________________________
- 157 -
140. Müller, I. A.; Kratz, F.; Jung, M.; Warnecke, A., Tetrahedron Lett. 2010, 51 (33), 4371-4374.
141. Maly, D. J.; Leonetti, F.; Backes, B. J.; Dauber, D. S.; Harris, J. L.; Craik, C. S.; Ellman, J. A., J. Org. Chem. 2002, 67, 910-915.
142. Wicks, Z. W., Prog. Org. Coat. 1975, 3, 73-99. 143. Linn, C. P.; Mize, P. D.; Hoke, R. A.; Quante, J. M.; Pitner, J. B., Anal.
Biochem. 1992, 200, 400-404. 144. Erez, R.; Shabat, D., Org. Biomol. Chem. 2008, 6 (15), 2669-2672. 145. Hammett, L. P., J. Am. Chem. Soc. 1937, 59 (1), 96-103. 146. Chapman, N. B.; Shorter, J., Correlation analysis in chemistry - recent
advances. Plenum Press: New York, 1978. 147. Hart, H.; Sedor, E. A., J. Am. Chem. Soc. 1967, 89 (10), 2342-2347. 148. Um, I. H.; Lee, J. Y.; Kim, H. T.; Bae, S. K., J. Org. Chem. 2004, 69, 2436-
2441. 149. Cordes, E. H.; Jencks, W. P., J. Am. Chem. Soc. 1962, 84 (5), 832-837. 150. Cordes, E. H.; Jencks, W. P., J. Am. Chem. Soc. 1963, 85 (18), 2843-2848. 151. Pitea, D.; Favini, G.; Grasso, D., J. Chem. Soc., Perkin Trans. 2 1974, 2,
1595-1600. 152. Carey, F. A.; Sundberg, R. J., Organische Chemie - Ein weiterführendes
Lehrbuch. 2. korr. Nachdruck; WILEY-VCH Verlag GmbH&Co.KGaA: Weinheim, 2004; 434-435.
153. Kragh-Hansen, U.; Chuang, V. T.; Otagiri, M., Biol. Pharm. Bull. 2002, 25 (6), 695-704.
154. Vallner, J. J., J. Pharm. Sci. 1977, 66 (4), 447-465. 155. Kratz, F., Int. J. Clin. Pharmacol. Ther. 2010, 48 (7), 453-455. 156. Veronese, F. M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.;
Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; Duncan, R., Bioconjugate Chem. 2005, 16 (4), 775-784.
157. Nuhn, P., Pharmazie 1975, 30 (8), 544-545. 158. Kolb, H. C.; Finn, M. G.; Sharpless, K. B., Angew. Chem. Int. Ed. Engl.
2001, 40 (11), 2004-2021. 159. Hein, C. D.; Liu, X.-M.; Wang, D., Pharm. Res. 2008, 25 (10), 2216-2230. 160. van Dijk, M.; Rijkers, D. T. S.; Liskamp, R. M. J.; van Nostrum, C. F.;
Hennink, W. E., Bioconjugate Chem. 2009, 20 (11), 2001-2016. 161. Dordunoo, S. K.; Burt, H. M., Int. J. Pharm. 1996, 133 (1-2), 191-201. 162. Elsadek, B., unveröffentlichte Ergebnisse. 163. Rahman, A.; Jonnalagadda, S. B., Catal. Lett. 2008, 123, 264-268. 164. Sander, M., Monatsh. Chem. 1964, 95 (2), 608-616. 165. Kratz, F.; Beyer, U., Drug Delivery 1998, 5, 281-299. 166. Warnecke, A., unveröffentlichte Ergebnisse. 167. Matsumura, Y., Jpn. J. Clin. Oncol. 2008, 38 (12), 793-802. 168. Musacchio, T.; Laquintana, V.; Latrofa, A.; Trapani, G.; Torchilin, V. P., Mol.
Pharm. 2009, 6 (2), 468-479. 169. Children's Hospital Boston: New oral angiogenesis inhibitor offers potential
nontoxic therapy for a wide range of cancers. ScienceDaily 30 June, 2008, <http://www.sciencedaily.com /releases/2008/06/080630114209.htm>.
170. de Groot, F. M. H.; Beusker, P. H.; Scheeren, J. W., Prodrugs built as multiple self-elimination-release spacers. WO 2004/043493 A1, 2004.

Anhang _________________________________________________________________
- 158 -
Anhang
A-1: Auftragungen zur Bestimmung der Geschwindigkeitskonstanten k1 und der
Halbwertszeiten t1/2 für die AMC-Freisetzung aus 1b und 4b–14b:*
pH 5.0 (Acetat-Puffer, 20 mM)
y = -0.0205x - 0.5578R2 = 0.9837
y = -0.0415x - 0.4088R2 = 0.9942
y = -0.0346x - 0.5242R2 = 0.9927
-3.2
-2.7
-2.2
-1.7
-1.2
-0.7
0 10 20 30 40 50 60t /min
ln[1
-(c/c
∞)]
1b
4b
5b
y = -0.026x - 0.3282R2 = 0.9918
y = -0.0245x - 0.6904R2 = 0.9778
y = -0.0114x - 0.1446R2 = 0.9949
-2.3
-1.8
-1.3
-0.8
-0.3
0 10 20 30 40 50 60t /min
ln[1
-(c/c
∞)]
6b
8b
9b
* c = Konzentration zum Zeitpunkt t; c∞ = extrapolierte Endkonzentration (pH 5: Messungen im 30 minütigen Abstand, bis sich die Konzentration erstmals um weniger als 0.1 % ändert; pH 7.4: Da die Messungen aufgrund der langsamen Hydrolysegeschwindigkeit vor Ende der Reaktion abgebrochen werden mussten, wurden die bei pH 5 ermittelten Konzentrationen für die Auswertung verwendet.)
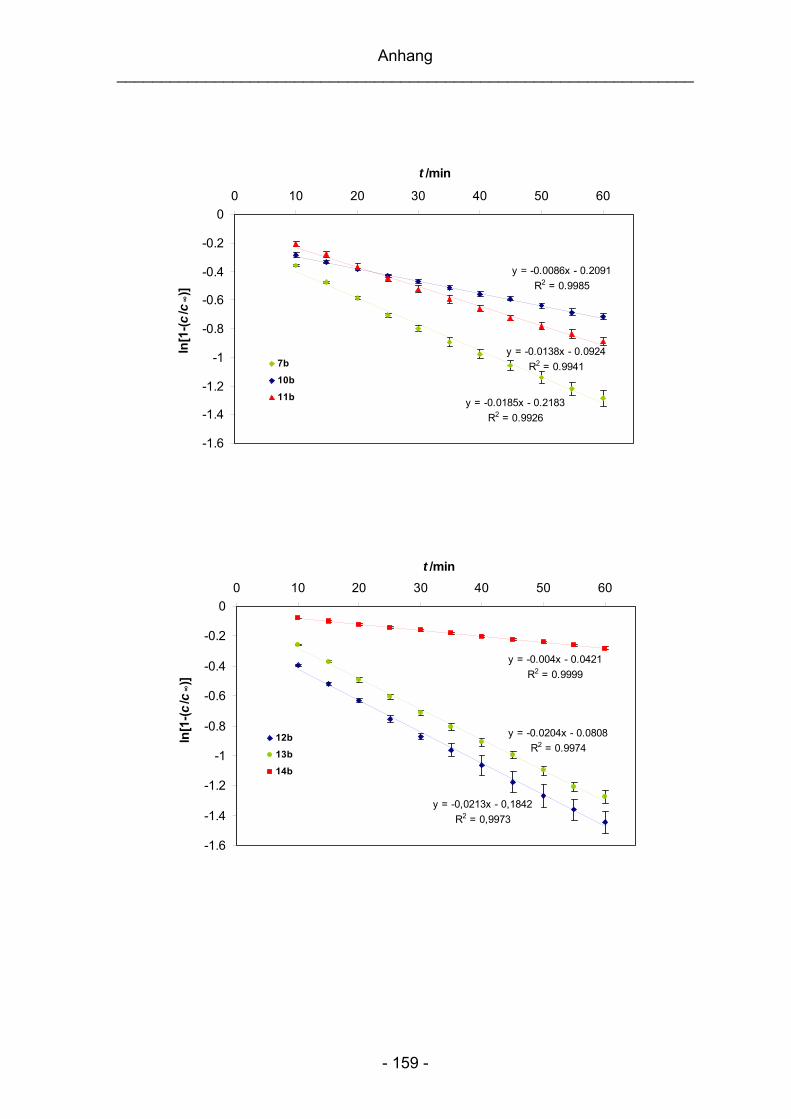
Anhang _________________________________________________________________
- 159 -
y = -0.0185x - 0.2183R2 = 0.9926
y = -0.0086x - 0.2091R2 = 0.9985
y = -0.0138x - 0.0924R2 = 0.9941
-1.6
-1.4
-1.2
-1
-0.8
-0.6
-0.4
-0.2
00 10 20 30 40 50 60
t /minln
[1-(c
/c∞)]
7b
10b
11b
y = -0,0213x - 0,1842R2 = 0,9973
y = -0.0204x - 0.0808R2 = 0.9974
y = -0.004x - 0.0421R2 = 0.9999
-1.6
-1.4
-1.2
-1
-0.8
-0.6
-0.4
-0.2
00 10 20 30 40 50 60
t /min
ln[1
-(c/c
∞)]
12b
13b
14b
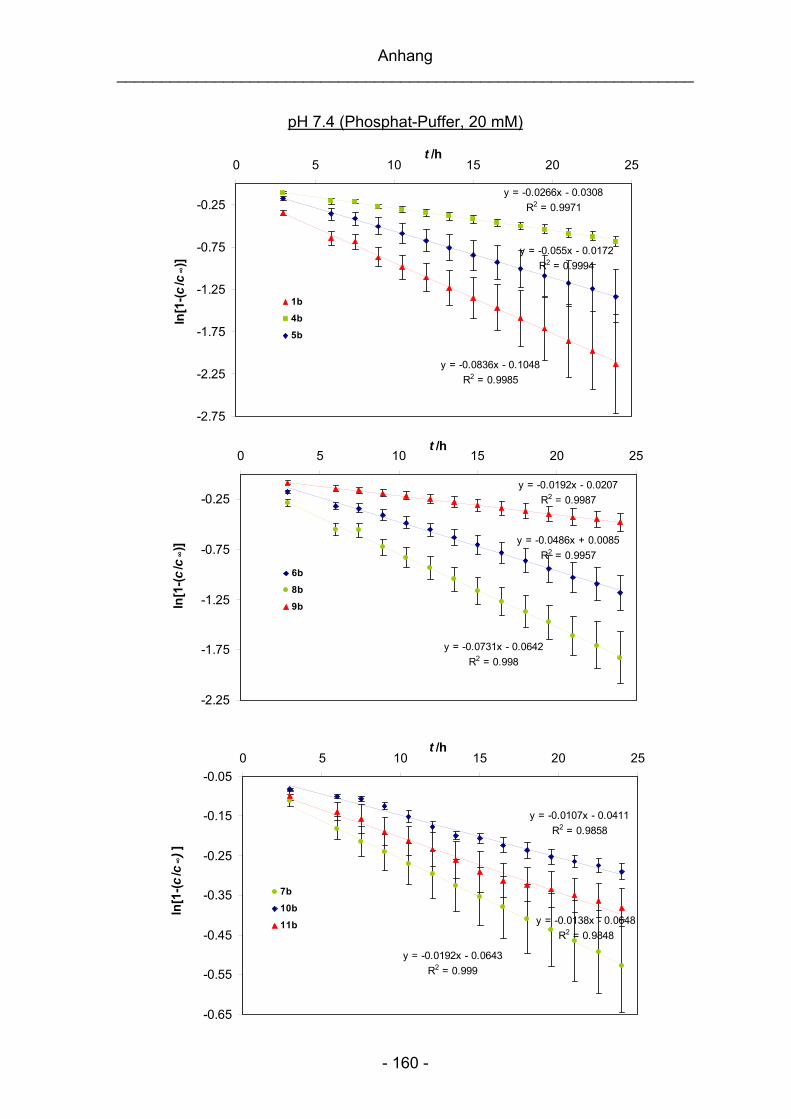
Anhang _________________________________________________________________
- 160 -
pH 7.4 (Phosphat-Puffer, 20 mM)
y = -0.0836x - 0.1048R2 = 0.9985
y = -0.0266x - 0.0308R2 = 0.9971
y = -0.055x - 0.0172R2 = 0.9994
-2.75
-2.25
-1.75
-1.25
-0.75
-0.25
0 5 10 15 20 25t /h
ln[1
-(c/c
∞)]
1b
4b
5b
y = -0.0486x + 0.0085R2 = 0.9957
y = -0.0731x - 0.0642R2 = 0.998
y = -0.0192x - 0.0207R2 = 0.9987
-2.25
-1.75
-1.25
-0.75
-0.25
0 5 10 15 20 25t /h
ln[1
-(c/c
∞)]
6b
8b
9b
y = -0.0192x - 0.0643R2 = 0.999
y = -0.0107x - 0.0411R2 = 0.9858
y = -0.0138x - 0.0648R2 = 0.9848
-0.65
-0.55
-0.45
-0.35
-0.25
-0.15
-0.050 5 10 15 20 25
t /h
ln[1
-(c/c
∞)]
7b
10b
11b

Anhang _________________________________________________________________
- 161 -
y = -0.0178x - 0.0408R2 = 0.9963
y = -0.0078x - 0.0648R2 = 0.9935
y = -0.0019x - 0.0525R2 = 0.9907
-0.6
-0.5
-0.4
-0.3
-0.2
-0.1
00 5 10 15 20 25
t /h
ln[1
-(c/c
∞)]
12b
13b
14b
A-2: Auftragung zur Bestimmung der Geschwindigkeitskonstanten k2 und der
Halbwertszeit t1/2 für die AMC-Freisetzung durch 1,4-Benzyleliminierung von 56
und 57 bei pH 7.4 (c∞ = extrapolierte Endkonzentration; Messungen im
30 minütigen Abstand, bis sich die Konzentration erstmals um weniger als 0.1 %
ändert):
y = -0.0533x + 0.0517R2 = 0.9961
y = -0.0141x - 0.0096R2 = 0.999
-0.8
-0.6
-0.4
-0.2
00 5 10 15 20 25 30 35
t /min
ln[1
-(c/c
∞)] 56
57


















