
Etablierung der Lasermikrodissektion zur proteomischen ... · DIGE differential in-gel...
218
Etablierung der Lasermikrodissektion zur proteomischen Analyse von humanem Filaminopathiegewebe und murinen Myoblasten Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften Doctor Rerum Naturalium (Dr. rer. nat.) angefertigt in der Abteilung Funktionelle Proteomik am Medizinischen Proteom-Center vorgelegt der Fakultät für Chemie und Biochemie an der Ruhr-Universität Bochum von Verena Theis, geb. Schwarz Bochum Juni, 2013
Transcript of Etablierung der Lasermikrodissektion zur proteomischen ... · DIGE differential in-gel...

Etablierung der Lasermikrodissektion zur proteomischen Analyse
von humanem Filaminopathiegewebe und murinen Myoblasten
Dissertation
zur Erlangung des Grades
eines Doktors der Naturwissenschaften
Doctor Rerum Naturalium (Dr. rer. nat.)
angefertigt
in der Abteilung Funktionelle Proteomik am Medizinischen Proteom-Center
vorgelegt
der Fakultät für Chemie und Biochemie
an der Ruhr-Universität Bochum
von
Verena Theis, geb. Schwarz
Bochum
Juni, 2013

Establishment of proteomic studies in patients with filaminopathy
and murine myoblasts
Dissertation
To obtain the degree
Doctor Rerum Naturalium (Dr. rer. nat.)
Dept. Functional Proteomics Medical Proteom Center
International Graduate School of Chemistry and Biochemistry,
Faculty of Chemistry and Biochemistry, Ruhr-University Bochum
Submitted by
Verena Theis, geb. Schwarz
Bochum
Juni 2013

Referent: Prof. Dr. Katrin Marcus
Funktionelle Proteomik
Ruhr Universität Bochum
Korreferent: Prof. Dr. Rolf Heumann
Molekulare Neurobiochemie
Ruhr Universität Bochum

Für Krümel und Cassian

i
Inhaltsverzeichnis
INHALTSVERZEICHNIS ............................................................................................................. I
ABKÜRZUNGSVERZEICHNIS..................................................................................................... I
1. EINLEITUNG ........................................................................................................................ 1
1.1. Die Skelettmuskulatur ......................................................................................................................... 1
1.1.1. Aufbau der Myofibrillen ..................................................................................................................... 2
1.1.2. Die Filamine....................................................................................................................................... 3
1.1.3. Die Bedeutung von Intermediärfilamenten ........................................................................................ 5
1.2. Myofibrilläre Myopathien (MFM) ........................................................................................................ 7
1.3. Die Filaminopathie ............................................................................................................................ 10
1.4. Lasermikrodissektion ......................................................................................................................... 12
1.5. Grundlagen der Proteomanalyse ....................................................................................................... 14
1.5.1. Proteomanalysen von Skelettmuskulatur ....................................................................................... 15
1.5.2. Auftrennung mittels 2D-Polyacrylamid-Gelelektrophorese (2D PAGE) ............................................ 17
1.5.3. Differentielle Proteomstudie und Quantifizierung von Proteinen ................................................... 17
1.6. Hochleistungsflüssigkeitschromatographie (HPLC) ............................................................................ 19
1.7. Massenspektrometrie........................................................................................................................ 19
1.7.1. Ionenquellen ................................................................................................................................. 21
1.7.2. Massenanalysatoren ...................................................................................................................... 23
1.8. Quantitative Massenspektrometrie ................................................................................................... 27
1.8.1. Isotopen-Labeling .......................................................................................................................... 27
1.8.2. Label-freie Quantifizierug mittels Spectral Index Calculation .......................................................... 29
2. ZIELSETZUNG DER ARBEIT ................................................................................................ 32
3. MATERIAL UND METHODEN ............................................................................................ 34
3.1. Verwendete Chemikalien .................................................................................................................. 34
3.2. Verwendete Geräte und Materialien ................................................................................................. 38

Inhaltsverzeichnis
ii
3.3. Präparation von Gewebeschnitten .................................................................................................... 43
3.4. Patientendaten .................................................................................................................................. 43
3.5. Immunfluorezenzfärbungen .............................................................................................................. 45
3.5.1. Immunfluoreszenzfärbungen auf Glasobjektträgern ....................................................................... 45
3.5.2. Immunfluoreszenzfärbung aufPET-Membran ................................................................................. 45
3.5.3. Validierung differentieller Proteine mittels Immunfluoreszenz ....................................................... 46
3.6. Herstellen von Muskellysaten ........................................................................................................... 46
3.7. Kultivierung von murinen Myoblasten .............................................................................................. 47
3.8. Zellbiologische Methoden ................................................................................................................. 48
3.8.1. Transformation von Konstrukten in E.coli-Zellen ............................................................................ 49
3.8.2. DNA-Extraktion und Überprüfung der Plasmid-DNA ....................................................................... 49
3.8.3. Herstellen von Glycerolstocks ........................................................................................................ 50
3.8.4. Transfektion der FLNc Konstrukte in murine Myoblasten ............................................................... 50
3.9. Lasermikrodissektion (LMD) .............................................................................................................. 51
3.10. Solubilisierung der Aggregate ............................................................................................................ 52
3.11. In-Lösung-Verdau .............................................................................................................................. 52
3.12. Aufreinigung der Proben nach dem In-Lösung-Verdau ...................................................................... 53
3.13. Differentielle In-Gel Elektrophorese des Proteoms von Patienten mit Filaminopathie ...................... 54
3.13.1. Markierung der Proteine durch CyDye™Farbstoffe ......................................................................... 54
3.13.2. Isoelektrische Fokussierung mit Trägerampholyten ........................................................................ 55
3.13.3. Proteinauftrennung nach Molekulargewicht .................................................................................. 57
3.13.4. Differentielle Bildanalyse ............................................................................................................... 58
3.13.5. Mini 2D Gel Elektrophorese ........................................................................................................... 59
3.14. Proteolytische Spaltung von Proteinen für LC-ESI-MS/MS und MALDI............................................... 60
3.14.1. Verdau der Spots ........................................................................................................................... 60
3.14.2. Extraktion der Peptide für MALDI................................................................................................... 60
3.14.3. Extraktion der Peptide für die LC-ESI-MS/MS ................................................................................. 61
3.15. Proteinidentifizierung mittels MALDI ................................................................................................ 61
3.15.1. Datenauswertung der MALDI-Messung .......................................................................................... 62
3.16. Proteinidentifizierung mittels LC-ESI-MS/MS ..................................................................................... 63
3.16.1. LTQ Orbitrap .................................................................................................................................. 64
3.16.2. LTQ Velos Pro ................................................................................................................................ 65
3.16.3. Datenauswertung der LC-ESI-MS/MS ............................................................................................. 65
3.17. Auswertung massenspektrometrischer Daten nach Datenabgleich ................................................... 67
3.17.1. Die pseudo-qualitative Auswertung ............................................................................................... 68
3.17.2. Spectral Index Calculation .............................................................................................................. 68
3.18. Western Blot ..................................................................................................................................... 70

Inhaltsverzeichnis
iii
4. ERGEBNISSE ..................................................................................................................... 72
4.1. Etablierung der LMD zur Label-freien Proteomanalyse ...................................................................... 73
4.1.1. Darstellbarkeit der Aggregate ........................................................................................................ 73
4.1.2. Solubilisierung der Aggregate ........................................................................................................ 74
4.1.3. Optimierung der Anzahl von Aggregaten ........................................................................................ 77
4.2.. Ergebnisse der Label-freien Proteomanalyse von Filaminopathiegewebe......................................... 80
4.2.1. Entwicklung einer geeigneten Auswertestrategie ........................................................................... 81
4.2.1.1. Vergleichende Analyse Aggregatproben - Patientenkontrollen ....................................................... 82
4.2.1.2. Vergleichende Analyse Patientenkontrollen - Negativkontrollen .................................................... 87
4.2.1.3. Vergleich der manuellen und automatisierten quantitativen Auswertung ...................................... 92
4.2.2. Validierung differentieller Proteine der Aggregatanalyse ................................................................ 97
4.3. Etablierung der 2D Differentiel In-Gel Elektrophorese mittels 2D DIGE.............................................. 98
4.3.1. Optimierung der Probenmenge ..................................................................................................... 98
4.3.2. Ergebnisse der differentiellen Proteomanalyse von Filaminopathiegewebe mittels 2D DIGE ......... 100
4.3.2.1. Ergebnisse der differentiellen Analyse mittels DeCyder ................................................................ 101
4.3.2.2. Identifizierung differentieller Proteins mittels MALDI ................................................................... 103
4.3.2.3. Überprüfen der Spectral Index Daten in Patientenkontrollen mittels 2D DIGE .............................. 104
4.3.3. Untersuchung einer posttranslationalen Modifikation an Desmin mittels 2D Blot ......................... 105
4.4. Murine Myoblasten als MFM-Zellkulturmodell ............................................................................... 110
4.4.1. Transfektion der p.W2710X-Mutation in murine Myoblasten ....................................................... 111
4.4.2. Optimierung der LMD .................................................................................................................. 113
4.4.3. Ergebnisse der Label-freien Aggregatanalyse im Zellkulturmodell ................................................. 117
5. DISKUSSION ....................................................................................................................120
5.1. Lasermikrodissektion ................................................................................................................... 121
5.2. Etablierung der differentiellen Proteomanalysen in Filaminopathien .............................................. 122
5.2.1. Label-freie Proteomanalyse ......................................................................................................... 123
5.2.1.1. Solubilisierung der Aggregate ...................................................................................................... 123
5.2.1.2. Optimierung der Probenmenge ................................................................................................... 124
5.2.1.3. Auswertestrategien ..................................................................................................................... 125
5.2.1.4. Die physiologische Bedeutung der differentiell exprimierten Proteine in den Aggregaten ............. 130
5.2.2. Gelbasierte differentielle Analyse ................................................................................................ 133
5.2.2.1.. Optimierung der Probenmenge ................................................................................................... 134
5.2.2.2. Detektion differentiell exprimierter Proteine ............................................................................... 135
5.2.2.3. Die physiologische Bedeutung der differentiell exprimierten Proteine in Patientenkontrollen ...... 137
5.2.2.4. Die biologische Relevanz der Desminphosphorylierung ................................................................ 143
5.2.2.5. Die Relevanz der Desminphosphorylierung in Filaminopathien ..................................................... 145
5.2.2.6. Die Detektion der Phosphorylierung in Desmin ............................................................................ 147
5.2.2.7. Lokalisation von Phosphorylierungen in der Aminosäuresequenz ................................................. 148
5.3. Proteomstudien im Zellkulturmodell ............................................................................................... 152
5.4. Fazit und Ausblick .......................................................................................................................... 157
6. ZUSAMMENFASSUNG .....................................................................................................159

Inhaltsverzeichnis
iv
7. SUMMARY ......................................................................................................................164
8. LITERATURVERZEICHNIS ..................................................................................................164
9. ABBILDUNGSVERZEICHNIS ..............................................................................................192
10. TABELLENVERZEICHNIS .................................................................................................192
11. LEBENSLAUF ..................................................................................................................192
12. DANKSAGUNG...............................................................................................................198
13. ANHANG .......................................................................................................................201

I
Abkürzungsverzeichnis
1D-PAGE Eindimensionale Polyacrylamidgelelektrophorese
2D PAGE Zweidimensionale Polyacrylamidgelelektrophorese
A Ampere
°A Anström
Abb. Abbildung
ABD Aktin-bindende Domäne
Agg
AK
Aggregatprobe
Antikörper
APEX absolute protein abundance index
APS Ammoniumpersulfat
ADP Adenosindiphosphat
ATP Adenosintriphosphat
AS Aminosäuren
AQUA absolute quantification of proteins
BAG3 BCL2-associated athanogene 3/ family molecular chaperone regulator 3
Bis-Tris Bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methan
Bp Basenpaare
BR Broad-range
BSA Bovine serum albumin (Rinderserumalbumin)
BT Bis-Tris
BVA Biological Variance Analysis
bzw. beziehungsweise
°C Grad Celsius
ca. circa
CBM Cytoplasmatische Body Myopathie
CHAPS
CID
3-[(Cholamidopropyl)-dimethylammonio]-propansulfonsäure
Collision induced dissociation
CK Creatin Kinase
cm Zentimeter
cm3 Kubikzentimeter
COX Cytochrome C Oxidase
CRYAB Alpha B Crystallin
Cy Cyanin Fluoreszenzfarbstoff
Da Dalton

Abkürzungsverzeichnis
II
DCM Dilatative Cardiomyopathie
DES Desmin-Gen
DIA Differential In-Gel Analysis
DIGE differential in-gel electrophoresis (Differentielle In-Gel Elektrophorese)
DHB 2,5-Dihydroxybenzoesäure
DMEM Dulbecco’s Modified Eagle Medium
DMF Dimethylformamid
DMSO Dimethylsulfoxid
DNA desoxyribonucleic acid (Desoxyribonukleinsäure)
ds double strain
DTT 1,4 – Dithiothreitol
Engl. englisch
EDTA Ethylendiamintetraacetat
EGFP Enhanced Green fluorescent Protein
emPAI exponentially modified protein abundance index
ESI electrospray-ionization (Elektrosprayionisation)
ETD Elektronentransferdissoziation
fl Full length
FLNC Filmain-Gen
FHL-1 Four and a half LIM domains protein 1
FA formic acid (Ameisensäure)
FCS fetal bovine serum (Fötales Kälberserum)
GFP Green fluorescent Protein
GGA Geranylgeranylaceton
GIST Global Internal Standard
h Stunde
HPLC high performance liquid chromatography
(Hochleistungsflüssigkeitschromatographie)
Hskm Humane Skelettmuskelzellen
HSP Heat Shock Protein
IAA Iodacetamid
ICAT isotope coded affinity tag
ICPL isotope coded protein label
IEF
IMAC
iTRAQ
kDa
Isoelelektrische Fokussierung
Metallaffinitätschromatographie
Isotope Tags For Relative and Absolute Quantification
Kilodalton
kV Kilovolt
LB-Medium Lysogeny Broth Medium (Nährmedium zur Kultivierung von Bakterien)
LC Liquid Chromatography (Flüssigkeitschromatographie)
LMD Lasermikrodissektion

Abkürzungsverzeichnis
III
M Mol
mA Milliampere
ml Milliliter
mM Millimol
mgf Mascot generic format
min Minute
m/z Masse-zu-Ladungsverhältnis
MOAC Metalloxidaffinitätschromatographie
MOPS Morpholinopropansulfonsäure
MS Massenspektrometrie
MS/MS Tandemmassenspektrometrie
Msec Millisekunde
Mut Mutante
MYL3 Myosin leichte Kette 3
MW molecular weight (Molekulargewicht)
n Anzahl unabhängiger Experimente
NADH Nicotinamidadenosindinukleotid
NEAA Non essential amino acids
NegKO Negativkontrolle
nL Nanoliter
nm Nanometer
Nmol Nanomol
NP-HPLC Normalphasen-Chromatographie
NRAP Nebulin-related-anchoring protein
Nr. Nummer
PAGE Polyacrylamidgelelektrophorese
PBS
PDA
PET
phosphate buffered saline (Phosphat gepufferte Kochsalzlösung)
Piperazin-Diacrylamid
Polyethylentetraphtalat
PEN Polyethylene Naphthalate
pI
PatKO
Isoelektrischer Punkt
Patientenkontrolle
PGM Phosphoglucomutase
PLEC1 Plectin 1-Gen
PMF
ppm
PYG
RP
rpm
peptide mass fingerprint (Peptidmassenfingerabdruck)
parts per million
Glykogen Phosphorylase
reversed phase
rounds per minute
RBM Reducing Body Myopathie
RT-HPLC Umkehrphasenchromatographie

Abkürzungsverzeichnis
IV
RT
SAX
SCX
Raumtemperatur
Anionaustauscher
Kationaustauscher
SDS
sec
sodiumdodecylsulfate (Natriumdodecylsulfat)
Sekunde
Seq Sequenz
SI Spectral Index
SILAC stable isotope labelling by amino acids in cell culture
SIMAC Sequential Elution from IMAC
SOC- Medium Super Optimal Broth (SOB) Nährmedium mit Zugabe von 20 mM
Glucose
TBS Tris gepufferte Kochsalzlösung
TCEP
TEMED
tris(2-carboxyethyl)phosphine
N,N,N',N'-Tetramethylethylendiamin
TE Tris/EDTA
TFA Trifluoressigsäure
TMT tandem mass tagging
TOF time of flight (Flugzeit)
Tris Base Tris(hydroxymethyl-)aminomethan
Tris HCl
TTID
Tween 20
UV
Tris(hydroxymethyl-)aminomethan Hydrochlorid
Myotilin-Gen
Polyoxyethylensorbitan-monolaurat
Ultraviolett
U Unit
V Volt
VDAC Voltage-dependent anion-selective Channel
VDCC Voltage-dependent calcium-selective Channel
Vers. Version
v/v volume per volume (Volumen pro Volumen)
w/v
Xin
Xirp2
weight per volume (Gewicht pro Volumen)
Xin actin-binding repeat-containing protein 1
Xin actin-binding repeat-containing protein 2
Z Ladungszahl
ZASP Z-band alternatively spliced PDZ motif containing protein
z.B. Zum Beispiel
z.T. Zum Teil
µg Mikrogramm
µl Mikroliter
µm Mikrometer
µm2 Quadratmikrometer

1
1. Einleitung
Myofibrilläre Myopathien (MFM) bilden eine genetisch wie klinisch heterogene
Gruppe von neuromuskulären Erkrankungen. Auf zellulärer Ebene zeichnen sie sich durch
fokale myofibrilliäre Zerstörung und intrazelluläre Proteinaggregation aus [Selcen et al.
2004]. Die Filaminopathie ist ein Subtyp der MFM und wird durch Mutationen im
codierenden Gen für Filamin C (FLNC) verursacht. Filamin C wird vorwiegend in Skelett- und
Herzmuskulatur exprimiert und ist innerhalb der Muskelzellen an der Z-Scheibe lokalisiert.
An den Z-Scheiben interagiert Filamin C mit diversen Proteinen, einschließlich Aktin und
Myotillin. Im Mittelpunkt der hier vorliegenden Dissertation stand die proteomische
Untersuchung der Proteinaggregate und des umliegenden Gewebes von 6
Filaminopathiepatienten mit 3 verschiedenen Mutationen im für Filamin C codierenden Gen.
Dafür war es zunächst notwendig entsprechende Protokolle zu etablieren, basierend auf der
Lasermikrodissektion (LMD). Für die Analyse der Proteinaggregate wurde ein Label-freier
Ansatz mit anschließender quantitativer Auswertung etabliert, zur Charakterisierung von
möglichen Frühmarkern in dem umliegenden Gewebe wurde die 2D DIGE Analyse gewählt.
Zusätzlich sollte ein Zellmodell etabliert werden, um zukünftige funktionelle Analysen zu
ermöglichen.
1.1. Die Skelettmuskulatur
Die Skelettmuskulatur umfasst die Muskeln des Bewegungsapparates sowie die
Muskeln in der Zunge, im Pharynx und im des oberen Anteils des Oesophagus [Schiebler
Anatomie 9. Auflage 2005]. Zusammen mit der Herzmuskulatur wird die Skelettmuskulatur
als quergestreift bezeichnet, da die kontraktilen Einheiten im Lichtmikroskop als helle und
dunkle Areale erscheinen. Ein Skelettmuskel ist hierarchisch aufgebaut und setzt sich aus
zahlreichen Muskelfaserbündeln zusammen. Die Muskelfaserbündel bestehen aus

Einleitung
2
Muskelfasern. Durch die Fusion vieler einkerniger Myoblasten in der Embryonalentwicklung
entsteht eine vielkernige Skelettmuskelzelle. Eine Zelle beinhaltet bis zu 100 randständige
Kerne, die unter dem Plasmalemm lokalisiert sind [Schiebler Anatomie 9. Auflage 2005].
1.1.1. Aufbau der Myofibrillen
Die Myofibrillen bilden die kleinste Untereinheit des Skelettmuskels. Die 1-2 µm
dicken Myofibrillen verlaufen parallel zur Muskellängsachse und bilden zylindrische
Strukturen. Sie werden durch die Z-Scheiben in Sarkomere unterteilt, welche die kleinsten
funktionellen Einheiten des Muskels bilden. Ein Sarkomer besteht neben dicken
Myosinfilamenten (10-15 nm) hauptsächlich aus dünnen Aktinfilamente (5-6 nm). Die
Myosinfilamente verschieben sich während der Muskelkontraktion gegeneinander [Schiebler
Anatomie 9. Auflage 2005]. Die Myosinmoleküle bestehen aus einem dünnen Schaft und
einem kugelförmigen Kopf. Die Myosinköpfchen verfügen über einen Myosin-ADP-Komplex
und weisen eine hohe ATPase-Aktivität auf. Sobald ATP in ADP und Phosphat gespalten wird,
bindet das Aktin an die Köpfchen und das Myosin gleitet entlang der Aktinfilamente. Die Z-
Scheiben nähern sich an und daraufhin verkürzt sich das Sarkomer aktiv.
Im Lichtmikroskop lassen sich einzelne Streifen als strukturelles Merkmal der
Myofibrillen erkennen (Abb. 1.1.). Die Streifen erscheinen aufgrund ihrer Lichtdurchlässigkeit
als helle und dunkle Zonen. Die I-Streifen werden im polarisierten Licht nur schwach
gebrochen und erscheinen deswegen hell. In diesen Streifen befinden sich ausschließlich
dünne Aktinfilamente. Im Gegensatz zu den I-Streifen erscheinen die A-Streifen im
polarisierten Licht dunkel, weil sie doppelt gebrochen werden. Sie werden von dicken
Myosinfilamenten mit zwischengelagerten dünnen Aktinfilamenten gebildet. In den A-
Streifen befindet sich die H-Zone, in dieser Zone befinden sich keine Aktinfilamente. Die
Mitte der H-Zone wird als M-Streifen bezeichnet. Hier sorgen Strukturproteine wie
Myomesin für die regelmäßige Anordnung der Myosinfilamente. Indirekt werden die dicken
Filamente mit den Z-Scheiben auch über Titin verbunden. Titin ist mit 3000-3800 kDa das
größte bekannte Molekül und ist in den Z-Scheiben wie auch in der M-Bande verankert.

Einleitung
3
Abbildung 1.1.: lichtmikroskopische Aufnahme und strukturelle Darstellung einer Myofibrille im Skelettmuskel,
modifiziert nach Schmidt et al. Physiologie des Menschen 29.Auflage 2005
Die hell erscheinende I-Bande wird durch die Z-Schreibe in zwei Hälften unterteilt und enthält
ausschließlich dünne Aktin-Filamente. An der Z-Scheibe sind die dünnen Filamente (Aktin) durch das
Gerüstprotein alpha-Aktinin befestigt. Die dicken Myosin-Filamente werden indirekt über Titin mit der
Z-Scheibe verbunden. Die Lokalisation von Myosinfilamenten beschränkt sich nur auf die im
Lichtmikroskop dunkel erscheinenden A-Streifen.
1.1.2. Die Filamine
Beim Menschen gibt es drei verschiedenen Filamine: A, B und C. Filamin A wurde
erstmals 1975 von Hartwig und Stossel beschrieben [Hartwig and Stossel 1975, Nakaumra et
al. 2011]. Die Struktur der verschiedenen Filamine ist sehr ähnlich, sie bestehen aus drei
strukturellen Komponenten. Eine schematische Darstellung eines Filamin Moleküls ist in
Abbildung 1.2. dargestellt. N-Terminal befindet sich die Aktin-bindende Domäne (ABD),
welche aus einer CH-Domäne und drei hydrophoben Aktinbindungstellen besteht [Pollard et
al. 1994]. An die ABD schließen sich 24 Wiederholungen aus Immunglobin-ähnlichen
Strukturen (Ig-like domäne) an [Xie et al. 1998]. Jede Domäne setzt sich aus 2 ß-Faltblätter

Einleitung
4
zusammen, die sich wiederum zu insgesamt 7 ß-Strängen zusammenlagern. Die Domänen 15
und 16 sowie 23 und 24 sind durch Gelenkregionen voneinander getrennt. Die sogenannten
„hinges“ verleihen der Quervernetzung Flexibilität. Die Domäne 24 ist essentiell für die
Dimerisierung von Filamin-Molekülen [Himmel et al. 2003]. Filamine gehören zu den Aktin-
bindenden Proteinen. Sie sind an der Quervernetzung von Aktinfilamenten, welche einen
Hauptbestandteil des Zytoskeletts bilden sowie an der Vernetzung von Aktinfilamienten mit
Zellmembranproteinen beteiligt. Indirekt spielen sie ebenfalls eine Rolle in der Zell-Zell und
Zell-Matrix-Verbindung. Die Sequenzhomologie zwischen den Filaminen A, B und C beträgt
60-80 % [Gorlin et al. 1990]. Filamin C ist die muskelspezifische Isoform der Filamine und
unterscheidet sich durch die Insertion von 81 Aminosäuren in die Domäne 20 (Abb. 1.2.,
grün dargestellt) von den anderen beiden Isoformen. Diese Modifizierung scheint die direkte
Verankerung mit den Z-Scheiben ermöglichen [Van der Veen et al. 2000]. Seine
Hauptaufgabe besteht darin, die Aktinfilamente mit den Z-Scheiben zu verankern und ist
daher entscheidend für die Stabilität der Myofibrillen. Im Laufe der Jahre wurden diverse
Bindungspartner des Filamin C beschrieben, u.a. γ-Sarkoglykan [Thompson et al. 2000],
Myotilin [van der Ven et al. 2000] und Xin [van der Ven et al. 2006]. Auf Grundlage des hier
etablierten Label-freien Ansatzes zur Proteomanalyse konnte kürzlich ein neuer
Bindungspartner beschrieben werden: Xirp2 [Kley et al. 2012]. Zusätzlich übernimmt das
Filamin C neben der mechanischen Funktion auch eine Rolle in der Signaltransduktion. Es
wird vermutet, dass durch die Bindung kleiner GTPasen der Rho-Familie an das C-Terminale
Ende das Filamin C direkt an Umbauprozesse des Aktinzyoskeletts beteiligt ist [Ohta et al.
1999].
Abbildung 1.2.: Schematische Darstellung des Filamin C Moleküls, modifiziert nach Duff et al. 2011
Das Filamin C Molekül besteht aus drei strukturellen Komponenten. N-Terminal befindet sich die
Aktin-bindende Domäne. Daran schließen sich 24 Immunogloblin-ähnliche Domänen an. Die Domäne
24 ist entscheidend für die Dimerisierung von Filamin C Molekülen. Im Gegensatz zu den Filaminen A
und B verfügt Filamin C über eine zusätzliche Domäne, die Domäne 20 (grün dargestellt).
Einleitung
5
1.1.3. Die Bedeutung von Intermediärfilamenten
Zusammen mit den Mikrofilamenten und den Mikrotubuli bilden die
Intermediärfilamente die Hauptkomponenten des Zytoskeletts. Die Hauptaufgabe der
Intermediärfilamente liegt in der mechanischen Stabilisierung der Myofibrillen [Lazarides
1980]. Die Bezeichnung Intermediärfilamente wurde aufgrund ihres Durchmessers (8-10 nm)
gewählt, da dieser zwischen dem Durchmesser der dünnen (Aktinfilamente) und der dicken
Filamente (Myosinfilamente) liegt. Im Vergleich zu den Aktinfilamenten und
Myosinfilamenten sind Intermediärfilamente flexibler und formbarer [Georgatos et al. 1996].
Die Familie der Intermediärfilamente setzt sich aus 6 Gruppen zusammen, welche in Tabelle
1.1. dargestellt sind [Stewart 1990, Kim and Couloombe 2007, Iwatsuki and Suda 2010].
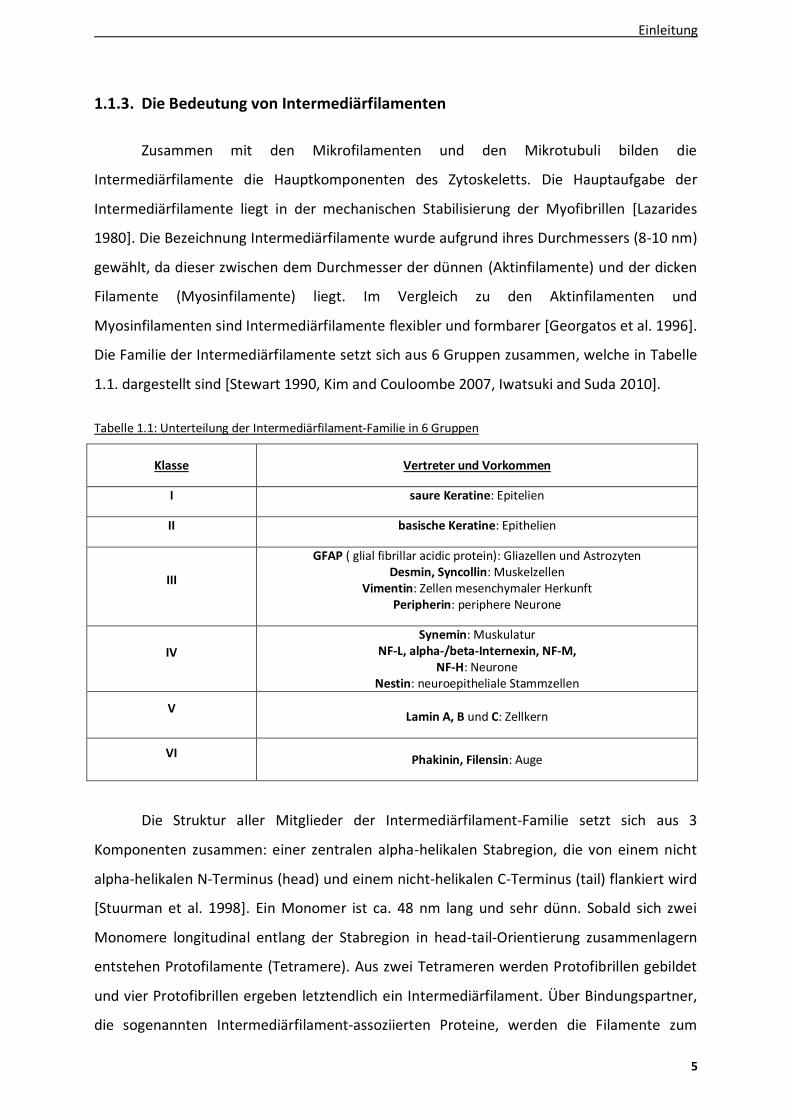
Tabelle 1.1: Unterteilung der Intermediärfilament-Familie in 6 Gruppen
Klasse Vertreter und Vorkommen
I saure Keratine: Epitelien
II basische Keratine: Epithelien
III
GFAP ( glial fibrillar acidic protein): Gliazellen und Astrozyten Desmin, Syncollin: Muskelzellen
Vimentin: Zellen mesenchymaler Herkunft Peripherin: periphere Neurone
IV
Synemin: Muskulatur NF-L, alpha-/beta-Internexin, NF-M,
NF-H: Neurone Nestin: neuroepitheliale Stammzellen
V
Lamin A, B und C: Zellkern
VI Phakinin, Filensin: Auge
Die Struktur aller Mitglieder der Intermediärfilament-Familie setzt sich aus 3
Komponenten zusammen: einer zentralen alpha-helikalen Stabregion, die von einem nicht
alpha-helikalen N-Terminus (head) und einem nicht-helikalen C-Terminus (tail) flankiert wird
[Stuurman et al. 1998]. Ein Monomer ist ca. 48 nm lang und sehr dünn. Sobald sich zwei
Monomere longitudinal entlang der Stabregion in head-tail-Orientierung zusammenlagern
entstehen Protofilamente (Tetramere). Aus zwei Tetrameren werden Protofibrillen gebildet
und vier Protofibrillen ergeben letztendlich ein Intermediärfilament. Über Bindungspartner,
die sogenannten Intermediärfilament-assoziierten Proteine, werden die Filamente zum

Einleitung
6
einen untereinander und zum anderen mit diversen Komponenten des Zytoskeletts vernetzt.
Das Intermediärfilament der Klasse III, Desmin, zählt zu den wichtigsten
Intermediärfilamenten in der Skelett-, Herz und glatten Muskulatur. Desmin spielt eine
entscheidende Rolle in der Aufrechterhaltung der zellulären Integrität. Durch den Ausbau
eines dreidimensionales Gerüstes werden die verschiedensten Kompartimente miteinander
vernetzt, z.B. Z-Scheiben untereinander und mit der Plasmamembran sowie die
Zellmembran mit cytosolischen Membranorganellen [Capetanaki et al. 1997]. Studien an
Desmin knock-out Mäusen zeigten neben der mechanischen Funktion eine zusätzliche
Vernetzung zwischen den Desminfilamenten und der Lokalisation, Form und Funktion von
Mitochondrien [Capetanaki 2002, Costa et al. 2004, Goldfarb et al. 2004]. Die gestörte
Desmin-Expression wird mit verschiedenen Myopathien in Verbindung gebracht [Goebel
1997]. 1998 wurde von Goldfarb et al. die erste Mutation im Desmin-Gen beschrieben,
seitdem wurden mehr als 60 verschiedene Mutationen im Desmin-Gen identifiziert
[Goldfarb et al. 1998, Maerkens et al. 2013]. Neben der proximalen und distalen
Muskelschwäche können bei dreiviertel aller Patienten mit Desminopathien auch
Kardiomyopathien beobachtet werden [Clemen et al. 2013].
Neben Desmin können auch weitere Intermediärfilamente mit neuromuskulären
Erkrankungen in Verbindung gebracht werden. Vimentin ist in reifen Muskelzellen nicht
nachweisbar [Goebel 1995], in der embryonalen Entwicklung hingegen ist es hoch abundant
und mit Desmin ko-lokalisiert. Die Expression von Vimentin konnte in regenerierenden
Muskelfasern, in inflammatorischen Myopathien und Muskeldystrophien sowie in atrophen
Fasern der spinalen Muskelatrophie Type I nachgewiesen werden [Bornemann et al. 1993].
Plectin zählt zu den Intermediärfilament-assoziierten Proteinen. Plectin wird im
Skelettmuskel vermehrt an den Z-Scheiben exprimiert. Zusätzlich lässt es sich im
Sarkolemma und im intermyofibrillären Netzwerk nachweisen [Banwell et al. 1999].
Wichtige Interaktionspartner des Plectin sind u.a. Desmin [Reipert et al. 1999], Vimentin
[Wiche et al. 1989] und Aktin [Steinbock et al. 1999]. Mutationen im Plectin1-Gen
verursachen schwere Formen der Hautkrankheit Epidermolysis Bullosa Simplex (EBS) mit
einhergehender Muskeldystrophie. Von Geburt an leiden die Patienten unter schmerzhafter
Blasenbildung auf der Haut, die proximale und distale Muskelschwäche entwickelt sich im
Laufe der späten Kindheit [Smith et al. 1996, Pulkkinen et al. 1996, Rezniczeck et al. 2009].

Einleitung
7
1.2. Myofibrilläre Myopathien
Die Myofibrillären Myopathien (MFM) bilden eine genetisch und phänotypisch
heterogene Gruppe neuromuskulärer Erkrankungen. Ein phänotypisches Merkmal der
Erkrankung ist die langsam progrediente Muskelschwäche der proximalen wie auch distalen
Skelettmuskulatur. Die Muskelschwäche macht sich vor allem im Bereich der Oberschenkel
und Arme deutlich. Das Hervorstechen der Schulterblätter ist typisch für einen Teil der
MFM-Patienten (Abb. 1.3. A und B). Zusätzlich können die Gesäß-, Waden- und
Unterarmmuskulatur von der Atrophie betroffen sein. Histologisch zeichnen sich die
Myofibrillären Myopathien durch die fokale Zerstörung der Myofibrillen und die Bildung von
Desmin-positiven Proteinaggregaten in den Muskelzellen aus [Schröder and Schoser 2009].
Das Auflösen der Struktur bewirkt einen Stabilitäts- und Funktionsverlust der Muskelzellen.
Elektronenmikroskopisch lässt sich der Zerfall der Myofibrillen deutlich nachweisen. Eine
vom Zerfall betroffene Myofibrille (Abb. 1.3. C, durch Pfeile gekennzeichnet) weist nicht
mehr die charakteristische Hell-Dunkel-Struktur auf. In direkter Nähe der zerstörten Z-
Scheiben lassen sich filamentäre Ablagerungen (Abb. 1.3. C, Sterne) erkennen. Im Vergleich
dazu ist eine intakte Myofibrille im oberen Teil des Bildes zu sehen (schwarzer Balken). Ein
weiteres histologisches Merkmal ist die Entstehung von Proteinaggregaten innerhalb der
Muskelzellen. In Abb. 1.3. sind zwei verschiedene Nachweistechniken dargestellt. Mittels
Gomori-Trichrom-Färbung und Immunfluoreszenzfärbung lassen sich die Proteinaggregate
spezifisch anfärben [Kley et al. 2007]. Myofibrilläre Myopathien werden in den meisten
Fällen autosomal dominant vererbt. Die Symptomatik und die klinische Manifestation sind
variabel. Erste Beschwerden treten allerdings meist ab dem Beginn der 4. Lebensdekade auf
[Selcen et al. 2004]. Die Lebenserwartung der Patienten kann durch das Auftreten einer
Kardiomyopathie sowie der Beeinträchtigung der Atemmuskulatur stark eingeschränkt sein.
Die Kardiomyopathie wird vor allem bei Patienten mit Desminopathie beobachtet [Levin et
al. 2010].
Einleitung
8
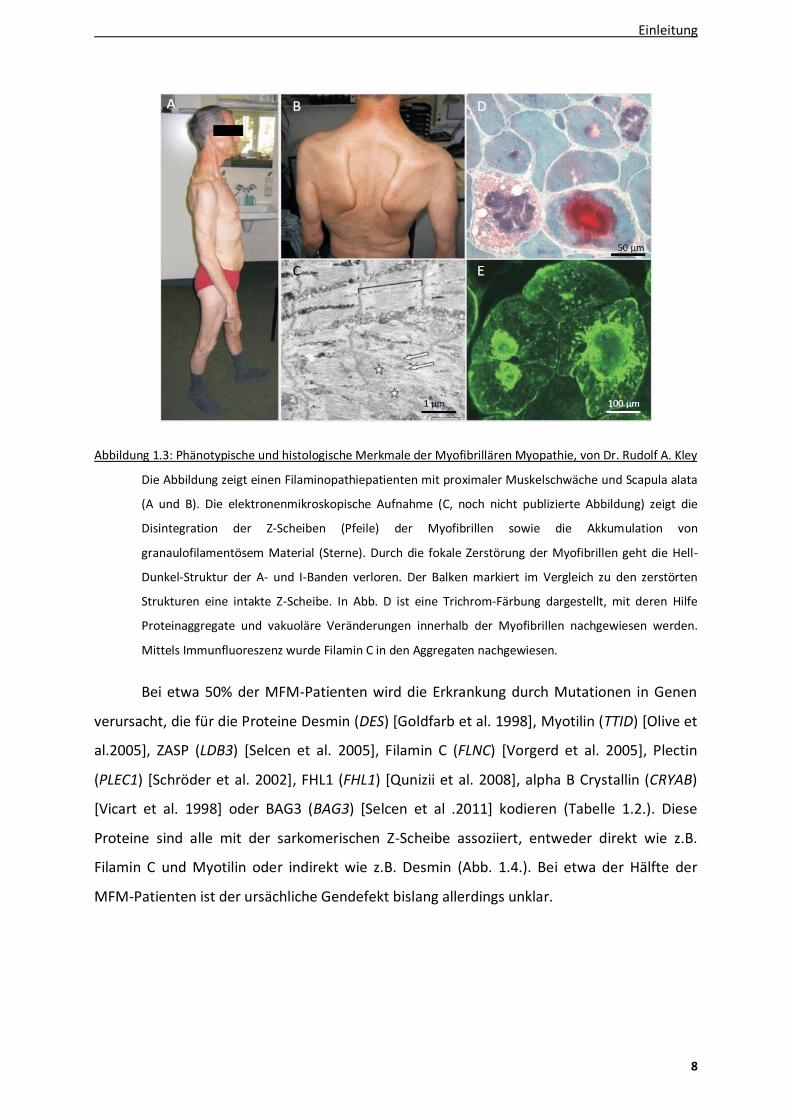
Abbildung 1.3: Phänotypische und histologische Merkmale der Myofibrillären Myopathie, von Dr. Rudolf A. Kley
Die Abbildung zeigt einen Filaminopathiepatienten mit proximaler Muskelschwäche und Scapula alata
(A und B). Die elektronenmikroskopische Aufnahme (C, noch nicht publizierte Abbildung) zeigt die
Disintegration der Z-Scheiben (Pfeile) der Myofibrillen sowie die Akkumulation von
granaulofilamentösem Material (Sterne). Durch die fokale Zerstörung der Myofibrillen geht die Hell-
Dunkel-Struktur der A- und I-Banden verloren. Der Balken markiert im Vergleich zu den zerstörten
Strukturen eine intakte Z-Scheibe. In Abb. D ist eine Trichrom-Färbung dargestellt, mit deren Hilfe
Proteinaggregate und vakuoläre Veränderungen innerhalb der Myofibrillen nachgewiesen werden.
Mittels Immunfluoreszenz wurde Filamin C in den Aggregaten nachgewiesen.
Bei etwa 50% der MFM-Patienten wird die Erkrankung durch Mutationen in Genen
verursacht, die für die Proteine Desmin (DES) [Goldfarb et al. 1998], Myotilin (TTID) [Olive et
al.2005], ZASP (LDB3) [Selcen et al. 2005], Filamin C (FLNC) [Vorgerd et al. 2005], Plectin
(PLEC1) [Schröder et al. 2002], FHL1 (FHL1) [Qunizii et al. 2008], alpha B Crystallin (CRYAB)
[Vicart et al. 1998] oder BAG3 (BAG3) [Selcen et al .2011] kodieren (Tabelle 1.2.). Diese
Proteine sind alle mit der sarkomerischen Z-Scheibe assoziiert, entweder direkt wie z.B.
Filamin C und Myotilin oder indirekt wie z.B. Desmin (Abb. 1.4.). Bei etwa der Hälfte der
MFM-Patienten ist der ursächliche Gendefekt bislang allerdings unklar.
Einleitung
9
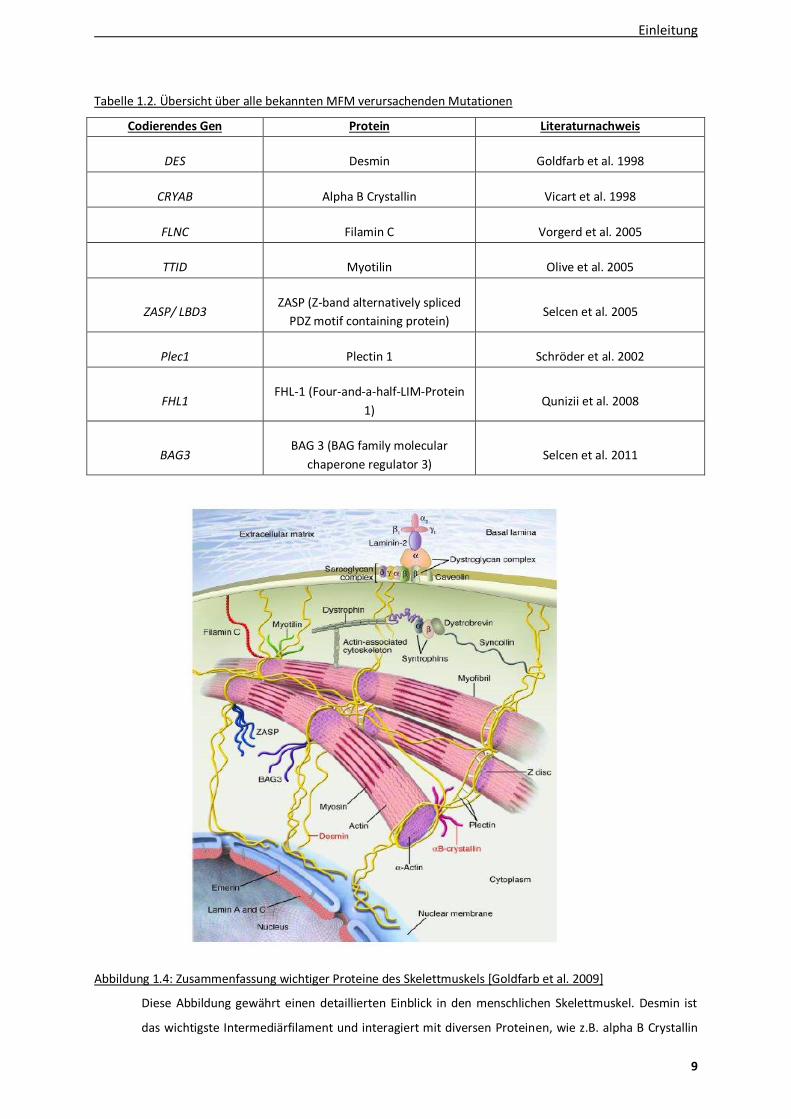
Tabelle 1.2. Übersicht über alle bekannten MFM verursachenden Mutationen
Codierendes Gen Protein Literaturnachweis
DES Desmin Goldfarb et al. 1998
CRYAB Alpha B Crystallin Vicart et al. 1998
FLNC Filamin C Vorgerd et al. 2005
TTID Myotilin Olive et al. 2005
ZASP/ LBD3 ZASP (Z-band alternatively spliced
PDZ motif containing protein) Selcen et al. 2005
Plec1 Plectin 1 Schröder et al. 2002
FHL1 FHL-1 (Four-and-a-half-LIM-Protein
1) Qunizii et al. 2008
BAG3 BAG 3 (BAG family molecular
chaperone regulator 3) Selcen et al. 2011
Abbildung 1.4: Zusammenfassung wichtiger Proteine des Skelettmuskels [Goldfarb et al. 2009]
Diese Abbildung gewährt einen detaillierten Einblick in den menschlichen Skelettmuskel. Desmin ist
das wichtigste Intermediärfilament und interagiert mit diversen Proteinen, wie z.B. alpha B Crystallin

Einleitung
10
und Plectin, und Strukturen (Z-Scheiben). Durch den Ausbau eines filamentären Netzwerks wird die
zelluläre Integrität der Zelle aufrechterhalten. Mutationen im Desmin führen zu einer Desorganisation
der Z-Scheiben, es bilden sich Proteinaggregate in den Myofibrillen, welche MFM verursachen.
Ähnliche Mechanismen werden für andere sarkomerische und cytoskeletale Proteine, wie Filamin C,
Plectin, BAG3, Myotilin und ZASP als krankheitsverursachend vermutet.
1.3. Die Filaminopathie
Die Filamin-C-assoziierten Myopathien werden durch Mutationen im Filamin C-Gen
(FLNC) verursacht. Es werden zwei Gruppen unterschieden: MFM-Filaminopathien und
Distale Filaminopathien [Fürst et al. 2013]. Die MFM-Filaminopathie ist eine progressive
Muskelerkrankung, die sich in den meisten Fällen zwischen der 4. und 6. Lebensdekade
manifestiert. Zu Beginn der klinischen Manifestation zeigen die Patienten eine proximal-
betonte Muskelschwäche, welche ihnen z.B. das Treppensteigen erschwert. Im Laufe der
Erkrankung wird es für die Patienten immer schwieriger lange Strecken zu laufen. Die
zunehmende Muskelschwäche kann schließlich zu einem Verlust der Gehfähigkeit führen.
Ein Teil der Patienten zeigt zusätzlich eine Beteiligung der Herz- und Atemmuskulatur [Kley
et al. 2007, Luan et al. 2010]. Insgesamt lässt sich sagen, dass die Patienten einen relativ
homogenen klinischen Phänotyp zeigen. Die erste Mutation p.W2710X (auch p.Tryp2710X, X
= Stopcodon) wurde 2005 von Vorgerd et al. beschrieben. Diese Punktmutation befindet sich
in der Dimerisierungsdomäne des Filamin C und wurde zunächst in einer großen deutschen
Familie nachgewiesen. Als Folge der Mutation wird das Filamin C um 16 Aminosäuren
verkürzt. Die p.W2710X-Mutation macht das Filamin C instabiler und anfälliger für die
Proteolyse [Vorgerd et al. 2005]. Die Dimerisierung des Filamin C wird so gestört und das
mutierte Protein bildet stattdessen Aggregate innerhalb der Myofibrillen [Vorgerd et al.
2005, Löwe et al. 2007]. Die p.W2710X- Mutation konnte kurz nach der ersten Beschreibung
in weiteren deutschen Familien, in Patienten der Majo MFM Kohorte und erst kürzlich in
jeweils einer mazedonischen und chinesischen Familie nachgewiesen werden [Kley et al.
2007, Selcen 2011, Kley et al. 2012]. Neben der p.W2710X-Mutation befindet sich eine
weitere Mutation in der Dimersierungsdomäne des Filamin C, die sogenannte d24Δ2-
Mutation (unpublizierte) Daten, mündliche Mitteilung durch Dr. Rudolf. A. Kley). Die
Mutation p.V930_T933del wurde 2009 in einer deutschen Familie (10 Mitglieder)

Einleitung
11
nachgewiesen. Hierbei handelt es sich um eine In-Frame Deletion in der lg-like-repeat-
Domäne 7 des Filamin C-Gens [Shatunov et al. 2009]. Die lg-ähnlichen-Domäne spielt eine
wichtige Rolle in der Protektion der zytoskeletalen Architektur. Während mechanischer
Belastung kann die Domäne entfaltet und im Anschluss wieder in ihren nativen Zustand
zurückgeführt werden. Dieser Mechanismus ist essentiell für die intrinsische Flexibilität des
Aktinnetzwerks [Furuike et al. 2001, Yamazaki et al. 2002]. Eine In-Frame Deletion in der Ig-
like-Domäne 7 des Filamin C bewirkt die Dysfunktion des kodierten Proteins [Shatunov et al.
2009]. Ebenfalls in dieser Domäne befindet sich die Mutation pK899-V964de/V899_C900ins
[Luan et al. 2010]. Die komplexe Deletion-Insertion-Mutation ist in der intermolekularen
Verbindung der 2 ß-Faltblätter lokalisiert. Der Zusammenhalt der Faltblätter wird
geschwächt und führt zu Dysfunktionen im Filamin C-Gen. Bei der p.Y1216N-Mutation
handelt es sich um eine Punktmutation in der Ig-ähnlichen Domäne 10 [Avila-Smirnov et al.
2010]. Die Mutation p.T2419M (Ig- ähnliche Domäne 22) wurde bislang in nur einem MFM-
Patienten nachgewiesen [Tasca et al. 2012]. Ergänzend zu den oben beschriebenen
Veränderungen im Filamin C-Gen konnten auch Mutationen identifiziert werden, die nicht
mit einer MFM-typischen Aggregatbildung einhergehen [Guergueltcheva et al. 2011, Duff et
al. 2011]. Die Mutation c.5160del_p.F1720LfsX63 befindet sich in der Ig-ähnlichen Domäne
15 und die Mutation p.A193T und p.M251T in der Aktin-bindenden Domäne (ABD) des Gens.
Die Mutationen in der ABD führt weder zur Bildung von Proteinaggregate noch konnten
andere MFM-spezifische Merkmale gefunden werden [Duff et al. 2011]. Diese beiden
Varianten der Erkrankung werden zu den distalen Filaminopathien gezählt. Sie manifestieren
sich um die 3. Lebensdekade und schädigen hauptsächlich die spezifischen Greifmuskeln in
der Hand. Erst später kommt zu es zur Beeinträchtigung der Beinmuskulatur.
Im Rahmen dieser Dissertation wurden insgesamt 6 Filaminopatihepatienten
analysiert. Bei 3 Patienten wurde die p.W2710X-Mutation nachgewiesen, bei 2 Patienten die
p.V930_T933del-Mutation und ein Patient zeigte die d24Δ2-Mutation (Abb. 1.5.).

Einleitung
12
Abbildung 1.5.: Zusammenfassung aller bekannten Mutationen im Filamin C-Gen, modifiziert nach Fürst et al.
2012
Insgesamt sind 9 Mutationen im Filamin C-Gen bekannt. Die p.W2710X-Mutation wurde 2005 erstmals
von Vorgerd et al. beschrieben und befindet sich, wie auch die d24Δ2-Mutation (noch nicht
veröffentlichte Daten), in der Dimersierungsdomäne des Gens. Einen gleichen Phäno- und Genotyp
wie die p.W2710X-Mutation zeigen die p.V930_T933del-Mutation [Shatunov et al. 2009] und die
pK899-V964de/V899_C900ins [Luan et al. 2010]. Mutationen in der Aktin-bindenden Domäne [Duff et
al. 2011] und die c.5160del_p.F1720LfsX63-Mutation [Guergueltcheva et al.2011] führen nicht zu einer
Aggregatbildung in Muskelzellen. Die Mutation p.T2419M wurde bislang in nur einem MFM-Patienten
mit spät einsetzender zerebellärer Ataxie nachgewiesen. Der Zusammenhang zwischen der Mutation
und den Symptomen ist noch ungeklärt. [Tasca et al. 2012]. In dieser Dissertation wurden 6
Filamimopathiepatienten mit 3 verschiedenen Mutationen (rot umrandet) analysiert.
1.4. Lasermikrodissektion
Die Lasermikrodissektion (LMD) ist ein Standardverfahren zum Isolieren von definierten
Zellstrukturen (z.B. Proteinaggregate) aus verschiedenen Gewebearten, das seit Mitte der
70er Jahre angewendet wird [Isenberg et al. 1976]. Im Laufe der Jahre erlangte die Methode
eine immer größer werdende Bedeutung in der Bioanalytik. Neben der manuellen
Mikrodissektion [Sitek et al. 2005, Poschmann et al. 2009] werden zwei laser-unterstütze
Systeme zur Mikrodissektion unterschieden. Die Laser Capture Mikrodissektion (LCM) wurde
Mitte der 90er Jahre von Michael R. Emmert-Buck etabliert [Emmert-Buck et al 1996] und
beruht auf der Verwendung einer thermoplastischen Membran, die von einem Infrarot- oder
Ultraviolettlaser geschmolzen wird. Dabei verbindet sich die inerte Membran mit dem zu
isolierenden Gewebe und kann im nächsten Schritt entfernt werden. Im Gegensatz dazu

Einleitung
13
wird bei der Laser Microbeam Mikrodissektion die gewünschte Region oder Zellpopulation
mithilfe eines Infrarot- oder Ultraviolettlasers aus dem Gewebe herausgeschnitten. Die
Probe wird dann entweder unter Einfluss der Schwerkraft in einem Reaktionsgefäß
gesammelt (Leica System) oder gegen die Schwerkraft in ein Reaktionsgefäß katapultiert
(PALM System). Die Proben werden mit größter Genauigkeit gesammelt. Das gewünschte
Areal wird exakt und schonend aus dem umliegenden Gewebe geschnitten. Dies bietet die
Möglichkeit, selbst kleinste Mengen an Probe zu untersuchen und z.B. mittels differentieller
Proteomanalyse Unterschiede zwischen krankem und gesundem Gewebe zu
charakterisieren [XU et al. 2010]. Ein weiterer Vorteil der LMD liegt darin, dass die Proben
mittels Gravitation in einem Reaktionsgefäß aufgefangen und somit Kontakt- und
Kontaminations-frei gesammelt werden können. Die Methode der LMD verbindet
(Fluoreszenz-)Mikroskopie mit Mikrodissektion mittels Laser-Technik. Das Mikroskop ist mit
einer Digitalkamera gekoppelt. Die Bilder der zu untersuchenden Probe werden direkt auf
einen angeschlossenen Computer übertragen. Eine spezielle Software sowie ein
Steuerelement sind ebenfalls direkt mit dem Mikroskop gekoppelt. So können die
gewünschten Areale direkt über den Bildschirm des Rechners markiert und ausgeschnitten
werden. Als Probe können Gewebe-Kryoschnitte auf eine spezielle Polyethylentetraphtalat-
(PET)-Membran (Leica, Wetzlar, Deutschland) übertragen oder Zellkulturen auf einer mit
Polyethylene Naphthalate-(PEN)-Membran in speziellen Petrischalen (WillCo-dish,
Amsterdam, Niederlande) kultiviert werden. Bei dem in dieser Arbeit verwendeten LMD
6500 (Leica, Wetzlar, Deutschland) war es möglich, die Proben sowohl im Durchlicht als auch
mit Fluoreszenzlicht zu mikroskopieren. Dies ermöglichte die eindeutige Identifizierung der
Proteinaggregate. In Abbildung 1.6. ist das Funktionsprinzip des LMD-Gerätes dargestellt.
Der gewünschte Bereich im Gewebe (hier ein Fluoreszenz markiertes Aggregat in einem
Filaminopathie-Kryoschnitt) wird markiert und anschließend ausgeschnitten. Das
mikrodissektierte Areal wird automatisch in einem sich darunter befindenden
Reaktionsgefäß aufgefangen. Die isolierten Proben können dann mit weiterführenden
Methoden wie z.B. solchen der Proteomanalyse weiter untersucht werden.

Einleitung
14
Abbildung 1.6.: Darstellung des LMD-Prozesses
In Abbildung A ist ein schematischer Ablauf der LMD, modifiziert nach Leica, dargestellt. Die 10 µm
dicken Gefrierschnitten befinden sich auf einem Membran-Objektträger. Der gewünschte Bereich
wird mit Hilfe eines Lasers, der mit dem Mikroskop gekoppelt ist, aus dem umliegenden Gewebe
isoliert. Die Probe fällt automatisch in ein sich darunter befindendes Einmal-Reaktionsgefäß.
Abbildung B spiegelt den Prozess anhand einer Mikrodissektion aus Filaminopathiegewebe wieder.
Das Aggregat wird umrandet und anschließend mittels Laser exakt aus dem umliegenden Gewebe
herausgeschnitten.
1.5. Grundlagen der Proteomanalyse
Proteomanalysen ermöglichen die globale Untersuchung der Gesamtheit der
Proteine von Zellen bzw. Geweben [Wilkins et al. 1996]. Die Analyse des Proteoms stellt eine
wichtige Ergänzung zur Genomanalyse dar, weil so auch Zustände, die auf der Ebene des
Genoms nicht zu ermitteln sind, erfasst werden können. Im Gegensatz zum Genom ist das
Proteom höchst dynamisch. Die Expression von Proteinen in einer Zelle variiert in
Abhängigkeit von physiologischen Funktionen, des Differenzierungsstatus und
Umweltfaktoren. Durch alternatives Spleißen der prä-mRNA sowie die zusätzliche
Einführung posttranslationaler Modifikationen (z.B. Phosphorylierungen, Glykosilierungen,
Ubiquitinierungen) entsteht ein komplexes Muster, das nur auf Proteinebene detektierbar

Einleitung
15
ist [Pandey and Mann 2000]. All diese Veränderungen werden in der Genomanalyse nicht
berücksichtigt und unterstreicht die enorme Bedeutung der Proteomanalytik [Patterson et
al. 2003]. Die Proteomanalyse gewährt einen Einblick in das Expressionsmuster von
Proteinen einer Zelle oder eines Gewebes und erlaubt es, Aussagen über qualitative und
quantitative Analyse der enthaltenen Proteine. Auf Genomebene ist dies nicht möglich, da
nur eine geringe Korrelation zwischen der mRNA-Menge und den entsprechenden
Genprodukten besteht.
1.5.1. Proteomanalysen von Skelettmuskulatur
In den vergangenen Jahren erwies sich die Proteomanalyse als vielversprechende
Methode zur Analyse von Skelettmuskulatur. Es wurden verschiedene Gel- und
Massenspektrometrie basierte Studien durchgeführt, die darauf abzielten die biochemische
Charakterisierung des Muskelproteoms von Nagern [Capitanio et al. 2009] oder zellulärer
Strukturen unter verschiedenen physiologischen Bedingungen zu beschreiben [Ferreira et al.
2010]. So wurden z.B. in Muskeln von Versuchstieren mit hohem Alter Veränderungen in der
Abundanz von Proteinen des Stoffwechsels, des kontraktilen Apparates, der
Myofibrillogenese und der zellulären Stressantwort gefunden [Ohlendieck et al. 2001]. Die
klinische Forschung nutzt proteomische Ansätze zur Identifizierung neuer Biomarker in
neuromuskulären Erkrankungen. Neben verschiedenen Tiermodellstudien z.B. Analyse von
Muskelgewebe einer Maus mit Duchenne Dystrophie [Lewis et al. 2009] wurden diverse
Proteomstudien an Patienten mit neuromuskulären Erkrankungen durchgeführt. 2005
wurden 19 Patienten mit Desminopatihe, Plectinopathie, Myotillinopathie und unbekannten
MFM-Formen analysiert. Dabei wurde die lösliche Fraktion aus Muskellysaten mittels 2D
PAGE (siehe 1.5.2.) aufgetrennt und anschließend mittels MALDI MS (siehe 1.6.2.2.)
identifiziert. Ein aus dieser Studie resultierendes Ergebnis, die Identifizierung von Hsp27,
ermöglichte die Differenzierung der primären Desminopathie von anderen MFM-Formen
[Clemen et al. 2005]. 2006 wurde von Palma et al. eine auf 2D PAGE basierender
differentieller Analyse von Dysferlinopathie-Patienten unter der Verwendung von
Muskellysaten durchgeführt. Es konnten 35 differentiell exprimierte Proteine identifiziert
werden, darunter befanden sich viele metabolische wie kontraktile Proteine [Palma et al.
2006]. Aufgrund dieser Ergebnisse wurde vermutet, dass eine Dysfunktion des Dysferlins zu
einem aktiven Umbau und Regeneration der Filamente führt. In allen bisher durchgeführten

Einleitung
16
Studien zur Proteomanalyse wurden komplette Muskellysate verwendet. Muskelgewebe ist
eine komplexe Mischung aus verschiedenen Zelltypen des Epi-, Endo- und Perimysium,
Blutgefäßen und Binde- bzw. Fettgewebe. Um die Komplexität der Proben zu reduzieren,
wurden neue Methoden eingeführt, gezielt definierte Bereiche aus dem Gewebe zu
isolieren. Dazu zählt die Mikrodissektion, welche manuell [Sitek et al. 2005, Poschmann et al.
2009] oder mit Unterstützung eines Lasers verwendet wird. Die erfolgreiche Kombination
von LMD und Massenspektrometrie wurde erstmals bei der Reducing Body Myopathie
beschrieben [Schessl et al. 2008]. Die Reducing Body Myopathie (RBM) ist eine hereditäre
Muskelerkrankung, die durch intracytoplasmatische Einschlüsse charakterisiert ist. Im
Rahmen der Studie wurden diese Einschlüsse mittels LMD aus dem Gewebe isoliert und
anschließend mittels MS analysiert. Das Protein FHL-1 wurde als das am höchsten abundante
Protein identifiziert. Parallel durchgeführte Mutationsanalysen bestätigten 4 verschiedenen
pathogene FHL-1-Mutationen. Diese Studie ist das erste Beispiel zum Nachweis einer
Krankheit-verursachenden Mutation in hereditären Myopathien. In 2012 veröffentlichten
Feldkirchner et al. eine Studie zur differentiellen Proteomanalyse von pathologischen
Proteinaggregaten in Skelettmuskel. Hier wurden Plaques aus 6 MFM-Patienten mit
verschiedenen Mutationen mittels LMD ausgelasert, welche mittels iTRAQ (isotope tags for
relative and absolute quantification) quantitativ ausgewertet wurden. Es wurde deutlich,
dass die Plaques der verschiedenen Patienten gewisse Gemeinsamkeiten in ihrer
Zusammensetzung aufwiesen und gleichzeitig wurden mutationsbedingte Unterschiede
deutlich wurden.
Die proteomische Analyse von Skelettmuskulatur hat sich im Laufe der Jahre von der
Analyse des kompletten Muskellysates hin zu einer spezifischen Analyse von definierten
Arealen entwickelt. Die in dieser Dissertation angewendete Methodik zur Analyse von
Filaminopathie gliedert sich optimal in die aktuelle Proteomanalyse von pathologischem
Muskelmaterial und trägt einen wichtigen Beitrag zum besseren Verständnis der MFM
zugrundeliegenden molekularen Mechanismen [Kley et al. 2012, Maerkens et al. 2013].

Einleitung
17
1.5.2. Auftrennung mittels 2D-Polyacrylamid-Gelelektrophorese (2D PAGE)
Die 2D-Polyacrylamid-Gelelektrophorse wurde in den 70er Jahren zeitgleich von
Klose und O’Farrell [Klose 1975, O’Farrell 1975] entwickelt. In der ersten Dimension werden
die Proteine entlang eines pH-Gradienten aufgetrennt. Hier ist zu erwähnen, dass es zwei
etablierte Verfahren gibt: Beim Trägerampholyt-System [Klose 1975] wird der pH-Gradient
während der isoelektrischen Fokussierung durch die Anwesenheit von Trägerampholyten
aufgebaut. Bei der Verwendung eines immobilisierten pH-Gradienten, IPG-System,
[Bjellqvist et al. 1982; Görg et al. 1988] ist der pH-Gradient in das Gel einpolymerisiert und
sehr stabil. In der zweiten Dimension erfolgt die Auftrennung der Proteine anhand ihres
Molekulargewichtes in einem SDS-Gel [Laemmli 1970]. Die 2D PAGE stellt eine exzellente
Methode zur Analyse komplexer Proteinproben dar. Durch die Auftrennung der Proteine ist
es möglich, eine Übersicht über das Proteom eines Organismus oder eines Gewebes zu
bekommen [Nesterenko et al. 1994]. Mit bis zu 10000 Proteinspots ist die Auflösung der 2D
PAGE extrem hoch [Klose et al. 1995]. Besonders gut lässt sich die 2D PAGE für hydrophile
Proben anwenden. Membranproteine und andere hydrophobe Proteine können aufgrund
ihrer Eigenschaften nur schwer dargestellt werden. Ähnliches gilt für die Darstellung gering
abundanter oder extrem saurer bzw. basischer Proteine. Besonders gering abundante
Proteine lassen sich nur schwer darstellen, da sie schnell von hoch abundanten Proteinen
überdeckt werden. Zum anderen ist die Empfindlichkeit gängiger Färbemethoden zu gering
um diese Proteine sichtbar zu machen. Die Einführung der Fluoreszenzfarbstoffe verbesserte
die Möglichkeit gering abundante Proteine zu detektieren.
1.5.3. Differentielle Proteomstudie und Quantifizierung von Proteinen
Mittels 2D PAGE war es bereits möglich, komplexe Proben zu analysieren und
Differenzen zwischen zwei Proben zu detektieren. Hierfür wurden die Proben über die erste
und zweite Dimension aufgetrennt und anschließend wurde die Intensität der Spots über
spezielle Bildprogramme ermittelt. Viele Proteinfärbemethoden, die eine Visualisierung der
Proteine im Gel ermöglichen, u.a. Silberfärbung und Kolloidal Coomassie, eignen sich
allerdings nur begrenzt für eine quantitative Analyse. Sie verfügen über einen zu geringen
linearen und auch dynamischen Bereich [Marcus et al. 2009]. Hinzu kommt, dass mit
herkömmlichen Methoden die Spots aus unterschiedlichen Gelen miteinander verglichen
werden müssen. Dies resultiert in einer geringen Reproduzierbarkeit und eine große

Einleitung
18
technische Streuung. Die Einführung der Zwei-Dimensionalen In-Gel Elektrophorese (2D
DIGE) erfolgte im Jahr 1997 und erhöhte die Reproduzierbarkeit und die Sensitivität der
Methode [Ünlü et al. 1997,Marcus et al. 2009]. Die zu vergleichenden Proben werden vor
der Auftrennung mit verschiedenen Fluoreszenzfarbstoffen (CyDyes™, GE Healthcare Europe
GmbH) gelabelt. Zusätzlich wird aus allen Proben ein interner Standard hergestellt, welcher
der Normalisierung der Spots über den Vergleich mit einem Referenzgel dient [Alban et al.
2003]. Anschließend werden die Proben mit dem internen Standard zeitgleich mittels 2D
PAGE aufgetrennt. So ist es möglich, die verschiedenen Gele miteinander zu vergleichen und
die Proteinspots anhand von Spotintensitäten-Verhältniswerte zu quantifizieren. Das DIGE-
System bietet zwei verschiedene Farbstoffe zur Auswahl: das CyDye™ minimal labeling und
das CyDye™ Saturation labeling. Beim minimal labeling reagieren die Farbstoffe mit den Ɛ-
Aminogruppen der Lysinreste durch Aminolyse der NHS-Ester-Bindungen. Mit dieser
Methode werden ca. 3-5 % aller Proteine markiert, wobei in jedem Protein nur ein Lysinrest
markiert wird. Es stehen drei Fluoreszenzfarbstoffe zur Verfügung. Ein Farbstoff wird für den
internen Standard verwendet, die anderen beiden werden zur Markierung der Proben
verwendet. Dies ermöglicht es, 2 Proben und einen internen Standard pro Gel aufzutrennen.
Beim Saturation labeling reagieren die Fluoreszenzfarbstoffe mit den Thiolgruppen der
Cysteinreste. Die Cysteine aller Proteine werden markiert, wodurch es möglich ist, Proben
mit sehr geringen Proteinkonzentrationen zu analysieren, die Nachweisgrenze liegt bei ca. 3
µg Protein [Sitek et al. 2005, Helling et al. 2006]. Wie beim minimal labeling gibt es auch hier
einen Farbstoff zum Markieren des internen Standards. Ein weiterer Farbstoff steht zur
Markierung einer Probe zur Verfügung. Das Spotmuster ist allerdings nicht mit einem
ungefärbten Spotmuster vergleichbar, da die Anzahl an Cysteinresten in den Aminosäure der
einzelnen Proteine variiert und somit die Proteinmasse unterschiedlich beeinflusst wird. Ein
Nachteil der Methode ist, dass Proteine, die keine Cysteine enthalten, nicht dargestellt
werden können und in einer differentiellen Studie nicht berücksichtigt werden. Im Anschluss
an die zweidimensionale Auftrennung werden die Gele über einen Fluoreszenzscanner
digitalisiert. Die Verwendung einer speziellen Auswertesoftware ermöglicht die Detektion
und Quantifizierung der einzelnen Spots.

Einleitung
19
1.6. Hochdruckflüssigkeitschromatographie (HPLC)
Es bestehen zwei Möglichkeiten ein Peptidgemisch zu analysieren. Zum einen kann
das Gemisch direkt in ein Massenspektrometer injiziert werden. Zum anderen ist es möglich,
das Gemisch zunächst über ein chromatographisches Verfahren aufzutrennen. In den
häufigsten Fällen wird hier die Umkehrphasen-Chromatographie (Reversed-Phase
Chromatography, RT-HPLC) verwendet [Steen et al. 2004]. Im Gegensatz zur Normalphasen-
Chromatographie (NP-HPLC), welche die Analytmoleküle aufgrund ihrer unterschiedlichen
Polarität auftrennt, ist hier die stationäre Phase der RT-HPLC apolar bzw. hydrophob. Die
Moleküle werden in umgekehrter Elutionsreihenfolge nach ihrer Hydrophobizität getrennt.
Die hydrophobsten Peptide werden als letztes von der Säule eluiert. In der Regel wird
Acetonitril mit ansteigendem Gradienten zur Elution der Peptide verwendet. In einem
nächsten Schritt werden die eluierten Peptide mittels Online-Kopplung ins
Massenspektrometer geleitet. Dieses Vorgehen ermöglicht die Konzentrierung niedrig-
abundanter Peptide sowie das Trennen unterschiedlicher Peptide.
1.7. Massenspektrometrie
Die Massenspektrometrie gehört heute zu den bedeutendsten Methoden in der
Peptid- und Proteinanalytik. Die Entwicklung dorthin dauerte allerdings viele Jahre, da es
lange lediglich möglich war kleine, organische Moleküle mittels Massenspektrometrie zu
analysieren. Dies änderte sich mit der Etablierung von Matrix-unterstützte Laser
Desorption/Ionisation (MALDI) und Elektrosprayionisation (ESI) als Ionisierungstechniken in
den 80er Jahren [Karas und Hillenkamp 1988, Fenn et a. 1989]. Von diesem Zeitpunkt an
gelang es große, organische Moleküle aus dem Festzustand (mittels MALDI) oder
Flüssigzustand (mittels ESI) in die Gasform zu überführen und anschließend zu ionisieren.
Ein Massenspektrometer besteht aus folgenden Hauptkomponenten: der
Ionenquelle, einem Massenanalysator und einem Detektor. Im Massenanalysator werden
die Massen nach ihrem Masse/Ladungsverhältnis (m/z) aufgetrennt. Der Detektor zeichnet
die Anzahl der entsprechenden Ionen auf. Am Ende einer Messung erhält man ein
Massenspektrum, das die Intensität der detektierten Ionen gegen den m/z-Wert aufzeigt

Einleitung
20
[Aebersold et al. 2003]. In den meisten Fällen sind Massenspektrometer, mit Ausnahme des
MALDI, mit Hochleistungsflüssigkeitschromatographen (high performance liquid
chromatography, HPLC) gekoppelt.
Abbildung 1.7.: Schematische Darstellung des allgemeinen Aufbaus von Massenspektrometern
In der vorliegenden Arbeit sollten Proteine aus flüssigem Material wie auch aus
Gelspots identifiziert werden. Die in Lösung vorliegenden Proben werden mittels In-Lösung-
Verdau für die massenspektrometrische Analyse vorbereitet. Zur Identifizierung
differentieller Proteinspots aus 2D-Gelen werden diese aus der Gelmatrix herausgestanzt
und im Gel verdaut. In beiden Fällen werden die Proteine mit Hilfe der am häufigsten
verwendeten Endoprotease Trypsin, welche spezifisch am C-terminalen Ende bevorzugt
Arginin und Lysin schneidet, in Peptide gespalten [Steen et al 2004]. Von den extrahierten
Peptiden werden in der massenspektrometrischen Analyse MS/MS-Spektren aufgenommen.
Um Peptidsequenzen aufnehmen zu können und diese anschließend einem Protein
zuordnen zu können, müssen die Peptide zunächst fragmentiert werden. Dafür werden
Ionenmassen eines bestimmten Massenbereichs ausgewählt. Diese Massen werden als
Precursor bezeichnet. Die Precursor werden dann einer Fragmentierung unterzogen, in den
meisten Fällen handelt es sich um die Fragmentierung mittels CID (collison induced
dissociation). Die Fragmentierung mittels ETD (electron transfer dissociation) wird vor allem
für die Charakterisierung von posttranslationalen Modifikationen verwendet. Die daraus
resultierenden Massenspektren lassen Rückschlüsse auf die Peptidsequenz zu. Die
generierten Massenspektren werden in einem nächsten Schritt in Proteindatenbänke
geladen, welche auf der Zusammenfassung aller bekannten und vorhersehbaren
Proteinsequenzen basieren. Die darin enthaltenden MS/MS-Spektren resultieren auf dem

Einleitung
21
theoretischen Verdau von Proteinen. Die Identifizierung der Proteine erfolgt durch den
Abgleich mit diesen theoretisch verdauten Proteinen und deren theoretisch generierten
MS/MS-Spektren. Mit Hilfe von speziellen Suchalgorithmen werden die Spektren
miteinander verglichen und deren Übereinstimmung errechnet. Zu den häufigsten
Algorithmen gehören MASCOT und Sequest [Yates et al. 1995, Perkins et al. 1999].
1.7.1. Ionenquellen
Die Ionenquelle ist ein elementarer Bestandteil eines Massenspektrometers. In der
Ionenquelle werden die Analyt-Moleküle durch Zufuhr von Energie in gasförmige Ionen
umgewandelt. Für die Ionisierung von Molekülen sind vor allem zwei Methoden von
Bedeutung: die Elektrospray-Ionisation (ESI) [Fenn et al. 1989] und die Matrix-assisted
Desorption/Ionisation [Karas et al. 1988].
1.7.1.1. Elektrospray-Ionisation (ESI)
Bei der Elektrospray-Ionisation werden die in Lösung vorliegenden Analyt-Moleküle
über eine Kapillare in ein elektrisches Feld geleitet [Fenn et al. 1989, Steen et al. 2004]. Die
Kapillare dient hier als Elektrode und der Massenanalysator wird als Gegenelektrode
eingesetzt. Es ist möglich, die Polaritäten der Elektroden anzupassen und je nach
Versuchsaufbau Anionen oder Kationen zu messen. Folglich ist es möglich, Messung in
einem Positiv- oder Negativmodus durchzuführen. In Abbildung 1.8. ist der Positivmodus
schematisch dargestellt. Die in Lösung vorliegenden Ionen bewegen sich aufgrund ihrer
Ladung im elektrischen Feld zur Gegenelektrode. An der Oberfläche des Flüssigkeitsstroms
entstehen positive Ladungen. Dies führt zur Bildung des sogenannten Taylor-Konus. In
ausreichendem Abstand zur Gegenelektrode wird der Taylor-Konus zunehmend instabil,
positiv geladene Tröpfchen entstehen. Durch den Einstrom eines neutralen Trägergases
(Stickstoff) werden die Lösungsmittelmoleküle verdampft. Dadurch erhöht sich die
Ladungsdichte an der Oberfläche der Tröpfchen. Die immer stärker werdende Abstoßung
zwischen den Teilchen führt zur Coulombexplosition. Die Tröpfchen zerfallen am Rayleigh-
Limit in viele kleine Tröpfchen mit positiver Nettoladung. Als Ergebnis weiterer
Desolvatisierungen entstehen letztendlich freie, gasförmige mehrfach geladene Ionen [Irbani
und Thompson 1976]. Im Anschluss an die ESI erfolgt meist die Weiterleitung an ein triple-
Quadrupol-Instrument oder eine Ionenfalle [Aebersold und Mann 2003]. Für die

Einleitung
22
massenspektrometrische Analyse des humanen Muskelgewebes sowie der murinen
Myoblasten wurde eine LTQ Orbitrap verwendet. Das Funktionsprinzip der der LTQ Orbitrap
im speziellen wird im Punkt 1.6.5. beschrieben.
Abbildung 1.8.: Schematische Darstellung der Elektrospray-Ionisation (ESI), modifiziert nach Cech und Enke
2001
Die Elektrospray-Ionisation wird dazu verwendet aus organischen und anorganischen Substanzen
Ionen zu erzeugen und diese mittels Massenspektrometrie zu analysieren. Die in der Lösung
vorhandenen Ionen bewegen sich aufgrund ihrer Ladung zu der entgegengesetzten Elektrode. Es bildet
sich ein Überschuss an Ionen mit gleicher Ladung. Dieser Zustand wird als Taylor-Konus bezeichnet.
Durch gezieltes Einströmen von Stickstoff wird die Verdampfung des Lösungsmittels verstärkt,
wodurch die Tröpfchengröße sich im gleichbleibenden elektrischen Feld verringert. Die Dichte des
elektrischen Feldes steigt auf der Oberfläche der Tröpfchen bis hin zur Stabilitätsgrenze (Raleigh-
Limit). Beim Überschreiten der Grenze zerfallen die Tröpfchen aufgrund ihrer Ladung in noch kleinere
Tröpfchen. Der Zerfall wird als Coulomb-Explosion bezeichnet. Letztendlich entstehen aus sehr kleinen
Tröpfchen (wenige Nanometer groß) Ionen, die dann in den Masseanalysator geleitet werden.
1.7.1.2. Matrix-assisted Desorption/Ionisation (MALDI)
MALDI beruht auf einer Entwicklung von Karas und Hillenkamp aus dem Jahre 1988
[Karas und Hillenkamp 1988]. Das Peptidgemisch wird auf einem metallischen Probenträger
mit einer im Überschuss vorliegenden speziellen Matrix ko-kristallisiert. In den meisten
Fällen wird für die Peptidanalystik als Matrix alpha-Cyano-4-Hydroyzimtsäure oder 2,5-
Dihydroxybenzoesäure (DHB) verwendet. Im Hochvakuum der Ionenquelle wird die Probe
Laserpulsen ausgesetzt. Die Energie wird von der Matrix absorbiert. Nach der Relaxation im
Kristallgitter werden explosionsartig Analyt- und Matrixmoleküle von dem Probenträger
abgelöst und ionisiert [Karas und Hillenkamp 1988]. Im Gegensatz zum ESI-Verfahren
entstehen hier hauptsächlich einfach geladene Ionen. Der Desorptions- und

Einleitung
23
Ionierungsprozess konnte bis heute nicht komplett aufgeklärt werden, die Optimierung der
analytischen Protokolle basiert zum größten Teil auf empirischen Daten [Karas et. al., 2000].
Die MALDI-Analyse eignet sich besonders gut zur Analyse wenig komplexer Proben, wie z.B.
2D Gelspots.
1.7.2. Masseanalysatoren
Zu den am häufigsten eingesetzten Masseanalysatoren gehören u.a. der Time-of-
Flight (TOF), die Ionenfalle und die Orbitrap. Zusätzlich werden auch noch Quadrupole und
das Fourier Transform Ion Cyclotron Resonance (FT-ICR) verwendet. Im Masseanalysator
werden die zuvor in der Ionenquelle erzeugten und beschleunigten Ionen nach
Masse/Ladungsverhältnis (m/z) getrennt. Die verschiedenen Masseanalysatoren werden in
der Regel mit den Ionenquellen gekoppelt. Die gängigste Kombination des MALDI ist der
TOF-Analysator. Bei der ESI existiert eine größere Vielfalt. In den meisten Fällen wird aber
eine Kombination aus ESI und Ionenfallen- oder Quadrupol-Analysatoren verwendet.
1.7.2.1. Time of Flight (TOF)
MALDI-Massenspektrometer werden in der Regel mit Flugzeit-Massenanalysatoren
(engl. Time-of-flight) gekoppelt. Die Analyt-Ionen durchlaufen eine feldfreie Strecke, in der
die sie gemäß ihres m/z-Verhältnis aufgetrennt werden. Es wird davon ausgegangen, dass
die Teilchen mit gleicher kinetischer Energie auf unterschiedlichen Geschwindigkeiten
beschleunigt werden, wenn sie unterschiedliche Massen besitzen [Abersold et al. 2003].
Sofern die Beschleunigungsspannung sowie die Länge der feldfreien Driftstrecke bekannt
sind, ist es möglich m/z für jedes Teilchen abhängig von der Flugzeit zu ermitteln.
1.7.2.2. Ionenfalle
Bei der Ionenfalle werden zwei Arten unterschieden: die 3D-Ionenfalle und die
lineare Ionenfalle (2D-Ionenfalle) [Aebersold et al. 2003]. In der 3D-Ionenfalle wird ein
zeitlich veränderliches elektromagnetisches Feld verwendet, um die Ionen festzuhalten und
anschließend gezielt auszustoßen. Die 3D-Ionenfalle besteht aus einer Ringelektrode, einer
Eintritts- und einer Endkappenelektrode. An die Ringelektrode wird eine hochfrequente
Wechselspannung angebracht, durch die ein elektrisches Feld im Inneren der Falle erzeugt
wird und die Ionen auf stabile Bahnen geleitet werden. Nach einer definierten Zeitspanne

Einleitung
24
wird die hochfrequente Wechselspannung variiert. Dies hat zur Folge, dass die Bahnen der
Ionen destabilisiert werden. Über die Endkappenelektrode werden die Ionen selektiv nach
ihrem m/z-Verhältnis aus der Ionenfalle zum Detektor geleitet. Die lineare Ionenfalle setzt
sich aus insgesamt vier hyperbolischen Metallstäben (Quadrupolstäbe) zusammen und durch
die Anordnung der Stäbe ergibt sich ein zweidimensionales elektrisches Feld. Die jeweils
gegenüberliegenden Stäbe bilden ein Paar und besitzen das gleiche Potential. An den
jeweiligen Paaren liegen eine entgegengesetzte Gleichspannung und eine um 180° versetzte
Wechselspannung an. In Abhängigkeit von der Amplitude und der Frequenz der
Wechselspannung bewegen sich Ionen mit definiertem m/z-Verhältnis auf stabilen Bahnen
[Paul 1989, Paul and Raether 1995]. Alle anderen Ionen bewegen sich auf instabilen Bahnen.
Die lineare Ionenfalle verfügt über zwei seitliche Öffnungen, über die Ionen bei Änderung
des elektrischen Potentials ausgschleust und zum Detektor geführt werden können [Mann et
al. 2001]. Im Gegensatz zu der 3D-Ionenfalle besitzt die lineare Ionenfalle eine höhere
Kapazität, welches es ermöglicht mehr Ionen zu fangen und zu speichern [Aebersold et al.
2003]. Aufgrund der beiden seitlichen Öffnungen ist es zusätzlich möglich, eine höhere
Anzahl an Ionen zu detektieren.
1.7.2.3. Orbitrap
Die Orbitrap gehört zu den modernsten Massenanalysatoren. Das Analysatorkonzept
basiert auf den Arbeiten von Alexander Makarov [Makarov 2000] und wurde vor 20 Jahren
entwickelt. Die Orbitrap ist eine Ionenfalle, in der die gasförmigen Ionen in einem
elektrostatischen Feld gefangen werden. In der Ionenfalle befindet sich zentral eine
spindelförmige Elektrode. Sobald der Ionenstrahl in die Orbitrap geleitet wird, bewegen sich
die Ionen aufgrund der elektrostatischen Anziehungskraft auf Kreisbahnen (Orbitalen) um
die Elektrode. Gleichzeitig wird eine z-axiale Oszillation der Ionen beobachtet, welche von
zwei äußeren Elektroden detektiert wird [Scigelova und Makarov, 2006]. Die Frequenz der
harmonischen Oszillation ist umgekehrt proportional zur Quadratwurzel von m/z [Hu et al.
2005, Yates et al. 2006]. Es ist möglich, die angelegten Spannungen an den Elektroden zu
ändern umso unterschiedliche Massen detektieren zu können. Die detektierten Signale
werden verstärkt und mittels Fourier Transformation in Frequenzspektren umgewandelt,
woraus letztendlich durch Zweipunktkalibrierung Massenspektren entstehen. Die Orbitrap
bietet eine hohe Massengenauigkeit, eine hohe Auflösung sowie einen hohen dynamischen

Einleitung
25
Bereich und eine hohe Sensitivität [Makarov et al. 2006 und Makarov et al. 2006].
Abbildung 1.9.: Querschnitt des Orbitrap-Massenanalysators, modifiziert nach Thermo Scientific
Die Ionen werden im elektrischen Feld gefangen. In der Ionenfalle befindet sich eine spindelförmige
Elektrode (A). Die Ionen gelangen radial in die Orbitrap und bewegen sich aufgrund der
elektrostatischen Anziehungskraft auf Kreisbahnen um die Zentralelektrode. Gleichzeitig oszillieren die
Ionen um die z-Achse. Die Außenelektrode (B) besteht aus zwei Teilen, die durch einen Keramikring (C)
isoliert werden.
In Abbildung 1.10. Ist der schematische Aufbau der LTQ Orbitrap Velos von Thermo Fisher
(Schwerte, Deutschland) dargestellt. Sie besteht aus einer linearen Ionenfalle (LTQ) und
einer C-Trap, die die LTQ und die Orbitrap miteinander verbindet. Das ionisierte
Peptidgemisch erreicht das Massenspektrometer in Gasform. Über mehrere Multipole
werden die Ionen transferiert und in einem Ionenstrahl fokussiert. Dabei durchqueren die
Ionen eine Pumpstrecke, in der stufenweise ein Hochvakuum aufgebaut wird. Dann
gelangen die Ionen in die lineare Ionenfalle. Daraufhin werden die Ionen aus der Falle
herausgeschleust und in die C-Trap geleitet. Die hier fokussierten Ionen werden gebündelt in
die Orbitrap überführt. In der Orbitrap werden dann die Precursor-Massen detektiert und
die MS-Spektren generiert. Die intensivsten Precursor-Massen werden über die C-Trap
zurück in die LTQ übermittelt. Durch Anlegen einer hochfrequenten Wechselspannung an
den Ringelektroden wird erreicht, dass sich die Ionen mit entsprechenden Precursor-Massen
aus dem ankommenden Ionenstrahl gesammelt werden und sich in stabilen Bahnen
bewegen. Nach einer definierten Zeit werden die elektrischen Potentiale an den
Ringelektroden geändert, die Bahnen der Ionen werden instabil. Die Ionen werden aus der
Ionenfalle herausgeschleust und über die C-Trap in die Kollisionszelle geleitet. Die dort
fragmentierten Ionen gelangen dann in die Orbitrap, in der die MS/MS-Spektren generiert
werden. Die Orbitrap verfügt über eine sehr hohe Massengenauigkeit und gleichzeitig eine
hohe Massenauflösung. Die Messung von Pepidionen bei hoher Auflösung benötigt längere

Einleitung
26
Zeit. Aufgrund der integrierten LTQ ist es möglich, zeitgleich zur MS-Generierung in der
Orbitrap MS/MS-Spektren in der LTQ zu erstellen.
Abbildung 1.10.: Schematischer Aufbau der LTQ Orbitrap Velos™, modifiziert nach Thermo Fisher
Nachdem die Ionen mittels ESI ionisiert worden sind, werden sie in die C-förmige C-Trap geleitet. Dort
werden die Ionen gesammelt. Anschließend werden die gebündelten Ionen in die Orbitrap geleitet,
um dort die Precursor Massen zu detektieren. Die Ionen werden tangential in das elektrische Feld
geleitet. Aufgrund des elektrischen Feldes können einige Ionen in festen ringförmigen Bahnen um die
innere Elektrode kreisen. Diese harmonischen Schwingungen sind umgekehrt proportional zu der
Quadratwurzel von m/Z. Die Änderung der angelegten elektrischen Spannung ermöglicht die
Detektion von Ionen unterschiedlicher Massen. Die Massen der einzelnen Precursor-Massen werden
ausgelesen und die am intensivsten gemessenen Massen werden über die C-Trap an die lineare
Ionenfalle geleitet. Die Ionen erreichen allerdings nicht alle gleichzeitig die LTQ, sondern werden nach
Masse getrennt und nacheinander in die LTQ abgegeben. Die in die LTQ geleiteten Massen werden
über den Einstrom vom Helium über CID fragmentiert. Die Massen der fragmentierten Ionen werden
gemessen und die entsprechenden MS/MS-Spektren generiert. Das Auswählen der Precursor-Massen
und das Fragmentieren der Ionen kann aufgrund der Kopplung von LTQ und Orbitrap parallel
durchgeführt werden.
1.7.2.4. Fragmentierung der Peptidonen
In der Massenspektrometrie werden zwei generelle Fragmentierungsarten
unterschieden. Die Fragmentierung der Peptide erfolgt mittels stoßinduzierter Kollision
(engl. Collison-induced dissociation, CID) durch niederenergetische Stöße mit einem
Kollisionsgas wie z.B. Helium, Argon oder Stickstoff [Stehen et al. 2004]. Die Kollision der
Moleküle ruft spezifische Bindungsbrüche im Peptidgerüst hervor. Durch den Bruch der
Amidbrücken entstehen hauptsächlich N-terminale y-Ionen und C-terminale b-Ionen
[Roepstorff and Fohlman. 1984, Biemann et al. 1990]. Eine Alternative zur CID stellt die

Einleitung
27
Elektronen Captur Dissoziation (ECD) dar. Die ECD wurde 1998 von Zubarev et al. eingeführt
[Zubarev et al. 1998]. Die ECD basiert auf der Freisetzung der elektrischen, potenziellen
Energie, die bei Aufnahme eines Elektrons entsteht [Zubarev et al. 1998]. Die
Fragmentierung erfolgt zwischen der Amidgruppe und des alpha-Kohlenstoffatoms der
Peptidbindung. So entstehen c- und z-Ionen. Anhand dieses Fragmentierungsmusters lassen
sich Phosphorylierungen lokalisieren [Stensballe et al. 2000]. Die Anregung der Elektronen
erfordert ein statisches Magnetfeld und beschränkt so die Anwendung der ECD auf FT-ICR-
Massenspektrometern [Thingholm et al. 2009]. Um eine ähnliche Fragmentierung auch für
andere Massenspektrometer nutzbar zu machen, wurde die ETD eingeführt. Die
Elektronentransferdissoziation (ETD) stellt eine Weiterentwicklung [Syka et al. 2004] der ECD
dar. Hier wird ein Anion mit geringer Elektronenaffinität zur Fragmentierung verwendet. Die
niedrigenergetische CID ist die am häufigsten angewendete Methode zur Fragmentierung
von Ionen. Allerdings eignet sie sich nur bedingt zur Charakterisierung von
posttranslationalen Modifikationen.
1.8. Quantitative Massenspektrometrie
Eine quantitative Auswertestrategie bietet die Möglichkeit Unterschiede in der
Synthese verschiedener Proteine zwischen zwei Proben zu ermitteln. Hierfür wird die
massenspektrometrische Analyse mit der quantitativen Auswertung kombiniert. Es gibt
verschiedene Ansätze, um dies zu erreichen, das Labeln der Proben durch Isotopen-
Markierung [Gygi et al. 1999, Ong et al. 2005] oder Label-freie Methoden [Neilson et al.
2011]. Generell wird zwischen relativer und absoluter Quantifizierung unterschieden.
1.8.1. Isotopen-Labelling
Das Isotopen-Labeling ist eine der am häufigsten angewendeten Methoden zur Gel-
freien Analyse. Es wird zwischen chemischen, enzymatischen und metabolischen Strategien
unterschieden. Die Verwendung des Isotopen-Labelings hat den Nachteil, dass es sehr teuer
ist und die Gefahr eines nicht vollständigen Labelns besteht [Hamacher et al. 2011]. 1999
wurde das sogenannte ICAT (isotope coded affinity tag) entwickelt [Gygi et al 1999]. Diese
Methode richtet sich spezifisch gegen die SH-Gruppen der Cysteine. Die Proben werden mit

Einleitung
28
einem Isotopen-gelabelten Affinitätstag gekoppelt, welches eine leichte (12C) oder schwere
(13C) C-Atome enthält. Um zwei Proben differentiell miteinander vergleichen zu können, wird
die eine Probe mit schweren bzw. leichten C-Atomen markiert. Anschließend werden die
Proben miteinander vermischt, enzymatisch verdaut und massenspektrometrisch analysiert.
Der größte Nachteil dieser Methode ist, dass nicht alle Proteine über Cysteinreste verfügen
und so nur ein Teil der Proteine quantifiziert werden können. Des Weiteren kann nur ein
geringer Teil der Sequenz ermittelt werden [Bantscheff et al. 2007]. Dies führt dazu, dass
Informationen über potentielle posttranslationale Modifikationen oder Proteinisoformen
verloren gehen. Ross und Thompson führten zwei weitere Methoden zum chemischen
Labeln ein: iTRAQ (isotope tags for relative and absolute quantification) und TMT (tandem
mass tagging) [Thompson et al. 2003, Ross et al. 2004]. Mit iTRAQ und TMT können Proteine
wie auch Peptide mit verschiedenen Tags, die die gleiche Masse besitzen und als
Reporterionen fungieren, markiert werden. iTraq sowie TMT bieten die Möglichkeit
gleichzeitig mehrere Proben relativ und absolut zu quantifizieren [Aggarwal et al. 2006,
Bantscheff et al. 2008]. Allerdings lassen sich mit diesen beiden Methoden keine komplexen
Proben analysieren. Hinzu kommt die Einschränkung bei der Wahl des
Massenspektrometers. Aufgrund der sehr geringen Masse der Reporterionen eignen sich nur
sehr sensitive Geräte (z.B. LTQ Orbitrap) [Bantscheff et al. 2008].
ICPL (isotope coded protein label) basiert auf dem isotopen Labeln aller freien
Aminosäuren in Proteinen [Schmidt et al. 2005]. Die Proteine in zwei verschiedenen Proben
werden zunächst extrahiert, alkyliert und dann entweder mit Isotopen-freien ICPL (leicht)
oder der dem ICPL-tag (schwer) markiert. Um die Komplexität des Proteingemisches zu
verringern wird dieses über eine 1D PAGE aufgetrennt. Nach dem enzymatischen Verdau der
Proteine können in der massenspektrometrischen Analyse aufgrund der schweren und
leichten Peptide Unterschiede detektiert werden. Mit dieser Methode ist es möglich, hoch
komplexe Proben zu analysieren und zu quantifizieren. Es besteht allerdings die Gefahr, dass
nicht alle Proteine vollständig markiert werden.
Die sogenannte 16O/18O-Methode beschreibt das enzymatische Labeln mit schwerem
Wasser und basiert auf dem Einbau von bis zu 2 O-Atomen während des tryptischen Verdaus
[Yao et al. 2001]. Die Proteine werden in Anwesenheit von schwerem (H218O) bzw. normalen
(H216O) Wasser verdaut [Staes et al. 2004]. Der Einbau von 18O-Atomen führt zu einem
Masseshift von 4 Da im Vergleich zu den Peptiden, die in Anwesenheit von normalem

Einleitung
29
Wasser (H216O) gespalten wurden. Die Methode ist einfach und eignet sich auch für
komplexe Proteingemische. Als nachteilig ist allerdings das Risiko zu erwähnen, dass schwere
O-Atome während der Prozessierung erneut gegen leichte ersetzt werden.
Beim GIST-Verfahren (global internal standard) werden die Proteine im Vorfeld der
Isotopenmarkierung enzymatisch in Peptide gespalten [Lottspeich, Bioanalytik, Elsevier
2006]. Diese Methode zieht allerdings gravierende Nachteile mit sich. Zum einen ist es nicht
möglich hoch komplexe Proben zu analysieren, da nur wenige Peptide detektiert und
quantifiziert werden können. Zum anderen gehen durch den vorzeitigen Verdau der Proteine
wichtige Informationen verloren.
SILAC (stable isotope labelling by amino acids in cell culture) ist ein metabolisches
Labeln basierend auf dem Einbringen von spezifischen Aminosäuren in vivo, die schwere
Isotope enthalten [Ong et al. 2005]. In der Regel erfolgt die Markierung in Zellkultur, es
lassen sich aber auch Proteine einfacher Organismen (z.B. Caenorhabditis elegans) oder
Mäusen [Krijgsveld et al. 2003, Kruger et al. 2008]. In höheren Mechanismen, wie z. B. dem
Menschen, ist diese Art der relativen Quantifizierung nicht durchführbar.
Im Rahmen dieser Dissertation sollten Filaminopathie-spezifische Proteinaggregate,
komplexe Proben aus humanem Gewebe, isoliert und quantitativ analysiert werden. Einige
der oben beschriebenen Methoden zur Isotopen-Markierung (SILAC, ICAT, GIST) eignen sich
nicht zur Analyse von komplexen Gewebeproben. Weitere Methoden mussten aufgrund der
Gefahr des unvollständigen Labelns ebenfalls für die hier durchgeführten Analysen
ausgeschlossen werden. Es wird vermutet, dass die Struktur der Proteine in gesundem und
erkranktem Muskelgewebe stark voneinander abweicht. Dies könnte direkte Auswirkungen
auf die Effizienz des Labeln haben und auch den Verdau der Proteine beeinflussen. Aus den
angeführten Gründen wurde ein Label-freier Ansatz für die Analyse der Proteinaggregate in
Filaminoapthiepatienten gewählt.
1.8.2. Label-freie Quantifizierung mittels Spectral Index Calculation
Die Entwicklung der Label-freien Quantifizierung sollte helfen, die folgenden
Nachteile des Isotopen-Labelings zu überwinden: unvollständiges Labeln, Verlust von
Proteinen durch Probenaufbereitung und hohe Kosten [Old et al. 2005, Mueller, L.N. et al.
2008]. Die Spectral Index Calculation ist ein von unserer Gruppe entwickeltes Label-freies
Verfahren zur semi-quantitativen Analyse, mit dem es möglich ist, unterschiedlich

Einleitung
30
abundante Proteine zwischen zwei Proben zu identifizieren [Müller et al. 2011]. Die
Methode basiert auf der Summierung aller in der massenspektrometrischen Analyse
generierten MS/MS-Spektren für jedes einzelne Peptid. Die aufsummierten Spektren werden
als Compounds bezeichnet. Die identifizierten Peptide werden mittels spezieller
Suchalgorithmen (z.B. Mascot™) entsprechenden Proteinen zugeordnet. Daraus ergibt sich
für jedes Protein eine unterschiedliche Anzahl an zugeordneten Peptiden und MS/MS-
Spektren, sogenannte Spectral Peptidcounts. Über den Vergleich der Summen aller Spectral
Peptide Counts ist es möglich, Rückschlüsse auf das relative Verhältnis der Menge des
Proteins in den Proben zu ziehen. Es wird davon ausgegangen, dass das Verhältnis von
Spectral Peptide Counts und die Abundanz eines Proteins proportional zueinander ist. So
zeigt ein Protein mit einer höheren Anzahl an Spectral Peptidcounts auch eine höhere
Abundanz im Vergleich zu der Kontrollprobe [Müller et. al 2011]. Bevor allerdings eine
eindeutige Aussage über die Abundanz eines Proteins in zu vergleichenden Proben getroffen
werden kann, ist es notwendig, die Werte zu normalisierten. Die Normalisierung beruht auf
der Tatsache, dass Unterschiede in der Probenbeschaffenheit, Probenaufarbeitung sowie in
der massenspektrometrischen Analyse Einfluss auf das Ergebnis haben und ausgeglichen
werden müssen. Alle Werte werden über die Gesamtzahl der Spectral Peptidcounts in den
jeweiligen Proben durch Bildung des Spectral Index normalisiert [Müller et al. 2011]. Der
Spectral Index bildet letztendlich die Grundlage, um Aussagen über die unterschiedliche
Abundanz von Proteinen in zu vergleichenden Proben treffen zu können.
Eine Alternative zum Spectral Counting stellt die vergleichende oder differentielle LC-
MS-Analyse dar [America and Cordewener 2008]. Hierbei werden zwei Proben (z.B. Zustand
A und B) mittels LC-MS/MS analysiert. Die gemessenen Intensitäten der einzelnen Peaks
werden dann miteinander verglichen, um Rückschlüsse auf die Abundanz zu ziehen. Um
sicherzugehen, dass es sich bei den verglichenen Peaks um die gleichen Moleküle handelt,
wird neben dem Masse-Ladungsverhältnis zusätzlich die Retentionszeit in den Vergleich
miteinbezogen. Im Vergleich zum Spectral Index Calculation ist die differentielle LC-MS
weniger sensitiv und zeigt Schwächen in der Reproduzierbarkeit [Old et al. 2005]. Die zu
untersuchenden Proben müssen hochgradig homolog sein müssen, da abweichende
Bestandteile Einfluss auf die Ionisierung anderer Moleküle haben können und so die
gemessenen Intensitäten beeinflussen können. Hinzu kommt der Nachteil, dass sich die
Methode nicht für die simultane Analyse mehrerer Proben eignet und sich Variationen

Einleitung
31
zwischen den verschiedenen Läufen nicht vermeiden lassen. Dennoch konnte die Methode
in einigen Studien erfolgreich angewendet werden [Lai et al. 2013]

32
2. Zielsetzung der Arbeit
Myofibrillären Myopathien stellen eine Gruppe von neuromuskulären Erkrankungen
dar, die sehr selten ist. Weltweit sind insgesamt 11 Familien bekannt, in denen die
Filaminopathie diagnostiziert werden konnte. In den letzten Jahren beschränkte sich die
Forschung größtenteils auf die genetische Analyse von Patienten sowie den
immunhistologischen Nachweis von Kandidatengenen im Skelettmuskel. Bis zum Beginn
dieser Dissertation war die Zusammensetzung der Proteinaggregate, die die MFM
kennzeichnen, bis auf wenige Komponenten unbekannt. Ziel der hier vorliegenden
Dissertation war es, ein Protokoll zur Identifizierung der Zusammensetzung der
Proteinaggregate zu etablieren. Dafür wurde eine Kombination aus LMD und Label-freier
massenspektrometrischer Analyse gewählt. Mittels LMD wurden eine definierte Fläche von
Proteinaggregate und Areale von nicht betroffenen Muskelzellen aus Filaminopathiegewebe
isoliert. Im Anschluss an die massenspektrometrische Analyse sollten die Daten mit einer
qualitativen Auswertung und 2 verschiedenen Ansätzen zur Quantifizierung analysiert
werden, um spezifisch akkumulierte Proteine in den Aggregaten zu identifizieren. Ein
weiteres Ziel stellte die Charakterisierung von Frühstadien in der Pathogenese der
Filaminopathie dar. Die Erkrankung manifestiert sich in den meisten Fällen ab dem 40.
Lebensjahr. Erst dann lassen sich Proteinaggregate im Muskel nachweisen. Eine differentielle
Proteomstudie von Filaminopathiegewebe sollte helfen, regulierte Proteine in Gewebe, das
(noch) keine Aggregate ausweist, zu identifizieren und deren Bedeutung in der frühen
Pathogenese zu klären. In Ergänzung zu den Analysen des humanen Materials sollte die
Proteomanalyse in die Zellkultur übertragen werden. In der hier vorliegenden Dissertation
wurde ein Vergleich von murinen Myoblasten und humanem Gewebe hinsichtlich ihrer
Aggregatzusammensetzung angestrebt. Es sollte untersucht werden, ob die Zellen bezüglich
der Aggregation von Proteinen ein geeignetes Modell für die Myofibrillären Myopathien
darstellen. Um eine Grundlage für nachfolgende Therapiestudien (z.B. durch Behandlung mit

Zielsetzung
33
Substanzen, die Aggregat-lösend wirken) zu ebnen, wurde Plasmid-DNA der p.W2710X-
Mutation in murine Myoblasten transfiziert. Im Rahmen einer Zeitreihe (24h, 48h und 72h
nach Transfektion) wurden positiv transfizierte Zellen, die Proteinaggregate gebildet hatten,
mittels LMD isoliert und analog zu den humanen Aggregaten massenspektrometrisch
analysiert.

34
3. Material und Methoden
3.1. Verwendete Chemikalien
Chemikalie Hersteller, Ort, Land
Aceton Riedel de Haën Sigma-Aldrich
Laborchemikalien GmbH, Seelze,
Deutschland
Acetonitril JT Baker, Deventer , Niederlande
Acrylamid (Elektrophoresegrade) Bio-Rad Laboratories, München,
Deutschland
Acrylamid-Bis Lösung
(30% Acrylamid, 0,4% Bis)
SERVA Electrophoresis GmbH,
Heidelberg, Deutschland
Agarose Merck KGaA, Darmstadt, Deutschland
Ammoniumacetat Biomol GmbH, Hamburg, Deutschland
Ammoniumhydrogencarbonat AppliChem, Darmstadt, Deutschland
Ammoniumpersulfat (APS) Bio-Rad Laboratories GmbH,
München, Deutschland
Ammoniumsulfat Bio-Rad Laboratories GmbH, München,
Deutschland
Amberlite IRN 150 Sigma-Aldrich Chemie GmbH, Steinheim,
Deutschland
Ampholin 3,5-10 GE Healthcare, München, Deutschland
Ameisensäure, 98% Carl Roth GmbH, Karlsruhe, Deutschland
BioRad Protein Assay Bio-Rad Laboratories GmbH,
München, Deutschland
Bis-Tris Sigma-Aldrich Chemie GmbH,
München, Deutschland
Bromphenolblau Bio-Rad Laboratories, München,
Deutschland
Cassettes 1,0 mm 25/Pk Life Technologies, Karlsruhe, Deutschland
BSA (bovine serum albumin) Carl Roth GmbH, Karlsruhe, Deutschland
CHAPS SERVA Electrophoresis GmbH,

Material und Methoden
35
Heidelberg, Deutschland
Complete REact® Buffer Set Invitrogen/Life Technologies, Karlsruhe,
Deutschland
Complete Mini EDTA-free Roche Applied Science, Mannheim,
Deutschland
α-Cyano-4-Hydroxyzimtsäure
(rekristallisiert)
Sigma-Aldrich Chemie GmbH,
München, Deutschland
DIGE Saturation Labelling System GE Healthcare, München, Deutschland
DMEM + GlutMAXTM – I,
Dilbecco’s Modified Eagle Medium
Gibco/Life Technologies, Darmstadt,
Deutschland
Di-Natriumhydrogenphosphat Merck KGaA, Darmstadt, Deutschland
Dimethylformamid (DMF) Sigma-Aldrich Chemie GmbH,
München, Deutschland
Dimethylsulfoxid (DMSO) Sigma-Aldrich Chemie GmbH,
München, Deutschland
1,4-Dithiothreitol (DTT) AppliChem, Darmstadt, Deutschland
EcoR I Invitrogen/Life Technologies, Darmstadt,
Deutschland
EDTA, 1% v/v in PBS Ca 2+ und Mg 2+ Biochrom AG, Berlin, Deutschland
E-Gel iBase Power System Invitrogen/Life Technologies, Darmstadt,
Deutschland
E-Gel 96 High Range DNA Marker Invitrogen/Life Technologies, Darmstadt,
Deutschland
E-Gel Safe Imager Real-Time TR Invitrogen/Life Technologies, Darmstadt,
Deutschland
E-Gel Sample Loading Puffer Invitrogen/Life Technologies, Darmstadt,
Deutschland
Essigsäure, 100% VWR International GmbH, Darmstadt,
Deutschland
Ethanol absolut Merck KGaA, Darmstadt, Deutschland
Ethylendiamin Merck KGaA, Darmstadt, Deutschland
FBS Sigma-Aldrich Chemie GmbH, Steinheim,
Deutschland
Iodacetamid AppliChem, Darmstadt, Deutschland
Glycerin JT Baker, Deventer, Niederlande
ß-Glycerophosphat Sigma-Aldrich Chemie GmbH,
München, Deutschland
Kaliumchlorid Merck KGaA, Darmstadt
Harnstoff Bio-Rad, München, Deutschland
Horse Serum, heat inactivated,
steril filtered
Sigma-Aldrich Chemie GmbH, Steinheim,
Deutschland

Material und Methoden
36
Kaliumhdrogenphosphat Merck KGaA, Darmstadt, Deutschland
Kanamycin 100x Invitrogen/Life Technologies,
Darmstadt, Deutschland
LB-Agar Invitrogen/Life Technologies,
Darmstadt, Deutschland
Lipofectamine LTX and Plus TM Reagents Invitrogen/Life Technologies,
Darmstadt, Deutschland
Morpholinopropansulfonsäure (MOPS) Sigma-Aldrich Chemie GmbH,
München, Deutschland
Natriumchlorid Merck KGaA, Darmstadt, Deutschland
Natriumhydrogenphosphat Merck KGaA, Darmstadt, Deutschland
Natriumdodecylsulfat (SDS) Biomol GmbH, Hamburg, Deutschland
Natriumdodecylsulfat (2D PAGE) Sigma-Aldrich Chemie GmbH,
München, Deutschland
Natriumorthovanadat Sigma-Aldrich Chemie GmbH,
München, Deutschland
Natriumfluorid Sigma-Aldrich Chemie GmbH,
München, Deutschland
Natrium-pyrophosphat Sigma-Aldrich Chemie GmbH, München,
Deutschland
Natriumpyruvat , 100 mM Invitrogen/Life Technologies, Darmstadt,
Deutschland
NEAA (non essentila amino acids) MEM,
100x
Gibco/ Invitrogen, Life Technologies,
Darmstadt, Deutschland
OneShot® Match1 TM-T1R Chemically
Competent E.coli
Invitrogen/Life Technologies, Darmstadt,
Deutschland
PBS pH 7,4, w/o Ca2+ und Mg2+ Biochrom AG, Berlin, Deutschland
PDA Bio-Rad Laboratories, München,
Deutschland
Plasmid Mini Kit Qiagen, Hilden, Deutschland
Plasmid Midi Kit Qiagen, Hilden, Deutschland
Pharmalyt 4,0-6,5 Amersham GE Healthcare, Freiburg,
Deutschland
Pharmalyt 5,0-8,0 GE Healthcare, München, Deutschland
o-Phosphorsäure JT Baker, Deventer, Niederlande
Phosphatgepufferte Kochsalzlösung (PBS) Invitrogen/Life Technologies, Darmstadt,
Deutschland
Quant iT ™ ds DNA BR Assay Invitrogen/Life Technologies, Darmstadt,
Deutschland
Quant iT ™ ds DNA BR Assay Fluorometer Invitrogen/Life Technologies, Darmstadt,
Deutschland

Material und Methoden
37
Qubit™ Assay Tubes Invitrogen/Life Technologies, Darmstadt,
Deutschland
Qubit™ Fluorometer Invitrogen/Life Technologies, Darmstadt,
Deutschland
Sal I Invitrogen/Life Technologies, Darmstadt,
Deutschland
SDS Bio-Rad Laboratories, München,
Deutschland
Servalyt 2,0-11 SERVA Electrophoresis GmbH, Heidelberg,
Deutschland
Servalyt 2,0-4,0 SERVA Electrophoresis GmbH, Heidelberg,
Deutschland
Servalyt 6,0-9,0 SERVA Electrophoresis GmbH, Heidelberg,
Deutschland
Sephadex G-75 GE Healthcare, München, Deutschland
SOC-Medium Invitrogen/Life Technologies, Darmstadt,
Deutschland
SYBR Safe DNA Gel Stain Invitrogen/Life Technologies, Darmstadt,
Deutschland
TE-Puffer Invitrogen/Life Technologies, Darmstadt,
Deutschland
TEMED Bio-Rad Laboratories, München,
Deutschland
Thiourea Merck KGaA, Darmstadt
Trifluoressigsäure, 99% Fluka (Sigma-Aldrich Chemie
GmbH),Steinheim, Deutschland
Tris(hydroxymethyl-)aminomethan (Tris
Base)
Sigma-Aldrich Chemie GmbH,
München, Deutschland
Tris(hydroxymethyl-)aminomethan
Hydrochlorid (Tris HCl)
Sigma-Aldrich Chemie GmbH,
München, Deutschland
Trypsin (aus Schweinepankreas) SERVA Electrophoresis GmbH, Heidelberg,
Deutschland
Trypsin 0,25& EDTA, 100 ml Invitrogen/Life Technologies, Darmstadt,
Deutschland
Urea (IEF Gele) Bio-Rad Laboratories GmbH,
München, Deutschland
Urea (IEF Puffer) Merck KGaA, Darmstadt, Deutschland

Material und Methoden
38
3.2. Verwendete Geräte und Materialien
Allgemein:
Analysenwaage BL 1500 S und BP 121 S Sartorius AG, Göttingen, Deutschland
AnchorChip-target (MTP 384-600) Bruker Corporation, Bremen, Deutschland
Binokular Olympus Deutschland GmbH,
Hamburg, Deutschland
Brutschrank Kendro Laboratory Products GmbH,
Langenselbold, Deutschland
Deckgläschen Menzel-Gläser (Thermo Fisher Scientific
Inc.), Braunschweig, Deutschland
Einfrierbehälter für Zellen Thermo Fisher Scientific Inc.,
Bonn, Deutschland
Einmal-Reaktionsgefäße (0,5 ml, 1,5 ml , 2,0
ml)
Eppendorf AG, Hamburg, Deutschland
Einmal-Reaktionsgefäße (0,2 ml)
Elektronenionisationsquelle Thermo Fisher Scientific Inc.,
Schwerte, Deutschland
Feinwaage Scaltec SBA 31 Sartorius, Göttingen, Deutschland
Fettstift Dako Deutschland GmbH, Hamburg,
Deutschland
Fluoreszenzmikroskop Olympus Deutschland GmbH, Hamburg,
Deutschland
Gewebe-Einbettmedium (Tissue Freezing
Medium®)
Jung (Leica Microsystems), Wetzlar,
Deutschland
Glasobjektträger SuperFrost® Menzel-Gläser (Thermo Fisher Scientific
Inc.), Braunschweig, Deutschland
Glyceringelatine Roti®-Mount FluorCare Carl Roth GmbH, Karlsruhe, Deutschland
Heizbad GFL, Burgwedel, Deutschland
HPLC-System Ultimate 3000 RSLCnano Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Kryostat CM 1950 Leica Microsystems, Wetzlar, Deutschland
LMDsgerät LMD 6500 Leica Microsystems, Wetzlar, Deutschland
Mikroskop CK X 31 Olympus Deutschland GmbH, Hamburg,
Deutschland
Muffelofen Carbolite CWF 1100,Watertown, USA
Omix C18, 100µl Varian Medical Systems, Zug, Schweiz
Bezeichnung Hersteller, Ort, Land

Material und Methoden
39
pH Indikatorstäbchen (pH 6,5-10,0 , nicht
blutend)
Knick, Berlin, Deutschland
pH-Meter Mettler-Toledo GmbH, Gießen, Deutschland
Pipetten (Eppendorf Pipetten Research
1000 µL, 200 µL, 20 µL, 10 µL)
Eppendorf GmbH, Hamburg, Deutschland
Polyethylentetraphtalat (PET-) Membran Leica Microsystems, Wetzlar, Deutschland
Qubit® Fluorometer Invitrogen/Life Technologies, Darmstadt,
Deutschland
Sterilbank Heraeus Holding GmbH, Hanau, Deutschland
Transferkapillare, metallbedampft Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Ultraschallbad Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Wasseranlage TKA Lab Tower Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Immunoblot:
Blottkammer (Nova Blot) GE Healthcare, München, Deutschland
Blottingmembran
(Nitrocellulose Protan 0,1 µm)
Whatman, Schleicher & Schüll GmbH
Dassel, Deutschland
Blottpapier Whatman, Schleicher & Schüll GmbH,
Dassel, Deutschland
Schüttler:
Schüttler für Gele (Typ 3005) GFL, Burgwedel, Deutschland
Schüttler für Reaktionsgefässe
(Thermomixer comfort)
Eppendorf GmbH, Hamburg, Deutschland
Vortex Mixer SA7 Bibby Scientific Limited, Staffordshire,
England
Zentrifugen:
Rotations-Vakuumkonzentrator (Speedvac)
RVC 2-25 CD plus
Martin Christ GmbH, Osterode am Harz,
Deutschland
Sprout Minizentrifuge Biozym Scientific GmbH, Hessisch
Oldendorf, Deutschland
Tischzentrifuge (Multifuge 3 S-R) Kendro Laboratory Products GmbH,
Langenselbold, Deutschland
Tischzentrifuge 5415 D Eppendorf GmbH, Hamburg, Deutschland

Material und Methoden
40
Tischzentrifuge 5415 R Eppendorf GmbH, Hamburg, Deutschland
Tischzentrifuge 5702 Eppendorf GmbH, Hamburg, Deutschland
HPLC Zubehör:
Gradientenpumpe Ultimate LC-Packings Dionex GmbH, Idstein,
Deutschland
Hauptsäule C18 Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Ladepumpe Switchos LC-Packings Dionex GmbH, Idstein,
Deutschland
PicoTip™ Emitter Nadeln New Objective Inc., Woburn, USA
Probennehmer Famos LC-Packings Dionex GmbH, Idstein,
Deutschland
Trennsäule, C18 Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Vorsäule C18 Thermo Fisher Scientific Inc., Dreieich,
Deutschland
Waters Multi λ Fluorescence Detector 2475 Waters GmbH, Eschborn, Deutschland
Massenspektrometer:
HPLC-System UltiMate 3000 RSLCnano Thermo Fisher Inc., Schwerte, Deutschland
LTQ Thermo Fisher Scientific Inc., Dreieich,
Deutschland
LTQ™ Orbitrap Velos Thermo Fisher Scientific Inc., Dreieich,
Deutschland
MALDI-TOF/TOF-MS/MS (Ultraflex™ II) Bruker Corporation, Bremen, Deutschland
Scanner:
Fluoreszenzscanner (Typhoon Trio™) GE Healthcare, München, Deutschland
Infrarotfluoreszenzscanner (Odyssey™) LI-COR Biosciences GmbH, Bad Homburg,
Deutschland
Netzteile:
Netzanschlussgerät (Consort E833) Sigma-Aldrich Chemie GmbH, München,
Deutschland
Netzanschlussgerät
(EPS 3501XL für Blotkammer)
GE Healthcare, München, Deutschland

Material und Methoden
41
Netzanschlussgerät (Consort EV231) Bio-Rad Laboratories GmbH, München,
Deutschland
Gelzubehör:
Abstandhalter, 1,5 x 10 x 300 mm Desaga GmbH, Wiesloch, Deutschland
Cellophanfolie Pütz GmbH & Co. Folien KG, Taunusstein,
Deutschland
E-Gele® mit SYBR Safe ™ Invitrogen/Life Technologies, Darmstadt,
Deutschland
Gelgießstand Desaga GmbH, Wiesloch, Deutschland
Gelkammer angefertigt
(für IEF nach Klose et al.)
Plexiglas Lehmann, Dortmund, Deutschland
Gelkammer Desaphor VA300
(für 2D PAGE 2. Dim.)
Desaga GmbH, Wiesloch, Deutschland
Gelkammer XCell-SureLock MidiCell Invitrogen GmbH, Karlsruhe, Deutschland
Gelkammer Xcell-SureLock MiniCell Invitrogen GmbH, Karlsruhe, Deutschland
GELoader™-Spitzen Eppendorf GmbH, Hamburg, Deutschland
Gelschienen, 250 mm angefertigt Plexiglas Lehmann, Dortmund, Deutschland
Geltrockner Bio-Rad Laboratories GmbH, München,
Deutschland
Glasplatten, 250 mm x 300 mm Desaga GmbH, Wiesloch, Deutschland
IEF Glasröhrchen, 1,5 x 250 mm Schott AG, Wertheim, Deutschland
Verbrauchsmaterial:
Kryoröhrchen TPP AG, Tragadingen, Schweiz
LMD Zellkulturschalen (30 mm PEN) WillCo-dish, Amsterdam, Niederlande
Parafilm Brand GmbH Co. KG, Wertheim,
Deutschland
Pipettenspitzen (für IEF, TipOne Extended
Length, Natural, 0,1-10 µL, 2-20 µL, 20
200 µL, 200-1000 µL
Starlab GmbH, Ahrensburg, Deutschland
Pipettenspitzen 100 mm lang Eppendorf GmbH, Hamburg, Deutschland
Pipettenspitzen 62 mm lang Eppendorf GmbH, Hamburg, Deutschland
BD Falcon Conical Centrifuge Tubes (15 ml
und 50 ml)
Thermo Fisher Scientific, Deutschland
Reaktionsgefässe 0,25 ml, 0,5 ml,
1,5 ml, 2 ml
Eppendorf GmbH, Hamburg, Deutschland

Material und Methoden
42
Computerprogramme:
Adobe Photoshop (Vers. 8.0) Adobe Systems Inc., Ratingen, Deutschland
Bildbearbeitungssoftware (IQ Tools™, Vers.
3.0)
GE Healthcare, General Electric Company,
München, Deutschland
Corel Graphics Suite (Vers. X4) Corel Corp., Unterschleißheim, Deutschland
DeCyder™ (Vers. 6.5) GE Healthcare, General Electric Company,
München, Deutschland
Image Quant (Vers. 5.2) GE Healthcare, General Electric Company,
München, Deutschland
Mascot 2.3.02 Matrixscience, London, England
Microsoft Office (Vers. 2007) Microsoft Office Corp., München,
Deutschland
Odyssey® (Vers. 3.0) LI-COR Biosciences GmbH, Bad Homburg,
Deutschland
ProteinScape™ Datenbank
(Vers. 1.4 und 2.1)
Bruker Corporation, Bremen, Deutschland
ProteomeDiscoverer 1.3™ Thermo Scientific Inc., Deutschland
Primärantikörper:
Antikörper Verdünnung Hersteller
Desmin, clone 33, monoclonal
Maus
1:500 Dako, Hamburg, Deutschland
Myotilin, monoclonal Maus 1:20 Novocastra/Leica
Microsystems, Wetzlar,
Deutschland
Phospho Serine, PSR-45,
monoclonal Maus
1:500 Abcam, UK
Rab35, polyclonal Hase
1:500 Abcam, UK
Xirp2, polyclonal Hase
1:1000 Biogenes, Berlin, Deutschland

Material und Methoden
43
Sekundärantikörper:
Antikörper Verdünnung Hersteller
Ziege-anti-Hase IgG, Dylight
488™ (grün)
1:1000 Dianova GmbH, Hamburg,
Deutschland
Ziege-anti-Maus IgG Texas
Red ™ (rot),
1:400 Dianova GmbH, Hamburg,
Deutschland
Maus IR680, Maus
1:15 000 LI-COR Biosciences, Bad
Homburg, Deutschland
Maus IR800, Maus
1:15 000 LI-COR Biosciences, Bad
Homburg, Deutschland
3.3. Präparation von Gefrierschnitten
Bei den hier verwendeten Gewebeproben handelte es sich um Skelettmuskel-
Biopsien von Filaminopathiepatienten, die im Rahmen eines operativen Eingriffs gewonnen
wurden. Die Biopsien wurden vorzugsweise aus dem Musculus medial gastrocnemius sowie
dem Musculus vastus lateralis entnommen. Das Gewebe wurde unmittelbar nach Entnahme
für die folgenden Analysen in 0,5 cm3 große Stücke geschnitten, in spezielles Eindeckmedium
(Tissue-Freezing Medium®, Leica Microsystems, Wetzlar, Deutschland) eingebettet und in
flüssigem Stickstoff eingefroren. Für die folgenden Analysen wurden von den Biopsien mit
Hilfe eines Kryostaten (Kryostat CM 1950 Leica Microsystems, Wetzlar, Deutschland)
Gefrierschnitte angefertigt. Für die Immunfluoreszenzfärbungen (siehe 3.5.1) wurden 6µm
dicke Gefrierschnitte auf Glasobjektträgern hergestellt, für die LMD der Proben 10µm dicke
Schnitte.
3.4. Patientendaten
Die Grundlage der Analysen bildeten Muskelbiopsien von insgesamt 6 verschiedenen
Filaminopathiepatienten. Die Genehmigung der Versuche erfolgte durch die Ethik-Komission
der Ruhr Universität Bochum [# 4078-11]. Bei allen 6 Patienten konnte mittels genetischen
Analysen sowie Immunfluorezenz-Studien von Skelettmuskelproben eine fortschreitende
Muskelschwäche diagnostiziert werden. Bei den hier beschriebenen Filaminopathie-
patienten handelt es sich um 6 Patienten mit 3 unterschiedlichen genetisch gesicherten
Material und Methoden
44
Mutationen im für Filamin C codierenden Gen FLNC. In den folgenden Tabellen sind
Mutation, Geschlecht, Alter und Herkunft der Skelettmuskelbiopsie aller Patienten
zusammengefasst. Es wurden Proben von 3 Patienten mit der Mutation p.W2710X in der
Dimerisierungsdomäne von Filamin C (d24Δ1) analysiert. Ein Patient zeigte eine weitere
Mutation in der Dimierisierungsdomäne (d24Δ2). Ergänzend dazu wurden Proben von 2
weiteren Patienten mit der Mutation p.V930_933del in der Ig-like-repeat-Domäne 7 des
Filamin C Gens analysiert. Alle Filaminopathiepatienten wurden nach Alter, Herkunft der
Muskelbiopsie und Geschlecht mit einer gesunden Kontrolle (Negativkontrolle) abgeglichen.
Die wichtigsten Parameter zum Abgleichen der Filaminopathiepatienten mit den
Negativkontrollen stellten das Alter bei Entnahme sowie die Herkunft der Biopsie. Das
Geschlecht spielte eine untergeordnete Rolle.
Tabelle 3.1.: Daten der analysierten Filaminopathiepatienten
Patient Alter
bei Biopsie
Herkunft
Muskelbiopsie FLNc-Mutation Geschlecht
1 52 Medial
gastrocnemius p.W2710X Weiblich
2 49 Vastus lateralis p.V930_930del Männlich
3 42 Vastus lateralis p.W2710X Männlich
4 46 Vastus lateralis p.W2710X Weiblich
5 46 Medial
gastrocnemius p.V930_930del Weiblich
6 41 Medial
gastrocnemius d24Δ2 weiblich
Tabelle 3.2.: Daten der gematchten Negativkontrollen
Gematchte Negativkontrolle
Alter bei Biopsie
Herkunft Muskelbiopsie Geschlecht
Zu Patient 1 50 Medial gastrocnemius männlich
Zu Patient 2 52 Vastus lateralis Männlich
Zu Patient 3 44 Vastus lateralis Männlich
Zu Patient 4 44 Vastus lateralis Weiblich
Zu Patient 5 42 Medial gastrocnemius Männlich
Zu Patient 6 42 Medial gastrocnemius Männlich

Material und Methoden
45
3.5. Immunfluoreszenzfärbungen
Im Rahmen der durchgeführten Analysen wurden zwei unterschiedliche
Färbeprotokolle erarbeitet. Zunächst wurden 6 µm dicke Gefrierschnitte auf
Glasobjektträgern angefertigt. Die Schnitte wurden mit einem Myotilin-Antikörper sowie
einem fluoreszenz-gekoppelten sekundären Antikörper gefärbt. Ziel war es, die
durchschnittliche Anzahl an für Myofibrilläre Myopathien (MFM) typische Proteinaggregaten
in den Proben zu ermitteln. Für die Lasermikrodissektion wurden 10 µm Gefrierschnitte auf
Polyethylenterephthalat(PET)-Membranen angefertigt. Zur Detektion der MFM-spezifischen
Proteinaggregate mittels Myotilin-Antikörper wurde ein optimiertes Färbeprotokoll
angewendet.
3.5.1. Immunfluoreszenzfärbungen auf Glasobjektträgern
Die Immunfluoreszenzfärbung auf Glasobjektträgern erfolgte mit 6 µm dicken Serien-
Gefrierschnitten der einzelnen Gewebeproben. Der Myotilin-Antikörper wurde in einer 1:20
Verdünnung verwendet, der sekundäre Antikörper (Ziege-anti-Maus IgG, Texas Red ™) in
einer 1:400 Verdünnung. Nach Anfertigung der Serienschnitte wurde das Gewebe zunächst
10 min bei RT mit reinem Aceton fixiert und anschließend getrocknet. Um das Verlaufen der
Lösungen auf den Objektträgern zu verhindern, wurden die Schnitte mit einem Fettstift
umrandet. Unspezifische Bindungsstellen wurden durch die Inkubation mit 2% (v/v) BSA
(bovine serum albumine)-Lösung geblockt. Anschließend erfolgte die Inkubation mit dem
primären Antikörper über Nacht bei 4°C. Nach zweimaligem Waschen mit PBS-Puffer für
2min wurden die Schnitte für 1h mit dem sekundären Antikörper bei RT im Dunkeln
inkubiert. Die Schnitte wurden erneut zweimal mit PBS (jeweils 2 min) gewaschen. Direkt im
Anschluss werden die Schnitte in Glyceringelatine unter Deckgläschen eingedeckt.
3.5.2. Immunfluoreszenzfärbungen auf PET-Membran
Um die MFM-spezifischen Proteinaggregate mittels LMD isolieren zu können, wurde
das in Punkt 3.5.1. beschriebene Protokoll modifiziert. Anstelle von Glasobjektträgern
wurden spezielle Objektträger mit PET-Membran verwendet. 10 µm dicke Kryoschnitte
wurden auf die Membranobjektträger übertragen und anschließend fluoreszenz-markiert.

Material und Methoden
46
Nach Anfertigung der Serienschnitte wurden die Schnitte ohne Fixierung mit dem ersten
Antikörper (Myotilin, 1:20) für 45 min im Dunkeln bei RT inkubiert. Nach dreimaligem
Waschen mit PBS-Puffer für jeweils 2 min wurden die Schnitte 45 min mit dem sekundären
Antikörper bei RT im Dunkeln inkubiert. In einem letzten Schritt wurden die Schnitte
getrocknet und bis zur Verwendung bei 4°C gelagert.
3.5.3. Validierung differentieller Proteine mittels Immunfluoreszenz
Für die Validierung von Proteinen, die als spezifisch in den Proteinaggregaten
identifiziert wurden, wurden Immunfluoreszenzfärbungen angefertigt. Hierfür wurden vom
Filaminopathiepatient 6 (s. Tabelle 3.4., Punkt 3.4) Serien-Kryoschnitte hergestellt. Die
Validierung erfolgte unter der Verwendung des Protokolls für Immunfluoreszenzen auf
Glasobjektträgern. Um die Proteine Xirp2 und Rab35 zu validieren wurden folgende
Verdünnungen der Antikörper eingesetzt: Xirp2 (polyklonal, Hase, 1:1000) und Rab35
(polyklonal, Hase, 1:500) und entsprechenden Sekundärantikörpern (Ziege-anti-Hase,
konjugiert mit DyLight 488™, 1:1000). Die Analyse der gefärbten Schnitte erfolgte mit Hilfe
eines Fluoreszenzmikroskops (Modell BX61, Olympus, Hamburg) bei 20facher Vergrößerung.
3.6. Herstellen von Skelett-Muskellysaten
Für die Herstellung von Muskellysaten wurden 10 µm dicke Kryoschnitte von
Filaminopathie- und Kontrollgewebe angefertigt. Je 5 komplette Schnitte wurden in ein
eisgekühltes Einmal-Reaktionsgefäß (1,5 ml) gegeben. Die Zugabe des ebenfalls eisgekühlten
DIGE-Lysepuffers erfolgte sofort. Um die Aktivität von Phosphatasen zu minimieren wurde
der Lysepuffer mit Phosphatase-Inhibitoren versetzt. Das Gewebe wurde in dem Puffer
sorgfältig resuspendiert und anschließend 10 min im Ultraschallbad behandelt. Die
Proteinkonzentration wurde mit Hilfe des BioRad Protein Assays nach Bradford [Bradford
1976] bestimmt. Die Proben wurden bis zur weiteren Verwendung bei -80°C gelagert.

Material und Methoden
47
Zusammensetzung Lysepuffer Chemikalie Endkonzentration Tris Base 1 M 30 mM Harnstoff 7 M Thioharnstoff 2 M CHAPS 4% (w/v) Natrium-orthovanadat 1 mM ß-Glycerophosphat 10 mM Natrium-Fluorid 9,5 mM Natrium-pyrophosphat 10 mM Complete Mini™ EDTA-free pH 8,0 mit HCl
3.7. Kultivierung von murinen Myoblasten
Alle in vitro Analysen wurden mit der Zelllinie C2C12 durchgeführt. C2C12-Zellen sind
murine Myoblasten, die aus isolierten Myoblasten eines Mausskelettmuskels kultiviert
werden und erstmals durch Yaffe et al. 1977 beschrieben wurden. Die Zellen besitzen die
Eigenschaft sehr schnell in Myotuben zu differenzieren und muskelspezifische Proteine zu
expremieren. Die Kryostabilate wurden Prof. Dr. Dieter O. Fürst, Leiter des Instituts für
Zellbiologie der Rheinischen Friedrich-Wilhelms-Universität Bonn, zur Verfügung gestellt. Zu
Beginn der Kultivierung wurden die Zellen bei 37°C langsam aufgetaut und mit 10 ml DMEM
(37°C) aufgefüllt. Die Zellsuspension wurde vorsichtig gemischt und anschließend bei 900
rpm 5 min zentrifugiert. Im Anschluss wurde der Überstand abgesaugt und dass Zellpellet in
5 ml Proliferationsmedium (37°C) resuspendiert. Die Zellsuspension wurde in eine 10 cm
Zellkulturschale, befüllt mit 5 ml Proliferationsmedium (37°C), gegeben. Die Kultivierung
erfolgte bei 5% Kohlenstoffdioxid und 37°C. Das Passagieren erfolgte alle 2-3 Tage, die Zellen
sollten eine Konfluenz von ca. 80 % aufweisen. Das Kulturmedium wurde zunächst
abgesaugt. Anschließend erfolgten zwei Waschschritte mit 0,02 % EDTA in PBS um
verbleibende Mediumreste zu lösen. Danach wurden 2 ml 0,05 % Trypsin in PBS auf die
adhärenten Zellen gegeben. Nach ca. 5 min Inkubation bei 37°C wurde die Reaktion durch
Zugabe von 4 ml frischem Proliferationsmedium gestoppt. Zur Kryokonservierung wurden
die konfluenten Zellen zweimal mit 0,02 % EDTA in PBS gewaschen. Anschließend wurden 2
ml Trypsin auf die adhärenten Zellen gegeben. Nach einer Inkubationszeit von ca. 5 min bei

Material und Methoden
48
37°C wurden 4 ml frisches Proliferationsmedium auf die Zellen gegeben. Die resuspendierten
Zellen wurden bei 900 rpm 5 min zentrifugiert. Der Überstand wurde abgesaugt und das
Pellet in FCS mit 10 % DMSO resuspendiert. Je 1 ml der Zellsuspension wurde in
Kryoröhrchen überführt und zunächst 4h bei -80°C gelagert. Danach erfolgte die Lagerung im
flüssigen Stickstoff.
Zusammensetzung Proliferationsmedium 500 ml Gesamtvolumen 410 ml DMEM + GlutMAXTM – I, Dilbecco’s Modified Eagle Medium 15 % FCS 2 %l Natrium-Pyruvat 1 %l NEAA
3.8. Zellbiologische Methoden
Die hier beschriebenen zellbiologischen Arbeiten basieren auf der Verwendung
zweier Vektoren: pEGFP N3 und pEGFP C2 (Clontech). Beide Vektoren eignen sich für die
Analyse der subzellulären Lokalisation von Proteinen. Im N3-Vektor ist das Zielprotein am C-
Terminus mit dem Fluoreszenzprotein EGFP fusioniert, im C2-Vektor sind Zielprotein und
EGFP am N-Terminus fusioniert. In die Vektoren wurde zum einen die humane Filamin C-
Wildtyp-Sequenz und zum anderen die humane Sequenz von Filamin C mit eingebrachter
Mutation in der Dimerisierungsdomäne hineinkloniert. Die Konstrukte wurden von der AG
Prof. Dr. Dieter O. Fürst, Institut für Zellbiologie der Rheinischen Friedrich-Wilhelms-
Universität Bonn, zur Verfügung gestellt. Die Klonierungsarbeiten wurden von Dipl. Biologin
Yvonne Leber, Doktorandin in der AG Prof. Dr. Fürst, durchgeführt.
Tabelle 3.3.: Übersicht über die verwendeten FLNC-Konstrukte
Bezeichnung Konstrukt
Mutation
FLNC fl C2 pEGFP wt Wildtyp
FLNC fl C2 pEGFP mut W2710X
FLNC fl N3 pEGFP wt Wildtyp
FLNC fl N3 pEGFP mut W2710X
FLNC fl N3 pEGFP Leervektor -

Material und Methoden
49
3.8.1. Transformation von Konstrukten in E. Coli-Zellen
Für die Transformation der DNA-Konstrukte wurden One Shot ®Mach1 ™ -T1R
chemisch kompetente E.Coli–Zellen (Invitrogen) verwendet. Die Zellen (50µl) wurden
langsam auf Eis aufgetaut. Sobald die Zellen aufgetaut waren, wurde 1µl DNA-Konstrukt
hinzugefügt und 30 min auf Eis inkubiert. Der Transformationsansatz wurde einem 30 sec
Hitzeschock unterzogen, dann folgte eine weitere Inkubation über 2 min auf Eis.
Anschließend wurden 250 µl SOC Medium (Invitrogen/Life Technologies, Darmstadt,
Deutschland) hinzugegeben und der Ansatz bei 37°C und leichtem Schütteln (225 rpm) 30
min inkubiert. 50µl des Transformationsansatzes wurden auf einer 10 cm Agarplatte
ausgestrichen. Die vereinzelten Zellen wuchsen über Nacht. Am nächsten Tag wurden 2 - 3
vereinzelte Kolonien gepickt und in 3 ml LB Medium (mit 15 µl Kanamycin) überführt. Die
Mini-Kultur schüttelte über Nacht bei 37°C. 2ml der Mini-Kultur wurden für eine Plasmid
Mini Präparation (Qiagen, siehe unten) verwendet. 1,5 ml der Kultur wurden in 100 ml LB-
Medium (mit 500 µl Kanamycin) beimpft, der Rest der Kultur wurde bei 4°C gelagert. Nach
ca. 12h Inkubation erfolgte eine Plasmid Midi-Präparation (Qiagen) nach Protokoll des
Herstellers.
3.8.2. DNA-Extraktion und Überprüfen der Plasmid-DNA
Die Extraktion der DNA erfolgte als Miniprep sowie als Midiprep nach
Herstellerprotokoll. Im Anschluss an die Midiprep wurden die DNA-Konzentrationen der
Proben gemessen. Hierfür wurde das Quant iT ™ ds DNA Assay Kit von Invitrogen nach
Herstellerprotokoll verwendet. Die DNA-Konzentrationsmessung erfolgte mit dem Qubit®
Fluorometer von Invitrogen. Zur Überprüfung der Plasmid-DNA wurde ein 1µg DNA im
Restriktionsverdau mit EcoR I und Sal I für 1h bei 37°C inkubiert. Im Anschluss wurden die
entstandenen Fragmente in einem AgaroseGel (E-Gel 1,2%, Invitrogen/Life Technologies,
Darmstadt, Deutschland) auf ihre Größe untersucht. Das Insert wies die erwartete Größe
von. 8,1 kb und der Vektor von 4,7 kb auf. 5 µl des Restriktionsansatzes wurden mit 15 µl
nuklease freiem H2O vermischt und auf ein E-Gel® mit SYBR SAFE ™ von Invitrogen gegeben.

Material und Methoden
50
Ansatz Restriktionsverdau, 20µl Menge Substanz 1 µg DNA 0,5 µl EcoR I (5U) 0,5 µl Sal I (5U) 2 µl Reaktionspuffer 16 µl Nukelase freies H2O
3.8.3. Herstellen von Glycerol Stocks
Hierfür wurden 3 ml LB-Medium mit 15 µl Kanamycin und 100 µl der Kultur versetzt.
Die Kulturen wurden über Nacht bei 37°C geschüttelt. Anschließend wurden 500 µl Glycerol
in ein 1,5, ml Einmal-Reaktionsgefäß vorgelegt. 500 µl der Über-Nacht-Kultur wurden dann
hinzugeben und beide Bestandteile wurden gut miteinander vermischt. Die Glycerol Stocks
wurden bei -80°C gelagert. Zur Herstellung einer Midi-Kultur wurden bei Bedarf 500 µl des
Glyercol Stocks in 100 ml LB-Medium (mit Kanamycin) bei 37°C über Nacht geschüttelt. Am
nächsten Tag wurde eine Plasmid Midi-Präparation (Qiagen) nach Protokoll des Herstellers
durchgeführt.
3.8.4. Transfektion muriner Myoblasten
Ziel der hier durchgeführten Transfektionen war es, die MFM-spezifischen
Aggregatbildung in C2C12-Zellen zu analysieren und mit dem humanen Proteom zu
vergleichen. In Anlehnung an die Analyse von humanem Material wurden die Aggregate
sowie die entsprechenden Kontrollflächen aus murinen Zellen mittels LMD isoliert. Die
C2C12-Zellen wurden in speziellen LMD-Zellkulturschalen (WillCo dish, PEN Membran 30 mm)
ausplattiert. Hierfür wurden 1000 µl Proliferationsmedium in die LMD-Schalen gegeben und
mit 250 µl Zellsuspension vermischt. Die Zellen wuchsen 24h bei 37°C bis zu einer Konfluenz
von ca. 70 %. Am nächsten Tag erfolgte die transiente Transfektion mit der Plasmid-DNA. Pro
Schale wurden 300 µl DMEM in ein Falcon vorgelegt. Anschließend wurden 2,25 µg der DNA
hinzugefügt und mit Hilfe des Vortexers gemischt. Als nächstes wurden 2,25 µl Plus ™
Reagent in das Falcon pipettiert. Zum Mischen wurde das Falcon vorsichtig geschwenkt. Es
folgte eine Inkubationszeit von 5 min bei RT. Im Anschluss wurden 4,5 µl Lipofectamin ™ LTX
zum Transfektionsansatz hinzugeben. Der Ansatz wurde mittels Vortexer gemischt und 30

Material und Methoden
51
min im Dunkeln bei RT inkubiert. Anschließend wurde der Transfektionsansatz
tröpfchenweise auf die C2C12-Zellen pipettiert. Die LMD erfolgte 24h, 48h und 72h nach
Transfektion.
3.9. Lasermikrodissektion (LMD)
Die LMD ist eine Methode, mit der es möglich ist, spezielle Areale aus dem Gewebe
zu isolieren und anschließend zu analysieren. Diese Methode wird hauptsächlich zur
Gewinnung von Proben eingesetzt, die durch ihr limitiertes Vorkommen charakterisiert sind.
Die dieser Arbeit zugrunde liegenden Muskelbiopsien wurden zum einen durch die geringe
Anzahl der Aggregate pro Patient limitiert, in einem gefärbten Kryoschnitt konnten ca. 10-15
Aggregate detektiert werden. Zum anderen stellten die wenigen vorhandenen
Filaminopathiepatienten eine Limitierung dar. Im Rahmen dieser Dissertation wurde das
LMD 6500 von Leica (Wetzlar, Deutschland) verwendet. Eine mit dem Mikroskop
verbundene Digitalkamera übertrug Bilder der 10 µm dicken Gefrierschnitte an einen
Rechner. Zur Erkennung und Isolierung der Aggregate wurde die 40fach Vergrößerung des
Mikroskops gewählt, im Falle des umliegenden Gewebes reichte die 20fache Vergrößerung
aus. Die Aggregate und die umliegenden Muskelfasern wurden über ein Steuerelement
markiert und anschließend automatisch mittels Laser ausgeschnitten. Die Gewebestücke
fielen senkrecht in ein leeres 0,5 µl Einmalgefäß. Im Anschluss an das Sammeln wurde der
Lysepuffer auf die Proben gegeben und für 30 min inkubiert. Anschließend wurden die
Proben 10 min bei 10000 rpm zentrifugiert. Die Proben wurden bei -80°C gelagert und vor
der Weiterverarbeitung 5 min im Ultraschallbad behandelt. Ziel der LMD war es die
Proteinaggregate hinsichtlich ihrer Zusammensetzung zu analysieren und Frühstadien der
Erkrankung anhand von aggregatfreiem Gewebe zu identifizieren. Um dies zu erreichen,
wurden drei verschiedene Gruppen an Skelettmuskelgewebe gesammelt:
(1) MFM-spezifische Proteinaggregate aus Filaminopathiegewebe
(2) umliegendes Gewebe ohne Proteinaggregate aus Filaminopathiegewebe
(3) gesundes Gewebe aus Kontrollgewebe

Material und Methoden
52
Im Vorfeld der Studien war es zunächst notwendig die Mengen an Probenmaterial zu
optimieren. Definierte Mengen an Gewebe wurden mittels Lasermikrodissektion ausgelasert.
Am Ende der Vortests stand fest, dass 100 Aggregate ≈ 250 000 µm2 für die Analyse optimal
zu sein scheint. Zur Charakterisierung von Frühmarkern erwiesen sich 6,7 mio µm2 als
optimal.
3.10. Solubilisierung der Aggregate
Zu Beginn der Arbeiten gab es in der Literatur keine Hinweise auf die Löslichkeit von
MFM-spezifischen Proteinaggregaten. Um die Zusammensetzung der Aggregate
aufschlüsseln zu können, musste im Anschluss an die LMD zunächst untersucht werden, in
welchem Puffer bzw. Lösungsmittel die Aggregate am besten solubilisiert werden können.
Für diese Vorstudien wurden 3 verschiedene Lösungen verwendet: in jeweils 20 µl 6M
Guanidin-HCl und 8M Urea und 20 bzw. 40 µl 98% Ameisensäure. 250 000 µm2
Aggregatfläche wurden aus Immunfluoreszenz-gefärbten Filaminopathiegewebe
ausgeschnitten. Nachdem die Proben gesammelt worden waren, wurden sie 30 min in den
Lösungen inkubiert und anschließend zentrifugiert. Bis zur massenspektrometrischen
Analyse wurden die Proben bei -80°C gelagert. Für die nachfolgende Studie erwies sich 98%
Ameisensäure als das Mittel der Wahl.
3.11. In-Lösung-Verdau
Für die massenspektrometrischen Analysen wurden die LMD-Proben einem
tryptischen Verdau unterzogen, um die Proteine in Peptide zu spalten. Zunächst wurden die
Proben 1h 30 min aufkonzentriert (Rotations-Vakuum-Konzentrator RVC 2-25 CD plus,
Martin Christ GmbH, Osterode am Harz). Anschließend wurden 74,25 µl 50 mM
Ammoniumbicarbonat (pH 7,8) auf die Proben gegeben. Durch die Zugabe und Inkubation
(20 min bei 56°C und 600 rpm) von 0,25 µl 2M DTT vorhandene Disulfidbrücken reduziert.
Um eine Reoxidation der Cysteinreste zu verhindern, wurden 2,7 µl 0,55 M Iodacetamid zu

Material und Methoden
53
den Proben gegeben und 15 min bei RT im Dunkeln inkubiert. Im nächsten Schritt wurde 1 µl
1 % (v/v) Trypsin Enhancer ProteaseMAX ™ Surfactant hinzugefügt. Vor der Zugabe von 1,8
µl (1µg/µl in 50 mM Essigsäure) wurde bei allen Proben der pH-Wert überprüft. Der pH-Wert
sollte bei 7,4–7,5 liegen um optimale Bedingungen für den tryptischen Verdau zu
gewährleisten. Der In-Lösung-Verdau erfolgte über Nacht bei 37°C. Am nächsten Morgen
wurde der Verdau durch die Zugabe von 5,25 µl 10 % TFA gestoppt.
3.12. Aufreinigung der Proben nach dem In-Lösung-Verdau
Für die Aufreinigung der Proben wurden spezielle Filter-Pipettenspitzen mit C18-
Membranen (OMIX C18-Tips, Varian Medical Systems, Schweiz) verwendet. Die hydrophoben
C18-Gruppen in den Filter-Pipettenspitzen binden spezifisch hydrophobe Peptide und
trennen diese von anderen Bestandteilen, wie z.B. unverdaute Proteine mit stark
hydrophilen Oberflächen und Salze des tryptischen Verdaus. Zu Beginn der Aufreinigung
wurden die OMIX C18-Tips zunächst zweimal mit 100 µl 50 %-igen Acetonitril-Lösung und
zweimal mit 100 µl einer 0,1 %-igen TFA-Lösung äqulibriert. Nachdem die OMIX C18-TIps
vorbereitet wurden, wurde jede einzelne Probe insgesamt 8-mal über die Säule pipettiert.
Anschließend wurde die C18-Membranen zweimal mit je 100 µl 0,1 % (v/v) TFA gewaschen
um Salze, unverdaute Proteine und andere nicht gebundene Bestandteile zu entfernen. Im
nächsten Schritt wurden die gebundenen Peptide durch zweimalige Zugabe einer Lösung aus
55% (v/v) Acetonitril und 0,1% (v/v) TFA eluiert. Die Eluate wurden 5 min im Rotations-
Vakuum-Konzentrator auf ein Gesamtvolumen von 18 µl eingeengt. Die eingeengten Eluate
wurden durch die Zugabe von 45 µl 0,1% (v/v) TFA-Lösung auf ein Gesamtvolumen von 63 µl
aufgefüllt. Für die massenspektrometrische Analyse wurden jeweils 16 µl Ansatz verwendet.

Material und Methoden
54
3.13. Differentielle In-Gel Elektrophorese des Proteoms von Patienten mit
Filaminopathie
Die Analyse des Proteoms von Filaminopathiepatienten erfolgte mittels differentieller
In-Gel Elektrophorese (DIGE). In der ersten Dimension erfolgt die Auftrennung nach dem
isoelektrischen Punkt (pI). Die Proteine wandern im elektrischen Feld entlang eines pH-
Gradienten bis zum dem pH-Wert, bei dem ihre Nettoladung gleich null ist. In der zweiten
Dimension erfolgt eine Auftrennung nach der elektrophoretischen Mobilität in einer SDS-
Polyacrylamid-Gelelektrophorese [Laemmli 1970].
3.13.1. Markierung der Proteine durch CyDye®-Farbstoffe
Im Vorfeld der Zweidimensionalen Auftrennung wurden die Proteine zunächst mit
Fluoreszenzfarbstoffen markiert [Marouga et al. 2005]. Für analytische Gele wurden 100
nMol Farbstoff bei 4°C mit 50 µl wasserfreiem Dimethylformamid (DMF) versetzt, für
präparative Gele wurden 400 nmol mit 20 µl DMF versetzt. Anschließend wurden die
Ansätze 30 sec gemischt und bei 12 000 x g 30 sec zentrifugiert. Die Endkonzentration der
Farbstoffe für analytische Gele betrug 2 mM und für präparative 20 mM. Aufgrund der
äußerst geringen Proteinkonzentration der Proben wurde das Saturation Labelling System
(GE Healthcare, München Deutschland) verwendet [Helling et al. 2006]. Hierfür wurden
zunächst die Proben mit 0,5 µl 2 mM Tris-2-carboxyethylphosphin-hydrochlorid (TCEP) bei
37°C für 1h im Dunkeln reduziert. Dieser Schritt diente der Entfaltung der Proteine sowie
dem Lösen der Disulfidbrücken. Die komplette Probe (ca. 5 µg) wurde anschließend mit 1µl
Farbstoff für 30 min bei 37°C inkubiert. Die optimale Menge von TCEP und Farbstoff wurde
in Vorstudien ermittelt. Um Farbstoff-spezifische Effekte zu verhindern, wurde ein
sogenannter „Color-Switch“ bei der Zugabe der Cy-Farbstoffe beachtet. Dies bedeutet, dass
die Patienten- und Negativkontrollen abwechselnd mit Cy3 und Cy5 markiert wurden. Zum
Abstoppen der Reaktion wurden die Proben direkt im Anschluss mit Dithiothreitol (DTT) 15
min bei RT inkubiert. Das eingesetzte DTT-Volumen richtete sich nach dem Gesamtvolumen
der Probe, es wurde jeweils 1/10 DTT zum Gesamtvolumen verwendet. Die Proben wurden
maximal 3 Tage vor dem Start der isoelektrischen Fokussierung markiert und bis zur
weiteren Verwendung wurden die Proben bei -80°C gelagert.

Material und Methoden
55
3.13.2. Isoelektrische Fokussierung mit Trägerampholyten
In der ersten Dimension wurden die Proben durch elektrische Fokussierung nach
Klose [Klose 1975] mittels Trägerampholyten in Rundgelen (20 cm, 1,5 cm Durchmesser mit
Beule) aus Polyacrylamid aufgetrennt. Die Gele bestanden aus einem Trenngel und dem
Abschlussgel an der Kathode. Die Probe wurde an der Anodenseite auf das Trenngel
aufgetragen und mit Schutzgel sowie Anodenpuffer überschichtet. Die Auftrennung erfolgte
in einem pH-Bereich von 2 bis 11. Zur Fokussierung der Proteine wurde der unten
beschriebene Spannungsgradient verwendet.
Spannungsgradient für die isoelektrische Fokussierung, 20 cm
Spannung (V) Dauer 100 1 h 200 1 h 400 17,5 h 650 1 h 1000 0,5 h 1500 10 min 2000 5 min
Trägerampholytgemisch pH 2 - 11, Stammlösung Menge/ Großansatz Ampholin, pH Bereich 8 ml Ampohlin pH 3,5 – 10 8 ml Servalyte pH 2 - 11 24 ml Pharmalyt pH 4 – 6,5 16 ml Pharmalyt pH 5 – 8 Trägerampholytgemisch, komplett Menge/ Großansatz Ampholin, pH Bereich 7 ml Trägerampholytgemisch Stammlösung 1 ml Servalyte pH 6 - 9

Material und Methoden
56
Separationsgel
EndKonzentration Substanz 9 M Harnstoff 5,1 % (w/v) Glycerin 3,59 % (w/v) Acrylamid 0,31 % (w/v) PDA 0,06 % (v/v) TEMED 4,1 % (v/v) Trägerampholytgemisch komplett Sephadexgemisch Menge Substanz 331 mg Harnstoff/Thioharnstoff-Gemisch (7 M/ 2M) 25 µL Trägerampholytmischung 270 mg Sephadexsuspension 25 µL DTT (1,08 g/ 5 mL) 2 M Abschlussgel, pH 2-11 Endkonzentration Substanz 12,3 % (w/v) Acrylamid 0,13 % (w/v) PDA 4,0 % (v/v) Trägerampholytmischung 9 M Harnstoff 0,5 % (w/v) Glycerin 0,06 % (v/v) TEMED Schutzlösung Endkonzentration Substanz 30 % (w/v) Harnstoff 5 % (w/v) Glycerin 2 % (v/v) Servalyte 2-4 Anodenpuffer
Konzentration Substanz 0,742 M Phosphorsäure 3,0 M Harnstoff Kathodenpuffer Konzentration Substanz 5 % (v/v) Ethylendiamin 5 % (v/v) Glycerin 9 M Harnstoff

Material und Methoden
57
Inkubationspuffer Endkonzentration Substanz 125 mM Tris Base-Lösung (1M, pH 6,8) 40 % Glycerin 65 mM DTT 104 mM SDS
3.13.3 Proteinauftrennung nach Molekulargewicht
Nach Beendigung der isoelektrischen Fokussierung wurden die Rundgele 15 min in
Inkubationspuffer inkubiert. Dieser Schritt war notwendig, um die Proteine mit SDS zu
beladen und die Gele für die zweite Dimension zu puffern. Anschließend wurden die
Rundgele mehrfach mit 1x SDS-Laufpuffer gewaschen. Im Anschluss wurden die Rundgele
luftblasenfrei auf die Geloberkante eines Polyacrylamidgels (15 %,250 mm x 300 mm x
15 mm) gelegt. Zur Fixierung wurden die Rundgele mit 0,4 % Agarose überschichtet. Der
Agarose wurde Bromphenolblau zugesetzt, welches als Markierung der Lauffront während
der zweiten Dimension diente. Zu Beginn der Auftrennung liefen die Proteine 15 min bei
75mA und 20°C in das Polyacrylamidgel ein. Anschließend wurde die Elektrophorese bei
einem Stromfluss von 100 mA pro Gel durchgeführt. Der Auftrennung wurde beendet als
sich die Lauffront ca. 2 cm vor der unteren Gelkante befand.
Trenngelpuffer, pH 8,8 EndKonzentration Substanz 15 % Acrylamid 0,2 % Bisacrylamid 0,75 M Tris HCl 0,06 % TEMED 0,2 % SDS Laufpuffer EndKonzentration Substanz 0,3 % Tris Base 1,44 % Glycin 0,1 % SDS

Material und Methoden
58
3.13.4. Differentielle Gelbildanalyse
Die Gele verblieben bis zur Bilderfassung zwischen den Glasplatten. Die Platten
wurden vor dem Scan gründlich mit 100 % Ethanol gereinigt. Anschließend wurden die Gele
im Typhoon Trio Fluorescence Imager (GE Healthcare) digitalisiert. Die Exitations- und
Emissionswellen wurden wie folgt eingestellt: Cy3 550 nm und Cy5 500nm. Bei der
Einstellung wurde darauf geachtet, dass die maximale Intensität aller Kanäle vergleichbar ist.
Der Vorscan wurde bei 1000 Microns durchgeführt, der Hauptscan bei 100 Microns.
Nach dem Erfassen der Bilder wurden diese mittels ImageQuant TL™ zurecht
geschnitten. Dies schloss das Abschneiden der Lauffront ein, da das Bromphenolblau die
Detektion der fluoreszierenden Spots stören und zu falsch positiven Signalen führen könnte.
Mit der DeCyder™ Software 6.5 wurden die Gele dann analysiert. Zunächst wurden alle Gele
über den Image Loader in die Software eingeladen und mit Hilfe der Differentiellen In-Gel
Analyse (DIA) ausgewertet. In diesem Schritt wurden die Rahmenbedingungen der Analyse
festgelegt. Die Spotanzahl pro Gel wurde auf maximal 5000 begrenzt und das Spotvolumen
musste einen Wert von 30 000 übersteigen. Am Ende des ersten Schrittes wurde das
qualitativ beste Gel ausgewählt, das sogenannte Mastergel. Mit der Biologischen Varianz
Analyse (BVA) wurden die Gele zunächst automatisch mit dem Mastergel verglichen. Bei
Bedarf wurden Spots manuell korrigiert. Für die nachfolgende Erfassung einzelner Spots in
die differentielle Analyse war es wichtig, dass die Spots in mindestens 80% der Gele
auffindbar waren. Die statistische Signifikanz der Daten wurde mittels Student’s t-test
überprüft. Spots mit einem p-Wert ≤ 0,05 wurden als signifikant angesehen. Um die
Änderung in der Spotintensität als signifikante Expressionserhöhung bewerten zu können,
galt generell, dass Änderungen in der Expression ab einem Wert von 1,5fach im Western Blot
validiert werden können. Aus diesem Grund wurden zur Beurteilung von signifikant
exprimierten Proteinen wie folgt festgelegt: Spots mit Werten ˂ - 1,5 wurden mit einer
reduzierten Expression in den Filaminopathieproben gedeutet, Werte > 1,5 mit einer
gesteigerten Expression im Vergleich zu den Kontrollen. Die Festlegung der Parameter wie
auch das manuelle Sichten der als differentiell angegebenen Spots bildeten die Grundlage
der Analyse und sollten Artefakte verhindern.

Material und Methoden
59
3.13.5. Mini 2D Gelelektrophorese
In den Punkten 3.12.1 – 3.12.3. wurde die Vorgehensweise zur 2D DIGE-Analyse von
großen Gelen (20cm) beschrieben. Analog dazu wurden kleine 2D-Gele (7cm) angefertigt.
Vor Beginn der Proteinauftrennung wurden 50 µg Muskellysat mit Fluorophoren markiert
und anschließend mittels Trägerampholyten auf Rundgelen (Länge 7 cm, Durchmesser
1,5mm) aus Polyacrylamid aufgetrennt. Die isoelektrische Fokussierung erfolgte nach dem
unten beschriebenen Spannungsgradienten.
Spannungsgradient für die isoelektrische Fokussierung, 7 cm
Spannung (V) DAUER 100 75 min 200 75 min 400 75 min 600 75 min 800 10 min 1000 5 min
Nach Beendigung der isoelektrischen Fokussierung wurden die Rundgele 15 min in
Inkubationspuffer inkubiert und im Anschluss auf selbst hergestellte 12 % Bis-Tris-Gele (8 cm
x 8 cm) aufgetragen. Zur Fixierung wurden die Gele mit 0,4 % Agarose überschichtet. Als
Elektrophorsepuffer wurde bei allen Gelen N-Morpholinopropansulfonsäure (MOPS)-Puffer
pH 7,3 verwendet. Der Einlauf der Proben erfolgte für 15 min bei 50 V, dann erfolgte die
Elektrophorese bei einer maximalen Spannung von 180 V für ca. 1h. Zur Überprüfung des
Spotmusters wurden die kleinen Gele nach Beendigung der 2. Dimension auf einem Typhoon
Trio Fluorescence Imager (GE Healthcare) digitalisiert.
MOPS-Puffer Konzentration Substanz 1 M MOPS 1 M Tris Base 2 % SDS 16 mM EDTA

Material und Methoden
60
3.14. Proteolytische Spaltung von Proteine für LC-ESI-MS/MS und MALDI
Die differentiellen Spots wurden für die Identifizierung aus dem Gel gestanzt und in
Verdauröhrchen überführt.
3.14.1. Verdau der Spots
Die Gelstücke wurden anschließend zur Entfernung möglicher Farbstoffreste und zur
Umpufferung des pH-Wertes dreimal im Wechsel mit je 20 µl Lösung A und B gewaschen.
Die Spots inkubierten für jeweils 10 min in den Lösungen. Im Anschluss an den dritten
Waschschritt in Lösung B wurden die Spots im Vakuumkonzentrator komplett eingeengt. Auf
die dehydratisierten Spots wurden dann je 2 µl Trypsinlösung gegeben (Trypsin in 10 mM
HCl gelöst, gemischt mit 100 mM Ammoniumhydrogencarbonat). Der Verdau erfolgte über
Nacht bei 37°C.
Lösung A
Konzentration Substanz 100 mM Ammoniumhydrogencarbonat Lösung B Konzentration Substanz 50 mM Ammoniumhydrogencarbonat 50 % (v/v) Acetonitril Trypsinlösung Konzentration Substanz 100 mM Ammoniumhydrogencarbonat 0,033 µg/µl Trypsin
3.14.2. Extraktion der Peptide fürs MALDI
Die Peptide wurden direkt im Anschluss an den tryptischen Verdau mit 10 µl 0,1 %
TFA aus den Gelstücken extrahiert. Die Extrakte wurden 30 min bei RT inkubiert und
anschließend auf ein AnchorChip-target von Bruker, Deutschland präpariert. Die α-Cyano-4-
hydroxy-zimtsäure-Matrix wurde mit 194 µl Acteon und 6 µl 0,1 % TFA vermischt, 5 min im
Ultraschallbad behandelt und weitere 5 min bei 16 000 g zentrifugiert. 10 µl des

Material und Methoden
61
Überstandes wurden auf die Ankerpositionen wie auch Zwischenräume des AnchorChip-
targets vorgelegt. Im nächsten Schritt wurden 3 µl des Peptidgemisches auf die
Ankerpositionen pipettiert. Nach 3 min wurde die Probe vorsichtig mit einer Pipette
abgezogen. Auf die Zwischenräume wurden 0,5 µl Standardlösung gegeben und komplett
eingetrocknet. Zum Abschluss wurden alle Positionen mit je 2,5 µl 0,1 % TFA gewaschen.
3.14.3. Extraktion der Peptide für LC-ESI-MS/MS
Direkt im Anschluss an den tryptischen Verdau wurden die Peptide mit 20 µl 0,1 %
TFA/ 100 % Acetonitril (1:1 Mischverhältnis) aus den Gelstücken extrahiert und 15 min im
Ultraschallbad behandelt. Der Überstand wurde in ein Verdauröhrchen überführt. Auf die
Gelstücken wurden erneut 20 µl der TFA/Acetonitril-Mischung gegeben und weitere 15 min
im Ultraschallbad behandelt. Nachdem der Überstand in das gleiche Verdauröhrchen wie
oben gegeben wurde, wurden die Proben in der SpeedVac ca. 45 min eingeengt. Die
eingetrockneten Peptide wurden mit 0,1 % TFA auf 20 µl aufgefüllt und bis zur Messung auf
der LTQ Velos Pro (Thermo) bei -80°C gelagert.
3.15. Proteinidentifizierung mittels MALDI
Die differentiellen Spots der DIGE-Analyse wurden mittels Matrix-assisted Laser
Desorption/Ionization (MALDI) identifiziert. MALDI eignet sich sehr gut zur Analyse von
wenig komplexen Proben und bietet zudem den Vorteil einer schnellen und sicheren
Identifizierung von Proteinen. Die Kalibrierung des MALDI-TOF/TOF (Ultaflex™, Bruker
Corporation Bremen, Deutschland) erfolgte zunächst durch Aufnahme von Spektren des
Peptidstandards. Im Anschluss wurden für jede Probe manuell mehrere Peptide Mass
Fingerprint (PMF) Spektren aufgenommen und aufsummiert. Die Spektren wurden im
Positivmodus des Geräts mit einer Targetspannung von 20 kV und einer gepulsten
Beschleunigungsspannung von 17,25 kV aufgenommen. Die Reflektorspannung betrug 21 kV
und die Detektorspannung 1,7 kV. Nach der Aufnahme der Spektren wurden diese intern
nachkalibriert mit 13 mono-isotopischen Massen von Autolyseprodukten von Trypsin und
Keratinfragmenten. Die generierten Spektren wurden durch das Erstellen von Massenlisten

Material und Methoden
62
der einzelnen Peptide ergänzt. Dies erfolgte durch den Gebrauch der Software FlexAnalysis
™ (Vers. 2.4, Bruker Corporation, Bremen, Deutschland). Bei der Erstellung der Listen
wurden folgende Parameter berücksichtigt: snap Peak Detektionslogarithmus, Signal-
Rausch-Verhältnis = 6, maximale Peak-Anzahl = 100, Grenzwert des Qualitätsfaktors = 50
und Subtraktion der Basislinie.
Matrixlösung
Menge Substanz α-Cyano-4-hydroxy-zimtsäure (gesättigt) 194 µl Aceton 6 µl Trifluoressigsäure Peptidstandard Konzentration Peptid m/z 2 mg/ mL Bradykinin 7,57 Da 10 mg/ mL Angiotensin 1296,66 Da 2 mg/ mL Bombesin 1619,82 Da 2 mg/ mL ACTH 1-17 2093,09 Da 2 mg/ mL Somatostatin 31477 Da
3.15.1. Datenauswertung der MALDI-Messung
Die mittels FlexAnalysis ™ (Vers. 2.4, Bruker Corporation, Bremen, Deutschland)
generierten Massenlisten wurden in ProteinScape 1.4 (Bruker Daltonics, Bremen,
Deutschland) geladen. Unter der Verwendung des Mascot Algorithmus wurden
Datenbanksuchläufe gegen die humane Decoy-Datenbank [Reidegeld et al. 2008]
(uniprot_sprot_decoy human) gestartet. Die folgenden Suchparameter wurden vor Start der
Suchen festgelegt: es wurde nur nach tryptischen Peptiden gesucht. Die Massentoleranz
zwischen dem theoretischen durchschnittlichen Wert und dem gemessenen Wert durfte
maximal 50 ppm betragen. Als feste Modifikation wurde Carbamidomethylierungen von
Cysteinen festgelegt, Methionin-Oxidationen sowie die Phosphorylierungen von Serin,
Tyrosin und Threonin wurden als variable Modifikationen gewählt. Zusätzlich wurde
festgelegt, dass eine übersprungene Schnittstelle akzeptiert und der Mascot™Score den
Wert 20 übertreffen sollte. Nach Beendigung der Suchläufe wurden die Ergebnisse in Form
einer Liste dargestellt. Mittels Mascot™Score wurde die Zuordnung der Peptide zu den

Material und Methoden
63
jeweiligen Spektren bewertet. Ein Protein wurde als identifiziert markiert, wenn mindestens
zwei Peptide dem Protein eindeutig zugeordnet werden konnten und wenn die theoretische
Masse des Proteins sowie der isoelektrische Punkt mit der Lokalisation des Spots im 2D-Gel
übereinstimmten.
Tabelle 3.4.: Zusammenfassung der Suchparameter in Protein Scape 1.4 für die Identifizierung von Gelspots
Suchparameter
Erlaubte fehlende Spaltungen 1
Feste/Variable Modifikationen Carbamidomethyl (C)
Oxidation (M)
Phospho (S,T,Y)
Massentoleranz (MS) 50 ppm
Parameter für die Datenbanksuche
Suchmaschine Mascot 2.3.02
Datenbank uniprot_sprot_decoy, human
Enzym Trypsin
MW Range 0.0 kDa
Kriterien für die Identifizierung von Proteinen
Mascot™ Score >20
3.16. Proteinidentifizierung mittels nanoLC-ESI-MS/MS
Für die Proteinidentifizierung mittels LC-ESI-MS/MS-Systeme wurden 2 verschiedene
Massenspektrometer verwendet: LTQ Orbitrap und LTQ Velos Pro (beide Thermo Fisher). In
Ergänzung zu der Identifizierung mittels MALDI wurden die differentiellen Spots der 2D DIGE
Analyse zusätzlich auf der LTQ VelosPro gemessen, um u.a. potentielle posttranslationale
Modifikationen zu detektierten. Hierfür wurden die Proteine in den Gelspots tryptsich
verdaut und anschließend extrahiert. Die mikrodissektierten Aggregatproben aus humanem
Gewebe und Zellkultur sowie der zugehörigen Kontrollen wurden auf der LTQ Orbitrap
gemessen. Die Proben wurden mittels In-Lösung-Verdau (beschrieben unter 3.10.) für die
massenspektrometrische Analyse vorbereitet.

Material und Methoden
64
3.16.1. LTQ Orbitrap
Die nanoLC wurde auf einem UltiMate 3000 RSLCnano LC-System (Thermo Fisher
Scientific Inc., Dreieich, Deutschland) durchgeführt. Zunächst wurden 15 µl der verdauten
Proben für 10 min auf eine Vorsäule (75 µm x 2 cm, C18-Säule, Partikelgröße 3 µm,
Porengröße 100 °A, Thermo Fisher Scientific Inc., Dreieich, Deutschland) injiziert und
aufkonzentriert. Der Lösungsmittelfluss betrug zu diesem Zeitpunkt 30 µl 0,1 % TFA/min. Die
Vorsäule wurde mit einer Trennsäule (300 µm x 25 cm für humanes Gewebe und 300 µm x
50 cm für Zellkultur, C18-Säule, Partikelgröße 3 µm, Porengröße 100 °A, Thermo Fisher
Scientific Inc., Dreieich, Deutschland) in Reihe geschaltet. Die Übertragung der Peptide von
der Vorsäule auf die Trennsäule erfolgte in einem Lösungsgemisch von 94 % Lösung A und
6 % Lösung B. Auf der C18-Trennsäule erfolgte die Auftrennung der Peptide mit einer
Flussrate von 400 nL/min. In einem ersten Gradienten über 95 min wurde der Anteil von
Lösung B auf 40 % erhöht und in einem zweiten Gradienten innerhalb von 2 min auf 95 %.
Durch den Anstieg des Acetonitril-Anteils erfolgte die Auftrennung der Peptide nach
steigender Hydrophobizität. In einem letzten Gradientenschritt wurde innerhalb von 5 min
der Anteil von Lösung B von 95 % auf 5 % verringert. Es folgte ein 60 minütiger Spülschritt.
Durch die Wahl unterschiedlicher Konzentrationen von Lösung B war es möglich Rückstände
der Peptidauftrennung von der Trennsäule zu waschen. Für die Elektrospray-Ionisation (ESI)
wurde die HPLC-Anlage direkt über eine entsprechende ESI-Quelle (Thermo Fisher Inc.,
Dreieich, Deutschland) mit der LTQ Orbitrap Velos™ (Thermo Fisher Inc., Dreieich,
Deutschland) gekoppelt. Die Peptide wurden für die ESI durch eine metallbedampfte
Glaskapillare (Pico™ Emitter, New Objective Inc., Woburn, USA) geleitet, an der eine
Spannung von 1,5 – 1,6 kV angelegt war. Die Temperatur betrug 275 °C. Durch das Anlegen
einer positiven Spannung wurden die Peptide in ein fein verteiltes Spray hochgeladener
Lösungsmitteltröpfchen überführt, aus denen ionisierte und positiv geladene
Analytmoleküle entstanden. Diese Moleküle wurden anschließend fragmentiert und
analysiert. Die Generierung der Spektren erfolgte in der Orbitrap mit einer Auflösung von R =
30 000 zwischen 300 und 2000 m/z. Die maximale Füllzeit betrug 500 ms. Für die interne
Kalibrierung wurde Polydimethylcyclosiloxan (m/z = 445.120) verwendet. Für die
anschließende MS/MS-Fragmentierung in der Ionenfalle LTQ wurden die 20 intensivsten
Massen ausgewählt. Die Füllzeit der LTQ betrug 50 ms. Die Fragmentierung der Peptide

Material und Methoden
65
erfolgte mittels low-energy collision-induced dissociation (CID) mit einer Kollisionsenergie
von 35 %. Alle gemessenen Massen wurden nach der Spektrenaufnahme für 35 sec auf eine
sogenannte Exklusionsliste gesetzt. So sollte verhindert werden, dass Peptide mehrfach
fragmentiert und gemessen werden.
3.16.2. LTQ Velos Pro
Die gewählten Parameter zur Auftrennung mittels nano LC und die
Elektrosprayionisation entsprachen generell der in Punkt 3.16.1. beschriebenen
Vorgehensweise. Die Messprotokolle unterscheiden sich allerdings in der Zusammensetzung
des Lösungsmittelgemisches während der Übertragung der Peptide auf die Trennsäule und
in der Wahl der Gradienten. Die Peptide wurden in einem in einem Lösungsmittelgemisch
von 96 % Lösung A und 4 % Lösung B übertragen. Zwischen der 6. und der 53. Minute wurde
der Anteil von Lösung B linear auf 40 % erhöht, innerhalb von 3 min auf 95 %. Ab der 59.
Minute wurde der Anteil von Lösung B wieder auf 4 % reduziert. Es folgte ein 5 minütiger
Waschschritt und die Re-Equilibrierung der Säule. Im Anschluss an die ESI wurden die
ionisierten Peptide in die LTQ Velos Pro geleitet. Der Scanbereich lag im Bereich von 300 bis
1500 m/z. Nach einem Übersichtscan wurden die 5 intensivsten Ionen ausgewählt und
mittels CID (365 – 2000 m/z, normalisierte Kollisionsenergie 35, Aktivierungszeit 10 ms) und
anschließend mittels ETD fragmentiert (50 – 2000 m/z, Aktivierungszeit 150 ms). Die m/z-
Werte von fragmentierten Ionen werden für 35 s auf eine Ausschlussliste gesetzt, um
mehrmaliges Messen desselben Ions zu verhindern.
Verwendete Lösungen für die HPLC
Lösung Zusammensetzung Lösung A 0,1 % (v/v) Ameisensäure Lösung B 80 % (v/v) Acetonitril, 0,1 % (v/v) Ameisensäure
3.16.3. Datenauswertung der nanoHPLC-ESI-MS/MS
Für die weitere Auswertung der massenspektrometrischen Analyse war es zunächst
notwendig die generierten Rohdaten (*raw-file-format) in das Mascot generic format (*mgf-
file) zu überführen. Dies erfolgte mit dem Programm Proteom Discoverer 1.3 von Thermo

Material und Methoden
66
Scientific, Deutschland. Die Rohdaten wurden erst in die Software importiert, dann wurden
die Massen der Precursor-Spektren zwischen 400 und 10000 Da gefiltert und am Ende
wurden die gefilterten Spektren in das *mgf-format überführt. Zur Identifizierung der
Proteine wurden die generierten mgf-files der einzelnen Proben in ProteinScape 2.1™
(Bruker Daltonics, Bremen, Deutschland) eingeladen und ein Abgleich mittels
Mascot™Suchalgorithmus (Matrixscience, London, England) gegen eine humane bzw. gegen
eine murine Protein-Datenbank durchgeführt. Zunächst wurde die IPI-decoy-Datenbank
verwendet. Im Laufe der Arbeiten wurde allerdings auf die uniprot_sprot-Datenbank
umgestellt. Die Suchkriterien für den Datenabgleich wurden wie folgt für beide Datenbanken
gewählt: Die Datenbanksuche wurde auf tryptisch verdaute Peptide beschränkt. Eine
überlesene Schnittstelle wurde akzeptiert. Insgesamt wurden 5 variable Modifikationen
berücksichtigt. Phosphorylierung von Serin, Tyrosin und Threonin wurden aufgrund von
posttranslationalen Modifikationen berücksichtigt. Carbamidomethylierung von Cysteinen
durch Acrylamid sowie die Methionin-Oxidation konnten während der Probenpräparation
beobachtet werden. Die Massentoleranz zwischen der theoretischen und der gemessenen
Masse durfte maximal 10 ppm betragen. In dem hier gewählten ESI-Verfahren war die
Wahrscheinlichkeit der Bildung mehrfach positiv geladener Ionen am höchsten. Dennoch
wurden in der Datenauswertung auch ein- und zweifach positiv geladene Ionen
berücksichtigt. Für die Peptididentifizierung wurden nur solche Peptide akzeptiert, die einen
minimalen Mascot™Score von 10 zeigten. Für die Proteinidentifikation wurden nur Peptide
bzw. MS/MS-Spektren zugelassen, die einen höheren Mascot™Score als 15 aufwiesen.
Diesen Peptiden werden Proteine zugeordnet, wenn für das jeweilige Protein mindestens 1
Peptid mit einem Mascot™Score von minimal 20 identifiziert wurde. Letztendlich können nur
Proteine als identifiziert angesehen werden, wenn die Summe aller Peptid-Mascot™Scores
den Wert 50 übersteigt. Nach Beendigung des Datenbankabgleichs wurden die
identifizierten Proteine in einer Liste zusammengefasst und sortiert.

Material und Methoden
67
Tabelle 3.5.: Zusammenfassung der Suchparameter in ProteinScape 2.1 zur Charakterisierung der
Proteinaggregatzusammensetzung
Suchparameter
Erlaubte fehlende Spaltungen 1
Variable Modifikationen Carbamidomethyl (C)
Oxidation (M)
Phospho (S,T,Y)
Massentoleranz (MS) 10 ppm
Massentoleranz (MS/MS) 0,5 Da Ladungen +1, +2, +3
Parameter für die Datenbanksuche
Suchmaschine Mascot 2.3.02
Datenbank uniprot_sprot_decoy, human
uniprot_sprot_decoy, Maus
Enzym Trypsin
Peptide score threshold 15
Minimum 1 Peptid mit Score >20
Kriterien für die Identifizierung von Proteinen
Mascot™ Score >50
Kriterien für die Identifizierung von Peptiden
Mascot™ Score >20
3.17. Auswertung massenspektrometrischer Daten nach Datenbankabgleich
Die Auswertung der Proteinlisten erfolgte in der hier vorliegenden Dissertation auf
drei verschiedene Weisen. Zu Beginn der Analysen von humanem Gewebe wurde ein
qualitativer Ansatz verfolgt. Im Anschluss an die massenspektrometrische Analyse wurden
alle identifizierten Proteine aus den Proben in einer separaten Liste zusammengefasst. An
die qualitative Auswertung schloss sich die manuell durchgeführte quantitative Auswertung
an. Ziel war es Proteine zu finden, die spezifische Bestandteile der Aggregatproben sowie der
Patientenkontrollen zu darstellten. Im Laufe der Arbeiten wurde die manuelle Auswertung
zu einer automatisierten quantitativen Strategie, basierend auf Spectral Index Calculation,
weiterentwickelt. Die Spectral Index Calculation ermöglichte es, differentiell exprimierte
Proteine herauszufiltern. Die Spectral Index Calculation für MFM-Proben wurde von M.Sc.
Alexandra Maerkens im Rahmen ihrer Masterarbeit etabliert und optimiert. Die optimierte
Auswertung wurde zur Auswertung der Analyse von Patientenkontrollen und

Material und Methoden
68
Negativkontrollen angewendet. Für den Datenbankabgleich wurden die in Punkt 3.15.2.
beschriebenen Parameter angewendet.
3.17.1. Die pseudo-quantitative Auswertung
Die hier angewendeten Parameter beruhten auf Erfahrungswerten früherer Analysen
sowie aus selbstgewählten Kriterien zur Identifizierung spezifischer Proteine in
Proteinaggregaten. Die Auswertungen basierten wie vorne beschrieben auf der Analyse von
6 Filaminopathiepatienten und 6 Kontrollen. Die in ProteinScape 2.1. generierte Proteinliste
wurde in Excel überführt. Nach Erstellung der Proteinlisten wurde wie folgt vorgegangen, um
spezifische Proteinkomponenten in den Aggregaten und dem umliegenden Gewebe der
Filaminopathiepatienten herauszufiltern. In einem ersten Schritt wurden jedem Protein die
durchschnittliche Anzahl an identifizierten Peptiden sowie der durchschnittliche
Mascot™Score zugeteilt. Es wurden nur Proteine akzeptiert, die mit mindestens 2 Peptiden
identifiziert werden konnten und gleichzeitig einen höheren Mascot™Score als 80 aufwiesen.
Innerhalb dieser Proteinliste wurden die Proteine hinsichtlich ihres Vorkommens in den
Patientenproben gruppiert. Jedes akzeptierte Protein wurde in mindestens 3 von 6 der
analysierten Proben identifiziert.
3.17.2. Spectral Index Calculation
Die Auswertung mittels Spectral Index Calculation basiert auf der Summierung aller
MS/MS-Spektren, die für ein Peptid in der massenspektrometrischen Analyse generiert
wurden [Müller et al. 2011. Der daraus resultierende Wert wird als Compound bezeichnet. In
der Datenbanksuche wurden die Peptide den entsprechenden Proteinen zugeordnet. Die
Spectral Peptide Counts (PC) drückten aus, wie viele Peptide bzw. MSM/MS-Spektren einem
Protein zugeordnet wurden. Für die Bildung des Spectral Index wurde zunächst der
Mittelwert (MW) der gesamten Spectral Peptidecounts (SP) aller Proben (Gleichung 1)
gebildet. In einem nächsten Schritt wurde der Ausgleichsfaktor (AF) gebildet. Hierzu wurde
der Quotient aus der Summe der SPs der jeweiligen Probe und dem Mittelwert der
gesamten SPs aus allen Proben errechnet. So wurden die SPs der einzelnen Proteine in jeder
Probe über die Gesamtanzahl der SPs in allen Proben normalisiert (Gleichung 2).
Die Berechnung des Spectral Index (SI) erfolgte für jedes einzelne Protein in jeder

Material und Methoden
69
Probe, indem der Quotient aus der nicht normalisierten Anzahl der Spectral Peptidecounts
eines Proteins und dem Ausgleichsfaktor gebildet wurde (Gleichung 3). Die Spectral Indices
dienten als Grundlage für die Identifizierung differentiell exprimierter Proteine in den
einzelnen Proben. Hierfür wurde die sogenannte Ratio (Verhältnis) gebildet. Die Ratio
beschrieb das Verhältnis für ein Protein zwischen den zu vergleichenden Proben. Die Ratio
wurde wie folgt berechnet: Es wurde der Mittelwert der Spectral Indices für ein einzelnes
Protein für alle Aggregat- und Patientenproben gebildet. Aus den gebildeten Mittelwerten
wurde dann für jedes einzelne Protein das Verhältnis Aggregatprobe zu Patientenkontrolle
(4a) und Patientenprobe zu Negativkontrolle gebildet (4b).
In allen Vergleichen wurden Proteine als differentiell exprimiert eingestuft, wenn für diese
ein mindestens 1,8fach (p-Wert ≤ 0.05 im Zweistichproben-t-Test) höherer Spectral Index im
erkrankten Gewebe im Verhältnis zu gesunden Kontrollen berechnet werden konnten.
Proteine, die eine Ratio von > 1,8 aufwiesen, wurden als akkumuliert im
(4b)
m = Anzahl der Proben in der Analyse PK = Patientenkontrolle NK = Negativkontrolle
(3)
m = Anzahl der Proben in der Analyse n = Anzahl Proteine innerhalb einer Probe Pc = peptidecounts
(2)
(1) m = Anzahl der Proben in der Analyse n = Anzahl Proteine innerhalb einer Probe Pc = peptidecounts
m = Anzahl der Proben in der Analyse n = Anzahl Proteine innerhalb einer Probe Pc = peptidecounts
m = Anzahl der Proben in der Analyse AG = Aggregatprobe PK = Patientenkontrolle
(4a)

Material und Methoden
70
Filaminopathiegewebe angesehen. Eine Ratio von ≤ 0,55 ließ auf eine verminderte
Expression der Proteine in den Filaminopathieproben schließen. Alle Werte wurden mittels
implementierten Zweistichproben-t-Test auf ihre statistische Signifikanz überprüft. Das
Signifikanzniveau wurde auf 5 % (p ≤ 0,05) festgelegt.
3.18. Western Blot
Für immunologische Nachweise wurden Mini 2D Blots angefertigt. Es wurden
Muskellysate von Filaminopathiepatienten sowie von gesundem Gewebe hergestellt. Die
Proteinmenge wurde mittels Bio-Rad Protein Assay nach Bradford bestimmt (beschrieben
unter Punkt 3.6.). Die Auftrennung der Proteine in der ersten Dimension erfolgte nach der
Methode von Klose [Klose 1975]. Die Vorgehensweise und die verwendeten Puffer bzw.
Lösungen zur isoelektrischen Fokussierung wurde in den Punkten 3.12.1. und 3.12.2.
beschrieben. Der Elektrotransfer der Proteine aus der Gelmatrix auf die Nitrocellulose-
Membran erfolgte mittels Semi-Dry-Blotting unter der Verwendung eines
diskontinuierlichen Puffersystems. Der Elektrotransfer erfolgte über 1h bei einer
Stromstärke von 2 mA/ cm2. Nach Beendigung des Elektrotransfers wurde die Membran in
Blocking Puffer für 2h bei RT und Schütteln inkubiert und anschließend mit Primäranitkörper
in TBS-T 5 % BSA inkubiert. Als Primärantikörper wurden PhosphoSerin und Desmin
verwendet. Die Antikörper wurden in einer 1: 500 Verdünnung verwendet und über Nacht
bei 4°C auf die Membran gegeben. Im Anschluss an die Primärantikörper-Inkubation wurde
die Membran 3x 10 min mit TBS gewaschen und dann mit Sekundärantikörper 2h inkubiert.
Als Sekundärantikörper wurden Infrarot-Fluoreszenzfarbstoff-gekoppelte Sekundäranti-
körper in einer Verdünnung von 1: 15000 in TBS (LI-COR Biosciences, Bad Homburg,
Deutschland) verwendet, die mit einen Infrarot-Fluoreszenz-Scanner (Odyssey™, LI-COR
BioSciences, Bad Homburg, Deutschland) detektiert wurden.

Material und Methoden
71
12 % Bis-Tris-Gele Konzentration Substanz 2,5 M Bis-Tris (BT)-Puffer 7fach, pH 6,5 Menge Substanz 20 ml Acrylamid, 30 % Lösung 7,1 ml BT 7fach 50 µl APS (40 %) 10 µl TEMED Anodenpuffer Konzentration Substanz 300 mM Tris 100 mM Tricin pH 8,7 – 8,8 Kathodenpuffer Konzentration Substanz 300 mM Aminocapronsäure 30 mM Tris pH 8,6 – 8,7 Blocking Puffer Konzentration Substanz 50 ml TBS 5 % BSA 0,1 % Tween TBS-Tween-Puffer, pH 7,4 Menge Substanz 73 mM Kaliumchlorid 2,7 mM Natriumchlorid 25 mM Tris 0,1 % Tween 20

72
4. Ergebnisse
Die Analyse von Muskelgewebe aus 6 Filaminopathiepatienten mit 3 verschiedenen
Mutationen erfolgte durch die Etablierung der Lasermikrodissektion in Kombination mit 2
proteomischen Ansätzen: eine 2D DIGE-Studie und die Label-freie massenspektrometrische
Analyse. Durch den Vergleich der Proteinzusammensetzung der Aggregate mit
entsprechenden nicht betroffenen Muskelzellen desselben Filaminopathiepatienten sollten
Rückschlüsse auf die Proteinzusammensetzung in den Aggregaten gezogen werden.
Zusätzlich sollte der Vergleich von (noch) nicht betroffenem Muskelgewebe
(Patientenkontrollen) aus Filaminopathiepatienten mit gesundem Kontrollgewebe die Frage
klären, ob es möglich ist, spezifische Marker für das Frühstadium der Filaminiopathie zu
identifizieren. In bereits veröffentlichten Transfektionsstudien konnte gezeigt werden, dass
sich das Zellkulturmodell sehr gut zur Charakterisierung von Filamin C-Mutationen eignet
[Vorgerd et al. 2008, Duff et al. 2011]. Nun sollte gezeigt werden, ob die Proteomanalyse in
eine murine Myoblasten-Zelllinie übertragen werden kann. Hierfür wurde die Plasmid-DNA
der von Vorgerd et al. 2005 erstmalig beschriebenen Mutation p.W2710X in die murinen
Myoblasten transfiziert, um die Zusammensetzung der Proteinaggregate in den Zellen mit
der Zusammensetzung der humanen Aggregate zu vergleichen. Ziel war es zu klären, ob sich
das gewählte Zellkultursystem hinsichtlich der Aggregation als Modellsystem eignet.

Ergebnisse
73
Im Rahmen dieser Dissertation wurden die Protokolle zur Proteomanalyse von
Muskelgewebe entwickelt. In Voranalysen war es notwendig, folgende Parameter zu
optimieren:
Solubilisierung der Proteinaggregate
Optimierung der Größe der mikrodissektierten Fläche von humanem Muskelgewebe
für die Label-freie Proteomanalyse und für die 2D DIGE Studie
Optimierung der mikrodissektierten Fläche muriner Myoblasten für die
Proteomanalyse
4.1. Etablierung der LMD zur Label-freien Proteomanalyse
Mittels LMD war es möglich, die Filaminopathie-spezifischen Aggregate exakt aus
dem umliegenden Gewebe zu isolieren. Die Proben konnten schonend gewonnen werden,
so dass die Aggregate trotz geringen Vorkommens für nachfolgende Downstream-Analysen
genutzt werden konnten. Als Kontrolle dienten zum einen das umliegende, augenscheinlich
keine Aggregate enthaltende Gewebe desselben Patienten sowie das Gewebe gesunder
Kontrollen. Ziel war es, die proteomische Zusammensetzung der Proteinaggregate zu
entschlüsseln. In einem weiteren Schritt sollten proteomische Analyse von kultivierten
Maus-Myoblasten durchgeführt werden.
4.1.1. Darstellbarkeit der Aggregate
Im Vorfeld der Analysen wurde zunächst die Anzahl der Proteinaggregate pro
Filaminopathiepatient ermittelt. Hierfür wurden 10 µm dicke fluoreszenzgefärbte Schnitte
auf Glasobjektträger gebracht, die Dokumentation der Aggregate erfolgte mittels des LMD
6500 (Leica, Wetzlar, Deutschland). Die Zahl und Größe der Aggregate in den Patienten
variierte zum Teil stark. In einigen Patienten konnten zahlreiche (>20) große Aggregate
gefunden werden. In anderen Patienten belief sich die Zahl auf ca. 10 – 12 Aggregate pro
Schnitt. Für die LMD mussten die Gefrierschnitte auf spezielle Membranobjektträger
übertragen werden. Aufgrund der Beschaffenheit der Polyethylentetraphthalat (PET)-
Membran wurde das Färbeprotokoll angepasst. Die Inkubationszeiten der Antikörper wie
auch die Waschschritte wurden auf ein Minimum reduziert (unter Punkt 3.5. beschrieben).
Der größte Unterschied zwischen den beiden Protokollen lag jedoch darin, dass die Schnitte
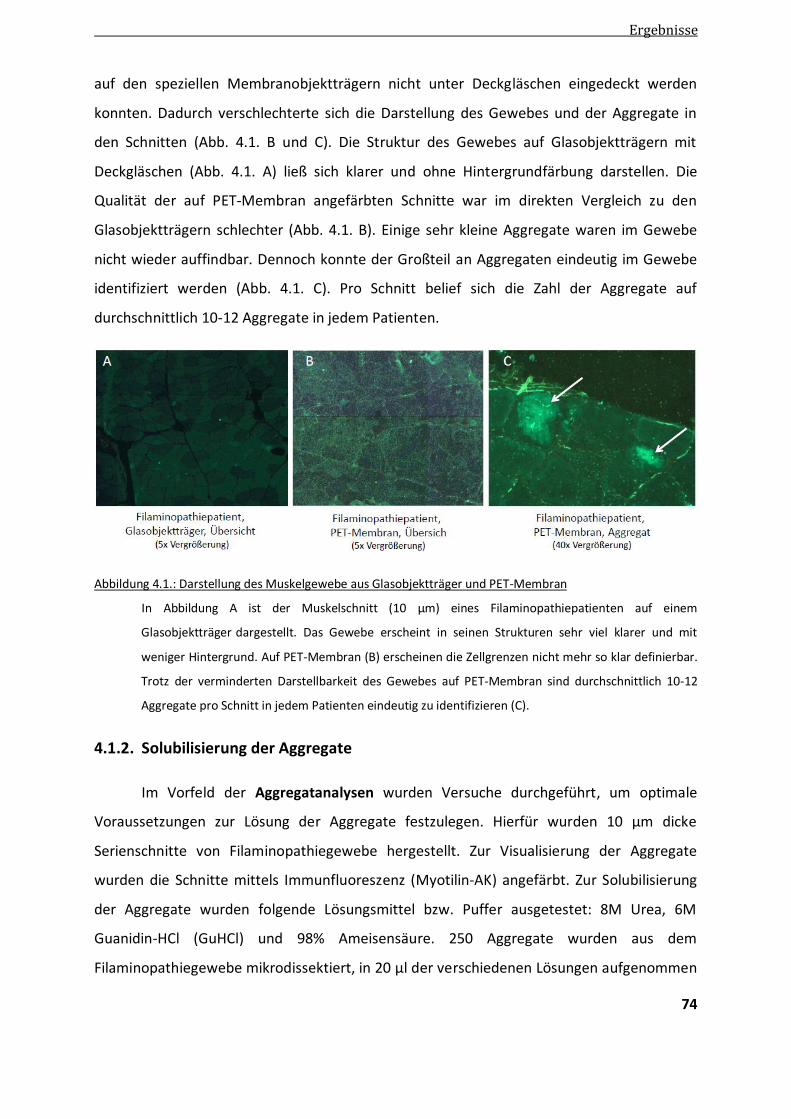
Ergebnisse
74
auf den speziellen Membranobjektträgern nicht unter Deckgläschen eingedeckt werden
konnten. Dadurch verschlechterte sich die Darstellung des Gewebes und der Aggregate in
den Schnitten (Abb. 4.1. B und C). Die Struktur des Gewebes auf Glasobjektträgern mit
Deckgläschen (Abb. 4.1. A) ließ sich klarer und ohne Hintergrundfärbung darstellen. Die
Qualität der auf PET-Membran angefärbten Schnitte war im direkten Vergleich zu den
Glasobjektträgern schlechter (Abb. 4.1. B). Einige sehr kleine Aggregate waren im Gewebe
nicht wieder auffindbar. Dennoch konnte der Großteil an Aggregaten eindeutig im Gewebe
identifiziert werden (Abb. 4.1. C). Pro Schnitt belief sich die Zahl der Aggregate auf
durchschnittlich 10-12 Aggregate in jedem Patienten.
Abbildung 4.1.: Darstellung des Muskelgewebe aus Glasobjektträger und PET-Membran
In Abbildung A ist der Muskelschnitt (10 µm) eines Filaminopathiepatienten auf einem
Glasobjektträger dargestellt. Das Gewebe erscheint in seinen Strukturen sehr viel klarer und mit
weniger Hintergrund. Auf PET-Membran (B) erscheinen die Zellgrenzen nicht mehr so klar definierbar.
Trotz der verminderten Darstellbarkeit des Gewebes auf PET-Membran sind durchschnittlich 10-12
Aggregate pro Schnitt in jedem Patienten eindeutig zu identifizieren (C).
4.1.2. Solubilisierung der Aggregate
Im Vorfeld der Aggregatanalysen wurden Versuche durchgeführt, um optimale
Voraussetzungen zur Lösung der Aggregate festzulegen. Hierfür wurden 10 µm dicke
Serienschnitte von Filaminopathiegewebe hergestellt. Zur Visualisierung der Aggregate
wurden die Schnitte mittels Immunfluoreszenz (Myotilin-AK) angefärbt. Zur Solubilisierung
der Aggregate wurden folgende Lösungsmittel bzw. Puffer ausgetestet: 8M Urea, 6M
Guanidin-HCl (GuHCl) und 98% Ameisensäure. 250 Aggregate wurden aus dem
Filaminopathiegewebe mikrodissektiert, in 20 µl der verschiedenen Lösungen aufgenommen

Ergebnisse
75
und anschließend mittels nanoLC-MS/MS analysiert. Eine Zusammenfassung der
massenspektrometrischen Analyse ist in Tabelle 4.1. dargestellt.
Die Analyse der in GuHCl gelösten Aggregate ergab keine Ergebnisse. In der
massenspektrometrischen Messung konnten keine Spektren aufgenommen werden. In den
Proben konnten somit keine Proteine identifiziert werden. Das Lösen der Aggregate in 8M
Urea ermöglichte die Identifizierung von insgesamt 16 Proteinen. Filamin C wurde dabei
eindeutig identifiziert. Ebenfalls unter den identifizierten Proteinen befanden sich 2
bekannte MFM-Proteine: Desmin und dessen Interaktionspartner Alpha-B-Crystallin
[Goldfarb et al. 2008]. Zusätzlich konnten verschiedene Isoformen von Myosin, einem
wichtigen Motorprotein in der Muskelkontraktion, detektiert werden. Das beste Ergebnis
ergab die Solubilisierung der Aggregate mit 98% Ameisensäure. Insgesamt wurden 40
Proteine identifiziert, darunter erneut Filamin C, das Z-Banden assoziierte Protein Desmin
und dessen Interaktionspartner Alpha-B-Crystallin. Xirp2, ein Interaktionspartner von Filamin
C [Kley et al. 2012] konnte zusätzlich identifiziert werde. Des Weiteren wurden Troponin und
Myosin-Isoformen sowie Strukturproteine wie Vimentin und Nebulin detektiert.
Fazit dieses ersten Vorversuchs war, dass durch Solubilisierung der Aggregate in
Ameisensäure mehr Proteine identifiziert wurden als mit Urea. Im Vergleich von 8M Urea
und 98% Ameisensäure konnte beobachtet werden, dass mit Ameisensäure 80 % aller mit
Urea identifizierten Proteinen nachgewiesen werden konnten. Generell ist zu sagen, dass es
mit Hilfe der angewendeten Methode möglich ist, wichtige Muskelproteine und MFM-
assoziierte Proteine zu identifizieren.
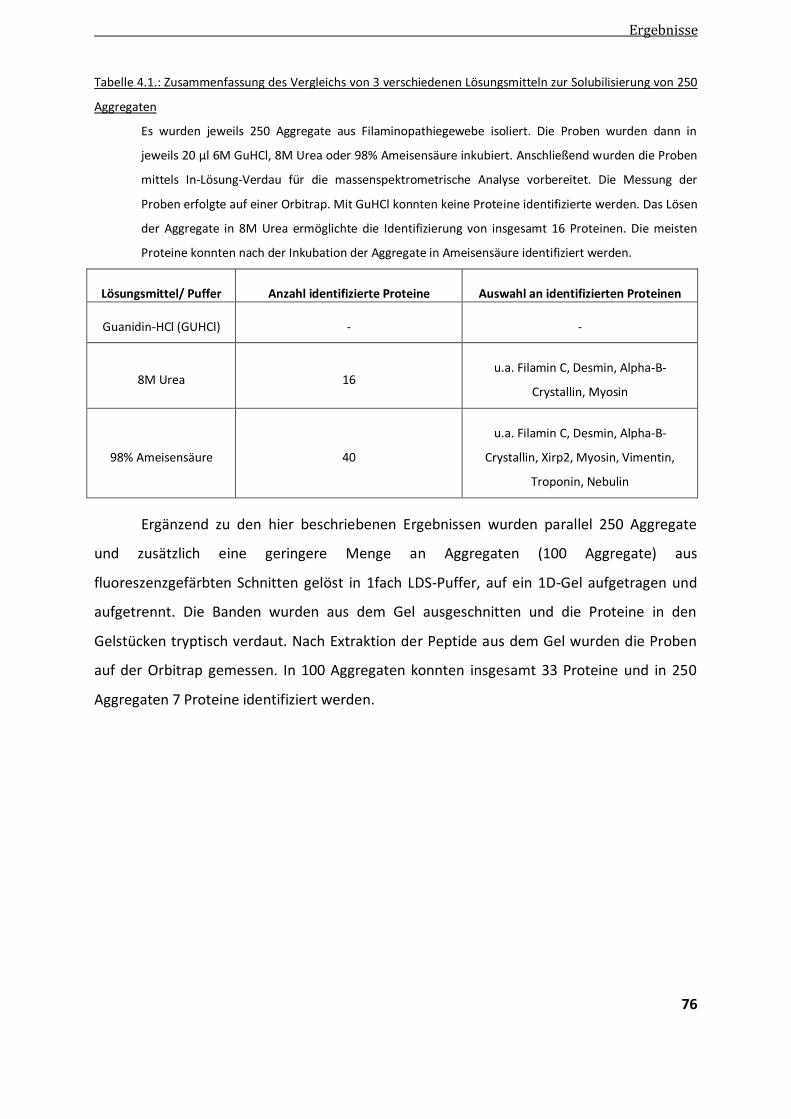
Ergebnisse
76
Tabelle 4.1.: Zusammenfassung des Vergleichs von 3 verschiedenen Lösungsmitteln zur Solubilisierung von 250
Aggregaten
Es wurden jeweils 250 Aggregate aus Filaminopathiegewebe isoliert. Die Proben wurden dann in
jeweils 20 µl 6M GuHCl, 8M Urea oder 98% Ameisensäure inkubiert. Anschließend wurden die Proben
mittels In-Lösung-Verdau für die massenspektrometrische Analyse vorbereitet. Die Messung der
Proben erfolgte auf einer Orbitrap. Mit GuHCl konnten keine Proteine identifizierte werden. Das Lösen
der Aggregate in 8M Urea ermöglichte die Identifizierung von insgesamt 16 Proteinen. Die meisten
Proteine konnten nach der Inkubation der Aggregate in Ameisensäure identifiziert werden.
Lösungsmittel/ Puffer Anzahl identifizierte Proteine Auswahl an identifizierten Proteinen
Guanidin-HCl (GUHCl) - -
8M Urea 16 u.a. Filamin C, Desmin, Alpha-B-
Crystallin, Myosin
98% Ameisensäure 40
u.a. Filamin C, Desmin, Alpha-B-
Crystallin, Xirp2, Myosin, Vimentin,
Troponin, Nebulin
Ergänzend zu den hier beschriebenen Ergebnissen wurden parallel 250 Aggregate
und zusätzlich eine geringere Menge an Aggregaten (100 Aggregate) aus
fluoreszenzgefärbten Schnitten gelöst in 1fach LDS-Puffer, auf ein 1D-Gel aufgetragen und
aufgetrennt. Die Banden wurden aus dem Gel ausgeschnitten und die Proteine in den
Gelstücken tryptisch verdaut. Nach Extraktion der Peptide aus dem Gel wurden die Proben
auf der Orbitrap gemessen. In 100 Aggregaten konnten insgesamt 33 Proteine und in 250
Aggregaten 7 Proteine identifiziert werden.
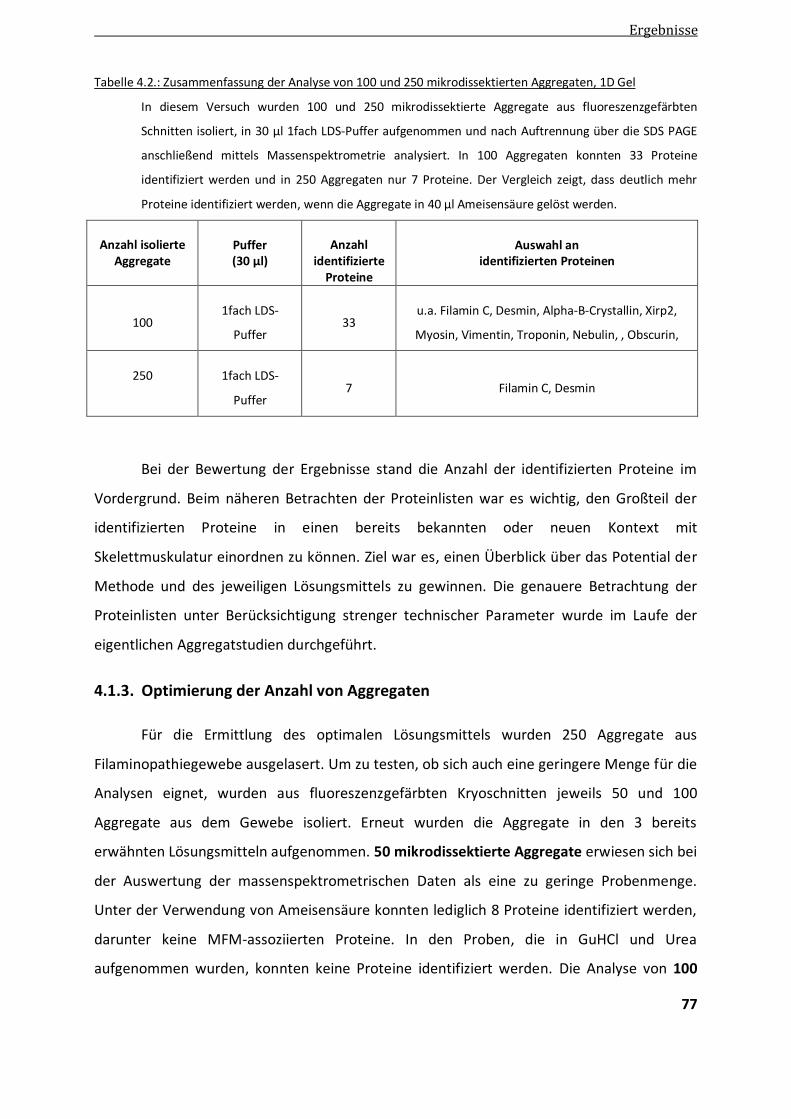
Ergebnisse
77
Tabelle 4.2.: Zusammenfassung der Analyse von 100 und 250 mikrodissektierten Aggregaten, 1D Gel
In diesem Versuch wurden 100 und 250 mikrodissektierte Aggregate aus fluoreszenzgefärbten
Schnitten isoliert, in 30 µl 1fach LDS-Puffer aufgenommen und nach Auftrennung über die SDS PAGE
anschließend mittels Massenspektrometrie analysiert. In 100 Aggregaten konnten 33 Proteine
identifiziert werden und in 250 Aggregaten nur 7 Proteine. Der Vergleich zeigt, dass deutlich mehr
Proteine identifiziert werden, wenn die Aggregate in 40 µl Ameisensäure gelöst werden.
Anzahl isolierte Aggregate
Puffer (30 µl)
Anzahl identifizierte
Proteine
Auswahl an identifizierten Proteinen
100 1fach LDS-
Puffer 33
u.a. Filamin C, Desmin, Alpha-B-Crystallin, Xirp2,
Myosin, Vimentin, Troponin, Nebulin, , Obscurin,
250 1fach LDS-
Puffer 7 Filamin C, Desmin
Bei der Bewertung der Ergebnisse stand die Anzahl der identifizierten Proteine im
Vordergrund. Beim näheren Betrachten der Proteinlisten war es wichtig, den Großteil der
identifizierten Proteine in einen bereits bekannten oder neuen Kontext mit
Skelettmuskulatur einordnen zu können. Ziel war es, einen Überblick über das Potential der
Methode und des jeweiligen Lösungsmittels zu gewinnen. Die genauere Betrachtung der
Proteinlisten unter Berücksichtigung strenger technischer Parameter wurde im Laufe der
eigentlichen Aggregatstudien durchgeführt.
4.1.3. Optimierung der Anzahl von Aggregaten
Für die Ermittlung des optimalen Lösungsmittels wurden 250 Aggregate aus
Filaminopathiegewebe ausgelasert. Um zu testen, ob sich auch eine geringere Menge für die
Analysen eignet, wurden aus fluoreszenzgefärbten Kryoschnitten jeweils 50 und 100
Aggregate aus dem Gewebe isoliert. Erneut wurden die Aggregate in den 3 bereits
erwähnten Lösungsmitteln aufgenommen. 50 mikrodissektierte Aggregate erwiesen sich bei
der Auswertung der massenspektrometrischen Daten als eine zu geringe Probenmenge.
Unter der Verwendung von Ameisensäure konnten lediglich 8 Proteine identifiziert werden,
darunter keine MFM-assoziierten Proteine. In den Proben, die in GuHCl und Urea
aufgenommen wurden, konnten keine Proteine identifiziert werden. Die Analyse von 100
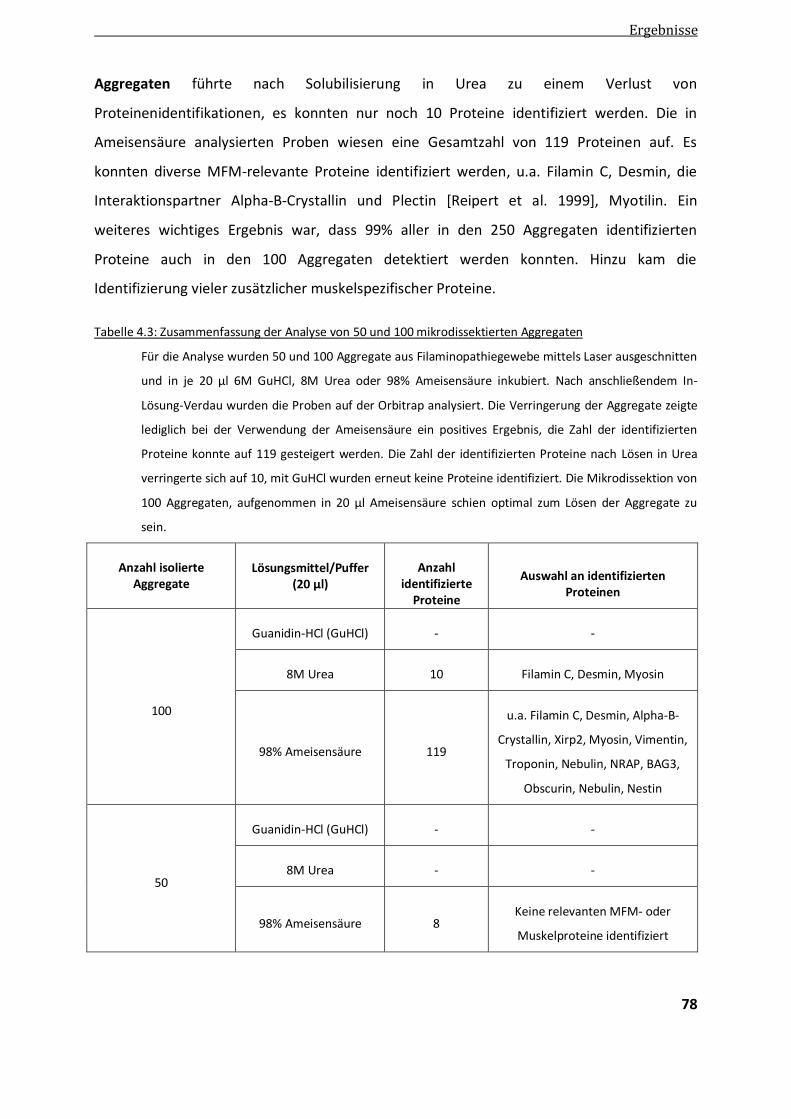
Ergebnisse
78
Aggregaten führte nach Solubilisierung in Urea zu einem Verlust von
Proteinenidentifikationen, es konnten nur noch 10 Proteine identifiziert werden. Die in
Ameisensäure analysierten Proben wiesen eine Gesamtzahl von 119 Proteinen auf. Es
konnten diverse MFM-relevante Proteine identifiziert werden, u.a. Filamin C, Desmin, die
Interaktionspartner Alpha-B-Crystallin und Plectin [Reipert et al. 1999], Myotilin. Ein
weiteres wichtiges Ergebnis war, dass 99% aller in den 250 Aggregaten identifizierten
Proteine auch in den 100 Aggregaten detektiert werden konnten. Hinzu kam die
Identifizierung vieler zusätzlicher muskelspezifischer Proteine.
Tabelle 4.3: Zusammenfassung der Analyse von 50 und 100 mikrodissektierten Aggregaten
Für die Analyse wurden 50 und 100 Aggregate aus Filaminopathiegewebe mittels Laser ausgeschnitten
und in je 20 µl 6M GuHCl, 8M Urea oder 98% Ameisensäure inkubiert. Nach anschließendem In-
Lösung-Verdau wurden die Proben auf der Orbitrap analysiert. Die Verringerung der Aggregate zeigte
lediglich bei der Verwendung der Ameisensäure ein positives Ergebnis, die Zahl der identifizierten
Proteine konnte auf 119 gesteigert werden. Die Zahl der identifizierten Proteine nach Lösen in Urea
verringerte sich auf 10, mit GuHCl wurden erneut keine Proteine identifiziert. Die Mikrodissektion von
100 Aggregaten, aufgenommen in 20 µl Ameisensäure schien optimal zum Lösen der Aggregate zu
sein.
Anzahl isolierte Aggregate
Lösungsmittel/Puffer (20 µl)
Anzahl identifizierte
Proteine
Auswahl an identifizierten Proteinen
100
Guanidin-HCl (GuHCl) - -
8M Urea 10 Filamin C, Desmin, Myosin
98% Ameisensäure 119
u.a. Filamin C, Desmin, Alpha-B-
Crystallin, Xirp2, Myosin, Vimentin,
Troponin, Nebulin, NRAP, BAG3,
Obscurin, Nebulin, Nestin
50
Guanidin-HCl (GuHCl) - -
8M Urea - -
98% Ameisensäure 8 Keine relevanten MFM- oder
Muskelproteine identifiziert
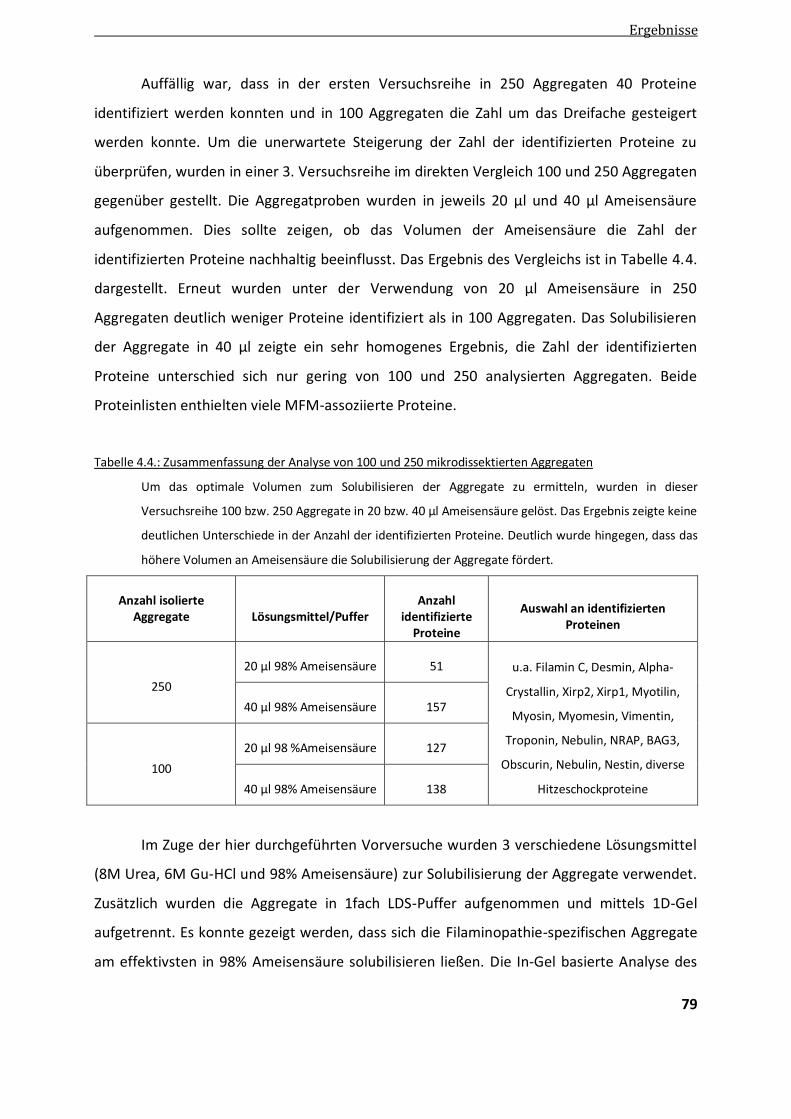
Ergebnisse
79
Auffällig war, dass in der ersten Versuchsreihe in 250 Aggregaten 40 Proteine
identifiziert werden konnten und in 100 Aggregaten die Zahl um das Dreifache gesteigert
werden konnte. Um die unerwartete Steigerung der Zahl der identifizierten Proteine zu
überprüfen, wurden in einer 3. Versuchsreihe im direkten Vergleich 100 und 250 Aggregaten
gegenüber gestellt. Die Aggregatproben wurden in jeweils 20 µl und 40 µl Ameisensäure
aufgenommen. Dies sollte zeigen, ob das Volumen der Ameisensäure die Zahl der
identifizierten Proteine nachhaltig beeinflusst. Das Ergebnis des Vergleichs ist in Tabelle 4.4.
dargestellt. Erneut wurden unter der Verwendung von 20 µl Ameisensäure in 250
Aggregaten deutlich weniger Proteine identifiziert als in 100 Aggregaten. Das Solubilisieren
der Aggregate in 40 µl zeigte ein sehr homogenes Ergebnis, die Zahl der identifizierten
Proteine unterschied sich nur gering von 100 und 250 analysierten Aggregaten. Beide
Proteinlisten enthielten viele MFM-assoziierte Proteine.
Tabelle 4.4.: Zusammenfassung der Analyse von 100 und 250 mikrodissektierten Aggregaten
Um das optimale Volumen zum Solubilisieren der Aggregate zu ermitteln, wurden in dieser
Versuchsreihe 100 bzw. 250 Aggregate in 20 bzw. 40 µl Ameisensäure gelöst. Das Ergebnis zeigte keine
deutlichen Unterschiede in der Anzahl der identifizierten Proteine. Deutlich wurde hingegen, dass das
höhere Volumen an Ameisensäure die Solubilisierung der Aggregate fördert.
Anzahl isolierte Aggregate Lösungsmittel/Puffer
Anzahl identifizierte
Proteine
Auswahl an identifizierten Proteinen
250
20 µl 98% Ameisensäure 51 u.a. Filamin C, Desmin, Alpha-
Crystallin, Xirp2, Xirp1, Myotilin,
Myosin, Myomesin, Vimentin,
Troponin, Nebulin, NRAP, BAG3,
Obscurin, Nebulin, Nestin, diverse
Hitzeschockproteine
40 µl 98% Ameisensäure 157
100
20 µl 98 %Ameisensäure 127
40 µl 98% Ameisensäure 138
Im Zuge der hier durchgeführten Vorversuche wurden 3 verschiedene Lösungsmittel
(8M Urea, 6M Gu-HCl und 98% Ameisensäure) zur Solubilisierung der Aggregate verwendet.
Zusätzlich wurden die Aggregate in 1fach LDS-Puffer aufgenommen und mittels 1D-Gel
aufgetrennt. Es konnte gezeigt werden, dass sich die Filaminopathie-spezifischen Aggregate
am effektivsten in 98% Ameisensäure solubilisieren ließen. Die In-Gel basierte Analyse des

Ergebnisse
80
mikrodissektierten Materials erwies sich im Vergleich zur Solubilisierung in Ameisensäure als
wenig erfolgreich, da sie mit hohen Verlusten an Proteinidentifizierungen einhergeht. In 100
Aggregaten konnten ca. 70% und in 250 Aggregaten ca. 95% weniger Proteine identifiziert
werden. Aufgrund der in dieser Arbeit durchgeführten Versuchsreihen und der daraus
resultierenden Ergebnisse lässt sich zusammenfassen, dass die Solubilisierung von 100
Aggregaten in 40 µl Ameisensäure in Bezug auf die anschließende massenspektrometrische
Analyse die besten Ergebnisse erzielte und zusätzlich einen sehr detaillierten Einblick in das
Proteom von Filaminopathie-Aggregaten gewährte. Da die Größe der Aggregate innerhalb
des Patientengewebes und auch zwischen den Patienten zum Teil enorm variierte, wurde
mit Hilfe des LMD 6500 (Leica, Wetzlar, Deutschland) die durschnittschliche Fläche von 100
Aggregaten ermittelt, diese betrug 250000 µm2. Für alle weiteren Analysen wurde diese
Fläche eingesetzt.
4.2. Ergebnisse der Label-freien Proteomanalyse von Filaminopathiegewebe
Zu Beginn dieser Dissertation waren lediglich vereinzelte Bestandteile der
Filaminopathie-spezifischen Proteinaggregate bekannt. Mittels Immunfluoreszenzstudien
konnten u.a. Filamin C, Desmin, Myotilin und Xin als feste Komponenten der Aggregate
charakterisiert werden [Kley et al. 2007]. Um einen detaillierteren Einblick in die
Zusammensetzung der Aggregate in Filaminopathien zu erlangen, wurde eine Fläche von 250
000 µm2 aus 6 Filaminopathiepatienten sowie aus 6 gesunden Kontrollen mittels LMD
isoliert. Aus dem Filaminopathiegewebe wurden einmal die spezifischen Proteinaggregate
(Aggregatproben) und einmal Muskelzellen ohne Aggregate (Patientenkontrolle, PatKO)
gesammelt, als Kontrolle dienten gesunde Muskelfasern (Negativkontrolle, NegKO). Alle
Proben wurden in Ameisensäure aufgenommen. Anschließend wurden die Proben mittels
Label-freier Proteomanalyse analysiert, die Kriterien zur Datenbanksuche sind unter Punkt
3.16.3. beschrieben.

Ergebnisse
81
4.2.1. Entwicklung einer geeigneten Auswertestrategie
Im Anschluss an die Datenbanksuche wurden die identifizierten Proteine in einer
Proteinliste zusammengefasst. Das Erstellen der Proteinlisten ermöglichte einen ersten,
qualitativen Eindruck von der Zusammensetzung der Proteinaggregate sowie des
umliegenden Gewebes und der Negativkontrollen. In den Aggregatproben konnten
durchschnittlich 415 Proteine identifiziert werden. In den zugehörigen Patienten- und
Negativkontrollen wurden 335 bzw. 324 Proteine identifiziert. In allen Proben wurden
diverse MFM-spezifische Proteine wie Filamin C, Desmin, BAG 3, Xin, Myotilin und N-Rap
identifiziert werden. Zusätzlich befanden sich zahlreiche Strukturproteine (z.B. Aktin,
Myosin) und Hitzeschockproteine in den generierten Proteinlisten. Im Vergleich zu den
Vorversuchen zum Optimieren der Parameter konnten in den Analysen deutlich mehr
Proteine analysiert werden. Dies lässt sich mit einem zwischenzeitlichen Umbau am
Orbitrap-Massenspektrometer und der daraus resultierenden höheren Sensitivität erklären.
Neben einer allgemeinen Charakterisierung der Zusammensetzung war ein quantitativer
Vergleich der verschiedenen Proben von großem Interesse. Hierfür wurden die Proteinlisten
der Aggregatproben mit denen der Patientenkontrollen sowie die der Patientenkontrollen
mit denen der Negativkontrollen verglichen. Ziel der hier durchgeführten Vergleiche war es,
spezifische Bestandteile der Filaminopathie-spezifischen Aggregate und des umliegenden
Gewebes zu charakterisieren. Hierfür war es wichtig, eine quantitative Auswertestrategie zu
entwickeln. Zu Beginn der Analysen konnte auf keine automatisierte Methode zur
quantitativen Auswertung zurückgegriffen werden. Aus diesem Grund wurden in einem
ersten Versuch die vorliegenden Daten mit Hilfe einer manuellen Strategie quantitativ
ausgewertet. Neben den unter Punkt 3.16.3. beschriebenen Parametern zur
Datenbanksuche wurden strenge, eigens gewählte technische Parameter herangezogen
(beschrieben unter Punkt 3.17.1.). Das Ansetzen der technischen Parameter diente der
Vorsortierung der Proteine. Zusätzlich wurden nur Proteine in die finale Proteinliste
aufgenommen, die ausschließlich in den Aggregaten bzw. im umliegenden
Filaminopathiegewebe und in keiner Kontrolle identifiziert wurden. Mittels dieses Kriteriums
sollten Rückschlüsse auf spezifische Bestandteile des erkrankten Gewebes gezogen werden.

Ergebnisse
82
4.2.1.1. Vergleichende Analyse Aggregatproben - Patientenkontrolle
Die Daten der Aggregatproben sowie die Daten der Patientenkontrollen wurden
zunächst in unabhängigen Excel-Tabellen zusammengefasst. Alle Proteine wurden
hinsichtlich ihres durchschnittlichen maximalen MascotScores sortiert. Alle Proteine mit
einem MascotScore über 50 wurden berücksichtigt. Als nächstes wurden diese Proteine nach
der Häufigkeit ihres Vorkommens in den Patienten sortiert. Es wurden die Proteine in der
Liste belassen, die in mindestens 3 von 6 Patienten zu identifizieren waren. Der
Datenbankabgleich der Aggregatproben ergab eine Gesamtzahl an 687 identifizierten
Proteinen. Nach Ansetzen der technischen Parameter verblieben 255 Proteine in der Liste.
Es Konnten 12 MFM-assoziierte Proteine identifiziert werden, u.a. Filamin C, Desmin, Plectin,
Obscurin, FHL1, Myotilin, BAG3, NRAP und Titin. Zusätzlich war es möglich diverse Motor-
und Strukturproteine des Skelettmuskels (z.B. Desmin, Myosin, Aktin, Tropomyosin),
Bindungspartner der Strukturproteine sowie ihre Isoformen zu detektierten. Die Analyse der
Patientenkontrollen zeigte ein vergleichbares Abbild des Proteoms. Die Zahl der
identifizierten Proteine belief sich auf 634, nach Sortieren der Proteine blieben 165 Proteine
erhalten. Auch hier wurden typische MFM-assoziierte Proteine und Strukturproteine des
Skelettmuskels identifiziert.
An die individuelle Analyse der beiden Probensätze schloss sich der Vergleich an. Die
Proteinlisten der Aggregatproben wurden den Daten der Patientenkontrollen (PatKO)
gegenübergestellt. In dieser finalen Proteinliste wurden nur die Proteine akzeptiert, die in
mindestens 3 der 6 Aggregatproben von Filaminopathiepatienten auftauchten und
gleichzeitig in keiner Patientenkontrolle. Mittels dieses Kriteriums sollten Rückschlüsse auf
spezifisch in den Aggregaten akkumulierte Proteine gezogen werden. Die Proteinlisten der
beiden Gruppen wurden zusammengefügt und alle doppelt identifizierten Proteine gelöscht.
Daraufhin wurden insgesamt 394 Proteine in den Vergleich miteinbezogen. 179 dieser
Proteine konnten ausschließlich den Aggregatproben zugeordnet werden, von diesen
wurden 111 in mindestens 3 der Patienten identifiziert. 202 Proteine wurden in beiden
Proben gefunden und 13 Proteine traten lediglich in den Patientenkontrollen auf. In Tabelle
4.5. sind alle Proteine aufgeführt, die als spezifische Charakteristika für Filaminopathien
gewertet werden könnten. Einige MFM-assoziierte Proteine konnten als spezifische
Bestandteile der Aggregate detektiert werden, darunter u.a. Xirp2, BAG3 und FHL1.
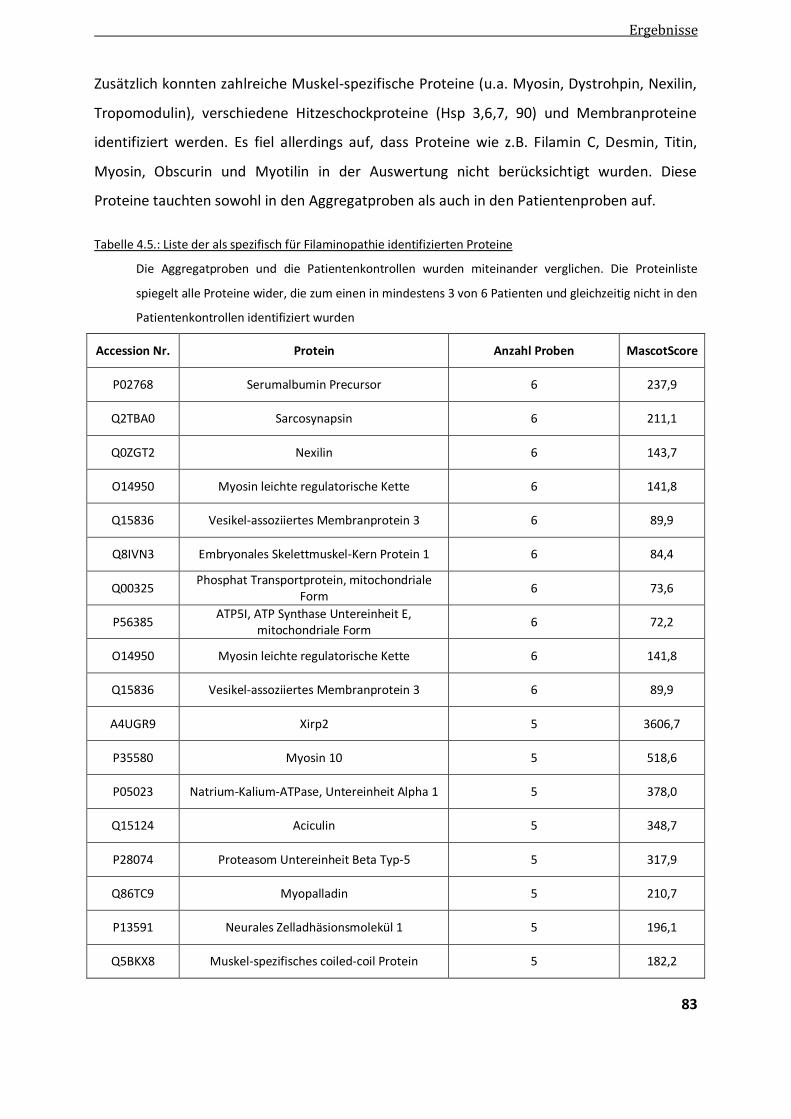
Ergebnisse
83
Zusätzlich konnten zahlreiche Muskel-spezifische Proteine (u.a. Myosin, Dystrohpin, Nexilin,
Tropomodulin), verschiedene Hitzeschockproteine (Hsp 3,6,7, 90) und Membranproteine
identifiziert werden. Es fiel allerdings auf, dass Proteine wie z.B. Filamin C, Desmin, Titin,
Myosin, Obscurin und Myotilin in der Auswertung nicht berücksichtigt wurden. Diese
Proteine tauchten sowohl in den Aggregatproben als auch in den Patientenproben auf.
Tabelle 4.5.: Liste der als spezifisch für Filaminopathie identifizierten Proteine
Die Aggregatproben und die Patientenkontrollen wurden miteinander verglichen. Die Proteinliste
spiegelt alle Proteine wider, die zum einen in mindestens 3 von 6 Patienten und gleichzeitig nicht in den
Patientenkontrollen identifiziert wurden
Accession Nr. Protein Anzahl Proben MascotScore
P02768 Serumalbumin Precursor 6 237,9
Q2TBA0 Sarcosynapsin 6 211,1
Q0ZGT2 Nexilin 6 143,7
O14950 Myosin leichte regulatorische Kette 6 141,8
Q15836 Vesikel-assoziiertes Membranprotein 3 6 89,9
Q8IVN3 Embryonales Skelettmuskel-Kern Protein 1 6 84,4
Q00325 Phosphat Transportprotein, mitochondriale
Form 6 73,6
P56385 ATP5I, ATP Synthase Untereinheit E,
mitochondriale Form 6 72,2
O14950 Myosin leichte regulatorische Kette 6 141,8
Q15836 Vesikel-assoziiertes Membranprotein 3 6 89,9
A4UGR9 Xirp2 5 3606,7
P35580 Myosin 10 5 518,6
P05023 Natrium-Kalium-ATPase, Untereinheit Alpha 1 5 378,0
Q15124 Aciculin 5 348,7
P28074 Proteasom Untereinheit Beta Typ-5 5 317,9
Q86TC9 Myopalladin 5 210,7
P13591 Neurales Zelladhäsionsmolekül 1 5 196,1
Q5BKX8 Muskel-spezifisches coiled-coil Protein 5 182,2
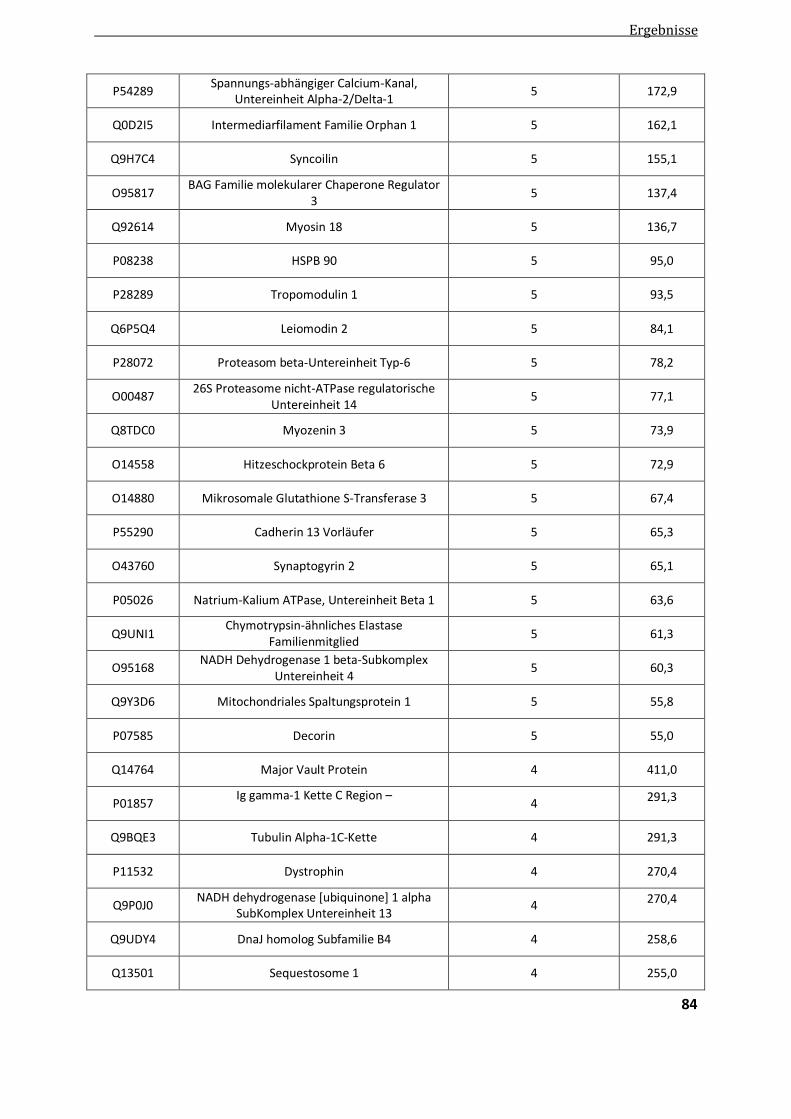
Ergebnisse
84
P54289 Spannungs-abhängiger Calcium-Kanal,
Untereinheit Alpha-2/Delta-1 5 172,9
Q0D2I5 Intermediarfilament Familie Orphan 1 5 162,1
Q9H7C4 Syncoilin 5 155,1
O95817 BAG Familie molekularer Chaperone Regulator
3 5 137,4
Q92614 Myosin 18 5 136,7
P08238 HSPB 90 5 95,0
P28289 Tropomodulin 1 5 93,5
Q6P5Q4 Leiomodin 2 5 84,1
P28072 Proteasom beta-Untereinheit Typ-6 5 78,2
O00487 26S Proteasome nicht-ATPase regulatorische
Untereinheit 14 5 77,1
Q8TDC0 Myozenin 3 5 73,9
O14558 Hitzeschockprotein Beta 6 5 72,9
O14880 Mikrosomale Glutathione S-Transferase 3 5 67,4
P55290 Cadherin 13 Vorläufer 5 65,3
O43760 Synaptogyrin 2 5 65,1
P05026 Natrium-Kalium ATPase, Untereinheit Beta 1 5 63,6
Q9UNI1 Chymotrypsin-ähnliches Elastase
Familienmitglied
5 61,3
O95168 NADH Dehydrogenase 1 beta-Subkomplex
Untereinheit 4 5 60,3
Q9Y3D6 Mitochondriales Spaltungsprotein 1 5 55,8
P07585 Decorin 5 55,0
Q14764 Major Vault Protein 4 411,0
P01857 Ig gamma-1 Kette C Region –
4 291,3
Q9BQE3 Tubulin Alpha-1C-Kette 4 291,3
P11532 Dystrophin 4 270,4
Q9P0J0 NADH dehydrogenase [ubiquinone] 1 alpha
SubKomplex Untereinheit 13 4 270,4
Q9UDY4 DnaJ homolog Subfamilie B4 4 258,6
Q13501 Sequestosome 1 4 255,0
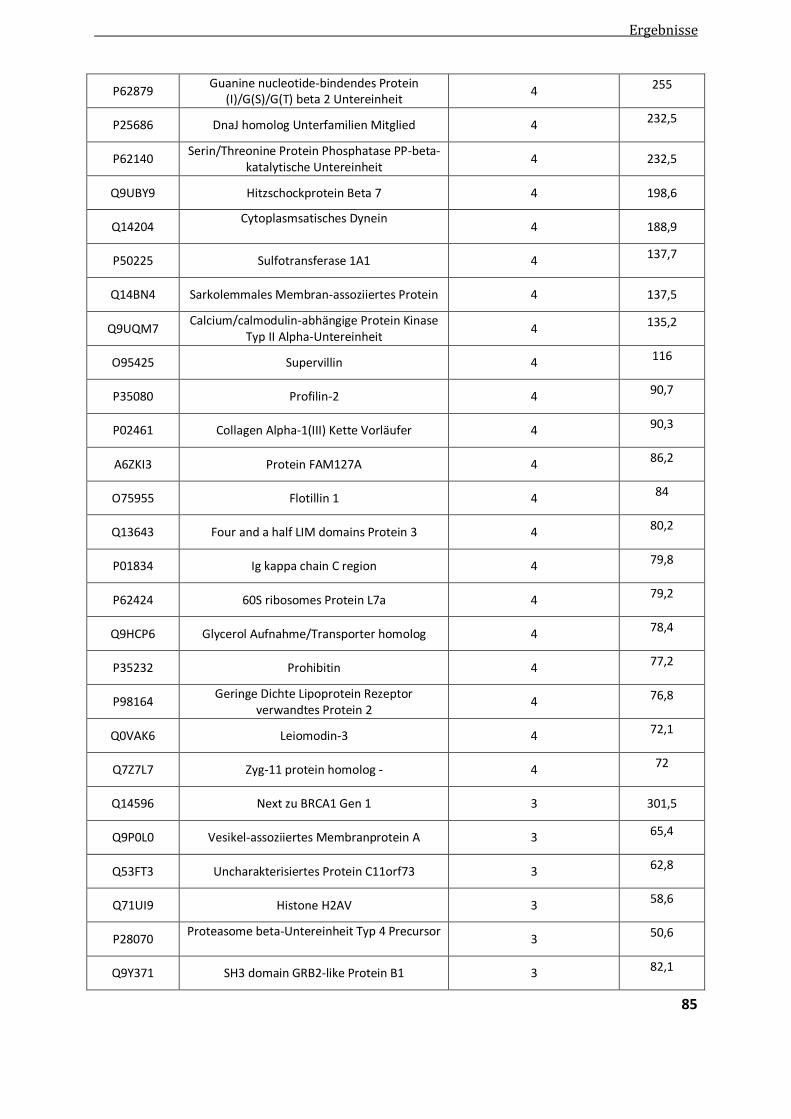
Ergebnisse
85
P62879 Guanine nucleotide-bindendes Protein
(I)/G(S)/G(T) beta 2 Untereinheit 4 255
P25686 DnaJ homolog Unterfamilien Mitglied 4 232,5
P62140 Serin/Threonine Protein Phosphatase PP-beta-
katalytische Untereinheit 4 232,5
Q9UBY9 Hitzschockprotein Beta 7 4 198,6
Q14204 Cytoplasmsatisches Dynein
4 188,9
P50225 Sulfotransferase 1A1 4 137,7
Q14BN4 Sarkolemmales Membran-assoziiertes Protein 4 137,5
Q9UQM7 Calcium/calmodulin-abhängige Protein Kinase
Typ II Alpha-Untereinheit 4 135,2
O95425 Supervillin 4 116
P35080 Profilin-2 4 90,7
P02461 Collagen Alpha-1(III) Kette Vorläufer 4 90,3
A6ZKI3 Protein FAM127A 4 86,2
O75955 Flotillin 1 4 84
Q13643 Four and a half LIM domains Protein 3 4 80,2
P01834 Ig kappa chain C region 4 79,8
P62424 60S ribosomes Protein L7a 4 79,2
Q9HCP6 Glycerol Aufnahme/Transporter homolog 4 78,4
P35232 Prohibitin 4 77,2
P98164 Geringe Dichte Lipoprotein Rezeptor
verwandtes Protein 2 4 76,8
Q0VAK6 Leiomodin-3 4 72,1
Q7Z7L7 Zyg-11 protein homolog - 4 72
Q14596 Next zu BRCA1 Gen 1 3 301,5
Q9P0L0 Vesikel-assoziiertes Membranprotein A 3 65,4
Q53FT3 Uncharakterisiertes Protein C11orf73 3 62,8
Q71UI9 Histone H2AV 3 58,6
P28070 Proteasome beta-Untereinheit Typ 4 Precursor
3 50,6
Q9Y371 SH3 domain GRB2-like Protein B1 3 82,1
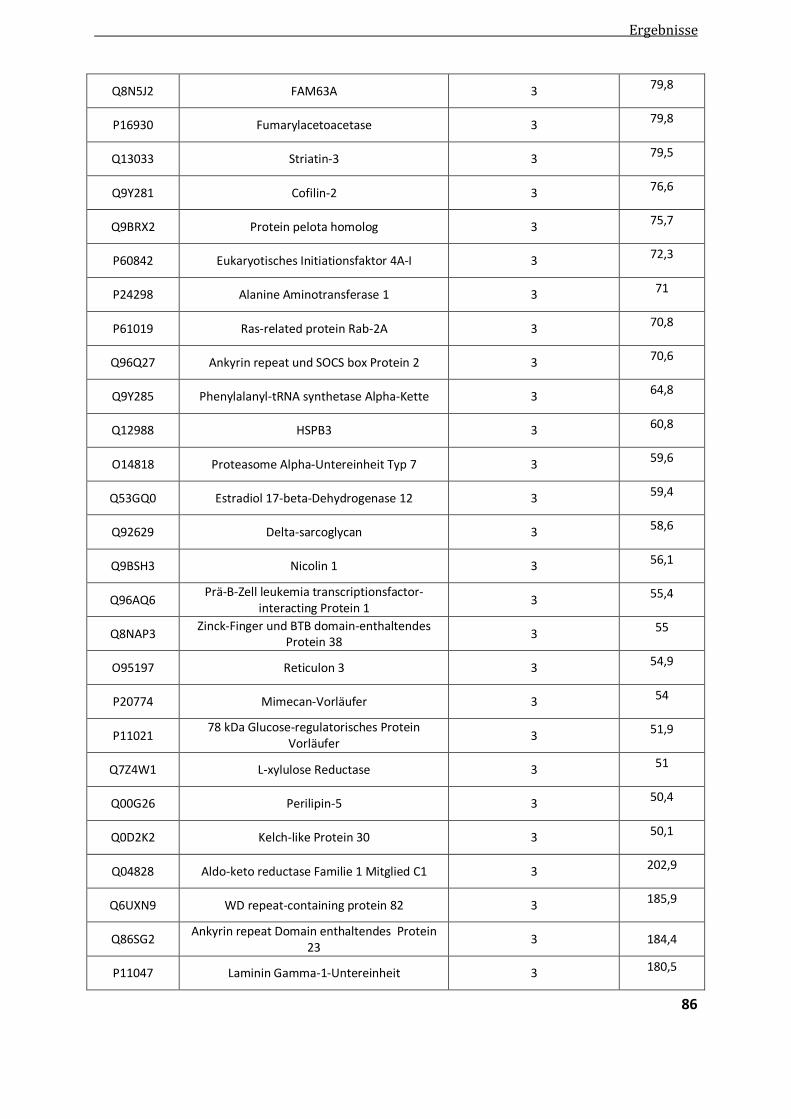
Ergebnisse
86
Q8N5J2 FAM63A 3 79,8
P16930 Fumarylacetoacetase 3 79,8
Q13033 Striatin-3 3 79,5
Q9Y281 Cofilin-2 3 76,6
Q9BRX2 Protein pelota homolog 3 75,7
P60842 Eukaryotisches Initiationsfaktor 4A-I 3 72,3
P24298 Alanine Aminotransferase 1 3 71
P61019 Ras-related protein Rab-2A 3 70,8
Q96Q27 Ankyrin repeat und SOCS box Protein 2 3 70,6
Q9Y285 Phenylalanyl-tRNA synthetase Alpha-Kette 3 64,8
Q12988 HSPB3 3 60,8
O14818 Proteasome Alpha-Untereinheit Typ 7 3 59,6
Q53GQ0 Estradiol 17-beta-Dehydrogenase 12 3 59,4
Q92629 Delta-sarcoglycan 3 58,6
Q9BSH3 Nicolin 1 3 56,1
Q96AQ6 Prä-B-Zell leukemia transcriptionsfactor-
interacting Protein 1 3 55,4
Q8NAP3 Zinck-Finger und BTB domain-enthaltendes
Protein 38 3 55
O95197 Reticulon 3 3 54,9
P20774 Mimecan-Vorläufer 3 54
P11021 78 kDa Glucose-regulatorisches Protein
Vorläufer
3 51,9
Q7Z4W1 L-xylulose Reductase 3 51
Q00G26 Perilipin-5 3 50,4
Q0D2K2 Kelch-like Protein 30 3 50,1
Q04828 Aldo-keto reductase Familie 1 Mitglied C1 3 202,9
Q6UXN9 WD repeat-containing protein 82 3 185,9
Q86SG2 Ankyrin repeat Domain enthaltendes Protein
23 3 184,4
P11047 Laminin Gamma-1-Untereinheit 3 180,5
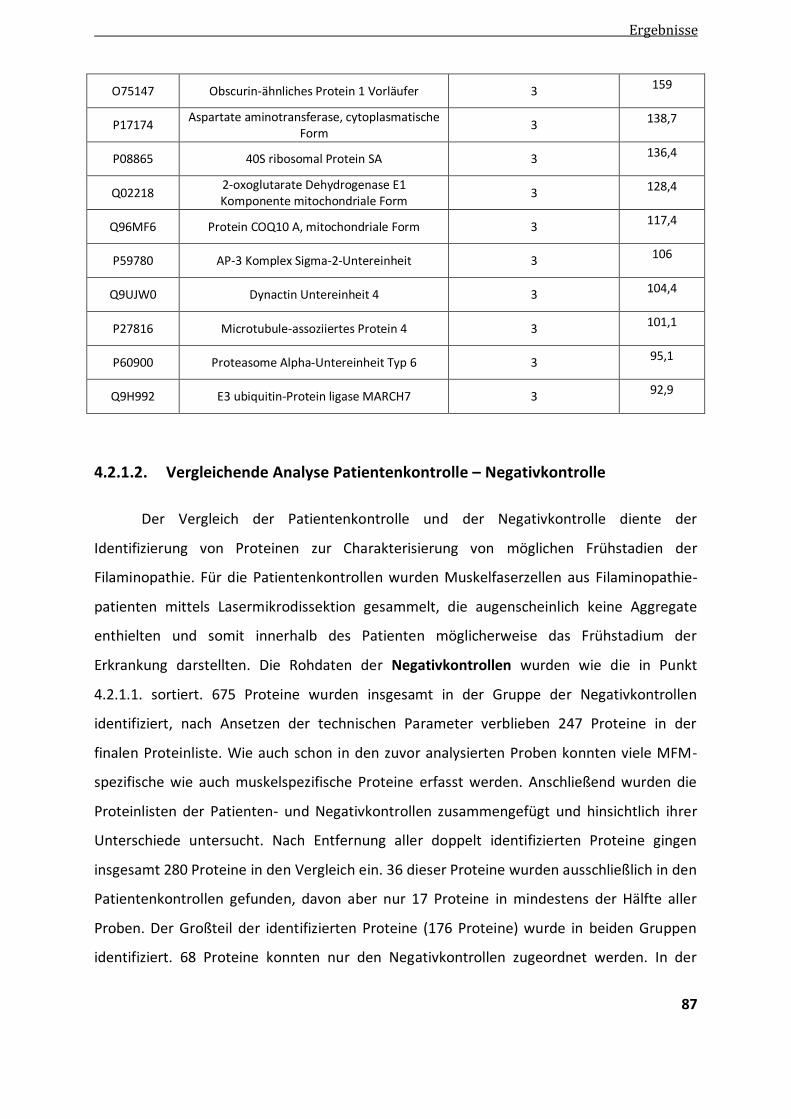
Ergebnisse
87
O75147 Obscurin-ähnliches Protein 1 Vorläufer 3 159
P17174 Aspartate aminotransferase, cytoplasmatische
Form 3 138,7
P08865 40S ribosomal Protein SA 3 136,4
Q02218 2-oxoglutarate Dehydrogenase E1 Komponente mitochondriale Form
3 128,4
Q96MF6 Protein COQ10 A, mitochondriale Form 3 117,4
P59780 AP-3 Komplex Sigma-2-Untereinheit 3 106
Q9UJW0 Dynactin Untereinheit 4 3 104,4
P27816 Microtubule-assoziiertes Protein 4 3 101,1
P60900 Proteasome Alpha-Untereinheit Typ 6 3 95,1
Q9H992 E3 ubiquitin-Protein ligase MARCH7 3 92,9
4.2.1.2. Vergleichende Analyse Patientenkontrolle – Negativkontrolle
Der Vergleich der Patientenkontrolle und der Negativkontrolle diente der
Identifizierung von Proteinen zur Charakterisierung von möglichen Frühstadien der
Filaminopathie. Für die Patientenkontrollen wurden Muskelfaserzellen aus Filaminopathie-
patienten mittels Lasermikrodissektion gesammelt, die augenscheinlich keine Aggregate
enthielten und somit innerhalb des Patienten möglicherweise das Frühstadium der
Erkrankung darstellten. Die Rohdaten der Negativkontrollen wurden wie die in Punkt
4.2.1.1. sortiert. 675 Proteine wurden insgesamt in der Gruppe der Negativkontrollen
identifiziert, nach Ansetzen der technischen Parameter verblieben 247 Proteine in der
finalen Proteinliste. Wie auch schon in den zuvor analysierten Proben konnten viele MFM-
spezifische wie auch muskelspezifische Proteine erfasst werden. Anschließend wurden die
Proteinlisten der Patienten- und Negativkontrollen zusammengefügt und hinsichtlich ihrer
Unterschiede untersucht. Nach Entfernung aller doppelt identifizierten Proteine gingen
insgesamt 280 Proteine in den Vergleich ein. 36 dieser Proteine wurden ausschließlich in den
Patientenkontrollen gefunden, davon aber nur 17 Proteine in mindestens der Hälfte aller
Proben. Der Großteil der identifizierten Proteine (176 Proteine) wurde in beiden Gruppen
identifiziert. 68 Proteine konnten nur den Negativkontrollen zugeordnet werden. In der
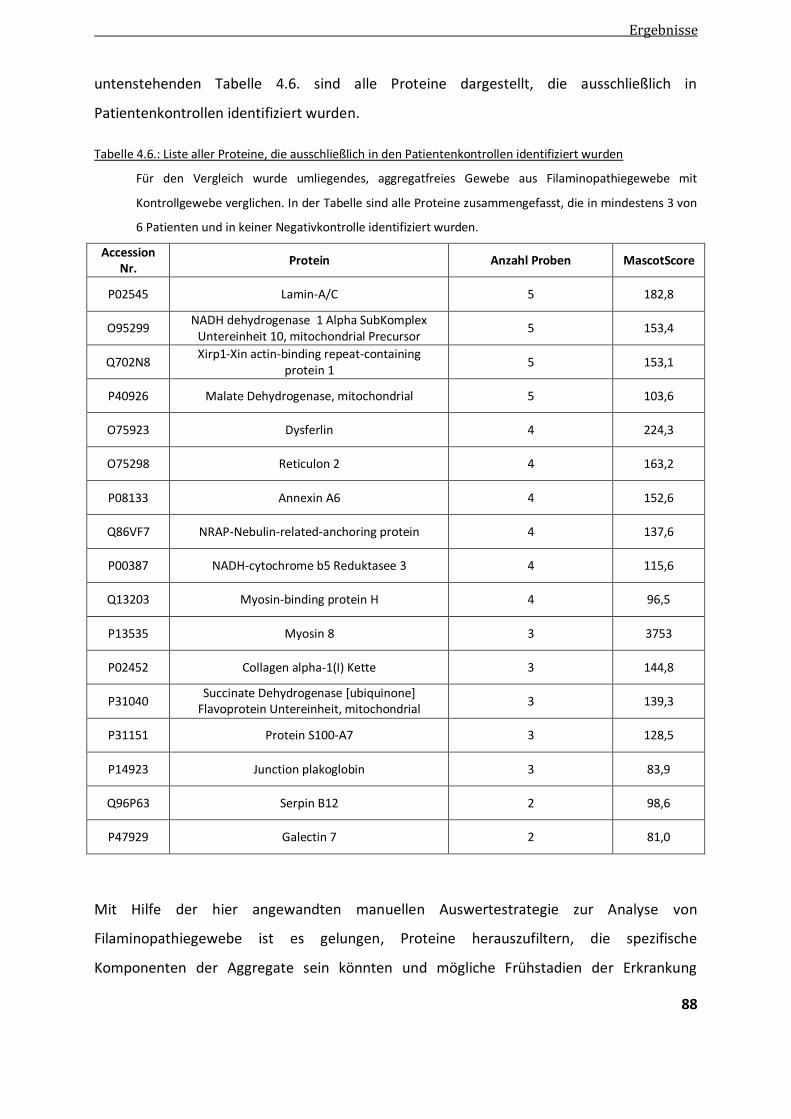
Ergebnisse
88
untenstehenden Tabelle 4.6. sind alle Proteine dargestellt, die ausschließlich in
Patientenkontrollen identifiziert wurden.
Tabelle 4.6.: Liste aller Proteine, die ausschließlich in den Patientenkontrollen identifiziert wurden
Für den Vergleich wurde umliegendes, aggregatfreies Gewebe aus Filaminopathiegewebe mit
Kontrollgewebe verglichen. In der Tabelle sind alle Proteine zusammengefasst, die in mindestens 3 von
6 Patienten und in keiner Negativkontrolle identifiziert wurden.
Accession Nr.
Protein Anzahl Proben MascotScore
P02545 Lamin-A/C 5 182,8
O95299 NADH dehydrogenase 1 Alpha SubKomplex
Untereinheit 10, mitochondrial Precursor 5 153,4
Q702N8 Xirp1-Xin actin-binding repeat-containing
protein 1 5 153,1
P40926 Malate Dehydrogenase, mitochondrial 5 103,6
O75923 Dysferlin 4 224,3
O75298 Reticulon 2 4 163,2
P08133 Annexin A6 4 152,6
Q86VF7 NRAP-Nebulin-related-anchoring protein 4 137,6
P00387 NADH-cytochrome b5 Reduktasee 3 4 115,6
Q13203 Myosin-binding protein H 4 96,5
P13535 Myosin 8 3 3753
P02452 Collagen alpha-1(I) Kette 3 144,8
P31040 Succinate Dehydrogenase [ubiquinone]
Flavoprotein Untereinheit, mitochondrial Precursor
3 139,3
P31151 Protein S100-A7 3 128,5
P14923 Junction plakoglobin 3 83,9
Q96P63 Serpin B12 2 98,6
P47929 Galectin 7 2 81,0
Mit Hilfe der hier angewandten manuellen Auswertestrategie zur Analyse von
Filaminopathiegewebe ist es gelungen, Proteine herauszufiltern, die spezifische
Komponenten der Aggregate sein könnten und mögliche Frühstadien der Erkrankung

Ergebnisse
89
charakterisieren könnten. Der größte Nachteil der hier beschriebenen Auswertestrategie lag
allerdings darin, dass in keiner analysierten Gewebegruppe Aussagen über die Abundanz der
Proteine innerhalb der Aggregate getroffen werden können. Die Wahl der technischen
Parameter ermöglichte es, die Auswahl an identifizierten Proteinen sinnvoll einzuschränken.
Dies gab aber keinerlei Auskunft über eine differentielle Expression von
Gewebekomponenten. Für die Entstehung der Filaminopathie wird es aber wichtig sein zu
sehen, welche Proteine angereichert, in ihrer Abundanz verringert oder abgebaut werden.
Aus diesem Grund war es wichtig, die manuell durchgeführte Strategie zur quantitativen
Auswertung hinsichtlich der quantitativen Aussagekraft weiter zu entwickeln, um eine
mögliche Regulation von Proteinen zu berücksichtigen. Im Rahmen einer bei uns
durchgeführten Masterarbeit von M.Sc. Alexandra Maerkens wurde eine automatisierte
Methode zur quantitativen Datenauswertung mittels Spectral Index Calculation entwickelt.
Die Spectral Index Calculation ist ein Label-freies Verfahren, basierend auf der Summierung
aller für ein Peptid aufgenommene MS/MS-Spektren. Im Rahmen dieses Projektes wurde
zwischen zwei Strategien unterschieden. Die allgemeine Auswertung bezog alle Peptide in
die Auswertung mit ein. Dies hatte allerdings den Nachteil, dass nicht-unique Peptide
mehreren Proteinen zugeordnet werden konnten. Somit war es nicht möglich, eindeutige
Aussagen zu der Zugehörigkeit eines Peptids zu einem Protein zu treffen. Daraus entwickelte
sich die zweite Strategie, die auf die Einbeziehung nur uniquer Peptiden abzielte. Unique
Proteine können eindeutig einem einzigen Protein zugeordnet werden. M.Sc. Alexandra
Maerkens optimierte die Spectral Index Calculation für unique Peptide im Zuge ihrer
Masterarbeit am Medizinischen Proteom-Center. Auf Grundlage der hier durchgeführten
massenspektrometrischen Analysen von mikrodissektierten Aggregaten aus 6
Filaminopathiepatienten führte sie die automatisierte Datenauswertung durch. Im Vergleich
zu meiner durchgeführten manuellen Auswertung war es mittels Spectral Index Calculation
möglich, differentiell exprimierte Proteine in den Aggregatproben zu detektieren. Hierfür
wurden zunächst innerhalb der Aggregatproben bzw. der Patientenkontrollen die Summen
aller Peptidcounts gebildet. Oft konnten deutliche Unterschiede der Summen innerhalb der
Gruppen beobachtet werden. Die Unterschiede entstanden aufgrund der Beschaffenheit
und Eigenschaften der Proben, Ungleichmäßigkeiten in der Probensammlung und in der
massenspektrometrischen Analyse. Um diese Unterschiede auszugleichen erfolgte eine

Ergebnisse
90
Normalisierung über die Gesamtzahl der Spectral Peptidcounts und dem Ausgleichsfaktor
(beschrieben unter 3.17.2.). Für die Berechnung der differentiell exprimierten Proteine
wurden zunächst die Spectral Indices berechnet und anschließend der durchschnittliche
Spectral Index für jedes einzelne Protein gebildet. Die Ratio beschrieb das Verhältnis
zwischen identifizierten Proteinen in Aggregaten und Patientenkontrollen. Ab einer Ratio
von ≥ 1,8 und einem p-Wert ≤ 0,05 wurden Proteine als höher abundant in den Aggregaten
bezeichnet, bei einer Ratio von ≤ 0,55 (p-Wert von ≤ 0,05) als geringer exprimiert im
Vergleich zu den Patientenkontrollen. Insgesamt konnten 69 Proteine als signifikant
hochreguliert identifiziert werden. Filamin C wies den höchsten durchschnittlichen Spectral
Index (SI) auf und stellte somit das am stärksten akkumulierte Protein in den Aggregaten dar.
Auf den nachfolgenden Rängen erschienen Desmin, Xirp2 und N-RAP. Die vollständigen
Proteinlisten sind im Anhang in den Tabellen 13.1. und 13.2. dargestellt.
Im Zuge der neu entwickelten automatisierten quantitativen Auswertung wurden die
generierten Daten der Patientenkontrollen und Negativkontrollen im Rahmen dieser
Dissertation neu ausgewertet. Der Vergleich von Patientenkontrollen und Negativkontrollen
wurde unter denselben Kriterien durchgeführt wie oben beschrieben. Insgesamt konnten 9
Proteine identifiziert werden, die in den Patientenkontrollen höher abundant sind als in den
Negativkontrollen (Tabellen 4.7, 4.8.). Unter den höher abundanten Proteinen befinden sich
2 Muskel-spezifische Proteine (LM 3 und Obscurin). Bei den anderen Proteinen handelt es
sich vorzugsweise um Enzyme und Bestandteile der Genexpression. Als niedrig abundante
Proteine in Patientenkontrollen wurden hauptsächlich Muskel-spezifische Proteine (u.a.
Aktin, Myosin, Myozenin, Troponin) identifiziert. Dies lässt vermuten, dass in Frühstadien
der Erkrankung vorzugsweise strukturgegebene Komponenten im Muskel zerstört werden.
Tabelle 4.7.: Liste der in Patientenkontrollen höher abundanten Proteine
Zusammenfassung der Proteine, die über Spectral Index Calculation als differentiell exprimiert in den
Patientenkontrollen identifiziert wurden. Die dargestellten Proteine weisen eine Ratio ≥ 1,8 mit einem
p-Wert ≤ 0,05 auf und gelten somit als höher abundant im Vergleich zu den Negativkontrollen. Für die
Patientenkontrollen (PatKO) wie auch die Negativkontrollen (NegKO) wurde für jedes einzelne Protein
der durchschnittliche Spectral Index (SI) aufgeführt. Die Liste wurde nach absteigendem SI in den
Patientenproben sortiert.
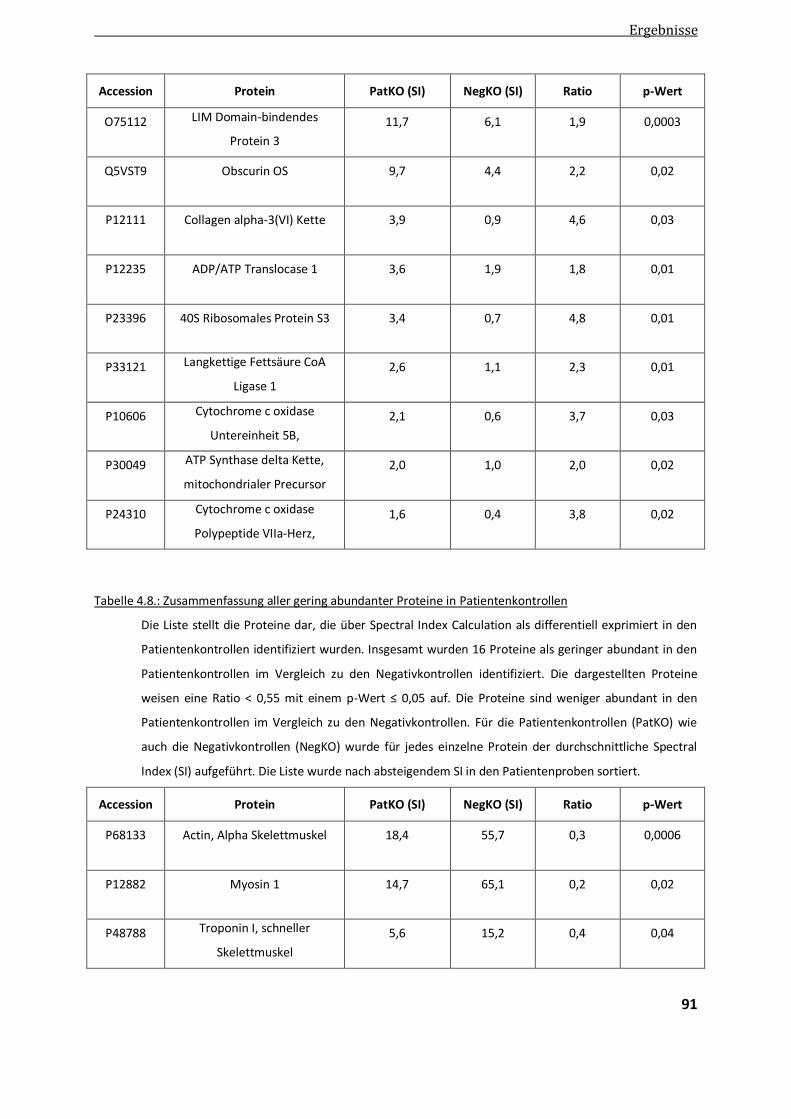
Ergebnisse
91
Accession Protein PatKO (SI) NegKO (SI) Ratio p-Wert
O75112 LIM Domain-bindendes
Protein 3
11,7 6,1 1,9 0,0003
Q5VST9 Obscurin OS 9,7 4,4 2,2 0,02
P12111 Collagen alpha-3(VI) Kette 3,9 0,9 4,6 0,03
P12235 ADP/ATP Translocase 1 3,6 1,9 1,8 0,01
P23396 40S Ribosomales Protein S3 3,4 0,7 4,8 0,01
P33121 Langkettige Fettsäure CoA
Ligase 1
2,6 1,1 2,3 0,01
P10606 Cytochrome c oxidase
Untereinheit 5B,
mitochondrialer Precursor
2,1 0,6 3,7 0,03
P30049 ATP Synthase delta Kette,
mitochondrialer Precursor
2,0 1,0 2,0 0,02
P24310 Cytochrome c oxidase
Polypeptide VIIa-Herz,
mitochondrialer Precursor
1,6 0,4 3,8 0,02
Tabelle 4.8.: Zusammenfassung aller gering abundanter Proteine in Patientenkontrollen
Die Liste stellt die Proteine dar, die über Spectral Index Calculation als differentiell exprimiert in den
Patientenkontrollen identifiziert wurden. Insgesamt wurden 16 Proteine als geringer abundant in den
Patientenkontrollen im Vergleich zu den Negativkontrollen identifiziert. Die dargestellten Proteine
weisen eine Ratio ˂ 0,55 mit einem p-Wert ≤ 0,05 auf. Die Proteine sind weniger abundant in den
Patientenkontrollen im Vergleich zu den Negativkontrollen. Für die Patientenkontrollen (PatKO) wie
auch die Negativkontrollen (NegKO) wurde für jedes einzelne Protein der durchschnittliche Spectral
Index (SI) aufgeführt. Die Liste wurde nach absteigendem SI in den Patientenproben sortiert.
Accession Protein PatKO (SI) NegKO (SI) Ratio p-Wert
P68133 Actin, Alpha Skelettmuskel 18,4 55,7 0,3 0,0006
P12882 Myosin 1 14,7 65,1 0,2 0,02
P48788 Troponin I, schneller
Skelettmuskel
5,6 15,2 0,4 0,04
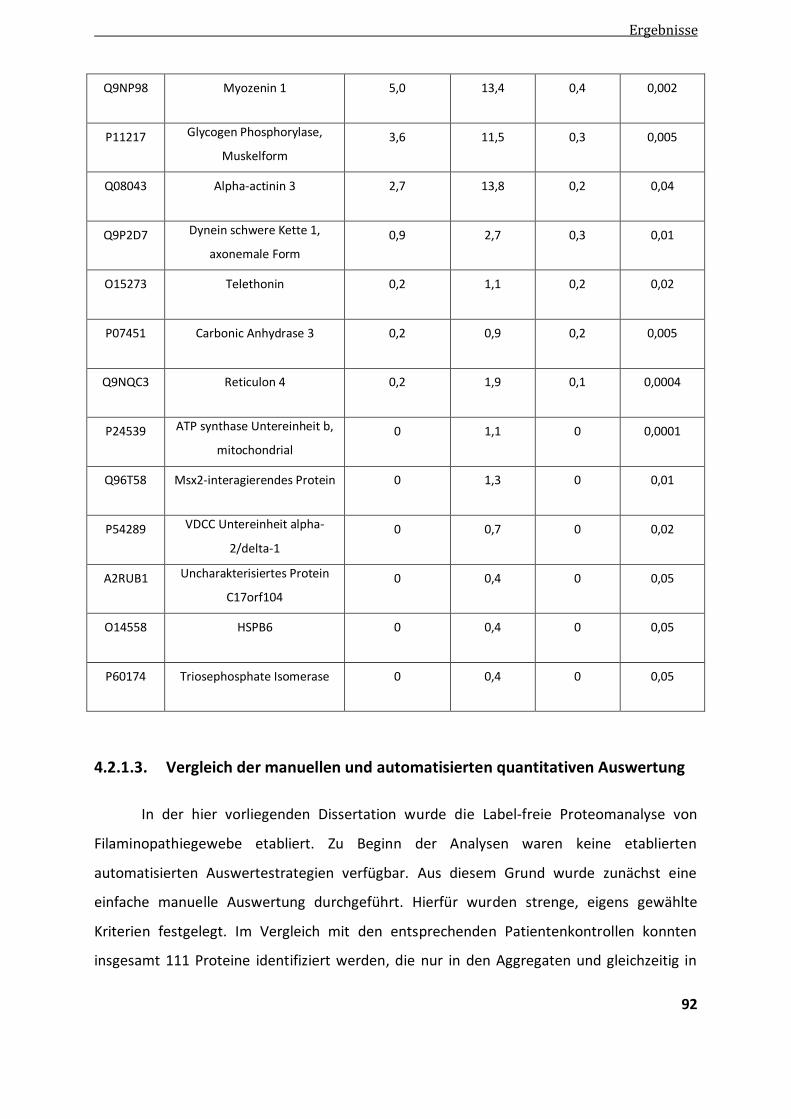
Ergebnisse
92
Q9NP98 Myozenin 1 5,0 13,4 0,4 0,002
P11217 Glycogen Phosphorylase,
Muskelform
3,6 11,5 0,3 0,005
Q08043 Alpha-actinin 3 2,7 13,8 0,2 0,04
Q9P2D7 Dynein schwere Kette 1,
axonemale Form
0,9 2,7 0,3 0,01
O15273 Telethonin 0,2 1,1 0,2 0,02
P07451 Carbonic Anhydrase 3 0,2 0,9 0,2 0,005
Q9NQC3 Reticulon 4 0,2 1,9 0,1 0,0004
P24539 ATP synthase Untereinheit b,
mitochondrial
0 1,1 0 0,0001
Q96T58 Msx2-interagierendes Protein 0 1,3 0 0,01
P54289 VDCC Untereinheit alpha-
2/delta-1
0 0,7 0 0,02
A2RUB1 Uncharakterisiertes Protein
C17orf104
0 0,4 0 0,05
O14558 HSPB6 0 0,4 0 0,05
P60174 Triosephosphate Isomerase 0 0,4 0 0,05
4.2.1.3. Vergleich der manuellen und automatisierten quantitativen Auswertung
In der hier vorliegenden Dissertation wurde die Label-freie Proteomanalyse von
Filaminopathiegewebe etabliert. Zu Beginn der Analysen waren keine etablierten
automatisierten Auswertestrategien verfügbar. Aus diesem Grund wurde zunächst eine
einfache manuelle Auswertung durchgeführt. Hierfür wurden strenge, eigens gewählte
Kriterien festgelegt. Im Vergleich mit den entsprechenden Patientenkontrollen konnten
insgesamt 111 Proteine identifiziert werden, die nur in den Aggregaten und gleichzeitig in
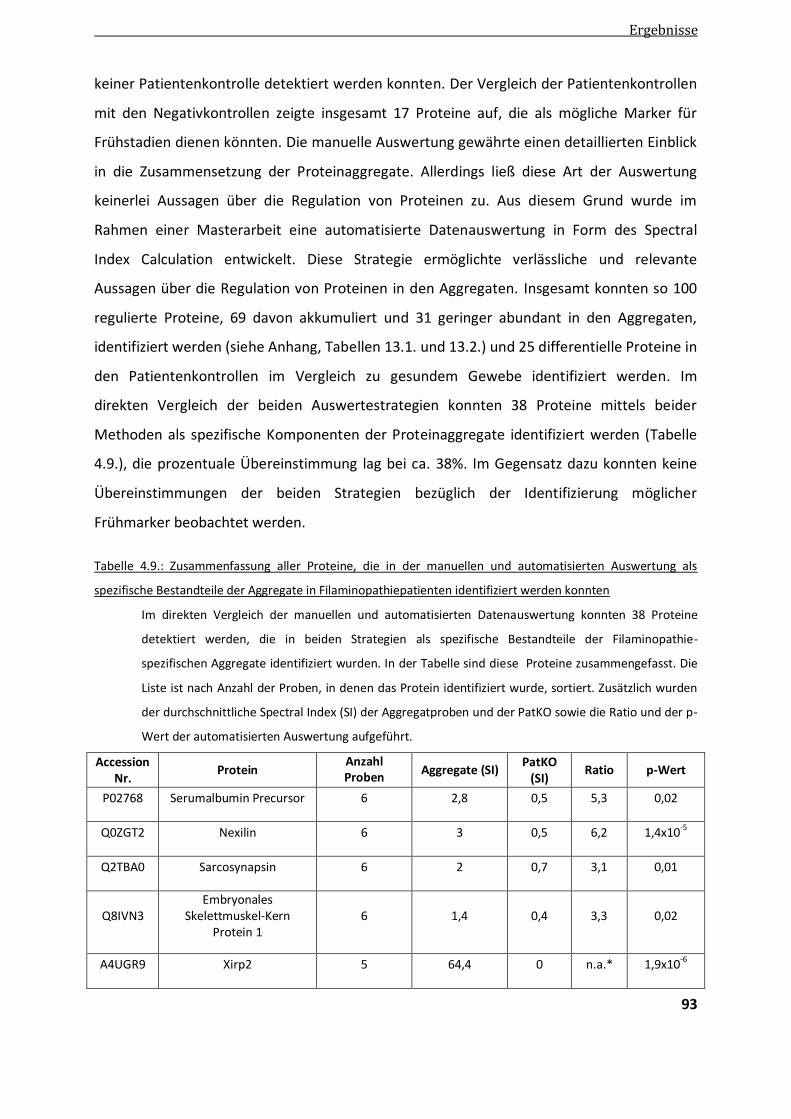
Ergebnisse
93
keiner Patientenkontrolle detektiert werden konnten. Der Vergleich der Patientenkontrollen
mit den Negativkontrollen zeigte insgesamt 17 Proteine auf, die als mögliche Marker für
Frühstadien dienen könnten. Die manuelle Auswertung gewährte einen detaillierten Einblick
in die Zusammensetzung der Proteinaggregate. Allerdings ließ diese Art der Auswertung
keinerlei Aussagen über die Regulation von Proteinen zu. Aus diesem Grund wurde im
Rahmen einer Masterarbeit eine automatisierte Datenauswertung in Form des Spectral
Index Calculation entwickelt. Diese Strategie ermöglichte verlässliche und relevante
Aussagen über die Regulation von Proteinen in den Aggregaten. Insgesamt konnten so 100
regulierte Proteine, 69 davon akkumuliert und 31 geringer abundant in den Aggregaten,
identifiziert werden (siehe Anhang, Tabellen 13.1. und 13.2.) und 25 differentielle Proteine in
den Patientenkontrollen im Vergleich zu gesundem Gewebe identifiziert werden. Im
direkten Vergleich der beiden Auswertestrategien konnten 38 Proteine mittels beider
Methoden als spezifische Komponenten der Proteinaggregate identifiziert werden (Tabelle
4.9.), die prozentuale Übereinstimmung lag bei ca. 38%. Im Gegensatz dazu konnten keine
Übereinstimmungen der beiden Strategien bezüglich der Identifizierung möglicher
Frühmarker beobachtet werden.
Tabelle 4.9.: Zusammenfassung aller Proteine, die in der manuellen und automatisierten Auswertung als
spezifische Bestandteile der Aggregate in Filaminopathiepatienten identifiziert werden konnten
Im direkten Vergleich der manuellen und automatisierten Datenauswertung konnten 38 Proteine
detektiert werden, die in beiden Strategien als spezifische Bestandteile der Filaminopathie-
spezifischen Aggregate identifiziert wurden. In der Tabelle sind diese Proteine zusammengefasst. Die
Liste ist nach Anzahl der Proben, in denen das Protein identifiziert wurde, sortiert. Zusätzlich wurden
der durchschnittliche Spectral Index (SI) der Aggregatproben und der PatKO sowie die Ratio und der p-
Wert der automatisierten Auswertung aufgeführt.
Accession Nr.
Protein Anzahl Proben
Aggregate (SI) PatKO
(SI) Ratio p-Wert
P02768 Serumalbumin Precursor 6 2,8 0,5 5,3 0,02
Q0ZGT2 Nexilin 6 3 0,5 6,2 1,4x10-5
Q2TBA0 Sarcosynapsin 6 2 0,7 3,1 0,01
Q8IVN3 Embryonales
Skelettmuskel-Kern Protein 1
6 1,4 0,4 3,3 0,02
A4UGR9 Xirp2 5 64,4 0 n.a.* 1,9x10-6
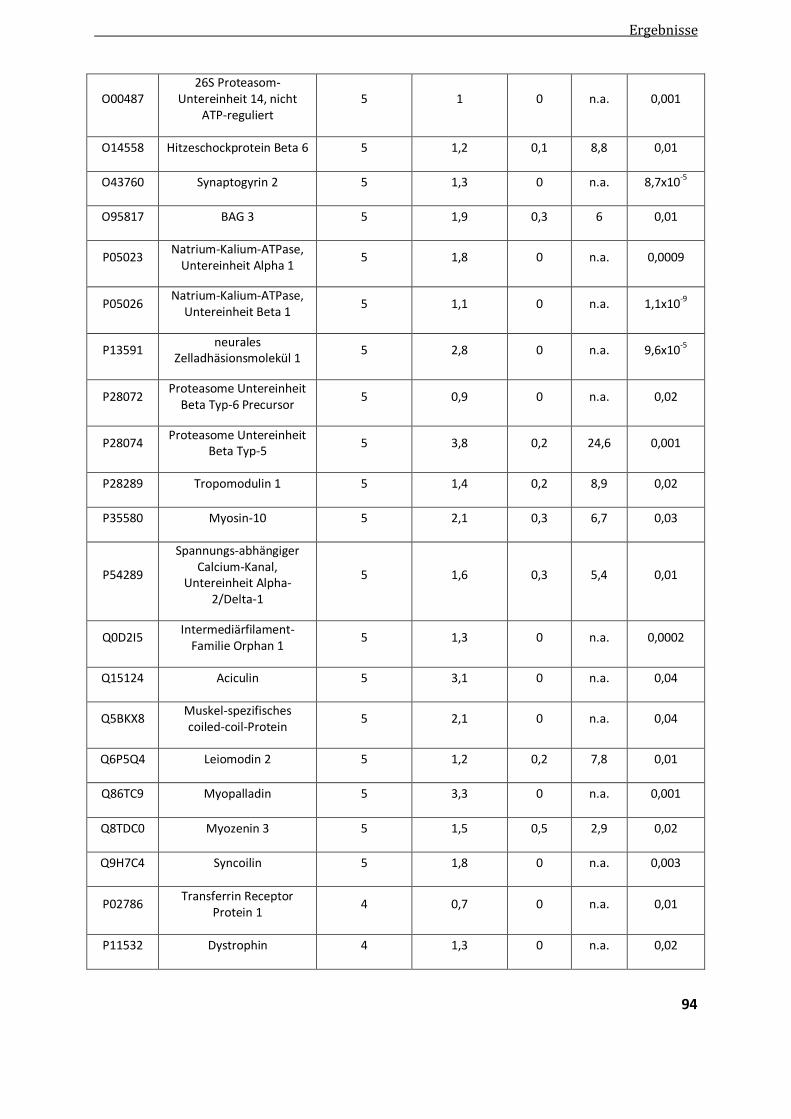
Ergebnisse
94
O00487 26S Proteasom-
Untereinheit 14, nicht ATP-reguliert
5 1 0 n.a. 0,001
O14558 Hitzeschockprotein Beta 6 5 1,2 0,1 8,8 0,01
O43760 Synaptogyrin 2 5 1,3 0 n.a. 8,7x10-5
O95817 BAG 3 5 1,9 0,3 6 0,01
P05023 Natrium-Kalium-ATPase,
Untereinheit Alpha 1 5 1,8 0 n.a. 0,0009
P05026 Natrium-Kalium-ATPase,
Untereinheit Beta 1 5 1,1 0 n.a. 1,1x10
-9
P13591 neurales
Zelladhäsionsmolekül 1 5 2,8 0 n.a. 9,6x10-5
P28072 Proteasome Untereinheit
Beta Typ-6 Precursor 5 0,9 0 n.a. 0,02
P28074 Proteasome Untereinheit
Beta Typ-5 5 3,8 0,2 24,6 0,001
P28289 Tropomodulin 1 5 1,4 0,2 8,9 0,02
P35580 Myosin-10 5 2,1 0,3 6,7 0,03
P54289
Spannungs-abhängiger Calcium-Kanal,
Untereinheit Alpha-2/Delta-1
5 1,6 0,3 5,4 0,01
Q0D2I5 Intermediärfilament-
Familie Orphan 1 5 1,3 0 n.a. 0,0002
Q15124 Aciculin 5 3,1 0 n.a. 0,04
Q5BKX8 Muskel-spezifisches coiled-coil-Protein
5 2,1 0 n.a. 0,04
Q6P5Q4 Leiomodin 2 5 1,2 0,2 7,8 0,01
Q86TC9 Myopalladin 5 3,3 0 n.a. 0,001
Q8TDC0 Myozenin 3 5 1,5 0,5 2,9 0,02
Q9H7C4 Syncoilin 5 1,8 0 n.a. 0,003
P02786 Transferrin Receptor
Protein 1 4 0,7 0 n.a. 0,01
P11532 Dystrophin 4 1,3 0 n.a. 0,02
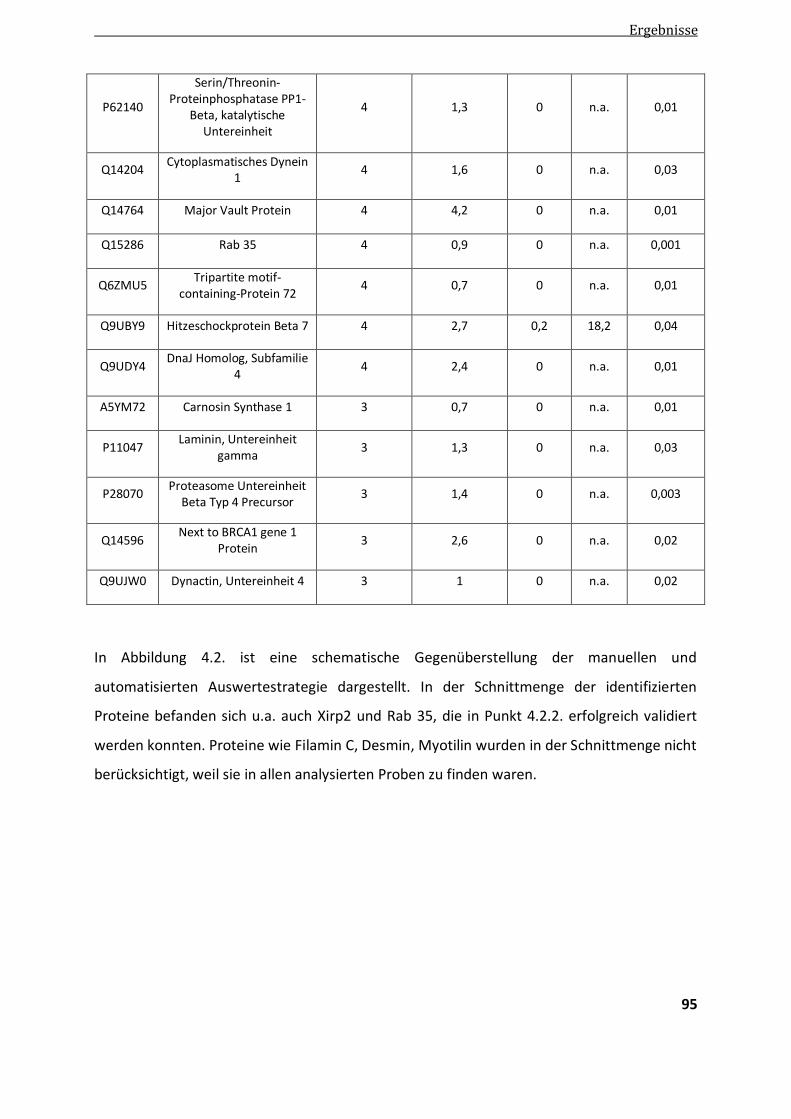
Ergebnisse
95
P62140
Serin/Threonin-Proteinphosphatase PP1-
Beta, katalytische Untereinheit
4 1,3 0 n.a. 0,01
Q14204 Cytoplasmatisches Dynein
1 4 1,6 0 n.a. 0,03
Q14764 Major Vault Protein 4 4,2 0 n.a. 0,01
Q15286 Rab 35 4 0,9 0 n.a. 0,001
Q6ZMU5 Tripartite motif-
containing-Protein 72 4 0,7 0 n.a. 0,01
Q9UBY9 Hitzeschockprotein Beta 7 4 2,7 0,2 18,2 0,04
Q9UDY4 DnaJ Homolog, Subfamilie
4 4 2,4 0 n.a. 0,01
A5YM72 Carnosin Synthase 1 3 0,7 0 n.a. 0,01
P11047 Laminin, Untereinheit
gamma 3 1,3 0 n.a. 0,03
P28070 Proteasome Untereinheit
Beta Typ 4 Precursor 3 1,4 0 n.a. 0,003
Q14596 Next to BRCA1 gene 1
Protein 3 2,6 0 n.a. 0,02
Q9UJW0 Dynactin, Untereinheit 4 3 1 0 n.a. 0,02
In Abbildung 4.2. ist eine schematische Gegenüberstellung der manuellen und
automatisierten Auswertestrategie dargestellt. In der Schnittmenge der identifizierten
Proteine befanden sich u.a. auch Xirp2 und Rab 35, die in Punkt 4.2.2. erfolgreich validiert
werden konnten. Proteine wie Filamin C, Desmin, Myotilin wurden in der Schnittmenge nicht
berücksichtigt, weil sie in allen analysierten Proben zu finden waren.

Ergebnisse
96
Abbildung 4.2.: Vergleich der gewählten Auswertestrategien
In der qualitativen (Schwarz-Weiß) Auswertung konnten insgesamt 111 Proteine identifiziert werden,
die ausschließlich in Filaminopathiegewebe vorkamen. 23 dieser Proteine, darunter auch Xirp2 und
Rab35, konnten ebenfalls in der Gesamtheit aller regulierten Proteine (31 akkumulierte und 10 niedrig
abundante Proteine) mittels Spectral Index Calculation identifiziert werden.
Es hat sich gezeigt, dass es sinnvoller ist, einen automatisierten Ansatz zur quantitativen
Analyse von Filaminopathiegewebe zu wählen. Das alleinige Wissen über die
Zusammensetzung der Aggregate war zu Beginn der Arbeiten hilfreich, führte im Weiteren
jedoch nicht zu den gewünschten Erkenntnissen. Erst die Möglichkeit differentielle Aussagen
zu den identifizierten Proteinen zu treffen, ermöglichte es vielversprechende Daten zu
generieren. Die Spectral Index Calculation bietet eine exzellente Grundlage differentiell
exprimierte Proteine zu identifizieren. Die Normalisierung über die Gesamtzahl der Spectral
Peptidcounts und das Einsetzen eines Ausgleichfaktors lässt es zu z.B. Proben mit
Unterschieden in der Beschaffenheit miteinander quantitativ vergleichen zu können.
Allerdings handelt es sich beim Spectral Index Calculation um eine relative Quantifizierung.
Für zukünftige Analysen wird es von enormer Bedeutung sein, eine Strategie zur absoluten
Quantifizierung zu etablieren (siehe Punkt 5.2.1.3.).
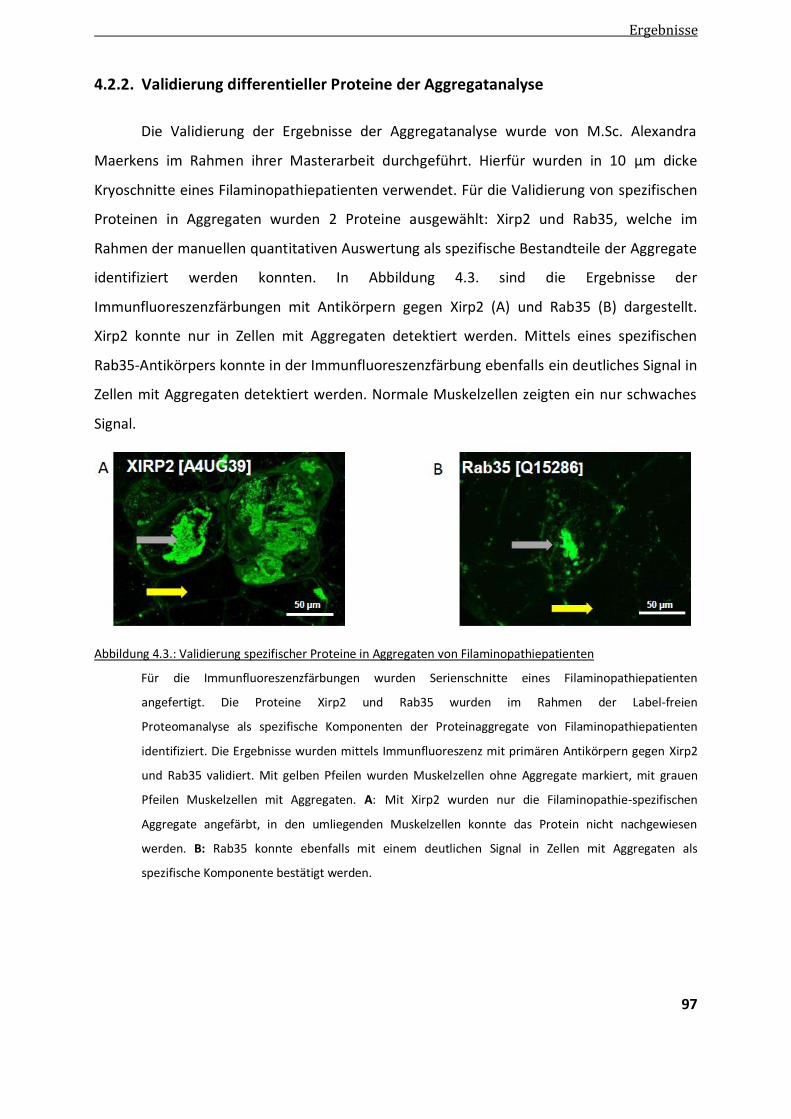
Ergebnisse
97
4.2.2. Validierung differentieller Proteine der Aggregatanalyse
Die Validierung der Ergebnisse der Aggregatanalyse wurde von M.Sc. Alexandra
Maerkens im Rahmen ihrer Masterarbeit durchgeführt. Hierfür wurden in 10 µm dicke
Kryoschnitte eines Filaminopathiepatienten verwendet. Für die Validierung von spezifischen
Proteinen in Aggregaten wurden 2 Proteine ausgewählt: Xirp2 und Rab35, welche im
Rahmen der manuellen quantitativen Auswertung als spezifische Bestandteile der Aggregate
identifiziert werden konnten. In Abbildung 4.3. sind die Ergebnisse der
Immunfluoreszenzfärbungen mit Antikörpern gegen Xirp2 (A) und Rab35 (B) dargestellt.
Xirp2 konnte nur in Zellen mit Aggregaten detektiert werden. Mittels eines spezifischen
Rab35-Antikörpers konnte in der Immunfluoreszenzfärbung ebenfalls ein deutliches Signal in
Zellen mit Aggregaten detektiert werden. Normale Muskelzellen zeigten ein nur schwaches
Signal.
Abbildung 4.3.: Validierung spezifischer Proteine in Aggregaten von Filaminopathiepatienten
Für die Immunfluoreszenzfärbungen wurden Serienschnitte eines Filaminopathiepatienten
angefertigt. Die Proteine Xirp2 und Rab35 wurden im Rahmen der Label-freien
Proteomanalyse als spezifische Komponenten der Proteinaggregate von Filaminopathiepatienten
identifiziert. Die Ergebnisse wurden mittels Immunfluoreszenz mit primären Antikörpern gegen Xirp2
und Rab35 validiert. Mit gelben Pfeilen wurden Muskelzellen ohne Aggregate markiert, mit grauen
Pfeilen Muskelzellen mit Aggregaten. A: Mit Xirp2 wurden nur die Filaminopathie-spezifischen
Aggregate angefärbt, in den umliegenden Muskelzellen konnte das Protein nicht nachgewiesen
werden. B: Rab35 konnte ebenfalls mit einem deutlichen Signal in Zellen mit Aggregaten als
spezifische Komponente bestätigt werden.
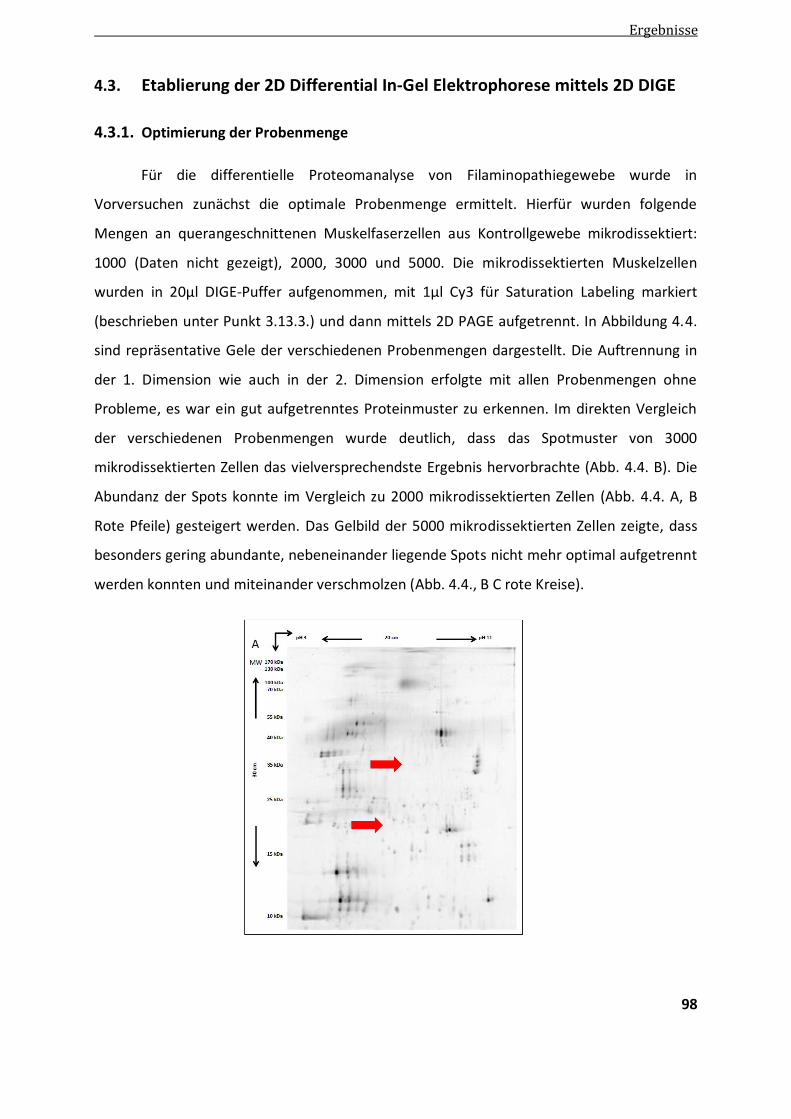
Ergebnisse
98
4.3. Etablierung der 2D Differential In-Gel Elektrophorese mittels 2D DIGE
4.3.1. Optimierung der Probenmenge
Für die differentielle Proteomanalyse von Filaminopathiegewebe wurde in
Vorversuchen zunächst die optimale Probenmenge ermittelt. Hierfür wurden folgende
Mengen an querangeschnittenen Muskelfaserzellen aus Kontrollgewebe mikrodissektiert:
1000 (Daten nicht gezeigt), 2000, 3000 und 5000. Die mikrodissektierten Muskelzellen
wurden in 20µl DIGE-Puffer aufgenommen, mit 1µl Cy3 für Saturation Labeling markiert
(beschrieben unter Punkt 3.13.3.) und dann mittels 2D PAGE aufgetrennt. In Abbildung 4.4.
sind repräsentative Gele der verschiedenen Probenmengen dargestellt. Die Auftrennung in
der 1. Dimension wie auch in der 2. Dimension erfolgte mit allen Probenmengen ohne
Probleme, es war ein gut aufgetrenntes Proteinmuster zu erkennen. Im direkten Vergleich
der verschiedenen Probenmengen wurde deutlich, dass das Spotmuster von 3000
mikrodissektierten Zellen das vielversprechendste Ergebnis hervorbrachte (Abb. 4.4. B). Die
Abundanz der Spots konnte im Vergleich zu 2000 mikrodissektierten Zellen (Abb. 4.4. A, B
Rote Pfeile) gesteigert werden. Das Gelbild der 5000 mikrodissektierten Zellen zeigte, dass
besonders gering abundante, nebeneinander liegende Spots nicht mehr optimal aufgetrennt
werden konnten und miteinander verschmolzen (Abb. 4.4., B C rote Kreise).
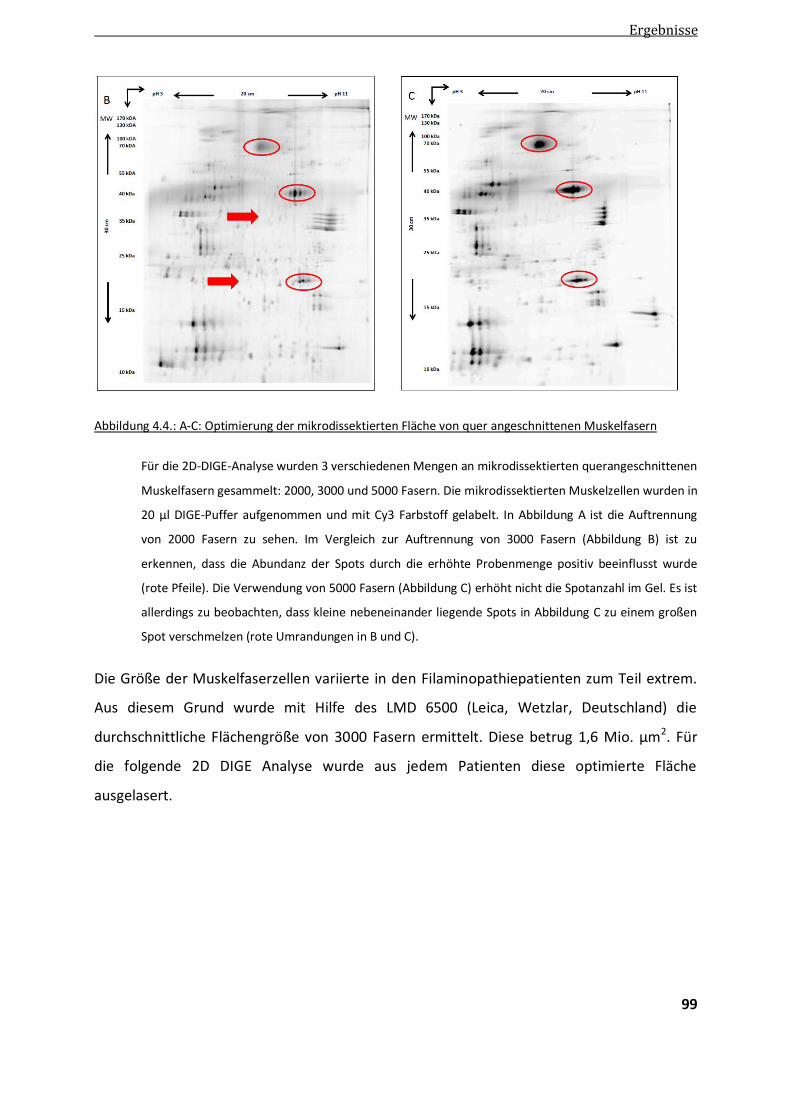
Ergebnisse
99
Abbildung 4.4.: A-C: Optimierung der mikrodissektierten Fläche von quer angeschnittenen Muskelfasern
Für die 2D-DIGE-Analyse wurden 3 verschiedenen Mengen an mikrodissektierten querangeschnittenen
Muskelfasern gesammelt: 2000, 3000 und 5000 Fasern. Die mikrodissektierten Muskelzellen wurden in
20 µl DIGE-Puffer aufgenommen und mit Cy3 Farbstoff gelabelt. In Abbildung A ist die Auftrennung
von 2000 Fasern zu sehen. Im Vergleich zur Auftrennung von 3000 Fasern (Abbildung B) ist zu
erkennen, dass die Abundanz der Spots durch die erhöhte Probenmenge positiv beeinflusst wurde
(rote Pfeile). Die Verwendung von 5000 Fasern (Abbildung C) erhöht nicht die Spotanzahl im Gel. Es ist
allerdings zu beobachten, dass kleine nebeneinander liegende Spots in Abbildung C zu einem großen
Spot verschmelzen (rote Umrandungen in B und C).
Die Größe der Muskelfaserzellen variierte in den Filaminopathiepatienten zum Teil extrem.
Aus diesem Grund wurde mit Hilfe des LMD 6500 (Leica, Wetzlar, Deutschland) die
durchschnittliche Flächengröße von 3000 Fasern ermittelt. Diese betrug 1,6 Mio. µm2. Für
die folgende 2D DIGE Analyse wurde aus jedem Patienten diese optimierte Fläche
ausgelasert.

Ergebnisse
100
4.3.2. Ergebnisse der differentiellen Proteomanalyse von Filaminopathiegewebe
mittels 2D DIGE
Die Grundlage für die differentielle Proteomstudie bildeten die unter Punkt 3.4.
beschriebenen Filaminopathiepatienten. Eine Patientin konnte in die Studie nicht mit
einbezogen werden, da das Gewebe durch die fortschreitende Erkrankung bereits zu sehr
geschädigt war. Ziel dieser Studie war es, durch die Identifizierung von differentiellen
Proteinen Rückschlüsse auf Frühstadien der Filaminopathie ziehen zu können. Hierfür
wurden aus 10 µm dicken Kryoschnitten von Filaminopathiepatienten mittels LMD
Muskelfaserzellen isoliert, die (noch) keine Aggregate enthielten. Als Kontrolle diente
gesunde Skelettmuskulatur. Für die Analyse der gering konzentrierten Gewebegruppen
wurde das 2D DIGE System in Kombination mit dem Saturation Labeling angewendet [Helling
et al. 2006]. Insgesamt wurden 10 2D-Gele angefertigt. Eine Patientenkontrolle bzw.
Negativkontrolle wurde zusammen mit dem internen Standard auf ein Gel aufgetragen. Der
interne Standard setzte sich aus allen mikrodissektierten Patientenproben und
Negativkontrollen zusammen. Im Wechsel wurden die Proben und der interne Standard mit
Cy3 bzw. Cy5 gelabelt. Der genannte Color-Switch führte zur Minimierung Farbstoff-
bedingter Differenzen. In Abb. 4.5. ist ein repräsentatives Gel der 2D DIGE Studie abgebildet.
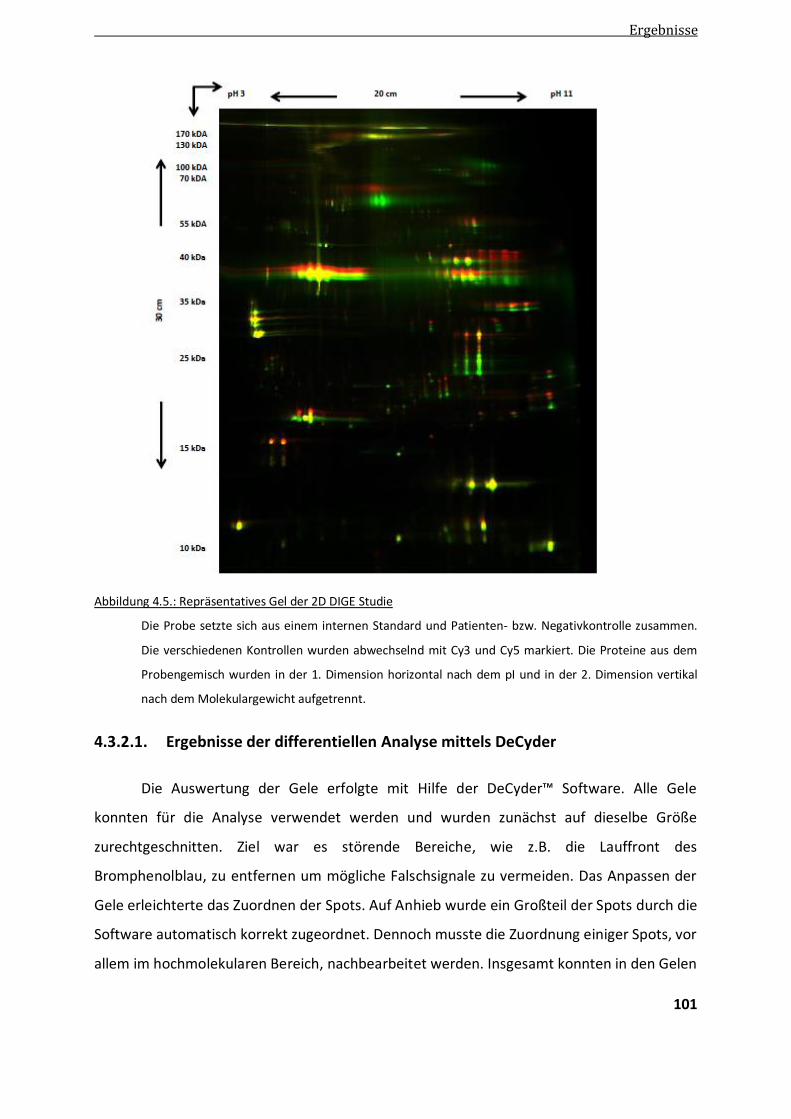
Ergebnisse
101
Abbildung 4.5.: Repräsentatives Gel der 2D DIGE Studie
Die Probe setzte sich aus einem internen Standard und Patienten- bzw. Negativkontrolle zusammen.
Die verschiedenen Kontrollen wurden abwechselnd mit Cy3 und Cy5 markiert. Die Proteine aus dem
Probengemisch wurden in der 1. Dimension horizontal nach dem pI und in der 2. Dimension vertikal
nach dem Molekulargewicht aufgetrennt.
4.3.2.1. Ergebnisse der differentiellen Analyse mittels DeCyder
Die Auswertung der Gele erfolgte mit Hilfe der DeCyder™ Software. Alle Gele
konnten für die Analyse verwendet werden und wurden zunächst auf dieselbe Größe
zurechtgeschnitten. Ziel war es störende Bereiche, wie z.B. die Lauffront des
Bromphenolblau, zu entfernen um mögliche Falschsignale zu vermeiden. Das Anpassen der
Gele erleichterte das Zuordnen der Spots. Auf Anhieb wurde ein Großteil der Spots durch die
Software automatisch korrekt zugeordnet. Dennoch musste die Zuordnung einiger Spots, vor
allem im hochmolekularen Bereich, nachbearbeitet werden. Insgesamt konnten in den Gelen
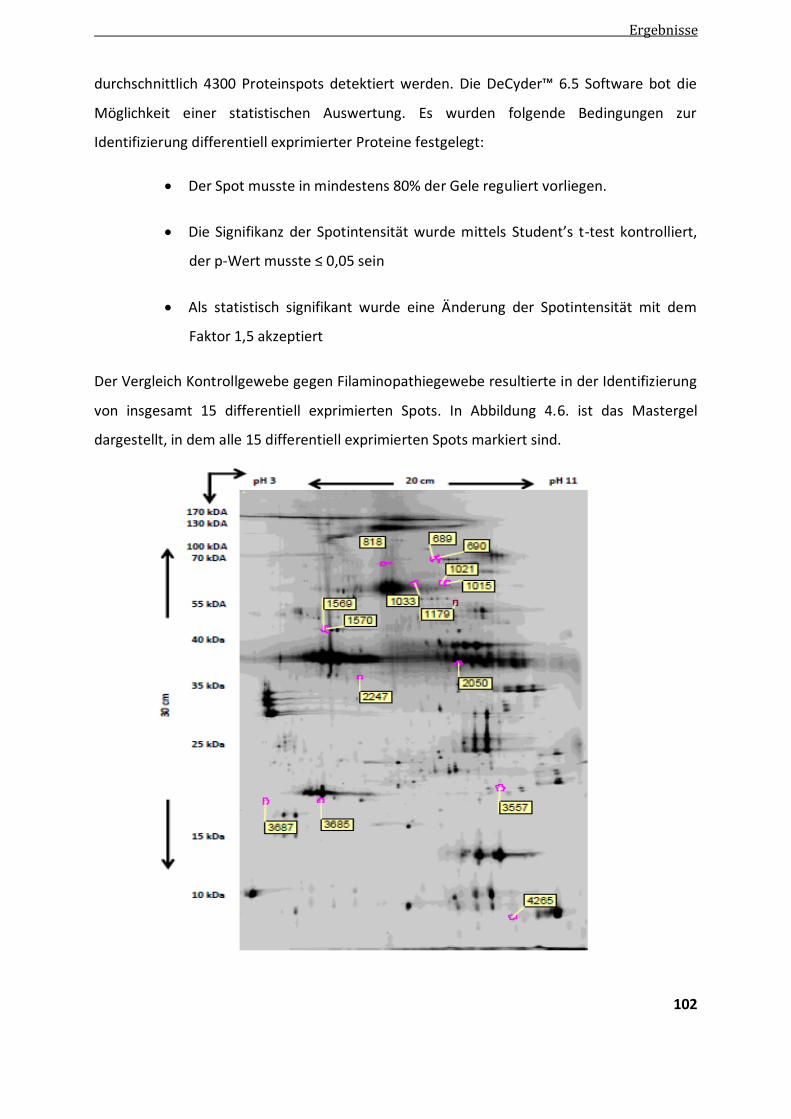
Ergebnisse
102
durchschnittlich 4300 Proteinspots detektiert werden. Die DeCyder™ 6.5 Software bot die
Möglichkeit einer statistischen Auswertung. Es wurden folgende Bedingungen zur
Identifizierung differentiell exprimierter Proteine festgelegt:
Der Spot musste in mindestens 80% der Gele reguliert vorliegen.
Die Signifikanz der Spotintensität wurde mittels Student’s t-test kontrolliert,
der p-Wert musste ≤ 0,05 sein
Als statistisch signifikant wurde eine Änderung der Spotintensität mit dem
Faktor 1,5 akzeptiert
Der Vergleich Kontrollgewebe gegen Filaminopathiegewebe resultierte in der Identifizierung
von insgesamt 15 differentiell exprimierten Spots. In Abbildung 4.6. ist das Mastergel
dargestellt, in dem alle 15 differentiell exprimierten Spots markiert sind.
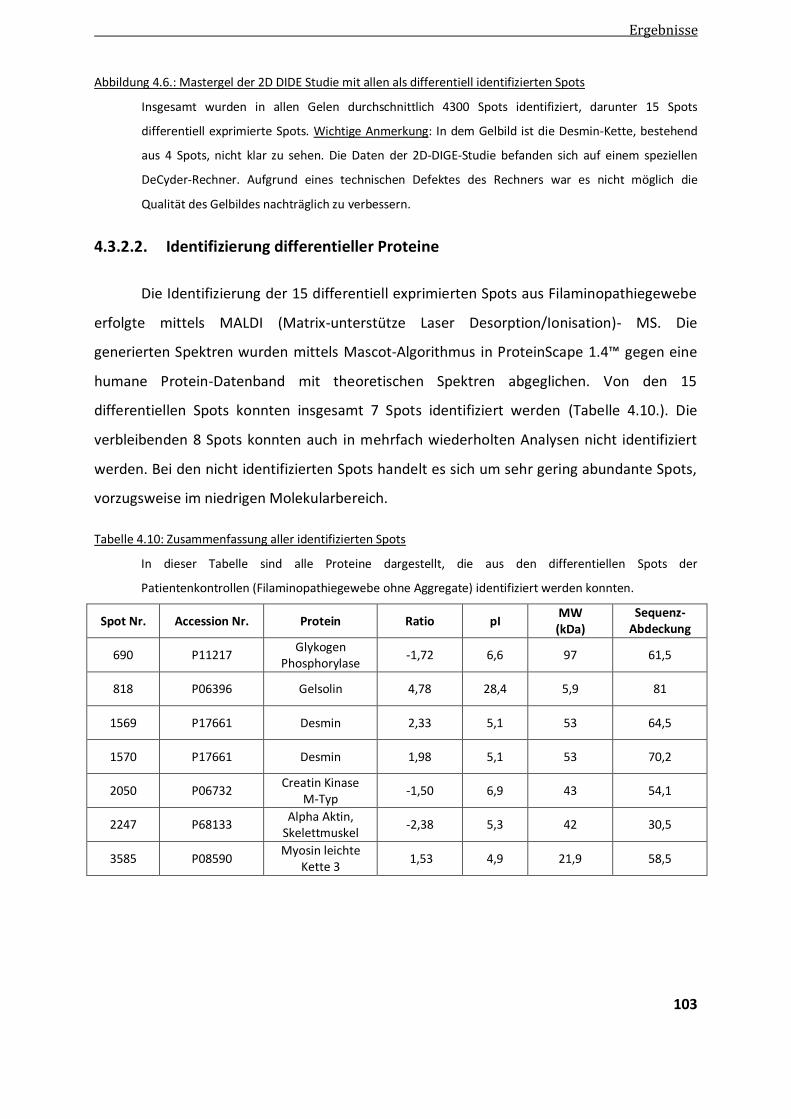
Ergebnisse
103
Abbildung 4.6.: Mastergel der 2D DIDE Studie mit allen als differentiell identifizierten Spots
Insgesamt wurden in allen Gelen durchschnittlich 4300 Spots identifiziert, darunter 15 Spots
differentiell exprimierte Spots. Wichtige Anmerkung: In dem Gelbild ist die Desmin-Kette, bestehend
aus 4 Spots, nicht klar zu sehen. Die Daten der 2D-DIGE-Studie befanden sich auf einem speziellen
DeCyder-Rechner. Aufgrund eines technischen Defektes des Rechners war es nicht möglich die
Qualität des Gelbildes nachträglich zu verbessern.
4.3.2.2. Identifizierung differentieller Proteine
Die Identifizierung der 15 differentiell exprimierten Spots aus Filaminopathiegewebe
erfolgte mittels MALDI (Matrix-unterstütze Laser Desorption/Ionisation)- MS. Die
generierten Spektren wurden mittels Mascot-Algorithmus in ProteinScape 1.4™ gegen eine
humane Protein-Datenband mit theoretischen Spektren abgeglichen. Von den 15
differentiellen Spots konnten insgesamt 7 Spots identifiziert werden (Tabelle 4.10.). Die
verbleibenden 8 Spots konnten auch in mehrfach wiederholten Analysen nicht identifiziert
werden. Bei den nicht identifizierten Spots handelt es sich um sehr gering abundante Spots,
vorzugsweise im niedrigen Molekularbereich.
Tabelle 4.10: Zusammenfassung aller identifizierten Spots
In dieser Tabelle sind alle Proteine dargestellt, die aus den differentiellen Spots der
Patientenkontrollen (Filaminopathiegewebe ohne Aggregate) identifiziert werden konnten.
Spot Nr. Accession Nr. Protein Ratio pI MW (kDa)
Sequenz- Abdeckung
(%) 690 P11217
Glykogen Phosphorylase
-1,72 6,6 97 61,5
818 P06396 Gelsolin 4,78 28,4 5,9 81
1569 P17661 Desmin 2,33 5,1 53 64,5
1570 P17661 Desmin 1,98 5,1 53 70,2
2050 P06732 Creatin Kinase
M-Typ -1,50 6,9 43 54,1
2247 P68133 Alpha Aktin,
Skelettmuskel -2,38 5,3 42 30,5
3585 P08590 Myosin leichte
Kette 3 1,53 4,9 21,9 58,5

Ergebnisse
104
4.3.2.3. Überprüfen der Spectral Index Daten aus Patientenkontrollen mittels 2D
DIGE
Das umliegende Gewebe ohne Aggregate aus Filaminopathiepatienten
(Patientenkontrolle) wurde über die Label-freie Proteomanalyse und parallel auch in einer
2D DIGE Studie analysiert. Von insgesamt 247 identifizierten Proteinen konnten in der Label-
freien Analyse mittels Spectral Index Calculation 10 abundante und 16 niedrig abundante
Proteine in den Patientenkontrollen ermittelt werden. In der 2D DIGE Studie konnten aus
insgesamt 15 differentiellen Spots 7 Proteine identifiziert werden. Nach genauer Durchsicht
aller mittels Spectral Index Calculation identifizierten Proteine konnten 2
Übereinstimmungen mit der 2D Studie beobachtet werden: Aktin und die Glykogen
Phosphorylase wurden in beiden Studien identifiziert. Für Aktin wie auch für die Glykogen
Phosphorylase, konnte in beiden Studien eine verringerte Exprimierung der Proteine
beobachten werden. Die leichte Kette des Myosins, die Creatin Kinase M-Typ, Desmin und
Gelsolin wurden nur in der 2D Studie als differentielle Proteine in den Patientenkontrollen
identifiziert. In der Label-freien Analyse konnte für diese Proteine keine Regulation
beobachtet werden. Die Ergebnisse der 2D DIGE Studie und der Label-freien Proteomstudie
mittels Spectral Counting überschnitten sich zum Teil. Nachfolgende Versuche müssen
genutzt werden, um die hier beschriebenen und weitere Proteine bzw. deren Regulation
eindeutig zu validieren. Hierfür würden sich der Western Blot oder die Immunhistochemie
mittels spezifischer Antikörper anbieten. Im Rahmen dieser Dissertation wurde bei den
Validierung der Fokus auf das Desmin gelegt (Punkt 4.4.3.), weil dem Intermediärfilament
eine entscheidende Rolle in der Pathogenese der Myofibrillären Myopathie zugeschrieben
wird [Caron and Chapon 1999, Janue et al. 2007] (siehe Punkt 5.2.2.4. und 5.2.2.5).
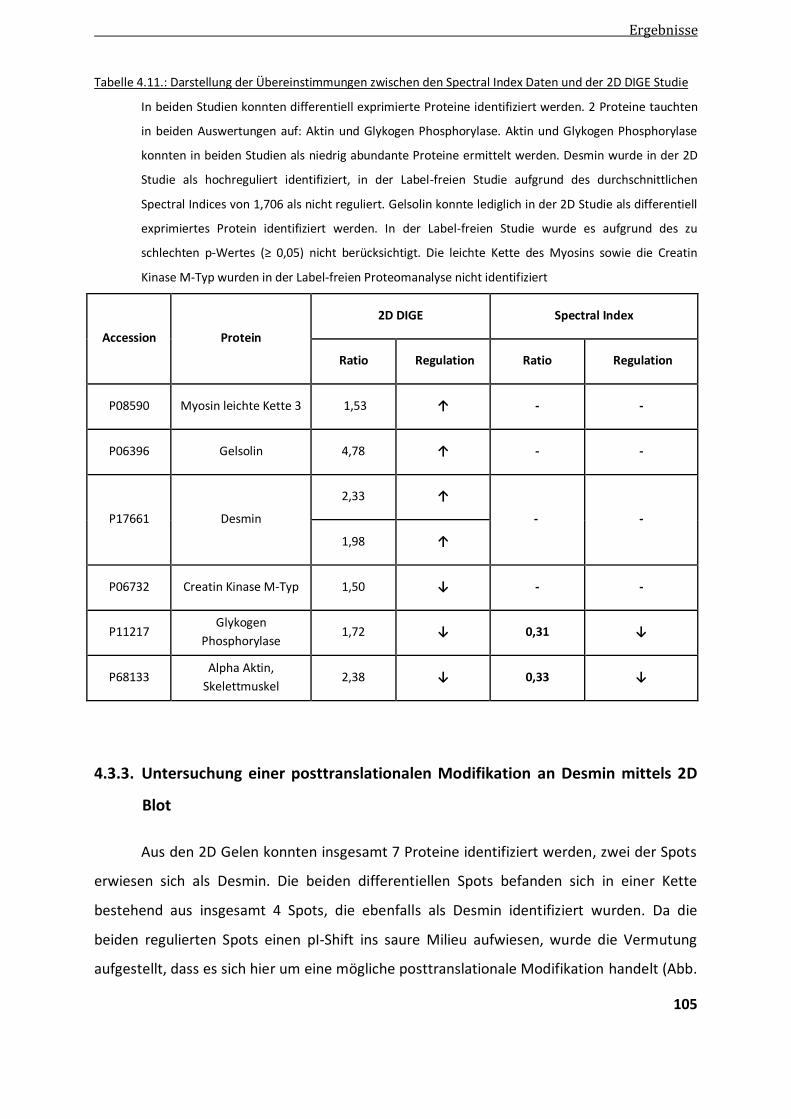
Ergebnisse
105
Tabelle 4.11.: Darstellung der Übereinstimmungen zwischen den Spectral Index Daten und der 2D DIGE Studie
In beiden Studien konnten differentiell exprimierte Proteine identifiziert werden. 2 Proteine tauchten
in beiden Auswertungen auf: Aktin und Glykogen Phosphorylase. Aktin und Glykogen Phosphorylase
konnten in beiden Studien als niedrig abundante Proteine ermittelt werden. Desmin wurde in der 2D
Studie als hochreguliert identifiziert, in der Label-freien Studie aufgrund des durchschnittlichen
Spectral Indices von 1,706 als nicht reguliert. Gelsolin konnte lediglich in der 2D Studie als differentiell
exprimiertes Protein identifiziert werden. In der Label-freien Studie wurde es aufgrund des zu
schlechten p-Wertes (≥ 0,05) nicht berücksichtigt. Die leichte Kette des Myosins sowie die Creatin
Kinase M-Typ wurden in der Label-freien Proteomanalyse nicht identifiziert
Accession Protein
2D DIGE Spectral Index
Ratio Regulation Ratio Regulation
P08590 Myosin leichte Kette 3 1,53 ↑ - -
P06396 Gelsolin 4,78 ↑ - -
P17661 Desmin
2,33 ↑
- -
1,98 ↑
P06732 Creatin Kinase M-Typ 1,50 ↓ - -
P11217 Glykogen
Phosphorylase 1,72 ↓ 0,31 ↓
P68133 Alpha Aktin,
Skelettmuskel 2,38 ↓ 0,33 ↓
4.3.3. Untersuchung einer posttranslationalen Modifikation an Desmin mittels 2D
Blot
Aus den 2D Gelen konnten insgesamt 7 Proteine identifiziert werden, zwei der Spots
erwiesen sich als Desmin. Die beiden differentiellen Spots befanden sich in einer Kette
bestehend aus insgesamt 4 Spots, die ebenfalls als Desmin identifiziert wurden. Da die
beiden regulierten Spots einen pI-Shift ins saure Milieu aufwiesen, wurde die Vermutung
aufgestellt, dass es sich hier um eine mögliche posttranslationale Modifikation handelt (Abb.

Ergebnisse
106
4.7.). Da es außer dem pI-Shift keine sichtbaren Veränderungen des Molekulargewichts gab,
schien lediglich eine Phosphorylierung als posttranslationale Modifikation in Frage zu
kommen. In der Literatur sind einige Studien bekannt, die sich mit posttranslationalen
Modifikationen, insbesondere der Phosphorylierung, am Desmin beschäftigt haben. Desmin
verfügt über zahlreiche Phosphorylierungsstellen, welche die Polymerisierung regulierten
[Costa et al. 2004]. Eine Erhöhung des phosphorylierten Anteils von Desmin führt zum Abbau
der Intermediärfilamente [Cohen et al. 2012]. In Myofibrillären Myopathien wurde die
zerstörende Wirkung von Phosphorylierungen 1999 erstmals beschrieben [Caron and
Chapon 1999]. Dieses Vorwissen rückte die Untersuchung der möglichen
Desminphosphorylierung in den Fokus der Validierungsarbeiten (siehe Punkt 5.2.2.5).
Abbildung 4.7.: pI-Shift der regulierten Desmin Spots in den sauren Bereich
Die DeCyder-Gelanalyse deckte die Regulierung von 2 Desmin Spots (Spot Nr. 1569 und 1570) auf. Die
beiden anliegenden Spots (Pfeile) wurden ebenfalls analysiert und als Desmin identifiziert. Die
Verschiebung der beiden differentiellen Spots in den sauren pI-Bereich ließ eine Phosphorylierung
vermuten. Wichtige Anmerkung: Die regulierten Spots sind in der Abbildung zu erahnen. Aufgrund des
technischen Defekts des DeCyder-Rechners war es nicht möglich, die Spots in besserer Qualität
darzustellen (siehe Abb. 4.6.).
Um die mögliche Phosphorylierung von Desmin zu untersuchen, wurde ein 2D Blot
durchgeführt. Es wurden je 50 µg Muskellysat aus Filaminopathiegewebe (Punkt 3.6.) und
gesundem Gewebe mit Fluorophoren markiert. Anschließend wurden die Proben mittels 2D
PAGE aufgetrennt und auf eine Nitrocellulosemembran transferiert. Jeweils direkt im
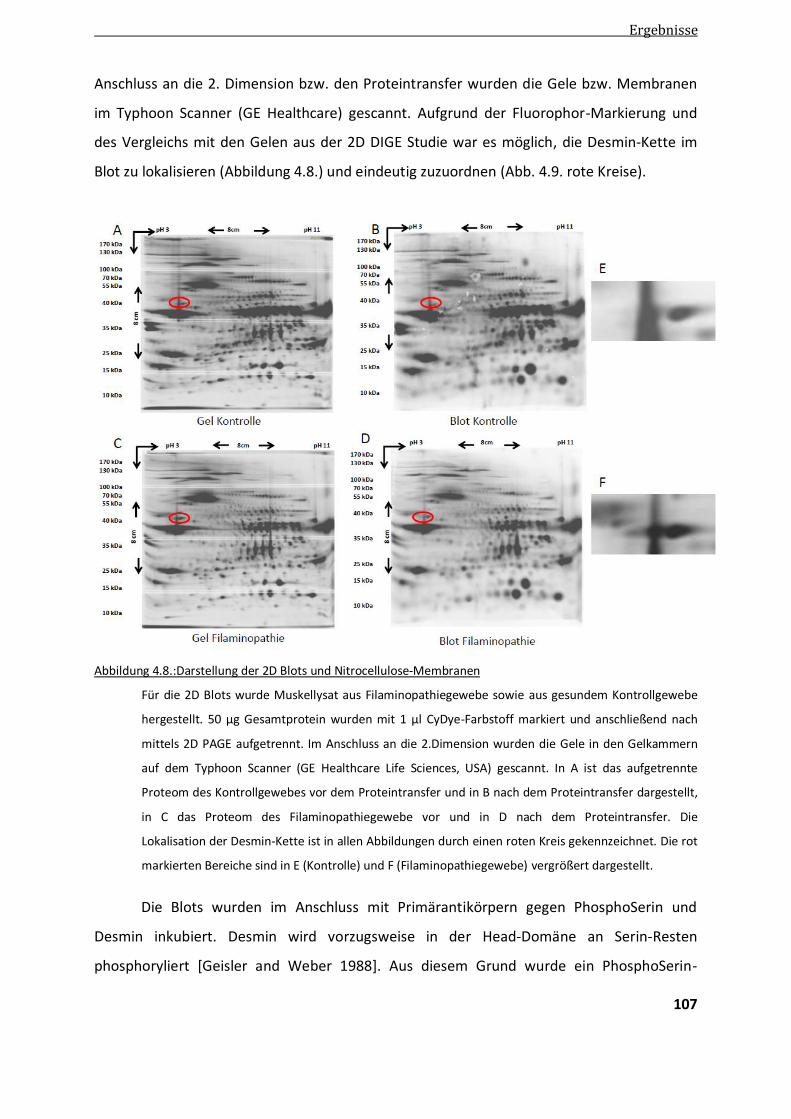
Ergebnisse
107
Anschluss an die 2. Dimension bzw. den Proteintransfer wurden die Gele bzw. Membranen
im Typhoon Scanner (GE Healthcare) gescannt. Aufgrund der Fluorophor-Markierung und
des Vergleichs mit den Gelen aus der 2D DIGE Studie war es möglich, die Desmin-Kette im
Blot zu lokalisieren (Abbildung 4.8.) und eindeutig zuzuordnen (Abb. 4.9. rote Kreise).
Abbildung 4.8.:Darstellung der 2D Blots und Nitrocellulose-Membranen
Für die 2D Blots wurde Muskellysat aus Filaminopathiegewebe sowie aus gesundem Kontrollgewebe
hergestellt. 50 µg Gesamtprotein wurden mit 1 µl CyDye-Farbstoff markiert und anschließend nach
mittels 2D PAGE aufgetrennt. Im Anschluss an die 2.Dimension wurden die Gele in den Gelkammern
auf dem Typhoon Scanner (GE Healthcare Life Sciences, USA) gescannt. In A ist das aufgetrennte
Proteom des Kontrollgewebes vor dem Proteintransfer und in B nach dem Proteintransfer dargestellt,
in C das Proteom des Filaminopathiegewebe vor und in D nach dem Proteintransfer. Die
Lokalisation der Desmin-Kette ist in allen Abbildungen durch einen roten Kreis gekennzeichnet. Die rot
markierten Bereiche sind in E (Kontrolle) und F (Filaminopathiegewebe) vergrößert dargestellt.
Die Blots wurden im Anschluss mit Primärantikörpern gegen PhosphoSerin und
Desmin inkubiert. Desmin wird vorzugsweise in der Head-Domäne an Serin-Resten
phosphoryliert [Geisler and Weber 1988]. Aus diesem Grund wurde ein PhosphoSerin-

Ergebnisse
108
Antikörper zur Validierung der vermuteten Phosphorylierung verwendet. Der Desmin-
Antikörper wurde verwendet, um zu zeigen, dass es sich bei den regulierten Spots
tatsächlich um Desmin handelt. Zur Visualisierung der Signale wurde als Sekundärantikörper
jeweils ein Maus IR800 (grün)- bzw. IR680 (rot)-Antikörper verwendet. Die Entwicklung der
Blots erfolgte im Odyssey Scanner (LI-COR Biosciences GmbH, Bad Homburg, Deutschland).
In Abbildung 4.9. ist das Ergebnis der Immunoblots zu sehen. In Filaminopathiegewebe wie
auch im Kontrollgewebe konnten die 4 im 2D Gel als Demin identifizierten Spots auch mittels
Desmin-Antikörper detektiert werden (Abb.4.9, B und D). Die Spots wurden in den
Abbildungen von 1-4 nummeriert. Die Spots 3 und 4 wurden in der 2D DIGE Studie als
reguliert identifiziert, die Spots 1 und 2 stellen die nicht regulierten Desmin-Spots dar. Mit
Hilfe des PhosphoSerin-Antikörpers konnten ebenfalls in beiden Gewebegruppen alle 4 als
Desmin identifizierten Spots detektiert werden. Dieses Ergebnis zeigt, dass Desmin stets
phosphoryliert vorliegt. Nachfolgende Studien müssen allerdings klären, ob der Anteil an
phosphoryliertem Desmin im Vergleich zum Kontrollgewebe in den Filaminopathiepatienten
erhöht ist. Zusätzlich zu den erwarteten Spots konnten unter der Verwendung des
PhosphoSerin-Antikörpers weitere Spots in den Blots detektiert werden. Zum einen wurde
im Filaminopathiegewebe und in der Kontrolle ein in der 2D Studie nicht erkannter 5.
Desminspot detektiert (Abb. 4.9., B, D), der nur im Filaminopathiegewebe phosphoryliert
vorlag (Abb. 4.9., A und C). Zum anderen tauchten insgesamt 4 weitere unbekannte Spots (a-
d) in den Blots auf. Die mit a und d gekennzeichneten Spots konnten in Filaminopathie- und
Kontrollgewebe detektiert werden, im kranken Gewebe lagen sie auch phosphoryliert vor.
Die Spots (b) und (c) wurden jedoch nur in Filaminopathiegewebe detektiert und könnten
spezifisch für Filaminopathiegewebe sein. Die Identifizierung und die Bedeutung der
zusätzlichen Desmin-Spots muss in weiteren Studien untersucht werden. Möglicherweise
können andere posttranslationale Modifikationen (höheres Molekulargewicht) oder
Abbauprodukte des Desmins (niedrigeres Molekulargewicht) identifiziert werden.

Ergebnisse
109
Abbildung 4.9: Ergebnis der Immunoblots zur Validierung der Desmin-Phosphorylierung
Die Blots wurden mit einem spezifischen PhosphoSerin-Antikörper inkubiert, als Sekundärantikörper
wurde ein Maus IR800 (grün) eingesetzt (Abb. A und C). Zur Validierung des in der 2D DIGE Studie
identifizierten Desmin wurde ein Desmin-Antikörper in Kombination mit einem Maus IR680 (rot) als
Sekundärantiköper verwendet (Abb. B und D). Die Visualisierung erfolgte mit Hilfe des Odyssey
Scanners. Die 4 bekannten Spots (1-4) aus der 2D DIGE Studie wurden in beiden Gewebegruppen als
Desmin detektiert (B, D). Zudem lagen sie phosphoryliert vor (A, C). Außerdem wurden zusätzliche
Spots von dem Desmin-Antikörper und dem PhosphoSerin-Antikörper detektiert. Der 5. Spot in der
Desminkette wurde in der 2D Studie nicht erkannt und lag auch nur im Filaminopathiegewebe
phosphoryliert vor (A, C). Die Spots a und d wurden in Filaminopathie-wie auch in Kontrollgewebe als
Desminspots detektiert (A, B, D). In krankem Gewebe liegen diese Spots auch phosphoryliert vor. Der
mit c gekennzeichnete Spot hingegen trat nur in Proben aus Filaminopathiegewebe auf (A, B).
Dies könnte auf eine Spezifität für das kranke Gewebe hindeuten. Weitere Studien müssen die
Bedeutung und die Identität der zusätzlichen Spots klären. Es wäre möglich, dass es sich um
Abbauprodukte des Desmin handelt und /oder weitere posttranslationale Modifikationen in den Spots
identifiziert werden können.

Ergebnisse
110
An den Nachweis der Phosphorylierung mittels spezifischer Antikörper schloss sich
die Lokalisation der phosphorylierten Aminosäuren mittels massenspektrometrischer
Analyse an. Hierfür wurden 50 µg Muskellysat aus Filaminopathiepatienten mit
Fluoreophoren markiert und anschließend mittels 2D PAGE aufgetrennt. Nach Beendigung
der 2. Dimension wurden die Gele im Typhoon Scanner (GE Healthcare) gescannt. Die
Fluorophor-Markierung und vergleichbare Gele aus der 2D Studie erleichterten die
Identifizierung der Desmin-Kette im Gel (siehe 4.3.2.1.). Die regulierten Spots wurden aus
dem Gel isoliert und anschließend wurden die Proteine über Nacht im Gel mit Trypsin
verdaut. Am nächsten Tag wurden die Peptide aus den Gelstücken verdaut und direkt auf
der LTQ (Thermo) gemessen. Um in der massenspektrometrischen Analyse bestmögliche
Voraussetzungen für die Identifizierung und Lokalisation der Serin-Phosphorylierung im
Desmin zu gewähren, wurden die Proben im Wechsel mittels CID und ETD gemessen (siehe
Punkt 5.2.2.7.). Trotz der Kombination der CID- und ETD-Fragmentierungstechnik konnten
keine Phosphorylierungen detektiert werden. Nach Durchsicht der UV-Spektren wurde
deutlich, dass die Proben zu gering konzentriert waren. Es konnten keine Spektren
aufgenommen werden.
4.4. Murine Myoblasten als MFM-Zellkulturmodell
In der hier vorliegenden Dissertation wurden erste Schritte zur Etablierung der
Proteomanalyse von Myofibrillären Myoblasten unternommen. Hierfür wurden C2C12-
Zellen verwendet. C2C12-Zellen sind Zellen, die aus isolierten Myoblasten eines
Mausskelettmuskels kultiviert und erstmals durch Yaffe et al. 1977 beschrieben wurden. Die
Zellen besitzen die Eigenschaft sehr schnell in Myotuben zu differenzieren und
muskelspezifische Proteine zu exprimieren. Das humane Material von
Filaminopathiepatienten ist limitiert. Um aber weitreichende Erkenntnisse über die
Pathomechnismen der Filaminopathie zu erlangen, wurde die Zellkultur als leicht
zugängliches System gewählt. Bereits 2008 konnten Vorgerd et al. (Kongressbeitrag) zeigen,
dass das Einbringen der humanen p.W2710X-Mutation in C2C12-Zellen zur Bildung von
Proteinaggregaten führt. Infolgedessen bestätigten andere Forschergruppen die C2C12-

Ergebnisse
111
Zelllinie als exzellentes Modellsystem für die Filaminopathie [Duff et al. 2011, Kley et al.
2012]. Ziel der hier durchgeführten Proteomanalysen in murinen Myoblasten war es, die
Zusammensetzung der Aggregate in den Zellen mit der Zusammensetzung der humanen
Aggregate zu vergleichen, um zu schauen, ob die Zellen hinsichtlich der Aggregation ein
geeignetes Modellsystem für die MFM darstellen.
4.4.1. Transfektion der p.W2710X Mutation in murine Myoblasten
Die kompletten Sequenzen des Filamin C-Wildtyps und der p.W2710X-Mutation
wurden in C2 pEGFP- und N3 pEGFP-Vektoren (Clonetech, USA) eingebracht. Beide Vektoren
eignen sich für die Analyse der subzellulären Lokalisation von Proteinen. Im N3-Vektor ist das
Zielprotein am C-Terminus mit dem Fluoreszenzprotein EGFP fusioniert, im C2-Vektor sind
Zielprotein und EGFP am N-Terminus fusioniert. Die Klonierungsarbeiten wurden von Dipl.
Biologin Yvonne Leber, AG Fürst, Institut für Zellbiologie, Rheinische Friedrich-Wilhelms-
Universität Bonn, durchgeführt. Kley et al. zeigten 2012, dass C2C12-Zellen nach
Transfektion der p.W2710X Mutante MFM-charakteristische intrazelluläre Proteinaggregate
ausbilden. Für die Transfektion wurden die C2C12-Zellen mit Plasmid-DNA der C2-und N3-
Konstrukte verwendet. Ein Vorversuch sollte zeigen, ob die Aggregatbildung ausschließlich
auf die eingebrachte Mutation zurückzuführen ist. Hierfür wurden die C2C12-Zellen zunächst
ausplattiert und nach 24h mit Plasmid-DNA der p.W2710X Mutation und des Wildtyps sowie
des Leervektors transfiziert. Die Zellen wurden nach 12h, 18h, 24h, 48h und 72h unter einem
Fluoreszenzmikroskop (Olympus, Hamburg, Deutschland) dokumentiert. Nach 12h und 18h
konnten weder transfizierte (grün leuchtende Zellen, weiße Pfeile) noch Zellen mit
Aggregaten beobachtet werden (Daten nicht gezeigt). Im Zeitraum zwischen 18h und 24h
setzte die Aggregatbildung in den Zellen ein. Die Aggregate befanden sich z.T. in den
Ausläufern der Zelle oder im Zellsoma. Die Transfektionseffizienz war beim Leervektor nach
24h mit ca. 75% am höchsten und blieb über die Zeitreihe konstant. Nach Transfektion mit
den Konstrukten, die den Wildtyp und die p.W2710X Mutation enthielten, konnten nur ca.
30% positiv transfizierte Zellen nach 24h beobachtet werden. Die Zahl der transfizierten
Zellen sank im Laufe der Zeitreihe bis auf ca. 20%. Wie in der letzten Spalte der Abbildungen
4.10. und 4.11. zu sehen ist, zeigten ausschließlich die Zellen Proteinaggregate, die mit der
Plasmid-DNA der Filamin C-Mutation transfiziert wurden. Die Anzahl variierte von einem
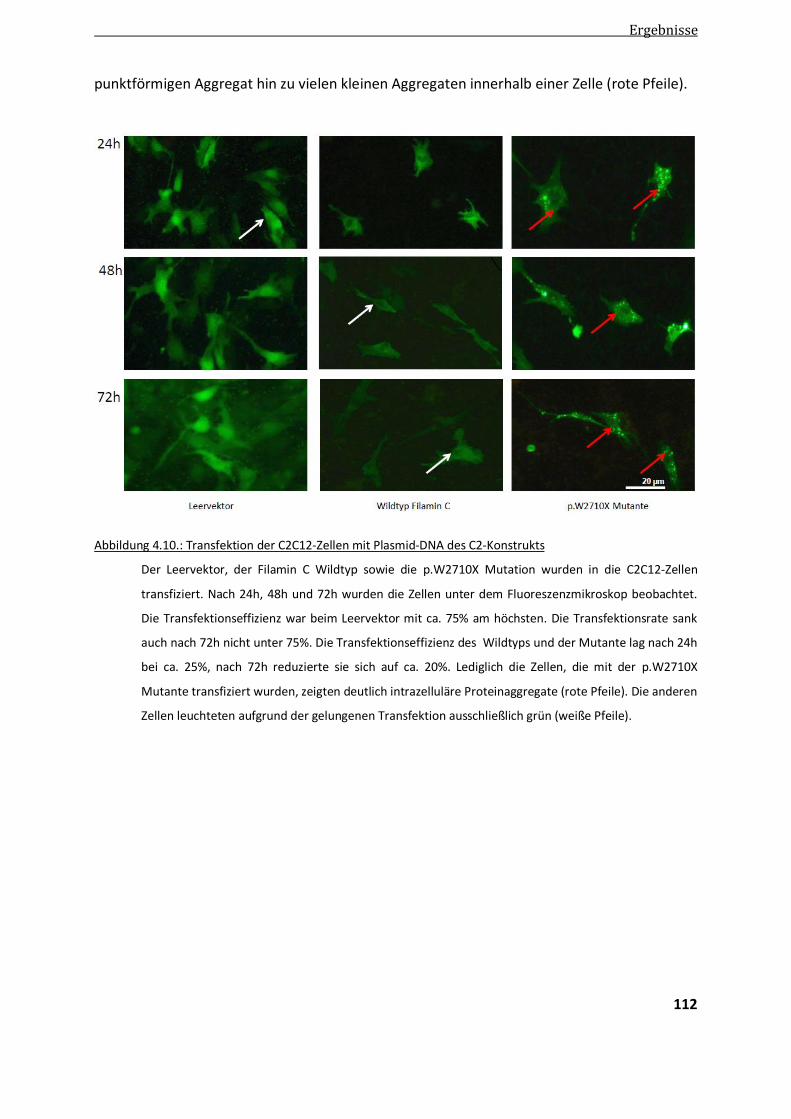
Ergebnisse
112
punktförmigen Aggregat hin zu vielen kleinen Aggregaten innerhalb einer Zelle (rote Pfeile).
Abbildung 4.10.: Transfektion der C2C12-Zellen mit Plasmid-DNA des C2-Konstrukts
Der Leervektor, der Filamin C Wildtyp sowie die p.W2710X Mutation wurden in die C2C12-Zellen
transfiziert. Nach 24h, 48h und 72h wurden die Zellen unter dem Fluoreszenzmikroskop beobachtet.
Die Transfektionseffizienz war beim Leervektor mit ca. 75% am höchsten. Die Transfektionsrate sank
auch nach 72h nicht unter 75%. Die Transfektionseffizienz des Wildtyps und der Mutante lag nach 24h
bei ca. 25%, nach 72h reduzierte sie sich auf ca. 20%. Lediglich die Zellen, die mit der p.W2710X
Mutante transfiziert wurden, zeigten deutlich intrazelluläre Proteinaggregate (rote Pfeile). Die anderen
Zellen leuchteten aufgrund der gelungenen Transfektion ausschließlich grün (weiße Pfeile).
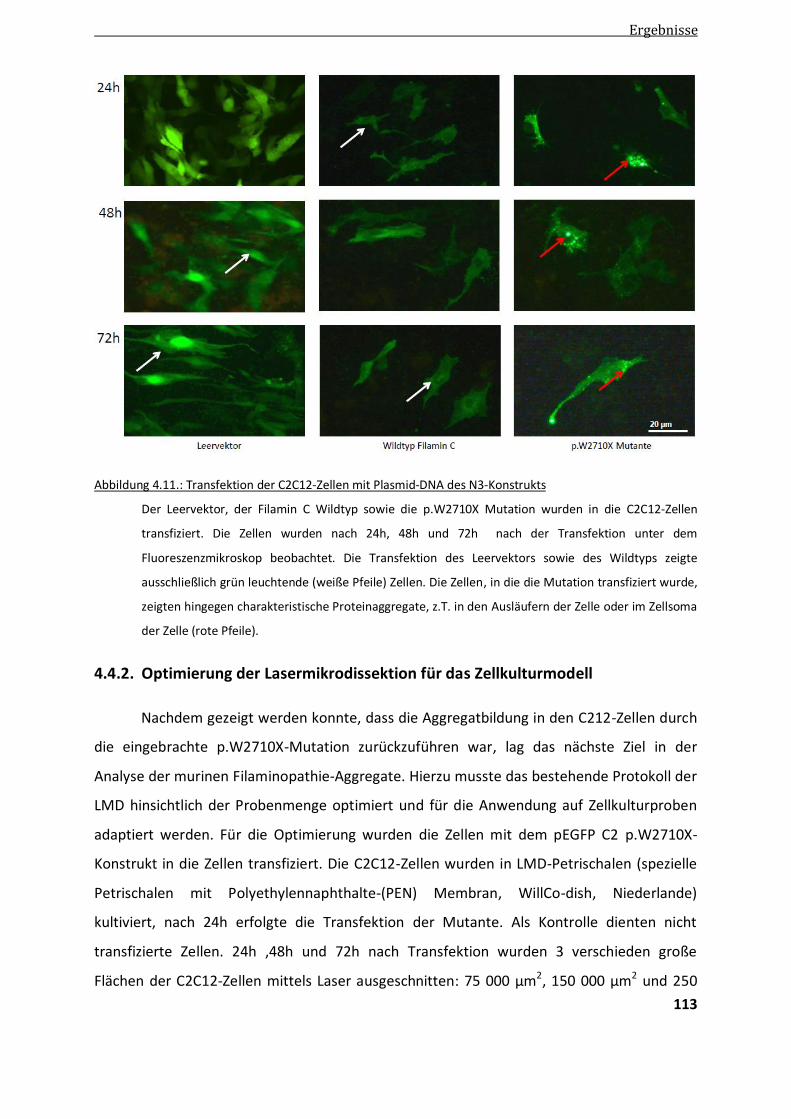
Ergebnisse
113
Abbildung 4.11.: Transfektion der C2C12-Zellen mit Plasmid-DNA des N3-Konstrukts
Der Leervektor, der Filamin C Wildtyp sowie die p.W2710X Mutation wurden in die C2C12-Zellen
transfiziert. Die Zellen wurden nach 24h, 48h und 72h nach der Transfektion unter dem
Fluoreszenzmikroskop beobachtet. Die Transfektion des Leervektors sowie des Wildtyps zeigte
ausschließlich grün leuchtende (weiße Pfeile) Zellen. Die Zellen, in die die Mutation transfiziert wurde,
zeigten hingegen charakteristische Proteinaggregate, z.T. in den Ausläufern der Zelle oder im Zellsoma
der Zelle (rote Pfeile).
4.4.2. Optimierung der Lasermikrodissektion für das Zellkulturmodell
Nachdem gezeigt werden konnte, dass die Aggregatbildung in den C212-Zellen durch
die eingebrachte p.W2710X-Mutation zurückzuführen war, lag das nächste Ziel in der
Analyse der murinen Filaminopathie-Aggregate. Hierzu musste das bestehende Protokoll der
LMD hinsichtlich der Probenmenge optimiert und für die Anwendung auf Zellkulturproben
adaptiert werden. Für die Optimierung wurden die Zellen mit dem pEGFP C2 p.W2710X-
Konstrukt in die Zellen transfiziert. Die C2C12-Zellen wurden in LMD-Petrischalen (spezielle
Petrischalen mit Polyethylennaphthalte-(PEN) Membran, WillCo-dish, Niederlande)
kultiviert, nach 24h erfolgte die Transfektion der Mutante. Als Kontrolle dienten nicht
transfizierte Zellen. 24h ,48h und 72h nach Transfektion wurden 3 verschieden große
Flächen der C2C12-Zellen mittels Laser ausgeschnitten: 75 000 µm2, 150 000 µm2 und 250
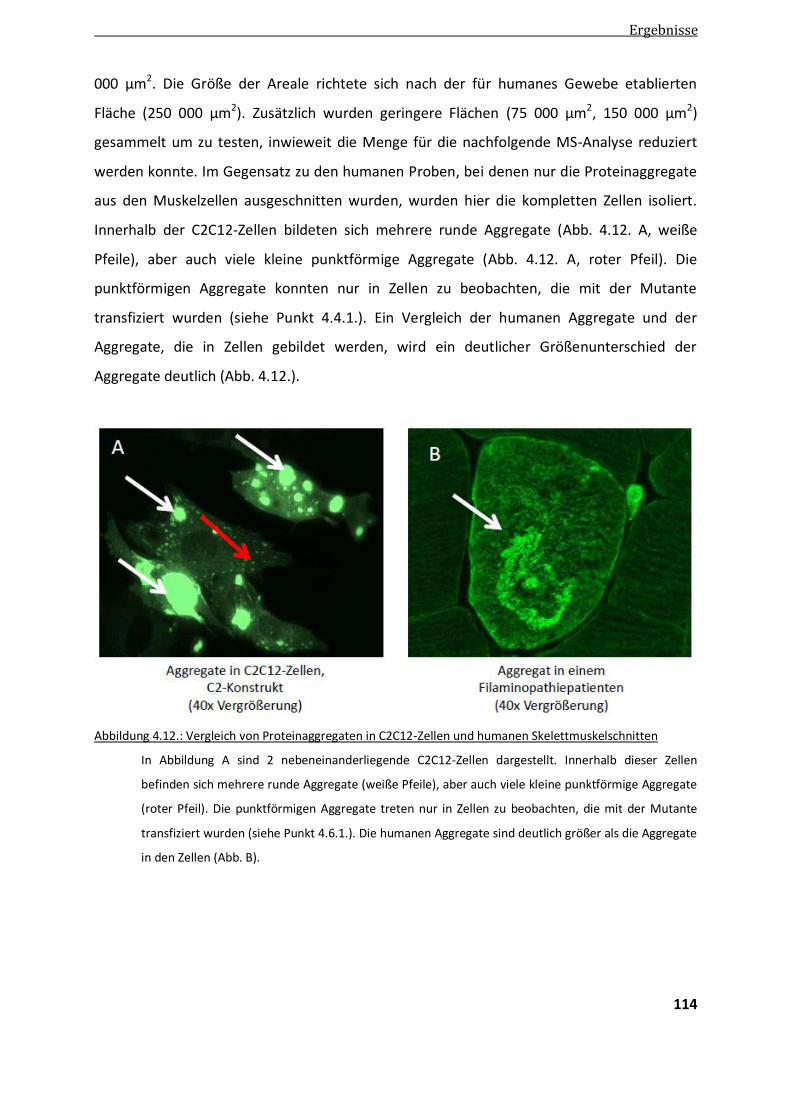
Ergebnisse
114
000 µm2. Die Größe der Areale richtete sich nach der für humanes Gewebe etablierten
Fläche (250 000 µm2). Zusätzlich wurden geringere Flächen (75 000 µm2, 150 000 µm2)
gesammelt um zu testen, inwieweit die Menge für die nachfolgende MS-Analyse reduziert
werden konnte. Im Gegensatz zu den humanen Proben, bei denen nur die Proteinaggregate
aus den Muskelzellen ausgeschnitten wurden, wurden hier die kompletten Zellen isoliert.
Innerhalb der C2C12-Zellen bildeten sich mehrere runde Aggregate (Abb. 4.12. A, weiße
Pfeile), aber auch viele kleine punktförmige Aggregate (Abb. 4.12. A, roter Pfeil). Die
punktförmigen Aggregate konnten nur in Zellen zu beobachten, die mit der Mutante
transfiziert wurden (siehe Punkt 4.4.1.). Ein Vergleich der humanen Aggregate und der
Aggregate, die in Zellen gebildet werden, wird ein deutlicher Größenunterschied der
Aggregate deutlich (Abb. 4.12.).
Abbildung 4.12.: Vergleich von Proteinaggregaten in C2C12-Zellen und humanen Skelettmuskelschnitten
In Abbildung A sind 2 nebeneinanderliegende C2C12-Zellen dargestellt. Innerhalb dieser Zellen
befinden sich mehrere runde Aggregate (weiße Pfeile), aber auch viele kleine punktförmige Aggregate
(roter Pfeil). Die punktförmigen Aggregate treten nur in Zellen zu beobachten, die mit der Mutante
transfiziert wurden (siehe Punkt 4.6.1.). Die humanen Aggregate sind deutlich größer als die Aggregate
in den Zellen (Abb. B).

Ergebnisse
115
Im Anschluss an die LMD wurden die Proben in 40 µl Ameisensäure aufgenommen. Nach
dem tryptischen Verdau der Proben wurden diese mittels nanoLC-MS/MS analysiert. Um die
optimale Fläche für die LMD von MFM-spezifischen Proteinaggregaten aus C2C12-Zellen
festzulegen, wurden die verschiedenen Zeitpunkte sowie die unterschiedlichen Flächen
miteinander vergleichen. Das Hauptaugenmerk wurde auf die Anzahl der identifizierten
Proteine gelegt. Die Zahl der identifizierten Proteine lag zwischen 227 und 813. Hier wurde
deutlich, dass die Zahl identifizierter Proteine nicht proportional zur Größe der ausgelaserten
Fläche anstieg (Tabelle 4.12.). In allen transfizierten Zellen konnten weniger Proteine
identifiziert werden als in den Kontrollen. Aufgrund der hier erzielten Ergebnisse wurde die
Fläche von 75 000 µm2 als optimal für die folgenden Aggregatanalysen in C2C12-Zellen
festgelegt. Generell konnten in allen Proben viele MFM- und Muskel-spezifische Proteine
identifiziert werden: u.a. Filamin C, Desmin, Myosin, Nestin, Tropomyosin, Aktin, Vimentin,
Lamin, diverse Hitzeschockproteine. Die meisten der identifizierten Proteine waren aus den
humanen Analysen bekannt. Dieses Ergebnis ließ darauf schließen, dass die beiden Systeme
prinzipiell miteinander vergleichbar sind. Weiterführende Analysen müssen allerdings noch
zeigen, ob und inwieweit eine quantitative Auswertung der Zellkulturdaten möglich ist und
ob sich ein ähnliches Akkumulierungsmuster zeigt wie in den humanen Muskelproben.
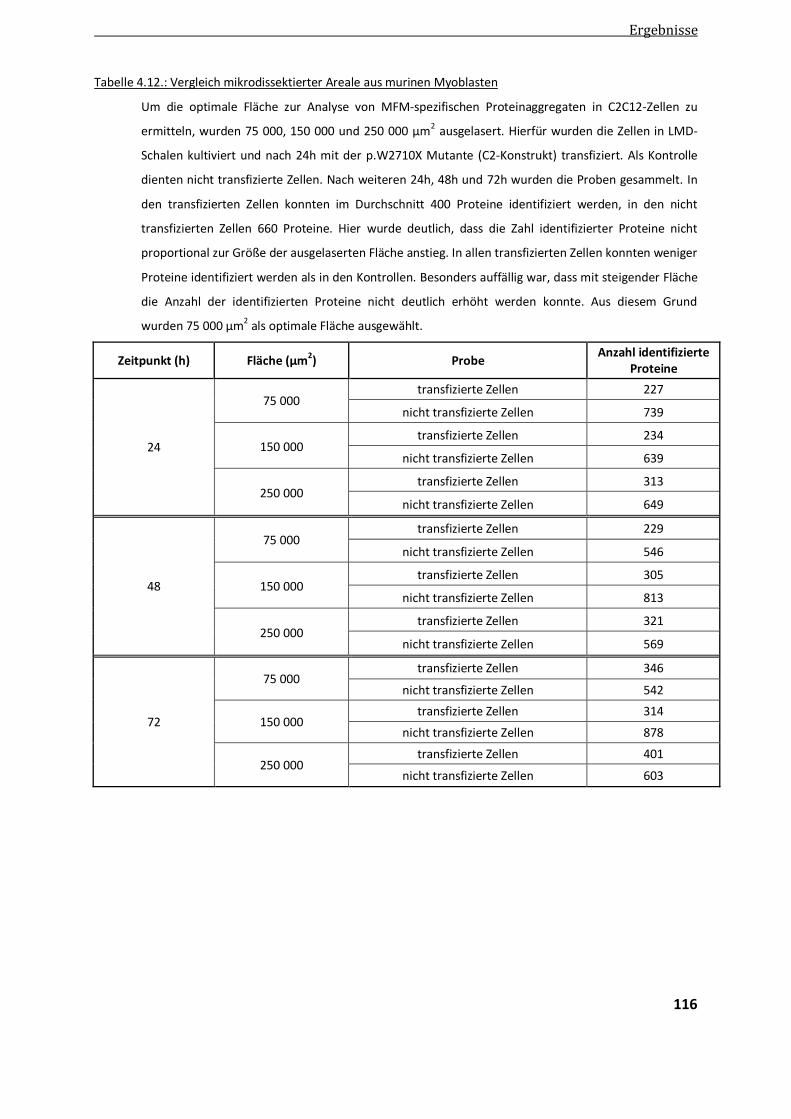
Ergebnisse
116
Tabelle 4.12.: Vergleich mikrodissektierter Areale aus murinen Myoblasten
Um die optimale Fläche zur Analyse von MFM-spezifischen Proteinaggregaten in C2C12-Zellen zu
ermitteln, wurden 75 000, 150 000 und 250 000 µm2 ausgelasert. Hierfür wurden die Zellen in LMD-
Schalen kultiviert und nach 24h mit der p.W2710X Mutante (C2-Konstrukt) transfiziert. Als Kontrolle
dienten nicht transfizierte Zellen. Nach weiteren 24h, 48h und 72h wurden die Proben gesammelt. In
den transfizierten Zellen konnten im Durchschnitt 400 Proteine identifiziert werden, in den nicht
transfizierten Zellen 660 Proteine. Hier wurde deutlich, dass die Zahl identifizierter Proteine nicht
proportional zur Größe der ausgelaserten Fläche anstieg. In allen transfizierten Zellen konnten weniger
Proteine identifiziert werden als in den Kontrollen. Besonders auffällig war, dass mit steigender Fläche
die Anzahl der identifizierten Proteine nicht deutlich erhöht werden konnte. Aus diesem Grund
wurden 75 000 µm2 als optimale Fläche ausgewählt.
Zeitpunkt (h) Fläche (µm2) Probe Anzahl identifizierte
Proteine
24
75 000 transfizierte Zellen 227
nicht transfizierte Zellen 739
150 000 transfizierte Zellen 234
nicht transfizierte Zellen 639
250 000 transfizierte Zellen 313
nicht transfizierte Zellen 649
48
75 000 transfizierte Zellen 229
nicht transfizierte Zellen 546
150 000 transfizierte Zellen 305
nicht transfizierte Zellen 813
250 000 transfizierte Zellen 321
nicht transfizierte Zellen 569
72
75 000 transfizierte Zellen 346
nicht transfizierte Zellen 542
150 000 transfizierte Zellen 314
nicht transfizierte Zellen 878
250 000 transfizierte Zellen 401
nicht transfizierte Zellen 603

Ergebnisse
117
4.4.3. Ergebnisse der Label-freien Aggregatanalyse im Zellkulturmodell
Im Anschluss an die Optimierungsarbeiten der LMD von Zellkulturmaterial wurde
eine Label-freie Aggregatanalyse mittels Spectral Index Calculation durchgeführt. Für die
Studie wurden die in Punkt 3.8. beschriebenen C2pEGFP- und N3pEGFP-Konstrukte transient
in die C2C12-Zellen eingebracht. Als Kontrolle wurden nicht transfizierte Zellen verwendet.
Um statistisch signifikante Ergebnisse zu erhalten, wurde pro Konstrukt 5mal dieselbe Fläche
(75 000 µm2) gesammelt. Für diese Analyse wurden komplette Zellen, die Aggregate
enthielten, mikrodissektiert. Nach 24h, 48h und 72h wurden die Proben gesammelt und in
40 µl Ameisensäure 30 min inkubiert. Die massenspektrometrische Analyse erfolgte analog
zum etablierten Protokoll zur Analyse von humanen Aggregaten. Ziel dieser Analyse sollte
sein, die Komposition der Proteinaggregate in Zellkultur zu charakterisieren und die
Zusammensetzung der Proteinaggregate im Zellmodell mit der Zusammensetzung der
humanen Aggregate abzugleichen. Die semi-quantitative Auswertung sollte zeigen, ob in
Zellkultur die gleichen Proteine hoch bzw. niedrig abundant vorliegen wie in humanen
Filaminopathie-Aggregaten. Die Ergebnisse der Label-freien Aggregatanalyse in C2C12-Zellen
wurden als Diagramm in Abbildung 4.13. zusammengefasst. Auf den ersten Blick wird
deutlich, dass die Zahl an identifizierten Proteinen in allen Gruppen extrem schwankt. In den
Kontrollen konnten die geringsten Schwankungen beobachtet werden. Trotz der extremen
Schwankungen konnten Gemeinsamkeiten in der Zusammensetzung der humanen
Aggregate und der Aggregate im Zellkulturmodell beobachtet werden. Es konnten diverse
Proteine identifiziert werden, die in beiden Studien als Bestandteile der Proteinaggregate
charakterisiert wurden. Dazu gehörten Filamin C und dessen Bindungspartner N-Rap, die
Intermediärfilamente Desmin und Vimentin, die Strukturproteine Aktin und Myosin sowie
diverse Hitzeschockproteine und andere Muskel-spezifische Proteine.
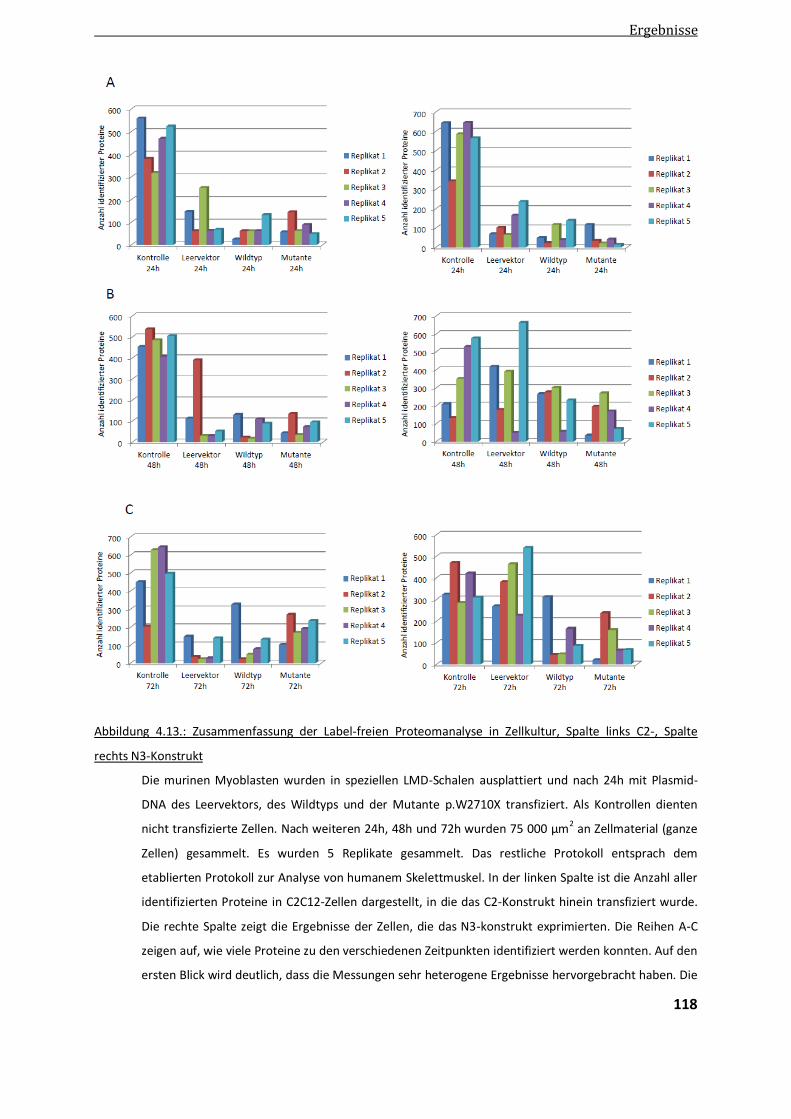
Ergebnisse
118
Abbildung 4.13.: Zusammenfassung der Label-freien Proteomanalyse in Zellkultur, Spalte links C2-, Spalte
rechts N3-Konstrukt
Die murinen Myoblasten wurden in speziellen LMD-Schalen ausplattiert und nach 24h mit Plasmid-
DNA des Leervektors, des Wildtyps und der Mutante p.W2710X transfiziert. Als Kontrollen dienten
nicht transfizierte Zellen. Nach weiteren 24h, 48h und 72h wurden 75 000 µm2 an Zellmaterial (ganze
Zellen) gesammelt. Es wurden 5 Replikate gesammelt. Das restliche Protokoll entsprach dem
etablierten Protokoll zur Analyse von humanem Skelettmuskel. In der linken Spalte ist die Anzahl aller
identifizierten Proteine in C2C12-Zellen dargestellt, in die das C2-Konstrukt hinein transfiziert wurde.
Die rechte Spalte zeigt die Ergebnisse der Zellen, die das N3-konstrukt exprimierten. Die Reihen A-C
zeigen auf, wie viele Proteine zu den verschiedenen Zeitpunkten identifiziert werden konnten. Auf den
ersten Blick wird deutlich, dass die Messungen sehr heterogene Ergebnisse hervorgebracht haben. Die
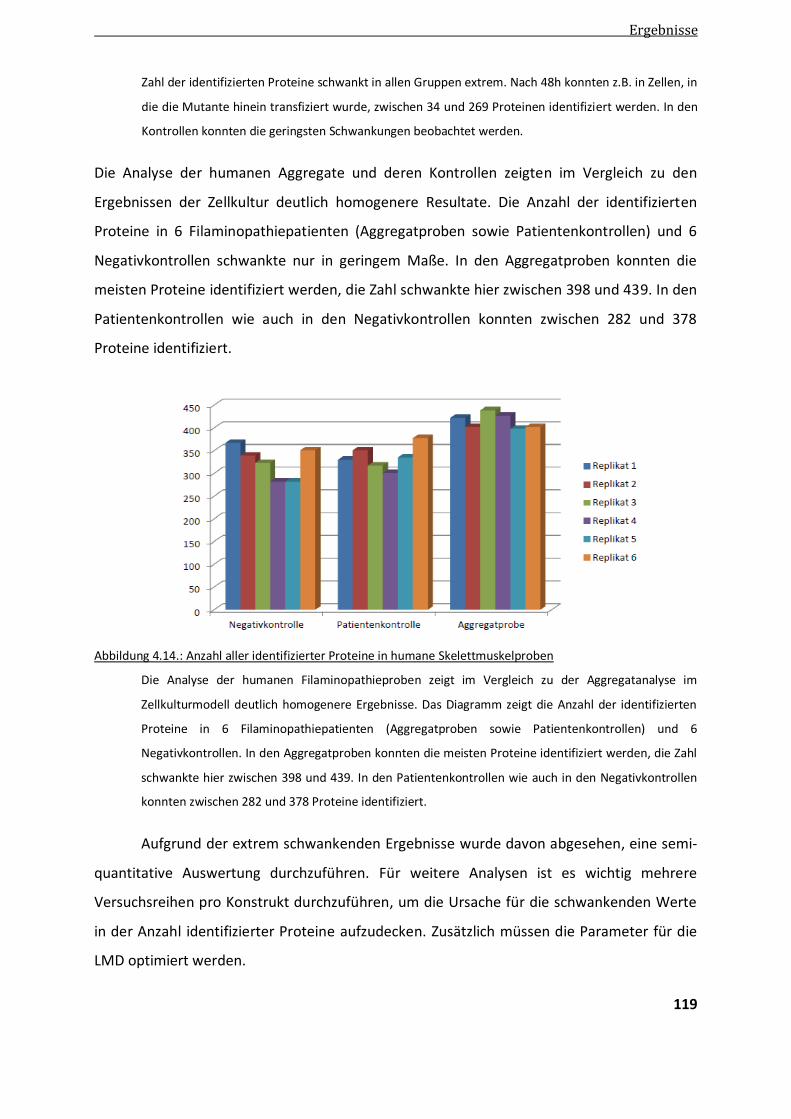
Ergebnisse
119
Zahl der identifizierten Proteine schwankt in allen Gruppen extrem. Nach 48h konnten z.B. in Zellen, in
die die Mutante hinein transfiziert wurde, zwischen 34 und 269 Proteinen identifiziert werden. In den
Kontrollen konnten die geringsten Schwankungen beobachtet werden.
Die Analyse der humanen Aggregate und deren Kontrollen zeigten im Vergleich zu den
Ergebnissen der Zellkultur deutlich homogenere Resultate. Die Anzahl der identifizierten
Proteine in 6 Filaminopathiepatienten (Aggregatproben sowie Patientenkontrollen) und 6
Negativkontrollen schwankte nur in geringem Maße. In den Aggregatproben konnten die
meisten Proteine identifiziert werden, die Zahl schwankte hier zwischen 398 und 439. In den
Patientenkontrollen wie auch in den Negativkontrollen konnten zwischen 282 und 378
Proteine identifiziert.
Abbildung 4.14.: Anzahl aller identifizierter Proteine in humane Skelettmuskelproben
Die Analyse der humanen Filaminopathieproben zeigt im Vergleich zu der Aggregatanalyse im
Zellkulturmodell deutlich homogenere Ergebnisse. Das Diagramm zeigt die Anzahl der identifizierten
Proteine in 6 Filaminopathiepatienten (Aggregatproben sowie Patientenkontrollen) und 6
Negativkontrollen. In den Aggregatproben konnten die meisten Proteine identifiziert werden, die Zahl
schwankte hier zwischen 398 und 439. In den Patientenkontrollen wie auch in den Negativkontrollen
konnten zwischen 282 und 378 Proteine identifiziert.
Aufgrund der extrem schwankenden Ergebnisse wurde davon abgesehen, eine semi-
quantitative Auswertung durchzuführen. Für weitere Analysen ist es wichtig mehrere
Versuchsreihen pro Konstrukt durchzuführen, um die Ursache für die schwankenden Werte
in der Anzahl identifizierter Proteine aufzudecken. Zusätzlich müssen die Parameter für die
LMD optimiert werden.

120
5. Diskussion
Die Myofibrillären Myopathien (MFM) bilden eine phänotypisch und genotypisch
heterogene Gruppe von erblichen Muskelerkrankungen. Charakterisiert werden sie durch
die fokale Zerstörung von Myofibrillen im Bereich der Z-Scheiben, die zum Strukturverlust
der Myofibrillen und Funktionseinschränkungen von Muskelfaserzellen führt. Ein weiteres
histologisches Merkmal der MFM ist die Bildung von intrazellulären Proteinaggregaten in
den Skelettmuskelzellen. [Selcen et al. 2004]. Verursacht werden MFM durch autosomal
dominante Mutationen in codierenden Genen von wichtigen Proteinen der Myofibrillen:
Filamin C, Desmin, Myotilin, Plectin, FHL-1, ZASP, alpha B-Crystallin und BAG 3. Entsprechend
der zugrunde liegenden Mutation setzt sich die Bezeichnung des jeweiligen Subtyps aus dem
Gennamen und der Endung –opathie, z.B. Filaminopathie, Desminopathie usw. zusammen.
In den meisten Fällen treten die ersten Symptome zu Beginn zwischen der 4. und 6.
Lebensdekade auf [Selcen et al. 2004, Kley et al. 2007]. Die Lebenserwartung der MFM-
Patienten ist durch Beteiligung der Atem- und Herzmuskulatur zum Teil drastisch reduziert
[Selcen and Engel 2003, Kley et al. 2007]. Die Filaminopathie ist ein Subtyp der
Myofibrillären Myopathien. Betroffen sind meist proximale Muskelgruppen. Der Verlauf der
Filaminopathie ist langsam progredient [Fürst et al. 2013]. Die Patienten haben zunächst
Schwierigkeiten, Treppen zu bewältigen, zum Ende hin können sie sich nur noch mit Hilfe
eines Rollstuhls fortbewegen.
In der hier vorliegenden Dissertation wurden Muskelproben von 6
Filaminopathiepatienten mit 3 verschiedenen Mutationen in der Stabregion des Filamin C
analysiert. Als Grundlage der Analysen dienten Gefrierschnitte von Muskelbiopsien der
Filaminopathiepatienten sowie von 6 gesunden Kontrollen. Ziel war es, die
Zusammensetzung der Proteinaggregate in den Muskelzellen zu entschlüsseln, sowie Marker
für das Frühstadium der Erkrankung zu identifizieren. Die Analyse der Aggregate und des
umliegenden Gewebes sollte einen neuen, detaillierten Einblick in die Pathogenese der

Diskussion
121
Filaminopathien geben. Es wurden 2 verschiedene auf LMD basierende Ansätze zur
Proteomanalyse der Filaminopathieproben und 3 unterschiedliche Auswertestrategien
etabliert. Mittels LMD wurden definierte Areale aus allen Proben isoliert. Für die Analyse der
Proteinaggregate wurde ein Massenspektrometrie-basierter Label-freier Ansatz gewählt. Im
Anschluss an die massenspektrometrische Messung wurden die generierten Daten mit einer
humanen Proteindatenbank abgeglichen. In einem ersten Schritt erfolgte eine qualitative
Auswertung der Daten. Daran schlossen sich eine manuelle sowie eine automatisierte
Quantifizierung an. Die automatisierte Quantifizierung erfolgte mittels Spectral Index
Calculation. So war es möglich diverse differentielle Proteine in den Aggregaten zu
identifizieren. In einer 2D DIGE Studie konnten insgesamt 6 differentielle Proteine
identifiziert werden, die als mögliche Frühmarker der Erkrankung in Frage kämen.
5.1. Lasermikrodissektion
In bereits veröffentlichten Studien konnten mittels immunhistologischen Färbungen
einzelne Bestandteile der Filaminopathie-spezifischen Proteinaggregate aufgedeckt werden.
Zu diesen Komponenten gehören u.a. Filamin C, Desmin, alpha B Crystallin, Myotilin, Xin
sowie diverse Hitzschockprotein [Kley et al. 2007, Kley et al. 2012]. Weitere Einblicke und
neue Rückschlüsse auf die Pathomechanismen der Erkrankung sollten proteomische
Analysen gewähren. Die Grundlage der hier durchgeführten Analysen bildeten humane
Muskelbiopsien von Filaminopathiepatienten. Das Gewebe der Patienten wurde im Rahmen
einer OP gewonnen. Die Anzahl und Größe der Proteinaggregate in den einzelnen Patienten
war äußerst limitiert. Um dennoch die Zusammensetzung der Proteinaggregate analysieren
zu können, wurde mit der der LMD eine Methode gewählt, die es ermöglicht, definierte
Zellstrukturen exakt und schonend aus dem umliegenden Gewebe zu isolieren. In der hier
vorliegenden Dissertation wurde das LMD 6500 von Leica verwendet. Unter Verwendung
des Systems von Leica wurden die Proteinaggregate präzise von einem Laser umfahren und
durch Schwerkraft kontakt- und kontaminationsfrei in einem Einmal-Reaktionsgefäß
gesammelt. Das Gewebe blieb dabei intakt. Bei älteren Systemen, wie der Laser Capture
Mikrodissektion (LCM), wird das Gewebe nicht herausgeschnitten sondern in einer Art
Raspelbewegung abgekratzt und mit einer Membran verschmolzen. Problematisch ist
hierbei die mögliche thermische Denaturierung der Proteine. Im Gegensatz zu älteren

Diskussion
122
Geräten bietet das LMD 6500 die Möglichkeit, die LMD in Kombination mit
Immunfluoreszenz einzusetzen. Während der Vorversuche stellte sich heraus, dass die
Filaminopathie-spezifischen Aggregate durch eine Immunfluoreszenzfärbung sehr effektiv im
Gewebe identifiziert werden konnten. Aus diesem Grund war die Fluoreszenzmikroskopie
essentiell für die Analysen. Ein zusätzlicher Vorteil des gewählten LMDs lag in der einfachen
und zeitsparenden Handhabung. Die LMD wurde stets bei Raumtemperatur durchgeführt
und um die Proteine vor vorzeitiger Denaturierung zu bewahren, war ein zügiges Arbeiten
von großem Vorteil.
5.2. Etablierung der differentiellen Proteomanalyse in Filaminopathien
Im Rahmen dieser Arbeit war es notwendig zwei Methoden zur differentiellen
Proteomanalyse von Proben aus Filaminopathiepatienten zu etablieren. Ziel der
differentiellen Proteomanalyse war es, spezifische Proteinkomponenten des
Filaminopathiegewebes und mögliche Frühstadien der Erkrankung zu charakterisieren. Für
die Charakterisierung von möglichen Frühstadien wurde das gut etablierte 2D DIGE System
gewählt. Diese Methode eignete sich allerdings aus zweierlei Gründen nicht für die
Identifizierung von spezifischen Proteinkomponenten des erkrankten Gewebes. Zum einen
erwiesen sich die Proteinaggregate als nur bedingt löslich in für die 2D-kompatiblen Urea
haltigen Puffern. Die äußerst geringe Größe und die limitierte Anzahl der Proteinaggregate in
den Patienten führten dazu, dass die Gesamtproteinkonzentration der Proben als extrem
niedrig einzustufen war. Obwohl es mit etablierten Methoden nicht möglich war, die
Proteinmenge in den Aggregaten zu bestimmen, kann davon ausgegangen werden, dass die
Proteinmenge deutlich unter 3 µg liegt, das ist die Nachweisgrenze des Saturation Labelings
[Helling et al. 2006]. Diese Umstände führten dazu, dass ein Label-freier Ansatz entwickelt
wurde, um die Proteinaggregate dennoch qualitativ und quantitativ analysieren zu können.

Diskussion
123
5.2.1. Label-freie Proteomanalyse
Die Analyse der Proteinzusammensetzung der Proteinaggregate erfolgte mittels einer
Label-freien Proteomanalyse. Die Filaminopathie-spezifischen Aggregate wurden mittels
LMD isoliert und massenspektrometrisch analysiert. Für die Auswertung der Daten wurden
eine qualitative und eine 2-stufige quantitative Strategie angewendet. Die Charakterisierung
neuer Proteinkomponenten der Aggregate und des umliegenden Gewebes sollte
Rückschlüsse auf die Pathogenese der Filaminopathie ermöglichen.
5.2.1.1. Solubilisierung der Aggregate
Für die Solubilisierung der Proteinaggregate wurden drei verschiedene Lösungsmittel
ausgetestet: 6 M Guanidin HCl (GuHCl), 8M Urea und 98 % Ameisensäure. Die chaotrophen
Substanzen 6 M GuHCl und 8 M Urea wurden verwendet, da sie bereits erfolgreich zum
Solubilisieren von Einschlusskörpern in Escherichia Coli (E.coli) verwendet wurden [Patra et
al. 2000, Tsumoto et al. 2003]. Es wurde vermutet, dass die hohe Konzentration der
chaotrophen Substanzen zu intra- und intermolekularen Interaktionen führt, infolgedessen
die Verklumpungen gelöst werden können. Die Verwendung hoch konzentrierter
Ameisensäure zum Lösen von Proteinaggregaten wurde ebenfalls mehrfach erfolgreich
durchgeführt [Klunk et al. 1994, Hazeki et al. 2000]. In der hier vorliegenden Arbeit wurden
250 Aggregate mittels LMD aus dem umliegenden Gewebe isoliert. Die
massenspektrometrische Analyse ergab, dass die Verwendung von Guanidin HCl sich nicht
zum Lösen der MFM-spezifischen Aggregate eignete. Es konnten keine Proteine identifiziert
werden. Auch die Verwendung von 8M Urea erwies sich als wenig erfolgreich. Es konnten
zwar 16 muskelspezifische Proteine identifiziert werden, doch die Zahl identifizierter
Proteine lag deutlich unter dem Ergebnis, welches mit Ameisensäure erzielt wurde.
Insgesamt konnten 119 Proteine nach Solubilisierung in hoch konzentrierter Ameisensäure
identifiziert werden. In der Literatur wurde eine mögliche Modifizierung der Proteine nach
Behandlung mit Ameisensäure in Form eines Esters an einem Serinrest beschrieben [Klunk et
al. 1994]. Diese mögliche Modifizierung wurde bei der Datenbanksuche berücksichtigt. Es
stellte sich heraus, dass keinerlei Einfluss auf die identifizierten Proteine beobachtet werden
konnte. Die Solubilisierung mit 98 % Ameisensäure erwies sich daher für die Analysen dieser
Arbeit als am erfolgversprechendsten. Dieses Protokoll für alle weiteren Analysen eingesetzt.

Diskussion
124
5.2.1.2. Optimierung der Probenmenge
Im vorangegangenen Punkt wurde beschrieben, dass sich die Aggregate am
erfolgreichsten in Ameisensäure lösen ließen. In einem nächsten Schritt sollte die optimale
Probenmenge für die massenspektrometrischen Analysen festgelegt werden. Ziel war es,
eine möglichst geringe Menge einzusetzen. In den ersten Analysen wurden 250 Aggregate
aus dem erkrankten Gewebe von Filaminopathiepatienten isoliert. Da die Zahl der Aggregate
in den einzelnen Patienten variierte und es nicht möglich war aus jedem Patienten 250
Aggregate zu isolieren, wurde versucht diese Menge zu reduzieren. Hierfür wurden 50 und
100 Aggregate mittels LMD gewonnen und anschließend massenspektrometrisch analysiert.
In 50 Aggregaten konnten lediglich 8 Proteine identifiziert werden, wobei keines der
Proteine in einen muskelspezifischen oder MFM-relevanten Kontext gebracht werden
konnte. Im Vergleich von 100 und 250 analysierten Aggregaten viel auf, dass trotz
verringerter Probenmenge die Zahl an identifizierten Proteinen in 100 Aggregaten gesteigert
werden konnte. Für eine weitere Optimierung wurden 250 und 100 Aggregate in
unterschiedlichen Volumina (20 und 40 µl) Ameisensäure aufgenommen und im direkten
Vergleich analysiert. Nach Solubilisierung von 100 Aggregaten in 20 µl Ameisensäure
konnten erneut mehr Proteine als in 250 Aggregaten identifiziert werden (siehe Tabelle 4.4.).
Die Durchsicht der UV-Spektren konnte ausschließen, dass Probleme während der
massenspektrometrischen Analyse verantwortlich für die unterschiedlichen Mengen an
identifizierten Proteinen waren. Der Grund für die extremen Schwankungen in der Zahl der
identifizierten Proteine, könnte in der Beschaffenheit der Aggregate zu finden sein. Es wird
davon ausgegangen, dass eine sehr hohe Proteinkonzentration in den Aggregaten vorliegt.
Zusätzlich könnten die Proteine sehr dicht aneinander oder ineinander gepackt sein. Umso
höher die mikrodissektierte Menge ist, umso höher ist auch die Dichte an akkumulierten
Proteinen. Das Volumen von 20 µl Ameisensäure könnte somit zu gering sein um die
akkumulierten Proteine ausreichend voneinander zu lösen. So würde nur ein minimaler
Anteil des Gesamtproteins in die massenspektrometrische Analyse mit eingehen. Unter der
Verwendung von 40 µl Ameisensäure wurden deutlich mehr Proteine identifiziert, was
suggeriert, dass mehr Proteine aus der Verklumpung gelöst wurden und für die
massenspektrometrischen Analyse zu Verfügung standen. Im direkten Vergleich zwischen
250 und 100 Aggregaten gelöst in 40 µl Ameisensäure unterschied sich die Anzahl an

Diskussion
125
identifizierten Proteinen nur gering. Aufgrund der Limitierung des Gewebes und der
Aggregatanzahl pro Schnitt erschienen 100 Aggregate, gelöst in 40 µl Ameisensäure, als
optimale Probenmenge. Die Größe der Aggregate variierte in den Patienten zum Teil extrem.
Um dennoch gewährleisten zu können, dass vergleichbare Mengen Gewebe isoliert werden,
wurden mit Hilfe des LMD 6500 die durchschnittliche Fläche von 100 Aggregaten ermittelt.
Diese belief sich auf 250 000 µm2 und wurde für alle weiteren Aggregatanalysen verwendet.
Eine zusätzliche Verbesserung der Zahl identifizierter Proteine konnte durch diverse
Aufrüstungen des Orbitrap-Massenspektrometers erreicht werden, welche die Sensitivität
des Gerätes deutlich verbesserten. Im Verlauf der Analysen konnte die Zahl der Proteine auf
durchschnittlich 450 erhöht werden.
5.2.1.3. Auswertestrategien
Im Rahmen dieser Dissertation wurden zwei verschiedene Ansätze zur Auswertung
der massenspektrometrischen Daten getestet. Zu Beginn des Projektes war die
Zusammensetzung der Filaminopathie-spezifischen Proteinaggregate sowie des umliegenden
Gewebes nahezu unklar. In Immunfluoreszenzstudien konnten zwar einige Bestandteile der
Aggregate aufgedeckt werden [Kley et al. 2007 und 2012], eine detaillierte Übersicht des
Proteoms von Filaminopathiegewebe lag jedoch nicht vor. Um eine detaillierte
Charakterisierung des Gewebes zu erreichen, wurden Aggregatproben und nicht betroffene
Muskelfasern (Patientenkontrolle) von 6 Filaminopathiepatienten mit 3 verschiedenen
Mutationen in der Stabregion des Filamin C analysiert. Als Kontrolle dienten Muskelfasern
aus gesunden Patienten (Negativkontrolle). Zwei Vergleiche wurden angestrebt: 1.
Aggregatproben gegen Patientenkontrollen, 2. Patientenkontrollen gegen Negativkontrollen.
Der Vergleich von Aggregatproben und Patientenkontrollen sollte helfen, Proteine zu
identifizierten, die ausschließlich in den Aggregaten vorkommen und somit spezifische
Bestandteile der Aggregate darstellen. Der 2. Vergleich (Patientenkontrollen gegen
Negativkontrollen) wurde gewählt, um Unterschiede in der Proteinzusammensetzung der
Patientenkontrolle zu den Negativkontrollen aufzudecken. Die Identifizierung spezifischer
Proteine in den Patientenkontrollen sollte neue Erkenntnisse über Frühstadien ermöglichen.
Mit den hier gewählten Vergleichen war es möglich intraindividuelle Unterschiede in den
Proben zu ermitteln und so spezifische Charakteristika im Gewebe der Patienten zu

Diskussion
126
identifizieren. In einem Vergleich von Aggregatproben aus Patienten und Negativkontrollen
aus gesundem Gewebe ist es nicht möglich intraindividuelle Unterschiede zu detektieren.
Aus diesem Grund wurde im Vorfeld der Analysen diese Art des Vergleichs ausgeschlossen.
Es wurde davon ausgegangen, dass generelle Unterschiede im Proteinmuster von
Filaminopathie-spezifischen Aggregaten und Normalgewebe bestehen.
Etablierung einer qualitativen und manuell durchgeführten
quantitativen Auswertestrategie
Für die qualitative Auswertung wurden aus den generierten MS/MS-Spektren mittels
Datenbankabgleich Proteinlisten erstellt. Es konnten durchschnittlich 415 Proteine in den
Aggregatproben der Filaminopathiepatienten identifiziert werden, von denen einige durch
bereits veröffentlichte Immunfluoreszenzstudien als feste Bestandteile der Filaminopathie-
spezifischen Aggregate bekannt waren. Der Großteil der generierten Proteinliste setzte sich
jedoch aus unbekannten Komponenten zusammen. Die Identifizierung von möglichen
Filaminopathie-spezifischen Proteinen in den Aggregaten, die nicht im umliegenden Gewebe
gefunden werden können, war von besonders großem Interesse. Um dies zu erreichen
musste zunächst eine manuell durchgeführte quantitative Auswertestrategie hinzugezogen
werden, da zu Beginn der Analyse auf keine etablierte automatisierte Auswertestrategie
zurückgegriffen werden konnte. Die Proteinlisten aus der qualitativen Auswertung wurden
zunächst nach stringenten Maßstäben sortiert (beschrieben unter Punkt 3.17.1.). Alle
Proteine, die in den Aggregatproben und in den Patientenkontrollen auftraten, wurden in
dieser Auswertung vernachlässigt. Insgesamt konnten durch diese Auswertestrategie 111
Proteine ausschließlich in den Aggregaten gefunden werden (siehe Tabelle 4.5.). Die
Proteinliste enthielt interessante Aggregat-spezifische Kandidaten, z.B. Xirp2, Rab35 und
DNAJB4. Im Laufe dieser Arbeit konnten die Proteine Xirp2 und Rab35 mittels
Immunfluoreszenfärbungen als neue Komponenten der Filaminopathie-spezifischen
Aggregate validiert werden. Zusätzlich befand sich das Protein DNAJB4 (Hitzeschockprotein
40 Homolog) unter den spezifischen Proteinen. DNAJB4 wird hauptsächlich im Herzen und
im Skelettmuskel exprimiert [Hoe et al. 1998]. Das Protein fungiert als Co-Chaperon und ist
in die Regulation des Hitzeschockproteins Hsp70-Aktivität involviert [Qiu et al. 2006]. In
Patienten mit Lungenkrebs konnte gezeigt werden, dass eine erhöhte Expression von

Diskussion
127
DNAJB4 die Apoptose in Zellen verstärkt [Wang et al. 2005 und 2007, Tsai et al. 2006, Lei et
al. 2011]. Die Sequenzhomologie zu anderen Mitgliedern der J-Proteinfamilie beträgt ca.
84% [Hoe et al. 1998]. Nach Durchsicht der MS/MS-Spektren konnte allerdings das
identifizierte Protein eindeutig als DNAJB4 bestätigt werden. Die Proteine DNAJB6 und
DNAJB1 wurden bereits in Verbindung mit Myofibrillären Myopathien gebracht: DNAJB6
[Sarparanta et al. 2012] und DNAJB1, welches erst kürzlich als Komponente der Aggregate
bestätigt werden konnte [Kley et al. 2013]. Die in früheren Immunfluoreszenzstudien als
Bestandteile der Aggregate identifizierten Proteine Filamin C, Desmin, Xin und Myotilin [Kley
et al. 2007] wurden unter der Verwendung der manuellen Auswertung nicht als spezifische
Merkmale der Aggregate identifiziert, sondern konnten wie zu erwarten ebenfalls in den
Patientenkontrollen identifiziert werden. Dies lässt sich mit der Funktion der erwähnten
Proteine begründen. Vor allem Desmin und Filamin C übernehmen eine wichtige Rolle zur
Aufrechterhaltung der intrazellulären Integrität. So ist es nicht verwunderlich, dass diese
Proteine in Aggregaten und Patientenkontrollen gefunden wurden. Bei dem Vergleich der
Patientenkontrollen mit den Negativkontrollen wurden mit der manuellen Strategie 17
Proteine als spezifische Bestandteile der Patientenkontrollen identifiziert, darunter
befanden sich u.a. die Filamin C-Bindungspartner Xin und N-Rap. 68 Proteine wurden nur in
den Negativkontrollen identifiziert, der Großteil der Proteine (176) konnte in den Patienten-
wie auch den Negativkontrollen identifiziert werden. Die massenspektrometrische Analyse
gekoppelt mit der hier gewählten Auswertestrategie ermöglichte neue Einblicke in die
Proteinzusammensetzung der Filaminopathie-spezifischen Aggregate. Dabei wurden viele
interessante Komponenten der Aggregate und mögliche Frühmarker im umliegenden
Gewebe der Patienten aufgedeckt, die eine entscheidende Rolle in der Filaminopathie
spielen könnten.
Die hier dargestellte Auswertung zur manuell durchgeführten quantitativen Analyse von
Filaminopathiegewebe stellte eine einfache Methode zur quantitativen Analyse des
Filaminopathiegewebes dar und war zu Beginn der Analysen die einzige Möglichkeit die
generierten Daten auszuwerten. In der Auswertung wurden lediglich die Proteine
berücksichtigt, die nur im Gewebe der Patienten identifiziert werden konnten. Diese
Einschränkung führte dazu, dass bereits durch Immunfluoreszenzstudien bestätigte
Komponenten der Aggregate in der finalen Proteinliste nicht berücksichtigt wurden. Es

Diskussion
128
wurde deutlich, dass sich physiologische Vorgänge nur bedingt über „Schwarz-Weiß“-Effekte
in der Proteinzusammensetzung beschreiben lassen. Es ist wichtig, die Regulation von
Proteinen miteinzubeziehen. Diese mögliche differentielle Expression von Proteinen in
unterschiedlichen Proben wurde durch die Entwicklung eines automatisierten Ansatzes
mittels Spectral Countings berücksichtigt.
Die relative Quantifizierung mittels Spectral Counting
Um differentielle Proteine in den Aggregaten bzw. in den Patientenkontrollen
identifizieren zu können, wurde im Rahmen der Masterarbeit von Alexandra Maerkens das
sogenannte Spectral Index Calculation als alternative Methode zur Analyse von
Filaminopathiegewebe etabliert. Die Spectral Index Calculation stellt eine Weiterentwicklung
der im Rahmen dieser Dissertation entwickelten manuellen quantitativen Auswertung. Unter
Verwendung des Spectral Countings konnten insgesamt 69 Proteine als statistisch signifikant
akkumuliert in den Aggregaten identifiziert werden. Unter den identifizierten Proteinen
befanden sich auch die Proteine, die in vorangegangen Studien als Bestandteile der
Proteinaggregate in Filaminopathien beschrieben wurden: Filamin C, Desmin, Xin, Myotilin,
alpha B Crystallin und diverse Hitzeschockproteine. In den Patientenkontrollen wurden im
Vergleich zu den Negativkontrollen 27 differentielle Proteine nachgewiesen (siehe Anhang
Tabellen 13.1. und 13.2.).
Bei dem Spectral Index Calculation handelt es sich um eine relative Quantifizierung
der identifizierten Proteine [Müller et al. 2011]. Die relative Quantifizierung erfasst
Unterschiede von Probe zu Probe ohne dabei die absolute Menge der darin enthaltenen
Proteine zu beachten. Die Spectral Index Calculation ist eine schnelle und kostengünstige
Methode, die vorzugsweise ihre Anwendung in der klinischen Forschung zur Identifizierung
neuer Biomarker findet [Zhou et al. 2010]. Die Ergebnisse und die daraus resultierende
Rangordnung der Proteine basieren auf der Summierung der Spektren detektierbarer
Peptide pro Proteine, welche proportional zur Konzentration eines Peptides ist [Liu et al.
2004, Müller et al. 2011]. Ein Nachteil des Spectral Index Calculation besteht darin, dass die
Anzahl der detektierbaren Peptide wahrscheinlich mit der Länge eines Proteins korreliert.
Dies bedeutet, je länger ein Protein ist, desto mehr Peptide können massenspektrometrisch
erfasst werden. Längere Proteine mit vielen Spaltstellen für Trypsin werden in dieser Analyse

Diskussion
129
bevorzugt detektiert. Kürzere, möglicherweise relevante Proteinen bleiben hingegen
unterrepräsentiert. Mittels absoluter Quantifizierung ist es möglich, über die Einbeziehung
der tatsächlich detektierten Peptide auch die theoretischen Peptide exaktere Informationen
über die Zusammensetzung von Proben zu gewinnen. Eine etablierte Methode zur absoluten
Quantifizierung stellt AQUA (absolute quantification of proteins) dar [Gerber et al. 2003].
AQUA erlaubt die direkte Quantifizierung von Differenzen in Proteinen und
posttranslationalen Modifikationen. Die Methode basiert auf der Verwendung chemisch
synthetisierter Isotopen-Peptide, welche unique für die Proteine der Wahl sind. Eine
definierte Menge der Peptide wird als interner Standard der Probe zugesetzt [Lil 2003,
Langenfeld et al. 2008]. Ein Nachteil der Methode ist, dass sie sehr aufwendig und kostspielig
ist. Alternative Methoden zur absoluten Quantifizierung sind: emPai (exponentially modified
protein abundance index) [Ishihama et al. 2005] und APEX (absolute protein abundance
index) [Lu et al. 2007]. Beide Methoden beruhen auf der Label-freien Quantifizierung. Im
Gegensatz zur Spectral Index Calculation werden bei diesen Methoden auch die Anzahl an
theoretisch entstehenden Peptiden berücksichtigt. Die Etablierung einer Methode zur
absoluten Quantifizierung könnte die Rangordnung an akkumulierten Proteinen in den
Filaminopathie-spezifischen Aggregaten (beschrieben im Anhang Tabellen 13.1. und 13.2.)
um weitere interessante Proteine ergänzen. Das Einbeziehen kleinerer Proteine und
Proteine, die über weniger Trypsinschnitt dahin verfügen, könnte zu einer noch
detaillierteren Aufklärung der Zusammensetzung der Aggregate führen. Daraus könnten sich
zusätzliche Hinweise auf die Pathomechanismen ergeben.
Vergleich der Auswertestrategien
Im Laufe dieser Arbeit entwickelte sich die Auswertestrategie von einer manuellen
(„pseudo-quantitativen“) Auswertung hin zu einer automatisierten relativen Quantifizierung.
Die manuelle Auswertung ermöglichte einen sehr interessanten Überblick über die
Zusammensetzung der Proteinaggregate und schien sehr gut geeignet um subtypische
Proteine zu identifizieren. 111 Proteine konnten mittels der einfachen und manuellen
Auswertung als Bestandteile der Proteinaggregate identifiziert werden. Zusätzlich konnten
17 Proteine in den Patientenkontrollen als mögliche Biomarker für Frühstadien der
Filaminopathie gefunden werden. Trotz dieser Erfolge musste die Aussagekraft dieses

Diskussion
130
Ansatzes als begrenzt eingeschätzt werden. Die manuelle Auswertung ermöglichte lediglich
Aussagen über das (Nicht-) Vorhandensein von Proteinen. Die Entstehung der
Filaminopathie unterliegt allerdings einem Entwicklungsprozess, erste Symptome der
Filaminopathie manifestieren sich erst zwischen dem 40. und 60. Lebensjahr [Vorgerd et al.
2005, Kley et al. 2007]. Die Entwicklung von gesundem zu erkranktem Gewebe wird mittels
der manuellen Auswertung nicht widergespiegelt. Durch die Weiterentwicklung der
Auswertestrategie hin zu einer automatisierten Analyse mittels Spectral Index Calculation
konnten Veränderungen während des Entwicklungsprozesses detektiert werden. Durch die
Spectral Index Calculation konnten regulierte Proteine dargestellt und eine Rangordnung der
identifizierten Proteine erstellt werden (siehe Anhang Tabellen 13.1. und 13.2.). 38 % (38
Proteine) der Proteine aus der manuellen Auswertung konnten auch mittels Spectral Index
Calculation identifiziert werden. In den Patientenkontrollen konnten dagegen keine
Gemeinsamkeiten beobachtet werden.
Zusammenfassend lässt sich sagen, dass beide Auswertestrategien sehr schnell und
einfach durchzuführen sind. Die Spectral Index Calculation stellt jedoch eine sinnvolle
Weiterentwicklung der hier manuell durchgeführten quantitativen Auswertung dar, um
differentiell regulierte Proteine zu identifizieren. Zusätzlich zeichnen sich beide Strategien als
einfache und kostengünstige Methoden aus. 38% aller Proteine, die ausschließlich in den
Aggregaten gefunden wurden, konnten mit beiden quantitativen Auswertestrategien
identifiziert werden. Die restlichen 62% der Proteine könnten sich gegenseitig ergänzen und
einen Schritt zur Aufklärung des Pathomechanismis beitragen. Beide Strategien bieten
ausreichend Potential für weiterführende (Validierungs-) Studien in den Aggregaten.
5.2.1.4. Die physiologische Bedeutung der differentiell exprimierten Proteine in
den Aggregaten
In der massenspektrometrischen Analyse konnten durchschnittlich 415 Proteine in
Filaminopathie-Aggregatproben und 335 Proteine in den entsprechenden
Patientenkontrollen identifiziert werden. Demnach konnten in den Aggregatproben ca. 20 %
mehr Proteine identifiziert werden als in den Patientenkontrollen. Dieses Ergebnis deckt sich
mit der Vermutung, dass die Proteinkonzentration in den Aggregaten erhöht ist. In der
qualitativen Proteomanalyse der Filaminopathie-Aggregatproben konnten viele Z-Scheiben

Diskussion
131
assoziierte Proteine, wie Filamin C und dessen Bindungspartner N-Rap, Obscurin, Myotilin
und Xin [Dalkilic et al. 2002, Lu et al. 2003, Van der Veen et al. 2000 und 2006] identifiziert
werden. Alle Proteine, bis auf N-Rap und Obscurin, wurden bereits früheren
Immunfluoreszenzstudien als Bestandteile der Aggregate verifiziert [Kley et al. 2007].
Mutationen im Filamin C können zu strukturellen Veränderungen führen, die sich direkt auf
die Z-Scheiben der Myofibrillen und Z-Scheiben assoziierte Proteine auswirken können. So ist
es nicht verwunderlich, dass neben Filamin C auch dessen Bindungspartner zu finden sind.
Dies sind allerdings nicht alle Proteine, die im direkten Kontakt zur Z-Scheibe stehen.
Proteine wie alpha Actinin und dessen Bindungspartner Myopodin [Faul et al. 2007,
Linnemann et al. 2010] konnten weder in der Label-freien Proteomstudie noch in
Immunfluoreszenzstudien als Bestandteile der Aggregate identifiziert werden [Kley et al.
2012]. Aufgrund dieser Erkenntnisse lassen sich zwei Gruppen von Z-Scheiben assoziierten
Proteine bilden: die erste Gruppe umschließt Filamin C und dessen Interaktionspartner, die
zweite Gruppe alpha Actinin samt Bindungspartner [Kley et al. 2012]. Diese Einteilung lässt
die Vermutung zu, dass Filamin C und dessen Bindungspartner eine bedeutende Rolle in der
Pathogenese der MFM, im speziellen der Filaminopathien, spielen. Actinin und dessen
Bindungspartner hingegen könnten in der Entwicklung anderer Myopathien involviert sein
[Claeys et al. 2009]. Als eine weitere Muskel-spezifische Proteingruppe wurden
Intermediärfilamente und einige ihrer Bindungspartner in den Aggregatproben identifiziert.
Desmin und dessen Interaktionspartner Vimentin, Nestin [Steinert et al. 1999], Syncollin
[Poon et al. 2002], Nexilin [Hassel et al. 2009] und alpha B Crystallin [Bennardini et al. 1992].
Die Intermediärfilamente spielen eine extrem wichtige Bedeutung in den Myofibrillen. Durch
die Verbindung anliegender Myofibrillen im Bereich der Z-Scheiben wird ein
dreidimensionales Netzwerk geschaffen, das die Stabilität der Myofibrillen gewährleistet.
In der manuell durchgeführten quantitativen Analyse konnten 111 Proteine als
spezifische Bestandteile der Aggregate identifiziert werden, darunter Xirp2 und Rab35. Xirp2
(Xin repeat protein 2) wurde ausschließlich in den Aggregatproben von
Filaminopathiepatienten identifiziert. 2004 wurde es von Pacholsky et al. erstmals
beschrieben. Xirp2 enthält eine sich wiederholende Sequenz bestehend aus 16
Aminosäuren, die sich auch in Xin wiederfindet [Wang et al. 1999]. Xirp2 wie auch Xin
verfügen über ein Aktin-bindendes Motiv und sind somit an der (Re-)Organisation des

Diskussion
132
Zytoskeletts beteiligt [Huang et al. 2006]. Erst kürzlich konnten wir zeigen, dass nicht nur Xin
ein Bindungspartner von Filamin C ist sondern auch Xirp2 [Kley et al. 2013]. Auch wenn beide
Proteine gleichzeitig mit Filamin C assoziieren können, konnte in Konkurrenzessays gezeigt
werden, dass die Bindung zwischen Xirp2 und Filamin C stärker ist als die Bindung zwischen
Filamin C und Xin [Kley et al. 2012]. Es bleibt zu klären, welche Rolle genau Xirp2, Xin und
Filamin C während der Proteinaggregation und der Destabilisierung der Myofibrillen spielen.
Rab35 (Ras related Protein 35) konnte als weiterer neuer Bestandteil von Filaminopathie-
spezifischen Aggregaten identifiziert werden. Rab35 ist eine kleine GTPase, dessen
Hauptaufgabe vor allem in der Beeinflussung der Aktin-Dynamik liegt [Chua et al. 2010]. Die
genaue Rolle in der Pathogenese der Filaminopathie und in anderen MFM-Subtypen muss in
Zukunft geklärt werden. Unter den 111 als spezifisch für Aggregate identifizierten Proteine
befanden sich allerdings weder Filamin C noch Desmin und deren Bindungspartner. Um
spezifische Proteinkomponenten der Aggregate identifizieren zu können, wurden strikte
Kriterien gewählt (beschrieben unter 3.17.1.). Es wurden nur die Proteine als spezifische
Bestandteile akzeptiert, die ausschließlich in den Aggregatproben und gleichzeitig in keiner
Patientenkontrolle gefunden wurden. Filamin C, Desmin und deren Bindungspartner sind
wichtige strukturgebende Proteine im gesunden Muskel, so dass es zu erwarten war, dass
diese Proteine keine spezifischen Komponenten der Aggregate darstellen können.
Durch die Weiterentwicklung der manuellen Auswertung zu einer automatisierten
Strategie war es möglich, differentielle Proteine in den Filaminopathie-spezifischen
Aggregaten zu identifizieren und eine Rangordnung der akkumulierten Proteine zu erstellen.
Proteine wie z.B. Filamin C, Desmin und deren Bindungspartner, die in der manuell
durchgeführten Analyse nicht berücksichtigt wurden, konnten mittels Spectral Index
Calculation als differentielle Proteine identifiziert werden. Die Spectral Index Calculation-
basierte Analyse der Proteinaggregate ergab ein sehr homologes Muster in der
Zusammensetzung. In allen Patienten konnte Filamin C, unabhängig von der vorliegenden
Mutation, als das am höchsten akkumulierte Protein nachgewiesen werden. Die
nachfolgenden Positionen waren ebenfalls mit Desmin, Xirp2 und N-Rap immer gleich
besetzt. Dieses homologe Muster in der Zusammensetzung steht in Einklang mit den
klinischen Merkmalen der Filaminopathie, welche unabhängig von der Mutation große
Ähnlichkeiten aufweisen [Kley et al. 2007].

Diskussion
133
Ziel der differentiellen Proteomanalyse von Proteinaggregaten war es, spezifische
Proteine zu identifizieren, die im Vergleich zum umliegenden Gewebe bevorzugt in die
Aggregate integriert bzw. eingebaut werden. Zusätzlich war es interessant zu sehen, ob
einige dieser Proteine als eindeutige Marker für die Filaminopathie dienen könnten. Das im
Rahmen dieser Arbeit validierte Protein Xirp2 konnte nur in den Aggregaten und nicht im
umliegenden Gewebe identifiziert werden. Allerdings zeigte eine erst kürzlich veröffentlichte
Proteomanalyse von Aggregaten aus Desminopathiepatienten, dass Xirp2 auch ein fester
Bestandteil der Desminopathie-Aggregate ist [Maerkens et al. 2012]. Rab35 hingegen konnte
ausschließlich in den Aggregaten der Filaminopathiepatienten als akkumuliertes Protein
identifiziert werden. In Zukunft müssen die Aggregate weiterer MFM-Subtypen analysiert
werden, um genau zu klären, welche Proteine subtypisch akkumulieren und inwieweit ein
gemeinsamer Pathomechanismus der verschiedenen Subtypen vorliegt.
5.2.2. Gel-basierte differentielle Analyse
Die zweidimensionale Polyacrylamid-Gelelektrophorese (2D PAGE) gehört zu der am
häufigsten angewandten Technik in der Proteomik [May et al. 2012]. Besonders die
Weiterentwicklung zur zweidimensionalen Differential-In-Gel-Elektrophorese (2D DIGE)
ermöglichte und vereinfachte den quantitativen Vergleich von mehreren Proben [Rabilloud
2012]. Doch in Bezug auf die hier durchgeführten Analysen der Proteinaggregate stieß die 2D
DIGE auf ihre Grenzen. In der Planungsphase der differentiellen Studien wurde die Analyse
der Aggregate und des umliegenden Gewebes (PatKO) mit beiden Ansätzen (DIGE und Label-
frei) in Betracht gezogen, um die erzielten Ergebnisse besser absichern zu können. Doch die
begrenzte Löslichkeit der Aggregate in Urea-haltigen Puffern verhinderte den Einsatz der 2D
DIGE als ergänzende Methode zum Label-freien Ansatz (siehe Punkt 4.1.2. und 4.1.3.). So
wurden lediglich Muskelzellen ohne Aggregate aus den Filaminopathiepatienten isoliert und
mittels der Label-freien Methode und mittels 2D DIGE analysiert, um diese auf mögliche
Marker für Frühstadien zu analysieren.
Essentiell für eine differentielle Proteomanalyse mittels 2D PAGE ist eine hohe
technische Reproduzierbarkeit. Technische Varianten entstehen z.B. während der
Probenpräparation und führen zu Unterschieden in der Spotanzahl sowie in der Abundanz
der Proteine. Daraus resultieren falsche Quantifizierungen. Besonders bei einer hohen

Diskussion
134
Probenzahl bzw. Gelanzahl ist es wichtig, die technischen Varianzen zu minimieren. Die
Weiterentwicklung der 2D PAGE zur zweidimensionalen 2D DIGE minimiert die technische
Variabilität [Ünlü et al. 1999]. Für die Analyse der mikrodissektierten Skelettmuskelproben
bietet sich die Verwendung des Saturation-Labeling-System an, weil es sich optimal für die
Markierung von gering konzentrierten Proben eignet [Helling et al .2006] Beim Saturation-
Labeling stehen zwei Farbstoffe zur Verfügung (Farbstoffe Cy3 und Cy5). Vor Beginn der
ersten Dimension werden die zu analysierenden Proben mit Fluoreszenzfarbstoffen
markiert. Der Einsatz der CyDye-Farbstoffe ermöglichte es, 2 Proben gleichzeitig auf ein Gel
aufzutragen und separieren zu können. Dadurch werden die Reproduzierbarkeit und auch
die Quantifizierbarkeit der Proteinspots verbessert [Klose 2009]. Durch die simultane
Auftrennung mehrerer Proben auf einem Gel wird die Gesamtzahl der Gele drastisch
reduziert [Tonge et al. 2001]. Durch den Einsatz des DIGE-Systems ist neben der
Minimierung technischer Variationen die Steigerung der relativen Quantifizierung [Tonge et
al. 2001] und Sensitivität durch Fluoreszenzdetektion möglich [Ünlü et al. 1997].
5.2.2.1. Optimierung der Probenmenge
Zu Beginn der Arbeiten war unklar, wie viele mikrodissektierte Muskelzellen für die
2D DIGE benötigt werden würde. Sitek et al. isolierten 1000 Zellen mittels LMD aus
Patienten mit Adenokarzinom und trennten diese nach Saturation Labeling über die 2D
PAGE auf [Sitek et al. 2005]. In der Studie konnte eine vergleichbare Menge an Proteinspots
wie in vorangegangen Studien detektiert werden, allerdings mit einem deutlich verringerten
Einsatz an Material. Es konnte gezeigt werden, dass eine Kombination aus LMD und 2D
Saturation Labeling sich optimal dazu eignet, gering konzentrierte Proben mittels 2D PAGE
aufzutrennen. Um in der hier vorliegenden Arbeit die optimale Menge an Muskelzellen
herauszufiltern, wurden 1000, 2000, 3000 und 5000 Zellen mittels LMD isoliert. 1000 (Daten
nicht gezeigt), 2000 und 5000 mikrodissektierte Zellen erwiesen sich als ungeeignet. Die
Auftrennung von 1000 mikrodissektierten Zellen erwies sich als zu wenig. Der direkte
Vergleich von 2000 und 3000 mikrodissektierten Zellen resultierte in einer vergleichbaren
Anzahl an detektierten Spots. Dennoch wurde das Ergebnis mit 3000 Zellen als am besten
eingestuft (siehe Punkt 4.3.1.). Es konnte eine sehr gute Auftrennung der Proteine erzielt
werden und die einzelnen Proteinspots distinkt dargestellt werden. Die Verwendung von

Diskussion
135
5000 mirkodissektierten Zellen verschlechterte das Proteinmuster. Zum einen verschmolzen
mehrere gering abundante Proteinspots zu einem großen Spot oder die gering abundanten
Spots wurde durch die erhöhte Proteingesamtkonzentration von anderen Spots überlagert.
Aufgrund der variierenden Größe der Muskelfasern wurde auch hier die
durchschnittliche Fläche von 3000 Zellen mit Hilfe des LMD 6500 (Leica, Wetzlar,
Deutschland) ermittelt. Diese betrug 1,6 Mio. µm2 und wurde für die folgenden Analysen
isoliert. Die Fläche war mehr als 6mal so groß wie die Fläche, die für die Label-freien
Aggregatanalysen (250 000 µm2) gesammelt wurde. Eine 2D DIGE Studie wurde aufgrund der
begrenzten Löslichkeit der Aggregate in Urea-haltigen Puffern im Vorfeld ausgeschlossen.
Die benötigte Fläche an Material von 1,6 Mio. µm2 hätte eine 2D Studie mit den ohnehin
limitierten Aggregaten zusätzlich erschwert.
5.2.2.2. Detektion differentiell exprimierter Proteine
Die 2D PAGE stellt eine exzellente Methode zur Auftrennung komplexer
Proteingemische (z.B. Zell- und Gewebelysate) dar. Besonders gut lassen sich Proteine mit
hydrophilen Eigenschaften darstellen. Membranproteine und andere hydrophobe Proteine
hingegen sind eher unterrepräsentiert [Santoni et al. 2000, Rabilloud 2002]. Die Bildanalyse
stellt einen elementaren Schritt zur Identifizierung differentieller Proteine dar. Nach der
Separation im Gel erfolgte die Quantifizierung durch einen Software-basierten Gelvergleich
(DeCyder™ Software). Vor Beginn der Analyse wurden die Gele zurechtgeschnitten. Es wurde
darauf geachtet, dass die Lauffront, bestehend aus Bromphenolblau, entfernt wurde, da
diese zu falsch positiven Detektionen führen kann Die verschiedenen Fluoreszenzbilder
wurden dann übereinander gelegt. Die Spotzuordnung erfolgte zunächst automatisch.
Allerdings musste die Zuordnung manuell nachbearbeitet werden. In früheren Studien
konnte gezeigt werden, dass eine falsche Spotzuordnung zu einer Abweichung von bis zu
einem Drittel bei der Quantifizierung führt [Schroder et. al. 2008]. Insgesamt wurden pro ca.
4300 Proteinspots pro Gel detektiert, darunter befanden sich 15 differentiell exprimierte
Spots. Die Proteine wurden tryptisch im Gel verdaut und mittels MALDI analysiert. Die
MALDI-MS [Karas und Hillenkamp 1988, Karas et al. 2000] ist eine schnelle und zuverlässige
Methode zur Hochdurchsatzanalyse. Die Identifizierung der Proteine erfolgt meist mittels
Peptidmassen-Fingerprint [Shevchenko et al. 1996]. 7 der 15 differentiell exprimierten Spots

Diskussion
136
konnten eindeutig identifiziert werden. In den restlichen Spots hingegen konnten auch nach
mehrfachen Wiederholungen keine Proteine identifiziert werden. Generell ist zu bemerken,
dass die Proteinmenge innerhalb der Spots als sehr gering einzuschätzen war. Somit könnte
dies eine Hauptursache für die fehlenden Proteinidentifizierungen sein. Um zu klären, ob die
geringe Proteinmenge das einzige Problem darstellt, wäre es in zukünftigen Versuchen
sinnvoll die Menge an mikrodissektierten Gewebe zu steigern. Allerdings wurde bereits in
den Vorstudien gezeigt, dass 2D Analysen mit 5000 mikrodissektierten Muskelzellen zu
keinem optimalen Ergebnis führen, da gering abundante Spots zu einem großen Spot
verschmolzen (beschrieben in Punkt 4.3.). In diesem Falle wäre es am sinnvollsten ein
sensitiveres Massenspektrometer zu wählen. Für die Wahl eines sensitiveren
Massenspektrometers sprechen auch die Arbeiten von Kondo et al. Kondo und dessen
Mitarbeiter führten diverse differentielle Studien unter Verwendung der LMD und
Saturation CyDyes durch [Fujii et al. 2005, Fujii et al. 2006; Kondo 2008]. Ihnen war es
möglich viele der als differentiell exprimierten Proteine zu identifizieren. Allerdings stellten
sie Probleme in der Identifizierung von fluoreszenz-markierten Proteinen mittels MALDI fest.
Sie vermuten, dass das Markieren der Proteine mit CyDyes die Ionisation der Peptide stört
und somit die Identifizierung verhindert. Im Weiteren wäre es sinnvoll die Effektivität des
tryptischen Verdaus zu überprüfen oder zu optimieren. Bao et al. beschrieben 2008 eine
alternatives Protokoll zum tryptischen Verdau. Darin wurden die Gelstückchen mit einer
Infrarot-Lampe behandelt. Die Dauer des Verdau konnte von 16h auf 5 min reduziert werden
[Bao et al .2008, Wang et al. 2008]. Die anschließende Analyse der Peptide mittels MALDI
resultierte in einer gesteigerten Sequenzabdeckung der identifizierten Proteine. Eine
ebenfalls von Wang et al. veröffentlichte Methode zur Optimierung des tryptischen Verdaus
beschreibt den beschleunigten In-Lösung-Verdau von Proteinen in einem elektrischen Feld
mit Wechselspannung [Wang et al. 2008]. Die Dauer des Verdaus konnte von 12h auf 5 min
reduziert werden. Die Tauglichkeit der Methode konnte auch für den In-Gel-Verdau bestätigt
werden.

Diskussion
137
5.2.2.3. Die physiologische Bedeutung der differentiell exprimierten Proteine in
Patientenkontrollen
Ziel der hier durchgeführten 2D DIGE Studie war es, differentiell exprimierte Proteine
in Filaminopathiegewebe zu identifizieren. Hierzu wurde Gewebe ohne Aggregate aus den
Patienten isoliert und gegen Gewebe von Gesunden verglichen. Das Aggregat-freie
Patientenmaterial stellte in der 2D Studie eine Art frühes Krankheitstadium dar. Die
Identifizierung von differentiellen Proteinen und deren Einordnung in den biologischen
Kontext sollte helfen, Rückschlüsse auf die Pathomechnismen der Filaminopathien zu
gewinnen. In der 2D DIGE konnten 15 differentielle Proteinspots in erkranktem Gewebe
detektiert werden. Die Gelspots wurden aus dem Gel ausgestanzt, tryptisch im Gel verdaut
und anschließend mittels MALDI analysiert. Insgesamt konnten 7 der 15 Spots eindeutig
identifiziert werden. Zwei der regulierten Spots wurden als Desmin identifiziert. Zusätzlich
wurden in den Spots die Proteine Glykogen Phosphorylase (muskelspezifische Form),
Creatine Kinase M-Typ (CKM), Aktin (alpha Aktin 1, Skelettmuskel), Gelsolin und die leichte
Kette 3 von Myosin. Desmin, CKM, Aktin und die Glykogen Phosphorylase konnten zusätzlich
in der Label-freien Proteomanalyse im Vergleich von Patienten- und Negativkontrollen als
differentielle Proteine identifiziert werden.
Desmin ist ein 53 kDa großes Intermediärfilament der Gruppe III. Es wird
hauptsächlich in Skelett- und Herzmuskulatur sowie in der glatten Muskulatur exprimiert.
Die Hauptaufgabe des Desmins liegt in der Ausbildung eines stabilen dreidimensionalen
Fasernetzwerks, dass die Integrität der Muskelzelle aufrecht erhält [Herrmann et al. 2004,
Olive et al. 2011]. Dieses Netz erstreckt sich durch die gesamte Zelle und vernetzt die
verschiedensten Kompartimente miteinander, z.B. Z-Scheiben untereinander und mit der
Plasmamembran sowie Zellmembran mit zytosolischen Membranorganellen [Herrmann et
al. 2004].Im Herzmuskel stellt Desmin die Hauptkomponente der Purkinje-Fasern dar und ist
besonders im Bereich der interkallierenden Scheiben vermehrt zu finden [Goldfarb 2004].
Bis heute sind über 60 Mutationen im für Desmin codierenden Gen bekannt, die u.a. MFM
verursachen können [Maerkens et al. 2013]. Die Desminopathie wurde erstmals von
Goldfarb 1998 beschrieben. Ähnlich wie in den Filaminopathien sind proximale aber auch
distale Muskelgruppen von der Erkrankung betroffen. Hinzu kommt, dass bei
Desminopathiepatienten auch Kardiomyopathien beobachtet werden können [Goldfarb et

Diskussion
138
al. 2004], die die Lebenserwartung der Patienten deutlich einschränken. Erst kürzlich
konnten wir zeigen, dass Desmin in Filaminopathie-spezifischen Aggregaten das am
zweithöchsten akkumulierte Proteinen ist [Kley et al. 2012]. In Desminopathien ist es das am
höchsten akkumulierte Protein in den Aggregaten [Maerkens et al. 2013].
Die Glykogen Phosphorylase (PYG) ist ein zytosolisches Enzym des
Glykogenstoffwechsels, welches die phosphorolytische Spaltung des Glykogens in Glukose-1-
Phosphat katalysiert. Es gibt drei verschiedene Isoformen: PYGB (Gehirn, Herz), PYGL (Leber)
und PYGM (Muskel). Die Glykogen Phosphorylase wird durch Glukose und ATP inhibiert und
durch AMP aktiviert [Berg, Tymoczko, Stryer, Biochemie. 5. Auflage. Spektrum Akademischer
Verlag, Heidelberg 2003]. 1951 wurde die Fehlfunktion der Glykogen Phosphorylase erstmals
mit einer Erkrankung der Muskulatur in Verbindung gebracht: McArdle-Krankheit [McArdle
1951]. Es handelt sich dabei um eine autosomal-rezessiv vererbbare Krankheit, in der der
Abbau von Glykogen in Glukose gestört ist. Dadurch kommt es zu einer Anhäufung von
Glykogen und zu einer Energieunterversorgung der Muskulatur. Die Folge sind
Muskelschmerzen und geringe Belastbarkeit der Muskeln. In schwerwiegenden Fällen kann
es zu akutem Nierenversagen kommen, da aufgrund der Störung der Integrität der
Muskelfasern große Proteine wie z.B. Myoglobin in den Blutkreislauf gelangen und die
Nieren schädigen können. 1993 wurde die erste Mutation von Tsujion et al. beschrieben.
Weitere 4 Mutationen konnten 1998 in 9 Patienten acht nicht verwandter Familien
identifiziert werden [Vorgerd et al. 1998]. Inzwischen sind über 50 Mutationen in der
Glykogen Phosphrylase bekannt. Bei ca. 70% aller Patienten tritt die R50X-Mutationen auf
[Deschauer et al. 2007]. Sie stellt ein Stopcodon dar, wodurch die Produktion des Enzyms
eingestellt wird. Als Folge wird ein verkürztes Enzym oder gar unwirksames Enzym
produziert. In der hier durchgeführten differentiellen Studie wurde die muskelspezifische
Isoform in den Patientenkontrollen als herunterreguliert identifiziert. Die Label-freie
Proteomanalyse bestätigte die Regulierung der Glykogen Phosphorylase. In der Literatur
wurde bis heute kein Zusammenhang zwischen einer verminderten Expression der Glykogen
Phosphorylase und den Filaminopathien bzw. MFM beschrieben. Es ist jedoch denkbar, dass
die verminderte Expression mit einer Typ-1-Muskelfaserdominanz in
Filaminopathiepatienten zu erklären ist [Kley et al. 2007]. Die Gründe für die Typ-1-
Faserdominanz in Patienten mit Filaminopathie sind noch unklar. Es ist anzunehmen, dass in

Diskussion
139
Typ-1-Muskelfasern generell weniger Glykogen Phosphorylase exprimiert wird [Schiaffiano
and Reggiani 2011]. Typ-1-Muskelfasern werden langsam zuckende Muskelfasern oder auch
oxidative Fasern genannt. Sie zeichnen sich durch eine hohe Anzahl an Mitochondrien und
eine hohe Aktivität an oxidativen Enzymen aus [Scherzmann et al. 1989, Jackmann and Willis
1996]. Die Energie gewinnen sie aerob und sind auf Dauerleistung mit begrenztem
Kraftauswand ausgelegt. Im Gegensatz dazu werden die Typ-2-Muskelfasern als schnell
kontrahierende Muskelfasern bezeichnet und dienen der schnellen Kraftentwicklung.
Dadurch benötigen sie sehr viel mehr Energie, welche überwiegend anaerob aus
Glykogenspeichern bereitgestellt wird.
Das Strukturprotein Aktin hat ein Molekulargewicht von 42 kDa und gehört zu den
hoch konservierten Proteinen in Eukaryonten. In Säugetieren gibt es 6 verschiedene
Isoformen [Vandekerckhove and Weber 1978]. Das in der 2D Studie identifizierte Alpha Aktin
1 stellt die skelettmuskelspezifische Form dar. In der Zelle bildet Aktin dynamische
Filamente, die Aktinfilamente oder auch dünne Filamente. Aktin ist ein essentieller
Bestandteil des kontraktilen Apparates und stabilisiert durch den Ausbau des
Filamentnetzwerkes die äußere Zellform. Veränderungen in dem für Alpha Aktin 1
codierenden Gen führen zu sogenannten Kongenitalen Myopathien. Die Gruppe der
Kongenitalen Myopathien beschreiben langsam progrediente Erkrankungen, die meist im
Neugeborenen- oder Kindesalter auftreten und durch proximale oder generalisierte
Muskelschwäche charakterisiert sind. In einigen Fällen wird auch eine Ateminsuffizienz
beobachtet [Sparrow et al. 2003]. In der Literatur wurden zusätzlich spät einsetzende
Formen der Erkrankung beschrieben [Niwa et. al. 2009]. Die Nemalin Myopathie (Typ 3) wird
zu den Kongenitalen Myopathien gezählt und wurde 1963 erstmals von Shy et al.
beschrieben. Betroffene Patienten leiden ab der Geburt unter Hypotonie der Muskulatur
und einer verzögerten motorischen Entwicklung. In den meisten Fällen erwerben die
Patienten im Laufe ihres Lebens die Fähigkeit zu laufen. Auf morphologischer Ebene wird die
Nemalin Myopathie durch stäbchen- und fadenförmige Strukturen charakterisiert. In der
hier durchgeführten 2D DIGE Studie wurde das Alpha Aktin 1 im Vergleich zu Kontrollgewebe
als niedrig abundantes Protein in Filaminopathiegewebe identifiziert. Aktin wird als dünnes
Filament bezeichnet. Cohen et al. stellten 2012 eine Theorie zu Auswirkungen einer
erhöhten Desminphosphorylierung auf. Sie gingen in ihrer Studie davon aus, dass durch die

Diskussion
140
verstärkte Phosphorylierung des N-Terminus die Stabilität der Myofibrillen verloren geht und
weitere Komponenten des Systems Schaden nehmen. Dazu wurden auch die dünnen
Filamente gezählt, welche in Folge der Phosphorylierung des Desmins ebenfalls
phosphoryliert und anschließend degradiert werden [Cohen et al. 2012]. Die verstärkte
Phosphorylierung des Desmins und das verminderte Vorkommen des Aktins in
Filaminopathiegewebe könnten im Einklang mit dieser Hypothese stehen.
Die Creatin Kinase (CK) ist ein Enzym, das eine N-Phosphoryl-Gruppe von Phospho-
Kreatin auf Adenosindiphosphat (ADP) überträgt. Dadurch entsteht Adenosintriphosphat als
universelle Energiequelle. Bei kurzfristigen Belastungen bietet die CK eine optimale
Möglichkeit des Gehirn oder den Muskel schnell mit Energie zu versorgen. Es sind 4
verschiedene Isoformen bekannt: CK-MM (Skelettmuskel), CK-MB (Myokard), CK-BB (Gehirn)
und CK-MiMi (Mitochondrien) [Göthert et al. 1975, Schlattner et al. 2005, Wallimann et al.
2007]. Eine Erhöhung der CK-Aktivität im Serum, welche als Folge einer Störung der
Faserintegrität und des Übertritts der CK in das Serum auftritt, ist oft ein Indiz für eine Herz-
oder Skelettmuskelerkrankung. Zu den häufigsten Skelettmuskelerkrankungen mit erhöhter
CK gehören die progressiven Muskeldystrophien, zu denen auch die Filaminopathien
gehören. In einer Studie von 2007 konnten bei 31 Patienten normale bis deutlich erhöhte
CK-Werte im Serum beobachtet werden [Kley et al. 2007]. In der im Rahmen dieser
Dissertation durchgeführten 2D Studie wurden mikrodissektierte Muskelfaserzellen
analysiert. Als Ergebnis wurde eine verringerte Expression der muskelspezifischen Form der
Creatin Kinase in den Filaminopathiepatienten beobachtet. In der Literatur ist keine
vergleichbare Studie an MFM-Gewebe durchgeführt worden um abschätzen zu können,
welche Gründe eine verminderte Expression der Creatin Kinase im Muskel haben könnte.
Eine 2011 veröffentlichte Studie beschreibt eine erniedrigte CK-Exprimierung im Muskel
aufgrund von dilatativer Kardiomyopathy [Diguet et al. 2011]. Als Folge der veränderten
Expression der CK konnten verringerte Mengen an Alpha Aktin und eine erhöhte Abundanz
von Desmin identifiziert werden. Die verminderte Exprimierung der CK wirkte sich negativ
auf die Stabilität des Aktinnetzwerkes aus und induzierte posttranslationale Modifizierungen
in Desmin, wodurch auch die Desorganisation der Intermediärfilamente hervorgerufen
wurde [Diguet et al. 2011]. Diese Ergebnisse ermöglichten neue Einblicke in die
Pathomechanismen der dilatativen Kardiomyopathie und könnten gleichzeitig interessante

Diskussion
141
Anstöße zur Aufdeckung der Pathogenese in Filaminopathien geben. Immerhin zeigt auch
ein großer Teil der Patienten mit Filaminopathie kardiale Symptome. Zusammenfassend lässt
sich sagen, dass in der 2D DIGE Analyse wie auch in der Studie von Diguet et al. die gleichen
Proteine (CK, Desmin und Alpha Aktin, ähnlich reguliert) detektiert werden konnten. Es
bleibt zu klären, inwieweit die von Diguet beschriebenen Abläufe auch für die Filaminopathie
relevant sein könnten.
Gelsolin ist Teil der Superfamilie der Aktin-bindenden und schneidenden Proteine
[Sun et al. 1999]. Das Protein hat ein Molekulargewicht von 81 kDa und liegt intrazellulär im
Zytosol und in den Mitochondrien sowie extrazellulär im Blutplasma vor, wo es die
Polymerisation von freiem Aktin aus zugrunde gegangen Zellen verhindert [Kwiatkowski
1999]. Im Skeletmuskel wird es als das Schlüsselprotein im Auf- und Abbau der
Aktinfilamente bezeichnet. Gelsolin bindet nach Ca2+ Aktivierung an Aktinfilamente [Hartwig
1992]. So wird eine Konformationsänderung des Aktins erreicht, die die Schwächung der
hydrophoben Wechselwirkungen zwischen den einzelnen Aktinmonomeren zur Folge hat
[McGough et al. 1998]. Dies führt zum Bruch der Filamentstruktur. Direkt im Anschluss an
das Zerschneiden der Aktinfilamente, setzt sich das Gelsolin an das schnellwachsende Ende
eines Monomers („Capping“) und blockiert so das Anlagern oder die weitere Dissoziation
von Monomeren. Durch das Ablösen des Gelsolins wird die erneute Polymerisation von
Aktinfilamenten gefördert. So ist es möglich, dass sich das Aktingerüst des Zytoskeletts
erneuern kann [Yin and Stull 1999.] Gelsolin wurde in der 2D Studie in
Filaminopathiegewebe als das am stärksten hoch regulierte Protein identifiziert (Ratio 4,3).
In der Label-freien Proteomanalyse von Muskelzellen ohne Aggregate aus
Filaminopathiepatienten wurde das Protein im Vergleich zu gesundem Gewebe aufgrund
eines zu schlechten p-Wertes (> 0,05, Ratio = 9,2) nicht als signifikant reguliert identifiziert.
In den MFM-spezifischen Proteinaggregaten hingegen befand sich Gelsolin unter den
signifikant akkumulierten Proteinen (Ratio 2,1, p-Wert ≤ 0,05). Im Hinblick der hier erzielten
Ergebnisse sollte die Validierung des Gelsolins in immunhistologischen Färbungen in einem
nächsten Schritt folgen. Die Rolle des Gelsolins innerhalb der Pathogenese der
Filaminopathie ist zurzeit unklar und sollte in nachfolgenden Studien geklärt werden. Es ist
jedoch denkbar, dass Gelsolin als eine Art protektives Protein für Aktin in Frühphasen der
Filaminopathie fungieren könnte. Die Bindung des Gelsolins an Aktin verhindert die

Diskussion
142
Verbindung von zwei Aktinmonomeren, gleichzeitig verhindert es aber auch den weiteren
Abbau des Monomers. Aktin wurde im Vergleich zu gesundem Gewebe in der 2D Studie als
signifikant geringer exprimiert in krankem Gewebe identifiziert, Gelsolin signifikant höher
abundant. Die Regulierung der Gelsolin-Expression könnte dem Zweck dienen, noch
bestehende Aktinfilamente zu spalten und durch das Capping die Aktinmonomere vor dem
Untergang zu bewahren.
Die leichte Kette 3 des Myosins (MYL3) ist neben den schweren Ketten Bestandteil
des funktionalen Myosins. Myosin bezeichnet die Gruppe von Motorproteinen in
eukaryontischen Zellen. Neben Aufgaben beim intrazellulären Transport von Vesikeln und
Zellorganellen ist Myosin ein wesentlicher Bestandteil des kontraktilen Apparates. Durch ein
Ineinanderschieben von Myosin- und Aktinfilamenten wird die Muskelkontraktion bewirkt.
Die Struktur des Myosins beinhaltet 2 schwere Ketten und 4 leichte Ketten. Es gibt zwei
Arten von leichten Ketten: essentielle leichte Ketten, die rein strukturelle Aufgaben
übernehmen und die regulatorischen leichten Ketten. Die leichte Kette 3 des Myosins wird
zu den regulatorischen Ketten gezählt. Sie binden an die schweren Ketten und können dort
die Myosinköpfchen während der Muskelkontraktion regulieren. Mutationen in der MYL3
führen zu hypertrophen Kardiomyopathien [Poetter et al. 1996, Richard et al. 2003]. In der
hier durchgeführten differentiellen Studie wurde das Protein MYL3 im Vergleich zu dem
Kontrollgewebe als geringer abundantes Protein in den Patientenkontrollen identifiziert.
Anderson et al. beschrieben 2012 eine neue Mutation in MYL3 in einem Patienten mit
hypertropher Kardiomyopathie, die mit einer verringerten Expression des Proteins
einhergeht [Andersen et al. 2012]. In der differentiellen 2D Studie von Lam et al. hingegen
konnte die MYL3 nicht als gering exprimiertes Protein identifiziert werden [Lam et al. 2010].
In Bezug auf MFM lässt sich sagen, dass bei diversen Patienten kardiale Beeinträchtigungen
beobachtet werden konnten. So zeigten Patienten mit der p.R120G Mutation in alpha B
Crystallin ebenfalls eine hypertrophe Kardiomyopathie [Vicart et al. 1998]. Auch bei
Patienten mit Desminopathien und Filaminopathien traten Komplikationen in Form von
Kardioyopathien auf [Goldfarb et al. 2004, Kley et al. 2007]. Die Ursache für die kardiale
Beteiligung bei MFM-Patienten sind nicht Mutationen im MYL3-Gen. Es wäre dennoch
interessant zu klären, inwieweit die Patienten der verschiedenen MFM-Subtypen eine
Mutation im MYL3-Gen verfügen und ob das Protein im aggregat-freien Gewebe differentiell

Diskussion
143
exprimiert vorliegt. Es wäre möglich, dass Mutationen im MYL3-Gen die Ausprägung der
kardialen Symptome beeinflusst.
Zusammenfassend lässt sich sagen, dass sich die Hälfte der hier durchgeführten 2D
Studie identifizierten Proteine in einen Kontext mit den MFM oder im speziellen mit der
Filaminopathie gebracht werden konnten. Für zukünftige Studien ist es wichtig, dass die
Proteine zu validieren und deren genaue Bedeutung in der Pathogenese zu klären. Auffällig
ist, dass 4 von 6 Proteine (CK, MYL3, Desmin und Aktin) in der Literatur mit kardialen
Symptomen in Verbindung gesetzt werden. Es scheint als könnte die kardiale Komponente in
Patienten mit Filaminopathie deutlich dominanter sein als bislang vermutet.
5.2.2.4. Die biologische Relevanz der Desminphosphorylierung
Die reversible Phosphorylierung von Proteinen spielt in der Regulation vieler
Prozesse, wie z.B. Metabolismus, Transduktion, Translation und Proteindegradation eine
entscheidende Rolle. Im Skelettmuskel ist die Phosphorylierung an Signalkaskaden beteiligt,
die den Substrat- und Energiemetabolismus, die Muskelkontraktion sowie die Muskelmasse
regulieren [Schiaffino et al. 2007]. Das generelle Verhältnis von Serin-, Threonin- und
Tyrosin-Phosphorylierungen liegt bei 1800:200:1. Es ist allerdings davon auszugehen, dass
das Verhältnis Gewebe-spezifische Unterschiede aufweist. So liegt in Skelettmuskel das
Verhältnis der Serin-, Threonin- und Tyrosin-Phosphorlylierungen bei 18:4:1 [Hojlund et al.
2009]. Im Skelettmuskel sind zwischen 2- und 5-fach so viele Threonine phosphoryliert wie in
anderen Geweben, so beträgt das Verhältnis z.B. in der Leber 90:9:1 [Han et al. 2008]. Das
Sarkomer bildet die wichtigste Untereinheit im Skelettmuskel. Durch eine optimierte
Anreicherung über Ionenaustauscher konnten Holjund et al. in allen wichtigen Komponenten
des Skelettmuskels Phosphorylierungen nachweisen. So konnten u.a. 26 verschiedene
Phosphorylierungsorte in Z-Scheiben assoziierten Proteinen, darunter befanden sich auch
Desmin, Filamin C, ZASP und Myotilin, identifiziert werden. Z-Scheiben-assoziierten
Proteinen, M-Banden Proteinen sowie Intermediärfilamente (Desmin) übernehmen wichtige
Rollen beim Zusammenbau, der Organisation und in der Funktion des kontraktilen
Apparates. In mehreren Arbeiten konnte gezeigt werden, dass reversible
Phosphorylierungen in zentralen Zellprozessen eine wichtige Rolle zu spielen scheinen
[Kawajiiri et al. 2003, Frank et al. 2006, Schiaffino et al. 2007]. Mutationen in für Z-Scheiben

Diskussion
144
codierenden Genen bilden die Ursache für erbliche Kardiomyopathien [Frank et al. 2006],
Muskeldystrophien [Cardamone et al. 2008] und Myopathien [u.a. Vorgerd et al. 2005, Kley
et al. 2007, Goldfarb et al. 2009]. Es ist wahrscheinlich, dass die Mutationen in den
jeweiligen Proteinen auch zu abnormalen Phosphorylierungen führen könnten. Die
Charakterisierung der Rolle von Phosphorylierungen in gesundem und krankem
Muskelgewebe könnte ein entscheidender Schritt zur Aufklärung der Pathomechnismen in
verschiedenen Muskelerkrankungen sein. Im Zuge der differentiellen Proteomanalyse von
Filaminopathiegewebe wurde in der vorliegenden Arbeit Desmin in zwei differentiellen Spots
sowie in zwei nicht regulierten Spots identifiziert (beschrieben unter Punkt 4.4.3.). Die Spots
befanden sich in einer Perlenketten-artigen Anordnung, wobei die regulierten Spots einen pI
Shift ins Saure aufwiesen. Da aber keine Änderungen des Molekulargewichts zu erkennen
waren, wurde die Phosphorylierung als wahrscheinlichste posttranslationale Modifikation im
Desmin als Ursache erachtet, welche als einzige einen solchen pI Shift zu verursachen.
Desmin ist das höchst abundante Intermediärfilament im menschlichen Skelettmuskel. Die
Hauptaufgabe der Intermediärfilamente liegt in der Aufrechterhaltung der Organisation des
Zytoplasmas [Lazarides 1982]. Durch die Verbindung verschiedener Membranstrukturen mit
den Z-Scheiben wird die Integration und Koordination des Zellwachstums der Myozyten
gewährleistet [Capetanaki et al. 2007]. Die Struktur des Desmins besteht aus 3 strukturellen
Komponenten: einer zentralen alpha-helikalen Stabregion, die von einem nicht alpha-
helikalen N-Terminus (head) und einem nicht helikalen C-Terminus (tail) flankiert wird
[Geißler and Weber 1982, Stuurman et al. 1998]. Durch das Zusammenlagern von zwei
Monomeren entlang der Stabregion entstehen die sogenannten Protofilamente. Aus zwei
Protofilamenten entstehen die Protofibrillen und vier Protofibrillen bilden letztendlich ein
Intermediärfilament. Der N-Terminus spielt bei der Polymerisation der einzelnen Filamente
und der Stabilität eine entscheidende Rolle [Nelson and Traub 1983, Kaufmann et al. 1985].
Kaufmann et al. konnte 1985 zeigen, dass ein Entfernen des N-Terminus von Desmin die
Polymerisation verhindert. Die Rolle des C-Terminus ist dagegen weniger klar definiert.
Während einige Studien davon ausgehen, dass das Fehlen einer intakten C-terminalen
Domäne keinen Einfluss auf den Ausbau des filamentären Netzwerkes hat [Kaufmann et al.
1985], schreiben andere der Domäne eine Aufgabe in der Kontrolle beim Zusammenbau der
Filamente zu [Birkenberger et al. 1990]. Der Prozess der reversiblen Phosphorylierung

Diskussion
145
reguliert im Desmin die (De-)Polymerisierung der Filamente sowie die Muskeldifferenzierung
und Mitose [Inagaki et al. 1988], der N-, wie auch der C-Terminus verfügen über
Phosphorylierungsstellen [Evans 1988].
5.2.2.5. Die Relevanz der Desminphosphorylierung in Filaminopathien
Die reversible Phosphorylierung des Desmins übernimmt eine wichtige Aufgabe in
der Organisation der Myofibrille, sie essentiell für den Ausbau des filamentären Netzwerkes
und für die Zellteilung. Im Zuge der Zellteilung depolymerisieren die Intermediärfilamente zu
Monomeren [Kawajiiri et al. 2003]. Während des gesamten Prozesses liegt Desmin in seiner
phosphorylierten Form vor [Geisler and Weber 1988, Inagaki et al. 1988]. Durch die
Dephosphorylierung vereinigen sich die einzelnen Monomere erneut zu
Intermediärfilamenten, so dass das filamentäre Netzwerk erneut ausgebaut werden kann. Es
ist zu vermuten, dass ein Ungleichgewicht in der Phosphorylierung und der
Dephosphorylierung zum Verlust des stabilisierenden Netzwerkes innerhalb der Muskelzelle
führt. Die MFM charakterisieren sich durch die fokale Zerstörung der Myofibrillen [Selcen et
al. 2004]. Ein zu hoher Anteil an phosphoryliertem Desmin könnte das Auflösen der
Myofibrillen begünstigen bzw. beschleunigen [Caron and Chapon 1999]. Diese These konnte
erst kürzlich in einer 2012 veröffentlichten Studie bekräftigt werden [Cohen et al. 2012]. In
der Studie wurde die Zusammensetzung der Skelettmuskulatur von Ratten nach
Nahrungsentzug untersucht. Hierbei wurde deutlich, dass als Folge der Unterernährung die
Phosphorylierung des Desmins zunahm und die Desorganisation der Filamente einsetzte.
Dieses Ergebnis steht im Einklang mit früheren Studien, die den Zusammenhang zwischen
Phosphorylierung und Demontage der Filamente zeigten [Chou et al. 1989, Sihag et al.
2007]. Allerdings wurde in diesen Studien die reversible Depolymerisation durch
Phosphorylierung als Folge zellulärer Prozesse (z.B. Mitose, Zelldifferenzierung, zellulärer
Proteintransport] beschrieben. Dephosphorylierte Filamente neigen zu einer
Wiedervereinigung, so dass kein Schaden für die Zelle entsteht [Geisler and Weber 1988,
Inagaki et al. 1988]. Im Gegensatz dazu zeigt die Studie von 2012, dass langanhaltende
Phosphorylierung die Struktur der Desminfilamente nachhaltig stört. Cohen et al. bieten
zwei Hypothesen für nachfolgende Mechanismen an. Zum einen werden die
phoshporylierten Desminfilamente ubiquitiniliert und anschließend degradiert. Die zweite

Diskussion
146
Hypothese besagt, dass die Modifizierung des Intermediärfilaments den myofibrillären
Untergang fördert. Die Desminfilamente spielen eine essentielle Rolle in der Stabilität der Z-
Scheiben und der anliegenden dünnen Filamente [Fuseler and Shay 1982, Conover et al.
2009]. Beim Verlust der Desminfilamente verliert das gesamte System an Stabilität und die
einzelnen Filamente werden zugänglicher für Ubiquitinilierungen. Dieser Mechanismus
könnte auch im Falle der in der Arbeit untersuchten Filaminopathie zum Tragen kommen.
Um die Hypothesen von Cohen et al. auf ihre Richtigkeit hin zu überprüfen würde es sich
anbieten, Muskellysat von Filaminopathiepatienten und von Negativkontrollen mittels 2D
PAGE aufzutrennen und im anschließenden Immunoblot mittels spezifischer Antikörper zu
überprüfen, ob die detektierten Desminspots in ubiquitinilierter Form vorliegen. Zusätzlich
zum Antikörper-basierten Nachweis von Ubiquitinilierungen könnte das Spotmuster im
Immunblot Hinweise auf mögliche Degradation des Desmins geben (siehe Punkt 5.2.2.6.).
Bislang konnte in der Literatur kein direkter Zusammenhang zwischen der
Filaminopathie und Desmin-Phosphorylierungen hergestellt werden. 1988 wurde von
Rappaport et al. das erste Mal von phosphoryliertem Desmin in erblichen Myopathien
berichtet. Auch in der zytoplasmatischen Body Myopathie (CBM) und der Desmin-
verwandten Myopathie (DRM) konnten 1999 phosphorylierte Isoformen von Desmin
identifiziert werden [Caron and Chapon 1999]. In beiden Fällen wurden die
Hyperphosphorylierung des Desmins mittels 2D PAGE nach O’Farrell [O’Farrell 1975] und
Western Blot charakterisiert. Als Erklärung für die Hyperphosphorylierung des Desmin in
CBM und DRM sowie die Akkumulation von Proteinen in anderen Myopathien wurde eine
gestörte Interaktion des Intermediärfilaments mit dem Chaperon alpha B Crystallin, welches
zur Stabilität der Intermediärfilamente beitragt [Bennardini et al. 1992], in Betracht gezogen.
Generell lässt sich vermuten, dass Störungen im Phosphorylierungsprozess diverser Proteine
zu erblich bedingten Myopathien führen. Dies konnte in Arbeiten belegt werden. 2005
beschrieben Clemen et al. die Hyperphosphorylierung von Hsp27, welche als spezifisches
Merkmal von Myopathien mit Desmin-Mutationen anzusehen ist. Ähnliche Ergebnisse
konnten bei der Analyse von verschiedenen alpha B Crystallin-Mutanten (R120G, Q151X,
464delCT) erzielt werden. Die Phosphorylierung dreier Mutanten in alpha B Crystallin
könnten Teil des Aggregierungsprozesses in Desminopathien sein [den Engelsman et al.
2005, Simon et al. 2007]. Ebenfalls 2007 konnten in Desmino- und Myotilinopathien weitere

Diskussion
147
posttranslationale Modifizierungen in Desmin beobachtet werden. Janue et al. untersuchten
5 Myotilinopathie-, und 3 Desminopathienpatienten. Sie konnten zeigen, dass Desmin in
diesen beiden Subtypen der Myofibrillären Myopathien oxidiert und nitriert vorliegt. Im
Vergleich zu gesundem Gewebe konnten signifikante Expressionsänderungen festgestellt
werden [Janue et al. 2007]. In zukünftigen Studien wäre es wichtig und interessant zu klären,
ob die Funktion von Desmin durch weitere posttranslationale Modifikationen (z.B.
Nitrierung, Oxidation, Ubiquitinilierung) in Filaminopathiepatienten beeinflusst wird.
5.2.2.6. Die Detektion der Phosphorylierung in Desmin
In der hier vorliegenden Dissertation wurde eine potentielle Phosphorylierung mittels
spezifischer Antikörper nachgewiesen. Für alle O-Phosphate gibt es spezifische Antikörper:
anti-Phospho-Serin (z.B. PSR-45, 3C171), anti-Phospho-Threonin (42H4) und anti-Phospho-
Tyrosin (z.B. 4G10, PY99). Desmin liegt stets phosphoryliert in der Muskelzelle vor [Costa et
al. 2004], dabei werden meist Serin-Reste im N-Terminus phosphoryliert [Geißler and Weber
1988, Inagaki et al. 1988, Cohen et al. 2012] Allerdings liegen nicht alle Serinreste komplett
phosphoryliert vor [Geisler and Weber 1988]. Zur Detektion der vermuteten Serin-
Phosphorylierung wurde ein spezifischer Phospho-Serin-Antikörpers (PSR-45) verwendet. Die
Phosphorylierung in Filaminopathiegewebe konnte eindeutig in allen Desminspots
nachgewiesen werden (beschrieben unter Punkt 4.4.3.). Auffällig war jedoch, dass mit einem
spezifischen Antikörper gegen Desmin zusätzliche als Desmin identifizierte Spots detektiert
wurden, die in der 2D Studie nicht als differentielle Spots detektiert werden konnten (Abb.
4.10., Spots a-d). Die mit a und d gekennzeichneten Spots konnten in Filaminopathie- und
Kontrollgewebe detektiert werden, im kranken Gewebe auch phosphoryliert. Die Spots (b)
und (c) wurden jedoch nur in Filaminopathiegewebe detektiert und könnten spezifisch für
Filaminopathiegewebe sein. Die Identifizierung und die Bedeutung der zusätzlichen Desmin-
Spots muss in weiteren Studien untersucht werden. Möglicherweise können andere
posttranslationale Modifikationen (höheres Molekulargewicht) oder Abbauprodukte des
Desmins (niedrigeres Molekulargewicht) identifiziert werden. Um mögliche Abbauprozesse
des Desmins näher untersuchen zu können, würde es sich anbieten Muskellysat aus
Filaminopathie- und Kontrollgewebe in einem 1D Gel aufzutrennen und die Banden mittels
eines spezifischen Antikörpers gegen Desmin sichtbar machen. Ein direkter Vergleich der

Diskussion
148
entstandenen Bandemuster könnte durch zusätzlich Banden im Filaminopathiegewebe erste
Hinweise darauf geben, ob Abbauprodukte des Desmins vorliegen. Ein weiterer Hinweis
wäre der Nachweis von ubiquitiniliertem Desmin in krankem Gewebe, da fehlgefaltete oder
nicht funktionstüchtige Proteine über eine Ubiquitinilierung für den Abbau im Proteasom-
System vorbereitet werden. Zusätzlich wäre es sinnvoll spezifische Antikörper gegen Tyrosin
und Threoin einzusetzen, um weitere posttranslationale Modifikationen im Desmin zu
detektieren. Alternativ wäre es möglich, die Spots a-d aus einem 2D Gel auszuschneiden und
mittels quantitativer Phosphoprotemics auf weitere potentielle posttranslationale
Modifikation zu analysieren. Hierfür wäre es jedoch unerlässlich die modifizierten
Aminosäuren anzureichern, um diese erfolgreich detektieren und lokalisieren zu können
(beschrieben in Punkt 5.2.2.7).
Die Detektion phosphorylierter Proteine mittels Western Blot ist eine hoch sensitive
Methode und erlaubt die Visualisierung von gering abundanten Proteinen. Dies könnte auch
der Grund dafür sein, dass die Spots a-d in der 2D Studie unentdeckt blieben. Der hier
durchgeführte 2D Western Blot eignet sich nicht zur Quantifizierung von Proteinen. Es kann
jeweils nur eine Probe auf ein Gel aufgetragen werden. Dies macht es schwierig die
verschiedenen Gele bzw. Membranen miteinander zu vergleichen. Eine quantitative
Auswertung von Immunoblots ist unter der Verwendung densitometrischer Methoden
dennoch möglich. Hier ist es jedoch nur sinnvoll ein 1D Gel als Versuchsgrundlage zu wählen.
Die zu vergleichenden Proben können zusammen auf ein Gel aufgetragen und verglichen
werden [Janue et al. 2007]. Die Verwendung eines Antikörpers für ein nicht reguliertes
Protein gewährt, dass für alle Proben die gleichen Mengen aufgetragen wurden. Alternativ
zur Detektion phosphorylierter Aminosäuren mittels Antikörper stehen weitere Methoden
zur Wahl.
5.2.2.7. Lokalisation von Phosphorylierungen in der Aminosäuresequenz
Die Phosphorylierung des Desmins in Filaminopathiegewebe konnte eindeutig
nachgewiesen werden (beschrieben unter Punkt 4.4.3). Wie unter Punkt 5.2.2.4. bereits
beschrieben, besteht Desmin aus drei strukturellen Komponenten. Besonders der N-
terminalen Region wird eine essentielle Rolle in der Aufrechterhaltung der Stabilität des
filamentären Netzwerkes zugeschrieben [Sihag et al. 2007]. Um die Lokalisation der mittels

Diskussion
149
Antikörper identifizierten Phosphorylierung von Desmin zu klären, wurde die LTQ (Thermo)
verwendet. Zur Aufklärung der patho-physiologischen Bedeutung der Phosphorylierung ist
es sehr wichtig die genaue(n) Phosphorylierungstellen zu lokalisieren. Die im Medizinischen
Proteom-Center Bochum verfügbare LTQ bietet die Möglichkeiten die Ionen mittels
Kollisionsdissoziation (CID) sowie Elektronentransferdissoziation (ETD) zu fragmentieren. Die
CID-Methode ist die am häufigsten angewandte Fragmentierungsmethode und führt in der
Analyse von Phosphoserin zu einem neutralen Verlust der HPO3 oder H2PO4-Gruppe (80 Da).
Die Fragmentierung mittels ETD ist schonender, labile Phosphorylierungen bleiben dabei
intakt. [Wiesner et al. 2008]. Um bestmögliche Voraussetzung für die Identifizierung und
Lokalisation der Serin-Phosphorylierung im Desmin zu gewähren, wurde eine Kombination
aus CID und ETD angewendet. In bereits veröffentlichten Studien konnte gezeigt werden,
dass die Kombination der beiden Fragmentierungsarten die Wahrscheinlichkeit, die
Phosphorylierung lokalisieren zu können, deutlich steigern konnte [Molina et al. 2007,
Tekirian et al. 2007, Lopez et al. 2011, Helling et al 2012]. Trotz der kombinierten
Fragmentierungen konnten in dem Filaminopathiegewebe die Serinphosphorylierung
detektiert nicht lokalisiert werden.
Die Voraussetzung für eine erfolgreiche Identifizierung der Phosphorylierung in
Filaminopathiegewebe ist eine ausreichende Menge an phosphorylierten Aminosäuren. Für
die generelle Identifizierung von Proteinen reichen in der Regel wenige beliebige Peptide aus
um diese eindeutig einem Protein zuordnen zu können. Für die Lokalisation einer
Phosphorylierung hingegen ist es notwendig die Sequenz zu detektieren, die die
Modifizierung enthält. Neben der kompletten Detektion der Sequenz ist die Abundanz der
Phosphorylierung entscheidend. Im Rahmen der hier vorliegenden Dissertation ist es nicht
gelungen die Phosphorylierung des Desmins zu lokalisieren. In der Regel können extreme
Konzentrationsunterschiede zwischen phosphorylierten und nicht-phosphorylierten
Proteinen beobachtet werden. Neben der sehr geringen Abundanz erschwert die
schlechtere Ionisationseffizienz phosphorylierter Proteine die Detektion mittels
Massenspektrometrie [Craig et al. 1994]. Die Charakterisierung von Phosphopeptiden kann
durch die Reduzierung der Komplexität der Proben und die Anreicherung der
Phosphoproteine bedeutend verbessert werden [Dunn et al. 2010]. In der Literatur sind
verschiedene Methoden beschrieben, um die Komplexität der Proben erfolgreich zu

Diskussion
150
reduzieren, u.a. 1D Gelelektrophorese mit anschließender Anfärbung von
Phosphorylierungen [Black et al. 2007], Ionenaustauschchromatographie [Gruhler et al 2005]
und Isoelektrische Fokussierung [Beranova-Giorgianni et al. 2006 und 2009, Chen et al.
2010]. Um die Phosphorylierung im Filaminopathiegewebe zu lokalisieren wäre es sinnvoll
die Komplexität des Muskellysates mittels Isoelektrischer Fokussierung und anschließender
2D PAGE-Auftrennung zu reduzieren, das zugehörige Protokoll wurde im Rahmen dieser
Dissertation etabliert. Für die erfolgreiche Anreicherung von Phosphopeptiden bieten sich
verschiedene Methoden an. Alle derzeitigen Methoden zur Anreicherung von
Phosphopeptiden basieren auf der Metallaffinitätschromatographie, die sich die Anziehung
verschiedener Metallionen oder Oxide zum Sauerstoffatom der Phosphatgruppe zunutze
macht. Die immobilisierte Metallaffinitätschromatographie (IMAC) stellt die am weitesten
verbreitete Methode zur Anreicherung von Phosphoproteinen dar. Das Prinzip der Methode
basiert auf der Affinität der Phosphatgruppe zu Metallionen, Fe3+-, Al3+, Ga3+, oder Co2+ Ionen
[Neville et al. 1997, Posewitz et al. 1999, Lopez et al. 2011]. Ein großer Nachteil der Methode
ist die Anreicherung von nicht phosphorylierten, negativ geladenen Peptide. Diese lassen
sich in der massenspektrometrischen Analyse besser ionisieren und können so die Signale
der phosphorylierten Peptide unterdrücken. Eine Weiterentwicklung des IMAC ist
Sequential Elution from IMAC (SIMAC) [Thingholm et al. 2009]. SIMAC ist eine schnelle und
einfache Methode, die gleichzeitig eine Verbesserung in der großflächigen Analyse von
mono- und multiphosphorylierten Peptiden aus hoch komplexen Proben darstellt. Auch hier
mindert die Anreicherung von nicht phosphorylierten Peptiden die Spezifität der Methoden.
Trotz der beschriebenen Schwächen konnte SIMAC in vielen Studien zur globalen
Phosphoanalytik angewendet werden [Lopez et al. 2011, Goldstrohm et al. 2011]. Die
Metalloxidaffinitätschromatographie (MOAC) sowie eine zugehörige 2D-LC-Strategie wurde
von Pinske et al. 2004 beschrieben. Am häufigsten wird eine Titanium-Oxid (TiO2)-Säule zur
Anreicherung verwendet [Gerrits and Bodenmiller 2010], welche die negativ geladenen
Phosphopeptide in wässriger Lösung bindet. Mit Hilfe des MOAC ist es möglich, sehr effektiv
einfach phosphorylierte Peptide anzureichern. Ein weiterer Vorteil ist, dass die Zahl der
Arbeitsschritte im Vergleich zu anderen Methoden reduziert wird und so der Verlust an
modifizierten Aminosäuren und Informationen verringert wird. Als nachteilig ist
anzumerken, dass die Elution multi-phosphorylierter Peptide mit einem hohen Aufwand

Diskussion
151
verbunden ist, da diese sehr stark an die Säule binden (Bahk et al. 2013, Review]. Zusätzlich
ist die Menge an nicht phosphorylierten Peptiden durch die Online-Messung relativ hoch.
Die Entwicklung einer Offline-Methode unter Verwendung der TiO2-Säule konnte die
Sensitivität und Spezifität der Anreicherung deutlich steigern [Jensen and Larsen 2007]. Die
Anreicherung TiO2 wurde bereits in vielen Studien erfolgreich angewendet [Helling et al
2012, Richardson et al. 2013]. Aufgrund der 2D-Kompatibilität und der verminderten
Arbeitsschritte im Vergleich zu anderen Methoden würde ich die Anreichung der
Phosphopeptide in Filaminopathiegewebe mittels TiO2 in zukünftigen Studien als Methode
der Wahl wählen. Die Anreicherung mittels Ionenaustausch-(Kation und Anion)
Chromatographie wurde bereits erfolgreich zur Anreicherung von Phosphoproteinen im
Skelettmuskel angewendet [Holjund et al. 2009]. Die Proteine wurden mittels tryptischen In-
Lösung-Verdau in Peptide gespalten und anschließend angereichert. Die Methode basiert
auf der negativ geladenen Phosphatgruppe PO4-. Unter der Verwendung von
Kationenaustauschern (SCX) werden positiv geladene Analyten auf eine negativ geladenen
stationäre Phase gegeben, bei Anionenaustauschern (SAX) sind die Ladungen genau
entgegensetzt. Die Kombination von SCX und IMAC erwies sich als erfolgreich in der
Identifizierung tausender phosphorylierter Peptide in komplexen biologischen Proben
[Nühse et al. 2003, Gruhler et al .2005]. Diese Art der Anreicherung wäre als Alternative zur
TiO2-Anreicherung denkbar.
Mit Hilfe der hier beschrieben Methoden zur Anreicherung von phosphorylierten
Aminosäuren in Kombination mit der massenspektrometrischen Analyse (Fragmentierung
durch CID/ETD) sollte es in Zukunft möglich sein, die Phosphorylierung im
Filaminopathiegewebe eindeutig zu lokalisieren.

Diskussion
152
5.3. Proteomstudien im Zellkulturmodell
Die Zellkultur ist ein leicht zugängliches System um Stoffwechselwege,
Signaltransduktion sowie die Proteinexpression zu untersuchen. Die Einteilung der
Zellkulturen erfolgt zum einen nach Ursprung der Zellen und zum anderen nach Art des
Wachstums. Als primäre Zelllinien (z.B. humane Skelettmuskelzellen, Hskm) werden Zellen
bezeichnet, die direkt aus dem Gewebe der Patienten entnommen und anschließend in
Kultur gebracht werden. Zu Beginn der Kultivierung ähneln diese Zellen noch am meisten der
in vivo Situation. Allerdings zeigen die Primärzellen nur eine begrenzte Teilungsfähigkeit und
sind nur über wenige Tage bis Wochen kultivierbar. In den hier durchgeführten Studien
wurden C2C12-Zellen als Zellmodell verwendet, welche zur Gruppe der immortalisierten
Zellen zählen [Yaffe et al. 1977, Blau et al. 1985]. Sie sind nahezu unbegrenzt teilbar. Ein
weiterer Vorteil gegenüber Primärzellen ist die große Robustheit, die das Kultivieren deutlich
vereinfacht. C2C12-Zellen gehören zur Gruppe der adhärenten Zelllinien, sie wachsen an
inerten Oberflächen und bilden einen einschichtigen Zellrasen (Monolayer). Im Gegensatz
dazu werden andere Zelllinien (z.B. Blutzellen oder Stammzellen) als Suspension im Medium
kultiviert. C2C12-Zellen gelten als ausgezeichnetes Modell zur Aufklärung der
Pathomechnismen in Filaminopathien und wurden bereits mehrfach in Transfektionsstudien
erfolgreich eingesetzt um das Verhalten mutierter Proteine im Skelettmuskel zu untersuchen
[Duff et al., 2011. Kley et al. 2012].
Ziel dieses Teils der Doktorarbeit war es, die C2C12-Zellen als geeignetes
Modellsystem für die Filaminopathie zu evaluieren und den Einfluss mutierter Proteine auf
die murinen Muskelzellen über einen Zeitspanne von bis zu 72h zu untersuchen. Um dies zu
erreichen wurde im Vorfeld der Arbeiten der Wildtyp wie auch die p.W2710X-Mutante des
humanen Filamin C in ihrer gesamten Länge (ca. 8,1 kb) in 2 Vektoren kloniert, die entweder
am N- oder am C-Terminus mit EGFP fusioniert waren. Beide Konstrukte wurden in die Zellen
eingebracht. Die Gesamtgröße des N3-, und C2-Konstrukts lag bei 13 kb. Die Transfektion
von derart großen Konstrukten ist erfahrungsgemäß problematisch und äußert sich in einer
geringeren Transfektionseffizienz. Dieser Effekt konnte auch in den hier durchgeführten
Studien beobachtet werden. Die Zellen, die mit isolierter Plasmid-DNA des Wildtyps oder der
Mutante transfiziert wurden, wiesen eine Transfektionseffizienz von ca. 30 % auf. Dieser
Wert ist aufgrund des extrem großen Konstrukts als sehr hoch anzusehen und entsprach den

Diskussion
153
Erwartungen aus vorangegangen Transfektionsstudien [Kley et al. 2012]. Als Kontrolle wurde
der Leervektor (4,7 kb) in die Zellen eingebracht, hier lag die Transfektionseffizienz bei ca.
70%. In den hier durchgeführten Transfektionsstudien konnten keine eindeutigen
Unterschiede der beiden transfizierten Konstrukte in Bezug auf Stabilität und
Transfektionsunterschiede beobachtet werden. Aufgrund einer Transfektionsstudie aus dem
Jahr 2012 ist allerdings anzunehmen, dass C212-Zellen nach Transfektion des C2-Konstruktes
deutlich mehr intrazelluläre Aggregate nach 24h bildeten als nach Transfektion des N3-
Konstruktes [Kley et al. 2012]. Im Rahmen der von Kley et al. durchgeführten
Transfektionsstudie wurden mehrere tausend Zellen systematisch ausgewertet.
Für die Label-freie Proteomanalyse wurden die C2C12-Zellen transient mit isolierter
Plasmid-DNA des Wildtyps, der Mutante p.W2710X und des Leervektors transfiziert. Um im
Vorfeld der Analysen auszuschließen, dass die Transfektion der pEGFP-Konstrukte zur
Aggregatbildung in Zellen führt, wurden die Zellen mit dem Leervektor, dem Filamin C-
Wildtyp und der p.W2710X-Mutante transfiziert. Im Einklang mit vorangegangen
Transfektionsstudien konnte gezeigt werden, dass lediglich die Zellen Aggregate ausbilden,
die mit der p.W2710X transfiziert wurden (beschrieben unter 4.4.1.). In den hier
beobachteten C2C12-Zellen, die mit Plasmid-DNA des Leervektors oder des Wildtyp
transfiziert wurden, konnten keine aggregat-ähnliche Strukturen nachgewiesen werden. Die
Zellen wurden nach 24h, 48h und 72h mittels LMD und analog zum humanen Protokoll
analysiert. Anstelle von 250 000 µm2 wurde aber eine Fläche von 75 000 µm2 Zellmaterial
(vollständige Zellen mit Aggregaten) ausgeschnitten. In Vorstudien hatte sich diese Fläche als
optimal für die Analysen erwiesen. Viele der identifizierten Proteine wurden auch in den
humanen Aggregaten identifiziert, darunter das Z-Scheiben assoziierte Protein Filamin C, die
Intermediärfilamente Vimentin und Desmin, dessen Interaktionspartner alpha B Crystallin,
Strukturproteine Tropomyosin und Aktin sowie diverse Hitzeschockproteine (Hsp 70, 71, 90).
Der erst kürzlich von uns charakterisierte neue Bindungspartner von Filamin C und
Bestandteil der humanen Aggregate, Xirp2, konnte nicht in den Aggregaten ausfindig
gemacht werden [Kley et al. 2012]. Es wäre möglich, dass diese Proteine erst nach >72h von
den murinen Myoblasten exprimiert werden. Dieses Ergebnis würde für ein Fortführen der
Zeitreihe (96h, 120h, usw.) sprechen. Allerdings muss erwähnt werden, dass nach
Ausdifferenzieren der Zellen das Konstrukt in den Myotuben abgebaut wird und so die Zahl

Diskussion
154
der transfizierten Zellen drastisch sinkt. Dieses Wissen beruht auf Transfektionsstudien, die
im Rahmen dieses Projektes durchgeführt wurden und deckt sich mit den Beobachtungen
der Transfektionseffizienz nach 72h. Die Zahl an aggregathaltigen Zellen verringerte sich
bereits nach 72h von 30 % auf ca. 20%. Es ist zu erwarten, dass sich die Zahl der
aggregathaltigen Zellen nach 96h, 120h usw. weiter verringert. Es könnte sich schwierig
gestalten genügend Zellen mit Aggregaten nach mehr als 72h zu isolieren. Um dennoch die
Zeitreihe ausbauen und testen zu können, ob sich weitere Komponenten der humanen
Aggregate, z.B. die Bindungspartner des Filamin C, würde es sich anbieten
Immunhistochemische Färbungen mit spezifischen Antikörpern in transfizierten Zellen
durchzuführen.
Die C2C12-Zellen wurden mit Konstrukten transfiziert, die die humane Form des
Filamin C enthielten. Filamin C wurde eindeutig als Bestandteil der Aggregate identifiziert.
Allerdings war es nicht möglich, das humane von dem murinen Filamin C zu unterscheiden.
Die Homologie der beiden Filamin C-Formen beträgt 98,4 %. Bei einer Gesamtlänge von
2725 (human) bzw. 2726 (murin) Aminosäuren unterschieden sich die beiden Formen in 45
Aminosäuren. Der theoretische Verdau der beiden Filamin C Proteine mit Trypsin (Protein
Prospector, Version 5.10.11.) zeigte, dass es möglich ist, durch die Identifizierung
spezifischer Peptide die jeweilige Form eindeutig zu bestimmen. Allerdings war es anhand
der identifizierten Peptide nicht möglich, in den durchgeführten massenspektrometrischen
Analysen eines dieser spezifischen Peptide zu identifizieren, so dass anhand der
identifizierten Peptide keine Unterschiede zwischen dem humanen und dem murinen
Filamin C detektiert werden konnten.
Ziel der Transfektionsstudien war es, analog zu den humanen Proben eine
quantitative Auswertung zu erstellen. Die daraus resultierende Proteinliste über die
Zusammensetzung der murinen Aggregate sollte dann mit den humanen Ergebnissen
verglichen werden. Im Rahmen dieser Arbeit wurde allerdings deutlich, dass eine
quantitative Auswertung der murinen Aggregatproben nicht möglich war. Die Anzahl an
identifizierten Proteinen schwankte in allen analysierten Gruppen extrem (beschrieben
unter Punkt 4.4.3.). Die Analyse der humanen Aggregatproben hingegen wies deutlich
geringere Schwankungen in der Anzahl an identifizierten Proteinen auf (Abb. 4.14.). Die
Gründe für die Unterschiede in der Zahl der identifizierten Proteine sind vielfältig. Für die

Diskussion
155
humanen Analysen wurden stets 10 µm dicke Gefrierschnitte aus Muskelbiopsien
verwendet. Dies gewährte eine gleichbleibende und vor allem vergleichbare Beschaffenheit
der Proben. Bei den Zellkulturproben konnten diese Voraussetzungen nicht geschaffen
werden. Die Zellen wurden 24h vor Transfektion ausplattiert und bei einer Konfluenz von ca.
60 – 70 % (empfohlene Konfluenz Protokoll Lipofectamin LTX, Invitrogen) transfiziert. Im
Laufe der Zeitreihe verweilten die Zellen in Proliferationsmedium und wuchsen somit weiter.
Während der LMD wurde nicht deutlich, ob und inwieweit die Zellen in mehreren Schichten
gewachsen sind. Aufgrund dieser Unsicherheit ist es möglich, an einigen Stellen deutlich
mehr Material ausgeschnitten zu haben und an anderen Stellen im Vergleich dazu deutlich
weniger. Dies würde die extremen Schwankungen erklären. Ein weiteres, generelles Problem
bei der Probensammlung ist, dass die mikrodissektierten Mengen extrem gering sind. Aus
diesem Grund ist weder bei den humanen noch bei den murinen Proben eine
Konzentrationsbestimmung durchführbar. Die homogene Beschaffenheit der humanen
Proben gewährleistet aber trotz fehlender Konzentrationsbestimmung eine vergleichbarere
Probenmenge. In nachfolgenden Experimenten mit Zellkulturmaterial müsste eine bessere
Kontrolle der Zellmenge praktiziert werden. Hierfür könnten z.B. weniger Zellen ausplattiert
werden und diese bei einer geringeren Konfluenz transfiziert werden. Eine andere
Möglichkeit wäre, die Zellen nach spätestens 48h auf Differenzierungsmedium umzustellen.
Die Umstellung des Mediums würde das Wachstum der Zellen und somit auch mögliche
Überlagerungen der Zellen zu verhindern. Bei diesem Ansatz müsste das Problem des
Plasmid-Abbaus in den Myotuben beachtet werden. Eine Testreihe müsste zeigen, ob eine
verstärkte Ausdifferenzierung der Myotuben die Stabilität der Konstrukte negativ
beeinflussen würde.
Der direkte Vergleich der humanen und murinen Proteinlisten wäre auch nach
erfolgreicher Quantifizierung mit Schwierigkeiten verbunden gewesen. Nach Transfektion
der Plasmid DNA der Mutanten bildeten sich in den C2C12-Zellen mehrere runde Aggregate.
Zusätzlich konnten punktförmige aggregat-ähnliche Strukturen beobachtet werden. Weder
im Leervektor noch im Wildtyp konnten vergleichbare Strukturen beobachtet werden
(beschrieben unter 4.4.2.). Um die Gesamtheit der Veränderungen innerhalb der C2C12-
Zellen, auch die sehr kleinen punktförmigen Strukturen, für die Analyse erfassen zu können,
mussten komplette Zellen ausgeschnitten werden. Die humanen Aggregate konnten mittels

Diskussion
156
Laser exakt aus dem umliegenden Gewebe isoliert werden. Die Unterschiede in der
Probengewinnung lassen auf Seiten der Zellkulturproben Proteine mit einfließen, die keine
direkten Komponenten der Aggregate darstellen. Es ist jedoch fraglich, ob das alleinige
Isolieren der Aggregate aus Zellkultur aufgrund ihrer sehr geringen Größe realisierbar ist. Die
Analyse der punktförmigen aggregat-ähnlichen Strukturen wäre auf jeden Fall unmöglich. In
hier durchgeführten Vorstudien konnte gezeigt werden, dass die Aggregatbildung in den
C2C12-Zellen ungefähr 18h - 24h nach Transfektion einsetzt (beschrieben unter Punkt
4.6.1.). Diese Zeitspanne könnte analog zu den nicht betroffenen Muskelzellen aus den
Filaminopathiepatienten als Frühstadium bezeichnet werden. Es wäre interessant zu
schauen, ob und welche, Proteine bzw. Proteingruppen an der Aggregatbildung maßgeblich
beteiligt sind. Hinsichtlich der punktförmigen Strukturen wären in einer zukünftigen Studie 2
Fragestellungen von Interesse: 1. Handelt es sich bei diesen Strukturen um Aggregat-
ähnliche Strukturen? und 2. vereinigen sich diese Strukturen im Zeitverlauf zu einem
größeren Aggregat?
Zusammenfassend lässt sich sagen, dass erste Schritte zur Proteomanalyse der
p.W2710X-Mutation in Zellkultur im Rahmen dieser Dissertation bewältigt werden konnten.
In nachfolgenden Studien müssen allerdings noch einige Versuchsreihen zur Optimierung
durchgeführt werden. Ein auf Zellkultur angepasstes Protokoll wäre eine optimale Grundlage
um neben der p.W2710X-Mutation auch weitere Filamin C-Mutationen auf Proteomebene
zu charakterisieren. Ein wichtiger Schritt in der Methodenoptimierung wird die Etablierung
einer stabil transfizierten Zelllinie sein. Der Nachteil von transienten Transfektionen liegt in
der geringen Lebensdauer der aufgenommenen DNA. Die Etablierung einer stabil
transfizierten Zelllinie würde die Erforschung vielfältiger Fragestellungen ermöglichen. Es
wäre möglich, die Proteinaggregate über einen längeren Zeitraum als 72h zu beobachten
und anschließend deren Zusammensetzung mit der der humanen Aggregate vergleichen. Auf
Basis der gemeinsamen Komponenten könnten gezielt therapeutische Ansätze in Zellkultur
(z.B. die Behandlung mit Aggregat-lösenden Substanzen) durchgeführt werden, um diese im
Anschluss für die Entwicklung von Therapien für die Patienten zu nutzen. Für die
Plectinopathie wie auch die desmin-verwandten Myopathien wurden bereits therapeutische
Studien im Zellkultur- bzw. Tiermodell durchgeführt. Im Falle der Plectinopathien wurden
Myocyten im Zellmodell mit 4-Phenylbutyrate, einem chemischen Chaperon, behandelt

Diskussion
157
[Yam et al. 2007]. Dies führte zu einer Verringerung der Proteinaggregate in den Zellen
(Vortrag von Lilli Winter, Wien, im Rahmen des DGNN Meeting in Erlangen 2012). Ein
ähnliches Ergebnis erzielte die Anwendung der Substanz Gernayl-Geranyl-Aceton (GGA),
welches die Expression kleiner Hitzeschockproteine veranlasst [Ooie et al. 2001]. Die R120G-
Missense-Mutation in alpha B Crystallin verursacht desmin-verwandte Kardiomyopathien
[Vicart et al. 1998]. R120G-transgene Mäuse wurden oral mit GGA behandelt. Neben der
erwarteten Hochregulierung der kleinen Hitzeschockproteine konnte zusätzlich ein Rückgang
der amyloider Aggregate beobachtet werden [Sanbe et al. 2009]. Die hier erwähnten
Substanzen zeigten im Zellkultur- bzw. Tiermodell eine Aggregat-auflösende Wirkung.
5.4. Fazit und Ausblick
In der hier vorliegenden Dissertation ist es gelungen, die LMD erfolgreich für die
Isolierung spezifischer Gewebeareale aus Filaminopathiepatienten zu etablieren. Ein
Massenspektrometrie-basierter Label-freier Ansatz wurde gewählt, um die
Zusammensetzung der Filaminopathie-spezifischen Proteinaggregate aufzuschlüsseln und
spezifische Bestandteile zu identifizieren. Die LMD erwies sich im Rahmen dieses Projektes
als äußerst erfolgreiche Methode zur Isolierung spezifischer Gewebeareale. Neben bereits
bekannten Proteinen wurden zahlreiche neue Komponenten der Proteinaggregate
aufgedeckt und validiert. Der Erfolg des Label-freien Ansatzes konnte durch die
Veröffentlichung von 2013 sowie die Übertragung auch auf einen weiteren Subtypen der
MFM, den Desminopathien, unterstrichen werden [Kley et al. 2013, Maerkens et al. 2013].
Ziel der hier durchgeführten 2D DIGE Studie war es, mögliche Marker für Frühstadien der
Filaminopathien zu identifizieren. Zusätzlich konnten 6 differentielle Proteine identifiziert
werden, die eine entscheidende Rolle im Frühstadium der Erkrankung spielen könnten. Im
Desmin wurde aufgrund der Perlenketten-artigen Anordnung der Spots eine potentielle
posttranslationale Modifikation vermutet, welche im weiteren Verlauf der Arbeiten als
Phosphorylierung validiert werden konnte. Die Phosphorylierung des Desmin könnte ein
entscheidendes Ereignis in der frühen Pathogenese der Filaminopathie sein, dass zur
Desorganisation der Myofibrillen beitragen könnte. Ob phosphoryliertes Desmin vor allem
eine Rolle im Frühstadium der Filaminopathie oder auch in der Aggregatbildung spielt,
müssen weitere Studien zeigen. Um dies zu untersuchen wäre es sinnvoll die mittels LMD

Diskussion
158
gesammelten Filaminopathie-spezifischen Proteinaggregate in 98 % Ameisensäure zu lösen,
tryptisch zu verdauen und anschließend die modifizierten Aminosäuren mittels TiO2-Säule
anzureichern. Die kombinierte Fragmentierung aus CID/ETD könnte in der
massenspektrometrischen Analyse zur Detektion potentieller posttranslationaler
Modifikationen führen. Ergänzend zu der Validierung der Desmin-Phosphorylierung müssten
die anderen als differentiell identifizierte Proteine mittels unabhängigen Methoden validiert
werden. Hier würden sich zum einen der 1D Western Blot zur Quantifizierung und zum
anderen die Immunhistochemie anbieten.
Murine Myoblasten stellten sich im Zuge diverser Transfektionsstudien als
exzellentes Modell zur Charakterisierung von Filamin C-Mutationen heraus. Die
Proteomanalyse in Zellkultur würde einen weiteren Beitrag zum besseren Verständnis der
Pathomechnismen der Filaminopathie leisten. Allerdings stellt die Etablierung der LMD von
C2C12-Zellen eine große Herausforderung dar. Das hier etablierte Protokoll für
Gewebeschnitte von humanen Filaminopathiepatienten konnte ohne weitere
Optimierungsarbeiten auf andere Subtypen der Myofibrillären Myopathien übertragen
werden. Die Übertragung des Protokolls auf die Zellkultur hingegen verlief nicht reibungslos.
Die Expression der humanen p.W2710X-Mutation führte in den C2C12-Zellen wie erwartet
zur Aggregatbildung innerhalb der Zellen, die in ihrer Zusammensetzung den humanen
Aggregaten ähnelten. Eine quantitative Analyse der Aggregatzusammensetzung war jedoch
nicht möglich, dies lässt sich vor allem vor allem durch die geringe Größe der Aggregate in
den Zellen und die schwer vergleichbare Probenbeschaffenheit erklären. Trotz dieser
Schwierigkeiten konnte gezeigt werden, dass sich die Zusammensetzung der
Proteinaggregate in Zellkultur und die der humanen Aggregate sehr ähnelt. In Zukunft wird
es wichtig sein, die Parameter zur differentiellen Proteomanalyse zu optimierten um einen
quantitativen Vergleich mit den humanen Aggregaten durchführen zu können. Auf Basis
dieses Vergleichs kann die Aggregat-lösende Wirkung diverser Substanzen ausgetestet
werden, welche die Entwicklung von Behandlungsmöglichkeiten von betroffenen Patienten
beschleunigen könnten.

159
6. Zusammenfassung
Die Filaminopathie ist ein Subtyp einer seltenen Gruppe hereditären
neuromuskulären Erkrankungen, den Myofibrillären Myopathien (MFM). Die Ursache für die
Filaminopathie liegt in Mutationen in dem für Filamin C codierenden Gen (FLNC). Die
Filaminopathie und die Myofibrillären Myopathien im Allgemeinen zeichnen sich durch die
fokale Zerstörung der Myofibrillen sowie die Bildung von intrazellulären Proteinaggregaten
in den Skelettmuskelzellen aus. Im Rahmen der vorliegenden Arbeite sollte erstmals die
Zusammensetzung der Filaminopathie-spezifischen Aggregate sowie des umliegenden
Gewebes analysiert werden, um 1. Filaminopathie-spezifische Bestandteile der Aggregate
und 2. mögliche Marker für Frühstadien zu identifizieren. Diese Daten sollten helfen, neue
Erkenntnisse bezüglich der Pathogenese der Filaminopathien zu gewinnen. Um dies zu
erreichen, wurden in dieser Arbeit zwei auf LMD basierende verschiedene proteomische
Ansätze mit drei verschiedenen Auswertestrategien etabliert. Für die Analysen wurden
mittels LMD aus Schnitten von Muskelbiopsien aus 6 Filaminopathiepatienten mit 3
unterschiedlichen Mutation und 6 gesunden Kontrollen gleich große Areale isoliert.
Die Aggregatanalyse erfolgte unter Verwendung eines Massenspektrometrie-
basierten Label-freien Ansatzes. Die Aggregate wurden aus dem Filaminopathiegewebe
isoliert und mit nicht betroffenen Muskelzellen desselben Patienten verglichen.
Anschließend erfolgte die massenspektrometrische Analyse. Im Rahmen einer qualitativen
Auswertung konnten durchschnittlich 415 Proteine in den Aggregatproben der
Filaminopathiepatienten identifiziert werden, von denen einige bereits aus früheren
Immunfluoreszenzanalysen bekannt waren. Den Großteil der identifizierten Proteine
machten jedoch unbekannte Komponenten der Aggregate aus. Zur Identifizierung von
möglichen Filaminopathie-spezifischen Proteinen in den Aggregaten, die nicht im
umliegenden Gewebe gefunden werden können, wurde eine manuell durchgeführte
quantitative Auswertestrategie hinzugezogen. Insgesamt konnten 111 Proteine als

Zusammenfassung
160
spezifische Bestandteile der Proteinaggregate identifiziert. 2 der nur in Aggregat-
vorkommenden Proteine konnten im Laufe der hier vorliegenden Arbeit validiert werden:
Xirp2 und Rab35. Xirp2 konnte erst kürzlich von uns als neuer Bindungspartner des Filamin C
charakterisiert werden. Allerdings ist Xirp2 kein spezifischer Marker für Filaminopathie, da es
auch in den Proteinaggregaten von Patienten mit Desminopathien gefunden wurde. Rab35
hingegen, dessen Rolle in der Aggregatbildung in der Filaminopathie noch ungeklärt ist,
könnte als ein spezifischer Marker der Filaminopathie dienen. Um Rab35 als eindeutigen
Marker für die Filaminopathie bestätigen zu können, müssen Aggregatanalysen anderer
MFM-Subtypen folgen. Die manuelle Auswertung ermöglichte einen sehr interessanten
Überblick über die Zusammensetzung der Proteinaggregate, ließ jedoch keine differentiellen
Aussagen zum Entwicklungsprozess der Filaminopathie zu. Mittels der weiter entwickelten
automatisierten Auswertestrategie in Form des Spectral Index Calculation konnten 69
akkumulierte und 31 niedrig abundante Proteine identifiziert werden. Die Übereinstimmung
zwischen den beiden quantitativen Auswertestrategien lag bei 38 % (38 Proteine). Beide
Auswertestrategien deckten viele neue, interessante Bestandteile der Aggregate auf, die in
zukünftigen Studien validiert werden müssen.
Zur Charakterisierung möglicher Frühstadien der Filaminopathie wurde Aggregat-
freies Gewebe aus den Patienten mittels einer Kombination aus LMD und 2D DIGE Analyse
untersucht. Insgesamt konnten 6 differentiell exprimierte Proteine identifiziert werden.
Darunter befand sich das Intermediärfilament Desmin, welches eine extrem wichtige Rolle in
der Aufrechterhaltung der zellulären Integrität übernimmt. Im Desmin wurde aufgrund der
Perlenketten-artigen Anordnung der Spots eine potentielle posttranslationale Modifikation
in Form einer Phosphorylierung vermutet. Meine Ergebnisse bestätigten diese Vermutung
und so könnte die Phosphorylierung des Desmin ein entscheidendes Ereignis in der frühen
Pathogenese der Filaminopathie darstellen. Die übermäßige Phosphorylierung des Desmins
führt zu einer Desorganisation des filamentären Netzwerks und könnte so zur Zerstörung der
Myofibrillen beitragen. Es bleibt zu klären, ob die Phosphorylierung des Desmin ein
spezifisches Merkmal für die Frühstadien der Filaminopathien darstellt. Generell scheint
Desmin in der Filaminopathie wie auch in anderen Subtypen der MFM eine Art Schlüsselrolle
in der Pathogenese zu übernehmen.

Zusammenfassung
161
In Ergänzung zu den Analysen des humanen Materials wurde in der hier vorliegenden
Dissertation ein Vergleich von murinen Myoblasten und humanem Gewebe hinsichtlich ihrer
Aggregatzusammensetzung angestrebt. Die p.W2710X-Mutation wurde in C2C12-Zellen
transfiziert. Die intrazelluläre Proteinaggregation, die in Muskelfasern von Filaminopathie-
patienten stattfindet, ließ sich durch Expression der Mutante in den kultivierten Zellen
reproduzieren. Die qualitative Analyse der murinen Zellaggregate zeigte, dass viele der
identifizierten Proteine auch als Bestandteile der humanen Aggregate detektiert werden
konnten. Eine quantitative Auswertung konnte aufgrund von stark schwankenden Zahlen an
identifizierten Proteinen nicht durchgeführt werden. Es bedarf aber noch einiger
Optimierungsarbeiten, um diese erfolgreich durchzuführen. Dennoch konnte in dieser
Dissertation gezeigt werden, dass die C2C21-Zellen ein geeignetes Modellsystem für
proteomische Studien darstellen.

162
7. Summary
The filaminopathy is a subtype of a rare group of hereditary neuromuscular diseases,
the myofibrilliar myopathies (MFM). The filaminopathies are caused by several mutations in
the gene coding for Filamin C (FLNC). The filaminopathy and myofibrilliar myopathies in
general are characterized by focal destruction of myofibrils and the formation of intracellular
protein aggregates in skeletal muscle cells. The most important aim of this work was to
elucidate the composition of the specific aggregates and the surrounding tissue in order to
identify 1. filaminopathy-specific components of the aggregates and 2 possible markers for
early stages of the disease. These data should help to gain new insights into the
pathogenesis of filaminopathies. Two different proteomic approaches, based on
lasermicrodissection, in combination with three different evaluation strategies were
established. The tissue of 6 filaminopathy patients with 3 different mutation and 6 healthy
controls was isolates by lasermicrodissection. The composition of the aggregates was
analyzed by a mass spectrometry analysis based label-free approach. The aggregates were
isolated from the muscle biopsies and compared to non-affected muscle of the same
patient. In a quantitative evaluation 415 proteins were identified in the aggregate, some of
which were already known from previous immunofluorescence analysis. However, the
majority of the proteins had not been identified as components of the aggregates. To
identify possible specific proteins which are specific markers for the aggregation process in
filaminopathy and could not be found in the surrounding tissue, a manually performed
quantitative evaluation strategy was consulted. A total of 111 proteins were identified as
specific components of protein aggregates. Two of these accumulated proteins in aggregates
could be validated: Xirp2 and Rab35. Recently we could characterize Xirp2 as a new binding
partner of the filamin C recently. However it is not a specific marker for filaminopathies
because of its presence in aggregates in patients with desminopathy. Whereas Rab35, whose
role in the formation of aggregates in filaminopathies is still unknown, might be considered

Summary
163
as a specific marker for filaminopathies. In order to confirm Rab35 as a unique marker for
filaminopathies, further studies of aggregates must follow. Manual analysis facilitated a very
interesting overview of the composition of the protein aggregates, but left no differential
statements to the development process of filaminopathy. By means of advanced automated
evaluation 69 accumulated and 31 low abundant proteins could be identified. The
quantitative agreement between the two evaluation strategies were (38 proteins) at 38%.
Both evaluation strategies revealed many new and interesting components of the
aggregates, which need to be validated in future studies.
To characterize early stages of aggregate-free tissue from patients with filaminopathy was
analyzed by a combination of LMD and 2D DIGE analysis. A total of 6 differentially expressed
proteins were identified. Among them we found the intermediate filament desmin, which
assumes an extremely important role in the maintenance of cellular integrity. In desmin a
potential post-translational modification has been suggested in the form of phosphorylation
due to the pearl chain-like arrangement of the spots. My results confirmed this assumption
and thus the phosphorylation of desmin could be a critical event in the pathogenesis of early
filaminopathy. An increased amount of phosphorylated desmin leads to disorganization of
the filamentous network and could thus contribute to the destruction of myofibrils. It
remains to be determined whether the phosphorylation of desmin represents a specific
feature of the early stages of filaminopathies. Generally desmin seems to play a key roll in
filaminopathies as well as in other subtypes of MFM. In addition to the analysis of human
muscle biopsies, a comparison of murine myoblasts and human tissue was intended in terms
of their aggregate composition. The p.W2710X mutation was transfected into C2C12 cells.
The intracellular protein aggregation that occurs in muscle fibers of patients with
filaminopathy was reproduced by expression of the mutant in the cultured cells. The
qualitative analysis of the murine cell aggregates showed that many of the identified
proteins could also be identified in the human aggregates. A quantitative analysis was not
performed due to fluctuating Pay nan identified proteins. It still requires some optimization
work to carry out the quantitative comparison successfully. Nevertheless, in this thesis it was
shown that the C2C21 cells represent a suitable model system for proteomic studies.

164
8. Literaturverzeichnis
Aebersold R. and Mann M. (2003). Mass spectrometry-based proteomics. Nature 422, 198–
207
Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I. (2003). A novel
experimental design for comparative two-dimensional gel analysis: two-dimensional
difference gel electrophoresis incorporating a pooled internal standard. Proteomics 3: 36-44.
Aggarwal K, Choe LH, Lee KH. (2006). Shotgun proteomics using the iTRAQ isobaric tags.
Brief Funct Genomic Proteomic. 5(2):112-20.
America AH, Cordewener JH. (2008). Comparative LC-MS: a landscape of peaks and valleys.
Proteomics. 8(4):731-49.
Andersen PS, Hedley PL, Page SP, Syrris P, Moolman-Smook JC, McKenna WJ, Elliott PM,
Christiansen M. (2012). A novel Myosin essential light chain mutation causes hypertrophic
cardiomyopathy with late onset and low expressivity. Biochem Res Int. 2012:685108.
Avila-Smirnow D, Béhin A, Gueneau L, Claeys K, Beuvin M, Goudeau B, Richard P, Ben Yaou
R, Romero NB, Mathis S, Voit T, Eymard B, Gil R, Fardeau M, Bonne G. (2010) A novel
missense FLNC mutation causes arrhythmia and late onset myofibrillar myopathy with
particular histopathology features. Neuromuscul Disord 20:623-624
Bahk YY, Mohamed B, Kim YJ. (2013). Biomedical application of phosphoproteomics in
neurodegenerative diseases. J Microbiol Biotechnol. 23(3):279-88.
Bantscheff M, Boesche M, Eberhard D, Matthieson T, Sweetman G, Kuster B. (2008). Robust
and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol Cell
Proteomics.7(9):1702-13.

Literaturverzeichnis
165
Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG. (1999). Myopathy,
myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J
Neuropathol Exp Neurol 58:832-46
Bao H, Liu T, Chen X, Chen G. (2008). Efficient in-gel proteolysis accelerated by infrared
radiation for protein identification. J Proteome Res. 7(12):5339-44.
Bennardini F, Wrzosck A, Chiesi M. (1992). aB-crystallin in cardiac tissue: association with
actin and desmin filaments. Circ Res 71:288–294
Beranova-Giorgianni S, Desiderio DM, Giorgianni F. (2009). Phosphoproteome analysis by in-
gel isoelectric focusing and tandem mass spectrometry. Methods Mol Biol. 519:383-96.
Beranova-Giorgianni S, Zhao Y, Desiderio DM, Giorgianni F. (2006). Phosphoproteomic
analysis of the human pituitary. Pituitary. 9(2):109-20.
Berg, Tymoczko, Stryer: Biochemie. 5. Auflage. Spektrum Akademischer Verlag, Heidelberg
2003
Biemann K. (1990). Peptides and proteins: overview and strategy. Methods Enzymol.
193:351–60
Birkenberger L, Ip W. (1990). Properties of the desmin tail domain: studies using synthetic
peptides and antipeptide antibodies. J Cell Biol. 111(5 Pt 1):2063-75.
Bjellqvist B, Ek K, Righetti PG, Gianazza E, Görg A, Westermeier R, Postel W. (1982).
Isoelectric focusing in immobilized pH gradients: principle, methodology and some
applications. J Biochem Biophys Methods 6: 317-39.
Black TM, Andrews CL, Kilili G, Ivan M, Tsichlis PN, Vouros P. (2007). Characterization of
phosphorylation sites on Tpl2 using IMAC enrichment and a linear ion trap mass
spectrometer. J Proteome Res. 6(6):2269-76.
Blau HM, Pavlath GK, Hardeman EC, Chiu C, Silberstein L, Webster SG, Miller SC and Webster
C. (1985). Plasticity of differentiated state. Science 230:758-766

Literaturverzeichnis
166
Bornemann A, Schmalbruch H. (1993). Anti-vimentin staining in muscle pathology.
Neuropathol Appl Neurobiol 19:414-9
Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-54.
Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. (2007). Muscle intermediate
filaments and their links to membranes and membranous organelles. Exp Cell Res.
313(10):2063-76.
Capetanaki Y, Milner DJ, Weitzer G. (1997). Desmin in muscle formation and maintenance:
Knockouts and consequences. Cell Struct Funct 22:103-16.
Capetanaki Y. (2002). Desmin cytoskeleton: a potential regulator of muscle mitochondrial
behavior and function. Trends Cardiovasc Med. 12(8):339-48. Review.
Capitanio, D., Vasso, M., Fania, C., Moriggi, M., Vigano, A., Procacci, P., Magnaghi, V., and
Gelfi, C. (2009). Comparative proteomic profile of rat sciatic nerve and gastrocnemius muscle
tissues in ageing by 2-D DIGE. Proteomics. 9, 2004–2020
Cardamone M, Darras BT, Ryan MM. (2008). Inherited myopathies and muscular
dystrophies. Semin Neurol. 28:250–259.
Caron A, Chapon F. (1999). Desmin phosphorylation abnormalities in cytoplasmic body and
desmin-related myopathies. Muscle Nerve.;22(8):1122-5.
Cech NB, Enke CG., (2001). Practical implications of some recent studies in electrospray
ionization fundamentals. Mass Spectrom Rev. 20(6):362-87.
Chen L, Giorgianni F, Beranova-Giorgianni S. (2010). Characterization of the
phosphoproteome in LNCaP prostate cancer cells by in-gel isoelectric focusing and tandem
mass spectrometry. J Proteome Res.;9(1):174-8.
Chou YH, Rosevear E, Goldman RD. (1989). Phosphorylation and disassembly of intermediate
filaments in mitotic cells. Proc Natl Acad Sci U S A. 86(6):1885-9.

Literaturverzeichnis
167
Chua CE, Lim YS, Tang BL. (2010). Rab35--a vesicular traffic-regulating small GTPase with
actin modulating roles. FEBS Lett. 584(1):1-6.
Claeys KG, van der Ven PF, Behin A, Stojkovic T, Eymard B, Dubourg O, Laforêt P, Faulkner G,
Richard P, Vicart P, Romero NB, Stoltenburg G, Udd B, Fardeau M, Voit T, Fürst DO.(2009).
Differential involvement of sarcomeric proteins in myofibrillar myopathies: a morphological
and immunohistochemical study. Acta Neuropathol 117, 293–307.
Clemen CS, Fischer D, Roth U, Simon S, Vicart P, Kato K, Kaminska AM, Vorgerd M, Goldfarb
LG, Eymard B, Romero NB, Goudeau B, Eggermann T, Zerres K, Noegel AA, Schröder R.
(2005). Hsp27-2D-gel electrophoresis is a diagnostic tool to differentiate primary
desminopathies from myofibrillar myopathies. FEBS Lett. 579(17):3777-82
Clemen CS, Herrmann H, Strelkov SV, Schröder R. (2013). Desminopathies: pathology and
mechanisms. Acta Neuropathol. 125(1):47-75.
Cohen S, Zhai B, Gygi SP, Goldberg AL. (2012). Ubiquitylation by Trim32 causes coupled loss
of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol. 198(4):575-89.
Conover GM, Henderson SN, Gregorio CC. (2009). A myopathy-linked desmin mutation
perturbs striated muscle actin filament architecture. Mol Biol Cell. 20(3):834-45.
Costa ML, Escaleira R, Cataldo A, Oliveira F, Mermelstein CS. (2004). Desmin: molecular
interactions and putative functions of the muscle intermediate filament protein. Braz J Med
Biol Res. 37(12):1819-30.
Craig AG, Hoeger CA, Miller CL, Goedken T, Rivier JE, Fischer WH. (1994). Monitoring protein
kinase and phosphatase reactions with matrix-assisted laser desorption/ionization mass
spectrometry and capillary zone electrophoresis: comparison of the detection efficiency of
peptide-phosphopeptide mixtures. Biol Mass Spectrom. 23(8):519-28.
Dalkilic, I., Thompson, T. G., Brossius, M. A., Puca, A. A., Feener, C., Muntoni, F., and Kunkel,
L. M. (2002). Journal of the Neurological Sciences. 199, 82

Literaturverzeichnis
168
Den Engelsman J, Gerrits D, de Jong WW, Robbins J, Kato K, Boelens WC. (2005). Nuclear
import of {alpha}B-crystallin is phosphorylation-dependent and hampered by
hyperphosphorylation of the myopathy-related mutant R120G. J Biol Chem. 280(44):37139-
48.
Deschauer M, Morgenroth A, Joshi PR, Gläser D, Chinnery PF, Aasly J, Schreiber H, Knape M,
Zierz S, Vorgerd M. (2007). Analysis of spectrum and frequencies of mutations in McArdle
disease. Identification of 13 novel mutations. J Neurol. 254(6):797-802.
Diguet N, Mallat Y, Ladouce R, Clodic G, Prola A, Tritsch E, Blanc J, Larcher JC, Delcayre C,
Samuel JL, Friguet B, Bolbach G, Li Z, Mericskay M. (2011). Muscle creatine kinase deficiency
triggers both actin depolymerization and desmin disorganization by advanced glycation end
products in dilated cardiomyopathy. J Biol Chem. 286(40):35007-19.
Duff RM, Tay V, Hackman P, Ravenscroft G, McLean C, Kennedy P, Steinbach A, Schöffler W,
van der Ven PF, Fürst DO, Song J, Djinović-Carugo K, Penttilä S, Raheem O, Reardon K,
Malandrini A, Gambelli S, Villanova M, Nowak KJ, Williams DR, Landers JE, Brown RH Jr, Udd
B, Laing NG, (2011). Mutations in the N-terminal actin-binding domain of filamin C cause a
distal myopathy. Am J Hum Genet. 88(6):729-40
Dunn JD, Reid GE, Bruening ML. (2010). Techniques for phosphopeptide enrichment prior to
analysis by mass spectrometry. Mass Spectrom Rev. 29(1):29-54.
Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA,
Liotta LA. (1996). Laser capture microdissection. Science. 274(5289):998-1001
Evans RM. (1988). The intermediate-filament proteins vimentin and desmin are
phosphorylated in specific domains. Eur J Cell Biol. 46(1):152-60.
Faul C, Dhume A, Schecter AD, Mundel P. (2007). Protein kinase A, Ca2+/calmodulin-
dependent kinase II, and calcineurin regulate the intracellular trafficking of myopodin
between the Z-disc and the nucleus of cardiac myocytes. Mol Cell Biol. 27(23):8215-27.

Literaturverzeichnis
169
Feldkirchner S, Schessl J, Müller S, Schoser B, Hanisch FG. (2012). Patient-specific protein
aggregates in myofibrillar myopathies: laser microdissection and differential proteomics for
identification of plaque components. Proteomics. 12(23-24):3598-609.
Fenn J.B., Mann M, Meng CK, Wong and SF. Whitehouse (CM). (1989). Electrospray
ionization for mass spectrometry of large biomolecules. Science 246: 64-71.
Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, Amado F. (2010).
Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose
functional specializations in skeletal muscle. Proteomics. 10, 3142–3154
Frank D, Kuhn C, Katus HA, Frey N. (2006). The sarcomeric Z-disc: a nodal point in signalling
and disease. J Mol Med (Berl). 84(6):446-68.
Fujii K, Kondo T, Yokoo H, Okano T, Yamada M, Yamada T, Iwatsuki K, Hirohashi S. (2006).
Database of two-dimensional polyacrylamide gel electrophoresis of proteins labeled with
CyDye DIGE Fluor saturation dye. Proteomics 6, 1640-53.
Fujii K, Kondo T, Yokoo H, Yamada T, Iwatsuki K, Hirohashi S. (2005). Proteomic study of
human hepatocellular carcinoma using two-dimensional difference gel electrophoresis with
saturation cysteine dye. Proteomics. 5(5):1411-22.
Fürst DO, Goldfarb LG, Kley RA, Vorgerd M, Olivé M, van der Ven PF. (2013). Filamin C-
related myopathies: pathology and mechanisms. Acta Neuropathol. 125(1):33-46
Furuike S, Ito T, Yamazaki M. (2001). Mechanical unfolding of single filamin A (ABP-280)
molecules detected by atomic force microscopy. FEBS Lett. 1;498(1):72-5.
Fuseler JW, Shay JW. (1982). The association of desmin with the developing myofibrils of
cultured embryonic rat heart myocytes. Dev Biol. 91(2):448-57.
Geisler N, Weber K. (1982). The amino acid sequence of chicken muscle desmin provides a
common structural model for intermediate filament proteins. EMBO J. 1(12):1649-56.

Literaturverzeichnis
170
Geisler N, Weber K. (1988). Phosphorylation of desmin in vitro inhibits formation of
intermediate filaments; identification of three kinase A sites in the aminoterminal head
domain. EMBO J. 7(1):15-20.
Georgatos SD, Maison C. (1996). Integration of intermediate filaments into cellular
organelles. Int Rev Cytol 164:91-138
Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. (2003). Absolute quantification of
proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A.
100(12):6940-5.
Gerrits B, Bodenmiller B. (2010). Mapping of phosphorylation sites by LC-MS/MS. Methods
Mol Biol. 658:127-36.
Goebel HH. (1997). Desmin-related myopathies. Curr Opin Neurol 10:426-9.
Goebel HH.(1995). Desmin-related neuromuscular disorders. Muscle Nerve 18:1306-20
Goldfarb LG, Park KY, Cervenáková L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW,
Semino-Mora C, Sivakumar K, Dalakas MC. (1998). Missense mutations in desmin associated
with familial cardiac and skeletal myopathy. Nat Genet 19:402-3
Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. (2004). Desmin myopathy, Brain 127 723–
734.
Goldfarb LG, Dalakas, MC. (2009). Tragedy in a heartbeat: malfunctioning desmin causes
skeletal and cardiac muscle disease. J. Clin. Invest 119, 1806–1813
Goldman RD, Chou YH, Dessev C, Dessev G, Eriksson J, Goldman A, Khuon S, Kohnken R,
Lowy M, Miller R. (1991). Dynamic aspects of cytoskeletal and karyoskeletal intermediate
filament systems during the cell cycle. Cold Spring Harb Symp Quant Biol. 56:629-42.
Goldstrohm DA, Broeckling CD, Prenni JE, Curthoys NP. (2011). Importance of manual
validation for the identification of phosphopeptides using a linear ion trap mass
spectrometer. J Biomol Tech. 22(1):10-20.

Literaturverzeichnis
171
Görg A, Postel W, Günther S. (1988). The current state of two-dimensional electrophoresis
with immobilized pH gradients. Electrophoresis 9(9): 531-46.
Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. (1990).
Human endothelial actin-binding protein (abp-280, nonmuscle, lamin): a molecular leaf
spring.J Cell Biol,111(3), 1089{1105
Göthert M, Bischoff D, Dreyer C. (1975). The influence of inhalation anaesthetics on
catecholamine secretion from the adrenal medulla in vivo. Anaesthesist. 24, Nr. 1, S. 19–26.
Graves JD, Krebs EG (1999) Protein phosphorylation and signal transduction. Pharmacol
Ther. 82 (2-3):111-21
Gruhler A, Olsen JV, Mohammed S, Mortensen P, Faergeman NJ, Mann M, Jensen ON.
(2005). Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway.
Mol Cell Proteomics. 4(3):310-27.
Guergueltcheva V, Peeters K, Baets J, Ceuterick-de Groote C, Martin JJ, Suls A, et al. (2011).
Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency.
Neurology 77: 2105–14
Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. (1999). Quantitative analysis of
complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 17(10):994-9.
Hamacher M, Eisenacher M, Stephan C. (2011). Data mining in Proteomics – From Standards
to applications. Springer Verlag, New York
Hamacher M, Hardt T, van Hall A, Stephan C, Marcus K, Meyer HE. (2008). Inside SMP
proteomics: six years German Human Brain Proteome Project (HBPP) - a summary.
Proteomics 8: 1118-28.
Han G, Ye M, Zhou H, Jiang X, Feng S, Jiang X, Tian R, Wan D, Zou H, Gu J. (2008). Large-scale
phosphoproteome analysis of human liver tissue by enrichment and fractionation of
phosphopeptides with strong anion exchange chromatography. Proteomics. 8:1346–1361.

Literaturverzeichnis
172
Hartwig JH, Stossel TP. (1975). Isolation and properties of actin, myosin and a new
actinbinding protein in rabbit alveolar. macrophages. J Biol Chem 250:5696-705
Hartwig JH. (1992). Mechanisms of actin rearrangements mediating platelet activation. J Cell
Biol. 118(6):1421-42.
Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, Just S, Hess A, Ehlermann P,
Weichenhan D, Grimmler M, Liptau H, Hetzer R, Regitz-Zagrosek V, Fischer C, Nürnberg P,
Schunkert H, Katus HA, Rottbauer W. (2009). Nexilin mutations destabilize cardiac Z-disks
and lead to dilated cardiomyopathy. Nat Med. 15(11):1281-8.
Hazeki N, Tukamoto T, Goto J, Kanazawa I. (2000). Formic acid dissolves aggregates of an N-
terminal huntingtin fragment containing an expanded polyglutamine tract: applying to
quantification of protein components of the aggregates. Biochem Biophys Res Commun.
277(2):386-93.
Helling S, Hüttemann M, Kadenbach B, Ramzan R, Vogt S, Marcus K. (2012). Discovering the
phosphoproteome of the hydrophobic cytochrome c oxidase membrane protein complex.
Methods Mol Biol. 893:345-58.
Helling S, Schmitt E, Joppich C, Schulenborg T, Müllner S, Felske-Müller S, Wiebringhaus T,
Becker G, Linsenmann G, Sitek B, Lutter P, Meyer HE, Marcus K. (2006). 2-D differential
membrane proteome analysis of scarce protein samples. Proteomics 6: 4506-13.
Herrmann H, Aebi U. (2004). Intermediate filaments: molecular structure, assembly
mechanism, and integration into functionally distinct intracellular Scaffolds. Annu Rev
Biochem. 73:749-89.
Himmel M, Van Der Ven PF, Stöcklein W, Fürst DO. (2003). The limits ofpromiscuity: isoform-
specic dimerization of lamins. Biochemistry 42(2), 430{439
Hoe KL, Won M, Chung KS, Jang YJ, Lee SB, Kim DU, Lee JW, Yun JH, Yoo HS. (1998). Isolation
of a new member of DnaJ-like heat shock protein 40 (Hsp40) from human liver. Biochim
Biophys Acta. 1383(1):4-8.

Literaturverzeichnis
173
Højlund K, Bowen BP, Hwang H, Flynn CR, Madireddy L, Geetha T, Langlais P, Meyer C,
Mandarino LJ, Yi Z.(2009). In vivo phosphoproteome of human skeletal muscle revealed by
phosphopeptide enrichment and HPLC-ESI-MS/MS. J Proteome Res. 8(11):4954-65.
Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R. (2005). The Orbitrap: a new
mass spectrometer. J Mass Spectrom. 40(4):430-43.
Huang HT, Brand OM, Mathew M, Ignatiou C, Ewen EP, McCalmon SA, Naya FJ. (2006).
Myomaxin is a novel transcriptional target of MEF2A that encodes a Xin-related alpha-
actinin-interacting protein. J Biol Chem 281 (51): 39370–9
Inagaki M, Gonda Y, Matsuyama M, Nishizawa K, Nishi Y, Sato C. (1988). Intermediate
filament reconstitution in vitro. The role of phosphorylation on the assembly-disassembly of
desmin. J Biol Chem. 263(12):5970-8.
Iribarne JV, Thomson BA. (1976). On the evaporation of small ions from charged droplets. J.
Chem. Phys. 64, 2287
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. (2005). Exponentially
modified protein abundance index (emPAI) for estimation of absolute protein amount in
proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics.
4(9):1265-72.
Iwatsuki H, Suda M. (2010). Seven kinds of intermediate filament networks in the cytoplasm
of polarized cells: structure and function. Acta Histochem Cytochem. 43(2):19-31.
Jackman MR, Willis WT. (1996). Characteristics of mitochondria isolated from type I and type
IIb skeletal muscle. Am J Physiol. 270(2 Pt 1):C673-8.
Janué A, Olivé M, Ferrer I. (2007). Oxidative stress in desminopathies and myotilinopathies:
a link between oxidative damage and abnormal protein aggregation. Brain Pathol. 17(4):377-
88.
Jensen SS, Larsen MR. (2007). Evaluation of the impact of some experimental procedures on
different phosphopeptide enrichment techniques. Rapid Commun Mass Spectrom.
21(22):3635-45.

Literaturverzeichnis
174
Karas M, Glückmann M, Schäfer J., (2000). Ionization in matrix-assisted laser
desorption/ionization: singly charged molecular ions are the lucky survivors. J Mass
Spectrom. 35(1):1-12.
Karas, M., Hillenkamp F. (1988). Laser desorption ionization of proteins with molecular
masses exceeding 10,000 daltons. Anal Chem 60: 2299-301.
Kaufmann E, Weber K, Geisler N.(1985). Intermediate filament forming ability of desmin
derivatives lacking either the amino-terminal 67 or the carboxy-terminal 27 residues. J Mol
Biol. 185(4):733-42.
Kaufmann H, Bailey JE, Fussenegger M., (2005). Use of antibodies for detection of
phosphorylated proteins separated by two-dimensional gel electrophoresis. Proteomics.
1(2):194-9.
Kawajiri A, Yasui Y, Goto H, Tatsuka M, Takahashi M, Nagata K, Inagaki M. (2003). Functional
significance of the specific sites phosphorylated in desmin at cleavage furrow: Aurora-B may
phosphorylate and regulate type III intermediate filaments during cytokinesis coordinatedly
with Rho-kinase. Mol Biol Cell. 14(4):1489-500.
Kim S, Coulombe PA. (2007). Intermediate filament scaffolds fulfill mechanical,
organizational, and signaling functions in the cytoplasm. Genes Dev. 21(13):1581-97. Review.
Kley RA, Hellenbroich Y, van der Ven PF, Fürst DO, Huebner A, Bruchertseifer V, Peters SA,
Heyer CM, Kirschner J, Schröder R, Fischer D, Müller K, Tolksdorf K, Eger K, Germing A,
Brodherr T, Reum C, Walter MC, Lochmüller H, Ketelsen UP, Vorgerd M. (2007). Clinical and
morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain.
130(Pt 12):3250-64.
Kley RA, Maerkens A, Leber Y, Theis V, Schreiner A, van der Ven PF, Uszkoreit J, Stephan C,
Eulitz S, Euler N, Kirschner J, Müller K, Meyer HE, Tegenthoff M, Fürst DO, Vorgerd M, Müller
T, Marcus K, (Epub 2012, 2013). A combined laser microdissection and mass spectrometry
approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy
patients. Mol Cell Proteomics. 12(1):215-27

Literaturverzeichnis
175
Kley RA, Serdaroglu-Oflazer P, Leber Y, Odgerel Z, van der Ven PF, Olivé M, Ferrer I, Onipe A,
Mihaylov M, Bilbao JM, Lee HS, Höhfeld J, Djinović-Carugo K, Kong K, Tegenthoff M, Peters
SA, Stenzel W, Vorgerd M, Goldfarb LG, Fürst DO. (2012). Pathophysiology of protein
aggregation and extended phenotyping in filaminopathy. Brain. 135(Pt 9):2642-60.
Kley RA, van der Ven PF, Olivé M, Höhfeld J, Goldfarb LG, Fürst DO, Vorgerd M. (2013).
Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C
mutations. Autophagy. 9(3):422-3
Klose, J. (1975). Protein mapping by combined isoelectric focusing and electrophoresis of
mouse tissues. A novel approach to testing for induced point mutations in mammals.
Humangenetik 26: 231-43.
Klose, J. (2009). From 2-D electrophoresis to proteomics. Electrophoresis 30 Suppl 1: S142-9.
Klose, J., U. Kobalz (1995). Two-dimensional electrophoresis of proteins: an updated
protocol and implications for a functional analysis of the genome. Electrophoresis 16: 1034-
59.
Klunk WE, Xu CJ, Pettegrew JW. (1994). NMR identification of the formic acid-modified
residue in Alzheimer's amyloid protein. J Neurochem. 62(1):349-54.
Kondo T. (2008). Tissue proteomics for cancer biomarker development: laser
microdissection and 2D-DIGE. BMB Rep. 41(9):626-34.
Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, Plasterk RH,
Heck AJ. (2003). Metabolic labeling of C. elegans and D. melanogaster for quantitative
proteomics. Nat Biotechnol. 21(8):927-31.
Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler
R, Mann M. (2008). SILAC mouse for quantitative proteomics uncovers kindlin-3 as an
essential factor for red blood cell function. Cell. 134(2):353-64.
Kwiatkowski DJ. (1999). Functions of gelsolin: motility, signaling, apoptosis, cancer. Curr
Opin Cell Biol. 11(1):103-8.

Literaturverzeichnis
176
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature 227: 680-5.
Lai X, Wang L, Witzmann FA. (2013). Issues and applications in label-free quantitative mass
spectrometry. Int J Proteomics2013:756039.
Lam L, Tsoutsman T, Arthur J, Semsarian C. (2010). Differential protein expression profiling
of myocardial tissue in a mouse model of hypertrophic cardiomyopathy. J Mol Cell Cardiol.
48(5):1014-22.
Langenfeld E, Meyer HE, Marcus K. (2008). Quantitative analysis of highly homologous
proteins: the challenge of assaying the "CYP-ome" by mass spectrometry. Anal Bioanal
Chem. 392(6):1123-34.
Lazarides E. (1980). Intermediate filaments as mechanical integrators of cellular space.
Nature. 283(5744):249-256.
Lazarides E. (1982). Intermediate filaments: a chemically heterogeneous, developmentally
regulated class of proteins. Annu Rev Biochem 51:219-50.
Lei JX, Cassone CG, Luebbert C, Liu QY. (2011). A novel neuron-enriched protein SDIM1 is
down regulated in Alzheimer's brains and attenuates cell death induced by DNAJB4 over-
expression in neuro-progenitor cells. Mol Neurodegener;6(1):9.
Levin J, Bulst S, Thirion C, Schmidt F, Bötzel K, Krause S, Pertl C, Kretzschmar H, Walter MC,
Giese A, Lochmüller H. (2010). Divergent molecular effects of desmin mutations on protein
assembly in myofibrillar myopathy. J Neuropathol Exp Neurol. 69(4):415-24.
Lewis, C., Carberry, S., Ohlendieck, K. (2009). Proteomic profiling of x-linked muscular
dystrophy. J.Muscle Res.Cell Motil. 30, 267–269
Lill J. (2003). Proteomic tools for quantitation by mass spectrometry. Mass Spectrom Rev.
22(3):182-94.

Literaturverzeichnis
177
Linnemann A, van der Ven PF, Vakeel P, Albinus B, Simonis D, Bendas G, Schenk JA, Micheel
B, Kley RA, Fürst DO. (2010). The sarcomeric Z-disc component myopodin is a multiadapter
protein that interacts with filamin and alpha-actinin. Eur J Cell Biol 89(9):681-92.
Liu H, Sadygov RG, Yates JR 3rd. (2004). A model for random sampling and estimation of
relative protein abundance in shotgun proteomics. Anal Chem. 76(14):4193-201.
López E, López I, Ferreira A, Sequí J. (2011). Clinical and technical phosphoproteomic
research. Proteome Sci. 2011 Jun 2;9:27.
Lottspeich, F. , Engels, J. Bioanalytik Elsevier GmbH, München, 2006.
Löwe T, Kley RA, van der Ven PF, Himmel M, Huebner A, Vorgerd M, Fürst DO.(2007). The
pathomechanism of filaminopathy: altered biochemical properties explain the cellular
phenotype of a protein aggregation myopathy. Hum Mol Genet 16: 1351–8.
Lu P, Vogel C, Wang R, Yao X, Marcotte EM. (2007). Absolute protein expression profiling
estimates the relative contributions of transcriptional and translational regulation. Nat
Biotechnol. 25(1):117-24.
Lu S, Carroll SL, Herrera AH, Ozanne B, Horowits R. (2003). New N-RAP-binding partners
alpha-actinin, filamin and Krp1 detected by yeast two-hybrid screening: implications for
myofibril assembly. J Cell Sci 116(Pt 11):2169-78.
Luan X, Hong D, Zhang W, Wang Z, Yuan Y. (2010). A novel heterozygous deletion-insertion
mutation (2695-2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy
in a large Chinese family. Neuromuscul Disord 20: 390–6.
Maerkens A, Kley RA, Olivé M, Theis V, van der Ven PF, Reimann J, Milting H, Schreiner A,
Uszkoreit J, Eisenacher M, Barkovits K, Güttsches AK, Tonillo J, Kuhlmann K, Meyer HE,
Schröder R, Tegenthoff M, Fürst DO, Müller T, Goldfarb LG, Vorgerd M, Marcus K. (2013).
Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J
Proteomics. 2013 Apr 29
Maerkens A. Masterarbeit. Analyse von Proteinaggregaten bei Myofibrilliären Myopathien

Literaturverzeichnis
178
Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, Strupat K, Horning S (2006).
Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem.
78 (7): 2113–20.
Makarov A, Denisov E, Lange O, Horning S (2006). Dynamic range of mass accuracy in LTQ
Orbitrap hybrid mass spectrometer". J. Am. Soc. Mass Spectrom. 17 (7): 977–82.
Makarov A. (2000). Electrostatic axially harmonic orbital trapping: a high-performance
technique of mass analysis. Anal Chem. 72(6):1156-62.
Mann M, Hendrickson RC and Pandey A. (2001). Analysis of proteins and proteomes by mass
spectrometry. Annu Rev Biochem 70: 437-73.
Marcus, K., Joppich C, May C, Pfeiffer K, Sitek B, Meyer HE, Stuehler K (2009). High-
resolution 2DE. Methods Mol Biol 519: 221-40.
Marouga R, David S, Hawkins E. (2005). The development of the DIGE system: 2D
fluorescence difference gel analysis technology. Anal Bioanal Chem 382: 669-78
May C, Brosseron F, Pfeiffer K, Meyer HE, Marcus K., (2012). Proteome analysis with classical
2D PAGE. Methods Mol Biol. 893:37-46.
McArdle (1951). Myopathy due to a defect in muscle glycogen breakdown. Clinical Science,
10: 13-33.
McGough AM, Staiger CJ, Min JK, Simonetti KD. (2003). The Gelsolin family factin regulatory
Proteins: modular structures,versatile functions.FEBSLett 552:7581
Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. (2007). Global proteomic profiling of
phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl
Acad Sci U S A. 104:2199–2204.
Mueller LN, Brusniak MY, Mani DR, Aebersold R. (2008). An assessment of software
solutions for the analysis of mass spectrometry based quantitative proteomics data. J
Proteome Res. 7(1):51-61.

Literaturverzeichnis
179
Müller T, Loosse C, Schrötter A, Schnabel A, Helling S, Egensperger R, Marcus K. (2011). The
AICD interacting protein DAB1 is up-regulated in Alzheimer frontal cortex brain samples and
causes deregulation of proteins involved in gene expression changes. Curr Alzheimer Res 8,
573– 582
Müller T, Schrötter A, Loosse C, Helling S, Stephan C, Ahrens M, Uszkoreit J, Eisenacher M,
Meyer HE, Marcus K. (2011). Sense and nonsense of pathway analysis software in
proteomics. J.Proteome.Res 10, 5398–5408 .
Nakamura F, Stossel TP, Hartwig JH. (2011). The filamins: organizers of cell structure and
function. Cell Adh Migr. 5(2):160-9. Epub 2011 Mar 1.
Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter
SC, Haynes PA. (2011). Less label, more free: approaches in label-free quantitative mass
spectrometry. Proteomics. 11(4):535-53.
Nelson WJ, Traub P. (1983). Proteolysis of vimentin and desmin by the Ca2+-activated
proteinase specific for these intermediate filament proteins. Mol Cell Biol. 3(6):1146-56.
Nesterenko MV., M. Tilley, S. J. Upton (1994). A simple modification of Blum's silver stain
method allows for 30 minute detection of proteins in polyacrylamide gels. J Biochem Biophys
Methods 28: 239-42.
Neville DC, Rozanas CR, Price EM, Gruis DB, Verkman AS, Townsend RR. (1997). Evidence for
phosphorylation of serine 753 in CFTR using a novel metalion affinity resin and matrix-
assisted laser desorption mass spectrometry. Protein Sci 6(11):2436-45.
Niwa F, Shiga K, Kimura M, Yamaguchi T, Kondo M, Nakagawa M. (2009). Adult-onset
nemaline myopathy with distal muscle atrophy--case report. Brain Nerve. 61(6):695-9.
Nühse TS, Stensballe A, Jensen ON, Peck SC. (2003). Large-scale analysis of in vivo
phosphorylated membrane proteins by immobilized metal ion affinity chromatography and
mass spectrometry. Mol Cell Proteomics. 2(11):1234-43. Epub 2003 Sep 22.

Literaturverzeichnis
180
O'Farrell PH. (1975). High resolution two-dimensional electrophoresis of proteins.. J Biol
Chem 250: 4007-21.
Ohlendieck K. (2011). Skeletal muscle proteomics: current approaches, technical challenges
and emerging techniques. Skelet.Muscle 1(1):6.
Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. (1999). The small GTPase RalA targets
filamin to induce filopodia. Proc Natl Acad Sci U S A. 96(5):2122-8.
Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA,
Ahn NG. (2005). Comparison of label-free methods for quantifying human proteins by
shotgun proteomics. Mol Cell Proteomics. 4(10):1487-502.
Olivé M, Odgerel Z, Martínez A, Poza JJ, Bragado FG, Zabalza RJ, Jericó I, Gonzalez-Mera L,
Shatunov A, Lee HS, Armstrong J, Maraví E, Arroyo MR, Pascual-Calvet J, Navarro C, Paradas
C, Huerta M, Marquez F, Rivas EG, Pou A, Ferrer I, Goldfarb LG. (2011). Clinical and
myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy.
Neuromuscul Disord. 21(8):533-42.
Olivé, M., Goldfarb, L. G., Shatunov, A., Fischer, D. & Ferrer, I. (2005). Myotilinopathy:
refining the clinical and myopathological phenotype. Brain 128, 2315–2326.
Ong SE, Mann M. (2005). Mass spectrometry-based proteomics turns quantitative. Nat
Chem Biol. 1(5):252-62.
Ooie T, Takahashi N, Saikawa T, Nawata T, Arikawa M, Yamanaka K, Hara M, Shimada T,
Sakata T. (2001). Single oral dose of geranylgeranylacetone induces heat-shock protein 72
and renders protection against ischemia/reperfusion injury in rat heart. Circulation
104(15):1837-43.
Pacholsky D, Vakeel P, Himmel M, Löwe T, Stradal T, Rottner K, Fürst DO, van der Ven PF.
(2004). Xin repeats define a novel actin-binding motif. J Cell Sci. 117(Pt 22):5257-68.
Palma, S. de, Morandi, L., Mariani, E., Begum, S., Cerretelli, P., Wait, R., and Gelfi, C. (2006).
Proteomic investigation of the molecular pathophysiology of dysferlinopathy. Proteomics. 6,
379–385

Literaturverzeichnis
181
Pandey, A., M. Mann (2000). Proteomics to study genes and genomes. Nature 405: 837-46
Patra AK, Mukhopadhyay R, Mukhija R, Krishnan A, Garg LC, Panda AK. (2000). Optimization
of inclusion body solubilization and renaturation of recombinant human growth hormone
from Escherichia coli. Protein Expr Purif. 2000 Mar;18(2):182-92.
Patterson SD, Aebersold RH. (2003). Proteomics: the first decade and beyond. Nat Genet 33
Suppl: 311-23
Paul W. (1989). Electromagnetic Traps for Charged and Neutral Particles (Nobel Lecture).
Angewandte Chemie International Edition in English 29: 739-748
Paul W, Raether M. (1995). Das elektrische Massenfilter. Zeitschrift für Physik A Hadrons and
Nuclei 140:262-273
Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. (1999). Probability-based protein
identification by searching sequence databases using mass spectrometry data.
Electrophoresis 20: 3551-67.
Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. (2004). Selective isolation at the
femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS
and titanium oxide precolumns. Anal Chem. 76(14):3935-43.
Poetter K., Jiang H., Hassanzadeh S., Master S.R., Chang A., Dalakas M.C., Rayment I., Sellers
J.R., Fananapazir L., Epstein N.D. (1996). Mutations in either the essential or regulatory light
chains of myosin are associated with a rare myopathy in human heart and skeletal muscle.
Nat. Genet. 13:63-69
Pollard, T. D., Almo, S., Quirk, S., Vinson, V., Lattman, E. E. (1994). Structure, of actin binding
proteins: insights about function at atomic resolution.Annu Rev Cell Biol,10, 207{249
Poon E, Howman EV, Newey SE, Davies KE. (2002). Association of syncoilin and desmin:
linking intermediate filament proteins to the dystrophin-associated protein complex. J Biol
Chem. 277(5):3433-9.

Literaturverzeichnis
182
Poschmann G, Sitek B, Sipos B, Ulrich A, Wiese S, Stephan C, Warscheid B, Klöppel G, Vander
Borght A, Ramaekers FC, Meyer HE, Stühler K. (2009). Identification of proteomic differences
between squamous cell carcinoma of the lung and bronchial epithelium.Mol.Cell Proteomics.
8, 1105–1116
Posewitz MC, Tempst P. (1999). Immobilized gallium(III) affinity chromatography of
phosphopeptides. Anal Chem 71(14):2883-92.
Pulkkinen L, Smith FJ, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WH,
Uitto J. (1996). Homozygous deletion mutations in the plectin gene (PLEC1) in patients with
epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol
Genet. 5(10):1539-46.
Qiu XB, Shao YM, Miao S, Wang L.(2006). The diversity of the DnaJ/Hsp40 family, the crucial
partners for Hsp70 chaperones. Cell Mol Life Sci 63:2560-2570.
Quinzii CM, Vu TH, Min KC, Tanji K, Barral S, Grewal RP, Kattah A, Camaño P, Otaegui D,
Kunimatsu T, Blake DM, Wilhelmsen KC, Rowland LP, Hays AP, Bonilla E, Hirano M. (2008). X-
linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-
and-a-half-LIM protein 1. Am J Hum Genet. 82(1):208-13.
Rabilloud T. (2012). The whereabouts of 2D gels in quantitative proteomics. Methods Mol
Biol. 893:25-35.
Rabilloud T. (2002). Two-dimensional gel electrophoresis in proteomics: old, old fashioned,
but it still climbs up the mountains. Proteomics 2: 3-10.
Rappaport L, Contard F, Samuel JL, Delcayre C, Marotte F, Tome´ F, Fardeau M. (1988).
Storage of phosphorylated desmin in a familial myopathy. FEBS Lett 1988;231:421–425.
Reidegeld KA, Eisenacher M, Kohl M, Chamrad D, Körting G, Blüggel M, Meyer HE, Stephan
C. (2008). An easy-to-use Decoy Database Builder software tool, implementing different
decoy strategies for false discovery rate calculation in automated MS/MS protein
identifications. Proteomics. 8(6):1129-37.

Literaturverzeichnis
183
Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G. (1999). Association of
mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res
252:479-91
Rezniczeck GA, Walko G, Wiche G. (2009). Plectin defects lead to various forms of
epidermolysis bullosa simplex. Dermatol Clin 28:33– 41
Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R.,
Dubourg O., Burban M., Gueffet J.-P., Millaire A., Desnos M., Schwartz K., Hainque B.,
Komajda M. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum
of mutations, and implications for a molecular diagnosis strategy. Circulation 107:2227-2232
Richardson BM, Soderblom EJ, Thompson JW, Moseley MA. (2013). Automated,
reproducible, titania-based phosphopeptide enrichment strategy for label-free quantitative
phosphoproteomics. J Biomol Tech. 24(1):8-16.
Roepstorff P, Fohlman J. (1984). Proposal for a common nomenclature for sequence ions in
mass spectra of peptides. Biomed. Mass Spectrom 11, 601
Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey
S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ.
(2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive
isobaric tagging reagents. Mol Cell Proteomics. 2004 Dec;3(12):1154-69.
Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, Yamauchi J, Tanonaka K, Glabe C, Tanoue
A. (2009). Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in
desmin-related cardiomyopathy. PLoS One. 4(4):e5351.
Santoni V, Kieffer S, Desclaux D, Masson F, Rabilloud T. (2000). Membrane proteomics: use
of additive main effects with multiplicative interaction model to classify plasma membrane
proteins according to their solubility and electrophoretic properties. Electrophoresis.
21(16):3329-44.

Literaturverzeichnis
184
Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM,
Mahjneh I, Vihola A, Raheem O, Penttilä S, Lehtinen S, Huovinen S, Palmio J, Tasca G, Ricci E,
Hackman P, Hauser M, Katsanis N, Udd B. (2012). Mutations affecting the cytoplasmic
functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet.
44(4):450-5, S1-2.
Schwerzmann K, Hoppeler H, Kayar SR, Weibel ER. (1989). Oxidative capacity of muscle and
mitochondria: correlation of physiological, biochemical, and morphometric characteristics.
Proc Natl Acad Sci U S A. 86(5):1583-7.
Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, Sewry C, Battini R, Hu Y, Cottle
DL, Rosenblatt M, Spruce L, Ganguly A, Kirschner J, Judkins AR, Golden JA, Goebel HH,
Muntoni F, Flanigan KM, Mitchell CA, Bönnemann CG.. (2008). Proteomic identification of
FHL1 as the protein mutated in human reducing body myopathy. J.Clin.Invest 118, 904–912
Schiaffino S, Reggiani C. (2011). Review Fiber types in mammalian skeletal muscles. Physiol
Rev. 91(4):1447-531.
Schiaffino S, Sandri M, Murgia M. (2007). Activity-dependent signaling pathways controlling
muscle diversity and plasticity. Physiology (Bethesda) 22:269–278.
Schiebler Anatomie 9.Auflage 2005
Schlattner U, Tokarska-Schlattner M, Wallimann T. (2006). Mitochondrial creatine kinase in
human health and disease. Biochim. Biophys. Acta. 1762, Nr. 2, S. 164–180.
Schmidt A, Kellermann J, Lottspeich F. (2005). A novel strategy for quantitative proteomics
using isotope-coded protein labels. Proteomics. 5(1):4-15.
Schmidt, Thews. Physiologie des Menschen 29. Auflage 2005
Schroder S, Zhang H, Yeung ES, Jansch L, Zabel C, Watzig H. (2008). Quantitative gel
electrophoresis: sources of variation. J Proteome Res 7: 1226-34.

Literaturverzeichnis
185
Schröder R, Kunz WS, Rouan F, Pfendner E, Tolksdorf K, Kappes-Horn K, Altenschmidt-
Mehring M, Knoblich R, van der Ven PF, Reimann J, Fürst DO, Blümcke I, Vielhaber S, Zillikens
D, Eming S, Klockgether T, Uitto J, Wiche G, Rolfs A. (2002). Disorganization of the desmin
cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex
with muscular dystrophy. J Neuropathol Exp Neurol. 61(6):520-30.
Schröder R, Schoser B. (2009). Myofibrillar myopathies: a clinical and myopathological guide.
Brain Pathol. 19(3):483-92
Scigelova M, Makarov A. (2006). Orbitrap mass analyzer--overview and applications in
proteomics. Proteomics. Suppl 2:16-21.
Selcen D. (2011). Myofibrillar myopathies. Neuromuscul Disord. 21(3):161-71.
Selcen D, Engel AG. (2005). Mutations in ZASP define a novel form of muscular dystrophy in
humans. Ann Neurol. 57(2):269-76.
Selcen D, Ohno K, Engel AG. (2004). Myofibrillar myopathy: clinical, mor-phological and
genetic studies in 63 patients. Brain 127, 439–451
Shatunov A, Olivé M, Odgerel Z, Stadelmann-Nessler C, Irlbacher K, van Landeghem F,
Bayarsaikhan M, Lee HS, Goudeau B, Chinnery PF, Straub V, Hilton-Jones D, Damian MS,
Kaminska A, Vicart P, Bushby K, Dalakas MC, Sambuughin N, Ferrer I, Goebel HH, Goldfarb
LG. (2009). In-frame deletion in the seventh immunoglobulin-like repeat of filamin C in a
family with myofibrillar myopathy. Eur.J.Hum.Genet 17, 656–663 .
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P,
Shevchenko A, Boucheriie H, Mann M. (1996). Linking genome and proteome by mass
spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc.
Natl. Acad. Sci. USA, (93): 14440-14445
Shy GM, Engel WK, Somers JE, Wanko T. Nemaline Myopathy. (1963). A New Congenital
Myopathy. Brain. 86:793-810.

Literaturverzeichnis
186
Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. (2007). Role of phosphorylation on the
structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res.
313(10):2098-109.
Simon S, Fontaine JM, Martin JL, Sun X, Hoppe AD, Welsh MJ, Benndorf R, Vicart P. (2007).
Myopathy-associated alphaB-crystallin mutants: abnormal phosphorylation, intracellular
location, and interactions with other small heat shock proteins. J Biol Chem. 282(47):34276-
87.
Sitek B, Lüttges J, Marcus K, Klöppel G, Schmiegel W, Meyer HE, Hahn SA, Stühler K. (2005).
Application of fluorescence difference gel electrophoresis saturation labelling for the
analysis of microdissected precursor lesions of pancreatic ductal adenocarcinoma.
Proteomics. 5(10):2665-79.
Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes
JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH,
Lane EB. (1996). Plectin deficiency results in muscular dystrophy with epidermolysis bullosa.
Nat Genet. 13(4):450-7.
Sparrow JC, Nowak KJ, Durling HJ, Beggs AH, Wallgren-Pettersson C, Romero N, Nonaka I,
Laing NG (2003). Muscle disease caused by mutations in the skeletal muscle alpha-actin gene
(ACTA1). Neuromuscul. Disord. 13 (7–8): 519–31.
Staes A, Demol H, Van Damme J, Martens L, Vandekerckhove J, Gevaert K. (2004). Global
differential non-gel proteomics by quantitative and stable labeling of tryptic peptides with
oxygen-18. J Proteome Res. 3(4):786-91.
Steen H, Mann M. (2004). The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell
Biol 5, 699–711.
Steinbock FA, Wiche G. (1999). Plectin: A cytolinker by design. Biol Chem 1999;380:151-8.

Literaturverzeichnis
187
Steinert PM, Chou YH, Prahlad V, Parry DA, Marekov LN, Wu KC, Jang SI, Goldman RD.
(1999). A high molecular weight intermediate filament-associated protein in BHK-21 cells is
nestin, a type VI intermediate filament protein. Limited co-assembly in vitro to form
heteropolymers with type III vimentin and type IV alpha-internexin. J Biol Chem.
274(14):9881-90.
Stensballe A, Jensen ON, Olsen JV, Haselmann KF, Zubarev RA. (2000). Electron capture
dissociation of singly and multiply phosphorylated peptides. Rapid Commun Mass Spectrom
14(19):1793-800.
Stewart M. (1990). Intermediate filaments: structure, assembly and molecular interactions.
Curr Opin Cell Biol. 2(1):91-100. Review.
Stuurman N, Heins S, Aebi U. (1998). Nuclear lamins: Their structure assembly, and
interactions. J Struct Biol 122:42-66
Sun HQ, Yamamoto M, Mejillano M, Yin HL (1999). Gelsolin, a multifunctional actin
regulatory protein. J. Biol. Chem. 274 (47): 33179–82.
Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. (2004). Peptide and protein
sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U
S A. 101(26):9528-33.
Tasca G, Odgerel Z, Monforte M, Aurino S, Clarke NF, Waddell LB, Udd B, Ricci E, Goldfarb
LG. (2012). Novel FLNC mutation in a patient with myofibrillar myopathy in combination with
late-onset cerebellar ataxia. Muscle Nerve. 46(2):275-82
Tekirian TL, Thomas SN, Yang A. (2007). Advancing signaling networks through proteomics.
Expert Rev Proteomics. 4(4):573-83.
Thingholm TE, Jensen ON, Larsen MR. (2009). Enrichment and separation of mono- and
multiply phosphorylated peptides using sequential elution from IMAC prior to mass
spectrometric analysis. Methods Mol Biol 2009, 527:67-78.

Literaturverzeichnis
188
Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R,
Mohammed AK, Hamon C. (2003). Tandem mass tags: a novel quantification strategy for
comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 75(8):1895-904.
Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S,
Kunkel LM. (2000). Filamin 2 ( FLNC2): A muscle-specificc sarcoglycan interacting protein. J
Cell iol,148(1), 115{126
Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I,
Davison M. (2001). Validation and development of fluorescence two-dimensional differential
gel electrophoresis proteomics technology. Proteomics. 2001 Mar;1(3):377-96.
Tsai MF, Wang CC, Chang GC, Chen CY, Chen HY, Cheng CL, Yang YP, Wu CY, Shih FY, Liu CC,
Lin HP, Jou YS, Lin SC, Lin CW, Chen WJ, Chan WK, Chen JJ, Yang PC. (2006). A new tumor
suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell
lung carcinoma. J Natl Cancer Inst 98:825-838.
Tsujino S, Shanske S, DiMauro S. (1993). Molecular genetic heterogeneity of
myophosphorylase deficiency (McArdle's disease). N Engl J Med. 329(4):241-5.
Tsumoto K, Umetsu M, Kumagai I, Ejima D, Arakawa T. (2003). Solubilization of active green
fluorescent protein from insoluble particles by guanidine and arginine. Biochem Biophys Res
Commun. 312(4):1383-6.
Ünlü M, Morgan ME, Minden JS. (1997). Difference gel electrophoresis: a single gel method
for detecting changes in protein extracts. Electrophoresis 18: 2071-7.
van der Ven PF, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Fürst DO. (2006).
Unusual splicing events result in distinct Xin isoforms that associate differentially with
filamin c and Mena/VASP. Exp Cell Res. 312(11):2154-67.
van der Ven PF, Obermann WM, Lemke B, Gautel M, Weber K, Fürst DO. (2000).
Characterization of muscle lamin isoforms suggests a possible role of gamma-lamin/abp-l in
sarcomeric z-disc formation. Cell Motil Cytoskeleton45(2),149-162

Literaturverzeichnis
189
van der Ven PF, Wiesner S, Salmikangas P, Auerbach D, Himmel M, Kempa S, Hayess K,
Pacholsky D, Taivainen A, Schröder R, Carpén O, Fürst DO. (2000). Indications for a novel
muscular dystrophy pathway. gamma-lamin, the muscle-specific lamin isoform, interacts
with myotilin. J Cell Biol,151(2), 235-248
Vandekerckhove J, Weber K. (1978). At least six different actins are expressed in a higher
mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic
peptide. J Mol Biol. 126:783–802.
Vicart P, Caron A, Guicheney P, Li Z, Prévost MC, Faure A, Chateau D, Chapon F, Tomé F,
Dupret JM, Paulin D, Fardeau M. (1998). A missense mutation in the alphaB-crystallin
chaperone gene causes a desmin-related myopathy. Nat Genet. 20(1):92-5.
Vorgerd M, Kubisch C, Burwinkel B, Reichmann H, Mortier W, Tettenborn B, Pongratz D,
Lindemuth R, Tegenthoff M, Malin JP, Kilimann MW. (1998). Mutation analysis in
myophosphorylase deficiency (McArdle's disease). Ann Neurol 43(3):326-31.
Vorgerd M, Kley RA, van der Ven PF, Fürst DO. (2008). Konferrenzbeitag: Molekulare
Pathomechanismen der Filaminopathie: funktionelle Charakterisierung der p.W2710X
Mutation. Akt Neurol 2008; 35 - V389
Vorgerd M, van der Ven PF, Bruchertseifer V, Löwe T, Kley RA, Schröder R, Lochmüller H,
Himmel M, Koehler K, Fürst DO, Huebner A.(2005). A mutation in the dimerization domain of
filamin c causes a novel type of autosomal dominant myofibrillar myopathy Am J Hum
Genet. 77(2):297-304.
Wallimann T, Tokarska-Schlattner M, Neumann D, Epand RM, Epand RF, Andres RH, Widmer
HR, Hornemann T, Saks VA, Agarkova I, Schlattner U (2007). The phospho-creatine circuit:
molecular and cellular physiology of creatine kinases, sensitivity to free radicals and
enhancement by creatine supplementation. Molecular Systems Bioenergetics: Energy for
Life, Basic Principles, Organization and Dynamics of Cellular Energetics (Saks, V.A., Editor),
195-264

Literaturverzeichnis
190
Wang CC, Tsai MF, Hong TM, Chang GC, Chen CY, Yang WM, Chen JJ, Yang PC. (2005). The
transcriptional factor YY1 upregulates the novel invasion suppressor HLJ1 expression and
inhibits cancer cell invasion. Oncogene 2005, 24:4081-4093.
Wang S, Bao H, Liu T, Zhang L, Yang P, Chen G. (2008). Accelerated proteolysis in alternating
electric fields for peptide mapping. Rapid Commun Mass Spectrom. 22(20):3225-32.
Wang S, Bao H, Zhang L , Yang P, Chen G. (2008). Infrared-Assisted On-Plate Proteolysis for
MALDI-TOF-MS Peptide Mapping. Anal. Chem., 80 (14), pp 5640–5647
Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ.
(1999). Requirement of a novel gene, Xin, in cardiac morphogenesis. Development.
126(6):1281-94.
Wiche G. (1989). Plectin: General overview and appraisal of its potential role as a subunit
protein of the cytomatrix. Crit Rev Biochem Mol Biol 1989;24:41-67
Wiesner J, Premsler T, Sickmann A. (2008). Application of electron transfer dissociation (ETD)
for the analysis of posttranslational modifications. Proteomics, 8(21):4466–83
Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I, Hochstrasser DF, Williams
KL. (1996). Progress with proteome projects: why all proteins expressed by a genome should
be identified and how to do it. Biotechnol. Genet. Eng. Rev 13, 19–50
Xie Z, Xu W, Davie EW, Chung DW. (1998). Molecular cloning of human ABPL, an actin-
binding protein homologue. Biochem Biophys Res Commun. 51(3):914-9.
Xu, BJ. (2010). Combining laser capture microdissection and proteomics: methodologies and
clinical applications. Proteomics.Clin.Appl 4, 116–123 .
Yaffe D, Saxel O. (1977). Serial passaging and differentiation of myogenic cells isolated from
dystrophic mouse muscle. Nature. 270(5639):725-7.
Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. (2007). Sodium 4-phenylbutyrate acts as a
chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress
and apoptosis. Invest Ophthalmol Vis Sci. 48(4):1683-90.

Literaturverzeichnis
191
Yamazaki M, Furuike S, Ito T. (2002). Mechanical response of single filamin A (ABP-280)
molecules and its role in the actin cytoskeleton. J Muscle Res Cell Motil. 23(5-6):525-34.
Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic (2001). 18O labeling for
comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem.
73(13):2836-42.
Yates JR, Cociorva D, Liao L, Zabrouskov V. (2006). Performance of a linear ion trap-Orbitrap
hybrid for peptide analysis. Anal Chem. 78(2):493-500.
Yates JR 3rd, Eng JK, McCormack AL, Schieltz D. (1995). Method to correlate tandem mass
spectra of modified peptides to amino acid sequences in the protein database. Anal Chem
67: 1426-36.
Yin HL, Stull JT. (1999). Proteins that regulate dynamic actin remodeling in response to
membrane signaling minireview series. J Biol Chem. 274(46):32529-30.
Zhu W, Smith JW, Huang CM. (2010). Mass spectrometry-based label-free quantitative
proteomics. J Biomed Biotechnol. 2010:840518.
Zubarev RA, Kelleher NL, McLafferty FW (1998). Electron capture dissociation of multiply
charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120 (13): 3265–66.

192
9. Abbildungsverzeichnis
Abbildung 1.1.: Strukturelle Darstellung einer Myofibrille__________________________________________ 3
Abbildung 1.2.: Schematische Darstellung eines Filamin C Moleküls _________________________________ 4
Abbildung 1.3: Phänotypische und histologische Merkmale der Myofibrillären Myopathie ________________ 8
Abbildung 1.4.: Zusammenfassung wichtiger Proteine des Skelettmuskels ____________________________ 9
Abbildung 1.5.: Zusammenfassung aller bekannten Mutationen im Filamin C-Gen _____________________ 12
Abbildung 1.6.: Schematische Darstellung Lasermikrodissektionsprozess ____________________________ 14
Abbildung 1.7.: Schematischer Aufbau eines Massenspektrometers ________________________________ 20
Abbildung 1.8.: Schematische Darstellung der Elektrospray-Ionisation_______________________________ 22
Abbildung 1.9.: Querschnitt durch den Orbitrap-Massenanalysator _________________________________ 25
Abbildung 1.10.: Schematische Darstellung der LTQ-Orbitrap ______________________________________ 26
Abbildung 4.1: Darstellung der Aggregate auf Glasobjektträgern und PET-Membran ___________________ 74
Abbildung 4.2: Vergleich der gewählten Auswertestrategien ______________________________________ 96
Abbildung 4.3.: Validierung spezifischer Proteine in Aggregaten von Filaminopathienpatienten __________ 97
Abbildung 4.4.: Optimierung der mikrodissektierten Fläche von Muskelfasernzellen ___________________ 99
Abbildung 4.5.: Repräsentatives Gel der 2D DIGE Studie _________________________________________ 101
Abbildung 4.6.: Mastergel der 2D DIDE Studie mit allen als differentiell identifizierten Spots ____________ 102
Abbildung 4.7.: pI-Shift der regulierten Desmin Spots in den sauren Bereich _________________________ 106
Abbildung 4.8.: Darstellung der 2D Blots und Nitrocellulose-Membranen ___________________________ 107
Abbildung 4.9.: Ergebnis Immunoblots zur Validierung der Desmin-Phosphorylierung _________________ 109
Abbildung 4.10.: Transfektion C2C12-Zellen mit C2-Konstrukt ____________________________________ 112
Abbildung 4.11.: Transfektion C2C12-Zellen mit N3-Konstrukt ____________________________________ 113
Abbildung 4.12.: Vergleich von Proteinaggregaten in C2C12-Zellen und humanen Skelettmuskelschnitten _ 114
Abbildung 4.13.: Zusammenfassung der Label-freien Proteomanalyse in Zellkultur ____________________ 118
Abbildung 4.14.: Darstellung aller identifizierter Proteine in humane Skelettmuskelproben _____________ 119

193
10. Tabellenverzeichnis
Tabelle 1.1.: Unterteilung der Intermediärfilament-Familie in 6 Gruppen _________________________ 5
Tabelle 1.2.: Übersicht über alle bekannten MFM verursachenden Mutationen ____________________ 9
Tabelle 3.1.: Daten der analysierten Filaminopathiepatienten _________________________________ 44
Tabelle 3.2.: Daten der gematchten Negativkontrollen _______________________________________ 44
Tabelle 3.3.: Übersicht über die verwendeten FLNC-Konstrukte ________________________________ 48
Tabelle 3.4.: Suchparameter für die Identifizierung von Gelspots _______________________________ 63
Tabelle 3.5.: Suchparameter für die Charakterisierung von spezifischer Aggregatbestandteile ________ 67 Tabelle 4.1.: Vergleichs von 3 verschiedenen Lösungsmitteln zur Solubilisierung von 250 Aggregaten __ 76
Tabelle 4.2.: Analyse von 100 und 250 mikrodissektierten Aggregaten, Auftrennung 1D Gel _________ 77
Tabelle 4.3.: Analyse von 50 und 100 mikrodissketierten Aggregaten____________________________ 78
Tabelle 4.4.: Analyse von 50 und 100 mikrodissketierten Aggregaten in 20 und 40 µl Ameisensäure ___ 79
Tabelle 4.5.: Liste der als spezifisch für Filaminopathie identifizierte Proteine in Aggregaten _________ 83
Tabelle 4.6.: Proteine, die ausschließlich in den Patientenkontrollen identifiziert wurden ___________ 88
Tabelle 4.7.: Liste der in Patientenkontrollen höher abundanten Proteinen_______________________ 91
Tabelle 4.8.: Liste der in Patientenkontrollen gering abundanten Proteine _______________________ 91
Tabelle 4.9.: Proteine, die in der manuellen und automatisierten Auswertung als spezifische Bestandteile
der Aggregate in Filaminopathiepatienten identifiziert werden konnten _________________________ 93
Tabelle 4.10: Zusammenfassung aller identifizierten Spots ___________________________________ 103
Tabelle 4.11.: Übereinstimmungen zwischen den Spectral Index Daten und der 2D DIGE Studie _____ 105
Tabelle 4.12.: Vergleich mikrodissektierter Areale aus murinen Myoblasten _____________________ 116

194
11. Lebenslauf
Persönliche Daten
Name Verena Theis, geb. Schwarz
Geburtsdatum, -ort 18. Mai 1982 in Bochum
Familienstand verheiratet, 1 Sohn
Nationalität deutsch
Hochschulbildung
02/2009 Beginn der Promotion bei Prof. Dr. Katrin Marcus und Prof. Dr.
Matthias Vorgerd/ Dr. Rudolf A. Kley
03/2009 Abschluss des Masterstudiums
10/2006 – 03/2009 Studium der Neurowissenschaften, Universität zu Köln
Masterarbeit: „Einfluss von miRNA auf die Lokalisation von
mRNA“ bei Qiagen GmbH, Hilden
10/2003 – 09/2006 Studium der Molekularen Medizin, Georg-August-Universität
Göttingen
Bachelorarbeit „Charakterisierung von Enzymmutanten, die mit
lysosomalen Speichererkrankungen einhergehen“, in der
Pädiatrie 2 des Universitätsklinikums Göttingen
10/2001 – 09/2003 Studium der Humanbiologie, Ernst-Moritz-Arndt Universität
Greifswald
Schulbildung
09/1990 – 06/2001 Städt. Helmholtz Gymnasium, Hilden
08/1988 – 02/1990 Gemeinschaftsgrundschule, Hilden

Lebenslauf
195
Lehrtätigkeiten
Vorlesungsnummer
Mitbetreuung des Praktikums der Biochemie / Molekularbiologie II 200512
(mit pathobiochemischen Bezügen):
Mitbetreuung des Praktikums der medizinischen Mikrobiologie: 200122
Expertise
Methoden
Elektrophorese und Chromatographie:
Ein- und zwei-dimensionale Gelelektrophorese (2D PAGE: Ampholine)
2D-DIGE-System („minimal“ und „saturation Labeling“)
Massenspektrometrie von Peptiden und Proteinen:
Orbitrap LTQ (Thermofisher), LTQ (Bruker)
MALDI-TOF-TOF-MS (Ultraflex™, Bruker Daltonik)
Sonstiges:
Lasermikrodissektion
Analyse und Charakterisierung von modifizierten Proteinen (Phosphorylierungen)
Immunoblotting (Westernblotting), Immunfluoreszenz
Zellkultur
EDV
MS Office
Bildbearbeitungsprogramme (Corel Draw, Corel Photo paint)
Auswertungsprogramme für Massenspektrometrie- und Proteomdaten
(ProteinScape, DeCyder™)
Veröffentlichungen
RA Kley, A. Maerkensᵻ, Y. Leber, V. Theis, A. Schreiner, PFM. van der Ven, J. Uszkoreit, C.
Stephan, S. Eulitz, N. Euler, J. Kirschner, K. Müller, HE. Meyer, M. Tegenthoff, DO.
Fürst, M. Vorgerd, T. Müller, K. Marcus . „A combined laser microdissection and mass
spectrometry approach reveals new disease relevant proteins accumulating in
aggregates of filaminopathy patients. Mol Cell Protoemics, Januar 2013

Lebenslauf
196
A Maerkens, RA Kley, M. Olivé M, V. Theis, PF van der Ven, J. Reimann, H. Milting, A.
Schreiner, J Uszkoreit, M. Eisenacher, K. Barkovits, AK. Güttsches, J. Tonillo, K.
Kuhlmann, HE Meyer, R. Schröder, M. Tegenthoff, DO Fürst, T Müller, LG Goldfarb, M
Vorgerd, K Marcus. Differential proteomic analysis of abnormal intramyoplasmic
aggregates in desminopathy. J Proteomics, April 2013
Ausgewählte Vorträge
November 2010:
DFG Forschergruppen Treffen FOR 1228, Bonn, Germany
“Label-free analysis of aggregates in patients with Filaminopathie”
Ausgewählte Posterpräsentationen
2009:
3th Summer School in Proteomic Basics, Brixen, Italy
“Establishment of laser microdissection and differential proteomic analysis
using samples from patients with myofibrilliar myopathy”
2010:
FoRUM Tagung 2010, Bochum, Germany
“Etablierung der Laser-Mikrodissektion und Proteomanalysen bei Patienten mit
Myofibrillären Myopathien (MFM) zur Identifikation neuer Kandidaten-Gene”
2012:
DGNN Kongress, Erlangen, Germany
“LMD-assisted Comparison of Muscle Proteome in Patients with
Filaminopathy and matched Controls”
Sonstige Kongresse
2009:
Arbeitstagung – Mikromethoden in der Proteinbiochemie, Martinsried, Deutschland
2011:
Jahrestagung MD-NET

Lebenslauf
197
Stipendien und Preise
08/2009 Reisestipendium Rektoratsprogramm der Ruhr Universität Bochum
für die Summer School in Brixen, Italien
08/2009 Reisestipendium der DGPF für die Summer School in Brixen, Italien
11/2010 Posterpreis bei der FoRUM Tagung 2010
12/2010 Posterpreis bei der Jahresabschlusstagung des MPC 2010
Zusatzqualifikationen
Leistungsnachweis in Gewerblicher Rechtsschutz: Patentwesen in den
Ingenieurwissenschaften (Fakultät für Ingenieurwissenschaften, Ruhr-Universität
Bochum 2011, Bochum, Germany)
Mitgliedschaften
Deutsche Gesellschaft für Proteomforschung (DGPF)

198
12. Danksagung
An dieser Stelle möchte ich bei den Menschen bedanken, die mich in all den Jahren
unterstützt und begleitet haben. Vor allem während und nach meiner Schwangerschaft
konnte ich auf die Unterstützung vieler zählen.
An erster Stelle möchte ich mich bei Prof. Dr. Katrin Marcus bedanken: Liebe Katrin, ich
danke dir für die Möglichkeit in deiner Gruppe promovieren zu können. In den vergangenen
Jahren durfte ich so viel lernen und konnte mich, dank deiner Hilfe, wissenschaftlich wie
auch persönlich weiterentwickeln.
Ein großer Dank gilt auch Dr. Rudolf Andre Kley und Prof. Dr. Matthias Vorgerd: Lieber Rudi,
lieber Matthias, ich möchte mich bei euch für die Unterstützung und das mir
entgegengebrachte Vertrauen in den letzten Jahren bedanken. Dank euch hatte ich die
Möglichkeit dieses interessante Thema zu bearbeiten.
Vielen Dank Alexandra Maerkens für die produktive und angenehme Zusammenarbeit!
Liebe Alex, ich danke dir, dass du während meiner Abwesenheit das Projekt so erfolgreich
weitergeführt hast.
Ich bedanke mich sehr herzlich bei Prof. Dr. Rolf Heumann für die freundliche Übernahme
des Korreferats.
Bei Herrn Prof. Dr. Helmut Meyer möchte ich mich für die Möglichkeit bedanken, am
Medizinischen Proteom Center zu arbeiten und die ausgezeichnet ausgestatteten
Laboratorien nutzen zu dürfen.

Danksagung
199
Ich möchte mich ganz herzlich bei der DFG-Forschergruppe FOR 1228 „Molekulare
Pathogenese der Myofibrillären Myopathien“, unter der Leitung von Prof. Dr. Rolf Schröder,
für die Unterstützung meiner Promotion bedanken.
Ich danke Dr. Thorsten Müller für die Einführung in die Proteomik zu Beginn meiner
Promotion und die Hilfestellung während der gesamten Zeit.
Ein großer Dank gilt auch Dr. Katja Rosenkranz. Ich möchte mich für die Hilfestellung bei der
Korrektur dieser Arbeit, die vielen Diskussionen und den guten Zuspruch bedanken.
Ein herzlicher Dank gilt Kathy Pfeiffer für ihre große Hilfsbereitschaft inner- und außerhalb
des Labors. Ohne sie und ihre Babysitterqualitäten wären viele Frühschichten im Labor nicht
denkbar gewesen!
Ich möchte mich bei allen anderen Mitarbeitern (teilweise ehemaligen Mitarbeitern) der
Abteilung Funktionelle Proteomik für das freundliche Arbeitsklima, die vielen Gespräche,
die fachlichen Diskussionen und die seelische Unterstützung bedanken. Besonders
hervorheben möchte ich Regina Küßner, Marie Lutz, Caroline May, Bettina Serschnitzki,
Frederic Brosseron, Peter Chartowski, Andre Przyborski und Christina Looße.
Ich möchte mich auch bei Anja Schreiner und Janine Mertens-Rill für die sehr angenehme
Zusammenarbeit im Muskellabor bedanken. Besonders Anja danke ich für die vielen
unterhaltsamen Stunden am LMD.
Thilo Lerari möchte ich für die unerschütterliche Geduld danken, mit er versucht hat mir die
LTQ und die Phosphoanalytik nahe zu bringen. Ohne den speziellen Phosphopinhibitor-
Cocktail hätte ich vielleicht nie die Phosphorylierung gefunden!
Christina John, Jason Tonillo und Heiner Falkenberg danke ich für die zuverlässige und
schnelle Messung meiner Proben.
Ein herzlicher Dank gilt auch allen anderen Mitarbeitern des Medizinischen Proteoms
Centers für die vier schönen Jahre und die angenehme Zusammenarbeit!

Danksagung
200
Bedanke möchte ich mich auch bei der Medizinischen Fakultät der Ruhr Universität, die im
Rahmen der „Forschungsförderung der Ruhr Universität Bochum“ (FoRUM) diese Arbeit mit
einer Anschubfinanzierung ermöglicht hat.
Zum Schluss möchte ich mich bei meiner Familie von Herzen danken:
Liebe Mama, lieber Papa! Ich möchte euch für die letzten 31 Jahre danken, in denen ihr
immer für mich da wart und mich auch in den schwierigsten Zeiten unterstützt habt. Ohne
euch wäre ich nie da, wo ich heute stehe. Ihr habt mir so viel ermöglicht, dafür werde ich
euch immer dankbar sein!
Lieber Christian! Als ich meine Masterarbeit 2008 schrieb, konntest du nur erahnen, welche
Naturgewalten während der Schreibphase der Dissertation auf dich zu kommen würden. Ich
bin mir sicher, dass ich deine Erwartungen deutlich übertreffen konnte. Ich danke dir, dass
du in den letzten Monaten jede erdenkliche Emotion mit mir durchlebt und mich zum
Durchhalten motiviert hast. Ich liebe dich!
Mein innigster Dank gilt Krümel und Cassian: Für euch schlägt mein Herz!
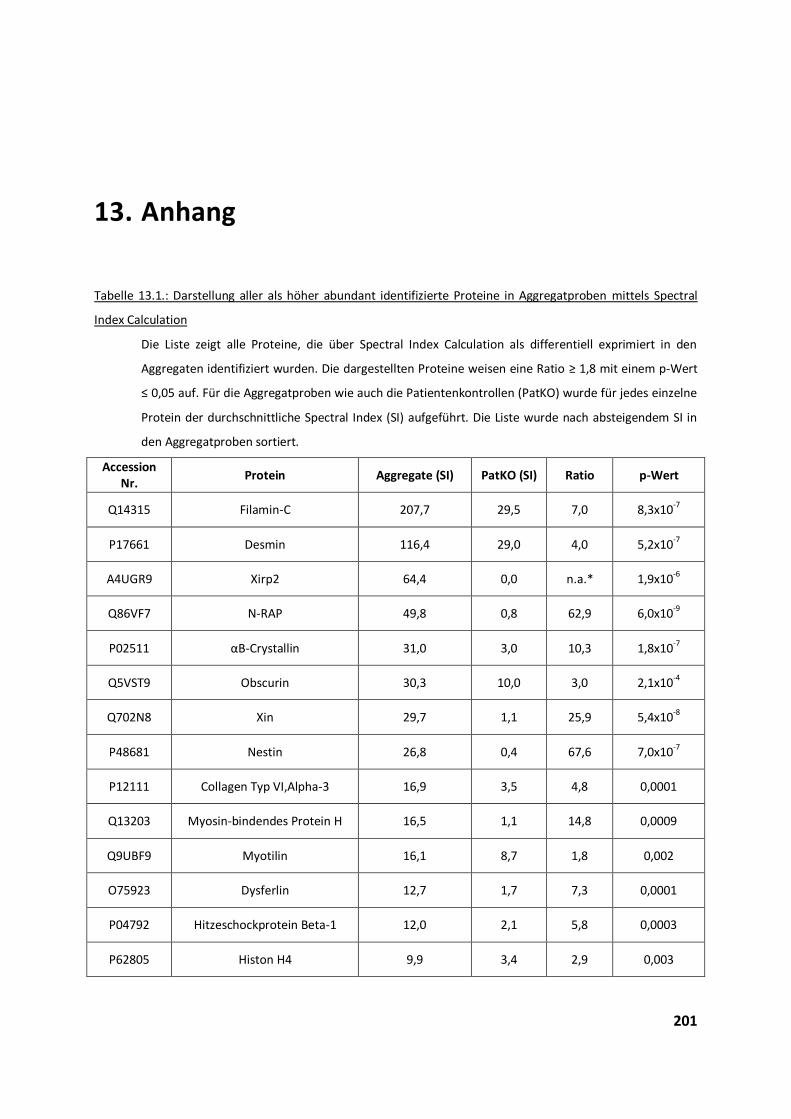
201
13. Anhang
Tabelle 13.1.: Darstellung aller als höher abundant identifizierte Proteine in Aggregatproben mittels Spectral
Index Calculation
Die Liste zeigt alle Proteine, die über Spectral Index Calculation als differentiell exprimiert in den
Aggregaten identifiziert wurden. Die dargestellten Proteine weisen eine Ratio ≥ 1,8 mit einem p-Wert
≤ 0,05 auf. Für die Aggregatproben wie auch die Patientenkontrollen (PatKO) wurde für jedes einzelne
Protein der durchschnittliche Spectral Index (SI) aufgeführt. Die Liste wurde nach absteigendem SI in
den Aggregatproben sortiert.
Accession Nr.
Protein Aggregate (SI) PatKO (SI) Ratio p-Wert
Q14315 Filamin-C 207,7 29,5 7,0 8,3x10-7
P17661 Desmin 116,4 29,0 4,0 5,2x10-7
A4UGR9 Xirp2 64,4 0,0 n.a.* 1,9x10-6
Q86VF7 N-RAP 49,8 0,8 62,9 6,0x10-9
P02511 αB-Crystallin 31,0 3,0 10,3 1,8x10-7
Q5VST9 Obscurin 30,3 10,0 3,0 2,1x10-4
Q702N8 Xin 29,7 1,1 25,9 5,4x10-8
P48681 Nestin 26,8 0,4 67,6 7,0x10-7
P12111 Collagen Typ VI,Alpha-3 16,9 3,5 4,8 0,0001
Q13203 Myosin-bindendes Protein H 16,5 1,1 14,8 0,0009
Q9UBF9 Myotilin 16,1 8,7 1,8 0,002
O75923 Dysferlin 12,7 1,7 7,3 0,0001
P04792 Hitzeschockprotein Beta-1 12,0 2,1 5,8 0,0003
P62805 Histon H4 9,9 3,4 2,9 0,003
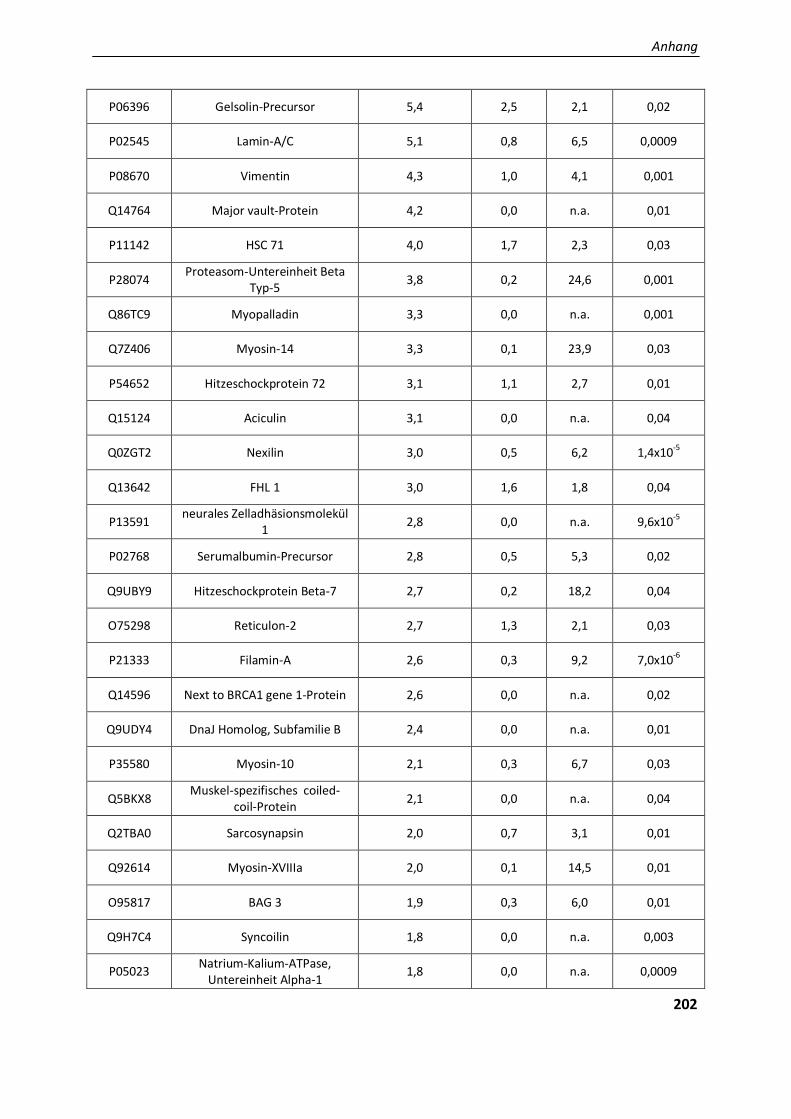
Anhang
202
P06396 Gelsolin-Precursor 5,4 2,5 2,1 0,02
P02545 Lamin-A/C 5,1 0,8 6,5 0,0009
P08670 Vimentin 4,3 1,0 4,1 0,001
Q14764 Major vault-Protein 4,2 0,0 n.a. 0,01
P11142 HSC 71 4,0 1,7 2,3 0,03
P28074 Proteasom-Untereinheit Beta
Typ-5 3,8 0,2 24,6 0,001
Q86TC9 Myopalladin 3,3 0,0 n.a. 0,001
Q7Z406 Myosin-14 3,3 0,1 23,9 0,03
P54652 Hitzeschockprotein 72 3,1 1,1 2,7 0,01
Q15124 Aciculin 3,1 0,0 n.a. 0,04
Q0ZGT2 Nexilin 3,0 0,5 6,2 1,4x10-5
Q13642 FHL 1 3,0 1,6 1,8 0,04
P13591 neurales Zelladhäsionsmolekül
1 2,8 0,0 n.a. 9,6x10
-5
P02768 Serumalbumin-Precursor 2,8 0,5 5,3 0,02
Q9UBY9 Hitzeschockprotein Beta-7 2,7 0,2 18,2 0,04
O75298 Reticulon-2 2,7 1,3 2,1 0,03
P21333 Filamin-A 2,6 0,3 9,2 7,0x10-6
Q14596 Next to BRCA1 gene 1-Protein 2,6 0,0 n.a. 0,02
Q9UDY4 DnaJ Homolog, Subfamilie B 2,4 0,0 n.a. 0,01
P35580 Myosin-10 2,1 0,3 6,7 0,03
Q5BKX8 Muskel-spezifisches coiled-
coil-Protein 2,1 0,0 n.a. 0,04
Q2TBA0 Sarcosynapsin 2,0 0,7 3,1 0,01
Q92614 Myosin-XVIIIa 2,0 0,1 14,5 0,01
O95817 BAG 3 1,9 0,3 6,0 0,01
Q9H7C4 Syncoilin 1,8 0,0 n.a. 0,003
P05023 Natrium-Kalium-ATPase,
Untereinheit Alpha-1 1,8 0,0 n.a. 0,0009
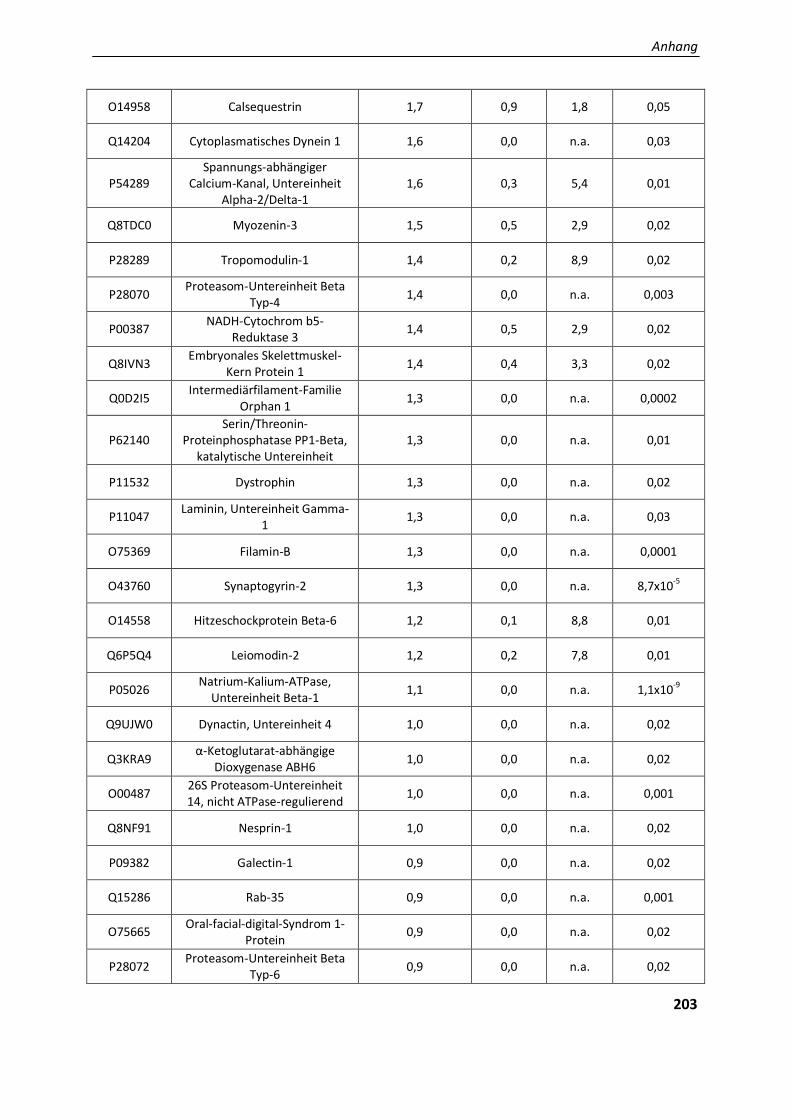
Anhang
203
O14958 Calsequestrin 1,7 0,9 1,8 0,05
Q14204 Cytoplasmatisches Dynein 1 1,6 0,0 n.a. 0,03
P54289 Spannungs-abhängiger
Calcium-Kanal, Untereinheit Alpha-2/Delta-1
1,6 0,3 5,4 0,01
Q8TDC0 Myozenin-3 1,5 0,5 2,9 0,02
P28289 Tropomodulin-1 1,4 0,2 8,9 0,02
P28070 Proteasom-Untereinheit Beta
Typ-4 1,4 0,0 n.a. 0,003
P00387 NADH-Cytochrom b5-
Reduktase 3 1,4 0,5 2,9 0,02
Q8IVN3 Embryonales Skelettmuskel-
Kern Protein 1 1,4 0,4 3,3 0,02
Q0D2I5 Intermediärfilament-Familie
Orphan 1 1,3 0,0 n.a. 0,0002
P62140 Serin/Threonin-
Proteinphosphatase PP1-Beta, katalytische Untereinheit
1,3 0,0 n.a. 0,01
P11532 Dystrophin 1,3 0,0 n.a. 0,02
P11047 Laminin, Untereinheit Gamma-
1 1,3 0,0 n.a. 0,03
O75369 Filamin-B 1,3 0,0 n.a. 0,0001
O43760 Synaptogyrin-2 1,3 0,0 n.a. 8,7x10-5
O14558 Hitzeschockprotein Beta-6 1,2 0,1 8,8 0,01
Q6P5Q4 Leiomodin-2 1,2 0,2 7,8 0,01
P05026 Natrium-Kalium-ATPase,
Untereinheit Beta-1 1,1 0,0 n.a. 1,1x10-9
Q9UJW0 Dynactin, Untereinheit 4 1,0 0,0 n.a. 0,02
Q3KRA9 α-Ketoglutarat-abhängige
Dioxygenase ABH6 1,0 0,0 n.a. 0,02
O00487 26S Proteasom-Untereinheit 14, nicht ATPase-regulierend
1,0 0,0 n.a. 0,001
Q8NF91 Nesprin-1 1,0 0,0 n.a. 0,02
P09382 Galectin-1 0,9 0,0 n.a. 0,02
Q15286 Rab-35 0,9 0,0 n.a. 0,001
O75665 Oral-facial-digital-Syndrom 1-
Protein 0,9 0,0 n.a. 0,02
P28072 Proteasom-Untereinheit Beta
Typ-6 0,9 0,0 n.a. 0,02
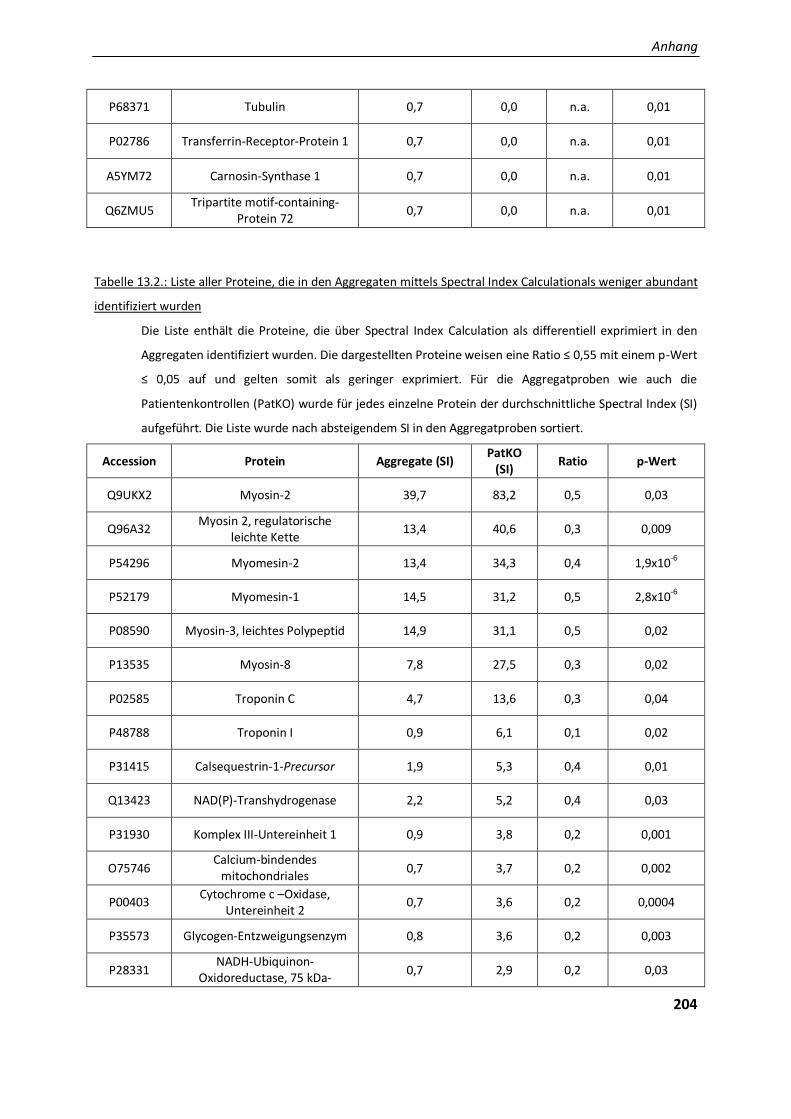
Anhang
204
P68371 Tubulin 0,7 0,0 n.a. 0,01
P02786 Transferrin-Receptor-Protein 1 0,7 0,0 n.a. 0,01
A5YM72 Carnosin-Synthase 1 0,7 0,0 n.a. 0,01
Q6ZMU5 Tripartite motif-containing-
Protein 72 0,7 0,0 n.a. 0,01
Tabelle 13.2.: Liste aller Proteine, die in den Aggregaten mittels Spectral Index Calculationals weniger abundant
identifiziert wurden
Die Liste enthält die Proteine, die über Spectral Index Calculation als differentiell exprimiert in den
Aggregaten identifiziert wurden. Die dargestellten Proteine weisen eine Ratio ≤ 0,55 mit einem p-Wert
≤ 0,05 auf und gelten somit als geringer exprimiert. Für die Aggregatproben wie auch die
Patientenkontrollen (PatKO) wurde für jedes einzelne Protein der durchschnittliche Spectral Index (SI)
aufgeführt. Die Liste wurde nach absteigendem SI in den Aggregatproben sortiert.
Accession Protein Aggregate (SI) PatKO
(SI) Ratio p-Wert
Q9UKX2 Myosin-2 39,7 83,2 0,5 0,03
Q96A32 Myosin 2, regulatorische
leichte Kette 13,4 40,6 0,3 0,009
P54296 Myomesin-2 13,4 34,3 0,4 1,9x10-6
P52179 Myomesin-1 14,5 31,2 0,5 2,8x10-6
P08590 Myosin-3, leichtes Polypeptid 14,9 31,1 0,5 0,02
P13535 Myosin-8 7,8 27,5 0,3 0,02
P02585 Troponin C 4,7 13,6 0,3 0,04
P48788 Troponin I 0,9 6,1 0,1 0,02
P31415 Calsequestrin-1-Precursor 1,9 5,3 0,4 0,01
Q13423 NAD(P)-Transhydrogenase 2,2 5,2 0,4 0,03
P31930 Komplex III-Untereinheit 1 0,9 3,8 0,2 0,001
O75746 Calcium-bindendes
mitochondriales Trägerprotein Aralar 1
0,7 3,7 0,2 0,002
P00403 Cytochrome c –Oxidase,
Untereinheit 2 0,7 3,6 0,2 0,0004
P35573 Glycogen-Entzweigungsenzym 0,8 3,6 0,2 0,003
P28331 NADH-Ubiquinon-
Oxidoreductase, 75 kDa-Untereinheit
0,7 2,9 0,2 0,03
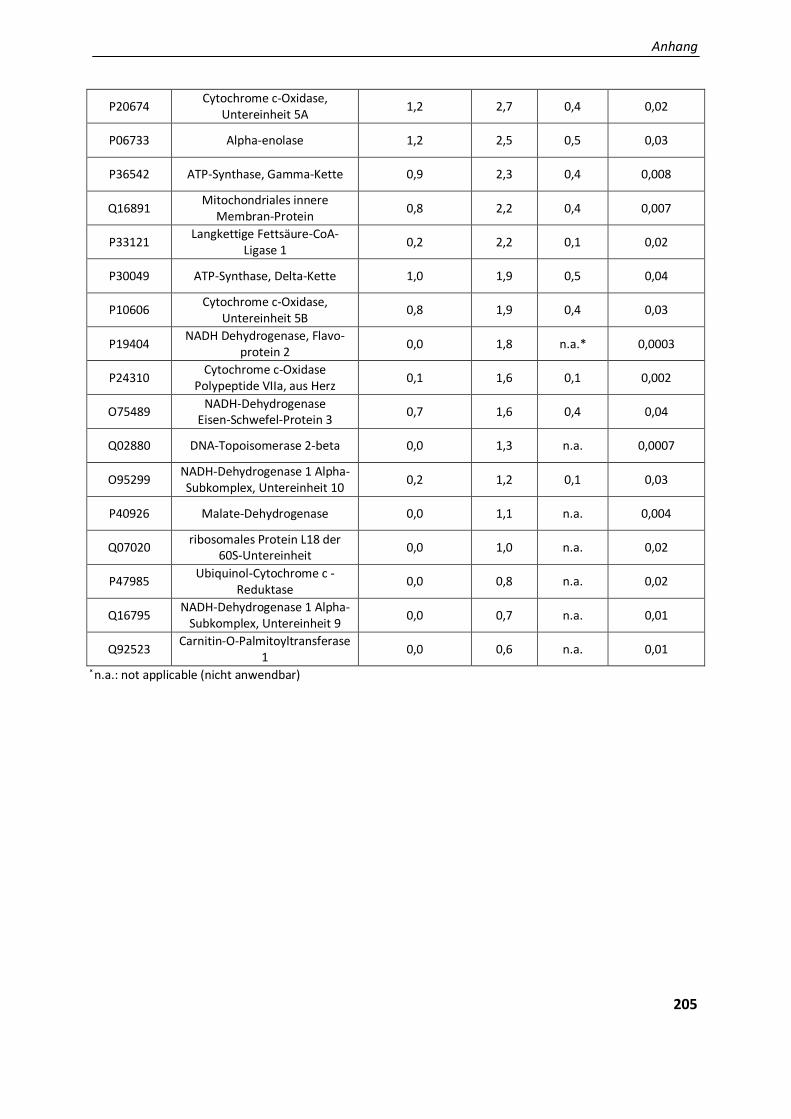
Anhang
205
P20674 Cytochrome c-Oxidase,
Untereinheit 5A 1,2 2,7 0,4 0,02
P06733 Alpha-enolase 1,2 2,5 0,5 0,03
P36542 ATP-Synthase, Gamma-Kette 0,9 2,3 0,4 0,008
Q16891 Mitochondriales innere
Membran-Protein 0,8 2,2 0,4 0,007
P33121 Langkettige Fettsäure-CoA-
Ligase 1 0,2 2,2 0,1 0,02
P30049 ATP-Synthase, Delta-Kette 1,0 1,9 0,5 0,04
P10606 Cytochrome c-Oxidase,
Untereinheit 5B 0,8 1,9 0,4 0,03
P19404 NADH Dehydrogenase, Flavo-
protein 2 0,0 1,8 n.a.* 0,0003
P24310 Cytochrome c-Oxidase
Polypeptide VIIa, aus Herz 0,1 1,6 0,1 0,002
O75489 NADH-Dehydrogenase
Eisen-Schwefel-Protein 3 0,7 1,6 0,4 0,04
Q02880 DNA-Topoisomerase 2-beta 0,0 1,3 n.a. 0,0007
O95299 NADH-Dehydrogenase 1 Alpha-Subkomplex, Untereinheit 10
0,2 1,2 0,1 0,03
P40926 Malate-Dehydrogenase 0,0 1,1 n.a. 0,004
Q07020 ribosomales Protein L18 der
60S-Untereinheit 0,0 1,0 n.a. 0,02
P47985 Ubiquinol-Cytochrome c -
Reduktase 0,0 0,8 n.a. 0,02
Q16795 NADH-Dehydrogenase 1 Alpha-
Subkomplex, Untereinheit 9 0,0 0,7 n.a. 0,01
Q92523 Carnitin-O-Palmitoyltransferase
1 0,0 0,6 n.a. 0,01
n.a.: not applicable (nicht anwendbar)

206
Erklärung
Hiermit erkläre ich, dass ich die Arbeit selbstständig verfasst und bei keiner anderen Fakultät
eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es
handelt sich bei der heute von mir eingereichten Dissertation um fünf in Wort und Bild völlig
übereinstimmende Exemplare.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in
keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, Juni 2013
Verena Theis






![Soventol Fibel 082017 - apothekenkanal.de · Soventol Gel 20 mg/1 g Gel. Wirkst.: Bamipin[(RS)-lactat. Zus.-setz.: 1 g Gel enth. 20 mg Bamipin[(RS)-lactat]. Sonst. Best.-teile: Gerein.](https://static.fdokument.com/doc/165x107/5e64f7ea3480624f8923b165/soventol-fibel-082017-soventol-gel-20-mg1-g-gel-wirkst-bamipinrs-lactat.jpg)











