GastroForum Bochum neu - drfalkpharma.de · Bochum Samstag, 21. März 2009 9.00 – 17.20 Uhr...
75
Gastroenterologie 2009: Neues aus Diagnostik und Therapie Bochum Samstag, 21. März 2009 9.00 – 17.20 Uhr Veranstaltungsort: RuhrCongress Bochum Stadionring 20 44791 Bochum Wissenschaftliche Leitung: Prof. Dr. W. Schmiegel, Bochum Abstracts Böblingen Böblingen 7. Februar 2009 7. Februar 2009 Braunschweig Braunschweig 25. April 2009 25. April 2009 Neustadt / Weinstraße Neustadt / Weinstraße 19. September 2009 19. September 2009 Bochum Bochum 21. März 2009 21. März 2009 Magdeburg Magdeburg 5. Dezember 2009 5. Dezember 2009 Gladbeck Gladbeck 13. Juni 2009 13. Juni 2009 Rostock Rostock 12. September 12. September 2009 München München 10. Oktober 2009 10. Oktober 2009
Transcript of GastroForum Bochum neu - drfalkpharma.de · Bochum Samstag, 21. März 2009 9.00 – 17.20 Uhr...

Gastroenterologie 2009:
Neues aus Diagnostik und Therapie
Bochum
Samstag, 21. März 2009
9.00 – 17.20 Uhr
Veranstaltungsort:
RuhrCongress Bochum
Stadionring 20
44791 Bochum
Wissenschaftliche Leitung:
Prof. Dr. W. Schmiegel, Bochum
Abstracts
Böblingen Böblingen 7. Februar 20097. Februar 2009
BraunschweigBraunschweig25. April 200925. April 2009
Neustadt / WeinstraßeNeustadt / Weinstraße19. September 200919. September 2009
BochumBochum21. März 200921. März 2009
MagdeburgMagdeburg5. Dezember 20095. Dezember 2009
GladbeckGladbeck13. Juni 200913. Juni 2009
RostockRostock12. September12. September 2009
MünchenMünchen10. Oktober 200910. Oktober 2009

1
Programm 9.00 Uhr
Begrüßung Prof. Dr. W. Schmiegel, Bochum
1. Hepatopankreatobiliäres System 1 Vorsitz: Dr. W.E. Schmidt, Bochum Prof. Dr. T. Sauerbruch, Bonn
9.05 Uhr Behandlung der akuten Pankreatitis Prof. Dr. M.M. Lerch, Greifswald
9.25 Uhr Autoimmune und hereditäre Pankreatitis � wann bedenken, wie behandeln? Prof. Dr. J. Mössner, Leipzig
9.45 Uhr Leitliniengerechte Therapie von Gallenwegserkrankungen: Kurzfassung der aktualisierten S3-Leitlinie der DGVS und DGAV zur Diagnostik und Behandlung von Gallensteinen Prof. Dr. F. Lammert, Homburg/Saar
2. Hepatopankreatobiliäres System 2 Vorsitz: Prof. Dr. G. Gerken, Essen Dr. T. Pietzsch, Bochum
10.10 Uhr Leberbiopsie � der Pathologe als letzte Instanz bei der Diagnostik diffuser Lebererkrankungen? Prof. Dr. A. Tannapfel, Bochum
10.25 Uhr Prävention und Management von Resistenzen bei der chronischen Hepatitis B Dr. M. Cornberg, Hannover
10.40 Uhr Individualisierung der Therapiestrategie bei chronischer Hepatitis C � neuer Standard Prof. Dr. S. Zeuzem, Frankfurt
10.55 Uhr Stadiengerechte Therapie der Varizenblutung Prof. Dr. T. Sauerbruch, Bonn
11.10�11.30 Uhr Kaffeepause

2
3. Darm 1 Vorsitz: Prof. Dr. W. Kruis, Köln PD Dr. G. Schmidt-Heinevetter, Bochum
11.30 Uhr Therapie des Reizdarmsyndroms Prof. Dr. P. Layer, Hamburg
11.50 Uhr Die pseudomembranöse Kolitis auf dem Vormarsch � Konzepte zur Diagnostik, Prophylaxe und Therapie Dr. C. Pox, Bochum
12.10 Uhr Bottom-up oder Top-down � welches Risiko bei welchem CED-Patienten? Prof. Dr. S. Schreiber, Kiel
12.30�13.25 Uhr Mittagspause
4. Darm 2 Vorsitz: Prof. Dr. M. Zeitz, Berlin Prof. Dr. J. Schölmerich, Regensburg
13.25 Uhr Divertikulitis � wann konservativ, wann interventionell, wann chirurgisch therapieren? Prof. Dr. W. Kruis, Köln
13.45 Uhr Ecksteine der Leitlinie Morbus Crohn Prof. Dr. M. Zeitz, Berlin
14.05 Uhr Die steroidrefraktäre Colitis ulcerosa � wie behandeln? Prof. Dr. J. Schölmerich, Regensburg
5. Gastroenterologische Onkologie Vorsitz: Dr. C. Mölleken, Bochum Prof. Dr. O.G. Opitz, Freiburg
14.30 Uhr Therapeutisches Vorgehen beim Ösophaguskarzinom Prof. Dr. O.G. Opitz, Freiburg
14.45 Uhr RFTA, SIRT oder TACE � lokalablative Therapieoptionen bei Lebertumoren (ohne Abstract) Prof. Dr. V. Nicolas, Bochum

3
15.00 Uhr (Neo-)Adjuvante und palliative Therapie des kolorektalen
Karzinoms Prof. Dr. W. Schmiegel, Bochum
15.15 Uhr Hereditäre Formen des kolorektalen Karzinoms � Was gilt es zu beachten? Dr. K. Schulmann, Bochum
15.30 Uhr Was bringen (Darm-)Krebszentren? (ohne Abstract) Prof. Dr. Dr. h.c. W. Hohenberger, Erlangen
15.45�16.00 Uhr Kaffeepause
6. Endoskopie Vorsitz: Prof. Dr. T. Rösch, Hamburg Dr. B. Viebahn, Bochum
16.00 Uhr Leitlinie Sedierung in der Endoskopie � was ändert sich? (ohne Abstract) Dr. A. Riphaus, Hannover
16.15 Uhr Kapselendoskopie des Kolons Dr. T. Brechmann, Bochum
16.30 Uhr Schneiden oder Brennen? Interventionelle Therapie des Barrett-Ösophagus (ohne Abstract) Prof. Dr. T. Rösch, Hamburg
16.45 Uhr Interventionelle Endosonografie � Möglichkeiten und Grenzen (ohne Abstract) Dr. S. Heringlake, Bochum
17.00 Uhr Schlusswort Prof. Dr. W. Schmiegel, Bochum
Anschriften der Referenten und Vorsitzenden siehe Seiten 75�76

5
Behandlung der akuten Pankreatitis
M.M. Lerch, M. Kraft, J. Mayerle
Klinik und Poliklinik für Innere Medizin A, Universitätsklinikum Greifswald
Einleitung
Die akute Pankreatitis zählt zu den häufigsten gastroenterologischen Erkrankungen.
Die Inzidenz der Neuerkrankungen einer akuten Pankreatitis liegt zwischen
10�46/100.000 Einwohner. Somit sind 2% des klinischen Krankenguts betroffen. In
den letzten Jahren wurde eine steigende Inzidenz beobachtet. Klinische Symptome
wie gürtelförmige Oberbauchbeschwerden und Erbrechen zusammen mit einem über
das 3-fache der Norm erhöhten Serumspiegel für Amylase oder Lipase führen zur
Diagnosestellung der akuten Pankreatitis. Die häufigste Ursache der Pankreatitis
sind eine Choledocholithiasis oder ein Alkoholabusus. In seltenen Fällen wird auch
eine medikamentös induzierte Pankreatitis beschrieben (Tab. 1). Im klinischen
Verlauf lassen sich für die akute Pankreatitis 2 Formen unterscheiden, deren
Auftreten unabhängig von der Ätiologie der Erkrankung ist: die akute interstitiell-
ödematöse Pankreatitis (75�85%) mit einer Letalität unter 1% und die akute
hämorrhagisch-nekrotisierende Pankreatitis (15�25%) mit einer Letalität zwischen
10�24%.
Volumen und Elektrolytsubstitution Die entscheidende therapeutische Maßnahme bei der Behandlung der akuten
Pankreatitis (und ebenso der häufigste Behandlungsfehler, wenn sie nicht erfolgt) ist
die ausreichende Substitution des Flüssigkeitsverlusts. In einer japanischen retro-
spektiven Analyse konnte gezeigte werden, dass die Mortalität einer Patientengruppe
mit akuter Pankreatitis 61,2% betrug, wenn weniger als 3,5 Liter Flüssigkeit in den
ersten 24 Stunden des Krankenhausaufenhalts infundiert wurden (1). Patienten mit
akuter Pankreatitis sequestrieren erhebliche Flüssigkeitsmengen, vor allem ins
Retroperitoneum, bei Vorliegen eines Ileus ins Darmlumen, in die Pleurahöhle und in
die freie Bauchhöhle (pankreatogener Aszites). Im Normalfall kann ein
Flüssigkeitsbedarf von mindestens 3�4 Litern pro Tag angenommen werden,
allerdings müssen in manchen Fällen mehr als 10 Liter in 24 h substituiert werden.
Eine Kontrolle des Flüssigkeitsbedarfs und der Substitution über den zentralvenösen
Druck, die stündliche Urinausscheidung (0,5 ml/kg KG/h) und die tägliche

6
Bestimmung des Hämatokrit ist in jedem Fall erforderlich. Als Richtwert für die
Absenkung des Hämatokrits durch ausreichende Flüssigkeitssubstitution gilt ein Wert
unter 35%. Der zentralvenöse Druck sollte auf Werte um 8�12 cm Wassersäule
angehoben werden. Ein nicht ausreichender Ersatz des Flüssigkeitsverlusts hat eine
Vasokonstriktion im Splanchnikusgebiet zur Folge, und hieraus kann eine Minder-
perfusion des Pankreas resultieren, was wiederum zur Progression der akuten
Pankreatitis beitragen würde.
Nahrungskarenz oder enterale Ernährung Nahrungskarenz hat einen positiven Einfluss auf den Verlauf des paralytischen Ileus,
der als Folge einer akuten Pankreatitis auftreten kann. Zudem empfinden viele
Patienten die Nahrungskarenz subjektiv als Erleichterung für ihre Übelkeit, ihr
Erbrechen und ihre Schmerzen. Auf den klinischen Verlauf oder die Prognose der
akuten Pankreatitis selbst hat die Nahrungskarenz nach neueren Studien keinen
positiven Einfluss. Vor allem die Vorstellung, dass durch Nahrungskarenz die Bauch-
speicheldrüse �ruhiggestellt� werden muss, gilt heute als obsolet. Sowohl in
experimentellen als auch in klinischen Studien wurde überzeugend belegt, dass im
Verlauf einer Pankreatitis die exokrine Sekretion blockiert ist und dass somit eine
Hemmung der Sekretion als therapeutisches Prinzip sinnlos ist. Eine therapeutische
Aufhebung der Sekretionsblockade bei der Pankreatitis wäre, zumindest aus
pathophysiologischen Überlegungen, ein vielversprechenderer Behandlungsansatz.
In 10 prospektiv randomisierten klinischen Studien (2�13) konnte inzwischen gezeigt
werden, dass eine enterale Ernährung der parenteralen Ernährung bei akuter
Pankreatitis überlegen ist. Die Gründe hierfür liegen nicht nur in den Kosten der
parenteralen Ernährung (6-mal so teuer wie die enterale Sondenernährung), sondern
vor allem in den Komplikationen der parenteralen Ernährung. Neben der Gefahr
einer zusätzlichen Infektionsquelle durch den zentralvenösen Katheter kommt es bei
ausschließlich parenteraler Ernährung innerhalb weniger Tage zu einer Zotten-
atrophie im Darm, die dann eine bakterielle Translokation in die umliegenden
parenchymatösen Organe erlaubt. Bei Patienten mit nekrotisierender Pankreatitis
siedeln sich die translozierten Bakterien bevorzugt in der Pankreasnekrose an und
können eine der gefürchtetsten Komplikationen der Pankreatitis � die infizierte
Nekrose oder den Pankreasabszess (s. unten) � verursachen. Eine enterale
Sondenernährung, die über eine tiefliegende Dünndarmsonde oder (neueste
Studien) mit gleicher Effektivität auch über eine Magensonde verabreicht wird, wirkt

7
der Translokation entgegen und hat sich als Alternative zur parenteralen Ernährung
bewährt (3, 14�16).
Analgetikatherapie Patienten mit akuter Pankreatitis leiden oft unter stärksten viszeralen Schmerzen.
Deshalb ist eine ausreichende Analgesie eines der wichtigsten und oft dringlichsten
Behandlungsziele. Die einst nur im deutschsprachigen Raum verbreitete Dauerin-
fusion des Lokalanästhetikums Procainhydrochlorid (Novocain, 2 g/24 h) zur
Schmerzbehandlung bei der Pankreatitis ist weder durch Studien noch durch
Fallberichte belegt. In einer klinischen Studie aus der Universitätsklinik Magdeburg
wurde gezeigt, dass die Novocain-Infusion für die Schmerzbehandlung bei akuter
Pankreatitis wirkungslos ist und den Bedarf an zusätzlich zu gebenden Opiat-
analgetika sogar noch erhöht (17, 18).
Auch das Argument einer möglichen Kontraktion der Duodenalpapille durch
Morphine und damit einer zusätzlichen Abflussbehinderung der Pankreassekretion
ist nach heutigem Wissensstand obsolet (19). Wir wissen heute, dass dieser Effekt
bei den meisten Analgetika dieser Gruppe nicht auftritt oder so gering ausgeprägt ist,
dass er klinisch keine Rolle spielt. Einige morphinanaloge Analgetika werden mit
Erfolg zur Schmerztherapie bei akuter Pankreatitis eingesetzt. Im angelsächsischen
Sprachraum wird überwiegend und mit gutem Erfolg Morphium zur Behandlung
starker Schmerzen bei akuter Pankreatitis eingesetzt. Das in Deutschland aus
betäubungsrechtlichen Gründen sehr gerne verordnete Tramadol (Tramal®) führt
nach persönlicher Erfahrung der Autoren bei Patienten mit akuter Pankreatitis
häufiger zu Übelkeit und Erbrechen, so dass andere Opiatanalgetika eher zu
verordnen sind (20,21).
Einige Zentren haben in zwischen gute Ergebnisse mit dem Einsatz der thorakalen
Periduralanalgesie erzielt. Diese führt nicht nur zur raschen Schmerzfreiheit der
Patienten, sondern verhindert oder therapiert zusätzlich einen paralytischen Ileus.
Voraussetzung für den Einsatz der PDA ist, dass der Patient weder analgosediert ist,
noch eine manifeste Gerinnungsstörung vorliegt (22, 23).
Behandlung mit Antibiotika und Probiotika Die Einstellung zur Behandlung der akuten Pankreatitis mit Antibiotika hat sich in den
letzten Jahren mehrfach gewandelt. In neueren Studien wurde überzeugend gezeigt,
dass eine generelle Antibiotikaprophylaxe keine Vorteile bietet und nur zur Selektion

8
resistenter Erreger beiträgt. Demgegenüber profitieren Patienten mit nachgewiesener
infizierter Pankreasnekrose von einer Antibiotikabehandlung erheblich. Die letzte
Metaanalyse zur prophylaktischen Antibiotikagabe, die auch die Daten der neuesten
Meropenem-Studie von (24) einschließt und damit 7 Studien mit insgesamt
467 Patienten in der Analyse berücksichtigt fand keinen Unterschied für die Rate an
infizierten Nekrosen. Auch die Gesamtmortalität war in der Antibiotikatherapiegruppe
nicht signifikant reduziert.
Probiotika sind lebende Mikroorganismen, die eine Reihe von positiven Effekten auf
die Gesundheit haben sollen. Olah und Kollegen haben in den letzten Jahren 2 RCT
Studien zur Prophylaxe einer infizierten Nekrose bei Patienten mit akuter Pankreatitis
durchgeführt. Beide Studien belegen, dass der Einsatz von Probiotika die Inzidenz
von infektiösen Komplikationen verminderte (6, 7). Umso mehr Aufsehen haben die
Ergebnisse der erst im letzten Jahr im Lancet veröffentlichten PROPATRIA-Studie
der Niederländischen Pankreatitis Studien Gruppe erregt. In einer doppelblinden
Plazebo-kontrollierten Studien an 298 Patienten mit schwerer akuter Pankreatitis
belegten die Autoren, dass die Probiotikagabe (Ecologic 641: Lactobacillus acido-
philus, Lactobacillus casei, Lactobacillus salivarius, Lactococccus lactis, Bifido-
bacterium bifidum und Bifidobacterium lactis) nicht zu einer signifikanten Abnahme
der infektiösen Komplikationen, sondern zu einer signifikanten Zunahme der
Mortalität, überwiegend verursacht durch Darmnekrosen in der Verumgruppe führte
(25, 26). Die Gabe von Probiotika zur Therapie der akuten Pankreatitis sollte somit
unbedingt unterbleiben bis weitere Studien die Hintergründe dieses Befunds klären.
Endoskopisches und operatives Vorgehen bei nekrotisierender Pankreatitis Ein operatives Vorgehen bei akuter nekrotisierender Pankreatitis ist nur bei nach-
gewiesener infizierter Nekrose und nicht bei einer sterilen Nekrose indiziert. Im
Verlauf der letzten 2 Jahrzehnte hat sich das therapeutische Konzept von einem
aggressiven operativen Vorgehen hin zu einem konservativen interventionellen
Management gewandelt. Ursprünglich wurde die Indikation zur Nekrosektomie bei
Auftreten eines Multiorganversagens gestellt. Dieses Vorgehen war mit einer
Mortalität von 65% verbunden, was den Nutzen des operativen Vorgehens in dieser
Situation infrage stellt. Noch im Jahr 2003 belief sich die Mortalität bei offener
Nekrosektomie auf 47% (27). Die offene Nekrosektomie sollte deshalb wo immer
möglich vermieden werden, da das operative Trauma ein schwer beherrschbares

9
SIRS induziert (28). Eine Studie von Mier und Kollegen aus dem Jahr 1997 belegt,
dass ein operatives Vorgehen innerhalb von 2 Wochen nach Krankheitsbeginn mit
einer signifikant höheren Mortalität behaftet ist (29). Wenn eine offene Nekros-
ektomie nicht vermeidbar ist, sollte sie durch konservative Maßnahmen wie eine
Drainageanlage und eine Resistogramm gerechte Antibiose bis zur 3. oder 4. Krank-
heitswoche hinausgezögert werden. Ein kombiniert konservatives und inter-
ventionelles Vorgehen ist auch bei infizierter Nekrose dem operativen Verfahren
gleichwertig (30). Eine Reihe von Studien haben in den letzten Jahren gezeigt, dass
minimalinvasive Therapieverfahren wie die perkutane Drainageanlage oder eine
laparoskopisch assistierte Nekrosektomie vielversprechende Ergebnisse liefern
(31, 32). Als neues und sehr wenig invasives Therapieverfahren gilt die trans-
gastrische oder transduodenale endoskopische Nekrosektomie. Bisher wurden in der
Literatur mehr als 100 Behandlungsfälle beschrieben. Die Indikation war entweder
eine nachgewiesene infizierte Nekrose oder ein Pankreasabszess. Die technische
Erfolgsrate bei diesen Patienten lag bei 92,1%, wobei es in 19,6% zu Komplikationen
wie Kolonfisteln, Blutung, Prothesendislokation, Schmerzen nach mehr als 24 h,
Perforationen oder Senkungsabszessen kommt. Die Mortalität in dieser Patienten-
gruppe betrug 5,6%, der Langzeiterfolg der Therapie lag bei 81,2% und die Anzahl
der Eingriffe bei im Median 2,3 (33�35). Insgesamt stellt dieses Verfahren bei
richtiger Indikationsstellung und frühestens 2�3 Wochen nach Krankheitsbeginn
einen vielversprechenden therapeutischen Ansatz dar und wird auch in unserer Klinik
inzwischen routinemäßig eingesetzt.
Korrespondenzadresse: Prof. Dr. Markus M. Lerch - Direktor - Klinik und Poliklinik für Innere Medizin A Ernst-Moritz-Arndt Universität Greifswald Friedrich-Loeffler-Str. 23A 17475 Greifswald Tel.: 03834 867230 Fax: 03834 867234 E-Mail: [email protected]

10
Tabelle 1: Medikamentöse Ursachen einer akuten Pankreatitis: Daten kumuliert aus Trivedi (36), Eland (37), Andersen (38), Lankisch (39) Medikamente
Anzahl der Fälle Re-Exposition Didanosin 883 9 Asparaginase 177 2 Azathioprin 101 22 Valproat 80 11 5-wertige Antimone (Leishmaniose-Therapie)
80 14
Pentamidin 79 2 Mercaptopurin 69 10 Mesalamin/Olsalazin 74 17 Östrogene 42 11 Opiate 42 5 Tetracyclin 34 2 Cytarabin 26 4 Stereoide 25 1 Sulfmethoxazol/ Trimethoprim
24 1
Sulfasalazin 23 5 Furosemid 21 3 Sulindac 21 8 Lamivudin 19 1 Octreotid 16 4 Acetaminophen 13 1 Phenformin 13 1 Interferon-α2b 13 3 Enalapril 12 2 Hydrochlorothiazid 12 1 Cisplatin 11 1 Erythromycin 11 1 Cimetidin 1 1 Methyldopa 2 2 Metronidazol 1 1 Oxyphenbutazon 1 1 Simvastatin 1 1
Literatur: 1. Mayumi T, Takada T, Kawarada Y, Hirata K, Yoshida M, Sekimoto M, Hirota M,
Kimura Y, Takeda K, Isaji S, Koizumi M, Otsuki M, Matsuno S. Management strategy for acute pancreatitis in the JPN Guidelines. J Hepatobiliary Pancreat Surg. 2006;13:61�7.

11
2. Working Party of the British Society of Gastroenterology; Association of Surgeons of Great Britain and Ireland; Pancreatic Society of Great Britain and Ireland; Association of Upper GI Surgeons of Great Britain and Ireland. UK guidelines for the management of acute pancreatitis. Gut. 2005;54(Suppl 3): iii1�9.
3. Eatock FC, Brombacher GD, Steven A, Imrie CW, McKay CJ, Carter R.
Nasogastric feeding in severe acute pancreatitis may be practical and safe. Int J Pancreatol. 2000;28:23�9.
4. Karamitsios N, Saltzman JR. Enteral nutrition in acute pancreatitis. Nutr Rev.
1997;55:279�82. 5. McClave SA, Greene LM, Snider HL, Makk LJ, Cheadle WG, Owens NA, Dukes
LG, Goldsmith LJ. Comparison of the safety of early enteral vs parenteral nutrition in mild acute pancreatitis. JPEN J Parenter Enteral Nutr. 1997;21: 14�20.
6. Oláh A, Belágyi T, Issekutz A, Olgyai G. [Combination of early nasojejunal
feeding with modern synbiotic therapy in the treatment of severe acute pancreatitis (prospective, randomized, double-blind study)]. Magy Seb. 2005;58:173�8.
7. Oláh A, Pardavi G, Belágyi T, Nagy A, Issekutz A, Mohamed GE. Early
nasojejunal feeding in acute pancreatitis is associated with a lower complication rate. Nutrition. 2002;18:259�62.
8. Powell JJ, Murchison JT, Fearon KC, Ross JA, Siriwardena AK. Randomized
controlled trial of the effect of early enteral nutrition on markers of the inflammatory response in predicted severe acute pancreatitis. Br J Surg. 2000;87:1375�81.
9. Pupelis G, Selga G, Austrums E, Kaminski A. Jejunal feeding, even when
instituted late, improves outcomes in patients with severe pancreatitis and peritonitis. Nutrition. 2001;17:91�4.
10. Sax HC, Warner BW, Talamini MA, Hamilton FN, Bell RH, Jr, Fischer JE, Bower
RH. Early total parenteral nutrition in acute pancreatitis: lack of beneficial effects. Am J Surg. 1987;153:117�24.
11. Windsor AC, Kanwar S, Li AG, Barnes E, Guthrie JA, Spark JI, Welsh F, Guillou
PJ, Reynolds JV. Compared with parenteral nutrition, enteral feeding attenuates the acute phase response and improves disease severity in acute pancreatitis. Gut. 1998;42:431�5.
12. Eckerwall GE, Axelsson JB, Andersson RG. Early nasogastric feeding in
predicted severe acute pancreatitis: A clinical, randomized study. Ann Surg. 2006;244:959�65; discussion 965�7.

12
13. Petrov MS, Kukosh MV, Emelyanov NV. A randomized controlled trial of enteral versus parenteral feeding in patients with predicted severe acute pancreatitis shows a significant reduction in mortality and in infected pancreatic complications with total enteral nutrition. Dig Surg. 2006;23:336�44; discussion 344�5.
14. Imrie CW, Carter CR, McKay CJ. Enteral and parenteral nutrition in acute
pancreatitis. Best Pract Res Clin Gastroenterol. 2002;16:391�7. 15. Kumar A, Singh N, Prakash S, Saraya A, Joshi YK. Early enteral nutrition in
severe acute pancreatitis: a prospective randomized controlled trial comparing nasojejunal and nasogastric routes. J Clin Gastroenterol. 2006;40:431�4.
16. Lecleire S, Antonietti M, Ben-Soussan E, Zenoni M, Savoye G, Goria O,
Ducrotté P, Lerebours E. Nasojejunal feeding in patients with severe acute pancreatitis: comparison of endoscopic and self-migration tube placement. Pancreas. 2007;35:376�8.
17. Kahl S, Zimmermann S, Pross M, Schulz HU, Schmidt U, Malfertheiner P.
Procaine hydrochloride fails to relieve pain in patients with acute pancreatitis. Digestion 2004;69:5�9.
18. Lerch MM. No more intravenous procaine for pancreatitis pain? Digestion.
2004;69:2�4. 19. Thompson DR. Narcotic analgesic effects on the sphincter of Oddi: a review of
the data and therapeutic implications in treating pancreatitis. Am J Gastro-enterol. 2001;96:1266�72.
20. Jakobs R, Adamek MU, von Bubnoff AC, Riemann JF. Buprenorphine or
procaine for pain relief in acute pancreatitis. A prospective randomized study. Scand J Gastroenterol. 2000;35:1319�23.
21. Staritz M. Pharmacology of the sphincter of Oddi. Endoscopy. 1988;20 Suppl
1:171�4. 22. Bernhardt A, Kortgen A, Niesel H, Goertz A. Anwendung der
Epiduralanästhesie bei Patienten mit akuter Pankreatitis � Prospektive Unter-suchung an 121 Patienten [Using epidural anesthesia in patients with acute pancreatitis � prospective study of 121 patients]. Anaesthesiol Reanim. 2002;27:16�22.
23. Niesel HC, Klimpel L, Kaiser H, Bernhardt A, al-Rafai S, Lang U. Epidurale
Blockade zur Analgesie und Behandlung der akuten Pankreatitis [Epidural blockade for analgesia and treatment of acute pancreatitis]. Reg Anaesth. 1991;14:97�100.
24. Dellinger EP, Tellado JM, Soto NE, Ashley SW, Barie PS, Dugernier T, Imrie
CW, Johnson CD, Knaebel HP, Laterre PF, Maravi-Poma E, Kissler JJ, Sanchez-Garcia M, Utzolino S. Early antibiotic treatment for severe acute necrotizing pancreatitis: a randomized, double-blind, placebo-controlled study. Ann Surg. 2007;245:674�83.

13
25. Besselink MG, van Santvoort HC, Buskens E, Boermeester MA, van Goor H, Timmerman HM, Nieuwenhuijs VB, Bollen TL, van Ramshorst B, Witteman BJ, Rosman C, Ploeg RJ, Brink MA, Schaapherder AF, Dejong CH, Wahab PJ, van Laarhoven CJ, van der Harst E, van Eijck CH, Cuesta MA, Akkermans LM, Gooszen HG; Dutch Acute Pancreatitis Study Group. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:651�9.
26. Sand J, Nordback I. Probiotics in severe acute pancreatitis. Lancet.
2008;371:634�5. 27. Nieuwenhuijs VB, Besselink MG, van Minnen LP, Gooszen HG. Surgical
management of acute necrotizing pancreatitis: a 13-year experience and a systematic review. Scand J Gastroenterol. Suppl 2003:111�6.
28. Connor S, Alexakis N, Raraty MG, Ghaneh P, Evans J, Hughes M, Garvey CJ,
Sutton R, Neoptolemos JP. Early and late complications after pancreatic necrosectomy. Surgery. 2005;137:499�505.
29. Mier J, León EL, Castillo A, Robledo F, Blanco R. Early versus late
necrosectomy in severe necrotizing pancreatitis. Am J Surg. 1997;173:71�5. 30. Runzi M, Niebel W, Goebell H, Gerken G, Layer P. Severe acute pancreatitis:
nonsurgical treatment of infected necroses. Pancreas. 2005;30:195�9. 31. Shankar S, vanSonnenberg E, Silverman SG, Tuncali K, Banks PA. Imaging
and percutaneous management of acute complicated pancreatitis. Cardiovasc Intervent Radiol. 2004;27:567�80.
32. Werner J, Feuerbach S, Uhl W, Büchler MW. Management of acute
pancreatitis: from surgery to interventional intensive care. Gut. 2005;54:426�36. 33. Raczynski S, Teich N, Borte G, Wittenburg H, Mössner J, Caca K.
Percutaneous transgastric irrigation drainage in combination with endoscopic necrosectomy in necrotizing pancreatitis (with videos). Gastrointest Endosc. 2006;64:420�4.
34. Seewald S, Groth S, Omar S, Imazu H, Seitz U, de Weerth A, Soetikno R,
Zhong Y, Sriram PV, Ponnudurai R, Sikka S, Thonke F, Soehendra N. Aggressive endoscopic therapy for pancreatic necrosis and pancreatic abscess: a new safe and effective treatment algorithm (videos). Gastrointest Endosc. 2005;62:92�100.
35. Seifert H, Wehrmann T, Schmitt T, Zeuzem S, Caspary WF. Retroperitoneal
endoscopic debridement for infected peripancreatic necrosis. Lancet. 2000;356:653�5.
36. Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin
Gastroenterol 2005;39:709�16. 37. Eland IA, van Puijenbroek EP, Sturkenboom MJ, Wilson JH, Stricker BH. Drug-
associated acute pancreatitis: twenty-one years of spontaneous reporting in The Netherlands. Am J Gastroenterol. 1999;94:2417�22.

14
38. Andersen V, Sonne J, Andersen M. Spontaneous reports on drug-induced pancreatitis in Denmark from 1968 to 1999. Eur J Clin Pharmacol. 2001;57:517�21.
39. Lankisch PG, Dröge M, Gottesleben F. Drug induced acute pancreatitis:
incidence and severity. Gut. 1995;37:565�7.

15
Autoimmune und hereditäre Pankreatitis � wann bedenken, wie behandeln?
J. Mössner
Medizinische Klinik für Gastroenterologie und Rheumatologie, Universitätsklinikum
Leipzig
Ätiologie und Pathogenese In den Industrienationen ist die chronische Pankreatitis in 70�80% aller Fälle alkohol-
induziert. In 20�30% der Fälle ist eine auslösende Ursache nicht erkennbar
(�idiopathische chronische Pankreatitis�). Die Aufklärung genetischer Verände-
rungen, die entweder die Krankheit verursachen oder bei zusätzlichen, zum Teil noch
unbekannten Kofaktoren, die Krankheitspenetranz beeinflussen, hat noch zu keinen
therapeutischen Konsequenzen geführt.
Angeborene Risikofaktoren der chronischen Pankreatitis Hereditäre Pankreatitis Erstbeschreibung und Klinik. Die klinische Beobachtung einer Familie, in der 6 von
36 Mitgliedern aus 4 Generationen eine rezidivierende Pankreatitis hatten, führte im
Jahr 1952 zur Erstbeschreibung der autosomal-dominant vererbten hereditären
Pankreatitis. Die Erkrankung beginnt überwiegend im Kindes- oder Jugendalter und
betrifft Jungen und Mädchen gleichermaßen. Meist treten wiederholte schmerzhafte
akute Schübe der chronischen Pankreatitis auf; selten ist eine klinische
Erstmanifestation aufgrund bereits entwickelter exokriner oder endokriner Insuf-
fizienz.
Genetik. Kationisches Trypsinogen ist das quantitativ wichtigste Trypsinogen im
Pankreassaft des Menschen. Einer nordamerikanisch-italienischen Arbeitsgruppe
gelang es im Jahr 1996, bei 5 Familien mit hereditärer Pankreatitis eine
krankheitsassoziierte Mutation in diesem Gen zu identifizieren. Bereits bei der
Publikation der R122H-Mutation wurde postuliert, dass die Aminosäure R122
aufgrund ihrer exponierten Lage als eine �Sollbruchstelle� anzusehen ist. Eine
Hydrolyse an dieser Position könnte zu einer Inaktivierung des aktiven Trypsins
führen. Im mutierten R122H-Molekül wäre diese Spaltung nicht mehr möglich,
sodass aktives Trypsin im Organ persistiert, was dann eine akute Pankreatitis

16
initiieren könnte. Dieses Konzept passt gut in die schon zuvor favorisierte Vorstellung
zur Pathogenese der Pankreatitis, welche von einer gesteigerten intrapankreatischen
Trypsinaktivität ausgeht. Die häufigsten krankheitsassoziierten Mutationen des
kationischen Trypsinogens, N29I und R122H, führen in der Tat zu einer erleichterten
Autoaktivierung und/oder erschwerten Inaktivierung der mutierten Trypsinmoleküle in
vitro.
In rascher Folge wurden weitere Varianten des kationischen Trypsinogens
identifiziert. Einige davon führten aufgrund ihrer Position im Molekül oder ihrer
biochemischen Eigenschaften zu interessanten pathogenetischen Konzepten.
Beispiele sind weitere Mutationen im Bereich der Trypsin-Inaktivierungsschnittstelle,
Mutationen direkt an der Trypsinogen-Aktivierungsschnittstelle oder durch Austausch
genetischen Materials entstandene Konversionsmutationen. Die subtil erhobene
Familienanamnese und ggf. das Hinzuziehen objektiver klinischer Befunde der
Familienangehörigen eines Indexpatienten ist Basis jeder sinnvollen genetischen
Diagnostik.
Hereditäre chronische Pankreatitis und Pankreaskarzinom Nachdem häufig bereits im jüngeren Erwachsenenalter eine Beruhigung der
Krankheitsaktivität eintritt, sind die Patienten über das Risiko eines Pankreas-
karzinoms besorgt. Die Datenlage zu dieser Frage ist uneinheitlich, da alle
bisherigen Untersuchungen nur sehr wenige Patienten mit hereditärer Pankreatitis
und Pankreaskarzinom umfassten. In der größten Kohorte mit 246 Patienten erlitten
8 Patienten ein Pankreaskarzinom; daraus wurde ein gegenüber der Normal-
bevölkerung 50-fach erhöhtes Pankreaskarzinomrisiko errechnet. In unserer eigenen
Kohorte fanden sich unter 101 Patienten 3 Fälle eines Pankreaskarzinoms mit einer
mittleren Manifestation 23 Jahre nach klinischem Beginn der Pankreatitis. In einer
älteren Untersuchung fand sich auch bei längerer Nachbeobachtung kein einziger
Fall bei 72 Patienten. Die einzige sinnvolle Konsequenz daraus ist die dringende
Empfehlung zum Nikotinverzicht und eine einmal jährlich durchgeführte nicht-
invasive bildgebende Untersuchung.
Idiopathische chronische Pankreatitis Mutationen des pankreatischen Trypsin-Inhibitor-Gens SPINK1. 4 Jahre nach der
Erstbeschreibung von Mutationen des kationischen Trypsinogens wurde eine
Assoziation von Mutationen des SPINK1-Gens (Serin-Proteasen-Inhibitor Typ Kasal)

17
mit der chronischen Pankreatitis berichtet. SPINK1 ist der wichtigste intrapan-
kreatische Gegenspieler vorzeitig aktivierten Trypsins; seine Mutationen scheinen
ebenfalls zu einer Destabilisierung des intrapankreatischen Proteasen-
Antiproteasen-Gleichgewichts beizutragen. Während Trypsinogen-Mutationen fast
ausschließlich bei Patienten mit autosomal-dominanter hereditärer chronischer
Pankreatitis gefunden wurden, finden sich SPINK1-Mutationen neben der
idiopathischen (20%) auch bei Patienten mit alkoholischer (6%) und tropischer (50%)
chronischer Pankreatitis in hoher Prävalenz. Die wichtigste genetische Veränderung
im SPINK1-Gen ist die Mutation N34S.
Mutationen des anionischen Trypsinogens. Wir analysierten das Gen des
anionischen Trypsinogens (PRSS2) sowohl in einer gesunden Kontrollgruppe als
auch bei Patienten mit chronischer Pankreatitis. Eine Variante des Codons 191
(G191R) war überraschenderweise bei Kontrollen statistisch signifikant über-
repräsentiert (3,4% vs. 1,3%; Odds-Ratio = 0,37; p = 1,1 x 10-8). In vitro zeigte
gereinigtes rekombinantes G191R-Protein einen vollständigen Verlust der
Aktivierbarkeit durch entweder Enterokinase oder Trypsin. Dies war durch die
Einführung einer neuen Region, die durch Trypsin gespalten werden kann, erklärt.
Diese Variante des anionischen Trypsins macht das Molekül hypersensitiv für eine
proteolytische Degradation. Die G191R-Variante des PRSS2 mitigiert somit die
intrapankreatische Trypsin-Aktivität und schützt somit vor der Entwicklung einer
Pankreatitis.
Mutationen des Chymotrypsin C. Chymotrypsin C (CTRC) degradiert Trypsin und
schützt somit ebenfalls das Pankreas vor seiner Autodigestion. Durch Analyse von
1320 Patienten mit chronischer Pankreatitis und 2888 Studienteilnehmern ohne
chronische Pankreatitis konnten wir eine Assoziation von genetischen Varianten des
CTRC mit der chronischen Pankreatitis nachweisen. Die gefundenen Varianten
zeigten funktionell eine verringerte Aktivität und/oder eine verminderte Sekretion.
Dieser Funktionsverlust stört das intrapankreatische Gleichgewicht von Proteasen
und Antiproteasen und führt � wie wir annehmen � über eine Autodigestion zur
Pankreatitis.

18
Chronische Pankreatitis und Mukoviszidose Epidemiologie. Die Mukoviszidose oder zystische (Pankreas-)Fibrose (CF) ist eine
autosomal-rezessiv vererbte Erkrankung, die durch Mutationen im CFTR-Gen (cystic
fibrosis transmembrane conductance regulator) verursacht wird. Die geschätzte
Inzidenz liegt in Europa bei 1:2500.
Pathophysiologie. Die Pankreasschädigung bei CF ist durch eine duktuläre
Obstruktion infolge eingedickten Sekrets charakterisiert, die zu einer Dilatation der
Pankreasgänge und zu einem Verlust der Azinuszellen mit interstitieller Fibrose und
Ersatz durch Fettgewebe führt. Die Pankreasbeteiligung variiert von einem
kompletten Verlust der exokrinen und endokrinen Funktion bis zu einer nahezu
normalen Pankreasfunktion. CFTR kodiert für ein Transmembranprotein, ist auf der
Oberfläche der meisten Epithelzellen nachweisbar und fungiert als cAMP-abhängiger
Chloridkanal. CFTR besitzt eine bedeutende Rolle in der duktulären Sekretion von
Bikarbonat in den Pankreassaft. CFTR-Mutationen verursachen einen defekten
Salztransport mit Bildung zäher Schleimsekrete in den betroffenen Organen.
Genetik. Bislang sind über 1000 verschiedene CFTR-Mutationen beschrieben. Bei
Patienten mit idiopathischer chronischer Pankreatitis finden sich CFTR-Mutationen
4-mal häufiger als erwartet. Aus dieser Beobachtung folgte die Hypothese, dass die
Kombination zweier milder bzw. einer schweren und einer milden CFTR-Mutation
den Phänotyp einer Pankreatitis verursacht, während die Kombination zweier
schwerer CFTR-Mutationen zu dem Phänotyp einer zystischen Fibrose führt.
Allerdings weist nur ein Bruchteil der Patienten mit idiopathischer Pankreatitis
2 CFTR-Mutationen auf. Warum lediglich heterozygote Mutationsträger ein erhöhtes
Risiko für eine chronische Pankreatitis besitzen, ist wenig verstanden. Neuere
Publikationen deuten darauf hin, dass die Kombination einer heterozygoten CFTR-
Mutation mit einem genetischen Defekt in einem anderen Gen, wie z. B. SPINK1, zur
chronischen Pankreatitis disponiert. Dieser Hypothese folgend, würde CFTR über
einen komplexen Erbgang zu einer Pankreatitis prädisponieren.
Pankreaskarzinomrisiko. Über das Pankreaskarzinomrisiko bei zystischer Fibrose ist
bislang wenig bekannt; insgesamt gibt es erst 9 publizierte Fälle weltweit. Von diesen
hatten 5 eine vorbestehende exokrine Pankreasinsuffizienz. Ob sich im Zuge der

19
deutlich verbesserten Prognose der pulmonalen Erkrankung zukünftig häufiger ein
Pankreaskarzinom finden wird, bleibt abzuwarten.
Autoimmunpankreatitis
Die Autoimmunpankreatitis (AIP) ist eine seltene Erkrankung und liegt bei maximal
10% der chronischen Pankreatitiden vor. Bei einem Teil der Patienten findet sich im
Serum ein erhöhtes IgG, Subtyp 4 und/oder Antikörper gegen Carboanhydrase.
Bildmorphologisch finden sich sowohl tumorartige Pankreasraumforderungen als
auch eine diffuse Organvergrößerung, sogenanntes Wurstpankreas. Histologisch
findet sich eine lymphoplasmazelluläre Infiltration. Die Erkrankung manifestiert sich
oftmals mit begleitenden Veränderungen im Gallengangssystem, bildmorphologisch
wie PSC.
Therapie
Hereditäre Pankreatitis Häufig kommt es im Verlauf zu einer Abschwächung der Krankheitsintensität. Auch
innerhalb einer Familie finden sich ausgeprägt variable Verläufe, die sich zum
heutigen Zeitpunkt weder vorhersagen noch spezifisch behandeln lassen. Die
Therapie orientiert sich bislang an den Leitlinien der akuten und chronischen
Pankreatitis anderer Genese. Chirurgische Therapieoptionen stehen eher im
Hintergrund.
Autoimmunpankreatitis Im Gegensatz zur PSC sprechen diese Gallenwegsentzündung und die Cholestase
gut auf Steroide an. Die AIP kann aufgrund eines Tumorverdachts fälschlicherweise
einer Operation (Resektion) zugeführt werden, die wiederum bei einem Teil der
Patienten zu einer deutlichen Exazerbation der Erkrankung mit ernstzunehmender
Verschlechterung führen kann. Das klinische Problem stellt die sichere Diagnose der
Erkrankung dar. Die AIP ist medikamentös gut mit Steroiden und Azathioprin
behandelbar.
Chronische Pankreatitis jedweder Ursache Die symptomatische Therapie der chronischen Pankreatitis ist stadiengerecht und
setzt daher eine Kenntnis des klinischen Bildes und der Komplikationsmöglichkeiten
voraus.

20
Stadium I: Präklinisches Stadium ohne manifeste Symptomatik mit bereits
chronisch entzündlichen Veränderungen des Organs
Stadium II: Klinische Symptome in Form von rezidivierenden akuten Schüben und
sekundären Komplikationen. Mit zunehmendem Untergang von Pankre-
asgewebe Nachlassen der Intensität der klinischen Symptome. Einige
Patienten zeigen auch ein chronisches Schmerzsyndrom und einen
Krankheitsverlauf ohne typische Schübe. Die häufigste Komplikation ist
die Entstehung von Pankreaspseudozysten mit unterschiedlichster
Symptomatik.
Stadium III: Progrediente exokrine und endokrine Insuffizienz mit zunehmender
Diarrhö und Steatorrhö, weiterem Gewichtsverlust sowie Symptomen
des Diabetes mellitus. Ca 10% aller Patienten werden aufgrund eines
primär schmerzlosen Verlaufs erst im Stadium III mit progredientem
Gewichtsverlust aufgrund ausgeprägter Maldigestion klinisch auffällig.
Leitsymptom ist der rezidivierende oft gürtelförmige Schmerz im Oberbauch sowie
Gewichtsverlust. Die Pathogenese der Schmerzen ist vielschichtig. Im Stadium II ist
der Gewichtsverlust durch unzureichende Kalorienzufuhr aufgrund nahrungs-
abhängiger Schmerzen erklärt, im Stadium III durch zunehmende Maldigestion. Bei
eingeschränkter exokriner Pankreasfunktion kommen Fettstuhl und andere Merkmale
der schweren Maldigestion, wie Folgeerkrankungen des Mangels fettlöslicher
Vitamine, hinzu. Das Spätstadium ist charakterisiert durch eine zunehmende
endokrine Insuffizienz mit Diabetes mellitus. Die Symptomatik kann erweitert werden
durch Folgeerkrankungen des Alkohol- und Nikotinabusus wie arterielle Verschluss-
krankheit, chronische Bronchitis, Lungenkarzinom, Fettleber, Leberzirrhose mit oder
ohne portale Hypertension. Die typische Trias von Gewichtsverlust mit oder ohne
Steatorrhö, Diabetes mellitus und Pankreaskalzifikationen findet sich bei einem
Drittel der Patienten, meist erst im Stadium III.
Literatur beim Verfasser. Prof. Dr. med. Joachim Mössner, Department für Innere Medizin und Dermatologie, Medizinische Klinik für Gastroenterologie und Rheumatologie, Universitätsklinikum Leipzig, AöR, Liebigstr. 20, 04103 Leipzig, Tel.: 0341 9712200, Fax: 0341 9712209, E-Mail: [email protected]

21
Die Arme der symptomatischen Therapie der chronischen Pankreatitis
Maßnahme Ziel Alkoholkarenz soziale Reintegration Verbesserung der Compliance Verzögerung des Krankheitsverlaufs? Reduktion der Komplikationen? Nikotinkarenz Verzögerung der Arteriosklerose Besserung der Schmerzen? Verzögerung des Krankheitsverlaufs Reduktion der Komplikationen? Medikamentöse Schmerztherapie Schmerzfreiheit
oral, sublingual, intravenös, transdermal, peridural, intrathekal, Plexus-coeliacus-Blockade
Interventionelle Endoskopie Gallengangdrainage Beseitigung einer Cholestase
Verhinderung einer sekundär biliären Leberzirrhose
Verhinderung einer Cholangitis Beseitigung des Pruritus
Pankreasgangdrainage Schmerzfreiheit Verzögerung der chronisch destruktiven Entzündung?
Pseudozystendrainage Schmerzfreiheit, Rupturverhinderung transkutan, endoskopisch transgastral, -duodenal, -papillär Pankreasgangsteinentfernung Schmerzfreiheit Verzögerung der Entzündung? ESWL + endoskopische Steinextraktion
Therapie der exokrinen Insuffizienz Beseitigung/Besserung der
Maldigestion Schweinepankreatin (säuregeschützte Mikrotabletten, -pellets) Konventionelles Pankreatin bei fehlender Magensäure
fettlösliche Vitamine, Diät Therapie der endokrinen Insuffizienz Therapie des pankreopriven Diabetes vorübergehend orale Antidiabetika, in der Regel Insulin Operation Schmerztherapie Therapie von Komplikationen Karzinomverdacht Verzögerung des Krankheitsverlaufs?

22
Leitliniengerechte Therapie von Gallenwegserkrankungen: Kurzfassung der aktualisierten S3-Leitlinie der DGVS und DGAV zur Diagnostik und Behandlung von Gallensteinen
F. Lammert
Klinik für Innere Medizin II, Universitätsklinikum des Saarlandes, Homburg/Saar
15�20% der Bevölkerung haben Gallensteine, von denen 20�30% Symptome
entwickeln, so dass jährlich in Deutschland mehr als 190.000 Cholezystektomien
durchgeführt werden. Prädisponierende Faktoren für Gallensteine sind höheres
Lebensalter, weibliches Geschlecht, hochkalorische, kohlenhydratreiche und ballast-
stoffarme Ernährung, Bewegungsmangel und genetische Faktoren.
Leitsymptome der Erkrankungen der Gallenblase und -wege sind kolikartige
Schmerzen im Oberbauch, Ikterus und Fieber. Die jährliche Komplikationsrate
symptomatischer Gallenblasensteine beträgt 1�3%, beim asymptomatischen
Steinträger jedoch nur 0,1�0,3%. Der natürliche Verlauf von Gallengangssteinen ist
nicht hinreichend geklärt. Das klassische klinische Bild der akuten Cholangitis mit
Fieber, Ikterus und rechtsseitigen Oberbauchschmerzen (Charcot-Trias) wird nur bei
etwa 25% der Patienten angetroffen.
Diagnostik
Der Nachweis oder Ausschluss einer Cholezystolithiasis und einer akuten
Cholezystitis erfolgt primär durch die transkutane Sonografie. Wenn der
sonografische Nachweis oder Ausschluss von Gallengangssteinen nicht gelingt,
bestimmen die klinischen Symptome und die Zeichen der biliären Abfluss-
behinderung den Einsatz weiterer diagnostischer Maßnahmen, der sich auch nach
ihrer Verfügbarkeit richtet: Bei hochgradigem Verdacht auf Gallengangssteine
(erweiterter Gallengang > 7�10 mm, Hyperbilirubinämie, erhöhte γ-GT/ALT)
ist die ERC indiziert. Bei mäßiggradig wahrscheinlicher Choledocholithiasis sollte die
Endosonografie � oder alternativ eine MRC � vorgeschaltet werden. Eine
Metaanalyse von randomisierten kontrollierten Studien (RCT) fand zwischen
Endosonografie und MRC keine signifikanten Unterschiede hinsichtlich der
Sensitivität (93% vs. 85%) und Spezifität (96% vs. 93%) für die Diagnose von
Gallengangssteinen.

23
Therapieprinzipien
Die asymptomatische Cholezystolithiasis ist in der Regel keine Indikation zur
Therapie. Ausnahmen:
� Patienten mit Porzellangallenblase oder Gallenblasenpolypen ≥ 1 cm sollten
wegen des Karzinomrisikos (bis 50%) unabhängig von der Symptomatik
cholezystektomiert werden. Insbesondere Porzellangallenblasen mit fleckförmigen
Wandverkalkungen tragen ein höheres Karzinomrisiko.
� Bei großen abdominellen Eingriffen (z. B. malabsorptive/restriktive Adipositas-
chirurgie, ausgedehnte Resektion bei Morbus Crohn, radikale Gastrektomie mit
Lymphadenektomie) kann eine simultane Cholezystektomie auch bei
asymptomatischen Steinen vorgenommen werden.
Die laparoskopische Cholezystektomie ist die Standardtherapie für die sympto-
matische Cholezystolithiasis. Bei Verdacht auf Gallenblasenkarzinom sollte eine
offene Cholezystektomie erfolgen. Die aktuelle Cochrane-Analyse ergab für die
laparoskopische Cholezystektomie identische Komplikationsraten bei einer im Mittel
um 3 Tage kürzeren Krankenhausverweildauer und einer um 3 Wochen kürzeren
Rekonvaleszenz. Die Gallengangsverletzungsrate liegt heute bei der laparo-
skopischen Cholezystektomie mit 0,2�0,4% nicht mehr höher als bei der offenen
Cholezystektomie.
Therapie der Choledocholithiasis bei Zustand nach Cholezystektomie
Symptomatische Gallengangssteine sind eine Behandlungsindikation. Die Daten zum
natürlichen Verlauf von Gallengangssteinen belegen, dass symptomatische Gallen-
gangssteine bei mehr als 50% der Patienten im Verlauf erneut Koliken verursachen
und bei einem Viertel auch Komplikationen nach sich ziehen. Asymptomatische
Gallengangssteine können ebenfalls behandelt werden. Auch wenn langfristige
prospektive Daten bisher fehlen, so lassen die vorliegenden Daten den Schluss zu,
dass weniger als die Hälfte der Patienten mit asymptomatischen Gallengangssteinen
symptomatisch werden und mehr als 20% der Steine spontan abgehen.
Bei cholezystektomierten Patienten mit symptomatischen Gallengangssteinen sollte
eine endoskopische Steinextraktion nach EPT vorgenommen werden. Bei Misslingen
(auch unter Einsatz der mechanischen Lithotripsie) der endoskopischen oder
perkutan-transhepatischen Steinextraktion werden als adjuvante Lithotripsie-
verfahren ESWL, intrakorporale Laserlithotripsie oder elektrohydraulische Lithotripsie
eingesetzt. Bei gleichzeitiger Cholezystolithiasis sollte die chirurgische Alternative
früh erwogen werden.

24
Therapie der simultanen Choledocho- und Cholezystolithiasis
Bei Patienten mit gleichzeitig vorliegenden Gallenblasen- und Gallengangssteinen
wird ein therapeutisches Splitting empfohlen. Bei hoher Wahrscheinlichkeit einer
gleichzeitigen Choledocholithiasis bleibt die präoperative EPT und Steinextraktion die
wichtigste Behandlungsoption und wird auch in > 85% der deutschen Kliniken
bevorzugt. Die EPT und die Cholezystektomie sollten nicht am selben Tag erfolgen,
um postinterventionelle Komplikationen vor Operationsbeginn ausschließen zu
können. Nach erfolgreicher Gallengangssanierung sollte bei Cholezystolithiasis unter
Risikoabwägung möglichst innerhalb einer Woche cholezystektomiert werden.
Verlaufsbeobachtungen bei Patienten mit funktionstüchtiger, steinfreier Gallenblase
erlauben den Schluss, dass eine funktionstüchtige steinfreie Gallenblase nach
erfolgreicher EPT und Steinextraktion aufgrund des sehr geringen Komplikations-
risikos nicht entfernt werden muss.
Therapie der akuten Cholezystitis
Die akute Cholezystitis ist eine Indikation zur frühelektiven laparoskopischen
Cholezystektomie (möglichst innerhalb von 72 h nach Diagnosestellung).
Metaanalysen von RCT (mit insgesamt lediglich 451 Patienten) stützen die Vorteile
der frühelektiven laparoskopischen Cholezystektomie innerhalb von 72 h bei der
akuten Cholezystitis: Die Krankenhausverweildauer war bei der Spätoperation
3 Tage länger, und es mussten 18% der Patienten während der präoperativen
Wartephase notfallmäßig operiert werden, während die Komplikationsraten des
Eingriffs identisch waren. Falls der Patient nicht frühelektiv operiert werden kann,
sollte die Cholezystektomie erst im Intervall nach 6 Wochen erfolgen.
Therapie der akuten Cholangitis
Die obstruktive akute Cholangitis sollte so rasch wie möglich (bei septischen Zeichen
notfallmäßig) durch endoskopische Beseitigung des Steins behandelt werden. Eine
antibiotische Begleittherapie ist indiziert. Gelingt die Steinentfernung nicht, müssen
eine nasobiliäre Sonde oder ein Stent eingelegt werden. Falls das transduodenale
Vorgehen misslingt, ist eine perkutane Drainage angezeigt.

25
Prävention von Gallensteinen
Bei Adipositas ist zur Primärprävention der Cholelithiasis eine langsame Reduktion
des Körpergewichts unter Vermeidung zyklischer Gewichtsschwankungen und langer
Fastenperioden sinnvoll. Mehrere RCT zeigen, dass das Steinrisiko in Situationen,
die infolge Gewichtsreduktion mit hohem Risiko zur Bildung von Gallenblasensteinen
einhergehen (z. B. Reduktionsdiät mit Gewichtsabnahme > 1,5 kg/Woche),
Adipositaschirurgie), durch UDCA vermindert werden kann. Die Dosis sollte
mindestens 500 mg/Tag betragen, und die Therapie sollte bis zur Gewichts-
stabilisierung fortgeführt werden.
Es gibt keine gesicherte medikamentöse Prävention der Entstehung von
Rezidivsteinen in den Gallengängen.
Literatur: 1. Gurusamy KS, Samraj K. Early versus delayed laparoscopic cholecystectomy
for acute cholecystitis. Cochrane Database Syst Rev. 2006:CD005440. 2. Lammert F, Neubrand MW, Bittner R, Feussner H, Greiner L, Hagenmüller F,
Kiehne KH, Ludwig K, Neuhaus H, Paumgartner G, Riemann JF, Sauerbruch T für die Teilnehmer der Konsensuskonferenz. S3-Leitlinie der Deutschen Gesellschaft für Verdauungs- und Stoffwechselkrankheiten und der Deutschen Gesellschaft für Viszeralchirurgie zur Diagnostik und Behandlung von Gallensteinen. AWMF-Register Nr. 021/008. Z Gastroenterol. 2007;45:971�1001 (http://www.dgvs.de/).
3. Martin DJ, Vernon DR, Toouli J. Surgical versus endoscopic treatment of bile
duct stones. Cochrane Database Syst Rev. 2006:CD003327. 4. Schiphorst AH, Besselink MG, Boerma D, Timmer R, Wiezer MJ, van Erpecum
KJ, Broeders IA, van Ramshorst B. Timing of cholecystectomy after endoscopic sphincterotomy for common bile duct stones. Surg Endosc. 2008;22:2046�50.
5. Yamashita Y, Takada T, Kawarada Y, Nimura Y, Hirota M, Miura F, Mayumi T,
Yoshida M, Strasberg S, Pitt HA, de Santibanes E, Belghiti J, Buchler MW, Gouma DJ, Fan ST, Hilvano SC, Lau JW, Kim SW, Belli G, Windsor JA, Liau KH, Sachakul V. Surgical treatment of patients with acute cholecystitis: Tokyo Guidelines. J Hepatobiliary Pancreat Surg. 2007;14:91�7.

26
Leberbiopsie � der Pathologe als letzte Instanz bei der Diagnostik diffuser Lebererkrankungen?
A. Tannapfel
Institut für Pathologie, Ruhr-Universität Bochum
Die Leberbiopsie ist eine wesentliche Maßnahme in der diagnostischen Abklärung
einer chronischen Lebererkrankung. Ihre klinische Relevanz ist trotz erheblicher
klinischer Fortschritte, auch in der bildgebenden Diagnostik und Molekularbiologie,
ungebrochen. Vor dem Hintergrund steigender und zunehmend differenzierter
therapeutischer Optionen ist jedoch eine kontinuierliche Anpassung an den aktuellen
Erkenntnisstand, eine Standardisierung der Befunde und vor allem eine Qualitäts-
sicherung erforderlich, um den diagnostischen Nutzen der Leberbiopsie zu
optimieren und ihren Einsatz zu rechtfertigen. Im Folgenden soll versucht werden,
den Wert der Leberbiopsie insbesondere bei den neueren Krankheitsentitäten zu
bewerten. Im Folgenden soll lediglich auf diese Krankheitsbilder eingegangen
werden, da der Wert der Leberbiopsie z. B. bei Raumforderungen in der Leber und
auch bei einer Hepatitis-Virusinfektion unstrittig ist.
Innerhalb der letzten Jahre hat sich insbesondere das Verständnis von autoimmunen
Lebererkrankungen sowie der Lebererkrankungen durch Alkohol und Fremdstoffe
gewandelt. Nach wie vor gilt es als unstrittig, dass über 60% aller chronischen
Lebererkrankungen in Deutschland durch Alkohol verursacht oder durch Alkohol
wesentlich verschlimmert werden. Insgesamt können 3 Krankheitsbilder der Alkohol-induzierten Leberschädigung abgegrenzt werden:
� Alkohol-bedingte Fettleber,
� Alkohol-Hepatitis,
� Alkohol-bedingte Leberzirrhose.
Die 3 Formen der Alkohol-bedingten Leberschädigung können sich nacheinander
entwickeln, wobei die Übergänge fließend sind. So kann eine Alkohol-bedingte
Fettleber mit Fibrose und granulozytärer Entzündungsreaktion (Fettleber- oder
Alkohol-Hepatitis) oder eine Zirrhose mit Fetteinlagerung als Primärmanifestation
auftreten. Auch bei Zirrhose kann eine Alkohol-Hepatitis beobachtet werden. Der
Goldstandard in der Diagnose einer Alkohol-bedingten Leberschädigung ist die
Leberbiopsie, die in mehr oder weniger regelhafter Ausprägung eine Fetteinlagerung

27
der Hepatozyten, Zelluntergänge und Fibrose (sogenannte Maschendrahtfibrose)
neben einer variablen Cholestase zeigt. Mallory-Körper können vorkommen, sind
jedoch nicht pathognomonisch, da sie auch bei anderen Lebererkrankungen
(Lebertumoren, Leberschädigung durch Fremdstoffe) beobachtet werden. Die
Leberbiopsie bei Alkohol-toxischer Schädigung liegt in der Möglichkeit der Erfassung
und Objektivierung der bereits eingetretenen Schädigung (Fibrose/Zirrhose) und in
der Aussage, ob noch eine floride Schädigung vorliegt.
Obwohl die überwiegende Mehrzahl der histopathologisch verifizierbaren
Änderungen durch Alkohol zumindest teilverursacht sind, weiß man heute, dass ein
nahezu gleichartiges histologisches Bild auch bei einer anderen Erkrankung
auftreten kann, die heute �nicht-alkoholische Steatohepatitis� (NASH) genannt
wird. Dieser Begriff wurde erstmals 1980 verwendet. Durch die Kenntnis dieses
neuen Krankheitsbildes glaubt man, dass mehr als 10% aller Lebererkrankungen
durch eine NASH verursacht sind. Die NASH wird heute als eigenständige
nosologische Einheit anerkannt. Unter NASH versteht man das gemeinsame
Auftreten eines Leberzellschadens in Form einer Verfettung in Zusammenhang mit
einer Entzündungszellinfiltration und einer (perivenulären) Fibrose. Damit lässt sich
das histopathologische Bild der NASH nicht immer von dem der Alkohol-
Steatohepatitis (ASH) unterscheiden. Synonyme der jetzt als NASH bezeichneten
Entität sind �Fettleber-Hepatitis�, �diabetische Hepatitis� oder �Pseudo-Alkohol-
toxische Hepatitis�. Die meisten Patienten sind Frauen, übergewichtig, im mittleren
Lebensalter, mit einer zum Teil schon manifesten Stoffwechselerkrankung (Diabetes
mellitus). Allerdings sind jetzt bereits Einzelfälle einer NASH dokumentiert, bei denen
es sich um normalgewichtige Männer handelt. Bei bis zu 75% der Patienten besteht
eine Insulinresistenz. Leberzirrhosen nach NASH sind beschrieben, allerdings
lediglich in 10�20%. Neben Adipositas und Diabetes mellitus scheinen Medikamente
und Stoffwechselstörungen bzw. Ernährungsstörungen eine weitere Rolle in der
Entstehung der NASH zu spielen. Insbesondere synthetische Östrogene, Amiodaron
als Antiarrhythmikum und lang dauernde Kortikosteroid-Gaben scheinen
ätiopathogenetisch relevant zu sein. Die gemeinsame Pathogenese dieser
leberschädigenden Agenzien ist der oxidative Zellstress, der bei der Entstehung von
NASH eine wichtige Rolle zu spielen scheint.
Aufgrund der zunehmenden Bedeutung von NASH sollte die früher häufig gestellte
Diagnose �Alkohol-induzierter Leberschaden� in der Biopsie daher etwas

28
zurückhaltender gestellt werden. Eine NASH kann auch bei (weitgehend) normalen
Transaminasen vorliegen. Der Wert der Leberbiopsie liegt in der Möglichkeit der
Diagnoseobjektivierung und dem Ausschluss möglicher zusätzlicher Schäden. Eine
standardisierte Befundübermittlung mittels eines Scores sollte angestrebt werden.
Eine neue Entität, die Chemotherapie-assoziierte Steatohepatitis (CASH) wird
dann diagnostiziert, wenn ein Patient die Zeichen einer NASH mit vorwiegender
medikamentös-toxischer Leberschädigung aufweist � und zusätzlich Endothelzell-
schäden aufgrund direkter Toxizität bei beispielsweise Oxaliplatin-Gabe.
Autoimmunerkrankungen und Overlaps Neben den 3 Hauptautoimmunerkrankungen der Leber, der Autoimmunhepatitis, der
primär biliären Zirrhose und der primär sklerosierenden Cholangitis sind eine ganze
Reihe von Überlappungssyndromen bekannt geworden, die allerdings bisher noch
nicht standardisiert definiert wurden. Es ist bis heute nicht klar, ob die Overlap-
Syndrome überhaupt distinkte Krankheitsentitäten darstellen oder Varianten von
Ausprägungsformen der etablierten autoimmunen Lebererkrankungen sind. Dennoch
sollten Overlap-Syndrome bei jeder Autoimmunerkrankung der Leber in die
differenzialdiagnostischen Erwägungen einbezogen werden, da erste Daten zeigen,
dass hier deutlich unterschiedliche Verläufe auftreten können.
Die Diagnose eines Overlap-Syndroms basiert auf einer typischen biochemischen
Serumkonstellation, darüber hinaus auf histologischen Faktoren, weniger auf der
klinischen Symptomatik.
Patienten mit Overlap-Syndromen zeigen meistens unspezifische Symptome
(Müdigkeit, Arthralgien, Myalgien), die eine Diagnostik in die eine oder andere
Richtung nicht zulassen. Erschwert wird die Problematik durch Übergangsformen
von einer in die andere autoimmune Hepatopathie (z. B. einer primär biliären
Zirrhose zu einer primär biliären Zirrhose-Autoimmunhepatitis nach langjährigem
Verlauf).
Der Begriff des Overlap-Syndroms sollte nicht benutzt werden, wenn Überlappungs-
syndrome zwischen einer autoimmunen und nicht autoimmunen Hepatopathie
bestehen, z. B. bei AIH und HCV. Patienten mit Autoimmunhepatitis und Hyper-
gammaglobulinämie haben darüber hinaus relativ häufig falsch-positive Anti-HCV-
Tests. Patienten mit HCV haben in bis zu 65% Autoantikörperphänomene.

29
AIH-PBC-Overlap Patienten, die klinische, biochemische, serologische und histologische Merkmale
beider Erkrankungen aufweisen, sind zunächst Anfang der 70er-Jahre beschrieben
worden. 8% aller Patienten mit AIH scheinen ein Overlap-Syndrom aufzuweisen.
Diese Patienten zeigen typische serologische Kennzeichen für eine PBC (AMA-M2-
positiv) und in der Histologie Gallengangsschädigungen (wie bei PBC), histologisch
allerdings zusätzlich ein mehr hepatitisches Bild, d. h. die Entzündung findet sich für
eine PBC unerwartet stark intraazinär ausgeprägt.
ANA-Autoantikörper sind zumeist ebenfalls vorhanden, allerdings aufgrund ihrer
häufigen falschen Positivität oder ihres Vorkommens in der gesunden Bevölkerung
nicht spezifisch. In der Literatur wird beschrieben, dass auch ein sequenzielles
Auftreten der Erkrankung (zunächst PBC, dann AIH) möglich ist. Eine Autoimmun-
hepatitis kann den Verlauf einer primär biliären Zirrhose deutlich verschlimmern.
Autoimmuncholangitis (AIC), AIC-AIH-Overlap-Syndrome Die Autoimmuncholangitis zeigt histologische Veränderungen (die mit der primär
biliären Zirrhose vergleichbar sind), ist aber serologisch AMA-negativ und wird daher
in einzelnen Publikationen auch als �AMA-negative PBC� bezeichnet. Die
Autoimmuncholangitis kommt häufiger bei Frauen vor und zeigt ein cholestatisches
Serumenzymprofil. Histologisch finden sich akute, floride Gallengangsläsionen. Der
Progress der Erkrankung hin zu einer Leberzirrhose ist langsam. Patienten mit AIC
sind AMA-negativ, zeigen aber relativ häufig ANAs oder andere Autoantikörper.
Der Verlauf der Erkrankung zeigt, dass es durchaus berechtigt erscheint, AIC und
PBC als Varianten einer einzelnen Erkrankung zu sehen, die lediglich in ihren
Autoantikörperprofilen differieren. Overlap-Syndrome im Sinne einer Autoimmun-
hepatitis und Autoimmuncholangitis sind beschrieben, gelten jedoch als sehr selten.
Sie werden in Analogie zur AIH-PBC-Overlap-Gruppe behandelt.
AIH-PSC-Overlap-Syndrome Während AIH-PBC-Overlap-Syndrome häufig bei Erwachsenen gefunden werden, ist
das Overlap-Syndrom der AIH-PSC häufig bei Kindern, Heranwachsenden und
jungen Erwachsenen beschrieben worden. Der AIH-Score zeigt in 8% der Patienten
mit PSC ein Overlap-Syndrom an. Die Diagnose eines AIH-PSC-Overlaps gilt dann
als wahrscheinlich, wenn der AIH-Score größer als 15 ist, ANAs oder ASMA-

30
Autoantikörper nachweisbar sind (Titer min. 1:40), und die Leberhistologie Motten-
fraßnekrosen, ein Lymphozyteninfiltrat sowie eine periportale oder periseptale
Inflammation aufweist � also ein Mischbild aus PSC und AIH. Patienten mit der AIH-
PSC-Overlap-Erkrankung scheinen häufiger schwere Verlaufsformen der CED zu
besitzen, zeigen positive ANCAs im Serum und erhöhte Transaminasen (höher als
bei AIH erwartet). Diagnostisch problematisch ist die Beobachtung, dass die AIH und
PSC nicht zusammen, sondern möglicherweise auch sequenziell vorkommen
können.
Patienten mit PSC-AIH zeigen einen deutlich schwereren Verlauf als die mit AIH
allein oder PBC-AIH. Patienten mit PSC-AIH sind generell jünger, häufig männlich.
Bei den hereditären Lebererkrankungen wurde nicht nur der Zusammenhang
zwischen Insulinresistenz, Diabetes mellitus und Eisenüberladung in letzter Zeit
immer deutlicher, sondern konnte letztendlich auch epidemiologisch jetzt zweifelsfrei
nachgewiesen werden. Bei Patienten, die histologisch eine Siderose der Leber, aber
keine hereditäre Hämochromatose aufwiesen, besteht überzufällig häufig eine
Insulinresistenz mit abnormer Glukosetoleranz, Adipositas und konsekutiver
Hyperlipidämie. Möglicherweise kann hier zukünftig eine neue Krankheitsentität
definiert werden. Allerdings ist deren Ursache und Pathogenese letztendlich noch
unklar.
Korrespondenzadresse: Prof. Dr. med. Andrea Tannapfel Institut für Pathologie Ruhr-Universität Bochum Bürkle-de-la Camp Platz 1 44789 Bochum Tel.: 0234 302-4800 Fax: 0234 302-4809 E-Mail: [email protected] Website: http://www.pathologie-bochum.de

31
Prävention und Management von Resistenzen bei der chronischen Hepatitis B
M. Cornberg
Klinik für Gastroenterologie, Hepatologie und Endokrinologie, Medizinische
Hochschule Hannover
Das Ziel der Therapie der chronischen Hepatitis B ist die Morbidität und Mortalität der
Hepatitis-B-Virus (HBV)-Infektion zu senken. Um dieses Ziel zu erreichen, müssen
Surrogatmarker während und nach der Behandlung zur Überprüfung des
Therapieerfolgs herangezogen werden. Die dauerhafte Serokonversion von HBs-
Antigenpositivität zur HBs-Antikörperpositivität wäre optimal, kann mit den heute zur
Verfügung stehenden Therapeutika aber nur in weniger als 5�10% der Fälle erreicht
werden. Kriterien eines Therapieansprechens sind: maximaler dauerhafter Abfall der
HBV DNA, dauerhafte HBe-Serokonversion bei der Wildtyp-Infektion, insbesondere
eine Abnahme des Fibrosestadiums in der Histologie, und letztendlich damit
verbunden Verhinderung von Zirrhose, hepatozellulärem Karzinom (HCC), Trans-
plantation und Tod.
Die Behandlungsoptionen der chronischen Hepatitis B haben sich in den letzten
Jahren zunehmend verbessert. Mittlerweile bestehen sie zum einen in der Gabe von
(pegyliertem) Interferon-α und auf der anderen Seite von Nukleosid- und
Nukleotidanaloga, die die HBV-Replikation hemmen. Da eine Interferon-Therapie
grundsätzlich zeitlich begrenzt und der Therapieerfolg in der Regel dauerhaft ist,
sollte zunächst geprüft werden, ob eine Interferon-Therapie möglich und sinnvoll ist
(1). Wird keine Interferon-Therapie begonnen oder der Patient hat nicht auf Interferon
angesprochen, kommen die Nukleos(t)idanaloga zum Einsatz. Mit diesen
Medikamenten ist es möglich bei fast allen Patienten die HBV DNA zu senken, ohne
dass die Therapie mit starken Nebenwirkungen verbunden ist. Die größte
Herausforderung ist aber die Vermeidung von Resistenzen. Daher ist es wichtig bei
der Auswahl von Nukleos(t)idanaloga das Stadium der Lebererkrankung sowie die
Höhe der HBV Virämie zu berücksichtigen. Bei einer Ausgangsviruslast > 1 Mio.
Kopien/ml ist die Wahrscheinlichkeit einer späteren Lamivudin-Resistenz sehr groß,
sodass bei höherer HBV DNA Lamivudin nicht eingesetzt werden sollte. Liegt eine
Leberzirrhose vor, sollte kein Risiko einer Resistenzentwicklung eingegangen
werden, daher ist eine Substanz mit hoher genetischer Resistenzbarriere oder primär

32
eine Kombinationstherapie zu bevorzugen (1, 2). Um Resistenzen zu verhindern, ist
die Kenntnis der antiviralen Effektivität (Tab. 2), der Resistenzbarriere (Tab. 1) und
des Resistenzprofils (Tab. 2) der zur Verfügung stehenden oralen antiviralen
Medikamente Voraussetzung für deren rationalen Einsatz. Wichtig im weiteren
Verlauf ist die Kontrolle der HBV DNA alle 3 Monate, um Resistenzen frühzeitig zu
erkennen (1).
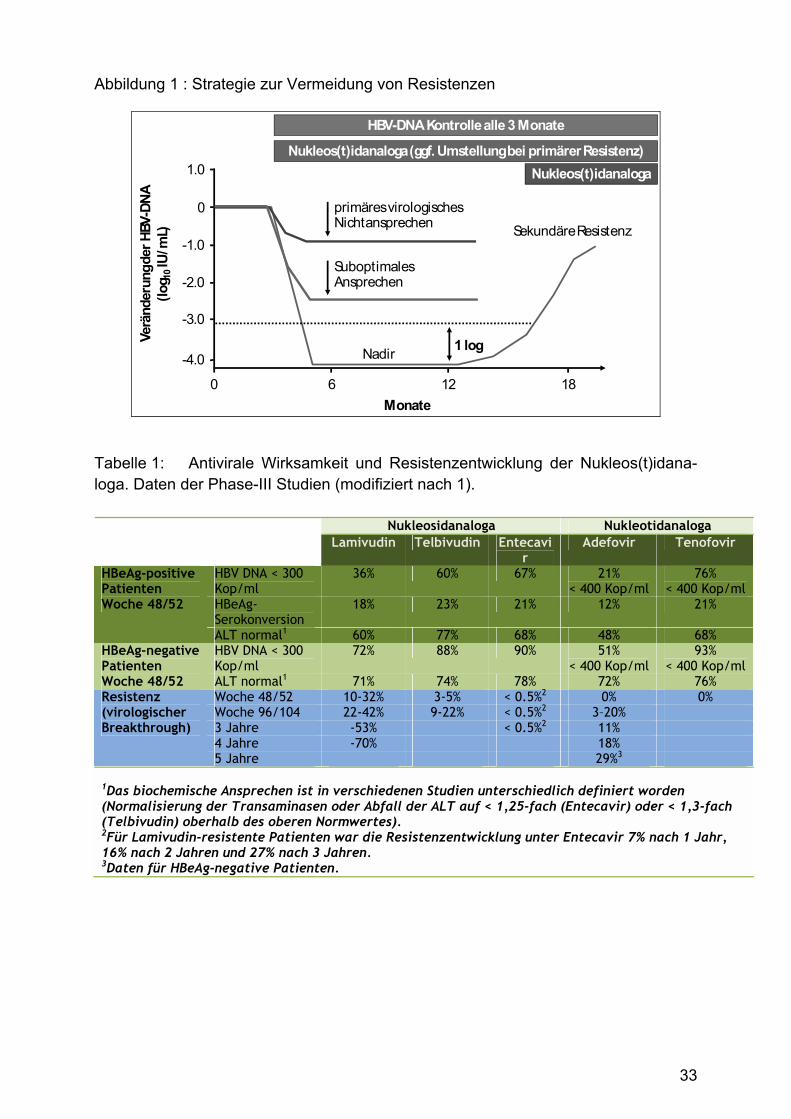
Grundsätzlich sind primäres und sekundäres Therapieversagen zu unterscheiden
(Abb. 1). Von einem primären virologischen Nichtansprechen wird ausgegangen,
wenn nicht mindestens 1-log-Abfall der HBV DNA nach 3-monatiger Therapie
vorliegt. Ein klinisch ausreichendes Ansprechen sollte allerdings nach 6 Monaten
eine Reduktion der HBV DNA unter 1000 Kopien/ml oder ein fortgesetzter Abfall der
HBV DNA bis Monat 12 vorliegen. Die Anforderungen an das Therapieansprechen
unterscheiden sich allerdings für die verschiedenen Substanzen, z. B. sollte die HBV
DNA beim Einsatz von Lamivudin nach 3 Monaten Therapie bereits unter der
Nachweisgrenze eines hochsensitiven Assays liegen.
Von einer sekundären Resistenz wird ausgegangen, wenn ein Anstieg der HBV DNA
um mindestens 1-log-Stufe über den Nadir unter fortgesetzter antiviraler Therapie
auftritt. Hier ist das Auftreten von Lamivudin-resistenten Mutanten (bis zu 20% pro
Jahr) zu erwähnen. In diesem Fall ist die Therapie möglichst frühzeitig anzupassen,
d. h. sobald sich ein virologischer Rückfall zeigt, auch wenn noch kein biochemischer
Rückfall erfolgt ist. Primär ist die zusätzliche Gabe (�add-on�) von Nukleotidanaloga
mit nicht-überlappendem Resistenzprofil (s. Tab. 2) zu empfehlen (1). Eine
unkontrollierte sequenzielle Therapie ist unbedingt zu vermeiden, da die Selektion
von Kreuzresistenzen erzeugt werden kann. Die Bestimmung von Polymerasegen-
Mutationen des Hepatitis-B-Virus ist möglich und sollte in bestimmten Fällen für die
Therapieplanung erwogen werden, insbesondere wenn bereits mehrere Medika-
mente eingesetzt wurden.
33
Abbildung 1 : Strategie zur Vermeidung von Resistenzen
1 logVerä
nder
ung d
er H
BV-D
NA(lo
g 10IU
/mL)
0
-1.0
-2.0
-3.0
-4.0
1.0
Nadir
Sekundäre Resistenz
Nukleos(t)idanaloga (ggf. Umstellung bei primärer Resistenz)
Monate60 12 18
primäres virologisches Nichtansprechen
Suboptimales Ansprechen
HBV-DNA Kontrolle alle 3 Monate
Nukleos(t)idanaloga
Tabelle 1: Antivirale Wirksamkeit und Resistenzentwicklung der Nukleos(t)idana-loga. Daten der Phase-III Studien (modifiziert nach 1).
Nukleosidanaloga Nukleotidanaloga Lamivudin Telbivudin Entecavi
r Adefovir Tenofovir
HBV DNA < 300 Kop/ml
36% 60% 67% 21% < 400 Kop/ml
76% < 400 Kop/ml
HBeAg-Serokonversion
18% 23% 21% 12% 21%
HBeAg-positive Patienten Woche 48/52
ALT normal1 60% 77% 68% 48% 68% HBV DNA < 300 Kop/ml
72% 88% 90% 51% < 400 Kop/ml
93% < 400 Kop/ml
HBeAg-negative Patienten Woche 48/52 ALT normal1 71% 74% 78% 72% 76%
Woche 48/52 10-32% 3-5% < 0.5%2 0% 0% Woche 96/104 22-42% 9-22% < 0.5%2 3�20% 3 Jahre -53% < 0.5%2 11% 4 Jahre -70% 18%
Resistenz (virologischer Breakthrough)
5 Jahre 29%3
1Das biochemische Ansprechen ist in verschiedenen Studien unterschiedlich definiert worden (Normalisierung der Transaminasen oder Abfall der ALT auf < 1,25-fach (Entecavir) oder < 1,3-fach (Telbivudin) oberhalb des oberen Normwertes). 2Für Lamivudin-resistente Patienten war die Resistenzentwicklung unter Entecavir 7% nach 1 Jahr, 16% nach 2 Jahren und 27% nach 3 Jahren. 3Daten für HBeAg-negative Patienten.

34
Tabelle 2: Resistenzprofile der Nukleos(t)idanaloga nach 2. HBV-Variante Lamivudin Telbivudin Entecavir Adefovir Tenofovir Wild-type S S S S S M204I R R I/R S S L180M + M204V R R I S S A181T/V I S S R S N236T S S S R I L180M + M204V/I±I169T±V173L±M250V
R R R S S
L180M + M204V/I±T184G±S202I/G
R R R S S
Literatur: 1. Cornberg M, Protzer U, Dollinger MM, Petersen J, Wedemeyer H, Berg T, Jilg
W, Erhardt A, Wirth S, Schirmacher P, Fleig WE, Manns MP. Prophylaxe, Diagnostik und Therapie der Hepatitis-B-Virus-(HBV-)Infektion � �Upgrade" der Leitlinie, AWMF-Register-Nr.: 021/011 [Prophylaxis, Diagnosis and Therapy of Hepatitis-B-Virus-(HBV-)Infection: upgrade of the guideline, AWMF-Register 021/011]. Z Gastroenterol. 2007;45:525�74.
2. European Association for the Study of the Liver. EASL Clinical Practice
Guidelines: Management of chronic hepatitis B. J Hepatol. 2009;50:227�42.

35
Individualisierung der Therapiestrategie bei chronischer Hepatitis C � neuer Standard
S. Zeuzem
Medizinische Klinik I, Universitätsklinikum Frankfurt
Als primäres Ziel einer antiviralen Therapie bei Patienten mit chronischer Hepatitis C
gilt ein dauerhaftes virologisches Ansprechen, definiert als fehlender Nachweis
Hepatitis-C-spezifischer RNA im Serum 6 Monate nach Therapieende. Wird dieses
Ziel erreicht, kommt es im weiteren Verlauf nur sehr selten (1�2%) zu einem späten
Rückfall mit erneutem Nachweis von HCV RNA im Serum.
Zur Beurteilung eines fehlenden Therapieansprechens nach Beginn der Behandlung
sollte bei Patienten mit einer HCV-Genotyp-1-Infektion eine Bestimmung der HCV
RNA im Blut zu Woche 12 und 24 herangezogen werden. Bei einem fehlenden Abfall
der Viruslast um 2 log-Stufen im Vergleich zur Viruslast vor Therapiebeginn bzw. bei
einer absoluten HCV-RNA-Konzentration über 30.000 IU/ml nach 12 Behandlungs-
wochen sowie einem qualitativen HCV-Nachweis nach 24 Behandlungswochen
(HCV RNA ≥ 10�50 IU/ml), wird ein Absetzen der Therapie empfohlen, da ein
dauerhaftes virologisches Ansprechen mit max. 1�2% praktisch nicht mehr möglich
ist (negativer prädiktiver Wert 98�100%). Zusätzlich kann unter besonderen
Voraussetzungen anhand einer Bestimmung der HCV RNA zu Therapiewoche 4 eine
Verkürzung der Therapiedauer vorgenommen werden (s. unten).
Therapie der chronischen Hepatitis C bei nicht vorbehandelten Patienten
HCV-Genotyp 1 Die Standardtherapie der chronischen Hepatitis C erfolgt mit einem pegylierten
Interferon in Kombination mit Ribavirin. Patienten mit einer HCV-Genotyp-1-Infektion
sollten über 48 Wochen behandelt werden. PEG-IFN wird einmal wöchentlich
subkutan injiziert. Die Gabe des Ribavirins erfolgt verteilt auf 2 Dosen pro Tag per os
mit einer Tagesgesamtdosis je nach Körpergewicht zwischen 800 und 1400 mg. Die
dauerhaften virologischen Ansprechraten für Genotyp-1-infizierte Patienten betragen
dabei 42�51%.

36
Eine Therapieverkürzung auf 24 Wochen ist für Patienten mit niedriger
Ausgangsviruslast (≤ 600.000 bzw. 800.000 IU/ml) zugelassen, wenn zusätzlich
unter der Kombinationstherapie mit PEG-IFN und Ribavirin bereits zu Woche 4 keine
HCV RNA mehr im Serum nachweisbar ist.
Patienten mit einem langsamen Abfall der HCV RNA profitieren von einer
Verlängerung der Therapiedauer. Verschiedene Studien zeigen, dass Patienten mit
einem verzögerten virologischen Ansprechen (Abfall der HCV RNA zu Woche 12 um
2 log-Stufen, aber noch HCV-RNA-positiv und Negativierung der HCV RNA
[< 50 IU/ml] im Verlauf zu Woche 24) nach einer 72-wöchigen Behandlung signifikant
höhere dauerhafte Ansprechraten aufweisen als nach einer 48-wöchigen Therapie.
HCV-Genotyp 2, 3 Die virologischen Ansprechraten mit einer PEG-IFN/Ribavirin-Kombinationstherapie
über 48 Wochen sind bei Patienten mit einer HCV-Genotyp-2- oder -3-Infektion
annähernd doppelt so hoch wie bei HCV-Genotyp 1 (76�82% vs. 42�52%). Viele
Studien haben gezeigt, dass eine Verkürzung der Therapiedauer von 48 auf
24 Wochen für Patienten mit einer Genotyp-2- oder -3-Infektion ohne Beeinträchti-
gung der dauerhaften virologischen Ansprechraten möglich ist. Basierend auf diesen
Ergebnissen wurde die 24-wöchige Therapie der Genotyp-2- und -3-Infektion als
Standard etabliert. Ebenfalls konnten mit der Kombinationstherapie aus PEG-IFN-α
mit einer fixen Ribavirin-Dosis von 800 mg pro Tag ähnlich hohe dauerhafte
Ansprechraten im Vergleich zu einer körpergewichtsadaptierten Dosierung
nachgewiesen werden. Basierend auf Subanalysen wurden in einzelnen Studien
unterschiedliche virologische Ansprechraten für Patienten mit Genotyp-2- und
-3-Infektion nachgewiesen (93% vs. 79%). Insbesondere Patienten mit Genotyp-3-
Infektion und hoher HCV-RNA-Konzentration vor Therapiebeginn (> 600.000 bzw.
800.000 IU/ml) zeigen signifikant häufiger einen Rückfall nach Therapieende auf als
Patienten mit Genotyp-2-Infektion oder Genotyp-3-Infektion mit niedriger Ausgangs-
viruslast.
In verschiedenen Studien wurde die Möglichkeit einer weiteren Reduktion der
Therapiedauer von 24 auf 16, 14 bzw. 12 Wochen mit der Gabe von PEG-IFN-α2a
bzw. -α2b und körpergewichtsadaptiertem Ribavirin untersucht. Patienten mit einer
niedrigen Ausgangsviruslast und einem raschen initialen Abfall der HCV-Virus-
konzentration (HCV RNA zu Woche 4 < 50�600 IU/ml) wiesen insgesamt hohe
dauerhafte virologische Ansprechraten von 80�95% (Genotyp 2) und 75�90%

37
(Genotyp 3) unter der verkürzten Therapiedauer auf, sodass bei diesen Patienten-
gruppen eine Reduktion der Therapiedauer ohne Verschlechterung der dauerhaften
virologischen Ansprechraten möglich scheint.
Patienten mit noch nachweisbarer HCV RNA zu Woche 4 zeigten deutlich niedrigere
dauerhafte virologische Ansprechraten auch unter der 24-wöchigen Therapie, sodass
in dieser Subgruppe in zukünftigen Studien sogar eine Verlängerung der gegen-
wärtigen 24-wöchigen Standardtherapiedauer geprüft werden muss.

38
Stadiengerechte Therapie der Varizenblutung
T. Sauerbruch
Medizinische Klinik und Poliklinik I, Universitätsklinikum Bonn
Leider gibt es in Deutschland keine guten epidemiologischen Daten zur Häufigkeit
der akuten Varizenblutung. Es ist aber der Eindruck der meisten Zentren, dass die
akute Varizenblutung abgenommen hat. Gleichzeitig ist die Klinikletalität der akuten
Blutung an Ösophagusvarizen auf etwa 20�30% zurückgegangen. Dies hat mehrere
mögliche Gründe:
- Erfolgreiche Therapie der Virus-induzierten chronischen Hepatitis
- Erfolgreiche Prophylaxe der ersten Varizenblutung
- Bessere therapeutische Konzepte der Behandlung der akuten Blutung
Nach wie vor gliedert sich die Behandlung der Varizenblutung in 3 Situationen:
Verhinderung der Erstblutung, Therapie der akuten Blutung und Verhinderung der
Rezidivblutung.
Hierfür stehen verschiedene Therapieansätze zur Verfügung:
Medikamentöse Senkung des Portaldrucks, Shunt-Verfahren, lokale endoskopische
Techniken sowie systemische Therapien zur Unterdrückung von Triggermechanis-
men (z. B. Zytokinausschüttung).
Zur Verhinderung der Erstblutung sind nicht-selektive Betablocker und die Ligatur
Standard. Die Behandlung der akuten Blutung umfasst die endoskopische
Hämostase sowie eine adjuvante medikamentöse Portaldrucksenkung zusammen
mit der Gabe von Antibiotika. Die Behandlung der Rezidivblutung gliedert sich in eine
Fortführung der Ligatur unter gleichzeitiger medikamentöser Therapie (nicht-selektive
Betablocker) oder die Anlage eines Shunts.

39
Therapie des Reizdarmsyndroms
P. Layer
Israelitisches Krankenhaus, Hamburg
Funktionelle Störungen des Verdauungstrakts sind weltweit verbreitet; auch in der
westlichen Hemisphäre zählen sie zu den häufigsten chronischen Erkrankungen,
wobei ca. mehr als 20% der Bevölkerung in unterschiedlicher Ausprägung betroffen
sind. Die wichtigste Rolle spielt hierbei das Reizdarmsyndrom (RDS).
Pathogenese und Pathophysiologie: Ätiologie und Pathogenese sind noch
weitgehend unklar, obwohl in den letzten Jahren viele wesentliche Erkenntnisse
gewonnen werden konnten. Es ist wahrscheinlich, dass verschiedene Patho-
mechanismen eine Rolle spielen können, die ihrerseits wieder für ein uneinheitliches
Symptombild veranwortlich sind. Von großer Bedeutung sind wahrscheinlich
Veränderungen der Schmerzwahrnehmung im Intestinum. Darüber hinaus werden
Störungen der Motilität, Unverträglichkeiten von Nahrung, immunologische oder
mikrobiologische Alterationen sowie psychosomatische Einflüsse mit unter-
schiedlichem Evidenzgrad vermutet. Bei einer Untergruppe der Patienten wird die
Erkrankung offenbar durch eine bakterielle Enteritis ausgelöst (�postinfektiöses
Reizdarmsyndrom�).
Symptomatik: Charakteristisch ist ein oft kombiniertes Auftreten unterschiedlicher
Symptome, insbesondere abdominale Schmerzen, wechselnde Stuhlkonsistenz
(Diarrhö, Obstipation) und Blähungen. Diese können mit verschiedenartigen weiteren
unspezifischen funktionellen oder vegetativen Symptomen assoziiert sein.
Diagnostik: Bei Vorhandensein der typischen Leitsymptome sowie bei Fehlen von
Beschwerden, die auf eine organische Ursache hinweisen (z. B. Fieber, sichtbares
Blut im Stuhl, Gewichtsverlust etc.) kann ein Reizdarmsyndrom vermutet werden. Die
Diagnosesicherung gelingt durch den Ausschluss wichtiger organischer Differen-
zialdiagnosen. Hierbei sollte diese Ausschlussdiagnostik nicht zu breit angelegt
werden, aber umfassend genug sein, um Patienten und behandelndem Arzt die
notwendige Sicherheit zu geben. Empfohlen wird hierbei: Basislabor, Abdomen-
sonografie und � in den meisten Fällen � die Koloskopie; bei Frauen sollte auch eine

40
gynäkologische Untersuchung erfolgen. Prinzipiell bewährt es sich, initial eine
einmalige, aber sorgfältige Abklärung durchzuführen, weil dies bereits selbst
therapeutisch effektiv ist. Eine wiederholte apparative Diagnostik ohne begründeten
Anlass ist aber unbedingt zu vermeiden.
Behandlung: Die unklare Ätiologie und Pathophysiologie bedingt, dass weder eine
kausale noch eine in die Pathomechanismen gezielt eingreifende Therapie verfügbar
ist. Ein wichtiges Fundament für jede Behandlung ist daher zunächst die solide
diagnostische Abklärung und Ausschluss der relevanten Differenzialdiagnosen,
welche die Basis jeder Behandlung darstellen muss; sie nimmt Patienten und Arzt
die Sorge vor einer versteckten gefährlichen Grundkrankheit und verbessert dadurch
das Ansprechen jeder weiteren Behandlung. Wichtig ist dann die klare Diagnose-
vermittlung mit anschaulicher, aber nicht bagatellisierender Aufklärung über das
Wesen und den langfristig benignen Verlauf der Erkrankung.
Die medikamentöse Therapie im weiteren Sinn erfolgt symptomorientiert und
konzentriert sich dementsprechend auf die Behandlung der wesentlichen
Beschwerden wie Schmerz, Diarrhö und Obstipation.
Ballaststoffe: Ballaststoffe werden versuchsweise empfohlen, da sie durch die
Absorption von Fetten, Bakterien und Gallensäuren die Transitzeit von Nahrungs-
mitteln verkürzen und den Darm schneller füllen und insgesamt einen positiven Effekt
auf das RDS sowohl vom Obstipationstyp als auch vom Diarrhötyp haben. Hierbei
sind Flohsamenpräparate (vom Typ des Mucofalk®) einfachen Kleieprodukten, die
bei vielen Patienten etwaige Blähungen und abdominale Missempfindungen
auslösen oder ungünstig beeinflussen, vorzuziehen.
Laxanzien: Sollten insbesondere die Obstipationsbeschwerden mithilfe von Ballast-
stoffen nicht beseitigt werden können oder werden diese nicht vertragen, kann ein
Therapieversuch mit Laxanzien angebracht sein. Hier sollten Laxanzien aus der
Gruppe der osmotisch wirksamen Laxanzien, in erster Linie PEG-Elektrolytlösungen
oder Lactulose/Lactitol (cave: Letztere können Blähungen verstärken!) bevorzugt
eingesetzt werden. Laxanzien wie Antrachinone oder Bisacodyl sollten erst Therapie
der zweiten Wahl sein.

41
Antidiarrhoika: Loperamid oder auch Diphenoxylat können beim Diarrhö-
dominanten Reizdarmsyndrom eingesetzt werden.
Anticholinergika wie Butylscopolamin oder in niedriger Dosierung auch trizyklische
Antidepressiva wirken bei rezidivierenden akuten krampfartigen Schmerzen. Die am
besten untersuchte Substanz aus der Gruppe der Muskelrelaxanzien gegen
chronisch rezidivierende Schmerzen ist das Mebeverin (Duspatal). Als Nebeneffekt
tritt neben der Normalisierung der Stuhlkonsistenz und -frequenz auch die Reduktion
etwaiger Blähungen ein.
Antidepressiva und SSRI: Durch die gezielte Beeinflussung der serotoninergen und
cholinergen Systeme mit diesen Substanzen lassen sich gerade Schmerzen beim
RDS gut beeinflussen, ihr Einsatz ist jedoch umstritten und nur in Zusammenarbeit
mit einem Psychiater zu empfehlen.
Ungesicherte Therapieansätze � Probiotika und Phytotherapeutika: Die
Datenlage für diese Therapieansätze ist derzeit noch unzureichend. Bei Obstipation
wurde über eine günstige Wirkung von E. coli-Stamm Nissle 1917 (Mutaflor®), und
bei Schmerzen und Blähungen von Lactobacillus-Präparaten berichtet; allerdings ist
die Studienlage uneinheitlich. Die Rolle verschiedener Phytotherapeutika (in der
Regel Gemische aus verschiedenen Heilpflanzen) ist derzeit noch unklar.
Neuere Therapieansätze Opiat-Kappa-Rezeptorantagonisten wie beispielsweise Fedotozin wirken über eine
pharmakologische Modulation der afferenten Opioid-Kappa-vermittelten
Schmerzwahrnehmung. Die Wirkungen in klinischen Studien waren aber
enttäuschend. Präparate sind in Deutschland derzeit nicht verfügbar.
Eine wesentlich größere Bedeutung dürfte demgegenüber die Beeinflussung der
Serotonin-3- bzw. -4-Rezeptoren durch selektive Antagonisten bzw. Agonisten
erlangen. Der selektive 5-HT3-Rezeptor-Antagonist Alosetron verlangsamt den
Kolontransit und reduziert die Schmerzen v. a. bei Diarrhö-prädominanter Form
signifikant wirksamer als Plazebo bzw. eine spasmolytische Standardmedikation.
Alosetron wurde wegen eines zwischenzeitlich erhobenen Verdachts auf potenzielle
relevante Nebenwirkungen zunächst zurückgezogen, inzwischen unter Auflagen

42
wieder eingeschränkt zugelassen. Analog wirksame Nachfolgesubstanzen (z. B.
Cilansetron) werden erwartet.
5-HT4-Rezeptor-Agonisten (z. B. Tegaserod, u. a. bereits in der Schweiz und in
Nordamerika zugelassen) beschleunigen demgegenüber den Kolontransit und wirken
gegen Obstipation und Schmerzen und verbessern so die Symptomatik beim
Obstipationstyp des RDS.
Mit der Erweiterung unserer Kenntnisse über die Pathomechanismen der Erkrankung
werden in absehbarer Zukunft neue medikamentöse Ansätze verfügbar werden, die
eine gezieltere, effektivere, pathophysiologisch begründete oder vielleicht sogar
kausale Therapie ermöglichen könnten.
Korrespondenzadresse: Prof. Dr. Peter Layer Israelitisches Krankenhaus in Hamburg Orchideenstieg 14 22297 Hamburg Tel.: 040 51125 5001 Fax: 040 51125 5009 E-Mail: [email protected]

43
Therapie des ReizdarmsyndromsTherapie des ReizdarmsyndromsZusammenfassungZusammenfassung
Ausschluß einer organischen ErkrankungÄrztliche Führung:
Aufklärung, Konzept, Diät

44
Die pseudomembranöse Kolitis auf dem Vormarsch � Konzepte zur Diagnostik, Prophylaxe und Therapie
C. Pox
Medizinische Klinik, Ruhr-Universität Bochum, Knappschaftskrankenhaus, Bochum
Die Clostridium-difficile-assoziierte Diarrhö wird durch die beiden Toxine von
C. difficile, einem Gram-positiven anaeroben sporenbildenden Keim hervorgerufen.
Sie ist für etwa 20% der Antibiotika-assoziierten Diarrhöen verantwortlich, kommt
aber in seltenen Fällen auch bei Patienten vor, die keine Antibiose erhalten haben.
Die Manifestation der Infektion verläuft klinisch variabel und reicht von asympto-
matischen Fällen bis zur fulminanten Kolitis mit septischem Verlauf. In der Regel
zeichnet sich die Infektion durch akute wässrige, selten blutige Diarrhöen aus
verbunden mit Bauchschmerzen und erhöhten Temperaturen. Häufig lässt sich eine
zum Teil ausgeprägte Leukozytose nachweisen. Die Infektion erfolgt in der Regel
nosokomial über die sehr umweltresistenten Sporen.
Die Rate an C. difficile-assoziierten Diarrhöen hat in den letzten Jahren deutlich
zugenommen. Von Bedeutung ist die Zunahme eines multiresistenten virulenten
Stamms NAP1/027, der für eine Reihe von Ausbrüchen verantwortlich war. Von
Interesse ist auch eine Zunahme des Nachweises einer C. difficile-Infektion bei
Patienten mit chronisch entzündlichen Darmerkrankungen.
Die Diagnose wird in der Regel über den Toxinnachweis im Stuhl gestellt. Der
eingesetzte Test sollte in der Lage sein beide Toxine nachweisen zu können. Eine
einmalige negative Testung schließt jedoch eine Infektion nicht aus und sollte bei
entsprechender Klinik wiederholt werden. Der kulturelle Nachweis hat eine höhere
Sensitivität und ermöglicht eine anschließende Genotypisierung, benötigt jedoch
aufgrund der erforderlichen Bakterienanzucht mehr Zeit. Zusätzlich ist auch hier der
Toxinnachweis erforderlich, da nur toxinbildende Stämme eine Clostridium-
assoziierte Diarrhö hervorrufen können. Eine Alternative stellt die endoskopische
Diagnostik mittels Sigmoidoskopie dar mit den pathognomonischen gelblichen
pseudomembranösen Schleimhautveränderungen. Bei leichten Fällen der Infektion
können die charakteristischen Schleimhautveränderungen fehlen, eine unauffällige
Endoskopie schließt also eine C. difficile-Infektion nicht aus.
Für die Behandlung werden neben einer symptomatischen Therapie mit Flüssigkeit
und Schmerzmitteln vor allem die Antibiotika Metronidazol und Vancomycin

45
eingesetzt. Hierbei ist Metronidazol sowohl oral als auch intravenös verabreicht
wirksam, wohingegen Vancomycin nur oral oder als Einlauf verabreicht wirksam ist.
Eine intravenöse Applikation ist unwirksam, da hierdurch keine ausreichenden
Spiegel im Kolon erreicht werden. Für leichte Formen sollte Metronidazol
(3 x 500 mg p.o.) eingesetzt werden. Bei schweren Erkrankungen war Vancomycin
(4 x 125 mg p.o.) in einer Studie Metronidazol überlegen und sollte in diesen Fällen
primär entweder als Monotherapie oder kombiniert mit Metronidazol i.v. eingesetzt
werden. Die Therapiedauer beträgt in der Regel 10 Tage. Die verursachende
Antibiose sollte sofern möglich abgesetzt oder zumindest umgestellt werden.
Bei etwa 20% der Betroffenen kommt es nach Absetzen der Therapie zu einem
Rezidiv. Interessanterweise findet man bei diesen Patienten nur sehr selten pseudo-
membranöse Veränderungen. Hier ist eine erneute Therapie für 14 Tage mit
Metronidazol oder Vancomycin erforderlich. Neue Therapiealternativen mit
z. B. Rifaximin, einem nicht resorbierbaren Antibiotikum, sind vielverspreichend,
müssen aber noch in weiteren Studien untersucht werden.
Bei Patienten mit vorangegegangener C. difficile-assoziierter Diarrhö kann eine
prophylaktische Therapie mit einem Probiotikum während einer erforderlichen
Antibiotikatherapie sinnvoll sein.
Wichtig zur Vermeidung einer Weiterverbreitung der Infektion ist eine strikte Isolation
von stationären Patienten im Krankenhaus mit nachgewiesener C. difficile-
assoziierter Diarrhö. Eine Kohortenisolation ist möglich. Die Sporen werden nicht
durch Händedesinfektionsmittel abgetötet. Hier ist zusätzlich sorgfältiges Hände-
waschen erforderlich.
Korrespondenzadresse: Dr. Christian Pox Medizinische Klinik Ruhr-Universität Bochum Knappschaftskrankenhaus In der Schornau 23-25 44892 Bochum E-Mail: [email protected]

46
Bottom-up oder Top-down � welches Risiko bei welchem CED-Patienten
S. Schreiber
Klinik für Allgemeine Innere Medizin und Institut für Klinische Molekularbiologie,
Christian-Albrechts-Universität, Kiel
Morbus Crohn und Colitis ulcerosa sind chronisch rezidivierende Erkrankungen, die
die Entwicklung langfristiger Therapiekonzepte benötigen. Nicht das Management
des einzelnen Schubes sondern die langfristige Reduktion der Krankheitsaktivität
und des Bedarfs an chronischer Medikation senkt Gesamtmorbidität und
Komplikationen. Neben der Reduktion der entzündlichen Aktivität ist daher die
Vermeidung einer Komorbidität durch chronische Therapien (z. B. durch eine
chronische Medikation mit Glukokortikoiden) notwendig.
Die Sekretion pro-entzündlicher Botenstoffe wie insbesondere TNF (Tumor-Nekrose-
Faktor) scheint ein Schlüsselelement der entzündlichen Pathophysiologie des M.
Crohn zu sein. Viele der klinisch eingesetzten Immunsuppresiva (z. B.
Glukokortikoide, Azathioprin) inhibieren breit die Freisetzung pro-entzündlicher
Botenstoffe. Die spezifische Hemmung des Tumor-Nekrose-Faktors (TNF) durch den
intravenösen Einsatz eines chimären Antikörpers (Infliximab) ist eine in den letzten
10 Jahren etablierte Alternative zur oralen Immunsuppression. Eine kurzfristige
Wirkung mit schnellem Wirkungseintritt aber auch eine langfristige Wirkung kann als
gut etabliert angesehen werden.
Das Nebenwirkungsspektrum der Immunsuppression ist beachtlich und reicht von
akuten Problemen (z. B. opportunistische Infektionen, Reaktivierung einer
Tuberkulose oder Sepsis oft aufgrund nicht erkannter Abszesse) zu chronischen
Erkrankungen (z. B. erhöhtes Lymphomrisiko, schnelleres Wachstum von
Karzinomen). Neuere Studien weisen darauf hin, dass hier sowohl den oralen
Immunsuppressiva als auch der anti-TNF-Therapie jeweils ein definiertes Spektrum
zukommt, das sich in der Kombinationstherapie (insbesondere wenn auch noch
Glukokortikoide chronisch eingesetzt werden) weiter erhöht.

47
Die Anforderungen an die Dauertherapie, Effizienz in ein ausgewogenes Verhältnis
zu den möglichen Nebenwirkungen zu stellen, sind daher erheblich. Gegenwärtige
Trends entwickeln sich von der Kombinationstherapie zu einer langfristigen
Monotherapie, wobei auch die Immunogenität der verwendeten Biologika eine
wichtige Rolle spielt.
Korrespondenzadresse: Prof. Dr. Stefan Schreiber Christian-Albrechts-Universität Kiel Universitätsklinikum Schleswig-Holstein Klinik für Allgemeine Innere Medizin Schittenhelmstr. 12 24105 Kiel Tel.: 0431 597 2350 Fax: 0431 597 1434 E-Mail: [email protected]

48
Divertikulitis � wann konservativ, wann interventionell, wann chirurgisch therapieren?
W. Kruis
Innere Medizin, Ev. Krankenhaus Kalk, Köln
Divertikel des Kolons sind ganz überwiegend Herniationen der Mukosa durch
Muskellücken und damit im eigentlichen Sinne Pseudodivertikel. Die Muskellücken
sind an der Durchtrittsstelle der Vasa recta im Bereich des Mesenterialansatzes zu
finden. Begünstigende Faktoren für eine Divertikelbildung sind Störungen der
Bindegewebsstruktur, erhöhter Darminnendruck infolge einer ballaststoffarmen Diät,
Motilitätsstörungen und höheres Lebensalter. Etwa 40% der über 70-Jährigen weist
Kolondivertikel auf, wobei kein Geschlechtsunterschied feststellbar ist.
Man unterscheidet die Divertikulose � zufällig gefundene Divertikel bei
asymptomatischen Menschen � von der Divertikelkrankheit. Die Divertikelkrankheit
kann unkompliziert verlaufen, d. h. mit einer einmaligen Attacke, seltenen Schüben,
aber auch chronisch rekurrierend. Im Fall von Attacken (Schüben) bestehen immer
Symptome wie Schmerzen und Stuhlunregelmäßigkeiten, die mit Zeichen einer
Entzündung (Divertikulitis, ggf. mit lokaler Abwehrspannung, Leukozytose, Erhöhung
von BSG/CRP) einhergehen können. Die Divertikelkrankheit kann sich auch
kompliziert entwickeln mit Peridivertikulitis und zunehmender Abszedierung. Im
ungünstigsten Fall kann es zu Komplikationen in Form von Stenosen, Fisteln,
Blutungen und Perforation kommen. Insgesamt werden jedoch nur 20�30% aller
Divertikelträger so symptomatisch, dass sie sich deswegen in ärztliche Behandlung
begeben.
Die Planung der Behandlung orientiert sich grundsätzlich an der Situation des
Betroffenen: Divertikulose mit dem Ziel einer Entwicklung zur Divertikelkrankheit
vorzubeugen, einmalige oder rekurrierende, aber unkomplizierte Attacken,
Sekundärprävention, komplizierte Divertikelkrankheit.
Primär- sowie Sekundärprävention bestehen in der Empfehlung zur körperlichen
Mobilität und einer ballaststoffreichen Mischkost. In der Regel wird man Quellstoff-
zusätze vor allem in Form von Plantago zugeben. Der BMI stellt einen aussage-
kräftigen Prognosefaktor dar � je höher, desto ungünstiger der Langzeitverlauf.

49
Die unkomplizierte Attacke wird je nach Schwere ambulant mit Flüssigkost,
Antibiotika (Metronidazol, Ciprofloxacin) per os und Spasmolytika oder stationär mit
Nulldiät, Elektrolyt- und Flüssigkeitsersatz sowie Daueranalgesie per infusionem und
i.v.-Antibiotika behandelt. Mesalazin hat in dieser Situation therapeutische Effekte.
Bei rekurrierenden Attacken muss zwischen elektiver Operation und medikamentöser
Dauertherapie entschieden werden. Letztere umfasst eine Basistherapie
(s. Prävention). Zusätzlich ist die 1-wöchige Antibiotikagabe per Monat und ggf. die
zusätzliche Verordnung von Mesalazin für einige Wochen häufig dazu in der Lage,
weitere Attacken, Komplikationen und eine Operation zu vermeiden.
Bei Komplikationen ist die eilige oder ggf. Notfalloperation das Verfahren der Wahl.
Während die Notfallindikation unbestritten ist, wird über die elektive Resektion bei
rekurrierender Divertikulitis diskutiert. Wahrscheinlich sollte bei der Entscheidung zur
Operation nicht die Zahl der vorausgehenden Attacken entscheidend sein, sondern
die Berücksichtigung der Komorbidität (Immunstatus!) und der Grad der entzündlich-
strukturellen extraluminalen Veränderungen (Sonografie, CT).
KolondivertikelDefinitionen
Divertikel
ohne Symptome mit Symptomen
Divertikulose Divertikelkrankheitmit Entzündung = Divertikulitis
Komplikationen
ja nein
(Stenose, Blutung, Abszess, Fistel, Perforation)
Surg Endosc. 1999;13:430-6

50
Kolondivertikel - Verlauf
Asymptomatisch
UnkomplizierteDivertikelkrankheitKomplikationen
70 %
30 %
5 %*
100% = alle Menschen mit Kolondivertikel�30%: irgendeine Form der Divertikelkrankheit� * 5% aller Divertikelträger haben Komplikationen
Dis Colon Rectum. 1989:32Dis Colon Rectum. 1996:39Clin Gastroenterol. 1975:4
Divertikelkrankheit � Therapie- Zusammenfassung -
Divertikelkrankheit/Divertikulitis
Akut Rekurrierend
Kompliziert Unkompliziert
Mild Moderat/Schwer
Operation Diät 0-Diätiv-Therapie/Subst
Analgesie Analgesie5-ASA Antibiotika
Mit OhneStrukturänderungen
Operation BallastAktivität
Intermitt.Antibiotika
5-ASAProbiotika

51
Ecksteine der Leitlinie Morbus Crohn
M. Zeitz
Medizinische Klinik I, Charité � Universitätsmedizin, Campus Benjamin Franklin
(CBF), Berlin
Die Leitlinienentwicklung für den Morbus Crohn in Deutschland ist auf einem sehr
aktuellen Stand: So wurde gerade die Überarbeitung der S3-Leitlinie �Diagnostik und
Therapie des Morbus Crohn� publiziert (Z. Gastroenterol. 2008; 46: 1094). Eine
europäische Leitlinie liegt aus dem Jahr 2006 vor.
Wichtige Definitionen in der aktuellen Leitlinie �Morbus Crohn� beziehen sich auf
unterschiedliche Verlaufsformen: Für die Therapieplanung sind folgende Begrifflich-
keiten wesentlich: Frührezidiv (Rezidiv innerhalb von 3 Monaten), steroidrefraktärer
Verlauf (aktive Erkrankung trotz adäquater Prednisolon-Gabe über mindestens
4 Wochen), steroidabhängiger Verlauf (keine Steroidfreiheit innerhalb von 4 Monaten
bzw. Rezidiv innerhalb von 3 Monaten nach Beendigung einer Steroidtherapie),
lokalisierte Erkrankung (Crohn-Befall unter 30 cm) und ausgedehnte Erkrankung
(Crohn-Befall über 100 cm). Die Erstdiagnostik eines Morbus Crohn umfasst zentral
die Ileokoloskopie mit Biopsieentnahme aus allen untersuchten Segmenten sowie
eine erweiterte Dünndarmdiagnostik (bevorzugt durch MRT-Enteroklysma). Eine
Endoskopie des oberen Gastrointestinaltrakts wird bei der Erstdiagnose bzw. bei
entsprechenden Symptomen empfohlen; der Kapselendoskopie wird derzeit wegen
meist fehlender therapeutischer Konsequenzen ein eher untergeordneter Wert
eingeräumt. In der Labordiagnostik ist insbesondere die Ausschlussdiagnostik von
infektiösen Komplikationen erwähnenswert (zunehmendes Problem durch
Clostridium-difficile- und Zytomegalievirus-Infektionen).
Die Therapie des akuten Schubs sollte entsprechend der aktuellen Empfehlungen
unverändert der klassischen �Step-up�-Strategie folgen: Steroide, bei komplexen
Verläufen zusätzlich Immunsuppressiva vom Typ des Azathioprins (bei fehlender
Wirksamkeit oder Unverträglichkeit Methotrexat), bei fehlendem Erreichen einer
Remission Antikörper gegen TNFα (Infliximab oder Adalimumab). Die Vorgehens-
weise bei akutem Schub hängt auch von der Lokalisation der Erkrankung ab: Bei
Ileozökalbefall kann ein Therapieversuch mit hoch dosiertem Mesalazin oder (besser
belegt) mit Budesonid erfolgen. Bei Crohn-Colitis sowie bei ausgedehntem Dünn-
darmbefall sollten frühzeitig Immunsuppressiva eingesetzt werden. Bei Befall des

52
oberen Gastrointestinaltrakts sollte ebenfalls frühzeitig an eine Immunsuppressiva-
therapie gedacht und bei Magen- bzw. Duodenumbefall Protonenpumpenhemmer
gegeben werden.
Der Einsatz von Antikörpern gegen TNFα ist entsprechend der derzeitigen
Empfehlungen noch als Drittlinientherapie nach Steroiden und Immunsuppressiva
anzusiedeln. Bei Persistenz einer hohen Krankheitsaktivität trotz adäquater Steroid-
dosis oder Kontraindikation für Glukokortikoide können TNFα-Antikörper auch vor
Immunsuppressiva eingesetzt werden. Bei steroidabhängigen oder steroidrefraktären
Verläufen sollte frühzeitig ein Einsatz von TNFα-Antikörpern diskutiert werden. Für
eine sogenannte �Top-down�-Strategie mit frühzeitigem Einsatz von TNFα-Anti-
körpern und Immunsuppressiva gibt es bisher keine ausreichende Literaturlage, um
eine entsprechende Empfehlung abzugeben.
Im Zentrum der Remissionserhaltung steht die Abstinenz von Tabakgebrauch bei
Rauchern. Eine generelle medikamentöse remissionserhaltende Therapie wird nicht
empfohlen, Steroide haben keinen Stellenwert in dieser klinischen Situation. Bei
komplexen Krankheitsverläufen stehen an erster Stelle Immunsuppressiva vom Typ
des Azathioprins bzw. Methotrexats und bei Wirkungslosigkeit Langzeittherapien mit
Antikörpern gegen TNFα. In allen klinischen Situationen sollten chirurgische
Optionen in Erwägung gezogen werden.
Operationsindikationen bestehen klar bei therapierefraktärem Subileus, Kolon-
stenosen unklarer Dignität, dem Nachweis hochgradiger intraepithelialer Neoplasien,
bei therapierefraktären Verläufen und bei isoliertem symptomatischem Ileozökalbefall
und fehlendem Ansprechen auf eine konservative Therapie.
Selbstverständlich bedürfen Abszesse einer interventionellen Therapie: initial Anti-
biotika plus perkutane oder chirurgische Drainage meist gefolgt von einer Resektion.
Eine generelle Empfehlung für eine postoperative Rezidivprophylaxe kann nicht
gegeben werden, Azathioprin kann perioperativ fortgesetzt werden ohne Nachteile
bezüglich der Komplikationsrate. Nur bei symptomatischen Fisteln steht eine
Therapieindikation, bei ausgedehntem Fistelleiden ist eine interdisziplinäre Abstim-
mung essenziell. Die Behandlung extraintestinaler Manifestationen stellt nach wie vor
eine Herausforderung dar. Bei Gelenkbeteiligung und Schmerzen ist vor NSAR zu
warnen, Cox-2-Inhibitoren sind bisher in ihrem intestinalen Nebenwirkungsprofil nicht
abschließend geklärt.

53
Definitionen � S3-Leitlinie Morbus Crohn� Aktive Erkrankung� Remission� Ansprechen: CDAI-Abfall > 100 Punkte� Rezidiv� Frührezidiv: innerhalb von 3 Monaten� Steroidrefraktärer Verlauf: aktiv trotz 0,75 mg/kg Prednisolon über 4 Wochen � Steroidabhängiger Verlauf: keine Steroidfreiheit innerhalb von 4 Monaten bzw.
Rezidiv 3 Monate nach Ende der Steroidtherapie� Lokalisierte Erkrankung: Crohn-Befall < 30 cm� Ausgedehnte Erkrankung: Crohn-Befall > 100 cm
Remission
Akuter Schub
Keine RemissionRemission
(i.v.) Glukokortikoide + Azathioprin
Glukokortikoide [Mesalazin mit begrenzter Wirksamkeit]
60-93% 7-40%
Remission Keine Remission2-8%6-36%
TNFα-Antikörper
Akuter Schub des Morbus Crohnklassische �Step-up�-Strategie

54
Akuter Schub des Morbus CrohnAbhängigkeit von der Lokalisation
Lokalisation AktivitätIleozökalbefall leicht / mäßig Mesalazin, Budesonid,
systemische SteroideIleozökalbefall hoch Steroide, Immunsuppressiva,
anti-TNFa (evtl. vor Aza), Chi (!)
Crohn-Colitis SASP, Steroide, Lokaltherapie, früh: Immunsuppressiva
ausgedehnter Dünndarmbefall
systemische Steroide, enteraleErnährung, früh: Immunsuppressiva
Ösophagus/ gastroduodenaler Befall
systemische Steroide, Magen/Duodenum: PPI, früh: Immunsuppressiva
Akuter Schub: Stellenwert von AK gegen TNFα
Bei Nichtansprechen auf Glukokortikoide und Immunsuppressivabzw. Nebenwirkungen - nach Ausschluss chirurgischer Therapieoptionen:AK gegen TNFα
Bei Persistenz der hohen Krankheitsaktivität trotz adäquater Steroiddosisoder Kontraindikationen für Glukokortikoide können TNFα-Antikörper vor Immunsuppressiva eingesetzt werden

55
Remissionserhaltung
Ziel der Langzeittherapie: Erhaltung der klinischen Remission
Bei komplexen Krankheitsverlauf: Azathioprin/6-MP
Keine ausreichende Basis für eine generelle medikamentöse remissionserhaltende
Therapie
Abstinenz von Tabakgebrauch
keine Steroide in der remissionserhaltenden Therapie
Bei Wirkungslosigkeit: Dosis? Alternativ MTX oder Antikörper gegen TNFα (in Kombination?)
- OperativesVorgehen besonders bei lokalisiertem Befall

56
Die steroidrefraktäre Colitis ulcerosa � wie behandeln?
J. Schölmerich
Klinik für Innere Medizin I, Universitätsklinikum Regensburg
Der natürliche Verlauf der Colitis ulcerosa in populationsbasierten Kohorten ist relativ
günstig. 60% der Patienten weisen nach dem initialen Schub in der Regel unter
Gabe von 5-Aminosalizylsäure einen benignen Verlauf auf. 30% zeigen immer
wieder Schübe trotz Rezidivprophylaxe, 10% haben einen chronisch aktiven Verlauf.
Diese Patienten bedürfen einer aggressiveren Therapie, um das Auftreten von
strukturellen Schäden bzw. die Kolektomie zu vermeiden, wo möglich. Das Konzept
einer aggressiven Therapie wird dadurch unterstrichen, dass in den letzten Jahren
deutlich wurde, dass genau die Patienten mit der chronischen, nie ganz zur Ruhe
kommenden Entzündung diejenigen sind, die überhaupt ein Risiko für die
Entwicklung eines kolorektalen Karzinoms aufweisen, während dies bei allen
anderen Patienten offenbar sehr selten der Fall ist.
Die Behandlung der Patienten mit diesen Formen der schweren, entweder
gegenüber Standardtherapie refraktären oder beispielsweise steroidabhängigen
Colitis ulcerosa bestand vor 40 Jahren in der hoch dosierten Steroidgabe und einer
parenteralen Ernährung. Letztere ist lange verlassen, die Steroidgabe ist nach wie
vor aktuell. Diese kann aber im besten Fall eine kurzzeitige Remissionsinduktion
bewirken und, wenn dauerhaft gegeben, den Zustand der Steroidabhängigkeit, der
mit einer Remission einhergeht, herbeiführen. Eine Langzeitsteroidtherapie ist aber
aufgrund der Nebenwirkungen heute obsolet. Im Wesentlichen sprechen die
Patienten, die in den ersten 3 Tagen bereits eine Verbesserung der Klinik aufweisen,
auch tatsächlich auf die Steroidgabe an, bei allen anderen kann man relativ früh auf
andere Therapieformen umstellen.
Bevor eine aggressivere Immunsuppression eingeleitet wird, sollte allerdings die
Gegenwart einer Superinfektion, beispielsweise mit Clostridium difficile oder CMV
ausgeschlossen werden. Die Häufigkeit solcher opportunistischen Infektionen,
besonders bei Patienten unter einer bereits laufenden Immunsuppression mit
Steroiden oder anderen Medikamenten, hat in den letzten Jahren drastisch
zugenommen; eine solche muss daher zwingend ausgeschlossen werden.

57
Ältere Studien zeigen, dass die Gabe von Azathioprin Patienten mit refraktärer Colitis
durchaus in Remission bringen kann, hier ist der Zeitablauf aber so langwierig, dass
dies heute den Patienten in der Regel nicht mehr zugemutet werden kann. Hier sei
angemerkt, dass auch Azathioprin durchaus nebenwirkungsträchtig ist, und gerade in
den letzten Jahren Berichte über nodulär regenerative Hyperplasie und Lymphome
dieses deutlich gemacht haben.
Bisheriger Standard bei Patienten mit schwerer refraktärer Colitis war die Gabe von
Ciclosporin intravenös, wobei initial 60�80% der Patienten ansprechen und
zumindest primär eine Kolektomie vermieden werden kann. Ein Ansprechen lässt
sich durch ein CRP unter 45 mg/l, eine Herzfrequenz unter 90 pro Minute und das
Fehlen von erhöhten Temperaturen wahrscheinlich machen. Das Fehlen schwerer
endoskopischer Veränderungen spricht ebenfalls eher für den Erfolg einer solchen
Therapie. Es ist allerdings nicht zu empfehlen, nur um hier eine Vorhersage zu
treffen, eine Endoskopie durchzuführen. Die Dosis von Ciclosporin kann wohl auf
2 mg/kg reduziert werden, ohne einen wesentlichen Effektivitätsverlust zu riskieren.
Hier sind die Nebenwirkungen beachtlich, immerhin 15% haben schwere
Nebenwirkungen, wobei die Nephrotoxizität, schwere Infektionen und Krämpfe im
Vordergrund stehen. Anschließend an die Ciclosporin-Therapie wird in der Regel mit
Azathioprin weiterbehandelt, wobei die Phase, in der neben den initial vorhandenen
Steroiden Ciclosporin und Azathioprin appliziert wird, besonders risikoreich ist. Hier
wird von etlichen Zentren eine präventive Antibiose empfohlen.
Neuerdings ist auch die Gabe von Infliximab bei diesen Patienten erprobt worden
und hat sich als effektiv erwiesen. Dies war zunächst in einer Studie aus Schweden
deutlich, wo die Patienten mit schwerer Colitis, nicht jedoch die mit fulminanter Colitis
wesentlich profitierten. Eine größere Patientengruppe in Belgien wies ein
Ansprechen bei 63% auf, was zu einer Kolektomierate von 18% in dieser
Untergruppe nach 2 Jahren führte. Hier wurden 6 schwere Infektionen und
4 Malignome beobachtet, sodass auch hier natürlich die Nebenwirkungen in Betracht
gezogen werden müssen. Vergleiche beider Therapiestrategien liegen bislang nicht
vor, verschiedene Ansätze zeigen, dass Therapieversager mit Ciclosporin mit
Infliximab und vice versa behandelt werden können. Ein Kohortenvergleich wies auf
eine geringe Überlegenheit der Ciclosporin-Therapie hin.
In allen Fällen muss mit dem Patienten auch die Option einer Kolektomie und Anlage
eines ileoanalen Pouches besprochen werden, die natürlich auch in der Lage ist, die
Krankheit zu beseitigen und damit auch das Karzinomrisiko zu nehmen. Inwieweit

58
Alternativen wie die Leukozytenapherese oder beispielsweise die Gabe von
Phosphatidylcholin bei steroidrefraktärer Colitis ulcerosa eine Rolle spielen werden,
ist offen; hier sind definitive Studien abzuwarten. Insgesamt scheint die Prognose
von Patienten mit schwerer Colitis ulcerosa durchaus günstig zu sein: Von 149
Episoden über 7 Jahre waren in einem Referenzzentrum 36 als Therapieversager
auf die Steroidtherapie klassifizierbar, von diesen sprachen 12 von 21 auf Ciclosporin
oder Infliximab an, 15 wurden primär, 24 insgesamt kolektomiert.

59
Therapeutisches Vorgehen beim Ösophaguskarzinom
O.G. Opitz
Tumorzentrum Ludwig Heilmeyer-Comprehensive Cancer Center, Freiburg
Ösophaguskarzinome unterscheiden sich im Wesentlichen in die 2 Entitäten des
Plattenepithelkarzinoms des Ösophagus und des Adenokarzinoms des Ösophagus,
welches in der Regel aus einem Barrett-Ösophagus entsteht. Diese 2 Tumorentitäten
verhalten sich in ihrer Epidemiologie, Pathogenese und Tumorepidemiologie gänzlich
unterschiedlich, dennoch sind sie bisher in Therapiestudien meistens gemeinsam
untersucht worden.
Plattenepithekarzinom des Ösophagus Das Plattenepithelkarzinom des Ösophagus ist die 6. häufigste Tumor-assoziierte
Todesursache, es hat eine durchschnittliche Inzidenz von ca. 2,5�5 pro 100.000
Einwohner, mit starken geographischen Schwankungen. Gemeinsam mit dem
Mundbodenkarzinom kommt es v. a. in Entwicklungsländern sehr häufig vor. An
Risikofaktoren sind, v. a. in der westlichen Welt der Tabak- und Alkoholkonsum zu
nennen, in Entwicklungsländern v. a. der Vitamin- und Mineralmangel sowie
chemische Karzinogene, wie die Nitrosamine. Weitere prädisponierende Faktoren
sind die Achalasie, Laugenverätzungen sowie eine stattgehabte Bestrahlung. Des
Weiteren ist das Plattenepithelkarzinom des Ösophagus mit einem niedrigen
sozioökonomischen Status assoziiert. Als familiäre Form gibt es die Tylose eine
Palmoplantarkeratose. Genetische Veränderungen sind v. a. im Zellzyklus
regulierenden Protein Cyclin D1, der Rezeptor-Tyrosinkinase, EGFR und den
Tumorsuppressorgenen p53 und p16 zu finden. Histologisch entwickelt sich das
Plattenepithelkarzinom des Ösophagus aus einem mehrschichtigen Plattenepithel
über die Dyplasie und das Carcinoma in situ zum invasiven Karzinom.
Adenokarzinom des Ösophagus Das Adenokarzinom des Ösophagus entsteht v. a. aus der intestinalen Metaplasie
dem sog. Barrett-Epithel. Dies wiederum entsteht im Wesentlichen durch einen
vermehrten gastroösophagealen Reflux im distalen Ösophagus. Das Adenokarzinom
des Ösophagus oder Barrett-Karzinom stellt die am stärksten zunehmende

60
Neoplasie der westlichen Welt dar. In den USA war über die letzen 2 Jahrzehnte
z. B. eine Zunahme von 450% zu verzeichnen. Während in Deutschland noch das
Plattenepithelkarzinom prädominant ist, ist das Verhältnis zwischen beiden Entitäten
in den USA inzwischen 1:1. Neben dem Barrett-Ösophagus gelten auch der
gastroösophageale Reflux selbst, sowie die Adipositas und das Rauchen als
Risikofaktoren. Prädisponierende Faktoren sind hier Zustände mit erhöhter
Säureexposition, wie das Zollinger-Ellison-Syndrom, Zustand nach Bougierung oder
Medikamente, die den unteren Ösophagussphinkter beeinflussen. 12% der Patienten
mit gastroösophagealer Refluxerkrankung entwickeln einen Barrett-Ösophagus. Aus
dem Barrett-Epithel kann sich dann in 4% eine leichtgradige intraepitheliale
Neoplasie bilden; diese kann sich entweder zurückentwickeln oder in insgesamt 1%
zu einer schwergradigen intraepithelialen Neoplasie foranschreiten. Hieraus
entstehen in bis zu 40% dann invasive Adenokarzinome des distalen Ösophagus.
Diagnostik Diagnostisch ist für beide Entitäten die Endoskopie federführend. Diese erfolgt zur
Diagnosestellung und histologischen Sicherung. Zum stadiengerechten Staging ist
zur Bestimmung der Eindringtiefe (T) die Endosonografie die beste Methode, zur
Bestimmung des Lymphknotenstatus die Endosonografie und die Computer-
tomografie (CT). Zur diagnostischen Evaluation einer etwaigen Fernmetastasierung
kann zusätzlich zum CT Thorax und Abdomen auch das PET herangezogen werden.
Beim Plattenepithelkarzinom des Ösophagus sollte aufgrund einer in bis zu 15%
auftretenden Koinzidenz mit Plattenepithelkarzinomen im Kopf-Hals-Bereich und der
Lunge noch ein CT Hals, eine HNO-Untersuchung sowie eine Bronchoskopie
durchgeführt werden. Vor einer neoadjuvanten Therapie beim Adenokarzinomen des
distalen Ösophagus sollte zusätzlich eine Laparoskopie durchgeführt werden.
Die Eindringtiefe im T-Stadium determiniert die lymphogene Metastasierung, wobei
schon im Stadium T1b bei ca. 20% der Adenokarzinome und bis zu 50% der
Plattenepithelkarzinome eine lymphogene Metastasierung vorliegen kann.
Therapeutische Optionen Die Therapie sollte dann stadienabhängig verlaufen, wobei hier die TNM-
Klassifikation wieder in klinische UICC-Stadien I�IV zusammengefasst werden. Für
die Therapieentscheidung sollen im Folgenden 3 Stadien exemplarisch diskutiert

61
werden: Das Frühkarzinom im UICC-Stadium 0�1, das lokal fortgeschrittene
Ösophaguskarzinom im UICC-Stadium II und das metastasierte Karzinom im UICC-
Stadium Vb.
Die Prognose richtet sich hier ebenso nach dem Tumorstadium, wobei
Frühkarzinome eine 5-Jahres-Überlebensrate von bis zu 80% aufweisen, lokal
fortgeschrittene Karzinome von 15% und metastasierte Karzinome von unter 1%. Die
Tumorresektion bleibt das einzig kurative Therapieverfahren, doch leider ist zum
Zeitpunkt der Diagnose nicht einmal die Hälfte der Patienten resezierbar. Über drei
viertel der Patienten mit Ösophaguskarzinom werden erst in einem mindestens lokal
fortgeschrittenen Stadium diagnostiziert.
Beim Frühkarzinom wird zunehmend, v. a. beim mukosalen Karzinom, d. h. im
Stadium T1a die endoskopische Mukosaresektion als Alternative durchgeführt. Dies
ist v. a. bei Barett-Frühkarzinomen erfolgreich untersucht worden worden. Bei
Eindringen in die Submucosa (T1b) ist die chirurgische Resektion die Methode der
Wahl, wegen der zunehmenden Gefahr einer lymphogenen Metastasierung. Auch
bei lokal begrenztem Tumorleiden steht natürlich die chirurgische Resektion im
Vordergrund. Eine adjuvante Chemotherapie ist bei resektablen Tumorstadien nicht
angezeigt.
Die Therapie des Ösophaguskarzinoms im lokal fortgeschrittenen Stadium ist eines
der am meist diskutierten Themen in der Onkologie überhaupt. Zum einen können
hier die Therapieempfehlungen zum Plattenepithelkarzinom und zum Adenokarzinom
unterschiedlich ausfallen, zum anderen gibt es für jede der Therapieoptionen eine
sehr unterschiedliche Studienlage. Im lokal fortgeschrittenen Stadium stellt sich die
Frage, ob die alleinige Operation, eine neoadjuvante Radiochemotherapie oder nur
eine neoadjuvante Chemotherapie oder gar eine definitive Radiochemotherapie unter
Weglassen einer chirurgischen Resektion die beste therapeutische Maßnahme
darstellt. Wenn man die heutige Studienlage subsumiert, kann man hier für die
jeweiligen Entitäten zu unterschiedlichen Empfehlungen kommen. Für das
Plattenepithelkarzinom haben mehrere Metaanalysen und eine Phase-III-Studie
gezeigt, dass die neoadjuvante Radiochemotherapie einer alleinigen Chirurgie
überlegen ist. Einzelstudien waren leider meist unterpowert oder haben ein
indifferentes Bild gezeigt. Für das Adenokarzinom des Ösophgus hat in
Metaanalysen und einigen Studien die neoadjuvante Radiochemotherapie ebenso

62
Vorteile gezeigt. Hier zeigen jedoch mehrere neuere Phase-III-Studien, die neben
dem Magenkarzinom auch distale Adenokarzinome des Ösophagus eingeschlossen
hatten, dass es einen klaren Überlebensvorteil für eine neoadjuvante Chemotherapie
mit nachfolgender Operation gibt. Dies stellt inzwischen auch die Empfehlung in den
meisten intersdiziplinären Krebszentren dar. Beim Plattenepithelkarzinom des
Ösophagus wird im lokal fortgeschrittenen Stadium derzeit der Stellenwert einer
definitiven Radiochemotherapie diskutiert, da es Hinweise gibt, dass in manchen
Situationen die nachfolgende Resektion zumindest bzgl. des Gesamtüberlebens
entbehrlich sein könnte. Derzeit wird jedoch die neoadjuvante Radiochemotherapie
mit anschließender chirurgischer Resektion empfohlen. Zudem gibt es zunehmend
Daten, die die Rolle des PET in der �Response Prediction� unter neoadjuvanten
Therapien nahelegt.
Im metastasierten Stadium steht v. a. bei den Plattenepithelkarzinomen eine
Symptom-orientierte Therapie im Vordergrund. Hier ist die Sicherung der
Nahrungspassage die wesentliche Aufgabe. Dies kann zum einen durch
endoskopische Verfahren, die Anlage von gecoverten und nicht-gecoverten Stents
erreicht werden, aber auch lokal ablative endoskopische Verfahren finden hier
Anwendung. Ebenso hat hier die palliative Radiotherapie, entweder perkutan oder
als Brachytherapie, ihren Stellenwert. Auch eine systemische Chemotherapie ist bei
einer Lebenserwartung > 6 Monaten möglich. Es stehen Chemotherapien mit
5FU/Cisplatin, Taxol/Carboplatin oder Vinorelbine zur Verfügung. Die palliative
Chemotherapie beim Adenokarzinom des Ösophagus richtet sich nach den
Therapieoptionen beim Magenkarzinom. Hier haben sich vor allem Cisplatin und
5FU-haltige Protokolle in der Erstlinientherapie durchgesetzt. Die besten
Ansprechraten hat hier das DCF-Protokoll aus Docetaxel, Cisplatin und 5FU, vor
dem ECF Protokoll mit Epirubicin, Cisplatin und 5FU. Hier konnte kürzlich gezeigt
werden, dass in diesem Protokoll ein Austausch von Cisplatin durch Oxaliplatin
oder/und 5FU durch Capecitabine bei gleicher Wirksamkeit aber etwas anderem
Nebenwirkungsprofil durchaus möglich ist. V. a. in der Zweitlinientherapie steht
zusätzlich das FOLFIRI-Protokoll zur Verfügung.
Fazit für die Praxis Das Ösophaguskarzinom teilt sich in 2 Entitäten, das Plattenepithelkarzinom und das
Ösophagus. Das Adenokarzinom ist hierbei die am stärksten zunehmende Neoplasie

63
der westlichen Welt. Beide Entitäten unterscheiden sich in ihren Risikofaktoren und
in Ihrer geografischen Verteilung. Eine stadiengerechte Diagnostik schließt die
Endoskopie, die Endosonografie, das CT Thorax und Abdomen sowie z. T. das PET
mit ein. Geeignete Tumormarker zu Diagnosestellung, Verlaufsbeurteilung gibt es
leider nicht. Die so diagnostizierten Stadien führen zu einer ebenso stadiengerechten
Therapie. Hier werden mukosale Frühkarzinome zunehmend endoskopisch Mukosa-
reseziert, lokal begrenzte Tumore reseziert und lokal fortgeschrittene Karzinome
neoadjuvant behandelt. Bei Plattenepithelkarzinomen empfiehlt man eine
neoadjuvante Radiochemotherapie bei Adenokarzinomen derzeit eher eine
neoadjuvante Chemotherapie. Im metastasierten Stadium steht der Erhalt der
Nahrungspassage im Vordergrund. Da die Therapie der Ösophaguskarzinome
zunehmend komplexer wird, sollten diese Tumore in interdisziplinären Zentren
behandelt werden. Außerdem ist zur Erweiterung der Therapieoptionen der
Einschluss dieser Patienten in Therapiestudien notwendig.
Literatur: Behrens A, May A, Gossner L, Günter E, Pech O, Vieth M, Stolte M, Seitz G, Ell C. Curative treatment for high-grade intraepithelial neoplasia in Barrett's esophagus. Endoscopy. 2005;37:999�1005. Gebski V, Burmeister B, Smithers BM, Foo K, Zalcberg J, Simes J; Australasian Gastro Intestinal Trials Group. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol. 2007;8:226�34 Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, Smith DB, Langley RE, Verma M, Weeden S, Chua YJ, MAGIC Trial Participants. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11�20. Stahl M, Stuschke M, Lehmann N, Meyer HJ, Walz MK, Seeber S, Klump B, Budach W, Teichmann R, Schmitt M, Schmitt G, Franke C, Wilke H. Chemoradiation with and without surgery in patients with locally advanced squamous cell carcinoma of the esophagus. J Clin Oncol. 2005;23:2310�7. Lordick F, Ott K, Krause BJ, Weber WA, Becker K, Stein HJ, Lorenzen S, Schuster T, Wieder H, Herrmann K, Bredenkamp R, Höfler H, Fink U, Peschel C, Schwaiger M, Siewert JR. PET to assess early metabolic response and to guide treatment of adenocarcinoma of the oesophagogastric junction: the MUNICON phase II trial. Lancet Oncol. 2007;8:797�805.

64
Korrespondenzadresse: Prof. Dr. Oliver G. Opitz Direktor Tumorzentrum Ludwig Heilmeyer � CCCF Universitätsklinikum Freiburg Albert-Ludwigs-Universität Tel.: 0761 270-7150/-7151 Fax: 0761 270-7152 E-Mail: [email protected] Website: http://www.tumorzentrum-freiburg.de

65
(Neo-)Adjuvante und palliative Therapie des kolorektalen Karzinoms
W. Schmiegel
Medizinische Klinik, Ruhr-Universität Bochum, Knappschaftskrankenhaus, Bochum
Jährlich erkranken in Deutschland 73.000 Menschen an einem kolorektalen
Karzinom (KRK); etwa 28.000 Patienten versterben jährlich an den Folgen eines
KRK. Die Prognose hängt entscheidend vom Tumorstadium bei Diagnose ab. Die
Therapie erfolgt entsprechend stadienadaptiert.
Im Stadium II mit einer 5-Jahres-Überlebensrate von 70�85% kann durch eine 5-FU-
basierte Chemotherapie ein geringer, aber statistisch signifikanter Überlebensvorteil
von knapp 4% gegenüber der alleinigen Operation erreicht werden. Eine adjuvante
5-FU-basierte Chemotherapie kann daher nach Beratung und Aufklärung des
Patienten empfohlen werden (�Kann�-Empfehlung). Bei Hochrisikopatienten (T4,
Tumor-Perforation, Ileus, G3 oder 4, venöse Infiltration [V1], < 10 Lymphknoten)
sollte eine adjuvante Chemotherapie erwogen werden, da der Überlebensvorteil hier
deutlicher ausgeprägt ist.
Im Stadium III mit einer 5-Jahres-Überlebensrate von 65�80% sollte eine adjuvante
Chemotherapie durchgeführt werden (�Soll�-Empfehlung). So wurde in zahlreichen
Studien ein signifikanter Überlebensvorteil für Patienten im Stadium III durch eine
adjuvante 5-FU-basierte Chemotherapie nachgewiesen. Eine Kombinationschemo-
therapie mit FOLFOX führt zu einem weiteren absoluten Überlebensvorteil
gegenüber 5-FU und gilt daher als Standard im Stadium III. Bei Kontraindikationen
gegen Oxaliplatin oder Komorbiditäten soll eine Monotherapie mit oralen Fluoro-
pyrimidinen durchgeführt werden.
Bei Rektumkarzinomen im Stadium II und III soll eine neoadjuvante Radiochemo-
therapie durchgeführt werden. Neben dem Downsizing und der höheren R0-Re-
sektionsrate ist eine höhere Rate an Sphinktererhalt durch eine neoadjuvante
Radiochemotherapie möglich. Durch die Chemotherapie-Komponente soll die Rate
an Fernmetastasierungen gesenkt werden. Durch eine Intensivierung der Chemo-
therapie im Rahmen von Studien kann diese ggf. optimiert werden.
In Bezug auf das therapeutische Vorgehen werden die Patienten mit einem meta-
stasierten KRK nach der klinischen Situation und dem Therapieziel in 3 Subgruppen
eingeteilt. Zu der Gruppe 1 gehören die Patienten mit primär resektablen Leber-
und/oder Lungenmetastasen. Bei Patienten der zweiten Gruppe wird eine intensi-

66
vierte systemische Therapie angestrebt. Zu dieser Gruppe gehören operable
Patienten mit potenziell resektablen Metastasen nach einer neoadjuvanten Chemo-
therapie sowie Patienten mit tumorbedingten Symptomen, raschem Progress oder
Organkomplikationen. Die Gruppe 3 hat eine weniger intensive Therapie zum Ziel,
welche bei Patienten mit multiplen Metastasen ohne Option für Resektion nach
Metastasenrückbildung, ohne tumorbezogene Symptome oder Organkomplikation
und/oder schwerer Komorbidität angewandt wird.
Eine Resektion von Metastasen (Gruppe 1) ist nur in 10�20% aller Patienten
möglich. Durch die erfolgreiche Entfernung von Lebermetastasen ist eine 5-Jahres-
Überlebensrate von 25�40% zu erreichen. Nach einer R0-Resektion von Lebermeta-
stasen kann eine adjuvante Chemotherapie erwogen werden. Im Einzelfall können
primär operable Lebermetastasen auch im Rahmen eines perioperativen (prä- und
postoperativen) Chemotherapieprotokolls behandelt werden.
Bei Patienten der zweiten Gruppe wird eine intensivierte systemische Therapie
angestrebt. Zu dieser Gruppe gehören operable Patienten mit potenziell resektablen
Metastasen nach einer neoadjuvanten Therapie sowie Patienten mit tumorbedingten
Symptomen, raschem Progress oder Organkomplikationen. Bei Leber- und/oder
Lungenmetastasen und im Einzelfall auch in anderen Lokalisationen ist daher bei
Diagnose und im Verlauf zu überprüfen, ob eine Resektion technisch möglich ist. Bei
primärer Irresektabilität der Metastasen soll mit einer neoadjuvanten systemischen
Chemotherapie begonnen werden, um ggf. eine sekundäre Metastasenresektion zu
erreichen. Derzeit stehen mehrere First-line-Therapien mit hohen Remissionsraten
zur palliativen Behandlung des metastasierten kolorektalen Karzinoms zur
Verfügung. Dabei ist die orale Gabe von Capecitabin der intravenösen Gabe von
5-FU/Folinsäure gleichwertig. Somit sollte aufgrund der höheren Ansprechrate bei
der Gruppe 2 eine 2-Fach-Kombinationstherapie (FOLFOX oder FOLFIRI) plus
monoklonalem Antikörper (Bevacizumab oder Cetuximab) oder eine 3-Fach-
Chemotherapie (FOLFOXIRI) gewählt werden.
Bei Patienten der Gruppe 3 mit Metastasen ohne Aussicht auf Resektabilität und
ohne wesentliche tumorbedingte Symptome kann als Erstlinientherapie auch eine
Monotherapie eingesetzt werden. So zeigten die CAIRO- und FOCUS-Studien
keinen signifikanten Unterschied beim Gesamtüberleben für Patienten, die primär mit
einer Monotherapie behandelt wurden.

67
Bei der Vielzahl der heute zur Verfügung stehenden Behandlungsoptionen steht die
Identifizierung von Biomarkern, die eine prognostische Einschätzung oder ein
Therapieansprechen vorhersagen können, im Fokus der translationalen Forschung.
Relevant ist hier derzeit bereits der Nachweis von K-ras-Mutationen im Tumor, da bei
Nachweis einer K-ras-Mutation kein Benefit für eine EGFR-inhibierende Therapie
besteht, sodass diese nicht indiziert (und dann auch nicht zugelassen) ist. Weitere
Bedeutung erlangen zunehmend der Nachweis einer Mikrosatelliteninstabilität oder
eines LOH18q in der adjuvanten Situation, oder der Nachweis einer Topoisomerase-
1-Expression bei Behandlung mit Irinotecan-haltigen Protokollen.

68
Hereditäre Formen des kolorektalen Karzinoms � Was gilt es zu beachten?
K. Schulmann
Medizinische Klinik, Ruhr-Universität Bochum, Knappschaftskrankenhaus, Bochum
In Deutschland stellt das kolorektale Karzinom (KRK) mit 73.000 Neuerkrankungen
pro Jahr die zweithäufigste Tumorerkrankung dar. In etwa 20% liegt eine positive
Familienanamnese vor. Ca. 2�5% aller KRK entstehen auf dem Boden einer im
engeren Sinne erblichen Krebsdisposition. Mit Abstand am häufigsten und ohne
charakteristischen Phänotyp ist das Lynch-Syndrom (HNPCC). Deutlich seltener und
aufgrund eines charakteristischen Phänotyps leichter zu erkennen sind die
Polyposis-Syndrome, wie die familiäre adenomatöse Polyposis und die sehr seltenen
hamartomatösen Polyposis-Syndrome (Peutz-Jeghers-Syndrom und familiäre
juvenile Polyposis). Einen Überblick gibt Tabelle 1. Neben erhöhten Risiken für ein
KRK besteht bei fast allen Formen ein erhöhtes Risiko weiterer Neoplasien.
Lynch-Syndrom Diagnosekriterien: Das Lynch-Syndrom ist eine autosomal dominant vererbte
Tumordisposition mit hoher Penetranz (bis 80%). 2�5% aller KRK entstehen auf dem
Boden eines Lynch-Syndroms. Charakteristisch ist das frühe Auftreten von über-
wiegend rechtsseitig lokalisierten KRK, das Auftreten von syn- und metachronen
KRK sowie von Karzinomen anderer Organlokalisationen, vor allem in Endometrium,
Nierenbecken/ableitenden Harnwegen, Dünndarm, aber auch in Magen, Ovarien,
Gallengang, Gehirn und Haut. Im Unterschied zu den hereditären Polyposis-
Syndromen mit auffälligem Phänotyp (s. u.) ist die klinische Diagnosestellung beim
Lynch-Syndrom meist nur in Zusammenschau mit der Familienanamnese des
Patienten möglich. Die Diagnose kann klinisch gestellt werden, wenn in der Familie
des Patienten die sog. Amsterdam-I- oder -II-Kriterien erfüllt sind. In den Amsterdam-
II-Kriterien werden neben den KRK gleichwertig auch Karzinome des Endometriums,
des Dünndarms und der oberen, ableitenden Harnwege dem Syndrom zugerechnet.
In populationsbasierten Studien erfüllt die Mehrzahl der Patienten mit Lynch-
Syndrom nicht die Amsterdam-Kriterien. Der Personenkreis, bei dem möglicherweise
ein Lynch-Syndrom vorliegen könnte, wird durch die revidierten �Bethesda-Kriterien"

69
definiert. Die Tumoren dieser Patienten sollten auf die für das Lynch-Syndrom
charakteristische hochgradige Mikrosatelliteninstabilität (MSI-H) untersucht werden.
In älteren Arbeiten wurde unter ausschließlicher Berücksichtigung von
Indexpatienten berichtet, dass KRK im Mittel im 44. Lebensjahr (LJ) auftreten. Bei
den erstgradig Verwandten der Indexpatienten, die ebenfalls ein KRK entwickelten,
lag das mittlere Diagnosealter bei 61 Jahren. Als Konsequenz muss bei jedem
Patienten mit KRK unabhängig vom Diagnosealter nach der Familienanamnese
gefragt werden.
Molekulargenetik: Ursache des Lynch-Syndroms ist ein Defekt in einem der sog.
mismatch-Reparatur (MMR)-Gene. Bisher wurden pathogene Keimbahnmutationen
in den MMR-Genen MSH2, MLH1, MSH6 und PMS2 identifiziert. Um die
Mutationssuche effizienter anzugehen, ist es sinnvoll, vorab MSI-H im Tumor
nachzuweisen (z. B. anhand von archiviertem Paraffin-eingebettetem Tumor-
gewebe). Nur bei Nachweis einer MSI-H im Tumor schließt sich eine
Keimbahnmutationssuche an, wobei der Mutationsnachweis in 50�90% der Fälle
gelingt. Die immunhistochemische Untersuchung der MMR-Proteinexpression in
Tumoren gibt einen Hinweis, in welchem Gen die Mutation lokalisiert ist.
Tumorrisiko: Mutationsträger haben ein Risiko von 60�80%, ein KRK zu entwickeln.
Das Risiko steigt ab dem 30. LJ an. Das Endometriumkarzinomrisiko wird mit
20�60% angegeben; dieses Risiko steigt ab dem 40. LJ an. Zusätzlich ist das Risiko
für Karzinome des Ovar (4�13%), des Magens (2�19%), der ableitenden Harnwege
(1�7%) und des Dünndarmes (1�4%) erhöht. Das Dünndarmkarzinomrisiko wird mit
4�7,5% angegeben. Dünndarm- und Magenkarzinome vor dem 30. LJ sind sehr
selten. Selten treten Hauttumoren, Hirntumoren, Pankreaskarzinome oder biliäre
Karzinome auf.
Früherkennung: Ab dem 25. LJ, bzw. 5 Jahre vor dem Erkrankungsalter des
jüngsten betroffenen Familienmitglieds, sollte jährlich eine Koloskopie, eine
Abdomensonografie und eine gynäkologische Untersuchung inklusive trans-
vaginalem Ultraschall durchgeführt werden. Ab dem 35. LJ wird eine jährliche
Ösophago-Gastro-Duodenoskopie (ÖGD) empfohlen (Richtlinie der Verbundstudie
�familiärer Darmkrebs� der Deutschen Krebshilfe). Mehrere Studien belegen die
mögliche Senkung der Inzidenz und Mortalität für Lynch-Syndrom-assoziierte KRK

70
durch regelmäßige Koloskopien und Polypektomien. Bei 3-jährlichen Koloskopien
werden jedoch Intervallkarzinome beobachtet, sodass derzeit in Deutschland
jährliche Koloskopien empfohlen werden. Die Daten zu Früherkennungs-
untersuchungen bez. der extrakolonischen Tumoren erlauben bisher keine
abschließende Einschätzung ihrer Effektivität. Aufgrund des hohen Endometrium-
karzinomrisikos ist bei Patientinnen mit abgeschlossener Familienplanung oder in der
Postmenopause die prophylaktische Hysterektomie (ggf. auch bilaterale Adnektomie)
als Alternative zur gynäkologischen Vorsorge zu diskutieren. Der Eingriff sollte nur
bei gesicherter genetischer Anlage (pathogene Mutation oder positive Lynch-
Syndrom-Tumoranamnese mit MSI-H-Nachweis) erfolgen.
Durch die Aufdeckung der genetischen Grundlagen erblicher KRK-Formen kann
heute in den betroffenen Familien mit molekulargenetischen Methoden festgestellt
werden, ob Angehörige die Tumordisposition geerbt haben noch bevor sich
Krankheitssymptome entwickeln. Damit kann für die einzelnen Familienmitglieder
individuell die entsprechend notwendigen Früherkennungsuntersuchungen
empfohlen werden. Aber auch für Patienten, die bereits an einer Tumorerkrankung
leiden, ist die Diagnosestellung einer erblichen KRK-Form wichtig. Bei ihnen ist das
Risiko einen Zweittumor (oder noch weiterer Tumoren) zu entwickeln deutlich erhöht.
Auch hier gilt, dass den Patienten entsprechend notwendige Früherkennungsunter-
suchungen empfohlen werden sollten.
Fazit: - Bei jedem Patienten mit KRK daran denken: individuelle Eigen- und
Familienanamnese hinsichtlich Tumoren und Indexläsionen, Erkrankungsalter. Nur
die Minderheit der Patienten mit Lynch-Syndrom erfüllt die Amsterdam-Kriterien.
Auch ältere Patienten können erstmals erkranken. Auch bei Ausschluss des
Lynch-Syndroms bei familiärer Häufung liegt ein erhöhtes Risiko vor. Vorsorge der
Angehörigen erforderlich.
- Bei Verdacht abklären: genetische Beratung, Vorstellung im Zentrum für
familiären Darmkrebs
- Bei Diagnose einer hereditären Form: bei den meisten Syndromen erhöhtes
Risiko für Zweitmalignome; entsprechende Vor-/Nachsorgeprogramme.
Erkrankung der Familie, keine nur Ihren Patienten betreffende Erkrankung.
Information des Patienten und Weitergabe der Information durch den Patienten an
die Angehörigen sind wesentlich.
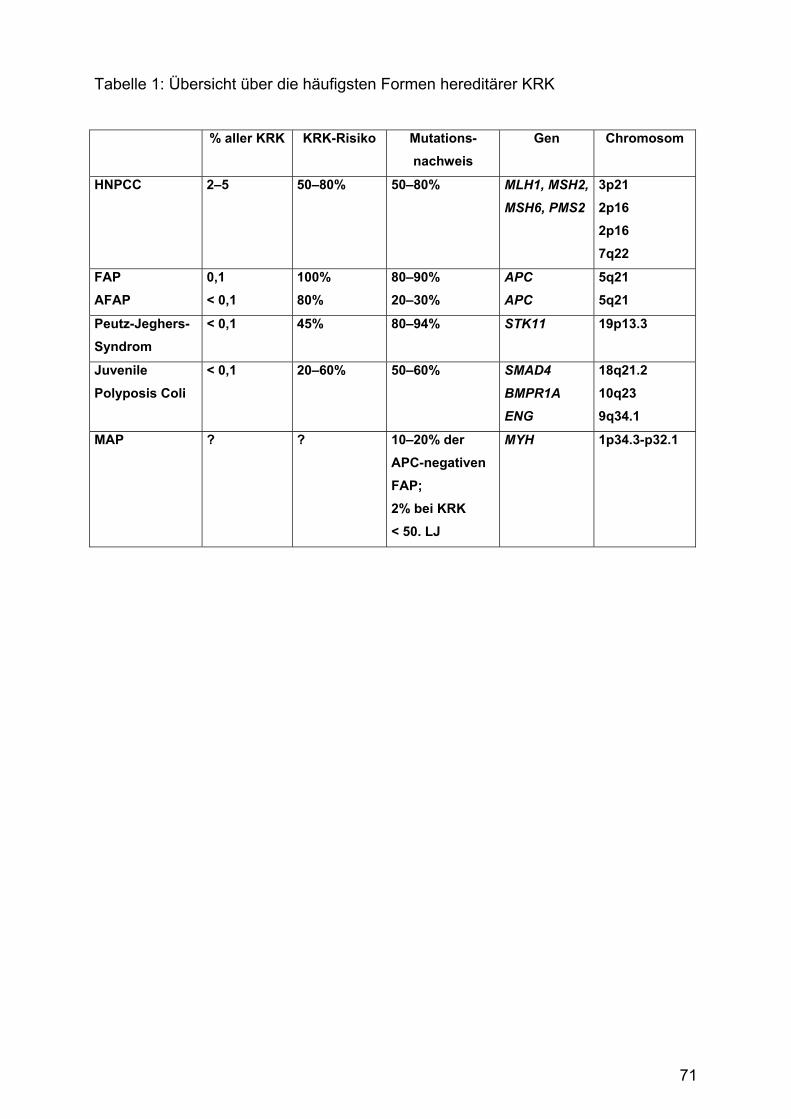
71
Tabelle 1: Übersicht über die häufigsten Formen hereditärer KRK
% aller KRK KRK-Risiko Mutations-
nachweis Gen Chromosom
HNPCC 2�5 50�80% 50�80% MLH1, MSH2,
MSH6, PMS2
3p21 2p16 2p16 7q22
FAP AFAP
0,1 < 0,1
100% 80%
80�90% 20�30%
APC
APC
5q21 5q21
Peutz-Jeghers-Syndrom
< 0,1 45% 80�94%
STK11
19p13.3
Juvenile Polyposis Coli
< 0,1 20�60%
50�60%
SMAD4
BMPR1A ENG
18q21.2 10q23 9q34.1
MAP ? ? 10�20% der APC-negativen FAP; 2% bei KRK < 50. LJ
MYH 1p34.3-p32.1

72
Kapselendoskopie des Kolons
T. Brechmann
Medizinische Klinik, Berufsgenossenschaftliches Universitätsklinikum Bergmanns-
heil, Bochum
Das kolorektale Karzinom liegt mit über 70.000 Neuerkrankungen und etwa 27.000
Todesfällen pro Jahr in Deutschland weit vorne auf der Rangliste der malignen
Tumorerkrankungen. Mit der vollständigen Koloskopie kann eine effektive Vorsorge-
untersuchung, die von den Krankenkassen ab dem 56. Lebensjahr übernommen
wird, angeboten werden. Leider ist die Akzeptanz in der Bevölkerung niedrig; pro
Jahr nehmen nur 2�3% der über 55-Jährigen die Möglichkeit einer vorsorglichen
Koloskopie zur Früherkennung eines kolorektalen Karzinoms wahr.
Alternative, weniger invasive Verfahren könnten die Rate verbessern. Mit der in
größerem Umfang untersuchten virtuellen CT-Kolonografie ist jedoch eine nicht
unerhebliche Strahlenbelastung verbunden, die die Anwendung im Rahmen einer
Vorsorge ebenso limitiert wie die im Vergleich zur Endoskopie geringere
Sensitivität/Spezifität und die ebenso notwendige Darmreinigung. Die telemetrische
Kapselendoskopie des Dickdarms ist ein neues Verfahren, das ohne ionisierende
Strahlung auskommt. Die Videokapselendoskopie des Dünndarms ist mittlerweile ein
gut etabliertes, minimalinvasives, intraluminal bildgebendes Verfahren, deren nahezu
einzige wesentliche Komplikation, je nach untersuchtem Patientenkollektiv, die
Kapselretention mit einer Rate von 0,7�2% ist. Die neu entwickelte Kolonkapsel ist
wenig größer dimensioniert und mit 2 Kameras ausgestattet. In ersten klinischen
Untersuchungen konnten der CT-Kolonografie vergleichbare Sensitivitäten und
Spezifitäten dokumentiert werden. Die Prozedur wurde gut toleriert, Komplikationen
wurden nicht gefunden. Der Reinigungsgrad und damit die Beurteilbarkeit der
Schleimhaut ist abhängig von dem zum Teil sehr intensiven Vorbereitungsprotokoll.
Allerdings wird insbesondere die Präparation des Darms bei der Koloskopie von den
Patienten als unangenehm empfunden. Bei einer anzunehmenden Polypen-
detektionsrate von 40% müsste nach dem Screening mittels Kapselendoskopie eine
erneute Darmreinigung für die Koloskopie zur Behandlung der detektierten Polypen
folgen. Damit wird eine wesentliche Verbesserung der Akzeptanz nicht zu erreichen
sein. Die Kombination von Kolonkapsel während der Nacht, Auswertung des aufge-
zeichneten Videos am nächsten Morgen mit Entscheidung bezüglich der Not

73
wendigkeit zur Koloskopie und ggf. Durchführung derselben (Nachtprozedur) wird
daher in einer Pilotstudie untersucht. Nach den ersten vorläufigen Ergebnissen von
20 Patienten kann zumeist eine gute Darmreinigung erreicht werden. Weitere Daten,
insbesondere der Vergleich der Polypendetektionsraten sind jedoch notwendig.

75
Anschriften der Referenten und Vorsitzenden
Dr. T. Brechmann Medizinische Klinik BG Universitätsklinikum Bergmannsheil Bürkle-de-la-Camp-Platz 1 44789 Bochum Dr. M. Cornberg Klinik für Gastroenterologie, Hepatologie und Endokrinologie Medizinische Hochschule Hannover Carl-Neuberg-Str. 1 30625 Hannover Prof. Dr. G. Gerken Gastroenterologie/Hepatologie Universitätsklinikum Essen Hufelandstr. 55 45147 Essen Dr. S. Heringlake Medizinische Klinik Ruhr-Universität Bochum Knappschaftskrankenhaus In der Schornau 23�25 44892 Bochum Prof. Dr. Dr. h.c. W. Hohenberger Chirurgie Universitätsklinikum Erlangen Krankenhausstr. 12 91054 Erlangen Prof. Dr. W. Kruis Innere Medizin Ev. Krankenhaus Kalk Buchforststr. 2 51103 Köln Prof. Dr. F. Lammert Klinik für Innere Medizin II Universitätsklinikum des Saarlandes Kirrberger Straße 66424 Homburg/Saar
Prof. Dr. P. Layer Innere Medizin Israelitisches Krankenhaus in Hamburg Orchideenstieg 14 22297 Hamburg Prof. Dr. M.M. Lerch Klinik und Poliklinik für Innere Medizin A Universitätsklinikum Greifswald Friedrich-Loeffler-Str. 23A 17475 Greifswald Dr. C. Mölleken Medizinische Klinik BG Universitätsklinikum Bergmannsheil Bürkle-de-la-Camp-Platz 1 44789 Bochum Prof. Dr. J. Mössner Medizinische Klinik für Gastroenterologie und Rheumatologie Universitätsklinikum Leipzig AöR Philipp-Rosenthal-Str. 27 04103 Leipzig Prof. Dr. V. Nicolas Radiologie und Nuklearmedizin BG Universitätsklinikum Bergmannsheil Bürkle-de-la-Camp-Platz 1 44789 Bochum Prof. Dr. O.G. Opitz Tumorzentrum Ludwig Heilmeyer Universitätsklinikum Freiburg Hugstetter Str. 55 79106 Freiburg Dr. T. Pietzsch Internist Zeppelinstr. 18 44791 Bochum

76
Dr. C. Pox Medizinische Klinik Ruhr-Universität Bochum Knappschaftskrankenhaus In der Schornau 23�25 44892 Bochum Dr. A. Riphaus Innere Medizin I Krankenhaus Siloah Klinikum Region Hannover Roesebeckstr. 15 30449 Hannover Prof. Dr. T. Rösch Interdisziplinäre Endoskopie Universitätsklinikum Eppendorf Martinistr. 52 20251 Hamburg Prof. Dr. T. Sauerbruch Medizinische Klinik und Poliklinik I Universitätsklinikum Bonn Sigmund-Freud-Str. 25 53105 Bonn Prof. Dr. W. Schmiegel Medizinische Klinik Ruhr-Universität Bochum Knappschaftskrankenhaus In der Schornau 23�25 44892 Bochum Prof. Dr. W.E. Schmidt Innere Medizin I St. Josef-Hospital Ruhr-Universität Bochum Gudrunstr. 56 44791 Bochum PD Dr. G. Schmidt-Heinevetter Internistin Kurt-Schumacher-Platz 4 44787 Bochum Prof. Dr. J. Schölmerich Klinik für Innere Medizin I Klinikum der Universität Regensburg 93042 Regensburg
Prof. Dr. S. Schreiber Klinik für Allgemeine Innere Medizin und Institut für Klinische Molekularbiologie Christian-Albrechts-Universität Schittenhelmstr. 12 24105 Kiel Dr. K. Schulmann Medizinische Klinik Ruhr-Universität Bochum Knappschaftskrankenhaus In der Schornau 23�25 44892 Bochum Prof. Dr. A. Tannapfel Institut für Pathologie Ruhr-Universität Bochum Bürkle-de-la-Camp-Platz 1 44789 Bochum Dr. B. Viebahn Internist Oskar-Hoffmann-Str. 153 44789 Bochum Prof. Dr. M. Zeitz Medizinische Klinik I Charité � Universitätsmedizin Campus Benjamin Franklin (CBF) Hindenburgdamm 30 12200 Berlin Prof. Dr. S. Zeuzem Medizinische Klinik I Klinikum der Johann Wolfgang Goethe-Universität Frankfurt Theodor-Stern-Kai 7 60590 Frankfurt