
Genetische Grundlagen erblicher Netzhauterkrankungen · Retinitis Pigmentosa ... Autosomal...
20
22.07.2009 22.07.2009 1 Genetische Grundlagen Genetische Grundlagen erblicher Netzhauterkrankungen erblicher Netzhauterkrankungen Genetische Grundlagen Genetische Grundlagen erblicher Netzhauterkrankungen erblicher Netzhauterkrankungen Bernd Wissinger Molekulargenetisches Labor Forschungsinstitut für Augenheilkunde Bernd Wissinger Molekulargenetisches Labor Forschungsinstitut für Augenheilkunde Department für Augenheilkunde Universitätsklinikum Tübingen Department für Augenheilkunde Universitätsklinikum Tübingen MoDe MoDe – Montags Montags-Demo zum Thema „Sehen“ Demo zum Thema „Sehen“ Studienzentrum für Sehgeschädigte, Universität Karlsruhe Studienzentrum für Sehgeschädigte, Universität Karlsruhe 13.07.2009 13.07.2009 Übersicht zum Vortrag Übersicht zum Vortrag Übersicht zum Vortrag Übersicht zum Vortrag Der 1.Sinn Der 1.Sinn Fallbeispiel Krankheitsbilder & Klassifikation Prävalenz erblicher NH-Erkrankungen Genetische Aspekte Fallbeispiel Krankheitsbilder & Klassifikation Prävalenz erblicher NH-Erkrankungen Genetische Aspekte Genetische Aspekte Funktionelle Genetik Blick in die Zukunft Genetische Aspekte Funktionelle Genetik Blick in die Zukunft
Transcript of Genetische Grundlagen erblicher Netzhauterkrankungen · Retinitis Pigmentosa ... Autosomal...

22.07.200922.07.2009
11
Genetische Grundlagen Genetische Grundlagen erblicher Netzhauterkrankungenerblicher Netzhauterkrankungen
Genetische Grundlagen Genetische Grundlagen erblicher Netzhauterkrankungenerblicher Netzhauterkrankungen
Bernd Wissinger
Molekulargenetisches LaborForschungsinstitut für Augenheilkunde
Bernd Wissinger
Molekulargenetisches LaborForschungsinstitut für Augenheilkundeg g
Department für AugenheilkundeUniversitätsklinikum Tübingen
g gDepartment für AugenheilkundeUniversitätsklinikum Tübingen
MoDeMoDe –– MontagsMontags--Demo zum Thema „Sehen“Demo zum Thema „Sehen“Studienzentrum für Sehgeschädigte, Universität KarlsruheStudienzentrum für Sehgeschädigte, Universität Karlsruhe
13.07.200913.07.2009
Übersicht zum VortragÜbersicht zum VortragÜbersicht zum VortragÜbersicht zum Vortrag
Der 1.Sinn Der 1.Sinn
Fallbeispiel
Krankheitsbilder & Klassifikation
Prävalenz erblicher NH-Erkrankungen
Genetische Aspekte
Fallbeispiel
Krankheitsbilder & Klassifikation
Prävalenz erblicher NH-Erkrankungen
Genetische AspekteGenetische Aspekte
Funktionelle Genetik
Blick in die Zukunft
Genetische Aspekte
Funktionelle Genetik
Blick in die Zukunft
22.07.200922.07.2009
22
RetinalRetinalpigmentpigmentepitheliumepithelium
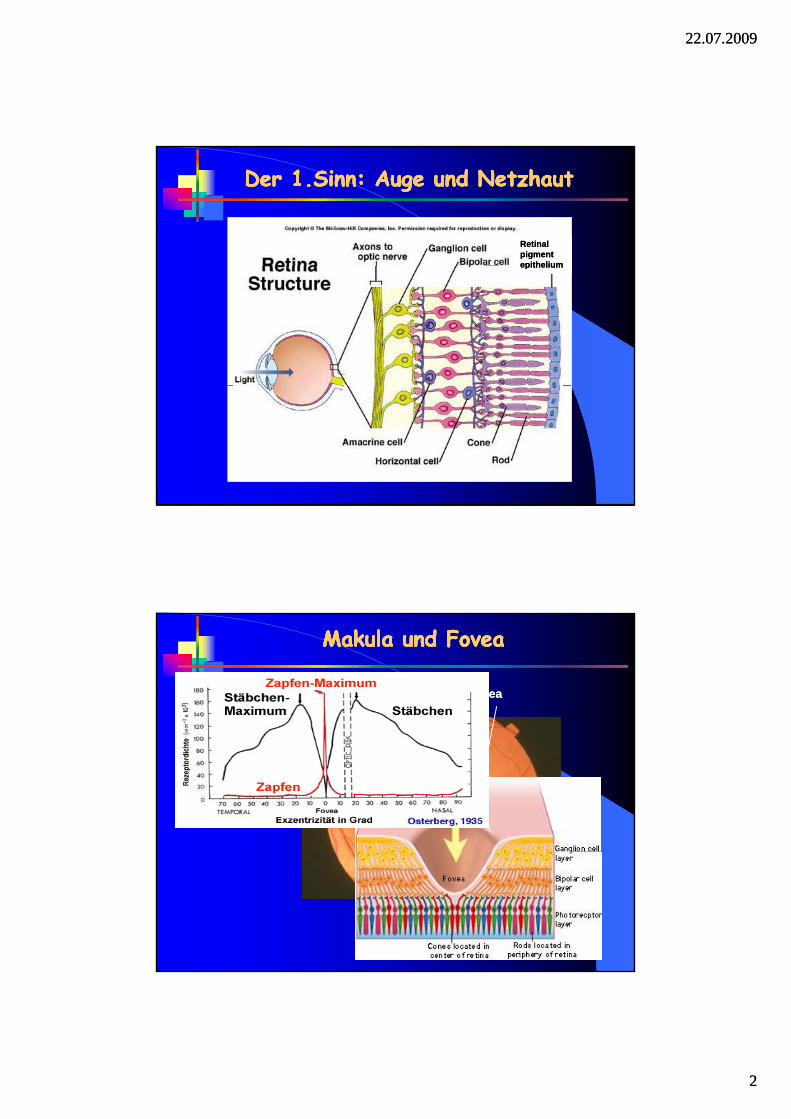
Der 1.Sinn: Auge und NetzhautDer 1.Sinn: Auge und NetzhautDer 1.Sinn: Auge und NetzhautDer 1.Sinn: Auge und Netzhaut
epitheliumepithelium
MaculaMacula lutealutea
MakulaMakula und und FoveaFoveaMakulaMakula und und FoveaFovea
FoveaFoveacentraliscentralis
22.07.200922.07.2009
33
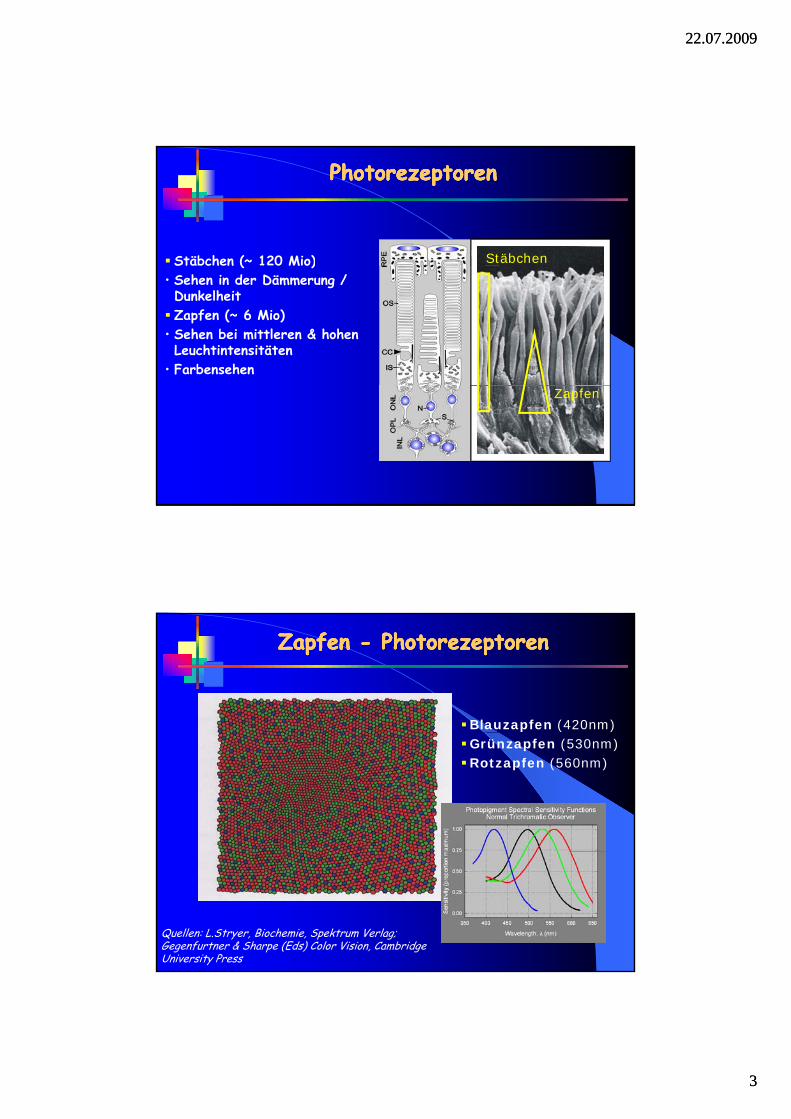
PhotorezeptorenPhotorezeptorenPhotorezeptorenPhotorezeptoren
Stäbchen (~ 120 Mio) Stäbchen( )• Sehen in der Dämmerung / DunkelheitZapfen (~ 6 Mio)
• Sehen bei mittleren & hohen Leuchtintensitäten
• Farbensehen
Zapfen
Zapfen Zapfen -- PhotorezeptorenPhotorezeptorenZapfen Zapfen -- PhotorezeptorenPhotorezeptoren
Blauzapfen (420nm)Grünzapfen (530nm)Rotzapfen (560nm)
Quellen: L.Stryer, Biochemie, Spektrum Verlag; Gegenfurtner & Sharpe (Eds) Color Vision, Cambridge University Press

22.07.200922.07.2009
44
PhototransduktionPhototransduktionPhototransduktionPhototransduktion
Patient L.M., 36 Jahre, SchlosserPatient L.M., 36 Jahre, Schlosser
Nachtblind seit später KindheitNachtblind seit später Kindheit
FallbeispielFallbeispielFallbeispielFallbeispiel
Nachtblind seit später KindheitNachtblind seit später Kindheitklagt über zunehmende Sehminderungklagt über zunehmende Sehminderunghat Sorge um Führerschein und Berufhat Sorge um Führerschein und Beruf
VisusVisus: 0.5 / 0.4: 0.5 / 0.4Gesichtsfeld: konzentrische EinengungGesichtsfeld: konzentrische EinengungFarbensehen: normalFarbensehen: normalElektroretinographieElektroretinographie: : ElektroretinographieElektroretinographie: :
skotopischskotopisch –– stark reduzierte Potentialestark reduzierte Potentialephotopischphotopisch –– in unteren Normbereichin unteren Normbereich
Fundus: knochenkörperförmige Fundus: knochenkörperförmige Pigmentablagerungen in der Peripherie, Pigmentablagerungen in der Peripherie, aufgehellte Papilleaufgehellte Papille
Leichte Linsentrübung Leichte Linsentrübung
22.07.200922.07.2009
55
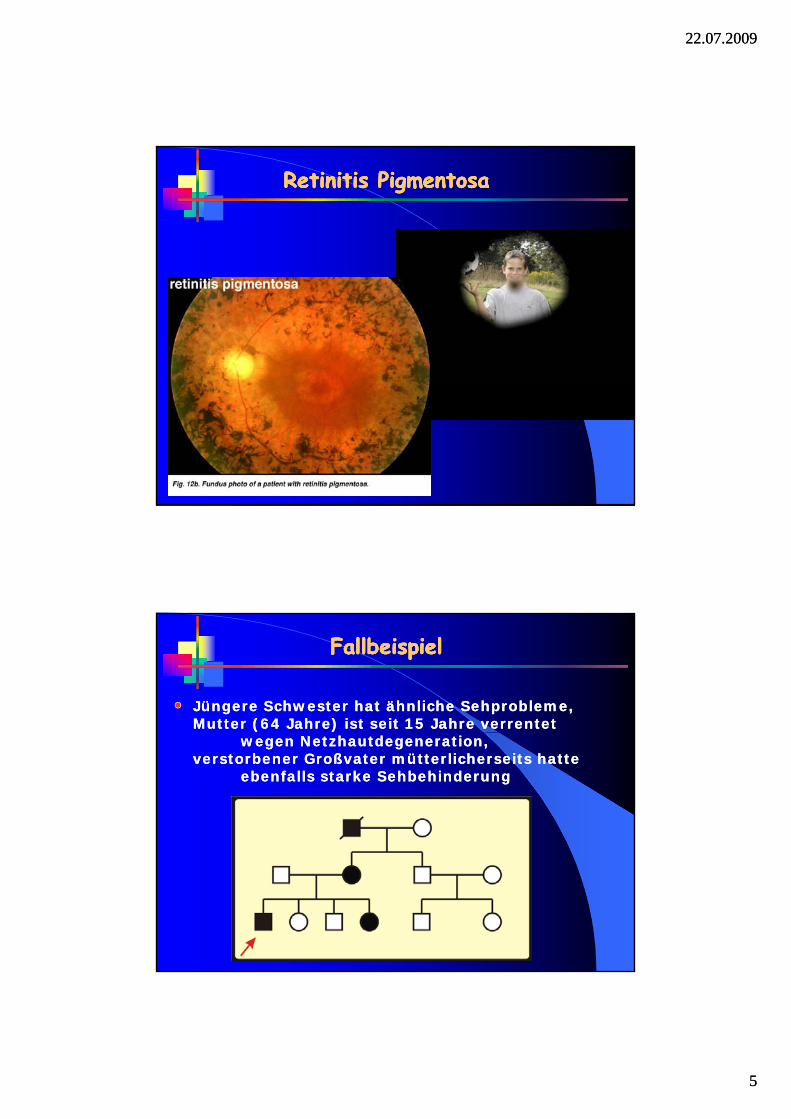
Retinitis Retinitis PigmentosaPigmentosaRetinitis Retinitis PigmentosaPigmentosa
Jüngere Schwester hat ähnliche Sehprobleme,Jüngere Schwester hat ähnliche Sehprobleme,Mutter (64 Jahre) ist seit 15 Jahre verrentet Mutter (64 Jahre) ist seit 15 Jahre verrentet
FallbeispielFallbeispielFallbeispielFallbeispiel
wegen Netzhautdegeneration, wegen Netzhautdegeneration, verstorbener Großvater mütterlicherseits hatte verstorbener Großvater mütterlicherseits hatte
ebenfalls starke Sehbehinderungebenfalls starke Sehbehinderung

22.07.200922.07.2009
66
Molekulargenetischer Befund: Molekulargenetischer Befund: „……Beim Patient liegt ein „……Beim Patient liegt ein heterozygoter heterozygoter B t hB t h C tidiC tidi Th iTh i N kl tid itiN kl tid iti
FallbeispielFallbeispielFallbeispielFallbeispiel
BasenaustauschBasenaustausch CytidinCytidin zu zu ThyminThymin an an NukleotidpositionNukleotidposition67 (c.67C>T) in 67 (c.67C>T) in ExonExon 1 des 1 des RhodopsingensRhodopsingens (RHO) (RHO) vor. vor. Dieser Austausch bedingt eine Dieser Austausch bedingt eine ProlinProlin zu Histidin zu Histidin SubstitutionSubstitution an an AminosäurepositionAminosäureposition 23 des Proteins 23 des Proteins (Pro23His)……“. (Pro23His)……“.
„…… Die Mutation ist bereits mehrfach in der Literatur „…… Die Mutation ist bereits mehrfach in der Literatur l h i l d i bli hl h i l d i bli hals Ursache einer autosomal dominant erblichen als Ursache einer autosomal dominant erblichen
RetintitisRetintitis pigmentosapigmentosa beschriebenbeschrieben und ist als pathogen und ist als pathogen klassifiziert. Der klassifiziert. Der klinische Verdacht einer erblichen klinische Verdacht einer erblichen NetzhauterkrankungNetzhauterkrankung wird somit durch die wird somit durch die molekulargenetische Untersuchung eindeutig molekulargenetische Untersuchung eindeutig bestätigtbestätigt. . ……“. ……“.
Molekulargenetischer Befund: Molekulargenetischer Befund: „…… Es ist in diesem Fall von einem „…… Es ist in diesem Fall von einem autosomal autosomal d i t E bd i t E b h d h b t ht i h d h b t ht i
FallbeispielFallbeispielFallbeispielFallbeispiel
dominanten Erbgangdominanten Erbgang auszugehen, d.h. es besteht ein auszugehen, d.h. es besteht ein 50%iges Risiko für jedes Kind eines 50%iges Risiko für jedes Kind eines Betroffenen/GenträgersBetroffenen/Genträgers. . Zur weiteren Diskussion der erblichen Aspekte und des Zur weiteren Diskussion der erblichen Aspekte und des Vererbungsrisikos empfehlen wir eine Vorstellung bei Vererbungsrisikos empfehlen wir eine Vorstellung bei einer genetischen Beratungsstelle.. ……“ einer genetischen Beratungsstelle.. ……“
„…… Es besteht derzeit „…… Es besteht derzeit keine wissenschaftlich gesicherte keine wissenschaftlich gesicherte Therapie oder BehandlungTherapie oder Behandlung bei der autosomal dominant bei der autosomal dominant erblichen Retinitis erblichen Retinitis pigmentosapigmentosa. . Wir empfehlen eine regelmäßige augenärztliche Wir empfehlen eine regelmäßige augenärztliche Kontrolluntersuchung und Vorstellung in unserer Kontrolluntersuchung und Vorstellung in unserer SehbehindertenambulanzSehbehindertenambulanz zwecks Anpassung optimierter zwecks Anpassung optimierter Sehhilfen ……“ Sehhilfen ……“

22.07.200922.07.2009
77
Klassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & Krankheitsbilder
Klassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & Krankheitsbilder
Verlauf: Stationäre <> ProgressivB i b ät if tVerlauf: Stationäre <> ProgressivB i b ät if tBeginn: angeboren <> spätmanifestLokalisation: zentral <> peripherFunktion: Stäbchen <> Zapfen,
Photorezeptor <> SynapseNeuroretina <> Ret.Pigmentepithel
Beginn: angeboren <> spätmanifestLokalisation: zentral <> peripherFunktion: Stäbchen <> Zapfen,
Photorezeptor <> SynapseNeuroretina <> Ret.Pigmentepithel
Morphologie: Degeneration <> DysfunktionSymptomatik: nichtsyndromal <> syndromalMorphologie: Degeneration <> DysfunktionSymptomatik: nichtsyndromal <> syndromal

22.07.200922.07.2009
88
Klassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & KrankheitsbilderKlassifikation & Krankheitsbilder
Periphere, progressive NH-ErkrankungenRetinitis PigmentosaZ t l i NH E k k
Periphere, progressive NH-ErkrankungenRetinitis PigmentosaZ t l i NH E k kZentrale, progressive NH-ErkrankungenStargardt‘sche MakuladystrophieGeneral., progressive NH-ErkrankungenZapfen-, Zapfen-Stäbchen-DystrophieKongenitale, stationäre NH-ErkrankungenCSNB A h t i
Zentrale, progressive NH-ErkrankungenStargardt‘sche MakuladystrophieGeneral., progressive NH-ErkrankungenZapfen-, Zapfen-Stäbchen-DystrophieKongenitale, stationäre NH-ErkrankungenCSNB A h t iCSNB, AchromatopsieTapetoretinale Dystrophien PigmentepitheliopathieSyndromale NH-Erkrankungen Usher-Syndrom, Bardet-Biedl-Syndrom
CSNB, AchromatopsieTapetoretinale Dystrophien PigmentepitheliopathieSyndromale NH-Erkrankungen Usher-Syndrom, Bardet-Biedl-Syndrom
Ursachen von Blindheit: Ursachen von Blindheit: Irland 1996; Sehschärfe < 6/60 oder GF < 20Irland 1996; Sehschärfe < 6/60 oder GF < 20°°Erwerbsalter:Erwerbsalter: n = 2196; Alter: 16n = 2196; Alter: 16--64 Jahre 64 Jahre
Prävalenz
Erwerbsalter:Erwerbsalter: n 2196; Alter: 16n 2196; Alter: 16 64 Jahre 64 Jahre
13%10%40%
RPAlbinismCataract cong.Cataract
9%8%
7%7%6%
Optic AtrophyMyopiaGlaucomaOthers

22.07.200922.07.2009
99
Prävalenz: Prävalenz: 1:1,4901:1,490 (Nordfrankreich) (Nordfrankreich) ((PuechPuech et al 1991 J Fr et al 1991 J Fr OphthalmolOphthalmol 14: 15314: 153--164)164)
Prävalenz
((PuechPuech et al., 1991, J Fr et al., 1991, J Fr OphthalmolOphthalmol 14: 15314: 153 164)164)
Prävalenz (Kindern): Prävalenz (Kindern): 1:10,0001:10,000 (Dänemark) (Dänemark) (Rosenberg, 1989, Doc (Rosenberg, 1989, Doc OphthalmolOphthalmol 73: 8173: 81--92.92.
InzidenzInzidenz (gesetzl. Blindheit, VA<1/50): (gesetzl. Blindheit, VA<1/50): 1:192 3001:192 300 WürttembergWürttemberg HollenzollernHollenzollern 1994 1994 1:192,3001:192,300 WürttembergWürttemberg--HollenzollernHollenzollern 1994 1994
((KrumpaszkyKrumpaszky et al., 1999, et al., 1999, OphthalmologicaOphthalmologica 213: 176213: 176--
182.)182.)
Retinitis Retinitis pigmentosapigmentosaPrävalenz: 1:2 500 Prävalenz: 1:2 500 –– 1:4 000 1:4 000
Prävalenz
Prävalenz: 1:2,500 Prävalenz: 1:2,500 1:4,000 1:4,000 ((BoughmanBoughman et al., 1980, Am J Hum Genet 32: 223et al., 1980, Am J Hum Genet 32: 223--235. 235. Haim et al., 1992, Acta Haim et al., 1992, Acta OphthalmolOphthalmol 70: 17870: 178--186.186.
Stargardt‘scheStargardt‘sche MakuladystrophieMakuladystrophiePrävalenz: ~1:10,000 Prävalenz: ~1:10,000 BlacharskiBlacharski, 1988, in , 1988, in NewsomeNewsome: : RetinalRetinal dystrophiesdystrophies andanddegenerationsdegenerations, Raven Press, NY, p.135, Raven Press, NY, p.135--159.159.
AchromatopsiaAchromatopsiaPrävalenz: ~ 1:30,000 Prävalenz: ~ 1:30,000 –– 1:50,000 1:50,000 ((Francois, 1961, Francois, 1961, HeredityHeredity in in OphthalmologyOphthalmology. CV Mosby). CV Mosby)

22.07.200922.07.2009
1010
Retinitis Retinitis pigmentosapigmentosa
~ 40~ 40--50% sporadisch50% sporadisch
Genetische Aspekte
~ 40~ 40 50% sporadisch50% sporadisch
~ 20~ 20--30% autosomal rezessiv30% autosomal rezessiv
~ 10~ 10--25% autosomal dominant25% autosomal dominant
~ 6~ 6--18% X18% X--chromosomalchromosomal rezessivrezessiv
Sehr selten: Sehr selten: maternalmaternal, , digenischdigenisch
Fishman 1978, Fishman 1978, ArchArch OphthalmolOphthalmol 96: 82296: 822--826 826 BoughmanBoughman et al., 1982, Am J Hum Genet 32: 223et al., 1982, Am J Hum Genet 32: 223--235 235 Jay 1982, Jay 1982, BrBr J J OphthalmolOphthalmol 66: 40566: 405--416 416 Hu 1982, Am J Hu 1982, Am J MedMed Genet 12: 51Genet 12: 51--56 56 Haim 2002, Acta Haim 2002, Acta OphthalmolOphthalmol ScandScand 233: 1233: 1--3434
Chromosomen sind Chromosomen sind die physikalische die physikalische
Physikalische Basis der Vererbung
Genetische Aspekte
die physikalische die physikalische VererbungseinheitenVererbungseinheiten
22 Paare22 PaareAutosomen (Chr.1Autosomen (Chr.1--22)22)
1 Paar 1 Paar GonosomenGonosomenGeschlechtschromosomenGeschlechtschromosomenXX XX bei Frauenbei FrauenXX XX –– bei Frauenbei FrauenXY XY –– bei Männern bei Männern

22.07.200922.07.2009
1111
Physikalische Basis der Vererbung
Genetische Aspekte
Erbsubstanz ist die DNA. Erbsubstanz ist die DNA. Ein Chromosomen ist Ein Chromosomen ist ein komplex gepacktes ein komplex gepacktes DNADNA--Molekül.Molekül.
Physikalische Basis der Vererbung
Genetische Aspekte
Ein Ein GenGen ist ein DNAist ein DNA--Abschnitt,Abschnitt,der ein funktionelles Produkt (Protein) kodiertder ein funktionelles Produkt (Protein) kodiertEin Ein AllelAllel ist eine von mehreren phänotypischen ist eine von mehreren phänotypischen Ausprägungsformen eines Gens (z.B. ABO Ausprägungsformen eines Gens (z.B. ABO Blutgruppen) Blutgruppen)

22.07.200922.07.2009
1212
Autosomal DominantAutosomal DominantDie Präsenz eines merkmalsgebenden Allels (Die Präsenz eines merkmalsgebenden Allels (aa) reicht zur ) reicht zur Ausprägung des Merkmals (der Erkrankung). Ausprägung des Merkmals (der Erkrankung).
aa ist dominant über ist dominant über AA
Genetische Aspekte: Erbgänge
aa ist dominant über ist dominant über AAbbetrifft Männer und Frauen gleichermaßen, etrifft Männer und Frauen gleichermaßen, ein Betroffener ist immer Kind eines Betroffenen, ein Betroffener ist immer Kind eines Betroffenen,
50%iges Risiko für jedes Kind eines Betroffenen50%iges Risiko für jedes Kind eines Betroffenen
Molekulargenetischer Befund: Molekulargenetischer Befund:
Die Mutation ist bereits mehrfach in der Literatur Die Mutation ist bereits mehrfach in der Literatur
Genetische Aspekte / Fallbeispiel
„…… Die Mutation ist bereits mehrfach in der Literatur „…… Die Mutation ist bereits mehrfach in der Literatur als Ursache einer autosomal dominant erblichen als Ursache einer autosomal dominant erblichen RetintitisRetintitis pigmentosapigmentosa beschriebenbeschrieben und ist als pathogen und ist als pathogen klassifiziert. Der klassifiziert. Der klinische Verdacht einer erblichen klinische Verdacht einer erblichen NetzhauterkrankungNetzhauterkrankung wird somit durch die wird somit durch die molekulargenetische Untersuchung eindeutig molekulargenetische Untersuchung eindeutig bestätigtbestätigt. . ……“. ……“.
„…… Es ist in diesem Fall von einem „…… Es ist in diesem Fall von einem autosomal autosomal dominanten Erbgangdominanten Erbgang auszugehen, d.h. es besteht ein auszugehen, d.h. es besteht ein 50%iges Risiko für jedes Kind eines 50%iges Risiko für jedes Kind eines Betroffenen/GenträgersBetroffenen/Genträgers. . Zur weiteren Diskussion der erblichen Aspekte und des Zur weiteren Diskussion der erblichen Aspekte und des Vererbungsrisikos empfehlen wir eine Vorstellung bei Vererbungsrisikos empfehlen wir eine Vorstellung bei einer genetischen Beratungsstelle.. ……“ einer genetischen Beratungsstelle.. ……“

22.07.200922.07.2009
1313
Autosomal dominante Retinitis Autosomal dominante Retinitis pigmentosapigmentosa::
RP4RP4 3q223q22 RHORHO 1515--25% 25%
Genetische AspekteLokusheterogenität
RP4RP4 3q223q22 RHORHO 1515 25% 25% RP7RP7 6p216p21 RDSRDS 33--5% 5% RP1RP1 8q128q12 RP1RP1 55--10% 10% RP13RP13 17q1317q13 PRPF8 PRPF8 55--10% 10% RP11RP11 19q1319q13 PRPF31 PRPF31 (21% in UK) (21% in UK) RP18RP18 1q211q21 PRPF3 PRPF3 3%3%RP10RP10 7q327q32 IMPDH1 IMPDH1 RP27RP27 14q1114q11 NRL NRL RP27RP27 14q1114q11 NRL NRL
19q1319q13 CRXCRX17q2517q25 FSCN2 FSCN2
RP17RP17 17q2317q23 CA4CA41q221q22 SEMA4ASEMA4A
RP9RP9 7p147p14 (PIM1K)?(PIM1K)?+ 2 + 2 GenlociGenloci (RP31, RP33), nicht kloniert(RP31, RP33), nicht kloniert
Molekulargenetischer Befund: Molekulargenetischer Befund: „……Beim Patient liegt ein „……Beim Patient liegt ein heterozygoter heterozygoter B t hB t h C tidiC tidi Th iTh i N kl tid itiN kl tid iti
Genetische Aspekte / Fallbeispiel
BasenaustauschBasenaustausch CytidinCytidin zu zu ThyminThymin an an NukleotidpositionNukleotidposition67 (c.67C>T) in 67 (c.67C>T) in ExonExon 1 des 1 des RhodopsingensRhodopsingens (RHO) (RHO) vor. vor. Dieser Austausch bedingt eine Dieser Austausch bedingt eine ProlinProlin zu Histidin zu Histidin SubstitutionSubstitution an an AminosäurepositionAminosäureposition 23 des Proteins 23 des Proteins (Pro23His)……“. (Pro23His)……“.
22.07.200922.07.2009
1414
Genetische Aspekte
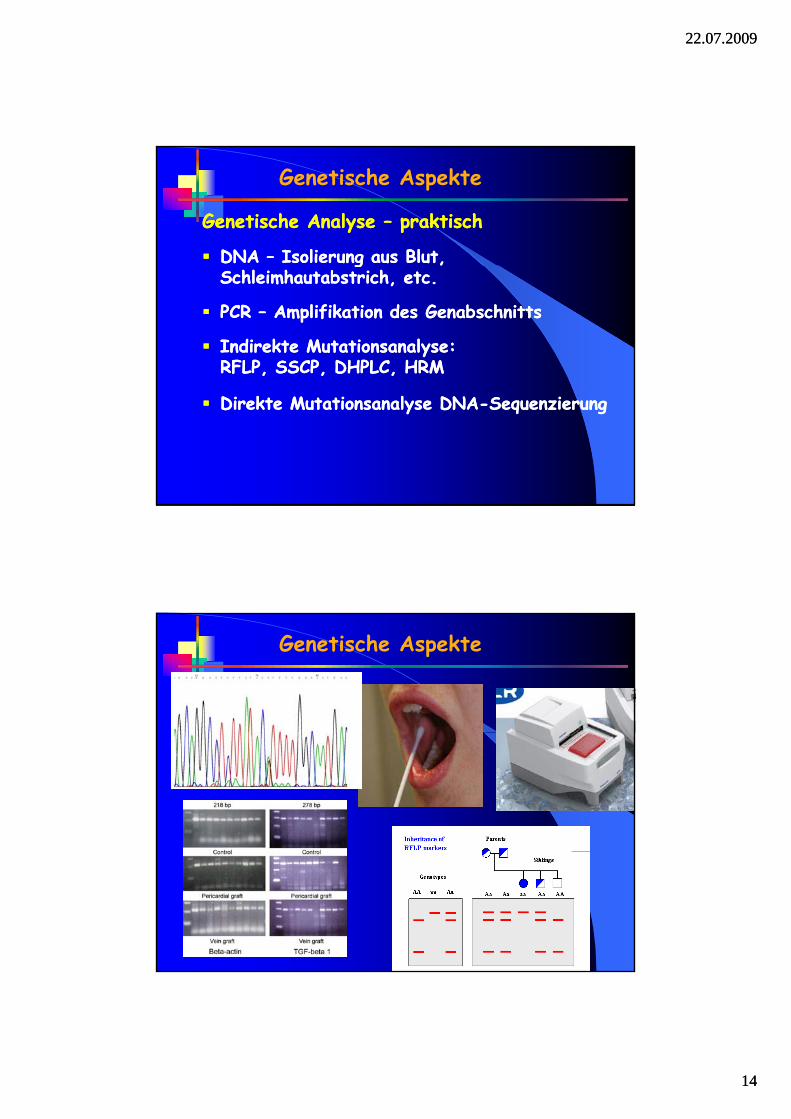
Genetische Analyse – praktisch
DNA – Isolierung aus Blut,
Genetische Analyse – praktisch
DNA – Isolierung aus Blut, gSchleimhautabstrich, etc.
PCR – Amplifikation des Genabschnitts
Indirekte Mutationsanalyse: RFLP, SSCP, DHPLC, HRM
gSchleimhautabstrich, etc.
PCR – Amplifikation des Genabschnitts
Indirekte Mutationsanalyse: RFLP, SSCP, DHPLC, HRM
Direkte Mutationsanalyse DNA-SequenzierungDirekte Mutationsanalyse DNA-Sequenzierung
Genetische Aspekte

22.07.200922.07.2009
1515
MutationenMutationen
MissenseMissense Mutation = Mutation = AminosäureaustauschAminosäureaustausch
Genetische Aspekte
MissenseMissense Mutation Mutation AminosäureaustauschAminosäureaustausch--GGGAGA-- --AAGAGA-- = = GlyGly ArgArg
NonsenseNonsense Mutation = Umwandlung in ein Mutation = Umwandlung in ein StoppcodonStoppcodon: : --GGGAGA-- --TTGAGA-- = = GlyGly StopStop
DeletionDeletion/Insertion = Leserasterverschiebung /Insertion = Leserasterverschiebung und/oder Verlust an Aminosäuren: und/oder Verlust an Aminosäuren: --GGGGA AGA GGA CA AGA GGA C-- --GAA GAG GACGAA GAG GAC--
GlyGly--ArgArg--GlyGly GluGlu--GluGlu--AspAsp
mRNAmRNA--ProzessierungsProzessierungs –– Mutationen Mutationen FehlsplicingFehlsplicing der der mRNAmRNA DeletionDeletion von von ExonsExons bzw. bzw. ExonteilenExonteilen, Insertionen, Insertionen
Molekulargenetischer Befund: Molekulargenetischer Befund: „……Beim Patient liegt ein „……Beim Patient liegt ein heterozygoter heterozygoter B t hB t h C tidiC tidi Th iTh i N kl tid itiN kl tid iti
Genetische Aspekte / Fallbeispiel
BasenaustauschBasenaustausch CytidinCytidin zu zu ThyminThymin an an NukleotidpositionNukleotidposition67 (c.67C>T) 67 (c.67C>T) in in ExonExon 1 des 1 des RhodopsingensRhodopsingens (RHO) (RHO) vor. vor. Dieser Austausch bedingt eine Dieser Austausch bedingt eine ProlinProlin zu Histidin zu Histidin SubstitutionSubstitution an an AminosäurepositionAminosäureposition 23 des Proteins 23 des Proteins (Pro23His)(Pro23His)……“. ……“.
22.07.200922.07.2009
1616
AllelAllel = Ausprägung eines Gens= Ausprägung eines Gens
ll l hll l h
Genetische Aspekte
AllelischeAllelische Heterogenität:Heterogenität:Es existieren in der Population verschiedene Es existieren in der Population verschiedene Ausprägungen eines Gens. Ausprägungen eines Gens. Im Zusammenhang mit Erkrankungen: Im Zusammenhang mit Erkrankungen: Die Erkrankungen wird (u.a.) durch verschiedene Die Erkrankungen wird (u.a.) durch verschiedene MutationenMutationen innerhalb eines Gens verursacht.innerhalb eines Gens verursacht.
AllelischeAllelische Heterogenität ist der Heterogenität ist der RegelfallRegelfall bei bei erblichen erblichen NetzhauterkankungenNetzhauterkankungen !!!!
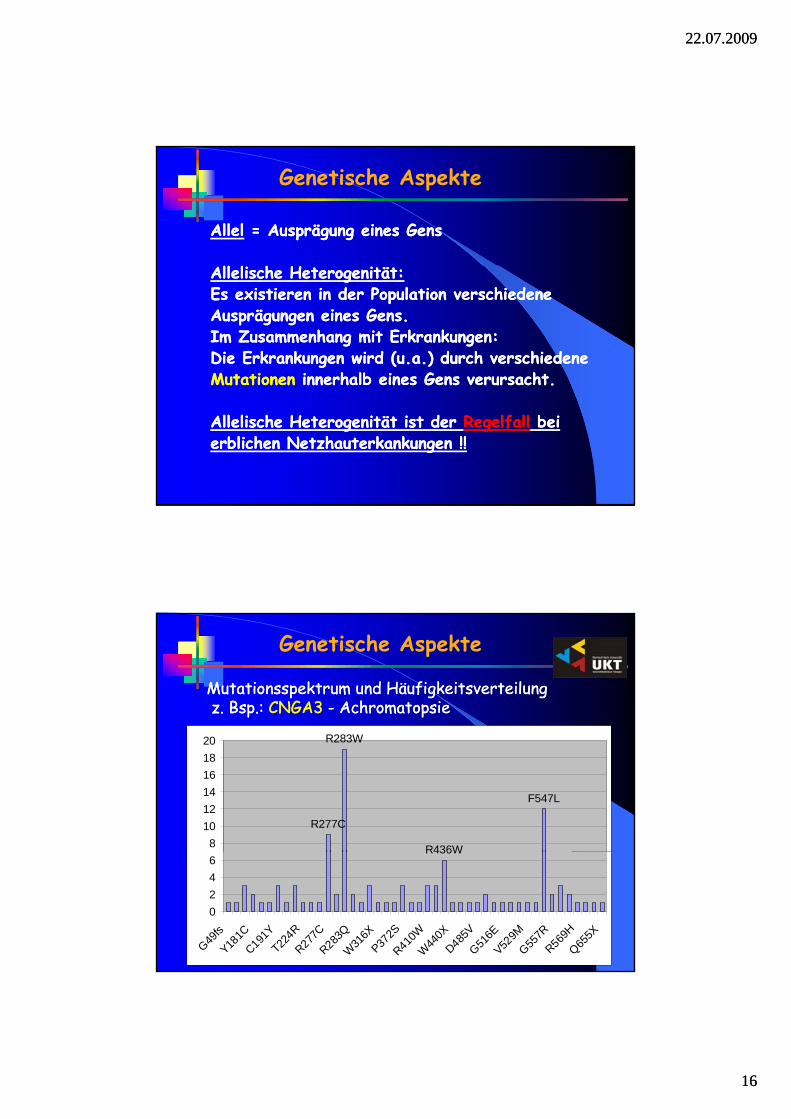
Mutationsspektrum und HäufigkeitsverteilungMutationsspektrum und Häufigkeitsverteilungz. Bsp.: z. Bsp.: CNGA3CNGA3 -- AchromatopsieAchromatopsie
Genetische Aspekte
R277C
R283W
R436W
F547L
8101214161820
R436W
0246
G49fs
Y181C
C191Y
T224R
R277C
R283Q
W31
6XP37
2S
R410W
W44
0XD485
V
G516E
V529M
G557R
R569H
Q655X

22.07.200922.07.2009
1717
CNGB3: CNGB3: PingelapPingelap BlindnessBlindnessCNGB3: CNGB3: PingelapPingelap BlindnessBlindness
Genetische Aspekte
Kohl et al., 2000, Hum Mol Genet 9: 2107-2116; Sundin et al. 2000, Nat Genet 25: 289-293
PhototransduktionskaskadePhototransduktionskaskade
RhodopsinRhodopsin RHORHO RP, CSNB RP, CSNB Z f iZ f i OPN1LWOPN1LW F b hd f kt F b hd f kt
Funktionelle Genetik
ZapfenopsineZapfenopsine OPN1LWOPN1LW Farbsehdefekte Farbsehdefekte TransducinTransducin GNAT1GNAT1 CSNB CSNB CC--TransducinTransducin GNAT2GNAT2 Achromatopsie Achromatopsie PhosphodiesterasePhosphodiesterase PDE6APDE6A RP RP
PDE6BPDE6B RP, CSNB RP, CSNB CNGCNG--KanalKanal CNGA1/B1CNGA1/B1 RP RP CC--CNGCNG--KanalKanal CNGA3/B3CNGA3/B3 Achromatopsie Achromatopsie CC--CNGCNG--KanalKanal CNGA3/B3CNGA3/B3 Achromatopsie Achromatopsie ArrestinArrestin SAGSAG RP, RP, M.OguchiM.OguchiRhodopsinkinaseRhodopsinkinase GRK1GRK1 M.OguchiM.OguchiGuanylatcyclaseGuanylatcyclase GUCY2DGUCY2D LCA, LCA, ConeCone--Rod Rod DysDys. . GCGC--AktivatorAktivator GUCA1AGUCA1A ConeCone DystrophyDystrophy

22.07.200922.07.2009
1818
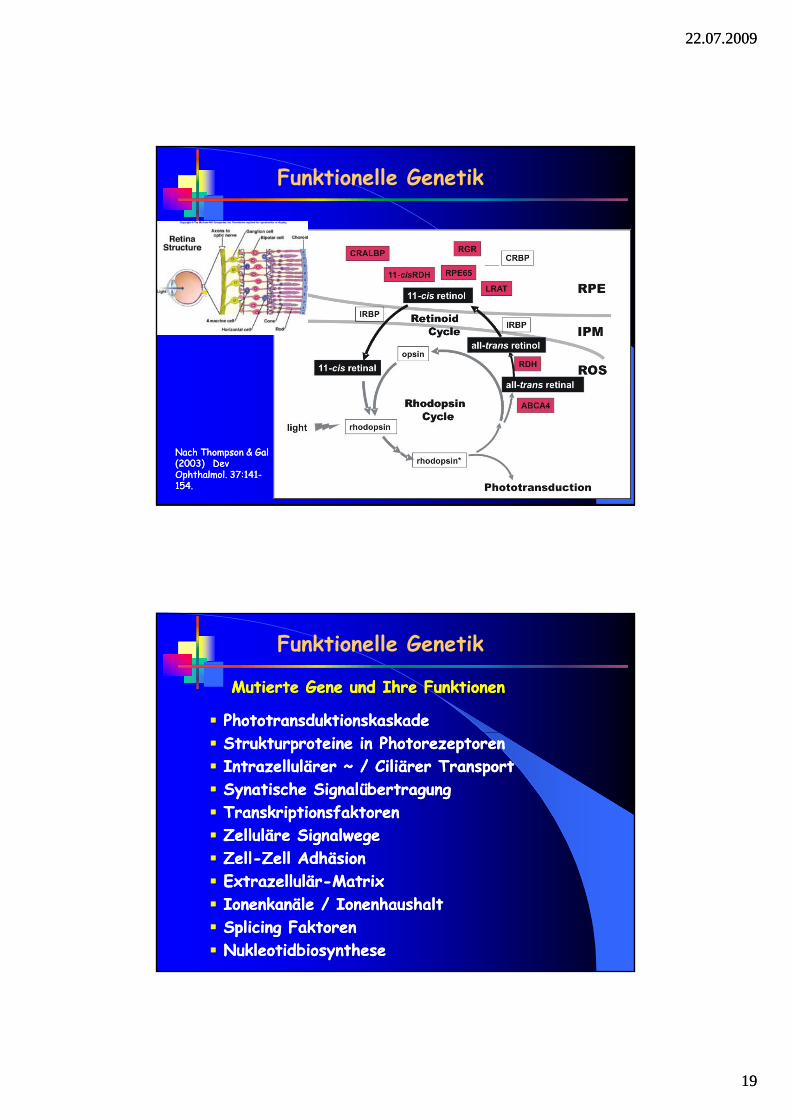
Funktionelle Genetik
Vitamin A Vitamin A –– Stoffwechsel und TransportStoffwechsel und Transport
Funktionelle Genetik
RERE--MetabolismusMetabolismus RPE65RPE65 LCA, RP LCA, RP R‘olR‘ol--Bindeprotein Bindeprotein RLBP1RLBP1 Ret. Ret. punctatapunctata alb. alb. RetinoldehydrogRetinoldehydrog. . RDH5RDH5 FdFd. . AlbipunctatusAlbipunctatusRetinoldehydrogRetinoldehydrog.. RDH12RDH12 LCA, RP LCA, RP Aktiv. GAktiv. G--Protein Protein RGRRGR RP RP Lec.Lec.--R‘olR‘ol Acyltr‘fseAcyltr‘fse LRATLRAT LCA LCA R ti lR ti l FliFli ABCA4ABCA4 St dtSt dt CRD RP CRD RP RetinolRetinol--FlippaseFlippase ABCA4ABCA4 StargardtStargardt, CRD, RP , CRD, RP R‘olR‘ol--Bindeprotein Bindeprotein RBP4RBP4 PEPE--PathiePathie
22.07.200922.07.2009
1919
Funktionelle Genetik
Nach Thompson & Gal Nach Thompson & Gal (2003) (2003) DevDevOphthalmolOphthalmol. 37:141. 37:141--154. 154.
Funktionelle Genetik
Mutierte Gene und Ihre Funktionen
Phototransduktionskaskade
Mutierte Gene und Ihre Funktionen
PhototransduktionskaskadeStrukturproteine in PhotorezeptorenIntrazellulärer ~ / Ciliärer TransportSynatische Signalübertragung TranskriptionsfaktorenZelluläre SignalwegeZ ll Z ll dh i
Strukturproteine in PhotorezeptorenIntrazellulärer ~ / Ciliärer TransportSynatische Signalübertragung TranskriptionsfaktorenZelluläre SignalwegeZ ll Z ll dh iZell-Zell AdhäsionExtrazellulär-MatrixIonenkanäle / IonenhaushaltSplicing Faktoren Nukleotidbiosynthese
Zell-Zell AdhäsionExtrazellulär-MatrixIonenkanäle / IonenhaushaltSplicing Faktoren Nukleotidbiosynthese

22.07.200922.07.2009
2020
Blick in aktuelle Zukunft
Verbesserte (?!) genetische Diagnostik RetChip (Uni Regensburg)
Verbesserte (?!) genetische Diagnostik RetChip (Uni Regensburg)RetChip (Uni Regensburg)Genomsequenz für 1.000 $
Genetische Risikofaktoren für Volkskrankheiten(z.Bsp. AMD, Glaukom)
Gentherapie für erbliche Netzhauterkrankungen
RetChip (Uni Regensburg)Genomsequenz für 1.000 $
Genetische Risikofaktoren für Volkskrankheiten(z.Bsp. AMD, Glaukom)
Gentherapie für erbliche Netzhauterkrankungen Gentherapie für erbliche Netzhauterkrankungen RPE65 – Versuche
Gentherapie für erbliche Netzhauterkrankungen RPE65 – Versuche
Powerpoint des VortragsPowerpoint des VortragsEE--Mail: Mail: wissinger@[email protected]
Weitere Informationen/Internetquellen
Webvision Webvision –– Online Online ResourceResource zu Netzhaut zu Netzhaut http://webvision.med.utah.edu/ http://webvision.med.utah.edu/
RetinalRetinal Information Network (RETNET) Information Network (RETNET) http://www.sph.uth.tmc.edu/Retnet/ http://www.sph.uth.tmc.edu/Retnet/
Online Online MendelianMendelian InheritanceInheritance in Man (OMIM) in Man (OMIM) ( )( )http:://www.ncbi.nlm.nih.gov/omim/ http:://www.ncbi.nlm.nih.gov/omim/
National Center National Center forfor Biotechnological Information (NCBI) Biotechnological Information (NCBI) http://www.ncbi.nlm.nih.gov/http://www.ncbi.nlm.nih.gov/
Pro Retina Deutschland Pro Retina Deutschland eV.eV. (Patientenselbsthilfe) (Patientenselbsthilfe) http://www.prohttp://www.pro--retina.de/retina.de/


















![TECHNISCHE UNIVERSITÄT MÜNCHEN Klinik und Poliklinik für ... · autosomal-dominat vererbte Neurofibromatose Typ Recklinghausen [34]. Einteilung der Weichteilsarkome Gemäß der](https://static.fdokument.com/doc/165x107/5dd0985cd6be591ccb61c0e3/technische-universitt-moenchen-klinik-und-poliklinik-fr-autosomal-dominat.jpg)