
¼hrt: Meinem Doktorvater, Herrn Prof. Dr. AntonioTogni, für die Ermöglichung dieser interessanten...
154
Research Collection Doctoral Thesis Cyclopropanierung mit Eisen-Arsoniummethylid-Komplexen und katalytische, asymmetrische Cyclopropanierung mit Trimethylsilyldiazomethan Author(s): Egli, Patrick Publication Date: 2001 Permanent Link: https://doi.org/10.3929/ethz-a-004276528 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of ¼hrt: Meinem Doktorvater, Herrn Prof. Dr. AntonioTogni, für die Ermöglichung dieser interessanten...

Research Collection
Doctoral Thesis
Cyclopropanierung mit Eisen-Arsoniummethylid-Komplexenund katalytische, asymmetrische Cyclopropanierung mitTrimethylsilyldiazomethan
Author(s): Egli, Patrick
Publication Date: 2001
Permanent Link: https://doi.org/10.3929/ethz-a-004276528
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Dissertation ETH Nr. 14473
Cyclopropanierung mit Eisen-Arsoniummethylid-Komplexen und
katalytische, asymmetrische Cyclopropanierungmit Trimethylsilyldiazomethan
Abhandlung zur Erlangung des Titels
DOKTOR DER NATURWISSENSCHAFTEN
der
EIDGENOESSISCHEN TECHNISCHEN HOCHSCHULE ZUERICH
vorgelegt von
Patrick Egli, Dipl. Chem. ETH
geboren am 7. Dezember 1972
von Bronschhofen (SG)
Angenommen auf Antrag von
Prof. Dr. A. Togni, Referent
Prof. Dr. H. Grützmacher, Korreferent
Zürich 2001

Für meine Eltern und Maureen

Dank gebührt:
Meinem Doktorvater, Herrn Prof. Dr. Antonio Togni, für die Ermöglichung dieser
interessanten Arbeit und seine stets entgegenkommende Hilfe bei der
Durchführung derselben. Sein Vertrauen und seine Offenheit ermöglichten mir
ein weitgehend selbständiges Arbeiten in äusserst angenehmer Atmosphäre.
Herrn Prof. Dr. Hansjörg Grützmacher für die freundliche Uebernahme des
Korreferats.
Der Givaudan Dübendorf AG für die finanzielle Unterstützung dieser Arbeit.
Meinem langjährigen Freund und Kollegen Rhony Aufdenblatten für die kurzen
und langen Stunden in- und ausserhalb des Labors.
Herrn Gionata Frasca für seinen Einsatz im Rahmen seiner Diplomarbeit und
den daraus resultierenden Forschungsergebnissen.
Den Herren Diego Broggini für die Ermittlung der Röntgenstrukturen, Luca Fadini
für die Unterstützung in Computerangelegenheiten und Mauro Perseghini für Rat
und Tat bei der GC-Messung und bei Computerfragen.
Allen, die mir während meiner Dissertationszeit in irgendeiner Weise behilflich
waren: meinen Kolleginnen und Kollegen der Arbeitsgruppe Togni: Andrea,
Antonio, Céline, Christoph, Claus, Diego, Francesca, Giorgio, Hans Martin,
Helen, Ingrid, Ivo, Luca, Lukas, Maria, Markus, Mauro, Nikolaus, Péter, Richard,
Robert, Romano, Stan, Stefan, Stephan, meinen Kollegen an der ETH: Achim,
Felix und Toni und denjenigen, mit denen ich angenehme Stunden in Zürich
verbringen durfte: Adrian, Georg, Gerd, Huwi, Jimmy, Roni und Simon und allen
anderen, die hier nicht namentlich erwähnt wurden.

Teile dieser Arbeit wurden in folgenden Artikeln veröffentlicht:
Synthesis, Structure, and Reactivity Towards Olefins of New Iron
Arsoniummethylide Complexes
Egli, P.; Togni, A. Chimia 2001, 55, 609.
Synthesis, Structure, and Reactivity Towards Olefins of New Iron
Arsoniummethylide Complexes
Egli, P.; Togni, A. Organometallics, eingereicht.

Inhaltsverzeichnis
Zusammenfassung
Abstract
1. Cyclopropanierung. 1
1.1 Simmons-Smith-Reaktion 1
1.2 Katalytische Zersetzung von Diazoestern 4
1.3 Pd- und Cu-katalysierte Cyclopropanierung mit Diazomethan 9
1.4 Cyclopropanierung mit Fischer-Carbenkomplexen 10
1.5 Das [CpFe(CO)2]+-Fragment in der Cyclopropanierung 11
1.6 Cyclopropanierung mit Yliden 13
1.7 Literatur 17
2. Eisen-Arsoniummethyl-Komplexe - Synthese, Struktur und Reaktivitätgegenüber
Olefinen 22
2.1 Einführung 22
2.2 Arsine 25
2.3 Arsoniummethyl-Komplexe 29
2.3.1 Achirale Eisen-Arsoniummethyl-Komplexe 29
2.3.2 Chirale Eisen-Arsoniummethyl-Komplexe 38
2.3.3 Molybdän- und Wolfram-Arsoniummethyl-Komplexe 40
2.4 Herstellung von Substraten und Referenz-Produkten 42
2.5 Cyclopropanierungsreaktionen 45
2.6 Zusammenfassung und Schlussfolgerungen 48
2.7 Literatur 50
3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung mit
Yliden 51
3.1 Einführung 51
3.2 Komplexsynthese 56
3.3 Synthese von Triarylmethylarsonium-Salzen und Yliden 61
3.4 Reaktionsversuche 65
3.5 Schlussfolgerungen 68
3.6 Literatur 68

4. Cu(I)-katalysierte, asymmetrische Cyclopropanierung mit
Trimethylsilyldiazomethan 70
4.1 Einführung 70
4.2 Liganden 75
4.3 Komplexe 77
4.4 Cyclopropanierungsreaktionen 79
4.5 Schlussfolgerungen 88
4.6 Literatur 88
5. Experimenteller Teil. 90
5.1 Allgemeines 90
5.2 Allgemeine Arbeitsvorschriften (AAV) 93
5.3 Triarylarsine 95
5.4 Diarylarsinchloride 98
5.5 Arsinferrocenyle 99
5.6 Komplexe 109
5.7 Arsoniumsalze 122
5.8 Olefinvorläufer, Olefine und Cyclopropane 124
5.9 Liganden 127
5.10 Reaktionsversuche 128
5.11 Literatur 131
6. Anhang 134
6.1 Abkürzungen 134
6.2 Kristallographische Daten 135
Lebenslauf 143

Zusammenfassung
Die Suche nach einer katalytischen, asymmetrischen Methylencyclo-
propanierungsreaktion steht im Zentrum dieser Arbeit. Sie ist eingeteilt in drei
Hauptteile, von denen jeder einem Kapitel entspricht.
A^^,
Me°H. 80 °C ^~ . +A ot=.
OC^FeN^CI + AsAr3 0C..x(Fe>v^As+Ar3BF4OC NaBF4 OC
-NaCI
CF3
K1,Ar=-Q K2,Ar=-^-F K3,Ar=-©-CI K4,Ar=-Q K5, Ar = -£^-CF3
Der erste Teil (Kapitel 2) behandelt die Synthese, die Struktur und die
Reaktivität gegenüber Olefinen von neuen Eisen-Arsoniummethyl-Komplexen.
Die Komplexe [CpFe(CO)2(CH2AsR3)]BF4 (mit R = Ph (K1), 4-F-Ph (K2), 4-CI-Ph
(K3), 3-CF3-Ph (K4) und 4-CF3-Ph (K5)) wurden aus [CpFe(CO)2(CH2CI)]f dem
entsprechenden Triaryiarsin und NaBF4 in MeOH hergestellt. Die
Kristallstrukturen von K2, K3 und K4 wurden bestimmt. Kristallisationsversuche
mit K5 ergaben das Zersetzungsprodukt [CpFe(CO)2As(4-CF3-Ph)3]BF4, dessen
Kristallstruktur bestimmt wurde. Die Komplexe K1-K5 reagieren mit Styrol (100
°C, 24 h) unter der Bildung von Cyclopropylbenzol in Ausbeuten von 14%, 47%,
76%, 84% bzw. 83%. Cyclopropanierungsexperimente mit den CpFe(CO)2-
Arsoniummethyl-Derivaten von zwei neuen, chiralen Ferrocenylarsinen und
einem prochiralen Olefin ergaben racemisches Cyclopropan. Die erhaltenen
Resultate deuten auf folgenden Mechanismus der Cyclopropanierungsreaktion:
Die reversible Dissoziation des Triarylarsins vom Eisen-Arsoniummethylid-
Komplex ergibt den Eisencarben-Komplex [CpFe(CO)2CH2]+, der mit dem Olefin
zum Cyclopropan reagiert.

Der zweite Teil (Kapitel 3) beinhaltet Reaktionsversuche zur Entwicklung einer
katalytischen Methylencyclopropanierung mit Yliden. Ausgehend von den
Resultaten im vorhergehenden Teil, in dem die Funktion von Eisen-
Arsoniummethylid-Komplexen als Cyclopropanierungsreagenzien entdeckt
wurde, sollte eine katalytische Cyclopropanierung entwickelt werden. Als
mögliche Katalysatorvorläufer wurden Komplexe mit Metallzentren, die sich
durch ihre bekannte Rolle in der Cyclopropanierung oder durch ihre Fähigkeit zur
Bildung von Ylidkomplexen auszeichnen, ausgewählt. Als Methylenquellen
sollten Sulfonium-, Sulfoxonium-, Arsonium- und Antimoniumylide dienen. Die
Reaktionsversuche ergaben keine Cyclopropane.
.—Ph < II II >—Ph
1 ph^^ + 1.2 TMSCHN2 + 0.05 CuCI + 0.12 V-N N-V+ 0.05 n-BuLi
THF/CICH2CH2CI, Ar
A.50 °C, 68 h Ph SiMe3
Ausbeute: 27%
trans/cis: 74.6
ee: 75% {eis)78% (frans)
Im dritten Teil (Kapitel 4) ist die Anwendung von chiralen Liganden in der Cu(l)-
katalysierten Cyclopropanierung mit Trimethylsilyldiazomethan beschrieben,
woraus die erste katalytische, asymmetrische Cyclopropanierung mit
Trimethylsilyldiazomethan resultierte. In der Cyclopropanierung von Styrol
wurden mit Bis(oxazolin)-Liganden als Träger der chiralen Information
Diastereoisomerenüberschüsse von bis zu 97% und Enantiomerenüberschüsse
von bis zu 80% beobachtet.

Abstract
The present thesis focuses on the search for a catalytic, asymmetric methylene
cyclopropanation reaction. It is organized in three major parts, of which each is
presented in a separate chapter.
^L„,
MeOH, 80 °C ~T^.
+A__
.
0C.*Fe^CI + AsAr3 oc..Fe^As+Ar3BF4OC NaBF4 OC
-NaCI
CF3
K1,Ar=-Q K2,Ar=-Q-F K3,Ar=-@-CI K4,Ar=-Q K5,Ar = -Q-CF3
The first part (chapter 2) deals with the synthesis, the structure, and the
reactivity towards olefins of new iron arsoniummethylide complexes. The
complexes [CpFe(CO)2(CH2AsR3)]BF4 (with R = Ph (K1), 4-F-Ph (K2), 4-CI-Ph
(K3), 3-CF3-Ph (K4) and 4-CF3-Ph (K5)) have been prepared from
[CpFe(CO)2(CH2CI)], the corresponding triarylarsine, and NaBF4 in MeOH. The
X-ray crystal structures of K2, K3 and K4 were determined. From crystallization
experiments with K5 the decomposition product [CpFe(CO)2As(4-CF3-Ph)3]BF4
was obtained and characterized by its X-ray crystal structure. The complexes K1-
K5 react with styrene (100 °C, 24 h) affording cyclopropylbenzene in 14%, 47%,
76%, 84%, and 83% yield, respectively. Cyclopropanation experiments with the
CpFe(CO)2 arsoniummethyl derivatives of two new chiral ferrocenylarsines and a
prochiral olefin yielded racemic cyclopropane. The results observed indicate the
following mechanism of the cyclopropanation reaction: The reversible
dissociation of the triarylarsine from the iron arsoniummethylide complex yields
the iron carbene complex [CpFe(CO)2CH2]+ that reacts with the olefin under
formation of the cyclopropane.

The second part (chapter 3) presents reaction experiments that aimed at the
development of a catalytic methylene cyclopropanation with ylides. Starting from
the results presented in the previous part, where the activity of iron
arsoniummethylide complexes as cyclopropanation reagents was observed, we
intended to develop a catalytic cyclopropanation reaction. Complexes with metal
centers that are known for their role in cyclopropanation or for their ability to form
ylide complexes were used as possible catalyst precursors. Sulfonium-,
sulfoxonium-, arsonium-, and antimonium-methylides were used as methylene
donors. In these reaction experiments no cyclopropanes were formed.
Ph+ 1.2 TMSCHN2 + 0.05 CuCI + 0.12 + 0.05 n-BuLi
THF / CICH2CH2CI, Ar
A
PrfSiMe350 °C, 68 h
yield: 27%
trans/eis: 74.6
ee: 75% (eis)78% (trans)
In the third part (chapter 4) the use of chiral ligands in the Cu(l)-catalyzed
cyclopropanation with trimethylsilyldiazomethane is described. These
experiments led to the first catalytic, asymmetric cyclopropanation with
trimethylsilyldiazomethane. In the cyclopropanation of styrene in the presence of
bis(oxazoline) ligands as carriers of the chiral information diastereomeric
excesses up to 97% and enantiomeric excesses up to 80% were observed.

1. Cyclopropanierung
1.1 Simmons-Smith-Reaktion
Forschungen auf dem Gebiet der Cyclopropanierung haben viele Methoden
hervorgebracht. Die Simmons-Smith-Reaktion ist eine sehr effiziente Methode zur
Methylencyclopropanierung. Sie beinhaltet die Umsetzung eines Zink-Kupfer-
Paares mit Diiodmethan zu einem Zinkcarbenoid-Reagens, das mit Olefinen zu
Cyclopropanen reagiert.1 Neben dem von Simmons und Smith ursprünglich zur
Aktivierung des metallischen Zinks verwendeten Cu kann die Aktivierung u.a. auch
mit Ag,2 TiCI43 und TMSCI/DBE (Knochel-Zink)4 erfolgen. Trotz verbesserter
Aktivierungsmethoden bleibt die Simmons-Smith-Reaktion eine heterogene
Reaktion mit den damit verbundenen Nachteilen. Zwei besonders gelungene
homogene Varianten der Simmons-Smith-Reaktion sind die Furakawa-Variante
(mit ZnEt2 und CH2I2)5 und die Sawada-Variante (mit EtZnl und CH2I2).6 Neben
anderen Vorteilen ist die homogene Reaktionsführung der heterogenen Variante
auch ausbeuteseitig in vielen Fällen überlegen.
Ueber die genaue Struktur des cyclopropanierenden Reagenzes in der Simmons-
Smith-Reaktion herrscht keine endgültige Klarheit.7 NMR-spektroskopische
Untersuchungen haben gezeigt, dass IZnCH2l mit Zn(CH2l)2 im Gleichgewicht steht
(Schema 1.1).6 Etherliganden verschieben das Gleichgewicht zugunsten von
Zn(CH2l)2.
Et20
2 CH2I2 + 2 Zn(Cu) 2 ICH2Znl ^ Zn(CH2l)2 + Znl2
Schema 1.1. Reaktion von Diiodmethan mit Zn/Cu.
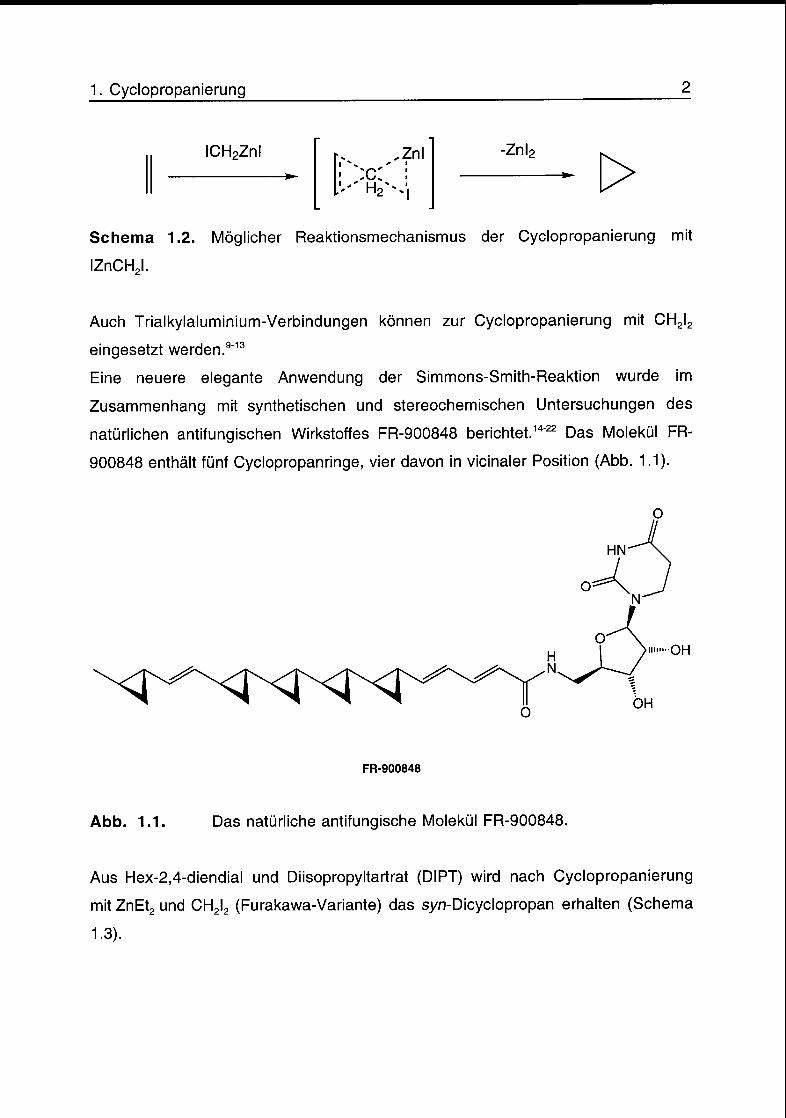
Ein möglicher Reaktionsmechanismus für die Cyclopropanierung mit IZnCH2l ist in
Schema 1.2 gezeigt. Theoretische Untersuchungen zur Addition eines Carbenoids
an ein Olefin unter Bildung des Cyclopropans sprechen für einen konzertierten
Mechanismus mit einem Uebergangszustand.8
1. Cyclopropanierung 2
ICH2Znl n .Znl
:^c' i•
'* H2 %«.i
-Znl;
D>
Schema 1.2. Möglicher Reaktionsmechanismus der Cyclopropanierung mit
IZnCH2l.
Auch Trialkylaluminium-Verbindungen können zur Cyclopropanierung mit CH2I2
eingesetzt werden.913
Eine neuere elegante Anwendung der Simmons-Smith-Reaktion wurde im
Zusammenhang mit synthetischen und stereochemischen Untersuchungen des
natürlichen antifungischen Wirkstoffes FR-900848 berichtet.14"22 Das Molekül FR-
900848 enthält fünf Cyclopropanringe, vier davon in vicinaler Position (Abb. 1.1).
OH
OH
FR-900848
Abb. 1.1. Das natürliche antifungische Molekül FR-900848.
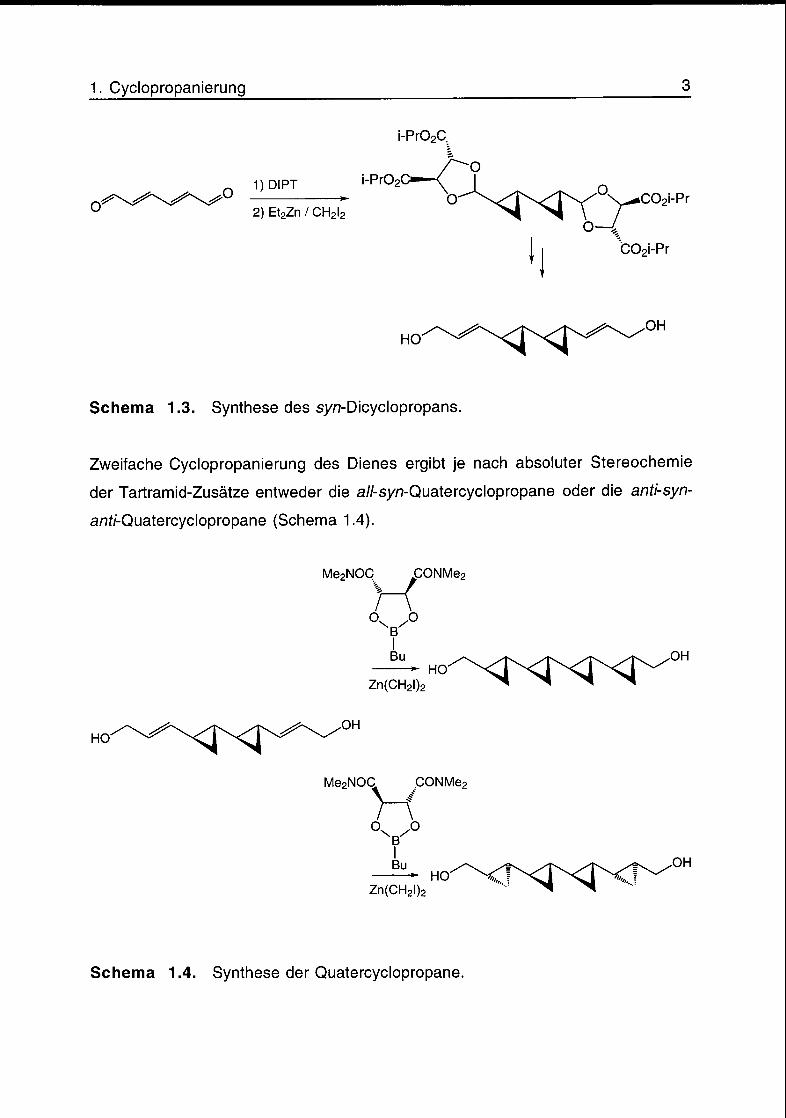
Aus Hex-2,4-diendial und Diisopropyltartrat (DIPT) wird nach Cyclopropanierung
mit ZnEt2 und CH2I2 (Furakawa-Variante) das syn-Dicyclopropan erhalten (Schema
1.3).
1. Cyclopropanierung 3
i-Pr02C.
1)DIPT^
2) Et2Zn / CH2I2
i-Pr020
C02i-Pr
Schema 1.3. Synthese des syn-Dicyclopropans.
Zweifache Cyclopropanierung des Dienes ergibt je nach absoluter Stereochemie
der Tartramid-Zusätze entweder die a//-syn-Quatercyclopropane oder die anti-syn-
antf-Quatercyclopropane (Schema 1.4).
Me2NOC CONMe2
H
IBu
Zn(CH2l)2ho^<JXJXIX^OH
Me2NOC
O.
UXB7
pONMe2
O
Bu
Zn(CH2l)2
Schema 1.4. Synthese der Quatercyclopropane.

1. Cyclopropanierung 4
Diese Untersuchungen zeigen das Potential der Simmons-Smith-Reaktion sowohl
bezüglich Stereoselektivität als auch in der Mehrfachcyclopropanierung und
beruhen auf Studien von Charette,23"27 Denmark2831 und Kobayashi.32"34
1.2 Katalytische Zersetzung von Diazoestern
Die zweite und wichtigste Methode für die Synthese von chiralen Cyclopropanen ist
die katalytische Zersetzung von Diazoestern in Gegenwart von Olefinen.35'38 Solche
Zersetzungen werden durch verschiedene Uebergangsmetalle katalysiert. Nach
wie vor schwer fassbare Carben-Spezies reagieren mit dem Olefin unter Bildung
des Cyclopropans. Der Mechanismus für die Zersetzung von Diazoverbindungen
mit Uebergangsmetallen wurde 1952 von Yates vorgeschlagen (Schema 1.5.).39
Nach dem nukleophilen Angriff der Diazoverbindung auf den Katalysatorkomplex
wird N2 freigesetzt. Der gebildete Carben-Komplex reagiert dann mit dem Olefin
unter Bildung des Cyclopropans und unter Wiederfreisetzung des Katalysators.
R-N2
C—N=N *- LnM=CR2
R
CH2
R III -r CH2
LnM—C:'+y -« Ln-M-C+R2
R
Schema 1.5. Uebergangsmetallkatalysierte Cyclopropanierung mit
Diazoverbindungen.
MLn
Ln"M-
R
MU
1. Cyclopropanierung 5
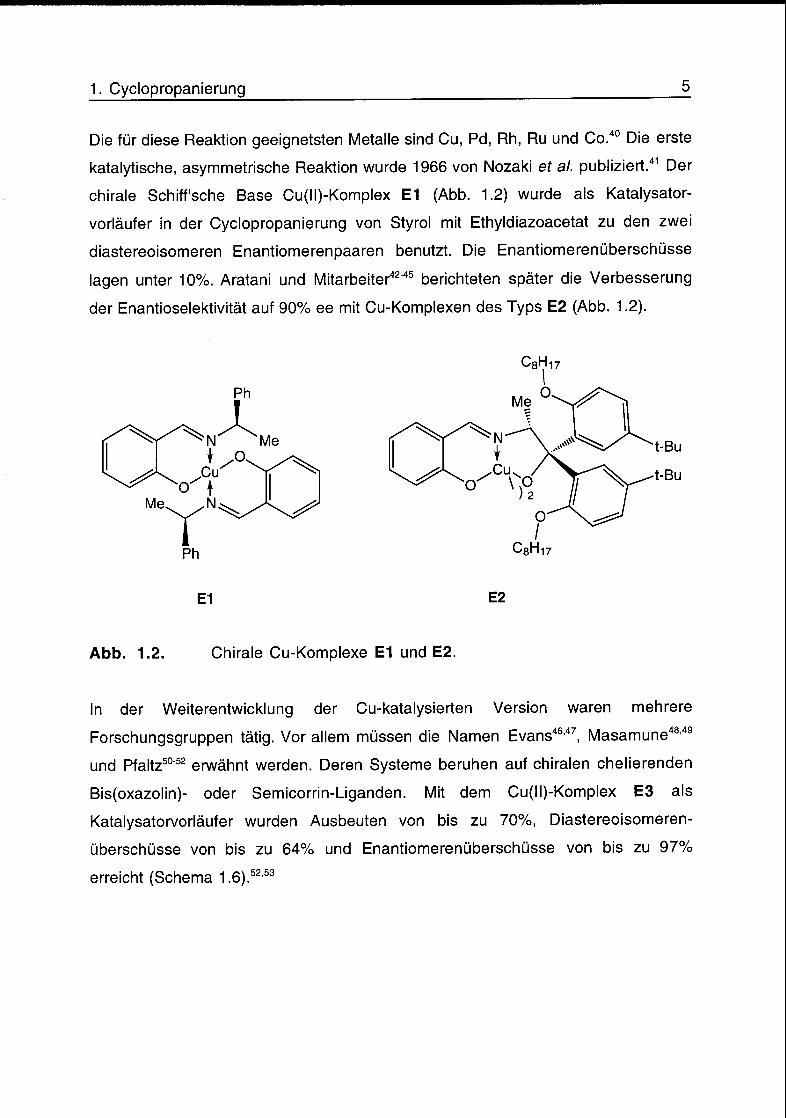
Die für diese Reaktion geeignetsten Metalle sind Cu, Pd, Rh, Ru und Co.40 Die erste
katalytische, asymmetrische Reaktion wurde 1966 von Nozaki er al. publiziert.41 Der
chirale Schiff'sche Base Cu(ll)-Komplex E1 (Abb. 1.2) wurde als Katalysator¬
vorläufer in der Cyclopropanierung von Styrol mit Ethyldiazoacetat zu den zwei
diastereoisomeren Enantiomerenpaaren benutzt. Die Enantiomerenüberschüsse
lagen unter 10%. Aratani und Mitarbeiter42"45 berichteten später die Verbesserung
der Enantioselektivität auf 90% ee mit Cu-Komplexen des Typs E2 (Abb. 1.2).
Ph C8H17
E1 E2
Abb. 1.2. Chirale Cu-Komplexe E1 und E2.
In der Weiterentwicklung der Cu-katalysierten Version waren mehrere
Forschungsgruppen tätig. Vor allem müssen die Namen Evans46,47, Masamune48,49
und Pfaltz50"52 erwähnt werden. Deren Systeme beruhen auf chiralen chelierenden
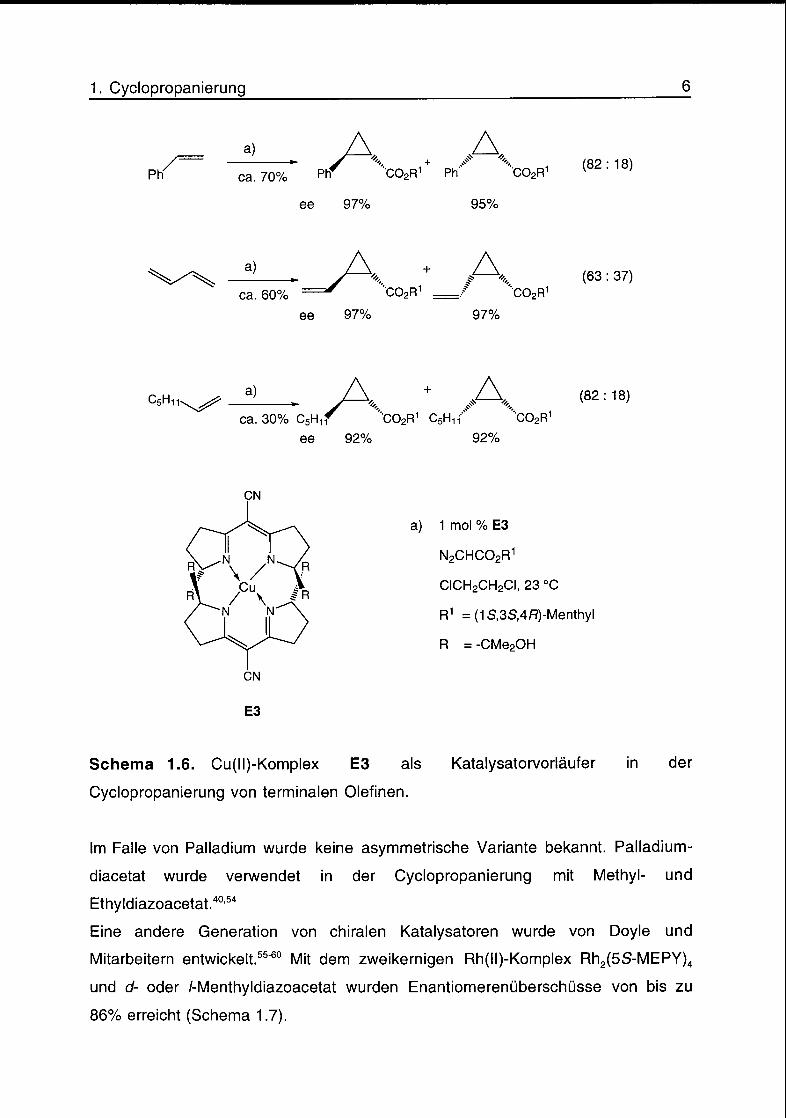
Bis(oxazolin)- oder Semicorrin-Liganden. Mit dem Cu(ll)-Komplex E3 als
Katalysatorvorläufer wurden Ausbeuten von bis zu 70%, Diastereoisomeren-
überschüsse von bis zu 64% und Enantiomerenüberschüsse von bis zu 97%
erreicht (Schema 1.6).52'53
1. Cyclopropanierung 6
PK/=
a) A''''' + **"* '''• (82 18^
ca. 70% PH" 'C02R1 Ph* 'C02R1 v° '
ee 97% 95%
%/%>a)
ca. 60%
AC02R1 C02R1
ee 97% 97%
(63 : 37)
CH ya) A + Aca. 30%C5H1f "C02R1 CgHn*' C02R1
ee 92% 92%
(82: 18)
a) 1 mol % E3
N2CHC02R1
CICH2CH2CI, 23 °C
R1 = (1 S,3S,4fî)-Menthyl
R = -CMe2OH
Schema 1.6. Cu(ll)-Komplex E3 als Katalysatorvorläufer in der
Cyclopropanierung von terminalen Olefinen.
Im Falle von Palladium wurde keine asymmetrische Variante bekannt. Palladium-
diacetat wurde verwendet in der Cyclopropanierung mit Methyl- und
Ethyldiazoacetat.4054
Eine andere Generation von chiralen Katalysatoren wurde von Doyle und
Mitarbeitern entwickelt.55"60 Mit dem zweikernigen Rh(ll)-Komplex Rh2(5S-MEPY)4
und d- oder /-Menthyldiazoacetat wurden Enantiomerenüberschüsse von bis zu
86% erreicht (Schema 1.7).
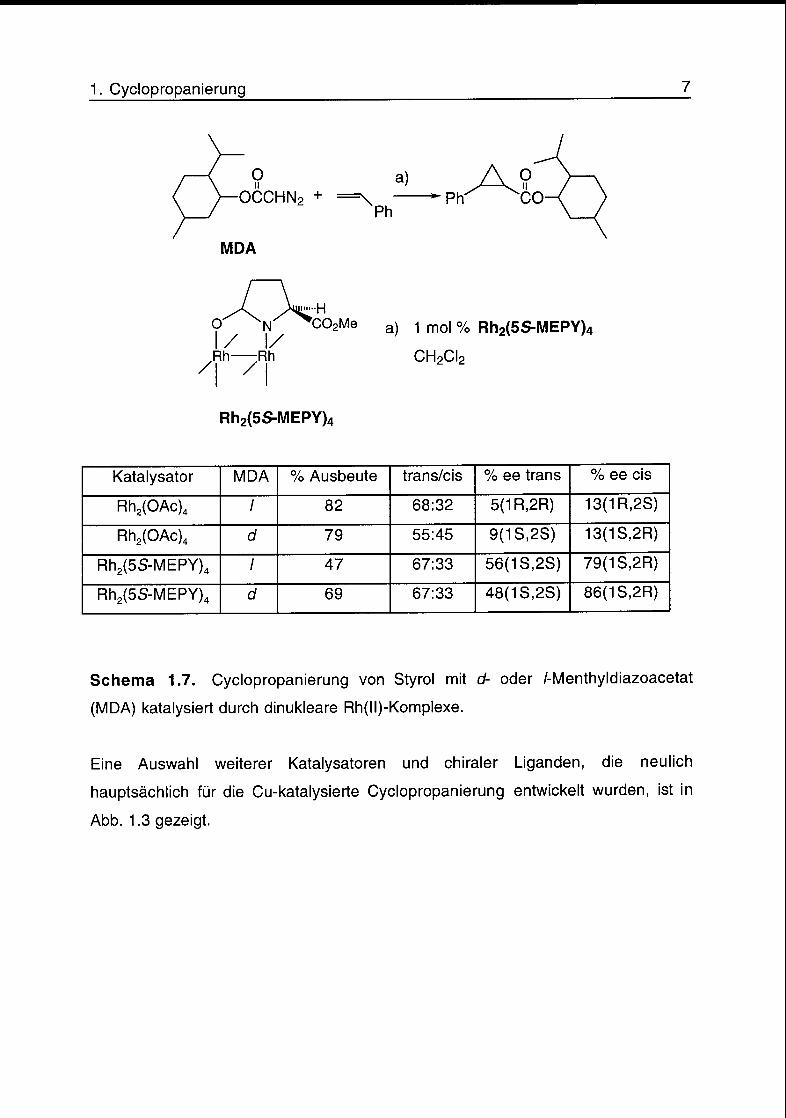
1. Cyclopropanierung 7
OCCHN2 + =
MDA
O
1/Rh Rh
iL»-H
/C02Me a) -1 m0| o/o Rh2(5S-MEPY)4
CH2CI2
Rh2(5S-MEPY)4
Katalysator MDA % Ausbeute trans/cis % ee trans % ee eis
Rh2(OAc)4 / 82 68:32 5(1R,2R) 13(1 R,2S)
Rh2(OAc)4 d 79 55:45 9(1S,2S) 13(1S,2R)
Rh2(5S-MEPY)4 1 47 67:33 56(1 S,2S) 79(1 S,2R)
Rh2(5S-MEPY)4 d 69 67:33 48(1 S,2S) 86(1 S,2R)
Schema 1.7. Cyclopropanierung von Styrol mit d- oder /-Menthyldiazoacetat
(MDA) katalysiert durch dinukleare Rh(ll)-Komplexe.
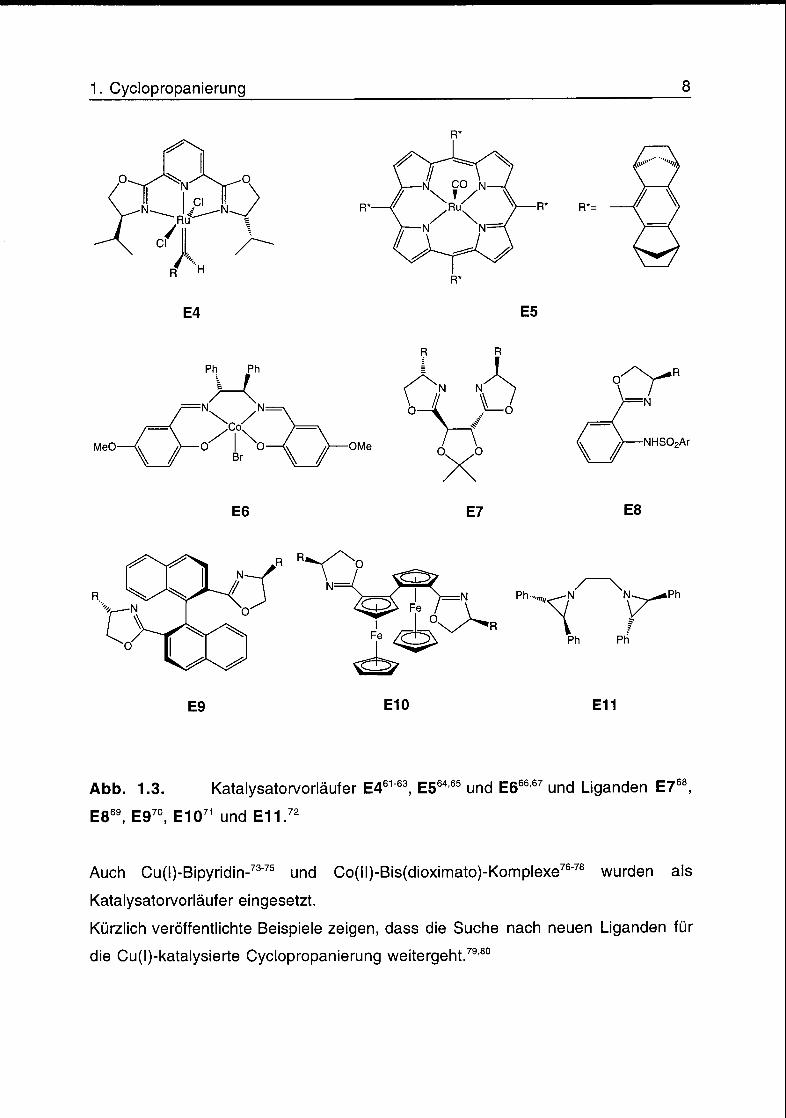
Eine Auswahl weiterer Katalysatoren und chiraler Liganden, die neulich
hauptsächlich für die Cu-katalysierte Cyclopropanierung entwickelt wurden, ist in
Abb. 1.3 gezeigt.
1. Cyclopropanierung 8
<S^
N-_ U N
/Cl
rf h
E4
R* R*
E5
MeO-
Ph Ph
M
-OMe
E6
N N
\U W
o^ ^o
E7
NHS02Ar
E8
E9 E10
/ V
Ph Ph
E11
Abb. 1.3. Katalysatorvorläufer E461"63, E564'65 und E666'67 und Liganden E768,
E869 E970 E1071 und E11 72
Auch Cu(l)-Bipyridin-73'75 und Co(ll)-Bis(dioximato)-Komplexe76'78 wurden als
Katalysatorvorläufer eingesetzt.
Kürzlich veröffentlichte Beispiele zeigen, dass die Suche nach neuen Liganden für
die Cu(l)-katalysierte Cyclopropanierung weitergeht.79,80
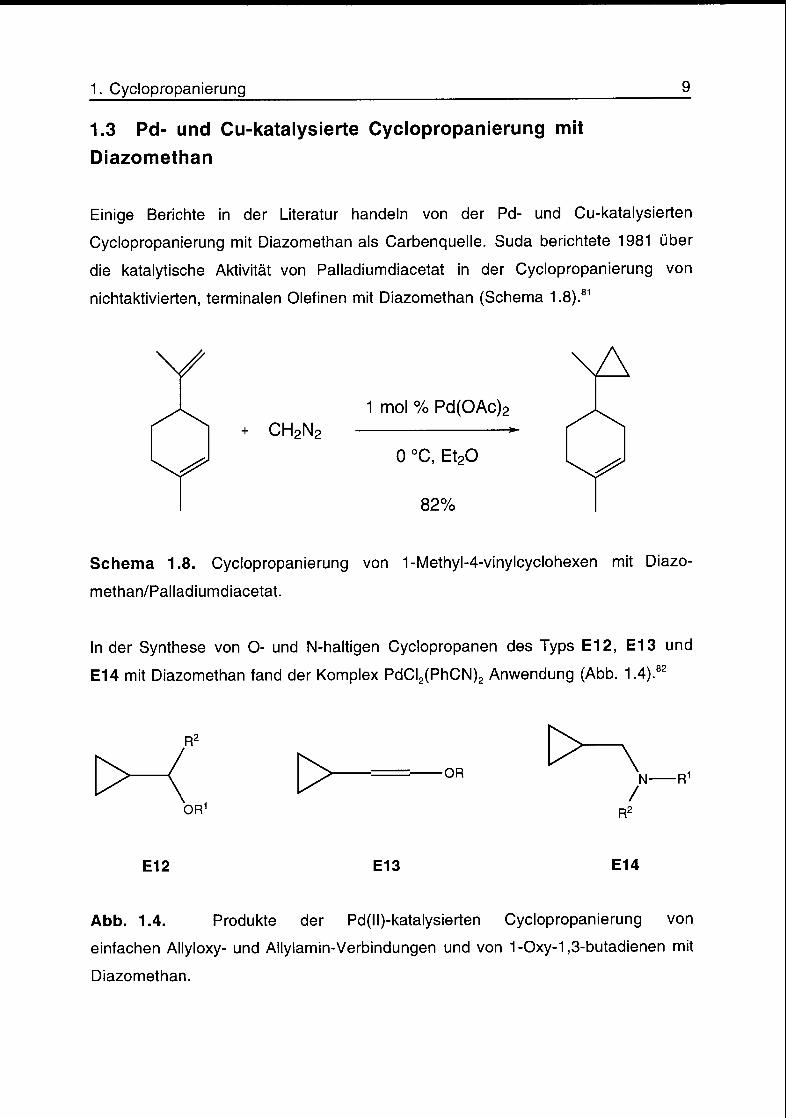
1. Cyclopropanierung 9
1.3 Pd- und Cu-katalysierte Cyclopropanierung mit
Diazomethan
Einige Berichte in der Literatur handeln von der Pd- und Cu-katalysierten
Cyclopropanierung mit Diazomethan als Carbenquelle. Suda berichtete 1981 über
die katalytische Aktivität von Palladiumdiacetat in der Cyclopropanierung von
nichtaktivierten, terminalen Olefinen mit Diazomethan (Schema 1.8).81
\/
+ CH2N2
\^
1 mol % Pd(OAc)2
0 °C, Et20
82%
\^
Schema 1.8. Cyclopropanierung von 1-Methyl-4-vinylcyclohexen mit Diazo-
methan/Palladiumdiacetat.
In der Synthese von O- und N-haltigen Cyclopropanen des Typs E12, E13 und
E14 mit Diazomethan fand der Komplex PdCI2(PhCN)2 Anwendung (Abb. 1.4).82
R2
OR1
•OR
D> \/
R2
N R1
E12 E13 E14
Abb. 1.4. Produkte der Pd(ll)-katalysierten Cyclopropanierung von
einfachen Allyloxy- und Allylamin-Verbindungen und von 1-Oxy-1,3-butadienen mit
Diazomethan.

1. Cyclopropanierung 10
Diese Methode wurde nicht wesentlich weiterentwickelt. Im Besonderen scheinen
keine Pd-Komplexe mit chiralen Liganden Anwendung gefunden zu haben.
Im Falle von Kupfer wurde die Anwendung des Bis(semicorrinato)-Cu(ll)-
Komplexes E3 (Schema 1.6) als Katalysatorvorläufer in der asymmetrischen
Methylencyclopropanierung von (£)-1-Phenylpropen zu 1-Methyl-2-phenylcyclo-
propan mit ca. 75% ee berichtet.83
1.4 Cyclopropanierung mit Fischer-Carbenkomplexen
Die Reaktion von heteroatom-stabilisierten Fischer-Carben-Komplexen (Cr, Mo,
W)84"87 mit elektronenreichen und elektronenarmen Olefinen unter der Bildung von
Cyclopropanen war eine der ersten synthetisch bedeutsamen Reaktionen mit
Komplexen dieser Klasse (Schema 1.9). Diese Cyclopropanierungsreaktionen sind
strikte stöchiometrisch, da die Fischer-Carbene in mehreren Syntheseschritten mit
verschiedenen Reagenzien hergestellt werden.
Zu den neueren Beispielen dieser Reaktion gehören die Arbeiten von
Söderberg/Hegedus88 und Harvey/Brown.89
R OMe
/OMe X
(CO)5Cr=<^ + =
R
R = Aryl, Vinyl, Heteroaryl X = CN, C02R\ S02R', OR'
Schema 1.9. Reaktion von stabilisierten Chrom-Carbenkomplexen mit Olefinen
zu Cyclopropanen.
Die Behandlung des Tetramethylammoniumacylat-Komplexes E15 mit
Pivaloylchlorid gefolgt von der Zugabe des ungesättigten Alkohols resultiert in der
Bildung des Chrom-Alkenyloxycarben-Komplexes E16. Nach Rühren dieses
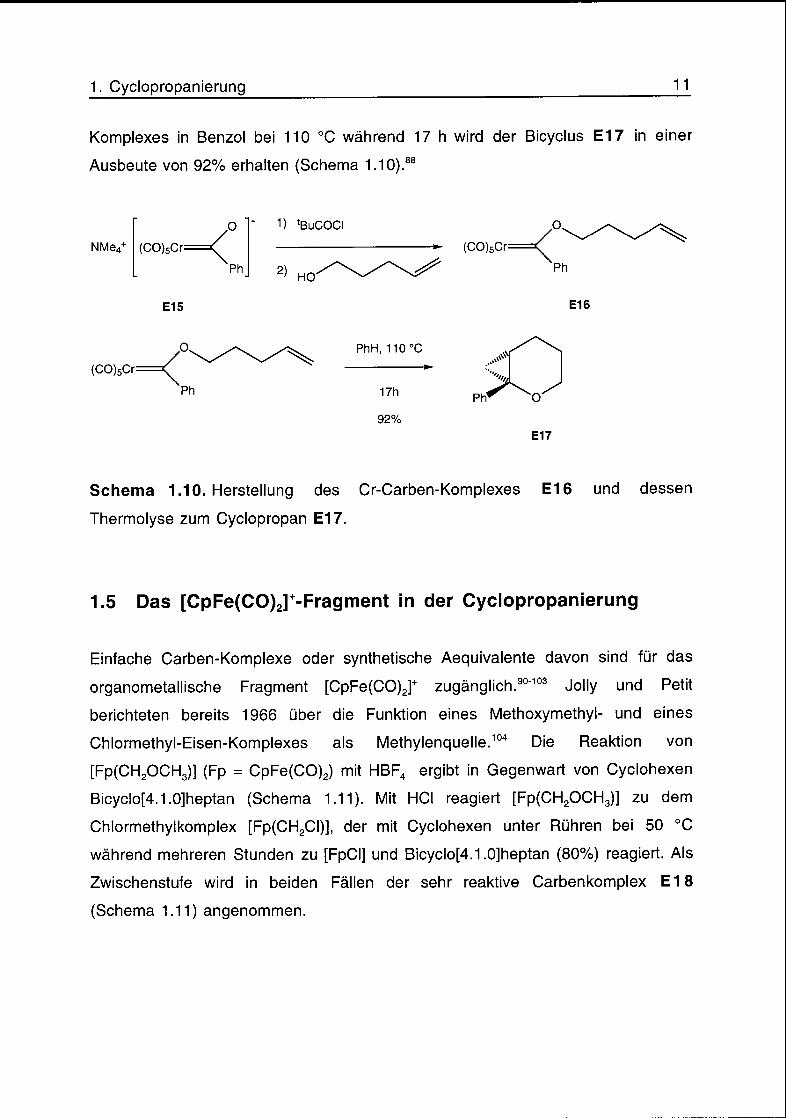
1. Cyclopropanierung 11
Komplexes in Benzol bei 110 °C während 17 h wird der Bicyclus E17 in einer
Ausbeute von 92% erhalten (Schema 1.10).88
NMe4+ (CO)5Cr=
E15
.O
\.Ph
1) 'BuCOCI
(CO)5Cr
E16
(CO)5Cr
PhH,110 °C
17h
92%
^
'""//3L
PrT O'
E17
Schema 1.10. Herstellung des Cr-Carben-Komplexes E16 und dessen
Thermolyse zum Cyclopropan E17.
1.5 Das [CpFe(CO)2]+-Fragment in der Cyclopropanierung
Einfache Carben-Komplexe oder synthetische Aequivalente davon sind für das
organometallische Fragment [CpFe(CO)2]+ zugänglich.90"103 Jolly und Petit
berichteten bereits 1966 über die Funktion eines Methoxymethyl- und eines
Chlormethyl-Eisen-Komplexes als Methylenquelle.104 Die Reaktion von
[Fp(CH2OCH3)] (Fp = CpFe(CO)2) mit HBF4 ergibt in Gegenwart von Cyclohexen
Bicyclo[4.1.0]heptan (Schema 1.11). Mit HCl reagiert [Fp(CH2OCH3)] zu dem
Chlormethylkomplex [Fp(CH2CI)], der mit Cyclohexen unter Rühren bei 50 °C
während mehreren Stunden zu [FpCI] und Bicyclo[4.1.0]heptan (80%) reagiert. Als
Zwischenstufe wird in beiden Fällen der sehr reaktive Carbenkomplex E18
(Schema 1.11) angenommen.

1. Cyclopropanierung 12
oc-yoc
..#ev
HBF4
"CH2OMe -MeOHoc/%
oc
E18
CH2
O
Schema 1.11. Reaktion von [Fp(CH2OCH3)] mit HBF4 in Gegenwart von
Cyclohexen.
1979 berichteten Brandt und Helquist die Synthese eines stabilen Eisen-
Sulfoniummethyl-Komplexes und dessen Anwendung in der Cyclopropanierung
von verschiedenen unfunktionalisierten Olefinen.91 Die Methylierung des Sulfides
E19 ergibt das Sulfoniumsalz E20, welches unter geeigneten Bedingungen mit
Olefinen unter der Bildung von Cyclopropanen reagiert (Schema 1.12).
oc-JFe>OC
[Me30]+BF4- ^oc-'fe-OC
/,S+ BF4
RCH=CHR'
E19 E20
Schema 1.12. Synthese und Anwendung des Fp-Sulfoniummethyl-Komplexes
E20.
Dieses System war die Grundlage für Untersuchungen in verschiedenen
Forschungsgruppen. 1998 publizierten McCarten und Barefield ihre Arbeit über
Kinetik und Mechanismus der Cyclopropanierung von Cycloocten mit
[Fp(CH2SPh2)]BF4.105 Der einzige Versuch jedoch, das System in eine katalytische
Version umzusetzen, wurde von Hossain und Mitarbeitern berichtet.98,99 Das
kationische THF-Addukt [Fp(THF)]BF4 (E21) katalysiert die Zersetzung von
Ethyldiazoacetat in Gegenwart von Olefinen unter Bildung von Cyclopropanen
(Schema 1.13).

1. Cyclopropanierung 13
Ph N2HC O
10mol%E21
».
12 h, 40 °C, CH2CI2
68%
Ph C02Et
85
PK* C02Et
15
oc-""|)Fe-OC
THF
BF4
E21
Schema 1.13. Anwendung von [Fp(THF)]BF4 E21 als Katalysator in der
Cyclopropanierung mit Ethyldiazoacetat.
1.6 Cyclopropanierung mit Yliden
Obwohl das erste Ylid schon zu Beginn des zwanzigsten Jahrhunderts beschrieben
wurde, wurden die Ylide erst nach der Geburt der Wittig-Reaktion 1953106 als
synthetisch nützliche Reagenzien erkannt.107 Die Cyclopropanierung mit Yliden ist
eine der ersten und am ausführlichsten studierten Ylidreaktionen.
Corey und Chaykovsky publizierten 1962 die Herstellung von Lösungen von
Dimethyloxosulfoniummethylid und deren Anwendung als Methylendonoren in der
Herstellung sowohl von Epoxiden aus Aldehyden und Ketonen als auch von 1-
Phenyl-2-benzoylcyclopropan aus Benzalacetophenon in 95% Ausbeute.108
Während das stabilere Dimethyloxosulfoniummethylid mit a,ß-ungesättigten
Ketonen zu Cyclopropylketonen reagiert, greift Dimethylsulfoniummethylid die
Carbonylgruppe an.109 Die Herstellung von (Dimethylamino)phenyloxosulfonium-
methylid und dessen Anwendung in der Epoxidierung von Aldehyden und der
Cyclopropanierung von a,ß-ungesättigten Ketonen und Carbonsäuren wurde 1968
veröffentlicht.110
Berichte über die Reaktivität von Dimethyloxosulfoniummethylid und
(Dimethylamino)phenyloxosulfoniummethylid als Nukleophil sind bekannt.110'113

1. Cyclopropanierung 1_4
Ylide können als spezielle Carbanionen mit einem positiven Heteroatom in
benachbarter Position angesehen werden. Die Reaktion eines Ylides mit einer oc,ß-
ungesättigten Carbonylverbindung ergibt die Zwischenstufe E22 (Schema 1.14).
Die Elimination der Gruppe mit dem Heteroatom führt zum Cyclopropan.
LnE CHR
n
LnE^=CHR
E = S, As, Te etc.
Schema 1.14. Cyclopropanierung mit Yliden über Zwischenstufe E22.
Die Anwendung von Dimethyloxosulfoniummethylid, (Dimethylamino)phenyloxo-
sulfoniummethylid und (A/-Methyl-A/-p-tolylamino)morpholinoxosulfoniummethylid
in der Cyclopropanierung von Triphenylvinylphosphoniumsalzen wurde 1995
berichtet.114
Neben Sulfonium- und Oxosulfoniumyliden werden auch Arsonium-115 und
Telluroniumylide116,117 zur Cyclopropanierung verwendet.
Viele Publikationen handeln von der asymmetrischen Cyclopropanierung von a,ß-
ungesättigten Carbonylverbindungen mit Yliden. Die chirale Induktion in diesen
Reaktionen stammt von den Substraten, den Reagenzien oder von chiralen
Liganden, die in Gegenwart einer Lewis-Säure eingesetzt werden. In der
reagenskontrollierten Version sind chirale Aminosulfoxoniummethylide,118,119
Sulfoniumylide,120"122 Sulfoxoniumylide,123125 Alkalisalze (Li, Na, K) von
Sulfoximinen126"128 und Arsoniumylide115 eingesetzt worden.
R2
R2 y—CH2
RCH—ELn
-LnE
E22

1. Cyclopropanierung 15
Aggarwal et al. verwendeten chirale und achirale Sulfide in der katalytischen
Cyclopropanierung von Enonen.129,130 Die in der Cyclopropanierung eingeführte
Phenylmethylengruppe stammt in dieser Reaktion allerdings vom explosiven
Phenyldiazomethan. Katalytische Mengen des dinuklearen Rh2(OAc)4 dienen der
Bildung des Sulfoniumylides aus Phenyldiazomethan und dem Sulfid. Das Ylid
reagiert dann mit dem Enon unter Freisetzung des Sulfides zum Cyclopropan. Der
katalytische Prozess ist in Schema 1.15 gezeigt.
Schema 1.15. Katalytische Cyclopropanierung von Enonen mit
Phenyldiazomethan über Sulfoniumylide als Cyclopropanierungsreagenzien.
Die Verwendung von substöchiometrischen anstelle von stöchiometrischen
Mengen eines chiralen Sulfides führte bei gleichen Bedingungen zu verminderten
Ausbeuten, während die Enantiomerenüberschüsse konstant blieben. Zugabe des
Phenyldiazomethans über längere Zeit führte zu einer Erhöhung der Ausbeuten.
Während die beobachteten Diastereoselektivitäten nur bescheiden waren, wurden
Enantiomerenüberschüsse von 98% erreicht. In aktuellen Anwendungen wird
Phenyldiazomethan in situ generiert.131,132
1. Cyclopropanierung 16
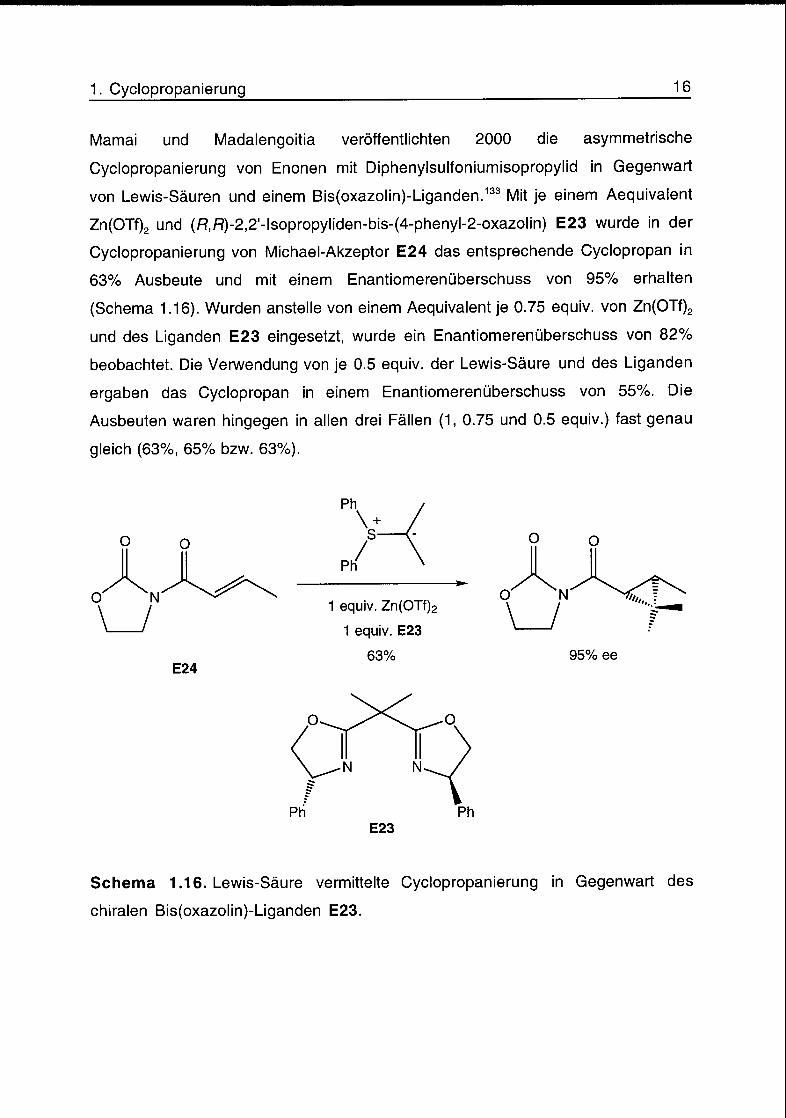
Marnai und Madalengoitia veröffentlichten 2000 die asymmetrische
Cyclopropanierung von Enonen mit Diphenylsulfoniumisopropylid in Gegenwart
von Lewis-Säuren und einem Bis(oxazolin)-Liganden.133 Mit je einem Aequivalent
Zn(OTf)2 und (fî,/:?)-2,2'-lsopropyliden-bis-(4-phenyl-2-oxazolin) E23 wurde in der
Cyclopropanierung von Michael-Akzeptor E24 das entsprechende Cyclopropan in
63% Ausbeute und mit einem Enantiomerenüberschuss von 95% erhalten
(Schema 1.16). Wurden anstelle von einem Aequivalent je 0.75 equiv. von Zn(OTf)2
und des Liganden E23 eingesetzt, wurde ein Enantiomerenüberschuss von 82%
beobachtet. Die Verwendung von je 0.5 equiv. der Lewis-Säure und des Liganden
ergaben das Cyclopropan in einem Enantiomerenüberschuss von 55%. Die
Ausbeuten waren hingegen in allen drei Fällen (1, 0.75 und 0.5 equiv.) fast genau
gleich (63%, 65% bzw. 63%).
Ph
E24
p/ \C}
1 equiv. Zn(OTf)2O ^N
\ /1 equiv. E23
63% 95% ee
E23
Schema 1.16. Lewis-Säure vermittelte Cyclopropanierung in Gegenwart des
chiralen Bis(oxazolin)-Liganden E23.

1. Cyclopropanierung 17
139
Das Ylid Diphenylsulfonium(ethoxycarbonyl)methylid kann als Ersatz für
Ethyldiazoacetat in der Cu(l)- und Rh(ll)-katalysierten, asymmetrischen
Cyclopropanierung von einfachen Olefinen eingesetzt werden.134 Während die
beobachteten Ausbeuten mit dem Ylid aber durchwegs signifikant kleiner sind als
mit der Diazoverbindung, änderten die Diastereoselektivitäten und
Enantioselektivitäten nur wenig. Diese Resultate beruhen auf Ergebnissen von
Trost,135,136 Cohen et a/.,137 Zhang und Schuster138 und Cimetière und Julia.1Î
1.7 Literatur
(1) Simmons, H. E.; Smith, R. D. J. Am. Chem. Soc. 1959, 81, 4256-4264.
(2) Denis, J. M.; Girard, C; Conia, J. M. Synthesis 1972, 549-551.
(3) Friedrich, E. C; Lunetta, S. E.; Lewis, E. J. J. Org. Chem. 1989, 54, 2388-
2390.
(4) Knöchel, P.; Singer, R. D. Chem. Rev. 1993, 93, 2117-2188.
(5) Furukawa, J.; Kawabata, N.; Nishimura, J. Tetrahedron Lett. 1966, 28, 3353-
3354.
(6) Sawada, S.; Inouye, Y. Bull. Chem. Soc. Jpn. 1969, 42, 2669-2672.
(7) Koert, U. Nachr. Chem. Tech. Lab. 1995, 43, 435-442.
(8) Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307-1370.
(9) Hoberg, H. Justus Liebigs Ann. Chem. 1962, 656, 1-17.
(10) Hoberg, H. Justus Liebigs Ann. Chem. 1966, 695, 1-15.
(11) Hoberg, H. Justus Liebigs Ann. Chem. 1967, 703, 1-16.
(12) Miller, D. B. Tetrahedron Lett. 1964, 17, 989-993.
(13) Maruoka, K.; Fukutani, Y.; Yamamoto, H. J. Org. Chem. 1985, 50, 4412-
4414.
(14) Barrett, A. G. M.; Doubleday, W. W.; Hamprecht, D.; Kasdorf, K.; Tustin, G. J.
Pure Appl. Chem. 1997, 69, 383-388.
(15) Barrett, A. G. M.; Doubleday, W. W.; Hamprecht, D.; Kasdorf, K.; Tustin, G. J.;
White, A. J. P.; Williams, D. J. J. Chem. Soc. Chem. Commun. 1997, 1693-
1700.
(16) Barrett, A. G. M.; Kasdorf, K. J. Chem. Soc. Chem. Commun. 1996, 325-326.
(17) Barrett, A. G. M.; Doubleday, W. W.; Tustin, G. J. Tetrahedron 1996, 52,
15325-15338.
(18) Barrett, A. G. M.; Doubleday, W. W.; Kasdorf, K.; Tustin, G. J. J. Org. Chem.
1996, 61, 3280-3288.
(19) Barrett, A. G. M.; Kasdorf, K.; Tustin, G. J.; Williams, D. J. J. Chem. Soc.
Chem. Commun. 1995, 1143-1144.
(20) Barrett, A. G. M.; Doubleday, W. W.; Kasdorf, K.; Tustin, G. J.; White, A. J. P.;
Williams, D. J. J. Chem. Soc. Chem. Commun. 1995, 407-408.
(21) Armstrong, R. W.; Maurer, K. W. Tetrahedron Lett. 1995, 36, 357-360.
(22) Theberge, C. R.; Zercher, C. K. Tetrahedron Lett. 1994, 35, 9181-9184.

1. Cyclopropanierung 18
(23) Charette, A. B.; Brochu, C. J. Am. Chem. Soc. 1995, 117, 11367-11368.
(24) Charette, A. B.; Prescott, S.; Brochu, C. J. Org. Chem. 1995, 60, 1081-1083.
(25) Charette, A. B.; Juteau, H. J. Am. Chem. Soc. 1994, 116, 2651-2652.
(26) Charette, A. B.; Marcoux, J.-F. J. Am. Chem. Soc. 1996, 118, 4539-4549.
(27) Charette, A. B.; Marcoux, J.-F. Syn/eff 1995, 1197-1207.
(28) Denmark, S. E.; O'Connor, S. P. J. Org. Chem. 1997, 62, 3390-3401.
(29) Denmark, S. E.; O'Connor, S. P. J. Org. Chem. 1997, 62, 584-594.
(30) Denmark, S. E.; Christenson, B. L.; O'Connor, S. P. Tetrahedron Lett. 1995,
36, 2219-2222.
(31) Denmark, S. E.; Christenson, B. L.; O'Connor, S. P.; Murase, N. Pure Appl.Chem. 1996, 68, 23-27.
(32) Imai, N.; Sakamoto, K.; Takahashi, H.; Kobayashi, S. Tetrahedron Lett. 1994,
35, 7045-7048.
(33) Imai, N.; Takahashi, H.; Kobayashi, S. Chem. Lett. 1994, 177-180.
(34) Imai, N.; Sakamoto, K.; Maeda, M.; Kouge, K.; Yoshizane, K.; Nokami, J.
Tetrahedron Lett. 1997, 38, 1423-1426.
(35) Singh, V. K.; DattaGupta, A.; Sekar, G. Synthesis 1997, 137-149.
(36) Doyle, M. P. In Comprehensive Organometallic Chemistry II; Abel, E. W.,
Stone, F. G. A., Wilkinson, G., Eds.; Elsevier: Oxford, 1995; Vol. 12.
(37) Brookhart, M.; Studabaker, W. B. Chem. Rev. 1987, 87, 411-432.
(38) Maas, G. Top. Curr. Chem. 1987, 137, 75.
(39) Yates, P. J. Am. Chem. Soc. 1952, 74, 5376-5381.
(40) Anciaux, A. J.; Hubert, A. J.; Noels, A. F.; Petiniot, N.; Teyssié, P. J. Org.Chem. 1980, 45, 695-702.
(41) Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 43,
5239-5244.
(42) Aratani, T.; Yoneyoshi, Y.; Nagase, T. Tetrahedron Lett. 1975, 21, 1707-
1710.
(43) Aratani, T.; Yoneyoshi, Y.; Nagase, T. Tetrahedron Lett. 1977, 30, 2599-
2602.
(44) Aratani, T.; Yoneyoshi, Y.; Nagase, T. Tetrahedron Lett. 1982, 23, 685-688.
(45) Aratani, T. Pure Appl. Chem. 1985, 57, 1839-1844.
(46) Evans, D. A.; Woerpel, K. A.; Hinman, M. M.; Faul, M. M. J. Am. Chem. Soc.
1991, 113, 726-728.
(47) Evans, D. A.; Woerpel, K. A.; Scott, M. J. Angew. Chem., Int. Ed. Engl. 1992,
31, 430-432.
(48) Lowenthal, R. E.; Abiko, A.; Masamune, S. Tetrahedron Lett. 1990, 31, 6005-
6008.
(49) Lowenthal, R. E.; Masamune, S. Tetrahedron Lett. 1991, 32, 7373-7376.
(50) Müller, D.; Umbricht, G.; Weber, B.; Pfaltz, A. Helv. Chim. Acta 1991, 74, 232-
240.
(51) Leutenegger, U.; Umbricht, G.; Fahrni, C; von Matt, P.; Pfaltz, A. Tetrahedron
1992, 48, 2143-2156.
(52) Pfaltz, A. Ace. Chem. Res. 1993, 26, 339-345.
(53) Fritschi, H.; Leutenegger, U.; Pfaltz, A. Helv. Chim. Acta 1988, 71, 1553-
1565.
(54) Anciaux, A. J.; Demonceau, A.; Noels, A. F.; Warin, R.; Hubert, A. J.; Teyssié,P. Tetrahedron 1983, 39, 2169-2173.

1. Cyclopropanierung 19
Doyle, M. P.; Pieters, R. J.; Martin, S. F.; Austin, R. E.; Oalmann, C. J.; Müller,
P. J. Am. Chem. Soc. 1991, 113, 1423-1424.
Doyle, M. P.; Winchester, W. R.; Hoorn, J. A. A.; Lynch, V.; Simonsen, S. H.;
Ghosh, R. J. Am. Chem. Soc. 1993, 115, 9968-9987.
Doyle, M. P. Aldrichimica Acta 1996, 29, 3-11.
Doyle, M. P. Ace. Chem. Res. 1986, 19, 348-356.
Doyle, M. P.; Brandes, B. D.; Kazala, A. P.; Pieters, R. J.; Jarstfer, M. B.;
Watkins, L. M.; Eagle, C. T. Tetrahedron Lett. 1990, 31, 6613-6616.
Doyle, M. P.; Bagheri, V.; Wandless, T. J.; Harn, N. K.; Brinker, D. A.; Eagle, C.
T.; Loh, K.-L. J. Am. Chem. Soc. 1990, 112, 1906-1912.
Nishiyama, H.; Itoh, Y.; Matsumoto, H.; Park, S.-B.; Itoh, K. J. Am. Chem. Soc.
1994, 116, 2223-2224.
Park, S.-B.; Nishiyama, H.; Itoh, Y.; Itoh, K. J. Chem. Soc. Chem. Commun.
1994, 1315-1316.
Nishiyama, H.; Itoh, Y.; Sugawara, Y.; Matsumoto, H.; Aoki, K.; Itoh, K. Bull.
Chem. Soc. Jpn. 1995, 68, 1247-1262.
Frauenkron, M.; Berkessel, A. Tetrahedron Lett. 1997, 38, 7175-7176.
Lo, W.-C; Che, C.-M.; Cheng, K.-F.; Mak, T. C. W. J. Chem. Soc. Chem.
Commun. 1997, 1205-1206.
Fukuda, T.; Katsuki, T. Tetrahedron 1997, 53, 7201-7208.
Fukuda, T.; Katsuki, T. Syn/eff 1995, 825-826.
Bedekar, A. V.; Andersson, P. G. Tetrahedron Lett. 1996, 37, 4073-4076.
Ichiyanagi, T.; Shimizu, M.; Fujisawa, T. Tetrahedron 1997, 53, 9599-9610.
Uozumi, Y.; Kyota, H.; Kishi, E.; Kitayama, K.; Hayashi, T. Tetrahedron:
Asymmetry\996, 7, 1603-1606.
Kim, S.-G.; Cho, C.-W.; Ahn, K. H. Tetrahedron: Asymmetry\997, 8, 1023-
1026.
Tanner, D.; Harden, A.; Johansson, F.; Wyatt, P.; Andersson, P. G. Acta
Chem. Scand. 1996, 50, 361-368.
Ito, K.; Katsuki, T. Synlett 1993, 638-640.
Ito, K.; Katsuki, T. Tetrahedron Lett. 1993, 34, 2661-2664.
Ito, K.; Yoshitake, M.; Katsuki, T. Heterocycles 1996, 42, 305-317.
Nakamura, A.; Konishi, A.; Tatsuno, Y.; Otsuka, S. J. Am. Chem. Soc. 1978,
100, 3443-3448.
Nakamura, A.; Konishi, A.; Tsujitani, R.; Kudo, M.; Otsuka, S. J. Am. Chem.
Soc. 1978, 100, 3449-3461.
Nakamura, A. Pure Appl. Chem. 1978, 50, 37-42.
Lötscher, D.; Rupprecht, S.; Stoeckli-Evans, H.; von Zelewsky, A.
Tetrahedron: Asymmetry 2000, 11, 4341-4357.
Sanders, C. J.; Gillespie, K. M.; Scott, P. Tetrahedron: Asymmetry 2001, 12,
1055-1061.
Suda, M. Synthesis 1981, 714.
Tomilov, Y. V.; Kostitsyn, A. B.; Shulishov, E. V.; Nefedov, O. M. Synthesis
1990, 246-248.
Fritschi, H. Dissertation ETH1989, Nr. 8951.
Fischer, E. O.; Dötz, K. H. Chem. Ber. 1970, 103, 1273-1278.
Dötz, K. H.; Fischer, E. O. Chem. Ber. 1972, 705, 1356-1367.
Fischer, E. O.; Dötz, K. H. Chem. Ber. 1972, 105, 3966-3973.
Cooke, M. D.; Fischer, E. O. J. Organomet. Chem. 1973, 56, 279-284.

1. Cyclopropanierung 20
88) Söderberg, B. C; Hegedus, L. S. Organometallics 1990, 9, 3113-3121.
89) Harvey, D. F.; Brown, M. F. J. Am. Chem. Soc. 1990, 112, 7806-7807.
90) Kerber, R. C. In Comprehensive Organometallic Chemistry II; Abel, E. W.,
Stone, F. G. A., Wilkinson, G., Eds.; Elsevier: Oxford, 1995; Vol. 7, pp 176-
181.
91) Brandt, S.; Helquist, P. J. Am. Chem. Soc. 1979, 101, 6473-6475.
92) Davidson, J. G.; Barefield, E. K.; Van Derveer, D. G. Organometallics 1985,
4, 1178-1184.
93) O'Connor, E. J.; Brandt, S.; Helquist, P. J. Am. Chem. Soc. 1987, 109, 3739-
3747.
94) Guerchais, V.; Lapinte, C; Thépot, J.-Y. Organometallics 1988, 7, 604-612.
95) Brookhart, M.; Liu, Y.; Goldman, E. W.; Timmers, D. A.; Williams, G. D. J. Am.
Chem. Soc. 1991, 113, 927-939.
96) Brookhart, M.; Liu, Y. J. Am. Chem. Soc. 1991, 113, 939-944.
97) Casey, C. P.; J., S. V. L. Organometallics 1992, 11, 738-744.
98) Seitz, W. J.; Saha, A. K.; Hossain, M. M. Organometallics 1993, 12, 2604-
2608.
99) Seitz, W. J.; Saha, A. K.; Casper, D.; Hossain, M. M. Tetrahedron Lett. 1992,
33, 7755-7758.
100) Brookhart, M.; Studabaker, W. B.; Humphrey, M. B. Organometallics 1989, 8,
132-140.
101) Guerchais, V.; Astruc, D.; Nunn, C. M.; Cowley, A. H. Organometallics 1990,
9, 1036-1041.
102) Vargas, R. M.; Theys, R. D.; Hossain, M. M. J. Am. Chem. Soc. 1992, 114,
777-778.
103) Roger, C; Lapinte, C. J. Chem. Soc. Chem. Commun. 1989, 1598-1600.
104) Jolly, P. W.; Pettit, R. J. Am. Chem. Soc. 1966, 88, 5044-5045.
105) McCarten, P.; Barefield, E. K. Organometallics 1998, 17, 4645-4648.
106) Wittig, G.; Geissler, G. Justus Liebigs Ann. Chem. 1953, 580, 44-57.
107) Li, A.-H.; Dai, L.-X. Chem. Rev. 1997, 97, 2341-2372.
108) Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1962, 84, 867-868.
109) Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1965, 87, 1353-1364.
110) Johnson, C. R.; Janiga, E. R.; Haake, M. J. Am. Chem. Soc. 1968, 90, 3890-
3891.
111) Nagao, Y.; Inoue, T.; Fujita, E.; Terada, S.; Shiro, M. Tetrahedron 1984, 40,
1215-1223.
112) Beautement, K.; Clough, J. M. Tetrahedron Lett. 1984, 25, 3025-3028.
113) Elkik, E.; Imbeaux-Oudotte, M. Bull. Soc. Chim. Fr. 1987, 861-866.
114) Okuma, K.; Ikari, K.; Ono, M.; Sato, Y.; Kuge, S.; Ohta, H.; Machiguchi, T. Bull.
Chem. Soc. Jpn. 1995, 68, 2313-2317.
115) Allen, D. G.; Wild, S. B. Organometallics 1983, 2, 394-399.
116) Guo, X.; Shen, W.; Shao, J.; Zhong, Q. Synth. Commun. 2000, 30, 3275-
3279.
117) Guo, X.; Shen, W.; Zheng, M.; Zhong, Q. Synth. Commun. 2000, 30, 3363-
3367.
118) Johnson, C. R. Ace. Chem. Res. 1973, 6, 341-347.
119) Johnson, C. R.; Schroeck, C. W. J. Am. Chem. Soc. 1973, 95, 7418-7423.
120) Trost, B. M.; Hammen, R. F. J. Am. Chem. Soc. 1973, 95, 962-963.
121) Tronchet, J. M. J.; Eder, H. J. Carbohydr. Chem. 1983, 2, 139-158.

1. Cyclopropanierung 21_
(122) Solladié-Cavallo, A.; Diep-Vohuule, A.; Isarno, T. Angew. Chem., Int. Ed.
Engl. 1998, 37, 1689-1691.
123) Johnson, C. R.; Schroeck, C. W. J. Am. Chem. Soc. 1968, 90, 6852-6854.
124) Furukawa, N.; Takahashi, F.; Yoshimura, T.; Oae, S. Tetrahedron Lett. 1977,
41, 3633-3636.
125) Furukawa, N.; Takahashi, F.; Yoshimura, T.; Oae, S. Tetrahedron 1979, 35,
317-322.
126) Johnson, C. R.; Kirchhoff, R. A.; Reischer, R. J.; Katekar, G. F. J. Am. Chem.
Soc. 1973, 95, 4287-4291.
127) Toda, F.; Imai, N. J. Chem. Soc, Perkin Trans. 11994, 2673-2674.
128) Pyne, S. G.; Dong, Z.; Skelton, B. W.; White, A. H. J. Org. Chem. 1997, 62,
2337-2343.
129) Aggarwal, V. K.; Smith, H. W.; Jones, R. V. H.; Fieldhouse, R. J. Chem. Soc.
Chem. Commun. 1997, 1785-1786.
130) Aggarwal, V. K.; Smith, H. W.; Hynd, G.; Jones, R. V. H.; Fieldhouse, R.; Spey,S. E. J. Chem. Soc, Perkin Trans. 1 2000, 3267-3276.
131) Aggarwal, V. K.; Alonso, E.; Fang, G.; Ferrara, M.; Hynd, G.; Porcelloni, M.
Angew. Chem., Int. Ed. Engl. 2001, 40, 1433-1436.
132) Aggarwal, V. K.; Alonso, E.; Hynd, G.; Lydon, K. M.; Palmer, M. J.; Porcelloni,
M.; Studley, J. R. Angew. Chem., Int. Ed. Engl. 2001, 40, 1430-1433.
133) Marnai, A.; Madalengoitia, J. S. Tetrahedron Lett. 2000, 41, 9009-9014.
134) Müller, P.; Fernandez, D.; Nury, P.; Rossier, J.-C. Helv. Chim. Acta 1999, 82,
935-945.
135) Trost, B. M. J. Am. Chem. Soc. 1966, 88, 1587-1588.
136) Trost, B. M. J. Am. Chem. Soc. 1967, 89, 138-142.
137) Cohen, T.; Herman, G.; Chapman, T. M.; Kuhn, D. J. Am. Chem. Soc. 1974,
96, 5627-5628.
138) Zhang, J.-J.; Schuster, G. B. J. Am. Chem. Soc. 1989, 111, 7149-7155.
139) Cimetière, B.; Julia, M. Synlett 1991, 271-272.

2. Eisen-Arsoniummethyl-Komplexe - Synthese, Struktur
und Reaktivität gegenüber Olefinen
2.1 Einführung
Die bis heute bedeutendsten Systeme zur Methylencyclopropanierung sind die
Simmons-Smith-Reaktion und die Pd- und Cu-katalysierte Cyclopropanierung mit
Diazomethan. Literatur zu diesen Reaktionen ist im ersten Kapitel zitiert.
Eine einfache Analyse der Simmons-Smith-Reaktion bezüglich Atom-Oekonomie
ergibt ein besonders schlechtes Resultat: Von den als Edukten eingesetzten
Substanzen, Zn und CH2I2, wird nur die Methylen-Gruppe in dem gewünschten
Produkt, dem Cyclopropan, wiedergefunden. Die Masse der Methylen-Gruppe
entspricht nur 4.2% der gesamten, zur Cyclopropanierung eingesetzten Edukt-
Masse. Nach dieser Ueberlegung ist eine effizientere, katalytische Version gefragt.
Eine auf Zn und Dihalogenmethanen beruhende katalytische Version der
Simmons-Smith Reaktion ist nicht möglich, da das Metall auch als "Halid-Senke"
dient und daher nicht in einen weiteren (katalytischen) Zyklus eintreten kann.
Ein sehr grosser Nachteil der katalytischen Cyclopropanierung mit CH2N2 ist die
Giftigkeit, Kanzerogenität und Explosivität dieser Methylenquelle.1 Es wird
empfohlen, Arbeiten mit Diazomethan nur in feuerpolierten Glaswaren, in einem
guten Abzug und hinter einem Schutzschild durchzuführen. Das Arbeiten mit
verdünnten Lösungen, bei tiefen Temperaturen (ca. 0 °C) und unter Schutz vor
starkem Licht verkleinert das Risiko. Von dem Lagern von Diazomethanlösungen
wird abgeraten.1'4
Weiter wurde eine grosse Zahl von Komplexen als Cyclopropanierungsreagenzien
verwendet.5 Das Salz [CpFe(CO)2(CH2SMe2)]BF4 ([Fp(CH2SMe2)]BF4) wurde von
Helquist und Mitarbeitern als Cyclopropanierungsmittel entwickelt und ausführlich
untersucht.6 Unter geeigneten Bedingungen setzt es Olefine in mittleren bis guten
Ausbeuten zu Cyclopropanen um. Kinetische Studien von McCarten und Barefield7
über die Cyclopropanierung von Cycloocten durch [Fp(CH2SPh2)]BF4 legen eine
Zwei-Schritt-Reaktion nahe.

2. Eisen-Arsoniummethyl-Komplexe 23
Nach der reversiblen Dissoziation von Diphenylsulfid, durch die das Carbenkation
[FpCH2]+ gebildet wird, erfolgt die kompetitive Umsetzung des Methylen-
Eisenkomplexes mit Cycloocten unter Bildung von Bicyclo[6.1.0]nonan (Schema
2.1).
[CpFe(CO)2(CH2SPh2)]+ t—^ [CpFe(CO)2CH2]+ + SPh2
[CpFe(CO)2CH2]+ + C8H14 ^ [CpFe(CO)2]+ + C9H16
Schema 2.1. Cyclopropanierung von Cycloocten mit [Fp(CH2SPh2)]+.
Erwartungsgemäss ist [Fp(CH2SMe2)]+ weniger reaktiv als das Diphenylsulfonium-
Analoge. Das Dimethylderivat benötigt hohe Temperaturen für die
Cyclopropanierung, während das Diphenyl-Analoge bereits bei Raumtemperatur
reagiert.7
Zur Entwicklung eines katalytischen Methylencyclopropanierungssystems ohne
Diazomethan sollten in einem ersten Schritt weitere Komplexe des Typs
[CpM(CO)n(CH2Z)]+X" hergestellt, charakterisiert und als Cyclopropanierungs¬
reagenzien getestet werden.
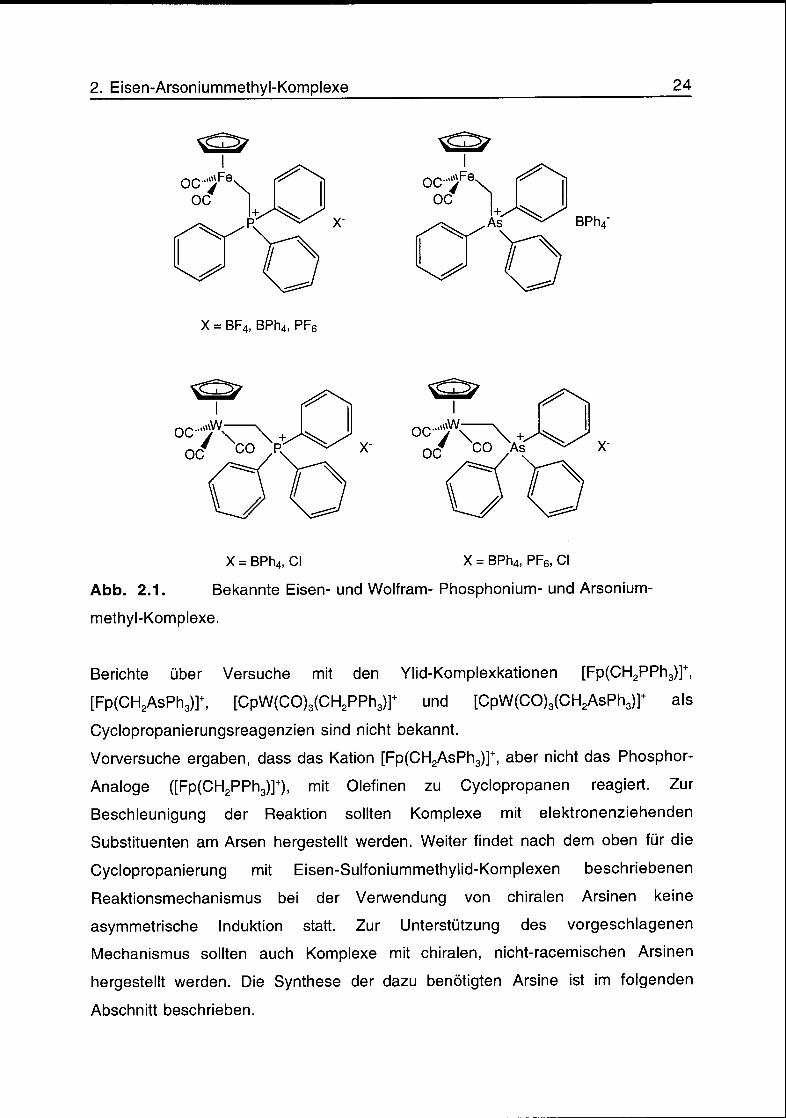
Reger und Culbertson berichteten 1977 die Synthese von [Fp(CH2PPh3)]BF4 aus
dem kationischen THF-Komplex [Fp(THF)]BF4 und Triphenylphosphoniummethylid
(CH2PPh3).8 Die Isolierung des Kations [Fp(CH2PPh3)]+ als BPh4-Salz wurde 1983
von Pelling, Botha und Moss veröffentlicht.9 Die Umsetzung von [Fp(CH2CI)] mit
PPh3 ergab nach Zugabe von NaBPh4 den formalen Ylid-Komplex. Nach dieser
Methode wurden ausgehend von [Fp(CH2CI)] oder [CpW(CO)3(CH2CI)] und PPh3
oder AsPh3 auch die Komplexe [Fp(CH2PPh3)]PF6, [Fp(CH2AsPh3)]BPh4 und
[CpW(CO)3(CH2PPh3)]+X" (X = BPh4, CI) erhalten. Im Jahr 1993 wurde von Friedrich
und Moss auch die Synthese der Wolfram-Arsoniummethylkomplexe
[CpW(CO)3(CH2AsPh3)]+X- (X = BPh4, PF6, CI) beschrieben.10 Die erwähnten Ylid-
Komplexe sind in Abb. 2.1 dargestellt. Die Umsetzung der Komplexe
[CpRu(CO)2(CH2CI)] und [CpMo(CO)3(CH2CI)] mit PPh3 zu den gewünschten Ylid-
Komplexen gelang nach Moss und Mitarbeitern hingegen nicht.
2. Eisen-Arsoniummethyl-Komplexe 24
BPh,
X = BF4, BPh4, PF6
X = BPh4, CI X = BPh4, PF6, CI
Abb. 2.1. Bekannte Eisen- und Wolfram- Phosphonium- und Arsonium-
methyl-Komplexe.
Berichte über Versuche mit den Ylid-Komplexkationen [Fp(CH2PPh3)]+,
[Fp(CH2AsPh3)]+, [CpW(CO)3(CH2PPh3)]+ und [CpW(CO)3(CH2AsPh3)]+ als
Cyclopropanierungsreagenzien sind nicht bekannt.
Vorversuche ergaben, dass das Kation [Fp(CH2AsPh3)]+, aber nicht das Phosphor-
Analoge ([Fp(CH2PPh3)]+), mit Olefinen zu Cyclopropanen reagiert. Zur
Beschleunigung der Reaktion sollten Komplexe mit elektronenziehenden
Substituenten am Arsen hergestellt werden. Weiter findet nach dem oben für die
Cyclopropanierung mit Eisen-Sulfoniummethylid-Komplexen beschriebenen
Reaktionsmechanismus bei der Verwendung von chiralen Arsinen keine
asymmetrische Induktion statt. Zur Unterstützung des vorgeschlagenen
Mechanismus sollten auch Komplexe mit chiralen, nicht-racemischen Arsinen
hergestellt werden. Die Synthese der dazu benötigten Arsine ist im folgenden
Abschnitt beschrieben.

2. Eisen-Arsoniummethyl-Komplexe 25
2.2 Arsine
Die Triarylarsine As(4-F-Ph)3 A1, As(4-CI-Ph)3 A2, As(3-CF3-Ph)3 A3, As(4-CF3-
Ph)3 A4, As(3,5-(F)2-Ph)3 A5, As(3,5-(CF3)2-Ph)3 A6, As(3-Br-Ph)3 A7 und As(3-CN-
Ph)3 A8 wurden aus Grignard-Reaktionen mit Arsentrichlorid erhalten. Die
Herstellung des Arsins A2 nach derselben Methode wurde bereits von McCortney
et al. ohne Angabe von Ausbeute und Analysedaten berichtet.11 Die Umsetzung
von As(3-Br-Ph)3 A7 zu As(3-CN-Ph)3 A8 gelang mit CuCN in Pyridin bei 250 °C.
Die hergestellten Triphenylarsine mit Substituenten an den Phenylringen sind in
Abb. 2.2 gezeigt. Die beobachteten Ausbeuten für die mittels Grignard-Reaktion
hergestellten Arsine A1-A7 lagen zwischen 79% und 96%. Das Arsin A8 wurde in
einer Ausbeute von 56% aus A7 erhalten.
CI As
A1 A2 A3 A4
A5 A6 A7 A8
Abb. 2.2. Triarylarsine A1-A8.
Chirale Arsinferrocenyle sollten aus (f?)-/V,/V-Dimethyl-1-ferrocenylethylamin und
Diarylchloroarsinen hergestellt werden. Letztere wurden aus der Umsetzung von
vier Aequivalenten des jeweiligen Grignardreagens mit einem Aequivalent
Arsenoxid (As203) nach Spaltung mit HCl conc. erhalten.

2. Eisen-Arsoniummethyl-Komplexe 26
Die nach dieser bereits für die Synthese von Diphenylarsinchlorid berichteten
Methode12 hergestellten Arsine AsCI(3-CF3-Ph)2 A9, AsCI(3,5-(CF3)2-Ph)2 A10 und
AsCI(4-CF3-Ph)2 A11 sind in Schema 2.2 abgebildet.
R1
As203 + 4 BrMg^^A—R2R3
v=<R3
r3~M ÄR2As'
As^^
R3^ V^
R2 R1
HCl conc.
R2 R1
As-Cl
R1 = CF3) R2 = H, R3 = H: A9
R1 = CF3, R2 = H, R3 = CF3: A10
R1 = H, R2 =CF3, R3 = H: A11
Schema 2.2. Herstellung der Diarylarsinchloride A9-A11.
Die (f?)-A/,A/-Dimethyl-{1-[(S)-2-(diarylarsino)ferrocenyl]ethyl}amine A12-A15
(siehe Schema 2.3) wurden nach der veröffentlichten Synthesevorschrift für (f?)-
A/,A/-Dimethyl-{1-[(S)-2-(diphenylphosphino)ferrocenyl]ethyl}amin hergestellt.13
Die Lithiierung von (R)-A/,A/-Dimethyl-1-ferrocenylethylamin mit Butyllithium ergab
nach Umsetzung mit dem entsprechenden Diarylarsinchlorid die Arsine
A12-A15.14 (fî)-A/,A7-Dimethyl-{1-[(S)-2-(diphenylarsino)ferrocenyl]ethyl}amin A15
wurde bereits von Butler, Cullen und Rettig beschrieben.15 Die Behandlung von
A12-A15 mit einem Ueberschuss Essigsäureanhydrid bei 100 °C ergab die (R)-
[(S)-(Diarylarsino)ferrocenyl]ethylacetate A16-A19. Diese Substitutionsreaktion
verläuft unter vollständiger Retention der absoluten Konfiguration des Chiralitäts-
zentrums am Kohlenstoff.16,17 Die planar-chiralen (S)-1-(Diarylarsino)-2-ethyl-
ferrocene A20-A23 wurden durch Reduktion der Ethylacetate A16-A19 mit
LAH/AICI3 erhalten. Die Syntheseschritte sind in Schema 2.3 gezeigt.

2. Eisen-Arsoniummethyl-Komplexe 27
Ein Phosphor-Analoges von (S)-A23, (fî)-1-Diphenylphosphino-2-ethylferrocen,
wurde mittels Rhodium-katalysierter Hydrierung aus (A?)-1-Diphenylphosphinoxid-2-
vinylferrocen und nachfolgender Reduktion des Phosphinoxids mit
Lithiumaluminiumhydrid hergestellt.14
H3C^ONMe2 nO
^"AsAr2 Ac2O,100°C <^"AsAr2 1)LAH,0°C ^T-AsAr2Fe Fe Fe
2) AICI3, RT
A12-A15 A16-A19 A20-A23
PF3
A12, A16, A20 Ar = -Çj A14, A18, A22 Ar = -{~}-CF3
PF3
A13, A17, A21 Ar = -Çj A15, A19, A23 Ar = -\J
CF3
Schema 2.3. Herstellung der Arsine A16-A23 aus den Arsinen A12-A15.
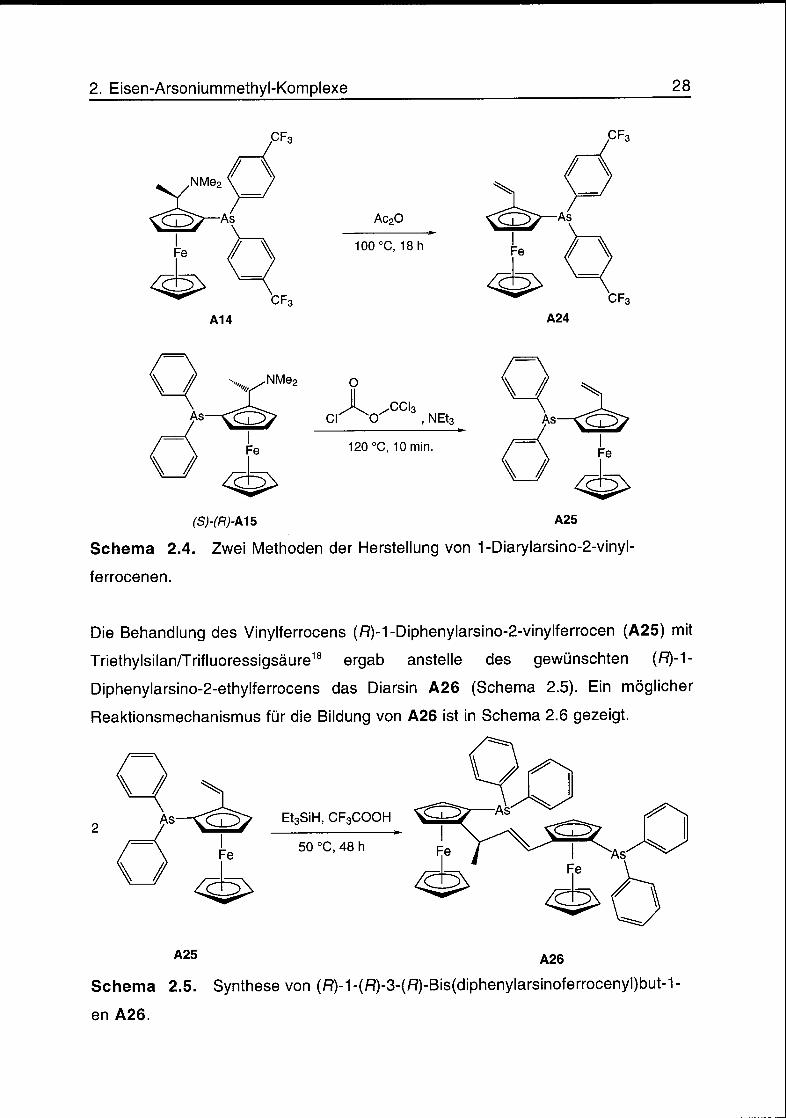
In der Synthese der (fî)-[(S)-(Diarylarsino)ferrocenyl]ethylacetate A16-A19 aus
den (fî)-A/,/V-Dimethyl-{1-[(S)-2-(diarylarsino)ferrocenyl]ethyl}aminen A12-A15
wurden nach einer Reaktionszeit von 3 h Ausbeuten zwischen 61% und 87%
erhalten. Bei einer Erhöhung der Reaktionszeit auf 18 h, erhielt man aus (R)-N,N-
Dimethyl-{ 1 -[(S)-2-(bis(4-trifluormethylphenyl)arsino)ferrocenyl]ethyl}amin (A14)
über den Ester A18 in einer Ausbeute von 80% das Eliminierungsprodukt (S)-1-
Bis(4-trifluormethylphenyl)arsino-2-vinylferrocen (A24). Das Vinylferrocen (/^-l-
Diphenylarsino-2-vinylferrocen (A25) wurde aus (S)-A/,/V-Dimethyl-{1-[(R)-2-
(diphenylarsino)ferrocenyl]ethyl}amin ((S)-(fî)-A15) in Gegenwart von
Chlorameisensäuretrichlormethylester und Triethylamin nach 10 min bei 120 °C in
einer Ausbeute von 88% erhalten. Die Herstellung der beiden 1-Diarylarsino-2-
vinylferrocene ist in Schema 2.4 dargestellt.
2. Eisen-Arsoniummethyl-Komplexe 28
A14
Ac20
100 °C, 18 h
.CFa
r\
As
XQCF3
A24
. ./CCI3
CI "O, NEt3
120 °C, 10 min.
\ /
As
IFe
(S)-(R)-M5 A25
Schema 2.4. Zwei Methoden der Herstellung von 1-Diarylarsino-2-vinyl-
ferrocenen.
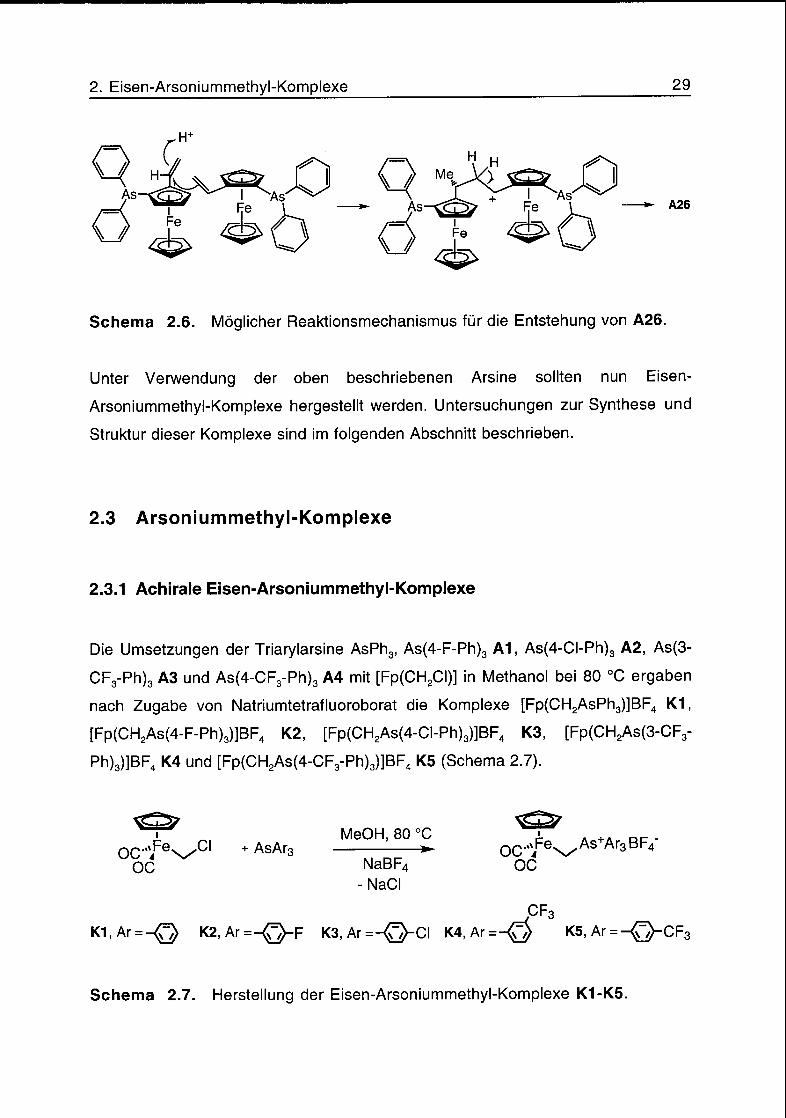
Die Behandlung des Vinylferrocens (fî)-1-Diphenylarsino-2-vinylferrocen (A25) mit
Triethylsilan/Trifluoressigsäure18 ergab anstelle des gewünschten (fî)-1-
Diphenylarsino-2-ethylferrocens das Diarsin A26 (Schema 2.5). Ein möglicher
Reaktionsmechanismus für die Bildung von A26 ist in Schema 2.6 gezeigt.
Et3SiH, CF3COOH)
50 °C, 48 h
^>
A25 A26
Schema 2.5. Synthese von (fî)-1-(f?)-3-(fî)-Bis(diphenylarsinoferrocenyl)but-1-
en A26.
2. Eisen-Arsoniummethyl-Komplexe 29
A26
Schema 2.6. Möglicher Reaktionsmechanismus für die Entstehung von A26.
Unter Verwendung der oben beschriebenen Arsine sollten nun Eisen-
Arsoniummethyl-Komplexe hergestellt werden. Untersuchungen zur Synthese und
Struktur dieser Komplexe sind im folgenden Abschnitt beschrieben.
2.3 Arsoniummethyl-Komplexe
2.3.1 Achirale Eisen-Arsoniummethyl-Komplexe
Die Umsetzungen der Triarylarsine AsPh3, As(4-F-Ph)3 A1, As(4-Cl-Ph)3 A2, As(3-
CF3-Ph)3 A3 und As(4-CF3-Ph)3 A4 mit [Fp(CH2CI)] in Methanol bei 80 °C ergaben
nach Zugabe von Natriumtetrafluoroborat die Komplexe [Fp(CH2AsPh3)]BF4 K1,
[Fp(CH2As(4-F-Ph)3)]BF4 K2, [Fp(CH2As(4-CI-Ph)3)]BF4 K3, [Fp(CH2As(3-CF3-
Ph)3)]BF4 K4 und [Fp(CH2As(4-CF3-Ph)3)]BF4 K5 (Schema 2.7).
± „,Me0H> 80 °C
CA +A on -
0C..Fe^CI +AsAr3 0C..4Fe^As+Ar3BF4OC NaBF4 OC
-NaCI
CF3
K1,Ar =-Q K2,Ar=-Q-F K3,Ar=-Q-CI K4,Ar=-^ K5,Ar = -Q-CF3
Schema 2.7. Herstellung der Eisen-Arsoniummethyl-Komplexe K1-K5.
2. Eisen-Arsoniummethyl-Komplexe 30
Die beobachteten Ausbeuten lagen zwischen 21% und 54%. Sämtliche Komplexe
wurden durch 1H- und 13C-NMR-Spektroskopie und Elementaranalyse
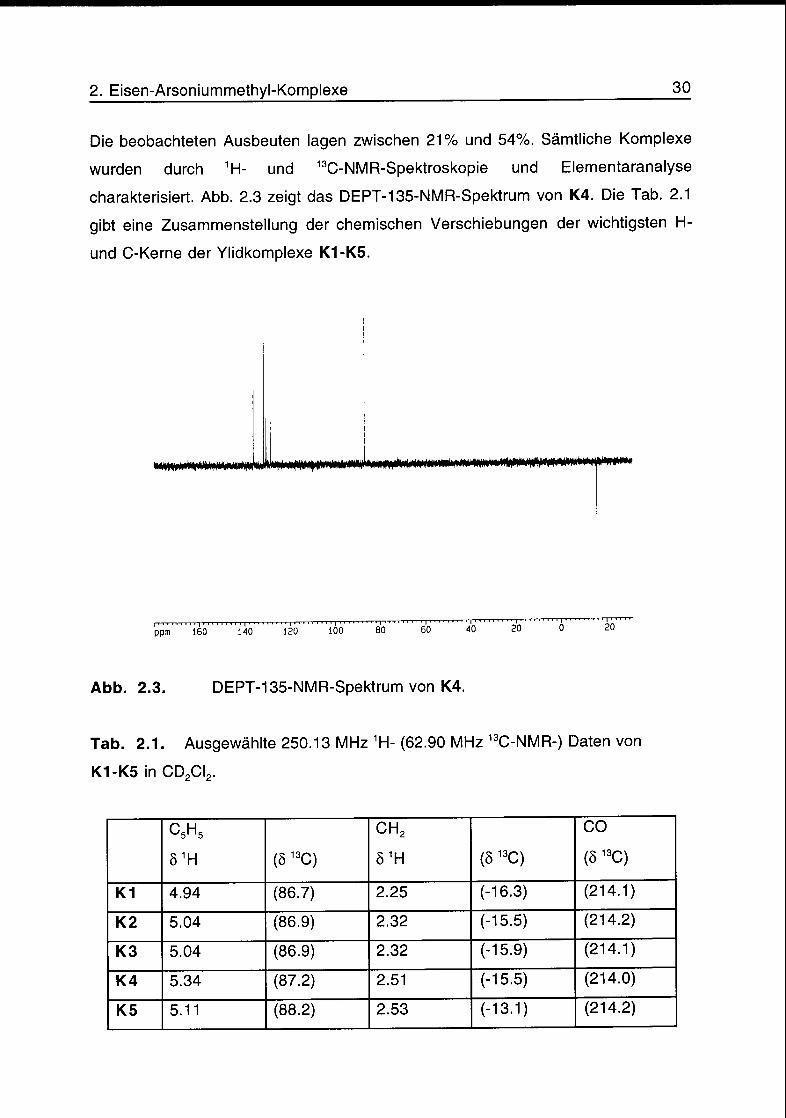
charakterisiert. Abb. 2.3 zeigt das DEPT-135-NMR-Spektrum von K4. Die Tab. 2.1
gibt eine Zusammenstellung der chemischen Verschiebungen der wichtigsten H-
und C-Kerne der Ylidkomplexe K1-K5.
M»»|WI^Hl|ltH»^^
ppm 160 140 120 100 80 60 40 20 -20
Abb. 2.3. DEPT-135-NMR-Spektrum von K4.
Tab. 2.1. Ausgewählte 250.13 MHz 1H- (62.90 MHz 13C-NMR-) Daten von
K1-K5 inCD2CI2.
C5H5
Ô1H (5 13C)
CH2
51H (8 13C)
CO
(Ô 13C)
K1 4.94 (86.7) 2.25 (-16.3) (214.1)
K2 5.04 (86.9) 2.32 (-15.5) (214.2)
K3 5.04 (86.9) 2.32 (-15.9) (214.1)
K4 5.34 (87.2) 2.51 (-15.5) (214.0)
K5 5.11 (88.2) 2.53 (-13.1) (214.2)

2. Eisen-Arsoniummethyl-Komplexe 31
Die Komplexe K2-K5 wurden zusätzlich mittels 19F-NMR-Spektroskopie und
Massenspektrometrie analysiert. Zusätzlich wurden aus Kristallisationsversuchen
mit K2 und K4 in Methanol und K3 in Dichloromethan/Pentan für die
Röntgenstrukturanalyse geeignete Kristalle erhalten. Im folgenden Teil wird die
Festkörperstruktur der Komplexkationen von K2-K4 vorgestellt und diskutiert.
Röntgenstrukturanalysen von K2, K3 und K4
Es wird angenommen, dass die Fe-C a-Bindungslänge ein Gradmesser für die
Reaktivität von Komplexen wie K2, K3 und K4 ist. Typische Fe-C(sp3)-Bindungen
haben eine Länge von 2.08-2.10 Â. Die Kristallstrukturen der Komplexe K2, K3
und K4 wurden bestimmt. ORTEP-Darstellungen der Kationen sind in den Abb. 2.4,
2.5 und 2.6 gezeigt. Kristalldaten, Mess- und Verfeinerungsparameter für K2, K3
und K4 finden sich im Anhang. Eine Auswahl von Bindungslängen und -winkeln für
die drei Komplexkationen ist in Tab. 2.2 gegeben.
Abb. 2.4. ORTEP-Darstellung des Kations von K2.
2. Eisen-Arsoniummethyl-Komplexe 32
C(3)
C(2)_
OasUBiCW
CH2)
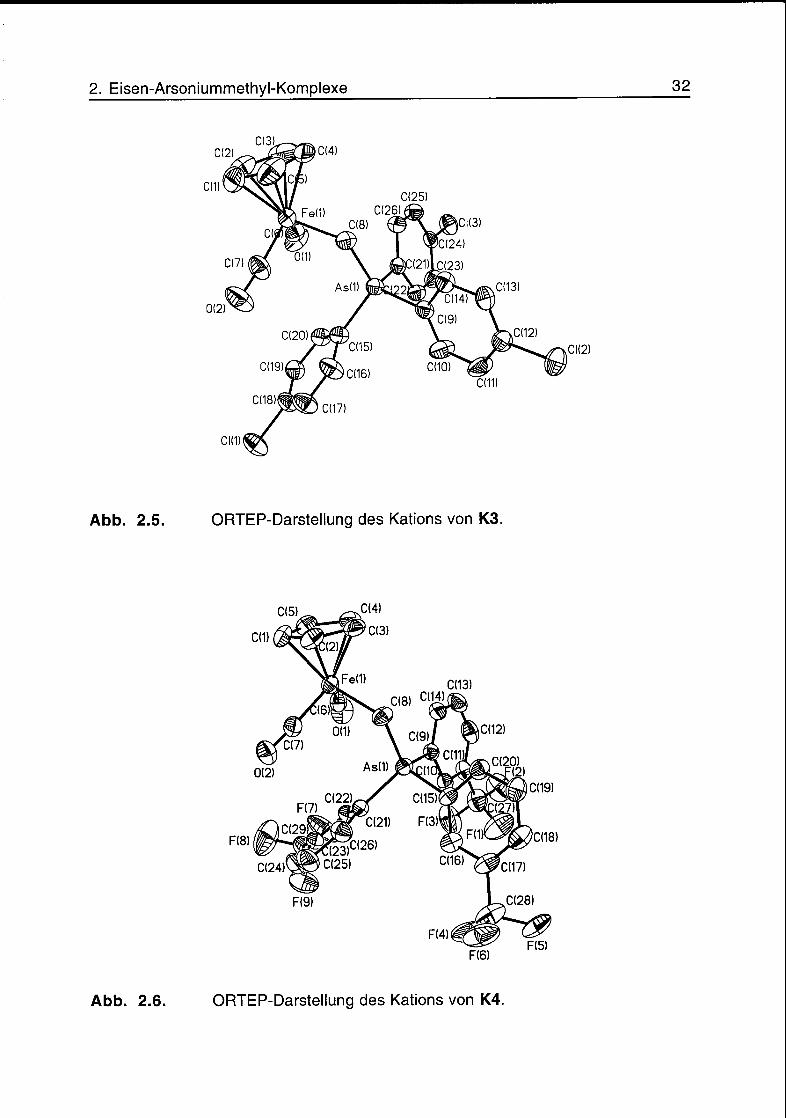
Abb. 2.5. ORTEP-Darstellung des Kations von K3.
C(5) .C(4)
C(19)
C(18)
Abb. 2.6. ORTEP-Darstellung des Kations von K4.
2. Eisen-Arsoniummethyl-Komplexe 33
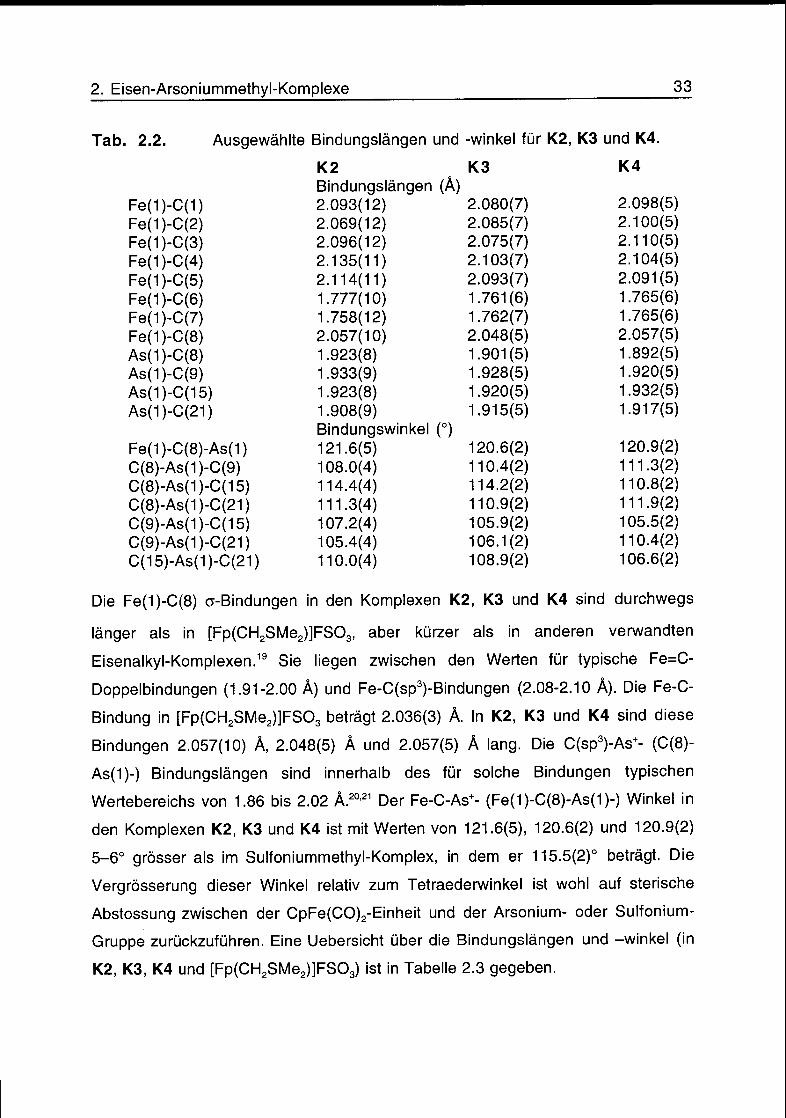
Tab. 2.2. Ausgewählte Bindungslängen und -Winkel für K2, K3 und K4.
K2 K3 K4
Bindungslängen (Â )
Fe(1)-C(1) 2.093(12) 2.080(7) 2.098(5
Fe(1)-C(2) 2.069(12) 2.085(7) 2.100(5
Fe(1)-C(3) 2.096(12) 2.075(7) 2.110(5
Fe(1)-C(4) 2.135(11) 2.103(7) 2.104(5
Fe(1)-C(5) 2.114(11) 2.093(7) 2.091(5
Fe(1)-C(6) 1.777(10) 1.761(6) 1.765(6
Fe(1)-C(7) 1.758(12) 1.762(7) 1.765(6
Fe(1)-C(8) 2.057(10) 2.048(5) 2.057(5
As(1)-C(8) 1.923(8) 1.901(5) 1.892(5
As(1)-C(9) 1.933(9) 1.928(5) 1.920(5
As(1)-C(15) 1.923(8) 1.920(5) 1.932(5
As(1)-C(21) 1.908(9)Bindungswinkel (°)
1.915(5) 1.917(5
Fe(1)-C(8)-As(1) 121.6(5) 120.6(2) 120.9(2
C(8)-As(1)-C(9) 108.0(4) 110.4(2) 111.3(2
C(8)-As(1)-C(15) 114.4(4) 114.2(2) 110.8(2]
C(8)-As(1)-C(21) 111.3(4) 110.9(2) 111.9(2,
C(9)-As(1)-C(15) 107.2(4) 105.9(2) 105.5(2]
C(9)-As(1)-C(21) 105.4(4) 106.1(2) 110.4(2'
C(15)-As(1)-C(21) 110.0(4) 108.9(2) 106.6(2]
Die Fe(1)-C(8) o-Bindungen in den Komplexen K2, K3 und K4 sind durchwegs
länger als in [Fp(CH2SMe2)]FS03, aber kürzer als in anderen verwandten
Eisenalkyl-Komplexen.19 Sie liegen zwischen den Werten für typische Fe=C-
Doppelbindungen (1.91-2.00 Â) und Fe-C(sp3)-Bindungen (2.08-2.10 Â). Die Fe-C-
Bindung in [Fp(CH2SMe2)]FS03 beträgt 2.036(3) Â. In K2, K3 und K4 sind diese
Bindungen 2.057(10) Â, 2.048(5) À und 2.057(5) Â lang. Die C(sp3)-As+- (C(8)-
As(1)-) Bindungslängen sind innerhalb des für solche Bindungen typischen
Wertebereichs von 1.86 bis 2.02 Â.2021 Der Fe-C-As+- (Fe(1)-C(8)-As(1)-) Winkel in
den Komplexen K2, K3 und K4 ist mit Werten von 121.6(5), 120.6(2) und 120.9(2)
5-6° grösser als im Sulfoniummethyl-Komplex, in dem er 115.5(2)° beträgt. Die
Vergrösserung dieser Winkel relativ zum Tetraederwinkel ist wohl auf sterische
Abstossung zwischen der CpFe(CO)2-Einheit und der Arsonium- oder Sulfonium-
Gruppe zurückzuführen. Eine Uebersicht über die Bindungslängen und -winkel (in
K2, K3, K4 und [Fp(CH2SMe2)]FS03) ist in Tabelle 2.3 gegeben.
2. Eisen-Arsoniummethyl-Komplexe 34
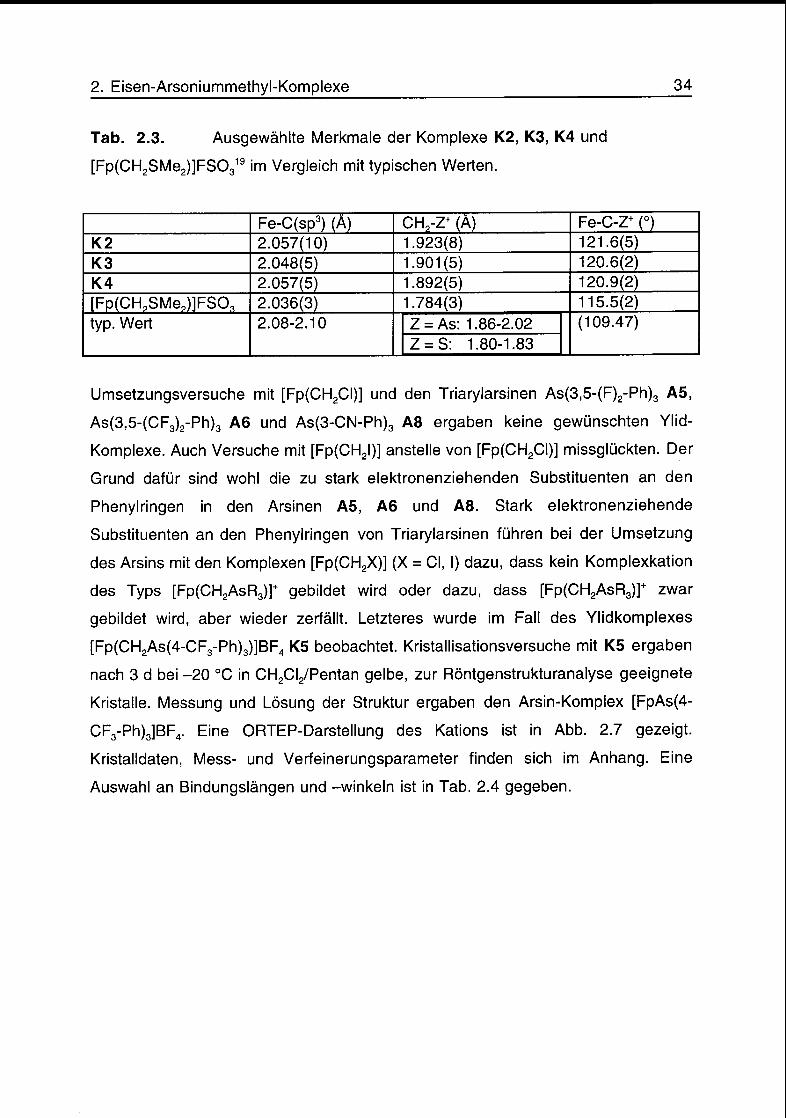
Tab. 2.3. Ausgewählte Merkmale der Komplexe K2, K3, K4 und
[Fp(CH2SMe2)]FS0319 im Vergleich mit typischen Werten.
Fe-C(sp3) (A) CH?-Z+ (À) Fe-C-Z+ OK2 2.057(10) 1.923(8) 121.6(5)K3 2.048(5) 1.901(5) 120.6(2)K4 2.057(5) 1.892(5) 120.9(2)
[FptCHpSMe^FSO, 2.036(3) 1.784(3) 115.5(2)
typ. Wert 2.08-2.10 Z = As: 1.86-2.02 (109.47)
Z = S: 1.80-1.83
Umsetzungsversuche mit [Fp(CH2CI)] und den Triarylarsinen As(3,5-(F)2-Ph)3 A5,
As(3,5-(CF3)2-Ph)3 A6 und As(3-CN-Ph)3 A8 ergaben keine gewünschten Ylid-
Komplexe. Auch Versuche mit [Fp(CH2l)] anstelle von [Fp(CH2CI)] missglückten. Der
Grund dafür sind wohl die zu stark elektronenziehenden Substituenten an den
Phenylringen in den Arsinen A5, A6 und A8. Stark elektronenziehende
Substituenten an den Phenylringen von Triarylarsinen führen bei der Umsetzung
des Arsins mit den Komplexen [Fp(CH2X)] (X = CI, I) dazu, dass kein Komplexkation
des Typs [Fp(CH2AsR3)]+ gebildet wird oder dazu, dass [Fp(CH2AsR3)]+ zwar
gebildet wird, aber wieder zerfällt. Letzteres wurde im Fall des Ylidkomplexes
[Fp(CH2As(4-CF3-Ph)3)]BF4 K5 beobachtet. Kristallisationsversuche mit K5 ergaben
nach 3 d bei -20 °C in CH2CI2/Pentan gelbe, zur Röntgenstrukturanalyse geeignete
Kristalle. Messung und Lösung der Struktur ergaben den Arsin-Komplex [FpAs(4-
CF3-Ph)3]BF4. Eine ORTEP-Darstellung des Kations ist in Abb. 2.7 gezeigt.
Kristalldaten, Mess- und Verfeinerungsparameter finden sich im Anhang. Eine
Auswahl an Bindungslängen und -winkeln ist in Tab. 2.4 gegeben.
2. Eisen-Arsoniummethyl-Komplexe 35
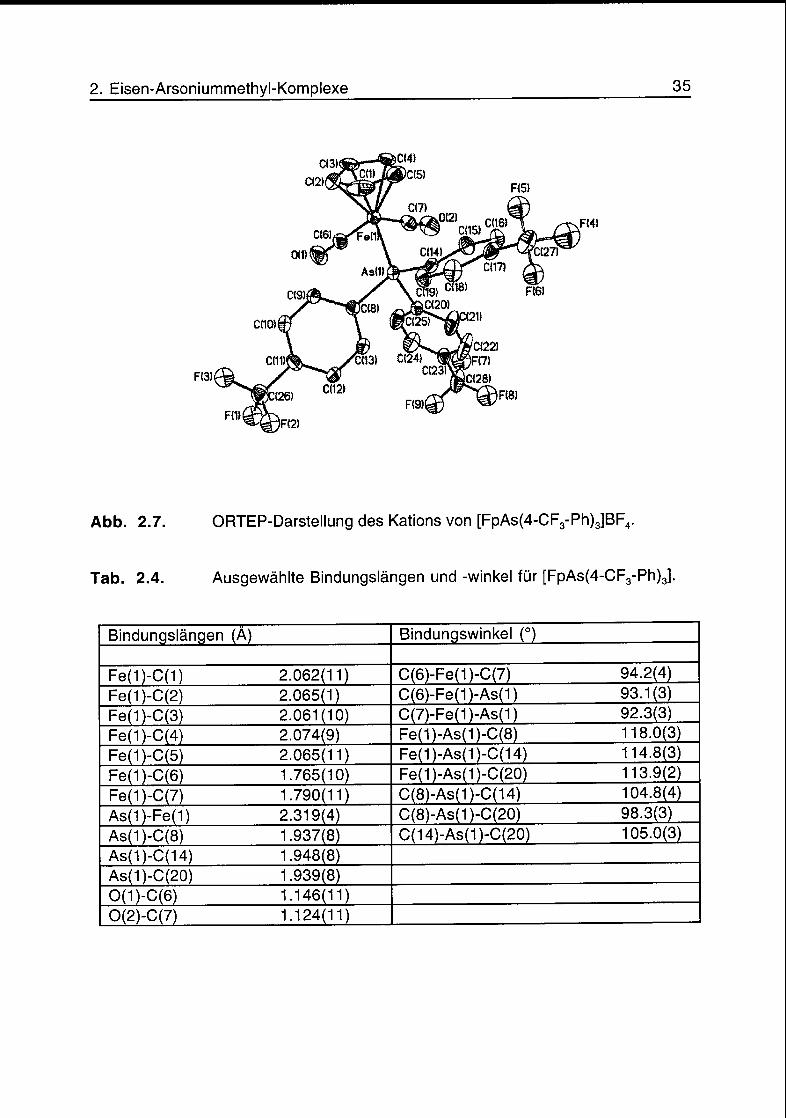
Abb. 2.7. ORTEP-Darstellung des Kations von [FpAs(4-CF3-Ph)3]BF4.
Tab. 2.4. Ausgewählte Bindungslängen und -winkel für [FpAs(4-CF3-Ph)3].
Bindungslängen (A) Bindungswinkel (°)
Fe(1)-C(1) 2.062(11) C(6)-Fe(1)-C(7) 94.2(4)
Fe(1)-C(2) 2.065(1) C(6)-Fe(1)-As(1) 93.1(3)
Fe(1)-C(3) 2.061(10) C(7)-Fe(1)-As(1) 92.3(3)
Fe(1)-C(4) 2.074(9) Fe(1)-As(1)-C(8) 118.0(3)
Fe(1)-C(5) 2.065(11) Fe(1)-As(1)-C(14) 114.8(3)
Fe(1)-C(6) 1.765(10) Fe(1)-As(1)-C(20) 113.9(2)
Fe(1)-C(7) 1.790(11) C(8)-As(1)-C(14) 104.8(4)
As(1)-Fe(1) 2.319(4) C(8)-As(1)-C(20) 98.3(3)
As(1)-C(8) 1.937(8) C(14)-As(1)-C(20) 105.0(3)
As(1)-C(14) 1.948(8)
As(1)-C(20) 1.939(8)
0(1)-C(6) 1.146(11)
0(2)-C(7) 1.124(11)
2. Eisen-Arsoniummethyl-Komplexe 36
Aufgrund der elektronenziehenden Eigenschaft der CF3-Gruppen in para-Position
der Phenylgruppen ist der Komplex K5 unter den zur Kristallisation angegebenen
Bedingungen nicht stabil. Scheinbar ist die Konzentration des
Eisencarbenkomplexes [FpCH2]+ genügend gross, so dass, unter irreversibler
Bildung von Ethen oder Polyethylen, der Arsinkomplex in grosser Menge entstehen
kann. In diesem Zusammenhang interessant ist, dass in den fast atom
bombardment (FAB) Massenspektren der Komplexe K2-K5 Signale vorhanden
sind, die den jeweiligen Eisen-Methylenkomplexen entsprechen. Die relativen
Intensitäten dieser Signale (m/z= 191) sind 0.7% (K2), 1.6% (K3), 0.6% (K4) und
16.5% (K5). Der grosse Wert im Falle von K5 kann als Hinweis auf dessen relative
Instabilität gedeutet werden. Eine Uebersicht über die massenspektrometrischen
Daten der Ylidkomplexe K2-K5 ist in Tab. 2.5 gegeben.
Tab. 2.5. Massenspektrometrische Daten m/z (rel. Intensität in %) für
K2-K5.
[M]+ [M-2CO]+ [MeAsAr3]+ [FpCH2]+
K2 551 (60) 495(100) 375(21) 1910)
K3 601 (97) 545(100) 425(19) 191 (2)
K4 701 (43) 645(100) 525 (28) 797(1)
K5 701 (66) 545(100) 525 (54) 797(17)
Als weitere Möglichkeit, alternative Komplexe zu erhalten, sollten Komplexe mit
verschiedenen Anionen hergestellt werden. O'Connor et al. beobachteten bei
Cyclopropanierungsexperimenten mit den Komplexen [Fp(CH2SMe2)]+X" (X = BF4,
FS03, I und BPh4) grosse Unterschiede, wobei mit BF4' als Gegenion die besten
Resultate erzielt wurden.5 Im Hinblick auf folgende Reaktionsversuche sollten
Arsoniummethylid-Komplexe mit anderen als den oben genannten Anionen
hergestellt werden.

2. Eisen-Arsoniummethyl-Komplexe 37
Reaktionsversuche zur Synthese von [Fp(CH2As(3-CF3-Ph)3)]+X" (X = PF6, AsF6 und
SbF6) nach der selben Methode wie für K4 mit KPF6, NaAsF6 und NaSbF6 anstelle
von NaBF4 ergaben den Komplex [Fp(CH2As(3-CF3-Ph)3)]SbF6 K6. Die
beobachtete Ausbeute betrug lediglich 4%. Die analogen Komplexe mit PF6" oder
AsF6" als Gegenionen konnten nicht isoliert werden.
Um stabilere Eisen-Carben-Komplexe zu erhalten, sollten weiter auch Komplexe
mit methylsubstituierten Cyclopentadieniden hergestellt werden. Sowohl der
Methylcyclopentadienid-Komplex [(C5H4Me)Fe(CO)2(CH2AsPh3)]BF4 K7 als auch
der Pentamethylcyclopentadienid-Komplex [(C5Me5)Fe(CO)2(CH2AsPh3)]BF4 K8
konnte erhalten werden. Die Synthese von K7 ist in Schema 2.8 dargestellt. Die
Synthese von K8 erfolgte analog zu der von K7 ausgehend von
Eisenpentacarbonyl und Pentamethylcyclopentadien.
[Fe2(C0)9] + 2
CeHß
\\ //RT, 24 h
[(C5H4Me)Fe(C0)2]2
+ 2 Na
+ 2 CICH20CH3
THF RT, 24 h
0C....*Fe CI
OC+ AsPh3+ NaBF4
65 °C, 12 h
64%
+ HCI
oc
K8 K7
Schema 2.8. Herstellung des Methylcyclopentadienid-Ylid-Komplexes
[(C5H4Me)Fe(CO)2(CH2AsPh3)]BF4 K7 und Darstellung von K8.
Während der Komplex K7 mit einer Ausbeute von 64% aus dem Chlormethyl-
Vorläufer erhalten wurde, ergab die analoge Umsetzung mit dem
Pentamethylcyclopentadienid-chlormethyl-Komplex das Produkt K8 in lediglich
10%. Beide Komplexe wurde durch 1H-, 13C- und 19F-NMR-Spektroskopie,
Massenspektrometrie und Elementaranalyse vollständig charakterisiert. Das 13C-
NMR-Spektrum von K7 ist in Abb. 2.8 dargestellt.
2. Eisen-Arsoniummethyl-Komplexe 38
iiiHMmmiu'iiiiiiWHnW M*MN|(IDM*W iHOh».
TTr
ppm 200T
150 100 50
" " " I "
0
Abb. 2.8. 13,C-NMR-Spektrum von K7.
Die Versuche, [Fp(CH2AsPh3)]BF4 aus Fp(THF)BF4 und Triphenylarsoniummethylid
(CH2AsPh3) analog zur Herstellung von [Fp(CH2PPh3)]BF48 zu erhalten, misslangen.
Anstelle des gewünschten Ylid-Komplexes wurde der Arsin-Komplex
[Fp(AsPh3)]BF4 isoliert.
2.3.2 Chirale Eisen-Arsoniummethyl-Komplexe
[Fp(CH2CI)] reagierte sowohl mit A25 als auch mit (S)-(fî)-A19 in MeOH und in
Gegenwart von NaBF4 zum chiralen Arsoniummethyl-Komplex K9 (Schema 2.9).
Die absolute Konfiguration S des Chiralitätszentrums in K9 ist postuliert. Es wird
angenommen, dass die Umsetzung der Acetoxyethylgruppe zum Methoxy-Derivat
unter Retention verläuft. Die Umsetzung eines Acetoxyethylferrocens zum Methoxy-
Derivat in Methanol wurde früher von Hayashi etat, berichtet.14 Diese Reaktion in
Gegenwart einer Base verlief ebenfalls unter Retention.
2. Eisen-Arsoniummethyl-Komplexe 39
OC -^Fe Cl
OCMeOH
NaBF4
65 °C,12h
MeOH
O. ^ NaBF4
ocft.Fe^ ^As+—^gl3> BF4
/ Ph2 '
OC Fe
^>65 °C, 12h K9
OC..-«Fe CI
OC
(S)-(fl)-A19
Schema 2.9. Herstellung des chiralen Eisen-Arsoniummethyl-Komplexes K9.
Wie für die achiralen Arsine führten die oben beschriebenen Experimente in
Komplexsynthese mit den chiralen Ferrocenylarsinen A20-A23 zu den Komplexen
K10 und K11 (Abb. 2.9). Mit A21 und A22 wurden die entsprechenden Komplexe
nicht beobachtet.
BF,
^^
BF,
K10 K11
Abb. 2.9. Chirale Arsoniummethyl-Komplexe K10 und K11.

2. Eisen-Arsoniummethyl-Komplexe 40
Die Komplexe K10 und K11 wurden in Ausbeuten von 20% und 37% erhalten. Sie
wurden durch 1H-, 13C- und 19F-NMR-Spektroskopie, Massenspektrometrie und
Elementaranalyse vollständig charakterisiert.
Bei der versuchten Umsetzung von [Fp(CH2CI)] mit dem Arsin (S)-(f?)-A19 zum
Arsoniumylid-Komplex entstand das Diarsin A26 (Schema 2.10). Eine mögliche
Erklärung für diese Reaktion ist die Bildung des Eliminationsproduktes A25
(Schema 2.4) mit nachfolgender Bildung des Diarsins wie in Schema 2.6 gezeigt.
(S)-(R)-A19 A26
Schema 2.10. Herstellung des Diarylarsins A26.
2.3.3 Molybdän- und Wolfram-Arsoniummethyl-Komplexe
Neben Eisenkomplexen sollte auch die Herstellung von anderen Metall-
Ylidkomplexen erprobt werden. [CpRu(CO)2(CH2CI)] reagierte nicht mit AsPh3 zum
gewünschten Ruthenium-Ylidkomplex [CpRu(CO)2(CH2AsPh3)]CI. Pelling, Botha
und Moss berichteten bereits 1983 den missglückten Versuch der Herstellung von
[CpRu(CO)2(CH2PPh3)]CI aus [CpRu(CO)2(CH2CI)] und PPh3.9
Einerseits sollte nun das bekannte Ylidkomplexkation [CpW(CO)3(CH2AsPh3)]+ als
BF4"-Salz isoliert werden und andererseits der analoge Molybdän-Komplex
[CpMo(CO)3(CH2AsPh3)]BF4 hergestellt werden. Beides gelang nach der in Schema
2.11 gezeigten Synthesefolge.
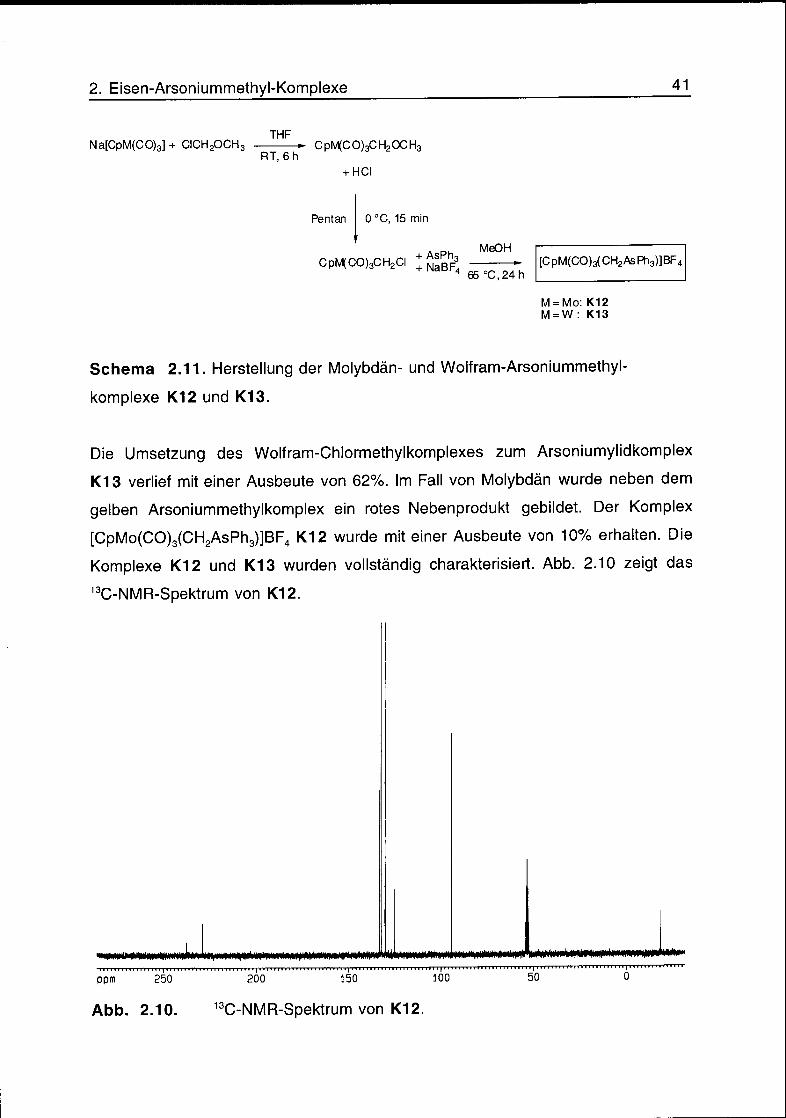
2. Eisen-Arsoniummethyl-Komplexe 41
Na[CpM(CO)3] + CICH2OCH3THF
RT, 6 hCpMtCO)3CH2OCH3
HCl
Pentan 0°C, 15 min
CpM(CO)3CH2CI ;JghF3MeOH
65 °C,24h
[CpM(CO)3(CH2AsPh3)]BF„
M = Mo:K12
M = W: K13
Schema 2.11. Herstellung der Molybdän- und Wolfram-Arsoniummethyl-
komplexe K12 und K13.
Die Umsetzung des Wolfram-Chlormethylkomplexes zum Arsoniumylidkomplex
K13 verlief mit einer Ausbeute von 62%. Im Fall von Molybdän wurde neben dem
gelben Arsoniummethylkomplex ein rotes Nebenprodukt gebildet. Der Komplex
[CpMo(CO)3(CH2AsPh3)]BF4 K12 wurde mit einer Ausbeute von 10% erhalten. Die
Komplexe K12 und K13 wurden vollständig charakterisiert. Abb. 2.10 zeigt das
13C-NMR-Spektrum von K12.
wmwmun»i»m»niH"»|imm mt*m*<mmkmm»**nimtiimi»mmMf¥ iiéH*iiwwi^>ww^^^
ppm 250 200 150 100 50
Abb. 2.10. 13,C-NMR-Spektrum von K12.

2. Eisen-Arsoniummethyl-Komplexe 42
Die in diesem Abschnitt vorgestellten Komplexe sollten nun als
Cyclopropanierungsreagenzien getestet werden. Für die Reaktionsversuche mit
chiralen Komplexen war es nötig, ein Substrat zu finden, dessen
Methylencyclopropanierungsderivat chiral ist. Ferner musste es möglich sein, die
Enantiomere der Produkte zu trennen.
2.4 Herstellung von Substraten und Referenz-Produkten
Als Modellsubstrat für die Cyclopropanierung mit den chiralen
Arsoniummethylkomplexen K9-K11 sollte ein geeignetes prochirales Olefin
gefunden werden. Die erste Wahl war das Cyclohexenderivat 1-Phenyl-1-
cyclohexen. Die Cyclopropanierung dieses Olefins mit 1.1 equiv. Diethylzink und
1.5 equiv. Diiodmethan (15 h bei 60 °C) ergab nach der Furakawa-Variante der
Simmons-Smith-Reaktion 1-Phenylbicyclo[4.1.0]heptan in einer Ausbeute von ca.
45% (nach FC und Destillation). Zur vollständigen Reinigung des Cyclopropans
war es nötig, restliches Olefin zum Alkohol umzusetzen und durch FC abzutrennen.
Das erhaltene Cyclopropan wurde durch 1H-NMR und MS(EI+) analysiert. In einem
nächsten Schritt sollte die Enantiomerentrennung mittels HPLC oder GC
durchgeführt werden.
HPLC ergab unter den in Tab. 2.6 angegebenen Bedingungen keine oder nur
unvollständige Enantiomerentrennung.
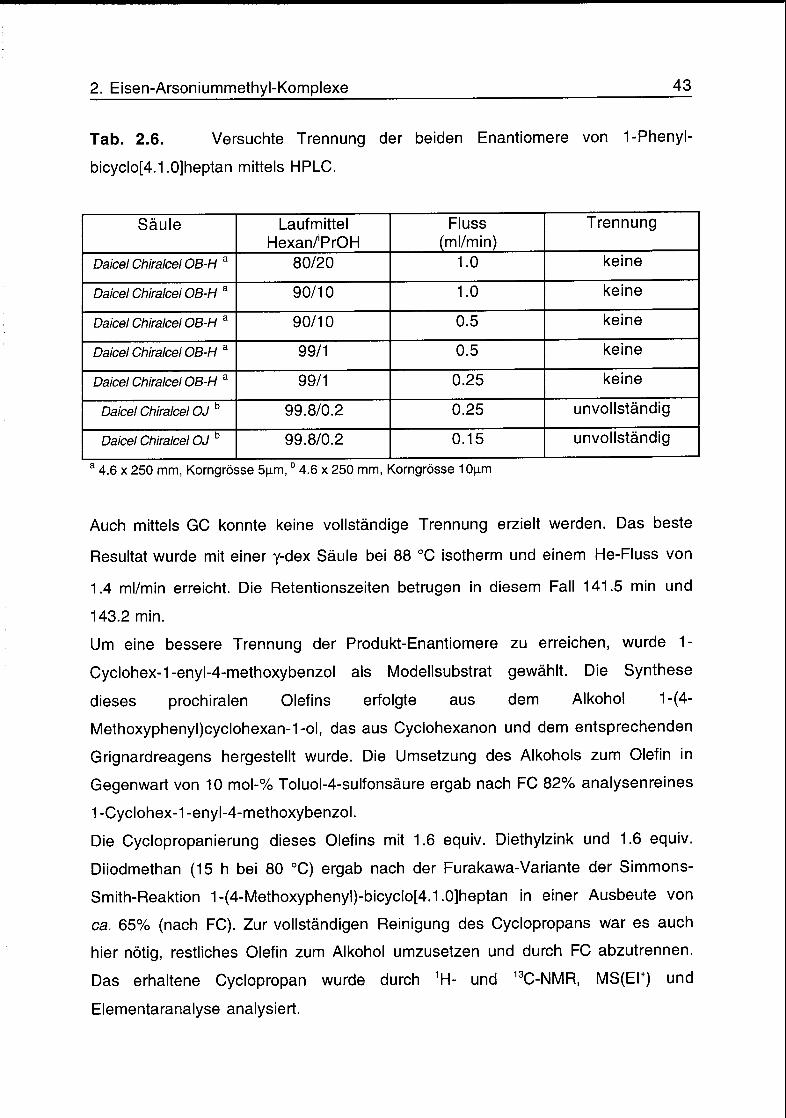
2. Eisen-Arsoniummethyl-Komplexe 43
Tab. 2.6. Versuchte Trennung der beiden Enantiomere von 1-Phenyl-
bicyclo[4.1.0]heptan mittels HPLC.
Säule Laufmittel
Hexan/'PrOH
Fluss
(ml/min)
Trennung
Daicel Chiralcel OB-Ha 80/20 1.0 keine
Daicel Chiralcel OB-Ha 90/10 1.0 keine
Daicel Chiralcel OB-Ha 90/10 0.5 keine
Daicel Chiralcel OB-Ha 99/1 0.5 keine
Daicel Chiralcel OB-Ha 99/1 0.25 keine
Daicel Chiralcel OJb 99.8/0.2 0.25 unvollständig
Daicel Chiralcel OJb 99.8/0.2 0.15 unvollständig
a4.6 x 250 mm, Korngrösse 5p,m,b 4.6 x 250 mm, Korngrösse 10jxm
Auch mittels GC konnte keine vollständige Trennung erzielt werden. Das beste
Resultat wurde mit einer y-dex Säule bei 88 °C isotherm und einem He-Fluss von
1.4 ml/min erreicht. Die Retentionszeiten betrugen in diesem Fall 141.5 min und
143.2 min.
Um eine bessere Trennung der Produkt-Enantiomere zu erreichen, wurde 1-
Cyclohex-1-enyl-4-methoxybenzol als Modellsubstrat gewählt. Die Synthese
dieses prochiralen Olefins erfolgte aus dem Alkohol 1-(4-
Methoxyphenyl)cyclohexan-1-ol, das aus Cyclohexanon und dem entsprechenden
Grignardreagens hergestellt wurde. Die Umsetzung des Alkohols zum Olefin in
Gegenwart von 10 mol-% Toluol-4-sulfonsäure ergab nach FC 82% analysenreines
1 -Cyclohex-1 -enyl-4-methoxybenzol.
Die Cyclopropanierung dieses Olefins mit 1.6 equiv. Diethylzink und 1.6 equiv.
Diiodmethan (15 h bei 80 °C) ergab nach der Furakawa-Variante der Simmons-
Smith-Reaktion 1-(4-Methoxyphenyl)-bicyclo[4.1.0]heptan in einer Ausbeute von
ca. 65% (nach FC). Zur vollständigen Reinigung des Cyclopropans war es auch
hier nötig, restliches Olefin zum Alkohol umzusetzen und durch FC abzutrennen.
Das erhaltene Cyclopropan wurde durch 1H- und 13C-NMR, MS(EI+) und
Elementaranalyse analysiert.
2. Eisen-Arsoniummethyl-Komplexe 44
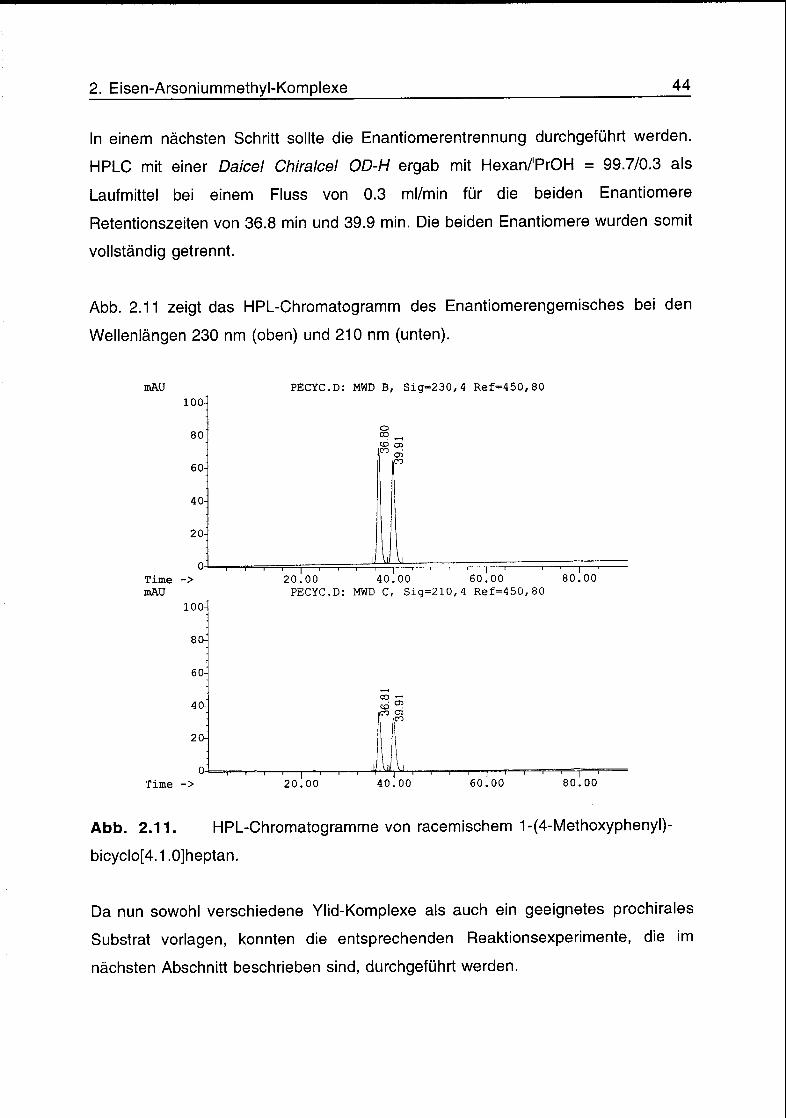
In einem nächsten Schritt sollte die Enantiomerentrennung durchgeführt werden.
HPLC mit einer Daicel Chiralcel OD-H ergab mit Hexan/PrOH = 99.7/0.3 als
Laufmittel bei einem Fluss von 0.3 ml/min für die beiden Enantiomere
Retentionszeiten von 36.8 min und 39.9 min. Die beiden Enantiomere wurden somit
vollständig getrennt.
Abb. 2.11 zeigt das HPL-Chromatogramm des Enantiomerengemisches bei den
Wellenlängen 230 nm (oben) und 210 nm (unten).
mAU
100-
80
60-
40-
20-1
Time ->
mAU
100-
80-
60-
40-
20-
0-
PECYC.D: MWD B, Sig=230,4 Ref=450,80
oOD^
œ
|CT3
20.00 40.00 60.00 80.00
PECYC.D: MWD C, Sig=210,4 Ref=450,80
CO —i
to°>
pO CT
iCO
Time -> 20.00
JlLU
40.00
-i 1 r
60.00 80.00
Abb. 2.11. HPL-Chromatogramme von racemischem 1-(4-Methoxyphenyl)-
bicyclo[4.1.0]heptan.
Da nun sowohl verschiedene Ylid-Komplexe als auch ein geeignetes prochirales
Substrat vorlagen, konnten die entsprechenden Reaktionsexperimente, die im
nächsten Abschnitt beschrieben sind, durchgeführt werden.

2. Eisen-Arsoniummethyl-Komplexe 45
2.5 Cyclopropanîerungsreaktionen
Zu Beginn sollte die Reaktivität der bekannten Eisenylid-Komplexe
[Fp(CH2PPh3)]BPh4 und [Fp(CH2AsPh3)]BPh4 gegenüber Styrol getestet werden.
Während im Fall des Phosphoniumylides keine Cyclopropanierung beobachtet
wurde, ergaben Reaktionsversuche mit [Fp(CH2AsPh3)]BPh4 (MeCN, 24 h, 100 °C)
Cyclopropylbenzol in kleinen Ausbeuten (< 5%).
Ein Vergleich der BPh/-, BF4- und Cl"-Salze des Kations [Fp(CH2AsPh3)]+ ergab die
mit Abstand besten Werte für [Fp(CH2AsPh3)]BF4. Nach 24 h bei 100 °C in MeCN
wurde mit dem BF4'-Salz eine Ausbeute von 14% beobachtet, während die
Ausbeuten mit dem BPh4- und dem Cl"-Salz unter 5% lagen.
Reaktionsversuche in verschiedenen Lösungsmitteln ergaben keine
Verbesserungen gegenüber Acetonitril. Während die Reaktion von
[Fp(CH2AsPh3)]BF4 mit Styrol in MeCN Cyclopropylbenzol in einer Ausbeute von
14% ergab, wurden in Methylenchlorid, Benzol und 1,4-Dioxan Ausbeuten unter
5% erhalten.
In einem zweiten Schritt wurden die Komplexe K1-K5 getestet. Die beobachteten
Ausbeuten von Cyclopropylbenzol waren nach 24 h bei 100 °C 14%, 47%, 76%,
84% und 83%. Diese Experimente zeigen, dass die Cyclopropanierungsreaktion
desto schneller verläuft, je elektronenziehender die Substituenten R am Arsen sind.
Die Tatsache, dass mit dem Komplex K5 eine kleinere Ausbeute erreicht wird als
mit K4, deutet darauf hin, dass, wenn die Konzentration des Methyleneisen-
Komplexes zu hoch ist, die Umsetzung zu Ethen oder Polymethylen und dem Arsin-
Komplex [FpAsR3]+ die Bildung von Cyclopropan vermindert. Dies wird angezeigt
durch die Bildung von [FpAs(4-CF3-Ph)3]BF4 in Kristallisationsversuchen mit K5 und
durch die grosse Intensität des Peaks m/z = 191 im Massenspektrum von K5, der
dem Methylenkomplex [FpCH2] entspricht (siehe Abschnitt 2.3.1).
Die Cyclopropanierung von Styrol mit dem SbF6"-Salz K6 ergab kleinere
Ausbeuten als mit dem analogen BF4'-Salz K4. Nach 12 h bei 100 °C in MeCN
wurden mit K4 63% mit K6 53% Cyclopropylbenzol erhalten.

2. Eisen-Arsoniummethyl-Komplexe 46
In der Cyclopropanierung von 1-Cyclohex-1-enyl-4-methoxybenzol durch K1, K7
und K8 wurden Ausbeuten von 38%, 48% und 18% beobachtet. Eine
Stabilisierung des Carben-Kations scheint die Ausbeute zu erhöhen. Eine
mögliche Erklärung für die kleinere Ausbeute mit K8 ist die Zersetzung des
entsprechenden Methyleneisen-Komplexes bei hohen Temperaturen.22
Reaktionsversuche mit den Molybdän- und Wolfram-Arsoniummethylkomplexen
K12 und K13 ergaben nach der selben Methode wie für die Komplexe K1-K5 kein
Cyclopropan.
Die Reaktionen der chiralen, nicht-racemischen Komplexe K9-K11 mit 1-
Cyclohex-1-enyl-4-methoxybenzol ergaben nach 5 d bei 100 °C 7%, 18% und 12%
racemisches 1-(4-Methoxyphenyl)bicyclo[4.1.0]heptan. Die Tatsache, dass
racemisches Produkt gebildet wurde, erklärt sich durch die Dissoziation des
Ferrocenylarsins, wodurch das reaktive, achirale Carben-Kation gebildet wird.
Weiter wurde auch hier beobachtet, dass je weniger elektronenstossend die
Substituenten am Arsen sind, desto schneller die Cyclopropanierung der Alkene
verläuft. Die wichtigsten Resultate sind in Tab. 2.7 zusammengefasst.
2. Eisen-Arsoniummethyl-Komplexe 47
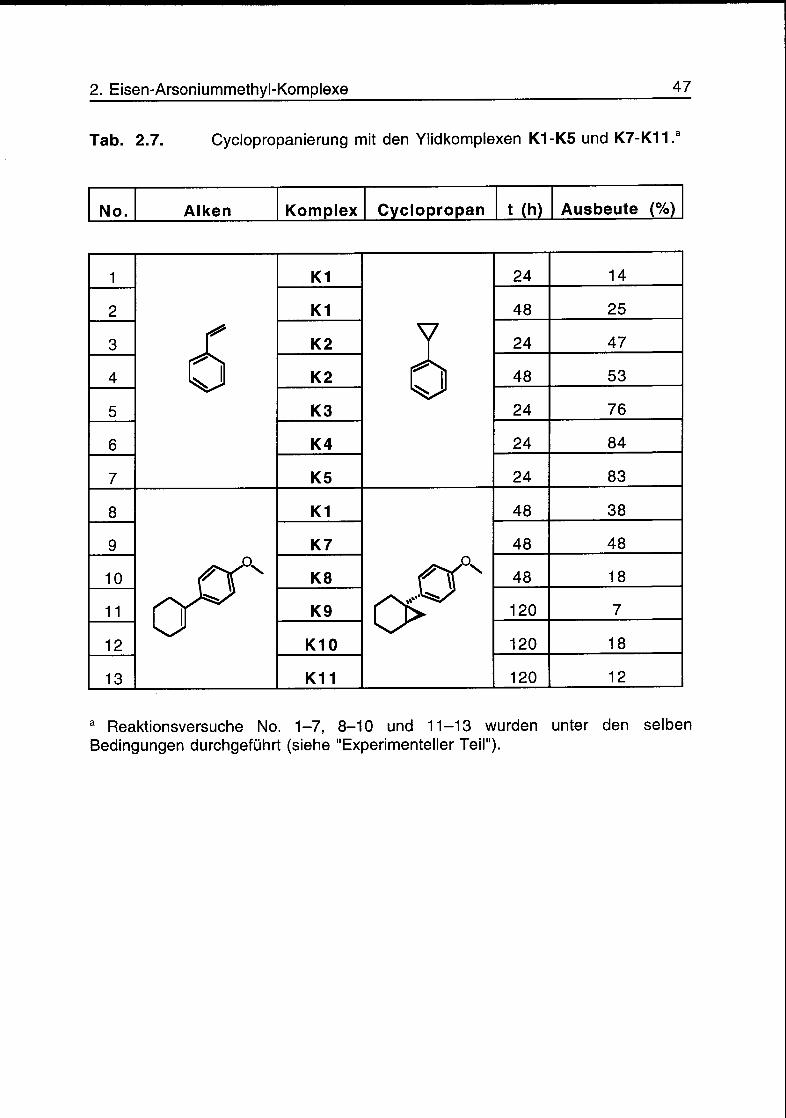
Tab. 2.7. Cyclopropanierung mit den Ylidkomplexen K1-K5 und K7-K11.a
No. Alken Komplex Cyclopropan t (h) Ausbeute (%)
1
6
K1
6
24 14
2 K1 48 25
3 K2 24 47
4 K2 48 53
5 K3 24 76
6 K4 24 84
7 K5 24 83
8
rv«
K1
0^
48 38
9 K7 48 48
10 K8 48 18
11 K9 120 7
12 K10 120 18
13 K11 120 12
a Reaktionsversuche No. 1-7, 8-10 und 11-13 wurden unter den selben
Bedingungen durchgeführt (siehe "Experimenteller Teil").

2. Eisen-Arsoniummethyl-Komplexe 48
2.6 Zusammenfassung und Schlussfolgerungen
Die Umsetzung von Styrol (CH3CN, 100 °C, 48 h) mit 1 equiv. [Fp(CH2AsPh3)]BF4
ergab 25% Cyclopropylbenzol. Neue Komplexe des Typs [Fp(CH2AsR3)]X wurden
synthetisiert und charakterisiert. Die Einführung von elektronenziehenden
Substituenten an den Phenylringen führte zu reaktiveren Komplexen. In der
Cyclopropanierung von Styrol wurden nach 24 h Ausbeuten von bis zu 84%
beobachtet. Wenn die Substituenten R am Arsen-Atom allerdings zu stark
elektronenziehend sind, scheint die unerwünschte Umsetzung des Methyleneisen-
Komplexes zu Polymethylen die Ausbeute an Cyclopropan zu vermindern.
Um stabilere kationische Carben-Komplexe zu erhalten, die vermutlich während
der Cyclopropanierungsreaktion gebildet werden, wurden die Komplexe K7 und
K8 hergestellt und charakterisiert. K7 und K8 wurden in der Cyclopropanierung
von 1-Cyclohex-1-enyl-4-methoxybenzol getestet. Während mit K7 eine
Verbesserung gegenüber dem Cyclopentadienid-Komplex erreicht wurde, zeigte
die Cyclopropanierung mit dem Pentamethylcyclopentadienid-Komplex K8 deutlich
kleinere Ausbeuten.
Chirale Arsoniummethyl-Komplexe wurden hergestellt. Reaktionsversuche mit
diesen und prochiralem 1-Cyclohex-1-enyl-4-methoxybenzol ergaben racemisches
1-(4-Methoxyphenyl)bicyclo[4.1.0]heptan. Die Chiralität am Arsonium induziert nicht
die Bildung von nicht-racemischen Cyclopropanen. Diese Resultate sind in
Uebereinstimmung mit den mechanistischen Studien von Barefield über die
Cyclopropanierung von Cycloocten durch [Fp(CH2SPh2)]BF4.7
Die erhaltenen Resultate der Reaktionsversuche deuten stark auf folgenden
Mechanismus der Cyclopropanierungsreaktion mit Komplexen des Typs
[Fp(CH2AsR3)]X hin: Das Triaryiarsin dissoziiert reversibel vom Ylidkomplex unter
Bildung des Eisencarbenkomplexes [FpCH2]+. Dieser Carbenkomplex reagiert mit
Olefinen zu Cyclopropanen, wobei das Kation [Fp]+ gebildet wird. Dieses Kation
kann nun mit dem Lösungsmittel oder mit freiem Arsin einen Komplex bilden. Eine
alternative Reaktion für den Carbenkomplex [FpCH2]+ ist die Reaktion mit einem
weiteren Eisencarben zum Ethen-Komplex [Fp(C2H4)]+ und zum Kation [Fp]+ oder zu
Polymethylen und zwei [Fp]+-Kationen.

2. Eisen-Arsoniummethyl-Komplexe 49
Eine katalytische Methylencyclopropanierung mit obigen Eisen-Arsoniummethyl-
Komplexen als direkte Vorläufer der cyclopropanierenden Spezies ist denkbar. Ein
vorgeschlagener Katalysezyklus ist in Schema 2.12 gezeigt.
[CpFe(CO)2(CH2AsR3)]X -^— [CpFe(CO)2CH2]X + AsR3
CH2AsR3 \ /
\/ AS AsR3
[CpFe(CO)2(S)]X -r— [CpFe(CO)2]X -t~— [CpFe(CO)2(AsR3)]X
Schema 2.12. Idee einer katalytischen Methylencyclopropanierung mit
Arsoniumyliden.
Verschiedene Voraussetzungen müssen allerdings erfüllt werden. Die wichtigste
darunter ist, dass der aus dem Carbenkomplex entstandene Komplex [Fp]X mit dem
Triarylarsoniummethylid zum Ylidkomplex reagiert. Weitere Bedingungen wären
eine nicht zu starke Koordination des Lösungsmittelmoleküls S oder des Arsins
AsR3 am Eisen(ll)-Zentrum, so dass [Fp]X überhaupt zur Verfügung steht.
Leider konnte [Fp(CH2AsPh3)]BF4 nicht aus [Fp(THF)]BF4 und dem
Arsoniummethylid erhalten werden.
Auf der Suche nach einer katalytischen Methylencyclopropanierung ohne
Diazomethan, sollten nun als mögliche Katalysatorvorläufer verschiedene
kationische Komplexe getestet werden. Die Metallzentren sollten aufgrund ihrer
bekannten Rolle in der Cyclopropanierung (wie Fe(ll), Ru(ll), Cu(l), Pd(ll), Rh(ll) und
Zn(ll)) oder aufgrund ihrer Fähigkeit zur Bildung von Ylidkomplexen (wie Mo(ll),
W(ll), Co(ll), Ni(ll) und Pt(ll)) ausgewählt werden. Als Methylenquellen wurden
Sulfonium-, Sulfoxonium-, Arsonium- und Stiboniumylide eingesetzt.

2. Eisen-Arsoniummethyl-Komplexe 50
2.7 Literatur
(1) Black, T. H. Aldrichimica Acta 1983, 16, 3-10.
(2) de Boer, T. J.; Backer, H. J. In Organic Syntheses; Wiley: New York, 1963;
Vol. Collect. Vol. 4, pp 250-253.
(3) Hudlicky, M. J. Org. Chem. 1980, 45, 5377-5378.
(4) Hudlicky, M. Org. Prep. Proc Int. 1982, 14, 354-356.
(5) O'Connor, E. J.; Brandt, S.; Helquist, P. J. Am. Chem. Soc 1987, 109, 3739-
3747.
(6) Mattson, M. N.; O'Connor, E. J.; Helquist, P. Org. Synth. 1992, 70, 177-185.
(7) McCarten, P.; Barefield, E. K. Organometallics 1998, 17, 4645-4648.
(8) Reger, D. L.; Culbertson, E. C. J. Organomet. Chem. 1977, 131, 297-300.
(9) Pelling, S.; Botha, C; Moss, J. R. J. Chem. Soc Dalton Trans. 1983, 1495-
1501.
(10) Friedrich, H. B.; Moss, J. R. J. Organomet. Chem. 1993, 453, 85-95.
(11) McCortney, B. A.; Jacobson, B. M.; Vreeke, M.; Lewis, E. S. J. Am. Chem.
Soc. 1990, 112, 3554-3559.
(12) Samaan, S. Methoden der organischen Chemie; 4 ed.; Thieme: Stuttgart,
1978; Vol. 13.
(13) Hayashi, T.; Kumada, M. Acc Chem. Res. 1982, 15, 395-401.
(14) Hayashi, T.; Mise, T.; Fukushima, M.; Kagotani, M.; Nagashima, N.; Hamada,
Y.; Matsumoto, A.; Kawakawi, S.; Konishi, M.; Yamamoto, K.; Kumada, M.
Bull. Chem. Soc. Jpn. 1980, 53, 1138-1151.
(15) Butler, I. R.; Cullen, W. R.; Rettig, S. J. Organometallics 1986, 5, 1320-1328.
(16) Gokel, G. W.; Marquarding, D.; Ugi, I. K. J. Org. Chem. 1972, 37, 3052-3058.
(17) Hayashi, T.; Mise, T.; Kumada, M. Tetrahedron Lett. 1976, 48, 4351-4354.
(18) Kursanov, D. N.; Parnes, Z. N.; Loim, N. M. Synthesis 1974, 633.
(19) O'Connor, E. J.; Helquist, P. J. Am. Chem. Soc. 1982, 104, 1869-1874.
(20) Allen, D. G.; Raston, C. L; Skelton, B. W.; White, A. H.; Wild, S. B. Aust. J.
Chem. 1984, 37, 1171-1181.
(21) Smit, F.; Stam, C. H. Acta Cryst. B1980, 36, 1254-1256.
(22) Guerchais, V.; Astruc, D. J. Chem. Soc Chem. Commun. 1985, 835-837.

3. Versuch der Entwicklung einer katalytischen
Methylencyclopropanierung mit Yliden
3.1 Einführung
Ausgehend von den Cyclopropanierungsreagenzien [Fp(CH2SR2)]X (mit R = Alkyl,
Aryl) und [Fp(CH2AsR3)]X (mit R = Aryl) sollte nun der zweite Schritt in der
Entwicklung einer katalytischen Methylencyclopropanierung unternommen werden.
[Fp(CH2SR2)]X und [Fp(CH2AsR3)]X sind formal Komplexe von Eisen(ll) mit einem
Ylid. Im ersten Fall ist das koordinierte Ylid ein Sulfonium- im zweiten Fall ein
Arsoniumylid. Neben kationischen Fe(ll)-Komplexen sollten - sowohl wegen der
bekannten Rolle von Ru-Komplexen in der Cyclopropanierung als auch wegen der
Zugehörigkeit von Ru zur Eisengruppe - auch kationische Ru(ll)-Komplexe als
Katalysatorvorläufer in der Cyclopropanierung mit Yliden getestet werden. Eine
Auswahl möglicher Fe(ll)- und Ru(ll)-Komplexe mit einem koordinierten
Lösungsmittelmolekül (S = THF, MeCN etc.) ist in Abb. 3.1 gezeigt. Diese Komplexe
wurden zu Beginn des Projektes vorgeschlagen.
I I
M = Fe, Ru
Abb. 3.1. Mögliche kationische Fe(ll)- und Ru(ll)-Komplexe als Katalysator¬
vorläufer in der Cyclopropanierung mit Methyliden.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 52
Als zu testende Katalysatorvorläufer wurden neben Fe(ll)- und Ru(ll)-Komplexen
Komplexe mit Metallzentren, die sich durch ihre bekannte Rolle in der
Cyclopropanierung (wie Cu(l), Pd(ll), Rh(ll) und Zn(ll)) oder durch ihre Fähigkeit zur
Bildung von Ylidkomplexen (wie Mo(ll), W(ll), Co(ll), Ni(ll) und Pt(ll)) auszeichnen,
ausgewählt. Neben Wolframarsoniumylid-Komplexen und dem im vorherigen
Kapitel vorgestellten Molybdänarsoniumylid-Komplex gibt es auch Ylidkomplexe
von Co, Ni, Pt und Cu. Bis(phosphoniummethylid)-Komplexe dieser Metalle sind
bekannt (Abb. 3.2). Besonders hervorzuheben ist, dass die Herstellung der Co(ll)-,
Ni(ll)- und Cu(l)-Ylidkomplexe direkt mit dem entsprechenden Phosphonium-
methylid gelingt. So wird aus Cobalt(ll)-chlorid und zwei Aequivalenten
Trimethylphosphoniummethylid der extrem luftempfindliche Komplex
Bis(chloro)bis(trimethylphosphoniummethylid)cobalt(ll) erhalten.1 Die Umsetzung
von Dichlorobis(trimethylphosphin)nickel(ll) mit zwei Aequivalenten
Trimethylphosphoniummethylid ergibt c/s-Chloro(trimethylphosphin)bis(trimethyl-
phosphoniummethylid)nickel(ll)-chlorid.2 Dieser gelbe Komplex ist oxidations- und
hydrolyseempfindlich jedoch thermisch stabil bis 125 °C. Die Behandlung von
Trimethylphosphinkupfer(l)-chlorid mit einer doppeltmolaren Menge Trimethyl¬
phosphoniummethylid ergibt den ebenfalls luftempfindlichen Komplex Bis(trimethyl-
phosphoniummethylid)kupfer(l)-chlorid.3 Im Falle der Platin-Ylid-Komplexe verläuft
die Synthese über Substitution an Haloalkyl-Komplexen. Sowohl c/'s-lodo-
(trimethylphosphin)bis(triphenylphosphoniummethylid)platin(ll)-iodid als auch cis-
Bis(trimethylphosphin)bis(triphenylphosphoniummethylid)platin(ll)-iodid wurden
aus (COD)Pt(CH2l)2 und dem entsprechenden Phosphin erhalten.4

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 53
©CI 2Q/CH2PMe3
CI XH2PMe3
©CI Q^CHgPMeg
Ni fà
Me3P CH2PMe3
©CI
© © ©Me3PCH2—Cu—CH2PMe3 CI
0
©
Ph3P^0 CH2PPh3
I CH2PPh3
©
©Me3P. CH2PPh3
/p\ ©Me3P CH2PPh3
21.©
Abb 3.2. Ylidkomplexe von Co(ll), Ni(ll), Pt(ll) und Cu(l).
Zusammen mit diesen Komplexen sollten Sulfonium-, Sulfoxonium-, Arsonium- und
Antimoniumylide als Methylenquellen in Reaktionsversuchen zur Entwicklung einer
katalytischen Cyclopropanierung eingesetzt werden. Wie in der Einführung erwähnt
(1.6 Cyclopropanierung mit Yliden) war die Cyclopropanierung eine der ersten
Ylidreaktionen. Die Cyclopropanierung mit Schwefelyliden wurde vorgestellt. Im
Folgenden soll nun näher auf die Arsonium- und Antimoniumylide eingegangen
werden.
Henry und Wittig veröffentlichten 1960 die Resultate ihrer Versuche der
Olefinierung von Benzophenon mit Triphenylarsoniummethylid und Triphenyl-
antimoniummethylid anstelle des analogen Phosphorylids.5 Die Ylide wurden dabei
aus dem entsprechenden lodid-Salz mit einem Aequivalent PhLi in Et20 hergestellt.
Die Reaktion des Arsoniummethylids mit einem Aequivalent Benzophenon ergab
ca. 65% Diphenylacetaldehyd und nur 25% Diphenylethylen. Im Falle des
Antimoniumylids wurden 92% Diphenylacetaldehyd und Spuren des Olefins
gefunden.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 54
Im Jahr 1968 berichteten Schmidbaur und Tronich die Herstellung und Isolierung
von Trimethylarsoniummethylid.67 Die Reaktion von Trimethylarsonium-
(trimethylsilyl)methylid mit Trimethylsilanol führte unter Desilylierung des Ylids und
unter Bildung von Hexamethyldisiloxan zum Arsoniummethylid. Trimethyl¬
arsoniummethylid ist eine farblose, kristalline Verbindung, ist luftempfindlich und
zersetzt sich langsam bei einer Temperatur über 38 °C. Die Hauptprodukte bei
einer thermischen Zersetzung sind Trimethylarsin und Ethylen.
Erst sieben Jahre später, 1975, veröffentlichten Yamamoto und Schmidbaur die
Herstellung und Isolierung von Triphenylarsoniummethylid.8 Die Umsetzung von
Methyltriphenylarsoniumbromid mit Natriumamid ergab nach Umkristallisation in
Et20 das Arsoniumylid als gelbe Kristalle in ca. 50% Ausbeute. Wie vorerst in einer
Fussnote angedeutet ("Kürzere oder längere Reaktionszeiten und tiefere oder
höhere Reaktionstemperaturen führen entweder zu viel kleineren Ausbeuten oder
einem totalen Misslingen der Synthese")8 oder später im Text vermerkt ("Es ist
bemerkenswert, dass die Synthese nur bei genauer Einhaltung bestimmter,
empirisch gefundener Bedingungen gelingt")9 ist die Synthese heikel.
Triphenylarsoniummethylid ist thermisch instabil. Die Zersetzungsprodukte
Triphenylarsin, Polymethylen und Ethylen wurden beobachtet. In späteren
Publikationen wurden Lösungen von Triphenylarsoniummethylid aus
Methyltriphenylarsonium-Iodid und Phenyllithium in Et2010 oder aus
Methyltriphenylarsonium-Tetrafluoroborat und Kalium-Bis(trimethylsilyl)amid in THF
eingesetzt.11
Im Jahr 1977 wurde die Herstellung von Triethylarsoniummethylid aus
Triethylmethylarsonium-bromid und Natriumamid veröffentlicht.9 Dieses Ylid
unterscheidet sich von den vorhergehenden durch seine grössere Stabilität. Die
farblose Flüssigkeit ist bei 37 °C/0.001 mbar unzersetzt destillierbar. Auch dieses
Ylid ist stark luftempfindlich.
Ein Ueberblick über Herstellung, Struktur und Reaktionen der Arsoniumylide wurde
1982 von Huang und Shen veröffentlicht.12

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 55
Mit den oben kurz vorgestellten Komplexen und Methylendonoren sind
grundsätzlich zwei Katalysezyklen für die Cyclopropanierung denkbar (siehe
Schema 3.1). Die Reaktion des Katalysatorvorläufers [M] mit dem Ylid ergibt den
Ylid-Komplex. Aus dem Ylid-Komplex kann nun ein Carben-Komplex entstehen,
der mit dem Olefin zum Cyclopropan reagiert. Eine andere Möglichkeit ist die
Koordination des Olefins am Ylid-Komplex mit nachfolgender Cyclopropanbildung.
In beiden Fällen beginnt mit der Koordination eines Ylids am Katalysator ein
weiterer Katalysezyklus.
[M]
M-CH2-ELn
LnE-CH2 LnE=CH2
[M-CH2-ELn]
[M=CH2]
Schema 3.1. Zwei mögliche Zyklen einer katalytischen Methylencyclo¬
propanierung mit Yliden (E = S, As, Sb) und Lewis-sauren Komplexen [M].
Die Synthese der Komplexe und der Ylide und deren Verwendung in
Reaktionsversuchen ist in den folgenden drei Abschnitten (3.2 - 3.4) dargestellt.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 56
3.2 Komplexsynthese
Viele Substitutionsreaktionen von Komplexen des Typs [CpFe(CO)2X] (X = CI, Br, I)
mit Phosphinen, Phosphiten, Aminen, Arsinen und Stibinen (ER3) zu Komplexen
des Typs [CpFe(CO)2ER3]X und [CpFe(CO)(ER3)X] sind bekannt.13"18 So ergibt die
Umsetzung von [CpFe(CO)2Br] mit einem Aequivalent Triphenylphosphin den
Komplex [CpFe(CO)(PPh )Br] in einer Ausbeute von 51 %.14 Bei Komplexen des
Typs [CpFe(CO)2X] steht also eine Koordinationsstelle zur Verfügung. Die
Komplexe [CpFe(CO)2X] (X = CI, Br, I) sollten daher hergestellt und als mögliche
Katalysatorvorläufer getestet werden. Neben den Pentamethylcyclopentadienyl-
Analogen sollten auch Cyclopentadienyl-Eisen(ll)-Komplexe, in denen ein oder
zwei Carbonyle durch einen Phosphinliganden substituiert sind, eingesetzt werden.
Eine Uebersicht der Komplexe ist in Abb. 3.3 dargestellt.
Die Herstellung der Komplexe [CpFe(CO)2X] (X = CI, Br, I) aus dem dinuklearen
[CpFe(CO)2]2 verläuft über die oxidative Spaltung der Metall-Metall-Bindung.19
Während die Bromo- und lodo-Derivate [CpFe(CO)2Br] und [CpFe(CO)2l] durch
direkte Oxidation mit Br2 bzw. I2 gewonnen werden, wird für die Synthese von
[CpFe(CO)2CI] die Oxidation des zweikernigen Fp2 mit Sauerstoff in Gegenwart von
HCl bevorzugt. Mit letzterer Methode wird relativ zu der direkten Umsetzung von Fp2
mit Cl2 die doppelte Ausbeute erhalten.
Eine völlig verschiedene Methode wurde für die Synthese des
Pentamethylcyclopentadienyl-Analogen [(C5Me5)Fe(CO) CI] angewandt. Hier führte
die oxidative Addition von 5-Chlor-1,2,3,4,5-pentamethyl-1,3-cyclopentadien an
Pentacarbonyleisen zum gewünschten Produkt.20 Während diese Reaktion selbst in
siedendem THF nicht eintritt, kann [(C5Me5)Fe(CO)2CI] bereits nach zwei Stunden in
guten Ausbeuten in Form eines hellroten Pulvers isoliert werden, wenn die
Reaktionslösung mit einer 150-W-Photolampe bestrahlt wird.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 57
Neben der am obigen Beispiel der Synthese von [CpFe(CO)(PPh3)Br] gezeigten
Substitution eines Carbonyls durch ein tertiäres Phosphin ist auch eine zweifache
Substitution zu einem carbonyl-freien Komplex möglich. Diese Herstellung doppelt¬
substituierter Komplexe durch direkte Substituierung ist allerdings schwieriger als
die Synthese einfach-substituierter Komplexe. Längere Reaktionszeiten, höhere
Reaktionstemperaturen oder UV-Bestrahlung sind oft nötig.21 Eine Methode zur
Herstellung von [CpFe(dppe)CI] ist dann auch die Umsetzung von [CpFe(CO)2CI]
mit dppe in Benzol unter UV-Bestrahlung.22 Hier wurde allerdings eine andere
Synthese bevorzugt, welche von der Herstellung von FeCI2(dppe) aus
wasserfreiem FeCI2 und dppe in Aceton/Chloroform ausgeht. Die Umsetzung von
FeCI2(dppe) mit einem Aequivalent TI(C5H5) in Benzol bei Raumtemperatur ergab
[CpFe(dppe)CI] als schwarzen Feststoff in bis zu 80% Ausbeute.23 Die analoge
Synthese des Pentamethylcyclopentadienyl-Komplexes wurde von Roger et al.
berichtet.24 Die Reaktion von FeCI2(dppe) mit Li(C5Me5) in THF ergab
[(C5Me5)Fe(dppe)CI] als dunkelgrünen Feststoff in 85% Ausbeute. Die Reaktivität
des Komplexes wiederspiegelt die Labilität der Fe-Cl-Bindung. Acetonitril
substituiert das Chlorid in weniger als einer Minute. Mit Lil und LiMe reagiert der
Komplex zu [(C5Me5)Fe(dppe)l] bzw. [(C5Me5)Fe(dppe)Me]. Behandelte man aber
(/V,/V'-Dicyclohexylethylendiimin)dichloroeisen(ll)25 mit einem Aequivalent
Thalliumcyclopentadienid entstand nicht das gewünschte [CpFe(A/,/V-
Dicyclohexylethylendiimin)CI]. Die Reaktion ergab neben einem Aequivalent TICI je
ein halbes Aequivalent Eduktkomplex, Ferrocen und N,N'-
Dicyclohexylethylendiimin.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 58
OC 4OC
X = CI
X = Br
X = l
Fe.
CSS
oc- 4 xi
oc
OC à Br
Ph3P
?SS
4 ^Cl ^Fe\ri
Ph2/P frh, Ph2p- /Cl
(yPPh2/ PPh2
Abb. 3.3. Cyclopentadienyl- und Pentamethylcyclopentadienyl-Eisen(ll)-
Halogenid-Komplexe.
Auch Komplexe von Fe(ll) und Ru(ll) mit einem oder mehreren koordinierten
Lösungsmittelmolekülen sollten hergestellt werden. Eine Uebersicht dieser
Komplexe ist in Abb. 3.4 gegeben. Für beide hypothetischen Katalysezyklen im
Schema 3.1 ist die Bildung eines Ylid-Komplexes Voraussetzung. Mit dem Komplex
[CpFe(CO)2(THF)]BF4 wurde im Falle von Tnphenylphosphoniummethylid der
gewünschte Ylid-Komplex isoliert.26 Die Komplexe dieses Typs waren als
Katalysatorvorläufer daher besonders vielversprechend.
Das THF-Addukt [CpFe(CO)2(THF)]BF4 wurde aus der Reaktion von [CpFe(CO)2l]
und AgBF4 in THF isoliert.27 Der rote Feststoff ist hygroskopisch und nur kurze Zeit
luftstabil. [CpFe(CO)2(THF)]BF4 reagiert mit vielen Olefinen durch Substitution des
Lösungsmittelmoleküls zum Olefin-Komplex.27,28 Der Styrol-Komplex
[CpFe(CO)2(Styrol)]BF4 wurde mit einer Ausbeute von 86% als gelber Feststoff
erhalten.28 Die Herstellung des Pentamethylcyclopentadienyl-Komplexes
[(C5Me5)Fe(CO)2(THF)]BF4 gelang analog aus [(C5Me5)Fe(CO)2l] und AgBF4 in
THF.29
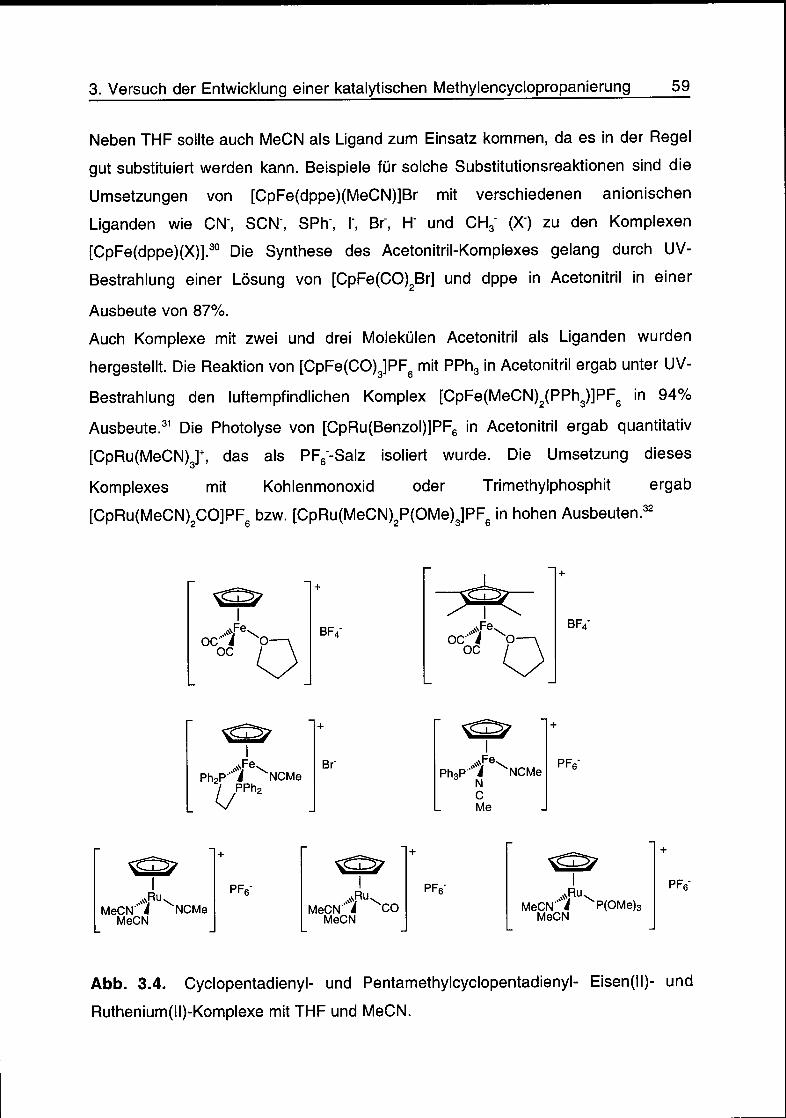
3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 59
Neben THF sollte auch MeCN als Ligand zum Einsatz kommen, da es in der Regel
gut substituiert werden kann. Beispiele für solche Substitutionsreaktionen sind die
Umsetzungen von [CpFe(dppe)(MeCN)]Br mit verschiedenen anionischen
Liganden wie CN", SCN-, SPh", I", Br, H' und CH3" (X) zu den Komplexen
[CpFe(dppe)(X)].30 Die Synthese des Acetonitril-Komplexes gelang durch UV-
Bestrahlung einer Lösung von [CpFe(CO)2Br] und dppe in Acetonitril in einer
Ausbeute von 87%.
Auch Komplexe mit zwei und drei Molekülen Acetonitril als Liganden wurden
hergestellt. Die Reaktion von [CpFe(CO)3]PF6 mit PPh3 in Acetonitril ergab unter UV-
Bestrahlung den luftempfindlichen Komplex [CpFe(MeCN)2(PPh3)]PF6 in 94%
Ausbeute.31 Die Photolyse von [CpRu(Benzol)]PF6 in Acetonitril ergab quantitativ
[CpRu(MeCN)3]+, das als PF6-Salz isoliert wurde. Die Umsetzung dieses
Komplexes mit Kohlenmonoxid oder Trimethylphosphit ergab
[CpRu(MeCN) CO]PFfi bzw. [CpRu(MeCN) P(OMe) 1PFR in hohen Ausbeuten.32
OC 4OC
BF4BF4
AFe>PhaP-'i
(/Ph2^NCMe
Br
I
Ph3P• i ^NCMeN
C
Me
PF6
MeCN' 4 NCMe
MeCN
PFfi>iRu\
MeCN' 4 CO
MeCN
PF6-I
,ARu\MeCN' 4 P(OMe)3
MeCN
PFfi
Abb. 3.4. Cyclopentadienyl- und Pentamethylcyclopentadienyl- Eisen(ll)- und
Ruthenium(ll)-Komplexe mit THF und MeCN.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 60
Da die Ylidkomplexe [CpM(CO)3(CH2AsPh3)]BF4 (M = Mo, W) zwar hergestellt
werden konnten, aber leider nicht als Cyclopropanierungsreagenzien aktiv waren,
sollten die Komplexe [CpM(CO)3(MeCN)]BF4 (M = Mo, W) hergestellt und als
mögliche Katalysatorvorläufer mit anderen als Arsoniumyliden getestet werden. Die
Hydrid-Komplexe [CpM(CO)3H] (M = Mo, W) wurden durch Reaktion der Anionen
[CpM(CO)3]" (M = Mo, W) mit Essigsäure erhalten.33 Sublimation der Rohprodukte
ergab die reinen Hydride. Während das Molybdän-Hydrid stark luftempfindlich ist,
kann der Wolfram-Komplex kurze Zeit an der Luft belassen werden. Hydrid-
Abstraktion mit Triphenylcarbenium-tetrafluoroborat ergibt in Gegenwart von
Acetonitril die Komplexe [CpM(CO)3(MeCN)]BF4 (M = Mo, W) in hohen
Ausbeuten.34 Der Syntheseweg ist in Schema 3.2 dargestellt.
CH3C02HTHF THF
M(CO)6 + NaC5H5 *- Na[CpM(CO)3] ^ CpM(CO)3H
65 °C, 24 h RT, 2 h
CH2CI2
CpM(CO)3H + Ph3C+BF4- + MeCN [CpM(CO)3(MeCN)]BF4-30 °C, 1 h
M = Mo: 87%
M = W: 88%
Schema 3.2. Herstellung der Komplexe [CpM(CO)3(MeCN)]BF4 aus M(CO)6
(M = Mo, W).

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 61
3.3 Synthese von Triarylmethylarsonium-Salzen und Yliden
Sulfonium-, Sulfoxonium-, Arsonium- und Antimoniumylide sollten als
Methylenquellen in den Cyclopropanierungsversuchen mit den oben vorgestellten
Komplexen zum Einsatz kommen. Die wichtigste Methode zur Synthese von Yliden
ist die sogenannte Salzmethode. Die Reaktion eines Methylsulfonium-,
Methylsulfoxonium-, Methylarsonium- oder Methylantimoniumsalzes mit einer
geeigneten Base ergibt das jeweilige Methylid. Aus diesem Grund wurden
vorgängig die entsprechenden Salze hergestellt.
Das Trialkylsulfoniumsalz Trimethylsulfonium-iodid wurde aus der Alkylierung von
Dimethylsulfid mit Methyliodid erhalten. Aus equimolaren Mengen der beiden
Edukte wird nach 12 h bei RT das Salz in quantitativer Ausbeute erhalten.35 Die
analoge Reaktion von Dimethylsulfoxid mit Methyliodid 3 d unter Rückfluss ergab
Trimethylsulfoxonium-iodid in 54% Ausbeute.36,37 Das Salz (Dimethylamino)-
(methyl)phenyloxosulfonium-tetrafluoroborat wurde in drei Stufen aus
(Methyl)phenylsulfoximin erhalten. Eine erste Alkylierung mit Trimethyl-oxonium-
tetrafluoroborat in Methylenchlorid ergab (Methylamino)(methyl)phenyl-
oxosulfonium-tetrafluoroborat, das mit Natronlauge zu N,S-Dimethyl S-Phenyl
Sulfoximin neutralisiert wurde. Eine zweite Methylierung ergab das gewünschte
(Dimethylamino)(methyl)phenyloxosulfonium-tetrafluoroborat.38
Die Umsetzung von Triphenylarsin mit Methylbromid und Methyliodid ergab die
Salze Methyltriphenylarsonium-bromid5'39 und -iodid.5 Da bei den Cyclo¬
propanierungsversuchen mit den Eisen-Triarylarsoniummethyl-Komplexen in
Kapitel 2 (2. Eisen-Arsoniummethyl-Komplexe - Synthese, Struktur und Reaktivität
gegenüber Olefinen) festgestellt wurde, dass elektronegative Substituenten an den
Phenylringen am Arsin die Cyclopropanierungsreaktion beschleunigen, sollten
auch Versuche mit den entsprechenden Yliden durchgeführt werden. Die
Arsoniumsalze [CH3As(4-F-Ph)3]l (S1), [CH3As(4-F-Ph)3]BF4 (S2), [CH3As(4-CI-
Ph)3]l (S3), [CH3As(4-CI-Ph)3]BF4 (S4) und [CH3As(3-CF3-Ph)3]BF4 (S5) wurden
durch Behandlung der entsprechenden Arsine mit lodmethan oder
Trimethyloxoniumtetrafluoroborat erhalten (Abb. 3.5).
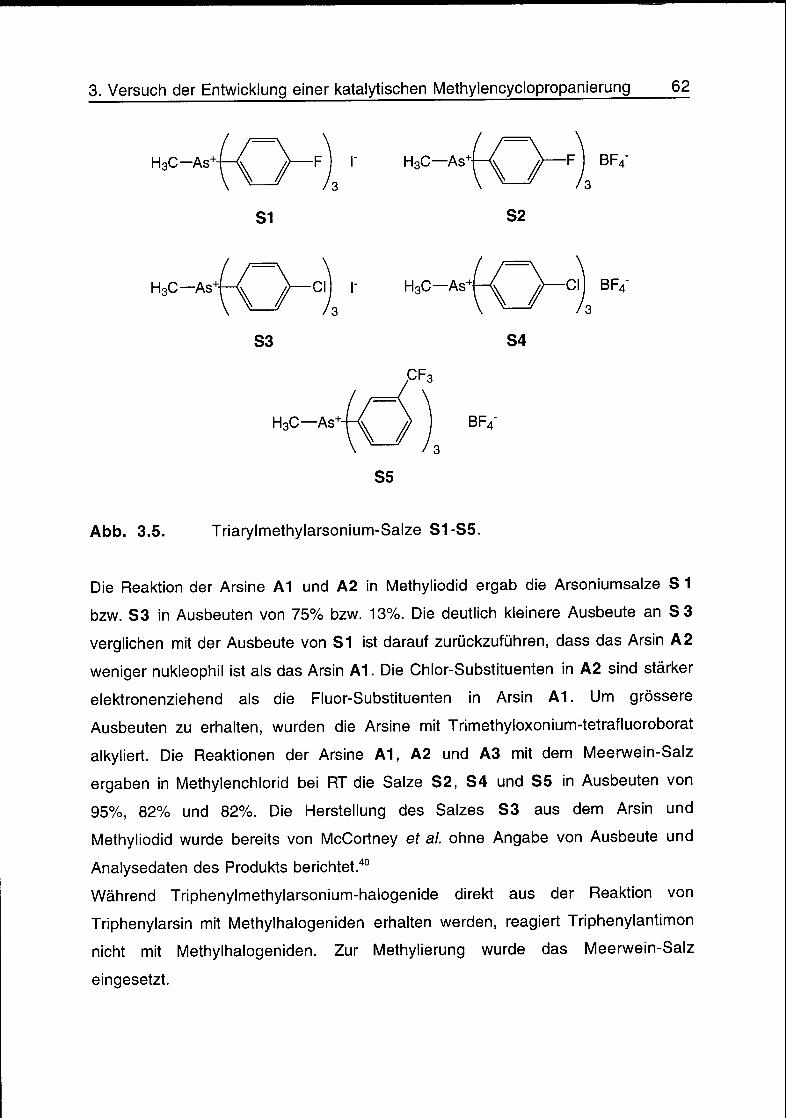
3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 62
H3C-As+-(—^ h—Fl I" H3C—As+-
S1 S2
Abb. 3.5. Triarylmethylarsonium-Salze S1-S5.
Die Reaktion der Arsine A1 und A2 in Methyliodid ergab die Arsoniumsalze S 1
bzw. S3 in Ausbeuten von 75% bzw. 13%. Die deutlich kleinere Ausbeute an S3
verglichen mit der Ausbeute von S1 ist darauf zurückzuführen, dass das Arsin A2
weniger nukleophil ist als das Arsin A1. Die Chlor-Substituenten in A2 sind stärker
elektronenziehend als die Fluor-Substituenten in Arsin A1. Um grössere
Ausbeuten zu erhalten, wurden die Arsine mit Trimethyloxonium-tetrafluoroborat
alkyliert. Die Reaktionen der Arsine A1, A2 und A3 mit dem Meerwein-Salz
ergaben in Methylenchlorid bei RT die Salze S2, S4 und S5 in Ausbeuten von
95%, 82% und 82%. Die Herstellung des Salzes S3 aus dem Arsin und
Methyliodid wurde bereits von McCortney et al. ohne Angabe von Ausbeute und
Analysedaten des Produkts berichtet.40
Während Triphenylmethylarsonium-halogenide direkt aus der Reaktion von
Triphenylarsin mit Methylhalogeniden erhalten werden, reagiert Triphenylantimon
nicht mit Methylhalogeniden. Zur Methylierung wurde das Meerwein-Salz
eingesetzt.
i

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 63
Die Reaktion von Triphenylantimon mit Trimethyloxonium-tetrafluoroborat in
Dichloromethan bei RT ergab das Tetrafluoroborat-Salz, das mit einem
Ueberschuss Natriumiodid in H20 unter Anionenaustausch zum gewünschten
Methyltriphenylantimonium-iodid5 reagierte. Das entsprechende [MeBiPh3]l konnte
auf keine der beiden geschilderten Arten erhalten werden.
Da nun die benötigten Salze vorhanden waren, konnte mit der Ylid-Synthese
begonnen werden.
Die Herstellung von Dimethylsulfoniummethylid (Me2S=CH2) gelang aus
equimolaren Mengen von Trimethylsulfoniumiodid und NaH in
Dimethylsulfoxid/THF bei 0 °C.41 Das Ylid zersetzt sich bei RT innerhalb weniger
Minuten unter der Bildung von Ethen. Bei 0 °C oder darunter ist es allerdings
deutlich stabiler.
Die entsprechende Umsetzung von Trimethylsulfoxonium-iodid mit Natriumhydrid
zu Dimethylsulfoxoniummethylid (Me2S(0)=CH2) wurde in Dimethylsulfoxid bei RT
durchgeführt.37,42 Lösungen des Ylids sind in einer inerten Atmosphäre bei 0 °C
während mehreren Monaten stabil. Bei RT wird die Zersetzung des Ylids nach etwa
einer Woche feststellbar. Lösungen von Dimethyloxosulfoniummethylid in
Dimethylsulfoxid, THF, Dichloromethan, Benzol und Acetonitril wurden hergestellt.
Die Synthese von (Dimethylamino)phenyloxosulfoniummethylid
((Me2N)(Ph)S(0)=CH2) aus (Dimethylamino)(methyl)phenyloxosulfonium-tetra-
fluoroborat mit Natriumhydrid wurde sowohl in THF als auch in Dimethylsulfoxid bei
RT beschrieben.38 Dieses Ylid ist in Dimethylsulfoxid unter Luft- und
Feuchtigkeitsausschluss bei RT über Monate stabil.
Die Analyse der hergestellten Sulfoxoniumylid-Lösungen erfolgte mittels Titration
(0.1 M HCl gegen Phenolphtalein) und Durchführung einer Referenzreaktion, der
Cyclopropanierung von (-)-Carvon ((fî)-5-lsopropenyl-2-methyl-2-cyclohexenon)
mit einem Aequivalent des Ylids. Sämtliche Ylidlösungen wurden in der glove-box
bei -20 °C gelagert.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 64
Triphenylarsoniummethylid (Ph3As=CH2) wurde sowohl in situ als auch isoliert
eingesetzt. Die Behandlung von Methyltriphenylarsonium-iodid oder -bromid
suspendiert in Diethylether mit einem Aequivalent Phenyllithium ergab eine gelbe
Suspension.5 Die Isolierung von Triphenylarsoniummethylid gelang nach der
Umsetzung von Methyltriphenylarsonium-bromid mit einem Aequivalent Natrium-
Bis(trimethylsilyl)amid in Diethylether bei RT. Die Reaktionsmischung wurde 20
Minuten gerührt. Sämtliche Syntheseschritte wurden in Dunkelheit ausgeführt.
Filtration, Einengen und Trocknen am HV ergaben das leuchtendgelbe Ylid, das
mittels 1H- und 13C-identifiziert wurde. Aufgrund seiner Wasser- und
Lichtempfindlichkeit wurde es in der glove-box bei -20 °C im Dunkeln aufbewahrt.
Die Herstellung der Ylide (4-F-Ph)3As=CH2, (4-CI-Ph)3As=CH2 und (3-CF3-
Ph)3As=CH2 erfolgte ausschliesslich in situ durch Behandlung der entsprechenden
Salze in Diethylether mit einer equimolaren Menge Natrium-Bis(trimethylsilyl)amid.
Ihre Bildung konnte nicht nachgewiesen werden. Elektronenziehende
Substituenten am Arsen destabilisieren die Arsoniummethylide. 13C-NMR
Untersuchungen von Yamamoto und Schmidbaur8 an Triphenylarsoniummethylid
haben ergeben, dass die Struktur des Ylids am ylidischen Kohlenstoffatom
pseudotetraedrisch ist, was mit einer sp3-Hybridisierung einhergeht. Demzufolge ist
der Beitrag der Resonanzstruktur II mit Ladungstrennung an die Gesamtstruktur des
Ylids grösser als der Beitrag der Resonanzstruktur I mit einer Doppelbindung (Abb.
3.6). Im letzteren Fall wäre das Ylid am ylidischen Kohlenstoffatom planar (sp2).
Elektronenziehende Substituenten am partiell positiv-geladenen Arsen führen auf
Kosten der Stabilität zu einer grösseren Polarisierung.
Ph3As=CH2 - - Ph3As—CH2
I II
®-H + -&Ph3As—C* Ph3As—C//-„.u
Abb. 3.6. Resonanzstrukturformeln von Triphenylarsoniummethylid.
Die in stfu-Synthese von Triphenylantimoniummethylid (Ph3Sb=CH2) gelang aus
dem lodid-Salz mit einem Aequivalent Phenyllithium in Et20 bei RT.5

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 65
3.4 Reaktionsversuche
In einer ersten Reihe von Reaktionsversuchen sollten die bei RT stabilen
Sulfoxoniummethylide Dimethylsulfoxoniummethylid und (Dimethylamino)phenyl-
oxosulfoniummethylid zum Einsatz kommen. Die eingesetzten Komplexe waren mit
Ausnahme des Styrol-Komplexes [CpFe(CO)2(Styrol)]BF4 Cyclopentadienyl- oder
Pentamethylcyclopentadienyl- Eisen(ll)- und Ruthenium(ll)- Komplexe mit einem
Halogenid oder einem Lösungsmittelmolekül als Ligand. Als Modellsubstrate
wurden Styrol und 1-Methyl-1-cyclohexen ausgewählt. Zu einer Lösung des Olefins
wurde in der glove-box bei RT eine equimolare Menge des Schwefelylids in
Lösung gegeben. Im Falle von Dimethylsulfoxoniummethylid wurde eine Lösung
des Ylids im jeweiligen Lösungsmittel, im Falle von (Dimethylamino)phenyloxo-
sulfoniummethylid eine Lösung in THF verwendet. Diese Reaktionsmischung
wurde mit 0.1 equiv. des Komplexes versetzt und in einem Schlenk mit Young-
Hahn 24 h bei RT gerührt. Die durchgeführten Reaktionsversuche sind in Tab. 3.1
zusammengefasst. Nach Abschluss der Reaktion wurde die Reaktionsmischung mit
Pentan versetzt und über Silicagel filtriert. Diese Lösung wurde dann mittels GC-
MS analysiert. In keinem der Reaktionsversuche wurde Cyclopropylbenzol oder 1-
Methyl-bicyclo[4.1.0]heptan gefunden.
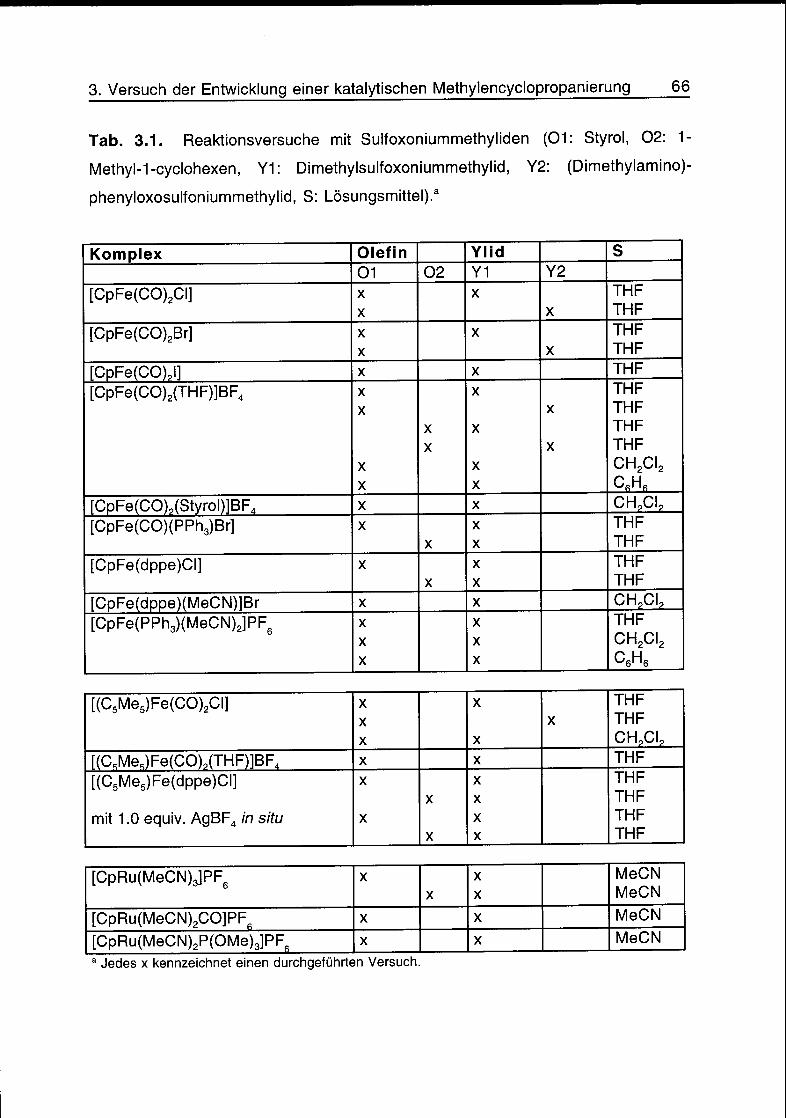
3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 66
Tab. 3.1. Reaktionsversuche mit Sulfoxoniummethyliden (01: Styrol, 02: 1-
Methyl-1-cyclohexen, Y1: Dimethylsulfoxoniummethylid, Y2: (Dimethylamino)-
phenyloxosulfoniummethylid, S: Lösungsmittel).3
Komplex Olefin Ylid S
01 02 Y1 Y2
[CpFe(CO)2CI] X
X
X
X
THF
THF
[CpFe(CO)2Br] X
X
X
X
THF
THF
[CpFe(CO)?l] X X THF
[CpFe(CO)2(THF)]BF4 X
X
X
X
X
X
X
X
X
X
X
X
THF
THF
THF
THF
CH2CI2CfiHR
[CpFe(CO)2(Styrol)]BF4 X X CHpClp
[CpFe(CO)(PPh3)Br] X
X
X
X
THF
THF
[CpFe(dppe)CI] X
X
X
X
THF
THF
[CpFe(dppe)(MeCN)]Br X X CH?CI?
[CpFe(PPh3)(MeCN)2]PF6 X
X
X
X
X
X
THF
CH2CI2C6H6
[(C5Me5)Fe(CO)2CI] x
x
X
X
X
X
THF
THF
CH?Clp
[(C,Me,)Fe(CO)9(THF)]BF4 X X THF
[(C5Me5)Fe(dppe)CI]
mit 1.0 equiv. AgBF4 in situ
X
X
X
X
X
X
X
X
THF
THF
THF
THF
[CpRu(MeCN)3]PF6 x
X
X
X
MeCN
MeCN
[CpRu(MeCN)2CO]PFfi x X MeCN
[CpRu(MeCN)2P(OMe)3]PFR x X MeCN
Jedes x kennzeichnet einen durchgeführten Versuch.
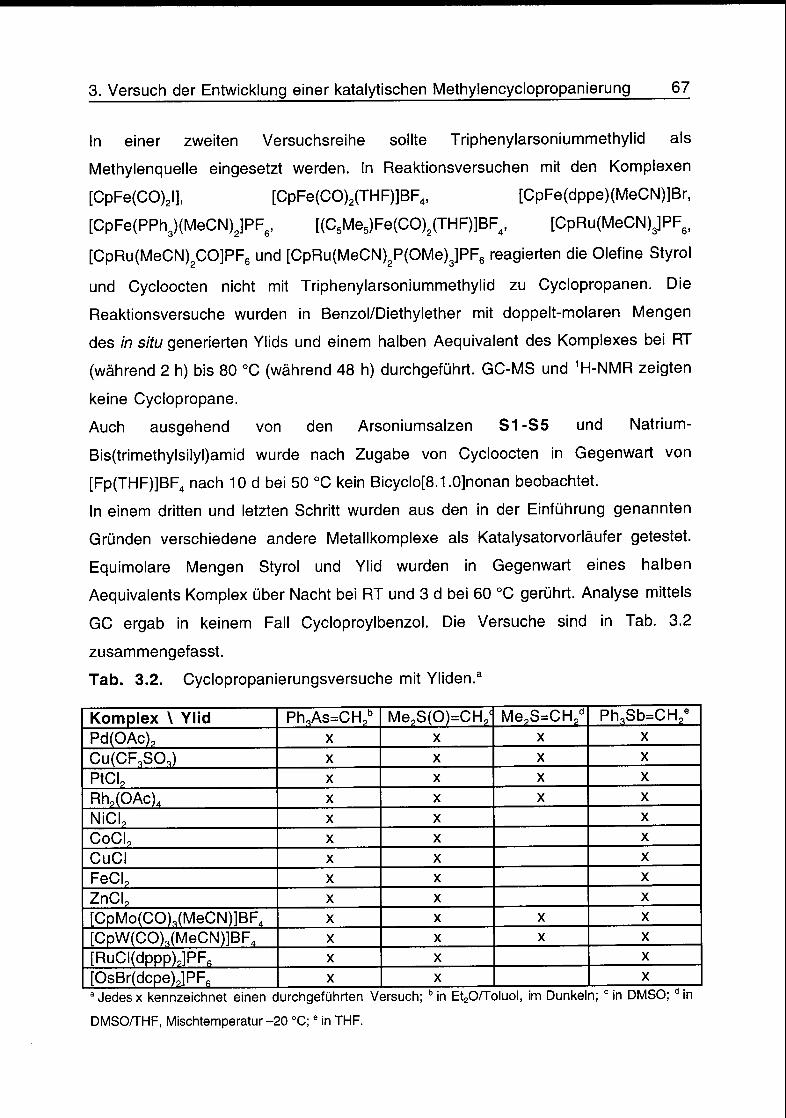
3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 67
In einer zweiten Versuchsreihe sollte Triphenylarsoniummethylid als
Methylenquelle eingesetzt werden. In Reaktionsversuchen mit den Komplexen
[CpFe(CO)2l], [CpFe(CO)2(THF)]BF4, [CpFe(dppe)(MeCN)]Br,
[CpFe(PPh3)(MeCN)2]PF6, [(C5Me5)Fe(CO)2(THF)]BF4, [CpRu(MeCN)3]PF6,
[CpRu(MeCN)2CO]PF6 und [CpRu(MeCN)2P(OMe)3]PF6 reagierten die Olefine Styrol
und Cycloocten nicht mit Triphenylarsoniummethylid zu Cyclopropanen. Die
Reaktionsversuche wurden in Benzol/Diethylether mit doppelt-molaren Mengen
des in situ generierten Ylids und einem halben Aequivalent des Komplexes bei RT
(während 2 h) bis 80 °C (während 48 h) durchgeführt. GC-MS und 1H-NMR zeigten
keine Cyclopropane.
Auch ausgehend von den Arsoniumsalzen S1-S5 und Natrium-
Bis(trimethylsilyl)amid wurde nach Zugabe von Cycloocten in Gegenwart von
[Fp(THF)]BF4 nach 10 d bei 50 °C kein Bicyclo[8.1.0]nonan beobachtet.
In einem dritten und letzten Schritt wurden aus den in der Einführung genannten
Gründen verschiedene andere Metallkomplexe als Katalysatorvorläufer getestet.
Equimolare Mengen Styrol und Ylid wurden in Gegenwart eines halben
Aequivalents Komplex über Nacht bei RT und 3 d bei 60 °C gerührt. Analyse mittels
GC ergab in keinem Fall Cycloproylbenzol. Die Versuche sind in Tab. 3.2
zusammengefasst.
Tab. 3.2. Cyclopropanierungsversuche mit Yliden.3
Komplex \ Ylid Ph3As=CH?b Me9S(0)=CH2c Me2S=CH,d Ph,Sb=CH9e
Pd(OAc)9 X X X X
Cu(CF9SOa) X X X X
PtCI9 X X X X
Rh9(0Ac)4 X X X X
Nid, X X X
CoCI, X X X
CuCI X X X
FeCI? X X X
ZnCI? X X X
[CpMo(CO)3(MeCN)]BF4 X X X X
[CpW(C0),(MeCN)]BF4 X X X X
[RuCI(dpppyPFR X X X
[OsBr(dcpe)?]PFR X X X
a Jedes x kennzeichnet einen d urchgeführten V<srsuch;b in Et20/Tcjluol, im Dunkel n;
c in DMSO;d in
DMSOATHF, Mischtemperatur -20 °C;e in THF.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 68
3.5 Schlussfolgerungen
Da aus keinem der Reaktionsversuche mit einer grossen Zahl an Komplexen und
Yliden des Schwefels, Arsens und Antimons in Gegenwart von Olefinen
Cyclopropane erhalten wurden, sollte ein anderes Cyclopropanierungsreagens
gefunden werden. Anstelle des gefährlichen Diazomethans (siehe dazu Abschnitt
2.1) sollte das käuflich erwerbliche Trimethylsilyldiazomethan eingesetzt werden.
Bis heute sind nur wenige Cyclopropanierungen mit Trimethylsilyldiazomethan
beschrieben worden. Es ist keine asymmetrische Version bekannt.
3.6 Literatur
(1) Karsch, H. H.; Klein, H.-F.; Kreiter, C. G.; Schmidbaur, H. Chem. Ber. 1974,
707, 3692-3696.
(2) Karsch, H. H.; Schmidbaur, H. Chem. Ber. 1974, 107, 3684-3691.
(3) Schmidbaur, H.; Adlkofer, J.; Heimann, M. Chem. Ber. 1974, 707, 3697-
3705.
(4) Hoover, J. F.; Stryker, J. M. Organometallics 1988, 7, 2082-2084.
(5) Henry, M. C; Wittig, G. J. Am. Chem. Soc. 1960, 82, 563-564.
(6) Schmidbaur, H.; Tronich, W. Inorg. Chem. 1968, 7, 168-169.
(7) Schmidbaur, H.; Richter, W.; Wolf, W.; Köhler, F. H. Chem. Ber. 1975, 708,
2649-2655.
(8) Yamamoto, Y.; Schmidbaur, H. J. Chem. Soc. Chem. Commun. 1975, 668-
669.
(9) Richter, W.; Yamamoto, Y.; Schmidbaur, H. Chem. Ber. 1977, 7 70, 1312-
1320.
(10) Shen, Y.; Gu, Z.; Ding, W.; Huang, Y. Tetrahedron Lett. 1984, 25, 4425-4428.
(11) Reetz, M. T.; Binder, J. Tetrahedron Lett. 1989, 30, 5425-5428.
(12) Huang, Y. Z.; Shen, Y. C. Adv. Organomet. Chem. 1982, 20, 115-157.
(13) Davison, A.; Green, M. L. H.; Wilkinson, G. J. Chem. Soc. 1961, 3172-3177.
(14) Treichel, P. M.; Shubkin, R. L.; Barnett, K. W.; Reichard, D. Inorg. Chem.
1966,5, 1177-1181.
(15) Fischer, E. O.; Moser, E. J. Organomet. Chem. 1966, 5, 63-72.
(16) Haines, R. J.; du Preez, A. L; Marais, I. L. J. Organomet. Chem. 1971, 28,
405-413.
(17) Cullen, W. R.; Patmore, D. J.; Sams, J. R.; Scott, J. C. Inorg. Chem. 1974, 13,
649-655.
(18) Alway, D. G.; Barnett, K. W. Inorg. Chem. 1978, 77, 2826-2831.
(19) Brauer, G., Ed. Handbuch der Anorganischen Chemie; 2 ed.; Enke F.:
Stuttgart, 1981; Vol. 3.
(20) Jutzi, P.; Mix, A. Chem. Ber. 1990, 723, 1043-1045.

3. Versuch der Entwicklung einer katalytischen Methylencyclopropanierung 69
(21) Nesmeyanov, A. N.; Chapovsky, Y. A.; Ustynyuk, Y. A. J. Organomet. Chem.
1967, 9, 345-353.
(22) King, R. B.; Houk, L. W.; Pannell, K. H. Inorg. Chem. 1969, 8, 1042-1048.
(23) Mays, M. J.; Sears, P. L. J. C. S. Dalton 1973, 1873-1875.
(24) Roger, C; Hamon, P.; Toupet, L; Rabaa, H.; Saillard, J.-Y.; Hamon, J.-R.;
Lapinte, C. Organometallics 1991, 70, 1045-1054.
(25) Dieck, H. t.; Diercks, R.; Stamp, L; Bruder, H.; Schuld, T. Chem. Ber. 1987,
720, 1943-1950.
(26) Reger, D. L.; Culbertson, E. C. J. Organomet. Chem. 1977, 737, 297-300.
(27) Reger, D. L.; Coleman, C. J. Organomet. Chem. 1977, 737, 153-162.
(28) Schmidt, E. K. G.; Thiel, C. H. J. Organometal. Chem. 1981, 220, 87-102.
(29) Hynes, M. J.; Mahon, M. F. T.; McArdle, P. J. Organomet. Chem. 1987, 320,
C44-C46.
(30) Treichel, P. M.; Molzahn, D. C. Synth. React. Inorg. Met. Org. Chem. 1979, 9,
21-29.
(31) Catheline, D.; Astruc, D. J. Organomet. Chem. 1984, 272, 417-426.
(32) Gill, T. P.; Mann, K. R. Organometallics 1982, 7, 485-488.
(33) King, R. B. Organometallic Syntheses; Academic Press: New York, 1965; Vol.
1.
(34) Beck, W.; Schloter, K. Z. Naturforsch. 1978, 33 b, 1214-1222.
(35) Tietze, L.-F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischenPraktikum; Georg Thieme Verlag: Stuttgart, New York, 1981, 61-62.
(36) Tietze, L.-F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischenPraktikum; Georg Thieme Verlag: Stuttgart, New York, 1981, 62.
(37) Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1965, 87, 1353-1364.
(38) Johnson, C. R.; Haake, M.; Schroeck, C. W. J. Am. Chem. Soc. 1970, 92,
6594-6598.
(39) Steinkopf, W.; Schwen, G. Chem. Ber. 1921, 54, 2969-2975.
(40) McCortney, B. A.; Jacobson, B. M.; Vreeke, M.; Lewis, E. S. J. Am. Chem.
Soc. 1990, 7 72, 3554-3559.
(41) Tietze, L.-F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischenPraktikum; Georg Thieme Verlag: Stuttgart, New York, 1981, 58-59.
(42) Tietze, L.-F.; Eicher, T. Reaktionen und Synthesen im organisch-chemischen
Praktikum; Georg Thieme Verlag: Stuttgart, New York, 1981, 215-216.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung
mit Trimethylsilyldiazomethan
4.1 Einführung
Katalytische Cyclopropanierungsreaktionen mit Diazomethan sind - wie in
Abschnitt 1.3 erwähnt wurde - bekannt. Als Katalysatorvorläufer traten die
Komplexe Pd(OAc)2 und [PdCI2(NCPh)2] in Erscheinung. Sie sind effizient in der
Katalyse der Cyclopropanierung von aktivierten Olefinen wie Styrol1, von oc,ß-
ungesättigten Carbonylverbindungen2 und von nicht-aktivierten, terminalen
Olefinen.3 Weder Cu- noch Rh-Komplexe erreichen deren Reaktivität in der
Cyclopropanierung mit Diazomethan.4 Ferner katalysieren weder Cu- noch Rh-
Komplexe die Cyclopropanierung von a,ß-ungesättigten Carbonylverbindungen mit
Diazomethan.
Obwohl mechanistische Details vollkommen unbekannt sind, wird angenommen,
dass Diazomethan mit Pd(ll) nicht unter der Bildung von Carbenkomplexen
reagiert.4 Die Hauptfunktion von Pd(ll) in der Cyclopropanierungsreaktion ist die
Bindung und damit einhergehende Aktivierung des Olefins, so dass Diazomethan
nukleophil angreifen kann. Die entstandene zwitterionische Zwischenstufe reagiert
in einem konzertierten Prozess unter Freisetzung des Stickstoffs und Bildung des
Cyclopropans. Die formale Oxidationsstufe des Metallzentrums bleibt unverändert.
Es ist keine asymmetrische Version der Pd-katalysierten Cyclopropanierung mit
Diazoverbindungen bekannt. (Die Verwendung von chiralen, nicht racemischen
Bis(oxazolin)-Verbindungen als Liganden für Palladium in der Cyclopropanierung
von Olefinen mit Diazomethan ergab racemische Produkte in guten bis sehr guten
Ausbeuten.5,6) Kationische, chirale Pd(ll)-Komplexe sollten auf ihre katalytische
Aktivität hin untersucht werden. Zur chiralen Modifikation dieser Komplexe sollten
Ferrocenylliganden dienen, da diese vielseitig variierbar sind. Drei Typen von
Pd(ll)-Komplexen, die hergestellt und als Katalysatoren getestet werden sollten,
sind in Abb. 4.1 gezeigt.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 71
^5
Fe
Cy2P
P—
Ph,
"Me _v/CI
(S)
Abb. 4.1. Mono- und dikationische, chirale Pd(ll)-Komplexe.
Diese Komplexe sollten aus den neutralen Chloro-Vorläufem durch selektive
Halogenidabstraktion hergestellt werden. Im Falle von zweizähnigen, chiralen
Liganden wird das Chlorid in fra/is-Position zum Fragment mit dem stärksten trans-
Effekt entfernt. Dadurch ist es möglich, die Koordinationsstelle des Olefins zu
kontrollieren.
Neben Pd- sollten auch Cu-Komplexe als Katalysatoren in der Cyclopropanierung
getestet werden. In der Kupfer-katalysierten Cyclopropanierung mit Diazoestern ist
Cu(l) - nicht Cu(ll) - die katalytisch aktive Spezies. Die Wirksamkeit von Cu(ll)-
Komplexen beruht auf der in situ Reduktion mit den Diazoverbindungen. Diese
Reduktion findet sowohl mit Diazomethan als auch mit Diazoestern statt.7,8 Es wird
angenommen, dass die Kupfer-katalysierte Cyclopropanierung mit Diazoestern
über einen Metall-Carben-Komplex verläuft (vgl. Abschnitt 1.2).9
Als chirale Liganden waren zweizähnige Ferrocenylliganden mit zwei Pyrazolyl-
Fragmenten vorgesehen (Abb. 4.2).
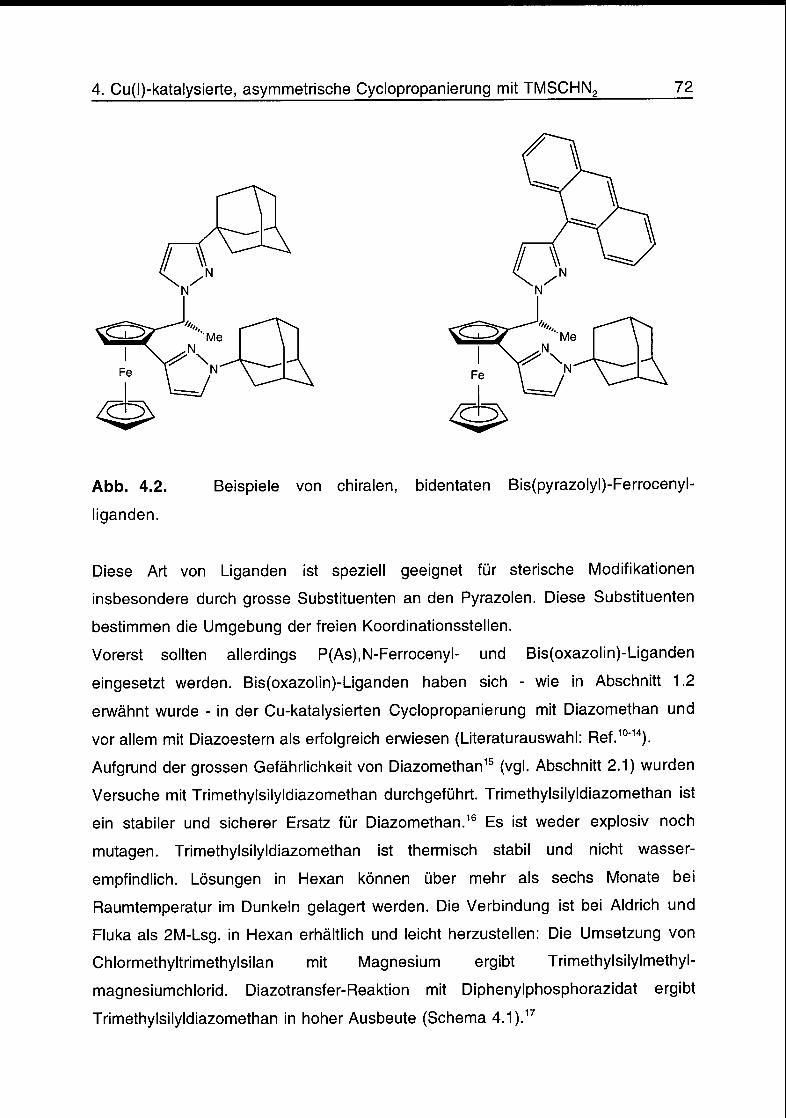
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 72
"X3\ ^XA
Abb. 4.2.
liganden.
Beispiele von chiralen, bidentaten Bis(pyrazolyl)-Ferrocenyl-
Diese Art von Liganden ist speziell geeignet für sterische Modifikationen
insbesondere durch grosse Substituenten an den Pyrazolen. Diese Substituenten
bestimmen die Umgebung der freien Koordinationsstellen.
Vorerst sollten allerdings P(As),N-Ferrocenyl- und Bis(oxazolin)-Liganden
eingesetzt werden. Bis(oxazolin)-Liganden haben sich - wie in Abschnitt 1.2
erwähnt wurde - in der Cu-katalysierten Cyclopropanierung mit Diazomethan und
vor allem mit Diazoestern als erfolgreich erwiesen (Literaturauswahl: Ref.10"14).
Aufgrund der grossen Gefährlichkeit von Diazomethan15 (vgl. Abschnitt 2.1) wurden
Versuche mit Trimethylsilyldiazomethan durchgeführt. Trimethylsilyldiazomethan ist
ein stabiler und sicherer Ersatz für Diazomethan.16 Es ist weder explosiv noch
mutagen. Trimethylsilyldiazomethan ist thermisch stabil und nicht wasser¬
empfindlich. Lösungen in Hexan können über mehr als sechs Monate bei
Raumtemperatur im Dunkeln gelagert werden. Die Verbindung ist bei Aldrich und
Fluka als 2M-Lsg. in Hexan erhältlich und leicht herzustellen: Die Umsetzung von
Chlormethyltrimethylsilan mit Magnesium ergibt Trimethylsilylmethyl-
magnesiumchlorid. Diazotransfer-Reaktion mit Diphenylphosphorazidat ergibt
Trimethylsilyldiazomethan in hoher Ausbeute (Schema 4.1 ).17

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 73
1.Mg, 2. DPPA
Et20, Hexan
Me3SiCH2CI Me3SiCHN2RT, 5 h
79 % in Hexan
DPPA = Phosphorsäure-diphenylester-azid, (PhO)2P(0)N3
Schema 4.1. Herstellung von Trimethylsilyldiazomethan aus Chlormethyl-
trimethylsilan.
Bereits ab 1968 wurde die Verwendung von Trimethylsilyldiazomethan in der Cu(l)-
katalysierten Cyclopropanierung von einfachen Olefinen berichtet.18"20 Shioiri und
Mitarbeiter publizierten 1989, dass die Behandlung von frans-Zimtsäureethylester
mit Trimethylsilyldiazomethan in Gegenwart von 5 mol-% Palladium(ll)-dichlorid
Silylcyclopropane in einer Ausbeute von 79% ergibt (Schema 4.2).21 Ohne
Katalysator findet keine Reaktion statt. Neben Palladium(ll)-dichlorid ist in dieser
Reaktion auch Palladium(ll)-diacetat katalytisch aktiv. Kupfer(l)-chlorid, Kupfersulfat
und Rhodium(ll)-diacetat katalysieren die Reaktion hingegen nicht.
5 mol % PdCI2 SiMe3
^COzEt C6H6,ArTMSCHN2
Ph'
-r | i ivi\jv~»ni^2
60 °C, 1h
Schema 4.2. Pd(ll)-katalysierte Cyclopropanierung eines Michael-Akzeptors
mit Trimethylsilyldiazomethan.
Umgekehrt verhält sich dagegen Styrol. Die Cyclopropanierung von Styrol wird
durch Kupfer(l)-chlorid, aber nicht durch Palladium(ll)-dichlorid katalysiert. In dieser
Reaktion mit 5 mol-% Katalysator wurde eine Ausbeute von 46% erreicht (Schema
4.3).

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 74
3 nV + 1 TMSCHN
5 mol % CuCI
C6H6, Ar
Ph'
RT, 1hPrf SiMe3
46%
Schema 4.3. Cu(l)-katalysierte Cyclopropanierung von Styrol mit
Trimethylsilyldiazomethan.
Nishiyama und Mitarbeiter veröffentlichten 1994 die Synthese des
Trimethylsilylcarben-Komplexes (S,S)-Bis(4-isopropyloxazolinyl)pyridin-(dichloro)-
ruthenium(ll)-trimethylsilylcarben.22 Dieser Komplex reagierte mit 25 equiv. Styrol
mit einer Ausbeute von lediglich 34% (2.5 h, 60 °C) zum Cyclopropan. Ueber die
Diastereoselektivität und Enantioselektivität der Reaktion wurde keine Aussage
gemacht. Der Carben-Komplex wurde allerdings als Katalysatorvorläufer in der
Cyclopropanierung mit Ethyldiazoacetat eingesetzt. Mit 2 mol-% des Komplexes
wurde eine Ausbeute von 60% und ein Diastereoisomerenverhältnis von 86:14
(trans/cis) beobachtet. Die Enantiomerenüberschüsse waren 80% (trans) und 47%
{eis).
Vor kurzem berichteten Maas und Seitz die katalytische Aktivität des polymeren
Ru(l)-Komplexes [Ru2(CO)4(OAc)2]n in der Cyclopropanierung mit
Trimethylsilyldiazomethan.23
Die folgenden drei Abschnitte geben Auskunft über die verwendeten Liganden und
Komplexe sowie die durchgeführten Reaktionsversuche. Die erste asymmetrische,
katalytische Cyclopropanierung von Olefinen mit Trimethylsilyldiazomethan wird
berichtet.
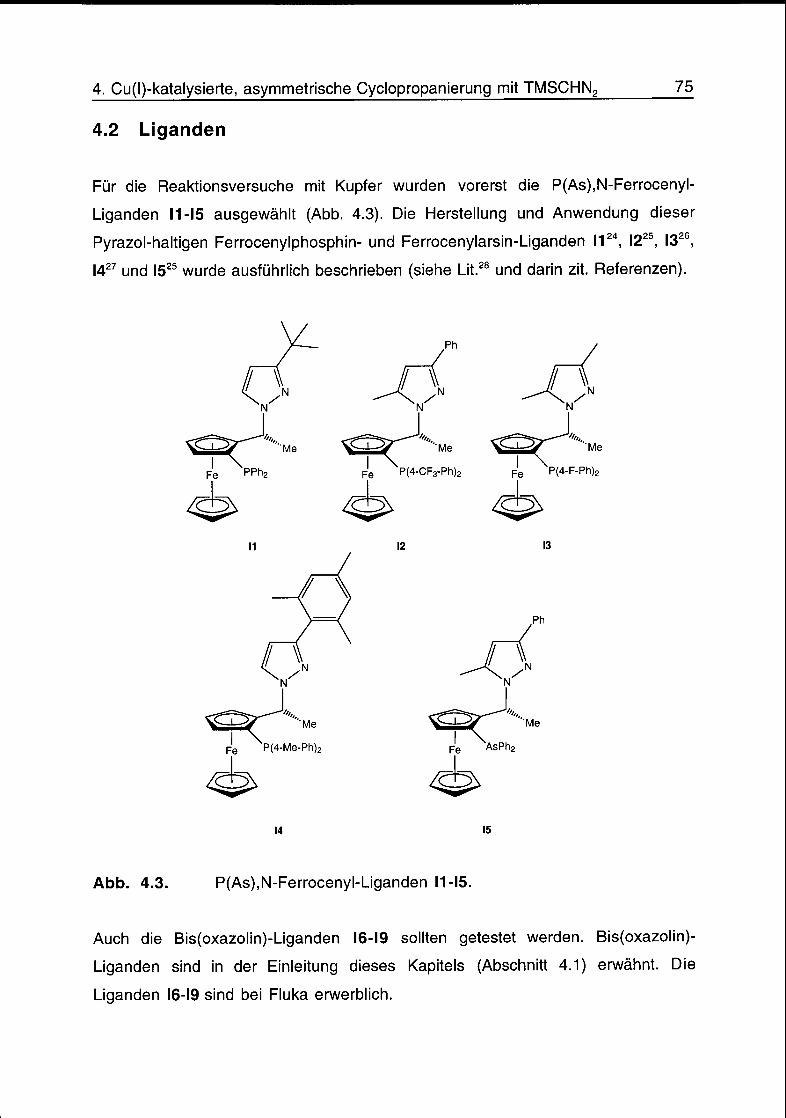
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 75
4.2 Liganden
Für die Reaktionsversuche mit Kupfer wurden vorerst die P(As),N-Ferrocenyl-
Liganden 11-15 ausgewählt (Abb. 4.3). Die Herstellung und Anwendung dieser
Pyrazol-haltigen Ferrocenylphosphin- und Ferrocenylarsin-Liganden 1124, I225, I326,
I427 und I525 wurde ausführlich beschrieben (siehe Lit.28 und darin zit. Referenzen).
I4 I5
Abb. 4.3. P(As),N-Ferrocenyl-Liganden 11-15.
Auch die Bis(oxazolin)-Liganden 16-19 sollten getestet werden. Bis(oxazolin)-
Liganden sind in der Einleitung dieses Kapitels (Abschnitt 4.1) erwähnt. Die
Liganden I6-I9 sind bei Fluka erwerblich.
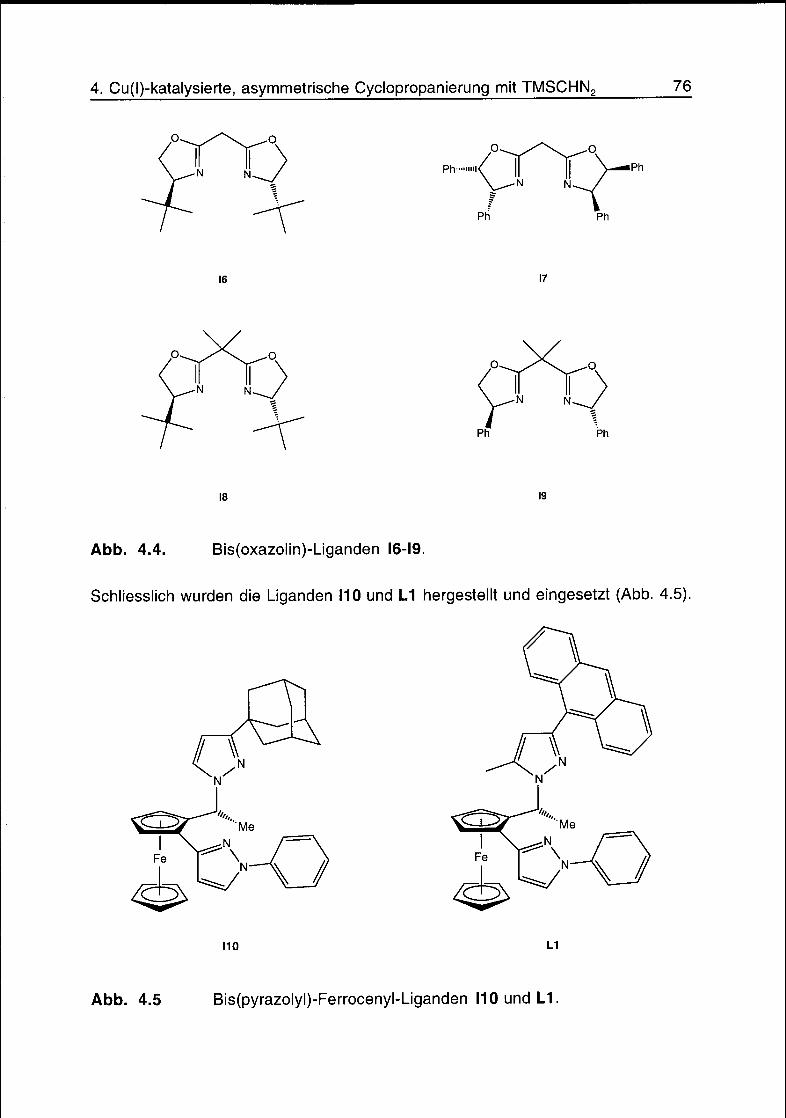
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 76
Ph
16 17
18
Abb. 4.4. Bis(oxazolin)-Liganden 16-19.
Schliesslich wurden die Liganden 110 und L1 hergestellt und eingesetzt (Abb. 4.5).
no L1
Abb. 4.5 Bis(pyrazolyl)-Ferrocenyl-Liganden 110 und L1.
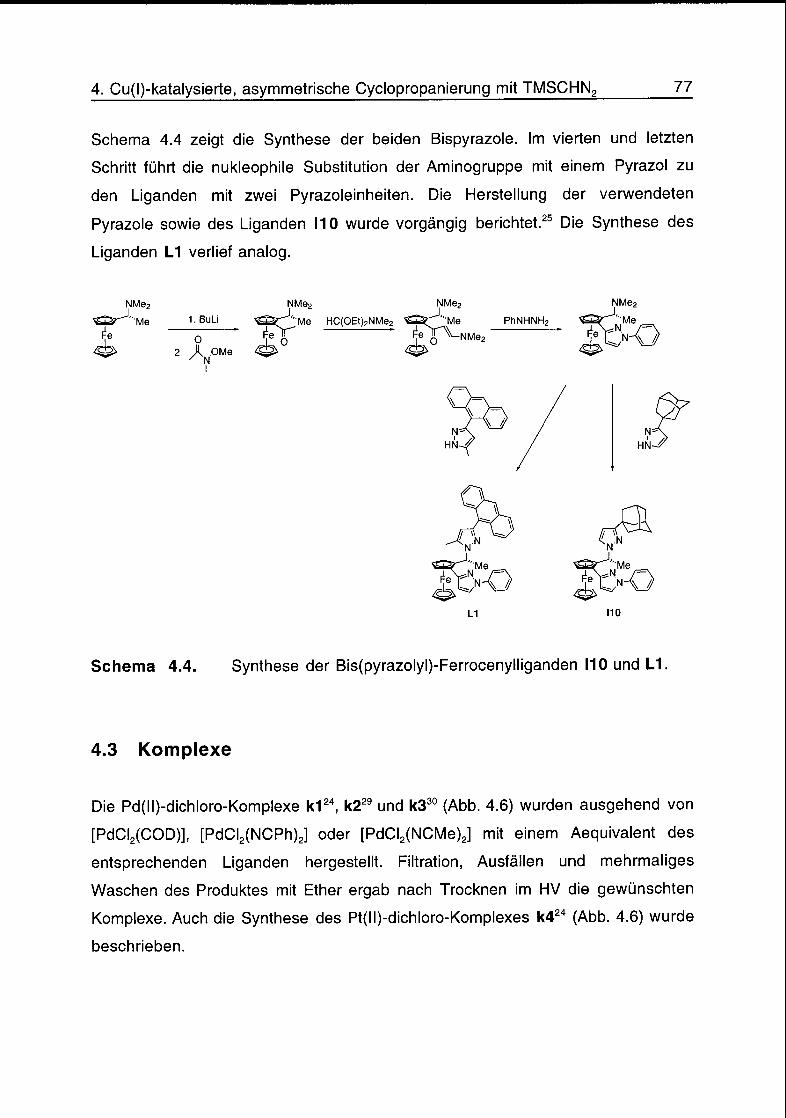
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 77
Schema 4.4 zeigt die Synthese der beiden Bispyrazole. Im vierten und letzten
Schritt führt die nukleophile Substitution der Aminogruppe mit einem Pyrazol zu
den Liganden mit zwei Pyrazoleinheiten. Die Herstellung der verwendeten
Pyrazole sowie des Liganden 110 wurde vorgängig berichtet.25 Die Synthese des
Liganden L1 verlief analog.
L1 110
Schema 4.4. Synthese der Bis(pyrazolyl)-Ferrocenylliganden 110 und L1.
4.3 Komplexe
Die Pd(ll)-dichloro-Komplexe k124, k229 und k330 (Abb. 4.6) wurden ausgehend von
[PdCI2(COD)], [PdCI2(NCPh)2] oder [PdCI2(NCMe)2] mit einem Aequivalent des
entsprechenden Liganden hergestellt. Filtration, Ausfällen und mehrmaliges
Waschen des Produktes mit Ether ergab nach Trocknen im HV die gewünschten
Komplexe. Auch die Synthese des Pt(ll)-dichloro-Komplexes k424 (Abb. 4.6) wurde
beschrieben.
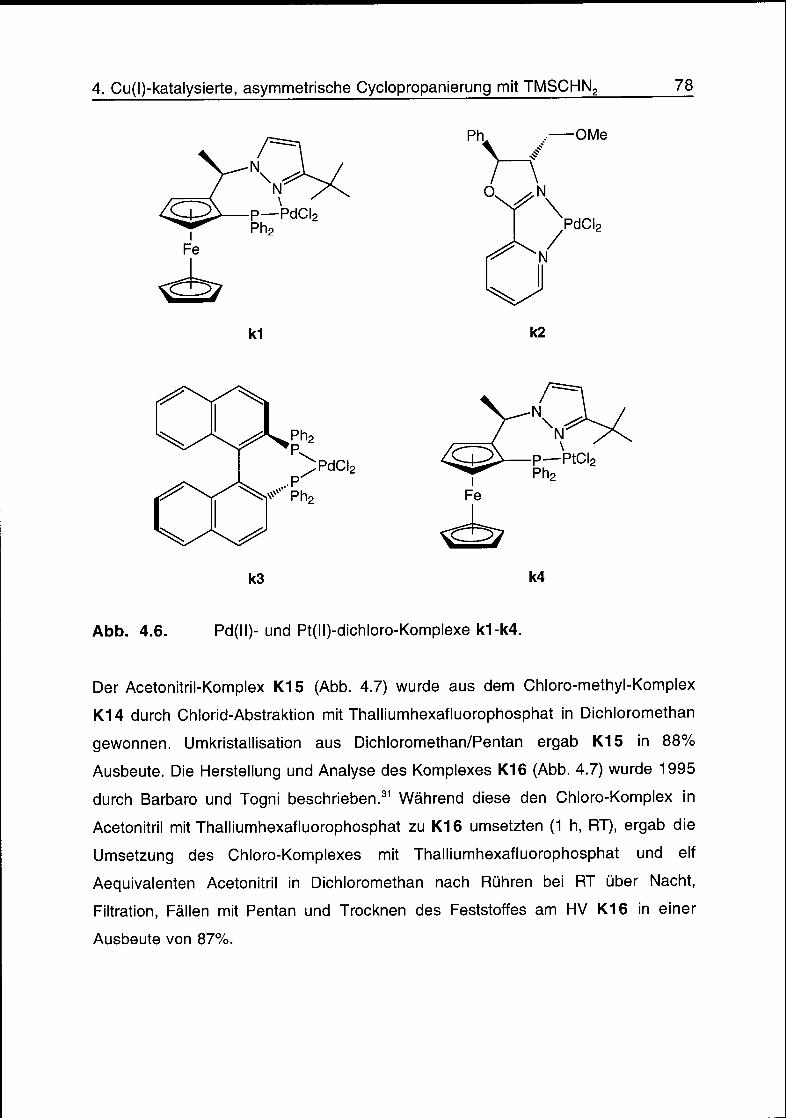
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 78
k1
-OMe
X^ /PdCI2
N
k2
PdCI2
k3 k4
Abb. 4.6. Pd(ll)- und Pt(ll)-dichloro-Komplexe k1-k4.
Der Acetonitril-Komplex K15 (Abb. 4.7) wurde aus dem Chloro-methyl-Komplex
K14 durch Chlorid-Abstraktion mit Thalliumhexafluorophosphat in Dichloromethan
gewonnen. Umkristallisation aus Dichloromethan/Pentan ergab K15 in 88%
Ausbeute. Die Herstellung und Analyse des Komplexes K16 (Abb. 4.7) wurde 1995
durch Barbara und Togni beschrieben.31 Während diese den Chloro-Komplex in
Acetonitril mit Thalliumhexafluorophosphat zu K16 umsetzten (1 h, RT), ergab die
Umsetzung des Chloro-Komplexes mit Thalliumhexafluorophosphat und elf
Aequivalenten Acetonitril in Dichloromethan nach Rühren bei RT über Nacht,
Filtration, Fällen mit Pentan und Trocknen des Feststoffes am HV K16 in einer
Ausbeute von 87%.
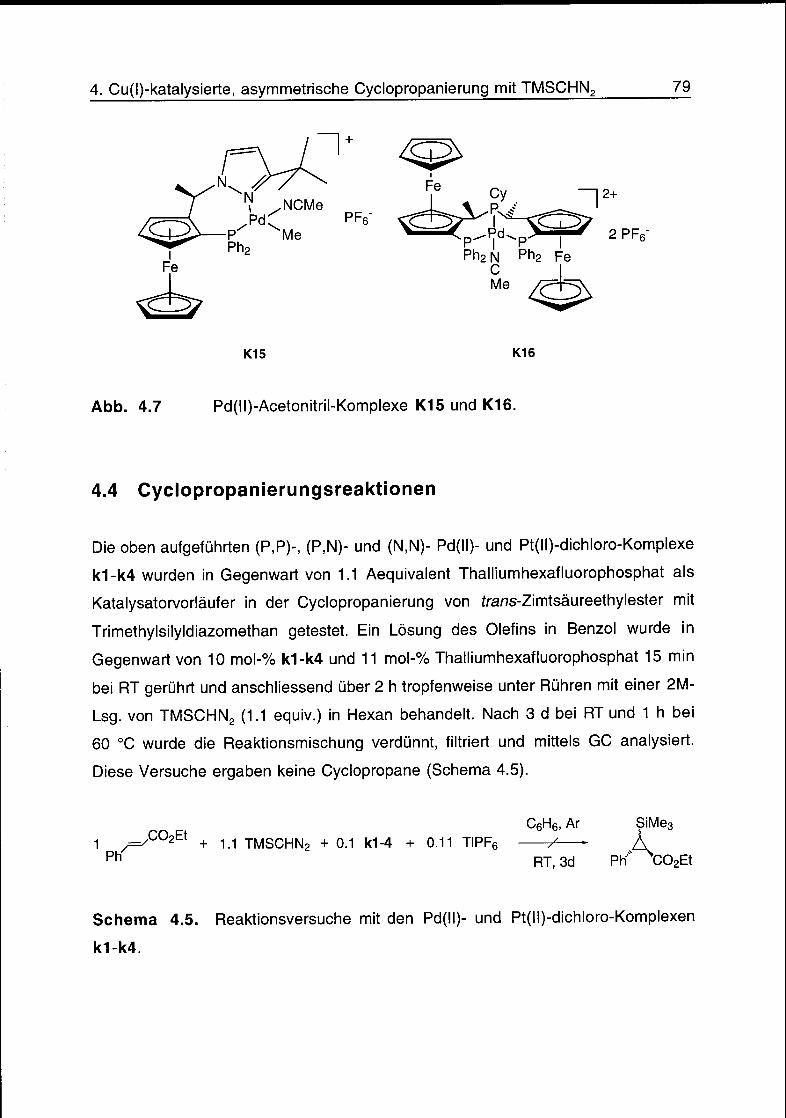
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 79
2 PF«
K15 K16
Abb. 4.7 Pd(ll)-Acetonitril-Komplexe K15 und K16.
4.4 Cyclopropanierungsreaktionen
Die oben aufgeführten (P,P)-, (P,N)- und (N,N)- Pd(ll)- und Pt(ll)-dichloro-Komplexe
k1-k4 wurden in Gegenwart von 1.1 Aequivalent Thalliumhexafluorophosphat als
Katalysatorvorläufer in der Cyclopropanierung von frans-Zimtsäureethylester mit
Trimethylsilyldiazomethan getestet. Ein Lösung des Olefins in Benzol wurde in
Gegenwart von 10 mol-% k1-k4 und 11 mol-% Thalliumhexafluorophosphat 15 min
bei RT gerührt und anschliessend über 2 h tropfenweise unter Rühren mit einer 2M-
Lsg. von TMSCHN2 (1.1 equiv.) in Hexan behandelt. Nach 3 d bei RT und 1 h bei
60 °C wurde die Reaktionsmischung verdünnt, filtriert und mittels GC analysiert.
Diese Versuche ergaben keine Cyclopropane (Schema 4.5).
1 /=/00^EX + 1.1 TMSCHN2 + 0.1 k1-4 + 0.11 TIPF6Ph
C6H6, Ar
^
RT, 3d
SiMe3
ÀPh C02Et
Schema 4.5. Reaktionsversuche mit den Pd(ll)- und Pt(ll)-dichloro-Komplexen
k1-k4.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 80
Als Nächstes sollten die kationischen Komplexe K15 und K16 getestet werden. Zu
einer Lösung von frans-Zimtsäureethylester und 10 mol-% des Komplexes in
Methylenchlorid wurde bei RT unter Rühren eine 2M-Lsg. von TMSCHN2 in Hexan
(1.1 equiv.) über 1 h getropft. Nach 1 d bei RT und 1 h bei 60 °C wurde die
Reaktionsmischung verdünnt, filtriert und mittels GC analysiert. Diese Versuche
ergaben keine Cyclopropane (Schema 4.6).
SiMe3
C02EtCH2CI2, Ar |
1 /=/ + 1.1 TMSCHN2 + 0.1 K15/K16 -/- *- /\Ph RT, 24 h p/ Ncc^Et
Schema 4.6. Reaktionsversuche mit den Pd(ll)-Acetonitril-Komplexen K15 und
K16.
Nun sollten Reaktionsversuche mit Cu durchgeführt werden. Rühren einer
Mischung von Cu(l)-triflat und Liganden 11-19 (Abb. 4.3 und 4.4) in CH2CI2 über
Nacht, Filtration dieser Mischung zu einer Lösung von Styrol und Dekan (als
interner GC-Standard) in CH2CI2 ergab nach langsamer Zugabe von TMSCHN2
mittels Spritzenpumpe und 48 h Rühren die 1-Phenyl-2-trimethylsilyl-cyclopropane
in Ausbeuten von 6-68% (Tab. 4.1). Die Ausbeuten und Selektivitäten wurden
mittels GC bestimmt (siehe Analysedaten im 5. Kapitel). Die beobachteten trans/cis-
Verhältnisse betrugen bis zu 29. Die grössten Enantiomerenüberschüsse waren
41% für das eis- und 63% für das frans-lsomer.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 81
Tab. 4.1. Erste bekannte katalytische, asymmetrische Cyclopropanierung
mit Trimethylsilyldiazomethan.
5 mol-% Cu(CF3S03)
1 Ph^~ + 1.2 TMSCHN2
6 mol-% I
CH2CI2, Ar
- APlf SiMe3
RT, 48 h
No. Ligand Ausbeute / % trans 1 eis ee / %a
eis trans
1 11 62 5.5 34 (2) 17 (1)
2 12 68 2.8 5 (2) 7 (1)
3 13 65 3.7 2 (2) 9 (1)
4 14 40 6.2 12 (2) 16 (1)
5 15 49 4.4 1 (2) 12 (1)
6 16 6 18.4 22 (1) 54 (2)
7 16 14 18.2 22 (1) 57 (2)b
8 17 34 16.3 26 (2) 50 (1)
9 17 35 16.3 27 (2) 50 (1)b
10 18 11 22.6 41 (1) 63 (2)b
11 19 57 29.4 35 (1) 61 (2)b
aDie Zahlen in Klammern sind die Nummern des jeweiligen Enantiomeren, das im Ueberschuss
gebildet wurde, wobei 1 jeweils das Enantiomer mit kürzerer und 2 das Enantiomer mit längerer
Retentionszeit im Gaschromatogramm bezeichnet.bZugabe von TMSCHN2 über 6 h, alle anderen: 3 h.
Die grössten Ausbeuten wurden mit den (P.N)-Liganden 11-13 erreicht (No. 1-3).
Die Verwendung des elektronenreicheren Liganden 14 führt zu kleineren
Ausbeuten (No. 4), was möglicherweise auf eine Stabilisierung des postulierten
intermediären Carben-Komplexes zurückzuführen ist. Diese Erklärung trifft auch auf
die beobachteten Ausbeuten mit den Bis(oxazolin)-Liganden I6-I9 zu (vgl. No. 6
mit 8, No. 7 mit 9 und No. 10 mit 11). Die besseren Selektivitäten wurden jedoch mit
den Bis(oxazolin)-Liganden erhalten.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 82
Besonders hervorzuheben ist der Reaktionsversuch mit dem Liganden 19 (No. 11),
mit dem nicht nur eine Ausbeute von 57%, sondern auch eine Diastereoselektivität
von 93% und Enantiomerenüberschüsse von 35% (eis) und 61% (trans) erreicht
wurden.
Die Ergebnisse der Reaktionsversuche mit den Liganden 110 und L1 sind in Tab.
4.2 gegeben. Um die Reaktionsversuche bei 50 °C durchführen zu können, wurde
der in situ gebildete Cu(l)-Komplex in Dichloromethan zu Styrol und Dekan in 1,2-
Dichloroethan filtriert.
Tab. 4.2. Katalytische, asymmetrische Cyclopropanierung mit Trimethyl¬
silyldiazomethan - Cu(l) /HO und L1.
5 mol% Cu(CF3S03)
1 Ph^~12 mo
+ 1.2 TMSCHN2Ar, 50
-% I10/L1
°C, 136 h Prf"SiMe3
Ligand Ausbeute / % trans 1 eis ee / %
eis trans
110 11 5.9 2 (1) 1 (2)
L1 13 3.9 11(1) 7 (2)
Die Reaktionsexperimente mit diesen zwei Bis(pyrazolyl)-Ferrocenylliganden
ergaben leider keine Verbesserungen. Es wurden kleine Ausbeuten und
Selektivitäten beobachtet.
Da bis zu diesem Zeitpunkt mit den Liganden 17 und 19 die besten Resultate
erhalten worden waren, wurden weitere Experimente mit diesen Liganden bei
verschiedenen Temperaturen und mit verschiedenen Ligand/Metall-Verhältnissen
durchgeführt.
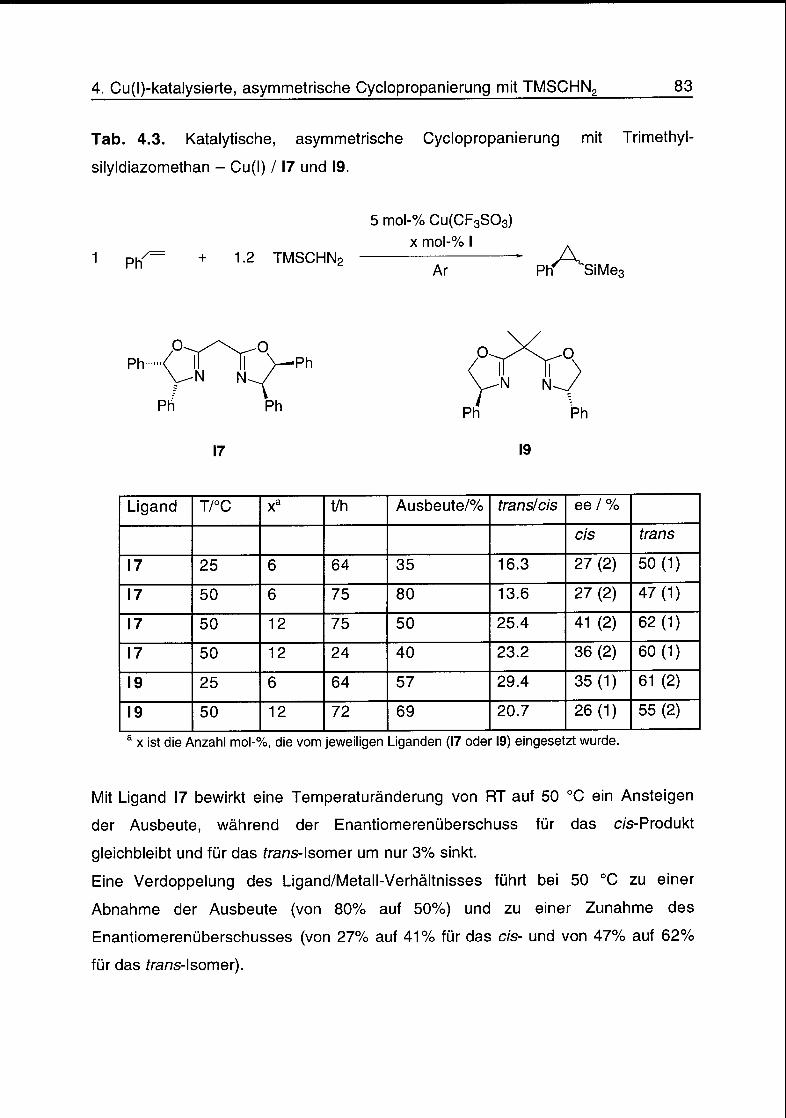
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 83
Tab. 4.3. Katalytische, asymmetrische Cyclopropanierung mit Trimethyl¬
silyldiazomethan - Cu(l) /17 und 19.
Ph+ 1.2 TMSCHN2
5 mol-% Cu(CF3S03)
x mol-% I
ArA
Prf SiMe3
Ph •/ N N ^PhV-N N~
Ph
17
Ph
VN N-V
Ph
19
Ph
Ligand T/°C xa t/h Ausbeute/% translcis ee / %
eis trans
17 25 6 64 35 16.3 27(2) 50(1)
17 50 6 75 80 13.6 27(2) 47(1)
17 50 12 75 50 25.4 41 (2) 62(1)
17 50 12 24 40 23.2 36(2) 60(1)
19 25 6 64 57 29.4 35(1) 61 (2)
19 50 12 72 69 20.7 26(1) 55(2)
x ist die Anzahl mol-%, die vom jeweiligen Liganden (17 oder 19) eingesetzt wurde.
Mit Ligand 17 bewirkt eine Temperaturänderung von RT auf 50 °C ein Ansteigen
der Ausbeute, während der Enantiomerenüberschuss für das c/s-Produkt
gleichbleibt und für das frans-lsomer um nur 3% sinkt.
Eine Verdoppelung des Ligand/Metall-Verhältnisses führt bei 50 °C zu einer
Abnahme der Ausbeute (von 80% auf 50%) und zu einer Zunahme des
Enantiomerenüberschusses (von 27% auf 41% für das eis- und von 47% auf 62%
für das frans-lsomer).

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 84
Diese Beobachtungen sind in Uebereinstimmung mit kürzlich veröffentlichten
Studien der Kinetik der Cu(l)-katalysierten Cyclopropanierung von Olefinen mit
Diazoestern.32 In diesen Studien wurde unter anderem auch das Cu-triflat-
Bis(oxazolin) als Katalysator eingesetzt, wobei der kationische, 14-Elektronen-
Komplex [(N-N)Cu]+ als aktiver Katalysator postuliert wurde. Da dieser 1:1-Komplex
aber mit dem 18-Elektronen-Komplex [(N-N)2Cu]+X" im Gleichgewicht steht, werden
zwei Aequivalente des Liganden benötigt, um die Existenz von freiem Cu-triflat zu
verhindern und daher die asymmetrische Induktion durch den Katalysator zu
maximieren. Kupfer(l)-triflat ist bekannt als aktiver Katalysator in der
Cyclopropanierung mit Diazoestern.7,33 Trifluormethylsulfonat ist ein sehr schwach
koordinierendes Anion. Die Röntgenstrukturanalyse des 1:1 Komplexes von (S,S)-
2,2'-Methylen-bis-(4-tert.-butyl-2-oxazolin) (16) mit Cu(l)-triflat ergab, dass das
Gegenion nicht an das Metallzentrum koordiniert ist. Weitere Untersuchungen
haben ergeben, dass das Gegenion auch in Lösung (CHCI3) völlig dissoziiert
vorliegt und dass der Ligand an das Metallzentrum koordiniert ist, aber schnellen
Ligandaustauschreaktionen unterliegt.14
Um die chirale Induktion zu erhöhen, sollte als Gegenion für das Cu(l)-Zentrum ein
Aequivalent des deprotonierten Liganden wirken. Die Umsetzung von 2 equiv. des
Bis(oxazolin)-Liganden mit 2 equiv. BuLi in THF und anschliessende Zugabe von 1
equiv. CuCI2 ergab nach Filtration über Alox, Einengen, Waschen mit Hexan und
Trocknen im HV einen violetten Feststoff, der mit einem Aequivalent
Phenylhydrazin als Katalysator-Mischung eingesetzt wurde (Schema 4.7). Die
Cyclopropane wurden in einer Ausbeute von 12% erhalten. Der
Diastereoisomerenüberschuss betrug 97% und die Enantiomerenüberschüsse
betrugen 74% für das eis- und 80% für das frans-lsomere. Die Produkteverteilung
ist in Abb. 4.8 dargestellt.
4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 85
200.5
159.1
117.8
(mVott)
76.4-
35.1
-6.3
C.-\GC-Daten\_PBGC4Daten\PE551 .dat
25.999 (min) 38.999
Sample name :PE551
1 1 ^
51.999 64.998
Abb. 4.8. Gaschromatogramm der Produktemischung.
Die Behandlung des postulierten Cu(ll)-Komplexes mit Phenylhydrazin bewirkt die
Reduktion des Cu(ll)-Metallzentrums zu Cu(l). Einer der zwei koordinierten
Liganden wird protoniert, wobei der neutrale Ligand 17 entsteht. (Die Synthese des
analogen Cu(ll)-Komplexes aus 2 mmol 16, 2 mmol n-BuLi und 1 mmol CuCI2 in
THF und dessen Anwendung als Katalysatorvorläufer unter Aktivierung mit
Phenylhydrazin in der Cyclopropanierung mit Ethyldiazoacetat wurde
beschrieben.10)

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 86
Ph Ph
2Ph
Ph
JD^-VCl THF, Ar RT, 12 h öS*Ph PhÔ'Uj X/-Ph + 2 n-BuL, + 1 CuCI2 «^V^^>h Ph Filtration über Alox y=^"^ ^N-f^
Waschen mit Hexan A. \ A c\
HV un/' PhPh'V-u
Ph Ph
17
ph/^ + 1 2 TMSCHN2 + 0 05 1 + 0 05 ,N-NH2H CH2CI2, Ar
ART.esh
Ph SlM8s
Ausbeute. 12%
trans/cis. 77.7
ee: 74% (eis 2)80% (transi)
Schema 4.7. In s/fu-Reaktionsversuch mit bester Selektivität.
Eine andere Möglichkeit, diese Cu(l)-Spezies mit einem neutralen und einem
deprotonierten, anionischen Liganden herzustellen, ist wohl die Behandlung von
Kupfer(l)-chlorid mit zwei Aequivalenten des neutralen Liganden und einem
Aequivalent Butyllithium. Die auf diese beiden Arten hergestellten
Katalysatorvorläufer zeigen in der Reaktion von Styrol mit Trimethylsilyldiazo¬
methan ungefähr die selben Produktverteilungen (Schema 4.8). Der Einfluss der
Reaktionstemperatur auf die Ausbeute und die Selektivitäten wurde bereits oben
(Tab. 4.3) erwähnt: Während eine Erhöhung der Temperatur keinen grossen
Einfluss auf die Selektivitäten hat, wird die Ausbeute deutlich vergrössert.
1 p\/== + 1.2 TMSCHN2 + 0.05 CuCI + 0.12 17 + 0.05 n-Buü
THF/c2H4Ci2,Ar A
Ausbeute: 27%
A trans/cis: 74.6~
Pi/^îiMea ee: 75 % (eis 2)50X,68h
378% (transi)
Schema 4.8. In s/fu-Reaktionsversuch bei erhöhter Temperatur.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 87
Anstelle von Butyllithium wurde nun zum Vergleich Silbertetrafluoroborat
eingesetzt, so dass also wieder ein achirales Gegenion vorhanden ist. Tatsächlich
sanken nun sowohl die Diastereoselektivität als auch die Enantioselektivitäten
deutlich (Schema 4.9). Die erhaltenen Selektivitäten sind ungefähr gleich den
Selektivitäten, die unter den gleichen Bedingungen mit Cu-triflat und zwei equiv.
des Liganden erhalten wurden.
1 Ph/= + 1-2 TMSCHN2 + 0.05 CuCI + 0.12 17 + 0.06 AgBF4
CH2ci2/C2H4ci2,Ar .
Ausbeute: 41%
/\ trans/cis: 27.6
PrT^SiMea ee: 39% (eis 2)50°c,68h 63% (transi)
Schema 4.9. In s/fcv-Reaktionsversuch mit BF4" als Gegenion.
Eine Verwendung des selben Ligand/Metall-Verhältnisses aber mit einem
zusätzlichen Gegenion anstelle eines Aequivalents des deprotonierten und daher
anionischen Liganden ergibt eine grössere Ausbeute und kleinere Enantiomeren¬
überschüsse. Eine mögliche Erklärung dafür ist, dass selbst mit 2.4 equiv. des
chiralen Liganden (bezüglich Cu(l)-Metall) in Gegenwart eines achiralen Anions
(BF4) ein achiraler, katalytisch aktiver Komplex vorliegt. Eine andere - wenn auch
völlig hypothetische - Erklärung ist, dass nicht nur der von Diaz-Requejo et al.32
postulierte 14-Elektronen-Komplex [(N-N)Cu]+ mit zwei freien Koordinationsstellen
katalytisch aktiv ist. Während eine der freien Koordinationsstellen von der Carben-
Einheit besetzt werden kann, ist immer noch eine Koordinationsstelle frei. Eine
einfache Koordination des deprotonierten, anionischen Bis(oxazolin)-Liganden
würde die grössere asymmetrische Induktion erklären. In diesem Zusammenhang
interessant ist, dass kürzlich das erste Beispiel einer enantioselektiven, Metall¬
katalysierten Reaktion mit lediglich einem chiralen Gegenion als Quelle der
Asymmetrie veröffentlicht worden ist.34

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 88
Es wurde postuliert, dass die Ionen-Paarung zwischen dem Cu+-Metallzentrum und
einem chiralen Gegenion für die asymmetrische Induktion sowohl in der
Aziridinierung mit [(p-Tolylsulfonyl)imino]phenyliodinan (Phl=NTs)35 als auch in der
Cyclopropanierung mit Ethyldiazoacetat verantwortlich ist.
4.5 Schlussfolgerungen
Eine asymmetrische Version der Cu(l)-katalysierten Cyclopropanierung mit
Trimethylsilyldiazomethan mit in der Cyclopropanierung sehr bekannten
Bis(oxazolin)-Liganden wurde gefunden. Die erreichten Selektivitäten sind
beachtlich. Während der maximale Diastereoisomerenüberschuss 97% betrug,
wurde für das frans-lsomere ein Enantiomerenüberschuss von 80% erreicht. Da
diese beiden Höchstwerte in der selben Reaktion erzielt wurden, enthielt das
Produkt also das Hauptenantiomer in 89%.
Sowohl Pfaltz und Mitarbeiter9 als auch Masamune und Mitarbeiter11 haben in der
Kupfer-katalysierten Cyclopropanierung mit Diazoestern beobachtet, dass die
translcis-Verhältnisse fast ausschliesslich von der Struktur des Olefins und dem
eingesetzten Diazoester, aber nicht von der Struktur des Katalysators abhängen. Im
Falle der oben gezeigten Cu(l)-katalysierten Cyclopropanierung mit
Trimethylsilyldiazomethan ist eine starke Abhängigkeit der Diastereoselektivitäten
von den eingesetzten Liganden zu beobachten.
Die bereits guten Selektivitäten dieser Reaktion und die Beobachtung, dass eine
Erhöhung der Reaktionstemperatur einen minimalen Einfluss auf die Selektivität
hat, aber die Ausbeuten deutlich verbessert, stimmt für diese Reaktion optimistisch.
4.6 Literatur
(1) Paulissen, R.; Hubert, A. J.; Teyssie, P. Tetrahedron Lett. 1972, 1465-1466.
(2) Mende, U.; Radüchel, B.; Skuballa, W.; Vorbrüggen, H. Tetrahedron Lett.
1975,0, 629-632.
(3) Suda, M. Synthesis Î96JI, 714.

4. Cu(l)-katalysierte, asymmetrische Cyclopropanierung mit TMSCHN2 89
(4) Doyle, M. P., Ed. Comprehensive Organometallic Chemistry II; Elsevier:
Oxford, 1995; Vol. 12.
(5) Denmark, S. E.; Stavenger, R. A.; Faucher, A.-M.; Edwards, J. P. J. Org.Chem. 1997, 62, 3375-3389.
(6) Denmark, S. E.; O'Connor, S. P. J. Org. Chem. 1997, 62, 584-594.
(7) Salomon, R. G.; Kochi, J. K. J. Am. Chem. Soc. 1973, 95, 3300-3310.
(8) Singh, V. K.; DattaGupta, A.; Sekar, G. Synthesis 1997, 137-149.
(9) Fritschi, H.; Leutenegger, U.; Pfaltz, A. Helv. Chim. Acta 1988, 71, 1553-
1565.
(10) Lowenthal, R. E.; Abiko, A.; Masamune, S. Tetrahedron Lett. 1990, 31, 6005-
6008.
Lowenthal, R. E.; Masamune, S. Tetrahedron Lett. 1991, 32, 7373-7376.
Evans, D. A.; Woerpel, K. A.; Hinman, M. M.; Faul, M. M. J. Am. Chem. Soc.
1991, 113, 726-728.
Müller, D.; Umbricht, G.; Weber, B.; Pfaltz, A. Helv. Chim. Acta 1991, 74, 232-
240.
Evans, D. A.; Woerpel, K. A.; Scott, M. J. Angew. Chem., Int. Ed. Engl. 1992,
31, 430-432.
Black, T. H. Aldrichimica Acta 1983, 16, 3-10.
Shioiri, T.; Aoyama, T. Advances in the Use of Synthons in Organic
Chemistry, JAY Press Ltd, 1993; Vol. 1.
Mori, S.; Sakai, I.; Aoyama, T.; Shioiri, T. Chem. Pharm. Bull. 1982, 30,
3380-3382.
Seyferth, D.; Dow, A. W.; Menzel, H.; Flood, T. C. J. Am. Chem. Soc. 1968,
90, 1080-1082.
Seyferth, D.; Menzel, H.; Dow, A. W.; Flood, T. C. J. Organomet. Chem. 1972,
44, 279-290.
Ashe, A. J. J. Am. Chem. Soc. 1973, 95, 818-821.
Aoyama, T.; Iwamoto, Y.; Nishigaki, S.; Shioiri, T. Chem. Pharm. Bull. 1989,
37, 253-256.
Park, S.-B.; Nishiyama, H.; Itoh, Y.; Itoh, K. J. Chem. Soc. Chem. Commun.
1994, 1315-1316.
Maas, G.; Seitz, J. Tetrahedron Lett. 2001, 42, 6137-6140.
Hintermann, L. Dissertation ETH 2000, Nr. 13892.
Burckhardt, U. Dissertation ETH 1997, Nr. 12167.
Schnyder, A. Dissertation ETH 1996, Nr. 11724.
Pioda, G. Dissertazione ETH 1999, Nr. 13405.
Baumann, M. Dissertation ETH2000, Nr. 13851.
Aeby, A. Dissertation ETH 1999, Nr. 13160.
Hayashi, T.; Matsumoto, Y.; Ito, Y. J. Am. Chem. Soc. 1988, 110, 5579-5581.
Barbara, P.; Togni, A. Organometallics 1995, 14, 3570-3573.
Diaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C; Prieto, F.; Perez, P. J.
Organometallics 1999, 18, 2601-2609.
Sanders, C. J.; Gillespie, K. M.; Scott, P. Tetrahedron: Asymmetry 2001, 12,
1055-1061.
Llewellyn, D. B.; Adamson, D.; Arndtsen, B. A. Org. Lett. 2000, 2, 4165-4168.
Yamada, Y.; Yamamoto, T.; Okawara, M. Chem. Lett. 1975, 361-362.

5. Experimenteller Teil
5.1 Allgemeines
Chemikalien und Lösungsmittel
Die verwendeten Lösungsmittel (Fluka, Riedel-de Haën, Merck, Baker, Scharlau)
waren von der Qualität puriss. p. a. und wurden so eingesetzt oder unter Argon
frisch über geeigneten Trocknungsmitteln destilliert (THF über K; Benzol, Toluol,
Et20, Pentan, Hexan, Heptan über Na, Benzophenon; CH2CI2, CH3CN über CaH2;
MeOH über Mg; 'PrOH über Na/Pr-Phthalat). Deuterierte Lösungsmittel stammten
von Dr. GlaserAG (CDCI3), ARMARAG (THF-d8) und Cambridge Isotope
Laboratories (alle anderen) und wurden wie obige gereinigt oder wie erhalten
eingesetzt.
Dimethylsulfid-boran (Acros), Arsentrichlorid, Arsentrioxid (AlfaAesar), 1-Brom-3,5-
difluorbenzol, Chlormethylmethylether, Diiodmethan, Natriumamid, 1-Phenyl-1-
cyclohexen, Trimethylsilyldiazomethan-Lsg. 2.0M in Hexan (Aldrich), Aluminium-
trichlorid, 1,3-Bis-(trifluormethyl)-5-brom-benzol, 1-Brom-4-chlorbenzol, 1-Brom-4-
fluorbenzol, 1-Brom-4-methoxybenzol, 1-Brom-3-trifluormethylbenzol, 1-Brom-4-
trifluormethylbenzol, Butyllithium-Lsg. ca. 1.6M in Hexan, Chlorameisensäure-
trichlormethylester, Cyclohexanon, 1,3-Dibrombenzol, Dieisennonacarbonyl,
Diethylzink, Essigsäureanhydrid, Kupfer(l)-chlorid, Kupfer(ll)-chlorid, Kupfercyanid,
Kupfer(l)-trifluormethansulfonat Benzol-Komplex, Lithiumaluminiumhydrid, Lithium-
bis(trimethylsilyl)amid, Methylbromid, Methylcyclopentadien dimer, Methyliodid,
Natrium-bis(trimethylsilyl)amid, Natriumhexafluoroantimonat, Natriumiodid,
Natriumtetrafluoroborat, Natriumtetraphenylborat, Pyridin, Styrol, Toluol-4-
sulfonsäure Monohydrat, Triethylamin, Triethylsilan, Trifluoressigsäure,
Trimethyloxonium-tetrafluoroborat (Fluka), Triphenylarsin (Merck), (R)-N,N-
Dimethyl-1-ferrocenylethylamin (Solvias) und Chlorwasserstoff (g, wasserfrei)
(PanGas) wurden wie erhalten eingesetzt. Die Konzentration der Butyllithium-Lsg.
in Hexan wurde vor Gebrauch bestimmt.1

5. Experimenteller Teil 91
Tri-(4-fluorphenyl)-arsin,2 Tri-(4-chlorphenyl)-arsin,2 Tri-(4-trifluormethylphenyl)-
arsin3 und Tri-(3-trifluormethylphenyl)-arsin3 wurden mittels Grignard-Reaktion
erhalten.
Folgende Substanzen wurden nach Literaturangaben hergestellt: As-Chlor-bis-(4-
trifluormethylphenyl)-arsin,4 As-Chlor-bis-(3-trifluormethylphenyl)-arsin,4 As-Chlor-
bis-(3,5-bis(trifluormethyl)phenyl)-arsin,4 Methyltriphenylarsonium-bromid,5
Methyltris(4-chlorphenyl)arsonium-iodid,6 Methyltriphenylantimonium-iodid,7
Methyltriphenylantimonium-tetrafluoroborat,7 N,/V-Dicyclohexylethylendiimin,81 -
Cyclohex-1-enyl-4-methoxybenzol,9 [FeCI2(/V,/V'-Dicyclohexylethylendiimin)],10
[(C5Me5)Fe(CO)2]2,11 [(C5H5)Fe(CO)2CI],12-14 [(C5H5)Fe(CO)2Br],14 [(C5H5)Fe(CO)2l],14
[(C5Me5)Fe(CO)2CI],15[(C5H5)Fe(CO)2(THF)]BF4,16[(C5H5)Fe(CO)2Styrol]BF4,17
[(C5Me5)Fe(CO)2(THF)]BF4,18[(C5H5)Fe(CO)(PPh3)Br],19[(C5H5)Fe(dppe)CI],20
[(C5Me5)Fe(dppe)CI],21[(C5H5)Fe(dppe)(MeCN)]Br,22[(C5H5)Fe(MeCN)2(PPh3)]PF6,2:
[(C5H5)Fe(CO)3]PF6,24 [(îi5-lndenyl)Fe(CO)2]2,25 [(ri5-lndenyl)Fe(CO)2l],26 [Rul33 H20],2
fA-ans-[RuCI2(PPh3)3],28[RuCI(A/,/\/'-Bis[o-(diphenylphosphin)benzyliden]-(1S,2S)-
diimincyclohexan)]PF6,29 frans-[RuCI2(dppp)2],30 [RuCI(dppp)2]PF6,30
[(C5H5)Ru(C6H6)]PF6,31[(C5H5)Ru(PPh3)2CI],32[(C5H5)Ru(MeCN)3]PF6,31
[(C5H5)Ru(MeCN)2CO]PF6,31[(C5H5)Ru(PPh3)2(MeCN)]BF4,33
[(C5H5)Ru(MeCN)2P(OMe)3]PF6,31[(C5H5)Mo(CO)3H],34[(C5H5)W(CO)3H],34
[(C5H5)Mo(CO)3(MeCN)]BF4,35[(C5H5)W(CO)3(MeCN)]BF4,35
[(C5H5)Fe(CO)2(CH2OCH3)],36[(C5H5)Fe(CO)2(CH2OCH2CH3)],37
[(C5H5)Fe(CO)2(CH2CI)],36[(C5H5)Ru(CO)2(CH2CI)],38
[(C5H5)Fe(CO)2(CH2PPh3)]BPh4,39[(C5H5)Fe(CO)2(CH2AsPh3)]CI,40
[(C5H5)Fe(CO)2(CH2AsPh3)]BPh4,39[(C5H5)Rh(PMe3)(C2H4)],41'42
[(C5H5)Rh(CH2l)(PMe3)l],43[(C5H5)Rh(CH2AsPh3)(PMe3)l]PF6,43
[(C5H5)Mo(CO)3(CH2CI)],36[(C5H5)W(CO)3(CH2CI)],36[PdCI2(COD)],44
[PdCIMe(COD)],45 [PtCI2(COD)],46 [Ptl2(COD)],47 [PdCI2((/:?)-(S)-JOSIPHOS)],48
3-terf-Butyl-1 -{(R)-1 -[(Sp)-2-(diphenylphosphanyl)ferrocenyl]ethyl}-1 H-pyrazol
(PPFPz'BuJ.^^PPFPz'BuJPdCy,49 1-Trimethylsilyl-2-phenylcyclopropan,50

5. Experimenteller Teil 92
3(5)-(9-Anthryl)-5(3)-methyl-1 H-pyrazol,51 3(5)-(1 -Adamantyl)-1 H-pyrazol,51
(R)-N, A/-Dimethyl-1 -[(S)-2-{3-(1 -phenyl)-1 H-pyrazolyl}ferrocenyl]ethylamin,51
1 -{(fî)-1 -[(S)-2-{3-(1 -Phenyl)-1 H-pyrazolyl}ferrocenyl]ethyl}-3-(1 -adamantyl)-1 H-
pyrazol.51
Arbeitstechniken, verwendete Geräte
Alle Reaktionen mit luft- oder feuchtigkeitsempfindlichen Substanzen wurden
mittels Standard-Schlenktechnik unter Argonatmosphäre oder in der glove-box
unter Stickstoffatmosphäre durchgeführt. Dünnschichtchromatographie (DC):
Kieselgel, Fertigfolien Kieselgel 60 mit Fluoreszenzindikator UV254 (Macherey-
Nagef), Laufmittel sind jeweils angegeben, Detektion unter UV-Licht (254 nm) oder
durch Besprühen mit einem geeigneten Reagens. Dickschichtchromatographie
(TLC): Kieselgel, PSC-Platten (20 cm, 20 cm, 2 mm), Kieselgel 60 F254 (Merck),
Laufmittel sind jeweils angegeben, Detektion unter UV-Licht (254 nm).
Säulenchromatographie ("flash"-Chromatographie, FC, Luft/N2-Ueberdruck ca. 0.2
bar): Kieselgel 60 (Fluka), Korngrösse 40-63 um; Alox Typ 507 C neutral (Fluka),
Korngrösse 100-125 mesh. Spezifische Drehung: Perkin-Elmer-341-Po\ar\rr\e\er,
10cm-Zelle, c in g/100 ml. Schmelzpunkte: Electrothermal, mit Metallheizblock. IR:
Perkin-Elmer-Paragon 7000-FT-IR-Spektrometer, Absorptionsmaxima in cm"1. 1H-
(250.13 MHz), 13C- (62.90 MHz), 19F- (188.31 MHz) und 31P- (101.26 MHz) NMR-
Spektren wurden mit einem Bruker DPX-250 Spektrometer aufgenommen. Die
chemischen Verschiebungen (<5) sind in ppm relativ zu TMS (1H, 13C) und CFCI3
(19F), Kopplungskonstanten (J) in Hz angegeben. Massenspektrometrische
Messungen wurden vom MS-Service des Laboratoriums für Organische Chemie
der ETHZ durchgeführt (Signallage m/z, Intensität in % des Basispeaks, [M\+ =
Molekülion). Die Elementaranalysen wurden vom Mikroelementaranalyse-Service
des Laboratoriums für Organische Chemie der ETHZ gemessen. Quantitative
gaschromatographische Analysen wurden auf einem Fisons Instruments GC 6000
mit einer SE 54 Säule (Macherey-Nagel, 25 m x 0.25 mm, 0.25 Jim Film)
durchgeführt.

5. Experimenteller Teil 93
Enantiomerentrennungen wurden mit einer ß-dex 120 Säule (Supelco, 30 m x 0.25
mm, 0.25 um Film) bei einem He-Fluss von 1.4 ml/min durchgeführt. Temperatur¬
programme und Retentionszeiten sind angegeben. HPLC: Hewlett-Packard Series
7050-Gerät mit UV-Detektor, chirale Säule: Daicel Chiralcel OD-H (4.6 x 250 mm,
Korngrösse 5 u.m), Elutionsbedingungen sind jeweils angegeben.
5.2 Allgemeine Arbeitsvorschriften (AAV)
Herstellung der Triarylarsine (AAV1)
In einem Dreihalskolben mit Rückflusskühler vorgelegtes Magnesiumpulver wurde
mit Et20 überdeckt und unter Rühren tropfenweise mit dem Bromaryl versetzt bis die
Reaktion unter Erwärmung startete. Diese Suspension wurde mit Et20 verdünnt und
tropfenweise mit einer Lösung des Bromaryls in Et20 versetzt. Nach Beendigung
der Zugabe wurde die Reaktionsmischung 1 h weitergerührt und dann tropfenweise
unter Rühren mit Arsentrichlorid versetzt. Nach Rühren unter Rückfluss über Nacht
wurde langsam 0.1 M wässrige HCI-Lsg. zugegeben. Die erhaltene Suspension
wurde dreimal mit Et,0 extrahiert. Die vereinigten organischen Phasen wurden mit
NaCI-Lsg. gewaschen, über MgS04 getrocknet und eingedampft. Nach dem
Trocknen im HV wurde das Rohprodukt wie beschrieben gereinigt.
Herstellung der (/?)-/V,/V-Dimethyl{1-[(S)-2-(diarylarsino)ferrocenyl]-
ethyl}amine, R*AsFA (AAV2)
Zu einer Lösung von (fî)-/V,A/-Dimethyl-1-ferrocenylethylamin (1 equiv.) in Et20
wurde nBuLi-Lsg. in Hexan (1.2 equiv.) zugegeben. Die erhaltene Lösung wurde 2
h bei RT gerührt und dann mit dem As-Chlor-diarylarsin (1.4 - 2.0 equiv.) in 10 ml
Et,0 versetzt. Nach 4 h Rühren unter Rückfluss wurde die Reaktionsmischung im
Eisbad abgekühlt und langsam mit wässriger NaHC03-Lsg. versetzt. Die
organische Phase wurde abgetrennt, über MgS04 getrocknet und eingeengt. Nach
dem Trocknen im HV wurde das Rohprodukt wie beschrieben gereinigt.

5. Experimenteller Teil 94
Herstellung der Acetoxy-Derivate von R*AsFA (AAV3)
Eine Lösung von R*AsFA in Essigsäureanhydrid wurde 3 h bei 100 °C gerührt.
Nach Abkühlen auf RT wurde H20 zugegeben. Die wässrige Lösung wurde dreimal
mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über MgS04
getrocknet und eingedampft. Nach dem Trocknen im HV wurde das Rohprodukt wie
beschrieben gereinigt.
Herstellung der (S)-1-(Diarylarsino)-2-ethylferrocene (AAV4)
Zu einer Suspension von LAH (4-5 equiv.) in THF wurde bei 0 °C 1 equiv. des
Acetoxy-Derivates zugegeben. Die erhaltene Mischung wurde 1 h bei RT gerührt
und dann mit Aluminiumtrichlorid (5-6 equiv.) versetzt. Nach 2 h Rühren bei RT
wurde ges. NaHC03-Lsg. zugegeben. Die organische Phase wurde abgetrennt,
eingedampft und der erhaltene Rückstand in Et,0 gelöst. Diese Lösung wurde mit
ges. NaHC03-Lsg. und H20 gewaschen, über MgS04 getrocknet und eingedampft.
Nach dem Trocknen im HV wurde das Rohprodukt wie beschrieben gereinigt.
Herstellung der Komplexe [Fp(CH2AsR3)]BF4 (AAV5)
[Fp(CH2CI)] und das Triaryiarsin wurden in MeOH suspendiert. Die
Reaktionsmischung wurde über Nacht bei 80 °C unter Rückfluss gerührt, auf RT
abgekühlt und zu Natriumtetrafluoroborat filtriert. Nach 15 min Rühren wurde die
Reaktionsmischung über Nacht bei -20 °C stehengelassen. Der gelbe
Niederschlag wurde abfiltriert und im HV getrocknet. Extraktion mit CH2CI2,
Abziehen des Lösungsmittels und Kristallisation aus CH2CI2/Pentan bei -20 °C
ergab reine Komplexe als gelbe Feststoffe.

5. Experimenteller Teil 95
5.3 Triarylarsine
Tri(4-fluorphenyl)arsin (A1). Aus 4.946 g (203.5
mmol) Mg, 35.430 g (202.4 mmol) 1-Brom-4-fluorbenzol j\s-
und 5.67 ml (67.2 mmol) AsCI3, gemäss AAV1. FC
(Hexan/Ethylacetat 10/1) ergab 20.232 g (84%) A1 als braunen Feststoff. 1H-NMR
(CDCI3): 5 7.32-7.24 (m, 6 H), 7.12-7.01 (m, 6 H). 19F-NMR (CDCI3): 5-112.8 (s).
MS (El+): m/z 360 (41, [M\+), 265 (9, [M-Ar]+), 190 (18, [Ar2]+), 171 (8, [Ar2-F]+), 170
(100, [AsAr]+), 95(1, [Ar]+).
Tri(4-chlorphenyl)arsin (A2). Aus 1.414 g (58.2
mmol) Mg, 10.571 g (55.2 mmol) 1-Brom-4-chlorbenzol As-
und 1.54 ml (18.3 mmol) AsCI3, gemäss AAV1. FC
(Hexan/Ethylacetat 10/1) ergab 6.801 g (91%) A2 als gelblichen Feststoff. DC
(Hexan/Ethylacetat 10/1): R, 0.74. 1H-NMR (CDCI3): 5 7.35-7.32 (m, 6 H), 7.25-7.21
(m, 6 H). 13C-NMR (CDCI3): 5137.2 (ar C), 135.3 (ar C), 134.8 (ar CH), 129.1 (ar
CH). MS (El+): m/z 408 (14, [M]+), 297 (8, [M-Ar]+), 261 (7, [M-Ar-Cl]+), 222 (13, [Ar2]+),
186 (100, [AsArf), 151 (31, [AsAr-CI]+), 111 (3, [Ar]+).
Tri[3-(trifluormethyl)phenyl]arsin (A3). Aus 2.981 g
(122.6 mmol) Mg, 27.440 g (122.0 mmol) 1-Brom-3-
trifluormethylbenzol und 3.33 ml (39.5 mmol) AsCI3, ^s . .
gemäss AAV1. FC (Hexan/Ethylacetat 10/1) ergab 18.331 \ \ //
g (91%) A3 als braunes Oel. DC (Hexan/Ethylacetat 10/1):
Ri 0.55. 1H-NMR (CDCI3): 5 7.69-7.62 (m, 6 H), 7.56-7.48 (m, 6 H). 13C-NMR
(CDCI3): 5139.5 (s, ar C), 136.8 (q, J(C,F) = 1.4, ar CH), 131.4 (q, J(C,F) = 32.4, ar
C), 130.3 (q, J(C,F) = 3.8, ar CH), 129.5 (s, ar CH), 126.1 (q, J(C,F) = 3.7, ar CH),
123.8 (q, J(C,F) = 272.7, CF3). 19F-NMR (CDCI3): 5-63.1 (s). MS (El+): m/z 510 (61,
[M\+), 491 (9, [M-F]+), 365 (8, [M-Ar]+), 290 (3, [Ar2]+), 271 (100, [Ar2-F]+), 220 (27,
[AsAr]+), 145 (2, [Ar]+), 126 (3, [Ar-F]+), 107 (14, [Ar-2F]+).
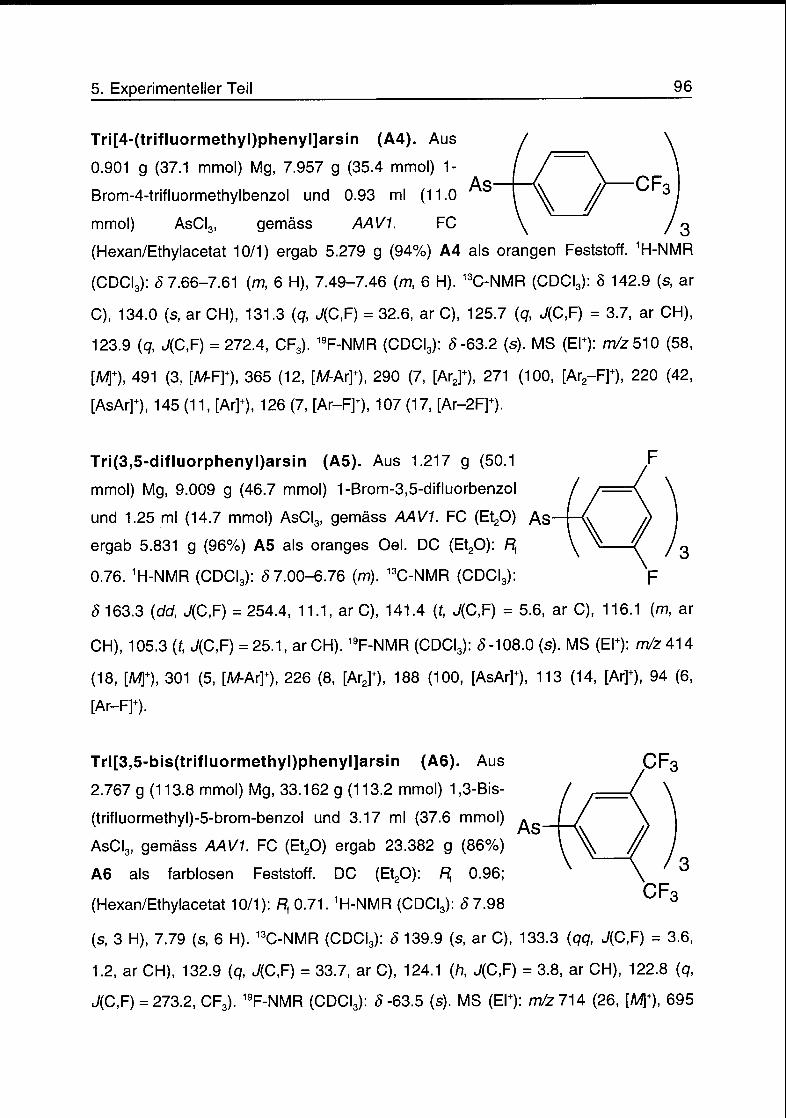
5. Experimenteller Teil 96
Tri[4-(trifluormethyl)phenyl]arsin (A4). Aus
0.901 g (37.1 mmol) Mg, 7.957 g (35.4 mmol) 1-
Brom-4-trifluormethylbenzol und 0.93 ml (11.0As_T\\ // CF3
mmol) AsCI3, gemäss AAV1. FC \' '
/o(Hexan/Ethylacetat 10/1) ergab 5.279 g (94%) A4 als orangen Feststoff. 1H-NMR
(CDCI3): 5 7.66-7.61 (m, 6 H), 7.49-7.46 (m, 6 H). 13C-NMR (CDCI3): Ô 142.9 (s, ar
C), 134.0 (s, ar CH), 131.3 (q, J(C,F) = 32.6, ar C), 125.7 (q, J(C,F) = 3.7, ar CH),
123.9 (q, J(C,F) = 272.4, CF3). 19F-NMR (CDCI3): 5-63.2 (s). MS (El+): m/zSîQ (58,
[M\+), 491 (3, [M-F]+), 365 (12, [M-Ar]+), 290 (7, [Ar2]+), 271 (100, [Ar2-F]+), 220 (42,
[AsAr]+), 145 (11, [Ar]+), 126 (7, [Ar-F]+), 107 (17, [Ar-2F]+).
Tri(3,5-difluorphenyl)arsin (A5). Aus 1.217 g (50.1
mmol) Mg, 9.009 g (46.7 mmol) 1-Brom-3,5-difluorbenzol
und 1.25 ml (14.7 mmol) AsCI3, gemäss AAV1. FC (Et20) As-
ergab 5.831 g (96%) A5 als oranges Oel. DC (Et20): /^
0.76. 1H-NMR (CDCI3): 5 7.00-6.76 (m). 13C-NMR (CDCI3):
5 163.3 (dd, J(C,F) = 254.4, 11.1, ar C), 141.4 (t, J(C,F) = 5.6, ar C), 116.1 (m, ar
CH), 105.3 (t, J(C,F) = 25.1, arCH). 19F-NMR (CDCI3): 5-108.0 (s). MS (El+): m/z 414
(18, [M\+), 301 (5, [M-Ar]+), 226 (8, [Ar2]+), 188 (100, [AsAr]+), 113 (14, [Ar]+), 94 (6,
[Ar-F]+).
Tri[3,5-bis(trifluormethyl)phenyl]arsin (A6). Aus
2.767 g (113.8 mmol) Mg, 33.162 g (113.2 mmol) 1,3-Bis-
(trifluormethyl)-5-brom-benzol und 3.17 ml (37.6 mmol)
AsCI3, gemäss AAV1. FC (Et20) ergab 23.382 g (86%)
A6 als farblosen Feststoff. DC (Et20): R^ 0.96;
(Hexan/Ethylacetat 10/1): fîf 0.71. 1H-NMR (CDCI3): 5 7.98
(s, 3 H), 7.79 (s, 6 H). 13C-NMR (CDCI3): 5 139.9 (s, ar C), 133.3 (qq, J(C,F) = 3.6,
1.2, ar CH), 132.9 (q, J(C,F) = 33.7, ar C), 124.1 (h, J(C,F) = 3.8, ar CH), 122.8 (q,
J(C,F) = 273.2, CF3). 19F-NMR (CDCI3): 5-63.5 (s). MS (El+): m/z 714 (26, [M\+), 695
As

5. Experimenteller Teil 97
(10, [M-FY), 501 (9, [M-Ar]+), 426 (4, [Ar2]+), 407 (100, [Ar2-F]+), 288 (56, [AsAr]+), 213
(11,[Ar]+), 194(13,[Ar-F]+).
Tri(3-bromphenyl)arsin (A7). Aus 1.689 g (69.5 Br
mmol) Mg, 16.353 g (69.3 mmol) 1,3-Dibrombenzol und
1.90 ml (22.6 mmol) AsCI3, gemäss AAV1. FC
(Hexan/Ethylacetat 20/1) ergab 9.652 g (79%) A7 als
gelbes Oel. DC (Hexan/Ethylacetat 20/1): R^ 0.65;
(Hexan/Ethylacetat 10/1): R, 0.69. 1H-NMR (CDCI3): 5 7.53-7.09 (m). 13C-NMR
(CDCI3): 5141.0(arC), 136.1 (arCH), 132.2 (ar CH), 132.0 (ar CH), 130.5 (ar CH),
123.5 (arC). MS (El+): m/z 542, 544 (1, [MY), 463 (1, [M-Br]+), 387 (2, [M-Ar]+), 230,
232 (28, [AsAr]+), 151 (100, [AsAr-Br]+), 76 (19, [C6H4]+).
Tri(3-cyanphenyl)arsin (A8). Eine Mischung von CN
4.278 g (7.9 mmol) A7 und 5.543 g (61.9 mmol)
Kupfercyanid in Pyridin wurde 44 h bei 240 °C gerührt. Die ^s
Reaktionsmischung wurde dreimal mit 50 ml Et20
extrahiert. Die vereinigten organischen Phasen wurden mit
wässriger NH3-Lsg., 0.1 M HCI-Lsg., ges. NaCI-Lsg. und H20 gewaschen, über
MgS04 getrocknet, eingeengt und im HV getrocknet. FC (Et20) ergab 1.696 g (56%)
A8 als gelben Feststoff. DC (Et20): fl; 0.70. 1H-NMR (CD2CI2): 5 7.79-7.38 (m). 13C-
NMR (CD2CI2): 5139.6 (CN), 138.1 (ar CH), 137.1 (ar CH), 133.4 (ar CH), 130.2 (ar
CH), 118.5 (ar C), 114.0 (ar C). MS (El+): m/z 381 (27, [MY), 279 (12, [M-Ar]+), 177
(100, [AsAr]+), 151 (8, [AsAr-CN]+), 102 (9, [Ar]+).
\ /

5. Experimenteller Teil 98
5.4 Diarylarsinchloride
Synthese von Bis[3-(trifluormethyl)phenyl]-
arsinchlorid (A9). Eine Grignard-Lösung aus
18.2g (81 mmol) 1-Brom-3-trifluormethylbenzol und qi as-
1.8 g (74 mmol) Mg in 10 ml Et20 wurde mit 35 ml
Benzol verdünnt und bei 0 °C unter starkem Rühren
rasch mit 3.4 g (17 mmol) Arsenoxid versetzt. Diese Reaktionsmischung wurde 17 h
unter Rückfluss gerührt und mit 20 ml HCl conc. versetzt. Nach 1 h Rühren unter
Rückfluss und Abkühlen auf RT wurde die organische Phase abgetrennt und die
wässrige Phase zweimal mit Benzol extrahiert. Die vereinigten organischen
Phasen wurden über CaCI2 getrocknet und eingeengt. Destillation des erhaltenen
Oels (120 °C, 3 mbar) ergab 8.0 g (59%) A9 als gelbes Oel. 1H-NMR (CDCI3): 5
7.68-7.65 (m, 4 H), 7.55-7.45 (m, 4 H). 13C-NMR (CDCI3): 5 139.4 (ar C), 136.7 (ar
CH), 131.3 (q, J(C,F) = 32.3, ar C), 130.2 (q, J(C,F) = 3.8, ar CH), 129.5 (ar CH),
126.1 (q, J(C,F) = 3.7, ar CH), 123.8 (q, J(C,F) = 272.9, CF3). MS (El+): m/z 400 (22,
[MY), 381 (10, [M-FY), 365 (6, [M-Cl]+), 290 (18, [Ar2]+), 271 (100, [Ar2-F]+), 255 (18,
[M-Ar]+), 220 (4, [M-Ar-Cl]+), 145 (10, [Ar]+), 107 (19, [Ar-2F]+).
Synthese von Bis[3,5-bis(trifluormethyl)-
phenyljarsinchlorid (A10). Eine Grignard-
Lösung aus 10.982 g (37 mmol) 1-Brom-3,5-p.
»
bis(trifluormethyl)benzol und 0.909 g (37 mmol) Mg
in 10 ml Et20 wurde mit 40 ml Benzol verdünnt.
Nach Zugabe von 1.6 g (8 mmol) Arsenoxid bei 0 °C
unter heftigem Rühren und 12 h Rühren unter Rückfluss wurde die Lösung mit 30
ml HCl conc. versetzt und 2 h unter Rückfluss gerührt. Die organische Phase wurde
abgetrennt und die wässrige Phase zweimal mit Benzol extrahiert. Die vereinigten
organische Phasen wurden über CaCI2 getrocknet und eingeengt. Destillation des
Rückstandes (120 °C, 1 mbar) ergab 8.024 g (94%) A10 als gelbes Oel. 1H-NMR
(CDCI3): 8.04 (s, 4 H), 7.98 (s, 2 H). 13C-NMR (CDCI3): 141.5 (s, ar C), 132.6 (q,
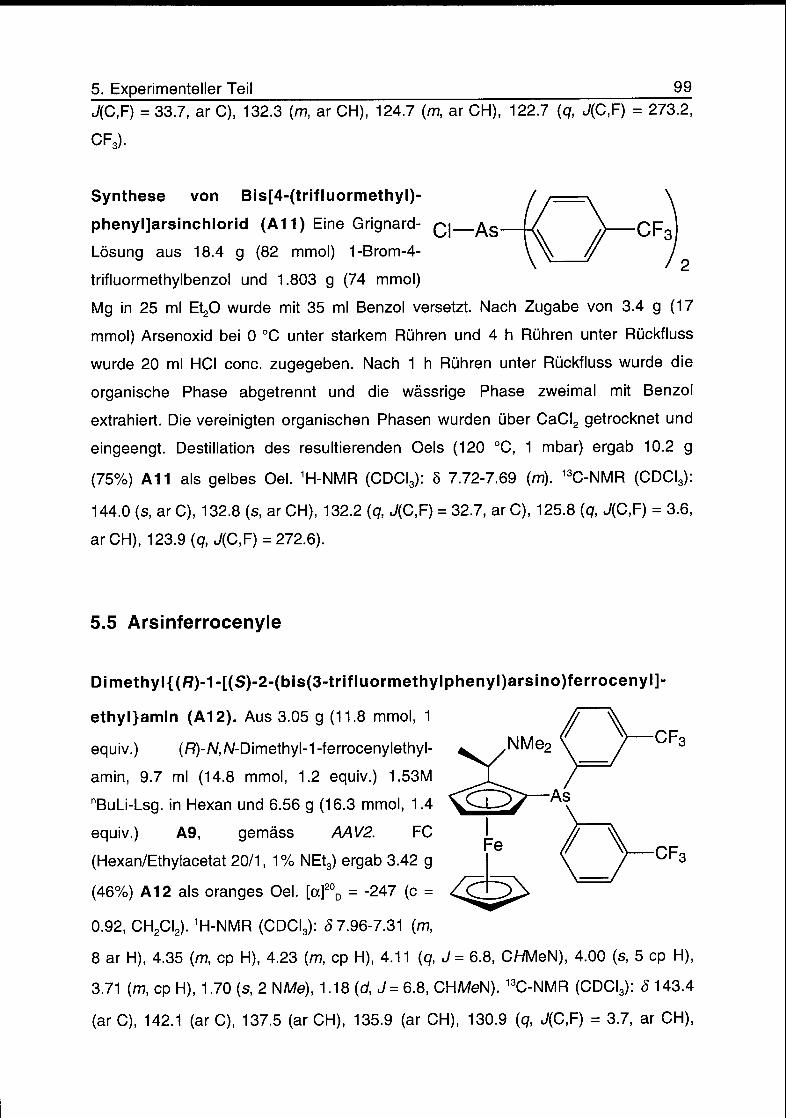
5. Experimenteller Teil 99
J(C,F) = 33.7, ar C), 132.3 (m, ar CH), 124.7 (m, ar CH), 122.7 (q, J(C,F) = 273.2,
CF3).
Synthese von Bis[4-(trifluormethyl)-
phenyljarsinchlorid (A11) Eine Grignard- pi Aq.
Lösung aus 18.4 g (82 mmol) 1-Brom-4-
trifluormethylbenzol und 1.803 g (74 mmol)
Mg in 25 ml Et,0 wurde mit 35 ml Benzol versetzt. Nach Zugabe von 3.4 g (17
mmol) Arsenoxid bei 0 °C unter starkem Rühren und 4 h Rühren unter Rückfluss
wurde 20 ml HCl conc. zugegeben. Nach 1 h Rühren unter Rückfluss wurde die
organische Phase abgetrennt und die wässrige Phase zweimal mit Benzol
extrahiert. Die vereinigten organischen Phasen wurden über CaCI2 getrocknet und
eingeengt. Destillation des resultierenden Oels (120 °C, 1 mbar) ergab 10.2 g
(75%) A11 als gelbes Oel. 1H-NMR (CDCI3): 5 7.72-7.69 (m). 13C-NMR (CDCI3):
144.0 (s, ar C), 132.8 (s, ar CH), 132.2 (q, J(C,F) = 32.7, ar C), 125.8 (q, J(C,F) = 3.6,
ar CH), 123.9 (q, J(C,F) = 272.6).
5.5 Arsinferrocenyle
Dimethyl{(/?)-1-[(S)-2-(bis(3-trifluormethylphenyl)arsino)ferrocenyl]-
ethyljamin (A12). Aus 3.05 g (11.8 mmol, 1
equiv.) (F?)-A/,A/-Dimethyl-1 -ferrocenylethyl-
amin, 9.7 ml (14.8 mmol, 1.2 equiv.) 1.53M
"BuLi-Lsg. in Hexan und 6.56 g (16.3 mmol, 1.4
equiv.) A9, gemäss AAV2. FC
(Hexan/Ethylacetat 20/1, 1% NEt3) ergab 3.42 g
(46%) A12 als oranges Oel. [a]20D = -247 (c = ^C5^
0.92, CH2CI2). 1H-NMR (CDCI3): 5 7.96-7.31 (m,
8 ar H), 4.35 (m, cp H), 4.23 (m, cp H), 4.11 (q, J = 6.8, CHMeN), 4.00 (s, 5 cp H),
3.71 (m, cpH), 1.70 (s, 2 NMe), 1.18 (d, J= 6.8, CH/WeN). 13C-NMR (CDCI3): 5143.4
(ar C), 142.1 (ar C), 137.5 (ar CH), 135.9 (ar CH), 130.9 (q, J(C,F) = 3.7, ar CH),
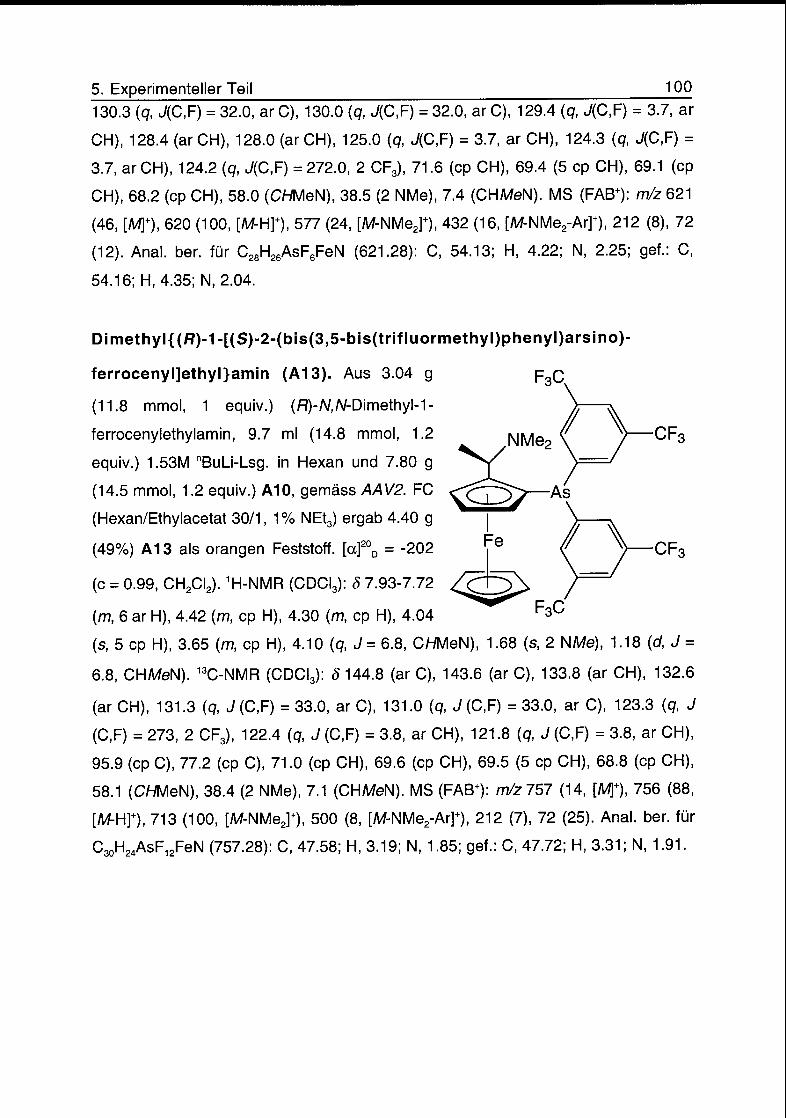
5. Experimenteller Teil 100
130.3 (q, J(C,F) = 32.0, ar C), 130.0 (q, J(C,F) = 32.0, ar C), 129.4 (q, J(C,F) = 3.7, ar
CH), 128.4 (arCH), 128.0 (arCH), 125.0 (q, J(C,F) = 3.7, ar CH), 124.3 (q, J(C,F) =
3.7, arCH), 124.2 (q, J(C,F) = 272.0, 2 CF3), 71.6 (cp CH), 69.4 (5 cp CH), 69.1 (cp
CH), 68.2 (cp CH), 58.0 (CHMeN), 38.5 (2 NMe), 7.4 (CHMeN). MS (FAB+): m/z 621
(46, [MY), 620 (100, [M-HY), 577 (24, [M-NMe2]+), 432 (16, [M-NMe2-Ar]+), 212 (8), 72
(12). Anal. ber. für C28H26AsF6FeN (621.28): C, 54.13; H, 4.22; N, 2.25; gef.: C,
54.16; H, 4.35; N, 2.04.
Dimethyl{(/?)-1-[(S)-2-(bis(3,5-bis(trifluormethyl)phenyl)arsino)-
ferrocenyl]ethyl}amin (A13). Aus 3.04 g p^Q
(11.8 mmol, 1 equiv.) (f?)-A/,A/-Dimethyl-1-
ferrocenylethylamin, 9.7 ml (14.8 mmol, 1.2
equiv.) 1.53M nBuLi-Lsg. in Hexan und 7.80 g
(14.5 mmol, 1.2 equiv.) A10, gemäss AAV2. FC
(Hexan/Ethylacetat 30/1, 1% NEt3) ergab 4.40 g
(49%) A13 als orangen Feststoff. [oc]20D = -202
(c = 0.99, CH2CI2). 1H-NMR (CDCI3): 5 7.93-7.72
(m, 6 ar H), 4.42 (m, cp H), 4.30 (m, cp H), 4.04
(s, 5 cp H), 3.65 (m, cp H), 4.10 (q, J = 6.8, CHMeN), 1.68 (s, 2 NMe), 1.18 (d, J =
6.8, CHMeN). 13C-NMR (CDCI3): 5 144.8 (ar C), 143.6 (ar C), 133.8 (ar CH), 132.6
(ar CH), 131.3 (q, J (C,F) = 33.0, ar C), 131.0 (q, J (C,F) = 33.0, ar C), 123.3 (q, J
(C,F) = 273, 2 CF3), 122.4 (q, J (C,F) = 3.8, ar CH), 121.8 (q, J (C,F) = 3.8, ar CH),
95.9 (cp C), 77.2 (cp C), 71.0 (cp CH), 69.6 (cp CH), 69.5 (5 cp CH), 68.8 (cp CH),
58.1 (CHMeN), 38.4 (2 NMe), 7.1 (CHMeN). MS (FAB+): m/z 757 (14, [MY), 756 (88,
[M-HY), 713 (100, [M-NMe2]+), 500 (8, [M-NMe2-Ar]+), 212 (7), 72 (25). Anal. ber. für
C30H24AsF12FeN (757.28): C, 47.58; H, 3.19; N, 1.85; gef.: C, 47.72; H, 3.31; N, 1.91.
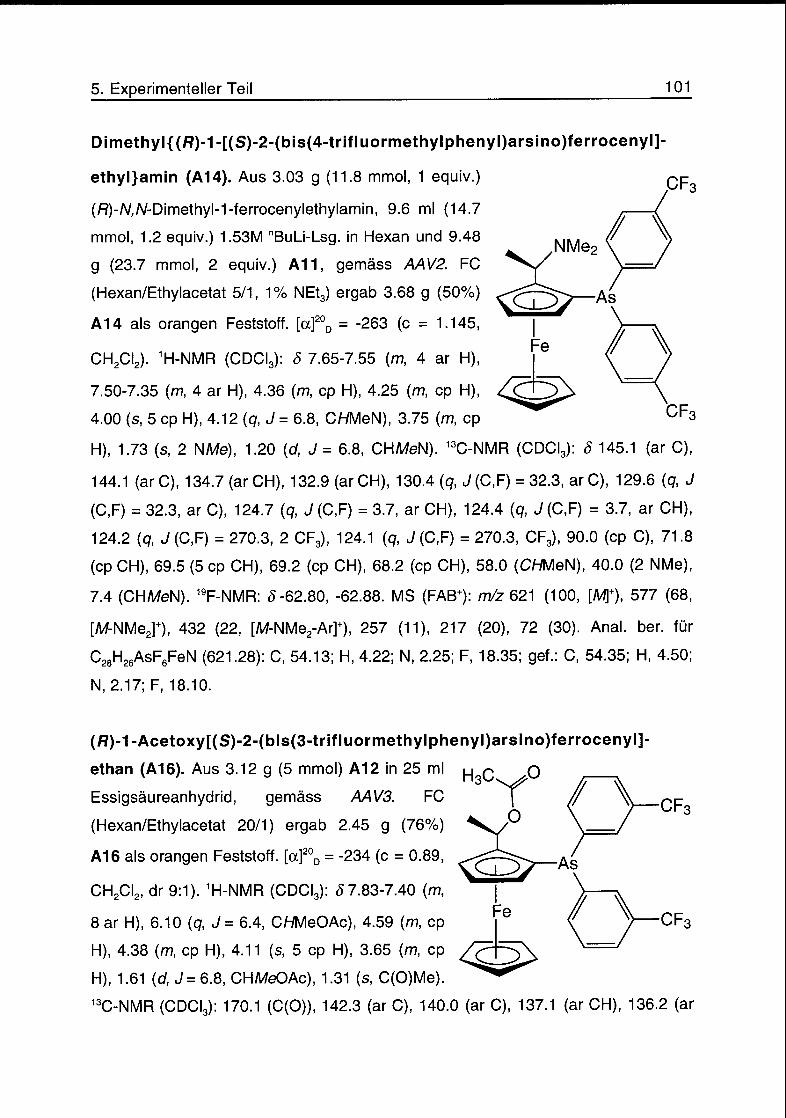
5. Experimenteller Teil 101
Dimethyl{(/?)-1-[(S)-2-(bis(4-trifluormethylphenyl)arsino)ferrocenyl]-
ethyl}amin (A14). Aus 3.03 g (11.8 mmol, 1 equiv.)
(fî)-A/,A/-Dimethyl-1-ferrocenylethylamin, 9.6 ml (14.7
mmol, 1.2 equiv.) 1.53M "BuLi-Lsg. in Hexan und 9.48
g (23.7 mmol, 2 equiv.) A11, gemäss AAV2. FC
(Hexan/Ethylacetat 5/1, 1% NEt3) ergab 3.68 g (50%)
A14 als orangen Feststoff. [a]20D = -263 (c = 1.145,
CH2CI2). 1H-NMR (CDCI3): 5 7.65-7.55 (m, 4 ar H),
7.50-7.35 (m, 4 ar H), 4.36 (m, cp H), 4.25 (m, cp H),
4.00 (s, 5 cp H), 4.12 (q, J = 6.8, CHMeN), 3.75 (m, cp
H), 1.73 (s, 2 NMe), 1.20 (d, J = 6.8, CHMeN). 13C-NMR (CDCI3): 5 145.1 (ar C),
144.1 (arC), 134.7 (arCH), 132.9 (arCH), 130.4 (q, J(C,F) = 32.3, arC), 129.6 (q, J
(C,F) = 32.3, ar C), 124.7 (q, J (C,F) = 3.7, ar CH), 124.4 (q, J (C,F) = 3.7, ar CH),
124.2 (q, J(C,F) = 270.3, 2 CF3), 124.1 (q, J (C,F) = 270.3, CF3), 90.0 (cp C), 71.8
(cp CH), 69.5 (5 cp CH), 69.2 (cp CH), 68.2 (cp CH), 58.0 (CHMeN), 40.0 (2 NMe),
7.4 (CHMeN). 19F-NMR: 5-62.80, -62.88. MS (FAB+): m/z 621 (100, [MJ+), 577 (68,
[M-NMe2]+), 432 (22, [M-NMe2-Ar]+), 257 (11), 217 (20), 72 (30). Anal. ber. für
C28H26AsF6FeN (621.28): C, 54.13; H, 4.22; N, 2.25; F, 18.35; gef.: C, 54.35; H, 4.50;
N,2.17;F, 18.10.
(/?)-1-Acetoxy[(S)-2-(bis(3-trifluormethylphenyl)arsino)ferrocenyl]-
ethan (A16). Aus 3.12 g (5 mmol) A12 in 25 ml|_| q ^q
Essigsäureanhydrid, gemäss AAV3. FC
(Hexan/Ethylacetat 20/1) ergab 2.45 g (76%)
A16 als orangen Feststoff. [a]20D = -234 (c = 0.89,
CH2CI2, dr 9:1). 1H-NMR (CDCI3): 5 7.83-7.40 (m,
8 ar H), 6.10 (q, J= 6.4, CHMeOAc), 4.59 (m, cp
H), 4.38 (m, cp H), 4.11 (s, 5 cp H), 3.65 (m, cp
H), 1.61 (d, J= 6.8, CHMeOAc), 1.31 (s, C(O)Me).
13C-NMR (CDCI3): 170.1 (C(O)), 142.3 (ar C), 140.0 (ar C), 137.1 (ar CH), 136.2 (ar
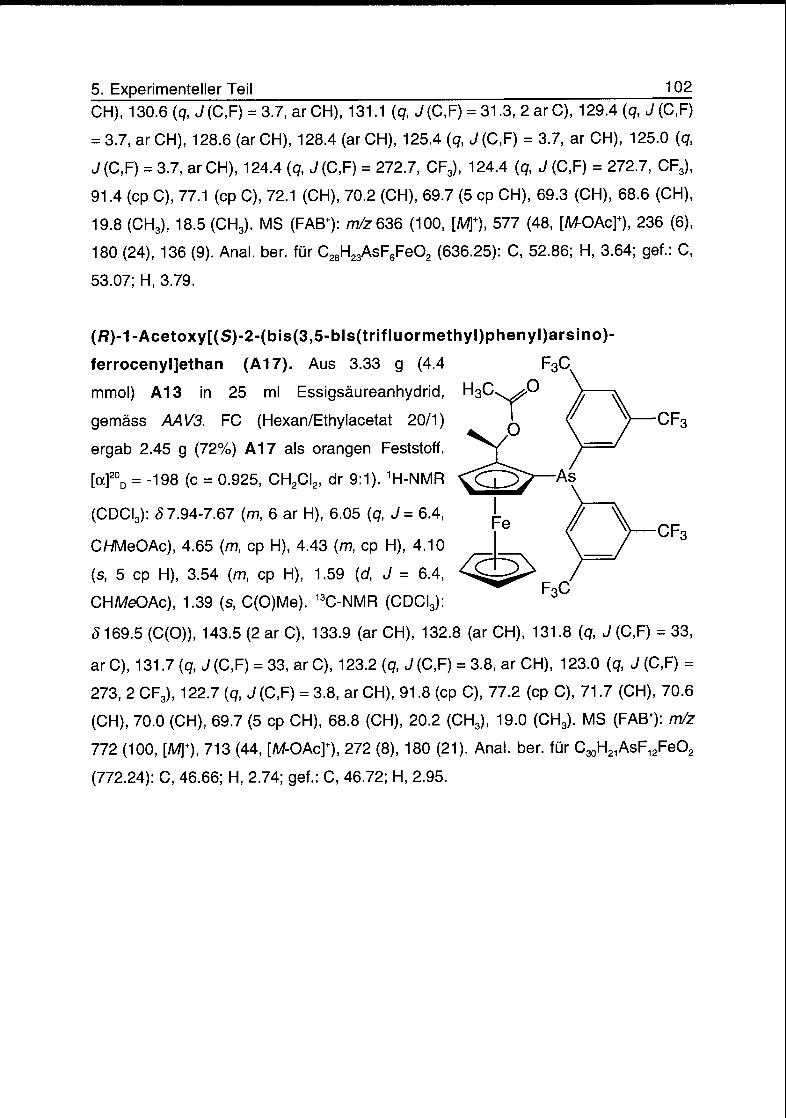
5. Experimenteller Teil 102
CH), 130.6 (q,J (C,F) = 3.7, arCH), 131.1 (qf, J(C,F) = 31.3, 2 arC), 129.4 (q, J (C,F)
= 3.7, arCH), 128.6 (arCH), 128.4 (arCH), 125.4 (q, J(C,F) = 3.7, ar CH), 125.0 (q,
J (C,F) = 3.7, ar CH), 124.4 (q, J (C,F) = 272.7, CF3), 124.4 (q, J (C,F) = 272.7, CF3),
91.4 (cp C), 77.1 (cp C), 72.1 (CH), 70.2 (CH), 69.7 (5 cp CH), 69.3 (CH), 68.6 (CH),
19.8 (CH3), 18.5 (CH3). MS (FAB+): m/z 636 (100, [MJ+), 577 (48, [M-OAc]+), 236 (6),
180 (24), 136 (9). Anal. ber. für C28H23AsF6Fe02 (636.25): C, 52.86; H, 3.64; gef.: C,
53.07; H, 3.79.
(/?)-1-Acetoxy[(S)-2-(bis(3,5-bis(trifluormethyl)phenyl)arsino)-
ferrocenyl]ethan (A17). Aus 3.33 g (4.4 F3C.
mmol) A13 in 25 ml Essigsäureanhydrid, H3C,
gemäss AAV3. FC (Hexan/Ethylacetat 20/1)
ergab 2.45 g (72%) A17 als orangen Feststoff.
[cc]20D = -198 (c = 0.925, CH2CI2, dr 9:1). 1H-NMR
(CDCI3): 5 7.94-7.67 (m, 6 ar H), 6.05 (q, J= 6.4,
CHMeOAc), 4.65 (m, cp H), 4.43 (m, cp H), 4.10
(s, 5 cp H), 3.54 (m, cp H), 1.59 (d, J = 6.4,
CHMeOAc), 1.39 (s, C(O)Me). 13C-NMR (CDCI3):
5169.5 (C(O)), 143.5 (2 ar C), 133.9 (ar CH), 132.8 (ar CH), 131.8 (q, J (C,F) = 33,
arC), 131.7 (q,J (C,F) = 33, arC), 123.2 (q, J(C,F) = 3.8, ar CH), 123.0 (q, J (C,F) =
273, 2 CF3), 122.7 (q, J (C,F) = 3.8, ar CH), 91.8 (cp C), 77.2 (cp C), 71.7 (CH), 70.6
(CH), 70.0 (CH), 69.7 (5 cp CH), 68.8 (CH), 20.2 (CH3), 19.0 (CH3). MS (FAB+): m/z
772 (100, [MJ+), 713 (44, [M-OAc]+), 272 (8), 180 (21). Anal. ber. für C30H21AsF12FeO2
(772.24): C, 46.66; H, 2.74; gef.: C, 46.72; H, 2.95.
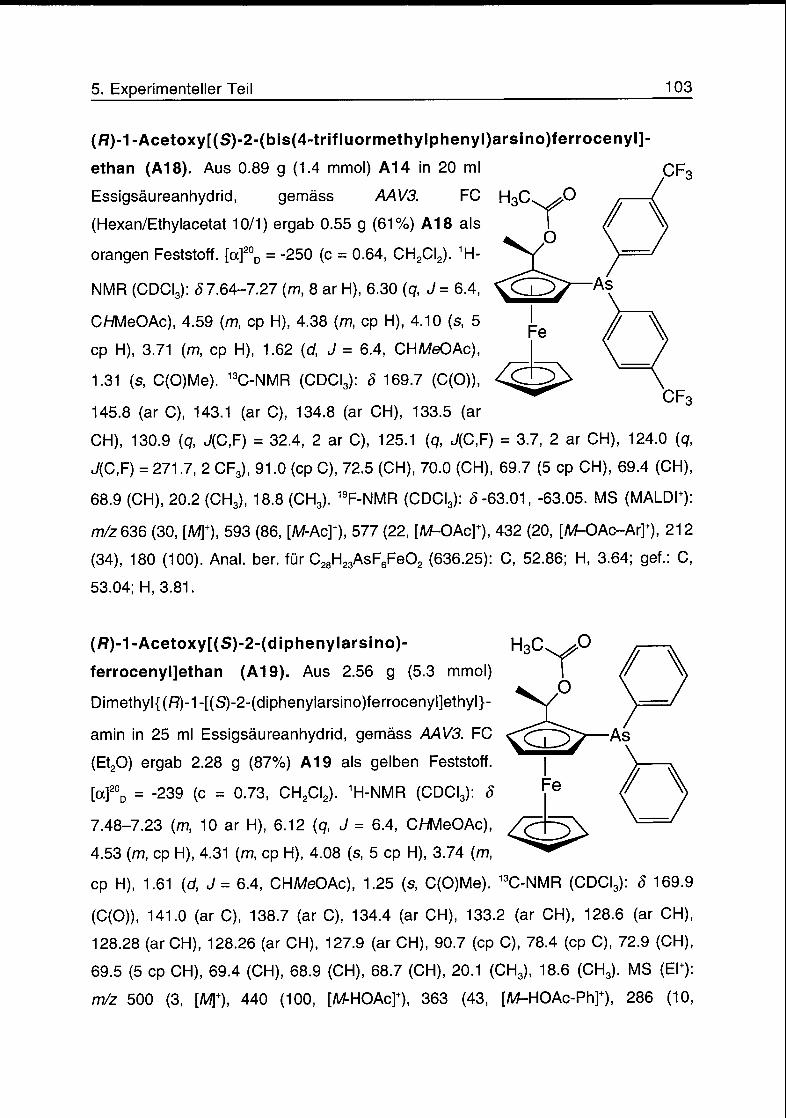
5. Experimenteller Teil 103
(/?)-1-Acetoxy[(S)-2-(bis(4-trifluormethylphenyl)arsino)ferrocenyl]-
ethan (A18). Aus 0.89 g (1.4 mmol) A14 in 20 ml
Essigsäureanhydrid, gemäss AAV3. FC H3C>
(Hexan/Ethylacetat 10/1) ergab 0.55 g (61%) A18 als
orangen Feststoff. [a]20D = -250 (c = 0.64, CH2CI2). 1H-
NMR (CDCI3): 57.64-7.27 (m, 8 ar H), 6.30 (q, J= 6.4,
CHMeOAc), 4.59 (m, cp H), 4.38 (m, cp H), 4.10 (s, 5
cp H), 3.71 (m, cp H), 1.62 (d, J = 6.4, CHMeOAc),
1.31 (s, C(O)Me). 13C-NMR (CDCI3): 5 169.7 (C(O)),
145.8 (ar C), 143.1 (ar C), 134.8 (ar CH), 133.5 (ar
CH), 130.9 (q, J(C,F) = 32.4, 2 ar C), 125.1 (q, J(C,F) = 3.7, 2 ar CH), 124.0 (q,
J(C,F) = 271.7, 2 CF3), 91.0 (cp C), 72.5 (CH), 70.0 (CH), 69.7 (5 cp CH), 69.4 (CH),
68.9 (CH), 20.2 (CH3), 18.8 (CH3). 19F-NMR (CDCI3): 5-63.01, -63.05. MS (MALDI+):
m/z 636 (30, [M]+), 593 (86, [M-Ac]+), 577 (22, [M-OAc]+), 432 (20, [M-OAc-Ar]+), 212
(34), 180 (100). Anal. ber. für C28H23AsF6Fe02 (636.25): C, 52.86; H, 3.64; gef.: C,
53.04; H,3.81.
(fl)-1-Acetoxy[(S)-2-(diphenylarsino)-
ferrocenyljethan (A19). Aus 2.56 g (5.3 mmol)
Dimethyl{(H)-1-[(S)-2-(diphenylarsino)ferrocenyl]ethyl}-
amin in 25 ml Essigsäureanhydrid, gemäss AAV3. FC
(Et20) ergab 2.28 g (87%) A19 als gelben Feststoff.
[cc]20D = -239 (c = 0.73, CH2CI2). 1H-NMR (CDCI3): 5
7.48-7.23 (m, 10 ar H), 6.12 (q, J = 6.4, CHMeOAc),
4.53 (m, cp H), 4.31 (m, cp H), 4.08 (s, 5 cp H), 3.74 (m,
cp H), 1.61 (d, J = 6.4, CHMeOAc), 1.25 (s, C(O)Me). 13C-NMR (CDCI3): 5 169.9
(C(O)), 141.0 (ar C), 138.7 (ar C), 134.4 (ar CH), 133.2 (ar CH), 128.6 (ar CH),
128.28 (arCH), 128.26 (ar CH), 127.9 (ar CH), 90.7 (cp C), 78.4 (cp C), 72.9 (CH),
69.5 (5 cp CH), 69.4 (CH), 68.9 (CH), 68.7 (CH), 20.1 (CH3), 18.6 (CH3). MS (El+):
m/z 500 (3, [MJ+), 440 (100, [M-HOAc]+), 363 (43, [M-HOAc-Ph]+), 286 (10,
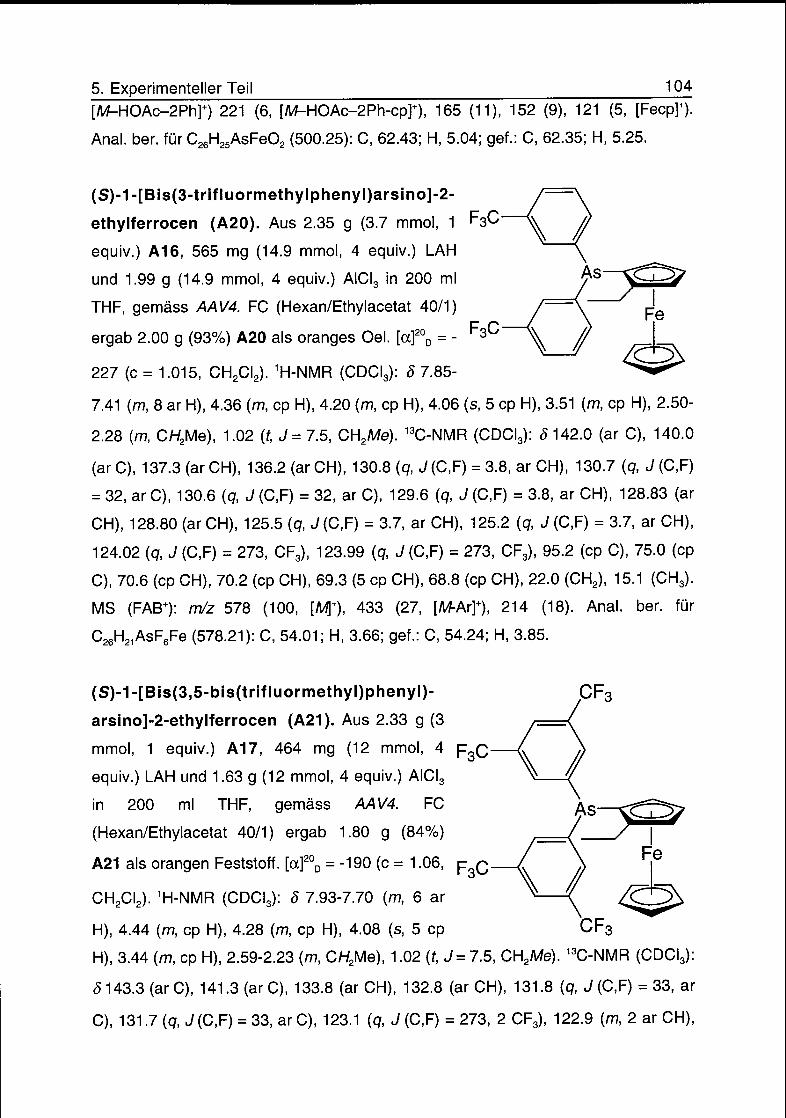
5. Experimenteller Teil 104
[M-HOAc-2Ph]+) 221 (6, [M-HOAc-2Ph-cp]+), 165 (11), 152 (9), 121 (5, [Fecpf).
Anal. ber. für C26H25AsFe02 (500.25): C, 62.43; H, 5.04; gef.: C, 62.35; H, 5.25.
(S)-1-[Bis(3-trifluormethylphenyl)arsino]-2-
ethylferrocen (A20). Aus 2.35 g (3.7 mmol, 1 F3C ^ /,
equiv.) A16, 565 mg (14.9 mmol, 4 equiv.) LAH
und 1.99 g (14.9 mmol, 4 equiv.) AICI3 in 200 ml
THF, gemäss AAV4. FC (Hexan/Ethylacetat 40/1)
ergab 2.00 g (93%) A20 als oranges Oel. [a]20D = -
F3C
227 (c = 1.015, CH2CI2). 1H-NMR (CDCI3): 5 7.85-
7.41 (m, 8 ar H), 4.36 (m, cp H), 4.20 (m, cp H), 4.06 (s, 5 cp H), 3.51 (m, cp H), 2.50-
2.28 (m, CH2Me), 1.02 (t, J= 7.5, CH2Me). 13C-NMR (CDCI3): 5 142.0 (ar C), 140.0
(arC), 137.3 (arCH), 136.2 (arCH), 130.8 (q, J(C,F) = 3.8, ar CH), 130.7 (q, J (C,F)
= 32, ar C), 130.6 (q, J (C,F) = 32, ar C), 129.6 (q, J (C,F) = 3.8, ar CH), 128.83 (ar
CH), 128.80 (ar CH), 125.5 (q, J (C,F) = 3.7, ar CH), 125.2 (q, J (C,F) = 3.7, ar CH),
124.02 (q, J(C,F) = 273, CF3), 123.99 (q, J (C,F) = 273, CF3), 95.2 (cp C), 75.0 (cp
C), 70.6 (cp CH), 70.2 (cp CH), 69.3 (5 cp CH), 68.8 (cp CH), 22.0 (CH2), 15.1 (CH3).
MS (FAB+): m/z 578 (100, [MJ+), 433 (27, [M-Ar]+), 214 (18). Anal. ber. für
C26H21AsF6Fe (578.21): C, 54.01; H, 3.66; gef.: C, 54.24; H, 3.85.
(S)-1-[Bis(3,5-bis(trifluormethyl)phenyl)-
arsino]-2-ethylferrocen (A21). Aus 2.33 g (3
mmol, 1 equiv.) A17, 464 mg (12 mmol, 4 p Q-
equiv.) LAH und 1.63 g (12 mmol, 4 equiv.) AICI3
in 200 ml THF, gemäss AAV4. FC
(Hexan/Ethylacetat 40/1) ergab 1.80 g (84%)
A21 als orangen Feststoff. [a]20D = -190 (c = 1.06, p q.
CH2CI2). 1H-NMR (CDCI3): 5 7.93-7.70 (m, 6 ar
H), 4.44 (m, cp H), 4.28 (m, cp H), 4.08 (s, 5 cp CF3
H), 3.44 (m, cp H), 2.59-2.23 (m, CH2Me), 1.02 (t, J= 7.5, CH2Me). 13C-NMR (CDCI3):
5143.3 (arC), 141.3(arC), 133.8 (ar CH), 132.8 (ar CH), 131.8 (q, J(C,F) = 33, ar
C), 131.7(q/, J(C,F) = 33, arC), 123.1 (q, J (C,F) = 273, 2 CF3), 122.9 (m, 2 ar CH),
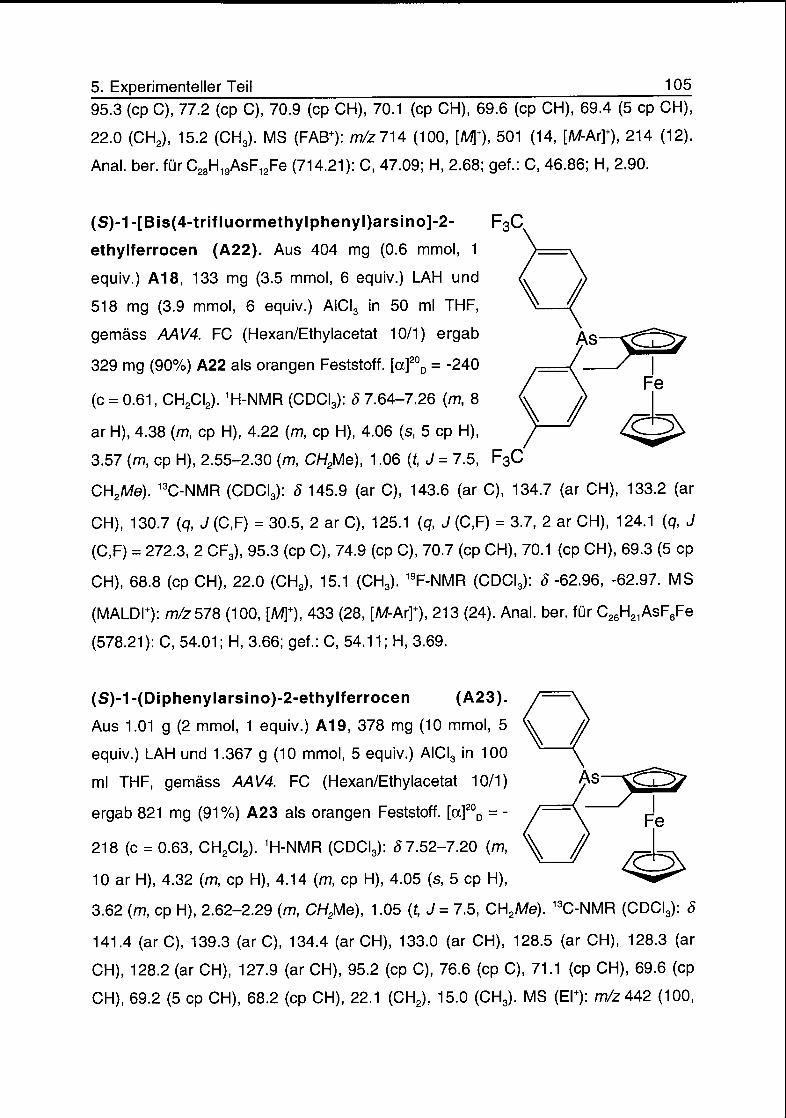
5. Experimenteller Teil 105
95.3 (cp C), 77.2 (cp C), 70.9 (cp CH), 70.1 (cp CH), 69.6 (cp CH), 69.4 (5 cp CH),
22.0 (CH2), 15.2 (CH3). MS (FAB+): m/z 714 (100, [MJ+), 501 (14, [M-Ar]+), 214 (12).
Anal. ber. für C28H19AsF12Fe (714.21): C, 47.09; H, 2.68; gef.: C, 46.86; H, 2.90.
(S)-1-[Bis(4-trifluormethylphenyl)arsino]-2- F3Cv
ethylferrocen (A22). Aus 404 mg (0.6 mmol, 1
equiv.) A18, 133 mg (3.5 mmol, 6 equiv.) LAH und
518 mg (3.9 mmol, 6 equiv.) AICI3 in 50 ml THF,
gemäss A41/4. FC (Hexan/Ethylacetat 10/1) ergab
329 mg (90%) A22 als orangen Feststoff. [cc]20D = -240
(c = 0.61, CH2CI2). 1H-NMR (CDCI3): 5 7.64-7.26 (m, 8
ar H), 4.38 (m, cp H), 4.22 (m, cp H), 4.06 (s, 5 cp H),
3.57 (m, cp H), 2.55-2.30 (m, CH2Me), 1.06 (t, J = 7.5, F3C
CH2Me). 13C-NMR (CDCI3): 5 145.9 (ar C), 143.6 (ar C), 134.7 (ar CH), 133.2 (ar
CH), 130.7 (q, J (C,F) = 30.5, 2 ar C), 125.1 (q, J (C,F) = 3.7, 2 ar CH), 124.1 (q, J
(C,F) = 272.3, 2 CF3), 95.3 (cp C), 74.9 (cp C), 70.7 (cp CH), 70.1 (cp CH), 69.3 (5 cp
CH), 68.8 (cp CH), 22.0 (CH2), 15.1 (CH3). 19F-NMR (CDCI3): 5 -62.96, -62.97. MS
(MALDI+): m/z 578 (100, [M]+), 433 (28, [M-Ar]+), 213 (24). Anal. ber. für C26H21AsF6Fe
(578.21): C, 54.01; H, 3.66; gef.: C, 54.11; H, 3.69.
(S)-1-(Diphenylarsino)-2-ethylferrocen (A23).
Aus 1.01 g (2 mmol, 1 equiv.) A19, 378 mg (10 mmol, 5
equiv.) LAH und 1.367 g (10 mmol, 5 equiv.) AICI3 in 100
ml THF, gemäss AAV4. FC (Hexan/Ethylacetat 10/1) A-S—^ÜZ^
ergab 821 mg (91%) A23 als orangen Feststoff. [oc]20D = - /===\'
p'
218 (c = 0.63, CH2CI2). 1H-NMR (CDCI3): 5 7.52-7.20 (m, \ //10 ar H), 4.32 (m, cp H), 4.14 (m, cp H), 4.05 (s, 5 cp H),
3.62 (m, cp H), 2.62-2.29 (m, CH2Me), 1.05 (t, J = 7.5, CH2Me). 13C-NMR (CDCI3): 5
141.4 (arC), 139.3 (ar C), 134.4 (ar CH), 133.0 (ar CH), 128.5 (ar CH), 128.3 (ar
CH), 128.2 (ar CH), 127.9 (ar CH), 95.2 (cp C), 76.6 (cp C), 71.1 (cp CH), 69.6 (cp
CH), 69.2 (5 cp CH), 68.2 (cp CH), 22.1 (CH2), 15.0 (CH3). MS (El+): m/z 442 (100,

5. Experimenteller Teil 106
[MJ+), 365 (47, [M-Ar]+), 290 (4), 227 (6), 212 (24), 195 (5), 165 (3), 152 (10), 121 (8),
84 (14). Anal. ber. für C24H23AsFe (442.21): C, 65.19; H, 5.24; gef.: C, 65.04; H, 5.25.
(S)-1-Bis(4-trifluormethylphenyl)arsino-2- F3C.
vinylferrocen (A24). Eine Lösung von 2.632 g (4.1
mmol) A14 in 25 ml Essigsäureanhydrid wurde 18 h
bei 100 °C gerührt, auf RT abgekühlt und mit H20
versetzt. Nach dreimaliger Extraktion mit Ethylacetat,
Vereinigung der organischen Phasen, Tocknen über
MgS04, Einengen und Trocknen am HV ergab FC
(Hexan/Ethylacetat 10/1) 1.907 g (80%) A24 als
oranges Pulver. DC (Hexan/Ethylacetat 10/1): R, 0.76.p q
[a]20D = + 140 (CH2CI2, c = 0.94). 1H-NMR (CDCI3): 5
7.68-7.30 (m, 8 ar H), 6.60 (dd, J= 17.4, 10.8, 1 H), 5.38 (dd, J= 17.4, 1.3, 1 H), 5.07
(dd, J= 10.8, 1.3, 1 H), 4.75 (m, 1 cp H), 4.35 (m, 1 cp H), 4.05 (s, 5 cp H), 3.65 (m, 1
cp H). 13C-NMR (CDCI3): 5 145.7 (ar C), 143.3 (ar C), 134.9 (ar CH), 133.0 (ar CH),
131.1 (q, J(C,F) = 32.5, ar C), 130.4 (q, J(C,F) = 32.3, ar C), 125.1 (CH), 124.0 (q,
J(C,F) = 272.3, arC), 124.0 (q, J(C,F) = 272.3, ar C), 113.1 (CH2), 88.1 (cp C), 77.2
(cp C), 72.2 (cp CH), 70.4 (cp CH), 70.1 (5 cp CH), 68.1 (cp CH). 19F-NMR (CDCI3): 5
-62.99 (s). MS (El+): m/z 576 (23, [Mj+), 557 (2, [M-F]+), 455 (30), 431 (14, [M-Ar]+),
356 (12), 337 (5), 309 (5), 291 (25), 271 (35), 233 (21), 220 (23), 215 (47), 201 (33),
183 (18), 165 (99), 152 (26), 127 (32), 121 (100, [Fecp]+), 107 (44), 89 (15), 56 (35).
Anal. ber. für C26H19AsF6Fe (576.20): C, 54.20; H, 3.32; gef.: C, 54.14; H, 3.53.

5. Experimenteller Teil 107
\ /
(/?)-1-Diphenylarsino-2-vinylferrocen (A25). Eine
Lösung von 3.115 g (6.4 mmol) Dimethyl{(S)-1-[(fl)-2- \ //
(diphenylarsino)ferrocenyl]ethyl}amin und 799 mg (7.9
mmol) Triethylamin in 20 ml Toluol wurde unter Rühren
tropfenweise mit 0.40 ml (3.3 mmol) Chlor-
ameisensäuretrichlormethylester (Diphosgen) versetzt. Die
Reaktionsmischung wurde 10 min bei 120 °C unter
Rückfluss gerührt, auf RT abgekühlt und dreimal mit 50 ml ges. NaHC03-Lsg., ges.
NaCI-Lsg. und H20 gewaschen. Die organische Phase wurde über Na2S04
getrocknet, eingeengt und im HV getrocknet. FC (Et20) ergab 2.463 g (88%) A25
als orangen Feststoff. DC (Hexan/Ethylacetat 20/1): R, 0.60; (Hexan/Ethylacetat
10/1): Fi, 0.68; (Hexan/Ethylacetat 1/1): Fl, 0.85; (Et20): R, 0.93. [oc]20D = - 240 (c =
0.85, CH2CI2). 1H-NMR (CD2CI2): 57.59-7.23 (m, 10 ar H), 6.71 (dd, J= 17.4, 10.8, 1
H), 5.41 (dd, J= 17.4, 1.5, 1 H), 5.08 (dd, J= 10.8, 1.5, 1 H), 4.76 (m, 1 cp H), 4.37
(m, 1 cp H), 4.09 (s, 5 cp H), 3.77 (m, 1 cp H). 13C-NMR (CD2CI2): 5 141.1 (ar C),
138.7 (arC), 134.3 (ar CH), 133.6 (ar CH), 132.4 (ar CH), 128.4 (ar CH), 128.1 (ar
CH), 128.0 (arCH), 127.7 (CH), 111.7 (CH2), 87.6 (cp C), 77.0 (cp C), 72.3 (cp CH),
69.8 (5 cp CH), 69.7 (cp CH), 67.3 (cp CH). MS (El+): m/z 440 (100, [MJ+), 363 (40,
[M-Ph]+), 288 (27), 286 (10, [M-2Ph]+), 220 (8), 195 (5), 165 (14), 152 (13), 121 (8,
[Fecp]+). Anal. ber. für C24H21AsFe (440.20): C, 65.49; H, 4.81 ; gef.: C, 65.56; H, 4.86.
(3/?)-1,3-Bis[(fl)-1-(diphenylarsino)-
ferrocen-2-yl]but-1-en (A26). Eine
Lösung von 249.8 mg (0.57 mmol) A25 in 5
ml CH2CI2 wurde tropfenweise mit 0.18 ml
(1.1 mmol) Triethylsilan und 0.15 ml (2.0
mmol) Trifluoressigsäure versetzt. Die
Reaktionsmischung wurde 48 h bei 50 °C unter Rückfluss gerührt, auf RT abgekühlt
und mit NaHC03-Lsg. neutralisiert. Nach dreimaliger Extraktion mit Et20 wurden die
organischen Phasen vereinigt, mit NaHC03-Lsg., NaCI-Lsg. und H20 gewaschen,
über MgS04 getrocknet und eingeengt. Trocknen des Rückstandes im HV und FC

5. Experimenteller Teil 108
(Hexan/Ethylacetat 20/1) ergab 332 mg (46%) A26 als gelben Feststoff. DC
(Hexan/Ethylacetat 20/1): R, 0.38; (Hexan/Ethylacetat 10/1): R, 0.53; (Et20): 0.90.
[a]20D = - 51 (c = 0.15, CH2CI2). 1H-NMR (CD2CI2): 5 7.58-7.14 (m, 20 ar H), 6.17
(dddd, J= 15.7, 1.3, 0.7, 0.5, 1 H), 5.62 (dd, J= 15.7, 7.2, 1 H), 4.19 (m, 2 H), 4.08
(m, 1 H), 3.96 (s, 5 cp H), 3.94 (m, 1 H), 3.83 (s, 5 cp H), 3.77 (m, 1 H), 3.59 (m, 1 H),
3.52 (dqd, J = 7.2, 7.0, 1.3, 1 H), 1.29 (d, J = 7.0, 3 H). 13C-NMR (CD2CI2): 5 142.0 (ar
C), 141.2(arC), 139.8 (ar C), 139.1 (ar C), 135.7 (ar CH), 134.8 (ar CH), 134.6 (ar
CH), 133.2 (ar CH), 132.9 (ar CH), 132.6 (ar CH), 128.58 (ar CH), 128.55 (ar CH),
128.4 (ar CH), 128.3 (ar CH), 128.2 (ar CH), 127.7 (ar CH), 127.2 (CH), 124.1 (CH),
98.3 (cp C), 88.4 (cp C), 76.1 (cp C), 76.0 (cp C), 71.8 (cp CH), 71.4 (cp CH), 69.7
(cp CH), 69.5 (cp CH), 69.1 (cp CH), 68.6 (cp CH), 68.1 (cp CH), 67.6 (cp CH), 36.3
(CH), 20.7 (CH3). MS (El+): m/z 880 (100, [Mj+), 651 (10, [M-AsPh2]+), 586 (3,
[M-AsPh2-cp]+), 440 (20, [M/2]+), 422 (17), 286 (6, [M/2-2 Ph]+), 199 (10), 154 (15),
121 (4, [Fecp]+). Anal. ber. für C48H42As2Fe2 (880.40): C, 65.49; H, 4.81; gef.: C,
65.40; H, 4.83. HPLC: Daicel Chiralcel OD-H, eluiert mit Hexan^PrOH = 99:1, Fluss
0.5 ml/min, Retentionszeit: 17.4 min, UV-Detektor (210 nm).
Eine Lösung von 266.1 mg (1.18 mmol) [Fp(CH2CI)] und 441.6 mg (0.88 mmol) (S)-
(R)-A19 in 10 ml CH3CN wurde über Nacht bei 65 °C gerührt und auf RT
abgekühlt. Die entstandene Suspension wurde filtriert und der feste Rückstand mit
MeOH und dann mit Pentan gewaschen. Extraktion mit CH2CI2, Einengen der
Extraktionslösung und Trocknen des Rückstandes im HV ergab 178 mg (45%) A26
als gelben Feststoff. [cc]20D = - 52 (c = 0.16, CH2CI2).
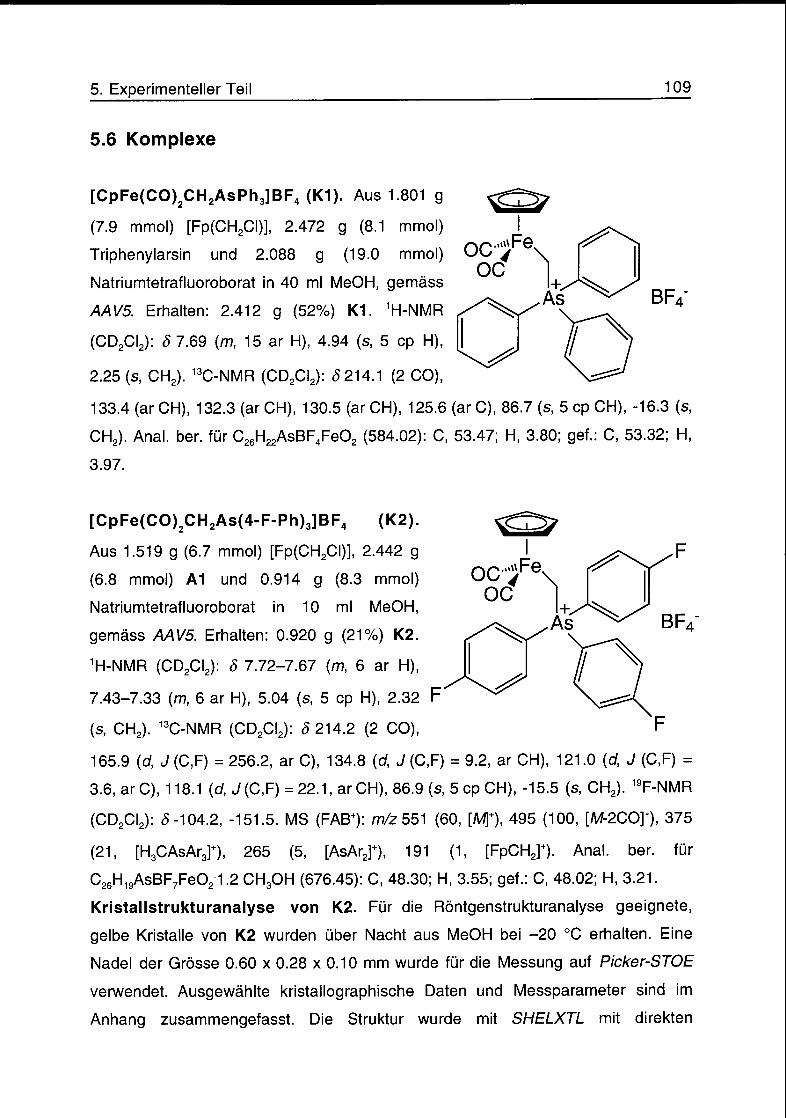
5. Experimenteller Teil 109
5.6 Komplexe
[CpFe(CO)2CH2AsPh3]BF4 (K1). Aus 1.801 g
(7.9 mmol) [Fp(CH2CI)], 2.472 g (8.1 mmol)...»\Fa
Triphenylarsin und 2.088 g (19.0 mmol) OC'yOC
Natriumtetrafluoroborat in 40 ml MeOH, gemäss i+
^ A4AAV5. Erhalten: 2.412 g (52%) K1. 1H-NMR
"^ '
^n
(CD2CI2): 5 7.69 (m, 15 ar H), 4.94 (s, 5 cp H),
2.25 (s, CH2). 13C-NMR (CD2CI2): 5 214.1 (2 CO),^^
BF,
133.4 (ar CH), 132.3 (ar CH), 130.5 (ar CH), 125.6 (ar C), 86.7 (s, 5 cp CH), -16.3 (s,
CH2). Anal. ber. für C26H22AsBF4Fe02 (584.02): C, 53.47; H, 3.80; gef.: C, 53.32; H,
3.97.
Q;
oc-JF\
[CpFe(CO)2CH2As(4-F-Ph)3]BF4 (K2).
Aus 1.519 g (6.7 mmol) [Fp(CH2CI)], 2.442 g
(6.8 mmol) A1 und 0.914 g (8.3 mmol)
Natriumtetrafluoroborat in 10 ml MeOH,
gemäss AAV5. Erhalten: 0.920 g (21%) K2.
1H-NMR (CD2CI2): 5 7.72-7.67 (m, 6 ar H),
7.43-7.33 (m, 6 ar H), 5.04 (s, 5 cp H), 2.32 F'
(s, CH2). 13C-NMR (CD2CI2): 5 214.2 (2 CO),
165.9 (d, J(C,F) = 256.2, ar C), 134.8 (d, J (C,F) = 9.2, ar CH), 121.0 (d, J (C,F) =
3.6, arC), 118.1 (d, J(C,F) = 22.1, arCH), 86.9 (s, 5 cp CH), -15.5 (s, CH2). 19F-NMR
(CD2CI2): 5-104.2, -151.5. MS (FAB+): m/z 551 (60, [MY), 495 (100, [M-2CO]+), 375
(21, [H3CAsAr3]+), 265 (5, [AsAr2]+), 191 (1, [FpCH2]+). Anal. ber. für
C26H19AsBF7Fe021.2 CH3OH (676.45): C, 48.30; H, 3.55; gef.: C, 48.02; H, 3.21.
Kristallstrukturanalyse von K2. Für die Röntgenstrukturanalyse geeignete,
gelbe Kristalle von K2 wurden über Nacht aus MeOH bei -20 °C erhalten. Eine
Nadel der Grösse 0.60 x 0.28 x 0.10 mm wurde für die Messung auf Pieker-STOE
verwendet. Ausgewählte kristallographische Daten und Messparameter sind im
Anhang zusammengefasst. Die Struktur wurde mit SHELXTL mit direkten
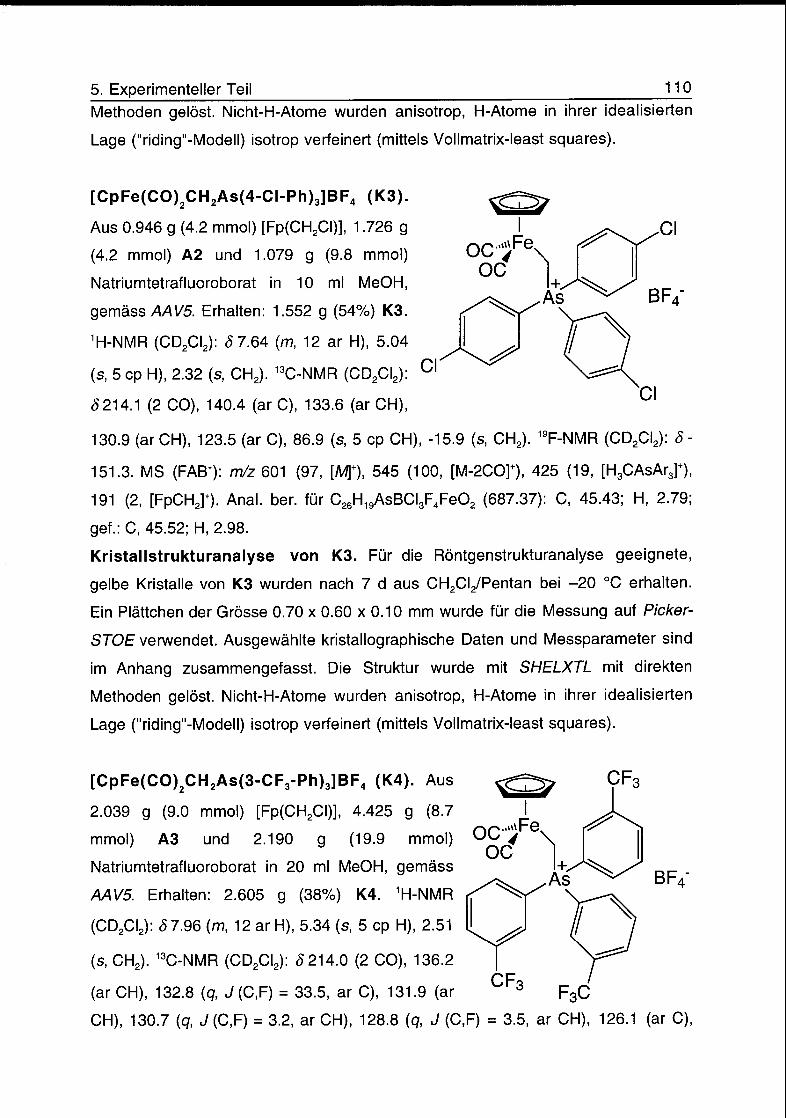
5. Experimenteller Teil 110
Methoden gelöst. Nicht-H-Atome wurden anisotrop, H-Atome in ihrer idealisierten
Lage ("riding"-Modell) isotrop verfeinert (mittels Vollmatrix-Ieast squares).
I
OC'f\
[CpFe(CO)2CH2As(4-CI-Ph)3]BF4 (K3).
Aus 0.946 g (4.2 mmol) [Fp(CH2CI)], 1.726 g
(4.2 mmol) A2 und 1.079 g (9.8 mmol)
Natriumtetrafluoroborat in 10 ml MeOH,
gemäss AAV5. Erhalten: 1.552 g (54%) K3.
1H-NMR (CD2CI2): 5 7.64 (m, 12 ar H), 5.04
(s, 5 cp H), 2.32 (s, CH2). 13C-NMR (CD2CI2):Cl
5214.1 (2 CO), 140.4 (ar C), 133.6 (ar CH),
130.9 (arCH), 123.5 (ar C), 86.9 (s, 5 cp CH), -15.9 (s, CH2). 19F-NMR (CD2CI2): 5-
151.3. MS (FAB+): m/z 601 (97, [MY), 545 (100, [M-2CO]+), 425 (19, [H3CAsAr3]+),
191 (2, [FpCH2]+). Anal. ber. für C26H19AsBCI3F4Fe02 (687.37): C, 45.43; H, 2.79;
gef.: C, 45.52; H, 2.98.
Kristallstrukturanalyse von K3. Für die Röntgenstrukturanalyse geeignete,
gelbe Kristalle von K3 wurden nach 7 d aus CH2CI2/Pentan bei -20 °C erhalten.
Ein Plättchen der Grösse 0.70 x 0.60 x 0.10 mm wurde für die Messung auf Pieker-
STOE verwendet. Ausgewählte kristallographische Daten und Messparameter sind
im Anhang zusammengefasst. Die Struktur wurde mit SHELXTL mit direkten
Methoden gelöst. Nicht-H-Atome wurden anisotrop, H-Atome in ihrer idealisierten
Lage ("riding"-Modell) isotrop verfeinert (mittels Vollmatrix-Ieast squares).
BF,
[CpFe(CO)2CH2As(3-CF3-Ph)3]BF4 (K4). Aus
2.039 g (9.0 mmol) [Fp(CH2CI)], 4.425 g (8.7
mmol) A3 und 2.190 g (19.9 mmol)
Natriumtetrafluoroborat in 20 ml MeOH, gemäss
AAV5. Erhalten: 2.605 g (38%) K4. 1H-NMR
(CD2CI2): 57.96 (m, 12 ar H), 5.34 (s, 5 cp H), 2.51
(s, CH2). 13C-NMR (CD2CI2): 5 214.0 (2 CO), 136.2
(ar CH), 132.8 (q, J (C,F) = 33.5, ar C), 131.9 (ar
CH), 130.7 (q, J(C,F) = 3.2, ar CH), 128.8 (q, J (C,F) = 3.5, ar CH), 126.1 (ar C),

5. Experimenteller Teil 111
123.1 (q, J(C,F) = 273.1, CF3), 87.2 (s, 5 cp CH), -15.5 (s, CH2). 19F-NMR (CD2CI2): 5
-63.6, -151.0. MS (FAB+): m/z 701 (43, [MY), 645 (100, [M-2CO]+), 525 (28,
[H3CAsAr3]+), 365 (4, [AsAr2]+), 191 (1, [FpCH2]+). Anal. ber. für C29H19AsBF13Fe02
(788.03): C, 44.20; H, 2.43; gef.: C, 44.38; H, 2.57.
Kristallstrukturanalyse von K4. Für die Röntgenstrukturanalyse geeignete,
gelbe Kristalle von K4 wurden über Nacht aus MeOH bei RT in einem offenen
Pillenglas erhalten. Eine Nadel der Grösse 0.80 x 0.10 x 0.08 mm wurde für die
Messung auf Pieker-STOE verwendet. Ausgewählte kristallographische Daten und
Messparameter sind im Anhang zusammengefasst. Die Struktur wurde mit
SHELXTL mit direkten Methoden gelöst. Nicht-H-Atome wurden anisotrop, H-Atome
in ihrer idealisierten Lage ("riding"-Modell) isotrop verfeinert (mittels Vollmatrix-Ieast
squares).
[CpFe(CO)2CH2As(4-CF3-Ph)3]BF4
(K5). Eine Mischung von 1.709 g (7.5
mmol) [Fp(CH2CI)], 3.563 g (7.0 mmol)
A4 und 1.400 g (12.8 mmol)
Natriumtetrafluoroborat in 30 ml MeOH
wurde 1h bei 65 °C unter Rückfluss
gerührt. Die Reaktionsmischung wurde F3C
eingeengt und im HV getrocknet. Der
Rückstand wurde mit Hexan gewaschen und mit CH2CI2 extrahiert. Nach Einengen
der Extraktionslösung wurde das Rohprodukt bei -20 °C mit MeOH gewaschen.
Kristallisation aus CH2CI2/Pentan bei -20 °C ergab 2.720 g (49%) K5 als gelben
Feststoff. 1H-NMR (CD2CI2): 5 7.92 (m, 12 ar H), 5.11 (s, 5 cp H), 2.53 (s, CH2). 13C-
NMR (CD2CI2): 5214.2 (2 CO), 135.5 (q, J (C,F) = 33.4, ar C), 133.5 (ar CH), 129.6
(arC), 128.1 (arCH), 123.2 (q, J(C,F) = 272.9, CF3), 88.2 (s, 5 cp CH), -13.1 (CH2).
19F-NMR (CD2CI2): 5-63.2, -150.3. MS (FAB+): m/z 701 (66, [MY), 645 (100, [M-
2CO]+), 525 (54, [H3CAsAr3]+), 365 (18, [AsAr2]+), 271 (91), 191 (17, [FpCH2]+). Anal,
ber. für C29H19AsBF13FeO20.1 CH2CI2 (796.52): C, 43.88; H, 2.43; gef.: C, 43.56; H,
2.39.

5. Experimenteller Teil 112
Kristallstrukturanalyse von [CpFe(CO)2As(4-CF3-Ph)3]BF4. Aus einer ges.
Lsg. von K5 in CH2CI2, die mit Pentan überschichtet worden war, wurden nach 3 d
bei -20 °C gelbe, zur Röntgenstrukturanalyse geeignete Nadeln von
[CpFe(CO)2As(4-CF3-Ph)3]BF4 erhalten. Ein Kristall der Grösse 0.58 x 0.14 x
0.08 mm wurde für die Messung auf Pieker-STOE verwendet. Ausgewählte
kristallographische Daten und Messparameter sind im Anhang zusammengefasst.
Die Struktur wurde mit SHELXTL mit direkten Methoden gelöst. Nicht-H-Atome
wurden anisotrop, H-Atome in ihrer idealisierten Lage ("riding"-Modell) isotrop
verfeinert (mittels Vollmatrix-Ieast squares).
SbFf
FaC
[CpFe(CO)2CH2As(3-CF3-Ph)3]SbF6 (K6).
Eine Mischung von 1.100 g (4.9 mmol)
[Fp(CH2CI)] und 2.396 g (4.7 mmol) A3 wurden OC'v^in 15 ml MeOH suspendiert. Die OC
Reaktionsmischung wurde 26 h bei 65 °C unter
Rückfluss gerührt, auf RT abgekühlt und zu
3.213 g (12.4 mmol) Natriumhexafluoro- "\x^
antimonat filtriert. Nach 4 h Rühren wurde diePF
Reaktionsmischung 3 d bei -20 °C stehen- or3
gelassen. Der Niederschlag wurde abfiltriert und im HV getrocknet. Aus Extraktion
mit CH2CI2 wurde nach Abziehen des Lösungsmittels kein Produkt erhalten. Das
Einengen der Mutterlauge ergab gelben Feststoff, der nach Trocknen im HV mit
CH2CI2 extrahiert wurde. Abziehen des Lösungsmittels, Tocknen im HV, Waschen
mit Pentan und Kristallisation aus CH2CI2/Pentan bei -20 °C ergab 175 mg (4%) K6
als gelben Feststoff. 1H-NMR (CD2CI2): 57.94 (m, 12 ar H), 5.06 (s, 5 cp H), 2.37 (s,
CH2). 13C-NMR (CD2CI2): 5 213.7 (2 CO), 135.8 (ar CH), 132.8 (q, J (C,F) = 33.4, ar
C), 131.9 (ar CH), 130.9 (q, J (C,F) = 3.4, ar CH), 128.6 (q, J (C,F) = 3.4, ar CH),
125.7 (ar C), 123.0 (q, J (C,F) = 273.1, CF3), 86.9 (s, 5 cp CH), -16.2 (s, CH2). 19F-
NMR (CD2CI2): 5-61.8 (s), -98 - -151.0 (m). MS (FAB+): m/z 701 (78, [MY), 645 (100,
[M-2COH, 525 (33, [H3CAsAr3]+), 365 (4, [AsAr2]+), 191 (7, [FpCH2]+). Anal. ber. für
C29H19AsF15Fe02Sb (936.97): C, 37.18; H, 2.04; gef.: C, 37.18; H, 2.22.

5. Experimenteller Teil 113
Sl2
oc;F\ r^N
BF,
[(C5H4Me)Fe(CO)2(CH2AsPh3)]BF4 (K7).
Zu 12.663 g (34.5 mmol) [Fe2(CO)9] in 80 ml
Benzol wurden unter Rühren 3 g (37 mmol)
Methylcyclopentadien getropft. Die erhaltene
Mischung wurde 24 h bei RT gerührt und
filtriert. Einengen der Filtrationslösung,
Waschen des Rückstandes mit Hexan und K^<^>Umkristallisation aus CH2CI2/Pentan ergab 723
m9 [(C5H4Me)Fe(CO)2]2 als schwarzen Feststoff. Eine Mischung von 597.6 mg
(1.56 mmol) [(C5H4Me)Fe(CO)2]2 und Na/Hg (7.8 mmol Na mit 2 ml Hg) in 100 ml
THF wurde 24 h bei RT gerührt. Die überstehende Lösung wurde abgetrennt, auf 0
°C abgekühlt und mit 0.35 ml (4.6 mmol) Chlormethylmethylether versetzt. Nach 12
h Rühren wurde die Lösung filtriert, eingeengt und der erhaltene Rückstand mit
Pentan extrahiert. Einengen der Extraktionslösung und Trocknen des Rückstandes
am HV ergab 369 mg [(C5H4Me)Fe(CO)2(CH2OMe)] als braunes Oel. DEPT-
NMR (CD2CI2): 5 85.9 (2 cp CH), 85.2 (2 cp CH), 66.8 (CH2), 60.3 (OCH3), 12.3
(CH3). Durch eine Lösung von 360 mg (1.52 mmol) [(C5H4Me)Fe(CO)2(CH2OMe)] in
5 ml Pentan wurde bei 0 °C während 5 min HCI-Gas geleitet. Einengen der
Reaktionsmischung ergab 358 mg [(C5H4Me)Fe(CO)2(CH2CI)] als braunen
Feststoff. DEPT-NMR (CD2CI2): 5 86.4 (2 cp CH), 85.2 (2 cp CH), 36.4 (CH2), 12.4
(CH3). Eine Suspension von 336 mg (1.4 mmol) [(C5H4Me)Fe(CO)2(CH2CI)] und 452
mg (1.5 mmol) Triphenylarsin in 10 ml MeOH wurde über Nacht unter Rückfluss
gerührt, auf RT gekühlt und zu 332 mg (3 mmol) Natriumtetrafluoroborat filtriert.
Nach 15 min Rühren wurde die Reaktionsmischung über Nacht bei -20 °C
stehengelassen. Der gelbe Niederschlag wurde abfiltriert und im HV getrocknet.
Kristallisation des Rohproduktes aus CH2CI2/Pentan bei -20 °C ergab 532 mg
(64%) K7 als gelben Feststoff. 1H-NMR (CD2CI2): 5 7.68 (m, 15 ar H), 4.85 (m, 2 cp
H), 4.77 (m, 2 cp H), 2.16 (s, CH2), 1.92 (s, CH3). 13C-NMR (CD2CI2): 5 214.5 (2 CO),
133.3 (ar CH), 132.2 (ar CH), 130.4 (ar CH), 125.7 (ar C), 104.4 (cp C), 86.8 (2 cp
CH), ,85.5 (2 cp CH), 12.6 (CH3), -15.2 (CH2). 19F-NMR (CD2CI2): 5 -152.5. MS
(FAB+): m/z511 (68, [MY), 455 (100, [M-2CO]+), 364 (24), 321 (18, [H3CAsPh3]+), 227

5. Experimenteller Teil 114
(11), 205 (1, [M-AsPh3]+). Anal. ber. für C27H24AsBF4FeO20.4 CH2CI2 (632.03): C,
52.07; H, 3.96; gef.: C, 51.90; H, 3.96.
0C.^Fe ^N
BF,
[(C5Me5)Fe(CO)2(CH2AsPh3)]BF4 (K8).
Eine Mischung von 294 mg (0.6 mmol)
[(C5Me5)Fe(CO)2]2 und Na/Hg (5 mmol Na mit 2
ml Hg) in 50 ml THF wurde 24 h bei RT gerührt.
Die überstehende Lösung wurde abgetrennt,
auf 0 °C abgekühlt und mit 0.2 ml (2.6 mmol)
Chlormethylmethylether versetzt. Nach 12 h
Rühren wurde die Lösung eingeengt und der
erhaltene Rückstand mit Heptan extrahiert.
Durch diese Extraktionslösung wurde bei 0 °C während 5 min ein langsamer Fluss
von HCI-Gas geleitet. Einengen der Reaktionsmischung ergab 145 mg eines
orangen Feststoffs. Eine Suspension dieses Feststoffes und 164 mg (0.5 mmol)
Triphenylarsin in 5 ml MeOH wurde über Nacht unter Rückfluss gerührt, auf RT
abgekühlt und zu 143 mg (1.3 mmol) Natriumtetrafluoroborat filtriert. Nach 15 min
Rühren wurde die Lösung eingeengt. Der erhaltene Rückstand wurde mit CH2CI2
extrahiert. Die Extraktionslösung wurde über Alox filtriert und eingeengt.
Kristallisation aus CH2CI2/Pentan bei -20 °C ergab 77 mg (10%) K8 als gelben
Feststoff. 1H-NMR (CD2CI2): 57.75-7.58 (m, 15 ar H), 1.77 (s, C5Me5), 1.53 (s, CH2).
13C-NMR (CD2CI2): 5 216.0 (2 CO), 133.6 (ar CH), 132.6 (ar CH), 130.7 (ar CH),
126.0 (arC), 97.5 (C5Me5), 10.0 (C5Me5), -5.4 (CH2). 19F-NMR (CD2CI2): 5-153.1. MS
(FAB+): m/z567 (100, [MY), 511 (76, [M-2CO]+), 420 (31), 321 (13, [H3CAsPh3]+), 261
(18, [M-AsPh3]+). Anal. ber. für C31H32AsBF4FeO20.3 CH2CI2 (679.65): C, 55.31; H,
4.83; gef.: C, 55.31; H, 5.11.

5. Experimenteller Teil 115
[FpCH2AsR3]BF4J AsR3 = (S)-1-Methoxy-[(/?)-2-(diphenylarsino)-
ferrocenyl]ethan (K9). Eine ^^^
Suspension von 644 mg (2.8 mmol)
[Fp(CH2CI)] und 1.297 g (2.6 mmol) (S)- ^S^
(f?)-A19 in 10 ml MeOH wurde über Nacht J_^^
nr...^Fe As C^T^> BF4unter Rückfluss gerührt, auf RT abgekühlt zLff N*^
und zu 674 mg (6.1 mmol) Natrium¬
tetrafluoroborat filtriert. Nach 15 min
Rühren wurde die Reaktionsmischung
über Nacht bei -20 °C stehengelassen. Der gelbe Niederschlag wurde abfiltriert
und im HV getrocknet. Nach Extraktion mit CH2CI2 und Einengen der erhaltenen
Lösung wurde der Rückstand mit Pentan gewaschen. Kristallisation aus
CH2CI2/Pentan bei -20 °C ergab 44 mg (2%) K9 als gelben Feststoff. [a]20D = + 70 (c
= 0.38, CH2CI2). 1H-NMR (CD2CI2): 5 7.80-7.23 (m, 10 ar H), 4.91 (s, 5 cp H), 4.72
(m, cp H), 4.66 (m, cp H), 4.44 (q, J= 5.6, CHMe), 4.31 (s, 5 cp H), 4.21 (m, cp H),
2.92 (s, OMe), 2.23 (m, CH2), 1.41 (d, J = 5.6, CHMe). 13C-NMR (CD2CI2): 5 214.0
(CO), 213.8 (CO), 132.78 (ar CH), 132.76 (ar CH), 132.1 (ar CH), 131.8 (ar CH),
129.74 (arCH), 129.72 (ar CH), 126.8 (arC), 126.2 (ar C), 69.0 (cp C), 68.9 (cp C),
86.3 (5 cp CH), 73.1 (cp CH), 72.0 (cp CH), 70.74 (5 cp CH), 70.71 (cp CH), 70.2
(CHMe), 52.7 (OMe), 16.1 (CHMe), -14.0 (CH2). 19F-NMR (CD2CI2): 5-152.5 (s). MS
(FAB+): m/z 663 (100, [MY), 607 (85, [M-2CO]+), 487 (10, [H3CAsPh2R]+), 472 (10,
[AsPh2R]+), 455 (17), 441 (13), 213 (15), 154 (14), 136 (13). Anal. ber. für
C33H32AsBF4Fe203 (750.04): C, 52.85; H, 4.30; gef.: C, 52.85; H, 4.36.
Eine Mischung von 576 mg (2.5 mmol) [Fp(CH2CI)] und 1.085 g (2.5 mmol) A25 in
15 ml MeOH wurde über Nacht unter Rückfluss gerührt. Die Reaktionsmischung
wurde auf RT abgekühlt und unter Rühren zu 507 mg (4.6 mmol)
Natriumtetrafluoroborat filtriert. Nach Stehenlassen bei -20 °C während 3 d wurde
der ausgefallene Feststoff abfiltriert, im HV getrocknet und mit CH2CI2 extrahiert.
Einengen der Filtrationslösung, Waschen des Rückstandes mit Pentan und
Trocknen im HV ergab 112.3 mg (6%) K9 als gelben Feststoff.
5. Experimenteller Teil 116
gemäss
a/->..ü«
K11. OC
BF,
[FpCH2AsR3]BF45 AsR3 = A20
(K10). Aus 249 mg (1.1 mmol)
[Fp(CH2CI)], 460 mg (0.8 mmol)
A20 und 520 mg (4.7 mmol)
Natriumtetrafluoroborat,aemäss
/^^.<^Fe
der Herstellung von
Kristallisation aus CH2CI2/Pentan
bei -20 °C ergab 139 mg (20%)
K10 als gelben Feststoff. [a]20D = -
31 (c = 0.13, CH2CI2). 1H-NMR (CD2CI2): 5 8.27-7.42 (m, 8 ar H), 5.06 (s, 5 cp H),
4.61 (m, cp H), 4.45 (m, cp H), 4.21 (s, 5 cp H), 4.10 (m, cp H), 2.50-2.36 (m, CH2),
2.15-2.03 (m, CH2), 1.08 (m, CH3). 13C-NMR (CD2CI2): 5 214.0 (CO), 213.7 (CO),
136.3 (2 ar CH), 132.0 (q, J(C,F) = 33.9, 2 ar C), 131.5 (2 ar CH), 130.7 (2 ar C),
129.9 (q, J(C,F) = 3.6, 2 ar CH), 128.6 (q, J(C,F) = 3.9, 2 ar CH), 122.9 (q, J(C,F) =
278.2, 2 CF3), 87.3 (5 cp CH), 72.0 (cp CH), 71.0 (cp CH), 70.9 (5 cp CH), 70.3 (cp
CH), 69.2 (cp C), 67.7 (cp C), 21.8 (CH2), 14.3 (CH3), -11.6 (CH2). 19F-NMR (CD2CI2):
5 -61.2, -150.4. MS (FAB+): m/z 769 (75, [MY), 713 (100, [M-2CO]+), 648 (3,
[M-2CO-cp]+), 593 (22), 578 (53), 433 (25), 387 (10), 288 (6), 227 (33), 212 (19),
121 (13). Anal. ber. für C^H.gAsBF^FeAO^ CH2CI2 (889.98): C, 46.43; H, 3.26;
gef.: C, 46.36; H, 3.10.
r"^
BF,
[FpCH2AsR3]BF4, AsR3 = A23
(K11). Eine Suspension von 188 mg
(0.8 mmol) [Fp(CH2CI)] und 322 mg
(0.7 mmol) A23 in 5 ml MeOH wurde
über Nacht unter Rückfluss gerührt, OC" v
auf RT abgekühlt und zu 471 mg (4.3OC
mmol) Natriumtetrafluoroborat filtriert.
Nach 15 min Rühren wurde die
Reaktionsmischung eingeengt und
der erhaltene Rückstand mit CH2CI2 extrahiert. Die Extraktionslösung wurde über

5. Experimenteller Teil 117
Alox filtriert und eingeengt. Das Rohprodukt wurde mit Pentan gewaschen.
Kristallisation aus CH2CI2/Pentan bei -20 °C ergab 195 mg (37%) K11 als gelben
Feststoff. [oc]20D = -16 (c = 0.18, CH2CI2). 1H-NMR (CD2CI2): 5 7.76-7.28 (m, 10 ar H),
4.92 (s, 5 cp H), 4.79 (m, cp H), 4.60 (m, cp H), 3.37 (m, cp H), 4.16 (s, 5 cp H),
2.31-1.99 (m, 2 CH2), 1.04 (t, J = 7.3, CH3). 13C-NMR (CD2CI2): 5 214.12 (CO),
214.09 (CO), 133.39 (ar CH), 133.35 (ar CH), 132.48 (ar CH), 132.45 (ar CH), 130.3
(ar CH), 130.2 (ar CH), 126.3 (ar C), 125.7 (ar C), 86.6 (5 cp CH), 71.9 (cp CH), 71.8
(cp CH), 70.50 (5 cp CH), 70.46 (cp, CH), 69.8 (cp C), 69.2 (cp C), 21.8 (CH2), 13.9
(CH3), -13.5 (CH2). 19F-NMR (CD2CI2): 5-152.35. MS (FAB+): m/z 633 (82, [MY), 577
(100, [M-2COD, 512 (9, [M-2CO-cp]), 457 (43), 442 (44), 437 (46), 381 (22), 365
(28), 212 (17), 154 (17). Anal. ber. für C32H30AsBF4Fe2O20.2 CH2CI2 (737.00): C,
52.48; H, 4.16; gef.: C, 52.49; H, 4.24.
lodomethyldicarbonyleisencyclopentadienyl. Zu einer^^_^.
Suspension von 24.229 g (161.6 mmol) Nal in 30 ml Aceton
wurde unter Rühren tropfenweise eine Lösung von 5.080 g (22.4 /-\p.»»\Fe A
mmol) [Fp(CH2CI)] in 25 ml Et20 filtriert. Die erhaltene Mischung OC
wurde 30 min bei RT gerührt, eingeengt und der Rückstand im HV getrocknet.
Dreimalige Extraktion mit Pentan, Einengen der Extraktionslösung und Trocknen
des Rückstandes im HV ergab 4.440 g (62%) [Fp(CH2l)]. 1H-NMR (CD2CI2): 5 5.07
(s, 5 cp H), 3.09 (s, 2 H). 13C-NMR (CD2CI2): ô 215.8 (2 CO), 88.0 (5 cp CH), -6.1
(CH2).
Umsetzungen von [Fp(THF)]BF4 mit Triphenylarsoniummethylid. Eine
Suspension von 632 mg (1.6 mmol) Methyltriphenylarsoniumbromid und 66 mg
(1.7 mmol) NaNH2 in THF wurde 1 h bei RT gerührt. Die Suspension wurde filtriert
und die erhaltene gelbe Filtrationslösung unter Rühren bei -78 °C zu einer Lösung
von 270 mg (0.8 mmol) [Fp(THF)]BF4 in THF getropft. Die Mischung wurde unter
Erwärmenlassen auf RT 65 h gerührt und eingeengt. Extraktion mit CH2CI2 und
Einengen der Extraktionslösung ergab einen braunen Feststoff.
Eine Mischung von 1.202 g (3.0 mmol) Methyltriphenylarsoniumbromid und 418 mg
(3.0 mmol) Lithium-bis(trimethylsilyl)amid in 20 ml Et20 wurde 30 min im Dunkeln

5. Experimenteller Teil 118
gerührt. Zu dieser Suspension wurde 733 mg (2.3 mmol) [Fp(THF)]BF4 suspendiert
in 20 ml Et,0 gegeben. Rühren im Dunkeln über 12 h, Filtration, Trocknen des
Rückstandes am HV, Extraktion mit 20 ml CH2CI2, Zugabe von 60 ml Et20 zur
gerührten Extraktionslösung, Filtration der Suspension und Trocknen des
Rückstandes am HV ergab braunen Feststoff. Die Analysedaten (1H-NMR (CD2CI2)
und IR (CH2CI2) waren in beiden Fällen übereinstimmend für [Fp(AsPh3)]BF4.52 Der
Komplex [Fp(CH2AsPh3)]BF4 (K1) wurde nicht beobachtet.
[CpMo(CO)3CH2AsPh3]BF4 (K12). Eine Mischung <^Z^yaus 3.050 g (10.3 mmol) [CpMo(CO)3(CH2CI)], 3.715 g oc..„Mo-CH2As+Ph3 BF4"
(12.1 mmol) Triphenylarsin und 2.361 g (21.5 mmol) OC co
Natriumtetrafluoroborat in MeOH wurde 24 h bei 65 °C gerührt, auf RT abgekühlt
und filtriert. Die Filtrationslösung wurde eingeengt und der erhaltene Rückstand mit
CH2CI2 extrahiert. Einengen der Extraktionslösung, Waschen mit Pentan und
Kristallisation aus CH2CI2/Pentan ergab 642 mg (10%) K12 als gelben Feststoff. 1H-
NMR (CD2CI2): 5 7.74-7.64 (m, 15 ar H), 5.55 (s, 5 cp H), 2.38 (s, CH2). 13C-NMR
(CD2CI2): 5 237.3 (CO), 228.9 (2 CO), 133.4 (ar CH), 132.4 (ar CH), 130.3 (ar CH),
125.3 (ar C), 94.1 (s, 5 cp CH), -18.2 (s, CH2). 19F-NMR (CD2CI2): 5 -151.9. MS
(FAB+): m/z 567 (82), 566 (100, [MY), 565 (78), 564 (86), 510 (56, [M-2CO]+), 321
(91, [MeAsPh3]+), 229 (25, [AsPh2]+). Anal. ber. für C27H22AsBF4MoO30.7 CH2CI2
(711.59): C, 46.76; H, 3.31 ; gef.: C, 46.85; H, 3.31.
[CpW(CO)3CH2AsPh3]BF4 (K13). Eine Mischung cg^p?aus 1.167 g (3.05 mmol) [CpW(CO)3(CH2CI)], 0.952 g 0C„w-CH2As+Ph3 BF4"
(3.11 mmol) Triphenylarsin und 0.832 g (7.58 mmol) OC c0
Natriumtetrafluoroborat in 20 ml MeOH wurde 24 h bei 65 °C gerührt, auf RT
abgekühlt und eingeengt. Der erhaltene Rückstand wurde mit CH2CI2 extrahiert.
Einengen der Extraktionslösung, Waschen mit Hexan und Kristallisation aus
CH2CI2/Hexan bei -20 °C ergab 1.410 g (62%) K13. 1H-NMR (CD2CI2): 5 7.74-7.64
(m, 15arH), 5.66 (s, 5 cp H), 2.56 (s, CH2). 13C-NMR (CD2CI2): 5 225.9 (CO), 219.3
(2 CO), 133.4 (ar CH), 132.5 (ar CH), 130.3 (ar CH), 125.3 (ar C), 92.7 (s, 5 cp CH),

5. Experimenteller Teil 119
-34.2 (s, CH2). 19F-NMR (CD2CI2): 5-151.7. MS (FAB+): m/z 655 (47), 653 (56), 652
(70, [MY), 596 (17, [M-2COD, 321 (100, [MeAsPh3]+), 229 (16, [AsPh2]+). Anal. ber.
für C27H22AsBF403W (740.04): C, 43.82; H, 3.00; gef.: C, 43.70; H, 3.23.
Versuchte Umsetzung von [CpRu(CO)2(CH2CI)] mit Triphenylarsin. Eine
Mischung von 664 mg (2.4 mmol) [CpRu(CO)2(CH2CI)] und 784 mg (2.6 mmol)
Triphenylarsin in 30 ml MeOH wurde 18 h unter Rückfluss gerührt. Nach Abkühlung
auf RT wurde unter Rühren eine Lösung von 1.937 g (5.7 mmol)
Natriumtetraphenylborat in 10 ml MeOH zugetropft. Einengen der orangen Lösung
ergab gelben Feststoff. Im 1H-NMR (CDCI3) wurden nur Eduktsignale beobachtet.
[(PPFPz'BuJPdMeCI] (K14). Eine Lösung von
181.0 mg (0.68 mmol) [(COD)Pd(Me)CI] und 373.5
mg (0.72 mmol) PPFPz'Bu in CH2CI2 wurde über
Nacht bei RT gerührt. Einengen und
Umkristallisation aus CH2CI2/Pentan bei -20 °C
ergab 384.2 mg (83%) K14 als orangen Feststoff.
1H-NMR (CDCI3): 5 7.73-7.65 (m, 2 H), 7.52-7.48
(m, 3 H), 7.34-7.25 (m, 3 H), 7.19-7.13 (m, 2 H), 6.70-6.63 (m, 2 H), 5.99 (d, J = 2.6,
H-C(4) Pz), 4.63 (m, 1 cp H), 4.38 (t, J= 2.6, 1 cp H), 3.94 (s, 5 cp H), 3.84 (m, 1 cp
H), 2.06 (d, J= 7.1, 3 H, CHMe), 1.29 (d, J(P,H) = 2.8, 3 H, PdMe), 1.00 (s, 9 H, 'Bu).
31P-NMR(CDCI3): 516.1 (s). MS (FAB+): m/z 1319 (16, [2M-CI]+), 641 (100, [M-Cl]+),
626 (16, [M-Cl-Me]+), 565 (10), 520 (12), 441 (6), 335 (9), 307 (9), 291 (19), 154
(19). Anal. ber. für C32H36CIFeN2PPd0.8 CH2CI2 (745.29): C, 52.86; H, 5.09; N, 3.76;
gef.: C, 52.98; H, 5.18; N, 3.67. (Aufgrund der Kopplungskonstanten 3J(P,H) = 2.8
Hz wurde auf die c/'s-Position der Methylgruppe am Palladium zum Phosphor
geschlossen.53,54)
5. Experimenteller Teil 120
[(PPFPz'BuJPdMefMeCNJJPFe (K15).
Eine Lösung von 296.8 mg (0.44 mmol)
[(PPFPz'Bu)PdMeCI] und 181 mg (4.4
mmol) MeCN in CH2CI2 wurde zu 181.5 mg
(0.52 mmol) Thalliumhexafluorophosphat
gegeben. Nach Rühren über Nacht wurde
die Suspension filtriert. Einengen der
Filtrationslösung und Umkristallisation aus
CH2CI2/Pentan bei -20 °C ergab 320.4 mg (88%) K15 als gelbes Pulver. 1H-NMR
(CD3CN): 57.76-7.24 (m, 8 ar H, H-C(5) Pz), 6.94 (q, J= 7.1, CHMe), 6.78-6.70 (m,
2 ar H), 6.19 (d, J= 2.7, H-C(4) Pz), 4.86 (m, 1 cp H), 4.55 (t, J= 2.6, 1 cp H), 4.02 (s,
5 cp H), 3.96 (m, 1 cp H), 2.14 (d, J = 7.1, 3 H, CHMe), 1.97 (s, MeCN), 1.10 (d,
J(P,H) = 1.9, 3 H, PdMe), 1.00 (s, 9 H, 'Bu). 31P-NMR (CD3CN): 5 17.3 (s), -143.9 (h,
J(P,F) = 706.4). Anal. ber. für C34H39F6FeN3P2Pd0.1 CH2CI2 (836.40): C, 48.97; H,
4.72; N, 5.02; gef.: C, 48.88; H, 4.93; N, 4.86.
[(PIGIPHOS)Pd(MeCN)](PF6)2 (K16).
Eine Mischung von 601.7 mg (2.11 mmol)
[(COD)PdCI2] und 2.048 g (2.25 mmol)
PIGIPHOS in CH2CI2 wurde über Nacht bei2 PF«
RT gerührt und filtriert. Die Filtrationslösung
wurde mit Hexan versetzt, wobei ein
oranger Feststoff ausfiel. Filtration,
Trocknen am HV und Umkristallisation aus CH2CI2/Pentan bei -20 °C ergab 2.261
g (99%) [(PIGIPHOS)PdCI]CI als orangen Feststoff. MS (FAB+): m/z 1049 (100,
[M-Cl]+), 1015 (11, [M-2CI]+), 929 (10), 397 (7), 212 (7). Anal. ber. für
C54H55Fe2P3PdCI20.1 CH2CI2 (1094.46): C, 59.37; H, 5.08; gef.: C, 59.21; H, 5.45.
Eine Suspension von 1.0802 g (0.99 mmol) [(PIGIPHOS)PdCI]CI und 370.7 mg
(1.05 mmol) Thalliumhexafluorophosphat in CH2CI2 wurde bei RT über Nacht
gerührt. Filtration, Einengen der Filtrationslösung und Umkristallisation aus
CH2CL/Pentan bei -20 °C ergab 1.105 g (93%) [(PIGIPHOS)PdCI]PF655 als
oranges Pulver. 1H-NMR (CD3CN): 5 7.96-7.79 (m, 4 ar H), 7.65-7.20 (m, 16 ar H),

5. Experimenteller Teil 121
5.01 (m, 1 cp H), 4.75 (m, 2 cp H), 4.66 (t, J= 2.4, 1 cp H), 4.56 (m, 1 cp H), 4.34 (m,
1 cp H), 3.97 (s, 5 cp H), 3.79 (s, 5 cp H), 3.70 (m, 1 H), 3.46 (m, 1 H), 2.96 (m, 1 H),
2.35 (m, 1 H), 2.16-1.78 (m, 7 H), 1.65 (dd, 3 H), 1.55-0.62 (m, 5 H). MS (FAB+): m/z
1051 (100, [MY), 1015 (5, [M-Cin, 1012 (10), 929 (9), 613 (6), 535 (9), 460 (17), 397
(13), 307 (58), 289 (25), 242 (10), 212 (11), 154 (75). Anal. ber. für
C54H55CIF6Fe2P4Pd0.5 CH2CI2 (1237.95): C, 52.88; H, 4.56; gef.: C, 52.63; H, 4.93.
Eine Mischung von 176.9 mg (0.50 mmol) Thalliumhexafluorophosphat und 233.9
mg (5.70 mmol) MeCN wurde mit einer Lösung von 521.8 mg (0.44 mmol)
[(PIGIPHOS)PdCI]PF6 in CH2CI2 versetzt. Die erhaltene Suspension wurde bei RT
über Nacht gerührt. Nach Filtration und Einengen der Filtrationslösung wurde der
Rückstand mit möglichst wenig CH2CI2 extrahiert. Die Extraktionslösung wurde mit
Pentan versetzt bis kein weiterer Niederschlag zu beobachten war. Rühren der
Suspension bei RT über 2 d, Filtration und Trocknen am HV ergab 518.2 mg (87%)
[(PIGIPHOS)Pd(MeCN)](PF6)255 als rotes Pulver. 1H-NMR (CD2CI2): 5 8.09-7.12
(m, 18 ar H), 6.85 (m, J= 7.1, 2 H), 5.05 (m, 1 cp H), 5.02 (m, 1 cp H), 4.86 (m, 1 cp
H), 4.81 (t, J= 2.5, 1 cp H), 4.74 (m, 1 cp H), 4.33 (m, 1 cp H), 4.20 (s, 5 cp H), 3.97
(m, 1 H), 3.90 (s, 5 cp H), 3.34 (m, 1 H), 2.75 (m, 1 H), 2.41 (m, 1 H), 2.14 (dd, 3 H),
1.88-1.70 (m, 6 H), 1.55-1.48 (m, 5 H), 1.05 (m, 1 H), 0.90 (m, 1 H), 0.50-0.26 (m, 2
H). Anal. ber. für C56H58F12Fe2NP5Pd (1346.05): C, 49.97; H, 4.34; N, 1.04; gef.: C,
50.04; H, 4.48; N, 0.93.
Zu 367.1 mg (1.04 mmol) Thalliumhexafluorophosphat wurde eine Mischung von
516.3 mg (0.48 mmol) [(PIGIPHOS)PdCI]CI und 238.1 mg (5.8 mmol) MeCN in
CH2CI2 gegeben. Die erhaltene Suspension wurde bei RT über Nacht gerührt und
filtriert. Einengen der Filtrationslösung und Umkristallisation des Rohproduktes aus
CH2CI2/Pentan bei -20 °C ergab 611.8 mg (95%)
[(PIGIPHOS)Pd(MeCN)](PF6)2 als oranges Pulver.

5. Experimenteller Teil 122
5.7 Arsoniumsalze
Methyltris(4-fluorphenyl)arsonium-
iodid (S1). Eine Mischung von 1.433 g (4.0 H3C—As+J
mmol) A1 und 10 ml (161 mmol) Methyliodid
wurde 4 d bei 40 °C gerührt. Der entstandene gelbe Feststoff wurde abfiltriert, mit
Et20 gewaschen und im HV getrocknet. Man erhielt 1.495 g (75%) S1 als
gelblichen Feststoff. 1H-NMR (CD2CI2): 5 7.89-7.80 (m, 6 ar H), 7.50-7.39 (m, 6 ar
H), 3.24 (s, CH3). 13C-NMR (CD2CI2): 5 166.4 (d, J (C,F) = 257.8, ar C), 135.4 (d, J
(C,F) = 9.3, ar CH), 117.6 (d, J (C,F) = 22.4, ar CH), 117.2 (d, J (C,F) = 3.3, ar C),
11.6 (CH3). 19F-NMR (CD2CI2): 5-102.4. Anal. ber. für C19H15AsF3l (502.15): C, 45.45;
H, 3.01 ; F, 11.35; gef.: C, 45.35; H, 3.08; F, 11.06.
Methyltris(4-fluorphenyl)-
arsoniumtetrafluoroborat (S2). u q Ac+|Eine Suspension von 1.427 g (4.0
mmol) A1 und 1.455 g (9.8 mmol)
Trimethyloxonium-tetrafluoroborat in 15 ml CH2CI2 wurde 4 d bei RT gerührt. Nach
Zugabe von 20 ml H20 wurde die organische Phase abgetrennt, über MgS04
getrocknet und eingeengt. Trocknen im HV ergab 1.755 g (95%) S2 als weissen
Feststoff. 1H-NMR (CD2CI2): 5 7.71-7.64 (m, 6 ar H), 7.48-7.39 (m, 6 ar H), 2.83 (s,
CH3). 13C-NMR (CD2CI2): 5 166.4 (d, J(C,F) = 257.7, ar C), 134.8 (d, J(C,F) = 9.4, ar
CH), 118.6 (d, J(C,F) = 22.4, ar CH), 117.2 (d, J(C,F) = 3.3, ar C), 8.5 (CH3). 19F-NMR
(CD2CI2): 5-102.4 (s), -151.1.
Methyltris(4-chlorphenyl)arsonium-
iodid (S3). Eine Mischung von 0.830 g u pa +.
(2.0 mmol) A2 und 10 ml (161 mmol)
Methyliodid wurde 3 d bei RT gerührt. Der
entstandene gelbe Feststoff wurde abfiltriert, gewaschen mit Et20 und im HV
getrocknet. Man erhielt 145 mg (13%) S3 als gelblichen Feststoff. 1H-NMR (CDCI3):

5. Experimenteller Teil 123
57.78-7.61 (m, 12 ar H), 3.25 (s, CH3). 13C-NMR (CDCI3): 5 141.5 (ar C), 134.0 (ar
CH), 131.2(arCH), 119.6(arC), 11.1 (CH3).
Methyltris(4-chlorphenyl)-
arsoniumtetrafluoroborat (S4).
Eine Suspension von 1.146 g (2.8 H3C—As+f—<^ ù Cil BF4
mmol) A2 und 1.375 g (9.3 mmol)
Trimethyloxonium-tetrafluoroborat in 20 ml CH2CI2 wurde 3 d bei RT gerührt. Nach
Zugabe von 20 ml H20 wurde die organische Phase abgetrennt, mit H20 und Et20
gewaschen, über MgS04 getrocknet und eingeengt. Man erhielt 1.192 g (82%) S 4
als weissen Feststoff. 1H-NMR (CDCI3): 57.73-7.57 (m, 12 ar H), 2.84 (s, CH3). 13C-
NMR(CDCI3):5141.5(arC), 133.5 (ar CH), 131.3 (ar CH), 119.6 (ar C), 8.1 (CH3).
Anal. ber. für C19H15AsBCI3F4 (511.41): C, 44.62; H, 2.96; gef.: C, 45.02; H, 3.13.
Methyltris(3-trifluormethylphenyl)-
arsoniumtetrafluoroborat (S5). Aus
5.494 g (10.8 mmol) A3 und 4.445 g (30 |_|3q As+-{-<1 /> l BF,
mmol) Trimethyloxonium-tetrafluoroborat
in 60 ml CH2CI2, gemäss Synthese von
S4. Kristallisation des Rohprodukts aus CH2CI2/Hexan ergab 5.423 g (82%) S5 als
weissen Feststoff. 1H-NMR (CD2CI2): 5 8.12-7.85 (m, 12 ar H), 3.05 (s, CH3). 13C-
NMR(CD2CI2):5136.1 (arCH), 133.1 (q, J (C,F) = 33.8, ar C), 132.1 (ar CH), 131.6
(q, J (C,F) = 3.5, ar CH), 128.7 (q, J (C,F) = 3.8, ar CH), 122.8 (q, J (C,F) = 273.2,
CF3), 122.2 (ar C), 8.4 (CH3). 19F-NMR (CD2CI2): 5 -63.6, -150.3. Anal. ber. für
C22H15AsBF13 (612.07): C, 43.17; H, 2.47; gef.: C, 43.35; H, 2.59.

5. Experimenteller Teil 124
5.8 Olefinvorläufer, Olefine und Cyclopropane
1-(4-Methoxyphenyl)cyclohexan-1-ol. OH
Eine Mischung von 26.179 g (140.0 mmol) 1-
Brom-4-methoxybenzol und 3.418 g (140.6
mmol) Mg in 250 ml THF wurde am Rückfluss
gekocht bis eine Lösung vorlag. Diese Lösung wurde rasch mit 13.658 g (139.2
mmol) Cyclohexanon versetzt. Nach 1 h Rühren bei RT wurde NH4CI-Lsg.
zugegeben und dreimal mit Et20 extrahiert. Vereinigung der organischen Phasen,
Trocknen über MgS04, Einengen, Trocknen im HV und FC (Et20) ergab 26.280 g
(91%) des Alkohols als farbloses Oel. DC (Hexan/Ethylacetat 20/1): R, 0.02;
(Hexan/Ethylacetat 10/1): R, 0.04; (Et20): 0.72. 1H-NMR (CDCI3): 5 7.38-7.32 (m, 2
H), 6.92-6.86 (m, 2 H), 3.80 (s, 3 H), 1.87-1.58 (m, 11 H). 13C-NMR (CDCI3): 5 158.3
(arC), 141.8 (ar C), 125.9 (ar CH), 113.5 (ar CH), 72.7 (C), 55.2 (CH3), 39.0 (CH2),
25.6 (CH2), 22.3 (CH2). MS (El+): m/z 206 (1, [MY), 188 (17, [M-H20]+), 173 (1,
[M-H20-CH3]+), 159 (8), 145 (2), 128 (3), 115 (3), 91 (2), 77 (2), 63 (1), 55 (1), 40
(2), 32 (24), 28 (100, [CO]+), 18 (3, [H20]+). Anal. ber. für C13H1802 (206.28): C, 75.69;
H, 8.79; gef.: C, 75.75; H, 8.74.
1-Cyclohex-1-enyl-4-methoxybenzol.9 ^^ O
Eine Lösung von 11.265 g (54.6 mmol) 1-(4- |
Methoxyphenyl)cyclohexan-1-ol und 0.988 g (5.2 ^^ /^v S
mmol) Toluol-4-sulfonsäure Monohydrat in 400 | |ml Benzol wurde 1 h bei 90 °C am Rückfluss L
.
gerührt, auf RT abgekühlt und mit ges. NaHC03-
Lsg. versetzt. Extraktion mit Et20, Trocknen über MgS04, Einengen, Trocknen im HV
und FC (Hexan/Ethylacetat 20/1) ergab 8.447 g (82%) des Olefins als weissen
Feststoff. DC (Hexan/Ethylacetat 20/1): R, 0.63. 1H-NMR (CDCI3): 5 7.38-7.32 (m, 2
ar H), 6.92-6.86 (m, 2 ar H), 6.08 (m, 1 H), 3.84 (s, 3 H), 2.46-2.38 (m, 2 H),
2.29-2.18 (m, 2 H), 1.88-1.75 (m, 2 H), 1.74-1.65 (m, 2 H). 13C-NMR (CDCI3): Ô
158.5 (arC), 136.0 (arC), 135.4(C), 126.0 (arCH), 123.2 (ar CH), 113.6 (CH), 55.3

5. Experimenteller Teil 125
(CH3), 27.5 (CH2), 25.9 (CH2), 23.2 (CH2), 22.3 (CH2). MS (El+): m/z 188 (100, [M]+),
173 (16, [M-CH3]+), 160 (40), 145 (14), 129 (13), 115 (12), 91 (7), 77 (5). Anal. ber.
fürC13H160 (188.27): C, 82.94; H, 8.57; gef.: C, 82.98; H, 8.61.
1-Phenylbicyclo[4.1.0]heptan. Zu einer Mischung von 4.716
g (29.8 mmol) 1-Phenylcyclohexen und Diethylzink in Hexan (33
mmol) in 30 ml Benzol wurde bei 60 °C unter Rühren
tropfenweise 3.7 ml (45.9 mmol) Diiodmethan zugegeben. Nach
15 h Rühren wurde die Reaktionsmischung auf RT abgekühlt und mit 50 ml 1 proz.
HCI-Lsg. versetzt. Die organische Phase wurde mit NaHC03-Lsg. gewaschen, über
MgS04 getrocknet und eingeengt. FC (Hexan/Ethylacetat 10/1) und Destillation (58
°C, 0.07 mbar) ergab 4.190 g einer farblosen Flüssigkeit, die durch 1H-NMR-
Spektroskopie als 1/1 Mischung von Cyclopropan/Olefin identifiziert wurde. Eine
Lösung von 1.033 g dieser Flüssigkeit in 10 ml THF wurde bei 0 °C langsam zu
Dimethylsulfid-boran in THF (3.6 mmol) getropft. Nach 2 h Rühren bei RT wurden 1
ml MeOH, 3 ml 2M KOH und 1 ml H202-Lsg. (30 proz. in H20) zugegeben. Die
Reaktionsmischung wurde 1 h bei 55 °C gerührt und mit 100 ml Et>0 versetzt. Die
organische Phase wurde abgetrennt, filtriert, mit H20 gewaschen, über MgS04
getrocknet und eingeengt. FC (Hexan) ergab 285 mg des Cyclopropans als
farblose Flüssigkeit. DC (Hexan/Ethylacetat 20/1): R, 0.80; (Hexan/Ethylacetat 10/1):
R, 0.84; (Et20): R, 0.87; (Hexan): R, 0.51. 1H-NMR (CDCI3): 5 7.30-7.26 (m, 5 H),
2.15-1.18 (m, 9 H), 0.95 (dd, J = 9.4, 4.4, 1 H), 0.65 (dd, J = 5.7, 4.4, 1 H). MS (El+):
m/z 172 (38, [M]+), 144 (40), 129 (100), 118 (25), 115 (71), 104 (63), 91 (64), 77 (34,
[Ph]+), 63 (29), 51 (34), 39 (43), 27 (23).
1-(4-Methoxyphenyl)bicyclo-[4.1.0]-heptan.
Zu einer Mischung von 7.39 g (39 mmol) 1-
Cyclohex-1-enyl-4-methoxybenzol und Diethylzink
in Hexan (62 mmol) in 30 ml Benzol wurden 5.1 ml
(63.3 mmol) Diiodmethan gegeben. Die
Reaktionsmischung wurde über Nacht bei 80 °C gerührt. Nach Zugabe von 100 ml
1 proz. HCI-Lsg. bei RT wurde die organische Phase mit NaHC03-Lsg. gewaschen,
über MgS04 getrocknet und eingeengt. FC (Hexan/Ethylacetat 20/1) ergab 6.729 g

5. Experimenteller Teil 126
einer farblosen Flüssigkeit, die durch 1H-NMR-Spektroskopie als 4/1 Mischung von
Cyclopropan/Olefin identifiziert wurde. Eine Lösung von 3.054 g dieser Flüssigkeit
in 20 ml THF wurde bei 0 °C zu Dimethylsulfid-boran in THF (5.0 mmol) gegeben.
Nach Rühren bei RT über Nacht wurden 2 ml MeOH, 6 ml 2M KOH und 2 ml H202-
Lsg. (30 proz. in H20) zugegeben. Die Reaktionsmischung wurde 1 h bei 50 °C
gerührt und mit 200 ml Et20 versetzt. Die organische Phase wurde abgetrennt,
filtriert, mit H20 gewaschen, über MgS04 getrocknet und eingeengt. FC
(Hexan/Ethylacetat 20/1) und Destillation ergaben 1.445 g des Cyclopropans als
farbloses, klares Oel. DC (Hexan/Ethylacetat 20/1): R, 0.52. 1H-NMR (CDCI3): 5
7.27-7.19 (m, 2 ar H), 6.87-6.81 (m, 2 ar H), 3.80 (s, 3 H), 2.15-1.85 (m, 3 H),
1.73-1.63 (m, 1 H), 1.59-1.15 (m, 5 H), 0.91 (dd, J=9.4, 4.4, 1 H), 0.65 (dd, J= 5.7,
4.4, 1 H). 13C-NMR (CDCI3): 5 157.4 (ar C), 142.0 (ar C), 128.5 (ar CH), 113.5 (ar
CH), 55.3 (CH3), 32.0 (CH2), 24.0 (CH2), 23.9 (C), 21.7 (2 CH2), 18.8 (CH), 18.1
(CH2). MS (El+): m/z 202 (100, [MY), 187 (13, [M-CH3]+), 173 (30), 159 (56), 148
(27), 134 (26), 129 (15), 121 (41), 115 (17), 108 (8), 91 (15), 77 (11). Anal. ber. für
C14H180 (202.30): C, 83.12; H, 8.97; gef.: C, 83.14; H, 8.91. HPLC-
Enantiomerentrennung: Chiralcel OD-H, eluiert bei 24.5 °C mit Hexan/'PrOH =
99.7/0.3, Fluss 0.3 ml/min Retentionszeiten: 36.8 min, 39.9 min.
1-Trimethylsilyl-2-phenylcyclopropan. Eine A
Suspension von 566.2 mg (5.4 mmol) Styrol und 257.3 mg J-—^(2.6 mmol) CuCI in 2 ml Toluol wurde bei RT unter Rühren Ph SiMe3
tropfenweise über 15 min mit 3.30 ml Trimethylsilyldiazomethan-Lsg. (2M in Hexan)
(6.6 mmol) versetzt. Nach Rühren bei RT über Nacht wurde die Reaktionsmischung
auf 60 °C erwärmt und 1 h gerührt. Nach Abkühlen auf RT wurde die
Reaktionsmischung eingeengt. FC (Hexan/Ethylacetat 10/1) ergab 388 mg eines
gelben Oels. DC (Hexan/Ethylacetat 10/1): R, 0.67. Eine Lösung dieser Flüssigkeit
in 2 ml THF wurde bei 0 °C langsam mit Dimethylsulfid-boran in THF (10 mmol)
versetzt. Nach 2 h Rühren bei RT wurden 1 ml MeOH, 3 ml 2M KOH und 1 ml H202-
Lsg. (30 proz. in H20) zugegeben. Die Reaktionsmischung wurde 1 h bei 55 °C
gerührt und mit 100 ml Et20 versetzt. Die organische Phase wurde abgetrennt,
filtriert, mit H20 gewaschen, über MgS04 getrocknet und eingeengt. FC
(Hexan/Ethylacetat 10/1) ergab 125.4 mg (12%) des Cyclopropans als farblose
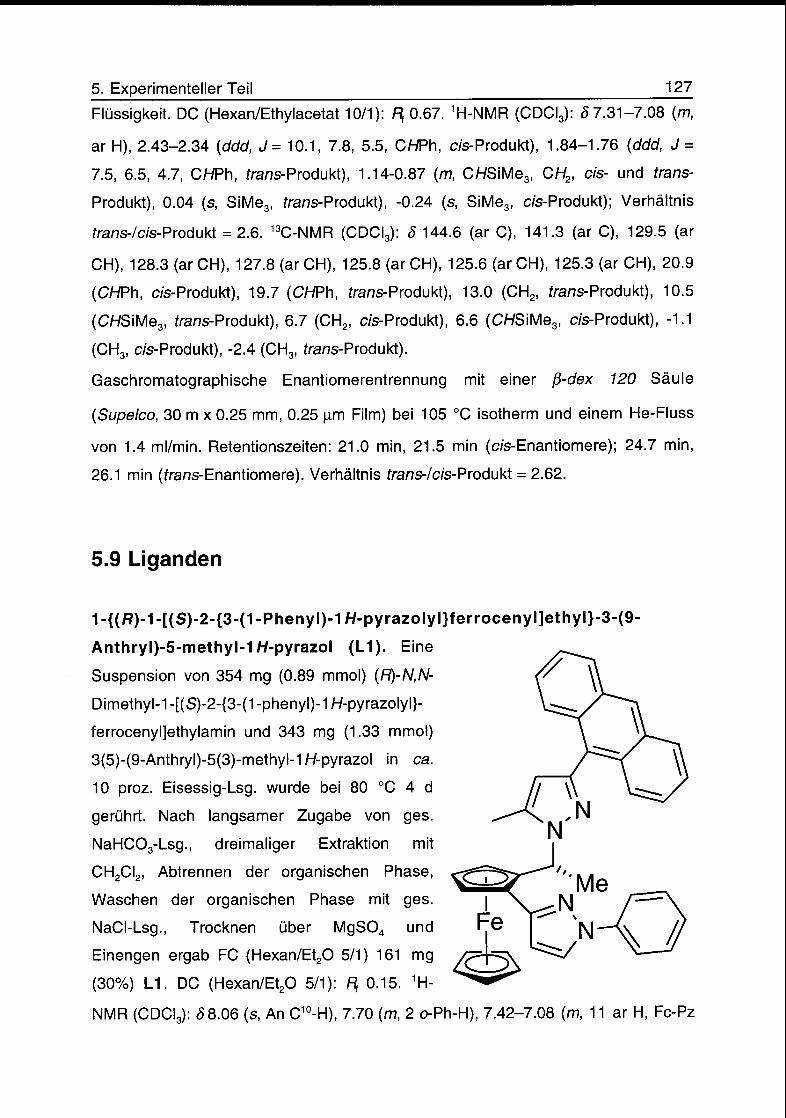
5. Experimenteller Teil 127
Flüssigkeit. DC (Hexan/Ethylacetat 10/1): R, 0.67. 1H-NMR (CDCI3): 5 7.31-7.08 (m,
ar H), 2.43-2.34 (ddd, J= 10.1, 7.8, 5.5, CHPh, c/'s-Produkt), 1.84-1.76 (ddd, J =
7.5, 6.5, 4.7, CHPh, trans-Produkt), 1.14-0.87 (m, CHSiMe3, CH2, eis- und trans-
Produkt), 0.04 (s, SiMe3, frans-Produkt), -0.24 (s, SiMe3, c/s-Produkt); Verhältnis
trans-/cis-Produk\ = 2.6. 13C-NMR (CDCI3): 5 144.6 (ar C), 141.3 (ar C), 129.5 (ar
CH), 128.3 (ar CH), 127.8 (ar CH), 125.8 (ar CH), 125.6 (ar CH), 125.3 (ar CH), 20.9
(CHPh, c/s-Produkt), 19.7 (CHPh, trans-Produkt), 13.0 (CH2, frans-Produkt), 10.5
(CHSiMe3, frans-Produkt), 6.7 (CH2, c/s-Produkt), 6.6 (CHSiMe3, c/s-Produkt), -1.1
(CH3, c/s-Produkt), -2.4 (CH3, trans-Produkt).
Gaschromatographische Enantiomerentrennung mit einer ß-dex 120 Säule
(Supeico, 30 m x 0.25 mm, 0.25 Lim Film) bei 105 °C isotherm und einem He-Fluss
von 1.4 ml/min. Retentionszeiten: 21.0 min, 21.5 min (c/s-Enantiomere); 24.7 min,
26.1 min (frans-Enantiomere). Verhältnis trans-lcis-Produkt = 2.62.
5.9 Liganden
1-{(fl)-1-[(S)-2-{3-(1-Phenyl)-1H-pyrazolyl}ferrocenyl]ethyl}-3-(9-
Anthryl)-5-methyl-1H-pyrazol (L1). Eine
Suspension von 354 mg (0.89 mmol) (R)-N,N-
Dimethyl-1 -[(S)-2-{3-(1 -phenyl)-1 H-pyrazolyl}-
ferrocenyl]ethylamin und 343 mg (1.33 mmol)
3(5)-(9-Anthryl)-5(3)-methyl-1H-pyrazol in ca.
10 proz. Eisessig-Lsg. wurde bei 80 °C 4 d
gerührt. Nach langsamer Zugabe von ges.
NaHC03-Lsg., dreimaliger Extraktion mit
CH2CI2, Abtrennen der organischen Phase,
Waschen der organischen Phase mit ges.
NaCI-Lsg., Trocknen über MgS04 und
Einengen ergab FC (Hexan/Et20 5/1) 161 mg
(30%) L1. DC (Hexan/Et20 5/1): R, 0.15. 1H-
NMR (CDCI3): 58.06 (s, An C10-H), 7.70 (m, 2 o-Ph-H), 7.42-7.08 (m, 11 ar H, Fc-Pz

5. Experimenteller Teil 128
C5-H), 6.00 (s, 1 An-Pz H), 5.87 (q, J= 7.1, CHMe), 5.69 (d, J = 2.5, Fc-Pz C4-H),
4.02 (m, 2 cp H), 3.83 (s, 5 cp H), 3.76 (m, 1 cp H), 2.47 (s, Pz-Me), 2.07 (d, J = 7.1,
CHMe). 13C-NMR (CDCI3): 5150.6 (C), 146.8 (C), 139.5 (C), 138.5 (C), 131.1 (C),
130.8 (C), 130.54 (C), 130.50 (C), 128.8 (ar CH), 127.8 (ar CH), 127.43 (ar CH),
127.37 (arCH), 126.3 (ar CH), 126.0 (ar CH), 125.4 (C), 125.1 (ar CH), 124.9 (ar
CH), 124.8 (arCH), 124.5 (ar CH), 117.5 (CH), 108.1 (CH), 105.5 (CH), 87.1 (cp C),
76.0 (cp C), 69.7 (cp CH), 68.0 (cp CH), 66.7 (cp CH), 66.3 (cp CH), 53.3 (CHMe),
22.5 (Pz-Me), 13.8 (CHMe). MS (FAB+): m/z 612 (100, [MY), 547 (12, [M-cp]+), 355
(33, [M-AnMeHPz]+), 289 (6, [M-cp-AnMeHPz]+), 154 (9), 137 (6). Anal. ber. für
C39H32FeN4 0.1 CH2CI2 (621.05): C, 75.62; H, 5.23; N, 9.02; gef.: C, 75.73; H, 5.51;
N, 8.87.
5.10 Reaktionsversuche
Cyclopropanierung von Styrol mit [FpCH2AsR3]X (X = CI, BPh4, BF4 oder
SbF6). Zu einer Mischung von Komplex (1.0 equiv.) und Styrol (1.0 equiv.) in
CH3CN/Pentan 9/2 (c = 0.10 M) wurde "Dekan (0.8 equiv.) als interner Standard für
die Gaschromatographie zugegeben. Reaktionstemperaturen und -zeiten sind
angegeben. Die Proben für die gaschromatographischen Analysen wurden direkt
der Reaktionsmischung entnommen. GC: Säule SE 54 (Macherey-Nagel, 25 m x
0.25 mm, 0.25 Lim Film), Temperaturprogramm: 50 °C während 5 min, mit 10 °C/min
bis 200 °C; Retentionszeit/min: Styrol: 8.9, "Dekan: 10.3, Cyclopropylbenzol: 12.0.
Cyclopropanierung von 1-Cyclohex-1-enyl-4-methoxybenzol mit
Komplexen K1, K7 und K8. Eine Mischung von Komplex (1.0 equiv.) und Olefin
(1.0 equiv.) in CH3CN (c = 0.10 M) wurde 48 h bei 100 °C gerührt. Nach Abkühlen
auf RT wurde die Reaktionsmischung eingeengt, extrahiert mit Et,0, filtriert über
Kieselgel und eingeengt. Die Analyse erfolgte mittels 1H-NMR-Spektroskopie.

5. Experimenteller Teil 129
Cyclopropanierung von Cyclohex-1-enyl-4-methoxybenzol mit den
chiralen Komplexen K9-K11. Eine Mischung aus Komplex (1 equiv.) und Olefin
(3 equiv.) in CH3CN/Benzol 2/1 (0.03 respektive 0.01 M) wurde 5 d bei 100 °C
gerührt. Die Reaktionsmischung wurde auf RT abgekühlt, eingeengt, extrahiert mit
Et20 und eingeengt. TLC (Hexan/Ethylacetat 20/1), Extraktion mit CH2CI2 und
Abziehen des Lösungsmittels ergab farbloses Oel. Diese Olefin/Cyclopropan-
Mischung wurde mittels 1H-NMR-Spektroskopie und HPLC analysiert. HPLC: Säule
Daicel Chiralcel OD-H (4.6 x 250 mm, Korngrösse 5 um), Hexan/PrOH 99.7/0.3, 0.3
ml/min, T = 24.5 °C, 230 nm; Retentionszeiten/min: 36.8, 39.9.
Versuchte Cyclopropanierung von Styrol mit K12 und K13. Eine
equimolare Mischung des Komplexes und des Olefins in CH3CN/Pentan 9/2 (c =
0.10 M) wurde mit "Dekan (0.8 equiv.) als interner Standard für die
Gaschromatographie versetzt und 24 h bei 100 °C gerührt. Nach Abkühlen auf RT
wurden die Proben für die gaschromatographische Analyse direkt der
Reaktionsmischung entnommen.
Versuchte Cyclopropanierung von Cycloocten mit Arsoniummethyliden.
Zu einer Mischung eines Methyltriarylarsonium-Salzes S1-S5 (1 equiv.) und
Natrium-bis(trimethylsilyl)amid (1 equiv.) in Benzol wurde [Fp(THF)]BF4 (1 equiv.)
und Cycloocten (1 equiv.) in CH3CN (c = 0.2 M) zugegeben. Die
Reaktionsmischung wurde 10 d bei 50 °C gerührt, auf RT abgekühlt, eingeengt bei
50 °C/100 mbar, extrahiert mit Et,0 und eingeengt. Die Analyse erfolgte mittels
GC/MS.
Katalytische Cyclopropanierung von Styrol mit
Trimethylsilyldiazomethan. Eine Mischung von Cu(CF3SO3)(C6H6)05 (5 mol-%)
und Ligand (11-110, L1) in einem 15ml-Pillenglas wurde in der glove-box mit
CH2CI2 versetzt und über Nacht bei RT gerührt. Diese Mischung wurde zu Styrol
(1.0 equiv.) und "Dekan (0.8 equiv.) als interner Standard für die
Gaschromatographie filtriert. Lösungsmittel, Reaktionstemperaturen und -zeiten
sind angegeben. Die Proben für die gaschromatographischen Analysen wurden

5. Experimenteller Teil 130
direkt der Reaktionsmischung entnommen. GC: Säule SE 54 (Macherey-Nagel, 25
m x 0.25 mm, 0.25 Lim Film), Temperaturprogramm: 50 °C während 5 min, mit 5
°C/min bis 200 °C; Retentionszeit/min: Styrol: 10.1, "Dekan: 14.2, c/s-1-Phenyl-2-
trimethylsilyl-cyclopropan: 23.7, frans-1-Phenyl-2-trimethylsilyl-cyclopropan: 24.4.
Gaschromatographische Enantiomerentrennung mit einer ß-dex 120 Säule
(Supeico, 30 m x 0.25 mm, 0.25 Lim Film) bei 105 °C isotherm und einem He-Fluss
von 1.4 ml/min. Retentionszeiten: 21.0 min, 21.5 min (c/s-Enantiomere); 24.7 min,
26.1 min (frans-Enantiomere).
Cyclopropanierung von Styrol mit Trimethylsilyldiazomethan mit einem
Cu(ll)-l7-Komplex. Eine Lösung von 259.2 mg (0.57 mmol) (4R,5S,4'R,5'S)-2,2'-
Methylen-bis-(4,5-diphenyl-2-oxazolin) in 5 ml THF wurde bei -78 °C tropfenweise
mit 0.36 ml einer 1.60M-Lsg. (0.57 mmol) von BuLi in Hexan versetzt. Nach 30 min
Rühren wurde die Reaktionsmischung auf RT erwärmt, weitere 30 min gerührt und
mit 38.3 mg (0.28 mmol) wasserfreiem CuCI2 versetzt. Nach Rühren über Nacht
wurde die erhaltene violette Lösung über Alox filtriert, eingeengt und im HV
getrocknet. Waschen des zurückgebliebenen Feststoffs bei -20 °C mit Hexan und
Trocknen im HV ergab 211.1 mg ("77%") eines blauen Feststoffs.
Zu einer Lsg. von 107.2 mg ("0.11 mmol") dieses Feststoffs in CH2CI2 wurde 0.22 ml
einer 0.50M-Lsg. (0.11 mmol) von Phenylhydrazin in CH2CI2 gegeben. Nach 5 min
Rühren wurde die Mischung mit 227.4 mg (2.18 mmol) Styrol und 180.5 mg (1.27
mmol) Dekan und tropfenweise mittels Spritzenpumpe mit 1.30 ml einer 2.0M-Lsg.
(2.6 mmol) von Trimethylsilydiazomethan in Hexan versetzt. Nach 65 h Rühren bei
RT wurde die Reaktionsmischung wie oben beschrieben analysiert.
Cyclopropanierung von Styrol mit Trimethylsilyldiazomethan mit einem
aus CuCI, 17 und BuLi in s/fu-generierten Cu(l)-l7-Komplex. Eine Lösung
von 91.1 mg (0.20 mmol) (4fî,5S,4'H,5'S)-2,2,-Methylen-bis-(4,5-diphenyl-2-
oxazolin) in THF wurde tropfenweise mit 0.05 ml einer 1.60M-Lsg. (0.08 mmol) von
BuLi in Hexan versetzt. Nach 5 min Rühren wurde diese Mischung zu 8.5 mg (0.08
mmol) wasserfreiem CuCI gegeben, über Nacht gerührt und zu 180.5 mg (1.73
mmol) Styrol und 105.1 mg (0.74 mmol) Dekan in 1,2-Dichloroethan filtriert. Die

5. Experimenteller Teil 131
erhaltene Lsg. wurde bei 50 °C tropfenweise mittels Spritzenpumpe über 3 h mit
1.0 ml einer2.0M-Lsg. (2.0 mmol) von Trimethylsilydiazomethan in Hexan versetzt.
Nach 68 h Rühren bei 50 °C wurde die Reaktionsmischung wie oben beschrieben
analysiert.
Cyclopropanierung von Styrol mit Trimethylsilyldiazomethan mit einem
aus CuCI, 17 und AgBF4 in s/fu-generierten Cu(l)-l7-Komplex. Eine
Mischung von 8.5 mg (0.08 mmol) wasserfreiem CuCI und 93.6 mg (0.20 mmol)
(4fl,5S,4'H,5'S)-2,2'-Methylen-bis-(4,5-diphenyl-2-oxazolin) in CH2CI2 wurde zu
22.2 mg (0.11 mmol) AgBF4 gegeben. Diese Suspension wurde über Nacht gerührt
und zu 178.0 mg (1.71 mmol) Styrol und 112.6 mg (0.79 mmol) Dekan in 1,2-
Dichloroethan filtriert. Die erhaltene Mischung wurde bei 50 °C tropfenweise mittels
Spritzenpumpe über 3 h mit 1.0 ml einer 2.0M-Lsg. (2.0 mmol) von
Trimethylsilydiazomethan in Hexan versetzt. Nach 68 h Rühren bei 50 °C wurde die
Reaktionsmischung wie oben beschrieben analysiert.
5.11 Literatur
(1) Kofron, W. G.; Baclawski, L. M. J. Org. Chem. 1976, 41, 1879-1880.
(2) De Ketelaere, R. F.; Delbeke, F. T.; van der Kelen, G. P. J. Organomet. Chem.
1971,28, 217-223.
(3) Cullen, W. R.; Yates, P. E. Can. J. Chem. 1963, 41, 1625-1627.
(4) Samaan, S. Methoden der organischen Chemie; 4 ed.; Thieme: Stuttgart,
1978; Vol. 13.
(5) Steinkopf, W.; Schwen, G. Chem. Ber. 1921, 54, 2969-2975.
(6) McCortney, B. A.; Jacobson, B. M.; Vreeke, M.; Lewis, E. S. J. Am. Chem.
Soc. 1990, 112, 3554-3559.
(7) Henry, M. C; Wittig, G. J. Am. Chem. Soc. 1960, 82, 563-564.
(8) Kliegman, J. M.; Barnes, R. K. Tetrahedron 1970, 26, 2555-2560.
(9) Trost, B. M.; Arndt, H. C. J. Am. Chem. Soc. 1973, 95, 5288-5298.
(10) Dieck, H. t.; Diercks, R.; Stamp, L; Bruder, H.; Schuld, T. Chem. Ber. 1987,
120, 1943-1950.
(11) Catheline, D.; Astruc, D. Organometallics 1984, 3, 1094-1100.
(12) Piper, T. S.; Cotton, F. A.; Wilkinson, G. J. Inorg. Nucl. Chem. 1955, 1, 165-
174.
(13) Fischer, E. O.; Moser, E. Inorg. Synth. 1970, 12, 36.
(14) Brauer, G., Ed. Handbuch der Anorganischen Chemie; 2 ed.; Enke F.:
Stuttgart, 1981; Vol. 3.
(15) Jutzi, P.; Mix, A. Chem. Ber. 1990, 123, 1043-1045.
(16) Reger, D. L; Coleman, C. J. Organomet. Chem. 1977, 131, 153-162.
(17) Schmidt, E. K. G.; Thiel, C. H. J. Organomet. Chem. 1981, 220, 87-102.

5. Experimenteller Teil 132
(18)
(19
(20(21
(22
(23(24
(25(26(27.
(28
(29(30(31(32(33(34
(35(36
(37
(38(39
(40
(41
(42
(43
(44
(45
(46
(47(48(49(50
Hynes, M. J.; Mahon, M. F. T.; McArdle, P. J. Organomet. Chem. 1987, 320,
C44-C46.
Treichel, P. M.; Shubkin, R. L; Barnett, K. W.; Reichard, D. Inorg. Chem.
1966,5, 1177-1181.
Mays, M. J.; Sears, P. L. J. Chem. Soc. Dalton 1973, 1873-1875.
Roger, C; Hamon, P.; Toupet, L.; Rabaa, H.; Saillard, J.-Y.; Hamon, J.-R.;
Lapinte, C. Organometallics 1991, 10, 1045-1054.
Treichel, P. M.; Molzahn, D. C. Synth. React. Inorg. Met. Org. Chem. 1979, 9,
21-29.
Catheline, D.; Astruc, D. J. Organomet. Chem. 1984, 272, 417-426.
Gill, U. S.; Lee, C. C; Sutherland, R. G. Synth. React. Inorg. Met. Org. Chem.
1984, 14, 953-956.
Forschner, T. C; Cutler, A. R. Inorg. Chim. Acta 1985, 102, 113-120.
Belmont, J. A.; Wrighton, M. S. Organometallics 1986, 5, 1421-1428.
Sarma, U. C; Poddar, R. K. Polyhedron 1988, 7, 2627-2633.
Komiya, S. Synthesis of Organometallic Compounds.; Wiley: Chichester,
1997.
Stoop, R. M. Dissertation E7H1999, Nr. 13394.
Bressan, M.; Rigo, P. Inorg. Chem. 1975, 14, 2286-2288.
Gill, T. P.; Mann, K. R. Organometallics 1982, 1, 485-488.
Bruce, M. I.; Windsor, N. J. Aust. J. Chem. 1977, 30, 1601-1604.
Fan, L; Einstein, F. W. B.; Sutton, D. Organometallics 2000, 19, 684-694.
King, R. B. Organometallic Syntheses; Academic Press: New York, 1965; Vol.
1.
Beck, W.; Schloter, K. Z Naturforsch. 1978, 33 b, 1214-1222.
Green, M. L. H.; Ishaq, M.; Whiteley, R. N. J. Chem. Soc. (A) 1967, 1508-
1515.
Crawford, E. J.; Lambert, C; Menard, K. P.; Cutler, A. R. J. Am. Chem. Soc.
1985, 107, 3130-3139.
Lin, Y. C; Milstein, D.; Wreford, S. S. Organometallics 1983, 2, 1461-1463.
Pelling, S.; Botha, C; Moss, J. R. J. Chem. Soc. Dalton Trans. 1983, 1495-
1501.
Bellinger, G. C. A.; Friedrich, H. B.; Moss, J. R. J. Organomet. Chem. 1989,
366, 175-186.
Werner, H.; Feser, R. J. Organomet. Chem. 1982, 232, 351-370.
Brookhart, M.; Hauptman, E.; Lincoln, D. M. J. Am. Chem. Soc. 1992, 114,
10394-10401.
Werner, H.; Paul, W.; Feser, R.; Zolk, R.; Thometzek, P. Chem. Ber. 1985,
118, 261-274.
Rettig, M. F.; Wing, R. M.; Wiger, G. R. J. Am. Chem. Soc. 1981, 103, 2980-
2986.
Rülke, R. E.; Emsting, J. M.; Spek, A. L.; Elsevier, C. J.; Van Leeuwen, P. W.
N. M.; Vrieze, K. Inorg. Chem. 1993, 32, 5769-5778.
Drew, D.; Doyle, J. R. Inorg. Synth. 1972, 13, 47'-52.
Al-Najjar, I. M. Inorg. Chim. Acte 1987, 128, 93-104.
Pioda, G. Dissertazione ETH 1999, Nr. 13405.
Hintermann, L. Dissertation ETH 2000, Nr. 13892.
Aoyama, T.; Iwamoto, Y.; Nishigaki, S.; Shioiri, T. Chem. Pharm. Bull. 1989,
37, 253-256.

5. Experimenteller Teil 133
(51) Burckhardt, U. Dissertation ETH 1997, Nr. 12167.
(52) Schumann, H.; Eguren, L. J. Organomet. Chem. 1991, 403, 183-193.
(53) Aeby, A. Dissertation ETH 1999, Nr. 13160.
(54) Gambs, C. Dissertation ETH 2001, Nr. 14156.
(55) Barbara, P.; Togni, A. Organometallics 1995, 14, 3570-3573.

6. Anhang
6.1 Abkürzungen
AAV Allgemeine Arbeitsvorschrift
Ac AcetylAcO AcetoxyAd 1 -AdamantylAn 9-AnthrylAr Arylar arylischBn BenzylnBu "Butyl
Cp r|5-Cyclopentadienyl
Cy CyclohexylCOD 1,5-Cyclooctadiend TagDBE DibutyletherDC Dünnschichtchromatographie
dcpe 1,2-Bis(dicyclohexylphosphin)ethanDMSO Dimethylsulfoxid
dppe 1,2-Bis(diphenylphosphin)ethan
dppp 1,3-Bis(diphenylphosphin)propanee Enantiomerenüberschuss
El Elektronenstossionisation
equiv. AequivalentFAB fast atom bombardment
FC "flash"-ChromatographieFe Ferrocenyl
Fp r(5-Cyclopentadienyleisendicarbonyl
g gasförmigGC Gaschromatographie
ges. gesättigth Stunde
HPLC Hochdruck-FlüssigkeitschromatographieHV Hochvakuum 0.1-0.001 mbar
LAH Lithiumaluminiumhydrid
Lsg. Lösungmin Minute
MS MassenspektrometrieNMR Kernspinresonanz
proz. prozentigRT RaumtemperaturTHF TetrahydrofuranTLC DickschichtchromatographieTMSCI Trimethylsilylchlorid
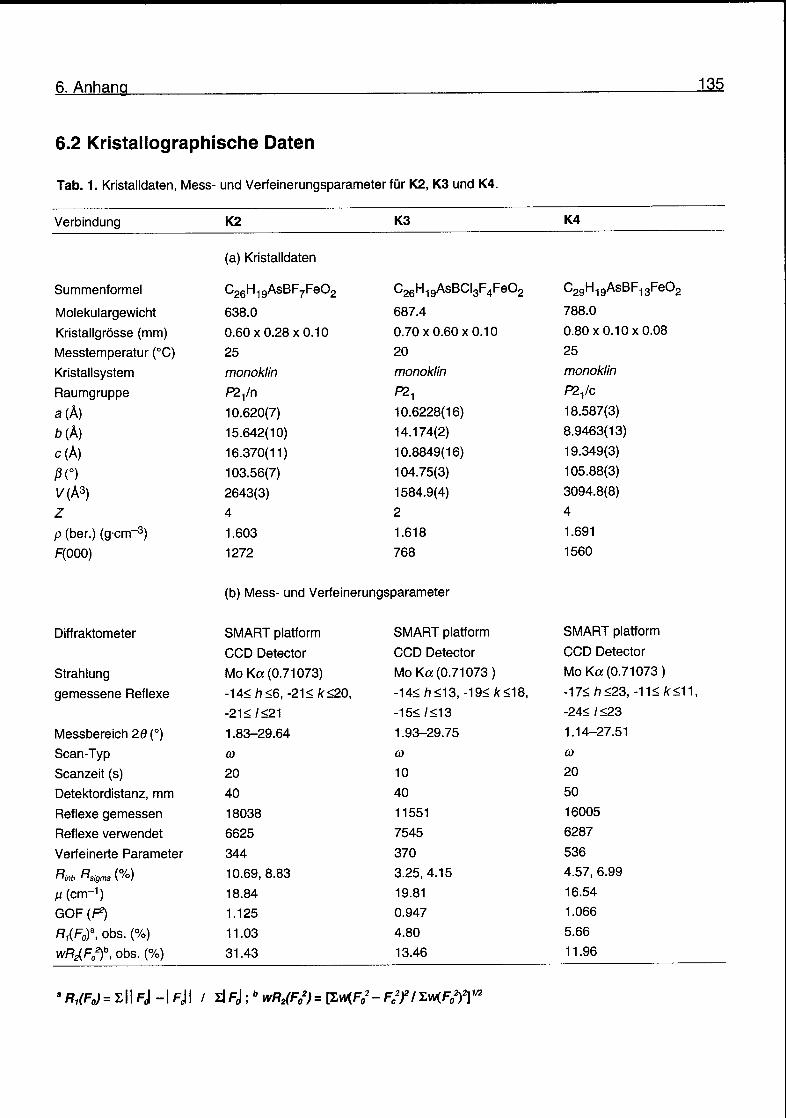
6. Anhang 135
6.2 Kristallographische Daten
Tab. 1. Kristalldaten, Mess- und Verfeinerungsparameter für K2, K3 und K4.
Verbindung K2 K3 K4
(a) Kristalldaten
Summenformel C26H19AsBF7Fe02 C26H19AsBCI3F4Fe02 C29H19AsBF13Fe02
Molekulargewicht 638.0 687.4 788.0
Kristallgrösse (mm) 0.60x0.28x0.10 0.70x0.60x0.10 0.80x0.10x0.08
Messtemperatur (°C) 25 20 25
Kristallsystem monoklin monoklin monoklin
Raumgruppe F2/n *2l P2^/c
a(Â) 10.620(7) 10.6228(16) 18.587(3)
b(Â) 15.642(10) 14.174(2) 8.9463(13)
c(k) 16.370(11) 10.8849(16) 19.349(3)
ßO 103.56(7) 104.75(3) 105.88(3)
l/(Â3) 2643(3) 1584.9(4) 3094.8(8)
Z 4 2 4
p(ber.) (gern-3) 1.603 1.618 1.691
F(000) 1272 768 1560
(b) Mess- und Verfeinerungsparameter
Diffraktometer SMART platform SMART platform SMART platform
CCD Detector CCD Detector CCD Detector
Strahlung Mo Ka (0.71073) Mo Ka (0.71073) Mo Ka (0.71073)
gemessene Reflexe -14</)<6, -21<^<20, -14< /7<13, -19</f<18, -17<rt<23,-11</f<11,
-21</<21 -15</<13 -24</<23
Messbereich 2 0(°) 1.83-29.64 1.93-29.75 1.14-27.51
Scan-Typ (0 CO CO
Scanzeit (s) 20 10 20
Detektordistanz, mm 40 40 50
Reflexe gemessen 18038 11551 16005
Reflexe verwendet 6625 7545 6287
Verfeinerte Parameter 344 370 536
"/nft "sigma ( '°l 10.69,8.83 3.25,4.15 4.57, 6.99
^(enr1) 18.84 19.81 16.54
GOF(F2) 1.125 0.947 1.066
RAF0r, obs. (%) 11.03 4.80 5.66
wRÄFo2)*, obs. (%) 31.43 13.46 11.96
aR,fFJ = 2;||Fj-|Fj| ' j\Fb;bwR2(F02)=\Zw(tV-Fc2//ï:iv(F/)<it 1/2
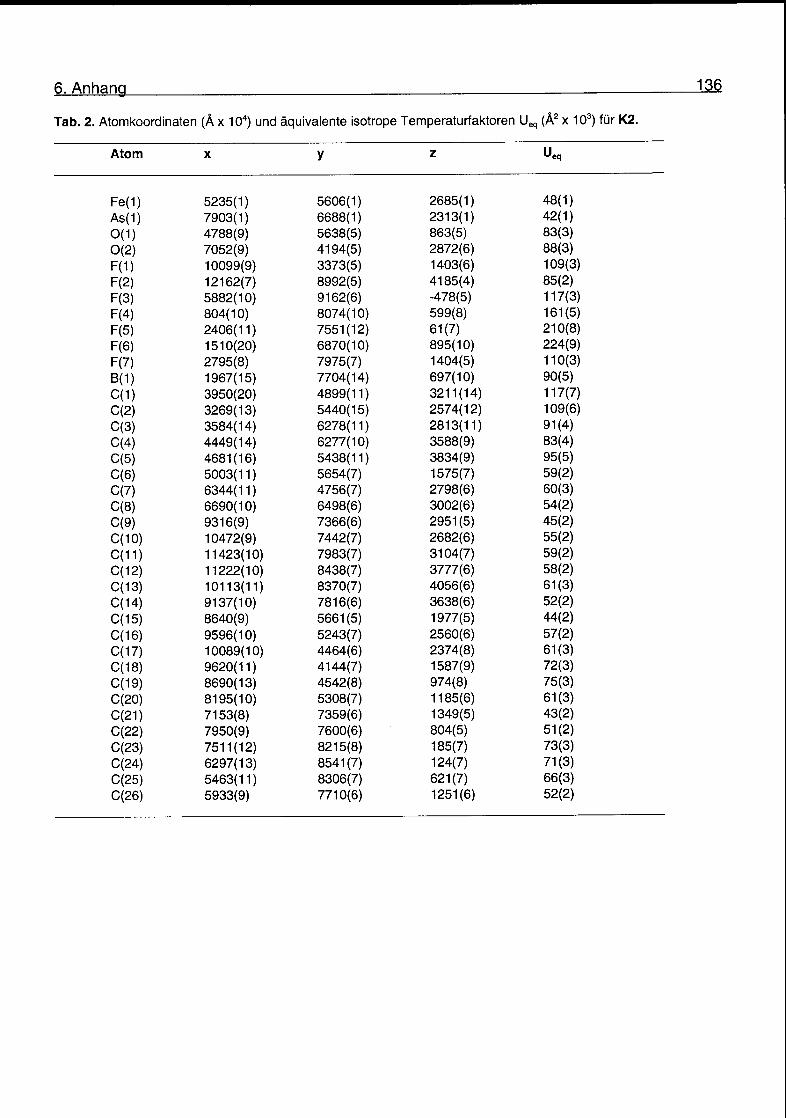
,~*.-^^^--
co
^~.com
oo
en
co^
Is»co*~*^~.^^^~.^^^~.^^~.^^^^^-,*~.^~.^^^^^^~.^.
^Kfc^^oIn^T-co^cM^o'^o^coinOTo^ininOT^^Kf^r^^Kfin^c^^^^cocM
^^coa3i-ocii-T-c\icviT-o)i-T-ffiooffiifi(Diß^ininincDin^-in(DNS(u^-ioNS(Din
t-
-i-
^-.co
co*
mco
,-3-
<M
T-^
,_
omoi-i-T-oio)SCD!DincDS(OCD<OlO
COOO-^LOOOCM-i-CM
T-opnsc»oinco
h-
CD
00
r^-m
co
co
oo
en^
co
h~
oo^
CD
LO—*
.-^-—-.—.co
ïnari£ttt~
oo**m*
i-m
-,
.._-(M
CO
If)m-i^—-—
•,..—
,_.—
-,.-„,.-_-_.-_,.
______
coi-coNOcoKo)C.u)OST-si-oonso)oincoosinns<ONoo'*co'*'*if)'5f'i-m
(oncooO'*i-ld.o)i-ci)'ito)wiflcoinooinNoo)CDT-so!Oo>in(ou)Ni-noa)wcüw
N«cowT-^Tu)(DcO'-(ococviwcocoT-cvicoc\iwcom'ii-nT-c\iiN'-o)T-T-coT-T-(OT-
.
^t
-i-m
t-o
^^^^^^^owo
..
_
C.Ci2.if2.ifÜf2-S£.C-C-C-t^.C*C-ZZ.lz.OC-t.|v~
*°
*°
|s*^
•*"^^£b£bSb£S£tb£
cd
oo
oo
**
co
evicm
•*^o^in'^oTo^a^i^oo'^co'oo'^cvfco'oö'Ö^cö''«-
co^T
^-
c\fafaTo
in
-i-
cdo
omcoo)NO)(ONU)NNOco^sNcoir)ino)o^cocoNi-cD'*(0't\foinOT-'*oi-
coœcoi-nmi-omaxDsoo^cMCM^cDN^n^œ^ncoœw^T-innncocviinnN
lOOin^cocofficosfDNS^iocotoifiin'tcoNNsmooNmifi't'i-^WNNooooooN
I-
-i-
a>
osa.w^o^^ÄC.^C.C.C.^^^^acj'cö'cvrcö'
mcocowo)cDwC(DO«)NOo>'*0)T-n'!to(os
coooomOT-oo-<to-r-CT)CDincooo-^-ooo-5j-c3)T-^-
c\iror^oocMooo-<tmh-a>o>cMin^-coocococoo
mt^^i-is~-i-i-inoocMi-cviT-cococo-^-'«tincocDa)i-
o^-.^-,,~.
COO^^CMCOi-^,
cûcXjT-^ocoooo'o^IîrcooT-rC'cÔ'co
^CM-i-co-^ojocMciOîinini-ajcûco
i-T-Oi-(DIOOCD(0'-'-0)lOW'>tO)
i-i-i-o)(oo)T-o)eo(DNSN(Dinin
cu
.
u-<
"
OO
C2i^£J,£2.S.J2.J£.ü-C-
o-^cNjco-^mcoi^
00
O)o
-i-
cm
co
t-
•>-
CM
CM
CM
C\J^m
co
CM
CM
CM
•*
in
co
r^
i-C-
£J-£2-
S*
!£i££.^
°°œ
_.-^
-
^_
^.-^-^—
ILitiLttlüüüüüOüüüüüüüüüüüüüüüüüüüü
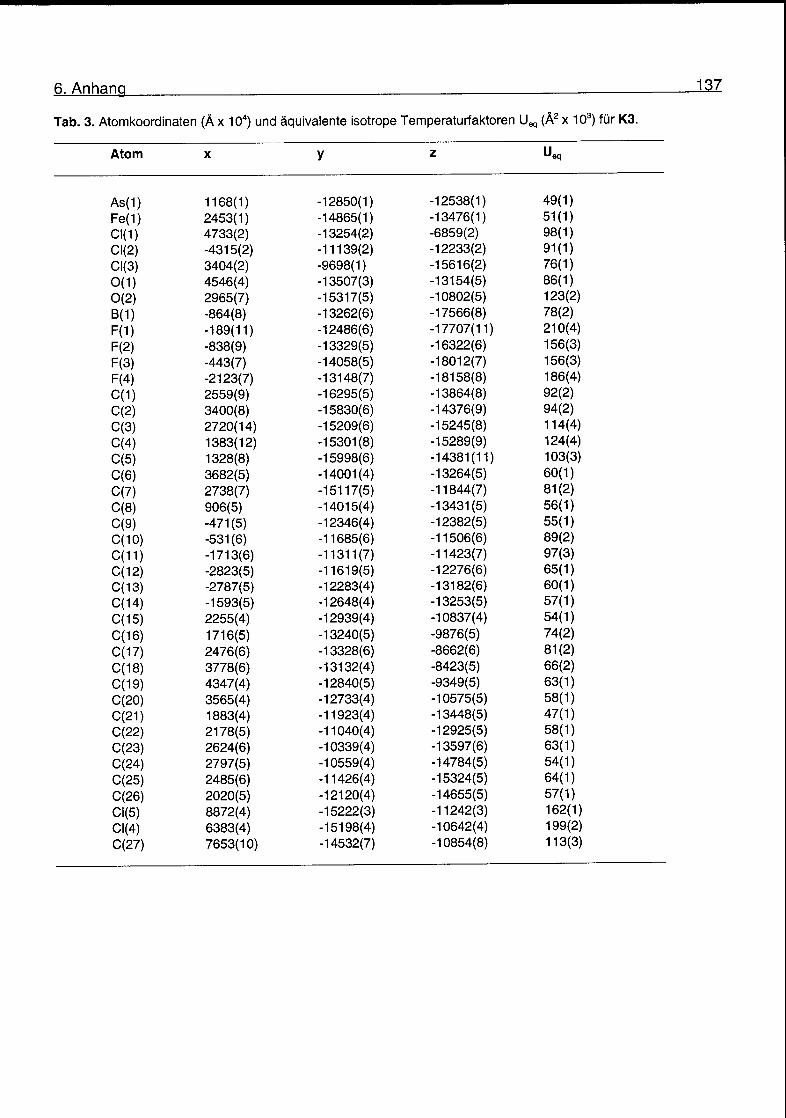
eo
COCD
^_^^^
._.^
CM^
"frCO
COt
.—.^-tf*
CO^^^-.^-..~._.^^^^^-..-^.—.^-.^^^^^^-.—.t-
CM
CO
o)i-eO'-(0(DWflOT-inmoo(M'ti-ojoO'-(oino)NmoN't'ti-<DncoNoocO't<tS(DO)i-
^mfflffiNooi-swT-T-i-cocci-T-i-tocoinmoocBiocommsMfocom^inQintumi-T-^
COt*—.
CO
1_
CDa.
ECD
HCDQ.
Oi_
•*—>
O<n
CD
«—«
c0CO>
N
T-i-^wcMininooi-iDsoocoocoro
00
CD
21-CO
CD^TCM
CO
^-~—
COSOICOt-IOOID
in^-incMcoi-oom
CMCOCOCMincOOI^
CD
ini^mmcot^cocoin^-—.^^^^^-minincoininmco-*oo
h~
CM
CM
00*
CO
in
en
1—*
**
OCM
mCD
1^
sfroo
oo
co
-a-1^
COO
T—
00
CO
CM
CM
CO
CM
00
t^-CD
00
co
CO*
LO
LO*a
»—
t-CVI(OCO(DWCOS
COCOOCMI^OOLOOOCOCMCOO)
«tcoin-^-CM-i-cMoo"
---
CM
CO
CO
h-
CO
CM*
oo
co
-^
co
O)
CO
00
CT)
.._...
t•*
inw
cm
•*
i^.'^-cMcncocMLn'^'^-Lo
m*
O)
in
r^.
OCO
CM
CO
•<*CO
CO
CM
CD
00
m*
>-oo
-i-T-cMCM^coincocomw
mcd
oo
co
in
co
om'*i3)CsNW(ûO)o!)ct)mo(j)T-ct)T-Nio(ûm'-fflnœo)0(0(Monnofl)0)iDOOJflow
incDLOCOCDO^CDCO<Nin^CI)COOOCJ)OT-^-^OOT-T-cO-*CO-*C\lCO-*COCM'tCOinCMCMCMO)CO
mmwT-cnnnw^noi-oji]ONcoo>Oi-o(0(Dco!ûw<oo)c\inr-ooso)oniO'*T-cvi'-in
cM^co^cocoincocMco^cocDLnininin^in^cM^^^c\j<Nc\icocr)cocMCM-r--^oo-i-CMLnin'^-
T-T-^T-CJ5'»--I-T-T-^T-T--l--I-'r-T-T--I--I--I-'I-T-T-'I-T-'r-T-'I-T-'r-T-'I-T-T-T-'I-'I-T-T--r-T-
cCO
c<CD
cr
:C0
oc3X
<
cCD
03
gT3l_OOEoCO
Eo<
PCMSS
CM*K
S*,
rx
£!-3
!_-cot-
en
t^;=r2.SÜCCS.
oococoi2^-coin^p
com
t-
**
t-
CM^T
co
o*
co
,co—
en
co
co
cmf£
—.^*
CM__—
cn
co
i-
-i-
oo
in
i^~
in
LOOjQo__crj^4-f\iLO
..
-tcvi'rT'rTvcMcocM
E-^£.ü2.iü^^lo
coS
ôôncôcïcoS-
~"cMoocn
TfNCOCOlDNO^-^^1^1"
t-
co
cm
en
LO
CM
CM
T-
^_^_^^^^.if2,cô];£!ï£iSS.2zi-
mcDCDcoMncoco^'s'mocjcocö'
lOi-NSTf(O(DNWO)00CüSC0U5
WN^Ncomcoi-toN^oconcD
CM-^CMCO-^COT-CMCMCMCMCMOOCOr--
'-'-C^'m^^'^'-^^^'---^'^'^-^'^-^'^—0,-WCO'*lOlDNCOO)0'-WC01-ir)(û^'5'S
-—--—-O^S-^cmt-^
cvj
co
^-
t-
cm
co^,
in
co
î^.°o
°)^,^.
*-
*-^^CC-CC0^
^^^
SS£L£L:—-5-£1-
<liüüüOOmiLiLiLiLÜÜÜÜÜÜÜÜÜÜÜÜÜÜÜÜOÜÜÜÜÜÜÜÜÜÜÜÜ
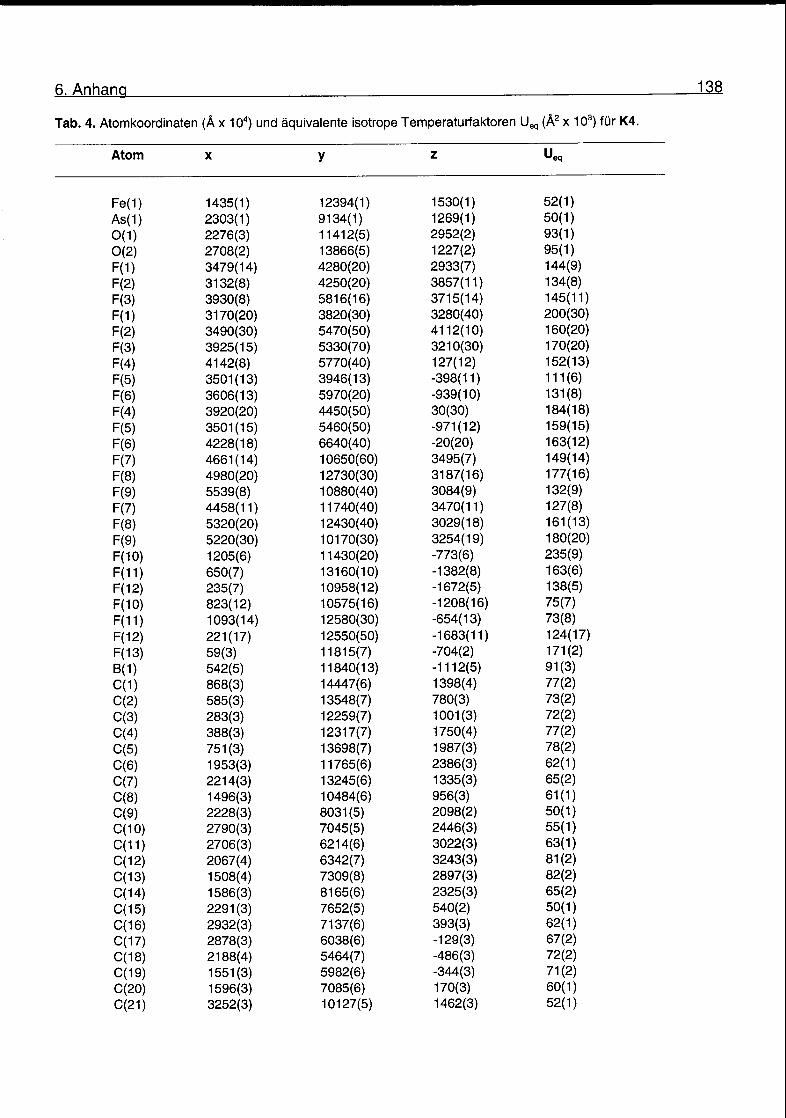
6. Anhang
Tab. 4. Atomkoordinaten (Â x 104) und äquivalente isotrope Temperaturfaktoren U^ (Â2 x 103) für K4.
Atom x y z U,
Fe(1) 1435(1) 12394(1) 1530(1) 52(1)
As(1) 2303(1) 9134(1) 1269(1) 50(1)
0(1) 2276(3) 11412(5) 2952(2) 93(1)
0(2) 2708(2) 13866(5) 1227(2) 95(1)
F(D 3479(14) 4280(20) 2933(7) 144(9)
F(2) 3132(8) 4250(20) 3857(11) 134(8)
F(3) 3930(8) 5816(16) 3715(14) 145(11)
F(1) 3170(20) 3820(30) 3280(40) 200(30)
F(2) 3490(30) 5470(50) 4112(10) 160(20)
F(3) 3925(15) 5330(70) 3210(30) 170(20)
F(4) 4142(8) 5770(40) 127(12) 152(13)
F(5) 3501(13) 3946(13) -398(11) 111(6)
F(6) 3606(13) 5970(20) -939(10) 131(8)
F(4) 3920(20) 4450(50) 30(30) 184(18)
F(5) 3501(15) 5460(50) -971(12) 159(15)
F(6) 4228(18) 6640(40) -20(20) 163(12)
F(7) 4661(14) 10650(60) 3495(7) 149(14)
F(8) 4980(20) 12730(30) 3187(16) 177(16)
F(9) 5539(8) 10880(40) 3084(9) 132(9)
F(7) 4458(11) 11740(40) 3470(11) 127(8)
F(8) 5320(20) 12430(40) 3029(18) 161(13)
F(9) 5220(30) 10170(30) 3254(19) 180(20)
F(10) 1205(6) 11430(20) -773(6) 235(9)
F(11) 650(7) 13160(10) -1382(8) 163(6)
F(12) 235(7) 10958(12) -1672(5) 138(5)
F(10) 823(12) 10575(16) -1208(16) 75(7)
F(11) 1093(14) 12580(30) -654(13) 73(8)
F(12) 221(17) 12550(50) -1683(11) 124(17)
F(13) 59(3) 11815(7) -704(2) 171(2)
B(1) 542(5) 11840(13) -1112(5) 91(3)
C(1) 868(3) 14447(6) 1398(4) 77(2)
C(2) 585(3) 13548(7) 780(3) 73(2)
C(3) 283(3) 12259(7) 1001(3) 72(2)
C(4) 388(3) 12317(7) 1750(4) 77(2)
C(5) 751(3) 13698(7) 1987(3) 78(2)
C(6) 1953(3) 11765(6) 2386(3) 62(1)
C(7) 2214(3) 13245(6) 1335(3) 65(2)
C(8) 1496(3) 10484(6) 956(3) 61(1)
C(9) 2228(3) 8031(5) 2098(2) 50(1)
C(10) 2790(3) 7045(5) 2446(3) 55(1)
C(11) 2706(3) 6214(6) 3022(3) 63(1)
C(12) 2067(4) 6342(7) 3243(3) 81(2)
C(13) 1508(4) 7309(8) 2897(3) 82(2)
C(14) 1586(3) 8165(6) 2325(3) 65(2)
C(15) 2291(3) 7652(5) 540(2) 50(1)
C(16) 2932(3) 7137(6) 393(3) 62(1)
C(17) 2878(3) 6038(6) -129(3) 67(2)
C(18) 2188(4) 5464(7) -486(3) 72(2)
C(19) 1551(3) 5982(6) -344(3) 71(2)
C(20) 1596(3) 7085(6) 170(3) 60(1)
C(21) 3252(3) 10127(5) 1462(3) 52(1)
£>SIS2SISI£
SISI
iNOKjlxSivjIxohoiNorx)
cooo~sicncn-£.GJiv>
> IX
3 Q
^.COCjOCO^.^.^.00
00UlCO4i-'UlCOC!)
Ul->100)CO-slCJlCO
CDCOOO-vICO-'-COCO
'SInoioS'Soo
co
co
-l^.-iO-'-^-^O
UC0O)O)^O)-'C0
C000-£.CD00CD-|i.CO
-'-'oœs^cBoi
co
,',co
oo--'
ro
ro
oÇ3o*.ocoo)Ui
-M^Çco-ncoocnoo
en.
Ul
*.
-&
co
co
COCOCO-JCOOOOU1
Tofororolororo^
CD

6. Anhang 140
Tab. 5. Kristalldaten, Mess- und Verfeinerungsparameter für [CpFe(CO)2As(4-CF3-Ph)3]BF4.
(a) Kristalldaten
Summenformel C28H17AsBF13Fe02
Molekulargewicht 774.0
Kristallgrösse (mm) 0.58x0.14x0.08
Messtemperatur (°C) -40
Kristallsystem monoklin
Raumgruppe P2:/c
a(A) 17.247(3)
b(k) 10.8544(16)
c(Â) 18.396(3)
ßO 114.05(15)
V(&) 3144.9(8)
Z 4
p(ber.) (gern-3) 1.694
F(000) 1584
(b) Mess- und Verfeinerungsparameter
Diffraktometer
Strahlung
gemessene Reflexe
Messbereich 20 (°)
Scan-Typ
Scanzeit (s)
Detektordistanz, mm
Reflexe gemessen
Reflexe verwendet
Verfeinerte Parameter
Rint> Rsigma \ '°)
^(cm~1)
G0F(F5)
RAF0)a, obs. (%)
wRAFo2?, obs. (%)
SMART platform
CCD Detector
Mo Ka (0.71073)
-20< A?<17, -12< /c<12, -20< /<21
1.29-24.74
CO
20
40
17262
5374
427
8.58, 8.84
16.32
1.042
7.79
22.95
'
RJFo) = £ 11 Fj -1 Fj I / El Fj ;* wR2(F02) = \Lw(F02 - Fc2f I Zm^F,,2)2] *
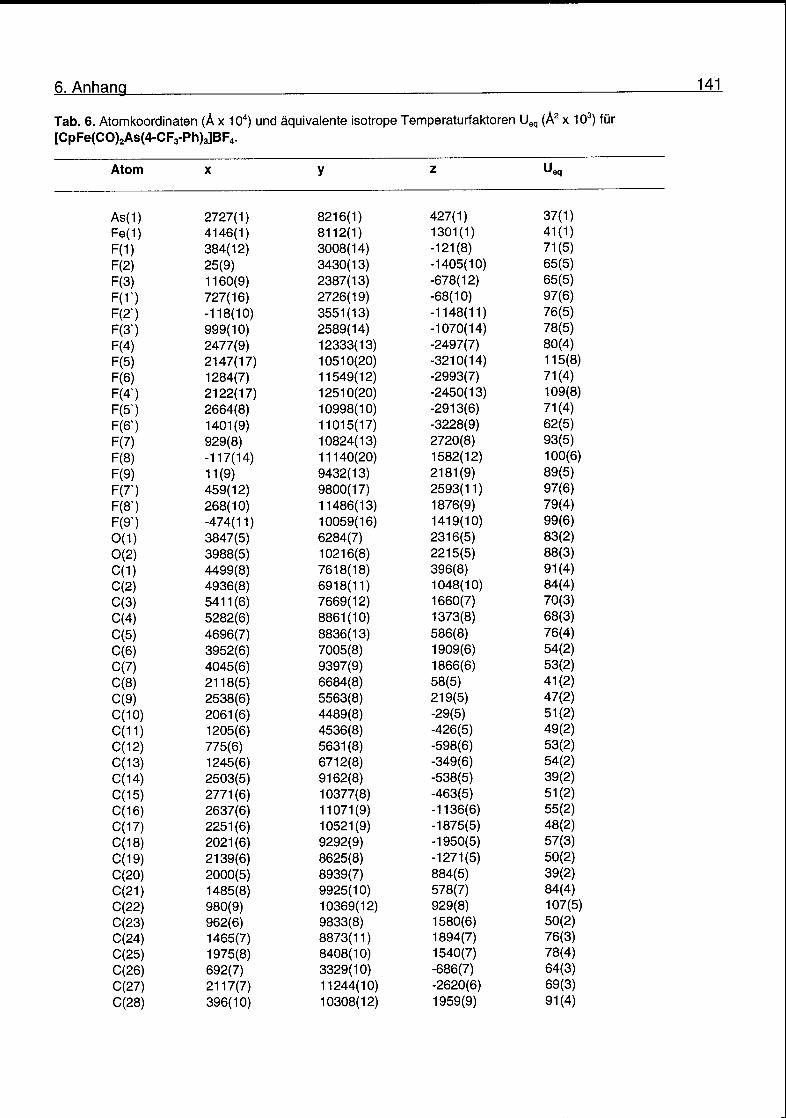
6. Anhang Hl
Tab. 6. Atomkoordinaten (À x 104) und äquivalente isotrope Temperaturfaktoren Ueq (Â2 x 103) für
[CpFe(CO)2As(4-CF3-Ph)3]BF4.
Atom x y z Ueq
As(1) 2727(1) 8216(1) 427(1) 37(1)
Fe(1) 4146(1) 8112(1) 1301(1) 41(1)
F(1) 384(12) 3008(14) -121(8) 71(5)
F(2) 25(9) 3430(13) -1405(10) 65(5)
F(3) 1160(9) 2387(13) -678(12) 65(5)
F(V) 727(16) 2726(19) -68(10) 97(6)
F(2-) -118(10) 3551(13) -1148(11) 76(5)
F(3-) 999(10) 2589(14) -1070(14) 78(5)
F(4) 2477(9) 12333(13) -2497(7) 80(4)
F(5) 2147(17) 10510(20) -3210(14) 115(8)
F(6) 1284(7) 11549(12) -2993(7) 71(4)
F(4~) 2122(17) 12510(20) -2450(13) 109(8)
F(5') 2664(8) 10998(10) -2913(6) 71(4)
F(6") 1401(9) 11015(17) -3228(9) 62(5)
F(7) 929(8) 10824(13) 2720(8) 93(5)
F(8) -117(14) 11140(20) 1582(12) 100(6)
F(9) 11(9) 9432(13) 2181(9) 89(5)
F(T) 459(12) 9800(17) 2593(11) 97(6)
F(8') 268(10) 11486(13) 1876(9) 79(4)
F(9-) -474(11) 10059(16) 1419(10) 99(6)
0(1) 3847(5) 6284(7) 2316(5) 83(2)
0(2) 3988(5) 10216(8) 2215(5) 88(3)
C(1) 4499(8) 7618(18) 396(8) 91(4)
C(2) 4936(8) 6918(11) 1048(10) 84(4)
C(3) 5411(6) 7669(12) 1660(7) 70(3)
C(4) 5282(6) 8861(10) 1373(8) 68(3)
C(5) 4696(7) 8836(13) 586(8) 76(4)
C(6) 3952(6) 7005(8) 1909(6) 54(2)
C(7) 4045(6) 9397(9) 1866(6) 53(2)
C(8) 2118(5) 6684(8) 58(5) 41(2)
C(9) 2538(6) 5563(8) 219(5) 47(2)
C(10) 2061(6) 4489(8) -29(5) 51(2)
C(11) 1205(6) 4536(8) -426(5) 49(2)
C(12) 775(6) 5631(8) -598(6) 53(2)
C(13) 1245(6) 6712(8) -349(6) 54(2)
C(14) 2503(5) 9162(8) -538(5) 39(2)
C(15) 2771(6) 10377(8) -463(5) 51(2)
C(16) 2637(6) 11071(9) -1136(6) 55(2)
C(17) 2251(6) 10521(9) -1875(5) 48(2)
C(18) 2021(6) 9292(9) -1950(5) 57(3)
C(19) 2139(6) 8625(8) -1271(5) 50(2)
C(20) 2000(5) 8939(7) 884(5) 39(2)
C(21) 1485(8) 9925(10) 578(7) 84(4)
C(22) 980(9) 10369(12) 929(8) 107(5)
C(23) 962(6) 9833(8) 1580(6) 50(2)
C(24) 1465(7) 8873(11) 1894(7) 76(3)
C(25) 1975(8) 8408(10) 1540(7) 78(4)
C(26) 692(7) 3329(10) -686(7) 64(3)
C(27) 2117(7) 11244(10) -2620(6) 69(3)
C(28) 396(10) 10308(12) 1959(9) 91(4)

6. Anhang 142
B(1) 4349(9) 12950(13) 950(8) 66(4)
F(10) 4891(13) 13725(16) 1210(14) 327(12)
F(11) 3871(11) 12890(20) 1298(10) 311(13)
F(12) 4817(10) 11986(14) 1050(9) 233(8)
F(13) 3924(7) 13165(12) 201(6) 183(5)
0(3) 3320(6) 6339(12) -1419(7) 129(4)
C(29) 3184(11) 5073(18) -1374(10) 125(6)

6. Anhang 143
Lebenslauf
Am 7. Dezember 1972 wurde ich als Sohn von Hugo und Ruth Egli-Stalder in Samen (OW)
geboren. Nach der Primarschule in Alpnach (1979-1985) trat ich 1985 in die Kantonsschule
Obwalden in Samen ein und schloss dort 1992 mit der Matura Typus B ab. Nach der
Rekrutenschule, der Unteroffiziersschule und dem Abverdienen begann ich im Herbst 1993 mit
dem Chemiestudium an der ETH Zürich, welches ich im Herbst 1997 abschloss.
Im April 1998 begann ich meine Doktorarbeit unter der Leitung von Prof. Dr. A. Togni.
Während meiner Promotionszeit war ich Assistent im Praktikum in Allgemeiner Chemie und im
Praktikum in Agrikulturchemie. Im Winter 1999/2000 betreute ich einen Studenten bei der
Diplomarbeit.
Am 1. Juni 2001 heiratete ich Maureen Rae O'Reilly aus Vernon, Connecticut.
Zürich, Oktober 2001
Patrick Egli


















