Immunvermittelte Erkrankungen der grauen ZNS … · KAPITEL Entzündliche und erregerbedingte...
15
KAPITEL Entzündliche und erregerbedingte Krankheiten Immunvermittelte Erkrankungen der grauen ZNS-Substanz sowie Neurosarkoidose Entwicklungsstufe: S1 Stand: September 2012 PDF Dow nload COI-Erklärung Federführend Prof. Dr. Christian Bien, Bielefeld [email protected] Was gibt es Neues? Autoantikörperdiagnostik ist zum diagnostisch entscheidenden Schritt bei immunvermittelten Erkrankungen der grauen ZNS-Substanz geworden. Die Prognose von Erkrankungen mit Antikörpern gegen Oberflächen-Antigene ist im Allgemeinen günstig. Die limbische Enzephalitis tritt auch bei Kindern und Jugendlichen auf. Die neu beschriebenen faziobrachialen dystonen Anfälle sind mit den ebenfalls neu entdeckten Antikörpern gegen das Leucine-rich Glioma Inactivated Protein 1 (LGI1) assoziiert. Sie münden unbehandelt in eine limbische Enzephalitis ein und eröffnen möglicherweise ein Zeitfenster, innerhalb dessen eine Immuntherapie diesen Übergang zu verhindern vermag. Die Diagnose einer Hashimoto-Enzephalopathie/Steroid-responsiven Enzephalopathie assoziiert mit Autoimmunthyreoiditis (SREAT) sollte nur gestellt werden, wenn spezifischere Autoantikörper, insbesondere solche gegen neurale Oberflächenproteine, ausgeschlossen sind. Das bildgebende (MRT) Spektrum der Neurosarkoidose-Manifestation weitet sich zunehmend aus. Die wichtigsten Empfehlungen auf einen Blick Bei mutm aßlich immunvermitte lten Erkrankungen der grauen ZNS-Substanz ist eine Antikörperdiagnostik erforderlich. Bei Nachweis von Antikörpern gegen Oberflächenproteine ist eine Immuntherapie erfolgversprechend. Rasmussen-Enzephalitis: Bei belastenden pharmakoresistenten epileptischen Anfällen sollte frühzeitig eine prächirurgische Diagnostik bezüglich einer Hemisphärektomie erfolgen. Kommt diese nicht in Betracht oder steht die Epilepsie nicht im Vordergrund der Beschwerden, bieten sich immunologische Langzeittherapien zur Verhinderung eines Gewebs- und Funktionsverlusts an. SREAT: Die Diagnose kann bei Ausschluss der spezifischen Antikörper nach einem Kriterienkatalog verdachtsweise gestellt werden und sollte Anlass zu einer Steroidtherapie sein, deren Erfolg die Diagnose bestätigt. Die Diagnose der Neurosarkoidose sollte, vor allem bei isoliertem ZNS-Befall, wegen der notwendigen Langzeittherapie immer auf einer Histologie basieren. Definition und Klassifikation Begriffs definition Leitlinien für Diagnostik und Therapie in der Neurologie 1
Transcript of Immunvermittelte Erkrankungen der grauen ZNS … · KAPITEL Entzündliche und erregerbedingte...

KAPITELEntzündliche und er regerbedingte Krankheiten
Immunvermittelte Erkrankungen dergrauen ZNS-Substanz sowie
NeurosarkoidoseEntw ick lungss tufe: S1Stand: September 2012
PDF Dow nloadCOI-Erklärung
Feder führendProf. Dr. Christian Bien, Bielefeld
Was gibt es Neues?
Autoantikörperdiagnostik ist zum diagnostisch entscheidenden Schritt bei immunvermittelten Erkrankungen dergrauen ZNS-Substanz geworden.Die Prognose von Erkrankungen mit Antikörpern gegen Oberflächen-Antigene ist im Allgemeinen günstig.Die limbische Enzephalitis tritt auch bei Kindern und Jugendlichen auf.Die neu beschriebenen faziobrachialen dystonen Anfälle sind mit den ebenfalls neu entdeckten Antikörperngegen das Leucine-rich Glioma Inactivated Protein 1 (LGI1) assoziiert. Sie münden unbehandelt in eine limbischeEnzephalitis ein und eröffnen möglicherweise ein Zeitfenster, innerhalb dessen eine Immuntherapie diesenÜbergang zu verhindern vermag.Die Diagnose einer Hashimoto-Enzephalopathie/Steroid-responsiven Enzephalopathie assoziiert mitAutoimmunthyreoiditis (SREAT) sollte nur gestellt werden, wenn spezifischere Autoantikörper, insbesonderesolche gegen neurale Oberflächenproteine, ausgeschlossen sind.Das bildgebende (MRT) Spektrum der Neurosarkoidose-Manifestation weitet sich zunehmend aus.
Die wichtigsten Empfehlungen auf einen Blick
Bei mutmaßlich immunvermitte lten Erkrankungen der grauen ZNS-Substanz ist eine Antikörperdiagnostikerforderlich. Bei Nachweis von Antikörpern gegen Oberflächenproteine ist eine Immuntherapieerfolgversprechend.Rasmussen-Enzephalitis: Bei belastenden pharmakoresistenten epileptischen Anfällen sollte frühzeitig eineprächirurgische Diagnostik bezüglich einer Hemisphärektomie erfolgen. Kommt diese nicht in Betracht oder stehtdie Epilepsie nicht im Vordergrund der Beschwerden, bieten sich immunologische Langzeittherapien zurVerhinderung eines Gewebs- und Funktionsverlusts an.SREAT: Die Diagnose kann bei Ausschluss der spezifischen Antikörper nach einem Kriterienkatalogverdachtsweise gestellt werden und sollte Anlass zu einer Steroidtherapie sein, deren Erfolg die Diagnosebestätigt.Die Diagnose der Neurosarkoidose sollte, vor allem bei isoliertem ZNS-Befall, wegen der notwendigenLangzeittherapie immer auf einer Histologie basieren.
Definition und KlassifikationBegriffs definition
Leitlinien für Diagnostik und Therapie in der Neurologie
1

Die hier abgehandelten Erkrankungen werden offenbar durch eine pathologische Interaktion von Elementen desImmunsystems mit dem ZNS verursacht. Es besteht noch wenig belastbare Evidenz hinsichtlich zu bevorzugenderTherapien.
Klassifikation
Die Erkrankungen werden in folgende Gruppen (die noch keinen Anspruch auf die Formierung von Klassen erhebenkönnen) unterteilt:
Autoantikörper-definierte ErkrankungenRasmussen-EnzephalitisNeurosarkoidose
Diagnostik
Autoantikörper-definierte Erkrankungen
Aus der Kombination klinischer und paraklinischer Befunde, den Resultaten einer Tumorsuche und dem Nachweisakzeptierter Autoantikörper im Serum oder Liquor ergeben sich Diagnosen, bestehend aus dem Syndrom mit Angabedes Tumor- und des Antikörperstatus. Vermutlich haben Antikörper gegen Oberflächen-Antigene einen direktenpathogenetisch-funktionellen Effekt auf das ZNS (Lai et al. 2009, Hughes et al. 2010, Lalic et al. 2011, Bien et al. 2012).
Klinisch-neuroradiologische Syndrome
Antikörper-definierte Erkrankungen der grauen ZNS-Substanz weisen nach gegenwärtigem Kenntnisstand typischeklinisch-radiologische Bilder auf. Dabei bestehen zwar charakteristische, aber keineswegs exklusiveÜberschneidungen mit bestimmten Autoantikörpern. Die meisten publizierten Informationen stammen auswissenschaftlich aktiven Antikörperlabors, deren Material auf einem nicht dokumentierbaren Selektionsprozess durcheinsendende Kliniker beruht. Man darf daher annehmen, dass in diesen Serien „klassische“ Syndromeüberrepräsentiert sind.
Außer typischen klinischen Bildern gibt es noch weitere Hinweise auf die immunvermittelte Genese von Erkrankungender grauen ZNS-Substanz (Zuliani et al. 2012):
(sub-)akute (< 12 Wochen) klinische Evolutionfokale hyperintense MRT-FLAIR/T2-Läsionen oder fokaler FDG-PET-Hypermetabolismus oder fokale SPECT-Hyperperfusion, die als am ehesten entzündlich eingeschätzt werdenim Liquor erhöhte Zellzahl oder oligoklonale Bandenhistopathologische Diagnose einer Enzephalitis
Autoantikörper
Die heute akzeptierten Antikörper werden unterteilt in solche gegen Antigene im Innern hirneigener Zellen(onkoneurale Antikörper und Antikörper gegen Glutamatdekarboxylase [GAD]) einerseits sowie die erst kürzlichentdeckten Antikörper gegen Antigene auf neuralen Oberflächen (Rezeptoren, Kanäle oder assoziierte Proteine)andererseits (Graus et al. 2010b). Eine Übersicht gibt ▶ Abb. 32.1.
Abb. 32.1 Systematische Übersicht über heute akzeptierte Autoantikörper.
Leitlinien für Diagnostik und Therapie in der Neurologie
2
In den meisten Fällen ist eine Serum-Antikörperuntersuchung ausreichend. In einzelnen Fällen – etwa bei Anti-N-Methyl-D-Aspartat-Rezeptor-(NMDAR-)Enzephalitis – mit peripher niedriger Antikörpermenge, aber intrathekalerSynthese können Antikörper auch nur im Liquor nachweisbar sein (Prüss et al. 2010).
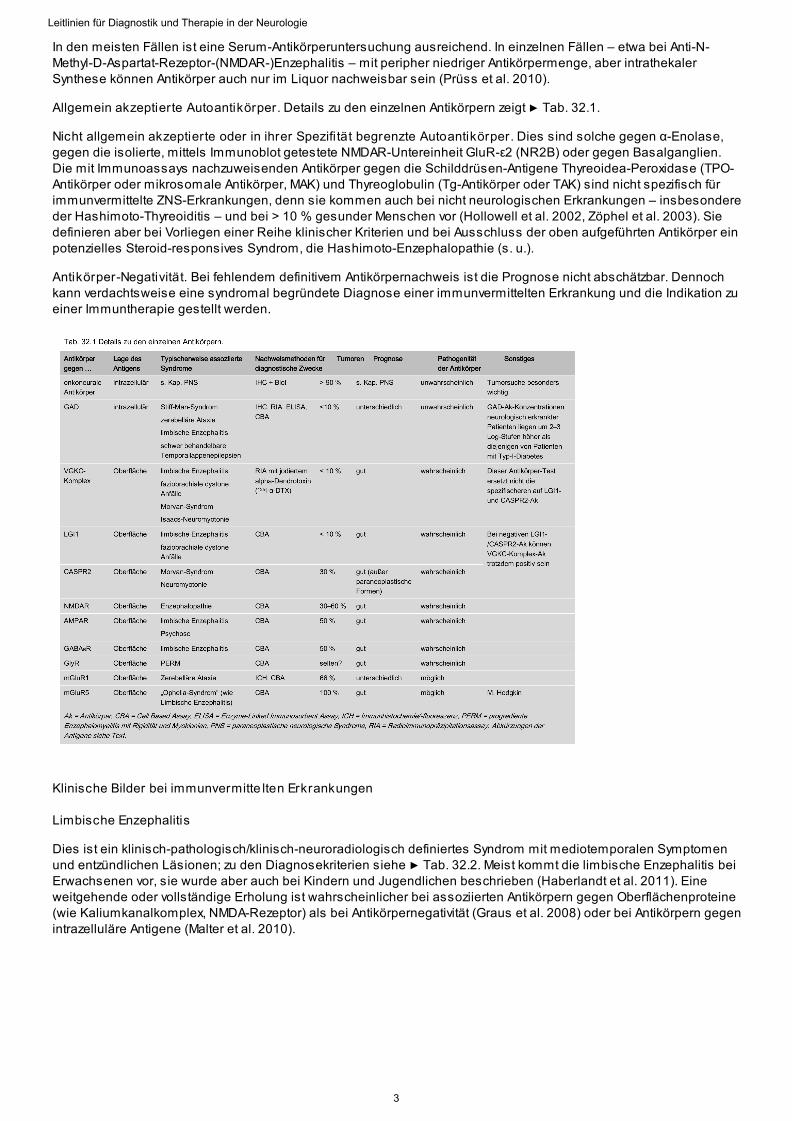
Allgemein akzeptierte Autoantikörper. Details zu den einzelnen Antikörpern zeigt ▶ Tab. 32.1.
Nicht allgemein akzeptierte oder in ihrer Spezifi tät begrenzte Autoantikörper. Dies sind solche gegen α-Enolase,gegen die isolierte, mittels Immunoblot getestete NMDAR-Untereinheit GluR-ε2 (NR2B) oder gegen Basalganglien.Die mit Immunoassays nachzuweisenden Antikörper gegen die Schilddrüsen-Antigene Thyreoidea-Peroxidase (TPO-Antikörper oder mikrosomale Antikörper, MAK) und Thyreoglobulin (Tg-Antikörper oder TAK) sind nicht spezifisch fürimmunvermittelte ZNS-Erkrankungen, denn sie kommen auch bei nicht neurologischen Erkrankungen – insbesondereder Hashimoto-Thyreoiditis – und bei > 10 % gesunder Menschen vor (Hollowell et al. 2002, Zöphel et al. 2003). Siedefinieren aber bei Vorliegen einer Reihe klinischer Kriterien und bei Ausschluss der oben aufgeführten Antikörper einpotenzielles Steroid-responsives Syndrom, die Hashimoto-Enzephalopathie (s. u.).
Antikörper-Negativität. Bei fehlendem definitivem Antikörpernachweis ist die Prognose nicht abschätzbar. Dennochkann verdachtsweise eine syndromal begründete Diagnose einer immunvermittelten Erkrankung und die Indikation zueiner Immuntherapie gestellt werden.
Klinische Bilder bei immunvermitte lten Erkrankungen
Limbische Enzephalitis
Dies ist ein klinisch-pathologisch/klinisch-neuroradiologisch definiertes Syndrom mit mediotemporalen Symptomenund entzündlichen Läsionen; zu den Diagnosekriterien siehe ▶ Tab. 32.2. Meist kommt die limbische Enzephalitis beiErwachsenen vor, sie wurde aber auch bei Kindern und Jugendlichen beschrieben (Haberlandt et al. 2011). Eineweitgehende oder vollständige Erholung ist wahrscheinlicher bei assoziierten Antikörpern gegen Oberflächenproteine(wie Kaliumkanalkomplex, NMDA-Rezeptor) als bei Antikörpernegativität (Graus et al. 2008) oder bei Antikörpern gegenintrazelluläre Antigene (Malter et al. 2010).
Leitlinien für Diagnostik und Therapie in der Neurologie
3

Faziobrachiale dystone Anfälle
Dabei handelt es sich um < 3 Sekunden dauernde Ereignisse mit Grimassieren und Dystonie des ipsilateralenArmes, zum Teil auch des ipsilateralen Beines. Der einzelne Anfall ist einseitig, aber es können seitenwechselndeAnfälle auftreten. Faziobrachiale dystone Anfälle treten 6–360 Mal pro Tag auf. Iktuale EEGs können rhythmischekontralateral-frontotemporale Spikes zeigen. Die Patienten haben hohe VGKC-Komplex-Antikörper-Titer, dasspezifische Antigen war in 90 % der Fälle LGI1. Unbehandelt gehen faziobrachiale dystone Anfälle in fast allen Fällenin eine limbische Enzephalitis mit kognitiven Beeinträchtigungen über.
Immunvermitte lte Enzephalopathie
Formelle Kriterien für eine Enzephalopathie sind in ▶ Tab. 32.3 genannt. Zwei spezielle Konstellationen sindbesonders erwähnenswert:
Anti-NMDAR-Enzephalitis. Betroffen sind überwiegend Mädchen oder junge Frauen. Initial stehen psychiatrischeStörungen bis hin zu Psychosen, kognitive Beeinträchtigungen oder Anfälle im Vordergrund. Binnen Tagen geht dieKrankheit über in Symptome wie Bewusstseinsstörung, abnorme Bewegungen, Mutismus, Hypoventilation undautonome Dysregulation mit Intensivpflichtigkeit. Bis zu 60 % der Patientinnen haben ein ovarielles Teratom; Kinder,Männer und bei ältere Patienten haben selten Tumoren (Irani et al. 2010, Dalmau et al. 2011) Das MRT ist häufig nichtinformativ. Eine in den ersten Tagen bestehende Pleozytose mit nachfolgender Normalisierung der Zellzahl undEntwicklung oligoklonaler Banden wurden beschrieben (Irani et al. 2010). Etwa 75 % der Patienten erholen sichweitgehend oder vollständig, oft nach monatelangen Aufenthalten auf Intensivstationen (Dalmau et al. 2011).
SREAT/Hashimoto-Enzephalopathie. Die SREAT manifestiert sich klinisch entweder mit Schlaganfall-ähnlicheinsetzenden fokalen, zum Teil multiplen Ausfällen (ca. 10–30 % der Fälle) oder aber als schleichend progredienteEnzephalopathie (bis hin zum Koma) mit zunehmenden epileptischen Anfällen oder einem Status epilepticus (ca. 70–90 % der Fälle) (Kothbauer-Margreiter et al. 1996, Chong et al. 2003, Chaudhuri u. Behan 2003). Die Diagnose„SREAT“ erfordert den Beleg einer Besserung auf Steroide (▶ Tab. 32.4) (Schäuble et al. 2003, Castillo et al. 2006).
Leitlinien für Diagnostik und Therapie in der Neurologie
4

Progrediente Enzephalomyelitis mit Rigidität und Myoklonien (PERM)
Kernsymptome sind Muskelsteifigkeit und Spasmen. Das volle klinische Spektrum wird soeben exploriert (Mas et al.2010). Es wird auf das Kapitel „Stiff-Man-Syndrom“ hingewiesen.
Andere
Ebenfalls autoimmunvermittelt können auftreten: das Opsoklonus-Myoklonus-Syndrom, die progredienteKleinhirnatrophie mit Ataxie, die Hirnstammenzephalitis, die chronische (Temporallappen-)Epilepsie, das Morvan-Syndrom (Schlafstörung, Psychose und periphere Nervenhyperexzitabilität mit Neuromyotonie und Schmerzen) sowie„Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids“ (CLIPPERS)(Pittock et al. 2010).
Diagnos tis ches Vorgehen
Die in ▶ Abb. 32.2 gegebenen Empfehlungen sollen eine Balance zwischen Über- und Unterdiagnostik halten. Ihnenzufolge kann auch bei negativem MRT oder normalen Liquor-Standardparametern eine solche Erkrankungdiagnostiziert werden. Andererseits reicht ein isolierter Antikörperbefund nicht zu einer solchen Diagnose aus.
Abb. 32.2 Vorgeschlagener Algorithmus für die diagnostische Aufarbeitung mutmaßlich Antikörper-assoziierterErkrankungen.
Leitlinien für Diagnostik und Therapie in der Neurologie
5

Diffe renzialdiagnostische Aufarbeitung:
Liquoruntersuchung zum Ausschluss einer infektiösen EnzephalitisHirn-MRT (später auch als Verlaufsparameter)
Bei Vorliegen der klinischen Voraussetzungen gemäß ▶ Abb. 32.2:
Antikörperdiagnostik im Serum (idealerweise auch im Liquor) hinsichtlich der in ▶ Abb. 32.2 genannten AntikörperTumorsuche (s. Kap. „Paraneoplastische Syndrome")Untersuchungen zur Dokumentation der Defizite des Patienten vor Therapiebeginn:
neurologische Untersuchung und psychopathologische ExplorationEigen-/Fremdanamnese zur Ermittlung der Anfallsfrequenz, idealerweise ergänzt durch Langzeit-EEGneuropsychologische Untersuchung (mindestens Bedside-Test)
Sollte sich trotz klinischer Annahme einer immunvermittelten Erkrankung der grauen ZNS-Substanz weder einAntikörper noch ein Tumor finden lassen, so bleiben als weitere diagnostische Möglichkeiten:
a. Hirnbiopsie, um eine entzündliche Genese zu belegenb. erneute Antikörper-Testung (vor allem bei innerhalb der ersten Krankheitstage gewonnenem ersten
Untersuchungsmaterial) und Tumorsuche im Intervallc. Tumorsuche auch bei nicht klassischem Syndromd. Serum- und Liquoraufbewahrung bei –20 ° C, um in der Zukunft auf dann neu entdeckte Antikörper testen zu
Leitlinien für Diagnostik und Therapie in der Neurologie
6
können (Prüss et al. 2010).
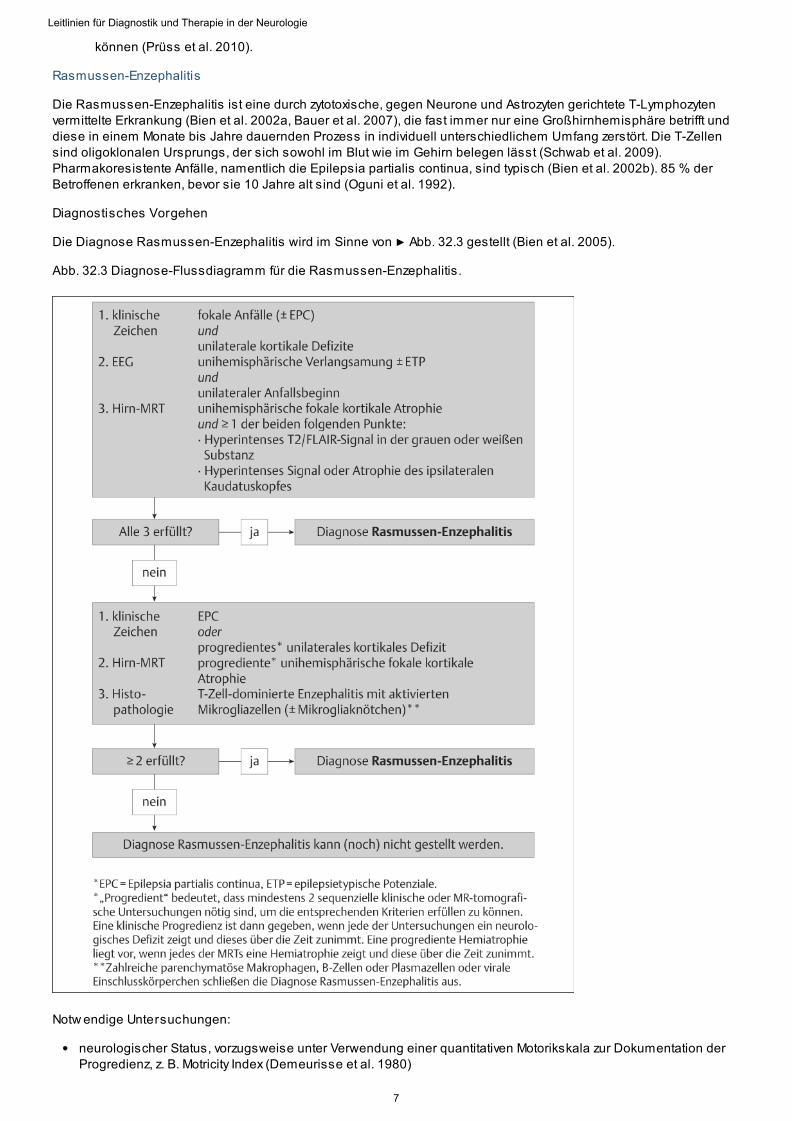
Rasmussen-Enzephalitis
Die Rasmussen-Enzephalitis ist eine durch zytotoxische, gegen Neurone und Astrozyten gerichtete T-Lymphozytenvermittelte Erkrankung (Bien et al. 2002a, Bauer et al. 2007), die fast immer nur eine Großhirnhemisphäre betrifft unddiese in einem Monate bis Jahre dauernden Prozess in individuell unterschiedlichem Umfang zerstört. Die T-Zellensind oligoklonalen Ursprungs, der sich sowohl im Blut wie im Gehirn belegen lässt (Schwab et al. 2009).Pharmakoresistente Anfälle, namentlich die Epilepsia partialis continua, sind typisch (Bien et al. 2002b). 85 % derBetroffenen erkranken, bevor sie 10 Jahre alt sind (Oguni et al. 1992).
Diagnostisches Vorgehen
Die Diagnose Rasmussen-Enzephalitis wird im Sinne von ▶ Abb. 32.3 gestellt (Bien et al. 2005).
Abb. 32.3 Diagnose-Flussdiagramm für die Rasmussen-Enzephalitis.
Notw endige Untersuchungen:
neurologischer Status, vorzugsweise unter Verwendung einer quantitativen Motorikskala zur Dokumentation derProgredienz, z. B. Motricity Index (Demeurisse et al. 1980)
Leitlinien für Diagnostik und Therapie in der Neurologie
7

neuropsychologische Verlaufsdokumentation, namentlich der sprachlichen Funktionen bei Befall der dominantenHemisphäreHirn-MRT zu diagnostischen Zwecken, später zur Dokumentation der Progredienz der Hemiatrophia cerebri imAbstand von 2–3 Monaten zu Erkrankungsbeginn, danach in 6–12-monatigen Abständen (Bien et al. 2004)EEG-Diagnostik, ggf. Langzeit-Video-EEG-Monitoring
Im Einzelfall erforderliche Untersuchungen:
Blut-/Liquoruntersuchungen zum Ausschluss von Differenzialdiagnosen (ausführliche Liste bei Bien et al. 2005)sowie bei Immunsuppression zum Therapiemonitoring (in Abhängigkeit vom verwendeten Regime)offene Hirnbiopsie bei frühen Verdachtsfällen, die die nicht invasiven Diagnosekriterien (noch) nicht erfüllen (▶Abb. 32.3)Wada-Test vor Indikationsstellung einer Hemisphärektomie
Neurosarkoidose
Die Sarkoidose ist definiert als multisystemische Granulomatose unbekannter Ätiologie, die vor allem jungeErwachsene befällt. Die charakteristische Histologie ist durch nicht verkäsende epitheloidzellige Granulomegekennzeichnet. Die Prävalenz klinisch manifester Fälle beträgt 5–20 pro 100.000, diejenige autoptisch verifizierterFälle 35 pro 100.000. Hauptmanifestationsorte sind die Lymphknoten der Lungenhili, das Lungenparenchym, die Hautund die Augen. Ein Befall des Nervensystems (Neurosarkoidose) findet sich klinisch manifest in 5 % (2–16 %) undautoptisch in bis zu 27 % der Fälle. Dies ist eher im frühen Verlauf einer systemischen Sarkoidose und eher beijüngeren Patienten (30–40-jährig) der Fall. Der Befall des Nervensystems stellt bei ca. 50 % die erste Manifestationder Sarkoidose überhaupt dar, bei Hirnnervenbefall sogar in 85 %. Bei über 90 % der Patienten mit Neurosarkoidosefinden sich auch andere Systemmanifestationen, Hiluslymphome bei über 80 % und Augenbefall (Uveitis,Keratokonjunktivitis) bei ca. 50 %.
Klinisches Bild
Am häufigsten ist ein Hirnnervenausfall (50–70 %), typischerweise multiple Hirnnerven betreffend (über 50 %) und oftrezidivierend. Zweithäufigste Manifestation ist eine aseptische Meningitis (18–26 %) vor allem der basalen Meningen,aber evtl. auch des Ependyms mit Kopfschmerzen, Erbrechen, Meningismus und Papillenödem. DritthäufigsteManifestation ist ein Hydrozephalus (9–17 %), entweder obstruktiv bei Granulomen im Bereich des III. oder IV.Ventrikels oder kommunizierend bei entzündlichem Verschluss der Pacchionischen Granulationen. Besonders häufigwerden Hypothalamus und Hypophyse (15–26 %) befallen, mit Diabetes insipidus, bulimischem Verhalten undHypersomnie. Zerebrale, subdurale und meningeale (en plaque) Massenläsionen können auch zu epileptischenAnfällen führen (ca. 20 %). Eine Myelopathie infolge komprimierender extramedullärer oder auch intramedullärerGranulome ist in 6–10 % zu beobachten, eine periphere Neuropathie (vor allem Typ Mononeuritis multiplex) in 4–14 %und eine Myopathie in 7–12 % (Stern 2004).
Der klinische Verlauf ist im Einzelfall nicht vorhersehbar, in zwei Drittel der Fälle ist er monophasisch, in einem Drittelrezidivierend. Die Prognose ist gut bei Hirnnervenläsionen, eher schlecht bei Hydrozephalus und zerebralenMassenläsionen.
Diagnostisches Vorgehen
Die Kernspintomografie hat die Diagnose des ZNS-Befalls wesentlich erleichtert. Der typische Befund ist eine knotigeoder auch flächige Verdickung der Meningen, speziell an der Schädelbasis mit starker Kontrastmittelanreicherung(Smith et al. 2004). Der Kveim-Test wäre zwar sensitiv und spezifisch, leider ist aber zuverlässiges Antigenmaterialkaum erhältlich. Die Gallium-Szintigrafie und die Bestimmung des Angiotensin-Konversions-Enzyms haben eine sehrgeringe Spezifität (Nowak u. Widenka 2001).
Je nach kernspintomografischem Befund ist die Diffe renzialdiagnose sehr breit:
bei parenchymatösen Massenläsionen: Lymphom, Gliom, Meningeom, Metastase, Tuberkulosebei vorwiegend periventrikulären herdförmigen Läsionen auch Multiple Sklerosebei vorwiegend menigealen Läsionen: bakterielle, mykotische oder tuberkulöse Infektionen, leukämische oderkarzinomatöse Infiltrationen
Notw endige Untersuchungen:
je nach klinischer Symptomatik: MRT von Kopf und/oder Rückenmarkzur Sicherung der Diagnose Sarkoidose: Suche nach Befall weiterer Systeme:
Thorax- und Abdomen-CTaugenärztliche UntersuchungUntersuchung von Haut und Lymphknotenstationen und ggf. Biopsie (vor allem Hiluslymphknoten)je nach Klinik: Muskelbiopsie oder Leberbiopsie
Leitlinien für Diagnostik und Therapie in der Neurologie
8
LungenfunktionstestLiquoranalyse inklusive Bakteriologie und Zytologie (zum Ausschluss infektiöser oder neoplastischerDifferenzialdiagnosen)
Im Einzelfall erforderliche Untersuchungen:
bei isoliertem ZNS-Befall (klinisch und bildgebend) – wenn immer möglich – meningeale und/oder zerebraleBiopsie je nach MRT-Befund
Therapie
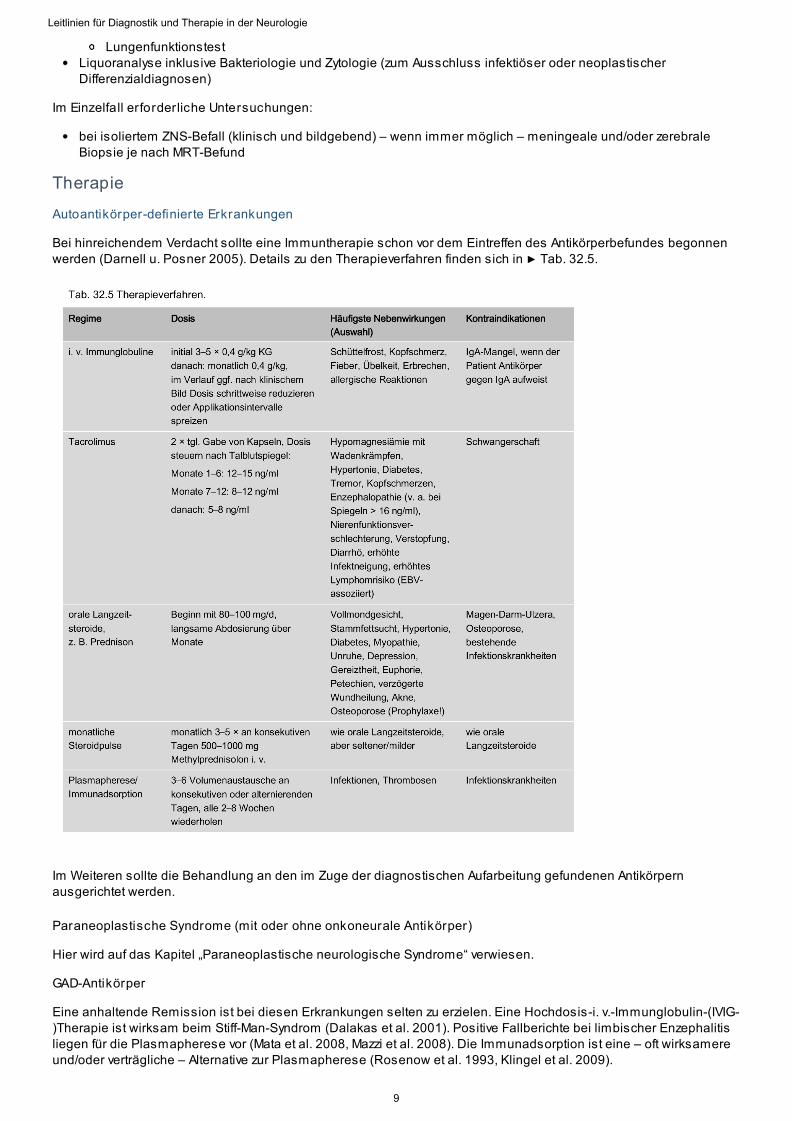
Autoantikörper-definierte Erkrankungen
Bei hinreichendem Verdacht sollte eine Immuntherapie schon vor dem Eintreffen des Antikörperbefundes begonnenwerden (Darnell u. Posner 2005). Details zu den Therapieverfahren finden sich in ▶ Tab. 32.5.
Im Weiteren sollte die Behandlung an den im Zuge der diagnostischen Aufarbeitung gefundenen Antikörpernausgerichtet werden.
Paraneoplastische Syndrome (mit oder ohne onkoneurale Antikörper)
Hier wird auf das Kapitel „Paraneoplastische neurologische Syndrome“ verwiesen.
GAD-Antikörper
Eine anhaltende Remission ist bei diesen Erkrankungen selten zu erzielen. Eine Hochdosis-i. v.-Immunglobulin-(IVIG-)Therapie ist wirksam beim Stiff-Man-Syndrom (Dalakas et al. 2001). Positive Fallberichte bei limbischer Enzephalitisliegen für die Plasmapherese vor (Mata et al. 2008, Mazzi et al. 2008). Die Immunadsorption ist eine – oft wirksamereund/oder verträgliche – Alternative zur Plasmapherese (Rosenow et al. 1993, Klingel et al. 2009).
Leitlinien für Diagnostik und Therapie in der Neurologie
9

VGKC-Komplex-Antikörper (LGI1-, CASPR2-Antikörper)
Eine frühzeitige Immuntherapie mit Steroiden, eventuell ergänzt durch i. v. Immunglobuline (IVIG) oderPlasmapherese/Immunadsorption beschleunigt den Heilungsverlauf vermutlich und verbessert möglicherweise auchdas Therapieergebnis. Der Therapieerfolg korreliert mit dem Absinken der Serum-Antikörperkonzentrationen (Vincentet al. 2004). Die Steroidtherapie bei LGI1-Antikörper-assoziierten faziobrachialen dystonen Anfällen verhindertmöglicherweise das Übertreten in eine limbische Enzephalitis (Irani et al. 2011). Nach Abklingen einer mit VGKC-Komplex-Antikörpern verbundenen Erkrankung können angesichts des fast immer monophasischenKrankheitsverlaufs zur symptomatischen Behandlung eingeführte Antikonvulsiva abdosiert werden.
NMDAR-Antikörper
Die Resektion eines eventuell vorhandenen Teratoms ist vordringlich. Ein früher Immuntherapiebeginn (≤ 40 Tage) istmit einem besseren Therapieergebnis assoziiert als ein später (> 40 Tage nach Krankheitsbeginn), wobei dieKombination von Steroiden mit mindestens einer weiteren Immuntherapie vorteilhaft ist (Irani et al. 2010). Dalmauberichtet über die Anwendung von Kortikosteroiden, IVIG oder Plasmapherese als First-Line-Therapien und dieZufügung von Cyclophosphamid und/oder Rituximab als intensivierter Behandlung, die vor allem bei verzögertemTherapiebeginn und Abwesenheit eines Tumors benötigt würde (Dalmau et al. 2011). Noch ist unklar, ob z. B.Azathioprin Rezidive verhindern kann.
AMPAR- und GABABR-Antikörper
Besserungen unter Immuntherapien wurden für beide Antikörper-definierten Erkrankungen unter Steroiden, zum Teilkombiniert mit IVIG, Plasmapherese oder Mycophenolatmofetil, beschrieben (Lai et al. 2009, Bataller et al. 2010,Graus et al. 2010a, Lancaster et al. 2010). Durch Azathioprin konnten AMPAR-Antikörper-assoziierte Rezidive offenbarverhindert werden (Lai et al. 2009).
Antikörper gegen den metabotropen Glutamatrezeptor
Antikörper gegen die Untereinheiten 1 und 5 wurden - meist paraneoplastisch - bei Patienten mit zerebellärer Ataxieoder limbischer Enzephalitis mit prädominanten Gedächtnisstörungen („Ophelia-Syndrom“) gefunden (Lancaster etal., 2011 mit weiteren Referenzen).
SREAT
Medikamente der ersten Wahl bei der SREAT sind Steroide, unter denen eine ausgezeichnete, oft innerhalb von Tageneinsetzende Wirkung beobachtet wird. Allerdings können auch bis zu 6 Wochen vergehen, ehe die Besserung eintritt(Kothbauer-Margreiter et al. 1996). Ein Therapie-Algorithmus ist in ▶ Abb. 32.4 dargestellt. Epileptische Anfälleerfordern eine antiepileptische Therapie, die nach Abheilen der Erkrankung und Normalisierung des EEG wiederausgeschlichen wird. Eine zweite Episode wird wie eine erste behandelt, möglicherweise mit noch langsameremAusschleichen der Steroide.
Abb. 32.4 Therapie-Algorithmus für die Steroid-responsive Enzephalopathie mit assoziierter Autoimmunthyroiditis(SREAT).
Bei weiteren Episoden oder einem wiederholten Aufflammen der Symptome nach Unterschreiten einer gewissenSteroiddosis sollten zusätzliche Steroid-sparende Immunsuppressiva zum Einsatz kommen. Einzelfallberichte/Mini-
Leitlinien für Diagnostik und Therapie in der Neurologie
10

Serien berichten über gute Effekte von Azathioprin, Methotrexat, Ciclosporin A, Hydroxychloroquin undMycophenolatmofetil. Pulstherapien mit intravenösen Immunoglobulinen und Cyclophyosphamid sowiePlasmapheresen wurden ebenfalls erfolgreich eingesetzt. Alle diese Therapien werden aber als zweite Wahlgegenüber Steroiden angesehen.
Antikörper-negative Syndrome
Bei der Diagnose CLIPPERS ist ein gutes Ansprechen auf Steroide, zum Teil ergänzt durch Immunsuppressiva(Dosierungen etwa wie in ▶ Tab. 32.5) beschrieben worden (Pittock et al. 2010, Gabilondo et al. 2011). Auch beianderen, mutmaßlich immunologisch verursachten Erkrankungen ohne Nachweis von Antikörpern oder einerentzündlichen Neuropathologie ist es vertretbar, für eine Zeitspanne von etwa 3 Monaten zur Besserung vordefinierterParameter z. B. eine Steroidtherapie durchzuführen. Wenn nach Ablauf dieser Zeit die angestrebte Besserungeingetreten ist, kann die Behandlung noch eine Zeitlang zur Stabilisierung fortgeführt werden. Andernfalls sollte dieImmuntherapie nur in begründeten Einzelfällen weitergeführt oder umgestellt werden.
Rasmussen-Enzephalitis
Die Hemisphärektomie in einer ihrer modernen Varianten ist hocheffektiv zur Beseitigung der pharmakoresistentenAnfälle bei Rasmussen-Enzephalitis (Bien u. Schramm 2009). Kein anderes Verfahren ist vergleichbar wirksam. EineLangzeitimmuntherapie kann den Gewebs- und Funktionsverlust verlangsamen oder stoppen, offenbar aber nicht dieAnfälle kontrollieren (Bien et al. 2004). Eine „erfolgreiche“ Immuntherapie bei Patienten mit noch erhaltener Funktionder betroffenen Hemisphäre, aber belastender Epilepsie kann zu einem Pyrrhussieg führen: Man bremst denFunktionsverlust, nicht aber die Epilepsie, sodass die Betroffenen nicht ohne Funktionsbeeinträchtigunghemisphärektomiert, aber auch nicht auf konservativem Wege anfallsfrei werden können (Bien u. Schramm 2009).
Für das therapeutische Vorgehen siehe das Flussdiagramm in ▶ Abb. 32.5 (Bien et al. 2005).
Abb. 32.5 Therapeutisches Vorgehen bei Rasmussen-Enzephalitis.
Pharmakotherapie
Die folgenden Immuntherapien werden vorgeschlagen, um die Hemisphärenzerstörung aufzuhalten (Granata et al.2003, Bien u. Schramm 2009):
Tacrolimus (Bien et al. 2004)i. v. Immunglobuline (Hart et al. 1994)orale Langzeitsteroide (Bahi-Buisson et al. 2007)Plasmapherese/Immunadsorption (Antozzi et al. 1998)
Für Details siehe ▶ Tab. 32.5.
Weitere, spezielle Therapieformen
Bei sehr umschriebener Epilepsia partialis continua kommt eine Botulinum-Toxin-Behandlung in Betracht (Lozsadi et
Leitlinien für Diagnostik und Therapie in der Neurologie
11

al. 2004, Browner et al. 2006). Wenn eine Hemisphärektomie nicht in Frage kommt, kann in Einzelfällen dieVagusnervstimulation die Anfallssituation bessern (Grujic et al. 2011).
Neurosarkoidose
Angesichts der anerkannten Morbidität und Mortalität der Neurosarkoidose empfehlen die meisten Autoren eine früheund aggressive Therapie. Aber es gibt zurzeit keine kontrollierte Studien als Grundlage von Therapieleitlinien (Hoitsmaet al. 2004).
Pharmakotherapie
Als Therapeutikum erster Wahl gelten orale Langzeit-Kortikosteroide in einer initialen Dosis von 1 mg/kg KG und Tagp. o. (▶ Tab. 32.5). In schweren Fällen können i. v. Steroide auch zu Beginn hochdosiert (Stoßtherapie) appliziertwerden. Bei ungenügender Wirkung oder raschem Rezidiv bei Dosisreduktion bzw. hoher notwendigerErhaltungsdosis werden verschiedene zytotoxische Immunsuppressiva empfohlen (Methotrexat, Azathioprin,Ciclosporin, Cyclophosphamid). Die größten Erfahrungen bestehen mit Methotrexat und Azathioprin. Einzelfällewurden auch erfolgreich mit Mycophenolatmofetil, Hydrochloroquin, TNF-α-Blockern (Infliximab) und monoklonalemAnti-CD20-Antikörpern (Rituximab) behandelt. Eine Langzeitbeobachtung legt nahe, dass bei schwerenneurologischen Symptomen der frühe Einsatz von Immunsuppressiva die Prognose verbessert (Scott et al. 2007).
Weitere, spezielle Therapieformen
Bei gegenüber den oben erwähnten immunsuppressiven Therapien therapieresistenten Einzelfällen oder beiintolerablen Nebenwirkungen wurde ein Entzündungsrückgang unter Strahlentherapie beobachtet (Menninger et al.2003). Hierbei kommt, je nach Entzündungslokalisation, sowohl die stereotaktische lokale Radiochirurgie(Sundaresan u. Jayamohan 2008) wie auch eine Low-Dose-Ganzhirnbestrahlung mit 20–25 Gy zur Anwendung (Brunset al. 2004). Bei Hydrozephalus ist die Shuntdrainage angezeigt. Zur Beurteilung der Therapieeffizienz dienen dieKlinik, das MRT und der Liquorbefund.
Versorgungskoordination
Autoantikörper-definierte Erkrankungen
Wegen der Schwere der Symptomatik und des differenzialdiagnostischen Aufwands ist mindestens initial bei allenhier abgehandelten Erkrankungen eine stationäre Untersuchung und Behandlungseinleitung erforderlich.
Rasmussen-Enzephalitis
Die initiale Diagnostik bedarf der Ausstattung eines stationären spezialisierten Settings. Für Verlaufsuntersuchungengilt dies, wenn die nachfolgenden Untersuchungen komplexere Untersuchungsprogramme erfordern oder aufgrundeiner hohen Anfallsfrequenz ein ambulantes Management nicht möglich ist.
Neurosarkoidose
Wegen der oft unspezifischen Klinik, der Schwere der Symptome und des differenzialdiagnostischen Aufwands, der –wenn immer möglich – eine Biopsie einschließen sollte, ist mindestens initial eine stationäre Untersuchung undBehandlungseinleitung erforderlich.
Redaktionskomitee
PD Dr. Stephan Rüegg, Abteilung für Klinische Neurophysiologie, Neurologische Klinik, Universitätsspital BaselUniv.-Prof. Dr. Erich Schmutzhard, Medizinische Universitätsklinik für Neurologie, InnsbruckProf. Dr. Matthias Sturzenegger, Inselspital, Neurologische Klinik, Universitätsspital Bern
Federführend: Prof. Dr. Christian Bien, Epilepsie-Zentrum Bielefeld-Bethel, Krankenhaus Mara, Maraweg 21, 33617BielefeldE-Mail: [email protected]
Entw icklungsstufe der Leitl inie: S1
Interessenkonflikte
Es bestehen keine für einzelne Mitglieder der Autorengruppe oder die ganze Leitliniengruppe bedeutsameInteressenkonflikte.
Finanzierung der Leitlinie
keine
Leitlinien für Diagnostik und Therapie in der Neurologie
12

Methodik der Leitlinienentwicklung
Zusammensetzung der Leitl iniengruppe, Beteiligung von Interessengruppen
s. Redaktionskomitee
Recherche und Auswahl der wissenschaftl ichen Belege
Es lagen keine bestehenden Leitlinien oder andere wichtige Quellen mit Leitliniencharakter (z.B. internationaleKonsensusstatements, IQWiG-Berichte) vor, ebensowenig wurden andere Quellen systematisch aufbereiteter Evidenz(Systematic Reviews, Metaanalysen, HTA-Berichte) empfohlen.
Verfahren zur Konsensfindung
Die Autoren haben sich überwiegend durch E-Mail-Korrespondenz während aller Schritte der LL-Erstellungverständigt. Hinzu kamen direkte persönliche Besprechungen nach Bedarfslage.
Literatur
Antozzi C, Granata T, Aurisano N et al. Long-term selective IgG immuno-adsorption improves Rasmussen'sencephalitis. Neurology 1998; 51: 302–305Bahi-Buisson N, Villanueva V, Bulteau C et al. Long term response to steroid therapy in Rasmussen encephalitis.Seizure 2007; 16: 485–492Bataller L, Galiano R, Garcia-Escrig M et al. Reversible paraneoplastic limbic encephalitis associated withantibodies to the AMPA receptor. Neurology 2010; 74: 265–267Bauer J, Elger CE, Hans VH et al. Astrocytes are a specific immunological target in Rasmussen's encephalitis.Ann Neurol 2007; 62: 67–80Bien CG, Bauer J, Deckwerth TL et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism inRasmussen's encephalitis. Ann Neurology 2002a; 51: 311–318Bien CG, Gleissner U, Sassen R et al. An open study of tacrolimus therapy in Rasmussen encephalitis. Neurology2004; 62: 2106–2109Bien CG, Granata T, Antozzi C et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: aEuropean consensus statement. Brain 2005; 128 (Pt 3): 454–471Bien CG, Schramm J. Treatment of Rasmussen encephalitis half a century after its initial description: Promisingprospects and a dilemma. Epilepsy Res 2009; 86: 101–112Bien CG, Vincent A, Barnett MH et al. Immunopathology of autoantibody-associated encephalitides: clues forpathogenesis. Brain 2012; 135: 1622–1638Bien CG, Widman G, Urbach H et al. The natural history of Rasmussen's encephalitis. Brain 2002b; 125 (Pt 8):1751–1759Browner N, Azher SN, Jankovic J. Botulinum toxin treatment of facial myoclonus in suspected Rasmussenencephalitis. Mov Disord 2006; 21: 1500–1502Bruns F, Pruemer B, Haverkamp U et al. Neurosarcoidosis: an unusual indication for radiotherapy. Br J Radiol2004; 77: 777–779Castillo P, Woodruff B, Caselli R et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol 2006; 63: 197–202Chaudhuri A, Behan PO. The clinical spectrum, diagnosis, pathogenesis and treatment of Hashimoto'sencephalopathy (recurrent acute disseminated encephalomyelitis). Curr Med Chem 2003; 10: 1945–1953Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003; 60: 164–171Dalakas MC, Fujii M, Li M et al. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med2001; 345: 1870–1876Dalmau J, Lancaster E, Martinez-Hernandez E et al. Clinical experience and laboratory investigations in patientswith anti-NMDAR encephalitis. Lancet Neurol 2011; 10: 63–74Darnell RB, Posner JB. A new cause of limbic encephalopathy. Brain 2005; 128: 1745–1746Demeurisse G, Demol O, Robaye E. Motor evaluation in vascular hemiplegia. Eur Neurol 1980; 19: 382–389Gabilondo I, Saiz A, Graus F et al. Response to immunotherapy in CLIPPERS syndrome. J Neurol 2011; 258:2090–2092Granata T, Fusco L, Gobbi G et al. Experience with immunomodulatory treatments in Rasmussen's encephalitis.Neurology 2003; 61: 1807–1810Graus F, Boronat A, Xifro X et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology2010a; 74: 857–859Graus F, Delattre JY, Antoine JC et al. Recommended diagnostic criteria for paraneoplastic neurologicalsyndromes. J Neurol Neurosurg Psychiatry 2004; 75: 1135–1140Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol 2010b; 257:509–517Graus F, Saiz A, Lai M et al. Neuronal surface antigen antibodies in limbic encephalitis: Clinical-immunologic
Leitlinien für Diagnostik und Therapie in der Neurologie
13

associations. Neurology 2008; 71: 930–936Grujic J, Bien CG, Pollo C et al. Vagus nerve stimulator treatment in adult-onset Rasmussen's encephalitis.Epilepsy Behav 2011; 20: 123–125Haberlandt E, Bast T, Ebner A et al. Limbic encephalitis in children and adolescents. Arch Dis Child 2011; 96:186–191Hart Y, Andermann F, Fish D et al. The medical treatment of chronic encephalitis and epilepsy. In: Wolf P, ed.Epileptic Seizures and Syndromes. London: John Libbey & Company; 1994: 399–404Hoitsma E, Faber CG, Drent M et al. Neurosarcoidosis: a clinical dilemma. Lancet Neurol 2004; 3: 397–407Hollowell JG, Staehling NW, Flanders WD et al. Serum TSH, T, and thyroid antibodies in the United Statespopulation (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin EndocrinolMetab 2002; 87: 489–499Hughes EG, Peng X, Gleichman AJ et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. JNeurosci 2010; 30: 5866–5875Irani SR, Bera K, Waters P et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical andparaclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133 (Pt 6):1655–1667Irani SR, Michell AW, Lang B et al. Faciobrachial dystonic seizures precede LGI1 antibody limbic encephalitis. AnnNeurol 2011; 69: 892–900Klingel R, Heibges A, Fassbender C. Plasma exchange and immunoadsorption for autoimmune neurologicdiseases - current guidelines and future perspectives. Atheroscler 2009; 10 (Suppl.): 129–132Kothbauer-Margreiter I, Sturzenegger M, Komor J et al. Encephalopathy associated with Hashimoto thyroiditis:diagnosis and treatment. J Neurol 1996; 243: 585–593Lai M, Hughes EG, Peng X et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location.Ann Neurol 2009; 65: 424–434Lalic T, Pettingill P, Vincent A et al. Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3pyramidal cell synaptic transmission. Epilepsia 2011; 52: 121–131Lancaster E, Lai M, Peng X et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: caseseries and characterisation of the antigen. Lancet Neurol 2010; 9: 67–76Lancaster E, Martinez-Hernandez E, Titulaer MJ et al. Antibodies to metabotropic glutamate receptor 5 in theOphelia syndrome. Neurology. 2011; 77:1698-1701Lozsadi DA, Hart IK, Moore AP. Botulinum toxin A improves involuntary limb movements in Rasmussen syndrome.Neurology 2004; 62: 1233–1234Malter MP, Helmstaedter C, Urbach H et al. Antibodies to glutamic acid decarboxylase define a form of limbicencephalitis. Ann Neurology 2010; 67: 470–478Mas N, Saiz A, Leite MI et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry2010; 82: 1399–1401Mata S, Muscas GC, Naldi I et al. Non-paraneoplastic limbic encephalitis associated with anti-glutamic aciddecarboxylase antibodies. J Neuroimmunol 2008; 199: 155–159Mazzi G, Roia DD, Cruciatti B et al. Plasma exchange for anti-GAD associated non paraneoplastic limbicencephalitis. Transfus Apher Sci 2008; 39: 229–233Menninger MD, Amdur RJ, Marcus RB jr. Role of radiotherapy in the treatment of neurosarcoidosis. Am J ClinOncol 2003; 26: e115–e118Nowak DA, Widenka DC. Neurosarcoidosis: a review of its intracranial manifestation. J Neurol 2001; 248: 363–372Oguni H, Andermann F, Rasmussen TB. The syndrome of chronic encephalitis and epilepsy. A study based on theMNI series of 48 cases. Adv Neurol 1992; 57: 419–433Pittock SJ, Debruyne J, Krecke KN et al. Chronic lymphocytic inflammation with pontine perivascular enhancementresponsive to steroids (CLIPPERS). Brain 2010; 133: 2626–2634Prüss H, Dalmau J, Harms L et al. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknownorigin. Neurology 2010; 75: 1735–1739Rosenow F, Haupt WF, Grieb P et al. Plasma exchange and selective adsorption in Guillain-Barré syndrome – acomparison of therapies by clinical course and side effects. Transfus Sci 1993; 14: 13–15Rüegg S. Schilddrüse und Epilepsie. Epileptologie 2011; 28: 30–37Schäuble B, Castillo PR, Boeve BF et al. EEG findings in steroid-responsive encephalopathy associated withautoimmune thyroiditis. Clin Neurophysiol 2003; 114: 32–37Schwab N, Bien CG, Waschbisch A et al. CD8+ T cell clones dominate brain infiltrates in Rasmussen encephalitisand persist in the periphery. Brain 2009; 132: 1236–1246Scott TF, Yandora K, Valeri A et al. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treatedpatients. Archs Neurology 2007; 64: 691–696Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. Am J Roentgenol 2004; 182:289–295Stern BJ. Neurological complications of sarcoidosis. Curr Opin Neurol 2004; 17: 311–316Sundaresan P, Jayamohan J. Stereotactic radiotherapy for the treatment of neurosarcoidosis involving the pituitarygland and hypothalamus. J Med Imaging Radiat Oncol 2008; 52: 622–626Vincent A, Buckley C, Schott JM etal. Potassium channel antibody-associated encephalopathy: a potentially
Leitlinien für Diagnostik und Therapie in der Neurologie
14

© Deutsche Gesellschaft für Neurologie
immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127 (Pt 3): 701–712Zöphel K, Saller B, Wunderlich G et al. Autoantibodies to thyroperoxidase (TPOAb) in a large population ofeuthyroid subjects: implications for the definition of TPOAb reference intervals. Clin Lab 2003; 49: 591–600Zuliani L, Graus F, Giometto B et al. Central nervous system neuronal surface antibody associated syndromes:review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83: 638–645
Aus: Hans-Christoph Diener, Christian Weimar (Hrsg.)Leitl inien für Diagnostik und Therapie in der NeurologieHerausgegeben von der Kommission "Leitlinien" der Deutschen Gesellschaft fürNeurologieThieme Verlag, Stuttgart, September 2012
Leitlinien für Diagnostik und Therapie in der Neurologie
15