
Inhaltsverzeichnis · Especially, inorganic-organic biohybrid NPs are considered to be important...
120
Transcript of Inhaltsverzeichnis · Especially, inorganic-organic biohybrid NPs are considered to be important...



Inhaltsverzeichnis
Inhalt des Abstactbandes
Grusswort
Unterstützer
Messestände
Vortragende Appetithäppchen
Abstracts der Teilnehmer
Teilnehmerliste nach Fachbereichen aufgeschlüsselt
Notizen
Programm (Rückseite)
Druck
Copier-Center Haase, Würzburg
Dieser Abstractband besitzt keine Seitenzahlen. Dafür sind jeweils am Rand die
Posternummern eingetragen. In eben dieser Reihenfolge sind die Poster auch während der
Posterpräsentation aufgestellt. Weitere Informationen können aus den Lageplänen
entnommen werden.


GRUSSWORT JCF
Liebe Teilnehmer der Chem-SyStM 2016, sehr geehrte Gäste,
vor ziemlich genau 10 Jahren hatten die damaligen, aktiven Mitglieder des
JungChemikerForums Würzburg die Idee ein Symposium an unserer Fakultät ins Leben zu
rufen und somit einen gewichtigen Beitrag zum wissenschaftlichen Austausch und zur
Vernetzung der einzelnen Fachbereiche zu leisten. Tragende Säule dieser Veranstaltung sollte,
damals wie heute, die Studentenschaft bilden und dadurch die Möglichkeit erhalten sich im
familiären Umfeld erste oder weiterführende Fertigkeiten im wissenschaftlichen Präsentieren
und Kommunizieren zu erlangen. Vor diesem Hintergrund und der Tatsache, dass sich dieses
Symposium auch zur 6. Auflage weiterhin großer Beliebheit erfreut, ist es dem JCF-Würzburg
ein besonderes Vergnügen Sie zur Chem-SyStM 2016 begrüßen zu dürfen.
Unter dem Motto „Großes vereint im Kleinsten“ bieten wir auch dieses Jahr wieder ein
abwechslungsreiches Programm, darunter die bekannten Poster-Appetithäppchen und die
Postersession, sowie einen Abendvortrag zum Thema „Chemie der Pyrotechnik“ gehalten von
Herrn Dr. Klein-Sommer. Darüber hinaus wird es erstmals bereits am Morgen einen Vortrag
geben, Vortragsgast ist Herr Christian Lange vom VAA (Verband angestellter Akademiker und
leitender Angestellter der Chemischen Industrie e.V.). Als abschließenden Höhepunkt dürfen
wir uns alle auf ein Feuerwerk auf dem Campusgelände freuen, bevor ein hoffentlich
ereignisreicher und wissenschaftlich ansprechender Tag bei geselliger Runde seinen
gebührenden Ausklang findet und vielleicht dem/der einen oder anderen Teilnehmer/in ein
zukünftiges Engagement im JungChemikerForum schmackhaft gemacht hat.
Wir wünschen Ihnen viel Freude, gute Unterhaltung und einen interessanten Austausch bei
der Chem-SyStM 2016.
Euer Sprecher-Team des JCF-Würzburg
Thomas Steffenhagen Sebastian Endres Domenik Schleier

GRUSSWORT OVV
Liebe Studierende,
Neugierde und Faszination sind gelebte Attribute, die uns in der Wissenschaft täglich Schritt
für Schritt auf dem steinigen Weg von der Idee zur Erkenntnis vorantreiben. Von den
Hochschulen werden Spitzenleistungen in Lehre und Forschung erwartet. Die Universität
Würzburg, im Speziellen die Fakultät für Chemie und Pharmazie, stellt sich diesen
Herausforderungen und forscht mit ihren Mitarbeitern und den Studierenden gemeinsam für
eine umfassende Perspektive auf die großen Zukunftsfragen wie Nachhaltigkeit und Gesundheit
oder neue Materialien und Bauelemente, führt aber auch wichtige Grundlagenforschung durch,
deren Relevanz sich vielleicht erst viel später zeigen wird.
Chemie – Großes vereint im Kleinsten kann natürlich vieles heißen. Großes schaffen mit
kleinen molekularen Einheiten. Es könnte aber auch bedeuten, die Ähnlichkeiten in großen
Gebilden, seien es biologische Zellen oder auf neuen Materialien basierende Bauelemente, sind
dadurch vereint, dass sie auf ähnlichen chemischen Prinzipien beruhen. Beides, das Große aber
auch die gemeinsamen Prinzipien bringt eine Komplexität der Fragestellungen mit sich, die
immer häufiger nur durch Netzwerke beantwortet werden können, an denen Wissenschaftler
aus vormals abgegrenzten Bereichen kooperieren müssen. Zentral für den Erfolg solcher
Netzwerke ist jedoch der kommunikative Austausch der einzelnen wissenschaftlichen
Disziplinen.
Das Chemie-Symposium der Studierenden Mainfrankens, kurz Chem-SyStM, die von dem
JCF-Regionalforum Würzburg in guter Tradition zum sechsten Mal organisiert wird,
ermöglicht eine solche Kommunikation, denn das grundlegende Ziel und die zentrale Aufgabe
dieses Symposiums liegen in der Präsentation und regen Diskussion wissenschaftlicher
Arbeiten von Studierenden sehr unterschiedlicher Fachrichtungen im familiären Umfeld der
eigenen Universität.
Ich freue mich auf interessante Forschungsarbeiten von Studierenden für Studierende und
wünsche allen Teilnehmerinnen und Teilnehmern einen spannenden und erkenntnisreichen
Tag.
Prof. Dr. Bernd Engels
Ortsverbandsvorsitzender
OV Unterfranken

GRUSSWORT Dekan
Liebe Mitglieder des Jungchemikerforums, liebe Kolleginnen und Kollegen, liebe
Studierende,
die ChemSyStM findet nun seit ihrer Etablierung im Jahre 2007 das sechste mal statt und
hat sich somit einen festen Platz im akademischen Jahr unserer Fakultät erarbeitet. Auch dieses
Jahr stehen wieder spannende Vorträge und eine Postersession an, in der junge
Nachwuchswissenschaftler ihre Forschung und damit einen Einblick in die Forschung unserer
Arbeitskreise vorstellen werden. Dies soll nicht nur dem wissenschaftlichen Austausch unter
den Arbeitskreismitgliedern dienen, sondern auch den Studierenden, sich über Arbeitskreise,
den dort bearbeiteten Forschungsschwerpunkten sowie über mögliche Bachelor- oder
Masterarbeitsthemen zu erkundigen. Aufgrund der stark gewachsenen Anzahl an
Arbeitskreisen haben wir und dieses Jahr dazu entschieden, die sonst übliche Präsentation der
Arbeitskreise durch Ultrakurzvorträge von den „Arbeitskreis-Chefs“ durch entsprechende
informative Poster zu ersetzen. Es würde mich freuen, wenn wir von Seiten der Teilnehmer
eine Rückmeldung über diese Vorgehensweise erhalten könnten, so dass wir Ihre Meinung bei
künftigen Planungen berücksichtigen können.
Für dieses Mal wünsche ich allen Teilnehmern eine spannende und nicht zuletzt auch
unterhaltsame ChemSyStM 2016!
Ihr
Christoph Lambert
Dekan


UNTERSTÜTZER
Bedanken möchten wir uns hiermit bei unseren finanziellen Unterstützern, den folgenden
Unternehmen und Institutionen:
Fakultät für Chemie und Pharmazie

MESSE
In diesem Jahr mit einem Informationsstand vertreten sind die folgenden Unternehmen und
Institutionen mit Ansprechpartnern:


Appetithäppchen
Appetithäppchen
# Name Vorname Arbeitskreis Kategorie
16 Paul Ursula Radius Anorganische Chemie und
Materialwissenschaften
22 Schmitt Hans-
Christian Fischer
Physikalische und Theoretische
Chemie
29 Lombe Blaise Bringmann Biochemie und Organische
Chemie
31 Schweeberg Sarah Krüger Biochemie und Organische
Chemie
38 Pres Sebastian Brixner Physikalische und Theoretische
Chemie
48 Warkentin Viktor Krüger Biochemie und Organische
Chemie
53 Griesbeck Stefanie Marder Anorganische Chemie und
Materialwissenschaften
66 Feizy Nilab Schatzschneider Anorganische Chemie und
Materialsynthese
67 Soberats Bartolome Würthner Biochemie und Organische
Chemie
72 Moustafa Ahmed Schatzschneider Anorganische Chemie und
Materialwissenschaften
80 Merz Julia Marder Anorganische Chemie und
Materialwissenschaften
83 Syamala Pradeep Würthner Biochemie und Organische
Chemie
86 Leonhardt Viktoria Beuerle Biochemie und Organische
Chemie
88 Wachtler Stefan Krüger Biochemie und Organische
Chemie
89 Nowak-Król Agnieszka Würthner Biochemie und Organische
Chemie



ABSTRACTS
ABSTRACTS


Anorganische Chemie und Materialwissenschaften
P1 AK Prof. Müller-Buschbaum
Hybrid Materials
Introduction and Overview of our Research
T. Schäfer, T. Wehner, S. Zottnick, J. Stangl, F. Mühlbach, A. Sedykh, J. Sorg, K. Müller-
Buschbaum
Institut für Anorganische Chemie, JMU-Würzburg, Am Hubland, 97074 Würzburg,
Germany
The research of our workgroup is focused on inorganic-organic hybrid materials based on solid-state
chemistry. This includes the field of coordination chemistry in particular complexes, coordination
polymers (CPs) and metal-organic frameworks (MOFs) as well as the functionalization of other
materials with our systems. For our syntheses, we employ various metal precursors and organic ligands
such as amines, amides, heterocyclic systems, ionic liquids and borates. Besides ordinary Schlenk-
techniques, various synthesis strategies are used, such as solvent-free melt synthesis and
mechanochemical synthesis, known from solid-state-chemistry. In addition, we are strongly interested
in the properties of our compounds with the synthesis of luminescent MOFs as well as fine-tuning of
their luminescence being one focus. The latter refers to maximize quantum-efficiencies as well as
accurate color adjustments, which is, among other things, achieved by co-doping of host lattices with
luminescent cations.
By combination of MOFs with other functional materials, new multifunctional hybrid materials and
composites with highly specialized properties can be obtained. Thus, we are interested in modification
of various substrates by deposition of defined thin films of MOFs on their surface. Furthermore, the
surface modification of luminescent or superparamagnetic micro- and nanoparticles with MOFs can lead
to unprecedented multifunctional sensor-type materials that combine the properties of both materials.
For the characterization of our materials, we use single crystal and powder X-Ray diffraction methods
assisted by a wide range of spectroscopic methods (IR, RAMAN, NMR, UV-VIS-NIR,
Photoluminescence spectroscopy and Fluorescence microscopy), elemental analysis methods like
CHNS and EDX and many other additional analytical methods such as simultaneous DTA/TG and BET.
For more information, please visit our website
http://www.mueller-buschbaum.anorganik.chemie.uni-wuerzburg.de/
or contact us via e-mail: [email protected]

Physikalische und Theoretische Chemie P2
AK Engel - höchst dynamisch!
Unser Ansatz:
Man nehme eine physikalisch-chemische Fragestellung und würze diese mit Mathematik, bis
sich ein Modell ergibt. Anschließend muss der Ansatz programmiert werden und hoffentlich
werden alle Bugs entfernt. Rechnungen mit Minuten oder Tagen an Rechenzeit liefern dann
mehr oder weniger erfreuliche Ergebnisse. Es fehlt dann noch deren Interpretation, die
eventuelle Modifizierung der Modelle und die publizierte Antwort auf alle Fragen.
In diesem Sinne werden auf unserem AK-Poster die grundlegenden Konzepte der
Quantendynamik und Auszüge aus der aktuellen Forschung vorgestellt.

Anorganische Chemie und Materialwissenschaften
P3 AK Mandel
Complex nanostructured particles by design
The Particle Technology Group (Karl Mandel)
Fraunhofer-Institut für Silicatforschung ISC Neunerplatz 2 97082 Würzburg
Lehrstuhl Chemische Technologie der Materialsynthese an der Julius-Maximilians-Universität
Würzburg, Röntgenring 11, 97070 Würzburg
The Particle Technology Group focuses on the synthesis and processing of nano- and microparticles.
The fundamental question is how complex, nanostructured particles can be synthesised and which novel
properties can be obtained from these (inorganic) particles.
In the recent years, the art of synthesising and tailoring nanoparticles with distinct properties has
attracted tremendous interest and has been explored very well.
The next step further is to consider these particles as nano-building blocks which shall be combined
bottom-up to form again particles, but now complex, nanostructured particles. The aim is to achieve
novel functional particles with interactive properties that can only be obtained from the smart assembly
of nano-building blocks to combined entities.
An example in the field of magnetic particles are nanocomposite particles which can be employed for
water purification[1,2] or as magnetically collectable, optical sensors for target substances in fluids[3] or
as unique hollow magnetic architectures.[4] Other nanoparticle building-blocks such as nano silica can
be assembled to nanostructured micro-raspberry particles which act as mechanically reactive container
structures, which can, for instance, be used in coatings with a refreshable surface functionality such as
an anti-bacterial activity.[5] Beyond that, many more unique properties, and from that, new applications
in the field of energy, environment or sustainability, can be targeted by building complex particle entities
from nano-building-blocks.
[1] ACS Applied Materials and Interfaces 2012, 4, 5633-5642.
[2] RSC Journal of Materials Chemistry A 2013, 1, 1840-1848.
[3] ACS Applied Materials and Interfaces 2016, 8, 5445-5452.
[4] ACS Nano, accepted, DOI: 10.1021/acsnano.6b06063
[5] ACS Applied Materials and Interfaces 2015, 7, 24909-24914.

Anorganische Chemie und Materialwissenschaften P4/5
AK Dembski
Multifunctional Nanoparticles for Medical Applications
S. Dembski, M. Straßer, C. Schneider, B. Christ, H. Walles
Fraunhofer Institute for Silicate Research, Neunerplatz 2, 97082 Wuerzburg, Germany
Translational Center Wuerzburg “Regenerative therapies in oncology and musculoskelettal diseases”
and Department Tissue Engineering and Regenerative Medicine (TERM), University Hospital
Wuerzburg, Roentgenring 11, 97070 Wuerzburg, Germany
The main emphasis of the competence team Theranostics at the Fraunhofer Institute for Silicate
Research ISC is, in cooperation with the Translational Center Würzburg “Regenerative therapies in
oncology and musculoskelettal diseases”, on products enabling highly efficient and personalized therapy
accompanying in vitro and in vivo diagnosis or even combine diagnosis and therapy in situ.
Well-tailored multifunctional nanoparticles (NPs), which are in focus of our R&D work, are playing
a major role in the development of future oriented advanced functional materials for life science
applications e.g. contrast agents in medical imaging, in vitro and in vivo diagnostics, drug delivery as
well as tissue engineering. Especially, inorganic-organic biohybrid NPs are considered to be important
for the development of smart materials and novel technologies for medical applications.
NPs can be prepared by wet-chemical methods: sol-gel, precipitation or hydrothermal synthesis. The
main focus of our R&D work lies on inorganic and hybrid materials e.g. silicate based materials, calcium
fluoride and phosphate, TiO2 and iron oxide. To ensure multifunctionality different approaches can be
applied e.g. labelling of NP matrix with organic dyes or lanthanoid ions as well as combination of
various materials by core-shell NP design. The resulting NPs are subsequently modified with various
chemical functionalities and biological moieties using conventional functionalization and
bioconjugation methods. The great potential of new developed NPs as immunodetection assay labels,
contrast agent for medical imaging or tool for tumor therapy is demonstrated by the different projects.

Organische Chemie und Biochemie
P6 AK Pöppler
Investigation of Polymeric Drug Delivery by NMR
Improvement of the drug delivery and bio-availability of active pharmaceutical ingredients (APIs) is
a huge and important field of research to which we would like to contribute on the basis of NMR
experiments in the solid-state, in gels and in solution (Figure 1).
For example, polymeric vehicles can reduce the toxicity of a
drug by shielding it, transport poorly soluble molecules to the
site of action or release molecules as response to an external
stimulus. A variety of systems such as different polymeric
architectures, microemulsions, liquid crystals or nanotubes can
be applied for this purpose.[1] Synthetic approaches can yield
tailored macromolecules with different functionalities[2] or
attached to biological molecules.[3]
The broad field of NMR Spectroscopy (Figure 2) provides a
variety of tools to study intermolecular interactions of the drug
molecules with each other as well as with the surrounding
polymeric environment. The poster will give a first impression
on three different steps, (i) the thorough analysis of the APIs by
NMR in the solid-state and in solution, (ii) the characterization
of polymer networks and hydrogels in general by anisotropic
NMR and (iii) the study of the interactions between the API and the polymer environment as well as the
drug release process (e.g. by diffusion, a stimulus, degradation, etc.). Furthermore, first NMR results
from a recently started study of curcumin in collaboration with the work group of Prof. Luxenhofer will
be shown.
Figure 2: Orientation dependent and orientation independent NMR interactions in solids and in liquids:
z = Zeemann, rf = radio frequency, D = diploar, CS = chemical shift, Q = quadrupolar and J = J coupling.
[1] N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno, O. C. Farokhzad, Chem. Soc. Rev.
2012, 41, 2971-3010.
[2] M. Danial, C. My-Nhi Tran, P. G. Young, S. Perrier, K. A. Jolliffe, Nat Commun 2013, 4.
[3] I. Cobo, M. Li, B. S. Sumerlin, S. Perrier, Nat Mater 2015, 14, 143-159.
Figure 1: Schematic representation of
the systems and NMR methods to
study drug delivery mediated by
polymeric systems.

Organische Chemie und Biochemie P7
AK Lambert
Projekte für Bachelorarbeiten
M. Moos1, C. Lambert1
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg
[email protected], [email protected]
Mehr Informationen erhalten Sie an Poster 7, wir freuen uns auf Ihr Kommen!

Pharmazie und Lebensmittelchemie
P8 Characterization of novel in-silico designed Hsp70 inhibitors
C. Plank1,2, A. Hofmann1, C. Grimm2, C. Sotriffer1
1 Institute of Pharmacy and Food Chemistry, University of Würzburg
2 Theodor Boveri Institute, Department of Biochemistry, University of Würzburg
Heat-shock protein 70 (Hsp70) has been shown to play a crucial role in the development of
Multiple Myeloma (MM), a neoplastic disease of terminally differentiated, antibody-producing
B-cells. Although various Hsp70 inhibitors have already been reported, these were mostly
directed against the nucleotide (ATP)-binding domain of Hsp70, rendering them likely to cause
selectivity problems. Our work focusses on identifying inhibitors that bind to the domain
interface of Hsp70, thus greatly enhancing their selectivity for Hsp70. Starting from structural
information about the Hsp70 protein, virtual screening identified compounds that displayed low
micromolar activities against MM cells. The most potent hit was expanded into a library of
derivatives, which led to a series of compounds active against MM cells without toxicity on
non-malignant peripheral blood mononuclear cells. The mode of action of these compounds is
now being investigated by an approach combining functional assays and structural studies via
X-ray crystallography. To this end, recombinantly purified truncated and double-mutated
bovine heat-shock cognate 70 (bHsc70 ED 1-554) and endogenous native full-length Hsc70
from pig brain have been screened for crystallization with these inhibitors. In parallel,
functional viability of the purified Hsc70 isoforms was ascertained with luciferase refolding
assays, demonstrating 30-50% chaperone activity. Inhibition assays are now being conducted
to quantify the potency of the inhibitors and characterize their inhibition mechanism. This might
help to develop more potent and selective inhibitors of Hsp70, thus springing open interesting
avenues in translational research for MM.

Anorganische Chemie und Materialwissenschaften
P9 LÖSUNGEN VON BRØNSTED-SÄUREN UND
MÜNZMETALLSALZEN IN NIEDRIG-VISKOSEN IONISCHEN
FLÜSSIGKEITEN
C. Kerpen1, J. A. P. Sprenger1, L. Herkert1, T. Ribbeck1, F. A. Brede1, N. V. Ignat'ev2,
K. Müller-Buschbaum1, M. Finze1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg;
2Berater, Merck KGaA, 64293 Darmstadt/D
[email protected], [email protected]
Ionische Flüssigkeiten, basierend auf Cyanoborat-Anionen, z. B. [BH4–n(CN)n]– (n = 2–4), sind
aufgrund ihrer hohen thermischen, elektrochemischen und chemischen Stabilität verbunden mit ihrer
meist sehr niedrigen Viskosität vielseitig einsetzbar.[1]
Gegenwärtig untersuchen wir ausgewählte physikalische und chemische Eigenschaften von
Hauptgruppen- und Übergangsmetallsalzen mit Cyanoborat-Anionen und ihre Lösungen in ionischen
Flüssigkeiten. Auf Cyanoborat-Anionen basierende Brønsted-Säuren stellen dabei zweckmäßige
Startmaterialien für eine Vielzahl von verschiedenen Cyanoboraten dar. Bisher wurden allerdings erst
wenige Protonen- und Oxonium-Salze mit Cyanoborat-Anionen beschrieben, beispielsweise
H[B(CN)4][2] und (H3O)2[B2(CN)6].[3]
Vor kurzem haben wir eine effiziente Synthese zu der bisher unbekannten Brønsted-Säure
H[BH2(CN)2] (Abb. 1) entwickelt,[4] die bisher beispiellose Eigenschaften in ihrem chemischen,
elektrochemischen und thermischen Verhalten zeigt. H[BH2(CN)2] besitzt eine außergewöhnlich hohe
Löslichkeit (ca. 35 Gew.% bei RT) in EMIm[BH2(CN)2].[5] Dies macht die Säure zu einem
ausgezeichneten Startmaterial für Synthesen, insbesonders von (Metall-)Salzen mit dem [BH2(CN)2]–-
Anion. Von großem Interesse sind die zu H+ isolobalen Münzmetall(I)-Ionen, wodurch sie für
Vergleiche mit Brønsted-Säuren interessant sind. Daher wurden die Ag+- und Cu+-Salze mit dem
[BH2(CN)2]–-Anion synthetisiert und ihre Eigenschaften mit denen des H+-Salzes verglichen.
Abbildung 1: Ausschnitt aus den Ketten von 1∞
H[BH2(CN)2]} (links) and 1∞
Ag(bpy)[BH2(CN)2]}
(rechts).
[1] N. V. Ignat'ev, M. Finze, J. A. P. Sprenger, C. Kerpen, E. Bernhardt, H. Willner, J. Fluorine
Chem. 2015, 177, 46-54.
[2] T. Küppers, E. Bernhardt, C. W. Lehmann, E. Willner, Z. Anorg. Allg. Chem. 2007, 633, 1666-
1672.
[3] J. Landmann, J. A. P. Sprenger, M. Hailmann, V. Bernhardt-Pitchougina, H. Willner, N.
Ignat'ev, E. Bernhardt, M. Finze, Angew. Chem. Int. Ed. 2015, 54, 11259-11264.
[4] M. Drisch, L. A. Bischoff, L. Herkert, J. A. P. Sprenger, M. Finze, N. Ignatyev, R. van Hal,
Merck Patent GmbH, WO2016074760A1, 2016.
[5] E. Bernhardt, V. Bernhardt-Pitchougina, H. Willner, N. Ignatyev, M. Schulte,
Merck Patent GmbH, WO2012163488, 2012.
Physikalische und Theoretische Chemie
P10 Photodissociation reactions of the ortho- & para-xylyl radical: A
Velocity Map Imaging study
K. Pachner1, I. Fischer1
1Institut für Physikalische und Theoretische Chemie, Julius-Maximilians-Universität Würzburg,
Am Hubland Süd, 97074 Würzburg, Deutschland
Xylyl radicals can be found as intermediates in combustion processes. Their parent molecules, the
xylenes, are used as additives in fuels to increase antiknock properties. The thermal decomposition of
the xylyl radicals has been explored recently by Hemberger et al. in a synchrotron experiment.[1] Based
on these studies, we investigated the photodissociation of the ortho- ¶-xylyl radical using velocity
map imaging. Xylyl radicals were formed via flash pyrolysis in a pulsed molecular beam using 2-
respectively 4-methylphenethyl nitrite as a precursor.
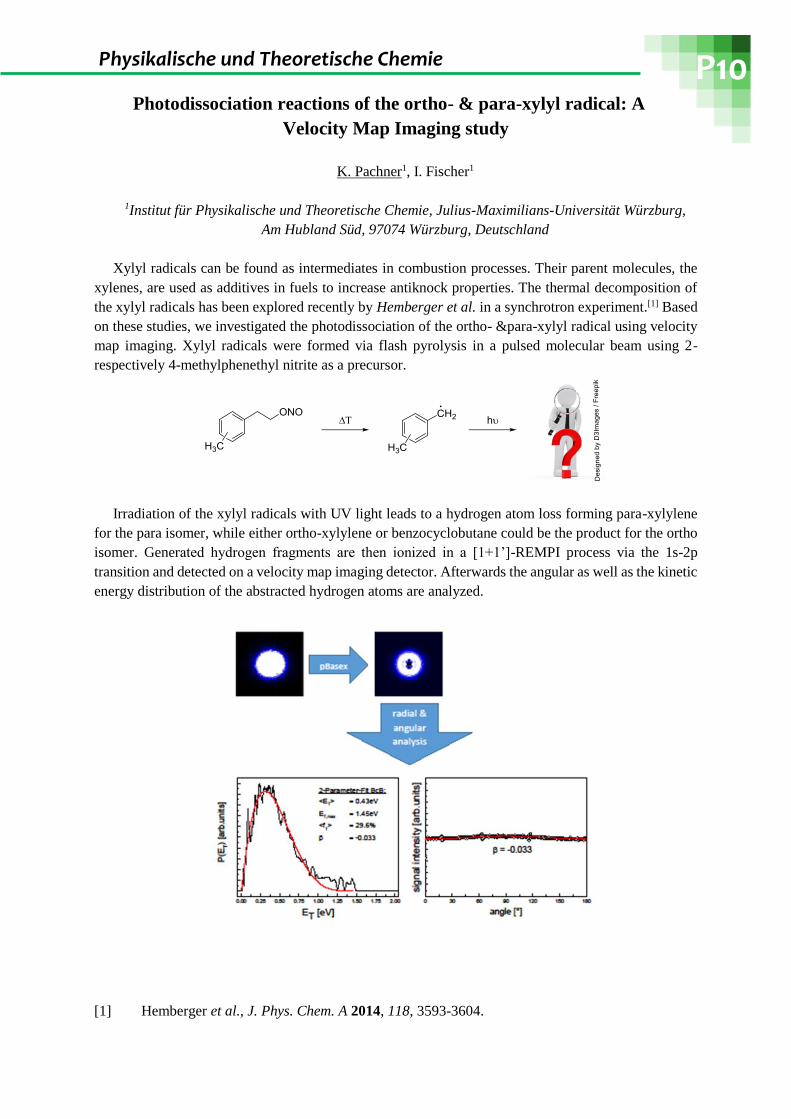
Irradiation of the xylyl radicals with UV light leads to a hydrogen atom loss forming para-xylylene
for the para isomer, while either ortho-xylylene or benzocyclobutane could be the product for the ortho
isomer. Generated hydrogen fragments are then ionized in a [1+1’]-REMPI process via the 1s-2p
transition and detected on a velocity map imaging detector. Afterwards the angular as well as the kinetic
energy distribution of the abstracted hydrogen atoms are analyzed.
[1] Hemberger et al., J. Phys. Chem. A 2014, 118, 3593-3604.

Organische Chemie und Biochemie P11 Star-shaped oligo(phenylenevinylene) mesogens for segregated
nanomaterials
M. Dechant, M. Hügel, M. Lehmann
Institut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg
Star-shaped oligo(p-phenylenevinylene) compounds are nonconventional shape-persistent
mesogens, which form liquid crystalline mesophases despite the large void space between their arms
(Figure 1).[1]
The four-armed phthalocyanine stars with terminal oligoethyleneoxy chains exhibit hexagonal
columnar LC-phases over broad temperature ranges by nanosegregation of the rigid core and the long
lateral chains. Phthalocyanines are highly interesting substances for the application in organic
photovoltaic cells due to their strong absorption in the red- and infrared range and their flat and broad
π-system, which allows efficient π-stacking along columnar assemblies.[2,3]
Analogous to previous work[4] fullerenes are covalently linked via short spacers to the stilbene arms.
This design should result in the segregation of fullerene and phthalocyanine/stilbene scaffolds and
consequently in quadruple helical columnar self-assemblies. The highly ordered, liquid-crystalline
structures are investigated by means of polarized optical microscopy, differential scanning calorimetry
and comprehensive X-ray scattering.
[1] M. Lehmann, B. Schartel, M. Hennecke, H. Meier, Tetrahedron 1999, 55, 13377-13394.
[2] R. F. Enes, J.-J. Cid, A. Hausmann, O. Trukhina, A. Gouloumis, P. Vázquez, J. A. S. Cavaleiro,
A. C. Tomé, D. M. Guldi, T. Torres, Chemistry – A European Journal 2012, 18, 1727-1736.
[3] P. Apostol, J. Eccher, M. E. R. Dotto, C. B. Costa, T. Cazati, E. A. Hillard, H. Bock, I. H.
Bechtold, Physical Chemistry Chemical Physics 2015, 17, 32390-32397.
[4] M. Lehmann, M. Hügel, Angewandte Chemie International Edition 2015, 54, 4110-4114.
Free space 1
R = O(C2H
4O)
3C
2H
5
n = 0,1,2
X = H,

Anorganische Chemie und Materialwissenschaften
P12 NHC-stabilisierte Derivate des Tricarbonylnitrosylkobalts
K. Lubitz, U. Radius*
Institut für Anorganische Chemie der Julius-Maximilians-Universität Würzburg, Am Hubland,
97074 Würzburg, E-Mail: [email protected]
Durch die Umsetzung des Tricarbonylnitrosylkobalts [Co(CO)3(NO)] mit N-Heterozyklischen
Carbenen (NHC´s) konnte eine Vielzahl einfach und zweifach substituierter Komplexe des Typs
[Co(CO)2(NO)(NHC)] bzw. [Co(CO)(NO)(NHC)2] isoliert und charakterisiert werden (NHC: iPr2Im,
nPr2Im, Cy2Im, Me2Im, iPr2ImMe, Me2ImMe, MeiPr2Im, MetBu2Im; R2Im = 1,3-di-alkyl-imidazolin-2-
ylidene).[1] Die Verwendung sterisch anspruchsvoller NHC´s führte dabei ausschließlich zu der Bildung
der einfach substituierten Komplexe [Co(Dipp2Im)(CO)2(NO)], [Co(Mes2Im)(CO)2(NO)] sowie
[Co(MecAAC)(CO)2(NO)]. Für die Umsetzung mit tBu2Im wurde eine „abnormale“ Koordination über
das Rückgrat beobachtet und der Komplex [Co(tBu2aIm)(CO)2(NO)] erhalten. Eine Untersuchung der
thermischen Eigenschaften ergab, dass sich die Komplexe leicht in die Gasphase überführen lassen,
sublimierbar sind und sich erst bei hohen Temperaturen zersetzen. Somit erfüllen sie die thermischen
Ansprüche, um zum Beispiel in der chemischen Gasphasenabscheidung Anwendung zu finden.[2,3]
[1] F. Hering, J. H. J. Bertel, Organometallics 2016, 35, 2806-2821.
[2] K. Gao, N. Yoshikai, Acc. Chem. Res. 2014, 47, 1208-1219.
[3] M. Leskalä, M. Ritala, Thin Solid Films 2002, 409, 138-146.

Anorganische Chemie und Materialwissenschaften P13
Copper(I)-catalyzed Suzuki-Miyaura type cross-coupling reactions
A. Eichhorn, U. Radius*, T. B. Marder*
Prof. Dr. Todd B. Marder, Chair of Inorganic Chemistry, Institut für Anorganische Chemie,
Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg
Email: [email protected]
We recently reported efficient systems for the borylation of arylhalides and alkylhalides using
copper(I) and phosphines as ligands.[1,2] In addition, we are interested in developing other Cu-catalyzed
B-C and C-C coupling processes, such as Suzuki-Miyaura type reactions.[3,4]
Proposed catalytic cycle for Cu(I) Discrete steps in Cu-catalyzed C-B and
catalyzed borylation. C-C coupling processes.
In order to obtain insight into reaction mechanisms and rates, we have begun a study of stoichiometric
reactions modelling discrete steps in likely catalytic pathways. To do this, we have prepared and
characterized a wide variety of (NHC)-Cu(I)-Y complexes wherein Y = halides, alkoxide, acetate, alkyl,
aryl and boryl. The synthesis and preliminary reactivity studies of such Cu(I) complexes will be
presented.
[1] C. Kleeberg, L. Dang, Z. Lin, T. B. Marder, Angew. Chem. Int. Ed. 2009, 48, 5350-5354.
[2] C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P.
G. Steel, T. B. Marder, L. Liu, Angew. Chem. Int. Ed. 2012, 51, 528-532.
[3] Y. Zhou, W. You, K. B. Smith, M. K. Brown, Angew. Chem. Int. Ed. 2014, 53, 3475-3479.
[4] S. K. Gurung, S. Thapa, A. Kafle, D. A. Dickie, R. Giri, Org. Lett. 2014, 16, 1264-1267.

Organische Chemie und Biochemie
P14 Living Supramolecular Polymerization of Dye Aggregates with
H- and J-Type Exciton Coupling
W. Wagner, M. Wehner, O. Soichiro, V. Stepanenko, F. Würthner*
Universität Würzburg, Center for Nanosystems Chemistry, Institut für Organische Chemie and
Bavarian Polymer Institute, Universität Würzburg, 97074 Würzburg, Germany
*e-mail: [email protected]
Supramolecular polymerization of π–conjugated has recently attracted attention in material science
as a concept to create well-defined architectures with controlled properties and functions.[1] Although
the thermodynamics of supramolecular polymerizations was studied in detail and analyzed by
mathematical models,the knowledge about the kinetics of aggregation processes is still not well-known.
Remarkably, kinetic effects in supramolecular systems are essential, playing an important role in the
structure and functions of the self-assembled aggregates. One strategy to control the kinetics of an
aggregation process is the seeded polymerization concept, which consists of the induction of
supramolecular polymerization of kinetically trapped building blocks by addition of seeds (small
aggregates acting as nuclei).[2]
Figure 1: Visualization of the concept of seeded polymerization and molecular structure of MeO-PBI.
In our previous studies we have successfully applied the concept of seeded supramolecular
polymerization in perylene bisimide (PBI) dye aggregates.[3] In the present work, we demonstrate that
this approach can be also employed to transform kinetically trapped non-fluorescent H-aggregates of a
dimethoxy-substituted PBI dye (MeO-PBI) into highly emissive J-aggregates by an innovative living
supramolecular polymerization mechanism. This is an unprecedented approach to control the kinetics
of supramolecular polymerization and to obtain out-of-equilibrium polymers with low polydispersity,
narrow size distribution and photofunctional properties.
[1] (a) T. Aida, E. W. Meijer, S. I. Stupp, Science 2012, 335, 813. (b) D. van der Zwaag, T. F. A.
de Greef, E. W. Meijer, Angew. Chem. Int. Ed. 2015, 54, 8334.
[2] S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu, M. Takeuchi, Nat. Chem. 2014, 6, 188.
[3] (a) S. Ogi, V. Stepanenko, J. Thein, F. Würthner, J. Am. Chem. Soc. 2016, 138, 670. (b) S. Ogi,
V. Stepanenko, K. Sugiyasu, M. Takeuchi, F. Würthner, J. Am. Chem. Soc. 2015, 137, 3300.

Anorganische Chemie und Materialwissenschaften P15
The Light at the End of the Cycle: Luminescent
Rhodacyclopentadienes and Rhodium 2,2 ՚ Biphenyl Complexes
C. Sieck1, M. Guan Tay2, M.-H. Thibault2, A. Steffen1,2, T. B. Marder1,2*
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg,
Am Hubland, 97074 Würzburg, Germany 2Department of Chemistry, Durham University, South Road, Durham DH1 3LE, UK
Reactions of [Rh(κ2-O,O-acac)(PMe3)2] (acac = acetylacetonato) and α,ω-bis-
(arylbutadiynyl)alkanes afford two isomeric types of MC4 metallacycles with very different
photophysical properties. As a result of a [2+2] reductive coupling at Rh, 2,5-
bis(arylethynyl)rhodacyclopentadienes (A) are formed, which display intense fluorescence (Φ
= 0.07-0.54, τ = 0.1-2.5 ns) despite the presence of the heavy metal atom. Rhodium biphenyl
complexes (B), which show exceptionally long-lived (hundreds of μs) phosphorescence (Φ =
0.02-0.29) at room temperature in solution, have been isolated as a second isomer originating
from an unusual [4+2] cycloaddition reaction and a subsequent β-H-shift. We attribute the
different photophysical properties of isomers A and B to a higher excited state density and a
less stabilized T1 state in the biphenyl complexes B, allowing for more efficient intersystem-
crossing S1→Tn and T1→S0. Control of the isomer distribution is achieved by modification of
the bis(diyne) linker length, providing a fundamentally new route to access photoactive metal
biphenyl compounds.[1]
[1] C. Sieck, M. G. Tay, M.-H. Thibault, R. M. Edkins, K. Costuas, J.-F. Halet, A. S. Batsanov, M.
Haehnel, K. Edkins, A. Lorbach, A. Steffen, T. B. Marder, Chem. Eur. J. 2016, 22, 1052.

Anorganische Chemie und Materialwissenschaften
P16 Eigenschaften und Reaktivität von zyklischen Alkylaminocarbenen
(CAACs) und deren Nickelcarbonylkomplexe
U. Paul, U. Radius*
Institut für Anorganische Chemie der Julius-Maximilians-Universität Würzburg
email: [email protected], [email protected]
Seit den bahnbrechenden Arbeiten von Arduengo et al. zur Synthese und Isolierung des stabilen 1,3-
Diadamantylimidazolin-2-ylidens[1] vor 25 Jahren hat die Chemie von N-Heterozyklischen Carbenen
(NHCs) enorme Beachtung erfahren, besonders durch deren Anwendung als Liganden in der
Übergangsmetallchemie.[2] Ihre Effizienz ist auf einen sterisch anspruchsvollen Aufbau und starke σ-
Donor-Eigenschaften zurückzuführen.[3] Durch Austausch eines der beiden elektronegativen
Aminosubstituenten in direkter Nachbarschaft zum Carbenkohlenstoffatom durch Alkylgruppen werden
sogenannte zyklische Alkylaminocarbene (CAACs) generiert. Diese sind folglich elektronenreicher und
somit nukleophiler als die zugrundeliegenden NHCs. Gleichzeitig resultiert dieser Austausch auch in
einer Absenkung des Carben π*-Orbitals, was zudem eine höhere Elektrophilie der CAACs bedingt.[4]
Im Fokus unserer Arbeiten standen CAAC-stabilisierte Nickelcarbonylkomplexe, welche primär
dazu dienen sollten, einen Einblick in elektronische und sterische Parameter, die diese Carbenklasse
auszeichnen, zu erlangen und sie mit denen der verwandten NHCs zu vergleichen.[5]
Desweiteren wollten wir die Reaktivität dieser neuartigen heteroleptischen Nickelcarbenkomplexe mit
Hauptaugenmerk auf CO-Substitution durch Neutralliganden und oxidativer Addition von Aryl- und
Allylhalogeniden untersuchen. Im Rahmen dieses Beitrags wollen wir die entsprechenden Ergebnisse
vorstellen und diskutieren.
[1] A. J. Arduengo III, R. L. Harlow, M. Kline, J. Am. Chem. Soc. 1991, 113, 361-363.
[2] a) P. de Frémont, N. Marion, S. P. Nolan, Coord. Chem. Rev. 2009, 253, 862-892; b) S. P. Nolan
(Ed.), N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley-VCH,
Weinheim, 2014.
[3] C. M. Crudden, D. P. Allen, Coord. Chem. Rev. 2004, 248, 2247-2273.
[4] a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu, G. Bertrand, Angew. Chem. Int. Ed. 2005,
44, 5705-5709; Angew. Chem. 2005, 117, 5851; b) M. Soleilhavoup, G. Bertrand, Acc. Chem.
Res. 2015, 48, 256-266.
[5] U. S. D. Paul, C. Sieck, M. Haehnel, K. Hammond, T. B. Marder, U. Radius, Chem. Eur. J.
2016, 22, 11005-11014.

Physikalische und Theoretische Chemie P17
Study of the self-reaction products of o-benzyne radicals
via IR/UV ion-dip-spectroscopy
F. Hirsch, P. Constantinidis, I. Fischer, A. M. Rijs
The self-reaction products of o-benzyne radicals produced by flash pyrolysis have been studied by ion-dip-
spectroscopy in a free jet. As recent studies suggest, these molecules might play an important role in the even
numbered growth of polycyclic aromatic hydrocarbons (PAH’s) and consequently soot, as they can serve as a
source of acetylene and diacetylene in combustion processes.
The spectroscopic method utilized in this study is capable of providing mass selective infrared spectra,
which can be used for unambiguous identification of the formed molecules. The radicals have been generated
by flash pyrolysis from the benzocyclobutendion precursor and ionized at fixed wavelengths at 265 and 275
nm. A tunable free electron laser provided infrared radiation in the range of 550-1750 cm-1. Subsequent
analysis of the differences in ion signals, with and without infrared excitation, resulted in the sought after
infrared spectra. Eventually comparison with theoretical and experimental data was performed for
identification of the various reaction products.
This poster will provide a fundamental overview of methodology and results of this study, executed at the
Free Electron Laser for Infrared eXperiments (FELIX) (Nijmegen, Netherlands).

Organische Chemie und Biochemie
P18
Defined Electron-Poor Nanographenes: One-Pot Synthesis and Single
Crystal Structure Analysis
S. Seifert, K. Shoyama, D. Bialas, D. Schmidt, F. Würthner*
Universität Würzburg, Institut für Organische Chemie and Center for Nanosystems Chemistry,
Am Hubland, 97074 Würzburg, Germany
*e-mail: [email protected]
Polycyclic aromatic hydrocarbons (PAHs) are an outstanding class of organic molecules that are
characterized by extended carbon-rich sp2-hybridized scaffolds with high thermal stability and interesting
electronic properties. Therefore, this type of compounds has attracted continuous interest during the last
decades as promising candidates for applications in organic electronics and photovoltaics. The synthesis of
electron rich PAHs is often realized by multistep procedures including C-C coupling reactions and oxidative
dehydrogenations[1] while the synthetic access to electron-poor systems is generally underdeveloped
presumably due to the instability of electron-deficient carbocations which are integral intermediates in
oxidative dehydrogenation reactions.
In our contribution we report the one-pot synthesis and full characterization of core extended pyrenes
bearing multiple dicarboximide substituents that can be regarded as electron-poor nanographenes.[2]
Figure 1: Solid-state molecular structure of an electron-poor nanographene (ellipsoids set at 50 % probability).
We efficiently combined palladium catalyzed Suzuki-Miyaura cross coupling and dehydrohalogenation to
synthesize large sized multiple imide containing chromophores with up to ten new C-C bonds formed in a
single reaction. The molecular structures of these π-extended systems have been elucidated by single-crystal
X-ray analysis confirming the formation of multiple C-C bonds and planar geometry of the scaffolds.
Moreover, the optical and electrochemical properties of these electron-poor PAHs have been characterized and
show their relevance for potential applications in (opto)electronic devices.
[1] a) A. Narita, X.-Y. Wang, X. Feng, K. Müllen, Chem. Soc. Rev. 2015, 44, 6616; b) M. Grzybowski,
K. Skonieczny, H. Butenschön, D. T. Gryko, Angew. Chem. Int. Ed. 2013, 52, 9900.
[2] a) S. Seifert, K. Shoyama, D. Schmidt, F. Würthner, Angew. Chem. Int. Ed. 2016, 55, 6390;
b) S. Seifert, D. Schmidt, F. Würthner, Org. Chem. Front. 2016, DOI: 10.1039/c6qo00421k.
Anorganische Chemie und Materialwissenschaften P19
Bismuth-based Luminescent Materials
J. R. Sorg, K. Müller-Buschbaum
Institut für Anorganische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg
This research projects aims at the synthesis and characterisation of new coordination polymers (CPs) and
metal-organic frameworks (MOFs) constructed from inorganic bismuth salts, such as bismuth halides, and
organic, multidentate, aromatic N-donor ligands. Various synthetic pathways have already been established
for the synthesis of such CPs, namely reactions in solution, in ligand- and salt-melts, under solvothermal
conditions, as well as mechanochemical reactions.
Bismuth-based CPs show extraordinary luminescence properties dominated by charge-transfer processes
between inorganic building units, e.g. bismuth/halide-units, and the π-system of the organic linkers.[1]
Additionally, Bi3+ and trivalent lanthanide cations Ln3+ prefer the same oxidation state +3 and exhibit similar
ionic radii, hence bismuth-based CPs are predestined as host-lattices for Ln3+ cations.[2] As lanthanides are
well-known emission centres in luminescent materials, these host/guest compounds promise applicability in
lighting and sensor technologies, as they combine the luminescence properties of the bismuth-based host-
lattices and the Ln3+ guests. Thus, colour-tunable emission by variation of the ratio of the different emission
centres becomes availbale.[3] Furthermore, these co-doped materials could be used as ratiometric sensors, in
which the emission of the host-lattice serves as internal standard to enable quantitative detection of small target
molecules.
The first examples of bismuth-based coordination polymers synthesised during this research project are the
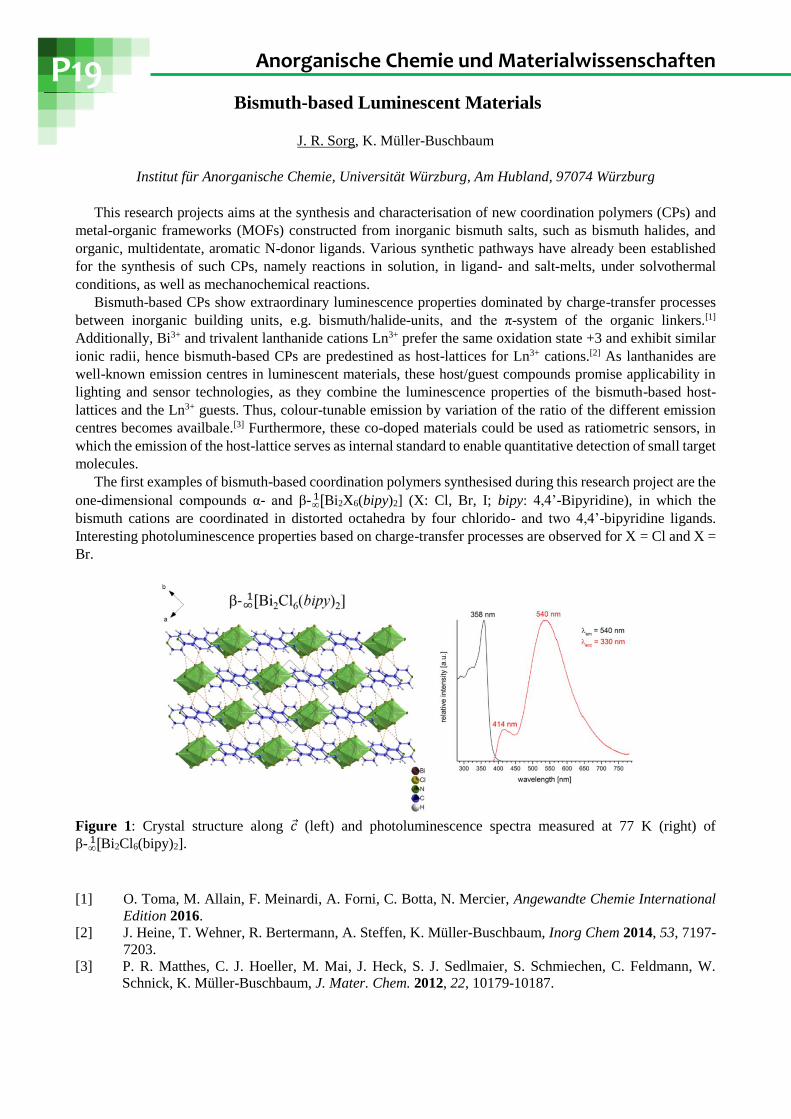
one-dimensional compounds α- and β- [∞1 Bi2X6(bipy)2] (X: Cl, Br, I; bipy: 4,4’-Bipyridine), in which the
bismuth cations are coordinated in distorted octahedra by four chlorido- and two 4,4’-bipyridine ligands.
Interesting photoluminescence properties based on charge-transfer processes are observed for X = Cl and X =
Br.
Figure 1: Crystal structure along 𝑐 (left) and photoluminescence spectra measured at 77 K (right) of
β- [∞1 Bi2Cl6(bipy)2].
[1] O. Toma, M. Allain, F. Meinardi, A. Forni, C. Botta, N. Mercier, Angewandte Chemie International
Edition 2016.
[2] J. Heine, T. Wehner, R. Bertermann, A. Steffen, K. Müller-Buschbaum, Inorg Chem 2014, 53, 7197-
7203.
[3] P. R. Matthes, C. J. Hoeller, M. Mai, J. Heck, S. J. Sedlmaier, S. Schmiechen, C. Feldmann, W.
Schnick, K. Müller-Buschbaum, J. Mater. Chem. 2012, 22, 10179-10187.

Pharmazie und Lebensmittelchemie
P20
Isotopically labelled mass tags as in vivo diagnostics
K. Dodt1, A. Schlosser2, J. Vanselow2, T. Lühmann1, L. Meinel1
1Institute of Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg,
Germany 2Rudolf-Virchow-Zentrum für Experimentelle Biomedizin, University of Würzburg,
Josef-Schneider- Str. 2, 97080 Würzburg, Germany
Profiling enzymatic activity of inflamed tissues is essential for patient stratification. In one scenario, each
patient is a priori categorized and directed to medicine one is going to respond to (personalized medicine). In
spite of this promise, proper tools for patient stratification within the context of inflamed joints (rheumatoid
arthritis - RA, osteoarthritis - OA) are yet to be found in the future. Therefore, we aim at developing protease-
cleavable linkers (PCL) conjugated to mass encoded peptides that are cleaved by upregulated matrix
metalloproteinases (MMP) in inflamed osteoarthritic joints, thereby reflecting the release of the drug in situ
(Figure 1).
Figure 1: Bioresponsive drug delivery system: protease-cleavable linkers are cleaved by up regulated MMP
and released mass tags are analysed by LC-MS/MS in body fluids.
To detect PCL cleavage by MMP activity in vivo, mass tags with isotopically labelled amino acids were
designed, allowing quantification by tandem mass spectrometry.[1] In order to create a suitable mass tag that
forms two major ion fragments, three amino acid sequences were synthesized by Fmoc based solid phase
peptide synthesis (SPPS). After RP-HPLC purification the mass tags were analysed by LC-MS/MS resulting
in the amino acid sequence SADGPGFR being the best fragmenting mass encoded peptide. Four different
PCLs and the respective mass tags are currently prepared by SPPS and are conjugated to a biopolymer by
conducting bio-orthogonal chemical reactions, such as copper-catalyzed azide–alkyne cycloaddition
(CuAAC).[2]
Future steps include the demonstration of the in vivo efficacy in relevant animal systems of RA and OA.
[1] G. A. Kwong, G. von Maltzahn, G. Murugappan, O. Abudayyeh, S. Mo, I. A. Papayannopoulos, D.
Y. Sverdlov, S. B. Liu, A. D. Warren, Y. Popov, D. Schuppan, S. N. Bhatia, Nat. Biotechnol. 2013,
31, 63.
[2] M. Gutmann, E. Memmel, A. C. Braun, J. Seibel, L. Meinel, T. Luhmann, Chembiochem 2016, 17,
866.
Mass
tag
Drug PCL Surfa
ce
MMP
Organische Chemie und Biochemie P21
Kinetic control of supramolecular polymerization by ultrasonication
M. Wehner, S. Ogi, W. Wagner, V. Stepanenko, F. Würthner*
Universität Würzburg, Center for Nanosystems Chemistry, Institut für Organische Chemie, and Bavarian
Polymer Institute, Universität Würzburg, 97074 Würzburg, Germany
*e-mail: [email protected]
Supramolecular polymerization has recently attracted particular attention for the development of well-
defined functional architectures.[1] While the thermodynamics of supramolecular polymerizations were studied
in detail by mathematical models, deep knowledge on the kinetics of aggregation processes is still lacking.
Perylene bisimide (PBI) dyes are suitable to study these phenomena since they possess favorable aggregation
behaviors and outstanding optical properties.[2] Recently, the kinetics of self-assembly processes of PBI
derivatives have been studied by seeded supramolecular polymerization.[3] In the present work we achieved
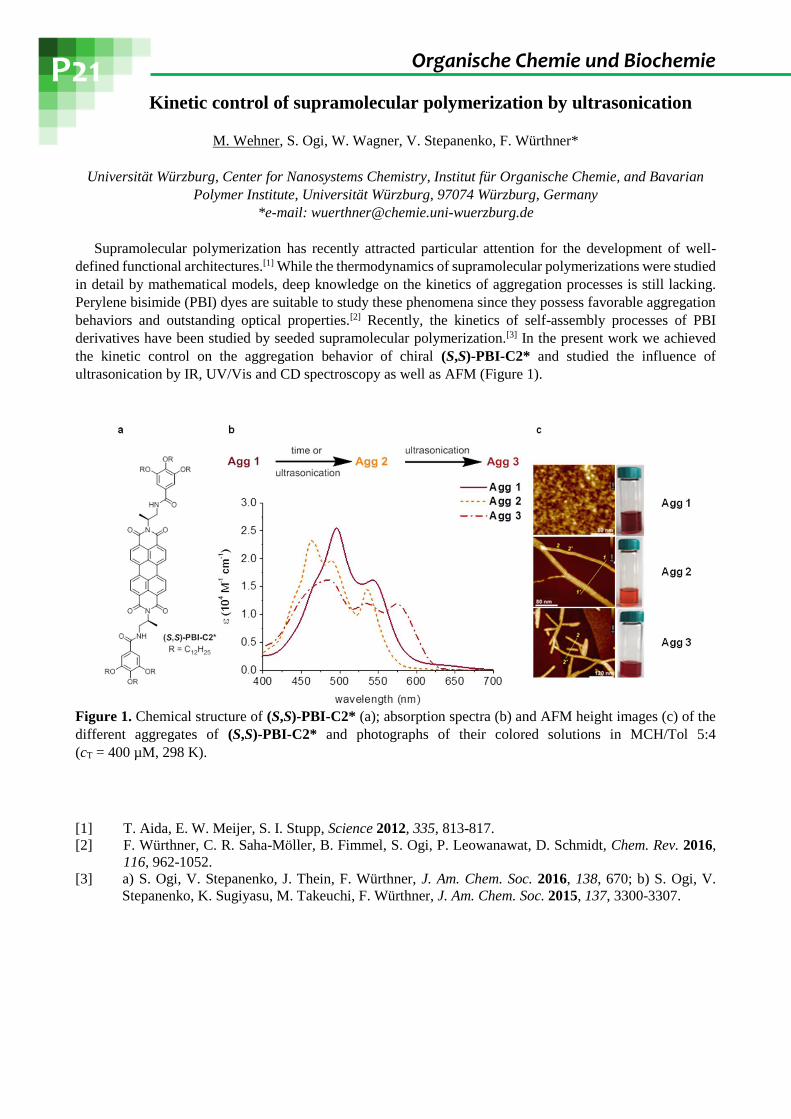
the kinetic control on the aggregation behavior of chiral (S,S)-PBI-C2* and studied the influence of
ultrasonication by IR, UV/Vis and CD spectroscopy as well as AFM (Figure 1).
Figure 1. Chemical structure of (S,S)-PBI-C2* (a); absorption spectra (b) and AFM height images (c) of the
different aggregates of (S,S)-PBI-C2* and photographs of their colored solutions in MCH/Tol 5:4
(cT = 400 µM, 298 K).
[1] T. Aida, E. W. Meijer, S. I. Stupp, Science 2012, 335, 813-817.
[2] F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat, D. Schmidt, Chem. Rev. 2016,
116, 962-1052.
[3] a) S. Ogi, V. Stepanenko, J. Thein, F. Würthner, J. Am. Chem. Soc. 2016, 138, 670; b) S. Ogi, V.
Stepanenko, K. Sugiyasu, M. Takeuchi, F. Würthner, J. Am. Chem. Soc. 2015, 137, 3300-3307.

Physikalische und Theoretische Chemie
P22
ps-zeitaufgelöste Photoionisation des S2(ππ*)-Zustands von Xanthon
M. Flock, H.-C. Schmitt, I. Fischer
Die Dynamik angeregter Zustände von Heterozyklen, die auch Carbonylgruppen enthalten, hängt stark von
der Konkurrenz der Desaktivierung über IC und ISC ab. Dies konnte bereits an den Molekülen 9-Fluorenon,[1]
NDCA[2] und Naphthalimid[3] gezeigt werden. Ein weiterer Vertreter dieser Substanzklasse ist Xanthon.
Mittels Photoinisations-Experimenten konnte die vibronische Struktur des S2-Zustands aufgelöst und die
Desaktivierung des Moleküls nach Anregung in diesen verfolgt werden. In einem gepulsten
Molekularstrahlexperiment wurden die Moleküle mit einem 325.5 nm Pump-Puls zunächst resonant in den S2-
Zustand angeregt. Anschließend erfolgte die Ionisation mit einem 351 nm Probe-Puls.
Zwei Zeitkonstanten sowie ein anschließender Offset wurden im Experiment beobachtet. Während die erste
Zeitkonstante unterhalb der zeitlichen Auflösung des Setups liegt, konnte für die zweite ein Wert von 24 ps
ermittelt werden. Der Offset kommt durch die Population eines im Rahmen der Zeitskala des Experiments
langlebigen Triplett-Zustands zustande, was in Einklang der literaturbekannten Phosphoreszenz steht.[4] Für
die Relaxation aus dem S2-Zustand können zwei verschiedene Modelle in Betracht gezogen werden. Es kann
zum einen zunächst ein IC-Übergang in den S1-Zustand und anschließend ein ISC-Prozess in die Triplett-
Umgebung stattfinden (Modell A). Andererseits kann es auch zuerst zu einem ultraschnellen ISC-Prozess in
die Triplett-Umgebung, gefolgt von anschließender Interner Konversion (IC) innerhalb dieser, kommen
(Modell B). Um dies aufklären zu können, sollen in Zukunft zeitaufgelöste Photoelektronen-Imaging-
Experimente durchgeführt werden.
[1] T. Gerbich, J. Herterich, J. Köhler, I. Fischer, The Journal of Physical Chemistry A 2014, 118, 1397-
1402.
[2] T. Gerbich, H.-C. Schmitt, I. Fischer, J. Petersen, J. Albert, R. Mitrić, The Journal of Physical
Chemistry A 2015, 119, 6006-6016.
[3] T. Gerbich, H.-C. Schmitt, I. Fischer, R. Mitrić, J. Petersen, The Journal of Physical Chemistry A 2016,
120, 2089-2095.
[4] H. J. Pownall, J. R. Huber, Journal of the American Chemical Society 1971, 93, 6429-6436.

Organische Chemie und Biochemie P23
MOLECULAR DESIGN OF METALLOSUPRAMOLECULAR
RUTHENIUM CATALYSTS FOR WATER OXIDATION
A. L. Meza, D. Schindler, V. Kunz, M. Schulze, F. Würthner
Universität Würzburg, Institut für Organische Chemie, Am Hubland, 97074 Würzburg, Germany
The development of new sustainable energy sources constitutes one of the most important challenges of
our generation. In this regard, the light-induced splitting of water into its elements has attracted particular
attention as both sunlight and water are widely available and inexpensive resources. The goal is to achieve a
cycle of clean energy production by using the elemental hydrogen obtained from the catalytic splitting of water
as an ecologically friendly fuel.
Currently, our research has focused on the half reaction of the oxidation of water to oxygen which is
considered to be the bottleneck of the entire water splitting process. So far we have been able to synthesize a
robust metallosupramolecular ruthenium catalyst, which has an activity comparable to that of the oxygen
evolving complex (OEC) of the photosystem II. Based on our mechanistic investigations on the catalytic water
oxidation our next goal is to introduce different substituents into the catalyst’s scaffold to tune its electronic
properties to obtain even higher catalytic activities.
Here we present the synthesis and characterization of novel molecularly designed metallo-supramolecular
ruthenium catalysts that are equipped with electron withdrawing or donating substituents. Investigations
regarding their activity toward chemical and light-induced water oxidation are currently being performed in
our laboratories.
[1] V. Balzani, A. Credi, M. Venturi, Chem. Sus. Chem. 2008, 1, 26-58.
[2] P. D. Frischmann, K. Mahata, F. Würthner, Chem. Soc. Rev. 2013, 42, 1847-1870.
[3] M. Schulze, V. Kunz, P. D. Frischmann, F. Würthner, Nat. Chem. 2016, 8, 576-583.

Organische Chemie und Biochemie
P24
Synthesis of a novel Donor – Sensitizer – Acceptor Triad for the
Investigation of Magnetic Field Effects
D. Mims, C. Schwarz, C. Lambert
Triads with small B1/2,hfc values, which are about one magnitude larger than the earth’s magnetic field have
already been investigated by Lambert et. al.[1] These triads comprise of triarylamine donors containing a
nitrogen atom with a nuclear spin of I = 1. Since the magnetic field effect is dependent on the nuclear magnetic
moment of the donor- and acceptor moiety, a substitution of these donors with the sulfur (I = 0) bearing
building block tetrathiafulvalene should lead to a charge separated state with a decreased effective magnetic
moment and thus should give rise to triads that are sensitive to the earth’s magnetic field.
Yet the synthesis of the desired complex proves to be difficult due to the electronrich tetrathiafulvalene
building block. Hence detailed information on planed and already realized synthesis is given in this study.
Figure 1: After absorption of visible light a charge separated state occurs. The triplet splitting of the cs-state
and its kinetics are dependent on the external magnetic field.[1]
[1] J. H. Klein, D. Schmidt, U. E. Steiner, C. Lambert, J. Am. Chem. Soc. 2015, 137, 11011-11021.

Organische Chemie und Biochemie P25
Functionalised precursors for extended π-systems with a
Tribenzotriquinacene core
R. Buschmann, A. Krueger
Institut für Organische Chemie, Am Hubland, 97074 Würzburg
Tribenzotriquinacene (TBTQ, 1) is a bowl-shaped aromatic hydrocarbon, whose curved structure derives
from the three fused five-membered rings at its centre. Theoretical studies have indicated that TBTQ can be
considered as a defect centre in a distorted nanographene.[1] The aim is to functionalise 1 by bridging the bay
regions (indicated in bold below) and thereby extend the carbon network to give PAH 2.
TBTQ synthesis can be achieved either using the Kuck synthesis pathway[2] or the triple cyclisation method
from Hopfet al.,[3] which allows access to ortho-functionalised derivatives, by using suitably substituted
starting materials. Examples of ortho-substituted TBTQ derivatives are limited,[4] due to increased steric strain
experienced at this position. TBTQs 3, 4 and 5 are synthetic aims of this work and progress made towards their
realisation will be presented and discussed.
[1] J. Tellenbröker, D. Kuck, Angew. Chem. Int. Ed. 1999, 38, 919.
[2] D. Kuck, Angew. Chem. Int. Ed. Engl. 1984, 23, 508.
[3] H. Hopf, G. Markopoulos, L. Henneicke, J. Shen, Y. Okamoto, P. G. Jones, Angew. Chem. Int. Ed.
2012, 51, 12884.
[4] Y. Kirchwehm, A. Damme, T. Kupfer, H. Braunschweig, A. Krueger, Chem. Comm. 2012, 48, 1502.

Physikalische und Theoretische Chemie
P26
Excitons and Polarons in Motion: Understanding Charge Generation
Pathways at the Interfaces of Organic Solar Cells
C. Brückner, B. Engels
Institut für Physikalische und Theoretische Chemie, Universität Würzburg, Germany
e-mail: [email protected]
In view of the need to exploit alternative energy resources, organic solar cells based on small organic
molecules were designed. While fullerene and derivatives are often employed as the n-type semiconductor,
the structural diversity of p-type semiconductors is larger.
For a selection of p-type molecular semiconductors in combination with fullerene C60, we generated model
structures of the organic-organic interfaces in organic solar cells using MM (molecular mechanics) techniques.
Careful benchmarking (DFT, semiempiric and wave-function based methods) was performed in order to
find quantum-mechanical methods that are both efficient and correct (to a reasonable extent) for the prediction
of geometric and electronic properties of the p-type semiconductors.
Energetic profiles of the bilayer interfaces were calculated using a dimer approach, electric fields and an
effective epsilon. The calculation of both charged and excited transport bands yields polaronic and excitonic
states in the vicinity of the organic-organic interface. From the energetic profiles of the interfaces, it can be
concluded that particularly interfacial trap states, i.e., geminate electron-hole pairs which are significantly
bound due to weakly shielded charge-charge interactions, could limit device efficiencies.
In order to model the charge generation route in organic solar cells, kinetic Monte Carlo simulations were
conducted. Resulting charge separation efficiencies highlighted the critical role of slow charge transport
processes around the organic-organic interface.

Pharmazie und Lebensmittelchemie P27
Personalisierte Dosisfindung in der Psychiatrie
A. Pospiech*1, M. Zilker*1, J. Scheiber2, U. Holzgrabe1, P. Högger1
*zu gleichen Teilen beigetragen
1 Institut für Pharmazie und Lebensmittelchemie, Universität Würzburg
2 BioVariance GmbH, Waldsassen
Einleitung
Die patientenindividuelle pharmakogenetische Ausstattung bestimmt die Pharmakokinetik von in der
Psychiatrie eingesetzten Arzneistoffen, weshalb Dosisanpassungen und Anwendungsbeschränkungen in der
psychiatrischen Praxis an der Tagesordnung sind. Das Ziel ist die Entwicklung eines Algorithmus, der
basierend auf Literaturdaten und einer experimentell angelegten Studie eine zuverlässige Aussage zur
Dosisfindung von Psychopharmaka liefert und für die Umsetzung einer Smartphone App geeignet ist.
Methoden
Zuerst wurde für die Arzneistoffklasse der trizyklischen Antidepressiva eine umfassende Datenerhebung
durchgeführt, da deren Metabolismus hochgradig von Polymorphismen beeinflusst wird. Parallel dazu wird
eine Genotypisierungsstudie durch das Labor für Therapeutisches Drug Monitoring des Zentrums für
psychische Gesundheit am Universitätsklinikum Würzburg organisiert. Die Literatursuche erfolgt nach einem
Rechercheleitfaden, der eine umfassende und systematische Datensammlung und –auswertung ermöglicht.
Ergebnisse
Für die trizyklischen Antidepressiva Amitriptylin, Nortriptylin, Clomipramin, Doxepin, Imipramin,
Desipramin und Trimipramin wurden detaillierte Informationen zu Polymorphismen relevanter CYP-Enzyme,
wie z.B. CYP 2D6 und CYP 2C19, Daten zur Pharmakokinetik sowie Häufigkeiten unerwünschter
Arzneimittelwirkungen in Abhängigkeit von CYP-Genvarianten gesammelt. Jede Quelle wurde mit Hilfe eines
Scoring Systems hinsichtlich ihrer Methodik und analytischen Qualität geprüft bevor die Daten aufbereitet
und strukturiert in die Datenbank eingetragen wurden.
Fazit
Am Ende soll ein Algorithmus etabliert werden, um eine intuitiv leicht bedienbare Datenbank zu
entwickeln, die alle Antidepressiva und Antipsychotika enthält. Dabei soll die Suche nach der optimalen
Dosierung basierend auf individuellen genetischen Daten ermöglicht und dadurch die Therapiesicherheit
erhöht werden.

Anorganische Chemie und Materialwissenschaften
P28
Conjugated Triarylborane Dendrimers
F. Rauch1, L. Ji1, T. B. Marder1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Würzburg, Germany
email: [email protected]
Triarylboranes are interesting acceptor moieties in the synthesis of π-conjugated materials and find
application in OLEDs, nonlinear optical materials and anion sensors as well as two-photon absorption (TPA)
materials.[1] Conjugated dendrimers also show promising properties for applications in OLEDs, TPA materials
and photovoltaics.[2]
Herein, we report the development of a new reaction sequence by which triarylboranes can be synthesized
using bench stable potassium trifluoroborate salts as precursors. Furthermore, we applied this reaction
sequence in the synthesis of a novel first generation borane dendrimer which exhibits a considerable amount
of conjugation throughout the π-system.
[1] For reviews see:
a) C. D. Entwistle, T. B. Marder, Angew. Chem. Int. Ed. 2002, 41, 2927-2931;
b) C. D. Entwistle, T. B. Marder, Chem. Mater. 2004, 16, 4574-4585.
[2] For reviews see:
a) D. Astruc, C. Ornelas, J. Ruiz, Acc. Chem. Res. 2008, 41, 841-856;
b) R. Hourani, A. Kakkar, Macromol. Rapid Commun. 2010, 31, 947-974.

Organische Chemie und Biochemie P29 Absolute Stereostructures and Possible Biosynthetic Origins of the
Novel Cage-like Naphthylisoquinoline Alkaloid Dimers
Cyclo-Mbandakamines A1 and A2
B. K. Lombe1,2, T. Bruhn1, D. Feineis1, V. Mudogo2, G. Bringmann1
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany 2Faculté des Sciences, Université de Kinshasa, B.P. 202, Kinshasa XI, Democratic Republic of the Congo
Naphthylisoquinoline alkaloids constitute a unique class of natural products, found so far only in the
Ancistrocladaceae and Dioncophyllaceae plant families.[1] They consist of two polyketide-derived molecular
portions, a naphthalene and an isoquinoline moiety, connected by a biaryl axis, which is, in most cases,
rotationally hindered. Besides this element of axial chirality, these alkaloids carry one or two stereogenic
centers in the isoquinoline part, hence making them stereochemically thrilling.[1] Even more stimulating are
the dimeric naphthylisoquinoline alkaloids, since some of them, depending on the individual structures, have
shown pronounced bioactivities[2,3] and a total number of seven stereogenic elements.[3,4]
From the leaves of a botanically yet undescribed Congolese Ancistrocladus species,[5] we have discovered
the very first oxygen-bridged naphthylisoquinoline alkaloids dimers, named cyclo-mbandakamines A1 (1) and
A2 (2). Their cage-like structures, displaying an unprecedented dihydrofuran-cyclohexenone-pyrane sequence,
reveal eight elements of chirality – the highest total number for all naphthyl isoquinoline alkaloids known so
far. In addition, one of their C,O-bonds (highlighted in yellow in the figure), arises presumably from a phenol
oxidation, thus making them the first representatives to result from four phenol-oxidative steps (three C,C- and
one C,O-bonds highlighted in the figure).
Our poster will provide more details on the structural elucidation of 1 and 2 and will also present the
proposed biosynthetic pathway leading to these unprecedented quateraryls.
[1] G. Bringmann, F. Pokorny, In The Alkaloids (Cordell, G. A., Ed.) 1995, 46, 127-271.
[2] M. R. Boyd et al., J. Med. Chem. 1991, 34, 3402-3405.
[3] G. Bringmann et al., Chem. Eur. J. 2013, 19, 916-923.
[4] G. Bringmann et al., Org. Lett. 2013, 15, 2590-2594.
[5] F. Turini et al., Taxon 2014, 63, 329-341.

Anorganische Chemie und Materialwissenschaften
P30
Reversible Oxidative Addition of Highly Polar Bonds to a
Transition Metal
J. Müssig, H. Braunschweig*
email: [email protected]
Department of Inorganic Chemistry, Julius-MaximiliansUniversitaetWuerzburg, 97074 Wuerzburg,
Germany
The reactivity of group 13 halides towards the well established metal Lewis base [Pt(PCy3)2] has
been studied in our group in the past years. We found that AlX3 (X = Cl, Br, I) and GaCl3 form
unsupported metal-only Lewis pairs (MOLPs) with [Pt(PCy3)2], whereas the reaction with BX3 (X =
Cl, Br, I), GaBr3 and GaI3 results in oxidative addition (OA) products: trans-(halo)(gally) and trans-
(halo)(boryl) complexes.[1-4] In the case of gallium halides the substitution of the halide plays an
important role to vary the reaction pathway. To gain better understanding of dative bonds between two
metals our current investigations are concentrated on the reactivity of InX3 (X = Cl, Br, I) towards
platinum(0) Lewis bases. The indium-platinum systems show well-defined equilibrium mixtures
between the products of oxidative additions of the highly polar In-X bonds (∆EN = 0.88-1.38) and their
MOLP counterparts (reductive elimination products).The results mark the first observation of an
equilibrium between MOLPs and OA isomers, as well as the most polar bond ever observed to undergo
reversible oxidative addition to a metal complex.
[1] H. Braunschweig, K. Gruss, K. Radacki, Angew. Chem. Int. Ed. 2007, 46, 7782-7784.
[2] H. Braunschweig, K. Gruss, K. Radacki, Inorg. Chem. 2008, 47, 8595-8597.
[3] H. Braunschweig, P. Brenner, A. Müller, K. Radacki, D. Rais, K. Uttinger, Chem. Eur. J. 2007, 13,
7171-7176.
[4] H. Braunschweig, K. Radacki, K. Uttinger, Inorganic Chemistry 2007, 46, 8796-8800.

Organische Chemie und Biochemie P31
Dispersability of Nanodiamonds in Physiological Media
S. Schweeberg1, S. Suliman2, M. Popa3, K. Mustafa2, A. Krueger1
1Institut für Organische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland,
97074 Würzburg 2Department of Clinical Dentistry, Center for Clinical Dental Research, University of Bergen, Norway
3KinN Therapeutics, Bergen, Norway
Nanotechnology is gaining increasing interest for a wide community of medical, chemical and physical
scientists.[1] Nanodiamond in particular has unique properties such as inertness, chemical reactivity if surface
functionalization is involved, hardness and biocompatibility.[2]
Nanodiamond particles tend to agglomerate in solutions with a high salt concentration so that physiological
media like PBS buffer are not suitable for the preparation of nanodiamond for biomedical applications.
Figure 1: Nanodiamond without dispersion and with dispersion in glucose solution.
Physiological glucose solution was used as an alternative for the preparation of stable, fully dispersed
colloids, and even after 20 days at room temperature the particles do not agglomerate, which is a very positive
indication of its suitability as a suitable, fully dispersed nanodiamond formulation.
With the nanodiamond solution in water, agglomeration takes places immediately just as the proteins of the
serum get in contact with the particles (figure 1). Due to the strong precipitation and agglomeration DLS
measurements were not successful. In glucose solution, no agglomeration takes place upon adding serum to
the nanodiamond solution.
Funding by Volkswagenstiftung (grant number: 88393) is gratefully acknowledged.
[1] V. N. Mochalin, Y. Gogotsi, Nat Nano 2012, 7, 11-23.
[2] A. M. Schrand, L. Dai, J. Phys. Chem. B 2007, 111, 2-7.

Organische Chemie und Biochemie
P32
A Supramolecular Cage linked by Boron Nitrogen Dative Bond and
Synthesis of a New Apically Functionalized Tribenzotriquinacene
Building Block
A. Dhara, F. Beuerle*
Universität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry, Am Hubland,
97074 Würzburg, Germany
*E-mail: [email protected]
Precise control of supramolecular self-assembly processes is fundamental requirement to construct a well-
defined architecture from a set of molecular building blocks by utilizing non-covalent interactions. Although
hydrogen bond, metal coordination and hydrophobic interactions are well-explored to form a number of
supramolecular structures[1] the bond strengths of these interactions are not so easy to tune systematically to
have the desired thermodynamic and kinetic stabilities of the self-assembled systems. In this respect BN
dative bonds which are formed by Lewis acid/base interactions between electrophilic boron center and nitrogen
nucleophiles can act as a suitable motif for supramolecular self-assembly.[2] Here we present a bipyramidal
[2+3] assembly formed by BN dative bond.[3] Thermodynamic equilibria of cage formation were studied by
isothermal titration calorimetry (ITC) and reversible cage opening/reassembly was investigated by variable
temperature 1H-NMR spectroscopy.
Also we report on the facile synthesis of a new tribenzotriquinacene (TBTQ) molecule possessing a
terminal alkyne group at the apical position which can be modified by the azide–alkyne Huisgen cyclo addition
and Sonogashira cross-coupling reaction to attach different functional groups as needed.[4] This can lead to
facile exohedral functionalization of organic cages formed by TBTQ building units. Also we describe a mild
and practically useful protocol to deprotect methoxy- groups in presence of terminal alkyne.
[1] M. Yoshizawa, J. K. Klosterman, M. Fujita, Angew. Chem. Int. Ed. 2009, 48, 3418-3438.
[2] K. Severin, Dalton Trans. 2009, 5254-5264.
[3] A. Dhara, F. Beuerle, Chem. Eur. J. 2015, 21, 17391-17396.
[4] A. Dhara, J. Weinmann, A-M. Krause, F. Beuerle, Chem. Eur. J. 2016, 22, 12473-12478.

Pharmazie und Lebensmittelchemie P33
Site-directed conjugation and bioresponsive delivery of IGF-I
A. Braun1, M. Gutmann1, R. Ebert2, F. Jakob2, T. Lühmann1, L. Meinel.1
1Institute for Pharmacy and Food Chemistry, University of Würzburg, Germany 2Orthopedic Center for Musculoskeletal Research, Würzburg, Germany
Human Insulin-like Growth Factor-I (IGF-I) is a 7.6 kDa peptide hormone with an anabolic function e.g.
in muscle growth and regeneration, therefore discussed as a potential treatment option for muscular atrophy.[1]
In this study we aimed at developing IGF-I delivery systems with superior safe and efficacy profiles by
directing the hormone’s release to inflamed muscle tissue and minimizing off-target activity.[1,2]
To this end, IGF-I was conjugated at position 68 to a protease-cleavable linker (PCL) responding to patho-
physiologically elevated matrix metalloproteinase (MMP)-9 as representative for the inflamed muscle.[3] In
the next step, this PCL-IGF-I conjugate was site-specifically coupled to DBCO-reactive PEG10kDa and
characterized before and after exposition to MMPs.[4] Characterization of the coupled IGF-I variants was
performed by MALDI-MS, HPLC and tricine gel electrophoresis. Bioactivity was determined by MG-63 cell
proliferation in comparison to wild type IGF-I.
Scheme 1: Principle of bioresponsive IGF-I release triggered by MMP-9 secretion of diseased tissue.
Site-directed conjugation of the PCL to IGF-I as well as response to MMP-9 was confirmed by HPLC and
MALDI-MS analysis. The PCL-IGF-I conjugate demonstrated comparable bioactivity as the wild type
analogue. The conjugate was successfully immobilized onto DBCO-functionalized agarose particles using
SPAAC click chemistry and labelled with monoclonal IGF-I antibody and Alexa 488 conjugated secondary
antibody. The soluble conjugate with PEG10kDa also responded to MMP-9 and is now profiled for systemic
delivery, thereby featuring site-directed conjugation of IGF-I to biomaterials as a promising approach for an
improved safety profile of anabolic growth factor delivery targeting muscle regeneration.
Acknowledgments: The financial support from the Bavarian Research Foundation (grant # AZ-1044-12
‘FORMOsA’) is gratefully acknowledged.
[1] I. Schultz, J. Wurzel, L. Meinel, European Journal of Pharmaceutics and Biopharmaceutics 2015, 97,
Part B, 329-337.
[2] T. Lühmann, L. Meinel, Current Opinion in Biotechnology 2016, 39, 35-40.
[3] A. C. Braun, M. Gutmann, R. Ebert, F. Jakob, H. Gieseler, T. Lühmann, L. Meinel, Pharmaceutical
Research 2016, 1-15.
[4] O. Germershaus, T. Lühmann, J. C. Rybak, J. Ritzer, L. Meinel, International Materials Reviews 2015,
60, 101-131.

Pharmazie und Lebensmittelchemie
P34
Site-directed functionalization of cell derived matrices by metabolic
glycol-engineering and click chemistry
M. Gutmann1, A. Braun1, E. Memmel2, J. Seibel2, L. Meinel1, T. Lühmann1
1Institute for Pharmacy and Food Chemistry, University of Würzburg, Germany
²Institute for Organic Chemistry,University of Würzburg, Germany
The extracellular matrix (ECM) is a complex and 3D-network that is secreted by various cell types. The ECM has
different essential roles in regulating the function, development and homeostasis of eukaryotic cells. It provides
mechanical support, regulates the abundance of signaling molecules (e.g. growth factors) and receptors as well as pH and
hydration status. Cell derived matrices (CDM) have been recently attracted attention as biocompatible scaffold material
for skeletal tissue engineering and cardiovascular/vascular tissue engineering.[1]
This study aims at modification of glyco-engineered ECM scaffolds derived from fibroblasts (NIH3T3) by site
directed chemistry deploying bioorthogonal azide-alkyne cycloadditions (CuAAC/SPAAC). For the synthesis of
glycoengineered CDMs, NIH3T3 fibroblasts were incubated with N-azido acetyl glucosamine[2] and stimulated with 50
µg/mL ascorbic acid and decellularized as previously described.[3,4] To analyze the presentation of the azide-modified
glycoproteins embedded in the CDM, we applied alkyne-azide click reactions using opposed fluorescent dyes (Sulfo-
Cy5-alkyne; DBCO-Sulfo-Cy5) in line with an anti-fibronectin antibody and a fluorescent-labeled second antibody for
ECM structure visualization. Site-directed immobilization of the fluorophor was investigated by confocal laser scanning
microscopy (CLSM) after different time points, displaying ECM like structures, which colocalized with the fibronectin
counterstaining (Figure 1).
Figure 1: Isolated modified ECM after 9 days. (A) Azido-sugar treated ECM stained with Sulfo-Cy5-Alkyne (1) and
Anti-Fibronectin-Alexa 488 (2). (B) Untreated ECM stained with Sulfo-Cy5-Alkyne (1) and Anti-Fibronectin-Alexa 488.
Ongoing experiments focus on the modification of glycoengineered material with respect to protein modification[5]
and on the characterization of the cell-derived material by western blotting procedures.
We successfully developed protocols enabling both CDM formation and the integration of functionalizable sugar-
moieties by metabolic glycolengineering. Engineering CDMs with functional cues is a promising strategy for scaffold
design allowing for high decoration versatility in tissue engineering applications.
[1] L. E. Fitzpatrick et al., Biomater. Sci. 2015, 3 12-24.
[2] E. Memmel et al., Chemical communications 2013, 49, 7301-7303.
[3] M. Gutmann et al., Chembiochem : a European journal of chemical biology 2016, 17, 866-875.
[4] R. Castello-Cros et al., Methods in molecular biology 2009, 522, 275-305.
[5] G. Wandrey et al., Journal of biological engineering 2016, 10, 11.

Organische Chemie und Biochemie P35
PBI-Cyclophane – Chromophormakrozyklen für die
Wirt/Gast-Chemie
M. Sapotta, A. Sieblist, P. Spenst, F. Würthner*
Universität Würzburg, Center for Nanosystems Chemistry, Institut für Organische Chemie,
Universität Würzburg, 97074 Würzburg, Germany
*e-mail: [email protected]
Seitdem die Entdeckung der Kronenether und deren Kationen-komplexierende Eigenschaft durch
Pedersen[1] in den 60er Jahren den Weg für die Supramolekulare Chemie ebnete, haben makrozyklische
Moleküle die Wissenschaft fasziniert. Eine ihrer bekanntesten Eigenschaften ist hierbei die Fähigkeit zur
Aufnahme von Gästen in ihrem Innenraum.[2] Eine besonders interessante Verbindungsklasse stellen
makrozyklische Verbände aus Chromophoren dar, in denen sich die optischen Eigenschaften des
Molekülverbands durch Aufnahme von Gastmolekülen verändern.[3]
Das kürzlich in unserem Arbeitskreis synthetisierte Cyclophan 1 illustriert dies in beeindruckender
Weise.[3] Durch die rigide Xylylenbrückeneinheit werden die Chromophore in einem Abstand von 6.5 Å
fixiert, der zwar noch schwache Wechselwirkungen zwischen den Chromophoren erlaubt, aber eine
vollständige Aggregation verhindert. Gleichzeitig wird eine Kavität aufgespannt, in der über π-π-
Wechselwirkungen eine Vielzahl aromatischer Kohlenwasserstoffe wie Naphthalin, Fluorenon oder Perylen
gebunden werden können. Bemerkenswert ist, dass hierbei die elektronische Situation des Gastes Einfluss auf
die Emissionseigenschaften des Cyclophans in Chloroform nimmt (Abbildung 1). Bei Komplexierung eines
elektronenreicheren Gastmoleküls kommt es zur Fluoreszenzlöschung des Wirts, wohingegen die Einlagerung
elektronenärmerer Gäste eine Fluoreszenzverstärkung hervorruft. Dadurch wird 1 gleichzeitig sowohl zu
einem turn on- als auch einem turn off-Fluoreszenzsensor für polycyclische Aromaten.
Abbildung 1: Struktur des Perylenbisimidcyclophans 1 und schematische Darstellung der bei der
Komplexierung von Gästen auftretenden Fluoreszenzverstärkung (links) oder Fluoreszenzlöschung (rechts).
[1] a) C. J. Pedersen, J. Am. Chem. Soc. 1967, 89, 7017−7036; b) C. J. Pedersen, J. Am. Chem. Soc. 1967,
2495–2496; c) C. J. Pedersen, Angew. Chem. Int. Ed. Engl. 1988, 27, 1021−1027.
[2] D. J. Cram, J. M. Cram, Science 1974, 183, 803−809.
[3] a) P. Spenst, F. Würthner, Angew. Chem. Int. Ed. 2015, 54, 10165−10168; b) P. Spenst, R. M. Young,
M. R. Wasielewski, F. Würthner, Chem. Sci. 2016, 7, 5428−5434.
Organische Chemie und Biochemie
P36
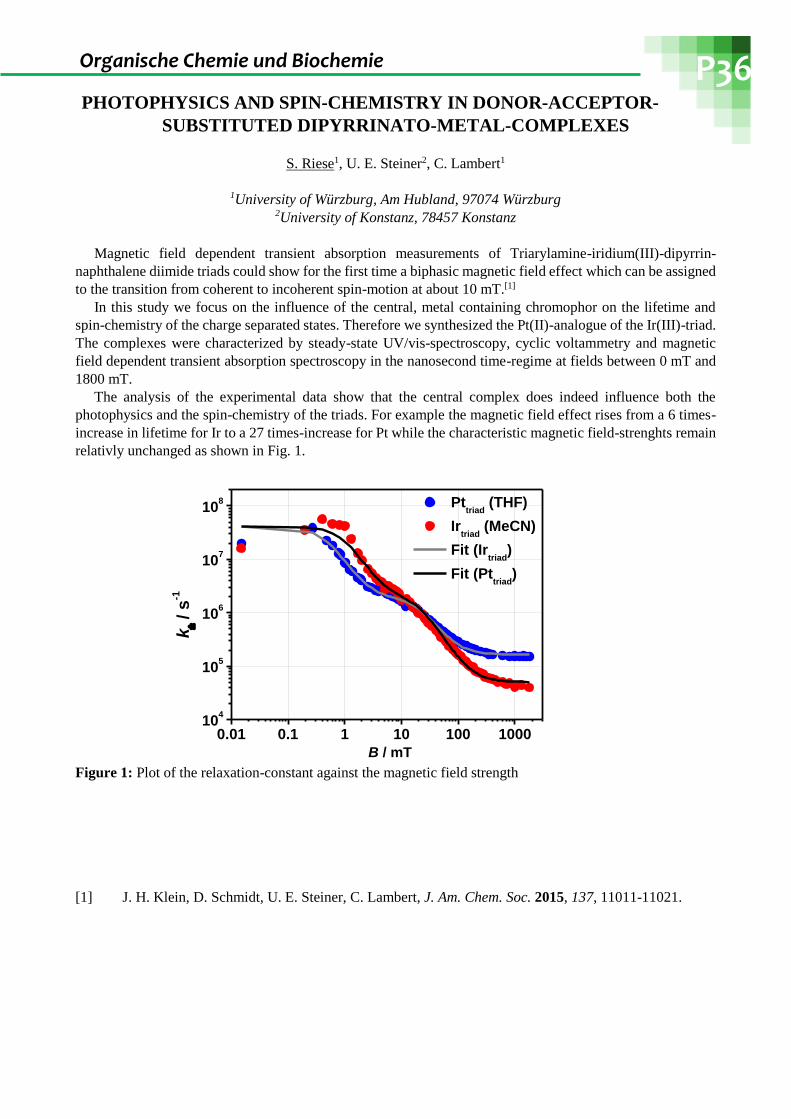
PHOTOPHYSICS AND SPIN-CHEMISTRY IN DONOR-ACCEPTOR-
SUBSTITUTED DIPYRRINATO-METAL-COMPLEXES
S. Riese1, U. E. Steiner2, C. Lambert1
1University of Würzburg, Am Hubland, 97074 Würzburg
2University of Konstanz, 78457 Konstanz
Magnetic field dependent transient absorption measurements of Triarylamine-iridium(III)-dipyrrin-
naphthalene diimide triads could show for the first time a biphasic magnetic field effect which can be assigned
to the transition from coherent to incoherent spin-motion at about 10 mT.[1]
In this study we focus on the influence of the central, metal containing chromophor on the lifetime and
spin-chemistry of the charge separated states. Therefore we synthesized the Pt(II)-analogue of the Ir(III)-triad.
The complexes were characterized by steady-state UV/vis-spectroscopy, cyclic voltammetry and magnetic
field dependent transient absorption spectroscopy in the nanosecond time-regime at fields between 0 mT and
1800 mT.
The analysis of the experimental data show that the central complex does indeed influence both the
photophysics and the spin-chemistry of the triads. For example the magnetic field effect rises from a 6 times-
increase in lifetime for Ir to a 27 times-increase for Pt while the characteristic magnetic field-strenghts remain
relativly unchanged as shown in Fig. 1.
Figure 1: Plot of the relaxation-constant against the magnetic field strength
[1] J. H. Klein, D. Schmidt, U. E. Steiner, C. Lambert, J. Am. Chem. Soc. 2015, 137, 11011-11021.
0.01 0.1 1 10 100 100010
4
105
106
107
108 Pt
triad (THF)
Irtriad
(MeCN)
Fit (Irtriad
)
Fit (Pttriad
)
B / mT
k
/ s
-1

Pharmazie und Lebensmittelchemie P37
Diagnostic chewing gums targeting the tongue as
24/7 available detector
T. Miesler1, J. Ritzer1, C. Rode2, M. C. Amstalden1, M. Pein3, T. Lühmann1, L. Meinel1
1Institute for Pharmacy and Food Chemistry, University of Würzburg, Germany 2 INNOVENT e.V., Technology Development Jena, Germany
3 Institute of Pharmaceutics and Biopharmaceutics, Heinrich-Heine-University Düsseldorf, Germany
Sore throat is a well perceptible symptom, but a lot of diseases have to be considered as the trigger.[1] On
the other hand, some infections are noted too late, because of a lack of specific symptoms, for example the
early stages of caries.[2] To this end, we (1) designed and synthesized specific systems reacting to the presence
of a respective pathogen by releasing a flavor (Fig. 1) and (2) formulated these systems into a chewing gum to
make use of the human tongue as a 24/7 detector.
Figure 1: The peptide bond of the system is cleaved by proteases from macrophages or bacteria, so the flavor
(F) is released from the nanoparticle (NP).
The basic concept consists of an optimized peptide sequence linking a flavor with a nanoparticle.
Denatonium was used as flavor component. Taste intensity was measured using an electronic tongue setup.
Systems were originally designed, reacting to aureolysin from staphylococcus aureus and human matrix
metalloproteinases 1, 8, 9 and 13. Numerous peptide sequences were synthesized and analyzed for their
cleavage rate and selectivity towards the named proteases. Several systems were formulated into a chewing
gum to examine the release of our compounds. With the information gathered, we are now addressing caries
by detection of the decay-causing bacterium streptococcus mutans.
First systems are now established, enabling the specific detection of certain pathogens by their unique
enzymatic properties. The development and closer investigation of such systems could lead to new forms of
diagnostics.
[1] B. Renner, C. A. Mueller , A. Shephard, Inflamm Res. 2012, 61, 1041-1052.
[2] B. Nyvad, Caries Res. 2004, 38, 192-198.

Physikalische und Theoretische Chemie
P38
A coherent two-dimensional nanoscopy setup
S. Pres1, T. Brixner1, L. Dietrich1, M. Hensen1, B. Huber1, V. Lisinetskii1, J. Lüttig1
1 Institut für Physikalische und Theoretische Chemie, Universität Würzburg, Am Hubland,
97074 Würzburg, Germany
How do energy transport processes between individual molecules or inside large heterogeneous structures
like light harvesting complexes occur? Is the transport dominated by step-by-step hopping of excitation or do
the individual molecules form a strongly coupled system in which the excitation is delocalized over all
constituents (quantum mechanical entanglement). How are these transport phenomena influenced by
interactions with the substrate, the environment and external light sources? Is it possible to control the outcome
of chemical reactions by nanostructuring the environment of molecules?
To investigate these questions the measurement signal of the prepared system has to be retrieved with high
temporal (femtosecond regime) and spatial resolution far below the optical diffraction limit (several
nanometers). Therefore, we bring ultra-fast optical 2D spectroscopy, which is usually used to investigate
resonances and couplings of molecular systems, to the nanoscale: Instead of detecting optical-diffraction
limited light fields, we measure non-optical observables, i.e. photoemitted electrons which allow a resolution
down to 1 nm at 1 eV kinetic energy.[1]
The optical excitation is generated by a noncollinear optical parametric amplifier (NOPA) featuring wide
spectral tuneability from the NIR to the UV range. The excited state population can then be probed as
photoemitted electrons which are detected with high spatial resolution (<10 nm) using an aberration corrected
photoemission electron microscope (AC-PEEM).
This setup offers many possibilities for thesis projects in the fields of (an)organic synthesis, sample
preparation and characterisation as well as electron microscopy and laser spectroscopy.
[1] M. Aeschlimann et al., Science 2011, 333, 1723-1726.

Physikalische und Theoretische Chemie P39
Theoretical Studies On The Water Oxidation Mechanism in
Ruthenium Macrocyclic Systems
J. Lindner, M. I. S. Röhr, R. Mitric
We used the QM/MM approach to simulate the spectroelectrochemistry of Ruthenium water oxidation
catalysts by calculating UV/VIS spectra in different catalyst oxidation states. As absorption spectra are highly
dependent on structural changes, averaged ensemble spectra were identified to be an appropriate model.
Furthermore, molecular dynamics simulations were used in order to give insights on the reaction
mechanism with priority to the outstanding turnover frequency of the macrocycle compared to the
corresponding monomer.[1] We found different reasons for the enhanced activity, including H-bonding
networks between the three catalytic moieties of the macrocycle.
Figure 1: H-bonding networks promoting the water nucleophilic attack of a water molecule to the Ru(V)=O
species.
[1] M. Schulze, V. Kunz, P. D. Frischmann, F. Würthner, Nat. Chem. 2016, 8, 576.

Pharmazie und Lebensmittelchemie
P40
Targeting Structural Differences in MIP Proteins and FKBPs
M. A. Kuhn1*, C. A. Sotriffer1
1Institute of Pharmacy and Food Chemistry, University of Würzburg, Würzburg, 97074, Germany
Bacterial Macrophage Infectivity Potentiator (MIP) proteins and human FK506-binding proteins (FKBPs)
both belong to the class of peptidyl-prolyl-isomerases and exhibit a highly conserved binding pocket. The MIP
proteins of Burkholderia pseudomallei and Legionella pneumophila are important for replication and full
virulence, rendering them potential targets for antimicrobial substances. Human FKBPs play miscellaneous
roles, as for example in the regulation of steroid hormone receptor function and the immune response.
Several small-molecule pipecolic acid derivatives are capable of binding to MIP proteins of B.
pseudomallei,[1,2] L. pneumophila[1,3] and other bacterial species as well as to human FKBPs. Analysing and
comparing the structure-activity relationships for these complexes is crucial for the development of new MIP
inhibitors with sufficient selectivity.
The impact of structural differences in two loops (called 50's and 80's loop in FKBP12) adjacent to the
binding pocket was investigated via molecular dynamics simulations. Substitutions at the phenyl ring of a
reference ligand (CJ168) positioned close to the 80's loop lead to differential effects on the ligand orientation
depending on the loop composition of the isoenzyme, thus modulating interaction pattern and binding affinity.
Furthermore, differences in the 50's loop between FKBPs and several MIP proteins should allow to
preferentially address the latter by modifying the ligand with an additional hydrogen bond acceptor.
[1] F. Seufert, M. Kuhn, M. Hein, M. Weiwad, M. Vivoli, I. Norville, M. Sarkar-Tyson, L. Marshall, H.
Bruhn, N. Harmer, C. Sotriffer, U. Holzgrabe, Bioorg. Med. Chem. 2016, 24, 5134-5147.
[2] D. Begley, D. Fox III, D. Jenner, C. Juli, P. Pierce, J. Abendroth, M. Muruthi, K. Safford, V. Anderson,
K. Atkins, S. Barnes, S. Moen, A. Raymond, R. Stacy, P. Myler, B. Staker, N. Harmer, I. Norville, U.
Holzgrabe, M. Sarkar-Tyson, T. Edwards, D. Lorimer, Antimicrob. Agents Chemother. 2014, 58, 1458-
1467.
[3] C. Juli, M. Sippel, J. Jäger, A. Thiele, M. Weiwad, K. Schweimer, P. Rösch, M. Steinert, C. Sotriffer,
U. Holzgrabe, J. Med. Chem. 2011, 54, 277-283.
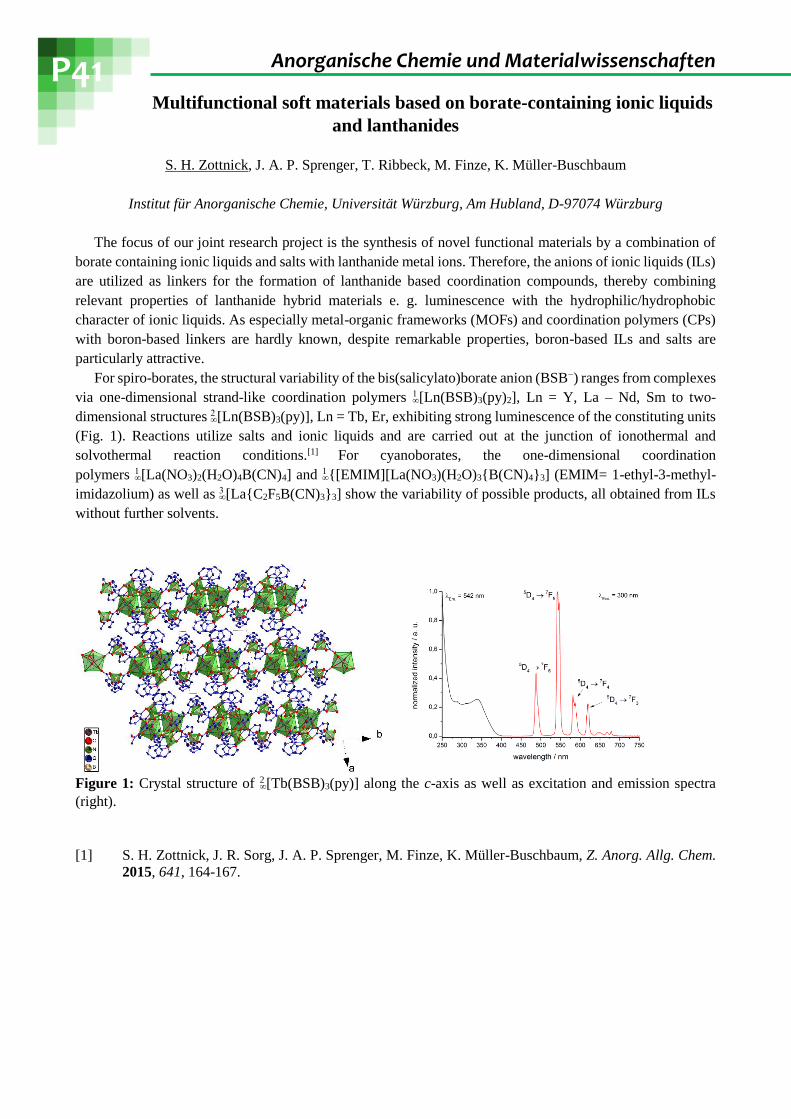
Anorganische Chemie und Materialwissenschaften P41
Multifunctional soft materials based on borate-containing ionic liquids
and lanthanides
S. H. Zottnick, J. A. P. Sprenger, T. Ribbeck, M. Finze, K. Müller-Buschbaum
Institut für Anorganische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg
The focus of our joint research project is the synthesis of novel functional materials by a combination of
borate containing ionic liquids and salts with lanthanide metal ions. Therefore, the anions of ionic liquids (ILs)
are utilized as linkers for the formation of lanthanide based coordination compounds, thereby combining
relevant properties of lanthanide hybrid materials e. g. luminescence with the hydrophilic/hydrophobic
character of ionic liquids. As especially metal-organic frameworks (MOFs) and coordination polymers (CPs)
with boron-based linkers are hardly known, despite remarkable properties, boron-based ILs and salts are
particularly attractive.
For spiro-borates, the structural variability of the bis(salicylato)borate anion (BSB−) ranges from complexes
via one-dimensional strand-like coordination polymers 1∞[Ln(BSB)3(py)2], Ln = Y, La – Nd, Sm to two-
dimensional structures 2∞[Ln(BSB)3(py)], Ln = Tb, Er, exhibiting strong luminescence of the constituting units
(Fig. 1). Reactions utilize salts and ionic liquids and are carried out at the junction of ionothermal and
solvothermal reaction conditions.[1] For cyanoborates, the one-dimensional coordination
polymers 1∞[La(NO3)2(H2O)4B(CN)4] and 1
∞{[EMIM][La(NO3)(H2O)3{B(CN)4}3] (EMIM= 1-ethyl-3-methyl-
imidazolium) as well as 3∞[La{C2F5B(CN)3}3] show the variability of possible products, all obtained from ILs
without further solvents.
Figure 1: Crystal structure of 2∞[Tb(BSB)3(py)] along the c-axis as well as excitation and emission spectra
(right).
[1] S. H. Zottnick, J. R. Sorg, J. A. P. Sprenger, M. Finze, K. Müller-Buschbaum, Z. Anorg. Allg. Chem.
2015, 641, 164-167.
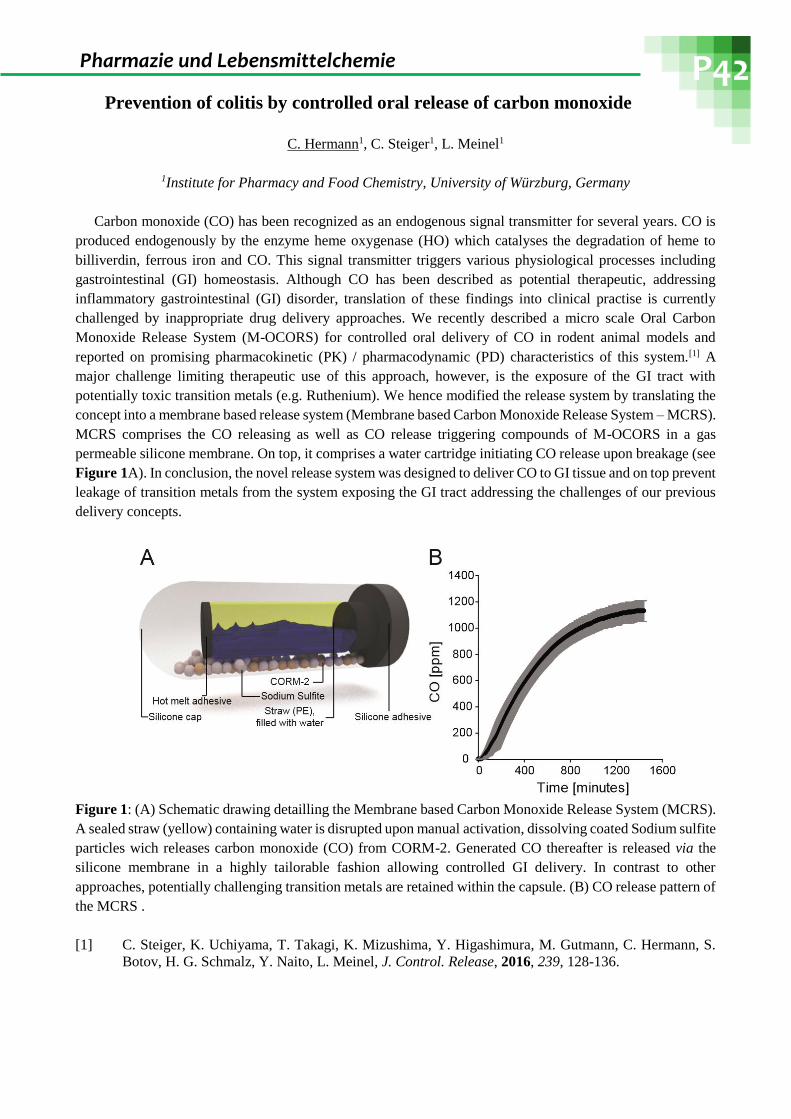
Pharmazie und Lebensmittelchemie
P42
Prevention of colitis by controlled oral release of carbon monoxide
C. Hermann1, C. Steiger1, L. Meinel1
1Institute for Pharmacy and Food Chemistry, University of Würzburg, Germany
Carbon monoxide (CO) has been recognized as an endogenous signal transmitter for several years. CO is
produced endogenously by the enzyme heme oxygenase (HO) which catalyses the degradation of heme to
billiverdin, ferrous iron and CO. This signal transmitter triggers various physiological processes including
gastrointestinal (GI) homeostasis. Although CO has been described as potential therapeutic, addressing
inflammatory gastrointestinal (GI) disorder, translation of these findings into clinical practise is currently
challenged by inappropriate drug delivery approaches. We recently described a micro scale Oral Carbon
Monoxide Release System (M-OCORS) for controlled oral delivery of CO in rodent animal models and
reported on promising pharmacokinetic (PK) / pharmacodynamic (PD) characteristics of this system.[1] A
major challenge limiting therapeutic use of this approach, however, is the exposure of the GI tract with
potentially toxic transition metals (e.g. Ruthenium). We hence modified the release system by translating the
concept into a membrane based release system (Membrane based Carbon Monoxide Release System – MCRS).
MCRS comprises the CO releasing as well as CO release triggering compounds of M-OCORS in a gas
permeable silicone membrane. On top, it comprises a water cartridge initiating CO release upon breakage (see
Figure 1A). In conclusion, the novel release system was designed to deliver CO to GI tissue and on top prevent
leakage of transition metals from the system exposing the GI tract addressing the challenges of our previous
delivery concepts.
Figure 1: (A) Schematic drawing detailling the Membrane based Carbon Monoxide Release System (MCRS).
A sealed straw (yellow) containing water is disrupted upon manual activation, dissolving coated Sodium sulfite
particles wich releases carbon monoxide (CO) from CORM-2. Generated CO thereafter is released via the
silicone membrane in a highly tailorable fashion allowing controlled GI delivery. In contrast to other
approaches, potentially challenging transition metals are retained within the capsule. (B) CO release pattern of
the MCRS .
[1] C. Steiger, K. Uchiyama, T. Takagi, K. Mizushima, Y. Higashimura, M. Gutmann, C. Hermann, S.
Botov, H. G. Schmalz, Y. Naito, L. Meinel, J. Control. Release, 2016, 239, 128-136.

Physikalische und Theoretische Chemie P43
Investigations on covalent-reversible inhibitors using QM and
QM/MM approaches
A. Heilos, T. A. Le, W. Waigel, B. Engels
Most drugs consist of ligands which interact with their target non-covalently. They have the advantage that
they are so unreactive that unintended reactions with DNA or proteins do not take place. However, they have
the drawback that their free energy of binding do generally not exceed 15 kcal/mol. Higher binding affinities
can only be achieved with ligands which form a covalent bond with their target. Despite famous examples as
Penicillin or Aspirin in the past the industry hesitated to develop new covalent drugs because they fear
unintended side reactions resulting from the reactivity of ligands. Since about 2005 covalent ligands undergo
an intensive renaissance in academia and industry, because various very selective drugs were detected in the
last few years. As for example covalent reversible protease inhibitors based on nitriles or a number of reversible
and irreversible kinase inhibitors. [1-4] In this work, several systems have been investigated in regard of possible
covalent inhibitors and inhibition mechanisms. Also calculations concerning the protonation state of active
site cysteine and histidine has been performed (Figure 1).
Figure 1: surface: CathB, crystal structure 1HUC; ball and stick: active site amino acids CYS29 and HIS199.
[1] Gütschow and Co-work., ChemMedChem 2013, 8, 1330.
[2] Schirmeister and Co-work., ChemMedChem 2013, 8, 967.
[3] Kwak et al., Proc. Natl. Acad. Sci. USA 2005, 102, 7665.
[4] Taunton and Co-work., J. Am. Chem. Soc. 2013, 135, 5298.
HIS19
CYS
?

Organische Chemie und Biochemie
P44
Controlling the Superstructure in Polymeric Squaraine Dyes
M. H. Schreck1, C. Lambert2
1,2Institut für Organische Chemie & Center for Nanosystems Chemistry, Universität Würzburg
Am Hubland 97074 Würzburg, Germany
E-mail: [email protected]
We recently prepared a polymer P1 based on a dicyanovinylene-substituted cis-indolenine squaraine dye
M1 via Ni-mediated Yamamoto homocoupling reaction.This homopolymer displays a bathochromic shift of
the absorption maximum but also a weaker band at higher energies. This observation can be explained by a
mixture of H- and J-type alignment of chromophores, which then exhibit hypsochromic (H) and bathochromic
(J) shifts. In this context, the solvent itself is significantly involved in the sort of superstructure (helical or zig-
zag-structures) formed by the polymer in solution.
The ongoing goal is to control the superstructure and the optical properties involved, and rule out the
superior role of the solvent by structurally modifying the parent squaraine monomer M1 which was realized
via two attempts: (1) Introducing bulky substituents into the indolenine moiety and (2) partially stiffening the
polymer backbone. Both new polymers P2 and P3 show a red-shifted absorption in comparison to their
corresponding monomers M2 and M3, respectively, indicating J-aggregate behavior and no dependence on
the solvent. Consequently, the polymers exclusively form zig-zag structures irrespective of the chosen solvent.
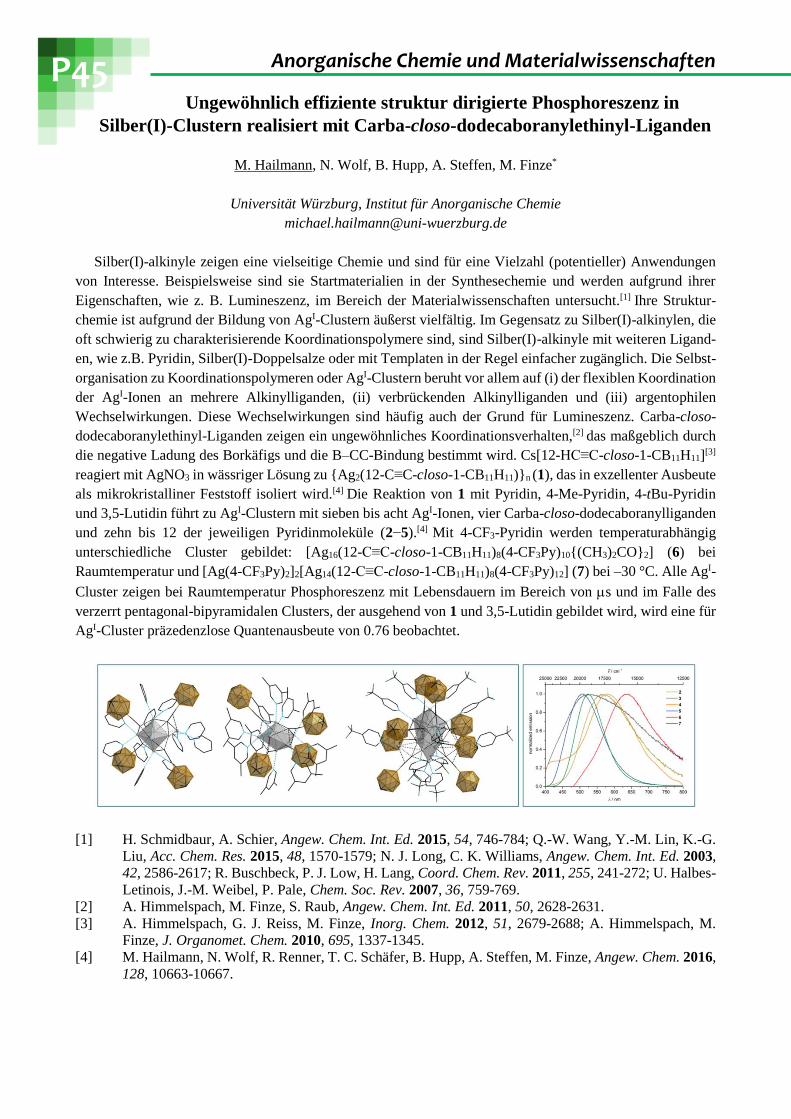
Anorganische Chemie und Materialwissenschaften P45
Ungewöhnlich effiziente struktur dirigierte Phosphoreszenz in
Silber(I)-Clustern realisiert mit Carba-closo-dodecaboranylethinyl-Liganden
M. Hailmann, N. Wolf, B. Hupp, A. Steffen, M. Finze*
Universität Würzburg, Institut für Anorganische Chemie
Silber(I)-alkinyle zeigen eine vielseitige Chemie und sind für eine Vielzahl (potentieller) Anwendungen
von Interesse. Beispielsweise sind sie Startmaterialien in der Synthesechemie und werden aufgrund ihrer
Eigenschaften, wie z. B. Lumineszenz, im Bereich der Materialwissenschaften untersucht.[1] Ihre Struktur-
chemie ist aufgrund der Bildung von AgI-Clustern äußerst vielfältig. Im Gegensatz zu Silber(I)-alkinylen, die
oft schwierig zu charakterisierende Koordinationspolymere sind, sind Silber(I)-alkinyle mit weiteren Ligand-
en, wie z.B. Pyridin, Silber(I)-Doppelsalze oder mit Templaten in der Regel einfacher zugänglich. Die Selbst-
organisation zu Koordinationspolymeren oder AgI-Clustern beruht vor allem auf (i) der flexiblen Koordination
der AgI-Ionen an mehrere Alkinylliganden, (ii) verbrückenden Alkinylliganden und (iii) argentophilen
Wechselwirkungen. Diese Wechselwirkungen sind häufig auch der Grund für Lumineszenz. Carba-closo-
dodecaboranylethinyl-Liganden zeigen ein ungewöhnliches Koordinationsverhalten,[2] das maßgeblich durch
die negative Ladung des Borkäfigs und die B–CC-Bindung bestimmt wird. Cs[12-HC≡C-closo-1-CB11H11][3]
reagiert mit AgNO3 in wässriger Lösung zu {Ag2(12-C≡C-closo-1-CB11H11)}n (1), das in exzellenter Ausbeute
als mikrokristalliner Feststoff isoliert wird.[4] Die Reaktion von 1 mit Pyridin, 4-Me-Pyridin, 4-tBu-Pyridin
und 3,5-Lutidin führt zu AgI-Clustern mit sieben bis acht AgI-Ionen, vier Carba-closo-dodecaboranylliganden
und zehn bis 12 der jeweiligen Pyridinmoleküle (2−5).[4] Mit 4-CF3-Pyridin werden temperaturabhängig
unterschiedliche Cluster gebildet: [Ag16(12-C≡C-closo-1-CB11H11)8(4-CF3Py)10{(CH3)2CO}2] (6) bei
Raumtemperatur und [Ag(4-CF3Py)2]2[Ag14(12-C≡C-closo-1-CB11H11)8(4-CF3Py)12] (7) bei –30 °C. Alle AgI-
Cluster zeigen bei Raumtemperatur Phosphoreszenz mit Lebensdauern im Bereich von s und im Falle des
verzerrt pentagonal-bipyramidalen Clusters, der ausgehend von 1 und 3,5-Lutidin gebildet wird, wird eine für
AgI-Cluster präzedenzlose Quantenausbeute von 0.76 beobachtet.
[1] H. Schmidbaur, A. Schier, Angew. Chem. Int. Ed. 2015, 54, 746-784; Q.-W. Wang, Y.-M. Lin, K.-G.
Liu, Acc. Chem. Res. 2015, 48, 1570-1579; N. J. Long, C. K. Williams, Angew. Chem. Int. Ed. 2003,
42, 2586-2617; R. Buschbeck, P. J. Low, H. Lang, Coord. Chem. Rev. 2011, 255, 241-272; U. Halbes-
Letinois, J.-M. Weibel, P. Pale, Chem. Soc. Rev. 2007, 36, 759-769.
[2] A. Himmelspach, M. Finze, S. Raub, Angew. Chem. Int. Ed. 2011, 50, 2628-2631.
[3] A. Himmelspach, G. J. Reiss, M. Finze, Inorg. Chem. 2012, 51, 2679-2688; A. Himmelspach, M.
Finze, J. Organomet. Chem. 2010, 695, 1337-1345.
[4] M. Hailmann, N. Wolf, R. Renner, T. C. Schäfer, B. Hupp, A. Steffen, M. Finze, Angew. Chem. 2016,
128, 10663-10667.
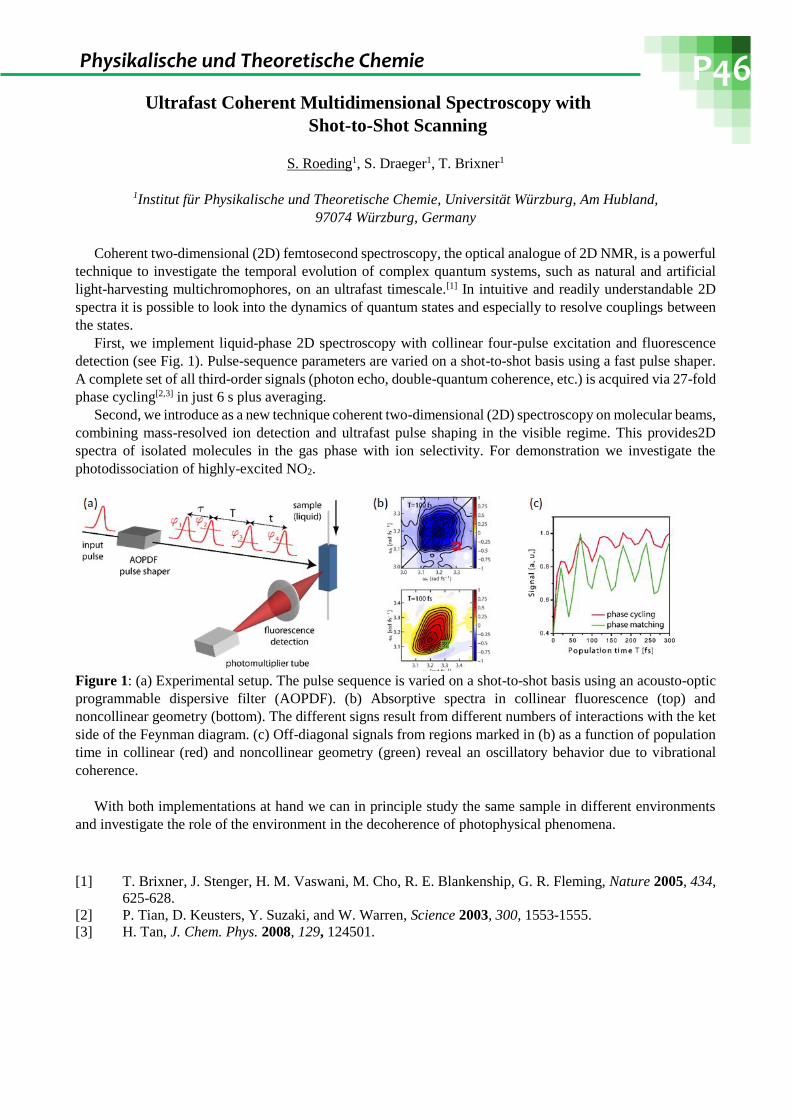
Physikalische und Theoretische Chemie
P46
Ultrafast Coherent Multidimensional Spectroscopy with
Shot-to-Shot Scanning
S. Roeding1, S. Draeger1, T. Brixner1
1Institut für Physikalische und Theoretische Chemie, Universität Würzburg, Am Hubland,
97074 Würzburg, Germany
Coherent two-dimensional (2D) femtosecond spectroscopy, the optical analogue of 2D NMR, is a powerful
technique to investigate the temporal evolution of complex quantum systems, such as natural and artificial
light-harvesting multichromophores, on an ultrafast timescale.[1] In intuitive and readily understandable 2D
spectra it is possible to look into the dynamics of quantum states and especially to resolve couplings between
the states.
First, we implement liquid-phase 2D spectroscopy with collinear four-pulse excitation and fluorescence
detection (see Fig. 1). Pulse-sequence parameters are varied on a shot-to-shot basis using a fast pulse shaper.
A complete set of all third-order signals (photon echo, double-quantum coherence, etc.) is acquired via 27-fold
phase cycling[2,3] in just 6 s plus averaging.
Second, we introduce as a new technique coherent two-dimensional (2D) spectroscopy on molecular beams,
combining mass-resolved ion detection and ultrafast pulse shaping in the visible regime. This provides2D
spectra of isolated molecules in the gas phase with ion selectivity. For demonstration we investigate the
photodissociation of highly-excited NO2.
Figure 1: (a) Experimental setup. The pulse sequence is varied on a shot-to-shot basis using an acousto-optic
programmable dispersive filter (AOPDF). (b) Absorptive spectra in collinear fluorescence (top) and
noncollinear geometry (bottom). The different signs result from different numbers of interactions with the ket
side of the Feynman diagram. (c) Off-diagonal signals from regions marked in (b) as a function of population
time in collinear (red) and noncollinear geometry (green) reveal an oscillatory behavior due to vibrational
coherence.
With both implementations at hand we can in principle study the same sample in different environments
and investigate the role of the environment in the decoherence of photophysical phenomena.
[1] T. Brixner, J. Stenger, H. M. Vaswani, M. Cho, R. E. Blankenship, G. R. Fleming, Nature 2005, 434,
625-628.
[2] P. Tian, D. Keusters, Y. Suzaki, and W. Warren, Science 2003, 300, 1553-1555.
[3] H. Tan, J. Chem. Phys. 2008, 129, 124501.

Pharmazie und Lebensmittelchemie P47 Fluorine Walk: The Role of Fluorine in Quinolone amides active
against T. b. brucei
M. Berninger1, A. Fuß2, E. Al-Momani3, I. Israel3, P. Guentzel1, M. Raschig1, S. Samnick3,
U. Holzgrabe1
1Institute of Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg,
Germany 2Medical Mission Institute Würzburg, Hermann-Schell-St. 7, 97074 Würzburg, Germany
3Institute of Nuclear Medicine, University Hospital of Würzburg, Josef-Schneider-Str. 2, 97080 Würzburg,
Germany
Human African Trypanosomiasis (HAT) is caused by an infection with Trypanosoma brucei, a vector-
borne parasite, which is transmitted by the bite of infected tsetse flies. Two clinically relevant stages can be
differentiated, i.e. stage I which is characterized by unspecific headache, fever and joint pains, and stage II in
which the parasites cross the blood brain barrier (BBB) and affect the central nervous system.[1] Previous
investigations identified novel 4-quinolone-3-carboxamides as a promising scaffold having a submicromolar
activity[2] and a confirmed in vivo efficacy against T. b. brucei.[3] As the ability of the quinolone amides to pass
the BBB should be investigated, 18F-labeled derivative of our most potent substance was synthesized and
subjected to autoradiography studies. Experiments using murine brain confirmed the ability of the respective
derivative 1 to pass the BBB 60 min after p.i. application. Besides utilizing fluorine for PET, we explored its
impact on toxicity, pharmacokinetic and pharmaco dynamics properties when being added in different position
of the quinolone scaffold. This “fluorine walk” led to structure 2 with an improved selectivity index >2000
due to lower cytotoxicity and consistent activity against T. b. brucei.
[1] WHO, www.who.int; fact sheet N°259; February 2016
[2] G. Hiltensperger et al., J. Med. Chem. 2012, 55, 2538-2548.
[3] G. Hiltensperger et al., Antimicrob Agents Chemother 2016, 60, 4442-4452.

Organische Chemie und Biochemie
P48
A Zwitterion a Day Keeps the Proteins Away
V. Warkentin, A. Krueger
Institut für Organische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg
Surface functionalization of nanoparticles (NPs) is an essential tool for the control of chemical, physical
and physiological behavior of these materials. Especially nanodiamond’s (ND’s) surface can easily be
modified by using their broad variety of functional moieties.[1] In addition, NDs have low to non-existing
toxicity, reliable production and chemical inertness which makes them attractive for biomedical applications.[2]
When NDs are exposed to bio-fluids, such as serum, proteins adsorb on the surface of the particles and
form in situ a “protein corona”. This corona masks all the desired functionalities on the surface and changes
the interaction with the surrounding fluid. Both, the colloidal stability and the physiological properties are not
controllable anymore.[3]
Here, we report zwitterionic moieties in combination with tetraethylene glycol (TEG) to improve the
colloidal dispersion in physiological media with strong ion background and the prevention of non-specific
interactions with proteins.[4]
Figure 1: Schematic illustration of a functionalized ND with zwitterionic head of a TEG chain.[5]
These properties play a central role for the bioapplication of nanomaterials since only a precise surface
functionalization leads to controlled behavior in biomedical application.
This project has received funding from the Volkswagenstiftung under Grant Agreement no. 88393.
[1] A. Krueger, D. Lang, Adv. Funct. Mater. 2012, 22, 890.
[2] A. Krueger, Chem. Eur. J. 2008, 14, 1382.
[3] W. C. W. Chan, C. D. Walkey, J. Am. Chem. Soc. 2012, 134, 2139.
[4] V. M. Rotello, D. F. Mayano, ACS Nano 2014, 8, 6748.
[5] S. Heywood, R. Adams, mAbs 2016, 8, 1336.

Organische Chemie und Biochemie P49
Coupled Chromophores for Studying Energy Transfer
N. A. Schopf, C. Lambert*
Institut für Organische Chemie & Wilhelm Conrad Röntgen Research Center for Complex Material
Systems, Universität Würzburg, Am Hubland, 97074 Würzburg
*E-mail: [email protected]
Energy transfer is a highly researched topic as it is not only a fundamental process in the
photosynthesis of plants and bacteria but also finds application in organic photovoltaics.[1]
Suitable model systems to study energy transfer processes indepth are needed. In such systems a
donor is excited and transfers energy to an acceptor, which then can fluoresce.[2] To get a better
understanding of the dependency of the energy transfer on the photophysical properties, different trimers
were synthesised by choosing from a pool of four different chromophores. With these chromophores a
broad spectral range of absorption and fluorescence wavelength can be covered. By varying the types
of chromophores, the amount of different chromophores, and the sequence several trimers can be
synthesised.
The energy transfer properties are investigated through steady-state absorption, fluorescence
spectroscopy and transient absorption spectroscopy.
[1] R. Ziessel, G. Ulrich, A. Haefele, A. Harriman, J. Am. Chem. Soc. 2013, 135, 1130-11344.
[2] G.-S. Jiao et al., Tetrahedron 2003, 59, 3109-3116.

Anorganische Chemie und Materialwissenschaften
P50
Superparamagnetic Luminescent MOF@Fe3O4/SiO2
Composite Particles
T. Wehner1, K. Mandel2, M. Schneider2, G. Sextl2, K. Müller-Buschbaum1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität, Am Hubland, 97074 Würzburg, Deutschland
2Fraunhofer-Institut für Silicatforschung ISC, Neunerplatz 2, 97082 Würzburg, Deutschland
Herein, we present the generation of multifunctional composite materials consisting of superparamagnetic
Fe3O4/SiO2 microparticles and different luminescent lanthanide-containing metal-organic frameworks (MOFs).
The modification could be achieved by various reaction conditions including mechanochemistry. The resulting
composites are core-shell materials with the microparticle as core and a MOF-containing shell and combine the
properties of both constituents: superparamagnetism and luminescence. If the magnetite particles are
functionalized with the 2D-MOF 2∞
[Eu2Cl6(Bipy)3]·2Bipy (Bipy = 4,4’-bipyridine), the hybrid material can be
used as potential water detector in fluids since the MOF luminescence is quenched by contact with low amounts
of water. Exploiting the magnetic properties of the composite, the signal can be augmented by gathering the
particles in one spot.
Figure 1: Detection of water and magnetic signal augmentation of the composite particles 2∞
[Eu2Cl6(Bipy)3]·2Bipy@Fe3O4/SiO2 (top) and principle of the water detector system (bottom).
A color mixture of red Eu3+ and green Tb3+ centers can be achieved via the composite 2∞
[EuxTb2-xCl6(Bipy)3]·2Bipy@Fe3O4/SiO2, which can therefore be used for color tuning and is potentially suitable
as ratiometric luminescent detector. The modification of Fe3O4/SiO2 with other MOF systems like 3∞
[Eu(Im)2]
(Im = imidazolate) and 3∞
[Eu2(BDC)3]·2DMF·2H2O (BDC = 1,4-benzendicarboxylate) leads to further variation
of the luminescence color and other properties of the composite.
Instead of spherical Fe3O4/SiO2 particles, magnetite microrods can be prepared, which exhibit intensive light
reflection in dependence of their orientation in an external magnetic field as additional property. Modification of
the microrods with 3∞
[Eu2(BDC)3]·2DMF·2H2O leads to a hybrid material, which combines the isotropic reflection
properties of the microparticles with the anisotropic luminescence of the MOF. Switching between both properties
is possible by variation of the excitation wavelength.
[1] T. Wehner, K. Mandel, M. Schneider, G. Sextl, K. Müller-Buschbaum, ACS Appl. Mater. Interfaces 2016,
8, 5445.
[2] K. Mandel, F. Hutter, C. Gellermann, G. Sextl, ACS Appl. Mater. Interfaces 2012, 4, 5633.
[3] C. J. Höller, M. Mai, C. Feldmann, K. Müller-Buschbaum, Dalton Trans. 2010, 39, 461.
[4] P. R. Matthes, C. J. Höller, M. Mai, J. Heck, S. J. Sedlmaier, S. Schmiechen, C. Feldmann, W. Schnick,
K. Müller-Buschbaum, J. Mater. Chem. 2012, 22, 10179.
[5] A. Zurawski, M. Mai, D. Baumann, C. Feldmann, K. Müller-Buschbaum, Chem. Commun. 2011, 47, 496.
[6] Z.-H. Zhang, S.-Y. Wan, T.-a. Okamura, W.-Y. Sun, N. Ueyama, Z. Anorg. Allg. Chem. 2006, 632, 679.

Pharmazie und Lebensmittelchemie P51
Bioresponsive interleukin-4 delivery system deploying silk-elastin-
like proteins for the treatment of osteoarthritis
V. Spieler1, C. Karavasili2, T. Lühmann1, L. Meinel1*
1Institute for Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg,
Germany 2Department of Pharmaceutical Technology, Aristotle University, Thessaloniki GR 54124, Greece
*Corresponding Author: Lorenz Meinel, [email protected]
Controlling macrophage polarization using a bioresponsive drug delivery system for rhIL-4 in
osteoarthritis holds potential in meeting challenges with the systemic administration of this anti-
inflammatory cytokine.[1,2] In this study, the silk-elastin-like protein (SELP) S2E8C, a genetically
engineered biomaterial, is used for immobilization of IL-4. SELPs consist of repeating units of silk and
elastin blocks and self-assemble into small micellar-like particles at body temperature.[3] S2E8C features
twelve cysteines allowing for bioorthogonal conjugation reactions. IL-4 modified with an unnatural
amino acid containing a functionality for copper(I)-catalyzed azide alkyne cycloaddition (CuAAC) at
position 42[4] and a protease cleavable linker (PCL) responding to proteases up regulated during
inflammation[5] modified with a sulfhydryl-reactive iodoacetyl group were used to decorate S2E8C.
Following intra articular injection, the system is designed to remain within the osteoarthritic joint, where
it deploys its sensory function for constant surveilling of the disease status only to respond to flare by
releasing IL-4, hence preventing disease progression and initiating tissue repair.
A new S2E8C version with thrombin cleavable His-tag was cloned and expressed in E. coli.
Purification was performed by precipitation at high temperature and low pH with subsequent re-
solubilization in water, thereby removing the potential need of a potentially immunogenic His-tag.
Conjugation of S2E8C to the PCL was with a succinimidyl iodo acetate crosslinker. CuAAC linking
S2E8C-PCL and IL-4 is currently under investigation.
S2E8C as a biomaterial for site-directed immobilization of IL-4 allows for creating a biocompatible
and injectable drug delivery system for targeting inflammation by controlling macrophage plasticity in
a timely and spatially regulated manner.[6]
[1] D. M. Mosser, J. P. Edwards, Nat. Rev. Immunol. 2008, 8, 958–969.
[2] T. Lühmann, L. Meinel, Curr. Opin. Biotechnol. 2016, 39, 35–40.
[3] X. X. Xia, Q. Xu, X. Hu, G. Qin, D. L. Kaplan, Biomacromolecules 2011, 12, 3844–3850.
[4] T. Lühmann, V. Spieler, V. Werner, M.-G. Ludwig, J. Fiebig, T. Müller, L. Meinel,
Chembiochem 2016.
[5] A. C. Braun, M. Gutmann, R. Ebert, F. Jakob, H. Gieseler, T. Lühmann, L. Meinel, Pharm. Res.
2016, 1–15.
[6] W. Huang, A. Rollett, D. L. Kaplan, Expert Opin. Drug Deliv. 2015, 12, 779–791.

Anorganische Chemie und Materialwissenschaften
P52
Reactivity of the Vicinal Biscarbenoid Bis(piperidyl)acetylene
H. Braunschweig1, S. Kachel1, H. Kelch1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität, Am Hubland, 97074 Würzburg,
Deutschland
We present the reactivity of the diaminoacetylene PipCCPip which exhibits strong biscarbenoid
behavior based on the extraordinarily high electron density of the acetylenic moiety.[1] The molecule
displays extensive bond activation chemistry beyond that of commonly employed monofunctional
carbenes. Treatment with moderately Lewis acidic and/or polar substrates leads to clean activation of
BB, BC and BN bonds under ambient conditions.[2] The resulting products feature intriguing
structural and electronical properties as exemplified in the first transition-metal-free synthesis of 1,4-
azaborinines – highly sought-after benzene analogs with potential applications in medicinal and material
science.[3] Cyclodimerization of PipCCPip with the aid of group XIV metals leads to the formation
of cyclobutadiene derivatives displaying a unique mode of stabilization based on charge separation and
Lewis pair formation.[4]
[1] A. R. Petrov, T. Bannenberg, C. G. Daniliuc, P. G. Jones, M. Tamm, Dalton Trans. 2011,
a 40, 10503.
[2] H. Kelch, S. Kachel, M. A. Celik, M. Schäfer, B. Wennemann, K. Radacki, A. R. Petrov, M.
Tamm, H. Braunschweig, Chem. Eur. J. 2016, 22, 13815.
[3] H. Lee, M. Fischer, B. K. Shoichet, S.-Y. Liu, J. Am. Chem. Soc. 2016, 138, 12021.
[4] R. Bertermann, H. Braunschweig, M. A. Celik, T. Dellermann, H. Kelch, Chem. Commun.,
DOI: 10.1039/C6CC07741B.

Anorganische Chemie und Materialwissenschaften P53 Water-Soluble 3-Coordinate Boron Chromophores for One- and
Two-Photon Excited Fluorescence Imaging of Mitochondria in Cells
S. Griesbeck1, Z. Zhang1, M. Gutmann2, T. Lühmann2, R. M. Edkins1, G. Clermont3, A. N. Lazar3,
M. Blanchard-Desce3, L. Meinel2, T. B. Marder1
email: [email protected] 1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Würzburg,
Germany 2Institut für Lebensmittelchemie und Pharmazie, Julius-Maximilians-Universität Würzburg,
Würzburg, Germany 3Institut de Sciences Moléculaires, Université de Bordeaux, Bordeaux, France
Triarylboranes have attracted a huge amount of interest due to their application in many different
fields such as anion sensing, OLEDs and non-linear optical materials.[1] Over the last few years, we have
studied the use of dimesitylboron-based -acceptors (A) in two-photon absorption (TPA) chromophores.
We designed dipolar, quadrupolar and octupolar compounds with exceptional TPA cross sections and
high fluorescence quantum yields.[2] Furthermore, we reported structure-TPA cross section relationships
for our quadrupolar A--A compounds.[3] Recently, we synthesized oligothiophene-BMes2
chromophores, with significantly enhanced TPA cross sections of up to 1930 GM in the near-infrared
region, the “biological transparent window”.[4] We present herein the further functionalization of such
chromophores with ammonium groups, an approach pioneered by Gabbaï,[5] in order to achieve
hydrophilicity and biocompatibility, and our initial results of both one- and two-photon excited
fluorescence (TPEF) microscopy allowing the imaging of mitochondria in cells with our chromophore.
[1] (a) C. D. Entwistle, T. B. Marder, Angew. Chem. Int. Ed. Engl. 2002, 41, 2927-2931. (b) C. D.
Entwistle, T. B. Marder, Chem. Mater. 2004, 16, 4574-4585.
[2] J. C. Collings, C. Katan, A. Beeby, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce, T. B.
Marder et al., Chem. Eur. J. 2009, 15, 198-208.
[3] (a) M. Charlot, L. Porrès, C. D. Entwistle, A. Beeby, T. B. Marder, M. Blanchard-Desce, Phys.
Chem. Chem. Phys. 2005, 7, 600-606. (b) C. D. Entwistle, J. C. Collings, A. Steffen, A. Beeby,
A. S. Batsanov, J. A. K. Howard, W.-Y. Wong, A. Boucekkine, J.-F. Halet, T. B. Marder et al.,
J. Mater. Chem. 2009, 19, 7532-7544.
[4] L. Ji, R. M. Edkins, A. Beeby, A. S. Batsanov, J. A. K. Howard, A. Boucekkine, Z. Liu, J.-F.
Halet, C. Katan, T. B. Marder et al., Chem. Eur. J. 2014, 20, 13618-13635.
[5] C.-W. Chiu, Y. Kim, F. P. Gabbaï, J. Am. Chem. Soc. 2009, 131, 60-61.

Anorganische Chemie und Materialwissenschaften
P54
Experimentelle und theoretische Untersuchungen sterischer und
elektronischer Parameter NHC-stabilisierter Nickel-Carbonylkomplexe
J. Berthel, U. Radius*
Julius-Maximilians-Universität Würzburg/Institut für Anorganische Chemie
[email protected], [email protected]
Freie NHCs finden seit ihrer erstmaligen Synthese und Isolierung[1] breitgefächerte Anwendung in
der Übergangsmetallchemie.[2-4] Dies gründet neben ihren starken σ-Donor- und schwachen π-
Akzeptoreigenschaften auch auf ihren mannigfaltigen Synthesemöglichkeiten bei struktureller
Variabilität und sterischer Flexibilität.[5-8] Mithilfe des sterischen Anspruchs und der elektronischen
Eigenschaftender NHCs kann dabei deren Einfluss auf die Struktur von Übergangsmetallkomplexen
bestimmt werden.[9]
Vorgestellt wird die Synthese neuer NHC-stabilisierter Nickel-Carbonylkomplexe und die
Untersuchungen bezüglich ihrer sterischen und elektronischen Eigenschaften.
Zur Unterstützung der Ergebnisse wurden quantenmechanische Berechnungen angefertigt und die
Bindungsbildungs- und Bindungsdissoziationsenergien ΔG der Komplexe abgeschätzt.
[1] A. J. Arduengo III, R. L. Harlow, M. Kline, J. Am. Chem. Soc. 1991, 113, 361.
[2] N-Heterocyclic Carbenes in Synthesis, S. P. Nolan (Hrsg.), Wiley-VCH, Weinheim, 2006.
[3] N-Heterocyclic Carbenes in Transition Metal Catalysis, F. Glorius (Hrsg.), Top. Organomet.
Chem., Vol. 21, 2007.
[4] N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, S. Díez-
González (Hrsg.), Catalysis Series No. 6, RSC, Cambridge, 2010.
[5] N. M. Scott, S. P. Nolan, Eur. J. Inorg. Chem. 2005, 1815.
[6] V. Lavallo, Y. Canac, A. DeHope, B. Donnadieu, G. Bertrand, Angew. Chem. 2005, 117, 7402;
Angew. Chem. Int. Ed. 2005, 44, 7236.
[7] E. Peris, R. H. Crabtree, Coord. Chem. Rev. 2004, 248, 2239.
[8] C. M. Crudden, D. P. Allen, Coord. Chem. Rev. 2004, 248, 2247.
[9] U. S. D. Paul, C. Sieck, M. Haehnel, K. Hammond, T. B. Marder, U. Radius, Chem. Eur. J.
2016, 22, 11005-11014.

Anorganische Chemie und Materialwisenschaften P55
„iClick“-Reaktionen von Ru- und Rh-Azid-Komplexen mit
elektronenarmen Alkinen:
Regioselektivität, Stabilität und Kinetik
L. Waag-Hiersch1, S. Eilbacher1, M. Voelkel1, J. Mößeler1, U. Schatzschneider1,*
1 Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg
Am Hubland, D-97074 Würzburg (Germany)
e-mail: [email protected]
Anorganische Click-Reaktionen (engl. inorganic click, „iClick“)[1-2] zwischen Metall-Azid-
Komplexen und elektronenarmen Alkinen stellen einen schnellen Zugang zu strukturell vielfältigen
Triazolatkomplexen dar. Die „iClick“-Reaktion von Ruthenium-Azid-Komplexen der allgemeinen
Formel [Ru(N3)(aren)(L-L)]+ mit bidentaten Stickstoffliganden sowie Rhodium-Azid-Komplexen der
allgemeinen Formel [Rh(Cp*)(N3)(bpyR,R)]+ mit unterschiedlich substituierten 2,2‘-Bipyridin-
Coliganden und elektronenarmen Alkinen wie Dimethylacetylendicaboxylat (DMAD) und 4,4,4-
Trifluorobut-2-insäureethylester wurde untersucht. Inbesondere wurde die Geschwindigkeit der
„iClick“-Reaktion von Ruthenium-Azid-Komplexen in Abhängigkeit von funktionellen Gruppen in 4-
und 4‘-Position am Bipyridin-Ligand sowie Variation des Aren-Liganden (Hexamethylbenzol vs. p-
Cymol) bestimmt und dafür verschiedene analytische Methoden (HPLC, IR-Spektroskopie in Lösung
und 19F NMR-Spektroskopie) verwendet. Da Triazolate prinzipiell über die N1-, N2- oder N3-
Stickstoffatome an ein Metallzentrum binden können, wurde ein besonderes Augenmerk auch auf die
Untersuchung der Regioselektivität gerichtet. Des Weiteren wurde die Stabilität der resultierenden
Verbindungen unter biorelevanten Bedingungen untersucht. Neben der Stabilität in saurem und schwach
basischem Milieu ist dabei insbesondere der mögliche Ligandenaustausch mit funktionellen Gruppen in
Aminosäureseitenketten von Bedeutung.
[1] T. J. D. Castillo, S. Sarkar, K. A. Abboud, A. S. Veige, Dalton Trans. 2011, 40, 8140-8144.
[2] L. Henry, C. Schneider, B. Mützel, P. V. Simpson, C. Nagel, K. Fucke, U. Schatzschneider,
Chem. Commun. 2014, 50, 15692-15695.

Anorganische Chemie und Materialwissenschaften
P56
Photocatalytic water splitting with [FeFe]-hydrogenase mimic in
aqueous micellar solution
M. Roos1, C. Lambert1, R. Luxenhofer2
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg
2Chair of chemical technology of material synthesis, University of Würzburg,
Röntgenring 11, 97074 Würzburg
Because of the continual growth of worldwide energy demand and against the background of the
climate change the interest in an environmental friendly energy carrier generation becomes more and
more important. One possible method is the photocatalytic water splitting, in which hydrogen is
generated due to sunlight irradiation. As catalyst a [FeFe]-hydrogenase mimic is used, which gets
reduced during the process by a light-excited photosensitizer. In order to archive a thermodynamic
favored electron cascade the redoxpotentials of the concerned components has to be adjusted to each
other.[1] Besides transition metal organic complexes like Tris(bipyridine)ruthenium(II) chloride also
pure organic compounds like the Xanthene dye Eosin Y could be use as photosensitizer.[1,2] As electron
source a sacrificial donor like triethylamine or the Vitamin C containing ascorbic acid is used.[1] To
perform the photocatalysis in pure water, the [FeFe]-catalyst, as a water insoluble compound, is
solubilized with poly(2-oxazoline) micelles.[3] Due to the dynamic character of the micelle formation
the exchange between the ambient solution and the [FeFe]-catalyst within the micelle is possible, so the
requirements for the photocatalytic activity of the three component system are fulfilled.
We synthesised in our group a series of [FeFe]-catalysts with different dithiolate bridging ligands
and characterised their photocatalytic properties. Thereby a maximum turnover number of 144 was
archived with the system shown above.
[1] M. Wang, L. Chen, X. Lia, L. Sun, Dalton Trans. 2011, 40, 12793-12800.
[2] C. Orain, F. Quentel, F. Gloaguen, ChemSusChem. 2014, 7, 638-643.
[3] Y. Han, Z. He, A. Schulz, T. K. Bronich, R. Jordan, R. Luxenhofer, A. V. Kabanov, Mol. Pharm.
2012, 9, 2302-2313.

Organische Chemie und Biochemie P57
Functionalised Diamond Particles for the Photocatalytic Conversion
of CO2
B. Kiendl, J. Fink, C. Heinz, A. Krueger1
1Institut für Organische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg
Carbon dioxide, one of the most alarming greenhouse gases, is usually reduced by transition metals
and their corresponding oxides.[1] Recently, synthetic diamond materials have been used to convert
carbon dioxide to carbon monoxide or even formaldehyde, either photocatalytically or
electrochemically.[2,3] Therefore, diamond based materials provide a cheap, antrophogenic and safe
alternative for the selective reduction of carbon dioxide.
Concerning diamond catalysed reduction processes, solvated electrons play an important role as
powerful reducing agent. In order to generate those electrons, boron-doped diamond materials have been
irradiated so far using high-energy UV light.[2] However, the electronic structure of diamond can not
only be influenced by dopants, but also by different surface terminations and functionalizations.
Figure 1: Immobilisation of transition metal complexes (TMC) by linker molecules.
In this work we present the synthesis of a bifunctionalised linker molecule and its subsequent grafting
onto diamond nanoparticles. Furthermore, the immobilisation of transition metal complexes on this
linker using the concept of click-chemistry is reported. The ability of these conjugates to reduce carbon
dioxide will be investigated in the context of photocatalysis.
This project has received funding from the European Unions´s Horizon 2020 Programme under Grant
Agreement no. 665085 (DIACAT).
[1] J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao,
J. Gu, Y. Yan, T. W. Shaw, E. Abelev, A. B. Bocarsly, Chem. Rev. 2015, 115, 12888-12935.
[2] L. Zhang, D. Zhu, G. M. Nathanson, R. J. Hamers, Angew. Chem., Int. Ed. 2014, 53, 9746-9750.
[3] K. Nakata, T. Ozaki, C. Terashima, A. Fujishima, Y. Einaga, Angew. Chem, Int. Ed. 2014, 53,
871-874.

Organische Chemie und Biochemie
P58
Synthesis and Characterization of Bay-tether-connected
Perylene Bisimide Dimers
C. Kaufmann, A. Nowak-Król, F. Würthner*
Universität Würzburg, Institut für Organische Chemie and Center for Nanosystems Chemistry,
Am Hubland, 97074 Würzburg, Germany
*e-mail: [email protected]
Perylenebisimides (PBI) form remarkable self-assembled structures induced by stacking of
monomeric units, yielding fascinating optical and electronic properties.[1] The aggregation behavior of
such organic dyes is often governed by a solvent, concentration and temperature dependence. This self-
assembly of perylenebisimide (PBI) dyes has been a subject of extensive investigations over more than
a decade. By developing a foldable system where single monomers are covalently fixed in a rigid
framework, the resulting PBI aggregate with defined geometry may shed light on the structure-property
relationship of this class of dyes.[2] Hence, we paid attention to covalent scaffolds that preorganize PBI
dyes in a suitable way with regard to intramolecular folding into specific three-dimensional
architectures. Therefore we present the synthesis and characterization of new covalently linked bis-
perylene-bisimids which were investigated by UV/Vis-, Fluorescence- and NMR-Spectroscopy to
determine their individual aggregation behavior – intermolecular aggregation into defined dye stacks
vs. self-assembly into larger aggregates.
Scheme 1: Synthetic route and molecular structures of bis-PBIs 1-3 (left) and structural model of bis-
PBI 1 dimers (right).
[1] a) X. Zhang, S. Rehm, M. M. Safont-Sempere, F. Würthner, Nature Chem. 2009, 1, 623-
629; b) Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P.
Marquetand, V. Engel, F. Würthner, Chem. Eur. J. 2007, 13, 436-449.
[2] B. Fimmel, M. Son, Y. M. Sung, M. Grüne, B. Engels, D. Kim, F. Würthner, Chem. Eur. J.
2015, 21, 615-630.

Anorganische Chemie und Materialwissenschaften P59
Rare earth metal coordination polymers with pyrene-2,7-
dicarboxylate linkers
A. E. Sedykh, J. Merz, L. Ji, T. B. Marder, K. Müller-Buschbaum
Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Würzburg, Germany
A coordination polymer (CP) is a coordination compound with repeating coordination entities
extending in 1, 2, or 3 dimensions.[1] Rare earth metals (RE) are outstanding functional metal centers
due to their coordination behavior[2] and luminescent properties.[3] The architecture of their coordination
polymers is directly connected with the linker structure.[4] Pyrene-2,7-dicarboxylic acid (H2PDC) is a
linear rigid molecule, and usage of it as a linker source might lead to the formation of coordination
polymers with potential voids – metal-organic frameworks (MOFs). Till now only a few examples of
CPs with pyrene-2,7-dicarboxylate are known, almost all of them with transition metals,[5,6] but one with
rare earth metal – terbium.[6]
Reaction between pyrene-2,7-dicarboxylic acid and rare earth metal chloridesleads to the formation
of desired products. Several different structures were obtained, determined by infrared spectroscopy and
powder X-ray diffraction, still yet only for one of them a single crystal was successfully acquired and
studied. Interesting results await in the luminescence properties studies: a combination of luminescent
lanthanides and linker leads to the complete absence of luminescence. This is most likely the result of
radiationless decay instead of emission from corresponding lanthanide and ligand energy levels.
Nonetheless, products of non-luminescent metals show ligand-based emission. Wherein, the ligand
emission band is shifted hypsochromically by 50 nm as result of deprotonation and coordination to the
metal. In addition, the intensity of some processes is increasing upon cooling.
[1] S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M.
O’Keeffe, M. P. Suh, J. Reedijk, Pure Appl. Chem. 2013, 85, 1715.
[2] S. A. Cotton, J. M. Harrowfield, “Lanthanides: Coordination Chemistry. Encyclopedia of
Inorganic and Bioinorganic Chemistry”, 2012.
[3] G. F. de Sa, O. L. Malta, C. de Mello Donega, A. M. Simas, R. L. Longo, P. A. Santa-Cruz, E.
F. da Silva Jr., Coord. Chem. Rev. 2000, 196, 165.
[4] G. Zhu, Coord. Chem. Rev. 2009, 253, 2891.
[5] M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O.M. Yaghi, Science 2002,
295, 469.
[6] N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O’Keeffe, O. M. Yaghi, J. Am. Chem. Soc. 2005,
127, 1504.

Pharmazie und Lebensmittelchemie
P60
Site-directed incorporation of propargyl-L-lysine into insulin-like
growth factor- I (IGF-I) for site specific decoration
F. Wu, J. Ritzer, T. Lühmann, L. Meinel
Institute for Pharmacy and Food Chemistry, University of Wuerzburg, Am Hubland, 97074,
Wuerzburg, Germany
Current decoration approaches, which typically follow random coupling strategies to present lysines
or cysteines on the protein-surface, have led to heterogeneous pharmaceutical products.[1] This impacts
their activity and challenges homogenous product outcome. Here we pursue a highly attractive strategy
for the controlled insulin-like growth factor- I (IGF-I) conjugates delivery.
We genetically engineered IGF-I variants (TrxplkIGF-I) containing a N-terminal thioredoxin[2] (Trx)
His6[3] tag-thrombin cleavage site and a non-natural amino acid, propargyl-protected lysine derivative
(plk) incorporated at position 3 of IGF-I, thereby allowing targeted, specific decoration of IGF-I with
other molecules. The expression of the TrxplkIGF-I fusion protein was compared in four different
Escherichia coli hosts (BL21 (DE3), shuffle T7, Rosetta DE3, and C321 delA.exp). Additionally, the
effects of different expression conditions (induction temperature, isopropyl β-D-1-
thiogalactopyranoside (IPTG) concentration, plk amount) were systematically investigated.
Figure 1: Expression of plkIGF-I. The propargyl-L-lysine (plk) is incorporated at the site of an amber
stop codon (UAG) of IGF-I by using the orthogonal pair PylRS/tRNAPyl from M. barkeri.
The TrxplkIGF-I fusion protein was expressed in all these four strains with different expression
levels, whereas shuffle T7 showed the highest soluble TrxplkIGF-I production. Furthermore,
TrxplkIGF-I production was correlated to plk concentrations and induction parameters including time
and IPTG amount. Purified plkIGF-I was successfully demonstrated by Western Blot. In conclusion, we
maximize the risk-benefit ratio of our IGF-I conjugate by directing these anabolic therapeutic to local
sites in need while minimizing systemic exposure.
[1] K. S. Masters, Macromolecular Bioscience 2011, 11, 1149-1163.
[2] D. Zhang, P. Wei, L. Fan, J. Lian, L. Huang, J. Cai, Z. Xu, Process Biochemistry 2010, 45,
1401-1405.
[3] E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentz, D. Stuber, Nat Biotech 1988, 6, 1321-1325.
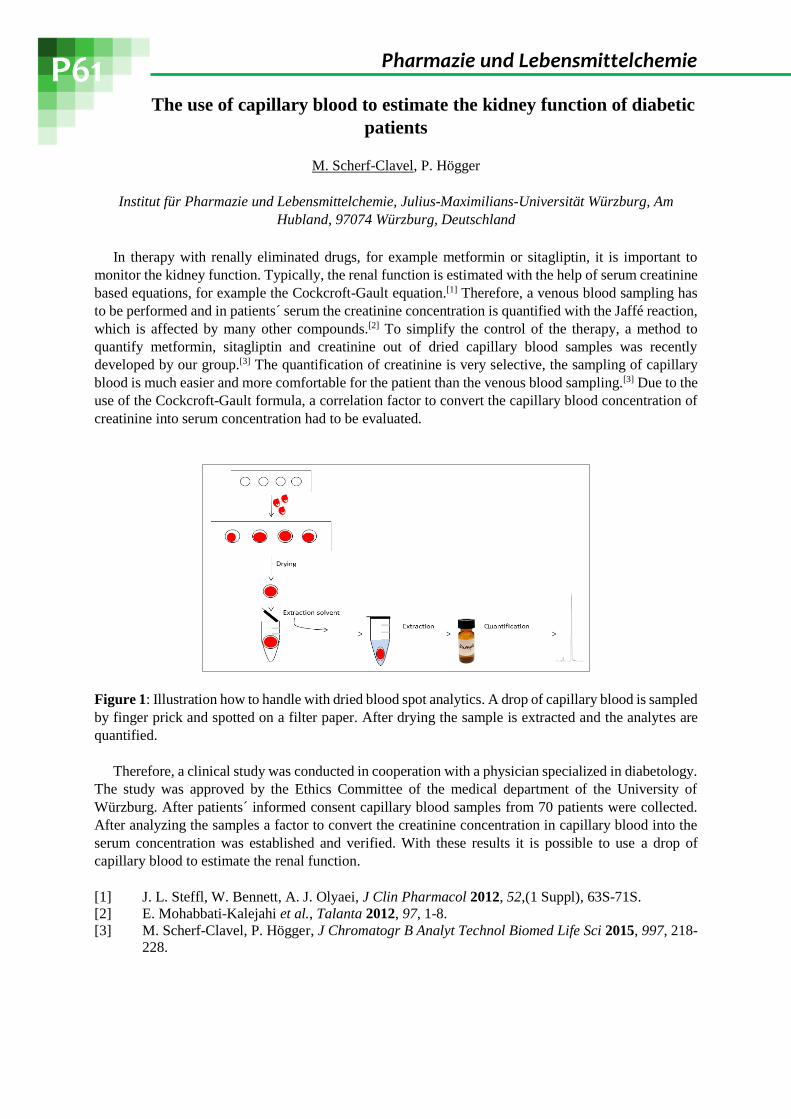
Pharmazie und Lebensmittelchemie P61
The use of capillary blood to estimate the kidney function of diabetic
patients
M. Scherf-Clavel, P. Högger
Institut für Pharmazie und Lebensmittelchemie, Julius-Maximilians-Universität Würzburg, Am
Hubland, 97074 Würzburg, Deutschland
In therapy with renally eliminated drugs, for example metformin or sitagliptin, it is important to
monitor the kidney function. Typically, the renal function is estimated with the help of serum creatinine
based equations, for example the Cockcroft-Gault equation.[1] Therefore, a venous blood sampling has
to be performed and in patients´ serum the creatinine concentration is quantified with the Jaffé reaction,
which is affected by many other compounds.[2] To simplify the control of the therapy, a method to
quantify metformin, sitagliptin and creatinine out of dried capillary blood samples was recently
developed by our group.[3] The quantification of creatinine is very selective, the sampling of capillary
blood is much easier and more comfortable for the patient than the venous blood sampling.[3] Due to the
use of the Cockcroft-Gault formula, a correlation factor to convert the capillary blood concentration of
creatinine into serum concentration had to be evaluated.
Figure 1: Illustration how to handle with dried blood spot analytics. A drop of capillary blood is sampled
by finger prick and spotted on a filter paper. After drying the sample is extracted and the analytes are
quantified.
Therefore, a clinical study was conducted in cooperation with a physician specialized in diabetology.
The study was approved by the Ethics Committee of the medical department of the University of
Würzburg. After patients´ informed consent capillary blood samples from 70 patients were collected.
After analyzing the samples a factor to convert the creatinine concentration in capillary blood into the
serum concentration was established and verified. With these results it is possible to use a drop of
capillary blood to estimate the renal function.
[1] J. L. Steffl, W. Bennett, A. J. Olyaei, J Clin Pharmacol 2012, 52,(1 Suppl), 63S-71S.
[2] E. Mohabbati-Kalejahi et al., Talanta 2012, 97, 1-8.
[3] M. Scherf-Clavel, P. Högger, J Chromatogr B Analyt Technol Biomed Life Sci 2015, 997, 218-
228.

Physikalische und Theoretische Chemie
P62
The CAST program – A tool for specialized potential energy surface
investigations
D. Weber1, D. Bellinger1, D. Kaiser1, M. Prem1, S. Sauer1, S. Wirsing1, B. Engels1
1Julius-Maximilians-Universität, Institut für Physikalische und Theoretische Chemie,
Emil-Fischer-Straße 42, D-97074 Würzburg, Germany
The CAST (Conformational Analysis and Search Tool)[1] program is a computational chemist’s
toolkit which enables users to investigate ground state chemistry problems like sampling, global
optimization and analysis in a sophisticated manner. The program-package is founded on the principle
to be independent of the underlying potential energy description. In this way, all implemented methods
can be performed via classical force field calculations as well as DFT or semi empirical methods.
CAST offers the Tabu-Search approach for finding the global minimum for a given system, e.g. an
optimal solvation shell. Multiple specially designed algorithms for the treatment of the solvation
problem are available, enhancing the speed and quality of optimizations of water shells. CAST is capable
to deal with complex chemical reactions through the use of the Pathopt[2] algorithm, as well as various
doubly nudged elastic band (NEB)[3] methods.
Within the framework of sampling of systems such as protein-inhibitor complexes CAST offers the
possibility to simulate changes in terms of free energy (FEP[4], modified FEP, Umbrella Sampling[5]). A
further but no less important issue is the analysis of data obtained from molecular simulations. Within
CAST this may be addressed through novel principal component analysis(PCA)[6] based approaches.
Acknowledgments: We are grateful to the Deutsche Forschungsgemeinschaft (GRK 1221, FOR 1809,
SPP 1355) for financial support.
[1] C. Grebner, J. Becker, D. Weber, D. Bellinger, M. Tafipolski, C. Brückner, B. Engels, J. Comp.
Chem. 2014, 35, 1801-1807.
[2] D. Weber, D. Bellinger, B. Engels, Methods in Enzymology 2016, 578, 145-167.
[3] G. Henkelman, B. P. Uberuaga, H. Jonsson, J. Chem. Phys. 2000, 113, 9901-9904.
[4] C. Chipot, A. Pohorille, Free energy calculations. Springer-Verlag Berlin, Heidelberg, 2007.
[5] G. M. Torrie, J. P. Valleau, J. Comp. Phys. 1977, 23, 187-199.
[6] H. Abdi, L. J. Williams, Wiley Interdisciplinary Reviews: Computational Statistics 2010, 2, 433-
459.
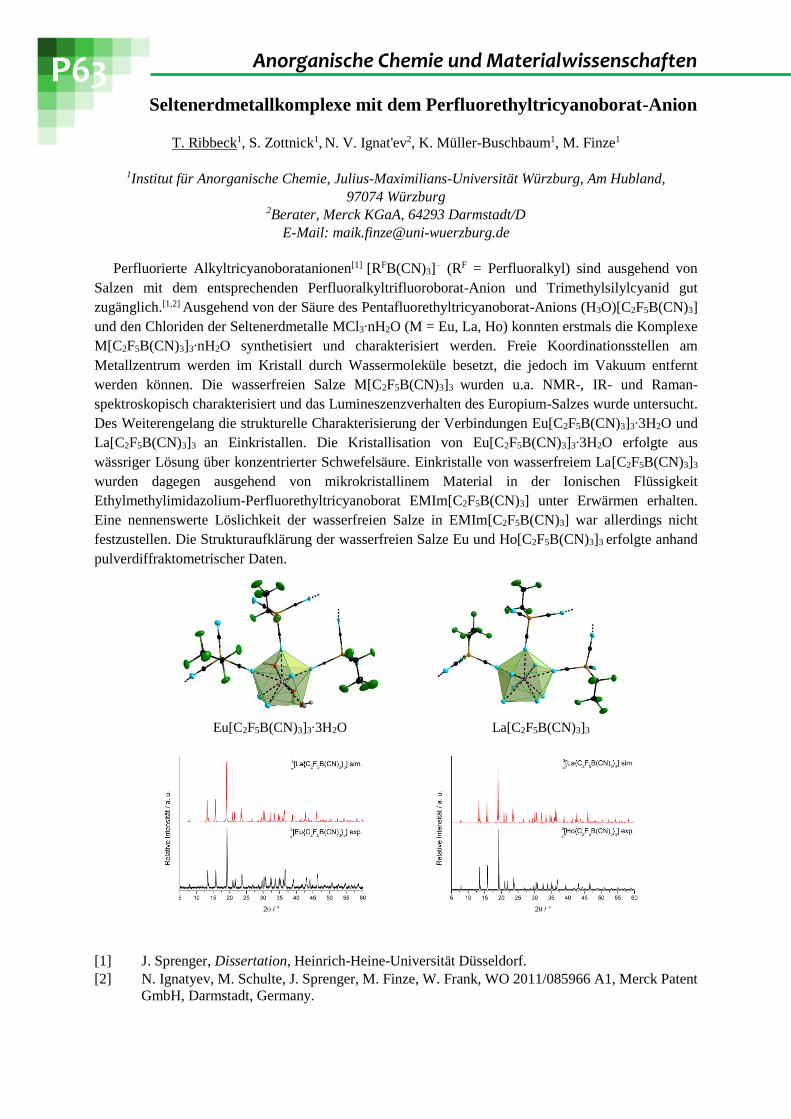
Anorganische Chemie und Materialwissenschaften P63
Seltenerdmetallkomplexe mit dem Perfluorethyltricyanoborat-Anion
T. Ribbeck1, S. Zottnick1, N. V. Ignat'ev2, K. Müller-Buschbaum1, M. Finze1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland,
97074 Würzburg 2Berater, Merck KGaA, 64293 Darmstadt/D
E-Mail: [email protected]
Perfluorierte Alkyltricyanoboratanionen[1] [RFB(CN)3]– (RF = Perfluoralkyl) sind ausgehend von
Salzen mit dem entsprechenden Perfluoralkyltrifluoroborat-Anion und Trimethylsilylcyanid gut
zugänglich.[1,2] Ausgehend von der Säure des Pentafluorethyltricyanoborat-Anions (H3O)[C2F5B(CN)3]
und den Chloriden der Seltenerdmetalle MCl3∙nH2O (M = Eu, La, Ho) konnten erstmals die Komplexe
M[C2F5B(CN)3]3∙nH2O synthetisiert und charakterisiert werden. Freie Koordinationsstellen am
Metallzentrum werden im Kristall durch Wassermoleküle besetzt, die jedoch im Vakuum entfernt
werden können. Die wasserfreien Salze M[C2F5B(CN)3]3 wurden u.a. NMR-, IR- und Raman-
spektroskopisch charakterisiert und das Lumineszenzverhalten des Europium-Salzes wurde untersucht.
Des Weiterengelang die strukturelle Charakterisierung der Verbindungen Eu[C2F5B(CN)3]3∙3H2O und
La[C2F5B(CN)3]3 an Einkristallen. Die Kristallisation von Eu[C2F5B(CN)3]3∙3H2O erfolgte aus
wässriger Lösung über konzentrierter Schwefelsäure. Einkristalle von wasserfreiem La[C2F5B(CN)3]3
wurden dagegen ausgehend von mikrokristallinem Material in der Ionischen Flüssigkeit
Ethylmethylimidazolium-Perfluorethyltricyanoborat EMIm[C2F5B(CN)3] unter Erwärmen erhalten.
Eine nennenswerte Löslichkeit der wasserfreien Salze in EMIm[C2F5B(CN)3] war allerdings nicht
festzustellen. Die Strukturaufklärung der wasserfreien Salze Eu und Ho[C2F5B(CN)3]3 erfolgte anhand
pulverdiffraktometrischer Daten.
Eu[C2F5B(CN)3]3∙3H2O La[C2F5B(CN)3]3
[1] J. Sprenger, Dissertation, Heinrich-Heine-Universität Düsseldorf.
[2] N. Ignatyev, M. Schulte, J. Sprenger, M. Finze, W. Frank, WO 2011/085966 A1, Merck Patent
GmbH, Darmstadt, Germany.
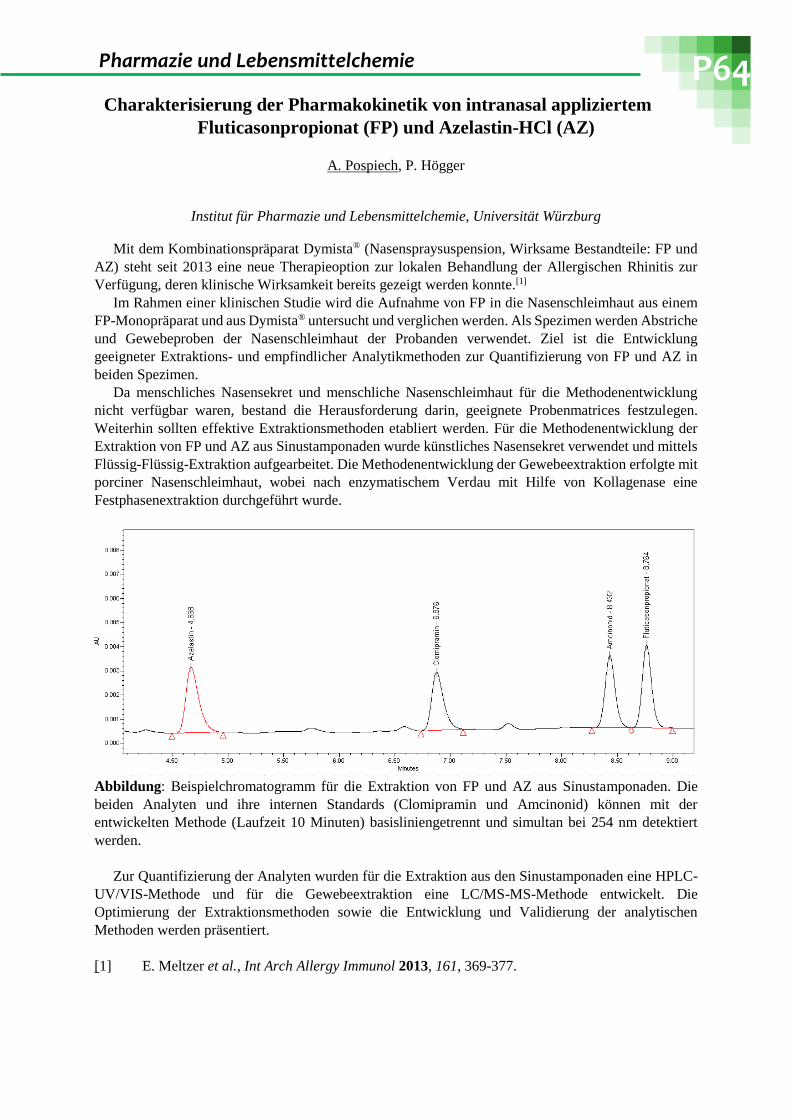
Pharmazie und Lebensmittelchemie
P64
Charakterisierung der Pharmakokinetik von intranasal appliziertem
Fluticasonpropionat (FP) und Azelastin-HCl (AZ)
A. Pospiech, P. Högger
Institut für Pharmazie und Lebensmittelchemie, Universität Würzburg
Mit dem Kombinationspräparat Dymista® (Nasenspraysuspension, Wirksame Bestandteile: FP und
AZ) steht seit 2013 eine neue Therapieoption zur lokalen Behandlung der Allergischen Rhinitis zur
Verfügung, deren klinische Wirksamkeit bereits gezeigt werden konnte.[1]
Im Rahmen einer klinischen Studie wird die Aufnahme von FP in die Nasenschleimhaut aus einem
FP-Monopräparat und aus Dymista® untersucht und verglichen werden. Als Spezimen werden Abstriche
und Gewebeproben der Nasenschleimhaut der Probanden verwendet. Ziel ist die Entwicklung
geeigneter Extraktions- und empfindlicher Analytikmethoden zur Quantifizierung von FP und AZ in
beiden Spezimen.
Da menschliches Nasensekret und menschliche Nasenschleimhaut für die Methodenentwicklung
nicht verfügbar waren, bestand die Herausforderung darin, geeignete Probenmatrices festzulegen.
Weiterhin sollten effektive Extraktionsmethoden etabliert werden. Für die Methodenentwicklung der
Extraktion von FP und AZ aus Sinustamponaden wurde künstliches Nasensekret verwendet und mittels
Flüssig-Flüssig-Extraktion aufgearbeitet. Die Methodenentwicklung der Gewebeextraktion erfolgte mit
porciner Nasenschleimhaut, wobei nach enzymatischem Verdau mit Hilfe von Kollagenase eine
Festphasenextraktion durchgeführt wurde.
Abbildung: Beispielchromatogramm für die Extraktion von FP und AZ aus Sinustamponaden. Die
beiden Analyten und ihre internen Standards (Clomipramin und Amcinonid) können mit der
entwickelten Methode (Laufzeit 10 Minuten) basisliniengetrennt und simultan bei 254 nm detektiert
werden.
Zur Quantifizierung der Analyten wurden für die Extraktion aus den Sinustamponaden eine HPLC-
UV/VIS-Methode und für die Gewebeextraktion eine LC/MS-MS-Methode entwickelt. Die
Optimierung der Extraktionsmethoden sowie die Entwicklung und Validierung der analytischen
Methoden werden präsentiert.
[1] E. Meltzer et al., Int Arch Allergy Immunol 2013, 161, 369-377.

Pharmazie und Lebensmittelchemie P65
Bacterial protease responsive antibiotic delivery
M. C. Amstalden1, A. A. Fayad2, R. Müller2, T. Lühmann1, L. Meinel1
1Institute of pharmacy and food chemistry, University of Würzburg, Am Hubland 97074 Würzburg
2Helmholtz-Institute for Pharmaceutical Research Saarland, Saarland University, 66123 Saarbrücken
One cause driving antibiotic resistance is untargeted systemic circulation, leading to higher doses as
well as impacting the commensal, desirable bacterial flora. In this study, we aimed at developing a
bioresponsive drug delivery system (DDS) responding to virulent Staphylococcus aureus (S. aureus)
with antibiotic discharge, thereby homing antibiotic activity to sites in need while minimizing off target
activity.
The DDI comprises a polymer, a peptide cleavable linker (PCL) and the antibiotic which is covalently
attached to the peptide’s C terminus. The PCL responds to the protease aureolysin, which is a virulence
factor of S. aureus strains,[1] while the remaining amino acids attached to the antibiotic are cleaved by
human aminopeptidases (Figure 1).
Figure 1: Schematic representation of the protease dependent DDS. The dual-gated mechanism
comprise cleavage of the index protease aureolysin followed by the human aminopeptidase, present in
human plasma, and release of the active drug compound at target sites
PCL prototypes were synthesized by Fmoc based solid phase peptide synthesis.[2] The PCLs were
analyzed for sensitivity towards aureolysin (1.4 µg/mL) and human proteases. The conjugation of the
PCL to the antibiotic was performed via EDC/NHS reaction.[3] Resulting peptide fragments after
aureolysin cleavage were further analyzed for aminopeptidase cleavage using a final concentration of
3.6 µg/mL. We identified two selective and sensitive PCLs for aureolysin. Aminopeptidase treatment
of theaureolysin-cleaved peptide-drug fragment resulted in the release of the free compound within 20
minutes. Ongoing work focuses on the construction of the entire conjugate and analysis of antibacterial
activity.
[1] A. J. Laarman et al., J. Immunol. 2011, 186, 6445-6453.
[2] J. L. T. Ritzer et al., Journal of controlled release (submitted).
[3] K. Sakurai, T. M. Snyder, D. R. Liu, J. Am. Chem. Soc. 2005, 127, 1660-1661.

Anorganische Chemie und Materialwissenschaften
P66
iClick reactions of Pt-and Pd-azide complexes with
electron-poor alkynes
N. Feizy1, U. Schatzschneider1,*
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, D-97074
Würzburg (Germany)
e-mail: [email protected]
The concept of "iClick" reactions was first introduced by Veige for the inorganic version of the
1,3-dipolar cycloaddition between a metal-coordinated azide such as triphenylphosphine gold(I) azide
with triphenylphosphine gold(I) phenylacetylide to form dinuclear triazolate-bridged gold complexes.[1]
Triazolates and their metal complexes are used in pharmaceutical and inorganic medicinal chemistry.[2]
For example, the treatment of human breast cancer cells with triazolate-linked Au(I)-peptide conjugates
led to a breakdown of essential cell functions and eventually cell death.[3] To extend the range of
applications of the iClick reaction,[4] in the present work, the synthesis of platinum(II) and palladium(II)
triazolate complexes through a [3+2]-cycloaddition of Pt or Pd azide complexes with
dimethylacetylenedicarboxylate (DMAD) was investigated. Thus, an anionic tridentate N,N,N-chelating
1,3-bis(2-pyridylimino)isoindoline ligand was prepared and coordinated to a palladium(II) unit to obtain
a neutral complex. The initial chloride ligand was then replaced by reaction with sodium azide. Finally,
the iClick reaction with DMAD lead to the desired triazolate product and thus opens up a new and easy
access to square-planar metal(II)triazolate complexes. As an alternative pathway to cationic complexes
of a similar core structure, substituted terpyridine derivatives are currently explored and the
corresponding platinum(II) compounds are also under preparation.
[1] T. J. Del Castillo, S. Sarkar, K. A. Abboud, A. S. Veige, Dalton Trans. 2011, 40, 8140-8144.
[2] T. M. Klapötke, J. Stierstorfer, J. Am. Chem. Soc. 2009, 131, 1122-1134.
[3] S. D. Köster, S. Can, I. Kitanovic, S. Wolfl, R. Rubbiani, I. Ott, P. Riesterer, A. Prokop, K.
Merz, N. Metzler-Notle, Chem. Sci. 2012, 3, 2062-2072.
[4] L. Henry, C. Schneider, B. Mützel, P. V. Simpson, C. Nagel, K. Fucke, U. Schatzschneider,
Chem. Commun. 2014, 50, 15692-15695.

Organische Chemie und Biochemie P67
Self-Assembled Bolamphiphilic Perylene Bisimides in Water
B. Soberats, D. Görl, P. Syamala, F. Würthner*
Universität Würzburg, Center for Nanosystems Chemistry, Institut für Organische Chemie, and
Bavarian Polymer Institute, Universität Würzburg, 97074 Würzburg, Germany
*e-mail: [email protected]
Self-assembly of dyes in aqueous media has attracted a great deal of attention for the development
of biocompatible sensors and stimuli responsive photoactive materials.[1] We recently focus on the
fundamental understanding of the thermodynamic aspects of self-assembly in water of bolamphiphilic
perylene bisimide (PBIs) dyes bearing oligoethylene glycol (OEG) groups (Figure 1a).[2] We found that
the entropic release of water from the OEG chains overcomes the enthalpic contribution from π- π
stacking interactions (Figure 1c), which has a determining influence on the thermodynamics of the
aggregation process.[3] This leads to an inverse temperature response which induces lower critical
solution temperature (LCST) phenomena, where the solution phase separates at a particular temperature
upon heating. In the present study we show a novel supramolecular approach to control the LCST
transitions in PBI 1/PBI 2 mixtures by adjusting their mixing ratio (Figure 1a).[4]
Figure 1: a) Schematic representation of the supramolecular approach applied to control the LCST
phase transition in bolamphiphilic PBI 1 and PBI 2 aggregates. b) Illustration of the LCST phase
transition in PBI1/PB2 hydrogels.
Furthermore, based on this concept, we prepared stimuli responsive hydrogels that undergo a
remarkable color change triggered by the LCST phase transition between a hydrogel state and a lyotropic
liquid-crystalline state (Figure 1b).[4] This study open new avenues to the development of photoactive
biocompatible sensors based on aqueous soft matter systems.
[1] E. Krieg, M. M. C. Bastings, P. Besenius, B. Rybtchinski, Chem. Rev. 2016, 116, 2414-2477.
[2] a) D. Görl, X. Zhang, V. Stepanenko, F. Würthner, Nat. Commun. 2015, 6, 7009. b) X. Zhang,
D. Görl, V. Stepanenko, F. Würthner, Angew. Chem. Int. Ed. 2014, 53, 1270-1274.
[3] D. Görl, F. Würthner, Angew. Chem. Int. Ed. 2016, 55, 12094-12098.
[4] D. Görl, B. Soberats, S. Herbst, V. Stepanenko, F. Würthner, Chem. Sci. 2016, 7, 6786-6790.
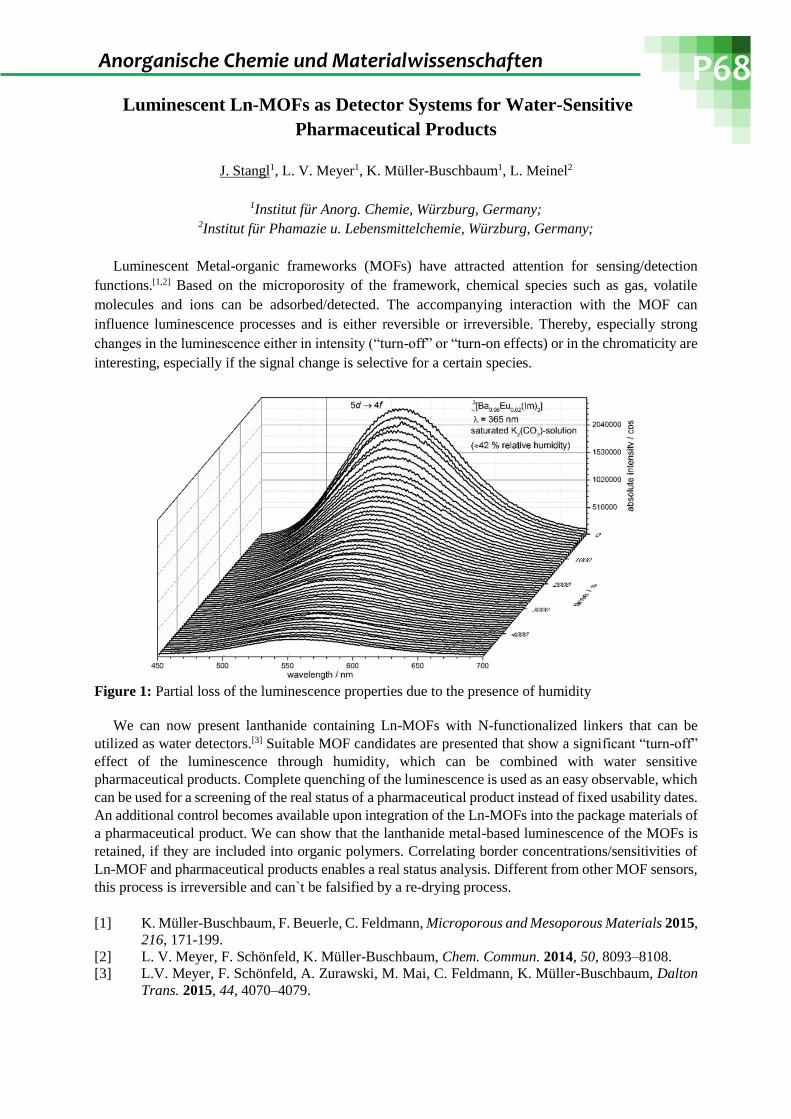
Anorganische Chemie und Materialwissenschaften
P68
Luminescent Ln-MOFs as Detector Systems for Water-Sensitive
Pharmaceutical Products
J. Stangl1, L. V. Meyer1, K. Müller-Buschbaum1, L. Meinel2
1Institut für Anorg. Chemie, Würzburg, Germany;
2Institut für Phamazie u. Lebensmittelchemie, Würzburg, Germany;
Luminescent Metal-organic frameworks (MOFs) have attracted attention for sensing/detection
functions.[1,2] Based on the microporosity of the framework, chemical species such as gas, volatile
molecules and ions can be adsorbed/detected. The accompanying interaction with the MOF can
influence luminescence processes and is either reversible or irreversible. Thereby, especially strong
changes in the luminescence either in intensity (“turn-off” or “turn-on effects) or in the chromaticity are
interesting, especially if the signal change is selective for a certain species.
Figure 1: Partial loss of the luminescence properties due to the presence of humidity
We can now present lanthanide containing Ln-MOFs with N-functionalized linkers that can be
utilized as water detectors.[3] Suitable MOF candidates are presented that show a significant “turn-off”
effect of the luminescence through humidity, which can be combined with water sensitive
pharmaceutical products. Complete quenching of the luminescence is used as an easy observable, which
can be used for a screening of the real status of a pharmaceutical product instead of fixed usability dates.
An additional control becomes available upon integration of the Ln-MOFs into the package materials of
a pharmaceutical product. We can show that the lanthanide metal-based luminescence of the MOFs is
retained, if they are included into organic polymers. Correlating border concentrations/sensitivities of
Ln-MOF and pharmaceutical products enables a real status analysis. Different from other MOF sensors,
this process is irreversible and can`t be falsified by a re-drying process.
[1] K. Müller-Buschbaum, F. Beuerle, C. Feldmann, Microporous and Mesoporous Materials 2015,
216, 171-199.
[2] L. V. Meyer, F. Schönfeld, K. Müller-Buschbaum, Chem. Commun. 2014, 50, 8093–8108.
[3] L.V. Meyer, F. Schönfeld, A. Zurawski, M. Mai, C. Feldmann, K. Müller-Buschbaum, Dalton
Trans. 2015, 44, 4070–4079.

Organische Chemie und Biochemie P69
Total Synthesis of the Antimalarial Naphthylisoquinolines Dionco-
phylline C, 5-epi-Dioncophylline C and Dioncophylline F
W. Shamburger, R. Seupel, S. K. Bischof, G. Bringmann
Institute of Organic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg
Dioncophylline C (3a), a 5,1’-coupled naphthylisoquinoline alkaloid isolated from the West African
liana Triphyophyllum peltatum, was found to show remarkable activity against Plasmodium falciparum
in vitro and against P. berghei in vivo.[1,2] Therefore 3a represents an attractive synthetic target. This
was further underlined by the outcome of Q(SAR) studies on structurally simplified analogs of 3a, such
as molecules 4, 5, and 6, which were all found to display distinct weaker antiplasmodial activities
compared to 3a.[3] Following a highly concise route, we now aim at the synthesis of compounds which
differ from 3a only in one single structural parameter like e.g., 5-epi-dioncophylline C (3b). Key step is
the Pd-catalyzed Suzuki-Miyaura cross-coupling of a naphthalene boronic acid ester (here 1) and an
isoquinoline bromine (here 2). This approach furnished 3a and its atropisomer 3b in a good chemical
yield and in stereochemically pure form after their resolution on a Symmetry-C18 column. This pathway
has also been successfully applied to the total synthesis of the first, and so far only, 5,8’-coupled
Dioncophyllaceae-type alkaloid dioncophylline F (7a and 7b), which is configurationally unstable at the
biaryl axis.
[1] G. Bringmann, Ann. Trop. Med. Parasitol. 1996, 90, 115-123.
[2] G. Bringmann, Antimicrob. Agents Chemother. 1997, 41, 2533-2539.
[3] G. Bringmann, Eur. J. Med. Chem. 2010, 45, 5370-5383.
OH
NH
Me
Me
R
N
Me
Me
5
8'
OMeOMe
PMe
S
Me
OH
NH
Me
Me
R
S N
Me
Me
5
8'
OMe
M
OMe
dioncophylline F ( + )7a 7b
fast
O Pri
NBn
Me
Me
Me
Br
R
R
Me
O Pri OMe
BPin
OH
NH
Me
Me
N
Me
Me
5
1'
OMeOH
MeP
OH
NH
Me
Me
N
Me
Me
5
1'
OMe
5
OH
Me
+
dioncophylline C ( )3a 5- -dioncophylline C ( )epi 3b
+
1 2
iPrO OMe
OH
NH
Me
OMe
N
Me
Cl
OH
NN
Me
R
Me
MeO
OH
NN
MeMe
4 5 6

Anorganische Chemie und Materialwissenschaften
P70
Pseudo-polypeptides forming thermo responsive, biocompatible and
injectable gels for multiple biomedical applications
T. Lorson1, T. Jüngst2, J. Groll2, T. Lühmann3, R. Luxenhofer1
1Functional Polymer Materials, Chair for Chemical Technology of Materials Synthesis, University of
Würzburg, Röntgenring 11, 97070 Würzburg, Germany 2Department for Functional Materials in Medicine and Dentistry, University of Würzburg,
Pleicherwall 2, D 97070 Würzburg, Germany 3Institute for Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg,
Germany
Stimuli-responsive polymers that form physical or chemical hydrogels find great interest among
engineers, scientist and clinicians.[1] With this development and the ever increasing visions on potential
and actual applications, the need of suitable and adjustable hydrogels is rapidly developing and can be
seen as a potential bottleneck. They have to meet various requirements like sufficient quantity, consistent
quality and tunable biological and physical properties.[2] Having this in mind, natural polymers as well
as synthetic polymers need to be taken into account.
In the last decades poly(2-oxazoline)s, a prominent member of pseudo-polypeptides, have been
intensely investigated, especially as thermo responsive materials[3] and for the usage as biomedical
applications.[4] However, to the best of our knowledge no reports on thermogelling poly(2-oxazoline)s
can be found through the literature. Here we report the synthesis of novel thermogelling pseudo-
polypeptides, their cytocompatibility and structure property relationships with respect to their
rheological properties. 3D-printing experiments revealed potential applicability as BioInk.
[1] T. Jüngst, W. Smolan, K. Schacht, T. Scheibel, J. Groll, Chem. Rev. 2015.
[2] S. Wang, J. M. Lee, W. Y. Yeong, Int. J. Bioprinting 2015.
[3] C. Weber, R. Hoogenboom, U. S. Schubert, Progress in Polymer Science 2012, 37, 686.
[4] R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov, R. Jordan, Macromolecular
rapid communications 2012, 33, 1613.

Anorganische Chemie und Materialwissenschaften P71
Reaktivität N-heterocyclischer Carbene gegenüber Hauptgruppen-
Element-Verbindungen der 14. und 15. Gruppe
H. Schneider, D. Schmidt, U. Radius*
Institut für Anorganische Chemie der Julius-Maximilians-Universität Würzburg
[email protected], [email protected]
In den letzten Jahren wurde durch verschiedenste Beispiele gezeigt, dass sich die Verwendung N-
heterocyclischer Carbene nicht nur auf den Einsatz als Coliganden in Übergangsmetall-Komplexen
beschränkt. Neuartige subvalente Hauptgruppen-Element-Verbindungen wie auch Verbindungen mit
Element-Element-Bindungen konnten durch die guten σ-Donor-Eigenschaften dieser Singulett-Carbene
stabilisiert werden. Überraschender Weise zeigte sich, dass NHCs neben der Rolle als unschuldige σ-
Donoren, ebenfalls zur Element-Element- bzw. Element-Wasserstoff-Bindungsaktivierung unter milden
Bedingungen fähig sind.[1] Weitere Untersuchungen belegten, dass die Addukte von NHCs mit Lewis-
sauren Verbindungen bzw. Bindungsaktivierungsprodukte gegebenenfalls bei höheren Temperaturen
eine Ringerweiterungsreaktion (RER) eingehen. Durch Umsetzung diverser NHCs mit Phenylsilanen
konnten wir zeigen, das eine Insertion der Silyl-Einheit in den fünfgliedrigen Heterocyclus des NHCs
stattfindet und durch Migration der zuvor an das Silan gebundenen Wasserstoffatome bzw. Reste die
entsprechende Diazasilinane gebildet werden.[2] Ähnliche Reaktivitäten wurden von Hill[3], Rivard[4],
Inoue[5], Stephan[6] und unserer Gruppe[7-8] für Beryllium- wie auch Borhydridverbindungen beobachtet
und es konnte damit veranschaulicht werden, dass es ein gänzlich unbekanntes Feld der Reaktivität N-
heterocyclischer Carbene zu entdecken gilt. Da sich die Untersuchungen der Reaktivität bis dato mit
wenigen Ausnahmen auf die Element-Hydrid-Verbindungen der 13. und 14. Gruppe konzentrieren,
präsentieren wir hier unsere Ergebnisse zur Reaktivität von NHCs gegenüber Phenylchlorsilanen[9]
sowie den deutlich Lewis-basischeren Arylphosphanen[10].
[1] G. D. Frey, J. D. Masuda, B. Donnadieu, G. Bertrand, Angew. Chem. Int. Ed. 2010, 49, 9444.
[2] D. Schmidt, J. H. J. Berthel, S. Pietsch, U. Radius, Angew. Chem. Int. Ed. 2012, 51, 8881.
[3] M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon, Angew. Chem.
Int. Ed. 2012, 51, 2098.
[4] S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson, E. Rivard, Chem. Eur. J. 2012, 18, 13810.
[5] D. Franz, S. Inoue, Chem. Asian J. 2014, 9, 2083.
[6] T. Wang, D. W. Stephan, Chem. Eur. J. 2014, 20, 3036.
[7] S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius, T. B. Marder, Chem. Eur. J. 2015, 21,
9018.
[8] S. Würtemberger-Pietsch, H. Schneider, T. B. Marder, U. Radius, Chem. Eur. J. 2016, 22,
13032.
[9] H. Schneider, D. Schmidt, U. Radius, Chem. Eur. J. 2015, 21, 2793.
[10] H. Schneider, D. Schmidt, U. Radius, Chem. Commun. 2015, 51, 10138.
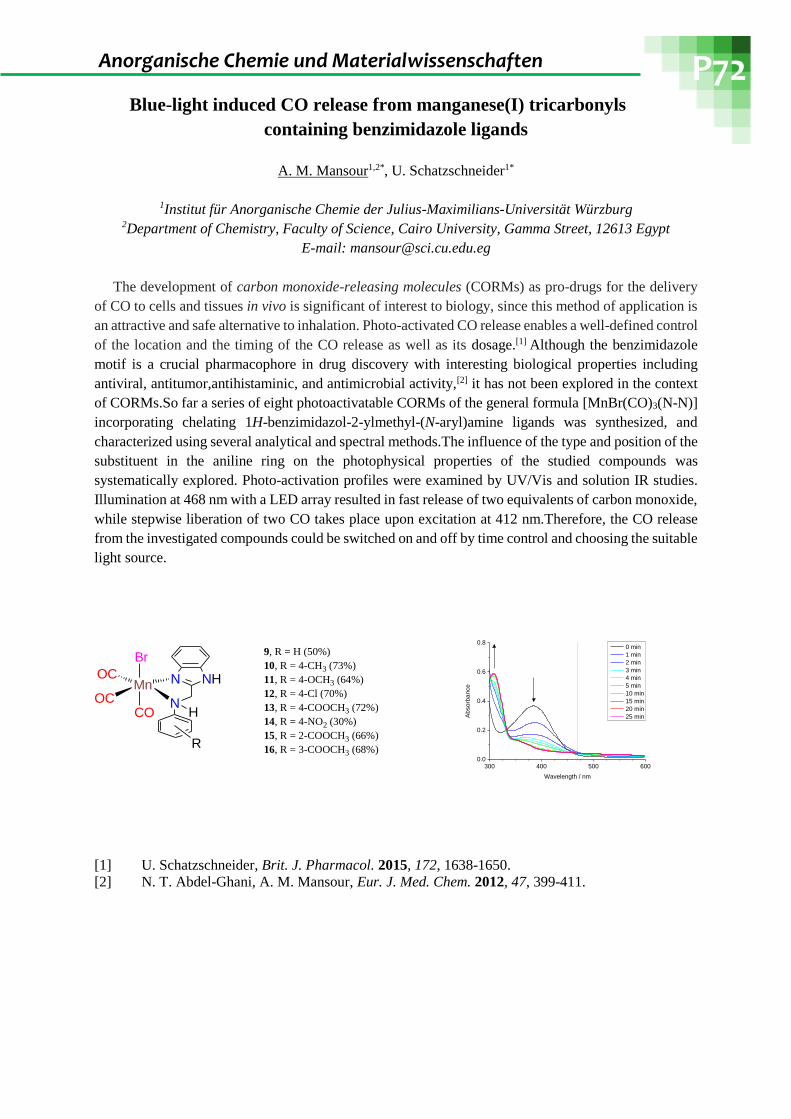
Anorganische Chemie und Materialwissenschaften
P72
Blue-light induced CO release from manganese(I) tricarbonyls
containing benzimidazole ligands
A. M. Mansour1,2*, U. Schatzschneider1*
1Institut für Anorganische Chemie der Julius-Maximilians-Universität Würzburg
2Department of Chemistry, Faculty of Science, Cairo University, Gamma Street, 12613 Egypt
E-mail: [email protected]
The development of carbon monoxide-releasing molecules (CORMs) as pro-drugs for the delivery
of CO to cells and tissues in vivo is significant of interest to biology, since this method of application is
an attractive and safe alternative to inhalation. Photo-activated CO release enables a well-defined control
of the location and the timing of the CO release as well as its dosage.[1] Although the benzimidazole
motif is a crucial pharmacophore in drug discovery with interesting biological properties including
antiviral, antitumor,antihistaminic, and antimicrobial activity,[2] it has not been explored in the context
of CORMs.So far a series of eight photoactivatable CORMs of the general formula [MnBr(CO)3(N-N)]
incorporating chelating 1H-benzimidazol-2-ylmethyl-(N-aryl)amine ligands was synthesized, and
characterized using several analytical and spectral methods.The influence of the type and position of the
substituent in the aniline ring on the photophysical properties of the studied compounds was
systematically explored. Photo-activation profiles were examined by UV/Vis and solution IR studies.
Illumination at 468 nm with a LED array resulted in fast release of two equivalents of carbon monoxide,
while stepwise liberation of two CO takes place upon excitation at 412 nm.Therefore, the CO release
from the investigated compounds could be switched on and off by time control and choosing the suitable
light source.
MnOC
OC
Br
CO
NHN
N
R
9, R = H (50%)
10, R = 4-CH3 (73%)
11, R = 4-OCH3 (64%)
12, R = 4-Cl (70%)
13, R = 4-COOCH3 (72%)
14, R = 4-NO2 (30%)
15, R = 2-COOCH3 (66%)
16, R = 3-COOCH3 (68%)
H
[1] U. Schatzschneider, Brit. J. Pharmacol. 2015, 172, 1638-1650.
[2] N. T. Abdel-Ghani, A. M. Mansour, Eur. J. Med. Chem. 2012, 47, 399-411.
300 400 500 6000.0
0.2
0.4
0.6
0.8
Ab
so
rba
nce
Wavelength / nm
0 min
1 min
2 min
3 min
4 min
5 min
10 min
15 min
20 min
25 min

Pharmazie und Lebensmittelchemie P73 Affinity Prediction of Protein-Ligand Complexes with Scoring
Functions Based on Clustered Linear Regression
L. P. Pason1*, C. A. Sotriffer1
1Institute of Pharmacy and Food Chemistry, University of Würzburg, Würzburg, 97074, Germany
email: [email protected]
The accurate prediction of binding affinities for protein-ligand complexes is a major challenge in
drug design.[1] For routine application in docking and virtual screening, methods like free energy
perturbation or thermodynamic integration are computationally still too demanding. Approximate but
much more rapid access to the desired quantity is provided by empirical scoring functions, which are
derived by relating structural features of protein-ligand complexes to the corresponding affinity values.
Classical linear regression techniques or machine leaning methods are applied for this purpose. The
circumvention of modelling assumptions in machine learning scoring functions has led to improved
performance in many cases,[2] but at the cost of reduced interpretability.
In this study, approaches of clustered linear regression [3] and clustered partial linear regression [4]
were followed to derive a set of new scoring functions based on the SFCscore descriptors and subsets
of the PDBbind refined set v2007 as training data.[5] The functions were tested against standard
benchmarks divided into subsets in a similar way as the training data. Comparing the performance to a
machine learning scoring function trained with the same experimental data and descriptors showed that
the clustered linear regression procedure approaches the predictive quality of the machine learning
function while maintaining full interpretability of the scoring functions.
[1] L. P. Pason, C. A. Sotriffer, Mol. Inf. 2016, DOI: 10.1002/minf.201600048.
[2] Q. U. Ain, A. Aleksandrowa, F. D. Roessler, P. J. Ballester, Wires Comput Mol Sci 2015,
5,405-424.
[3] B. Ari, H. A. Güvenir, Knowl-Based Syst 2002, 15, 169-175.
[4] L. Torgo, J. P. Da Costa, Mach Learn 2003, 50, 303-319.
[5] T. Cheng, X. Li, Y. Li, Z. Liu, R. Wang, J Chem Inf Model 2009, 49, 1079-1093.

Organische Chemie und Biochemie
P74
Entwicklung neuer Biokatalysatoren zur Synthese von
Oligosacchariden und Saccharose-Analoga
C. Possiel, M. E. Ortiz-Soto, M. Timm, J. Görl, J. Seibel
Institut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Deutschland
Die Levansucrase von B. megaterium ist eine bakterielle Fruktosyltransferase und gehört zu der
Enzymfamilie der Glykosidhydrolasen 68 (GH 68). Dieses Enzym katalysiert hauptsächlich die
Hydrolyse von Saccharose in die Monomer-Bausteine Glukose und Fruktose (90% Hydrolyse-
Aktivität), aber auch den Fruktosyl-Transfer auf weitere Zucker-Moleküle (Saccharose), was zur
Bildung von kurz- und langkettigen β(2→6)-verknüpften Fructanen (Levan)[1]sowie in Gegenwart eines
Glykosid-Akzeptors zu (1→2)-glykosidisch verknüpften Saccharose-Analoga führt.[2]
Durch die geringe Spezifizität entsteht in Gegenwart von Saccharose oder Saccharose-Analoga
jedoch eine große Bandbreite von verschiedenen Oligo- und Polysacchariden. Aus diesem Grund sollen
durch gezielte Mutagenese effizientere Biokatalysatoren für die Synthese der einzelnen Zucker-
Verbindungen entwickelt werden, die spezifisch (durch Verminderung des Produktspektrums oder
höherer Fruktosyl-Transferrate) kurzkettige Di- bzw. Oligosaccharide wie z. B. Blastose, 1-Kestose,
6-Kestose und Neokestose produzieren können.
Des Weiteren liegt der Fokus auf der Optimierung der Saccharose-Analoga-Synthese. Im Rahmen
eines EU-Projektes werden neue Varianten der Levansucrase entwickelt und so Biokatalysatoren für die
Synthese maßgeschneidert, um möglichst selektiv neuartige (1→2)-glykosidisch verknüpfte
Saccharose-Analoga herzustellen. Die erhaltenen Saccharose-Analoga können durch die Anwendung
zweier weiterer Enzyme gespalten, aktiviert und auf Aglykone (nicht-Zucker-Komponenten wie z. B.
Wirkstoffe) übertragen werden, wodurch u. a. die Löslichkeit sowie die biologische Aktivität der
Verbindungen erhöht werden können.[3,4]
[1] G. Meng, K. Fütterer, Nat. Struct. Biol. 2003, 10, 935-941.
[2] A. Homann, J. Seibel, Appl. Microbiol. Biotechnol. 2009, 83, 209-216.
[3] S. G. Withers et al., Annu. Rev. Biochem. 2008, 77, 521-555.
[4] Y. Zheng et al., J. Biol. Chem. 2011, 286, 36108-36118.

Anorganische Chemie und Materialwissenschaften P75 Nickel-Catalyzed Borylation of Polyfluoroarenes via C-F Bond
Cleavage
M. W. Kuntze-Fechner1, U. Radius1
email: [email protected], [email protected] 1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg.
The selective synthesis of fluoroarene compounds has become a subject of growing interest due to the
prominent role such species play in many modern pharmaceuticals, agrochemicals and other industrially important
products.[1] An attractive route for the selective substitution of fluoroarenes is based on the functionalization of
activated aromatic C–F bonds derived from readily available perfluoroarenes. The presentation covers the
synthesis and reactivity of NHC-stabilized nickel complexes, which transfer the complex fragments [Ni(R2Im)2]
(R2Im = 1,3-Di(organyl)imidazole-2-ylidene) in stoichiometric and catalytic reactions under mild conditions, and
their use in the functionalization to polyfluorinated arenes.[2] We are currently developing convenient
methodologies to generate and use suitable, partially fluorinated organic precursors. These strategies employ, for
example, (i) C-F functionalization of polyfluoroaromatics or (ii) use of the polyfluoroaromatics or fluoroaryl
boronic ester in subsequent Suzuki-Miyaura coupling reactions. A smart way to achieve this goal would employ
fluoroaromatic boronic acids or boronate esters. Arylboronic acid esters are versatile reagents in organic synthesis,
especially in substituent conversions or in the widely employed Suzuki-Miyaura cross-coupling reaction.
Transition-metal-catalyzed direct C-H borylation of arenes[3] and borylation of aryl halides[4] has emerged as one
of the most important mild and attractive routes for the synthesis of aryl boronates in recent years. The conversion
of fluoroaromatics into arylboronic esters via C-F bond activation, however, is relatively unexplored and was
restricted to noble metal catalysts until recently. A focus of the contribution will be thus on the use of NHC nickel
complexes in carbon fluorine activation and the use of these processes in catalytic borylation.[5]
This work was supported in part by Deutsche Forschungsgemeinschaft (DFG) and the Julius-Maximilians-
Universität Würzburg.
[1] (a) H. Amii, K. Uneyama, Chem. Rev. 2009, 109, 2119; (b) A. D. Sun, J. A. Love, Dalton Trans. 2010,
39, 10362; (c) T. Braun, D. Lenz, Angew. Chem. Int. Ed. 2013, 52, 3328; (d) T. Ahrens, J. Kohlmann, M.
Ahrens, T. Braun, Chem. Rev. 2015, 115, 931.
[2] (a) T. Schaub, U. Radius, Chem. Eur. J. 2005, 11, 5024; (b) T. Schaub, M. Backes, U. Radius, J. Am.
Chem. Soc. 2006, 128, 15964; (c) T. Schaub, M. Backes, U. Radius, Eur. J. Inorg. Chem. 2008, 2680; (d)
T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius, A. Mix, J. Am. Chem. Soc. 2008, 130, 9304; (e)
T. Schaub, P. Fischer, T. Meins, U. Radius, Eur. J. Inorg. Chem. 2011, 3122; (f) T. Zell, M. Feierabend,
B. Halfter, U. Radius, J. Organomet. Chem. 2011, 696, 1380; (g) P. Fischer, K. Götz, A. Eichhorn, U.
Radius, Organometallics 2012, 31, 1374.
[3] (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy, J. F. Hartwig, Chem. Rev. 2009, 110,
890; (b) A. Ros, R. Fernandez, J. M. Lassaletta, Chem. Soc. Rev. 2014, 43, 3229.
[4] (a) M. Murata, Heterocycles 2012, 85, 1795; (b) W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P.
Lau, W. T. Wong, F. Y. Kwong, RSC Advances 2013, 3, 12518.
[5] J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B.
Marder, U. Radius, J. Am. Chem. Soc. 2016, 138, 5250-5253.

Anorganische Chemie und Materialwissenschaften
P76
iClick reactions for a modular access to Pt/Pd- complexes with high
anticancer activity
K. Peng1, P. Simpson2, U. Schatzschneider1*
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg,
Am Hubland, D-97074 Würzburg (Germany) 2Nanochemistry Research Institute–Department of Chemistry, Curtin University
Kent Street, Bentley 6102 WA(Australia)
e-mail: [email protected]
Along with the widespread success of cisplatin in the clinical treatment of various types of
malignancies, the chemistry of metal-based drugs has received enormous attention over the last few
decades.[1] However, the effectiveness of these metal-based compounds as antitumor agents is often
hampered by their poor solubility, low bioavailability, and lack of target specificity. Therefore, synthetic
technology which quickly generates high molecular diversity and has great reaction efficiency is
required to optimize compounds for specific biological properties and activities.
Taking this perspective into consideration, inorganic Click (iClick) reactions, which will allow to
easily tune the bioavailability and ADMET (absorption, distribution, metabolism, excretion, toxicity)
properties of the novel compounds for bioactivity studies, were applied for a modular access to quickly
generate molecular diversity in the synthesis of transition metal complexes with high antitumor activity
based on novel structural motifs.[2] Consequently, two Pt/Pd- triazole complexes were synthesized by
this approach (Scheme 1).
Scheme 1: Synthetic routes to (a) Pt complexes and (b) Pd complexes by iClick reactions.
With a certain amount of pure Pt/Pd- iClick products at hand, we are now trying to investigate their
cytotoxicities, and their speciation and interaction with bio(macro)molecules such as amino acids, DNA
nucleobases, and double-stranded DNA to deeply understand antitumor activities of the two compounds.
[1] C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato, C. Marzano, Chem. Rev. 2014, 114, 815.
[2] L. Herny, C. Schneider, B. Mützel, P. Simpson, C. Negal, K. Fucke, U.Schatzschneider, Chem.
Commun. 2014, 50, 15692.
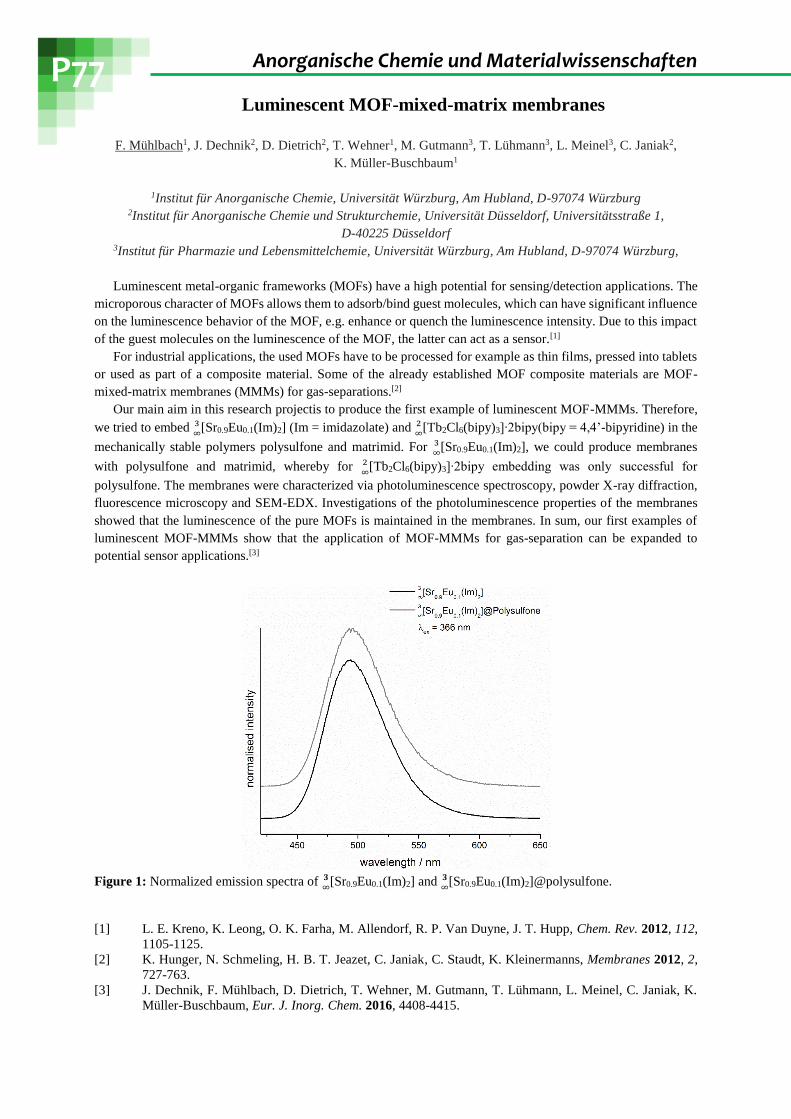
Anorganische Chemie und Materialwissenschaften P77 Luminescent MOF-mixed-matrix membranes
F. Mühlbach1, J. Dechnik2, D. Dietrich2, T. Wehner1, M. Gutmann3, T. Lühmann3, L. Meinel3, C. Janiak2,
K. Müller-Buschbaum1
1Institut für Anorganische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg
2Institut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, Universitätsstraße 1,
D-40225 Düsseldorf 3Institut für Pharmazie und Lebensmittelchemie, Universität Würzburg, Am Hubland, D-97074 Würzburg,
Luminescent metal-organic frameworks (MOFs) have a high potential for sensing/detection applications. The
microporous character of MOFs allows them to adsorb/bind guest molecules, which can have significant influence
on the luminescence behavior of the MOF, e.g. enhance or quench the luminescence intensity. Due to this impact
of the guest molecules on the luminescence of the MOF, the latter can act as a sensor.[1]
For industrial applications, the used MOFs have to be processed for example as thin films, pressed into tablets
or used as part of a composite material. Some of the already established MOF composite materials are MOF-
mixed-matrix membranes (MMMs) for gas-separations.[2]
Our main aim in this research projectis to produce the first example of luminescent MOF-MMMs. Therefore,
we tried to embed 3∞
[Sr0.9Eu0.1(Im)2] (Im = imidazolate) and 2∞
[Tb2Cl6(bipy)3]∙2bipy(bipy = 4,4’-bipyridine) in the
mechanically stable polymers polysulfone and matrimid. For 3∞
[Sr0.9Eu0.1(Im)2], we could produce membranes
with polysulfone and matrimid, whereby for 2∞
[Tb2Cl6(bipy)3]∙2bipy embedding was only successful for
polysulfone. The membranes were characterized via photoluminescence spectroscopy, powder X-ray diffraction,
fluorescence microscopy and SEM-EDX. Investigations of the photoluminescence properties of the membranes
showed that the luminescence of the pure MOFs is maintained in the membranes. In sum, our first examples of
luminescent MOF-MMMs show that the application of MOF-MMMs for gas-separation can be expanded to
potential sensor applications.[3]
Figure 1: Normalized emission spectra of 𝟑∞
[Sr0.9Eu0.1(Im)2] and 𝟑∞
[Sr0.9Eu0.1(Im)2]@polysulfone.
[1] L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne, J. T. Hupp, Chem. Rev. 2012, 112,
1105-1125.
[2] K. Hunger, N. Schmeling, H. B. T. Jeazet, C. Janiak, C. Staudt, K. Kleinermanns, Membranes 2012, 2,
727-763.
[3] J. Dechnik, F. Mühlbach, D. Dietrich, T. Wehner, M. Gutmann, T. Lühmann, L. Meinel, C. Janiak, K.
Müller-Buschbaum, Eur. J. Inorg. Chem. 2016, 4408-4415.

Organische Chemie und Biochemie
P78
FULLERENE-MEROCYANINE DYADS FOR SINGLE
COMPONENT ORGANIC SOLAR CELLS
R. Hecht, D. Bialas, M. Stolte, F. Würthner*
Institut für Organische Chemie & Center for Nanosystems Chemistry, Universität Würzburg,
Am Hubland, 97074 Würzburg, Germany, *E-Mail: [email protected]
R. Wagener: [email protected]
Dyads of fullerene and donor molecules are interesting components for organic photovoltaics,
because the fabrication of the cells is simplified and the morphology within the solar cell is more stable
compared to bulk heterojunction (BHJ) solar cells. Additionally, the light absorption and the charge
separation take place within the same molecule, so that short exciton diffusion length should not be a
limiting factor.[1]
A set of new dyad molecules was synthesized (Figure 1a). The dyads consist of a phenyl-C61-
propionic acid (PC61PM), which is covalently bound to three different merocyanine dyes. We prepared
solar cells from solution of these dyads (Figure 1b) and succeeded with first optimization experiments,
reaching power conversion efficiencies (PCE) of up to 0.53 % in air. For comparison we investigated
BHJ solar cells, using the merocyanines MD302, HB366[2] and CS254 as donor and PC61BM as
acceptor in a molar ratio of 1:1. To these reference systems we added dyad molecules as mediator, to
stabilize the donor-acceptor interface. The cells were characterized by measuring current-voltage
characteristics, UV/Vis absorption, EQE and AFM.
Figure 1: New dyad molecules and corresponding merocyanine dyes (a), studied in single component
solar cells in the following device architecture (b).
[1] J. Roncali, Advanced Energy Materials 2011, 1, 147-160.
[2] H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger,
M. Stolte, K. Meerholz, F. Würthner, Angewandte Chemie International Edition 2011, 50,
11628-11632.

Anorganische Chemie und Materialwissenschaften P79 Microbubbles loaded with platinum complexes for ultrasound-
mediated treatment of brain cancer
V. Mawamba1, C. Hagemann2, M. Löhr2, V. Sturm2, U. Schatzschneider1
1Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, D-97074
Würzburg 2Tumor biologisches Labor, Neurochirugische Klinik und Polyklinik, Universitätsklinikum Würzburg,
Joseph-Schneider Str.11/B1, D-97080 Würzburg
e-mail: [email protected]
Tumors of the central nervous system are among the most disabling and lethal types of cancer.[1]
Because of their depth and location in functional inoperable areas, some brain cancers are surgically
unreachable. Therefore, only standard treatment such as radio- and chemotherapy or a combination of
both are possible. Chemotherapy has gained a lot of attention in the past two decades. Anti-cancer drugs
are designed to kill malignant cells, but when administrated, only a small fraction of the injected dose
reaches the target site, with the rest circulating through healthy tissue, resulting in dose-limiting side
efffets. Encapsulation of drugs within microbubbles combined with local release triggered by focused
ultrasound is thought to increase the local concentration of a chemotherapeutic agent at the site of disease
while minimizing side effects on healthy tissue.[2,3]
Figure 1: Fluorescence microscopy picture of octafluoropropane-filled lipid microbubbles stained with
1,1‘-dioctadecyl-3,3,3‘,3‘-tetramethylindocarbocyanine perchlorate (left) and general structure of
membrane-affine cytotoxic platinum complex (right).
Microbubbles are gas-filled colloidal particles with a size range between 1-8 µm. In medicine, they
are well-known as ultrasound contrast agents for imaging and diagnosis.[4] Our current work to be
presented here is focused on the encapsulation of different platinum-based drug candidates within
microbubbles, the study of their stability, and payload delivery in vitro under application of ultrasound
as an external stimulus.
[1] S. A. Khuder, A. B. Mutgi, E. A. Schaub, Am. J. Ind. Med. 1998, 34, 252-260
[2] Y. Liu, H. Miyoshi, M. Nakamura, J. Control. Release 2006, 114, 89-99
[3] R. Deckers, C. T. W. Moonen, J. Control. Release 2010, 148, 25-33
[4] S. Tinkov, R. Bekeredjian, G. Winter, C. Coester, J. Pharm. Sci. 2009, 98, 1935-1961

Anorganische Chemie und Materialwissenschaften
P80
Pyrene MO Shuffle
Adjusting the HOMO-LUMO gap by changing the frontier MO ordering
J. Merz, L. Ji, J. Fink, A. Friedrich, I. Krummenacher, H. Al Mamari, S. Lorenzen, M. Hähnel,
A. Eichhorn, T. B. Marder*
Institut für Anorganische Chemie, Julius-Maximilians Universität Würzburg
E-Mail: [email protected]
Pyrene is a polycyclic aromatic hydrocarbon (PAH) that has very interesting photophysical
properties which makes it suitable for a broad range of applications.[1] The 2,7-positions of pyrene are
situated on nodal planes in both the HOMO and LUMO. Hence, electrophilic reactions take place at the
1-, 3-, 6-, and 8-positions. We developed a selective method to substitute directly the 2,7-positions by
an iridium-catalyzed C-H borylation.[2] First investigations demonstrated that pyrene derivatives
functionalized at the 2-position have different photophysical properties compared to the “traditional”
functionalized pyrenes at the 1-position.[3] We report a series of new donor-acceptor and donor-donor
pyrene systems with remarkable properties. Donors can raise HOMO-1 above the pyrene HOMO
whereas acceptors can lower LUMO+1 below the pyrene LUMO which facilitates strong
communication through the pyrene bridge. Further we could demonstrate the effectiveness of our pyrene
bridge in comparison with biphenyl by photophysical and electrochemical measurements that are
supported by DFT calculations and contrast with previous reports.
Figure 1: General numbering of pyrene (left) and our three target molecules (right).
[1] T. M. Figueira-Duarte, K. Mullen, Chem. Rev. 2011, 111, 7260
[2] A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen,
J. C. Collings, A. S. Batsanov, J. A. K. Howard, T. B. Marder, Chem. Eur. J. 2012, 18, 5022.
[3] A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer, T. B.
Marder, J. Am. Chem. Soc. 2011, 133, 13349.

Anorganische Chemie und Materialwissenschaften P81 Formulation of extremely hydrophobic drugs using amphiphilic
pseudo-polypeptide block copolymers
M. Lübtow1, B. Christ2, S. Dembski3, L. Persano4, R. Luxenhofer1
1Functional Polymer Materials, Chair for Chemical Technology of Material Synthesis, University of Würzburg, Röntgenring
11, 97070 Würzburg (Germany) 2Translational Center “Regenerative therapies in oncology and musculoskeletal diseases”, Fraunhofer IGB, Röntgenring 11,
97070 Würzburg (Germany) 3Fraunhofer Institute for Silicate Research ISC, Neunerplatz 2, 97082 Würzburg (Germany)
4National Nanotechnology Laboratory of InstitutoNanoscienze-CNR, Università del Salento, via Arnosano, I-73100 Leece
(Italy)
Modern techniques like high-throughput combinatorial screenings[1] can quickly assay the biological or biochemical
activity of a large number of potential drugs, but when it comes to formulation, one major issue remains. More than 40% of all
NCEs (new chemical entities) developed in the pharmaceutical industry are practically insoluble in water.[2] Taking into
account, that 90% of all compounds in today’s drug delivery pipelines are reported to be poorly water-soluble, this poses a
major challenge for the pharmaceutical industry.[3] The demand for excipients, which increase the water solubility without
influencing the bioactivity of such hydrophobic drugs is enormous. Motivated by the high drug loadings of poly(2-
oxazoline)sPOx based micelles for paclitaxel (PTX) of up to 45 wt%,[4] this work presents a preliminary study on the structure
property relationships of pseudo-polypeptide based amphiphiles on the solubilization capacity for the non-water soluble
compound curcumin (CUR).The extraordinary large loading capacity for CUR of more than 50 wt% active drug as well as high
water solubility of the resulting formulation emphasize the use of these drug-loaded polymeric micelles for detailed in vitro
and in vivo studies with regard of their anticancer and anti-inflammatory potential. Besides the formulation in aqueous solution,
core-shell fibers comprised of a drug-loaded polymeric micelles core encapsulated in a biodegradable polycaprolactone (PCL)
shell[5] as well as silica fibers loaded with polymeric micelles were prepared to enable a more sustained drug-release.
Amphiphilic Block-Copolymers Extremely hydrophobic drugs
Figure 1: Solubilization of extremely hydrophobic drugs with amphiphilic poly(2-oxazoline) (POx) based block-copolymers.
[1] A. Persidis, Nat. Biotech. 1998, 16, 488-489.
[2] E. M. Merisko-Liversidge, G. G. Liversidge, Toxicologic Pathology 2008, 36, 43-48.
[3] V. A. V. Vharti, A. Munde, A. Birajdar, S. Bais, JIPBS 2015, 2, 482-494.
[4] a) R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich, E. V. Batrakova, R. Jordan, A. V. Kabanov,
Biomaterials 2010, 31, 4972-4979; b) A. Schulz, S. Jaksch, R. Schubel, E. Wegener, Z. Di, Y. Han, A. Meister, J.
Kressler, A. V. Kabanov, R. Luxenhofer, C. M. Papadakis, R. Jordan, ACS Nano 2014, 8, 2686-2696; c) Y. Seo, A.
Schulz, Y. Han, Z. He, H. Bludau, X. Wan, J. Tong, T. K. Bronich, M. Sokolsky, R. Luxenhofer, R. Jordan, A. V.
Kabanov, Polymers for Advanced Technologies 2015, 26, 837-850; d) Z. He, X. Wan, A. Schulz, H. Bludau, M. A.
Dobrovolskaia, S. T. Stern, S. A. Montgomery, H. Yuan, Z. Li, D. Alakhova, M. Sokolsky, D. B. Darr, C. M. Perou,
R. Jordan, R. Luxenhofer, A. V. Kabanov, Biomaterials 2016, 101, 296-309.
[5] L. Romano, A. Camposeo, R. Manco, M. Moffa, D. Pisignano, Molecular Pharmaceutics 2016, 13, 729-736.

Pharmazie und Lebensmittelchemie
P82
Chemistry Meets Cancer Immunotherapy: Synthesis of Hapten-like
Compounds to Redirect the Specificity of Programmable Antibodies
and T Cells
P. A. Nagl1, L. Wallstabe2, M. Hudecek2, U. Holzgrabe1
1 University of Würzburg, Institute of Pharmacy and Food Chemistry, Am Hubland, 97074 Würzburg,
Germany 2Universitätsklinikum Würzburg, Medizinische Klinik und Poliklinik II,
Oberdürrbacherstraße 6, 97080 Würzburg, Germany
Immunotherapies are evolving as highly promising strategies for the treatment of cancer.
An essential component of these approaches are reactive, tumor-specific T lymphocytes, which are rare for
many malignancies and are difficult to isolate. However, they can be generated by genetic modification using
genes encoding chimeric antigen receptors (CARs) or T cell receptors (TCRs). CAR-modified T cells are
endowed with the specificity of tumor-specific antibodies.[1] Chemical programming of the used single-chain
variable fragment (scFv) should be possible in the same way as performed by chemically programmed
antibodies (cpAbs).[2] With this approach, the scope of potential tumor-specific targets could be enlarged
vastly while the production of effective T lymphocytes could be simplified.
The aim of this work is to contribute to the development of chemically programmed T cells for cancer
immunotherapy. Therefore, different hapten-like compounds should be synthesized. All of these compounds
have reactive groups, which selectively bind to the lysine residue of the antigen receptor, and ether a targeting
or labeling module (see figure 1).[3]
Figure 1: Different reactions for reprogramming monoclonal antibodies or CAR T cells.
The test results of the first molecules, equipped with biotin as labeling module, proofed the concept by
showing that the compounds are not binding to reactive groups on the cell surface, but selectively react with
the lysine residue of the receptor. Current work includes the synthesis of an Arg-Gly-Asp (RGD)
peptidomimetic motif as targeting module for integrin αvβ3, which is overexpressed in solid tumor cells [4].
[1] C. J. Turtle, M. Hudecek, M. C. Jensen, S. R. Riddell, Curr. Opin. Immunol. 2012, 24, 633-639.
[2] C. Rader, Trends Biotechnol. 2014, 32, 186-197.
[3] J. I. Gavrilyuk, U. Wuellner, C. F. Barbas, Bioorg. Med. Chem. Lett. 2009, 19, 1421-1424.
[4] L. Li, C. Rader, M. Matsushita, S. Das, C. F. Barbas, R. a Lerner, S. C. Sinha, J. Med. Chem. 2004,
47, 5630-5640.
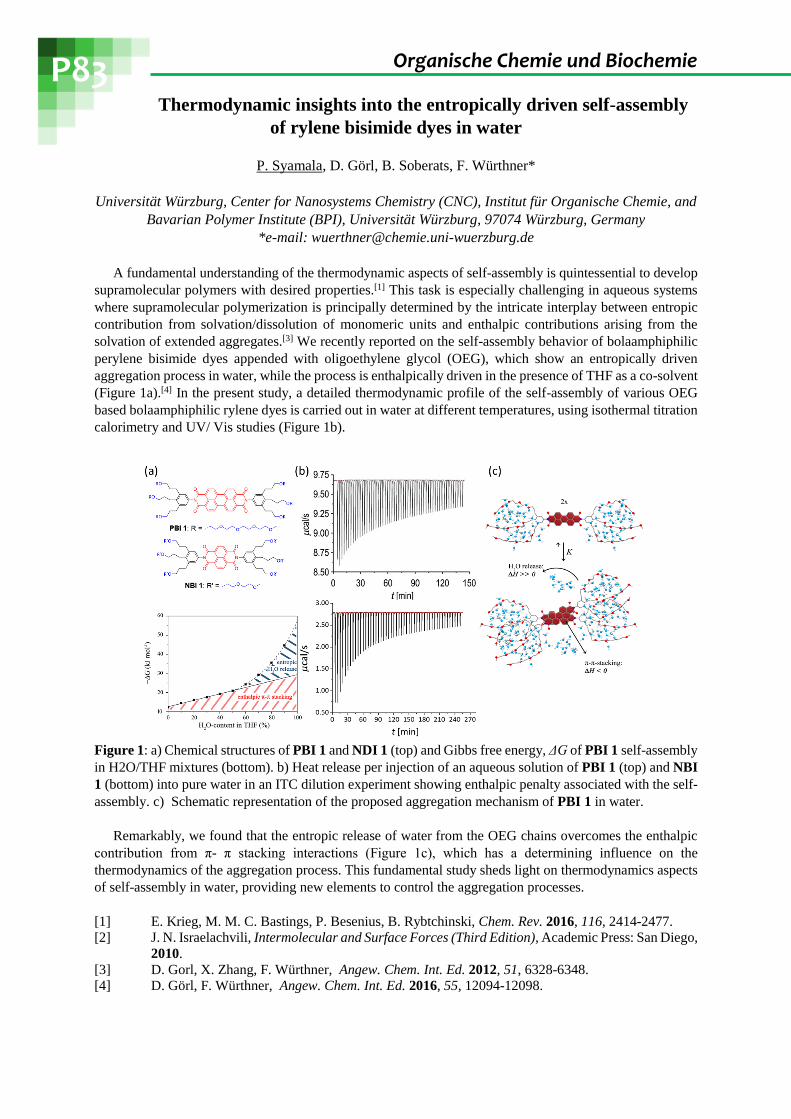
Organische Chemie und Biochemie P83 Thermodynamic insights into the entropically driven self-assembly
of rylene bisimide dyes in water
P. Syamala, D. Görl, B. Soberats, F. Würthner*
Universität Würzburg, Center for Nanosystems Chemistry (CNC), Institut für Organische Chemie, and
Bavarian Polymer Institute (BPI), Universität Würzburg, 97074 Würzburg, Germany
*e-mail: [email protected]
A fundamental understanding of the thermodynamic aspects of self-assembly is quintessential to develop
supramolecular polymers with desired properties.[1] This task is especially challenging in aqueous systems
where supramolecular polymerization is principally determined by the intricate interplay between entropic
contribution from solvation/dissolution of monomeric units and enthalpic contributions arising from the
solvation of extended aggregates.[3] We recently reported on the self-assembly behavior of bolaamphiphilic
perylene bisimide dyes appended with oligoethylene glycol (OEG), which show an entropically driven
aggregation process in water, while the process is enthalpically driven in the presence of THF as a co-solvent
(Figure 1a).[4] In the present study, a detailed thermodynamic profile of the self-assembly of various OEG
based bolaamphiphilic rylene dyes is carried out in water at different temperatures, using isothermal titration
calorimetry and UV/ Vis studies (Figure 1b).
Figure 1: a) Chemical structures of PBI 1 and NDI 1 (top) and Gibbs free energy, ΔG of PBI 1 self-assembly
in H2O/THF mixtures (bottom). b) Heat release per injection of an aqueous solution of PBI 1 (top) and NBI
1 (bottom) into pure water in an ITC dilution experiment showing enthalpic penalty associated with the self-
assembly. c) Schematic representation of the proposed aggregation mechanism of PBI 1 in water.
Remarkably, we found that the entropic release of water from the OEG chains overcomes the enthalpic
contribution from π- π stacking interactions (Figure 1c), which has a determining influence on the
thermodynamics of the aggregation process. This fundamental study sheds light on thermodynamics aspects
of self-assembly in water, providing new elements to control the aggregation processes.
[1] E. Krieg, M. M. C. Bastings, P. Besenius, B. Rybtchinski, Chem. Rev. 2016, 116, 2414-2477.
[2] J. N. Israelachvili, Intermolecular and Surface Forces (Third Edition), Academic Press: San Diego,
2010.
[3] D. Gorl, X. Zhang, F. Würthner, Angew. Chem. Int. Ed. 2012, 51, 6328-6348. [4] D. Görl, F. Würthner, Angew. Chem. Int. Ed. 2016, 55, 12094-12098.

Anorganische Chemie und Materialwissenschaften
P84
Synthesis of Neutral Boron-Centered Lewis Bases Derived from
Terminal Arylborylene-Complexes
M. Nutz, C. W. Tate, H. Braunschweig
Julius-Maximilians-Universitaet-Wuerzburg, Department of Inorganic Chemistry, 97074 Wuerzburg,
Germany
email: [email protected]
A variety of neutral electron-rich monovalent boron compounds has been synthesized (1−3).[1] The
synthetic strategy used – liberation of a borylene ligand from a transition metal – is broadly applicable,
leading to a number of unprecedented monovalent boron species bearing different Lewis basic
groups.[1,2] Depending on the steric demand of the base, homoleptic (1, 3) or heteroleptic (2) compounds
are formed, which all show a trigonal planar geometry at the boron atom. Analysis of the frontier orbitals
indicates that the HOMO level consists of a three-centered π-bonding interaction between the boron and
the attached ligands. Further computational investigations show an extensive boron-to-ligand
backbonding in these molecules
[1] H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vergas, Q.
Ye, Nature 2015, 522, 327-330.
[2] H. Braunschweig. R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas, Q. Ye, Angew. Chem.
Int. Ed. 2015, 54, 1-6.

Anorganische Chemie und Materialwissenschaften P85 Reactivity of N-heterocyclic Carbenes towards Chloro- and
Hydrostannanes
M. J. Krahfuß, H. Schneider, U. Radius
Institut für Anorganische Chemie der Julius-Maximilians-Universität Würzburg,
[email protected], [email protected]
Since the isolation of the first N-heterocyclic carbene in 1991 through Arduengo[1] a multitude of
novel NHC-metal-complexes had been synthesized and characterized. In main-group element chemistry
these small organic molecules open up a variety of reaction pathways, which fundamentally differ from
their use as ligands in the coordination sphere of metals.[2,3]
Besides the deprotonation of Brønsted-acids using carbenes our group could observe reductive
dehydrocoupling of primary and secondary phosphines. Carbenes are also capable of oxidative addition
reactions with bond activation in e.g. boranes, silanes and phosphines. Furthermore, ring expansion
reactions of E-H-substrates of the groups 2, 13 and 14, as well as adduct formation followed by
rearrangement in silanes are known.[4]
Considering the significantly different reaction patterns of silicon hydrides and chlorides with NHCs
we were interested in their reactivity towards the heavier homologues of tin (RmSnHn/Xn). Studies of
Wesemann et al. with the sterically demanding stannanes trip2SnH2 and trip3SnH showed solely the
formation of the distannanes via reductive dehydrocoupling.[5] On this account we studied tin
compounds with more simple substituents like methyl and phenyl to investigate their behavior towards
the NHCs iPr2Im and iPr2ImMe.[6]
[1] A. J. Arduengo, R. L. Harlow, M. Kline, J. Am. Chem. Soc. 1991, 113, 361-363.
[2] H. Braunschweig, R. D. Dewhurst, K. Ferkinghoff, Chem. Commun. 2016, 52, 183-185.
[3] P. Ai, A. A. Danopoulos, P. Braunstein, Dalton Trans. 2016, 45, 4771-4779.
[4] a) D. Schmidt, J. H. J. Berthel, S. Pietsch, U. Radius, Angew. Chem. Int. Ed. 2012, 51, 8881-
8885; b) H. Schneider, D. Schmidt, U. Radius, Chem. Commun. 2015, 51, 10138-10141; c) H.
Schneider, D. Schmidt, U. Radius, Chem. Eur. J. 2015, 21, 2793-2797.
[5] a) C. P. Sindlinger, L. Wesemann, Chem. Sci. 2014, 5, 2739-2746; b) C. P. Sindlinger, W.
Grahneis, F. S. W. Aicher. L. Wesemann, Chem. Eur. J. 2016, 22, 7554-7566.
[6] H. Schneider, M. J. Krahfuß, U. Radius, Z. Anorg. Allg. Chem. 2016, ahead of print.

Organische Chemie und Biochemie
P86
Shape-controlled Synthesis of Covalent Organic Cage Compounds
V. Leonhardt, S. Klotzbach, F. Beuerle*
Universität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry,
Am Hubland, Würzburg, Germany
*E-mail: [email protected]
Porous functional materials are promising candidates for applications in the areas of heterogeneous
catalysis, sensing, gas storage and separation, or membranes.[1] For example, organic cage compounds
can be constructed via the concept of dynamic covalent chemistry.[2]
Previously, we reported on the synthesis of molecular cubes,[3] tetrahedra and bipyramids by
crosslinking the catechol units of tribenzotriquinacene (TBTQ) with various diboronic acids.
Currently we elaborate on the self-assembly of planar, catechol functionalized hexahydroxytri-
phenylene (HHTP) with rigid diboronic acids possessing different bite angles into molecular cages.
Thereby, the geometry of the diboronic acids is transferred to structure and shape of the self-assembled
nanosized objects. Synthesis of the cage compounds can be monitored by 1H-NMR spectroscopy and
size and shape is analyzed by 2D-NMR as well as DOSY-NMR spectroscopy.
[1] G. Férey, Chem Soc. Rev. 2008, 37, 191-214.
[2] M. Mastalerz, Chem. Eur. J. 2012, 18, 10082-10091.
[3] S. Klotzbach, T. Scherpf, F. Beuerle, Chem. Comm. 2014, 50, 12454-12457.
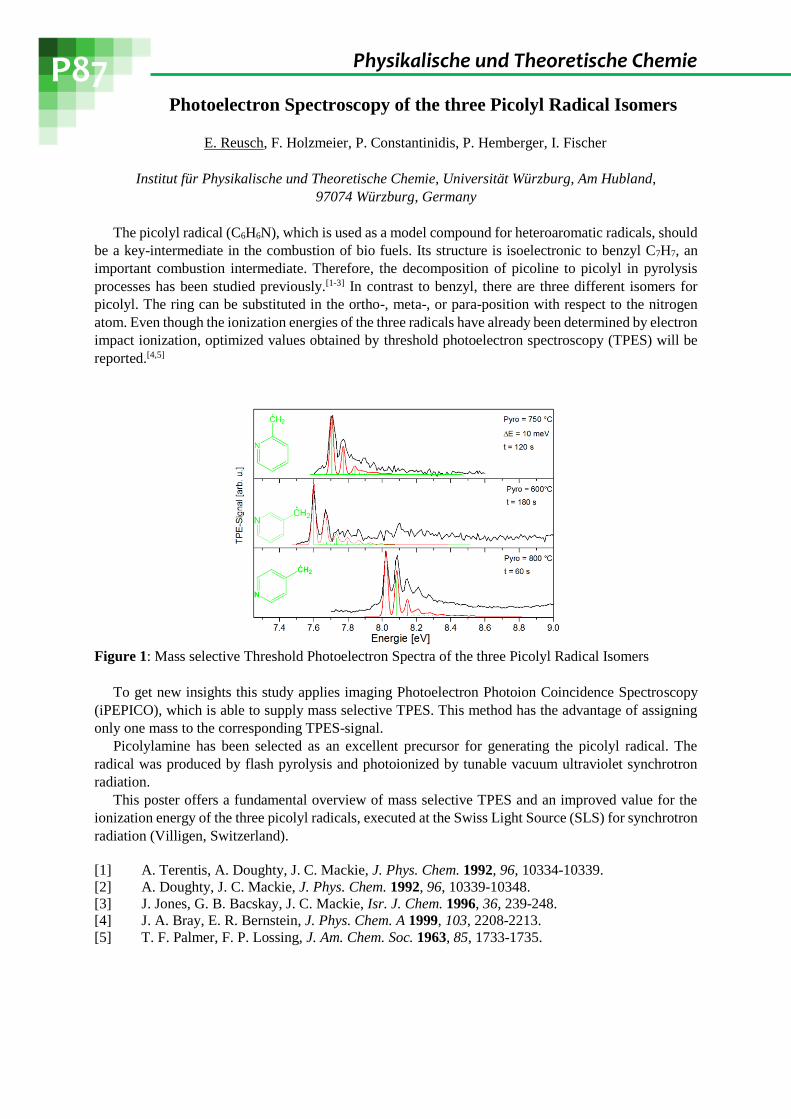
Physikalische und Theoretische Chemie P87 Photoelectron Spectroscopy of the three Picolyl Radical Isomers
E. Reusch, F. Holzmeier, P. Constantinidis, P. Hemberger, I. Fischer
Institut für Physikalische und Theoretische Chemie, Universität Würzburg, Am Hubland,
97074 Würzburg, Germany
The picolyl radical (C6H6N), which is used as a model compound for heteroaromatic radicals, should
be a key-intermediate in the combustion of bio fuels. Its structure is isoelectronic to benzyl C7H7, an
important combustion intermediate. Therefore, the decomposition of picoline to picolyl in pyrolysis
processes has been studied previously.[1-3] In contrast to benzyl, there are three different isomers for
picolyl. The ring can be substituted in the ortho-, meta-, or para-position with respect to the nitrogen
atom. Even though the ionization energies of the three radicals have already been determined by electron
impact ionization, optimized values obtained by threshold photoelectron spectroscopy (TPES) will be
reported.[4,5]
Figure 1: Mass selective Threshold Photoelectron Spectra of the three Picolyl Radical Isomers
To get new insights this study applies imaging Photoelectron Photoion Coincidence Spectroscopy
(iPEPICO), which is able to supply mass selective TPES. This method has the advantage of assigning
only one mass to the corresponding TPES-signal.
Picolylamine has been selected as an excellent precursor for generating the picolyl radical. The
radical was produced by flash pyrolysis and photoionized by tunable vacuum ultraviolet synchrotron
radiation.
This poster offers a fundamental overview of mass selective TPES and an improved value for the
ionization energy of the three picolyl radicals, executed at the Swiss Light Source (SLS) for synchrotron
radiation (Villigen, Switzerland).
[1] A. Terentis, A. Doughty, J. C. Mackie, J. Phys. Chem. 1992, 96, 10334-10339.
[2] A. Doughty, J. C. Mackie, J. Phys. Chem. 1992, 96, 10339-10348.
[3] J. Jones, G. B. Bacskay, J. C. Mackie, Isr. J. Chem. 1996, 36, 239-248.
[4] J. A. Bray, E. R. Bernstein, J. Phys. Chem. A 1999, 103, 2208-2213.
[5] T. F. Palmer, F. P. Lossing, J. Am. Chem. Soc. 1963, 85, 1733-1735.

Organische Chemie und Biochemie P88 Sugar Coated Nanodiamonds
S. Wachtler1, C. Fessele2, V. Chandrasekaran2, C. Stiller1, T. K. Lindhorst2, A. Krueger1
1Institut für Organische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg
[email protected] 2Institut für Organische Chemie, Universität Kiel, Otto-Hahn-Platz 3–4,
D-24118 Kiel, [email protected]
Nanodiamonds have proven to be an interesting material for biological and biomedical applications
because of their low toxicity.[1] Several studies have also shown promising results of saccharide
functionalized nanodiamonds for detecting specific pathogens and creating antiadhesive surfaces. One
of these pathogens is type 1 fimbriated E. Coli bacteria which possess lectin FimH on their surface and
therefore show a specific binding to α-D-mannosides.[2]
Figure 1: Functionalization of nanodiamond with saccharides using thiourea-bridging.
In order to link the saccharides to the nanodiamond particles, a new binding method using the
formation of thiourea from an amino group and an isothiocyanate has been developed. All three
synthesized samples show affinity for bacterial lectin FimH and their binding strength is in accordance
with the expected interaction between the mannose derivatives and the lectin FimH binding pocket.[3]
This project has received funding from the European commision and the German Bundesministerium
für Bildung und Forschung (BMBF) (contract NanoDiaMed grant number 13N12255 in
EuroNanoMed).
[1] A. Krueger, Chem. Eur. J. 2008, 14, 1382-1390.
[2] M. Hartmann, P. Betz, Y. Sun, S. N. Gorb, T. K. Lindhorst, A. Krüger, Chem. Eur. J. 2012, 18,
6485-6492.
[3] C. Fessele, S. Wachtler, V. Chandrasekaran, C. Stiller, T. K. Lindhorst, A. Krüger, Eur. J. Org.
Chem. 2015, 25, 5519-5525.

Organische Chemie und Biochemie P89 Synthesis and Properties of Methoxy-Bay-Substituted Perylene
Bisimides
A. Nowak-Król, P. Leowanawat, F. Würthner
Universität Würzburg, Institut für Organische Chemie, Am Hubland, D-97074 Würzburg, Germany
Universität Würzburg, Center for Nanosystems Chemistry (CNC) & Bavarian Polymer Institute (BPI),
Theodor-Boveri-Weg, 97074 Würzburg, Germany
Perylene bisimides (PBIs) feature high molar absorption coefficients and mirror image fluorescence
with fluorescence quantum yields typically above 90% in a wide range of solvents. A combination of
these favorable properties along with an exceptional thermal and photostability accounts for an extensive
exploration of PBIs in a large variety of fields spanning from commercial colorant chemistry,
supramolecular polymerization, liquid crystals to complex supramolecular architectures.[1]
Core-non-substituted PBIs bearing a large variety of imide substituents and tetraphenoxy-PBIs were
commonly prepared and investigated within this group of compounds, whereas tetra-bay-substituted
alkoxy-PBIs were not accessible. Recently, we have developed an efficient protocol for the synthesis of
tetra-methoxy-bay-substituted PBIs via copper-mediated cross-coupling of 1,6,7,12-tetrabromo-
perylene tetraester with sodium methoxide, which gave rise to the corresponding PBIs.[2] The
introduction of four methoxy groups into bay positions of the PBI core entailed a significant modulation
of the electronic band gap. This new class of compounds display interesting photophysical properties,
such as bathochromically shifted absorption and emission (beyond 600 nm), and most notably high
fluorescence quantum yields despite electron-donating bay substituents. We have also identified
interesting features of the solid packing which have implications on the general understanding of
dynamic processes occurring in bay-substituted perylene bisimides. These aspects as well as
photophysical properties of the new class of compounds will be discussed and compared with the
previously studied bay-substituted-PBIs.
[1] F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat, D. Schmidt, Chem. Rev.
2016, 116, 962-1052.
[2] P. Leowanawat, A. Nowak-Król, F. Würthner, Org. Chem. Front. 2016, 3, 537-544.

Organische Chemie und Biochemie P90 Anti-tumoral Naphthoquinones: Isolation from Plants
and Total Synthesis
C. Froschgeiser1, J. Maier1, G. Zhang1, A. Irmer1, S. Rüdenauer1, R. Bargou2, M. Chatterjee2,
G. Bringmann1
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
2Department of Internal Medicine II, Comprehensive Cancer Center Mainfranken, Hospital of
Würzburg, Versbacher Straße 5, 97078 Würzburg, Germany
The West African liana Triphophyllum peltatum (Dioncophyllaceae) has proven to be a rich source
of natural products. Besides the plant itself, callus cultures were also found to produce distinct amounts
of secondary metabolites, in particular naphthoquinones like plumbagin and droserone. By solidification
of the medium, these cell cultures were exposed to aerobic and thus oxidative conditions, which favored
the formation of further, structurally related new naphthoquinones like dioncoquinones A (1) and B (2).
These new compounds (especially 2) show excellent - and specific - antitumoral activities against the
multiple-myeloma (MM) cell line INA-6 without any significant toxicity on normal blood cells.[1]
An efficient total synthesis of dioncoquinone B (2) was established to provide this lead structure in
sufficient amounts for a more-in-depth biological evaluation. A first series of structural analogs of
dioncoquinone B (2) was obtained by semi and total synthesis for structure-activity relationship (SAR)
studies hopefully leading to new agents with enhanced anti-MM activities and low cytotoxicities.[2]
Moreover, a synthetic route to a dioncoquinone B derivative was developed, equipped with a probe
(e.g., biotin like in 3) suited for affinity labeling experiments to investigate the interactions of 3 with its
cellular target(s) and/or binding partner(s).
[1] G. Bringmann et al., Phytochemistry 2008, 69, 2501-2509.
[2] G. Bringmann et al., Eur. J. Med. Chem. 2011, 46, 5778-5

Organische Chemie und Biochemie P91 Quality of Antimalarial Drugs in Africa: Data on Artemisinin-Based
Combination Therapy Products (Artemether-Lumefantrine) from
the Democratic Republic of the Congo
J. P. Mufusama1,2, K. Ndjoko Ioset1, U. Holzgrabe3, G. Bringmann1
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany 2Faculté des Sciences Pharmaceutiques, Université de Kinshasa, B.P. 212, Kinshasa XI,
Democratic Republic of the Congo 3Institute of Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg,
Germany
Malaria, with 90% of prevalence in Sub-Saharan Africa, is the most dangerous tropical disease with
more than two million people dying each year. Nearly half of the world population is exposed to the
disease; pregnant women and children under the age of five years are the main victims.[1]
Combination therapies in which an artemisinin (1) derivative like artemether (2) is associated to another
class of antimalarial drugs like lumefantrine (3), the so-called artemisinin-based combination therapies
(ACT), are nowadays recommended by the World Health Organization (WHO) as first-line treatment
for uncomplicated falciparum malaria.[2] The poster will address quality issues of these essential
antimalarial medicines within the African continent including findings of a nationwide survey in the
Democratic Republic of the Congo. Results were obtained based on packaging, thin-layer
chromatography (TLC), and high-performance liquid chromatography (HPLC) analyses.
[1] WHO (2015) World Malaria Report 2015. WHO, Geneva.
[2] WHO (2015) Guidelines for the treatment of malaria, 3rd Edition. WHO, Geneva.

Organische Chemie und Biochemie P92 Korundamines and Mbandakamines, Unsymmetric
NaphthylisoquinolineDimers from the Congolese Liana
Ancistrocladus ealaensis
D. T. Tshitenge1,2, D. Feineis1, B. K. Lombe1, G. Bringmann1
1Institute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
2Faculty of Pharmaceutical Sciences, University of Kinshasa, P.O. Box 212 Kinshasa XI,
Democratic Republic of the Congo
Naphthylisoquinoline alkaloids (NIQs) are thrilling natural products occurring in the two paleotropic
plant families of Ancistrocladaceae and Dioncophyllaceae.[1] They are unique due to their unprecedented
biosynthetic origin, their unusual structural features, and their high anti-infective activities.[1]
Ancistrocladus ealaensis is a widespread Central African liana mainly located in the Northwestern
part of the Democratic Republic of the Congo.[2] Most recent phytochemical studies have shown that the
plant is a rich source of structurally intriguing, highly unsymmetric dimeric NIQs. More than twelve
new dimers, among them new korundamine-type dimers, have of two mbandakamine-type dimers,[3]
viz., 1-epi-mbandakamine A (2) and 1-epi-cyclombandakamine A (3).
The korundamines are the first fully elucidated NIQ ‘mixed’ dimers consisting of a 5,8’- and a 7,8’-
linked monomeric halves, connected by a freely rotating central biaryl axis.The new dimer 2 combines
in the highest known steric hindrance at the central chiral axis between the monomers, with the scarce
cis-relative configuration in one molecular half. Most exciting is the fact that dimers like 2 can undergo
further intramolecular cyclization reactions, due to the extreme steric constrains, leading to
unprecedented cage-like dimers, like e.g. 3.
The detection of dimers of a huge steric demand at the central axis, together with dimers possessing
a freely rotating central axis indicates the unique synthetic potential of A. ealaensis. Moreover, these
compounds exhibit remarkable antimalarial activities against Plasmodium falciparum.
[1] G. Bringmann et al., Progress in the Chemistry of Organic Natural Products, Springer-Verlag,
Wien, New York, 2001, 82, pp. 111-123.
[2] J. Léonard, Bull. Soc. Bot. Belg. 1949, 82, 27-40.
[3] G. Bringmann et al., Org.Lett. 2013, 15, 2590-259

Abstract Abendvortrag Dr. Klein-Sommer
Dr. Günther Klein-Sommer
Chemie der Pyrotechnik – Theorie und Effektdemonstration
Im Gegensatz zur Sprengtechnik werden in der Pyrotechnik die Explosionsstoffe für
Vergnügungs- oder technische Zwecke eingesetzt. Unter Ausnutzung der nach der Zündung
freigesetzten Energie können Licht-, Schall-, Rauch-, Nebel- oder Bewegungswirkungen
erzeugt werden. Von besonderem Interesse, wie die Umsätze der Pyrotechnischen Industrie
zum Jahresende zeigen, ist die Pyrotechnik im Zusammenhang mit der “Feuerwerkerei“.
Im Rahmen des Vortrages wird einleitend die historische Entwicklung der Feuerwerkskunst
vorgestellt. Aus der Erfahrung heraus resultiert die Entwicklung der heute verwendeten
pyrotechnische Zubereitungen und pyrotechnischen Gegenstände “Feuerwerkskörper“ die zur
Erzeugung von Licht-, Schall-, und Raucheffekten oder von Bewegungswirkungen verwendet
werden. Im Anschluss an den Vortrag erfolgt eine Demonstration von Effekten aus den
Bereichen Bühnenpyrotechnik, Spezialeffekte und Feuerwerk.

TEILNEHMER
TEILNEHMER


TEILNEHMER – Anorganische Chemie/Materialwissenschaften
# Name Vorname Arbeitskreis
54 Berthel Johannes Radius
13 Eichhorn Antonius Radius
66 Feizy Nilab Schatzschneider
53 Griesbeck Stefanie Marder
45 Hailmann Michael Finze
52 Kachel/ Kelch Stephanie u. Hauke Braunschweig
9 Kerpen Christoph Finze
85 Krahfuß Mirjam Radius
75 Kuntze-Fechner Maximilian Radius
70 Lorson Thomas Luxenhofer
12 Lubitz Katharina Radius
81 Lübtow Michael Luxenhofer
79 Mawamba Kemo Viviane Schatzschneider
80 Merz Julia Marder
72 Moustafa Mansour Ahmed Schatzschneider
77 Mühlbach Friedrich Müller-Buschbaum
30 Müssig Jonas Braunschweig
84 Nutz Marco Braunschweig
16 Paul Ursula Radius
76 Peng Kun Schatzschneider
28 Rauch Florian Marder
63 Ribbeck Tatjana Finze
71 Schneider Heidi Radius
59 Sedykh Alexander Müller-Buschbaum
15 Sieck Carolin Marder
19 Sorg Jens Müller-Buschbaum
68 Stangl Johannes Müller-Buschbaum
55 Waag-Hiersch Luisa Schatzschneider
50 Wehner Tobias Müller-Buschbaum
41 Zottnick Sven Müller-Buschbaum

TEILNEHMER – Organische Chemie/Biochemie
# Name Vorname Arbeitskreis
25 Buschmann Rachel Krüger
11 Dechant Moritz Lehmann
32 Dhara Ayan Beuerle
90 Froschgeiser Christina Bringmann
78 Hecht Reinhard Würthner
58 Kaufmann Christina Würthner
57 Kiendl Benjamin Krüger
86 Leonhardt Viktoria Beuerle
29 Lombe Blaise Kimbadi Bringmann
23 Meza Ana Lucia Würthner
24 Mims David Lambert
91 Mufusama Jean-Pierre Bringmann
89 Nowak- Król Agnieszka Würthner
74 Possiel Christian Seibel
36 Riese Stefan Lambert
56 Roos Markus Lambert
35 Sapotta Meike Würthner
49 Schopf Nina Lambert
44 Schreck Maximilian Lambert
31 Schweeberg Sarah Krüger
18 Seifert Sabine Würthner
69 Shamburger William Bringmann
67 Soberats Bartolome Würthner
83 Syamala Peethambaran Nair Würthner
92 Tshitenge Dieudonné Bringmann
88 Wachtler Stefan Krüger
14 Wagner Wolfgang Würthner
48 Warkentin Viktor Krüger
21 Wehner Marius Würthner

TEILNEHMER – Pharmazie/Lebensmittelchemie
# Name Vorname Arbeitskreis
65 Amstalde Cecilia Meinel
47 Berninger Michael Holzgrabe
33 Braun Alexandra Meinel
20 Dodt Katharina Meinel
34 Gutmann Marcus Meinel
42 Hermann Cornelius Meinel
40 Kuhn Maximilia Sotriffer
37 Miesler Tobias Meinel
82 Nagl Patrick Holzgrabe
73 Pason Lukas Sotriffer
8 Plank Christina Sotriffer
64 Pospiech Andreas Högger
27 Pospiech/Zilker Andreas u. Markus Högger/Holzgrabe
61 Scherf-Clavel Maike Högger
51 Spieler Valerie Meinel
60 Wu Fang Meinel

TEILNEHMER – Physikalische Chemie/Theoretische Chemie
# Name Vorname Arbeitskreis
62 Bellinger Daniel Engels
26 Brückner Charlotte Engels
22 Flock/Schmitt Marco u. Hans-
Christian Fischer
17 Hirsch Florian Fischer
43 Le Thien Anh Engels
39 Lindner Joachim Mitric
10 Pachner Kai Fischer
38 Pres Sebastian Brixner
87 Reusch Engelbert Fischer
46 Roeding Sebastian Brixner

NOTIZEN

Notizen

NOTIZEN

Organisationskomitee
Das Jungchemikerforum Würzburg im Februar 2016.


www.jcf-wuerzburg.de
JungChemikerForumWürzburg
Programm
ChemSyStM 2016
11.00 Begrüßung durch das JCF Würzburg und den
GDCh Ortsverband (HS B)
11.15 Gastvortrag des Verbandes angestellter
Akademiker und leitender Angestellter (VAA) (HS A)
12.00 Mittagspause
13.00 Posterappetizer (HS A)
14.00 Postersession
17.00 Sektempfang
17.15 Abendvortrag der Firma Sommer-Feuerwerk:
„Chemie der Pyrotechnik“ (HS A)
18.00 Preisverleihung (HS A)
18.30 Abschlussfeuerwerk im Freien (Sommer-Feuerwerk)
…gemütlicher Ausklang bei Abendessen und Bier


















