
Isolierung des Protein VII aus Adenovirus II und ... · desweiteren lösen sie auch lokale...
89
Aus der Klinik und Poliklinik für Innere Medizin des Universitätsklinikums Hamburg-Eppendorf Direktor: Prof. Dr. H. Greten Isolierung des Protein VII aus Adenovirus II und Expression in E.coli- Stämmen – Virusproteine als Transfermoleküle für menschliche DNA DISSERTATION zur Erlangung des Grades eines Doktors der Medizin dem Fachbereich Medizin der Universität Hamburg vorgelegt von Raphaela Augustine Barbara Basdekis aus Hamburg Hamburg, 2001
Transcript of Isolierung des Protein VII aus Adenovirus II und ... · desweiteren lösen sie auch lokale...

Aus der Klinik und Poliklinik für Innere Medizin
des Universitätsklinikums Hamburg-Eppendorf
Direktor: Prof. Dr. H. Greten
Isolierung des Protein VII aus Adenovirus II und Expression in E.coli-
Stämmen – Virusproteine als Transfermoleküle für menschliche DNA
DISSERTATION
zur Erlangung des Grades eines Doktors der Medizin
dem Fachbereich Medizin der Universität Hamburg vorgelegt von
Raphaela Augustine Barbara Basdekis
aus Hamburg
Hamburg, 2001

Angenommen von dem Fachbereich Medizin
der Universität Hamburg am: 8. Januar 2002
Tag der mündlichen Prüfung: 26. Februar 2002
Gedruckt mit Genehmigung des Fachbereichs
Medizin der Universität Hamburg
Dekan: Prof. Dr. C. Wagener
Referent: Priv. Doz. Dr. D. Ameis
Korreferent: Prof. Dr. H. Greten

1
INHALTSVERZEICHNIS
A. Einleitung......................................................................................................................... 4
1. Einführung in die Thematik...............................................................................................................4
2. Gentransfer...........................................................................................................................................52.1. Bedeutung des Gentransfers...........................................................................................................................5
2.2. Entwicklung des Gentransfers........................................................................................................................52.3. Methoden des Gentransfers............................................................................................................................6
2.4. Das adenovirale Vektorsystem.......................................................................................................................82.5. Neuere Entwicklung des adenoviralen Vektorsystems .................................................................................9
3. Molekularbiologischer Aufbau des Adenovirus ..............................................................................93.1. Chemische und physikalische Eigenschaften der Adenoviren ...................................................................10
3.2. Das Virusgenom ...........................................................................................................................................12
4. Das Virus-Kapsid: Gliederung und Zusammensetzung ...............................................................134.1. Das Hexon.....................................................................................................................................................144.2. Das Penton ....................................................................................................................................................15
4.3. Weitere mit dem Kapsid verbundene Proteine............................................................................................16
4.4. Der Aufbau des Kapsids...............................................................................................................................17
5. Der Viruskern.....................................................................................................................................185.1. Der Beweis für die Existenz einer Kernhülle ..............................................................................................18
5.2. Der Aufbau des DNA-Protein-Komplexes (Nukleokapsid) .......................................................................19
5.3. Das Versuchsmodell des Adenovirus-Nukleokapsids.................................................................................20
6. Das Körpermodell des Adenovirus..................................................................................................25
7. Vektoren..............................................................................................................................................267.1. Plasmide........................................................................................................................................................267.2. Bakteriophagen .............................................................................................................................................27
7.3. pJM17-Aufbau..............................................................................................................................................27
7.4. pQE30-Aufbau..............................................................................................................................................28
8. Konstruktaufbau pVII-pQE30.........................................................................................................298.1. Aufbau...........................................................................................................................................................29
8.2. Zielvorstellungen ..........................................................................................................................................30
9. Expression in SG13009 und M15.....................................................................................................30
10. Fragestellung der vorliegenden Arbeit .........................................................................................31
B. Material und Methoden.................................................................................................. 32
1. Materialien..........................................................................................................................................321.1. Chemikalien ..................................................................................................................................................321.2. Molekulargewichtsstandards und Proteine ..................................................................................................33
1.3. Puffer, Medien, Agarplatten und Antibiotika..............................................................................................331.4. Sonstige Pufferlösungen...............................................................................................................................34
1.5. Wasser...........................................................................................................................................................35
1.6. Bakterienstämme ..........................................................................................................................................361.7. Reagenzienkits..............................................................................................................................................36
1.8. Weitere Materialien ......................................................................................................................................361.9. Geräte ............................................................................................................................................................36

2
2. Allgemeine Arbeitsmethoden ...........................................................................................................372.1. Sterilisation ...................................................................................................................................................37
2.2. Absorptionsmessung.....................................................................................................................................37
3. Präparation des Plasmids pJM17...................................................................................................373.1. Prinzip ...........................................................................................................................................................373.2. Durchführung................................................................................................................................................37
4. Quantifizierung der DNA-Menge durch Spektralphotometrie ...................................................384.1. Prinzip ...........................................................................................................................................................38
4.2. Durchführung der OD-Messung ..................................................................................................................38
5. Schneiden des Plasmids pJM17 und des Vektors pQE30 mit SalI.............................................395.1. Prinzip ...........................................................................................................................................................395.2. Durchführung................................................................................................................................................39
6. Amplifikation der dsDNA von Protein VII durch die Polymerase-Kettenreaktion (PCR) .....406.1. Prinzip ...........................................................................................................................................................40
6.2. Verwendete Oligonukleotide .......................................................................................................................416.3. Vorbereitung des PCR-Reaktions-Mix........................................................................................................44
6.4. Durchführung der PCR.................................................................................................................................45
6.5. Modifikation der PCR nach dem PCR-Optimization-Kit ...........................................................................45
7. Analytische Agarosegelelektrophorese ...........................................................................................487.1. Prinzip ...........................................................................................................................................................48
7.2. Vorbereitung und Beladen des Agarosegels................................................................................................48
8. Subklonierung des PCR-Produktes.................................................................................................498.1. Prinzip ...........................................................................................................................................................49
9. Verdau des Protein VII und des Vektors pQE30 mit BamHI und HindIII ...............................509.1. Prinzip ...........................................................................................................................................................509.2. Vorbereitung .................................................................................................................................................50
9.3. Durchführung des Verdaus...........................................................................................................................51
10. Ligation der Verdau-Produkte ......................................................................................................5110.1. Prinzip .........................................................................................................................................................51
10.2. Vorbereitung ...............................................................................................................................................5110.3. Durchführung..............................................................................................................................................51
11. Transformation des Ligationsproduktes in XL 1-blue ...............................................................5211.1. Prinzip .........................................................................................................................................................52
11.2. Vorbereitung ...............................................................................................................................................5211.3. Durchführung..............................................................................................................................................52
12. DNA-Sequenzierung nach Sanger .................................................................................................5312.1. Reinigung der PCR-Produkte.....................................................................................................................53
12.2. Bestimmung der DNA-Sequenz.................................................................................................................5412.3. Auswertung der Sequenzen........................................................................................................................55
13. Transformation des Klonierungsproduktes in M15....................................................................5513.1. Vorbereitungen ...........................................................................................................................................55
13.2. Durchführung..............................................................................................................................................56
14. Probe-Expression des Protein VII in M15 (Small-scale Expression).......................................5614.1. Prinzip .........................................................................................................................................................5614.2. Vorbereitung ...............................................................................................................................................57

3
14.3. Durchführung..............................................................................................................................................57
15. Polyacrylamidgele (SDS-PAGE) zur quantitativen Analyse der Expression ..........................5815.1. Prinzip .........................................................................................................................................................5815.2. Vorbereitung ...............................................................................................................................................58
15.3. Durchführung..............................................................................................................................................59
15.4. Coomassie-Blue-Färbung...........................................................................................................................59
C. Ergebnisse ...................................................................................................................... 60
1. Klonierung und Subklonierung von Protein VII...........................................................................601.1. Isolierung aus pJM17 durch PCR ................................................................................................................601.2. Verdau von pVII und pQE30 mit BamHI und HindIII ...............................................................................61
1.3. Ligation von pVII und pQE30 .....................................................................................................................611.4. Transformation des Ligationsproduktes in XL-1BLUE-E.coli-Stämme....................................................62
2. Analyse der Sequenzierung ..............................................................................................................632.2. Zusammenhang zwischen DNA-Sequenz von Protein VII und Mutation .................................................64
3. Expressionsversuch............................................................................................................................643.1. Transformation des Ligationsproduktes in M15 .........................................................................................65
3.2. Expression des Protein VII...........................................................................................................................653.3. Ergebnis der Expression...............................................................................................................................66
D. Diskussion...................................................................................................................... 67
1. Das adenovirale Vektormodell .........................................................................................................67
2. Probleme des adenoviralen Vektormodells ....................................................................................68
3. Ein Protein als Transfermolekül für DNA –Modifizierung des adenoviralen Vektormodells ................................................................................68
4. Grundlegendes zur Verwendung von Virusproteinen als Transportermoleküle......................694.1. Ziel dieser Arbeit ..........................................................................................................................................694.2. Das Expressionsmodell ................................................................................................................................69
4.3. Probleme bei der Proteinexpression.............................................................................................................70
5. Vergleich mit anderen Modellen für die Fremdgenexpression....................................................715.1. Retrovirale Vektoren ....................................................................................................................................725.2. Adenovirale Vektoren ..................................................................................................................................72
6. Sicherheit und Ethik..........................................................................................................................72
E. Zusammenfassung.......................................................................................................... 74
F. Verzeichnis der Abkürzungen ........................................................................................ 76
G. Verzeichnis der Abbildungen und Tabellen ................................................................... 77
H. Literaturverzeichnis ....................................................................................................... 78

4
A. EINLEITUNG
Die vorliegende Arbeit beschreibt die experimentellen Untersuchungen zur Isolierungund Klonierung des Protein VII von Adenovirus Typ 2. Hierbei wurde erstmalsversucht, das Protein in kompetenten E. coli Stämmen zu exprimieren. Dieses Proteinwird, aufgrund seines hohen Gehaltes an Arginin und Alanin und seiner darausresultierenden guten Bindungsfähigkeit an DNA, für die künftige Molekularbiologieund so auch für die Medizin von Bedeutung sein. Im Laufe der Klonierung ergabensich Schwierigkeiten mit der Virus-DNA, die sich in wiederholten Mutationen zeigten.
1. Einführung in die ThematikAdenoviren sind der häufigste Grund für eine Erkrankung des Menschen und somitverantwortlich für eine ungeheure finanzielle Belastung der Gesellschaft. Hieraus läßtsich erklären, weshalb der Anreiz zur Isolierung des Virus von der Medizin ausging(Rowe et al., 1953; Hilleman und Werner, 1954; Huebner et al.,1954). Als Erreger fürInfektionskrankheiten sind sie seit den 50er Jahren bekannt. 1953 wurde dieBeobachtung gemacht, daß Zellkulturen aus menschlichen Tonsillen scheinbar spontanlysierten (Rowe et al., 1953). Im folgenden Jahr wurde die Isolation eines ähnlichenErregers beschrieben, der für fieberhafte Atemwegsinfektionen in einer Kaserneverantwortlich war (Hilleman et al., 1954). 1956 wurde der einheitliche Name„Adenovirus“ für diese Erreger festgelegt (Enders et al., 1956). Adenoviren sindhäufig Ursache für fieberhafte Infektionen der Atemwege (Dingle et al., 1968),desweiteren lösen sie auch lokale Infektionen aus, wie die Keratokonjunktivitisepidemica (Jawetz, 1959), Erkrankungen des Gastrointestinaltraktes, wie dieGastroenteritis (Wigand et al., 1983), seltener auch eine akute hämorrhagische Zystitis(Numazaki et al., 1973) oder eine Meningoenzephalitis (Kelsey, 1978). 1962 wurdeerstmals entdeckt, daß manche Adenoviren auch eine onkogene Potenz besitzen(Trentin et al., 1962); Adenovirus Typ 2 und 5 hingegen, die als Vektoren für dieGentherapie genutzt werden, besitzen keine Onkogenität (Ali et al., 1994). Seitherwurden 41 verschiedene Antigen-Typen gefunden, die sowohl nach ihrenphysikalischen als auch chemischen Eigenschaften klassifiziert wurden (Matthews,1982). Eine immunologische Verwandtschaft vereinigt diese Virustypen, die dieSäugetiere durch eine kreuzreagierende Antigen-Determinante auf den freien Hexoneninfizieren, nämlich das sog. major core protein (Matthews, 1982, Ginsberg, 1979). DerInfektionsweg umschließt eine gut geordnete Serie von Ereignissen, die durch dieVerknüpfung eines Adenoviruspartikels mit einer empfänglichen Zelle mit Hilfe seinerFiberproteine ausgelöst wird und mit einer Ansammlung von nahezu 104 infektiösenVirions pro Zelle gipfelt ( entspricht Beobachtungen bei Adenovirus Typ 2 oder 5).Als Folge dieser Infektion können die infizierten Zellen zerstört werden und absterbenund so Krankheiten hervorrufen; sie können auch in ihrem Genom verändert werden,so daß sie Tumore induzieren, oder die infizierten Zellen bewirken eine latente

5
Infektion der Lymphozyten, so wie es bei der ersten Entdeckung der Adenoviren durchRowe et al., 1953 gezeigt wurde.
2. Gentransfer
2.1. Bedeutung des Gentransfers
Das Verständnis menschlicher Organe war vor Entwicklung moderner biochemischerund molekularbiologischer Methoden in der Grundlagenforschung ein gänzlichanderes: ihnen wurde lediglich eine Reaktionsfähigkeit auf nervale oder hormonelleSignale zugesprochen. Im Gegensatz dazu steht heute ihre Synthese- undRegulationsfähigkeit im Vordergrund. Mit Hilfe der Molekularbiologie gelang dieCharakterisierung vieler Gene, die diese Mechanismen steuern. Im gleichen Zugewurden auch Gene identifiziert, die kausal an Krankheitsprozessen beteiligt sind. Aufdieser Basis ermöglichten es verschiedene molekularbiologische Gentransfer-techniken, unterschiedliche Konzepte zur Analyse und Therapie von Erkrankungen zuentwickeln (von der Leyen et al., 1995).
2.2. Entwicklung des Gentransfers
Das Konzept des Gentransfers und der Gentherapie baut auf den Erkenntnissen derMolekularbiologie, der Biochemie und der Medizin auf (Leiden, 1995; Blau et al.,1995; Haddada et al., 1995).In den 60er Jahren entdeckte man, daß das Genmaterial der Papovaviren SV 40 undPolyoma während der neoplastischen Umwandlung ihrer Wirtszelle stabil undvererbbar in deren Genom integriert wurde (Sambrook et al., 1968). Hieraus entstanddie Idee zur Transformation von Zellen mit Hilfe der Einschleusung von exogenemgenetischen Material (Rogers et al., 1968). SV 40 diente dabei als transduzierenderVektor zur Übertragung, zum Einbau und zur Vermehrung von Fremdgenen inSäugetierzellen (Subramani et al., 1983; Jackson et al.,1972). In den 70er Jahrenbegannen die ersten Versuche von Gentransfer in humane Zellen mit einemtherapeutischen Hintergrund, leider ohne Erfolg (Rogers et al., 1973). Erst durch dieEntwicklung der Technik der rekombinanten DNA konnten größere Fortschritte erzieltwerden; durch sie wurde es möglich, DNA-Sequenzen zu verändern, neuzusammenzusetzen und größere Mengen an DNA, wie sie für den Gentransfer benötigtwerden, herzustellen. Grundlegende Voraussetzungen für einen effektiven in vivoGentransfer waren/sind: klonierte, rekombinante Gene und effiziente Methoden ihrerÜbertragung.Diese Entwicklung im Bereich des Gentransfers und der Technologie derrekombinanten DNA hat zu einer weltweiten experimentellen Anwendung derGenexpression geführt, mit Hilfe derer krankhafte Prozesse wie Enzymdefektedaraufhin untersucht wurden, ob sie einer Behandlung mit Gentherapie zugänglichsind. Die Aufmerksamkeit richtet sich nicht nur auf Einzeldefekte, sondern auch auf

6
Krankheiten mit einer komplexeren Pathogenese, wie zum Beispiel Erkrankungen deskardiovaskulären und hämatopoetischen Systems, hier insbesondere die Leberbetreffend, die eine zentrale Rolle bei der Mehrheit der metabolischen Erkrankungenspielt. In folgender Tabelle sind einige mögliche Ziele der Gentherapiezusammengestellt (Strauss, 1994):
Tab. 1: Genetisch bedingte Erkrankungen
Erkrankung Defizientes Genprodukt
Genetisch vererbte Erkrankungen
Familiäre Hypercholesterinämie Low-density-Lipoproteinrezeptor
Fettstoffwechselstörungen Apolipoproteineα1-Antitrypsin-Mangel α1-Antitrypsin
Phenylketonurie Phenylalanin-HydroxylaseHämophilie A und B Faktor VIII und IXLysosomale Speicherkrankheit VerschiedeneOrnithin-Transcarbamylase-Mangel OTC
Hereditäre Tyrosinämie Fumaryl-Azetoazetat-Hydroxylase-Mangel
Maligne Tumoren
Hepatozelluläres Karzinom VerschiedeneMetastasen Nicht relevant
Infektionskrankheiten
Virus-Hepatitis (A, B, C) Epitope virale Genexpression
Die Methoden sind noch nicht genügend ausgereift, und viele Probleme sind noch zulösen. Eine Aufgabe der Zukunft wird es sein, die Effektivität des Gentransfers durchbessere Transfersysteme zu steigern, Nebenwirkungen zu reduzieren und dieZielgerichtetheit in das zu therapierende Gewebe zu optimieren.
2.3. Methoden des Gentransfers
Die einfachsten Methoden zum DNA-Transfer sind die Calciumphosphat- und dieDiethylaminoethydextran (DEAE-)-Methode, die aber in vivo nur begrenzt einsetzbarund in vitro nur wenig effizient und stabil sind (Sambrook et al., 1989; Felgner et al.,1991. Siehe auch Tabelle 4 ). Dies sind auch die limitierenden Faktoren für eineerfolgreiche Gentherapie. Im allgemeinen haben virale Vektoren die höchste Effizienzund werden deshalb bevorzugt gegenüber physikalischen Methoden verwendet. Unterdiesen spielen retrovirale und adenovirale Vektoren eine große Rolle in demHepatozyten-gerichteten Gentransfer in vitro und in vivo.

7
Tab.2: Methoden des Gentransfers
Methode Stabilität Applikationex vivo in vivo
chemisch
Kalzium-Phospat S ++ −
physikalisch
Mikroinjektion S +++ −
Jet-Injektion T ++ +
viral
Retroviren S +++ +
Adenoviren T +++ +++Die Stabilität der transferierten DNA und ihre Expression wurden als stabil (S), basierend auf ihrer Integration, und
vergänglich (T), basierend auf dem Verlust an nicht-integrierter DNA, klassifiziert. −, nicht applizierbar; +, geringe Effizienz
oder wenig geeignet; ++, mäßige Effizienz oder gut geeignet; +++, hohe Effizienz oder besonders geeignet (nach Strauss,
1994).
2.3.1. Versuche mit retroviralen Vektoren
Retrovirale Vektoren sind die am besten untersuchten Transportmittel für DNA(Mulligan, 1993; Grossman und Wilson, 1993), woraus sich folgendeSchlußfolgerungen ergaben:
1. amphotrope retrovirale Vektoren haben ein breites Wirtsspektrum bezüglich derSpezies und Gewebe,
2. sie können bis zu 7 kb fremder DNA unterbringen,3. eine Kopie des Virusgenoms wird in das Wirtsgenom integriert ohne Spezifität
bezüglich der Zielsequenz und4. das Virusgenom wird nur in sich replizierende Zellen integriert.
Während die ersten drei Punkte den retroviralen Vektor für eine Gentherapiegebräuchlich erscheinen lassen, begrenzt der vierte Punkt diese Möglichkeit auf sichteilende Zellen, welche man in vivo nur selten findet. Es wurde versucht, Hepatozytenin vitro zu transduzieren und anschließend zu transplantieren (Wilson et al., 1988;Grossman et al., 1991 und 1992; Chowdhury et al., 1991). Hierbei stellte sich heraus,daß nur 2-5 % der Leber durch funktionsfähige transduzierte Hepatozyten ersetztwerden konnte.Eine Alternative ergab sich mit Hilfe der Hepatektomie, welche eineregenerationsinduzierende Wirkung hat und somit die Zellen zur Teilung anregt. Dieretroviralen Vektoren wurde in das portale Gefäßsystem nach der Hepatektomieeingeführt. Doch auch hier konnten nicht mehr als 5 % der Hepatozyten transduziertwerden (Ferry et al.,1991; Kay et al., 1992).

8
2.3.2. Adenovirale Vektoren
Adenovirale Vektoren haben bezüglich verschiedener Gewebe eine ähnlicheEffektivität wie Retroviren, ihr entscheidener Vorteil liegt darin, daß man sie auch inruhende Zellen transduzieren kann (Stratford-Perricaudet et al., 1990; Levrero et al.,1991). Im Gegensatz zu Retroviren integrieren Adenoviren ihr Genom nicht stabil indas Wirtsgenom, es geht nach einigen Zellteilungszyklen verloren. AdenoviraleVektoren können effizient transferiert werden, sowohl in vitro als auch in vivo(Stratford-Perricaudet et al., 1990; Levrero et al.;1991; Jaffe et al., 1992 und Li et al.,1993), wobei ein Verhältnis von Adenoviren/Zellen von 100/1 bestehen muß. Unterdiesen Bedingungen werden über 95 % der Zellen transduziert (Li et al., 1993), dieExpressionsrate aber sinkt nach ca. vier Wochen auf 0,5-10 % ab. Durch diese Aspektewerden adenovirale Vektoren interessant für eine Kurztherapie bei akutenErkrankungen, ein einziges Problem entsteht bei wiederholter Anwendung durch dieBildung von Antikörpern gegen Adenoviren (Ginsberg et al., 1991).
2.4. Das adenovirale Vektorsystem
Zur Konstruktion von adenoviralen Vektoren wird fremde DNA in das Virusgenomeingebaut, nachdem zuvor entsprechende DNA-Sequenzen aus dem viralen Genomentfernt wurden. Dies ist notwendig, da das adenovirale Kapsid max. 105 % desWildtyp-Genoms aufnehmen kann (Bett et al., 1993). Bei den Vektoren der erstenGeneration wurden Sequenzen aus der E1-oder E3-Region ersetzt, es wurden auchkombinierte Deletionen durchgeführt; so konnten bis zu 7,5 kb fremder DNA in dasVirusgenom integriert werden. Da die E1-Region des Adenovirusgenoms essentiell istfür die Replikation des Virus in der Wirtszelle (Ali et al., 1994), wurde diese Regionvor der Transfektion entfernt, das Virus wurde sozusagen replikationsdefizient für alleZellen außer 293-Helferzellen, die diese Region ersetzen (Graham et al., 1977). ZurGewährleistung der Sicherheit bezüglich unkontrollierter Virusreplikationen werdenzur Zeit nur Viren eingesetzt, die diese Deletion aufweisen. Die E3-Region beeinflußtdie Replikationsfähigkeit des Virus überhaupt nicht, so daß die zu ersetzende DNA andieser Stelle in das Virusgenom eingebaut werden kann, ohne eine Einschränkung derReplikation in der Wirtszelle zur Folge zu haben (Ali et al., 1994). Da dieherkömmlichen adenoviralen Vektoren starke immunologische Reaktionenhervorgerufen haben (Yang et al., 1994), wurden diese dahingehend verändert, daßdurch eine zusätzliche Deletion der E2A-Region, die für adenovirale Proteine kodiert,die immunologische Abwehrreaktion verhindert wurde. Diese Deletion verbessert diePersistenz der transferierten DNA und führt zu einer Reduktion der Infiltration desinfizierten Gewebes mit zytotoxischen CD8+-T-Zellen, wie man es sonst beobachtenwürde (Engelhardt et al., 1994). Einige Proteine, für die die E3-Region kodiert,können die immunologische Abwehrmechanismus des Organismus auf infizierteZellen herabsetzen (Wold et al., 1991), wie z.B. das Protein gp19k (Molekulargewicht:19 kD), welches den Transport von MHC I (major histocompatibility complex class I)-

9
Proteinen an die Zelloberfläche verhindert, welche für die Erkennung von infiziertenZellen durch CD8+-T-Zellen notwendig sind (Williams et al., 1990; Cox et al., 1990).Tatsächlich verlängert das Protein gp19k bei einem E1/E3-deletierten adenoviralenVektors die Persistenz der eingeführten DNA (Lee et al., 1995).
2.5. Neuere Entwicklung des adenoviralen Vektorsystems
Im Laufe der Entwicklung molekularbiologischer Methoden zum Gentransfer stelltesich die Frage nach Vereinfachung des DNA-Transportes und der Möglichkeit, DNAauf natürlichere Art und Weise in die Zellen einzuschleusen. Eine der am meistenerprobten Transfektionsmethode ist die Einbindung von DNA in einen Ca2+-Phosphat-Komplex (Mandel und Higa, 1970; Graham und van der Eb, 1973).Ein natürlicher Weg, fremde DNA in den Kern von eukaryoten Zellen einzubringen,ist der während einer DNA-Virus-Infektion. Eine andere Art ergibt sich aus demVersuch, diesen Infektionsweg zu imitieren, indem man fremde DNA mit einemviralen Kernprotein, das eng an Virus-DNA gebunden ist, koppelt. Dies wurde mitHilfe von Adenoviren in zahlreichen Studien versucht. Der Kern des Adenovirusbesteht (wie im folgenden beschrieben) aus dem Virus-DNA-Molekül – 35.937 bp fürAdenovirus Typ 2 (Roberts et al., 1986) – und aus den Proteinen V und VII (Laver etal., 1968; Maizel et al., 1968; Prage et al., 1968; Laver, 1970; Prage et al., 1970;Russel et al., 1971; Brown et al., 1975; Vayda et al., 1983). Der Kern enthält ebenfallsdas terminale Virus-Protein, welches kovalent an das 5´-Ende der Virus-DNAgebunden ist (Robinson et al., 1973), und das sehr basische µ-Protein (Hosokawa undSung, 1976; Sung et al., 1977). Es konnte gezeigt werden, daß sich Adenovirus-DNAund das Kernprotein VII in Lösung zu Strukturen, die viralem Chromatin ähneln,wiederverbinden (Sato und Hosokawa, 1984). Das Protein VII hat einen hohen Gehaltan Arginin (23 %) und Alanin (18 %) (Prage und Pettersson, 1971; Russel et al., 1971;Sung et al., 1977) und bindet aufgrund seiner eigenen positiven Ladung gut mit dennegativen Ladungen von DNA. In den Versuchen wurde die Möglichkeit vonAdenovirus-DNA und dem Kernprotein VII, sich zu bestimmten strukturellenEinheiten wieder zusammenzuschließen, genutzt.
3. Molekularbiologischer Aufbau des Adenovirus
Es gibt drei Ebenen, auf denen der strukturelle Aufbau von Adenoviren untersuchtwerden kann:• Die Molekülstruktur – zunächst durch die Tertiärstruktur von Makromolekülen.• Die Struktur des Makromoleküls – hauptsächlich die Quartärstruktur, d.h. die
Morphologie von isolierten Makromolekülen, wie Fiberproteine, Spikes.• Makromolekulare Verbindungen – d.h. die Struktur von Makromolekülkomplexen
und ihren wechselseitigen Verbindungen im Inneren des Virions.

10
Die Elektronenmikroskopie betrachtet die Makromoleküle (Fiberproteine, Hexone)und ihre Komplexe (Kapsid, Kern). Die ersten Studien begannen vor mehr als 20Jahren (Nermut, 1980a). Sie zeigten, daß das Virus die Form eines Ikosaeders(Zwanzigflächners) hat und aus zwei strukturellen Hauptkomplexen besteht: demKapsid – einer ikosaedrischen Proteinhülle – und dem Kern – einem inneren Körper,der die Virus-DNA und zwei Major-Proteine einschließt. Die meisten Daten, die vonAdenoviren bekannt sind, stammen von Adenovirus Typ 2 und 5 (Ad2 und 5).
3.1. Chemische und physikalische Eigenschaften der Adenoviren
Das Adenovirus enthält 11,6-13,5 % lineare doppelsträngige DNA (dsDNA), der Restbesteht aus Protein (Hitt et al., 1994). Es gibt keine Lipide, und nur 1 % istglykosyliert (Fasern). Das Molekulargewicht von Säugetier-Adenovirus-DNA beträgt0,20-0,25 x 108 Da [ 36,000 bp], wohingegen Vogel-Adenovirus-DNA einMolekulargewicht von 0,30 x 108 Da besitzt. Die Sedimentationskonstante von Ad5-DNA beträgt 31 S, die von Ad2-DNA 32 S (Black und Center, 1979). Die DNA istlinear angeordnet und ca. 11-13 µm lang (M. Green et al.,1967a), im Inneren aberkann eine zirkuläre Form bestehen, vermittelt durch das sog. terminale DNA-Protein(Robinson et al.,1973; Rekosh et al., 1977; Keegstra et al., 1977). Außerdemexistieren in dem Virion elf Arten von Polypeptiden mit Molekulargewichtenzwischen 3000 (3 K) bis 110 K (Tab. 1) (Philipson et al., 1975; Ginsberg 1979; Flint,1980; Referenzen: Akusjävi und Persson, 1981). Das Molekulargewicht des gesamtenVirions beträgt 1,75-1,85 x 108 Da (M. Green et al., 1967b; Devaux et al., 1983), derDichtegradient in Cäsiumchlorid (CsCl) 1,33-1,35. Bei der Berechnung der Größe eineAdenovirion muß man die ikosaedrische Form berücksichtigen, da ihr Ausmaß sichvon ihren Kanten, d.h. dem Abstand zwischen zwei Pentonen, ableitet. Die Größe wirdentweder als Begriff eines Eck-zu-Eck-Abstandes (P= Kante x 1,902), als Kante-zu-Kante-Abstand (E= Kante x 1,618) oder als Durchmesser (D= Kante 1,7) desViruspartikels in einer fünffachen Symmetrieorientierung angegeben (Mattern, 1969).Für Ad5 ergibt sich elektronenmikroskopisch eine Kantenlänge von 430 Å; darausergeben sich folgende Werte: P≈ 820 Å, E= 696 Å und D= 731 Å. Ein Wert von 736 Åfür D ergab sich nach Gefriertrocknen und Verdunkelung (Nermut, 1975). Schließlichwurde die Kantenlänge aufgrund der Daten der Neutronendiffraktion auf einen Wertvon 520 Å berechnet. Dies zeigte, daß der Durchmesser D eines vollständig hydriertenViruspartikels ungefähr 880 Å beträgt (Devaux et al., 1983).Über eine Bindung der Fiberproteine der Viruskapsel an einen Rezeptor auf derZelloberfläche dringt das Adenovirus in die Wirtszelle ein (Defer et al., 1990). DerEintritt in das Zellinnere erfolgt durch rezeptorvermittelte Endozytose (Verga et al.,1991). Eine Integration der adenoviralen DNA in das Wirtszellgenom ist äußerst selten(Ali et al., 1994), dennoch wies die DNA adenoviraler Vektoren eine langandauerndeStabilität in transfizierten Zellkulturen auf, was mittels der Polymerase-Kettenreaktion(PCR) nachgewiesen werden konnte (Merklein 1998). Bei der Betrachtung des

11
Infektionsweges ist es notwendig, Adenoviren mit und ohne onkogenem Potential zuunterscheiden. Die für den Gentransfer eingesetzten Adenoviren des Serotyps 2 und 5besitzen kein onkogenes Potential. Andere hingegen haben durchaus die Fähigkeit, dasGenom der Wirtszelle zu transformieren, hierfür sind vor allem die Adenovirustypen12, 18 und 31 bekannt (Horwitz 1990). Diese Fähigkeit ist hauptsächlich in derTatsache begründet, daß diese Adenoviren ihre DNA in das Wirtszellgenomintegrieren (Doerfler et al., 1995).
Adenovirus-Infektion
Zelle
ChromosomaleDNA
Virus-DNA
Kern
Abb. 1: Schema: Adenovirus-Infektion
Tab. 3: Proteine des Adenovirus1
Nr. Mol.gew.2Anzahld. Kopien/Virion
Name Stellung(Ort)
Bemerkungen
II 108.000 720 Hexon Kapsid drei Polypeptide/Hexon
III 85.000 36 (?) Penton-Basis Kapsid,Horizontale
wahrscheinlich drei Po-lypeptide/Penton-Basis
IIIa 66.000 60 (?) − Peripento-nale Region
phosphoryliert
IV 62.000 24 (?) Faser Kapsid, Ho-
rizontale
glykosyliert
1 Die Tabelle wurde aus verschiedenen Quellen zusammengestellt und basiert hauptsächlich auf Daten von
Adenovirus Typ 2 und 5.2 Die Asteriske markieren Durchschnittswerte.

12
Nr. Mol.gew.2Anzahld. Kopien/Virion
Name Stellung(Ort)
Bemerkungen
IVa2 50.000
55.000
− (?)
2
−
terminalesDNA-Protein
Kern-gebun-denes 5´-Ende derDNA
DNA-gebunden
−
V 48.000 180 Kern-Protein I
Kernhülle schwach basisch, wenigphosphoryliert, DNA-gebunden
VI 23.4000 420 (?) − Hexon-gebunden
phosphoryliert, DNA-gebunden
VII 18.000 1,070 Kern-Protein II
Nukleo-kapsid
basisch, DNA-gebunden
VIII 13.000 − − Hexon-ge-bunden
−
IX 11.500 300 − gebunden an9er-Gruppen
−
X 7000* 50 − Intern −
XI 4500* 125 − Intern wahrscheinlich ein
Fragment von VII oderidentisch mit dem µ
Protein
XII 3000* − − Intern wahrscheinlich einFragment von VIII
3.2. Das Virusgenom
Das adenovirale Genom läßt sich funktionell in eine frühe (E1-E4) und eine späteTranskriptionsregion (L1-L5) unterteilen (Haddada et al., 1995). Die E1-Region wirdsofort nach Eintritt des viralen Genoms in den Kern der Wirtszelle aktiv; sie kodiertfür Regulatorproteine, die in der frühen Infektionsperiode benötigt werden (Grand,1987). Die Proteine für die Virusreplikation, wie z.B. die DNA-Polymerase, sind inder E2-Region kodiert. Die E3-Region kodiert für Proteine, die der Herabsetzung derWirtszellreaktion auf die Infektion dienen und die Viruselimination verlangsamen. DieE4-Region beinhaltet Gene für Proteine, die der bevorzugten Expression von viralenGenen zum Nachteil der Wirtszellgene dienen. Die späten Transkriptionsregionenkodieren für Polypeptide, die die Viruskapsel aufbauen (Horwitz, 1990).

13
4. Das Virus-Kapsid: Gliederung und Zusammensetzung
240 Hexone und 12 Pentone, die jeweils aus der Penton-Basis und einer Faserbestehen, bilden die ikosaedrische Proteinhülle des Adenovirus (Valentine und Pereira,1965). Eine Besonderheit dieses Kapsid besteht darin, daß es nicht unter bestimmterBehandlung in die dreieckigen Facetten zerfällt (siehe Abb.2) – wie es zu erwartenwäre -,sondern in 9er-Gruppen von Hexonen, die entweder an Pentone gebunden (sog.peripentonale Hexone) oder frei sind. Elektronenmikroskopisch imponieren diese 9er-Gruppen auf zwei Arten: entweder linkshändig (LH) oder rechtshändig (RH),entsprechend der Definition von Pereira und Wrigley (1974).
Abb.4: Modell der Kernhülle,
bestehend aus 240 ringförmigenwahrscheinlich pseudosechs-eckigen Proteinmolekülen. DieVertikalen sind von den Kapsid-Pentonen oder einem anderenVirusprotein (eventuell IIIa)
besetzt Aus Nermut 1980a
Abb. 2: Aufbau einer triangulären Facettedes Kapsids. P Pentone; Dreiecke – Spitzender Hexone; Sechsecke – Seiten der Hexone.Aus: Nermut und Perkins (1979).
Abb. 3: Gefriertrocknung eines Avian -Adenovirus. Die Pfeile bezeichnen diedreieckige Form einiger Hexone. Aus:Hayat 1977.

14
4.1. Das Hexon
Das Hexon ist ein Kapsomer und somit Hauptbestandteil der Proteinhüllenoberfläche.Die Aufgabe der Hexone besteht darin, gelöste Substanzen oder größere Moleküle,wie z.B. Enzyme, entlang des Kapsids zu transportieren; desweiteren spielen sie eineRolle für das Virus bezüglich seiner Interaktionen mit der Umwelt und den zellulärenMembranen. Ein Hexon besteht aus drei identischen Polypeptiden (Grütter undFranklin, 1974) und besitzt ein Molekulargewicht von 360 K. Die Basis ist rund undhat eine axiale Öffnung (25-30 Å); der Diameter beträgt 75-80 Å. Die Mitte erscheinthexagonal mit einem Durchmesser von 105 Å. Die Spitze ist dreieckig mit einem Y-förmigen Spalt, die Länge zwischen den Ecken und Kanten beträgt 95 Å. Sie ist inbezug auf die Mitte um 30 ° rotiert. Das Hexon hat eine Gesamthöhe von 105-110 Å.
Abb. 5: Adenovirus-Kapsomere nachNegativfärbung. a) Hexone in verschiedenerAusrichtung, sowohl einzeln als auch in„Neuner-Gruppen“. b) links- (L) undrechtsdrehende (R) Neuner-Gruppen. AusNermut 1980a. c) Ad5-Pentone. d) Pentone
mit zwei Fibern eines Avian-Adenovirus; dielängere Fiber ist oft stark gebogen. Von Dr.
N. G. Wrigley.

15
Es besteht weiterhin eine Polarität innerhalb des Hexons: die Spitze ist hydrophil undvorwiegend negativ geladen, wohingegen die Basis hydrophobe Eigenschaftenaufweist (Nermut und Perkins, 1979). Die hydrophobe Seite interagiert mit derKernoberfläche, welche hauptsächlich durch das Polypeptid V gebildet wird,außerdem bestimmt sie die Gruppenspezifität (Norrby, 1969). Die hydrophile Seite desHexons trägt die Typ-spezifischen Antigeneigenschaften. Die Masse der Hexone proVirion wurde als 0,778 x 108 Da berechnet, was ungefähr 46 % der Gesamtmasse einesVirions entspricht (Devaux et al.,1982).
4.2. Das Penton
Die Kapsomere der Vertikalen des Ikosaeders werden als Pentone bezeichnet, da sievon fünf (peripentonalen) Hexonen umgeben sind (Ginsberg et al.,1966). Es wird auseiner Penton-Basis und einem dünnen, antenngleichen Anhängsel, der sog. Faser,gebildet.
Abb. 6: H e x o n - M o d e l l,entwickelt aus elektronenmikro-
skopischen Abbildungen undcomputergestützter Modelierung(Nermut und Perkins, 1979). a)dreieckige Spitze, b) rundeGrundpartie mit der axialenÖffnung, c) die Seitenansicht undd) die Übersicht. Die pseudohexa-gonale Form ist hauptsäch-lichdurch die dritte Lage von unten
bedingt.

16
Abb. 7: Diagramm einer Fiber des Ad2. Die Dimensionen werden in Ångström angegeben.(aus Nermut 1984).
Über das Molekulargewicht herrscht noch einige Ungewissheit: Das Molekulargewichtder Basis wurde mit 245 K berechnet, was hieße , daß es sich um ein Trimer handelte,das Gesamtgewicht mit 365 K. Das bedeutete, daß das Molekulargewicht der Faser120 K betragen müßte, sie könnte also ein Dimer sein. Messungen mittels derNeutronenstreuung haben aber ein Gewicht von 160 K ermittelt (Devaux et al., 1982),was eher für einen trimeren Aufbau spricht. Einige elektronenmikroskopische Aufnah-men zeigten einen pentagonalen Aufbau der Penton-Basis (Pettersson und Höglund,1969; Boudin et al., 1979), der Aufbau aus den drei Untereinheiten konnte aber nochnicht geklärt werden. Die Veröffentlichung der DNA-Sequenz half dabei, dasMolekulargewicht der Faser zu berechnen (62,294). Anhand dieser Sequenz wurde einModel der Faser konstruiert, woraus ersichtlich wird, daß der Schaft aus zweiPolypeptidketten in gekreuzter β-Konfiguration besteht; die hydrophobe Grundflächensind einander entgegengesetzt angeordnet (Earnshaw et al., 1979; Green et al., 1983).
4.3. Weitere mit dem Kapsid verbundene Proteine
Das Virus-Polypeptid (VP) IIIa, verbunden mit dem Scheitelpunkt des Kapsids, hat dieFunktion eines Mediators zwischen der Penton-Basis – angenommen, daß es sich umein Trimer handelt – und den fünf peripentonalen Hexonen inne (Everitt et al.,1975;Devaux et al.,1982). VP IIIa liegt in fünf Kopien pro Scheitelpunkt vor (Boudin etal.,1980). Desweiteren hat man herausgefunden, daß VP IVa2, V, VI und VII an DNAgebunden sind (Russell und Precious, 1982), wobei die Aufgabe von IVa2 im Kernnoch unbekannt ist. Protein VI ist an das Hexon gebunden und löst sich von demViruskern unter der Behandlung mit Pyridin oder Desoxycholat (DOC) im Rahmeneiner Gradientenzentrifugation (Everitt et al., 1973; Nermut, 1979). Das Polypeptid IXist an die 9er-Gruppen gebunden, wahrscheinlich in einem Verhältnis von 15 Kopienpro 9er-Gruppe (Boulanger et al., 1979; Pereira und Wrigley, 1974; Colby und Shenk,1981).

17
4.4. Der Aufbau des Kapsids
Die Anordnung des Adenoviruskapsids richtet sich nach den Erfordernissen der 5-, 3-,2-Faltsymmetrie und der Forderung nach der größtmöglichen Anzahl von Bindungen,d.h. einem Minimum an freier Energie, zwischen den Kapsomeren. Das Kapsid gehörtzur P-1-Klasse, mit einer Triangulationszahl T= 25. Die dreieckigen Spitzen derHexone im Inneren einer 9er-Gruppe haben einen konstanten Scheitelkreis von 60 °nahe der Kante der Dreicksfacette, was erklären könnte, warum das Kapsid bevorzugtin 9er-Gruppen zerfällt und nicht in Dreiecke oder wahllose Teile. DieWechselwirkungen im Innern der 9er-Gruppen sind stärker als zwischen zweibenachbarten (Nermut und Perkins, 1979); eine alternative Erklärung dafür wäre dieExistenz eines Verbindungsproteins innerhalb der 9er-Gruppen (Boulanger etal.,1979). Die Ausrichtung der peripentonalen Hexone und ihr Zusammenspiel mit denPentonen ist nur eine Annahme. Es kann sich auch um elektrostatische Anziehungenzwischen Hexonen und Pentonen handeln. Die Folgerungen aus dem Modell vonStruktur und Funktion des Virus-Kapsids lauten also:
1) Das Kapsid ist stabil genug, um auch ohne Inhalt, d.h. den Kern, zu existieren(Pereira und Wrigley, 1974; Philipson 1979).
2) Der enge Kontakt zwischen Hexonen bedeutet, daß das Kapsid für größereMoleküle, wie z.B. Nukleasen mit einem Molekulargewicht von 16 K,vollkommen verschlossen ist, und so nur die Passage für gelöste Stoffe in das
Abb. 8: Adenoviruskerne –verschiedene Arten der Präparation.a) Gefriertrocknung mit Negativ-färbung; die Kerne liegen hier nahe
an den Kapsiden. b) 0,5 % DOCbei 56°C, Negativfärbung mitAmmonium-Molybdat; die Ober-fläche ist bedeckt von ringförmigenUntereinheiten. Aus Nermut 1984.

18
Virusinnere ermöglicht wird. Das Kapsid wirkt also als Permeabilitätsbarriere,wahrscheinlich zusammen mit der darunterliegenden Kernhülle.
5. Der Viruskern
Abgesehen von dem ikosaedrischen Kapsid werden die Virusbestandteile als Kernbezeichnet; diese Bezeichnung ist seit 1968 allgemein gebräuchlich (Laver et al.,1968; Russel et al., 1971), aber die Definitionen und Vorstellungen über die Strukturdieser Kerne ist uneinheitlich. Die biochemische Definition (z.B. Mirza und Weber,1977) besagt, daß der Kern aus Virus-DNA und zwei Kern-Proteinen (Protein V undVII) besteht. Sie sagt aber nichts über die Beschaffenheit des Komplexes aus, ob ervon Natur aus gegliedert ist oder nicht. Die morphologische Definition beschreibt denKern als einen dichten, wahrscheinlich ikosaedrisch geformten Körper, der aus eineroberflächlichen Proteinhülle und dem inneren DNA-Protein-Komplex, demeigentlichen Nukleokapsid, besteht.3 Trotzdem herrscht noch viel Unklarheit über denAufbau und die Funktion der zwei Hauptproteine V und VII (siehe auch Everitt et al.,1973; Mirza und Weber, 1982; Nermut, 1979).
5.1. Der Beweis für die Existenz einer Kernhülle
Mit Hilfe der Gefrierschnitt-Technik gefolgt von einer Negativfärbung konntenEinzelheiten der Kernoberfläche sichtbar gemacht (siehe Abb.9) (Brown et al., 1975;Nermut, 1975 und 1978), und so gezeigt werden, daß der Kern eine oberflächlicheHülle besitzt. Die Frage, die sich nun ergab, war die, welches der beiden Kernproteinediese Hülle bildet. Da sich gezeigt hatte, daß das Protein VII eng mit der Virus-DNAverbunden ist, blieb nur die Schlußfolgerung, daß das Protein V die Hülle bilden muß.Dieses Polypeptid hat ein Molekulargewicht von 48 K und liegt in 180 Kopien proVirion vor (Everitt et al., 1973). Aufnahmen des Kerns mit einer hohen Auflösungzeigten ein ringförmiges Molekül von 50-80 Å im Durchmesser und einemMolekulargewicht zwischen 45 und 60 K (Nermut, 1980a). Bedingt durch die Nähevon Kern und Kapsid muß die Hülle wie ein Ikosaeder geformt sein mit einem T= 25,aber aus kleineren Proteinmolekülen als das Hexon. Die Scheitelpunkte der Hüllescheinen durch die Penton-Basis gut verschlossen zu sein. Dies zeigte die Entfernungder Basis durch Dialyse; hierdurch wurde die Hülle, und somit das Virion, durchlässigfür Nukleasen, so daß sie in das Virusinnere gelangen konnten. Das alles erlaubte dieAnnahme, daß die Kernhülle zum einen das Virusgenom schützt, zum anderenzusammen mit dem Kapsid den Transport von Wasser und Salzen in das Innere undauch nach außen reguliert.
3 Gemäß der Definition des Virus-Nukleokapsids des International Committee for Nomenclature of Viruses
(Wildy, 1971).

19
5.2. Der Aufbau des DNA-Protein-Komplexes (Nukleokapsid)
Verschiedene Methoden (siehe Ginsberg, 1979) haben gezeigt, daß dieser Komplexaus dem Protein VII und der an dieses gebundenen DNA besteht. Das Protein bestehtzu ca. 22 % aus Arginin, was die negative Ladung der DNA vollkommen neutralisiert(Laver, 1970). Der exakte Aufbau dieses Komplexes wurde mit Hilfe derElektronenmikroskopie und verschiedener biochemischer Versuche analysiert.
5.2.1. Elektronenmikroskopische Beobachtungen des Viruskerns
Es wurde beobachtet, daß die verschiedenen Untersuchungsmethoden zuverschiedenen Graden der Entspannung oder sogar zum Zerfall der DNA führen;oftmals wirkte dann die DNA wie „Spinnen“: der dichte Kern war mit Schleifen vonDNA umgeben (Brown et al.,1975; Nermut et al., 1975). Unter Behandlung mit DOC(z.B. 0,4 % DOC bei 56 °C für 40 sek) konnte die DNA kontrolliert relaxiert undmittels der Elektronenmikroskopie betrachtet werden. Bei einer höheren Konzentrationvon Ethylenglykol-bis-(β-amino-ethyl-ether) N,N´-tetraessigsaurer Säure (EGTA), pH7,5 , kam es zu einer Bandbildung des Kerns, oft mit einem perlschnurartigenAussehen, mit einer Länge von ungefähr 100 Å (Nermut, 1979). Bei der Behandlungmit hohen Salzkonzentrationen (0,5-2,0 M NaCl) oder hohen pH-Werten (z.B. 10)kam es zu ähnlichen Erscheinungen; darüber hinaus sah man auch anstelle der Bänderstäbchenförmige Elemente mit einer Dicke von ca. 150 Å und einer Länge bis zu100 Å. Die Begründung für dieses Verhalten der DNA in beiden Fällen liegt wohl indem Entzug von Calcium, welches Brücken zwischen den DNA-Phosphatresten bildetund so die stäbchenförmigen Elemente in dem natürlichen Kern zusammenhält. Wirddieses Calcium nun entfernt, so trennen die Abstoßungskräfte die „Stäbchen“voneinander und machen sie sichtbar.Zusammenfassend läßt sich sagen, daß hohe Salzkonzentrationen zu einerproteinfreien DNA führen, da es wahrscheinlich zu einem Zusammenbruch derelektrostatischen Wechselwirkungen zwischen DNA und Protein VII kommt.
5.2.2. Biochemische Versuche mit „relaxierten“ Kernen
Mit [3H]-Arginin markierte Virusbestandteile (Vayda et al., 1983) bestätigten, daß mitPyridin vorbehandelte Kerne Protein V, VII und µ enthalten, wie es schon vonHokosawa und Sung 1976 beschrieben wurde. Aber dennoch wurde nur Protein VII inden Kernen, die ,mit hochkonzenrierten Salzlösungen behandelt wurden, gefunden.Durch UV-Bestrahlung wurde versucht, die Virus-DNA mit dem assoziierten Proteinzu verbinden. Man fand heraus, daß nur das Protein VII einen Komplex mit der DNAbildet, und beide Monomere und Dimere des Protein VII konnten auf einem SDS-Gelnachgewiesen werden (Sato und Hokosawa, 1981). Die Dimere wurden ebenfalls nachFixierung des Viruskerns mit Glutaraldehyd nachgewiesen (Mirza und Weber, 1982).Weitere Aussagen über die Stellung der DNA in dem DNA-Protein-Komplex konntenmittels der zirkulären Dichroismus -(CD-) Versuche gemacht werden (Tate, 1976;

20
Nermut, 1979; Boulanger und Loucheux-Lefevbre, 1982). Die Ergebnisse zeigten, daßdie DNA-Zusammensetzung durch die Kraft ihrer Wechselwirkungen mit denKernproteinen verändert werden kann, und daß der DNA-Protein-Komplex einegeordnete Struktur bildet (Cowman und Fasman, 1978).
5.3. Das Versuchsmodell des Adenovirus-Nukleokapsids
Das Hauptproblem der Viruszusammensetzung bestand in der Frage, wie dasverhältnismäßig große DNA-Molekül in dem zur Verfügung stehenden Raumkonfiguriert sein konnte. Denn jede dichte Verpackung von DNA-Molekülen erfordertdie Überwindung zweier spezifischer Eigenschaften der DNA: zum einen die Steifheitvon DNA, die sich in Lösung wie ein starrer Stab von 625 Å Länge verhält (Hays etal., 1969). Zum anderen die starke negative Ladung der Phosphatgruppen an derOberfläche der DNA. Daraus folgt, daß die DNA mit der höchstmöglichenVerdichtung gegliedert sein muß. Dies kann nur durch eine helikale Windung derDNA um einen Proteinkern mit einem minimalen Durchmesser von 80 Å verwirklichtwerden. Energieberechnungen zeigten, daß DNA, ohne zu knicken (nonkinking), biszu einem Krümmungsradius von 40-50 Å gebeugt werden kann (Finch et al., 1977;Sussman und Trifonov, 1978). Die zu einer Helix gebogene DNA benötigt Energie,um diese Form halten zu können. Es wurde vermutet, daß die Energie derelektrostatischen Bindung zwischen einem basischen Protein wie Protein VII und denDNA-Phosphatgruppen diesen Sachverhalt erfüllt. Wenn dem so wäre, wäre die DNAum einen Proteinkern gewunden, der entweder die Form einer Helix oder eineroligomeren Einheit hätte. Die noch vorhandenen oberflächlichen Phosphatrestekönnten entweder durch Kationen oder – für den Fall eines engen physikalischenKontaktes – durch Überbrückung von divalenten Kationen oder Polyaminenneutralisiert werden. Der Aufbau des Proteinkerns hängt von den Eigenschaften desDNA-gebundenen Proteins ab. Schon vor mehr als 40 Jahren wurde gezeigt (Crane,1950), daß eine lineare Struktur durch identische, asymmetrische Einheiten, diemiteinander auf eine identische Art und Weise interagieren, einen helikalen Aufbauhaben. Existieren zwei oder mehr Proteine, werden diese identischen Untereinheitenwie die „Perlen auf einer Kette“ oder als eine superhelikale Struktur, wenn es weiterverdichtet würde, angeordnet. Für das Modell des Adenovirus-Nukleokapsids lagenfolgende Daten vor: Die DNA hat eine Länge von 11-12 µm, was 34.000-36.000 bpentspricht. Das Protein VII liegt in ca. 1100 Kopien vor. Eventuell sind noch andereProteine (z.B. µ, X oder andere mit niedrigem Molekulargewicht) beteiligt. DerVerdau der DNA mit einer Nuklease aus Staphylococcus aureus legt nahe, daß ein 150bp langes sich wiederholendes DNA-Fragment existiert. Der Kern beinhaltet einelineare Struktur von fast 150 Å im Durchmesser und einer Länge bis zu 0,3 µm(Nermut, 1979). DNA-Filamente mit sog. „Perlen“ wurden nach Pyridin-Behandlungbeobachtet (Mirza und Weber, 1982; Vayda et al., 1983). Sechs stäbchenförmigeElemente mit einer Dicke von 150 Å und einer Länge von 400-500 Å füllen das

21
Innere des ikosaedrischen Raumes des Virions aus. Das Volumen dieses Raumesbeträgt 71.000 nm3 (basierend auf einer inneren Kantenlänge von 320 Å). DasVolumen einer sphärischen Perle beträgt bei einem Durchmesser von 90 Å 380 nm3.Es existieren prinzipiell zwei Möglichkeiten, wie der DNA-Protein-Komplex desAdenovirus aufgebaut sein könnte:
a) als kontinuierliche Helixoder
b) als diskontinuierliche Helix.
Der entscheidende Faktor ist hier der Proteinbestandteil. Gäbe es nur ein DNA-assoziiertes Protein, müßte der Komplex wie eine kontinuierliche Helix konfiguriertsein. Existierte noch ein anderes Protein, würde der kontinuierliche Aufbauunterbrochen werden, es würden sich wiederholende Untereinheiten bilden und dieDNA-Superhelix wäre diskontinuierlich. Im folgenden sollen beide Modellebesprochen werden, obwohl heutzutage mehr Beweise für das Nukleosomen-Modellvorliegen (siehe Kap. 4.3.2.).
5.3.1. Das Modell der kontinuierlichen Helix
Dieses Modell geht – wie oben erwähnt – von der Existenz nur eines DNA-assoziierten Proteins aus, nämlich des Protein VII. Wie bereits erläutert wurde(Nermut, 1980a), würde dieses Protein ein lineares Filament als „Kern“ der DNAbilden, die darum herum spiralig gewunden wäre (siehe Abb. 9a+b). Dieses Filamentwäre 80-90 Å dick und würde bei einer Länge von 1µm 1134 Kopien des Protein VIIund 11,5 µm DNA (= 36.000 bp) enthalten. Solche Filamente wurden beobachtet(Nermut, 1980a); sie könnten superhelikale Strukturen bilden – die Stäbchen (Abb.9c+d). Die eng gepackten DNA-Protein-Filamente würden durch Kalziumbrückenzwischen zwei gegenüberliegenden Phosphatgruppen zusammengehalten. EineWindung der DNA umschlösse 86 bp (= 280 Å) und diese Form wäre verantwortlichfür das Fehlen der typischen Nukleosomenmuster bei „eingekapselten“Nukleokapsiden (Tate und Philipson, 1979; Brown und Weber, 1980; Mirza undWeber, 1982). Ein Proteinmolekül würde ca. 20 bp schützen, zwei Proteine bis zu 50bp, wenn man relaxierte Kerne gefunden hat (Nermut, 1980b).
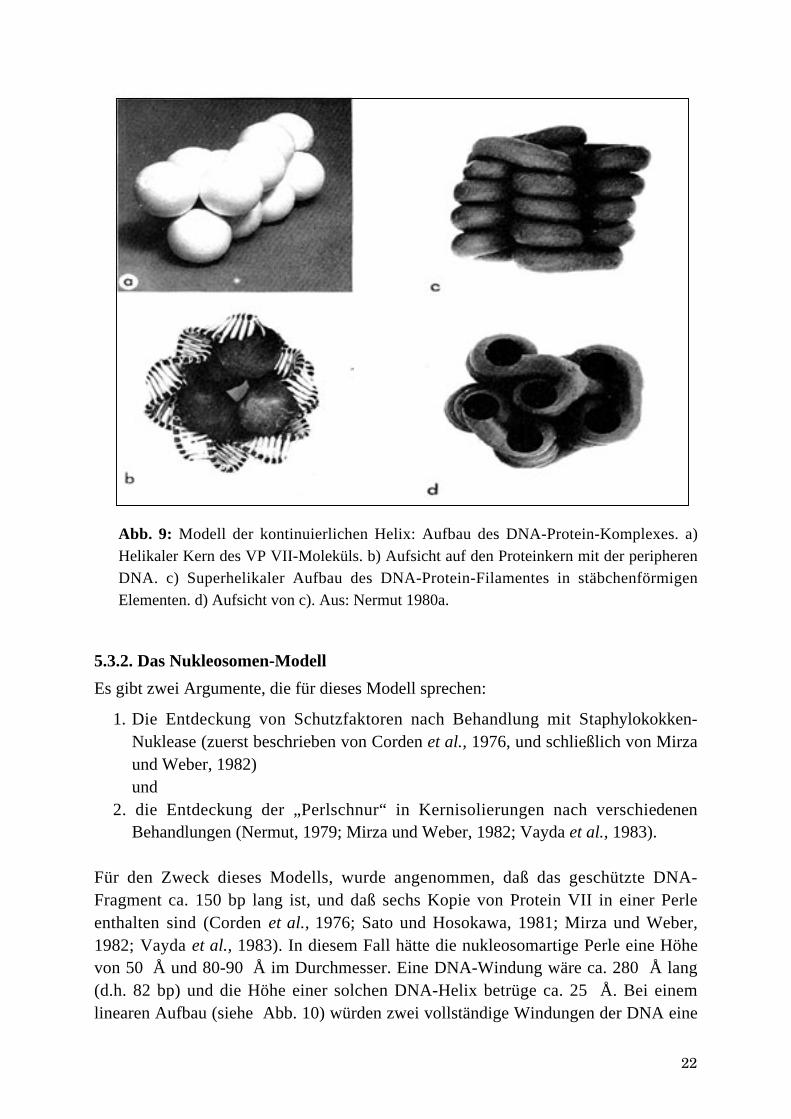
22
Abb. 9: Modell der kontinuierlichen Helix: Aufbau des DNA-Protein-Komplexes. a)Helikaler Kern des VP VII-Moleküls. b) Aufsicht auf den Proteinkern mit der peripherenDNA. c) Superhelikaler Aufbau des DNA-Protein-Filamentes in stäbchenförmigenElementen. d) Aufsicht von c). Aus: Nermut 1980a.
5.3.2. Das Nukleosomen-Modell
Es gibt zwei Argumente, die für dieses Modell sprechen:
1. Die Entdeckung von Schutzfaktoren nach Behandlung mit Staphylokokken-Nuklease (zuerst beschrieben von Corden et al., 1976, und schließlich von Mirzaund Weber, 1982)und
2. die Entdeckung der „Perlschnur“ in Kernisolierungen nach verschiedenenBehandlungen (Nermut, 1979; Mirza und Weber, 1982; Vayda et al., 1983).
Für den Zweck dieses Modells, wurde angenommen, daß das geschützte DNA-Fragment ca. 150 bp lang ist, und daß sechs Kopie von Protein VII in einer Perleenthalten sind (Corden et al., 1976; Sato und Hosokawa, 1981; Mirza und Weber,1982; Vayda et al., 1983). In diesem Fall hätte die nukleosomartige Perle eine Höhevon 50 Å und 80-90 Å im Durchmesser. Eine DNA-Windung wäre ca. 280 Å lang(d.h. 82 bp) und die Höhe einer solchen DNA-Helix betrüge ca. 25 Å. Bei einemlinearen Aufbau (siehe Abb. 10) würden zwei vollständige Windungen der DNA eine

23
Perle umgeben, ein DNA-Stück mit einer Länge von 100 Å (= 30 bp) könnte zweiPerlen miteinander verbinden. Bei einem superhelikalen, magnetspulartigen Aufbau(siehe Abb. 11) würden nur 1,8 Windungen der DNA (= 148 bp und 504 Å) mit demProteinkern in Verbindung stehen, die Verbindungs-DNA wäre 20 bp lang. Mehrerersolcher Knäuel von Superhelices würden eine lineare Struktur mit einer Dicke von 150Å bilden. Hier ergeben sich zwei Probleme: zum einen – wenn man eine Höhe dereinzelnen Windung von 25 Å annimmt und fünf Perlen eine superhelikale Windungbilden – könnte es nur zwei Windungen und zehn Perlen pro 500 Å-Länge einesStäbchens geben (bei 60 Perlen pro Kern). Zum anderen gäbe es viel Raum zwischender ersten und der fünften Perle. Diese Probleme könnten gelöst werden, wenn manannähme, daß die perlschnurartigen Filamente eine antiparallele Helix bilden, wobeidie Rille mit der aufsteigenden Superhelix gefüllt wird (siehe Abb.12). In diesem Fallewürde sich die Zahl der Perlen verdoppeln, und wenn man die Verbindungsstückemitzählte, käme man auf ca. 150 Perlen pro Virion. Dieses Modell ist für dieBerechnungen der DNA-Länge und der Zahl der Proteinkopien pro Virion maßgebendgewesen. Mehrere Autorengruppen berechneten die Anzahl der Perlen anhand derLänge der DNA und der Länge der Nuleosom-DNA-Fragmente, so z.B. Mirza undWeber (1982): sie fanden heraus, daß es 200 Perlen pro Virion geben müßte, um36.000 bp DNA unterbringen zu können. Aber schon 187 Perlen würden den zur Ver-fügung stehenden Raum des Kerninnern vollständig ausfüllen, vorausgesetzt, daß die90 Å-Perlen möglichst dicht beieinander liegen. Daraus folgte, daß die Anzahl derPerlen unter 200 liegen mußte, man könnte sie auf 150-180 schätzen, abhängig vonihrer Größe (siehe Tabelle 4). Diese wurde in den Berechnungen mit 90 Å imDurchmesser und 50 Å in der Höhe angenommen, was ungefähr in der Größenordnungvon Nukleosomen tierischen Chromatins liegt; so ergaben eselektronenmikroskopische Aufnahmen und solche mittels Röntgen-Kristallographie(Richmond et al., 1981). So ergab sich, daß ein Oktamer von vier Histon-Molekülenden Proteinkern bilden, und das H1-Histon, welches zwischen den Nukleosomen liegt,hat die Funktion eines „Organisators“ (Vayda et al., 1983). Dieses Modell wurdedurch die Ergebnisse der Elektronenmikroskopie unterstützt; trotzdem sind noch vieleFragen offen. Eines bleibt allerdings gewiß: der Adenovirus Nukleokapsid – der DNA-Protein(e)-Komplex hat eine helikale Struktur, was mittels der Röntgen-Diffraktigraphie bestätigt wurde (Devaux et al., 1983).

24
Tab. 4: Der DNA- und Proteingehalt pro Adenovirion, berechnet für 150 und 180nukleosomartige Perlen (Nermut 1984)
Perlen/Virion
Basenpaare/Perle +Abstands-DNA
Gesamtlänge derDNAbp µm
VII-Ko-pien/Virion
Länge der perlförmigenFilamente4
CTC= 90 Å CTC= 100 Å
150 170
180
25.500
27.000
8,7
9,2
900
900
1,35 µm 1,5 µm
80 170
180
30.600
32.400
10,5
12
1080
1080
1,6 µm 1,8 µm
Abb. 10: Diagrammförmige Darstellung des DNA-Verlaufes in dem „Nukleosomen-Modell“.
In diesem linearen Arrangement verläuft die DNA in zwei Windungen um die Perlen herum;die Verbindungs-DNA wäre 26 bis 30 bp lang. Aus Nermut 1984.
4 CTC: Abstand von Zentrum zu Zentrum (Center-to-center) aneinander angrenzender Perlen.

25
Abb. 11a + b: Superhelikaler Aufbau der Nukleosomen; es wird eine helikale Windunggezeigt, hier interagieren 1.8 Windungen der DNA mit sechs Kopien des VP VII, derZwischenraum wird von ca. 20 bp geformt.
Abb. 12: Modell eines stäbchenförmigen Elementes, gebildet aus zwei antiparallelen Helicesder Nukleosome. Die weißen Kugeln repräsentierren eine aufsteigende Helix, die schwarzeneine absteigende. Der Pfeil bezeichnet die Stelle, an der sie sich treffen. Aus Nermut 1984.
6. Das Körpermodell des Adenovirus
In diesem Modell sind nur die bekannten Strukturen beinhaltet, wie das Kapsid, dieKernhülle und der Nukleoprotein-Komplex. So faßt dieses Modell die letztenJahrzehnte der Adenovirusforschung zusammen: die ikosaedrische Form und dieExistenz von zwei verschiedenen Kapsomeren wurde schon sehr früh erkannt (Horne,1962; Valentine und Pereira, 1965), die genaueren Einzelheiten des Hexons und desViruskerns erst durch die computervermittelte Elektronenmikroskopie, die Röntgen-Kristallographie, die Neutronendiffraktion, Gefriertechniken und die Fortschritte in derBiochemie. Dieses Modell gewährt einen Einblick entlang der doppeltenSymmetrieachse (siehe Abb. 13a) mit einem Ausschnitt des Inneren des Adenovirion(siehe Abb. 13b). Man erkennt. daß die 9er-Gruppe von Hexonen auf der linken undrechten Seite einer dreieckigen Facette an einer Kante des Ikosaederszusammenkommen, so daß zwei Hexone einander gegenüber zu liegen kommen übereinen Eck-zu-Eck-Kontakt, und nicht wie die 9er-Gruppen über einen Ecke-zu-
Abb 12Abb 11

26
Kanten-Kontakt. Betrachtet man das Innere des Modells, sieht man den innerenNukleoprotein-Komplex in der Form eines Stäbchens, bedingt durch das alsSuperhelix konfigurierte DNA-Protein-Filament.
Abb. 13 a+b: Maßstabsgerechtes Modell des Adenovirion. a) Sicht entlang der doppeltenSymmetrieachse; 9er Gruppen treffen an der Ecke zusammen, im Kapsid befinden sich diese9er Gruppen in linksdrehender Orientierung. b) Offene Sicht auf die Kernhülle unterhalb desKapsids und drei der superhelikal angeordneten Stäbchen des eigentlichen Nukleokapsids. (C)Kapsid, (CS) Kernhülle, (NP) DNA-Protein-Komplex. Aus Nermut 1984.
7. Vektoren
Es werden bestimmte Voraussetzungen von einem DNA-Molekül verlangt, daß alsKlonierungsvektor dienen soll: die wichtigste davon ist seine Replikationsfähigkeit,damit viele Kopien des DNA-Moleküls an die Tochterzellen des Wirtsorganismusweitergegeben werden können. Desweiteren ist die Größe von besonderer Wichtigkeit:relativ kleine Moleküle sind bezüglich ihrer Präparation einfacher zu handhaben, imIdealfall liegt ihre Größe unter zehn Kilobasen (kb). Außerdem sollte ein Vektor überwenigstens eine nur einmal vorkommende Restriktionsschnittstelle verfügen, um fürdie Herstellung von Rekombinanten fremde DNA einfügen zu können. Einselektierbarer Marker wie z.B. eine Antibiotikaresistenz erleichtert die Identifizierungder rekombinanten Klone. In Bakterienzellen wurden zwei DNA-Moleküle entdeckt,die diesen Anforderungen genügen: Plasmide und Bakteriophagen.
7.1. Plasmide
Bei Plasmiden handelt es sich um ringförmige extrachromosomale DNA-Moleküle,die zumeist für Antibiotikaresistenzen kodieren. Sie enthalten außerdem einen

27
Replikationsstartpunkt (origin of replication, ori), der es ihnen ermöglicht, sichunabhängig von dem Bakterienchromosom vermehren zu können. Sie kommen inGrößen zwischen 1 und 250 kb vor, ihre Kopienzahl, d.h. die Zahl an Plasmiden, diesich normalerweise in einer Bakterienzelle findet, schwankt zwischen eins und 50 undmehr: für die Klonierung ist also ein Plasmid ideal, das bis zu 10 kb groß ist und eineKopienzahl von mindestens 50 hat. Um dies zu erreichen bzw. sie den verschiedenenErfordernissen anzupassen, mußten die natürlich vorkommenden Plasmide verändertwerden.
7.2. Bakteriophagen
Bei den Bakteriophagen ( kurz: Phagen) handelt es sich um Viren, die sich nur mitHilfe von Bakterien vermehren können, d.h. spezifisch Bakterien infizieren. Siebestehen zumeist aus einem DNA-Molekül und einer Schutzhülle aus Protein, demKapsid. Man unterscheidet Phagen mit Kopf und Schwanz, ohne Schwanz undfilamentöse Phagen. Für die Molekularbiologie sind sie deshalb von Interesse, da diemeisten lebenden Organismen von Viren infiziert werden und so die Möglichkeitgegeben ist, Viren als Klonierungsvektoren für höhere Organismen einzusetzen.
7.3. pJM17-Aufbau
Bei pJM17 handelt es sich um ein von McGrory et al. konstruiertes Plasmid, welchesdie gesamte DNA von Adenovirus Typ 5 enthält, mit einem Insert in der E1 Region,das die Verpackungszwänge(?) des Adenoviruskapsids übersteigt.

28
Abb. 14: Schnittstellenkarte des pJM17
7.4. pQE30-Aufbau
Die von der Firma Qiagen bereitgestellten pQE-Vektoren gehören zu den pDS-Plasmiden (Bujard et al., 1987). Sie wurden aus den Plasmiden pDS56/RBSII undpDS871/RBSII-DHFRS entwickelt (Stüber et al., 1990), haben eine Größe von ca.3500 bp und enthalten folgende Elemente:
• A: Ein optimiertes, regulierbares Promotor-/Operator-Element, bestehend aus demT5-Promotor des E.coli Phagen und zwei lac-Operator-Sequenzen, welches durchden lac I-Repressor unterdrückt und durch Zugabe von IPTG induziert wird (Bujardet al., 1987).
• B: Eine synthetische Ribosomen-Bindungsstelle (RBSII), für eine optimalemRNA-Erkennung und –Bindung.
• 6 x His: Eine optimierte Kodierungssequenz für das 6xHis-Affinitäts-Ende.• Eine mehrfache Klonierungsstelle (Polylinker).• Ein Translations-Stop-Codon in allen Leserahmen.• C: Den Transkriptions-Terminator 't0' des λ-Phagen (Schwarz et al., 1987).• D: Eine Promotor-freies Gen für die Chloramphenicol-Acetyltransferase mit
seinem eigenen Translationssignal (Marcoli et al., 1980).

29
• E: Den Transkriptions-Terminator T1 des E.coli-rrnB-Operons (Brosius et al.,1981).
• Die Replikations-Region und das Gen für die β-Lactamase des Plasmids pBR322(Sutcliffe, 1979).
Der in diesen Experimenten benutzte Vektor pQE30 trägt sein 6xHistidin-Affinitätsschwanz am 5'-Ende der multiplen Klonierungsstelle (Abb. 16a und b–Vektor-Schema). Der zu verwendende Vektor muß nur mit den dazu notwendigenRestriktionsendonukleasen verdaut werden, die lineare Form isoliert und dieKodierungsregion mit dem pQE-Vektor im Leserahmen ligiert werden.
Abb. 15: Aufbau und Schnittstellen des Vektors pQE30. Aus The QIAexpressionist 1992.
8. Konstruktaufbau pVII-pQE30
Für diese Verbindung – im Handbuch der Firma Qiagen auch Typ IV-Konstruktgenannt – muß der benötigte Vektor nur mit den entsprechendenRestriktionsendonukleasen verdaut, die lineare Form isoliert und die zu kodierendeRegion in Leserichtung ligiert werden. Zu diesem Zweck befindet sich das 6 xHistidin-Ende am 5'-Ende der multiplen Klonierungsstelle.
8.1. Aufbau
In der folgenden Tabelle wird das Vektorkonstrukt pQE30 mit der Insertionsstelle fürdie Sequenz des Protein VII dargestellt, wie es von der Firma Qiagen geliefert wird.

30
Tab. 5: Sequenz von pQE30 mit Markierung der Insertionsstelle für pVII (violetteund rosa Markierung)5
1 CTCGAGAAAT CATAAAAAAT TTATTTGCTT TGTGAGCGGA TAACAATTAT
51 AATAGATTCA ATTGTGAGCG GATAACAATT TCACACAGAA TTCATTAAAG
101 AGGAGAAATT AACTATGAGA GGATCGCATC ACCATCACCA TCACGGATCC
151 GCATGCGAGC TCGGTACCCC GGGTCGACCT GCAGCCAAGC TTAATTAGCT
201 GAGCTTGGAC TCCTGTTGAT AGATCCAGTA ATGACCTCAG AACTCCATCT
251 GGATTTGTTC AGAACGCTCG GTTGCCGCCG GGCGTTTTTT ATTGGTGAGA
usw.
8.2. Zielvorstellungen
Die pQE-Vektoren der Firma Qiagen sind für eine Expression in verschiedenen E.coli-Stämmen geeignet. Durch das regulierbare Promotor-/Operator-Element kann einegezielte und kontrollierte Expression erfolgen, die durch Zugabe von IPTG gesteuertwird, welches des Repressor in- und den Promotor aktiviert. Mit Hilfe des 6 xHistidin-Endes wird eine Reinigung des Expressionsproduktes vereinfacht, da dieseran speziellem Nickel-NTA-Harz-beschichteten Säulen bindet, und das Protein so scho-nend aus den Zellen entfernt werden kann.
9. Expression in SG13009 und M15
Die ebenfalls von der Firma Qiagen bereitgestellten E.coli-Stämme SG13009(Gottesmann et al., 1981) und M15 (Villarejo und Zabin 1974) gewähren eine hoheExpressionsrate und sind einfach in ihrer Handhabung. Sie enthalten das PlasmidpREP4, welches sowohl das Gen für die Neomycin-Phosphotransferase kodiert (NEO,Beck et al., 1982), die für die Kanamycin-Resistenz dieser Bakterienstämmeverantwortlich ist, als auch das lac I-Gen, welches für den lac-Repressor kodiert(Farabaugh 1978). Das führt dazu, daß diejenigen Zellen, die den transformiertenVektor enthalten, leicht mit Hilfe von Kanamycin-enthaltendem Nährmedium selek-tiert werden können. Die Expression wird über die Inaktivierung des Repressorsgesteuert. Darüber hinaus enthält dieses Plasmid eine Region des PlasmidspACYC184, welches alle Informationen für die Replikation und die Retention vonPlasmiden beinhaltet (Chang und Cohen 1978).
5 rot: M13-Primer für die Sequenzierung; grün: Startcodon; blau: 6 x Histidin-Ende; violett: Schnittstelle für
BamHI; rosa: Schnittstelle für HindIII; braun: Biotinilierter Primer für die Sequenzierung.

31
10. Fragestellung der vorliegenden Arbeit
Molekularbiologische Methoden des Gentransfers ermöglichten es, verschiedeneKonzepte zur Analyse und Therapie genetisch bedingter Erkrankungen zu entwickeln.Eines davon besteht darin, durch Übertragung von Genen neue Therapieansätzeherauszuarbeiten. Es ist bekannt, daß virale Vektoren als effektive Transporter fürGene in den menschlichen Organismus eingebracht werden können, so auch viraleKernproteine, die aufgrund ihrer Ladungseigenschaften gut an DNA binden. Bislangwurden diese Proteine aus in humanen HeLa-Zellen angezüchteten Virusstämmen(Doerfler, 1969) mit aufwendigen Methoden isoliert (Hosokawa und Sung, 1976; Sunget al., 1977).In der vorliegenden Arbeit sollte daher versucht werden, die adenovirale dsDNA, diefür das Kernprotein VII kodiert, mittels der Polymerase-Kettenreaktion (PCR) zuamplifizieren und nach dem Einbau in einen geeigneten Vektor in superkompetenteE.coli-Stämmen zu transferieren. Diese Zellen sollen das Protein nach vorgenommenerInduktion exprimieren. Das exprimierte Protein sollte über Nickel-beschichtete Säulengereinigt werden. Da sich im Verlauf der Arbeit herausstellte, daß sich das Proteineben wegen seiner eigenen stark positiven Ladung nur schwer mittels der PCRamplifizieren läßt, und daß es außerdem zu wiederholten Mutationen im Bereich desLeserahmens kam, konnte das Protein in den dafür vorgesehenen Zellen nichtexprimiert werden.

32
B. MATERIAL UND METHODEN
1. Materialien
1.1. Chemikalien
Alle Chemikalien wurden in der höchsten erhältlichen Qualität verwendet. Nichtaufgeführte Chemikalien wurden in p.A. Qualität von der Firma Merck (Darmstadt)bezogen.Acrylamid BioRad, MünchenAgarose (Seakem GTG) BioRad, MünchenAmmoniumpersulfatBactoagar Difco, DetroitBactotrypton Difco, DetroitBorsäure Merck, DarmstadtBromphenolblau Sigma, DeisenhofenCoomassie (Willis Reagenz)Dimethylsulfoxid (DMSO) Sigma, DeisenhofenDynabeads M-280 Streptavidin Dynal, OsloEisessig Merck, DarmstadtEssigsäure Merck,DarmstadtEthanol Merck, DarmstadtEthidiumbromid Sigma, DeisenhofenEthylendiaminintetraesigsäure (EDTA) BioRad, MünchenGlukose Merck, DarmstadtGlyzerin, wasserfrei BioRad, MünchenIsopropanol Merck, DarmstadtIsopropyl-β-D-thiogalactopyranosid (IPTG) Sigma, DeisenhofenKaliumcarbonat Sigma, DeisenhofenKaliumchlorid Sigma, DeisenhofenKaliumhydroxid Sigma, DeisenhofenKalziumchlorid Sigma, DeisenhofenKOAcMagnesiumchlorid Merck, DarmstadtMagnesiumsulfat Merck, DarmstadtManganchlorid Merck, Darmstadtβ- Mercaptoethanol BioRad, MünchenMethanol Merck, DarmstadtMOPSNatriumacetat Merck, DarmstadtNatriumchlorid Merck, DarmstadtNatriumdodecyl-Sulfat (SDS) BioRad, München

33
Natriumhydroxid (wasserfrei) Merck, DarmstadtRubidiumchlorid Merck, DarmstadtSalzsäure Merck, DarmstadtTEMEDTris[Hydroxylmethyl]aminomethan(TRIS) BioRad, MünchenTriton X-100 BioRad, MünchenYeast-Extrakt Difco, Detroit
1.2. Molekulargewichtsstandards und Proteine
AmpliTaqDNA-Polymerase Perkin Elmer,Weiterstadt
Bovines Serum Albumin Stratagene, GermanyDeoxyribonukleotidtriphosphate(dATP, dCPT, dGPT, dTTP) Boehringer, MannheimDNA-Marker III Boehringer, MannheimDNA.Marker VI Boehringer, MannheimOligonukleotide Genset, Paris
TIB MOLBIOL,BerlinPerfect MatchDNA polymerase enhancer Stratagene, GermanyRestriktionsenzym Sal I Boehringer, MannheimRestriktionsenzym Bam HI Boehringer, MannheimRestriktonsenzym Hind III Boehringer, MannheimTaq-DNA-Polymerase Boehringer, MannheimT4-Ligase Boehringer, MannheimVektor pQE30 QIAGEN, DüsseldorfVent-DNA-Polymerase Bio Labs, USA
1.3. Puffer, Medien, Agarplatten und Antibiotika
1.3.1. Medien und Agarplatten
Broth-Agar: 1,5 % Bactoagar, 1 % Bactotrypton, 0,5 % Yeast-Extrakt, 0,5 % NaCl, 1 % 1M Tris-HCl (pH 7,5),1 % 1M MgSO4.
L-Broth-Medium: 1 % Bactotrypton, 0,5 % Yeast-Extrakt, 0,5 % NaCl,1 % 1M Tris-HCl (pH 7,5), 1 % 1M MgSO4.
Lennox-L-Broth-Medium: 1 % Bactotrypton, 0,5 % Yeast-extrakt, 0,5 % NaCl,0,1 % Glukose, pH auf 7,5 eingestellt mit1 N NaOH.
SOC-Medium: 2% Bactotrypton, 0,5 % Yeast-Extrakt, 0,05 %NaCl, 0,4 % Glukose, 10 mM MgCl2, 10 mM SO4.

34
1.3.2. Puffer und Lösungen für die Kompetentmachung von SG13009 und M15
TFB 1: 100 mM RbCl, 50 mM MnCl2, 30 mM KOAc,10 mM CaCl, 15 % Glycerin, pH 5,8 , steril filtriert.
TFB 2: 10 mM MOPS, 10 mM RbCl, 75 mM CaCl2,15 % Glyzerin, pH 8,0 , autoklaviert.
1.3.3. Antibiotika
Ampicillin Sigma, DeisenhofenKanamycin GIBCO BRL, EggensteinMethicillin Sigma, Deisenhofen
1.4. Sonstige Pufferlösungen
1 x Bindungs- und Wasch(B&W) –Puffer : 5 mM Tris-HCl (pH 7,5), 5 mM EDTA (pH 8,0),
1 M NaCl.1 x TAE-Puffer : 40 mM Tris-HCl (pH 8,0), 2 mM Essigsäure,
1 mM EDTA (pH 8,0).1 x TBE-Puffer : 90 mM Tris-HCl (pH 8,0), 89 mM Borsäure,
20 mM EDTA (pH 8,0).1 x TNE-Puffer : 100 mM NaCl, 50 mM Tris-HCl (pH 7,5),
1 mM EDTA.1 x TE : 10 mM Tris-HCl (pH 7,5), 1 mM EDTA ( pH 8,0).6.6.2.-Puffer : 6 mM Tris-HCl (pH 7,2), 6 mM NaCl,
0,2 mM EDTA (pH 8,0).P1-Puffer : 50 mM Tris-HCl (pH 8,0), 10 mM EDTA, 100 µg/ml RNAse A.P2-Puffer : 200 mM NaOH, 1 % SDS.P3-Puffer : 3 M Kaliumazetat (pH 5,5).QBT-Puffer : 750 mM NaCl, 50 mM MOPS (pH 7,0),
15 % Ethanol, 0,15 % Triton-X-100.QC-Puffer : 1 M NaCl, 50 mM MOPS (pH 7,0), 15 % Ethanol.QF-Puffer : 1,25 M NaCl, 50 mM Tris-HCl (pH 8,5),
15 % Ethanol.QX1, QX2 und QX3 : Die Zusammensetzung wird von der Firma
QIAGEN nicht bekanntgegeben.B-Puffer(für Restriktionsenzyme): 10 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2,
1 mM 2-Merkaptoethanol, pH 8,0.H-Puffer(für Restriktionsenzyme):

35
B-Puffer (Expression): 8 M Harnstoff, 0,1 M Na-Phosphat, 0,01 M Tris-HCl (pH 8,0).
C-Puffer (Expression): 8 M Harnstoff, 0,1 M Na-Phosphat, 0,01 M Tris-HCl (pH 6,3)
PAGE-Proben-Puffer: 15 % β -Merkaptoethanol, 15 % SDS, 1,5 %Bromphenolblau,
50 % GlyzerinT4-Ligase-Puffer : 100 mM Tris-HCl (pH 7,6), 50 mM MgCl2,
50 mM DTE, 3 mM ATP50xMaster MixPuffer 20 mM Tris-HCl (pH 8,0), 250 nM EDTA
Opti-PrimePuffer 1-12:Puffer #1 100 mM Tris-HCl (pH 8,3), 15 mM Mg2Cl,
250 mM KClPuffer #2 100 mM Tris-HCl (pH 8,3), 15 mM Mg2Cl,
750 mM KClPuffer #3 100 mM Tris-HCl (pH 8,3), 35 mM Mg2Cl,
250 mM KClPuffer #4 100 mM Tris-HCl (pH 8,3), 35 mM Mg2Cl,
750 mM KCLPuffer #5 100 mM Tris-HCl (pH 8,8), 15 mM Mg2Cl,
250 mM KClPuffer #6 100 mM Tris-HCl (pH 8,8), 15 mM Mg2Cl,
750 mM KClPuffer #7 100 mM Tris-HCl (pH 8,8), 35 mM Mg2Cl,
250 mM KClPuffer #8 100 mM Tris-HCl (pH 8,8), 35 mM Mg2Cl,
750 mM KClPuffer #9 100 mM Tris-HCl (pH 9,2), 15 mM Mg2Cl,
250 mM KClPuffer #10 100 mM Tris-HCl (pH 9,2), 15 mM Mg2Cl,
750 mM KClPuffer #11 100 mM Tris-HCl (pH 9,2), 35 mM Mg2Cl,
250 mM KClPuffer #12 100 mM Tris-HCl (pH 9,2), 35 mM Mg2Cl,
750 mM KCl
1.5. Wasser
Das Wasser wurde mit einem Destillator Bi-Dest 2304 der Firma GFL Burgwedeldoppelt destilliert und autoklaviert oder sterilfiltriert verwendet.

36
1.6. Bakterienstämme
Epicurian ColiXL1-BLUE MRF’ KAN competent cells Stratagene,Heidelberg
SG13009 QIAGEN,Düsseldorf
M15 QIAGEN,Düsseldorf
1.7. Reagenzienkits
ABI PRISM Dye Primer Cycle Sequenzing Perkin Elmer,Weiterstadt
Ready Reaction KitNi-NTA Spin Kit QIAGEN, DüsseldorfOPTI-PRIME PCR Optimization Kit Stratagene, HeidelbergPCR-Script SK (+) Cloning Kit Stratagene, HeidelbergPlasmid Preparation Kit QIAGEN, DüsseldorfQIAexpress Type IV Kit QIAGEN, DüsseldorfQIAquick Gel Extraction Kit QIAGEN, Düsseldorf
1.8. Weitere Materialien
Sterile Labormaterialien wurden von den Firmen Nunc (Wiesbaden-Biebrecht) undFalcon (Cockeysville, MD) bezogen.
Nalgene-Einweg-Filtrationssysteme0,2 µm und 0,45 µm Nalge, RochesterPCR-Reaktionsgefäße Sarstedt, NümbrechtUltrazentrifugenröhrchen Beckman, München
1.9. Geräte
Autoklav „Bioclav“ Schütt, GöttingenGelkammern für Agarosegele BioRad, MünchenGelkammern für Acrylamidgele BioRad, MünchenMagnetrührerPCR-Thermocycler Perkin-E., MünchenpH-Meter 537 A WTW, WeilheimSpektrophotometer DU-62 Beckman, MünchenUltrazentrifuge L5-50 B Beckman, MünchenWaageWasserbadZentrifugen Minifuge RF und Biofuge A02 Heraeus, Hannover

37
2. Allgemeine Arbeitsmethoden
2.1. Sterilisation
Hitzestabile Geräte und Lösungen wurden in einem Autoklaven in feuchter Hitze bei121 °C und 2 bar für 30 min sterilisiert. Lösungen mit hitzeempfindlichen Substanzenwurden mit einem 0,2 µm Filter sterilisiert.
2.2. Absorptionsmessung
Absorptionsmessungen wurden mit einem Spektralphotometer durchgeführt. Imultravioletten Bereich wurden Glasküvetten (Quarzglas Suprasil) mit einerSchichtdicke von 1 mm und 0,5 ml Volumen verwendet.
3. Präparation des Plasmids pJM17
3.1. Prinzip
Die Methode zur Präparation von DNA aus einer Bakterienkultur kann in vier Schritteunterteilt werden:
1. Die Bakterienkultur wird herangezüchtet und dann geerntet.2. Die Zellen werden aufgebrochen, damit ihr Inhalt frei wird.3. Der Zellextrakt wird gereinigt.4. Die entstandene DNA-Lösung wird angereichert.
3.2. Durchführung
Das Plasmid pJM17, welches die gesamte DNA des Adenovirus Typ 2 enthält(McGrory et al.,1987), trägt sowohl eine Ampicillin- als auch eine Tetrazyklin-Resistenz. Für das Wachsen der Bakterienkultur ist es allerdings günstiger, demNährmedium – in diesem Fall Lennox-L-Broth-Medium - nur Ampicillin in einerKonzentration von 20 µg/ml zuzufügen, da es sonst öfter zu Neuanordnungeninnerhalb des Plasmids kommt (Rudy et al., 1994). Es wurde mit der Inkubation einer2-5 ml-Kultur aus einer Kolonie oder 0,1 ml einer eingefrorenen Kultur bei 37 °C imBrutschrank begonnen. Ca. 8 Stunden später wurde diese Kultur mit einem LiterMedium verdünnt und über Nacht unter starkem Schütteln im Brutschrank bei 37 °Cinkubiert. Die Plasmid-Präpararion erfolgte nach dem QIAGEN Plasmid PurificationHandbook, und zwar nach dem Maxi-Protokoll. Die 100 ml Bakterienkulturenwurden für 15 min bei 6000 Upm und 4 °C zentrifugiert. Der Überstand wurdeentfernt und das Sediment in 10 ml des RNAse A enthaltenden P1-Pufferresuspendiert. Dieses visköse Gemisch wurde unter Zugabe von 10 ml P2-Puffer für 5min bei Raumtemperatur inkubiert, wobei das Reaktionsröhrchen 4-6 Mal gewendetwurde. Durch eine weitere Zugabe von 10 ml eiskalten P3-Puffer wurde dieFlüssigkeit noch visköser und trüber, sie mußte unmittelbar nach Zugabe gemischt

38
für 15-20 min auf Eis inkubiert werden . Daraufhin wurde das Gemisch für 30 min bei15000Upm und 4 °C zentrifugiert. In der Zwischenzeit wurden die QIAEX-Säulen(QIAGEN-tip 500) durch den Durchfluß von jeweils 10 ml QBT-Puffer equilibriert.Der Überstand der zentrifugierten Proben wurde abgenommen und die Säulenaufgetragen. Der Durchfluß durch die Säulen wurde nicht manuell beschleunigt, son-dern erfolgte allein durch die Schwerkraft. Die nun an das Säulenmaterial (speziellbehandeltes Harz, das eine genaue Selektion von DNA gegenüber RNA und anderenZellbestandteilen erlaubt) gebundene DNA wurde zweimal mit jeweils 30 ml QC-Puffer gewaschen. Die Elution erfolgte durch Auswaschen der DNA mit.15 ml QF-Puffer in frische, sterile 30 ml Glasröhrchen (Fa. GIBCO). Anschließendwurde die DNA in 0,7 Volumeneinheiten (10,5 ml) raumtemperiertem Isopropanolpräzipitiert und für 30 min bei 9500 Upm und 4 °C zentrifugiert. Das die DNAenthaltende Sediment wurde nach Abzug des Isopropanols mit 15 ml eiskaltem 70%igem Ethanol gewaschen und hierzu erneut für 10 min zentrifugiert. Der Überstandwurde entfernt und vollständig luftgetrocknet. Anschließend wurde die gereinigteDNA in 10-30 µl TE-Puffer aufgenommen. Die Quantifizierung der DNA-Mengeerfolgte mittels Spektralphotometrie wie unter 3.3.2. beschrieben.
4. Quantifizierung der DNA-Menge durch Spektralphotometrie
Die Bestimmung der DNA-Konzentration erfolgte durch Absorptionsmessung beiverschiedenen Wellenlängen im ultravioletten Bereich (Sambrook, 1989).
4.1. Prinzip
Licht einer definierten Wellenlänge wird beim Durchstrahlen einer probenhaltigenKüvette im Vergleich zu einer lösungsmittelhaltigen Küvette in seiner Lichtintensitätverändert. Diese Schwächung oder Extinktion einer Lösung ist proportional derKonzentration der gelösten lichtabsorbierenden Substanz, ihrem Absorptions-koeffizienten und der Schichtdicke der Lösung. Diesen Zusammenhang beschreibt dasLambert-Beer-Gesetz, mit dessen Hilfe in diesem Fall die Konzentration der Lösungerrechnet werden kann, gemäß OD = -log10 (Intensität des durchgelassenenLichtes)/(Intensität des eingestrahlten Lichtes).
4.2. Durchführung der OD-Messung
Zunächst wurden 5 µl der isolierten und in kleiner Menge TE-Puffer gelösten DNAmit 495 µl TE-Puffer versetzt (1:100 Verdünnung). Zum Abgleich des Photometerswurde der Leerwert von 500 µl TE-Puffer bestimmt und das Gerät mit TE-Puffer beiden Wellenlängen 260, 270, 280 und 320 nm kalibriert. Die Proben wurden gemischtund die Extinktion in Quarzküvetten von 1 cm Schichtdicke bei den oben genanntenWellenlängen gemessen. Zwischen der Messung der verschiedenen DNA-Probenwurde die verwendete Küvette mit TE-Puffer gespült. Die Werte gaben in

39
aufsteigender Reihenfolge den DNA-, Phenol- und Proteingehalt an. Mit denExtinktionswerten bei 320 nm läßt sich die Löslichkeit der DNA abschätzen; derQuotient aus den Meßwerten bei 260 nm und bei 280 nm läßt Rückschlüsse auf dieReinheit der DNA zu, bzw. wie stark die Verunreinigung mit Proteinresten ist. EinQuotient von 1,6 – 1,8 läßt eine ausreichende Reinheit der DNA vermuten. Aus derExtinktion bei 260 nm läßt sich die Gesamtkonzentration der DNA in der Lösungberechnen, dabei entspricht eine A260 von 1,0 einer Konzentration an dsDNA von 50µg/ml.Für die Plasmidpräparation von pJM17 ergab sich eine Konzentration von 0,13 µg/µl,die Proben von pQE30 eine Konzentration von 0,80 µg/µl.
5. Schneiden des Plasmids pJM17 und des Vektors pQE30 mit SalI
5.1. Prinzip
Das Schneiden des Plasmids pJM17 mit der Restriktionsendonuklease SalI diente derFraktionierung, damit die DNA des Adenovirus Typ 2 bzw. letztlich die DNA desProtein VII kloniert werden konnte. Zur Konstruktion des Expressionsplasmids wurdeder Vektor pQE30 mittels der gleichen Restriktionsendonuklease linearisiert, um dannfür die weitere Klonierung vorbereitet werden zu können. DieseRestriktionsendonuklease SalI stammt aus dem Bakterium Streptomyces albus G undschneidet das Plasmid pJM17 an vier Stellen (Abb.), die DNA von pQE30 nur aneiner; die entstehenden Enden bezeichnet man als klebrig, da die Schnitte der DNA-Stränge versetzt sind bzw. überstehend (siehe auch 5. Kapitel, Vorbereitungen zurSubklonierung – Verdau mit BamHI und HindIII).
5.2. Durchführung
Für diesen Vorgang, der auch als Verdau bezeichnet wird, wurden Plasmid-Präparationen von pJM17 und pQE30, welches auf die gleiche Weise hergestelltwurde (siehe Kapitel 3), eingesetzt. Mit Hilfe der angegebenen Units/ml, d.h. derEnzymaktivität, die 1 µg λDNA in 1 h bei 37 °C komplett spaltet, der Anzahl derSchnittstellen des jeweiligen Enzyms in der λDNA und dem Verhältnis der Größe derλDNA zu der zu schneidenden DNA, konnte die einzusetzende Enzymmengeerrechnet werden :
SalI : 10 U/µlSchnittstellen in λDNA : 7Größe der λDNA : 50 kbGröße von pJM17 : 40 kbGröße von pQE30 : 3,5 kb

40
λDNA (kb)/ Vektor (kb) x 1/Schnittstellen in λDNA x 46 =U/µl/h/10 = Enzymmenge (µl).
Desweiteren benötigt SalI einen Inkubations-Puffer (H-Puffer, Fa. Boehringer), der ineiner Endkonzentration von 10 % (5 µl) eingesetzt wurde. Die Einsatzmengen derDNA lagen zwischen 1 µg (pQE30) und 5 µg (pJM17), das Gesamtvolumen je Probebetrug 50 µl. Die Proben wurden gemischt und bei 37 °C für 1 h im Wasserbadinkubiert. Zur Dokumentation wurden die Proben auf ein1 %iges Agarosegel geladen und elektrophoretisch aufgetrennt (siehe Kapitel 6). DasGel wurde mittels Rotfilter unter UV-Licht auf Polaroidfilm photographiert.
6. Amplifikation der dsDNA von Protein VII durch die Polymerase-Kettenreaktion (PCR)
Die DNA-Amplifikation durch die Polymerase-Kettenreaktion (polymerase chainreaction, PCR) wurde nach einer von White beschriebenen Methode durchgeführt(White et al., 1989).
6.1. Prinzip
Mit der Polymerase-Kettenreaktion kann ein DNA-Segment, welches zwischen zweiRegionen bekannter Sequenzen liegt, um ein Vielfaches amplifiziert werden. Dazubenötigt man zwei Oligonukleotidprimer, die zu den amplifizierenden DNA-Abschnitten homolog sind, wobei beide unterschiedliche Sequenzen besitzen müssen.Die DNA wird nun zunächst durch Spaltung ihrer Wasserstoff-Brückenbindungendurch Hitze (94 °C) reversibel in Einzelstrang-DNA überführt (Denaturierung). Esschließt ein Schritt bei geringerer Temperatur an ( 55 °C) der es den Oligonukleotidenermöglicht, an die Zielsequenz auf der DNA zu binden (Hybridisierung bzw.Annealing). Anschließend wird unter Anwesenheit einer DNA-Polymerase I ausThermus aquaticus (Taq-Polymerase), die sehr hitzestabil ist, und den vierDesoxyribonukleotid-Triphosphaten (dATP, dCTP, dGTP und dTTP),die sich an diejeweils komplementären Basen des Matrizenstranges anlagern, die neue DNA beieiner Temperatur von 72 °C synthetisiert (Extension). Diese Dreier-Sequenzentspricht einem Verdopplungszyklus, der aus den oben genannten drei Phasen derDenaturierung, Annealing und DNA-Synthese besteht. Der folgende Zyklus beginntwieder mit der Denaturierung in Einzelstrang-DNA. Da bei jedem neuen Zyklus derbereits neu synthetisierte DNA-Abschnitt als Matrize dient, kommt es zu einerexponentiellen Amplifikation der Ziel-DNA (Saiki et al., 1988).
6 Um zu gewährleisten, daß eine ausreichende Menge des Enzyms eingesetzt wird, multipliziert man mit dem
Faktor 4.

41
5´ 3´ 3´ 5´
doppelsträngige DNA
Denaturierung5´ 3´
Primer
3´ 5´Primer
5´ 3´Taq Polymerase
3´ 5´Synthese
Zyklus wirdwiederholt
Taq Polymerase 4
816
etc.
Abb. 16: Die Polymerase-Kettenreaktion (PCR). Der Zyklus der Denaturierung, Priming undDNA-Synthese wird viele Male wiederholt. Die Zahlen an den Pfeilen geben die Anzahl derDNA-Stränge an: Die Reaktion startet mit zwei, nach dem ersten Zyklus sind es vier, nachdem zweiten acht, nach dem dritten 16 usw. Theoretisch sind nach 30 Zyklen 2,15 x 109
Stränge entstanden (aus Nicholl DST).
6.2. Verwendete Oligonukleotide
Für die Amplifikation der kompletten kodierenen Region des Protein VII aus pJM17wurden die Oligonukleotide ADVII 16464UHind und ADVII 15945DBamHIverwendet (Tab.). Für die Analyse der Ligationsprodukte wurden die in Tab.angegebenen Oligonukleotide eingesetzt. Sie wurden basierend auf der DNA-Sequenzdes Vektors pQE 30 synthetisiert. Es wurde angestrebt, die Primer aus 17 bis 25 Basenmit einem Cytosin-Guanin-Gehalt von maximal 50 % auszuwählen, um optimaleAmplifikationsbedingungen zu gewährleisten. In den PCR-Amplifikationen, die derSequenzierung unterzogen werden sollten, wurde ein am 5'-Ende biotinyliertesOligonukleotid zur Reinigung an Streptavidin-beschichteten Dynabeads undFestphasen-Sequenzierung verwendet. Der zweite PCR-Primer enthielt eine 17-Basen-5'-Extension mit der Erkennungssequenz des Bakteriophage M 13-Sequenzierprimers.Hierdurch wurde eine halbautomatische DNA-Sequenzierung mit fluorochrom-markierten Oligonukleotiden ermöglicht. Die Sequenzen der verwendeten Primer sindin Tab.6 zusammengefaßt.

42
Tab.6: Synthetische Oligonukleotide für die DNA-Amplifikation und Sequenzanalyse
I. Primer für die DNA-Amplifikation
AdVII16464UHindIII 5´-GGG CAA GCT TTT TTC TTG CAA TCT AGT TGC GCG G-3´
AdVII15945DBamHI 5´-TAA AGG ATC CGC AAA GAA GCG CTC CGA CCA ACA C-3´
II. Primer für die DNA-Sequenzbestimmung
AdVII16451Ubio 5´-Bio-TCT TGC AAT CTA GTT GCG CG-3´
AdVII15957DM13 5´-TGT AAA ACG ACG GCC AGT AAA GAA GCG CTC CGA CCA ACA-3´
pQE30U1bio 5´-Bio-TCA TTA CTG GAT CTA TCA AC-3´
pQE30U2bio 5´-Bio-TAT ATC AAC GGT GGT ATA TC-3´
pQE30DM13 5´-TGT AAA ACG ACG GCC AGT TGT GAG CGG ATA ACA ATT TC-3´
D und U kennzeichnen Plusstrang und Minusstrang
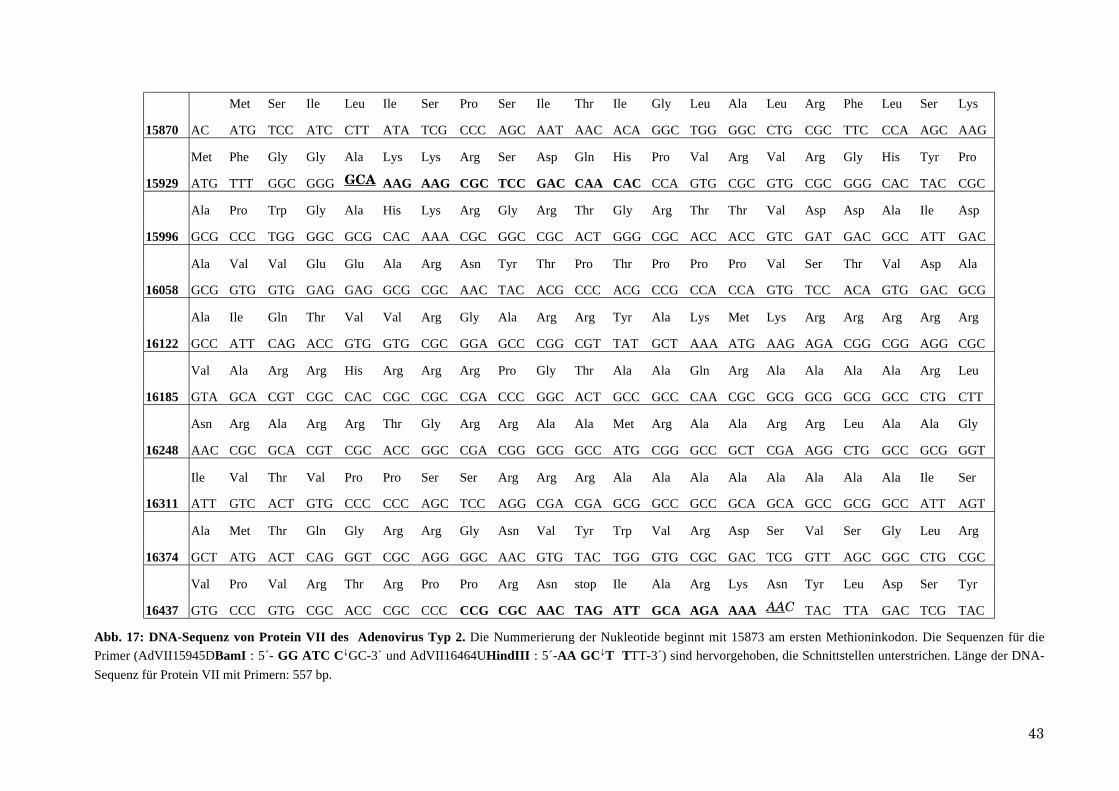
43
Met Ser Ile Leu Ile Ser Pro Ser Ile Thr Ile Gly Leu Ala Leu Arg Phe Leu Ser Lys
15870 AC ATG TCC ATC CTT ATA TCG CCC AGC AAT AAC ACA GGC TGG GGC CTG CGC TTC CCA AGC AAG
Met Phe Gly Gly Ala Lys Lys Arg Ser Asp Gln His Pro Val Arg Val Arg Gly His Tyr Pro
15929 ATG TTT GGC GGG GCA AAG AAG CGC TCC GAC CAA CAC CCA GTG CGC GTG CGC GGG CAC TAC CGC
Ala Pro Trp Gly Ala His Lys Arg Gly Arg Thr Gly Arg Thr Thr Val Asp Asp Ala Ile Asp
15996 GCG CCC TGG GGC GCG CAC AAA CGC GGC CGC ACT GGG CGC ACC ACC GTC GAT GAC GCC ATT GAC
Ala Val Val Glu Glu Ala Arg Asn Tyr Thr Pro Thr Pro Pro Pro Val Ser Thr Val Asp Ala
16058 GCG GTG GTG GAG GAG GCG CGC AAC TAC ACG CCC ACG CCG CCA CCA GTG TCC ACA GTG GAC GCG
Ala Ile Gln Thr Val Val Arg Gly Ala Arg Arg Tyr Ala Lys Met Lys Arg Arg Arg Arg Arg
16122 GCC ATT CAG ACC GTG GTG CGC GGA GCC CGG CGT TAT GCT AAA ATG AAG AGA CGG CGG AGG CGC
Val Ala Arg Arg His Arg Arg Arg Pro Gly Thr Ala Ala Gln Arg Ala Ala Ala Ala Arg Leu
16185 GTA GCA CGT CGC CAC CGC CGC CGA CCC GGC ACT GCC GCC CAA CGC GCG GCG GCG GCC CTG CTT
Asn Arg Ala Arg Arg Thr Gly Arg Arg Ala Ala Met Arg Ala Ala Arg Arg Leu Ala Ala Gly
16248 AAC CGC GCA CGT CGC ACC GGC CGA CGG GCG GCC ATG CGG GCC GCT CGA AGG CTG GCC GCG GGT
Ile Val Thr Val Pro Pro Ser Ser Arg Arg Arg Ala Ala Ala Ala Ala Ala Ala Ala Ile Ser
16311 ATT GTC ACT GTG CCC CCC AGC TCC AGG CGA CGA GCG GCC GCC GCA GCA GCC GCG GCC ATT AGT
Ala Met Thr Gln Gly Arg Arg Gly Asn Val Tyr Trp Val Arg Asp Ser Val Ser Gly Leu Arg
16374 GCT ATG ACT CAG GGT CGC AGG GGC AAC GTG TAC TGG GTG CGC GAC TCG GTT AGC GGC CTG CGC
Val Pro Val Arg Thr Arg Pro Pro Arg Asn stop Ile Ala Arg Lys Asn Tyr Leu Asp Ser Tyr
16437 GTG CCC GTG CGC ACC CGC CCC CCG CGC AAC TAG ATT GCA AGA AAA AAC TAC TTA GAC TCG TAC
Abb. 17: DNA-Sequenz von Protein VII des Adenovirus Typ 2. Die Nummerierung der Nukleotide beginnt mit 15873 am ersten Methioninkodon. Die Sequenzen für die
Primer (AdVII15945DBamI : 5´- GG ATC C↓GC-3´ und AdVII16464UHindIII : 5´-AA GC↓T TTT-3´) sind hervorgehoben, die Schnittstellen unterstrichen. Länge der DNA-
Sequenz für Protein VII mit Primern: 557 bp.

44
6.2.1. Berechnung der Oligonukleotidprimer
Die Primer wurden in einer Menge von 2,5 bis 20 pmol in die PCR eingesetzt undnach folgender Formel berechnet:
Anzahl der Basen eines Oligonukleotids x 308 – 61 = Molmasse des Primers in g/mol.
Mit Hilfe der optischen Dichtewerte (OD) der Primerstocks, konnten die Volumina derbenötigten Oligonukleotide berechnet werden:
Menge des benötigten Oligonukleotids in µg : OD des Primers in µg/µl.
Hierdurch ergab sich das für einen PCR-Ansatz benötigte Stammvolumen einesPrimers. Dieses Verfahren wurde auf alle Oligonukleotide angewandt. DieStammlösung hatte eine Konzentration von 100 pmol/µl, für den jeweiligen PCR-Ansatz wurde die gewünschte Konzentration in einem Gesamt-volumen von 20 µlH2O dd bzw. TE-Puffer verdünnt.
6.3. Vorbereitung des PCR-Reaktions-Mix
6.3.1. PCR-Puffer
Ansatz des 10 x PCR-Reaktionspuffers Endkonzentration
500 µl 1 M KCl 500 mM100 µl 1 M Tris-HCl (pH8,3) 100 mM
30 µl 1 M MgCl2 30 mM50 µl Tween-20 0,5 %50 µl Triton X-100 0,5 %
mit H2O dd auf 1000 µl aufgefüllt.
6.3.2. Desoxynukleotidtriphosphate
Ansatz des 10 x dNTP-Mixes Endkonzentration
200 µl 10 mM dATP 2 mM200 µl 10 mM dCTP 2 mM200 µl 10 mM dGTP 2 mM200 µl 10 mM dTTP 2mM
mit 10 mM Tris-HCl (pH 8,0) auf 1000 µl aufgefüllt.
6.3.3. Reaktionsgemisch
Jeder Probe eines PCR-Ansatzes wurde ein Reaktionsgemisch nach folgendem Ansatzhinzugegeben:
• 10 µl 10 x PCR-Puffer

45
• 10 µl 10 x dNTP-Mix• 39,5 µl H2O dd• 0,5 µl (2U) Taq-DNA-Polymerase• ggf. 6,5 µl Glyzerin (wasserfrei).
6.3.4. Temperaturprofil für die PCR
Zyklen Zeit / Temperaturen
1 3 min 94 °C35 1 min 94 °C / 1 min 55 °C / 2 min 72 °C
1 10 min 72 °Cüber Nacht 4 °C
6.4. Durchführung der PCR
Grundsätzlich wurden bei den Vorbereitungen und der Durchführung der PCR dieProben bis zum Einsetzen in den Thermozykler bei 4 °C auf Eis gekühlt. Zunächstwurde der Reaktionsmix ohne die Polymerase angesetzt und auf. Bei derErstamplifikation wurden einmal 10 ng und einmal 100 ng der mit SalI verdauten undgereinigten DNA von pJM17 eingesetzt. Bei den PCR-Amplifikationen, die der DNA-Sequenzierung unterzogen werden sollten, wurden 50 ng gereinigte Plasmid-DNAeingesetzt. Das Volumen wurde mit H2O dd auf 20 µl aufgefüllt. Zu diesen 20 µlwurden 20 µl Primermix aus zwei Primern gegeben, wobei die Primerkonzentrationbei der Amplifikation aus pJM17 50 pmol/Primer entsprach, bei der Amplifikation vongereinigter Plasmid-DNA zum Zwecke der Sequenzierung jedoch 5 pmol/Primer. Eswurde nun bei ständiger Kühlung auf Eis die DNA-Polymerase zu dem Reaktionsmixgegeben, alles gemischt und 60 µl des fertigen Reaktionsgemisches zu den 40 µlDNA-Primer-Gemisch gegeben. Eine Überschichtung mit Mineralöl war bei derErstamplifikation notwendig, entfiel aber bei der Sequenzierung durch entsprechendeReaktionsgefäße (Fa. Sarstedt), die durch einen im Deckel integrierten Stempel einefest verschlossene Reaktionskammer bildeten und somit ein Verdampfen vonFlüssigkeit verhinderten. Die Proben wurden in den programmierten PCR-Thermalzykler gestellt. Nach Erreichen der Ausgangstemperatur des 1. Zyklus von 94°C wurden je Probe noch 4,1 µl 25 mM Magnesiumazetat hinzugegeben. Dieser alsHot-Start bezeichnete Vorgang verhindert die unspezifische Amplifikation von DNA-Segmenten beim Hochheizen. Es folgte die ca. 3-4 stündige Amplifikation bei denbeschriebenen Temperaturen im Thermozykler.
6.5. Modifikation der PCR nach dem PCR-Optimization-Kit
Aufgrund von Unterschieden bezüglich der Reinheit, des Guanin-Cytosin-Gehaltesund der Anzahl der Sekundärstrukturen, sowie chemischer Modifikationen oderanderen Charakteristika der zu untersuchenden DNA, kann es zu einer nicht-

46
spezifischen oder zu geringen Amplifikation kommen. Da dies bei der DNA desProtein VII der Fall war, wurde eine Versuchsreihe zur Verbesserung der PCR mittelsdes Opti-PrimePCR-Optimization Kit der Firma Stratagene eingeleitet.
6.5.1. Prinzip
Durch eine Veränderung der Pufferbestandteile und des pH-Wertes derReaktionspuffer kann die Amplifikationsrate erhöht werden. Desweiteren existiert dieMöglichkeit, durch Zugabe von verschiedenen Zusätzen die Spezifität bzw. dieAmplifikationsrate zu erhöhen. Der Kit enthält 12 10xPCR-Puffer mit pH-Wertenzwischen 8,3 und 9,2, einer Magnesiumchloridkonzentration zwischen 1,5 und3,5 mM und einer Natriumchloridkonzentration zwischen 25 und 75 mM. Desweiterensind folgende sechs Zusätze enthalten:
• Formamid 50 % (v/v), welches die Annealing-Reaktion erleichtert undwahrscheinlich die Denaturierungstemperatur von schmelzresistenter DNAerniedrigt (Sarkar et al., 1990).
• Dimethylsulfoxid (DMSO) 100 % (v/v), das ebenso wie Glycerol dieDenaturierung von Guanin-Cytosin-reicher DNA verbessert und hilft,Schwierigkeiten der Polymerase-Extension aufgrund von Sekundärstrukturen zuüberwinden (DMSO 10 % als Zusatz zur PCR konnte als essentiell für dieAmplifikation des Retinoblastomgens nachgewiesen werden (Hung et al., 1990),ebenso bei der Amplifikation von HSV-Sequenzen; Glycerol verbessert die Mengean Amplifikationsprodukt und wirkt als Enzymstabilisator (Smith et al., 1990)).
• Glycerol 100 % (v/v).• Rinderserumalbumin (BSA), dieses fungiert als nicht-spezifischer
Enzymstabilisator, der auch an bestimmte PCR-Inhibitoren bindet (Paabo et al.,1988).
• Ammoniumsulfat [(NH4)2SO4] 750 mM.• Perfect MatchDNA polymerase enhancer der Firma Stratagene, der die Spezifität
und die Menge des PCR-Produktes durch Destabilisierung fehlerhafter Primer-Matrix-Komplexe erhöht und Sekundärstrukturen entfernt, die die normaleExtension behindern könnten (Nielson und Mathur 1990).
Desweiteren enthält der Kit einen sog. 50xMaster MixPuffer, der die DNA-Matrixund die Primer stabilisieren soll. Die Konzentration von EDTA in diesem Puffer ist zugering, um die Amplifizierungsreaktion behindern zu können.
6.5.2. Durchführung
Es wurden 12 Reaktionsgefäße mit 5 µl eines jeden 10xPuffers vorbereitet. Einer jedenProbe wurden 45 µl eines Reaktionsgemisches (Master Mix) mit folgendem Ansatzzugegeben:

47
• 12,5 µl 50xMaster Mix Puffer (= 400 nM Tris-HCl (pH 8,0) und 5 nM EDTA)• 12,5 µl 10 mM dNTP• 2,5 µg von jedem Oligonukleotidprimer (= 8,48 µl)• 2,5 µg der genomischen DNA (= 200 ng/Reaktion) oder• 1,2 µg der Plasmid-DNA (= 100 ng/Reaktion)• 30.0 U der Taq DNA Polymerase
dH2O bis zu einem Endvolumen von ~565 µl (= 528,56 µl dH2O).
Abschließend wurde das Reaktionsgemisch mit jeweils 25 µl Mineralöl versiegelt.Die PCR erfolgte im Thermozycler mit folgenden Temperaturprofil:
PCR-Schritte Temperatur Zeitdauer
a. Denaturierung 94°C 3 Minutenb. Annealing 50 °C 2 Minuten
c. Wiederholung der folgenden Zyklen 20-30 mal
i. Extension 72°C 1,5 Minutenii. Denaturierung 94°C 1 Minuteiii. Annealing 50°C 1 Minute
d. Extension 72°C 8 Minuten
Im Folgenden wurden die Proben bzw. die PCR-Produkte mittels Elektro-phoresetechnik auf einem 6 %igem Agarosegel auf ihre korrekte Größe und ihreMasse hin untersucht. Diejenigen Proben, die die größte Menge an PCR-Produkthatten, wurden durch Zugabe der verschiedenen Zusätze einer weiteren Optimierungs-PCR unterzogen. Es wurden 7 Reaktionsgefäße mit jeweils 37,5 µl desjenigen Puffersvorbereitet, der in der ersten PCR das beste Ergebnis erzielt hatte, desweiteren wurdenjeweils 45 µl des folgenden Reaktionsgemisches zugegeben:
• 7,5 µl der 10 mM dNTP• 1,5 µg eines jeden Primers• 1,5 µg der genomischen DNA (= 200 ng/Reaktion) oder• 7,5 ng der Plasmid-DNA (= 100 ng/Reaktion)• 18.0 U Taq DNA Polymerase• dH2O bis zu einem Endvolumen von 340 µl.
Ebenso wurden diese Proben mit jeweils 25 µl Mineralöl versiegelt, bei dem gleichenTemperaturprofil zur Reaktion gebracht und ebenfalls mittels eines 6 %igenAgarosegels sichtbar gemacht.

48
7. Analytische Agarosegelelektrophorese
7.1. Prinzip
Die DNA-Agarosegelelektrophorese bietet die Möglichkeit, eine Analyse von PCR-Produkten bezüglich ihrer Größe durchzuführen, sie wurde nach der Methode vonSambrook et al., 1989 durchgeführt. Die DNA wird hierzu auf einer Agarosematrixim elektrischen Feld elektrophoretisch aufgetrennt. Durch Hinzugabe vonEthidiumbromid, einem planaren Aromaten, der zwischen die Basenpaare der DNAinterkaliert, ohne dabei die Wasserstoff-Brückenbindungen zu zerstören, kann dieDNA im ultravioletten Licht sichtbar gemacht werden. Dies geschieht mit der Fluo-reszenzfarbe, die bei ultravioletter Lichteinstrahlung von Wellenlängen zwischen 253nm und 366 nm das Licht bei einer Wellenlänge von 590 nm des sichtbarenSpektrums, also im rot-orangenen Bereich, zurückstrahlt. So kann bei einem Vergleichmit einem DNA-Marker bekannter Größe abgeschätzt werden, wie groß deramplifizierte DNA-Abschnitt ist, und ob der richtige DNA-Abschnitt amplifiziertwurde. Zur Photodokumentation wurden ein roter Interferenzfilter und Polaroidfilmeverwendet.
7.2. Vorbereitung und Beladen des Agarosegels
Mit 100 ml 1 x TBE-Puffer wurden Gelansätze mit Agarosekonzentrationen zwischen0,8 und 1,2 % hergestellt, wobei die Trennleistung eines höherprozentigen Gelsaufgrund seiner sehr viel kleineren Poren für kleine DNA-Fragmente größer ist undumgekehrt. Dieses Gemisch wurde nach kurzem Aufkochen mit dem Magnetrührerauf 50 °C im vorgeheizten Wasserbad abgekühlt. Nach Zugabe von 5 µl einer 10mg/ml Ethidiumbromidlösung (Endkonzentration 0,5 µg/ml) wurde das noch flüssigeGel in die vorher abgeklebte Gelkammer gegossen. Nach ca. 30 min Abkühlung wardas Agarosegel soweit polymerisiert, daß es für die Beladung der Proben zurVerfügung stand. Es wurde nun in die mit 1 x TBE-Puffer, der mit 0,5 µg/mlEthidiumbromidlösung versetzt war, gefüllte Gelkammer gesetzt, so daß es vollständigmit Pufferlösung bedeckt war. Von den PCR-Produkten wurden jeweils 10 µlabpipettiert und mit 2 µl einer 0,25 %igen Bromphenolblaulösung BPB (25 µl 10%iges Bromphenolblau, 300 µl wasserfreies Glyzerin, aufgefüllt mit H2O dd ad1000µl) versetzt. Es folgte das Laden der Proben mit sterilen Einmalpipetten von obendirekt in die Geltaschen hinein. Zur Größenmarkierung wurden an beiden Seiten derGele der DNA-Marker Boehringer III (Gemisch aus EcoRI-Verdau und HindIII-Verdau von λ-DNA) und Boehringer VI (Gemisch aus BglI-Verdau und HinfI-Verdauvon pBR 328) in einer Endkonzentration von 0,35 µg/ 10 µl verwendet. DieElektrophorese erfolgte bei einer Spannung von 70-80 V für ca. 60-90 min. Danachwurde das Gel auf einem UV-Leuchtkasten bei UV-Illumination von 280 nm unterVerwendung eines Rotfilters auf Polaroidfilm photographiert.

49
8. Subklonierung des PCR-Produktes
8.1. Prinzip
Zur Analyse der Sequenz des Protein VII mußte die DNA aus der DNA des pJM17präpariert (siehe Kapitel 2) ,geschnitten (siehe Kapitel 4) und kloniert (siehe Kapitel 9und 10) werden. Um ein bestimmtes DNA-Fragment in E. coli zu klonieren, wird einPlasmidvektor benötigt. Dieser Vektor wird genau wie das betreffende DNA-Fragmentmit Restriktionsendonukleasen des Typs II (Restriktionsenzyme) inkubiert. Daswichtigste Merkmal dieser Enzyme besteht darin, daß es für jedes Enzym einebestimmte Erkennungssequenz gibt, an der es DNA spaltet. Desweiteren sind fast alleErkennungssequenzen Palindrome, d.h. es ergibt sich in beiden Leserichtungen diegleiche Sequenz. Man unterscheidet bei den Restriktionsendonukleasen solche, diesogenannte glatte Enden erzeugen durch Schneiden beider DNA-Stränge in der Mitteder Erkennungssequenz (blunt ends), oder solche, die die DNA-Stränge versetztschneiden, so daß die entstehenden DNA-Fragmente an ihren Enden kurzeüberstehende Einzelstrangabschnitte besitzen, sogenannte klebrige Enden (sticky ends)(Tab.7). Werden ein Vektor und das betreffende DNA-Segment mit den gleichenRestriktionsenzymen inkubiert, so entstehen einander komplementäre, einzelsträngigeEnden, die nach einer Re-Hybridisierung mit einer DNA-Ligase, isoliert aus mitBakteriophagen T4 infizierten E.coli-Bakterien, miteinander verknüpft werdenkönnen. Es folgt damit eine Ligation zwischen dem Vektor und dem DNA-Segment.Um eine Selektion derjenigen Vektoren, die das DNA-Segment aufgenommen haben,zu erreichen – dieses ist nicht bei allen der Fall – gibt es verschiedene Methoden, dierekombinanten Vektoren zu isolieren und zu vermehren. Die hier verwendete Methodeberuht auf einer Antibiotikaresistenz sowohl gegen Ampicillin als auch Kanamycin.Da der Vektor die kodierende Region für die Ampicillin-Resistenz trägt und in den E.coli Strang M15 transformiert wird, welcher das Repressor-Plasmid pREP4 mit seinerKanamycin-Resistenz - vermittelt durch das Gen für die Neomycin-Phosphotransferase – trägt, kommt es bei rekombinanten Vektoren zu einerzweifachen Antibiotikaresistenz. Die Selektion erfolgte durch Ausplattieren dertransformierten Zellen auf Ampicillin- (100 µg/ml) und Kanamycin- (25 µg/ml)haltigen Platten und einer Inkubation über Nacht. Die gewachsenen Kolonienenthalten den rekombinanten Vektor und konnten so isoliert und weiter vermehrtwerden. Die Klonierung wurde nach dem Protokoll des Skriptes The QIAexpressionist(Fa. QIAGEN) und dem PCR-ScriptSK(+)Cloning System (Fa. Stratagene)durchgeführt.

50
Tab. 7: Erkennungssequenzen der verwendeten Restriktionsendonukleasen
Enzym Organismus Erkennungs- Schnittstellen Endensequenz
BamHI Bacillus 5´-GGATTC-3´ G↓GATC C 5´C CTAG↑Gamylolique-faciens
HindIII Haemophilus 5´-AAGCTT-3´ A↓AGCT T 5´T TCGA↑Ainfluenzae Rd
SalI Streptomyces 5´-GTCGAC-3´ G↓TCGA C 5´C AGCT↑Galbus G
Anmerkung: Die Erkennungssequenzen sind einsträngig in 5´→ 3´ Richtung angegeben und als Palindromeerkennbar, die Schnittstellen als Doppelstrang, um die vom jeweiligen Enzym erzeugte Form der Enden zucharakterisieren; 5´und 3´beziehen sich auf die überstehenden 5´-und 3´-Enden.
9. Verdau des Protein VII und des Vektors pQE30 mit BamHI und HindIII
9.1. Prinzip
Das in der PCR amplifizierte Protein VII wurde gereinigt und mit den Re-striktionsendonukleasen BamHI und HindIII inkubiert. Auf eben diese Weise wurdemit dem Expressionsvektor pQE30 verfahren.
9.2. Vorbereitung
9.2.1. Reinigung und Konzentrierung der PCR-Produkte
Nach einer gelelektrophoretischen Auftrennung der gesammelten PCR-Produkte ineinem Agarose-/TAE-Puffer-Gel in einer TAE-Puffer gefüllten Kammer erfolgte dieExzision der Protein VII enthaltenden DNA-Banden unter UV-Licht. Hierbei wurdedie Größe der Bande des Protein VII anhand der bekannten Sequenz errechnet und dieHöhe anhand der Marker-Banden abgeschätzt. Die Exzision erfolgte mit einem sterilenSkalpell. Anschließend wurde das Gewicht der einzelnen Gelstücke bestimmt. DieExtraktion aus dem Gel erfolgte anhand des QIAEX DNA Gel-Extraktions Protokolls.hierbei wurde pro 300 mg ausgeschnittenen Gels 300 µl QX 1-Puffer zugegeben. DieQIAEX-Partikel-Suspension wurde für 30 sec gemischt und danach jeweils 10 µlQIAEX II in jede Probe gegeben. Die Proben wurden für 10 min bei 50 °C inkubiert,und dabei alle 2 min erneut durchmischt. Nach dieser Zeit wurden die Proben für 30sec bei 12000 Upm zentrifugiert. Der Überstand wurde verworfen. Das DNA-Präzipitat wurde je zweimal mit 500 µl QX 2-Puffer und QX 3-Puffer gewaschen. DasPräzipitat wurde in 500 µl PE-Puffer resuspendiert, zentrifugiert und der Überstandentfernt. Anschließend wurde das die DNA und die QIAEX-Partikel enthaltende

51
Präzipitat für 10-15 min luftgetrocknet. Zur Elution der DNA von den Partikeln wurdedas Präzipitat in 20 µl 10 mM Tris-HCl (pH 8,5) aufgenommen und für 30 sec bei12000 Upm zentrifugiert. Der die DNA enthaltende Überstand wurde abgenommenund wie folgt konzentriert: Je 30 µl des Gemisches wurden mit 100 µl 95 %igemEthanol und 2 µl 3 M Natriumacetat versetzt und für 15 min auf Eis inkubiert. Nacheiner 20 minütigen Zentrifugation bei 12000 Upm und 4 °C wurde das Präzipitat mit70 %igem Ethanol gewaschen. Nach dem Abziehen des Überstandes wurde dasPräzipitat in 10 µl 6.6.2.-Puffer aufgenommen.
9.3. Durchführung des Verdaus
Die Vorgehensweise ist identisch mit der in Kapitel 4 beschriebenen. Es wurdenlediglich andere Enzyme, und zwar BamHI und HindIII, und eine anderer Inkubations-Puffer, nämlich B-Puffer verwendet. Die Proben wurden auf ein 1,2 %iges Agarosegelgeladen und elektrophoretisch aufgetrennt. Zur Photodokumentation wurde das Gelunter UV-Licht mittels eines roten Interferenzfilters auf Polaroidfilm photographiert.
10. Ligation der Verdau-Produkte
10.1. Prinzip
Die Ligation der beiden Fragmente (Vektor-Protein VII) wurde in einer sog.Eintopfreaktion durchgeführt, wobei die Fragmente in einem molaren Verhältnis vonVektor : Protein VII 1:5 eingesetzt wurden. Da die beiden Fragmente vorher wie inKapitel 8 beschrieben durch Restriktionsendonukleasen vorbereitet worden waren,konnten sie nur folgendermaßen ligiert werden : Vektor (BamHI) – (BamHI) ProteinVII (HindIII) – (HindIII) Vektor.
10.2. Vorbereitung
Die Verdau-Produkte wurden mittels Plasmidpräparation (Maxi-Präparation nachQIAGEN-Protokoll) gereinigt.
10.3. Durchführung
Zu der gereinigten DNA wurde zugegeben :
• 4 µl 5 x T4-Ligase-Puffer• 1 µl T4-Ligase (1U/µl)• 1 µl H2O dd,
das Endvolumen betrug 20 µl. Die Proben wurden gemischt und bei 14 °C für 18 hinkubiert. Anschließend wurde 1U T4-Ligase zugefügt und für weitere 6 h ligiert. ZurDokumentation wurde ein 1,2 %iges Agarosegel beladen und photographiert.

52
11. Transformation des Ligationsproduktes in XL 1-blue
11.1. Prinzip
Als Transformation bezeichnet man die Aufnahme von DNA-Molekülen durch Zellenjeglichen Typs. Da dieser Vorgang unter normalen Bedingungen in nur begrenztemUmfang auftritt, muß man die Zellen einer physikalisch und/oder chemischenBehandlung unterziehen, die die Fähigkeit, DNA aufzunehmen, verstärkt. Zellen, dieauf eine solche Art behandelt wurden, bezeichnet man als kompetent. Es erfolgte hierzunächst die Transformation in XL 1-blue, um zu gewährleisten, daß nur erfolgreichligierte DNA gereinigt und in die kompetenten Zellen für die Expression transformiertwurde. Die Selektion erfolgte durch die mittels des Vektors eingebrachte Ampicillin-Resistenz, das Methicillin verhinderte das Wachsen von Satellitenkolonien.
11.2. Vorbereitung
Es wurden LB-Agarplatten mit einer Konzentration von 20 µl/ml Ampicillin und 80µl/ml Methicillin gegossen. Die dafür benötigten Bestandteile wurden autoklaviert,Ampicillin nach Abkühlen auf ca. 50 °C hinzugegeben und in 25 cm2 Schalengegossen. Des weiteren wurde SOC-Medium hergestellt.
11.3. Durchführung
Die Transformation erfolgte in kompetente Zellen des Stammes XL 1-blue und wurdenach dem PCR-ScriptSK(+) Cloning Kit - Protokoll der Fa. Stratagene durchgeführt.Die Zellen, die bei –80 °C gelagert wurden, wurden langsam auf Eis aufgetaut undgemischt. 40 µl dieser Zellen je Probe wurden in ein vorgekühltes Falcon 2059-Reaktionsgefäß gegeben. Hierzu wurden pro Röhrchen 0,7 µ l 1,44 M β-Merkaptoethanol (Endkonzentration 25 mM) gegeben. Dieses Gemisch wurde für 10min auf Eis inkubiert und dabei alle 2 min durch Wenden des Röhrchens gemischt.Nach der Zugabe von 2 µl DNA aus der Ligation wurden die Proben erneut gemischtund für 30 min auf Eis gestellt. Zum Einbringen der nun z.T. Protein VII-Inserttragenden Vektoren in die Zellen erfolgte ein Hitzeimpuls von 42 °C für genau 50-55sec im Wasserbad. Nach einer anschließenden 2-minütigen Inkubation auf Eis wurdenje Probe 450 µl auf 42 °C vorgewärmtes SOC-Medium hinzugegeben. Hiernachwurden die Proben für 1 h bei 37 °C im Brutschrank geschüttelt. Nach dieser Inkuba-tion wurden jeweils zwei Platten pro Probe mit 100 µ l bzw. 400 µ l einesTranformationsansatzes bestrichen. Die Platten wurden an der unter dem Abzug kurzgetrocknet und dann für ca. 12-18 h bei 37 °C umgedreht im Brutschrank inkubiert.Nach Isolierung mehrerer Klone und Anlegen von Über-Nacht-Kulturen erfolgte einScreening mit den schon in die PCR von der DNA des Protein VII eingesetztenPrimern AdVII16464UHINDIII und AdVII15945DBAMHI. Aus den positiven Klonen,das sind die Klone, bei denen die Screening-PCR zeigte, daß sie den rekombinantenVektor enthalten, wurde je Probe 10 Klone isoliert. Nach dem Plasmid-Präparations-

53
protokoll von QIAGEN (siehe auch Kapitel 3) wurden für diese Klone Maxi-Präparationen durchgeführt (Sambrook et al.,1989). Diese wiederum waren Grund-lage für die folgende Sequenzierungsreaktion (Siehe Kapitel 11).
12. DNA-Sequenzierung nach Sanger
Die DNA-Sequenzierung erfolgte nach der von Sanger et al., 1977 beschriebenenKettenabbruchmethode. Zur DNA-Sequenzbestimmung der gesamten DNA desProtein VII wurde die in Abb. gezeigte Strategie eingesetzt. Die hierzu verwendetenOligonukleotide sind in Tab.6 zusammengestellt.
12.1. Reinigung der PCR-Produkte
12.1.1. Prinzip
In den für die Sequenzierung vorgesehenen PCR-Reaktionen wurden, wie unter 6.2.erwähnt, ein am 5'-Ende biotinyliertes Oligonukleotid und ein zweiter Primer miteiner 17-Basen-5'-Extension mit der Erkennungssequenz des Bakteriophagen M13-Sequenzierprimers verwendet. Der biotinylierte Primer dient hierbei als Hilfe für dieReinigung des PCR-Produktes. Nach der Inkubation des PCR-Produktes mitStreptavidin beschichteten Magnetpartikeln (Dynabeads, Fa. Dynal) binden diebiotinylierten DNA-Stränge an diese, es kommt somit zu einer Bindung an dieFestphase. Mit Hilfe eines Magnetständers kann die Reinigung und Auftrennung inEinzelstrang-DNA erfolgen, ohne daß es zu einem Verlust des biotinylierten DNA-Stranges kommt.
12.1.2. Durchführung
Da für ca. 100 µl PCR-Produkt 100 µl Dynabeads (5 µg/µl) benötigt werden, wurden50 µl Dynabeads (10 µl/µg) entnommen und nach zweimaligem Waschen in je 100 µlB&W-Puffer in 100 µl B&W-Puffer aufgenommen. Zu den so verdünnten Dynabeadswurde das gesamte PCR-Produkt gegeben, gemischt und für ca. 30 min untermehrmaligem Aufschütteln bei Raumtemperatur inkubiert. Danach wurden die Probenfür 30 sec in einen Magnetständer gestellt und der Überstand entfernt. Nachzweimaligem Waschen der Magnetpartikel mit 100 µl B&W-Puffer wurden dieProben mit je 25 µl frisch angesetztem 0,1 M NaOH versetzt. Nach dem Mischenerfolgte die Inkubation für 10 min bei Raumtemperatur. Der den abgelösten DNA-Einzelstrang enthaltene Überstand wurde verworfen. Der biotinylierte, an dieMagnetpartikel gebundene Einzelstrang wurde mit 125 µl 0,1 M NaOH, danach mit100 µl B&W-Puffer und zuletzt mit 125 µl H2O dd gewaschen. Nach Abziehen desH2O dd erfolgte die Elution in 5,5 µl H2O dd. Dieses H2O-Magnetpartikel-Gemischwurde in die Sequenzierreaktion eingesetzt.

54
12.2. Bestimmung der DNA-Sequenz
12.2.1. Prinzip
Das Prinzip der von Sanger entwickelten Kettenabbruchmethode ist eineAmplifikation, bei der neben Desoxynukleotiden (dNTP) auch Didesoxynukleotide(ddNTP) eingesetzt werden. Des weiteren enthält der Mix einen spezifischen M13Primer (Tab.8) und AmpliTaq-DNA-Polymerase FS. Wird in den entstehendenPolynukleotidstrang ein Didesoxynukleotid (ddNTP) eingebaut, bricht die DNA-Synthese an dieser Stelle ab, da die molekulare Veränderung im Didesoxynukleotidkeine weitere Verknüpfung zum nächsten Nukleotid zuläßt. Das Verhältnis dernormalen Desoxynukleotide zum Didesoxynukleotid wird so gewählt, daß statistischhinter jeder Base der Sequenz die Reaktion einmal abbrechen kann. Es entsteht so einGemisch aus DNA-Fragmenten aller möglichen Größen. Für jede Basenart wird eineseparate Reaktion angesetzt, wobei jeweils nur ein Didesoxynukleotid vorliegt. Jedesunterschiedliche Didesoxynukleotid ist dabei auch unterschiedlichfluoreszenzmarkiert. Nach der Amplifikationsreaktion werden die vier Ansätzevereinigt, und das Gemisch aus DNA-Fragmenten mit einerPolyacrylamidgelelektrophorese aufgetrennt. An der anodischen Seite des Gels werdendie austretenden DNA-Fragmente anhand ihrer Fluoreszenz laseroptisch detektiert.Mittels rechnervermittelter Analyse erhält man die DNA-Sequenz des PCR-Produktes.
Tab. 8: Primer für die DNA-Sequenzbestimmung
AdVII16451Ubio 5´-Bio-TCT TGC AAT CTA GTT GCG CG-3´
AdVII15957DM13 5´-TGT AAA ACG ACG GCC AGT AAA GAA GCGCTC CGA CCA ACA-3´
pQE30U1bio 5´-Bio-TCA TTA CTG GAT CTA TCA AC-3´
pQE30U2bio 5´-Bio-TAT ATC AAC GGT GGT ATA TC-3´
pQE30DM13 5´-TGT AAA ACG ACG GCC AGT TGT GAG CGGATA ACA ATT TC-3´
D und U kennzeichnen Plusstrang und Minusstrang.
12.2.2. Durchführung der Sequenzierreaktion
Die Sequenzierung wurde mit dem PRISM Ready Reaction Dye Primer CycleSequenzing System mit der AmpliTaq-DNA-Polymerase FS (Fa. Applied Biosystems)durchgeführt.Zunächst wurden für jede Probe vier Reaktionsgefäße angelegt, wobei eines 4 µl A-d/ddNTP-Mix, eines 4 µl C-d/ddNTP-Mix, eines 8 µl G-d/ddNTP-Mix und das letzte 8µl T-d/ddNTP-Mix enthielt. Von der im Schritt der Reinigung des PCR-Produktesgewonnenen gereinigten, an Dynabeads gekoppelten DNA wurden je 1 µl in die A-und C-Gefäße und je 2 µl in die G- und T-Gefäße gegeben und gemischt. Da PCR-

55
Reaktionsgefäße von Sarstedt verwendet wurden, entfiel die Überschichtung derProben mit Mineralöl. Die Proben wurden im Temperaturzykler wie folgt inkubiert:
Verwendetes Temperaturprofil
Zyklen Zeit / Temperaturen
1 5 min 94 °C15 10 sec 94 °C / 40 sec 55 °C / 1 min 68 °C15 10 sec 94 °C1 4 °C
Nach der PCR wurde der Inhalt der vier Einzelgefäße pro Probe wiederzusammengeführt, und der Überstand nach 30 sec im Magnetständer von denMagnetpartikeln getrennt. Die Fällung der DNA aus dem Überstand erfolgte mit 100µl 95 %igem Ethanol und 2 µl 4 M Ammoniumazetat für 30 min auf Eis. Durch 20minütige Zentrifugation bei 14000 Upm und 4 °C konnte das DNA-Präzipitatgewonnen werden. Nach 10 minütigem Waschen mit 70 %igem und –20 °C kaltemEthanol bei 14000 Upm wurde der Ethanol abgezogen. Das Präzipitat wurdeluftgetrocknet. Die gelelektrophoretische Auftrennung und die Analyse erfolgte aufeinem Applied Biosystems 373A Sequenator im Institut für Zellbiochemie(Universitäts-Krankenhaus-Eppendorf).
12.3. Auswertung der Sequenzen
Die vom Institut für Zellbiochemie sowohl auf Diskette als auch als farbiger Ausdruckerhaltenen Sequenzen wurden per Hand vom Ausdruck gelesen und mit der bekanntenSequenz des Protein VII einschließlich des Leserahmens des Vektors pQE 30verglichen.Dieser Vergleich ergab eine Punktmutation im Bereich des Leserahmens des VektorspQE 30, die sich auch in darauf folgenden Sequenzierungen bestätigte.
13. Transformation des Klonierungsproduktes in M15
13.1. Vorbereitungen
Es wurden LB-Agarplatten mit einer Konzentration von 100 µg/ml Ampicillin und 25µg/ml Kanamycin gegossen. Hierzu wurden die angegebenen Bestandteileautoklaviert, Ampicillin und Kanamycin nach Abkühlung auf ca. 50 °C hinzugegebenund in 25 cm2 Schalen gegossen.Des weiteren wurde Psi-broth-Medium hergestellt.Für die Präparation der kompetenten E. coli Stränge M15[pREP4] (Villarejo undZabin, 1974) bzw. SG13009 (Gottesmann et al.,1981) wurden zunächst LB-Agarplatten mit einer Konzentration von 25 µg/ml Kanamycin gegossen – Vorgehen

56
wie oben beschrieben. Die Bakterienstämme wurden ausplattiert und über Nacht bei37 °C inkubiert. Anschließend wurden einzelne Klone isoliert und zunächst in 1 mldes kanamycinhaltigen LB-Mediums inokuliert. Danach wurde die 1 ml Bakte-rienkultur in 100 ml LB-Medium mit 25 µg/ml Kanamycin bei37 °C im Brutschrank-Schüttler inkubiert, bis eine Dichte (OD600) von 0,5 erreichtwurde. Die Bakterienkultur wurde für 5 min bei 2000 Upm und 4 °C in sterilenReaktionsröhrchen zentrifugiert. Der Überstand wurde entfernt und das Sediment inkaltem (4 °C) TFB1-Puffer (30 ml/100 ml Kultur) resuspendiert und für 90 min aufEis gestellt. Dieses Gemisch wurde erneut für 5 min bei 2000 Upm und 4 °Czentrifugiert und der Überstand entfernt. Das Sediment wurde in eiskaltem TFB2-Puffer (4 ml/100 ml Kultur) resuspendiert und in 500 µl-Portionen in sterilen Reak-tionsröhrchen als Dauerkulturen angelegt. Für die eigentliche Transformationverwendete ich nur die M15-Stränge.
13.2. Durchführung
Die Durchführung erfolgte prinzipiell nach dem gleichen unter Kapitel 10aufgeführtem Schema, lediglich einige Parameter bzw. Medien wurden entsprechendden Erfordernissen der anderen Zellinien von E. coli angepaßt. Es wurden 125 µl derZellen je Probe in ein 5 ml-Reaktionsröhrchen zu dem gesamten Ligationsproduktgegeben, gemischt und für 20 min auf Eis gestellt. Es folgte ein Hitzeimpuls bei 42 °Cfür 90 sec im Wasserbad. Anschließend wurden je Probe 500 µ l auf 42 °Cvorgewärmtes Psi-Broth-Medium hinzugegeben und für 90 min bei 37 °C und 250Upm im Brutschrank geschüttelt. Nach der Inkubation wurden je zwei Platten proProbe mit 100 µl bzw. 400 µl des Transformationsansatzes bestrichen. Die Plattenwurden kurz unter dem Abzug getrocknet und dann für ca. 18 h bei 37 °C umgedrehtim Brutschrank inkubiert. Als Negativkontrolle wurde TNE-Puffer anstelle desLigationsmixes eingesetzt, als Positivkontrolle 1 ng (entsprechend 2 µl auf 18 µl TNE-Puffer) des Plasmids pDHFRS-(0/0)-6xHis.
14. Probe-Expression des Protein VII in M15 (Small-scale Expression)
14.1. Prinzip
Die Expression erfolgte anhand des QIAGEN-Protokolls The QIAexpressionist, dasauf der Ligation zwischen dem zu exprimierenden Protein bzw. seiner DNA undeinem Expressionsplasmid beruht, welches an seinem N- oder C-Ende einen Anhangbestehend aus sechs Histidin-Resten enthält (6 x His-tag). Desweiteren enthalten diezu verwendenden E. coli Stränge (M15) ein Repressor-Plasmid (pREP4) – es kodiertden lac-Repressor -, welches an das Promotor/Operator-Element N250PSN250P29 (A)(M.Lanzer and H. Bujard, unpublished) des Plasmids bindet, das die Expressionsteuert. Die Promotor-Aktivität kann durch die Zugabe von IPTG wiederhergestelltwerden, das den Repressor inaktiviert und den Promotor reinigt. Außerdem enthält das

57
Expressionsplasmid eine ribosomale Bindungsstelle (RBS II) (B) der 5´-nicht-übersetzten-RNA, welche eine optimale Wiedererkennung und Bindung gewährleistet,den Transkriptions-Terminator „to“ (C) des Phagen λ (Schwarz et al., 1987), der dieFreigabe der RNA-Polymerase kontrolliert, das Promotor-freie Gen für dieChloramphenicol-Acetyltransferase (D) mit ihren eigenen Translationssignalen(Marcoli et al., 1980), den Transkriptions-Terminator T1 des E. coli-rrnB-Operon (E)(Brosius et al., 1981) und den Replikationsort und das Gen für die β-Lactamase desPlasmids pBR322 (Sutcliffe 1979), welche verändert wurde durch die Zerstörung derSpaltungsstellen der Restriktionsendonukleasen HincII und PstI, ohne daß dieAminosäuresequenz selbst beeinträchtigt wurde. Es gibt vier Gruppen von Plasmidenfür die Expression von Proteinen, für diesen Versuch wurde das Modell Nr. IVgewählt, welches den 6 x His tag am N-Ende enthält, d.h. 6 x His – Protein. Die Probe-Expression soll als Kontrolle dienen, ob Protein exprimiert wurde oder nicht. Um einZeitdiagramm der Expression zu erstellen, wurden zu den Zeitpunkten t(h) 0,1, 2, 3und 4 jeweils 1 ml der Proben entnommen.
Abb. 18: pQE 30 – Schnittstellenkarte.
14.2. Vorbereitung
Es wurde L-Broth-Medium, wie in Kapitel 12 beschrieben, vorbereitet, welches 100µg/ml Ampicillin und 25 µg/ml Kanamycin enthielt. Desweiteren wurde ein 12,5%iges Polyacrylamidgel vorbereitet (siehe Kapitel 14).
14.3. Durchführung
Es wurden einzelne Klone der Transformation (Kapitel 12) in 1,5 ml Mediumaufgenommen. Ebenso wurde mit dem Kontroll-Plasmid pQE16 verfahren, welchesDHFR exprimiert. Ein Klon wurde als nicht-induzierte (Negativ-) Kontrolle belassen.Diese Kulturen wurden für 18 h bei 37 °C im Brutschrank inkubiert. Jeweils 1,5 mlvorgewärmtes Medium wurde mit 500 µl dieser Kulturen beimpft und für 30 min bei37 °C im Brutschrank schüttelnd inkubiert, bis die Kulturen eine Dichte (A600) von0,7-0,9 erreicht hatten. Dieser Vorgang gewährleistete, daß alle Kulturen eine fastgleiche Dichte vor Beginn der Induktion besaßen. Die Expression wurde durch Zugabevon IPTG (finale Konzentration: 2 mM) induziert, mit Ausnahme natürlich derNegativkontrolle. Die Kulturen wurden weitere 5 h bei 37 °C im Brutschrank

58
inkubiert, wobei jede Stunde 1 ml einer Probe entnommen wurde, um dieExpressionsrate zu bestimmen. Anschließend wurden die Proben für 3 min bei 3000Upm und Raumtemperatur zentrifugiert und der Überstand entfernt. Das Sedimentwurde in 200 µl B-Puffer (8 M Harnstoff, 0,1 M Na-Phosphat, 0,01 M Tris-HCl (pH8,0)) resuspendiert und die Lösung solange gemischt, bis sie klar war. Das Lysatwurde für 10 min bei 15000 Upm und Raumtemperatur zentrifugiert, um dieZellüberreste zu präzipitieren, der Überstand wurde in ein frisches Reaktionsröhrchenüberführt. Hierzu wurden 50 µl des Ni-NTA-Harzes gegeben, der an den His-Rest amexprimierten Protein bindet, und so die Reinigung über die Ni-NTA-Säulenermöglicht, und für 30 min bei Raumtemperatur gemischt. Hieran schloß sich eineZentrifugation für 10 sec bei 15000 Upm, um das Gemisch zu präzipitieren, derÜberstand wurde verworfen. Das Präzipitat wurde dreimal mit1 ml C-Puffer ( 8 M Harnstoff, 0,1 M Na-Phosphat, 0,01 M Tris-HCl (pH 6,3))gewaschen. Danach wurden jeder Probe 20 µl C-Puffer/100 mM EDTA zugegebenund für 2 min unter Mischen bei Raumtemperatur inkubiert. Das EDTA bindet dieNi2+-Ionen des NTA-Harzes und wäscht so das Protein aus. Die Proben wurdenwiederum für 10 sec bei 15000 Upm und Raumtemperatur zentrifugiert und 20 µl desÜberstandes in ein neues Reaktionsröhrchen überführt. Hieran schloß sich dieElektrophorese auf einem Polyacrylamidgel.
15. Polyacrylamidgele (SDS-PAGE) zur quantitativen Analyse derExpression
15.1. Prinzip
Mit Hilfe des Polyacrylamidgels kann das Proteinprodukt des klonierten DNA-Fragmentes elektrophoretisch sichtbargemacht werden (SDS-Page). Diese enthaltenHarnstoff, ein Denaturierungsmittel, das die Effekte der DNA-Sekundärstrukturverringert. Außerdem sind diese Gele sehr dünn (0,5 mm oder weniger) und laufen beieiner so großen Spannung, daß sie sich auf60 – 70 °C aufheizen, was weiterhin zur Aufrechterhaltung der denaturierendenBedingungen beiträgt. Hier wurde ein 12,5 %iges SDS-Polyacrylamidgel verwendet(Ameis et al.,1991).
15.2. Vorbereitung
Zwischen zwei Glasscheiben wurde zunächst das 4 %ige Sammelgel gegossen (4 %Acrylamid/0,107 % PDA; 0,125 M Tris-HCl (pH 6,8); 0,2 % SDS; =,0375 %Ammoniumpersulfat; 0,125 % (v/v) TEMED) (Takacs, 1979), welches dazu dient , dieProben auf eine schmale Lauffront zu bringen. Daran schloß sich das 12,5 %igeTrenngel an (12,5 % Acrylamid/0,334 % PDA; 0,375 M Tris-HCl (pH 8,8); 0,2 %SDS; 0,0625 % (v/v) TEMED) (Takacs, 1979), in dem die Proben nach Größe getrenntwurden. Zu beachten war bei der Zubereitung, daß man die Ansätze für die Gele ca. 10

59
min entgasen ließ, bevor TEMED dazugegeben werden konnte. Kathoden- undAnodenpuffer bestanden aus 50 mM Tris-HCl (pH 8,3), 380 mM Glyzin, 0,2 % SDS.
15.3. Durchführung
Die Proben wurden bei 95 °C für 7 min in 5x PAGE-Probenpuffer denaturiert (15 %β-Mercaptoethanol; 15 % SDS; 1,5 % Bromphenolblau; 50 % Glyzerin). Pro Fachwurden 21 µl der Proben geladen, das Gel lief bei 200 V für ca. 2h. Das Gel wurde mitder Coomassie-blue-Methode gefärbt (siehe Kapitel 15, Abschnitt 4).
15.4. Coomassie-Blue-Färbung
Nach der Elektrophorese wurde das Gel für 30 min in einer Lösung bestehend aus 40% Ethanol und 10 % Eisessig entfärbt und anschließend für 30 min in 50 % Methanolfixiert. Für die Färbung wurde das Gel für 60 min in Coomassie Lösung (0,1 %Coomassie Brillant Blue R250, 40 % Methanol, 10 % Essigsäure) gefärbt.Anschließend wurde es dreimal mit Wasser gewaschen und der überschüssigeFarbstoff mit Entfärbepuffer (40 % Methanol, 10 % Eisessig) entfernt (Chrambach etal.,1967).

60
C. ERGEBNISSE
Aufgabe der vorliegenden Dissertation war es, das Protein VII aus Adenovirus Typ 2mittels der Methode der Klonierung bzw. Subklonierung zu isolieren und inkompetenten E. coli-Stämmen zu exprimieren. Dieses Protein ist aufgrund seinerhohen Bindungskraft zu DNA, bedingt durch seinen hohen Gehalt an Arginin, undseiner Fähigkeit, in menschliche Zellen einzudringen, von besonderem Interesse fürdie Molekularbiologie, vor allem in dem Bereich der Gentherapie. Das PlasmidpJM17, welches die DNA des Adenovirus enthielt, wurde freundlicherweise von Dr.F. L. Graham (McMaster Universität, Ontario, Kanada) zur Verfügung gestellt.
1. Klonierung und Subklonierung von Protein VII
Zur Expression des isolierten Protein VII war es notwendig, dieses zunächst mit Hilfeder Polymerase-Kettenreaktion nach Isolierung aus pJM17 zu amplifizieren. Nacheiner Reinigung und dem Verdau durch Restriktionsendonukleasen wurde das ProteinVII mit einem Vektor ligiert, der ebenfalls mit denselben Restriktionsenzymen verdautworden war und zudem bestimmte Eigenschaften erfüllt, die ihn geeignet machen füreine gesteuerte Expression. Vor der endgültigen Expression wurde dasLigationsprodukt in Bakterienstämme transformiert, mit Hilfe bestehenderAntibiotikaresistenz selektioniert und anschließend sequenziert, um eine fehlerloseKlonierung gewährleisten zu können.
1.1. Isolierung aus pJM17 durch PCR
Nach großen Schwierigkeiten konnte die Sequenz für das Protein VII amplifiziertwerden; da die Menge sehr gering war – ca. 13 ng/µl – wurde diese DNA mit Hilfe derGelextraktion isoliert und gereinigt.
517 bp
M M
Abb. 19/ Photo 1: Protein VII nach Gelextraktion (ca. 20 ng); Marker (M): Boehringer VI

61
1.2. Verdau von pVII und pQE30 mit BamHI und HindIII
Beide Liganden wurden mit identischen Mengen an Restriktionsenzymen verdaut undim Anschluß daran elektrophoretisch aufgetrennt. Die Elektrophorese zeigte zwei klareBanden, einmal bei ~ 560 bp entsprechend der zu erwartenden Größe von Protein VII(557 bp inklusive Primern), und zum anderen bei ~ 3500 bp entsprechend der Größevon pQE30 (3462 bp).
Abb. 20/ Photo 2: Verdau von P VII (ca. 20 ng) und pQE30 (ca. 12 ng) mit BamHI undHindIII
1.3. Ligation von pVII und pQE30
Protein VII und pQE30 wurden über 24 Stunden einmal in einem Verhältnis 1:1 undauch 5:1 inkubiert. In der Aufnahme der Gelelektrophorese erkennt man eineschwache Bande bei ~ 4100 bp, was der zu erwartenden Größe des Ligationsproduktesentspricht, die bei der Probe der 1:1-Ligation deutlich stärker hervortritt. Danebenlassen sich noch zwei weitere Banden finden, eine bei ∼ 1200 bp, hier könnte es sicheventuell um zirkuläre DNA des Protein VII handeln, oder auch um Fraktionen desVektors pQE30. Eine zweite Bande sieht man bei ∼ 560 bp, was der DNA des ProteinVII entspricht. Trotz mehrfacher Versuche ist es nicht gelungen, die Ergebnisqualitätbzw. -quantität der Ligation zu optimieren, so daß mit diesen Produktenweitergearbeitet wurde.
560 bp
1200 bp
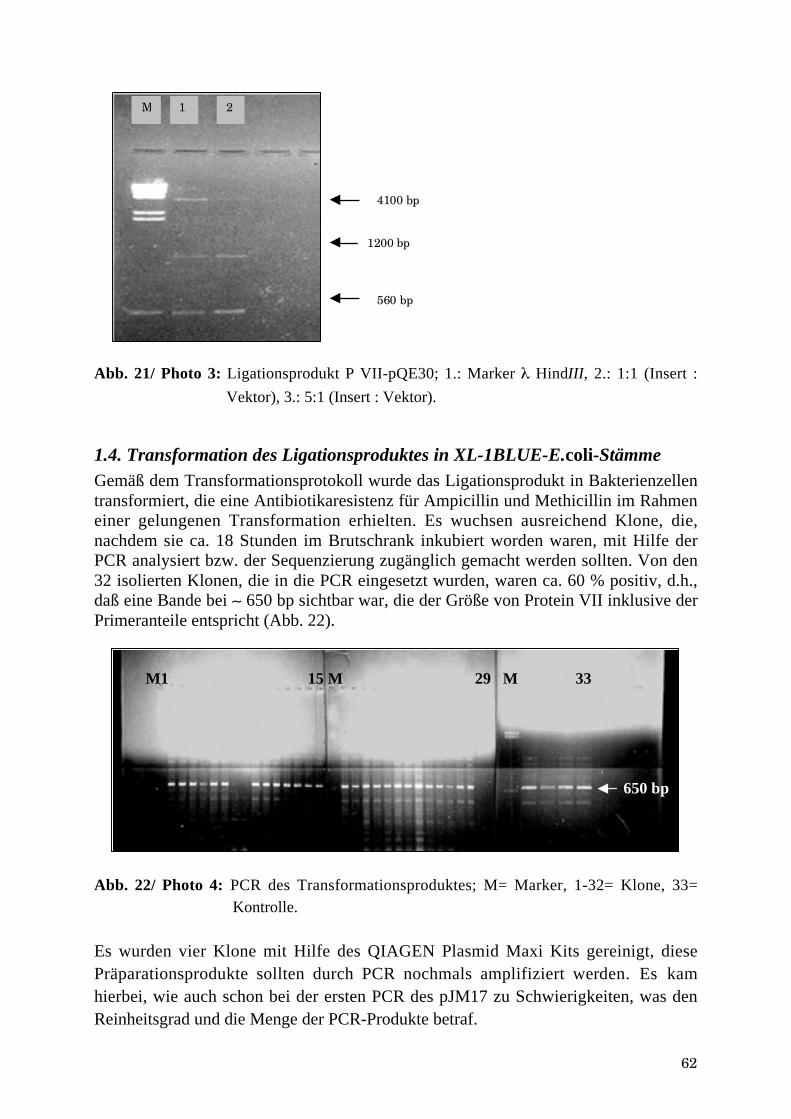
62
M 1 2
4100 bp
1200 bp
560 bp
Abb. 21/ Photo 3: Ligationsprodukt P VII-pQE30; 1.: Marker λ HindIII, 2.: 1:1 (Insert :
Vektor), 3.: 5:1 (Insert : Vektor).
1.4. Transformation des Ligationsproduktes in XL-1BLUE-E.coli-StämmeGemäß dem Transformationsprotokoll wurde das Ligationsprodukt in Bakterienzellentransformiert, die eine Antibiotikaresistenz für Ampicillin und Methicillin im Rahmeneiner gelungenen Transformation erhielten. Es wuchsen ausreichend Klone, die,nachdem sie ca. 18 Stunden im Brutschrank inkubiert worden waren, mit Hilfe derPCR analysiert bzw. der Sequenzierung zugänglich gemacht werden sollten. Von den32 isolierten Klonen, die in die PCR eingesetzt wurden, waren ca. 60 % positiv, d.h.,daß eine Bande bei ∼ 650 bp sichtbar war, die der Größe von Protein VII inklusive derPrimeranteile entspricht (Abb. 22).
M1 15 M 29 M 33
Abb. 22/ Photo 4: PCR des Transformationsproduktes; M= Marker, 1-32= Klone, 33=Kontrolle.
Es wurden vier Klone mit Hilfe des QIAGEN Plasmid Maxi Kits gereinigt, diesePräparationsprodukte sollten durch PCR nochmals amplifiziert werden. Es kamhierbei, wie auch schon bei der ersten PCR des pJM17 zu Schwierigkeiten, was denReinheitsgrad und die Menge der PCR-Produkte betraf.
650 bp

63
Es folgte eine Versuchsreihe zur Optimierung der PCR anhand des OPTI-PRIMEPCR-Optimization-Kits der Fa. Stratagene. Resultat dieser Versuchsreihe war, daß einpH-Wert des PCR-Puffers um 8,3 und eine Konzentration von Mg2Cl von 1,5 mMoptimal war, die KCl-Konzentration spielte keine so große Rolle. Desweiteren konnteeindeutig ein positiver Effekt durch den Zusatz von Glycerol oder DMSO bewiesenwerden (Abb.23 Photo 5). In den folgenden PCR-Untersuchungen wurde daraufhinimmer ein Zusatz von Glycerol 15 % verwendet.
Abb. 23/ Photo 5: M= Boehringer VII; 1-4= Probennummern; 5= Kontrolle.
2. Analyse der Sequenzierung
Die Sequenzierreaktion wurde mit dem PRISM Ready Reaction Kit der Fa. AppliedBiosystems durchgeführt. Als Kontrolle wurde die in dem Kit enthaltende DNA desPlasmids pGEM-3Zf(+) verwendet. Nach Alkoholfällung und anschließenderZentrifugation und Waschen der DNA bei Raumtemperatur wurde die zuuntersuchende DNA (Ausschnitt der in pQE30 ligierten DNA des Protein VII) inWasser gelöst. Die Analyse der Sequenzierung des Ligationsproduktes ergab einePunktmutation im Bereich des Leserahmens bei fehlerfrei abgelaufener Reaktion – diePositivkontrolle war fehlerfrei.
M 1 2 3 4 5 M

64
2.1. Mutation im Bereich des Leserahmens7
Die Sequenz beginnt mit der M13-Primer pQE30FM13 (oliv), es folgt die Sequenz desHistidinrestes, von BamHI und schließlich die des Protein VII. Abschließend folgt dieSequenz des biotinilierten Primers AdVII16464Ubio (oliv).
Tab 9: Sequenz des ausgeschnittenen Ligationsproduktes
TAANNNGGATAACAATTTTCACACAGAATTCATTAAAGAGGAGAAATTAACTATGAGAG
GATCTCACCATCACCATCACCATACGGATCCGCAAAGAAGCGCTCCGACCAACACCCAGTGCGCGTGCGCGGGCACTACCGCGCGCCCTGGGGCCACAAACGCGGCCGCACTGGGCGCACCACCGTCGATGACGCCATCGACGCGGTGGTGGAGGAGGCGCGCAACTACACGCCCACGCCGCCACCAGTGTCCACAGTGGACGCGGCCATTNNAACCGTGGTGCGCGGACCCGGCGCTATGCTAAAATGAAGAGACGGCGGAGGCGCGTANACGTCGCCACCGCCGNCGACCCGGCACTGCCGCCCAACGCGCGGCGGCGACCCTGCTTAACCGCGCACGTCGCACCGGCCGACGGGCGGCCATGCGGGCCGCTCGAAGGCTGGCCGCGGGTATTGTCACTGTGCCCCCCAGGTCCAGGCGACGAGCGGCCGCCGCAGCAGCCGCGGCCATTAATTGCTATGACTCAGGGTCGCAGGGGCAACGTGTATTTGGTGCGCGATTCGGTTANCGGCTTGCGCGTGCCCGTGCGCAACCGCCCCCCGCGNAANTAGTTGNNNNAGN
NAANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNT
2.2. Zusammenhang zwischen DNA-Sequenz von Protein VII und Mutation
Für diese Punktmutation im Bereich des Leserahmens können viele Ursachengefunden werden. Zum einen käme in Betracht, daß es aufgrund der hohenBindungskraft zwischen den einzelnen DNA-Strängen zu einer nur unzureichendenAblösung kommt, die wiederum zu einer fehlerhaften Amplifizierung führt. Insgesamtsprechen die vielen negativ verlaufenen Versuche einer PCR für diese Annahme.Weiter ist an eine Spontanmutation während der Ligation zu denken bzw. an eineDeletion einer Base während des Verdaus mit den Restriktionsendonukleasen. Da eshierbei um die Erzeugung von sog. klebrigen Enden ging, ist es durchaus möglich, daßwährend dieses Vorgangs eine Base deletiert wurde, aber dennoch eine Ligationmöglich war.
3. Expressionsversuch
Trotz der Mutation im Leserahmen, die im wesentlichen die Region der sechsHistidinreste betrifft, die erst für den Schritt der Reinigung relevant werden, wurde ein
7 grün: Startkodon; hellgrün: Stopkodon; rosa Primer; rot: Mutationsstellen bzw. Deletionsstelle: T→G, C→T,AC→CAC; blau: Beginn von BamHI; fett: Protein VII. Die Folge CAC CAT usw. steht für die sechs Histidin-Reste. Aufgrund der zweiten Mutation ist es zu einer Verschiebung der Reihenfolge der einzelnen Triplets, diefür diesen Histidinrest kodieren. Resultat ist, daß zwar für 6 x Histidin kodiert wird, – das siebte Triplet bleibtunvollständig (AC) – aber statt CAC CAT am Ende dieses Histidinrestes steht (eigentliche Sequenz: 5´ GATCCAT CAC CAT CAC CAT CAC G 3´).
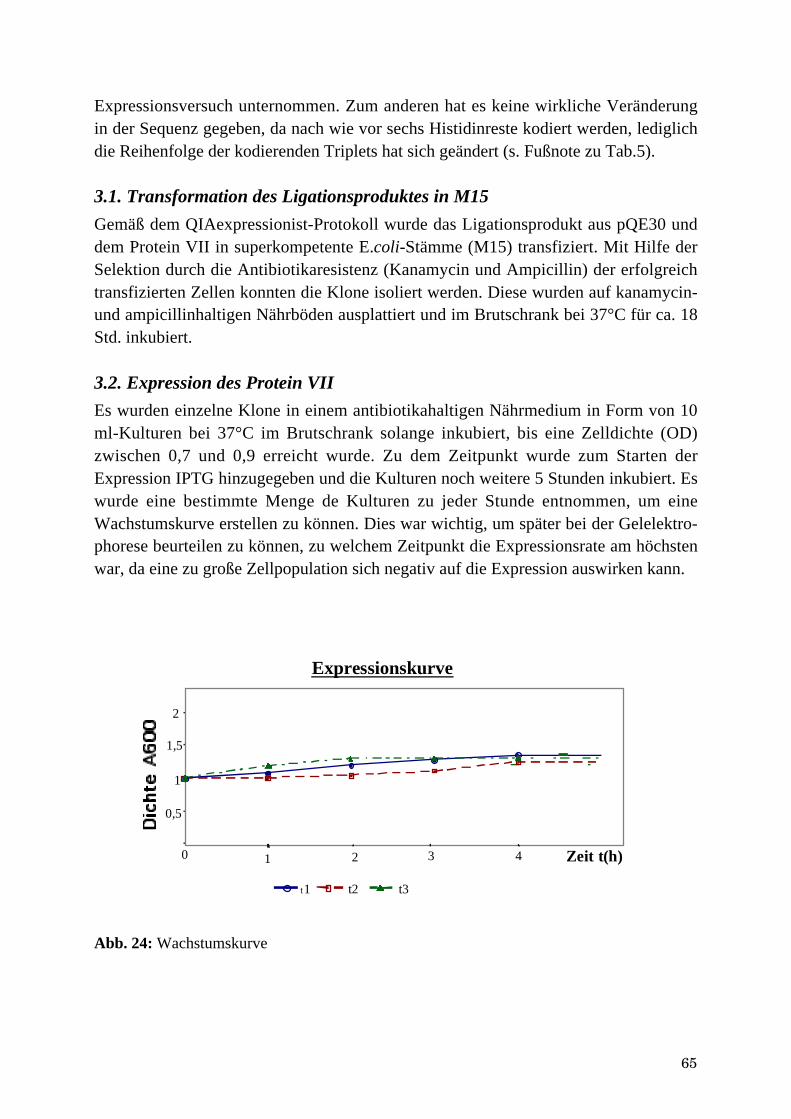
65
Expressionsversuch unternommen. Zum anderen hat es keine wirkliche Veränderungin der Sequenz gegeben, da nach wie vor sechs Histidinreste kodiert werden, lediglichdie Reihenfolge der kodierenden Triplets hat sich geändert (s. Fußnote zu Tab.5).
3.1. Transformation des Ligationsproduktes in M15
Gemäß dem QIAexpressionist-Protokoll wurde das Ligationsprodukt aus pQE30 unddem Protein VII in superkompetente E.coli-Stämme (M15) transfiziert. Mit Hilfe derSelektion durch die Antibiotikaresistenz (Kanamycin und Ampicillin) der erfolgreichtransfizierten Zellen konnten die Klone isoliert werden. Diese wurden auf kanamycin-und ampicillinhaltigen Nährböden ausplattiert und im Brutschrank bei 37°C für ca. 18Std. inkubiert.
3.2. Expression des Protein VII
Es wurden einzelne Klone in einem antibiotikahaltigen Nährmedium in Form von 10ml-Kulturen bei 37°C im Brutschrank solange inkubiert, bis eine Zelldichte (OD)zwischen 0,7 und 0,9 erreicht wurde. Zu dem Zeitpunkt wurde zum Starten derExpression IPTG hinzugegeben und die Kulturen noch weitere 5 Stunden inkubiert. Eswurde eine bestimmte Menge de Kulturen zu jeder Stunde entnommen, um eineWachstumskurve erstellen zu können. Dies war wichtig, um später bei der Gelelektro-phorese beurteilen zu können, zu welchem Zeitpunkt die Expressionsrate am höchstenwar, da eine zu große Zellpopulation sich negativ auf die Expression auswirken kann.
Expressionskurve
0,5
1
1,5
2
Zeit t(h)
t1 t2 t3
0 1 2 3 4
Abb. 24: Wachstumskurve

66
3.3. Ergebnis der Expression
Mit Hilfe der Elektrophorese auf einem Polyacrylamidgel und der anschließendenFärbung des Gels sollte das exprimierte Protein VII sichtbar gemacht werden. Dochtrotz wiederholter Expressionsversuche kam es zu keinem Zeitpunkt zu einerExpression. Man muß davon ausgehen, daß die Mutation bzw. Deletion im Bereichdes Leserahmens doch von essentieller Bedeutung für die Expression ist. Es kam zwarzu einer ausreichend wachsenden Dichte der Zellpopulation, jedoch konnte nachInduktion in keinem Fall eine Expression des Protein VII nachgewiesen werden.

67
D. DISKUSSION
1. Das adenovirale Vektormodell
Das adenovirale Vektormodell hat sich bis jetzt als leistungsfähige Methode zurÜbertragung und Expression verschiedenster DNA in vitro erwiesen. Ein wichtigesProblem, das bei dem Gentranfer bzw. bei der Fremdexpression in vivo mit Hilfe desAdenovirus als solchem auftrat, war die Immunreaktion des Wirtes (Yang et al.,1994). Durch diese Immunabwehr wird die Effektivität des Gentransfers und damit derFremdexpressionsrate gemindert, die transformierte DNA „überlebt“ nicht langegenug in der Wirtszelle, um eine wirklich dauerhafte Therapie gewährleisten zukönnen. Diese Immunreaktion kann zu einer vorzeitigen Eliminierung derrekombinanten DNA führen (Yang et al., 1994), was auf eine erhöhte Produktion vonTumor-Nekrosefaktor (TNF-α) zurückgeführt werden konnte, welche ihrerseits durchdie Infektion mit Adenovirus induziert wurde. Dieser Vorgang wird so erklärt, daßTNF-α zu einer Erhöhung der Expression eines 19 kD Glykoproteins (gp19k) führt,welches in der E3-Region des Adenovirus kodiert wird (Korner et al., 1992). DiesesProtein behindert den Transport von MHC I-Molekülen an die Zelloberfläche; das istjedoch eine entscheidende Bedingung für das Erkennen adenoviraler Proteine durchzytotoxische Zellen (Cox et al., 1990; Williams et al., 1990). Durch Integration desGens für gp19k in das Genom eines E3-deletierten adenoviralen Vektors konnte diezytotoxische Immunreaktion der Wirtszelle reduziert werden (Lee et al., 1995).Desweiteren bewahren andere Genprodukte der E3-Region die infizierten Zellen vordem zytolytischen Effekt des TNF-α (Gooding et al., 1991), so daß man sagen kann,daß die E3-Region eine protektive Wirkung auf die infizierten Zellen bezüglich derzytotoxischen Reaktionen der Wirtszellen hat und somit Lebensdauer des zutransferierenden rekombinanten Gens erhöht (Smith et al., 1993; Poller et al., 1996).Es gelang, das adenovirale Genom dahingehend zu verändern, daß dessen eigeneMöglichkeiten, die Immunabwehr der Wirtszelle abzuschwächen, durch Integrationfremder DNA gestärkt werden konnte (Wold et al., 1991; Lee et al., 1995).Dahingegen bieten adenovirale Vektorsysteme einige Vorteile gegenüber anderenMethoden des Gentransfers. So z.B. deren Unabhängigkeit von derWirtszellproliferation zur Fremdgenexpression, wie das unter anderem bei retroviralenVektoren gefordert ist, was bis dahin ein Hauptproblem bei der Verwendung vonVektorsystemen war (Rosenfeld et al., 1991).Um den Sicherheitsstandards zu genügen, werden für den Gentransfer meist nurreplikationsdefiziente adenovirale Vektoren verwendet. Es wurde bei diesen Vektorendie für die Replikation essentielle E1-Region ausgeschaltet, so daß es in der Wirtszellenicht mehr zu einer unkontrollierten Vermehrung des Vektors kommen kann (Ali etal., 1994). Dieses Risiko kann man jedoch in bezug auf einen therapeutischenGentransfer beim Menschen nicht verantworten. Die Replikationsdefizienz führtjedoch, wie mehrere Versuche zeigten, zu einer verminderten Expressionsrate des

68
rekombinanten Proteins. Hingegen konnte gezeigt werden, daß die Lebensdauer derWirtszelle im Gegensatz zu replikationsfähigen Vektoren nicht verkürzt war (Merklein1998).Ein weiterer Vorteil der adenoviralen Vektorsystem liegt in seinem weitenWirtsspektrum; so konnten bisher die unterschiedlichsten Organe auf ihreInfektionsfähigkeit durch Adenoviren untersucht werden, wie z.B. Muskulatur undLeber (Stratford-Perricaudet et al., 1990 und 1992), Lunge (Rosenfeld et al., 1992),Niere (Moullier et al., 1994) und Gehirn (Akli et al., 1993; LeGal-LaSalle et al.,1993). Es konnten auch schon in jüngerer Zeit Tumorerkrankungen mit Hilfe vonadenoviralem Gentransfer behandelt werden (Cordier et al., 1995).Die Effizienz des Adenovirus-vermittelten Gentransfers ist proportional zu dereingesetzten Vektordosis, d.h., daß die Genexpression nach Transfer bei hohenVektordosen besonders effizient ist. Auch bei diesen hohen Dosen konntelichtmikroskopisch keine Schädigung der Wirtszellen festgestellt werden. Zubemerken wäre weiterhin, daß sich die meisten adenoviralen Vektoren auf AdenovirusTyp 2 oder 5 zurückführen lassen, für die kein onkogenes Potential bekannt ist (Ali etal., 1994); Adenoviren mit einer solchen Potenz – z.B. Typ 12, 18 und 31 (Horwitz1990) – integrieren ihr Genom in das der Wirtszelle (Doerfler et al., 1995), was auf dieTypen 2 und 5 nicht zutrifft.
2. Probleme des adenoviralen Vektormodells
Da es in den meisten Fällen nicht zu einer Integration der Virus-DNA in dasWirtsgenom kommt, ist natürlich die Stabilität der Expression auf nur wenige Wochenbegrenzt. Dies zusammen mit der Immunabwehr der Wirtszelle schränkt diePraktikabilität dieser Gentransfer-Methode doch erheblich ein. Ein weiterer Aspekt,der die Anwendungsbreite der klassischen adenoviralen Vektorsysteme betrifft, liegtdarin, daß es immer schwer ist, Patienten davon zu überzeugen, sich mit „infektiösem“Material, in diesem Fall mit Adenoviren behandeln zu lassen. Die Angst in derBevölkerung – und auch in den einzelnen Ethikkommissionen – ist doch zu groß, alsdaß man sich vorstellen könnte, daß ein solches Verfahren zu einer Routinemethodewerden kann. Bisher blieb es nur den sog. hoffnungslosen Fällen vorbehalten, beidenen eine jede Infektionsgefahr immer in Kontrast zu dem sonst bald zu erwartendenTod stand.
3. Ein Protein als Transfermolekül für DNA – Modifizierung desadenoviralen VektormodellsAufgrund dieser oben genannten Problematik, wie Infektionsgefahr, immunologischeAbwehrreaktion und zu geringe Expressionsdauer, wurden einige Veränderungen andiesem Modell vorgenommen.Der vorliegenden Arbeit liegt die Überlegung zu Grunde, anstelle des gesamten Virus– abgesehen von der Eliminierung der E1-Region – nur dasjenige Protein zu

69
transferieren, welches die Fähigkeit hat, in die Wirtszelle einzudringen, und darüberhinaus noch eine hohe Affinität zu DNA hat; also das „perfekte“ Transportermolekülfür DNA. Ein solches Protein hätte verschiedene Vorteile gegenüber dem gesamtenVirus: zum einen wäre eine Infektion ausgeschlossen, da nur ein isoliertes Proteinübertragen werden würde. Ebenso ist keine immunologische Abwehrreaktion zuerwarten, die die Überlebenszeit der rekombinanten DNA verkürzen könnte. Auch dieGefahr einer onkogenen Potenz wäre gleich null gesetzt. Ein Protein des Adenovirusdas die erforderten Eigenschaften erfüllt, ist das sog. maior core protein, das ProteinVII. Durch seinen hohen Gehalt an Arginin besitzt dieses Protein eine hohe Affinitätzu der negativ geladenen DNA. Außerdem besitzt es die Fähigkeit, in Zelleneinzudringen, es hat eine sog. Schlüsselrolle im Infektionsweg der Adenoviren. Somitstellt das Protein VII den idealen Transporter für DNA in humane Zellen dar.
4. Grundlegendes zur Verwendung von Virusproteinen alsTransportermoleküle
Das Protein VII wurde schon frühzeitig als mögliches Vehikel für DNA im Rahmeneiner Gentherapie entdeckt. Jedoch war die Isolierung bzw. die Herstellung von einergenügenden Menge dieses Proteines sehr umständlich. Eine entsprechend großeAnzahl von Adenoviren mußte spezifisch behandelt werden, um aus jedem einzelnendas Protein VII isolieren zu können.
4.1. Ziel dieser Arbeit
Die Überlegungen, die die Grundlage dieser Arbeit bildeten, bezogen sich auf ebendiese herkömmlichen Verfahren zur Isolierung des Protein VII aus den Viren. Es wardie Idee, die kodierende Sequenz für das Protein mit Hilfe der Polymerase-Kettenreaktion zu amplifizieren, dieses Produkt mittels eines Expressionsvektors inBakterienzellen zu transferieren, in denen dann das Protein in hoher Anzahl exprimiertwird. Dieses Verfahren würde den Einsatz von Protein VII für den Gentransfer inhohem Maße erleichtern, da man neben der Vereinfachung, eine große Menge desProteins zu erhalten, also einer höheren Quantität, auch eine Stabilität bezüglich derQualität garantieren könnte. Die Expression von Proteinen in Bakterienzellen ist einebewährte und schon viel eingesetzte Methode.
4.2. Das Expressionsmodell
Voraussetzung für die Expression von Fremdgenen in Bakterienzelle wie z.B. E.coliist der Einbau in einen geeigneten Expressionsvektor, der unter dem Einfluß vonE.coli-Expressionssignalen steht. Hierbei ist zum einen die stärke des Promotors vonBedeutung, denn er steuert die Effizienz der Expression durch eine hoheTranskriptionsgeschwindigkeit. Außerdem muß man bei der Konstruktion einesExpressionsvektors noch die Art der Steuerungsfähigkeit bedenken; hier gibt es zweiMöglichkeiten: Induktion und Repression, d.h. durch Zugabe einer bestimmten

70
chemischen Verbindung wird die Transkription entweder ein- oder abgeschaltet. EineRegulationsmöglichkeit der Expression ist in jedem Fall sehr wichtig, da einigeProteine eine schädliche Wirkung auf die Bakterien haben können, und man soverhindern kann, daß dieses sich in einer toxischen Konzentration in derBakterienzelle ansammelt. Abgesehen davon kann eine dauerhaft starke Transkriptiondie Replikationsfähigkeit der rekombinanten DNA nachteilig beeinflussen.In diesem Versuch wurde das ein starker und gut steuerbarer Promoter vomInduktionstyp gewählt, nämlich der lac-Promotor. Diese Sequenz reguliert dieTranskription der β-Galaktosidase, für die das lacZ-Gen kodiert – M13-Vektorentragen hiervon nur einen Abschnitt, das lacZ´-Genfragment. Dieser Promotor wird vonIPTG induziert, wird dieses dem Nährmedium zugesetzt, so wird die Expressionangeschaltet (siehe Abb. 25).
Der lac-Promotor
-35 -10
lac Z IPTG
Transkription
Abb. 25: Der lac-Promotor (aus: Brown TA)
Weitere für die Transkription bzw. Translation wichtige Signale sind zum einen derTerminator, der das Ende des zu transkribierenden Gens beschreibt, und dieRibosomenbindungsstelle, an der das mRNA-Molekül anheften soll.Expressionsvektoren, die alle diese Signale (Promotor, Terminator undRibosomenbindungsstelle) enthalten, nennt man Kassettenvektoren, da sie diese inForm einer Kassette enthalten sind, in die die rekombinante DNA in eine nur einmalvorhandene Restriktionsstelle eingebaut wird. In dieser Arbeit wurde ein solcherKassettenvektor pQE30 verwendet, in dessen einmalig vorkommendenRestriktionstellen BamHI und HindIII die Sequenz des Protein VII eingebaut wurde.
4.3. Probleme bei der Proteinexpression
Trotz der großen Fortschritte und der Optimierung bei der Herstellung vonKassettenvektoren gibt es immer noch zahlreiche Schwierigkeiten bei der Expressionvon Fremdgenen in großem Maßstab:
• bei der Transkription: die fremde DNA kann Sequenzen enthalten, die in denBakterienzellen als Terminatoren wirken und somit die Expression vorzeitigbeenden.
• zwischen Transkription und Translation: es ist essentiell vor Einbau der fremdenDNA deren Introns zu entfernen, da diese von den Bakterienzellen nicht aus dem

71
Transkript herausgeschnitten werden können. Dies ist allerdings nur möglich, wennman über eine cDNA verfügt, die mit Hilfe von mRNA konstruiert wurde unddeshalb keine Introns enthält.
• bei der Translation: Es gibt für eine Aminosäure meist mehrere Codons, mannennt dies Codonnutzung. Es kommt aber häufig vor, daß bestimmte möglicheCodons von der Transfer-RNA (tRNA) bevorzugt werden. Dies spiegelt sich ineiner Überzahl der entsprechenden tRNA wider; enthält aber die rekombinanteDNA genau von diesen wenig genutzten Codons mehr, so kann die Translationbehindert sein durch die zu geringe Anzahl der erforderlichen tRNA.
• nach der Translation: hier existieren zwei Probleme, zum einen kann es zu einemschnellen Abbau durch Proteasen, die Fremdgene erkennen und abbauen; dies kanndurch veränderte E.coli-Stämme verhindert werden, denen diese Proteasen fehlen.Zum anderen durch die Unterschiede in der Weiterverarbeitung (processing);meistens werden Proteine nach der Translation durch Anfügen von chemischenGruppen o.ä. weiterverarbeitet, was für ihre Funktion essentiell ist Dies verläuft beiden einzelnen Organismen auf unterschiedliche Art und Weise, so daß eineVeränderung hier die Funktionsfähigkeit des Proteins erheblich einschränken kann.
• in der Zelle: ein exprimiertes Protein kann die Wirtszelle auf verschiede Weiseschädigen. Zum einen durch Toxizität, zum anderen durch einen hohen Energie-und Materialverbrauch, der das Bakterium hinsichtlich seinerWachstumsgeschwindigkeit und seiner Zellstabilität im Gegensatz zu den nichttransfizierten Zellen benachteiligt, so daß die Rekombinanten in einer Kultur leichtüberwuchert werden können, und so das protein nicht mehr exprimiert wird.
Bei der vorliegenden Arbeit stehen mehrere Probleme im Vordergrund: erstens dieSchwierigkeiten bei dem Versuch der Amplifizierung mittels der Polymerase-Kettenreaktion als einer Voraussetzung für die Klonierung,. Zweitens die hoheBindungkraft der DNA-Stränge aufgrund des hohen Gehaltes an Arginin. Drittenswahrscheinlich die verschiedene Codonnutzung bezüglich der Histidinreste, die hiervor allem durch CAT und CAC codiert wurden und viertens die Mutation imLeserahmen, die allerdings kein Problem der Expression im eigentlichen Sinne ist,sondern aus der Beschaffenheit der DNA resultiert.
5. Vergleich mit anderen Modellen für die Fremdgenexpression
Die einfachsten Methoden, fremde DNA zu transferieren, sind chemischer oderphysikalischer Art, wie z.B. die Ca2+-Phosphat-Methode oder die Mikroinjektion,welche aber in vivo nur begrenzt einsatzfähig sind. So sind diese auch durch dieEntdeckung und Nutzbarmachung viraler Vektoren stark verdrängt worden; hierwiederum stehen vor allem retrovirale und adenovirale Vektoren im Vordergrund.

72
5.1. Retrovirale Vektoren
Die Tatsache, daß Retroviren für so geeignete Vektoren erklärt, liegt darin begründet,daß diese ihr genetisches Material in das Wirtsgenom integrieren. Bei diesen Vektorenhandelt es sich um Retroviren, bei denen die Signalsequenzen des Virusgenoms für dieKapselbildung entfernt wurden; so produzieren diese Vektoren alle für dieVirusreplikation und -zusammensetzung notwendigen Proteine, ohne das Gefahrgelaufen wird, daß sich infektiöse Partikel bilden können.Retrovirale Vektoren bergen allerdings den Nachteil, daß sie auf sich replizierendeZellen angewiesen sind, deren Zahl sich in vivo stark begrenzt hält.
5.2. Adenovirale Vektoren
Wie schon weiter oben erwähnt haben adenovirale Vektoren ein ähnlich breitesWirtsspektrum wie Retroviren, sie sind jedoch nicht auf die Replikationsfähigkeit ihrerWirtszellen angewiesen, was ihren Einsatz in vivo besonders bevorzugt.Hier ist jedoch der Nachteil, daß diese Vektoren innerhalb der Wirtszelle nicht sehrstabil sind, und deshalb auch eine Expression durch sie nur von begrenzter Dauer ist.Dies läßt sich darauf zurückführen, daß Adenoviren ihr Genom nicht in das derWirtszelle integrieren.
6. Sicherheit und EthikWie bei jeder neuen klinischen Technik müssen auch bei der Gentechnik diemöglichen Risiken der Methode dem möglichen Erfolgen gegenübergestellt werden.Bezüglich der Verwendung von retroviralen Vektoren muß hier an die Möglichkeit derKontamination mit replikationskompetenten Viren gedacht werden. Bei adenoviralenVektoren muß man die onkogene Potenz einzelner Stämme bedenken, sowie an dieHerstellung replikationsdefizienter Viren. Beide Faktoren können zu einer Induktionbzw. Infektion mit folgendem Tumorwachstum führen. Es konnte erklärt werden, daßein replikationskompetentes Virus pathogen sein kann, wenn es eine chronischeVirämie bei einem immungeschädigten Wirt auslöst.Zu den Sicherheitsbestimmungen in der humanen Gentherapie kommen auch noch dieethischen Gesichtspunkte. Die Gentherapie wie sie heutzutage betrieben wird,behandelt ausschließlich Körperzellen und keine Keimzellen. Der Stellenwert dieserArt von Gentherapie ist ähnlich dem von Transplantationen oder anderenZellbehandlungsmethoden. So bleiben zwei Aspekte offen: einmal die Keimzell-Gentherapie und der Punkt, der eine Verstärkung der Technologie begrenzt. Was füreine Keimzell-Gentherapie spräche, wäre die Tatsache, daß viele genetische bedingteErkrankungen nur effektiv während der Embryogenese behandelt werden können. Aufder anderen Seite müssen die Sicherheitsstandards hoch angesetzt und eineAusweitung der Möglichkeiten der Keimzell-Gentherapie auf schwerwiegende, d.h.lebensbedrohliche Erkrankungen beschränkt bleiben.

73
Für die zukünftige Entwicklung der Gentherapie müssen drei Hindernisse überwundenwerden:
1. Die für den Gentransfer verwendeten Vektoren sollen direkt in den Patienteninjizierbar seien. Dies setzt ebenso voraus, daß der Vektor die Zielzelle erkenntund das entsprechende Organ penetrieren kann.
2. Befindet sich der Vektor in der Zielzelle, sollte er sicher an einerunproblematischen Stelle des Chromosoms integriert werden, oder das defizienteGen ersetzen. Dies würde die Gefahr einer malignen Entartung rapideherabsetzen. Bis jetzt konnte nur gezeigt werden, daß adenoassoziierte Viren inder Lage sind, ihr Genom in eine kleine Region des kurzen Arms vonChromosom 19 zu integrieren. Werden die biochemischen Mechanismen, die fürdiese Integration verantwortlich sind, von den adenoassoziierten Viren isoliert,könnte es möglich sein, diese mit den bisherigen Vektormodellen zukombinieren, und so deren Effizienz zu erhöhen.
3. Es sollte möglich werden, daß die induzierten Gene die Fähigkeit haben, aufphysiologische Veränderungen im Blut oder von zellulären Metaboliten zureagieren; so z.B. in der Gentherapie des Diabetes mellitus die stetigenVeränderungen des Blutzuckerspiegels.
Die Zukunft für die Gentherapie verspricht viel, und man soll hoffen, daß dieseTechnologie soweit ausreifen wird, daß eine große Bandbreite von Erkrankungen imnächsten Jahrhundert behandelt werden kann.

74
E. ZUSAMMENFASSUNG
Mit Hilfe der Entwicklungen auf dem Gebiet der Molekularbiologie und derBiochemie entstanden die Techniken wie Amplifikation, Ligation, Transfektion undExpression, mit denen in der vorliegenden Arbeit ein Protein aus dem Adenovirus Typ2 isoliert und in E.coli-Zellen exprimiert werden sollte. Adenoviren stellen eine guteVoraussetzung für den Transfer von Genen dar. Sie haben ein sehr breitesWirtsspektrum und sie benötigen keine sich replizierenden Zellen. Nachteile ergebensich durch die Tatsache, daß das Virusgenom nicht stabil in das Wirtsgenom integriertwird, und somit die Expressionszeit durch Elimination der Virus-DNA nach einigenZellzyklen beschränkt ist; ein weiteres Problem ergibt sich bei wiederholterAnwendung durch Bildung von Antikörpern gegen die Adenoviren. Ziel dieser Arbeitwar es zum einen, die oben genannten Probleme bei der Verwendung vonadenoviralen Vektoren dadurch zu umgehen, daß nicht das gesamte Virus, sonder nurein Protein als Vektor dient. Zum anderen, dieses Protein nicht auf herkömmlichenWege durch Züchtung großer Viruskulturen und Herausschneiden des Proteins ausdenselben zu isolieren, sondern es sollte versucht werden, die für das gesuchte Proteinkodierende Sequenz mit Hilfe der Polymerase-Kettenreaktion zu amplifizieren, dasProdukt mit einem vorbereiteten Vektor zu ligieren, dies wiederum in E.coli-Zellen zutransfizieren und so das Protein zu exprimieren. Diese adenovirale Protein faßt dieVorzüge de adenoviralen Vektorsystems zusammen, während die Nachteile ausge-klammert werden. Es handelt sich bei diesem um das Protein VII, einem sog.Kernprotein, mit einem Molekulargewicht von ca. 18.500 Dalton und einem hohenGehalt an Arginin (ca. 23 %) und Alanin (ca. 20 %). Dieses Protein liegt gebunden anDNA im Nukleokapsid. Aufgrund seines Aufbaus, hat das Protein VII die MöglichkeitDNA zu binden; außerdem konnte gezeigt werden, daß es die Zielzelle finden undpenetrieren kann. Demgegenüber besteht bei einem Gentransfer mit diesem Proteinnicht die Gefahr einer Immunreaktion und sie besitzen eine größere Stabilität, was derExpressionszeit zu gute kommt.Bei der Expression bzw. Amplifikation ergaben sich verschiedene Schwierigkeiten.Erstens konnte nur eine geringe Menge der DNA-Sequenz amplifiziert werden; es kamimmer wieder zu Störungen bei der Polymerase-Kettenreaktion in den verschiedenenPhasen des Experimentes. Zweitens ergab sich, daß durch die Klonierung der Protein-sequenz in den vorbereiteten Vektor eine Punktmutation im Bereich des Leserahmensentstanden war. Aufgrund der Probleme bei der Polymerase-Kettenreaktion konnte dieKlonierung leider nicht reproduziert werden. Die Expressionsversuche in den E.coli-Zellen waren ebenfalls negativ, was zum einen auf die Mutation, als auch auf generelleSchwierigkeiten bei Expressionen wie die Codonnutzung, der hohen Bindungskraft derDNA-Stränge und den daraus resultierenden Schwierigkeiten bei der Polymerase-Kettenreaktion zurückzuführen ist.Es ist also leider nicht gelungen, die für das Protein VII kodierende DNA mit Hilfevon Kassettenvektoren und Expressionszellen zu klonieren. Nichtsdestoweniger muß

75
die Forschung der Gentherapie dieses Protein im Auge behalten. Nicht nur, daß esviele Vorteile gegenüber den bisher verwendeten Gentransfermodellen hat, es birgtauch weniger Gefahren für den Empfänger, d.h. den zu behandelnden Patienten, dakeine Gefahr für Pathogenität geschweige denn Onkogenität besteht. Es berücksichtigtsomit alle wichtigen Aspekte der modernen Gentherapie: Effektivität, im Sinne vonQuantität und Qualität, Sicherheit und den ethischen Anspruch unserer Gesellschaft.

76
F. VERZEICHNIS DER ABKÜRZUNGEN
bp BasenpaarDa DaltonDMSO DimethylsulfoxidEDTA Ethylendiamin-TetraessigsäureNaCl NatriumchloridPBS phosphatgepufferte SalzlösungSDS Sodiumdodecyl-SulfatTEMED N,N,N’,N’-TetramethylethylendiaminTRIS tris[Hydroxymethyl]aminomethan

77
G. VERZEICHNIS DER ABBILDUNGEN UND TABELLEN
Abb.1: Schema der Adenovirusinfektion..................................................... 12Abb.2: Aufbau des Kapsids (aus Nermut und Perkins 1979).................... 14Abb.3: Gefriertrocknung des Avian-Adenovirus (aus Nermut 1980)........ 14Abb.4: Modell der Kernhülle (aus Nermut 1977)........................................ 14Abb.5: Negativfärbung von Adenovirus-Kapsomeren
(aus Nermut 1980a und Wrigley).....................................................15Abb.6: Hexon-Modell (aus Nermut und Perkins 1979)............................... 16Abb.7: Diagramm einer Fiber von Ad2 (aus Nermut 1984)........................17Abb.8: Adenoviruskerne – verschiedene Arten der
Präparation (Nermut 1984).............................................................. 18Abb.9: Modell der kontinuierlichen Helix (Nremut 1980a)........................ 23Abb.10: Diagrammförmige Darstellung des DNA-Verlaufes im
„Nukleosomen-Modell“ (Nermut 1984).............................................25Abb.11a+b: Superhelikaler Aufbau der Nukleosomen................................ 26Abb.12: Modell eines Stäbchenelementes der Nukleosomen
(Nermut 1984)..................................................................................26Abb.13a+b: Modell des Adenovirion (Nermut 1984)....................................27Abb.14: Schnittstellenkarte des pJM17....................................................... 28Abb.15: Aufbau und Schnittstellenkarte des Vektors pQE30.................... 29Abb.16: Polymerase-Kettenreaktion (Schema)............................................ 41Abb.17:DNA-Sequenz von Ad2......................................................................43Abb.18: pQE30-Schnittstellenkarte.............................................................. 57Abb.19: Photo 1: pVII nach Gelextraktion....................................................60Abb.20: Photo 2: Verdau von pVII und pQE30 mit
BamHI und HindIII......................................................................... 61Abb.21: Photo 3: Ligationsprodukt pVII-pQE30.......................................... 62Abb.22: Photo 4: PCR des Transformationsproduktes................................ 62Abb.23: Photo 5: PCR-Optimization............................................................. 63Abb.24: Expressionskurve............................................................................ 65Abb.25: Der lac-Promotor............................................................................. 69
Tab.1: Genetisch bedingte Erkrankungen (Strauss 1994).............................7Tab.2: Methoden des Gentransfers (Strauss 1994)........................................8Tab.3: Proteine des Adenovirus................................................................12/13Tab.4: DNA- und Proteingehalt pro Adnovirion (Nermut 1984)................ 25Tab.5: Sequenz von pQE30............................................................................ 30Tab.6: Synthetische Oligonukleotide für die
DNA-Amplifikation und Sequenzanalyse......................................... 42Tab.7: Erkennungssequenzen der verwendeten
Restriktionsendonukleasen...............................................................50Tab.8: Primer für die DNA-Sequenzbestimmung........................................54Tab.9: Sequenz des Ligationsproduktes.......................................................64

78
H. LITERATURVERZEICHNIS
Akusjävi G, Perrson H (1981) gene and mRNA for precursor polypeptide VI from adenovirustype 2. J Virol 38:469
Akli S, Caillaud C, Vigne E, Stratford-Perricaudet L, Poenaru L, Perricaudet M, Kahn A undReschanski M (1993) Transfer of a foreign gene into the brain using adenovirus vectors.Nat Gen 3: 224-234
Ali M, Lemoine N und Ring C (1994) The use of DNA viruses for gene therapy. GeneTherapy 1: 367-384
Ameis D, Greeve J (1996) Gentherapeutische Ansätze bei Stoffwechselerkrankungen.Internist 37:360-368
Beck E, Ludwig G, Auerswald EA, Reiss B und Schaller H (1982) Nucleotide sequence andexact localization of the neomycin phosphotransferase gene from transposon T5. Gene 19:327-336
Bett A, Prevec L und Graham F (1993) Packaging capacity and stability of human adenovirustype 5 vectors. J Virol 67: 5911-5921
Black BC, Center MS (1979) DNA-binding properties of the major core protein of adenovirus2. Nucleic Acids Res 6:2339
Blau H und Springer M (1995) Molecular medicine. Gene Therapy – A novel form of drugdelivery. N Engl J Med 333(18): 1204-1207
Boudin ML, D`Halluin J-C, Cousin C, Boulanger PA (1980) Human adenovirus type 2protein IIIa. II. Maturation and encapsidation. Virology 101:144
Boudin ML, Moncany M, D`Halluin J-C, Boulanger PA (1979) Isolation and characterizationof adenovirus type 2 vertex capsomer (penton base). Virology 92:125
Boulanger PA, Lemay P, Blair GE, Russell WC (1979) Characterization of adenovirus proteinIX. J Gen Virol 44:783
Boulanger PA, Loucheux-Lefevbre M-H (1982) structure of adenovirus nucleoprotein corestudied by circular dichroism and selective radiochemical labeling. Biochem Biophys ResCommun 107:470
Brosius J, Dull TJ, Sleeter DD und Noller HF (1981) Gene organization and primary structureof ribosomal RNA operon from E.coli. J Mol Biol 148:107-127
Brown M, Weber J (1980) Virion core-like organization of intranuclear adenovirus chromatinlate in infection. Virology 107: 306
Brown PT, Westphal M, Burlingham BT, Winterhof U, Doefler W (1975) Structure andcomposition of adenovirus type 2 core. J Virol 16:366
Brown TA (1993) Gentechnologie für Einsteiger: Grundlagen, Methoden, Anwendungen.Spektrum, Akad, Verl., Heidelberg, Berlin, Oxford
Bujard H, Gentz R, Lanzer M, Stüber D, Müller M, Ibrahimi I, Häuptle MT und DobbersteinB (1987) A T5 promotor based transcription-translation system for the analysis of proteinsin vivo and in vitro. Meth Enzym 155: 416-433
Chang ACY und Cohen SN (1978) Phenotypic expression in E.coli of a DNA sequencecoding for mouse dihydrofolate reductase. J Bactriol 134: 1141-1156
Chowdhury RJ, Grossman M, Gupta S, Chrowdhurym NR, Baker JR und Wilson JM (1991)Long-term improvement of hypercholesterolemia after ex vivo gene therapy in LDLR-deficient rabbits. Science 254: 1802-1805
Colby WW, Shenk T (1981) Adenovirus type 5 virions can be assembled in vivo in theabsence of detectable polypeptide IX. J Virol 39:977

79
Corden J, Engelking M, Pearson GD (1976) Chromatin-like organization of the adenoviruschromosome. Proc Natl Acad Sci U.S.A 73:401
Cordier L, Duffour MT, Sabourin JC, Lee M, Cabannes J, Ragot T, Perricaudet M undHaddada H (1995) Complete recovery of mice from a pre-established tumor by direktintratumoral delivery of an adenovirus vector harboring the murine IL-2 gene. Gene Ther2: 16-21
Cowman MK, Fasman GD (1978) Circular dichroism analysis of mononucleosome DNAconformation. Proc Natl Acad Sci U.S.A. 75:4759
Cox J, Yewdell J, Eisenlohr L, Johnson P und Bennink L (1990) Antigen presentationrequires transport of MHC class I molecules from the endoplasmic reticulum. Science 247:715-718
Crane HR (1950) Principles and problems of biological growth. Sci Mon 70:376de Duve C (1975) Exploring cells with a centrifuge. Science 189:186-194Defer C, Belin MT, Caillet-Boudin ML und Boulanger P (1990) Human adenovirus-host
interactions: comparative study with members of subgroups B and C. J Virol 64: 3661-3673
Devaux C, Timmins PA, Berthet-Colominas C (1983) Structural studies of adenovirus type 2by neutron
Devaux C, Zulauf M, Boulanger PA, Jacrot B (1982) Molekular weight of adenovirusserotype 2. J Mol Biol 68:181
Dingle J und Langmuir A (1968) Epidemiology of acute respiratory disease in militaryrecruits. Am Rev Respir D 97: 1-65
Earnshaw WC, Goldberg EB, Crowther RA (1979) The distal half of the tail fibre ofbacteriophage T4. J Mol Biol 132:101
Enders J, Bell J, Dingle J, Francis T, Hilleman M, Huebner R und Payne A (1956)Adenoviruses: Group name proposed for new respiratory tract viruses. Science 124: 119-120
Engelhardt J, Ye X, Doranz B und Wilson J (1994) Ablation of E2A in recombinantadenoviruses improves transgene persistence and decreases inflammatory response inmouse liver. Proc Natl Acad Sci U.S.A. 91: 6196-6200
Everitt E, Lutter L, Philipson L (1975) Structural proteins of adenovirus. XII. Location andneighbor relationship among proteins of adenovirus type 2 as revealed by enzymaticiodination, immunoprecipitation and chemical cross-linking. Virology 67:197
Everitt E, Sundquist B, Pettersson NU, Philipson L (1973) Structural proteins ofadenoviruses. X. Isolation and topography of low molekular weight antigens from thevirion of adenovirus type 2. Virology 52:130
Farabaugh PJ (1978) Sequence of the lac I gene. Nature 274: 765-769Felgner P und Rhodes G (1991) Gene therapeutics. Nature 394: 351-352Felgner P, Gader T und Holm M (1987) Lipofectin: A highly efficient, lipid mediated DNA-
transfection procedure. Proc Natl Acad Sci U.S.A. 84: 7413-7417Ferry N, Duplessis O, Houssin D, Danos O und Heard J-L (1991) Retroviral-mediated gene
transfer into hepatocytes in vivo. Proc Natl Acad Sci U.S.A. 88: 8377-8381Finch JF, Lutter LC, Rhodes D, Brown RS, Rushton B, Levitt M, Klug A (1977) Structure of
nucleosome core particles of chromatin. Nature (London) 269:29Flint SJ (1980) Structure and genomic organization of adenoviruses. In: Tooze J (ed) The
Molecular Biology of Tumor Viruses, Part 2, DNA Tumor Viruses, 2nd rev. edn. ColdSpring Harbor, New York, pp. 383-442

80
Ginsberg HS (1979) Adenovirus structural proteins. In: Fraenkel-Conrat H,Wagner RR (eds)Comprehensive Virology, Plenum, New York, Vol. 13, pp. 409-457
Ginsberg JS, Moldawer LL, Sehgal PB, Redington M, Kilian PL, Chanock RM und PrinceGA (1991) A mouse model for investigating the molecular pathogenesis of adenoviruspneumonia. Proc Natl Acad Sci U.S.A. 88: 1651-1655
Ginsberg HS, Pereira HG, Valentine RC, Wilcox WC (1966) A proposed terminology for theadenovirus antigens and virion morphological subunits. Virology 28:728
Gooding L, Ranheim T, Tollefson A, Aquino L, Duerksen-Hughes P, Horton T und Wold W(1991) The 10,400-dalton proteins encoded by region E3 of adenovirus function togetherto protect many but not all mouse cell lines against lysis by tumor necrosis factor. J Virol65: 4114-4123
Gottesmann S, Halpern E und Trisler P (1981) J Bacteriol 148: 265-273Graham F, Smiley J, Russel W und Nairn R (1977) Characteristics of a human cell line
transformed by DNA from human adenovirus type 5. J Gen Virol 36: 59-77Graham FL und van der Eb AJ (1973) Transformation of rat cells by DNA of human
adenovirus 5. Virology 54: 536-539Grand R (1987) The structure and function of the adenovirus early region 1 proteins. J
Biochem 241: 25-38Green M, Piña M, Kimes RC (1967b) Biochemical studies on adenovirus multiplication. XII.
Plaquing efficiencies of purified human adenoviruses. Virology 31:562Green M, Piña M, Kines RC, Wensid PC, MacHattie LA, Thomas CA Jr. (1967a)
Adenovirus-DNA. I. Molekular weight and conformation. Proc Natl Acad Sci U.S.A.57:1302
Green NM, Wrigley NG, Russel WC, Martin SR, McLachlan AD (1983) Evidence for arepeating cross-beta sheet structure in the adenovirus fibre. Eur Mol Biol Org J 2:1357
Grossman M, Raper SE und Wilson JM (1991) Towards liver-directed gene therapy:retrovirus-mediated gene transfer into human hepatocytes. Somat Cell Mol Genet 17: 601-607
Grossman M, Raper SE und Wilson JM (1992) Transplantation of genetically modifiedautologous hepatocytes into nonhuman primates: feasibility an short-term toxicity. HumGene Ther 3: 501-510
Grossman M und Wilson JM (1993) Retroviruses: delivery vehicel to the liver. Curr OpinGenet Dev 3: 110-114
Grütter M, Franklin RM (1974) Studies on the molekular weight of the adenovirus type 2hexon and its subunits. J Mol Biol 89:163
Haddada H, Cordier L und Perricaudet M (1995) Gene therapy using adenovirus vectors. In:Doerfler W und Böhm P (eds) The molecular repertoire of adenoviruses, Springer, Berlin -Heidelberg – New York, 3: 297-306
Hays JB, Magar ME, Zimm BH (1969) Persistence length of DNA. Biopolymers 8:531Hilleman MR,Werner JR (1954) Recovery of a new agent from patients with acute respiratory
illness. Proc Soc Exp Biol Med 85;183-188Hitt M, Bett A, Prevec L und Graham F (1994) Construction and propagation of human
adenovirus vectors. In: Cell Biology: A Laboratory Handbook. Academic Press Inc: 479Horne RW (1962) The comparative structure of adenoviruses. Ann NY Acad Sci 101:475Horwitz M (1990) Adenoviridae and their replication. In: Fields B und Knipe D (eds)
Virology, Raven Press, New York: 1679-1721

81
Hosokawa K, Sung MT (1976) Isolation and characterization of an extremly basic proteinfrom adenovirus type 5. J Virol 17:924
Huebner RJ,Rowe WP, Ward TG Parrott RH, Bell JA (1954) Adenoidal-pharyngeal-conjunctival agents: A newly recognized group of common viruses of the respiratorysystem. N Engl J Med 251:1077-1086
Hung T, Mak K und Fong K (1990) Nucleic Acids Res 18(16):4953Jackson D, Symons R und Berg P (1972) Biochemical method for inserting new genetic
information into DNA of simian virus 40: Circular SV 40 DNA molecules containinglambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad SciU.S.A. 69: 2904-2909
Jaffe JA, Daniel C, Longenecker M et al. (1992) Adenovirus-mediated in vivo gene transferand expression in normal rat liver. Nat genet 1: 372-378
Jawetz E (1959) The story of shipyard eye. B Med J 1: 873-878Kay MA, Li Q, Liu TJ, Leland F, Toman C, Finegold M und Woo SL (1992) Hepatic gene
therapy: persistent expression of human alpha 1-antitrypsin in mice after direct genedelivery in vivo. Hum Gene Ther 3: 641-647
Keegstra W, Van Wielink PS, Sussenbach JS (1977) The visualization of a circular DNA-protein complex from adenovirions. J Virol 76:444
Kelsey D (1978) Adenovirus meningoencephalitis. Pediatrics 61: 291-293Korner H, Fritzsche U und Burgert HG (1992) Tumor necrosis factor stimulates expression of
adenovirus early region 3 proteins: implications for viral persistence. Proc Natl Acad SciU.S.A. 89: 11857-11861
LeGal-LaSalle G, Robert J, Berrard S, Ridoux V, Stratford-Perricaudet L, Perricaudet M undMallet J (1993) An adenovirus vector for gene transfer into neurons and glia in the brain.Science 259: 988-990
Laver WG (1970) Isolation of an arginine-rich protein from particles of adenovirus type 2.Virology 41:488
Laver WG, Pereira HG, Russell WC, Valentine RC (1968) Isolation of an internal componentfrom adenovirus type 5. J Mol Biol 37:379
Lee M, Abina M, Haddada H und Perricaudet M (1995) The constitutive expression of theimmunomodulatory gp19k protein in E1-, E3-adenoviral vectors strongly reduces the hostcytotoxic T cell response against the vector. Gen Ther 2(4): 256-262
Leiden J (1995) Gene therapy – promise, pitfalls and prognosis. N Engl J Med 333(13): 871-873
Levrero M, Barban V, Manteca S et al. (1991) Defective and nondefective adenovirus vectorsfor expressing foreign genes in vitro and in vivo. Gene 101: 195-202
Li QT, Kay MA, Finegold M, Stratford-Perricaudet LD und Woo SLC (1993) Assessment ofrecombinant adenoviral vectors for hepatic gene therapy. Hum Gene Ther 4: 403-409
Maizel JV Jr, White DO und Scharff MD (1968) The polypeptides of adenovirus. I. Evidencefor multiple protein components in the virion and a comparison of types 2, 7A und 12.Virology 36: 115-125
Mandel M und Higa A (1970) Calcium-dependent bacteriophage 2 DNA infection. J Mol Biol53: 159-162
Marcoli R, Iida S und Brickle TA (1980) The DNA sequence of an IS1-flanked transposoncoding for resistance to chloramphenicol and fusidic acid. FEBS Letters 110: 11-14

82
Mattern CFT (1969) Virus architecture as determined by X-ray diffraction and electronmicroscopy. In; Levy HB (ed) Biochemistry of Viruses, Marcel Dekker, New York andLomdon, pp. 55-100
Matthews REF (1982) Classification and nomenclature of viruses. Intervirology 17:59-61McGrory WJ, Bautista DS und Graham FL (1988) Short Communications: A simple
technique for the rescue of early region 1 mutations into infectious human adenovirus type5. Virology 163: 614-617
Merklein F (1998) Adenovirus-vermittelter Gentransfer und Expression von normalen undmutagenisierten Antiproteasen, Gerinnungsfaktor IX und Firefly Luciferase inPrimärkulturen humaner vaskulärer Endothelzellen. Würzburg, Univ Diss 1998
Mirza MA, Weber J (1977) Genetic analysis of adenovirus type 2. VII. Cleavage-modifiedaffinity for DNA of internal virion proteins. Virology 80:83
Mirza MA, Weber J (1982) Structure of adenovirus chromatin. Biochem Biophys Acta 696:76Moullier P, Friedlander G, Calise D, Ronco P, Perricaudet M und Ferry N (1994) Adenoviral-
mediated gene transfer to renal tubular cells in vivo. Kid Int 45: 1220-1225Mulligan RC (1993) The basic science of gene therapy. Science 260: 926-932Nermut MV (1975) Fine structure of adenovirus type 5. Virology 65:480Nermut MV (1977) Freeze-drying for electron microscopy. In: Hayat MA (ed.) Principles and
techniques of electron microscopy VI, 79-117. New York: Van Nostrand Reinhold 1977Nermut MV (1978) Strutural elements in adenovirus cores: Studies by means of freeze-
fracturing and ultrathin sectioning. Arch Virol 57:323Nermut MV (1980a) The architecture of Adenoviruses: Recent views and problems. Arch
Virol 64:175Nermut MV (1980b) Effects of micrococcal nuclease on "relaxed" cores of adenovirus type 5
on molecular biology of adenoviruses. (Abstr.) EMBO-Workshop, Peebles, p.7Nermut MV (1984) The architecture of addenoviruses. In: Ginsberg HS (ed) The Adenovirus.
Plenum Press, New York and London, 1984Nermut MV, Perkins WJ (1979) Consideration of the three dimensional structure of the
adenovirus hexon from electron microscopy and computer modelling. Micron 10:247Nicholl DST (1995) Gentechnische Methoden. Spektrum: Akad. Verl. Heidelberg, Berlin,
OxfordNielson K und Mathur EJ (1990) Strategies 3(2): 17-22Norrby E (1969) The structural and functional diversity of adenovirus capsid components. J
Gen Virol 5:221Numazaki Y, Kumasaka T, Yano N, Yamanaka M, Miyazawa T, Takai S und Ishida N (1973)
Further study of acute hemorrhagic cystitis due to adenovirus type 11. N Engl J Med 189:344-347
Paabo S, Gifford JA und Wilson AC (1988) Nucleic Acids Res 16(20): 9775-9787Pereira HG, Wrigley NG (1974) In vitro reconstitution, hexon bonding and handedness of
incomplete adenovirus capsid. J Mol Biol 85:617Pettersson U, Höglund S (1969) Structural proteins of adenovirus. III. Purification and
characterization of the adenovirus type 2 penton antigen. Virology 39:90Philipson L (1979) Adenovirus proteins and their messenger RNAs. Adv Virus Res 25:357Philipson L,Pettersson U, Lindberg U (1975) Molekular biology of adenoviruses. III.
Purification and characterization of the adenovirus type 2 penton antigen. Virology 39:90

83
Prage L und Pettersson U (1971) Structural proteins of adenoviruses VII. Purification andproperties of an arginine-rich core protein from adenovirus type 2 and type 3. Virology 45:364.373
Prage L, Pettersson U und Philipson L (1968) Internal basic proteins in adenovirus. Virology36: 508-511
Prage L, Pettersson U, Höglund S, Lonbergholm K und Philipson L (1970) Structural proteinsof adenoviruses IV. Sequential degradation of the adenovirus type 2 virion. Virology 42:341-358
Ragot T, Vincent N, Chafey P, Vigne E, Gilgenkrantz H, Couton D, Cartaud J, Briand P,Kaplan J, Perricaudet M und Kahn A (1993) Efficient adenovirus-mediated transfer of ahuman minidystrophin gene to skeletal muscle of mdx mice. Nature 361: 647-650
Rekosh DMK, Russel WC, Bellét AJD, Robinson AJ (1977) Identification of a protein linkedto the ends of adenovirus DNA. Cell 11:283
Richmond TJ, Klug A, Finch JT, Lutter LC (1981) The organization of DNA in thenucleosome core particle. In: Sarma RH (ed) Biomolecular Stereodynamics, Ademine,New York, Vol II, pp.109-159
Roberts RJ, Akusjärvi G, Aleström P, Gelinas RE, Gingeras TR, Sciaky D und Pettersson U(1986) A consensus sequence for the adenovirus-2 genome. In: Doerfler W (ed)Developments in Molecular Virology, Martinus Nijhoff, Boston, vol. 8: 1-51
Robinson AJ, Younghusband HB, Bellét AJD (1973) A circular DNA-Protein complex fromadenoviruses. Virology 56:54
Rogers S und Pfuderer P (1968) Use of viruses as carriers of added genetic information.Nature 219: 749-751
Rogers S, Lowenthal A, Terheggen H und Colombo J (1973) Induction of arginase activitywith Shope papilloma virus in tissue culture cells from argininemic patient. J Exp Med137: 1091-1096
Rosenfeld M, Siegfried W, Yoshimura K, Yoneyama K, Fukayama M, Stier L, Pääkkö P,Gilardi P, Stratford-Perricaudet L, Perricaudet M, Jallat S, Pavirani A, Leococq L undCrystal R (1991) Adenovirus-mediated transfer of a recombinant α1-antitrypsin gene to thelung epithelium in vivo. Science 252: 431-434
Rosenfeld M, Yoshimura K, Trapnell B, Yoneyama K, Rosenthal E, Dalemans W, FukayamaM, Bargon J, Stier L, Stratford-Perricaudet L, Perricaudet M, Guggino W, Pavirani A,Leococq L und Crystal R (1992) In vivo transfer of the human cystic fibrosistransmembrane conductance regulator gene to the airway epithelium. Cell 68: 143-155
Rowe WP, Huebner AJ, Gilmore LK, Parrot RH, Ward TG (1953) Isolation of acytopathogenetic agent from human adenoids undergoing spontaneous degeneration intissue culture. Proc Soc Exp Biol Med 84:570-573
Russell WC, McIntosh K, Skehel JJ (1971) The preparation and properties of adenoviruscores. J Gen Virol 11:35
Russell WC, Precious B (1982) Nucleic acid-binding properties of adenovirus structuralproteins. J Gen Virol 63:69
Sarkar G, Kapelner S und Sommer S (1990) Nucleic Acids Res 18(24): 7465Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold
Spring Harbor, Cold Spring Harbor LaboratorySambrook J, Westphal H, Srivansan P und Dulbecco R (1968) The integrated state of viral
DNA in SV 40-transformed cells. Proc Natl Acad Sci U.S.A. 59: 1288-1293

84
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors.Proc Natl Acad Sci USA 74:5463-5467
Sato K, Hosokawa K (1981) The structure of adenovirus chromatin revealed by ultravioletlight-induced cross-linking. Biochem Biophys Res Commun 101:1318
Sato K und Hosokawa K (1984) Analysis of the interaction between DNA and major coreprotein in adenovirus chromatin by circular dichroism and ultraviolet light-inducedcrosslinking. J Biochem 95: 1031-1039
Schwarz E, Scherrer G, Hobom G und Kössel H (1987) Nucleotide sequence of cro, cll andpart of the 0 gene in phage lambda DNA. Nature 272: 410-414
Smith K, Long C, Bowman B und Manos M (1990) Amplifications 5: 16-17Stratford-Perricaudet LD, Levrero M, Chasse JF, Perricaudet M und Briand P (1990)
Evaluation of the transfer and expression in mice of an enzyme-encoding gene using ahuman adenovirus vector. Hum Gene Ther 1: 241-256
Stratford-Perricaudet L, Makeh I, Perricaudet M und Briand P (1992) Widespread long-termgene transfer to mouse skeletal muscles and heart. J Cli Inv 90: 626-630
Strauss M (1994) Liver-directed gene therapy: prospects and problems. Gene Therapy 1: 156-164
Stüber D, Matile H und Garotta G (1990) System for high-level production in Eschericia coliand rapid purificaton of recombinant proteins: Application to epitope mapping, preparationof antibodies, and structure-function analysis. In: Lefkovits I und Pernis B (eds)Immunological Methods, Vol. IV: 121-152
Sung MT, Lischwe MA, Richards JC und Hosokawa K (1977) Adenovirus chromatin. I.Isolation and characterization of the major core protein VII and precursor pro-VII. J BiolChem 252: 4981-4987
Sussman JC, Trifonov EN (1978) Possibility of nonkinked packing of DNA in chromatin.Proc Natl Acad Sci U.S.A. 75:103
Sutcliffe JG (1979) Complete nucleotide sequence of the E.coli plasmid pBR322. Cols SpringHarbor Symp. Quant. Biol. 43: 77-90
Tate VE (1976) Structural and functional studies on DNA and nucleoprotein components ofadenovirus type 5. Ph D thesis, Council for National Academic Rewards London
Tate VE, Philipson L (1979) Parental adenovirus DNA accumulates in nucleosome-likestructures in infected cells. Nucleic Acids Res 6:2769
Trentin J, Yabe Y und Taylor G (1962) The quest for human cancer viruses. Science 137:835-849
Valentine RC, Pereira HG (1965) Antigens and structure of the adenovirus. J Mol Biol 13:13Vayda ME, Rogers AE, Flint SJ (1983) The structure of nucleoprotein cores released from
adenovirions. Nucleic Acids Res 11:441-460Verga M, Weibull C und Everitt E (1991) Infectious entry pathway of adenovirus type 2. J
Virol 65: 6061-6070Villarejo MR und Zabin I (1974) β-galactosidase from termination and deletion mutant
strains. J Bacteriol 110: 171-178von der Leyen H, Gibbons G, Morishita R, Lewis N, Zhang L, Nakajima M, Kaneda Y,
Cooke J und Dzau V (1995) Gene therapy inhibiting neointimal vascular lesion: In vivotransfer of endothelian cell nitric oxide synthase gene. Proc Natl Acad Sci U.S.A. 92:1137-1141
White TJ, Arnheim N, Ehrlich AH (1989) The polymerase chain reaction. Trends Genet5:185-189

85
Wigand R, Baumeister H, Maas G, Kuhn J und Hammer H (1983) Isolation and identificationof enteric adenovirus. J Med Virol 11: 233-240
Wildy P (1971) Classification and nomenclature of viruses. In: Melwick JL (ed) Monographsin Virology, S. Karger, Basel, Vol 5, pp. 1-81
Williams J (1990) Lymphoid specific gene expression of the adenovirus early region 3promotor is mediated by NF-kappaB binding motifs. EMBO J 9: 4435-4442
Wilson J, Johnston D, Jefferson D und Mulligan R (1988) Correction of the genetic defect inhepatocyte from the Watanabe heritable hyperlipidemic rabbit. Proc Natl Acad Sci U.S.A.84:4421-4425
Wold W und Goodling L (1991) Region E3 of adenovirus: a cassette of genes involved inhost immunosurveillance and virus-cell interactions. Virology 184: 1-8
Yang Y, Nunes F, Berencsi K, Furth E, Gönczöl E und Wilson J (1994) Cellular immunity toviral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci U.S.A.91: 4407-4411

DANKSAGUNG
Ich möchte mich bei der gesamten Arbeitsgruppe des Lipidlabors bedanken. Herrn PD. Dr. med. D. Ameis und
Frau Dr. Anne Tilkorn danke ich für ihre fachliche Betreuung bei der Erstellung dieser Arbeit. Bei Frau Julia
Rethmeier möchte ich mich für die praktische Unterstützung in der Durchführung der Experimente bedanken.
Für Kritik und Unterstützung gilt mein Dank Herrn Dipl. Chem. Oliver Zschenker, Dipl. Chem. Georg Josza,
Nikola, Gerald, meinen Eltern und vor allem Paul.
LEBENSLAUF VON RAPHAELA BASDEKIS-JOZSA
Geburtstag und Ort: 16.02.1972 in Hamburg
Schulbildung: 1978 – 1991 Grundschule, Orientierungsstufe, Gymnasium (Herzogliches Wilhelm-
Gymnasium zu Braunschweig), Abschluss: Abitur
Studium: 1992 – 1999 Medizinstudium an der Universität Hamburg,
Abschluss: Drittes Staatsexamen
Beruf: 1999 – 2001 Ärztin im Praktikum in der Klinik und Poliklinik für Psychiatrie und
Psychotherapie des Universitätskrankenhauses Hamburg-Eppendorf.
Seit 2001 wissenschaftliche Mitarbeiterin im Zentrum für Interdisziplinäre
Suchtforschung (ZIS) der Universität Hamburg, c/o: Universitätsklinikum Hamburg-
Eppendorf (UKE).
Seit 11/2002 zusätzlich Mitarbeiterin in der Ambulanz für chronisch mehrfach
Abhängige (CMA-Ambulanz) der Psychiatrischen Klinik des UKE.

ERKLÄRUNG
Ich versichere ausdrücklich, dass ich die Arbeit selbständig und ohne fremde Hilfe verfasst, andere als die von
mir angegebenen Quellen und Hilfsmittel nicht benutzt und die aus den benutzten Werken wörtlich oder
inhaltlich entnommenen Stellen einzeln nach Ausgabe (Auflage und Jahr des Erscheinens), Band und Seite des
benutzten Werkes kenntlich gemacht habe, und dass ich die Dissertation bisher nicht einem Fachvertreter an
einer anderen Hochschule zur Überprüfung vorgelegt oder mich anderweitig um Zulassung zur Promotion
beworben habe.


















