Kapitel 4 Regler, Leittechnik Arbeits- und Kapitel 5 Preisbuch
Kapitel KMolekülspektren
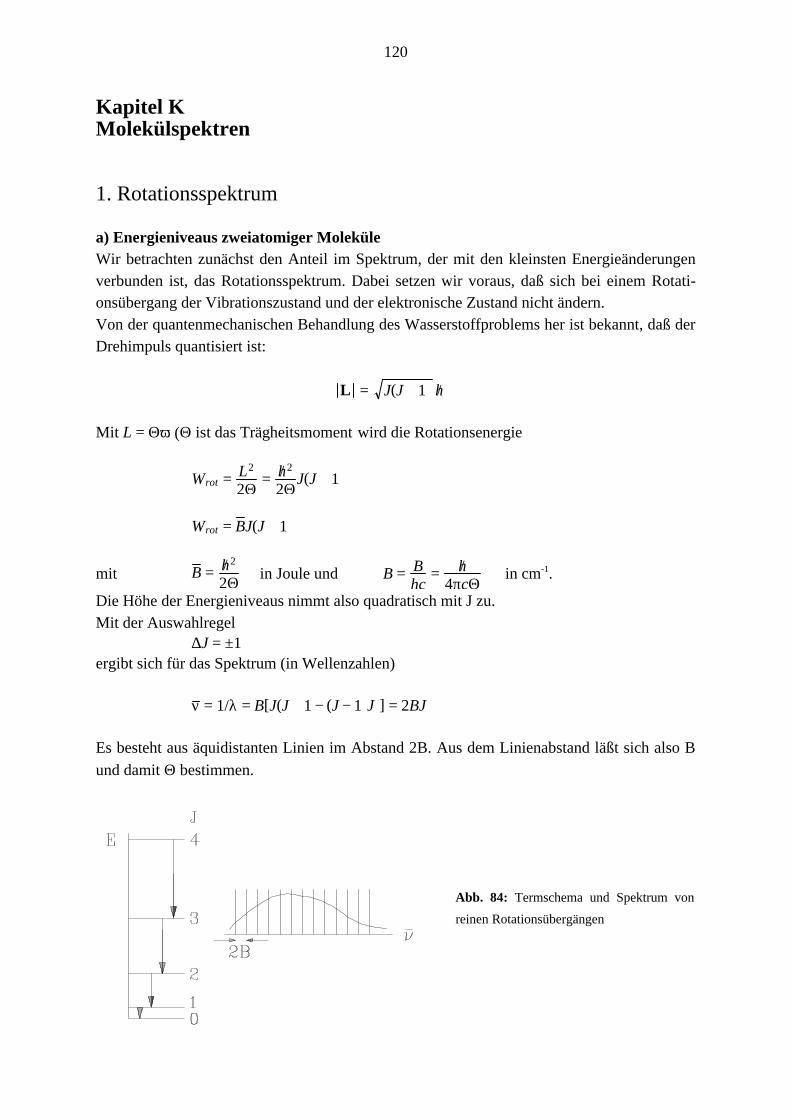
1. Rotationsspektrum
a) Energieniveaus zweiatomiger MoleküleWir betrachten zunächst den Anteil im Spektrum, der mit den kleinsten Energieänderungenverbunden ist, das Rotationsspektrum. Dabei setzen wir voraus, daß sich bei einem Rotati-onsübergang der Vibrationszustand und der elektronische Zustand nicht ändern.Von der quantenmechanischen Behandlung des Wasserstoffproblems her ist bekannt, daß derDrehimpuls quantisiert ist:
L = J(J + 1) h/
Mit wird die RotationsenergieL = Θω (Θ ist das Tragheitsmoment)
Wrot = L2
2Θ= h/ 2
2ΘJ(J + 1)
Wrot = BJ(J + 1)
mit in Joule und in cm-1.B = h/ 2
2Θ B = Bhc
= h/4πcΘ
Die Höhe der Energieniveaus nimmt also quadratisch mit J zu.Mit der Auswahlregel
∆J = ±1ergibt sich für das Spektrum (in Wellenzahlen)
ν = 1/λ = B[J(J + 1 − (J − 1)J)] = 2BJ
Es besteht aus äquidistanten Linien im Abstand 2B. Aus dem Linienabstand läßt sich also B
und damit Θ bestimmen.
120
Abb. 84: Termschema und Spektrum von
reinen Rotationsübergängen

b) Intensitätenα) Innerhalb einer RotationsstrukturDie Intensität ist wie bei Atomen gegeben durch
I = h/ωN1A12I
bei Emission, d.h. im wesentlichen durch die Besetzungszahl des Ausgangsniveaus, (bei Ab-sorptionsspektroskopie durch die Besetzung des unteren Niveaus) und die Übergangswahr-scheinlichkeit. Die Übergangswahrscheinlichkeit kann man in erster Näherung innerhalb derRotationsstruktur als konstant ansehen. Die Besetzungszahlen hängen von den statistischenGewichten der Zustände gJ = 2J + 1 und ihrer Energie ab.
N1
N0=
g j
g0e−E/kT ∼ (2J + 1)e−BJ(J+1)/kT
Dies ergibt wie xe-x2 eine Kurve mit einem Maximum bei Jmax . Jmax hängt von der Temperaturab.
Aus der Bestimmung der Quantenzahl Jmax, bei der die Intensität ein Maximum besitzt, läßtsich die Temperatur ermitteln.
β) InfrarotaktivitätRotierende Moleküle können nach dem klassischen Bild nur strahlen, wenn mit der Bewe-gung eine Beschleunigung von Ladung verbunden ist, d.h. wenn sie ein permanentes Dipol-moment besitzen. Das gleiche gilt für Vibration. D.h. Moleküle ohne permanentes Dipolmo-ment zeigen kein Rotations- und Vibrationsspektrum. Man sagt, sie sind infrarot inaktiv.Hierzu gehören alle zweiatomigen Moleküle aus gleichen Atomen (homonukleare Atome)wie H2, N2, O2 und mehratomige lineare Moleküle mit Inversionszentrum wie CO2. Das Rota-tionsspektrum dieser Moleküle ist aber bei der Raman-Streuung oder im elektronischen Spek-trum beobachtbar.
γ) Einfluß des Kernspins (IK)Der Kernspin führt zu einer Hyperfeinstrukturaufspaltung, die mit genügend hoher Auflösungbeobachtet werden kann. Es gibt allerdings Einflüsse des Kernspins über statistische Effekte,die auch mit geringer Auflösung leicht beobachtbar sind. Es handelt sich um einen der
121
Abb. 85: Intensitätsverteilung in einer Rotationsbande

Effekte, bei denen quantenmechanische Regeln zu makroskopischen Auswirkungen führen,ähnlich wie das Fehlen des 1s Grundzustandes im Triplettsystem des Helium. Dies wird amWasserstoffmolekül H2 erörtert.Der Kernspin der beiden Protonen im H2 kann parallel ausgerichtet sein, man spricht dannvon Orthowasserstoff oder o - H2, oder antiparallel beim Parawasserstoff p - H2. Die Über-
gangswahrscheinlichkeit zwischen beiden Modifikationen ist gering (Auswahlregel ∆IK= 0).Im thermischen Gleichgewicht hat man stets eine Mischung von o- und p-Modifikation. DasVerhältnis wird durch die statistischen Gewichte bestimmt. Beim o - H2 ist IK = 1, bei p - H2 IK = 0, die statistischen Gewichte und damit die Konzentrationen verhalten sich wie 3 : 1.
Der Kernspin beeinflußt nun über seine Parität die Parität der Gesamtwellenfunktion und dadiese für Fermionen nach dem Pauliprinzip -1 sein muß, ist die Parität vom Produkt alle üb-rigen beteiligten Funktionen wie der elektronischen, der Vibration, der Rotation zusammenvorherbestimmt.Im elektronischen Grundzustand (s) ist die Parität der Elektronenfunktion gerade. Ebenso hatder Grundzustand bezüglich Vibration eine gerade Parität, denn das System ist symmetrischgegenüber Austausch der beiden Kerne. Die Parität der Gesamtwellenfunktion ist also durch
Pges = Prot · PKern = - 1 bestimmt. Da - wie wir für das H-Atom gezeigt haben - die Rotationsniveaus eine Parität
P = (-1)J
besitzen, sind die einzigen möglichen Rotationsniveaus für
o - H2: J = 1,3,5,...p - H2: J = 0,2,4,..
Mit der Auswahlregel ist damit überhaupt kein Übergang erlaubt. H2 strahlt∆J = ±1, ;∆IK = 0aber sowieso nicht bei Rotation, da es infrarot inaktiv ist. Die Rotationsstruktur läßt sich abermit der Ramanstreuung oder im elektronischen Spektrum beobachten. Im Ramanspektrumgilt die Auswahlregel . Das Ramanspektrum besteht also aus einer Überlagerung der∆J = ±2Spektren des o-H2 und p-H2, wobei die Linien abwechseln und einen Intensitätsunterschiedvon 3:1 entsprechend der Konzentrationen der beiden Modifikationen aufweisen.Da o-H2 als tiefsten Zustand den metastabilen Zustand mit J = 1 hat, zerfällt o-H2 in p-H2. DieZerfallszeit ist allerdings in einem Gas von nicht zu hohem Druck von der Größenordnung
122
Abb. 86: Mögliche Übergänge beim Ortho- und Parawasserstoff

eines Jahres. Man kann den Zerfallsprozeß katalytisch beschleunigen und sich so p-H2 her-stellen und lagern.Deuterium hat IK = 1, ist damit ein Boson. Für ein D2-Molekül führt daher die obige Betrach-tung zu dem umgekehrten Ergebnis, da gegenüber Vertauschung von Bosonen die Gesamt-wellenfunktion gerade sein muß. Folgende Rotationszustände sind also erlaubt
p - D2: J = 1, 3, 5o - D2: J = 0, 2, 4
In diesem Fall ist also die o-Modifikation stabil.Ein ähnliches Ergebnis wie für H2 erhält man für 14N2, aber wegen unterschiedlicher statisti-scher Gewichte ist hier das Intensitätsverhältnis der Linien 1/2. Bei 14N 15N, also einem Stick-stoffmolekül aus Isotopen mit unterschiedlicher Masse ist kein Intensitätswechsel in der Ro-tationsbande zu beobachten, da die Symmetrie des Atoms durch die unterschiedlichen Mas-sen verlorengegangen ist. Bei 16O2 ist der Kernspin IK = 0. Daher kommen im Rotationspek-trum nur Terme mit ungeradem J vor.
c) Rotations-RamanspektrumInfrarotspektroskopie wird im allgemeinen in Absorption betrieben (Abb. 87). Die Probe wirdentweder mit einer kontinuierlichen Lichtquelle bestrahlt und mit einem Monochromator ana-lysiert, oder mit schmalbandiger, abstimmbarer Laserstrahlung beleuchtet und die Absorptiongemessen. Probleme gibt es außer mit empfindlichen Detektoren mit Streulicht, das im Infra-roten besonders schwer zu unterdrücken ist. Monochromatoren werden daher häufig als Dop-pelmonochromatoren oder in Littrow-Aufstellung betrieben.Bei der Ramanstreuung strahlt man mit sichtbarem Licht ein und beobachet das unter 90° ge-
streute Licht in der Umgebung der Rayleigh-Linie.
Wird dem eingestrahlten Licht Energie zur Anregung von Rotation entnommen, erniedrigtsich die Frequenz der Streustrahlung. Man spricht von Stokes-Linien.
h/ωs = h/ωL − h/ωrot
Gibt ein rotierendes Molekül Energie an das Streulicht ab, spricht man von Antistokeslinien.
h/ωs = h/ωL + h/ωrot
123
Abb. 87: Anordnug für Infrarot- und
Ramanspektroskopie

Da die Abregung von einem höheren Niveau aus erfolgt, das nach Boltzmann schwächer be-setzt ist, sind die Antistokeslinien weniger intensiv als die Stokeslinien.Man erreicht mit dem Raman-Effekt eine Transformation des Rotationspektrums in denSpektralbereich des Lasers, also z.B. in das Sichtbare.Ein anderer Nutzen des Ramaneffektes ist die Möglichkeit, die veränderten Auswahlregelnauszunutzen. Da die Ramanstreuung ein Zweiphotonenprozeß ist, gilt hier . Außer-∆J = ±2
dem zeigen viele Moleküle, die infrarot inaktiv sind, ein Ramanspektrum, da hierfür kein per-manentes Dipolmoment vorhanden sein muß, sondern sich nur die Polarisierbarkeit mit demKernabstand ändern muß (s. Kap. L).
Im H2 sieht man also das Rotations-Ramanspektrum: 1. da H2 Raman aktiv ist und 2. da
jetzt erlaubte Übergänge darstellen. Das Spektrum ist eine Überlagerung des p - H2∆J = ±2
und o - H2 Spektrums, d.h. die mit den geraden Quantenzahlen verbundenen Übergänge, diezum p - H2 gehören sind um einen Faktor 1/3 weniger intensiv als die, die zu den geraden ge-hören. Insgesamt wechseln sich also Linien mit dem Intensitätsverhältnis 3/1 ab.
d) Der nichtstarre RotatorDas Modell der starren Hantel ist nicht streng gültig. Wegen der Elastizität der Bindung er-wartet man eine Erhöhung des Abstandes und damit des Trägheitsmomentes bei stärkerer Ro-tation. Rechnet man dies für gegebene Federkonstante k klassisch durch und ersetzt zumSchluß L durch , so erhält man für die RotationsenergienJ(J + 1) h/
Wrot = BJ(J + 1) − D[J(J + 1)]2
wobei D ~ 1/k ist. Diese Abnahme von ∆ zwischen den einzelnen Linien einer Rotations-νbande bei hohen J läßt sich tatsächlich beobachten und mit den Federkonsten, die genauerausdemVibrationsspektrum gemessen werden, korrelieren.
124
Abb. 88: So etwa sieht ein Rotations-Ramanspektrum
aus
Abb. 89: Rotations-Ramanspektrum des H2

e) Isotopie-EffektDie verschiedenen Massen von Isotopen machen sich im Rotationsspektrum bemerkbar.Nimmt man an, daß die Potentialkurve V(Rab) nicht von der Atommasse abhängt, so verhaltensich die Trägheitsmomente wie die Massen und damit B umgekehrt wie die Massen. DieserEffekt ist besonders groß bei D2 und H2, wo BD = 1/2 BH. f) Mehratomige MoleküleMehratomige Moleküle besitzen immer drei Hauptträgheitsachsen, die senkrecht aufeinanderstehen. Wir legen das Koordinatenkreuz so, daß x, y, z mit den drei Trägheitsachsen, zu de-
nen die drei Hauptträgheitsmomenten Θx, Θy, Θz gehören, zusammenfallen. Die Gesamtener-gie der Rotation läßt sich dann schreiben
W =Lx
2
2Θx+
Ly2
2Θy+
Lz2
2Θz
Bei einem unsymmetrischen Kreisel - wie etwa H2O - sind alle Trägheitsmomente unter-schiedlich. In diesem Fall sind Lx, Ly, Lz nicht gequantelt. Jedes Molekül muß individuell be-handelt werden.Wenn zwei der Hauptträgheitsmomente gleich sind, etwa Θx = Θy Θz , spricht man vom≠symmetrischen Kreisel (Beispiel NH3). Dann hat man zwei Quantisierungsbedingungen
L = J(J + 1) h/Lz = Kh/
K = 0, ±1, ±2, ...,±J
K hat also eine ähnliche Funktion wie die magnetische Quantenzahl m im Atom, die Projekti-on von L bezieht sich allerdings nicht auf eine außen vorgegebene Richtung, etwa ein Feld,sondern auf die Molekülachse. Die Energie schreibt sich dann
Wrot = BJ(J + 1) + CK2
mit B = h/4πcΘz
, C= h/4πc
1Θy
− 1Θx
Bei einem prolaten Molekül, d.h. einem zigarrenförmigen, ist C > 0 und damit wächst dieGesamtenergie mit steigendem K, während bei einem oblaten Molekül, also einem diskusför-
migen etwa wie Benzol C < 0 und damit ∆W mit steigendem K abnimmt.
125

2. Schwingungsspektren
a) Potential eines zweitomigen Moleküls
Abb. 90 zeigt den qualitativen Verlauf des Potentials in Abhängigkeit vom Abstand R derbeiden Atome, die das Molekül bilden. Für viele Zwecke ausreichend läßt sich die Formdurch ein Morsepotential annähern
V(R) = De[1 − e−a(R−Re)]2
Hierin ist De die Dissoziationsenergie vom Minimum der Potentialkurve aus gerechnet, Re istder Gleichgewichtsradius des Moleküls und a ist ein Maß für die Krümmung im Potentialmi-nimum und damit für die Rückstellkraft. Bei Ionenbindung ist manchmal eine Näherungdurch eine Art Lennard-Jones Potential günstiger
V(R) = −e2
4πε0R+ 1
R9
Wie bei der Rotation ist die Vibration im Infrarotspektrum nur beobachtbar, wenn das Mole-kül ein permanentes elektrisches Dipolmoment besitzt. Homonukleare zweiatomige Molekülezeigen also kein Vibrations- (d.h. Rotations-Vibrations) Spektrum.
b) Harmonische NäherungIm Potentialminimum kann V(R) entwickelt werden. Die einfachste nichttriviale Näherungdes Potentials ist ein parabelförmiges Potential, also das Potential eines linearen Oszillators:
V(R) = k2
(R − Re)2
Klassisch ergibt sich als Schwingungsfrequenz
ω02 = k
µ
wobei k die Rückstellkraft und µ die reduzierte Masse ist.
126
Abb. 90: Potential eines zweiatomigen Moleküls in
Abhängigkeit vom Kernabstand

1µ = 1
m1+ 1
m2
Quantenmechanisch erhält man äquidistante Energieniveaus
Wvib = h/ω0v + 1
2
die durch die Vibrationsquantenzahl v = 0, 1, 2, .... charakterisiert werden. In Wellenzahlen-einheiten schreibt man üblicherweise
Wvib = ϖev + 1
2
wobei die Dimension einer Wellenzahl hat und nicht mit einer Kreisfrequenzϖe = h/ω0
hc= ν
verwechselt werden darf.
Das tiefste Niveau liegt nicht bei V(Re) = 0, sondern bei . V0 ist die Nullpunkt-V0 = 12
h/ω0
senergie. Die Auswahlregel heißt , woraus folgt, daß nur eine einzige Frequenz∆ v= ±1beobachtet wird: ω = ω0.
c) Der anharmonische OszillatorDas Schrödingerproblem mit Morsepotential ist vollständig lösbar. In der Umgebung des Po-tentialminimums geht natürlich die Lösung in die des harmonischen Oszillators über, wobeidie Rückstellkraft
k = 2Dea2 ist.
Die Tatsache, daß das Potential nicht harmonisch ist, hat zwei Konsequenzen:α) Die Energieniveaus sind nicht mehr äquidistant. Sie können approximiert werden durch
Wvib = h/ω0v + 1
2 + h/ω0
4De
v + 1
2
2
= h/ω0v + 1
2 + xeh/ω0
v + 1
2
2
xe hat die Größenordnung 0,01. Für genauere Vergleiche mit Messungen werden auch höherePotenzen von (v + 1/2) mitgenommen. Die Abstände zwischen den Niveaus nehmen also mitwachsendem v ab.
127
Abb. 91: Potential, Energieniveaus und Übergänge
beim harmonischen Osszillator

β) Die Auswahlregeln lassen nun außer auch und zu. Dabei∆v= ±1 ∆v= ±2 ∆v= ±3schwingt der Oszillator mit den Frequenzen ω = ω0 , ω = 2ω0 . also den Oberschwingungenzur Grundfrequenz. Man spricht auch von Obertönen. Die Obertöne nehmen mit steigenderOrdnung an Intensität ab.Die Anzahl der Vibrationsniveaus, vmax, die bis zur Dissoziationsgrenze in die Potentialmuldepassen, ist beim Morsepotential endlich.
h/ω0vmax + 1
2 + xe
vmax + 1
2
2 = De
für HCl mit νν0 = 2900 cm-1, xe = 0,017 ergibt sich z.B. vmax = 22.Bild
Die Dissoziationsenergie D0 ist nicht durch den Abstand gegeben,V(R → ∞) − V(Re) = De;sondern durch den Abstand zur Nullpunktsenergie D0. Die Nullpunktsenergie
De − D0 = 12
h/ω0 = h/2
km
hängt von der Masse der beteiligten Atome ab. Nimmt man an, daß die Bindungskräfte unddamit die Potentialkurve nicht von der Masse der beteiligten Atome abhängt, so ergibt sichaus den gemessenen Dissoziationsenergien die Nullpunktsenergie.Z.B. sind die Grundschwingungen des Wasserstoffs (1H2) und Deuteriums (2D2)
ν0H = 4115cm−1
ν0D = 2990cm−1
Die Differenz der Nullpunktsenergien wird damit
12
(v0H − ν0D) = 584cm−1
Dies muß der Differenz der Dissoziationsenergien entsprechen
(De − D0)D − (De − D0)H
Für den Fall gleicher Potentialkurven von H und D wird dies DeH - DeD . der gemessene Wert für die Differenz der Dssotiationsenergien ist: ∆νν = 621cm-1.
128
Abb. 92: Die Dissoziationsenergie D0 und die Tiefe der
Potentialmulde De unterscheiden sich

Das Bild ist also im großen und ganzen korrekt und der obige Vergleich kann als Nachweisder Nullpunktsenergie angesehen werden.Die Differenz der Ionisierungsenergie von Isotopen ist die Grundlage für Isotopentrennungmit Laserstrahlung.
d) Rotation-Schwingungsspektrumα) Ohne KopplungGenaugenommen sind Rotations- und Schwingungsenergie nicht unabhängig voneinander,denn z.B. bei Anregung von Vibration vergrößert sich der mittlere Abstand der Atome unddamit das Trägheitsmoment. Wenn man von dieser Kopplung absieht, ergeben sich die Ener-gieniveaus zu
W = Wvib + Wrot
W = h/ω0v + 1
2 + xeh/ω0
v + 1
2
2
+ BJ(J + 1)
Mit den Auswahlregeln und resultiert das bekannte Vibrati-∆J = ±1 ∆v= ±1, ; ± 2, ; ± 3, ...;onsspektrum, das in der unmittelbaren Umgebung eine Rotationsstruktur aufweist. Die Bedin-gung besagt, daß Vibrationsspektren alleine nicht vorkommen, sondern daß mit je-∆J = ±1der Vibration auch eine Rotation angeregt wird.
Abb. 93 zeigt einen Ausschnitt aus dem Termschema der Rotations-Vibrationsniveaus für 2unterschiedliche Vibrationsquantenzahlen und drei Rotationsquantenzahlen. Den unterenTerm versieht man normalerweise mit zwei Strichen.
Den Zweig mit ∆J = -1, d.h. J' = J'' - 1 nennt man den P-Zweig, den mit ∆J = +1 , d.h.J' = J'' + 1 den R-Zweig. Es gibt Ausnahmen, in denen der Q-Zweig mit ∆J = 0 ebenfalls er-laubt ist, dann weist das Spektrum an der Stelle des reinen Vibrationsüberganges eine Linieauf. Sonst fehlt diese sogenannte Null-Linie.
129
Abb. 93: Übergänge im Rotations-Schwingungsspektrum
Abb. 94: Rotations-Vibrationsstruktur
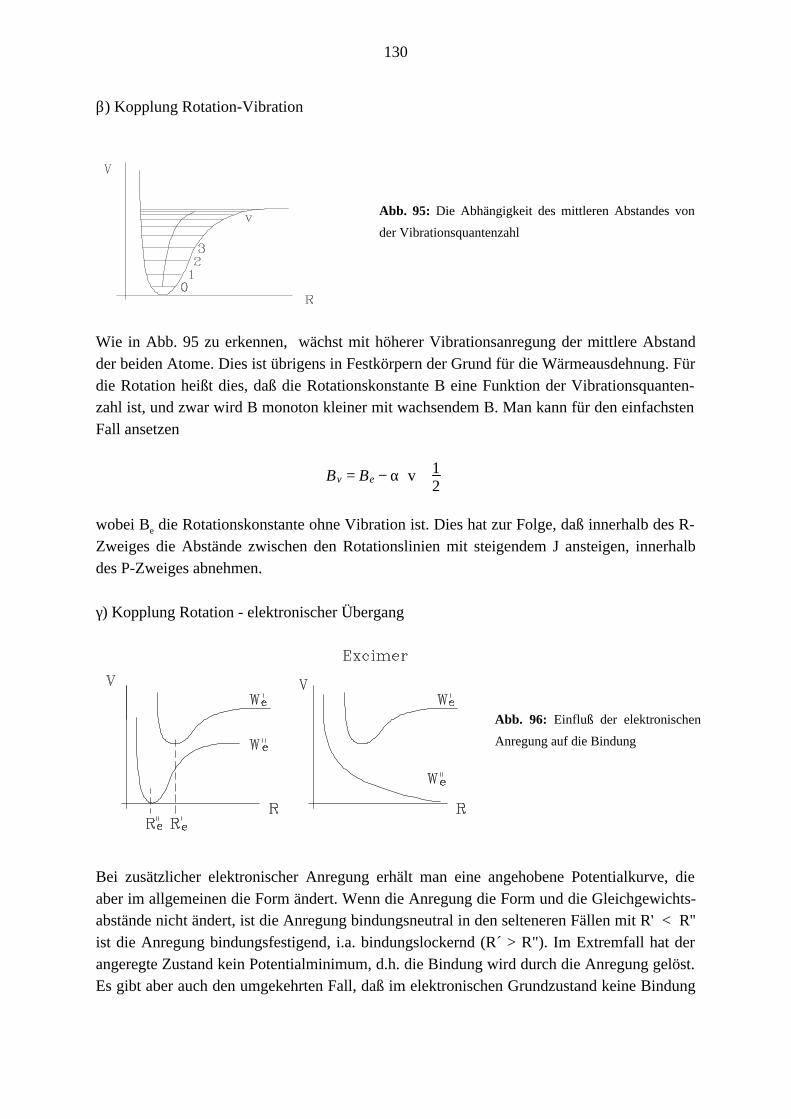
β) Kopplung Rotation-Vibration
Wie in Abb. 95 zu erkennen, wächst mit höherer Vibrationsanregung der mittlere Abstandder beiden Atome. Dies ist übrigens in Festkörpern der Grund für die Wärmeausdehnung. Fürdie Rotation heißt dies, daß die Rotationskonstante B eine Funktion der Vibrationsquanten-zahl ist, und zwar wird B monoton kleiner mit wachsendem B. Man kann für den einfachstenFall ansetzen
Bv = Be − αv + 1
2
wobei Be die Rotationskonstante ohne Vibration ist. Dies hat zur Folge, daß innerhalb des R-Zweiges die Abstände zwischen den Rotationslinien mit steigendem J ansteigen, innerhalbdes P-Zweiges abnehmen.
γ) Kopplung Rotation - elektronischer Übergang
Bei zusätzlicher elektronischer Anregung erhält man eine angehobene Potentialkurve, dieaber im allgemeinen die Form ändert. Wenn die Anregung die Form und die Gleichgewichts-abstände nicht ändert, ist die Anregung bindungsneutral in den selteneren Fällen mit R' < R''ist die Anregung bindungsfestigend, i.a. bindungslockernd (R´ > R"). Im Extremfall hat derangeregte Zustand kein Potentialminimum, d.h. die Bindung wird durch die Anregung gelöst.Es gibt aber auch den umgekehrten Fall, daß im elektronischen Grundzustand keine Bindung
130
Abb. 95: Die Abhängigkeit des mittleren Abstandes von
der Vibrationsquantenzahl
Abb. 96: Einfluß der elektronischen
Anregung auf die Bindung

vorliegt, durch eine Anregung aber ermöglicht wird. Solche Stoffe nennt man Excimere.Edelgas-Halegonid-Excimere spielen als Lasermedium eine Rolle.Wir gehen vom Fall aus, daß beide Potentialkurven ein Minimm besitzen, dieses sich aberdurch die Anregung verschiebt. Als Folge ändert sich der mittlere Abstand und damit die Ro-tationskonstante. Die Wellenzahlen ergeben sich aus
νν = ν0(v'v'') + B'J'(J+1) - B''J''(J''+1)
Da B' ≠ B'', hebt sich der quadratische Term nicht mehr wie im reinen Rotationsspektrum her-aus. Man erhält bei konstantem ∆J, das die Zweige charakterisiert, für νν(J) Parabelbögen. Au-ßer ∆J = ±1 ist hier auch der Q-Zweig ∆J = 0 häufig erlaubt und der O- und S-Zweig(∆J = ±2). νν(J) ist das sogenannte Fortrat-Diagramm (Abb.97 ), das es gestattet, eine gewisseÜbersicht in ein Bandenspektrum zu bringen.
Aus der Richtung der Abschattierung läßt sich erkennen, ob die Anregung die Bindung för-dert oder lockert. Bei B' = B'' hat die Rotationsstruktur das gleiche Aussehen wie imRotations-Schwingungsspektrum.
e) Mehratomige MoleküleNormalschwingungenBei einer Normalschwingung schwingen alle Atome mit konstanter Amplitude, Frequenz undgegenseitiger Phasendifferenz. Bekannt sind die Normalschwingungen zweier gekoppelterPendel. Hier gibt es genau 2 Normalschwingungen: Bei der einen schwingen beide Pendel ingleicher Richtung, bei der anderen gegeneinander. Auch die Eigenschwingungen von Saitenkann man als Normalschwingungen auffassen. Wie bei Saiten haben auch in allen anderenschwingenden Systemen die Normalschwingungen die Eigenschaft, daß sich alle möglichenSchwingungsformen aus Linearkombinationen der Eigenschwingungen zusammensetzen las-sen. (Bei Saiten wird dies durch den Satz von Fourier begründet).Ein Molekül, das aus n Atomen besteht, hat 3n Freiheitsgrade. Zieht man hiervon die 3 Frei-heitsgrade der Translation und die 3 der Rotation ab, bleiben im allgemeinen 3n - 6 Freiheits-grade für Schwingungen übrig. Bei einem linearen Atom entfällt der Freiheitsgrad der Rotati-on um diese Achse. Ein Molekül hat also im allgemeinen 3n - 6 Normalschwingungen, ein li-neares Atom 3n - 5.
131
Abb. 97: Das Fortrat
- Diagrammm

Beispiel:Bei zweiatomigen Molekülen ist die Anzahl der Normalschwingungen 6 - 5 = 1.Wasser ist nicht linear, n = 3 also gibt es 3 Moden. CO2 ist ein lineares Molekül aus 3 Ato-men und hat daher 4 Normalschwingungen (s.Abb. 99) Die beiden Biegeschwingungen sinddabei entartet.
Die Symmetrie der Normalschwingungen läßt sich auf die Symmetrie der irreduziblen Dar-stellungen der Gruppe, denen das Molekül angehört, zurückführen.Als Beispiel wird das H2O-Molekül betrachtet. Es gehört, wie wir aus Kapitel K wissen, derPunktgruppe C2v an. Die Charaktertafel mit dem linearen Teil der Basis ist in Tabelle XIII wiedergegeben.
Unterziehen wir die in Abb. 98 angegebenen Normalschwingungen den verschiedenen Sym-metrieoperationen der Gruppe, sehen wir, daß ν1 und ν2 wie A1 transformieren, ν3 wie B1. Man kann diese Symmetrien auch systematisch ermitteln, ohne daß die Form der Normal-schwingungen bekannt ist.
Dazu zeichnet man an jedes Atom ein Koordinatenkreuz und überlegt sich, wie der Vektor
132
Abb. 98: Die Normalschwingungen
vom Wassermolekül
Abb. 99: Die Normalschwingungen bei
CO2
Tabelle XIII: Charaktertafel der
Punktgruppe C2v

bei den verschiedenen Symmetrieoperationen transformiert (s. Abb. 79 und 80). Man schreibtdie zugehörigen 9 x 9 Matrizen hin und bestimmt deren Spur. Man erhält damit die Charakte-re der reduziblen Darstellung. Bei H2O ergibt sich:
Die Dimension ist gleich der Anzahl der Freiheitsgrade 9. Durch Ausprobieren mit der Cha-raktertafel der Gruppe findet man, daß sich diese Darstellung zusammensetzt aus denirreduziblen
Γtot = 3a1 + a2 + 3b1 + 2b2
Hiervon zieht man noch die irreduziblen Darstellungen ab, die die Translationen (x, y, z) undRotationen (Rx, Ry, Rz) als Basis haben.
Γvib = Γrot - Γtrans - Γrot
= (3a1+a2+3b1+2b2) - (a1+b1+b2) - (a2+b1+b2) = 2a1 + b1
Es gibt also zwei Normalschwingungen mit der Symmetrie a1 und eine mit der Symmetrie b1.
133

3. Elektronisches Spektrum
a) Terme zweiatomiger Moleküle mit einem ValenzelektronDie elektronischen Zustände von Molekülen werden an den einfachsten Objekten erörtert:den zweiatomigen Molekülen, wobei zunächst davon ausgegangen wird, daß das Molekül we-der vibriere noch rotiere. Die Zustände sind dann ähnlich wie die im Atom im starken Feld,da die Molekülachse eine Vorzugsrichtung darstellt. Die Molekülachse wird als z-Richtunggewählt. Zur Beschreibung des Zustandes dienen folgende Quantenzahlen:
n: die Hauptquantenzahl bezeichnet die Elektronenschale wie früherl: der Bahnmdrehimpuls des Elektrons ist keine gute Quantenzahl mehr, da der Dreh- impuls um die Molekülachse präzediert. l = 0, 1, ..., n-1lz: statt l ist die Projektion von ll auf die Molekülachse eine geeignete Quantenzahllz = mlh, ml = l, l - 1, ..., l
ml hat also die Funktion der magnetischen Quantenzahl. Im Unterschied zum Atom im Ma-gnetfeld haben Zustände mit ±ml die gleiche Energie, da die Energie ohne Aufhebung der
Entartung, etwa durch eine Störung, unabhängig von der Umlaufrichtung des Elektrons umdie Molekülachse ist. Man führt daher eine neue Quantenzahl ein:
λ = |ml|mit folgender Zuordnung
Die Termsymbole entsprechen den lateinischen Buchstaben, S, P, D, F,... Dabei sindσ-Zustände bezüglich ml einzeln, können aber wegen der zwei möglichen Spineinstellungen
des Elektrons ms = ±1 mit zwei Elektronen besetzt werden. Alle anderen Zustände mit λ ≠ 0sind doppelt wegen ml = ±λ und können daher mit 4 Elektronen besetzt werden.
Die Quantenzahlen zur Beschreibung des Zustandes in einem Molekül sind also (n, (l), λ, ms).Um die Herkunft der Terme aus denen der Einzelatome anzugeben, werden diese durchNachstellung der Bezeichnungen für die Einzelatome gekennzeichnet, z.B.
σ1sA;oder;σ2pB
Man kann auch die Termbezeichnung des vereinigten Atoms vorstellen.Durch den Index g oder u wird angegeben, ob es sich um gerade oder ungerade Zustände be-züglich Inversion handelt, wobei wir wie früher durch geeignete Kombination gleicher Einze-latomzustände gerade oder ungerade Zustände bilden können:
134

σg1s = N(σ1sA + σ1sB)σu1s = N(σ1sA − σ1sB)
N ist die Normierungkonstante. Nicht bindende Zustände werden mit einem Stern versehen.Abb.100 zeigt die beiden tiefsten Zustände mit den niedrigsten Energien beim H2-Molekül.Rechts und links sind die Einzelatomzustände (AO = Atomare Orbitale) gezeichnet, dazwi-schen die des durch die Vereinigung entstandenen Moleküls.
Die Zustände werden wie beim Atom von kleinsten Energien anfangend unter Beachtung desPauliverbots besetzt. Beim H2 ist also der bindende Zustand vollständig mit 2 Elektronen be-
setzt. Beim He2 ist auch der nichtbindende Zustand 1σu* mit zwei Elektronen besetzt. Die An-
zahl der Elektronen in bindenden Zuständen minus der in nichtbindenden geteilt durch 2,nennt man auch die Bindungsordnung; in H2 ist sie 1, in He2 0. An ihr kann man die Stärkeder Bindung erkennen. H2 ist stabil, He2 instabil. Durch Anregung kann beim He2 ein Elek-tron aus einem nicht bindenden Niveau in ein bindendes übergehen. Dies ist der Grund fürExcimerbildung.
Abb. 101 zeigt die 18 tiefsten Niveaus der leichten homonuklearen Moleküle und ihre Auffül-lung bei N2 und O2. Die drei Elektronenpaare in den stabilen 2p Niveaus bei N2 entsprechen
der Dreifachbindung. Beim O2 kommen zwei Elektronen im nichtbindenden πg* 2p Orbital
hinzu, die die Bindung der Elektronen im σg2p - Orbital kompensieren, so daß eine Zwei-fachbindung übrigbleibt.
135
Abb. 100: Die tiefsten elektronischen
Zustände von zweiatomigen
Molekülen
Abb. 101: Die Besetzung der tiefsten Niveaus bei
Stickstoff und Sauerstoff

Die Lage der einzelnen Niveaus in einer Schale kann von Atom zu Atom variieren. EinenÜberblick verschafft das Korrelationsdiagramm Abb. 102
Hier sind links die Niveaus des vereinigten Atoms, rechts die der Einzelatome aufgeführt. Zu-stände gleicher Symmetrie rechts und links sind so verbunden, daß sich Linien, die von Zu-ständen gleicher Symmetrie ausgehen, nicht kreuzen. Die Lage unterschiedlicher Moleküle indiesem Diagramm ist angegeben. Anhand der Lage der Verbindungslinien läßt sich die relati-ve Lage der Terme ermitteln.
b) Die Kopplung mehrerer Elektronen im zweiatomigen MolekülBei mehreren Elektronen muß man beachten, wie die verschiedenen Drehimpulse zum Ge-samtdrehimpuls des Systems koppeln. Entsprechend der LS und jj-Kopplung im Atom gibt esverschiedene extreme Kopplungsfälle. Nach Hund unterscheidet man zwischen 4 Kopp-lungsfällen, von denen wir zwei angeben. Sie unterscheiden sich im wesentlichen in dem Ein-fluß der Molekülachse.Im Hundschen Kopplungsfall A sind s und ll an die Molekülachse gekoppelt.Die Projektionen der Drehimpulse auf die Achse addieren sich algebraisch zur Projektion desGesamtdrehimpulses
Lz = ±Λh/Λ = Σ (±λ i)
Die Zuordnung zwischen der Termbezeichnung und dem Wert von Λ ist dabei wie im Atom,nur daß statt lateinischer Buchstaben griechische verwendet werden:
Die Spins der einzelnen Elektronen addieren sich zum Gesamtspin S. S addiert sich mit Λ Λ , dem Vektor der Länge Λ, der in z-Richtung zeigt, vektoriell, so daß die Projektionen von S
136
Abb. 102: Korrelationsdiagramm

auf die Achse, Σ, ganzzahlige Differenzen haben. Σ und Λ addieren sich algebraisch zur z-Komponente des gesamten Elektronendrehimpulses Ω.
Ω = Σ + ΛDie Verhältnisse sind ähnlich wie beim Paschen-Back-Effekt.Der Gesamtdrehimpuls des ganzen Moleküls J ergibt sich dann aus einer vektoriellen Additi-on des Impulses der Kernrotation (Hantelimpuls) N und der Elektronen
J = N + ΩΩ
Die Multiplizität der Feinstruktur ist wie bei Atomen durch 2S+1 gegeben.
Übergänge gehorchen den Auswahlregeln
∆Λ = 0, ±1∆S = 0
d.h. Interkombinationslinien sind i.a. wie bei Atomen verboten. Bei schwereren Atomen kom-men sie vor. Beispiele sind die atmosphärische Sauerstoffbande, die durch einen Übergang3Σ→1Σ des O2 entsteht und die Cameron Bande von CO mit dem Übergang 3Π - 1Σ:Beispiel:Für drei Valenzelektronen sind die Quantenzahlen
n = 2 ll = 1 λ = 1
n = 3 ll = 0 λ = 0
n = 3 ll = 0 λ = 1
Damit ergeben sich für Λ die beiden Möglichkeiten
oder 0 (Σ)Λ = λ1 ± λ2 = 2 (∆)
137
Abb. 103: Addition der Drehimpulse beim Hundschen
Kopplungsfall A
Abb. 104: Vektorgerüst für die Addition von S und ΛΛ im
Hundschen Kopplungsfall A

Die Komponente des Spins auf die Molekülachse kann für drei Elektronen 1/2 und 3/2 betra-gen. Mit den beiden möglichen Vorzeichen ergeben sich also insgesamt 4 mögliche Termeder Spinaufspaltung. Die Multiplizität ist also wie beim Atom durch 2S + 1 gegeben. Für
Λ = 2 ergeben sich also die vier möglichen Terme
4∆7/2, 4∆5/2, 4∆3/2, 4∆1/2
Beim Hundschen Kopplungsfall B ist S schwach an die Achse gekoppelt.
Hier addieren sich zunächst N und ΛΛ vektoriell zu einem Vektor K, der dann mit dem Spin-
vektor den Gesamtdrehimpuls J ergibt.
c) IntensitätenDie Intensitäten im Vibrationsspektrum werden durch das Franck-Condon-Prinzip geregelt.Dies stützt sich auf die Born-Oppenheimer Näherung, die für die Darstellung der elektroni-schen Übergänge zwischen zwei Potentialkurven wie in Abb. 106 besagt, daß die Übergängein diesem Bild immer auf senkrechten Geraden erfolgen. Um herauszufinden, zwischen wel-chen Schwingungsquantenzahlen v die intensivsten Übergänge stattfinden, wählt man nachFranck-Condon solche Werte für R, bei denen im oberen und im unteren Niveau die größteBesetzung vorliegt.
Maßgeblich für die Intensität ist also der Grad der Überlappung der Wellenfunktionen im
oberen und unteren Zustand bei gleichem R.
∫ ψ(R)ψ”(R)dr
138
Abb. 105: Hundscher Kopplungsfall B
Abb. 106: Zur Veranschaulichung des Franck-
Condon Prinzips

Im Beispiel von Abb. 106a wird angenommen, daß das Molekül anfangs im Grundzustand
vorliegt. Da hier ψ2 in der Mitte ein Maximum besitzt, während in den angeregten Niveausdem klassischen Bilde entsprechend am Rand die größte Aufenthaltswahrscheinlichkeit vor-liegt, werden die intensivesten Übergänge von v'' = 0 nach v' = 5 oder die benachbartenQuantenzahlen zu beobachten sein, während im Falle der Bindungsneutralität (Abb. 106b) dieÜbergänge hauptsächlich zwischen Niveaus mit gleichem v stattfinden.Um sich einen Überblick über die Lage der beiden Potentialkurven zu verschaffen, ist das so-genannte Kantenschema nützlich. Hier zeichnet man in ein Diagramm v'/v'' die Lage der hell-sten Linien ein. Im Falle der Bindungsneutralität (Re' = Re'') erhält man eine Gerade, im FalleRe' ≠ Re'' eine Parabel. (Abb. 107)
d) PhotodissoziationDas Franck-Condon Prinzip macht auch verständlich, warum direkte Dissoziation aus demGrundzustand durch einen optischen Prozeß nicht möglich ist. Da die höheren Niveaus in derMitte praktisch keine Aufenthaltswahrscheinlichkeit besitzen, ist ein Übergang entlang einervertikalen Linie beliebig unwahrscheinlich. Ein Übergang mit Änderung des Atomabstandesist aber verboten. Die Dissoziation erfolgt also über Stöße oder optisch über Anregung einesZwischenzustandes (Abb. 108). Durch Ermittlung der Konvergenzgrenze im oberen Niveauund der Anregungsenergie aus der Ausstrahlung der Zerfallsprodukte läßt sich die Dissoziati-onsenergie bestimmen.
139
Abb.107: Kantenschema
Abb. 108: Möglichkeit der Photodissoziotion

4. Ramanspektrum
a) Die klassische Beschreibung des Schwingungs-Ramaneffektes
Bei der Ramanstreuung werden die Moleküle in einer Probe der Strahlung einer Welleausgesetzt
E0 = cosω0tE
Dadurch wird im Molekül ein Dipolmoment p induziert
p = αE = αE cos ω0t
Wenn nun die Polarisierbarkeit α vom Atomabstand im Molekül abhängt, und das Molekülmit der Frequenz ωv vibriert, ergibt sich für das Dipolmoment mit R = Re + R cos ωvt;
p(t) = α(Re) + dα
dRR cos ωvt
cos ω0t
Nach dem Additionstheorem des Kosinus kann man die Schwingung von p(t) und damit dasgestreute Licht auffassen als die Überlagerung zweier Wellen mit
(Antistokesstrahlung)ωs = ω0 + ωv
(Stokesstrahlung)ωs = ω0 − ωv
b) RamanaktivitätMoleküle sind also ramanaktiv, wenn die Polarisierbarkeit vom Abstand der Atome abhängt.
dαdR
≠ 0
Zu diesen Molekülen gehören alle zweiatomigen homonuklearen Moleküle und lineare mitInversionszentrum. Letztere zeigen eine gewisse Komplementarität bezüglich Infrarot undRamanaktivität.So ist z.B. in CO2 die symmetrische Streckschwingung infrarot inaktiv, da mit dem symmetri-schen Molekül kein Dipolmoment verbunden ist, aber Raman aktiv, da sich die Polarisierbar-keit mit dem Abstand der O-Atome vom C-Atom ändert. Andererseits ist die asymmetrischeStreckschwingung des CO2 infrarot aktiv, da mit der asymmetrischen Verschiebung der Ato-me die Erzeugung eines Dipolmomentes verbunden ist. Die Änderung der Polarisierbarkeit
140
Abb. 109: Der Raman Effekt wird im Streulicht
beobachtet

kompensiert sich aber gerade in den beiden Hälften des Moleküls, da an einer Seite eine Stau-chung stattfindet, wenn die andere gestreckt wird.
b) Quantenmechanische BeschreibungNach der quantenmechanischen Beschreibung wird bei dem Stokesanteil des gestreuten Lich-tes dem einfallenden Licht ein Quant zur Anregung einer Vibration entnommen, bei dem An-tistokesanteil ein bei der Abregung einer Schwingung frei gewordenes Lichtquant der einfal-lenden Strahlung zugefügt.
c) Kohärente RamanstreuungBei der Überlagerung zweier Wellen tritt neben der Mischung im Frequenzraum eine Mi-schung im Wellenzahlraum auf. Wenn die Eingangswellen den Verlauf sin(ω1t - k1r) undsin(ω2t - k2r) haben, enthält das Ausgangssignal Wellen mit
ks = k1 + k2
ks = k1- k2
Im Quantenbild ist die Summierung von ω und k der Energie und Impulssatz
h/ωs = h/ω1 + h/ω2
h/ks = h/k1 + h/k2
Bei der kohärenten Ramanstreuung strahlt man zwei Wellen so ein, daß die Differenzfre-quenz mit einer Molekülschwingung zusammenfällt. Da außer dieser Frequenzanpassung eineWellenzahlanpassung stattfinden muß, wird die Streustrahlung in einen kleinen Raumwinkelum den Wellenvektor ks
gestrahlt, der die Dreiecksbeziehung der k erfüllt. Der Vorteil ist ei-ne beträchtliche Intensitätserhöhung im Streulicht verglichen zur inkohärenten Ramannstreu-ung, da dort das Licht in alle Richtungen gestreut wird.Ein anderer Vorteil kann durch die bei Zweiphotonenprozessen geänderten Auswahlregelnentstehen.Die verschiedenen Techniken der kohärenten Ramanspektroskopie benutzen verschiedeneArten der Frequenzmischung.
141
Abb. 110: Übergänge bei Stokes- und Antistokes
Strahlung
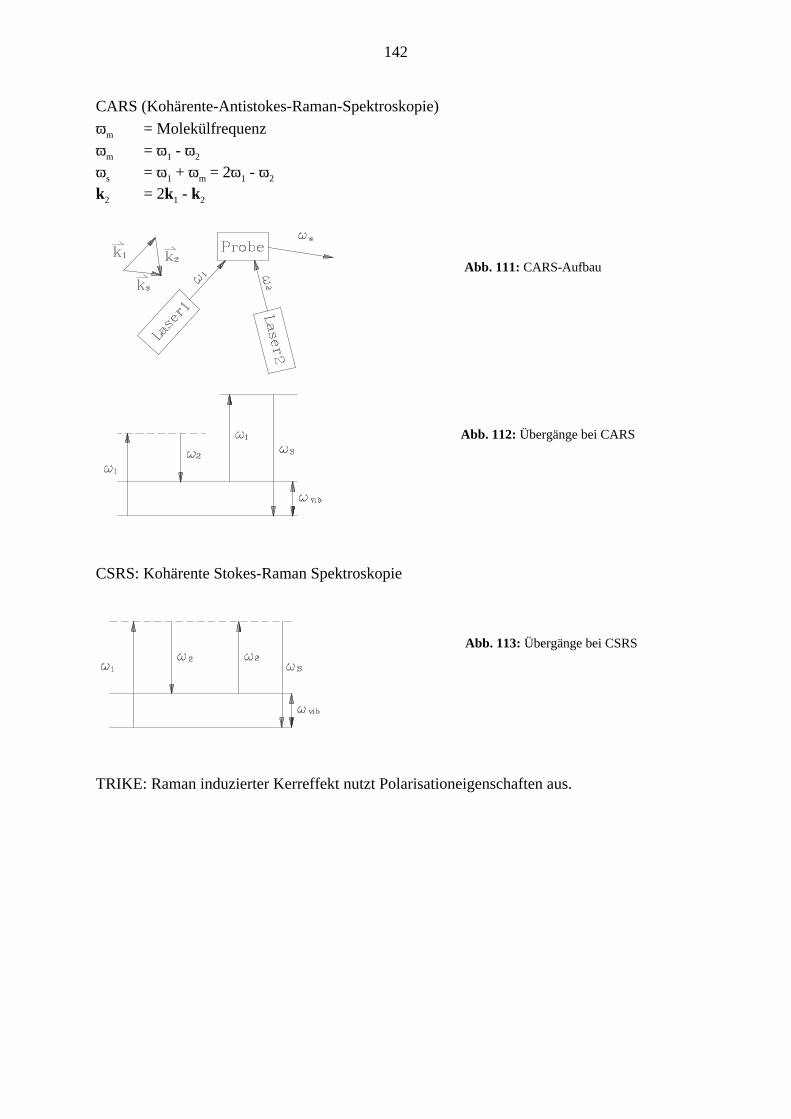
CARS (Kohärente-Antistokes-Raman-Spektroskopie)
ωm = Molekülfrequenzωm = ω1 - ω2
ωs = ω1 + ωm = 2ω1 - ω2
k2 = 2k1 - k2
CSRS: Kohärente Stokes-Raman Spektroskopie
TRIKE: Raman induzierter Kerreffekt nutzt Polarisationeigenschaften aus.
142
Abb. 111: CARS-Aufbau
Abb. 113: Übergänge bei CSRS
Abb. 112: Übergänge bei CARS