
Klinikleitfaden Hämatologie Onkologie - shop.elsevier.de · 1 Spezielles in der Onkologie und...
22
Klinikleitfaden Hämatologie Onkologie K. Possinger A. C. Regierer J. Eucker (Hrsg.) onkologiewelt.de
Transcript of Klinikleitfaden Hämatologie Onkologie - shop.elsevier.de · 1 Spezielles in der Onkologie und...

KlinikleitfadenHämatologieOnkologie
K. Possinger A. C. Regierer J. Eucker (Hrsg.)
onkologiewelt.de

Inhaltsverzeichnis
1 Spezielles in der Onkologie und Hämatologie 11.1 Allgemeines 21.2 Befundmitteilung – Sprechende Medizin 21.3 Onkologie und Psyche 31.4 Ausgangssituationen für eine Behandlung 81.5 Therapieerfolgs- und Toxizitätsbeurteilung 91.6 Therapieplanung und -durchführung 10
2 Supportive Therapien 312.1 Übelkeit und Erbrechen 322.2 Schmerztherapie 362.3 Maligne Ergüsse 412.4 Fertilitätserhaltende Maßnahmen 45
3 Stammzelltransplantation (SZT) 473.1 Einleitung 483.2 Autologe Stammzelltransplantation 483.3 Allogene Stammzelltransplantation 493.4 Transplantatabstoßung 563.5 Rezidiv und Immuntherapie mit
Spender-Lymphozyten 60
4 Infektionen bei Patienten mit hämatologisch-onkologischen Erkrankungen 61
4.1 Prädisposition 624.2 Defi nitionen 624.3 Risikoeinteilung 624.4 Spezielle Risikofaktoren 644.5 Typische klinische Syndrome 654.6 Erregerspektrum 654.7 Klinik 664.8 Diagnostik 674.9 Therapie 704.10 Hygienemaßnahmen bei granulozytopenischen
Patienten (< 1 000 Leukozyten/μl) 75
5 Tumor- und therapieassoziierte Notfälle 795.1 Tumorassoziierte Notfälle 805.2 Therapieassoziierte Notfälle 89
6 Anämien 956.1 Allgemeine Grundlagen 966.2 Eisenmangelanämie 1006.3 Anämie bei chronischer Erkrankung 1046.4 Thalassämien 1056.5 Isolierte Aplasie der Erythropoese (Pure Red Cell
Aplasia) 106
+24296_Possinger.indb XIX96_Possinger.indb XIX 22.06.2018 09:40:4122.06.2018 09:40:41

XX Inhaltsverzeichnis
6.6 Sideroachrestische Anämien 1066.7 Megaloblastäre Anämien 1076.8 Vitamin-B12-Mangelanämie 1086.9 Folsäuremangelanämie 1116.10 Hämolytische Anämien – Allgemeines 1126.11 Hereditäre Sphärozytose 1156.12 Glukose-6-Phosphat-Dehydrogenase-
(G-6-PDH-)Defi zienz und Pyruvatkinase-(PK-)Mangel 116
6.13 Sichelzellanämie 1176.14 Paroxysmale nächtliche Hämoglobinurie (PNH) 1196.15 Mikroangiopathische hämolytische Anämien
(MAHA) 1206.16 Autoimmunhämolytische Anämien (AIHA) 1226.17 Medikamenteninduzierte Immunhämolysen 124
7 Aplastische Anämien 1277.1 Aplastische Anämie 1287.2 Isolierte aplastische Anämie (PRCA) 135
8 Akute Leukämien 1378.1 Defi nition 1388.2 Akute lymphatische Leukämie (ALL) 1388.3 Akute myeloische Leukämie (AML) 145
9 Myelodysplastische Syndrome (MDS) 153
10 Chronische myeloproliferative Neoplasien (CMPN) 161
10.1 Defi nition 16210.2 Chronische myeloische Leukämie (CML) 16210.3 Polycythaemia vera (PV) 16810.4 Primäre Myelofi brose (PMF) 17010.5 Essenzielle Thrombozythämie (ET) 17110.6 Chronische eosinophile Leukämie (CEL),
hypereosinophiles Syndrom (HES) 17310.7 Systemische Mastozytose (SM) 175
11 Hodgkin-Lymphom (HL) 177
12 Indolente (niedrigmaligne) Non-Hodgkin-Lymphome 185
12.1 Non-Hodgkin-Lymphome (NHL) 18612.2 Follikuläres Lymphom (FL) 18612.3 Mantelzell-Lymphom (MCL) 19112.4 Chronische lymphatische Leukämie (CLL) 19312.5 Lymphoplasmozytisches Lymphom (LPL) und
Waldenström-Makroglobulinämie 20012.6 Marginalzonen-B-Zell-Lymphom (MZL) 20312.7 Haarzellleukämie 205
+24296_Possinger.indb XX96_Possinger.indb XX 22.06.2018 09:40:4122.06.2018 09:40:41

13 Aggressive (hochmaligne) Non-Hodgkin-Lymphome (NHL) 209
13.1 Grundlagen 21013.2 Diffuse großzellige B-Zell-Lymphome (DLBCL) 21313.3 Burkitt-Lymphom (BL) 21813.4 Periphere T-Zell-Lymphome (PTCL) und natürliche
Killerzell-Lymphome/ T-Zell-Lymphome (NKTCL) 21913.5 Lymphoblastisches Lymphom 220
14 Plasmazellneoplasien 22314.1 Monoklonale Gammopathie 22414.2 Monoklonale Gammopathie unklarer Signifi kanz
(MGUS) 22414.3 Multiples Myelom (MM) 226
15 Malignome des Respirationstrakts 23715.1 Lungenkarzinome 23815.2 Kleinzellige Lungenkarzinome (SCLC) 24115.3 Nicht kleinzellige Lungenkarzinome (NSCLC) 24515.4 Tumoren des Mediastinums 25615.5 Pleuramesotheliom 261
16 Kopf-Hals-Karzinome 267
17 Malignome des Gastrointestinaltrakts (GIT) 27517.1 Ösophaguskarzinom 27617.2 Magenkarzinom 27917.3 Pankreaskarzinom 28417.4 Cholangiozelluläres und Gallenblasen karzinom 30117.5 Hepatozelluläres Karzinom (HCC) 31517.6 Dünndarmtumoren 33217.7 Kolorektales Karzinom (CRC) 33517.8 Analkarzinom 350
18 Malignome des Urogenitaltrakts 35718.1 Nierenzellkarzinom 35818.2 Malignome der ableitenden Harnwege 36318.3 Prostatakarzinom 36918.4 Hodentumoren 37918.5 Peniskarzinom 390
19 Mammakarzinome 39719.1 Mammakarzinom – Allgemeines 39819.2 Invasives Karzinom: Stadium I–III 40519.3 Lokalrezidiv/lokoregionäres Rezidiv 41119.4 Metastasierte Erkrankung: Stadium IV 41119.5 Infl ammatorisches Mammakarzinom 41419.6 Auswahl von Behandlungsschemata 415
+24296_Possinger.indb XXI96_Possinger.indb XXI 22.06.2018 09:40:4122.06.2018 09:40:41

XXII Inhaltsverzeichnis
20 Gynäkologische Tumoren 42120.1 Ovarialtumoren 42220.2 Zervixkarzinom 43020.3 Endometriumkarzinom 436
21 Tumoren der Haut 44121.1 Melanom 44221.2 Basalzellkarzinom 44821.3 Merkelzellkarzinom 449
22 Malignome der Weichteile und der Knochen 45122.1 Weichteilsarkome 45222.2 Gastrointestinaler Stromatumor (GIST) 45622.3 Knochentumoren 459
23 Malignome endokriner Organe 46523.1 Schilddrüsenkarzinom 46623.2 Karzinome der Nebenschilddrüsen 47223.3 Tumoren der Nebenniere (NN) 47323.4 Neuroendokrine Neoplasien (NEN) des
gastroenteropankreatischen Systems 479
24 Malignome unbekannter Primärlokalisation 501
25 Malignome des zentralen Nervensystems (ZNS) 51325.1 ZNS-Tumoren allgemein 51425.2 Gliome 52525.3 Kindliche ZNS-Tumoren 52725.4 Tumoren im Sellabereich 52825.5 Meningeome 52925.6 Tumoren der Hirnnerven und peripheren Nerven 53025.7 Hirnmetastasen und Meningeosis neoplastica 531
26 HIV-assoziierte Malignome 53926.1 Allgemeines 54026.2 HIV-assoziiertes Kaposi-Sarkom 54126.3 HIV-assoziierte hochmaligne Non-Hodgkin-Lymphome
(NHL) 54426.4 HIV-assoziiertes Zervixkarzinom 54826.5 Stammzelltransplantation bei HIV-Infektion 54926.6 Antiinfektive Therapie während der
Krebsbehandlung 550
27 Hämorrhagische Diathesen 55127.1 Das Hämostasesystem 55227.2 Spezielle hämorrhagische Diathesen 558
+24296_Possinger.indb XXII96_Possinger.indb XXII 22.06.2018 09:40:4122.06.2018 09:40:41

28 Thrombophilie, thromboembolische Erkrankungen und antithrombotische Therapie 579
28.1 Thrombophilie (Hyperkoagulabilität) 58028.2 Venöse thromboembolische Erkrankungen (VTE) 58528.3 Arterielle thromboembolische Erkrankungen 58928.4 Antikoagulationstherapie 59028.5 Fibrinolytika 594
Index 597
+24296_Possinger.indb XXIII96_Possinger.indb XXIII 22.06.2018 09:40:4122.06.2018 09:40:41

8
8.1 Defi nition 1388.2 Akute lymphatische Leukämie
(ALL) 1388.3 Akute myeloische Leukämie
(AML) 145
Akute LeukämienMarkus Schaich
+24296_Possinger.indb 13796_Possinger.indb 137 22.06.2018 09:40:5922.06.2018 09:40:59

138 8 Akute Leukämien
8
8.1 Defi nition• Maligne Erkrankungen mit klonaler Vermehrung hämatopoetischer Vor-
läuferzellen.• Vermehrung von lymphatischen Vorläuferzellen des KM, des lymphatischen
Systems oder des Th ymus Akute lymphatische Leukämie (ALL).• Vermehrung von myeloischen Vorläuferzellen des KM Akute myeloische
Leukämie (AML).• I.d.R. plötzliches Auft reten, rasche Progredienz und unbehandelt rascher
Tod.• Durch Expansion des leukämischen Zellklons (Blasten) im KM Verdrängung
der normalen Hämatopoese und Ausschwemmung der Blasten in das peri-phere Blut.
8.2 Akute lymphatische Leukämie (ALL)Epidemiologie • Ca. 20 % der akuten Leukämien des Erw. und ca. 80 % der akuten Leukämien
im Kindesalter.• Höchste Inzidenz im Kindesalter: ca. 7/100 000. Kontinuierlicher Abfall mit
zunehmendem Alter: ca. 0,5/100 000 zwischen 30. und 40. Lj. Zweiter Häufi g-keitsgipfel im Alter > 80 J. mit ca. 2/100 000.
• Verhältnis Männer : Frauen: ca. 1,4 : 1.Ätiologie• Weitgehend unbekannt.• Faktoren mit erhöhter ALL-Inzidenz:
– Seltene hereditäre Erkrankungen, z. B. Down-Syndrom, Ataxia teleangiec-tatica, Bloom-Syndrom, angeborene Immunmangelerkrankungen.
– Strahlenexposition.– Myelotoxische Substanzen, z. B. Benzol oder Zytostatika wie Melphalan,
Busulfan oder Cyclophosphamid.• Seit einiger Zeit – wahrscheinlich durch den Einsatz aggressiver Th er.-Proto-
kolle in der Hämatologie/Onkologie – Zunahme der Fälle von sek. ALL nach Chemotherapie oder Bestrahlung anderer Tumorerkrankungen.
Klassifi kationen • WHO-Klassifi kation (▶ Tab. 8.1): Einordnung der ALL zusammen mit den
lymphoblastischen Lymphomen bei den lymphatischen Vorläuferzellneopla-sien. KM-Befall < 25 %: lymphoblastisches Lymphom; KM-Befall ≥ 25 %: Leukämie.
• Immunologische Klassifi kation (▶ Tab. 8.2): für die klin. Praxis entschei-dend.
Klinik • Verdrängung der Hämatopoese: mit häufi g akutem Beginn:
– Symptome der Anämie: Müdigkeit, verminderte Leistungsfähigkeit, Dys-pnoe, Tachykardie, blasses Aussehen.
– Symptome der Granulozytopenie: Fieber bei Infektionen, v. a. von Lun-ge, Rachen, Haut und perianal, bis hin zur Sepsis. Häufi ge Keime: Staphy-lococcus spp., Pseudomonas spp., HSV, Candida spp.
– Symptome der Th rombozytopenie: Blutungen (Petechien, Ekchymosen, Menorrhagien, Epistaxis).
+24296_Possinger.indb 13896_Possinger.indb 138 22.06.2018 09:40:5922.06.2018 09:40:59

139 8.2 Akute lymphatische Leukämie (ALL)
8• Extramedullärer Befall: häufi g.
– Lymphadenopathie: 55 %.– Splenomegalie: 50 %.– Hepatomegalie: 45 %.– ZNS-Befall (6 %) mit Hirnnervenlähmungen (häufi g N. VII), Kopfschmer-
zen, Übelkeit, Erbrechen, Meningismus.– Mediastinale Lymphome: 15 %; bes. bei T-ALL, evtl. mit Zeichen einer
oberen Einfl ussstauung.– Prinzipiell Befall aller anderen Organe möglich (Retina, Haut, Hoden,
Ovarien, Niere, Lunge etc.).
Tab. 8.1 WHO-Klassifi kation der ALL (2016)
B-lymphoblastische Leukämie/Lymphom mit spezifi schen genetischen Verän-derungen
• B-lymphoblastische Leukämie/Lymphom mit t(9;22)(q34.1;q11.2); BCR-ABL1
• B-lymphoblastische Leukämie/Lymphom mit t(v;11q23.3); KMT2A rearranged
• B-lymphoblastische Leukämie/Lymphom mit t(12;21)(p13.2;q22.1); ETV6-RUNX1
• B-lymphoblastische Leukämie/Lymphom mit Hyperdiploidie• B-lymphoblastische Leukämie/Lymphom mit Hypodiploidie• B-lymphoblastische Leukämie/Lymphom mit t(5;14)
(q31.1;q32.3); IL3-IGH• B-lymphoblastische Leukämie/Lymphom mit t(1;19)
(q23;p13.3); TCF3-PBX1• Provisorische Entität: B-lymphoblastische Leukämie/Lymphom, BCR-ABL1-like• Provisorische Entität: B-lymphoblastische Leukämie/Lymphom mit iAMP21
Nicht anderweitig klassifi zierbare B-lymphoblastische Leukämie/Lymphom
T-lymphoblastische Leukämie/Lymphom
• Provisorische Entität: Frühe T-Zell-Vorläufer lymphoblastische Leukämie• Provisorische Entität: NK Zell lymphoblastische Leukämie/Lymphom
Tab. 8.2 Klassifi kation der ALL in den GMALL-Studien
Subtyp Marker Häufi gkeit (%)
B-Linien-ALL HLA-DR+, TdT+, CD19+1, CD79a+1, CD22+1 76
Pro-B-ALL CD10−, keine zusätzlichen Differenzierungsmarker 11
Common ALL CD10+ 49
Prä-B-ALL CD10+/−, cyIgM+ 12
Reife B-ALL CD10+/−, sIgM+ 4
T-Linien-ALL TdT+, cyCD3+, CD7+ 24
Early T-ALL CD2−, sCD3−, CD1a− 6
Thymische T-ALL sCD3+/−, CD1a+ 12
Reife T-ALL sCD3+, CD1a− 6
1 Mind. 2 von 3 müssen pos. sein.cy = zytoplasmatisch; s = Oberfl äche
+24296_Possinger.indb 13996_Possinger.indb 139 22.06.2018 09:40:5922.06.2018 09:40:59

140 8 Akute Leukämien
8
• Leukostase (häufi g bei Leukos > 100 000/μl):– Hypoxie.– Diff use pulmonale Verschattungen.– Retinale Einblutungen.– Verwirrtheit oder andere neurologische Auff älligkeiten.– Art. Verschlüsse (selten).! Leukostatische Symptome: Ind. zur Notfalltherapie der ALL.
Diagnostik Labordiagnostik: • BB: Hb , Leukos , Th rombos .! Auch aleukämischer Verlauf mit Leukopenie möglich!• Diff -BB: Neutrophile , Blastenpopulation nachweisbar.• Gerinnungsanalyse (Quick-Wert, PTT, Fibrinogen, AT III, Fibrinogenspalt-
produkte): Ausschluss gravierender Gerinnungsstörungen, z. B. einer Ver-brauchskoagulopathie.
• Blutgruppenbestimmung und AK-Screening mit Coombs-Test und Bestim-mung thrombozytärer AK zum Ausschluss von Auto- und HLA-AK, mit dem Ziel, Komplikationen notwendiger Transfusionen zu vermeiden.
• Klin.-chemische Untersuchung: Leber- und Nierenfunktionsparameter, Elek-trolyte zur Beurteilung der Th er.-Fähigkeit, LDH als Marker für Tumormasse und -lyse.
• Schwangerschaft stest bei Frauen im gebärfähigen Alter.• Mikrobiologische Basisdiagnostik zum Ausschluss von Infekten: Untersu-
chung von Rachen-, Nasenabstrich, Sputum, Mittelstrahlurin.• Virusserologie: Hepatitisviren A, B und C sowie HIV, CMV, HSV und VZV
zum Ausschluss akuter Viruserkrankungen bzw. um Möglichkeiten der Vi-rusreaktivierung vor Th er.-Beginn zu erkennen.
• HLA-Typisierung des Pat. und möglicher Geschwister zur Evaluierung der Option einer späteren allogenen SZT.
• Liquorpunktion zum Nachweis/Ausschluss einer ZNS-Beteiligung im Sinne einer Meningeosis leucaemica mit atypischen unreifen lymphatischen Zellenim Liquor.
! Vorher Ausschluss von Hirndruck, bei Th rombos < 50 000/μl Th rombozyten-transfusion!
Apparative Diagnostik:• Rö-Th orax und NNH zum Infektausschluss• EKG• Echo bei kardialer Vorerkrankung• CT Th orax und Abdomen wegen möglicher mediastinaler und abdominaler
LymphadenopathieZytologie und Histologie:• KM-Ausstrich: ≥ 25 % blastäre Zellen (▶ Abb. 8.1). Morphologie und Zyto-
chemie ( Myeloperoxidase-[MPO-], Sudan-Schwarz-B-, Chloracetat- und α-Naphthyl-Acetat-Esterase neg.).
• KM-Histologie: insbes. bei Punctio sicca zum Nachweis von KM-Infi ltratenunreifer lymphatischer Blasten.
Immunphänotypisierung:• Entscheidende Untersuchung für Diagnose und Risikoeinteilung.• Untersuchung der leukämischen Zellen auf ihre Oberfl ächenantigenexpressi-
on:– Abgrenzung zur minimal diff erenzierten AML.– Zuordnung zur B- oder T-Zell-Reihe.
+24296_Possinger.indb 14096_Possinger.indb 140 22.06.2018 09:40:5922.06.2018 09:40:59

141 8.2 Akute lymphatische Leukämie (ALL)
8
– Charakterisierung des Diff erenzierungsgrads.– Einordnung in eine immunologische Subgruppe der ALL.
• Empfohlenes Panel: AK gegen die beschriebenen Oberfl ächenmarker (▶ Tab. 8.2).
Zytogenetik:
Zytogenetische Untersuchung bei ALL obligat.
• Ziel: Identifi kation unabhängiger Prognosefaktoren, insbes. von t(9;22) und t(4;11).
• Nachweis zytogenetischer Aberrationen bei bis zu 85 % der Pat.– Am häufi gsten (ca. 25 %): t(9;22), sog. Philadelphia-Chromosom.– t(4;11) in ca. 6 % aller Fälle, aber bei 70 % der Pat. mit Pro-B-ALL nach-
weisbar.• Alle anderen klonalen Aberrationen: ca. 10 %.Molekularbiologie:• Additiver Einsatz zum sehr sensitiven Nachweis der Fusionstranskripte klo-
naler Translokationen• Insbes. wichtig zum Nachweis der minimalen Resterkrankung (MRD) im
Th er.-VerlaufRisikofaktoren Wichtige RF: Alter, Leukozytenzahl, Zeit bis zum Erreichen einer CR, Immunphänotyp, Zyto- bzw. Molekulargenetik und MRD.Risikogruppen für Pat. bis zu einem Alter von 55 J.:• Standardrisiko (SR):
– B-Vorläufer-ALL, wenn folgende Bedingungen vorliegen: CR nach Induk-tion I und Leukos < 30 000/μl, keine Pro-B-ALL bzw. t(4;11)/ALL1-AF4-pos. ALL, keine t(9;22)/BCR-ABL-pos. ALL.
– Th ymische T-ALL.• Hochrisiko (HR):
– B-Vorläufer-ALL, wenn folgende Bedingungen vorliegen: CR erst nach Induktion II oder Leukos > 30 000/μl oder Pro-B-ALL bzw. t(4;11)/ALL1-AF4-pos. ALL, keine t(9;22)/BCR-ABL-pos. ALL.
– Early T-ALL.– Reife T-ALL.
• t(9;22)/BCR-ABL-positive ALL.
Abb. 8.1 Lymphatische Blastenpopulation [M927]
+24296_Possinger.indb 14196_Possinger.indb 141 22.06.2018 09:40:5922.06.2018 09:40:59

142 8 Akute Leukämien
8
Mithilfe der MRD weitere Unterteilung der SR-Pat. im Krankheitsverlauf (Wo. 16):• MRD-Standardrisiko: MRD-Niveau < 10–4 vor Konsolidation I (Tag 71) und
Konsolidation II (Wo. 16).• MRD-Hochrisiko: MRD-Niveau > 10–4 an Tag 71 und Wo. 16.
Pat., die keine molekulare Remission erreichen, haben ein hohes Rückfallri-siko Bestimmung der MRD bei der ALL unverzichtbar.
Th erapie Zu aktuellen Studienprotokolle, Th er.-Empfehlungen, Behandlungsschemata und Kontaktadressen der GMALL (German Multicenter ALL-Studiengruppe) siehe Homepage des Kompetenznetzes „Akute und chronische Leukämien“ unter http://www.kompetenznetz-leukaemie.de.Spezifi sche Th erapie:
Wenn möglich, Th er. der ALL im Rahmen des ALL-Registers oder einer lau-fenden Th er.-Studie und an einem hämatologisch-onkologisch erfahrenen Zentrum. Pat. im fortpfl anzungsfähigen Alter vor Beginn der Th er. immer über fertilitätserhaltende Maßnahmen und Notwendigkeit der Antikonzep-tion aufk lären.
Systemische Th erapie:• B-Vorläufer-/T-Linien-ALL: 3 Phasen:
– Induktion:– Ziel: komplette hämatologische Remission (CR; normale KM-Zellulari-tät mit < 5 % Blasten und Normalisierung des peripheren BB) und gute Remissionsqualität auf molekularer Ebene (molekulare CR).– Besteht meist aus einer Vorphase und 2 Phasen einer Kombinationsche-motherapie mit Dexamethason, Vincristin, einem Anthrazyklin und As-paraginase. Zur frühen Intensivierung weiterhin Cyclophosphamid, Ara-C und Mercaptopurin.– Durch parallele Gabe von G-CSF höhere Dosisintensität mit höherer Remissionsrate und niedriger Frühmortalität.– Remissionskontrolle im KM nach den Induktionen I und II (prognosti-scher Faktor) sowie MRD-Kontrolle!
– Konsolidierung:– Ziel: nach Erreichen der CR weitere Reduktion der Leukämielast mit Verbesserung des leukämiefreien Überlebens.– Verminderung der Resistenzentwicklung durch zyklische Gabe wechseln-der Polychemotherapien mit in der Induktionstherapie noch nicht verwen-deten Substanzen wie Vindesin, Etoposid, Hochdosis-MTX oder Hochdo-sis-Ara-C und modifi zierter Einsatz der Induktionstherapie (Re-Induktion).– Risikoadaptierte Konsolidierungsstrategie mit allogener Familien- oder Fremdstammzelltransplantation für Pat. mit HR- oder Philadelphia-Chromosom-pos. (Ph+) ALL und für Pat. mit SR und einem molekularen Th er.-Versagen (MRD-Hochrisiko) in Wo. 16. Ist kein Spender vorhan-den, autologe SZT in Betracht ziehen. SR-Pat. nur mittels Chemotherapie wie oben beschrieben konsolidieren.– Bei Pat. mit Ph+ ALL während der Induktion und Konsolidierung zu-sätzlich Imatinib deutliche Prognoseverbesserung. Derzeit Überprüfung
+24296_Possinger.indb 14296_Possinger.indb 142 22.06.2018 09:40:5922.06.2018 09:40:59

143 8.2 Akute lymphatische Leukämie (ALL)
8
von Th yrosinkinaseinhibitoren (TKI) der 2. Generation wie Dasatinib oder Nilotinib zur Th er.-Optimierung.– Zusätzliche Gabe des Anti-CD20-AK Rituximab zur Chemotherapie während Induktion und Konsolidierung bei der CD20+-B-Vorläufer-ALL.
– Erhaltungstherapie:– Ziel: Verhinderung von Spätrezidiven.– Nur für Pat., die keine SZT erhalten, also insbes. SR-Pat. mit molekula-rer CR in Wo. 16.– Standard: Kombination von MTX und Mercaptopurin bis zu einer Ge-samttherapiedauer von 2,5 J.
• Reife B-ALL: konventionelle ALL-Th er. weitgehend unwirksam. Wegen ra-scher Progredienz und großer Tumorzellmasse hohe Dosisintensität in Form kurzer Chemotherapiezyklen über 6 Mon. ohne Erhaltungstherapie, v. a. mit Hochdosis-MTX, Hochdosis-Ara-C, Cyclophosphamid und Ifosfamid. Wei-tere Intensivierung und Verbesserung der Th er.-Ergebnisse durch zusätzliche Gabe von Rituximab.
ZNS-Prophylaxe: integraler Bestandteil der ALL-Th erapie. Intrathekale Th er. mit 15 mg MTX absolut allein oder in Kombination mit 40 mg Ara-C und 4 mg Dexa-methason (jeweils absolut), parallel zur systemischen Chemotherapie, ergänzt durch die prophylaktische Schädelbestrahlung.Stammzelltransplantation: Empfehlung einer allogenen SZT für Pat. mit HR- oder Ph+ ALL sowie für SR-Pat. mit molekularem Th er.-Versagen möglichst früh-zeitig in 1. CR und einer möglichst guten molekularen Remission. Der Chemothe-rapie im Hinblick auf das leukämiefreie Überleben überlegen.Besonderheiten der Th erapie des älteren Patienten: Die ALL des älteren Men-schen weist im Gegensatz zur Erkrankung des jüngeren Menschen folgende Be-sonderheiten auf:• Häufi ger prognostisch ungünstige Subgruppen.• Höhere Chemotherapieresistenz.• Höhere Komorbidität.
Schlechtere Durchführbarkeit intensiver Chemotherapiezyklen mit geringeren Remissionsraten und kürzerem Überleben!• Prinzipien:
– Kurative Ansätze meist nur mit verkürzten, dosisreduzierten Chemothe-rapien möglich.
– Rituximab kann zur nebenwirkungsarmen Intensivierung eingesetzt werden.– Zugewinn an Lebenszeit durch kurative Th er. bedingt meist längere Hos-
pitalisierungsphasen.– Bei älteren HR-Pat. in gutem AZ kann trotzdem eine dosisreduzierte allo-
gene SZT erwogen werden.! Sorgfältige Abwägung zwischen AZ, möglichem Zugewinn an Lebenszeit
und der damit einhergehenden Lebensqualität bei Pat. > 55 Jahren. Geriat-risches Assessment hilfreich.
Notfalltherapie:• Neutropene Sepsis: sofortige empirische Breitspektrumantibiose, Intensiv-
therapie, ggf. Reanimation und Beatmung.• Hyperleukose oder leukostatische Symptome: Leukapherese. Ist diese nicht
möglich, rasche Senkung der peripheren Blastenanzahl durch Vorphasenthe-rapie (im Rahmen der Studien meist standardisiert; ▶ Tab. 8.3).
• Meningismus: Liquorpunktion und intrathekale Th er.! Vorher Ausschluss von Hirndruck, ggf. Th rombozytentransfusion.
+24296_Possinger.indb 14396_Possinger.indb 143 22.06.2018 09:40:5922.06.2018 09:40:59

144 8 Akute Leukämien
8
• Tumorlysesyndrom: Auft reten i. d. R. akut 12–24 h nach Beginn der Chemo-therapie: Übelkeit, Erbrechen, Hyperkaliämie, Hyperphosphatämie, Hyperuri-kämie, Hypokalzämie engmaschiges Monitoring (EKG, ZVD, Laborwerte), Hydratation, Ausgleich der Elektrolytstörungen, Senkung der Harnsäure i.S.
Supportive Th erapie:! Immer aktuelle Zulassung und Fachinformation der einzelnen Medikamente
beachten.• Haut- und Schleimhautpfl ege, insbes. der Mundschleimhaut und im Anoge-
nitalbereich.• Infektionsprophylaxe: etablierte Protokolle zur selektiven oralen Infektions-
prophylaxe:– Chinolon (Levofl oxacin 1 × 500 mg/d p. o. oder Ciprofl oxacin 2 × 250 mg/d
p. o.) und Fluconazol 2 × 200 mg/d p. o.– Colistin 4 × 6 Tbl. (= 600 mg)/d p. o. und Amphotericin B 6 × 4 ml
(= 2 400 mg)/d p. o.– Nur Amphotericin B 6 × 4 ml (= 2 400 mg)/d p. o.– Auch Verzicht auf Prophylaxe möglich höhere Rate dokumentierter In-
fektionen ohne Zunahme der Mortalitätsrate bei adäquater Infektionsthe-rapie.
! Während Th er.-Phasen mit Vinca-Alkaloiden wegen möglicher Interaktionen und erhöhter Gefahr von Neurotoxizität keine Gabe von Azolen zur Prophylaxe.
• Hyperurikämieprophylaxe: Allopurinol 1 × 300 mg/d p. o. oder Rasburicase 1 × 0,2 mg/kg KG/d i. v. während der Induktionsphase. Später bei geringerer Leukämiezellmasse nur Überwachung des Harnsäure i.S.
• Konjunktivitisprophylaxe bei Hochdosis-Ara-C-Th er.: 2-stündliche Augen-spülungen mit kortikoidhaltigen und NaCl-Augentropfen im Wechsel.
• Substitution von Erythrozyten- und Th rombozytenkonzentraten sowie vonGerinnungsfaktoren.
• Menstruationsprophylaxe bei prämenopausalen Frauen, z. B. mit Lynestre-nol (bis 3 × 5 mg/d p. o).
• Zentraler Venenzugang.Prognose Abhängig von Alter, Subgruppe und entsprechender Risikogruppe (▶ Tab. 8.4).
Tab. 8.3 Vorphasetherapie der ALL
Zytostatika Dosierung Applikation Zeitpunkt
Dexamethason 10 mg/m2 KOF p. o. Tage 1–5
Cyclophosphamid 200 mg/m2 KOF i. v. über 1 h Tage 3–5
Bei Pat. mit initialer Granulozytopenie < 500 Granulozyten/μl ab Tag 1 G-CSF
Tab. 8.4 Prognostische Kennzahlen der ALL
Alter > 55 J. Alter < 55 J.
B-Vorläufer-/T-Linien-ALL B-ALL
SR HR Ph+-ALL
Remissionsrate (%) 70–80 85–95 80–90 80–90 80–95
5-Jahres-OS (%) 20–30 60–70 30–50 50–60 70–80
+24296_Possinger.indb 14496_Possinger.indb 144 22.06.2018 09:41:0022.06.2018 09:41:00

145 8.3 Akute myeloische Leukämie (AML)
8
Nachsorge • Während der gesamten Dauer der Erhaltungstherapie in 1- bis 2-monatigen
Intervallen.• Anamnese, klin. Untersuchung, BB, KM- und MRD-Diagnostik nach Erfor-
dernissen der jeweiligen Studien.• Augenmerk insbes. auf Spätfolgen der Th er. und der Erkrankung legen: Kar-
dio-, Neurotoxizität, Sekundärneoplasien, grauer Star, Infertilität, Fatigue, hormonelle Störungen, psychische Erkrankungen.
8.3 Akute myeloische Leukämie (AML)Epidemiologie • Ca. 75–80 % der akuten Leukämien des Erw., ca. 15–20 % der akuten Leuk-
ämien im Kindesalter.• Inzidenz: altersspezifi sch. Deutliche Zunahme mit dem Alter: 3/100 000/J.
(bis 45 J.) bis 35/100 000/J. (mit 90 J.).• Männer : Frauen: ca. 1,4 : 1.• Medianes Erkrankungsalter: 63 J.Ätiologie• Ätiologie weitgehend unklar.• RF: Exposition gegenüber Chemikalien, bes. Benzol, ionisierender Strahlung
und Chemotherapeutika wie Alkylanzien oder Topoisomerase-II-Hemmer.• Bei Rauchern gering erhöhtes Risiko.• Erhöhte Inzidenz bei verschiedenen angeborenen und erworbenen Erkran-
kungen, z.B. Fanconi-Anämie, Down-Syndrom, myelodysplastische oder my-eloproliferative Syndrome.
• Sek. AML (sAML): durch Transformation eines MDS oder einer anderen KM-Erkrankung bzw. nach Chemotherapie oder Bestrahlung.
• Alle anderen Fälle: De-novo- AML.Klassifi kation WHO-Klassifi kation (▶ Tab. 8.5): Einteilung in biologische Sub-gruppen durch Korrelation morphologischer, genetischer und klin. Kriterien. Klinik • Verdrängung der Hämatopoese mit häufi g akutem Beginn:
– Symptome der Anämie: Müdigkeit, verminderte Leistungsfähigkeit, Dys-pnoe, Tachykardie, blasses Aussehen.
– Symptome der Granulozytopenie: Fieber bei Infektionen, v. a. von Lun-ge, Rachen, Haut und perianal, bis hin zur Sepsis. Häufi ge Keime: Staphy-lococcus spp., Pseudomonas spp., HSV, Candida spp.
– Symptome der Th rombozytopenie: Blutungen (Petechien, Ekchymosen, Menorrhagien, Epistaxis).
! Blutungen aber auch durch disseminierte intravasale Gerinnung (DIC) und Hyperfi brinolyse möglich (insbes. bei der APL).
• Extramedullärer Befall (insbes. bei monozytären/monoblastären AML): He-patosplenomegalie, Hautinfi ltrate, Gingivahyperplasie, ZNS-Befall. Insgesamt bei der AML des Erw. aber selten.
• Leukostase, häufi g bei Leukos > 100 000/μl: Hypoxie, diff use pulmonale Ver-schattungen, retinale Einblutungen, Verwirrtheit oder andere neurologische Auff älligkeiten, selten art. Verschlüsse.
! Leukostatische Symptome: Ind. zur Notfalltherapie der AML.
+24296_Possinger.indb 14596_Possinger.indb 145 22.06.2018 09:41:0022.06.2018 09:41:00
146 8 Akute Leukämien
8
Diagnostik Labordiagnostik:• BB: Hb-Wert , Leukos , Th rombos .! Auch aleukämischer Verlauf mit Leukopenie möglich.• Diff -BB: Neutrophile , Blastenpopulation nachweisbar.• Gerinnungsanalyse zum Ausschluss einer DIC bzw. einer Hyperfi brinolyse;
diese kann insbes. bei APL auft reten.• Blutgruppenbestimmung und AK-Screening mit Coombs-Test und Bestim-
mung thrombozytärer AK zum Ausschluss von Auto- und HLA-AK, mit dem Ziel, Komplikationen notwendiger Transfusionen zu vermeiden.
• Klin.-chemische Diagnostik: Leber-/Nierenfunktionsparameter und Elektro-lyte zur Beurteilung der Th er.-Fähigkeit, LDH als Marker für Tumormasse und Tumorlyse.
• Schwangerschaft stest bei Frauen im gebärfähigen Alter.• Mikrobiologische Basisdiagnostik zum Ausschluss von Infekten: Rachenab-
strich, Nasenabstrich, Sputum, Mittelstrahlurin.
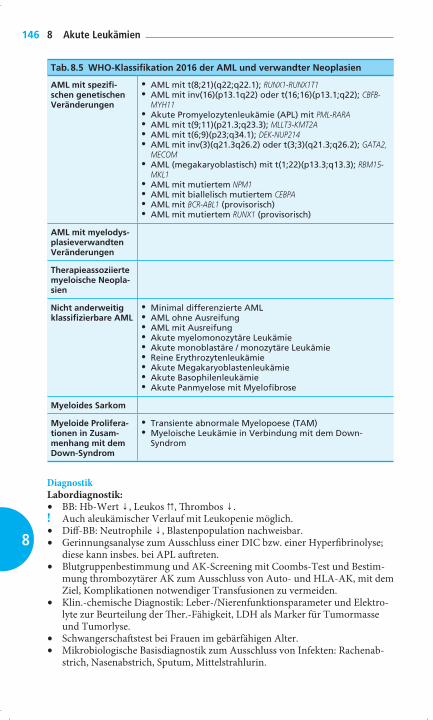
Tab. 8.5 WHO-Klassifi kation 2016 der AML und verwandter Neoplasien
AML mit spezifi -schen genetischen Veränderungen
• AML mit t(8;21)(q22;q22.1); RUNX1-RUNX1T1• AML mit inv(16)(p13.1q22) oder t(16;16)(p13.1;q22); CBFB-
MYH11• Akute Promyelozytenleukämie (APL) mit PML-RARA• AML mit t(9;11)(p21.3;q23.3); MLLT3-KMT2A• AML mit t(6;9)(p23;q34.1); DEK-NUP214• AML mit inv(3)(q21.3q26.2) oder t(3;3)(q21.3;q26.2); GATA2,
MECOM• AML (megakaryoblastisch) mit t(1;22)(p13.3;q13.3); RBM15-
MKL1• AML mit mutiertem NPM1• AML mit biallelisch mutiertem CEBPA• AML mit BCR-ABL1 (provisorisch)• AML mit mutiertem RUNX1 (provisorisch)
AML mit myelodys-plasieverwandten Veränderungen
Therapieassoziierte myeloische Neopla-sien
Nicht anderweitig klassifi zierbare AML
• Minimal differenzierte AML• AML ohne Ausreifung• AML mit Ausreifung• Akute myelomonozytäre Leukämie• Akute monoblastäre / monozytäre Leukämie• Reine Erythrozytenleukämie• Akute Megakaryoblastenleukämie• Akute Basophilenleukämie• Akute Panmyelose mit Myelofi brose
Myeloides Sarkom
Myeloide Prolifera-tionen in Zusam-menhang mit dem Down-Syndrom
• Transiente abnormale Myelopoese (TAM)• Myeloische Leukämie in Verbindung mit dem Down-
Syndrom
+24296_Possinger.indb 14696_Possinger.indb 146 22.06.2018 09:41:0022.06.2018 09:41:00

147 8.3 Akute myeloische Leukämie (AML)
8
• Virusserologie: Hepatitisviren A, B und C, HIV, CMV, HSV und VZV zum Ausschluss akuter Viruserkrankungen bzw. um Möglichkeiten der Virusreak-tivierung vor Th er.-Beginn zu kennen.
• HLA-Typisierung des Pat. und möglicher Geschwister zur Evaluierung der Option einer späteren allogenen SZT.
Apparative Diagnostik:• Rö-Th orax und -NNH zum Infektausschluss• EKG• Echo bei kardialer VorerkrankungZytologie:• KM-Ausstrich: ≥ 20 % blastäre Zellen. Morphologie und Zytochemie ( MPO-,
Sudan-Schwarz-B-, Chloracetat- und α-Naphthyl-Acetat-Esterase- Nachweis; PAS-Nachweis).
• KM-Histologie, insbes. bei Punctio sicca, zum Nachweis der KM-Infi ltrationdurch unreife myeloische Blasten.
Immunphänotypisierung: zur Abgrenzung der minimal diff erenzierten AML ge-genüber der akuten Leukämie ohne eindeutige Linienzugehörigkeit oder der ALL und zur diagnostischen Sicherheit bei Vorliegen einer reinen Erythroleukämie oder einer akuten Megakaryozytenleukämie.Zytogenetik:
Zytogenetische Untersuchung der Blasten in KM oder peripherem Blut bei jedem Pat. mit AML obligat.
• Wann immer möglich, komplette Analyse des Karyotyps (liefert unabhängige prognostische Informationen, wichtig für Th er.-Entscheidung).
• Ist die konventionelle Zytogenetik nicht möglich, Fluoreszenz-In-situ-Hybri-disierung (FISH).
Molekularbiologie:• Additiver Einsatz der (sehr sensitiven) molekularen Analyse (Reverse-Tran-
skriptase-Polymerasekettenreaktion, RT-PCR) bei Erstdiagnose (bei nicht aussagekräft iger konventioneller Zytogenetik) und zur Verlaufskontrolle (MRD-Diagnostik) mit Nachweis spezifi scher genetischer Rearrangements: z. B. PML-RARA, RUNX1-RUNX1T1, CBFB-MYH11.
• Nachweis anderer molekularer Veränderungen wie z.B. FLT3-, NPM1- oder CEBPA-Mutationen , die Einfl uss auf die Prognose der AML haben.
Risikofaktoren • Wichtige RF: klin. Faktoren wie Alter oder Ansprechen auf die 1. Induktions-
therapie. Zytogenetik wichtig zur Prognoseabschätzung und Risikoeinteilung.• Risikogruppen ▶ Tab. 8.6. Th erapie Aktuelle Studienprotokolle, Behandlungsschemata und Kontaktadres-sen der deutschen AML-Studiengruppen siehe http://www.kompetenznetz- leukaemie.de.Spezifi sche Th erapie:
Wenn möglich, Th er. der AML im Rahmen eines Registers oder in einer lau-fenden Th er.-Studie und an einem hämatologisch-onkologisch erfahrenen Zentrum. Pat. im fortpfl anzungsfähigen Alter vor Beginn der Th er. immer über fertilitätserhaltende Maßnahmen und die Notwendigkeit der Antikon-zeption aufk lären.
+24296_Possinger.indb 14796_Possinger.indb 147 22.06.2018 09:41:0022.06.2018 09:41:00

148 8 Akute Leukämien
8
Systemische Th erapie: kurative Intention, 2 Phasen:• Induktion:
– Ziel: Erreichen einer kompletten hämatologischen Remission (CR; nor-male Zellularität des KM mit < 5 % Blasten und Normalisierung des peri-pheren BB). Hämatologische CR = Reduktion der Blasten um 3 Log-Stu-fen.
– Bei Pat. ≤ 60 J. 2 Zyklen einer Kombinationschemotherapie, typischerwei-se mit Ara-C und einem Anthrazyklin (▶ Tab. 8.7).
– Bei Pat. > 60 J. mit gleicher Dosierung wie für Pat. bis 60 J. (▶ Tab. 8.7).2. Kurs nur, wenn KM an Tag 15 noch ≥ 5 % Blasten enthält.
! Während der Aplasie nach 1. Induktionstherapie KM-Punktion zur Ab-schätzung des Th er.-Ansprechens (sog. Tag-15-Punktion).
– Bei fehlendem Ansprechen Salvage-Th er., meist bestehend aus einer Poly-chemotherapie, deren wichtigstes Element Hochdosis-Ara-C darstellt.
• Konsolidierung:– Ziel: nach Erreichen der CR weitere Reduktion der Leukämielast und Ver-
ringerung des Rezidivrisikos.– Optimaler Umfang und Zusammensetzung der Konsolidierung unklar.
Standard für Pat. ≤ 60 J.: 3 Th er.-Zyklen, bestehend aus einer Chemothera-pie mit Hochdosis-Ara-C (Einzeldosis 3 g/m2, ▶ Tab. 8.7).
– Pat. ≤ 60 J.: i. d. R. risikostratifi zierte Konsolidierung: bei NR Hochdosis-Ara-C (▶ Tab. 8.7), bei SR mit HLA-kompatiblem Familienspender alloge-ne SZT in 1. CR (ohne Familienspender: Hochdosis-Ara-C-Th er.; ▶ Tab. 8.7), bei HR allogene SZT – möglichst früh in 1. CR von einem Fa-milien- oder Fremdspender.
Tab. 8.6 Risikogruppen der AML gemäß des European LeukemiaNet (ELN)
Risikogruppe Genetische Veränderungen
Niedrigrisiko (NR)
• t(8;21)(q22;q22.1); RUNX1-RUNX1T1• inv(16)(p13.1q22) oder t(16;16)(p13.1;q22); CBFB-MYH11• Mutiertes NPM1 ohne FLT3-ITD oder mit FLT3-ITD (Ratio niedrig1)• Biallelisch mutiertes CEBPA
Standard-risiko (SR)
• Mutiertes NPM1 und FLT3-ITD (Ratio hoch1)• Wildtyp NPM1 ohne FLT3-ITD oder mit FLT3-ITD (Ratio niedrig1) (ohne
gleichzeitige zytogenetische Hochrisikoveränderungen)t(9;11)(p21.3;q23.3); MLLT3-KMT2AZytogenetische Veränderungen, die nicht als Nierigrisiko oder Hoch-risiko klassifi ziert sind
Hochrisiko (HR)
• t(6;9)(p23;q34.1); DEK-NUP214• t(v;11q23.3); KMT2A rearranged• t(9;22)(q34.1;q11.2); BCR-ABL1• inv(3)(q21.3q26.2) oder t(3;3)(q21.3;q26.2); GATA2, MECOM• -5 oder del(5q); -7; -17/abn(17p)• Komplexer Karyotyp2, Monosomaler Karyotyp3
• Wildtyp NPM1 und FLT3-ITD (Ratio hoch1)• Mutiertes RUNX1• Mutiertes ASXL1• Mutiertes TP53
1 Allelische Ratio von FLT3-ITD zu FLT3-Wildtyp, niedrig < 0,5, hoch ≥ 0,5.2 3 oder mehr chromosomale Aberrationen.3 1 Monosomie und 1 weitere Monosomie oder 1 weitere strukturelle Aberration.
+24296_Possinger.indb 14896_Possinger.indb 148 22.06.2018 09:41:0022.06.2018 09:41:00
149 8.3 Akute myeloische Leukämie (AML)
8
– Pat. > 60 J.: intensive Konsolidierungsprotokolle oft nicht möglich (▶ 8.3„Besonderheiten der Th erapie von Pat. > 60 J.“). Im Vergleich zu Pat. bis 60 J. reduzierte Einzeldosis für Ara-C, nur 2 Kurse Ara-C (▶ Tab. 8.7).
Erhaltungstherapie: Wird außerhalb von Studien nicht mehr durchgeführt. Aus-nahmen: APL (▶ 8.3 „Th erapie der akuten Promyelozytenleukämie“) oder ältere AML-Pat., denen eine intensive Konsolidierung nicht zugemutet werden kann.Stammzelltransplantation (▶ 3):• Autologe SZT: In einer prädiktiv angelegten Untersuchung zu Konsolidie-
rungsstrategien bei AML-Pat. ≤ 60 J. profi tierte eine Gruppe von SR-Pat. bes. von der autologen SZT. Welcher Pat. in 1. CR in diese Gruppe fällt, kann mit-hilfe des Post-Remissions-Th erapie-(PRT-)Scores aus Alter, Zytogenetik, Krankheitsstatus, FLT3-Ratio und Anteil der CD34-pos. Blasten online be-rechnet werden (http://www.kompetenznetz-leukaemie.de/content/aerzte/scores/prognose).
• Allogene SZT: Bei HLA-kompatiblem Familienspender und in 1. CR durch-geführte Konsolidierung der 1. Wahl bei SR- und HR-Pat.: höchstes antileuk-ämisches Potenzial aller bekannten Konsolidierungsstrategien, zur Verringe-rung der Toxizität so früh wie möglich in der Konsolidierung durchführen, also möglichst ohne vorgeschalteten Hochdosis-Ara-C-Th erapieblock. Bei HR-Pat. ohne passenden Familienspender Fremdspender-Tx anstreben möglichst frühzeitige Spendersuche.
Besonderheiten der Th erapie von Pat. > 60 J. im Vergleich zu jüngeren Pat.:• Häufi ger sek. Erkrankungen.• Häufi ger numerische zytogenetische Aberrationen und Hochrisikoaberratio-
nen.• Seltener balancierte Translokationen und NR-Aberrationen.• Häufi ger Resistenzgenexpression (MDR1).! Dies führt zu Th er.-Refraktärität und hoher Rezidivrate.• Komorbiditäten häufi g, die die Durchführung einer intensiven Th er. unmög-
lich machen. • Höhere therapieassoziierte Toxizität Vorgesehene Th er. kann oft nicht oder
nur unvollständig durchgeführt werden Prognoseverschlechterung.
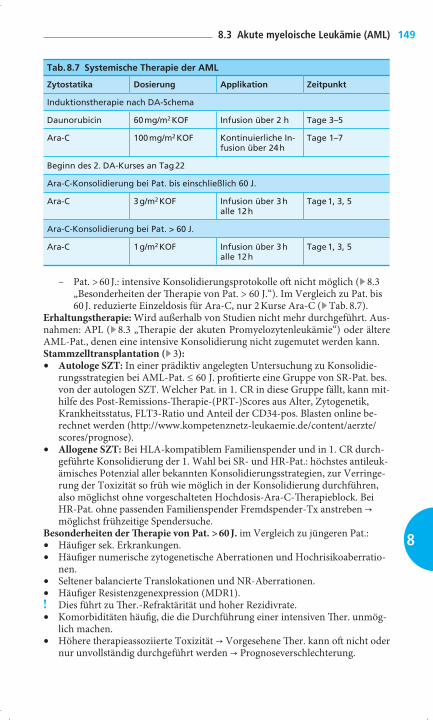
Tab. 8.7 Systemische Therapie der AML
Zytostatika Dosierung Applikation Zeitpunkt
Induktionstherapie nach DA-Schema
Daunorubicin 60 mg/m2 KOF Infusion über 2 h Tage 3–5
Ara-C 100 mg/m2 KOF Kontinuierliche In-fusion über 24 h
Tage 1–7
Beginn des 2. DA-Kurses an Tag 22
Ara-C-Konsolidierung bei Pat. bis einschließlich 60 J.
Ara-C 3 g/m2 KOF Infusion über 3 h alle 12 h
Tage 1, 3, 5
Ara-C-Konsolidierung bei Pat. > 60 J.
Ara-C 1 g/m2 KOF Infusion über 3 h alle 12 h
Tage 1, 3, 5
+24296_Possinger.indb 14996_Possinger.indb 149 22.06.2018 09:41:0022.06.2018 09:41:00

150 8 Akute Leukämien
8
Stratifi ziertes therap. Vorgehen nach zytogenetischen und molekularbiologischen Risikogruppen:• SR- und NR-Pat.: konventionelle Induktionstherapie.• HR-Pat.: wahrscheinlich kein Ansprechen auf konventionelle Th er. alterna-
tive Th er.-Verfahren im Rahmen von Studien, demethylierende Substanzen oder andere palliative Th erapien.
Zur Unterstützung des Aufk lärungsgesprächs Entwicklung des sog. AML-Scores zur Abschätzung von CR-Rate und Frühmortalität eines individuellen älteren Pat. mit AML unter Induktionstherapie bei Diagnosestellung (www.aml-score.org).Th erapie mit demethylierenden Substanzen: bei älteren Pat. der konventionel-len, intensiven Chemotherapie nicht unterlegen. Th er. nur für Pat., die nicht in-tensiv behandelbar sind (trotz Remissionen keine Heilung):• Azacytidin für AML Pat., die für eine Tx hämatopoetischer Stammzellen
nicht geeignet sind• Decitabin für AML Pat. ≥ 65 J., für die eine Standard-Induktionstherapie
nicht infrage kommtTh erapie der akuten Promyelozytenleukämie (APL):• Promyelozytenleukämie, FAB M3 mit t(15;17): therap. Ausnahme und AML-
Subentität mit der besten Prognose.• Allein mit All-trans-Retinolsäure (ATRA), 45 mg/m2 KOF/d p. o., Ausdiff e-
renzierung der unreifen Zellen und damit Remission möglich.• Mit Kombination ATRA + Anthrazyklin Remissionsraten von 85–95 %.• Standardtherapie vor Einführung von Arsentrioxid (ATO): 3 Konsolidie-
rungszyklen mit ATRA + Anthrazyklin bzw. Mitoxantron sowie 2-jährige ATRA-haltige Erhaltungstherapie.
• Heutzutage risikoadaptierte Th er. üblich.• NR- und Intermediärrisiko-Pat.: Th er. mit der chemotherapiefreien Kombi-
nation von ATRA und ATO.• HR-Pat. mit Leukos > 10 000/μl: Standardtherapie mit ATRA + Anthrazyklin/
Mitoxantron + Ara-C.Palliative Th erapie:• Ziel: Lebensverlängerung bei nicht kurativ behandelbaren Pat. mit Erhalt der
Lebensqualität, möglichst im ambulanten Setting.• Vermeidung von Komplikationen durch zytoreduktive Th er. in Kombination
mit supportiven Maßnahmen (Transfusionen, Infektprophylaxe).• Zytostatika:
– Hydroxyharnstoff : 500–3 000 mg/d absolut p. o.– Melphalan: 2 mg/d absolut p. o.– Ara-C: 40–100 mg/d absolut s. c.– Etoposid (VP-16): 50–200 mg/d absolut p. o.– Mitoxantron: 10–15 mg 1 ×/Wo. absolut i. v.
Notfalltherapie:• Neutropene Sepsis: sofortige empirische Breitspektrumantibiose, Intensiv-
therapie, ggf. Reanimation und Beatmung.• Hyperleukose oder leukostatische Symptome: Leukapherese. Ist diese nicht
möglich, rasche Senkung der peripheren Blastenanzahl mit Hydroxyharnstoff .• DIC: phasenspezifi sche Th erapie. Eine DIC kann insbes. bei APL auft reten.• Tumorlysesyndrom (▶ 5.2.2):
– Auft reten i. d. R. akut 12–24 h nach Beginn der Chemotherapie.– Symptome: Übelkeit, Erbrechen, Hyperkaliämie, Hyperphosphatämie,
Hyperurikämie, Hypokalzämie.
+24296_Possinger.indb 15096_Possinger.indb 150 22.06.2018 09:41:0022.06.2018 09:41:00

151 8.3 Akute myeloische Leukämie (AML)
8
– Engmaschiges Monitoring (EKG, ZVD, Laborwerte), Hydratation, Aus-gleich der Elektrolytstörungen, Senkung der Harnsäurewerte.
• ATRA- Syndrom:– Sofortiger Beginn einer Th er. mit Dexamethason.! Absetzen von ATRA meist ohne Eff ekt.– Tritt schnell und lebensbedrohlich unter Th er. der APL mit ATRA auf.– Symptome: Niereninsuffi zienz, Blutdruckabfall, Lungenödem, Capillary
Leak, Atemnot, Fieber.Supportive Th erapie:• Wie bei ALL (▶ 8.1).• Spezifi ka bei der AML:
– Infektionsprophylaxe: Posaconazol 3 × 200 mg/d p. o. zugelassen zur Pro-phylaxe bei AML unter Induktionstherapie.
– Substitution von Blutprodukten: Th rombozytensubstitutionsgrenzwert für die prophylaktische Th rombozytengabe bei AML-Th er.: 10 000/μl. Gilt nur für Pat. ohne Fieber, ohne plasmatische Gerinnungsstörung, ohne Hyperleukose und ohne Blutungen. Bei Vorliegen einer DIC oder einer APL Th rombos > 50 000/μl halten.
Prognose Abhängig von Alter und entsprechender Risikogruppe (▶ Tab. 8.8). Nachsorge • In Studien nach den jeweiligen Erfordernissen geregelt. • Nach Th er.-Abschluss: KM-Punktion zur Kontrolle des Th er.-Erfolgs.• Vorliegen einer CR: anschließend engmaschige klin. und BB-Kontrollen:
– Im ersten halben Jahr monatlich, dann alle 3 Mon.– Nach 2 J. und bis zum 5. J. alle 6 Mon.– Bei Beschwerden sofort.
• KM-Punktion jeweils bei auff älligem BB.
Tab. 8.8 Prognostische Kennzahlen der AML
Alter > 60 J. Alter ≤ 60 J.
Niedrigrisiko Standardrisiko Hochrisiko
Remissionsrate (%) 30–50 80–95 70–80 40–50
5-JÜR (%) 10–15 60–70 45–55 15–20
+24296_Possinger.indb 15196_Possinger.indb 151 22.06.2018 09:41:0022.06.2018 09:41:00

Erhältlich in jeder Buchhandlung oder im Elsevier Webshop
Klinikleitfaden Hämatologie Onkologie2018. 632 Seiten. ISBN 978-3-437-24296-0€ [D] 52,- / € [A] 53,50 / SFr 70,-
Irrtü
mer
und P
reisän
deru
ngen
vorb
ehalt
en. S
tand 0
8/201
8
Der Leitfaden gibt Ihnen Sicherheit im Umgang mit den Besonderheiten der Hämatologie und Onkologie. Konkret und praxisnah vermittelt er Ihnen Wissen über alle wichtigen Krankheitsbilder inkl. deren Diagnostik und Therapie.
Darüber hinaus werden Sie mit verschiedenen Supportivtherapien vertraut gemacht und erhalten praktische Handlungsanleitungen im Umgang mit Notfällen in der Hämatologie/Onkologie.
*Angebot freibleibend


















