
Klinische Chemie und Laboratoriumsdiagnostik Teil 12 · • Thrombozytopathien (hereditär und...
73
Klinische Chemie und Laboratoriumsdiagnostik Teil 12 Hämostaseologie Prof. Dr. Ralf Lichtinghagen Medizinische Hochschule Hannover Klinische Chemie Tel.: 0511-5323940
Transcript of Klinische Chemie und Laboratoriumsdiagnostik Teil 12 · • Thrombozytopathien (hereditär und...
Klinische Chemie und Laboratoriumsdiagnostik
Teil 12Hämostaseologie
Prof. Dr. Ralf LichtinghagenMedizinische Hochschule Hannover
Klinische ChemieTel.: 0511-5323940

Hämostase
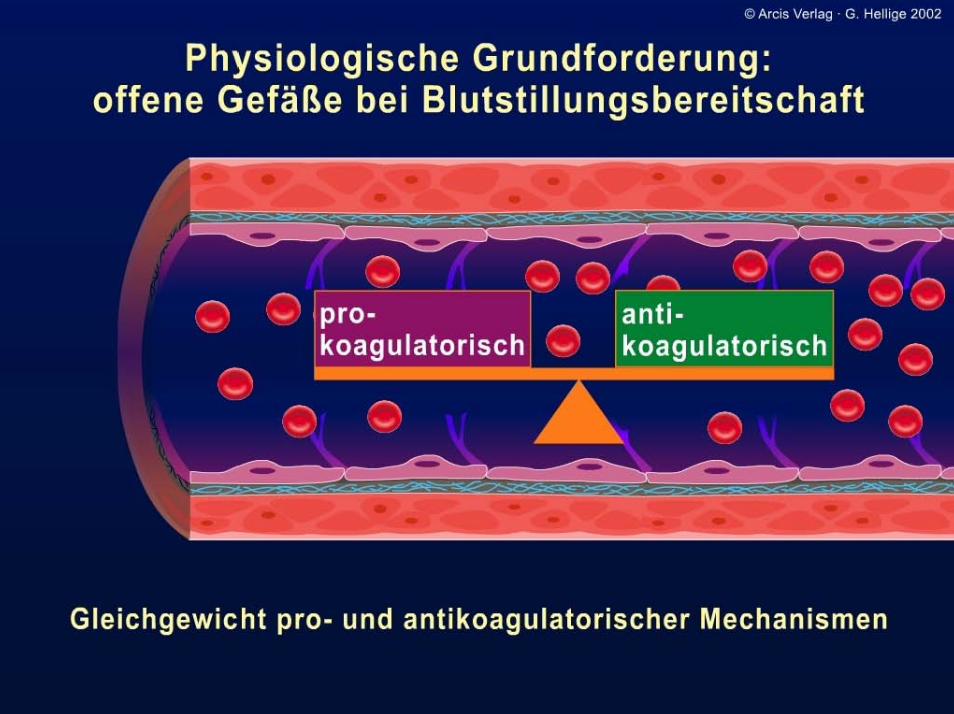
Bedeutung: Blutstillung (Organismus schützt sich bei Gewebs-verletzungen bei Eröffnung kleiner Gefäße gegen Blut-verlust)
Vorgänge gewährleisten
• Fließfähigkeit des Blutes innerhalb der Blutgefäße.• Abdichtung der Gefäße sowie Drosselung bzw. Beendigung von
Blutaustritten.• Wiederherstellung der Gefäßstruktur bzw. eine Narbenbildung.
Gefäßsystem Thrombozyten(Blutplättchen)
plasmatische Gerinnungsfaktorenplasmatische Fibrinolysefaktoren(Reparaturvorgänge)

Hauptaufgabe: Wundheilung

Komponenten des Hämostasesystems
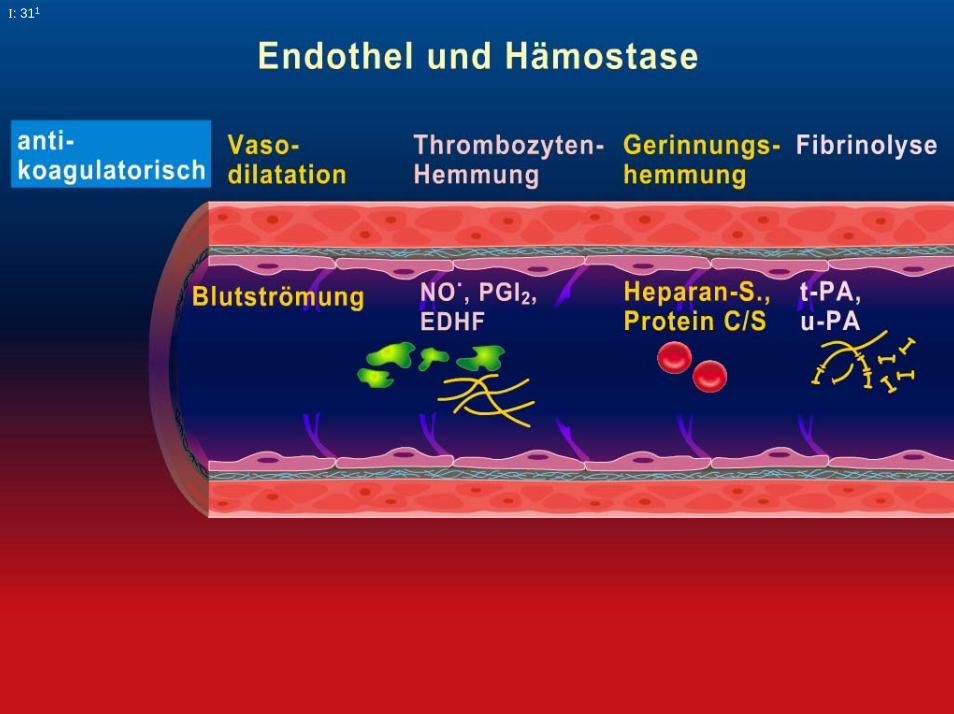
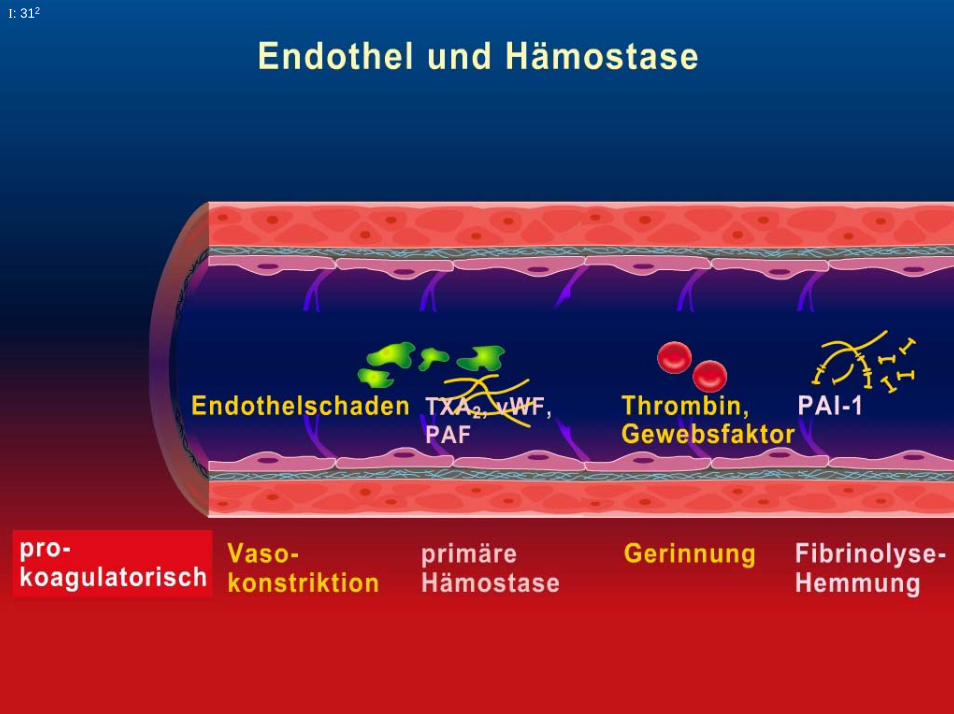
- vaskuläre Komponente Endothel, Subendothel,Gefäßmuskulatur
- zelluläre Komponente Thrombozyten, Erythrozyten,Leukozyten
- plasmatische Komponente Gerinnungsfaktoren+ Inhibitoren
- fibrinolytische Komponente Fibrinolysefaktoren+ Inhibitoren
I: 311
I: 312
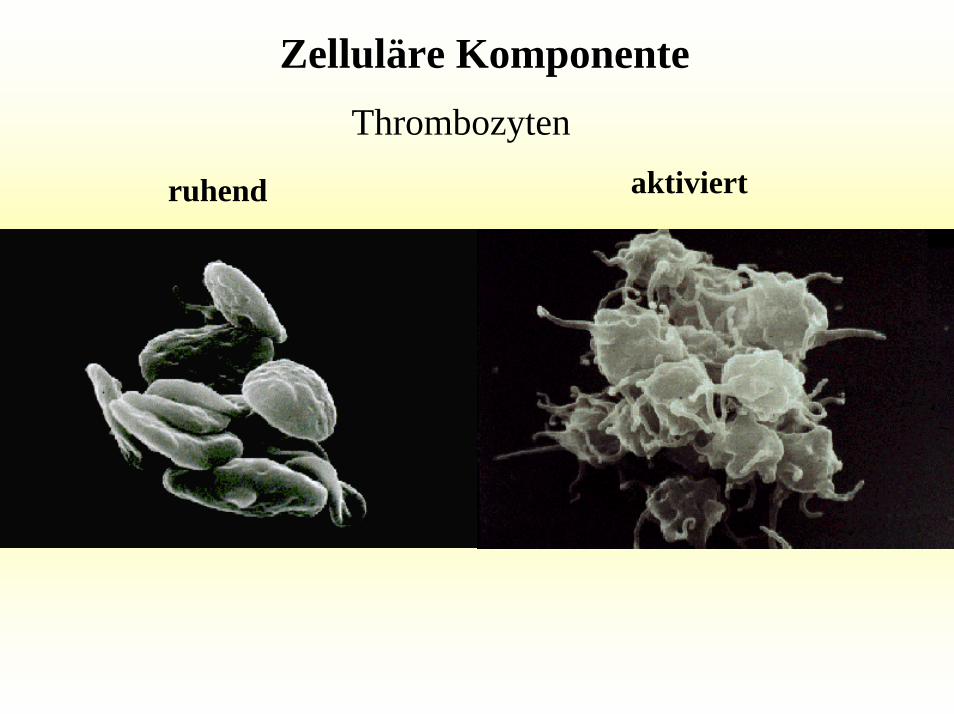
Zelluläre KomponenteThrombozyten
aktiviertruhend
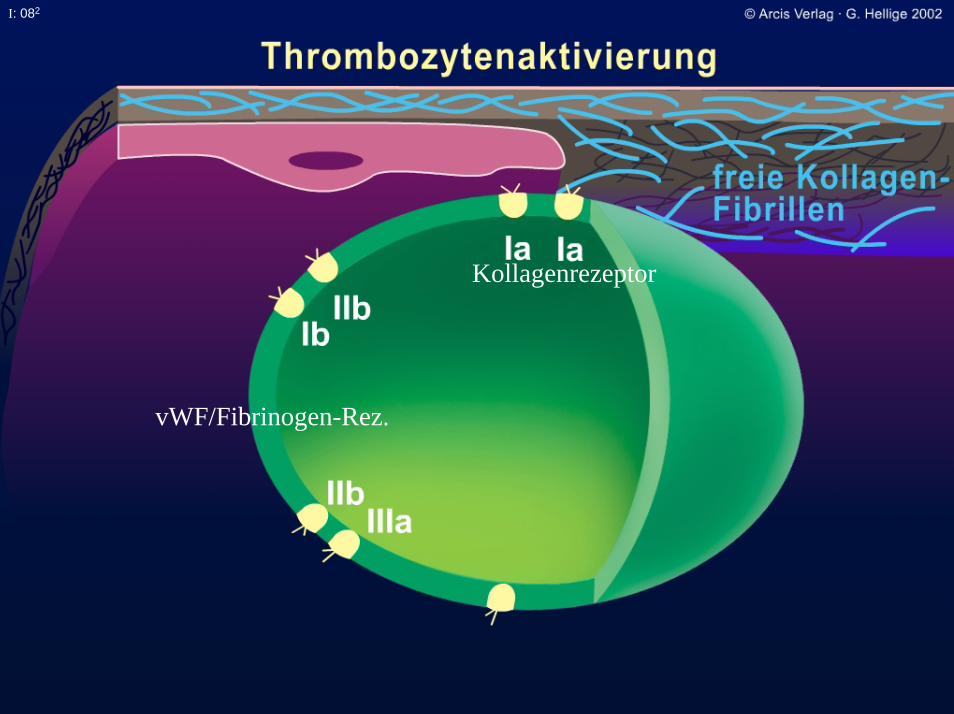
I: 082
Kollagenrezeptor
vWF/Fibrinogen-Rez.
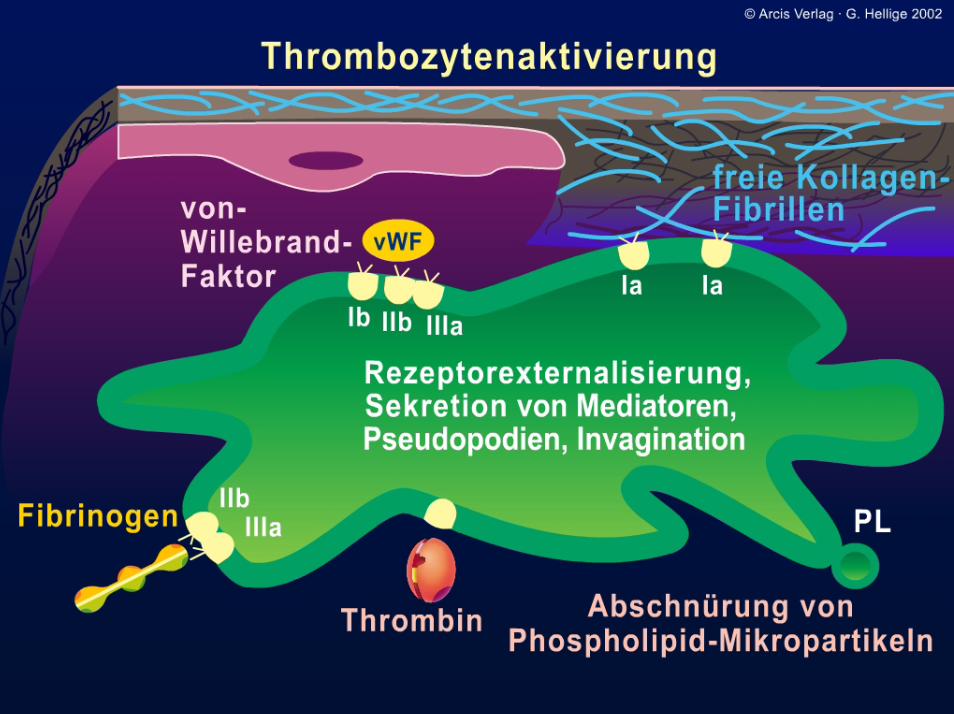

I: 09


Plasmatische Komponente

VitK
VitKVitK
VitK
VitKVitK
VitK: Vitamin K-abhängige Bildung
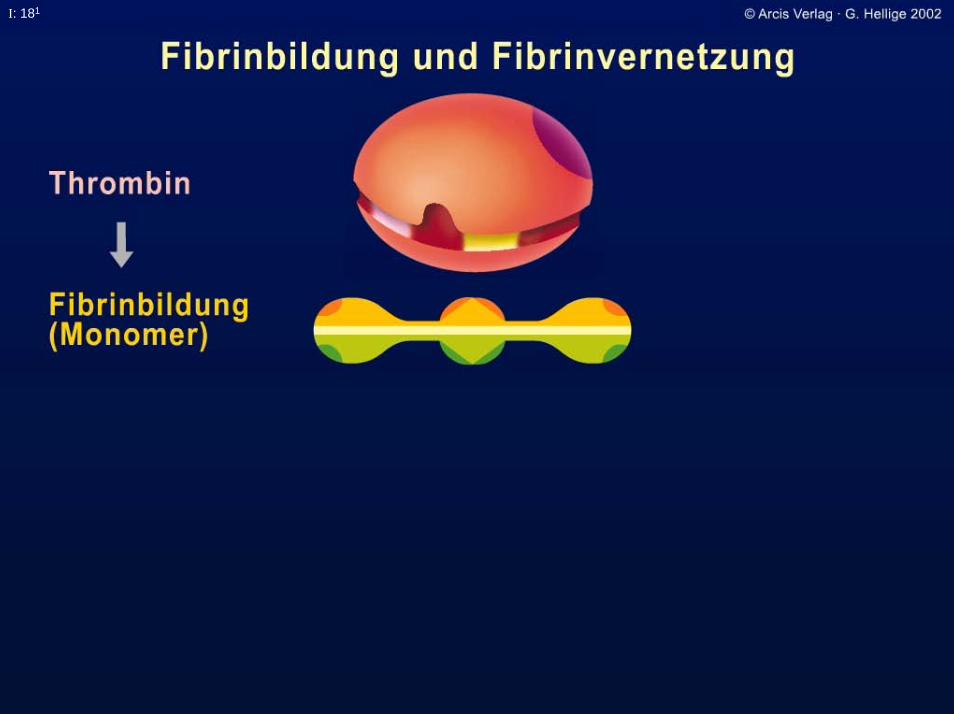
I: 181

I: 182

I: 184
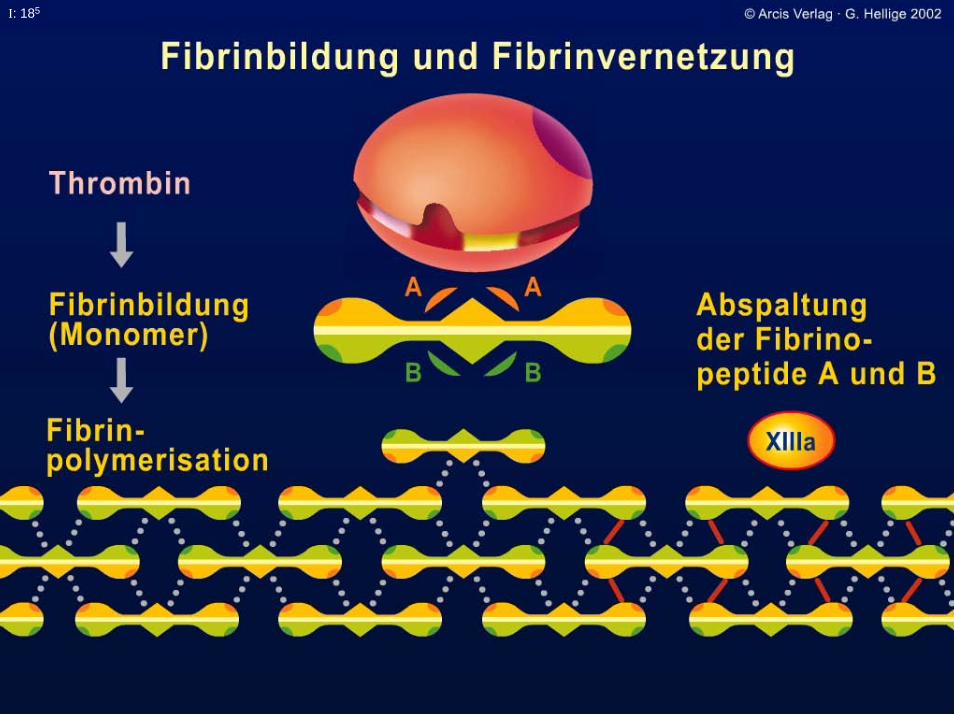
I: 185

I: 18
Fibrin-Stabilisierungsfaktor
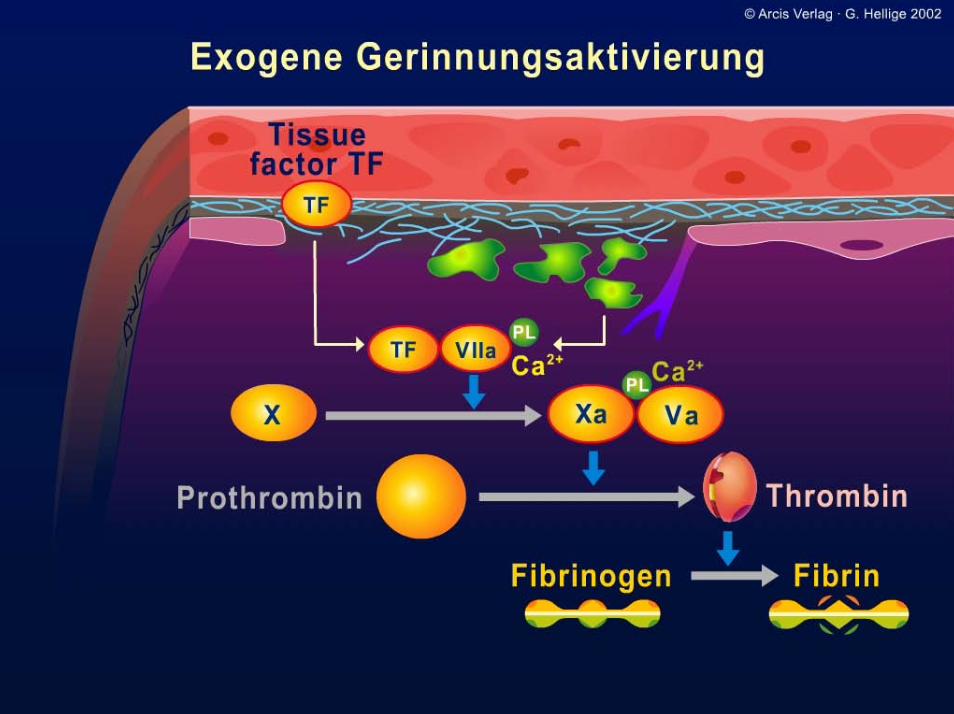


I: 19

Synthese aus Kaskade und Netzwerk
ExogenesSystemInitiation
Amplifikation
Propagation
Gerinnselbildung
Endogenes System
Thrombozyt
Aktiv. Thrombozyt

und Protein S
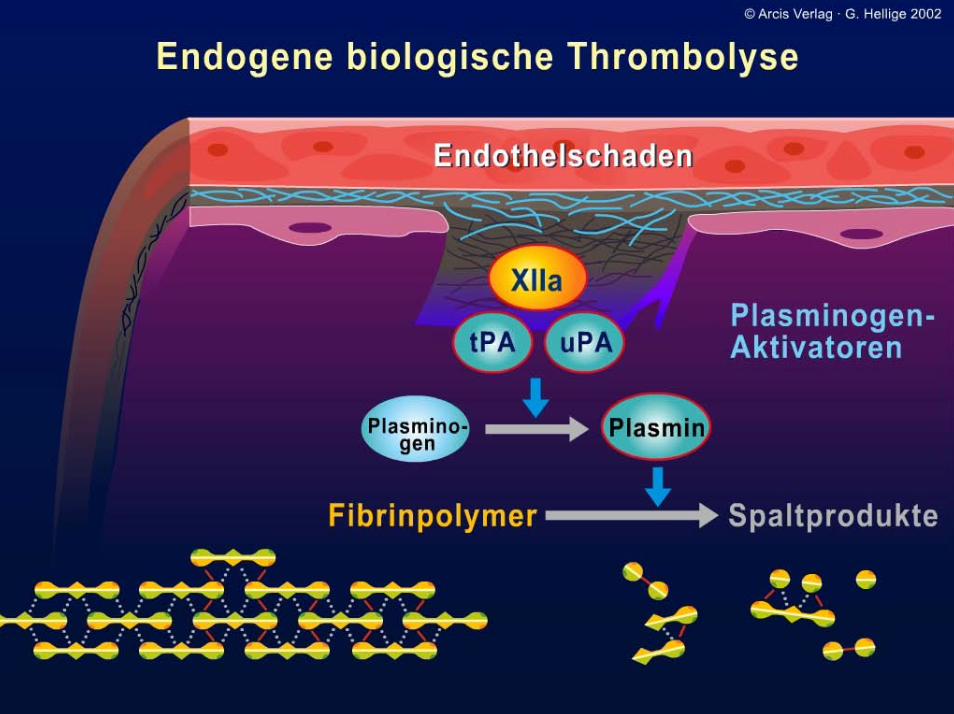

Störungen der Hämostase: Einteilungsmöglichkeiten
• Hereditär
• Erworben
• Vaskulär
• Thrombozytär
• Plasmatisch
• Störungen mit Blutungsneigung
• Störungen mit Thrombose

Plasmatisch bedingte Blutungsneigungen
• Hämophilie A & B
• Von Willebrand-Syndrom• Andere Einzelfaktorenmängel (FI bis FXIII, nicht FXII)
• Dys-/Hypo-/Afibrinogenämien
• Hyperfibrinolyse
• Plasmininhibitor-Mangel

Hämophilie A & B = The „Royal“ Disease
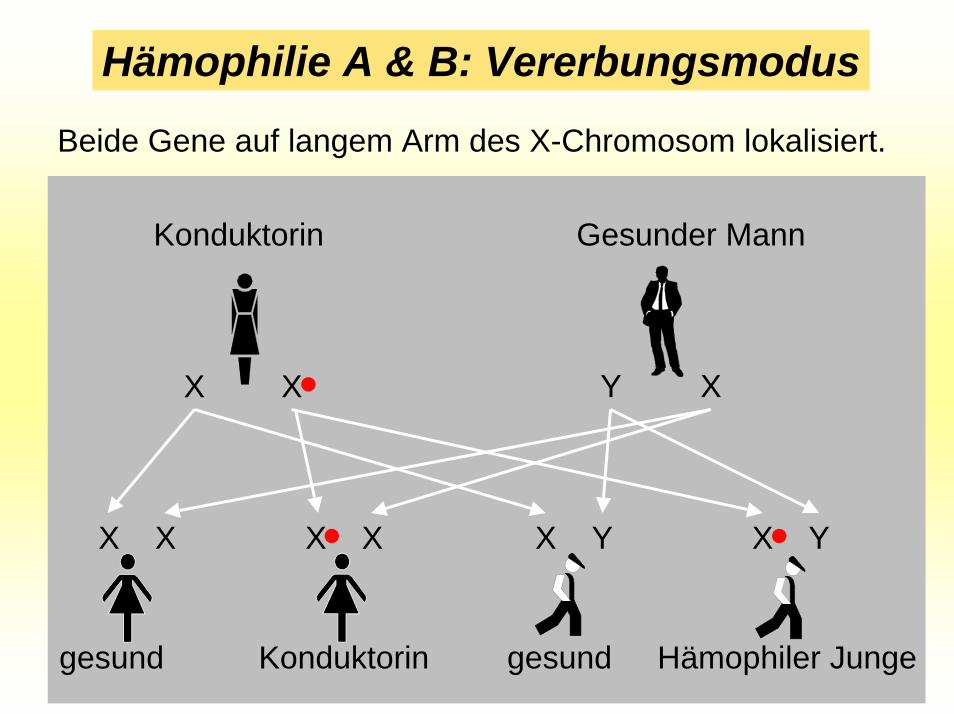
Hämophilie A & B: Vererbungsmodus
Beide Gene auf langem Arm des X-Chromosom lokalisiert.
X X XY
Gesunder MannKonduktorin
•
•X X X X
gesund Konduktorin
X Y X Y•
gesund Hämophiler Junge

Hämophilie A & B: Vererbungsmodus
X X
Gesunde Frau
XY
Hämophiler Mann
•
X Y X Y
gesund gesund
•X X X X
Konduktorin Konduktorin
•

Hämophilie: Epidemiologie
Inzidenz:• Hämophilie A: 2:10.000 Jungen• Hämophilie B: 1:25.000 JungenPrävalenz:• Hämophilie A: 5:100.000• Hämophilie B: 1:100.000
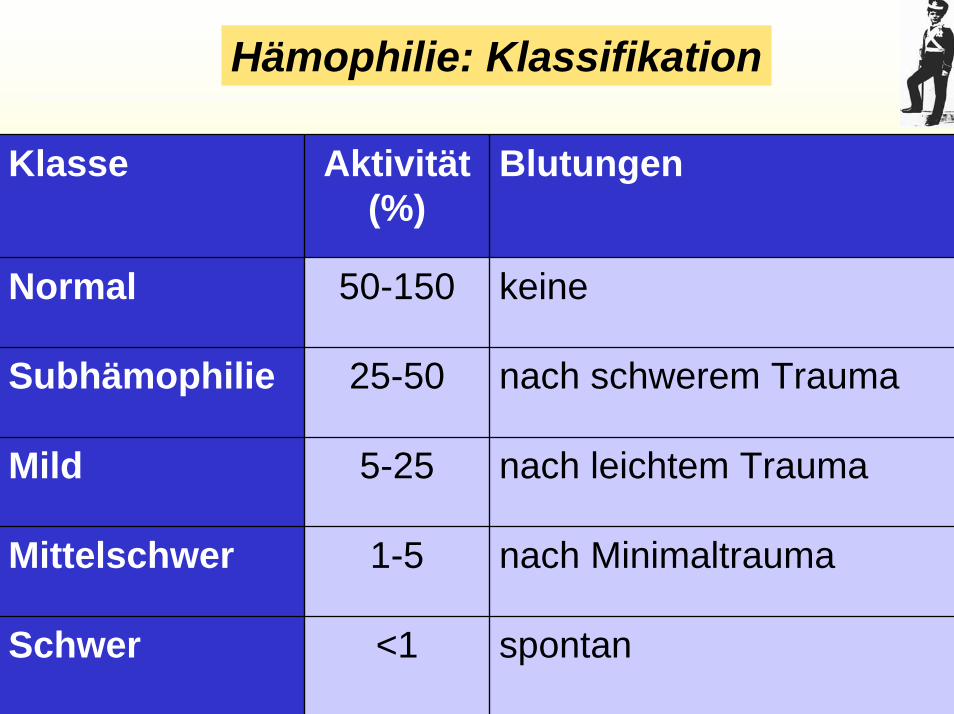
Hämophilie: Klassifikation
Klasse Aktivität (%)
Blutungen
Normal
Subhämophilie
Mild
Mittelschwer
Schwer
50-150 keine
25-50 nach schwerem Trauma
5-25 nach leichtem Trauma
1-5 nach Minimaltrauma
<1 spontan

Hämophilie: Klinik
• Gelenkblutungen => hämophile Arthropathie(Blutergelenke)
• Blutungen nach Verletzungen, Zahnextraktion,aus gastrointestinalen Läsionen
• Weichteilblutungen:MuskulaturPsoas (Lendenmuskel)-Blutung!Mundbodenblutung!Hirnblutung!
• Wundheilungsstörungen



Hämatom nach i.m.-Injektion

Hämophilie: Therapie
• Substitution mit Faktor VIII (Hämophilie A) Faktor IX (Hämophilie B)
• Eine Einheit (=> historisch gewonnen aus 1 ml Plasma)/ kg Körpergewicht führt zu einer Erhöhung des Faktors um 1-2%
• Faktorenkonzentrate (aus humanem Plasma) rekombinante Faktoren
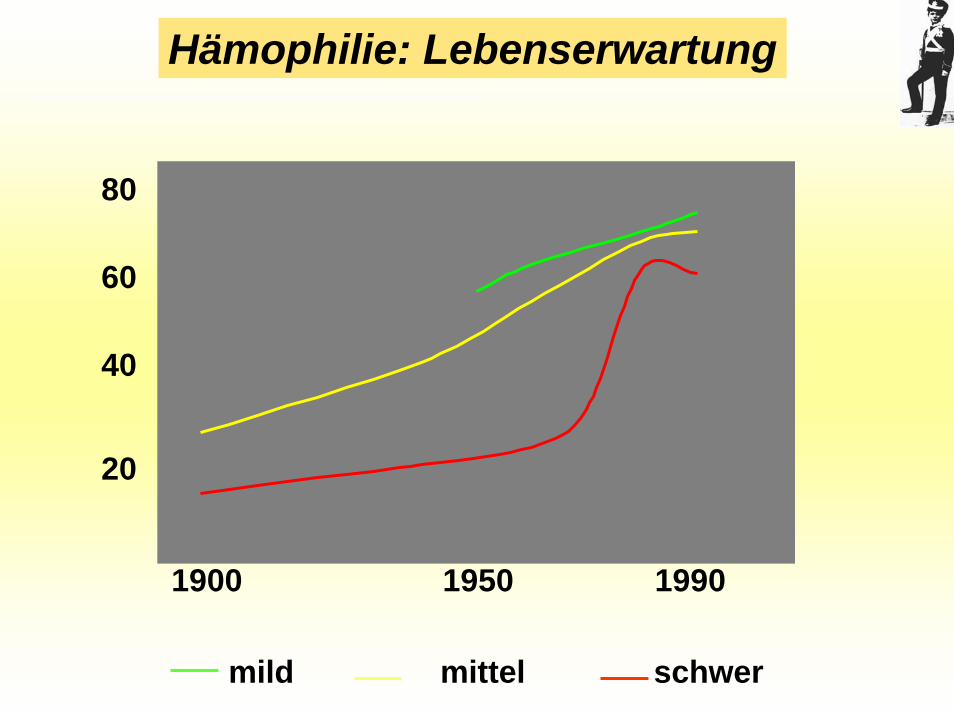
Hämophilie: Lebenserwartung
1900 1950 1990
80
60
40
20
mild mittel schwer

Hepatitis C
Ca. 70% aller Bluter sind in Deutschland über Blutprodukte mit HCV infiziert worden.Ca. 80% davon sind replikativ und damit infektiös!
Hepatozelluläres KarzinomLeber-Zirrhose
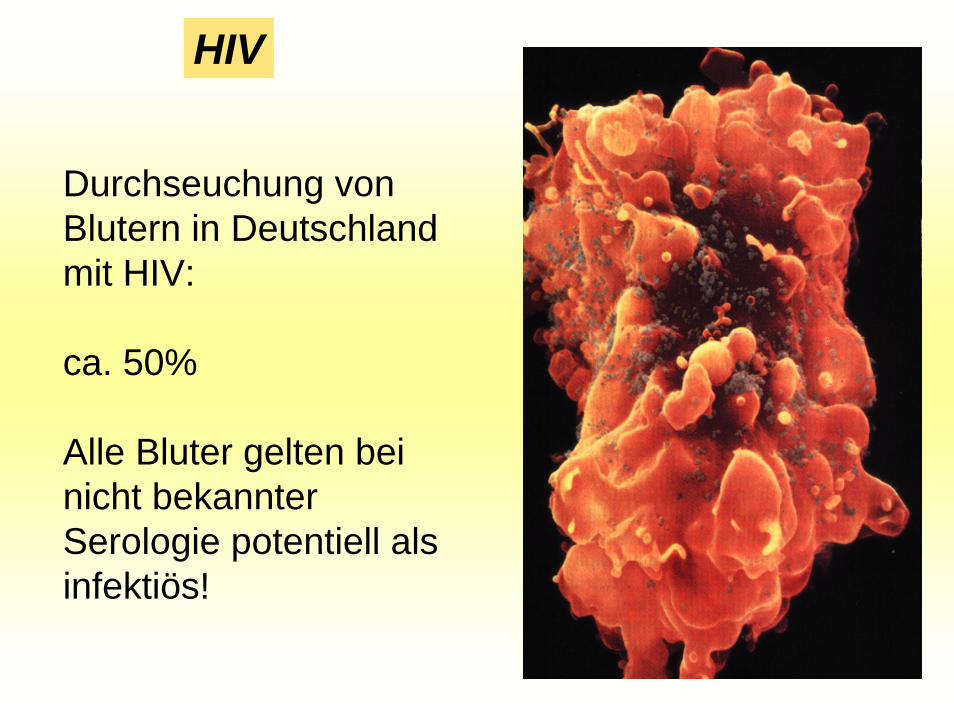
HIV
Durchseuchung von Blutern in Deutschland mit HIV:
ca. 50%
Alle Bluter gelten bei nicht bekannter Serologie potentiell als infektiös!

Von Willebrand Syndrom
• Willebrand Faktor: Molekül 309 kD; Gen auf Chomosom 12
• Produziert von den Endothelzellen der Gefäße, Megakaryozyten• Rolle bei der Thrombozytenadhäsion• Faktor VIII Synthese, Sekretion und Stabilität von vWF reguliert• Faktor VIII bei Typ 1-3 der Erkrankung vermindert
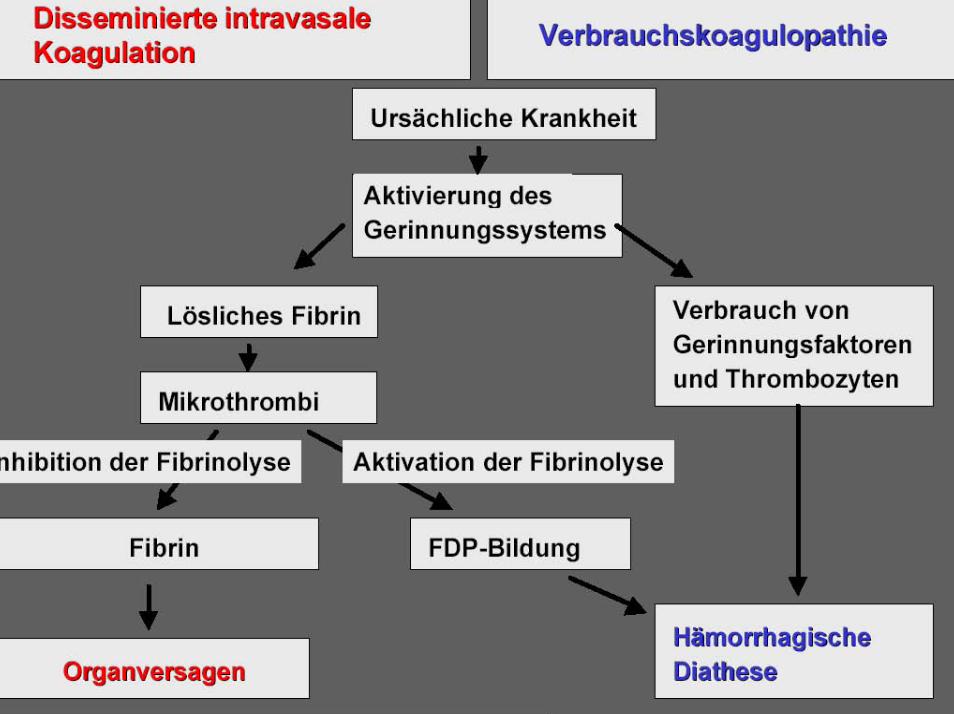

Gerinnungsdefekte
• Koagulopathien:Hämophilie A und BVon Willebrand-Syndrom
• Thrombopenie durch Plättchenabbau:Thrombotisch thrombozytopenische PurpuraIdiopathische thrombozytopenische Purpura (ITP)Heparin-induzierte Thrombopenie (HIT) Typ II
• Thrombozytopathien (hereditär und erworben)
• Vaskulär bedingte hämorrhagische Diathesen:Schoenlein-Hennoch PurpuraMorbus OslerAngiodysplasie

Wichtige Tests bei Hämostasestörungen
Globaltests:Thromboplastinzeit (Quick-Wert)bei Störungen des exdogenen Aktivierungsweges
(aktivierte) partielle Thromboplastinzeitbei Störungen im endogenen Aktivierungsweg
Thrombozytenzahl
Bestimmung von Einzelfaktoren

Thromboplastinzeit (Quick-Wert)
Globaler Suchtest bei hämorrhagischen Diathesen (Blutungsneigung) zur Erkennung von Störungen im exogenen Aktivierungsweg des plasmatischenGerinnungssystems (Faktoren:VII, X, II, V, I (Fibrinogen))
Erforderlich vor jedem größeren operativen Eingriff
Überwachung der oralen Antikoagulanzientherapie
Bei Verdacht auf Vitamin K-Mangel (wichtig: Faktoren: II, VII, X)
Methode: In einer Plasmaprobe wird die Fibrinbildung durch Zugabe von Gewebsthromboplastin und Ca2+ ausgelöst. Die Gerinnungszeit hängt von den Faktoren (s.o.) ab.
Angabe des Ergebnissesin Prozent der Norm (70-130%)
als International Normalized Ratio (INR)(weitgehend unabhängig vom verwendeten Reagenz, der eingesetzten Methode, bessere Vergleichbarkeit der Werte)geeignet für Kontrolle der Antikoagulanzientherapie
Lebersyntheseleistung spiegelt sich außerdem auch in Ergebnis wieder, da erfasste Faktoren dort synthetisiert werden.

(Aktivierte) partielle ThromboplastinzeitGlobaler Suchtest bei hämorrhagischen Diathesen (Blutungsneigung) zur Erkennung von Störungen im endogenen Aktivierungsweg des plasmatischen Gerinnungssystems (Faktoren: XII, XI, X, IX, VIII, V, II, I (Fibrinogen))
Erforderlich vor jedem größeren operativen Eingriff
Kontrolle der Heparintherapie (nur mit ATIII wirksam)
Methode: In einer Plasmaprobe wird die Fibrinbildung durch Zugabe von partiellem Thromboplastin (Phosphoplipid) und Ca2+ ausgelöst. Die Gerinnungszeit hängt von den Faktoren (s.o.) ab.
Angabe des Ergebnissesin Sekunden (ca 35-40 sec.)(abhängig von verwendeten Geräten und Reagenzien.)
Nur Verlängerungen der PTT bzw. APTT sind klinisch relevant.Erfasst wird eine Verminderung oben angegebener Faktoren, Methode der Wahl zur Erkennung der Hämophilie A (Mangel an Faktor VIII), Hämophilie B (Mangel an Faktor IX), auch beim von Willebrand-Syndrom verlängert (Faktor VIII vermindert).
Lebersyntheseleistung spiegelt sich außerdem auch in Ergebnis wieder, da die meisten erfassten Faktoren dort synthetisiert werdenSubcutane low-dose Heparin-Gabe bedarf keiner Laborkontrolle

Angeborene oder erworbene Thrombophilie
Ungleichgewicht zwischen
Antikoagulation Prokoagulation
bedingt durch:Mangel an Inhibitoren (AT, PC, PS u.a.)erhöhte Aktivität verschiedener Faktoren (FII, FVIII u.a.)Moleküldefekte (FV:Q506-Mutation u.a.)andere Mechanismen (verminderte Fibrinolyse u.a.)

Risikofaktoren (hereditär)
• APC-Resistenz (Faktor V:Q506-Mutation)• Prothrombin G20210A-Mutation• FII- und FVIII-Erhöhung• Antithrombin-Mangel/Defekt• Protein C-Mangel/Defekt• Protein S-Mangel/Defekt• Hyperhomocysteinämie• Lipoprotein (a)-Erhöhung• Dysfibrinogenämie• Plasminogen-Mangel• andere (Faktor XII-Mangel, PAI-1, HC II, etc.)

Erworbene Risikofaktoren
• Antiphospholipid-Antikörpersyndrom• Malignome (Trousseau-Phänomen)• Heparin-induzierte Thrombozytopenie Typ II• Asparaginase-Therapie (z.B. bei ALL)• Lebersynthesestörung• Nephrotisches Syndrom• u.v.m.

FV:Q506-Mutation
• Prävalenz in der Normalbevölkerung:5-9%
• Ursache der APC-Resistenz:>95%
• Bei Patienten mit Thrombose nachweisbar:ca. 40%

FV:Q506-Mutation
Thromboserisiko (VTE):
• 5-7x bei Heterozygotie
• 30-50x bei Heterozygotie + 1 RF
• 50-100x bei Homozygotie
• 100-200x bei Homozygotie + 1 RF

Prothrombinmutation G20210A
• Prävalenz in der Normalbevölkerung:1-3% (3% n=397)
• Prävalenz bei Patienten mit Thrombose:18% (7,5% n=517)
v. Depka et al., Blood ‘98

Antithrombin
• Synthese in der Leber• inaktiviert FIIa und FXa durch Komplexbildung• Komplexbildung durch Heparin 1000x schneller
UFH bindet auch an IIa
NMH bindet nicht an IIa
5
10
ATXa
5
13
ATXa
Konformationsänderung
5
13
AT IIa
5
10
AT IIa
Konformationsänderung
A
B

Lipoprotein (a)
• Assoziation zw. Lp(a)-Phänotyp und KHK (Berg et al., 1978)
• Atherogenität wohl Folge des Apolipoprotein (a)
• cDNA durch McLean 1987: hohe Analogie zu Plasminogen
• enge Lokalisation der Gene Lp(a) und Plasminogen
Q: NCBH/NIH, Molecular Modelling Database

Thrombophilie-Diagnostik: Was?
• APC-R ggf. Gentypisierung FV:Q506
• Prothrombin G20210A-Genanalyse• Protein C-Aktivität• freies Protein S• Antithrombin-Aktivität• Lipoprotein (a)• Homocystein• FII- und FVIII-Aktivität

Thrombophilie-Diagnostik: Wann?
• in der stabilen Phase 6-8 Wochen nach Ereignis bzw. nach Absetzen der Cumarintherapie(PC, PS, FII)
• sofort bei bedrohlichen thromboembolischenEreignissen, die ggf. eine zusätzliche Substitutionstherapie erfordern (AT, PC)

Thrombophilie-Diagnostik: Wen?
• Patienten mit familiärer Thromboseneigung• Erstmanifestation vor dem 50. Lebensjahr
(juvenile Thrombophilie)• Ungewöhnliche Lokalisation
(cerebral, Splanchnicusregion, Arm u.a.)• Thrombose ohne exogenen Trigger

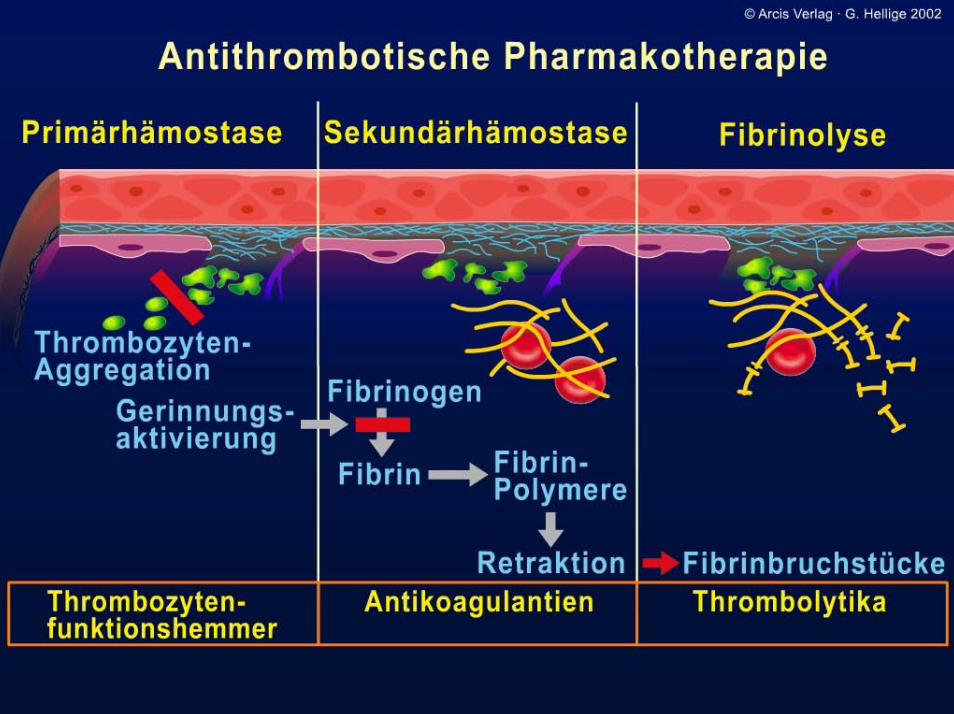



Die Cumarine
Kompetitive Hemmung der Vitamin-K-Epoxidreduktase

Die Ku(h)marine
WarfarinCoumadin®



II: 12

II: 18

II: 20



II: 31
= Refludan; Revasc
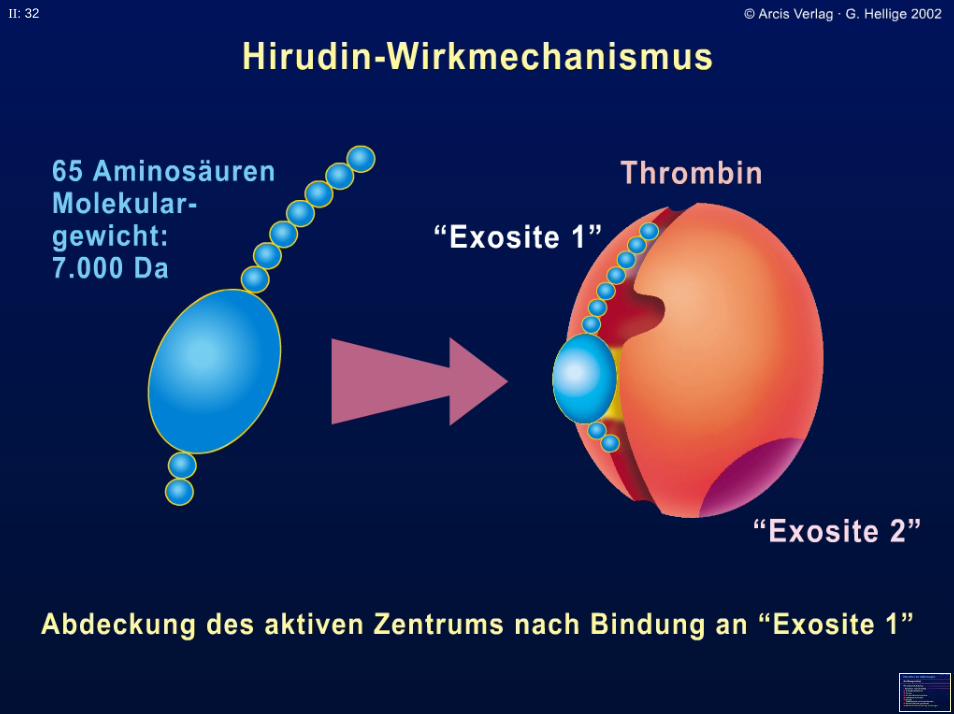
II: 32








![TECHNISCHE UNIVERSITÄT MÜNCHEN Neurologische Klinik des ... · hereditär und symptomatisch (z. B. medikamentös bedingt) [Markham 1992]. Eine weitere Einteilung erfolgt nach Alter](https://static.fdokument.com/doc/165x107/5e15e0639466844bb878cb1d/technische-universitt-moenchen-neurologische-klinik-des-hereditr-und-symptomatisch.jpg)



