
L2B: DNA Origami - · PDF fileBiophysik Fortgeschrittenenpraktikum - Versuch L2B DNA Origami...
14
Biophysik Fortgeschrittenenpraktikum - Versuch L2B DNA Origami Arbeitsgruppe von Prof. Tim Liedl Sommersemester 2014 Vorbemerkungen Die DNA Origami Technik nutzt ein langes, virales, einzelsträngiges DNA Molekül als Ge- rüststrang (der Scaffold), welcher durch hunderte kurze DNA Stücke (die Staples) in eine gewünschte Form gebracht wird. Das Ziel dieses Praktikumsversuches ist es, einen Einblick in die DNA Origami Technik zu geben. Hierfür wird jeder StudentInnen die Scaffold DNA mit Hilfe von Bakterien selbst herstellen, eine Struktur am Computer konstruieren und diese im Labor anfertigen und anschließend mit Hilfe der Elektronenmikroskopie analysieren. Vorbereitung Zur Vorbereitung auf diesen Versuch ist diese Anleitung bitte sorgfältig zu lesen. Die ein- zelnen Schritte der Versuchsdurchführung werden zum jeweiligen Zeitpunkt im Labor von der betreuenden Person noch genauer erklärt. Außerdem soll die zum Strukturdesign genutzte Software caDNAno (http://cadnano.org/legacy; v0.2.3 honeycomb lattice) heruntergeladen und ausprobiert werden (es gibt inzwischen eine neuere Version unter http://cadnano.org welche aber schon wiederholt Probleme bei der Installation auf Windows Rechner bereitet hat). Folgende zwei Tutorial Videos geben eine sehr verständliche Übersicht darüber, wie die Geometrie der DNA genutzt werden kann, um dreidimensionale Strukturen zu bauen: • http://www.youtube.com/watch?v=cwj-4Wj6PMc&feature=youtu.be • http://www.youtube.com/watch?v=EabqNaYAI7o&feature=youtu.be Zur weiterführenden Vorbereitung sollten noch folgende Veröffentlichungen angesehen werden: • Paul Rothemund: Folding DNA to Create Nanoscale Shapes and Patterns (Nature 2006) • Shawn Douglas et al.: Self-assembly of DNA into nanoscale three-dimensional shapes (Nature 2009) • Tim Liedl et al.: Self-assembly of three-dimensional prestressed tensegrity structures from DNA (Nature Nanotechnology 2010) 1
Transcript of L2B: DNA Origami - · PDF fileBiophysik Fortgeschrittenenpraktikum - Versuch L2B DNA Origami...

Biophysik Fortgeschrittenenpraktikum - Versuch L2B
DNA Origami
Arbeitsgruppe von Prof. Tim Liedl
Sommersemester 2014
Vorbemerkungen
Die DNA Origami Technik nutzt ein langes, virales, einzelsträngiges DNA Molekül als Ge-rüststrang (der Scaffold), welcher durch hunderte kurze DNA Stücke (die Staples) in einegewünschte Form gebracht wird. Das Ziel dieses Praktikumsversuches ist es, einen Einblick indie DNA Origami Technik zu geben. Hierfür wird jeder StudentInnen die Scaffold DNA mitHilfe von Bakterien selbst herstellen, eine Struktur am Computer konstruieren und diese imLabor anfertigen und anschließend mit Hilfe der Elektronenmikroskopie analysieren.
Vorbereitung
Zur Vorbereitung auf diesen Versuch ist diese Anleitung bitte sorgfältig zu lesen. Die ein-zelnen Schritte der Versuchsdurchführung werden zum jeweiligen Zeitpunkt im Labor vonder betreuenden Person noch genauer erklärt. Außerdem soll die zum Strukturdesign genutzteSoftware caDNAno (http://cadnano.org/legacy; v0.2.3 honeycomb lattice) heruntergeladenund ausprobiert werden (es gibt inzwischen eine neuere Version unter http://cadnano.orgwelche aber schon wiederholt Probleme bei der Installation auf Windows Rechner bereitethat). Folgende zwei Tutorial Videos geben eine sehr verständliche Übersicht darüber, wie dieGeometrie der DNA genutzt werden kann, um dreidimensionale Strukturen zu bauen:
• http://www.youtube.com/watch?v=cwj-4Wj6PMc&feature=youtu.be
• http://www.youtube.com/watch?v=EabqNaYAI7o&feature=youtu.be
Zur weiterführenden Vorbereitung sollten noch folgende Veröffentlichungen angesehen werden:
• Paul Rothemund: Folding DNA to Create Nanoscale Shapes and Patterns (Nature 2006)
• Shawn Douglas et al.: Self-assembly of DNA into nanoscale three-dimensional shapes(Nature 2009)
• Tim Liedl et al.: Self-assembly of three-dimensional prestressed tensegrity structures fromDNA (Nature Nanotechnology 2010)
1

L2B: DNA Origami Sommersemester 2014
Eine gute Übersicht über die verschiedenen vielversprechenden Anwendungsmöglichkeiten desDNA Origami mit einer guten Einführung in die Methode ist folgender Artikel:
• Thomas Tørring et al.: DNA origami: a quantum leap for self-assembly of complex struc-tures (Chemical Society Reviews 2011)
Bei Fragen zum Versuch kann natürlich auch jederzeit im Vorfeld die betreuende Person kon-taktiert werden.
Versuchsablauf
Tag 1
Beginn: 9:00 Uhr - Dauer: ca. 10 Stunden
• Mündliche Abfrage über das Verständnis der Thematik
• Herstellung der Scaffold DNA
• Computergestütztes Design einer DNA Origami Struktur
• Faltung von DNA Origami Strukturen mit dem hergestellten Scaffold über Nacht
Tag 2
Beginn: 9:00 Uhr - Dauer: ca. 3 Stunden
• Färbung der gefalteten Proben
• TEM Analyse der gefärbten Proben
2

L2B: DNA Origami Sommersemester 2014
1 Theoretische Grundlagen
1.1 Das DNA-Molekül
Desoxyribonukleinsäure (DNS, im folgenden immer mit der weit gebräuchlicheren englischenAbkürzung DNA bezeichnet) ist als Speicher und Überträger der Erbinformation von einerGeneration zur nächsten ein essentieller Bestandteil jedes bekannten Lebewesens. DNA istein polymerisches Makromolekül und besteht aus sich wiederholenden Untereinheiten, denNukleotiden. Jedes Nukleotid besteht aus dem Monosaccharid Desoxyribose, einer der vierNukleobasen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T) und einem Mono-, Di-oder Triphosphat. Die Nukleobase ist mit dem 1’ C-Atom der Desoxyribose verbunden undzusammen bilden sie ein sogenanntes Nukleosid. Das Phosphat ist an das 5’ C-Atom desZuckerringes im Nukleosid gebunden und komplettiert somit das Nukleotid. Nukleotidmole-küle spielen eine zentrale Rolle im Stoffwechsel von allen lebenden Organismen: sie dienen alsEnergieträger (z.B. Adenosintriphosphat ATP), partizipieren an Signalkaskaden (z.B. cycli-sches Adenosinmonophosphat cAMP) und sind Teile von wichtigen Cofaktoren enzymatischerReaktionen (z.B. Flavinmononukleotid FMN).
O
O
O
OO
O
O
O O
O
O
O
O
O
O
O
OO
O
O
O
OO
O
O
O
O
O
O
O
O
OO
O
O
O
OO
N
N
N
N
N
N
N
N
N
N
N
NN
N
N
NN
N
NN
O _
O _
O _
O _
O _
_ O
_ O
_ O
_ O
_ O
P
P
P
P
P
P
P
P
NH 2
OH
OH
NH
H 2 N
HN
NH 2
H 2 N
HN
H 2 N
NH
NH 2
3'
5'
3'
5'
Adenine Thymine
CytosineGuanine
Phosphate-deoxyriboseBackbone
2 nm
1 turn = 10.5 bp =
3.4nm
Major Groove
Minor Groove
Abbildung 1: Chemische Struktur der DNA Doppelhelix (links) und ein Kalottenmodell derB-Form DNA (rechts)
Jedes der vier möglichen Monophosphat-Nukleotide kann über das Phosphat mit dem 3’ C-Atom des Zuckerringes eines anderen Nukleotides verbunden werden. Dies bedeutet, dass langePolymere mit einer beliebigen Zusammensetzung von Nukleotiden und somit einer beliebigenSequenz entstehen können. Diese Moleküle nennt man dann Polynukleotide oder einzelsträngi-ge DNA (single-stranded DNA (ssDNA)). Als Konsequenz der asymetrischen PhosphodiesterBindung zwischen zwei benachbarten Zuckerringen kann dem Polynukleotid nun eine Rich-tung zugewiesen werden. Ein Ende der ssDNA endet mit einer Phosphatgruppe am 5’ C-Atom(5’-Ende) und das entgegengesetzte Ende mit einer Hydroxygruppe am 3’ C-Atom (3’-Ende)der Desoxyribose (siehe Abbildung 1). Diese Richtungsinformation spielt eine wichtige Rolle in
3

L2B: DNA Origami Sommersemester 2014
der enzymatische Amplifikation der DNA und somit hat jeder DNA-Strang eine Leserichtung(so werden DNA Sequenzen üblicherweise vom 5’-Ende zum 3’-Ende angegeben).
Hybridisierung
Die vier Basen können über Wasserstoffbrücken Basenpaare bilden. A und T formen zusam-men zwei, G und C zusammen drei Wasserstoffbrücken. Somit ist die G-C Paarung energetischstabiler. In wässrigen Lösungen bilden die vier Basen auch Wasserstoffbrücken mit den Was-sermolekülen. Da die aromatischen Ringe der Nukleotide fast senkrecht zur Länge des DNAStranges positioniert sind, überlappen sich die π-Orbitale benachbarter Ringe. Die Kohlen-stoffringe richten sich zueinander aus und Wassermoleküle werden aus dem Raum zwischenden Basen verdrängt. Dieser Prozess wird als base stacking bezeichnet und die Summe die-ser stacking Interaktionen stabilisiert die DNA Doppelhelix. Zwei ssDNA Moleküle mit einerentgegengesetzten komplementären Sequenz können sich somit zusammenfügen. Dieser Pro-zess wir Hybridisierung genannt und die zwei antiparallelen Stränge formen zusammen eineDoppelhelix. Unter physiologischen Bedingungen liegt die rechtsgängige B-Form DNA vor.Eine volle Umdrehung der Doppelhelix entspricht 10,5 Basen und erstreckt sich über 3.4 nm.Der Durchmesser beträgt 2 nm und die vertikale Distanz zwischen zwei benachbarten Basen0.34 nm. Andere Formen sind z.B. die A-Form und die Z-Form DNA, welche aber für dasVerständniss dieses Praktikumsversuches keine Rolle spielen.Die Hybridisierung zweier Einzelstränge zu einem doppelsträngigen Molekül kann durch Zu-fuhr von Wärme wieder rückgängig gemacht werden. Dieser Prozess wird als Schmelzen be-zeichnet. Jedes DNA-Molekül hat eine charakteristische Schmelztemperatur, welche von derSequenz und besonders der Länge des Stranges abhängig ist: lange Moleküle habe eine höhereSchmelztemperatur als kurze Moleküle.
Optische Eigenschaften
Die Nukleobasen haben ein Absorpstionsmaximum von UV-Licht bei 260 nm Wellenlänge. Diegesamte Absorption eines DNA Moleküls hängt von der Summe der Absorption der einzelnenNukleotide und den Interaktionen zwischen benachbarten Basen ab. Aufgrund dieser Interak-tionen absorbiert ein ssDNA-Molekül weniger Licht als die pure Summe der einzelnen Basenund ein dsDNA-Molekül weniger Licht als die Summe der zwei enthaltenen Einzelstränge.Der Unterschied in der Absorption von ssDNA und dsDNA wird unter anderem zur ex-perimentellen Bestimmung der Schmelztemperatur von DNA-Molekülen mit Hilfe von UV-Photometrie ausgenutzt. Auch die Konzentrationsbestimmung von DNA in Lösungen wirdmit Hilfe der UV-Photometrie durchgeführt: Hierfür wird die Absorption bei 260 nm gemes-sen und mit Hilfe des Gesetztes von Lambert-Beer die Konzentration errechnet (Mehr hierzuspäter).
1.2 DNA als Baumaterial in der Nanotechnologie
DNA hat sich in letzter Zeit als vielversprechendes Material in der Nanobiotechnologie entwi-ckelt. Folgende Eigenschaften machen sie zu einem idealen Baumaterial: DNA ist in wässrigenLösungen und einer Vielzahl von Puffern stabil und einfach zu handhaben. Sie ist für einenrelativ geringen Preis kommerziell verfügbar und Sequenzen bis zu einer Länge von 100 Basenkönnen einfach chemisch synthetisiert werden. DNA ist biokompatibel, modular und hat eineprogrammierbare Sequenz.
4

L2B: DNA Origami Sommersemester 2014
Gentechniker benutzen schon seit den siebziger Jahren dsDNA-Moleküle mit komplementä-ren überhängenden einzelsträngigen Enden (sogenannte sticky ends), um neue lineare DNAKonstrukte herzustellen. Der Chemiker Nadrian Seeman von der New York University pos-tulierte schon Anfang der achtziger Jahre, dass es möglich ist, periodische dreidimensionaleNetzwerke aus DNA herzustellen. Anfang der neunziger Jahre stellte seine Arbeitsgruppe dieerste DNA-basierte Supramolekulare Struktur vor: einen 3D Würfel aus sechs einzelsträngigenDNA Schleifen (siehe Abbildung 2a).
a b
Abbildung 2: (a) Ned Seemans DNA Würfel, bestehend aus 6 einzelsträngigen DNA Schleifen(b) Holliday Junction: links zeigt die molekulare Struktur wie die Einzelsträngevon einer Doppelhelix zur anderen übergehen, keine der Basen bleibt ungepaart,rechts ist die selbe Struktur noch einmal schematisch darsgestellt.
Ein zentrales Motif in der strukturellen DNA Nanotechnologie ist die sogenannte HollidayJunction (benannt nach dem australischen Wissenschaftler Robin Holliday). Holliday Juncti-ons sind kovalente Phosphatbindungen zwischen zwei DNA Doppelsträngen. In Organismenspielen diese Kreuzungen eine wichtige Rolle für die Rekombination von homologen Sequenzenwährend der Zellteilung. Hier kann die Holliday Junction kann frei zwischen zwei verbundenenDoppelhelizes entlang gleiten. Eine immobilisierte Junction hingegen kann durch Asymmetrieder Sequenzen erreicht werden. Solche Junctions können genutzt werden um eine räumlichfixierte Verbindung zwischen zwei Doppelhelizes zu schaffen.
1.3 DNA Origami
Die DNA Origami Technik, von Paul Rothemund im Jahr 2006 veröffentlicht, nutzt einelanges einzelsträngiges, virales DNAMolekül (der sogenannte Scaffold), um gewünschte Musterund Formen auf der Nanometerskala herzustellen. Der Scaffold wird durch hunderte kurzeOligonukleotide (die sogenannten Staples) in die gewünsche Form gefaltet (eine schematischeDarstellung dieses Prozesses ist in Abbildung 3 zu sehen). Rothemund nutzte als Scaffoldeine 7249 Basen lange modifizierte Version des Genoms des M13mp18 Bakteriophagen (mehrInformationen zu diesem Virus in 1.4). Die Sequenz dieses Scaffolds ist bekannt und er kannentweder kommerziell erworben oder relativ einfach im Labor mit Hilfe von E.coli Bakterienhergestellt werdenIm Gegensatz zu der japanischen Kunst des Papierfaltens basiert die hier beschriebene DNAOrigami Technik auf molekularer Selbstassemblierung. Der Scaffold wird mit den Staples undzweiwertigen Kationen (in der Regel Mg2+) in einer Pufferlösungen zusammen gemischt. Diese
5

L2B: DNA Origami Sommersemester 2014
Abbildung 3: Derlange einzelsträngige Scaffold (links) wird durch den kurzen Staple, welcherzwei komplementäre Stellen auf dem Scaffold hat, zusammengehalten (mitte).Wenn alle Staples hinzugefügt werden, wird der Scaffold in die vorher definierteForm gefaltet (links). Die nebeneinanderliegenden antiparallelen Doppelhelizeswerden durch Staple Doppelübergänge in der Form von immobilisierten Hollidayjunctions in der gewünschten Form gehalten.
Mischung wird dann auf eine Temperatur aufgeheizt, bei der alle DNA Moleküle einzelsträn-gig, also geschmolzen, vorliegen und über einen Zeitraum von Stunden bis Tagen langsamauf Raumtemperatur abgekühlt. Während der Abkühlung binden die Staples an ihre komple-mentären Stellen auf dem Scaffold und eine Anordnung von antiparallelen Doppelhelizes wirdgeformt. In einer gefalteten Struktur ist die Position jeder einzelnen Base bekannt. ChemischeModifikation der Basen wie zum Beispiel das Anbringen von Fluoreszenzmolekülen oder vonBiotin erlauben nun die Adressierung jeder einzelnen Base innerhalb der Struktur mit einertheoretischen Auflösung von 0.34 nm (der Abstand zwischen zwei benachbarten Basen in derDoppelhelix). Diese Eigenschaft zusammen mit der hohen Parallelisierung der Methode (etwa100 Milliarden Objekte entstehen gleichzeitig per Selbstassemblierung in einem 50µL TropfenProbenlösung) sorgen für die erstaunlichen Einsatzmöglichkeiten dieser Technik. Die Abbil-dung 4 zeigt eine der zweidimensionalen Strukturen, die Rothemund in seiner Veröffentlichungvorgestellt hat.
Abbildung 4: Darstellung eines 2D DNA Origami Rechteckes. Es misst 70 x 100 nm und ist2 nm hoch (entspricht dem Durchmesser der Doppelhelix). Links: schematischeDarstellung des Designs: es besteht aus 24 antiparallelen Doppelhelizes, welcheals Zylinder dargestellt sind. Mitte: Ausschnitt aus dem Designschema. Es zeigtden Pfad des Scaffolds durch die Struktur (in blau), welcher durch die Staples (inrot) durch Holliday Junctions zwischen benachbarten Helizes in der gewünschtenForm gehalten wird. Das 5’-Ende der Staples ist durch Rechtecke markiert, das3’-Ende durch Dreiecke. Rechts: AFM Aufnahme von gefalteten Objekten.
6

L2B: DNA Origami Sommersemester 2014
Im Jahr 2009 zeigte die Arbeitsruppe von William Shih in Harvard, dass die DNA OrigamiMethode auch auf drei Dimensionen erweitert werden kann. Die Abbildung 5 zeigt an demBeispiel eines Bündels aus sechs Doppelhelizes (Six Helix Bundle ) das Konzept: die Doppel-helizes werden in einem hexagonalen Bienenwabengitter (honeycomb lattice) mit Übergängenzu benachbarten Helizes angeordnet. Die B-Form Geometrie der DNA mit 10,5 Basenpaarenpro Umdrehung erlaubt Übergänge mit 120◦ Winkeln zueinander.
0
12345
3
05
4
1
2120°
Abbildung 5: Das Six Helix Bundle (6HB) kann vereinfacht durch sechs benachbarte Helizes,die zu einem Hexagon gefaltet wurden, dargestellt werden. In dieser Anordnunghat jede Helix 2 direkte Nachbarn und Übergänge zu diesen sind in Winkelnvon 120◦ möglich. Der Auschnitt aus dem Desigschema zeigt ein ausgerolltes6HB mit dem Scaffold in blau und den Staples in rot. Übergange des Scaffoldsund der Staples sind nur zwischen Helizes möglich, welche auch in dem Hexagonnebeneinander liegen. Die Elektronenmikroskopaufnahme rechts unten zeigt ein428 nm langes 6HB mit einem Durchmesser von 6 nm.
Vom Strukturdesign zum fertigen Objekt
Das Design von DNA Origami Strukturen hat sich durch die Entwicklung eines CAD Soft-warepacketes namens caDNAno1 extrem vereinfacht. Im Grunde kann zwischen vier Schrittenwährend des Strukturdesigns unterschieden werden. Im ersten Schritt wird ein geometrischesModell definiert, das ungefähr die gewünschte Struktur wiedergibt. Dieses geometrische Mo-dell wird dann mit antiparallelen Doppelhelizes gefüllt und ein Scaffold Pfad durch die ganzeStruktur entlang antiparallelen Übergängen an erlaubten Positionen wird gewählt. Im nächs-ten Schritt werden zum Scaffold komplementäre Staples automatisch erstellt. Diese Staplesmüssen dann in Segmente zwischen 18 und 49 Basen länge unterteilt werden (üblicherweisestrebt man eine durchschnittliche Staplelänge von 32 bis 42 Basen Länge an). Im letzten Desi-gnschritt wir der Scaffold Pfad mit der Sequenz des vorgesehenen Scaffolds bevölkert und somitsind die Staple Sequenzen determiniert. Abhängig vom Design und der Länge des Scaffoldsmüssen zwischen 200 und 300 verschiedene Staples pro Design synthetisiert werden.Diese synthetisierten Staples werden dann mit dem Scaffold in einem Verhältnis von 10:1in einer Pufferlösung mit MgCl2 als Quelle für zweiwertige Kationen gemischt. Die Probewor danach auf 80◦C aufgeheizt und langsam über einen Zeitraum von einer Stunde bis hin
1Die Software als auch Video Tutorials und Design Beispiele sind unter http://cadnano.org zu finden.
7

L2B: DNA Origami Sommersemester 2014
zu mehreren Tage, je nach Geometrie der Struktur, auf Raumtemperatur abgekühlt. DieserProzess wird als Annealing bezeichnet. Die Konzentration von MgCl2 und die Dauer des Anne-alings variieren von Design zu Design und müssen individuell für jede Geometrie experimentellbestimmt werden.
1.4 Herstellung des Scaffold-Stranges
Um flexibler im Design von DNA Origami Strukturen zu sein, ist es wünschenswert, auchverschieden lange Scaffolds zur Hand zu haben. Zu diesem Zweck wurden PCR-amplifizierteFragmente der Bakteriophagen λ-DNA in das M13mp18 RF Plasmid eingebaut (siehe http://www.neb.com/nebecomm/products/productn4018.asp für mehr Informationen zu dem M13Vektor).Zur Zeit haben wir in unserer Arbeitsgruppe verschieden Lange Versionen des urspünglich 7249Basen langen M13mp18 Vektors: 7308, 7560, 8064 und 8634 Basen lange Scaffolds. Währenddes Praktikumsversuches wird jeder Student einen dieser Scaffolds selbst herstellen.Die Produktion des Scaffolds erfolgt mit Hilfe von E.coli Bakterien. Eine Bakterienflüssigkul-tur wird mit den Viren infiziert, die Bakterien verfielfältigen den Virus und geben ihn wiederan das umgebende Flüssigmedium ab. Die Virenpartikel werden von den E.coli Zellen unddem Medium getrennt und die genomische DNA wird aus den Viren isoliert: diese DNA wirdnoch aufgereinigt und dient dann als Scaffold. Im folgenden sind einige Informationen überden M13 Bakteriophagen und seinen Replikationszyklus.
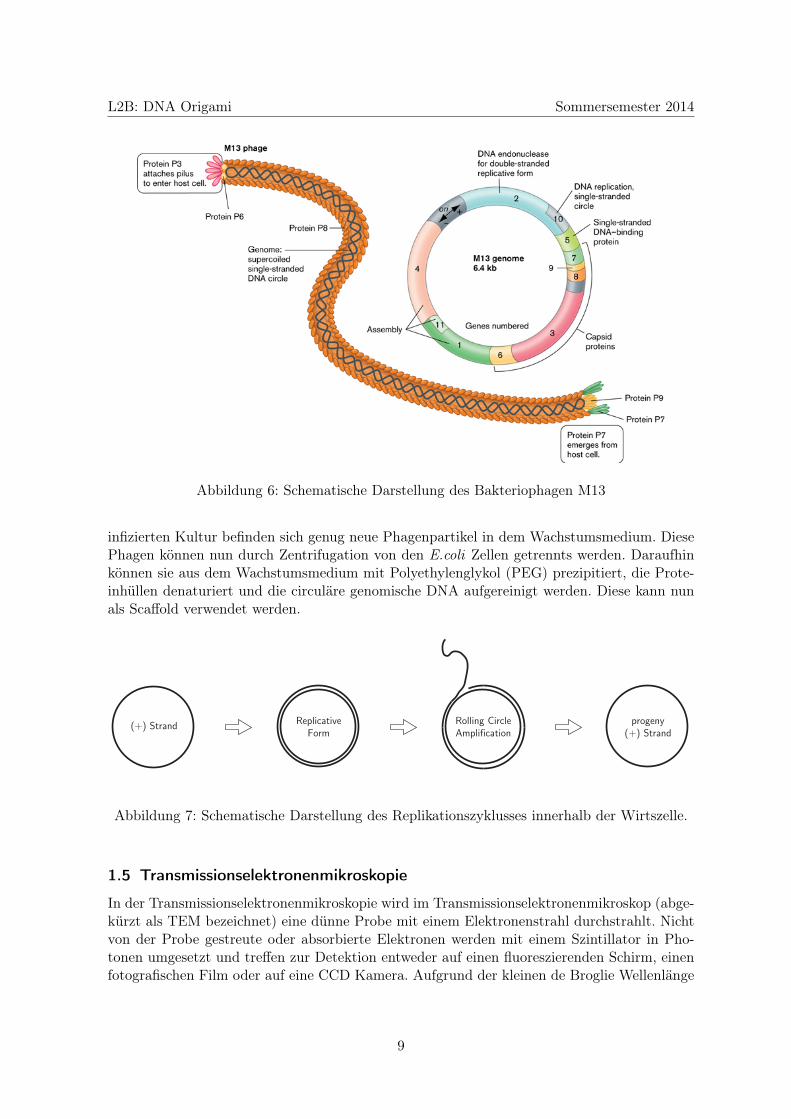
Der Bakteriophage M13
Der Bakteriophage M13 (Abbildung 6) ist ein filamentöser Virus, der E.coli Bakterien befällt.Die native Form des Virus ist ungefähr 900 nm lang und 6.5 nm breit. Die DNA erstrecktsich innerhalb der Proteinhülle entlang des Phagen und die Länge des Genomes bestimmt dieLänge des Viruspartikels. So ist ein Phagenpartikel für den 8634 Basen langen Scaffold deutlichlänger als ein Phagenpartikel für den 7249 Basen langen Scaffold. Der M13 Phage befälltnur E.coli mit einem sogenannten F-Pilus. Dieser F-Pilus dient in der Welt der Bakterienzum horizontalen Gentransfer also zum Austausch von genetischer Information in Form vonPlasmiden zwischen zwei Zellen der gleichen Generation. Somit ist es für die Scaffoldherstellungwichtig, einen geeigneten E.coli Stamm zu verwenden. Der Viruspartikel dockt mit Hilfe einesOberflächenproteins an seinem einen Ende an den F-Pilus an und die infektiöse einzelsträngigeVirus-DNA wird in die Wirtszelle eingeschleust.
Replikationszyklus
Sobald sich die infektiöse DNA ((+) Strang) in der Wirtszelle befindet, beginnt der Repli-kationszyklus (Abbildung 7). der (+) Strang wird in die doppelsträngige, replikative Form(RF DNA) prozessiert und diese darauf vervielfältigt. Der (-) Strang (entspricht dem Teil desDoppelstranges, der komplementär zum (+) Strang ist) der RF DNA wird in virale mRNA’stranskribiert und diese in die Phagenproteine translatiert. Die Amplifikation des Nachkommen(+) Strangs findet mittels einer sogenannten Rolling Circle Amplification statt. Die neu ent-standenen (+) Stränge werden in die Proteinhülle gepackt und die folgende Virusgenerationwird in das umgebende Medium freigegeben. Die Wirtszelle wird bei diesem Prozess nichtgetötet und wächst weiter, allerdings mit circa der halben Rate. Pro Generation der Wirtszel-le werden einige hundert neue Phagen produziert. Nach einigen Stunden Inkubationszeit der
8
L2B: DNA Origami Sommersemester 2014
Abbildung 6: Schematische Darstellung des Bakteriophagen M13
infizierten Kultur befinden sich genug neue Phagenpartikel in dem Wachstumsmedium. DiesePhagen können nun durch Zentrifugation von den E.coli Zellen getrennts werden. Daraufhinkönnen sie aus dem Wachstumsmedium mit Polyethylenglykol (PEG) prezipitiert, die Prote-inhüllen denaturiert und die circuläre genomische DNA aufgereinigt werden. Diese kann nunals Scaffold verwendet werden.
(+) Strand ReplicativeForm
Rolling CircleAmplification
progeny(+) Strand
Abbildung 7: Schematische Darstellung des Replikationszyklusses innerhalb der Wirtszelle.
1.5 Transmissionselektronenmikroskopie
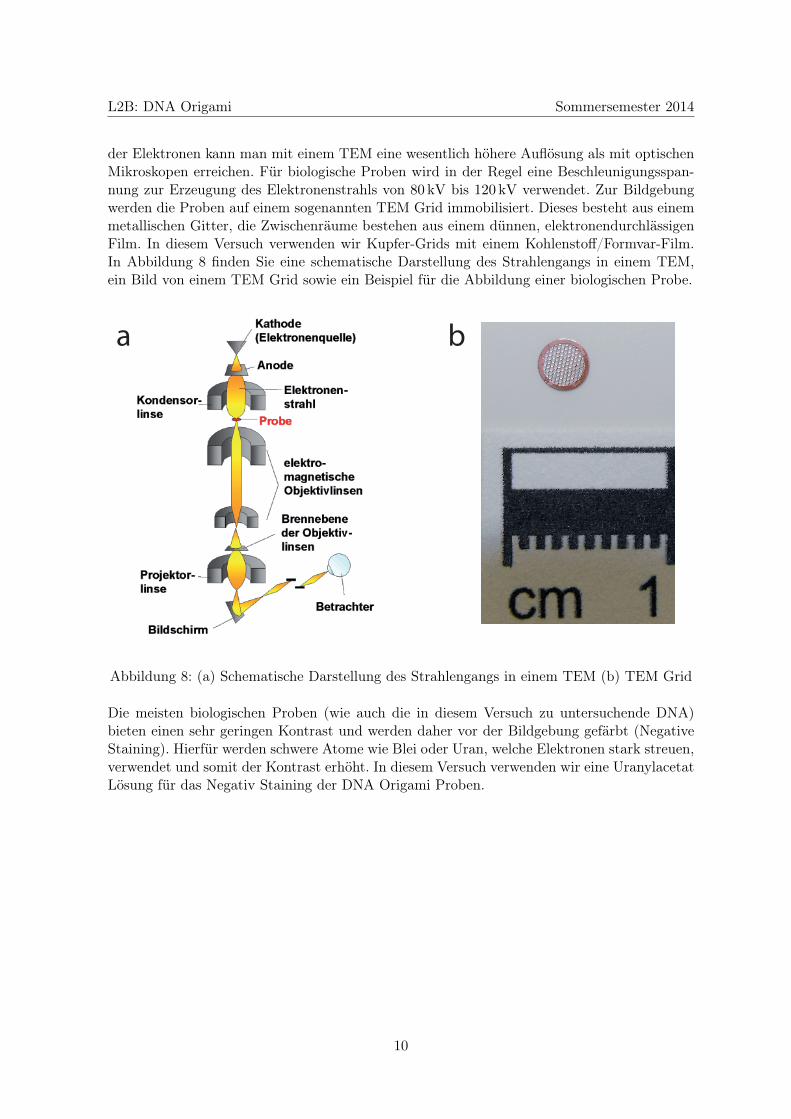
In der Transmissionselektronenmikroskopie wird im Transmissionselektronenmikroskop (abge-kürzt als TEM bezeichnet) eine dünne Probe mit einem Elektronenstrahl durchstrahlt. Nichtvon der Probe gestreute oder absorbierte Elektronen werden mit einem Szintillator in Pho-tonen umgesetzt und treffen zur Detektion entweder auf einen fluoreszierenden Schirm, einenfotografischen Film oder auf eine CCD Kamera. Aufgrund der kleinen de Broglie Wellenlänge
9
L2B: DNA Origami Sommersemester 2014
der Elektronen kann man mit einem TEM eine wesentlich höhere Auflösung als mit optischenMikroskopen erreichen. Für biologische Proben wird in der Regel eine Beschleunigungsspan-nung zur Erzeugung des Elektronenstrahls von 80 kV bis 120 kV verwendet. Zur Bildgebungwerden die Proben auf einem sogenannten TEM Grid immobilisiert. Dieses besteht aus einemmetallischen Gitter, die Zwischenräume bestehen aus einem dünnen, elektronendurchlässigenFilm. In diesem Versuch verwenden wir Kupfer-Grids mit einem Kohlenstoff/Formvar-Film.In Abbildung 8 finden Sie eine schematische Darstellung des Strahlengangs in einem TEM,ein Bild von einem TEM Grid sowie ein Beispiel für die Abbildung einer biologischen Probe.
a b
Abbildung 8: (a) Schematische Darstellung des Strahlengangs in einem TEM (b) TEM Grid
Die meisten biologischen Proben (wie auch die in diesem Versuch zu untersuchende DNA)bieten einen sehr geringen Kontrast und werden daher vor der Bildgebung gefärbt (NegativeStaining). Hierfür werden schwere Atome wie Blei oder Uran, welche Elektronen stark streuen,verwendet und somit der Kontrast erhöht. In diesem Versuch verwenden wir eine UranylacetatLösung für das Negativ Staining der DNA Origami Proben.
10

L2B: DNA Origami Sommersemester 2014
2 Versuchsdurchführung
Tag 1
Amplifikation der Bakteriophagen
Die Vermehrung des Phagen erfolgt in einer E.coli Flüssigkultur. Hierfür benötigt man einer-seits einen geeigneten Stamm von E.coli Bakterien, welcher von dem Phagen infiziert werdenkann (die Zellen müssen also einen F-Pilus besitzen). Wir verwenden hierfür E.coli K91endA-Zellen. Diese werden in einem Tiefkühler bei -80◦C gelagert und sind nach dem Auftauenlebensfähig und beginnen sich zu teilen. Außerdem braucht man ein geeignetes Wachstums-medium. Das am häufigsten genutzte Medium zur Kultivierung von E.coli Bakterien ist dasLB Medium (siehe http://de.wikipedia.org/wiki/LB-Medium). Um eine optimale Phagen-ausbeute zu bekommen, verwenden wir ein verändertes LB Medium: das 2xYT Medium. Esenthält 16 g Trypton, 10 g Hefeextrakt und 5 g NaCl pro Liter.
Zu Beginn des ersten Versuchstages steht schon eine Flüssigkultur zur Verfügung. Hierfür wer-den am Tag vorher einige tiefgefrorene Zellen in 50mL LBMedium gegeben und über Nacht bei37◦C (entspricht der optimalen Wachstumstemperatur für die Zellen) in einem Schüttelinku-bator (sorgt für einen hohen Sauerstoffgehalt in der Flüssigkeit) gestellt. Die Zellen vermehrensich nun exponentiell und am Morgen des ersten Versuchstages ist eine Übernachtkultur mithoher Zelldichte entstanden. Diese wird dann von der betreuenden Person in 200mL 2xYTMedium mit 5mM MgCl2 verdünnt und wiederum bei 37◦C inkubiert.
Diese Vorbereitete Flüssigkultur wird nun bis zu einer optischen Dichte bei 650 nm von 0,5inkubiert.
• Bestimmung des Zellwachstums alle 15 Minuten durch die Messung der OD650 bis zueinem Wert von 0,5 (dies entspricht einer Zelldichte von 4× 108 Zellen/mL)
• Zugabe des Phagen in einem Phagen-zu-Zell Verhältnis von 1
• inkubieren für 3− 5 hrs bei 37◦C, 300 rpm, danach Entnahme aus dem Schüttelinkubator
• überführen in Zentrifugengefäße und zentrifugieren bei 4000 rcf, 4◦C für 15min
Durch diesen Zentrigutationsschritt bildet sich ein festes Pellet aus Zellen am Boden desGefäßes, die von den Bakterien produzierten Phagen bleiben allerdings in der Mediumslösung.Diese Mediumslösung wird nun weiter verwendet, die Bakterienzellen können entsorgt werden.
PEG Präzipitation der Viruspartikel
Die sich in der Lösung befindenden Phagen werden nun mit Hilfe von PEG-8000 und NaClprezipitiert und können dann abzentrifugiert werden. Es bildet sich wiederum ein festes Pelletam Boden, diesmal enthält es allerdings die prezipitierten Phagen und der Überstand kannentsorgt werden.
• Zum Überstand, 8 g PEG 8000 und 6 g NaCl hinzufügen
• die Lösung mit einem Magnetrührer 10-15min durchmischen
11

L2B: DNA Origami Sommersemester 2014
• auf Eis 15 − 30min inkubieren
• bei 10 000 rcf, 4◦C für 20min zentrifugieren
• Überstand verwerfen
• restliche Flüssigkeit mit Pipette aus dem Zentrifugengefäß entfernen
• Phagenpellet in 5mL 10mM Tris (pH8.5) resuspendieren
Extraktion der viralen einzelsträngigen Genom-DNA
Nun haben wir den Phagen aufgereinigt in einer Pufferlösung vorliegen. Da wir aber nur dieDNA benötigen, die sich innerhalb der Proteinhülle des Phagen befindet, werden nun dieHüllenproteine denaturiert.
• zu 5mL Phagenlösung werden hinzugegeben:
• 5mL PPB2 (0.2M NaOH, 1% SDS), leicht mischen durch Schwenken, 3min warten
• 3.75mL PPB3 (3M KOAc pH 5,5 titriert mit Eisessig), leicht mischen durch invertieren
• im Eiswasserbad 10min inkubieren
• mit 16.500 rcf bei 4◦C für 30min zentrifugieren
Nach dem zentrifugieren hat sich ein Pellet aus den denaturierten Proteinen gebildet und dieDNA befindet sich gelöst im Überstand: das Pellet wird also entsorgt und der Überstand fürdie weiteren Schritte gesammelt.
Ethanol Präzipitation der gewonnenen einzelsträngigen Genom-DNA
Nun wollen wir noch die gewonnene DNA aufreinigen. Dies geschieht durch die Ethanol Pre-zipitation. Durch die Zugabe von 2 bis 3 mal dem Volumen der Lösung Ethanol wird diePolarität der Lösung reduziert und somit die Hydrathüllen, welche die negative Ladung derDNA abschirmen, aufgebrochen. Die elektrostatischen Wechselwirkungen zwischen dem nega-tiv geladenen Phosphatrückgrat der DNA und positiven Ionen in der Lösung (durch die Zugabevon Natriumacetat) werden nun so stark, das die DNA prezipitiert. Durch Zentrifugation kannnun wieder ein Pellet gebildet werden.
• 1/10 des Probenvolumens 3M Natriumacetat zugeben
• 2 mal das Probenvolumen gekühlten 100% Ethanol zugeben
• 30min auf Eis inkubieren
• zentrifugieren: 16.500 rcf, 4◦C, 30min
• vorsichtig den Überstand entfernen
• 20mL gekühlten 70% Ethanol zugeben
• zentrifugieren: 16.500 rcf, 4◦C, 10min
12

L2B: DNA Origami Sommersemester 2014
• sofort den Überstand entfernen
• das Pellet trocknen lassen
• Pellet in 1mL TE resuspendieren
Analyse der gewonnenen DNA
Nun haben wir die gewonnene DNA aufgereinigt und wollen die Konzentration und die Rein-heit bestimmen. Dies geschieht mit Hilfe eines UV/Vis-Photometers, dem Nanodrop. Hierfürgenügen 2µL Probenvolumen. Die Absorption bei 260 nm wird gemessen und daraus die Kon-zentraion in mol/L errechnet. Die Software des Nanodrops benutzt einen Standard Absorp-tionskoeffizienten und gibt die Konzentration in ng/µL aus. Diese gemessene Konzentrationkann nun über die Anzahl der Basen des DNA Moleküls sowie der durchschnittlichen moleku-laren Masse der einzelnen Basen (= 330 g/mol) umgerechnet werden.
Faltung der DNA Origami Strukturen
Als DNA Origami Struktur wird ein 6HB gefaltet. Im der Probenlösung sollen am Ende 10nMScaffold, 100nM Staples, 1xTE Puffer und 14mM MgCl2 vorhanden sein. Für einen 20µLAnsatz wird folgendes in einem 200µL PCR-Tube zusammenpipettiert:
• 10µL Staples (200nM)
• 6µL H2O
• 2µL 10xTE, 140mM MgCl2
• 2µL des hergestellten Scaffolds (100nM)
Die Probe wird nun im Thermocycler platziert, auf 80◦C aufgeheizt und innerhalb einer Stundeauf Raumtemperatur abgekühlt.
Tag 2
Färbung der gefalteten Proben
Um eine optimale Verteilung der Probe auf dem TEM Grid zu ermöglichen, sollte die Ober-fläche des Grids möglichst hydrophil sein. Dies wird durch eine Plasmabehandlung der Gridserreicht: hierfür werden die Grids vor dem Aufbringen der Probe für 60 s bei 240V mit einemArgon Plasma behandelt.
Für die Färbung der Probe mit Uranylacetat müssen unbedingt Nitrilhandschuhe und einLaborkittel getragen werden!
• 2µL Probe werden auf das Grid gegeben, 1 Minute warten
• zwei 7µL Tropfen 1% Uranylacetat werden auf Parafilm platziert
• Probenflüssigkeit auf dem Grid wird mit Filterpapier abgezogen
13

L2B: DNA Origami Sommersemester 2014
• Grid wird in ersten Uranylacetat Tropfen getaucht und die Flüssigkeit mit Filterpapierabgezogen
• Grid wird in zweiten Uranylacetat Tropfen getaucht und 10 s gewartet
• die Flüssigkeit wird mit Filterpapier abgezogen
Nach 30min Trocknung können die Proben im TEM analysiert werden
TEM Analyse der gefärbten Proben
Details zur TEM Analyse der Proben werden am Gerät von der betreuenden Person gegeben.
14


















