
MANAGEMENT OF NEUROMUSCULAR DISEASES - dgm.org · ren. Die nukleären Lamine (Lamin A/C, B1) sind...
12
MANAGEMENT OF NEUROMUSCULAR DISEASES LETTER NR. 28 Zusammenfassung Erst seit einigen Jahren ist bekannt, dass Veränderungen an Kernhüllen- proteinen gewebsspezifische, häufig neuromuskuläre Veränderungen zur Folge haben können. Zurzeit sind zwei Kernhüllenproteine bekannt, das Lamin A/C auf dem LMNA-Gen und Emerin auf dem STA-Gen, bei denen Mutationen zu phänotypisch definier- baren Erkrankungen führen. Die Emery-Dreifuss-Muskeldystrophie mit X-chromosomalem Erbgang (XR- EDMD) ist klinisch neben den mus- kulären Symptomen durch Kontraktu- ren und eine später einsetzende kar- diale Beteiligung (Herzrhythmus- störungen) gekennzeichnet. Die kardia- le Beteiligung ist für die Prognose von entscheidender Bedeutung. Die klinischen Manifestationsformen einer Mutation auf dem LMNA-Gen umfassen die autosomal-dominant vererbte Hauptmann-Thannhauser- Muskeldystrophie (HTMD, häufig auch als autosomal-dominante Form der Emery-Dreifuss-Muskeldystrophie bezeichnet), die Gliedergürtel-Mus- keldystrophie 1B (LGMD 1B), die dila- tative Kardiomyopathie 1A (DCM1A) und die familiäre partielle Lipodystro- phie Typ Dunnigan (FPLD Dunnigan), die autosomal-rezessiv vererbte here- ditäre sensomotorische Neuropathie Typ 2B (HSMN 2B) und die mandibulo- Kernhüllenerkrankungen (Emery-Dreifuss-Muskeldystrophie, Hauptmann-Thannhauser-Muskeldystrophie, hereditäre sensomotorische Neuropathie Typ 2B) Frank Hanisch und Stephan Zierz
Transcript of MANAGEMENT OF NEUROMUSCULAR DISEASES - dgm.org · ren. Die nukleären Lamine (Lamin A/C, B1) sind...

MANAGEMENT OF NEUROMUSCULAR DISEASES
LETTER NR. 28
Zusammenfassung
Erst seit einigen Jahren ist bekannt,dass Veränderungen an Kernhüllen-proteinen gewebsspezifische, häufigneuromuskuläre Veränderungen zurFolge haben können. Zurzeit sind zweiKernhüllenproteine bekannt, dasLamin A/C auf dem LMNA-Gen undEmerin auf dem STA-Gen, bei denenMutationen zu phänotypisch definier-baren Erkrankungen führen. Die Emery-Dreifuss-Muskeldystrophiemit X-chromosomalem Erbgang (XR-EDMD) ist klinisch neben den mus-kulären Symptomen durch Kontraktu-ren und eine später einsetzende kar-diale Beteiligung (Herzrhythmus-störungen) gekennzeichnet. Die kardia-
le Beteiligung ist für die Prognose vonentscheidender Bedeutung. Die klinischen Manifestationsformeneiner Mutation auf dem LMNA-Genumfassen die autosomal-dominantvererbte Hauptmann-Thannhauser-Muskeldystrophie (HTMD, häufig auchals autosomal-dominante Form derEmery-Dreifuss-Muskeldystrophiebezeichnet), die Gliedergürtel-Mus-keldystrophie 1B (LGMD 1B), die dila-tative Kardiomyopathie 1A (DCM1A)und die familiäre partielle Lipodystro-phie Typ Dunnigan (FPLD Dunnigan),die autosomal-rezessiv vererbte here-ditäre sensomotorische NeuropathieTyp 2B (HSMN 2B) und die mandibulo-
Kernhüllenerkrankungen(Emery-Dreifuss-Muskeldystrophie,
Hauptmann-Thannhauser-Muskeldystrophie,
hereditäre sensomotorische Neuropathie Typ 2B)Frank Hanisch und Stephan Zierz

akrale Dysplasie sowie verschiedeneProgerieformen (Tabelle 1). X-chromo-somal rezessive Emery-Dreifuss-Mus-keldystrophie und autosomal-domi-nante Hauptmann-Thannhauser-Mus-keldystrophie sind klinisch kaum unter-scheidbar. Es ist bislang noch unge-klärt, wie Art und Lokalisation einerMutation auf dem LMNA-Gen den Phä-notyp beeinflussen und worauf dieFunktionsstörung des Lamin A/C-Pro-teins beruht [1].
Emerin-Protein und STA-Gen
Emerin ist ein transmembranales, inder inneren Kernhüllenmembran ver-ankertes und ins Nukleoplasma rei-chendes Protein mit einem Molekular-gewicht von 34 kDa und wird durchdas auf dem distalen Xq28-Chromo-som lokalisierte STA-Gen kodiert [2].Die Mehrzahl der über das gesamteGen verteilten Mutationen stellenPunktmutationen (47,3 %) oder zueiner Verschiebung des Leserastersführende kleinere Deletionen (32,5 %)
2
Abbildung 1:
Schematische Darstellung der Kernhülle mit ausgewählten Kernhüllen-
proteinen. LAP – laminassoziiertes Protein, LBR – Lamin-B-Rezeptor.
(Abbildung erstellt von Thorsten Schmidt)

oder Insertionen (10,9 %) dar, die zueinem Abbruch der Translation und inFolge dessen zu einem trunkierten Pro-tein führen. In ca. 95 % der Fälle kanndie Diagnose durch das Fehlen einer34 kDa Bande im Western-Blot oderreduzierte bzw. fehlende Immunreakti-vität für Emerin in der Haut- oder Mus-kelbiopsie gestellt werden [3] (Abbil-dung 1, 2a, Tabelle 2).
Lamin A/C-Proteine und
LMNA-GenDas LMNA-Gen befindet sich auf demLocus 1q21.2-1q21.3, umfasst 24 kbund besteht aus 12 Exons [4]. Durchalternatives Spleißen innerhalb desExon 10 entstehen mRNAs, die LaminA (80 kDa) und Lamin C (65 kDa) kodie-
ren. Die nukleären Lamine (Lamin A/C,B1) sind netzartig angeordnete, derinneren Kernmembran aufliegendeIntermediärfilament-Proteine.Bisher wurden etwa 40 Punktmutatio-nen in allen Exons, eine Nonsense-Mutation, fünf Deletionen und eineMutation am Ort des Spleißensbeschrieben. Nur vier Mutationenhaben ein trunkiertes, nicht funktions-fähiges Protein zur Folge [4,5]. Bei denanderen Mutationen vermutet man alsMechanismus eine Haploinsuffizienz:Die Menge des vom normalen Allelexprimierten Proteins scheint nichtausreichend, ein funktionsfähigesLaminnetzwerk zu gewährleisten. Indiesen Fällen stellt sich das Lamin A/C-Protein jedoch immunhistologisch
3
Krankheit Abkürzung Vererbungsmodus Gen GenproduktEmery-Dreifuss-Muskeldystrophie XR-EDMD XR STA EmerinHauptmann-Thannhauser-Muskeldystrophie AD-EDMD AD, (AR) LMNA Lamin A/CGliedergürtel-Muskeldystrophie Typ 1B LGMD 1B1 AD LMNA Lamin A/CDilatative Kardiomyopathie 1A DCM 1A AD LMNA Lamin A/CFamiliäre partielle Lipodystrophie FPLD AD LMNA Lamin A/CTyp Dunnigan Typ DunniganHereditäre sensomotorische Neuropathie HSMN 2B AR LMNA Lamin A/CTyp 2BMandibuloakrale Dysplasie MAD AR LMNA Lamin A/CHutchinson-Gilbert-Progerie-Syndrom HGPS AD LMNA Lamin A/CWerner-Syndrom2 AD LMNA Lamin A/C
1 - (engl.) Limb Girdle Muscular Dystrophy2 - progerieähnliches SyndromAD – autosomal-dominant, AR – autosomal-rezessiv, XR – X-chromosomal-rezessiv
Tabelle 1:
Übersicht über Krankheiten, die auf Defekten der Kernhülle beruhen.

unauffällig dar. Bei klinischem Ver-dacht auf eine Störung des Lamin A/C-Proteins ist das Sequenzieren desgesamten LMNA-Gens erforderlich(Abbildung 1, 2b, Tabelle 2).
Aufgrund der Lokalisation von Emerinund Lamin A/C wird eine Funktion imAufbau und der Erhaltung der Kern-hülle, aber auch zur Chromosomenpo-sition vermutet.Bislang ist es nicht möglich, aus derLokalisation oder Art der zugrunde lie-genden Mutation eine Vorhersage deszu erwartenden Phänotyps oder desVerlaufs zu treffen. Mögliche, den Phä-notyp modifizierende Mechanismensind bislang ebenso unbekannt. Häu-fig besteht bei allen im Folgendengeschilderten Erkrankungen einegroße inter- und intrafamiliäre Variabi-lität [1,8]
Emery-Dreifuss-Muskel-dystrophie
(XR-EDMD)1966 beschrieben Emery und Dreifussmännliche Mitglieder einer amerikani-schen Familie mit Gliedergürtelsyn-drom, Kontrakturen, kardialer Beteili-gung und Anhalt für einen X-chromo-somalen Erbmodus [3]. Die Emery-Dreifuss-Muskeldystrophiemanifestiert sich überwiegend im Kin-desalter, jedoch selten vor dem zwei-ten Lebensjahr mit Gangstörungenund häufigen Stürzen. Im Verlauf derzweiten Lebensdekade treten skapulo-humeral betonte Atrophien, im Glie-dergürtel bzw. humeroperoneal beton-te Paresen, Kontrakturen im Ellbogen-und Sprunggelenk, seltener im Hüft-oder Kniegelenk und eine verminderteWirbelsäulenbeweglichkeit auf. Leich-
4
Abbildung 2:
Regelrechte immunhistochemische
Darstellung der Zellkerne im Muskel-
gewebe mit Antikörpern gegen Eme-
rin (a) und Lamin A/C (b). Nur in
wenigen Fällen hat eine Mutation im
Lamin A/C-Gen eine verminderte
Darstellung des Proteins zur Folge.

5
te Gehbehinderungen sind ab der 4.Lebensdekade häufig, selbst im höhe-ren Lebensalter ist die notwendigeBenutzung eines Rollstuhls seltengegeben. Verlaufsformen mit einerraschen Progredienz muskulärer Symp-tome und einem schweren Behinde-rungsgrad sind jedoch möglich.Wadenhypertrophie oder Fußdefor-mitäten können vorkommen. Die Krea-tinkinase ist normal bis mäßig erhöht.Histologisch sind gering- bis mäßig-gradige myopathische Veränderungennachweisbar. Die Achillessehnenver-kürzung kann operativ-orthopädischversorgt werden [3,7].Die kardialen Symptome sind progno-sebestimmend (AV-Block, Vorhofflim-mern- und -flattern). Die Penetranz derkardialen Symptome liegt ab dem 30.Lebensjahr bei > 95 %. Nach Diagnose-stellung sollten mindestens jährlich einRuhe-EKG, ein 24-h-Holter-EKG undeine transthorakale Echokardiographiedurchgeführt werden. Wegen derGefahr eines plötzlichen Herztodessollte schon bei asymptomatischenPatienten mit beginnenden Auffällig-
keiten im EKG ein Schrittmacher mitzusätzlicher Defibrillatorfunktion im-plantiert werden. Beim Nachweissupraventrikulärer Überleitungsstö-rungen ist eine Antikoagulanzienthera-pie indiziert.Im Gegensatz zu den anderen X-chro-mosomal rezessiven Muskeldystrophi-en vom Typ Duchenne und Becker fal-len Konduktorinnen nur selten durcheine erhöhte Kreatinkinase auf. Mitzunehmendem Lebensalter nimmt beiKonduktorinnen das Risiko atrioventri-kulärer Überleitungsstörungen zu.Deshalb gelten die oben genanntenEmpfehlungen für asymptomatischemännliche Mutationsträger als auchKonduktorinnen [3,8].
Hauptmann-Thannhauser-
Muskeldystrophie
(HTMD, häufig auch als autosomal-dominante Form der Emery-Dreifuss-Muskeldystrophie bezeichnet) Die zwei aus Deutschland emigriertenÄrzte Hauptmann und Thannhauserbeschrieben 1941 neun Mitgliedereiner Familie mit "muscular shortening
Histochemie Emerinopathie LaminopathieImmunreaktivität in ~ 95 % fehlend in ~ 95 % normalWestern-Blot in ~ 95 % fehlende in ~ 95 % normale
34 kDa Bande 70 kDa BandeGen-Sequenzierung in 5 % erforderlich in 95 % erforderlich
Tabelle 2:
Protein- und Gendiagnostik bei Verdacht auf Kernhüllenerkrankung.

and dystrophy". Im anglophonenSprachgebrauch ist jedoch die Bezeich-nung autosomal-dominante Emery-Dreifuss-Muskeldystrophie üblich [9].Phänotypisch sind die X-chromosomal
rezessive Emery-Dreyfuss-Muskeldys-trophie und die Hauptmann-Thann-hauser-Muskeldystrophie nicht sicherunterscheidbar [3] (Tabelle 3, Abbil-dung 3). Ab der 2., meist jedoch im Verlauf der3. Lebensdekade werden, zunächstüberwiegend asymptomatisch, Herz-rhythmusstörungen (Bradykardie,atrioventrikulärer Block I-III) auffällig.Das Risiko, im mittleren Lebensaltereinen plötzlichen Herztod zu erleiden,ist hoch. Parallel zu den Überleitungs-störungen kann sich eine dilatativeKardiomyopathie manifestieren. [5].Da infolge einer progredienten dilatati-ven Kardiomyopathie das Risiko für einSick-Sinus-Syndrom oder ventrikuläreTachykardien besteht, sollten die Pati-enten mit einem Schrittmacher mitzusätzlicher Defibrillatorfunktion ver-sorgt werden. Aus den oben genann-ten Gründen ist die Anlage eines Herz-schrittmachers schon bei asymptoma-tischen Patienten bei Auftreten vonSinus- oder av-Knotenstörungen zuempfehlen. Bei einem Teil der Patien-ten wird eine Herztransplantationerforderlich. Es erscheint sinnvoll, bei allen Mutati-onsträgern ab der 2. Lebensdekademindestens jährlich eine 24-h-Holter-EKG-Untersuchung und eine transtho-rakale Echokardiographie durchzu-führen. Beim Nachweis supraventri-kulärer Überleitungsstörungen ist eineAntikoagulanzientherapie indiziert[4,5,8,10]. Bisher sind mehr als 20 über das
6
Abbildung 3 a, b:
16-jähriger Patient mit Hauptmann-
Thannhauser-Muskeldystrophie
infolge der Missense-Mutation
R401C. Auffällig sind Lordose,
Beugekontrakturen der Ellbogen-
und Hüftgelenke, humeroperoneal
betonte Atrophien und beidseitige
Scapula alata.
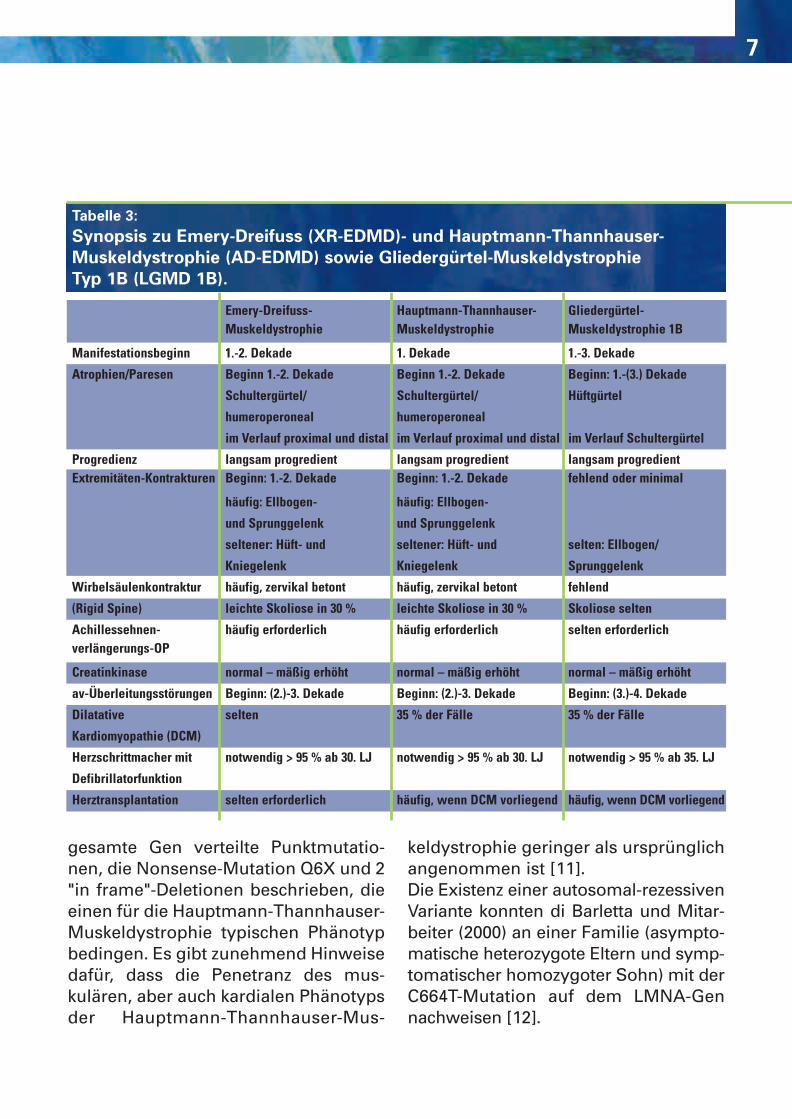
gesamte Gen verteilte Punktmutatio-nen, die Nonsense-Mutation Q6X und 2"in frame"-Deletionen beschrieben, dieeinen für die Hauptmann-Thannhauser-Muskeldystrophie typischen Phänotypbedingen. Es gibt zunehmend Hinweisedafür, dass die Penetranz des mus-kulären, aber auch kardialen Phänotypsder Hauptmann-Thannhauser-Mus-
keldystrophie geringer als ursprünglichangenommen ist [11].Die Existenz einer autosomal-rezessivenVariante konnten di Barletta und Mitar-beiter (2000) an einer Familie (asympto-matische heterozygote Eltern und symp-tomatischer homozygoter Sohn) mit derC664T-Mutation auf dem LMNA-Gennachweisen [12].
7
Emery-Dreifuss- Hauptmann-Thannhauser- Gliedergürtel-Muskeldystrophie Muskeldystrophie Muskeldystrophie 1B
Manifestationsbeginn 1.-2. Dekade 1. Dekade 1.-3. Dekade
Atrophien/Paresen Beginn 1.-2. Dekade Beginn 1.-2. Dekade Beginn: 1.-(3.) Dekade
Schultergürtel/ Schultergürtel/ Hüftgürtel
humeroperoneal humeroperoneal
im Verlauf proximal und distal im Verlauf proximal und distal im Verlauf Schultergürtel
Progredienz langsam progredient langsam progredient langsam progredientExtremitäten-Kontrakturen Beginn: 1.-2. Dekade Beginn: 1.-2. Dekade fehlend oder minimal
häufig: Ellbogen- häufig: Ellbogen-
und Sprunggelenk und Sprunggelenk
seltener: Hüft- und seltener: Hüft- und selten: Ellbogen/
Kniegelenk Kniegelenk Sprunggelenk
Wirbelsäulenkontraktur häufig, zervikal betont häufig, zervikal betont fehlend
(Rigid Spine) leichte Skoliose in 30 % leichte Skoliose in 30 % Skoliose selten
Achillessehnen- häufig erforderlich häufig erforderlich selten erforderlichverlängerungs-OP
Creatinkinase normal – mäßig erhöht normal – mäßig erhöht normal – mäßig erhöht
av-Überleitungsstörungen Beginn: (2.)-3. Dekade Beginn: (2.)-3. Dekade Beginn: (3.)-4. Dekade
Dilatative selten 35 % der Fälle 35 % der Fälle
Kardiomyopathie (DCM)
Herzschrittmacher mit notwendig > 95 % ab 30. LJ notwendig > 95 % ab 30. LJ notwendig > 95 % ab 35. LJ
Defibrillatorfunktion
Herztransplantation selten erforderlich häufig, wenn DCM vorliegend häufig, wenn DCM vorliegend
Tabelle 3:
Synopsis zu Emery-Dreifuss (XR-EDMD)- und Hauptmann-Thannhauser-
Muskeldystrophie (AD-EDMD) sowie Gliedergürtel-Muskeldystrophie
Typ 1B (LGMD 1B).

Gliedergürtel-Muskel-
dystrophie 1B (limb girdle muscular dystrophy 1B –LGMD 1B)Bei der Gliedergürtel-Muskeldystro-phie 1B (LGMD 1B) mit autosomal-dominantem Erbgang konnte entwe-der eine Missense-Mutation, eine "inframe"-Deletion oder eine "splicedonor site"-Mutation auf dem LMNA-Gen identifiziert werden. Nur bei letzte-rer Mutation war ein trunkiertes LaminA/C-Protein nachweisbar [13,14]. In der Hälfte der Fälle manifestiertsich die Erkrankung im Kindesalter,bei den anderen in der 2. oder 3.Lebensdekade. Paresen betreffenzunächst den Hüftgürtel und dieOberarmmuskulatur. Typisch ist dieAussparung der tibialen und pero-nealen Muskeln sowie eine Atrophiedes M. biceps brachii ab der 4. Deka-de. Gelegentlich ist eine Wadenhy-pertrophie auffällig.Die Erkrankung verläuft hinsichtlichder skelettmuskulären Beteiligunggering progredient. Klinisch scheinteine Abgrenzung zur Hauptmann-Thannhauser-Muske ldyst rophieberechtigt, da Bewegungseinschrän-kungen im Wirbelsäulenbereich bis-lang nicht beschrieben wurden undKontrakturen im Ellbogen- oderSprunggelenk sich erst spät manifes-tierten oder nur gering ausgeprägtsind. Die Kreatinkinase kann normwer-tig oder gering erhöht sein (Tabelle 3). Kardiale Symptome, in der Regel atrio-ventrikuläre Überleitungsstörungen,
selten eine dilatative Kardiomyopathie,treten im Verlauf der 3. – 4. Lebensde-kade auf. Auch bei dieser Erkrankungs-form sollten nach Diagnosestellungwegen des Risikos eines plötzlichenHerztodes in jährlichen Abständen ein24-h-Holter-EKG und eine transthora-kale Echokardiographie durchgeführtwerden und großzügig die Indikationfür eine Schrittmacherimplantation mitzusätzlicher Defibrillatorfunktion ge-stellt werden. Beim Nachweis supra-ventrikulärer Überleitungsstörungenist eine Antikoagulanzientherapie indi-ziert [8].
Dilatative Kardiomyopathie 1A
(DCM 1A)Die dilatative Kardiomyopathie vomTyp 1A (DCM 1A) ist die erste familiä-re DCM mit autosomal-dominantemErbgang, bei der Genort und zugrundeliegendes Genprodukt identifiziertwurden [15]. Typisch ist die sich über-wiegend in der 3. – 4. Lebensdekademanifestierende Kombination ausdilatativer Kardiomyopathie und atrio-ventrikulären Überleitungsstörungen.Eine Skelettmuskelbeteiligung ist häu-fig nicht nachweisbar. Sie kann sichjedoch in einer asymptomatischenKreatinkinaseerhöhung oder demgering ausgeprägten Phänotyp einerHauptmann-Thannhauser-Muskeldys-trophie bzw. einer Gliedergürtel-Mus-keldystrophie manifestieren [5,8,15,16].
8

Hereditäre sensomotorische Neuropathie Typ 2B (HSMN 2B; englisch: Charcot-Marie-Tooth Type 2B)Die genetisch unterschiedlichen, selte-nen hereditären sensomotorischenNeuropathien mit axonaler Schädi-gung und autosomal-rezessivem Erb-gang wurden bislang meist in maghre-binischen Ländern aufgrund der häufi-gen Ehen zwischen Blutsverwandtenidentifiziert. Die HSMN 2B wurde inzehn algerischen, teilweise blutsver-wandten, Familien beschrieben.Ursächlich war ausschließlich diehomozygote C892T-Mutation. Die Erkrankung manifestiert sich amEnde der 1. oder im Verlauf der 2.Lebensdekade mit Steppergang infol-ge Parese und Atrophie der Mm. pero-nei longus et tibiales anteriores. EinPes cavus ist in allen Fällen nachweis-bar. Im Verlauf breiten sich Schwächeund Atrophie auf die distale Muskula-tur der oberen Extremität, aber auchdie Hüftmuskulatur aus. Eine sensibleStörung mit strumpfförmiger Vertei-lung ist klinisch weniger bedeutend.Bei ca. 50 % der Betroffenen führt dieMutation schon während der 3. und 4.Lebensdekade zu erheblichen Gehbe-hinderungen und einer deutlichenBeeinträchtigung der Handfunktion intäglichen Aufgaben. Der Krankheits-verlauf ist von Fall zu Fall jedoch rechtheterogen [17].Eine Fettverteilungsstörung oder einekardiale Mitbeteiligung wurde bislangbei keinem Patienten festgestellt.
Neuromuskuläre oder kardialeSymptome bei anderen
Krankheiten mit LMNA-MutationNeben den Erkrankungen mit primärneuromuskulärer oder kardialer Betei-ligung sind weitere Syndromebekannt, die auf LMNA-Mutationenberuhen. Dazu gehören die familiärepartielle Lipodystrophie Typ Dunnigan(FPLD), die mandibuloakrale Dysplasie(MAD), das Hutchinson-Gilbert-Proge-rie-Syndrom und das Werner-Syndrom(Tabelle 1). Die FPLD ist einerseits durch eine Atro-phie des subkutanen Fettgewebes inExtremitäten und Rumpf, andererseitsdurch eine Lipidakkumulation inGesicht und Nacken gekennzeichnet.Klinisch stehen die Folgen einer Insu-linresistenz und Dyslipidämie mit densich daraus ergebenden atheroskleroti-schen Komplikationen im Vordergrund[18]. Bei drei Familien und einem Einzelfallmit FPLD konnten zusätzliche Sympto-me im Sinne einer kardialen Mitbeteili-gung oder Gliedergürtel-Muskeldystro-phie nachgewiesen werden [13,19].Auch die MAD kann mit partieller odergeneralisierter Lipodystrophie undGelenkkontrakturen einhergehen [20].
9

Literatur
1. Hanisch F, Zierz S (2003) Kernhüllen-erkrankungen – Eine neue Gruppe neu-romuskulärer, kardiologischer undendokrinologischer Erkrankungen.Aktuelle Neurologie 30:369-742. Bione S, Maestrini E, Rivella S, et al.(1994) Identification of a novel X-linkedgene responsible for Emery-Dreifussmuscular dystrophy. Nat Genet 8:323-3273. Toniolo D, Silvia B, K Arahata (1998)Emery-Dreifuss Muscular Dystrophy.In: Emery AEH. Neuromuscular Disor-ders: Clinical and Molecular Genetics.John Wiley & Sons LTD4. Bonne G, DiBarletta MR, Varnous S,et al. (1999) Mutations in the geneencoding lamin A/C cause autosomaldominant Emery-Dreyfuss musculardystrophy. Nat Genet 21:285-85. Bonne G, Mercuri E, Muchir A, et al.(2000) Clinical and molecular geneticspectrum of autosomal dominantEmery-Dreifuss muscular dystrophydue to mutations of the lamin A/Cgene. Ann Neurol 48:170-1806. Nagano A, Arahata K (2000) Nuclearenvelope proteins and associateddiseases. Curr Opin Neurol 13: 533-397. Merlini L, Granata C, Dominici P, etal. (1986) Emery-Dreifuss musculardystrophy: report of five cases in afamily and a review of the literature.Muscle Nerve 9:481-58. Bushby K, Muntoni F, Bourke JP(2003) 107th ENMC internationalworkshop: the management of cardiacinvolvement in muscular dystrophy
and myotonic dystrophy. NeuromusculDisord 13:166-729. Krasnianski M, Ehrt U, Neudecker S,et al. (2004) Alfred Hauptmann, Sieg-fried Thannhauser, and an endangeredmuscular disorder. Arch Neurol (imDruck)10. Hanisch F, Neudecker S, Wehnert M,et al. (2002) Die Hauptmann-Thann-hauser-Muskeldystrophie und Diffe-rentialdiagnosen von Myopathien mitKontrakturen. Nervenarzt 73:1004-1111. Vytopil M, Ricci E, Dello Russo A, etal. (2003) Frequent low penetrancemutations in the lamin A/C gene, caus-ing Emery Dreifuss muscular dystro-phy. Neuromuscul Disord 12:958-6312. Di Barletta M, Ricci E, Galluzzi G, etal. (2000) Different mutations in theLMNA gene cause autosomal domi-nant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am JHum Genet 66:1407-1213. Van der Kooi AJ, Bonne G, EymardB, et al. (2002) Lamin A/C mutationswith lipodystrophy, cardiac abnormali-ties, and muscular dystrophy. Neurolo-gy 59:620-2314. Van der Kooi AJ, Ledderhof TM, deVoogt WG et al. (1996) A newly recog-nized autosomal dominant limb girdlemuscular dystrophy with cardiac invol-vement. Ann Neurol 39:636-4215. Fatkin D, MacRae C, Sasaki T, et al.(1999) Missense mutations in the roddomain of the lamin A/C gene as caus-es of dilated cardiomyopathy and con-duction system disease. N Engl J Med
10

341:1715-2416. Brodsky GL, Muntoni F, Miocic S, etal. (2000) Lamin A/C gene mutationassociated with dilated cardiomyopa-thy with variable skeletal muscle invol-vement. Circulation 101:473-617. Tazir M, Azzedine H, Assami S, et al.(2004) Phenotypic variability in auto-somal recessive axonal Charcot-Marie-Tooth disease due to R298Cmutation in lamin A/C. Brain 127:154-6318. Garg A, Vinaitheerthan M, Weath-wall PT, et al. (2001) Phenotypic hete-rogeneity in patients with familial par-tial lipodystrophy (Dunnigan type)related to the site of missense mutati-ons in lamin A/C gene. J Clin Endocri-nol Metab 86:59-6519. Garg A, Speckamn RA, BowcockAM (2002) Multisystem dystrophy syn-drome due to novel missense muta-tions in the amino-terminal head andalpha-helical rod domains in the laminA/C gene. Am J Med 112:549-5520. Simha V, Agarwal AK, Oral EA, et al.(2003) Genetic and phenotypic hetero-geneity in patients with mandibulo-acral dysplasia-associated lipodystro-phy. J Clin Endocrinol Metab 88: 2821-4
11

Impressum:
DGM · Deutsche Gesellschaft für Muskelkranke e.V.Im Moos 4 · 79112 FreiburgTel.: 07665/9 44 70
Anschrift der Verfasser:
Prof. Dr. med. Stephan ZierzKlinik und Poliklinik für Neurologie Martin-Luther-Universität Halle-WittenbergErnst-Grube-Str. 4006097 Halle/Saale
Kontaktadresse:
Dr. med. F. HanischKlinik und Poliklinik für Neurologie Martin-Luther-Universität Halle-WittenbergErnst-Grube-Str. 4006097 Halle/SaaleE-Mail: [email protected]
Telefon: 0345/557-2934Fax: 0345/557-2934
Herausgeber der Schriftenreihe:
Prof. Dr. med. R. Dengler · HannoverProf. Dr. med. D. Pongratz · München
Verantwortlich für den Inhalt
dieser Ausgabe:
Prof. Dr. med. D. Pongratz · München
Aventis Pharma Deutschland GmbHGeschäftseinheit:Innovation Praxis & Klinik Königsteiner Straße 1065812 Bad Soden am TaunusTel.: 069/305 220 44
Management of Neuromuscular Diseases Kernhüllenerkrankungen
ARCIS Verlag GmbH · München ISSN 0949-15039. Jahrgang
12
313
407










