Metabolic Engineering von - Rostocker...
143
Metabolic Engineering von Clostridium acetobutylicum zur Produktion von 1,4-Butandiol Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Universität Rostock vorgelegt von Markus Klipp Rostock, 23.03.2016
Transcript of Metabolic Engineering von - Rostocker...

Metabolic Engineering von
Clostridium acetobutylicum zur Produktion von
1,4-Butandiol
Dissertation
zur
Erlangung des akademischen Grades
doctor rerum naturalium (Dr. rer. nat.)
der Mathematisch-Naturwissenschaftlichen Fakultät
der Universität Rostock
vorgelegt von
Markus Klipp
Rostock, 23.03.2016
zef007
Schreibmaschinentext
urn:nbn:de:gbv:28-diss2016-0087-1
zef007
Schreibmaschinentext

Gutachter: Prof. Dr. Hubert Bahl
Universität Rostock,
Mathematisch-Naturwissenschaftliche Fakultät,
Institut für Biowissenschaften, Abteilung Mikrobiologie
Prof. Dr. Bernd Kreikemeyer
Universitätsmedizin Rostock,
Medizinische Fakultät,
Institut für Medizinische Mikrobiologie, Virologie und Hygiene
Datum der Einreichung: 23. März 2016
Datum der Verteidigung: 24. Juni 2016

Inhaltsverzeichnis I
Inhaltsverzeichnis
Inhaltsverzeichnis ....................................................................................................................... I
Abkürzungsverzeichnis .............................................................................................................. V
Abbildungsverzeichnis ............................................................................................................... X
Tabellenverzeichnis ................................................................................................................ XIII
1. Einleitung ............................................................................................................................ 1
1.1 Zielstellung ................................................................................................................... 1
1.2 1,4-Butandiol ............................................................................................................... 2
1.3 Clostridium acetobutylicum ......................................................................................... 4
1.4 4-Hydroxybutyryl-CoA-Dehydratase/Vinylacetyl-CoA-∆-Isomerase ........................... 6
2. Material und Methoden ................................................................................................... 10
2.1 Organismen und Plasmide ......................................................................................... 10
2.2 Oligonukleotide ......................................................................................................... 15
2.3 Nährmedien ............................................................................................................... 16
2.3.1 Nährmedium für E. coli ................................................................................... 16
2.3.2 Medien für C. acetobutylicum ........................................................................ 16
2.3.3 Medienzusätze ................................................................................................ 19
2.4 Stammhaltung ........................................................................................................... 20
2.5 Zellanzucht ................................................................................................................. 20
2.5.1 Zellanzucht von E. coli ..................................................................................... 20
2.5.2 Zellanzucht von C. acetobutylicum ................................................................. 20
2.5.2.1 Zellanzucht von C. acetobutylicum auf Festmedium ........................ 21
2.5.2.2 Zellanzucht von C. acetobutylicum in statischer Kultur .................... 21
2.5.2.3 pH-kontrollierte Batch-Fermentation ............................................... 21
2.6 Bestimmung physiologischer Parameter ................................................................... 22
2.6.1 Optische Dichte ................................................................................................ 22
2.6.2 Bestimmung der Wachstumsrate und Verdopplungszeit ............................... 22
2.6.3 Bestimmung des externen pH-Wertes ............................................................ 23
2.6.4 Optisch-enzymatische Glukosebestimmung ................................................... 23
2.6.5 Optisch-enzymatische Laktatbestimmung ...................................................... 24
2.6.6 Gaschromatografie .......................................................................................... 25

Inhaltsverzeichnis II
2.6.6.1 Gaschromatografische Analyse der Gärungsprodukte von
C. acetobutylicum .............................................................................. 25
2.6.6.2 Gaschromatografische Analyse von 1,4-Butandiol und
γ-Hydroxybutyrat .............................................................................. 26
2.7 Visualisierung von 1,4-Butandiol ............................................................................... 27
2.8 Arbeiten mit Nukleinsäuren ...................................................................................... 28
2.8.1 Isolierung von Nukleinsäuren ......................................................................... 28
2.8.1.1 Isolierung von Plasmid-DNA aus E. coli.............................................. 28
2.8.1.2 Isolierung chromosomaler DNA aus Clostridien ................................ 29
2.8.2 PCR-Techniken ................................................................................................ 31
2.8.2.1 Oligonukleotid-Design....................................................................... 31
2.8.2.2 Standard-PCR .................................................................................... 32
2.8.2.3 „Colony“-PCR .................................................................................... 32
2.8.3 Enzymatische Modifikation von DNA ............................................................. 32
2.8.3.1 Restriktion von DNA .......................................................................... 32
2.8.3.2 Dephosphorylierung von DNA-Fragmenten ..................................... 33
2.8.3.3 Ligation von DNA-Fragmenten.......................................................... 33
2.8.3.4 in vivo Methylierung von Plasmid-DNA ............................................ 34
2.8.4 Agarosegelelektrophorese .............................................................................. 34
2.8.5 Reinigung von Nukleinsäuren ......................................................................... 35
2.8.5.1 Extraktion von DNA aus Agarosegelen ............................................. 35
2.8.5.2 Kit-basierte DNA-Aufreinigung ......................................................... 35
2.9 Erzeugung rekombinanter Organismen .................................................................... 35
2.9.1 DNA-Transfer in E. coli .................................................................................... 35
2.9.1.1 CaCl2-vermittelte Transformation in E. coli ...................................... 35
2.9.1.2 Transformation von E. coli durch Elektroporation ........................... 36
2.9.2 DNA-Transfer in C. acetobutylicum ................................................................. 36
2.9.2.1 Transformation von C. acetobutylicum durch Elektroporation ........ 36
2.10 Arbeiten mit Proteinen ............................................................................................. 37
2.10.1 Zellaufschluss von C. acetobutylicum mittels Ultraschall ............................. 37
2.10.2 Proteinaufreinigung mittels Affinitätschromatografie ................................. 38
2.10.3 Konzentrationsbestimmung von Proteinen (Bradford, 1976) ...................... 39

Inhaltsverzeichnis III
2.10.4 Eindimensionale SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE)........... 39
2.10.5 Kolloidale Coomassie-Färbung ...................................................................... 41
2.10.6 Transfer von Proteinen auf Membranen (Western Blot) .............................. 41
2.10.7 Detektion von Strep-tag II Fusionsproteinen................................................. 42
2.11 Bezugsquellen ........................................................................................................... 43
3. Ergebnisse ......................................................................................................................... 46
3.1 1,4-Butandioltoleranz von C. acetobutylicum ATCC 824 ........................................... 46
3.2 Nachweismethoden für 1,4-Butandiol ...................................................................... 49
3.2.1 Dünnschichtchromatografie-basierte Nachweismethode ............................. 49
3.2.2 GC/MS-basierte Nachweismethode ............................................................... 50
3.3 Generierung von rekombinanten C. acetobutylicum-Stämmen zur Produktion von
1,4-Butandiol ............................................................................................................. 52
3.4 Wachstumsphysiologische Charakterisierung von generierten 1,4-BDO-Produktions-
stämmen .................................................................................................................... 56
3.4.1 Wachstumsverlauf und Gärungsprodukte ...................................................... 57
3.4.2 1,4-Butandiolproduktion ................................................................................ 61
3.4.3 Zeitlicher Verlauf der 1,4-BDO-Produktion..................................................... 63
3.5 Optimierungsstrategien ............................................................................................. 65
3.5.1 pH-regulierte Batch-Fermentation von C. acetobutylicum
pTc::P(thlA)cbei_2100 ...................................................................................... 65
3.5.2 Verwendung verschiedener Kohlenhydratquellen .......................................... 67
3.5.3 Modifikation der 4HBD-Expression durch Verwendung unterschiedlicher
Promotoren ..................................................................................................... 70
3.5.3.1 Vektorgenerierung .............................................................................. 71
3.5.3.2 1,4-BDO-Produktion rekombinanter C. acetobutylicum-Stämme ...... 73
3.5.4 Stoffwechseldefektmutanten von C. acetobutylicum ATCC 824 .................... 75
3.5.4.1 C. acetobutylicum-Mutanten mit Defekten in der Säurebildung ...... 75
3.5.4.2. C. acetobutylicum-Mutanten mit Defekten in der
Lösungsmittelbildung ......................................................................... 77
4. Diskussion ......................................................................................................................... 80
4.1 1,4-Butandioltoleranz ................................................................................................ 81
4.2 1,4-Butandiolproduktion ........................................................................................... 82

Inhaltsverzeichnis IV
4.2.1 Limitierung durch die 4HBD ............................................................................ 83
4.2.2 Limitierung durch die Aldehyd-/Alkoholdehydrogenase (AdhE) .................... 84
4.2.3 Codon Usage ................................................................................................... 85
4.3 GHB-Produktion ......................................................................................................... 86
4.4 1,4-BDO-Produktion unter Verwendung verschiedener Kohlenhydratquellen ........ 88
4.4.1 Pentosen ......................................................................................................... 88
4.4.1.1 Xylose .................................................................................................. 88
4.4.1.2 Arabinose ............................................................................................ 90
4.4.2 Hexosen ........................................................................................................... 91
4.4.2.1 Galaktose ............................................................................................ 91
4.4.2.2 Mannose ............................................................................................. 91
4.4.3 Kohlenhydratgemisch ...................................................................................... 92
4.5 C. acetobutylicum pT::P(ptb)ckl_3020 ....................................................................... 94
4.6 Stoffwechseldefektmutanten .................................................................................... 95
4.6.1 Defekte in der Lösungsmittelproduktion ......................................................... 95
4.6.2 Defekte in der Säureproduktion ...................................................................... 97
4.7 Fazit ............................................................................................................................ 98
5. Zusammenfassung .......................................................................................................... 100
6. Literaturverzeichnis ........................................................................................................ 101
7. Anhang ............................................................................................................................ 113
Danksagung ............................................................................................................................ 126
Selbstständigkeitserklärung .................................................................................................. 128

Abkürzungsverzeichnis V
Abkürzungsverzeichnis
α alpha
A Adenin, Ampere
A. Aqua
ABC “ATP binding cassette”
AbfD 4HBD aus Clostridium aminobutyricum
abfD 4hbd-Gen aus Clostridium aminobutyricum
ad auffüllen auf
adc Acetoacetadecarboxylase-Gen
adhE Aldehyd-Alkoholdehydrogenase-Gen
Amp Ampicillin
AP alkalische Phosphatase
AS Aminosäure
ATCC „American Type Culture Collection”
ATP Adenosintriphosphat
β beta
BCIP 5-Bromo-4-chloro-3-indolyl-phosphat
BDH Butanol-Dehydrogenase
BDO Butandiol
Bp Basenpaar
BSA Rinderserumalbumin
Buk Butyratkinase
BYDH Butyraldehyd-Dehydrogenase
γ gamma
c Konzentration, centi (10-2)
C Kohlenstoff, Cytosin
C- Carboxy-
C. Clostridium
°C Grad Celsius
ca. zirka

Abkürzungsverzeichnis VI
Cbei_2100 4HBD aus Clostridium beijerinckii
cbei_2100 4hbd-Gen aus Clostridium beijerinckii
CCR „carbon catabolite repression”
CGM „Clostridial Growth Medium”
Ckl_3020 4HBD aus Clostridium kluyveri
ckl_3020 4hbd-Gen aus Clostridium kluyveri
Cm Chloramphenicol
CoA Coenzym A
ctfA Acetoacetyl-CoA:Acyl-CoA-Transferase-Gen
d desoxy
D „dextro“ (rechts)
Da Dalton
DC Dünnschichtchromatografie
dest. destiliert
DNA Desoxyribonukleinsäure
dNTP Desoxyribonukleosid-5-triphosphat
E Extinktion
E. Escherichia
EDTA Ethylendiamintetraessigsäure
Ery Erythromycin
et al. „et alteri“ (lat., und andere)
FAD Flavinadenindinukleotid
FID Flammenionisationsdetektor
Form. Formel
fw forward
g Gramm, Erdbeschleunigung
G Guanin
GC Gaschromatograph(ie)
GHB γ-Hydroxbutyrat
h Stunde
HBD 4-Hydroxybutyryl-CoA-Dehydratase/Vinylacetyl-CoA-∆-Isomerase

Abkürzungsverzeichnis VII
hbd 4-Hydroxybutyryl-CoA-Dehydratase/Vinylacetyl-CoA-∆-Isomerase-Gen
IS interner Standard
k kilo (103)
K Kontrolle
konz. konzentriert
l Liter, Länge
L „levo“ (links)
LB Luria-Bertani
ldh Laktatdehydrogenase-Gen
LDH Laktatdehydrogenase
ln natürlicher Logarithmus
log dekadischer Logarithmus
Lsg. Lösung
LW Leloir-Weg
µ Mikro (10-6)
m Meter, milli (10-3), „messenger“
M Molar, Marker
MBDSTFA N-Methyl-N-tert-butyldimethylsilyl-tifluoracetamid
MES 2-(N-Morpholino)ethansulfonsäure
min Minute
MMfvK Minimalmedium für Vorkulturen
mod. modifiziert
MS „medium-synthetique“
MW Molekulargewicht
n Nano (10-9), Anzahl der Versuche
N Stickstoff, Normal
N- Amino-
NAD(P)+ Nicotinamidadenindinucleotid-(phosphat), oxidiert
NAD(P)H Nicotinamidadenindinucleotid-(phosphat), reduziert
NaOH Natriumhydroxid
NBT Nitroblautetrazoliumchlorid

Abkürzungsverzeichnis VIII
NCBI „National Center for Biotechnology Information”
NCIMB „National Collection of Industrial, Marine and Food Bacteria”
NEB „New England Biolabs“
Nr. Nummer
nt Nukleotid(e)
NTA Nitrilotriessigsäure
OD optische Dichte
ORF offener Leserahmen
P Promotor, Phosphat
PABA Para-aminobenzoesäure
PAGE Polyacrylamidgelelektrophorese
PCR Polymerase-Kettenreaktion
PDO Propandiol
pH negativer Logarithmus der Protonenkonzentration
pI isoelektrischer Punkt
PKW Phosphoketolaseweg
PPW Pentosephosphatweg
pta Phosphotransacetylase-Gen
ptb Phosphotransbutyrylase-Gen
PTS Phosphoenolpyruvat-Transferasesystem
R Resistenz
RCA „Reinforced Clostridial Agar”
rev reverse
RNA Ribonukleinsäure
RNase Ribonuklease
RT Raumtemperatur
s Sekunde
SDS Natriumdodecylsulfat
SIM „Selected Ion Mode“
t Zeit
T Thymin

Abkürzungsverzeichnis IX
T6PW Tagatose-6-Phosphatweg
Tab. Tabelle
TAE Tris-Acetat-EDTA
Tc Tetracyclin
thlA Thiolase A-Gen
Tm Thiamphenicol
Tm Schmelztemperatur
U Units
UE Untereinheit
ü. N. über Nacht
Upm Umdrehungen pro Minute
UV Ultraviolett
V Volt
VF Verdünnungsfaktor
Vol. Volumen
v/v Volumen pro Volumen
W Watt
WT Wildtyp
w/v Masse pro Volumen

Abbildungsverzeichnis X
Abbildungsverzeichnis
Abb. 1.1: Strukturformel von 1,4-Butandiol. ............................................................................. 2
Abb. 1.2: Produktderivatkette von 1,4-Butandiol ..................................................................... 3
Abb. 1.3: Biphasischer Gärungsstoffwechsel von C. acetobutylicum. ....................................... 5
Abb. 1.4: Reaktionen der 4HBD ................................................................................................. 6
Abb. 1.5: Vorausgesagter Reaktionsmechanismus der 4HBD ................................................... 7
Abb. 1.6: Strukturelle Darstellung der 4HBD aus C. aminobutyricum. ...................................... 9
Abb. 3.1: Wachstum von C. acetobutylicum ATCC 824 in Anwesenheit von unterschiedlichen
1,4-BDO-Konzentrationen. ....................................................................................... 47
Abb. 3.2: Phasenkontrastmikroskopische Aufnahmen von C. acetobutylicum bei
verschiedenen 1,4-BDO-Konzentrationen. .............................................................. 48
Abb. 3.3: Dünnschichtchromatogramm von 1,4-BDO. ............................................................ 49
Abb. 3.4: Schematische Darstellung von 1,4-BDO und GHB nach Silylierungsreaktion mit
MBDSTFA. ................................................................................................................. 51
Abb. 3.5: SIM-Chromatogramm von 1,4-BDO und GHB. ......................................................... 51
Abb. 3.6: Erzeugung der 4HBD-kodierenden Gene cbei_2100, ckl_3020 und abfD. .............. 52
Abb. 3.7: Vektorkarten. ............................................................................................................ 53
Abb. 3.8: Kontrollrestriktion der Plasmide pTc::P(thlA)cbei_2100, pTc::P(thlA)ckl_3020 und
pTc::P(thlA)abfD. ....................................................................................................... 55
Abb. 3.9: Verifizierung von rekombinanten C. aetobutylicum-Klonen. ................................... 56
Abb. 3.10: Kultivierungsexperimente von C. acetobutylicum pTc::P(thlA)cbei_2100,
C. acetobutylicum pTc::P(thlA)ckl_3020 und C. acetobutylicum pTc::P(thlA)abfD
vergleichend zum C. acetobutylicum ATCC 824 Typstamm und C. acetobutylicum
Kontrollvektorstamm ............................................................................................. 57

Abbildungsverzeichnis XI
Abb. 3.11: Gärungsprodukte der Kultivierungsexperimente C. acetobutylicum
pTc::P(thlA)cbei_2100, C. acetobutylicum pTc::P(thlA)ckl_3020 und
C. acetobutylicum pTc::P(thlA)abfD vergleichend zum C. acetobutylicum ATCC
824 Wildtypstamm und C. acetobutylicum pTc-Kontrollvektorstamm ................. 59
Abb. 3.12: 1,4-BDO- und GHB-Produktion der Expressionsstämme nach 120 h Kultivierung in
200 ml MS-MES. ...................................................................................................... 62
Abb. 3.13: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion. .......................................... 64
Abb. 3.14: Kultivierungsexperiment von C. acetobutylicum pTc::P(thlA)cbei_2100 in 1,5 l
Kulturvolumen......................................................................................................... 66
Abb. 3.15: 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100 in
1,5 l Kulturvolumen in Abhängigkeit verschiedener Medien. ................................ 67
Abb. 3.16: Kultivierungsexperimente von C. acetobutylicum pTc::P(thlA)cbei_2100 auf
verschiedenen C-Quellen. ....................................................................................... 68
Abb. 3.17: 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100 auf
verschiedenen C-Quellen. ....................................................................................... 69
Abb. 3.18: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum
pTc::P(thlA)cbei_2100. ............................................................................................ 70
Abb. 3.19: Schematische Darstellung der Vektorherstellung mit unterschiedlichen
Promotoren. ............................................................................................................ 72
Abb. 3.21: Gärungsstoffwechsel von C. acetobutylicum. ........................................................ 76
Abb. 3.22: 1,4-BDO- und GHB-Produktion rekombinanter C. acetobutylicum-Mutanten. ..... 77
Abb. 3.23: Gärungsstoffwechsel von C. acetobutylicum. ........................................................ 78
Abb. 3.24: 1,4-BDO- und GHB-Produktion rekombinanter C. acetobutylicum-Mutanten. ..... 79
Abb. 4.1: Schematische Darstellung der potentiellen Bildungswege von 4-Hydroxbutyrat
(GHB). ........................................................................................................................ 87
Abb. 4.2: Schematische Darstellung der Kohlenhydratverwertung in C. acetobutylicum ...... 89

Abbildungsverzeichnis XII
Abb. 4.3: Schematische Darstellung der Lignocellulose-Zusammensetzung .......................... 93
Abb. A1: Dünnschichtchromatogramm von 1,4-BDO ............................................................ 113
Abb. A2: Dünnschichtchromatogramm von 1,4-BDO mit C. acetobutylicum Kulturüberstand
................................................................................................................................................ 113
Abb. A3: Analysenfunktion für die Kalibrierung der 1,4-BDO-Messung ............................... 114
Abb. A4: Analysenfunktion für die Kalibrierung der GHB-Messung ...................................... 114
Abb. A5: Aminosäure-Alignment der 4HBDs ......................................................................... 115
Abb. A6: Vektorkarte vom pT-Vektor .................................................................................... 116
Abb. A7: Nukleotidsequenz von cbei_2100 nach Sequenzierung positiver Plasmide ........... 116
Abb. A8: Nukleotidsequenz von ckl_3020 nach Sequenzierung positiver Plasmide ............. 117
Abb. A9: Nukleotidsequenz von abfD nach Sequenzierung positiver Plasmide ................... 117
Abb. A10: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum
pTc::P(thlA)cbei_2100 auf verschiedenen Kohlenstoffquellen ............................ 118
Abb. A11: Vektorkarten der generierten Vektoren zur Variation der Promotoren .............. 120
Abb. A12: Vektorkarten pT::P(ptb)pp2, pT::P(adc)pp2 und pT::P(fac)pp2 ............................ 121
Abb. A13: Colony-PCR auf die 4HBD-kodierenden Gene ...................................................... 122
Abb. A14: SDS-PAGE aufgereinigter 4HBDs ........................................................................... 122
Abb. A15: Western Blot der aufgetrennten Proteinproben .................................................. 123
Abb. A16: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum
pT::P(ptb)ckl_3020. ............................................................................................... 125

Tabellenverzeichnis XIII
Tabellenverzeichnis
Tab. 1.1: Bifunktionelle 4-Hydroxybutyryl-CoA Dehydratasen/Vinylacetyl-CoA-∆-Isomerasen
(4HBDs) aus verschiedenen Clostridien-Spezies.......................................................... 8
Tab. 2.1: Organismen ............................................................................................................... 10
Tab. 2.2: Vektoren .................................................................................................................... 13
Tab. 2.3: rekombinante Plasmide ............................................................................................ 14
Tab. 2.4: Oligonukleotide ......................................................................................................... 15
Tab. 2.5: Medienzusätze .......................................................................................................... 19
Tab. 2.6: Restriktionsendonukleasen ....................................................................................... 33
Tab. 2.7: SDS-PAGE-Gellösungen ............................................................................................. 40
Tab. 2.8: Bezugsquellen für Chemikalien ................................................................................. 43
Tab. 2.9: Bezugsquellen für Geräte und Materialien ............................................................... 44
Tab. 3.1: Wachstumsraten (µ) und Verdopplungszeiten (td) von C. acetobutylicum bei
unterschiedlichen 1,4-BDO-Konzentrationen............................................................ 47
Tab. 3.2: 1,4-BDO-Produktivitäten der Expressionsstämme. .................................................. 63
Tab. A1: Kalibrierung der 1,4-BDO- und GHB-Nachweismethode ......................................... 114
Tab. A2: Wachstumsparameter und Gärungsprodukte der 200-ml Kultivierungsexperimente
................................................................................................................................................ 118
Tab. A3: Wachstumsparameter und Gärungsprodukte von rekombinanten C. acetobutylicum
Stämmen .................................................................................................................. 123
Tab. A4: Vergleich der Häufigkeiten der verwendeten Codons in C. acetobutylicum,
Cbei_2100, Ckl_3020 und AbfD ............................................................................... 124
Tab. A5: Gärungsprodukte transformierter C. acetobutylicum-Mutantenstämmen in 200 ml
MS-MES nach 120 h ................................................................................................. 125

Einleitung 1
1. Einleitung
1.1 Zielstellung
Die Möglichkeit der Nutzung von fossilen Ressourcen, wie z. B. Erdöl, Kohle und Gas führten
zur Entwicklung der heutigen Gesellschaft und ihres Wohlstands. Aus diesem Grund lag bis
ins 20. Jahrhundert hinein der Schwerpunkt vieler Forschungen auf der Entwicklung der
erdölbasierten Produktion von Chemikalien und anderen Gütern (Demirbas, 2006; Chandra
et al., 2012). Dies resultierte jedoch in einer Abhängigkeit der heutigen Weltwirtschaft von
diesen nicht-erneuerbaren Energieträgern, sodass ca. 90 % der genutzten Energiequellen
fossilen Ursprungs sind (Chandra et al., 2012). Aufgrund exzessiven Gebrauchs jener Quellen
kommt es durch Emission von Kohlenstoffdioxid neben Luftverschmutzungen in urbanen
Ballungsgebieten vor allem zu einem Anstieg der Treibhausgase in der Erdatmosphäre
(Sarkar et al., 2012), welche maßgeblich mit der Erderwärmung assoziiert sind (Chandra et
al., 2012). Durch die zunehmende Weltbevölkerung und dem damit einhergehenden
steigenden Energiebedarf wird die Situation in Hinblick auf Nachhaltigkeit und Klimawandel
erheblich verschlechtert. Aus diesem Grund ist die Entwicklung von alternativen Strategien
zur Nutzung und Optimierung von erneuerbaren Energiequellen unabdingbar. Ein
entsprechender Ansatz beinhaltet die Erzeugung von bisher auf Erdölbasis hergestellten
Grundchemikalien für die chemische Industrie aus nachwachsenden Rohstoffen. Hierzu zählt
unter anderem die biotechnologische Produktion von Diolen, die als Plattformchemikalien
für viele verschiedene industrielle Anwendungen eingesetzt werden (Zeng und Sabra, 2011).
Zu den wichtigsten industriell genutzten Diolen gehören 1,3-Propandiol (1,3-PDO),
2,3-Butandiol (2,3-BDO) und 1,4-Butandiol (1,4-BDO). Dabei werden 1,3-PDO und 2,3-BDO
bereits im industriellen Großmaßstab biotechnologisch produziert (Zeng und Sabra, 2011).
Im Gegensatz zu diesen Diolen kommt 1,4-BDO in der Natur nicht vor, sondern wird
ausschließlich über chemische Synthesen auf Erdölbasis erzeugt (Yim et al., 2011). Daher ist
es von Interesse auch diese Verbindung unabhängig von fossilen Energiequellen aus
erneuerbaren Rohstoffen zu gewinnen. Vor wenigen Jahren gelang es Yim et al. (2011) einen
1,4-BDO-Biosyntheseweg in Escherichia coli zu etablieren. Hierfür waren jedoch komplexe
und umfangreiche genetische Veränderungen des Wirtsorganismus notwendig. Im
Gegensatz dazu beinhaltete die in dieser Arbeit verfolgte Strategie die Generierung eines

Einleitung 2
1,4-BDO-Biosyntheseweges durch eine einzige genetische Veränderung des Wirtsorganismus
Clostridium acetobutylicum. Durch das Zusammenspiel einer heterolog exprimierten 4HBD
(1.4) und einem nativen Enzym (AdhE) sollte der Stoffwechsel des Wirtsorganismus
erweitert und so die 1,4-BDO-Bildung ermöglicht werden. Demnach bestand das vorrangige
Ziel dieser Arbeit darin, C. acetobutylicum durch Bereitstellung der genetischen
Voraussetzungen zu einem 1,4-BDO-Produzenten zu entwickeln. Bei erfolgreicher 1,4-BDO-
Produktion sollte weiterhin durch erste Optimierungsversuche die Produktbildung
verbessert und der Einfluss verschiedener Kohlenhydratquellen auf die Biosynthese
untersucht werden.
1.2 1,4-Butandiol
1,4-Butandiol ist ebenso wie 2,3-Butandiol ein Isomer des Butandiols und wird aufgrund der
beiden Hydroxylgruppen (OH-) der Gruppe der Diole zugeordnet (Zeng und Sabra, 2011). Die
OH-Gruppen sind dabei an den Kohlenstoffatomen 1 und 4 gebunden (Abb. 1.1). 1,4-BDO ist
eine farb- und geruchslose Flüssigkeit und leicht biologisch abbaubar (OECD 301C: 100 %
nach 14 Tagen).
Abb. 1.1: Strukturformel von 1,4-Butandiol.
Von den vier Kohlenstoff-basierten Diolen in der Industrie wird 1,4-BDO am häufigsten im
Großmaßstab verwendet (Haas et al., 2005). Die jährliche chemische Produktion beträgt ca.
1,3 Mio. Tonnen pro Jahr (Yim et al., 2011; Zeng und Sabra, 2011) mit einem Marktpreis von
ca. 1850 €/t (van Haveren et al., 2008). Analysen bescheinigen dem globalen 1,4-BDO-Markt
eine jährliche Wachstumsrate von 4,8 % bis 2020 (in: 1,4-Butanediol market analysis and
segment forecasts to 2020; www.hexareports.com/report/1-4-butanediol-market-analysis-
and-segment-forecasts-to-2020/details, Stand: 25.02.2016). Die Produktderivatkette des
1,4-BDO umfasst vier Hauptzweige: Tetrahydrofurane (THF), γ-Butyrolactone (GBL),
Polybutylenterephthalate (PTB) und Polyurethane (PUR) (Abb. 1.2). Daraus gehen wiederum
jährlich 2,5 Mio. Tonnen Plastik, Polyester und Elastanfasern hervor (Yim et al., 2011), die

Einleitung 3
unter anderem Anwendung in der Automobil-, Textil- und Kosmetikindustrie finden (Haas et
al., 2005). In Form von Funktionsbekleidung, Schuhsohlen, Schutzgehäusen für
Haushaltskleingeräte und technisches Plastik für Armaturenbretter sind diese Erzeugnisse
aus dem heutigen Alltag nicht mehr wegzudenken. Aufgrund dieser zahlreichen
verschiedenen Anwendungsmöglichkeiten wird 1,4-BDO infolgedessen auch als eine „Top-
Mehrwert“-Chemikalie bezeichnet (Yim et al., 2011).
Abb. 1.2: Produktderivatkette von 1,4-Butandiol (1,4-BDO). THF, Tetrahydrofuran; GBL, γ-Butyrolacton; PTB,
Polybutylenterephthalat; PUR, Polyurethan.
Die Produktion von 1,4-BDO erfolgt bisher rein synthetisch aus erdölbasierten Rohstoffen,
wie Acetylen, Butan, Propylen oder Butadien (Haas et al., 2005). Damit unterliegt die
Produktion der wichtigen Plattformchemikalie den allgemeinen wirtschaftlichen Problemen,
wie Schwankungen in den Erdölpreisen und verknappenden Erdölreserven. Um eine vom
Erdöl unabhängige Produktion von 1,4-BDO zu gewährleisten, wurde für die
biotechnologische Erzeugung dieser Chemikalie der Modellorganismus C. acetobutylicum als
Wirtsstamm eingesetzt.
1,4-BDO
OH
HO
THF GBL PTB PUR
Lösungsmittel
Thermoplastik
Highperformance-
Polyester
Lösungsmittel
(Co-)Polymer
Kosmetik
Thermoplastik
Plastikgehäuse
Kleinelektronik
Automobilindustrie
Verpackungsmaterial
Bauschaum
Elastanfasern

Einleitung 4
1.3 Clostridium acetobutylicum
Nach Isolierung und Erstbeschreibung durch Chaim Weizmann zwischen 1912 und 1914
wurde C. acetobutylicum vorwiegend für die Aceton-Butanol-Ethanol-(ABE) Fermentation
eingesetzt (Jones und Woods, 1986). Vor allem während des 1. Weltkrieges erlangte die
ABE-Fermentation eine enorme Bedeutung, insbesondere in Großbritannien, da Aceton für
die Herstellung von Kordit, einem rauchfreien Schießpulver, benötigt wurde (Dürre, 2008).
Mit der raschen Ausbreitung der Automobilindustrie in den USA nach dem Krieg wurde das
als Nebenprodukt angesehene Butanol immer wichtiger. Ab den 1950er Jahren kam es
jedoch aufgrund gesunkener Rohölpreise und der Entwicklung von wirtschaftlicheren und
effizienteren petrochemischen Verfahren zu einem rapiden Rückgang der industriellen ABE-
Fermentation (Dürre, 2008). Gestiegene Erdölpreise in den 70er Jahren und ein
zunehmendes Interesse an der Erzeugung von Biokraftstoffen führten dann zu einem
erneuten Aufschwung der ABE-Fermentation (Dürre, 2008). Besonders in China wurden
seitdem alte ABE-Fermentationsanlagen wieder in Stand gesetzt und neue eingerichtet,
wodurch bis heute mehrere 100.000 t Butanol pro Jahr erzeugt werden (Chiao und Sun,
2007). Somit rückte C. acetobutylicum in den Fokus aktueller Forschungen (Lütke-Eversloh
und Bahl, 2011) und entwickelte sich so zu einem Modellorganismus apathogener
Clostridien (Bahl und Dürre, 1993).
Bei C. acetobutylicum handelt es sich um ein Gram-positives, sporenbildendes und strikt
anaerobes Bakterium (Madigan et al., 2013), welches in der Lage ist, eine Vielzahl an
verschiedenen Kohlenhydraten, wie z. B. Arabinose, Galaktose, Glukose, Mannose und
Xylose, zu verwerten (Qureshi et al., 2006; Ezeji und Blaschek, 2008; Servinsky et al., 2010).
Mit Blick auf die Verwendung als biotechnologische Produktionsplattform ist dies ein
wichtiger ökonomischer Vorteil für die Umwandlung von preisgünstigen Substraten. Denn
diese Zucker sind die Hauptkomponenten von Hemicellulose, einem wesentlichen
Bestandteil der Lignocellulose (Mussatto und Teixeira, 2010), einem günstigen und
nachwachsenden Rohstoff, der aktuell in Form von Weizenstroh, Getreidestroh, Reisstroh
und Bagasse (Sarkar et al., 2012) jedes Jahr in sehr großen Mengen verfügbar ist
(10-50 x 109 Tonnen; Kuhad und Singh, 1993). Lignocellulose-Hydrolysate würden somit ein
optimales Substrat für die biotechnologische Fermentation darstellen, so auch für die in
dieser Arbeit angestrebte 1,4-BDO-Synthese. Die Metabolisierung der Kohlenhydratquellen

Einleitung 5
erfolgt in einem biphasischen Gärungsstoffwechsel (Abb. 1.3), welcher für C. acetobutylicum
charakteristisch ist und in eine säure- (acidogene) und lösungsmittelbildende (solventogene)
Phase gegliedert wird. Während des exponentiellen Wachstums werden die
aufgenommenen Kohlenhydrate über die Glykolyse zu den Säuren Acetat und Butyrat
umgesetzt, welche in das umgebende Medium abgegeben werden. Dies führt zu einem
Absinken des externen pH-Wertes (Hartmanis und Gatenbeck, 1984). Durch die zunehmende
Akkumulation der Säuren kommt es beim Übergang von der exponentiellen in die stationäre
Wachstumsphase zu einer Veränderung im Genexpressionsprofil von C. acetobutylicum
(Alsaker und Papoutsakis, 2005) und zur Einleitung des „Lösungsmittelshifts“. Infolgedessen
werden Acetat und Butyrat assimiliert und in die pH-neutralen Lösungsmittel Aceton,
Butanol und Ethanol im Verhältnis von 3:6:1 umgewandelt (Mitchell, 1998).
Abb. 1.3: Biphasischer Gärungsstoffwechsel von C. acetobutylicum. Der Abzweig des 1,4-BDO-
Biosynthesewegs sowie die veranwortliche rekombinante 4HBD sind in blau, native Enzyme in rot dargestellt.
Gestrichelte Linien weisen auf die Reassimilation der Säuren während der Solventogenese hin. Abkürzungen:
Ldh, Lactat-Dehydrogenase; Pfor, Pyruvat-Ferredoxin-Oxidoreduktase; Hyd, Hydrogenase; Fnor, Ferredoxin-
NAD(P)+-Oxidoreduktase; Pta, Phosphotransacetylase, Ack, Acetatkinase; AdhE, Aldehyd/ Alkohol-
Dehydrogenase; CtfAB, Acetoacetyl-CoA:Acyl-CoA-Transferase; Adc, Acetoacetat-Decarboxylase; Thl, Thiolase;
Hbd, 3-Hydroxybutyryl-CoA-Dehydrogenase; Crt, Crotonase; 4HBD, 4-Hydroxybuytryl-CoA-Dehydratase; Bcd,
Butyryl-CoA-Dehydrogenase-Komplex; Ptb, Phosphotransbutyrylase; Buk, Butyratkinase; Acetyl-P,
Acetylphosphat; Ac-CoA, Acetyl-CoA; AcAld, Acetaldehyd; AcAc-CoA, Acetoacetyl-CoA; AcAc, Acetoacetat; 3HB-
CoA, 3-Hydroxybutyryl-CoA; 4HB-CoA, 4-Hydroxybutyryl-CoA; Butyryl-P, Butyrylphosphat; BuAld, Butyraldehyd.

Einleitung 6
Der C4-Stoffwechsel von C. acetobutylicum bietet die besondere Möglichkeit durch die
Einbringung einer 4HBD-Aktivität einen Weg zur 1,4-BDO-Bildung zu eröffnen (Abb. 1.3).
1.4 4-Hydroxybutyryl-CoA-Dehydratase/Vinylacetyl-CoA-∆-
Isomerase
Eine weitverbreitete Reaktion in biochemischen Stoffwechselwegen ist die Dehydratation
(Martins et al., 2004). Derzeit sind mehr als 100 Dehydratasen (Hydrolyasen [EC 4.2.1.-])
bekannt (Martins et al., 2007). Die meisten dieser Enzyme katalysieren dabei die
Eliminierung von Wasser unter Ausbildung einer Doppelbindung, wobei das α-Wasserstoff
an Position C2 durch die Elektronen-ziehende Wirkung der benachbarten funktionellen
Gruppe (z. B. CoA-Thioestergruppe) aktiviert und dann als Proton entfernt werden kann.
Daraufhin wird die Hydroxylgruppe in β-Position (C3) freigegeben (Martins et al., 2007). In
anaeroben Mikroorganismen gibt es jedoch die Möglichkeit durch Radikal-vermittelte
Reaktionen ein Wasserstoffatom von einem nicht-aktivierten β-Kohlenstoff (C3) zu
entfernen (Martins et al., 2004). Die 4-Hydroxybutyryl-CoA-Dehydratase/Vinylacetyl-CoA-∆-
Isomerase (4HBD; EC 4.2.1.120/ EC 5.3.3.3) ist solch eine atypische Dehydratase und
katalysiert sowohl die reversible, sauerstoffempfindliche Dehydrierung von
4-Hydroxybutyryl-CoA (4HB-CoA) zu Crotonyl-CoA als auch die irreversible, Sauerstoff-
unempfindliche Isomerisierung von Vinylacetyl-CoA zu Crotonyl-CoA (Scherf und Buckel,
1993) (Abb. 1.4). Die Gesamtreaktion verläuft dabei trotz eines nicht-kovalent gebundenen
FADs ohne Netto-Redoxveränderungen ab (Müh et al., 1996), was zu der Annahme eines
Radikal-basierten Reaktionsmechanismus führte (Martins et al., 2004) (Abb. 1.5).
Abb. 1.4: Reaktionen der 4HBD (Martins et al., 2007; mod.).
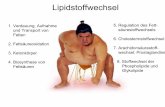
4-Hydroxybutyryl-CoA Vinylacetyl-CoA Crotonyl-CoA
Einleitung 7
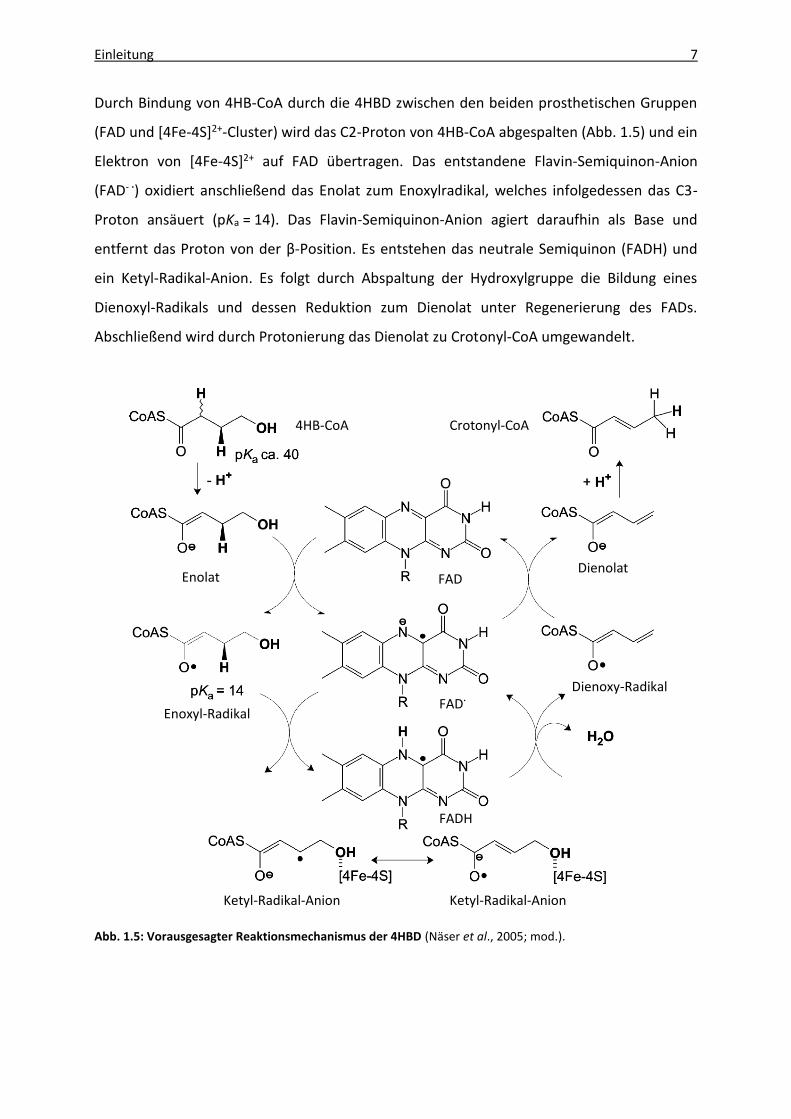
Durch Bindung von 4HB-CoA durch die 4HBD zwischen den beiden prosthetischen Gruppen
(FAD und [4Fe-4S]2+-Cluster) wird das C2-Proton von 4HB-CoA abgespalten (Abb. 1.5) und ein
Elektron von [4Fe-4S]2+ auf FAD übertragen. Das entstandene Flavin-Semiquinon-Anion
(FAD- ·) oxidiert anschließend das Enolat zum Enoxylradikal, welches infolgedessen das C3-
Proton ansäuert (pKa = 14). Das Flavin-Semiquinon-Anion agiert daraufhin als Base und
entfernt das Proton von der β-Position. Es entstehen das neutrale Semiquinon (FADH) und
ein Ketyl-Radikal-Anion. Es folgt durch Abspaltung der Hydroxylgruppe die Bildung eines
Dienoxyl-Radikals und dessen Reduktion zum Dienolat unter Regenerierung des FADs.
Abschließend wird durch Protonierung das Dienolat zu Crotonyl-CoA umgewandelt.
Abb. 1.5: Vorausgesagter Reaktionsmechanismus der 4HBD (Näser et al., 2005; mod.).
4HB-CoA
Enolat
Enoxyl-Radikal
Crotonyl-CoA
Dienolat
Dienoxy-Radikal
Ketyl-Radikal-Anion Ketyl-Radikal-Anion
FAD
FAD·
FADH
Einleitung 8
Die Dehydratation von 4-Hydroxyacyl-CoA-Derivaten zu Crotonyl-CoA ist zwar von
anaeroben Bakterien, vorzugsweise bei Vertretern der Fusobacteriales und Clostridiales
bekannt (Buckel et al., 2005), C. acetobutylicum verfügt jedoch über keine 4HBD. Allerdings
kommen 4HBD homologe Enzyme aus relativ nah verwandten Arten, wie beispielsweise
C. aminobutyricum (AbfD), C. beijerinckii (Cbei_2100) und C. kluyveri (Ckl_3020) (Tab. 1.1), in
denen sie eine wesentliche Rolle bei der Aminobutyrat- bzw. Succinat-Fermentation
einnehmen, vor (Scherf und Buckel, 1993; Scherf et al., 1994; Buckel, 2001). Aufgrund der
Verwandtschaftsbeziehung der Donorstämme zum Wirtsorganismus galten diese Enzyme als
erste Kandidaten, um eine 1,4-BDO-Synthese in C. acetobutylicum zu ermöglichen. Denn
durch die 4HBD sollte es möglich sein, das im Stoffwechsel von C. acetobutylicum gebildete
Crotonyl-CoA zu 4HB-CoA umzuwandeln, welches dann weiter zu 1,4-BDO umgesetzt wird
(Abb. 1.3).
Tab. 1.1: Bifunktionelle 4-Hydroxybutyryl-CoA Dehydratasen/Vinylacetyl-CoA-∆-Isomerasen (4HBDs) aus
verschiedenen Clostridien-Spezies.
Organismus ORF annotierte Funktion
Gen [Bp]
Protein [AS]
MW [kDa]1
pI [pH]
C. beijerinckii cbei_2100 Vinylacetyl-CoA-∆-Isomerase
EC [5.3.3.3]
1455 484 53,6 6,61
C. kluyveri a) ckl_3020 4-Hydroxybutyryl-CoA Dehydratase
EC [4.2.1.120]
1455 484 53,7 5,84
C. aminobutyricum b) abfD 4-Hydroxybutyryl-CoA Dehydratase
EC [4.2.1.120]
1473 490 54,4 5,78
1 Theoretisch ermitteltes Molekulargewicht über die AS-Sequenz (http://web.expasy.org/compute_pi/).
Legende: ORF, offener Leserahmen; Bp, Anzahl der Basenpaare; AS, Anzahl der Aminosäuren; MW,
Molekulargewicht der Proteine; kDa, Kilodalton; pI, isoelektrischer Punkt. a) Scherf et al., 1994, b) Scherf und
Buckel, 1993.
Die katalytisch aktive Form der 4HBD ist eine homotetramere Struktur mit einem
Gesamtmolekulargewicht von 232 kDa, welche sich aus vier identischen Untereinheiten
(MW ca. 54 kDa) zusammensetzt (Scherf und Buckel, 1993) (Abb. 1.6, A). Dabei sind beide
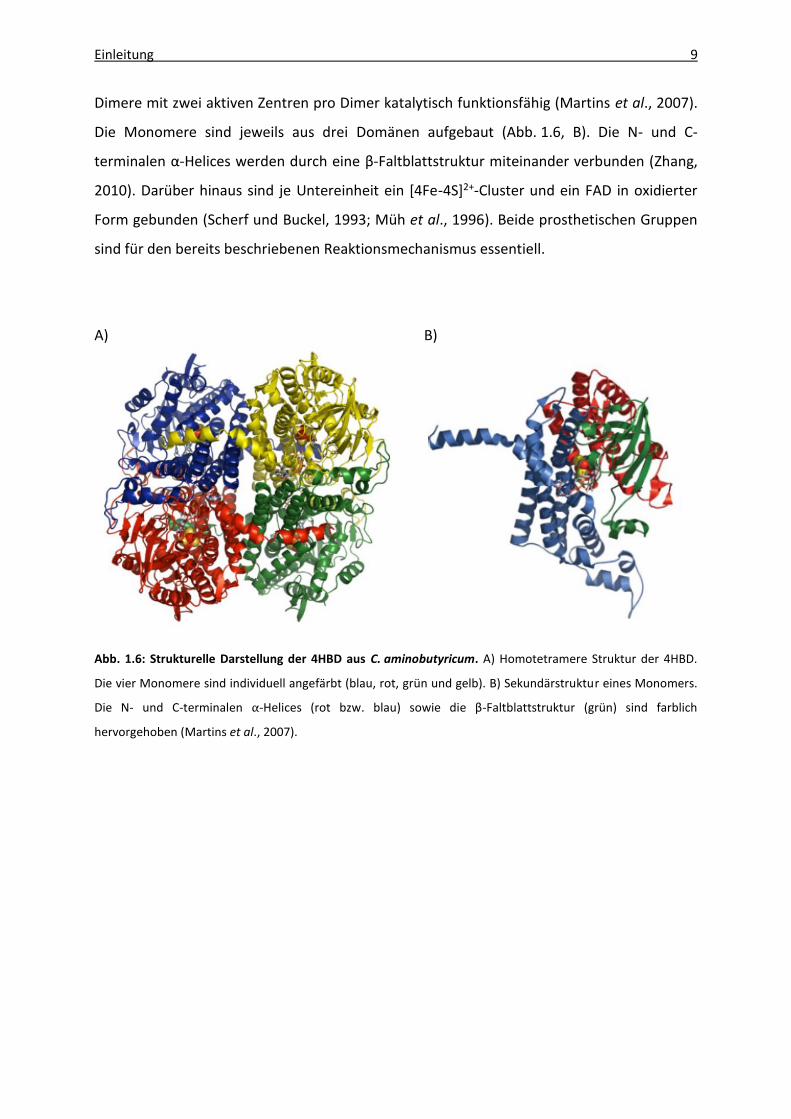
Einleitung 9
Dimere mit zwei aktiven Zentren pro Dimer katalytisch funktionsfähig (Martins et al., 2007).
Die Monomere sind jeweils aus drei Domänen aufgebaut (Abb. 1.6, B). Die N- und C-
terminalen α-Helices werden durch eine β-Faltblattstruktur miteinander verbunden (Zhang,
2010). Darüber hinaus sind je Untereinheit ein [4Fe-4S]2+-Cluster und ein FAD in oxidierter
Form gebunden (Scherf und Buckel, 1993; Müh et al., 1996). Beide prosthetischen Gruppen
sind für den bereits beschriebenen Reaktionsmechanismus essentiell.
A) B)
Abb. 1.6: Strukturelle Darstellung der 4HBD aus C. aminobutyricum. A) Homotetramere Struktur der 4HBD.
Die vier Monomere sind individuell angefärbt (blau, rot, grün und gelb). B) Sekundärstruktur eines Monomers.
Die N- und C-terminalen α-Helices (rot bzw. blau) sowie die β-Faltblattstruktur (grün) sind farblich
hervorgehoben (Martins et al., 2007).

Material und Methoden 10
2. Material und Methoden
2.1 Organismen und Plasmide
Die in dieser Arbeit verwendeten Organismen (Tab. 2.1), Vektoren (Tab. 2.2) und
rekombinanten Plasmide (Tab. 2.3) sind nachfolgend aufgeführt.
Tab. 2.1: Organismen
Organismen Genotyp Herkunft/ Referenz
C. acetobutylicum ATCC 824
Typstamm ATCC, Laborsammlung Nr. 258
C. acetobutylicum pTc
C. acetobutylicum ATCC 824 pTc-Vektor ohne Insert, AmpR, CmR/TmR
Schulz, 2013, Laborsammlung Nr. 356
C. acetobutylicum pTc::P(thlA)cbei_2100
C. acetobutylicum ATCC 824 pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 648
C. acetobutylicum pT::P(ptb)cbei_2100
C. acetobutylicum ATCC 824 pT::P(ptb)cbei_2100; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 649
C. acetobutylicum pT::P(adc)cbei_2100
C. acetobutylicum ATCC 824 pT::P(adc)cbei_2100; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 650
C. acetobutylicum pT::P(fac)cbei_2100
C. acetobutylicum ATCC 824 pT::P(fac)cbei_2100; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 651
C. acetobutylicum pTc::P(thlA)ckl_3020
C. acetobutylicum ATCC 824 pTc::P(thlA)ckl_3020; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 652
C. acetobutylicum pT::P(ptb)ckl_3020
C. acetobutylicum ATCC 824 pT::P(ptb)ckl_3020; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 653
C. acetobutylicum pT::P(adc)ckl_3020
C. acetobutylicum ATCC 824 pT::P(adc)ckl_3020; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 654
C. acetobutylicum pT::P(fac)ckl_3020
C. acetobutylicum ATCC 824 pT::P(fac)ckl_3020; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 655
C. acetobutylicum pTc::P(thlA)abfD
C. acetobutylicum ATCC 824 pTc::P(thlA)abfD; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 656
Material und Methoden 11
Tab. 2.1: Organismen (Fortsetzung)
Organismen Genotyp Herkunft/ Referenz
C. acetobutylicum pT::P(ptb)abfD
C. acetobutylicum ATCC 824 pT::P(ptb)abfD; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 657
C. acetobutylicum pT::P(adc)abfD
C. acetobutylicum ATCC 824 pT::P(adc)abfD; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 658
C. acetobutylicum pT::P(fac)abfD
C. acetobutylicum ATCC 824 pT::P(fac)abfD; C-terminaler Strep-tag II, EryR, AmpR
diese Arbeit, Laborsammlung Nr. 659
C. acetobutylicum pta::int(80)
Gruppe II Intron integrierte zw. 80./81. Bp von pta, EryR
Lehmann, 2012a; Laborsammlung Nr. 328
C. acetobutylicum pta::int(80) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 80./81. Bp von pta, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 660
C. acetobutylicum ptb::int(87)
Gruppe II Intron integrierte zw. 87./88. Bp von ptb, EryR
Lehmann, 2012a; Laborsammlung Nr. 329
C. acetobutylicum ptb::int(87) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 87./88. Bp von ptb, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 661
C. acetobutylicum ldh1::int(93)
Gruppe II Intron integrierte zw. 93./94. Bp von ldh1, EryR
Lehmann, 2012a; Laborsammlung Nr. 326
C. acetobutylicum ldh1::int(93) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 93./94. Bp von ldh1, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 662
C. acetobutylicum ldh2::int(231)
Gruppe II Intron integrierte zw. 231./232. Bp von ldh2, EryR
Lehmann, 2012a; Laborsammlung Nr. 327
C. acetobutylicum ldh2::int(231) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 231./232. Bp von ldh2, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 663
C. acetobutylicum adhE1::int(158)
Gruppe II Intron integrierte zw. 158./159. Bp von adhE1, EryR
Lehmann, 2012a; Laborsammlung Nr. 332
C. acetobutylicum adhE1::int(158) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 158./159. Bp von adhE1, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 664
C. acetobutylicum adhE2::int(114)
Gruppe II Intron integrierte zw. 114./115. Bp von adhE2, EryR
Lehmann, 2012a; Laborsammlung Nr. 333
Material und Methoden 12
Tab. 2.1: Organismen (Fortsetzung)
Organismen Genotyp Herkunft/ Referenz
C. acetobutylicum adhE2::int(114) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 114./115. Bp von adhE2, EryR; pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 665
C. acetobutylicum adc::int(180)
Gruppe II Intron integrierte zw. 180./181. Bp von adc, EryR
Lehmann, 2012a; Laborsammlung Nr. 330
C. acetobutylicum adc::int(180) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 180./181. Bp von adc, EryR;pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 666
C. acetobutylicum ctfA::int(352)
Gruppe II Intron integrierte zw. 352./353. Bp von ctfA, EryR
Lehmann, 2012a; Laborsammlung Nr. 331
C. acetobutylicum ctfA::int(352) pTc::P(thlA)cbei_2100
Gruppe II Intron integrierte zw. 352./353. Bp von ctfA, EryR;pTc::P(thlA)cbei_2100; C-terminaler Strep-tag II, CmR/TmR, AmpR
diese Arbeit, Laborsammlung Nr. 667
C. beijerinckii NCIMB 8052
Typstamm NCIMB, Laborsammlung Nr. 107
C. kluyveri Typstamm Laborsammlung
E. coli DH5α supE44, ∆lacU169, hsdR17, (φ80lacZ∆M15), recA1, endA1, gyrA96, thi-1, relA1
Grant et al., 1990; Laborsammlung Nr. 272
E. coli DH5α pTc::P(thlA)cbei_2100
pTc::P(thlA)cbei_2100, C-terminaler Strep-tag II, AmpR, CmR/TmR
diese Arbeit, Laborsammlung Nr. 668
E. coli DH5α pT::P(ptb)cbei_2100
pT::P(ptb)cbei_2100, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 669
E. coli DH5α pT::P(adc)cbei_2100
pT::P(adc)cbei_2100, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 670
E. coli DH5α pT::P(fac)cbei_2100
pT::P(fac)cbei_2100, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 671
E. coli DH5α pTc::P(thlA)ckl_3020
pTc::P(thlA)ckl_3020, C-terminaler Strep-tag II, AmpR, CmR/TmR
diese Arbeit, Laborsammlung Nr. 672
E. coli DH5α pT::P(ptb)ckl_3020
pT::P(ptb)ckl_3020, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 673
E. coli DH5α pT::P(adc)ckl_3020
pT::P(adc)ckl_3020, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 674
Material und Methoden 13
Tab. 2.1: Organismen (Fortsetzung)
Organismen Genotyp Herkunft/ Referenz
E. coli DH5α pT::P(fac)ckl_3020
pT::P(fac)ckl_3020, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 675
E. coli DH5α pTc::P(thlA)abfD
pTc::P(thlA)abfD, C-terminaler Strep-tag II, AmpR, CmR/TmR
diese Arbeit, Laborsammlung Nr. 676
E. coli DH5α pT::P(ptb)abfD
pT::P(ptb)abfD, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 677
E. coli DH5α pT::P(adc)abfD
pT::P(adc)abfD, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 678
E. coli DH5α pT::P(fac)abfD
pT::P(fac)abfD, C-terminaler Strep-tag II, AmpR, EryR
diese Arbeit, Laborsammlung Nr. 679
E. coli DH5α pT::P(ptb)pp2
pT::P(ptb)pp2, C-terminaler Strep-tag II, AmpR, EryR
Schulz, 2013; Laborsammlung Nr. 233
E. coli DH5α pT::P(adc)pp2
pT::P(adc)pp2, C-terminaler Strep-tag II, AmpR, EryR
Schulz, 2013; Laborsammlung Nr. 212
E. coli DH5α pT::P(fac)pp2
pT::P(fac)pp2, C-terminaler Strep-tag II, AmpR, EryR
Schulz, 2013; Laborsammlung Nr. 349
E. coli DH5α pASK-IBA3+::abfD
pASK-IBA3+::abfD, C-terminaler Strep-tag II, AmpR
diese Arbeit, Laborsammlung Nr. 680
E. coli ER2275 trp-31, his-1, tonA2, rpsL104, supE44, xyl-7,
mtl-2, metB1, el4-, ∆(lac)U169, endA1, recA1,
R(zbg10::Tn10) Tcs, ∆(mcr-hsd-mrr)114::1510,
[F´, proAB, laqlqZ∆2.15zzf::mini-Tn10 (Km
r)]
NEB, Laborsammlung Nr. 271
Tab. 2.2: Vektoren
Vektor Relevantes Merkmal Herkunft/ Referenz
pANII TcR , Φ3tI; p15A oriR Heap et al., 2007; Laborsammlung Nr. 271
pTc::hydA thlA Promotor, hydA, Strep-tag II, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression in E. coli
Schulz, 2013; Laborsammlung Nr. 397
pASK-IBA3+::abfD ColE1 origin, F1 origin, MCS, AmpR, tet-Repressor, cytoplasm. Express. in E. coli
Gerhardt et al., 2000
Material und Methoden 14
Tab. 2.2: Vektoren (Fortsetzung)
Vektor Relevantes Merkmal Herkunft/ Referenz
pT::P(ptb)pp2 ptb Promotor, pp2, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression in E. coli
Schulz, 2013 Laborsammlung Nr. 233
pT::P(adc)pp2 adc Promotor, pp2, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression in E. coli
Schulz, 2013; Laborsammlung Nr. 212
pT::P(fac)pp2 fac Promotor, pp2, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression in E. coli
Schulz, 2013; Laborsammlung Nr. 349
Tab. 2.3: rekombinante Plasmide
Plasmid Relevantes Merkmal Insert Herkunft
pTc::P(thlA)cbei_2100 thlA-Promotor, 5´BamHI, 3´Cfr9I, cbei_2100, Strep-tag II, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Exp.
1455 Bp diese Arbeit
pT::P(ptb)cbei_2100 ptb-Promotor, 5´BamHI, 3´Cfr9I, cbei_2100, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pT::P(adc)cbei_2100 adc-Promotor, 5´BamHI, 3´Cfr9I, cbei_2100, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pT::P(fac)cbei_2100 fac-Promotor, 5´BamHI, 3´Cfr9I, cbei_2100, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pTc::P(thlA)ckl_3020 thlA-Promotor, 5´NheI, 3´Cfr9I, ckl_3020, Strep-tag II, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pT::P(ptb)ckl_3020 ptb-Promotor, 5´NheI, 3´Cfr9I, ckl_3020, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pT::P(adc)ckl_3020 adc-Promotor, 5´NheI, 3´Cfr9I, ckl_3020, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pT::P(fac)ckl_3020 fac-Promotor, 5´NheI, 3´Cfr9I, ckl_3020, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Expression
1455 Bp diese Arbeit
pTc::P(thlA)abfD thlA-Promotor, 5´NheI, 3´Cfr9I, abfD, Strep-tag II, AmpR, CmR/TmR, repL, ColE1 ori, cytoplasmatische Expression
1473 Bp diese Arbeit

Material und Methoden 15
Tab. 2.3: rekombinante Plasmide (Fortsetzung)
Plasmid Relevantes Merkmal Insert Herkunft
pT::P(ptb)abfD ptb-Promotor, 5´NheI, 3´Cfr9I, abfD, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Exp.
1473 Bp diese Arbeit
pT::P(adc)abfD adc-Promotor, 5´NheI, 3´Cfr9I, abfD, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Exp.
1473 Bp diese Arbeit
pT::P(fac)abfD fac-Promotor, 5´NheI, 3´Cfr9I, abfD, Strep-tag II, AmpR, EryR, repL, ColE1 ori, cytoplasmatische Exp.
1473 Bp diese Arbeit
2.2 Oligonukleotide
Die in dieser Arbeit verwendeten Oligonukleotide sind in der Tabelle 2.4 aufgelistet.
Tab. 2.4: Oligonukleotide
Bezeichnung Sequenz (5´ 3´)* Verwendung
cbei_2100_fw_BamHI CCCCGGATCCATGCCATTAAAAACAAAGGAAC Klonierung
cbei_2100_rev_Cfr9I GGGGCCCGGGTTCTTTAATTCTAGCTATTTTCTTAGC cbei_2100
ckl_3020_fw_NheI_BamHI GGGGGGATCCGCTAGCATGTCTTTAATGACTGGGGA Klonierung
ckl_3020_rev_Cfr9I GGGGCCCGGGATCTTCTATTCTAGCTATCTTCTTTG ckl_3020
abfD_fw_NheI CCCCGCTAGCATGTTAATGACAGCAGAACAG Klonierung
abfD_rev_Cfr9I GGGGCCCGGGTTTAATTCCAGCGATTGCC abfD
pT_fw_SacI CCCCGAGCTCTGATAAATATGAACATGA Schnittstellen-
pT_rev_NheI_Cfr9I GGGGCCCGGGGCTAGCTCTAACTAACCTCCTTGATCC anpassung
Strep-tag_rev_KasI TTTTGGCGCCTTATTTTTCAAATTGAGGATGTGAC Klonierung Strep-tagII
Padc_fw_SacI GGGGGAGCTCTATTTATTTTTTGTATTGGAATTGTTTATAGT Klonierung
Padc_rev_NheI_BamHI TTTTGGATCCGCTAGCTTCATCCTTTAACATAAAAGTCAC adc-Promotor
Pptb_fw_SacI GGGGGAGCTCTATGAAGGATACAGTAAGCAGT Klonierung
Pptb_rev_NheI_BamHI AAAAGGATCCGCTAGCAATCACTGGTCGTACACT ptb-Promotor
Pfac_fw_SacI GGGGGAGCTCTCTTTAAATATAGATAAAGTTATAGAAGCAA Klonierung
Pfac_rev_NheI_BamHI TTTTGGATCCGCTAGCCATATGAAATACACCTCCTTAAAATT fac-Promotor
pT_seq_fw GGGATAAACTATGGAACTTATGAAA Sequenzierung
pT_seq_rev TGCAAGAATGTGAGAGCTAGAAA der 4hbd
pT_seq_fw_P GAGCCGATTTCAAAGATATTATCATG Sequenzierung
pT_seq_rev_P TGCAAGAATGTGAGAGCTAGAAA Promotoren * unterstrichene Basen symbolisieren entsprechende Restriktionsschnittstellen.

Material und Methoden 16
2.3 Nährmedien
Hitzestabile Nährlösungen und Medien wurden im Anschluss an die Herstellung für 20 min
bei 121 °C autoklaviert. Hitzelabile Lösungen wurden mittels Einwegfiltern (Porengröße
0,2 µm; Sarstedt) sterilfiltriert.
2.3.1 Nährmedium für E. coli
Zellen von E. coli wurden entweder in LB-Flüssigmedium oder auf LB-Agarplatten bei 37 °C
kultiviert. Medienzusätze wurden bei Bedarf zugegeben (2.3.3).
LB-Medium (Luria Bertani) (Sambrook und Russel, 2001)
Hefeextrakt 5,0 g
Trypton 10,0 g
NaCl 10,0 g
A. dest. ad 1000 ml
Zur Herstellung von LB-Festmedien wurden dem Medium vor dem Autoklavieren 1,5 % (w/v)
Agar-Agar zugegeben.
2.3.2 Medien für C. acetobutylicum
Anaerobe Nährmedien für die Anzucht von C. acetobutylicum wurden nach Vorschrift von
Breznak und Costilow (1994) hergestellt. Die Medien wurden in einer Mikrowelle zur
Entfernung des Luftsauerstoffs aufgekocht und unter kontinuierlicher Stickstoffbegasung
abgekühlt. Die Anaerobität der Medien konnte optisch anhand des Redoxindikators
Resazurin (0,0001 % [w/v]) überprüft werden. Die Kulturgefäße wurden im Anschluss
luftdicht verschlossen und autoklaviert. Etwaiger Restsauerstoff wurde durch die Zugabe von
Titan-(III)-Nitrilotriessigsäure (NTA) (2.3.3) vor Verwendung der Nährmedien reduziert.

Material und Methoden 17
CGM (clostridial growth medium) (Wiesenborn et al., 1988; mod.)
Glukose x H2O (50 %, [w/v])* 50,0 ml
Hefeextrakt 5,0 g
Asparagin 2,0 g
(NH4)2SO4 2,0 g
NaCl 1,0 g
KH2PO4 0,75 g
K2HPO4 0,75 g
MgSO4 x 7 H2O 0,71 g
MnSO4 x H2O 10,0 mg
FeSO4 x 7 H2O 10,0 mg
Resazurin (0,1 % [w/v]) 1,0 ml
A. dest. ad 1000 ml
* Nach dem Autoklavieren und vor dem Beimpfen wurde die Glukose aus einer sauerstofffreien, sterilen
Stammlösung (50 %, [w/v]) zugegeben.
MS-MES Medium (medium synthetique) (Monot et al., 1982; mod.)
Glukose x H2O 60,0 g
KH2PO4 0,55 g
K2HPO4 0,55 g
MgSO4 x 7 H2O 0,22 g
FeSO4 x 7 H2O 11,0 mg
Eisessig 2,3 ml
MES* 21,3 g
PABA (0,8 mg/l)** 10,0 ml
Biotin (0,08 mg/l)** 1,0 ml
Resazurin (0,1 % [w/v]) 1,0 ml
A. dest. ad 1000 ml
* Vor der Zugabe von MES wurde der pH-Wert mit NH4OH auf 6,6 eingestellt.
** Vor dem Autoklavieren wurde PABA aus einer 100-fachen und Biotin aus einer 1000-fachen sterilen
Stammlösung hinzugegeben.

Material und Methoden 18
Bei Verwendung verschiedener Kohlenstoffquellen (C-Quellen) wurde anstelle der Glukose
die entsprechende C-Quelle aus einer anaeroben, sterilfiltrierten Stammlösung (Mannose
50 % [w/v], Galaktose 50 % [w/v], Arabinose 50 % [w/v], Xylose 50 % [w/v]) zugegeben.
MMfvK (Minimalmedium für Vorkulturen) (Fischer et al., 2006)
Glukose (50 % [w/v])* 40,0 ml
CaCO3 1,0 g
K2HPO4 x 3 H2O 1,0 g
KH2PO4 1,0 g
MgSO4 x 7 H2O 0,1 g
(NH4)2SO4 2,0 g
NaCl** 10,0 mg
Na2MoO4 x 2 H2O** 10,0 mg
CaCl2 x 2 H2O* * 10,0 mg
MnSO4 x H2O* * 15,0 mg
FeSO4 x 7 H2O** 15,0 mg
PABA** 2,0 mg
Thiamin-HCl** 2,0 mg
Biotin** 0,1 mg
Resazurinlösung (0,1 % [w/v]) 1,0 ml
Na2S2O4 35,0 mg
A. dest. ad 1000 ml
* Nach dem Autoklavieren wurde die Glukose direkt vor dem Beimpfen des Mediums aus einer sauerstofffreien,
sterilen Stammlösung (50 % [w/v]) zugesetzt.
** Diese Komponenten wurden zusammen als 100-fache Stammlösung angesetzt, aliquotiert und bei -20 °C
gelagert.

Material und Methoden 19
RCA (reinforced clostridial agar)*
Glukose x H2O 5,0 g
Hefeextrakt 3,0 g
Trypton 10,0 g
NaCl 5,0 g
Fleischextrakt 10,0 g
Na-Acetat 3,0 g
Cystein-HCl 0,5 g
Stärke 1,0 g
Agar-Agar 15,0 g
A. dest. ad 1000 ml
* RCA wurde als kommerzielles Komplettmedium von der Firma Oxoid (Wesel) erworben. Der pH-Wert
entsprach ohne Einstellung 6,8.
2.3.3 Medienzusätze
Den Nährmedien wurden im Bedarfsfall nach dem Autoklavieren und Abkühlen auf etwa
50 °C bzw. direkt vor dem Animpfen nachstehende Medienzusätze (Tab. 2.5) beigefügt.
Tab. 2.5: Medienzusätze
Mediumzusatz Stammkonzentration Arbeitskonzentration
Ampicillin (Amp) 50 mg/ml in A. dest. (sterilfiltriert)* 100 µg/ml
Chloramphenicol (Cm) 34 mg/ml in Ethanol (96 % [v/v])** 25 µg/ml
Tetracyclin (Tc) 10 mg/ml in Ethanol (96 % [v/v])** 10 µg/ml
Erythromycin (Ery) 50 mg/ml in Ethanol (96 % [v/v])** 20 µg/ml
Thiamphenicol (Tm) 15 mg/ml in Ethanol (96 % [v/v])** 15 µg/ml
* Porengröße 0,2 µm (Sterilfilter, Schleicher & Schuell)
** in Ethanol gelöste Substanzen bedürfen keiner Sterilfiltration

Material und Methoden 20
Titan-(III)-NTA-Lösung
Die Herstellung der Titan-(III)-Nitrilotriessigsäure erfolgte wie bei Lehmann (2012 a)
beschrieben.
2.4 Stammhaltung
E. coli Zellen wurden vorübergehend auf LB-Agarplatten bis zu vier Wochen bei 4 °C gelagert.
Für eine dauerhafte Konservierung wurden 1 ml einer exponentiell wachsenden E. coli-
Kultur zu 0,5 ml LB-Glycerin-Lösung (Glycerin 60 % [v/v], LB-Medium 40 % [v/v]) gegeben,
gründlich durchmischt und anschließend bei -70 °C gelagert.
Zur Stammhaltung von C. acetobutylicum wurden zu 1-ml exponentiell wachsenden Zellen in
CGM (2.3.2) 0,5 ml CGM-Glycerin-Lösung (Glycerin 60 % [v/v], CGM 40 % [v/v]) zugegeben
und anschließend nach einer halbstündigen Inkubationszeit (37 °C, Anaerobenbox) bei -70 °C
konserviert.
Vitalitäts- und Reinheitskontrollen wurden durch erneutes Ausstreichen auf Antibiotika-
haltigen Agarplatten und mittels Mikroskopie durchgeführt.
2.5 Zellanzucht
2.5.1 Zellanzucht von E. coli
Die Anzucht der E. coli-Stämme (Tab. 2.1) erfolgte unter aeroben Bedingungen in LB-
Flüssigkulturen unter Schütteln (180 Upm) bzw. auf LB-Festmedien (2.3.1) im Brutschrank
bei 37 °C unter Verwendung entsprechender Medienzusätze (2.3.3).
2.5.2 Zellanzucht von C. acetobutylicum
C. acetobutylicum Stämme wurden zum einen auf RCA-Festmedien und zum anderen in
Flüssigmedien (CGM und MS-MES) (2.3.2) kultiviert.

Material und Methoden 21
2.5.2.1 Zellanzucht von C. acetobutylicum auf Festmedium
Für die Anzucht auf Festmedien wurden die aerob angefertigten Platten zur Minimierung
des Sauerstoffgehaltes mindestens 24 h vor Verwendung in eine Anaeroben-Werkbank
(MACS-MG-1000-anaerobic work station, Meintrup DWS, Lähden-Holte) eingeschleust. Die
anschließende Kultivierung von C. acetobutylicum erfolgte ebenda bei 37 °C unter
Stickstoffatmosphäre. Zur Reduktion von eingetragenem Sauerstoff wurden maximal 5 %
[v/v] Wasserstoff zugegeben.
2.5.2.2 Zellanzucht von C. acetobutylicum in statischer Kultur
Die Kultivierung von C. acetobutylicum in statischer Kultur (Batch-Kultur) erfolgte anaerob
unter einer abgeschlossenen N2-Atmosphäre bei 37 °C in Hungate-Röhrchen (Ochs GmbH)
oder in Müller&Krempel-Serumflaschen (Müller & Krempel AG). Batch-Kulturen (10 ml)
wurden mit 0,1 Vol. einer MS-MES Sporensuspension bzw. mit 1 ml einer CGM-Glycerin-
Stammkultur (2.4) inokuliert. Im Falle einer Sporensuspension wurde die Sporenkeimung
durch Pasteurisierung (80 °C, 10 min) induziert. Anschließend erfolgte die Inkubation bei
37 °C. Vorkulturen dienten in der Regel der Anzucht von Batch-Hauptkulturen in
Müller&Krempel-Serumflaschen mit unterschiedlichen Volumina (50 - 500 ml), die aufgrund
der Gasentwicklung und des damit einhergehenden Druckanstieges nur bis maximal 60 %
des Fassungsvermögens befüllt wurden. Die Anzucht von Vorkulturen erfolgte in
Komplexmedium (CGM; 2.3.2). Wachstumsversuche wurden stets in 200-ml MS-MES
durchgeführt. Im Falle eines Medienwechsels war eine weitere Vorkultur in MS-MES
Medium notwendig. Hierfür wurden 0,1 Vol. der CGM-Vorkultur in 10-ml-MS-MES-
Vorkulturen überimpft, die bei Bedarf mit verschiedenen C-Quellen (Mannose, Galaktose,
Arabinose und Xylose) versetzt wurden. Hauptkulturen wurden in der Regel mit einer
optischen Dichte (OD600) von 0,1 inokuliert.
2.5.2.3 pH-kontrollierte Batch-Fermentation
Für höhere Kulturvolumina wurde ein pH-regulierter „BIOSTAT B“ Chemostat (BBI,
Melsungen) mit einem Arbeitsvolumen von 1,5 l eingesetzt. Die Steuereinheit ermöglichte
eine konstante Temperatur von 37 °C und eine Regulation des pH-Wertes durch die Zufuhr

Material und Methoden 22
von 2 M KOH. Das Kulturgefäß wurde mit 1,3 l MS-MES bzw. MMfvK (2.3.2) gefüllt,
autoklaviert und während des Abkühlens mit N2 durchgast. Vor dem Beimpfen der
Hauptkultur wurde zur Reduzierung des Restsauerstoffs 0,5 ml Titan-(III)-NTA-Lösung (2.3.3)
zugegeben. CGM-Vorkulturen (2.5.2.2) dienten der Inokulation von 10 ml-Vorkulturen (MS-
MES bzw. MMfvK), die nach einer Inkubation von 12 h bei 37 °C für die Inokulation von
200 ml Kulturmedium eingesetzt wurden. Die Hauptkultur wurde über einen sterilen
Beimpfungsschlauch mit einer gut gasenden, exponentiell wachsenden 200-ml-Vorkultur
inokuliert. Die Anwachsphase der Kultur erfolgte bei einer pH-Wertkontrolle von 5,2 und
einer Durchmischung bei 50 Upm. Nach Erreichen einer OD600 von ca. 1,0 wurde die
Durchgasung mit N2 eingestellt und die Rührgeschwindigkeit erhöht (200 Upm). Bei einer
OD600 von ca. 2,0 wurde die pH-Wertkontrolle auf 4,7 gesetzt, um die Lösungsmittelphase
einzuleiten (Fontaine et al., 2002).
2.6 Bestimmung physiologischer Parameter
2.6.1. Optische Dichte
Das Wachstum der Batchkulturen wurde photometrisch bei einer Wellenlänge von 600 nm
in einem Spektralphotometer (Photometer WPA Biowave II, Biochrom, Cambridge) gegen
A.dest. in einer Plastikküvette mit 1 cm Schichtdicke ermittelt. Bei einer Extinktion über 0,3
erfolgte die Verdünnung der Zellsuspension mit A. dest.
2.6.2 Bestimmung der Wachstumsrate und Verdopplungszeit
Die Wachstumsrate µ gibt die masseabhängige Verdopplung pro Stunde an und lässt sich mit
folgender Formel berechnen (Formel 2.1). Über die Wachstumsrate kann weiterhin die
Verdopplungszeit td bestimmen werden (Formel 2.2), welche das Zeitintervall der
Zellmasseverdopplung pro Stunde angibt (Madigan et al., 2013).
µ =𝑙𝑛𝑥2−𝑙𝑛𝑥1
(𝑡2−𝑡1)
Formel 2.1: Berechnung der spezifischen Wachstumsrate (µ). x1/x2, Messwerte der optischen
Dichte während des exponentiellen Wachstums zu den Zeitpunkten t1 und t2.

Material und Methoden 23
𝑡𝑑 =𝑙𝑛2
µ
Formel 2.2: Berechnung der Verdopplungszeit (td). µ, spezifische Wachstumsrate
2.6.3 Bestimmung des externen pH-Wertes
Die Veränderung des externen pH-Wertes in Batch-Kulturen wurde in zellfreien Überständen
nach Sedimentation (13000 Upm, 4 °C, 5 min) mit Hilfe eines pH-Meters (pH-Meter
SevenEasy) verfolgt.
2.6.4 Optisch-enzymatische Glukosebestimmung
Zur Bestimmung des Glukoseverbrauchs während des Wachstums von C. acetobutylicum
diente ein optisch-enzymatischer Test (Bergmeyer, 1983, mod.). Dabei wird D-Glukose in
einer ersten Reaktion durch die Hexokinase [EC 2.7.1.1] unter ATP-Verbrauch zu Glukose-6-
Phosphat und ADP umgewandelt. Anschließend erfolgt die Umwandlung von Glukose-6-
Phosphat unter NADP+-Verbrauch durch die Glukose-6-Phosphat-Dehydrogenase
[EC 1.1.1.49] zu 6-Phospho-D-Glukonat und NADPH+H+. Die gebildete NADPH-Menge ist
dabei direkt proportional zur Glukosemenge und kann photometrisch durch Bestimmung der
Extinktionsänderung bei einer Wellenlänge von 340 nm (WPA Biowave II) gemessen werden.
Die Berechnung der Glukosekonzentration erfolgte anschließend mit der Formel 2.3.
cGlukose = ∆E ∗ V ∗ MWGlukose
ɛ ∗ 𝑑 ∗ 𝜈∗ VF [
g
l]
Formel 2.3: Berechnung der Glukosekonzentration. c, Konzentration; ∆E, Extinktionsänderung; V,
Gesamtvolumen des Ansatzes (ml); MW, Molekulargewicht (180,16 g/mol); ɛ, Extinktionskoeffizient
von NADPH bei 340 nm (6,3 l * mmol-1 * cm-1); d, Schichtdicke der Küvette (1 cm); v, Probenvolumen.
Für die Bestimmung der Glukosekonzentration wurde folgender Reaktionsansatz in einer
Halbmikroliterplastikküvette durchgeführt:
0,2 M Tris-HCl + 0,002 M MgSO4 (pH 7,6) 900 µl
NADP+ (44 mg/ml) 10 µl
ATP (96 mg/ml) 10 µl
Probe (zellfreier Überstand) 10 µl

Material und Methoden 24
Nach dem Mischen des Ansatzes wurde die Extinktion des Leerwertes (E1) bei 340 nm
gemessen. Durch Zugabe von 10 µl des Hexokinase/Glucose-6-Phosphat-Dehydrogenase
Enzymgemisches (3 mg/ml, Roche) wurde die zuvor beschriebene Reaktion eingeleitet. Der
Reaktionsansatz wurde erneut gemischt und für 5 min bei Raumtemperatur inkubiert. Es
folgte die erneute Messung der Extinktion der Probe (E2) bei 340 nm.
2.6.5 Optisch-enzymatische Laktatbestimmung
Die Bestimmung des während des Wachstums produzierten Laktats wurde in einem optisch-
enzymatischen Test mit Hilfe des „D-/L-Laktat“-Kits von Megazyme (Megazyme
International, Irland) durchgeführt. Die Quantifizierung des Laktats erforderte zwei
Enzymreaktionen. Während der ersten Reaktion wird das D- bzw. L-Laktat in Anwesenheit
von NAD+ zu Pyruvat über die D-Laktatdehydrogenase (D-LDH) [EC 1.1.1.27] bzw. L-
Laktatdehydrogenase (L-LDH) [EC 1.1.1.27] oxidiert. In einer zweiten Reaktion, die der
Rückreaktion zum D-/L-Laktat entgegenwirkt, wird das gebildete Pyruvat über die D-
Glutamat-Pyruvat-Transaminase (D-GPT) [EC 2.6.1.2] zu D-Alanin und 2-Oxoglutarat
umgewandelt. Dabei ist die Menge des gebildeten NADH aus der ersten Reaktion
stöchiometrisch zu der Laktatmenge. Die Bestimmung der Laktatkonzentration erfolgte
photometrisch in einer Halbmikroliter-Plastikküvette bei einer Wellenlänge von 340 nm. Für
die Berechnung der Konzentration wurde die Formel 2.4 verwendet.
cLaktat = V ∗ MWLaktat
ɛ ∗ 𝑑 ∗ 𝜈∗ ∆E [
g
l]
Formel 2.4: Berechnung der Gesamtlaktatkonzentration. c, Konzentration; V, Gesamtvolumen des
Ansatzes (ml); MW, Molekulargewicht von Laktat (90,1 g/mol); ɛ, Extinktionskoeffizient von NADH
bei 340 nm (6,3 l * mmol-1 * cm-1); d, Schichtdicke der Küvette (1 cm); v, Probenvolumen (ml); ∆E,
Extinktionsänderung.
Der Reaktionsansatz umfasste 750 µl A. dest., 250 µl Glycylglycin-Puffer, 50 µl NAD+-Lösung
und 10 µl D-GPT. Anschließend wurden 50 µl der zu untersuchenden Probe, nach zu voriger
Sedimentation (13000 Upm, 4 °C, 10 min), dem Ansatz zugeführt, gründlich durchmischt und
die Extinktion des Blindwertes gegen A. dest. und der Leerwerte bei 340 nm gemessen. Die
zuvor beschriebenen Reaktionen wurden durch Zugabe von jeweils 10 µl D-LDH und L-LDH

Material und Methoden 25
gestartet. Nach 10 minütiger Inkubation der Reaktionsansätze bei RT wurde erneut die
Extinktion gemessen.
2.6.6 Gaschromatografie
2.6.6.1 Gaschromatografische Analyse der Gärungsprodukte von C. acetobutylicum
Die Analyse und Quantifizierung der Gärungsprodukte Acetat, Butyrat, Aceton, Butanol und
Ethanol erfolgte mit einem Agilent 7890A Gaschromatographen (Agilent Technologies,
Böblingen) unter Verwendung eines Flammenionisationsdetektors (FID). Als Trägergas
wurde über Feuchtigkeits- und Sauerstofffilter gereinigtes N2 eingesetzt, welches der
Zurückhaltung von Wasser, Sauerstoff und schwefeligen und chlorierten Substanzen diente.
Die FID-Brenngase, synthetische Luft und Wasserstoff, wurden zur Entfernung von
organischen Substanzen über einen Aktivkohlefilter geführt. Für die Analyse von Alkoholen
und Carbonsäuren wurde eine mit Porapak P (80-100 mesh) gepackte Säule eingesetzt.
Nach Sedimentation der zu analysierenden Zellsuspension bei 13000 Upm und 4 °C für
10 min wurde der zellfreie Überstand in ein steriles 1,5-ml Eppendorfreaktionsgefäß
überführt und bis zur Verwendung bei -20 °C eingefroren. Für die Bestimmung des
Produktspektrums wurden 100 µl des zellfreien Überstandes zu 900 µl destilliertem Wasser
und 100 µl internem Standard (IS; 55 mM Isobutanol in 2 M HCl und 0,5 M Acetoin) in ein
Rollrandgefäß gegeben. Anschließend wurden die Rollrandgefäße mittels Bördelkappen
gasdicht verschlossen. Über einen automatisierten Probengeber wurden 0,5 µl der Probe zur
Analyse in das System injiziert.
Die Quantifizierung der Gärungsprodukte erfolgte über eine Eichlösung bestehend aus 5 mM
der zu analysierenden Produkte einschließlich des internen Standards, woraus ein
Eichchromatogramm erstellt werden konnte. Über das Programm EZChrom Elite (Agilent
Tehnologies, Böblingen) erfolgte sowohl die Steuerung des Gaschromatographen und des
Probengebers als auch die Auswertung der Signale.
Die Analysebedingungen für die Detektion der Substanzen Acetat, Butyrat, Aceton, Butanol
und Ethanol sind nachfolgend dargestellt.

Material und Methoden 26
Chromatografiesäule: INNOSteel-GC-Säule (2 m x 1/8“AD x 2 mm, Porapak P
80/100 mesh)
Säulentemperatur: 155-197 °C; 9 °C/min
Trägergas: N2 (30 ml/min)
Injektortemperatur: 195 °C
Detektor: FID; 230 °C
2.6.6.2 Gaschromatografische Analyse von 1,4-Butandiol und γ-Hydroxybutyrat
Die Analyse und Quantifizierung von 1,4-BDO und GHB erfolgte mit einem
Gaschromatographen mit massenselektivem Detektor (GC/MS) bestehend aus einem Agilent
GC 6890 Gaschromatographen (Agilent Technologies, Böblingen) und einem
massenselektiven Detektor (MSD 593). Als Trägergas diente Helium.
Die zu analysierenden Zellsuspensionen wurden bei 13000 Upm und 4 °C für 10 min
sedimentiert und der zellfreie Überstand in ein steriles 1,5-ml Eppendorfreaktionsgefäß
überführt. Bis zur Weiterverwendung wurden die Proben bei -20 °C gelagert. Für die Analyse
der 1,4-BDO- und GHB-Konzentration wurden 50 µl des Silylierungsreagenz N-Methyl-N-tert-
butyldimethylsilyl-tifluoracetamid (MBDSTFA, Macherey und Nagel, Düren) in ein
Rollrandgefäß mit Insert vorgelegt. Anschließend wurden je 1 µl der internen Standards (8-
fach deuteriertes 1,4-BDO [100 µg/ml] und 6-fach deuteriertes GHB [100 µg/ml]) und 1 µl
der Probe zugegeben, gut vermischt und das Probengefäß luftdicht verschlossen. Zur
Beschleunigung der Silylierung der Proben und der internen Standards mit MBDSTFA folgte
eine Inkubation des Reaktionsansatzes bei 80 °C für 20 min. Im Anschluss daran wurde 1 µl
des Reaktionsansatzes über einen automatisierten Probengeber (Autosampler 7683) in das
System injiziert.
Für die Quantifizierung von 1,4-BDO und GHB wurde eine isotopenverdünnte
Nachweismethode mit dem „Selected Ion Mode“ (SIM) angewandt. Die SIM-Ionen für 1,4-
BDO/ 1,4-BDO-d8 und GHB/ GHB-d6 sind 219/221 bzw. 317/323. Die Analysebedingungen
von 1,4-BDO und GHB sind nachfolgend dargestellt.

Material und Methoden 27
Chromatografiesäule: VF-5ms (25 m x 0,2 mm; 0,33 µm)
Säulentemperatur: Initial 80 °C (3 min halten), + 20 °C/min bis 150 °C,
+ 10 °C/min bis 300 °C (8,5 min halten)
Trägergas: Helium (0,9 ml/min)
Injektionsmodus: splitless
Injektortemperatur: 290 °C
Detektor: MSD
2.7 Visualisierung von 1,4-Butandiol
Für einen schnellen, präzisen und kostengünstigen Nachweis von 1,4-Butandiolproduzenten
wurde eine auf Dünnschichtchromatografie-basierte Methode evaluiert. Als stationäre
Phase diente eine ALUGRAM SIL G DC-Fertigfolie (Macherey-Nagel, Düren) mit Kieselgel
(Schichtdicke 0,2 mm) als Sorptionsmittel. Diese wurde zur Aktivierung bei 90 °C für 30 min
erhitzt und nach dem Abkühlen mit den Proben beladen. Hierfür wurde der zellfreie
Überstand von C. acetobutylicum bzw. 1,4-BDO (Sigma Aldrich Chemie GmbH, Taufkirchen)
ca. 1 cm vom unteren Rand tropfenweise aufgetragen. Nach Lufttrocknung der Proben
wurde die beladene Kieselgelplatte aufrecht in eine mit Fließmittel enthaltene Trennkammer
überführt. Dabei galt es zu beachten, dass die Probenfront oberhalb des Fließmittels lag. Als
mobile Phase wurde ein Gemisch aus Chloroform und Methanol im Verhältnis 80:20
gewählt. Damit eine mit dem Fließmittel gesättigte Atmosphäre entstand, wurde die
Trennkammer geschlossen. Durch Kapillarkräfte stieg das Fließmittel vom unteren zum
oberen DC-Plattenrand und transportierte so die aufgetragenen Proben mit. Sobald die
Lauffront annähernd den oberen Rand der Platte erreichte, wurde die Platte aus der
Trennkammer entnommen und an der Luft getrocknet. Es folgte das Besprühen der DC-
Platte mit einer Vanillin-Färbelösung. Hierfür wurde in 98 ml Ethanol (96 %, reinst) 2 ml
konzentrierte Schwefelsäure gelöst und mit 1 g Vanillin versetzt. Die Entwicklung der Spots
erfolgte bei 100 °C bis eine Farbreaktion zu beobachten war. Anschließend konnten die
Platte der Kammer entnommen und fotodokumentiert werden.

Material und Methoden 28
2.8 Arbeiten mit Nukleinsäuren
Zur Vermeidung von Verunreinigungen bei Arbeiten mit Nukleinsäuren wurden alle
hitzestabilen Lösungen und Materialien bei 121 °C für 20 min autoklaviert. Hitzelabile
Lösungen hingegen wurden sterilfiltriert (Porengröße 0,2 µm, Sarstedt).
2.8.1 Isolierung von Nukleinsäuren
2.8.1.1 Isolierung von Plasmid-DNA aus E. coli
Die Isolierung von Plasmid-DNA aus E. coli erfolgte nach den Prinzipien der alkalischen Lyse
von Birnboim und Doly (1979). In der Regel wurden 5 ml einer Übernachtkultur (2.5.1) für
die Plasmid-Isolierung eingesetzt und nach folgender Prozedur behandelt.
1. Sedimentation der Zellen durch Zentrifugation (13000 Upm, 4 °C, 2 min)
2. Suspendieren des Zellpellets mit 300 µl Puffer P1
3. Zugabe von 300 µl Puffer P2, vorsichtiges Schwenken und Inkubation für 5 min bei RT
4. Zugabe von 300 µl Puffer P3, vorsichtiges Schwenken und Inkubation für 5 min bei RT
5. Zentrifugation für 20 min bei 13000 Upm und 4 °C
6. Überführung des Überstandes in ein neues 1,5-ml Eppendorfreaktionsgefäß
7. Zentrifugation für 10 min bei 13000 Upm und 4 °C
8. Überführung des Überstandes in ein neues 1,5-ml Reaktionsgefäß unter Zugabe von 0,8 Vol. Isopropanol
9. Zentrifugation für 20 min bei 13000 Upm und 4 °C
10 . Überstand verwerfen und Waschen des Pellets mit 500 µl eiskaltem Ethanol (70 % [v/v], reinst]
11. Zentrifugation für 5 min bei 13000 Upm und 4 °C
12. Dekantieren des Überstandes, Trocknung des Pellets und Aufnahme in 15 µl A. dest.
Die Qualität der isolierten Plasmid-DNA wurde in einem 0,8 %igen Agarosegel (2.8.4)
überprüft.
Puffer P1
50 mM Tris-HCl (pH 8,0) 10 ml (1 M Stammlösung)
10 mM EDTA (pH 8,0) 4 ml (0,5 M Stammlösung)
100 µg/ml RNaseA 2 ml (10 mg/ml Stammlösung)
A. dest. ad 200 ml
Die Zugabe der RNaseA erfolgte nach dem Autoklavieren. Der Puffer wurde bei 4 °C gelagert.

Material und Methoden 29
Puffer P2
1 % [w/v] SDS 10 ml (10 % Stammlösung)
200 mM NaOH 2 ml (1 M Stammlösung)
A. dest. ad 100 ml
Der Puffer wurde nicht autoklaviert. Die Lagerung erfolgte bei RT.
Puffer P3
Kaliumacetat 58,88 g
A. dest. ad 100 ml
Der pH-Wert wurde mit Eisessig auf 5,5 eingestellt. Anschließend wurde der Puffer
autoklaviert.
2.8.1.2 Isolierung chromosomaler DNA aus Clostridien
Zur Isolierung von chromosomaler DNA aus C. acetobutylicum, C. beijerinckii und C. kluyveri
wurden jeweils 50 ml CGM (2.3.2) mit 0,1 Vol. einer entsprechenden Vorkultur inokuliert
und bis zu einer OD600 von 1,0 bei 37 °C inkubiert. Im Anschluss folgte die Sedimentation der
Zellen (5000 Upm, 10 min, 4 °C), das Waschen der Zellpellets mit 10 ml Waschpuffer und die
Überführung in 2-ml-Eppendorfreaktionsgefäße. Bis zur Weiterverwendung wurden die
Zellen bei -20 °C gelagert. Die Isolierung der chromosomalen DNA erfolgte nach der
Methode von Bertram (1989):
1. Resuspension der gefrorenen Zellpellets in 1 ml Waschpuffer
2. Zentrifugation bei 6000 g und 4 °C für 5 min
3. Suspendieren des Zellpellets in 1 ml Lysispuffer
4. Zugabe von 100 µl Lysozymlösung (200 mg/ml) und 5 µl RNase-A-Lösung (10 mg/ml)
5. Ansatz schwenken und Inkubation für 30 min bei 37 °C
6. Zugabe von 30 µl SDS-Lösung (20 % [w/v]) und 30 µl Proteinase K (20 mg/ml)
7. Inkubation des Ansatzes für 30-60 min bei 37 °C
8. Zugabe von 1 Vol. Phenol-Chloroform-Isoamylalkohol (PCI, 25:24:1 [v/v])
9. Zentrifugation bei 8000 Upm, 4 °C für 10 min
10. Überführung der oberen, wässrigen Phase in ein neues Eppendorfreaktionsgefäß
11. Zweimalige Wiederholung der Schritte 8-10

Material und Methoden 30
12. Extraktion mit 1 Vol. Chloroform-Isoamylalkohol (24:1 [v/v]) zur Phenolentfernung
13. Zentrifugation bei 8000 Upm, 4 °C für 5 min
14. Abnehmen der oberen, wässrigen Phase und Fällung der DNA mit 0,1 Vol. 3 M Na-
Acetat (pH 5,2) und 2,5 Vol. Ethanol (96 % [v/v], reinst)
15. Inkubation für 5 min bei RT
16. Zentrifugation bei 8000 Upm, 4 °C für 5 min
17. Dekantieren des Überstandes und Trocknung des Pellets
18. Aufnahme des Pellets in 60 µl TE-Puffer
19. Zugabe von 30 µl RNase-A-Lösung (10 mg/ml)
20. Inkubation für 15 min bei 37 °C
21. Zugabe von 30 µl Proteinase K (20 mg/ml)
22. Inkubation über Nacht bei 37 °C
23. Erhöhung des Volumens mit A. dest. auf 400 µl
24. Zugabe von 60 µl 3 M Na-Acetat (pH 5,2)
25. Extraktion mit 1 Vol. Phenol-Chloroform-Isoamylalkohol (Schritte 8-10)
26. Extraktion mit 1 Vol. Chloroform-Isoamylalkohol (Schritte 12 und 13)
27. Fällung der DNA mit 1 Vol. 3 M Na-Acetat (pH 5,2) und 2,5 Vol. Ethanol (96 % [v/v],
reinst)
28. Zentrifugation bei 13000 Upm, 4 °C für 15 min
29. Zweimaliges Waschen des Pellets mit eiskaltem Ethanol (70 % [v/v], reinst)
30. Zentrifugation bei 6000 Upm, 4 °C für 15 min
31. Trocknung und Aufnahme des Pellets in 50 µl TE-Puffer bzw. A. dest.
Die Qualität der DNA wurde anschließend in einem 0,8 % igen Agarosegel (2.8.4) überprüft.
Waschpuffer Lysispuffer
EDTA (0,5 M; pH 8,0) 40 ml NaCl (5 M) 4 ml
Tris-HCl (1 M) 10 ml EDTA (0,5 M; pH 8,0) 20 ml
KCl (1 M) 25 ml A. dest. ad 200 ml
A. dest. ad 200 ml Der pH wurde mit HCl auf 7,5 eingestellt.

Material und Methoden 31
RNase-A-Lösung Lysozym-Lösung
RNase-A 10 mg Lysozym 200 mg
Tris-HCl (1 M; pH 8,0) 0,1 ml A. dest. ad 1 ml
NaCl 9 mg
A. dest. ad 1 ml
Der pH wurde mit HCl auf 7,5 eingestellt.
1 ml Aliquots wurden bei -20 °C gelagert.
Proteinase K TE-Puffer
Protinase K 20 mg EDTA (0,5 M; pH 8,0) 1 ml
A. dest. ad 1 ml Tris-HCl (1 M; pH 8,0) 0,2 ml
Aliquots zu je 30 µl wurden bei -20 °C gelagert. Der pH wurde mit HCl auf 8,0 eingestellt.
2.8.2 PCR-Techniken
Zur Erzeugung von DNA-Fragmenten, die für weitere Klonierungsschritte (2.8.3) benötigt
wurden und zur Verifikation positiver Klone wurde die Polymerase-Kettenreaktion (PCR)
eingesetzt. Die Amplifikation fand in Thermocyclern mit Deckelbeheizung (PCR-Cycler,
Biometra) statt. Standard-PCR-Analysen wurden mit der Pwo DNA-Polymerase (2,5 U/µl)
(Genaxxon biosciene GmbH, Ulm) durchgeführt.
2.8.2.1 Oligonukleotid-Design
Zur Amplifizierung von DNA-Fragmenten wurden spezifisch an die entsprechenden
Zielsequenzen angepasste Oligonukleotide (Tab. 2.4) abgeleitet. Darüberhinaus wurden die
Oligonukleotide derart konstruiert, dass die Amplifikate am 5´- und 3´-Ende über
entsprechende Restriktionsschnittstellen für eine spätere Klonierung verfügten. Zur
Berechnung der Schmelztemperaturen und zur Überprüfung der Ausbildung von Homo- und
Heterodimeren der Oligonukleotide wurden diese mit dem Oligo-Analyzer von IDT
(http://eu.idtdna.com/calc/analyzer) überprüft. Bei Oligonukleotid-Paaren wurde darauf
geachtet, dass sich die Schmelztemperaturen nicht mehr als 3 °C unterschieden.

Material und Methoden 32
2.8.2.2 Standard-PCR
Ein Standard-PCR Reaktionsansatz setzte sich wie folgt zusammen:
Template DNA 10-100 ng
dNTPs (10 mM) 1 µl
Fw-Primer (10 µM) 1 µl
Rev-Primer (10 µM) 1 µl
10 x Pwo PCR-Puffer (komplett) 5 µl
Pwo DNA-Polymerase (2,5 U/µl) 1 µl
A. dest. ad 50 µl
Nachfolgendes PCR-Programm wurde durchgeführt:
Denaturierung 94 °C 2 min 1x
Denaturierung 94 °C 30 s
Annealing Tm(Primer)-3 °C 30 s 30x
Elongation 72 °C 1 min/kBp
Elongation 72 °C 5 min 1x
Lagerung 4 °C ∞ unendlich
2.8.2.3 „Colony“-PCR
Zur Verifikation von positiven C. acetobutylicum-Klonen wurden entsprechende
Einzelkolonien mit einer sterilen Impföse von RCA-Platten entnommen und in 25 µl sterilem
A. dest. resuspendiert, bei 99 °C für 10 min aufgekocht und die Zelltrümmer sedimentiert
(13000 Upm, 1 min, 4 °C). Anschließend konnte 1 µl des zellfreien Überstands für einen
25 µl-Standard-PCR-Reaktionsansatz (2.8.2.2) eingesetzt werden.
2.8.3 Enzymatische Modifikation von DNA
2.8.3.1 Restriktion von DNA
Zur Erzeugung von DNA-Fragmenten mit definierten Enden wurde die DNA mit
nachfolgenden Restriktionsendonukleasen (Tab. 2.6) behandelt. Die erzeugten linearen
DNA-Fragmente wurden zum einen für eine Agarosegelelektrophorese (2.8.4) und zum

Material und Methoden 33
anderen für Ligationsreaktionen (2.8.3.3) eingesetzt. Die Verwendung der empfohlenen
Puffer und Inkubationszeiten erfolgte nach Herstellerangaben (Thermo Scientific,
Braunschweig). Das Mindestvolumen des Restriktionsansatzes für den Verdau von 1 µg DNA
betrug 10 µl.
Tab. 2.6: Restriktionsendonukleasen
Restriktionsendonuklease Erkennungssequenz1 Puffersystem
BamHI 5'-G˄GATCC-3' 3'-CCTAG˅G-5'
1x BamHI-Puffer, 1x TangoTM
Cfr9I (XmaI) 5'-C˄CCGGG-3' 3'-GGGCC˅C-5'
1x Cfr9I-Puffer, 1x TangoTM
NheI 5'-G˄CTAGC-3' 3'-CGATC˅G-5'
1x TangoTM
KasI (SspDI) 5'-G˄GCGCC-3' 3'-CCGCG˅G-5'
1x TangoTM
SacI 5'-GAGCT˄C-3'
3'-C˅TCGAG-5' 1x SacI-Puffer, 1x TangoTM
SatI (Fnu4HI) 5'-GC˄NGC-3'
3'-CGN˅CG-5' 1x Puffer G, 1x TangoTM
1 N = A, T, C oder G; ˄˅ Restriktionsschnittstellen
2.8.3.2 Dephosphorylierung von DNA-Fragmenten
Zur Verminderung der Selbstligation enzymatisch hydrolysierter Vektor-DNA wurde diese
mit einer alkalischen Phosphatase (FastAP, Thermo Scientific, Braunschweig) behandelt, was
zu einer Dephosphorylierung der freien 5´-Ende führte. Hierfür wurden 2 U FastAP (1 U/µl)
zu dem Restriktionsansatz gegeben und für 20 min bei 37 °C inkubiert. Im Anschluss daran
wurde die Phosphatase hitzeinaktiviert (75 °C, 5 min) und die DNA mittels „Plasmid PLUS
DNA Purification Mini Prep Kit“ (Genaxxon BioScience GmbH, Ulm) aufgereinigt (2.8.5.2).
2.8.3.3 Ligation von DNA-Fragmenten
Die Ligation von DNA-Fragmenten erfolgte unter Verwendung der T4-DNA-Ligase (1 U/µl)
(Thermo Scientific, Braunschweig) in einem 20 µl-Standardvolumen mit 1x T4-Ligase-Puffer
bei 22 °C für 1 h oder bei 16 °C über Nacht. Für eine optimale Ligationseffizienz wurde ein
molares Verhältnis von Vektor zu Insert von 1:3 gewählt. Der Ligationsansatz konnte
anschließend für die Transformation in CaCl2-kompetente E. coli-Zellen verwendet werden.

Material und Methoden 34
2.8.3.4 in vivo Methylierung von Plasmid-DNA
Für eine Transformation von Plasmid-DNA in C. acetobutylicum müssen die Plasmide zuvor
spezifisch methyliert werden, da diese andernfalls als Fremd-DNA erkannt und abgebaut
werden. Hierfür wurden die entsprechenden Plasmide in E. coli ER2275 pANII transformiert
(2.9.1.2). Dieser Stamm verfügt über eine auf dem Plasmid pANII kodierte Methyltransferase
Φ3T I (B. subtilis Phage), die das gleiche Methylierungsmuster wie C. acetobutylicum
(Cac824I) aufweist (Mermelstein et al., 1992). Die Plasmide wurden erneut isoliert (2.8.1.1),
auf Methylierung mittels Restriktionsverdau mit SatI kontrolliert (2.8.3.1) und anschließend
für die Transformation in C. acetobutylicum (2.9.2.1) eingesetzt.
2.8.4 Agarosegelelektrophorese (Sambrook und Russell, 2001)
Zur qualitativen und quantitativen Beurteilung der DNA diente die horizontale
Agarosegelelektrophorese. Dabei variierten in Abhängigkeit von den DNA-Fragmenten die
Agarosekonzentrationen von 0,8 - 2,0 % (w/v) in 1x TAE-Puffer. Zum Beschweren der Proben
und um eine Markierung der Lauffront zu gewährleisten, wurden die Ansätze mit 0,2 Vol. 6x
Loading Dye versetzt. Weiterhin wurden zur Größenabschätzung linearisierter DNA die
Längenstandards GeneRulerTM 1 kb Ladder bzw. MassRulerTM DNA Ladder Mix (Thermo
Scientific, Braunschweig) mitgeführt. Letzterer ermöglichte zudem eine
Konzentrationsabschätzung der DNA. Die Auftrennung der DNA-Fragmente erfolgte in 1x
TAE-Laufpuffer bei einer konstanten Spannung von 90 V (Power Pack P 25; Biometra,
Göttingen) für ca. 45 min. Zur Visualisierung der DNA kamen zwei Techniken zum Einsatz:
einerseits wurden die Gele in einem Ethidiumbromidbad (1 µg/ml in A. dest.) für 15-30 min
inkubiert und anschließend die Nukleinsäuren mit einer Photodokumentationsanlage
(Gelprint 2000; MWG-Biotech, Ebersberg) bei einer Wellenlänge von 254 nm visualisiert und
dokumentiert; andererseits konnten die Nukleinsäuren durch die Anwendung des
Farbstoffes „GelRed“ (Genaxxon BioScience GmbH, Ulm) im Loading Dye direkt bei einer
Wellenlänge von 312 nm mittels einer Photodokumentationsanlage (DarkHood DH-50;
Biostep GmbH, Barkhardtsdorf) sichtbar gemacht und dokumentiert werden.

Material und Methoden 35
50x TAE-Puffer
Tris 242 g
Eisessig (konz.) 57 ml
EDTA (0,5 M, pH 8,0) 100 ml
A. dest. ad 1000 ml
Der pH-Wert wurde mit 0,1 M HCl auf 7,5 eingestellt.
2.8.5 Reinigung von Nukleinsäuren
2.8.5.1 Extraktion von DNA aus Agarosegelen
Für die Extraktion von DNA-Fragmenten aus Agarosegelen wurde das „Gel extraction Mini
Prep Kit“ (Genaxxon BioScience GmbH, Ulm) verwendet. Die gewünschten DNA-Banden
wurden nach gelelektrophoretischer Auftrennung (2.8.4) unter UV-Licht mit einem Skalpell
ausgeschnitten und nach Herstellerangaben unter Verwendung der entsprechenden Puffer
behandelt. Es folgte die Qualitätsüberprüfung der DNA in einem 0,8 %igem Agarosegel
(2.8.4).
2.8.5.2 Kit-basierte DNA-Aufreinigung
Die Aufreinigung von DNA-Fragmenten aus einem PCR-Ansatz oder nach einem
Restriktionsverdau erfolgte mit dem „Plasmid PLUS DNA Purification Mini Prep Kit“
(Genaxxon BioScience GmbH, Ulm) gemäß den Herstellervorgaben.
2.9 Erzeugung rekombinanter Organismen
2.9.1 DNA-Transfer in E. coli
2.9.1.1 CaCl2-vermittelte Transformation in E. coli
Für die CaCl2-vermittelte Transformation von Vektor-DNA in E. coli-Zellen (Tab. 2.1) wurden
10 µl eines Ligationsansatzes (2.8.3.3) zu 50 µl auf Eis aufgetauter, kompetenter Zellen
gegeben und für 30 min auf Eis inkubiert. Es folgte ein Hitzeschock für 90 s bei 42 °C, eine
anschließende Inkubation für 2 min auf Eis und die Zugabe von 500 µl LB-Medium (2.3.1).
Die Regenerierung der Zellen erfolgte für 1 h bei 37 °C unter Schütteln (180 rpm).

Material und Methoden 36
Abschließend wurden je 50 µl, 100 µl und 150 µl des Transformationsansatzes zur Selektion
auf Antibiotika-haltigen LB-Agarplatten ausplattiert und über Nacht bei 37 °C inkubiert.
2.9.1.2 Transformation von E. coli durch Elektroporation
Die Elektroporation von E. coli Zellen (Tab. 2.1) erfolgte nach der Methode von Dower et al.
(1988) unter Verwendung eines GenePulserIITM (Bio-Rad Laboratories, München). Während
die elektrokompetenten E. coli Zellen auf Eis aufgetaut wurden, wurden Elektroporations-
küvetten mit einem Elektrodenabstand von 0,2 cm bei -20 °C vorgekühlt und die Plasmide
entsprechend ihrer Konzentration 1:5 bzw. 1:10 verdünnt. Die verdünnten Plasmide (2 µl)
wurden zu 40 µl kompetenter Zellen gegeben und der Ansatz in die Elektroporations-
küvetten überführt. Die Elektroporation erfolgte bei einer Kapazität von 25 µF, einem
Widerstand von 200 Ω und einer Spannung von 2,5 kV. Ideale Zeitkonstanten lagen im
Bereich von 4,5 - 5,5 ms. Nach dem Elektroporationsvorgang wurden die Zellen zur
Regeneration in 500 µl LB-Medium aufgenommen und für 1 h bei 37 °C inkubiert. Jeweils
10 µl, 20 µl und 50 µl des Transformationsansatzes wurden auf LB-Agarplatten mit
entsprechenden Medienzusätzen (2.3.3) ausplattiert und über Nacht bei 37 °C inkubiert.
2.9.2 DNA-Transfer in C. acetobutylicum
2.9.2.1 Transformation von C. acetobutylicum durch Elektroporation
Die Transformation methylierter Vektor-DNA (2.8.3.4) in C. acetobutylicum erfolgte nach der
Methode von Mermelstein et al. (1992) unter zu Hilfenahme eines GenePulserIITM (Bio-Rad
Laboratories, München) unter anaeroben Bedingungen in einer Anaerobenwerkbank (MACS-
MG 1000; Meintrup dws, Lähden-Holte). Für die Elektroporation wurden stets frisch
kultivierte Zellen eingesetzt. Hierfür wurden 50 ml CGM mit einer exponentiell wachsenden
CGM-Vorkultur inokuliert (2.5.2) und bis zu einer OD600 von 1,0 bei 37 °C inkubiert. Die
nachfolgenden Schritte wurden auf Eis durchgeführt:
1. Inkubation der Hauptkultur für 30 min auf Eis
2. Zentrifugation für 5 min bei 5000 x g und 4 °C
3. Waschen der Zellen mit 10 ml vorgekühltem Elektroporationspuffer
4. Zentrifugation für 5 min bei 5000 x g und 4 °C
5. Resuspendieren des Zellpellets in 0,5-2,0 ml Elektroporationspuffer

Material und Methoden 37
6. Überführung von 400 µl kompetenter Zellen und 25 µl Plasmid-DNA in vorgekühlte Elektroporationsküvetten (0,4 cm Elektrodenabstand)
7. Inkubation des Ansatzes für 5 min auf Eis
8. Elektroporation: 50 µF, 600 Ω und 1,8 kV (Zeitkonstanten: 10 - 20 ms)
9. Zugabe von 1 ml CGM und Überführung des Ansatzes in 2-ml Schraubdeckelröhrchen
10. Regeneration der Zellen für ca. 4 h bei 37 °C
11. Ausplattieren von 300 µl Zellsuspension und Auftropfen von max. 500 µl auf RCA-Platten mit entsprechenden Medienzusätzen
12. Inkubation für mindestens 48 h bei 37°C
Natriumphosphat-Puffer (pH 7,4)1 Elektroporationspuffer2
NaH2PO4 (200 mM) 22,6 ml Saccharose-Lsg. (270 mM) 10 ml
Na2HPO4 (200 mM) 77,4 ml Natriumphosphat-Puffer (pH 7,4) 150 µl
1 Der Puffer wurde autoklaviert. 2 Der Elektroporationspuffer wurde steril filtriert.
2.10 Arbeiten mit Proteinen
2.10.1 Zellaufschluss von C. acetobutylicum mittels Ultraschall
Für den Nachweis der heterologen Proteinexpression wurden die Zellen der rekombinanten
C. acetobutylicum Stämme mittels Ultraschall (UP200S Hielscher, Teltow) aufgeschlossen.
Dazu wurde eine 200-ml CGM-Hauptkultur mit 0,1 Vol. einer entsprechenden 10-ml CGM-
Übernachtkultur inokuliert und bei 37 °C bis zu einer OD600 von 2,0 - 3,0 inkubiert. Die Zellen
wurden unter Antibiotika-Zusatz (2.3.3) kultiviert. Die Zellernte erfolgte durch Zentrifugation
(Sorvall RC6+, Rotor F12S-6x500 LEX) für 10 min bei 10000 Upm und 4 °C. Anschließend
wurde das Zellpellet in 5 ml Puffer W (2.10.2) aufgenommen und bis zur weiteren
Verwendung bei -20 °C gelagert. Nach dem Auftauen der Zellen wurden diese in einen
Glasmessbecher überführt, auf Eis gelagert und für die Ultraschallbehandlung eingesetzt.
Der Aufschluss der Zellen erfolgte mittels Ultraschall und einer S2 bzw. S3 Sonotrode bei
einem Zyklus von 3 min Beschallung (Amplitude: 50 %, cycle: 0,5) und 1 min Pause für eine
Dauer von 30 min. Der Erfolg des Aufschlusses wurde mittels Lichtmikroskopie überprüft. Es
folgte die Sedimentation der Zelltrümmer (13000 Upm, 4 °C, 10 min). Der geklärte
Überstand wurde anschließend für eine Affinitätschromatographie eingesetzt (2.10.2).

Material und Methoden 38
2.10.2 Proteinaufreinigung mittels Affinitätschromatografie
Die Aufreinigung rekombinanter Strep-tagII Fusionsproteine aus geklärten Lysaten von
C. acetobutylicum (2.10.1) erfolgte chromatografisch über eine mit Strep-Tactin
immobilisierte Sepharose-Säule (IBA, Göttingen). Der aus dem Ultraschallaufschluss (2.10.1)
gewonnene Überstand wurde auf die Säule gegeben und der daraus resultierende
Durchfluss aufgefangen. Es folgte zur Entfernung überschüssiger Proteine das fünfmalige
Waschen der Säule mit jeweils 1 ml Puffer W und das Sammeln der einzelnen
Waschfraktionen in 1,5-ml Eppendorfreaktionsgefäßen. Die aufgrund des Strep-tagII
zurückgehaltenen Fusionsproteine wurden durch Zugabe von 6x 0,5 ml Puffer E von der
Säule eluiert und in 1,5-ml Eppendorfreaktionsgefäßen aufgefangen. Die Proteinfraktionen
wurden bei -20 °C gelagert und zur Konzentrationsbestimmung (2.10.3) und für die SDS-
PAGE (2.10.4) eingesetzt. Zur Regeneration der Säule wurde selbige mit 10 ml Puffer R
behandelt, erkennbar durch einen Farbumschlag der Säule von weiß zu orange-rot. Die
anschließende Zugabe von 10 ml Puffer W führte zu einer Entfärbung der Säule. Die so
behandelte Säule konnte mit 1 ml Puffer W beschichtet bei 4 °C bis zur weiteren
Verwendung gelagert werden.
Puffer W1 Puffer E
Tris 100 mM Tris 100 mM
NaCl 100 mM NaCl 100 mM
EDTA (pH 8,0) 0,5 mM EDTA (pH 8,0) 0,5 mM
A. dest. ad 300 ml Desthiobiotin 2,5 mM
1 Der pH wurde mit 1 M HCl auf 8,0 eingestellt. Der Puffer wurde anschließend autoklaviert
und bei RT gelagert.
Puffer R
Tris 100 mM
NaCl 100 mM
EDTA (pH 8,0) 0,5 mM
HABA 1,0 mM

Material und Methoden 39
2.10.3 Konzentrationsbestimmung von Proteinen (Bradford, 1976)
Für die Bestimmung der Proteinkonzentration in wässrigen Lösungen wurde die Methode
nach Bradford (1976) angewandt. Es wurden 1 ml Bradford-Reagenz mit 50 µl Proteinprobe
(gegebenenfalls verdünnt) versetzt, gut durchmischt und für 5 min bei RT inkubiert.
Anschließend erfolgte die Messung der Absorption bei 595 nm in einer Plastikküvette
(Schichtdicke: 1 cm) in einem Photometer (WPA Biowave II) gegen einen Blindwert (1 ml
Bradford-Reagenz + 50 µl A. dest.). Die Berechnung der Proteinkonzentration erfolgte mit
Hilfe einer mit Rinderserumalbumin (BSA) erstellten Eichreihe (0 - 0,15 mg/ml).
Bradford-Reagenz
Brilliant-Blau G-250 70 ml
Ethanol (96 % [v/v]) 50 ml
H3PO4 (85 % [v/v]) 100 ml
A. dest. ad 1000 ml
Das Reagenz wurde lichtgeschützt bei RT gelagert.
2.10.4 Eindimensionale SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Zur Auftrennung von Proteinen in Abhängigkeit von ihrem Molekulargewicht wurde die
denaturierende SDS-PAGE nach Laemmli (1970, mod.) durchgeführt. Die Bindung von
negativ geladenen SDS-Molekülen („Sodium Dodecyl-Sulfate“) an die hydrophoben Regionen
der Proteine führt zu einer Überlagerung der nativen Proteinladung, sodass alle Proteine
eine negative Ladung erhalten. Die Durchführung der SDS-PAGE erfolgte in entsprechenden
Gelkammern (Biometra, Göttingen) unter Verwendung der in Tabelle 2.7 dargestellten
Gellösungen. Das Sammelgel, mit einem Acrylamidgehalt von 4 % [w/v] und neutralem pH-
Wert, diente der Bündelung der Proteine, welche anschließend im Trenngel
(Acrylamidgehalt 12 % [w/v]) separiert wurden.
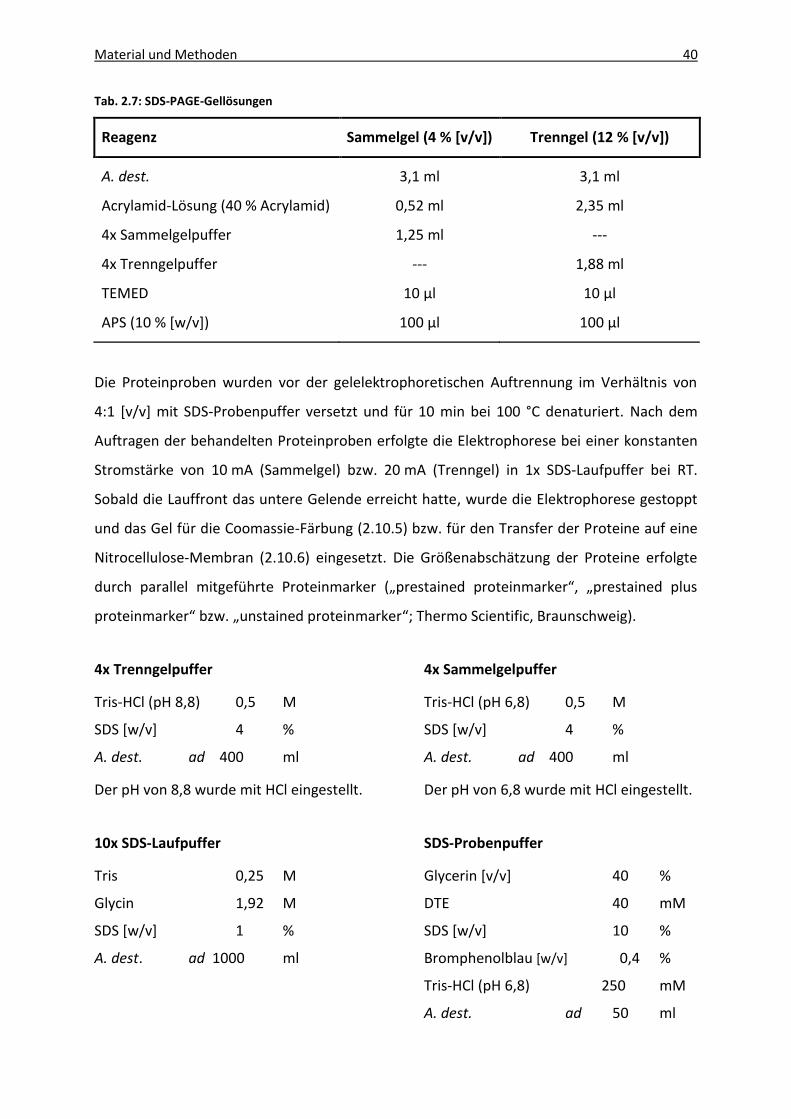
Material und Methoden 40
Tab. 2.7: SDS-PAGE-Gellösungen
Reagenz Sammelgel (4 % [v/v]) Trenngel (12 % [v/v])
A. dest. 3,1 ml 3,1 ml
Acrylamid-Lösung (40 % Acrylamid) 0,52 ml 2,35 ml
4x Sammelgelpuffer 1,25 ml ---
4x Trenngelpuffer --- 1,88 ml
TEMED 10 µl 10 µl
APS (10 % [w/v]) 100 µl 100 µl
Die Proteinproben wurden vor der gelelektrophoretischen Auftrennung im Verhältnis von
4:1 [v/v] mit SDS-Probenpuffer versetzt und für 10 min bei 100 °C denaturiert. Nach dem
Auftragen der behandelten Proteinproben erfolgte die Elektrophorese bei einer konstanten
Stromstärke von 10 mA (Sammelgel) bzw. 20 mA (Trenngel) in 1x SDS-Laufpuffer bei RT.
Sobald die Lauffront das untere Gelende erreicht hatte, wurde die Elektrophorese gestoppt
und das Gel für die Coomassie-Färbung (2.10.5) bzw. für den Transfer der Proteine auf eine
Nitrocellulose-Membran (2.10.6) eingesetzt. Die Größenabschätzung der Proteine erfolgte
durch parallel mitgeführte Proteinmarker („prestained proteinmarker“, „prestained plus
proteinmarker“ bzw. „unstained proteinmarker“; Thermo Scientific, Braunschweig).
4x Trenngelpuffer 4x Sammelgelpuffer
Tris-HCl (pH 8,8) 0,5 M Tris-HCl (pH 6,8) 0,5 M
SDS [w/v] 4 % SDS [w/v] 4 %
A. dest. ad 400 ml A. dest. ad 400 ml
Der pH von 8,8 wurde mit HCl eingestellt. Der pH von 6,8 wurde mit HCl eingestellt.
10x SDS-Laufpuffer SDS-Probenpuffer
Tris 0,25 M Glycerin [v/v] 40 %
Glycin 1,92 M DTE 40 mM
SDS [w/v] 1 % SDS [w/v] 10 %
A. dest. ad 1000 ml Bromphenolblau [w/v] 0,4 %
Tris-HCl (pH 6,8) 250 mM
A. dest. ad 50 ml

Material und Methoden 41
Ammoniumpersulfatlösung (APS)
Ammoniumpersulfat 10 %
A. dest. ad 100 ml
2.10.5 Kolloidale Coomassie-Färbung
SDS-Polyacrylamidgele (2.10.4) wurden nach der Methode von Neuhoff et al. (1988)
behandelt und zunächst für mindestens 1 h in einer Fixierlösung unter Schwenken inkubiert.
Im Anschluss daran wurde die Fixierlösung entfernt und durch die kolloidale Coomassie-
Lösung ersetzt, in welcher die Gele über Nacht bei RT unter Schwenken inkubiert wurden.
Durch mehrmaliges Waschen mit destilliertem Wasser konnten die Gele anschließend
entfärbt werden, bis ein ausreichender Kontrast zwischen Hintergrund und Probe vorlag.
Kolloidales Coomassie Fixierlösung
Coomassie Brilliant Blue G-250 0,75 g Essigsäure 10 %
o-Phosphorsäure (85 %) 15 ml Ethanol (96 % [v/v], reinst) 50 %
Ammoniumsulfat 75 g A. dest. ad 1000 ml
Methanol 250 ml
A. dest. ad 1000 ml
2.10.6 Transfer von Proteinen auf Membranen (Western Blot)
Bei einem Western Blot (Towbin et al., 1979) werden die Proteine von einem
Polyacrylamidgel auf eine Nitrocellulose-Membran übertragen, um diese mit
immunologischen Methoden spezifisch detektieren zu können. Der Transfer der
aufgereinigten Proteinproben (2.10.2) auf eine Nitrocellulose-Membran wurde nach einer
gelelektrophoretischen Auftrennung (2.10.4) in einer Semi-Dry-Blottingapparatur (Biometra,
Göttingen) durchgeführt. Hierfür wurden zuvor zwei Lagen extra dickes Blottingpapier (Bio-
Rad Laboratories, Hercules) und die Nitrocellulose-Membran (0,2 µm, Biometra) auf die
Gelgröße (abzüglich des entfernten Sammelgels) zugeschnitten und mit Transferpuffer
befeuchtet. Auf die Anodenseite der Blottingapparatur wurde luftblasenfrei eine Lage
Blottingpapier gelegt, gefolgt von der Nitrocellulose-Membran, dem ebenfalls mit

Material und Methoden 42
Transferpuffer benetztem Polyacrylamidgel und der zweiten Lage Blottingpapier.
Überschüssiger Puffer wurde mit einem saugfähigen Papier entfernt. Durch Auflegen der
Kathodenplatte wurde die Blotting-Kammer geschlossen und der Transfer bei einer
konstanten Stromstärke von 5 mA/m2 durchgeführt. Sobald die Spannung auf die Hälfte
ihres Ausgangswertes absank, wurde der Blotvorgang beendet.
Transferpuffer
Tris 125 mM
Glycin 192 mM
Ethanol (96 % [v/v], reinst) 20 %
A. dest. ad 1000 ml
Der Puffer wurde bei 4 °C gelagert.
2.10.7 Detektion von Strep-tag II Fusionsproteinen
Die Detektion der rekombinanten Fusionsproteine erfolgte über einen Strep-Tactin-
Antikörper, welcher gegen das Strep-tag Antigen der Fusionsproteine gerichtet und an eine
alkalische Phosphatase gekoppelt ist. Durch die Umwandlung der Substrate
Nitroblautetrazoliumchlorid (NBT) und 5-Bromo-4-Chloro-3-Indoyl-Phosphat (BCIP) kommt
es aufgrund der gekoppelten alkalischen Phosphatase zu einer Farbreaktion.
Nach Beendigung des Blotvorgangs (2.10.6) wurde die Membran in 50 ml Blocking-Puffer
über Nacht bei 4 °C inkubiert. Im Anschluss daran wurde die Membran zweimal mit 1x TGST-
Puffer für jeweils 10 min gewaschen und mit 50 ml 1x TGST-Puffer und 5 µl Strep-Tactin-AP-
Konjugatlösung (IBA, Göttingen) für 45 min bei RT unter Schwenken inkubiert. Zur
Entfernung von ungebundenen Antikörpern wurde die Membran erneut dreimal mit 50 ml
1x TGST-Puffer für jeweils 10 min gewaschen. Anschließend wurde die Membran mit 5 ml
1x AP-Puffer und 45 µl NBT/BCIP-Lösung (Roche, Mannheim) im Dunkeln und bei RT
gelagert, bis die Signale erkennbar waren. Die Farbreaktion konnte bei ausreichender
Intensität der Signale durch Spülen der Membran mit destilliertem Wasser abgestoppt und
die Membran getrocknet und fotodokumentiert werden.

Material und Methoden 43
10x TGST-Puffer 10x AP-Puffer
Tris 1,21 g Tris 1,21 g
NaCl 3,55 g NaCl 5,7 g
Tween20* 0,5 g MgCl2 0,47 g
A. dest. ad 100 ml A. dest. ad 100 ml
*Zugabe nach dem Autoklavieren Der pH wurde mit HCl auf 9,5 eingestellt.
Der pH wurde mit HCl auf 8,0 eingestellt.
Blocking-Puffer
Magermilchpulver 2,5 g
BSA 0,5 g
1x TGST-Puffer ad 50 ml
Dem Puffer wurde eine Spatelspitze Avidin zugegeben.
2.11 Bezugsquellen
Chemikalien ohne zusätzlichen Vermerk wurden mit dem Reinheitsgrad „reinst“ oder „zur
Analyse“ von den Firmen AppliChem (Darmstadt), Carl Roth GmbH & Co.KG (Karlsruhe),
Diagonal (Münster), Merck KG (Darmstadt) und Sigma-Aldrich (Taufkirchen) bezogen. Gase
wurden von der Westfalen AG (Münster) geliefert.
Tab. 2.8: Bezugsquellen für Chemikalien
Bezugsquelle Produkt
AppliChem GmbH, Darmstadt
Acrylamid, Agarose, Ammoniumsulfat,
Ammoniumhydroxid, Ampicillin, Asparagin,
Avidin, β-NADH, BSA, DNase I, Erythromycin,
Ethidiumbromid, Glukose, Glycin, Lysozym,
Magermilchpulver, Mannose, MES, Phenol-
Chloroform-Isoamylalkohol, Proteinase K,
RNase A, SDS, TEMED, Tris,
Biolab Inc., Lawrenceville, USA Hefeextrakt, Trypton
Carl Roth GmbH & Co. KG, Karlsruhe Brilliant Blue G-250, Tris, Vanillin, Xylose
Chemos GmbH, Regenstauf Thiamphenicol

Material und Methoden 44
Tab 2.8: Bezugsquellen für Chemikalien (Fortsetzung).
Bezugsquelle Produkt
Difco Laboratories, Hamburg Agar-Agar
Genaxxon Bioscience GmbH, Ulm GelRed
IBA GmbH, Göttingen Desthiobiotin, Strep-Tactin-Sepharose,
Strep-Tactin AP-Konjugatlösung
Merck KGaA, Darmstadt Chloramphenicol, Magnesiumsulfat,
Mangansulfat, PABA
Oxoid GmbH, Wesel Reinforced Clostridial Agar (RCA)
Peqlab Biotechnologie GmbH, Erlangen Pwo-Polymerase I
Roche Diagnostics GmbH, Mannheim NBT/ BCIP, Hexokinase/ Glucose-6-
Phosphat-Dehydrogenase
Sigma-Aldrich Chemie GmbH, Taufkirchen 1,4-Butandiol; 2,3-Butandiol,Oligonukleotide
1,4-Butandiol-d8, γ-Hydroxybutyrat-d6
Tab. 2.9: Bezugsquellen für Geräte und Materialien
Bezugsquelle Geräte/ Materialien
Agilent Technologies GmbH, Böblingen Agilent 6890, Agilent 7890A, EZ Chrom Elite,
Amersham Buchler GmbH, Braunschweig Photometer Ultrospec 3000
Biochrom, Camebridge, UK Photometer WPA Biowave II
Biometra GmbH, Göttingen Agarosegelelektrophorese-Kammern,
Blotting-Apparatur, Nitrocellulosemembran,
PCR-Cykler, Power Pack P25,
Biorad GmbH, München Gene PulserTM II, Blottingpapier
Biostep GmbH, Jahnsdorf Geldokumentationsanalge Dark Hood DH+50
Braun AG, Melsungen sterile Kanülen
Eppendorf Research AG, Hamburg Reaktionsgefäße, Thermomixer Comfort
Genaxxon Bioscience GmbH, Ulm Gel Extraction Kit, Plasmid Plus DNA
Purification Mini Prep Kit
Heraeus-Holding GmbH, Hanau Tischzentrifuge Biofuge fresco
Hielscher Ultrasonic GmbH, Teltow Hielscher UP200S
Kendro Laboratory Products GmbH,
Langenselbold
Sorvall RC 6C Plus Zentrifuge

Material und Methoden 45
Tab. 2.9: Bezugsquellen für Geräte und Materialien (Fortsetzung).
Bezugsquelle Geräte/ Materialien
Macherey-Nagel GmbH & Co. KG, Düren ALUGRAM SIL G DC-Fertigfolie, Kieselgel
0,2 mm
Megazyme International Ltd., Wicklow Laktat-Kit
Meintrup DWS Laborgeräte GmbH, Lähden-
Holte
MACS-MG-500/MG-1000-
anaerobic workstation
Memmert GmbH & Co.KG, Schwabach Brutschrank
Mettler-Toledo GmbH, Giessen pH Meter SevenEasy
Müller&Krempel AG, Bülach Müller&Krempel Serumflaschen
MWG-Biotech AG, Ebersberg Geldokumentationsanalage TFP-M/WL
Ochs Laborfachhandel e. K., Bovenden Hungateröhrchen
Olympus Deutschland GmbH, Hamburg Mikroskop CH20
Peqlab Biotechnologie GmbH, Erlangen Elektroporationsküvetten, peqGold Gel
Extraction Kit
Sartorius AG, Göttingen Biostat B Plus Twin Fermenter
Sarstedt AG & Co., Nürnberg 50-ml Röhrchen, 0,2 µm Sterilfilter,
Plastikküvetten, Einmal-Impföse, Einmal-
Spatel
Schütt Labortechnik GmbH, Göttingen Wasserbad
Thermo Scientific Inc., Braunschweig dNTPs, Fast-AP, GeneRuler 1kB DNA Ladder,
Mass Ruler (SM403), Restriktionsenzyme,
Protein Molecular Weight Marker (SM0431,
SM0672), T4-DNA-Ligase

Ergebnisse 46
3. Ergebnisse
Zur Erzeugung und Optimierung biotechnologischer Produktionsstämme werden
unterschiedliche Strategien angewandt, welche Veränderungen in enzymatischen und
regulatorischen Funktionen im jeweiligen Produktionsstamm bewirken (Steinbüchel & Lütke-
Eversloh, 2003; Atsumi et al., 2008; Keasling, 2010). Diese sind auch unter dem Begriff
„Metabolic Engineering“ zusammengefasst (Stephanopoulos et al., 1998). Dementsprechend
war das Ziel dieser Arbeit die Veränderung des Metabolismus des obligaten Anaerobiers
C. acetobutylicum zur Produktion von 1,4-Butandiol (1,4-BDO).
Die Etablierung des neuen Biosyntheseweges von 1,4-BDO erfolgte durch eine
plasmidbasierte, heterologe Expression der bifunktionellen 4-Hydroxybutyryl-CoA
Dehydratasen/Vinylacetyl-CoA-∆-Isomerasen (4HBD) aus C. aminobutyricum, C. beijerinckii
bzw. C. kluyveri im Wirtsorganismus C. acetobutylicum. Neben unterschiedlichen Strategien
zur Produktionssteigerung konnte ebenso die Verwertung verschiedener Kohlenstoffquellen
für die 1,4-BDO-Produktion gezeigt werden. So ist der Einsatz von
Lignocellulosehydrolysaten als kostengünstiges, nicht mit der Lebensmittelindustrie
konkurrierendes Substrat für die 1,4-BDO-Produktion möglich. Darüber hinaus erfolgte
neben dem GC/MS-Nachweis von 1,4-BDO auch die Validierung mittels einer auf
Dünnschichtchromatografie basierten Nachweismethode.
3.1 1,4-Butandioltoleranz von C. acetobutylicum ATCC 824
Zur Vorbereitung dieser Arbeit erfolgte die Überprüfung der Toleranz von C. acetobutylicum
gegenüber der zu produzierenden Verbindung 1,4-BDO. Chemische Verbindungen, die von
biotechnologischen Produktionswirten heterolog erzeugt werden, können einen
inhibierenden Effekt auf den jeweiligen Produktionsstamm ausüben und dadurch die
Ausbeuten negativ beeinflussen (Maiorella et al., 1983).
Bisher ist nicht bekannt, dass die hochreduzierte Verbindung 1,4-BDO natürlich synthetisiert
wird (Yim et al., 2011). Da die Produkttoleranz ein wesentlicher Faktor bei der
Realisierbarkeit von biotechnologischen Produktionsprozessen ist (Burk, 2010), wurde das
Wachstum des Wirtsstamms bei verschiedenen Konzentrationen von 1,4-BDO dokumentiert.
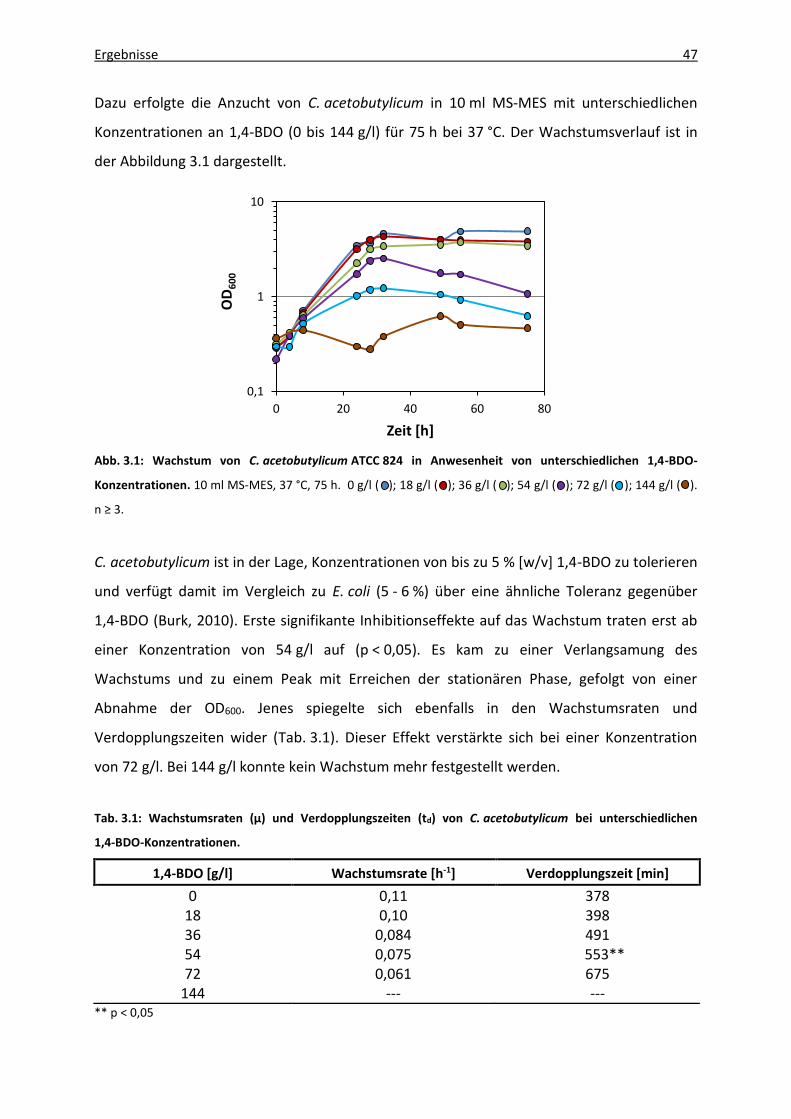
Ergebnisse 47
Dazu erfolgte die Anzucht von C. acetobutylicum in 10 ml MS-MES mit unterschiedlichen
Konzentrationen an 1,4-BDO (0 bis 144 g/l) für 75 h bei 37 °C. Der Wachstumsverlauf ist in
der Abbildung 3.1 dargestellt.
Abb. 3.1: Wachstum von C. acetobutylicum ATCC 824 in Anwesenheit von unterschiedlichen 1,4-BDO-
Konzentrationen. 10 ml MS-MES, 37 °C, 75 h. 0 g/l ( ); 18 g/l ( ); 36 g/l ( ); 54 g/l ( ); 72 g/l ( ); 144 g/l ( ).
n ≥ 3.
C. acetobutylicum ist in der Lage, Konzentrationen von bis zu 5 % [w/v] 1,4-BDO zu tolerieren
und verfügt damit im Vergleich zu E. coli (5 - 6 %) über eine ähnliche Toleranz gegenüber
1,4-BDO (Burk, 2010). Erste signifikante Inhibitionseffekte auf das Wachstum traten erst ab
einer Konzentration von 54 g/l auf (p < 0,05). Es kam zu einer Verlangsamung des
Wachstums und zu einem Peak mit Erreichen der stationären Phase, gefolgt von einer
Abnahme der OD600. Jenes spiegelte sich ebenfalls in den Wachstumsraten und
Verdopplungszeiten wider (Tab. 3.1). Dieser Effekt verstärkte sich bei einer Konzentration
von 72 g/l. Bei 144 g/l konnte kein Wachstum mehr festgestellt werden.
Tab. 3.1: Wachstumsraten (µ) und Verdopplungszeiten (td) von C. acetobutylicum bei unterschiedlichen
1,4-BDO-Konzentrationen.
1,4-BDO [g/l] Wachstumsrate [h-1] Verdopplungszeit [min]
0 0,11 378 18 0,10 398 36 0,084 491 54 0,075 553** 72 0,061 675
144 --- --- ** p < 0,05
0,1
1
10
0 20 40 60 80
OD
60
0
Zeit [h]
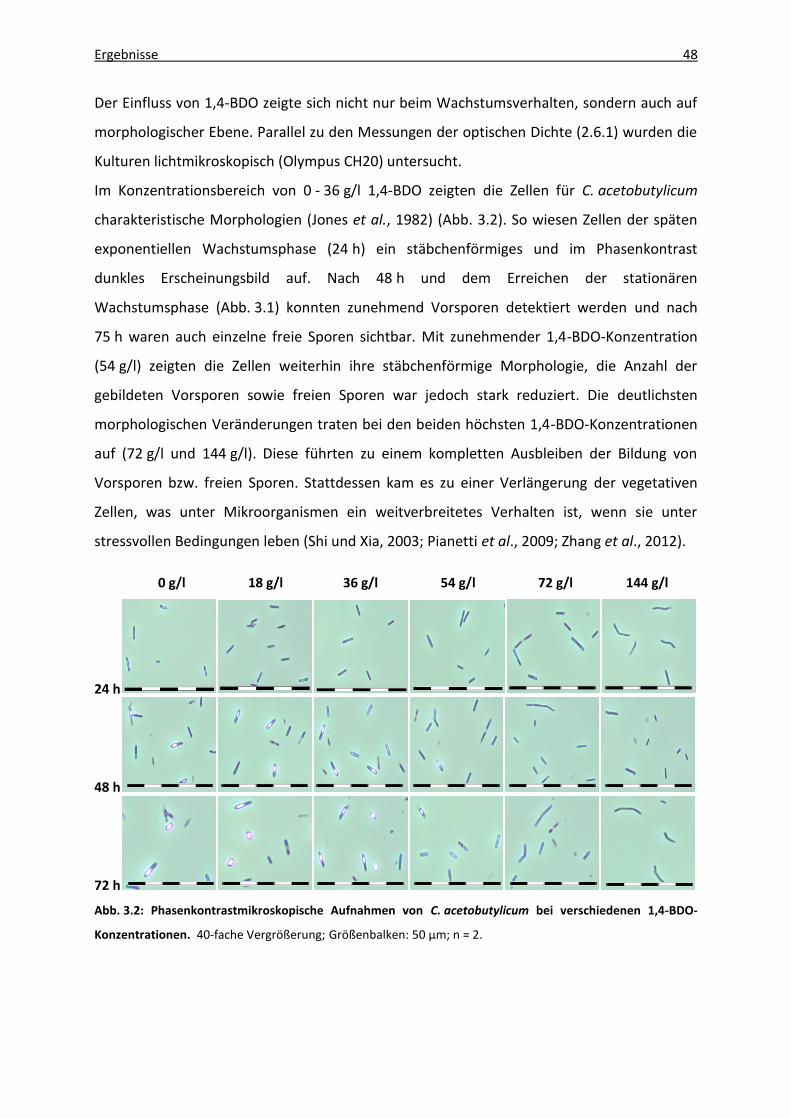
Ergebnisse 48
Der Einfluss von 1,4-BDO zeigte sich nicht nur beim Wachstumsverhalten, sondern auch auf
morphologischer Ebene. Parallel zu den Messungen der optischen Dichte (2.6.1) wurden die
Kulturen lichtmikroskopisch (Olympus CH20) untersucht.
Im Konzentrationsbereich von 0 - 36 g/l 1,4-BDO zeigten die Zellen für C. acetobutylicum
charakteristische Morphologien (Jones et al., 1982) (Abb. 3.2). So wiesen Zellen der späten
exponentiellen Wachstumsphase (24 h) ein stäbchenförmiges und im Phasenkontrast
dunkles Erscheinungsbild auf. Nach 48 h und dem Erreichen der stationären
Wachstumsphase (Abb. 3.1) konnten zunehmend Vorsporen detektiert werden und nach
75 h waren auch einzelne freie Sporen sichtbar. Mit zunehmender 1,4-BDO-Konzentration
(54 g/l) zeigten die Zellen weiterhin ihre stäbchenförmige Morphologie, die Anzahl der
gebildeten Vorsporen sowie freien Sporen war jedoch stark reduziert. Die deutlichsten
morphologischen Veränderungen traten bei den beiden höchsten 1,4-BDO-Konzentrationen
auf (72 g/l und 144 g/l). Diese führten zu einem kompletten Ausbleiben der Bildung von
Vorsporen bzw. freien Sporen. Stattdessen kam es zu einer Verlängerung der vegetativen
Zellen, was unter Mikroorganismen ein weitverbreitetes Verhalten ist, wenn sie unter
stressvollen Bedingungen leben (Shi und Xia, 2003; Pianetti et al., 2009; Zhang et al., 2012).
0 g/l 18 g/l 36 g/l 54 g/l 72 g/l 144 g/l
24 h
48 h
72 h
Abb. 3.2: Phasenkontrastmikroskopische Aufnahmen von C. acetobutylicum bei verschiedenen 1,4-BDO-
Konzentrationen. 40-fache Vergrößerung; Größenbalken: 50 µm; n = 2.

Ergebnisse 49
3.2 Nachweismethoden für 1,4-Butandiol
Nachdem gezeigt werden konnte, dass C. acetobutylicum in der Lage ist, 1,4-BDO-
Konzentrationen von mindestens 54 g/l zu tolerieren, sollte im nächsten Schritt eine
geeignete Nachweismethode für 1,4-BDO etabliert werden. Hierfür wurden zwei
verschiedene Ansätze erprobt.
3.2.1 Dünnschichtchromatografie-basierte Nachweismethode
Zu Beginn der Arbeiten sollte eine möglichst schnelle, einfache und kostengünstige Methode
zum Nachweis von 1,4-BDO entwickelt werden. Hierfür wurde eine auf
Dünnschichtchromatografie-basierende Nachweismethode evaluiert (2.7). Die auf eine mit
Kieselgel beschichtete DC-Fertigfolie (Macherey-Nagel) aufgetragenen Proben wurden nach
entsprechender Auftrennung mit einem Vanillinreagenz zur Visualisierung der Spots
besprüht und zeigten eine für 1,4-BDO spezifische Pinkfärbung (Abb. 3.3). Ähnliche
Moleküle, wie das 1,3-Propandiol und 2,3-Butandiol, wiesen unter gleichen Bedingungen
eine Lila- bzw. Blaufärbung auf (Anand und Saxena, 2012).
(A) (B)
Abb. 3.3: Dünnschichtchromatogramm von 1,4-BDO. Zur Visualisierung wurde ein Vanillin-Sprühreagenz
angewandt. (A) 1,4-BDO und 2,3-BDO (Sigma-Aldrich). (B) 1,4-BDO (Sigma-Aldrich) gemischt mit
C. acetobutylicum-Kulturüberstand (48 h). K, Kontrolle, Kulturüberstand ohne Zusatz von 1,4-BDO.
1 10 100 1 10 mM mM mM mM mM
10 100 1000 K mM mM mM
1,4-BDO
2,3-BDO
1,4-BDO

Ergebnisse 50
Mit Hilfe der Dünnschichtchromatografie (DC) war es also möglich, 1,4-BDO aus einem
Gemisch aufzutrennen und zu visualisieren. Dabei ließ sich 1,4-BDO durch eine Vanillin-
Färbelösung spezifisch anfärben (rosa Spots), während strukturell ähnliche Moleküle, wie
2,3-BDO, eine charakteristische Blaufärbung zur Folge hatten. Die Intensität der 1,4-BDO-
Färbung war von der eingesetzten Konzentration abhängig (Abb. 3.3, A; A1 und A2). Bis zu
10 mM waren die Spots mit bloßen Augen noch feststellbar, bei niedrigeren Konzentrationen
(< 10 mM) erschwerte die Hintergrundfärbung die optische Beurteilung.
Weiterhin wurde die Anwendbarkeit dieser Methode zur Detektion von 1,4-BDO in
Kulturüberständen überprüft. Dazu wurden die Überstände einer 48-stündigen
C. acetobutylicum Kultur mit unterschiedlichen Konzentrationen 1,4-BDO versetzt und über
DC aufgetrennt. Die Visualisierung erfolgte ebenfalls über die Vanillin-Färbelösung. Es zeigte
sich, dass die im Überstand enthaltenen Stoffwechselprodukte (Aceton, Butanol und
Ethanol) keinen Einfluss auf die Auftrennung oder die Anfärbung hatten (Abb. 3.3, B; A2).
Da in den Arbeiten von Yim et al. (2011) und Hwang et al. (2014) eingangs deutlich geringere
1,4-BDO-Konzentrationen (0,6 mM bzw. 0,3 mM) erzielt wurden, ist davon auszugehen, dass
die nachzuweisenden Konzentrationen in C. acetobutylicum in einem ähnlichen Bereich sein
werden. Aus diesem Grund musste eine sensitivere Nachweismethode etabliert werden.
3.2.2 GC/MS-basierte Nachweismethode
1,4-BDO ist aufgrund seiner endständigen Hydroxylgruppen ein sehr polares Molekül, was
seine gaschromatografische Auftrennung deutlich erschwert (pers. Mitteilung, Dr. Rentsch).
Aus diesem Grund wurde 1,4-BDO zunächst mittels N-Methyl-N-tert-butyldimethylsilyl-
tifluoracetamid (MBDSTFA, Macherey-Nagel) silyliert (Abb. 3.4) (2.6.6.2). Durch den
Austausch der endständigen Hydroxylgruppen durch Silylgruppen wechselt der polare
Charakter des Moleküls zu einem apolaren, gleichzeitig wird die Molekülstabilität erhöht und
so eine bessere gaschromatografische Auftrennung ermöglicht (Corey und Venkateswarlu,
1972).

Ergebnisse 51
Abb. 3.4: Schematische Darstellung von 1,4-BDO (links) und GHB (rechts) nach Silylierungsreaktion mit
MBDSTFA. Erläuterungen siehe Text.
Die Normierung der Proben erfolgte über einen isotopenmarkierten internen Standard (IS),
einem 8-fach-deuterierten 1,4-BDO (1,4-BDO-d8, Sigma-Aldrich), in dem alle
Wasserstoffatome im Molekül durch Deuteriumatome ersetzt sind. Die chemischen
Eigenschaften wurden dabei jedoch nicht verändert, sodass sich der IS analytisch ähnlich zu
dem zu untersuchenden 1,4-BDO verhielt. Über einen massenselektiven Detektor konnte
zwischen deuterierten und undeuterierten Verbindungen unterschieden werden. Die
Unterscheidung beider Verbindungen erfolgte über die Auswerteionen 219 für 1,4-BDO und
221 für 1,4-BDO-d8. Vorversuche ergaben, dass in den Kulturproben der rekombinanten
Stämme zusätzlich γ-Hydroxybutyrat (GHB) detektiert werden konnte. Für dessen qualitative
und quantitative Analyse wurde es ebenso wie 1,4-BDO silyliert (Abb. 3.4) und über einen
entsprechenden isotopenmarkierten internen Standard (GHB-d6, Sigma-Aldrich) normiert.
Die für die Auswertung herangezogenen SIM-Ionen waren 317 für GHB und 323 für GHB-d6.
Über ein entsprechendes SIM-Chromatogramm (Abb. 3.5) konnte durch das Verhältnis der
Fläche des Analyten zur Fläche des dazugehörigen IS und mit Hilfe von erstellten
Eichgeraden (Tab. A1; Abb. A3 und A4) die Konzentrationsbestimmung der jeweiligen
Verbindungen erfolgen.
Abb. 3.5: SIM-Chromatogramm von 1,4-BDO und GHB. Erläuterungen siehe Text.
219
221
323
317

Ergebnisse 52
3.3 Generierung von rekombinanten C. acetobutylicum-Stämmen
zur Produktion von 1,4-Butandiol
Die in der Arbeit verwendeten und eingangs beschriebenen 4HBDs aus C. aminobutyricum
(AbfD), C. beijerinckii (Cbei_2100) und C. kluyveri (Ckl_3020) zeigten für die Aminosäure-
Sequenzen in in silico-Analysen (Abb. A5) eine Übereinstimmung von 73 - 79 % und besaßen
das für 4HBDs charakteristische [4Fe-4S]2+-Cluster-Motiv (CX3CX179-183HX6C). Dabei sind die
katalytisch relevanten Aminosäuren in den aktiven Zentren (Martins et al., 2004)
konserviert.
Die jeweiligen 4HBD-kodierenden Gene wurden mittels PCR (2.8.2) amplifiziert (Abb. 3.6, C)
und über entsprechende Oligonukleotide (Tab. 2.4) mit Restriktionsschnittstellen am 5´- und
3´- Ende für eine Insertion in den pTc-Shuttlevektor (Abb. 3.7) versehen. Als PCR-Matrize für
die Gene cbei_2100 und ckl_3020 diente die zuvor isolierte chromosomale DNA (2.8.2.2) aus
C. beijerinckii bzw. C. kluyveri (Abb. 3.6, A). Für die Amplifizierung des abfD-Gens aus
C. aminobutyricum wurde der Vektor pASK-IBA3+::abfD (Gerhardt et al., 2000) herangezogen
(Abb. 3.6, B).
(A) (B) (C) M1 1 2 M2 3 M2 4 5 6
Abb. 3.6: Erzeugung der 4HBD-kodierenden Gene cbei_2100, ckl_3020 und abfD. 0,8%ige Agarosegele. (A)
Chromosomale DNA (1 µg pro Spur) von C. beijerinckii (Spur 1) und C. kluyveri (Spur 2). Spur M1: MassRulerTm
DNA-Ladder Mix. (B) Plasmid-DNA pASK-IBA3+::abfD (4 µg Spur 3). Spur M2: 1kb-GeneRulerTM DNA-Ladder. (C)
PCR-Amplifikate der Gene cbei_2100 (Spur 4), ckl_3020 (Spur 5) und abfD (Spur 6).
ca. 1455 Bp
1,0 kBp
1,5 kBp
3,0 kBp 5,0 kBp 6,0 kBp
10,0 kBp
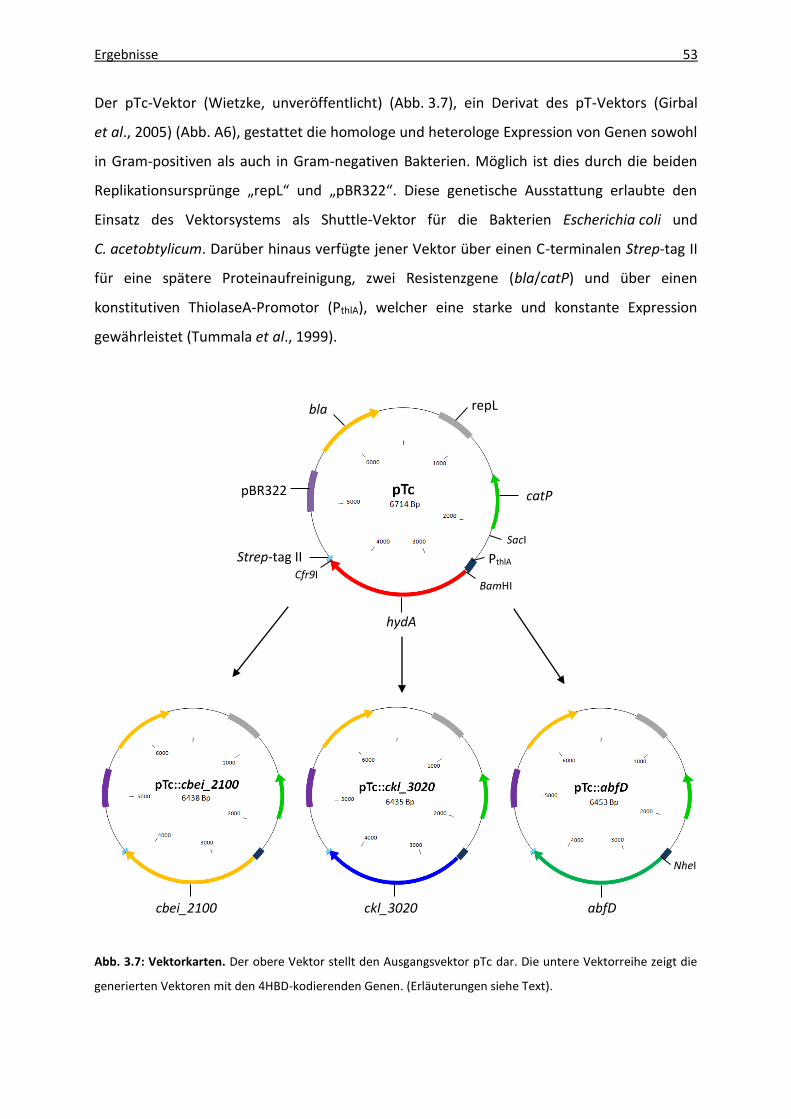
Ergebnisse 53
Der pTc-Vektor (Wietzke, unveröffentlicht) (Abb. 3.7), ein Derivat des pT-Vektors (Girbal
et al., 2005) (Abb. A6), gestattet die homologe und heterologe Expression von Genen sowohl
in Gram-positiven als auch in Gram-negativen Bakterien. Möglich ist dies durch die beiden
Replikationsursprünge „repL“ und „pBR322“. Diese genetische Ausstattung erlaubte den
Einsatz des Vektorsystems als Shuttle-Vektor für die Bakterien Escherichia coli und
C. acetobtylicum. Darüber hinaus verfügte jener Vektor über einen C-terminalen Strep-tag II
für eine spätere Proteinaufreinigung, zwei Resistenzgene (bla/catP) und über einen
konstitutiven ThiolaseA-Promotor (PthlA), welcher eine starke und konstante Expression
gewährleistet (Tummala et al., 1999).
Abb. 3.7: Vektorkarten. Der obere Vektor stellt den Ausgangsvektor pTc dar. Die untere Vektorreihe zeigt die
generierten Vektoren mit den 4HBD-kodierenden Genen. (Erläuterungen siehe Text).
repL
catP
PthlA
bla
pBR322
Strep-tag II
hydA
Cfr9I BamHI
SacI
cbei_2100 ckl_3020 abfD
NheI

Ergebnisse 54
Durch Behandlung des pTc-Vektors mit den Restriktionsenzymen BamHI und Cfr9I wurde das
Hydrogenase-Gen (hydA) entfernt (2.8.3.1) und nach einer anschließenden Ligationsreaktion
(2.8.3.3) durch die entsprechenden 4HBD-kodierenden Gene ersetzt. Für die Insertion der
beiden Gene cbei_2100 und ckl_3020 in den Vektor waren keine weiteren Anpassungen der
Restriktionsschnittstellen notwendig, jedoch für die Insertion des Gens abfD. Hierfür musste
die BamHI-Restriktionsschnittstelle des pTc-Vektors ausgetauscht werden, da diese auch im
abfD-Gen vorhanden war. Zu diesem Zweck wurde der Vektorbereich zwischen den
Schnittstellen SacI und BamHI (Abb. 3.7) mit spezifischen Oligonukleotiden (Tab. 2.4)
amplifiziert. Die verwendeten Oligonukleotide verfügten am 5´-Ende anstelle der BamHI-
eine NheI-Schnittstelle mit einer nachfolgenden Cfr9I-Erkennungsstelle. Infolgedessen wurde
die BamHI- durch eine NheI-Schnittstelle ersetzt. Die NheI- und Cfr9I-Erkennungsstellen
waren nun direkt aufeinanderfolgend. Anschließend wurde sowohl der pTc::hydA-Vektor als
auch das PCR-Amplifikat mit den Restriktionsenzymen SacI und Cfr9I verdaut. Es ergab sich
für den Verdau des Vektors ein 4515 Bp- und ein 2199 Bp-Fragment, welches den ThiolaseA-
Promotor und das hydA-Gen enthielt. Das 2199 Bp-Fragment wurde nachfolgend durch den
zuvor amplifizierten Vektorbereich ersetzt (2.8.3.3). Der neu generierte Vektor verfügte nun
über den ThiolaseA-Promotor, aber nicht mehr über das hydA-Gen. Es folgten die Restriktion
des angepassten Vektors und des abfD-Gens mit den Restriktionsenzymen NheI und Cfr9I
sowie die anschließende Klonierung des abfD-Gens in den Vektor.
Zur Kontrolle der korrekten Integration der 4HBD-kodierenden Gene wurden die Konstrukte
aus transformierten E. coli-Zellen (Tab. 2.1) isoliert und einem Kontrollverdau unterzogen
(Abb. 3.8). Desweiteren wurden die Genbereiche mittels Sequenzierung (LGC Genomics,
Berlin) auf mögliche Veränderungen in den Nukleotidsequenzen überprüft (Abb. A7-9).

Ergebnisse 55
M 1 2 3 4 5 6 7 8
Abb. 3.8: Kontrollrestriktion der Plasmide pTc::P(thlA)cbei_2100, pTc::P(thlA)ckl_3020 und pTc::P(thlA)abfD.
0,8%iges Agarosegel. Restriktionsenzyme BamHI/SmaI bzw. NheI/SmaI. Spur M: MassRulerTm DNA-Ladder Mix,
Spuren 1-3: pTc::P(thlA)cbei_2100 (Klone 1 [5 µg], 2 [3 µg] und 6 [4 µg]), Spuren 4-6: pTc::P(thlA)ckl_3020
(Klone 8 [3 µg], 15 [3 µg] und 18 [2 µg]), Spuren 7 und 8: pTc::P(thlA)abfD (Klone 22 [4 µg] und 24 [4 µg]).
Vektoren, welche die erwarteten Restriktionsfragmente (ca. 4980 Bp und 1455 Bp)
aufwiesen (Abb. 3.8), wurden anschließend für die Transformation von C. acetobutylicum
eingesetzt (2.9.2.1) und die Klone mittels Colony-PCR (2.8.2.3), SDS-PAGE (2.10.4) und
Western Blot-Analysen (2.10.6) verifiziert (Abb. 3.9). Da die Gene cbei_2100, ckl_3020 und
abfD nicht im Genom von C. acetobutylicum vorkommen und somit keine Falsch-positiven
Signale verursachen, konnte ihr Nachweis mittels „Colony“-PCR (2.8.2.3) erfolgen. Nur Klone,
die das entsprechende Plasmid aufgenommen haben, zeigten Banden auf Höhe von 1,5 kBp
im Agarosegel (Abb. 3.9, A). Jeweils einer der positiven Klone, welche ein entsprechendes
Plasmid aufgenommen haben, wurde in 200 ml CGM kultiviert und für die
Proteinaufreinigung via Affinitätschromatografie eingesetzt (2.10.2). Elutionsfraktionen mit
den höchsten Proteinkonzentrationen wurden für eine SDS-PAGE mit anschließendem
Western Blot verwendet. Im SDS-Polyacrylamidgel (Abb. 3.9, B) und im Western Blot
(Abb. 3.9, C) sind entsprechende Banden bzw. Signale der aufgereinigten Proteine
erkennbar.
Demnach haben die transformierten C. acetobutylicum-Stämme die jeweiligen Plasmide
aufgenommen und die auf den Vektoren kodierten Gene exprimiert.
5,0 kBp
1,5 kBp 4hbd, linear (ca. 1455 Bp)
pTc, linear (ca. 4980 Bp)
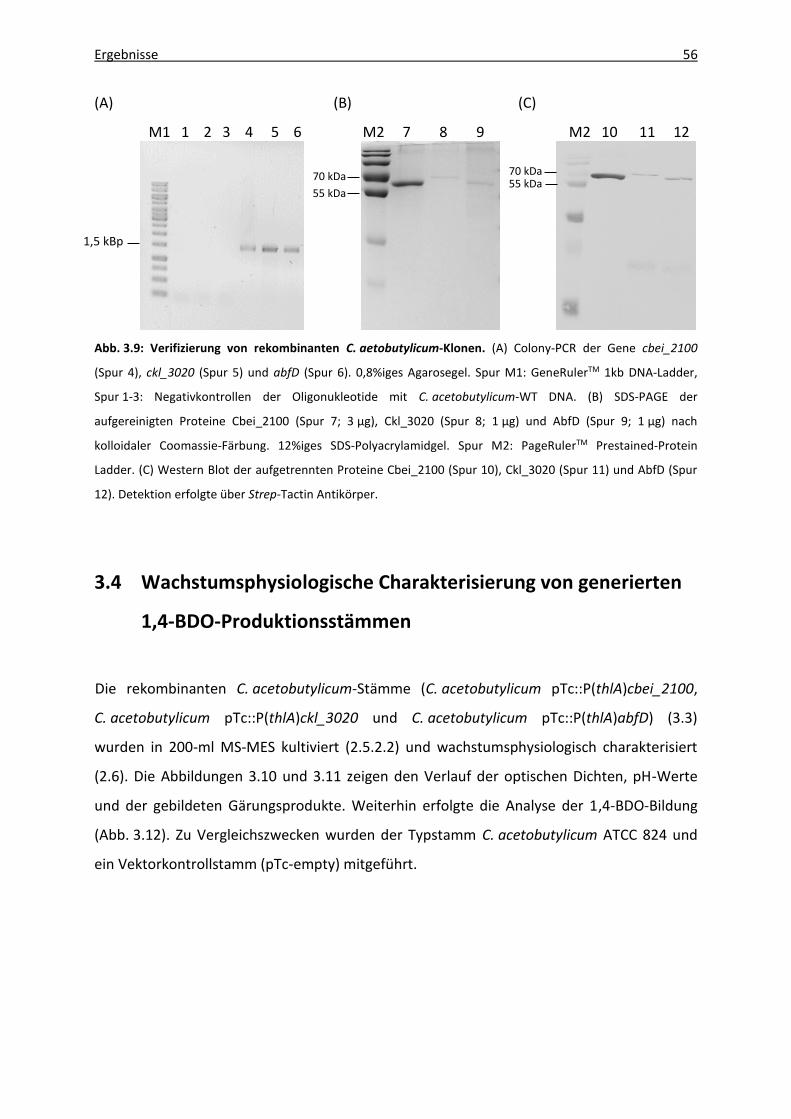
Ergebnisse 56
(A) (B) (C)
M1 1 2 3 4 5 6 M2 7 8 9 M2 10 11 12
Abb. 3.9: Verifizierung von rekombinanten C. aetobutylicum-Klonen. (A) Colony-PCR der Gene cbei_2100
(Spur 4), ckl_3020 (Spur 5) und abfD (Spur 6). 0,8%iges Agarosegel. Spur M1: GeneRulerTM 1kb DNA-Ladder,
Spur 1-3: Negativkontrollen der Oligonukleotide mit C. acetobutylicum-WT DNA. (B) SDS-PAGE der
aufgereinigten Proteine Cbei_2100 (Spur 7; 3 µg), Ckl_3020 (Spur 8; 1 µg) und AbfD (Spur 9; 1 µg) nach
kolloidaler Coomassie-Färbung. 12%iges SDS-Polyacrylamidgel. Spur M2: PageRulerTM Prestained-Protein
Ladder. (C) Western Blot der aufgetrennten Proteine Cbei_2100 (Spur 10), Ckl_3020 (Spur 11) und AbfD (Spur
12). Detektion erfolgte über Strep-Tactin Antikörper.
3.4 Wachstumsphysiologische Charakterisierung von generierten
1,4-BDO-Produktionsstämmen
Die rekombinanten C. acetobutylicum-Stämme (C. acetobutylicum pTc::P(thlA)cbei_2100,
C. acetobutylicum pTc::P(thlA)ckl_3020 und C. acetobutylicum pTc::P(thlA)abfD) (3.3)
wurden in 200-ml MS-MES kultiviert (2.5.2.2) und wachstumsphysiologisch charakterisiert
(2.6). Die Abbildungen 3.10 und 3.11 zeigen den Verlauf der optischen Dichten, pH-Werte
und der gebildeten Gärungsprodukte. Weiterhin erfolgte die Analyse der 1,4-BDO-Bildung
(Abb. 3.12). Zu Vergleichszwecken wurden der Typstamm C. acetobutylicum ATCC 824 und
ein Vektorkontrollstamm (pTc-empty) mitgeführt.
55 kDa
70 kDa
1,5 kBp
55 kDa 70 kDa
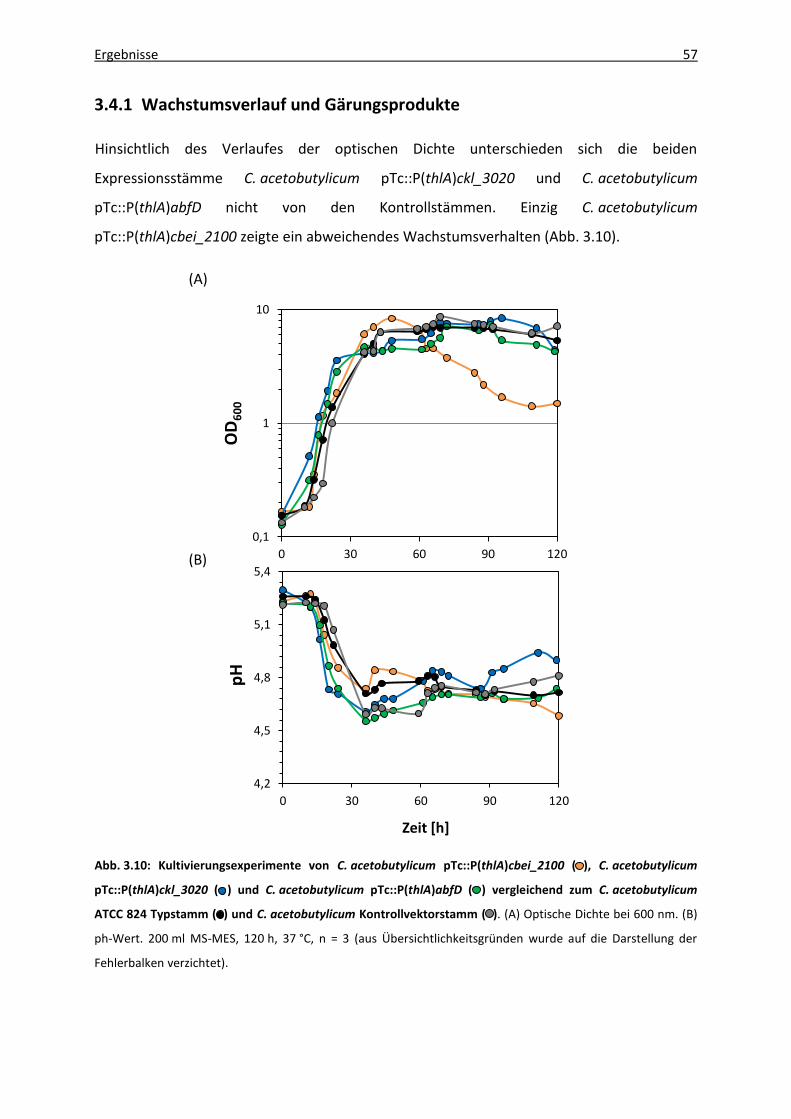
Ergebnisse 57
3.4.1 Wachstumsverlauf und Gärungsprodukte
Hinsichtlich des Verlaufes der optischen Dichte unterschieden sich die beiden
Expressionsstämme C. acetobutylicum pTc::P(thlA)ckl_3020 und C. acetobutylicum
pTc::P(thlA)abfD nicht von den Kontrollstämmen. Einzig C. acetobutylicum
pTc::P(thlA)cbei_2100 zeigte ein abweichendes Wachstumsverhalten (Abb. 3.10).
(A)
Abb. 3.10: Kultivierungsexperimente von C. acetobutylicum pTc::P(thlA)cbei_2100 ( ), C. acetobutylicum
pTc::P(thlA)ckl_3020 ( ) und C. acetobutylicum pTc::P(thlA)abfD ( ) vergleichend zum C. acetobutylicum
ATCC 824 Typstamm ( ) und C. acetobutylicum Kontrollvektorstamm ( ). (A) Optische Dichte bei 600 nm. (B)
ph-Wert. 200 ml MS-MES, 120 h, 37 °C, n = 3 (aus Übersichtlichkeitsgründen wurde auf die Darstellung der
Fehlerbalken verzichtet).
0,1
1
10
0 30 60 90 120
OD
60
0
4,2
4,5
4,8
5,1
5,4
0 30 60 90 120
pH
Zeit [h]
(B)

Ergebnisse 58
Nach einer ca. 12-stündigen lag-Phase gingen die Kulturen in die exponentielle
Wachstumsphase über (Abb. 3.10, A). Dabei zeigten die Expressionsstämme
C. acetobutylicum pTc::P(thlA)ckl_3020 (µ = 0,13 h-1) und C. acetobutylicum pTc::P(thlA)abfD
(µ = 0,129 h-1) eine ähnliche Wachstumsgeschwindigkeit wie der Vektorkontrollstamm
(µ = 0,134 h-1). Der Expressionsstamm C. acetobutylicum pTc::P(thlA)cbei_2100 (µ= 0,146 h-1)
besaß die höchste ermittelte Wachstumsrate (Tab. A2). Dieser Stamm erreichte bereits nach
48 h als erstes die maximale optische Dichte von 8,35. Im Vergleich zu den weiteren
Stämmen war die Wachstumsrate des Wildtyps (µ = 0,116 h-1) am geringsten. Die
exponentielle Wachstumsphase wurde anschließend nach etwa 40 h von der stationären
Wachstumsphase abgelöst. Hierbei zeigte ebenfalls einzig C. acetobutylicum
pTc::P(thlA)cbei_2100 ein abweichendes Verhalten, indem nach Erreichen eines Peaks die
optische Dichte deutlich abnahm. Die finale OD lag nach 120 h bei 1,5.
Zusätzlich zu der optischen Dichte wurde der extrazelluläre pH-Wert der Kulturen gemessen
(Abb. 3.10, B). Der Ausgangswert von ca. 5,2 sank mit Eintreten der Kulturen in die
exponentielle Wachstumsphase aufgrund der Produktion der Säuren Acetat und Butyrat
(Abb. 3.11, A und B) bis auf einen Minimalwert von pH 4,6 (C. acetobutylicum
pTc::P(thlA)abfD). Die pH-Werte des Wildtyps und von C. acetobutylicum
pTc::P(thlA)cbei_2100 sanken indes nur bis jeweils 4,7. Mit dem Übergang in die stationäre
Wachstumsphase setzte der für C. acetobutylicum charakteristische „Lösungsmittelshift“ ein.
Dieser ging mit der Assimilierung der Säuren und deren Umwandlung in die pH-neutralen
Lösungsmittel Aceton, Butanol und Ethanol einher (Abb. 3.11, C, D und E). Dies wiederum
führte zu einem erneuten, leichten Anstieg des pH-Werts.
Auch im Hinblick auf das Produktspektrum zeigte der Stamm C. acetobutylicum
pTc::P(thlA)cbei_2100 ein abweichendes Verhalten im Vergleich zu allen anderen
untersuchten Stämmen (Abb. 3.11). Deutliche Unterschiede traten vor allem beim
„Lösungsmittelshift“ und somit bei der Assimilation der Säuren Acetat und Butyrat auf
(Abb. 3.11, A und B).
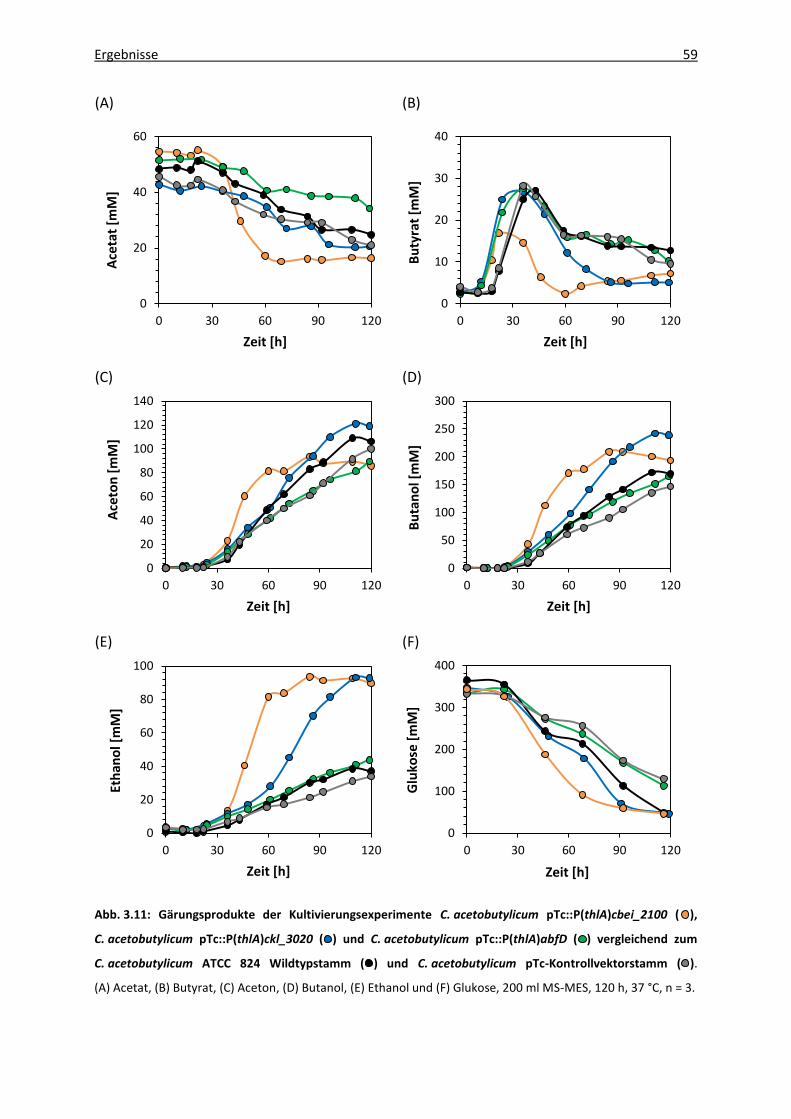
Ergebnisse 59
(A) (B)
(C) (D)
(E) (F)
Abb. 3.11: Gärungsprodukte der Kultivierungsexperimente C. acetobutylicum pTc::P(thlA)cbei_2100 ( ),
C. acetobutylicum pTc::P(thlA)ckl_3020 ( ) und C. acetobutylicum pTc::P(thlA)abfD ( ) vergleichend zum
C. acetobutylicum ATCC 824 Wildtypstamm ( ) und C. acetobutylicum pTc-Kontrollvektorstamm ( ).
(A) Acetat, (B) Butyrat, (C) Aceton, (D) Butanol, (E) Ethanol und (F) Glukose, 200 ml MS-MES, 120 h, 37 °C, n = 3.
0
20
40
60
0 30 60 90 120
Ace
tat
[mM
]
Zeit [h]
0
10
20
30
40
0 30 60 90 120
Bu
tyra
t [m
M]
Zeit [h]
0
20
40
60
80
100
120
140
0 30 60 90 120
Ace
ton
[m
M]
Zeit [h]
0
50
100
150
200
250
300
0 30 60 90 120
Bu
tan
ol [
mM
]
Zeit [h]
0
20
40
60
80
100
0 30 60 90 120
Eth
ano
l [m
M]
Zeit [h]
0
100
200
300
400
0 30 60 90 120
Glu
kose
[m
M]
Zeit [h]

Ergebnisse 60
Die Acetatassimilation der Expressionsstämme C. acetobutylicum pTc::P(thlA)ckl_3020 und
C. acetobutylicum pTc::P(thlA)abfD sowie der beiden Kontrollstämme erstreckte sich über
einen längeren Zeitraum und führte zu einer stetigen Abnahme der Acetatkonzentration
über den gesamten Wachstumsverlauf hinweg. Infolgedessen wurde bis zum Ende der
Kultivierungsexperimente ca. 50 % (C. acetobutylicum pTc::P(thlA)ckl_3020 und beide
Kontrollstämme) bzw. 34 % (C. acetobutylicum pTc::P(thlA)abfD) Acetat assimiliert
(Abb. 3.11, A). Im Gegensatz dazu sank die Acetatkonzentration bei C. acetobutylicum
pTc::P(thlA)cbei_2100 nach 22 h Kultivierung während der exponentiellen Wachstumsphase
deutlich und erreichte nach weiteren 38 h Wachstum einen Minimalwert von ca. 17 mM.
Dies entsprach einer Acetatassimilation von 67,5 %.
Der Butyratstoffwechsel von C. acetobutylicum pTc::P(thlA)cbei_2100 wies ebenfalls einige
Unterschiede zu den anderen Stämmen auf (Abb. 3.11, B). Während des exponentiellen
Wachstums kam es zum Anstieg der Butyratkonzentration, welche nach Erreichen eines
Peaks aufgrund der Butyratreassimilation wieder sank. Dabei betrug die maximale
Butyratkonzentration bei C. acetobutylicum pTc::P(thlA)cbei_2100 17 mM und entsprach
damit lediglich 63 % der Butyratkonzentrationen der weiteren Stämme (ca. 27 mM). Darüber
hinaus wurde das Maximum bereits nach 22 h und somit 14 h eher im Vergleich zu den
anderen Stämmen erreicht. Ebenfalls auffällig war die stärkere Abnahme der
Butyratkonzentration bei C. acetobutylicum pTc::P(thlA)ckl_3020, welche bis auf 5 mM
herabsank und damit die niedrigste Endkonzentration einnahm.
Mit Übergang in die stationäre Wachstumsphase werden die Säuren assimiliert und die
Lösungsmittel Aceton, Butanol und Ethanol gebildet (Jones und Woods, 1986), (Abb. 3.11, C,
D und E). Die Lösungsmittelproduktion setzte bei C. acetobutylicum pTc::P(thlA)cbei_2100
(22 h) deutlich früher ein als bei allen anderen Stämmen (36 h), diese stagnierte jedoch
bereits nach ca. 80 h. Es zeigte sich, dass die Acetonkonzentration trotz früher einsetzender
Produktion durch C. acetobutylicum pTc::P(thlA)cbei_2100 am geringsten war (85,7 mM) und
damit C. acetobutylicum pTc::P(thlA)abfD (89,3 mM) ähnelte. Im Gegensatz dazu produzierte
C. acetobutylicum pTc::P(thlA)ckl_3020 das meiste Aceton nach 120 h (119,0 mM),
(Abb. 3.11, C). Die Ausbeuten an Butanol erhöhten sich bei C. acetobutylicum
pTc::P(thlA)cbei_2100 (193,7 mM) und deutlich bei C. acetobutylicum pTc::P(thlA)ckl_3020
(238,6 mM) im Vergleich zu den beiden Kontrollstämmen (ATCC 824 [169,2 mM] und pTc
[147,1 mM]), (Abb. 3.11, D). Die größte Zunahme konnte jedoch bei der Produktion von

Ergebnisse 61
Ethanol durch die beiden Stämme C. acetobutylicum pTc::P(thlA)cbei_2100 (89,7 mM) und
C. acetobutylicum pTc::P(thlA)ckl_3020 (92,7 mM) detektiert werden. Die Menge des
produzierten Ethanols betrug ca. das 2,5-fache der Konzentration der beiden
Kontrollstämme (ATCC 824 [37,0 mM] und pTc [33,8 mM]), (Abb. 3.11, E).
Die höchste Gesamtkonzentration an Lösungsmitteln (28,9 g/l) konnte bei C. acetobutylicum
pTc::P(thlA)ckl_3020 detektiert werden. Dieser Stamm produzierte 112 % mehr Aceton,
141 % mehr Butanol (p < 0,01) und 250 % mehr Ethanol (p < 0,001) als die Kontrollstämme.
Es bleibt festzuhalten, dass im Hinblick auf das Wachstumsverhalten und das
Produktspektrum der Stamm C. acetobutylicum pTc::P(thlA)abfD ein ähnliches
Wachstumsverhalten wie die Kontrollstämme aufwies. Der Stamm C. acetobutylicum
pTc::P(thlA)cbei_2100 zeigte neben einer früher einsetzenden Assimilation der Säuren auch
eine früher einsetzende Lösungsmittelproduktion, wohingegen C. acetobutylicum
pTc::P(thlA)ckl_3020 sich durch eine deutlich erhöhte Lösungsmittelbildung auszeichnete.
Bei beiden letztgenannten Stämmen konnte zudem eine gesteigerte Ethanolproduktion
festgestellt werden. Einen genauen Überblick über die Wachstumsparameter und
Gärungsprodukte liefert die Tabelle A2 (Anhang).
3.4.2 1,4-Butandiolproduktion
Nachdem die Expressionsstämme C. acetobutylicum pTc::P(thlA)cbei_2100,
C. acetobutylicum pTc::P(thlA)ckl_3020 und C. acetobutylicum pTc::P(thlA)abfD über eine
entsprechende 4HBD verfügten und diese auch exprimierten (3.3), galt es anschließend die
Produktion von 1,4-BDO in Kultivierungsexperimenten nachzuweisen. Aus diesem Grund
wurde neben der gaschromatografischen Analyse der herkömmlichen Gärungsprodukte
(3.4.1) auch die Produktion von 1,4-BDO mittels GC/MS-Analysen überprüft.
Wie aus der Abbildung 3.12 hervorgeht, waren alle drei Expressionsstämme in der Lage
1,4-BDO zu synthetisieren.
Dabei produzierte der Stamm C. acetobutylicum pTc::P(thlA)cbei_2100 mit 0,46 mM
(41,5 mg/l) 77 % mehr 1,4-BDO als C. acetobutylicum pTc::P(thlA)ckl_3020 (0,26 mM,
[23,7 mg/l]) und 3,5 mal so viel wie C. acetobutylicum pTc::P(thlA)abfD (0,13 mM,
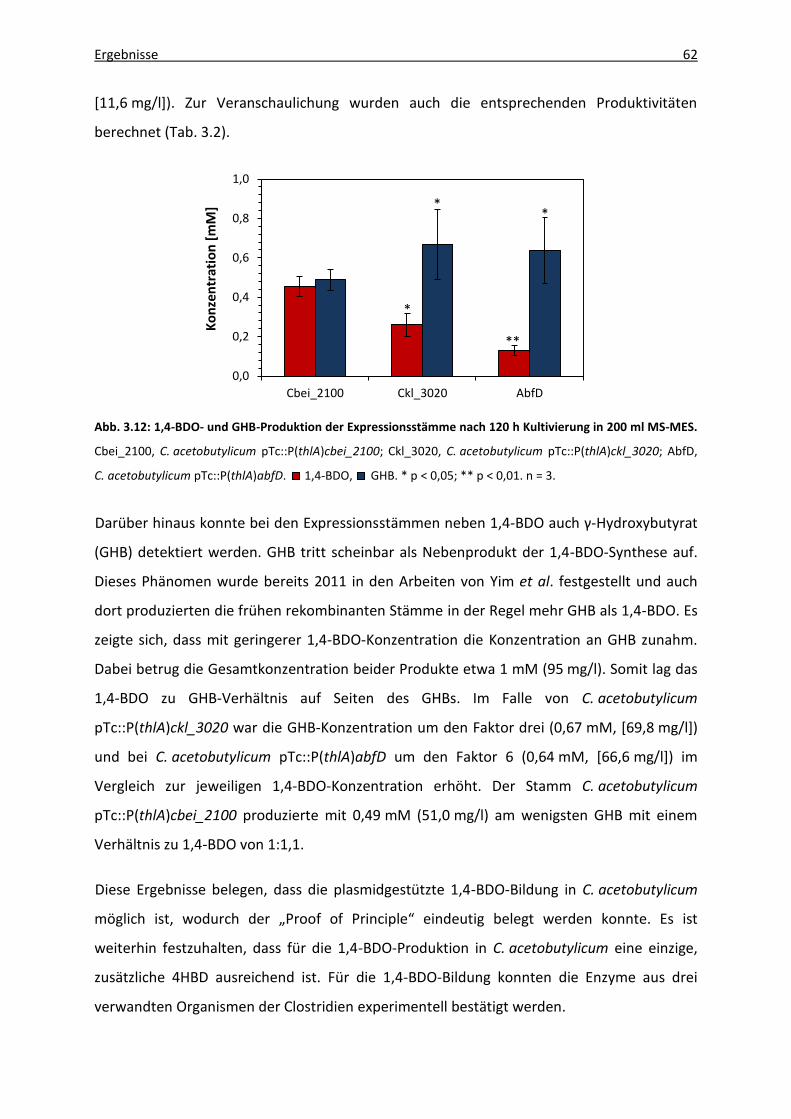
Ergebnisse 62
[11,6 mg/l]). Zur Veranschaulichung wurden auch die entsprechenden Produktivitäten
berechnet (Tab. 3.2).
Abb. 3.12: 1,4-BDO- und GHB-Produktion der Expressionsstämme nach 120 h Kultivierung in 200 ml MS-MES.
Cbei_2100, C. acetobutylicum pTc::P(thlA)cbei_2100; Ckl_3020, C. acetobutylicum pTc::P(thlA)ckl_3020; AbfD,
C. acetobutylicum pTc::P(thlA)abfD. 1,4-BDO, GHB. * p < 0,05; ** p < 0,01. n = 3.
Darüber hinaus konnte bei den Expressionsstämmen neben 1,4-BDO auch γ-Hydroxybutyrat
(GHB) detektiert werden. GHB tritt scheinbar als Nebenprodukt der 1,4-BDO-Synthese auf.
Dieses Phänomen wurde bereits 2011 in den Arbeiten von Yim et al. festgestellt und auch
dort produzierten die frühen rekombinanten Stämme in der Regel mehr GHB als 1,4-BDO. Es
zeigte sich, dass mit geringerer 1,4-BDO-Konzentration die Konzentration an GHB zunahm.
Dabei betrug die Gesamtkonzentration beider Produkte etwa 1 mM (95 mg/l). Somit lag das
1,4-BDO zu GHB-Verhältnis auf Seiten des GHBs. Im Falle von C. acetobutylicum
pTc::P(thlA)ckl_3020 war die GHB-Konzentration um den Faktor drei (0,67 mM, [69,8 mg/l])
und bei C. acetobutylicum pTc::P(thlA)abfD um den Faktor 6 (0,64 mM, [66,6 mg/l]) im
Vergleich zur jeweiligen 1,4-BDO-Konzentration erhöht. Der Stamm C. acetobutylicum
pTc::P(thlA)cbei_2100 produzierte mit 0,49 mM (51,0 mg/l) am wenigsten GHB mit einem
Verhältnis zu 1,4-BDO von 1:1,1.
Diese Ergebnisse belegen, dass die plasmidgestützte 1,4-BDO-Bildung in C. acetobutylicum
möglich ist, wodurch der „Proof of Principle“ eindeutig belegt werden konnte. Es ist
weiterhin festzuhalten, dass für die 1,4-BDO-Produktion in C. acetobutylicum eine einzige,
zusätzliche 4HBD ausreichend ist. Für die 1,4-BDO-Bildung konnten die Enzyme aus drei
verwandten Organismen der Clostridien experimentell bestätigt werden.
0,0
0,2
0,4
0,6
0,8
1,0
Cbei_2100 Ckl_3020 AbfD
Ko
nze
ntr
atio
n [
mM
]
*
**
* *
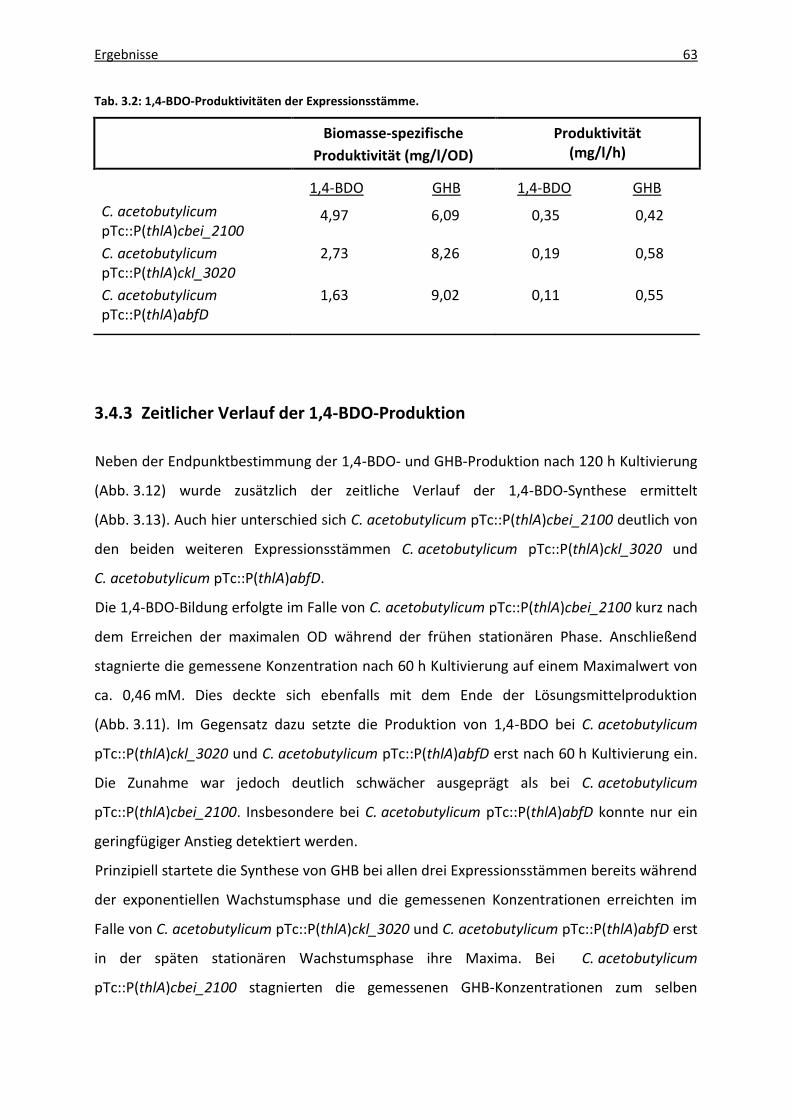
Ergebnisse 63
Tab. 3.2: 1,4-BDO-Produktivitäten der Expressionsstämme.
Biomasse-spezifische
Produktivität (mg/l/OD)
Produktivität (mg/l/h)
1,4-BDO GHB 1,4-BDO GHB
C. acetobutylicum pTc::P(thlA)cbei_2100
4,97 6,09 0,35 0,42
C. acetobutylicum pTc::P(thlA)ckl_3020
2,73 8,26 0,19 0,58
C. acetobutylicum pTc::P(thlA)abfD
1,63 9,02 0,11 0,55
3.4.3 Zeitlicher Verlauf der 1,4-BDO-Produktion
Neben der Endpunktbestimmung der 1,4-BDO- und GHB-Produktion nach 120 h Kultivierung
(Abb. 3.12) wurde zusätzlich der zeitliche Verlauf der 1,4-BDO-Synthese ermittelt
(Abb. 3.13). Auch hier unterschied sich C. acetobutylicum pTc::P(thlA)cbei_2100 deutlich von
den beiden weiteren Expressionsstämmen C. acetobutylicum pTc::P(thlA)ckl_3020 und
C. acetobutylicum pTc::P(thlA)abfD.
Die 1,4-BDO-Bildung erfolgte im Falle von C. acetobutylicum pTc::P(thlA)cbei_2100 kurz nach
dem Erreichen der maximalen OD während der frühen stationären Phase. Anschließend
stagnierte die gemessene Konzentration nach 60 h Kultivierung auf einem Maximalwert von
ca. 0,46 mM. Dies deckte sich ebenfalls mit dem Ende der Lösungsmittelproduktion
(Abb. 3.11). Im Gegensatz dazu setzte die Produktion von 1,4-BDO bei C. acetobutylicum
pTc::P(thlA)ckl_3020 und C. acetobutylicum pTc::P(thlA)abfD erst nach 60 h Kultivierung ein.
Die Zunahme war jedoch deutlich schwächer ausgeprägt als bei C. acetobutylicum
pTc::P(thlA)cbei_2100. Insbesondere bei C. acetobutylicum pTc::P(thlA)abfD konnte nur ein
geringfügiger Anstieg detektiert werden.
Prinzipiell startete die Synthese von GHB bei allen drei Expressionsstämmen bereits während
der exponentiellen Wachstumsphase und die gemessenen Konzentrationen erreichten im
Falle von C. acetobutylicum pTc::P(thlA)ckl_3020 und C. acetobutylicum pTc::P(thlA)abfD erst
in der späten stationären Wachstumsphase ihre Maxima. Bei C. acetobutylicum
pTc::P(thlA)cbei_2100 stagnierten die gemessenen GHB-Konzentrationen zum selben
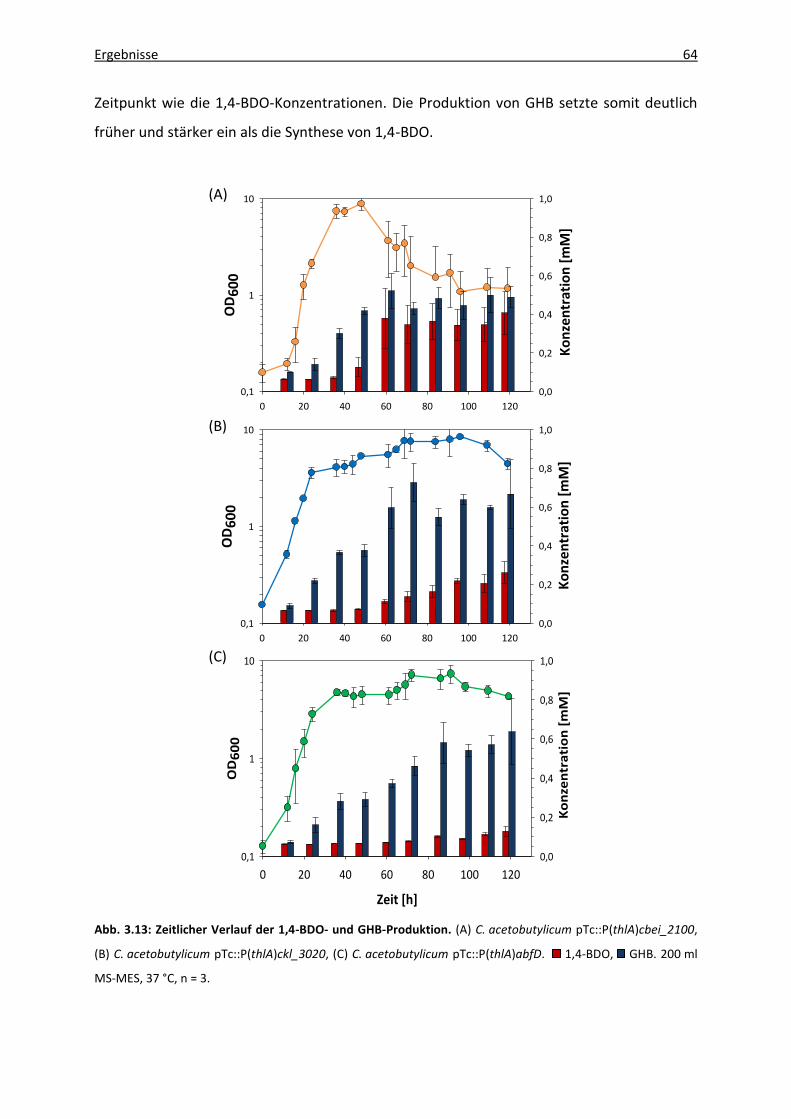
Ergebnisse 64
Zeitpunkt wie die 1,4-BDO-Konzentrationen. Die Produktion von GHB setzte somit deutlich
früher und stärker ein als die Synthese von 1,4-BDO.
Abb. 3.13: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion. (A) C. acetobutylicum pTc::P(thlA)cbei_2100,
(B) C. acetobutylicum pTc::P(thlA)ckl_3020, (C) C. acetobutylicum pTc::P(thlA)abfD. 1,4-BDO, GHB. 200 ml
MS-MES, 37 °C, n = 3.
0 20 40 60 80 100 120
OD
60
0
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Zeit [h]
0 20 40 60 80 100 120
OD
60
0
0,1
1
10
Ko
nze
ntr
ati
on
[m
M]
0,0
0,2
0,4
0,6
0,8
1,0
0 20 40 60 80 100 120
OD
60
0
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0(A)
(B)
(C)

Ergebnisse 65
3.5 Optimierungsstrategien
Da nachgewiesen werden konnte, dass die generierten Stämme in der Lage waren, 1,4-BDO
zu synthetisieren (3.4), lag das Interesse weiterer Arbeiten auf ersten Versuchen zur
Optimierung der 1,4-BDO-Produktion. Hierfür sollte zunächst der Einfluss einer Erhöhung
des Kulturvolumens („Scale-up“) und die Anwendung eines anderen Minimalmediums
untersucht werden (3.5.1). Darüber hinaus wurden verschiedene Kohlenstoffquellen für die
Fermentation von C. acetobutylicum und deren Einfluss auf die 1,4-BDO-Produktion geprüft,
um mögliche Alternativen zur Glukose aufzuzeigen (3.5.2). Weiterhin wurden
unterschiedliche Promotoren für die Expression der 4HBD-kodierenden Gene (3.5.3) und
diverse Stoffwechseldefektmutanten von C. acetobutylicum (3.5.4) als Rezipientenstämme
ausgetestet.
3.5.1 pH-regulierte Batch-Fermentation von C. acetobutylicum
pTc::P(thlA)cbei_2100
Für die industrielle Nutzung von mikrobiell erzeugten Verbindungen stellt die Überführung
vom Labormaßstab (0,1 – 10 l) zum Industriemaßstab (10.000 – 500.000 l) einen wichtigen
Aspekt dar (Takors, 2012). Denn mit Erhöhung des Kulturvolumens können Veränderungen
in physiologischen und physikalischen Parametern auftreten, welche zu beeinträchtigenden
Reaktionsbedingungen und infolgedessen zu verringerten Produktausbeuten führen
(Schmidt, 2005). Daher wurde die experimentelle Erhöhung des Kultivierungsmaßstabes von
0,2 l- auf 1,5 l-Batch-Kulturen vorgenommen. Aufgrund der höchsten erzielten 1,4-BDO-
Konzentrationen wurden diese Versuche mit dem Stamm C. acetobutylicum
pTc::P(thlA)cbei_2100 durchgeführt.
In einem 2-l-Bioreaktor erfolgte die Kultivierung unter Regulation des pH-Wertes. Diese
wurde während der Anwachsphase konstant bei pH 5,2 gehalten und erst nach Erreichen
einer OD600 von 2,0 auf pH 4,7 herabgesetzt. Neben MS-MES wurde dabei ein weiteres
Medium (MMfvK, 2.3.2) eingesetzt.
Hinsichtlich des Wachstumsverhaltens (Abb. 3.14) konnte ein Unterschied zwischen den
beiden Medien festgestellt werden. Während der Stamm C. acetobutylicum
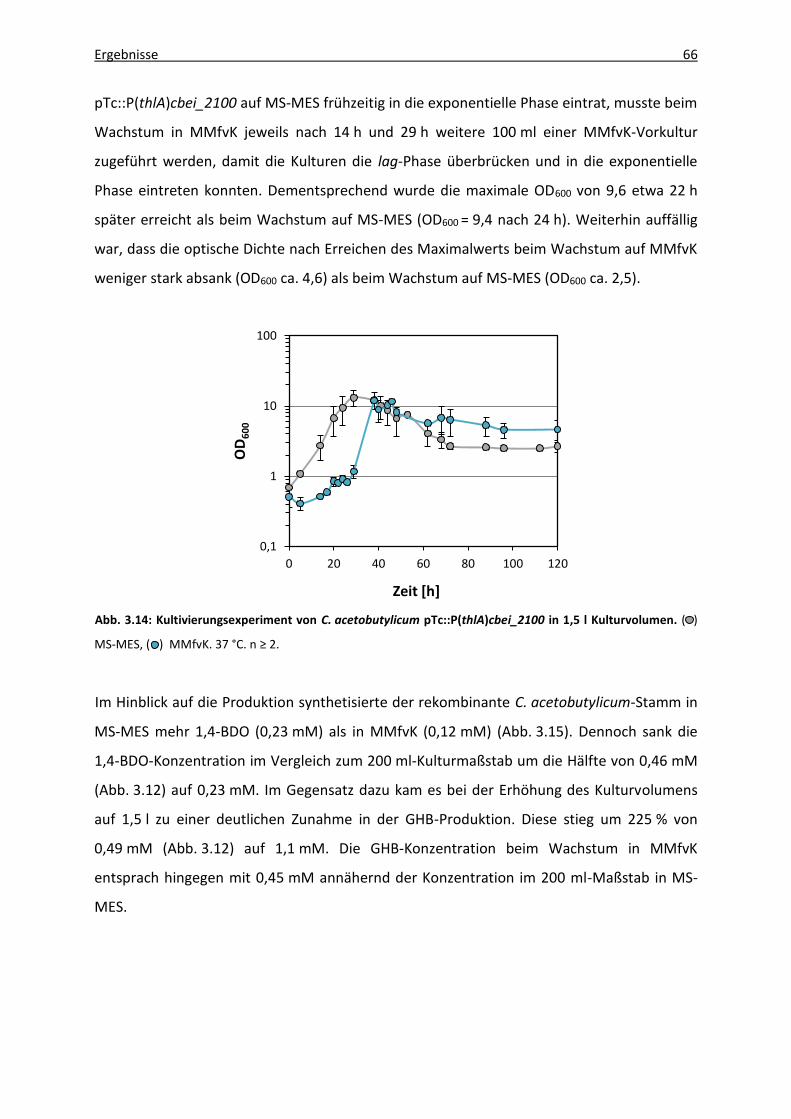
Ergebnisse 66
pTc::P(thlA)cbei_2100 auf MS-MES frühzeitig in die exponentielle Phase eintrat, musste beim
Wachstum in MMfvK jeweils nach 14 h und 29 h weitere 100 ml einer MMfvK-Vorkultur
zugeführt werden, damit die Kulturen die lag-Phase überbrücken und in die exponentielle
Phase eintreten konnten. Dementsprechend wurde die maximale OD600 von 9,6 etwa 22 h
später erreicht als beim Wachstum auf MS-MES (OD600 = 9,4 nach 24 h). Weiterhin auffällig
war, dass die optische Dichte nach Erreichen des Maximalwerts beim Wachstum auf MMfvK
weniger stark absank (OD600 ca. 4,6) als beim Wachstum auf MS-MES (OD600 ca. 2,5).
Abb. 3.14: Kultivierungsexperiment von C. acetobutylicum pTc::P(thlA)cbei_2100 in 1,5 l Kulturvolumen. ( )
MS-MES, ( ) MMfvK. 37 °C. n ≥ 2.
Im Hinblick auf die Produktion synthetisierte der rekombinante C. acetobutylicum-Stamm in
MS-MES mehr 1,4-BDO (0,23 mM) als in MMfvK (0,12 mM) (Abb. 3.15). Dennoch sank die
1,4-BDO-Konzentration im Vergleich zum 200 ml-Kulturmaßstab um die Hälfte von 0,46 mM
(Abb. 3.12) auf 0,23 mM. Im Gegensatz dazu kam es bei der Erhöhung des Kulturvolumens
auf 1,5 l zu einer deutlichen Zunahme in der GHB-Produktion. Diese stieg um 225 % von
0,49 mM (Abb. 3.12) auf 1,1 mM. Die GHB-Konzentration beim Wachstum in MMfvK
entsprach hingegen mit 0,45 mM annähernd der Konzentration im 200 ml-Maßstab in MS-
MES.
0,1
1
10
100
0 20 40 60 80 100 120
OD
60
0
Zeit [h]
Ergebnisse 67
Abb. 3.15: 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100 in 1,5 l Kulturvolumen
in Abhängigkeit verschiedener Medien. 120 h. 1,4-BDO, GHB. n ≥ 2.
3.5.2 Verwendung verschiedener Kohlenhydratquellen
Für die Entwicklung neuer Biosynthesewege wird im Allgemeinen auf das bevorzugte
Substrat des jeweiligen Produktionsstammes zurückgegriffen. Häufig handelt es sich dabei
um Glukose, da viele heterotrophe Mikroorganismen jene Kohlenhydratquelle präferieren
(Servinsky et al., 2010). Um mit Blick auf eine mögliche, zukünftige 1,4-BDO-Synthese durch
C. acetobutylicum einen größeren Handlungsspielraum im Hinblick auf die Substratnutzung
zu bekommen, wurden während dieser Arbeit verschiedene Kohlenhydratquellen
(Arabinose, Xylose, Galaktose und Mannose) als Substrate für die 1,4-BDO-Fermentation
eingesetzt. Aufgrund der höchsten 1,4-BDO-Produktion wurde dazu der Stamm
C. acetobutylicum pTc::P(thlA)cbei_2100 verwendet (Abb. 3.12).
Dieser Stamm zeigte auf allen untersuchten C-Quellen ein ähnliches Wachstumsverhalten
(Abb. 3.16). Nach einer etwa 12-stündigen lag-Phase erfolgte das exponentielle Wachstum.
Bei den Pentosen Arabinose und Xylose wurde die maximale OD600 von 7,7 bzw. 7,55 nach
ca. 60 h erreicht. Bei den Hexosen erfolgte dies bereits nach 48 h Kultivierung und somit 12 h
früher. Beim Wachstum auf Galaktose wurde nur eine maximale OD600 von 5,5 erzielt.
Jedoch verblieb der Stamm am längsten in der stationären Wachstumsphase. Ein
gegensätzliches Bild zeigte sich beim Wachstum auf Mannose. Die maximale OD600 von 8,4
entsprach in etwa der beim Wachstum auf Glukose (8,35), es folgte jedoch die deutlichste
Absenkung der OD600 bis zum Ende des Kultivierungsexperimentes auf 0,7.
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
MS-MES MMfvK
Ko
nze
ntr
atio
n [
mM
]
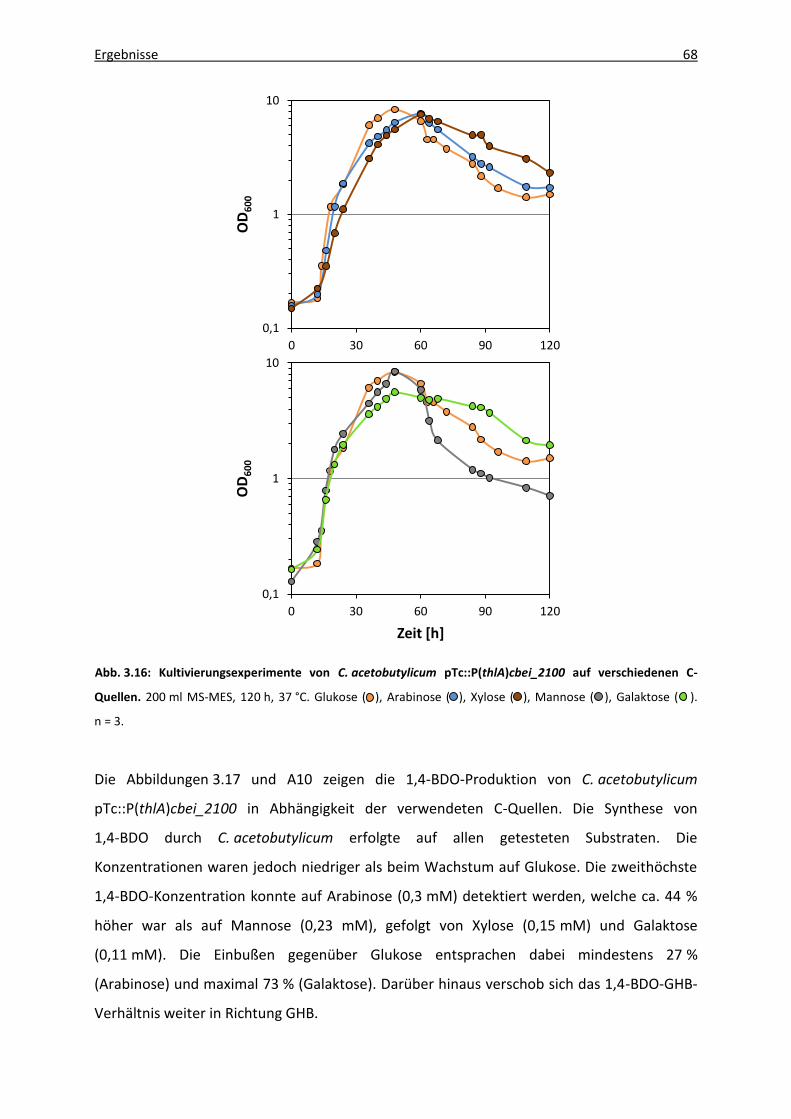
Ergebnisse 68
Abb. 3.16: Kultivierungsexperimente von C. acetobutylicum pTc::P(thlA)cbei_2100 auf verschiedenen C-
Quellen. 200 ml MS-MES, 120 h, 37 °C. Glukose ( ), Arabinose ( ), Xylose ( ), Mannose ( ), Galaktose ( ).
n = 3.
Die Abbildungen 3.17 und A10 zeigen die 1,4-BDO-Produktion von C. acetobutylicum
pTc::P(thlA)cbei_2100 in Abhängigkeit der verwendeten C-Quellen. Die Synthese von
1,4-BDO durch C. acetobutylicum erfolgte auf allen getesteten Substraten. Die
Konzentrationen waren jedoch niedriger als beim Wachstum auf Glukose. Die zweithöchste
1,4-BDO-Konzentration konnte auf Arabinose (0,3 mM) detektiert werden, welche ca. 44 %
höher war als auf Mannose (0,23 mM), gefolgt von Xylose (0,15 mM) und Galaktose
(0,11 mM). Die Einbußen gegenüber Glukose entsprachen dabei mindestens 27 %
(Arabinose) und maximal 73 % (Galaktose). Darüber hinaus verschob sich das 1,4-BDO-GHB-
Verhältnis weiter in Richtung GHB.
0,1
1
10
0 30 60 90 120
OD
60
0
0,1
1
10
0 30 60 90 120
OD
60
0
Zeit [h]
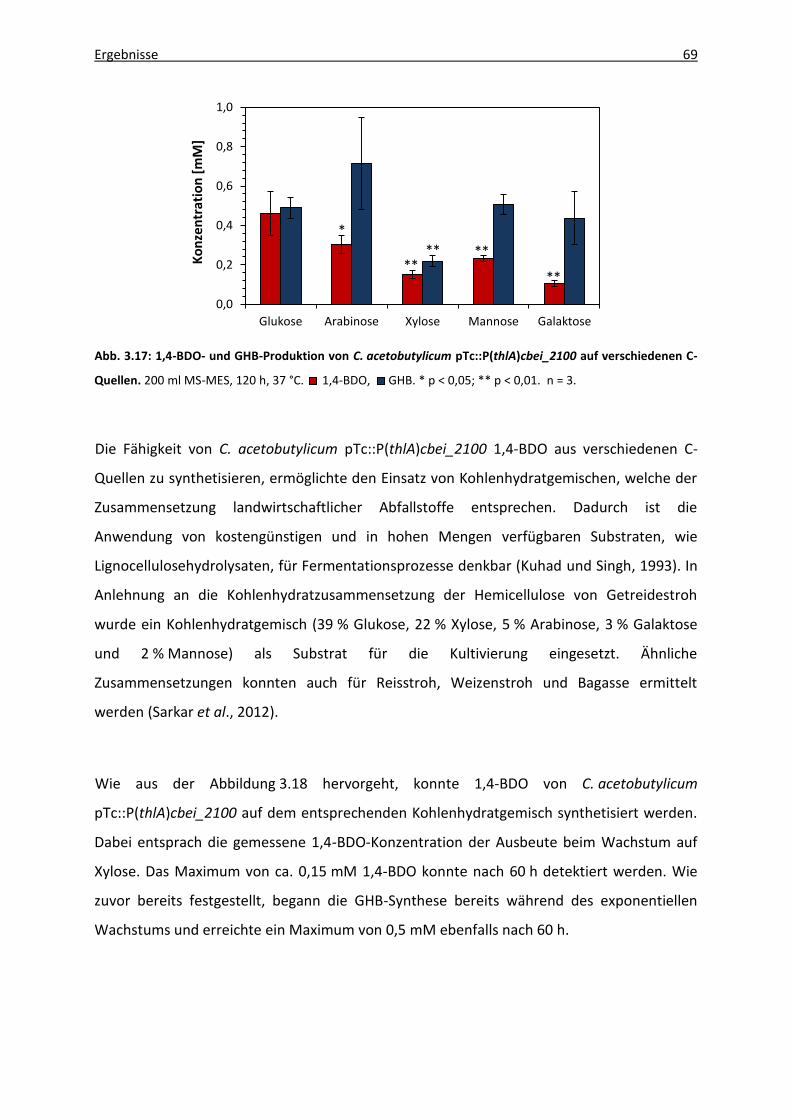
Ergebnisse 69
Abb. 3.17: 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100 auf verschiedenen C-
Quellen. 200 ml MS-MES, 120 h, 37 °C. 1,4-BDO, GHB. * p < 0,05; ** p < 0,01. n = 3.
Die Fähigkeit von C. acetobutylicum pTc::P(thlA)cbei_2100 1,4-BDO aus verschiedenen C-
Quellen zu synthetisieren, ermöglichte den Einsatz von Kohlenhydratgemischen, welche der
Zusammensetzung landwirtschaftlicher Abfallstoffe entsprechen. Dadurch ist die
Anwendung von kostengünstigen und in hohen Mengen verfügbaren Substraten, wie
Lignocellulosehydrolysaten, für Fermentationsprozesse denkbar (Kuhad und Singh, 1993). In
Anlehnung an die Kohlenhydratzusammensetzung der Hemicellulose von Getreidestroh
wurde ein Kohlenhydratgemisch (39 % Glukose, 22 % Xylose, 5 % Arabinose, 3 % Galaktose
und 2 % Mannose) als Substrat für die Kultivierung eingesetzt. Ähnliche
Zusammensetzungen konnten auch für Reisstroh, Weizenstroh und Bagasse ermittelt
werden (Sarkar et al., 2012).
Wie aus der Abbildung 3.18 hervorgeht, konnte 1,4-BDO von C. acetobutylicum
pTc::P(thlA)cbei_2100 auf dem entsprechenden Kohlenhydratgemisch synthetisiert werden.
Dabei entsprach die gemessene 1,4-BDO-Konzentration der Ausbeute beim Wachstum auf
Xylose. Das Maximum von ca. 0,15 mM 1,4-BDO konnte nach 60 h detektiert werden. Wie
zuvor bereits festgestellt, begann die GHB-Synthese bereits während des exponentiellen
Wachstums und erreichte ein Maximum von 0,5 mM ebenfalls nach 60 h.
0,0
0,2
0,4
0,6
0,8
1,0
Glukose Arabinose Xylose Mannose Galaktose
Ko
nze
ntr
atio
n [
mM
]
*
** **
**
**
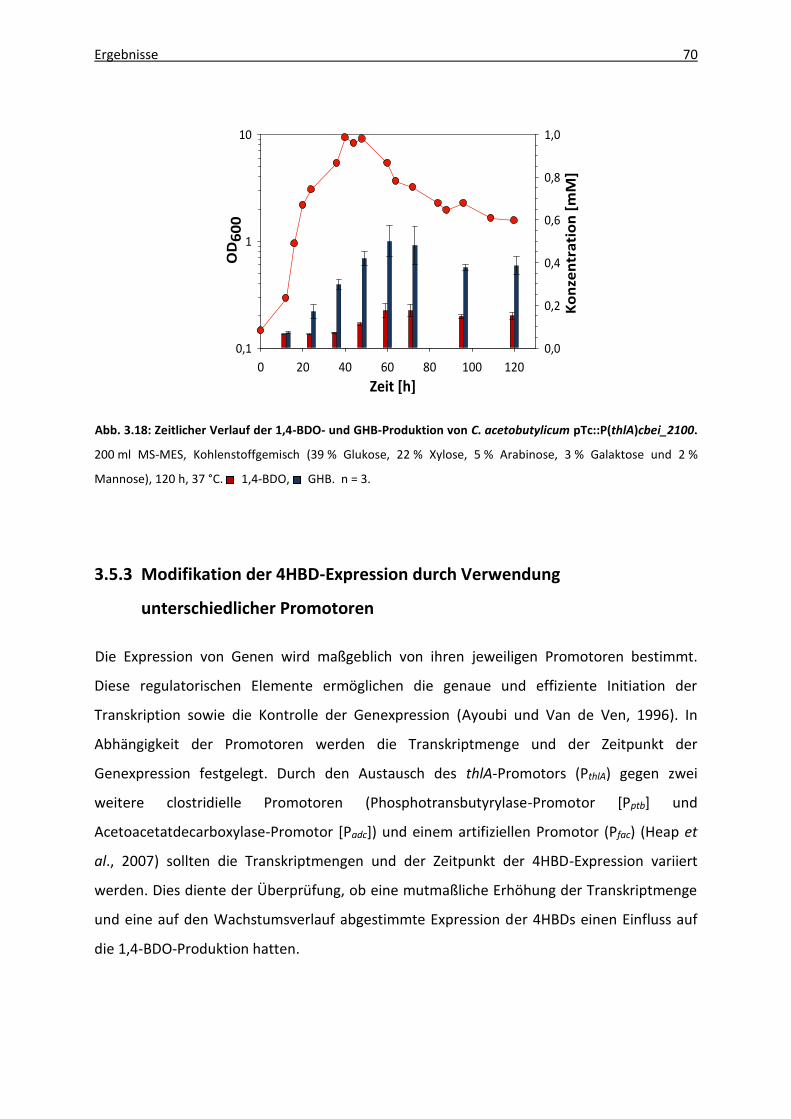
Ergebnisse 70
Abb. 3.18: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100.
200 ml MS-MES, Kohlenstoffgemisch (39 % Glukose, 22 % Xylose, 5 % Arabinose, 3 % Galaktose und 2 %
Mannose), 120 h, 37 °C. 1,4-BDO, GHB. n = 3.
3.5.3 Modifikation der 4HBD-Expression durch Verwendung
unterschiedlicher Promotoren
Die Expression von Genen wird maßgeblich von ihren jeweiligen Promotoren bestimmt.
Diese regulatorischen Elemente ermöglichen die genaue und effiziente Initiation der
Transkription sowie die Kontrolle der Genexpression (Ayoubi und Van de Ven, 1996). In
Abhängigkeit der Promotoren werden die Transkriptmenge und der Zeitpunkt der
Genexpression festgelegt. Durch den Austausch des thlA-Promotors (PthlA) gegen zwei
weitere clostridielle Promotoren (Phosphotransbutyrylase-Promotor [Pptb] und
Acetoacetatdecarboxylase-Promotor [Padc]) und einem artifiziellen Promotor (Pfac) (Heap et
al., 2007) sollten die Transkriptmengen und der Zeitpunkt der 4HBD-Expression variiert
werden. Dies diente der Überprüfung, ob eine mutmaßliche Erhöhung der Transkriptmenge
und eine auf den Wachstumsverlauf abgestimmte Expression der 4HBDs einen Einfluss auf
die 1,4-BDO-Produktion hatten.
Zeit [h]0 20 40 60 80 100 120
OD
60
0
0,1
1
10
Ko
nze
ntr
ati
on
[m
M]
0,0
0,2
0,4
0,6
0,8
1,0

Ergebnisse 71
Der thlA-Promotor, welcher standardmäßig im pTc-Vektorsystem vorlag, ermöglichte eine
konstitutive Expression der 4HBDs und zeigt eine konstant hohe Aktivität (Girbal et al., 2003;
Schulz, 2013). Im Gegensatz dazu besitzt der adc-Promotor neben einem Aktivitätsmaximum
in der späten exponentiellen Wachstumsphase auch ein konstantes Aktivitätslevel während
der solventogenen Phase (Girbal et al., 2003). Als gegensätzliche Strategie wurde der ptb-
Promotor verwendet, welcher vor allem mit der frühen Acidogenese assoziiert ist (Tummala
et al., 1999; Girbal et al., 2003). Bei dem dritten eingesetzten Promotor handelte es sich um
den artifiziellen fac-Promotor (Heap et al., 2007), bestehend aus einem lacO-Operator aus
E. coli und dem fdx-Promotor des Ferredoxingens aus C. pasteurianum. Der fac-Promotor
zeichnete sich unter anderem durch ein hohes Expressionslevel von heterologen Genen in
Clostridien aus (Heap et al., 2007).
3.5.3.1 Vektorgenerierung
Als Ausgangspunkt für die Generierung der Plasmide pT::P(ptb)cbei_2100,
pT::P(adc)cbei_2100, pT::P(fac)cbei_2100, pT::P(ptb)ckl_3020, pT::P(adc)ckl_3020,
pT::P(fac)ckl_3020, pT::P(ptb)abfD, pT::P(adc)abfD und pT::P(fac)abfD (Abb. A11) dienten die
von Schulz (2013) erzeugten Vektoren pT::P(ptb)pp2, pT::P(adc)pp2, pT::P(fac)pp2
(Abb. A12).
Die notwendigen Anpassungsschritte bei der Vektorherstellung sind in der Abbildung 3.19
dargestellt.
Für die Klonierung der Gene cbei_2100 und ckl_3020 wurden die Ausgangs-Vektoren (Schulz,
2013) mit den Restriktionsenzymen BamHI und KasI verdaut (I). Dies führte zur Entfernung
des Reporterproteingens pp2. Parallel dazu wurden die Vektoren pTc::P(thlA)cbei_2100 und
pTc::P(thlA)ckl_3020 ebenfalls mit BamHI und KasI hydrolysiert (II). Nach Auftrennung der
Fragmente in einem 2 %igem Agarosegel und einer anschließenden Gelelution wurden die
beiden 4HBD-kodierenden Gene (cbei_2100 und ckl_3020) in einer nachfolgenden
Ligationsreaktion (2.8.3.3) in die vorbereiteten Vektoren (I) ligiert (III).
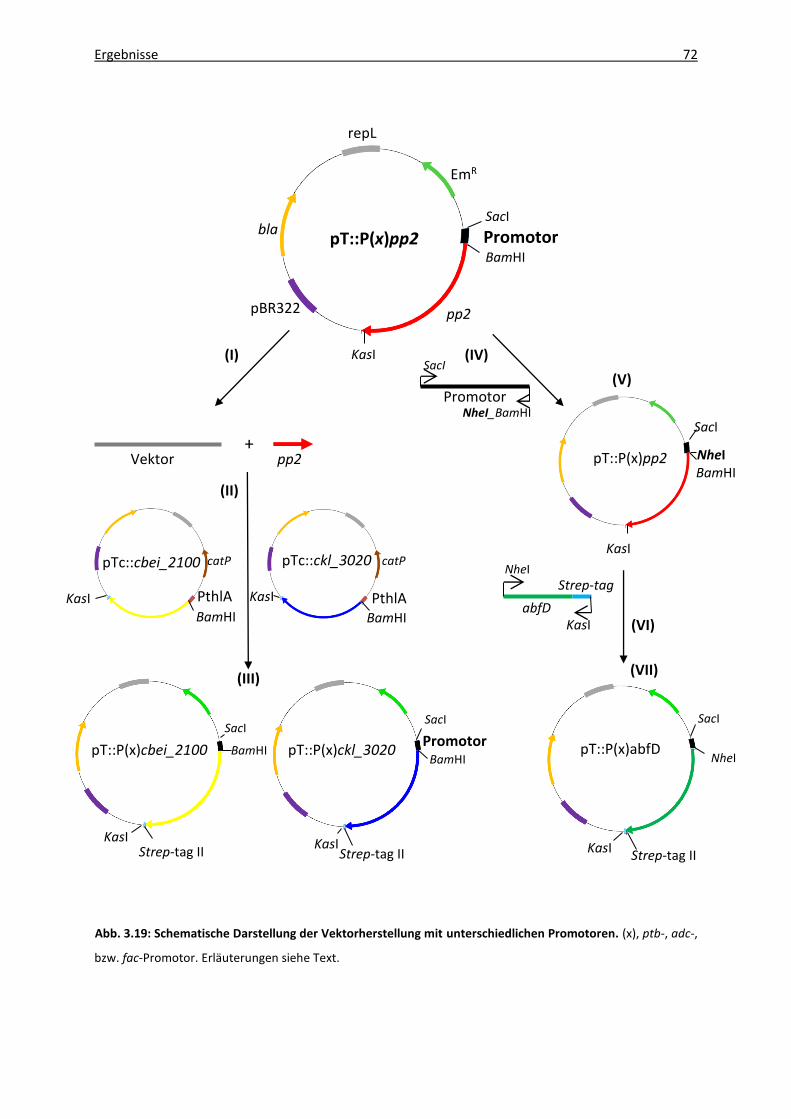
Ergebnisse 72
Abb. 3.19: Schematische Darstellung der Vektorherstellung mit unterschiedlichen Promotoren. (x), ptb-, adc-,
bzw. fac-Promotor. Erläuterungen siehe Text.
+ Vektor pp2
pT::P(x)pp2
EmR
bla
repL
SacI
pp2
Promotor
pBR322
BamHI
KasI
catP
PthlA KasI
BamHI
pTc::cbei_2100
KasI PthlA
catP
BamHI
NheI_BamHI
SacI
Promotor
pT::P(x)ckl_3020 Promotor
BamHI
KasI Strep-tag II
SacI
pT::P(x)cbei_2100 BamHI
KasI Strep-tag II
SacI
Strep-tag II KasI
SacI
NheI
SacI
NheI BamHI
KasI
NheI
KasI abfD
Strep-tag
pTc::ckl_3020
(I)
(II)
(IV)
(V)
pTc::ckl_3020
pT::P(x)pp2
pT::P(x)abfD
(III)
(VI)
(VII)

Ergebnisse 73
Da der Vektor pTc::P(thlA)abfD über eine BamHI-Schnittstelle innerhalb des Genbereichs
verfügte, wurden die entsprechenden Promotorbereiche der Vektoren pT::P(ptb)pp2,
pT::P(adc)pp2 und pT::P(fac)pp2 über eine PCR amplifiziert (IV). Die verwendeten
Oligonukleotide (Tab. 2.4) waren mit einer zusätzlichen NheI-Erkennungsstelle ausgestattet,
sodass die beiden Schnittsequenzen für BamHI und NheI in den amplifizierten
Promotorbereichen direkt aufeinander folgend waren. Die so veränderten
Promotorbereiche wurden ebenso wie die jeweiligen Vektoren (pT::P(ptb)pp2, pT::P(adc)pp2
und pT::P(fac)pp2) mit den Restriktionsenzymen BamHI und SacI verdaut und in einer
anschließenden Ligationsreaktion in die Vektoren kloniert (V). Die resultierenden Vektoren
entsprachen somit den Ausgangsvektoren pT::P(ptb)pp2, pT::P(adc)pp2 und pT::P(fac)pp2
mit der Ausnahme einer zusätzlichen NheI-Erkennungsstelle. Mittels PCR wurde das abfD-
Gen mit zugehöriger Strep-tag II-Sequenz des Vektors pTc::P(thlA)abfD amplifiziert (VI).
Anschließend folgten die Restriktionen des amplifizierten Genbereiches und der zuvor
veränderten Vektoren mit NheI und KasI und die Klonierung des abfD-Gens in die
vorbereiteten Vektoren (VII).
Die korrekte Integration der Promotoren und Gene in die jeweiligen Vektoren wurde durch
Sequenzierung (LGC Genomics) mit Promotor- und genspezifischen Oligonukleotiden
(Tab. 2.4) bestätigt.
Die generierten Vektoren wurden nach einer in vivo Methylierung in E. coli ER2275 (2.8.3.4)
für die Transformation von C. acetobutylicum (2.9.2) eingesetzt. Die Verifizierung
rekombinanter Klone erfolgte im Anschluss mittels Colony-PCR, SDS-PAGE und Western Blot-
Analysen (Abb. A13-15). Weiterhin wurden die rekombinanten C. acetobutylicum-Stämme
wachstumsphysiologisch charakterisiert (Tab. A3) und im Hinblick auf die 1,4-BDO-
Produktion analysiert (3.5.3.2).
3.5.3.2 1,4-BDO-Produktion rekombinanter C. acetobutylicum-Stämme
Anhand der Abbildung 3.20 ist ersichtlich, dass die unter 3.5.3 generierten
C. acetobutylicum-Stämme in der Lage waren, 1,4-BDO zu synthetisieren.
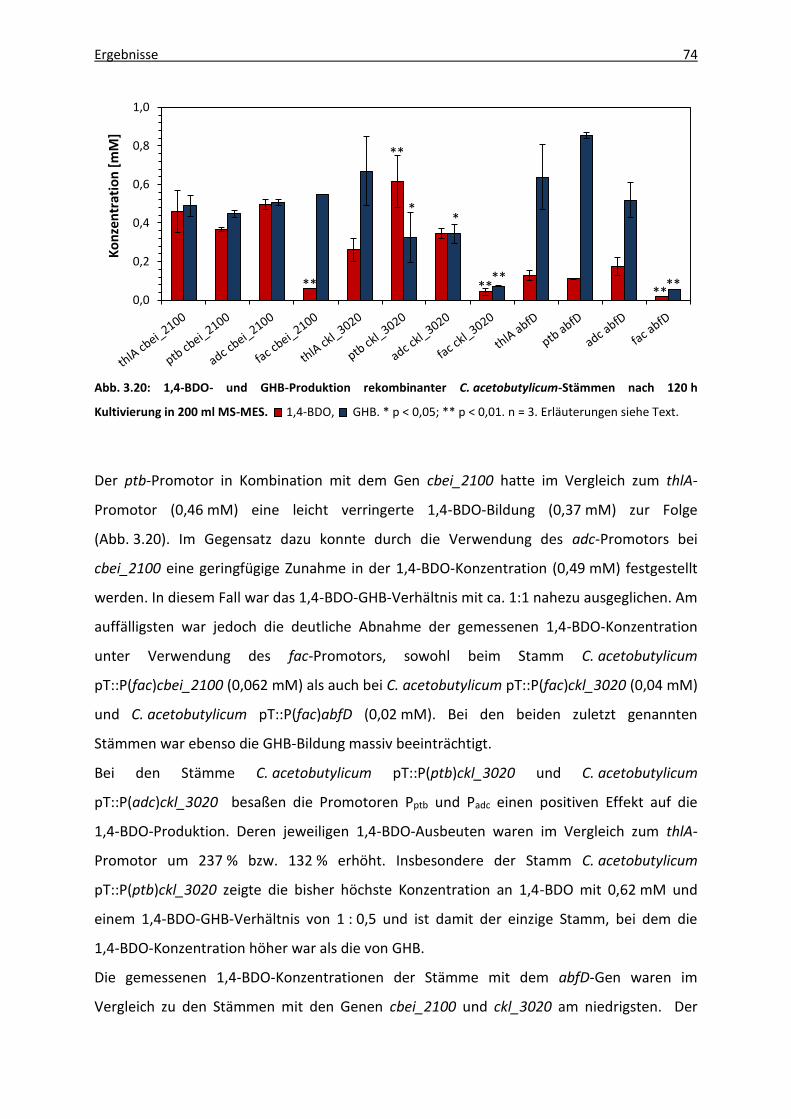
Ergebnisse 74
Abb. 3.20: 1,4-BDO- und GHB-Produktion rekombinanter C. acetobutylicum-Stämmen nach 120 h
Kultivierung in 200 ml MS-MES. 1,4-BDO, GHB. * p < 0,05; ** p < 0,01. n = 3. Erläuterungen siehe Text.
Der ptb-Promotor in Kombination mit dem Gen cbei_2100 hatte im Vergleich zum thlA-
Promotor (0,46 mM) eine leicht verringerte 1,4-BDO-Bildung (0,37 mM) zur Folge
(Abb. 3.20). Im Gegensatz dazu konnte durch die Verwendung des adc-Promotors bei
cbei_2100 eine geringfügige Zunahme in der 1,4-BDO-Konzentration (0,49 mM) festgestellt
werden. In diesem Fall war das 1,4-BDO-GHB-Verhältnis mit ca. 1:1 nahezu ausgeglichen. Am
auffälligsten war jedoch die deutliche Abnahme der gemessenen 1,4-BDO-Konzentration
unter Verwendung des fac-Promotors, sowohl beim Stamm C. acetobutylicum
pT::P(fac)cbei_2100 (0,062 mM) als auch bei C. acetobutylicum pT::P(fac)ckl_3020 (0,04 mM)
und C. acetobutylicum pT::P(fac)abfD (0,02 mM). Bei den beiden zuletzt genannten
Stämmen war ebenso die GHB-Bildung massiv beeinträchtigt.
Bei den Stämme C. acetobutylicum pT::P(ptb)ckl_3020 und C. acetobutylicum
pT::P(adc)ckl_3020 besaßen die Promotoren Pptb und Padc einen positiven Effekt auf die
1,4-BDO-Produktion. Deren jeweiligen 1,4-BDO-Ausbeuten waren im Vergleich zum thlA-
Promotor um 237 % bzw. 132 % erhöht. Insbesondere der Stamm C. acetobutylicum
pT::P(ptb)ckl_3020 zeigte die bisher höchste Konzentration an 1,4-BDO mit 0,62 mM und
einem 1,4-BDO-GHB-Verhältnis von 1 : 0,5 und ist damit der einzige Stamm, bei dem die
1,4-BDO-Konzentration höher war als die von GHB.
Die gemessenen 1,4-BDO-Konzentrationen der Stämme mit dem abfD-Gen waren im
Vergleich zu den Stämmen mit den Genen cbei_2100 und ckl_3020 am niedrigsten. Der
0,0
0,2
0,4
0,6
0,8
1,0K
on
zen
trat
ion
[m
M]
**
**
* *
** ** **
**

Ergebnisse 75
Einsatz verschiedener Promotoren führte bei jenen Stämmen zu keiner wesentlichen
Erhöhung der 1,4-BDO-Produktion. Im Gegensatz dazu war das 1,4-BDO-GHB-Verhältnis
deutlich auf Seiten des GHBs. Weiterhin wurden bei diesen Stämmen die höchsten
Konzentrationen an GHB gemessen, insbesondere beim Stamm C. acetobutylicum
pT::P(ptb)abfD mit 0,86 mM.
Im Wesentlichen veränderte sich die 1,4-BDO-Konzentration mit den Genen cbei_2100 und
abfD trotz verschiedener Promotoren (Pptb und Padc) nicht. Im Gegensatz dazu führten diese
beiden Promotoren in Kombination mit dem Gen ckl_3020 zu einer Steigerung der 1,4-BDO-
Bildung. Insbesondere mit dem ptb-Promotor konnte eine deutliche Zunahme von 238 % an
1,4-BDO im Vergleich zum Referenzstamm C. acetobutylicum pTc::P(thlA)ckl_3020
verzeichnet werden. Einen deutlichen negativen Effekt konnte bei allen drei Synthesegenen
unter Einfluss des fac-Promotors beobachtet werden, sodass dieser für zukünftige Arbeiten
ausgeschlossen wurde. Desweiteren ist die Verwendung des abfD-Gens aufgrund der
geringen 1,4-BDO-Bildung ebenfalls als ungeeignet einzustufen.
3.5.4 Stoffwechseldefektmutanten von C. acetobutylicum ATCC 824
Aufgrund der Nebenproduktbildung, welche die Gesamtausbeute an 1,4-BDO verringern
kann, wurde im Folgenden auf bereits zuvor erzeugte Stoffwechseldefektmutanten
(Tab. 2.1) (Lehmann, 2012 a) zurückgegriffen. Diese zeichneten sich unter anderem dadurch
aus, dass sowohl die Produktion der Säuren Acetat und Butyrat als auch die Bildung der
Lösungsmittel Aceton, Butanol und Ethanol durch den Funktionsverlust bestimmter Gene
nahezu ausgeschaltet werden konnte (Lehmann et al., 2012 b und c).
Da der Vektor pTc::P(thlA)cbei_2100 (Abb. 3.7) die bisher höchste 1,4-BDO-Produktion
ermöglichte (Abb. 3.12), wurde dieser für die Transformation der nachfolgenden
Stoffwechseldefektmutanten eingesetzt.
3.5.4.1 C. acetobutylicum-Mutanten mit Defekten in der Säurebildung
Die Eingangsenzyme für die Produktion der Säuren Acetat und Butyrat sind die
Phosphotransacetylase (Pta) und die Phosphotransbutyrylase (Ptb) (Abb. 3.21). Durch

Ergebnisse 76
Ausschalten der entsprechenden Gene konnte die Acetat- bzw. Butyrat-Bildung stark
verringert werden (Lehmann et al., 2012 b und c).
Darüber hinaus wurden neben den Pta- und Ptb-Defektmutanten (C. actobutylicum
pta::int(80) und C. acetobutylicum ptb::int(87)) auch die Ldh1- und Ldh2-Mutanten
(C. acetobutylicum ldh1::int(93) und C. acetobutylicum ldh2::int(231)), (Lehmann, 2012 a) für
die Untersuchungen herangezogen (Abb. 3.21). Die Laktatdehydrogenasen sind für die
Laktatbildung unter Verbrauch von NADH+H+ verantwortlich und stehen somit in Konkurrenz
zur 1,4-BDO-Synthese.
Abb. 3.21: Gärungsstoffwechsel von C. acetobutylicum. Inaktivierte Enzyme werden durch ein rotes X
gekennzeichnet. Ldh, C. acetobutylicum ldh1::int(93) bzw. C. acetobutylicum ldh2::int(231); Pta,
C. acetobutylicum pta::int(80) und Ptb, C. acetobutylicum ptb::int(87). Erläuterungen siehe Text. Abkürzungen:
Ac-CoA, Acetyl-CoA; Acetyl-P, Acetyl-Phosphat; AcAld, Acetaldehyd; AcAC-CoA, Acetoacetyl-CoA; AcAc,
Acetoacetat; 3HB-CoA, 3-Hydroxybutyryl-CoA; 4HB-CoA, 4-Hydroxybutyryl-CoA; BuAld, Butyraldehyd; Butyryl-P,
Butyryl-Phosphat, Ack, Acetatkinase; Buk, Butyratkinase; 4-HBD, 4-Hydroxybutyryl-CoA Dehydratase; AdhE,
Aldehyd/Alkoholdehydrogenase.
Die Abbildung 3.22 zeigt die 1,4-BDO-Produktion transformierter Rezipientenstämme. Es ist
ersichtlich, dass die 1,4-BDO-Konzentration durch Verwendung von Stoffwechselmutanten
mit Defekten in der Säurebildung nicht gesteigert werden konnte. Stattdessen kam es zu
einer Verringerung der 1,4-BDO-Produktion, im deutlichsten Fall um bis zu 81 %
(C. acetobutylicum ptb::int(87) pTc::P(thlA)cbei_2100: 0,079 mM) im Vergleich zum
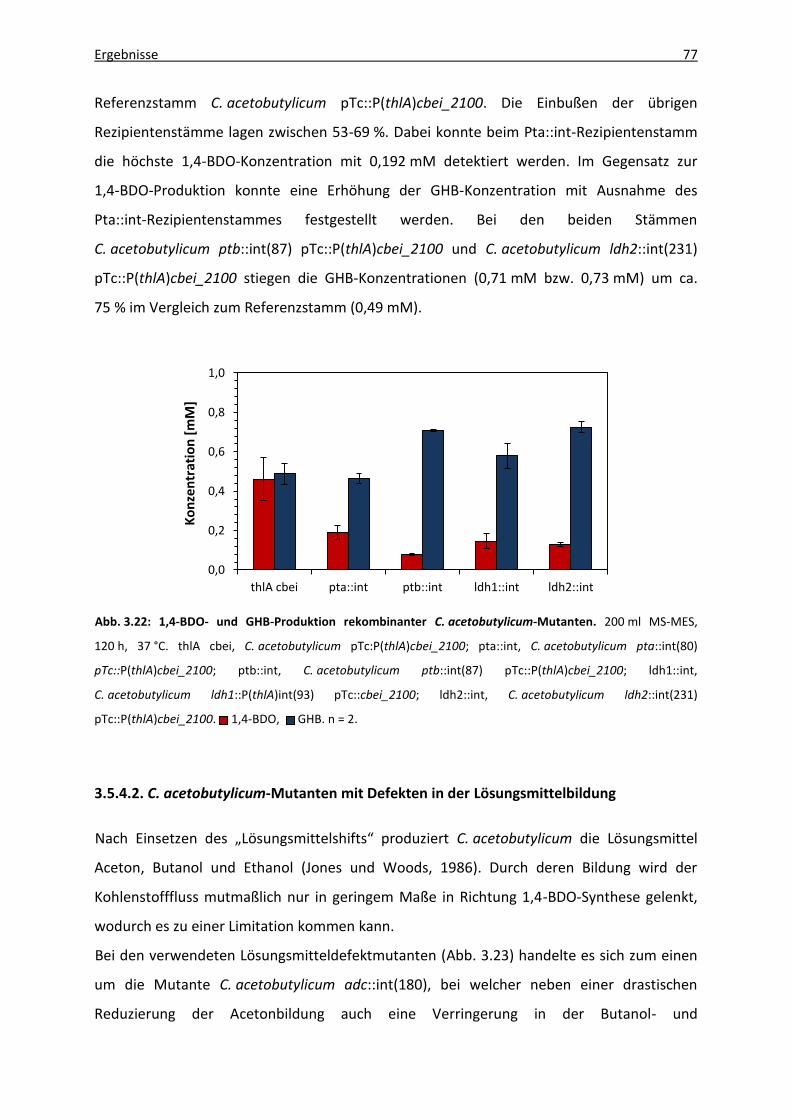
Ergebnisse 77
Referenzstamm C. acetobutylicum pTc::P(thlA)cbei_2100. Die Einbußen der übrigen
Rezipientenstämme lagen zwischen 53-69 %. Dabei konnte beim Pta::int-Rezipientenstamm
die höchste 1,4-BDO-Konzentration mit 0,192 mM detektiert werden. Im Gegensatz zur
1,4-BDO-Produktion konnte eine Erhöhung der GHB-Konzentration mit Ausnahme des
Pta::int-Rezipientenstammes festgestellt werden. Bei den beiden Stämmen
C. acetobutylicum ptb::int(87) pTc::P(thlA)cbei_2100 und C. acetobutylicum ldh2::int(231)
pTc::P(thlA)cbei_2100 stiegen die GHB-Konzentrationen (0,71 mM bzw. 0,73 mM) um ca.
75 % im Vergleich zum Referenzstamm (0,49 mM).
Abb. 3.22: 1,4-BDO- und GHB-Produktion rekombinanter C. acetobutylicum-Mutanten. 200 ml MS-MES,
120 h, 37 °C. thlA cbei, C. acetobutylicum pTc:P(thlA)cbei_2100; pta::int, C. acetobutylicum pta::int(80)
pTc::P(thlA)cbei_2100; ptb::int, C. acetobutylicum ptb::int(87) pTc::P(thlA)cbei_2100; ldh1::int,
C. acetobutylicum ldh1::P(thlA)int(93) pTc::cbei_2100; ldh2::int, C. acetobutylicum ldh2::int(231)
pTc::P(thlA)cbei_2100. 1,4-BDO, GHB. n = 2.
3.5.4.2. C. acetobutylicum-Mutanten mit Defekten in der Lösungsmittelbildung
Nach Einsetzen des „Lösungsmittelshifts“ produziert C. acetobutylicum die Lösungsmittel
Aceton, Butanol und Ethanol (Jones und Woods, 1986). Durch deren Bildung wird der
Kohlenstofffluss mutmaßlich nur in geringem Maße in Richtung 1,4-BDO-Synthese gelenkt,
wodurch es zu einer Limitation kommen kann.
Bei den verwendeten Lösungsmitteldefektmutanten (Abb. 3.23) handelte es sich zum einen
um die Mutante C. acetobutylicum adc::int(180), bei welcher neben einer drastischen
Reduzierung der Acetonbildung auch eine Verringerung in der Butanol- und
0,0
0,2
0,4
0,6
0,8
1,0
thlA cbei pta::int ptb::int ldh1::int ldh2::int
Ko
nze
ntr
atio
n [
mM
]

Ergebnisse 78
Ethanolkonzentration festgestellt werden konnte (Lehmann et al., 2012 b). Zum anderen
wurde die Mutante C. acetobutylicum ctfA::int(352) eingesetzt, da diese sich ebenfalls durch
eine deutlich verringerte Lösungsmittelproduktion auszeichnete (Lehmann et al., 2012 b).
Desweiteren wurden Mutanten mit Defekten in der Aldehyd/Alkoholdehydrogenase (AdhE,
C. acetobutylicum adhE1::int(158) und C. acetobutylicum adhE2::int(114)), (Lehmann 2012 a)
ausgewählt, um zu überprüfen, inwieweit die AdhE2 essentiell für die 1,4-BDO-Bildung ist
(Yim et al., 2011).
Abb. 3.23: Gärungsstoffwechsel von C. acetobutylicum. Inaktivierte Enzyme werden durch ein rotes X
gekennzeichnet. Adc, C. acetobutylicum adc::int(180), AdhE, C. acetobutylicum adhE1::int(158) bzw.
C. acetobutylicum adhE2::int(114) ; CtfAB, C. acetobutylicum ctfA::int(352). Erläuterungen siehe Text.
Abkürzungen: Ac-CoA, Acetyl-CoA; Acetyl-P, Acetyl-Phosphat; AcAld, Acetaldehyd; AcAC-CoA, Acetoacetyl-CoA;
AcAc, Acetoacetat; 3HB-CoA, 3-Hydroxybutyryl-CoA; 4HB-CoA, 4-Hydroxybutyryl-CoA; BuAld, Butyraldehyd;
Butyryl-P, Butyryl-Phosphat, 4-HBD, 4-Hydroxybutyryl-CoA Dehydratase.
Die Abbildung 3.24 gibt Aufschluss über die 1,4-BDO- und GHB-Produktion transformierter
C. acetobutylicum-Defektmutanten. Es zeigte sich, dass die 1,4-BDO-Konzentrationen in den
Lösungsmitteldefektmutanten im Vergleich zum Referenzstamm verringert waren. Die
Einbußen lagen dabei im Bereich von 66 - 86 %. Der Stamm C. acetobutylicum adc::int(180)
pTc::P(thlA)cbei_2100 produzierte mit 0,14 mM am meisten 1,4-BDO. Bei den weiteren
Mutantenstämmen lagen die 1,4-BDO-Konzentrationen unter 0,1 mM. Die gemessenen
GHB-Konzentrationen waren ebenfalls verringert, welche jedoch weniger deutlich
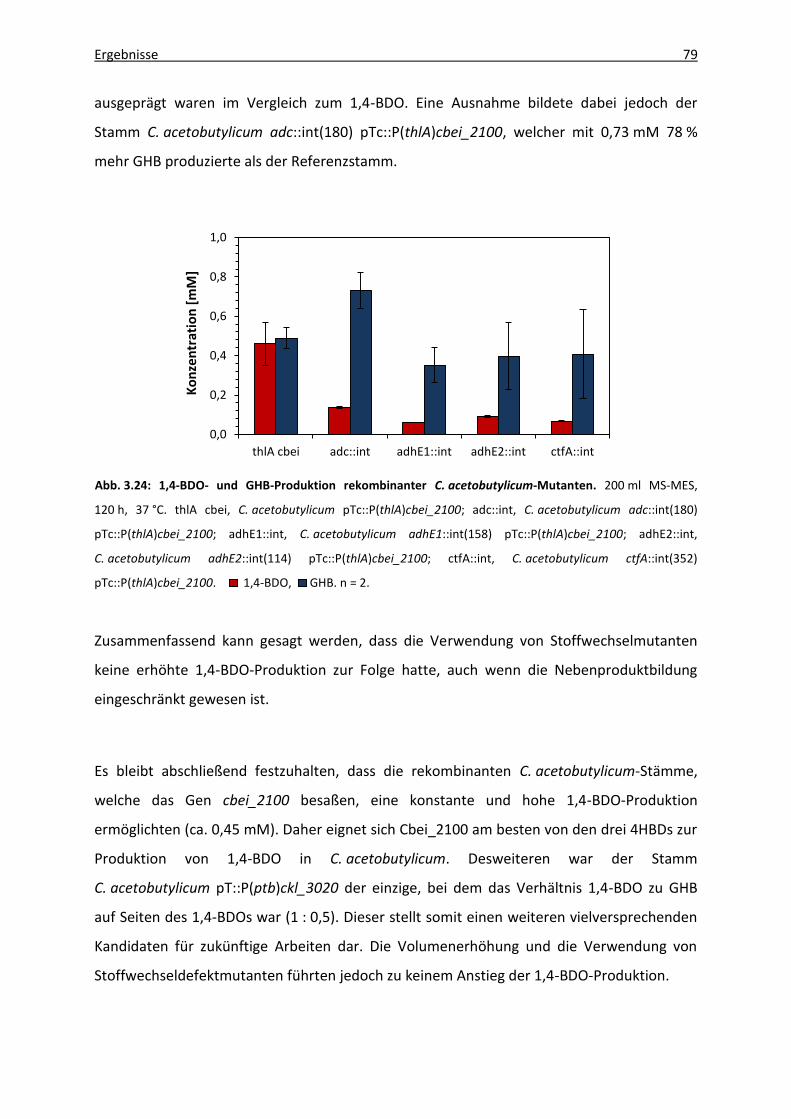
Ergebnisse 79
ausgeprägt waren im Vergleich zum 1,4-BDO. Eine Ausnahme bildete dabei jedoch der
Stamm C. acetobutylicum adc::int(180) pTc::P(thlA)cbei_2100, welcher mit 0,73 mM 78 %
mehr GHB produzierte als der Referenzstamm.
Abb. 3.24: 1,4-BDO- und GHB-Produktion rekombinanter C. acetobutylicum-Mutanten. 200 ml MS-MES,
120 h, 37 °C. thlA cbei, C. acetobutylicum pTc::P(thlA)cbei_2100; adc::int, C. acetobutylicum adc::int(180)
pTc::P(thlA)cbei_2100; adhE1::int, C. acetobutylicum adhE1::int(158) pTc::P(thlA)cbei_2100; adhE2::int,
C. acetobutylicum adhE2::int(114) pTc::P(thlA)cbei_2100; ctfA::int, C. acetobutylicum ctfA::int(352)
pTc::P(thlA)cbei_2100. 1,4-BDO, GHB. n = 2.
Zusammenfassend kann gesagt werden, dass die Verwendung von Stoffwechselmutanten
keine erhöhte 1,4-BDO-Produktion zur Folge hatte, auch wenn die Nebenproduktbildung
eingeschränkt gewesen ist.
Es bleibt abschließend festzuhalten, dass die rekombinanten C. acetobutylicum-Stämme,
welche das Gen cbei_2100 besaßen, eine konstante und hohe 1,4-BDO-Produktion
ermöglichten (ca. 0,45 mM). Daher eignet sich Cbei_2100 am besten von den drei 4HBDs zur
Produktion von 1,4-BDO in C. acetobutylicum. Desweiteren war der Stamm
C. acetobutylicum pT::P(ptb)ckl_3020 der einzige, bei dem das Verhältnis 1,4-BDO zu GHB
auf Seiten des 1,4-BDOs war (1 : 0,5). Dieser stellt somit einen weiteren vielversprechenden
Kandidaten für zukünftige Arbeiten dar. Die Volumenerhöhung und die Verwendung von
Stoffwechseldefektmutanten führten jedoch zu keinem Anstieg der 1,4-BDO-Produktion.
0,0
0,2
0,4
0,6
0,8
1,0
thlA cbei adc::int adhE1::int adhE2::int ctfA::int
Ko
nze
ntr
atio
n [
mM
]

Diskussion 80
4. Diskussion
Die heutige Weltwirtschaft ist überwiegend von verschiedenen fossilen Energiequellen, wie
Öl, Kohle und Gas abhängig (Sarkar et al., 2012). In Anbetracht der steigenden
Weltbevölkerung und dem damit einhergehenden Anstieg des Energiebedarfs und der
Ausbeutung nicht-erneuerbarer Energiequellen wird ein kritischer Punkt im Hinblick auf
Nachhaltigkeit und Klimawandel erreicht. Alternative Strategien, wie die Nutzung
erneuerbarer Ressourcen für die biotechnologische Produktion von Treibstoffen und
Basischemikalien, könnten dieser Situation entgegenwirken.
In den letzten Jahrzehnten wurde eine Vielzahl verschiedenster Forschungen auf dem Gebiet
der mikrobiellen Produktion von Chemikalien durchgeführt, welche zukünftig mit der
Petrochemie konkurrieren könnten (u.a. Biebl et al., 1999; Nakamura und Whited, 2003; Ma
et al., 2009; Zhang et al., 2010). Dabei wurden oft gut erforschte Mikroorganismen wie
Saccharomyces cerevisiae, Escherichia coli oder Klebsiella oxytoca durch genetische
Veränderungen des Genoms zu Produktionsstämmen modifiziert und beständig optimiert
(Nissen et al., 2000; Ji et al., 2010; Clomburg und Gonzalez, 2011). Aufgrund der Fähigkeit
zur Bildung der Lösungsmittel Aceton, Butanol und Ethanol rückte ebenso C. acetobutylicum
wieder in den Fokus und bedingte so die Entwicklung neuer genetischer
Manipulationstechniken (Desai & Papoutsakis, 1999; Papoutsakis, 2008; Lütke-Eversloh &
Bahl, 2011). Diese verbesserten das Verständnis über den Metabolismus von
C. acetobutylicum und führten zu „Metabolic Engineering“-Strategien, die gezielt den
Stoffwechsel veränderten (Lehmann et al., 2012 b und c). So kann auch die Synthese von
Verbindungen, welche natürlicherweise nicht in einem Organismus vorkommen, in Betracht
gezogen werden. Hierzu zählt unter anderem das bisher ausschließlich petrochemisch
erzeugte 1,4-Butandiol, welches im industriellen Großmaßstab mit 1,3 Mio. Tonnen pro Jahr
für die Produktion von Plastik und Polyester eingesetzt wird (Yim et al., 2011). Durch die
zusätzliche genetische Information zur heterologen Expression der 4HBD und der damit
einhergehenden Veränderung des Stoffwechsels von C. acetobutylicum sollte diese
Verbindung auf biologische Weise und damit unabhängig von den bisher genutzten
petrochemischen Verfahren erzeugt werden.

Diskussion 81
4.1 1,4-Butandioltoleranz
Alkohole und andere organische Lösungsmittel, wie Aromate und Phenole, besitzen eine
toxische Wirkung auf die Zellen, sodass die mikrobielle Produktion dieser Chemikalien
limitiert wird (Isken und de Bont, 1998; Tomas et al., 2003). Die Akkumulation von
organischen Lösungsmitteln führt unter anderem zu einer Permeabilisierung der
Zellmembran und zu einem passiven Flux von ATP, Protonen und Ionen (Sikkema et al.,
1995). Dadurch kommt es zum Zusammenbruch der Protonenmotorischen Kraft sowie des
Protonengradienten, was wiederum einen Einbruch im Energieaushalt der Zelle nach sich
zieht (Tomas et al., 2003).
Ähnliche Effekte konnten auch bei C. acetobutylicum in Gegenwart von Butanol beobachtet
werden (Bowles und Ellefson, 1985). Butanol bewirkt aufgrund seiner chaotrophen Wirkung
auf die Zellmembran eine Änderung in der Phospholipidzusammensetzung (Ingram, 1976;
Vollherbst-Schneck et al., 1984; Baer et al., 1987) und führt letztlich zu ihrer Zerstörung.
Dadurch ist die Zelle nicht mehr in der Lage, den intrazellulären pH-Wert sowie das
Membranpotential aufrechtzuerhalten (Tomas et al., 2003). Die Aktivitäten der
membrangebundenen ATPase, der Glukose-Aufnahmesysteme und Nährstofftransporter
kommen zum Erliegen (Bowles und Ellefson, 1985). Aufgrund dessen ist die Produktion von
Butanol bei C. acetobutylicum auf etwa 13 g/l limitiert (Jones und Woods, 1986).
Im Gegensatz dazu zeigte sich, dass C. acetobutylicum in der Lage ist, bis zu 5 % an
extrazellulärem 1,4-BDO zu tolerieren. Erste wachstumsinhibierende Effekte traten ab einer
Konzentration von 54 g/l auf (3.1). Damit ist die Toleranzgrenze gegenüber 1,4-BDO ca.
viermal höher als gegenüber Butanol, wenn 1,4-BDO der alleinige Stressfaktor ist. Ein
ähnliches Toleranzverhalten gegenüber 1,4-BDO konnte auch bei E. coli beobachtet werden.
Dessen natürliche Toleranz liegt ebenfalls bei 5 - 6 %, welche durch entsprechende
Adaptationsversuche auf > 10 % angehoben werden konnte (Burk, 2010). Die hohen
biotechnologischen Produktionen anderer Diole deuten auf ebenso hohe Toleranzwerte der
entsprechenden Organismen hin (Zeng und Sabra, 2011). So ist beispielsweise ein
rekombinanter E. coli-Stamm in der Lage, über 130 g/l 1,3-Propandiol (1,3-PDO) zu
produzieren (Nakamura und Whited, 2003). In vergleichbaren Konzentrationen wird
2,3-Butandiol (2,3-BDO) von Klebsiella pneumoniae (130 g/l) bzw. Serratia marcescens
(152 g/l) synthetisiert (Ma et al., 2009; Zhang et al., 2010).

Diskussion 82
Die Toleranz von Mikroorganismen gegenüber Diolen scheint dabei deutlich höher zu sein
als gegenüber Alkoholen. Somit sind Diole im Vergleich zum chemisch ähnlichen Butanol
vermeintlich weniger toxisch für die Zellen. Die Toxizität einer Verbindung korreliert dabei
mit dem Oktanol-Wasser-Koeffizienten (log Pow) (De Bont, 1998). Dieser beschreibt das
Verhältnis der Konzentration einer Substanz in einem Zwei-Phasen-System bestehend aus
n-Oktanol und Wasser (Sangster, 1989) und ermöglicht Rückschlüsse über das Verhalten von
Verbindungen in lipophilen bzw. wässrigen Lösungen. Positive log Pow-Werte deuten auf eine
lipophile Verbindung hin, negative Werte auf eine hydrophile. Generell gilt, je geringer der
log Pow-Wert, desto weniger toxisch ist die Verbindung (Siemerink et al., 2011).
Verbindungen mit einem log Pow-Wert zwischen 1 und 5 sind stark toxisch für
Mikroorganismen (Isken und de Bont, 1998). Butanol mit einem log Pow-Wert von 0,8 (Inoue
und Horikoshi, 1989) besitzt einen positiven Wert und kann dementsprechend gut mit der
lipophilen Zellmembran interagieren, wodurch die zuvor beschriebenen Effekte auf die
Membran hervorgerufen werden. Diole, wie 1,3-PDO, 2,3-BDO und 1,4-BDO, hingegen
besitzen negative log Pow-Werte (-1,0; -0,92 bzw. -0,8) (https://pubchem.ncbi.nlm.nih.gov).
Diese sind bis zu einer Einheit niedriger als beim Butanol. Dies bedeutet, dass Diole folglich
schlechter mit der Membran interagieren können und so ein toxischer Effekt dieser
Verbindungen erst bei deutlich höheren Konzentrationen auftritt.
4.2 1,4-Butandiolproduktion
Die heterologe Expression der 4-Hydroxybutyryl-CoA Dehydratase/Vinylacetyl-CoA-∆-
Isomerase (4HBD) generierte einen Abzweig im Stoffwechsel von C. acetobutylicum
(Abb. 1.3) und befähigt die rekombinanten Stämme, 1,4-BDO auf biologischem Wege unter
Nutzung von nachwachsenden Rohstoffen zu produzieren. Dabei lagen die Ausbeuten in
Abhängigkeit der drei in dieser Arbeit heterolog in C. acetobutylicum exprimierten 4HBD-
kodierenden Gene (abfD, cbei_2100 und ckl_3020) im Bereich von ca. 0,1 - 0,5 mM
(9 - 45 mg/l) (Abb. 3.12). Ähnliche Konzentrationen wurden bei den frühen rekombinanten
E. coli-Stämmen von Hwang et al. (2014) (29 mg/l) und Yim et al. (2011) (54 mg/l) erzielt. Im
Falle von E. coli konnten durch Anpassungen und Veränderungen die Produktionen auf
0,66 g/l (Hwang et al., 2014) bzw. 18 g/l (Yim et al., 2011) erhöht werden. Um gegenüber der

Diskussion 83
chemischen Erzeugung von 1,4-BDO wettbewerbsfähig zu sein, wird eine biotechnologische
Produktionsleistung von > 100 g/l, wie bei 1,3-PDO und 2,3-BDO, vorausgesetzt (Zeng und
Sabra, 2011). Die hier bisher erzielten Konzentrationen sind noch deutlich geringer. Sie
zeigten aber erstmalig die prinzipielle Realisierbarkeit der anaeroben 1,4-BDO-Synthese
durch C. acetobutylicum und stellen damit die Grundlage für die weitere Optimierungen dar.
Allerdings ist die Produktion von 1,4-BDO von verschiedenen Faktoren beeinflussbar, welche
die Gesamtausbeute limitieren können. Da sie für die weitere Strategie essentiell sind, sollen
die Faktoren im Folgenden erörtert werden.
4.2.1 Limitierung durch die 4HBD
Zu den limitierenden Faktoren zählt unter anderem das Eingangsenzym der 1,4-BDO-
Synthese, die 4HBD. Dieses Enzym katalysiert die reversible Reaktion von 4-Hydroxybutyryl-
CoA (4-HB-CoA) zu Crotonyl-CoA (Willadsen und Buckel, 1990; Scherf und Buckel, 1993;
Scherf et al., 1994; Müh et al., 1996; Martins, 2004). Dabei liegt die Gleichgewichtskonstante
der Reaktion mit K = 4,2 deutlich auf Seiten von Crotonyl-CoA (Scherf und Buckel, 1993). Die
Bevorzugung der Crotonyl-CoA-Bildung spiegelt sich ebenfalls in der Kinetik der reversiblen
Dehydratation von 4-HB-CoA wider. Während ca. 80 % des 4-HB-CoA durch die 4HBD zu
Crotonyl-CoA umgewandelt wird, beträgt der Anteil der Rückreaktion (Crotonyl-CoA zu
4-HB-CoA) gerade einmal 20 % (Scherf und Buckel, 1993). Eine Ursache für die Begünstigung
der Crotonyl-CoA-Bildung könnte die höhere Stabilität von Crotonyl-CoA im Vergleich zu
4-HB-CoA sein (Müh et al., 1996).
Darüber hinaus ist die spezifische Aktivität der 4HBD für 4-HB-CoA mit 209 nkat/mg relativ
niedrig (Scherf und Buckel, 1993). In der Regel liegt die spezifische Aktivität von Enzymen
zumeist zwischen 1 - 2 µkat/mg (Scherf und Buckel, 1993), wie z. B. die am
Primärstoffwechsel von C. acetobutylicum beteiligte Thiolase (3,6 µkat/mg; Wiesenborn et
al., 1988). Die Aktivität der 4HBD ist somit um den Faktor 5 bis 10 niedriger. Um dies zu
kompensieren, ist die 4HBD das dominierende Enzym im Cytosol mit einem Anteil von
20 - 25 % in C. aminobutyricum (Scherf und Buckel, 1993). Durch die in dieser Arbeit
erzielten heterologen Expressionen der 4HBDs in C. acetobutylicum (Abb. 3.9 und Abb. A14)
ist dieser hohe Anteil nicht gegeben, sodass die geringe spezifische Aktivität des Enzyms
nicht durch die Proteinmenge ausgeglichen werden konnte.

Diskussion 84
Das bedeutet, eine geringe Enzymmenge und die Bevorzugung der Crotonyl-CoA-Bildung
durch die 4HBD lassen in direkter Konsequenz auch nur wenig 4-HB-CoA erwarten und in der
Folge auch nur minimale Mengen an 1,4-BDO.
4.2.2 Limitierung durch die Aldehyd-/Alkoholdehydrogenase (AdhE)
Desweiteren können die beiden nachfolgenden Reduktionsreaktionen von 4-HB-CoA über
4-Hydroxybutyraldehyd zum 1,4-BDO, welche durch die bifunktionelle Aldehyd-/Alkohol-
Dehydrogenase (AdhE) von C. acetobutylicum katalysiert werden, limitierend wirken.
Bereits 1986 stellten Jewell et al. fest, dass der Wildtyp von C. acetobutylicum in der Lage ist,
externes 4-Hydroxybutyrat (4-HB bzw. GHB) zu 1,4-BDO zu verstoffwechseln. Die
verantwortlichen Gene bzw. Enzyme waren jedoch noch nicht identifiziert. Erst 2011 wurde
durch Yim et al. die Beteiligung der AdhE2 von C. acetobutylicum an der Bildung von 1,4-BDO
nachgewiesen. Dies stellte bisher das einzige Enzym dar, welches unter in vivo Bedingungen
dazu befähigt war. Entsprechende Untersuchungen hinsichtlich der Enzymaktivität zeigten
eine spezifische Aktivität von 2,5 - 4,8 U/mg (Yoo et al., 2015). Bei der Nutzung eines nicht-
natürlichen Substrates wird die Umwandlungseffizienz jedoch deutlich herabgesetzt.
Darüber hinaus wird die AdhE2 im Gegensatz zur AdhE1 spezifisch in alkohologenen Kulturen
exprimiert, welche sich durch die Bildung von Ethanol und Butanol, aber nicht von Aceton,
auszeichnen (Fontaine et al., 2002). Dieses metabolische Stadium wird beim Wachstum bei
neutralem pH-Wert und unter hoher NAD(P)H-Verfügbarkeit eingeleitet und unterscheidet
sich somit vom solventogenen Zustand, in dem die Lösungsmittel Aceton, Butanol und
Ethanol in der Regel bei saurem pH-Wert produziert werden (Fontaine et al., 2002), welches
den Kulturbedingungen in dieser Arbeit entspricht. Untersuchungen konnten zeigen, dass
während des solventogenen Zustands der Proteinanteil der AdhE2 in der Zelle
verschwindend gering ist und der Anteil an der Butanolbildung gerade einmal 5 - 9 % beträgt
(Yoo et al., 2015). Ein anderes Bild zeigt sich jedoch beim alkohologenen Stadium. Dieses
wird ausschließlich mit 94 - 100 % Beteiligung an der Butanolbildung von der AdhE2
dominiert (Yoo et al., 2015). Das bedeutet, dass die Verfügbarkeit des mutmaßlich
hauptverantwortlichen Enzyms (AdhE2) für die beiden finalen Syntheseschritte der 1,4-BDO-
Bildung unter den solventogen Bedingungen dieser Arbeit massiv eingeschränkt gewesen ist.
Dadurch wurde die 1,4-BDO-Synthese vermutlich negativ beeinflusst. In diesem Fall könnte

Diskussion 85
sich die parallele Überexpression der AdhE2 in C. acetobutylicum anbieten, da auch Yim
et al. (2011) eine Zunahme der 1,4-BDO-Produktion bei erhöhter AdhE2-Expression
verzeichnen konnten. Desweiteren ist es möglich den alkohologenen Zustand der Kulturen
zu induzieren, indem einerseits der pH-Wert auf pH 6,5 reguliert und andererseits Glycerol
als zusätzliches Substrat eingesetzt wird. Glycerol ist im Vergleich zur Glukose deutlich
stärker reduziert und setzt bei der Umwandlung zum Pyruvat doppelt so viel NADH frei
(Vasconcelos et al., 1994), welches über die NADH-Aufnahmewege (Butanol-/ Ethanol- bzw.
1,4-BDO-Bildung) oxidiert werden muss.
4.2.3 Codon Usage
Wie in Abschnitt 3.4.2 dargestellt, variierte die Produktion von 1,4-BDO der rekombinanten
C. acetobutylicum-Stämme in Abhängigkeit der 4HBD-kodierenden Gene (Abb. 3.12). Dabei
konnte unter Verwendung des Enzyms Cbei_2100 die höchste Konzentration an 1,4-BDO mit
0,46 mM detektiert werden, gefolgt von Ckl_3020 mit 0,26 mM und AbfD mit 0,13 mM.
Obwohl die Aminosäuren im katalytischen Zentrum bei allen drei 4HBDs konserviert sind
(Abb. A5), weichen die 1,4-BDO-Ausbeuten doch erheblich voneinander ab. Dies ist somit
nicht auf das katalytische Zentrum zurückzuführen. Da die 4HBDs untereinander nur bis zu
73 - 79 % übereinstimmen, scheinen die nicht-konservierten Aminosäuren einen Effekt auf
die Bildung der jeweiligen 4HBDs und damit auch auf die erzielbaren 1,4-BDO-
Konzentrationen zu haben. Zusätzlich zu den Aminosäuresequenzen unterscheiden sich die
4HBDs hinsichtlich ihres GC-Gehalts sowohl untereinander als auch zum Wirtsorganismus.
Dieser ist mit ca. 25 % (Cornillot et al., 1997) deutlich niedriger als die GC-Gehalte der 4HBDs
(Cbei_2100: 33,3 %, Ckl_3020: 35,4 % und AbfD: 41,5 %; http://www.bioinformatics.org/sms
2/codon_usage.html). Der GC-Gehalt beeinflusst dabei die Codon-Verteilung eines
Organismus (Gustafsson et al., 2004), welches wiederum einen Einfluss auf das Codon Usage
der 4HBDs hat.
Während bei der Translation von Cbei_2100 überwiegend die gleichen Codons wie in
C. acetobutylicum verwendet werden, variiert das Codon Usage bei Ckl_3020 etwas und bei
AbfD deutlich (Tab. A4). So werden bei Ckl_3020 die drei Aminosäuren Glutamin (Gln),
Aspartat (Asp) und Lysin (Lys) neben den mit dem Wirtsorganismus übereinstimmenden
Codons auch durch weitere synonyme Codons kodiert. Diese zweiten synonymen Codons

Diskussion 86
(CAG, GAC bzw. UGC) werden in C. acetobutylicum wesentlich seltener verwendet.
Besonders deutlich werden die Unterschiede beim Codon Usage zwischen AbfD und
C. acetobutylicum. Hier werden die Aminosäuren Phenylalanin (Phe), Isoleucin (Ile), Prolin
(Pro), Histidin (His), Glutamin (Gln), Asparagin (Asn), Lysin (Lys), Cystein (Cys) und Glycin
(Gly) bevorzugt von anderen Codons kodiert, die in C. acetobutylicum deutlich
unterrepräsentiert sind. Darüber hinaus werden die synonymen Codons der Aminosäuren
Serin (Ser), Tyrosin (Tyr) und Aspartat (Asp) mit ähnlich hoher Häufigkeit verwendet, wie die
bevorzugten in C. acetobutylicum.
Die Häufigkeit, in welcher die unterschiedlichen Codons verwendet werden, variiert dabei
deutlich zwischen den verschiedenen Organismen (Gustafsson et al., 2004, Hershberg und
Petrov, 2008), sodass ein unterschiedliches Codon Usage zum Teil gravierende Auswirkungen
auf die heterologe Proteinexpression haben kann (Kane, 1995; Kurland und Gallant, 1995;
Plotkin und Kudla, 2011). Dabei können Fehler im Ablauf der Proteinsynthese durch
Abstoppen der Ribosomen an Tripletts, welche bestimmte, seltene tRNA-Moleküle
benötigen, auftreten. Dies kann dazu führen, dass entweder die Synthese abbricht, eine
falsche Aminosäure eingebaut wird (Substitution) oder es zum „in-frame hopping“ kommt,
wodurch der Leserahmen um eine Position nach vorne oder hinten verschoben und damit
die Codonabfolge verändert wird (Kurland, 1991; Kurland und Gallant, 1995). Somit ist die
quantitative und qualitative Produktion des zu synthetisierenden Proteins reduziert (Kane,
1995). Die unterschiedliche Verwendung der Codons insbesondere bei AbfD im Vergleich
zum Wirtsorganismus kann daher die Proteinsynthese derart beeinträchtigen, dass
schlussendlich die finale 1,4-BDO-Konzentration am geringsten war. Abhilfe könnte der
Einsatz von synthetischen Genen, die auf den entsprechenden Wirtsorganismus abgestimmt
sind, bringen. Das „Protein-Engineering“ ist heutzutage eine gängige Strategie beim
„Metabolic-Engineering“. So konnte im Vergleich zum nativen Enzym durch eine Codon-
optimierte Variante die 1,4-BDO-Produktion in E. coli gesteigert werden (Yim et al., 2011).
4.3 GHB-Produktion
Neben 1,4-BDO konnte in den rekombinanten C. acetobutylicum-Stämmen GHB (0,49 mM,
0,66 mM bzw. 0,63 mM) nachgewiesen werden (Abb. 3.12). Dabei wurden höhere
Konzentrationen an GHB als an 1,4-BDO gemessen, wodurch das 1,4-BDO-GHB-Verhältnis

Diskussion 87
auf Seiten des GHB lag. Bei niedrigeren 1,4-BDO-Konzentrationen waren erhöhte GHB-
Konzentrationen feststellbar. Die GHB-Bildung konnte bereits während der exponentiellen
Wachstumsphase detektiert werden, wohingegen 1,4-BDO erst in der stationären Phase
vermehrt produziert wurde (Abb. 3.13).
GHB als Nebenprodukt wurde ebenso bei den Arbeiten von Yim et al., (2011) festgestellt. Die
GHB-Konzentrationen des frühen rekombinanten E. coli-Stammes waren mit 2,1 mM etwa
um den Faktor 3 höher als die des 1,4-BDOs (0,6 mM). Es wird angenommen, dass das GHB
spontan durch Abspaltung der CoA-Gruppe aus 4-HB-CoA entsteht (Yim et al., 2011)
(Abb. 4.1) und somit nur indirekt durch den Einsatz einer effizienteren Umwandlung von
4-HB-CoA zu 1,4-BDO beeinflusst werden kann.
Abb. 4.1: Schematische Darstellung der potentiellen Bildungswege von 4-Hydroxbutyrat (GHB).
Darüber hinaus könnte in C. acetobutylicum eine weitere theoretische Möglichkeit der GHB-
Bildung existieren. Ein bereits im Wirtsorganismus vorhandener und somit natürlicher
Stoffwechselweg ist die Umwandlung von Butyryl-CoA über Butyryl-Phosphat zu Butyrat
durch die beiden Enzyme Phosphotransbutyrylase (Ptb) und Butyratkinase (Buk)
(Wiesenborn et al., 1989). Das vielfältige Substratspektrum der Ptb (Wiesenborn et al., 1989)
beinhaltet unter anderem auch 4-HB-CoA (Liu und Steinbüchel, 2000). Dieses wird mit einer
spezifischen Aktivität von 21,7 U/mg Protein zu 4-HB-Phosphat umgewandelt, was einer
Restaktivität von 3 % im Vergleich zum Butyryl-CoA entspricht (Liu und Steinbüchel, 2000).
Analog zum Butyryl-Phosphat könnte 4-HB-Phosphat durch die Buk zu GHB verstoffwechselt

Diskussion 88
werden (Abb. 4.1). Letzterer Schritt ist theoretisch denkbar, da die Buk auch die
entgegengesetzte Reaktion – die Phosphorylierung von GHB – mit einer Restaktivität von 7 %
im Vergleich zum Butyrat katalysiert (Liu und Steinbüchel, 2000). Es ist vorstellbar, dass die
Dephosphorylierungsreaktion von 4-HB-Phosphat mit einer höheren Aktivität abläuft, da
dies mit der eigentlichen Funktion der Buk (Dephosphorylierung von Butyryl-Phosphat)
übereinstimmt. Demnach könnten die beiden in C. acetobutylicum natürlich vorkommenden
Enzyme (Ptb und Buk) zu einem weiteren Stoffwechselweg führen, welcher die Umwandlung
von 4-HB-CoA zu GHB beinhaltet. Durch die Synthese eines zusätzlichen Moleküls ATP kann
dieser unter Umständen vom Organismus bevorzugt werden.
4.4 1,4-BDO-Produktion unter Verwendung verschiedener
Kohlenhydratquellen
Solventogene Clostridien, wie C. acetobutylicum, sind in der Lage, die Lösungsmittel Aceton,
Butanol und Ethanol aus einer Vielzahl verschiedener Kohlenhydratquellen zu generieren
(Ounine et al., 1985; Qureshi et al., 2006; Zverlov et al., 2006; Ren et al., 2010). Dabei
können sowohl Hexosen, wie z. B. Mannose und Galaktose, als auch Pentosen, wie z. B.
Arabinose und Xylose von C. acetobutylicum verstoffwechselt werden (Servinsky et
al., 2010). Dieses weitgefächerte Substratspektrum ermöglicht dem Bakterium einen
Wettbewerbsvorteil gegenüber anderen Mikroorganismen, da unter anderem die Fähigkeit
zur Fermentation von Pentosen bei Mikroorganismen eingeschränkt ist (Ounine et al., 1985).
Desweiteren gestattet die Substratvielfalt eine größere Handlungsfreiheit bei der
industriellen Nutzung von C. acetobutylicum, da bis zu 79 % der Kosten durch Substratkosten
verursacht werden (Green, 2011). Daher wird nachfolgend die Verwertung ausgewählter
Pentosen, wie Xylose und Arabinose aber auch der Hexosen Galaktose und Mannose
erörtert.
4.4.1 Pentosen
4.4.1.1 Xylose
Die Verwendung von D-Xylose als alleinige C-Quelle führte bei C. acetobutylicum
pTc::P(thlA)cbei_2100 zu einem langsameren Wachstum (µ = 0,11 h-1) im Vergleich zum
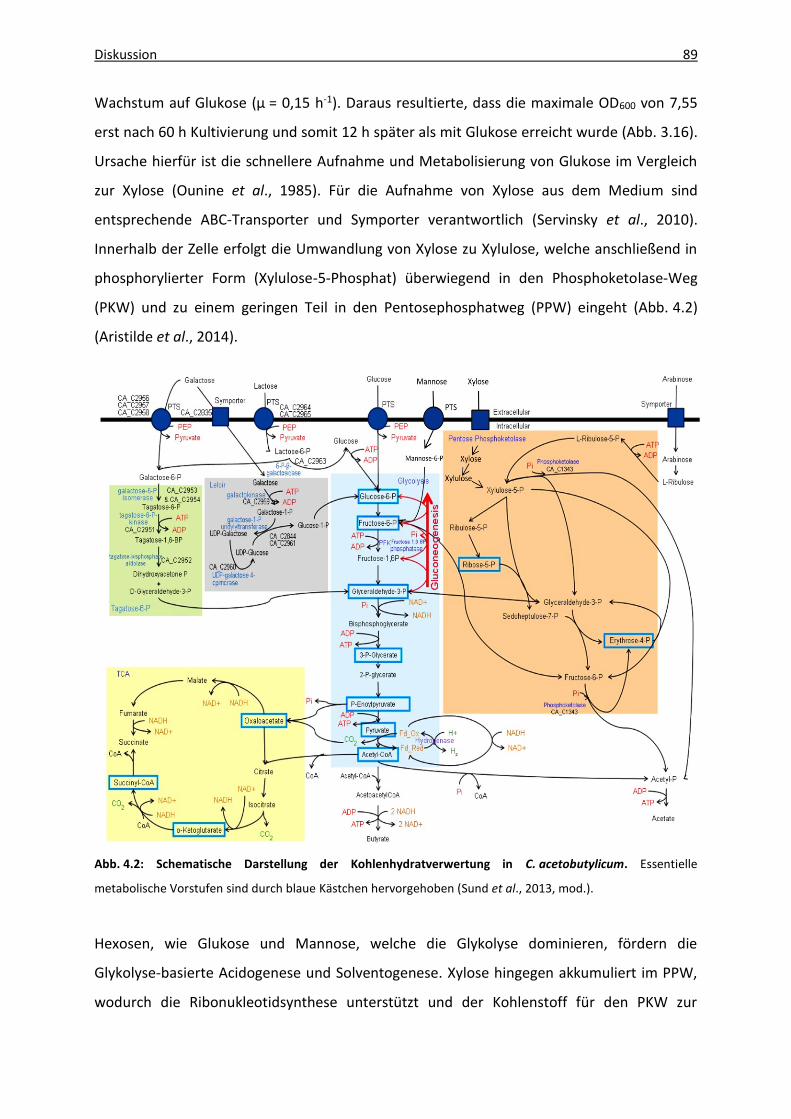
Diskussion 89
Wachstum auf Glukose (µ = 0,15 h-1). Daraus resultierte, dass die maximale OD600 von 7,55
erst nach 60 h Kultivierung und somit 12 h später als mit Glukose erreicht wurde (Abb. 3.16).
Ursache hierfür ist die schnellere Aufnahme und Metabolisierung von Glukose im Vergleich
zur Xylose (Ounine et al., 1985). Für die Aufnahme von Xylose aus dem Medium sind
entsprechende ABC-Transporter und Symporter verantwortlich (Servinsky et al., 2010).
Innerhalb der Zelle erfolgt die Umwandlung von Xylose zu Xylulose, welche anschließend in
phosphorylierter Form (Xylulose-5-Phosphat) überwiegend in den Phosphoketolase-Weg
(PKW) und zu einem geringen Teil in den Pentosephosphatweg (PPW) eingeht (Abb. 4.2)
(Aristilde et al., 2014).
Abb. 4.2: Schematische Darstellung der Kohlenhydratverwertung in C. acetobutylicum. Essentielle
metabolische Vorstufen sind durch blaue Kästchen hervorgehoben (Sund et al., 2013, mod.).
Hexosen, wie Glukose und Mannose, welche die Glykolyse dominieren, fördern die
Glykolyse-basierte Acidogenese und Solventogenese. Xylose hingegen akkumuliert im PPW,
wodurch die Ribonukleotidsynthese unterstützt und der Kohlenstoff für den PKW zur

Diskussion 90
Verfügung gestellt wird (Aristilde et al., 2014). Da durch die Verwendung von Xylose als C-
Quelle andere Stoffwechselwege gefördert werden, kam es im Vergleich zum Wachstum auf
Glukose zur Verringerung der 1,4-BDO-Synthese um 67 % (Abb. 3.17). Ein weiterer wichtiger
Aspekt, welcher die 1,4-BDO-Bildung beeinträchtigen kann, ist die höhere Anfälligkeit des
Xylose-Metabolismus gegenüber Butanol, aufgrund der Inhibierung der Xylose-Permease ab
einer Butanolkonzentration von 8 g/l (Ounine et al., 1985). Demgegenüber wird der
Stoffwechsel beim Wachstum auf Glukose erst ab einer Butanolkonzentration von 13 g/l
inhibiert (Jones und Woods, 1986).
4.4.1.2 Arabinose
Unter Verwendung von L-Arabinose als Substrat war das Wachstum von C. acetobutylicum
pTc::P(thlA)cbei_2100 im Vergleich zur Glukose (µ = 0,15 h-1) minimal verlangsamt
(µ = 0,13 h-1), erreichte jedoch wie auf Xylose eine maximale OD600 von 7,7 nach 60 h
Kultivierung (Abb. 3.16).
Ähnlich wie Xylose wird Arabinose über Symporter in die Zelle aufgenommen (Servinsky et
al., 2010) und über L-Ribulose und L-Ribulose-5-Phosphat dem Stoffwechsel zugeführt
(Abb. 4.2). Für die Metabolisierung zeigt sich ebenfalls der Phosphoketolaseweg (PKW)
hauptverantwortlich (Aristilde et al., 2014). Darüber hinaus wird der PKW durch den
Pentosephosphatweg unterstützt (Sund et al., 2013). Der metabole Flux in die Glykolyse ist
hingegen nur minimal (Aristilde et al., 2014), was sich ebenfalls auf die 1,4-BDO-Produktion
auswirkt und zu einer Verminderung um 27 % führte (Abb. 3.17). Trotz ähnlich genutzter
Stoffwechselwege ist die 1,4-BDO-Produktion auf Arabinose (0,3 mM) höher als auf Xylose
(0,15 mM). Als Grund ist die Bevorzugung von Arabinose von C. acetobutylicum zu nennen
(Qureshi et al., 2006; Aristilde et al., 2014). Das liegt einerseits daran, dass in Anwesenheit
von Arabinose die mRNA-Expression von Enzymen des PKW um das 100-fache gesteigert
wird (Servinsky et al., 2010). Andererseits gilt der Xylose-Metabolismus als ineffizienter
durch die ratenlimitierenden Schritte beim Transport, bei der Isomerisierung und
Phosphorylierung von Xylose (Xiao et al., 2011), was sich ebenso in der niedrigeren
Wachstumsrate widerspiegelte (Ounine et al., 1983).

Diskussion 91
4.4.2 Hexosen
4.4.2.1 Galaktose
Der rekombinante Stamm C. acetobutylicum pTc::P(thlA)cbei_2100 erreichte beim
Wachstum auf Galaktose die geringste maximale OD600 von 5,5. Dafür konnte sich dieser am
längsten in der stationären Phase halten (Abb. 3.16).
Für die Metabolisierung von Galaktose stehen C. acetobutylicum zwei verschiedene Wege
zur Verfügung (Abb. 4.2): zum einen der Leloir-Weg (LW) und zum anderen der Tagatose-6-
Phosphatweg (T6PW), (Servinsky et al., 2010; Sund et al., 2013). Die Aufnahme von
Galaktose aus dem Medium erfolgt einerseits durch Symporter und andererseits durch das
Phosphoenolpyruvat-Transferase-System (PTS), (Sund et al., 2013). Dabei wird in
Abhängigkeit der Aufnahmesysteme Galaktose entweder über den bevorzugten Leloir-Weg
(Symporter-assoziiert) oder über den T6P-Weg (PTS-assoziiert) metabolisiert (Sund et al.,
2013). Deren Intermediate können anschließend in die Glykolyse eingehen. Das bedeutet,
dass für das Wachstum auf Galaktose zwei verschiedene Stoffwechselwege, die einer
komplexen Regulation unterliegen (Servinsky et al., 2010), aufrechterhalten werden müssen.
Dies bedeutet jedoch höhere Kosten für den Organismus. Infolgedessen ist die
Lösungsmittelproduktion auf Galaktose verringert (Yu & Saddler, 1982) und führte letztlich
zu einer verminderten 1,4-BDO-Synthese, die mit 0,11 mM am geringsten von den
eingesetzten Kohlenhydraten war (Abb. 3.17).
4.4.2.2 Mannose
Mannose zählt zu den bevorzugten Kohlenhydratquellen von C. acetobutylicum (Servinsky et
al., 2010), was sich auch im ähnlichen Wachstumsverhalten des rekombinanten Stammes
auf Glukose und Mannose widerspiegelte (Abb. 3.16). Es wurde eine ähnlich hohe maximale
OD600 von 8,4 nach 48 h Kultivierung verglichen mit Glukose (OD600 = 8,35) erreicht.
Wie bei Hexosen üblich, erfolgt die Aufnahme von Mannose aus dem Medium über das PTS
(Servinsky et al., 2010; Voigt et al., 2014). In der Zelle wird das gebildete Mannose-6-
Phosphat zu Fructose-6-Phosphat umgewandelt, welches anschließend in die Glykolyse
eingeht (Abb. 4.2). Das bedeutet, für den Mannose-Stoffwechsel wird im Gegensatz zu den
anderen getesteten Kohlenhydratquellen nur ein einziges weiteres Enzym (Mannose-6-

Diskussion 92
Phosphat-Isomerase) benötigt (Servinsky et al., 2010), wodurch die entstehenden Kosten für
den Organismus niedrig gehalten werden. Desweiteren werden die Gene, welche für die
Aufnahme und den Metabolismus von Mannose verantwortlich sind, konstitutiv exprimiert
(Servinsky et al., 2010), wodurch eine schnelle und effiziente Umwandlung ermöglicht wird.
Dennoch stellte sich heraus, dass die 1,4-BDO-Produktion (0,23 mM) im Vergleich zur
Glukose (0,46 mM) verringert war (Abb. 3.17). Es lässt daraus schließen, dass in Anwesenheit
von Glukose die am Stoffwechsel beteiligten Enzyme effektiver stimuliert werden und
dementsprechend effizienter sind.
4.4.3 Kohlenhydratgemisch
In dieser Arbeit konnte gezeigt werden, dass der rekombinante C. acetobutylicum-Stamm
prinzipiell in der Lage ist, 1,4-BDO durch Fermentation verschiedener, einzelner C-Quellen zu
synthetisieren. Dies eröffnet die Möglichkeit auch Lignocellulose-Hydrolysate, in denen diese
Kohlenhydrate vorkommen, für die 1,4-BDO-Produktion einzusetzen.
Lignocellulose ist ein Bestandteil der pflanzlichen Zellwand und gehört demzufolge zu den
nahezu unbegrenzt auf der Erde verfügbaren, nachwachsenden Rohstoffen (Kuhad & Singh,
1993; Lee, 1997; Ezeji et al., 2007), welche jedoch größtenteils ungenutzt bleiben (Sarkar et
al., 2012). Der Einsatz von Forstabfällen oder landwirtschaftlichen Abfallstoffen, wie
Getreidestroh, Reisstroh und Weizenstroh bieten das Potential, die Kosten für
Fermentationssubstrate deutlich zu senken (Lee, 1997). Die Zusammensetzung der
Lignocellulose umfasst drei Hauptgruppen von Polymeren: Cellulose, Hemicellulose und
Lignin. Cellulose stellt den größten Anteil mit 33 - 51 %, gefolgt von Hemicellulose (19 - 34 %)
und Lignin (21 - 32 %) dar (Ibraheem und Ndimba, 2013) und besteht aus β-1,4-glykosidisch-
verknüpften α-D-Glukosemolekülen. Die Hemicellulose ist hingegen ein sehr heterogenes
Polymer bestehend aus den Zuckern L-Arabinose, D-Galaktose, D-Glukose, D-Mannose und
D-Xylose (Mussatto und Teixeira, 2010). Durch Hydrolyse der Lignocellulose können die
Zuckermoleküle anschließend für Fermentationsprozesse eingesetzt werden (Abb. 4.3).

Diskussion 93
Abb. 4.3: Schematische Darstellung der Lignocellulose-Zusammensetzung. Die Pfeile deuten auf die
Hauptzucker der einzelnen Lignocellulose-Fraktionen hin. (Ibraheem und Ndimba, 2013; mod.).
Die Fermentation von simulierten Lignocellulose-Hydrolysaten basierend auf der
Hemicellulose-Zusammensetzung von Getreidestroh (Sarkar et al., 2012) durch
C. acetobutylicum pTc::P(thlA)cbei_2100 erbrachte eine 1,4-BDO-Produktion von 0,15 mM
(Abb. 3.17). Im Vergleich zum Wachstum auf Glukose verringerte sich dabei die 1,4-BDO-
Konzentration um 67 % und entsprach damit der Ausbeute mit Xylose.
C. acetobutylicum ist jedoch nicht in der Lage, die verschiedenen Kohlenhydrate simultan zu
verwerten, da die im Medium enthaltene Glukose die Aufnahme der anderen Kohlenhydrate
inhibiert (= C-Katabolit-Repression, CCR), (Servinsky et al., 2010). Erst wenn die Glukose
nahezu aufgebraucht ist, wird die CCR aufgehoben und die weiteren Kohlenhydrate können
aufgenommen und verstoffwechselt werden. Die CCR limitiert infolgedessen die
Fermentation von Lignocellulose-Hydrolysaten (Ren et al., 2010). Die geringere Glukose-
Konzentration im Gemisch (30 g/l) im Vergleich zum Medium mit Glukose als alleinige
C-Quelle (60 g/l) und die Verwertung der weniger bevorzugten Xylose (Aristilde et al., 2014)
begünstigten die verminderte 1,4-BDO-Bildung.
Nichtsdestotrotz stellt der Einsatz von Lignocellulose-Hydrolysaten aufgrund der
Verfügbarkeit und Kostenersparnis einen vielversprechenden Ansatz für
Fermentationsstrategien dar. Insbesondere da Ren et al. (2010) eine ccpA-Mutante von
C. acetobutylicum generierten, in welcher die CCR unterdrückt wird.

Diskussion 94
4.5 C. acetobutylicum pT::P(ptb)ckl_3020
Der Zeitpunkt der Expression eines Enzyms kann einen starken Effekt auf die Produktbildung
haben (Sillers et al., 2009). Daher sah die Vorgehensweise dieser Arbeit die Verwendung von
drei unterschiedlichen Promotoren für die Expression der 4HBDs vor, die gut untersucht
waren und eine starke Expression in C. acetobutylicum erwarten ließen (3.5.3).
Der Effekt der Promotoren PthlA, Pptb und Padc auf die 1,4-BDO-Produktion mit Cbei_2100 war
jedoch trotz unterschiedlicher Expressionszeitpunkte minimal. Anders verhielt es sich mit
der 1,4-BDO-Synthese unter Verwendung von Ckl_3020. Hier führten die Promotoren Pptb
und Padc zu einer Steigerung der 1,4-BDO-Bildung im Vergleich zum PthlA und infolgedessen zu
einer Verminderung der GHB-Konzentration (Abb. 3.20). Insbesondere C. acetobutylicum
pT::P(ptb)ckl_3020 zeichnete sich durch eine deutliche Zunahme der 1,4-BDO-Produktion
aus (0,62 mM). Analysen der 1,4-BDO-Synthese über die Zeit (Abb. A16) offenbarten, dass
die 1,4-BDO-Bildung nun mehr dem Verhalten von C. acetobutylicum pTc::P(thlA)cbei_2100
(Abb. 3.13, A) und weniger dem von C. acetobutylicum pTc::P(thlA)ckl_3020 (Abb. 3.13, B)
entsprach. Durch die mutmaßlich frühere Expression von Ckl_3020 durch Pptb stieg die
Konzentration von 1,4-BDO bereits nach 48 h Kultivierung an und erreichte bereits nach 60 h
die Maximalkonzentration. Im Vergleich dazu begann die 1,4-BDO-Produktion bei
C. acetobutylicum pTc::P(thlA)ckl_3020 erst nach 60 h. Diesen Effekt konnten auch Sillers
et al. (2009) feststellen: die frühere Expression von Lösungsmittel-assoziierten Enzymen in
C. acetobutylicum führte ebenfalls zu einer früher einsetzenden und vermehrten
Alkoholbildung. Ein Aspekt weiterer fortführender Arbeiten sollte deshalb eine detailliertere,
zeitliche Analyse der Expression der einzelnen Gene beinhalten. Quantitative RT-PCR-
Analysen können dabei Aufschluss über die mRNA-Transkriptmengen geben.
Immunologische Arbeiten mittels entsprechender Antikörper ermöglichen weiterhin eine
Auskunft darüber, inwieweit gesteigerte Konzentrationen von 1,4-BDO mit erhöhten
Enzymmengen korrelieren.

Diskussion 95
4.6 Stoffwechseldefektmutanten
Die Produktion der Lösungsmittel Aceton, Butanol und Ethanol führte zu einer Verringerung
der 1,4-BDO-Produktion, da der Kohlenstofffluss vom neuen Syntheseweg weggeführt
wurde. Zuvor generierte Stoffwechseldefektmutanten von C. acetobutylicum (Lehmann,
2012 a) zeichneten sich zum Teil durch ein stark verändertes Produktspektrum aus (Lehmann
et al., 2012 b und c). Aus diesem Grund wurden für die weitere Herangehensweise dieser
Arbeit verschiedene C. acetobutylicum-Mutanten (Tab. 2.1) für die 1,4-BDO-Synthese
eingesetzt.
4.6.1 Defekte in der Lösungsmittelproduktion
Die rekombinanten Stämme produzierten ebenso wie der Wildtyp die Lösungsmittel Aceton,
Butanol und Ethanol. Dabei konnten jedoch bei C. acetobutylicum pTc::P(thlA)cbei_2100 und
C. acetobutylicum pTc::P(thlA)ckl_3020 erhöhte Konzentrationen an Butanol und Ethanol
detektiert werden (Abb. 3.11). Die für die Lösungsmittelbildung verantwortlichen Enzyme
sind unter anderem die Acetoacetatdecarboxylase (Adc) und Aldehyd-
/Alkoholdehydrogenase (AdhE). Beide Enzyme sind in den entsprechenden
Mutantenstämmen (Lehmann, 2012 a) ausgeschaltet (Tab. 2.1).
Die transformierte Adc-Defektmutante zeigte einen ähnlichen Phänotyp wie die Adc-
Mutante (Lehmann et al., 2012 b). Die Acetonproduktion erfolgte nur noch durch die
spontane Decarboxylierung von Acetoacetat (Han et al., 2011) und wurde dementsprechend
ohne Enzymunterstützung drastisch reduziert (Endkonzentration ca. 5 mM). Infolgedessen
kam es zu einer deutlichen Akkumulation von Acetat (ca. 67 mM), mutmaßlich
hervorgerufen durch einen Rückstau von Acetoacetat, welches wiederum die Enzymaktivität
der Acetoacetyl-CoA:Acyl-CoA-Transferase (CtfAB) durch Feedback-Inhibierung reduzierte
und somit die Acetatassimilation verringerte. Die Reduktion der Acetonbildung führte jedoch
nicht zu einer erhöhten 1,4-BDO-Synthese. Stattdessen war der gesamte Stoffwechsel der
Mutante derart beeinflusst, dass auch die 1,4-BDO-Bildung deutlich reduziert war
(0,14 mM), (Abb. 3.24). In gleicher Weise wirkte sich dieser Effekt auch auf die
Butanolsynthese aus. Diese ist um etwa die Hälfte (ca. 90 mM) im Vergleich zum
Wildtypstamm gesunken und konnte ebenfalls bei Lehmann et al. (2012 b) festgestellt

Diskussion 96
werden. Durch Akkumulation und verringerter Assimilation des Acetats stand der
verwendete Kohlenstoff nicht mehr für die Bildung der Lösungsmittel und insbesondere für
die 1,4-BDO-Synthese zur Verfügung, sodass eine verringerte 1,4-BDO-Produktion die Folge
war.
Auf ähnliche Weise wirkte sich der Defekt im ctfA-Gen der entsprechenden Mutante auf die
1,4-BDO-Bildung aus. Die Coenzym A-Transferase ist für die Assimilierung der Säure Acetat
verantwortlich (Hartmanis et al., 1984). Der Defekt verursachte eine Akkumulation des
Acetats und führte aufgrund der „Kohlenstoffspeicherung“ infolgedessen zu einer
verminderten Lösungsmittel- (Lehmann et al., 2012 b) sowie 1,4-BDO-Produktion
(Abb. 3.24).
Yim et al. zeigten 2011, dass die AdhE2 von C. acetobutylicum in der Lage ist, die Bildung von
1,4-BDO zu katalysieren. Weitere getesteten Aldehyd- und Alkoholdehydrogenasen waren
dazu nicht befähigt. Da C. acetobutylicum weitere Gene für Aldheyd- und
Alkoholdehydrogenasen im Genom besitzt (Nölling et al., 2001), galt es zu überprüfen, ob
diese Fähigkeit der AdhE2 ein Alleinstellungsmerkmal und somit essentiell für die 1,4-BDO-
Bildung ist oder ob eine andere entsprechende Dehydrogenase ebenso die Funktion
ausführen kann. Aus diesem Grund wurden die beiden Aldehyd-/Alkohohldehydrogenasen
AdhE1 und AdhE2 von C. acetobutylicum für Untersuchungen herangezogen. Wenn die
AdhE2 essentiell für die 1,4-BDO-Bildung ist, müsste die Produktion im
Rezipientenmutantenstamm ausbleiben. Bei der transformierten AdhE1-Mutante hingegen
dürfte der Defekt keine Auswirkungen auf die 1,4-BDO-Produktion haben. Es zeigte sich
jedoch, dass sowohl bei der AdhE1-Mutante als auch bei der AdhE2-Mutante die Bildung von
1,4-BDO im Vergleich zum Referenzstamm C. acetobutylicum pTc::P(thlA)cbei_2100 deutlich
sank, nicht aber komplett eingestellt wurde (0,06 mM bzw. 0,09 mM; Abb. 3.24). Dies
könnte bedeuten, dass die AdhE2 nicht hauptverantwortlich für die 1,4-BDO-Bildung ist,
sondern, dass die finalen Syntheseschritte durch beide AdhEs katalysiert werden können.
Inwieweit weitere Aldehyd- und Alkoholdehydrogenasen, wie die
Butyraldehyddehydrogenase (BYDH) und Butanoldehydrogenase (BDH), daran beteiligt sind,
bleibt weiterhin zu überprüfen.
Obwohl die Lösungsmittelbildung bei den Defektmutanten reduziert werden konnte
(Tab. A5), spiegelt sich der veränderte Kohlenstofffluss nicht in einer erhöhten 1,4-BDO-

Diskussion 97
Konzentration wider. Damit blieb die 1,4-BDO-Bildung der Mutantenstämme hinter den
Erwartungen zurück. „Metabolic Engineering“-Strategien, wie z. B. das Ausschalten von
bestimmten Genen, ermöglichen eine Vielzahl von Veränderungen im Stoffwechsel und
bieten ein großes Potential für zukünftige Arbeiten. Es ist jedoch zu berücksichtigen, dass
einzelne Veränderungen zum Teil drastische Auswirkungen auf den Stoffwechsel haben
können und dementsprechend komplex sind. Desweiteren fehlen noch genaue Angaben zum
Transkriptom und zur biochemischen Charakterisierung der jeweiligen Mutantenstämme.
Das Verhalten jener Mutanten auf molekularer Ebene ist somit noch unklar, weshalb weitere
Analysen folgen müssen. Aus diesem Grund ist eine finale Aussage bezüglich der 1,4-BDO-
Produktion der Mutantenstämme auf diesem Stand schwierig und noch nicht eindeutig.
4.6.2 Defekte in der Säureproduktion
Die Produktion der Säuren Acetat und Butyrat dient C. acetobutylicum der
Energiegewinnung (Hartmanis und Gatenbeck, 1984; Wiesenborn et al., 1989) und stellt
somit einen wichtigen Aspekt im Stoffwechsel dar. Die Eingangsenzyme für die jeweiligen
Biosynthesewege sind die Phosphotransacetylase (Pta) und Phosphotransbutyrylase (Ptb).
Letztere steht, wie zuvor bereits beschrieben, im Verdacht die GHB-Produktion durch die
Umwandlung von 4-HB-CoA zu 4-HB-Phosphat (Liu und Steinbüchel, 2000) zu begünstigen.
Um dies näher zu beleuchten, wurde die Ptb-Rezipientenmutante (Tab. 2.1) hinsichtlich der
1,4-BDO- und GHB-Bildung untersucht. Bei einer Beteiligung der Ptb an der GHB-Produktion
müsste die GHB-Konzentration im Vergleich zum Referenzstamm C. acetobutylicum
pTc::P(thlA)cbei_2100 verringert sein, da der mutmaßliche GHB-Syntheseweg ausgeschaltet
ist. Die Ergebnisse zeigten jedoch, dass es zu einem deutlichen Anstieg der GHB- (0,71 mM)
und zu einer Abnahme der 1,4-BDO-Konzentration (0,08 mM) kam (Abb. 3.22). Dies
bedeutet, dass die Bildung von GHB höchstwahrscheinlich überwiegend spontan abläuft und
somit unabhängig von Enzym-katalysierten Reaktionen ist. Um diesen Sachverhalt weiter zu
untermauern und um auszuschließen, dass die Pta, welche eine ähnliche Reaktion
(Acetyl-CoA zu Acetyl-Phosphat) wie die Ptb katalysiert (Boynton et al., 1996), an der GHB-
Bildung beteiligt ist, wurde zusätzlich die Pta-Mutante näher untersucht. Es stellte sich dabei
heraus, dass die GHB-Konzentration im Vergleich zum Ptb-Rezipientenstamm sank
(0,46 mM) und gleichzeitig die 1,4-BDO-Konzentration anstieg (0,19 mM). Vergleichend zum

Diskussion 98
Referenzstamm C. acetobutylicum pTc::P(thlA)cbei_2100 konnte jedoch kein Effekt des Pta-
Defekts auf die GHB-Produktion festgestellt werden.
Die Hinweise deuten somit verstärkt auf eine spontane Bildung von GHB hin und weniger auf
eine enzymkatalysierte. Dass dies im Konzentrationsbereich von ca. 1 mM möglich ist, zeigt
die nicht-enzymatische Decarboxylierung von Acetaldehyd zu Aceton in entsprechenden
Defektmutanten (Lehmann et al., 2012 b). Nichtsdestotrotz sollte dies durch eine Pta/Ptb-
Doppel-„Knock down“ Mutante überprüft werden. Bei dieser wäre ein Überleben der Zellen
noch möglich und der theoretische Einfluss beider Enzyme auf die GHB-Bildung minimal.
4.7 Fazit
Der globale Bedarf und die Bedeutung von 1,4-BDO wächst Jahr für Jahr (in: 1,4-Butanediol
market analysis and segment forecasts to 2020; www.hexareports.com; Stand: 25.02.2016),
sodass die Produktion von 1,4-BDO immer wichtiger wird. Zukunftsorientierte Forschungen
setzen dabei vermehrt auf Erdöl-unabhängige Produktionen von Plattformchemikalien (Zeng
und Sabra, 2011).
Die in dieser Arbeit vorgestellte Strategie ermöglichte durch die Bereitstellung einer 4HBD-
Aktivität eine Erweiterung des zentralen Stoffwechsels und des Produktspektrums von
C. acetobutylicum und die erstmalige anaerobe biotechnologische Erzeugung von 1,4-BDO.
Somit konnte der „Proof-of-Principle“ gezeigt und ein erster, wichtiger Schritt für eine
petrochemisch unabhängige Synthese von 1,4-BDO erfolgreich genommen werden. Die
1,4-BDO-Produktion in C. acetobutylicum wurde durch drei verschiedene 4HBDs ermöglicht.
Da die erzielten 1,4-BDO-Konzentrationen (11 - 45 mg/l) bisher noch deutlich unter den
wirtschaftlichen 100 g/l lagen, wurden bereits erste Optimierungsansätze angedeutet.
Aufgrund der Komplexität des Wirtsorganismus hatten diese bisher jedoch noch keine
1,4-BDO-Steigerung zur Folge, sodass noch weitere wichtige Arbeiten, wie z. B. die Analyse
und Veränderung der Kultivierungsparameter im „Scale-up“ sowie die Aufklärung der
zeitlichen Expression und mRNA-Menge der 4HBD durch immunologische Methoden und
qRT-PCR, notwendig sind. Ein vielversprechender Ansatz, welcher weiterhin verfolgt werden
sollte, zeigte sich durch die Verwertbarkeit von verschiedenen Kohlenhydratquellen für die
1,4-BDO-Synthese, wodurch Lignocellulose-Hydrolysate als kostengünstiges Substrat

Diskussion 99
denkbar sind. Es müssen jedoch noch weitere Untersuchungen, insbesondere mit
Lignocellulose-Hydrolysaten, folgen. Dabei sollte der Einfluss von inhibierenden
Verbindungen, wie z. B. Furfural, Hydroxymethylfurfural, Ferulasäure, Coumarinsäure und
Glucoronsäure, die bei der Aufarbeitung von Lignocellulose entstehen (Ezeji et al., 2007), auf
C. acetobutylicum genauer untersucht werden.
Es bleibt abschließend festzuhalten, dass C. acetobutylicum das Potential durch
Weiterentwicklung und Modifikation besitzt, sich als zukünftiger 1,4-BDO-Produktionsstamm
zu etablieren.

Literaturverzeichnis 100
5. Zusammenfassung
1. Das nicht-natürlich vorkommende 1,4-Butandiol konnte durch einen neuen
anaeroben Stoffwechselweg in Clostridium acetobutylicum auf biologische Weise
erzeugt werden.
2. Für die Erzeugung von 1,4-BDO in C. acetobutylicum wird nur ein zusätzliches Enzym,
die 4HBD, benötigt.
3. Für den 1,4-BDO-Nachweis wurden zwei verschiedene Methoden etabliert: zum
einen eine schnelle und kostengünstige Methode basierend auf der
Dünnschichtchromatografie und zum anderen ein sensitiver Nachweis via GC/MS-
Analysen.
4. Die 1,4-BDO-Ausbeuten variierten in Abhängigkeit der verwendeten 4HBDs:
Cbei_2100 (0,46 mM), Ckl_3020 (0,26 mM) und AbfD (0,13 mM).
5. Als Nebenprodukt der 1,4-BDO-Synthese konnte γ-Hydroxybutyrat (GHB) detektiert
werden. Die Konzentrationen an GHB waren in den meisten rekombinanten
Stämmen höher als 1,4-BDO.
6. Die 1,4-BDO-Produktion mit Hilfe von C. acetobutylicum war unter Verwendung
verschiedener Kohlenhydratquellen (Arabinose, Xylose, Mannose, Galaktose und
Glukose) möglich. Dadurch ist der Einsatz von Lignocellulose-Hydrolysaten für die
Produktion von 1,4-BDO denkbar.
7. Der Austausch des ThiolaseA-Promotors durch den Phosphotransbutyrylase-
Promotor führte bei dem rekombinanten Stamm C. acetobutylicum
pT::P(ptb)ckl_3020 zu einer erhöhten 1,4-BDO-Produktion (0,62 mM) und zu einer
Verschiebung des 1,4-BDO-GHB-Verhältnisses zum 1,4-BDO.
8. Weitere erste Optimierungsansätze, wie der „Scale-up“ und die Verwendung von
Stoffwechseldefektmutanten, führten bisher noch zu keiner Steigerung der 1,4-BDO-
Produktion.

Literaturverzeichnis 101
6. Literaturverzeichnis
Alsaker, K. V., & Papoutsakis, E. T. 2005. Transcriptional program of early sporulation and
stationary-phase events in Clostridium acetobutylicum. J. Bacteriol. 187: 7103-7118.
Anand, P., & Saxena, R. K. 2012. A novel thin-layer chromatography method to screen 1,3-
propanediol producers. J Ind Microbiol Biotechnol. 39: 1713-1718.
Aristilde, L., Lewis, I. A., Park, J. O., & Rabinowitz, J. D. 2014. Hierarchy in Pentose sugar
metabolism in Clostridium acetobutylicum. Appl. Environ. Microbiol.
doi:10.1128/AEM.03199-14.
Atsumi, S., Cann, A. F., Connor, M. R., Shen, C. R., Smith, K. M., Brynildsen, M. P., Chou, K.
J. Y., Hanai, T., & Liao, J. C. 2008. Metabolic engineering of Escherichia coli for 1-butanol
production. Metabol Engineer. 10: 305-311.
Ayoubi, T. A. Y., & Van de Ven, W. J. M. 1996. Regulation of gene expression by alternative
promoters. FASEB J. 10: 453-460.
Baer, S. H., Blaschek, H. P., & Smith, T. L. 1987. Effect of butanol challenge and temperature
on lipid composition and membrane fluidity of butanol tolerant Clostridium acetobutylicum.
Appl. Environ. Microbiol. 53: 2854-2861.
Bahl, H., & Dürre, P. 1993. Clostridia, S. 285-323. In H. Sahm (ed.), Biotechnology, 2nd ed.,
vol. 1. VCH-Verlag GmbH, Weinheim, Germany.
Bergmeyer, H. U. 1983. Methods in enzymatic analysis. Verlag Chemie Weinheim, Germany.
Bertram, J. 1989. Entwicklung eines Systems zum DNA-Transfer und zur Transposon-
Mutagenese für Clostridium acetobutylicum. Dissertation, Georg-August-Universität
Göttingen.
Biebl, H., Menzel, K., Zeng, A.-P., & Deckwer, D.-W. 1999. Microbial production of 1,3-
propanediol. Appl. Microbiol. Biotechnol. 52: 289-297.
Birnboim, H. C., & Doly, J. 1979. A rapid alkaline extraction procedure for screening
recombinant plasmid DNA. Nucl Acids Research 7: 1513-1523.

Literaturverzeichnis 102
Bowles, L. K., & Ellefson, W., L. 1985. Effects of butanol on Clostridium acetobutylicum. Appl.
Environ. Microbiol. 50: 1165-1170.
Boynton, Z. L., Bennett, G. N., & Rudolph, F. B. 1996. Cloning, sequencing, and expression of
genes encoding Phosphotransacetylase and Acetate Kinase from Clostridium acetobutylicum
ATCC 824. Appl. Environ. Microbiol. 62: 2758-2766.
Bradford, M. 1976. A Rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-
254.
Breznak, J. A. & Costilow, R. N. 1994. Physiochemical factors of growth. In: Methods of
general and molecular bacteriology. (Gerhardt, Ed.) Washington DC, USA: American Society
for Microbiology. 137-154.
Buckel, W. 2001. Unusual enzymes involved in five pathways of glutamate fermentation.
Appl. Microbiol. Biotechnol. 57: 263-273.
Buckel, W., Martins, B. M., Messerschmidt, A., & Golding, B. T. 2005. Radical-mediated
dehydration reactions in anerobic bacteria. Biol. Chem. 386: 951-959.
Burk, M. J. 2010. Sustainable production of industrial chemicals from sugars. Int. Sugar J.
112: 30-35.
Chandra, R., Takeuchi, H., & Hasegawa, T. 2012. Methane production from lignocellulosic
agricultural crop wastes: A review in context to second generation of biofuel production.
Ren. Sust. Ener. Rev. 16: 1462-1476.
Chiao, J.-s., & Sun, Z.-h. 2007. History of the acetone-butanol-ethanol fermentation industry
in China: Development of continuous production technology. J. Mol. Microbiol. Biotechnol.
13: 12-14.
Clomburg, J. M., & Gonzalez, R. 2011. Metabolic engineering of Escherichia coli for the
production of 1,2-propanediol from glycerol. Biotechnol. Bioeng. 108: 897-879.
Corey, E. J., & Venkateswarlu, A. 1972. Protection of hydroxyl groups as tert-
butyldimethylsilyl derivatives. J. Am. Chem. Soc. 94: 6190-6191.

Literaturverzeichnis 103
Cornillot, E., Croux, C., & Soucaille, P. 1997. Physical and genetic map of the
Clostridium acetobutylicum ATCC 824 chromosome. J. Bacteriol. 179: 7426-7434.
De Bont, J. A. M. 1998. Solvent-tolerant bacteria in biocatalysis. TIBTECH. 16: 493-499.
Demirbas, M.F. 2006. Current technologies for biomass conversion into chemicals and fuels.
Energy Sources, Part A: recovery, utilization, and environmental effects. 28: 1181-1188.
Desai, R. P., & Papoutsakis, E. T. 1999. Antisense RNA strategies for metabolic engineering
of Clostridium acetobutylicum. Appl. Environ. Microbiol. 65: 936-945.
Dower, W. J., Miller, J. F., & Ragsdale, C. W. 1988. High efficiency transformation of E. coli
by high voltage electroporation. Nucl. Acids Research 16: 6127-6145.
Dürre, P. 2008. Fermentative Butanol production – Bulk chemical and biofuel. Ann. N.Y.
Acad. Sci. 1125: 353-362.
Ezeji, T., Qureshi, N., & Blaschek, H. P. 2007. Butanol production from agricultural residues:
Impact of degradation products on Clostridium beijerinckii growth and butanol fermentation.
Biotechnol. Bioeng. 97: 1460-1469.
Ezeji, T., & Blaschek, H. P. 2008. Fermentation of dried distillers´grains and solubles (DDGS)
hydrolysates to solvents and value-added products by solventogenic clostridia. Bior. Tech.
99: 5232-5242.
Fischer, R. J., Oehmcke, S., Meyer, U., Mix, M., Schwarz, K., Fiedler, T., & Bahl, H. 2006.
Transcription of the pst operon of Clostridium acetobutylicum is dependent on phosphate
concentration and pH. J. Bacteriol. 188: 5469-5478.
Fontaine, L., Meynial-Salles, I., Girbal, L., Yang, X., Croux, C., & Soucaille, P. 2002. Molecular
characterization and transcriptional analysis of adhE2, the gene encoding the NADH-
dependent Aldehyde/ Alcohol Dehydrogenase responsible for Butanol production in
alcoholgenic cultures of Clostridium acetobutylicum ATCC 824. J. Bacteriol. 184: 821-830.
Gerhardt, A., Cinkaya, I., Lindner, D., Huisman, G., & Buckel, W. 2000. Fermentation of 4-
aminobutyrate by Clostridium aminobutyricum: cloning oft wo genes involved in the
formation and dehydration of 4-hydroxybutyryl-CoA. Arch. Microbiol. 174: 189-199.

Literaturverzeichnis 104
Girbal, L., Mortier-Barrière, I., Raynaud, F., Rouanet, C., Croux, C., & Soucaille, P. 2003.
Development of a Sensitive Gene Expression Reporter System for
Clostridium acetobutylicum. Appl. Environ. Microbiol. 69: 4985-4988.
Girbal, L., von Abendroth, G., Winkler, M., Benton, P. M. C., Meynial-Salles, I., Croux, C.,
Peters, J. P., Happe, T., & Soucaille , P. 2005. Homologous and Heterologous Overexpression
in Clostridium acetobutylicum and Characterization of purified clostridial and algal Fe-only
hydrogenases with high specific activities. App. Environ. Microbiol. 5: 2777-2781.
Grant, S. G. N., Jessee, J., Bloom, F. R., & Hanahan, D. 1990. Differential plasmid rescue
from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc.
Natl. Acad. Sci. 87: 4645-4649.
Green, E. M. 2011. Fermentative production of butanol – the industrial perspective. Curr.
Opin. Biotechnol. 22: 337-343.
Gustafsson, C., Govindarajan, S., & Minshull, J. 2004. Codon bias and heterologous protein
expression. Trends Biotechnol. 22: 346-353.
Haas, T., Jaeger, B., Weber, R., Mitchell, F. S., & King, C. F. 2005. New diol processes: 1,3-
propanediol and 1,4-butanediol. Appl. Catalys. 280: 83-88.
Han, B., Gopalan, V., & Ezeji, T. C. 2011. Acetone production in solventogenic Clostridium
species: new insights from non-enzymatic decarboxylation of acetoacetate. Appl. Microbiol.
Biotechnol. 91: 565-576.
Hartmanis, M. G. N, & Gatenbeck, S. 1984. Intermediary metabolism in Clostridium
acetobutylicum: Levels of enzymes involved in the formation of acetate and butyrate. App.
Environ. Microbiol. 47: 1277-1283.
Hartmanis, M., Klason, T., & Gatenbeck, S. 1984. Uptake and activation of acetate and
butyrate in Clostridium acetobutylicum. App. Microbiol. Biotechnol. 20: 66-71.
Heap, J. T., Pennington, O. J., Cartman, S. T., Carter, G. P., & Minton, N. P. 2007. The
ClosTron: A universal gene knock-out system for the genus Clostridium. J. Microbiol. Meth.
70: 452-464.

Literaturverzeichnis 105
Hershberg, R., & Petrov, D. A. 2008. Selection on codon bias. Annu. Rev. Genet. 42: 287-299.
Hwang, H. J., Park, J. H., Kim, J. H., Kong, M. K., Kim, J. W., Park, J. W., Cho, K. M., & Lee, P.
C. 2014. Engineering of a butyraldehyde dehydrogenase of
Clostridium saccharoperbutylacetonicum to fit an engineered 1,4-butanediol pathway in
Escherichia coli. Biotechnol. Bioeng. 111: 1374-1384.
Ibraheem, O., & Ndimba, B. K. 2013. Molecular adaptation mechanisms employed by
ethanologenic bacteria in response to lignocelluloses-derived inhibitory compounds. Int. J.
Biol. Sci. 9: 598-612.
Ingram, L. O. 1976. Adaption of membrane lipids to alcohols. J. Bacteriol. 125: 670-678.
Inoue, A., & Horikoshi, K. 1989. A Pseudomonas thrives in high concentrations of toluene.
Nat. 338: 264-266.
Isken, S., & de Bont, J. A. M. 1998. Bacteria tolerant to organic solvents. Extremophiles. 2:
229-238.
Jewell, J. B., Coutinho, J. B., & Kropinski, A. M. 1986. Bioconversion of Propionic, Valeric,
and 4-Hydroxybutyric acids into the corresponding alcohols by Clostridium acetobutylicum
NRRL 527. Curr. Microbiol. 13: 215-219.
Ji, X.-J., Huang, H., Zhu, J.-G., Ren, L.-J., Nie, Z.-K., Du, J., & Li, S. 2010. Engineering Klebsiella
oxytoca for efficient 2,3-butanediol production through insertional inactivation of
acetaldehyde dehydrogenase gene. Appl. Microbiol. Biotechnol. 85: 1751-1758.
Jones, D. T., van der Westhuizen, A., Long, S., Allock, E. R., Reid, S. J., & Woods, D. R. 1982.
Solvent production and morphological changes in Clostridium acetobutylicum. Appl. Environ.
Microbiol. 43: 1434-1439.
Jones, D.T, & Woods, D. R. 1986. Acetone-Butanol fermentation revisited. Microbiol. Rev.
50: 484-524.
Kane, J. F. 1995. Effects of rare codon clusters on high-level expression of heterologous
proteins in Escherichia coli. Curr. Opin. Biotechnol. 6: 494-500.

Literaturverzeichnis 106
Keasling, J. D. 2010. Manufacturing molecules through metabolic engineering. Science. 330:
1355-1358.
Kuhad, R. C., & Singh, A. 1993. Lignocellulose Biotechnology: Current and future prospects.
Crit. Rev. Biotechnol. 2: 151-172.
Kurland, C. G. 1991. Codon bias and gene expression. FEBS. 285: 165-169.
Kurland, C. G., & Gallant, J. 1995. Errors of heterologous protein expression. Curr. Opin.
Biotechnol. 7: 489-493.
Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nat. 227: 680-685.
Lee, J. 1997. Biological conversion of lignocellulosic biomass to ethanol. J. Biotechnol. 56: 1-
24.
Lehmann, D. 2012 a. Neue Einblicke in den Gärungsstoffwechsel von
Clostridium acetobutylicum. Dissertation. Universität Rostock.
Lehmann, D., Hönicke, D., Ehrenreich, A., Schmidt, M., Weuster-Botz, D., Bahl, H., & Lütke-
Eversloh, T. 2012, b. Modifying the product pattern of Clostridium acetobutylicum –
Physiological effects of disrupting the acetate and acetone formation pathways. Appl.
Microbiol. Biotechnol. 94: 743-754.
Lehmann, D., Radomski, N., & Lütke-Eversloh, T. 2012 c. New insights into the butyric acid
metabolism of Clostridium acetobutylicum. Appl. Microbiol. Biotechnol. 96: 1325-1339.
Liu, S.-J., & Steinbüchel, A. 2000. Exploitation of butyrate kinase and
phosphotransbutyrylase from Clostridium acetobutylicum for the in vitro biosynthesis of
poly(hydroxyalkanoic acid). Appl. Microbiol. Biotechnol. 53: 545-552.
Lütke-Eversloh, T., & Bahl, H. 2011. Metabolic engineering of Clostridium acetobutylicum:
recent advances to improve butanol production. Curr. Opin. Biotechnol. 22: 634-647.
Ma, C., Wang, A., Qin, J., Li, L., Ai, X., Jiang, T., Tang, H., & Xu, P. 2009. Enhanced 2,3-
butanediol production by Klebsiella pneumoniae SDM. Appl. Microbiol. Biotechnol. 82: 49-57.

Literaturverzeichnis 107
Madigan, M. T., Martinko, J. M., Stahl, D. A., & Clark, D. P. 2013. Brock - Biology of
Microorganisms. 13. überarbeitete Auflage. Pearson Studium, San Francisco, USA .
Maiorella, B., Blanch, H. W., & Wilke, C. R. 1983. By-product inhibition effects on ethanolic
fermentation by Saccharomyces cerevisiae. Biotechnol. Bioeng. 25: 103-121.
Martins, B. M., Dobbek, H., Cinkaya, I., Buckel, W., & Messerschmidt, A. 2004. Crystal
structure of 4-hydroxybutyryl-CoA dehydratase: radical catalysis involving a [4Fe-4S] cluster
and flavin. PNAS. 101: 15645-15649.
Martins, B. M., Messerschmidt, A., Friedrich, P., Zhang, J., & Buckel, W. 2007. 4-
Hydroxybutyryl-CoA dehydratase. Handbook Metalloproteins. 1-11.
Mermelstein, L. D., Welker, N. E., Bennett, G. N., & Papoutsakis, E. T. 1992. Expression of
Cloned Homologous Fermentative Genes in Clostridium acetobutylicum ATCC 824.
Bio/Technology 10: 190-195.
Mermelstein, L. D., & Papoutsakis, E. T. 1993. In Vivo Methylation in Escherichia coli by the
Bacillus subtilis Phage φ3T I Methyltransferase To Protect Plasmids from Restriction upon
Transformation of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 59: 1077-
1081.
Mitchell, W. J. 1998. Physiology of carbohydrate to solvent conversion by clostridia. Adv.
Microb. Physiol. 39: 31-130.
Monot, F. J.-R. M., Petitdemange, H. & Gay, R. 1982. Acetone and Butanol Production by
Clostridium acetobutylicum in a Synthetic Medium. Appl. Environ. Microbiol. 44: 1318-1324.
Mussatto, S. I., & Teixeira, J. A. 2010. Lignocellulose as raw material in fermentation
processes. Curr. Research. 2: 897-907.
Müh, U., Cinkaya, I., Albracht, S. P. J., & Buckel, W. 1996. 4-Hydroxybutyryl-CoA
Dehydratase from Clostridium aminobutyricum: Characterization of FAD and Iron-Sulfur
Clusters involved in an overall non-redox reaction. Biochem. 35: 11710-11718.
Nakamura, C. E., & Whited, G. M. 2003. Metabolic engineering for the microbial production
of 1,3-propanediol. Curr. Opin. Biotechnol. 14: 454-459.

Literaturverzeichnis 108
Näser, U., Pierik, A. J., Scott, R., Cinkaya, I., Buckel, W., & Golding, B. T. 2005. Synthesis of
13C-labeld γ-hydroxybutyrates for EPR studies with 4-hydroxybutyryl-CoA dehydratase.
Bioorg. Chem. 33: 53-66.
Neuhoff, V., Arold, N., Taube, D., & Ehrhardt, W. 1988. Improved staining of proteins in
polyacrylamide gels including isoelectric focusing gels with clear background at nanogram
sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9: 255-262.
Nissen, T. L., Kielland-Brandt, M. C., Nielsen, J., & Villadsen, J. 2000. Optimization of
ethanol production in Saccharomyces cerevisiae by metabolic engineering of the ammonium
assimilation. Metabol. Engineer. 2: 69-77.
Nölling, J., Breton, G., Omelchenko, M. V., Makarova, K. S., Zeng, Q., Gibson, R., Lee, H. M.,
Dubois, J., Qiu, D., Hitti, Y., et al. 2001. Genome sequence and comparative analysis of the
solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183: 4823-4838.
Ounine, K., Petitdemange, H., Raval, G., & Gay, R. 1983. Acetone-butanol production from
pentoses by Clostridium acetobutylicum. Biotechnol. L. 5: 605-610.
Ounine, K., Petitdemange, H., Raval, G., & Gay, R. 1985. Regulation and Butanol inhibition
of D-Xylose and D-Glucose uptake in Clostridium acetobutylicum. Appl. Environ. Microbiol.
49:874-878.
Papoutsakis, E. T. 2008. Engineering solventogenic clostridia. Curr. Opin. Biotechnol. 19: 420-
429.
Pinaetti, A., Battistelli, M., Citterio, B., & Bruscolini, F. 2009. Morphological changes in
Aeromonas hydrophila in response to osmotic stress. Micron. 40: 426-433.
Plotkin, J. B., & Kudla, G. 2011. Synonymous but not the same: the causes and
consequences of codon bias. Nat. 12: 32-42.
Qureshi, N., Li, X.-L., Hughes, S., Saha, B. C., & Cotta, M. A. 2006. Butanol Production from
Corn Fiber Xylan Using Clostridium acetobutylicum. Biotechnol. Prog. 22: 673-680.

Literaturverzeichnis 109
Ren, C., Gu, Y., Hu, S., Wu, Y., Wang, P., Yang, Y., Yang, C., Yang, S., & Jiang, W. 2010.
Identification and inactivation of pleitropic regulator CcpA to eliminate glucose repression of
xylose utilization in Clostridium acetobutylicum. Metabol. Engineer. 12: 446-454.
Sambrook, J., & Russell, D. 2001. Molecular cloning: a laboratory manual, 3rd edn. Cold
Spring Harbor, NY: Cold Spring Harbor Laboratory.
Sangster, J. 1989. Octanol-Water Partition Coefficients of simple organic compounds. J. Phys.
Chem. Ref. 18: 1111-1227.
Sarkar, N., Ghosh, S. K., Bannerjee, S., & Aikat, K. 2012. Bioethanol production from
agricultural waste: an overview. Ren. Ener. 37: 19-27.
Scherf, U., & Buckel, W. 1993. Purification and properties of an iron-sulfur and FAD
containing 4-hydroxybutyryl-CoA dehydratase/ vinylacetyl-CoA ∆3-∆2- isomerase from
Clostridium aminobutyricum. Eur. J. Biochem. 215: 421-429.
Scherf, U., Söhling, B., Gottschalk, G., Linder, D., & Buckel, W. 1994. Succinate-ethanol
fermentation in Clostridium kluyveri: purification and chracterisation of 4-hydroxybutyryl-
CoA dehydratase/ vinylacetyl-CoA ∆3-∆2- isomerase. Arch. Microbiol. 161: 239-245.
Schmidt, F. R. 2005. Optimization and scale up of industrial fermentation processes. App.
Microbiol. Biotechnol. 68: 425-435.
Schulz, F. 2013. Fluoreszenzproteine in Clostridium acetobutylicum- Ein neues in vivo
Reportersystem. Dissertation, Universität Rostock.
Servinsky, M. D., Kiel, J. T., Dupuy, N. F., & Sund, C. J. 2010. Transcriptional analysis of
differential carbohydrate utilization by Clostridium acetobutylicum. Microbiol. 156: 3478-
3491.
Shi, B., & Xia, X. 2003. Morphological changes of Pseudomonas pseudoalcaligenes in
response to temperature selection. Curr. Microbiol. 46: 120-123.
Sikkema, J., de Bont, J. A. M., & Poolman, B. 1995. Mechanisms of membrane toxicity of
hydrocarbons. Microbiol. Rev. 59: 201-222.

Literaturverzeichnis 110
Sillers, R., Al-Hinai, M. A., & Papoutsakis, E. T. 2009. Aldehyde-Alcohol Dehydrogenase and/
or Thiolase Overexpression Coupled with CoA Transferase Downregulation Lead to Higher
Alcohol Titers and Selectivity in Clostridium acetobutylicum Fermentations. Microbiol.
Bioeng. 102: 38-49.
Siemerink, M. A. J., Kuit, W., Lopez Contreras, A. M., Eggink, G., van der Oost, J., & Kengen,
S. W. M. 2011. D-2,3-Butanediol Production due to heterologous expression of an acetoin
reductase in Clostridium acetobutylicum. Appl. Environ. Microbiol. 77: 2582-2588.
Steinbüchel, A., & Lütke-Eversloh, T. 2003. Metabolic engineering and pathway construction
for biotechnological production of relevant polyhydroxyalkanoatesin microorganisms.
Biochem. Engineer. J. 16: 81-96.
Stephanopoulos, G. N., Aristidou, A. A., & Nielsen, J. 1998. Metabolic Engineering.
Principles and Methodologies. Academic Press, San Diego. S. 1 ff.
Sund, C. J., Servinsky, M. D., & Gerlach, E. S. 2013. Differing roles for
Clostridium acetobutylum´s Galactose utilization pathways. Advan. Microbiol. 3: 490-497.
Takors, R. 2012. Scale-up of microbial processes: Impacts, tools and open questions. J.
Biotechnol. 160: 3-9.
Tomas, C. A., Welker, N. E., & Papoutsakis, E. T. 2003. Overexpression of groESL in
Clostridium acetobutylicum results in increased solvent production and tolerance, prolonged
metabolism, and changes in the cell´s transcriptional program. Appl. Environ. Microbiol. 69:
4951-4965.
Towbin, H., Staehelin, T., & Gordon, J. 1979. Electrophoretic transfer of proteins from
polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proceedings
of the National Academy of Sciences of the United States of America. 76: 4350-4354.
Tummala, S. B., Welker, N. E., & Papoutsakis, E. T. 1999. Development and characterization
of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl.
Environ. Microbiol. 65: 3793-3799.
Van Haveren, J., Scott, E. L., & Sanders, J. 2008. Bulk chemicals from biomass. Biofuels,
Bioprod. Bioref. 2: 41-57.

Literaturverzeichnis 111
Vasconcelos, I., Girbal., L, & Soucaille, P. 1994. Regulation of carbon and electron flow in
Clostridium acetobutylicum grown in chemostat culture at neutral pH on mixtures of glucose
and glycerol. J. Bacteriol. 176: 1443-1450.
Voigt, C., Bahl, H., & Fischer, R.-J. 2014. Identification of PTSFru as the major fructose uptake
system of Clostridium acetobutylicum. App. Microbiol. Biotechnol. 98: 7161-7172.
Vollherbst-Schneck, K., Sands, J. A., & Montenecourt, B. S. 1984. Effect of butanol on lipid
composition and fluidity of Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol.
47: 193-194.
Wiesenborn, D. P., Rudolph, F. B., & Papoutsakis, E. T. 1988. Thiolase from
Clostridium acetobutylicum ATCC 824 and Its Role in the Synthesis of Acids and Solvents.
Appl. Environ. Microbiol. 54: 2717-2722.
Wiesenborn, D. P., Rudolph, F. B., & Papoutsakis, E. T. 1989. Phosphotransbutyrylase from
Clostridium acetobutylicum ATCC 824 and Its role in acidogenesis. Appl. Environ. Microbiol.
55: 317-322.
Willadsen, P., & Buckel, W. 1990. Assay of 4-Hydroxybutyryl-CoA dehydratase from
Clostridium aminobutyricum. FEMS Microbiol. Le. 70: 187-192.
Xiao, H., Gu, Y., Ning, Y., Yang, Y., Mitchell, W. J., Jiang, W., & Yang, S. 2011. Confirmation
and elimination of xylose metabolism bottlenecks in Glucose-Phosphoenolpyruvate-
dependent Phosphotransferase system-deficient Clostridium acetobutylicum for
simultaneous utilization of glucose, xylose and arabinose. Appl. Environ. Microbiol. 77: 7886-
7895.
Yim, H., Haselbeck, R., Niu, W., Pujol-Baxley, C., Burgard, A., Boldt, J., Khandurina, J.,
Trawick, J. D., Osterhout, R. E., Stephen, R., Estadilla, J., Teisan, S., Schreyer, H. B., Andrae,
S., Yang, T. H., Lee, S. Y., Burk, M. J., & Van Dien, S. 2011. Metabolic engineering of
Escherichia coli for direct production of 1,4-butanediol. Nat. Chem. Biol. 7: 445-452.
Yoo, M., Bestel-Corre, G., Croux, C., Riviere, A., Meynial-Salles, I., & Soucaille, P. 2015. A
quantitative system-scale characterization of the metabolism of Clostridium acetobutylicum.
mBio. 6: 1-12.

Literaturverzeichnis 112
Yu, E. K. C., & Saddler, J. N. 1982. Enhanced acetone-butanol fermentation by Clostridium
acetobutylicum grown on D-Xylose in the presence of acetic or butyric acid. FEMS Microbiol.
L. 18: 103-107.
Zeng, A.-P., & Sabra, W. 2011. Microbial production of diols as platform chemicals: recent
progresses. Curr. Opin. Biotechnol. 22: 749-757.
Zhang, J. 2010. On the enzymatic mechanism of 4-hydroxybuytyryl-CoA dehydratase and 4-
hydroxybutyrate-CoA transferase from Clostridium aminobutyricum. Dissertation. Philipps-
Universität Marburg.
Zhang, L., Sun, J., Hao, Y., Zhu, J., Chu, J., Wie, D., & Shen, Y. 2010. Microbial production of
2,3-butanediol by a surfactant (serrawettin)- deficient mutant of Serratia marcescens H30. J.
Ind. Microbiol. Biotechnol. 37: 857-862.
Zhang, H., Chong, H., Ching, C. B., Song, H., & Jiang, R. 2012. Engineering global
transcription factor cyclic AMP receptor protein of Escherichia coli for improved 1-butanol
tolerance. Appl. Microbiol. Biotechnol. 94: 1107-1117.
Zverlov, V. V., Berezina, O., Velikodvorskaja, G. A., & Schwarz, W. H. 2006. Bacterial
acetone and butanol production by industrial fermentation in the Soviet Union: use of
hydrolyzed agricultural waste for biorefinery. Appl. Microbiol. Biotechnol. 71: 587-597.
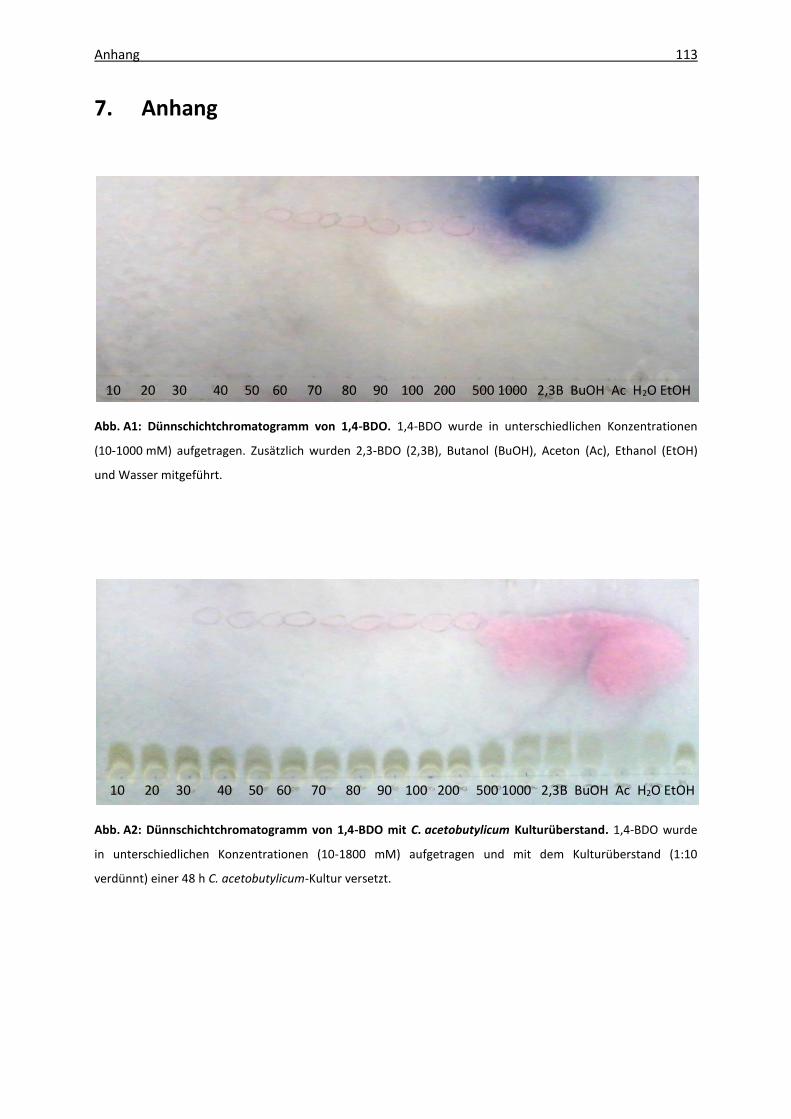
Anhang 113
7. Anhang
Abb. A1: Dünnschichtchromatogramm von 1,4-BDO. 1,4-BDO wurde in unterschiedlichen Konzentrationen
(10-1000 mM) aufgetragen. Zusätzlich wurden 2,3-BDO (2,3B), Butanol (BuOH), Aceton (Ac), Ethanol (EtOH)
und Wasser mitgeführt.
Abb. A2: Dünnschichtchromatogramm von 1,4-BDO mit C. acetobutylicum Kulturüberstand. 1,4-BDO wurde
in unterschiedlichen Konzentrationen (10-1800 mM) aufgetragen und mit dem Kulturüberstand (1:10
verdünnt) einer 48 h C. acetobutylicum-Kultur versetzt.
10 20 30 40 50 60 70 80 90 100 200 500 1000 2,3B BuOH Ac H2O EtOH
10 20 30 40 50 60 70 80 90 100 200 500 1000 2,3B BuOH Ac H2O EtOH
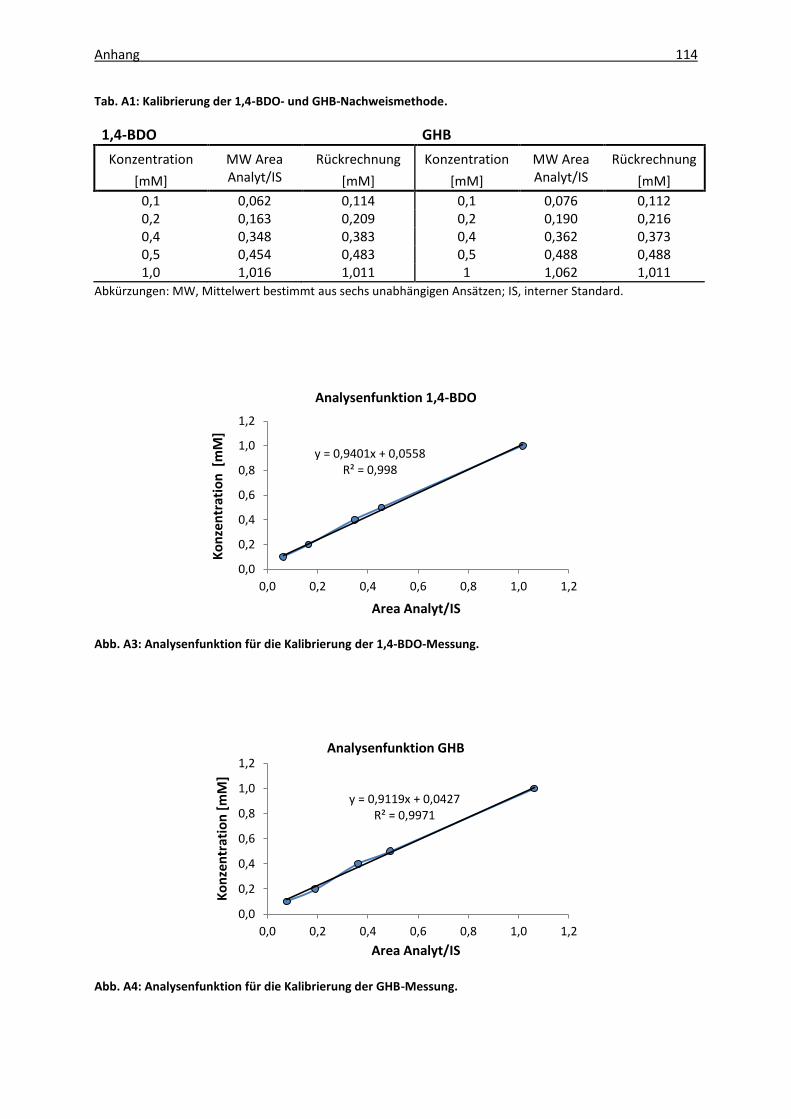
Anhang 114
Tab. A1: Kalibrierung der 1,4-BDO- und GHB-Nachweismethode.
1,4-BDO GHB
Konzentration
[mM]
MW Area Analyt/IS
Rückrechnung
[mM]
Konzentration
[mM]
MW Area Analyt/IS
Rückrechnung
[mM]
0,1 0,062 0,114 0,1 0,076 0,112 0,2 0,163 0,209 0,2 0,190 0,216 0,4 0,348 0,383 0,4 0,362 0,373 0,5 0,454 0,483 0,5 0,488 0,488 1,0 1,016 1,011 1 1,062 1,011
Abkürzungen: MW, Mittelwert bestimmt aus sechs unabhängigen Ansätzen; IS, interner Standard.
Abb. A3: Analysenfunktion für die Kalibrierung der 1,4-BDO-Messung.
Abb. A4: Analysenfunktion für die Kalibrierung der GHB-Messung.
y = 0,9401x + 0,0558R² = 0,998
0,0
0,2
0,4
0,6
0,8
1,0
1,2
0,0 0,2 0,4 0,6 0,8 1,0 1,2
Ko
nze
ntr
atio
n [
mM
]
Area Analyt/IS
Analysenfunktion 1,4-BDO
y = 0,9119x + 0,0427R² = 0,9971
0,0
0,2
0,4
0,6
0,8
1,0
1,2
0,0 0,2 0,4 0,6 0,8 1,0 1,2
Ko
nze
ntr
atio
n [
mM
]
Area Analyt/IS
Analysenfunktion GHB

Anhang 115
AbfD 1 M-LMTAEQYIESLRKLNTRVYMFGEKIENWVDHPMIRPSINCVAMTYELAQDPQYADLMTTKSNLIGKTINRFANLHQST 79
Ckl_3020 1 MSLMTGEEYVESLRKLKLNVYYLGEKIDNPVDNPVLRPSLNSVKMTYDLAQEAEYEDLMTTTSNITGEKINRFTNLHQSS 80
Cbei_2100 1 MPLKTKEQYIESLRKLNLKVYMFGKPVDNVVDNPIIRPSLNSVAMTYELAQMPEYEDLMTATSNLTGEKVNRFAHLHQST 80
AbfD 80 DDLRKKVKMQRLLGQKTASCFQRCVGMDAFNAVFSTTYEIDQKYGTNYHKNFTEYLKYIQENDLIVDGAMTDPKGDRGLA 159
Ckl_3020 81 EDLVKKVKMQRLCGQKTAACFQRCVGMDSFNAVYSTTFEIDKEYNTNYFENFKKFLTYVQKNDLTVDGAMTDPKGDRGLS 160
Cbei_2100 81 DDLIKKVKMQRLLGQKTASCFQRCVGMDAFNALYSSTYEIDKACGTNYHENFNKFLKYVQENDLTVDGAMTDPKGDRGLS 160
AbfD 160 PSAQKDPDLFLRIVEKREDGIVVRGAKAHQTGSINSHEHIIMPTIAMTEADKDYAVSFACPSDADGLFMIYGRQSCDTRK 239
Ckl_3020 161 PSKQADPDLYLRVVERREDGVVVRGAKAHQTGICNSHEVLVMPTIAMRPDDKDYAIAFSVPTDAEGITMIIGRQSCDTRK 240
Cbei_2100 161 PSKQADGDLYLRVVERREDGVVVRGAKCHQTGMLNSHEVVVMPTIALTPNDKDWAISFAVPTDAEGIIHIYGRQSCDTRK 240
AbfD 240 MEEGADIDLGNKQFGGQEALVVFDNVFIPNDRIFLCQEYDFAGMMVERFAGYHRQSYGGCKVGVGDVVIGAAALAADYNG 319
Ckl_3020 241 MEKDADIDVGNKEFGGVEALVVFDDVFVPNDRIFLNGETEYAGMLVERFAGYHRQSYGGCKVGVGDVLIGAAAVAADYNG 320
Cbei_2100 241 LEPGADIDLGNAEFGGQETLTIFDNVFVPNERIFLNGETDFAGMIVERFAGYHRQSYGGCKVGVGDVLIGAAAVAADYNG 320
AbfD 320 AQKASHVKDKLIEMTHLNETLYCCGIACSAEGYPTAAGNYQIDLLLANVCKQNITRFPYEIVRLAEDIAGGLMVTMPSEA 399
Ckl_3020 321 AAKASHIKDKLIEMMHLNETLYACGIACSAEGHATKAGNYQIDLLLANVCKQNITRFPYEIVRLAEDIAGGLMVTMPSEK 400
Cbei_2100 321 AHKASHVKDKLIEMTHLNETLFSCGIACSAMGSKTEAGNYYIDNLLANVCKQNVTRFPYEICRLAEDIAGGIMVTMPSEA 400
AbfD 400 DFKS[8]ETIGDFCNKFFAAAPTCTTEERMRVLRFLENICLGASAVGYRTESMHGAGSPQAQRIMIARQGNINAKKELAK 484
Ckl_3020 401 DYKS PEVGKYVEKYLVGVASVPVEDRMKILRLLENICLGTAAVGYRTESMHGAGSPQAQRIMISRQGNLAHKKKLAK 477
Cbei_2100 401 DFKD EKIGPYIDKYLRGVNSVSTENRMRILRLIENICLGTAAVGYRTESMHGAGSPQAQRIMIARQGNLAAKKKIAK 477
AbfD 485 AIAGIK- 490
Ckl_3020 478 KIARIED 484
Cbei_2100 478 KIARIKE 484
Abb. A5: Aminosäure-Alignment der 4HBDs. Schwarz markierte AS sind konserviert, grün hervorgehobene AS
sind verantwortlich für die katalytische Funktion der Enzyme. Blau unterlegte AS bilden das Eisen-Schwefel-
Cluster-Motiv CX3CX179-183HX6C. Abkürzungen: AS, Aminosäuren; AbfD, 4HBD aus C. aminobutyricum; Ckl_3020,
4HBD aus C. kluyveri; Cbei_2100, 4HBD aus C. beijerinckii.

Anhang 116
Abb. A6: Vektorkarte vom pT-Vektor (Girbal et al., 2005; mod.). Der pT-Vektor verfügt über zwei
Replikationsursprünge („repL“ und „pBR322“), welche den Einsatz des Vektorsystems in Gram-positiven und
Gram-negativen Bakterien ermöglicht. Darüber hinaus besitzt dieser Vektor zwei Resistenzgene- bla
(Ampicillin-Resistenzgen) und EmR (Erythromycin-Resistenzgen), eine C-terminalen Strep-tag II Sequenz und
einen ThiolaseA-Promotor.
CAAGGAGGTTAGTTAGAATGGGATCCATGCCATTAAAAACAAAGGAACAATATATTGAAAGCCTTAGAAAGTTAAACCTA
AAAGTGTACATGTTTGGAAAACCAGTTGACAATGTTGTTGATAATCCAATCATAAGACCATCTCTTAATTCAGTTGCTATGA
CTTATGAATTAGCACAAATGCCAGAGTATGAAGATTTAATGACAGCTACTTCTAATTTAACAGGGGAAAAAGTAAATAGAT
TTGCTCATTTACATCAAAGCACAGATGATCTTATTAAAAAAGTAAAGATGCAAAGATTACTTGGTCAAAAAACCGCATCAT
GTTTCCAAAGGTGCGTTGGTATGGATGCTTTTAATGCATTATACAGTTCAACATACGAAATAGATAAAGCGTGTGGAACAA
ATTATCATGAAAACTTTAATAAGTTTTTAAAATATGTTCAAGAAAACGACTTAACCGTTGATGGTGCAATGACTGATCCTAA
AGGGGACAGAGGATTATCGCCAAGTAAACAAGCTGATGGAGATTTATACTTAAGGGTTGTTGAAAGAAGAGAAGATGGG
GTAGTTGTTAGAGGAGCAAAATGTCACCAAACTGGAATGTTAAATTCTCATGAGGTAGTAGTAATGCCAACAATAGCATTA
ACTCCAAATGATAAGGATTGGGCAATATCTTTTGCAGTGCCTACAGATGCTGAAGGAATAATTCATATATATGGAAGGCAA
TCTTGTGATACAAGAAAATTGGAGCCAGGTGCTGATATAGATCTTGGAAATGCTGAATTTGGTGGACAAGAAACATTAAC
AATATTTGATAATGTATTTGTACCAAATGAAAGAATTTTCTTAAATGGAGAAACTGATTTTGCTGGAATGATAGTTGAAAG
ATTTGCAGGATATCATAGACAAAGTTATGGTGGATGCAAAGTCGGAGTTGGAGATGTTCTAATTGGAGCTGCTGCAGTAG
CTGCTGATTATAATGGAGCTCACAAAGCATCTCATGTTAAAGATAAATTAATAGAAATGACTCACTTAAATGAAACTTTATT
CTCTTGTGGTATTGCATGTTCAGCAATGGGTTCTAAAACAGAAGCTGGAAATTATTATATTGATAATCTGCTAGCAAATGTA
TGTAAGCAAAATGTTACAAGATTCCCTTATGAAATATGTAGACTTGCAGAAGATATTGCTGGAGGAATCATGGTGACTATG
CCATCAGAAGCAGATTTTAAAGATGAAAAGATAGGTCCATATATAGATAAATATTTGAGAGGTGTAAACTCAGTTTCAACA
GAAAATAGAATGAGAATATTAAGATTAATAGAAAATATTTGTTTAGGGACTGCAGCTGTCGGATATAGAACAGAATCAAT
GCATGGAGCTGGATCACCACAAGCTCAAAGAATTATGATAGCTAGACAAGGAAATTTAGCAGCTAAAAAGAAAATAGCTA
AGAAAATAGCTAGAATTAAAGAACCCGGGTGGTCACATCCTCAATTTGAAAAATAAGGCGCCA
Abb. A7: Nukleotidsequenz von cbei_2100 nach Sequenzierung positiver Plasmide. Unterstrichene Sequenzen
signalisieren die Restriktionsschnittstellen. Grün markierte Nukleotide zeigen den Beginn und das Ende des
Gens. Die Nukleotidsequenz des Strep-tag II ist blau hervorgehoben.

Anhang 117
TAGTTAGAGCTAGCATGTCTTTAATGACTGGGGAAGAATATGTAGAAAGTTTACGTAAATTAAAATTAAACGTTTATTATCT
GGGAGAGAAAATAGATAATCCAGTGGATAACCCTGTACTTCGTCCATCTTTAAATTCTGTTAAGATGACTTATGACTTGGC
CCAAGAAGCAGAATATGAGGATTTAATGACAACAACATCAAATATAACAGGTGAAAAAATAAATAGATTTACAAATTTACA
TCAAAGCAGTGAAGATTTGGTAAAAAAAGTTAAAATGCAGAGATTGTGCGGACAAAAAACTGCAGCCTGTTTCCAAAGAT
GCGTTGGTATGGACTCATTTAATGCAGTGTACAGTACTACTTTTGAAATAGATAAGGAATATAATACAAATTATTTTGAAAA
TTTCAAGAAATTTTTAACTTATGTTCAAAAGAACGATTTAACAGTAGATGGAGCTATGACAGATCCAAAAGGAGACAGAG
GATTGTCACCAAGCAAACAGGCTGATCCAGATTTATATTTAAGAGTTGTAGAAAGAAGAGAAGACGGTGTAGTTGTAAGA
GGAGCAAAGGCTCACCAGACAGGTATATGTAATTCTCATGAAGTATTGGTTATGCCAACTATTGCTATGAGACCGGATGAT
AAAGATTATGCAATAGCTTTTTCTGTTCCTACAGATGCAGAAGGAATAACTATGATAATTGGAAGACAGTCCTGTGATACT
AGAAAAATGGAAAAAGATGCGGACATAGATGTTGGTAATAAAGAATTTGGCGGAGTAGAAGCATTAGTAGTATTTGACG
ATGTATTTGTTCCAAATGACAGGATATTCTTAAATGGTGAAACTGAATATGCAGGAATGTTAGTAGAGAGATTTGCAGGAT
ATCATAGACAAAGTTATGGTGGATGCAAAGTAGGAGTGGGAGATGTATTGATAGGCGCTGCTGCAGTGGCTGCAGACTA
TAATGGAGCTGCAAAGGCATCTCACATAAAGGATAAATTAATAGAAATGATGCATTTAAATGAAACTCTTTACGCTTGCGG
AATTGCGTGCTCAGCAGAAGGACATGCAACAAAAGCTGGAAATTACCAAATAGACTTGCTTCTTGCAAATGTATGTAAGCA
AAATATAACAAGATTCCCTTATGAAATTGTGAGATTAGCAGAAGATATAGCAGGAGGATTAATGGTTACTATGCCTTCTGA
AAAGGATTATAAGAGTCCAGAGGTTGGAAAATATGTAGAGAAGTACTTGGTGGGAGTTGCATCCGTACCTGTTGAAGACA
GAATGAAGATATTAAGATTATTAGAAAATATATGTCTTGGAACGGCTGCAGTAGGATATAGAACTGAATCCATGCATGGA
GCAGGTTCACCTCAAGCACAGAGAATAATGATATCAAGACAGGGAAACCTAGCACATAAGAAGAAACTTGCAAAGAAGA
TAGCTAGAATAGAAGATCCCGGGTGGTCACATCCTCAATTTGAAAAATAAGGCGCCAC
Abb. A8: Nukleotidsequenz von ckl_3020 nach Sequenzierung positiver Plasmide. Unterstrichene Sequenzen
signalisieren die Restriktionsschnittstellen. Grün markierte Nukleotide zeigen den Beginn und das Ende des
Gens. Die Nukleotidsequenz des Strep-tag II ist blau hervorgehoben.
CCGTAGGATCAAGGAGGTTAGTTAGAGCTAGCATGTTAATGACAGCAGAACAGTACATTGAGAGTCTAAGAAAGCTAAA
CACAAGAGTTTATATGTTTGGTGAAAAAATCGAGAATTGGGTGGATCATCCAATGATCAGACCTTCCATCAACTGCGTAGC
AATGACTTATGAATTAGCTCAGGATCCTCAGTACGCTGACTTAATGACTACAAAGTCAAACTTAATAGGTAAAACTATCAAC
AGATTTGCAAATCTACACCAGAGCACAGATGACCTTAGAAAAAAGGTTAAGATGCAGAGACTTCTTGGACAGAAGACCGC
ATCATGCTTCCAGAGATGTGTAGGTATGGACGCTTTCAATGCAGTTTTCTCAACTACATATGAAATCGACCAGAAATATGG
AACAAACTATCACAAGAACTTTACTGAATACTTAAAGTATATACAGGAAAATGACCTTATTGTTGACGGTGCAATGACTGA
CCCTAAGGGTGACAGAGGACTTGCTCCATCCGCACAGAAGGATCCAGATCTTTTCTTGAGAATCGTTGAAAAAAGAGAAG
ATGGTATCGTTGTAAGAGGAGCTAAGGCTCACCAGACTGGTTCCATCAACTCCCACGAACACATCATCATGCCTACAATCG
CTATGACAGAAGCTGATAAGGATTATGCAGTATCATTTGCTTGTCCTTCCGATGCTGATGGTCTATTCATGATCTACGGCAG
ACAGTCATGTGACACAAGAAAGATGGAAGAAGGCGCTGACATTGACCTTGGTAACAAGCAGTTCGGCGGACAGGAAGCT
TTAGTCGTATTCGATAACGTATTTATTCCAAATGACAGAATCTTCCTTTGCCAAGAATATGATTTCGCTGGCATGATGGTAG
AAAGATTTGCTGGATACCACAGACAGTCATACGGCGGATGTAAGGTTGGAGTAGGCGACGTTGTAATCGGTGCTGCTGCT
TTAGCTGCTGACTACAATGGAGCTCAGAAGGCTTCTCACGTTAAAGATAAGCTTATCGAAATGACTCACTTAAATGAAACT
TTATATTGCTGCGGTATTGCTTGTTCAGCAGAAGGTTATCCAACTGCTGCTGGTAACTATCAGATTGACCTTCTTCTTGCAA
ATGTATGTAAGCAGAACATCACTAGATTCCCTTACGAAATCGTAAGACTAGCTGAAGATATCGCTGGTGGATTAATGGTTA
CTATGCCTTCAGAAGCTGACTTTAAGTCAGAAACAGTTGTTGGTAGAGATGGCGAAACTATTGGAGATTTCTGCAATAAGT
TCTTCGCTGCTGCTCCTACTTGCACAACAGAAGAAAGAATGAGAGTTCTTAGATTCTTAGAAAACATCTGCTTAGGTGCATC
CGCTGTAGGTTACAGAACTGAATCCATGCATGGTGCAGGTTCCCCTCAGGCTCAGAGAATCATGATCGCTCGTCAGGGCA
ACATCAACGCTAAGAAAGAATTAGCTAAGGCAATCGCTGGAATTAAACCCGGGTGGTCACATCCTCAATTTGAAAAATAA
GGCGCCAC
Abb. A9: Nukleotidsequenz von abfD nach Sequenzierung positiver Plasmide. Unterstrichene Sequenzen
signalisieren die Restriktionsschnittstellen. Grün markierte Nukleotide zeigen den Beginn und das Ende des
Gens. Die Nukleotidsequenz des Strep-tag II ist blau hervorgehoben.
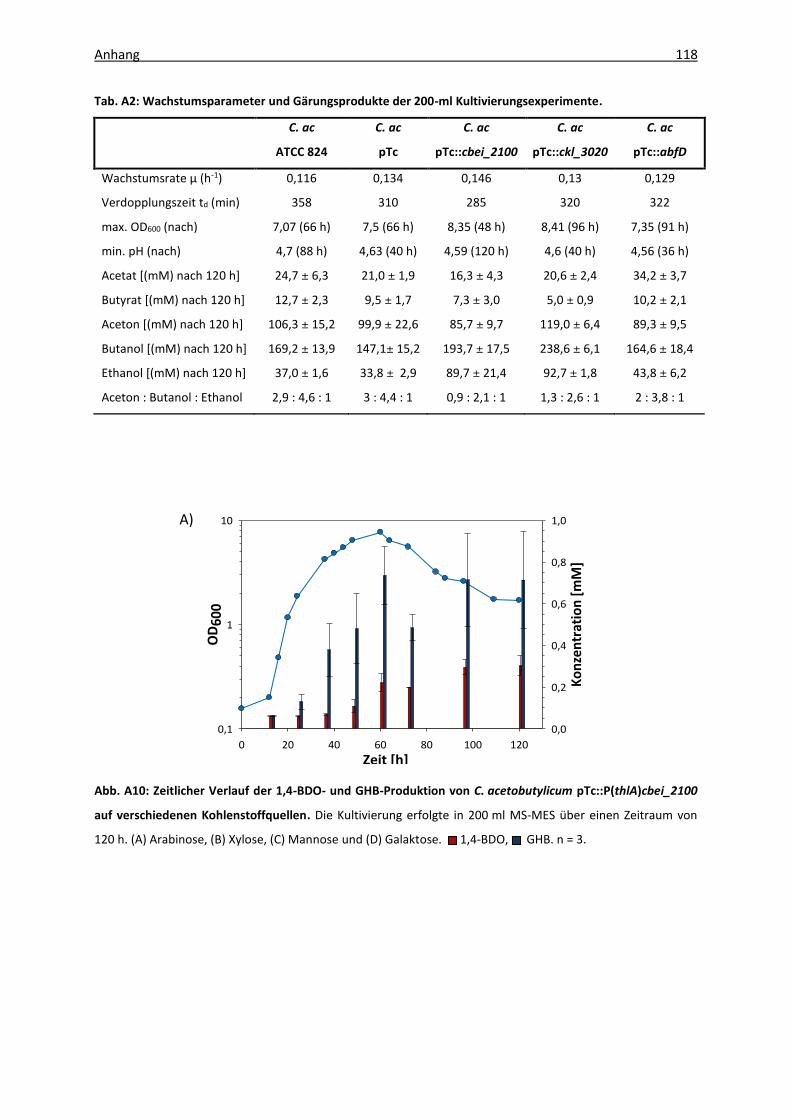
Anhang 118
Tab. A2: Wachstumsparameter und Gärungsprodukte der 200-ml Kultivierungsexperimente.
C. ac
ATCC 824
C. ac
pTc
C. ac
pTc::cbei_2100
C. ac
pTc::ckl_3020
C. ac
pTc::abfD
Wachstumsrate µ (h-1) 0,116 0,134 0,146 0,13 0,129
Verdopplungszeit td (min) 358 310 285 320 322
max. OD600 (nach) 7,07 (66 h) 7,5 (66 h) 8,35 (48 h) 8,41 (96 h) 7,35 (91 h)
min. pH (nach) 4,7 (88 h) 4,63 (40 h) 4,59 (120 h) 4,6 (40 h) 4,56 (36 h)
Acetat [(mM) nach 120 h] 24,7 ± 6,3 21,0 ± 1,9 16,3 ± 4,3 20,6 ± 2,4 34,2 ± 3,7
Butyrat [(mM) nach 120 h] 12,7 ± 2,3 9,5 ± 1,7 7,3 ± 3,0 5,0 ± 0,9 10,2 ± 2,1
Aceton [(mM) nach 120 h] 106,3 ± 15,2 99,9 ± 22,6 85,7 ± 9,7 119,0 ± 6,4 89,3 ± 9,5
Butanol [(mM) nach 120 h] 169,2 ± 13,9 147,1± 15,2 193,7 ± 17,5 238,6 ± 6,1 164,6 ± 18,4
Ethanol [(mM) nach 120 h] 37,0 ± 1,6 33,8 ± 2,9 89,7 ± 21,4 92,7 ± 1,8 43,8 ± 6,2
Aceton : Butanol : Ethanol 2,9 : 4,6 : 1 3 : 4,4 : 1 0,9 : 2,1 : 1 1,3 : 2,6 : 1 2 : 3,8 : 1
Zeit [h]0 20 40 60 80 100 120
OD
600
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Abb. A10: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100
auf verschiedenen Kohlenstoffquellen. Die Kultivierung erfolgte in 200 ml MS-MES über einen Zeitraum von
120 h. (A) Arabinose, (B) Xylose, (C) Mannose und (D) Galaktose. 1,4-BDO, GHB. n = 3.
A)
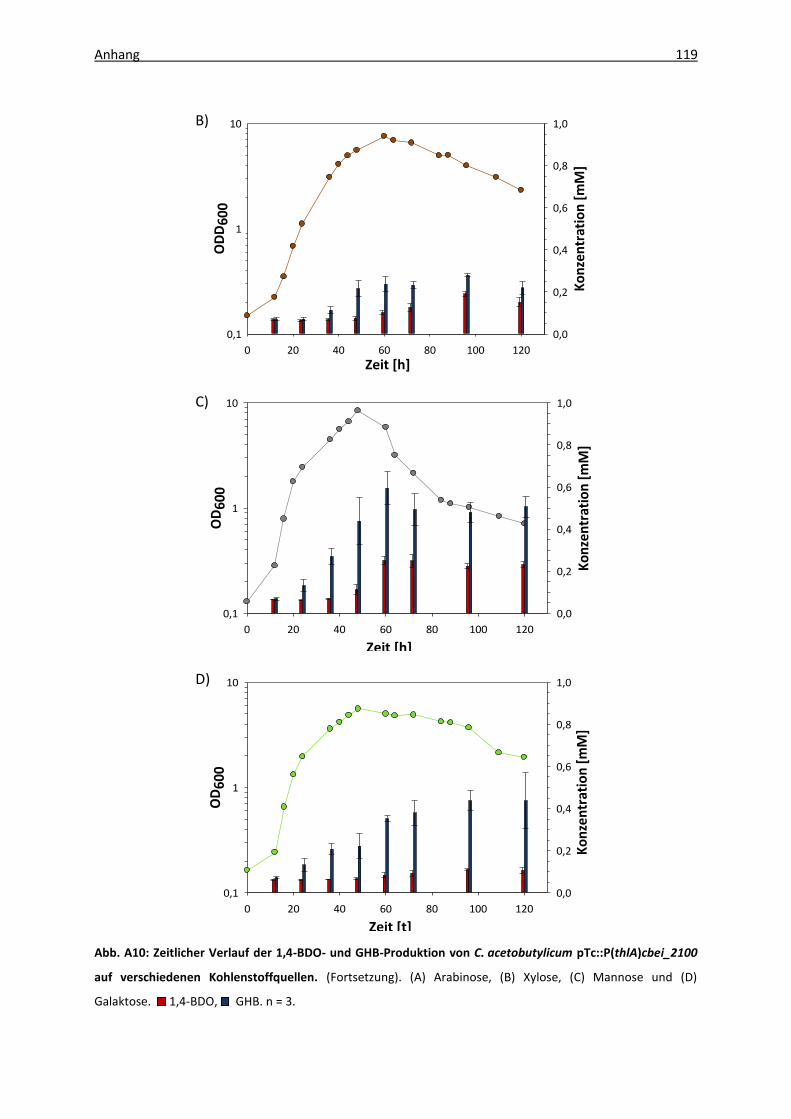
Anhang 119
Zeit [h]0 20 40 60 80 100 120
OD
D60
0
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Zeit [h]
0 20 40 60 80 100 120
OD
600
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Zeit [t]
0 20 40 60 80 100 120
OD
600
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Abb. A10: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum pTc::P(thlA)cbei_2100
auf verschiedenen Kohlenstoffquellen. (Fortsetzung). (A) Arabinose, (B) Xylose, (C) Mannose und (D)
Galaktose. 1,4-BDO, GHB. n = 3.
B)
C)
D)
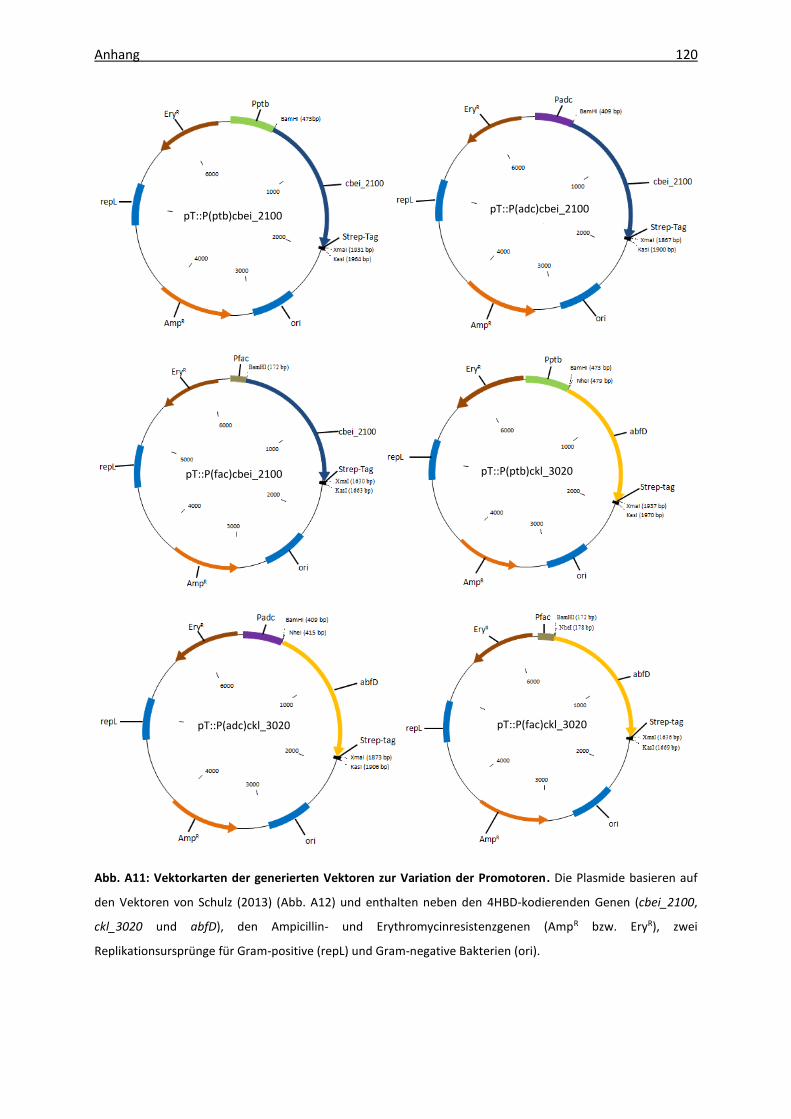
Anhang 120
Abb. A11: Vektorkarten der generierten Vektoren zur Variation der Promotoren. Die Plasmide basieren auf
den Vektoren von Schulz (2013) (Abb. A12) und enthalten neben den 4HBD-kodierenden Genen (cbei_2100,
ckl_3020 und abfD), den Ampicillin- und Erythromycinresistenzgenen (AmpR bzw. EryR), zwei
Replikationsursprünge für Gram-positive (repL) und Gram-negative Bakterien (ori).
pT::P(ptb)ckl_3020
pT::P(adc)ckl_3020 pT::P(fac)ckl_3020
pT::P(ptb)cbei_2100 pT::P(adc)cbei_2100
pT::P(fac)cbei_2100
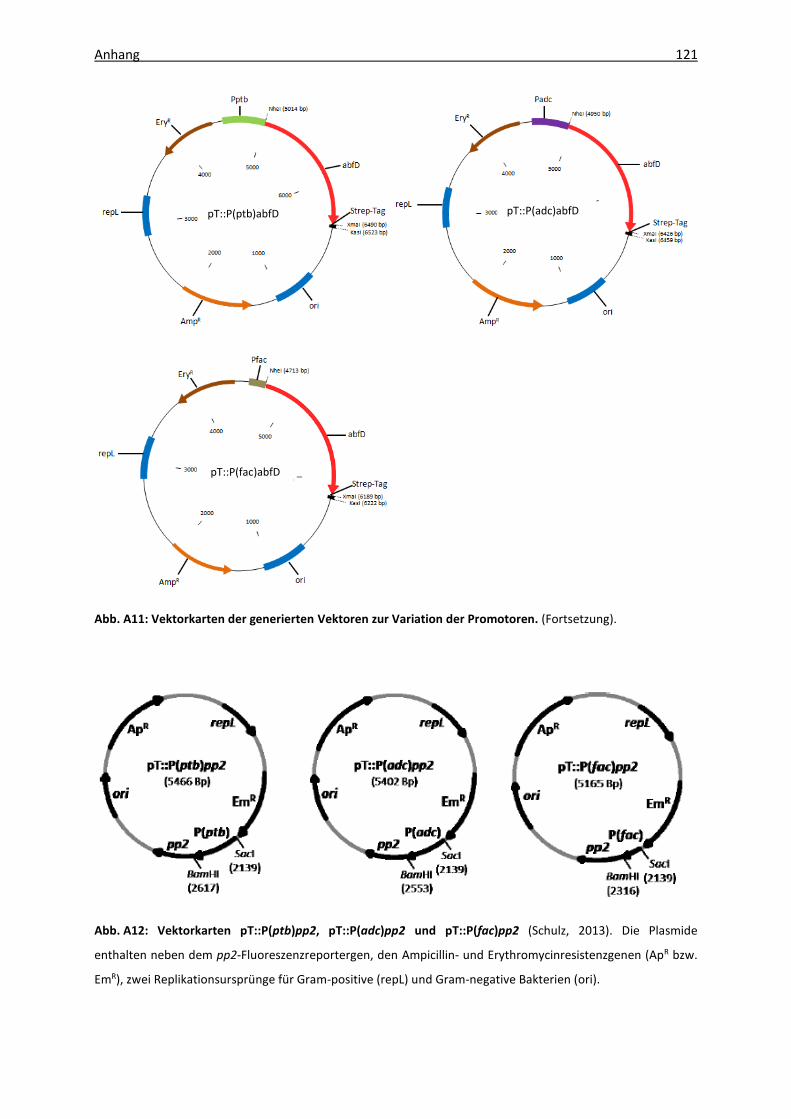
Anhang 121
Abb. A11: Vektorkarten der generierten Vektoren zur Variation der Promotoren. (Fortsetzung).
Abb. A12: Vektorkarten pT::P(ptb)pp2, pT::P(adc)pp2 und pT::P(fac)pp2 (Schulz, 2013). Die Plasmide
enthalten neben dem pp2-Fluoreszenzreportergen, den Ampicillin- und Erythromycinresistenzgenen (ApR bzw.
EmR), zwei Replikationsursprünge für Gram-positive (repL) und Gram-negative Bakterien (ori).
pT::P(ptb)abfD pT::P(adc)abfD
pT::P(fac)abfD
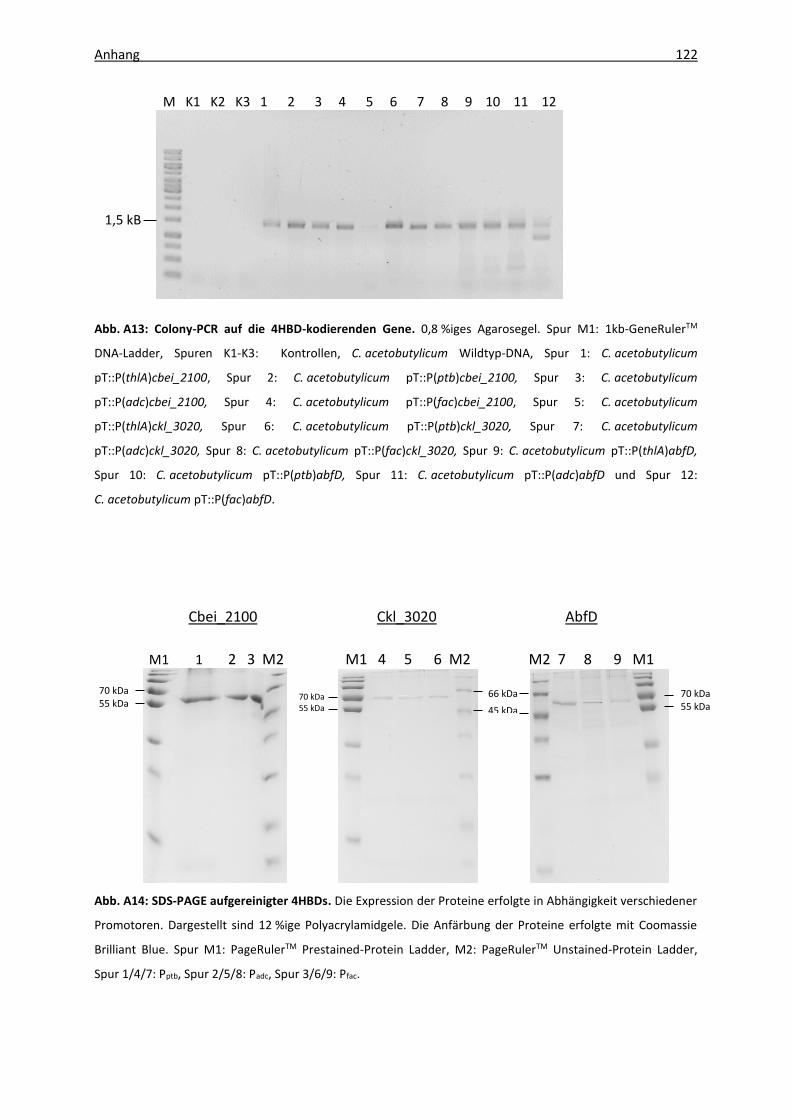
Anhang 122
M K1 K2 K3 1 2 3 4 5 6 7 8 9 10 11 12
Abb. A13: Colony-PCR auf die 4HBD-kodierenden Gene. 0,8 %iges Agarosegel. Spur M1: 1kb-GeneRulerTM
DNA-Ladder, Spuren K1-K3: Kontrollen, C. acetobutylicum Wildtyp-DNA, Spur 1: C. acetobutylicum
pT::P(thlA)cbei_2100, Spur 2: C. acetobutylicum pT::P(ptb)cbei_2100, Spur 3: C. acetobutylicum
pT::P(adc)cbei_2100, Spur 4: C. acetobutylicum pT::P(fac)cbei_2100, Spur 5: C. acetobutylicum
pT::P(thlA)ckl_3020, Spur 6: C. acetobutylicum pT::P(ptb)ckl_3020, Spur 7: C. acetobutylicum
pT::P(adc)ckl_3020, Spur 8: C. acetobutylicum pT::P(fac)ckl_3020, Spur 9: C. acetobutylicum pT::P(thlA)abfD,
Spur 10: C. acetobutylicum pT::P(ptb)abfD, Spur 11: C. acetobutylicum pT::P(adc)abfD und Spur 12:
C. acetobutylicum pT::P(fac)abfD.
Cbei_2100 Ckl_3020 AbfD
M1 1 2 3 M2 M1 4 5 6 M2 M2 7 8 9 M1
Abb. A14: SDS-PAGE aufgereinigter 4HBDs. Die Expression der Proteine erfolgte in Abhängigkeit verschiedener
Promotoren. Dargestellt sind 12 %ige Polyacrylamidgele. Die Anfärbung der Proteine erfolgte mit Coomassie
Brilliant Blue. Spur M1: PageRulerTM Prestained-Protein Ladder, M2: PageRulerTM Unstained-Protein Ladder,
Spur 1/4/7: Pptb, Spur 2/5/8: Padc, Spur 3/6/9: Pfac.
1,5 kB
70 kDa 55 kDa
66 kDa
45 kDa
70 kDa 55 kDa
70 kDa 55 kDa
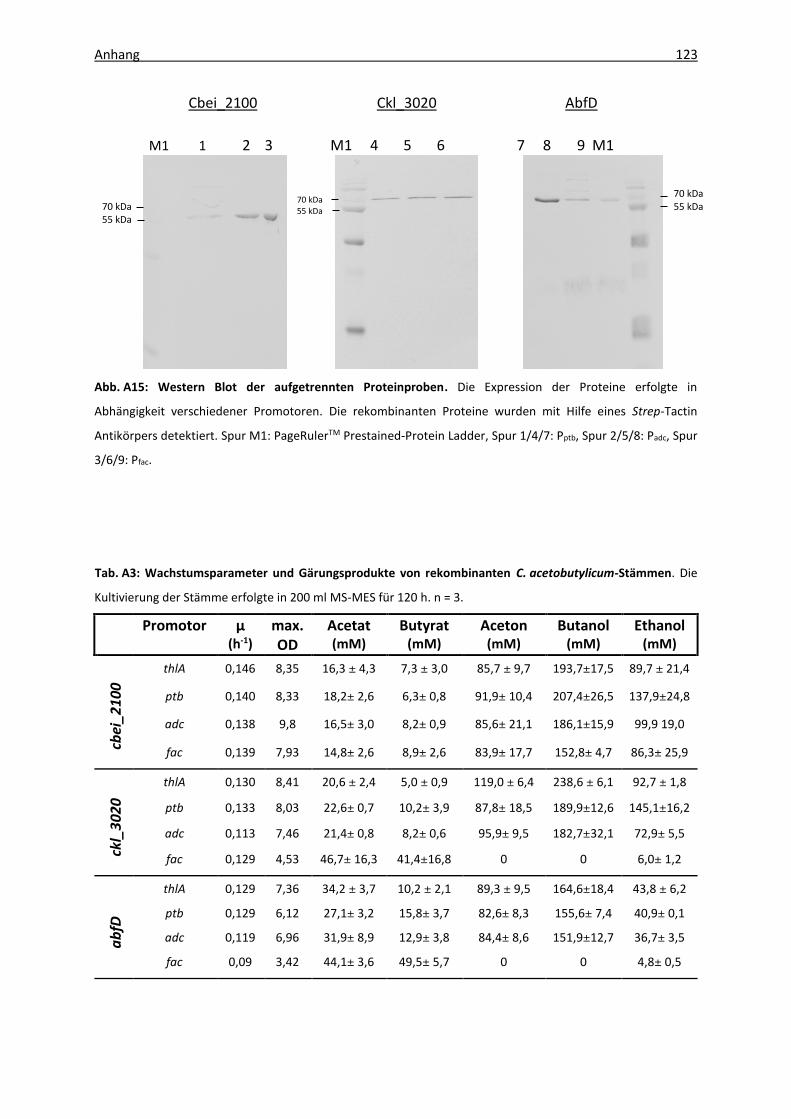
Anhang 123
Cbei_2100 Ckl_3020 AbfD
M1 1 2 3 M1 4 5 6 7 8 9 M1
Abb. A15: Western Blot der aufgetrennten Proteinproben. Die Expression der Proteine erfolgte in
Abhängigkeit verschiedener Promotoren. Die rekombinanten Proteine wurden mit Hilfe eines Strep-Tactin
Antikörpers detektiert. Spur M1: PageRulerTM Prestained-Protein Ladder, Spur 1/4/7: Pptb, Spur 2/5/8: Padc, Spur
3/6/9: Pfac.
Tab. A3: Wachstumsparameter und Gärungsprodukte von rekombinanten C. acetobutylicum-Stämmen. Die
Kultivierung der Stämme erfolgte in 200 ml MS-MES für 120 h. n = 3.
Promotor µ (h-1)
max. OD
Acetat (mM)
Butyrat (mM)
Aceton (mM)
Butanol (mM)
Ethanol (mM)
cb
ei_2
100
thlA 0,146 8,35 16,3 ± 4,3 7,3 ± 3,0 85,7 ± 9,7 193,7±17,5 89,7 ± 21,4
ptb 0,140 8,33 18,2± 2,6 6,3± 0,8 91,9± 10,4 207,4±26,5 137,9±24,8
adc 0,138 9,8 16,5± 3,0 8,2± 0,9 85,6± 21,1 186,1±15,9 99,9 19,0
fac 0,139 7,93 14,8± 2,6 8,9± 2,6 83,9± 17,7 152,8± 4,7 86,3± 25,9
ck
l_3
020
thlA 0,130 8,41 20,6 ± 2,4 5,0 ± 0,9 119,0 ± 6,4 238,6 ± 6,1 92,7 ± 1,8
ptb 0,133 8,03 22,6± 0,7 10,2± 3,9 87,8± 18,5 189,9±12,6 145,1±16,2
adc 0,113 7,46 21,4± 0,8 8,2± 0,6 95,9± 9,5 182,7±32,1 72,9± 5,5
fac 0,129 4,53 46,7± 16,3 41,4±16,8 0 0 6,0± 1,2
ab
fD
thlA 0,129 7,36 34,2 ± 3,7 10,2 ± 2,1 89,3 ± 9,5 164,6±18,4 43,8 ± 6,2
ptb 0,129 6,12 27,1± 3,2 15,8± 3,7 82,6± 8,3 155,6± 7,4 40,9± 0,1
adc 0,119 6,96 31,9± 8,9 12,9± 3,8 84,4± 8,6 151,9±12,7 36,7± 3,5
fac 0,09 3,42 44,1± 3,6 49,5± 5,7 0 0 4,8± 0,5
70 kDa 55 kDa
70 kDa 55 kDa 70 kDa
55 kDa
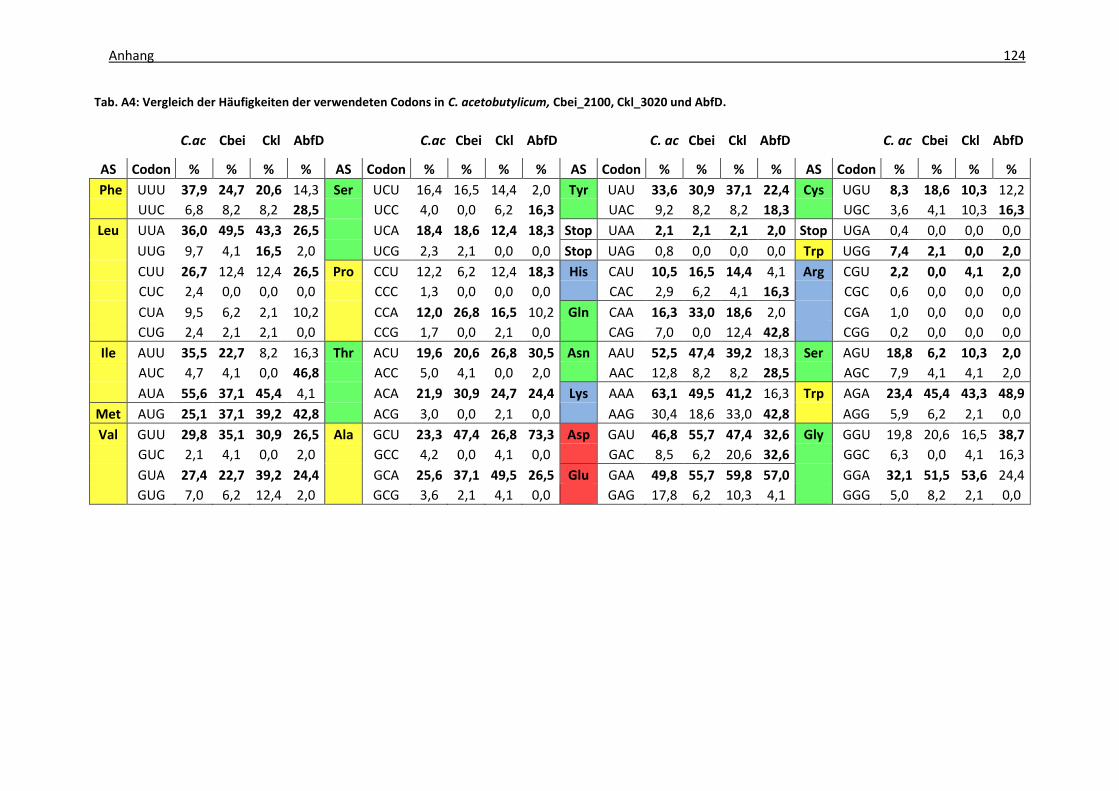
Anhang 124
Tab. A4: Vergleich der Häufigkeiten der verwendeten Codons in C. acetobutylicum, Cbei_2100, Ckl_3020 und AbfD.
C.ac Cbei Ckl AbfD C.ac Cbei Ckl AbfD C. ac Cbei Ckl AbfD C. ac Cbei Ckl AbfD
AS Codon % % % % AS Codon % % % % AS Codon % % % % AS Codon % % % %
Phe UUU 37,9 24,7 20,6 14,3 Ser UCU 16,4 16,5 14,4 2,0 Tyr UAU 33,6 30,9 37,1 22,4 Cys UGU 8,3 18,6 10,3 12,2
UUC 6,8 8,2 8,2 28,5 UCC 4,0 0,0 6,2 16,3 UAC 9,2 8,2 8,2 18,3 UGC 3,6 4,1 10,3 16,3
Leu UUA 36,0 49,5 43,3 26,5 UCA 18,4 18,6 12,4 18,3 Stop UAA 2,1 2,1 2,1 2,0 Stop UGA 0,4 0,0 0,0 0,0
UUG 9,7 4,1 16,5 2,0 UCG 2,3 2,1 0,0 0,0 Stop UAG 0,8 0,0 0,0 0,0 Trp UGG 7,4 2,1 0,0 2,0
CUU 26,7 12,4 12,4 26,5 Pro CCU 12,2 6,2 12,4 18,3 His CAU 10,5 16,5 14,4 4,1 Arg CGU 2,2 0,0 4,1 2,0
CUC 2,4 0,0 0,0 0,0 CCC 1,3 0,0 0,0 0,0 CAC 2,9 6,2 4,1 16,3 CGC 0,6 0,0 0,0 0,0
CUA 9,5 6,2 2,1 10,2 CCA 12,0 26,8 16,5 10,2 Gln CAA 16,3 33,0 18,6 2,0 CGA 1,0 0,0 0,0 0,0
CUG 2,4 2,1 2,1 0,0 CCG 1,7 0,0 2,1 0,0 CAG 7,0 0,0 12,4 42,8 CGG 0,2 0,0 0,0 0,0
Ile AUU 35,5 22,7 8,2 16,3 Thr ACU 19,6 20,6 26,8 30,5 Asn AAU 52,5 47,4 39,2 18,3 Ser AGU 18,8 6,2 10,3 2,0
AUC 4,7 4,1 0,0 46,8 ACC 5,0 4,1 0,0 2,0 AAC 12,8 8,2 8,2 28,5 AGC 7,9 4,1 4,1 2,0
AUA 55,6 37,1 45,4 4,1 ACA 21,9 30,9 24,7 24,4 Lys AAA 63,1 49,5 41,2 16,3 Trp AGA 23,4 45,4 43,3 48,9
Met AUG 25,1 37,1 39,2 42,8 ACG 3,0 0,0 2,1 0,0 AAG 30,4 18,6 33,0 42,8 AGG 5,9 6,2 2,1 0,0
Val GUU 29,8 35,1 30,9 26,5 Ala GCU 23,3 47,4 26,8 73,3 Asp GAU 46,8 55,7 47,4 32,6 Gly GGU 19,8 20,6 16,5 38,7
GUC 2,1 4,1 0,0 2,0 GCC 4,2 0,0 4,1 0,0 GAC 8,5 6,2 20,6 32,6 GGC 6,3 0,0 4,1 16,3
GUA 27,4 22,7 39,2 24,4 GCA 25,6 37,1 49,5 26,5 Glu GAA 49,8 55,7 59,8 57,0 GGA 32,1 51,5 53,6 24,4
GUG 7,0 6,2 12,4 2,0 GCG 3,6 2,1 4,1 0,0 GAG 17,8 6,2 10,3 4,1 GGG 5,0 8,2 2,1 0,0
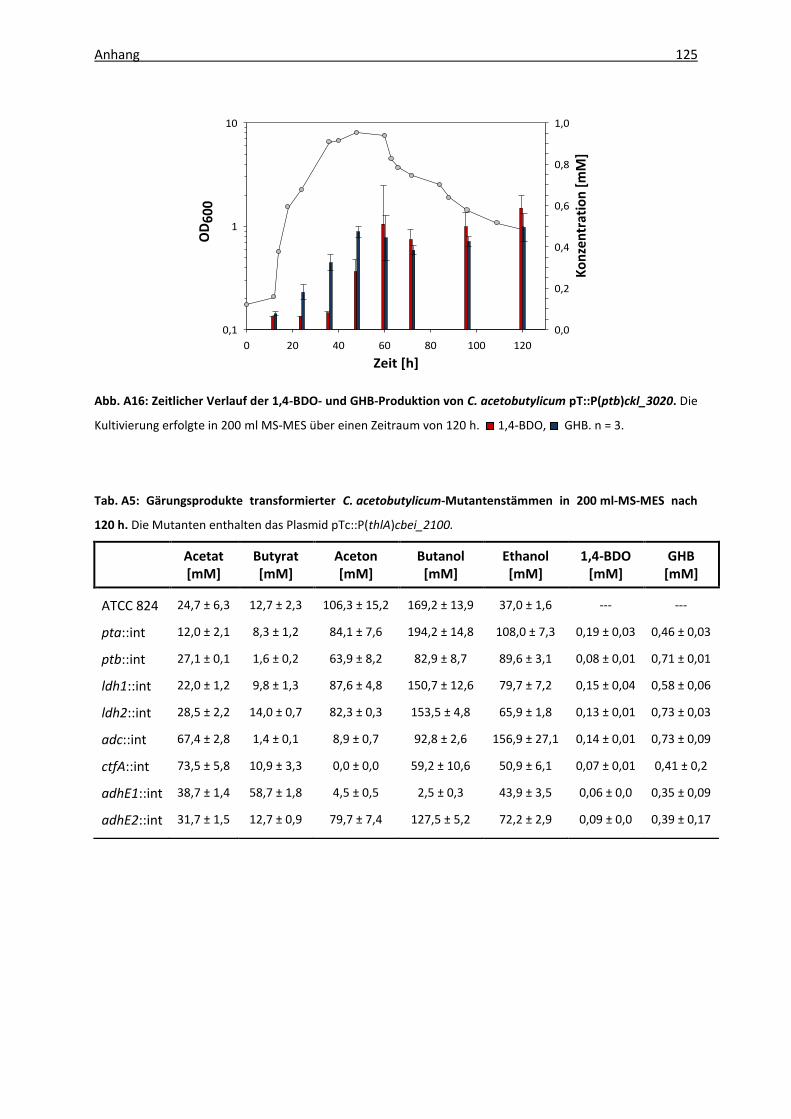
Anhang 125
Zeit [h]
0 20 40 60 80 100 120
OD
600
0,1
1
10
Ko
nze
ntr
atio
n [
mM
]
0,0
0,2
0,4
0,6
0,8
1,0
Abb. A16: Zeitlicher Verlauf der 1,4-BDO- und GHB-Produktion von C. acetobutylicum pT::P(ptb)ckl_3020. Die
Kultivierung erfolgte in 200 ml MS-MES über einen Zeitraum von 120 h. 1,4-BDO, GHB. n = 3.
Tab. A5: Gärungsprodukte transformierter C. acetobutylicum-Mutantenstämmen in 200 ml-MS-MES nach
120 h. Die Mutanten enthalten das Plasmid pTc::P(thlA)cbei_2100.
Acetat [mM]
Butyrat [mM]
Aceton [mM]
Butanol [mM]
Ethanol [mM]
1,4-BDO [mM]
GHB [mM]
ATCC 824 24,7 ± 6,3 12,7 ± 2,3 106,3 ± 15,2 169,2 ± 13,9 37,0 ± 1,6 --- ---
pta::int 12,0 ± 2,1 8,3 ± 1,2 84,1 ± 7,6 194,2 ± 14,8 108,0 ± 7,3 0,19 ± 0,03 0,46 ± 0,03
ptb::int 27,1 ± 0,1 1,6 ± 0,2 63,9 ± 8,2 82,9 ± 8,7 89,6 ± 3,1 0,08 ± 0,01 0,71 ± 0,01
ldh1::int 22,0 ± 1,2 9,8 ± 1,3 87,6 ± 4,8 150,7 ± 12,6 79,7 ± 7,2 0,15 ± 0,04 0,58 ± 0,06
ldh2::int 28,5 ± 2,2 14,0 ± 0,7 82,3 ± 0,3 153,5 ± 4,8 65,9 ± 1,8 0,13 ± 0,01 0,73 ± 0,03
adc::int 67,4 ± 2,8 1,4 ± 0,1 8,9 ± 0,7 92,8 ± 2,6 156,9 ± 27,1 0,14 ± 0,01 0,73 ± 0,09
ctfA::int 73,5 ± 5,8 10,9 ± 3,3 0,0 ± 0,0 59,2 ± 10,6 50,9 ± 6,1 0,07 ± 0,01 0,41 ± 0,2
adhE1::int 38,7 ± 1,4 58,7 ± 1,8 4,5 ± 0,5 2,5 ± 0,3 43,9 ± 3,5 0,06 ± 0,0 0,35 ± 0,09
adhE2::int 31,7 ± 1,5 12,7 ± 0,9 79,7 ± 7,4 127,5 ± 5,2 72,2 ± 2,9 0,09 ± 0,0 0,39 ± 0,17

Danksagung 126
Danksagung
Zunächst bedanke ich mich recht herzlich bei Herrn Prof. Dr. Hubert Bahl für das
entgegengebrachte Vertrauen und die Gelegenheit der selbstständigen Anfertigung dieser
Arbeit, sowie für die Freiheiten, die ich er mir bei der Bearbeitung ließ.
Desweiteren möchte ich mich bei Frau Dr. Tina Lütke-Eversloh für die Bereitstellung dieses
Themas, für die wissenschaftliche Betreuung sowie für die interessanten Gespräche und
Anregungen bedanken.
Darüber hinaus möchte ich auch Herrn Dr. Ralf-Jörg Fischer danken, der vor allem im letzten
Jahr mit konstruktiver Kritik und Ratschlägen meine Arbeit voranbrachte. Ebenso danke ich
ihm für das Korrekturlesen dieser Arbeit.
Weiterhin gilt einen großen Dank allen derzeitigen und ehemaligen Mitarbeitern der
Abteilung Mikrobiologie der Universität Rostock, die mich während meiner Zeit begleitet
haben. Insbesondere Ilona Boldt, Hella Goschke, Regina Karstädt und Monika Timm möchte
ich für die vielen großen und kleinen Hilfestellungen während des Laboralltags danken.
Darüber hinaus bedanke ich mich bei Igor Bill, Tina Drenckhan, Franziska Jenzen, Dr. Katja
Kriebel, Maria Lehmann, Anne Lunze, Dr. Antje May, Christoph Prohaska und Dr. Christine
Voigt für Hilfestellungen jeglicher Art und für die gute Arbeitsatmosphäre sowohl im Labor
als auch im Büro.
Ganz besonders bedanke ich mich bei Ronny Uhlig und Dr. Daniela Wetzel, die mich vor
allem mit Ratschlägen und Arbeitseifer stets unterstützt haben und immer ein offenes Ohr
für meine Probleme hatten. Insbesondere die fröhlichen und unterhaltsamen Mittagspausen
werde ich vermissen. Ebenso gilt ein großer Dank Katja Zimmermann, die viel Arbeit und
Mühe in das Korrekturlesen dieser Arbeit gesteckt und es immer wieder geschafft hat, mich
zu motivieren.
Ich danke ebenfalls Dajana Domik für das super Arbeitsklima über Abteilungsgrenzen
hinweg und für den Spaß während langer Labornächte.
Dr. Daniel Rentsch, aus der Abteilung forensische Toxikologie, gebührt ebenfalls ein sehr
großes Dankeschön, da ohne sein Einsatz und Interesse an diesem Thema eine

Danksagung 127
Nachweismethode für das 1,4-Butandiol nicht zustande gekommen und somit die
Anfertigung dieser Arbeit fraglich gewesen wäre.
Der Landesgraduiertenförderung Mecklenburg-Vorpommern danke ich für die finanzielle
Förderung dieser Promotion.
Bei meinen Freunden, besonders bei Sebastian und Gyöngyver Foth und Rene Kaiser,
möchte ich mich für die vielen schönen Zeiten und Ablenkungen außerhalb des Labors
bedanken.
Schließlich gilt jedoch der größte Dank meiner Familie, meinen Eltern Michael und Doris
Klipp und meiner Schwester Carolin Klipp, die mich bei allen meinen Vorhaben unterstützt
haben und ständig für mich da sind. Der Dank, den ich meiner Verlobten Julia Ruttmann
entgegenbringe, ist kaum in Worte zu fassen, da sie immer für mich da ist, mich aufbaut,
mich motiviert, mich beruhigt und mich zum Lachen bringt. Ohne sie wäre es ein viel
beschwerlicherer Weg gewesen. Vielen Dank!

Selbstständigkeitserklärung 128
Selbstständigkeitserklärung
Ich versichere hiermit an Eides statt, dass ich die vorliegende Arbeit selbstständig verfasst
und keine anderen als die angegeben Quellen und Hilfsmittel verwendet habe. Die aus
fremden Quellen direkt oder indirekt übernommenen Gedanken sind als solche
gekennzeichnet
Rostock, den 23.03.2016 Markus Klipp







![Kapitel 6) Ctitratcyklus+Atmungskette [Kompatibilitätsmodus]biochemietrainingscamp.de/stoff/ci/Ctitratcyklus.pdf · TPP Liponsäure NAD FAD CoA ( Vergleiche die Pyruvat-Dehydrogenase)](https://static.fdokument.com/doc/165x107/5e1287ca50baaa02a71d88e6/kapitel-6-ctitratcyklusatmungskette-kompatibilittsmodusbi-tpp-liponsure.jpg)