
Molekülspektroskopisches Praktikum SS 2014 - Institut · Bitte bringen Sie zum Praktikum neben...
25
Molekülspektroskopisches Praktikum SS 2014 Infrarot- und UV-Spektroskopie Flammen–Atomabsorptionsspektrometrie NMR-Spektroskopie Massenspektrometrie
Transcript of Molekülspektroskopisches Praktikum SS 2014 - Institut · Bitte bringen Sie zum Praktikum neben...

Molekülspektroskopisches Praktikum
SS 2014
Infrarot- und UV-Spektroskopie Flammen–Atomabsorptionsspektrometrie NMR-Spektroskopie Massenspektrometrie

Hinweise zum Protokoll Jede Gruppe gibt ein gemeinsames schriftliches Protokoll zu allen 4 Stationen des Praktikums im TA, Zi. 604, bei Frau Katrin Steinke ab. Die elektronische Version (pdf-Format) senden Sie bitte an [email protected] bis 24.00 Uhr am Abgabetermin. Die gedruckte Version muss entweder am gleichen Tag bis 16.00 Uhr vorliegen oder am folgenden Werktag bis 12.00 Uhr.
Abgabetermin gruppenspezifisch, siehe Praktikumsplan http://analytik.chemie.uni-leipzig.de/lehre-teaching-ss/ Das Protokoll behandelt die Ergebnisse der 4 Stationen und geht auf die dort gestellten Fragen ein. Die Lösung der Struktur der unbekannten Verbindung wird aus Erkenntnissen aller drei molekülspektroskopischen Stationen erarbeitet. Das Protokoll für AAS wird dem Gesamtprotokoll hinzugefügt. Die Vorlage für das Deckblatt ist auf der folgenden Seite zu finden. Formatvorschriften:
- Bitte heften Sie das Protokoll so in einen A4-Hefter, dass es fortlaufend lesbar ist. (Bitte verwenden Sie keine Klarsichthüllen, die nur oben offen sind.)
- Blocksatz / 1,5-zeilig / Nutzung der Silbentrennung - Vorlage für Deckblatt komplett ausfüllen - Inhaltsverzeichnis - durchgängige Nummerierung der Abschnitte vom ersten bis zum letzten
Versuch - Beschriftung der Graphiken und Tabellen - Seitennummerierung - Quellenverzeichnis - Anlagen nummerieren und beschriften - alle Spektren und Originalmessdaten in den Anhang aufnehmen

Praktikum „Molekülspektroskopie“ SS 2014
Gruppe: …….
Versuch Praktikums-Datum
Verantwortlicher für Protokoll Matrikel-Nr.
IR / UV
NMR
MS
AAS

Infrarot-Spektroskopie 1. Vorbereitung
Voraussetzung für die Durchführung des Praktikums ist die Grundlagenkenntnis von UV/Vis - und IR-Spektroskopie sowie von den Prinzipien der angeführten Geräte und Methoden. Insbesondere wird auf die folgenden Punkte Wert gelegt: - elektromagnetische Wellen und ihre Messgrößen - Wechselwirkung elektromagnetischer Wellen mit Materie - praktikumsrelevante physikalische Modelle (z.B. harmonischer Oszillator,
starrer Rotator) - Herleitung und Besonderheiten des Lambert-Beerschen Gesetzes - Aufbau und Funktionsweise eines optischen Spektrometers (dispersive
Spektrometer und Fourier-Transform-Spektrometer mit Michelson-Interferometer)
- praktische Durchführung einer IR-Messung (Probenvorbereitung, Küvettenmaterial, Lösungsmittel, Messbedingungen)
- ATR-Messprinzip (Abgeschwächte Totalreflexion) - Auswertung von Schwingungs- und Rotationsschwingungs-Spektren Literaturempfehlung: [1] C.N. Banwell, E. M. McCash
"Molekülspektroskopie: Ein Grundkurs" R. Oldenbourg Verlag, 1999
[2] M. Hesse, H. Meier, B. Zeeh, "Spektroskopische Methoden in der Organischen Chemie", 8. überarbeitete Aufl., Thieme, Stuttgart, 2011 [3] W. Gottwald, G. Wachter "IR-Spektroskopie für Anwender" Wiley-VCH Weinheim, 1997 u.a.
Bitte bringen Sie zum Praktikum neben Schreibmaterial auch Schutzkleidung (Kittel, lange Hose, geschlossenes Schuhwerk) und Schutzbrille mit. Praktikumsort ist das Technikum/Analytikum Linnéstr. 3, Raum 606. Benutzen Sie bitte das hintere Treppenhaus am Hofeingang.

2. Aufgabenstellungen für das Praktikum 2.1. UV/Vis-Spektren der zu identifizierenden Substanz und der gemeinsamen
Substanz
Nehmen Sie von der zu identifizierenden Substanz ihrer Gruppe ein UV/Vis-Spektrum im Bereich 200 bis 700 nm auf. Stellen Sie dazu eine geeignete Verdünnung der Probe her. Als Lösungsmittel steht abs. Ethanol (in UV/Vis-reiner Qualität) bereit. Das UV/Vis-Spektrum der gemeinsamen Substanz für alle Gruppen bekommen Sie als Kopie ausgehändigt. Berechnen Sie daran für die angegebenen Wellenlängen jeweils den molaren Extinktionskoeffizienten (in der Einheit L/(mol*cm). Alle benötigen Angaben finden Sie auf dem Spektrum. Beide Spektren sind bezüglich des Gehalts an Strukturinformationen zu diskutieren.
2.2. IR-Spektren von unbekannten Substanzen
Nehmen Sie die IR-Spektren von einer unbekannten flüssigen Substanz und der zu identifizierenden Gruppensubstanz im Bereich von 400 bis 4000 cm-1 auf. Verwenden Sie die Dünnfilmtechnik (Flüssigkeitsfilm zwischen zwei KBr-Platten) für flüssige Proben. Von Feststoffen ist ein KBr-Pressling herzustellen. Das IR-Spektrum der gemeinsamen Substanz für alle Gruppen bekommen Sie als Kopie ausgehändigt. Klassifizieren Sie die Substanzen mit Hilfe von IR-Absorptionstabellen, z.B. in [2]. Diskutieren Sie die gefundenen Absorptionsbanden aller Spektren und geben Sie einen Strukturvorschlag für die unbekannte flüssige Substanz und die zu identifizierende Gruppensubstanz an.
2.3. Wirkung von Massenänderung auf die Lage von Schwingungsbanden
Untersuchen Sie den Einfluss der Masse auf die C-H-Valenzschwingung am Bei-spiel von Aceton.
• Vergleichen Sie dazu die Lagen der Schwingungsbanden der C-H- bzw. der C-D- und der C=O-Valenzschwingung von „normalem“ Aceton (Aceton-D0, CH3-CO-CH3) und Deuterium-substitutierten Aceton (Aceton-D6).
• Berechnen Sie die Verhältnisse der Wellenzahlen der C-H- und der C-D-Valenzschwingung nach dem Modell des harmonischen Oszillators (unter Annahme von gleichen Kraftkonstanten).
• Vergleichen Sie Ihr Ergebnis mit dem Verhältnis der gemessenen Wellenzahlen. Ist die Anwendung des einfachen Modells sinnvoll?

2.4. IR-Spektren von amorphen Feststoffen – ATR-Technik Messen Sie die IR-Spektren zweier Polymere. Die Proben liegen als feste Folien/Streifen vor, bei denen übliche Transmissionsmessungen nicht möglich sind. Die IR-Spektren sind mit der ATR-Technik aufzunehmen. Identifizieren Sie anhand des IR-Spektrums charakteristische Banden und nehmen Sie eine möglichst genaue Bestimmung der vorliegenden Kunststoffe vor.
2.5. Rotationsschwingungsspektren von Gasen
Es soll das Rotationsschwingungsspektrum von Kohlenmonoxid ausgewertet werden.
• Befüllen Sie eine Gasküvette mit dem Rauch einer Filterzigarette. Dazu stehen Ansatzstücken bereit, mit denen die brennende Zigarette und eine Saugvorrichtung an den Küvettenstutzen angesetzt werden können. Das Gasspektrum muss mit hoher Auflösung (1 cm-1) aufgenommen werden. Die Valenzschwingungsbande von CO ist bei 2143 cm-1 gut sichtbar.
• Bestimmen Sie am Spektrum die Rotationskonstante B.
• Berechnen Sie die Bindungslänge der CO-Bindung anhand des Modells des starren Rotators und die Kraftkonstante anhand des Modells des harmonischen Oszillators. Die Bindungslänge läßt sich aus dem Trägheitsmoment bestimmen, wenn man ein einfaches Hantelmodell annimmt. Das Trägheitsmoment I des zweiatomigen Moleküls ist dann
mit r Abstand zwischen beiden (Punkt-)Massen (d. h. die gesuchte
Bindungslänge) M reduzierte Masse
Das Trägheitsmoment I selbst kann man aus der gemessenen Rotationskonstante B ermitteln mit:
mit: h PLANCKsches Wirkungsquantum c Lichtgeschwindigkeit Hinweis: Die Rechnung wird wesentlich erleichtert, wenn man konsequent SI-Einheiten verwendet und alle Zahlen in Zehnerpotenz-Schreibweise einsetzt. Protokollieren Sie Ihren Rechenweg so, daß er nachvollziehbar ist.
2rMI ⋅=
IchB
⋅⋅=
28π

• Beurteilen Sie die Bindungsstärke von Kohlenmonoxid im Vergleich zu anderen linearen Molekülen anhand der berechneten Kraftkonstante. Im Folgenden sind beispielhaft einige Moleküle und ihre Kraftkonstanten zum Vergleich angeführt:
HCl 481 Nm-1 SO2 1.001 Nm-1 O2 1.141 Nm-1 N2 2.242 Nm-1
Bedenken Sie auch die elektronische Struktur der betrachteten Moleküle. Welche Rolle spielen induktive und mesomere Effekte?
2.6. Quantitative IR-Spektroskopie
Ein kommerzieller Farbverdünner besteht aus n-Hexan als Hauptkomponente. Als einer der Nebenkomponenten im einstelligen Vol.-%-Bereich ist auch Aceton enthalten. Bestimmen Sie den Volumenanteil einer Lösung von Aceton in n-Hexan.
• Stellen Sie dazu sieben Kalibrierlösungen mit Volumenanteilen von 1,0 – 4,0% (v/v) Aceton (in 0,5%-Schritten) in n-Hexan her, indem Sie das Aceton mit einer Bürette genau abmessen und dann in einem 10-ml-Messkolben mit n-Hexan auffüllen.
• Nehmen Sie die IR-Spektren mit Standardauflösung (4 cm-1) im Transmissions-Modus im Bereich 2000 – 1000 cm-1 auf. Das Leer-Spektrum (Background) muss gegen Luft (d.h. ganz ohne Küvette) aufgenommen werden, weil einerseits eine Aufnahme mit Lösungsmittel eine sehr starke Absorption und damit ein schwaches Signal ergibt, was Probleme bei der Quotientenbildung (I / I0) verursacht, und sich andererseits die Aufnahme einer Leerküvette wegen Interferenzbildung verbietet (die Schichtdicke ist hier in der Größenordnung der Wellenlänge des Lichtes). Damit steht allerdings für die quantitative Auswertung die benötigte I0-Intensität nicht zur Verfügung.
• Die gut erkennbaren Keton-typischen Aceton-Banden liegen bei 1719 cm-1 und 1213 cm-1, die beide einzeln auszuwerten sind. Um die Intensität I0 zu korrigieren, legt man als Grundlinie eine Gerade durch den linken und rechten Fußpunkt der jeweiligen Bande. Als Messwerte werden die Intensität I an der Spitze der Bande und die Intensität I0 am Schnittpunkt des Lots mit der Grundlinie entnommen.
• Tragen Sie für die sieben Lösungen die Extinktionen beider Banden in eine Tabelle und berechnen Sie je eine Ausgleichsgerade (lineare Regression). Aus dieser bestimmen Sie nach Messung des Farbverdünners dessen Acetongehalt. Schätzen Sie die Fehlergröße Ihrer Messung in Hinblick auf Präzision und Richtigkeit ab (Größtfehler-Rechnung) und geben Sie das Ergebnis unter Beachtung der abgeschätzten Genauigkeit an. Diskutieren Sie mögliche Ursachen des Fehlers und die Auswirkung verschiedener Fehlerquellen auf die Richtigkeit der Kalibrierung. Inwiefern lässt sich anhand der abschätzbaren Fehler eine Aussage über die Richtigkeit des Ergebnisses treffen?
Weitere relevante Moleküle sollen aus eigenständiger Literatur-recherche dem Vergleich zugeführt werden.

3. Anmerkungen zur Protokollführung
• Das Protokoll sollte durchgehend lesbar und übersichtlich formatiert sein. Alle Betrachtungen sollten möglichst in Text ausformuliert werden. Resultate müssen in einem erkennbaren Abschlußsatz zu finden sein.
• Protokolle sollten eine Rekonstruktion der Experimente durch dritte Personen ermöglichen. Prüfen Sie bitte die Erfüllung dieser Anforderung. Aufgabenstellung und Durchführung sollten in Kurzform zu Beginn des jeweiligen Teilversuchs wiedergegeben werden.
• Grundlegende theoretische Einführungen können vorausgesetzt werden. Die Einleitung sollte lediglich kurze physikalische und technische Grundlagen bereitstellen, die für Verständnis und Durchführung der jeweiligen Versuche notwendig sind
• Zu allen ausgedruckten Spektren wird eine Spektrendiskussion im Rahmen der jeweiligen Interpretierbarkeit erwartet. Spekulationen sollten ggf. durch zusätzliche Informationen gestützt werden.
• Auf eine sorgfältige Kenntlichmachung von benutzten Quellen wird Wert gelegt.

Flußdiagramm zur Polymeren-Bestimmung
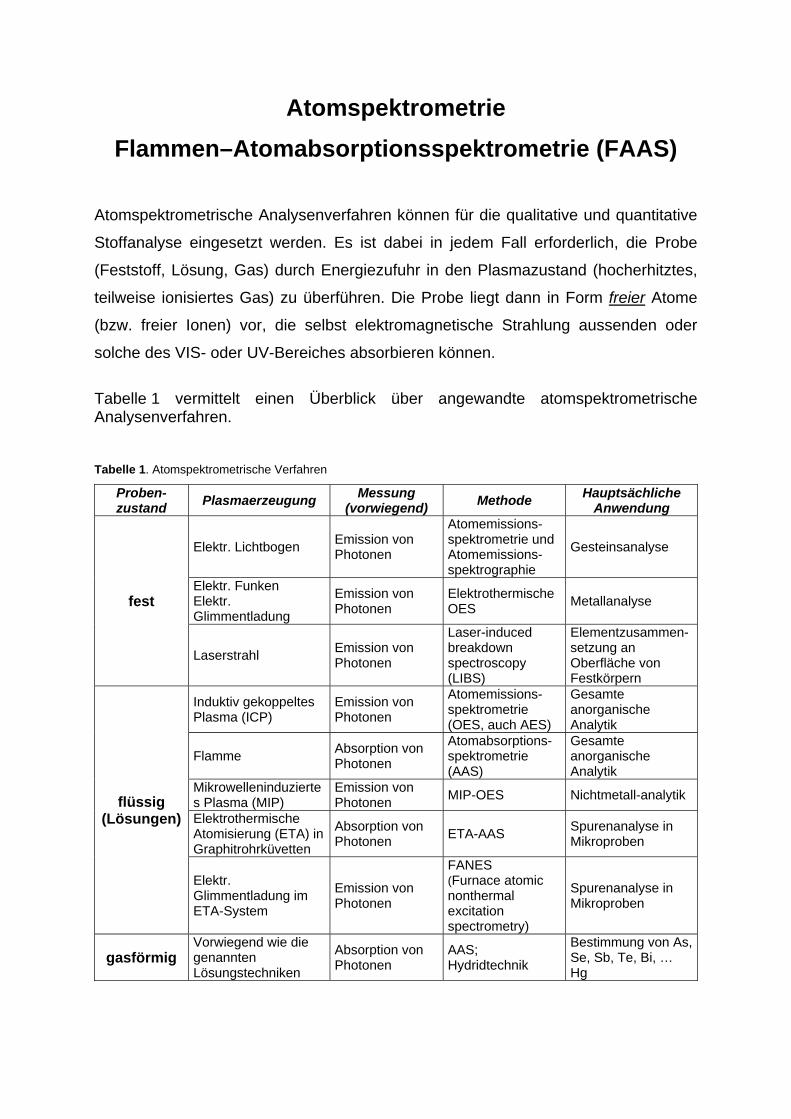
Atomspektrometrie Flammen–Atomabsorptionsspektrometrie (FAAS)
Atomspektrometrische Analysenverfahren können für die qualitative und quantitative
Stoffanalyse eingesetzt werden. Es ist dabei in jedem Fall erforderlich, die Probe
(Feststoff, Lösung, Gas) durch Energiezufuhr in den Plasmazustand (hocherhitztes,
teilweise ionisiertes Gas) zu überführen. Die Probe liegt dann in Form freier Atome
(bzw. freier Ionen) vor, die selbst elektromagnetische Strahlung aussenden oder
solche des VIS- oder UV-Bereiches absorbieren können.
Tabelle 1 vermittelt einen Überblick über angewandte atomspektrometrische Analysenverfahren. Tabelle 1. Atomspektrometrische Verfahren
Proben-zustand Plasmaerzeugung Messung
(vorwiegend) Methode Hauptsächliche Anwendung
fest
Elektr. Lichtbogen Emission von Photonen
Atomemissions-spektrometrie und Atomemissions-spektrographie
Gesteinsanalyse
Elektr. Funken Elektr. Glimmentladung
Emission von Photonen
Elektrothermische OES Metallanalyse
Laserstrahl Emission von Photonen
Laser-induced breakdown spectroscopy (LIBS)
Elementzusammen-setzung an Oberfläche von Festkörpern
flüssig (Lösungen)
Induktiv gekoppeltes Plasma (ICP)
Emission von Photonen
Atomemissions- spektrometrie (OES, auch AES)
Gesamte anorganische Analytik
Flamme Absorption von Photonen
Atomabsorptions-spektrometrie (AAS)
Gesamte anorganische Analytik
Mikrowelleninduziertes Plasma (MIP)
Emission von Photonen MIP-OES Nichtmetall-analytik
Elektrothermische Atomisierung (ETA) in Graphitrohrküvetten
Absorption von Photonen ETA-AAS Spurenanalyse in
Mikroproben
Elektr. Glimmentladung im ETA-System
Emission von Photonen
FANES (Furnace atomic nonthermal excitation spectrometry)
Spurenanalyse in Mikroproben
gasförmig Vorwiegend wie die genannten Lösungstechniken
Absorption von Photonen
AAS; Hydridtechnik
Bestimmung von As, Se, Sb, Te, Bi, … Hg

1. AnforderungenandasAntestat
Theoretische Grundlagen der Atomspektrometrie
Aufbau eines Atomabsorptionsspektrometers
Vor- und Nachteile im Vergleich Flammenatomisierung versus elektrother-
mischer Arbeitsweise
Interferenzerscheinungen in der AAS und Möglichkeiten zu deren Korrektur
2. Versuchsbeschreibung:
QuantitativeAnalysedurchFlammen‐AAS
2.1 Aufgabe Mittels Flammen-AAS (Acetylen/Luft) soll die Konzentrationsbestimmung eines
Elementes in Realproben (Mineralwasser, Ca2+-Konzentration) unter Anwendung
einer externen Kalibration bzw. des Standardadditionsverfahrens erfolgen.
2.2 Geräte Atomabsorptionsspektrometer AAS vario 6 (Analytik Jena AG)
Abbildung 1. AAS-Gerät der Fa. Analytik Jena AG
2.3 Durchführung Die Bestimmung der Konzentration eines ausgewählten Elementes in dieser Lösung
soll durch Anwendung verschiedener analytischer Auswerteverfahren erfolgen.
Zunächst wird für das betreffende Element ausgehend von einer Standardlösung
(c = 1000 mg/l) eine Kalibrationskurve aufgenommen (externe Kalibration). Die
einzelnen Kalibrierlösungen werden jeweils dreimal vermessen, die Analysenlösung
fünfmal. Anhand des Extinktionswertes (Mittelwert) der Probe wird mit der

Kalibrationskurve der Elementgehalt in der Probe ermittelt. Es wird geprüft, in
welchem Konzentrationsbereich eine lineare Regression möglich ist. Diese wird zur
Bestimmung der Empfindlichkeit und des Blindwertes durchgeführt. Mit der inversen
Analysenfunktion wird das Ergebnis berechnet und anschließend die
Standardabweichung des Anlaysenergebnisses ermittelt.
Als ein weiteres Kalibrationsverfahren wird die Standard-Additions-Methode zur
Konzentrationsbestimmung herangezogen.
2.4 Auswertung/Protokoll Das Protokoll sollte übersichtlich formatiert sein (Zeilenabstand 1,5; Blocksatz; ect.)
und alle Betrachtungen sollten – unabhängig von sonstigen übersichtlichen
Darstellungsformen – in ausformuliertem Text wiederzufinden sein.
Ermittlung der Konzentration und Diskussion der Ergebnisse, die aus beiden
Kalibrierverfahren erhalten wurden, sollten durch Dritte nachvollziehbar sein.
Es ist eine ordentliche Fehlerbetrachtung durchzuführen!!!
Quellen sind kenntlich zu machen.

Einführung in die hochauflösende NMR‐Spektroskopie
1 Übersicht Dieser Versuch soll die Grundlagen der Flüssig‐NMR‐Spektroskopie sowie die praktischen Aspekte bei der Aufnahme von ein‐ und zweidimensionalen 1H‐ und 13C‐Spektren behandeln. Vor der Durchführung der Versuche findet ein Antestat statt, in dem folgende Schwerpunkte besprochen werden:
◦ physikalischen Grundlagen der magnetischen Kernresonanz (Resonanzbedingung, Zeeman‐Aufspaltung/Energie‐Niveaus, gyromagnetisches Verhältnis, Magnetisierungs‐Vektor‐Modell, Messprinzip, Relaxationsmechanismen)
◦ Aufbau eines FT‐NMR‐Spektrometers und Durchführung der Messung (wichtige Bau‐teile, Locken, Shimmen)
◦ Spektrale Parameter eindimensionaler NMR‐Spektren und ihre Bedeutung (chem. Verschiebung, skalare Kopplung, Linienform) am Beispiel der 1H‐ und 13C‐NMR
◦ Prinzip der zweidimensionalen NMR‐Spektroskopie (allgemeiner Ablauf, Informa‐tionsgehalt grundlegender 2D‐Spektren)
In dieser Praktikumsanleitung werden die wichtigsten anwendungsbezogenen Themen er‐klärt. Informieren Sie sich im Vorfeld anhand des Vorlesungsstoffs und/oder entsprechender Literatur bitte auch über die theoretischen Sachverhalte! Sie sollten ebenfalls in der Lage sein, die Fragen in dieser Vorschrift zu beantworten. Die während des Versuchs aufgenommenen Spektren erhalten Sie sowohl in gedruckter Form sowie digital, während Ihnen die im Vorfeld aufgenommenen Spektren nur digital zur Verfügung gestellt werden. Die Dateien finden Sie auf dem Spektrenserver des Instituts für Analytische Chemie; für den Download ist ein FTP‐Programm erforderlich (Adresse des Spektrenservers: http://spekserv.chemie.uni‐leipzig.de, die Zugangsdaten werden Ihnen während des Praktikums mitgeteilt). Die zur Prozessierung und Darstellung der Spektren benötigte Software MNOVA kann unter folgendem Link heruntergeladen werden: http://www.uni‐leipzig.de/~nmr/MNOVA . Dieses Programm ist ebenfalls an einem der Rechner im PC‐Pool installiert. Literatur: [1] H. Friebolin, Ein‐ und zweidimensionale NMR‐Spektroskopie ‐ Eine Einführung;
5. Auflage Wiley‐VCH, Weinheim, 2013. [2] M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen Chemie;
8. Auflage,Thieme, Stuttgart, 2012. [3] M. Findeisen, S. Berger, 50 and More Essential NMR Experiments, 1. Auflage, Wiley‐
VCH, Weinheim, 2014.

2 Theoretische Grundlagen / Experimentelle Anforderungen 2.1 Der prinzipielle Aufbau eines FT‐NMR‐Spektrometers Ein NMR‐Spektrometer ist aus den folgenden, wesentlichen Komponenten aufgebaut:
Supraleitender Magnet Probenkopf (beinhaltet u.a. die Sender‐ und Empfängerspulen für 1H‐ und 13C‐Kerne, temperierbar, für 5 mm Proberöhrchen) HF‐Sender mit Synthesizer (1H‐Sender 300 MHz, 13C‐Sender 75 MHz) HF‐Empfänger & Verstärker Steuer‐, Bedien‐ und Ausgabeeinheiten
1. Welche Funktion haben die einzelnen Komponenten?
Abb. 1: Schematische Darstellung des Aufbaus eines NMR‐Spektrometers (links) und eines Flüssig‐NMR‐
Probenkopfs.
2.2 Probenvorbereitung/Vorbereitung des Spektrometers
In diesem Praktikum erfolgt die NMR‐Messung stets in Lösung, wobei ausschließlich deuterierte Lösungsmittel verwendet werden. Zur Referenzierung der chemischen Verschie‐bung ist diesem Lösungsmittel ein interner Standard zugesetzt. 2. Welche Standards werden in der Flüssig‐NMR verwendet? 3. Bei welcher chemischen Verschiebung (in ppm) erscheint das 1H‐Signal von CHCl3, wenn bei einem 400 MHz‐Spektrometer eine Frequenzdifferenz von 2908 Hz zum TMS gemessen wurde?

Das NMR‐Röhrchen wird mit einem sauberen Tuch abgewischt und in dem sog. Spinner platziert, wobei die korrekte Höhe mithilfe des Sample‐Racks festgelegt wird. Anschließend wird das NMR‐Röhrchen im Spinner über den sog. air‐lift in den Probenkopf des Spektrometers überführt. Bevor das gewünschte NMR‐Experiment durchgeführt wird, erfolgt das Locken und Shimmen des Spektrometers, um die zeitliche bzw. örtliche Homogenität des externen B0‐Feldes zu gewährleisten. 4. Erklären Sie kurz, wie Locken und Shimmen funktionieren! 2.3 Das Impuls‐NMR‐Experiment/Aufnahme der Spektren
Beim Impuls‐Verfahren werden alle magnetischen Momente einer Kernsorte durch einen Hochfrequenzimpuls (HF‐Puls) gleichzeitig angeregt. Die Pulslänge liegt im Bereich von ei‐nigen Mikrosekunden. Die Frequenz ist durch die unveränderliche Magnetfeldstärke Bo des statischen Magnetfelds und durch das gyromagnetische Verhältnis γ der zu untersuchenden Kernsorte bestimmt. Der HF‐Puls bewirkt eine Auslenkung des makroskopischen Magnetisierungsvektors M0 in die xy‐Ebene um einen bestimmten Winkel α. Der Pulswinkel (Flip‐Winkel) α ist dabei der Pulsdauer (p1) proportional. Der Flip‐Winkel ist zusätzlich von der Feldstärke des Magnetfelds des Pulses (B1) sowie dem gyromagnetischen Verhältnis (γ) der Kernsorte abhängig. Die Pulswinkel 90° sowie 180° sind für viele Impulsexperimente von besonderer Bedeutung. Nach Abschalten des Impulses kehrt das Spinsystem wieder in den Gleichgewichtszustand zurück. Dieser Vorgang heißt Relaxation. Während des Relaxationsvorgangs wird in der Empfängerspule, welche sich in der x,y‐Ebene befindet, die Abnahme der Quermagnetisierung My in Abhängigkeit von der Acquisitionszeit (aq) gemessen. Dieses Signal wird als freier Induktionsabfall (free induction decay, FID) bezeichnet. Durch die Fourier‐Transformation wird das Interferogramm (d.h. der FID) aus der Zeitdomäne in die Frequenzdomäne umgerechnet.
Abb. 2: Schematische Darstellung des Impuls‐NMR‐Experiments.

Tab. 1: Wichtige Acquisitionsparameter.
Parameter Bedeutungpulprog Pulsprogrammtd Anzahl der aufgenommenen Datenpunkted1 [s] Prescan‐Delayns Anzahl der Scansp1 [µs] Pulsdauer des Pulses im 1. Kanal (F1)pldb1 [dB] Leistung des HF‐Pulses im 1. Kanalo1p [ppm]rg
Transmitter Offset („Mitte“ des Spektrums)Receiver Gain (Verstärkung des FIDs im Empfänger)
sw [ppm] Spektrale Breite Zur Durchführung eines NMR‐Experiments müssen die jeweiligen Messparameter (Acquisitionsparameter) festgelegt werden, um die gewünschten Ergebnisse zu erzielen. Die wichtigsten Acquisitionsparameter sind in Tab. 1 zusammengefasst. 5. Wie hängt das Signal‐zu‐Rausch‐Verhältnis S/N von der Anzahl der Scans ab? 6. Wie viele Signale erwarten Sie für das 1H‐NMR‐Spektrum von Pyridin? Warum sieht man keine 13C‐13C‐Kopplung im 13C‐Spektrum der Verbindung? 7. Welche Informationen können aus den Experimenten APT, DEPT sowie dem 1H‐gekoppelten 13C‐NMR‐Spektrum entnommen werden? 2.4 Relaxation Durch den HF‐Anregungspuls wird das thermische Gleichgewicht des Spinsystems gestört, was eine Änderung der Besetzungsverhältnisse der Kernspin‐Niveaus sowie die Ausbildung der Quermagnetisierung zur Folge hat. Nach der Beendigung dieser Störung relaxiert das Spinsystem wieder in den Gleichgewichtszustand, wobei zwei Relaxationsmechanismen un‐terschieden werden. Die longitudinale Relaxation (Spin‐Gitter‐Relaxation) beschreibt die Rückkehr zur Gleichgewichtsmagnetisierung M0 und wird anhand der T1‐Relaxationszeit cha‐rakterisiert. Die transversale Relaxation (Spin‐Spin‐Relaxation) beschreibt das Auffächern der Spins in der xy‐Ebene und ist durch die Relaxationszeit T2 angegeben. Die Spin‐Gitter‐Relaxa‐tionszeit richtet neben Temperatur und Solventsumgebung vor allem nach Molekülgröße und Bindungssituation und liegt bei Protonen im üblicherweise im Bereich von wenigen Sekunden, während sie bei 13C‐Kernen Werte zwischen einigen Sekunden bis hin zu über 100 Sekunden annehmen kann. 8. Welche Experimente werden zur Bestimmung von T1 bzw. T2 genutzt? 9. Welche Größenbeziehung besteht zwischen der Dauer von T1 und T2 und warum?

2.5 Zweidimensionale NMR‐Spektroskopie Zur Aufklärung unbekannter Strukturen komplexer organischer Moleküle reichen die herkömmlichen, eindimensionalen 1H‐und 13C‐Spektren häufig nicht aus, sodass die Anwendung zweidimensionaler Techniken erforderlich ist. Jedes zweidimensionale NMR‐Spektrum beinhaltet eine direkte (meist horizontale Achse, F2) und eine indirekte Dimension (vertikale Achse, F1). Während sich die direkte Dimension analog zu den eindimensionalen Experimenten aus der Aufnahme des FIDs während der Acquisitionszeit (t2) ergibt, muss für die Erzeugung der indirekten Dimension ein Zeitabschnitt in der gewählten Pulsfolge (Evolutionszeit, t1) inkrementiert werden (Abb. 3). Dazu werden viele 1D Spektren mit systematisch veränderter t1‐Zeit aufgenommen. Anschließend erfolgt eine zweidimensionale Fourier‐Transformation der so erhaltenen Spektrenmatrix nach den Acquisitions‐ und der Evolutionszeit (Abb. 4), welche das zweidimensionale Frequenzspektrum liefert, die dritte Dimension ist die Signalintensität.
Abb. 3: Schematischer Ablauf eines 2D‐NMR‐Experiments am Beispiel des COSY‐Experiments.
Abb. 4: Schematische Darstellung der zweidimensionalen Fourier‐Transformation.
Die in den gebräuchlichen zweidimensionalen Spektren abgebildeten Zusammenhänge basieren auf skalaren oder räumlichen Wechselwirkungen zwischen benachbarten Kernen einer oder verschiedener Kernsorten (z.B. 1H/1H bzw. 1H/13C). In der nachfolgenden Tabelle sind die wichtigsten zweidimensionalen NMR‐Experimente zusammengefasst.
FT (t2) FT (t1)
F2 F2F1
t1
t2
t1

Tab. 2: Übersicht über die wichtigsten 2D‐Spektren
2D‐Experiment InformationsgehaltCOSY skalare Kopplung zwischen benachbarten 1H‐Kernen NOESY Wechselwirkung von 1H‐Kernen über den Raum (KEINE
skalare Kopplung!) HSQC od. HMQC Wechselwirkung von 13C mit dem direkt an dieses C‐Atom
gebundenen 1H‐Kernen (1JCH) HMBC Wechselwirkung von 13C mit 1H‐Kernen über 2,3 oder
mehr Bindungen (2JCH und 3JCH) 10. Was ist der „organische Spektrensatz“ und was bedeuten die Abkürzungen der entsprechenden Pulssequenzen? Welche Signale werden zur Auswertung der zweidimensionalen Spektren benutzt? 3 Versuchsdurchführung Nach dem Antestat erfolgt zunächst eine Einweisung in die Handhabung des Spektrometers und die Betriebssoftware Topspin®. Anschließend werden Sie selbständig die Pulsdauer des 90°‐1H‐Pulses bestimmen und eine T1‐Relaxationszeitmessung für zwei Protonensignale des Zimtsäure‐n‐Propylesters (Abb. 5) durchführen. Die dazu verwendeten Pulssequenzen und Messparameter werden Ihnen bei der Durchführung mitgeteilt. Abschließend sollen sie jeweils das 1H‐Spektrum von dieser Verbindung sowie von Ihrer unbekannten Substanz aufnehmen. Die restlichen Spektren des organischen Spektrensatzes bekommen Sie für beide Verbin‐dungen bereits gemessen als Dateien zur Verfügung gestellt. Danach erfolgt eine Einweisung in die Prozessierung der aufgenommenen ein‐ und zwei‐dimensionalen Spektren mittels Topspin®. Die Prozessierung der FID‐Dateien sollen Sie für das Protokoll zu Hause mithilfe der Software MNOVA selbständig durchführen (Hinweise zum Download der Spektren und Installation der Software siehe Abschnitt 1).
Abb. 5: Struktur von Zimtsäure‐n‐propylester.

Zur Bestimmung der Pulsdauer wird bei gegebener Leistung (pl1) die Dauer des Pulses (p1) systematisch variiert, sodass man einen sinusartigen Verlauf der Signalintensitäten erhält (Abb. 6). Aus dieser Darstellung wird die Dauer des 360°‐Pulses anhand des zweiten Null‐durchgangs der Sinuskurve bestimmt. Um den 90°‐Puls zu erhalten, muss die so bestimmte Pulsdauer durch vier dividiert werden.
Abb. 6: Sinusartiger Verlauf der Signalintensitäten bei der Bestimmung der Pulsdauer.
Die Bestimmung der T1‐Relaxationszeit erfolgt mit dem Inversion‐Recovery‐Experiment, dessen allgemeine Pulsfolge in Abb. 7 dargestellt ist.
Abb. 7: Pulsfolge des Inversion‐Recovery‐Experiments zur Bestimmung der T1‐Zeit.
Die Gleichgewichts‐Magnetisierung M0 wird durch den 180°‐Puls zunächst in –z‐Richtung ausgelenkt. Während der variablen Delay‐Zeit τ relaxiert diese z‐Magnetisierung mit der Ge‐schwindigkeitskonstante k = T1‐1, wobei die nach Ende dieser Zeit noch vorhandene z‐Mag‐netisierung Mz durch den 90°‐Puls auf die y‐Achse gedreht wird und somit ein messbares Signal ergibt. Die Intensität des Signals ändert sich dabei mit τ in charakteristischer Weise. In Abhängigkeit der Delay‐Zeit ergibt sich ein exponentieller Anstieg der Signalintensitäten, welcher mit der folgenden Gleichung beschrieben wird:
⎟⎟⎠
⎞⎜⎜⎝
⎛−=
τ−
1210zT
eMM (1)
Aus den bei verschiedenen τ‐Werten bestimmten Signalintensitäten bzw. Integralen kann die gesuchte Relaxationszeit mithilfe des T1/T2‐Relaxationsmoduls der Software Topspin® nach Gleichung (1) ermittelt werden.
p1 in µs

4 Hinweise zur Anfertigung des Protokolls 4. 1 Aufbau des Protokolls Das Protokoll soll wie folgt aufgebaut sein: 1 Einleitung: schriftliche Beantwortung der Fragen 1‐10 aus dieser Versuchsvorschrift 2 Pulsdauerbestimmung und T1‐Messung:
kurze Angabe der vermessenen Proben und verwendeten Lösungsmittel mit dem Pulsprogramm und den Acquisitionsparametern des durchgeführten NMR‐Experiments(in tabellarischer Form möglich) Auswertung und Interpretation der Messdaten
3. Auswertung der Spektren von Zimtsäure‐n‐propylester: Hinweise siehe 4.2 4. Auswertung der Spektren Ihrer unbekannten Substanz: Hinweise zur Angabe der Signale siehe 4.2
Beschreiben Sie zusätzlich kurz, wie Sie zur Lösung der Struktur gekommen sind. Sind die aus den NMR‐Spektren enthaltenen Informationen ausreichend? Wie müssen die anderen Methoden des Praktikums hier mit einbezogen werden?
4. 2 Hinweise zur Spektreninterpretation Jedes Spektrum sollte vollständig interpretiert werden, d.h. alle relevanten Informationen im Spektrum (chemische Verschiebung, Signalintensität, Multiplizität, Kopplungskonstante, Zu‐ordnung) müssen aufgelistet werden. Die korrekte Auswertung für 1D 1H‐ bzw. 13‐C‐Spekt‐rum soll so erfolgen:
1H‐NMR (CDCl3, int. TMS, 300 MHz): δ= 2.95, (s, 3H, N‐CH3); 3.47 (dd, 2H,
3JHH = 7.6 Hz, 2JHH = 8.7 Hz, CH2‐NH); 3.94 (dd, 2H, 3JHH = 7.6 Hz,
2JHH = 8.7 Hz, CH2‐NH); 5.48 (dd, 1H, 3JHH = 8.1 Hz, Ph‐CH); 7.40, (m, 5H, Ph).
Bei der Angabe der C‐ und H‐Atome ist eine entsprechende Nummerierung bzw. Kennzeichnung der Atome in der Zielstruktur möglich (z.B. HA, CA oder C1, H1). Für 2D‐Spektren ist die Verwendung von Tabellen denkbar.

Organische Massenspektrometrie (MS) 1 Ziel des Praktikums (1) Kennenlernen eines Massenspektrometers, eines Sektorfeldgerätes mit Elektronenstoß‐
Ionisation (EI) (2) Aufnahme von EI‐ Massenspektren unter Anleitung (4) Übungen zur EI‐ Spektreninterpretation.
Das Praktikum wird unter Zuhilfenahme Ihrer Vorlesungsmitschriften und der unten angegebenen Literatur selbstständig vorbereitet. In einem Antestat wird Ihr Wissen zu folgenden Sachverhalten geprüft:
• Grundlagen der Massenspektrometrie, Aufbau eines Massenspektrometers • Aussehen eines Massenspektrums, Begriffe • häufig verwendete Ionisationsarten und Analysatoren • Ausführlich: Electron Impact Ionisation, Aufbau der Quelle • Sektorfeld, Sekundärelektronenvervielfacher • Spektreninterpretation: Isotopie und akkurate Masse • Entstehende Spezies, Regeln der Fragmentierung
Dabei dienen die gestellten Aufgaben als Orientierung. Die Lösungen der im Skript gestellten Übungsaufgaben sind gleichzeitig Bestandteil des Protokolls. 2 Einleitung Das Grundprinzip der EI‐ Technik ist der Beschuss der im Hochvakuum (10‐5‐10‐7 mbar) isolierten, gasförmigen Moleküle mit Elektronen hoher kinetischer Energie (meist 70 eV). Dabei wird aus dem Molekül ein Elektron herausgeschlagen und das Molekülion M∙+. erzeugt, ein Radikalkation (open‐shell ion, odd‐electron ion).
EI ist eine „harte“ (energiereiche) Ionisationsmethode. Zur Erzeugung von Radikalkationen aus organischen Molekülen reichen prinzipiell Energien von 8‐14 eV. Der Energieüberschuss führt deshalb bereits in der Ionenquelle zur Fragmentierung der Radikalkationen:
M+. → A+ + B. Fragmentierung in Ion (closed‐shell) und Radikal M+. → A. + B+ M+.
→ C+. + D; A+ → F+ + G Neutralverlust (open‐shell ion)
Radikalische Spaltungen treten nur aus Radikalkationen auf (open‐shell‐Ionen), daraus entstandene Kationen (closed‐shell‐Ionen, odd‐electron) fragmentieren unter Neutralabspaltung (even‐electron rule), so dass auch Folgefragmentierungen möglich sind. Neutralabspaltungen treten auch direkt aus dem Molekülradikalkation auf. 3 Grundlagen der Spektreninterpretation 3.1 Isotopie der Elemente Informieren Sie sich über Einteilung der Elemente nach Häufigkeit und Isotopenabstand.

Informationen aus den Isotopenpeaks Die Häufigkeit der Moleküle mit einem Molekulargewicht über dem Molekulargewicht des monoisotopischen Moleküls hängt von der Anzahl der vorhandenen Atome und von der relativen Häufigkeit der Isotope in den beteiligten Elementen ab. (Orientierungsfrage: welche Häufigkeit tragen 6 C‐Atome auf der M+1 Stelle bei?) Daher kann man aus der Häufigkeitsverteilung der Isotope mit entsprechenden Massenabstand (M+1, M+2;...) auf Art und Anzahl der im Molekül vorhandenen Elemente schließen. Tabelle 1: Isotopenverteilung in der MS wichtiger Elemente, bezogen auf das häufigste Isotop.
Element M M+1 M+2 Masse % Masse % Masse %H 1 100 2 0.015 ‐ ‐C 12 100 13 1.1 ‐ ‐N 14 100 15 0.37 ‐ ‐O 16 100 17 0.04 18 0.21F 19 100 ‐ ‐ ‐ ‐Si 28 100 29 5.1 30 3.4S 32 100 33 0.8 34 4.5Cl 35 100 ‐ ‐ 37 32.0Br 79 100 ‐ ‐ 81 98I 127 100 ‐ ‐ ‐ ‐
Berechnung der Isotopenverteilung Die Isotopenverteilung kann nach einem vereinfachten Ausdruck berechnet werden: (a + b)n
a ist die relative Häufigkeit des leichten Isotops b ist die relative Häufigkeit des schweren Isotops n ist die Anzahl der Atome des betrachteten Elements im Molekül
Die Verteilung ergibt sich dabei aus den Summanden. Bei Si und S können Überlagerungen mit dem 13C‐ oder anderen Mustern leicht zum Verwischen führen.
Aufgabe 1: Berechnen Sie mit Hilfe der Gleichung (a + b)n das Isotopenmuster von Cl4.
3.2 Fragmentierungsreaktionen Ein Massenspektrum ist das Ergebnis einer Reihe von Zerfallsreaktionen eines Ions. In der nachfolgenden Aufgabe werden wichtige Fragmentierungs‐ und Umlagerungsreaktionen genannt, die nach der Elektronenstoß‐ Ionisation auftreten. Bitte erläutern Sie diese in 1‐2 Sätzen und anhand des angegebenen Substanzbeispiels.
Aufgabe 2: Versuchen Sie die unten aufgeführten Fragmentierungsreaktionen für die in Klammern angegebenen Verbindungen nachzuvollziehen und geben Sie möglichst alle mesomeren Grenzstrukturen der entstehenden Produkte an (stabilstes Produkt): Radikal induzierte Spaltungen
- (Alkyl‐) �−Spaltung (1‐methylethyl‐Cyclohexan) - α‐Spaltung aktivierter Bindungen:
Heteroatom (Cyclohexylethylketon)

Allylspaltung (4‐Methyl‐2‐hepten) Benzylspaltung (2‐Benzyl‐propan)
- McLafferty‐Umlagerung, McL (Pentansäuremethylester) - Retro‐Diels‐Alder‐Reaktion, RDA (2,2,2‐Bicycloocten‐2)
Ladungsinduzierte Spaltungen - Induktive Spaltung (1‐Chlorhexan) - Eliminierung, H‐Umlagerung (6‐Hydroxy‐2‐hexanon) - Onium‐Reaktion (Butyldiethylamin) - Neutralverlust, keine H‐Umlagerung (2,5‐Cyclohexadien‐1,4‐dion)
Aufgabe 3: Welche Fragmentierungen sind für dieses Molekül möglich? Bitte geben Sie ausführliche Reaktionsgleichungen an und benennen die entsprechende Fragmentierung.
OHO
O
Cl
3.3 Herangehensweise an die Spektreninterpretation Im Nachfolgenden wird die allgemeine Herangehensweise bei der Interpretation eines Massenspektrums skizziert. Bei der Spektreninterpretation wird bereits vorhandenes Wissen über den Analyten immer mit einbezogen.
1. Charakterisieren Sie den Molekülionenpeak. Welche Heteroatome vermuten Sie bzw. können Sie ausschließen (Isotopenmuster!)?
2. Erstellen Sie einen oder mehrere Strukturvorschläge durch Interpretation großer und charakteristischer Schlüsselbruchstücke bzw. Massendifferenzen, besonders für den Basispeak. Fertigen Sie Modellfragmentierungen von den Strukturvorschlägen an, vergleichen Sie mit dem Spektrum, und achten Sie besonders auf „fehlende“ Ionen.
Regeln für die EI‐ Spektreninterpretation
Regel 1: Zwischen Molekülion und Fragmentionen müssen als Folge der Fragmentierung des Molekülions chemisch sinnvolle Massendifferenzen bestehen.
Regel 2: Einmal gebildete closed‐shell‐ Ionen A+ oder B+ gehen keine erneute Radikalspaltung mehr ein, sondern zeigen nur noch Neutralabspaltungen. (even electron rule)
Regel 3: Bei konkurrierenden Homolysen bestimmt meist die Produktstabilität den bevor‐zugten Reaktionsweg.
Regel 4: Enthält ein Molekül 1, 3, 5, ... Stickstoffatome, ist seine Molmasse nicht geradzahlig. Enthält ein Molekül 0, 2, 4,... Stickstoffatome, ist seine Molmasse geradzahlig (Stickstoffregel).
Regel 5: Homolysen (Radikalabspaltungen) führen zur Bildung von Primärfragmenten mit nicht geradzahliger Massendifferenz zum Ausgangsion. Verluste intakter Moleküle (Umlagerungen, Neutralabspaltungen) führen zu geradzahligen Massendifferenzen zum Molekülion. Zusammen mit einer ungeradzahligen Anzahl Stickstoffatome im

Molekül kann eine Umkehrung dieser Regel eintreten (durch H2NR. bzw. NH3‐ Verluste).
Regel 6: Man berechnet Doppelbindungs‐ und Ringäquivalente, sobald man die Summen‐formel einer Verbindung zu kennen glaubt. Diskrepanzen deuten auf Fehler in der Summenformel oder auf Elemente mit anderer Oxidationsstufe als angenommen. Die Berechnung der Doppelbindungsäquivalente erfolgt für eine Substanz der allgemeinen Summenformel CmHnOqNrHals mit Hilfe der folgenden Formel:
F = (2m + 2 − (n − r + s)) /2 Beispiel C13H20O: (26 +2 ‐20)/2 = 4
Regel 7: Eine korrekte Zuordnung charakteristischer Ionen im Spektrum ist zur Absicherung des Strukturvorschlags und der Summenformel unerlässlich.
Aufgabe 4 und 5: Es sind relative Intensitäten aus dem Peaklisting zweier EI‐Massenspektren gezeigt. Welches Molekül könnte sich hinter dem Spektrum verbergen? Erklären und zeigen Sie das Zustandekommen der charakteristischen Fragmente (mind. 2 davon anhand einer Abb., Tabelle reicht nicht)!
m/z rel. Int. [%] m/z rel. Int. [%] 51 12 73 10052 0.5 74 8.477 45.3 75 3.478 2.7 131 2479 0.2 132 3.77112 100 133 1.63113 6.9 146 11.4114 33 147 1.92 148 0.78
4 Versuchsbeschreibung Zunächst wird in einem Antestat (50 % der Praktikumsnote) Ihr Wissen über die Grundlagen der Massenspektrometrie überprüft. Die im Skript gestellten Aufgaben sollten zur Vorbereitung des Antestats gelöst werden. Sie sind Bestandteil des Protokolls und können bei Problemen im Anschluss an das Antestat besprochen werden. Im zweiten Teil werden EI‐Massenspektren einer bekannten sowie einer unbekannten Verbindung an einem Sektorfeldgerät mit EI‐Ionisation (MAT 8230) unter Anleitung aufgenommen. Notieren Sie sich alle relevanten experimentellen Parameter für Ihre Dokumentation.
Experimenteller Teil Probenvorbereitung Probe in den Probentiegel einfüllen und den Tiegel auf den Schubstangenkopf aufstecken.
Vorbereitung der Messung: - Anschalten der Schubstangenkühlung, Ablesen der Ionenquellentemperatur - Anwählen der Ionisierungstechnik, Anschalten der Kathode, Emission auf 0,5 mA
einstellen - Hochspannung und SEV anschalten - Öffnen der Software zur Datenaufnahme und –auswertung (MASPECII)

- Vorbereitung der Datenaufnahme ‐ Erstellen eines Mess‐Files (Eintragen aller relevanten Parameter): Methode: Low‐resolution Magnet Scan, Centroid (d.h. Strichspektren), Auflösung: 1000, Scanrate: 5 sec/decade für EI‐ Schubstangenbetrieb, Massebereich (Hochspannung) 2100 amu (3 kV), Interscan delay: 1 sec für EI‐Schubstangenbetrieb
- File: Description: für Ihre Kommentare - Destination File: File‐ Namen vergeben, unter dem Ihre Messung abgespeichert wird - Scan: geeigneten Massenbereich eintragen - Cal: geeigneten Kalibrierfile unterlegen (wird Ihnen mitgeteilt)
Durchführung der Messung: - „Start Acquisition“ - Ansetzen der Schubstange - Öffnen des Vorvakuumventils, Evakuieren der Schleuse - Schließen des VV‐ Ventils, Öffnen des Hochvakuumventils - Einführen der Schubstange - Beobachtung der Peakintensitäten ‐ gegebenenfalls manuelles Heizen der
Schubstange unter Kontrolle der Signalintensitäten (zwischen 1 und 10 V) - Herausziehen der Schubstange, Schließen des HV‐Ventils - „Stop“ und „Close Acquisition“ - Entfernen der Schubstange, Entfernung des Probentiegels
Hinweise zur Anfertigung des Versuchsprotokolls Erstellen Sie bitte in jeder Gruppe ein Protokoll über den durchgeführten Versuch. Beginnen Sie mit einer kurzen Darstellung der Aufgabe, keine theoretische Einleitung. Achten Sie auf eine systematische und übersichtliche Formatierung (Inhaltsverzeichnis, Legenden, Überschriftenhierarchie).
- Lösen Sie schriftlich die im Skript gestellten und im Antestat besprochenen Aufgaben. - Interpretieren Sie ausführlich die Spektren, die von der bekannten Verbindung
aufgenommen wurden (Sie erhalten von jedem erzeugten Spektrum einen Ausdruck). Orientieren Sie sich an Punkt 3.3. „Herangehensweise an die Spektreninterpretation“. Ordnen Sie prägnante Fragmentionen anhand von Zerfallsgleichungen entsprechenden Strukturen zu. Für das Zeichnen chemischer Strukturen können Sie z.B. die im Netz frei herunterladbaren Programme Accelrys Draw, Marvin Sketch, ACD/ChemSketch, oder BKChem benutzen.
- Diskutieren Sie ebenso ausführlich die Massenspektren Ihrer Verbindung. Nutzen Sie dazu den Strukturvorschlag, den Sie sich aus allen Analysen abgeleitet haben. Waren die mit der Massenspektrometrie erhaltenen Informationen zur Strukturaufklärung ausreichend oder benötigten Sie weitere Methoden, und wenn ja, welche?


















