MUSKEL- DYSTROPHIE DUCHENNE (DMD) · verleiht der Muskelfaser und der Membran während der...
8
MUSKEL- DYSTROPHIE DUCHENNE (DMD) Was Sie wissen müssen
Transcript of MUSKEL- DYSTROPHIE DUCHENNE (DMD) · verleiht der Muskelfaser und der Membran während der...

MUSKEL- DYSTROPHIEDUCHENNE (DMD)
Was Sie wissen müssen

WAS IST DUCHENNE-MUSKELDYSTROPHIE (DMD)?
DMD ist eine schwere und lebensbedrohende genetische Erkrankung. Sie betrifft vorwiegend Jungen (X-chromosomale Vererbung) und ist selten: 1 von 3.600 bis 6.000 lebendgeborenen Jungen erkrankt.1–4 Kinder mit DMD verlieren fortschreitend Muskelgewebe. Dabei tritt in der frühen Kindheit, beginnend im Alter von 2 bis 3 Jahre, eine Muskelschwäche in Erscheinung.1–5
DMD wird durch Mutationen des Gens verursacht, das Dystrophin kodiert. Dystrophin ist ein wichtiges Protein, das zur Stabilität und Struktur von Muskelfasern beiträgt.1,6 Mutationen im Dystrophin-Gen bewirken einen Mangel oder eine Schädigung des Dystrophin-Proteins.1,2 In der Folge wird nach und nach Muskel durch Fett- und Bindegewebe ersetzt.6,7
Der Muskelverlust im Rahmen der DMD führt dazu, dass die Kinder Meilensteine ihrer motorischen Entwicklung, z. B. Gehen, verspätet erreichen. Defizite in der Sprachentwicklung sind ebenfalls gut dokumentiert.2 Dem natürlichen Krankheitsverlauf entsprechend verlieren Kinder mit DMD ihre Gehfähigkeit und werden im frühen Teenager-Alter rollstuhlpflichtig.1,2 Letzten Endes führt der Verlust an Muskelgewebe zu respiratorischen, orthopädischen und kardialen Komplikationen. Häufig tritt der Tod im dritten Lebensjahrzehnt ein.1,4
Funktion, Lebensqualität, Gesundheit und Lebenserwartung können durch eine Behandlung mit Kortikosteroiden sowie respiratorische, kardiale, orthopädische Interventionen und Rehabilitationsmaßnahmen verbessert werden.2,8 Damit kann es möglich sein, die Therapieergebnisse zu verbessern und das Überleben bis in das vierte Lebensjahrzehnt zu verlängern.2,4
Durch Identifizierung der frühen Zeichen der Muskelschwäche sowie anderer Frühindikatoren und dann rasche Überweisung der Patienten an Spezialisten zur weiteren Diagnose und Behandlung kann dazu beigetragen werden, dass Jungen und junge Männer mit DMD ihre Chancen optimieren.4

DEMOGRAPHIE UND EPIDEMIOLOGIE DER DMD
• DMD betrifft vor allem Jungen, obwohl rund 10 % der weiblichen Überträgerinnen von Dystrophin-Mutationen Symptome der Erkrankung aufweisen2
• Das mittlere Alter bei Diagnosestellung liegt üblicherweise zwischen 4,1 und 4,5 Jahren. Die Erkrankung wird Schätzungen zufolge erst 19,2 bis 30 Monate nach Auftreten der ersten Symptome diagnostiziert4
• In 30 % der Fälle gibt es keine familiäre Veranlagung9
• Die Inzidenz liegt schätzungsweise zwischen 1 : 3.600 bis 1 : 6.000 männlicher Lebendgeborener2
PRÄVALENZ DER DMD IN DEN USA: CA. 15,00010 PRÄVALENZ DER DMD IN DER EU: CA. 12,500–31,00011,12

Bei den weitaus meisten Patienten mit DMD mangelt es an Dystrophin-Protein.13 Dieses Fehlen funktionalen Dystrophins bewirkt eine Degeneration von Muskelzellen. Bindegewebe und Fett ersetzen dann die verlorengegangenen Muskelfasern.6
MAGNETRESONANZ-BILDER DES MUSKELS BEI PROGREDIENTER DMD
DYSTROPHIN IM GESUNDEN KÖRPER UND BEI KRANKHEIT
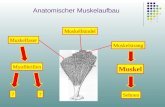
Dystrophin spielt eine Rolle bei der Vernetzung des internen Zytoskeletts mit der extrazellulären Matrix von Muskelzellen.13 Es verleiht der Muskelfaser und der Membran während der Kontraktion Stabilität und Struktur.6,14 In anderen Worten wirkt Dystrophin als molekularer Stoßdämpfer, indem es Muskelzellen erlaubt, nach einem Stressereignis zu ihrem ursprünglichen Zustand zurückzukehren.14
DER DYSTROPHIN-ASSOZIIERTE PROTEINKOMPLEX IM MUSKEL
Nach Sweeney HL.15Modifiziert nach Nowak KJ und Davies KE.13 Nur zur Illustration
6–7 JAHRE5 JAHRE 8–9 JAHRE
12–13 JAHRE10–11 JAHRE 14 JAHRE
Sarkolemm DystroglykanBiglykan Sarkospan
Caveolin-3
Aktin Zytoskelett
N-Terminus
C-TerminusDystrophinSarkoglykane
Syntropine
Dystobrevin
SyncoilinDysbindin
Desmin
Extrazelluläre Matrix Kollagen Laminin-2extrazellular
intrazellular
Fett- und BindegewebeMuskelgewebe

MUTATIONEN BEI DMD
Das Dystrophin-Gen ist das längste Gen des humanen Genoms. Es umfasst 2,6 Millionen DNA-Basenpaare und enthält 79 Exons.13
Bisher wurden mehr als 7.000 unterschiedliche Mutationen identifiziert. Sie fallen, grob gesagt, in zwei Kategorien: große Mutationen (umfassen 1 Exon oder mehr), die für die Mehrzahl der Mutationen verantwortlich sind, und kleine Mutationen wie z. B. Nonsense-Mutationen (umfassen weniger als 1 Exon).16
~65% ~10%
~10–15%
~10%
Große Mutationen Deletionen Duplikationen
Kleine Mutationen Nonsense-Mutationen Andere
Modifiziert nach: Bladen CL, et al.,16 Pichavant C, et al.17 and Kalman L, et al.18
Mit der Methode, mit der große Mutationen detektiert werden (Multiplex-ligationsabhängige Sondenamplifizierung), können keine kleinen Mutationen, wie z. B. Nonsense-Mutationen, detektiert werden. Dazu ist eine Gen-Sequenzierung erforderlich.18
Deletion, Duplikation und Nonsense-Mutationen können zu DMD führen.16–18 Deletionen und Duplikationen können ein einzelnes Codon oder auch ein vollständiges Exon oder mehr betreffen.16,17 Nonsense-Mutationen betreffen ein einzelnes Codon.16,17
WILDTYP UUA CAU GGU GUG AAG
Normale Sequenz ohne Mutation
DELETION UUA GUG AAG Ein Teil der Gensequenz fehlt
DUPLIKATION UUA CAU GGU GUG CAU GGU GUG AAG
Ein Teil der Gensequenz ist wiederholt vorhanden
NONSENSE UAA CAU GGU GUG AAG
Das veränderte Codon ist ein Stopp-Codon, das vorzeitig der Zelle signalisiert, die Bildung von Protein zu stoppen
Modifiziert nach: Genetics Home Reference.19

TYPISCHER NATÜRLICHER VERLAUF DER DMD*
Auswirkungen auf den Alltag2,20,24
• Braucht länger, um Alltagsaktivitäten wie Anziehen, einen Raum queren oder in die Schule gehen zu absolvieren
• Aktivitäten wie Spielen, Schwimmen oder Radfahren dürfen nicht zu lange dauern und müssen durch Ruhephasen ausgeglichen werden, da eine Ermüdung schneller eintritt
• Schwierigkeiten bei der Teilnahme an sozialen Aktivitäten mit anderen Kindern und Familien führt zu sozialer Isolation
• Verlust der Unabhängigkeit; Betroffener ist zunehmend auf Hilfe durch Pflegepersonen angewiesen. Benötigt Unterstützung beim Waschen, Anziehen, Gehen, Essen und bei der Körperpflege
• Benötigt Betreuung rund um die Uhr
BIS ZUM 2. LEBENSJAHR
• Ab Geburt: stark erhöhte Kreatinkinase• Leicht verzögerte motorische Meilensteine, evtl.
einschließlich Gehfähigkeit• Verzögerte Sprachentwicklung
AB DEM 3.–4. LEBENSJAHR
• Schwierigkeiten beim Springen, Rennen und Treppensteigen• Reduzierte körperliche Ausdauer (gegenüber Gleichaltrigen)• Gowers-Zeichen†
• Wadenhypertrophie‡
AB DEM 5.–8. LEBENSJAHR
• Plateauphase der motorischen Entwicklung• Abnorme Bewegungen: betonter Armschwung, breit
watschelnder Gang, Zehenspitzengang
VOR DEM 13. LEBENSJAHR • Verlust der eigenständigen Gehfähigkeit
NACH DEM VERLUST DER
GEHFÄHIGKEIT
• Muskelschwäche (rumpfnahe Muskulatur, Arme, Hände)• Neuromuskuläre Skoliose• Schwäche der Atemmuskulatur• Kardiomyopathie mit/ohne Arrhythmie• Verschlechterung des Sprechens und Schluckens
Modifiziert nach: Annexstad et al.20 *Beachten Sie, dass es große Unterschiede im Hinblick auf die Geschwindigkeit der Krankheitsprogression gibt. †Zwischen dem 3. und 4. Lebensjahr können Kinder das klassische Gowers-Zeichen zeigen. Hierbei können Betroffene nur durch Abstützen der Hände an Boden bzw. Oberschenkeln in den Stand gelangen.20 Ursache ist eine Schwäche der rumpfnahen Muskeln.21 ‡Pseudohypertrophie, bei der die Wadenmuskulatur aufgrund von Fett- und Bindegewebsablagerungen an Umfang zunimmt.

Der Verlust der Gehfähigkeit ist ein wesentlicher Prädiktor (Vorhersagefaktor) der natürlichen Krankheits-progression, einschließlich der schweren Ateminsuffizienz und dem Tod aufgrund von respiratorischem Ver-sagen.22 Daraus folgt, dass eine Verlangsamung der Verschlechterung der körperlichen Funktion noch geh fähiger Kinder die Krankheitsprogression verzögern kann.23 Dies bedeutet, dass das Kind so lange wie möglich körperlich aktiv und unabhängig bleiben kann, sowie länger ohne Rollstuhl und ohne Atemunterstützung leben kann.
KORRELATION ZWISCHEN ALTER BEI VERLUST DER GEHFÄHIGKEIT UND RESPIRATORISCHER INSUFFIZIENZ
VERLUST DER GEHFÄHIGKEIT: EIN SCHLÜSSELEREIGNIS IM NATÜRLICHEN VERLAUF DER DMD
Über
leben
sver
teilu
ngsfu
nktio
n
Alter zum Zeitpunkt der respiratorischen Insuffizienz (vorhergesagte FVC <30 %; Jahre)
1.00
0.75
0.50
0.25
0.00
0 10 20 30 40 50
Verlust der Gehfähigkeit <8 Jahre (n=43) Verlust der Gehfähigkeit 8–11 Jahre (n=140) Verlust der Gehfähigkeit 11–16 Jahre (n=86)
p<0,0001 zwischen allen 3 Gruppen
Modifiziert nach: Humbertclaude et al.22 FVC, forced vital capacity.

DMD: WICHTIGE PUNKTE
• DMD ist eine selten, X-chromosomal vererbte, progrediente, lebensbedrohende, muskelabbauende Erkrankung1–4
• Sie wird durch Mutationen des Gens verursacht, das für Dystrophin codiert 1
• Kinder mit DMD zeigen einen progredienten Verlust von Muskelgewebe, das durch Fett- und Bindegewebe ersetzt wird7
• Die Erkrankung führt letzten Endes zum Tod. Dennoch können Krankheitsverlauf und Lebenserwartung durch eine Therapie mit Kortikosteroiden sowie durch respiratorische, kardiale, orthopädische und rehabilitative Maßnahmen verbessert werden2,4
• Frühzeitige Erkennung ist wichtig, um dem Patienten eine spezialisierte Betreuung zu ermöglichen4
Referenzen: 1. Goemans N, et al. Eur Neurol Rev. 2014;9:78–82. 2. Bushby K, et al. Lancet Neurol. 2010;9:77–93. 3. McDonald CM, et al. Muscle Nerve. 2013;48:343–356. 4. Van Ruiten HJ, et al. Arch Dis Child. 2014;99:1074–1077. 5. Ciafoloni E, et al. J Pediatr. 2009;155:380-385. 6. Amato AA and Brown RH Jr. Muscular Dystrophies and other muscle diseases. In: Longo DL, Fauci AS, Kaspar DL, et al., eds., Harrison’s Principles of Internal Medicine, 19th Ed. Access Medicine McGraw-Hill. http://accessmedicine.mhmedical.com/book.aspx?bookId=331. 7. Blake DJ, et al. Physiol Rev. 2002; 82:291–329. 8. Matthews E, et al. Cochrane Database Syst Rev. 2016;5:CD003725. 9. Garcia S, et al. Int J Med Sci. 2014;11:988–993. 10. McDonald CM, et al. Muscle Nerve. 2013;48:357–368. 11. Theadom A, et al. Neuroepidemiology. 2014;43:259-268. 12. Worldometers. Population of Europe; 2016. Accessed at: http://www.worldometers.info/world-population/europe-population/. 13. Nowak KJ and Davies KE. EMBO
Rep. 2004;5:872–876. 14. Ervasti JM. Biochim Biophys Acta. 2007;1772:108–117. 15. Sweeney HL. Developing skeletal muscle MRI/MRS as a biomarker for DMD therapeutic development. 2014 Annual Connect Conference, Chicago, IL. Accessed at: http://www.parentprojectmd.org/site/DocServer/Session_8_-_Sweeney_b.pdf?docID=15384. 16. Bladen CL, et al. Hum Mutat. 2015;36:395–402. 17. Pichavant C, et al. Mol Ther. 2011;19:830–840. 18. Kalman L, et al. J Mol Diagn. 2011;13:167–174. 19. National Institutes of Health/Genetics Home Reference: What kinds of gene mutations are possible? Updated September 2016. Accessed at: https://ghr.nlm.nih.gov/primer/mutationsanddisorders/possiblemutations. 20. Annexstad EJ, et al. Tidsskr Nor Laegeforen. 2014;134:1361–1364. 21. Mah JK. Neuropsychiatr Dis Treat. 2016;12 1795–1807. 22. Humbertclaude V, et al. Eur J Paediatr Neurol. 2012;16:149–160. 23. McDonald CM, et al. Muscle Nerve. 2013;48:32-54. 24. A guide for parents: Duchenne muscular dystrophy. Accessed at www.musculardystrophyuk.org.
Dieses Dokument wurde von PTC Therapeutics als medizinischer Service erstellt.
PTC
1704
CW
015