
Neuroendokrine Tumoren -...
52
14 Neuroendokrine Tumoren Oliver Blankenstein, Marianne Pavel 14.1 Endokrine Tumoren – 894 14.2 Medulläres Schilddrüsenkarzinom – 896 14.3 Phäochromozytom – 896 14.4 NET im Kindes und Jugendalter – 897 14.5 Fallbeispiele – 900 14.6 Literatur – 944
-
Upload
nguyendien -
Category
Documents
-
view
236 -
download
0
Transcript of Neuroendokrine Tumoren -...

14
Neuroendokrine TumorenOliver Blankenstein, Marianne Pavel
14.1 Endokrine Tumoren – 894
14.2 Medulläres Schilddrüsenkarzinom – 896
14.3 Phäochromozytom – 896
14.4 NET im Kindes und Jugendalter – 897
14.5 Fallbeispiele – 900
14.6 Literatur – 944

14
894 Kapitel 14 · Neuroendokrine Tumoren
14.1 Endokrine Tumoren
Epidemiologie Endokrine Tumoren im engeren Sinn umfassen nach Definition der WHO (2000) Tumoren des diffusen neuroendokrinen Zellsystems einschließlich endokriner Pankreastumoren, im weiteren Sinn auch Phäochromozytome, Paragangliome und das medulläre Schilddrüsenkarzinom.
Neuroendokrine Tumoren (NET) des diffusen neuroendokrinen Systems treten zu 75 % im gastroenteropankreatischen System (GEPNET) auf, zu 20 % im bronchopulmonalen System und sehr selten an anderen Lokalisationen wie Thymus, Ovar oder Haut.
Die Inzidenz der NET ist steigend und wird nach den umfangreichsten epidemiologischen Daten (Surveillance Epidemiology and End Results [SEER]Databank der USA) mit 2,50–5,25/100 000 Einwohner angegeben [1], [2]. Insbesondere nimmt die Häufigkeit von NET der Lunge, des Intestinums und Rektums zu.
Klinische Präsentation Klinisch werden funktionelle von nichtfunktionellen NET unterschieden. Funktionell aktive Tumoren, die ein Drittel aller NET ausmachen, führen zu verschiedenen klinischen Syndromen in Abhängigkeit von Primärtumorsitz und Sekretionsprodukt. So führen in der Regel die im Dünndarm angesiedelten NET infolge der Sekretion von Serotonin zum Karzinoidsyndrom, einem Symptomenkomplex, der durch anfallartige Flushanfälle, Diarrhöen und gelegentlich durch asthmatische Beschwerden geprägt ist. Ein Syndrom tritt i. Allg. nur auf, wenn der Tumor metastasiert ist. Die Leber ist Hauptmetastasierungsort. Der Primärtumor im Dünndarm kann oft klein sein (< 1 cm) bei nicht selten größeren Lymphknotenmetastasen. Charakteristisch ist eine Lebermetastasierung in über 80 % der Fälle bei Erstdiagnose. Manipulationen am
Tumor (z. B. Embolisation, Operation) können eine Karzinoidkrise auslösen.
Endokrine Pankreastumoren dagegen metastasieren in Abhängigkeit von der Primärtumorgröße. Auch ohne eine Metastasierung liegt bei Hormonhypersekretion ein klinisches Syndrom vor (. Tab. 14.1). Die häufigsten endokrin aktiven Tumoren sind das Insulinom und das Gastrinom. VIPome sind selten; Glukagonome und Somatostatinome sind sehr seltene Tumoren. Die Mehrheit der endokrinen Pankreastumoren ist nichtfunktionell und wird durch unspezifische Beschwerden oder zufällig anhand konventioneller Bildgebung detektiert. Endokrine Pankreastumoren (gehäuft Gastrinome) treten in bis zu 20–30 % der Fälle im Rahmen eines hereditären Syndroms (Multiple Endokrine Neoplasie Typ 1 = MEN 1) in Assoziation mit Hypophysentumor und Nebenschilddrüsenhyperplasie auf.
Klassifikation Histopathologisch werden hochdifferenzierte neuroendokrine Tumoren bzw. neuroendokrine Karzinome von schlecht differenzierten neuroendokrinen Karzinomen unterschieden (WHOKlassifikation von 2000). Bei der Mehrzahl der NET handelt es sich um hochdifferenzierte Tumoren bzw. Karzinome (90 %), die oft einen langsam progredienten Verlauf aufweisen; schlecht differenzierte NET verhalten sich dagegen hochmaligne.
Neuerdings wird auch die TNMKlassifikation verwendet [3]. Diese ist mit einem GradingSystem verbunden, basierend auf der Proliferationsrate der NET, und erlaubt eine prognostische Einschätzung bei Erstdiagnose.
Die Histologie ist entscheidend für die Diagnosesicherung und die weitere Auswahl der Diagnostik und Therapie. Neben einem typischen lichtmikroskopischen Bild sind der immunhistochemische Nachweis von Chromo
. Tab. 14.1 Klinische Präsentation und biologisches Verhalten von NE
Tumor/Syndrom Symptom Sekretionsprodukt Malignität
Nichtfunktionell Unspezifisch Chromogranin A, PP, a/ß-HCG, Kalzitonin 60–80 %
Karzinoidsyndrom Flush, Diarrhö, Bronchialobstruktion Serotonin 100 %
Insulinom Hypoglykämie Insulin, Proinsulin 5–10 %
Gastrinom/ZES Ulzera, Diarrhö Gastrin 60–80 %
VIPom Wässrige Diarrhö, Hypokalämie Vasoaktives intestinales Peptid 40–75 %
Glukagonom Nekrolytisches migratorisches Erythem, Diabetes mellitus
Glukagon 50–80 %
Somatostatinom Diabetes mellitus, Steatorrhö, Cholelithiasis Somatostatin 50 %
GHRHomCRHom, ACTHom
AkromegalieCushing-Syndrom
GHRHCRH, ACTH
100 %90–100 %

1489514.1 · Endokrine Tumoren
granin und Synaptophysin maßgebend für die Diagnosesicherung und die Bestimmung der Proliferationsrate (Ki67) für die Klassifikation.
14.1.1 Diagnostik
Die biochemische Diagnostik ist gewöhnlich der spezifischen Bildgebung vorangestellt und umfasst Marker der funktionellen Aktivität und allgemeine Tumormarker.
Zur Sicherung des Karzinoidsyndroms dient die Bestimmung der 5Hydroxyindolessigsäure (5HIES) im 24hUrin. Dagegen erfolgt bei endokrin aktiven Tumoren mit Ursprung im Pankreas oder Duodenum die Bestimmung des Peptids im Plasma, beispielsweise Gastrin oder vasoaktives intestinales Peptid (VIP). Die Diagnose des Insulinoms wird in der Regel im 1 bis 3tägigen Fastentest mit inappropriater Erhöhung von Insulin bzw. Proinsulin (bzw. fehlender Supprimierbarkeit) bei gleichzeitigem Nachweis einer Hypoglykämie gestellt.
Chromogranin A ist ein Tumormarker mit hoher Sensitivität (~ 90 %), wobei zahlreiche andere Ursachen, die eine Erhöhung dieses Markers bewirken können, Berücksichtigung finden sollten; in erster Linie die Einnahme von Protonenpumpeninhibitoren (PPI) oder die chronisch atrophische Gastritis [4].
Hochdifferenzierte NET weisen in der Regel einen hohen Besatz an Somatostatinrezeptorsubtypen auf, dies ist bei schlecht differenzierten NET selten der Fall. Dementsprechend sind erstere mit der Somatostatinrezeptorszintigraphie (Octreoscan) und der 68GaDOTATOCPET/CT visualisierbar. Der Vorzug der PET/CT liegt in der höheren Sensitivität gegenüber dem Octreoscan [5]. Unbekannte Primariuslokalisationen, die der konventionellen Bildgebung oft entgehen, insbesondere bei NET des Dünndarms, können auf diese Weise häufiger visualisiert werden. Die Überlegenheit der PET/CT liegt auch in der häufigeren Detektion von Metastasen (z. B. im Skelettsystem). Die 68GaDOTATOCPET/CT ist vorwiegend das PETVerfahren der Wahl, bei negativem Befund kann die Durchführung einer 18FDOPAPET/CT weiterführend sein. Bei schlecht differenzierten NET kann die FDGPET/CT alternativ zur GanzkörperCT eingesetzt werden, vorzugsweise bei unbekanntem Primarius oder kurativchirurgischem Ansatz.
Bei endokrinen Pankreastumoren, insbesondere wenn diese einen Durchmesser < 1 cm aufweisen bzw. durch konventionelle Bildgebung nicht detektiert wurden, ist die Endosonographie das Verfahren der Wahl bei hoher Sensitivität (~ 95 %). Speziell bei Patienten mit hereditärem Syndrom wie der MEN1Erkrankung sind die Pankreastumoren oft sehr klein (< 5 mm) und entgehen der konventionellen Bildgebung [6].
Gerade bei funktionellen pankreatischen Tumoren wie den Insulinomen und Gastrinomen kommt neben der Endosonographie der PET/CTDiagnostik (mit 68GaDOTATOC oder 18FDOPA) bei unklaren Befunden eine besondere Bedeutung zu, da mit dem Ziel der kurativen Resektion die genaue anatomische Lokalisation wichtig für das operative Vorgehen ist. In 15–25 % bleibt der Primarius unbekannt. Die PET/CT kann hier die Rate unbekannter Primärtumorlokalisationen reduzieren. Das ist von hoher Relevanz, da sich die therapeutische Strategie bei Tumoren des Pankreas bzw. der Lunge von denen des Dünndarms unterscheidet.
In der bildgebenden Verlaufskontrolle kommen vor allem MRT und CT des Abdomens zum Einsatz. Intervalle für Nachsorgeuntersuchungen variieren zwischen 3 und 12 Monaten und sind u. a. durch Differenzierungsgrad und Grading, Wachstumsverhalten, Dauer der Erkrankung, Tumorstadium und Ausmaß der Funktionalität bedingt [7]. Da sich die Erkrankung oftmals auf die Leber bzw. das Abdomen beschränkt, gehört eine Bildgebung des Thorax nicht regelhaft zur Nachsorge.
Bei steigendem Tumormarker, Chromogranin A und/oder spezifischem Mediator ist eine Ausdehnung der Bildgebung indiziert, wobei die PET/CT den Vorzug einer Abbildung aller potenziellen Tumorlokalisationen hat und insbesondere lymphatische, pulmonale und ossäre Metastasen erfasst werden. In größeren Intervallen, alle 2–3 Jahre, dient sie dem Ausschluss einer fortschreitenden Metastasierung, auch wenn der Tumormarker nicht richtungweisend verändert ist.
14.1.2 Therapie
Primäres Ziel ist die kurative Resektion von Primärtumor und/oder Metastasen. Da die Diagnosestellung oft sehr verzögert erfolgt, ist dies in der Mehrheit der Fälle nicht möglich.
Die Therapie nicht kurativ resektabler NET umfasst antisekretorische und antiproliferative Ansätze. Somatostatinanaloga (SSA) und αInterferon (IFN) sind die einzigen zugelassenen Therapieverfahren bei funktionellen Tumoren, insbesondere dem Karzinoidsyndrom. Sie inhibieren die Mediatorausschüttung und sorgen für eine bessere Symptomkontrolle. Für beide Substanzen sind auch antiproliferative Eigenschaften beschrieben. Bei besserer Verträglichkeit wird den SSA oft der Vorzug gegeben [8], [9]. In erster Linie treten Wachstumsstabilisierungen auf, während Tumorrückbildungen selten sind. SSA sind auch das Medikament der Wahl, periinterventionell bzw. perioperativ zur Prävention einer Karzinoidkrise. Lokoregionale Verfahren (Embolisation, Chemoembolisation und andere) können zu einer Reduktion der Tumormasse in der Leber beitragen [10].

14
896 Kapitel 14 · Neuroendokrine Tumoren
Nach Versagen von SSA und/oder IFN kommt bei progredienten Tumoren mit Primarius im Darm eine Somatostatinrezeptorvermittelte Radiorezeptortherapie (PRRT) in Betracht. Diese Therapie kann auch im Verlauf bei Tumoren mit Ursprung im Pankreas gewählt werden. Vorrangig ist hier jedoch bei progredienten NET die systemische Chemotherapie mit Streptozotocin im Einsatz. Neuer dings werden auch molekularzielgerichtete Therapien (Everolimus, Sunitinib) im Rahmen von klinischen Studien evaluiert.
Für die Auswahl der PRRT ist die Bildgebung mit 68GaDOTATOCPET/CT bzw. Octreoscan maßgeblich. Bei entsprechend hohem Uptake in den Tumorläsionen erfolgt eine Dosimetrie, um die Eignung für dieses Verfahren zu prüfen [11].
14.2 Medulläres Schilddrüsenkarzinom
Das medulläre Schilddrüsenkarzinom entsteht aus den parafollikulären Zellen der Schilddrüse und macht 4 % aller Schilddrüsenkarzinome aus. Charakteristisch ist die Synthese und Sekretion von Kalzitonin. Diese ist z. T. mit den klinischen Symptomen der Diarrhö oder des Flush assoziiert. In diesen Fällen liegt dann üblicherweise ein metastasiertes Tumorleiden vor.
Die meisten medullären Schilddrüsenkarzinome entstehen sporadisch; diese machen etwa 80 % aller Fälle aus. Das typische Manifestationsalter ist das 5. bis 6. Lebensjahrzehnt. Familiär gehäuft treten sie im Rahmen eines endokrinen Syndroms, der multiplen endokrinen Neoplasie Typ 2 (MEN 2), vergesellschaftet mit primärer Nebenschilddrüsenhyperplasie und Phäochromozytom und dann in jüngerem Alter auf. Zugrundeliegend ist eine Mutation im RETProtoOnkogen.
Das medulläre Schilddrüsenkarzinom manifestiert sich am häufigsten mit einem isolierten Schilddrüsenknoten und wird zytologisch/histologisch gesichert. Bei den meisten Patienten liegt bereits eine Metastasierung in die Lymphknoten, seltener in andere Lokalisationen zum Zeitpunkt der Diagnose vor, obwohl die Einführung des Kalzitoninscreenings im Serum in Europa zu einer früheren Diagnosestellung beigetragen hat.
Neben Kalzitonin ist CEA ein zirkulierender Tumormarker. Zur bildgebenden Diagnostik gehört die Sonographie der Halsregion zur Abklärung eines zervikalen LKBefalls. Die Leitlinien der American Thyroid Association (2009) empfehlen konventionelle Bildgebung mit CT des Halses und des Thorax sowie abdominell die 3PhasenCT oder KMverstärkte LeberMRT bei erhöhtem Kalzitoninspiegel und/oder lokalem LKBefall.
In einzelnen Fällen wurde ein positiver Befund in der 111InOctreotidSzintigraphie detektiert, auch bei unauffälligem CTBefund. Aufgrund der oftmals kleinen Tumor
absiedlungen könnte die 68GaPET/CT dem Octreoscan überlegen sein. Die Sensitivität der FDGPET zum Nachweis einer metastasierten Erkrankung ist variabel und liegt zwischen 20 und 78 % in Abhängigkeit von der Höhe des Kalzitoninspiegels. Bei der Auswertung der Bildgebung ist zu beachten, dass eine NNRaumforderung im Rahmen eines MENSyndroms vorliegen könnte.
14.3 Phäochromozytom
Das Phäochromozytom ist ein vom Nebenierenmark ausgehender Katecholaminproduzierender Tumor, der überwiegend benigne ist. Etwa 10 % der Phäochromozytome sind maligne. Es handelt sich um einen seltenen Nebennierentumor, der in weniger als 0,5 % der Fälle bei Patienten mit Hypertonus Ursache desselbigen ist.
Dem Phäochromozytom verwandte Tumoren mit extra adrenalem Ursprung in den sympathischen oder parasympathischen Ganglien werden als Paragangliome (ca. 20 %) bezeichnet. Ein Auftreten im Rahmen verschiedener genetischer Syndrome (MEN 2, VonHippelLindauSyndrom [VHL], Neurofibromatose Typ 1, Mutationen im SuccinatDehydrogenase [SDH]Gen [B, C oder D]) ist beschrieben. Mutationen im SDHBGen sind in bis zu 70 % mit malignen Tumoren assoziiert.
Richtungweisend für das Phäochromozytom sind eine typische klinische Symptomatik und der Nachweis eines Nebennierentumors. Zu den klassischen Symptomen gehören Kopfschmerzen (bis 90 %), Schwitzen (60–70 %) und Tachykardie. Charakteristisch ist das anfallartige Auftreten der Symptome (ca. 50 %). Eine Präsentation mit normalem Blutdruck schließt ein Phäochromozytom nicht aus.
Die Diagnose wird zunächst biochemisch gesichert durch den Nachweis von Plasma oder UrinMetanephrinen und Katecholaminen. MRT und CT sind sensitive Methoden (98–100 %), die Spezifität liegt bei 70 %. In der MRT erscheint das Phäochromozytom typischerweise hyperintens im T2gewichteten Bild.
Seit 1981 wird die 123I und 131IMetaiodbenzylguanidin (MIBG)Szintigraphie zur Lokalisationsdiagnostik eingesetzt. Grundlage hierfür ist die Aufnahme des MIBG über den gleichen Transporter wie das Noradrenalin. Die Sensitivität der 123IMIBGSzintigraphie wird mit 83–100 %, die Spezifität mit 95–100 % angegeben. Die Sensitivität des Verfahrens ist jedoch geringer bei extraadrenalen und malignen Prozessen. Da verschiedene Faktoren wie Tumorgröße, Lokalisation, Dedifferenzierung und die Einnahme von Medikamenten, die mit dem MIBGUptake interferieren, zu falschnegativen Befunden führen können, wurden andere diagnostische Verfahren evaluiert.
Die 111InPentetreotideSzintigraphie (Octreoscan), GanzkörperMRT oder PET stellen weitere bildgebende

1489714.4 · NET im Kindes und Jugendalter
Methoden dar. PETScanning mit 18FFluorodeoxyglucose, 11CHydroxyephedrin oder 6[18F]Fluorodopamin können hilfreich für das Auffinden metastatischer Absiedlungen sein und sind z. T. der MIBGSzintigraphie überlegen.
14.4 NET im Kindes und Jugendalter
Neuroendokrine Tumoren treten im Kindesalter nur sehr selten und selbst in großen Kliniken nur vereinzelt auf. In den USA wurde die Inzidenz mit < 1 auf 100 000 Einwohner unter 16 Jahren angegeben [12], [13]. Zu den isolierten oder sporadisch auftretenden neuroendokrinen Tumoren des Kindesalters zählen: Schilddrüsenkarzinome, Nebennierenkarzinome, Phäochromozytome und maligne Karzinoide. Alle anderen neuroendokrinen Tumoren sind als extrem selten anzusehen. Generell sind die Erfahrungen mit der PET/CTDiagnostik auf wenige Einzelfälle oder kleine Serien beschränkt, systematische Studien liegen nicht vor. In der Auswahl der Tracer orientiert man sich daher an den vergleichbaren Tumoren des Erwachsenenalters [14].
z Multiple endokrine Neoplasien (MEN)Die multiplen endokrinen Neoplasien sind familiär auftretende Erkrankungen, bei denen es gehäuft zur Ausbildung von Tumoren der endokrinen Organe kommt, die bei den Betroffenen z. T. bereits im Kindesalter auftreten können [15], [16]. In der Literatur wird die Inzidenz der MENSyndrome mit 1 auf 25 000 angegeben. Da die MENbedingten Tumoren der endokrinen Organe unter den endokrinen Tumoren bei Kindern und Jugendlichen eine relativ häufige Ursache darstellen, sollte bei jeder Erstdiagnostik auch das zu dem jeweiligen Tumor passende MENSyndrom genetisch untersucht werden (. Tab. 14.2).
z Multiple endokrine Neoplasie Typ 1 (MEN 1)Die multiple endokrine Neoplasie vom Typ 1 ist ein seltenes vererbtes Tumorleiden, bei dem Tumoren der Nebenschilddrüse (85–88 %), der Inselzellen des Pankreas (30–70 %) und der Hirnanhangdrüse (15–90 %) auftreten können. In einigen Familien sind auch Karzinoide des Thymus beschrieben. Der Erbgang ist autosomal dominant, Ursache
sind inaktivierende Mutationen in einem TumorSuppressorGen (Menin/MEN1Gen). Die Art der Tumoren sowie der Zeitpunkt des Auftretens variiert in den verschiedenen MEN1Familien, wobei die Tumoren in der Regel erst nach dem 10. Lebensjahr auftreten [17].
Die besondere Problematik der bildgebenden Verfahren bei MEN 1 besteht in der Vielfalt der – insbesondere gastrointestinal – auftretenden Tumoren, die zu einer völlig unterschiedlichen Visualisierung durch die verschiedenen PETTracersubstanzen führen können. Aus den eigenen Erfahrungen heraus hat sich 68GaDOTATOC hier als Tracer mit der höchsten Detektionsrate erwiesen. Dies ist auf den hohen Anteil Somatostatinrezeptorexprimierender Tumoren zurückzuführen. Wie auch bei den insulinproduzierenden Tumoren des Erwachsenenalters erreicht die Detektionsrate bei den endokrin aktiven Tumoren nicht die Größenordnung wie beim kongenitalen Hyperinsulinismus [18]. Unabhängig von der Darstellung verschiedener Tumoren kann man jedoch bei keinem der verwendeten Tracer davon ausgehen, dass tatsächlich alle oder die gesuchten Tumoren dargestellt werden. Dies liegt insbesondere an der Tumorgröße < 1 cm bei der Mehrheit der Läsionen. Überwiegend bei metastasierten Tumoren kann bei erfolgreicher Darstellung der Metastasen von einer Anreicherung des Primärtumors mit der entsprechenden Methodik ausgegangen werden (. Tab. 14.3).
z Kongenitaler HyperinsulinismusDer kongenitale Hyperinsulinismus ist mit einer Inzidenz von 1 auf 30–40 000 Neugeborene eine seltene Erkrankung, gleichzeitig aber die häufigste Ursache für die Entwicklung von bleibenden Hypoglykämien im Neugeborenen und Säuglingsalter [19]–[21] Im Gegensatz zu den neuroendokrinen Tumoren zählt der kongenitale Hyperinsulinismus nicht zu den Tumorerkrankungen, sondern ist eine spezielle Form der Pankreasentwicklungsstörung. Klinisches Hauptmerkmal ist eine unkontrollierte Insulinsekretion der βZelle des endokrinen Pankreas, die zu einer oft direkt nach der Geburt auftretenden Neigung schwerer und schwerster Unterzuckerungen führt. Die besondere Gefahr besteht darin, dass es durch die besondere Anfälligkeit des Gehirns in der Neugeborenenphase zu
. Tab. 14.2 Prävalenz des spezifischen Syndroms bei Vorliegen eines der aufgeführten neuroendokrinen Tumore
Syndrom Gen Neuroendokrine Tumoren Andere Tumoren Spezifische Prävalenz
Multiple endokrine Neoplasie Typ 1 (MEN 1)
MEN 1 Pankreas, Nebenschilddrüse, Hypophyse, Nebenniere, Karzinoide
U. a. Haut (Angiofibrome) Schilddrüse
bis 30 %
Multiple endokrine Neoplasie Typ 2 (MEN 2)
RET Schilddrüse (MTC), Nebenniere (Phäochromo-zytom), Nebenschilddrüse
Bei MEN 2B: Neurome bis 30 %

14
898 Kapitel 14 · Neuroendokrine Tumoren
schweren und bleibenden neurologischen und statomotorischen Störungen kommen kann [22]–[24] Histomorphologisch unterscheidet man zwischen einer diffusen Form und einer fokalen Form (. Abb. 14.1). Während bei der diffusen Form – z. B. durch eine Keimbahnmutation – alle insulinproduzierenden Zellen der Bauchspeicheldrüse betroffen sind, kommt es bei der fokalen Form auf der Basis einer heterozygoten Mutation in der Fetalphase zu einem Verlust des zweiten (gesunden) Allels mit gleichzeitiger Aktivierung von (in der Fetalzeit als Wachstumsfaktor relevantem) IGF2, die zu einem klonalen Wachstum der so betroffenen Zelle führt. Die so betroffenen Zellen sind dann nur in einem mehr oder weniger umschriebenen Abschnitt der Bauchspeicheldrüse zu finden und bilden den sog. Fokus.
Insbesondere für die Therapie hat diese Unterscheidung erhebliche Relevanz, da bei fokalen Formen durch eine chirurgische Entfernung des funktionsgestörten Fo
kus eine komplette Heilung erreicht werden kann, während bei diffusen Formen eine schwierige und langfristige medikamentöse Einstellung des Blutzuckers oder sogar eine subtotale Entfernung des Organs erforderlich sein können, um den Blutzucker zu kontrollieren und bedrohliche Hypoglykämien zu vermeiden [25]–[30].
Die Unterscheidung dieser beiden Formen ist anhand der klinischen Symptome, durch Sonographie, CT oder MRT nicht möglich – auch nicht durch invasivere Methoden wie die Endosonographie oder den intraarteriellen Kalziumstimulationstest. Seit 2003 gibt es erste Erfahrungen mit der 18FDOPAPET zur Lokalisationsdiagnostik bei kongenitalem Hyperinsulinismus. In diesem Patientengut ist es mit dem hochselektiven Tracer erstmalig möglich, die Aktivität der βZellen des Pankreas darzustellen und mit hoher Sicherheit zwischen einer fokalen Form und einer diffusen Form zu unterscheiden [31]–[38].
Durch die Anwendung der hochauflösenden PET/CT können auch Läsionen unter 5 mm Größe sicher erkannt und durch Imageguidedsurgery das intraoperative Auffinden des Fokus beschleunigt sowie das Operationstrauma verringert werden. Die schwierigen Lagebeziehungen erfordern für die intraoperative Lokalisation umfangreiche Erfahrungen und ein eingespieltes Team von Kinderchirurgen, Pädiatrischen Endokrinologen, Nuklearmedizinern und Radiologen. Durch die jetzt bestehende Möglichkeit der PET/CT kann die Therapie gezielt für die jeweilige Form geplant und dadurch zusätzliche Schäden und Risiken vermieden werden. Erste Versuche mit anderen Tracern wie 68GaDOTATOC zeigen, dass zumindest ein Teil der fokalen Läsionen auch damit darstellbar ist. Hierzu liegen bis auf Einzelfälle aber noch keine systematischen Untersuchungen vor.
. Abb. 14.1 Fokale und diffuse Form des CHI
. Tab. 14.3 Nachweis endokrin aktiver Tumoren bei 4 pädiatrischen Patienten mit neuroendokrinen gastrointestinalen Tumoren mit den verschiedenen bildgebenden Verfahren
Stufenschema zur bildgebenden Diagnostik bei MEN 1
P 1 P 2 P 3 P 4
Intra-arterieller Ca-Stimulationstest + +
68Ga-DOTATOC-PET/CT + (+)* - +
18F-DOPA-PET/CT - - - -
Endosonographie - - - -
Nachgewiesene MEN-1-Mutation + + - +

14
900 Kapitel 14 · Neuroendokrine Tumoren
14.5 Fallbeispiele
14.5.1 Patient 1 – Medulläres Schilddrüsen-karzinom
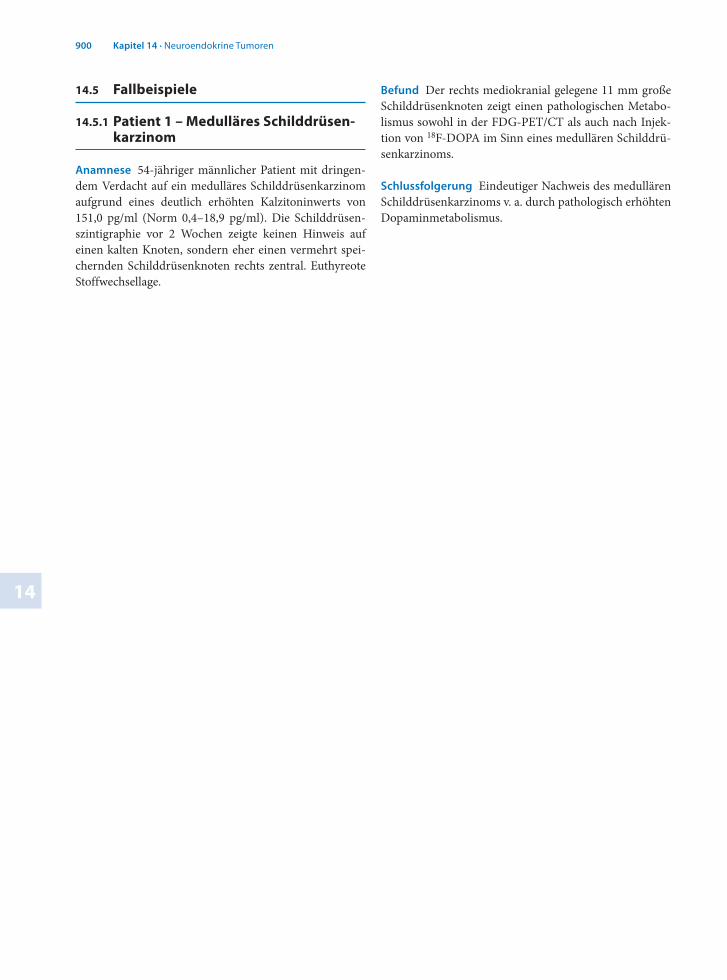
Anamnese 54jähriger männlicher Patient mit dringendem Verdacht auf ein medulläres Schilddrüsenkarzinom aufgrund eines deutlich erhöhten Kalzitoninwerts von 151,0 pg/ml (Norm 0,4–18,9 pg/ml). Die Schilddrüsenszintigraphie vor 2 Wochen zeigte keinen Hinweis auf einen kalten Knoten, sondern eher einen vermehrt speichernden Schilddrüsenknoten rechts zentral. Euthyreote Stoffwechsellage.
Befund Der rechts mediokranial gelegene 11 mm große Schilddrüsenknoten zeigt einen pathologischen Metabolismus sowohl in der FDGPET/CT als auch nach Injektion von 18FDOPA im Sinn eines medullären Schilddrüsenkarzinoms.
Schlussfolgerung Eindeutiger Nachweis des medullären Schilddrüsenkarzinoms v. a. durch pathologisch erhöhten Dopaminmetabolismus.
14.5 · Fallbeispiele14901
. Abb. 14.2 Pathologisch erhöhter Dopaminmetabolismus in einem 11 mm großen Schilddrüsenknoten bei medullärem Schild-drüsenkarzinom, koronaler Schnitt
. Abb. 14.4 Pathologisch erhöhter Dopaminmetabolismus in einem 11 mm großen Schilddrüsenknoten bei medullärem Schild-drüsenkarzinom, Spätaufnahme, transaxialer Schnitt
. Abb. 14.3 Pathologisch erhöhter Dopaminmetabolismus in einem 11 mm großen Schilddrüsenknoten bei medullärem Schild-drüsenkarzinom, Spätaufnahme, koronaler Schnitt

14
902 Kapitel 14 · Neuroendokrine Tumoren
14.5.2 Patient 2 – Medulläres Schilddrüsen-karzinom
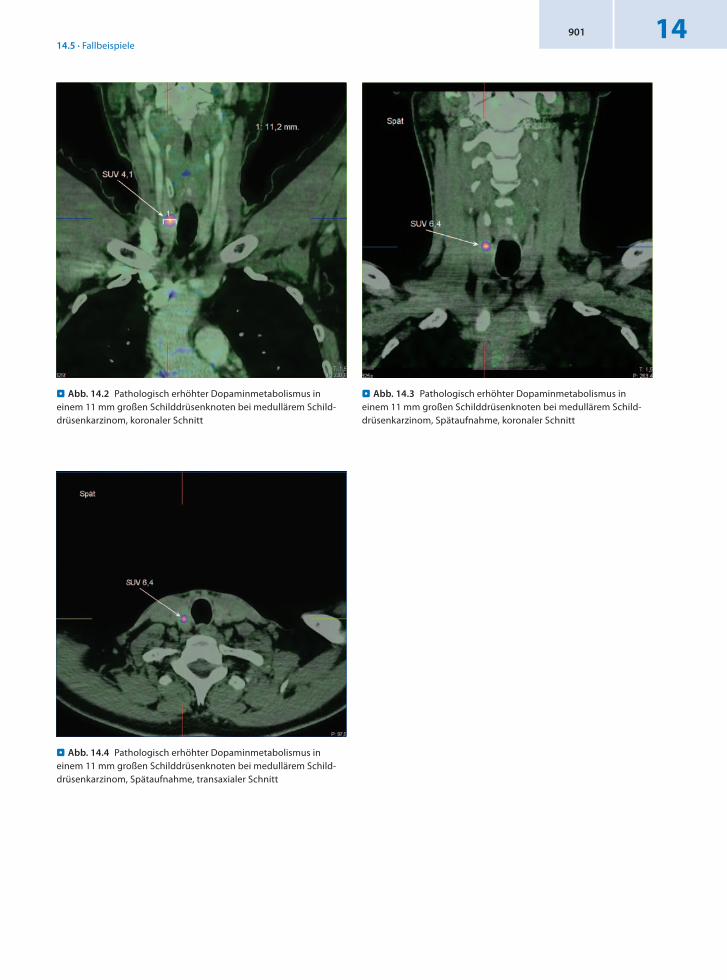
Anamnese 88jähriger männlicher Patient mit bekannter Struma seit 15 Jahren. Zustand nach Radiojodtherapie bei Hyperthyreose vor 5 Jahren. Szintigraphisch kalter Bezirk rechts kaudozentral bei sonographisch knotig umgewandelter Schilddrüse beidseits. Seit 3 Jahren leichte Kalzitoninerhöhung. Im Pentagastrintest vor 2 Monaten war ein deutlicher Anstieg des Kalzitoninwerts auf 288 pg/ml
zu verzeichnen. Somit besteht der Verdacht auf ein medulläres Schilddrüsenkarzinom.
Befund In Verbindung mit dem erhöhten Kalzitoninwert bestätigt sich in der 68GaDOTATOCPET/CT der Verdacht auf ein medulläres Schilddrüsenkarzinom rechts, insbesondere retrosternal.
Schlussfolgerung Nachweis des medullären Schilddrüsenkarzinoms durch 68GaDOTATOCPET/CT.
14.5 · Fallbeispiele14903
. Abb. 14.5 Medulläres Schilddrüsenkarzinom mit retrosternalem Anteil, koronaler Schnitt
. Abb. 14.7 Medulläres Schilddrüsenkarzinom mit retrosternalem Anteil, Spätaufnahme, koronaler Schnitt
. Abb. 14.6 Medulläres Schilddrüsenkarzinom mit retrosternalem Anteil, transaxialer Schnitt

14
904 Kapitel 14 · Neuroendokrine Tumoren
14.5.3 Patient 3 – Rezidiv eines medullären Schilddrüsenkarzinoms mit Lymphknotenmetastasen
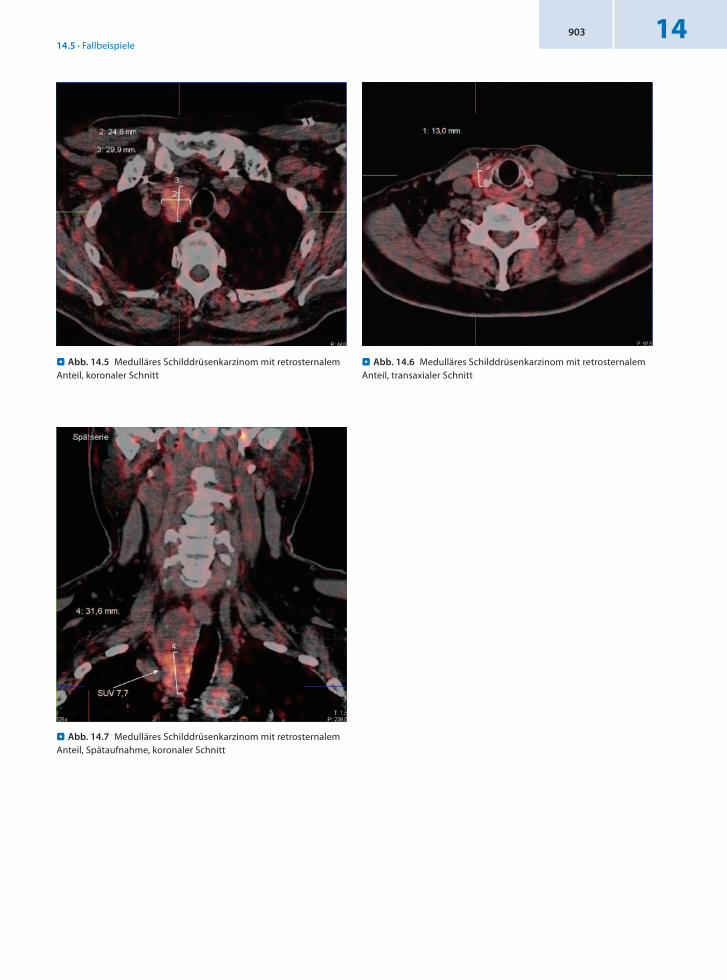
Anamnese 44jährige Patientin mit bekannter Struma, seit 10 Jahren ohne Behandlung. Chirurgische Primärtherapie eines Schilddrüsenknotens mit einer Größe von 20 mm vor 4 Jahren. Histologie: bifokales CZellKarzinom im Rahmen einer MEN 2a mit Mutation im RETProtoonkogen Exon 13, Codon 780. Vor 3 Jahren radikale Lymphknotenresektion. Daraufhin sank der basale Kalzitoninwert von 4 000 auf 115 pg/ml. Im Rahmen der Tu
mornachsorge langsamer Anstieg des Kalzitoninspiegels auf 200–300 pg/ml.
Befund Malignomtypischer Metabolismus in der Schilddrüsenloge links paratracheal im Sinn eines Lokalrezidivs. Darüber hinaus Nachweis von 2 Lymphknotenmetastasen ipsilateral.
Schlussfolgerung Nachweis eines Rezidivs und von 2 LKMetastasen im Rahmen der 18FDOPAGanzkörperPET/CT.
14.5 · Fallbeispiele14905
. Abb. 14.8 Rezidiv eines medullären Schilddrüsenkarzinoms links paratracheal
. Abb. 14.10 Lymphknotenmetastase eines medullären Schild-drüsenkarzinoms links zervikal
. Abb. 14.9 Lymphknotenmetastase eines medullären Schild-drüsenkarzinoms links zervikal submandibulär

14
906 Kapitel 14 · Neuroendokrine Tumoren
14.5.4 Patient 4 – Lymphknotenmetastasen nach OP eines medullären Schilddrüsenkarzinoms
Anamnese 49jährige Patientin mit Erstdiagnose eines sporadischen CZellKarzinoms links vor 3 Jahren. Zustand nach Thyreoidektomie und selektiver Lymphknotendissektion. Vor 2 Jahren wurde eine systematische Mikrodissektion der Lymphknotenkompartimente zervikozentral und zervikolateral beidseits bei Lymphknotenrezidiv durchgeführt. Danach Normalisierung des
Kalzitoninwerts. Seit eineinhalb Jahren unklarer progredienter Kalzitoninanstieg.
Befund In der DOPAPET/CT Nachweis von 2 Lymphknotenmetastasen in Höhe des Kehlkopfs sowie paraösophageal in Höhe von HWK 7.
Schlussfolgerung Klärung des Kalzitoninanstiegs und präzise Lokalisation von 2 LKMetastasen durch 18FDOPAPET/CT.
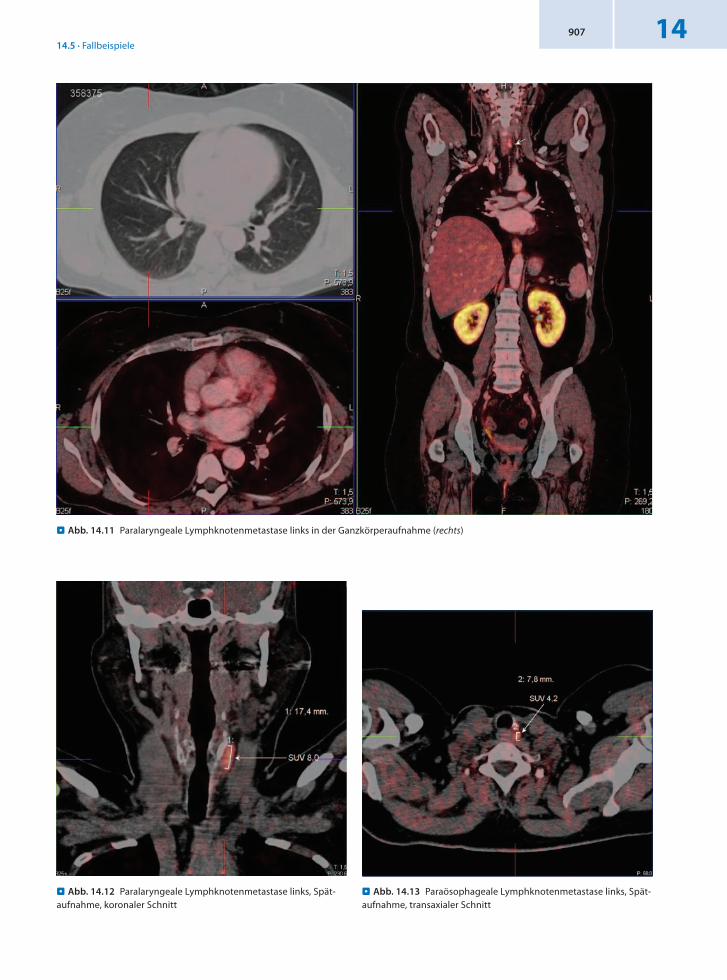
14.5 · Fallbeispiele14907
. Abb. 14.11 Paralaryngeale Lymphknotenmetastase links in der Ganzkörperaufnahme (rechts)
. Abb. 14.13 Paraösophageale Lymphknotenmetastase links, Spät-aufnahme, transaxialer Schnitt
. Abb. 14.12 Paralaryngeale Lymphknotenmetastase links, Spät-aufnahme, koronaler Schnitt

14
908 Kapitel 14 · Neuroendokrine Tumoren
14.5.5 Patient 5 – Lymphknotenmetastasen bei Zustand nach medullärem Schilddrüsenkarzinom
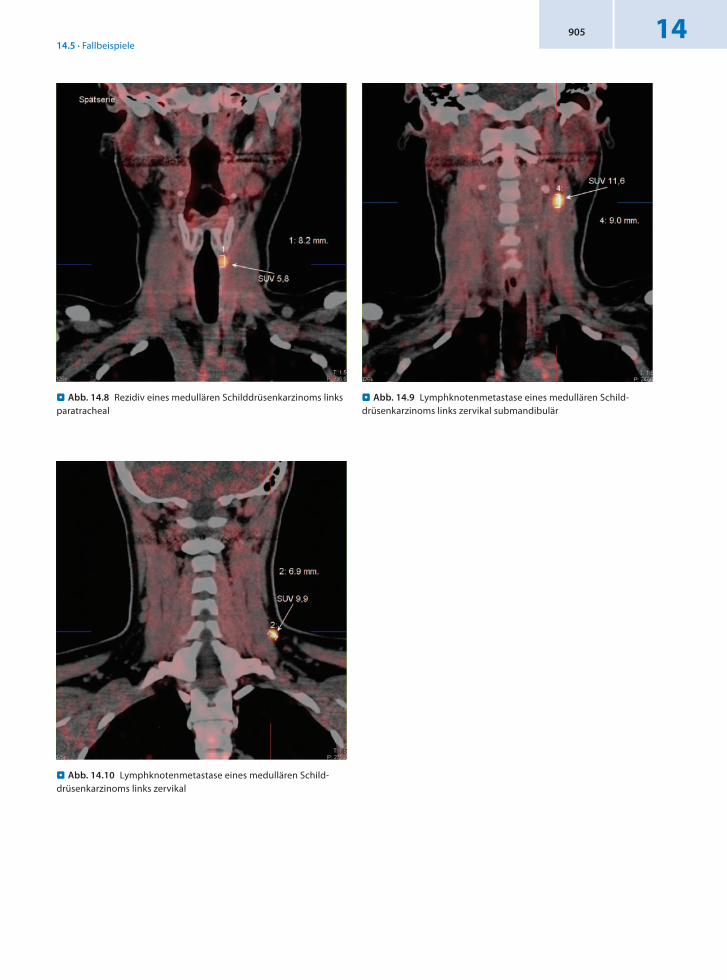
Anamnese 43jähriger männlicher Patient mit Schluckbeschwerden seit 8 Monaten. Daraufhin Schilddrüsendiagnostik mit Szintigraphie und Pentagastrintest. Vor 5 Monaten wurden eine Thyreoidektomie und systematische Lymphknotendissektion Kompartment 1 A+B, Kompartment 3 durchgeführt. Diagnose: medulläres Karzinom der Schilddrüse im linken Schilddrüsenlappen, Tumorgröße 20 mm. Zervikale Reexploration rechts bei
Verdacht auf Lymphknotenmetastase vor 6 Wochen, histologisch kein Nachweis von Anteilen eines MTC. Der Kalzitoninwert stieg binnen 2 Monaten von 1 660 mg/l auf zuletzt 2 052 mg/l an.
Befund In der DOPAPET/CT Nachweis von 2 Lymphknotenmetastasen zervikal, oberhalb der rechten Klavikula medial gelegen sowie oberhalb des Schildknorpels links.
Schlussfolgerung Klärung des Kalzitoninanstiegs und präzise Lokalisation von 2 LKMetastasen durch 18FDOPAPET/CT.
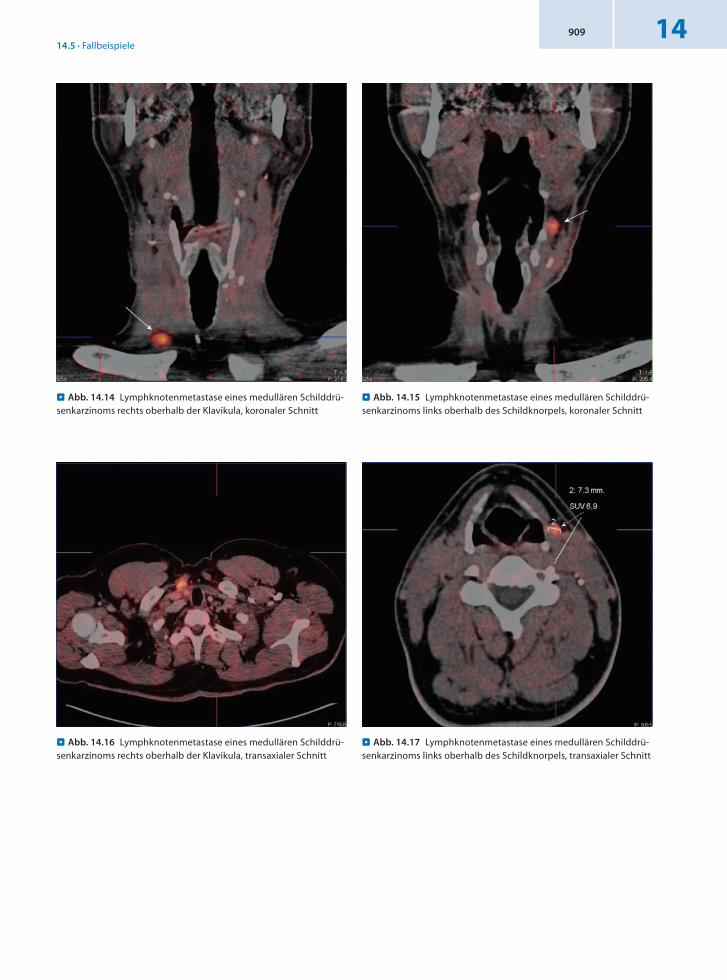
14.5 · Fallbeispiele14909
. Abb. 14.14 Lymphknotenmetastase eines medullären Schilddrü-senkarzinoms rechts oberhalb der Klavikula, koronaler Schnitt
. Abb. 14.17 Lymphknotenmetastase eines medullären Schilddrü-senkarzinoms links oberhalb des Schildknorpels, transaxialer Schnitt
. Abb. 14.16 Lymphknotenmetastase eines medullären Schilddrü-senkarzinoms rechts oberhalb der Klavikula, transaxialer Schnitt
. Abb. 14.15 Lymphknotenmetastase eines medullären Schilddrü-senkarzinoms links oberhalb des Schildknorpels, koronaler Schnitt
14
910 Kapitel 14 · Neuroendokrine Tumoren
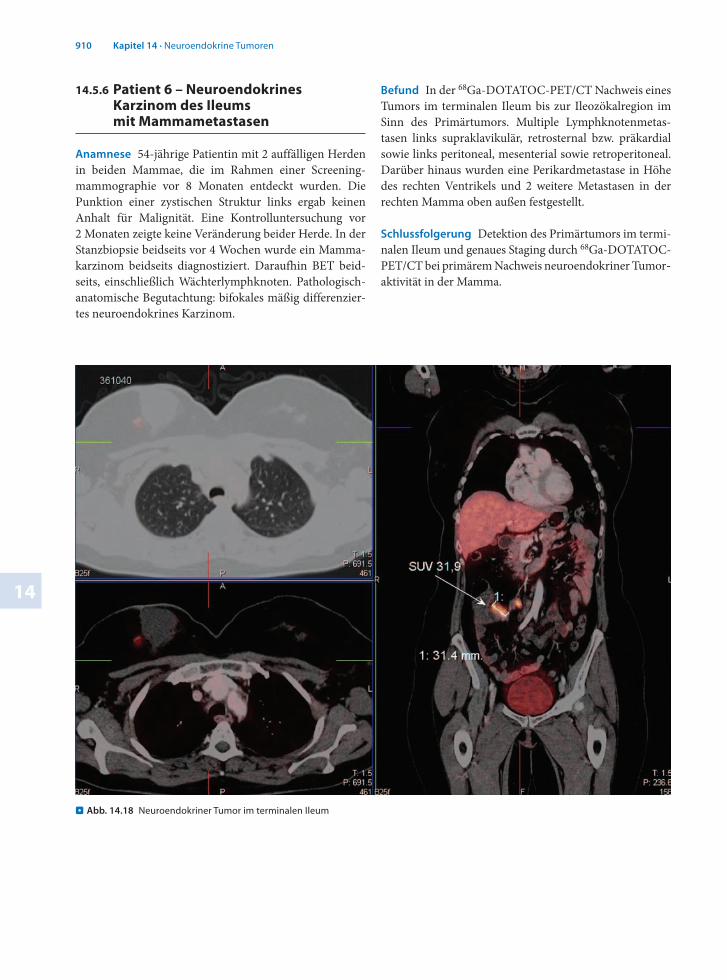
14.5.6 Patient 6 – Neuroendokrines Karzinom des Ileums mit Mammametastasen
Anamnese 54jährige Patientin mit 2 auffälligen Herden in beiden Mammae, die im Rahmen einer Screeningmammographie vor 8 Monaten entdeckt wurden. Die Punktion einer zystischen Struktur links ergab keinen Anhalt für Malignität. Eine Kontrolluntersuchung vor 2 Monaten zeigte keine Veränderung beider Herde. In der Stanzbiopsie beidseits vor 4 Wochen wurde ein Mammakarzinom beidseits diagnostiziert. Daraufhin BET beidseits, einschließlich Wächterlymphknoten. Pathologischanatomische Begutachtung: bifokales mäßig differenziertes neuroendokrines Karzinom.
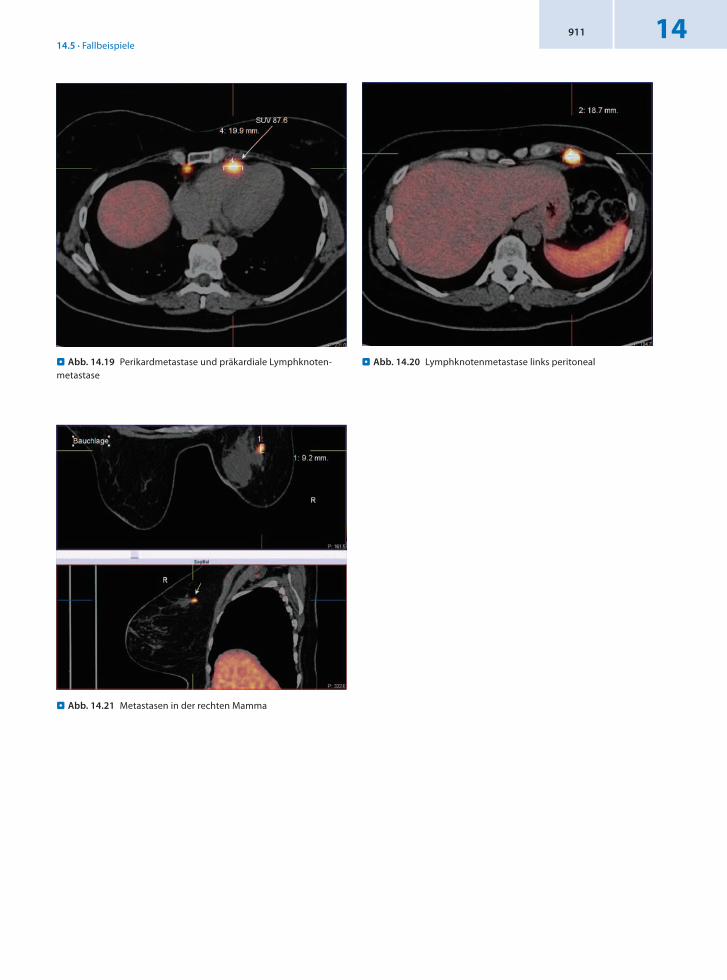
Befund In der 68GaDOTATOCPET/CT Nachweis eines Tumors im terminalen Ileum bis zur Ileozökalregion im Sinn des Primärtumors. Multiple Lymphknotenmetastasen links supraklavikulär, retrosternal bzw. präkardial sowie links peritoneal, mesenterial sowie retroperitoneal. Darüber hinaus wurden eine Perikardmetastase in Höhe des rechten Ventrikels und 2 weitere Metastasen in der rechten Mamma oben außen festgestellt.
Schlussfolgerung Detektion des Primärtumors im terminalen Ileum und genaues Staging durch 68GaDOTATOCPET/CT bei primärem Nachweis neuroendokriner Tumoraktivität in der Mamma.
. Abb. 14.18 Neuroendokriner Tumor im terminalen Ileum
14.5 · Fallbeispiele14911
. Abb. 14.19 Perikardmetastase und präkardiale Lymphknoten-metastase
. Abb. 14.21 Metastasen in der rechten Mamma
. Abb. 14.20 Lymphknotenmetastase links peritoneal

14
912 Kapitel 14 · Neuroendokrine Tumoren
14.5.7 Patient 7 – Lokalrezidiv eines Phäochromozytoms
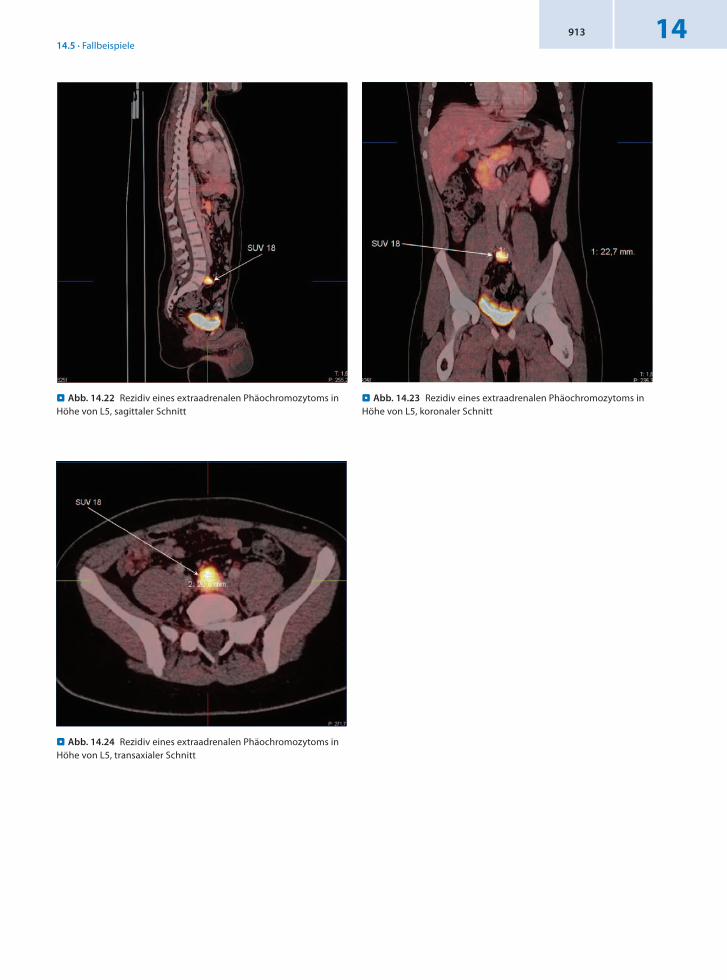
Anamnese 15jähriger männlicher Patient mit Zustand nach Exstirpation eines extraadrenalen Phäochromozytoms links paraaortal vor 2 Jahren. Der Tumor wurde in toto entfernt. Ein Lymphknotenbefall wurde intraoperativ nicht festgestellt. Jetzt seit Monaten erhöhte InvitroParameter. Daher Verdacht auf Rezidiv.
Befund In der DOPAPET/CT Nachweis eines Lokalrezidivs, DD Lymphknotenmetastase unterhalb der Aortengabel bzw. in Höhe von L5.
Schlussfolgerung Nachweis eines Lokalrezidivs und Ausschluss eines weiteren Befalls durch 18FDOPAPET/CT.
14.5 · Fallbeispiele14913
. Abb. 14.22 Rezidiv eines extraadrenalen Phäochromozytoms in Höhe von L5, sagittaler Schnitt
. Abb. 14.24 Rezidiv eines extraadrenalen Phäochromozytoms in Höhe von L5, transaxialer Schnitt
. Abb. 14.23 Rezidiv eines extraadrenalen Phäochromozytoms in Höhe von L5, koronaler Schnitt
14
914 Kapitel 14 · Neuroendokrine Tumoren
14.5.8 Patient 8 – Metastasiertes Phäochromozytom
Anamnese 18jährige Patientin seit 9 Monaten mit Kopfschmerzen und erhöhtem Blutdruck, zunächst als Migräne diagnostiziert und behandelt.
Sonographie: Tumor in der linken Nebenniere mit erhöhten Katecholaminwerten im Urin, damit Verdacht auf Phäochromozytom.
Adrenalektomie links und PE der Leber aus den Segmenten III und IV bei suspekten Herdbefunden in der Sonographie.
Histologie: malignes Phäochromozytom mit einer Größe von 8,5 cm mit Lebermetastasen.
MIBGSzintigraphie: Detektion einer Lebermetastase – Teilresektion der Leber geplant.
CTDiagnostik: diffuser Befall der Leber – Lebertransplantation geplant.
Vor 5 Monaten Laparotomie mit Lymphadenektomie aortokaval, im Bereich des Ligamentum hepatoduodenale, suprapankreatisch und trunchal aus dem Mesenterium.
Histologie: Lymphknotenmetastasen des bekannten malignen Phäochromozytoms. Damit wurde die geplante
Lebertransplantation abgesagt und stattdessen eine MIBGTherapie durchgeführt.
Die MRTKontrolle des Abdomens vor 7 Wochen zeigte ein unverändertes Fortbestehen diffuser Lebermetastasen. Lymphknotenmetastasen wurden nicht nachgewiesen.
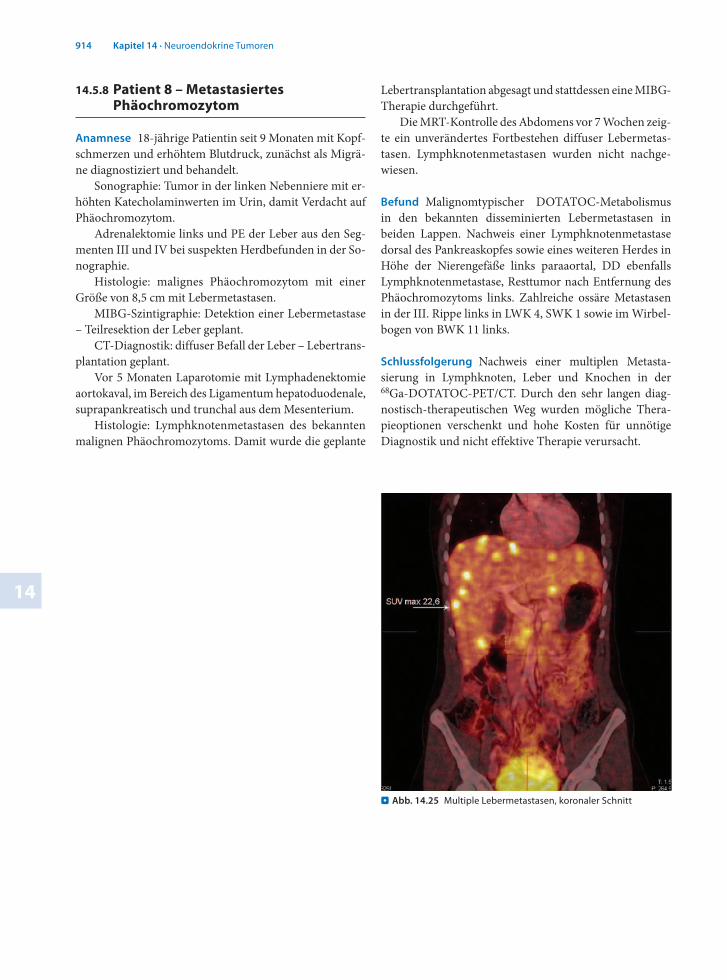
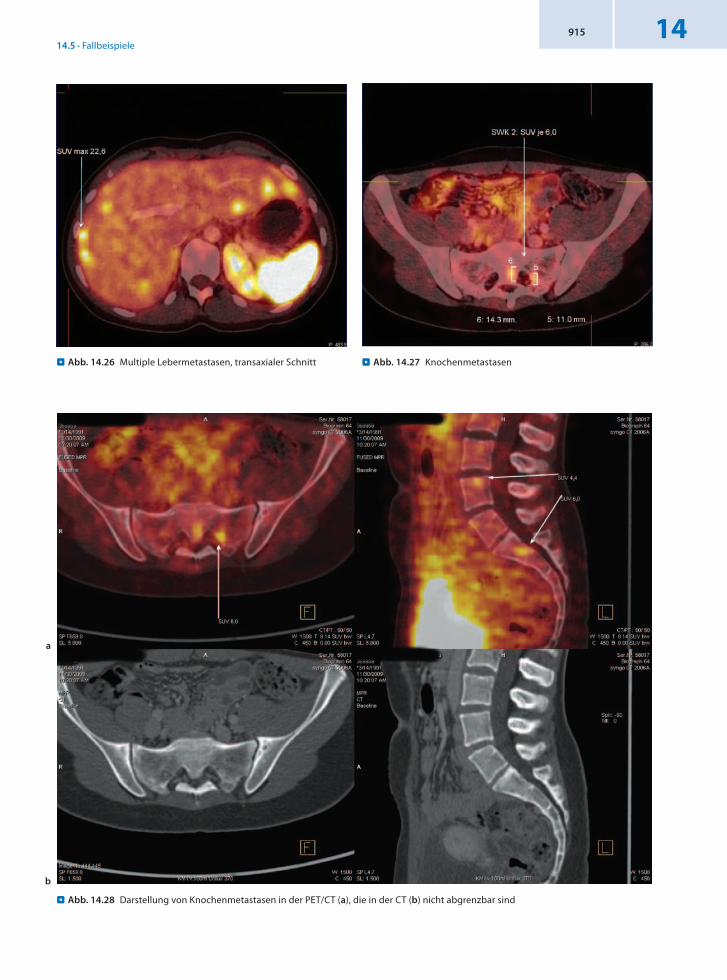
Befund Malignomtypischer DOTATOCMetabolismus in den bekannten disseminierten Lebermetastasen in beiden Lappen. Nachweis einer Lymphknotenmetastase dorsal des Pankreaskopfes sowie eines weiteren Herdes in Höhe der Nierengefäße links paraaortal, DD ebenfalls Lymphknotenmetastase, Resttumor nach Entfernung des Phäochromozytoms links. Zahlreiche ossäre Metastasen in der III. Rippe links in LWK 4, SWK 1 sowie im Wirbelbogen von BWK 11 links.
Schlussfolgerung Nachweis einer multiplen Metastasierung in Lymphknoten, Leber und Knochen in der 68GaDOTATOCPET/CT. Durch den sehr langen diagnostischtherapeutischen Weg wurden mögliche Therapieoptionen verschenkt und hohe Kosten für unnötige Diagnostik und nicht effektive Therapie verursacht.
. Abb. 14.25 Multiple Lebermetastasen, koronaler Schnitt
14.5 · Fallbeispiele14915
. Abb. 14.26 Multiple Lebermetastasen, transaxialer Schnitt . Abb. 14.27 Knochenmetastasen
. Abb. 14.28 Darstellung von Knochenmetastasen in der PET/CT (a), die in der CT (b) nicht abgrenzbar sind
a
b

14
916 Kapitel 14 · Neuroendokrine Tumoren
14.5.9 Patient 9 – Kongenitaler Hyperinsulinismus: DOPA- vs. DOTATOC-PET/CT
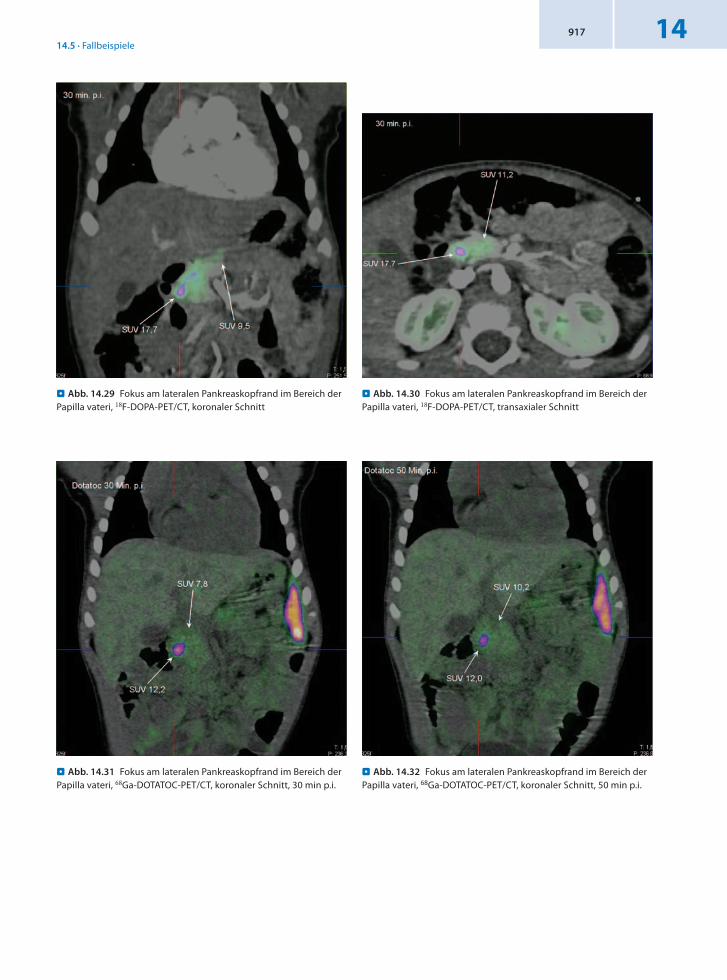
Anamnese 16monatiges männliches Kleinkind mit weiter bestehender Hypoglykämie bei Zustand nach Resektion eines Fokus aus dem Pankreaskopfbereich vor 6 Monaten. Zuvor war eine DOPAPET/CTUntersuchung durchgeführt worden, bei der ein Fokus von 6 mm Durchmesser im Pankreaskopfbereich diagnostiziert wurde.
Befund Die KontrollPET/CTUntersuchung mit 18FDOPA und 68GaDOTATOC ergibt weiterhin das Vorliegen einer fokalen Form des kongenitalen Hyperinsulinismus. Der Fokus hat einen Größendurchmesser von ca. 6 × 4 mm. Er liegt am lateralen Pankreaskopfrand im Bereich der Papilla vateri, angrenzend an die Pars descendens duodeni. Die Lage entspricht der bei der Voruntersuchung angesprochenen Fokuslokalisation. Die Distanz zur medialen Begrenzung des oberen Gallenblasenbereichs beträgt heute nur 10 mm und zum Confluens 26 mm.
Schlussfolgerung Präzise Lokalisation für eine gezielte therapeutische Behandlung mit sowohl 18FDOPA als auch 68GaDOTATOCPET/CT.
14.5 · Fallbeispiele14917
. Abb. 14.29 Fokus am lateralen Pankreaskopfrand im Bereich der Papilla vateri, 18F-DOPA-PET/CT, koronaler Schnitt
. Abb. 14.32 Fokus am lateralen Pankreaskopfrand im Bereich der Papilla vateri, 68Ga-DOTATOC-PET/CT, koronaler Schnitt, 50 min p.i.
. Abb. 14.31 Fokus am lateralen Pankreaskopfrand im Bereich der Papilla vateri, 68Ga-DOTATOC-PET/CT, koronaler Schnitt, 30 min p.i.
. Abb. 14.30 Fokus am lateralen Pankreaskopfrand im Bereich der Papilla vateri, 18F-DOPA-PET/CT, transaxialer Schnitt

14
918 Kapitel 14 · Neuroendokrine Tumoren
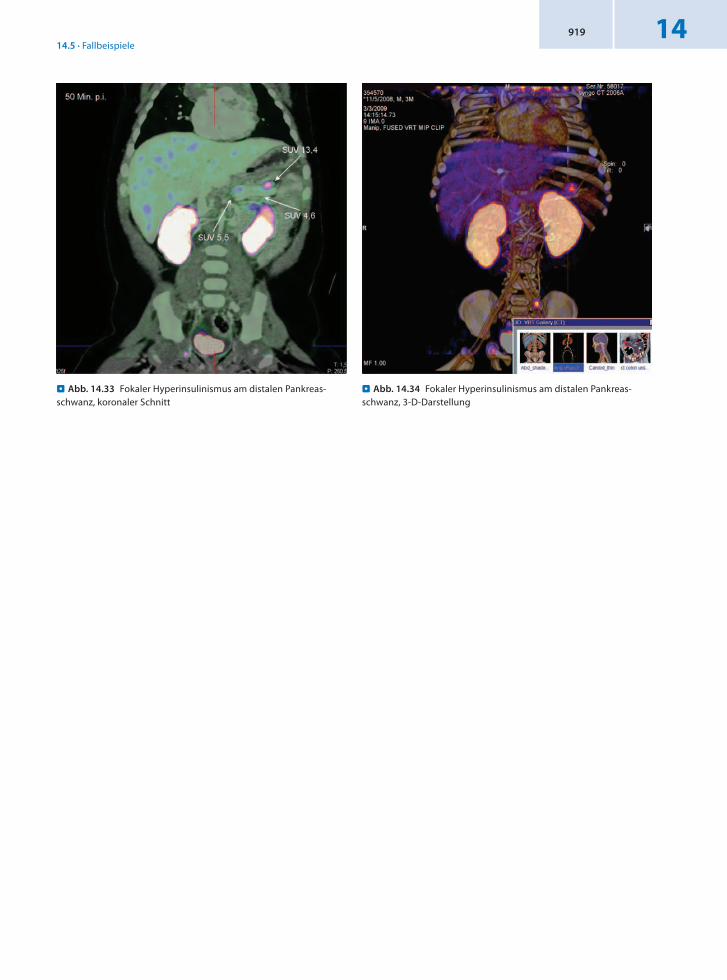
14.5.10 Patient 10 – Kongenitaler Hyperinsulinismus
Anamnese 4monatiger männlicher Säugling mit kongenitalem Hyperinsulinismus.
Befund In der 18FDOPAPET/CT Nachweis einer fokalen Form des kongenitalen Hyperinsulinismus im distalen Pankreasschwanz. Die Größe des Fokus beträgt
5 × 5 × 4 mm. Für die intraoperative Lokalisation sind folgende Bezugsparameter anzugeben:1. Der Fokus liegt unmittelbar am Pankreasschwanzende an der kranialen Begrenzung der Cauda pancreatis.2. Die Distanz zwischen Fokuszentrum und Confluens beträgt 42 mm.
Schlussfolgerung Präzise Lokalisation eines Fokus mit 18FDOPAPET/CT für eine effektive Therapie.
14.5 · Fallbeispiele14919
. Abb. 14.33 Fokaler Hyperinsulinismus am distalen Pankreas-schwanz, koronaler Schnitt
. Abb. 14.34 Fokaler Hyperinsulinismus am distalen Pankreas-schwanz, 3-D-Darstellung
14
920 Kapitel 14 · Neuroendokrine Tumoren
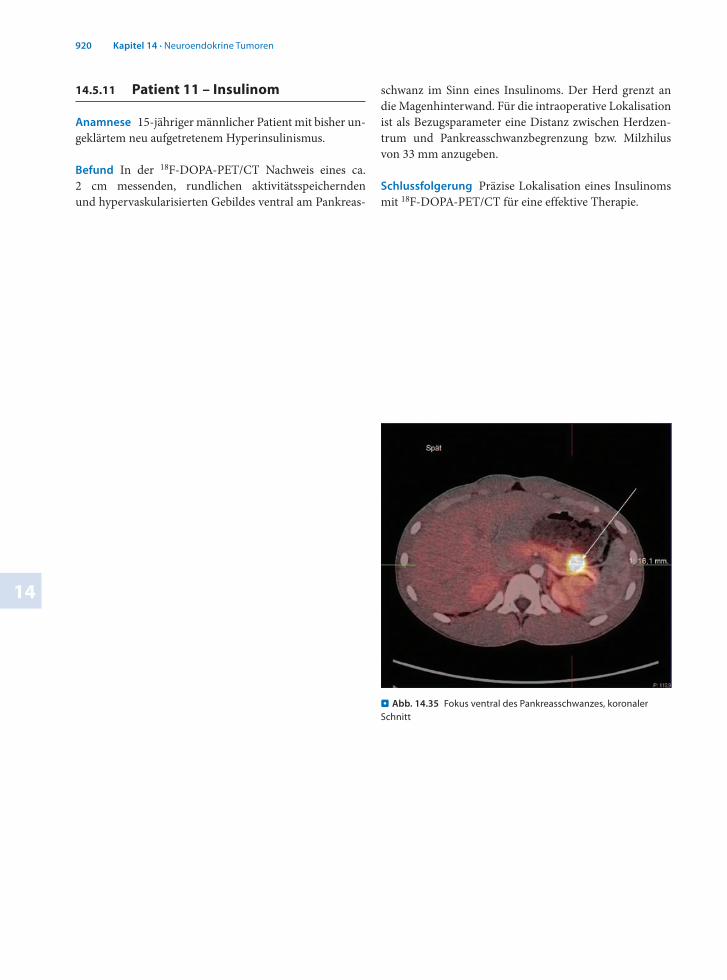
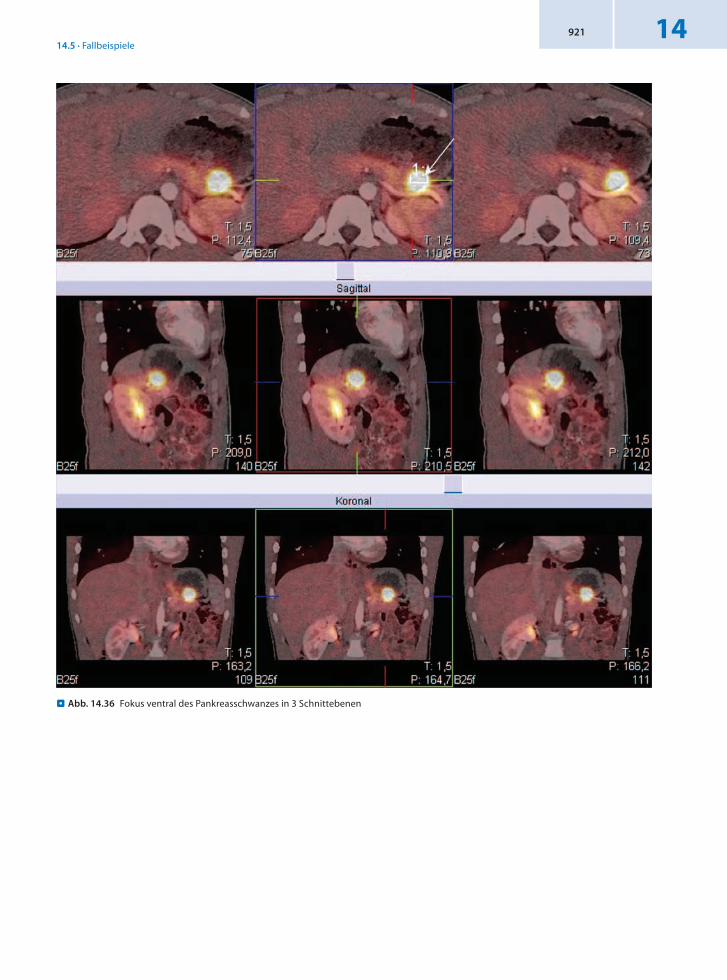
14.5.11 Patient 11 – Insulinom
Anamnese 15jähriger männlicher Patient mit bisher ungeklärtem neu aufgetretenem Hyperinsulinismus.
Befund In der 18FDOPAPET/CT Nachweis eines ca. 2 cm messenden, rundlichen aktivitätsspeichernden und hypervaskularisierten Gebildes ventral am Pankreas
schwanz im Sinn eines Insulinoms. Der Herd grenzt an die Magenhinterwand. Für die intraoperative Lokalisation ist als Bezugsparameter eine Distanz zwischen Herdzentrum und Pankreasschwanzbegrenzung bzw. Milzhilus von 33 mm anzugeben.
Schlussfolgerung Präzise Lokalisation eines Insulinoms mit 18FDOPAPET/CT für eine effektive Therapie.
. Abb. 14.35 Fokus ventral des Pankreasschwanzes, koronaler Schnitt
14.5 · Fallbeispiele14921
. Abb. 14.36 Fokus ventral des Pankreasschwanzes in 3 Schnittebenen

14
922 Kapitel 14 · Neuroendokrine Tumoren
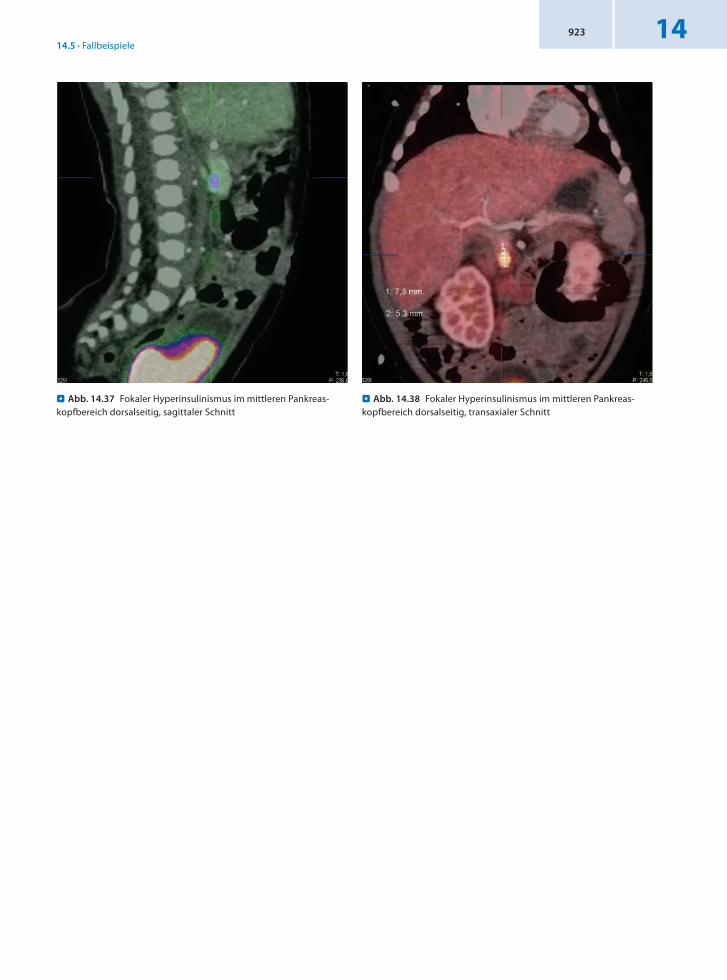
14.5.12 Patient 12 – Kongenitaler Hyperinsulinismus
Anamnese 5wöchiger weiblicher Säugling mit kongenitalem Hyperinsulinismus.
Befund In der 18FDOPAPET/CT Nachweis der fokalen Form des kongenitalen Hyperinsulinismus im mittleren Pankreaskopfbereich dorsalseitig mit einer Größe von 5 × 5 × 8 mm.
Für die intraoperative Lokalisation sind folgende Bezugsparameter anzugeben:1. Der Fokus liegt rechts lateral des Verlaufs der Vena mesenterica superior.2. Der Abstand zwischen kranialer Fokusbegrenzung und der Vena portae in Höhe des Confluens beträgt 10 mm.3. Der Fokus überragt dorsalseitig deutlich die Pankreaskontur.
Schlussfolgerung Präzise Lokalisation eines Fokus mit 18FDOPAPET/CT für eine effektive Therapie.
14.5 · Fallbeispiele14923
. Abb. 14.37 Fokaler Hyperinsulinismus im mittleren Pankreas-kopfbereich dorsalseitig, sagittaler Schnitt
. Abb. 14.38 Fokaler Hyperinsulinismus im mittleren Pankreas-kopfbereich dorsalseitig, transaxialer Schnitt
14
924 Kapitel 14 · Neuroendokrine Tumoren
14.5.13 Patient 13 – Kongenitaler Hyperinsulinismus
Anamnese 6monatiger männlicher Säugling mit kongenitalem Hyperinsulinismus.
Befund In der 18FDOPAPET/CT Nachweis der fokalen Form des kongenitalen Hyperinsulinismus im Pankreaskorpusbereich, mehr kaudal. Der Herd liegt unmittelbar ventral der Einmündungsstelle der Vena lienalis in den
Confluens. Er hat eine Größe von 10 × 7 × 8 mm. Für die intraoperative Lokalisation ist als Bezugsparameter anzugeben, dass die Distanz zwischen dem Fokuszentrum und der ventralen Begrenzung der Vena lienalis in Höhe der Einmündung zum Confluens nur 4 mm beträgt mit Einbeziehung des Ductus pancreaticus.
Schlussfolgerung Präzise Lokalisation eines Fokus mit 18FDOPAPET/CT für eine effektive Therapie.
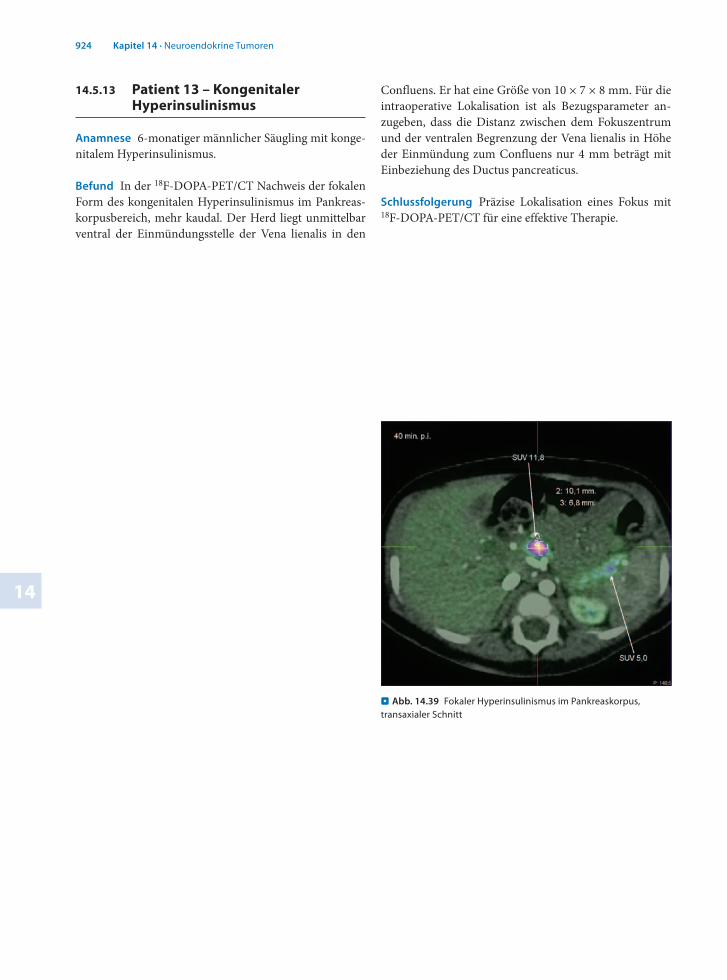
. Abb. 14.39 Fokaler Hyperinsulinismus im Pankreaskorpus, transaxialer Schnitt
14.5 · Fallbeispiele14925
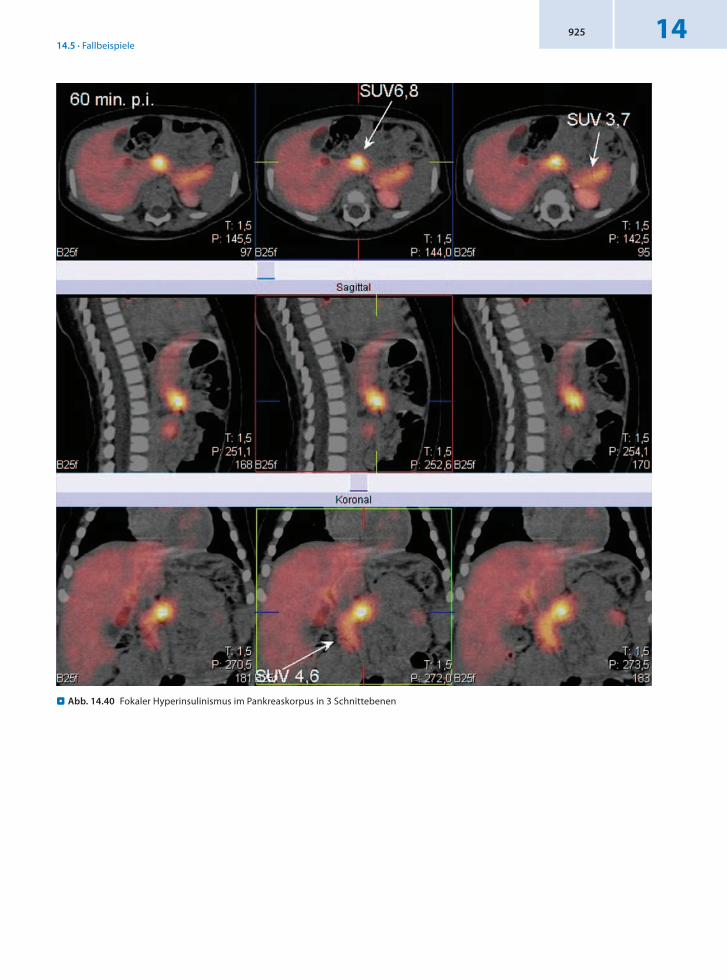
. Abb. 14.40 Fokaler Hyperinsulinismus im Pankreaskorpus in 3 Schnittebenen
14
926 Kapitel 14 · Neuroendokrine Tumoren
14.5.14 Patient 14 – Fokaler kongenitaler Hyperinsulinismus
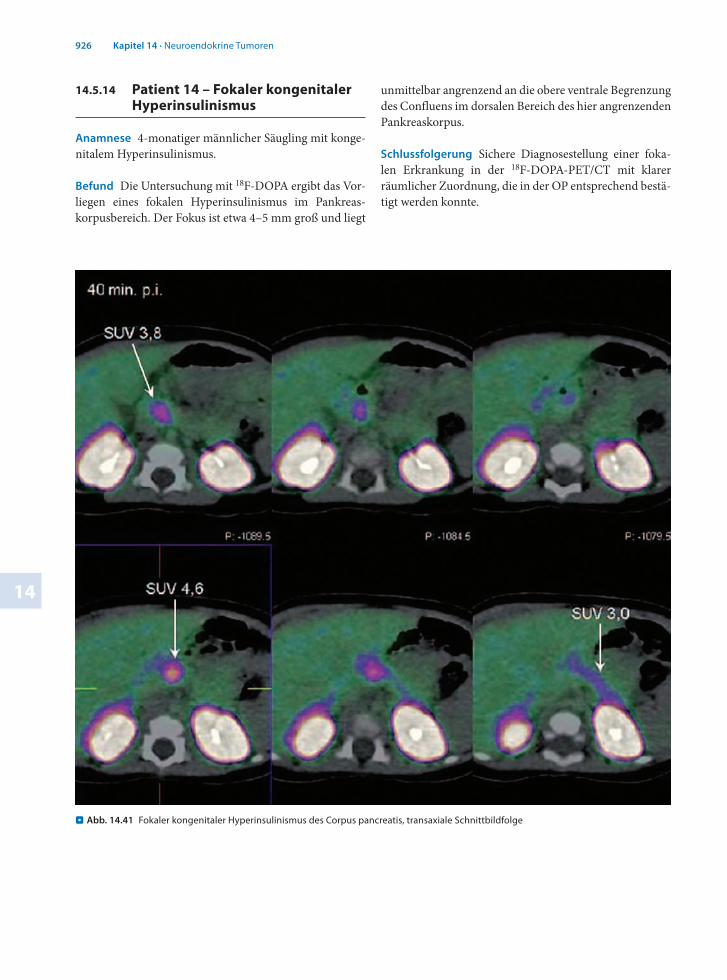
Anamnese 4monatiger männlicher Säugling mit kongenitalem Hyperinsulinismus.
Befund Die Untersuchung mit 18FDOPA ergibt das Vorliegen eines fokalen Hyperinsulinismus im Pankreaskorpusbereich. Der Fokus ist etwa 4–5 mm groß und liegt
unmittelbar angrenzend an die obere ventrale Begrenzung des Confluens im dorsalen Bereich des hier angrenzenden Pankreaskorpus.
Schlussfolgerung Sichere Diagnosestellung einer fokalen Erkrankung in der 18FDOPAPET/CT mit klarer räumlicher Zuordnung, die in der OP entsprechend bestätigt werden konnte.
. Abb. 14.41 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis, transaxiale Schnittbildfolge
14.5 · Fallbeispiele14927
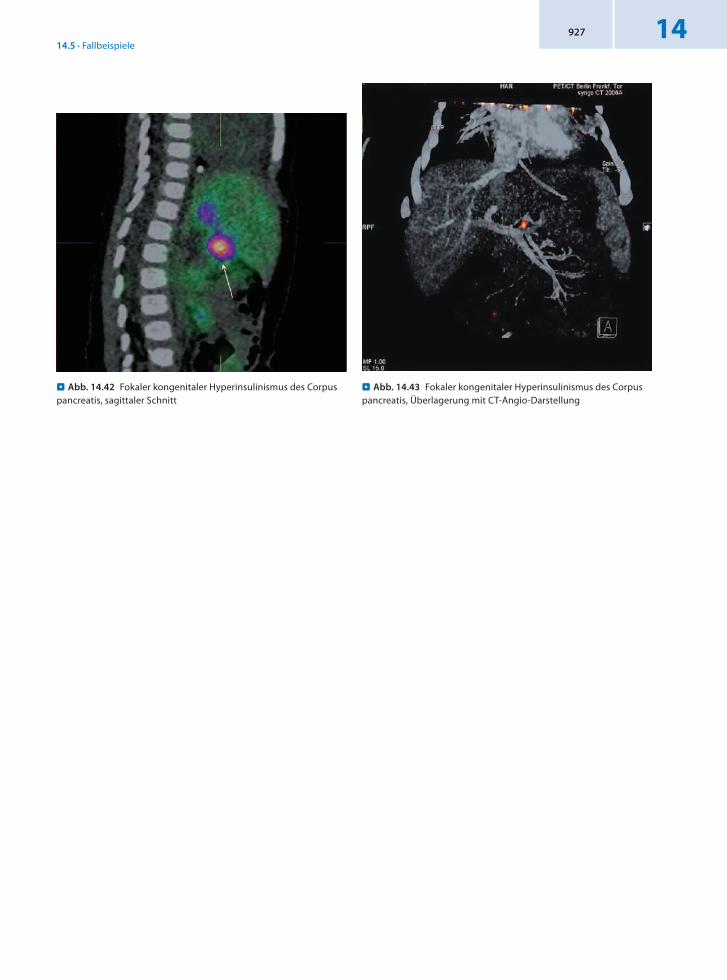
. Abb. 14.42 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis, sagittaler Schnitt
. Abb. 14.43 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis, Überlagerung mit CT-Angio-Darstellung
14
928 Kapitel 14 · Neuroendokrine Tumoren
14.5.15 Patient 15 – Fokaler kongenitaler Hyperinsulinismus
Anamnese Verdacht auf kongenitalen Hyperinsulinismus (CHI).
Befund Die Untersuchung mit 18FDOPA ergibt das Vorliegen eines Fokus im Sinn des fokalen kongenitalen Hyperinsulinismus. Der Fokus liegt an der ventralen Begren
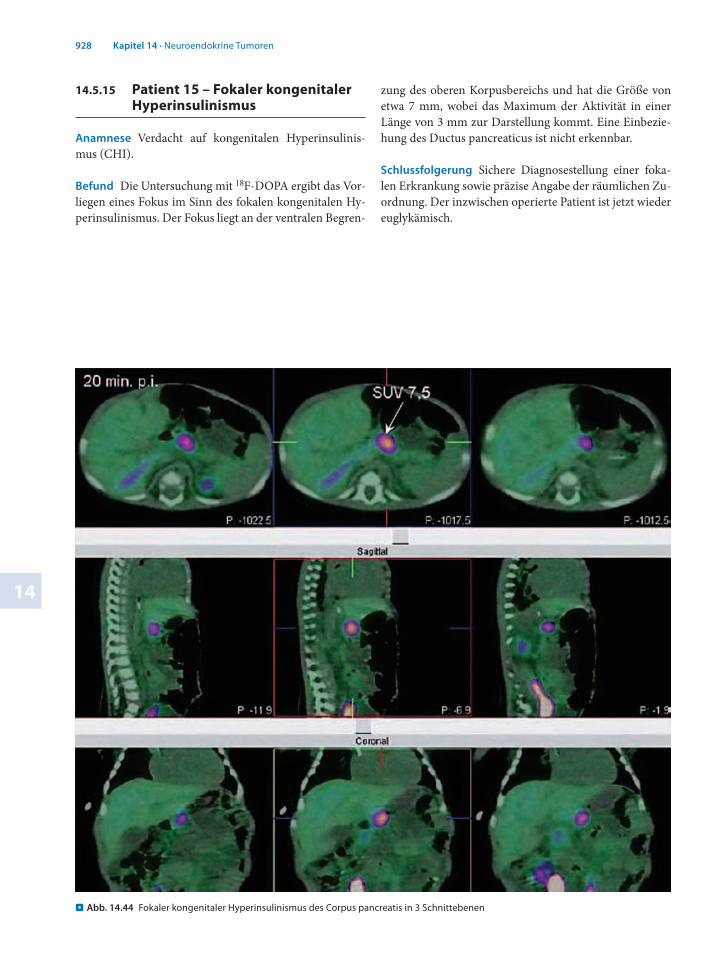
zung des oberen Korpusbereichs und hat die Größe von etwa 7 mm, wobei das Maximum der Aktivität in einer Länge von 3 mm zur Darstellung kommt. Eine Einbeziehung des Ductus pancreaticus ist nicht erkennbar.
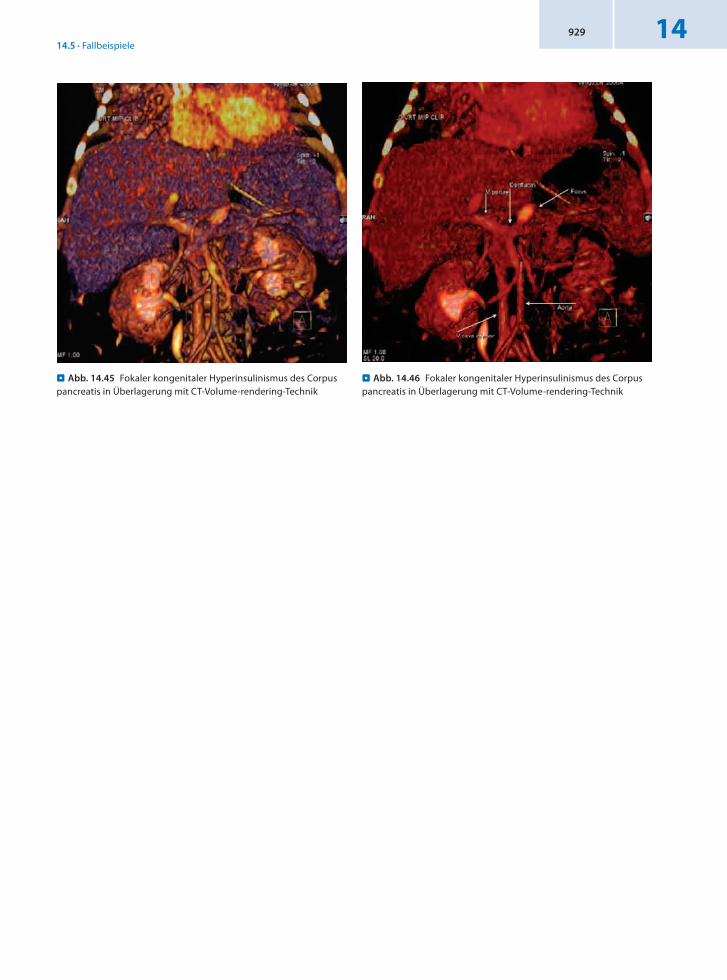
Schlussfolgerung Sichere Diagnosestellung einer fokalen Erkrankung sowie präzise Angabe der räumlichen Zuordnung. Der inzwischen operierte Patient ist jetzt wieder euglykämisch.
. Abb. 14.44 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis in 3 Schnittebenen
14.5 · Fallbeispiele14929
. Abb. 14.45 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis in Überlagerung mit CT-Volume-rendering-Technik
. Abb. 14.46 Fokaler kongenitaler Hyperinsulinismus des Corpus pancreatis in Überlagerung mit CT-Volume-rendering-Technik

14
930 Kapitel 14 · Neuroendokrine Tumoren
14.5.16 Patient 16 – Neuroendokriner Tumor
Anamnese 68jähriger männlicher Patient mit Erstdiagnose einer diffusen Lebermetastasierung (sonographisch und computertomographisch) vor 5 Wochen. Bisher keine Sicherung eines Primarius. Inappetenz, Schwäche und Gewichtsverlust von 10 kg innerhalb von 6 Monaten. Bisherige Diagnose: Colon irritabile 5–8 Stühle/die.
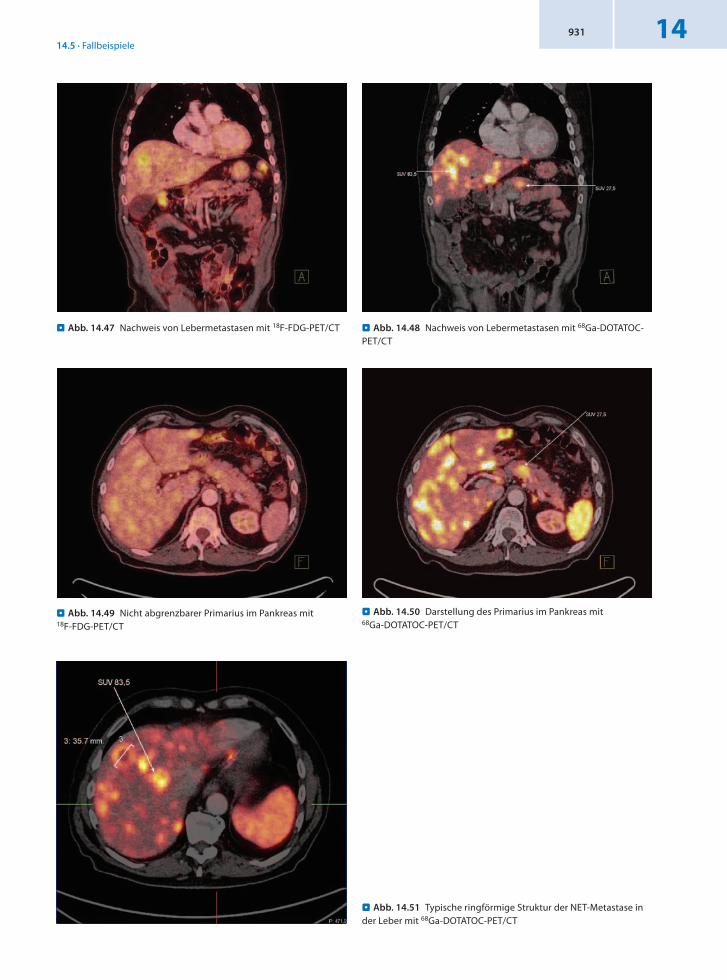
Befund Malignomtypischer Glukosemetabolismus im Bereich der diffus mit Metastasen durchsetzten Leber. Der Primärtumor konnte mit FDG nicht diagnostiziert werden. Mittels 68GaDOTATOCPET/CT Nachweis eines neuroendokrinen Tumors im Pankreaskorpus.
Der Primärtumor im Pankreas ist offensichtlich differenziert (somit keine Darstellung mit 18FFDG), während die Lebermetastasen weitgehend entdifferenziert sind und somit bereits in der FDGPET sichtbar werden.
Schlussfolgerung Sicherung des Primarius nur mit 68GaDOTATOCPET/CT, Lebermetastasen auch mit 18FFDGPET/CT.
14.5 · Fallbeispiele14931
. Abb. 14.47 Nachweis von Lebermetastasen mit 18F-FDG-PET/CT
. Abb. 14.51 Typische ringförmige Struktur der NET-Metastase in der Leber mit 68Ga-DOTATOC-PET/CT
. Abb. 14.50 Darstellung des Primarius im Pankreas mit 68Ga-DOTATOC-PET/CT
. Abb. 14.49 Nicht abgrenzbarer Primarius im Pankreas mit 18F-FDG-PET/CT
. Abb. 14.48 Nachweis von Lebermetastasen mit 68Ga-DOTATOC-PET/CT

14
932 Kapitel 14 · Neuroendokrine Tumoren
14.5.17 Patient 17 – Neuroendokriner Tumor im rechten Mittelbauch
Anamnese 83jährige Patientin mit gynäkologischer TotalOP wegen eines Unterbauchtumors vor 49 Jahren. Vor 19 Jahren Mammakarzinom rechts mit Ablatio mammae, vor 15 Jahren Lymphknotenrezidiv ausgehend von der rechten Mamma. Seit 2 Jahren gesichertes Plasmozytom. Aktuell akute abdominelle Beschwerdesymptomatik (Übelkeit, Erbrechen). Endosonographisch sowie zytologisch besteht der Verdacht auf einen neuroendokrinen Tumor unterhalb der Pankreaskopfregion.
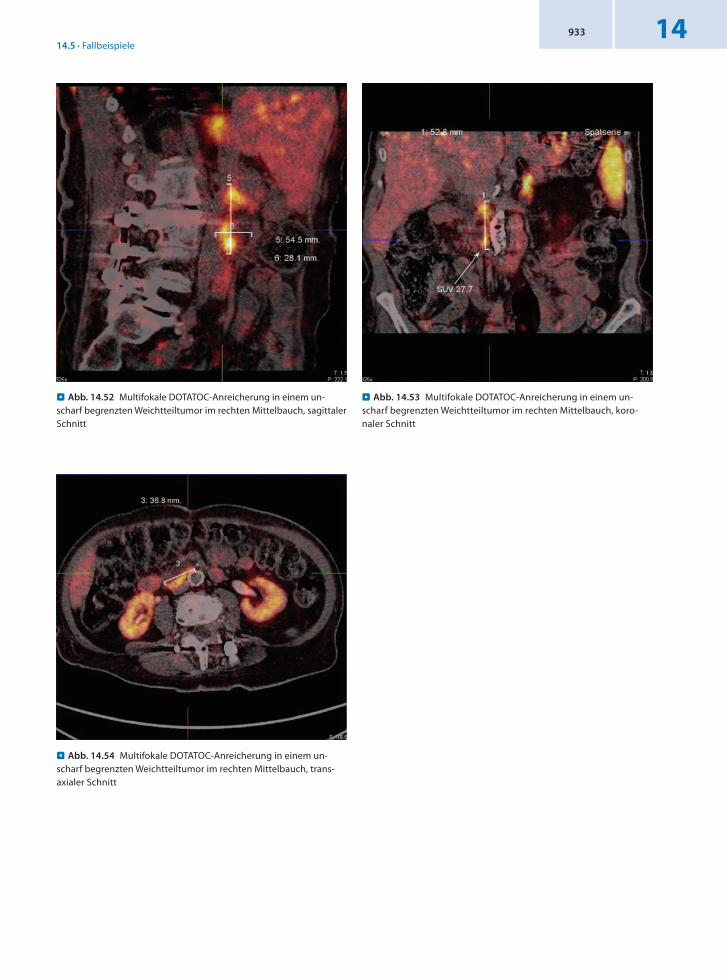
Befund Nachweis einer DOTATOCAnreicherung im Bereich einer insgesamt schwer abgrenzbaren 3,7 × 2,8 × 5,5 cm großen Weichteilraumforderung im rechten Mittelbauch mit inhomogen randständig betonter DOTATOCMehrbelegung als Ausdruck des neuroendokrinen Tumors.
Schlussfolgerung Verdachtsbestätigung und Nachweis des NET im Dünndarm mit Lymphknotenmetastasen.
14.5 · Fallbeispiele14933
. Abb. 14.52 Multifokale DOTATOC-Anreicherung in einem un-scharf begrenzten Weichtteiltumor im rechten Mittelbauch, sagittaler Schnitt
. Abb. 14.54 Multifokale DOTATOC-Anreicherung in einem un-scharf begrenzten Weichtteiltumor im rechten Mittelbauch, trans-axialer Schnitt
. Abb. 14.53 Multifokale DOTATOC-Anreicherung in einem un-scharf begrenzten Weichtteiltumor im rechten Mittelbauch, koro-naler Schnitt
14
934 Kapitel 14 · Neuroendokrine Tumoren
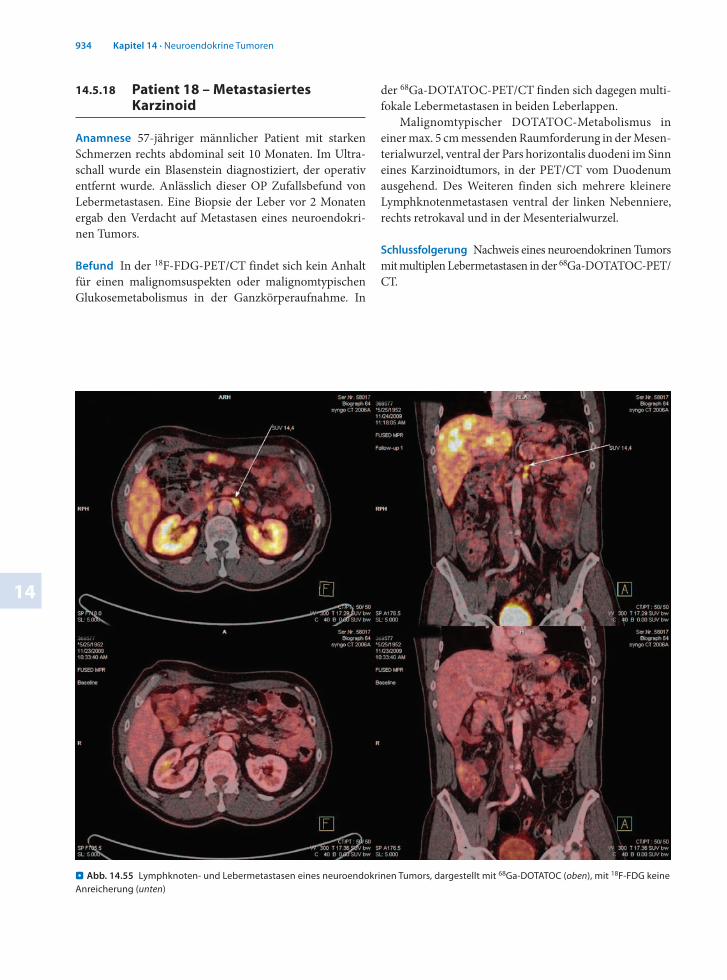
14.5.18 Patient 18 – Metastasiertes Karzinoid
Anamnese 57jähriger männlicher Patient mit starken Schmerzen rechts abdominal seit 10 Monaten. Im Ultraschall wurde ein Blasenstein diagnostiziert, der operativ entfernt wurde. Anlässlich dieser OP Zufallsbefund von Lebermetastasen. Eine Biopsie der Leber vor 2 Monaten ergab den Verdacht auf Metastasen eines neuroendokrinen Tumors.
Befund In der 18FFDGPET/CT findet sich kein Anhalt für einen malignomsuspekten oder malignomtypischen Glukosemetabolismus in der Ganzkörperaufnahme. In
der 68GaDOTATOCPET/CT finden sich dagegen multifokale Lebermetastasen in beiden Leberlappen.
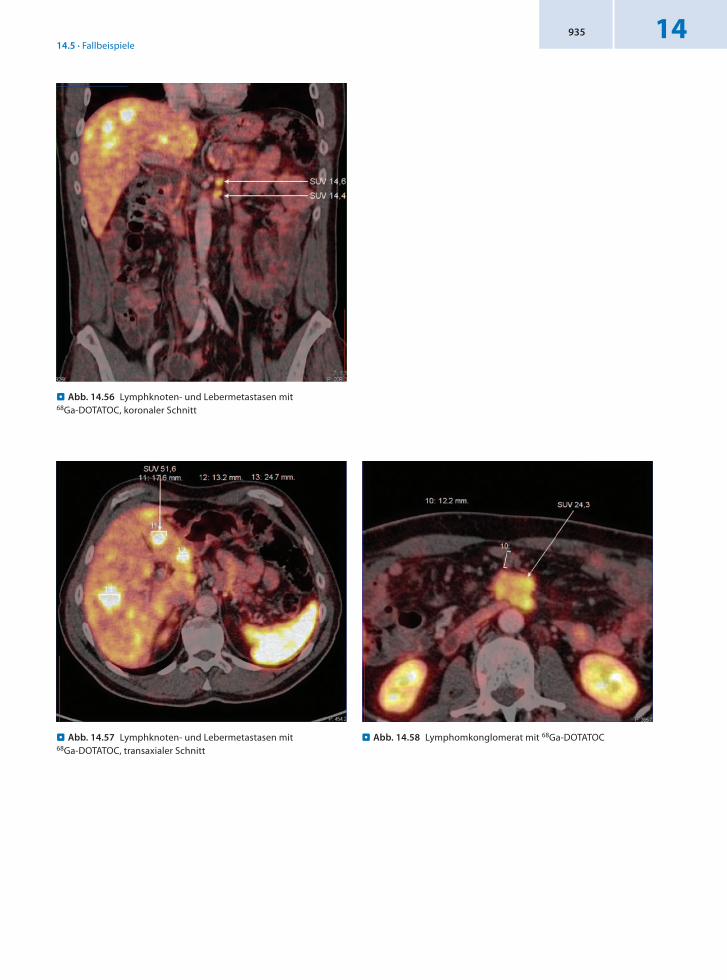
Malignomtypischer DOTATOCMetabolismus in einer max. 5 cm messenden Raumforderung in der Mesenterialwurzel, ventral der Pars horizontalis duodeni im Sinn eines Karzinoidtumors, in der PET/CT vom Duodenum ausgehend. Des Weiteren finden sich mehrere kleinere Lymphknotenmetastasen ventral der linken Nebenniere, rechts retrokaval und in der Mesenterialwurzel.
Schlussfolgerung Nachweis eines neuroendokrinen Tumors mit multiplen Lebermetastasen in der 68GaDOTATOCPET/CT.
. Abb. 14.55 Lymphknoten- und Lebermetastasen eines neuroendokrinen Tumors, dargestellt mit 68Ga-DOTATOC (oben), mit 18F-FDG keine Anreicherung (unten)
14.5 · Fallbeispiele14935
. Abb. 14.56 Lymphknoten- und Lebermetastasen mit 68Ga-DOTATOC, koronaler Schnitt
. Abb. 14.58 Lymphomkonglomerat mit 68Ga-DOTATOC . Abb. 14.57 Lymphknoten- und Lebermetastasen mit 68Ga-DOTATOC, transaxialer Schnitt

14
936 Kapitel 14 · Neuroendokrine Tumoren
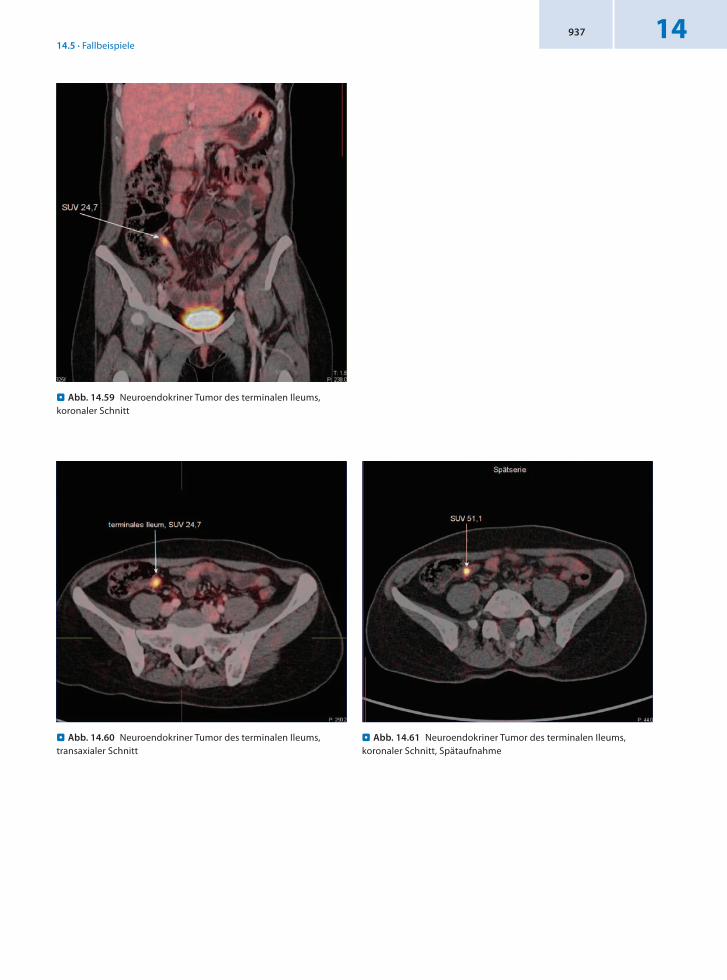
14.5.19 Patient 19 – Karzinoid des terminalen Ileums
Anamnese 41jährige Patientin mit einem neuroendokrinen Tumor des terminalen Ileums und einer Größe von 0,8 cm. Histologie: hochdifferenziertes neuroendokrines Karzinom.
Seit 2–3 Jahren postprandial Darmgeräusche, Kopfschmerzen und Druckgefühl im Kopf. Vor 2 Monaten starke Unterbauchbeschwerden links. Daraufhin wurde eine Koloskopie mit Nachweis eines Polypen im termina
len Ileum durchgeführt. Multiple Biopsien aus dem Befund, der etwa 3 cm proximal der Bauhin‘schen Klappe liegt.
Befund Malignomtypischer DOTATOCMetabolismus in dem koloskopisch und histologisch gesichertem polypösen Tumorsubstrat im terminalen Ileum.
Schlussfolgerung Befundbestätigung und präzise Lokalisation mit 68GaDOTATOCPET/CT.
14.5 · Fallbeispiele14937
. Abb. 14.59 Neuroendokriner Tumor des terminalen Ileums, koronaler Schnitt
. Abb. 14.61 Neuroendokriner Tumor des terminalen Ileums, koronaler Schnitt, Spätaufnahme
. Abb. 14.60 Neuroendokriner Tumor des terminalen Ileums, transaxialer Schnitt

14
938 Kapitel 14 · Neuroendokrine Tumoren
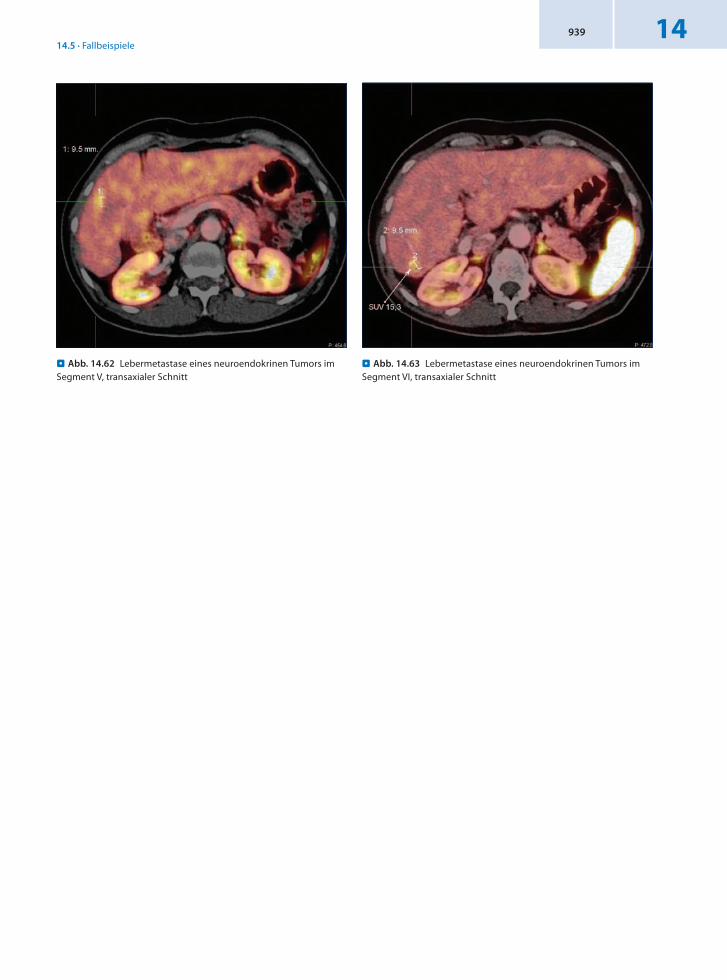
14.5.20 Patient 20 – Hepatische Metastasierung eines NET des Ileums
Anamnese 58jährige Patientin mit Erstdiagnose eines neuroendokrinen Karzinoms des terminalen Ileums mit primärer hepatischer Filialisierung mit anfangs funktioneller Aktivität (Hitzewallungen und Durchfälle) sowie Leberkapselschmerz vor 2 Jahren. Zustand nach Hemikolektomie rechts und Teilhepatektomie rechts. Die Untersuchungen mit MRT und Sonographie ergaben den Verdacht auf 2 neue Lebermetastasen.
Befund Malignomtypischer DOTATOCMetabolismus im Bereich zweier Lebermetastasen. Darüber hinaus findet sich eine inhomogene Nuklidverteilung in der Leber mit kleineren in Ausbildung begriffenen Leberfiliae.
Schlussfolgerung Verdachtsbestätigung und Befunderweiterung der Metastasierung eines NET mit 68GaDOTATOCPET/CT.
14.5 · Fallbeispiele14939
. Abb. 14.62 Lebermetastase eines neuroendokrinen Tumors im Segment V, transaxialer Schnitt
. Abb. 14.63 Lebermetastase eines neuroendokrinen Tumors im Segment VI, transaxialer Schnitt

14
940 Kapitel 14 · Neuroendokrine Tumoren
14.5.21 Patient 21 – Hepatische Metastasierung eines NET unter Therapie
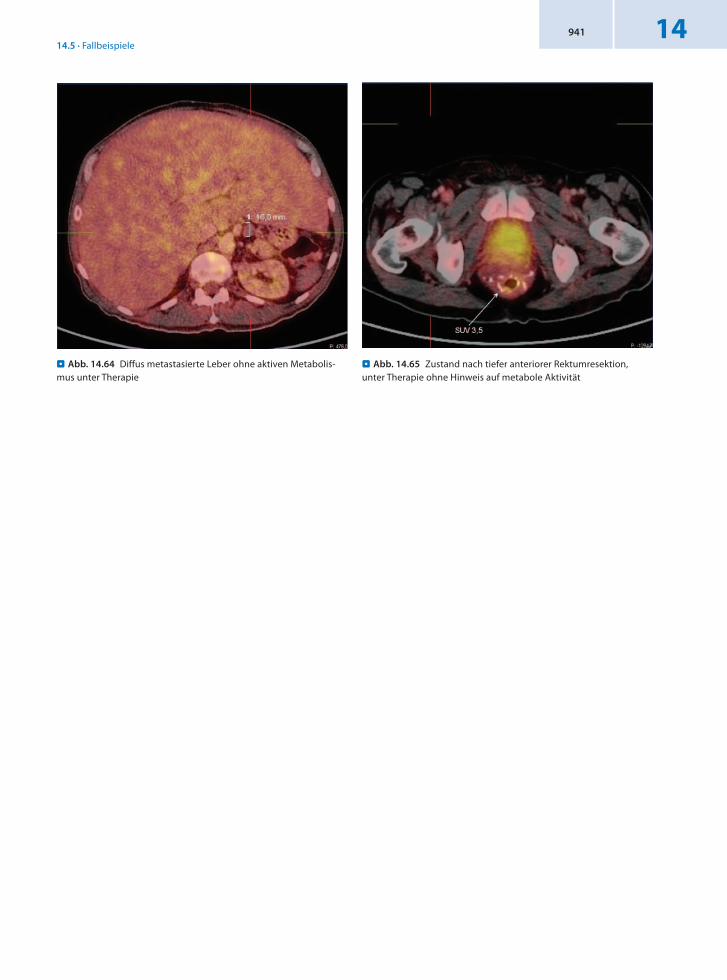
Anamnese 59jähriger männlicher Patient mit Zustand nach tiefer anteriorer Rektumresektion vor 4 Monaten wegen eines NET des Rektums. Diffuse Metastasierung der Leber. Szintigraphisch (Octreoscan) findet sich eine Herdläsion im BWK 10, LWK 3, im Os ilium links, in der Schädelkalotte dorsal sowie in der linken Lungenspitze. Anlage eines Ileostoma. Bisphosphonat und Somatostatinanalogatherapie. In der PET/CT vor 2 Monaten fand sich in den metastatischen Bereichen nur ein gering gesteigerter Metabolismus. Weiterführung der Therapie.
Befund Bei bekanntem metastasierendem NET des Rektums findet sich unter der laufenden Somatostatinanalogatherapie kein wesentlich erhöhter Glukosemetabolismus im Bereich der bekannten Knochenmetastasen des Schädels, der Wirbelsäule sowie des linken Os iliums und im Bereich der Lymphknotenmetastasen des Ligamentum hepatoduodenale. Weitere Vergrößerung der diffus metastasierten Leber. Insgesamt ist eine Suppression des Metastasenstoffwechsels unter laufender Therapie dargestellt.
Schlussfolgerung Nachweis eines guten Therapieansprechens in der 18FFDGPET/CT.
14.5 · Fallbeispiele14941
. Abb. 14.64 Diffus metastasierte Leber ohne aktiven Metabolis-mus unter Therapie
. Abb. 14.65 Zustand nach tiefer anteriorer Rektumresektion, unter Therapie ohne Hinweis auf metabole Aktivität

14
942 Kapitel 14 · Neuroendokrine Tumoren
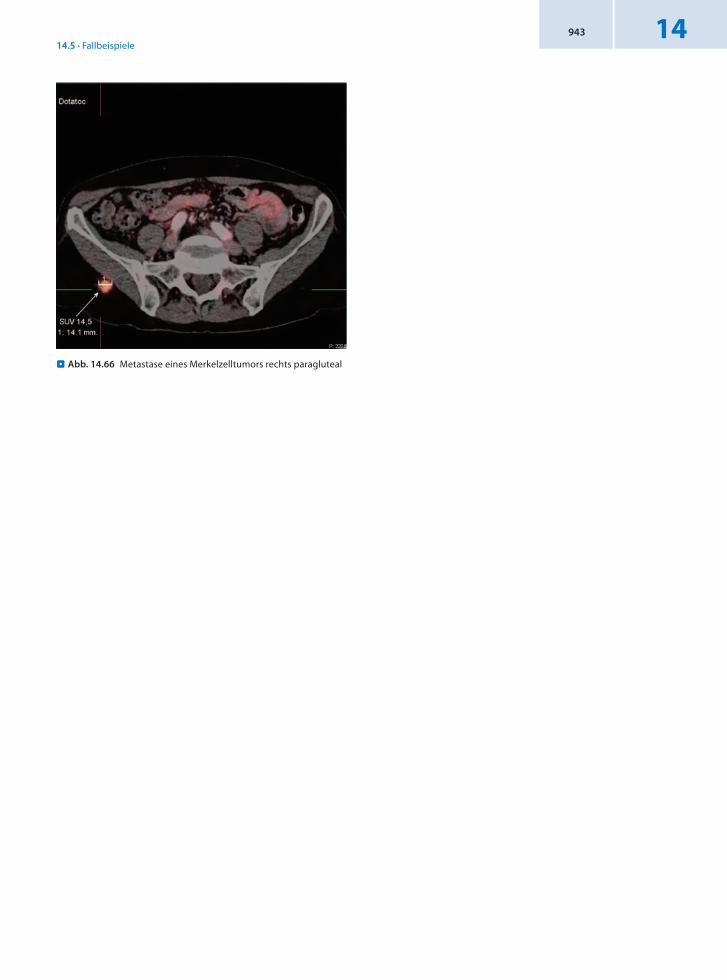
14.5.22 Patient 22 – Merkelzelltumor
Anamnese 62jährige Patientin mit Erstdiagnose eines Merkelzellkarzinoms der Nase (Nasenrücken), Stadium II nach PE vor 2 Jahren. Exzision mit R1Resektion. Nachweis von 2 zytologisch als befallen gesicherten submentalen rechts und links gelegenen Lymphknoten. Daraufhin NeckDissection I–III beidseits bei Lymphknotenmetastasen Regio I–II zervikal beidseits, teils kapselüberschreitend. Bis vor eineinhalb Jahren wurde eine Strahlentherapie durchgeführt. Vor 6 Wochen Entfernung eines unklaren Tumors im rechten Kieferwinkel retroaurikulär,
histologisch passend zu einem Merkelzellkarzinom, DD Metastase eines neuroendokrinen Tumors anderer Organlokalisationen.
Befund Nachweis einer nodulären Struktur rechts paragluteal mit malignomtypischem 68GaDOTATOCMetabolismus im Sinn einer weiteren Absiedlung eines Merkelzellkarzinoms.
Schlussfolgerung Nachweis einer Absiedlung des Merkelzelltumors in der 68GaDOTATOCPET/CT.
14.5 · Fallbeispiele14943
. Abb. 14.66 Metastase eines Merkelzelltumors rechts paragluteal

14
944 Kapitel 14 · Neuroendokrine Tumoren
14.6 Literatur
1. Modlin IM, Lye KD, Kidd M (2003) A 5-decade analysis of 13,715 carcinoid tumors. Cancer 97: 934–959
2. Yao JC, Hassan M et al. (2008) One hundred years after »carci-noid«: epidemiology of and prognostic factors for neuroendo-crine tumors in 35,825 cases in the United States. J Clin Oncol 26(18): 3063–3072
3. Rindi G, Klöppel G, Ahlmann H et al. (2006) TNM staging of fore-gut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 449(4): 395–401
4. Modlin IM, Gustafsson BI, Moss SF, Pavel M et al. (2010) Chromo-granin A--biological function and clinical utility in neuro endo-crine tumor disease. Ann Surg Oncol 17: 2427–2443
5. Gabriel M, Decristoforo C, Kendler D et al. (2007) 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with soma-tostatin receptor scintigraphy and CT. J Nucl Med 48: 508–18
6. Kann PH, Balakina E, Ivan D et al. (2006) Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Endocr Relat Cancer 13(4): 1195–1202
7. Sundin A, Vullierme MP, Kaltsas G et al. (2009) ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological examinations. Neuroendocrinology 90(2): 167–183
8. Eriksson B, Klöppel G, Krenning E et al. (2008) Consensus guide-lines for the management of patients with digestive neuroendo-crine tumors – well-differentiated jejunal-ileal tumor/carcinoma. Neuroendocrinology 87(1): 8–19
9. Modlin M, Pavel M, Kidd M, Gustadsson BI (2010) Review article: somatostatin analogues in the treatment of gastroenteropancre-atic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 31(2): 169–188
10. Steinmüller T, Kianmanesh R, Falconi M et al. (2008) Consensus guidelines for the management of patients with liver metastases from digestive (neuro)endocrine tumors: foregut, midgut, hind-gut, and unknown primary. Neuroendocrinology 87(1): 47–62
11. Kwekkeboom DJ, de Herder WW, van Eijck CHJ, Kam BL, van Essen M, Teunissen JJM, Krenning EP (2010) Peptide receptor radionuclide therapy in patients with gastroenteropancreatic neuroendocrine tumors. Semin Nucl Med 40(2): 78–88
12. Halfdanarson TR et al. (2008) Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol 19(10): 1727–1733
13. Zhuge Y et al. (2009) Pediatric intestinal foregut and small bowel solid tumors: a review of 105 cases. J Surg Res 156(1): 95–102
14. Rufini V, Calcagni ML, Baum RP (2006) Imaging of neuroendo-crine tumors. Semin Nucl Med 36(3): 228–247
15. Machens A. et al. (2007) Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multi-centre study of 258 gene carriers. Clin Endocrinol (Oxf ) 67(4): 613–622
16. Marini F et al. (2006) Multiple endocrine neoplasia type 1. Or-phanet J Rare Dis 1: 38
17. Pieterman CR et al. (2009) Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf ) 70(4): 575–581
18. Tessonnier L, Sebag F, Ghander C et al. (2010) Limited value of 18F-F-DOPA PET to localize pancreatic insulin-secreting tumors in adults with hyperinsulinemic hypoglycemia. J Clin Endocrinol Metab 95(1): 303–307
19. Kapoor RR et al. (2009) Hyperinsulinaemic hypoglycaemia. Arch Dis Child 94(6): 450–457
20. Hussain K et al. (2007) Hyperinsulinaemic hypoglycaemia: bio-chemical basis and the importance of maintaining normoglycae-mia during management. Arch Dis Child 92(7): 568–570
21. Hussain K (2007) Insights in congenital hyperinsulinism. Endocr Dev 11: 106–121
22. Pieterman CR et al. (2009) Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf ) 70(4): 575–581
23. Kuijpers SC, Noordam C, Boelen C, Wijnen R (2004) [Congenital hyperinsulinism in 15 infants, 1981-1999; experiences and new insights]. Ned Tijdschr Geneeskd 148(3): 140–143
24. Meissner T et al. (2003) Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol 149(1): 43–51
25. Kapoor RR, James C, Hussain K (2009) Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab 5(2): 101–112
26. James C et al. (2009) The genetic basis of congenital hyperinsu-linism. J Med Genet 46(5): 289–299
27. De León DD, Stanley CA (2007) Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab 3(1): 57–68
28. Giurgea I et al. (2006) The Knudson‹s two-hit model and timing of somatic mutation may account for the phenotypic diversity of focal congenital hyperinsulinism. J Clin Endocrinol Metab 91(10): 4118–4123
29. Suchi M, MacMullen CM, Thornton PS et al. (2006) Molecular and immunohistochemical analyses of the focal form of congenital hyperinsulinism. Mod Pathol 19(1): 122–129
30. Sempoux C et al. (2004) Focal and diffuse forms of congenital hy-perinsulinism: the keys for differential diagnosis. Endocr Pathol 15(3): 241–246
31. Capito C et al. (2009) Value of 18F-fluoro-L-dopa PET in the pre-operative localization of focal lesions in congenital hyperinsulin-ism. Radiology 253(1): 216–222
32. Mohnike K et al. (2008) [18F]-DOPA positron emission tomogra-phy for preoperative localization in congenital hyperinsulinism. Horm Res 70(2): 65–72
33. Barthlen W et al. (2008) Evaluation of [18F]fluoro-L-DOPA posi-tron emission tomography-computed tomography for surgery in focal congenital hyperinsulinism. J Clin Endocrinol Metab 93(3): 869–875
34. Kapoor RR, Gilbert C, Mohnike K, Blankenstein O et al. (2008) Congenital hyperinsulinism: [18F]DOPA PET/CT scan of a focal lesion in the head of the pancreas. Arch Dis Child Fetal Neonatal Ed 93(2): F166
35. Ribeiro MJ, Boddaert N et al. (2007) The added value of [18F]fluoro-L-DOPA PET in the diagnosis of hyperinsulinism of infancy: a retro-spective study involving 49 children. Eur J Nucl Med Mol Imaging 34(12): 2120–2128
36. Ribeiro MJ, Boddaert N et al. (2007) Functional imaging of the pancreas: the role of [18F]fluoro-L-DOPA PET in the diagnosis of hyperinsulinism of infancy. Endocr Dev 12: 55–66
37. Mohnike K, Blankenstein O et al. (2006) Proposal for a standard-ized protocol for 18F-DOPA-PET (PET/CT) in congenital hyperin-sulinism. Horm Res 66(1): 40–42
38. Otonkoski T et al. (2006) Noninvasive diagnosis of focal hyperin-sulinism of infancy with [18F]-DOPA positron emission tomogra-phy. Diabetes 55(1): 13–18

![Neuroendokrine Tumoren - Charakterisierung der Rolle von ... · WHO-Klassifikation [Solcia E. et al, International Histological Classification of Tumours 2000], basierend auf der](https://static.fdokument.com/doc/165x107/5e05e79ca4943d5ab916f9c9/neuroendokrine-tumoren-charakterisierung-der-rolle-von-who-klassifikation.jpg)
















