Neuroprotektiver Effekt von Ribavirin bei Borna Disease ... · abhängigen RNA-Polymerase von...
125
Aus dem Institut für Virologie und Immunbiologie der Universität Würzburg Lehrstuhl für Virologie Vorstand: Professor Dr. med. A. Rethwilm Neuroprotektiver Effekt von Ribavirin bei Borna Disease Virus infizierten Lewis Ratten Inaugural – Dissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Bayerischen Julius-Maximilians-Universität zu Würzburg vorgelegt von Robert Schlaberg aus Konstanz Würzburg, Dezember 2003
Transcript of Neuroprotektiver Effekt von Ribavirin bei Borna Disease ... · abhängigen RNA-Polymerase von...

Aus dem Institut für Virologie und Immunbiologie
der Universität Würzburg
Lehrstuhl für Virologie
Vorstand: Professor Dr. med. A. Rethwilm
Neuroprotektiver Effekt von Ribavirin bei Borna Disease Virus infizierten Lewis Ratten
Inaugural – Dissertation
zur Erlangung der Doktorwürde der
Medizinischen Fakultät
der
Bayerischen Julius-Maximilians-Universität zu Würzburg
vorgelegt von
Robert Schlaberg
aus Konstanz
Würzburg, Dezember 2003

Referent: Professor Dr. med. V. ter Meulen
Koreferent: Professor Dr. med. P. Rieckmann
Dekan: Professor Dr. med. S. Silbernagl
Tag der mündlichen Prüfung: 09.07.2004
Der Promovend ist Arzt.

Inhaltsverzeichnis
Inhaltsverzeichnis 1 Einleitung ............................................................................................................... 1
1.1 Borna Disease Virus (BDV) ............................................................................ 1 1.1.1 Klassifikation................................................................................................ 1 1.1.2 Struktur und allgemeine Eigenschaften ....................................................... 2 1.1.3 Genom ......................................................................................................... 2 1.1.4 Replikation und Transkription ...................................................................... 4 1.1.5 Proteine ....................................................................................................... 5 1.1.6 Wirtsspektrum.............................................................................................. 6
1.2 Bornasche Krankheit (Borna Disease, BD)..................................................... 7 1.2.1 Geschichte................................................................................................... 7 1.2.2 Krankheitsverlauf und Klinik natürlicher Infektionen .................................... 7 1.2.3 Viruslast ....................................................................................................... 8 1.2.4 Reservoir und Übertragungsweg ................................................................. 8
1.3 Experimentelles Modell für BD: adulte Lewis Ratte ........................................ 8 1.3.1 Klinik ............................................................................................................ 8 1.3.2 Pathogenese und Histopathologie............................................................... 9 1.3.3 Zelluläre Immunantwort ............................................................................. 11 1.3.4 Humorale Immunantwort ........................................................................... 12 1.3.5 Zytokinprofile ............................................................................................. 12 1.3.6 Beeinflussung neuronaler Funktionen ....................................................... 13 1.3.7 Diagnostik und Virusnachweis................................................................... 13 1.3.8 Therapieversuche ...................................................................................... 14
1.4 Ribavirin ........................................................................................................ 16 1.4.1 Chemische Struktur ................................................................................... 16 1.4.2 Wirkung und Wirkmechanismen ................................................................ 16 1.4.3 Wirkspektrum............................................................................................. 16 1.4.4 Pharmakokinetik ........................................................................................ 17 1.4.5 Nebenwirkungen........................................................................................ 18
1.5 Fragestellung und Design der Studie............................................................ 19 2 Materialien............................................................................................................ 21
2.1 Verbrauchsmaterialien .................................................................................. 21 2.2 Chemikalien .................................................................................................. 22 2.3 Lösungen und Puffer..................................................................................... 24 2.4 Radiochemikalien.......................................................................................... 27

Inhaltsverzeichnis
2.5 Kits ................................................................................................................ 27 2.6 Nukleotide, Primer und Plasmide.................................................................. 28 2.7 Enzyme und Proteine.................................................................................... 28 2.8 Sera und Antikörper ...................................................................................... 28 2.9 Zelllinien........................................................................................................ 29 2.10 Virus.............................................................................................................. 29 2.11 Versuchstiere ................................................................................................ 29 2.12 Geräte ........................................................................................................... 30 2.13 Software........................................................................................................ 32
3 Methoden.............................................................................................................. 33 3.1 Toxikologie und Pharmakokinetik ................................................................. 33
3.1.1 Randomisierung......................................................................................... 33 3.1.2 Körpergewicht und Überlebensrate ........................................................... 33 3.1.3 Wirkstoffzubereitung .................................................................................. 33 3.1.4 Intrazerebroventrikuläre Injektion .............................................................. 33 3.1.5 Gewebepräparation ................................................................................... 36 3.1.6 Konzentrationsbestimmung – RIA ............................................................. 36 3.1.7 Objektträger-Vorbereitung ......................................................................... 36 3.1.8 Gefrierschnitte ........................................................................................... 37 3.1.9 HE Färbung ............................................................................................... 37 3.1.10 Gram-Färbung ........................................................................................... 37
3.2 Randomisierung, Infektion und Applikation................................................... 37 3.2.1 Virusinokulation ......................................................................................... 37 3.2.2 Randomisierung......................................................................................... 38 3.2.3 Ribavirinapplikation.................................................................................... 38
3.3 Bestimmung klinischer Parameter ................................................................ 38 3.3.1 Körpergewicht............................................................................................ 38 3.3.2 Verhalten und klinische Scores ................................................................. 38
3.4 Bestimmung viraler Parameter ..................................................................... 39 3.4.1 Gewebspräparation ................................................................................... 39 3.4.2 RNase freie Bedingungen.......................................................................... 40 3.4.3 Gefrierschnitt ............................................................................................. 40 3.4.4 In situ Hybridisierung ................................................................................. 41 3.4.5 Quantitative Taqman PCR......................................................................... 47 3.4.6 Virustitrierung............................................................................................. 50

Inhaltsverzeichnis
3.4.7 Western Immunoblot.................................................................................. 51 3.5 Bestimmung immunologischer Parameter .................................................... 54
3.5.1 Gewebspräparation und Gefrierschnitt ...................................................... 54 3.5.2 Immunhistochemie (IHC) ........................................................................... 54 3.5.3 Mikrophotographie ..................................................................................... 55 3.5.4 ELISA......................................................................................................... 56 3.5.5 RNase Protektionsassay (RPA)................................................................. 56
4 Ergebnisse ........................................................................................................... 60 4.1 Toxikologie und Pharmakokinetik ................................................................. 60
4.1.1 Überlebensrate .......................................................................................... 60 4.1.2 Körpergewicht............................................................................................ 61 4.1.3 Gewebespiegel von Ribavirin .................................................................... 62 4.1.4 HE Färbung ............................................................................................... 63 4.1.5 Gram Färbung ........................................................................................... 63
4.2 Therapeutische Wirksamkeit - klinische Parameter ...................................... 63 4.2.1 Körpergewicht............................................................................................ 63 4.2.2 Verhalten und klinische Scores ................................................................. 65
4.3 Therapeutische Wirksamkeit - virale Parameter ........................................... 67 4.3.1 Quantität und Verteilung viraler RNA – in situ Hybridisierung ................... 67 4.3.2 Quantitative Taqman PCR......................................................................... 71 4.3.3 Virustitrierung............................................................................................. 72 4.3.4 Western Immunoblot.................................................................................. 73
4.4 Therapeutische Wirksamkeit - immunologische Parameter.......................... 74 4.4.1 Immunhistochemie..................................................................................... 74 4.4.2 RNase Protektions Assay (RPA) ............................................................... 81 4.4.3 ELISA......................................................................................................... 82
5 Diskussion ........................................................................................................... 83 5.1 Toxikologie und Pharmakokinetik ................................................................. 83 5.2 Virostatischer Effekt ...................................................................................... 87 5.3 Immunmodulatorischer Effekt (Neuroprotektion) .......................................... 91
6 Zusammenfassung.............................................................................................. 99 7 Abkürzungen...................................................................................................... 101 8 Literaturangaben ............................................................................................... 103

1- Einleitung
1
1 Einleitung
1.1 Borna Disease Virus (BDV) 1.1.1 Klassifikation
Das Borna Disease Virus (BDV) ist Mitglied der Ordnung Mononegavirales und weist
eine einsträngige, unsegmentierte RNA negativer Polarität auf. Die Organisation des
viralen Genoms entspricht im wesentlichen der anderer Vertreter seiner Ordnung. Vor
wenigen Jahren ist BDV daher als bisher einziger Vertreter des Genus Bornavirus
innerhalb der taxonomisch neuen Familie Bornaviridae klassifiziert worden. Grund
hierfür ist die Kombination einer Vielzahl spezieller Replikationsstrategien, die nukleäre
Lokalisation von Transkription und Replikation - einzigartig unter den tierpathogenen
Vertretern der Ordnung Mononegavirales - sowie das deutlich kürzere und kompaktere
Genom.
Ordnung Familie Unterfamilie Genus Spezies Mononegavirales
Bornaviridae Bornavirus Borna Disease Virus (BDV) Filoviridae "Ebola-like Viruses" Ebola Virus "Marburg-like Viruses" Marburg Virus Paramyxoviridae Paramyxovirinae Respirovirus Human Parainfluenzavirus 1 Morbillivirus Masern Virus Rubulavirus Mumps Virus Pneumovirinae Pneumovirus Human Respiratory Syncytial Virus (RSV) Metapneumovirus Turkey Rhinotracheitis Virus Rhabdoviridae Vesiculovirus Vesicular Stomatitis Indiana Virus Lyssavirus Rabies Virus Ephemerovirus Bovine Ephemeral Fever Virus Novirhabdovirus Infectious Haematopoetic Necrosis Virus Cytorhabdovirus Lettuce Necrotic Yellows Virus Nucleorhabdovirus Potato Yellow Dwarf Virus
Tabelle 1. Übersicht über die taxonomische Zuordnung von BDV (nach [1]).
1- Einleitung
2
1.1.2 Struktur und allgemeine Eigenschaften
Frühe Filtrationsstudien, die Hinweise auf eine virale Ätiologie der Bornaschen
Krankheit gaben, deuteten auf einen Durchmesser des Erregers von 85-125 nm hin [2,
3]. Elektronenmikroskopisch stellt sich BDV als sphärisches Partikel mit 90 nm
Durchmesser dar [4, 5]. Das Molekulargewicht des Virions ist nicht bekannt, seine
Sedimentationsdichte beträgt 1,16-1,22 g/cm3 in CsCl, 1,22 g/cm3 in Sukrose und 1,13
g/cm3 in Renografin. Bei 37°C besitzt es eine große Umweltstabilität und verliert bei
24stündiger Seruminkubation nur minimal an Infektiosität. Dagegen lässt sich das
Virion durch Erhitzen auf 56°C, saures Milieu (pH 5), organische Lösungsmittel,
Detergentien, Chlor, Formaldehyd und UV-Strahlung rasch inaktivieren [6]. BDV
zeichnet sich durch einen ausgeprägten Neurotropismus und einen nicht-lytischen
Replikationszyklus aus, der in viraler Persistenz mündet.
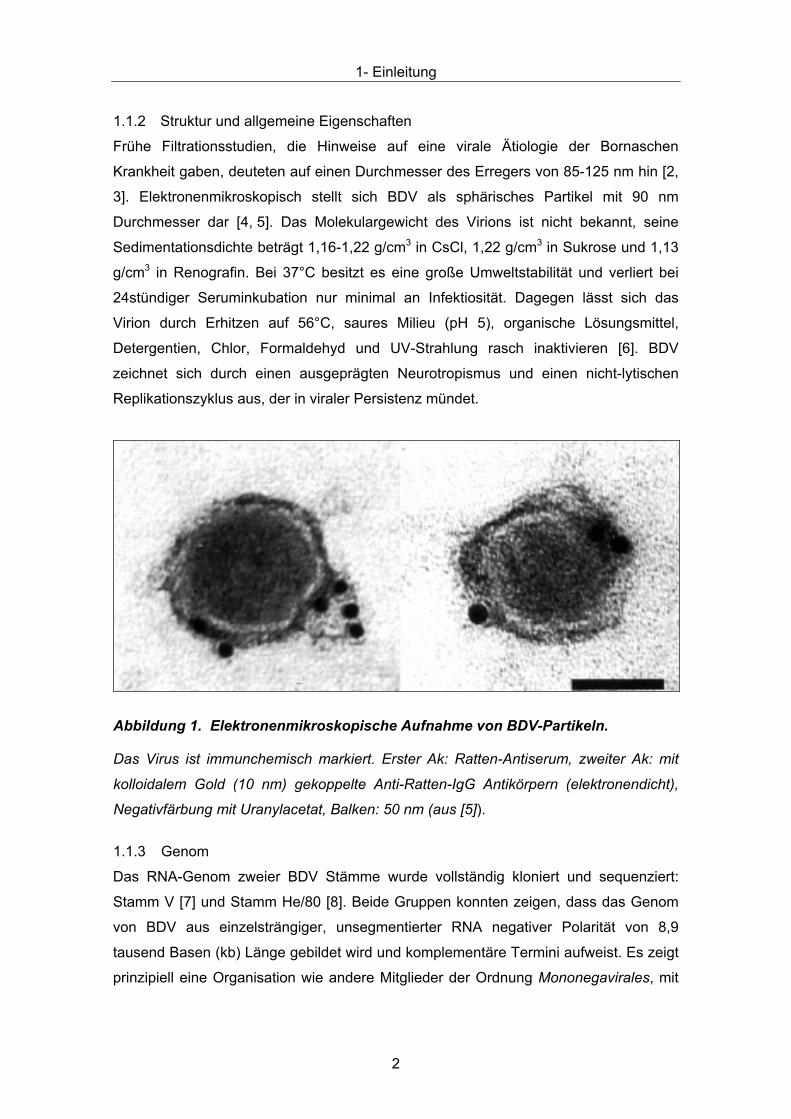
Abbildung 1. Elektronenmikroskopische Aufnahme von BDV-Partikeln.
Das Virus ist immunchemisch markiert. Erster Ak: Ratten-Antiserum, zweiter Ak: mit
kolloidalem Gold (10 nm) gekoppelte Anti-Ratten-IgG Antikörpern (elektronendicht),
Negativfärbung mit Uranylacetat, Balken: 50 nm (aus [5]).
1.1.3 Genom
Das RNA-Genom zweier BDV Stämme wurde vollständig kloniert und sequenziert:
Stamm V [7] und Stamm He/80 [8]. Beide Gruppen konnten zeigen, dass das Genom
von BDV aus einzelsträngiger, unsegmentierter RNA negativer Polarität von 8,9
tausend Basen (kb) Länge gebildet wird und komplementäre Termini aufweist. Es zeigt
prinzipiell eine Organisation wie andere Mitglieder der Ordnung Mononegavirales, mit

1- Einleitung
3
nur 8,9 kb ist es jedoch deutlich kompakter als das der Rhabdoviridae (etwa 11-15 kb),
Paramyxoviridae (etwa 16 kb) oder Filoviridae (etwa 19 kb). Ermöglicht wird dies durch
den Gebrauch einer Reihe von Transkriptions- und Replikationsstrategien, wie sie in
ihrer Kombination einzigartig innerhalb der Ordnung ist. Dazu zählen Überlappung von
Transkriptionseinheiten, Transkriptionssignalen und Open Reading Frames (ORFs),
unterschiedlicher Gebrauch von Translationsstart- und -stopsignalen, sowie RNA-
Splicing [9].
Die sechs großen ORFs von BDV sind in 3 Transkriptionseinheiten angeordnet
(s. Abbildung 2). Nur die erste Transkriptionseinheit ist monocistronisch und codiert für
das Nukleoprotein (N), von dem die zwei Isoformen p40 und p38 exprimiert werden.
Auf der bicistronischen zweiten Transkriptionseinheit werden die Proteine X (p10) und
das Phosphoprotein (P, p23) von überlappenden ORFs exprimiert. Das Matrixprotein
(M, atypisches Glykoprotein, gp 18), das glykosylierte Membranprotein (G, Typ 1
Membranprotein, p57, gp94) und die RNA abhängige RNA-Polymerase (L, p190)
werden von der dritten Transkriptionseinheit exprimiert. Dabei werden wie erwähnt
RNA-Splicing, Überlesen von Transkriptionsstops sowie unterschiedliche Transkrip-
tionsstarts ausgenutzt [9, 10]. Die Variation des Leserasters ist eine zusätzliche
Strategie zur Expression der sechs viralen Proteine von dem kurzen Genom. Die ORFs
von N, P, M and L befinden sich im selben Leseraster, während X im +2/-1 und G im
+1/-2 Raster kodiert sind [7].
Eine geringe genetische Variabilität mit einer Übereinstimmung von circa 95% auf
Nukleotidniveau zwischen den beiden Stämmen V und He/80 ist ein weiteres Merkmal
von BDV [7, 8].
Kürzlich wurde ein potentiell neuer Subtyp von BDV sequenziert, der sich von den
beiden genannten Stämmen auf Nukleotidebene um mehr als 15% unterscheidet [11].
Abbildung 2. Schematische Darstellung der relativen Anordnung der 6 ORF (aus [12])
Startcodons (S1-4) und Terminationssignale (E1-4) werden unterschiedlich kombiniert.

1- Einleitung
4
1.1.4 Replikation und Transkription
Als einziger tierpathogener Vertreter der Ordnung Mononegavirales wird BDV im
Zellkern infizierter Zellen repliziert und transkribiert [13-14]. Diese Eigenschaft hat BDV
mit den pflanzenpathogenen Nukleorhabdoviridae (Ordnung Mononegavirales) [15]
und segmentierten negativ-strängigen RNA Viren (Familie Orthomyxoviridae) gemein.
Dabei nutzt BDV wie bereits erwähnt eine nicht-lytische Replikationsstrategie.
Replikation und Transkription werden bei anderen Vertretern der Ordnung
Mononegavirales von Promotoren im Bereicht der 3’- und 5’-untranskribierten Nukleo-
tide initiiert. Auch von den 42 bzw. 54 untranskribierte Nukleotide des 3’-, bzw. 5’-
Terminus des BDV Genoms wird angenommen, dass sie diese Funktion besitzen [9].
Mindestens sechs vollständig polyadenylierte und mit 5’-Cap modifizierte mRNA
Transkripte von 0,8 kb, 1,2 kb, 1,9 kb, 2,8 kb, 3,5 kb und 7,1 kb Länge werden von
BDV in einem von 3’ nach 5’ abnehmenden Verhältnis exprimiert. Dies resultiert in
einem niedrigen Expressionslevel der viralen Polymerase, dem am weitesten 5’
lokalisierten ORF [9]. Durch alternatives Splicing dreier Introns mittels zellulärer
Enzyme werden die 2,8 kb sowie die 7,1 kb mRNA posttranskriptionell verändert und
die Expression der viralen Proteine M, G und L ermöglicht und reguliert [16-18]. Das
Splicing von mRNA kommt innerhalb der Ordnung Mononegavirales nur bei BDV vor.
Durch die Kombination dieser unterschiedlichen Replikationsstrategien ist eine
Regulation auf mehreren Stufen der Genexpression möglich. Möglicherweise steht das
in ursächlichem Zusammenhang mit Neurotropismus und Persistenz.
Abbildung 3. Transkriptionskarte von BDV (aus [12]).
Transkription und posttranskriptionelles Splicing der mRNA.

1- Einleitung
5
1.1.5 Proteine
Die auf drei Transkriptionseinheiten verteilten ORFs von BDV kodieren für sechs
Proteine [9, 10]:
Auf der ersten Transkriptionseinheit wird ein Protein von 40 kDa kodiert. Obwohl ein
detaillierter Nachweis noch aussteht, wird es aufgrund seiner Position, Größe und
hohen Konzentration als virales Nukleoprotein (N) eingeteilt. Es kommt in zwei
Isoformen vor (p38 und p40), die beide im nichtinfektiösen Überstand perforierter,
infizierter Zellen als Komplex mit P gefunden werden. Dieser wird als lösliches Antigen
(S-Antigen) bezeichnet.
Die bicistronische zweite Transkriptionseinheit kodiert für die Proteine X (p10) und P
(p23). Für X wird die Funktion eines Kernexportproteins diskutiert, während P
voraussichtlich als Kofaktor für die virale Transkription und Replikation dient. P wird auf
komplexer Weise hauptsächlich durch den Isotyp ε der zellulären Proteinkinase C
(PKCε) und in geringerem Maße durch die Casein Kinase II (CK II) phosphoryliert und
bildet Komplexe mit anderen viralen Proteinen. Interessanterweise findet man in
Regionen mit hohen Konzentrationen der PKCε auch eine hohe Viruslast [9].
Nach posttranskriptionellem Splicing des dritten Transkripts werden die drei Proteine M
(gp18), G (gp97) und L (p190) synthetisiert:
Das atypische Glykoprotein M formt in vitro stabile Tetramere und besitzt
hydrophobische Sequenzen, wie sie für Transmembranproteine charakteristisch sind.
Die Tatsachen, dass Antisera gegen M neutralisierende Funktion besitzen und
Inkubation empfänglicher Zellen mit M eine anschließende Infektion zu verhindern
vermag, sprechen für die Lokalisation von M auf der Virusoberfläche und eine Funktion
als Matrixprotein [9].
Das Typ 1 Membranprotein G (57 kDa) wird posttranslationell N-glykosyliert (p97) und
anschließend durch die zelluläre Protease Furin gespalten. Sowohl das ungeschnittene
Protein wie auch ein C-terminales Fragment von 43kDa (gp43) werden in das Virion
integriert. Nur gp43 wird anschließend in die Zellmembran transloziert [12]. Beide, G
und gp43 sind wahrscheinlich für frühe Schritte des Adsorption bzw. Penetration
bedeutend [9].
Der größte ORF auf dem Genom von BDV, der in homologer Lokalisation zu den RNA
abhängigen RNA-Polymerase von Rhabdoviridae, Paramyxoviridae and Filoviridae
lokalisiert ist, wird posttranskriptionell gespliced und dient anschließend der Synthese
der viralen Polymerase (L, p190) [10].

1- Einleitung
6
1.1.6 Wirtsspektrum
Natürliche Infektionen. Klassischerweise sind von der Infektion mit BDV Pferde,
Rinder und Schafe vor allem in Endemiegebieten Südostdeutschlands, sowie im
Bereich des oberen Rheintals in Regionen Österreichs, der Schweiz und des
Fürstentums Liechtenstein betroffen. Hier wird die Infektion jährlich bei annähernd 100
Pferden und 100 Schafen diagnostiziert [19].
In letzter Zeit häufen sich Berichte von natürlich Infektionen in anderen Teilen Europas
(Schweden [20], Österreich [21]) sowie in Nordamerika (USA [22]) und Asien (Iran [23],
Japan [24-26]). Zusätzlich zu den klassischen Wirten werden Infektionen von
Kaninchen [27], Straußen [28], Ziegen [29], Katzen [20], einem Hund [30], sowie einem
Luchs [31] beschrieben. Es muss also sowohl eine weltweite Ausbreitung, wie auch ein
weiteres Wirtsspektrum angenommen werden. Ob für die zunehmenden Berichte eine
Ausbreitung von BDV oder verbesserte diagnostische Möglichkeiten verantwortlich
sind, ist unklar.
Experimentelle Wirte. Das Spektrum experimentell infizierbarer Spezies reicht von
Vögeln bis zu Primaten [19]. Man geht davon aus, dass die meisten, wenn nicht alle
Warmblüter potentielle Wirte für BDV darstellen. Krankheitsverlauf und Inkubationszeit
unterliegen dabei einer großen Schwankung, die von Virusstamm, Wirtsspektrum,
Immunstatus des Wirts und Infektionsweg abhängig sind [12]. Das am besten studierte
Tiermodell ist die Ratte.
Humane Infektionen. Das breite Wirtsspektrum einschließlich nicht-humaner
Primaten, die Infizierbarkeit menschlicher Zellen in Kultur und Parallelen einiger
Aspekte klinischer Symptome bei infizierten Tieren zu menschlichen Erkrankungen wie
uni- und bipolare affektive Störungen, Schizophrenie und Autismus haben zu
Spekulationen über eine ätiologische Beteiligung von BDV an den genannten
Erkrankungen geführt. Des weiteren wird von reaktiven Antikörpertests und
molekularbiologischem Nachweis viraler Nukleinsäure mit erhöhter Prävalenz bei
neuropsychiatrischen Patienten, sowie von humanen Virusisolaten berichtet [9, 33, 34].
Die niedrig produktive Replikation von BDV, sein ausgesprochener Neurotropismus,
die Anfälligkeit hochsensitiver Nachweisverfahren gegenüber Kontamination und der
Mangel an einheitlichen Verfahren für eine Diagnose intra vitam sind Hindernisse, die
momentan die Interpretation der Ergebnisse und Beantwortung dieser Frage
erschweren. Ein Konsens über eine prinzipielle Suszeptibilität des Menschen scheint
zu bestehen, eine ätiologische Beteiligung dagegen ist Gegendstand kontroverser
Diskussion [9, 12, 35].

1- Einleitung
7
1.2 Bornasche Krankheit (Borna Disease, BD)
1.2.1 Geschichte
Die erste Beschreibung des Krankheitsbildes reicht zurück ins Jahr 1767 [9]. Die
zunächst als „Verrückte Kopfkrankheit der Pferde“ bekannt gewordene Erkrankung
erhielt ihren heutigen Namen nach der sächsischen Stadt Borna, wo 1895/96 einer
Epidemie die sächsische Kavallerie dezimierte [9]. Ihre Erforschung begann am Anfang
des 20. Jahrhunderts. Ernst Joest and Kurt Degen entdeckten 1909 die nach ihnen
benannte Einschlusskörperchen in Zellkernen infizierter, hippokampaler Ganglienzellen
[12]. Wilhelm Zwick gelang 1924 die Übertragung der Erkrankung auf ein Kaninchen,
indem er neuronales Gewebe eines erkrankten Pferdes intrakraniell injizierte [35].
Dadurch wurde der Verdacht auf eine infektiöse Genese bekräftigt. In
Filtrationsexperimenten ergab sich dann eine Größe des infektiösen Agens von 85 bis
125 nm, was erste Hinweise auf einen viralen Erreger lieferte [2, 3].
1.2.2 Krankheitsverlauf und Klinik natürlicher Infektionen
Die Inkubationszeit bei Pferden beträgt ca. 6 Wochen [36]. Frühe Zeichen des
Syndroms sind unspezifisch und beinhalten agitiertes oder gehemmtes Verhalten,
Fieber, Anorexie, Ikterus, Obstipation und Koliken. Ein bis zwei Wochen später treten
die klassischen Zeichen von BD auf. Dazu zählen eine aufrechte, breitbasige Haltung
mit dorsalflektiertem Kopf, repetitives Verhalten und später Retinopathie mit
reduziertem Visus [9].
In Verlaufskontrollen von Herde mit spontanen Fällen von BD konnte gezeigt werden,
dass ein asymptomatischer Trägerstatus zumindest vorübergehend existiert [12, 37].
Für die Frequenz asymptomatischer Verläufe liegen allerdings keine gesicherten Daten
vor. Neben diesen klinisch inapperenten Verläufen reicht das Spektrum der
hervorgerufenen Symptomatik von subtilen Veränderungen von Lern- und
Gedächtnisleistungen, schweren Verhaltens- und Bewegungsabnormalitäten bis hin
zur klassischen Form der potentiell letalen Meningoenzephalomyelitis mit schwerer
neurologischer Symptomatik [9]. Die Mortalität in der akuten Phase beträgt 80 bis
100% [38].
Die Krankheitsverläufe anderer natürlicher Wirte zeigen ebenfalls eine große
Schwankungsbreite von Symptome ähnlich derer von Pferden [39-41], einige Spezies
zeigen keine Symptome und bleiben unauffällige Virusträger [42, 38].

1- Einleitung
8
1.2.3 Viruslast
Hohe Virustiter werden im ZNS infizierter Pferde gemessen. Besonders betroffen sind
der Neokortex, der piriforme Kortex, Bulbus olfactorius und der Hippocampus [43-45].
1.2.4 Reservoir und Übertragungsweg
Reservoir und Übertragungsweg von BDV sind bislang unbekannt. Berichtet wurde
sowohl von horizontaler wie auch von vertikaler Übertragung [38, 39, 46]. Als
potentielles Reservoir sowie als Vektor werden Nagetiere diskutiert [20, 35]. Virale
Genprodukte wurden in Speichel, Urin und Fezes infizierter Nager gefunden [34]. Die
Infektion scheint sowohl über das olfaktorische Neuroepithel, als auch über eine orale
Route möglich zu sein [12]. Infektionen treten meist sporadisch und besonders im
Frühling auf, Epidemien sind selten [19].
1.3 Experimentelles Modell für BD: adulte Lewis Ratte 1.3.1 Klinik
Die experimentelle Infektion adulter Lewis Ratten ist das am besten charakterisierte
Modell der Bornaschen Krankheit. Lewis Ratten zeichnen sich durch eine hohe
Empfänglichkeit gegenüber BDV aus und entwickeln eine klinische Symptomatik, die
der natürlicher Infektionen ähnelt [12, 47]. Nach einer Inkubationszeit von ca. drei
Wochen (intranasale Inokulation [48]), etabliert BDV eine persistente Infektion des
ZNS, die klinisch einen biphasischen Verlauf zeigt. In der akuten Phase, die
histologisch mit einer intensiven nicht-purulenten Meningoenzephalomyelitis einher-
geht, zeigen die Tiere Hyperaktivität, Schreckhaftigkeit, aggressives Verhalten und
Desorientierung. Verhaltensabnormalitäten wie Hyperaktivität und verstärkte
Schreckhaftigkeit treten simultan mit der Nachweisbarkeit viraler RNA in limbischen
Strukturen und erster Immunzellinfiltrate im ZNS auf [34]. Zusätzlich werden hohe
Virustiter und der Untergang neuronaler Zellen in der Retina beobachtet, was zu einer
Erblindung führen kann. Der Höhepunkt der klinischen Symptomatik ist 30 bis 40 Tage
p.i. erreicht [12]. Die akute Phase weist eine erhöhte Mortalität auf [49, 50].
Eine Zwischenphase ist durch Auftreten stereotyper Verhaltensweisen gekennzeichnet
[51]. Bis zu zehn Prozent der Tiere neigen in dieser Phase zu extremen Formen von
Adipositas [6]. In der chronischen Phase fallen die Tiere dann durch Passivität,
Lethargie und Blindheit auf. Die Symptomatik der Entzündungsreaktion geht zurück,
wobei die Virustiter auf unverändert hohen Werten verbleiben [12].

1- Einleitung
9
1.3.2 Pathogenese und Histopathologie
Experimentell ist jeder Infektionsweg, bei dem das Virus Zugang zu peripher- oder
zentralnervösen Nervenendigungen erhält möglich: intramuskulär, Injektion in die
Fußsohle, intranasal, oral, intraperitoneal oder intrazerebral. Im Falle einer intranasalen
Inokulation erfolgt zunächst die Infektion von neuronalen Rezeptorzellen des
olfaktorischen Epithels. Von hier werden über den Bulbus olfactorius und die
nachfolgenden Stationen der Riechbahn große Teile des ZNS infiziert [12]. Die
Ausbreitung erfolgt höchst wahrscheinlich axonal und transsynaptisch über
vorhandene neuronale Vernetzungen [52]: Eine zentripetale Ausbreitung viraler
Proteine ist nach intranasaler, intraokulärer oder intraperitonealer Inokulation
nachweisbar [46, 48]. Die Inkubationszeit nimmt dabei mit zunehmend peripherem
Inokulationsort zu. Bei neurektomierten Ratten kann sich nach Inokulation in die
Fußsohle keine Infektion etablieren [48]. Ob BDV dabei wie das verwandte Rabies
Virus als Ribonukleoprotein-Komplex (RNP) streut ist nicht sicher. Das Fehlen reifer
Viruspartikel während der Ausbreitung von BDV, wie der Nachweis der Infektiosität von
RNP sprechen dafür [12].
Wie bereits erwähnt, zeichnet sich BDV durch einen ausgesprochenen Neurotropismus
aus. Neben neuronalen Zellen werden auch Astrozyten, Oligodendrozyten,
Schwann’sche Zellen und Ependymzellen infiziert [53, 54]. Die Infektion betrifft in der
akuten Phase das ZNS und die Retina [55, 56], später auch das PNS. In dieser Phase
kann es zur Streuung in Tränen-, Speichel- und Talgdrüsen, das Epithel der
Gesichtshaut und den Intestinaltrakt [52] kommen. In immunkompromitierten Tieren
breitet sich die Infektion über Zellen des PNS zentrifugal in andere, parenchymatöse
Organe aus [57, 58]. Auch aus peripheren mononukleären Blutzellen wurde
virusspezifische RNA amplifiziert [59, 60]. Die Viruslast bleibt dabei aber zentralnervös
stets deutlich höher als außerhalb des ZNS [61] und erreicht die höchsten Werte im
limbischen System (bes. Hippocampus und Corpora amygdaloidea), in kortikalen
Strukturen (Präfrontalkortex, enterorhinaler Kortex), dem Thalamus und dem
Cerebellum [62, 12].
Mehrere Mechanismen werden für die Neurodestruktion diskutiert: Eine wesentliche
Rolle spielen zytotoxische T-Lymphozyten. Durch die Reduktion von CD8+ T-Lympho-
zyten kann ein starker Rückgang der Neurodestruktion erreicht werden [63]. Allerdings
findet die Infiltration von CD8+ T-Lymphozyten in der akuten Phase statt, während die
stärkste Zerstörung neuronalen Gewebes erst in der chronischen Phase auftritt [64], so
dass zusätzlich andere Mechanismen eine Rolle spielen müssen.

1- Einleitung
10
Ebenfalls einen Anteil an der Pathogenese könnte Stickstoffmonoxid (NO) als toxischer
Faktor haben: mRNA Level für die induzierbare NO-Synthetase (iNOS) sind im Hirn
normaler Ratten nicht nachweisbar. In Hirnen infizierter Ratten lässt sich mRNA für
iNOS 14 Tage p.i. nachweisen, wobei maximale Werte 21 Tage p.i. gefunden werden.
Dieser Effekt korreliert zeitlich also mit dem Auftreten klinischer Symptome [65, 66].
Wie die Anwesenheit von IgG und Komponenten des Komplementsystems [64, 67]
zeigt, könnten auch diese im Rahmen einer komplementmediierten oder antikörper-
abhängigen zellmediierten Zytotoxizität an der progredienten Neurodestruktion beteiligt
sein. Unspezifisch kann es durch das entzündungsbedingte Hirnödem und den
entstehenden Hirndruck zu einer Schädigung des Gehirns kommen.
Histopathologisch ist die akute Phase der Bornaschen Erkrankung geprägt von einer
meningealen, perivaskulären sowie parenchymatösen Immunzellinfiltration, die in der
chronischen Phase regressiv verläuft und in einen progredienten Verlust von Neuronen
mündet. Die anfängliche Entzündungsreaktion ist besonders auf die graue Substanz
des Cortex, Bulbus olfactorius, Thalamus, Hippocampus und der Basalganglien
konzentriert. Das zelluläre Infiltrat wird dabei vorwiegend von CD4+ und CD8+ T-
Lymphozyten, natürlichen Killerzellen (NK Zellen) und Makrophagen gebildet, wobei
CD4+ T-Lymphozyten gegenüber CD8+ überwiegen [68]. CD4+ T-Lymphozyten sind
fast ausschließlich perivaskulär und nur in geringer Zahl im Hirnparenchym
anzutreffen. CD8+ T-Lymphozyten dagegen finden sich in der akuten Phase
gleichermaßen perivaskulär wie parenchymal [68, 69].
Zusätzlich ist morphologisch eine Aktivierung von Mikroglia und Astrozyten zu
beobachtet, die besonders prominent in Nachbarschaft von Immunzellinfiltraten ist [53,
64]. Bei der Retinopathie BDV infizierter Ratten spielen Mikroglia wie Makrophagen
eine Rolle. Hier konnte gezeigt werden, dass beide Zelltypen phagozytotische Aktivität
gegenüber neuronalen Zellen besitzen [50]. Eine BDV bedingte Mikrogliose konnte bei
neonatal infizierten Lewis Ratten beobachtet werden [70-72]. Sie findet sich
kolokalisiert mit neuropathogene Foci und scheint kausal an der Pathogenese beteiligt
zu sein [70], potentiell durch die Synthese proinflammatorischer Zytokine [72].
Zusätzlich fällt eine Expression von CD4 und CD8 Molekülen auf der Oberfläche
aktivierter Mikroglia auf [70]. Ein funktionelles Korrelat ist jedoch noch nicht bekannt.
In infizierten Ganglienzellen fanden Ernst Joest and Kurt Degen intranukleäre
Einschlusskörperchen (Joest-Degen-Einschlusskörperchen) [73], die vermutlich BDV-

1- Einleitung
11
Partikeln entsprechen [74]. Trotz der ausgeprägten Meningoenzephalomyelitis bleibt
die Blut-Hirn-Schranke unbeeinträchtigt [64, 68, 75].
Im Laufe der Erkrankung und mit dem Eintritt in die chronische Phase geht die zelluläre
Entzündungsreaktion zurück [76] während die virale Infektion persistiert. Dieser
Rückgang der zellulären Immunreaktion erfolgt simultan mit der Zunahme der Zahl von
NK Zellen, B-Lymphozyten und aktivierter Mikroglia und kann mit einem Shift von einer
Th1- zu einer Th2-betonten Immunreaktion beschrieben werden [68]. Diese
Beobachtungen stehen im Einklang mit der Veränderung des Zytokinprofils beim
Übertritt in die chronische Phase (s. 1.3.5).
In der chronische Phase verläuft die Zerstörung neuronalen Gewebes progredient und
führt zur Ausbildung eines Hydrozephalus ex vacuo [76, 77]. Der Mechanismus, mit
Hilfe dessen BDV einer Clearance durch die intensive Immunreaktion des
Wirtsorganismus entgeht ist unbekannt.
1.3.3 Zelluläre Immunantwort
Trotz der immunologisch privilegierten Stellung des ZNS induziert die Infektion im
immunkompetenten Wirt eine intensive zelluläre und humorale Immunantwort, die in
zeitlichem Zusammenhang mit dem Auftreten von Symptomen steht [43]. Für die
Pathogenese der Bornaschen Krankheit bedeutend ist die Anwesenheit einer intakten
zellulären Immunreaktion, wie Experimente mit splenektomierten Rhesusaffen und
pharmakologisch (Cyclophosphamid, Cyclosporin A) oder genetisch (homozygot
athymische Tiere) immunkompromitierten Lewis Ratten gezeigt werden konnte. Die
Tiere in allen drei Ansätzen zeigen eine verlängerte Inkubationszeit und reduzierte
Symptomatik, bzw. einen asymptomatischen Verlauf simultan mit einer persistierenden
Infektion [58, 78-81, 12].
Sowohl CD8+, als auch CD4+ T-Lymphozyten sind an der Ausprägung von BD beteiligt:
Durch den Transfer einer BDV spezifischen CD4+ T-Zellinie konnten klassische
Symptome in immunsupprimierten Ratten induziert werden [82, 83]. Dabei ist die
Infiltration von intakten CD8+ T-Lymphozyten in das ZNS für das Auftreten der
Symptomatik nötig [84]. Abwesenheit oder funktioneller Blockade von CD8+ T-
Lymphozyten resultieren in Reduktion sowohl von histopathologischen Veränderungen,
als auch von klinischer Symptomatik [12, 58, 63, 69, 85, 86].
CD8+ T-Lymphozyten aus Hirnen infizierter Ratten zeigten in Kultur eine MHC-I
abhängige, lytische Aktivität gegenüber infizierten Zellen [85]. Neuronale Zellen
exprimieren unter normalen Bedingungen kaum MHC-I Moleküle. In BDV infizierten

1- Einleitung
12
Tieren dagegen findet eine vermehrte Expression statt [12]. In das Hirnparenchym
eingewanderte CD8+ T-Lymphozyten könnten somit infizierte Neurone MHC-I abhängig
lysieren und den bei BD beobachtete Verlust neuronaler Zellen, der in der chronischen
Phase in Hirnatrophie mit Hydrozephalus ex vacuo [77] endet, auslösen. Als ein
mögliches Epitop wurde ein Fragment von N in Assoziation mit einem Ratten MHC-I
Molekül beobachtet [87].
CD4+ T-Lymphozyten spielen als Mediatoren der Entzündungsreaktion pathogenetisch
eine bedeutende Rolle [82, 83, 86]. Transferiert man virusspezifische CD4+ T-Lympho-
zyten vor der Infektion mit BDV in Lewis Ratten, so kommt es zur Clearance durch
Induktion zytotoxischer CD8+ T-Lymphozyten [88].
Zusammenfassend scheinen CD4+ T-Zellen für die Induktion der Entzündungsreaktion
essentiell zu sein, während diese ohne die Anwesenheit intakter CD8+ T-Zellen nicht zu
Neurodestruktion und Hirnatrophie mit den Folgen der BD typischen neurologischen
Symptomatik führt. Den CD8+ T-Zellen kommt damit die Bedeutung von Effektorzellen
zu, die für die progrediente Neurodestruktion verantwortlich sind.
1.3.4 Humorale Immunantwort
Erwachsene Lewis-Ratten reagieren nach der BDV Infektion mit eine intensiven
humoralen Immunantwort [89, 90]. Diese scheint allerdings zumindest für die
Auslösung der Symptome in der akuten Phase keine bedeutende Rolle zu spielen [57,
61, 81, 91]. Die hohen Antikörpertiter werden für die Restriktion der Infektion auf das
ZNS mitverantwortlich gemacht [92] und könnten eine Modulation der viralen
Genexpression bewirken [9, 90].
1.3.5 Zytokinprofile
Aktivierte CD4+ T-Lymphozyten des Subtyps Th1 produzieren proinflammatorische
Zytokine wie IFNγ, TNFα und IL2, die eine Rekrutierung und Aktivierung von
zytotoxischen T-Lymphozyten, sowie antigenpräsentierenden Zellen (APC) bewirken
und somit eine zelluläre Immunreaktion induzieren [68]. Weitere potentielle
Produzenten proinflammatorischer Zytokine sind aktivierte Makrophagen, aber auch
ortständige Zellen wie Neurone, Astrozyten und Mikroglia [12, 93]. Th2 Zellen dagegen
induzieren eine vermehrt humorale Immunreaktion über die Produktion von unter
anderem IL4 und TGFβ. Ergebnisse von “RNase Protektion Assays (RPA)“ zur

1- Einleitung
13
Bestimmung der mRNA-Level verschiedener Zytokine in Gehirnen infizierter Lewis
Ratten sprechen für einen Shift von einer Th1 dominierten Antwort in der akuten Phase
zu einer Th2 induzierten humoralen Immunantwort im späten Stadium der Erkrankung:
mRNA-Level für die Zytokine IFNγ, TNFα, IL1α, IL2 und IL6 erreichen in der akuten
Phase maximale Werte und gehen mit der Ausnahme von IFNγ in der chronischen
Phase wieder stark zurück. Dagegen bleiben mRNA Levels für IL4 und TGFβ, die einer
zellulären Immunantwort entgegenwirken, auch in der chronischen Phase erhöht, bzw.
erreichten hier ihre maximalen Werte (IL4) [68, 94].
1.3.6 Beeinflussung neuronaler Funktionen
Störungen von Motorik und Verhalten wie sie während des gesamten Verlaufs der
Bornaschen Erkrankung auftreten, werden mit Funktionsstörungen in monoaminergen
Transmittersystemen – dopaminerg wie serotoninerg – in Verbindung gebracht [51, 95-
98]. Des weiteren wird eine Beeinflussung des cholinergen Systems vermutet [99].
Zusätzlich scheint die Expression von neuromodulatorischen Substanzen, beziehungs-
weise ihre Synthese beeinflussende Hormone und Enzyme wie Somatostatin,
Cholezystokinin und Glutamatdecarboxylase in der akuten Phase stark reduziert zu
sein [100].
1.3.7 Diagnostik und Virusnachweis
Intra vitam Diagnostik natürlicher Infektionen erweist sich als wenig zuverlässig.
Anhand von BD typischen, insgesamt aber unspezifischen Verhaltensauffälligkeiten
kann ein Verdacht geäußert werden. Der Nachweis von Antikörpern in Serum und
Liquor mittels indirektem Immunofluoreszenzassay (IFA) [101] und enzyme-linked
immunosorbent assay (ELISA) [102] sind die zuverlässigsten Methoden [19]. In einigen
Studien konnte virale Nukleinsäure aus Zellen des peripheren Blutes mittels nested
reverse Transkriptase Polymerasekettenreaktion (nested RT-PCR) nachgewiesen
werden [103].
Post mortem kann zusätzlich Immunhistologie bevorzugt gegen die viralen Antigene
mit der höchsten Gewebekonzentration (Proteine N und P) durchgeführt werden.
Virusisolierung mit anschließendem Focus-Immunoassay [104], in situ Hybridisierung
sowie Nachweis viraler RNA mittels RT-PCR gelingen bei experimenteller Infektion.
(Elektronen-) Mikroskopisch können Joest-Degen-Einschlusskörperchen [73] und
mittels Gold-konjugierter Antisera spezifisch ca. 90 nm große zellfreie Viruspartikel
nachgewiesen werden [5].

1- Einleitung
14
1.3.8 Therapieversuche
Immunisierung. Aktiven Immunisierungsversuchen steht die Problematik gegenüber, dass durch eine
forcierte Immunreaktion eine Reduktion der Viruslast ermöglicht werden kann, diese
aber mit intensivierter Klinik und vermehrten neuropathologischen Läsionen einhergeht
[105]. In einer Studie gelang durch eine hochdosierte Inokulation von attenuiertem
Virus eine Induktion hoher Antikörpertiter mit Clearance einer nachfolgenden BDV
Infektion. Dieser Effekt ist dosisabhängig und führt bei zu niedriger Dosierung zu
klassischer BD und schweren neuropathologischen Veränderungen [106].
Andere Ansätze wie der Transfer von virusspezifischen CD4+ T-Lymphozyten oder die
Applikation monoklonaler Antikörper gegen CD4+ und CD8+ Moleküle haben eher
experimentellen Charakter [86, 88].
Interferon. Der antivirale Effekt von Interferon in vitro ist stark von der verwendeten Zelllinie und
einem frühen Behandlungsbeginn abhängig [107, 108].
Immunsuppressiva. Der positive Effekt von Immunsuppressiva wie Cyclosporin A, Cyclophosphamid und
Corticoiden auf die Pathologie und Klinik von BD beruht auf der Reduktion der für die
Pathogenese entscheidenden Funktion des Immunsystems. Immunsupprimierte Tiere
zeigen geringere bis fehlende histopathologische Veränderungen und klinische
Symptome [49, 81, 109, 110]. Systemische Nebenwirkungen einer allgemeinen
Immunsuppression, die Notwendigkeit einer zum Zeitpunkt der Infektion vorbestehen-
den Suppression [81] und eine fehlende Beeinflussung der viralen Replikation sind die
entscheidenden Nachteile dieses Therapieansatzes.
Amantadin. Amantadin, zugelassen für die Therapie von Influenza A, zeigte einen antiviralen Effekt
in vitro sowie einen antidepressiven und antiviralen Effekt in zwei klinischen Studien
[111, 112]. Verschiedene Versuche, diese Ergebnisse in vitro und in vivo zu
reproduzieren und zu quantifizieren blieben bisher erfolglos: in vitro Ergebnisse zeigten
keinen nachweisbaren Effekt auf virale Parameter in verschiedenen Zelllinien und
Konzentrationen [113-115]. Auch in vivo gelang bei unterschiedlicher Applikationsart
und in verschiedenen Tiermodellen keine Reproduktion der positiven klinischen
Ergebnisse [113, 114].

1- Einleitung
15
Ribavirin. Zwei unabhängige Studien konnten zeigen, dass Transkription und Replikation von
BDV in persistent infizierten Zellen in vitro durch das Guanosinanalog Ribavirin
inhibiert werden kann [116, 117]. Dieser inhibitorische Effekt auf zwei verschiedene
Virusstämme besteht in drei unterschiedlichen Zelllinien bei Ribavirinkonzentrationen
von 2-200 µM [116, 117]. Virustranskripte wurden dabei zelllinien- und virusstamm-
abhängig um 60-80%, Virustiter um 90 bis 99% reduziert [116]. Die Inhibition ist
charakterisiert durch eine hohe Dynamik: ein maximaler Effekt ist bereits nach 24
Stunden beobachtbar. Nach Entfernen von Ribavirin aus dem Kulturmedium kommt es
innerhalb von zwei Tagen zu einem Anstieg von Virustiter und Transkripten auf die
Ausgangswerte [116].

1- Einleitung
16
1.4 Ribavirin
1.4.1 Chemische Struktur
Ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamid), erstmals 1972 von Robins
und Witkowski synthetisiert, ist ein Guanosinanalog, bei dem die Base des Nukleosids
verändert ist (s. ). Die farblose Reinsubstanz ist wasserlöslich und hat ein
Molekulargewicht von 244,2 [118].
Abbildung 4
Abbildung 4. Strukturformeln von Ribavirin und Guanosin (aus [118])
Ribavirin Guanosin
1.4.2 Wirkung und Wirkmechanismen
Bereits 1972 wurde erstmals ein direkter antiviraler Effekt beschrieben. Daneben ist
erst seit neuerem eine immunmodulatorische Wirkung bekannt. Zusätzlich weist
Ribavirin nach Inkorporation in virale RNA einen mutagenen Effekt auf, wobei Cytosin
und Uracil mit gleicher Effizienz als komplementäre Base dienen können.
1.4.3 Wirkspektrum
Ribavirin zeigt antiviralen Effekt gegenüber einem breiten Spektrum von RNA- und
DNA-Viren (s. ). Tabelle 2
1- Einleitung
17
Virus in vitro in vivo
(Tiermodell) klinisch
experimentell klinischer
Einsatz BDV [116, 117] RSV
[119] Baumwollratte
[120] bei schweren
Verläufe (Aerosol) [121, 122]
HCV + IFNα [144, 145, 146]
HBV Einzeltherapie [126, 127],
+ IFNα [128, 129]
West Nile Virus [130] Influenza Viren
[131] Maus
[131, 132] akute schwere
Influenza (Aerosol [133])
Parainfluenza Viren
[134] Maus [134, 135]
Kinder mit SCIS [136]
Masern Virus [120]
Baumwollratte, Hamster: SSPE
[120, 137]
+ IFNα bei 2 Kindern mit SSPE
[138]
Adenoviren [139] bei Immunsuppres-sion: haemorrha-
gische Zystitis [159, 160, 161],
Pneumonie [143], Nephritis [144]
Coxsackie A Viren
[118]
Rhinoviren [118] Coronaviren [118]
Lassa Affe [145] [146] bei frühem Therapiebeginn
[147] Hanta Virus [148] HFRS [149] Chimean-
Congo Hemorrhagic
Fever
[150]
Maus [151]
[152]
Rift Valley Maus [153] [154] HPV [155, 156]
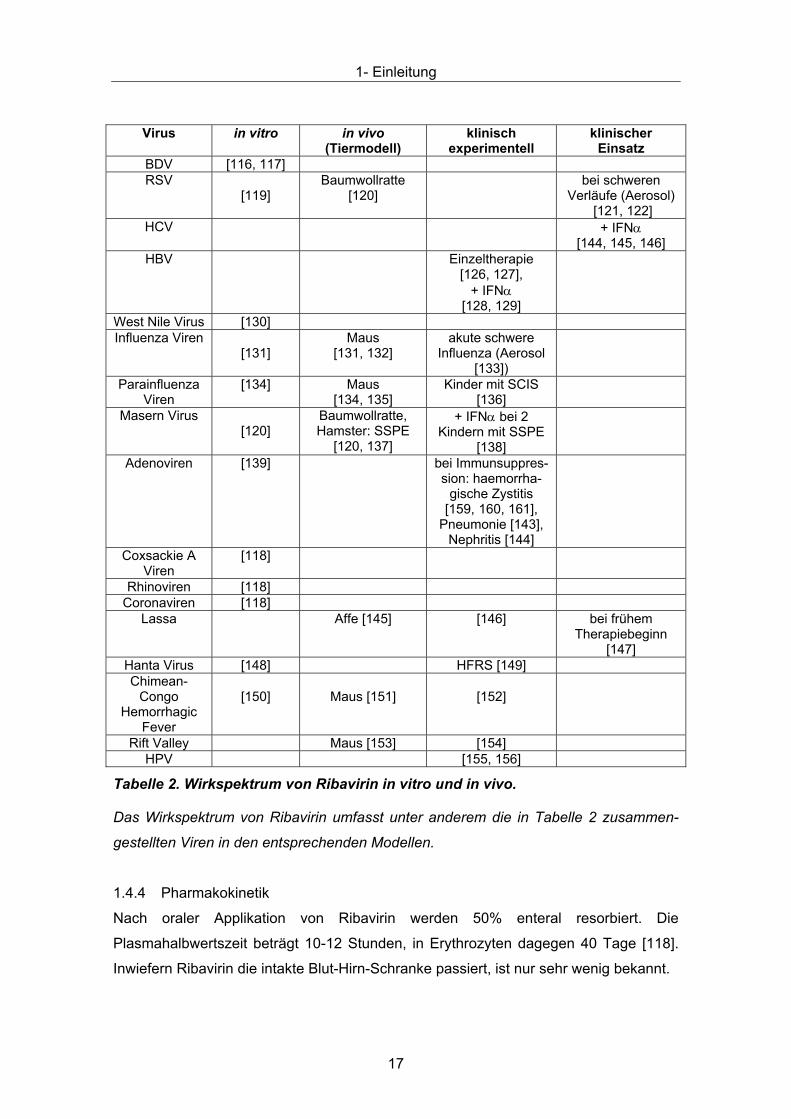
Tabelle 2. Wirkspektrum von Ribavirin in vitro und in vivo.
Tabelle 2Das Wirkspektrum von Ribavirin umfasst unter anderem die in zusammen-
gestellten Viren in den entsprechenden Modellen.
1.4.4 Pharmakokinetik
Nach oraler Applikation von Ribavirin werden 50% enteral resorbiert. Die
Plasmahalbwertszeit beträgt 10-12 Stunden, in Erythrozyten dagegen 40 Tage [118].
Inwiefern Ribavirin die intakte Blut-Hirn-Schranke passiert, ist nur sehr wenig bekannt.

1- Einleitung
18
1.4.5 Nebenwirkungen
Bei oraler oder intravenöser (i.v.) Applikation kann Ribavirin eine milde reversible
Anämie, sowie transiente Erhöhung von Serum-Bilirubinspiegeln auslösen [118]. Bei
inhalativer Therapie sind keine spezifischen unerwünschten Wirkungen bekannt. Bei
RSV infizierten Kindern wurden Bronchiospasmen, Veränderungen der Lungen-
funktion, Apnoe, Herzstillstand, Hypotension, Digitalis-Intoxikation und bei intubierten
Kindern Pneumothoraces beobachtet [118].

1- Einleitung
19
1.5 Fragestellung und Design der Studie
Weder immunmodulatorische [81, 86, 88, 105-110, 157] noch antivirale [111, 112]
Verfahren konnten bisher ein klinisch anwendbaren Therapieansatz für BD liefern. In
vitro Versuche mit Ribavirin zeigten reproduzierbare Hinweise auf einen virostatischen
Effekt gegenüber BDV [116, 117]. Neben der bereits seit Anfang der Siebzigerjahre
bekannten, virostatischen Wirkung ist seit kürzerer Zeit auch ein immunmodulato-
rischer Effekt von Ribavirin bekannt. In verschiedenen Systemen konnte ein Verschie-
ben der Balance von T-Helferzell-Subpopulationen in Richtung Th1 und somit eine
verstärkte Induktion einer zellulären Immunantwort nachgewiesen werden [158-161].
In dieser Studie soll der Frage nachgegangen werden, ob die Applikation von Ribavirin
bei einer akuten BDV Infektion ausgewachsener Lewis Ratten einen positiven Einfluss
auf den Krankheitsverlauf hat. Zusätzlich soll der Effekt von Ribavirin auf die
Virusreplikation und die Immunantwort untersucht werden. Als Tiermodell wird die
Lewis-Ratte gewählt, da das Krankheitsbild der natürlichen Infektion stark ähnelt und
es das am besten untersuchte Modell ist. Zusätzlich ist in der Lewis-Ratte die
Immunantwort nach BDV Infektion eingehend untersucht worden. Es handelte sich
hierbei um die erste Anwendung von Ribavirin bei einer BDV Infektion in vivo.
Da über die Liquorgängigkeit von Ribavirin bei intakter Blut-Hirn-Schranke nur wenig
Informationen vorliegen, wird der Wirkstoff mittels implantiertem Katheter direkt in das
Ventrikelsystem injiziert. Um zunächst Informationen über die Pharmakokinetik und
Toxizität von Ribavirin zu sammeln, wird in einem Vorversuch gesunden männlichen
Lewis-Ratten Ribavirin in Konzentrationen von 0, 1,25, 2,5, 5, 10 und 20 mg/kg
Körpergewicht appliziert und anschließend die Gewebskonzentration im ZNS
quantifiziert.
In einem anschließenden pharmakodynamischen Versuch werden Lewis-Ratten mit
BDV infiziert und die Applikation von Ribavirin nach drei Wochen, simultan mit dem
Auftreten einer BD-typischen Symptomatik [34] begonnen. Dieses zeitlich verzögerte
Vorgehen birgt das Risiko, einer zum Zeitpunkt des Therapiebeginns bereits etablierten
Infektion eines Großteils des ZNS. Auf der anderen Seite ist ein Therapiebeginn zum
Zeitpunkt einer möglichen Diagnosestellung realistischer und aussagekräftiger für die
klinische Situation.
Der Einfluss der Therapie auf den klinischen Verlauf der Erkrankung wird anhand
zweier Parameter beurteilt: Zum einen durch die tägliche Aufzeichnung des
Körpergewichts der Tiere und zum anderen durch einen klinischen Score, der am

1- Einleitung
20
letzten Tag des Experiments zugewiesen wird. Das Körpergewicht dient als
unspezifischer Parameter des Gesundheitszustandes, in den klinischen Score gehen
die Ausprägung klassischer Symptome einer BDV Infektion im akuten Stadium ein.
Anhand der Quantifizierung infektiöser Partikel, virale RNA sowie Proteine werden der
antivirale Effekt von Ribavirin und die vier bekannten virostatischen Mechanismen
untersucht (s. 1.4.2 und 5.2). Dazu dienen die Titrierung infektiöser Viruspartikel aus
dem Hippocampus, die Quantifizierung viraler RNA mittels PCR und in situ
Hybridisierung, sowie die Bestimmung der Gewebekonzentration des viralen
Phosphoproteins. Durch eine regionale Auswertung der ISH wird zusätzlich eine
qualitative Auswertung und Beurteilung eines potentiell veränderten Infektionsmusters
ermöglicht.
Die Untersuchung des immunmodulatorischen Effekts erfolgt mittels immunhisto-
chemischer Methoden, der Quantifizierung der Antikörperproduktion in einem ELISA,
sowie der Beurteilung des Zytokinmusters mittels RPA.

2 - Materialien
2 Materialien
2.1 Verbrauchsmaterialien Aluminiumfolie UCI Storehouse
Autoradiographie Film Kodak BioMax MR Film
Deckgläser Fisher Scientific
Diafilm Kodak
Filterpapier Whatman International
Führungskanüle Small Parts (Single Guide Cannula,
C313G/SPC), 22 Gauge, 4,8 mm Pene-
trationstiefe
Glasglocke Fisher Scientific, für Inhalations-
anästhesie
Handschuhe Diamond Grip
Injektionskanüle Small Parts (Single Internal Cannula,
C313I/SPC) 28 Gauge, 4,9 mm Pene-
trationstiefe
Injektionskanülen, Standard Fisher Scientific
Kim Whipes Kimberly-Clark
Masken Fisher Scientific
Microspin 200 HR Säulen Pharmacia
Mikrofilter Fisher Scientific, auf Spritze aufsteckbar
Mikrofilter Nalgene, für Sukrosesterilisation
Mikrotiterplatten (96 Loch für Taqman) MicroAmp Optical 96-Well Reaction
Plate, Perkin Elmer Applied Biosystems
mit MicroAmp Optical Caps, Perkin
Elmer Applied Biosystems
Mikrotiterplatten (96 Loch) Costar
Nitrozellulosemembranen Schleicher & Schuell
Objektträger Fisher Scientific (Superfrost plus und
Standard)
Papier-Abdecktücher Fisher Scientific
Papierhandtücher Scott
Parafilm American National Can
Petrischalen Fisher Scientific (100x15 mm)
21

2 - Materialien
Plastikbeutel, konisch Harvard Apparatus, für Guillotine
Rasierklingen Fisher Scientific
Röhrchen 0,5 ml Fisher Scientific
Röhrchen 1,5 ml Fisher Scientific
Röhrchen 2,0 ml Fisher Scientific
Röhrchen 50 ml Fisher Scientific
Schutzoveralls VWR
Spritze (50 µl) Hamilton, für i.c.v. Injektionen
Stößel Kimble/Kontes (749520-0090), RNase
frei, für Gewebehomogenisation
Szintillationsflüssigkeit Fisher Scientific
Szintillationspapier Whatman International
Verbindungsschlauch Plastics One (Tygon Tube TGY-010-100)
0,03’’ äußerer Durchmesser; 0,01’’ inner-
er Durchmesser, für i.c.v. Injektionen
Verschlussstab Small Parts (Dummy Cannula, C313DC)
Zellkulturflaschen 25 ml Fisher Scientific
Zellkulturflaschen 75 ml Fisher Scientific
2.2 Chemikalien 2-Mercaptoethanol Sigma
Acrylamid Bio-Rad
AEC Sigma
Agarose Fisher Scientific
Americlear Stephens Scientific
Ammonium Acetat Fisher Scientific
Autoradiographie Emulsion Typ NTB2, Kodak Scientific Imaging
Systems
Borsäure Fisher Scientific
Brilliantblau R Sigma
Chloroform ACROS Organics
4-chloro-1-naphthol (HRP-Substrat) Pierce
Chromkaliumsulfat Fisher Scientific
DEPC Sigma
Dextran Sulfat Sigma
22

2 - Materialien
DMEM Irvine Scientific
DMSO Fisher Scientific
DTT Sigma
Dulbecco's PBS Sigma
EDTA Fisher Scientific
Eisessigsäure Fisher Scientific
Entwickler Kodak, Dektol D19 Developer
Eosin Fisher Scientific (SE 22-475)
Ethanol (RNase free, 200 proof) Goldshield Chemical
Ethanol UCI Storehouse
Ethidium Bromid Sigma
FCS Irvine Scientific
Ficoll 400 Pharmacia
Fixierer Kodak Fix, Kodak
Formaldehyd Fisher Scientific
Formamid EM Science
Gelatine Sigma
Harnstoff Fisher Scientific
HCl Fisher Scientific
Hämatoxylin Sigma
HEPES Fisher Scientific
Immersionsöl Fisher
Ionenaustauscher Bio-Rad AG 501-X8
Isopropanol Fisher Scientific
L-Glutamine Sigma
Methanol Fisher Scientific
Metofan Pitman-Moore Pharmaceuticals
Milchpulver (fettfrei) Carnation
Na Acetate Sigma
Na Citrat Fisher Scientific
NaCl Fisher Scientific
NaH2PO4•H2O Fisher Scientific
NaOH Fisher Scientific
Paraformaldehyd Fisher Scientific
PBS Sigma
23

2 - Materialien
Penicillin Sigma
Permount Fisher Scientific
Phenol Fisher Scientific
Photochemikalien (für Automat) Kodak Scientific Imaging Systems
PIPES Fisher Scientific
Ponceau S Sigma
Ribavirin ICN Pharmaceuticals
RNase freies Wasser ICN Pharmaceuticals
Salpetersäure RNase freie Glasgefäße Schwefelsäure Sigma, ¼ konzentriert
SDS Fisher Scientific
Streptomycin Sigma Sukrose Fisher Scientific
TEMED Sigma
Tissuetec Sakuraus Finetek
TriReagent Molecular Research Center
Tris HCl Boehringer Mannheim
Tris Hydroxymethylaminomethan Fisher Scientific
Triton Sigma
Tween 20 Fisher Scientific
Wasserstoffperoxyd Fisher Scientific
2.3 Lösungen und Puffer Acetat/Citrat-Puffer 8,2 g/l Na Acetat
21,01 g/l Zitronensäure-1-hydrat
pH 6,0
Acrylamidgel 5% Acrylamid
8 M Harnstoff
1 X TBE
Ammoniumacetat 3 M
Ammoniumchlorid-Lysepuffer 0,15 M Ammoniumchlorid
1 mM Kaliumbicarbonat
pH 7,2 – 7,4
24

2 - Materialien
Borat Puffer (pH 8,4-8,5) 100 mM Borsäure
25 mM Na2B4O7
75 mM NaCl in H2O
Coomassie Blau 0,2% Coomassie Brilliant Blau R
50% Methanol
10% Eisessigsäure
Denhard 100X 10 g Ficoll 400
10 g Polyvinylpyrolidon
10 g BSA
H2O auf 500 ml
Dextransulfat 50%ig in H2O
ELISA Blocklösung 0,05% Tween 20
0,5% BSA in PBS
ELISA TMB Substratmix 9,9 ml Acetat/Citrat Puffer
0,1 ml TMB/DMSO
1,5 µl 30% H2O2
ELISA Waschpuffer 0,05% Tween 20 in PBS
ETS (pH 7,4) 10 mM EDTA
10 mM Tris
0,2% SDS
Kulturmedium - Zellkultur 500 ml DME
5 ml L-Glutamin (200 mM)
5 ml Penicillin
5 ml Streptomycin
50 ml FCS (bei 4°C ÜN getaut
und 15-30 min bei 56°C hitzeinaktiviert)
Natrium Acetate Puffer (3M) 3 M Ausgangslösung mittels Eisessig-
säure auf pH 5,2 titriert
Paraformaledyd 4% in PB 1000 ml PB (55-60°C)
40 g Paraformaldehyd
bei 4°C bis zu einer Woche lagern
PBS (pH 7,4; 10X) 80 g/l NaCl
14,4 g/l Na2HPO4 •2H2O
2 g/l KH2PO4
25

2 - Materialien
Phosphate Puffer (pH 7,4; 2X) 7,7 g/l NaOH
33,65 g/l NaH2PO4•H2O
Ponceau S Farbstoff (10X) 2% Ponceau S
30% wt/vol Trichloressigsäure
30% wt/vol Sulfosalicylsäure
in 1% Essigsäure zu 1X verdünnen
RNase Digestionspuffer III Ambion RPA III Kit
RT Puffer Perkin Elmer Taqman RT Kit
saurer Alkohol 0,5% 995 ml 80% Ethanol
5 ml konz. HCl
SP6 Transkriptions-Puffer Promega
SSC (pH 7,0) 3 M NaCl
0,3 M Na Citrat
Substrat-Mix (Virustitrierung) 0,2 mg/ml AEC
10% DMSO
0,003% H2O2 in Natrium Acetat Puffer
H2O2 wird direkt vor Gebrauch zugefügt
Sukrose 20%/30% (RNase frei) in PBS 200/300 g Sukrose
100 ml PBS (RNase frei)
auf 1000 ml
steril filtrieren
T7 Transkriptions-Puffer PharMingen
TBE Elektrophoresepuffer (pH 8,0) 89 mM Trisbase
89 mM Borsäure
20 mM EDTA
TBS (Tris balanced saline, pH 7,5) 50 mM Tris-HCl
150 mM NaCl
TE 10 mM Tris HCl
1 mM EDTA
Tris Puffer (IHC) 0,125 M Tris Base
0,375 M Tris HCl
1% FCS
pH 7,6
Triton 1% X-100 in PBS
26

2 - Materialien
Western - Elektrophoresepuffer (pH 8,3) 25 mM Tris Base
192 mM Glycine
0,1% SDS
Western - Probenladepuffer 20 mM Tris HCl (pH 6,8)
4% SDS
5 mM EDTA
20% Glycerol
10% β-Mercaptoethanol
0,5 mg/ml Bromphenolblau
Western - Transferpuffer (pH 8,3) 25 mM Tris Base
192 mM Glycin
20% Methanol
Western - Waschpuffer 1% Milchpulver und 0,05% Tween 20 in
TBS
2.4 Radiochemikalien
α-32P-UTP New England Nuclear (NEN), 800-1200
Ci/mMol
α-35S-CTP New England Nuclear (NEN), 800-1200
Ci/mMol
C14 Standards (ISH) American Radiolabeled Chemicals (ARC
146 und 146B), 0,02 mCi Carbone-14
2.5 Kits DAB Substrat Kit Vector Laboratories
ECL Western Blotting Detection System Amersham Biosciences
Gram-Färbung (3 Schritte) Becton Dickinson Microbiology Systems
MAXIscript Kit Ambion
RiboQuant Multi-Probe Template Set rCK-1 PharMingen
RPA III Kit Ambion
Taqman Reverse Transkription Kit Perkin Elmer Applied Biosystems
Taqman Universal PCR Master Mix Perkin Elmer Applied Biosystems
Vectastain Elite ABC Kit Vector Laboratories
27

2 - Materialien
2.6 Nukleotide, Primer und Plasmide dNTP’s Promega
Hefe tRNA Sigma
Lachssperma DNA Sigma
rNTP’s Promega
Borna Standard-Plasmide pBSKBVcore = BDV Nukleotide 1-8897
in pBluescript SK II (Stratagene), kloniert
in Not I nach Sal I
Plasmid ISH kompletter P ORF (610 nt, nt 1270 bis
nt1880 des BDV Genoms, Verwendung
des Sp6 Promotors für Transkription von
RNA in genomischer Orientierung)
p40 (N) forward Primer p40bobe – 187F
5’ – CAG TCA CGG CGC GAT ATG T
p40 (N) reverse Primer p40bobe – 286R
5’ – GCA CCC CTC CGT GAA CAA
p40 (N) TaqMan probe p40bobe – 247T
5' -6FAM- ATC CCA GGA CTG CAC
GCT GCG -XT-TAMRA
2.7 Enzyme und Proteine pankreatische RNase (RNase A) Calbiochem
Proteinase K Boehringer Mannheim
RNase A/T1 Ambion
RNase Inhibitor Promega, Perkin Elmer
RQ1 DNase Promega, Ambion
SP6 Polymerase Promega
T7 Polymerase PharMingen
Trypsin Sigma
BDV N Protein recp40 [102]
BDV P Protein recp23 [102]
2.8 Sera und Antikörper NRtS normales Rattenserum, Negativkontrolle
Ratte 1.1 Positivkontrolle
28

2 - Materialien
anti-N anti-recp40 [102]
anti-P anti-recp23 [102]
Ox-8 [162], s. [70]
Ox-42 [163], s. [70]
W3/25 [164], s. [70]
Ziege-anti-Kaninchen-HRP Sigma
Ziege-anti-Maus-HRP Sigma
Ziege-anti-Ratte-HRP Sigma
2.9 Zelllinien Fetale Rattengliazellen:
Nach steriler Entnahme werde Hirngewebe neugeborener Ratten zerkleinert, mit
0,25% Trypsin gemischt und durch eine Spritze passagiert, um ein Einzelzellpräparat
zu erhalten. Anschließend werden die Zellen in Zellkulturplatten mit EDM (5% FCS)
Medium angezogen. Nach 24 Stunden werden nicht adhärente Zellen entfernt und
adhärente Zellen nach Trypsinisierung in neue Zellkulturplatten gesplitet [104, 116].
2.10 Virus Borna Disease Virus (BDV) Stamm He/80, Isolat von einem natürlich
infizierten Pferde aus dem Jahr 1980,
anschließend mehrmals in Kaninchen,
Ratten und mehreren Zelllinien in Kultur
passagiert (s. [116])
2.11 Versuchstiere 200-225 g schwere und 56-60 Tage alte männliche Lewis Ratten wurden von Charles
River Laboratories (Wilmington, MA, USA) bezogen. Die Gewichtsentwicklung während
der Wachstumsphase läuft wie folgt ab:
29
2 - Materialien
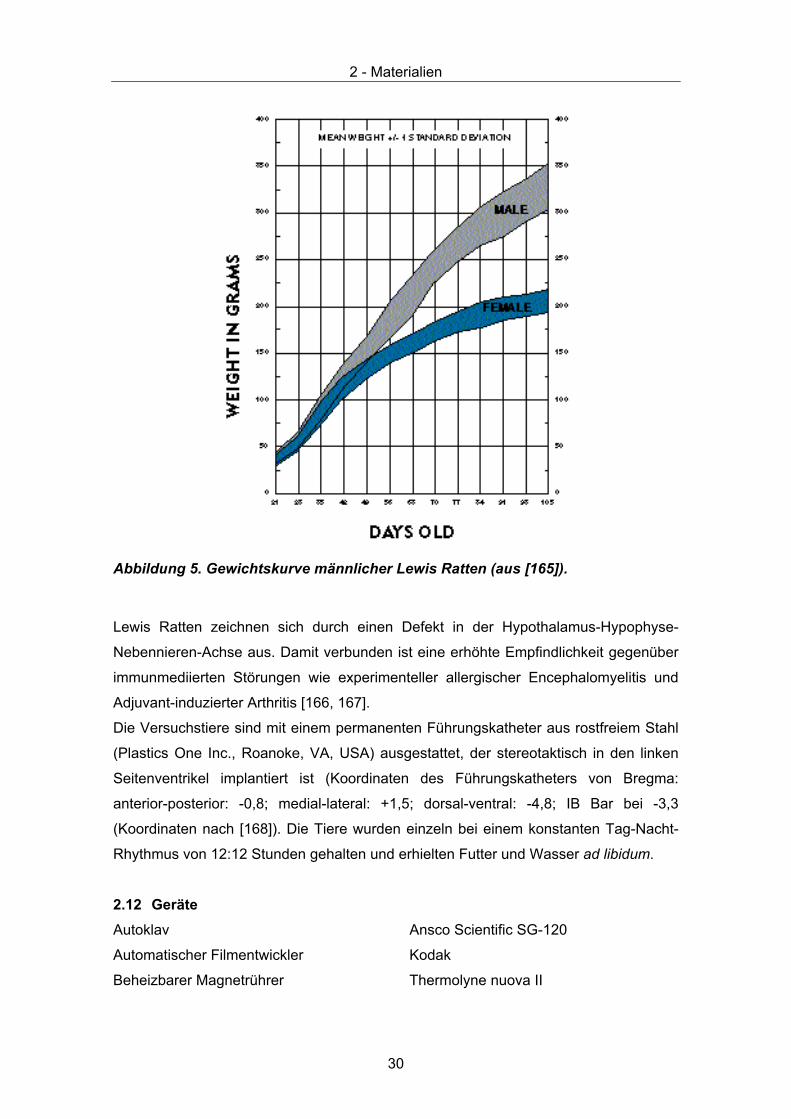
Abbildung 5. Gewichtskurve männlicher Lewis Ratten (aus [165]).
Lewis Ratten zeichnen sich durch einen Defekt in der Hypothalamus-Hypophyse-
Nebennieren-Achse aus. Damit verbunden ist eine erhöhte Empfindlichkeit gegenüber
immunmediierten Störungen wie experimenteller allergischer Encephalomyelitis und
Adjuvant-induzierter Arthritis [166, 167].
Die Versuchstiere sind mit einem permanenten Führungskatheter aus rostfreiem Stahl
(Plastics One Inc., Roanoke, VA, USA) ausgestattet, der stereotaktisch in den linken
Seitenventrikel implantiert ist (Koordinaten des Führungskatheters von Bregma:
anterior-posterior: -0,8; medial-lateral: +1,5; dorsal-ventral: -4,8; IB Bar bei -3,3
(Koordinaten nach [168]). Die Tiere wurden einzeln bei einem konstanten Tag-Nacht-
Rhythmus von 12:12 Stunden gehalten und erhielten Futter und Wasser ad libidum.
2.12 Geräte Autoklav Ansco Scientific SG-120
Automatischer Filmentwickler Kodak
Beheizbarer Magnetrührer Thermolyne nuova II
30

2 - Materialien
Chirurgische Instrumente Roboz Surgical Instruments
Diascanner Nikon
Durchlicht UV Stratagene Eagleeye II
Eismaschine Scotsman
Elektrophoresegeltrockner BioRad, Model 583
Filmkassetten Fisher Scientific
Gefriermikrotom Mikrom 505EP (Cryostat)
Gefrierschrank (-20°C) Puffer Hubbard FUF1821A
Gefrierschrank (-70°C) NuAire Nu-66176
Gelelektrophoresekammer für RPA Life Technologies, Model S2
Gelelektrophoresekammer Werkstatt
Glaswaren (Histologie) Fisher Scientific
Guillotine Cervical Dislocators
Hand UV Lampe Fisher Scientific
Inkubationsbox (Histologie) Werkstatt
Inkubator (Bakterien, 37°C) Blue M
Inkubator (Histologie, 37°C) Fisher Isotemp 500 Serie
Inkubator (RNase freie Glaswaren, 80°C) Fisher Scientific
Inkubator (Zellkultur, 37°C) Haereus Kamera für Mikrophotographie Nikon Fx 35A
Kühlraum Kolpak
Kühlschrank Fisher Scientific
Laminar Flow Hood (Radioisotope) Hamilaton Safeaire
Laminar Flow Hood (Zellkultur) NuAire
Magnetrührer Nuova Stirrer
MCID M5 Microcomputer Imaging Devices
Metallständer (Histologie) Fisher Scientific
Mikroskop (Mikrophotographie) Nikon Eclipse E600
Mikroskop (Zellkultur) Nikon TE-200
Mikrowelle Amana Radarange
PCR Geräte MJ Research PTC-200
pH-Meter Corning pH-Meter 240
Phosphoimager Molecular Dynamics
Photometer (automatisiert, ELISA) Molecular Devices
Pinzette Roboz Surgical Instruments
31

2 - Materialien
Pipette (automatisch) Drummond
Pipette (Mehrkanal) Fisher Scientific
Pipette Gilson
Spannungstransformator (Elektrophorese) EC Apparatus Corp. EC-105
Spektralphotometer Beckman Coulter DU 640BIO
Stoppuhr Fisher
Szintillationszähler Beckman LS 6500
Taqman ABI Prism 7700 Sequence Detector,
Perkin Elmer
Thermometer Fisher Scientific
Tierwaage Fisher
Ultraschallhomogenisator Braun (Braun-Sonic H)
Vortex Baxter Scientific Products
Waage (µg Bereich) Mettler Balance (01-909-379)
Waage (Chemikalien) Mettler Balance (01-913-507C)
Wasserbad Blue M
Wasserfilter Barnstead Nanopure Zentrifuge VWR Mini-Zentrifuge
Zentrifuge (kühlbar) Baxter 17R
Zentrifuge (Tischzentrifuge) Eppendorff 5415C
Zentrifuge (Zellkultur) Beckman Alegra Bk 366816
2.13 Software Densitometrie (ELISA - Photometer) Soft max 2.35 Software (Molecular
Devices)
Densitometrie (Phosphoimager) IQMac Software (Molecular Dynamics)
Primerdesign (Taqman) Primer Express (Perkin Elmer)
RIA RIA smartTM Software (Packard
Instrument Company)
Quantifizierung (Taqman) Real-Time Sequence Detection Software
(Perkin Elmer)
Statistik t-Test in Microsoft Excel (Office XP)
ANOVA
32

3 - Methoden
3 Methoden
3.1 Toxikologie und Pharmakokinetik
3.1.1 Randomisierung
Die Tiere werden zufällig nach vorheriger Nummerierung den experimentellen Gruppen
zugeteilt.
3.1.2 Körpergewicht und Überlebensrate
Die Tiere werden jeden Morgen vor der Applikation von Ribavirin gewogen. Vor dem
Tag 6 verstorbene Tiere gehen nicht in die Bestimmung der Gewebekonzentration von
Ribavirin und die histologische Auswertung mit ein.
3.1.3 Wirkstoffzubereitung
Ribavirin wird bei Raumtemperatur ungelöst, in Reinform gelagert. Die Ribavirin
Lösung in sterilem PBS wird täglich unmittelbar vor der Injektion hergestellt. Die
Ausgangskonzentration beträgt 0,125 mg/µl (1X). Dies entspricht ungefähr einer
0,5 molaren Lösung. Für die Injektionen der Tiere, die 1,25 mg/kg erhalten, wird die
Ausgangslösung 1:2 in sterilem PBS verdünnt.
3.1.4 Intrazerebroventrikuläre Injektion
Die intrazerebroventrikuläre Injektion erfolgt über permanent implantierte
Führungskanülen, die stereotaktisch in den linken Seitenventrikel der Tiere implantiert
sind (Koordinaten s. 2.11, s. auch Abbildung 6 und Abbildung 7).
Abbildung 6. Injektionssystem während der Injektion.
33
3 - Methoden
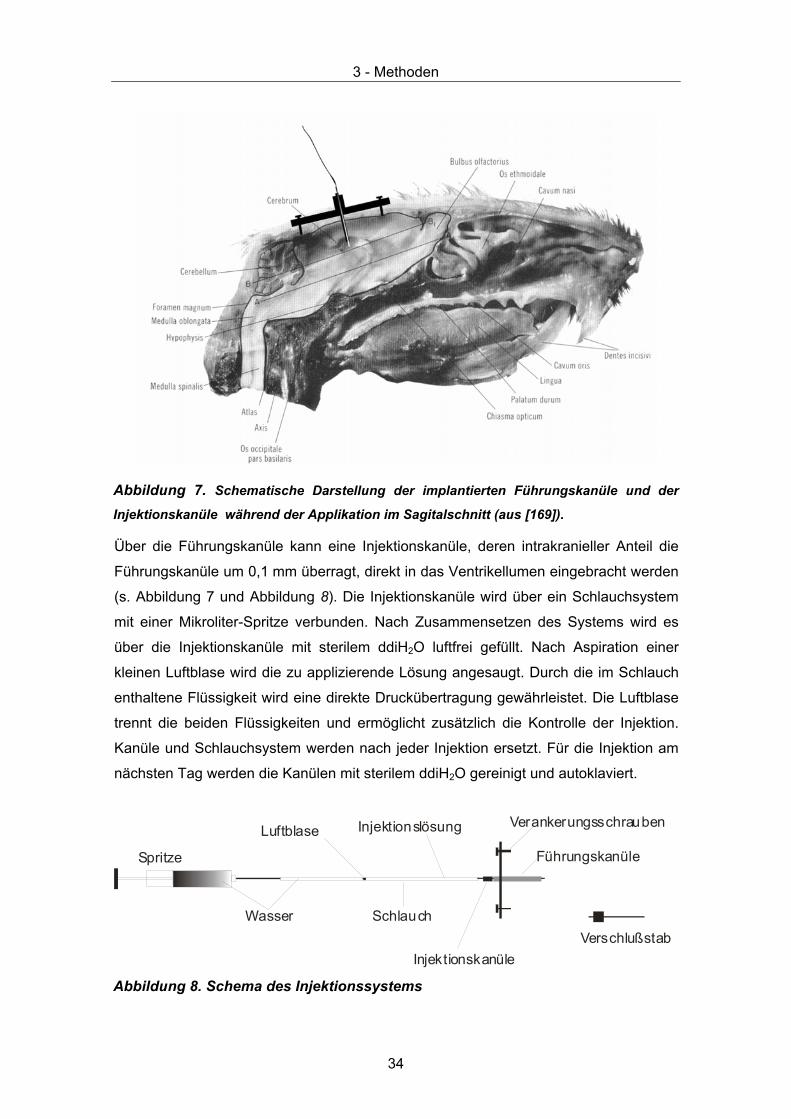
Abbildung 7. Schematische Darstellung der implantierten Führungskanüle und der
Injektionskanüle während der Applikation im Sagitalschnitt (aus [169]).
Über die Führungskanüle kann eine Injektionskanüle, deren intrakranieller Anteil die
Führungskanüle um 0,1 mm überragt, direkt in das Ventrikellumen eingebracht werden
(s. Abbildung 7 und Abbildung 8). Die Injektionskanüle wird über ein Schlauchsystem
mit einer Mikroliter-Spritze verbunden. Nach Zusammensetzen des Systems wird es
über die Injektionskanüle mit sterilem ddiH2O luftfrei gefüllt. Nach Aspiration einer
kleinen Luftblase wird die zu applizierende Lösung angesaugt. Durch die im Schlauch
enthaltene Flüssigkeit wird eine direkte Druckübertragung gewährleistet. Die Luftblase
trennt die beiden Flüssigkeiten und ermöglicht zusätzlich die Kontrolle der Injektion.
Kanüle und Schlauchsystem werden nach jeder Injektion ersetzt. Für die Injektion am
nächsten Tag werden die Kanülen mit sterilem ddiH2O gereinigt und autoklaviert.
Luftblase Injektionslösung
Wasser Schlauch
Injektionskanüle
Spritze
Verankerungsschrauben
Führungskanüle
Verschlußstab
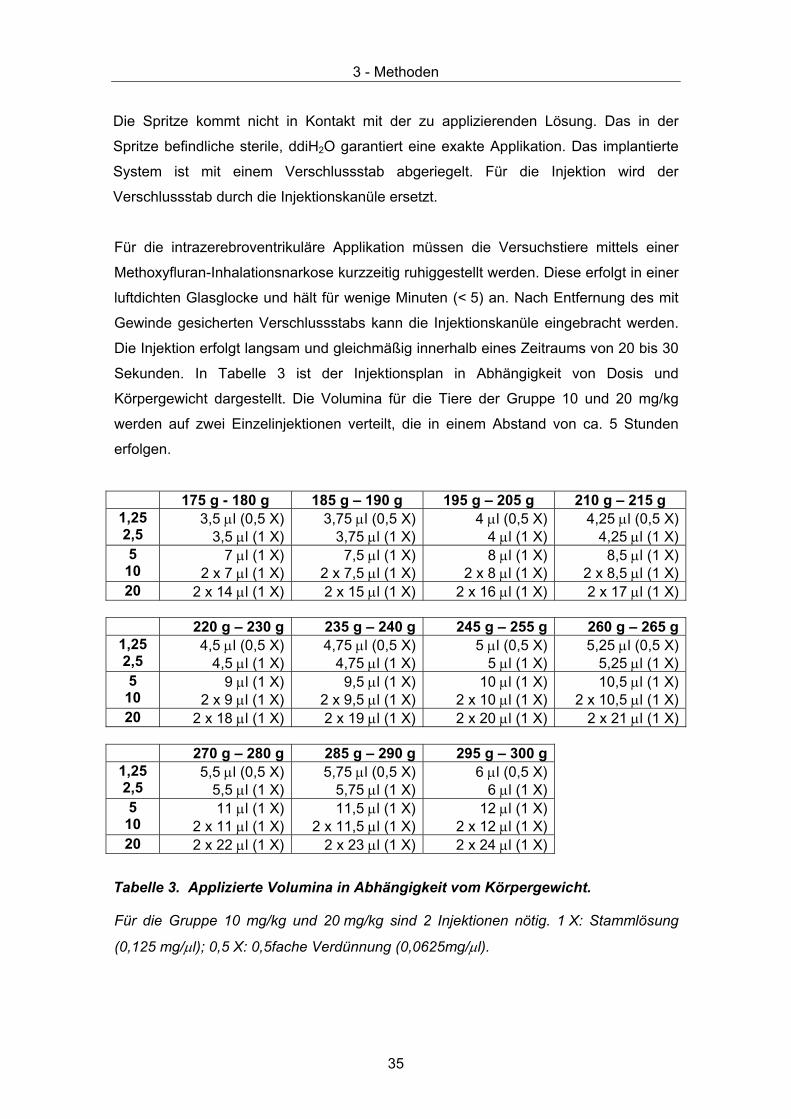
Abbildung 8. Schema des Injektionssystems
34
3 - Methoden
Die Spritze kommt nicht in Kontakt mit der zu applizierenden Lösung. Das in der
Spritze befindliche sterile, ddiH2O garantiert eine exakte Applikation. Das implantierte
System ist mit einem Verschlussstab abgeriegelt. Für die Injektion wird der
Verschlussstab durch die Injektionskanüle ersetzt.
Für die intrazerebroventrikuläre Applikation müssen die Versuchstiere mittels einer
Methoxyfluran-Inhalationsnarkose kurzzeitig ruhiggestellt werden. Diese erfolgt in einer
luftdichten Glasglocke und hält für wenige Minuten (< 5) an. Nach Entfernung des mit
Gewinde gesicherten Verschlussstabs kann die Injektionskanüle eingebracht werden.
Die Injektion erfolgt langsam und gleichmäßig innerhalb eines Zeitraums von 20 bis 30
Sekunden. In Tabelle 3 ist der Injektionsplan in Abhängigkeit von Dosis und
Körpergewicht dargestellt. Die Volumina für die Tiere der Gruppe 10 und 20 mg/kg
werden auf zwei Einzelinjektionen verteilt, die in einem Abstand von ca. 5 Stunden
erfolgen.
175 g - 180 g 185 g – 190 g 195 g – 205 g 210 g – 215 g 1,25 2,5
3,5 µl (0,5 X) 3,5 µl (1 X)
3,75 µl (0,5 X)3,75 µl (1 X)
4 µl (0,5 X)4 µl (1 X)
4,25 µl (0,5 X)4,25 µl (1 X)
5 10
7 µl (1 X) 2 x 7 µl (1 X)
7,5 µl (1 X)2 x 7,5 µl (1 X)
8 µl (1 X)2 x 8 µl (1 X)
8,5 µl (1 X)2 x 8,5 µl (1 X)
20 2 x 14 µl (1 X) 2 x 15 µl (1 X) 2 x 16 µl (1 X) 2 x 17 µl (1 X) 220 g – 230 g 235 g – 240 g 245 g – 255 g 260 g – 265 g
1,25 2,5
4,5 µl (0,5 X) 4,5 µl (1 X)
4,75 µl (0,5 X)4,75 µl (1 X)
5 µl (0,5 X)5 µl (1 X)
5,25 µl (0,5 X)5,25 µl (1 X)
5 10
9 µl (1 X) 2 x 9 µl (1 X)
9,5 µl (1 X)2 x 9,5 µl (1 X)
10 µl (1 X)2 x 10 µl (1 X)
10,5 µl (1 X)2 x 10,5 µl (1 X)
20 2 x 18 µl (1 X) 2 x 19 µl (1 X) 2 x 20 µl (1 X) 2 x 21 µl (1 X) 270 g – 280 g 285 g – 290 g 295 g – 300 g
1,25 2,5
5,5 µl (0,5 X) 5,5 µl (1 X)
5,75 µl (0,5 X)5,75 µl (1 X)
6 µl (0,5 X)6 µl (1 X)
5 10
11 µl (1 X) 2 x 11 µl (1 X)
11,5 µl (1 X)2 x 11,5 µl (1 X)
12 µl (1 X)2 x 12 µl (1 X)
20 2 x 22 µl (1 X) 2 x 23 µl (1 X) 2 x 24 µl (1 X)
Tabelle 3. Applizierte Volumina in Abhängigkeit vom Körpergewicht.
Für die Gruppe 10 mg/kg und 20 mg/kg sind 2 Injektionen nötig. 1 X: Stammlösung
(0,125 mg/µl); 0,5 X: 0,5fache Verdünnung (0,0625mg/µl).
35

3 - Methoden
Nach abgeschlossener Injektion erfolgt eine Pause von ca. 30 Sekunden, um ein
Abströmen zu ermöglichen, anschließend wird die Injektionskanüle langsam entfernt
und die Führungskanüle wieder verschlossen. Die komplette Applikation erfolgt unter
bestmöglicher Einhaltung kontaminationsfreier Bedingungen. Die Tiere werden
unmittelbar nach Beendigung der Injektion wieder aktiv.
3.1.5 Gewebepräparation
Die Versuchstiere werden nach einer kurzen Anästhesie dekapitiert und das Gehirn
möglichst schnell freipräpariert. Auf einer mit Eis gefüllten Petrischale wird ein 3 mm
breiter sagitaler Gewebsschnitt des lateralen Anteils der rechten Hemisphäre
abgetrennt, gewogen und in 1 ml sterilem PBS homogenisiert. Nach einer
Zentrifugation von 20 min. bei 4000 rpm. wird der Überstand abgenommen und für das
anschließendes Radioimmunoassay (RIA) verwendet. Zusätzlich wird die linke
Hirnhälfte abgetrennt, über Nacht in 4% PFA fixiert, 2mal über Nacht in 20% Sukrose
inkubiert, in Tissuetec eingebettet und bei -70°C bis zur histologischen Aufarbeitung
gelagert.
3.1.6 Konzentrationsbestimmung – RIA
Das RIA wird von der Pediatric Pharmacology Assay Laboratory der University of
California in San Diego durchgeführt. Für die Standardkurve wird ein Four Parameters
Logistic Curve Fit (4PL) verwendet. Ihr Korrelationskoeffizient beträgt 1,0. 0% Bindung
der Gesamtzähler entspricht 23% und die unspezifische Bindung beträgt 2,7%. Die
Sensitivitätsgrenze beträgt 1 mg/ml. Aus den errechneten Werten in µg/ml Homo-
genisat wird auf eine Konzentration in µg/g Hirngewebe umgerechnet.
3.1.7 Objektträger-Vorbereitung
Ein Liter DEPC behandeltes, ddiH2O wird in einem RNase freien Erlenmeierkolben auf
50 bis 60°C erhitzt. Unter ständigem Rühren werden 10 g Gelatine vollständig in
Lösung gebracht. Anschließend werden 0,4 g Chromkaliumsulfat zugegeben. Die
Lösung nimmt eine leicht bläuliche Farbe an. Nach Abkühlen auf ca. 35°C werden die
RNase freie Glas-Objektträger für 30 Sekunden vollständig in die Gelatinelösung
getaucht. Anschließend werden die Objektträger aus der Lösung herausgehoben und
für mehrere Stunden unter Alufolie abgedeckt bei 37°C getrocknet.
36

3 - Methoden
3.1.8 Gefrierschnitte
20 µm dicke, sagitale Paramedianschnitte werden angefertigt, auf Gelatine-
beschichtete Objektträger transferiert und bei -70°C gelagert.
3.1.9 HE Färbung
Die in Tissuetec eingebetteten und gefriergeschnittenen, histologischen Schnitte
werden in einer Ethanolreihe absteigender Konzentration (2x100%, 95%, 70%, 50%,
2xdiH2O) je 2 min rehydriert. Die anschließende Färbung mit Hämatoxylin dauerte in
Abhängigkeit von der Farbintensität 90 bis 120 Sekunden. Danach werden die
Präparate jeweils 5 bis 10 Sekunden in diH2O, sowie saurem Ethanol entfärbt und
anschließend 7 bis 10 Minuten unter fließendem Leitungswasser gespült. Nach
Färbung mit Eosin für 12 Sekunden wird anschließend in diH2O gewaschen und in
aufsteigender Ethanolreihe dehydriert. Nach 10minütiger Lufttrocknung werden Reste
des Einbettungsmaterials in zweimaliger, je 5minütiger Inkubation in Americlear
beseitigt. Die luftdichte Versiegelung erfolgte mittels Permount und Abdeckung mit
Deckglas.
3.1.10 Gram-Färbung
Es wird eine Gram Stain Kit für eine dreistufige Gram-Färbung verwendet. Die 20 µm
dicken Gewebeschnitte werden 1 min mit Gram Kristallviolett gefärbt. Anschließend
wird der Farbstoff mit kaltem Leitungswasser entfernt und mit stabilisiertem Gram Jod
für 1 min überschichtet. Nach Abwaschen mit Gram Safranin wird 20-50 s mit frischem
Gram Safranin gegengefärbt. Abschließend wird mit kaltem Leitungswasser
gewaschen, luftgetrocknet und das Gewebe mit einem Deckglas abgedeckt.
3.2 Randomisierung, Infektion und Applikation
3.2.1 Virusinokulation
Unter Metofan-Inhalationsnarkose werden den Tieren in einem Volumen von 60 µl
1,6x104 ffu/zellkulturinfektiöse Dosiseinheiten inokuliert. Ein 10%iges (wt/vol) Hirn-
homogenisat infizierter Ratten in PBS dient hierbei als Stammlösung [48]. Die
erfolgreiche Infektion wird durch Auftreten typischer Symptomatik (Hyperaktivität,
übermäßige Schreckhaftigkeit) bestätigt.
37

3 - Methoden
3.2.2 Randomisierung
s. 3.1.1
3.2.3 Ribavirinapplikation
s. 3.1.3 und 3.1.4
3.3 Bestimmung klinischer Parameter 3.3.1 Körpergewicht
s. 3.1.2
3.3.2 Verhalten und klinische Scores
Scores werden anhand 6 klinischer Symptome, die bei einer akuten BDV Infektion von
männlichen, ausgewachsenen Lewisratten beobachtet werden können [50, 170],
zugeteilt. Die Beurteilung der Tiere findet am letzten Tag des Experiments durch
2 Personen statt. Die maximal erreichbare Punktzahl ist 22, der minimaler Score 6. Im
Folgenden sind die 6 Symptome mit den jeweiligen Ausprägungsformen aufgeführt:
hämorrhagische Augen:
4 sehr hämorrhagisch, periorbital blutiges Fell
3 sehr hämorrhagisch
2 hämorrhagisch
1 nicht hämorrhagisch
hämorrhagische Nostrilen:
4 sehr hämorrhagisch, um die Nostrilen blutiges Fell
3 sehr hämorrhagisch
2 hämorrhagisch
1 nicht hämorrhagisch
Respiration:
4 schwere Dyspnoe
2 Tachypnoe
1 normal
38

3 - Methoden
Aktivität:
4 keine Reaktion oder spontane Bewegung
3 geringe, ataktische Spontanbewegungen
2 ataktische Spontanbewegungen
1 normal
äußere Erscheinung:
2 ungepflegtes Fell
1 normal
Gewichtsverlust:
4 >31% Tag 22 bis Tag 28
3 21-30% "
2 11-20% "
1 0-10% "
3.4 Bestimmung viraler Parameter
3.4.1 Gewebspräparation
ISH. Nach Präparation (s. 3.1.3) wird die rechte Hemisphäre über Nacht mit 4%igem
Paraformaldehyd in PB fixiert, zweimal über Nacht mit RNase freier 20%iger Sukrose-
Lösung in PB inkubiert und bis zur Aufarbeitung für die ISH bei -70°C gelagert. Für die
Hybridisierung werden paramediane Schnitte, die den Hippocampus enthalten,
verwendet.
PCR. Ein 3 mm breiter, mit dem Frontalpol der linken Hemisphäre beginnender
coronarer Schnitt, der den Präfrontalkortex einschließt, wird in TriReagent mit Hilfe
eines Stößels homogenisiert und bei -70°C für spätere Taqman-RT-PCR gelagert.
IHC. Anschließend werden drei aufeinanderfolgende je 2 mm dicke coronare Schnitte,
die Cortex und Striatum enthalten hergestellt, für 1-1,5 Stunden in 4%igem
Paraformaldehyd fixiert, zweimal über Nacht mit 20%iger Sukrose-Lösung inkubiert
und bis zur histologischen Aufarbeitung für die Immunhistochemie bei -70°C gelagert.
Virustitrierung. Caudal hiervon wird aus einem 3 mm breiten coronaren Schnitt
Hippocampus präpariert und in Dulbecco’s modified Eagle Medium (DMEM) mittels
39

3 - Methoden
Ultraschall homogenisiert, 10 min bei 1000 x g zentrifugiert und der Überstand bei
-20°C aufbewahrt.
3.4.2 RNase freie Bedingungen
Arbeitsplatz. Um unter möglichst RNase armen Bedingungen arbeiten zu können wird
die Arbeitsfläche unmittelbar vor Arbeitsbeginn 2mal mit 70%igem Ethanol gereinigt
und anschließend mit einseitig kunststoffbeschichteten Papiertüchern abgedeckt.
Glasgefäße. RNase freie Glasgefäße für histologische Färbungen werden durch
Inkubation in 10%iger Salpetersäure über Nacht und anschließendes Waschen mit
ddiH2O vorbereitet. Glasbehälter für wässrige Lösungen und RNase freies Wasser
entstehen bei der Behandlung von Wasser mit DEPC.
Metall. Metallständer für Objektträger werden im Autoklaven mit Programm „feste
Güter“ sterilisiert und anschließend im Inkubator bei 80°C für 24 h inkubiert.
Pipettenspitzen. Für alle RNA-Arbeiten werden aerosolresistente Einmalspitzen
verwendet.
Lösungen. RNase freies Wasser wird mittels DEPC hergestellt. ddiH2O wird über
Nacht mit 1 V‰ DEPC nach starkem Schütteln auf einem Magnetrührer unter
ständigem Rühren inkubiert und anschließend autoklaviert. Feste und flüssige
Ausgangssubstanzen für RNase freie Lösungen entstammen speziell als solche
markierten Vorratsbehältern, die nur für RNA-Arbeiten verwendet werden.
Objektträger. Objektträger für in situ Hybridisierung werden vom Hersteller RNase frei
geliefert. Die anschließende Gelatine-Beschichtung erfolgt mit RNase freien Lösungen
(s. 3.1.7).
3.4.3 Gefrierschnitt
Auf Trockeneis werden die Gewebeproben in Tissue Tec eingebettet, für in situ
Hybridisierung und HE Histologie 20 µm dicke Schnitte angefertigt und anschließend
auf Gelatine-beschichtete Objektträger transferiert. Für in situ Hybridisierung wird dabei
die Klinge des Cryotoms nach jedem Präparat mit 70%igem Ethanol gereinigt und
unter möglichst strenger Einhaltung RNase freier Bedingungen gearbeitet. Für IHC
werden 14 µm dicke Schnitte angefertigt.
Auf jeden Objektträger wird Gewebe je eines Tieres pro Gruppe übertragen, um
mögliche Abweichungen in späteren Vorgängen von Objektträger zu Objektträger auf
alle Untersuchungsgruppen gleichmäßig zu verteilen. Anschließend werden die
Objektträger bei -70°C gelagert.
40

3 - Methoden
3.4.4 In situ Hybridisierung
Bei der in situ Hybridisierung wird eine radioaktiv markierte RNA-Sonde, die
komplementär zu einer Teilsequenz der zu detektierenden ⊕-Strang RNA ist, mit
dieser hybridisiert. Der Prozess findet in histologischen Gewebeschnitten statt, so dass
neben der quantitativen Beurteilung der Signalintensität auch eine Aussage über die
Lokalisation des Signals bezüglich anatomischer Strukturen, über die Verteilung auf
zellulärer Ebene sowie deren Bezug zu histopathologischen Veränderungen gemacht
werden kann. Die Visualisierung erfolgt zum einen über die Belichtung eines
strahlensensiblen Filmmaterials, das für quantitative Analysen verwendet werden kann.
Zum anderen wird eine photosensible Emulsion direkt auf das Gewebe aufgebracht
und nach Entwicklung und Gegenfärbung mikroskopisch beurteilt. Bei dieser in situ
Autoradiographie entstehen bei der Belichtung der Emulsion Silberkörner, die
mikroskopisch als lichtundurchlässige, körnige Strukturen auffallen und eine Aussage
über die Lokalisation der hybridisierten Sonde auf zellulärer Ebene zulassen.
Im Einzelnen werden folgende Schritte durchgeführt:
1. Synthese der Sonde
2. Vorbereitung des Gewebes für die Hybridisierung
3. Hybridisierung mit der markierten Sonde
4. Waschen und Trocknen der Schnitte
5. Visualisierung
Alle Prozeduren bis zum RNase Verdau in den Waschschritten (s. 3.4.4.5) müssen
unter streng RNase freien Bedingungen durchgeführt werden, da es ansonsten zu
Verlusten von Sonde und Ziel-RNA kommt. Der Versuch wird im mehrfach Ansatz
durchgeführt wobei fünf benachbarte Schnitte je Tier eingesetzt und ausgewertet
werden.
3.4.4.1 Synthese der radioaktiv markierten Sonde mittels in vitro Transkription
Für die Synthese der radioaktiv markierten Sonde werden folgende Reagenzien in der
angegebenen Reihenfolge pipetiert:
41

3 - Methoden
6,5 µl DEPC behandeltes H2O
5 µl 5X Transkriptionspuffer
1 µl 50 mM DTT
2 µl linearisiertes Plasmidtemplate (0,5µg/µl)
1 µl RNase Inhibitor
3 µl 3,3 mM rNTP’s
4,5 µl 35S CTP
2 µl SP6 Polymerase (10-20 U/µl)
25 µl Gesamtvolumen
Nach einer einstündigen Transkription bei 37°C im Wasserbad wird 1 µl DNase 1
(1 U/µl) zugegeben, und das Plasmidtemplate für 10 Minuten bei 37°C verdaut.
Anschließend wird für die Extraktion der synthetisierten RNA der Ansatz mit 100 µl
Phenol/Chloroform/Isoamylalkohol versetzt, 10 Minuten bei 14.000 rpm in einer Tisch-
zentrifuge zentrifugiert und die wässrige Phase abpipetiert.
Zur späteren Berechnung der Inkorporationsrate wird 1 µl entnommen und auf einem
Szintillationspapier zum Trocknen gebracht. Eine Microsin 200 HR Säule wird 1 Minute
bei 14.000 rpm in einer Tischzentrifuge zentrifugiert, um die Equilibrationslösung zu
entfernen. Anschließend wird die wässrige Phase auf die Säule pipetiert und das Eluat
gesammelt. Nichtinkorporiertes 35S CTP, rNTP's sowie verdautes Template verbleiben
in der Säule, die radioaktiv markierte Sonde befindet sich im Eluat. 1 µl des Eluats wird
auf einem Szintillationspapier getrocknet und die Inkorporation als Quotient von
radioaktivem Signal vor und nach Säulenchromatographie berechnet.
Die Sonde wird in kleine Aliquots abgefüllt und kann bis zu einer Woche lang bei -70°C
gelagert werden.
3.4.4.2 Herstellung der Prähybridisierungslösung
Die Prähybridisierungslösung setzt sich aus den folgende Reagenzien in den
angegebenen Konzentrationen zusammen:
42

3 - Methoden
Volumen (ml) Reagenz Endkonzentration
20 deionisiertes Formamid 50%
6 5 M NaCl 750 mM
0,8 1 M PIPES (pH 7) 20 mM
0,8 0,5 M EDTA 10 mM
8 50% Dextransulfat (60°C) 10%
2 100X Denhardts 5 X
2 1 M DTT 50 mM
0,4 20% SDS 0,2%
0,4 10 mg/ml Lachssperma DNA 100 µg/ml
0,4 10 mg/ml Hefe tRNA 100 µg/ml
Lachssperma DNA und Hefe tRNA werden vor dem Pipetieren für 5 Minuten in einem
15 ml Falcon Röhrchen im Wasserbad zum Kochen gebracht. Für die Deionisierung
von Formamid werden 100 ml Formamid mit 5 g Ionenaustauscher inkubiert. Das
Dextransulfat wird vor dem pipetieren im Wasserbad auf 60°C erhitzt um den
Pipetiervorgang zu präzisieren und zu erleichtern.
Pro Objektträger werden 0,5 bis 1 ml Prähybridisierungslösung benötigt. Zusätzlich
werden 100 µl der Prähybridisierungslösung pro Objektträger für die spätere
Hybridisieirung entnommen und bei 60°C im Wasserbad gelagert.
3.4.4.3 Prähybridisierungsschritte
Die Objektträger werden unter einer Laminar Flow Hood langsam auf Raumtemperatur
erwärmt und in ein RNase freien Metallträger transferiert. Hier verbleiben sie für die
nachfolgenden Inkubationsschritte. Der Verdau mit Proteinase K findet im Wasserbad
bei 37°C, die Waschschritte in jeweils frischem PBS statt.
43

3 - Methoden
Dauer (min) Lösung Temperatur
5 4% Formaldehyd in PBS RT
2 x 2 PBS RT
7,5 Proteinase K 37°C
2 x 2 PBS RT
7,5 0,05 N HCl RT
2 x 2 PBS RT
2 4% Formaldehyd in PBS RT
2 x 2 PBS RT
5 60% Ethanol/0,3M Ammoniumacetat RT
5 80% Ethanol/0,3M Ammoniumacetat RT
5 95% Ethanol/0,3M Ammoniumacetat RT
Anschließend werden die Objektträger bei Raumtemperatur unter einer Laminar Flow
Hood zum Trocknen gebracht. Das Gewebe auf dem Objektträger wird dann komplett
mit Prähybridisierungslösung bedeckt (0,5 - 1 ml). Um ein Verdunsten der Lösung zu
verhindern werden die Objektträger in eine Plexiglasbox transferiert und in einem
Inkubator bei 48-50°C für 1-3 Stunden inkubiert.
3.4.4.4 Hybridisierung
Die Prähybridisierungslösung wird entfernt und pro Objektträger werden etwa 10 ng
Sonde zu 100 µl Prähybridisierungslösung pipetiert, gemischt, auf die Gewebeschnitte
gleichmäßig verteilt, diese mit einem Deckglas abgedeckt und anschließend über
Nacht bei 48-50°C inkubiert. Für die Hybridisierung verbleiben die Objektträger in der
Plexiglasbox.
3.4.4.5 Posthybridisierungsschritte
Nach Entfernen der Deckgläser in 4X SSC/70 mM 2-Mercaptoethanol finden folgende
Waschschritte statt.
44

3 - Methoden
Dauer (min) Lösung Temperatur
15 4X SSC/70 mM 2-ME RT
15 4X SSC RT
15 20 µg/ml pankreatische RNase 37°C
in 0,5 M NaCl/1X TE
15 0,5 M NaCl/1X TE 37°C
15 2X SSC 56°C
Anschließend wird in einer aufsteigenden Ethanol/Ammoniumacetat Reihe dehydriert
(s. 3.4.4.3), die Schnitte für 2 Stunden luftgetrocknet und dann für die Belichtung der
Autoradiographie-Filme verwendet.
3.4.4.6 Filmbelichtung
Die Belichtung der Autoradiographie-Filme erfolgt für 12-16 Stunden in einer lichtun-
durchlässigen Filmkassette. Gleichzeitig wird jeder Film mit 2 Objektträgern, auf die
radioaktive C14-Standards in aufsteigender Intensität aufgebracht sind, belichtet. Diese
dienen später der Erstellung einer Standardkurve und als Referenz zwischen den
Filmen.
3.4.4.7 Filmentwicklung
Die Entwicklung der Autoradiographiefilme erfolgt in einem vollautomatischen
Filmentwickler. Die belichteten Filme werden in völliger Dunkelheit in den Automaten
eingelegt.
3.4.4.8 MCID-Auswertung
Die quantitative, densitometrische Auswertung der entwickelten Filme erfolgt mittels
MCID M5 System. Hierbei werden die Filme auf einen Leuchtschirm gelegt, mittels
Kamera digitalisiert und stark vergrößert, sowie farbkodiert am Bildschirm dargestellt.
Nach Standardisierung von Kamera-Film-Abstand (57 cm), Blende (5,6), Zoomfaktor
und Intensität des Leuchtschirms (226) werden die Dichtemesswerte für die
C14-Standards erhoben und eine Standardkurve erstellt.
Die hirnregionenspezifische Auswertung der Versuchstiere erfolgt mittels
standardisierter Messfelder, die über ausgewählte Regionen hoher Signalintensität
projiziert werden können. Die Software liefert einen Durchschnittswert für die
ausgewählte Fläche. Acht Hirnregionen, für die BDV einen Tropismus zeigt, werden
45

3 - Methoden
ausgewählt (s. ): Gesamtkortex, Präfrontalkortex, Kortex regional, Striatum,
Nucleus reticularis des Thalamus, sowie die hippocampalen Strukturen Gyrus dentalis
und CA3. Die Werte für Gesamtcortex, Gyrus dentalis und CA3 werden ermittelt, indem
die komplette Struktur in jedem Schnitt individuell markiert und ein Durchschnittswert
errechnet wird. Der Wert für das Striatum wird aus zwei Einzelmessungen im ventralen
Bereich gemittelt. Aus den Messwerten des Fünffachansatzes wird anschließend ein
Mittelwert errechnet, der den für die gemessene Struktur und das Versuchstier
repräsentiert. Die Gruppenwerte ergeben sich als Mittelwert aller Individualwerte.
Abbildung 9
Abbildung 9. Für die Quantifizierung ausgewählte Strukturen.
Die Markierungen entsprechen den Arealen, die standardisiert in jedem Schnitt
quantifiziert werden. Zusätzlich werden individuell der Kortex, sowie die hippocampalen
Strukturen Gyrus dentalis und CA3 markiert und densitometrisch ausgewertet (nach
[168]). PFK: Präfrontalkortex, K1 und K2: regionale Auswertung des Kortex, S1 und
S2: Striatum, T: Thalamus (Nucleus reticularis).
3.4.4.9 Autoradiographie
Herstellung der Autoradiographieemulsion. Der gesamte Prozess findet unter
Dunkelkammerbedingungen statt. Der strahlenempfindliche Feststoff wird in einem
46

3 - Methoden
Volumenverhältnis von 1:1 mit 6 M Ammoniumacetat gemischt und im Wasserbad bei
41°C während 30 Minuten unter vorsichtigem Mischen in Emulsion gebracht.
Belichtung. Die Objektträger werden 2x langsam in die Emulsion getaucht und
daraufhin in senkrechter Position für 3-4 Stunden getrocknet. Anschließend werden die
Schnitte, durch jeweils einen leeren Objektträger voneinander getrennt, um eine
Belichtung benachbarter Schnitte zu vermeiden und 3-5 Tage bei 4°C gelagert.
Entwicklung. Die Entwicklung erfolgt bis einschließlich des Fixierungsschrittes unter
Dunkelraumbedingungen. Kodak D19 Entwickler wird im Verhältnis 1:1 mit ddiH2O
gemischt und filtriert. Entwickler, ddiH2O für Waschschritte und Fixierer werden im
Eisbad auf 15°C abgekühlt. Jetzt werden die Schnitte für 4 min im Entwickler, 30 s in
ddiH2O und 8 min in Fixierer inkubiert und anschließend 15 min in Leitungswasser
gewaschen. Anschließend werden die Schnitte mit Hämatoxylin/Eosin gegengefärbt (s.
3.1.9).
3.4.5 Quantitative Taqman PCR
Die Taqman PCR beruht auf einer biphasichen Amplifikation von Template DNA (oder
cDNA), bei der die entstehenden Produkte simultan amplifiziert und quantifiziert
werden. Neben einem Primer-Paar wird eine DNA Sonde eingesetzt, die am 5'-Ende
mit einem Reporter- (FAM: 6-Carboxy-fluorescein oder JOE: 2,7-Dimethoxy-4,5-
dichloro-6-carboxyfluorecein) und 3'-Ende mit einem Quencher-Fluoreszenzfarbstoff
(TAMRA: 6-Carboxy-tetramethylrhodamin) markiert ist. In diesem Zustand wird die
Fluoreszenz des Reporter-Farbstoffs durch die räumliche Nähe zum Quencher-
Farbstoff unterdrückt. Die Sonde bindet komplementär an einen DNA-Abschnitt der
vom Primer-Paar flankiert wird. Die AmpliTaq Gold Polymerase besitzt
Exonukleaseaktivität und spaltet bei der Amplifikation die hybridisierte Sonde. Dadurch
entfällt die räumliche Kopplung von Reporter und Quencher und die vom Reporter
nach Anregung durch einen Argon-Laser emittierte Fluoreszenz wird als Maß für das
entstandene Amplifikationsprodukt quantifiziert (s. ). Da die Sonde zum
Zeitpunkt der Amplifikation hybridisiert haben muss, besteht der Amplifikationszyklus
aus nur zwei Phasen: einer Denaturierungsphase und einer Annealing-Amplifikations-
Phase. Durch den biphasische Verlauf ergeben sich strenge Anforderungen an Primer-
und Sondendesign, das durch Primer Express Software unterstützt wird.
Abbildung 10
47

3 - Methoden
3.4.5.1 Leukozyten-Präparation
Bei der Dekapitation wird ca. 1 ml Blut in EDTA-Röhrchen aufgefangen, 5 min bei 20°C
und 1500 x g (2500 rpm in Beckman Alegra) zentrifugiert und das Plasma abpipetiert.
Das Zellpallet wird in 5 ml Ammoniumchlorid-Lysepuffer resuspendiert, 10 min bei 4°C
inkubiert und erneut 5 min bei 4°C und 1500 x g zentrifugiert. Falls nötig wird der Lyse-
Schritt wiederholt. Anschließend werden die Leukozyten mit 3 ml Dulbecco's PBS
gewaschen und 5 min bei 4°C und 700 x g (1700 rpm in Beckman Alegra) zentrifugiert.
Nach Entfernen des Waschpuffers werden die Leukozyten in TRI Reagent
resuspendiert und RNA extrahiert (s. 3.4.5.2).
3.4.5.2 RNA Extraktion
50-100 mg Hirngewebe werden in 1 ml TRI Reagent mit einem Einmalstößel
homogenisiert und mindestens 5 Minuten bei Raumtemperatur inkubiert. RNA aus
Leukozyten wird durch Zugabe von 1 ml TRI Reagent und einer Inkubation von
5 Minuten bei RT extrahiert. Eine längere Aufbewahrung kann bei -70°C erfolgen. Zu
dem Homogenisat werden 200 µl Chloroform pipetiert und durch starkes Schütteln
gemischt, 10 min bei Raumtemperatur inkubiert und dann 15 min bei 4°C und
14.000 rpm in einer Tischzentrifuge zentrifugiert. Die wässrige Phase wird in ein neues
Eppendorf Cap transferiert, die RNA durch Inkubation für 10 min mit 500 µl Isopropanol
gefällt und mittels Zentrifugation für 8 min bei 4°C und 14.000 rpm in einer
Tischzentrifuge präzipitiert. Nach Entfernen des Überstandes wird das RNA-Pallet mit
1 ml 75%igem Ethanol gewaschen und erneut 5 min zentrifugiert. Anschließend
entfernt man das Ethanol, trocknet das RNA-Pallet bis es beginnt, glasig zu werden
und löst es in RNase freiem Wasser. Die RNA wird anschließend mittels Spektral-
photometer quantifiziert und bei -70°C gelagert.
3.4.5.3 Reverse Transkription
500 ng der aus dem rechten Präfrontalkortex und den periphere mononukleäre
Blutzellen (PBMC) extrahierten RNA je Versuchstier wird revers transkribiert. Dabei
werden Multiscribe RT (PE) und Random Primers (PE) verwendet.
3.4.5.4 Taqman PCR
Die entstandene cDNA wird anschließend für die Taqman real-time PCR verwendet.
Das verwendete Primer-Paar p40bobe-187F und p40bobe-286R amplifiziert ein ca.
100 bp langes Segment aus dem für N kodierenden ersten ORF. Mittels der 5' FAM
48

3 - Methoden
und 3' TAMRA markierten Sonde p40bobe-247T kann das Amplifikationsprodukt
quantifiziert werden.
3’5’ 3’
5’
R
QPol
3’5’ 3’
5’R Q
PolPrimer1
Primer2
Sonde
3’5’ 3’
5’
RQ
Pol
Abbildung 10. Quantifikation des Amplifikationsprodukts bei der Taqman PCR.
Durch die Exonukleaseaktivität der AmpliTaq Gold Polymerase wird die während der
Amplifikation hybridisierte Sonde hydrolysiert. Dabei kommt es zur räumlichen
Entkopplung von Quencher- und Reporter-Fluoreszenzfarbstoff, der jetzt durch einen
Argon-Laser angeregt werden kann. Die vom Reporter emittierte Fluoreszenzstrahlung
dient als Maß für die Menge entstandenen Amplifikationsprodukts (nach [171]).
Pol AmpliTaq GoldTM DNA Polymerase
R Reporter Fluoreszenzfarbstoff
Q Quencher Fluoreszenzfarbstoff
Für die Erzeugung einer Standardkurve wird eine Verdünnungsreihe von 0 bis
2,5 x 105 Kopien des Plasmids pBSKBVcore eingesetzt. Es besteht aus einem
kommerziellen pBluescript SK II-Plasmid in das von der Not I- bis zur Sal I-
Restriktionsenzym-Schnittstelle die BDV Nukleotide 1-8897 kloniert sind. Als
Negativkontrolle dient RNA, die parallel aus einem nicht infizierten Tier extrahierte
wurde.
49

3 - Methoden
Die Reaktionen finden in einer speziellen für die optische Auswertung geeigneten 96
Loch Mikrotiterplatte (MicroAmp Optical 96-Well Reaction Plate) in einem
Reaktionsvolumen von 25µl statt. Die Amplifikationsparameter sind: Präinkubation bei
50°C für 2 min, initiale Denaturierung bei 95°C für 10 min sowie anschließend 45
Zyklen mit Denaturierung bei 95°C für 15 s und Annealing/Elongation bei 60°C für 1
min. Ramp-Zeiten werden auf den voreingestellten Werten belassen.
Der Reaktionsansatz besteht aus 6,25µl H2O, 12,5µl Taqman Universal PCR Master
Mix (enthält AmpliTaq Gold® DNA Polymerase, AmpErase® UNG, dNTPs (mit dUTP),
sowie Puffer), je 1,25µl Primer p40bobe – 187F, p40bobe – 286R (jeweils 4 mM) und
Sonde p40bobe – 247T (2 mM) sowie 2,5µl cDNA.
Die cDNA wird in die MicroAmp Mikrotiterplatte vorpipetiert und eine Mastermix aus
den restlichen Reagenzien dazugegeben. Nach Mischen durch mehrmaliges auf und
ab Pipetieren wird die Mikrotiterplatte mit den Deckeln verschlossen und die PCR
gestartet. Sowohl Standards, als auch Proben werden im Doppelansatz bestimmt und
die Ergebnisse gemittelt. Die Auswertung erfolgt mittels Real-Time Sequence
Detection Software (Perkin Elmer). Für die Standardkurve werden die Zahl der
Ausgangskopien der Standards gegen den Threshhold Cycle geplottet (Amplifikations-
zyklus, bei dem es erstmals zu einem signifikanten Anstieg des Fluoreszenzsignal
kommt). Mit Hilfe der Standardkurve kann anschließend für jeden einzelnen
Reaktionsansatz die Ausgangskopienzahl extrapoliert werden.
3.4.6 Virustitrierung
Die Quantifizierung aus Hippocampus isolierter, infektiöser Viruspartikel erfolgt mittels
Focus Immunoassay [104]. Dabei werden in Kultur befindliche, empfängliche Zellen mit
Gewebshomogenisat in einer Verdünnungsreihe infiziert und nach 3-4 Tagen mittels
Anti-N und Anti-P Antikörpern auf Anwesenheit virale Antigene untersucht. Die
Visualisierung erfolgt mit einem zweiten, HRP-gekoppelten Antikörper und
anschließender Enzym-Substrat-Reaktion (3-Amino-9-Ethyl-Carbazol, H2O2). Mikro-
skopisch können Focus Forming Units (ffu) ausgezählt und durch Multiplikation mit der
entsprechenden Verdünnung der Virustiter im Ausgangsgewebe berechnet werden.
3.4.6.1 Zellkultur
In Kultur befindliche fötale Rattengliazellen werden ca. 1:2 in einem Volumen von
100µl/Loch in 96 Loch Mikrotiterplatten ausgesät, so dass nach 24 Stunden ein
annähern konfluierender Zellrasen entsteht. Als Kulturmedium wird DMEM mit einem
50

3 - Methoden
Zusatz von 1V% L-Glutamine (200 mM), 1V% Penicillin/Streptomycin und 10V% FCS
verwendet.
3.4.6.2 Titrierung
Aus dem 10%igen Hirnhomogenisat wird eine Verdünnungsreihe mit 1:100 als
Startverdünnung und schrittweise 10facher Weiterverdünnung in DMEM hergestellt.
100µl pro Verdünnungsstufe werden auf den annähernd konfluierenden Zellrasen
pipetiert und 3-4 Tage bei 37°C und bei 5% CO2 kultiviert. Die Titrierung erfolgt im
Doppelansatz.
Volumen (µl) Reagenz Inkubationszeit (min) Temperatur
200 3% PFA 15 RT
400 1% Triton/PBS 10-15 RT
200 PBS 4 °C
100 1% FCS/PBS 10 RT
100 anti-N und anti-P 1:3000 in 90 37°C
1% FCS/PBS
200 1% FCS/PBS (3 x Waschen) RT
100 Ziege-anti-Kaninchen-Ak 1:2000 in 60 37°
1% FCS/PBS
200 1% FCS/PBS (3 x Waschen) RT
100 Substrat-Mix * RT
200 PBS (Waschen) RT
* Die Enzym-Substrat-Reaktion wird bei Erscheinen eines rötlichen Farbumschlags mit
PBS gestoppt, das anschließend auf den Zellen verbleibt. Die Mikrotiterplatten können
bis zur mikroskopischen Auswertung bei 4°C gelagert werden.
3.4.7 Western Immunoblot
Für die Quantifizierung viraler Proteine wird ein Western Immunoblot durchgeführt.
Dabei wird Hippocampus-Homogenisat elektrophoretisch aufgetrennt und auf eine
Nitrozellulosemembran transferiert. Antikörper gegen das Phosphoprotein (P) von BDV
und Aktin als zelluläres Referenzprotein markieren diese selektiv. Durch einen zweiten,
fluoreszenzmarkierten Antikörper werden die Proteinbanden dargestellt und
51
3 - Methoden
anschließend fluorometrisch quantifiziert. Die Konzentration von P wird als Anteil der
Konzentration von Aktin angegeben.
3.4.7.1 Elektrophoresegel
Es wird ein 12%iges Acrylamidgel verwendet, das mit einem 6%igen Gel überschichtet
wird, in dessen Vertiefungen die Proben geladen werden.
Für die beiden Gele wird wie folgt pipetiert: Die Reagenzien 1-3 werden zusammen-
pipetiert und gut gemischt. Die Lösungen 4-6 werden direkt vor dem Gießen des Gels
zugegeben, da unmittelbar danach die Polymerisation beginnt.
12%iges Gel 6%iges Gel
1
2a
2b
3
4
5
6
dH20
1,5 M Tris HCl pH 8,8
0,5 M Tris HCl pH 6,8
10% SDS
30% Acrylamid 30:0,8
10% APS
TEMED
1,7 ml
1,25 ml
50 µl
2 ml
25 µl
5 µl
1,33 ml
625 µl
25 µl
500 µl
15 µl
5 µl
Nach Gießen des 12%igen Elektrophoresegels wird dieses mit ddiH20 überschichtet
und die vollständige Polymerisation abgewartet (ca. 15-20 min). Anschließend wird
nach Entfernen des Wassers das 6%ige Gel über das bereits polymerisierte Gel
gegossen und mit einem Kamm die Vertiefungen für das spätere Laden der Proben
freigehalten. Nach abgeschlossener Polymerisation (ca. 5-10 min) wird der Kamm
entfernt, das Gel in den dafür vorgesehenen Behälter gegeben, mit Elektrophorese-
puffer bedeckt und die Vertiefungen mit Hilfe einer Spritze mit Elektrophoresepuffer
ausgewaschen.
3.4.7.2 Laden des Gels und Elektrophorese:
10 µl Probe werden mit dem gleichen Volumen Probenladepuffer gemischt,
gemeinsam mit dem Marker 3 Minuten bei 85°C erhitzt und auf Eis gekühlt.
Anschließend werden 10-15 µl des Gemischs in die Vertiefung des Gels pipetiert und
die Elektrophorese bei einer Stromstärke von 40 mA gestartet. Sobald der Marker das
andere Ende des Gels erreicht hat, wird die Elektrophorese gestoppt.
52

3 - Methoden
3.4.7.3 Transfer
Vier Lagen Whatman Papier werden mit Transferpuffer befeuchtetes und in die
Transferbox gelegt. Auf diese wird das aus dem Elektrophoreseapparat gelöste Gel
gelegt, mit der ebenfalls in Transferpuffer befeuchteten Nitrozellulose-Membran und
anschließend weiteren vier Lagen Whatman Papier bedeckt, wobei darauf geachtet
wird, dass sich keine Luftblasen zwischen dem Gel und der Nitrozellulosemembran
befinden. Nach Zusammenbau der Box wird der Transfer bei 3 mA/cm2 gestartet. Nach
einstündigem Transfer und Markierung der Proteinseite wird die Membran vorsichtig
entnommen.
3.4.7.4 Blockieren unspezifischer Bindung
Die Membran wird ca. 10 min mit Ponceau Farbstoff gefärbt und anschließend 5 min
mit ddiH20 gewaschen. Um spätere unspezifische Bindungen zu vermeiden wird für 1 h
mit einer 1%igen Milchpulverlösung in Waschpuffer bei 4°C inkubiert.
3.4.7.5 Markierung der Proteine
Die Nitrozellulosemembran wird nun in einer 1:5000 Verdünnung von anti-P Antikörper
in Waschpuffer, bzw. Maus mAb gegen Aktin für 1 h bei RT inkubiert. Anschließend
wird drei Mal mit Waschpuffer gewaschen und dann für 1 h bei RT mit dem zweiten
Antikörper (HRP-konjugierter Ziege-anti-Kaninchen IgG) in einer 1:2000 Verdünnung in
Waschpuffer inkubiert. Für Aktin wird HRP-konjugierter Ziege-anti-Maus IgG Antikörper
in einer 1:2000 Verdünnung Waschpuffer verwendet. Anschließend wird erneut drei
Mal mit Waschpuffer gewaschen.
3.4.7.6 Detektion
Der Detektionsmix (Wasserstoffperoxid und 4-Chloro-1-naphthol) wird wie vom
Hersteller angegeben vorbereitet und die Membran mit 1-2 ml des Mixes beschichtet.
Nach 5minütiger Inkubation bei RT wird der Detektionsschirm des Phosphoimmagers
exponiert.
3.4.7.7 Auswertung
Die Quantifizierung des Fluoreszenzsignals erfolgt mittels Phosphoimager und IQMac
Software.
53

3 - Methoden
3.5 Bestimmung immunologischer Parameter
3.5.1 Gewebspräparation und Gefrierschnitt
s. 3.4.1 und 3.4.3
3.5.2 Immunhistochemie (IHC)
Für die selektive Darstellung bestimmter Antigene im histologischen Schnitt werden
monoklonale Antikörper (hier aus der Maus) mit dem Gewebe inkubiert und mit ihrem
spezifischen Epitop zur Bindung gebracht. Nach Entfernung nicht-gebundener
Antikörper wird mit einem zweiten, biotinylierten Anti-Maus-IgG-Antikörper inkubiert,
der an den monoklonalen Antikörper bindet. Anschließend werden makromolekulare
Avidin - biotynilierte HRP - Komplexe (Vectastain Elite ABC Kit) über die hochaffine
Avidin-Biotin-Bindung gekoppelt und über eine Enzym-Substrat-Reaktion dargestellt (s.
Abbildung 11).
Gewebe
Biotin
monokl. Ak
biotinyl. 2. Ak
Avidin
biotynil. HRP
Substrat
Produkt
Abbildung 11. Prinzip der IHC mittels mAb und ABC Elite Kit.
Detektion spezifischer Epitope durch monoklonale Antikörper und Avidin-Biotin-
Komplexe. Die Visualisierung erfolgt mittels DAB-Reaktion (nach: [172]).
54

3 - Methoden
Hierfür werden die histologischen Schnitte 30 min in Tris IHC-Puffer angefeuchtet und
mit dem unverdünnten ersten, monoklonalen Antikörper (Ox-42, Ox-8 oder W3/25) bei
4°C für 12-16 Stunden beschichtet [70]. Nach dreimaligem Waschen in Tris IHC-Puffer
wird mit dem HRP gekoppelten Ziege-anti-Maus-IgG-Antikörper in einer 1:200
Verdünnung erneut 12-16 Stunden bei 4°C inkubiert. Anschließend wird dreimal
gewaschen und durch eine 20-30minütige Inkubation in einer 20%igen H2O2
Verdünnung in Methanol unspezifische Peroxidaseaktivität im Gewebe abgepuffert.
Nach drei weiteren Waschschritten erfolgt die Inkubation mit dem makromolekularen
Avidin-biotinylierte HRP-Komplex (Avidin und biotynilierte HRP bilden innerhalb von
30 min Vorinkubation makromolekulare Komplexe). Nach 90 min bei RT wird erneut
dreimal gewaschen und die Enzym-Substrat-Reaktion zur Visualisierung gestartet. Als
Substrat dient Diaminobenzidin-tertahydrochlorid (DAB), das in ein rotbraunes
Reaktionsprodukt umgewandelt wird. Anschließend wird mit Hämatoxylin gegengefärbt
(s. 3.1.9).
Für die Beurteilung der mikroglialen Reaktion, sowie der T-Lymphozyteninfiltrate
werden korrespondierende Schnitte, die Cortex und Striatum enthalten, verwendet. Auf
jedem Objektträger befindet sich Gewebe von einem Tier je Gruppe.
Ox-42 bindet den Complement-Rezeptor Typ 3 [163] und dient der Detektion
mikroglialer Zellen und Makrophagen [70], während Ox-8 und W3/25 das CD8 alpha-
bzw. CD4-Molekül binden und Marker für CD8+- bzw. CD4+-T-Lymphozyten darstellen
[70, 162, 164].
In 5 Gesichtsfelder bei 100X Vergrößerung je Versuchstier werden gefärbte CD4(+)-
bzw. CD8(+)-T-Lymphozyten ausgezählt und ein Mittelwert gebildet. Die Werte werden
anschließend kategorisiert und für jede Gruppe der Median ermittelt.
+ < 20 Zellen
++ 20-40 Zellen
+++ > 40 Zellen
Die Auswertung der auf Mikroglia gefärbten Schnitte erfolgt qualitativ. Dabei werden
Unterschiede in Aktivierungsgrad und Zahl gefärbter Mikrogliazellen beurteilt.
3.5.3 Mikrophotographie
Sämtliche mikrophotographischen Aufnahmen werden an einem Nikon Eclipse E600
Mikroskop und einer Nikon Fx 35A Kamera hergestellt. Dabei werden 100 ASA
55

3 - Methoden
Diafilme von Kodak verwendet und die entstandenen Diapositive mit einem Dia-
Scanner digitalisiert.
3.5.4 ELISA
3.5.4.1 Beschichtung der Mikrotiterplatten
96-Loch Mikrotiterplatten werden mit Antigen p40(N) bzw. p23(P) beschichtet. Dafür
wird eine Antigenlösung in Borat Puffer mit einer Konzentration von 0,5 ng Antigen/µl
hergestellt und jedes Loch der Mikrotiterplatte mit in einem Volumen von 100 µl gefüllt.
Anschließend werden die Mikrotiterplatten 12-16 Stunden bei 37°C in feuchtem Milieu
inkubiert.
3.5.4.2 ELISA [102]
Nach der Beschichtung werden die Vertiefungen der Mikrotiterplatte drei Mal mit ELISA
Waschlösung gewaschen. Um unspezifische Bindungen an der Mikrotiterplatte zu
verhindern wird bei 37°C für 45 min mit 150 µl ELISA Blocklösung inkubiert und erneut
drei Mal gewaschen. Als erster Antikörper wird jetzt 100 µl einer Serumverdünnungs-
reihe beginnend mit einer Verdünnung von 1:50 in ELISA Blocklösung dazupipetiert
und 60 min bei 37°C inkubiert. Nach drei weiteren Waschschritten wird für 60 min bei
37°C mit dem zweiten Antikörper, 1:4000 in ELISA Blocklösung verdünnter HRP-
gekoppelter Ziege-anti-Ratte-IgG-Antikörper inkubiert. Nach erneutem dreimaligem
Waschen wird die Enzym-Substrat-Reaktion durch Zugabe des ELISA TMB
Substratmixes gestartet und nach 60 min durch Zugabe von ¼-konzentrierter
Schwefelsäure gestoppt
Als positiv Kontrolle dient Referenzserum "Ratte 1.1" einer BDV infizierten Ratte (Labor
Standard, Startverdünnung 1:100 (P) bzw. 1:1000 (N)) und als negativ Kontrolle NRtS
(normales Rattenserum).
3.5.4.3 optische Auswertung
Die Farbintensität wird mit einem automatisierten Photometer bei 450 nm gemessen
und die Werte mittels Soft max 2.35 Software (Molecular Devices) ausgewertet.
3.5.5 RNase Protektionsassay (RPA)
Das RNase Protektionsassay basiert auf folgendem Mechanismus: Eine radioaktiv
markierte RNA-Sonde, die komplementär zu dem zu quantifizierenden RNA-Abschnitt
56

3 - Methoden
ist, wird mit der extrahierten RNA hybridisiert (Abbildung 12 #1 und 2). In einem
anschließenden Verdau mit einer einzelstrangspezifischen RNase wird nur nicht-
hybridisierte Proben-RNA und Sonde hydrolysiert (Abbildung 12 #3 und 4).
Hybridisierte Abschnitte hingegen werden nicht verdaut (RNase Protektion, Abbildung
12 #5). Nach denaturierender elektrophoretischer Auftrennung wird der Anteil unver-
dauter Sonde als Maß für die komplementäre Proben-RNA anhand der emittierten
Strahlung quantifiziert (Abbildung 12 #6).
Das RPA wird hier im Multiprobe-Ansatz durchgeführt. Dabei kommen Sonden für eine
Reihe verschiedener Zielsequenzen gleichzeitig zum Einsatz, die sich nach
Elektrophorese anhand ihrer unterschiedlichen Länge unterscheiden lassen.
Alle Prozeduren bis zum RNase-Verdau laufen streng unter RNase-freien
Bedingungen ab.
isolierte RNA
komplementärer Anteil derzu quanti fizierenden mRNA
einzelstrangspezifische RNaseRNA Sonde
2
3 6
5
1 4
Abbildung 12. Prinzip des RNase Protektionsassays (RPA).
Beim RNase Protektions Assay werden komplementäre radioaktiv-markierte Sonden
zu RNA hybridisiert und nach einem Verdau mit einzelstrangspezifischer RNase und
elektrophoretischer Auftrennung die verbliebene Sonde quantifiziert. Die
Signalintensität dient als Maß für die stattgefundene Hybridisierung und damit bei
Sondenüberschuß für die in der Ausgangs-RNA enthaltene Menge zur Sonde
komplementärer Nukleinsäure.
57

3 - Methoden
3.5.5.1 Synthese radioaktiv markierter Sonden
Die radioaktiv markierten Sonden für die Hybridisierung werden durch Transkription
eines RiboQuant Multi-Probe Template Set (rCK-1 + rCK3) mittels MAXIscript Kit
synthetisiert. Dafür werden die Reagenzien in der folgenden Reihenfolge pipetiert und
eine Stunde bei 37°C inkubiert.
Volumen (µl) Reagenz
2 Nuklease freies Wasser
1 RiboQuant Multi-Probe Template Set rCK-1
2 10X Transkriptionspuffer
je 1 10mM ATP, UTP, GTP
10 α-35S-CTP
2 T7 RNA Polymerase und Nukleaseinhibitor
Für den Verdau der DNA-Templates wird 1 µl DNase I (2 U/µl) dazupipetiert und
30 min bei 37°C inkubiert und anschließend 80 µl RNase freies H2O zugefügt. Für die
folgende RNA-Extraktion werden 100 µl Phenol/Chloroform/Isoamylalkohol zugegeben,
zentrifugiert und die wässrige Phase entnommen. Jetzt wird für die Präzipitation der
synthetisierten RNA 10 µl (oder 1/10 Volumen) Ammoniumacetat sowie 275 µl (2,5
Volumina) Ethanol dazupipetiert, 30 min bei -20°C inkubiert und die RNA durch 15 min
Zentrifugation bei 4°C und 14.000 rpm (Baxter 17R) zum Präzipitieren gebracht. Nach
Abpipetieren des Überstandes wird die RNA in 50 µl RPA III Hybridisierungslösung
resuspendiert. Mit Hilfe zweier Aliquots à 1 µl, deren Radioaktivität im
Szintillationszähler bestimmt wird, wird die Inkorporationsrate berechnet und die Sonde
verdünnt. Pro Ansatz werden 2 µl verdünnter Sonde benötigt.
3.5.5.2 RNase Präparation und Hybridisierung
10 µg RNA je Versuchstier, extrahiert aus dem linken Präfrontalkortex, sowie zwei
Hefe-RNA-Kontrollen werden in der Zentrifuge vakuumgetrocknet und die RNA in 8 µl
RPA III Hybridisierungslösung resuspendiert. Je Ansatz werden 2 µl Sonde zupipetiert,
2-3 min auf 95°C erhitzt und bei 56°C über Nacht hybridisiert
58

3 - Methoden
3.5.5.3 RNase Verdau
Einzelstrangspezifischer RNase A/T1 Mix wird 1:1000 in RNase Digestions-Puffer
verdünnt und 100 µl zu jedem Ansatz pipetiert. Eine der Hefe-RNA-Kontrollen erhält
reinen RNase Digestions-Puffer ohne RNase. Sie dient als Kontrolle für den RNase-
Verdau. Nach Mischen wird 45 min bei 30°C inkubiert und die Reaktion anschließend
durch Zugabe von 150 µl Inaktivierungs-/Präzipitationslösung III gestoppt und die RNA
für 15 min präzipitiert. Nach einer Zentrifugation für 15 min bei 4°C und 14.000 rpm
(Baxter 17R) wird der Überstand entfernt und die präzipitierte RNA luftgetrocknet.
3.5.5.4 Auftrennung und Visualisierung unverdauter RNA
Die RNA-Pallets werden in 5 µl Gel-Loading Puffer resuspendiert, 3-5 min bei 95°C
erhitzt und auf Eis abgekühlt. Je Ansatz werden 4 µl auf ein denaturierendes 5%
Acrylamidgel (8 M Harnstoff/1XTBE) geladen und durch Elektrophorese aufgetrennt.
Als Laufpuffer wird 5X TBE verwendet. Anschließend wird das Gel vakuumgetrocknet
und ein Phosphoimager-Schirm über Nacht belichtet.
3.5.5.5 Auswertung
Die densitometrische Analyse erfolgt mittels IQMac software. Anschließend werden die
Ergebnisse anhand einer internen Kontrolle (GAPDH Kontrollsonde) normalisiert.
59
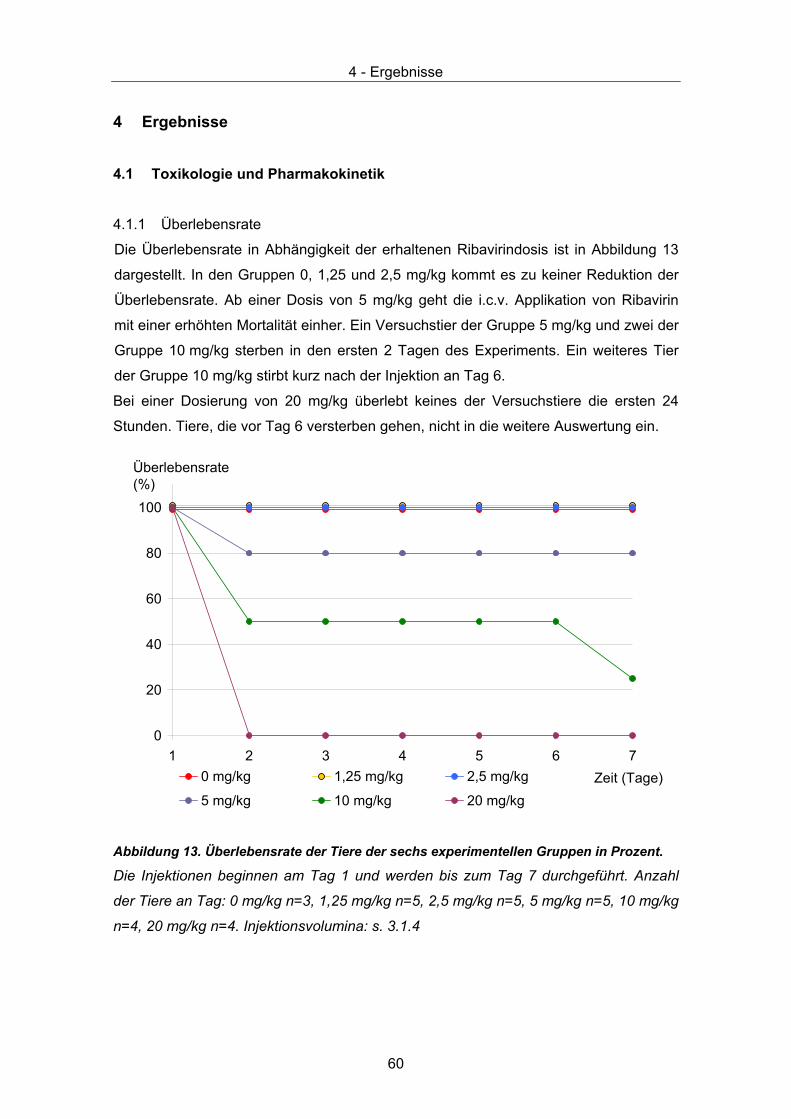
4 - Ergebnisse
4 Ergebnisse
4.1 Toxikologie und Pharmakokinetik
4.1.1 Überlebensrate
Die Überlebensrate in Abhängigkeit der erhaltenen Ribavirindosis ist in Abbildung 13
dargestellt. In den Gruppen 0, 1,25 und 2,5 mg/kg kommt es zu keiner Reduktion der
Überlebensrate. Ab einer Dosis von 5 mg/kg geht die i.c.v. Applikation von Ribavirin
mit einer erhöhten Mortalität einher. Ein Versuchstier der Gruppe 5 mg/kg und zwei der
Gruppe 10 mg/kg sterben in den ersten 2 Tagen des Experiments. Ein weiteres Tier
der Gruppe 10 mg/kg stirbt kurz nach der Injektion an Tag 6.
Bei einer Dosierung von 20 mg/kg überlebt keines der Versuchstiere die ersten 24
Stunden. Tiere, die vor Tag 6 versterben gehen, nicht in die weitere Auswertung ein.
0
20
40
60
80
100
1 2 3 4 5 6 7Zeit (Tage)
Überlebensrate (%)
0 mg/kg 1,25 mg/kg 2,5 mg/kg
5 mg/kg 10 mg/kg 20 mg/kg
Abbildung 13. Überlebensrate der Tiere der sechs experimentellen Gruppen in Prozent.
Die Injektionen beginnen am Tag 1 und werden bis zum Tag 7 durchgeführt. Anzahl
der Tiere an Tag: 0 mg/kg n=3, 1,25 mg/kg n=5, 2,5 mg/kg n=5, 5 mg/kg n=5, 10 mg/kg
n=4, 20 mg/kg n=4. Injektionsvolumina: s. 3.1.4
60
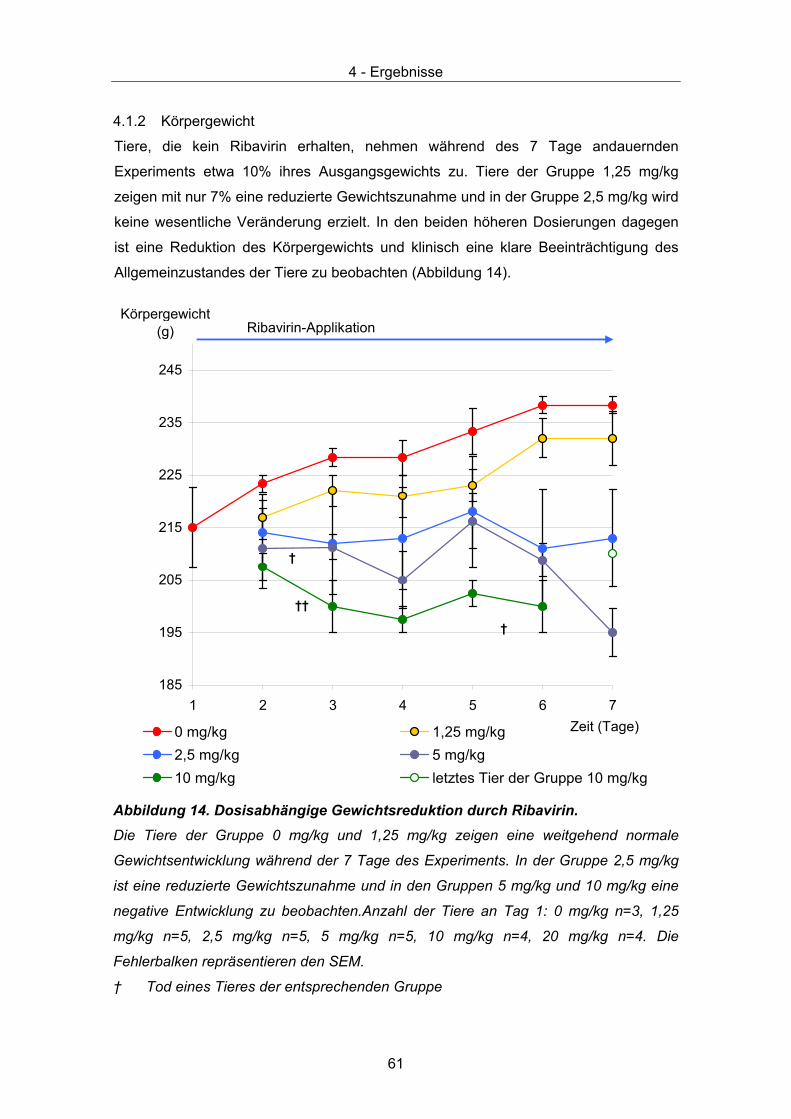
4 - Ergebnisse
4.1.2 Körpergewicht
Tiere, die kein Ribavirin erhalten, nehmen während des 7 Tage andauernden
Experiments etwa 10% ihres Ausgangsgewichts zu. Tiere der Gruppe 1,25 mg/kg
zeigen mit nur 7% eine reduzierte Gewichtszunahme und in der Gruppe 2,5 mg/kg wird
keine wesentliche Veränderung erzielt. In den beiden höheren Dosierungen dagegen
ist eine Reduktion des Körpergewichts und klinisch eine klare Beeinträchtigung des
Allgemeinzustandes der Tiere zu beobachten (Abbildung 14).
185
195
205
215
225
235
245
1 2 3 4 5 6 7Zeit (Tage)
Körpergewicht (g)
0 mg/kg 1,25 mg/kg2,5 mg/kg 5 mg/kg10 mg/kg letztes Tier der Gruppe 10 mg/kg
Ribavirin-Applikation
†
††
†
Abbildung 14. Dosisabhängige Gewichtsreduktion durch Ribavirin. Die Tiere der Gruppe 0 mg/kg und 1,25 mg/kg zeigen eine weitgehend normale
Gewichtsentwicklung während der 7 Tage des Experiments. In der Gruppe 2,5 mg/kg
ist eine reduzierte Gewichtszunahme und in den Gruppen 5 mg/kg und 10 mg/kg eine
negative Entwicklung zu beobachten.Anzahl der Tiere an Tag 1: 0 mg/kg n=3, 1,25
mg/kg n=5, 2,5 mg/kg n=5, 5 mg/kg n=5, 10 mg/kg n=4, 20 mg/kg n=4. Die
Fehlerbalken repräsentieren den SEM.
† Tod eines Tieres der entsprechenden Gruppe
61
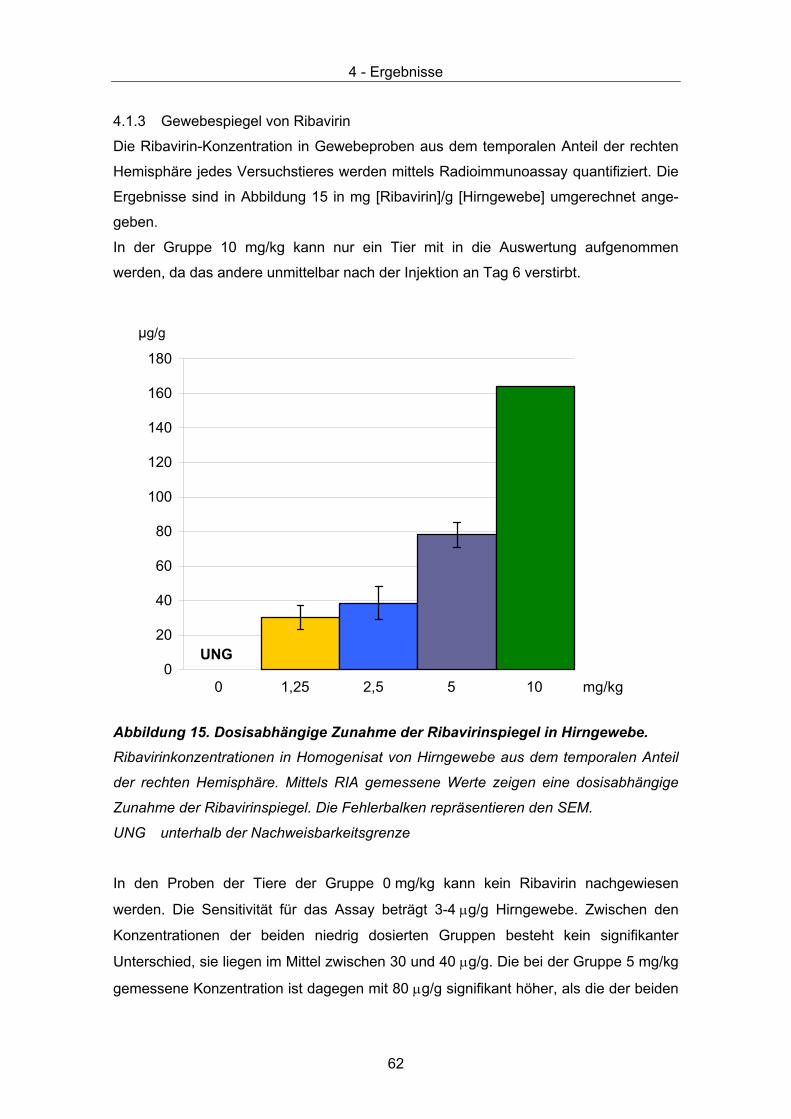
4 - Ergebnisse
4.1.3 Gewebespiegel von Ribavirin
Die Ribavirin-Konzentration in Gewebeproben aus dem temporalen Anteil der rechten
Hemisphäre jedes Versuchstieres werden mittels Radioimmunoassay quantifiziert. Die
Ergebnisse sind in Abbildung 15 in mg [Ribavirin]/g [Hirngewebe] umgerechnet ange-
geben.
In der Gruppe 10 mg/kg kann nur ein Tier mit in die Auswertung aufgenommen
werden, da das andere unmittelbar nach der Injektion an Tag 6 verstirbt.
0
20
40
60
80
100
120
140
160
180
UNG
µg/g
1052,51,250 mg/kg
Abbildung 15. Dosisabhängige Zunahme der Ribavirinspiegel in Hirngewebe. Ribavirinkonzentrationen in Homogenisat von Hirngewebe aus dem temporalen Anteil
der rechten Hemisphäre. Mittels RIA gemessene Werte zeigen eine dosisabhängige
Zunahme der Ribavirinspiegel. Die Fehlerbalken repräsentieren den SEM.
UNG unterhalb der Nachweisbarkeitsgrenze
In den Proben der Tiere der Gruppe 0 mg/kg kann kein Ribavirin nachgewiesen
werden. Die Sensitivität für das Assay beträgt 3-4 µg/g Hirngewebe. Zwischen den
Konzentrationen der beiden niedrig dosierten Gruppen besteht kein signifikanter
Unterschied, sie liegen im Mittel zwischen 30 und 40 µg/g. Die bei der Gruppe 5 mg/kg
gemessene Konzentration ist dagegen mit 80 µg/g signifikant höher, als die der beiden
62

4 - Ergebnisse
niedriger dosierten Gruppen (p < 0,05, bzw. p < 0,005). Bei dem einzelnen Tier der
Gruppe 10 mg/kg ist eine Ribavirinkonzentration von 160 µg/g gemessen worden.
Die bei dem an Tag 6 verstorbenen Tier gemessene Konzentration beträgt ca.
1300 µg/g Hirngewebe (Daten nicht gezeigt).
4.1.4 HE Färbung
Zum Ausschluß potentieller histopathologischer Veränderungen, die durch die i.c.v.
Injektion verursacht sein könnten, werden histologische Schnitte paramedian in der
Sagitalebene der linken Hemisphäre hergestellt und nach Übersichtsfärbung mit
Hämatoxylin und Eosin mikroskopisch beurteilt. Hierbei können keine direkten,
injektionsbedingten Veränderungen festgestellt werden.
In Schnitten einer Vielzahl von Tieren, die 2,5 mg/kg oder mehr Ribavirin erhalten
haben, ist eine zunehmende entzündliche Infiltration zu beobachten. Diese prominiert
meningeal und ependymal. Auch intravasal lassen sich in zahlreichen
Gefäßanschnitten Entzündungszellen nachweisen. Dagegen sind bei den Tieren der
Gruppen 0 mg/kg und 1,25 mg/kg keine Veränderungen gegenüber einem Kontrolltier
ohne Katheter feststellbar.
4.1.5 Gram Färbung
Bei der Beurteilung der Gramfärbungen im Hinblick auf eine potentiell während der
Injektionen übertragene, bakterielle Infektion zeigen sich keine anfärbbaren Erreger.
Ein direkter Hinweis auf eine bakterielle Besiedelung kann nicht gefunden werden.
4.2 Therapeutische Wirksamkeit - klinische Parameter
4.2.1 Körpergewicht
Die Tiere werden am Tag 1 des Experiments nach Randomisierung intranasal mit BDV
inokuliert. Ab dem Tag 22 beginnt die Applikation von Ribavirin bzw. PBS für 7
aufeinanderfolgende Tage. Die Durchschnittswerte des Körpergewichts der Tiere sind
in Abbildung 16 gegen die Zeit aufgetragen. Ihr Verlauf während der Tage 22 bis 28
differiert dosisabhängig (p = 0,0015). Ribavirin inhibiert den krankheitsbedingten
Gewichtsverlust mit zunehmender Dosierung. Am prominentesten ist die Differenz am
letzten Tag der Applikation, Tag 28 (p = 0,001).
63
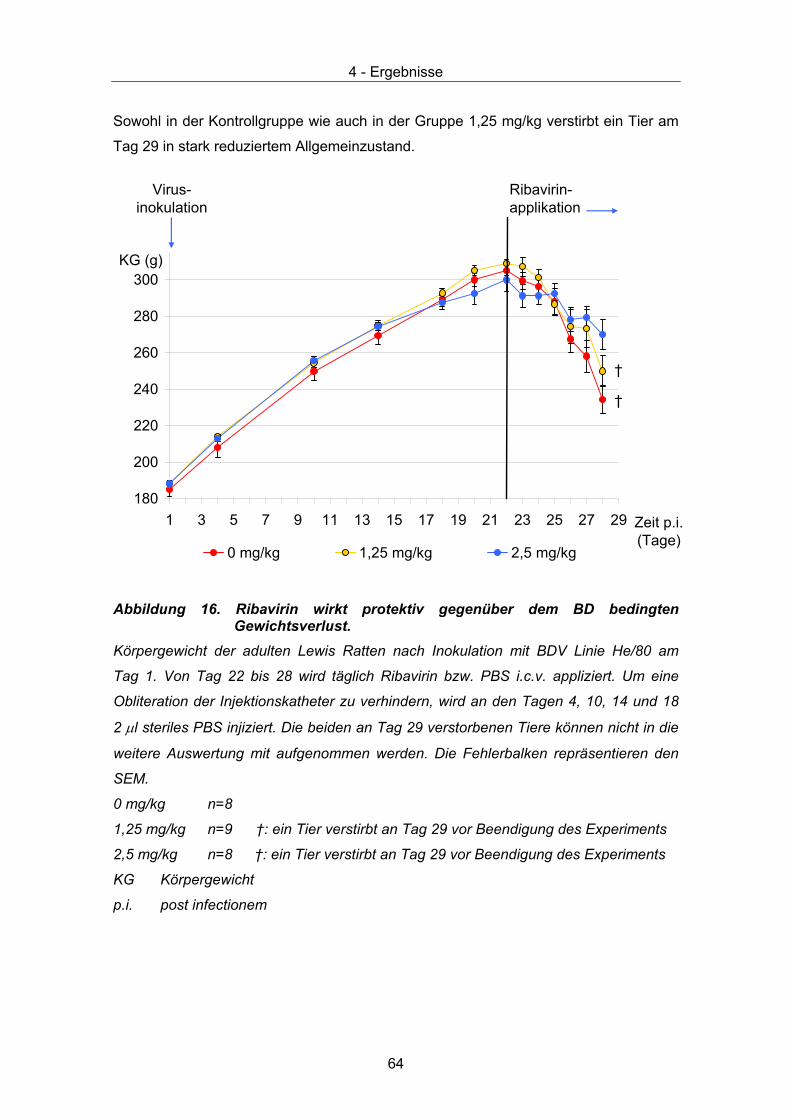
4 - Ergebnisse
Sowohl in der Kontrollgruppe wie auch in der Gruppe 1,25 mg/kg verstirbt ein Tier am
Tag 29 in stark reduziertem Allgemeinzustand.
180
200
220
240
260
280
300
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 Zeit p.i. (Tage)
KG (g)
0 mg/kg 1,25 mg/kg 2,5 mg/kg
Ribavirin-applikation
Virus- inokulation
†
†
Abbildung 16. Ribavirin wirkt protektiv gegenüber dem BD bedingten Gewichtsverlust.
Körpergewicht der adulten Lewis Ratten nach Inokulation mit BDV Linie He/80 am
Tag 1. Von Tag 22 bis 28 wird täglich Ribavirin bzw. PBS i.c.v. appliziert. Um eine
Obliteration der Injektionskatheter zu verhindern, wird an den Tagen 4, 10, 14 und 18
2 µl steriles PBS injiziert. Die beiden an Tag 29 verstorbenen Tiere können nicht in die
weitere Auswertung mit aufgenommen werden. Die Fehlerbalken repräsentieren den
SEM.
0 mg/kg n=8
1,25 mg/kg n=9 †: ein Tier verstirbt an Tag 29 vor Beendigung des Experiments
2,5 mg/kg n=8 †: ein Tier verstirbt an Tag 29 vor Beendigung des Experiments
KG Körpergewicht
p.i. post infectionem
64
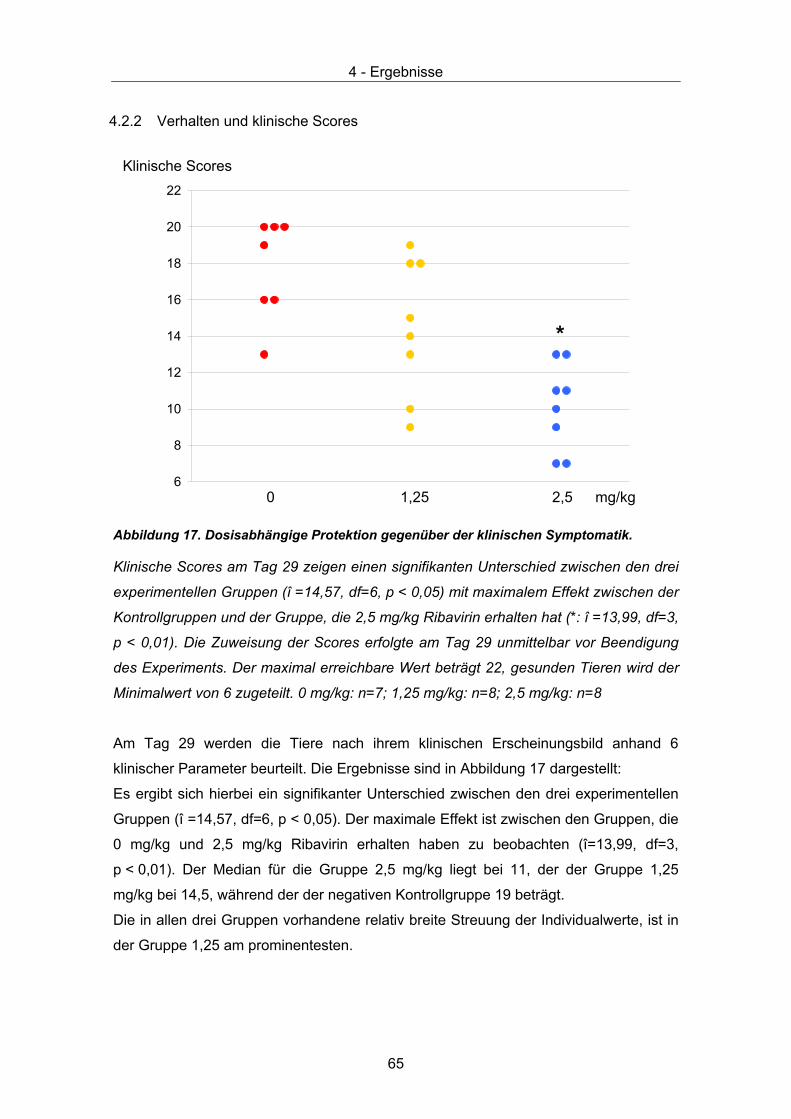
4 - Ergebnisse
4.2.2 Verhalten und klinische Scores
6
8
10
12
14
16
18
20
22
*
Klinische Scores
0 1,25 2,5 mg/kg
Abbildung 17. Dosisabhängige Protektion gegenüber der klinischen Symptomatik.
Klinische Scores am Tag 29 zeigen einen signifikanten Unterschied zwischen den drei
experimentellen Gruppen (î =14,57, df=6, p < 0,05) mit maximalem Effekt zwischen der
Kontrollgruppen und der Gruppe, die 2,5 mg/kg Ribavirin erhalten hat (*: î =13,99, df=3,
p < 0,01). Die Zuweisung der Scores erfolgte am Tag 29 unmittelbar vor Beendigung
des Experiments. Der maximal erreichbare Wert beträgt 22, gesunden Tieren wird der
Minimalwert von 6 zugeteilt. 0 mg/kg: n=7; 1,25 mg/kg: n=8; 2,5 mg/kg: n=8
Am Tag 29 werden die Tiere nach ihrem klinischen Erscheinungsbild anhand 6
klinischer Parameter beurteilt. Die Ergebnisse sind in Abbildung 17 dargestellt:
Es ergibt sich hierbei ein signifikanter Unterschied zwischen den drei experimentellen
Gruppen (î =14,57, df=6, p < 0,05). Der maximale Effekt ist zwischen den Gruppen, die
0 mg/kg und 2,5 mg/kg Ribavirin erhalten haben zu beobachten (î=13,99, df=3,
p < 0,01). Der Median für die Gruppe 2,5 mg/kg liegt bei 11, der der Gruppe 1,25
mg/kg bei 14,5, während der der negativen Kontrollgruppe 19 beträgt.
Die in allen drei Gruppen vorhandene relativ breite Streuung der Individualwerte, ist in
der Gruppe 1,25 am prominentesten.
65
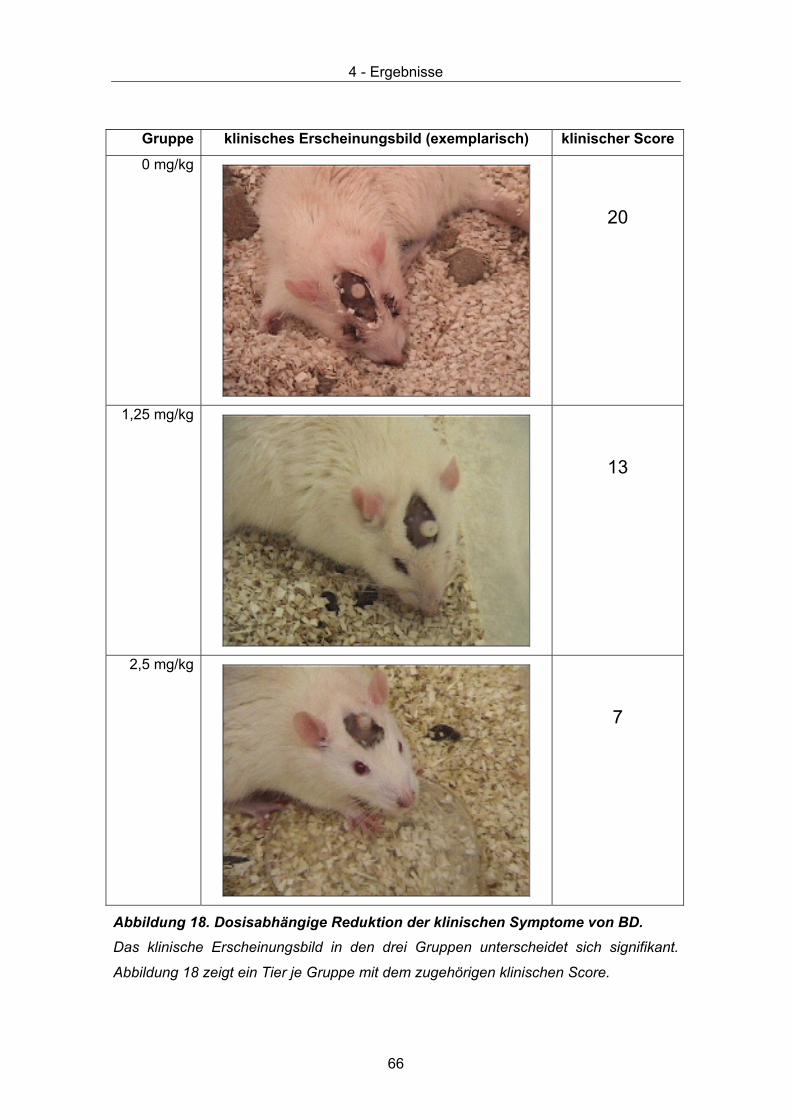
4 - Ergebnisse
Gruppe klinisches Erscheinungsbild (exemplarisch) klinischer Score
0 mg/kg
20
1,25 mg/kg
13
2,5 mg/kg
7
Abbildung 18. Dosisabhängige Reduktion der klinischen Symptome von BD. Das klinische Erscheinungsbild in den drei Gruppen unterscheidet sich signifikant.
Abbildung 18 zeigt ein Tier je Gruppe mit dem zugehörigen klinischen Score.
66
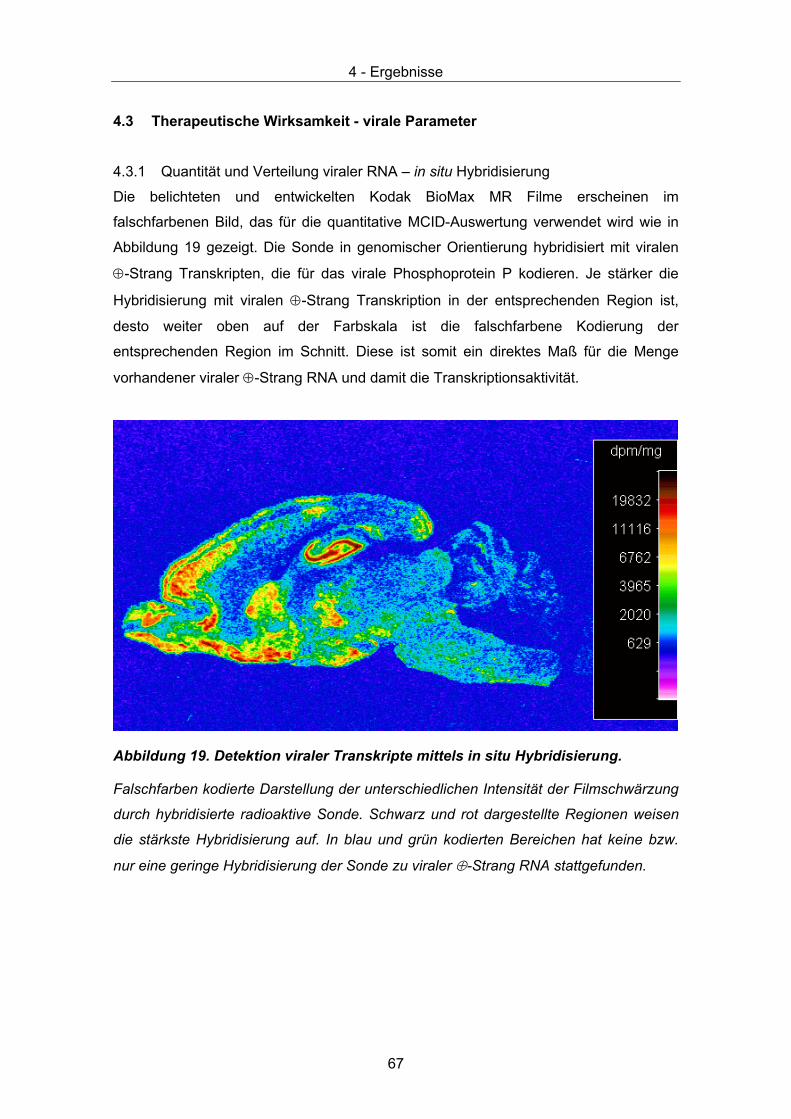
4 - Ergebnisse
4.3 Therapeutische Wirksamkeit - virale Parameter
4.3.1 Quantität und Verteilung viraler RNA – in situ Hybridisierung
Die belichteten und entwickelten Kodak BioMax MR Filme erscheinen im
falschfarbenen Bild, das für die quantitative MCID-Auswertung verwendet wird wie in
Abbildung 19 gezeigt. Die Sonde in genomischer Orientierung hybridisiert mit viralen
⊕-Strang Transkripten, die für das virale Phosphoprotein P kodieren. Je stärker die
Hybridisierung mit viralen ⊕-Strang Transkription in der entsprechenden Region ist,
desto weiter oben auf der Farbskala ist die falschfarbene Kodierung der
entsprechenden Region im Schnitt. Diese ist somit ein direktes Maß für die Menge
vorhandener viraler ⊕-Strang RNA und damit die Transkriptionsaktivität.
Abbildung 19. Detektion viraler Transkripte mittels in situ Hybridisierung.
Falschfarben kodierte Darstellung der unterschiedlichen Intensität der Filmschwärzung
durch hybridisierte radioaktive Sonde. Schwarz und rot dargestellte Regionen weisen
die stärkste Hybridisierung auf. In blau und grün kodierten Bereichen hat keine bzw.
nur eine geringe Hybridisierung der Sonde zu viraler ⊕-Strang RNA stattgefunden.
67

4 - Ergebnisse
Abbildung 20. Strukturen, in denen virale Transkripte quantifiziert werden.
Anatomische Strukturen, in denen die Transkriptionsaktivität für virale Transkripte
quantifiziert wird. Nicht eingezeichnet sind Gesamtkortex, Gyrus dentalis und CA3, die
in jedem Schnitt individuell komplett markiert und ausgewertet werden (nach [168]).
Auffällig hierbei ist eine inhomogene Verteilung des radioaktiven Signals: Strukturen
mit starker Hybridisierung sind der Kortex (bes. Präfrontalkortex), das Striatum,
thalamische Strukturen und der Hippocampus. Auch innerhalb des Kortex deutet sich
eine inhomogene, geschichtete Verteilung des Signals an. Im Hippocampus werden
sehr selektiv und außerordentlich stark der Gyrus dentalis und die Region CA3 infiziert.
Ebenfalls stark betroffen sind alle Strukturen der Riechbahn. Eine nur relativ geringe
Hybridisierung zu viraler RNA hat im Marklager, dem Hirnstamm und dem Kleinhirn
stattgefunden.
Makroskopisch fallen keine Unterschiede in der Verteilung oder Intenstität der
hybridisierten radioaktiven Sonde zwischen den experimentellen Gruppen auf. Um
einen quantitativen Vergleich der Viruslast in anatomisch entsprechenden Strukturen
und eine Veränderung der Verteilung zwischen den Versuchsgruppen anstellen zu
können, werden anschließend Strukturen mit prominenter Viruslast standardisiert
densitometrisch ausgewertet (Abbildung 20 und 3.4.4.8). Die Daten ermöglichen einen
Vergleich der Verteilung, sowie der absoluten Intensität der Hybridisierung. Die
Ergebnisse sind in Abbildung 21 dargestellt:
68
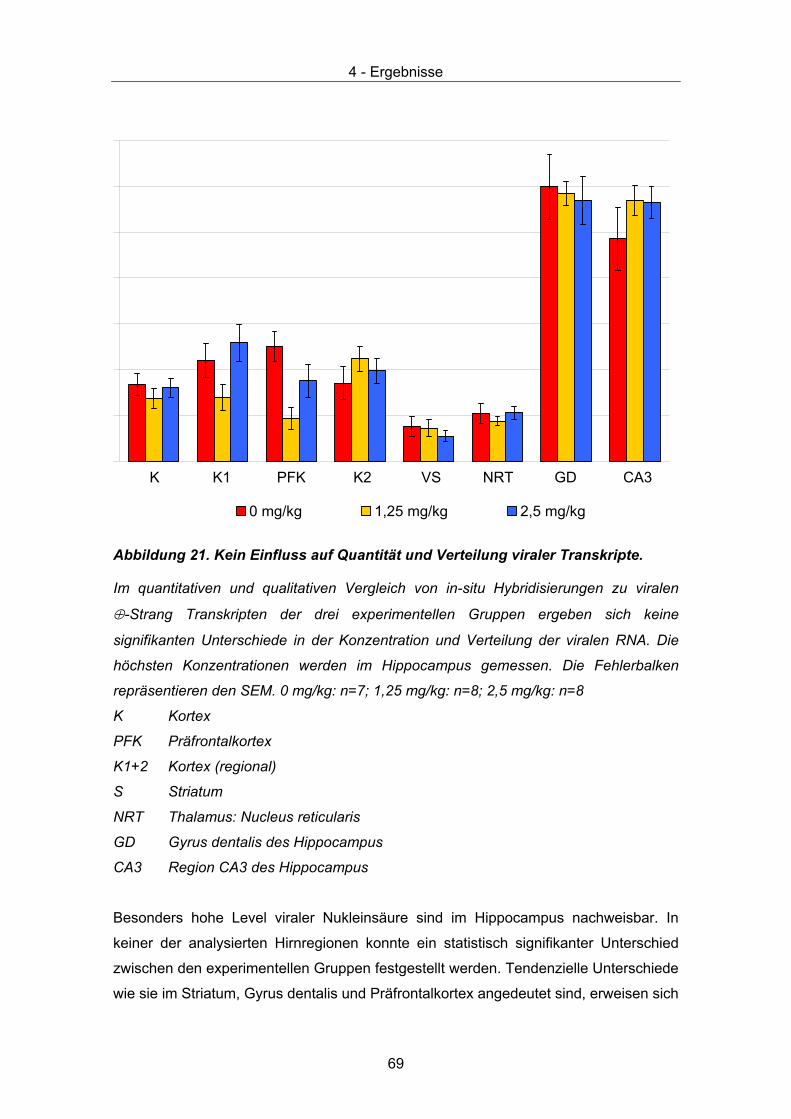
4 - Ergebnisse
0
2000
4000
6000
8000
10000
12000
14000
0 mg/kg 1,25 mg/kg 2,5 mg/kg
K K1 PFK K2 VS NRT GD CA3
Abbildung 21. Kein Einfluss auf Quantität und Verteilung viraler Transkripte.
Im quantitativen und qualitativen Vergleich von in-situ Hybridisierungen zu viralen
⊕-Strang Transkripten der drei experimentellen Gruppen ergeben sich keine
signifikanten Unterschiede in der Konzentration und Verteilung der viralen RNA. Die
höchsten Konzentrationen werden im Hippocampus gemessen. Die Fehlerbalken
repräsentieren den SEM. 0 mg/kg: n=7; 1,25 mg/kg: n=8; 2,5 mg/kg: n=8
K Kortex
PFK Präfrontalkortex
K1+2 Kortex (regional)
S Striatum
NRT Thalamus: Nucleus reticularis
GD Gyrus dentalis des Hippocampus
CA3 Region CA3 des Hippocampus
Besonders hohe Level viraler Nukleinsäure sind im Hippocampus nachweisbar. In
keiner der analysierten Hirnregionen konnte ein statistisch signifikanter Unterschied
zwischen den experimentellen Gruppen festgestellt werden. Tendenzielle Unterschiede
wie sie im Striatum, Gyrus dentalis und Präfrontalkortex angedeutet sind, erweisen sich
69

4 - Ergebnisse
entweder als nicht signifikant, oder zeigen keine Korrelation zur erhaltenen Ribavirin-
Dosis. Auch in der Verteilung viraler Nukleinsäure ist kein Unterschied zwischen den
Gruppen erkennbar.
Neben dieser quantitativen und qualitativen Auswertung ist in der HE gegengefärbten
in-situ Autoradiographie eine Aussage über die Lokalisation und Verteilung der viralen
RNA auf zellulärer Ebene sowie der Bezug zu histopathologischen Veränderungen
möglich. Beurteilt werden Lokalisation und Typus infizierter Zellen, sowie die
Korrelation zu histopathologischen Foci.
Hierbei sind keine qualitativen Unterschiede zwischen den Tieren der drei
experimentellen Gruppen beobachtbar: Das Infektionsmuster bleibt durch die Ribavirin-
applikation unbeeinflußt. Auch ist in der HE-Gegenfärbung keine veränderte
Wirtsreaktion auf die Virusreplikation zu beobachten.
Die makroskopisch angedeutete Inhomogenität des Signals innerhalb des Kortex findet
ihr histologische Korrelat in einer besonders hohen Signalintensität über
Pyramidenzellen. Eine starke Hybridisierung zeigen daher die Schichten 3 und 5 des
Neokortex (Mikrograph nicht gezeigt).
70
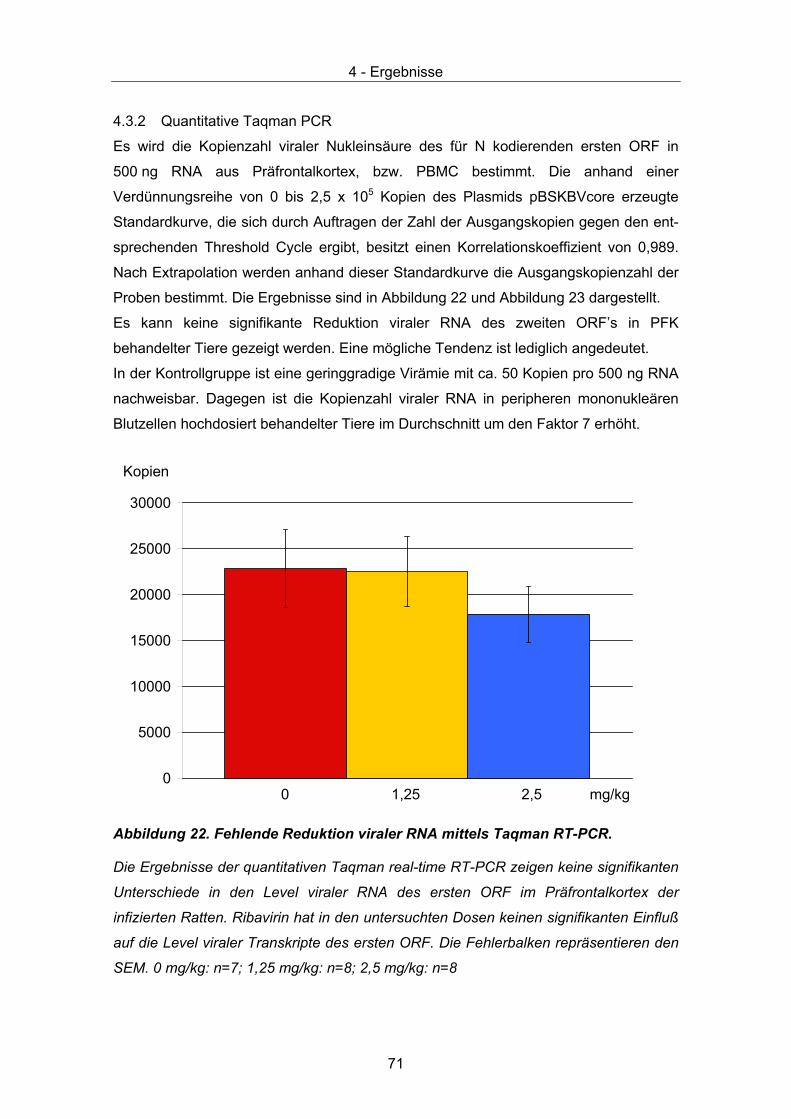
4 - Ergebnisse
4.3.2 Quantitative Taqman PCR
Es wird die Kopienzahl viraler Nukleinsäure des für N kodierenden ersten ORF in
500 ng RNA aus Präfrontalkortex, bzw. PBMC bestimmt. Die anhand einer
Verdünnungsreihe von 0 bis 2,5 x 105 Kopien des Plasmids pBSKBVcore erzeugte
Standardkurve, die sich durch Auftragen der Zahl der Ausgangskopien gegen den ent-
sprechenden Threshold Cycle ergibt, besitzt einen Korrelationskoeffizient von 0,989.
Nach Extrapolation werden anhand dieser Standardkurve die Ausgangskopienzahl der
Proben bestimmt. Die Ergebnisse sind in Abbildung 22 und Abbildung 23 dargestellt.
Es kann keine signifikante Reduktion viraler RNA des zweiten ORF’s in PFK
behandelter Tiere gezeigt werden. Eine mögliche Tendenz ist lediglich angedeutet.
In der Kontrollgruppe ist eine geringgradige Virämie mit ca. 50 Kopien pro 500 ng RNA
nachweisbar. Dagegen ist die Kopienzahl viraler RNA in peripheren mononukleären
Blutzellen hochdosiert behandelter Tiere im Durchschnitt um den Faktor 7 erhöht.
0
5000
10000
15000
20000
25000
30000
0 1,25 2,5
Kopien
mg/kg
Abbildung 22. Fehlende Reduktion viraler RNA mittels Taqman RT-PCR.
Die Ergebnisse der quantitativen Taqman real-time RT-PCR zeigen keine signifikanten
Unterschiede in den Level viraler RNA des ersten ORF im Präfrontalkortex der
infizierten Ratten. Ribavirin hat in den untersuchten Dosen keinen signifikanten Einfluß
auf die Level viraler Transkripte des ersten ORF. Die Fehlerbalken repräsentieren den
SEM. 0 mg/kg: n=7; 1,25 mg/kg: n=8; 2,5 mg/kg: n=8
71
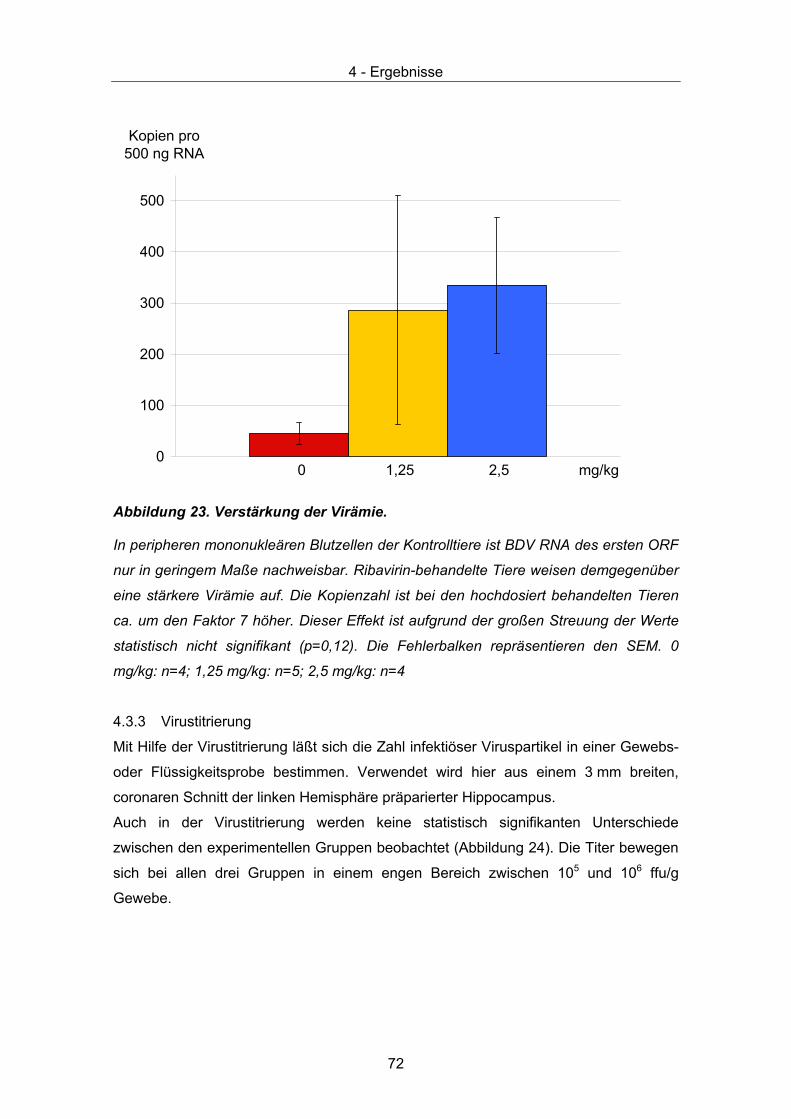
4 - Ergebnisse
0
100
200
300
400
500
Kopien pro 500 ng RNA
0 1,25 2,5 mg/kg
Abbildung 23. Verstärkung der Virämie.
In peripheren mononukleären Blutzellen der Kontrolltiere ist BDV RNA des ersten ORF
nur in geringem Maße nachweisbar. Ribavirin-behandelte Tiere weisen demgegenüber
eine stärkere Virämie auf. Die Kopienzahl ist bei den hochdosiert behandelten Tieren
ca. um den Faktor 7 höher. Dieser Effekt ist aufgrund der großen Streuung der Werte
statistisch nicht signifikant (p=0,12). Die Fehlerbalken repräsentieren den SEM. 0
mg/kg: n=4; 1,25 mg/kg: n=5; 2,5 mg/kg: n=4
4.3.3 Virustitrierung
Mit Hilfe der Virustitrierung läßt sich die Zahl infektiöser Viruspartikel in einer Gewebs-
oder Flüssigkeitsprobe bestimmen. Verwendet wird hier aus einem 3 mm breiten,
coronaren Schnitt der linken Hemisphäre präparierter Hippocampus.
Auch in der Virustitrierung werden keine statistisch signifikanten Unterschiede
zwischen den experimentellen Gruppen beobachtet (Abbildung 24). Die Titer bewegen
sich bei allen drei Gruppen in einem engen Bereich zwischen 105 und 106 ffu/g
Gewebe.
72
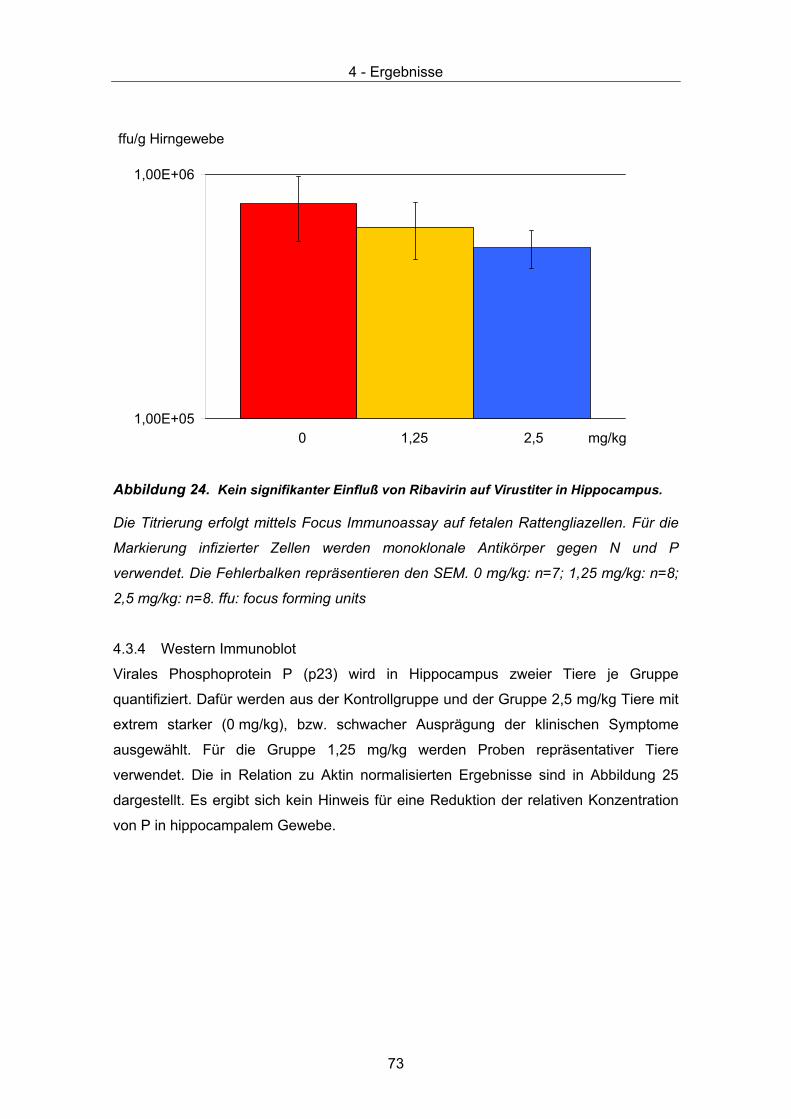
4 - Ergebnisse
1,00E+05
1,00E+06
ffu/g Hirngewebe
0 1,25 2,5 mg/kg
Abbildung 24. Kein signifikanter Einfluß von Ribavirin auf Virustiter in Hippocampus.
Die Titrierung erfolgt mittels Focus Immunoassay auf fetalen Rattengliazellen. Für die
Markierung infizierter Zellen werden monoklonale Antikörper gegen N und P
verwendet. Die Fehlerbalken repräsentieren den SEM. 0 mg/kg: n=7; 1,25 mg/kg: n=8;
2,5 mg/kg: n=8. ffu: focus forming units
4.3.4 Western Immunoblot
Virales Phosphoprotein P (p23) wird in Hippocampus zweier Tiere je Gruppe
quantifiziert. Dafür werden aus der Kontrollgruppe und der Gruppe 2,5 mg/kg Tiere mit
extrem starker (0 mg/kg), bzw. schwacher Ausprägung der klinischen Symptome
ausgewählt. Für die Gruppe 1,25 mg/kg werden Proben repräsentativer Tiere
verwendet. Die in Relation zu Aktin normalisierten Ergebnisse sind in Abbildung 25
dargestellt. Es ergibt sich kein Hinweis für eine Reduktion der relativen Konzentration
von P in hippocampalem Gewebe.
73
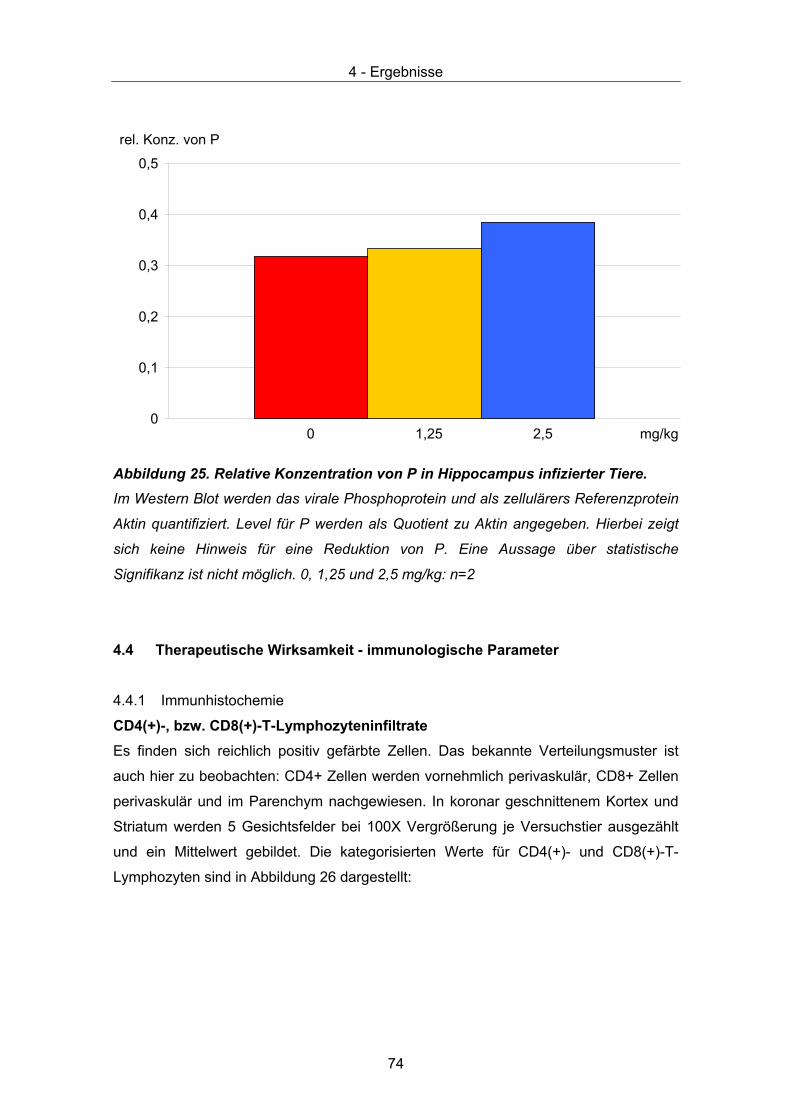
4 - Ergebnisse
0
0,1
0,2
0,3
0,4
0,5
rel. Konz. von P
0 1,25 2,5 mg/kg
Abbildung 25. Relative Konzentration von P in Hippocampus infizierter Tiere. Im Western Blot werden das virale Phosphoprotein und als zellulärers Referenzprotein
Aktin quantifiziert. Level für P werden als Quotient zu Aktin angegeben. Hierbei zeigt
sich keine Hinweis für eine Reduktion von P. Eine Aussage über statistische
Signifikanz ist nicht möglich. 0, 1,25 und 2,5 mg/kg: n=2
4.4 Therapeutische Wirksamkeit - immunologische Parameter
4.4.1 Immunhistochemie
CD4(+)-, bzw. CD8(+)-T-Lymphozyteninfiltrate Es finden sich reichlich positiv gefärbte Zellen. Das bekannte Verteilungsmuster ist
auch hier zu beobachten: CD4+ Zellen werden vornehmlich perivaskulär, CD8+ Zellen
perivaskulär und im Parenchym nachgewiesen. In koronar geschnittenem Kortex und
Striatum werden 5 Gesichtsfelder bei 100X Vergrößerung je Versuchstier ausgezählt
und ein Mittelwert gebildet. Die kategorisierten Werte für CD4(+)- und CD8(+)-T-
Lymphozyten sind in Abbildung 26 dargestellt:
74
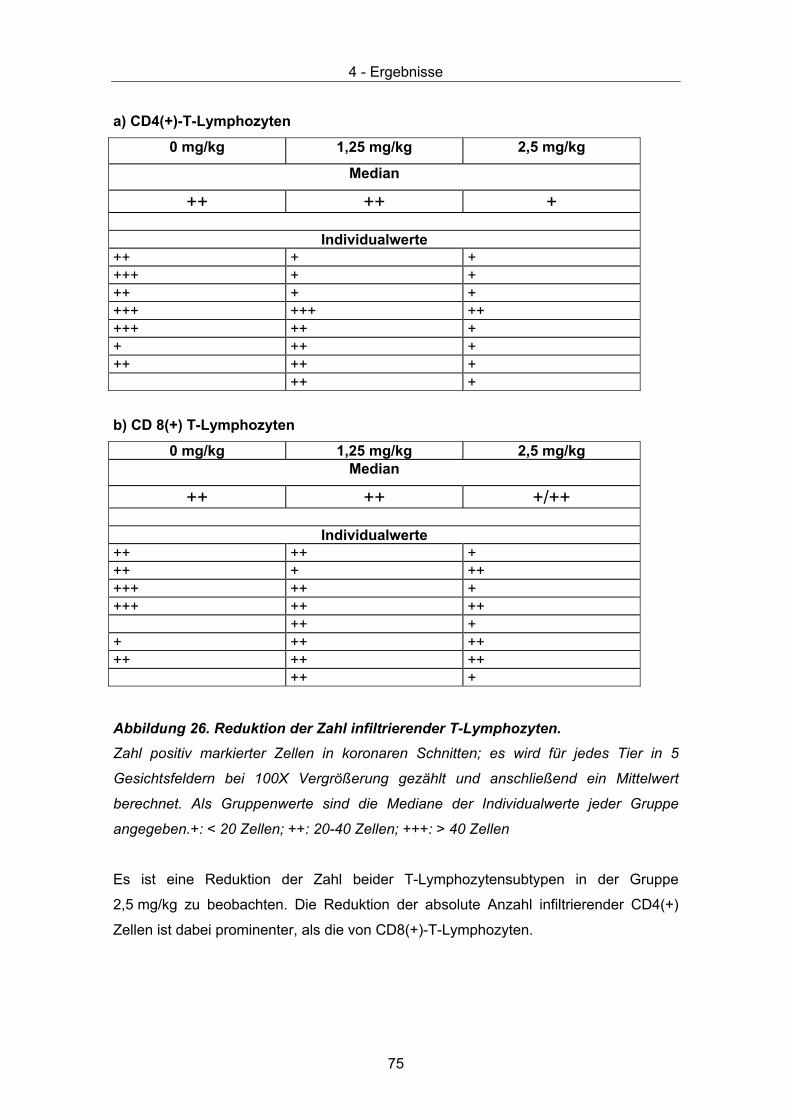
4 - Ergebnisse
a) CD4(+)-T-Lymphozyten
0 mg/kg 1,25 mg/kg 2,5 mg/kg
Median
++ ++ +
Individualwerte ++ + + +++ + + ++ + + +++ +++ ++ +++ ++ + + ++ + ++ ++ + ++ +
b) CD 8(+) T-Lymphozyten
0 mg/kg 1,25 mg/kg 2,5 mg/kg Median
++ ++ +/++
Individualwerte ++ ++ + ++ + ++ +++ ++ + +++ ++ ++ ++ + + ++ ++ ++ ++ ++ ++ +
Abbildung 26. Reduktion der Zahl infiltrierender T-Lymphozyten. Zahl positiv markierter Zellen in koronaren Schnitten; es wird für jedes Tier in 5
Gesichtsfeldern bei 100X Vergrößerung gezählt und anschließend ein Mittelwert
berechnet. Als Gruppenwerte sind die Mediane der Individualwerte jeder Gruppe
angegeben.+: < 20 Zellen; ++: 20-40 Zellen; +++: > 40 Zellen
Es ist eine Reduktion der Zahl beider T-Lymphozytensubtypen in der Gruppe
2,5 mg/kg zu beobachten. Die Reduktion der absolute Anzahl infiltrierender CD4(+)
Zellen ist dabei prominenter, als die von CD8(+)-T-Lymphozyten.
75
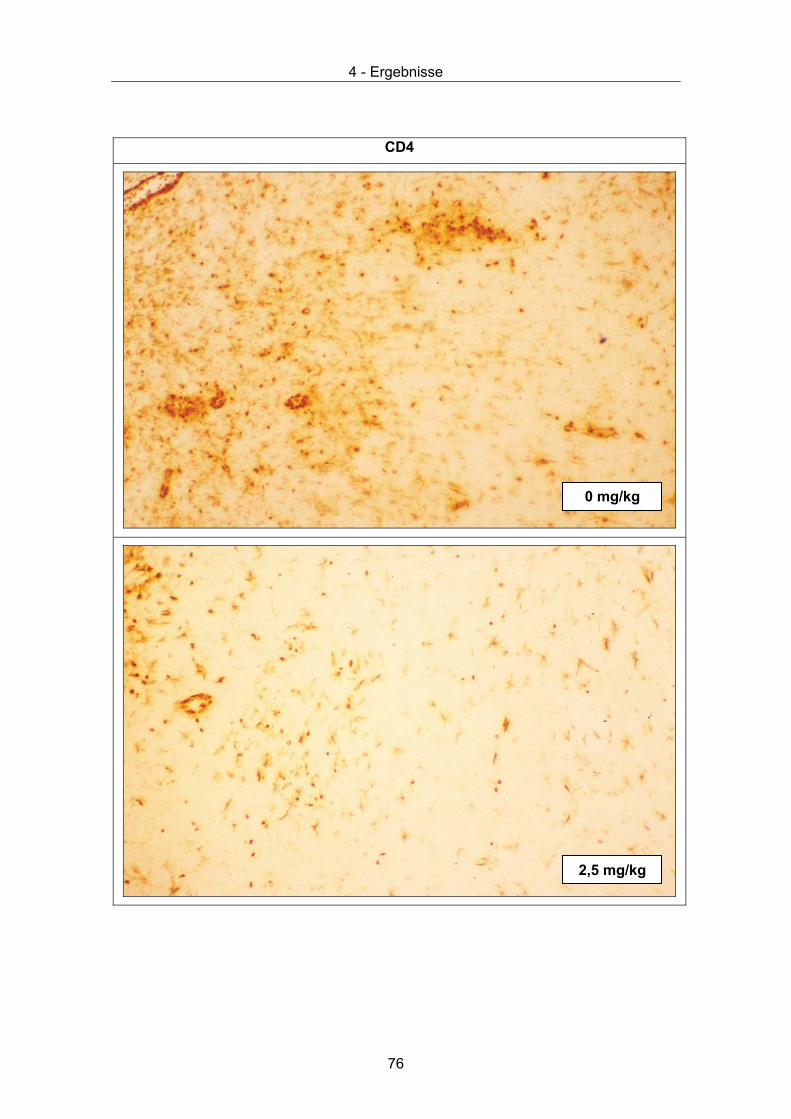
4 - Ergebnisse
CD4
0 mg/kg
2,5 mg/kg
76
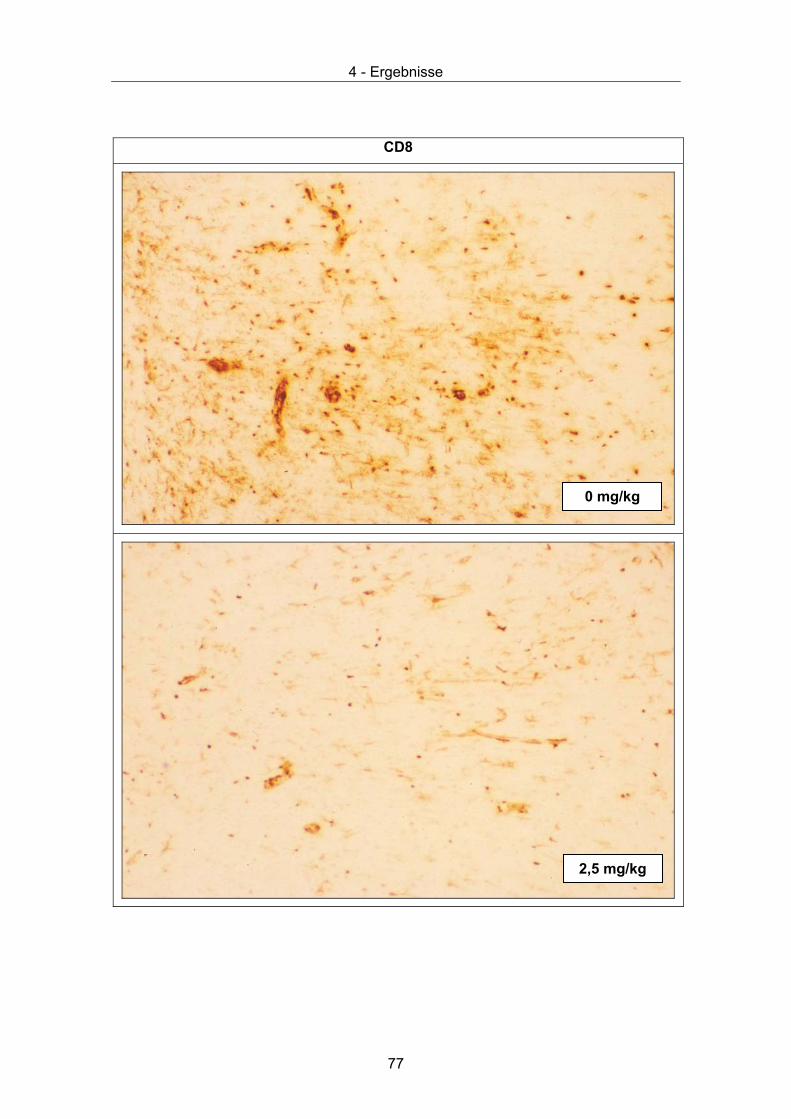
4 - Ergebnisse
CD8
0 mg/kg
2,5 mg/kg
77

4 - Ergebnisse
Abbildung 27. Reduktion von CD4+- und CD8+-T-Lymphozyten bei hochdosierter
Ribavirinapplikation.
CD4+- und CD8+-T-Lymphozyten finden sich in coronaren Schnitten bei Tieren der
Gruppe 2,5 mg/kg in reduzierter Zahl. In Abbildung 27 sind repräsentative Schnitte
(100X Vergrößerung) dargestellt. Zusätzlich ist die Anfärbbarkeit von Zellen
mikroglialer Morphologie durch beide mAb erkennbar.
Sowohl in der Färbung auf CD4, als auch auf CD8 fällt eine Anfärbung auch von Zellen
mikroglialer Morphologie auf.
Mikroglia Der qualitative Vergleich mittels Ox-42 detektierter Mikrogliazellen erfolgt in Bezug auf
Zeichen von Aktivierung. Maß hierfür sind unter anderem eine Kontraktion der
mikroglialen Fortsätze, einhergehend mit einer Vergrößerung und Abkugelung der
Perikarien [50]. Wie in Abbildung 28 in kortikalen Ausschnitten exemplarisch gezeigt,
sind keine Unterschiede im Aktivierungsgrad gefärbter Mikrogliazellen in Gehirnen mit
Ribavirin behandelter Tiere gegenüber denen der Gruppe 0 mg/kg feststellbar. Im
Schnitt eines nicht-infizierten Kontrolltiers sind Mikrogliazellen in nicht-aktiviertem
Zustand gezeigt. Hier sind die Fortsätzen der Mikrogliazellen gut erkennbar. Im Schnitt
der Tiere der Gruppen 0 mg/kg und 2,5 mg/kg wirkt der Bereich zwischen den
Mikrogliazellen aufgehellt, es sind deutlich weniger Zellfortsätze angefärbt.
Demgegenüber ist die absolute Zahl angefärbter Zellen in den Schnitten behandelter
Tiere im Vergleich mit unbehandelten, infizierten Ratten deutlich reduziert. Die
Applikation von Ribavirin resultiert also in einer Reduktion der Zahl der Mikrogliazellen
in infizierten Tieren. Der beschriebene Effekt ist besonders prominent zwischen der
Gruppe 2,5 mg/kg und der Kontrollgruppe. Das Bild bei den Tieren der Gruppe
1,25 mg/kg reiht sich zwischen diesen beiden Extremen ein (nicht gezeigt).
78
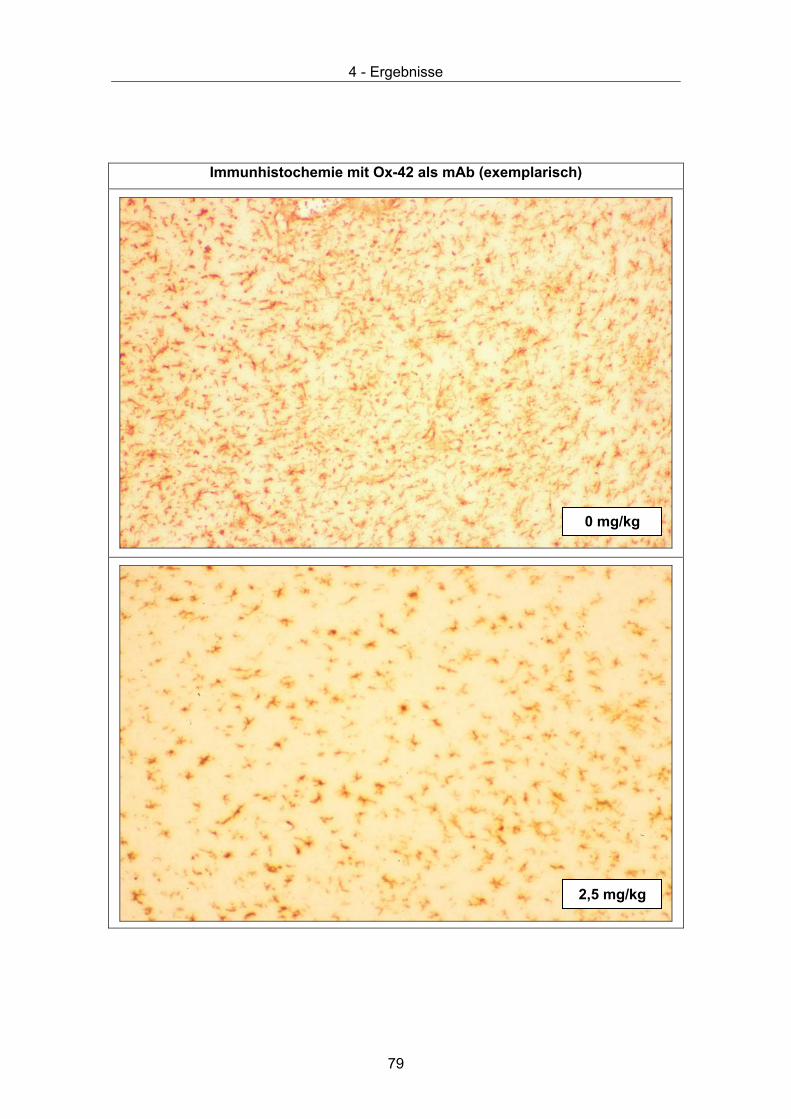
4 - Ergebnisse
Immunhistochemie mit Ox-42 als mAb (exemplarisch)
0 mg/kg
2,5 mg/kg
79
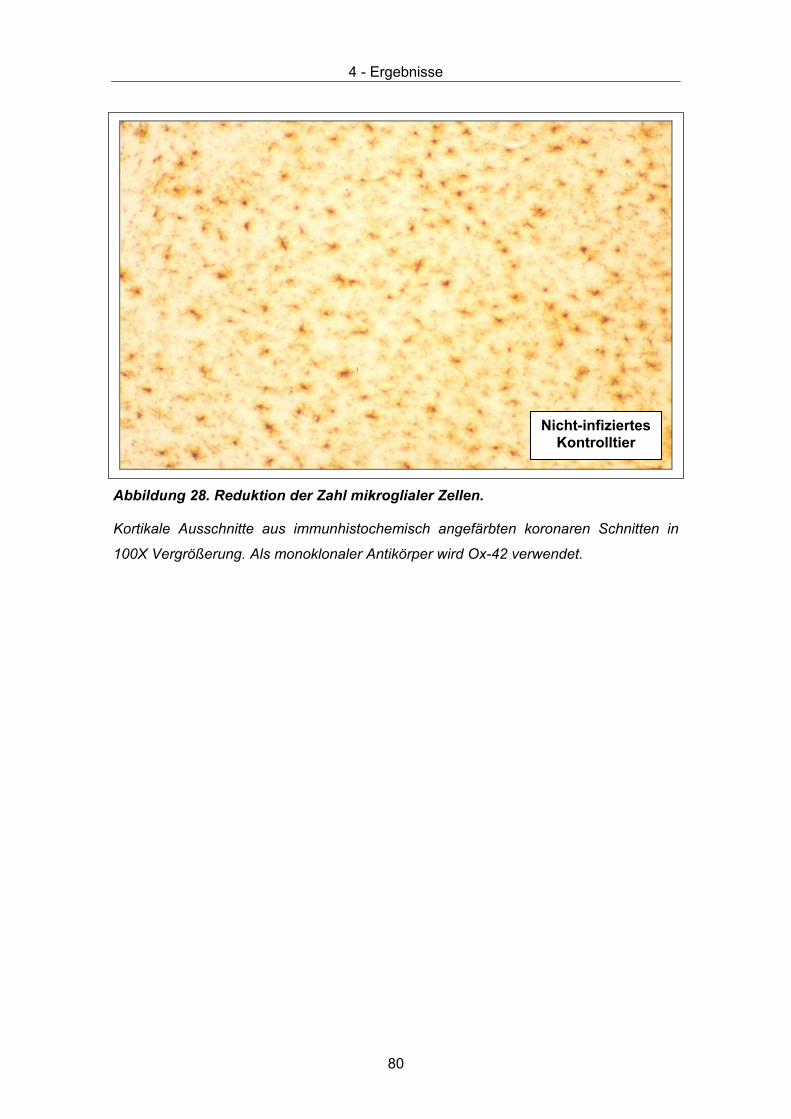
4 - Ergebnisse
Nicht-infiziertes Kontrolltier
Abbildung 28. Reduktion der Zahl mikroglialer Zellen.
Kortikale Ausschnitte aus immunhistochemisch angefärbten koronaren Schnitten in
100X Vergrößerung. Als monoklonaler Antikörper wird Ox-42 verwendet.
80
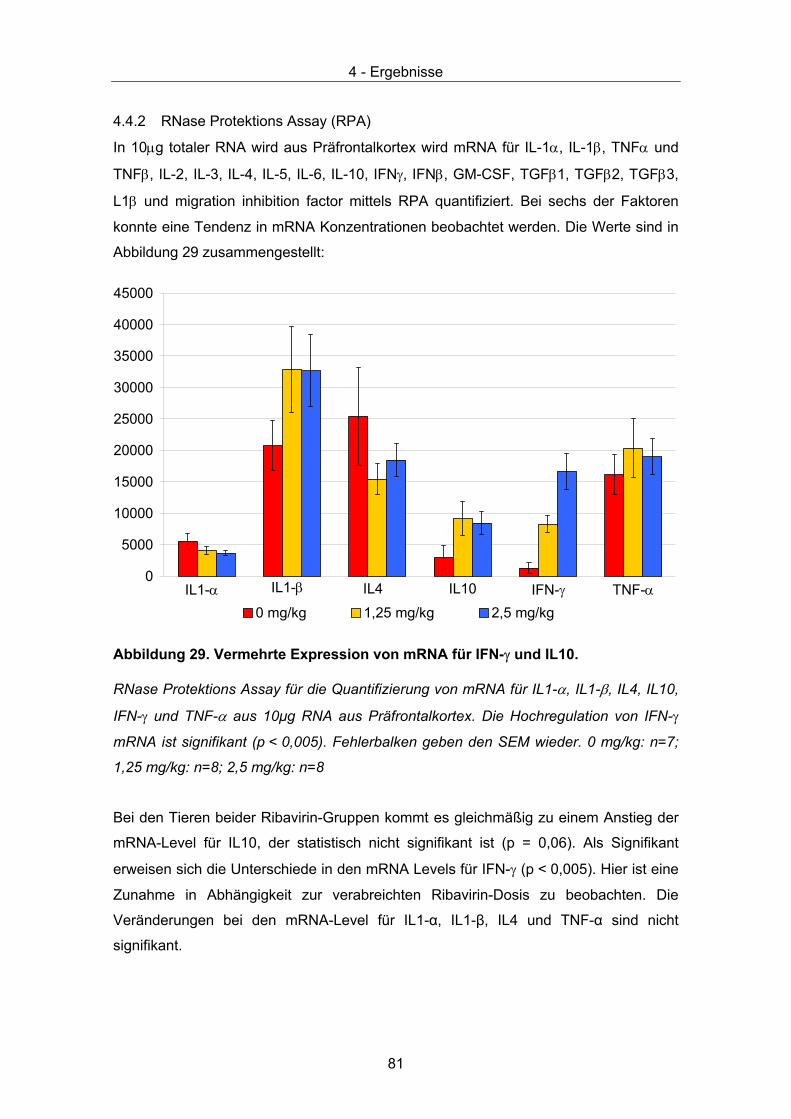
4 - Ergebnisse
4.4.2 RNase Protektions Assay (RPA)
In 10µg totaler RNA wird aus Präfrontalkortex wird mRNA für IL-1α, IL-1β, TNFα und
TNFβ, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, IFNγ, IFNβ, GM-CSF, TGFβ1, TGFβ2, TGFβ3,
L1β und migration inhibition factor mittels RPA quantifiziert. Bei sechs der Faktoren
konnte eine Tendenz in mRNA Konzentrationen beobachtet werden. Die Werte sind in
zusammengestellt: Abbildung 29
Abbildung 29. Vermehrte Expression von mRNA für IFN-γ und IL10.
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
0 mg/kg 1,25 mg/kg 2,5 mg/kgIL1-α IL1-β IL4 IL10 IFN-γ TNF-α
RNase Protektions Assay für die Quantifizierung von mRNA für IL1-α, IL1-β, IL4, IL10,
IFN-γ und TNF-α aus 10µg RNA aus Präfrontalkortex. Die Hochregulation von IFN-γ
mRNA ist signifikant (p < 0,005). Fehlerbalken geben den SEM wieder. 0 mg/kg: n=7;
1,25 mg/kg: n=8; 2,5 mg/kg: n=8
Bei den Tieren beider Ribavirin-Gruppen kommt es gleichmäßig zu einem Anstieg der
mRNA-Level für IL10, der statistisch nicht signifikant ist (p = 0,06). Als Signifikant
erweisen sich die Unterschiede in den mRNA Levels für IFN-γ (p < 0,005). Hier ist eine
Zunahme in Abhängigkeit zur verabreichten Ribavirin-Dosis zu beobachten. Die
Veränderungen bei den mRNA-Level für IL1-α, IL1-β, IL4 und TNF-α sind nicht
signifikant.
81
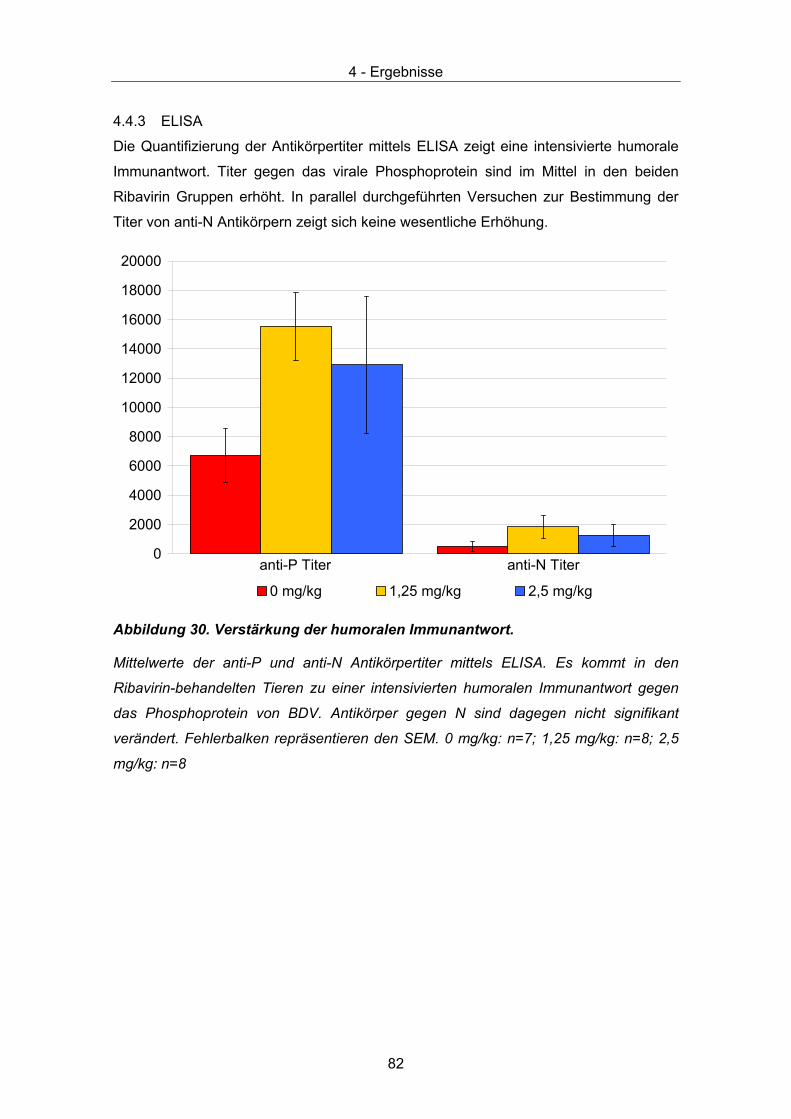
4 - Ergebnisse
4.4.3 ELISA
Die Quantifizierung der Antikörpertiter mittels ELISA zeigt eine intensivierte humorale
Immunantwort. Titer gegen das virale Phosphoprotein sind im Mittel in den beiden
Ribavirin Gruppen erhöht. In parallel durchgeführten Versuchen zur Bestimmung der
Titer von anti-N Antikörpern zeigt sich keine wesentliche Erhöhung.
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
0 mg/kg 1,25 mg/kg 2,5 mg/kg
anti-P Titer anti-N Titer
Abbildung 30. Verstärkung der humoralen Immunantwort.
Mittelwerte der anti-P und anti-N Antikörpertiter mittels ELISA. Es kommt in den
Ribavirin-behandelten Tieren zu einer intensivierten humoralen Immunantwort gegen
das Phosphoprotein von BDV. Antikörper gegen N sind dagegen nicht signifikant
verändert. Fehlerbalken repräsentieren den SEM. 0 mg/kg: n=7; 1,25 mg/kg: n=8; 2,5
mg/kg: n=8
82

5 - Diskussion
83
5 Diskussion
5.1 Toxikologie und Pharmakokinetik In der vorliegenden Studie wird Ribavirin zum ersten Mal auf seinen therapeutischen
Nutzen gegenüber einer Infektion mit BDV in vivo getestet. Da nur wenig über die
Passagefähigkeit von Ribavirin über die intakte Blut-Hirn-Schranke (BHS) bekannt ist,
wird der Wirkstoff direkt in das ZNS appliziert. Bezüglich der Pharmakokinetik von
Ribavirin bei intrakranieller Applikation liegen bisher nur Daten aus einem Hamster-
modell vor, wo Ribavirin subarachnoidal in Volumina von 50 µl und Dosen von 5, 10,
und 20, 40 und 80 mg/kg KG/Tag injiziert wurde. Die maximal erreichten Konzen-
trationen stellten sich dabei an Tag 6-7 ein [173].
Auf der Grundlage dieser Ergebnisse wurde für die Charakterisierung der
Pharmakokinetik in dem vorliegenden Modell Dosen von 5, 10 und 20 mg/kg KG/Tag
ausgewählt. Augrund der Tatsache, dass das Gehirn eines Hamsters einen Anteil von
ca. 2,8%, das einer Ratte dagegen nur 0,7% des Gesamtgewichts ausmacht [174],
wurden zusätzlich Dosen von 2,5 und 1,25 mg/kg extrapoliert. Die experimentell
eingesetzten männliche Lewis Ratten erhielten somit Ribavirin i.c.v. in Dosierungen
von 0; 1,25; 2,5; 5; 10 und 20 mg/kg über einen Zeitraum von 7 Tagen.
Dosen von 1,25 und 2,5 mg/kg wurden von den Lewis Ratten gut toleriert: Die Tiere
erholten sich schnell von der täglichen Narkose und unterschieden sich hinsichtlich
ihres Putz- und Fressverhaltens kaum von den Kontrolltieren. Als Maß für den
allgemeinen Gesundheitszustand der Tiere wurde täglich das Körpergewicht bestimmt.
Mit einem Alter von 56-60 Tage befinden sich männliche Lewis Ratten noch in der
Wachstumsphase und steigern ihr Gewicht innerhalb der nächsten Lebenswoche im
Mittel um ca. 10% (s. 2.11). Die Kontrolltiere unterscheiden sich von dem
Normalkollektiv, das diesen Werte zugrunde liegt durch die wenige Tage
zurückliegende Katheterimplantation, die täglich Narkose sowie die intrakranielle
Volumenbelastung. Dennoch weisen die Durchschnittswerte der Kontrollgruppe
(Injektion von PBS) von Tag 1 zu Tag 7 eine Zunahme des Körpergewichts von 10,9%
auf. Die Implantation sowie die tägliche Manipulation zeigt also keinen negativen
Einfluß auf die Zunahme des Körpergewichts der betroffenen Tiere. Eine leicht
verzögerte Gewichtsentwicklung und eine Überlebensrate von 100% in den Gruppen
1,25 und 2,5 mg/kg (s. ) sprechen für eine allgemein gute Verträglichkeit
von Ribavirin in dieser Dosierung. Dagegen geht die Applikation von 5, 10 und 20
mg/kg mit einer zunehmenden Toxizität (deutlicher Gewichtsverlust in den Gruppen 5
Abbildung 14

5 - Diskussion
84
und 10 mg/kg) und erhöhten Mortalität (5, 10 und 20 mg/kg) einher: Die LD50 in diesem
Modell beträgt etwa 10 mg/kg KG/Tag.
Die gemessenen Gewebespiegel von Ribavirin weisen eine positive Korrelation mit
den verabreichten Dosen auf (s. ) und liegen zwischen 30 und 160µg/g
Hirngewebe. Die Applikation von 10 mg/kg KG/Tag resultiert bei dem Tier, das bis zum
Tag 8 überlebt, in einer Konzentration von 160 µg/g. Die Konzentration von etwa 1300
µg/g, die bei dem an Tag 6 verstorbenen Tier gemessen wird, ist mit der unmittelbar
vorher erfolgten Injektion zu erklären und spiegelt vermutlich nicht die reale
Konzentration im Gehirn wider. Trotz des abweichenden Applikationswegs (i.c.v. vs.
subarachnoidal), den unterschiedlichen applizierten Volumina (5-20 µl vs. 50 µl) und
dem verwendeten Versuchstier Ratte decken sich diese Werte mit den Ergebnissen
des Hamstermodells: Hier liegen die maximal verträglichen Gewebespiegel zwischen
50 und 150 µg/g, die toxische Dosis wird zwischen 250 und 400 µg/g angegeben. Dass
die maximal tolerierten Spiegel im vorliegenden Modell bereits bei 5 bis 10 mg/kg KG
erreicht werden (im Vergleich mit bis zu 20 mg/kg KG beim Hamster), ist vermutlich mit
dem geringeren relativen Gewicht des Gehirns der Ratte zu erklären.
Abbildung 15
I.c.v. applizierte Volumina bis etwa 5 µl werden erfahrungsgemäß gut vertragen
(M. V. Solbrig, personal communications). Dieses geringe Volumen kann aber
aufgrund der Löslichkeit von Ribavirin mit maximal 125 mg/ml PBS (ca. 0,5 M) bei
einer Applikation pro Tag nicht für alle experimentellen Gruppen eingehalten werden.
Um die notwendigen Dosen zu erzielen müssen Volumina und/oder Zahl der
Injektionen erhöht werden. Im vorliegenden Modell kommen Volumina zwischen
einmalig 5 µl und zweimalig 15-20 µl zur Anwendung (s. ). Für die hoch-
dosiert behandelten Tiere addiert sich hierdurch der toxische Effekt von Ribavirin mit
einer Steigerung des Hirndrucks durch die größere intrathekale Volumenbelastung. Die
ermittelte toxische Dosis für Ribavirin bei i.c.v. Injektion ist somit an die verwendeten
Volumina gekoppelt.
Tabelle 3
Um durch die Injektionstechnik verursachte, histopathologisch fassbare
Veränderungen zu beurteilen wurde anschließend Hirngewebe unter Einschluss des
linken Seitenventrikels in HE gefärbten histologischen Schnitte untersucht. Hierbei
konnten keine injektionsbedingten Alterationen festgestellt werden. Es zeigte sich
jedoch bei den hochdosiert behandelten Tieren meningeale und perivaskuläre

5 - Diskussion
85
Entzündungsinfiltrate (s. 4.1.4). Zur orientierenden Beurteilung einer applikations-
bedingten bakteriellen Infektion wurde eine Gramfärbung angeschlossen, die keinen
Hinweis auf eine bakterielle Besiedelung erbrachte. Eine bakterielle Genese kann nicht
ausgeschlossen werden, ist jedoch unwahrscheinlich, da sich die beschriebenen
Veränderungen dosisabhängig verhalten und bei den Tieren der Gruppen 0 und
1,25 mg/kg nicht zu beobachten sind. Vermutlich handelt es sich bei den beobachteten
Infiltraten um die lokale Reaktion auf eine toxische Schädigung.
Für eine weitere Testung in BDV infizierten Lewis-Ratten wurden auf Grundlage dieser
Ergebnisse Dosen von 1,25 und 2,5 mg/kg ausgewählt: die toxischen Nebenwirkungen
sind in beiden Gruppen vernachlässigbar, die zu applizierenden Volumina für beide
Dosierungen gleich und ausreichend gering.
An Tag 1 des Experiments wurden 25 Tiere intranasal mit BDV inokuliert. Die Infektion
über das olfaktorische Epithel entspricht dem vermuteten natürlichen Infektionsweg
[12]. Die verwendeten Lewis-Ratten zeigten bereits zwei Wochen später erste Zeichen
der Infektion: sowohl verstärkte Schreckreaktionen als auch Hyperaktivität waren
beobachtbar und bestätigten klinisch die Infektion aller Tiere [48]. Nach
Randomisierung der Tiere (0 mg/kg: n=8, 1,25 mg/kg: n=9, 2,5 mg/kg: n=8) wurde ab
Tag 22 mit der 24stündigen Applikation von Ribavirin oder PBS begonnen. Die Therpie
setzt also zu einem Zeitpunkt ein, der eine klinische Diagnose zulässt.
In der folgenden Woche kommt es bei den Tieren aller drei experimentellen Gruppen
zu einem BD-bedingten Gewichtsverlust. Dieser korelliert jedoch negativ mit der
verabreichten Ribavirinmenge: In der Kontrollgruppe kommt es zu einer
Gewichtsreduktion von 23%, bei einer Dosierung von 1,25 beträgt sie 18% und in der
Gruppe 2,5 mg/kg 10% (Tag 22 bis 28, s. Abbildung 16). Diese günstige Beeinflussung
des Gewichtsverlaufes ist signifikant (p=0,0015) mit einem maximalen Effekt an Tag 28
(p=0,001). Hierbei dient das Körpergewicht als einfach und objektiv zu ermittelnder
Übersichtswert für den körperlichen Zustand der Tiere und den Schweregrad der
Erkrankung. Auch die an Tag 29 ermittelten klinischen Scores spiegeln diesen
protektiven Effekt von Ribavirin wider. 24 Stunden nach der letzten Injektion weisen die
mit Ribavirin behandelten Tiere eine weit weniger schwere Ausprägung von BD auf:
Bei der Beurteilung äußerlicher Zeichen der Erkrankung, des Putzverhaltens, der
motorischen Aktivität, der Atmung und des Ernährungszustands zeigen sich
signifikante Unterschiede zwischen den Gruppen (î =14,57, df=6, p < 0,05) mit einem
maximalen Effekt zwischen der Kontrollgruppe und der hochdosierten Gruppe

5 - Diskussion
86
(î=13,99, df=3,p < 0,01). Die Mediane der individuellen Scores liegen bei 19, 14,5 und
11 auf einer Scala von 6-22. Dabei repräsentiert ein Punktwert von 22 die stärkste
Ausprägung der beurteilten Symptome (s. Abbildung 17. Dosisabhängige Protektion
gegenüber der klinischen Symptomatik.).
Durch die intrathekale Therapie mit Ribavirin im akuten Stadium von BD gelingt bei
Lewis-Ratten eine dosisabhängige, signifikante und erhebliche Reduktion der
klinischen Symptomatik. Besonders die sehr schweren Krankheitsverläufe werden
durch eine hochdosierte Ribavirintherapie verhindert. Ein weiteres Indiz für den
Therapieerfolg gerade bei schweren Verläufen ist eine Überlebensrate von 100% in
der hochdosiert behandelten Gruppe. In den beiden anderen Gruppen verstirbt
dagegen je ein Tier an Tag 29 in stark reduziertem Allgemeinzustand (s. Abbildung
16).
Zwei verschiedene Mechanismen können potentiell für diesen protektiven Einfluss von
Ribavirin verantwortlich sein:
a) Zum einen inhibiert Ribavirin in vitro die Replikation von BDV [116, 117]. Eine
reduzierte virale Replikation könnte einen schwächeren Stimulus für die
Immunantwort des Wirts darstellen, was wiederum in eine mildere Symptomatik
bewirken würde (Immunpathogenese von BD s. [12]).
b) Alternativ oder additiv könnte die Immunantwort des Wirtstieres direkte moduliert
oder supprimiert werden. Dadurch kann eine Verbesserung der klinischen Situation
auch ohne Reduktion der Viruslast erreicht werden [58, 63, 69, 78-81, 85, 86, 12].
Um beide Mechanismen auf ihre Bedeutung in diesem Modell zu prüfen, werden im
nächsten Schritt die Virusreplikation im ZNS der infizierten Ratten quantifiziert und die
hierdurch induzierte Immunreaktion charakterisiert.
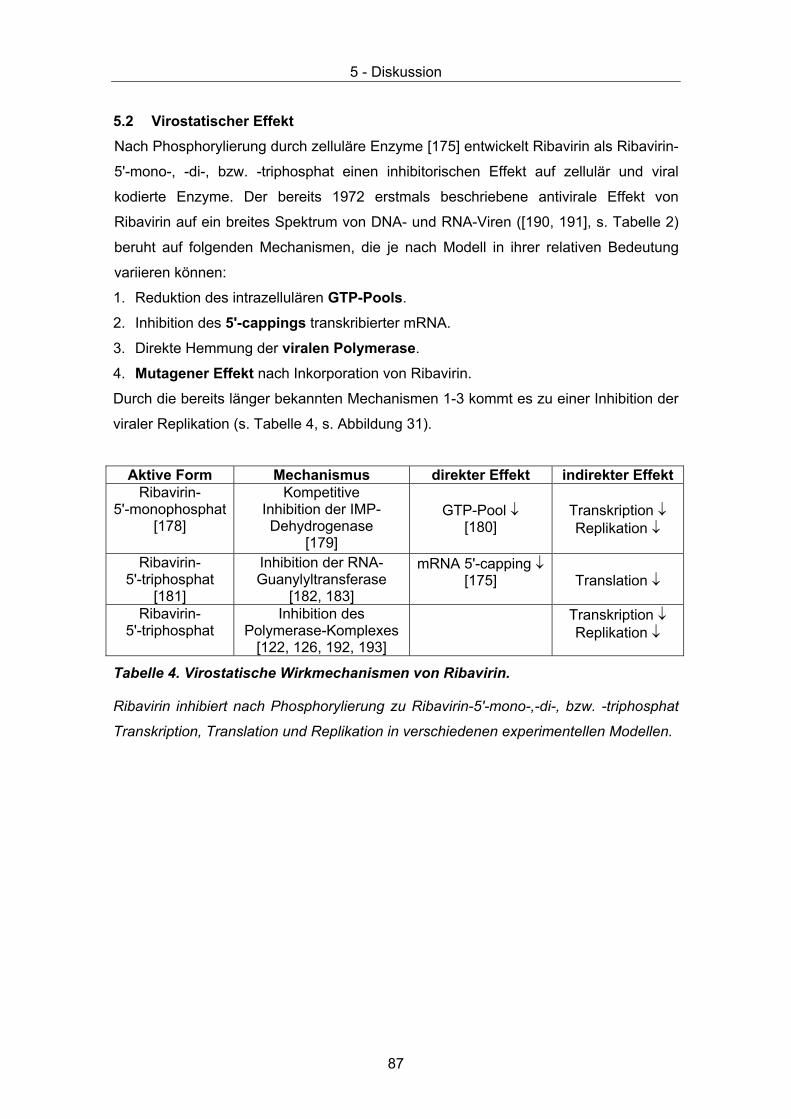
5 - Diskussion
87
5.2 Virostatischer Effekt Nach Phosphorylierung durch zelluläre Enzyme [175] entwickelt Ribavirin als Ribavirin-
5'-mono-, -di-, bzw. -triphosphat einen inhibitorischen Effekt auf zellulär und viral
kodierte Enzyme. Der bereits 1972 erstmals beschriebene antivirale Effekt von
Ribavirin auf ein breites Spektrum von DNA- und RNA-Viren ([190, 191], s. Tabelle 2)
beruht auf folgenden Mechanismen, die je nach Modell in ihrer relativen Bedeutung
variieren können:
1. Reduktion des intrazellulären GTP-Pools.
2. Inhibition des 5'-cappings transkribierter mRNA.
3. Direkte Hemmung der viralen Polymerase.
4. Mutagener Effekt nach Inkorporation von Ribavirin.
Durch die bereits länger bekannten Mechanismen 1-3 kommt es zu einer Inhibition der
viraler Replikation (s. Tabelle 4, s. Abbildung 31).
Aktive Form Mechanismus direkter Effekt indirekter Effekt Ribavirin-
5'-monophosphat [178]
Kompetitive Inhibition der IMP- Dehydrogenase
[179]
GTP-Pool ↓
[180]
Transkription ↓ Replikation ↓
Ribavirin-
5'-triphosphat [181]
Inhibition der RNA- Guanylyltransferase
[182, 183]
mRNA 5'-capping ↓ [175]
Translation ↓
Ribavirin- 5'-triphosphat
Inhibition des Polymerase-Komplexes
[122, 126, 192, 193]
Transkription ↓ Replikation ↓
Tabelle 4. Virostatische Wirkmechanismen von Ribavirin.
Ribavirin inhibiert nach Phosphorylierung zu Ribavirin-5'-mono-,-di-, bzw. -triphosphat
Transkription, Translation und Replikation in verschiedenen experimentellen Modellen.

5 - Diskussion
88
Abbildung 31. Inhibition der IMP-DH, Reduktion der Synthese von GTP (aus
[118]).
Ribavirin führt über eine kompetitive Hemmung der IMP-Dehydrogenase zu einem
reduzierten GTP-Pool und dadurch einer geringeren Transkription und Replikation.
Desweiteren konnte kürzlich ein mutagener Effekt von inkorporiertem Ribavirin auf die
Polymerase von RNA-Viren, im Modell der Poliovirus 3Dpol, nachgewiesen werden
(Mechanismus 4). Nach Inkorporation von Ribavirin wird Cytidin und Uridin in gleicher
Häufigkeit als Partnerbase von Ribavirin gefunden [187]. In dem genannten Modell
resultiert daraus in Zellkultur eine Reduktion infektiöser Polioviruspartikel auf
0,00001%, wobei der antivirale Effekt direkt mit der mutagenen Aktivität korreliert. Er
wird ausgelöst über Induktion letaler Mutationen (error catastrophe) und Reduktion der
Synthese viraler RNA [187, 188]. Inwieweit dieser mutagene Effekt auch bei
Polymerasen anderer RNA-Viren von Bedeutung ist, muss noch gezeigt werden.
Transkriptionsaktivität: Kommt es in diesem in vivo Modell aufgrund der Mechanismen 1,3 und/oder 4 zu einer
Transkriptionsinhibition, lässt sich dies durch Quantifizierung viraler RNA nachweisen:
In Strukturen mit gewöhnlich hoher viraler Transkriptionsaktivität wird mittels in situ
Hybridisierung und Taqman-RT-PCR virale RNA quantifiziert. Die in situ Hybridisierung

5 - Diskussion
89
liefert zusätzlich Information über die Verteilung viraler Nukleinsäure in verschiedenen
Hirnregionen. Als Target für die radioaktive Sonde in der in situ Hybridisierung wird
⊕-Strang RNA ausgewählt, die für das Phosphoprotein von BDV kodiert. P zeichnet
sich durch eine hohe Abundanz in Gewebe mit intensiver viraler Transkriptionsaktivität
aus [189]. Quantifiziert wird in Hippocampus, Striatum und Thalamus der rechten
Hemisphäre. Neben einer hohen Viruslast zeichnen sich diese Strukturen durch eine
enge Lagebeziehung zum inneren Liquorraum und somit eine kurze Diffusionsstrecke
für das applizierte Virostatikum aus. Für die quantitative RT-PCR wird Nukleinsäure
verwendet, die aus dem linken Präfrontalkortex isoliert wird. Auch für diese Struktur
weist BDV einen hohen Tropismus auf. Weder in situ Hybridisierung noch RT-PCR
können eine signifikante Reduktion viraler Nukleinsäure zeigen (s. Abbildung 21 und
Abbildung 22). Auch in der Verteilung der Sonde lässt sich kein Unterschied zwischen
den experimentellen Gruppen nachweisen. Unabhängig von der Entfernung zum
Applikationsort zeigt Ribavirin in diesem Modell keinen inhibitorischen Effekt auf die
Transkription von BDV. Auch auf mikroskopischer Ebene lässt sich kein Ribavirin-
induzierter Effekt auf das Infektionsmuster und die Verteilung viraler RNA nachweisen.
Das hier beobachtete Verteilungsmuster der viralen Transkripte spiegelt den für BDV
typischen und mehrfach beschriebenen Tropismus für das limbischen System, den
Kortex, Thalamus und das Cerebellum wider ([46, 48, 54, 89] reviewed [62] und [12]).
Weder bei der Quantifizierung absoluter viraler Nukleinsäurespiegel in den drei
experimentellen Gruppen noch bei der Analyse der relativen Verteilung der Virus-
transkripte konnte ein signifikanter Unterschied zwischen den Gruppen nachgewiesen
werden. Daher muss davon ausgegangen werden, dass in dem untersuchten Modell
keiner der Mechanismen 1, 3 oder 4 für den klinisch protektiven Effekt einer i.c.v.
Applikation von Ribavirin verantwortlich ist.
Translationseffizienz: Reduzierte Translation viraler mRNA zeigt sich in einer geringeren Konzentration
viraler Proteine. Zur orientierenden Beurteilung dieses Mechanismus wird virales
Nukleoprotein in Hippocampus der infizierten Tiere mittels Western-Immunoblot
quantifiziert. Dafür wurden je zwei Vertreter pro Gruppe ausgewählt: in der
Kontrollgruppe Tiere mit klinisch schwere Ausprägung und für die Gruppe 2,5 mg/kg
Tiere, bei denen der protektive Effekt von Ribavirin besonders prominent ist. Zusätzlich
wird Gewebe zweier repräsentativer Tiere der Gruppe 1,25 mg/kg verwendet. Es zeigt
sich keine Korrelation zwischen reduzierter Proteinkonzentration und verringerter

5 - Diskussion
90
klinischer Symptomatik. Somit ist auch Mechanismus 2 als kausaler Faktor für den
positiven Effekt von Ribavirin wenig wahrscheinlich. Beobachtet wird eher eine
Zunahme der Konzentration des Nukleoproteins mit steigender Ribavirindosis. Die
Bestimmung erfolgt jedoch lediglich orientierend. Ein prominenter Effekt kann nicht
nachgewiesen werden (s. Abbildung 25). Da nur zwei Werte je Gruppe vorliegen, kann
keine Aussage über statistische Signifikanz gemacht werden.
Virusreplikation: Alle vier Mechanismen, die für den virostatischen Effekt von Ribavirin bekannt sind
führen letztendlich zu einer Inhibition der viralen Replikation. Um die Konzentration
infektiöser BDV-Partikel bei den drei Therapiegruppen zu quantifizieren, wird eine
Virustitrierung mittels Focus Immunoassay durchgeführt. Die Titer liegen in allen Fällen
in einem engen Bereich zwischen 105 und 106 ffu/g Hippocampusgewebe (s. Abbildung
24), einer Größenordnung, die regelmäßig in adulten BDV-infizierten Lewis-Ratten
gemessen wird [170]. Tendenziell lässt sich eine dosisabhängige Reduktion der Titer
erkennen. Die Unterschiede sind jedoch nicht signifikant und kommen auch wegen der
geringen Differenz nicht als kausaler Faktor für den beobachteten positiven klinischen
Effekt in Frage.

5 - Diskussion
91
5.3 Immunmodulatorischer Effekt (Neuroprotektion) Neben dem antiviralen Effekt von Ribavirin sind zusätzlich immunmodulatorische
Mechanismen beschrieben worden: bei chronisch viraler B und C Hepatitis kann mit
einer Ribavirintherapie ein positiver Effekt auf klinische Parameter (Serum-ALT-
Spiegel) erzielt werden [126, 127, 190-194]. Dabei kommt es jedoch nur selten zu einer
Clearance des Virus, so dass die Frage nach dem Mechanismus zunächst offen bleibt.
Ebenso wurde in einigen Fällen eine Reduktion histopathologischer Veränderungen
beobachtet [192-194]. Bei Analyse der Immunreaktion fällt auf, dass es zu einer
absolut oder relativ erhöhten Produktion der Zytokine IL-2, IFN-γ sowie TNF-α kommt.
Dies hat eine Polarisation der T-Zell-Antwort in Richtung Typ 1 mit einer aggravierten
zellulären Immunantwort zur Folge [158-161]. Dieser Effekt wird für die beobachtete
Normalisierung von Laborparametern, Leberhistologie und Klinik bei Patienten mit
chronisch viralen Hepatitiden verantwortlich gemacht.
Als Pathogenesefaktoren bei BD sind wie erwähnt verschiedene Komponenten der
Immunreaktion des Wirtsorganismus auf die Infektion mit BDV identifiziert worden:
Bedeutend ist eine funktionierende zelluläre Immunantwort, wobei T-Helfer- wie
zytotoxische T-Zellen von Bedeutung sind (s. 1.3.2). Zusätzlich wurde in mehreren
Modellen eine reaktive Mikrogliose kolokalisiert mit neuropathologischen Foci sowie
eine Aktivierung von Mikroglia besonders in Nachbarschaft von Immunzellinfiltraten
[53, 64] beobachtet und eine kausale Beteiligung postuliert [70-72]. Als Mechanismus
kommt einerseits die Synthese proinflammatorischer Zytokine [72] in Frage. Bei der
Untersuchung der BDV induzierten Retinopathie konnte andererseits eine
phagozytotische Aktivität von Mikroglia und Makrophagen gegenüber neuronalen
Zellen nachgewiesen werden [50]. Mikroglia und/oder Makrophagen scheinen auch an
der gesteigerten Expression von mRNA für die induzierbare NO-Synthetase (iNOS)
während der akuten Phase von BD beteiligt zu sein. Diese korreliert zeitlich mit dem
Auftreten klinischer Symptome und liefert weitere Hinweise für eine pathogenetische
Beteiligung dieser Zellpopulation [65, 66].
Für die Beurteilung eines potentiellen Einflusses von Ribavirin auf die Immunantwort
der infizierten Tiere werden folgende immunologischen Parameter untersucht:
a) Infiltration von T-Lymphozyten als Maß für die zelluläre Immunantwort.
b) Zahl und Aktivierungsgrad der Mikroglia.
c) Zytokinprofil. d) Antikörpertiter gegen virale Proteine (humorale Immunreaktion).

5 - Diskussion
92
T-Lymphozyten und Mikroglia: Sowohl CD4+, wie auch CD8+ T-Lymphozyten sind in den Infiltraten der akuten Phase
von BD in großer Zahl nachweisbar [68]. Hier werden die beiden Subtypen quantifiziert,
um einen potentiellen Einfluß von Ribavirin auf die Intensität der zellulären
Immunantwort beurteilen zu können. Dabei wird eine reduzierte Zahl infiltrierender T-
Lymphozyten beobachtet. Diese Effekt ist prominenter für CD4+ Zellen (s.
). Die typische Verteilung der Subtypen lässt sich auch in dieser Studie nachweisen:
CD4+ Zellen finden sich hauptsächlich in perivaskulären Cuffs, während die CD8+
T-Lymphozyten auch das Hirnparenchym infiltrieren. Ob die beobachtete quantitative
Reduktion auf eine verminderte Extravasation zirkulierender Zellen und/oder eine
Hemmung der Proliferation bereits im Parenchym befindlicher Zellen zurückzuführen
ist, lässt sich mit dieser Methode nicht klären. Denkbar ist einerseits eine reduzierte
Infiltration durch verminderte chemotaktische Aktivität oder Stabilisierung der Blut-Hirn-
Schranke. Andererseits wäre auch ein direkt antiproliferativer Effekt von Ribavirin auf
aktivierte T-Lymphozyten möglich.
Abbildung
26
Ebenso stellt sich beim Vergleich der Zahl und des Aktivierungsgrads mikroglialer
Zellen in den Hirnen unbehandelter Tiere eine deutlich größere Zellzahl dar. Besonders
auffällig ist dies beim Vergleich der Gruppe 2,5 mg/kg mit der Kontrollgruppe (s.
Abbildung 28). Das Bild bei den Tieren der Gruppe 1,25 mg/kg reiht sich zwischen den
beiden gezeigten ein. Die bei neonatal infizierten Tieren beobachtete Expression von
CD4 und CD8 Molekülen auf der Oberfläche aktivierter Mikroglia [70] tritt auch in
diesem Modell auf (s. Abbildung 27). Sie erscheint in den Verum-Gruppen reduziert,
ein funktionelles Korrelat ist jedoch nicht beschrieben.
Es kommt somit zu einer Reduktion von zwei Zellenpopulationen, die einen
entscheidenden Anteil an der Pathogenese von BD haben. Für T-Lymphozyten ist
bekannt, dass ihre funktionelle Blockade zu asymptomatischen Krankheitsverläufen
führt [63, 69, 85, 58, 86]. Die Kenntnisse über die kausale Beteiligung von Mikroglia an
der Pathogenese sind noch unvollständig. Im Modell der neonatal infizierten Lewis-
Ratten und bei der BDV induzierten Retinopathie wurden jedoch Beobachtungen
gemacht, die für eine pathogenetische Beteiligung sprechen. Die Reduktion von
T-Lymphozyteninfiltraten und reaktiver Mikrogliose durch Ribavirin kann somit einen
protektiven Einfluss auf den Verlauf von BD erklären.

5 - Diskussion
93
Zytokinprofil: Wie bereits erwähnt wurde in letzter Zeit in mehreren expermientellen Ansätzen eine
Polarisation des Zytokinprofils in Richtung einer Typ 1 Antwort beschrieben [158-161].
Bei der experimentellen BD in Lewis-Ratten wird während der akuten Phase der
Erkrankung eine intensive zelluläre Entzündungsreaktion im ZNS beobachtet. Mit dem
Übertritt in die chronische Phase verschiebt sich das Gleichgewicht deutlich in
Richtung einer humoralen Immunantwort. Das Zytokinprofil in der akuten Phase kann
als Typ 1-, das der chronischen Phase als Typ 2-dominiert beschrieben werden [68,
94]. Eine Polarisation der Immunantwort in Richtung Th1 sollte daher mit einer
intensivierten zellulären Immunreaktion, verstärkten akuten Krankheitsphase und
prominenteren Symptomatik einhergehen. Um diesen Mechanismus im vorliegenden
Modell zu untersuchen, werden die mRNA-Level von Zytokinen der Th1 wie der Th2
Reihe in RNA aus dem Präfrontalkortex der infizierten Tiere mittels RPA quantifiziert.
Dabei wird eine vermehrte Expression von mRNA für IL10, IL1β und IFN-γ beobachtet
(s. ). Erhöhte mRNA Spiegel für IFN-γ, einem proinflammatorischen
Zytokin, sowie für IL1β, ebenfalls einem Typ 1 Zytokin sind charakteristisch für die
akute Phase von BD. IL10 gehört dagegen zur Reihe der Zytokine des Typ 2 Profils, so
dass die beobachteten Veränderungen der Zytokinspiegel keine eindeutige Tendenz in
Hinblick auf eine Veränderung der Th1/Th2 Balance erkennen lassen. IFN-γ wird mit
einer Hochegulation der MHC Klasse I Expression auf der Oberfläche von Neuronen
mit erloschener elektrophysiologische Aktivität in Verbindung gebracht [195, 196].
Exprimieren funktionell defiziente Zellen MHC Klasse I auf ihrer Oberfläche, können sie
von zytotoxischen T-Lymphozyten erkannt und über die Ausschüttung von Perforin in
die Nekrose getrieben werden [197]. Die durch Ribavirin erhöhte Konzentratione von
IFN-γ könnte über die Induktion der Expression von MHC Klasse I Antigen zu einer
verbesserten Erkennung und Ausschaltung infizierter Zellen führen. Gegen diesen
Mechanismus spricht jedoch die nicht messbar reduzierte virale Replikation, sowie die
beobachtete generelle Neuroprotektion. Es scheint nicht zu einer effizienteren
Elimination infizierter Zellen und besseren Kontrolle der Virusreplikation zu kommen.
Abbildung 29
Humorale Immunantwort: Bei der Charakterisierung der humoralen Immunantwort der infizierten Tiere werden
erhöhte Antikörpertiter in den Ribavirin-Gruppen gemessen. Möglicherweise werden
diese durch die verstärkte Virämie induziert. Eine intensivierte humorale Immun-
reaktion spricht gegen eine generelle unspezifische Immunsuppression.

5 - Diskussion
94
Die intrathekale Applikation von Ribavirin hat also einen deutlichen klinisch protektiven
Effekt auf Lewis-Ratten mit akuter BD. Dieser ist dosisabhängig und besonders
ausgeprägt bei Verabreichung von 2,5 mg/kg KG/Tag. Bei nicht infizierten Tieren ist
eine Dosierung von 5 mg/kg bereits mit Zeichen für Toxizität und einer gesteigerter
Mortalität verbunden. 2,5 mg/kg werden dagegen ohne ausgeprägte toxische Wirkung
toleriert. Die therapeutische Breite von Ribavirin bei i.c.v. Applikation zur Therapie der
akuten BD ist also gering, der therapeutische Nutzen jedoch ausgeprägt: Hochdosiert
behandelte Tiere zeigen sowohl eine stark reduzierte Symptomatik, als auch eine
reduzierte Mortalität. Bei der Präparation der Gehirne fällt bei unbehandelten Tieren
eine verminderte Konsistenz auf. Die Präparation ist durch diese Erweichung erheblich
erschwert. Diese Beobachtung wird bei den hochdosiert behandelten Tieren nicht
gemacht, was als makroskopisches Korrelat für die inhibierte Entzündungsaktivität
gewertet werden kann. Die viralen Parameter lassen allenfalls tendenziell eine
Inhibition der Virusreplikation erkennen. Der stark antireplikative Effekt auf BDV, der in
vitro beobachtet werden konnte, läßt sich in diesem Modell mittels vier unabhängiger
Verfahren zur Quantifizierung viraler Nukleinsäure, Proteine und Viruspartikel nicht
nachweisen. Eine Abschwächung der klinischen Symptomatik durch einen
virostatischer Effekt ist somit unwahrscheinlich.
Große Unterschiede fallen demgegenüber bei der Charakterisierung der
virusinduzierten Immunreaktion zwischen der Kontrollgruppe und den beiden Ribavirin-
Gruppen auf. Es kommt zu einer deutlichen, quantitativen Reduktion von T-
Lymphozyten in perivaskulären Cuffs und im Hirnparenchym der hochdosiert
behandelten Tiere. Zusätzlich ist die reaktive Mikrogliose quantitativ erheblich
reduziert. Beide Beobachtungen können den milderen Verlauf der Erkankung in den
behandelten Tieren erklären. Zeichen der supprimierten zellulären Immunreaktion im
ZNS der infizierten Tiere sind auch die verstärkte Virämie, sowie die intensivierte
humorale Immunantwort. Dieser Einfluß auf die Aktivität der Entzündungszellen ist
vermutlich nicht auf eine Beeinflussung des Zytokinprofils zurückzuführen. Die
beobachtete Induktion von IFN-γ und IL10 lassen keine Polarisation der Immunantwort
erkennen, wie sie in anderen Studien beschrieben wurde. Über welchen Mechanismus
Ribavirin zu einer Immunmodulation führt, bleibt daher zunächst unklar. Die bei
Ribavirintherapie von BD beobachtete Reduktion von T-Lymphozyten und Mikroglia ist
jedoch konsistent mit einem antiproliferativem Einfluß auf aktivierte Lymphozyten in
anderen Ansätzen: Nachgewiesen wurde eine Inhibition der in vitro Proliferation

5 - Diskussion
95
humaner Lymphozyten [198]. Dosisabhängig wurde eine Reduktion der Zahl
stimulierter T-Lymphozyten um bis zu 94% beobachtet [199]. Die simultan
quantifizierte Zytokinproduktion blieb dabei unverändert [199]. Bei Patienten mit
chronisch viraler C Hepatitis konnte ebenfalls ein antiproliferativer Effekt auf
mononukleäre Zellen und T-Lymphozyten nachgewiesen werden [200]. Zusätzlich ist
ein antiproliferativer Effekt auf verschiedene, maligne transformierte Zelllinien bekannt
(humane Ovarial-Carzinom-Zellen [201], humane Melanomzellen IGR39 [202] [180],
Adenovirus-12 induzierte CBA Maus-Tumor-Zellen [203], murine Erythroleukämie-
Zellen [204]). Für diesen Effekt wird unter anderem die Inhibition der IMP-
Dehydrogenase und die resultierende Erschöpfung des intrazellulären GTP-Pools
verantwortlich gemacht [180]. Beobachtet wird ein Zellarrest in der G1 Phase [201,
202]. Auch andere Inhibitoren der IMP-DH besitzen antiproliferative Eigenschaften:
Tiazofurin, ein Strukturanalogon von Ribavirin besitzt einen zytostatischen Effekt und
induziert Differenzierung und Apoptose in verschiedenen Zelllinien [202, 205]. Ein
weiterer, nichtkompetitiver Inhibitor der IMP-DH, Mycophenolat Motefil (aktiver
Metabolit MPA) wird für die Immunsuppression nach Nieren- und Herztransplantation
eingesetzt [179]. Es ist zu vermuten, dass die hier beobachtete Neuroprotektion auf
einen antiproliferativen Mechanismus zurückzuführen ist.
Wiederholt wurde eine Detektion von BDV RNA auch im peripheren Blut infizierter
Lewis-Ratten beschrieben [59, 60, 103]. Besonders bei Insuffizienz der Immunantwort
kommt es zur Virusreplikation außerhalb des Nervensystems [46, 57]. Die Frage der
Nachweisbarkeit von BDV RNA im peripheren Blut ist besonders auch wegen einer
diagnostischen Nutzung von Bedeutung: In mehreren Studien wurde BDV RNA auch in
PBMCs humanen Ursprungs amplifiziert [206-210] und Granulozyten als mögliche
Wirtszellen identifiziert [211]. Diese Ergebnisse werden jedoch kontrovers diskutiert
[19]. In dem vorliegenden Modell wurde bei je vier bis fünf Tieren der drei
experimentellen Gruppen versucht, virale RNA des zweiten ORF mittels Taqman RT-
PCR in peripheren mononukleären Blutzellen nachzuweisen. Dies gelang in allen drei
Gruppen (s. Abbildung 23). In den beiden Verum-Gruppen wurden dabei höhere
Kopienzahlen als in den Tieren der Kontrollgruppe gemessen. Die Applikation von
Ribavirin scheint mit der Restriktion der viralen Replikation auf das ZNS zu interferieren
und einer Streuung der Infektion mit verstärkter Virämie Vorschub zu leisten. Im
Vergleich mit der absoluten Zahl der Kopien viraler RNA im ZNS sind die im Blut
gemessenen Werte im Mittel um den Faktor 50 bis 100 niedriger. Die hier beobachtete

5 - Diskussion
96
periphere Replikation von BDV steht im Einklang mit oben genannten Ergebnissen
anderer Studien und spricht für eine mögliche Nutzung des Nukleinsäurenachweises in
peripherem Blut als weitere diagnostischer Methode zum in vivo Nachweis einer BDV
Infektion.
Wie ist die fehlende Reproduzierbarkeit des in vitro beobachteten virostatischen
Effekts von Ribavirin in dieser ersten Anwendung gegen BDV in vivo zu erklären?
In anderen Ansätzen konnte regelmäßig mit einer Suppression der Immunreaktion eine
klinische Verbesserung erzielt werden. Gleichzeitig ging diese jedoch mit einer
Erhöhung der Viruslast einher [49]. Das Immunsystem, das zum einen für die
Neurodestruktion und damit klinische Symptomatik verantwortlich ist, garantiert zum
anderen auch eine gewisse Inhibition der Virusreplikation. Wird sie in ihrer Intensität
reduziert, sind beide - sowohl der neuropathogene wie auch der replikations-
inhibierende Effekt - betroffen und die Folgen beider als Symptomreduktion und
erhöhten Viruslast meßbar. Bei der i.c.v. Applikation von Ribavirin kommt es dagegen
zu einer deutlichen Reduktion der klinischen Symptomatik ohne vermehrte
Virusreplikation. Im Gegenteil, die in dem vorliegenden Modell gemessenen Virustiter
deuten eher eine Reduktion als eine Verstärkung der viralen Replikation an. Dass kein
virostatische Effekt meßbar ist, könnte somit durch die Überlagerung zweier
gegensätzlicher Einflüsse erklärt werden: Durch die Inhibition der Immunreaktion
verschiebt sich das Gleichgewicht zwischen viraler Replikation und deren Kontrolle
durch das Immunsystem zugunsten der Virusreplikation. Gleichzeitig wird diese durch
den virostatischen Effekt von Ribavirin jedoch gedrosselt. Effektiv resultiert die
Überlagerung beider gegenläufiger Effekte in keiner signifikanten Veränderung der
Viruslast.
Eine weitere Ursache könnte darin bestehen, dass postmitotischen Zellen wie den
Neuronen, in denen die Virusreplikation stattfindet, effektive Mechanismen für die
Aufnahme von Ribavirin fehlen.

5 - Diskussion
97
Verschiedene Ansätze für eine Therapie von BD wurden bereits in vivo getestet. Bei
aktiver Immunisierung gelang eine Reduktion der Viruslast, gleichzeitig wurde aber
durch Stimulation der Immunabwehr die klinische Symptomatik intensiviert [105]. Der
antivirale Effekt von Interferon ist stark von der verwendeten Zellinie und einem frühen
Behandlungsbeginn abhängig [107, 108]. Amantadin versprach in therapeutischen
Studien antivirale Aktivität, diese war jedoch bisher weder in vitro noch in vivo
reproduzierbar [111-115]. Unter Immunsuppression kann zwar ein asymptomatischer
Verlauf der Erkankung induziert werden, allerdings nur bei präexpositionell
bestehender Suppression. Sie geht zudem mit den bekannten Nebenwirkungen einher.
Simultan kommt es zu erhöhten Virustitern, was auch für eine potentielle
Ausscheidung infektiöser Partikel und deren Weiterverbreitung Implikationen besitzt.
Die intrathekale Applikation von Ribavirin ist somit der erste pharmakotherapeutische
Ansatz, der zu einer Suppression der klinischen Symptomatik führt, ohne gleichzeitig
eine unkontrollierte Virusreplikation zuzulassen.
Zukünftige Therapieansätze könnten von einer Kombination von Ribavirin mit
zusätzlicher immunmodulatorischer Beeinflussung profitieren. Antivirale Therapie plus
Immunstimulation unterstützt die Clearance von andernfalls persistierenden LCMV
Infektionen in Mäusen. Ein Ähnlicher positiver Einfluß wird auch für andere nicht-
lytische Virusinfektionen postuliert [212]. Dabei wäre zu prüfen, inwiefern sich die
invasive zentrale Applikation durch eine schonendere inhalative Therapie ersetzen
ließe. Ribavirin kumuliert bei Applikation als Aerosol in Hirnen behandelter Mäuse zu
Konzentrationen von ca. 15 µM. Dabei wurden Ribavirinlösungen mit Konzentrationen
von 20 mg/ml, bzw. 60 mg/ml im Reservoir des Aerosolgenerators für 12, bzw. 2 oder
4 h eingesetzt [213, 214]. Der erzielte Gewebespiegel liegt zwar unter dem bei i.c.v.
Applikation, es bliebe aber eine weitere Akkumulation zu prüfen. Ein nicht-invasives
Vorgehen ist der entscheidende Vorteil dieser Applikationsart.
Die BDV Infektion in Lewis-Ratten wird zusätzlich als Modell für immunmediierte,
neurodegenerative Erkankungen verwendet. Der protektive Effekt von Ribavirin in
diesem Modell legt eine Prüfung in anderen Erkrankungen mit kausaler Beteiligung
einer zellulären Immunreaktion nahe. Mikroglia scheinen auch in anderen
neurodegenerativen Erkrankungen wie der HIV Demenz, Multipler Sklerose und
Alzheimer’schen Erkrankung eine entscheidende pathogenetische Rolle zu spielen
[215]. Der beobachtete Effekt von Ribavirin auf die Entwicklung neuropathogener
Prozesse in Hirnen BDV infizierter Lewis-Ratten und die verbundene Inhibition

5 - Diskussion
98
klinischer schwerer Verläufe liefern weitere Hinweise für einen weit komplexeren
Wirkmechanismus des seit etwa drei Jahrzehnten bekannten antiviralen Wirkstoffs
Ribavirin. Der klinisch Einsatzbereich von Ribavirin ist verglichen mit der antiviralen
Wirkung auf ein breites Spektrum von DNA- wie RNA-Viren in verschiedensten
experimentellen Settings gering (s. ). Zur breiteren Anwendung kommt
Ribavirin seit wenigen Jahren in Kombination mit IFN-α bei der Therapie chronischer
Virushepatitien. Hier korreliert der therapeutische Nutzen jedoch nur bedingt mit der
antiviralen Therapie. Eine weitere Untersuchung immunmodulatorischer, wie antiproli-
ferativer Mechanismen ist daher besonders wichtig und könnte zu einer noch deutlich
größeren therapeutischen Nutzbarkeit von Ribavirin führen.
Tabelle 2

6 - Zusammenfassung
6 Zusammenfassung
Die Infektion mit dem Borna Disease Virus (BDV) ruft bei einem weiten Spektrum von
Warmblütern ein teilweise progredientes, immunmediiertes neurologisches Syndrom
mit außerordentlicher Symptomvarianz hervor. Der Erreger zeichnet sich durch ein
einzelsträngiges RNA Genom negativer Polarität, ausgeprägten Neurotropismus und
einen nicht-lytischen Replikationszyklus aus, der in viraler Persistenz mündet.
Seroreaktivität, virale RNA und Proteine wurden mit erhöhter Prävalenz in Blut und
zentralnervösem Gewebe neuropsychiatrischer Patienten nachgewiesen. Diese
Korrelation sowie eine potentielle ätiologische Beteiligung bei affektiven Störungen,
Schizophrenie und chronischem Müdigkeitssyndrom (CFS) werden jedoch kontrovers
diskutiert.
In vitro Experimente zeigten kürzlich einen virostatischen Effekt des Guanosinanalogs
Ribavirin in persistent BDV infizierten Zellen. Ziel der vorliegenden Studie war es, den
therapeutischen Nutzen von intrathekaler Ribavirinapplikation bei akuter Bornascher
Erkrankung (BD) in vivo im Rattenmodell zu charakterisieren. Vorausgehende
toxikologische und pharmakokinetische Untersuchungen ergaben eine maximal
verträgliche tägliche Dosis von 2,5 mg/kg KG in einer Bolusinjektion von ca. 5 µl PBS.
Drei Wochen nach intranasaler Virusinokulation und simultan mit dem Auftreten erster
Symptome akuter BD wurde Ribavirin in einer Dosis von 0, 1,25 und 2,5 mg/kg KG/Tag
für 7 Tage intrathekal appliziert. Hierdurch gelang es, die klinische Symptomatik akuter
BD dosisabhängig zu reduzieren. Die Bestimmung der Konzentration viraler RNA,
Proteine, sowie infektiöser Partikel in zentralnervösem Gewebe mittels in situ
Hybridisierung, quantitativer real-time RT-PCR, Western Blot und Virustitrierung ergab
jedoch keine signifikante Reduktion viraler Parameter bei den behandelten Tieren.
Anschließende immunhistologische Untersuchungen zeigten eine quantitative
Reduktion von T-Lymphozyten und mikroglialen Zellen in Hirnparenchym hochdosiert
behandelter Tiere, sowie eine Beeinflussung der mRNA Level für die Zytokine IL10,
IL1β und IFN-γ.
T-Lymphozyten sind wesentlich an der progressiven Neurodestruktion im Rahmen von
BD sowohl als zytotoxische Effektorzellen, wie auch als T-Helferzellen beteiligt. Auch
für Mikrogliazellen wird eine kausale Beteiligung an der Pathogenese von BD
postuliert. Mikroglia, ortständige immunkompetente Zellen des ZNS, sind wichtige
intrazerebrale Produzenten proinflammatorischer Zytokine. Der klinisch protektive
Effekt der zentralen Ribavirinapplikation scheint in dem vorliegenden Modell nicht auf
99

6 - Zusammenfassung
Inhibition der Virusreplikation, sondern auf die Suppression der neuropathogenen
Immunantwort des Wirtsorganismus zurückzuführen zu sein. Eine antiproliferative
Wirkung von Ribavirin auf aktivierte Lymphozyten, sowie verschiedene maligne
transformierte Zelllinien ist mehrfach beschrieben. Erklärt wird dieser Effekt durch die
Inhibition der IMP-Dehydrogenase mit Depletion des intrazellulären GTP-Pools. Auch
andere IMP-DH-Inhibitoren weisen einen antiproliferativen Effekt auf. Dieser wird zum
Teil für die immunsuppressive Therapie nach Transplantation parenchymatöser
Organe genutzt (Bsp. Mycophenolat Motefil).
Wie in anderen Studien wiederholt beschrieben, konnte virale RNA im peripheren Blut
infizierter Lewis-Ratten detektiert werden. Die Virämie bei hochdosiert behandelten
Tieren ist dabei deutlich ausgeprägter. Dies ist im Einklang mit Beobachtungen einer
generalisierten Virusreplikation unter insuffizienter Immunkontrolle durch den
Wirtsorganismus. Hieraus ergeben sich auch Implikationen für die diagnostische
Nutzbarkeit des peripheren Nukleinsäurenachweises zur Erkennung von BDV
Infektionen, die in vivo bisher nicht zuverlässig möglich ist.
Das Ausbleiben eines messbaren virostatischen Effekts von Ribavirin gegen BDV im
vorliegenden Tiermodell könnte durch die Überlagerung zweier gegensätzlicher
Einflüsse erklärt werden: Einerseits begünstigt die Immunsuppression eine
unkontrollierte Virusreplikation. Gleichzeitig kommt es durch den virostatischen Effekt
von Ribavirin jedoch zu deren Drosselung.
Die intrathekale Ribavirinapplikation ist der erste therapeutische Ansatz, der zu einer
Linderung des akuten Krankheitsbildes ohne gleichzeitige verstärkte Virusreplikation
führt. In zukünftige Studien bleibt die Kombination von Ribavirin mit zusätzlicher
immunmodulatorischer Beeinflussung zu prüfen, um eine Elimination des Virus zu
erreichen. Dabei wäre zu untersuchen, ob sich die intrathekale Applikation durch eine
Inhalation eines Ribavirinaerosols ersetzen ließe.
Mikroglia scheinen auch in anderen neurodegenerativen Erkrankungen wie der HIV
Demenz, Multipler Sklerose und Alzheimer’schen Erkrankung eine entscheidende
pathogenetische Rolle zu spielen. Eine weitere Untersuchung immunmodulatorischer,
wie antiproliferativer Mechanismen von Ribavirin ist daher besonders wichtig und
könnte zu einem noch deutlich größeren klinischen Nutzen führen.
100

7 - Abkürzungen
101
7 Abkürzungen
’’ Inch
Ak Antikörper
APC antigenpräsentierende Zelle
b Basen
BD Borna Disease
BDV Borna Disease Virus
bp Basenpaare
BSA bovines Serumalbumin
DEPC Diethylpyrocarbonat
diH2O deionisiertes Wasser
ddiH2O doppelt deionisiertes Wasser
DMSO Dimethylsulfoxid
ELISA Enzyme-linked Immunosorbent Assay
FCS foetal calf serum (fetales Kälberserum)
g Erdbeschleunigung
GAPDH Glycerinaldehyd-3-phosphat-Dehydrogenase
GM-CSF Granulocyte-Macrophage Colony-Stimulating Factor
HBV Hepatitis B Virus
HCV Hepatitis C Virus
HEPES n-2-Hydroxyethylpiperazin-N-Ethansulfonsäure
HFRS Haemorrhagic Fever with Renal Syndrome
HIV Human Immunodeficiency Virus
HPV Human Papilloma Virus
HRP Horseradish Peroxidase
i.c.v. intracerebroventrikulär
IFN Interferon
IHC Immunhistochemie
IL Interleukin
ISH in situ Hybridisierung
kb Kilobasen
kDa Kilodalton
konz. konzentriert
MHC-I Major Histocompatibility Complex Class I

7 - Abkürzungen
102
min Minute(n)
mM millimolar
M molar
nt Nukleotide
ORF Open Reading Frame
PB Phosphate Buffer
PBS Phopshate Buffered Saline
PCR Polymerase Chain Reaction
PFA Paraformaldehyd
pH pH-Wert
RNase Ribonuklease
RNP Ribonukleoprotein-Komplex
RPA RNase Protection Assay
rpm Umdrehungen pro Minute
RT Raumtemperatur
RT Reverse Transkriptase
s Sekunde(n) SCID schweres kombiniertes Immundefektsyndrom SDS Natrium-Dodecylsulfat
SEM Standard Error of the Mean
SSC Natriumchlorid/Natriumcitrat
SSPE subakute sklerosierende Panenzephalopathie
TBE Tris/Borat/EDTA
TEMED NNNN-Tetramethylethylendiamin
TGF Transforming Growth Factor
U Unit
ÜN über Nacht
WHO World Health Organisation
wt/vol Weight per Volume
V% Volumenprozent
V‰ Volumenpromill
vs. versus

8 - Literaturangaben
8 Literaturangaben
1. Virus Taxonomy: Seventh Report of the International Committee on Taxonomy
of Viruses, ed. D.H.L.B. H.V. Van Regenmortel, M. H. Van Regenmortel, Claude
M. Fauquet (Eds).
2. Nicolau, S.G., I.A., Borna disease and enzootic encephalomyelitis of sheep and
cattle. Privy Council, Med. Res. Council, Spec. Rep. Ser. 1-115., 1928.
3. Elford, W.J.G., I.A., Filtration of the virus of Borna disease through graded
collogion membranes. Brit. J. Exp. Biochem., 1933. 132: p. 6-13.
4. Lipkin, W.I., T. Briese, and J.C. de la Torre, Borna disease virus: molecular
analysis of a neurotropic infectious agent. Microb Pathog, 1992. 13(3): p. 167-
70.
5. Zimmermann, W., et al., Borna disease virus: immunoelectron microscopic
characterization of cell-free virus and further information about the genome. J
Virol, 1994. 68(10): p. 6755-8.
6. Ludwig, H., L. Bode, and G. Gosztonyi, Borna disease: a persistent virus
infection of the central nervous system. Prog Med Virol, 1988. 35: p. 107-51.
7. Briese, T., et al., Genomic organization of Borna disease virus. Proc Natl Acad
Sci U S A, 1994. 91(10): p. 4362-6.
8. Cubitt, B., C. Oldstone, and J.C. de la Torre, Sequence and genome
organization of Borna disease virus. J Virol, 1994. 68(3): p. 1382-96.
9. Jordan, I. and W.I. Lipkin, Borna disease virus. Rev Med Virol, 2001. 11(1): p.
37-57.
10. Walker, M.P., et al., Expression and characterization of the Borna disease virus
polymerase. J Virol, 2000. 74(9): p. 4425-8.
11. Nowotny, N., et al., Isolation and characterization of a new subtype of Borna
disease virus. J Virol, 2000. 74(12): p. 5655-8.
12. Gonzalez-Dunia, D., C. Sauder, and J.C. de la Torre, Borna disease virus and
the brain. Brain Res Bull, 1997. 44(6): p. 647-64.
13. Briese, T., et al., Borna disease virus, a negative-strand RNA virus, transcribes
in the nucleus of infected cells. Proc Natl Acad Sci U S A, 1992. 89(23): p.
11486-9.
14. Cubitt, B. and J.C. de la Torre, Borna disease virus (BDV), a nonsegmented
RNA virus, replicates in the nuclei of infected cells where infectious BDV
ribonucleoproteins are present. J Virol, 1994. 68(3): p. 1371-81.
103

8 - Literaturangaben
15. Martins, C.R., et al., Sonchus yellow net rhabdovirus nuclear viroplasms contain
polymerase-associated proteins. J Virol, 1998. 72(7): p. 5669-79.
16. Cubitt, B., et al., RNA splicing contributes to the generation of mature mRNAs of
Borna disease virus, a non-segmented negative strand RNA virus. Virus Res,
1994. 34(1): p. 69-79.
17. Schneider, P.A., A. Schneemann, and W.I. Lipkin, RNA splicing in Borna
disease virus, a nonsegmented, negative-strand RNA virus. J Virol, 1994. 68(8):
p. 5007-12.
18. Cubitt, B., C. Ly, and J.C. de la Torre, Identification and characterization of a
new intron in Borna disease virus. J Gen Virol, 2001. 82(Pt 3): p. 641-6.
19. Staeheli, P., et al., Epidemiology of Borna disease virus. J Gen Virol, 2000. 81
Pt 9: p. 2123-35.
20. Lundgren, A.L., et al., Natural Borna disease in domestic animals others than
horses and sheep. Zentralbl Veterinarmed [B], 1993. 40(4): p. 298-303.
21. Weissenbock, H., et al., Borna disease in Austrian horses. Vet Rec, 1998.
143(1): p. 21-2.
22. Kao, M., et al., Detection of antibodies against Borna disease virus in sera and
cerebrospinal fluid of horses in the USA. Vet Rec, 1993. 132(10): p. 241-4.
23. Bahmani, M.K., et al., Varied prevalence of Borna disease virus infection in
Arabic, thoroughbred and their cross-bred horses in Iran. Virus Res, 1996.
45(1): p. 1-13.
24. Nakamura, Y., et al., Demonstration of Borna disease virus RNA in peripheral
blood mononuclear cells from healthy horses in Japan. Vaccine, 1995. 13(12): p.
1076-9.
25. Nakamura, Y., et al., Demonstration of borna disease virus RNA in peripheral
blood mononuclear cells derived from domestic cats in Japan. J Clin Microbiol,
1996. 34(1): p. 188-91.
26. Hagiwara, K., et al., High prevalence of Borna disease virus infection in healthy
sheep in Japan. Clin Diagn Lab Immunol, 1997. 4(3): p. 339-44.
27. Metzler, A., F. Ehrensperger, and R. Wyler, [Natural borna virus infection in
rabbits]. Zentralbl Veterinarmed [B], 1978. 25(2): p. 161-4.
28. Malkinson, M., et al., Borna disease in ostriches [letter; comment]. Vet Rec,
1993. 133(12): p. 304.
29. Caplazi, P., et al., [Borna disease in Switzerland and in the principality of
Liechtenstein]. Schweiz Arch Tierheilkd, 1999. 141(11): p. 521-7.
104

8 - Literaturangaben
30. Weissenbock, H., et al., Borna disease in a dog with lethal meningoencephalitis.
J Clin Microbiol, 1998. 36(7): p. 2127-30.
31. Degiorgis, M.P., et al., Borna disease in a free-ranging lynx (Lynx lynx). J Clin
Microbiol, 2000. 38(8): p. 3087-91.
32. Bode, L., et al., First isolates of infectious human Borna disease virus from
patients with mood disorders [see comments]. Mol Psychiatry, 1996. 1(3): p.
200-12.
33. Nakamura, Y., et al., Isolation of Borna disease virus from human brain tissue. J
Virol, 2000. 74(10): p. 4601-11.
34. Hatalski, C.G., A.J. Lewis, and W.I. Lipkin, Borna disease. Emerg Infect Dis,
1997. 3(2): p. 129-35.
35. Gellert, M., ["In the beginning the horse is sad"--a historical abstract
of Borna disease]. Tierarztl Prax, 1995. 23(3): p. 207-16.
36. Katz, J.B., et al., Clinical, serologic, and histopathologic characterization of
experimental Borna disease in ponies. J Vet Diagn Invest, 1998. 10(4): p. 338-
43.
37. Metzler, A., et al., [Borna disease, a veterinary problem of regional significance].
Schweiz Arch Tierheilkd, 1979. 121(4): p. 207-13.
38. Rott, R. and H. Becht, Natural and experimental Borna disease in animals. Curr
Top Microbiol Immunol, 1995. 190: p. 17-30.
39. Waelchli, R.O., et al., Borna disease in a sheep. Vet Rec, 1985. 117(19): p. 499-
500.
40. Bode, L., R. Durrwald, and H. Ludwig, Borna virus infections in cattle associated
with fatal neurological disease. Vet Rec, 1994. 135(12): p. 283-4.
41. Caplazi, P., et al., Borna disease in naturally infected cattle. J Comp Pathol,
1994. 111(1): p. 65-72.
42. Herzog, S., K. Frese, and R. Rott, Studies on the genetic control of resistance of
black hooded rats to Borna disease. J Gen Virol, 1991. 72(Pt 3): p. 535-40.
43. Zwick, W., Bornasche Krankheit und Enzephalomyelitis der Tiere., in Handbuch
der Viruskrankheiten., E.H.O.W. E. Gildenmeister, Editor. 1939, Fischer: Jena.
p. S. 254-354.
44. Gosztonyi, G. and H. Ludwig, Borna disease of horses. An immunohistological
and virological study of naturally infected animals. Acta Neuropathol, 1984.
64(3): p. 213-21.
105

8 - Literaturangaben
45. Lebelt, J. and K. Hagenau, [Distribution of Borna disease virus in naturally
infected animals with clinical disease]. Berl Munch Tierarztl Wochenschr, 1996.
109(5): p. 178-83.
46. Morales, J.A., et al., Axonal transport of Borna disease virus along olfactory
pathways in spontaneously and experimentally infected rats. Med Microbiol
Immunol, 1988. 177(2): p. 51-68.
47. Richt, J.A., et al., Borna disease virus infection in animals and humans. Emerg
Infect Dis, 1997. 3(3): p. 343-52.
48. Carbone, K.M., et al., Pathogenesis of Borna disease in rats: evidence that intra-
axonal spread is the major route for virus dissemination and the determinant for
disease incubation. J Virol, 1987. 61(11): p. 3431-40.
49. Morimoto, K., et al., Intrinsic responses to Borna disease virus infection of the
central nervous system. Proc Natl Acad Sci U S A, 1996. 93(23): p. 13345-50.
50. Kacza, J., et al., Neuron-glia interactions in the rat retina infected by Borna
disease virus. Arch Virol, 2000. 145(1): p. 127-47.
51. Solbrig, M.V., J.H. Fallon, and W.I. Lipkin, Behavioral disturbances and
pharmacology of Borna disease. Curr Top Microbiol Immunol, 1995. 190: p. 93-
101.
52. Gosztonyi, G. and H. Ludwig, Borna disease--neuropathology and
pathogenesis. Curr Top Microbiol Immunol, 1995. 190: p. 39-73.
53. Carbone, K.M., et al., Astrocytes and Schwann cells are virus-host cells in the
nervous system of rats with Borna disease. J Neuropathol Exp Neurol, 1989.
48(6): p. 631-44.
54. Carbone, K.M., T.R. Moench, and W.I. Lipkin, Borna disease virus replicates in
astrocytes, Schwann cells and ependymal cells in persistently infected rats:
location of viral genomic and messenger RNAs by in situ hybridization. J
Neuropathol Exp Neurol, 1991. 50(3): p. 205-14.
55. Narayan, O., et al., Pathogenesis of Borna disease in rats: immune-mediated
viral ophthalmoencephalopathy causing blindness and behavioral abnormalities.
J Infect Dis, 1983. 148(2): p. 305-15.
56. Krey, H., H. Ludwig, and R. Rott, Spread of infectious virus along the optic nerve
into the retina in Borna disease virus-infected rabbits. Arch Virol, 1979. 61(4): p.
283-8.
57. Herzog, S., et al., Replication of Borna disease virus in rats: age-dependent
differences in tissue distribution. Med Microbiol Immunol, 1984. 173(4): p. 171-7.
106

8 - Literaturangaben
58. Stitz, L., D. Schilken, and K. Frese, Atypical dissemination of the highly
neurotropic Borna disease virus during persistent infection in cyclosporine A-
treated, immunosuppressed rats. J Virol, 1991. 65(1): p. 457-60.
59. Rubin, S.A., et al., Hematologic consequences of Borna disease virus infection
of rat bone marrow and thymus stromal cells. Blood, 1995. 85(10): p. 2762-9.
60. Sierra-Honigmann, A.M., et al., Borna disease virus in peripheral blood
mononuclear and bone marrow cells of neonatally and chronically infected rats.
J Neuroimmunol, 1993. 45(1-2): p. 31-6.
61. Hirano, N., M. Kao, and H. Ludwig, Persistent, tolerant or subacute infection in
Borna disease virus-infected rats. J Gen Virol, 1983. 64(Pt 7): p. 1521-30.
62. Briese, T., M. Hornig, and W.I. Lipkin, Bornavirus immunopathogenesis in
rodents: models for human neurological diseases. J Neurovirol, 1999. 5(6): p.
604-12.
63. Bilzer, T. and L. Stitz, Immune-mediated brain atrophy. CD8+ T cells contribute
to tissue destruction during borna disease. J Immunol, 1994. 153(2): p. 818-23.
64. Deschl, U., et al., Determination of immune cells and expression of major
histocompatibility complex class II antigen in encephalitic lesions of
experimental Borna disease. Acta Neuropathol, 1990. 81(1): p. 41-50.
65. Zheng, Y.M., et al., Severity of neurological signs and degree of inflammatory
lesions in the brains of rats with Borna disease correlate with the induction of
nitric oxide synthase. J Virol, 1993. 67(10): p. 5786-91.
66. Koprowski, H., et al., In vivo expression of inducible nitric oxide synthase in
experimentally induced neurologic diseases. Proc Natl Acad Sci U S A, 1993.
90(7): p. 3024-7.
67. Dietzschold, B., The role of nitric oxide in the pathogenesis of virus-induced
encephalopathies. Curr Top Microbiol Immunol, 1995. 196: p. 51-6.
68. Hatalski, C.G., W.F. Hickey, and W.I. Lipkin, Evolution of the immune response
in the central nervous system following infection with Borna disease virus. J
Neuroimmunol, 1998. 90(2): p. 137-42.
69. Sobbe, M., et al., Induction of degenerative brain lesions after adoptive transfer
of brain lymphocytes from Borna disease virus-infected rats: presence of CD8+
T cells and perforin mRNA. J Virol, 1997. 71(3): p. 2400-7.
70. Weissenbock, H., et al., Microglial activation and neuronal apoptosis in
Bornavirus infected neonatal Lewis rats. Brain Pathol, 2000. 10(2): p. 260-72.
107

8 - Literaturangaben
71. Hornig, M., et al., An infection-based model of neurodevelopmental damage.
Proc Natl Acad Sci U S A, 1999. 96(21): p. 12102-7.
72. Sauder, C. and J.C. de la Torre, Cytokine expression in the rat central nervous
system following perinatal Borna disease virus infection. J Neuroimmunol, 1999.
96(1): p. 29-45.
73. Joest, E.D., K., Über eigentümliche Kerneinschlüsse der Ganglienzellen bei der
enzootischen Gehirn-Rückenmarksentzündung der Pferde. Z. Inf. krankh.
Haustiere, 1909. 6: p. 348-356.
74. Sasaki, S. and H. Ludwig, In borna disease virus infected rabbit neurons 100 nm
particle structures accumulate at areas of Joest-Degen inclusion bodies.
Zentralbl Veterinarmed [B], 1993. 40(4): p. 291-7.
75. Ludwig, H. and P. Thein, Demonstration of specific antibodies in the central
nervous system of horses naturally infected with Borna disease virus. Med
Microbiol Immunol (Berl), 1977. 163(4): p. 215-26.
76. Narayan, O., et al., Behavioral disease in rats caused by immunopathological
responses to persistent borna virus in the brain. Science, 1983. 220(4604): p.
1401-3.
77. Irigoin, C., et al., Immunocytochemical study of the subcommissural organ of
rats with induced postnatal hydrocephalus. Exp Brain Res, 1990. 82(2): p. 384-
92.
78. Cervos-Navarro, J., et al., [The encephalitic reaction in Borna disease virus
infected rhesus monkeys]. Verh Dtsch Ges Pathol, 1981. 65: p. 208-12.
79. Stitz, L., H. Krey, and H. Ludwig, Borna disease in rhesus monkeys as a models
for uveo-cerebral symptoms. J Med Virol, 1981. 6(4): p. 333-40.
80. Herzog, S., et al., Effect of Borna disease virus infection on athymic rats. J Gen
Virol, 1985. 66(Pt 3): p. 503-8.
81. Stitz, L., et al., Inhibition of immune-mediated meningoencephalitis in
persistently Borna disease virus-infected rats by cyclosporine A. J Immunol,
1989. 143(12): p. 4250-6.
82. Richt, J., et al., Borna disease virus-induced meningoencephalomyelitis caused
by a virus-specific CD4+ T cell-mediated immune reaction. J Gen Virol, 1990.
71(Pt 11): p. 2565-73.
83. Richt, J.A., et al., Borna disease, a progressive meningoencephalomyelitis as a
model for CD4+ T cell-mediated immunopathology in the brain. J Exp Med,
1989. 170(3): p. 1045-50.
108

8 - Literaturangaben
84. Planz, O., T. Bilzer, and L. Stitz, Immunopathogenic role of T-cell subsets in
Borna disease virus-induced progressive encephalitis. J Virol, 1995. 69(2): p.
896-903.
85. Planz, O., et al., Lysis of major histocompatibility complex class I-bearing cells in
Borna disease virus-induced degenerative encephalopathy. J Exp Med, 1993.
178(1): p. 163-74.
86. Stitz, L., M. Sobbe, and T. Bilzer, Preventive effects of early anti-CD4 or anti-
CD8 treatment on Borna disease in rats. J Virol, 1992. 66(6): p. 3316-23.
87. Planz, O., et al., A naturally processed rat major histocompatibility complex
class I-associated viral peptide as target structure of borna disease virus-
specific CD8+ T cells. J Biol Chem, 2001. 276(17): p. 13689-94.
88. Noske, K., et al., Virus-specific CD4+ T cells eliminate borna disease virus from
the brain via induction of cytotoxic CD8+ T cells. J Virol, 1998. 72(5): p. 4387-
95.
89. Ludwig, H., et al., [Borna virus infection (Borna disease) in naturally and
experimentally infected animals: its significance for research and practice].
Tierarztl Prax, 1985. 13(4): p. 421-53.
90. Hatalski, C.G., W.F. Hickey, and W.I. Lipkin, Humoral immunity in the central
nervous system of Lewis rats infected with Borna disease virus. J
Neuroimmunol, 1998. 90(2): p. 128-36.
91. Stitz, L., B. Dietzschold, and K.M. Carbone, Immunopathogenesis of Borna
disease. Curr Top Microbiol Immunol, 1995. 190: p. 75-92.
92. Stitz, L., et al., A functional role for neutralizing antibodies in Borna disease:
influence on virus tropism outside the central nervous system. J Virol, 1998.
72(11): p. 8884-92.
93. Rothwell, N.J. and P.J. Strijbos, Cytokines in neurodegeneration and repair. Int J
Dev Neurosci, 1995. 13(3-4): p. 179-85.
94. Shankar, V., et al., Kinetics of virus spread and changes in levels of several
cytokine mRNAs in the brain after intranasal infection of rats with Borna disease
virus. J Virol, 1992. 66(2): p. 992-8.
95. Solbrig, M.V., et al., Tardive dyskinetic syndrome in rats infected with Borna
disease virus. Neurobiol Dis, 1994. 1(3): p. 111-9.
96. Solbrig, M.V., et al., A neural substrate of hyperactivity in borna disease:
changes in brain dopamine receptors. Virology, 1996. 222(2): p. 332-8.
109

8 - Literaturangaben
97. Solbrig, M.V., et al., Prefrontal cortex dysfunction in Borna disease virus (BDV)--
infected rats. Biol Psychiatry, 1996. 40(7): p. 629-36.
98. Solbrig, M.V., G.F. Koob, and W.I. Lipkin, Cocaine sensitivity in Borna disease
virus-infected rats. Pharmacol Biochem Behav, 1998. 59(4): p. 1047-52.
99. Gies, U., et al., Disturbance of the cortical cholinergic innervation in Borna
disease prior to encephalitis. Brain Pathol, 1998. 8(1): p. 39-48.
100. Lipkin, W.I., et al., Neurotransmitter abnormalities in Borna disease. Brain Res,
1988. 475(2): p. 366-70.
101. Waltrip, R.W., 2nd, et al., Borna disease virus and schizophrenia. Psychiatry
Res, 1995. 56(1): p. 33-44.
102. Briese, T., et al., Enzyme-linked immunosorbent assay for detecting antibodies
to Borna disease virus-specific proteins. J Clin Microbiol, 1995. 33(2): p. 348-51.
103. Vahlenkamp, T.W., H.K. Enbergs, and H. Muller, Experimental and natural
borna disease virus infections: presence of viral RNA in cells of the peripheral
blood. Vet Microbiol, 2000. 76(3): p. 229-44.
104. Pauli, G., J. Grunmach, and H. Ludwig, Focus-immunoassay for Borna disease
virus-specific antigens. Zentralbl Veterinarmed [B], 1984. 31(7): p. 552-7.
105. Lewis, A.J., et al., Effect of immune priming on Borna disease. J Virol, 1999.
73(3): p. 2541-6.
106. Oldach, D., et al., Induction of protection against Borna disease by inoculation
with high-dose-attenuated Borna disease virus. Virology, 1995. 206(1): p. 426-
34.
107. Hallensleben, W. and P. Staeheli, Inhibition of Borna disease virus multiplication
by interferon: cell line differences in susceptibility. Arch Virol, 1999. 144(6): p.
1209-16.
108. von Rheinbaben, F., L. Stitz, and R. Rott, Influence of interferon on persistent
infection caused by Borna disease virus in vitro. J Gen Virol, 1985. 66(Pt 12): p.
2777-80.
109. Gierend, M. and H. Ludwig, Influence of immunosuppressive treatment of Borna
Disease in rabbits. Arch Virol, 1981. 67(3): p. 217-28.
110. Krey, H., H. Ludwig, and M. Gierend, Borna disease virus-induced retinouveitis
treated with immunosuppressive drugs. Albrecht Von Graefes Arch Klin Exp
Ophthalmol, 1981. 216(2): p. 111-9.
110

8 - Literaturangaben
111. Bode, L., et al., Amantadine and human Borna disease virus in vitro and in vivo
in an infected patient with bipolar depression. Lancet, 1997. 349(9046): p. 178-
9.
112. Ferszt, R., et al., Amantadine revisited: an open trial of amantadinesulfate
treatment in chronically depressed patients with Borna disease virus infection.
Pharmacopsychiatry, 1999. 32(4): p. 142-7.
113. Stitz, L., O. Planz, and T. Bilzer, Lack of antiviral effect of amantadine in Borna
disease virus infection. Med Microbiol Immunol (Berl), 1998. 186(4): p. 195-200.
114. Hallensleben, W., M. Zocher, and P. Staeheli, Borna disease virus is not
sensitive to amantadine. Arch Virol, 1997. 142(10): p. 2043-8.
115. Cubitt, B. and J.C. de la Torre, Amantadine does not have antiviral activity
against Borna disease virus. Arch Virol, 1997. 142(10): p. 2035-42.
116. Jordan, I., et al., Inhibition of Borna disease virus replication by ribavirin. J Virol,
1999. 73(9): p. 7903-6.
117. Mizutani, T., et al., Inhibition of Borna disease virus replication by ribavirin in
persistently infected cells. Arch Virol, 1998. 143(10): p. 2039-44.
118. Galasso, Antiviral Agents and Human Viral Diseases. Vol. 4. Edition: Lippincott -
Raven.
119. Hruska, J.F., et al., Effects of ribavirin on respiratory syncytial virus in vitro.
Antimicrob Agents Chemother, 1980. 17(5): p. 770-5.
120. Wyde, P.R., et al., Use of cotton rats to evaluate the efficacy of antivirals in
treatment of measles virus infections. Antimicrob Agents Chemother, 2000.
44(5): p. 1146-52.
121. Taber, L.H., et al., Ribavirin aerosol treatment of bronchiolitis associated with
respiratory syncytial virus infection in infants. Pediatrics, 1983. 72(5): p. 613-8.
122. Hall, C.B., et al., Ribavirin treatment of respiratory syncytial viral infection in
infants with underlying cardiopulmonary disease. Jama, 1985. 254(21): p. 3047-
51.
123. Davis, G.L., et al., Interferon alfa-2b alone or in combination with ribavirin for the
treatment of relapse of chronic hepatitis C. International Hepatitis Interventional
Therapy Group. N Engl J Med, 1998. 339(21): p. 1493-9.
124. McHutchison, J.G., et al., Interferon alfa-2b alone or in combination with ribavirin
as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy
Group. N Engl J Med, 1998. 339(21): p. 1485-92.
111

8 - Literaturangaben
125. Pianko, S. and J.G. McHutchison, Treatment of hepatitis C with interferon and
ribavirin. J Gastroenterol Hepatol, 2000. 15(6): p. 581-6.
126. Fried, M.W., et al., Therapy of chronic hepatitis B with a 6-month course of
ribavirin. J Hepatol, 1994. 21(2): p. 145-50.
127. Galban-Garcia, E., et al., Efficacy of ribavirin in patients with chronic hepatitis B.
J Gastroenterol, 2000. 35(5): p. 347-52.
128. Kakumu, S., et al., Pilot study of ribavirin and interferon-beta for chronic hepatitis
B. Hepatology, 1993. 18(2): p. 258-63.
129. Cotonat, T., et al., Pilot study of combination therapy with ribavirin and interferon
alfa for the retreatment of chronic hepatitis B e antibody-positive patients.
Hepatology, 2000. 31(2): p. 502-6.
130. Jordan, I., et al., Ribavirin inhibits West Nile virus replication and cytopathic
effect in neural cells. J Infect Dis, 2000. 182(4): p. 1214-7.
131. Stephen, E.L., et al., Therapeutic effects of ribavirin given by the intraperitoneal
or aerosol route against influenza virus infections in mice. Antimicrob Agents
Chemother, 1976. 10(3): p. 549-54.
132. Wyde, P.R., et al., Efficacy of high dose-short duration ribavirin aerosol in the
treatment of respiratory syncytial virus infected cotton rats and influenza B virus
infected mice. Antiviral Res, 1987. 7(4): p. 211-20.
133. Knight, V. and B.E. Gilbert, Ribavirin aerosol treatment of influenza. Infect Dis
Clin North Am, 1987. 1(2): p. 441-57.
134. Sidwell, R.W., et al., In vitro and in vivo effect of 1-beta-D-ribofuranosyl-1,2,4-
triazole-3-carboxamide (ribavirin) on types 1 and 3 parainfulenza virus
infections. Chemotherapy, 1975. 21(3-4): p. 205-20.
135. Larson, E.W., E.L. Stephen, and J.S. Walker, Therapeutic effects of small-
particle aerosols of ribavirin on parainfluenza (sendai) virus infections of mice.
Antimicrob Agents Chemother, 1976. 10(4): p. 770-2.
136. Gelfand, E.W., et al., Ribavirin treatment of viral pneumonitis in severe
combined immunodeficiency disease. Lancet, 1983. 2(8352): p. 732-3.
137. Takahashi, T., et al., The cooperative effect of interferon-alpha and ribavirin on
subacute sclerosing panencephalitis (SSPE) virus infections, in vitro and in vivo.
Antiviral Res, 1998. 37(1): p. 29-35.
138. Tomoda, A., et al., Combined treatment with interferon-alpha and ribavirin for
subacute sclerosing panencephalitis. Pediatr Neurol, 2001. 24(1): p. 54-9.
112

8 - Literaturangaben
139. Scheffler, P., D. Haghchenas, and R. Wigand, The effect of purine and
pyrimidine analogues and virazole on adenovirus replication. Acta Virol, 1975.
19(2): p. 106-15.
140. Murphy, G.F., et al., Adenovirus-associated hemorrhagic cystitis treated with
intravenous ribavirin. J Urol, 1993. 149(3): p. 565-6.
141. Miyamura, K., et al., Successful ribavirin therapy for severe adenovirus
hemorrhagic cystitis after allogeneic marrow transplant from close HLA donors
rather than distant donors. Bone Marrow Transplant, 2000. 25(5): p. 545-8.
142. Jurado, M., et al., Adenovirus-associated haemorrhagic cystitis after bone
marrow transplantation successfully treated with intravenous ribavirin. Bone
Marrow Transplant, 1995. 15(4): p. 651-2.
143. Shetty, A.K., et al., Intravenous ribavirin therapy for adenovirus pneumonia.
Pediatr Pulmonol, 2000. 29(1): p. 69-73.
144. Liles, W.C., et al., Severe adenoviral nephritis following bone marrow
transplantation: successful treatment with intravenous ribavirin. Bone Marrow
Transplant, 1993. 12(4): p. 409-12.
145. Jahrling, P.B., et al., Lassa virus infection of rhesus monkeys: pathogenesis and
treatment with ribavirin. J Infect Dis, 1980. 141(5): p. 580-9.
146. WHO. . in WHO International Commission to Zaire 1978.
147. McCormick, J.B., et al., Lassa fever. Effective therapy with ribavirin. N Engl J
Med, 1986. 314(1): p. 20-6.
148. Murphy, M.E., et al., In vitro antiviral activity of lactoferrin and ribavirin upon
hantavirus. Arch Virol, 2000. 145(8): p. 1571-82.
149. Huggins, J.W., et al., Prospective, double-blind, concurrent, placebo-controlled
clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal
syndrome. J Infect Dis, 1991. 164(6): p. 1119-27.
150. Watts, D.M., et al., Inhibition of Crimean-Congo hemorrhagic fever viral
infectivity yields in vitro by ribavirin. Am J Trop Med Hyg, 1989. 41(5): p. 581-5.
151. Tignor, G.H. and C.A. Hanham, Ribavirin efficacy in an in vivo model of
Crimean-Congo hemorrhagic fever virus (CCHF) infection. Antiviral Res, 1993.
22(4): p. 309-25.
152. Fisher-Hoch, S.P., et al., Crimean Congo-haemorrhagic fever treated with oral
ribavirin. Lancet, 1995. 346(8973): p. 472-5.
113

8 - Literaturangaben
153. Kende, M., et al., Enhanced efficacy of liposome-encapsulated ribavirin against
Rift Valley fever virus infection in mice. Antimicrob Agents Chemother, 1985.
27(6): p. 903-7.
154. Canonico, P.G., Efficacy, toxicology and clinical applications of ribavirin against
virulent RNA viral infections. Antiviral Res, 1985. Suppl(1): p. 75-81.
155. Morrison, G.A., B. Kotecha, and J.N. Evans, Ribavirin treatment for juvenile
respiratory papillomatosis. J Laryngol Otol, 1993. 107(5): p. 423-6.
156. McGlennen, R.C., et al., Pilot trial of ribavirin for the treatment of laryngeal
papillomatosis. Head Neck, 1993. 15(6): p. 504-12; discussion 512-3.
157. Richt, J.A., et al., Borna disease virus-specific T cells protect against or cause
immunopathological Borna disease. J Exp Med, 1994. 179(5): p. 1467-73.
158. Hultgren, C., et al., The antiviral compound ribavirin modulates the T helper (Th)
1/Th2 subset balance in hepatitis B and C virus-specific immune responses. J
Gen Virol, 1998. 79(Pt 10): p. 2381-91.
159. Liu, M.F., et al., Resistance of naive mice to murine hepatitis virus strain 3
requires development of a Th1, but not a Th2, response, whereas pre-existing
antibody partially protects against primary infection. Adv Exp Med Biol, 1998.
440: p. 415-23.
160. Ning, Q., et al., Ribavirin inhibits viral-induced macrophage production of TNF,
IL-1, the procoagulant fgl2 prothrombinase and preserves Th1 cytokine
production but inhibits Th2 cytokine response. J Immunol, 1998. 160(7): p.
3487-93.
161. Tam, R.C., et al., Ribavirin polarizes human T cell responses towards a Type 1
cytokine profile. J Hepatol, 1999. 30(3): p. 376-82.
162. Johnson, P., et al., Purification, chain separation and sequence of the MRC OX-
8 antigen, a marker of rat cytotoxic T lymphocytes. Embo J, 1985. 4(10): p.
2539-45.
163. Robinson, A.P., T.M. White, and D.W. Mason, Macrophage heterogeneity in the
rat as delineated by two monoclonal antibodies MRC OX-41 and MRC OX-42,
the latter recognizing complement receptor type 3. Immunology, 1986. 57(2): p.
239-47.
164. White, R.A., et al., T-lymphocyte heterogeneity in the rat: separation of
functional subpopulations using a monoclonal antibody. J Exp Med, 1978.
148(3): p. 664-73.
165. Laboratories, C.R., Charles River Laboratories Online-Katalog. 2001.
114

8 - Literaturangaben
166. Sternberg, E.M., et al., A defect in the central component of the immune system-
-hypothalamic-pituitary-adrenal axis feedback loop is associated with
susceptibility to experimental arthritis and other inflammatory diseases. Ann N Y
Acad Sci, 1990. 594: p. 289-92.
167. Cizza, G. and E.M. Sternberg, The role of the hypothalamic-pituitary-adrenal
axis in susceptibility to autoimmune/inflammatory disease. Immunomethods,
1994. 5(1): p. 73-8.
168. Watson, G.P.C., "The Rat Brain". .
169. Zeman, W., Craigie's Neuroanatomy of the Rat.
170. Hatalski, C.G., Alterations in the immune response within the central nervous
system of rats infected with Borna disease virus: potential mechanisms for viral
persistence, in Department of Microbiology. 1996, University of California Irvine:
Irvine.
171. Elmer, P., Handbuch zu ABI Prism 7700 Sequenze Detector.
172. Laboratories, V., Instruction For Immunohistochemical Staining.
173. Ishii, T., et al., Effective ribavirin concentration in hamster brains for antiviral
chemotherapy for subacute sclerosing panencephalitis. Antimicrob Agents
Chemother, 1996. 40(1): p. 241-3.
174. Davies, B. and T. Morris, Physiological parameters in laboratory animals and
humans. Pharm Res, 1993. 10(7): p. 1093-5.
175. Gilbert, B.E. and V. Knight, Biochemistry and clinical applications of ribavirin.
Antimicrob Agents Chemother, 1986. 30(2): p. 201-5.
176. Sidwell, R.W., et al., Broad-spectrum antiviral activity of Virazole: 1-beta-D-
ribofuranosyl-1,2,4-triazole-3-carboxamide. Science, 1972. 177(50): p. 705-6.
177. Huffman, J.H., et al., In vitro effect of 1-beta-D-ribofuranosyl-1,2,4-triazole-3-
carboxamide (virazole, ICN 1229) on deoxyribonucleic acid and ribonucleic acid
viruses. Antimicrob Agents Chemother, 1973. 3(2): p. 235-41.
178. Streeter, D.G., et al., Mechanism of action of 1- -D-ribofuranosyl-1,2,4-triazole-3-
carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc Natl Acad
Sci U S A, 1973. 70(4): p. 1174-8.
179. Sintchak, M.D. and E. Nimmesgern, The structure of inosine 5'-monophosphate
dehydrogenase and the design of novel inhibitors. Immunopharmacology, 2000.
47(2-3): p. 163-84.
115

8 - Literaturangaben
180. Fouchier, F., et al., The effects of ribavirin on the GTP level and the VIP receptor
dynamic of human IGR39 cells. J Recept Signal Transduct Res, 1996. 16(1-2):
p. 39-58.
181. Wray, S.K., B.E. Gilbert, and V. Knight, Effect of ribavirin triphosphate on primer
generation and elongation during influenza virus transcription in vitro. Antiviral
Res, 1985. 5(1): p. 39-48.
182. Sharma, O.K., B.B. Goswami, and E. Borek, Inhibition of mRNA methylation: an
approach to specific inhibition of viral replication. Princess Takamatsu Symp,
1982. 12: p. 109-16.
183. Goswami, B.B., et al., The broad spectrum antiviral agent ribavirin inhibits
capping of mRNA. Biochem Biophys Res Commun, 1979. 89(3): p. 830-6.
184. Eriksson, B., et al., Inhibition of influenza virus ribonucleic acid polymerase by
ribavirin triphosphate. Antimicrob Agents Chemother, 1977. 11(6): p. 946-51.
185. Toltzis, P., K. O'Connell, and J.L. Patterson, Effect of phosphorylated ribavirin
on vesicular stomatitis virus transcription. Antimicrob Agents Chemother, 1988.
32(4): p. 492-7.
186. Cassidy, L.F. and J.L. Patterson, Mechanism of La Crosse virus inhibition by
ribavirin. Antimicrob Agents Chemother, 1989. 33(11): p. 2009-11.
187. Crotty, S., et al., The broad-spectrum antiviral ribonucleoside ribavirin is an RNA
virus mutagen. Nat Med, 2000. 6(12): p. 1375-9.
188. Crotty, S., C.E. Cameron, and R. Andino, RNA virus error catastrophe: direct
molecular test by using ribavirin. Proc Natl Acad Sci U S A, 2001. 98(12): p.
6895-900.
189. Conzelmann, K.K., Nonsegmented negative-strand RNA viruses: genetics and
manipulation of viral genomes. Annu Rev Genet, 1998. 32: p. 123-62.
190. Galban Garcia, E., et al., [Role of ribavirin in the treatment of chronic hepatitis
B]. Gastroenterol Hepatol, 2000. 23(4): p. 165-9.
191. Camps, J., et al., Ribavirin in the treatment of chronic hepatitis C unresponsive
to alfa interferon. J Hepatol, 1993. 19(3): p. 408-12.
192. Hoofnagle, J.H., et al., Prolonged therapy of chronic hepatitis C with ribavirin. J
Viral Hepat, 1996. 3(5): p. 247-52.
193. Dusheiko, G., et al., Ribavirin treatment for patients with chronic hepatitis C:
results of a placebo-controlled study. J Hepatol, 1996. 25(5): p. 591-8.
194. Bodenheimer, H.C., Jr., et al., Tolerance and efficacy of oral ribavirin treatment
of chronic hepatitis C: a multicenter trial. Hepatology, 1997. 26(2): p. 473-7.
116

8 - Literaturangaben
195. Neumann, H., et al., Major histocompatibility complex (MHC) class I gene
expression in single neurons of the central nervous system: differential
regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J
Exp Med, 1997. 185(2): p. 305-16.
196. Neumann, H., et al., Induction of MHC class I genes in neurons. Science, 1995.
269(5223): p. 549-52.
197. Rensing-Ehl, A., et al., Neurons induced to express major histocompatibility
complex class I antigen are killed via the perforin and not the Fas (APO-1/CD95)
pathway. Eur J Immunol, 1996. 26(9): p. 2271-4.
198. Joksic, G., et al., Influence of ribavirin on the micronucleus formation and in vitro
proliferation of human lymphocytes. Neoplasma, 2000. 47(5): p. 283-7.
199. Shiffman, M.L., S.B. Verbeke, and P.M. Kimball, Alpha interferon combined with
ribavirin potentiates proliferative suppression but not cytokine production in
mitogenically stimulated human lymphocytes. Antiviral Res, 2000. 48(2): p. 91-9.
200. Martin, J., et al., Effects of the ribavirin-interferon alpha combination on cultured
peripheral blood mononuclear cells from chronic hepatitis C patients. Cytokine,
1998. 10(8): p. 635-44.
201. Li, W., F. Shen, and G. Weber, Ribavirin and quercetin synergistically
downregulate signal transduction and are cytotoxic in human ovarian carcinoma
cells. Oncol Res, 1999. 11(5): p. 243-7.
202. Vallee, S., et al., Ribavirin-induced resistance to heat shock, inhibition of the
Ras-Raf-1 pathway and arrest in G(1). Eur J Pharmacol, 2000. 404(1-2): p. 49-
62.
203. Potter, C.W., et al., Antiviral, immunosuppressive and antitumour effects of
ribavirin. Nature, 1976. 259(5543): p. 496-7.
204. Kerr, S.J., Ribavirin induced differentiation of murine erythroleukemia cells. Mol
Cell Biochem, 1987. 77(2): p. 187-94.
205. Tricot, G., et al., Tiazofurin: biological effects and clinical uses. Int J Cell
Cloning, 1990. 8(3): p. 161-70.
206. Kishi, M., et al., Sequence variability of Borna disease virus open reading frame
II found in human peripheral blood mononuclear cells. J Virol, 1996. 70(1): p.
635-40.
207. Kishi, M., et al., Prevalence of Borna disease virus RNA in peripheral blood
mononuclear cells from blood donors. Med Microbiol Immunol (Berl), 1995.
184(3): p. 135-8.
117

8 - Literaturangaben
208. Kishi, M., et al., Demonstration of human Borna disease virus RNA in human
peripheral blood mononuclear cells. FEBS Lett, 1995. 364(3): p. 293-7.
209. Bode, L., et al., Borna disease virus genome transcribed and expressed in
psychiatric patients [see comments]. Nat Med, 1995. 1(3): p. 232-6.
210. Planz, O., et al., Persistence of Borna disease virus-specific nucleic acid in
blood of psychiatric patient. Lancet, 1998. 352(9128): p. 623.
211. Planz, O., et al., Pathogenesis of borna disease virus: granulocyte fractions of
psychiatric patients harbor infectious virus in the absence of antiviral antibodies.
J Virol, 1999. 73(8): p. 6251-6.
212. Seiler, P., et al., Additive effect of neutralizing antibody and antiviral drug
treatment in preventing virus escape and persistence. J Virol, 2000. 74(13): p.
5896-901.
213. Gilbert, B.E., et al., Aerosol and intraperitoneal administration of ribavirin and
ribavirin triacetate: pharmacokinetics and protection of mice against
intracerebral infection with influenza A/WSN virus. Antimicrob Agents
Chemother, 1991. 35(7): p. 1448-53.
214. Gilbert, B.E. and P.R. Wyde, Pharmacokinetics of ribavirin aerosol in mice.
Antimicrob Agents Chemother, 1988. 32(1): p. 117-21.
215. Gonzalez-Scarano, F. and G. Baltuch, Microglia as mediators of inflammatory
and degenerative diseases. Annu Rev Neurosci, 1999. 22: p. 219-40.
118

Danksagung
Ganz besonders bedanken möchte ich mich bei Prof. Dr. V. ter Meulen, der sich bereit
erklärt hat, meine Doktorarbeit zu betreuen und mich während der gesamten
Bearbeitungszeit unterstützt hat. Einen ganz besonderen Dank für die Vermittlung des
Platzes im Labor von Prof. Dr. W. I. Lipkin und die Betreuung bei alle Fragen und
Problemen nicht nur im direkten Zusammenhang mit meiner Doktorarbeit! Große Hilfe
habe ich von ihm auch bei der Planung und Durchführung des gesamten
Auslandsaufenthaltes erhalten.
Ohne die besonders großzügige Gastfreundschaft von Prof. Dr. W. Ian Lipkin wäre der
Aufenthalt an der University of California in Irvine nicht realisierbar gewesen. Hierfür
und für die Überlassung des Themas, sowie die intensive persönliche Betreuung und
freundliche Aufnahme in der Arbeitsgruppe bin ich sehr dankbar. Ebenfalls bedanken
möchte ich mich bei ihm und Dr. Mady Hornig für die liebenswürdige Gastfreundschaft.
Bei Frau Dr. Marylou V. Solbrig möchte ich mich für die herzliche und lehrreiche
Betreuung und Unterstützung während des gesamten Projektes bedanken. Besonders
bei der Arbeit mit den Versuchstieren und der histopathologischen Auswertung habe
ich von ihrer großen Erfahrung besonders profitiert.
Dr. Ingo Jordan hat mir insbesondere während der Einarbeitungszeit im Labor und bei
unzähligen organisatorischen Problemen geholfen. Seine geduldigen und äußerst
einprägsamen Erklärungen haben mir das Verständnis zahlreicher molekular-
biologischer Methoden ermöglicht. Unter der Leitung von Dr. Thomas Briese haben die
Versuche zu Virustitrierung, Taqman-Analysen, ELISA und Westernblot stattgefunden.
Durch seine verständlichen und geduldigen Erklärungen konnte ich hierbei sehr viel
lernen. Außerdem möchte ich mich bei ihm für das Überlassen von Primer, Probes und
Kontrollen für die Taqman-Assays bedanken.
Dr. Nigel Horscroft bin ich für die Unterstützung bei den RNase Protektions Assays
dankbar. Für die freundliche Überlassung der Transkriptionsplasmide bei der in situ
Hybridisierung möchte ich mich bei Dr. Dale Carpenter bedanken.
Dr. Benedikt Weissbrich und Dr. Jörg Schubert möchte ich für die Betreuung bei
meinem ersten Kontakt mit der Virologie während eines Praktikums in der
Virusdiagnostik danken. Nach meiner Rückkehr aus den USA fand ich hier einen Platz,
um das Gelernte weiter zu vertiefen und neue Erfahrungen zu sammeln.

Lebenslauf Name: Robert Schlaberg
Anschrift: Laufergasse 6
D - 97082 Würzburg
Geburtsdatum: 11.09.1975
Schulische Laufbahn: 1982 - 1986 Grund- und Hauptschule Dettingen/Wallhausen
1986 - 1995 Heinrich-Suso-Gymnasium Konstanz, Abitur 1995
1995 - 1996 Zivildienst im Erzbischöflichen Studienheim St. Konrad in Konstanz
Studium: 1996 - 1997 Medizinstudium an der Friedrich-Schiller-Universität Jena
1997 - 1999 Medizinstudium an der Bayrischen-Julius-Maximilians-Universität
Würzburg
09/1998 Physikum
08/1999 1. Staatsexamen
10/1999 – 9/2000 Forschungsaufenthalt bei Prof. W. Ian Lipkin, MD, Emerging
Diseases Laboratory, University of California Irvine (USA)
seit 10/2000 Medizinstudium an der Bayrischen-Julius-Maximilians-Universität
Würzburg
09/2002 2. Staatsexamen
10/2002-09/2003 Praktische Jahr:
10/02-01/03 Innere Medizin, Universitätsspital Zürich (Schweiz)
02/03-05/02 Pathologie, Columbia University, College of
Physicians and Surgeons, New York (USA)
06/03-09/03 Chirurgie, University of Cape Town (Südafrika)
11/2003. 3. Staatsexamen