
Prognostische Bedeutung von KRAS- und BRAF-Mutationen in ... · DALMs Dysplasia associated lesion...
83
Aus der Medizinischen Klinik des Knappschaftskrankenhauses Bochum Langendreer - Universitätsklinik - der Ruhr-Universität Bochum Direktor: Prof. Dr. med. Wolff Schmiegel Prognostische Bedeutung von KRAS- und BRAF-Mutationen in der Erstlinien-Chemotherapie des metastasierten kolorektalen Karzinoms mit Oxaliplatin und Fluoropyrimidinen Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen Medizinischen Fakultät der Ruhr-Universität Bochum vorgelegt von Nina Mareike Bruns aus Dortmund 2012
Transcript of Prognostische Bedeutung von KRAS- und BRAF-Mutationen in ... · DALMs Dysplasia associated lesion...

Aus der
Medizinischen Klinik
des Knappschaftskrankenhauses Bochum Langendreer
- Universitätsklinik -
der Ruhr-Universität Bochum
Direktor: Prof. Dr. med. Wolff Schmiegel
Prognostische Bedeutung von KRAS- und BRAF-Mutationen in der
Erstlinien-Chemotherapie des metastasierten kolorektalen Karzinoms
mit Oxaliplatin und Fluoropyrimidinen
Inaugural-Dissertation
zur
Erlangung des Doktorgrades der Medizin
einer
Hohen Medizinischen Fakultät
der Ruhr-Universität Bochum
vorgelegt von
Nina Mareike Bruns
aus Dortmund
2012

Dekan: Prof. Dr. med. K. Überla
Referent: Prof. Dr. med. A. Reinacher-Schick
Korreferent: PD Dr. med. K.-H. Bauer
Tag der Mündlichen Prüfung: 11.12.2012

Abstract
Bruns, Nina Mareike
Prognostische Bedeutung von KRAS- und BRAF-Mutationen in der Erstlinien-Chemotherapie des
metastasierten kolorektalen Karzinoms mit Oxaliplatin und Fluoropyrimidinen
Problem: Das kolorektale Karzinom (KRK) ist derzeit in Europa die zweithäufigste tumorbedingte
Todesursache. Bei Erstdiagnose weisen etwa 25% der Patienten eine Fernmetastasierung auf. Durch die
Einführung neuer Chemotherapeutika konnte die Prognose des mKRK deutlich verbessert werden. Dabei
kommt der Identifikation von Biomarkern eine entscheidende Bedeutung zu.
Methode: Die hier untersuchten Gewebeproben stammen von Patienten aus einer prospektiv randomisierten
Phase-III-Studie der AIO zur Erstlinientherapie des mKRK mit Oxaliplatin und Fluoropyrimidinen (Oxaliplatin
und Capecitabin (CAPOX) oder Oxaliplatin und infusionales 5-Fluorouracil (5-FU, FUFOX)). Das untersuchte
Teilkollektiv unterschied sich in Bezug auf Alter, Geschlecht und Therapie nicht vom Gesamtkollektiv aller
Studienpatienten. Die DNA wurde aus pseudonymisierten, formalinfixierten, Paraffin-eingebetteten
Tumorgewebeproben (FFPE) extrahiert. Mit Hilfe des QIAmp DNA Mini Kits wurden 7 KRAS-Mutationen in
den Kodons 12 und 13 detektiert. Im BRAF-Gen wurde gezielt die Hotspot-Mutation V600E analysiert. Nur
Tumoren mit Wildtyp-KRAS-Status wurden auf eine BRAF-Mutation hin untersucht. Der Mutationsstatus der
Gene wurde mit dem Remissionsverhalten, progressionsfreien Überleben und Gesamtüberleben korreliert.
Ergebnis: Eine KRAS-Mutation wurde in 75 der 205 (36,6%) verfügbaren Tumoren nachgewiesen. Eine BRAF
V600E-Mutation wurde in 13 von 130 (10,0%) Tumoren mit Wildtyp-KRAS detektiert. 87 der 205 Patienten
(42,4%) wiesen entweder eine KRAS- oder eine BRAF-Mutation auf. Patienten mit KRAS-Mutationen zeigten
eine signifikant geringere Remissionsrate als Patienten mit KRAS-Wildtyp-Status (44,4% vs. 63,0%, p=0,012).
Patienten mit KRAS-Wildtyp zeigten ein tendenziell besseres medianes Gesamtüberleben (18,9 Monate,
95%-KI: 14,7-23,2) als Patienten mit einer KRAS-Mutation (16,1 Monate, 95%-KI: 14,7-17,6). War eine KRAS-
oder BRAF-Mutation vorhanden, fiel das Gesamtüberleben signifikant kürzer aus (15,6 Monate, 95%-KI: 12,4-
18,8; p=0,013). Das mediane PFS war bei Patienten mit KRAS- und BRAF-Wildtyp (7,6 Monate, 95%-KI: 6,5-
8,6) und Patienten mit KRAS- oder BRAF-Mutation (7,5 Monate, 95%-KI: 4,9-10,0) nicht unterschiedlich
(p=0,89). 136 der 205 Patienten erhielten eine Folgetherapie. 80% der Patienten wurden mit einer Irinotecan-
haltigen Zweitlinientherapie behandelt. 30% der Patienten erhielten den Anti-EGFR-Antikörper Cetuximab. Bei
Patienten ohne Folgetherapie zeigte sich bzgl. des Postprogressionsüberlebens kein wesentlicher
Unterschied. Bei den mit Irinotecan behandelten Patienten zeigte sich ein Trend zu einem günstigeren
Postprogressionsüberleben in der KRAS-Wildtyp-Gruppe (p=0,072). Insbesondere KRAS-Wildtyp-Patienten
mit einer Cetuximab-haltigen Folgetherapie profitierten erwartungsgemäß hinsichtlich des Gesamtüberlebens
(p=0,087).
Diskussion: Patienten mit einer KRAS- oder einer BRAF-Mutation weisen unter einer Oxaliplatin- und
Fluoropyrimidin-haltigen Erstlinientherapie eine signifikant geringere Ansprechrate auf als Patienten mit
Wildtyp-Tumoren. Auch das Gesamtüberleben der Patienten mit Mutation ist signifikant kürzer. Beim PFS
zeigt sich hingegen kein Unterschied. Die potenzielle Oxaliplatin-Resistenz bei der Ansprechrate von
mutierten Tumoren ist für die weitere Therapie von KRK von großer Bedeutung. Möglicherweise könnte dies
mit der Expression des Reparaturproteins ERCC-1 zusammenhängen, das eine Platinresistenz bei
verschiedenen Tumoren vermitteln kann. In Bezug auf eine Folgetherapie und das Gesamtüberleben
profitierten Patienten besonders von einer Cetuximab-haltigen Therapie, wenn ein KRAS-Wildtyp vorlag, was
den bekannten prädiktiven Wert einer KRAS-Mutation für eine Anti-EGFR-Therapie bestätigt.

Meiner Familie,
Else und Heinz Adamik
und Dominik

- 1 -
Inhaltsverzeichnis 1. Einleitung .............................................................................................. - 6 -
1.1. Epidemiologie und Risikofaktoren des kolorektalen Karzinoms ........... - 6 - 1.2. Pathologie und Klassifikation ............................................................... - 7 - 1.3. Molekulares Progressionsmodell des kolorektalen Karzinoms ............ - 9 - 1.4. Klinik, Diagnostik und Früherkennung ............................................... - 10 - 1.5. Therapie ............................................................................................ - 10 - 1.6. Etablierte und potenzielle Biomarker beim kolorektalen Karzinom .... - 15 -
1.6.1. Mikrosatelliteninstabilität ............................................................. - 15 - 1.6.2. p53 .............................................................................................. - 16 - 1.6.3. SMAD4 ........................................................................................ - 16 - 1.6.4. Die Rolle der KRAS- und BRAF-Mutationen beim kolorektalen Karzinom ............................................................................................... - 17 - 1.6.4.1. KRAS und Prognose ............................................................. - 19 - 1.6.4.2. BRAF und Prognose ............................................................. - 23 -
2. Zielsetzung ......................................................................................... - 26 - 3. Material und Methoden ................................................................... - 27 -
3.1. Material .............................................................................................. - 27 - 3.2. Methodik ............................................................................................ - 29 - 3.3. Statistik .............................................................................................. - 30 -
4. Ergebnisse .......................................................................................... - 31 - 4.1. Beschreibung des Patientenkollektivs ............................................... - 31 - 4.2. Häufigkeit von KRAS- und BRAF-Mutationen .................................... - 31 - 4.3. Korrelation mit klinischen Variablen ................................................... - 32 - 4.4. Korrelation mit dem Therapieansprechen .......................................... - 32 - 4.5. Korrelation mit dem Überleben .......................................................... - 34 -
4.5.1. Progressionsfreies Überleben ..................................................... - 34 - 4.5.2. Gesamtüberleben........................................................................ - 36 - 4.5.3. Möglicher Einfluss von Folgetherapien ....................................... - 37 -
5. Diskussion .......................................................................................... - 41 - 6. Zusammenfassung .......................................................................... - 54 - 7. Literaturverzeichnis ......................................................................... - 56 - 8. Danksagung 9. Lebenslauf

- 2 -
Verzeichnis der Abkürzungen
AAPC attenuierte adenomatöse Polyposis coli
AIO Arbeitsgemeinschaft Internistische Onkologie
APC Adenomatosis polyposis coli
AS Aminosäure
BMI Body-mass-index
BRAF Rapidly growing fibrosarcoma type B
CAPOX Capecitabin + Oxaliplatin
CEA Carcino-embryonales Antigen
CIMP CpG-Island-Methylatorphänotyp
CIN Chromosomale Instabilität
DALMs Dysplasia associated lesion or mass
DNA Desoxyribonukleinsäure
EGFR Epidermal-Growth-Factor-Receptor
ELK-1 Ets Like gene 1
ERCC-1 Excision repair cross-complementing rodent repair
deficiency, complementation group 1
Et al. Et alii
FAP Familiäre Adenomatosis polyposis Koli
FFPE Formalin-fixierte, Paraffin eingebettete Gewebeproben
FOLFIRI Folinsäure + Fluorouracil + Irinotecan, 14-tägiges Protokoll
FOLFOXIRI Folinsäure + Fluorouracil + Irinotecan + Oxaliplatin
FOLFOX Folinsäure + Fluorouracil + Oxaliplatin, 14-tägiges Protokoll
FOS FBJ murine osteosarcoma viral oncogene homolog
FS Folinsäure
5-FU Fluorouracil
FUFIRI Fluorouracil + Irinotecan + Folinsäure, wöchentliches
Protokoll
FUFOX Fluorouracil + Folinsäure + Oxaliplatin, wöchentliches
Protokoll
HNPCC Hereditäres nicht-polypöses kolorektales Krebssyndrom
(Lynch-Syndrom)
HR Hazard ratio
IFL Irinotecan + Fluorouracil + Leucovorin

- 3 -
ILCT International Lung Cancer Trial
kDa Kilodalton
KI Konfidenzintervall
KRAS Kirsten Rat Sarcoma
KRK Kolorektales Karzinom
LOH Loss of heterozygosity
LV Leukovorin
MAP Mitogen-activated protein
MEK MAP-Kinase-Kinase
MIN Mikrosatelliteninstabilität
MSI Mikrosatelliteninstabilität
N Numerus/Anzahl
NSCLC non-small cell lung carcinoma
N-Kategorie Lymphknoten-Kategorie
M Männlich
Meta Metachrone Metastasierung
mKRK Metastasiertes kolorektales Karzinom
MMR-System Mismatch repair system
M-Kategorie Metastasenkategorie
Mut Mutation
OS Overall Survival
P Probability-value
P53 Protein 53
PFS Progressionsfreies Überleben
SMAD Sma and Mad related protein
S-Phase Synthesephase
Syn Synchrone Metastasierung
T-Kategorie Tumor-Kategorie
ÜLR Überlebensrate
UICC Union internationale contre le cancer
VEGF Vascular Endothelial Growth Factor
Vs. Versus
W Weiblich
WT Wildtyp

- 4 -
Verzeichnis der Tabellen
Tab. 1 Stadieneinteilung nach UICC modifiziert nach Wittekind
et al., 2003. Prognose: O’Connell, J Natl Cancer Inst. 2004………- 8 -
Tab. 2 Geräte und Materialien ………………………………………………- 28 -
Tab. 3 Korrelation mit klinischen Variablen ………………………………..- 32 -
Tab. 4 Darstellung des Therapieansprechens …………………………….- 33 -
Tab. 5 Korrelation zwischen Remissionsverhalten und KRAS- und
BRAF-Mutation ……………………………………………………….- 34 -

- 5 -
Verzeichnis der Abbildungen
Abb. 1 Progressionsfreies Überleben nach KRAS-Mutations-Status …...- 35 -
Abb. 2 Progressionsfreies Überleben nach KRAS-/BRAF-Status ………- 35 -
Abb. 3 Gesamtüberleben nach KRAS-Mutationsstatus ………………….- 36 -
Abb. 4 Gesamtüberleben nach KRAS-/BRAF-Mutationsstatus …………- 37 -
Abb. 5 Postprogressionsüberleben nach KRAS-Mutationsstatus
bei Patienten, die keine Zweitlinientherapie
erhielten (n=82) ……………………………………………………….- 38 -
Abb. 6 Postprogressionsüberleben nach KRAS-Mutationsstatus
bei Patienten, die eine Irinotecan-haltige Zweitlinientherapie
erhielten (n=116) ……………………………………………………..- 39 -
Abb. 7 Postprogressionsüberleben nach KRAS-Mutationsstatus
bei Patienten, die eine Cetuximab-haltige Folgetherapie
erhielten (n=41) ……………………………………………………….- 40 -
Abb. 8 Postprogressionsüberleben nach KRAS-Mutationsstatus
bei Patienten, die eine Irinotecan-haltige Zweitlinientherapie
ohne eine Cetuximab-Gabe erhielten (n=75) ……………………..- 40 -

- 6 -
1. Einleitung
1.1. Epidemiologie und Risikofaktoren des kolorektalen Karzinoms
Das kolorektale Karzinom (KRK) ist der häufigste maligne Tumor des
Gastrointestinaltraktes. Mit mehr als 73000 Neuerkrankungen pro Jahr in
Deutschland gilt es bei Männern nach dem Prostata- und bei Frauen nach dem
Mammakarzinom als das am zweithäufigsten diagnostizierte Karzinom
(www.rki.de, 30.09.2008). Das KRK ist bei beiden Geschlechtern die
zweithäufigste krebsbedingte Todesursache. Insgesamt versterben pro Jahr in
Deutschland circa 28000 Patienten an den Folgen eines KRKs. Die relative 5-
Jahres-Überlebensrate liegt für Männer bei 52% und für Frauen bei 55% (Krebs in
Deutschland, 2008). Das individuelle Lebenszeitrisiko für die Erkrankung an einem
KRK liegt in Deutschland bei 4-6% (Nelson et al., 1999). Das mittlere
Erkrankungsalter beträgt für Männer 69 Jahre und für Frauen 79 Jahre. Nach dem
50. Lebensjahr ist eine Verdopplung der Neuerkrankungen und Mortalität pro
Lebensdekade zu beobachten (Nelson et al., 1999). Ein deutlich erhöhtes
Erkrankungsrisiko besteht bei der familiären Adenomatosis polyposis Koli (FAP)
und dem hereditären nicht-polypösen kolorektalen Krebssyndrom (HNPCC;
Lynch-Syndrom).
Circa 90-95% der KRK sind sporadischen Ursprungs und entstehen vermutlich
multifaktoriell. Entscheidend bei den Risikofaktoren ist insbesondere ein
„westlicher“ Lebensstil (Fuchs et al., 1999; Almendingen et al, 2001; Bingham et
al., 2003; Kim JH et al, 2007; Zandonai et al., 2012). Das Risiko für ein KRK ist
ebenso durch regelmäßigen Alkohol- und Nikotinkonsum erhöht (Takeshita et al.,
2000; Almendingen et al., 2001; Pedersen et al., 2003; Tiemersma et al., 2003).
Des Weiteren zählen Bewegungsmangel und Übergewicht (BMI über 25 kg/m²) zu
den Risikofaktoren (Giacosa et al., 1999; Friedenreich et al., 2002; Giovannucci et
al., 2003; Wei et al., 2004).
Verwandte ersten Grades von KRK-Patienten über bzw. unter 50 Jahren gehören
ebenfalls einer Risikogruppe an, da sie mit einem Karzinomrisiko von 10% bzw.
30% überdurchschnittlich häufig erkranken (Fuchs et al., 1994).
Zudem sind Patienten mit einer Colitis ulcerosa gefährdet, an einem KRK zu
erkranken. Das Risiko beträgt 18% nach 30 Jahren Erkrankungsdauer (Eaden,
2004).

- 7 -
Ebenfalls ist bei einem Morbus Crohn von einem erhöhten Risiko auszugehen.
Allerdings ist die Datenlage noch spärlich und die Meinungen schwanken
zwischen keinem erhöhten Risiko und einem erhöhten Risiko für ein KRK von 3,5-
bis 7-fach (Greenstein et al., 1981; Persson et al., 1994; Rubio et al., 2008).
1.2. Pathologie und Klassifikation
Als KRK werden alle malignen epithelialen Primärtumoren des Kolons und des
Rektums bezeichnet. Fast ausschließlich handelt es sich um eine drüsige
Differenzierung im Sinne eines Adeno-Karzinoms. Nichtepitheliale Tumoren im
Kolon sind selten. Ungefähr die Hälfte aller Fälle hat ihren Ursprung im Rektum
und circa 25% der Fälle treten im Sigma auf. Die übrigen Karzinome entstehen
annähernd ähnlichen Anteils im Caecum, Colon ascendens, Transversum und
Descendens. Zudem können Mehrfachkarzinome auftreten, wobei metachrone
von synchronen Karzinomen unterschieden werden. Unter einer metachronen
Metastasierung versteht man Metastasen, die später als 4 Monate nach der
Diagnosestellung des Primärtumors auftreten.
In etwa 70% liegt ein Adenokarzinom intestinalen Typs vor, wobei verschiedene
Differenzierungsgrade (G1-G3) unterschieden werden. In etwa 20% der
Adenokarzinome liegt ein muzinöses Karzinom und in weniger als 10% liegen
seltenere histologische Varianten vor.
Basis der Entstehung eines KRKs ist in weit über 90% der Fälle die sog. Adenom-
Karzinom-Sequenz mit der sukzessiven Akkumulation genetischer Abberationen.
Aufgrund einer mehrstufigen Sequenz genetischer Ereignisse, bei der
Tumorsuppressorgene inaktiviert und Onkogene aktiviert werden, entsteht aus
einem Mikroadenom über ein makroskopisch erkennbares Adenom das Karzinom.
Ein erhöhtes Entartungsrisiko weisen Adenome größer 1 cm, villöse Adenome und
Adenome mit höhergradigen Epitheldysplasien auf. Aus diesem Grund werden
diese Polypen als „advanced adenoma“ bezeichnet. Allgemein wird akzeptiert,
dass die Tumorprogression entlang der Adenom-Karzinom-Sequenz mehr als 10
Jahre dauert.
Weitere präkanzeröse Vorläuferläsionen stellen flache Adenome („flat adenomas“)
(Soetikno et al., 2008) und flach-erhabene und flache Epitheldysplasien bei Kolitis-

- 8 -
assoziierten Karzinomen dar, die aber mittels moderner Endoskopietechniken
zunehmend besser erkennbar werden.
Die Prognose von Patienten mit KRK ist wesentlich abhängig von der Eindringtiefe
des Tumors in die Darmwand (T-Kategorie), dem Befall regionaler Lymphknoten
(N-Kategorie) und dem Vorhandensein von Fernmetastasen (M-Kategorie).
Die klinische Einteilung und die Beschreibung der anatomischen
Tumorausbreitung (Staging) erfolgt nach der TNM-Klassifikation (Wittekind et al.,
2003) und der daraus abgeleiteten Stadieneinteilung der UICC. In der unten
aufgeführten Tabelle 1 sieht man die TNM-Klassifikation der 6. Auflage, wie sie
bisher verwendet wurde. Aktuell wurde die 7. Auflage der TNM-Klassifikation
eingeführt, in welcher einige Änderungen vorgenommen wurden. Überlebensraten
sind bei der neuen Klassifikation allerdings noch nicht vorhanden (www.uicc.org).
Tabelle 1: Stadieneinteilung nach UICC modifiziert nach Wittekind et al., 2003.
Prognose: O’Connell, J Natl Cancer Inst. 2004
UICC-Stadium TNM-System Anteil 5-Jahres-Überlebensrate
UICC 0 Tis, N0, M0
UICC I T1-2, N0, M0 15% 80-100%
UICC II A T3, N0, M0 20-30%
85%
UICC II B T4, N0, M0 72%
UICC III A T1-2, N1, M0
30-40%
83%
UICC III B T3-4, N1, M0 64%
UICC III C jedes T, N2, M0 44%
UICC IV jedes T, jedes N, M1 20-25% 5-7%; bei resektablen Leber-/ Lungenmetastasen 25-40%
Bereits bei Diagnosestellung finden sich bei ungefähr 25% der Patienten
Metastasen, die insbesondere in der Leber lokalisiert sind. Bei Lebermetastasen
liegt in 80-90% eine nicht-resektable Situation vor. 50% aller Patienten mit einem
kurativ resezierten KRK der Stadien I bis III entwickeln im Laufe ihrer Erkrankung
Metastasen (Geoghegan, 1999). Bei Patienten eines resezierten Karzinoms mit
dem Stadium II treten in 15-30% der Fälle Metastasen auf und bei einem Stadium
III-Karzinom entwickeln sich in 16-60% Metastasen. Bei dem UICC-Stadium I sind
Metastasen in der Folge selten.

- 9 -
1.3. Molekulares Progressionsmodell des kolorektalen Karzinoms
Die Basis der Entstehung des KRKs ist entlang der sog. molekularen Adenom-
Karzinom-Sequenz, auch genannt das Progressions-Modell. Hiernach entsteht die
Neoplasie nicht aufgrund einer einzelnen genetischen Veränderung, sondern
resultiert aus einer sukzessiven Akkumulation unterschiedlicher Alterationen
verschiedener Tumorsuppressor- bzw. Onkogene (Kinzler und Vogelstein, 1998).
Der Karzinomentstehung liegt eine genetische Instabilität zugrunde.
Unterschieden werden heute hierbei die chromosomale Instabilität (CIN) und die
Mikrosatelliteninstabilität (MIN). Bei 85% aller sporadischen KRK lässt sich ein
CIN-Phänotyp nachweisen. Dies bedeutet, dass chromosomale Verluste oder ein
Zugewinn genetischen Materials vorliegen. Am Anfang der Kaskade der
genetischen Alterationen beim KRK beim CIN-Typ steht die biallelische
Inaktivierung des APC-Gens. Durch diese Inaktivierung werden über verschiedene
Zwischenstationen die Zellproliferation, Zellzykluskontrolle und Apoptoseinhibition
beeinflusst (Heppner und Groden, 2000). Nachfolgend sind aktivierende
Mutationen des KRAS-Onkogens und Alterationen auf dem Chromosom 18, auf
welchem sich insbesondere die Gene SMAD4, SMAD2 und DCC befinden,
nachweisbar. Für die Progression zu einem Karzinom sind letztendlich Mutationen
im Gen des Tumorsuppressorgens p53 auf dem Chromosom 17p verantwortlich.
In 15% der KRK findet man hingegen keinen CIN-Phänotyp, sondern eine
Mikrosatelliteninstabilität (MIN/MSI) (Aaltonen et al., 1993).
Neben der chromosomalen Instabilität und der Mikrosatelliteninstabilität kann als
weiterer Phänotyp der CpG-Island-Methylatorphänotyp (CIMP) auftreten. Dieser
zeigt eine epigenetische Inaktivierung von Tumorsuppressorgenen, welche bei
familiären Karzinomsyndromen typischerweise Keimbahnmutationen aufweisen
wie Rb, BRCA, VHL oder p16. Häufig gehen CIMP-positive KRK mit einer MSI
einher, was auf eine Promotormethylierung des Mismatch-Reparatur-Gens MLH1
zurückzuführen ist (Issa, 2008).
Die Entstehung einer Mikrosatelliteninstabilität beruht auf einem Defekt im MMR-
System. Dieser geht entweder aus einer somatischen/ hereditären Mutation in
einem MMR-Gen oder einer Hypermethylierung des MLH1-Promotors hervor
(Grandy, 2004). Ein Expressionsverlust des MLH1-Gens liegt in 70% aller
sporadischen KRK vor (Cunningham et al., 1998). Bei HNPCC-Fällen liegt mit
einer Häufigkeit von 90% eine Mikrosatelliteninstabilität vor, somit kann bei einer

- 10 -
unauffälligen Mikrosatellitenanalyse eine HNPCC weitestgehend ausgeschlossen
werden.
1.4. Klinik, Diagnostik und Früherkennung
Aufgrund der unspezifischen Symptome und seines oft langen klinisch stummen
Vorhandenseins bleibt das KRK häufig zunächst unbemerkt. Da es keine
zuverlässigen Frühsymptome gibt, wird eine Krebsfrüherkennung empfohlen.
Diese ist seit 2002 in das gesetzliche Vorsorgeprogramm aufgenommen. Als
Warnzeichen gelten Stuhlunregelmäßigkeiten, Ileus und Blut im Stuhl. Jede
plötzliche Änderung der Stuhlgewohnheit im Alter von über 40 Jahren,
beispielsweise eine veränderte Stuhlbeschaffenheit, Wechsel von Obstipation und
Diarrhö, Ileuserscheinungen oder Blutbeimischung zum Stuhl sowie eine
ungeklärte Gewichtsabnahme, Anämie und Schmerzsymptome können einen
Hinweis auf ein KRK darstellen.
Im Rahmen der Krebsfrüherkennung wird ab dem 50. Lebensjahr für Nicht-
Risikopersonen ein fäkaler Okkultblut-Test (FOBT/ Guaiak-Test) mit einer digital-
rektalen Untersuchung empfohlen. Zudem sollte ab dem 55. Lebensjahr eine
Koloskopie durchgeführt werden, die bei unauffälligem Befund und fehlenden
Risikofaktoren im Abstand von zehn Jahren wiederholt wird. Im Vergleich zu allen
anderen Maßnahmen der Früherkennung besitzt die Koloskopie die höchste
Sensitivität und Spezifität und wird somit als Goldstandard angesehen. Studien
zeigten, dass durch eine Polypektomie bei einer Koloskopie die Inzidenz von
kolorektalen Karzinomen um 66-90% gesenkt wird (Winawer et al., 1993; Citarda
et al., 2001). Bei unklarer Symptomatik mit Tumorverdacht ist eine Koloskopie
unerlässlich.
1.5. Therapie
Die Therapie beim KRK erfolgt nach histologischer Sicherung stadienabhängig.
Anzustreben ist grundsätzlich die kurative Resektion. Das Standardverfahren in
der Chirurgie ist die elektive radikale En-bloc-Resektion des befallenen
Darmabschnittes mit systematischer Entfernung des Lymphabflussgebietes.

- 11 -
Perikolische Lymphknotenmetastasen treten in der Regel maximal circa zehn
Zentimeter entfernt vom Tumorrand auf, die Lymphknotendissektion sollte bis zum
zentralen Gefäßabgang erfolgen. Eine adjuvante Chemotherapie ist bei Patienten
mit einem Tumor im UICC-Stadium I nicht indiziert. Im Stadium II kann eine
adjuvante 5-FU-basierte Chemotherapie nach Beratung und Aufklärung des
Patienten empfohlen werden („Kann“-Empfehlung) (Schmiegel et al., 2008), da
durch Studien ein Überlebensvorteil bei dieser Therapie von 3-5% belegt wurde
(Quasar Collaborative G., 2007).
Bei Hochrisikopatienten (T4, Tumor-Perforation, Ileus, <10 Lymphknoten) sollte
eine adjuvante Chemotherapie erwogen werden (Schmiegel et al., 2008).
Im UICC-Stadium III soll eine adjuvante Chemotherapie durchgeführt werden
(„Soll“-Empfehlung) (Schmiegel et al., 2008). Empfohlen wird derzeit das
FOLFOX-Schema (LV5FU2+Oxaliplatin) aufgrund einer Studie, welche eine
signifikante Überlegenheit für die FOLFOX-Chemotherapie gegenüber einer 5-FU-
Chemotherapie zeigte (Andre et al., 2004; DeGramont et al., 2007; Andre et al.,
2009). Besteht eine Unverträglichkeit gegenüber Oxaliplatin oder ist der
Allgemeinzustand reduziert, soll eine Monotherapie mit oralen Fluoropyrimidinen
durchgeführt werden. Neoadjuvante Therapieverfahren spielen in erster Linie bei
Rektumkarzinomen eine Rolle. Insbesondere bei den UICC-Stadien II und III soll
eine neoadjuvante Radio- oder Radiochemotherapie durchgeführt werden. In
Studien konnte gezeigt werden, dass eine präoperative Radiotherapie eine
bessere Wirksamkeit erzielt als eine postoperative Therapie (Sauer et al., 2004).
In Bezug auf das therapeutische Vorgehen werden die Patienten mit einem
metastasierten KRK nach der klinischen Situation und dem Therapieziel in drei
klinische Gruppen eingeteilt. Zu der Gruppe 1 gehören die Patienten mit primär
resektablen Leber- und/oder Lungenmetastasen. Bei Patienten der zweiten
Gruppe wird eine intensivierte systemische Therapie angestrebt. Zu dieser Gruppe
gehören operable Patienten mit potenziell resektablen Metastasen nach einer
Konversionstherapie sowie Patienten mit tumorbedingten Symptomen, raschem
Progress oder Organkomplikationen. Bei der Gruppe 3 kann eine weniger
intensive Therapie appliziert werden, welche bei Patienten mit multiplen
Metastasen ohne Option für Resektion nach Metastasenrückbildung, ohne
tumorbezogene Symptome oder Organkomplikation und/oder schwerer
Komorbidität angewandt wird. Auch bei Patienten über 75 Jahren kann mit einer
Monotherapie begonnen werden.

- 12 -
Eine Resektion von Metastasen (Gruppe 1) ist nur bei 10-20% aller Patienten
möglich (Adson, 1987; Goldberg et al., 1998). Durch die erfolgreiche Entfernung
von Lebermetastasen ist eine 5-Jahres-Überlebensrate von 25-40% zu erreichen
(Nagorney, 1987). Die Datenlage für eine perioperative Therapie bei resektablen
Lebermetastasen ist nicht eindeutig. Vermutlich verbessert die perioperative
Therapie mit FOLFOX das rezidivfreie Überleben. Eine Verbesserung des
Gesamtüberlebens konnte bislang nicht gezeigt werden (Wieser et al., 2010). Bei
Patienten der zweiten Gruppe wird eine intensivierte systemische Therapie
angestrebt. Bei Leber- und/oder Lungenmetastasen und im Einzelfall auch bei
anderen Lokalisationen ist daher bei Diagnose und im Verlauf zu überprüfen, ob
eine Resektion technisch möglich ist. Bei primärer Irresektabilität der Metastasen
soll mit einer neoadjuvanten systemischen Chemotherapie begonnen werden, um
ggf. eine sekundäre Metastasenresektion zu erreichen (Konversionstherapie).
Derzeit stehen mehrere Erstlinientherapien mit hohen Remissionsraten zur
palliativen Behandlung des metastasierten KRKs zur Verfügung. Dabei ist das
orale Capecitabine dem intravenösen 5-FU/Folinsäure gleichzusetzen. Die
Kombinationen von 5-FU/FS mit Oxaliplatin als auch mit Irinotecan sind einer
Monotherapie mit 5-FU überlegen. (De Gramont et al., 2000; Douillard et al., 2000;
Seymour et al., 2007). Oxaliplatin ist ein Dritt-Generations-Platinanalogon,
welches durch Derivate, die bei der Biotransformation von Oxaliplatin entstehen,
zu Inter- und Intrastrang-Quervernetzungen in der DNA führt. Die Folge ist ein
Abbruch der DNA-Synthese und somit eine antitumorale Wirkung. Irinotecan ist
ein Camptothecinderivat, welches die Topoisomerase 1 hemmt. Dadurch kommt
es zu einem Abbruch der DNA-Replikation und damit letztendlich zur Apoptose.
Somit ist Irinotecan pharmakologisch ein Zytostatikum. Durch die synergistische
Wirkung von 5-FU/FS und Oxaliplatin beispielsweise werden Remissionsraten von
bis zu 50% erreicht (Giacchetti et al., 2000).
Somit sollte aufgrund der höheren Ansprechrate bei der Gruppe 2 eine
Kombinationstherapie (2-fach Chemotherapie + monoklonaler Antikörper oder 3-
fach Chemotherapie (FOLFOXIRI)) gewählt werden. Als monokolonale Antikörper
können beispielsweise Cetuximab oder Bevacizumab zum Einsatz kommen.
Cetuximab bindet an den EGF-Rezeptor, welcher auf den Tumorzellen vermehrt
exprimiert wird. Cetuximab führt in einer Kombinationstherapie vor allem zu einer
Erhöhung der Remissionsraten, während das progressionsfreie Überleben nur
moderat verbessert wird (van Cutsem et al., 2009; Bokemeyer et al., 2009).

- 13 -
Cetuximab scheint ferner das Gesamtüberleben zu verbessern (van Cutsem,
2010). Bei Patienten der Gruppe 3 mit Metastasen ohne Aussicht auf Rückbildung
kann als Erstlinientherapie auch eine Monotherapie eingesetzt werden. Bei
Progress unter einer Monotherapie sollte eine weitere Substanz hinzu genommen
werden. Bei einem Großteil der Patienten wird im Verlauf der Erkrankung
Irinotecan oder Oxaliplatin eingesetzt. Ob eine Irinotecan- oder Oxaliplatin-
Kombination gewählt wird, sollte anhand des Toxizitätsspektrums festgemacht
werden. Bei den Irinotecan-Kombinationen sollten insbesondere Diarrhön
Beachtung finden und bei der Oxaliplatin-Kombination muss an die kumulative
Neurotoxizität (PNP) gedacht werden. Vergleicht man die 5-FU-Protokolle, ist ein
Vorteil bei den 14-tägigen FOLFIRI- und FOLFOX-Therapien gegenüber den
wöchentlichen FUFIRI- und FUFOX-Protokollen erkennbar.
Ebenfalls Anwendung in der Erstlinientherapie finden antiangiogenetische
Substanzen. Bei Bevacizumab handelt es sich um einen monoklonalen Antikörper
gegen den Vascular Endothelial Growth Factor (VEGF). In Studien konnte gezeigt
werden, dass Bevacizumab in Kombination mit einer Chemotherapie das
progressionsfreie Überleben und zum Teil auch das Gesamtüberleben der
Patienten verbessern kann (Hurwitz et al., 2003; Kabbinavar et al., 2005; Saltz et
al., 2008). Die Ansprechrate wird weniger deutlich verbessert (Hurwitz et al., 2003;
Saltz et al., 2008). Bevacizumab kann sowohl in Kombination mit Oxaliplatin als
auch in Irinotecan-haltigen Protokollen eingesetzt werden.
Ebenfalls für die Erstlinien-Chemotherapie zugelassen ist die Integration von
Cetuximab in eine Kombinationschemotherapie. In der so genannten CRYSTAL-
Studie wurde der Effekt der Zugabe von Cetuximab zum FOLFIRI-Protokoll
untersucht. Es wurden 599 Patienten mit einem mKRK mit dem FOLFIRI-Protokoll
behandelt und weitere 599 Patienten erhielten FOLFIRI plus Cetuximab (van
Cutsem et al., 2009). Es zeigte sich bei der alleinigen Gabe von FOLFIRI ein
signifikant kürzeres PFS (8,0 vs. 8,9 Monate, p=0,048). Zu der EGFR-Antikörper-
Therapie liegt bereits eine randomisierte Phase III Studie vor. Es wurde gezeigt,
dass bei einer Vortherapie mit Fluoropyrimidinen, Irinotecan oder Oxaliplatin oder
einer Kontrainidikation gegen diese Medikamente bei Patienten mit
immunhistochemisch nachweisbarem EGFR durch Cetuximab eine Verbesserung
des Gesamt- oder PFS erreicht werden kann (Jonker et al., 2007).

- 14 -
Bei Patienten mit Wildtyp-KRAS zeigte sich ein verlängertes PFS (9,9 vs. 8,7
Monate; HR=0,68; 95%-Konfidenzintervall (KI): 0,50-0,94) und ein verlängertes
Gesamtüberleben (24,9 vs. 21,0 Monate, HR=0,84; 95%-KI: 0,64-1,11) bei der
Zugabe von Cetuximab im Vergleich zu Patienten mit KRAS-Mutationen (PFS:
FOLFIRI+Cetuximab vs. FOLFIRI=7,6 vs. 8,1 Monate; p=0,07; OS: FOLFIRI
+Cetuximab vs. FOLFIRI=17,5 vs. 17,7 Monate; p=0,44).
In der sogenannten OPUS-Studie wurde der Effekt von Cetuximab in Kombination
mit einer FOLFOX-Therapie betrachtet (FOLFOX, n=168 vs. FOLFOX+Cetuximab,
n=169) (Bokemeyer et al., 2009). Es zeigte sich bei der Zugabe von Cetuximab
zur FOLFOX-Therapie eine Verbesserung der Ansprechrate (61% vs. 37%,
p=0.011) und des PFS (7,7 Monate vs. 7,2 Monate, HR=0,57; p=0,163) bei KRAS-
Wildtyp-Tumoren. Bei den Karzinomen mit einer KRAS-Mutation hingegen zeigte
sich im Therapiearm mit Cetuximab+FOLFOX ein tendenziell schlechteres
Ansprechen (33% vs. 49%, p=0,106) und ein verringertes PFS (5,5 vs. 8,6
Monate; HR=1,830; p=0,192). Wie aus den beiden Studien ersichtlich wird, kommt
es durch den monoklonalen Antikörper Cetuximab nur bei KRAS-Wildtyp-Tumoren
zu einem gewünschten positiven Effekt. Aus diesem Grund ist die Zulassung
beschränkt auf Patienten mit einem KRAS-Wildtyp-Status des Tumorgewebes.
Hingegen konnte die Verbesserung des Überlebens durch Cetuximab bei
Patienten mit Wildtyp-KRAS exprimierendem Tumor sowohl in der COIN-Studie
(Maughan et al., 2010) als auch in der Nordic-Studie (Tveit et al., 2011) nicht
bestätigt werden. Eine Ursache dafür können verschiedene
Chemotherapiekombinationen sein. Cetuximab scheint mit infusionalem 5-FU und
mit Irinotecan besser synergistisch zu wirken als mit oralem 5-FU und/oder
Oxaliplatin (Maughan et al., 2010; Tveit et al., 2011).
Zudem wurde die Addition von Cetuximab zu einer Kombination aus Capecitabine,
Oxaliplatin und Bevacizumab als Erstlinientherapie beim KRK untersucht. Die
Kombination beider Antikörper hat in der CAIRO2-Studie
(CAPOX/Bevacizumab±Cetuximab) zu einer signifikanten Verschlechterung des
PFS geführt (Punt et al., 2008), so dass die Kombination zweier monoklonaler
Antikörper beim KRK derzeit nicht zu empfehlen ist.
Bei Progress unter einer Erstlinientherapie sollte zu einem alternativen
Therapieprotokoll gewechselt werden. Für die Auswahl der Zweitlinientherapie
sind die chemotherapeutische Vorbehandlung, die therapiefreie Zeit, das

- 15 -
Therapieziel sowie die individuelle Patientensituation entscheidend. Grundsätzlich
stehen aufgrund der zunehmenden Vielfalt an Medikamenten mehrere Optionen
zur Auswahl. Die Therapie sollte bis zum Progress der Erkrankung durchgeführt
werden. Infolge einer Erstlinientherapie gemäß dem Irinotecan-haltigen Protokoll
kann im Anschluss mit einer Oxaliplatin-haltigen Therapie behandelt werden und
umgekehrt (Wong et al., 1999). Durch dieses Verfahren kann in 30-70% eine
Wachstumskontrolle erreicht werden (Grothey et al., 2004; Tournigand et al.,
2004). Auf eine 5-FU-Monotherapie sollte eine Irinotecan-Monotherapie oder die
FOLFOX- Therapie folgen (Cunningham et al., 1998; Rothenberg et al., 2003). Die
Zweitlinientherapie mit Irinotecan nach Versagen einer Fluorouracil-Monotherapie
erbrachte im Vergleich mit BSC oder 5FU/FS einen deutlichen Vorteil im
Gesamtüberleben (Cunningham et al., 1998; Rougier et al., 1998).
Ebenfalls von den Vorläufertherapien bestimmt werden die Drittlinientherapien.
1.6. Etablierte und potenzielle Biomarker beim kolorektalen Karzinom
1.6.1. Mikrosatelliteninstabilität
Wie bereits erwähnt, kann in 15% der KRK eine Mikrosatelliteninstabilität
aufgefunden werden, zu welcher es aufgrund einer Schädigung des zellulären
DNA-Mismatch-Reparatur-Systems gekommen ist. In Bezug auf die Prognose bei
Vorliegen einer Mikrosatelliteninstabilität ist die Datenlage divergent. Einige
zunächst publizierte Studien konnten zeigen, dass bei Patienten mit einer MSI im
Stadium II und III die Prognose nach einer 5-FU-basierten Chemotherapie besser
war als bei mikrosatellitenstabilen Tumoren (Hemminiki et al., 2000; Watanabe et
al., 2001). In anderen Studien jedoch wiesen Patienten mit MSI-positiven Tumoren
keinen Benefit von einer 5-FU-basierten Chemotherapie auf (Carethers et al.,
2004). So untersuchten auch Ribic et al. den Benefit einer 5-FU-basierten
adjuvanten Chemotherapie bei Patienten mit einem KRK im Stadium II oder III.
Insgesamt wurden 570 Patienten in die Studie eingeschlossen, von denen 287
Patienten keine adjuvante Therapie erhielten. Patienten mit einer hochfrequenten
MSI zeigten eine bessere 5-Jahres-Überlebensrate als Patienten mit einer
niedrigfrequenten MSI bzw. ohne MSI (hazard ratio for death 0,31; 95%-KI: 0,14-
0,72; p=0,004). In Bezug auf eine adjuvante 5-FU-basierte Chemotherapie wurde
das Gesamtüberleben bei Patienten mit einer niedrigfrequenten MSI bzw. ohne

- 16 -
eine MSI verlängert (hazard ratio for death 0,72; 95%-KI: 0,53-0,99; p=0,04), bei
Patienten mit einer hochfrequenten MSI zeigte sich jedoch kein Benefit (Ribic et
al., 2003). In der sogenannten FOCUS-Studie von Braun et al., welche die größte
Studie zu dieser Fragestellung darstellt, wurden 1628 Patienten, die adjuvant mit
Irinotecan behandelt wurden, bezüglich progressionsfreiem und Gesamtüberleben
untersucht. Eine MSI konnte nur selten nachgewiesen werden (4,4%). Es zeigte
sich keine prognostische Relevanz bezüglich der Veränderungen der
Mikrosatelliten (Braun et al., 2008).
1.6.2. p53
Das Tumorsuppressorprotein p53 ist als Transkriptionsfaktor an der Expression
von Genen beteiligt, die für die Kontrolle des Zellzyklus, der Induktion der
Apoptose oder der DNA-Reparatur verantwortlich sind. Auf diese Weise inhibiert
es das Wachstum genetisch alterierter und somit potentiell neoplastischer Zellen.
Es hat eine Molekularmasse von 53 kDa. Das zugehörige TP-53-
Tumorsuppressorgen ist auf Chromosom 17p13.1 lokalisiert. Ein Verlust der
Heterozygotät (loss of heterozygosity; LOH) dieses Chromosoms kann in 75%
aller KRK nachgewiesen werden, eine somatische Mutation ist in 40-60%
vorhanden (Cunningham et al., 1992; Forslund et al., 2001). Bezüglich der
Prognose bei einer Mutation oder einer LOH ist die Datenlage in der Literatur
kontrovers. Morrin et al. wiesen nach, dass eine Korrelation zwischen einer
Mutation in p53 weder mit dem UICC-Stadium, noch der Tumordifferenzierung
oder dem 5-Jahres-Überleben besteht (Morrin et al., 1994). In anderen Studien
hingegen wurde belegt, dass eine p53-Mutation einen negativen Effekt auf die
Prognose beim KRK erzielt. In der Untersuchung von Pricolo et al. beispielsweise
betrug das 5-Jahres-Überleben bei Patienten ohne Mutation 75% und bei einer
vorhandenen TP53-Mutation 21% (p=0.01) (Pricolo et al., 1996).
1.6.3. SMAD4
Wie bereits erwähnt, ist ein weiteres wichtiges Tumorsuppressorgen das SMAD4,
welches auf dem langen Arm des Chromosoms 18 an der Stelle 18q21.1
lokalisiert ist. Es sind acht verschiedene SMAD-Gene und Proteine bekannt,

- 17 -
welche in drei verschiedenen funktionellen Gruppen klassifiziert werden: Die
Rezeptor regulierten R-SMADs, die inhibitorischen I-SMADs sowie die ‚common-
mediator‘ Co-SMADs, dessen einziger Vertreter das SMAD4 ist. Dieses spielt eine
zentrale Rolle im TGF-beta-Signalweg. SMAD4 hat so einen Einfluss auf
Zellproliferation, Zelldifferenzierung, Zellmigration, Zellzykluskontrolle, Apoptose,
Angiogenese, Immunmodulation sowie Zell-Zell-Kontakte und die extrazelluläre
Matrix. Eine Reihe von genetischen und zellbiologischen Arbeiten konnten SMAD4
als Tumorsuppressorprotein im Kolon etablieren (Schwarte-Waldhoff, 2003). Einer
Studie von Salovaara et al. zufolge war keine SMAD4-Expression in 20 von 53
unselektierten KRK auffindbar (38%). Eine reduzierte Expression fand in weiteren
15 KRK statt (28%). Somit liegt der Schluss nahe, dass eine Abwesenheit von
SMAD4 bzw. eine reduzierte Expression eine entscheidende Rolle bei der
Entstehung von KRK spielt (Salovaara et al., 2002). Auch im menschlichen
Kolongewebe konnte gezeigt werden, dass der SMAD4-Verlust entlang der
Progression hin zu invasiven und metastasierten Stadien zunimmt (Miyaki et al.,
1999; Maitra et al., 2000; Reinacher-Schick et al., 2004).
Alazzouzi et al. untersuchten 86 Patienten mit einem KRK im Stadium UICC III.
Patienten mit einer hohen SMAD4-Expressionsrate zeigten ein signifikant längeres
Überleben als Patienten mit einer niedrigen Expression (p˂0,025) (Alazzouzi et al.,
2005). In Bezug auf die adjuvante Therapie konnte gezeigt werden, dass
Patienten mit einer normalen SMAD4-Expression bei einer 5-Fluorouracil-
basierten Chemotherapie sowohl beim Gesamtüberleben als auch beim
progressionsfreien Überleben mehr profitierten (Boulay et al., 2002).
1.6.4. Die Rolle der KRAS- und BRAF-Mutationen beim kolorektalen Karzinom
Zu der Familie der RAS-Protoonkogene gehören KRAS, NRAS und HRAS. Die
RAS-Protoonkogene kodieren für kleine, monomere Guaninnucleotid-bindende
Proteine (G-Proteine) mit einem Molekulargewicht von 21 kDa. RAS-Onkogene
sind wichtig für das Zellwachstum und die Zelldifferenzierung (Wittinghofer und
Pai, 1991; Bazan et al., 2002). Die RAS-Proteine sind eine zentrale Komponente
bestimmter mitogener Signalkaskaden, besonders für die Zellproliferation, die an
Rezeptoren (insbesondere EGFR) zur Wachstumsstimulation beginnen und im
Zellkern ihr Ende finden (Egan et al., 1993). Zunächst wird nach

- 18 -
Aktivierungsschritten durch Wachstumsfaktoren RAS in einen aktiven Zustand
gewandelt. Das aktive GTP-RAS aktiviert die MAP-Kaskade: Die RAF-Kinase wird
durch Bindung an das GTP-RAS aktiv und aktiviert durch Phosphorylierung die
MAP-Kinase-Kinase (MEK). Diese wiederum phosphoryliert die MAP-Kinase,
welche in dem nun aktiven Zustand für den Übergang in den Zellkern
verantwortlich ist. ELK-1, ein Transkriptionsfaktor, wird aktiviert und das FOS-Gen
transkribiert, wodurch das FOS-Protein entsteht. Dieses bildet mit dem JUN-
Protein einen Komplex, der als AP1 bezeichnet wird. Durch den AP1-Komplex
können weitere Gene, die für die Zellproliferation wichtig sind, aktiviert werden. In
erster Linie sind diese in der Mitose bei dem Übergang der G1- in die S-Phase von
Bedeutung.
Im Allgemeinen treten Mutationen im RAS-Gen bei circa 30% aller humanen
Tumoren auf, jedoch mit einer ziemlich variablen Inzidenz bei den verschiedenen
Krebsarten. Im KRK liegt eine KRAS-Gen-Mutation mit einer Häufigkeit von circa
30-40% vor (Kampman et al., 2000; Bleeker et al., 2001; Esteller et al., 2001;
Bazan et al., 2002; Di Nicolantonio et al., 2008). Bei Adenomen unter einem
Zentimeter treten in weniger als 10% KRAS-Mutationen auf und bei größeren
Adenomen und Karzinomen konnten in bis zu 50% der Fälle Veränderungen im
RAS-Gen nachgewiesen werden (Vogelstein et al., 1988). Über 90% der
Mutationen wurden im Kodon 12 und 13 gefunden, seltener sind Mutationen im
Kodon 61 (Bazan et al., 2002). Die Mutationen haben eine Veränderung des
Proteins mit einer reduzierten GTPase-Aktivität und einer verringerten Interaktion
mit GTPase-aktivierendem Protein (GAP) zur Folge, was zu einer vermehrten
Aktivität der Signalkaskade führt. In Bezug auf die Adenom-Karzinom-Sequenz
wird die RAS-Mutation in die frühe Phase der Adenomentstehung eingeordnet und
ist in erster Linie für die Progression zum größeren Adenom verantwortlich
(Vogelstein et al., 1988).
Eine weitere wichtige Rolle in dem RAS/RAF/MEK/MAP-Signalweg kommt beim
KRK dem BRAF-Gen zu. Bei den RAF-Proteinen (rapidly growing fibrosarcoma)
handelt es sich um Serin-Threonin-Proteinkinasen. Es gibt die Isoformen ARAF,
BRAF und CRAF. Davies hat als Erstbeschreiber 2002 berichtet, dass eine BRAF-
missense-Mutation mit einer Häufigkeit von 66% in malignen Melanomen und mit
einer geringeren Häufigkeit in einer Vielzahl anderer Tumoren vorkommt (Davies
et al., 2002).

- 19 -
In der Literatur wird das Vorkommen einer BRAF-Mutation mit einer Häufigkeit von
circa 5-10% beschrieben (Davies et al., 2002; Fransen et al., 2004; Di
Nicolantonio et al., 2008; Lièvre et al., 2010). So explorierten Fransen et al. in
einer Studie das Vorkommen einer BRAF-Mutation in 130 kolorektalen Tumoren.
Es zeigte sich eine Häufigkeit von 11,5% (Fransen et al., 2004). In der Studie von
Di Nicolantonio et al. wurden 113 Patienten mit einem mKRK untersucht. Mit einer
Häufigkeit von 9,7% aller 113 Patienten konnte eine BRAF-Mutation
nachgewiesen werden. Ein mutiertes BRAF trat in 11 von 79 Patienten mit einem
KRAS-Wildtyp auf (13,9%) (Di Nicolantonio et al., 2008).
Es konnte gezeigt werden, dass eine Mutation im BRAF- und KRAS-Gen nicht
gleichzeitig vorkommt (Fransen et al., 2004). So fand man in einer Studie in 4 von
43 Tumoren (9,3%) eine BRAF-Mutation und in 13 weiteren Tumoren (30,2%) eine
KRAS-Mutation (Yoshitake et al., 2007). Ebenso scheint es eine Korrelation mit
einer Mikrosatelliteninstabilität zu geben. Samowitz et al. zeigten, dass in 5%
mikrosatellitenstabiler Tumoren und in 51,8% mikrosatelliteninstabiler Karzinome
eine BRAF-Mutation vorhanden war. Auch Fransen et al. fanden eine signifikante
Korrelation zwischen einer BRAF-Mutation und einer Mikrosatelliteninstabilität
(Fransen et al., 2004). In mikrosatellitenstabilen Tumoren korreliert diese BRAF-
Mutation mit einem schlechten Gesamtüberleben und dem Hyperme-
thylierungsphänotyp (CIMP, CpG Island Methylator Phenotype) (Samowitz et al.,
2005). Bezugnehmend auf den Hypermethylierungsphänotypen konnte auch in
einer anderen Untersuchung gezeigt werden, dass CIMP mit einer hohen Rate an
Mikrosatelliteninstabilität sowie BRAF-Mutationen assoziiert ist (Ogino et al.,
2007).
1.6.4.1. KRAS und Prognose
Ob KRAS-Mutationen mit einer schlechten Prognose, der Invasionstiefe, dem
Tumorstadium, der Lokalisation und dem Gesamtüberleben bei Patienten mit KRK
korreliert sind, ist nicht vollständig geklärt (Finkelstein et al., 1993; Troungos et al.,
1997; Cerottini et al., 1998; Samowitz et al., 2000). Der Zusammenhang von
Tumorstadium und KRAS-Mutation wurde schon zu früher Zeit untersucht. So
führten Oliva et al. beispielsweise bereits 1990 eine Studie bei 58 Patienten mit
einem KRK durch. Es wurde eine KRAS-Mutation in 24,1% (14 Fälle)

- 20 -
nachgewiesen. Ein Zusammenhang zwischen einer Mutation und dem
Tumorstadium konnte nicht gezeigt werden (Oliva et al., 1990). 1997 wurde in
einer Studie von Troungos et al. zwar eine positive Korrelation zwischen dem
Vorhandensein einer KRAS-Mutation und der Invasionstiefe des Tumors
nachgewiesen, zwischen der Mutation und den regionalen Lymphknoten-
metastasen aber kein Zusammenhang gefunden (Troungos et al., 1997).
Bezüglich der Prognose kamen viele Studien zu dem Ergebnis, dass das
Vorhandensein einer KRAS-Mutation mit einem verkürzten Überleben verbunden
ist und somit der KRAS-Status ein möglicher Prognosefaktor sein könnte.
In einer Studie von Lee et al. wiesen Patienten mit mutierten KRAS-Genen im
UICC-Stadium I und II ein verkürztes Gesamtüberleben im Vergleich zu Patienten
ohne Mutationen auf. Das relative Mortalitätsrisiko bei diesen Stadien betrug
ungefähr 95%. Aus diesem Grund ist möglicherweise besonders in den frühen
Stadien das Vorliegen einer KRAS-Mutation ein Prognosefaktor bezüglich des
Überlebens (Lee et al., 1996). Auch Ahnen et al. konnten nachweisen, dass
KRAS-Mutationen im UICC-Stadium II mit einer schlechten Prognose
einhergehen. So betrug das 7-Jahres-Überleben in dieser Studie bei Patienten mit
Mutationen 58% und bei der Kontrollgruppe 86%. Die Hazard ratio (HR) in Bezug
auf das Sterben betrug 4,5 (95%-KI: 1,7-12,1; p=0,012) (Ahnen et al., 1998).
Samowitz et al. untersuchten in einer großen Studie 1413 Patienten mit einem
kolorektalen Karzinom und konnten in 32% der Tumoren eine KRAS-Mutation
nachweisen. Eine KRAS-Mutation im Kodon 13 war verbunden mit einer erhöhten
Mortaliät von 40% (95%-KI: 0,95-2,0) (Samowitz et al., 2000). Winder et al.
untersuchten 342 Patienten mit einem KRK. Eine KRAS-Mutation wurde mit einer
Häufigkeit von 28% nachgewiesen und es konnte gezeigt werden, dass Patienten
mit einer Mutation im Kodon 12 (G12V-Mutation) ein signifikant kürzeres
Gesamtüberleben aufwiesen als Patienten mit einem Wildtypen (HR=2,56 (1,15-
5,69)). Bei Nachweis anderer Mutationen hingegen zeigte sich ein verbessertes
Gesamtüberleben im Vergleich zu Wildtyppatienten (HR=0,44-0,99) (Winder et al.,
2009).
Auch in Hinblick auf den Einfluss von KRAS auf den Therapieerfolg gehen die
Meinungen in der Literatur auseinander.
Interessanterweise profitieren sowohl Patienten mit Tumoren im Stadium II als
auch Patienten mit einer KRAS-Mutation im Stadium III gemäß einer Studie von

- 21 -
Ahnen et al. nicht von einer adjuvanten 5-FU-basierten Chemotherapie (Ahnen et
al., 1998). In einer neueren Arbeit der Quasar Collaborative Group hingegen
wurde bei 3239 Patienten mit einem KRK im Stadium II, die entweder mit
Fluorouracil und Folinsäure (n=1622) behandelt oder der reinen Beobachtung
(n=1617) unterzogen wurden, eine signifikant höhere Sterberate bei der reinen
Beobachtung gesehen (370 Todesfälle vs. 311 Todesfälle, 95%-KI: 0,70-0,95,
p=0,008). Auch die Rezidivrate war bei den Patienten, die keine Chemotherapie
erhielten, signifikant höher (359 Rezidive vs. 293 Rezidive, 95%-KI: 0,67-0,91,
p=0,001) (Quasar Collaborative Group, 2007). In der Studie von Ahnen et al.
wurde als Weiteres die Prognose in Abhängigkeit von dem KRAS-Status
untersucht. Dabei wurde gezeigt, dass das 7-Jahres-Überleben beim Wildtyp-
KRAS im Stadium III allerdings durch die 5-FU-basierte Chemotherapie positiv im
Vergleich zu KRAS-mutierten Tumoren beeinflusst wird. In anderen Studien wurde
hingegen kein Zusammenhang zwischen dem klinischen Benefit und einer KRAS-
Mutation bei adjuvant behandelten KRK gezeigt (Westra et al., 2005; Fuchs et al.,
2009).
Bezüglich des Zusammenhangs einer adjuvanten FOLFOX-Therapie und der
Prognose bei KRAS-Mutationen sind derzeit keine Studien publiziert.
Beim mKRK ist die Datenlage bei der Frage der Korrelation des KRAS-Status und
des Therapieansprechens ebenso nicht eindeutig. Während in der Studie von
Rosty et al. keine signifikante Korrelation zwischen einem Ansprechen auf die
palliative Chemotherapie mit 5-FU und KRAS-Mutationen gesehen wurde (Rosty
et al., 2001) und ebenso in der Studie von Ince et al. bei Mutationen im KRAS-Gen
beim mKRK keine Lebensverlängerung durch die Zugabe von Bevacizumab zu
IFL (Irinotecan+5-FU+Leucovorin) erreicht wurde (Ince et al., 2005) und auch in
einer Studie von Ogino et al. keine signifikante Korrelation zwischen dem KRAS-
Status und dem Ansprechen auf eine Therapie mit Gefitinib gezeigt werden konnte
(Ogino et al., 2005), war in einer anderen Studie hingegen bei Patienten, die eine
palliative Therapie mit CPT-11 (Irinotecan) nach Therapieversagen von 5-FU-
Kombinationen erhielten, bei KRAS-positiven Tumoren eine signifikant schlechtere
Prognose zu beobachten (Nemunaitis et al., 1997).
Einen wichtigen Beitrag zum Therapieverhalten bei KRAS-Mutationen konnten die
OPUS- und die CRYSTAL-Studie leisten, welche ebenfalls zeigen, dass eine
KRAS-Mutation mit einem schlechteren Therapieansprechen assoziiert ist.

- 22 -
In der OPUS-Studie wurde der Effekt von Cetuximab in Kombination mit einer
FOLFOX-Therapie betrachtet (FOLFOX, n=168 vs. FOLFOX+Cetuximab, n=169)
(Bokemeyer et al., 2009). Bei Kombination von FOLFOX und Cetuximab zeigte
sich bei KRAS-Wildtyp-Tumoren eine Verbesserung der Ansprechrate (61% vs.
37%, p=0,011) und des PFS (7,7 Monate vs. 7,2 Monate, HR=0,57; p=0,163) im
Vergleich zur alleinigen FOLFOX-Therapie. Bei Karzinomen mit einer KRAS-
Mutation hingegen zeigte sich im Therapiearm mit Cetuximab+FOLFOX ein
tendenziell schlechteres Ansprechen im Vergleich zur alleinigen Chemotherapie
(33% vs. 49%, p=0,106). Ein gleiches Ergebnis zeigte sich in der neueren OPUS-
Studie, bei der neue Gewebeproben untersucht wurden (Bokemeyer et al., 2011).
Durch die Zugabe von Cetuximab zu FOLFOX-4 ergab sich bei Patienten mit
KRAS-Wildtyp ein verlängertes progressionsfreies Überleben (HR=0,567;
p=0,0064) und eine Verbesserung der Ansprechrate (OR=2,551, p=0,0027).
In der CRYSTAL-Studie wurden 599 Patienten mit einem mKRK mit dem
FOLFIRI-Protokoll behandelt und 599 Patienten mit FOLFIRI plus Cetuximab (van
Cutsem et al., 2009). Bei Patienten mit einem Wildtyp-KRAS zeigte sich ein
verlängertes progressionsfreies Überleben bei der Zugabe von Cetuximab (9,9 vs.
8,7 Monate; HR=0,68; 95%-KI: 0,50-0,94) und ein tendentiell verlängertes
Gesamtüberleben (24,9 vs. 21,0 Monate, HR=0,84; 95%-KI: 0,64-1,11). Werden
nur Patienten mit KRAS-Mutationen betrachtet, besteht sowohl beim
progressionsfreien Überleben (FOLFIRI+Cetuximab: 7,6 vs. FOLFIRI: 8,1 Monate;
p=0,07) als auch bei dem Gesamtüberleben (17,5 vs. 17,7 Monate bei FOLFIRI
alleine; p=0,44) kein Vorteil durch die Addition von Cetuximab. Zu einem ähnlichen
Ergebnis kam auch die wesentlich kleinere Studie von Lièvre et al., in welcher bei
13 von 30 Patienten mit einem mKRK Mutationen im KRAS-Gen gefunden werden
konnten. Es erfolgte eine Therapie mit Cetuximab, wobei bei den 11 Respondern
bei keinem Patienten eine KRAS-Mutation vorhanden war. Bei 19 Patienten mit
einer Resistenz gegen Cetuximab hingegen konnte in 68,4% der Fälle eine
Mutation nachgewiesen werden (Lièvre et al., 2006). Es wurde ebenso in den
nächsten beiden genannten Studien gezeigt, dass bei Vorhandensein einer
KRAS-Mutation das Therapieansprechen oder die Prognose schlechter sind. In
der Studie von Di Nicolantonio et al. wiesen 30% von 113 untersuchten Patienten
eine KRAS-Mutation auf. Eine Resistenz bezüglich der Therapie mit Cetuximab
oder Panitumumab konnte signifikant nachgewiesen werden (p=0,011) (Di
Nicolantonio et al., 2008). In der Studie von Amado et al. wurde eine

- 23 -
Panitumumab-Therapie in Abhängigkeit von dem KRAS-Status untersucht. Es
stellte sich heraus, dass das progressionsfreie Überleben bei Patienten mit einem
KRAS-Wildtyp-Status signifikant besser war als bei einem mutierten KRAS (95%-
KI: 0,34-0,59 vs. 95%-KI: 0,73-1,36, p˂0,0001) (Amado et al., 2008).
Studien, die den Zusammenhang zwischen dem KRAS-Status und dem
Ansprechen auf eine oxaliplatinhaltige Therapie bei einem KRK im UICC Stadium
IV untersuchen, waren bislang rar. Im Verlauf der Erstellung dieser Arbeit wurde
die MRC FOCUS Studie publiziert. Diese zeigte, dass eine KRAS- oder BRAF-
Mutation zwar mit einem schlechteren Gesamtüberleben (95%-KI: 1,20-1,65,
p˂0,0001) und einem verringerten progressionsfreien Überleben (95%-KI: 1,00-
1,36, p=0,05) zusammenhänge, durch eine Mutation das Ansprechen auf eine
Irinotecan- oder Oxaliplatintherapie allerdings nicht beeinflusst werde. Somit
profitierten Patienten mit einem mutierten KRAS oder BRAF ebenso von einer
solchen Therapie (Richman et al., 2009). Eine weitere Studie zu einer
Oxaliplatintherapie ist die CAIRO-Studie, welche untersuchte, ob es bei einer
sequentiellen oder kombinierten Gabe von Capecitabine, Irinotecan und
Oxaliplatin einen Unterschied im Therapieansprechen gibt. Es konnte gezeigt
werden, dass die Art der Gabe das Gesamtüberleben nicht wesentlich beeinflusst
(Koopman et al., 2007).
1.6.4.2. BRAF und Prognose
In Bezug auf die Prognose eines KRK liegen bisher weniger Studienergebnisse in
Korrelation mit BRAF als für KRAS vor, die verschiedenen Ergebnisse divergieren
jedoch auch hier. Hinsichtlich der Inzidenz und dem prognostischen Einfluss von
Biomarkern beim KRK untersuchten Roth et al. 1564 Gewebeproben der
adjuvanten PETACC 3-Studie von Van Cutsem et al. mit 3278 Patienten. Für den
Tumormarker BRAF wurden keine signifikanten Unterschiede bezüglich des
Vorkommens in den einzelnen Tumorstadien gefunden. Eine Mutation in BRAF
wurde sowohl im Stadium II als auch im Stadium III mit 8% evaluiert. Auch in
Bezug auf die Prognose zeigte sich kein signifikanter Unterschied (BRAF: St. II:
p=0,89, St. III: p=0,26) (Roth et al., 2010). Demgegenüber ist allerdings eine
Studie publiziert, in welcher Patienten mit einer BRAF-Mutation ein signifikant
schlechteres Gesamtüberleben hatten als Patienten mit BRAF-Wildtyp (median

- 24 -
10,4 Monate vs. 34,7 Monate, p˂0,0001). Eine BRAF-Mutation wurde in 11%
(57/524) nachgewiesen (Tran et al., 2010). In einer anderen Studie wird eine
signifikante Korrelation einer BRAF-Mutation sowohl mit dem T-Stadium (p=0,016)
als auch mit dem Grading (p=0,002) beschrieben (Aust et al., 2010). Material von
493 Patienten aus der PETACC2-Studie wurde retrospektiv untersucht. Dabei
konnte eine BRAF-Mutation kein Mal in dem T1-Stadium (0/13, 0%) und zwei Mal
in dem T2-Stadium (2/39, 5,1%) nachgewiesen werden. Die Anzahl der BRAF-
Mutationen nahm im T3- und T4-Stadium zu (T3: 32/347, 9,2%, T4: 17/84, 20,2%).
Ebenfalls konnte eine zunehmende Anzahl der BRAF-Mutationen mit
ansteigendem Grading-Stadium nachgewiesen werden (G1: 1/35, 1,9%, G2:
26/316, 8,2%; G3: 24/128, 18,8%). Bezüglich des Profits einer 5-FU-haltigen
Chemotherapie je nach BRAF-Status konnte jedoch keine Korrelation
nachgewiesen werden. Nach drei Jahren unter einer adjuvanten Therapie mit 5-
FU/FA lebten noch 68% der Patienten mit KRAS/BRAF-Wildtyp (HR=0,89) und
65% der Patienten mit einer BRAF-Mutation (HR=1,08) (Aust et al., 2010).
In einer Studie von Ince et al. konnte ebenfalls keine signifikante Beziehung
zwischen Mutationen im BRAF-Gen und dem medianen Überleben bei einer
Addition von Bevacizumab zu IFL beim mKRK gezeigt werden (Ince et al., 2005).
In einigen Studien wurden BRAF-Mutationen mit Resistenzen gegenüber anti-
EGFR-Strategien in Verbindung gebracht. Die Meinungen in der Literatur gehen
jedoch auseinander. In einer Studie von Finocchiaro et al. wurden 85 Patienten mit
einem mit Cetuximab therapierten metastasierten kolorektalen Karzinom, bei
denen der EGFR- und KRAS-Status bekannt war, unter anderem auf eine
Mutation des BRAF untersucht. Eine BRAF-Mutation wurde bei 5% der Patienten
gefunden und keiner dieser Patienten profitierte von der Therapie mit Cetuximab
im Vergleich zum Wildtyp (Progressionszeit 1,2 vs. 4,6 Monate, p=0,09; OS 5,4 vs.
9,8 Monate, p=0,3) (Finocchiaro et al., 2008). In einer Studie von Di Nicolantonio
et al. sprach keiner der Patienten mit einer BRAF-Mutation auf eine Therapie mit
Cetuximab oder Panitumumab an, im Vergleich dazu befand sich unter den
Therapierespondern kein Patient mit einer BRAF-Mutation (p=0,029). Patienten
mit einer BRAF-Mutation zeigten ein signifikant kürzeres progressionsfreies
Überleben (p=0,011) und ein signifikant kürzeres Gesamtüberleben (p<0,0001) (Di
Nicolantonio et al., 2008). Ebenso sprach keiner der Patienten mit einer BRAF-
Mutation (8 von 50) in der Studie von Weickhardt et al. auf eine Cetuximab-haltige
Therapie an (Weickhardt et al., 2010).

- 25 -
In einer relativ neuen Studie von Maughan et al. wurden 1630 Patienten entweder
mit Oxaliplatin und Fluoropyrimidinen oder noch zusätzlich mit Cetuximab
behandelt. Eine BRAF-Mutation wurde bei 102 Patienten nachgewiesen (8%). Es
konnte jedoch kein signifikanter Unterschied bei der Addition von Cetuximab
aufgezeigt werden (OS: HR 1,04, 95%-KI: 0,87-1,23, p=0,67; PFS: HR: 0,96, 95%-
KI: 0,82-1,12, p=0,60) (Maughan et al., 2010). Zu einem ähnlichen Ergebnis kam
es in der Analyse der OPUS- und CRYSTAL-Studien von Bokemeyer et al., nach
welcher der BRAF-Status kein entscheidender prädiktiver Marker bezüglich des
Ansprechens auf Cetuximab sei. Diese Aussage wird jedoch in dem Artikel
insofern eingeschränkt, als dass die Anzahl der BRAF-Mutationen für eine
definitive Aussage möglicherweise zu gering sei (Bokemeyer et al., 2010). Auch
gemäß den Ergebnissen der CAIRO2-Studie sei eine BRAF-Mutation zwar
korreliert mit einem verringerten progressionsfreien Überleben, ein besseres oder
schlechteres Ergebnis bei Cetuximab-Gabe konnte jedoch nicht aufgezeigt
werden (PFS: 5,9 vs. 12,2 Monate in dem Arm Chemotherapie+Bevacizumab,
p=0,003; 6,6 vs. 10,4 Monate in dem Arm Chemotherapie+
Bevacizumab+Cetuximab, p=0,010) (Tol et al., 2009).

- 26 -
2. Zielsetzung
Das KRK ist in Europa die zweithäufigste tumorbedingte Todesursache. Bei
Erstdiagnose weisen etwa 25% der Patienten eine Fernmetastasierung und 30-
60% der Patienten eine Metastasierung im Verlauf auf.
Durch die Einführung neuer Chemotherapeutika konnte die Prognose des mKRK
deutlich verbessert werden. Allerdings ist das Ansprechen auf die Therapie nicht
vorhersagbar, zudem können schwerwiegende Toxizitäten bei der Therapie
auftreten und die Behandlungskosten nehmen durch die Einführung neuer
Substanzen zu. Daher kommt der Identifikation von Biomarkern zur Prädiktion des
Therapieansprechens und zur Prognoseabschätzung eine entscheidende
Bedeutung zu.
Der am besten etablierte Biomarker bei KRK ist der Nachweis von KRAS-
Mutationen hinsichtlich der Prädiktion eines Ansprechens auf eine anti-EGFR-
Therapie mit monoklonalen Antikörpern alleine oder in Kombination mit einer
konventionellen Chemotherapie. Ein weiterer potenzieller prädiktiver oder
prognostischer Marker bei KRK sind BRAF-Mutationen. Bei KRK liegt eine KRAS-
Mutation in circa 30-40% vor. Eine BRAF-Mutation kommt mit einer Häufigkeit von
5-10% vor. Dabei konnte gezeigt werden, dass Mutationen im BRAF- und KRAS-
Gen nicht gleichzeitig vorkommen.
KRAS hat sich mittlerweile als ein wichtiger prognostischer Marker beim KRK
etabliert, Ergebnisse divergieren jedoch noch immer. Insbesondere in Bezug auf
das Ansprechen auf eine anti-EGFR-Antikörpertherapie scheint ein
Zusammenhang zu bestehen. Mutationen in BRAF scheinen ähnliche
Auswirkungen zu haben, der genaue prognostische und prädiktive Nutzen ist
jedoch noch nicht geklärt. Insbesondere der Effekt von KRAS- oder BRAF-
Mutationen auf eine Oxaliplatin-Therapie ist derzeit noch unklar. Aus diesem
Grund sind weitere Nachforschungen auf diesem Gebiet unerlässlich.
In dieser Promotionsarbeit soll die prognostische Bedeutung von KRAS- und
BRAF-Mutationen bei Patienten mit mKRK unter einer Kombinations-
chemotherapie mit Fluoropyrimidinen und Oxaliplatin evaluiert werden. Die
Gewebeproben sowie die klinischen Verlaufsdaten entstammen einer Sub-
population von 205 Patienten mit verfügbaren Gewebeproben aus einer bereits
abgeschlossenen prospektiven, randomisierten, multizentrischen Phase-III-
Therapiestudie mit insgesamt 474 Patienten.

- 27 -
3. Material und Methoden
3.1. Material
Die untersuchten Gewebeproben stammen von Patienten aus einer prospektiv
randomisierten zweiarmigen Phase-III-Studie (FUFOX versus CAPOX) der AIO
zur Erstlinientherapie des metastasierten KRK. Die Patienten des Armes A wurden
mit 5-Fluorouracil/Folinsäure plus Oxaliplatin (FUFOX) therapiert und die
Patienten des Armes B erhielten Capecitabin plus Oxaliplatin (CAPOX). Die
Details zu den Ein- und Ausschlusskriterien, der Behandlungsprotokolle und der
Ergebnisse der klinischen Studie mit 474 Patienten sind publiziert (Porschen et al.,
2007). Hauptendpunkt war die Nicht-Unterlegenheit im progressionsfreien
Überleben (PFS). Als Hauptergebnis konnte in dieser Studie gezeigt werden, dass
eine Erstlinientherapie mit CAPOX bei Patienten mit einem mKRK der Gabe von
FUFOX in Bezug auf das progressionsfreie Überleben gleichwertig ist. Die
sekundären Endpunkte beinhalteten den Vergleich der Remissionsraten, der
Gesamtüberlebenszeiten, der Toxizitäten sowie die Beurteilung der Lebens-
qualität.
Es waren Gewebeproben von 207 Patienten verfügbar, wobei der KRAS- und
BRAF-Status erfolgreich in 205 Tumoren analysiert werden konnte.
Das untersuchte Teilkollektiv unterscheidet sich in Bezug auf Alter, Geschlecht,
Therapie (FUFOX bzw. CAPOX) nicht vom Gesamtkollektiv aller Studienpatienten.
Es wurde bei der Ethik-Kommission der Ruhr-Universität Bochum am 8. Juli 2002
der Antrag mit dem Thema „5-Fluorouracil/Folinsäure plus Oxaliplatin (FUFOX)
versus Oxaliplatin/Capecitabin (CAPOX) beim fortgeschrittenen kolorektalen
Karzinom“ vorgelegt. Es wurde diesem Antrag am 12. Juli 2002 unter der
Registrier-Nr. 1923 durch ein positives Votum der Ethik-Kommission zugestimmt.
Das translationale Begleitforschungsprogramm (die u.a. die Analyse von KRAS
und BRAF in diesem Kollektiv beinhaltet) wurde im Studienprotokoll erläutert und
in der Patientenaufklärung aufgeführt. Es fand eine freiwillige Teilnahme am
translationalen Forschungsprogramm von Seiten der Patienten statt. Es wurde von
jedem Patienten zur Analyse der Proben und Teilnahme an der Studie eine
Einwilligung eingeholt.
- 28 -
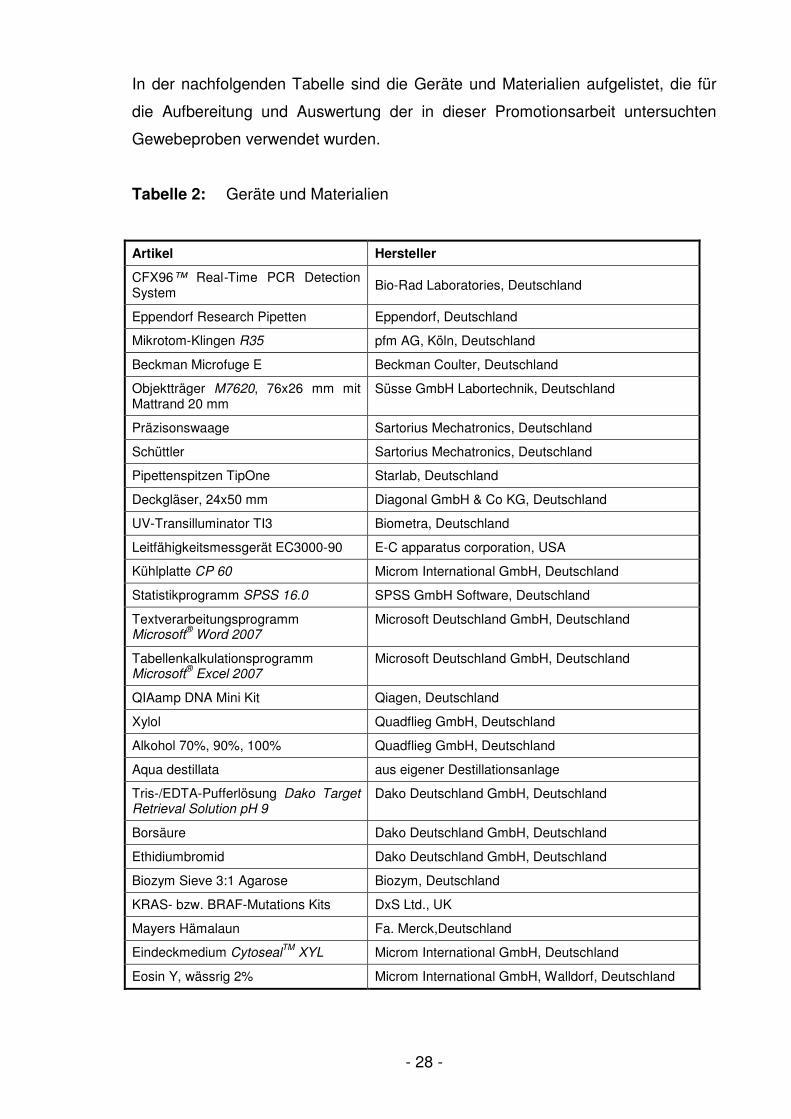
In der nachfolgenden Tabelle sind die Geräte und Materialien aufgelistet, die für
die Aufbereitung und Auswertung der in dieser Promotionsarbeit untersuchten
Gewebeproben verwendet wurden.
Tabelle 2: Geräte und Materialien
Artikel Hersteller
CFX96™ Real-Time PCR Detection System
Bio-Rad Laboratories, Deutschland
Eppendorf Research Pipetten Eppendorf, Deutschland
Mikrotom-Klingen R35 pfm AG, Köln, Deutschland
Beckman Microfuge E Beckman Coulter, Deutschland
Objektträger M7620, 76x26 mm mit Mattrand 20 mm
Süsse GmbH Labortechnik, Deutschland
Präzisonswaage Sartorius Mechatronics, Deutschland
Schüttler Sartorius Mechatronics, Deutschland
Pipettenspitzen TipOne Starlab, Deutschland
Deckgläser, 24x50 mm Diagonal GmbH & Co KG, Deutschland
UV-Transilluminator TI3 Biometra, Deutschland
Leitfähigkeitsmessgerät EC3000-90 E-C apparatus corporation, USA
Kühlplatte CP 60 Microm International GmbH, Deutschland
Statistikprogramm SPSS 16.0 SPSS GmbH Software, Deutschland
Textverarbeitungsprogramm Microsoft
® Word 2007
Microsoft Deutschland GmbH, Deutschland
Tabellenkalkulationsprogramm Microsoft
® Excel 2007
Microsoft Deutschland GmbH, Deutschland
QIAamp DNA Mini Kit Qiagen, Deutschland
Xylol Quadflieg GmbH, Deutschland
Alkohol 70%, 90%, 100% Quadflieg GmbH, Deutschland
Aqua destillata aus eigener Destillationsanlage
Tris-/EDTA-Pufferlösung Dako Target Retrieval Solution pH 9
Dako Deutschland GmbH, Deutschland
Borsäure Dako Deutschland GmbH, Deutschland
Ethidiumbromid Dako Deutschland GmbH, Deutschland
Biozym Sieve 3:1 Agarose Biozym, Deutschland
KRAS- bzw. BRAF-Mutations Kits DxS Ltd., UK
Mayers Hämalaun Fa. Merck,Deutschland
Eindeckmedium CytosealTM
XYL Microm International GmbH, Deutschland
Eosin Y, wässrig 2% Microm International GmbH, Walldorf, Deutschland

- 29 -
3.2. Methodik
Die DNA wurde aus pseudoanonymisierten Formalin-fixierten, Paraffin
eingebetteten Gewebeproben (FFPE) extrahiert. Pro Patient wurden fünf 10 µm
dicke Schnitte angefertigt. Ein zusätzlicher 1 µm dicker Schnitt wurde in H&E-
Färbung gefärbt. Regionen mit mehr als 70% Tumorgewebe wurden markiert und
makrodisseziiert. Für die DNA-Extraktion wurde der QIAamp DNA Mini Kit
(Qiagen, Hilden, Germany) nach den Herstellerangaben verwandt. Die
Makrodissektion erfolgte im Institut für Pathologie der Berufsgenossenschaftlichen
Kliniken Bergmannsheil. Für die real-time PCR, welche ebenfalls in den Räumen
des Instituts für Pathologie der Berufsgenossenschaftlichen Kliniken
Bergmannsheil durchgeführt wurde, wurden 20 ng DNA eingesetzt. Für die
Analytik wurden kommerzielle Kits der DxS Ltd. (Manchester, UK) nach den
Herstellerangaben eingesetzt. Mit Hilfe des KRAS-Mutations-Kits wurden sieben
KRAS-Mutationen detektiert: Mutationen an den Aminosäuren 12Aspartat,
13Aspartat, 12Valin, 12Alanin, 12Cystein, 12Serin und 12Arginin. Durch dieses
Verfahren wurden mehr als 95% der bekannten KRAS-Mutationen nachgewiesen.
Generell dient die Polymerase-Kettenreaktion (PCR) dazu, die DNA zu
vervielfältigen und auf diesem Wege die o.g. Mutationen detektieren zu können.
Die PCR besteht aus mehreren Zyklen, welche in einem Thermocycler
durchgeführt werden. Jeder Zyklus besteht aus drei Schritten. Zunächst werden
durch eine Erhitzung der DNA auf 94-96 Grad Celcius die
Wasserstoffbrückenbindungen gelöst und die DNA somit denaturiert, bis nur noch
Einzelstränge vorliegen. Der nächste Schritt ist die Primerhybridisierung, welche
die Anlagerung der zugegebenen Primer an den gewünschten DNA-Abschnitt
darstellt. Darauf folgt die Elongation durch die zugegebenen DNA-Polymerasen.
Durch die Zugabe von verschiedenen Primern kann somit bestimmt werden,
welcher DNA-Abschnitt aufgefunden und somit amplifiziert wird, beispielsweise die
oben beschriebenen KRAS-Mutationen.
Bzgl. BRAF wurde gezielt die Hotspot-Mutation V600E-Mutation detektiert. Da
gezeigt wurde, dass KRAS- und BRAF-Mutationen nicht gleichzeitig in einem
Tumor vorkommen (Yoshitake et al., 2007), untersuchten wir nur diejenigen
Tumoren auf eine BRAF V600E-Mutation, in denen keine KRAS-Mutation
nachgewiesen werden konnte. Es fand eine Verblindung zwischen dem
Laborpersonal und den klinischen Daten statt.

- 30 -
3.3. Statistik
Für die statistische Auswertung wurden der Fisher’s-Exact-Test für den
Zusammenhang zwischen KRAS bzw. BRAF und anderen dichotomen Variablen,
der Student-T-Test für die Prüfung der Signifikanz der Beziehung zwischen
kontinuierlichen Variablen sowie der Kruskal-Wallis-Test für die Identifikation der
signifikanten Unterschiede einer kontinuierlichen Variable zwischen verschiedenen
Gruppen angewandt.
Des Weiteren wurde der Mann-Withney-U-Test verwendet, um intervallskalierte
Variablen zwischen zwei verschiedenen Gruppen zu vergleichen.
Das Gesamtüberleben und das progressionsfreie Überleben wurden mit Hilfe von
Kaplan-Meier-Kurven graphisch dargestellt (Kaplan und Meier, 1958) und das
Gesamtüberleben sowie das progressionsfreie Überleben der Subgruppen mittels
Log-Rank-Test verglichen.
Als statistisch signifikant wurden p-Werte kleiner als 0,05 in 2-seitigen Tests
bewertet. Als Remissionsrate sei der prozentuale Anteil erfolgreich behandelter
Patienten zu verstehen, bei denen eine Tumorverkleinerung (partielle Remission)
oder die vollständige Rückbildung des Tumors (komplette Remission) erzielt
wurde. Unter der Krankheitskontrollrate versteht man den Prozentsatz der
Patienten mit komplettem bzw. partiellem Ansprechen oder Stabilisierung der
Erkrankung.

- 31 -
4. Ergebnisse
4.1. Beschreibung des Patientenkollektivs
Bei den 205 Patienten mit verfügbaren und erfolgreich analysierbaren
Gewebeproben handelte es sich um 124 (60,5%) männliche und 81 (39,5%)
weibliche Patienten mit einem mittleren Alter von 64,2 ± 8,6 Jahren mit einer
unteren Grenze von 35 Jahren und einem maximalen Alter von 82 Jahren.
105 (51,2%) Patienten wurden im FUFOX-Arm und 99 (48,3%) Patienten im
CAPOX-Arm der Studie therapiert. Die Therapie eines Patienten konnte nicht
nachvollzogen werden. Bei 134 (65,4%) Personen war das Kolon befallen und bei
61 (29,8%) Patienten das Rektum. Bei 10 (4,9%) Patienten war die Lokalisation
nicht bekannt. 104 (50,7%) Tumoren wiesen synchrone und 82 (40,0%)
Karzinome metachrone Metastasen auf. Bei 19 (9,3%) Patienten war dies nicht
bekannt.
Das hier analysierte Subkollektiv unterschied sich bzgl. der o.g. Variablen nicht
vom Gesamtkollektiv aller 474 Patienten.
4.2. Häufigkeit von KRAS- und BRAF-Mutationen
Der KRAS-Wildtyp wurde in 130 (63,4%) der 205 verfügbaren Tumoren
nachgewiesen und eine KRAS-Mutation lag in 75 Fällen vor (36,6%). Die häufigste
Mutation mit 25 Fällen (12,2%) wurde im Kodon 12 nachgewiesen und führte zu
einem Aminosäure (AS)-Austausch zu Aspartat. 15 Mutationen (7,3%) führten in
Kodon 13 zu einem AS-Austausch zu Aspartat und 13 Mutationen (6,3%) in Kodon
12 zu einem AS-Austausch zu Valin. 7 Mutationen (3,4%) führten in Kodon 12 zu
einem AS-Austausch zu Alanin und mit einer Häufigkeit von jeweils 6 Mutationen
(jeweils 2,9%) traten Mutationen in Kodon 12 auf, die zu einem AS-Austausch zu
Cystein und Serin führten. 1 Mutation (0,5%) führte in Kodon 12 zu einem AS-
Austausch zu Arginin. Ein Tumor (0.5%) wies zwei verschiedene KRAS-
Mutationen auf.
Die BRAF V600E-Mutation wurde in 13 Tumoren von 130 Tumoren (10,0%) mit
Wildtyp-KRAS nachgewiesen, entsprechend 6,3% der gesamten untersuchten 205

- 32 -
Tumorpatienten. 88 der 205 Patienten (42,9%) wiesen entweder eine KRAS- oder
eine BRAF-Mutation auf.
4.3. Korrelation mit klinischen Variablen
Es bestand keine signifikante Korrelation zwischen einer KRAS-Mutation und dem
Geschlecht. Der Mittelwert des Alters betrug bei dem KRAS- und dem BRAF-
Wildtyp 64,5 ± 8,7 Jahre, während das mittlere Alter bei den KRAS- und BRAF-
Mutationen bei 64,0 ± 8,5 Jahren lag (p=0,94). Ebenso bestand kein signifikanter
Unterschied hinsichtlich des Therapiearms, der Lokalisation und der Art der
Metastasierung (s. Tabelle 3).
Tabelle 3: Korrelation mit klinischen Variablen
KRAS-
WT
KRAS-
Mut p
BRAF-
WT
BRAF-
Mut p
Geschlecht M 82/124 42/124
0,37 72/78 5/78
0,1 W 48/81 33/81 36/44 8/44
Therapiearm FUFOX 65/105 40/105
0,69 53/62 9/62
0,24 CAPOX 64/99 35/99 54/59 4/59
Lokalisation Kolon 85/134 49/134
0,42 70/78 7/78
0,55 Rektum 35/61 26/61 29/34 5 /34
Metastasierung Syn 69/104 35/104
0,27 54/60 6/60
0,52 Meta 48/82 34/82 44/49 4/49
4.4. Korrelation mit dem Therapieansprechen
Die Evaluation des Therapieansprechens war bei 199 Patienten möglich. Bei
Patienten mit KRAS-Wildtyp-Tumoren wurden 6 komplette Remissionen, 74
Patienten mit partiellen Remissionen, 31 stabile Erkrankungen und 16
progrediente Erkrankungen beobachtet. Bei den Patienten mit KRAS-Mutationen
wurden 3 komplette Remissionen, 29 partielle Remissionen, 25 stabile Er-
krankungen und 15 progrediente Tumoren festgestellt. Beim BRAF-Wildtyp lagen

- 33 -
5 komplette Remissionen, 60 partielle Remissionen, 26 stabile Erkrankungen und
14 progrediente Erkrankungen vor. Bei der BRAF-Mutation hatte kein Patient eine
komplette Remission. Bei 8 Patienten wurde eine partielle Remission und bei 4
Patienten eine Stabilisierung beobachtet. Ein Patient mit BRAF-Mutation war
progredient. Die Unterschiede in der globalen Verteilung waren statistisch nicht
signifikant (p=0,68).
Tabelle 4: Darstellung des Therapieansprechens
KRAS-WT KRAS-Mut BRAF-WT BRAF-Mut
Komplette Remission
6 3 5 0
Partielle Remission
74 29 60 8
Stabile Erkrankung
31 25 29 4
Progrediente Erkrankung
16 15 14 1
Nicht evaluierbar 3 3 0 0
Summe
130 75 108 13
Tumoren mit KRAS-Mutationen wiesen eine signifikant geringere Remissionsrate
als Tumoren ohne KRAS-Mutation auf (44,4% vs. 63,0%, p=0,012) (Tabelle 5). Bei
Hinzunahme der BRAF-Mutationen wurde der signifikante Unterschied beim
Vergleich von Tumoren mit einer KRAS- oder BRAF-Mutation gegenüber Tumoren
mit einem KRAS- und BRAF-Wildtyp beibehalten (46,4% vs. 63,5%, p=0,021). Bei
der Krankheitskontrollrate konnte kein signifikanter Unterschied zwischen
Tumoren mit einer KRAS- oder BRAF-Mutation im Vergleich zu Tumoren mit
Wildtyp für beide Gene festgestellt werden (80.9 vs. 87,0, p=0,323). KRAS-
Mutationen allein betrachtet zeigten ebenfalls keinen statistisch signifikanten
Unterschied bezüglich des Stabilisierungsgrades (p=0,155). BRAF-Mutationen
allein waren weder mit einer unterschiedlichen Remission noch mit einer
unterschiedlichen Krankheitskontrollrate im Vergleich zu BRAF-Wildtyp-Tumoren
assoziiert. Hier muss bei der Interpretation jedoch die geringe Zahl von BRAF-
Mutationen berücksichtigt werden.
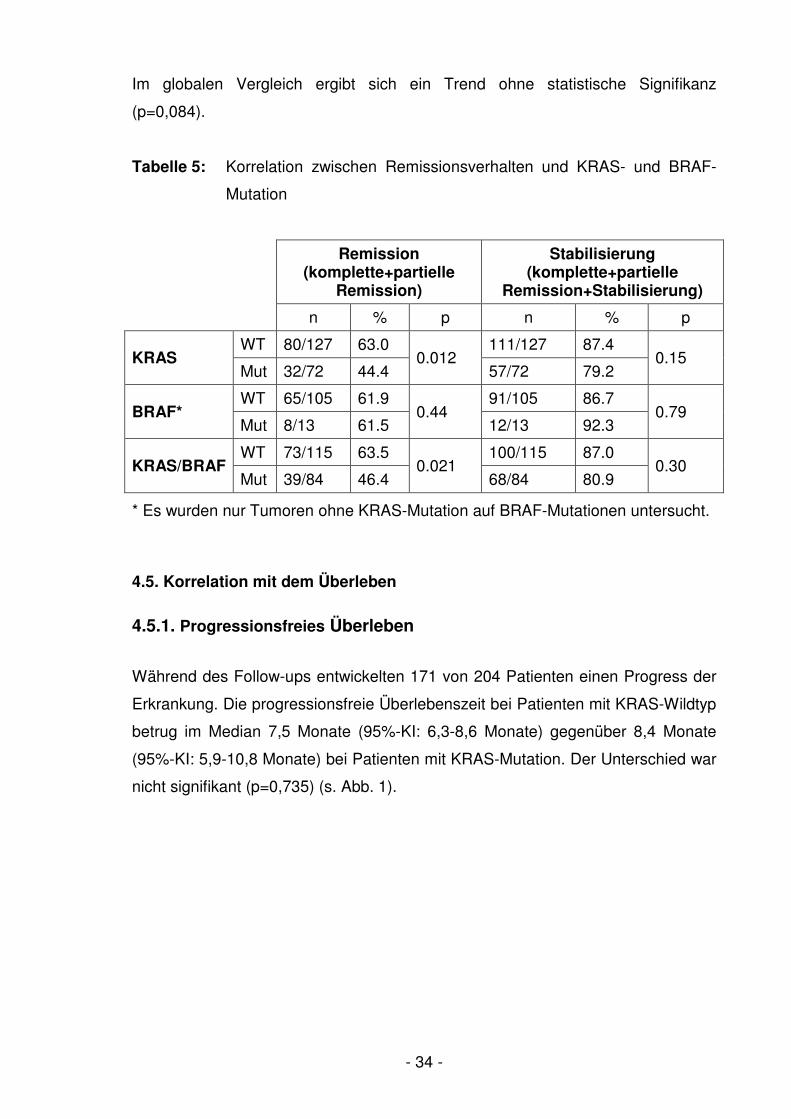
- 34 -
Im globalen Vergleich ergibt sich ein Trend ohne statistische Signifikanz
(p=0,084).
Tabelle 5: Korrelation zwischen Remissionsverhalten und KRAS- und BRAF-
Mutation
Remission (komplette+partielle
Remission)
Stabilisierung (komplette+partielle
Remission+Stabilisierung)
n % p n % p
KRAS WT 80/127 63.0
0.012 111/127 87.4
0.15 Mut 32/72 44.4 57/72 79.2
BRAF* WT 65/105 61.9
0.44 91/105 86.7
0.79 Mut 8/13 61.5 12/13 92.3
KRAS/BRAF WT 73/115 63.5
0.021 100/115 87.0
0.30 Mut 39/84 46.4 68/84 80.9
* Es wurden nur Tumoren ohne KRAS-Mutation auf BRAF-Mutationen untersucht.
4.5. Korrelation mit dem Überleben
4.5.1. Progressionsfreies Überleben
Während des Follow-ups entwickelten 171 von 204 Patienten einen Progress der
Erkrankung. Die progressionsfreie Überlebenszeit bei Patienten mit KRAS-Wildtyp
betrug im Median 7,5 Monate (95%-KI: 6,3-8,6 Monate) gegenüber 8,4 Monate
(95%-KI: 5,9-10,8 Monate) bei Patienten mit KRAS-Mutation. Der Unterschied war
nicht signifikant (p=0,735) (s. Abb. 1).

- 35 -
Abbildung 1: Progressionsfreies Überleben nach KRAS-Mutations-Status
Patienten mit einem KRAS- und BRAF-Wildtyp hatten ein medianes
progressionsfreies Überleben von 7,6 Monaten (95%-KI: 6,5-8,6 Monate)
gegenüber 7,5 Monaten (95%-KI: 4,9-10,0 Monate) für Patienten mit KRAS- oder
BRAF-Mutation (p=0,89) (s. Abb. 2).
Abbildung 2: Progressionsfreies Überleben nach KRAS-/BRAF-Status
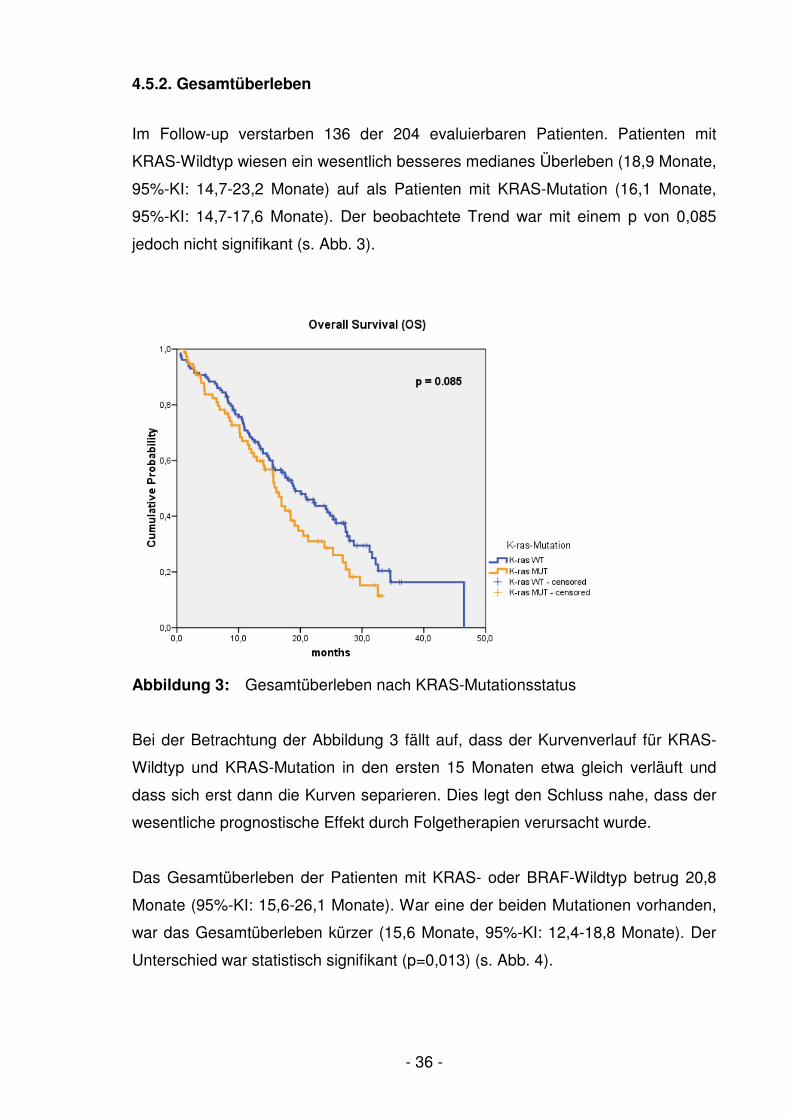
- 36 -
4.5.2. Gesamtüberleben
Im Follow-up verstarben 136 der 204 evaluierbaren Patienten. Patienten mit
KRAS-Wildtyp wiesen ein wesentlich besseres medianes Überleben (18,9 Monate,
95%-KI: 14,7-23,2 Monate) auf als Patienten mit KRAS-Mutation (16,1 Monate,
95%-KI: 14,7-17,6 Monate). Der beobachtete Trend war mit einem p von 0,085
jedoch nicht signifikant (s. Abb. 3).
Abbildung 3: Gesamtüberleben nach KRAS-Mutationsstatus
Bei der Betrachtung der Abbildung 3 fällt auf, dass der Kurvenverlauf für KRAS-
Wildtyp und KRAS-Mutation in den ersten 15 Monaten etwa gleich verläuft und
dass sich erst dann die Kurven separieren. Dies legt den Schluss nahe, dass der
wesentliche prognostische Effekt durch Folgetherapien verursacht wurde.
Das Gesamtüberleben der Patienten mit KRAS- oder BRAF-Wildtyp betrug 20,8
Monate (95%-KI: 15,6-26,1 Monate). War eine der beiden Mutationen vorhanden,
war das Gesamtüberleben kürzer (15,6 Monate, 95%-KI: 12,4-18,8 Monate). Der
Unterschied war statistisch signifikant (p=0,013) (s. Abb. 4).

- 37 -
Abbildung 4: Gesamtüberleben nach KRAS-/BRAF-Mutationsstatus
Zusammenfassend zeigte sich, dass KRAS-Mutationen oder die Kombination von
KRAS- und BRAF-Mutationen mit einem schlechteren Therapieansprechen und
einem schlechteren Gesamtüberleben assoziiert sind, während sich beim
progressionsfreien Überleben kein Unterschied beobachten ließ.
4.5.3. Möglicher Einfluss von Folgetherapien
136 der 205 Patienten erhielten eine Folgetherapie. Von diesen wiesen 83
Patienten einen KRAS-Wildtyp und 53 Patienten eine KRAS-Mutation auf. 80%
der Patienten erhielten eine Zweitlinientherapie mit Irinotecan, 30% der Patienten
wurden mit einer Drittlinientherapie mit Cetuximab behandelt.
Bei Patienten ohne Folgetherapie (n=82) zeigte sich bezüglich des
Gesamtüberlebens ab Progress kein wesentlicher Unterschied (s. Abb. 5).

- 38 -
Abbildung 5: Postprogressionsüberleben nach KRAS-Mutationsstatus bei
Patienten, die keine Zweitlinientherapie erhielten (n=82)
116 Patienten erhielten eine Irinotecan-haltige Zweitlinientherapie. In dieser
Gruppe zeigte sich ein Trend zu einem günstigeren Postprogressionsüberleben in
der KRAS-Wildtyp-Gruppe (p=0,072) (s. Abb. 6).

- 39 -
Abbildung 6: Postprogressionsüberleben nach KRAS-Mutationsstatus bei
Patienten, die eine Irinotecan-haltige Zweitlinientherapie erhielten
(n=116)
Insbesondere die Subgruppe der Patienten mit KRAS-Wildtyp und einer
Folgetherapie mit Cetuximab (n=41) profitierten beim Gesamtüberleben von dieser
Therapie, während Patienten ohne Cetuximab-haltige Folgetherapie keinen
Unterschied im Postprogressionsüberleben in Abhängigkeit vom KRAS-Status
aufwiesen (s. Abb. 7 und 8) .
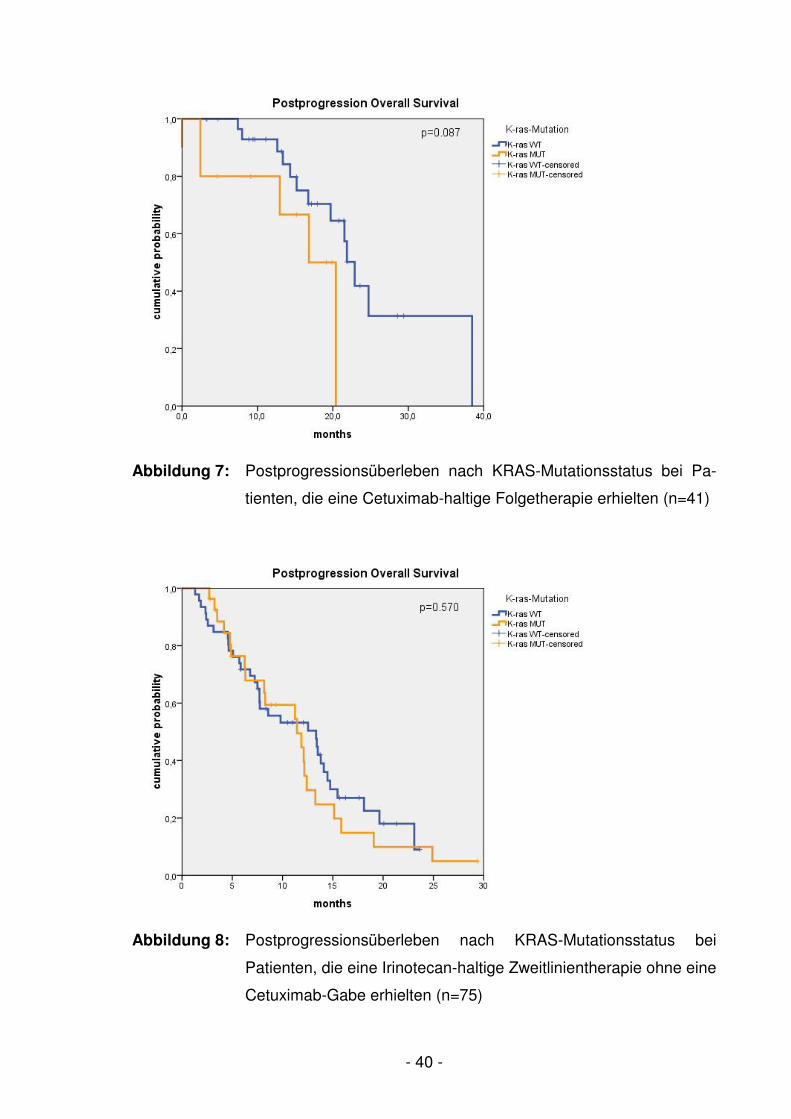
- 40 -
Abbildung 7: Postprogressionsüberleben nach KRAS-Mutationsstatus bei Pa-
tienten, die eine Cetuximab-haltige Folgetherapie erhielten (n=41)
Abbildung 8: Postprogressionsüberleben nach KRAS-Mutationsstatus bei
Patienten, die eine Irinotecan-haltige Zweitlinientherapie ohne eine
Cetuximab-Gabe erhielten (n=75)

- 41 -
5. Diskussion
In den vergangenen Jahren konnte die Behandlung und die Prognose der
Patienten mit einem KRK aufgrund neuer Chemotherapeutika und zielgerichteter
Substanzen deutlich verbessert werden. In erster Linie profitieren Patienten mit
einem metastasierten Karzinom von neuen Kombinationen der Medikamente, da
dadurch das mediane Überleben von nur sechs Monaten durch beste supportive
Behandlung auf über zwei Jahre verlängert werden kann, wenn alle Mittel
ausgeschöpft werden. Allerdings ist das Ansprechen auf die Therapie derzeit nicht
vorhersagbar, d.h. nicht alle Patienten profitieren gleichermaßen von den
verschiedenen Medikamenten, das Auftreten schwerwiegender Toxizitäten ist
möglich und nur bedingt vorhersagbar. Da bereits circa 25% der Patienten bei
Erstdiagnose eine Fernmetastasierung aufweisen und bei 30-60% der Patienten
das KRK im Verlauf metastasiert und die Behandlungskosten durch die Einführung
neuer Substanzen erheblich steigen, besteht eine deutliche gesundheits-
ökonomische Dimension bzgl. der Identifikation von prädiktiven Biomarkern, die
eine Individualisierung der Therapie hinsichtlich Therapieansprechen und
Toxizitäten erlauben.
In der vorliegenden Promotionsarbeit wurden Proben von 207 der 474 Patienten,
die in eine prospektive randomisierte zweiarmige Phase-II/III-Studie (FUFOX
versus CAPOX) der AIO zur Erstlinientherapie des mKRK eingeschlossen wurden
(Porschen et al., 2007), auf das Vorliegen von KRAS- und BRAF-Mutationen
untersucht. Für die Untersuchung lagen Formalin-fixierte paraffin-eingebettete
Gewebeproben vor. Damit wurde ein Subkollektiv von 43,2% des
Gesamtkollektivs der klinischen Studie retrospektiv evaluiert, womit für eine
multizentrische Studie mit vielen beteiligten Studienzentren und Primärpathologien
eine gute Quote erreicht wurde. Die Patientenpopulation mit verfügbaren
Gewebeproben war repräsentativ in Bezug auf das Gesamtpatientenkollektiv aller
474 Patienten. 205 der vorliegenden 207 Gewebeproben konnten erfolgreich
analysiert werden (99%). Die technische Durchführbarkeit erbrachte ein sehr
gutes Ergebnis, wenn man bedenkt, dass die Proben der Primärtumoren zum Teil
bis zu zehn Jahre alt waren.
In 75 Fällen konnte eine KRAS-Mutation nachgewiesen werden (36,6%), wobei
die häufigste Mutation mit 25 Fällen im Kodon 12 gefunden wurde, welche zu

- 42 -
einem Aminosäurewechsel zu Aspartat führte (33,3% der KRAS-Mutationen).
Dieses Ergebnis entspricht der publizierten Literatur, wonach bei KRK eine KRAS-
Gen-Mutation mit einer Häufigkeit von circa 30-40% vorliegt (Bleeker et al., 2001;
Esteller et al., 2001; Bazan et al., 2002; Fransen et al., 2004). Auch in der Studie
von Roth et al. mit 1564 Patienten mit einem KRK wurde eine KRAS-Mutation im
Stadium II mit einer Häufigkeit von 36% und im Stadium III mit einer Häufigkeit von
37% nachgewiesen (Roth et al., 2010). Zudem sind bei KRK über 90% der KRAS-
Mutationen im Kodon 12 und 13 lokalisiert (Bazan et al., 2002).
Bei der Frage, ob eine KRAS-Mutation mit dem Tumorstadium, der Lokalisation
oder dem Grading korreliert ist, ist die Datenlage kontrovers (Cerottini et al., 1998;
Samowitz et al., 2000).
In einer frühen Studie von Oliva et al. wurde kein Zusammenhang zwischen einer
Mutation und dem Tumorstadium gesehen (Oliva et al., 1990). Troungos et al.
wiesen hingegen eine Korrelation zwischen einer vorhandenen KRAS-Mutation
und der Invasionstiefe des Tumors nach. Ein Zusammenhang zwischen einer
Mutation und regionalen Lymphknotenmetastasen wurde aber auch nicht gesehen
(Troungos et al., 1997). In einer relativ neuen Studie von Roth et al. wurden
wiederum keine signifikanten Unterschiede bezüglich des Vorkommens in den
einzelnen Tumorstadien gefunden. Ein mutiertes KRAS kam im Stadium II mit
einer Häufigkeit von 36% und im Stadium III mit 37% bei 1564 Gewebeproben der
adjuvanten PETACC3-Studie von van Cutsem et al. mit 3278 Patienten vor (Roth
et al., 2010).
Eine Reihe von Studien deuten auf einen negativen prognostischen Wert einer
KRAS-Mutation hin. So untersuchten Ahnen et al. 66 Patienten mit einem
Karzinom im UICC-Stadium II und 163 Patienten mit einem UICC-Stadium III-
Karzinom. Sie wiesen ein verkürztes 7-Jahres-Gesamtüberleben bei vorhandenen
KRAS-Mutationen im UICC-Stadium II nach (58% bei KRAS-Mutationen vs. 86%
in der KRAS-Wildtyp-Gruppe). Die Hazard ratio (HR) bezüglich des Sterbens lag
bei 4,5 (95%-KI: 1,7-12,1; p=0,012) (Ahnen et al., 1998). Samowitz et al.
untersuchten 1413 Patienten mit einem KRK und wiesen in 32% der Tumoren eine
KRAS-Mutation nach. Eine KRAS-Mutation im Kodon 13 war verbunden mit einer
erhöhten Mortalität von 40% (95%-KI: 0,95-2,0) (Samowitz et al., 2000). Cerottini
et al. analysierten bei 98 Patienten den Zusammenhang einer KRAS-Mutation und
der Prognose. Sowohl das progressionsfreie (p=0,02) als auch das

- 43 -
Gesamtüberleben (p=0,03) waren signifikant kürzer bei Vorhandensein einer
KRAS-Mutation.
Im Vergleich zu den oben genannten Studien liegen jedoch auch Studien vor, die
keinen oder nur teilweise einen Zusammenhang zwischen dem KRAS-Status und
der Prognose sehen. Beispielsweise wurden in einer Studie von Bouzourene et al.
122 Patienten mit einem sporadischen KRK des Stadiums UICC II auf das
Vorliegen einer KRAS- oder p53-Mutation untersucht (Bouzourene et al., 2000).
57 Patienten (38%) wiesen eine KRAS-Mutation im Kodon 12 oder 13 auf. Das
Vorliegen oder die Abwesenheit einer KRAS-Mutation war nicht mit der Prognose
korreliert (p=0,703). In einer Studie von Winder et al. wurden 342 Patienten mit
einem KRK untersucht. Eine KRAS-Mutation wurde mit einer Häufigkeit von 28%
nachgewiesen und es konnte gezeigt werden, dass Patienten mit einer Mutation
im Kodon 12 (G12V-Mutation) ein signifikant kürzeres Gesamtüberleben
aufwiesen als Patienten mit einem Wildtyp (HR=2,56 (1,15-5,69)). Bei Nachweis
anderer Mutationen als der Mutation G12V hingegen zeigte sich ein verbessertes
Gesamtüberleben im Vergleich zu Wildtyp-Patienten (HR=0,44 (0-0,99)) (Winder
et al., 2009). Lee et al. untersuchten 64 Patienten mit einem KRK. 4 dieser
Patienten hatten ein Karzinom im UICC Stadium I, 34 Patienten wiesen das UICC
Stadium II auf, 21 Patienten das Stadium III und in 5 KRK wurde das Stadium IV
nachgewiesen. 13 Tumoren (20%) zeigten eine KRAS-Mutation. In dieser Studie
konnte ein Zusammenhang bei den Stadien I und II zwischen einer KRAS-
Mutation und einem verkürzten Gesamtüberleben aufgezeigt werden (p=0,003).
Das relative Mortalitätsrisiko bei den Stadien I und II betrug 7,74 (95%-KI: 1,72-
34,89). Bei den UICC-Stadien III und IV war hingegen keine Korrelation zum
Gesamtüberleben nachzuweisen (Lee et al., 1996).
In Bezug auf den prädiktiven Nutzen einer KRAS-Mutation gibt es in der Literatur
ebenfalls unterschiedliche Meinungen. In der vorliegenden Promotionsarbeit
konnte das Therapieansprechen in Bezug auf FUFOX oder CAPOX bei 199
Patienten evaluiert werden. Da in dem klinischen Teil der zugrunde liegenden
Studie von Porschen et al. als Hauptergebnis gezeigt werden konnte, dass eine
Erstlinientherapie mit CAPOX bei Patienten mit einem mKRK der Gabe von
FUFOX in Bezug auf das progressionsfreie Überleben nicht unterlegen ist,
konnten für die vorliegende Auswertung beide Therapiearme zusammen
ausgewertet werden.

- 44 -
So gibt es in der Literatur kontroverse Ergebnisse bezüglich des Ansprechens auf
eine adjuvante 5-FU-basierte Chemotherapie bei Vorliegen einer KRAS-Mutation.
Während in einigen Studien keine Assoziation zwischen dem klinischen Benefit
und einer KRAS-Mutation bei adjuvant behandelten KRK gefunden wurde (Bleeker
et al., 2001; Westra et al., 2005; Fuchs et al., 2009), zeigten andere Autoren einen
deutlich besseren Profit der Tumoren mit KRAS-Wildtyp im Vergleich zu einer
KRAS-Mutation (Ahnen et al., 1998), was auch dem Ergebnis dieser
Promotionsarbeit entspricht. So untersuchten Ahnen et al. 66 KRK des Stadiums II
und 163 Stadium III-Tumoren, welche mit Levamisol oder 5-FU plus Levamisol
adjuvant therapiert wurden. In 41% der Tumoren konnte eine KRAS-Mutation
gefunden werden. Im Tumorstadium III beeinflusste eine adjuvante Therapie mit
Levamisol und 5-FU das 7-Jahres-Überleben bei Patienten mit Wildtyp-KRAS
positiv im Vergleich zu KRAS-mutierten Tumoren (76 vs. 44%, HR: 0,4; 95%-KI:
0,2-0,8). Patienten mit einer KRAS-Mutation sowie Patienten mit einem Tumor im
Stadium II profitierten nicht von einer adjuvanten Therapie. In einer neueren Arbeit
der Quasar Collaborative Group wurden 3239 Patienten mit einem KRK im
Stadium II generell in Hinblick auf das Ansprechen einer Therapie mit Fluorouracil
und Folinsäure untersucht. Sie erhielten entweder Fluorouracil und Folinsäure
(n=1622) oder wurden der reinen Beobachtung (n=1617) unterzogen. Es zeigte
sich eine signifikant höhere Sterberate bei der reinen Beobachtung (370
Todesfälle vs. 311 Todesfälle, 95%-KI: 0,70-0,95, p=0,008) sowie eine signifikant
höhere Rezidivrate bei den Patienten, die keine Chemotherapie erhielten (359
Rezidive vs. 293 Rezidive, 95%-KI: 0,67-0,91, p=0,001) im Vergleich zu Patienten,
die mit Fluorouracil und Folinsäure behandelt wurden (Quasar Collaborative
Group, 2007). Wird in der CRYSTAL-Studie das Gesamtüberleben betrachtet,
sieht man einen ähnlichen Tenor wie in der Studie von Ahnen et al. Es zeigte sich
bei einer KRAS-Mutation und einer alleinigen FOLFIRI-Therapie ein verkürztes
Gesamtüberleben im Vergleich zu Patienten mit KRAS-Wildtyp (17,7 vs. 21,0
Monate) (van Cutsem et al., 2009). Zhang et al. führten eine Studie mit 173
Patienten durch, die in einer Phase II-Studie mit Oxaliplatin und 5-FU behandelt
wurden. Patienten mit einem Wildtyp-KRAS-Status wiesen eine Ansprechrate von
27% auf, Patienten mit einer KRAS-Mutation zeigten nur eine Ansprechrate von
7% (p=0,26). Das mediane progressionsfreie Überleben betrug bei den Patienten
mit KRAS-Wildtyp 5,2 Monate und bei Patienten mit KRAS-Mutation 3,1 Monate
(p=0,007). Eine Korrelation zwischen dem KRAS-Mutationsstatus und dem

- 45 -
Gesamtüberleben bestand hingegen nicht (p=0,18) (Zhang et al., 2009). In dieser
Promotionsarbeit konnte bei den Tumoren mit KRAS-Mutation ebenfalls eine
signifikant geringere Ansprechrate als bei Tumoren ohne eine KRAS-Mutation
nachgewiesen werden (44,4% vs. 63,0%, p=0,012). Auch wenn die BRAF-
Mutationen zusammen mit den KRAS-Mutationen betrachtet werden, bleibt der
signifikante Unterschied etwas abgeschwächt weiterhin erhalten (46,4% vs.
63,5%, p=0,021). Jedoch sollte man berücksichtigen, dass eine BRAF V600E-
Mutation nur in 13 Tumoren von 130 Tumoren (10,0%) mit Wildtyp-KRAS
nachgewiesen werden konnte. Gemäß der CRYSTAL-Studie, allerdings entgegen
der Studie von Zhang et al., zeigte sich in dieser Promotionsarbeit auch ein
tendenziell besseres medianes Gesamtüberleben bei Patienten mit KRAS-Wildtyp
(18,9 Monate, 95%-KI: 14,7-17,6 Monate; p=0,085). Bei allen Patienten, deren
Tumoren weder eine KRAS- noch eine BRAF-Mutation aufzeigten, betrug das
Gesamtüberleben 20,8 Monate (95%-KI: 15,6-26,1). War eine der beiden
Mutationen vorhanden, fiel das Gesamtüberleben signifikant kürzer aus (15,6
Monate, 95%-KI: 12,4-18,8; p=0,013). Somit zeigte sich, dass sowohl in den oben
genannten Studien als auch in dieser Promotionsarbeit der KRAS-Status als ein
wichtiger prognostischer Marker in Bezug auf eine Fluoropyrimidin-haltige bzw.
Oxaliplatin-haltige Therapie heraus gearbeitet werden konnte.
Die sog. OPUS-Studie spiegelt ein anderes Ergebnis als diese Promotionsarbeit
wider. Wird in der OPUS-Studie nur der Therapiearm mit FOLFOX betrachtet (73
Patienten der 134 KRAS-Wildtyp-Patienten und 47 Patienten mit einer KRAS-
Mutation erhielten eine FOLFOX-Therapie), profitierten Patienten mit einer KRAS-
Mutation bezüglich des progressionsfreien Überlebens mehr von der alleinigen
Therapie als Patienten mit KRAS-Wildtyp (KRAS mutiert: 8,6 Monate vs. KRAS
Wildtyp: 7,2 Monate) (Bokemeyer et al., 2009). Auch das Therapieansprechen
zeigte bei Patienten mit einer KRAS-Mutation mit 49% (95%-KI: 34-64%) einen
besseren Trend als bei Patienten mit KRAS-Wildtyp (37%; 95%-KI: 26-49%).
Jedoch muss man auch bei dieser Studie, genau wie bei den Untersuchungen
dieser Promotionsarbeit, die relativ geringe Anzahl an untersuchten Fällen
berücksichtigen. Mögliche Ursachen für die heterogenen Ergebnisse könnten die
Größe der Studien, die verschiedenen Patientenpopulationen bei Einschluss und
unterschiedliche Folgetherapien sein.

- 46 -
136 der 205 Patienten dieser Promotionsarbeit erhielten eine Folgetherapie,
wovon 83 Patienten einen KRAS-Wildtyp und 53 Patienten eine KRAS-Mutation
aufwiesen. 80% der Patienten erhielten eine Irinotecan-haltige Folgetherapie und
30% der Patienten erhielten eine Cetuximab-haltige Drittlinientherapie. Es fiel auf,
dass das Gesamtüberleben zunächst sowohl bei KRAS-Wildtyp-Patienten und der
Patientengruppe mit mutiertem KRAS annähernd ähnlich war, sich jedoch nach
circa 15 Monaten zugunsten der KRAS-Wildtyp-Tumoren separierte. Durch diese
Ergebnisse liegt der Schluss nahe, dass der wesentliche prognostische Effekt der
KRAS- und BRAF-Mutationen durch Folgetherapien verursacht wird und das
Vorhandensein- bzw. Nichtvorhandensein einer KRAS-Mutation einen Einfluss auf
die Folgetherapie nehmen könnte.
Bei Patienten ohne eine Folgetherapie zeigte sich bezüglich des
Gesamtüberlebens ab Progress der Erstlinientherapie innerhalb der Studie kein
wesentlicher Unterschied. Bei Patienten, die mit der Irinotecan-haltigen
Zweitlinientherapie behandelt wurden, zeigte sich ein Trend zu einem günstigeren
Postprogressionsüberleben in der KRAS-Wildtyp-Gruppe (p=0,072). Ein signi-
fikanter Unterschied konnte jedoch nicht nachgewiesen werden.
In der PETACC 3-Studie von van Cutsem et al. wurde der Effekt von der Zugabe
von Irinotecan zu der adjuvanten Therapie mit Fluorouracil und Leucovorin
(LV5FU2) bei 2094 Patienten mit KRK des Stadiums III untersucht. Es zeigte sich
kein signifikanter Unterschied beim 5-Jahres-progressionsfreien Überleben
(Irinotecan/LV5FU2: 56,7%, LV5FU2 alleine: 54,3%; p=0,106). Bezüglich des 5-
Jahres-Gesamtüberlebens zeigten sich ebenfalls keine signifikanten Unterschiede
(Irinotecan/LV5FU2: 73,6%, LV5FU2 alleine: 71,3%; p=0,94) (Van Cutsem et al.,
2009).
Zahlreiche neuere Studien nutzten molekularbiologische Verfahren, um
verschiedene Gene und ihre Rolle als prognostische und prädiktive Marker in
mKRK zu evaluieren. Hierdurch konnte der Mutationsstatus von KRAS als
wichtiger prädiktiver Marker für das Ansprechverhalten auf eine anti-EGFR-
Antikörper-Behandlung retrospektiv ermittelt werden (Amado et al., 2008; van
Cutsem et al., 2009; Bokemeyer et al., 2009). Dies konnte auch im Rahmen dieser
Promotionsarbeit, wie im Ergebnisteil beschrieben, bestätigt werden, da sich
zeigte, dass insbesondere Patienten mit KRAS-Wildtyp im Rahmen einer Dritt-
linientherapie von der anti-EGFR-Antikörper-Behandlung mit Cetuximab profi-

- 47 -
tierten. 30% der Patienten erhielten vermutlich aufgrund eines Nichtansprechens
auf eine Irinotecan-haltige Therapie im Rahmen einer Drittlinientherapie
Cetuximab. Bei Patienten ohne eine Folgetherapie zeigte sich bezüglich des
Gesamtüberlebens ab Progress der Erstlinientherapie innerhalb der Studie kein
wesentlicher Unterschied. Insbesondere Patienten mit KRAS-Wildtyp und einer
Cetuximab-haltigen Folgetherapie profitierten hinsichtlich des Gesamtüberlebens
(p=0,087).
So wiesen Amado et al. in ihrer Studie eine 0%ige Ansprechrate auf eine anti-
EGFR-Antikörper-Behandlung mit Panitumumab bei Patienten mit einem mutierten
KRAS-Status nach, während beim KRAS-Wildtyp eine Ansprechrate von 17% zu
beobachten war. Patienten mit einem KRAS-Wildtyp profitierten auch in Bezug auf
das progressionsfreie Überleben bei einer Behandlung mit Panitumumab
gegenüber Patienten mit einem mutierten KRAS (p<0,000199) (Amado et al.,
2008).
In der OPUS-Studie zeigte sich sowohl eine Verbesserung der Ansprechrate (61%
vs. 37%, p=0,011) als auch eine Verbesserung des progressionsfreien Überlebens
(7,7 Monate vs. 7,2 Monate, HR=0,57; p=0,163) bei KRAS-Wildtyp-Tumoren,
wenn Cetuximab zu der FOLFOX-Therapie hinzugefügt wurde (FOLFOX, n=168
vs. FOLFOX+Cetuximab, n=169) (Bokemeyer et al., 2009). Bei den Karzinomen
mit einer KRAS-Mutation hingegen zeigte sich im Therapiearm mit
Cetuximab+FOLFOX ein tendenziell schlechteres Ansprechen im Vergleich zur
alleinigen Chemotherapie (33% vs. 49%, p=0,106).
Zu einem ähnlichen Ergebnis gelangte man in der CRYSTAL-Studie, in welcher
599 Patienten mit einem mKRK mit dem FOLFIRI-Protokoll und weitere 599
Patienten mit FOLFIRI plus Cetuximab behandelt wurden (van Cutsem et al.,
2009). Die Patienten profitierten auch hier von der Gabe von Cetuximab. So zeigte
sich bei der alleinigen Gabe von FOLFIRI ein signifikant kürzeres
progressionsfreies Überleben gegenüber dem FOLFIRI plus Cetuximab-Arm (8,0
vs. 8,9 Monate, p=0,048). In Bezug auf das Gesamtüberleben ergab sich kein
signifikanter Unterschied (19,9 vs. 18,6 Monate, p=0,31). Auch in dieser Studie
war der Therapieerfolg abhängig von dem KRAS-Status. Bei Patienten mit einem
Wildtyp-KRAS zeigte sich ein verlängertes progressionsfreies Überleben bei der
Zugabe von Cetuximab (9,9 vs. 8,7 Monate; HR=0,68; 95%-KI: 0,50-0,94). Auch
bezüglich des Gesamtüberlebens profitierten Patienten mit einem KRAS-Wildtyp-
Tumor tendenziell von der Therapie FOLFIRI plus Cetuximab (24,9 vs. 21,0

- 48 -
Monate, HR=0,84; 95%-KI: 0,64-1,11). Werden nur Patienten mit KRAS-
Mutationen betrachtet, bestand sowohl beim progressionsfreien Überleben
(FOLFIRI+Cetuximab: 7,6 Monate vs. FOLFIRI: 8,1 Monate; p=0,07) als auch bei
dem Gesamtüberleben (17,5 vs. 17,7 Monate bei FOLFIRI alleine; p=0,44) kein
Vorteil durch die Addition von Cetuximab. Wird das progressionsfreie und
Gesamtüberleben in Bezug auf den KRAS-Status verglichen, fällt auf, dass
Patienten mit einem KRAS-Wildtyp mehr von der Zugabe von Cetuximab
profitierten als Patienten mit einer KRAS-Mutation (PFS: KRAS-mutiert: 7,6
Monate vs. KRAS-Wildtyp: 9,9 Monate; OS: KRAS mutiert: 17,5 Monate vs.
KRAS-Wildtyp: 24,9 Monate).
Aus diesen Studien wird ersichtlich, dass nur KRAS-Wildtyp-Patienten von einer
zusätzlichen Therapie mit einem anti-EGFR-Antikörper profitieren (Dempke et al.,
2010). Hingegen besteht bei einer vorhandenen KRAS-Mutation kein Vorteil in
Bezug auf das progressionsfreie oder Gesamtüberleben durch die Zugabe von
Cetuximab oder Panitumumab. In neueren Studien allerdings konnten die bisher
aufgeführten Ergebnisse und somit der nachgewiesene Profit von der
Drittlinientherapie der Patienten dieser Promotionsarbeit nicht bestätigt werden.
Sowohl in der COIN-Studie (Maughan et al., 2010) als auch in der Nordic-Studie
(Tveit et al., 2011) wurde keine Verbesserung des Überlebens durch die Zugabe
von Cetuximab bei Patienten mit Wildtyp-KRAS erreicht. Eine Ursache dafür
können verschiedene Chemotherapiekombinationen sein. Cetuximab scheint mit
infusionalem 5-FU und mit Irinotecan besser synergistisch zu wirken als mit
oralem 5-FU und/oder Oxaliplatin (Maughan et al., 2010; Tveit et al., 2011).
Dieses sehr aktuelle Thema wird derzeit von einer Reihe von Studien adressiert.
Neben dem KRAS-Mutationsstatus wurden die Tumoren auf BRAF-Mutationen
untersucht. In dieser Doktorarbeit wurden ausschließlich BRAF V600E-Mutationen
untersucht, da in Studien gezeigt werden konnte, dass die V600E-Mutation eine
sogenannte Hotspot-Mutation ist, d.h., dass das BRAF in der überwiegenden Zahl
der Fälle an dieser Stelle mutiert ist (Kadiyska et al., 2007; Tan et al., 2008).
Es wurden ausschließlich Tumoren mit einem KRAS-Wildtyp auf eine BRAF
V600E-Mutation untersucht, da nachgewiesen werden konnte, dass eine KRAS-
und BRAF-Mutation nicht gleichzeitig vorkommen (Yoshitake et al., 2007). Im
Rahmen dieser Promotionsarbeit wurde in 13 Tumoren von 130 Tumoren (10,0%)
mit Wildtyp-KRAS eine V600E-BRAF-Mutation nachgewiesen, somit trat eine

- 49 -
V600E-BRAF-Mutation mit einer Häufigkeit von 6,3% aller 205 Patienten auf.
Dieses Ergebnis ist mit den bisherigen veröffentlichten Ergebnissen, die ein
ähnliches Patientenkollektiv untersuchten, zu vergleichen. In der Literatur wird ein
Vorkommen einer BRAF-Mutation bei KRK von 5 bis 10% beschrieben (Davies et
al., 2002; Fransen et al., 2004; Di Nicolantonio et al., 2008; Roth et al., 2010). In
der Studie von Di Nicolantonio et al. wurde bei 113 Patienten mit einem mKRK in
9,7% eine BRAF-Mutation nachgewiesen (Di Nicolantonio et al., 2008). Ein
mutiertes BRAF trat in 11 von 79 Patienten mit einem KRAS-Wildtyp auf (13,9%).
In einer Studie von Yoshitake et al. wurde eine BRAF-Mutation in 9,3% (4 von 43)
der Tumoren nachgewiesen. 87 der 205 Patienten (42,4%) wiesen entweder eine
KRAS- oder eine BRAF-Mutation auf (Yoshitake et al., 2007).
Bezüglich des BRAF-Status zeigte sich in dieser Promotionsarbeit keine
Korrelation mit der Remission oder der Krankheitskontrollrate. Allerdings wurden
Studien veröffentlicht, bei denen eine Beziehung zwischen einem mutierten BRAF
und einem schlechteren Gesamtüberleben (Tran et al., 2010) oder einem höheren
T-Stadium oder Grading-Stadium (Aust et al., 2010) dargestellt wurde. Auch
gemäß den Ergebnissen der CAIRO2-Studie sei eine BRAF-Mutation mit einem
verringerten progressionsfreien Überleben korreliert (Tol et al., 2009).
In der FOCUS-Studie zeigte sich, dass eine KRAS- oder BRAF-Mutation zwar mit
einem schlechteren Gesamtüberleben (95%-KI: 1,20-1,65, p˂0,0001) und einem
verringerten progressionsfreien Überleben (95%-KI: 1,00-1,36, p=0,05) zusammen
hänge, durch eine Mutation das Ansprechen auf eine Irinotecan- oder
Oxaliplatintherapie allerdings nicht beeinflusst werde. Somit profitierten Patienten
mit einem mutierten KRAS oder BRAF ebenso von einer solchen Therapie
(Richman et al., 2009). Auch in der Studie von Aust et al. konnte keine Korrelation
zwischen dem Profit einer 5-FU-haltigen Chemotherapie und dem BRAF-Status
nachgewiesen werden (Aust et al., 2010).
Zu derart unterschiedlichen Ergebnissen kann es aufgrund verschieden großer
Kollektive in den Studien und unterschiedlicher Arbeitsbedingungen kommen.
BRAF hat sich mittlerweile als ein bedeutender Prognosefaktor bezüglich des
Gesamtüberlebens im Stadium IV etabliert. Dass wir in dieser Promotionsarbeit zu
einem anderen Ergebnis kommen, hängt mit den geringen Zahlen zusammen. Da
die BRAF-Mutation, wie bereits erwähnt, in der Regel mit einer Häufigkeit von 5-
10% vorkommt, ist es erstrebenswert, insbesondere Studien mit vielen Patienten
durchzuführen, um die Aussagekraft der Ergebnisse weiter zu verbessern. Dass

- 50 -
Ergebnisse bezüglich einer BRAF-Mutation und der Prognose in eine ähnliche
Richtung gehen wie bei KRAS-Mutationen, ist durchaus erklärbar, da mutiertes
BRAF ebenso den onkogenen Weg RAS/RAF/MEK/ERK aktiviert.
In einigen Studien wurden BRAF-Mutationen mit Resistenzen gegenüber anti-
EGFR-Strategien in Verbindung gebracht. Finocchiaro et al. untersuchten
Tumoren von 85 Patienten mit einem mit Cetuximab behandelten mKRK, bei
denen der EGFR- und KRAS-Status bekannt war, unter anderem auf eine BRAF-
Mutation. Eine BRAF-Mutation wurde bei 5% der Patienten nachgewiesen und
keiner dieser Patienten profitierte von der Therapie mit Cetuximab im Vergleich
zum Wildtyp (Progressionszeit 1,2 vs. 4,6 Monate, p=0,09; OS 5,4 vs. 9,8 Monate,
p=0,3) (Finocchiaro et al., 2008). Nicolantonio et al. untersuchten 113 Patienten,
die mit Cetuximab oder Panitumumab behandelt wurden. Keiner der Patienten mit
einer BRAF-Mutation sprach auf eine Therapie mit Cetuximab oder Panitumumab
an, im Vergleich dazu befand sich unter den Therapierespondern kein Patient mit
einer BRAF-Mutation (p=0,029). Patienten mit einer BRAF-Mutation zeigten ein
signifikant kürzeres progressionsfreies Überleben (p=0,011) und ein signifikant
kürzeres Gesamtüberleben (p<0,0001) (Di Nicolantonio et al., 2008). Allerdings
sollte berücksichtigt werden, dass in der Studie nur wenige Fälle untersucht
wurden und dass keine Randomisierung statt fand. Zu einem anderen Ergebnis
gelangten Maughan et al., Bokemeyer et al. und Tol et al., nach welchen der
BRAF-Status kein entscheidender prädiktiver Marker bezüglich des Ansprechens
auf Cetuximab sei (Tol et al., 2009; Bokemeyer at al., 2010; Maughan et al.,
2010). Diese Aussage wird jedoch in dem Artikel von Bokemeyer et al. insofern
eingeschränkt, als dass die Anzahl der BRAF-Mutationen für eine definitive
Aussage möglicherweise zu gering sei. Im Vergleich zu Di Nicolantonio wurde
allerdings eine Randomisierung durchgeführt.
Da sich Mutationen im KRAS-Gen und im BRAF-Gen ausschließen, wird bei
Patienten mit schlechtem Benefit einer anti-EGFR-Therapie und bereits
ausgeschlossener KRAS-Mutation derzeit eine BRAF-Mutationsanalyse
empfohlen, um die Prognose eines Therapieansprechens auf eine anti-EGFR-
Antikörpertherapie beim KRK zu verbessern.

- 51 -
Die potenzielle, in der vorliegenden Arbeit berichtete Oxaliplatin-Resistenz von
KRAS- und BRAF-mutierten Tumoren ist bei Bestätigung für weitere adjuvante
und palliative Therapien von KRK von weitreichender Bedeutung. Oxaliplatin
gehört in die Gruppe der Platin-Derivate. Metabolite der Platin-Derivate
interagieren mit der DNA und bilden Quervernetzungen. Die Platin-DNA-
Verbindungen inhibieren die DNA-Polymerase und wirken somit antineoplastisch.
Es konnte gezeigt werden, dass das ‚Nucleotide Excision Repair Protein‘ ERCC-1
durch mutiertes Ras hochreguliert werden kann. Hochreguliertes ERCC-1 kann
Platin-induzierte DNA-Schädigungen reparieren und die antineoplastische
Wirkung von Platin-Präparaten abschwächen (Youn et al, 2004).
Viguier et al. untersuchten in ihrer Studie von 2005 bei 91 Patienten die
Mechanismen des ERCC-1 und zeigten, dass die Ansprechrate von mKRK auf
Oxaliplatin bei einem herunterregulierten ERCC-1 höher ist als bei einem normal
funktionierenden ERCC-1. Die Ansprechrate auf eine Kombination aus Oxaliplatin
und 5-FU war bei Patienten mit herunter reguliertem ERCC-1 signifikant höher als
bei den anderen Patientengruppen (61,9% vs. 42,3% und 21,4%; p=0,018).
Hingegen zeigte sich kein signifikanter Unterschied bei Patienten, die nur mit 5-FU
oder in Kombination mit Irinotecan behandelt wurden (Viguier et al., 2005).
Ebenso konnten Shirota et al. zeigen, dass das Protein ERCC-1 einen
entscheidenden Einfluss auf das Ansprechen einer Oxaliplatin-Therapie nimmt.
Für die Studie wurden die Patienten mit einer 5-FU/Oxaliplatin-haltigen
Zweitlinientherapie therapiert. Patienten mit einer ERCC-1-Expression ≤4,9*0,001
(40 von 50 Patienten) hatten ein medianes Überleben von 10,2 Monaten.
Patienten mit einer ERCC-1-Expression ≥4,9*0,001 zeigten ein medianes
Überleben von 1,9 Monaten (p˂0,001) (Shirota et al., 2001).
Im International Lung Cancer Trial (ILCT) wurden retrospektiv verschiedene
prognostische und prädiktive Biomarker in der adjuvanten Therapie des NSCLC
mit einer Platin-basierten 2er-Kombinationstherapie analysiert. 1867 Patienten mit
einem Zustand nach Resektion eines nichtkleinzelligen Lungenkarzinoms wurden
entweder mit einer Kombination aus Cisplatin und einem anderen
Chemotherapeutikum behandelt (932 Patienten) oder der reinen therapeutischen
Beobachtung unterzogen. Patienten, die eine Chemotherapie erhielten, hatten ein
signifikant längeres 5-Jahres-Überleben (44,5% vs. 40,4%, 95%-KI: 0,76- 0,98,
p˂0,03) und ein höheres progressionsfreies Überleben (39,4% vs. 34,3%, 95%-KI:
0,74-0,94, p=0,003) als Patienten ohne eine Chemotherapie (ILCT, 2004). In einer

- 52 -
darauf folgenden Studie von Olaussen et al. (Olaussen et al., 2006) wurde als
Grundlage die zuvor genannte Studie verwendet, um den Einfluss der Expression
von ERCC-1 auf die Prognose beim resezierten nichtkleinzelligen
Lungenkarzinom zu evaluieren. In 335 von 761 untersuchten Tumoren (44%)
wurde eine ERCC-1-Produktion nachgewiesen. Ein Benefit einer Cisplatin-
basierten Chemotherapie korrelierte mit einer Abwesenheit von ERCC-1
(Olaussen et al., 2006).
In weiteren Studien wurde ebenfalls gezeigt, dass bei Patienten mit einem
nichtkleinzelligen Bronchialkarzinom und einer erfolgten Therapie mit Cisplatin
oder anderen Platin-Derivaten das Gesamtüberleben oder das mediane
progressionsfreie Überleben bei ERCC1-positiven Tumoren im Vergleich zu
ERCC1-negativen Tumoren verringert ist (Lord et al., 2002; Ceppi et al., 2006;
Hwang et al., 2008; Schettino et al., 2008; Ota et al., 2009; Azuma et al., 2009;
Lee et al., 2009).
Die vorliegenden Ergebnisse sind die Basis für weitere Untersuchungen, die den
Mechanismus der potenziellen Oxaliplatinresistenz weiter untersuchen sollten. Es
wurde daher eine immunhistochemische Analyse der ERCC1-Expression an
einem Pilotkollektiv durchgeführt. Ein Unterschied bezüglich der ERCC1-
Expression und des Therapieansprechens wurde jedoch nicht gesehen.
Interessant wäre darüber hinaus der Vergleich von mit Irinotecan-behandelten
Patienten (z.B. FOLFIRI), um zu klären, ob der beobachtete Effekt Oxaliplatin-
spezifisch oder Ausdruck einer höheren biologischen Aggressivität von KRAS-
mutierten KRK ist.
KRAS und BRAF sind mittlerweile wichtige prognostische Marker beim KRK.
BRAF hat sich inzwischen als ein bedeutender Prognosefaktor bezüglich des
Gesamtüberlebens im Stadium IV beim KRK etabliert. Der Stellungswert von
KRAS als Prognosefaktor unter einer reinen Kombinationschemotherapie ist noch
nicht abschließend geklärt. Insbesondere bei einer anti-EGFR-Antikörpertherapie
scheint eine Korrelation zwischen mutiertem KRAS und einem schlechteren
Ansprechen zu bestehen. Ergebnisse divergieren jedoch noch immer. Es stellt
sich insbesondere die Frage, ob es bezüglich verschiedener Mutationen in KRAS
Unterschiede gibt bzw. wodurch es zu verschiedenen Interaktionen mit den
Chemotherapien kommt.

- 53 -
Es wird ersichtlich, dass die Forschung bezüglich des KRK und dessen Therapie
noch lange nicht abgeschlossen und von großer Bedeutung ist. Um sowohl die
Studiendurchführbarkeit zu erleichtern als auch die Ergebnisse zu verbessern, ist
für zukünftige Studien die standardisierte, sorgfältige und umfassende Sammlung
von Gewebe- und Blutproben aller Studienpatienten in sog. Biobanken eine
wesentliche Anforderung. Um eine qualitativ hochwertige translationale Forschung
zu ermöglichen, die Kosten für die Durchführung von Studien senken zu können
und um eine bessere Versorgung der Krebspatienten zu gewährleisten, sind gut
geführte Biobanken das wesentliche Werkzeug in der zukünftigen Gegenwart. Auf
diesem Wege kann eine individualisierte Krebsmedizin erreicht werden.

- 54 -
6. Zusammenfassung
Das kolorektale Karzinom ist in Europa die zweithäufigste tumorbedingte
Todesursache. Bei Erstdiagnose weisen etwa 25% der Patienten eine
Fernmetastasierung und 30-60% eine Metastasierung im Verlauf auf. Die
Prognose des mKRK konnte durch die Einführung neuer Chemotherapeutika
deutlich gebessert werden. Eine große Bedeutung hat dabei die Identifikation von
Biomarkern. Die hier untersuchten Gewebeproben stammen von Patienten aus
einer prospektiv randomisierten zweiarmigen Phase-III-Studie der AIO zur
Erstlinientherapie des mKRK. Es wurde die Gabe eines oralen Fluoropyrimidins
(Capecitabin) in Kombination mit Oxaliplatin mit der Applikation von infusionalem
5-FU verglichen und eine Nicht-Unterlegenheit aufgezeigt. Das untersuchte
Teilkollektiv unterschied sich in Bezug auf Alter, Geschlecht, Therapie (FUFOX
bzw. CAPOX) nicht vom Gesamtkollektiv aller Studienpatienten.
Es erfolgte eine Extraktion von DNA aus pseudonymisierten, formalinfixierten,
Paraffin-eingebetteten Tumorgewebeproben (FFPE). Mit Hilfe des QIAmp DNA
Mini Kits wurden sieben KRAS-Mutationen in den Kodons 12 und 13 detektiert. Im
BRAF-Gen wurde gezielt die Hotspot-Mutation V600E-Mutation nachgewiesen. Da
in Studien gezeigt wurde, dass KRAS- und BRAF-Mutationen nicht gleichzeitig in
einem Tumor vorkommen, wurden nur Tumoren ohne eine KRAS-Mutation auf
eine BRAF-Mutation hin untersucht. Es erfolgte eine Korrelation von
Mutationsstatus mit Remissionsverhalten, progressionsfreiem Überleben und
Gesamtüberleben.
Eine KRAS-Mutation war in 75 der 205 (36,6%) verfügbaren Tumoren vorhanden.
Eine BRAF V600E-Mutation wurde in 13 von 130 (10,0%) Tumoren mit Wildtyp-
KRAS nachgewiesen. 87 der 205 Patienten (42,4%) wiesen entweder eine KRAS-
oder eine BRAF-Mutation auf. Patienten mit KRAS-Mutationen zeigten eine
signifikant geringere Remissionsrate als Tumoren ohne KRAS-Mutation (44,4%
vs. 63,0%, p=0,012). Patienten mit KRAS-Wildtyp wiesen ein tendenziell besseres
medianes Gesamtüberleben (18,9 Monate, 95%-KI: 14,7-23,2 Monate) als
Patienten mit einer KRAS-Mutation (16,1 Monate, 95%-KI: 14,7-17,6 Monate)
(p=0,085) auf. Das Gesamtüberleben der Patienten mit KRAS- oder BRAF-Wildtyp
betrug 20,8 Monate (95%-KI: 15,6-26,1). War eine KRAS- oder BRAF-Mutation
vorhanden, war das Gesamtüberleben signifikant kürzer (15,6 Monate, 95%-KI:
12,4-18,8; p=0,013). Das mediane PFS zeigte bei Patienten mit KRAS- und

- 55 -
BRAF-Wildtyp (7,6 Monate, 95%-KI: 6,5-8,6) und Patienten mit KRAS- oder
BRAF-Mutation (7,5 Monate, 95%-KI: 4,9-10,0) keinen signifikanten Unterschied
(p=0,89). 136 der 205 Patienten erhielten eine Folgetherapie. 80% der Patienten
wurden mit einer Irinotecan-haltigen Zweitlinientherapie behandelt und 30% der
Patienten erhielten im Rahmen einer Drittlinientherapie Cetuximab. Bei Patienten
ohne Folgetherapie zeigte sich bezüglich des Postprogressionsüberlebens kein
wesentlicher Unterschied. Bei den mit Irinotecan behandelten Patienten zeigte
sich ein Trend zu einem günstigeren Postprogressionsüberleben in der KRAS-
Wildtyp-Gruppe (p=0,072). Insbesondere KRAS-Wildtyp-Patienten mit einer
Cetuximab-haltigen Folgetherapie profitierten hinsichtlich des Gesamtüberlebens
(p=0,087).
Patienten mit einer KRAS- oder einer BRAF-Mutation wiesen unter einer
Oxaliplatin- und Fluoropyrimidin-haltigen Erstlinientherapie eine signifikant
geringere Ansprechrate als Patienten mit Wildtyp-Tumoren auf. Auch das
Gesamtüberleben der Patienten mit Mutation war signifikant kürzer. Beim
progressionsfreien Überleben zeigte sich hingegen kein Unterschied.
Die potenzielle Oxaliplatin-Resistenz bei der Ansprechrate von mutierten Tumoren
ist für die weitere Therapie von KRK von großer Bedeutung. Möglicherweise
könnte dies mit der Expression des Reparaturproteins ERCC-1 zusammen-
hängen, das eine Platinresistenz bei verschiedenen Tumoren vermitteln kann. In
Bezug auf eine Folgetherapie und das Gesamtüberleben profitierten Patienten
insbesondere von einer Cetuximab-haltigen Therapie, wenn ein KRAS-Wildtyp
vorlag. Dies bestätigt den bekannten prädiktiven Wert einer KRAS-Mutation für
eine Anti-EGFR Therapie.

- 56 -
7. Literaturverzeichnis
Aaltonen, L.A., Peltomäki, P., Leach, F.S., Sistonen, P., Pylkkänen, L., Mecklin,
J.P., Järvinen, H., Powell, S.M., Jen, J., Hamilton, S.R. (1993). Clues to the
pathogenesis of familial colorectal cancer. Science 260(5109), 812-816
Adson, M.A. (1987). Resection of liver metastases: when is it worth-while? World J
Surg 11(4), 511-520
Ahnen, D.J., Feigl, P., Quan, G., Fenoglio-Preiser, C., Lovato, L.C., Bunn, P.A. Jr.,
Stemmerman, G., Wells, J.D., Macdonald, J.S., Meyskens, F.L. Jr. (1998). Ki-ras
mutation and p53 overexpression predict the clinical behaviour of colorectal
cancer: a Southwest Oncology Group study. Cancer Res 58(6), 1149-1158
Alazzouzi, H., Alhopuro, P., Salovaara, R., Sammalkorpi, H., Järvinen, H., Mecklin,
J.P., Hemminki, A., Schwartz, S. Jr., Aaltonen, L.A., Arango, D. (2005). SMAD4 as
a prognostic marker in colorectal cancer. Clin Cancer Res. 11(7), 2606-11
Almendingen, K., Hofstad, B., Trygg, K., Hoff, G., Hussain, A., Vatn, M. (2001).
Current diet and colorectal adenoms: a case-control study including different sets
of traditionally chosen control groups. Eur J Cancer Prev 10(5), 395-406
Amado, R.G., Wolf, M., Peeters, M., Van Cutsem, E., Siena, S., Freeman, D.J.,
Juan, T., Sikorski, R., Suggs, S., Radinsky, R., Patterson, S.D., Chang, D.D.
(2008). Wild-type KRAS is required for panitumumab efficacy in patients with
metastatic colorectal cancer. JCO 26(10), 2626-34
André, T., Boni, C., Mounedji-Boudiaf, L., Navarro, M., Tabernero, J., Hickish, T.,
Topham, C., Zaninelli, M., Clingan, P., Bridgewater, J., Tabah-Fisch, I., de
Gramont, A. (2004). Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment
for colon cancer. N Engl J Med 350(23), 2343-2351
André, T., Boni, C., Navarro, M., Tabernero, J., Hickish, T., Topham, C., Bonetti,
A., Clingan, P., Bridgewater, J., Rivera, F., de Gramont, A. (2009). Improved
Overall Survival With Oxaliplatin, Fluorouracil, and Leucovorin As Adjuvant

- 57 -
Treatment in Stage II or III Colon Cancer in the MOSAIC Trial. JCO 27(19), 3109-
3116
Aust, D.E., Lutz, M.P., Mauer, M., Popov, I., Baretton, G.B., Bedenne, L., Carrato,
A., Kohne, C. (2010). Lessons from PETACC 2: No prognostic impact of KRAS-
/BRAF-status in stage III colon cancer treated with adjuvant 5-FU monotherapy.
JCO 28(15s), abstr. 3591
Azuma, K., Sasada, T., Kawahara, A., Hattori, S., Kinoshita, T., Takamori, S.,
Ichiki, M., Imamura, Y., Ikeda, J., Kage, M., Kuwano, M., Aizawa, H. (2009).
Expression of ERCC1 and class III beta-tubulin in non-small cell lung cancer
patients treated with a combination of cisplatin/docetaxel and concurrent thoracic
irradiation. Cancer Chemother Pharmacol. 64(3), 565-73
Bazan, V., Migliavacca, M., Zanna, I., Tubiolo, C., Grassi, N., Latteri, M.A., La
Farina, M., Albanese, I., Dardanoni, G., Salerno, S., Tomasino, R.M., Labianca,
R., Gebbia, N., Russo, A. (2002). Specific codon 13 KRAS mutations are
predicitve of clinical outcome in colorectal cancer patients, whereas codon 12
KRAS mutations are associated with mucinous histotype. Annals of Oncology
13(9), 1438-1446
Bingham, S.A., Day, N.E., Luben, R., Ferrari, P., Slimani, N., Norat, T., Clavel-
Chapelon, F., Kesse, E., Nieters, A., Boeing, H., Tjønneland, A., Overvad, K.,
Martinez, C., Dorronsoro, M., Gonzalez, C.A., Key, T.J., Trichopoulou, A., Naska,
A., Vineis, P., Tumino, R., Krogh, V., Bueno-de-Mesquita, H.B., Peeters, P.H.,
Berglund, G., Hallmans, G., Lund, E., Skeie, G., Kaaks, R., Riboli, E. (2003).
Dietary fibre in food and protection against colorectal cancer in the European
Prospective Investigation into Cancer and Nutrition (EPIC): an observational study.
Lancet 361(9368), 1496-501
Bleeker, W.A., Hayes, V.M., Karrenbeld, A., Hofstra, R.M., Verlind, E., Hermans,
J., Poppema, S., Buys, C.H., Plukker, J.T. (2001). Prognostic significance of
KRAS and TP53 mutation in the role of adjuvant chemotherapy on survival in
patients with Dukes C colon cancer. Dis Colon Rectum 44(3), 358-363

- 58 -
Bokemeyer, C., Bondarenko, I., Hartmann, J.T., de Braud, F., Schuch, G., Zubel,
A., Celik, I., Schlichting, M., Koralewski, P. (2011). Efficacy according to biomarker
status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal
cancer: the OPUS study. Ann Oncol 22(7), 1535-46
Bokemeyer, C., Bondarenko, I., Makhson, A., Hartmann, J.T., Aparicio, J., de
Braud, F., Donea, S., Ludwig, H., Schuch, G., Stroh, C., Loos, A.H., Zubel, A.,
Koralewski, P. (2009). Fluorouracil, eucovorin, and oxaliplatin with and without
cetuximab in the first-line treatment of metastatic colorectal cancer. JCO 27(5),
663-71
Bokemeyer, C., Kohne, C., Rougier, P., Stroh, C., Schlichting, M., Van Cutsem, E.
(2010). Cetuximab with chemotherapy (CT) as first-line treatment for metastatic
colorectal cancer (mCRC): Analysis of the CRYSTAL and OPUS studies according
to KRAS and BRAF mutation status. JCO 28(15s), abstr. 3506
Boulay, J.L., Mild, G., Lowy, A., Reuter, J., Lagrange, M., Terracciano, L., Laffer,
U., Herrmann, R., Rochlitz, C. (2002). SMAD4 is a predictive marker for 5-
fluorouracil-based chemotherapy in patients with colorectal cancer. Br J Cancer.
87(6), 630-4
Bouzourene, H., Gervaz, P., Cerottini, J.P., Benhattar, J., Chaubert, P., Saraga,
E., Pampallona, S., Bosman, F.T., Givel, J.C. (2000). P53 and Ki-ras as prognostic
factors for Dukes’ stage B colorectal cancer. Eur J Cancer 36(8), 1008-15
Braun, M.S., Richman, S.D., Quirke, P., Daly, C., Adlard, J.W., Elliott, F., Barrett,
J.H., Selby, P., Meade, A.M., Stephens, R.J., Parmar, M.K., Seymour, M.T.
(2008). Predictive Biomarkers of Chemotherapy Efficacy in Colorectal Cancer:
Results From the UK MRC FOCUS Trial. JCO 26(16), 2690-2698
Carethers, J.M., Smith, E.J., Behling, C.A., Nguyen, L., Tajima, A., Doctolero,
R.T., Cabrera, B.L., Goel, A., Arnold, C.A., Miyai, K., Boland, C.R. (2004). Use of
5-fluorouracil and survival in patients with microsatellite-unstable colorectal
cancer. Gastroenterology 126(2), 394-401

- 59 -
Ceppi, P., Volante, M., Novello, S., Rapa, I., Danenberg, K.D., Danenberg, P.V.,
Cambieri, A., Selvaggi, G., Saviozzi, S., Calogero, R., Papotti, M., Scagliotti, G.V.
(2006). ERCC1 and RRM1 gene expression but not EGFR are predictive of
shorter survival in advanced non small cell lung cancer treated with cisplatin and
gemcitabine. Ann. Oncol. 17(12), 1818-1825
Cerottini, J.P., Caplin, S., Saraga, E., Givel, J.C., Benhattar, J. (1998). The type of
KRAS mutation determines prognosis in colorectal cancer. Am J Surg 175(3), 198-
202
Citarda, F., Tomaselli, G., Capocaccia, R., Barcherini, S., Crespi, M. (2001).
Efficacy in standard clinical practice of colonoscopic polypectomy in reducing
colorectal cancer incidence. Gut 48(6), 812-815
Cunningham, J.M., Christensen, E.R., Tester, D.J., Kim, C.Y., Roche, P.C.,
Burgart, L.J., Thibodeau, S.N. (1998). Hypermethylation of the hMLH1 promoter in
colon cancer with microsatellite instability. Cancer Res 58(15), 3455-3460
Cunningham, D., Humblet, Y., Siena, S., Khayat, D., Bleiberg, H., Santoro, A.,
Bets, D., Mueser, M., Harstrick, A., Verslype, C., Chau, I., Van Cutsem, E. (2004).
Cetuxmab monotherapy and cetuximab plus irinotecan in irinotecan-refractory
metastatic colorectal cancer. N EnglJ Med 351(4), 337-45
Cunningham, J., Lust, J.A., Schaid, D.J., Bren, G.D., Carpenter, H.A., Rizza, E.,
Kovach, J.S., Thibodeau, S.N. (1992). Expression of p53 and 17p allelic loss
incolorectal carcinoma. Cancer Res 52(7), 1974-1980
Cunningham, D., Pyrhönen, S., James, R.D., Punt, C.J., Hickish, T.F., Heikkila, R.,
Johannesen, T.B., Starkhammar, H., Topham, C.A., Awad, L., Jacques, C., Herait,
P. (1998). Randomised trial of irinotecan plus supportive care versus supportive
care alone after fluorouracil failure for patients with metastatic colorectal cancer.
Lancet 352(9138), 1413-1418
Davies, H., Bignell, G.R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J.,
Woffendin, H., Garnett, M.J., Bottomley, W., Davis, N., Dicks, E., Ewing, R., Floyd,

- 60 -
Y., Gray, K., Hall, S., Hawes, R., Hughes, J., Kosmidou, V., Menzies, A., Mould,
C., Parker, A., Stevens, C., Watt, S., Hooper, S., Wilson, R., Jayatilake, H.,
Gusterson, B.A., Cooper, C., Shipley, J., Hargrave, D., Pritchard-Jones, K.,
Maitland, N., Chenevix-Trench, G., Riggins, G.J., Bigner, D.D., Palmieri, G.,
Cossu, A., Flanagan, A., Nicholson, A., Ho, J.W., Leung, S.Y., Yuen, S.T., Weber,
B.L., Seigler, H.F., Darrow, T.L., Paterson, H., Marais, R., Marshall, C.J., Wooster,
R., Stratton, M.R., Futreal, P.A. (2002). Mutations of the BRAF gene in human
cancers. Nature 417(6892), 906-7
De Gramont, A., Boni, C., Navarro, M., Tabernero, J., Hickish, T., Topham, C.,
Bonetti, A., Clingan, P., Lorenzato, C., André, T. (2007). Oxaliplatin/5FU/LV in
adjuvant colon cancer: Updated efficacy results of the MOSAIC trial, including
survival, with a median follow-up of six years. Journal of Clinical Oncology, No.
18S, abstr. 4007
De Gramont, A., Figer, A., Seymour, M., Homerin, M., Hmissi, A., Cassidy, J.,
Boni, C., Cortes-Funes, H., Cervantes, A., Freyer, G., Papamichael, D., Le Bail,
N., Louvet, C., Hendler, D., de Braud, F., Wilson, C., Morvan, F., Bonetti, A.
(2000). Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment
in advanced colorectal cancer. J Clin Oncol 18(16), 2938-2947
Dempke, W.C. und Heinemann, V. (2010). Ras mutational status is a biomarker
for resistance to EGFR inhibitors in colorectal carcinoma. Anticancer Res. 30(11),
4673-7
Di Nicolantonio, F., Martini, M., Molinari, F., Sartore-Bianchi, A., Arena, S., Saletti,
P., De Dosso, S., Mazzucchelli, L., Frattini, M., Siena, S., Bardelli, A. (2008). Wild-
type BRAF is required for response to panitumumab or cetuximab in metastatic
colorectal cancer. JCO 26(35), 5105-12
Douillard, J.Y., Cunningham, D., Roth, A.D., Navarro, M., James, R.D., Karasek,
P., Jandik, P., Iveson, T., Carmichael, J., Alakl, M., Gruia, G., Awad, L., Rougier,
P. (2000). Irinotecan combined with fluorouracil compared with fluorouracil alone
as first-line treatment for metastatic colorectal cancer: a multicentre randomised
trial. Lancet 355(9209), 1041-1047

- 61 -
Eaden, J. (2004). Review article: colorectal carcinoma and inflammatory bowel
disease. Aliment Pharmacol Ther. 20(Suppl 4), 24-30
Egan, S.E., Giddings, B.W., Brooks, M.W., Buday, L., Sizeland, A.M., Weinberg,
R.A. (1993). Association of Sos Ras exchange protein with Grb2 is implicated in
tyrosine kinase signal transduction and transformation. Nature 363(6424), 45-51
Esteller, M., González, S., Risques, R.A., Marcuello, E., Mangues, R., Germà,
J.R., Herman, J.G., Capellà, G., Peinado, M.A. (2001). KRAS and p16 Aberrations
Confer Poor Prognosis in Human Colorectal Cancer. J Clin Oncol. 19(2), 299-304
Finkelstein, S.D., Sayegh, R., Bakker, A., Swalsky P. (1993). Determination of 64
tumor aggressiveness in colorectal cancer by KRAS-2 analysis. Arch Surg 128(5),
526-531
Finocchiaro, G., Cappuzzo, F., Rossi, E., Toschi, L., Janne, P.A., Roncalli, M.,
Ligorio, C., Rimassa, L., Santoro, A., Varella-Garcia M. (2008). Insuline like growth
factor receptor-1 (IGFR-1), MET, and BRAF and primary resistance to cetuximab
therapy in colorectal cancer patients. ASCO Vol 26(15s), abstr. 4135
Forslund, A., Lonnroth, C., Andersson, M., Brevinge, H., Lundholm, K. (2001).
Mutations and allelic loss of p53 in primary tumor DNA from potentially cured
patients with colorectal carcinoma. J Clin Oncol 19(11), 2829-2836
Fransén, K., Klintenäs, M., Osterström, A., Dimberg, J., Monstein, H.J.,
Söderkvist, P. (2004). Mutation analysis of the BRAF, ARAF and RAF-1 genes in
human colorectal adenocarcinomas. Carcinogenesis 25(4), 527-33
Friedenreich, C.M. und Orenstein, M.R. (2002). Physical activity and cancer
prevention: etiologic evidence and biological mechanisms. J Nutr 132(11), 3456-
3464

- 62 -
Fuchs, C.S., Giovannucci, E.L., Colditz, G.A., Hunter, D.J., Speizer, F.E., Willett,
W.C. (1994). A prospective study of family history and the risk of colorectal cancer.
N Engl J Med 331(25), 1669-74
Fuchs, C.S., Giovannucci, E.L., Colditz, G.A., Hunter, D.J., Stampfer, M.J.,
Rosner, B., Speizer, F.E., Willett, W.C. (1999). Dietry fiber and the risk of
colorectal cancer and adenoma in woman. N Engl J Med 340(3), 169-179
Fuchs, C., Ogino, S., Meyerhardt, J.A., Irahara, N., Niedzwiecki, D., Hollis, D.,
Saltz, L.B., Mayer, R.J., Bertagnolli; M.M. (2009). KRAS mutation, cancer
recurrence, and patient survival in stage III colon cancer: Findings from CALGB
89803. JCO 27(15s), abstr. 4037
Gesellschaft der epidemiologischen Krebsregister in Deutschland e.V. (2008).
Krebs in Deutschland Häufigkeiten und Trends
Geoghegan, J.G. und Scheele, J. (1999). Treatment of colorectal liver metastases.
British Journal of Surgery 86(2), 158-169
Giacchetti, S., Perpoint, B., Zidani, R., Le Bail, N., Faggiuolo, R., Focan, C.,
Chollet, P., Llory, J.F., Letourneau, Y., Coudert, B., Bertheaut-Cvitkovic, F.,
Larregain-Fournier, D., Le Rol, A., Walter, S., Adam, R., Misset, J.L., Lévi, F.
(2000). Phase-III multicenter randomized trial of oxaliplatin added to
chronomodulated fluorouracilleucovorin as first-line treatment of metastatic
colorectal cancer. J Clin Oncol 18(1), 136-147
Giacosa, A., Franceschi, S., La Vecchia, C., Favero, A., Andreatta, R. (1999).
Energy intake, overweight, physical exercise and colorectal cancer risk. Eur J
Cancer Prev 8(Suppl 1), 53-60
Giantonio, B.J., Catalano, P.J., Meropol, N.J., O'Dwyer, P.J., Mitchell, E.P.,
Alberts, S.R., Schwartz, M.A., Benson, A.B. (2005). High-dose bevacizumab
improves survival when combined with FOLFOX4 in previously treated advanced
colorectal cancer: Results from the Eastern Cooperative Oncology Group (ECOG)
study E3200. JCO 23(16s), abstr. 2

- 63 -
Giovannucci, E. (2003). Diet, body weight, and colorectal cancer: a summary of
the epidemiologic evidence. J Womens Health (Larchmt) 12(2), 173-182
Glimelius, B., Hoffman, K., Graf, W., Påhlman, L., Sjödén, P.O. (1994). Quality of
life during chemotherapy in patients with symptomatic advanced colorectal cancer.
Cancer 73(3), 556-562
Goldberg, R.M., Fleming, T.R., Tangen, C.M., Moertel, C.G., Macdonald, J.S.,
Haller, D.G., Laurie, J.A. (1998). Surgery for recurrent colon cancer: strategies for
identifying resectable recurrence and success rates after resection. Ann Intern
Med 129(1), 27-35
Grandy, W.M. (2004). Genomic instability and colon cancer. Cancer and
Metastasis Reviews 23(1-2), 11-27
Greenstein, A.J., Sachar, D.B., Smith, H., Janowitz, H.D., Aufses, A.H. Jr. (1981).
A comparison of cancer risk in Crohn’s disease and ulcerative colitis. Cancer
48(12), 2742-2745
Grothey, A., Sargent, D., Goldberg, R.M., Schmoll, H.J. (2004). Survival of
patients with advanced colorectal cancer improves with the availability of
fluorouracil-leucocorin, irinotecan, and oxaliplatin in the course of treatment. J Clin
Oncol 22(7), 1209-1214
Hemminki, A., Mecklin, J.P., Järvinen, H., Aaltonen, L.A., Joensuu, H. (2000).
Microsatellite instability is a favourable prognostic indicator in patients with
colorectal cancer receiving chemotherapie. Gastroenterology 119(4), 921-928
Heppner, K. und Groden, J. (2000). Biology of the Adenomatous Polyposis Coli
Tumor Supressor. J Clin Oncology 18(9), 1967-1979
Hurwitz, H., Fehrenbacher, L., Cartwright, T., Hainsworth, J., Heim, W., Berlin, J.,
Griffing, S., Novotny, W., Holmgren, E., Kabbinavar, F. (2003). Bevacizumab (a
monoconal antibody to vascular endothelial growth factor) prolongs survival in first

- 64 -
line colorectal cancer (CRC): results of a phase 3 trial of bevacizumab in
combination with bolus IFL (irinotecan, 5-fluorouracil, leucovorin) as first line
therapy in subjects with metatsatic CRC. Proc Am Soc Clin Oncol 22, abstr. 3646
Hwang, I.G., Ahn, M.J., Park, B.B., Ahn, Y.C., Han, J., Lee, S., Kim, J., Shim,
Y.M., Ahn, J.S., Park, K. (2008). ERCC1 expression as a prognostic marker in
N2(+) nonsmall-cell lung cancer patients treated with platinum-based neoadjuvant
concurrent chemoradiotherapy. Cancer 113(6), 1379-86
Ince, W.L., Jubb, A.M., Holden, S.N., Holmgren, E.B., Tobin, P., Sridhar, M.,
Hurwitz, H.I., Kabbinavar, F., Novotny, W.F., Hillan, K.J., Koeppen, H. (2005).
Association of KRAS, b-raf, and p53 status with the treatment effect of
bevacizumab. J Natl Cancer Inst 97(13), 981-989
Issa, J.P. (2008). Colon cancer: it’s CIN or CIMP. Clin Cancer Res. 14(19), 6005-
13
Jonker, D.J., Maroun, J.A. und Kocha, W. (2000). Survival benefit of
chemotherapy in metastatic colorectal cancer: a meta-analysis of randomised
controlled trials. Br J Cancer 82(11), 1789-1794
Jonker, D.J., O'Callaghan, C.J., Karapetis, C.S., Zalcberg, J.R., Tu, D., Au, H.J.,
Berry, S.R., Krahn, M., Price, T., Simes, R.J., Tebbutt, N.C., van Hazel, G.,
Wierzbicki, R., Langer, C., Moore, M.J. (2007). Cetuximab for the treatment of
colorectal cancer. N Engl J Med 357(20), 2040-8
Kabbinavar, F.F., Schulz, J., McCleod, M., Patel, T., Hamm, J.T., Hecht, J.R.,
Mass, R., Perrou, B., Nelson, B., Novotny, W.F. (2005). Addition of bevacizumab
to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results
of a randomized phase III trial. J Clin Oncol. 23(16), 3697-705
Kadiyska, T.K., Konstantinova, D.V., Atanasov, V.R., Kremensky, I.M., Mitev, V.I.
(2007). Frequency and application of the hot spot BRAF gene mutation (p.V600E)
in the diagnostic strategy for Hereditary Nonpolyposis Colorectal Cancer. Cancer
Detect Prev. 31(3), 254-6

- 65 -
Kampman, E., Voskuil, D.W., van Kraats, A.A., Balder, H.F., van Muijen, G.N.,
Goldbohm, R.A., van't Veer, P. (2000). Animal products and KRAS codon 12 and
13 mutations in colon carcinomas. Carcinogenesis 21(2), 307-309
Kim, J.H., Lim, Y.J., Kim, Y.H., Sung, I.K., Shim, S.G., Oh, S.O., Park, S.S., Yang,
S., Son, H.J., Rhee, P.L., Kim, J.J., Rhee, J.C., Choi, Y.H. (2007). Cancer
Epidemiol Biomarkers. Prev. 16(8), 1543-6
Koopman, M., Antonini, N.F., Douma, J., Wals, J., Honkoop, A.H., Erdkamp, F.L.,
de Jong, R.S., Rodenburg, C.J., Vreugdenhil, G., Loosveld, O.J., van Bochove, A.,
Sinnige, H.A., Creemers, G.J., Tesselaar, M.E., Slee, P.H., Werter, M.J., Mol, L.,
Dalesio, O., Punt, C.J. (2007). Sequential versus combination chemotherapy with
capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a
phase III randomised controlled trial. Lancet 370(9582), 135-142
Lee, H.W., Choi, Y.W., Han, J.H., Kim, J.H., Jung, J.H., Jeong, S.H., Kang, S.Y.,
Choi, J.H., Oh, Y.T., Park, K.J., Hwang, S.C., Sheen, S.S. (2009). Expression of
excision repair cross-complementation goup 1 protein predicts poor outcome in
advanced non-small cell lung cancer patients treated with platinum-based doublet
chemotherapy. Lung Cancer 65(3), 377-82
Lee, J.C., Wang, S.T., Lai, M.D., Lin, Y.J., Yang, H.B. (1996). KRAS gene
mutation is a useful predictor of the survival of early stage colorectal cancers.
Anticancer Res. 16(6B), 3839-44
Lièvre, A., Bachet, J.B., Le Corre, D., Boige, V., Landi, B., Emile, J.F., Côté, J.F.,
Tomasic, G., Penna, C., Ducreux, M., Rougier, P., Penault-Llorca, F., Laurent-
Puig, P. (2006). KRAS Mutation Status is Predictive of Response to Cetuximab
Therapy in Colorectal Cancer. Cancer Res 66(8), 3992-3995
Lièvre, A., Rouleau, E., Buecher, B., Mitry, E. (2010). Clinical significance of BRAF
mutations in colorectal cancer. Bull Cancer 97(12), 1441-1452
Lord, R.V., Brabender, J., Gandara, D., Alberola, V., Camps, C., Domine, M.,
Cardenal, F., Sánchez, J.M., Gumerlock, P.H., Tarón, M., Sánchez, J.J.,

- 66 -
Danenberg, K.D., Danenberg, P.V., Rosell, R. (2002). Low ERCC1 expression
correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in
non-small cell lung cancer. Clin Cancer Res. 8(7), 2286-2291
Maitra, A., Molberg, K., Albores-Saavedra, J., Lindberg, G. (2000). Loss of Dpc4
expression in colonic adenocarcinomas correlates with the presence of metastatic
disease. Am J Pathol. 157(4), 1105-11
Maughan, T.S., Adams, R., Smith, C.G., Seymour, M.T., Wilson, R.H., Meade,
A.M., Fisher, D., Madi, A., Cheadle, J., Kaplan, R.S. (2010). Oxaliplatin and
fluoropyrimidine chemotherapy plus or minus cetuximab: The effect of infusional 5-
FU or capecitabine on the outcomes of the MRC COIN trial in advanced colorectal
cancer (ACRC). ASCO, abstr. 402
Mikami, T., Ookawa, K., Shimoyama, T., Fukuda, S., Saito, H., Munakata, A.
(2001). KAI1, CAR, and Smad4 expression in the progression of colorectal tumor.
J Gastroenterol. 36(7), 465-9
Miyaki, M., Iijima, T., Konishi, M., Sakai, K., Ishii, A., Yasuno, M., Hishima, T.,
Koike, M., Shitara, N., Iwama, T., Utsunomiya, J., Kuroki, T., Mori, T. (1999).
Higher frequency of Smad4 gene mutation in human colorectal cancer with distant
metastasis. Oncogene. 18(20), 3098-103
Morrin, M., Kelly, M., Barret, N., Delaney, P. (1994). Mutations of Ki-ras and p53
genes in colorectal cancer and their prognostic significance. Gut. 35(11),1627-31
Nagorney, D.M. (1987). Hepatic resection for metastases from colorectal cancer.
Probl Gen Surg 4, 83-92
Nelson, R.L., Persky, V., Tuyk, M. (1999). Determination of factors responsible fort
he declining incidence of colorectal cancer. Dis Colon Rectum 42(6), 741-752
Nemunaitis, J., Cox, J., Meyer, W., Courtney, A., Mues, G. (1997). Irinotecan
hydrochloride (CPT-11) resistance identified by KRAS mutation in patients with

- 67 -
progeressive colon cancer after treatment with 5-fluorouracil (5-FU). Am J Klin
20(5), 527-529
O'Connell, J.B., Maggard, M.A., Ko, C.Y. (2004). Colon cancer survival rates with
the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer
Inst. 96(19), 1420-5
Ogino, S., Kawasaki, T., Kirkner, G.J., Yamaji, T., Loda, M., Fuchs, C.S. (2007).
Loss of nuclear p27 (CDKN1B/KIP1) in colorectal cancer is correlated with
microsatellite instability and CIMP. Mod Pathol. 20(1), 15-22
Ogino, S., Meyerhardt, J.A., Cantor, M., Brahmandam, M., Clark, J.W., Namgyal,
C., Kawasaki, T., Kinsella, K., Michelini, A.L., Enzinger, P.C., Kulke, M.H., Ryan,
D.P., Loda, M., Fuchs, C.S. (2005). Molecular Alterations in Tumors and
Response to Combination Chemotherapy with Gefitinib for Advanced Colorectal
Cancer. Clin Cancer Res 11(18), 6650-6656
Olaussen, K.A., Dunant, A., Fouret, P., Brambilla, E., André, F., Haddad, V.,
Taranchon, E., Filipits, M., Pirker, R., Popper, H.H., Stahel, R., Sabatier, L.,
Pignon, J.P., Tursz, T., Le Chevalier, T., Soria, J.C. (2006). DNA repair by ERCC1
in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J
Med. 355(10), 983-991
Oliva, M.R., Cabrera, T., Esquivias, J., Perez-Ayala, M., Redondo, M., Ruiz-
Cabello, F., Garrido, F. (1990). KRAS mutations (codon 12) are not involved in
down-regulation of MHC class-I genes in colon carcinomas. Int J Cancer 46(3),
426-31
Ota, S., Ishii, G., Goto, K., Kubota, K., Kim, Y.H., Kojika, M., Murata, Y.,
Yamazaki, M., Nishiwaki, Y., Eguchi, K., Ochiai, A. (2009). Immunohistochemical
expression of BCRP and ERCC1 in biopsy specimen predicts survival in in
advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy.
Lung Cancer 64(1), 98-104

- 68 -
Persson, P.G., Karlén, P., Bernell, O., Leijonmarck, C.E., Broström, O., Ahlbom,
A., Hellers, G. (1994). Crohn's disease and cancer: a population-based cohort
study. Gastroenterology 107(6), 1675-1679
Porschen, R., Arkenau, H.T., Kubicka, S., Greil, R., Seufferlein, T., Freier, W.,
Kretzschmar, A., Graeven, U., Grothey, A., Hinke, A., Schmiegel, W., Schmoll,
H.J. (2007). Phase III study of capecitabine plus oxaliplatin compared with
fluorouracil and leucovorin plus oxaliplatin in metastatic colorectal cancer: a final
report of the AIO Colorectal Study Group. J Clin Oncol. 25(27), 4217-23
Pricolo, V.E., Finkelstein, S.D., Wu, T.T., Keller, G., Bakker, A., Swalsky, P.A.,
Bland, K.I. (1996). Prognostic value of TP53 and KRAS-2 mutational analysis in
stage 3 carcinoma of the colon. Am J Surg. 171(1), 41-6
Quasar Collaborative G. (2007). Adjuvant chemotherapy versus observation in
patients with colorectal cancer: a randomised study. Lancet 370(9604), 2020-2029
Reinacher-Schick, A., Baldus, S.E., Romdhana, B., Landsberg, S., Zapatka, M.,
Mönig, S.P., Hölscher, A.H., Dienes, H.P., Schmiegel, W., Schwarte-Waldhoff, I.
(2004). Loss of Smad4 correlates with loss of the invasion suppressor E-cadherin
in advanced colorectal carcinomas. J Pathol 202(4), 412-20
Ribic, C.M., Sargent, D.J., Moore, M.J., Thibodeau, S.N., French, A.J., Goldberg,
R.M., Hamilton, S.R., Laurent-Puig, P., Gryfe, R., Shepherd, L.E., Tu, D., Redston,
M., Gallinger, S. (2003). Tumor microsatellite-instability status as a predictor of
benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J
349(3), 247-57
Richman, S.D., Seymour, M.T., Chambers, P., Elliott, F., Daly, C.L., Meade, A.M.,
Taylor, G., Barrett, J.H., Quirke, P. (2009). KRAS and BRAF mutations in
advanced colorectal cancer are associated with poor prognosis but do not
preclude benefit from oxaliplatin or irinotecan:results from the MRC FOCUS trial.
JCO 27(35), 5866-7

- 69 -
Rosty, C., Chazal, M., Etienne, M.C., Letoublon, C., Bourgeon, A., Delpero, J.R.,
Pezet, D., Beaune, P., Laurent-Puig, P., Milano, G. (2001). Determination of
microsatellite instability, p53 and KRAS mutations in hepatic metastases from
patients with colorectal cancer: relationship with response to 5-flourouracil and
survival. Int J Cancer. 95(3), 162-167
Roth, A.D., Tejpar, S., Delorenzi, M., Yan, P., Fiocca, R., Klingbiel, D., Dietrich, D.,
Biesmans, B., Bodoky, G., Barone, C., Aranda, E., Nordlinger, B., Cisar, L.,
Labianca, R., Cunningham, D., Van Cutsem, E., Bosman, F. (2010). Prognostic
role of KRAS and BRAF in stage II and III resected colon cancer: results of the
translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin
Oncol 28(3), 466-74
Rothenberg, M.L., Oza, A.M., Bigelow, R.H., Berlin, J.D., Marshall, J.L.,
Ramanathan, R.K., Hart, L.L., Gupta, S., Garay, C.A., Burger, B.G., Le Bail, N.,
Haller, D.G. (2003). Superiority of oxaliplatin and fluorouracil-leucovorin compared
with either therapy alone in patients with progressive colorectal cancer after
irinotecan and fluorouracil-leucovorin: interim results of a phase 3 trial. J Clin
Oncol 21(11), 2059-2069
Rougier, P., Van Cutsem, E., Bajetta, E., Niederle, N., Possinger, K., Labianca, R.,
Navarro, M., Morant, R., Bleiberg, H., Wils, J., Awad, L., Herait, P., Jacques, C.
(1998). Randomised trial of irinotecan versus fluorouracil by continuous infusion
after fluorouracil failure in patients withmetastatic colorectal cancer. Lancet
352(9138), 1407-1412
Rubio, C.A. und Befrits, R. (2008). Colorectal cancer in Crohn's disease-review of
a 56-year experience in Karolinska Institute University Hospital. J Environ Pathol
Toxicol Oncol. 27(4), 257-66
Salovaara, R., Roth, S., Loukola, A., Launonen, V., Sistonen, P., Avizienyte, E.,
Kristo, P., Järvinen, H., Souchelnytskyi, S., Sarlomo-Rikala, M., Aaltonen, L.A.
(2002). Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 51(1),
56-9

- 70 -
Saltz, L.B., Clarke, S., Díaz-Rubio, E., Scheithauer, W., Figer, A., Wong, R., Koski,
S., Lichinitser, M., Yang, T.S., Rivera, F., Couture, F., Sirzén, F., Cassidy, J.
(2008). Bevacizumab in combination with oxaliplatin-based chemotherapy as first-
line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin
Oncol. 26(12), 2013-9
Samowitz, W.S., Curtin, K., Schaffer, D., Robertson, M., Leppert, M., Slattery, M.L.
(2000). Relationship of Ki-Ras mutations in colon cancers to tumor location, stage,
and survival: a population-based study. Cancer Epidemiol Biomarkers Prev. 9(11),
1193-7
Samowitz, W.S., Sweeney, C., Herrick, J., Albertsen, H., Levin, T.R., Murtaugh,
M.A., Wolff, R.K., Slattery, M.L. (2005). Poor survival associated with the BRAF
V600E Mutation in microsatellite-stable colon cancers. Cancer Res 65(14), 6063-
6070
Sauer, R., Becker, H., Hohenberger, W., Rödel, C., Wittekind, C., Fietkau, R.,
Martus, P., Tschmelitsch, J., Hager, E., Hess, C.F., Karstens, J.H., Liersch, T.,
Schmidberger, H., Raab, R. (2004). Preoperative versus postoperative
chemoradiotherapy for rectal cancer. N Engl J Med. 351(17), 1731-40
Schettino, C., Bareschino, M.A., Maione, P., Rossi, A., Ciardiello, F., Gridelli, C.
(2008). The potential role of pharmacogenomic and genomic in the adjuvant
treatment of early stage non small cell lung cancer. Curr Genomics 9(4), 252-62
Schmiegel, W., Reinacher-Schick, A., Arnold, D., Graeven, U., Heinemann, V.,
Porschen, R., Riemann, J., Rodel, C., Sauer, R., Wiesner, M., Schmitt, W.,
Schmoll, H.J., Seufferlein, T., Kopp, I., Pox, C. (2008). Update S3-guideline
“colorectal cancer“ 2008. Z Gastroenterol 46(8), 799-840
Schwarte-Waldhoff, I. (2003). Funktionen des Tumorsuppressorgens Smad4 in
der mehrstufigen Karzinogenese. BIOspektrum 3, 260-264
Seymour, M.T., Maughan, T.S., Ledermann, J.A., Topham, C., James, R.,
Gwyther, S.J., Smith, D.B., Shepherd, S., Maraveyas, A., Ferry, D.R., Meade,

- 71 -
A.M., Thompson, L., Griffiths, G.O., Parmar, M.K., Stephens, R.J. (2007). Different
strategies of sequential and combination chemotherapy for patients with poor
prognosis advanced colorectal cancer (MRC FOCUS): a randomised controlled
trial. Lancet 370(9582), 143-152
Shirota, Y., Stoehlmacher, J., Brabender, J., Xiong, Y.P., Uetake, H., Danenberg,
K.D., Groshen, S., Tsao-Wei, D.D., Danenberg, P.V., Lenz, H.J. (2001). ERCC1
and Thymidylate Synthase mRNA Levels Predict Survival for Colorectal Cancer
Patients Receiving Combination Oxaliplatin and Fluorouracil Chemotherapy. J Clin
Oncol 19(23), 4298-4304
Soetikno, R.M., Kaltenbach, T., Rouse, R.V., Park, W., Maheshwari, A., Sato, T.,
Matsui, S., Friedland, S. (2008). Prevalence of Nonpolypoid (Flat and Depressed)
Colorectal Neoplasms in Asymptomatic and Symptomatic Adults. JAMA 299(9),
1027-1035
Takeshita, T., Morimoto, K., Yamaguchi, N., Watanabe, S., Todoroki, I., Honjo, S.,
Nakagawa, K., Kono, S. (2000). Relationships between cigarette smoking, alcohol
drinking, the ALDH2 genotype and adenomatous types of colorectal polyps in
male self-defense force officials. J Edidemiol 10(6), 366-371
Tan, Y.H., Liu, Y., Eu, K.W., Ang, P.W., Li, W.Q., Salto-Tellez, M., Lacopetta, B.,
Soong, R. (2008). Detection of BRAF V600E mutation by pyrosequencing.
Pathology 40(3), 295-8
The International Adjuvant Lung Cancer Trial Collaborative Group. (2004).
Cisplatin-based adjuvant chemotherapy in patients with completely resected non-
small-cell lung cancer. N. Engl. J. Med. 350(4), 351-360
Tiemersma, E.W., Wark, P.A., Ocké, M.C., Bunschoten, A., Otten, M.H., Kok, F.J.,
Kampman, E. (2003). Alcohol consumption, alcohol dehydrogenase 3
polymorphism, and colorectal adenomas. Cancer Epidemiol Biomarkers Prev
12(5), 229-237

- 72 -
Tol, J., Dijkstra, J.R., Vink-Borger, M.E., Koopman, M., Vincent, A.D., van Krieken,
J.H.J.M., Ligtenberg, M.J.L., Nagtegaal, I.D., Punt, C.J.A. (2009). BRAF mutation
is associated with a decreased outcome in patients (pts) with advanced colorectal
cancer (ACC) treated with chemotherapy and bevacizumab with or without
cetuximab. Eur J Cancer Suppl. Vol. 7(No. 3), abstr. O-6002
Tol, J., Koopman, M., Rodenburg, C.J., Cats, A., Creemers, G.J., Schrama, J.G.,
Erdkamp, F.L., Vos, A.H., Mol, L., Antonini, N.F., Punt, C.J. (2008). A randomized
phase III study on capecitabine, oxaliplatin and bevacizumab with or without
cetuximab in first-line advanced colorectal cancer, the CAIRO2 study of the Dutch
Colorectal Cancer Group (DCCG). An interim analysis of toxicity. Ann Oncol 19(4),
734-8
Tournigand, C., Louvet, C., Quinaux, E., André, T., Lledo, G., Flesch, M., Ganem,
G., Landi, B., Colin, P., Denet, C., Mery-Mignard, D., Risse, M.L., Buyse, M., and
DeGramont, A. (2001). FOLFIRI followed by FOLFOX versus FOLFOX followed by
FOLFIRI in metastatic colorectal cancer: final results of a phase-III study. Proc
ASCO, abstr. 494
Tran, B., Kopetz, S., Tie, J., Gibbs, P., Jiang, Z., Lieu, C.H., Agarwal, A., Maru, D.,
Sieber, O., Desai, J. (2010). Differences in sites of metastatic disease and
outcomes observed in patients with BRAF mutant colorectal cancer. JCO 28(15s),
abstr. 3592
Tveit, K., Guren, T., Glimelius, B., Pfeiffer, P., Sorbye, H., Pyrhonen, S., Kure, E.,
Ikdahl, T., Skovlund, E., Christoffersen, T. (2011). Randomized phase III study of
5-fluorouracil/folinate/oxaliplatin given continuously or intermittently with or without
cetuximab, as first-line treatment of metastatic colorectal cancer: The NORDIC VII
study (NCT00145314), by the Nordic Colorectal Cancer Biomodulation Group.
JCO 29(Suppl.4), abstr. 365
Van Cutsem, E., Köhne, C.H., Hitre, E., Zaluski, J., Chang Chien, C.R., Makhson,
A., D'Haens, G., Pintér, T., Lim, R., Bodoky, G., Roh, J.K., Folprecht, G., Ruff, P.,
Stroh, C., Tejpar, S., Schlichting, M., Nippgen, J., Rougier, P. (2009). Cetuximab

- 73 -
and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J
Med 360(14), 1408-17
Van Cutsem, E., Labianca, R., Bodoky, G., Barone, C., Aranda, E., Nordlinger, B.,
Topham, C., Tabernero, J., André, T., Sobrero, A.F., Mini, E., Greil, R., Di
Costanzo, F., Collette, L., Cisar, L., Zhang, X., Khayat, D., Bokemeyer, C., Roth,
A.D., Cunningham, D. (2009). Randomized Phase III Trial Comparing Biweekly
Infusional Fluorouracil/ Leukovorin Alone or with Irinotecan in the Adjuvant
Traetment of Stage III Colon Cancer : PETCAA-3. J Clin Oncol 27(19), 3117-3125
Van Cutsem, E., Lang, I. Folprecht, G., Nowacki, M., Cascinu, S., Shchepotin, I.,
Maurel, J., Cunningham, D., Celik, I., Kohne, C. (2010). Cetuximab plus FOLFIRI
in the treatment of metastatic colorectal cancer (mCRC): The influence of KRAS
and BRAF biomarkers on outcome: Updated data from the CRYSTAL trial. ASCO,
abstr. 281
Viguier, J., Boige, V., Miquel, C., Pocard, M., Giraudeau, B., Sabourin, J.C.,
Ducreux, M., Sarasin, A., Praz, F. (2005). ERCC-1 Codon 118 Polymorphism Is a
Predictive Factor for the Tumor Response to Oxaliplatin/5-Fluorouracil
Combination Chemotherapy in Patients with Advanced Colorectal Cancer. Clin
Cancer Res. 11(17), 6212-6217
Vogelstein, B., Fearon, E.R., Hamilton, S.R., Kern, S.E., Preisinger, A.C., Leppert,
M., Nakamura, Y., White, R., Smits, A.M., Bos, J.L. (1988). Genetic alterations
during colorectal tumor development. N Engl J Med 319(9), 525-532
Watanabe, T., Wu, T.T., Catalano, P.J., Ueki, T., Satriano, R., Haller, D.G.,
Benson, A.B. 3rd, Hamilton, S.R. (2001). Molecular Predictors of Survival After
Adjuvant Chemotherapy for Colon Cancer. N Engl J Med 344(16), 1196-1205
Wei, E.K., Giovannucci, E., Wu, K., Rosner, B., Fuchs, C.S., Willett, W.C., Colditz,
G.A. (2004). Comparison of risk factors for colon and rectal cancer. Int J Cancer
108(3), 433-442

- 74 -
Weickhardt, A.J., Price, T.J., Pavlakis, N., Skrinos, E., Dobrovic, A., Salemi, R.,.
Scott, A.M., Mariadason, J., Chong G. and Tebbutt, N.C. (2010). DUX study: A
phase II study of evaluating dual targeting of the EGFR using the combination of
cetuximab and erlotinib in patients with chemotherapy refractory metastatic
colorectal cancer. JCO 28(15s), abstr. 3533
Westra, J.L., Schaapveld, M., Hollema, H., de Boer, J.P., Kraak, M.M., de Jong,
D., Elster, A., Mulder, N.H., Buys, C.H., Hofstra, R.M., Plukker, J.T. (2005).
Determination of TP53 mutation is more relevant than microsatellite instability
status for the prediction of disease-free survival in adjuvant treated stage 3 colon
cancerpatients. JCO 23(34), 5635-43
Wieser, M., Sauerland, S., Arnold, D., Schmiegel, W., Reinacher-Schick, A.
(2010). Peri-operative chemotherapy for the treatment of resectable liver
metastases from colorectal cancer: A systematic review and meta-analysis of
randomized trials. BMC Cancer 10, 309
Winawer, S.J., Zauber, A.G., Gerdes, H., O'Brien, M.J., Gottlieb, L.S., Sternberg,
S.S., Bond, J.H., Waye, J.D., Schapiro, M., Panish, J.F. (1996). Risk of colorectal
cancer in the families of patients with adenomatous polyps. N Engl J Med 334(2),
82-87
Winawer, S.J., Zauber, A.G., Ho, M.N., O'Brien, M.J., Gottlieb, L.S., Sternberg,
S.S., Waye, J.D., Schapiro, M., Bond, J.H., Panish, J.F. (1993). Prevention of
colorectal cancer by colonoscopic polypectomy. N Engl J Med 329(27), 901-906
Winder, T., Mündlein, A., Rhomberg, S., Dirschmid, K., Hartmann, B.L., Knauer,
M., Drexel, H., Wenzl, E., De Vries, A., Lang, A. (2009). Different types of KRAS
mutations are conversely associated with overall survival in patients with colorectal
cancer. Oncol Rep. 21(5), 1283-7
Wittekind, C., Greene, F., Henson, D., Hutter, R., Sobin, L. (2003). TNM
Supplements 3rd edition. A commentary on uniform use. John Wiley and Sons.
New York

- 75 -
Wittekind, C. und Tannapfel, A. (2003). Regression grading of colorectal
carcinoma after preoperative radiochemotherapy. An inventory. Pathologe. 24(1),
61-5
Wittinghofer, A. und Pai, E.F: (1991). The structure of Ras protein: a model for a
universal molecular switch. TIBS 16(10), 382-387
Wong, J.H., Severino, R., Honnebier, M.B., Tom, P., Namiki, T.S. (1999). Number
of nodes examined and staging accuracy in colorectal carcinoma. J Clin Oncol
17(9), 2896-2900
Yoshitake, N., Fujii, S., Mukawa, K., Tominaga, K., Fukui, H., Ichikawa, K., Tomita,
S., Ono, Y., Imai, Y., Terano, A., Hiraishi, H., Fujimori, T. (2007). Mutational
analysis of the BRAF genein colorectal mucinous carcinoma in association with
with histological configuration. Oncol Rep 17(1), 9-15
Youn, C.K., Kim, M.H., Cho, H.J., Kim, H.B., Chang, I.Y., Chung, M.H., You, H.J.
(2004). Oncogenic H-Ras up-regulates expression of ERCC1 to protect cells from
platinum-based anticancer agents. Cancer Res. 64(14), 4849-57
Zandonai, A.P., Sonobe, H.M., Sawada N.O. (2012). The dietary risk factors for
colorectal cancer related to meat consumption. Rev Esc Enferm USP. 46(1), 234-
239
Zhang, W., El-Khoueiry, A., Yang, D., Pohl, A., Ning, Y., Lurje, G., Manegold, P.,
Iqbal, S., Lenz, H. (2009). KRAS mutation status associated with clinical outcome
in metastatic colorectal cancer patients treated with 5-fluorouracil/oxaliplatin.
ASCO, abstr. 340

8. Danksagung
An dieser Stelle möchte ich mich ganz herzlich bei Prof. Dr. med. Anke Reinacher-
Schick für die Bereitstellung des interessanten Themas dieser Promotionsarbeit
sowie für die ausgezeichnete Beratung und Betreuung bei der Verfassung der
Arbeit bedanken.
Ich danke Prof. Dr. med. Wolff Schmiegel als Direktor der Medizinischen
Universitätsklinik Knappschaftskrankenhaus Bochum für die Möglichkeit, in seiner
Klinik diese Dissertation verfassen zu können.
Ein ganz besonderer Dank gilt Dr. med. Karsten Schulmann für die gesamte
Betreuung der Doktorarbeit, für die vielen Treffen, die Ratschläge und
Korrekturen.
Aus dem Institut für Pathologie der Berufsgenossenschaftlichen Kliniken
Bergmannsheil gilt mein Dank Frau Prof. Dr. med. Andrea Tannapfel für die
Möglichkeit, uneingeschränkt in den Räumen des Instituts für Pathologie arbeiten
und die Materialien für die Versuche nutzen zu dürfen. Des Weiteren danke ich
Frau Dr. rer. nat. Malgorzata Jaworska und Herrn Dipl. Biol. Markus Vogt für die
gute Betreuung und die Hilfe im Labor sowie Frau Dr. rer. nat. Berenike Flott-
Rahmel für die Betreuung im Rahmen der Doktorandengruppe.
Mein weiterer Dank gilt Frau Dipl.-Stat. Renate Klaaßen-Mielke für das
Kontrollieren meiner statistischen Auswertung sowie die Beantwortung meiner
vielen Fragen.
Vielmals möchte ich mich bei den Patientinnen und Patienten bedanken, die in die
Verwendung der Daten und Materialien für die Studie eingewilligt haben. Mein
weiterer Dank ist an die zuständigen Prüfärzte sowie an die Arbeitsgemeinschaft
Internistische Onkologie gerichtet.
Danken möchte ich auch Frank Zurmühlen, Petra Fischer, Nikolai Senekowitch
und Jutta Dreier für das Durchsehen meiner Promotionsarbeit in Hinblick auf

grammatikalische Fehler sowie Frau Ulrike Eberhardt für das Hinweisen auf
formelle Fehler.
Von ganzem Herzen danken möchte ich meinen Eltern, meiner Schwester Alessa
und Heinz und Else Adamik für die liebevolle und immer vorhandene
Unterstützung bei meinem Studium, der Promotionsarbeit und in allen
Lebenslagen. Ebenso möchte ich Dominik Meier für den nicht endenden Rückhalt,
das viele Aufmuntern und den Austausch von Ratschlägen und Tipps danken.
Natürlich möchte ich mich auch ganz herzlich bei meinen engsten Freunden für
die Hilfe und Nachsicht aber auch für das gelegentliche Ablenken bedanken.

9. Lebenslauf
Nina Mareike Bruns
_________________________________________________________________
Geburtsdatum/ -ort: 23. April 1985 in Dortmund
Konfession: evangelisch
Familienstand: ledig
Staatsangehörigkeit: deutsch
Schulausbildung: 1991-1995: Grundschule im Sande, Bramsche
1995-1997: Orientierungsstufe Gartenstadt, Bramsche
1997-2004: Greselius-Gymnasium, Bramsche
Abschluss: Abitur (1,8)
1999: zweiwöchiger Schüleraustausch nach
Harfleur, Frankreich
Studium: 2004-2010: Ruhr-Universität Bochum,
Humanmedizin
2004-2006: Vorklinik
Abschluss: 1. Staatsexamen (2,5)
2006-2010: Klinik
Abschluss: 2. Staatsexamen (2,0)
Gesamtnote Ärztliche Prüfung: 2,17
08/09-12/09: 1. PJ-Tertial: Innere Medizin,
Marienhospital Herne
12/09-04/10: 2. PJ-Tertial: Chirurgie,
Bürgerspital Solothurn, Schweiz
04/10-08/10: 3. PJ-Tertial: Dermatologie
St. Josef-Hospital Bochum

Beruflicher Werdegang: seit 04/11: Assistenzärztin in der Derma-
tologischen Abteilung des St. Josef-
Hospitals, Klinikum der
Ruhr-Universität Bochum
Bisherige Praktika: 09/2001: 2-wöchiges Betriebspraktikum in
der Zahnarztpraxis Dr. Simon,
Bramsche
03/2005-04/2005: Krankenpflegepraktikum,
Johanniter-Krankenhaus,
Bramsche
08/2005-09/2005: Krankenpflegepraktikum,
Johanniter-Krankenhaus,
Bramsche
02/2007-03/2007: Famulatur, Chirurgische
Notfallambulanz,
Johanniter-Krankenhaus,
Bramsche
08/2007-09/2007: Famulatur, Kinderchirurgie, Hopital
Lenval, Nizza, Frankreich
09/2007-10/2007: Famulatur, Innere Medizin,
Knappschaftskrankenhaus Bochum
03/2008: Famulatur, Gynäkologie, Bezirks-
krankenhaus Schwaz, Österreich
09/2008: Famulatur, Gynäkologie, Augusta-
Kranken-Anstalt, Bochum
Besondere Leistungen: Diplôme d'Etudes en langue française 1 (DELF 1)
Diplôme d'Etudes en langue française 2 (DELF 2)
Stipendium des Bildungsfonds der RUB im Rahmen
des NRW-Stipendienprogramms (2009/2010)
1999-2004: ehrenamtliche Mitarbeiterin in der
St. Johannis-Kirche Bramsche


















