![Aus der Medizinischen Klinik und Poliklinik I der ... · kinase pathway) [8], welcher als gemeinsam genutzter Signalweg der beiden ischämieprotektiven Interventionen Prä- und Postkonditionierung](https://static.fdokument.com/doc/165x107/5d5e435688c99346098bb801/aus-der-medizinischen-klinik-und-poliklinik-i-der-kinase-pathway-8-welcher.jpg)
RAS RAF MEK ERK Der MAPK-Signalweg. Übersicht Der MAPK-Signalweg: –Übersicht...
30
RAS RAF MEK ERK Der MAPK-Signalweg
-
Upload
guenter-vogt -
Category
Documents
-
view
226 -
download
0
Transcript of RAS RAF MEK ERK Der MAPK-Signalweg. Übersicht Der MAPK-Signalweg: –Übersicht...
RAS
RAF
MEK
ERK
Der MAPK-Signalweg

Übersicht• Der MAPK-Signalweg:
– Übersicht– Signaltransduktion
• Dysregulierter MAPK-Signalweg– Bei Tumorkrankungen allgemein– Beim Melanom
• BRAF-Inhibition beim Melanom mit BRAFV600-Mutation• Resistenzmechanismen gegen die BRAF-Inhibition beim Melanom mit
BRAFV600-Mutation– Mechanismen der Reaktivierung des MAPK-Signalwegs– Resistenzmechanismen gegen BRAF-Inhibitoren unabhängig von
der Reaktivierung des MAPK-Signalwegs• Paradoxe Aktivierung des MAPK-Signalwegs durch BRAF-Inhibitoren• Kombination aus BRAF- und MEK-Inhibitoren beim metastasierten
Melanom• Glossar
2
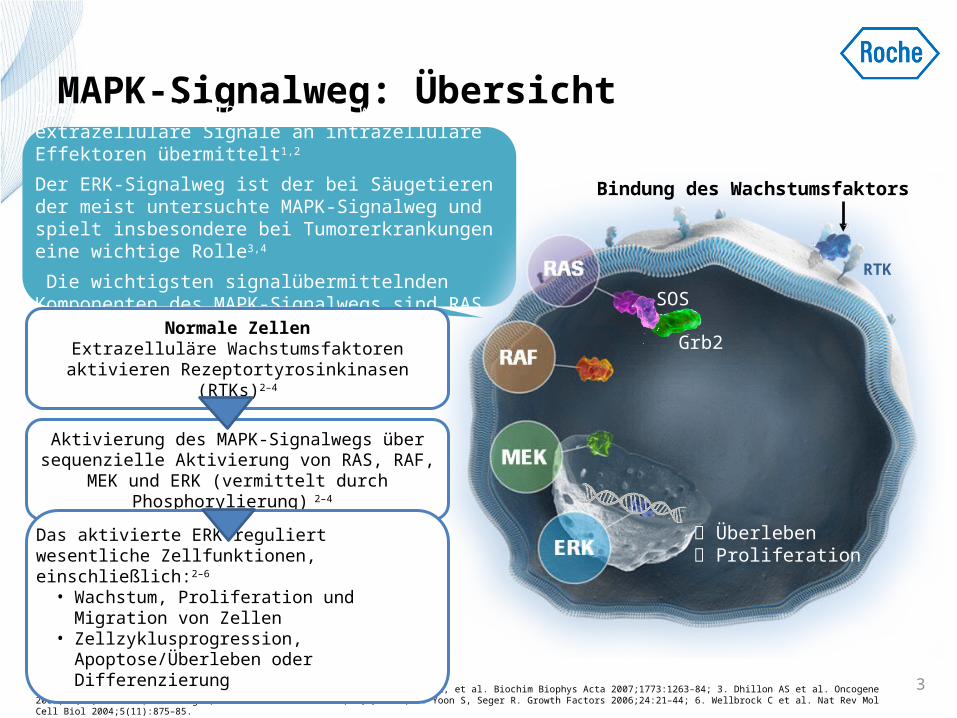
MAPK-Signalweg: Übersicht
1. Robinson MJ, Cobb MH. Curr Opin Cell Biol 1997;9:180–6; 2. McCubrey JA, et al. Biochim Biophys Acta 2007;1773:1263–84; 3. Dhillon AS et al. Oncogene 2007;26(22):3279–90; 4. Zhang W, Liu HT. Cell Res 2002;12(1):9–18; 5. Yoon S, Seger R. Growth Factors 2006;24:21–44; 6. Wellbrock C et al. Nat Rev Mol Cell Biol 2004;5(11):875–85.
Durch die MAPK-Signalkaskade werden extrazelluläre Signale an intrazelluläre Effektoren übermittelt1,2
Der ERK-Signalweg ist der bei Säugetieren der meist untersuchte MAPK-Signalweg und spielt insbesondere bei Tumorerkrankungen eine wichtige Rolle3,4
Die wichtigsten signalübermittelnden Komponenten des MAPK-Signalwegs sind RAS, RAF, MEK, ERK1-3
Bindung des Wachstumsfaktors
Überleben Proliferation
RTK
Grb2
SOSNormale Zellen
Extrazelluläre Wachstumsfaktoren aktivieren Rezeptortyrosinkinasen (RTKs)2–4
Aktivierung des MAPK-Signalwegs über sequenzielle Aktivierung von RAS, RAF, MEK
und ERK (vermittelt durch Phosphorylierung) 2–4
Das aktivierte ERK reguliert wesentliche Zellfunktionen, einschließlich:2–6 • Wachstum, Proliferation und Migration von
Zellen• Zellzyklusprogression,
Apoptose/Überleben oder Differenzierung3
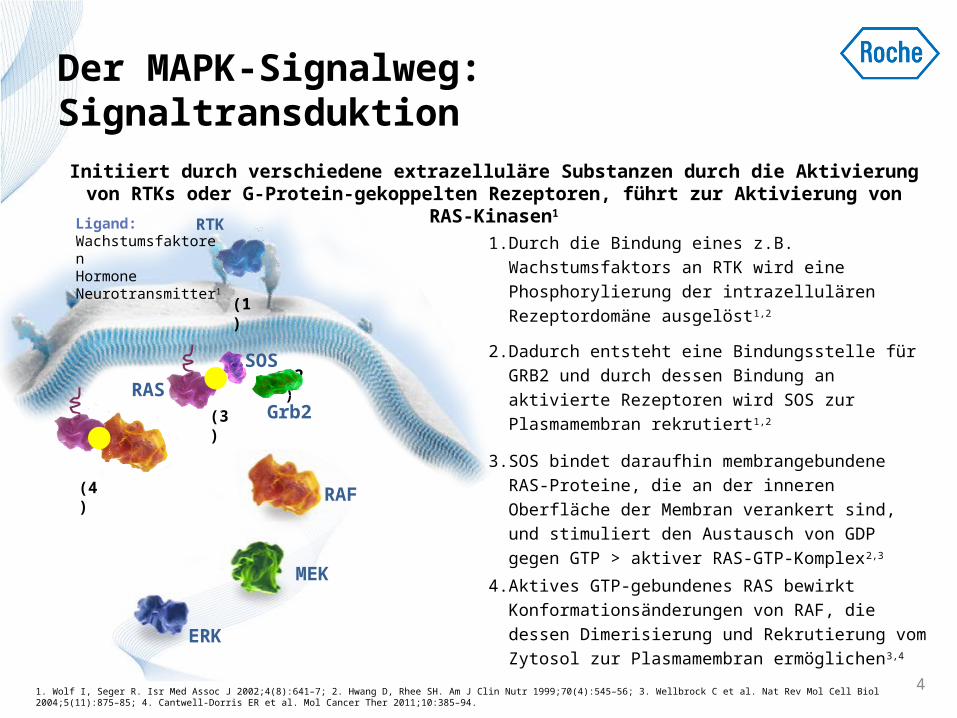
Der MAPK-Signalweg: Signaltransduktion
1. Durch die Bindung eines z.B. Wachstumsfaktors an RTK wird eine Phosphorylierung der intrazellulären Rezeptordomäne ausgelöst1,2
2. Dadurch entsteht eine Bindungsstelle für GRB2 und durch dessen Bindung an aktivierte Rezeptoren wird SOS zur Plasmamembran rekrutiert1,2
3. SOS bindet daraufhin membrangebundene RAS-Proteine, die an der inneren Oberfläche der Membran verankert sind, und stimuliert den Austausch von GDP gegen GTP > aktiver RAS-GTP-Komplex2,3
4. Aktives GTP-gebundenes RAS bewirkt Konformationsänderungen von RAF, die dessen Dimerisierung und Rekrutierung vom Zytosol zur Plasmamembran ermöglichen3,4
Initiiert durch verschiedene extrazelluläre Substanzen durch die Aktivierung von RTKs oder G-Protein-gekoppelten Rezeptoren, führt zur Aktivierung von RAS-
Kinasen1
41. Wolf I, Seger R. Isr Med Assoc J 2002;4(8):641–7; 2. Hwang D, Rhee SH. Am J Clin Nutr 1999;70(4):545–56; 3. Wellbrock C et al. Nat Rev Mol Cell Biol 2004;5(11):875–85; 4. Cantwell-Dorris ER et al. Mol Cancer Ther 2011;10:385–94.
(1)
(2)
(3)
(4)
Grb2
SOS
RAF
MEK
ERK
RAS
Ligand:WachstumsfaktorenHormone Neurotransmitter1
RTK

Der MAPK-Signalweg: Signaltransduktion
5. Die RAS-vermittelte Rekrutierung von RAF zur Membran begünstigt Phosphorylierungsänderungen bei RAF, die dessen Serin-/Threoninkinasedomäne aktivieren2
6. Aktiviertes RAF phosphoryliert und aktiviert MEK1 und MEK2 (dualspezifische Proteinkinasen)2–9
7. Aktiviertes MEK1 und MEK2 aktivieren ERK1 und ERK2 (Serin-/Threoninkinasen)2–9
8. ERK1 und ERK2 regulieren Wachstumsfaktor-responsive Zielmoleküle und Transkriptionsfaktoren,4,7–9 so dass letztlich eine Wachstumsfaktor-abhängige Regulation von Transkription, Metabolismus und zytoskelettalem Rearrangement erfolgen kann7
RAS aktiviert ein konserviertes dreistufiges Kinase-Signalmodul bestehend aus RAF, MEK und ERK,
die durch sequenzielle aktivierende Phosphorylierungsereignisse das Signal übertragen2,3
51. Wolf I, Seger R. Isr Med Assoc J 2002;4(8):641–7; 2. Cantwell-Dorris ER et al. Mol Cancer Ther 2011;10:385–94; 3. Dhillon AS et al. Oncogene 2007 ;26(22):3279–90; 4. Michaloglou C et al. Oncogene 2008;27:877–95; 5. Sharma A et al. Cancer Res 2005;65:2412–21; 6. Cseh B et al. FEBS Lett 2014;588:2398-406; 7. Wellbrock C et al. Nat Rev Mol Cell Biol 2004;5(11):875–85; 8. Schulze A et al. Mol Biol Cell 2004;15:3450–63; 9. Roskoski R, Jr. Pharmacol Res 2012;66:105–43; 10. Jilaveanu LB et al. Clin Cancer Res 2009; 15(18):5704–13.
Ligand:WachstumsfaktorenHormone Neurotransmitter1
(1)
(2)
(3)
(4)
RTK
Grb2
SOS
RAF
MEK
ERK
(5)
(6)
PP
P P
(7)
(8)
RAS
Zytoplasmatische, zytoskelettale und nukleäre
Zielstrukturen
• Es gibt drei Isoformen von RAF: ARAF, BRAF und CRAF2,4,7
• Die Isoformen von RAF haben unterschiedliche Funktionen und eine unterschiedliche zelluläre Verteilung6,10
• BRAF ist der stärkste MEK-Aktivator aller drei Isoformen10

(1)
(2)
(3)
(4)
Grb2
SOS
BRAF
MEK
ERK
(5)
(6) P
P
P P
(7)
(8)
RAS
Dysregulierter MAPK-Signalweg bei Tumoren
• Schätzungen zufolge ist der RAS/BRAF/MEK/ERK-Signalweg bei 30 % aller Tumorarten mutiert2
• Mutationen des BRAF-Gens finden sich bei rund 8 % aller Tumorarten3
• Eine fehlregulierte ERK-Aktivierung kann Proliferation, Migration, Überleben und Invasion von Zellen sowie Metastasen begünstigen4,5
Eine Fehlregulation der MAPK-Signalübertragung wird mit mehreren Tumorarten in Verbindung gebracht2,4,5
BRAF-MutationMelanom, Schilddrüsen- und Kolorektalkarzinom6,9
MEK-MutationMelanom13
RAS-MutationPankreas-, Schilddrüsen-, Bronchial-, Leber-, Blasen-, Nieren- und Kolorektalkarzinom, AML, MDS und Melanom6–8
Rezeptorveränderungen (z. B. Genamplifikation und darauf folgende Überexpression und aktivierende Mutationen)6,10–12
61. Wolf I, Seger R. Isr Med Assoc J 2002;4(8):641–7; 2. Garnett M, Marais R. Cancer Cell 2004;6:313–9; 3. Holderfield M et al. Nat Rev Cancer 2014;14(7):455–67; 4. Smalley KS. Int J Cancer 2003;104(5):527–32; 5. .Inamdar GS et al. Biochem Pharmacol 2010;80(5):624–37; 6. Dhillon AS et al. Oncogene 2007;26:3279–90; 7. Pylayeva-Gupta Y, et al. Nature Rev Cancer 2011;11:761–72; 8. Prenen H et al. Clin Cancer Res 2010;16(11):2921–6; 9. Rahman MA, et al. Exp Mol Pathol 2013;95:336–42; 10. Peters S, Adjei AA. Nature Rev Clin Oncol 2012;9:314–26; 11. Christensen JG et al. Cancer Lett 2005;225(1):1–26; 12. Yewale C, et al. Biomaterials 2013;34:8690–707; 13. Emery CM et al. Proc Natl Acad Sco USA 2009;106:20411–6.
Zytoplasmatische, zytoskelettale und nukleäre
Zielstrukturen
Ligand:WachstumsfaktorenHormone Neurotransmitter1
RTK

Fehlregulierter MAPK-Signalweg beim Melanom: BRAF-Mutationen• BRAF-Mutationen finden sich in ~40–67 % aller Melanome1–5; diese Zahl ist
höher als bei anderen Tumorarten1
• Dies könnte mit der Rolle von BRAF in Melanozyten zusammenhängen1
– Das Vorliegen von BRAF-Mutationen ist mit einer schlechten Prognose assoziiert6,7
• Bei Melanomen mit BRAF-Mutation ist die häufigste Mutation die Substitution eines einzelnen Nukleotids, bei der Valin (V) in Codon 600 durch Glutaminsäure (E) ersetzt wird (V600E). Dies wird in 92 % der Fälle beobachtet1
– V600K-, V600R- oder V600D-Substitutionen treten an dieser Stelle deutlich seltener auf8
• BRAFV600-Mutationen verleihen BRAF eine unabhängige, konstitutive Kinaseaktivität, ohne dass es durch GTP-gebundenes RAS zur Plasmamembran rekrutiert werden muss1,9–11
1. Davies H et al. Nature 2002;417:949–54; 2. Shinozaki M et al.Clin Cancer Res 2004;10:1753–57; 3. Libra M et al. Cell Cycle 2005;4:1382–84; 4. Omholt K et al. Clin Cancer Res 2003;9:6483–88; 5. Maldonado JL et al. J Natl Cancer Inst 2003;95:1878–90; 6. Long GV et al. J Clin Oncol. 2011;29(10):1239–46; 7. Mann GJ et al. J Invest Dermatol 2013;133(2):509–17; 8. Heinzerling L et al. Br J Cancer 2013;108(10):2164–71; 9. Cantwell-Dorris ER et al. Mol Cancer Ther 2011;10:385–94; 10. Wan PT et al. Cell 2004;116:855–67; 11. Mercer KE, Pritchard CA. Biochim Biophys Acta 2003;1653:25–40.
7
GlutaminsäureAsparaginsäureLysinArginin
Konservierte Regionen („conserved regions“, CR)Ras-bindende Domäne („RAS-binding domain“, RBD)Zystein-reiche Domäne („cysteine-rich domain“, CRD)
N-RegionATP-bindende DomäneAktivierungssegment
Substitution von Valin durch:

Fehlregulierter MAPK-Signalweg beim Melanom: BRAF-Mutationen
• Eine hohe Frequenz von BRAF-Mutationen wurde bei benignen Nävi, primären Melanomen und Metastasen identifiziert, was darauf schließen lässt, dass diese Veränderungen zur Tumorprogression beitragen2–6
• Die Onkogenität von BRAFV600E wurde in zahlreichen Mausmodellen für verschiedene Krebsarten bestätigt7
• Eine Signalübertragung durch onkogenes BRAFV600E war assoziiert mit erhöhter und unkontrollierter Zellproliferation und Apoptoseresistenz in Melanomzelllinien und humanen Melanozyten, vermittelt durch die Regulation des Transkriptionsfaktors MITF8
• Die BRAFV600-Mutation spielt bei verschiedenen der Tumorgenese des Melanoms zugrunde liegenden Mechanismen eine Rolle.9–11 Die meisten hiervon sind mit der fehlregulierten, von vorgeschalteten Ereignissen des MAPK-Signalwegs unabhängigen Aktivierung von MEK1 und MEK2 verbunden9,12,13
BRAFV600-Mutationen sind ein maßgeblicher Onkogenitätsfaktor beim Melanom1,10
1. Wang AX, Qi XY. Life. 2013;65:748–58; 2. Shinozaki M et al.Clin Cancer Res 2004;10:1753–57; 3. Libra M et al. Cell Cycle 2005;4:1382–84; 4. Pollock PM et al. Nat Genet 2003;33:19–20; 5. Uribe P et al. Am J Dermatopathol 2003;25:365–70; 6. Kumar R et al. J Invest Dermatol 2004;122:342–48; 7. Holderfield M et al. Nat Rev Cancer. 2014 ;14(7):455–67; 8. Wellbrock C et al. PLoSOne 2008;3(7):e2734; 9. Cantwell-Dorris ER et al. Mol Cancer Ther 2011;10:385–94; 10. Davies H et al. Nature 2002;417:949–54; 11. Gray-Schopfer VC et al. Cancer Metastasis Rev 2005;24:165–83; 12. Michaloglou C et al. Oncogene 2008;27:877–95; 13. Sharma A et al. Cancer Res 2005;65:2412–21.
8
RTK
BRAF
MEK
ERK
RAS
Bei aktivierenden BRAF-Mutationen ist keine Wachstumsfaktor-aktivierung erforderlich10

Neben BRAF-Mutationen führen auch Rezeptorveränderungen zu einer Fehlregulation der MAPK-Signalübertragung beim Melanom • Der MAPK-Signalweg wird primär durch Mutationen der BRAF- und
RAS-Gene aktiviert. Bei Melanomen, die diese Mutationen nicht aufweisen,1 können Melanomwachstum, -invasion und/oder -metastasen auf folgendes zurückzuführen sein:
– Überexpression,2,3 chromosomale Zugewinne4 und aktivierende Mutationen von cMET, einem Rezeptor für den Hepatozyten-Wachstumsfaktor (HGF)5
– Überexpression des epidermalen Wachstumsfaktorrezeptors (EGFR), verbunden mit chromosomalen Zugewinnen6
– Expressionsänderungen, Mutation und Aktivierung mehrerer anderer RTKs7,8
91. Inamdar GS et al. Biochem Pharmacol 2010;80(5):624–37; 2. Natali PG et al. Br J Cancer 1993;68:746–50; 3. Cruz J et al. Oncology 2003;65:72–82; 4. VanBrocklin et al. Pigment Cell Melanoma Res 2009;22(4):454–60; 5. Puri N et al. Clin Cancer Res 2007;13:2246–53; 6. Bastian BC et al. Cancer Res 1998;58:2170–75; 7. Easty DJ et al. Pigment Cell Melanoma Res 2011;24(3):446–61; 8. Schlegel J et al. J Clin Invest 2013;123(5):2257–67.

Neben BRAF-Mutationen führen auch MEK-Mutationen zu einer Fehlregulation der MAPK-Signalübertragung beim Melanom• MEK-Mutationen sind bei humanen Tumorarten selten1
• Die Inzidenz somatischer MEK-Mutationen beim humanen Melanom ist niedrig (3–8 %)2,3
• Einige MEK-Mutationen bewirken eine konstitutive Phosphorylierung von ERK3 und können zur Resistenz gegen die BRAF-Inhibition führen4,5
• MEK-Inhibitoren haben sich bei Melanompatienten mit BRAFV600-Mutation als wirksame Therapiestrategie erwiesen6,7
• Allerdings können Mutationen von MEK14,5,8 und MEK23 auch mit einer Resistenz gegen die MEK-Inhibition verbunden sein
101. Wangari-Talbot J, Chen S. Front Genet. 2013;3:330; 2. Murugan AK et al. Cell Cycle. 2009 ; 8(13):2122–4; 3. Nikolaev SI et al. Nat Genet 2011; 44(2):133–9; 4. Wagle N et al. J Clin Oncol 2011;29:3085–96; 5. Emery CM et al. Proc Natl Acad Sci U S A 2009;106:20411–16; 6. Gilmartin AG, et al. Clin Cancer Res. 2011; 17(5):989–1000; 7. Kim K. Et al J Clin Oncol 2013;31(4):482-9. 8. Trunzer K et al. J Clin Oncol 2013;31(14):1767–74.

Neben BRAF-Mutationen führen auch RAS-Mutationen zu einer Fehlregulation der MAPK-Signalübertragung beim Melanom • Bestimmte Isoformen von RAS sind bei bestimmten Tumorarten besonders häufig
mutiert1
• Die meisten RAS-Mutationen bei Melanomen sind NRAS-Mutationen2,3 – konstitutiv aktivierende Mutationen bei NRAS finden sich bei 15–25 % der metastasierten Melanome4
– KRAS-und HRAS-Mutationen werden nur selten festgestellt und haben in der Regel nur eine schwache onkogene Wirkung beim Melanom2
• Onkogene RAS-Mutationen führen zur Stabilisierung der GTP-Bindung und zur konstitutiven Aktivierung von RAS und den nachgeschalteten Kinasen5
• Der NRAS-Mutationsstatus hat sich als unabhängiger Prädiktor für ein kürzeres Überleben nach Diagnose eines Melanoms im Stadium IV erwiesen6
• Alle Versuche, Moleküle zu entwickeln, die auf die biologische Aktivität von mutiertem RAS einwirken, waren bislang erfolglos7
1. Castellano E, Santos E. Genes Cancer 2011;2(3):216–31; 2. Platz A et al. Mol Oncol 2008;1(4):395–405; 3. Tsao H et al. Genes Dev 2012;26(11):1131–55; 4. Thumar J et al. Mol Cancer 2014;13:45; 5. Samatar AA, Poulikakos PI. Nat Rev Drug Discov 2014;13(12):928–42; 6. Jakob JA et al. Cancer 2012;118(16):4014–23; 7. Gysin S et al. Genes Cancer 2011;2(3):359–72; 8. Prior IA et al. Cancer Res 2012;72(10):2457–67.
11
Schematische Darstellung der Isoformen des RAS-Proteins und häufiger Mutationen. Die wichtigsten onkogenen Mutationen befinden sich in der Region, die bei den 3 Isoformen identisch ist. Nahezu alle der 44 Punktmutationen, die bei den Isoformen von RAS beschrieben wurden, treten in den Codons 12, 13 und 61 auf.8
Sequenzidentität der Isoformen Funktionelle Domänen
Häufige Mutationen
Mg2+/Nukleotidbindung
Effektorbindung
Membranbindung/-trafficking
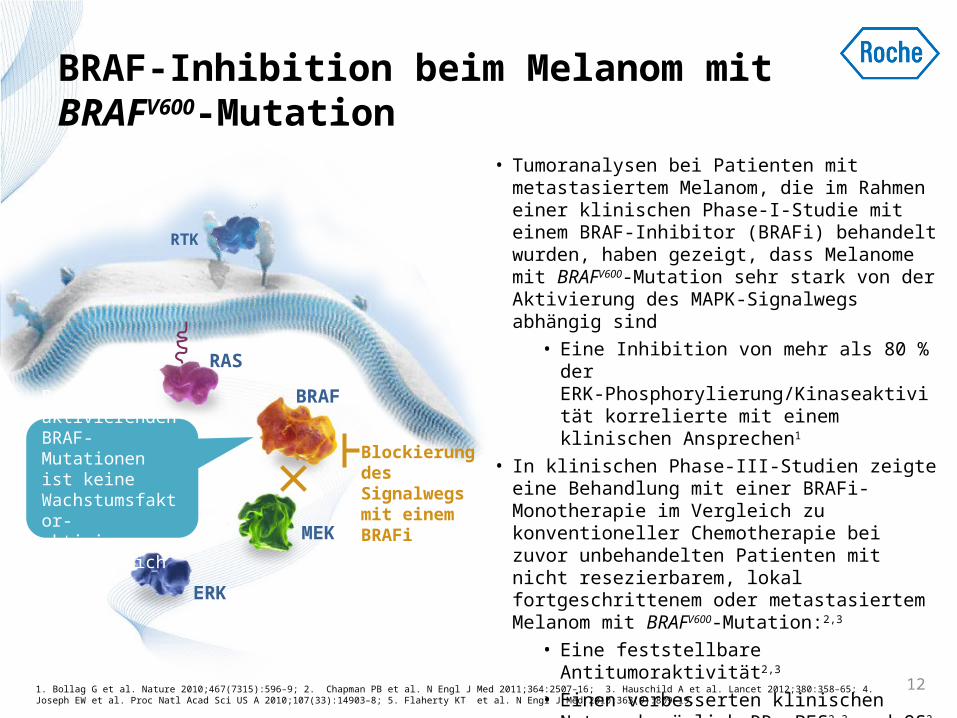
BRAF-Inhibition beim Melanom mit BRAFV600-Mutation
• Tumoranalysen bei Patienten mit metastasiertem Melanom, die im Rahmen einer klinischen Phase-I-Studie mit einem BRAF-Inhibitor (BRAFi) behandelt wurden, haben gezeigt, dass Melanome mit BRAFV600-Mutation sehr stark von der Aktivierung des MAPK-Signalwegs abhängig sind
• Eine Inhibition von mehr als 80 % der ERK-Phosphorylierung/Kinaseaktivität korrelierte mit einem klinischen Ansprechen1
• In klinischen Phase-III-Studien zeigte eine Behandlung mit einer BRAFi-Monotherapie im Vergleich zu konventioneller Chemotherapie bei zuvor unbehandelten Patienten mit nicht resezierbarem, lokal fortgeschrittenem oder metastasiertem Melanom mit BRAFV600-Mutation:2,3
• Eine feststellbare Antitumoraktivität2,3 • Einen verbesserten klinischen Nutzen
bezüglich RR, PFS2,3 und OS2
• BRAFi hat nur bei Melanompatienten mit BRAFV600-Mutation einen klinischen Nutzen4,5
1. Bollag G et al. Nature 2010;467(7315):596–9; 2. Chapman PB et al. N Engl J Med 2011;364:2507–16; 3. Hauschild A et al. Lancet 2012;380:358–65; 4. Joseph EW et al. Proc Natl Acad Sci US A 2010;107(33):14903–8; 5. Flaherty KT et al. N Engl J Med 2010;363(9):809–19.
12
RTK
BRAF
MEK
ERK
RAS
Blockierung des Signalwegs mit einem BRAFi
Bei aktivierenden BRAF-Mutationen ist keine Wachstumsfaktor-aktivierung erforderlich

Resistenzmechanismen gegen BRAFi beim Melanom mit BRAFV600-Mutation
• Letztendlich entwickelt sich bei Melanompatienten mit BRAFV600-Mutation eine Resistenz, die zur Krankheitsprogression führt1–3
• Resistenzmechanismen4–6
1. Reaktivierung des MAPK-Signalwegs2. Erhöhte Signalübertragung durch den
PI3K/AKT-Signalweg
Eine Reaktivierung des MAPK-Signalwegs wird in 70 % der BRAFV600-Melanome beobachtet,
die eine Resistenz gegen BRAFi entwickeln6
1. Chapman PB et al. N Engl J Med 2011;364:2507–16; 2. Hauschild A et al. Lancet 2012;380:358–65; 3. Roche. Zelboraf Summary of product characteristics. 2012; 4. Nazarian R et al. Nature 2010;468:973–7; 5. Paraiso KH et al. Cancer Res 2011;71:2750–60; 6. Shi H et al. Cancer Discov 2014;4:80–93.
Mechanismen bei progressiven Melanomen6
52%
18%4%
26%
Nur MAPK-AktivierungMAPK-Aktivierung und erhöhte AKT-SignalübertragungNur erhöhte PI3K-PTEN-AKT-SignalübertragungUnbekannt
13
RTK
BRAF
MEK
ERK
RAS
PI3K-AKT-Signalweg
CRAF
Resistenz gegen BRAFi durch Reaktivierung über CRAF oder Aktivierung des PI3K-AKT-Signalwegs4-6
Resistenz gegen BRAFi
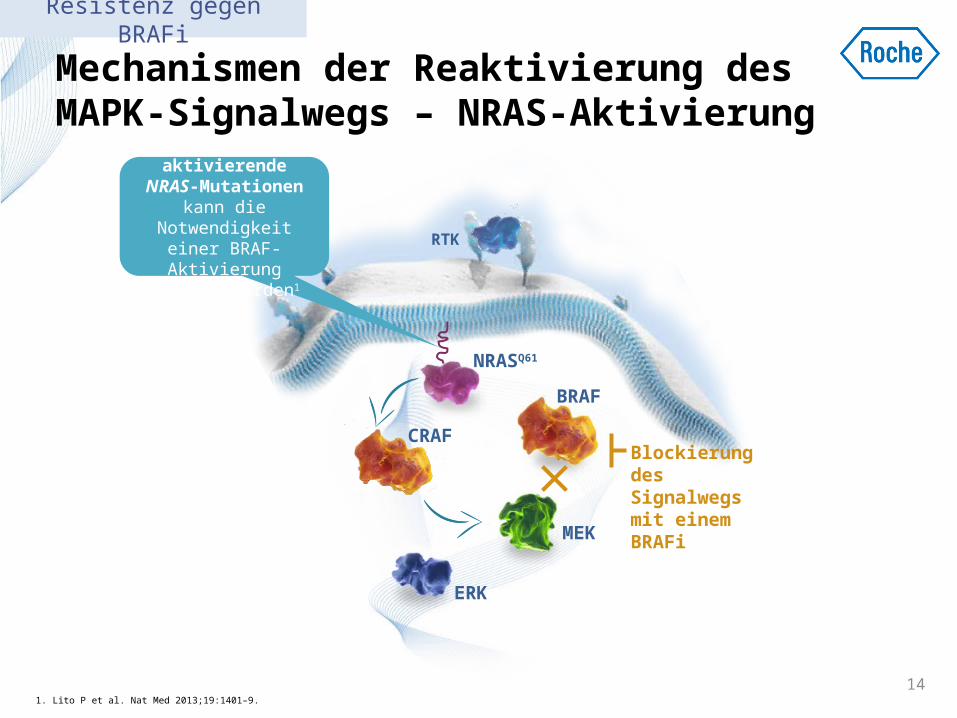
Mechanismen der Reaktivierung des MAPK-Signalwegs – NRAS-Aktivierung
1. Lito P et al. Nat Med 2013;19:1401–9. 14
RTK
BRAF
MEK
ERK
NRASQ61
Durch aktivierende
NRAS-Mutationen kann die
Notwendigkeit einer BRAF-Aktivierung
umgangen werden1
CRAFBlockierung des Signalwegs mit einem BRAFi
Resistenz gegen BRAFi
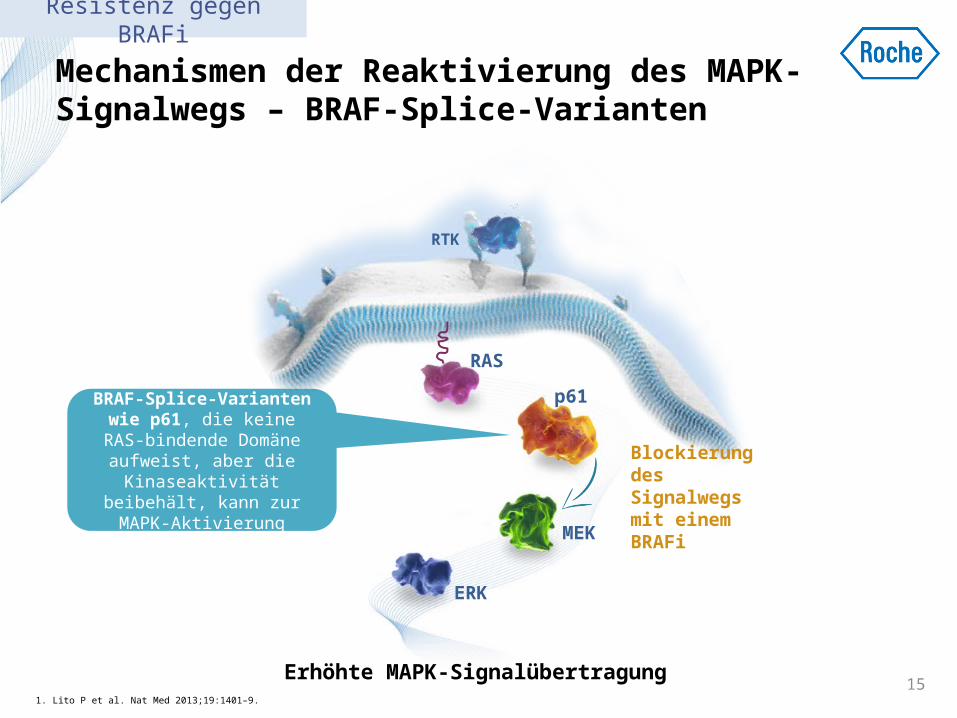
Mechanismen der Reaktivierung des MAPK-Signalwegs – BRAF-Splice-Varianten
Eine Expression von BRAF-Splice-Varianten wie
p61, die keine RAS-bindende Domäne aufweist, aber die
Kinaseaktivität beibehält, kann zur MAPK-Aktivierung
führen1
1. Lito P et al. Nat Med 2013;19:1401–9. 15
RTK
p61
MEK
ERK
RAS
Erhöhte MAPK-Signalübertragung
Blockierung des Signalwegs mit einem BRAFi
Resistenz gegen BRAFi
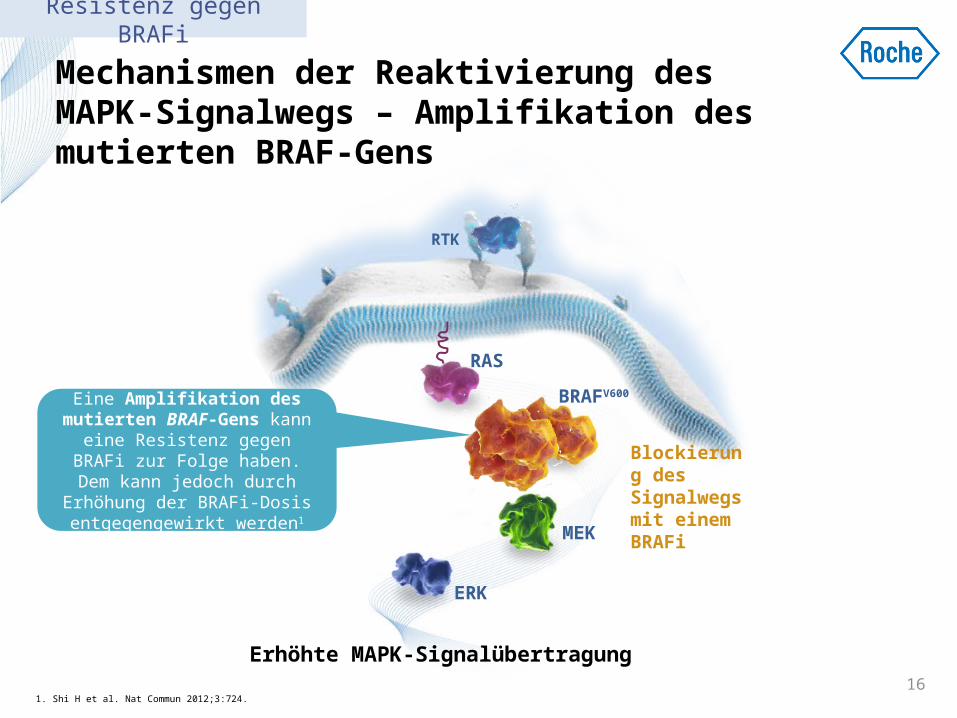
Mechanismen der Reaktivierung des MAPK-Signalwegs – Amplifikation des mutierten BRAF-Gens
1. Shi H et al. Nat Commun 2012;3:724.16
RTK
BRAFV600
MEK
ERK
RAS
Erhöhte MAPK-Signalübertragung
Blockierung des Signalwegs mit einem BRAFi
Eine Amplifikation des mutierten BRAF-Gens kann eine Resistenz gegen BRAFi zur Folge haben. Dem kann jedoch durch Erhöhung der
BRAFi-Dosis entgegengewirkt werden1
Resistenz gegen BRAFi
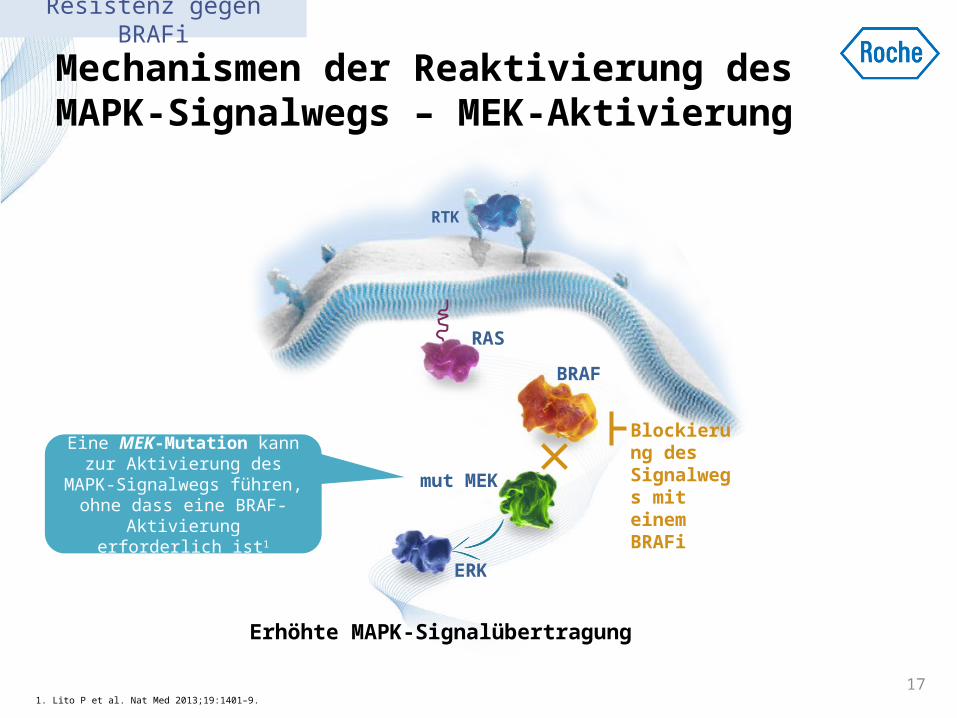
Mechanismen der Reaktivierung des MAPK-Signalwegs – MEK-Aktivierung
1. Lito P et al. Nat Med 2013;19:1401–9. 17
RTK
BRAF
mut MEK
ERK
RAS
Erhöhte MAPK-Signalübertragung
Blockierung des Signalwegs mit einem BRAFi
Eine MEK-Mutation kann zur Aktivierung des MAPK-Signalwegs führen, ohne
dass eine BRAF-Aktivierung erforderlich ist1
Resistenz gegen BRAFi
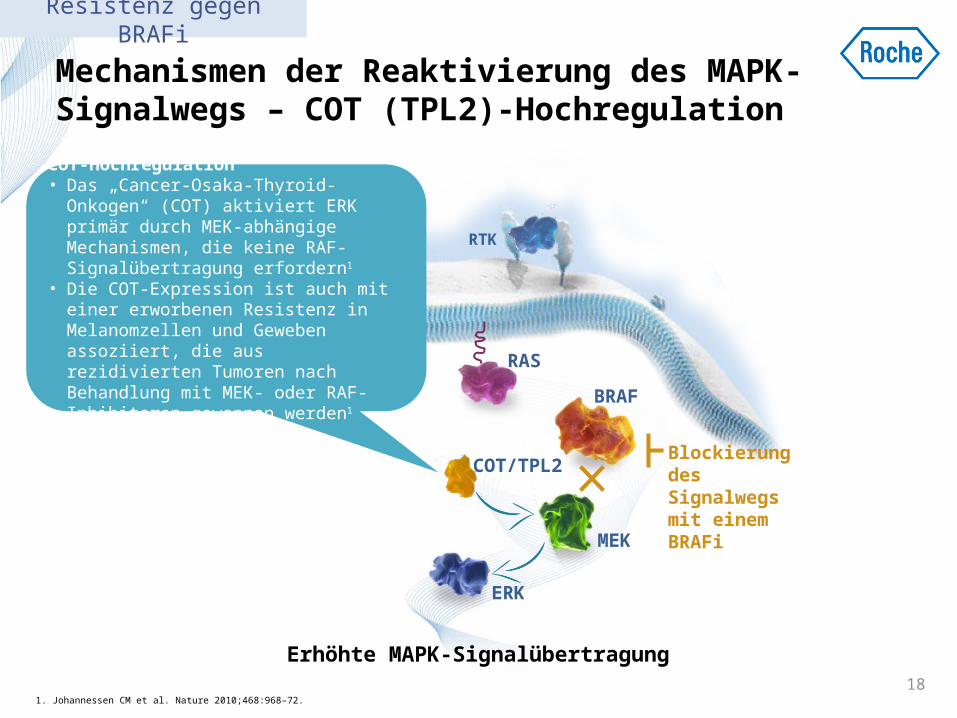
Mechanismen der Reaktivierung des MAPK-Signalwegs – COT (TPL2)-Hochregulation
1. Johannessen CM et al. Nature 2010;468:968–72.18
RTK
BRAF
MEK
ERK
RAS
Erhöhte MAPK-Signalübertragung
Blockierung des Signalwegs mit einem BRAFi
COT/TPL2
COT-Hochregulation • Das „Cancer-Osaka-Thyroid-Onkogen“
(COT) aktiviert ERK primär durch MEK-abhängige Mechanismen, die keine RAF-Signalübertragung erfordern1
• Die COT-Expression ist auch mit einer erworbenen Resistenz in Melanomzellen und Geweben assoziiert, die aus rezidivierten Tumoren nach Behandlung mit MEK- oder RAF-Inhibitoren gewonnen werden1
Resistenz gegen BRAFi
RTK
BRAF
MEK
ERK
RAS
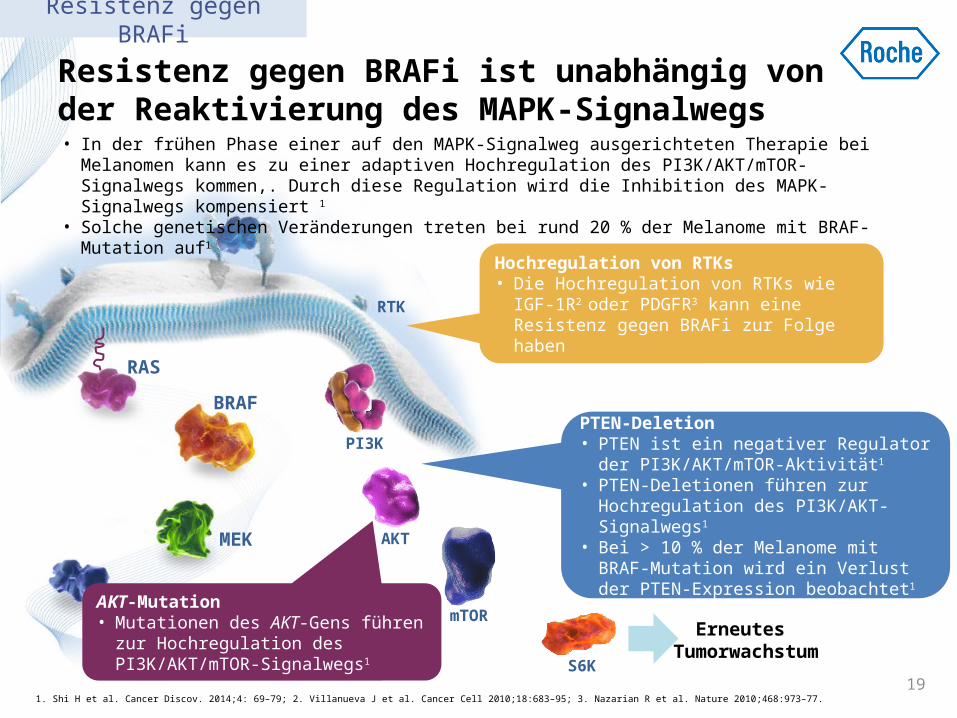
Resistenz gegen BRAFi ist unabhängig von der Reaktivierung des MAPK-Signalwegs
Erneutes Tumorwachstu
m 1. Shi H et al. Cancer Discov. 2014;4: 69–79; 2. Villanueva J et al. Cancer Cell 2010;18:683–95; 3. Nazarian R et al. Nature 2010;468:973–77.
PI3K
AKT
mTOR
S6K
Hochregulation von RTKs • Die Hochregulation von RTKs wie IGF-1R2
oder PDGFR3 kann eine Resistenz gegen BRAFi zur Folge haben
• In der frühen Phase einer auf den MAPK-Signalweg ausgerichteten Therapie bei Melanomen kann es zu einer adaptiven Hochregulation des PI3K/AKT/mTOR-Signalwegs kommen,. Durch diese Regulation wird die Inhibition des MAPK-Signalwegs kompensiert 1
• Solche genetischen Veränderungen treten bei rund 20 % der Melanome mit BRAF-Mutation auf1
PTEN-Deletion • PTEN ist ein negativer Regulator der
PI3K/AKT/mTOR-Aktivität1
• PTEN-Deletionen führen zur Hochregulation des PI3K/AKT-Signalwegs1
• Bei > 10 % der Melanome mit BRAF-Mutation wird ein Verlust der PTEN-Expression beobachtet1
AKT-Mutation • Mutationen des AKT-Gens führen
zur Hochregulation des PI3K/AKT/mTOR-Signalwegs1
19
Resistenz gegen BRAFi
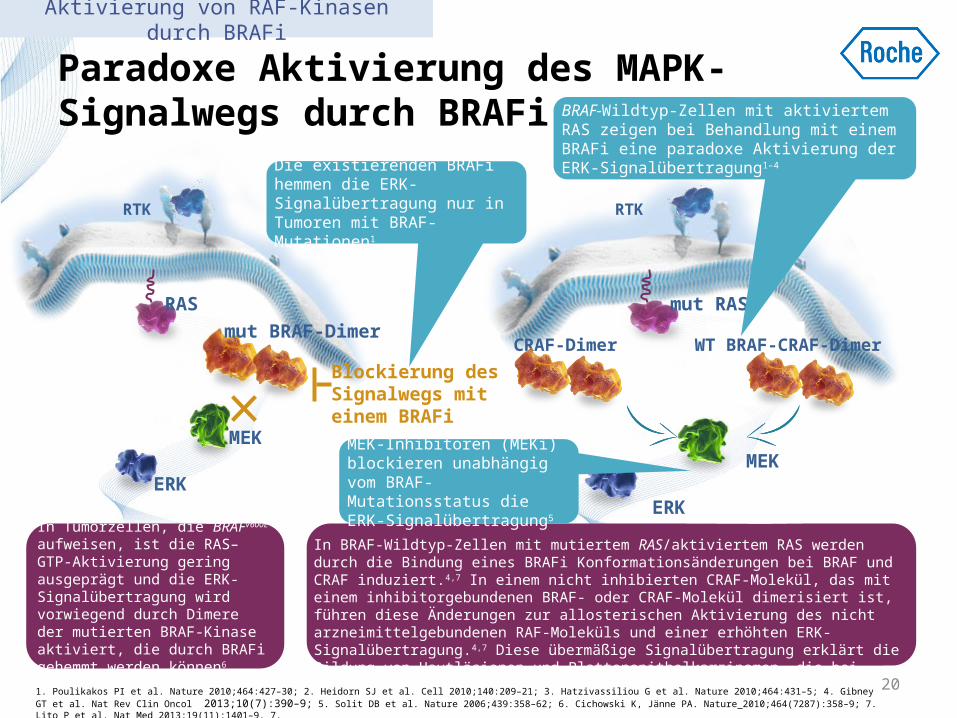
Paradoxe Aktivierung des MAPK-Signalwegs durch BRAFi
20
RTK
mut BRAF-Dimer
MEK
ERK
RAS
In Tumorzellen, die BRAFV600E aufweisen, ist die RAS–GTP-Aktivierung gering ausgeprägt und die ERK-Signalübertragung wird vorwiegend durch Dimere der mutierten BRAF-Kinase aktiviert, die durch BRAFi gehemmt werden können6
RTK
mut RAS
CRAF-Dimer
MEK
ERK
WT BRAF-CRAF-Dimer
In BRAF-Wildtyp-Zellen mit mutiertem RAS/aktiviertem RAS werden durch die Bindung eines BRAFi Konformationsänderungen bei BRAF und CRAF induziert.4,7 In einem nicht inhibierten CRAF-Molekül, das mit einem inhibitorgebundenen BRAF- oder CRAF-Molekül dimerisiert ist, führen diese Änderungen zur allosterischen Aktivierung des nicht arzneimittelgebundenen RAF-Moleküls und einer erhöhten ERK-Signalübertragung.4,7 Diese übermäßige Signalübertragung erklärt die Bildung von Hautläsionen und Plattenepithelkarzinomen, die bei Patienten mit Melanom beobachtet werden6
Blockierung des Signalwegs mit einem BRAFi
Die existierenden BRAFi hemmen die ERK-Signalübertragung nur in Tumoren mit BRAF-Mutationen1
BRAF-Wildtyp-Zellen mit aktiviertem RAS zeigen bei Behandlung mit einem BRAFi eine paradoxe Aktivierung der ERK-Signalübertragung1–4
MEK-Inhibitoren (MEKi) blockieren unabhängig vom BRAF-Mutationsstatus die ERK-Signalübertragung5
1. Poulikakos PI et al. Nature 2010;464:427–30; 2. Heidorn SJ et al. Cell 2010;140:209–21; 3. Hatzivassiliou G et al. Nature 2010;464:431–5; 4. Gibney GT et al. Nat Rev Clin Oncol 2013;10(7):390–9; 5. Solit DB et al. Nature 2006;439:358–62; 6. Cichowski K, Jänne PA. Nature 2010;464(7287):358–9; 7. Lito P et al. Nat Med 2013;19(11):1401–9. 7.
Aktivierung von RAF-Kinasen durch BRAFi
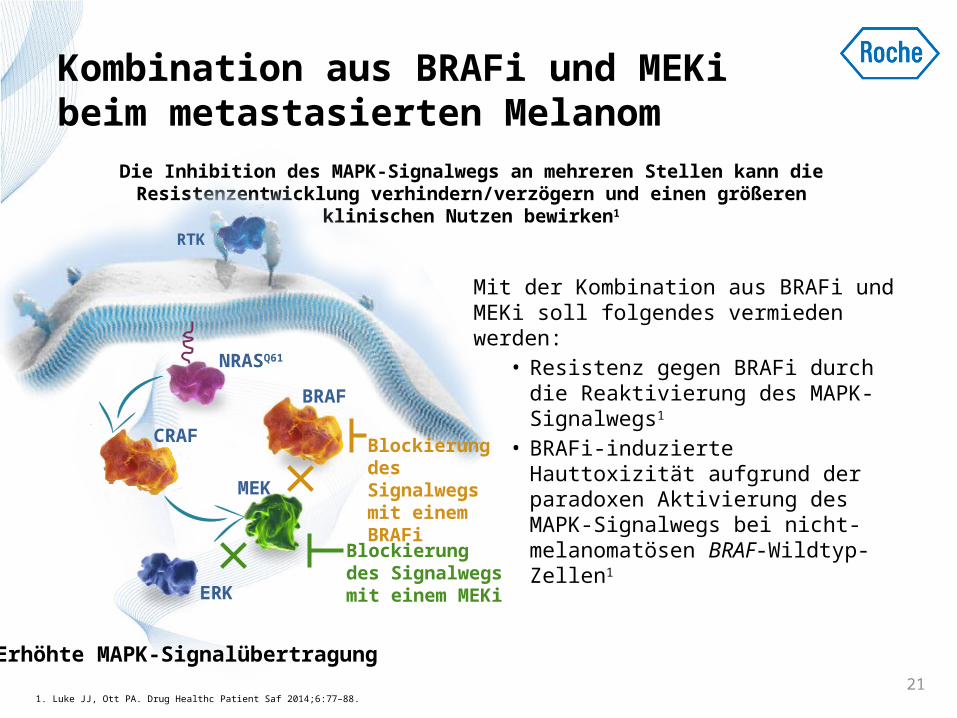
Kombination aus BRAFi und MEKi beim metastasierten Melanom
Mit der Kombination aus BRAFi und MEKi soll folgendes vermieden werden:
• Resistenz gegen BRAFi durch die Reaktivierung des MAPK-Signalwegs1
• BRAFi-induzierte Hauttoxizität aufgrund der paradoxen Aktivierung des MAPK-Signalwegs bei nicht-melanomatösen BRAF-Wildtyp-Zellen1
1. Luke JJ, Ott PA. Drug Healthc Patient Saf 2014;6:77–88.
Die Inhibition des MAPK-Signalwegs an mehreren Stellen kann die Resistenzentwicklung verhindern/verzögern und einen größeren
klinischen Nutzen bewirken1
21
RTK
BRAF
MEK
ERK
NRASQ61
Erhöhte MAPK-Signalübertragung
CRAF Blockierung des Signalwegs mit einem BRAFi
Blockierung des Signalwegs mit einem MEKi
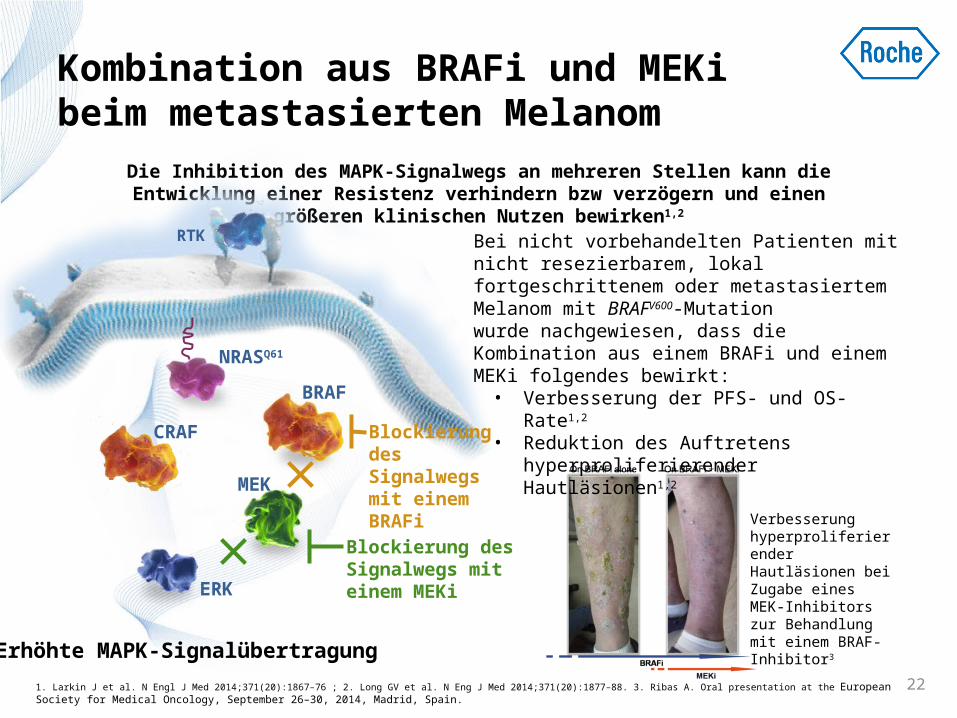
Kombination aus BRAFi und MEKi beim metastasierten Melanom
1. Larkin J et al. N Engl J Med 2014;371(20):1867–76 ; 2. Long GV et al. N Eng J Med 2014;371(20):1877–88. 3. Ribas A. Oral presentation at the European Society for Medical Oncology, September 26–30, 2014, Madrid, Spain.
Die Inhibition des MAPK-Signalwegs an mehreren Stellen kann die Entwicklung einer Resistenz verhindern bzw verzögern und einen
größeren klinischen Nutzen bewirken1,2
Bei nicht vorbehandelten Patienten mit nicht resezierbarem, lokal fortgeschrittenem oder metastasiertem Melanom mit BRAFV600-Mutation wurde nachgewiesen, dass die Kombination aus einem BRAFi und einem MEKi folgendes bewirkt: • Verbesserung der PFS- und OS-Rate1,2
• Reduktion des Auftretens hyperproliferierender Hautläsionen1,2
22
RTK
BRAF
MEK
ERK
NRASQ61
Erhöhte MAPK-Signalübertragung
CRAF Blockierung des Signalwegs mit einem BRAFi
Blockierung des Signalwegs mit einem MEKi
Verbesserung hyperproliferierender Hautläsionen bei Zugabe eines MEK-Inhibitors zur Behandlung mit einem BRAF-Inhibitor3

GlossarBegriff Beschreibung
Apoptose Eine Art des Zelltods, der durch eine Reihe molekularer Schritte in der betreffenden Zelle ausgelöst wird. Dies ist der normale Weg des Körpers, nicht benötigte oder abnorme Zellen zu beseitigen. Der Prozess der Apoptose kann in Tumorzellen blockiert sein. Wird auch als programmierter Zelltod bezeichnet.National Cancer Institute. Krebs-Wörterbuch. http://www.cancer.gov/dictionary. Aufgerufen am 28. Juli 2014.
BRAF BRAF gehört zur RAF-Familie der Serin-/Threoninkinasen und bildet einen wesentlichen Bestandteil des MAPK-Signalwegs.
ERK Die Extracellular-Signal-Regulated-Kinase (durch extrazelluläre Signale regulierte Kinase, ERK) ist eine Proteinkinase, die einen Bestandteil des Ras-Raf-Signalwegs bildet. Die ERK ist ein Typ der MAPK. Die Aktivierung von ERK führt zur Aktivierung von Transkriptionsfaktoren, die die Expression von Genen bewirken. Diese Gene regulieren die Proliferation und das Überleben von Zellen.
Glutaminsäure (E), Valin (V), Asparaginsäure (D), Lysin (K), Arginin (R)
Proteinogene Aminosäuren, also Aminosäuren, die Vorläufer von Proteinen sind und während der Translation in Proteine eingebaut werden.
GDP Guanosindiphosphat (GDP) ist ein Nukleosiddiphosphat, das bei der Dephosphorylierung von GTP durch GTPasen entsteht, z. B. die an der Signaltransduktion beteiligten G-Proteine; GDP wird mithilfe von Pyruvatkinase und Phosphoenolpyruvat in GTP umgewandelt.
GTP Guanosin-5'-triphosphat (GTP) ist ein Purin-Nukleosidtriphosphat. GTP ist essentiell für die Signaltransduktion, insbesondere bei Beteiligung von G-Proteinen an Second-Messender-Kaskaden, wobei es durch GTPasen in GDP umgewandelt wird
MAPK Mitogenaktivierte Proteinkinasen (MAPKs) sind Enzyme mit Serin-/Threoninkinaseaktivität (z. B. ERK). MAPKs regulieren über nachgeschaltete regulierende zelluläre Zielmoleküle infolge extrazellulärer Stimuli verschiedene zelluläre Prozesse wie Zellproliferation und -differenzierung.
23
GlossarBegriff Beschreibung
Melanozyt Melanozyten sind Zellen in der Basalschicht der Epidermis1. Sie produzieren das Pigment Melanin,1–4 vorwiegend in einer als Melanosom bezeichneten zytoplasmatischen Organelle.1,3 Melanin wird infolge von UVR-Einstrahlung in Keratinozyten transportiert4 und über die „sonnenexponierte“ Seite der Zellkerne verteilt, um die Haut vor Strahlenschäden zu schützen3
MEK MEK ist eine Proteinkinase, die einen Bestandteil der MAPK-Signalkaskade bildet. Diese reguliert die Expression einer großen Zahl von Proteinen, die an der Kontrolle der Proliferation, Differenzierung und Apoptose von Zellen beteiligt sind.
MITF Der Mikrophthalmie-assoziierte Transkriptionsfaktor (MITF) ist an der Regulation der zelllinienspezifischen Signalwege vieler Zelltypen einschließlich der Melanozyten beteiligt
PFS Progressionsfreies Überleben (PFS) – gibt in einer klinischen Studie die Länge des Zeitraums an, in dem der Krebs oder Tumor nicht fortschreitet; ist in vielen klinischen Studien ein primärer Endpunkt.
RAF Raf ist eine Proteinkinasefamilie, die bei verschiedenen für die Tumorgenese relevanten zellulären Reaktionen, einschließlich Zellproliferation, Invasion, Überleben und Angiogenese, eine Rolle spielt. Die Raf-Familie besteht aus den drei Mitgliedern ARAF, BRAF und CRAF, die alle eine unterschiedliche Funktion haben und auf verschiedenen Ebenen unterschiedlich reguliert werden.
RAS Ras ist ein GTP-bindendes Protein, das infolge der Bindung von Wachstumsfaktoren, Zytokinen und Hormonen an Rezeptoren auf der Zelloberfläche aktiviert wird. Die GTP-gebundenen Formen von Ras gehen eine direkte Bindung mit Raf ein und rekrutieren es zur Plasmamembran.
RR Ansprechrate („response rate“) – der prozentuale Anteil der Patienten, deren Tumoren nach der Behandlung kleiner werden (partielles Ansprechen) oder ganz verschwinden (komplettes Ansprechen).
241. Cichorek M et al. Postepy Dermatol Alergol. 2013;30(1):30–4; 2. Yaar M, Gilchrest BA. Clin Exp Dermatol. 2001;26(7):583–91; 3. Lin JY, Fisher DE. Nature 2007;445(7130):843–50; 4. Tarafder AK et al J Invest Dermatol 2014;134(4):1056–66.

GlossarBegriff Beschreibung
RTKs Rezeptortyrosinkinasen sind membrandurchspannende Proteine an der Zelloberfläche, die eine extrazelluläre Ligandenbindungsdomäne am N-Terminus und eine intrazelluläre Tyrosinkinasedomäne am C-Terminus aufweisen.McKay MM, Morrison DK. Integration von Signalen von den RTKs zu ERK/MAPK. Oncogene. 2007;26:3113-3121.
OS Gesamtüberleben („overall survival“) – der prozentuale Anteil der Patienten einer Studie, die nach einer bestimmten Zeit im Anschluss an die Studie noch immer am Leben sind.
25

Weiterführende Literatur
In den folgenden Artikeln erhalten Sie weitere Informationen über den MAPK-Signalweg und seine Rolle beim Melanom
• Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014 Jul;14(7):455–467.
• Pritchard AL, Hayward NK. Molecular pathways: mitogen-activated protein kinase pathway mutations and drug resistance. Clin Cancer Res. 2013 May 1;19(9):2301–9. Epub 2013 Feb 13
• Spagnolo F, Ghiorzo P, Flaherty KT. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget. 2014;5(21):10206–10221.
• Sullivan RJ1, Flaherty K. MAP kinase signalling and inhibition in melanoma. Oncogene. 2013 May 9;32(19):2373–9. Epub 2012 Sep 3
• Lito P et al. Tumour adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401–9.
Literatur
27
Bastian BC et al. Cancer Res 1998;58:2170–75 Evans SS et al. Dermatol Surg 2014;40(12):1385–9
Bollag G et al. Nature 2010;467(7315):596–9 Finn L et al. BMC Med 2012;10:23
Cantwell-Dorris ER et al. Mol Cancer Ther 2011;10:385–94
Flaherty KT et al. N Engl J Med 2010;363(9):809–19
Castellano E, Santos E. Genes Cancer 2011;2(3):216–31
Garbe C et al. Eur J Cancer 2012;48(15):2375–90
Chapman PB et al. N Engl J Med 2011;364:2507–16 Garnett M, Marais R. Cancer Cell 2004;6:313–9
Christensen JG et al. Cancer Lett 2005;225(1):1–26Gilmartin AG, et al. Clin Cancer Res. 2011; 17(5):989–1000
Cichowski K, Jänne PA. Nature 2010;464(7287):358–9Gibney GT et al. Nat Rev Clin Oncol 2013;10(7):390–9
Cruz J et al. Oncology 2003;65:72–82 Gray-Schopfer V et al. Nature 2007;445(7130):851–7
Cseh B et al. FEBS Lett 2014;588:2398–406Gray-Schopfer VC et al. Cancer Metastasis Rev 2005;24:165–83
Davies H et al. Nature 2002;417:949–54 Gysin S et al. Genes Cancer 2011;2(3):359–72
Davis MJ, Schlessinger JJ Cell Biol 2012;199:15–19 Hatzivassiliou G et al. Nature 2010;464:431–5
Dhillon AS et al. Oncogene 2007 ;26(22):3279–90 Hauschild A et al. Lancet 2012;380:358–65Diepgen TL, Mahler V. Br J Derm 2002;146 Suppl 61:1–6 Heidorn SJ et al. Cell 2010;140:209–21Easty DJ et al. Pigment Cell Melanoma Res 2011;24(3):446–61
Heinzerling L et al. Br J Cancer 2013;108(10):2164–71
Emery CM et al. Proc Natl Acad Sco USA 2009;106:20411–6
Holderfield M et al. Nat Rev Cancer. 2014 ;14(7):455–67
Hsu M et al. J Cell Sci 2000;113:1535–42 Lemech C et al. Ther Adv Med Oncol 2012;4:61–73Hwang D, Rhee SH. Am J Clin Nutr 1999;70(4):545–56 Lens MB, Dawes M. Br J Derm 2004;150(2):179–85Inamdar GS et al. Biochem Pharmacol 2010;80(5):624–37
Li G, Quian H. Cell Biochem Biophys 2003;39(1):45–59
Jacobs JF et al. Lancet Oncol 2012;13(1):e32–42 Libra M et al. Cell Cycle 2005;4:1382–4Jakob JA et al. Cancer. 2012;118(16):4014–23 Linos E et al. J Inv Dermatol 2009 ;129(7):1666–74Jarkowski A 3rd et al. Ann Pharmacother 2014;48(11):1456–68 Lito P et al. Nat Med 2013;19:1401–9Jilaveanu LB et al. Clin Cancer Res 2009; 15(18):5704–13 Lo JA, Fisher DE. Science. 2014;346(6212):945–9
Johannessen CM et al. Nature 2010;468:968–72 Long GV et al. J Clin Oncol. 2011;29(10):1239–46 Joseph EW et al. Proc Natl Acad Sci US A 2010;107(33):14903–8 Long GV et al. N Eng J Med 2014;371(20):1877–88
Kim K. J et al. Clin Oncol 2013;31(4):482-9 Luke JJ, Ott PA. Drug Healthc Patient Saf 2014;6:77–88
Koprowski H et al. Somat Cell MolGenet 1985;11:297–302
Maldonado JL et al. J Natl Cancer Inst 2003;95:1878–90
Korn EL et al. J Clin Oncol 2008;26(4):527–34 Mann GJ et al. J Invest Dermatol 2013;133(2):509–17
Kumar R et al. J Invest Dermatol 2004;122:342–48 McArthur GA, Ribas A. J Clin Oncol 2013;31(4):499–506
Kwong LN, Davies MA. Oncogene 2014;33:1–9 McCubrey JA, et al. Biochim Biophys Acta 2007;1773:1263–84
Larkin J et al. N Engl J Med 2014 ;371(20):1867–76 Mercer KE, Pritchard CA. Biochim Biophys Acta 2003;1653:25–40
Literatur
28
Michaloglou C et al. Oncogene 2008;27:877–95 Puri N et al. Clin Cancer Res 2007;13:2246–53
Mielke LA et al. J Immunol 2009;183(12):7984–93 Pylayeva-Gupta Y, et al. Nature Rev Cancer 2011;11:761–72
Miller AJ, Mihm MC Jr. N Engl J Med 2006;355(1):51–65 Rahman MA, et al. Exp Mol Pathol 2013;95:336–42Murphy LO, Blenis J.Trends Biochem Sci 2006;31: 268–75 Ribas A et al. Clin Cancer Res 2012;18:336–41
Murugan AK et al. Cell Cycle. 2009 ; 8(13):2122–4 Roberts PJ, Der CJ. Oncogene 2007;26(22):3291–310
Natali PG et al. Br J Cancer 1993;68:746–50 Robinson MJ, Cobb MH. Curr Opin Cell Biol 1997;9:180–6
Nazarian R, et al. Nature 2010;468:973–977 Roche. Zelboraf Summary of product characteristics. 2012
Nikolaev SI et al. Nat Genet. 2011; 44(2):133–9 Roskoski R, Jr. Pharmacol Res 2012;66:105–43
Omholt K et al. Clin Cancer Res 2003;9:6483–8 Samatar AA, Poulikakos PI. Nat Rev Drug Discov 2014;13(12):928–42
Paraiso KH et al. Cancer Res 2011;71:2750–60 Schlegel J et al. J Clin Invest 2013;123(5):2257–67Peters S, Adjei AA. Nature Rev Clin Oncol 2012;9:314–26 Schulze A et al. Mol Biol Cell 2004;15:3450–63
Platz A et al. Mol Oncol 2008 ;1(4):395–405 Sharma A et al. Cancer Res 2005;65:2412–21Pollock PM et al. Nat Genet 2003;33:19–20 Sharma A et al. Cancer Res 2005;65:2412–21Poulikakos PI et al. Nature 2010;464:427–30 Shi H et al. Cancer Discov 2014;4:80–93Prenen H et al. Clin Cancer Res 2010;16(11):2921–6 Shi H, et al. Cancer Discov. 2014; 4: 69–79
Literatur
29
Literatur
30
Shi H, et al.2012 Nat Commun 2012;3:724 Wan PT et al. Cell 2004;116:855–67Shinozaki M et al. Clin Cancer Res 2004;10:1753–1757 Wang AX, Qi XY. Life. 2013;65:748–58
Smalley KS. Int J Cancer 2003;104(5):527–32 Wangari-Talbot J, Chen S. Front Genet. 2013 Jan 25;3:330
Solit DB, Rosen N. N Engl J Med 2011; 364:772–74 Wellbrock C et al. Nat Rev Mol Cell Biol 2004;5(11):875–85
Tarafder AK et al J Invest Dermatol 2014;134(4):1056–66 Wellbrock C et al. PLoSOne 2008;3(7):e2734
Thumar J et al. Mol Cancer 2014;13:45 Wiltshire RN et al. Cancer Res 1995;55:3954–57Tronnier M, Mitteldorf C. Cancer Manag Res 2014;6:349–56 Wolf I, Seger R. Isr Med Assoc J 2002;4(8):641–7
Trunzer K et al. J Clin Oncol 2013;31(14):1767–74 Wortzel I, Seger R. Genes Cancer. 2011 Mar;2(3):195–209
Tsao H et al. Genes Dev 2012;26(11):1131–55 Yamamoto T et al. Curr Biol 2006;16: 1171–82Uribe P et al. Am J Dermatopathol 2003;25:365–70 Yewale C, et al. Biomaterials 2013;34:8690–707Villanueva J, et al. Cancer Cell 2010;18:683–95 Yoon S, Seger R. Growth Factors 2006;24:21–44Wagle N et al. J Clin Oncol 2011;29:3085–96 Zhang W, Liu HT. Cell Res 2002;12(1):9–18


















