
Reinigung, Faltung und Charakterisierung von GFP · großes Protein, welches 1962 von Shimomura...
20
Reinigung, Faltung und Charakterisierung von GFP Erster Praktikumsteil zum Modul Proteinchemie 2 ifmb Institut für Mikrobiologie der Leibniz-Universität Hannover
Transcript of Reinigung, Faltung und Charakterisierung von GFP · großes Protein, welches 1962 von Shimomura...

Reinigung, Faltung undCharakterisierung von GFP
Erster Praktikumsteil zum Modul
Proteinchemie 2
ifmbInstitut für Mikrobiologie
der Leibniz-Universität Hannover

Inhaltsverzeichnis
1 Einleitung 4
2 Theorie 4
2.1 GFP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2 Expressionssystem . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.3 Proteinfaltung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2.3.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2.3.2 Inclusion bodies . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.3.3 IB–Präparation und Faltung von GFP . . . . . . . . . . . . . . 8
2.4 Spektroskopie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.4.1 CD–Spektroskopie . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.4.2 Fluoreszenzspektroskopie . . . . . . . . . . . . . . . . . . . . . . 9
3 Praktikum 11
3.1 Lösungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
3.2 Zellanzucht und –ernte . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Anzucht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Ernte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
3.3 Zellaufschluss und IB-Präparation . . . . . . . . . . . . . . . . . . . . . 12
Aufschluss . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
IB–Präparation . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
3.4 Ni–IMAC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
Äquilibrierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
Beladen und Waschen . . . . . . . . . . . . . . . . . . . . . . . 13
Elution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
Dialyse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
3.5 Proteinkonzentrationsbestimmung . . . . . . . . . . . . . . . . . . . . . 14
3.6 SDS–PAGE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Herstellung der Gele . . . . . . . . . . . . . . . . . . . . . . . . 15
Beladung und Lauf . . . . . . . . . . . . . . . . . . . . . . . . . 15
Gelscan und –färbung . . . . . . . . . . . . . . . . . . . . . . . 15
2

3.7 CD–Spektroskopie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Messung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.8 Fluoreszenzspektroskopie . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4 Protokoll 17
Generelles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
Einleitung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
Materialien und Methoden . . . . . . . . . . . . . . . . . . . . . 17
Ergebnisse und Diskussion . . . . . . . . . . . . . . . . . . . . . 17
Literaturverzeichnis . . . . . . . . . . . . . . . . . . . . . . . . . 18
Format . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Einheiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Gene und Proteine . . . . . . . . . . . . . . . . . . . . . . . . . 18
Rechtschreibung . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Abbildungen, und: Präsens oder Perfekt? . . . . . . . . . . . . . 19
3

1 Einleitung
Die Produktion großer Proteinmengen ist sowohl für die grundlagenorientierten mo-
lekularen Biowissenschaften als auch die Biotechnologie von immenser Wichtigkeit.
Erstere benötigen hochreines Protein zur Aufklärung von Strukturen und Wirkmecha-
nismen, während letztere große Menge für die Herstellung von Medikamenten (Insulin)
oder Enzymen (in Waschmitteln etc.) benötigt. In einem Großteil aller Fälle wird dabei
auf ein bakterielles Produktionssystem zurückgegriffen, welches Proteine einfach und
effizient produzieren kann.
Damit erlangt auch die Charakterisierung von Proteineigenschaften große Bedeutung:
Welche Struktur besitzt mein Protein? Wie stabil ist es? Sind seine Eigenschaften unter
der Bedingung X die gleichen wie unter Bedingung Y?
In diesem Praktikum wird anhand des Proteins GFP gezeigt, wie Proteine überpro-
duziert werden können, wie sie von einer aggregierten Form in ihre native und damit
funktionsfähige Form überführt werden können und mit welchen Methoden die Struk-
tur und Stabilität von Proteinen analysiert werden kann.
2 Theorie
2.1 GFP
Das Grün Fluoreszierende Protein (green fluorescent protein, GFP) ist ein etwa 27 kDa
großes Protein, welches 1962 von Shimomura Osamu aus der Qualle Aequorea victo-
ria isoliert werden konnte. In seiner natürlichen, zellulären Umgebung absorbiert es
blaues Licht, welches vom Protein Aequorin produziert wird und strahlt es als grünes
Licht wieder ab (Fluoreszenz). Nachdem Anfang der 1990er Jahre die Klonierung des
Gens und seine rekombinante Expression in E. coli gelang, zeigte sich, dass GFP (ne-
ben blauem Licht) keine weiteren Cofaktoren benötigt und stabil aus dem Bakterium
gereinigt werden kann. Darüberhinaus zeigte die Aufklärung der Kristallstruktur, dass
es die Form einer aus 12 β–Faltblättern aufgebaute „Tonne“ einnimmt (sog. β–barrel,
Abb.2.1), in deren Mitte ein Chromophor mittels zweier kurzer α–Helices „eingespannt“
vorliegt. Dieser Chromophor entsteht autokatalytisch durch die Zyklisierung des Pro-
teinrückgrats der Serin 65, Tyrosin 66 und Glycin 67, wodurch die Absorption blauen
4

Lichts mit anschließender Fluoreszenz ermöglicht wird (Abb.2.1). N– und C–Terminus
des Proteins liegen zugänglich auf der Außenseite des Proteins, sodass sich GFP auch
für Fusionsexperimente sehr gut eignet. Durch geringe Veränderungen der Aminosäure-
sequenz konnten in anderen Wellenlängenbereichen fluoreszierende Varianten von GFP
hergestellt werden, unter anderem die blau, gelb und rot fluoreszierenden Varianten
CFP, YFP und RFP. Für die Entdeckung, Klonierung und Charakterisierung erhiel-
ten Martin Chalfie, Shimomura Osamu und Roger Y. Tsien 2008 den Nobelpreis für
Chemie.
Abbildung 2.1: Struktur des Grün Fluoreszierenden Proteins (GFP). Links: Darstellung der Ober-fläche des Proteins. Rechts: Darstellung der β–barrel–Sekundärstruktur mit dem innenliegenden Chro-mophor (grün).
2.2 Expressionssystem
Für die Reinigung und Analyse von GFP bietet sich die rekombinante Expression des
entsprechenden Gens in E. coli an. Im Praktikum wird der E. coli–Stamm BL21(DE3)
verwendet. Er ist ein für hohe Proteinproduktion optimierter Stamm, welcher in der hier
verwendeten Form noch ein auf dem Genom eingefügtes Phagen–RNA–Polymerasegen
(für die T7–Polymerase) trägt. Kontrolliert wird die Expression dieses zusätzlichen
Gens durch Voranstellung des lac–Promotors, welcher bekanntlichermaßen durch Zuga-
be von Laktose (oder ihres nicht abbaubaren Analogons IPTG) induziert werden kann.
Wird nun ein Plasmid in die Zelle eingebracht, welches ein Gen mit T7–Polymerase–
Bindesequenz enthält, wird dieses von der sehr effizienten T7–Polymerase erkannt und
die entsprechende mRNA mit einer hohen Transkriptionsrate produziert.
5

Abbildung 2.2: Detailansicht des durch zwei Helices mit der β–barrel–Struktur verknüpften GFP–Chromophors. Die durch Autokatalyse entstandene Bindung ist mit einem Pfeil markiert.
Im Rahmen des Praktikums wird ein solches Konstrukt, gfp–H6 im pET–Vektorsystem,
verwendet. Das Gen besitzt einen 3’–Anhang an der gfp–Basensequenz, welcher im fer-
tigen Protein zur Anfügung von sechs Histidinen führt (Hexahistidin-Tag, His-Tag),
was für die Reinigung des Proteins von entscheidender Bedeutung sind.
2.3 Proteinfaltung
2.3.1 Grundlagen
Alle Proteine müssen eine gefaltete Konformation einnehmen, um ihre zelluläre Aufgabe
übernehmen zu können. Die Faltungsreaktion läuft unter physiologischen Bedingungen
spontan ab, und führt von einer größtenteils ungefalteten Polypeptidkette über die
Ausbildung kleinerer Strukturelemente bis zu einem funktionalen Protein.
Dieser Weg lässt sich aus dem natürlichen Bestreben des Systems (Protein + Umwelt)
verstehen, die Freie Reaktionsenthalpie (Gibbs–Energie, G) zu minimieren. Jede Ver-
änderung, welche G verringert (also Gvorher −Gnachher =∆G= negativ), findet spontan
statt. Der korrekt gefaltete Zustand eines Proteins muss folglich also jener Zustand
6

sein, welcher bei physiologischen Verhältnissen unter allen möglichen Konformationen
des Proteins die geringste Freie Reaktionsenthalpie besitzt und damit den größten ∆G
zum ungefalteten Zustand.
Welche Faktoren spielen nun bei der Proteinfaltung eine Rolle? Aus der Gibbs–Gleichung
∆G=∆H - T∆S wird ersichtlich, dass sowohl die Reaktionsenthalpie (H) als auch die
Entropie des Systems (S, Maß für die „Unordnung“) die Gibbs–Energie beeinflussen.
Die Faltung selbst wird unter anderem dadurch vorangetrieben, dass hydrophobe Sei-
tenketten durch den Hydrophoben Effekt zusammengelagert und vom polaren Medium
abgeschirmt werden. Dadurch nimmt die Entropie des Systems teilweise ab (da gefal-
tete Polypeptidketten höher geordnet sind als frei bewegliche), aber auch zu (da vorher
um hydrophobe Seitenketten strukturiert vorliegende Wassermoleküle frei werden). Zu
diesem Einfluss von ∆S auf ∆G kommen noch Veränderungen von ∆H hinzu, die
durch die Ausbildung von z.B. Wasserstoffbrücken und Interaktionen geladener Res-
te (Salzbrücken) einen negativen Wert aufweisen, sodass ∆G für die Faltungsreaktion
schlussendlich negativ wird und die Reaktion somit spontan abläuft.
2.3.2 Inclusion bodies
Inclusion bodies (IBs) sind große Aggregate aus falsch– bzw. ungefalteten Proteinen.
Sie entstehen spontan unter bestimmten zellulären Bedingungen. Da zu IBs aggregierte
Proteine von den Organismen nicht mehr genutzt werden können, und für sie damit
ausschließlich einen Verlust an Aminosäuren und Energie bedeuten, sind sowohl die
Proteinproduktion als auch die Proteine selbst evolutionär so entwickelt, dass in nicht–
manipulierten Organismen keine nennenswerte IB–Bildung auftritt.
IBs entstehen aus einer Konkurrenzsituation zweier Reaktionswege: 1. der korrekten
Faltung eines Proteins und 2. Interaktion eines ungefalteten bzw. falschgefalteten Pro-
teins mit einem anderen. Proteine falten größtenteils spontan in eine korrekte Form,
wobei die Reaktion einer Kinetik 1. Ordnung folgt (Reaktionsgeschwindigkeit hängt nur
von der Proteinkonzentration ab). Aggregationsreaktionen sind aber immer von der In-
teraktion der Proteine abhängig, und sind damit mindestens Reaktionen 2. Ordnung
(Geschwindigkeit abh. vom Quadrat der Konzentration des ungefalteten Proteins).
Das bedeutet im Organismus, dass IB–Bildung immer dann stattfinden kann, wenn die
Geschwindigkeit der Aggregationsreaktion größer ist als die der korrekten Faltung. Um-
7
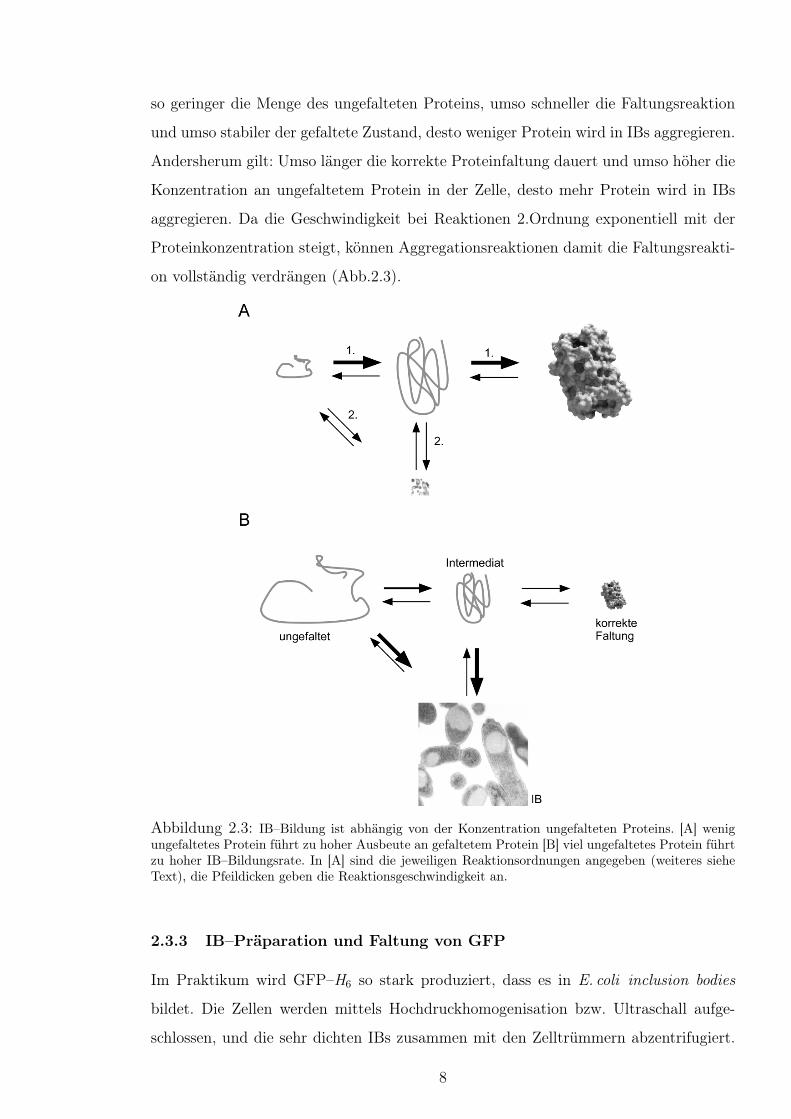
so geringer die Menge des ungefalteten Proteins, umso schneller die Faltungsreaktion
und umso stabiler der gefaltete Zustand, desto weniger Protein wird in IBs aggregieren.
Andersherum gilt: Umso länger die korrekte Proteinfaltung dauert und umso höher die
Konzentration an ungefaltetem Protein in der Zelle, desto mehr Protein wird in IBs
aggregieren. Da die Geschwindigkeit bei Reaktionen 2.Ordnung exponentiell mit der
Proteinkonzentration steigt, können Aggregationsreaktionen damit die Faltungsreakti-
on vollständig verdrängen (Abb.2.3).
Abbildung 2.3: IB–Bildung ist abhängig von der Konzentration ungefalteten Proteins. [A] wenigungefaltetes Protein führt zu hoher Ausbeute an gefaltetem Protein [B] viel ungefaltetes Protein führtzu hoher IB–Bildungsrate. In [A] sind die jeweiligen Reaktionsordnungen angegeben (weiteres sieheText), die Pfeildicken geben die Reaktionsgeschwindigkeit an.
2.3.3 IB–Präparation und Faltung von GFP
Im Praktikum wird GFP–H6 so stark produziert, dass es in E. coli inclusion bodies
bildet. Die Zellen werden mittels Hochdruckhomogenisation bzw. Ultraschall aufge-
schlossen, und die sehr dichten IBs zusammen mit den Zelltrümmern abzentrifugiert.
8

Daraufhin werden die Proteine aus den IBs in Lösung gebracht. Dies geschieht durch
Zugabe großer Mengen Harnstoff, was dazu führt, dass die Lösung der Proteine im Puf-
fer energetisch günstiger wird als die Aggregationsreaktion, sodass sich die IBs spontan
auflösen und die Proteine damit in Lösung vorliegen.
Aus dieser Protein–Harnstofflösung können die Proteine nun mittels einer Nickelchelat–
Affinitätschromatographie (Ni–IMAC für immobilized metal ion affinity chromatogra-
phy) gereinigt werden. Der His–Tag des rekombinant produzierten GFP komplexiert
die an die Säulenmatrix gebundenen Nickelionen, wodurch das Protein auf der Säule
zurückgehalten wird. Während das Protein auf der Säule gebunden bleibt, wird die
Harnstoffkonzentration des Säulenpuffers sukzessive verringert, sodass das Protein auf
der Säulenmatrix gebunden seine native Konformation einnehmen kann.
Durch Zugabe großer Mengen Imidazol, welches strukturell mit dem für die Komple-
xierung verantwortlichen Ringsystem des Histidins übereinstimmt, wird das Protein
von den Nickelionen verdrängt und somit von der Säulenmatrix getrennt.
2.4 Spektroskopie
2.4.1 CD–Spektroskopie
Die in der Proteinchemie eingesetzte Circulardichroismus–Spektroskopie (CD-Spektroskopie)
macht sich die Eigenschaft von Proteinen zunutze, dass rechts– und links–circular pola-
risiertes Licht vom Proteinrückgrat unterschiedlich stark absorbiert wird. Dabei ist die
Absorption abhängig von der Orientierung der Peptidbindungen zueinander, wodurch
sich die regelmäßigen Sekundärstrukturelemente wie α–Helix und β–Sheet anhand ihres
CD–Spektrums unterscheiden lassen (Abb.2.4).
2.4.2 Fluoreszenzspektroskopie
Fluoreszenz ist die Eigenschaft eines Moleküls, Licht einer gewissen Wellenlänge zu
absorbieren (also Photonen mit einer definierten Enerige), und mit geringer Verzöge-
rung (Sekundenbruchteile) langwelligeres Licht wieder abzustrahlen. Die Absorption
der Photonen führt dabei zu einer Energetisierung des Fluorophors, dessen Hüllelek-
tronen damit vom Grundzustand in einen angeregten Zustand überführt werden. In
kürzester Zeit verliert das angeregte Molekül jedoch Energie durch Vibration und an-
dere molekulare Effekte, sodass das beim Zurückfallen in den Grundzustand abgegebene
9
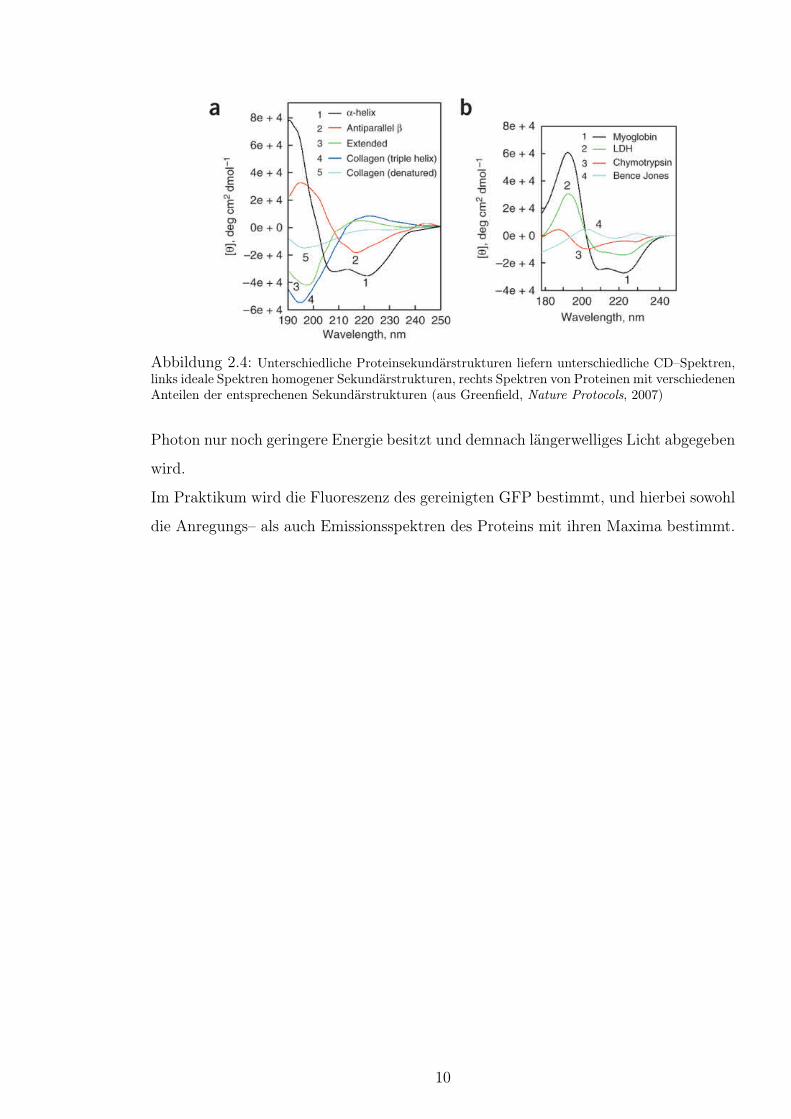
Abbildung 2.4: Unterschiedliche Proteinsekundärstrukturen liefern unterschiedliche CD–Spektren,links ideale Spektren homogener Sekundärstrukturen, rechts Spektren von Proteinen mit verschiedenenAnteilen der entsprechenen Sekundärstrukturen (aus Greenfield, Nature Protocols, 2007)
Photon nur noch geringere Energie besitzt und demnach längerwelliges Licht abgegeben
wird.
Im Praktikum wird die Fluoreszenz des gereinigten GFP bestimmt, und hierbei sowohl
die Anregungs– als auch Emissionsspektren des Proteins mit ihren Maxima bestimmt.
10

3 Praktikum
3.1 Lösungen
Folgende Puffer werden benötigt:
Aufschlusspuffer (200ml)
20mM TRIS/HCl pH 8,0
Renaturierungspuffer (100ml)
20mM TRIS/HCl pH 8,0, 8M Harnstoff, 2,5mM DTT, 20mM Imidazol
Waschpuffer A (10ml)
20mM TRIS/HCl pH 8,0, 100mM NaCl, 8M Harnstoff, 20mM Imidazol
Waschpuffer B (10ml)
20mM TRIS/HCl pH 8,0, 100mM NaCl, 4M Harnstoff, 20mM Imidazol
Waschpuffer C (10ml)
20mM TRIS/HCl pH 8,0, 100mM NaCl, 20mM Imidazol
Elutionspuffer (10ml)
20mM TRIS/HCl pH 8,0, 100mM NaCl, 250mM Imidazol
Dialysepuffer (1 l für 2 Gruppen
50mM Natriumphosphatpuffer (pH 8,0), 100mM NaCl
Vorhandene Stammlösungen:
1M TRIS/HCl pH 8,0
1M NaCl
100mM DTT (Handschuhe tragen)
1M Natriumphosphatpuffer pH 8,0
1M Imidazol (Handschuhe tragen)
11

3.2 Zellanzucht und –ernte
Anzucht Die Anzucht der Zellen erfolgt in LB-Medium (10 g NaCl, 10 g Trypton,
5 g Hefeextrakt pro Liter H2O). Ein Liter LB–Medium wird mit einer Übernachtkultur
des Stammes E. coli BL21(DE3) pET–H6–gfp inokuliert und bei 37 °C bis zum Erreichen
von OD600=0,6 schüttelnd inkubiert. Daraufhin erfolgt die Induktion der Genexpression
mit IPTG (Endkonzentration 1mM im Medium) und weiteres Wachstum der Zellen
für 3 h.
Unter dem Fluoreszenzmikroskop werden Bakterien nach der Induktion mit denen einer
nichtinduzierten Kultur verglichen.
Ernte Ein Mililiter der Zellkulturen wird in einem 1,5ml–Eppendorf–Tube in der
Tischzentrifuge bei maximaler Drehzahl für 5min abzentrifugiert, der Überstand ver-
worfen, und das Zellpellet in 100µl SDS-PAGE-Probenpuffer resuspendiert (B SDS–
Probe „NI“). Die Zellen werden in 1 l–Zentrifugenbechern für 10min in der Cryofuge
6–6 bei 3000U/min abzentrifugiert. Der Überstand wird in die leeren Kolben zurück-
geschüttet (nicht mehr als 1 l pro Kolben, da sonst beim Autoklavieren die Gefahr des
Überlaufens besteht) und autoklaviert. Die Zellpellets werden in den Bechern bei -20 °C
über Nacht gelagert, bzw. mittels Löffeln aus den Bechern in 50ml–Greiner–Röhrchen
überführt und dann bei -20 °C über Nacht gelagert (je nach Absprache).
3.3 Zellaufschluss und IB-Präparation
I Die Lagerung der Zellen, Zellfraktionen und Puffer erfolgt durchgehend
auf Eis.
Aufschluss Die Zellpellets werden aufgetaut und im 5–fachen Volumen (w/v)1 eis-
kalten Aufschlusspuffers (20mM TRIS/HCl pH 8,0, eine Spatelspitze DNAse zugeben)
mit einer Plastikpipette vollständig resuspendiert. Die Zellen werden dann entweder
mittels Hochdruckhomogenisation (French Press, zwei Durchgänge bei 1250 psi) oder
Ultraschall aufgeschlossen (Einteilung nach Absprache).1weight per volume, d.h. pro 1 g Zellgewicht 1ml Puffer
12

IB–Präparation Die Zellsuspension wird bei 15000U/min für 20min zentrifugiert
(Sorvall RC5C, SS34–Rotor, austarieren auf ±0,1 g, Zentrifuge vorkühlen). Dadurch
werden die Zelltrümmer sowie inclusion bodies von den Membranvesikeln und den
löslichen Bestandteilen des Cyto– und Periplasmas getrennt (B SDS–Probe „ÜS“ des
Überstandes). Der Überstand wird verworfen.
Das Zelltrümmerpellet wird zweimal mit Aufschlusspuffer (5–faches Volumen) gewa-
schen (d.h. resuspendiert und wie oben abzentrifugiert). Nach dem zweiten Wasch-
schritt wird das Pellet in Renaturierungspuffer (20mM TRIS/HCl pH 8,0, 8M Harn-
stoff, 2,5mM DTT, 20mM Imidazol) resuspendiert und 1 h auf Eis rührend inkubiert.
Die Zelltrümmer werden nun von den gelösten Proteinen der inclusion bodies durch
Zentrifugation getrennt. Die Zentrifugation erfolgt bei 15000U/min für 15min (Sor-
vall RC5C, SS34–Rotor, austarieren auf ±0,1 g, Zentrifuge vorkühlen). Der Überstand
wird in ein neues 50ml–Greiner–Röhrchen überführt und für die Ni-IMAC verwendet.
Das Zelltrümmerpellet wird nochmals in 5–fachem Puffervolumen (Aufschlusspuffer)
aufgenommen und eine SDS–PAGE–Probe genommen (B SDS–Probe „ZT“ des Zell-
trümmerpellets)
3.4 Ni–IMAC
Äquilibrierung In eine Säule werden etwa 1ml einer 50%igen Ni–NTA–Lösung pi-
pettiert (Lösung vorher gut schütteln), sodass durch das Abfließen der Lagerlösung eine
Ni-NTA-Säule mit einem Säulenvolumen von etwa 0,5ml entsteht. Die Säule wird nun
mit 10 Säulenvolumen Renaturierungspuffer (20mM TRIS/HCl pH 8,0, 8M Harnstoff,
2,5mM DTT, 20mM Imidazol) äquilibriert (entsprechendes Volumen mit Greiner–
Röhrchen abmessen und auf die Säule geben, Durchfluss in Abfallbecher auffangen).
Das Austrocknen der Säule muss durch Verschließen der oberen Öffnung mit dem Säu-
lendeckel verhindert werden (Unterdruck hält Lösung zurück).
Beladen und Waschen Den Überstand der Zelltrümmerzentrifugation wird nun auf
die Säule gegeben, und der Durchlauf mit einem frischen Becherglas aufgefangen (B
SDS–Probe „D“ des Durchlaufs) und nach vollständigem Durchfluss in ein Greiner–
Röhrchen überführt. Die Säule wird nun zuerst mit 2 Säulenvolumen Waschpuffer A
(20mM TRIS/HCl pH 8,0, 100mM NaCl, 8M Harnstoff, 20mM Imidazol) gewaschen
(im Abfallbecher auffangen), und dann durch Waschen mit 2 Säulenvolumen Wasch-
13

puffer B (20mM TRIS/HCl pH 8,0, 100mM NaCl, 4M Harnstoff, 20mM Imidazol)
und 2 Säulenvolumen Waschpuffer C (20mM TRIS/HCl pH 8,0, 100mM NaCl, 20mM
Imidazol) die Harnstoffkonzentration auf der Säule veringert (Waschpuffer C separat
auffangen, B SDS–Probe„W“ des Waschens mit Waschpuffer C).
Elution Die Elution des Proteins erfolgt in 7 Schritten: Elutionen 1 bis 4 mit 1/2
Säulenvolumen, Elutionen 5 bis 7 mit 1 Säulenvolumen des Elutionspuffers (20mM
TRIS/HCl pH 8,0, 100mM NaCl, 250mM Imidazol). Jeder Elutionsschritt wird se-
parat in einem 1,5ml–Eppendorf–Tube aufgefangen und auf Eis gelagert (B SDS–
Probe„E1–E7“ der Elutionsfraktionen). Die Säule wird daraufhin mit 10 Säulenvolu-
men Waschpuffer C gewaschen und die Ni-NTA-Agarose aller Praktikumsgruppen in
einem Greiner–Röhrchen gesammelt (Lagerung im Kühlschrank).
Dialyse Zur Dialyse wird die am stärksten leuchtende Elutionsfraktion verwendet.
Diese wird in einem Dialyseschlauch zweimal gegen 1 l Phosphatpuffer (50mM Natri-
umphosphatpuffer (pH 8,0), 100mM NaCl) bei 4 °C rührend dialysiert (erst für 2 h,
dann nach Pufferwechsel über Nacht). I Für’s Protokoll: Warum wird für spektro-
skopische (Temperaturstabilitäts-)Messungen Phosphatpuffer statt z.B. TRIS–Puffer
verwendet?
3.5 Proteinkonzentrationsbestimmung
Die Menge bzw. Konzentration des gereinigten GFPs (dialysierte Elutionsfraktionen)
muss vor den weiteren Schritten bestimmt werden. Die Bestimmung erfolgt mittels
Roti–Nanoquant.
Die Roti–Nanoquant Stammlösung wird in einer 1:5–Verdünnung verwendet. Der ganze
Kurs stellt nur eine Eichreihe her, die BSA-Stammlösung steht in einer Konzentration
von 100µg/ml zur Verfügung. Die Eichreihe wird wie in Tabelle 1 beschrieben pipet-
tiert. Die Proben des eigenen, dialysierten GFPs werden von jeder Gruppe 1:20 und
1:100 mit H2O verdünnt (Endvolumen: je 200µl).
Alle Eichlösungen sowie alle verdünnten GFP-Proteinlösungen werden mit 800µl Roti-
Nanoquant-Lösung gemischt und nacheinander die OD bei 590 und 450 nm bestimmt.
Der Quotient von OD590 zu OD450 der Eichreihenlösungen wird dann gegen die BSA-
Endkonzentration (BSA-Konzentration auf x-Achse) aufgetragen. Über die resultieren-
14

de Geradengleichung einer linearen Regression kann der Quotient der GFP-Proteinlösungen
einer Proteinkonzentration zugeordnet werden (Verdünnungsfaktoren nicht vergessen!).
Tabelle 1: BSA–Eichreihe
BSA–Endkonz. (µg/ml) µl BSA-Stammlsg. µl H2O
0 - 200
10 20 180
20 40 160
30 60 140
40 80 120
50 100 100
60 120 80
70 140 60
80 160 40
90 180 20
100 200 0
3.6 SDS–PAGE
Herstellung der Gele Die Gele werden nach den während des Praktikums bespro-
chenen Angaben hergestellt. Alle hierzu benötigten Lösungen und Materialien werden
bereitgestellt.
Beladung und Lauf Je 10µl der SDS–PAGE–Proben (gut mischen, evtl. noch ein-
mal kurz vortexen und anzentrifugieren, NICHT erhitzen! I Für’s Protokoll: Warum?)
werden in folgender Reihenfolge auf das Gel aufgetragen: M, VI, NI, ÜS, ZT, D, W,
E1–7.
Gelscan und –färbung Die Gele werden auf dem Typhoon–Gelscanner mit passen-
dem Anregungs– und Detektionsfilter eingescannt. Daraufhin werden sie in Coomassie–
Färbelösung für mindestens 1 h inkubiert und das überschüssige Coomassie mit Ent-
färberlösung entfernt (über Nacht).
15

3.7 CD–Spektroskopie
Für die CD–Spektroskopie werden 500µl einer 100µg/ml GFP-Lösung angesetzt (abh.
von der gemessenen Proteinkonzentration). Zur Verdünnung wird der schon für die
Dialyse verwendete Phosphatpuffer (50mM Natriumphosphatpuffer (pH 8,0), 100mM
NaCl) verwendet.
Messung Die CD–Spektren werden im Bereich von 250 nm bis 195 nm aufgenommen.
Die Scangeschwindigkeit beträgt 50 nm/min mit einer Signalintegrationszeit (D.I.T)
von 125ms, die Temperatur 20 °C. Jeweils drei Durchgänge werden gemittelt.
Mit einer GFP–Probe wird exemplarisch die Temperaturstabilität von GFP gemes-
sen. Hierzu wird das CD–Signal einer GFP–Probe bei einer konstanten Wellenlänge
von 218 nm in einem Temperaturbereich von 20 °C bis 90 °C gemessen. Die Tempera-
turrampe wird mit einem Anstieg von 1 °C/min gemessen, und danach wiederum ein
Spektrum bei 20 °C gemessen.
3.8 Fluoreszenzspektroskopie
Für diese Messungen wird ein Teil der dialysierten GFP-Lösung mit Phosphatpuf-
fer(50mM Natriumphosphatpuffer (pH 8,0), 100mM NaCl) auf eine Endkonzentration
von 50µg/ml (Endvolumen: 2ml) verdünnt. Verwendet werden Suprasil–Quarzglasküvetten
mit einer Schichtdicke von 1 cm. Angaben zu den Einstellungen wie Scangeschwindig-
keit und Temperatur, sowie der Aufnahme von Anregungs– und Emissionsspektren
werden vor der Messung besprochen.
16

4 Protokoll
Das Protokoll sollte innerhalb von 14 Tagen nach Beendigung des Praktikums fertig-
gestellt und persönlich oder per E-Mail ([email protected]) beim
Betreuer abgeliefert worden sein. Als Anreiz: Wenn das Protokoll innerhalb einer Wo-
che (ordentlich) angefertigt und beim Betreuer abgegeben wurde, kann die Erwähnung
und Beschreibung einer wissenschaftlichen Veröffentlichung zu GFP (siehe weiter un-
ten) weggelassen werden.
Generelles Alle im Skript mit „I Für’s Protokoll:“ markierten Fragen sollten an
einer geeigneten Stelle im Protokoll beantwortet werden. Ein Inhaltsverzeichnis ist nicht
erforderlich.
Einleitung Es sollte hier eine kurze Einführung in das bearbeitete Thema gegeben
werden. Dazu gehören einige Worte zu Punkten wie z.B. dem verwendeten Organis-
mus, dem gereinigten Protein und den Funktionsweisen der verwendeten Methoden.
Es ist hier keine vollständige Nacherzählung des Skripts notwendig. In der Einleitung
sollte mit einer kurzen Zusammenfassung (wenige Sätze!) auf eine Veröffentlichung zu
GFP eingegangen werden (Suche unter: http://www.ncbi.nlm.nih.gov/pubmed/), um
auf die Einsatzmöglichkeiten von GFP einzugehen. Dabei ist unerheblich, in welchem
Zusammenhang GFP (oder andere Varianten wie z.B. CFP) verwendet wird, und ob
es Hauptgegenstand oder nur Mittel zum Zweck ist.
Materialien und Methoden Auch hier ist keine Nacherzählung notwendig. Ein
Satz als Hinweis auf das Skript reicht aus, nur eventuelle Veränderungen sollten aufge-
führt werden.
Ergebnisse und Diskussion Diese beiden — eigentlich streng getrennten — Tei-
le einer wissenschaftlichen Arbeit sollten hier zusammen behandelt werden. Alle im
Verlauf des Praktikums erhaltenen Ergebnisse sollten aufgeführt und diskutiert wer-
den (Was wurde gemacht? Was kam raus? Welche Fehler sind aufgetreten? etc.). Um
die Nachvollziehbarkeit für den Leser zu erhöhen, kann zur Verbindung der einzelnen
Ergebnisse kurz auf den Ablauf des Experiments eingegangen werden. Beispiel: „Die
17

Elutionsfraktion mit dem höchsten Proteingehalt wurde daraufhin für die nachfolgen-
den kalorimetrischen Messungen eingesetzt.“
Literaturverzeichnis Alle verwendeten Literaturstellen und Veröffentlichungen sind
hier anzugeben.
Format Grundsätzlich gilt wie bei allen anderen wissenschaftlichen Arbeiten: Schrift
mit Serifen (also Times, Garamond usw. aber nicht Arial, Calibri usw.), Schriftgröße
12, 1,5–facher Zeilenabstand, Blocksatz, einseitiger Druck, Seitennummerierung (bis
auf Deckblatt). Variabler sind meist Angaben zu Seitenrändern, hier bitte: links 4 cm,
rechts 1,5 cm, oben 2,5 cm, unten 2,0 cm.
Einheiten Auch hier gilt analog zu anderen wissenschaftlichen Arbeiten: Zwischen
Zahl und Einheit wird ein geschütztes Leerzeichen gesetzt (unter Word: Strg+ Um-
schalt+ Leertaste) um Zeilenumbrüche und Vergrößerung des Zwischenraumes im Block-
satz zu verhindern. Das Prozentzeichen wird ebenso behandelt (also: 5% SDS). Bei
Angaben in Grad Celsius wird zwischen Zahl und °C ein geschütztes Leerzeichen ge-
setzt (also: 5 °C). Als Einheitenzeichen für Liter wird das kleine „l“ verwendet (also
auch ml und nicht mL). Damit wird der älteren Definition des Einheitenzeichens der
Vorzug vor dem alternativen Zeichen „L“ gegeben, und damit dem weit überwiegenden
Teil der internationalen Fachpresse (u.a. Science und Nature) gefolgt.
Gene und Proteine In der Bakteriologie werden Proteinnamen generell groß ge-
schrieben (TorA), die entsprechenden Gennamen klein und kursiv (torA). Für nicht
aus Bakterien stammende Proteine/Gene weicht die Benennung oft ab, z.B. bei GFP
(Protein komplett groß geschrieben) und gfp. Da die Schreibweise den Bezug zum Pro-
tein oder Gen eindeutig determiniert, kann auf redundante Begriffe wie „das TorA-
Protein“ oder „das torA-Gen“ verzichtet werden. Die Protein-/Genbezeichnungen wer-
den zu Eigennamen, d.h. man sagt und schreibt „MscL (mechanosensitive channel of
large conductance) ist ein interessantes Protein“ und nicht „Der MscL ist ein interessan-
tes Protein“, ebenso wie „BMW (Bayerische Motoren Werke) baut schnelle Autos“ statt
„Die BMW bauen schnelle Autos“.
18

Rechtschreibung Da es sich um einen sehr häufig auftretenden Fehler handelt: Die
deutsche Sprache kennt sog. Nominalkomposita (zusammengesetzte Substantive). Un-
zulässig ist daher generell das einfache Aneinanderreihen von Substantiven, z.B. „Tryp-
tophan Fluoreszenz Spektroskopie“, auch wenn dies im Englischen („tryptophane fluo-
rescence spectroscopy“) der Norm entspricht. Es heißt also in diesem Fall korrekt „Tryp-
tophanfluoreszenzspektroskopie“. Um die Lesbarkeit zu erhöhen, kann ein Bindestrich
zur Gliederung verwendet werden, also: „Tryptophan-Fluoreszenzspektroskopie“. Auch
Zusammensetzungen, die Abkürzungen/Zahlen enthalten, sind von dieser Regel nicht
ausgeschlossen: „TorA-Enzym“ statt „TorA Enzym“, „6-fach“ statt „6 fach“. Englische
Fachbegriffe, die (noch) nicht eingedeutscht wurden, sollten im Text durch Kursivset-
zung hervorgehoben werden, also z.B. „coiled coil “. Kursivsetzung gilt ebenfalls für
alle Artennamen. Wird der Begriff in einem zusammengesetzten Substantiv verwen-
det, wird das gesamte Wort mit Bindestrichen durchgekoppelt und groß geschrieben:
„Das Coiled-coil–Protein“. Dies gilt ebenso für „E.–coli–Stämme“.
Abbildungen, und: Präsens oder Perfekt? Als Richtwert gilt: Alle erhaltenen
Ergebnisse sind in der Vergangenheitsform zu präsentieren. Dabei wird niemals auf
Abbildungen beschreibend eingegangen (also nicht: „Auf dem Gel in Abbildung 2“
oder „Die Kurven in Abbildung 2“ etc.) sondern nur auf sie hingewiesen. Abbildun-
gen dienen nur dem Leser als Hilfe zum Nachvollziehen der erhaltenen Ergebnisse. Der
Autor selbst wertet seine Ergebnisse aus, nicht Bilder, die seine Ergebnisse für die
Nachwelt dokumentieren. Demnach wäre also z.B. richtig: „Mittels SDS–PAGE konn-
te GFP in allen Elutionsfraktionen nachgewiesen werden (Abb.2, Spur E1 bis E7).“
oder „Es zeigte sich eine deutliche Verschiebung des Intensitätsmaximums auf 300 nm
(Abb.2, gestrichelte Kurve)“. Alle Abbildungen werden durchnummeriert und müs-
sen eine unterhalb des Bildes stehende Legende besitzen, die für sich stehend Aufbau
und Inhalt des Bildes verständlich macht. Dazu gehört eine kurze Abbildungsbeschrei-
bung (Inhalt) sowie eine Erklärung aller Abkürzungen, z.B. bei einer SDS–PAGE „
Abb.2: Reinigung von rekombinantem TorA mittels Ni-IMAC. SDS–PAGE der Reini-
gungsfraktionen, Coomassiefärbung. M = Marker, W = Waschschritt usw.). Tabellen
werden ebenfalls durchnummeriert, jedoch oberhalb der eigentlichen Tabelle mit ei-
ner Überschrift versehen (also: „Tab.1: Verwendete Plasmide“), die Legende steht hier
aber wieder unter der Tabelle. Sobald von den erhaltenen Ergebnissen ausgehend ei-
19

ne theoretische Aussage zu einem Sachverhalt getroffen wird, deren Aussagekraft über
das eigentliche Experiment hinaus Gültigkeit besitzt, wird von der Vergangenheits–
in die Gegenwartsform gewechselt. Der Autor beschreibt nun Allgemeingültigkeiten —
oder zumindest hofft er das. Hier kann als Faustregel gelten, dass all jene Sätze im
Präsens geschrieben werden können, welche Aussagen betreffen, in die man „im Allge-
meinen“ einfügen kann (ohne den Satz logisch zu verfälschen). Beispiel: „Somit konnte
gezeigt werden, dass GFP (im Allgemeinen) bei einem pH-Wert von 5,0 fluoresziert“
oder „Demnach ist PspA (im Allgemeinen) ein membranassoziiertes Protein“.
20








