Reiter-Fink E, Deutinger J - kup.at · 18 27. Jahrgang, 4/2009 Die Mikrozephalie E. Reiter-Fink, J....
8
Geburtshilfe ∕ Frauen-Heilkunde ∕ Strahlen-Heilkunde ∕ Forschung ∕ Konsequenzen Homepage: www.kup.at/speculum Online-Datenbank mit Autoren- und Stichwortsuche P.b.b. 02Z031112 M, Verlagsort: 3003 Gablitz, Linzerstraße 177A/21 Krause & Pachernegg GmbH • Verlag für Medizin und Wirtschaft • A-3003 Gablitz Reiter-Fink E, Deutinger J Die Mikrozephalie Speculum - Zeitschrift für Gynäkologie und Geburtshilfe 2009; 27 (4) (Ausgabe für Österreich), 18-22 Speculum - Zeitschrift für Gynäkologie und Geburtshilfe 2009; 27 (4) (Ausgabe für Schweiz), 18-18
Transcript of Reiter-Fink E, Deutinger J - kup.at · 18 27. Jahrgang, 4/2009 Die Mikrozephalie E. Reiter-Fink, J....

Geburtshilfe ∕ Frauen-Heilkunde ∕ Strahlen-Heilkunde ∕ Forschung ∕ Konsequenzen
Homepage:
www.kup.at/speculum
Online-Datenbank mit Autoren-
und Stichwortsuche
P.b.b. 02Z031112 M, Verlagsort: 3003 Gablitz, Linzerstraße 177A/21
Krause & Pachernegg GmbH • Verlag für Medizin und Wirtschaft • A-3003 Gablitz
Reiter-Fink E, Deutinger J
Die Mikrozephalie
Speculum - Zeitschrift für Gynäkologie und Geburtshilfe 2009; 27 (4)(Ausgabe für Österreich), 18-22
Speculum - Zeitschrift für Gynäkologie und Geburtshilfe 2009; 27 (4)(Ausgabe für Schweiz), 18-18

T h o m a s S t a u d i n g e r
M a u r i c e K i e n e l
ECMO
für die Kitteltasche
Copyright 2018
Thomas Staudinger - Herausgeber
2. Auflage
Ab sofort in unserem Verlag
Krause & PacherneggGmbH
Bestellen Sie noch heute Ihr Exemplar aufwww.kup.at/cd-buch/75-bestellung.html
Thomas Staudinger Maurice Kienel
ECMOfür die Kitteltasche
2. Auflage Jänner 2019ISBN 978-3-901299-65-078 Seiten, div. Abbildungen19.80 EUR

18
27. Jahrgang, 4/2009
Die Mikrozephalie
E. Reiter-Fink, J. Deutinger
Pränatale Aspekte der Mikro-zephalie
Die Mikrozephalie ist eine Fehlbildung, beiwelcher der Kopf eine abnorm kleine Grö-ße aufweist. Die Inzidenz beträgt ca. 1 auf8500 Geburten. Eine viel größere Häufig-keit, nämlich 1,6 auf 1000 tritt auf, wennman das erste Lebensjahr miteinbezieht.
Der Ausdruck Mikrozephalie bedeutetgenau übersetzt „kleiner Kopf“. In Wirk-lichkeit sollte die Definition lauten „Mikro-enzephalie“ (kleines Gehirn und geistigeRetardierung).
Die Relation zwischen Kopfgröße undGehirnmasse ist gut dokumentiert und er-forscht. Fehler in der Proliferation von Ner-venzellen können entweder zu einer Mikro-oder Makrozephalie führen, jedoch wederein zu kleiner noch ein zu großer Kopfmuss unbedingt anormal sein.
Während die Makrozephalie pränatal rela-tiv selten diagnostiziert wird, stellt die Dia-gnostik der Mikrozephalie eine stetige Her-ausforderung für den Pränatalmedizinerdar; die Differenzialdiagnose zwischen ab-normer Biometrie bei normaler Entwick-lung und abnormer Biometrie bei krank-hafter Entwicklung des Feten benötigt vielErfahrung und kann sehr schwierig sein.
Die Problematik der Diagnostik der Mi-krozephalie liegt im Zeitpunkt des Auftre-tens: Zum Zeitpunkt des traditionellen Or-ganscreenings (20.–22. Schwangerschafts-woche) ist die Diagnose einer Mikrozepha-lie meist nicht möglich; diese wird erst umdie 26.–28. Woche (je nach Schweregrad) ma-nifest und damit diagnostizierbar (Abb. 1–
4). Häufig ist es in der Pränataldiagnostikschwierig herauszufinden, welche Formder Mikrozephalie vorliegt. Wie unten an-geführt, gibt es zahlreiche Ursachen undzahlreiche Formen der Mikrozephalie, vonder erzielten Diagnose (chromosomale Di-agnostik, Ultraschall) hängt auch die ent-sprechende Prognose ab. In den vergan-genen Jahren hat die Miteinbeziehung desMRI entscheidende Fortschritte in die prä-(und post-) natale Beurteilung der Mikro-zephalie gebracht.
Prinzipiell unterscheiden wir eine pri-märe und eine sekundäre Mikrozephalie.Die primäre Mikrozephalie entsteht durcheine Störung im Bereich der neuralen Pro-liferation und Differenzierung zwischendem 2. und 5. Monat der Schwangerschaft.Die sekundäre Mikrozephalie entsteht z. B.durch eine pränatale Infektion mit Toxo-plasmose oder Röteln, Hypoxie, durch dasFetal-Alcohol-Syndrome oder Kokainabusus.Aber auch eine unbehandelte mütterlichePhenylketonurie kann zu einem Mikroze-phalus führen.
Wenn Infektionen, Stoffwechselerkrankun-gen und Syndrome ausgeschlossen sind,bleibt für die übrigen Fälle empirisch einWiederholungsrisiko von 15 % und damitdie Wahrscheinlichkeit, dass eine geneti-sche Ursache dahintersteckt. Für die Prog-nose der Mikrozephalie ist nicht nur dieGröße der Reduktion des Kopfumfangs,sondern natürlich die Ätiologie von großerBedeutung. Es gibt auch eine autosomalrezessiv vererbbare Form bei Konsanguini-tät.
Für die Diagnose der Mikrozephalie istder biparietale Durchmesser keineswegsausreichend, entscheidend ist der Kopfum-fang! Die Diagnose der Mikrozephalie istauch durch eine einzelne Untersuchungnicht möglich. Entscheidend sind serielleUntersuchungen. Von einer Mikrozephaliewird pränatal dann gesprochen, wenn beider Sonographie der Kopfumfang unterhalbder 3-fachen Standardabweichung bzw. un-terhalb der 3. Perzentile liegt.
In die Diagnostik miteinbezogen wird dieRelation von Kopfumfang zur Rumpfgrößeund zur Biometrie der Extremitätenkno-chen; ein gesichertes Gestationsalter ist Vor-aussetzung. Die Differenzialdiagnose zwi-schen Mikrozephalie und Kraniosynostosiskann pränatal nicht mit Sicherheit gestelltwerden.
For personal use only. Not to be reproduced without permission of Krause & Pachernegg GmbH.
19
27. Jahrgang, 4/2009
Beim Geburtsmodus ist eine vaginale Ge-burt durchaus anzustreben und möglich.Ist jedoch bekannt, dass durch die fehlendeKopfgröße eine mangelnde Eröffnung desMuttermundes stattfinden kann, muss so-mit an die Möglichkeit einer Schulter-dystokie gedacht werden.
1. Der Großteil der mikrozephalen Kinderist geistig retardiert, die meisten schwer.
2. Allgemein gilt: je kleiner der Kopf, um-so schlechter die Prognose.
3. Ist die Mikrozephalie Teil eines bestimm-ten genetischen Syndroms (z. B. Meckel-Syndrom), ist der langfristige Outcomebesonders schlecht.
4. Aber: Es gibt auch Kinder, die mit einemsehr kleinen Kopf mit normaler Intelli-genz geboren werden.
Die Mikrozephalie ist eine nicht behan-delbare Erkrankung. Entscheidend ist dieDiagnostik assoziierter Anomalien; Stufe 2,Stufe 3, Ultraschall, chromosomale Diag-nostik und MRI sind obligat.
Postnatale Aspekte der Mikro-zephalie
Mikrozephalie wird variabel definiert alsKopfumfang mehr als 2 Standardabwei-chungen unterhalb des Durchschnittes(= 50. Perzentile) bezogen auf Alter undGeschlecht einerseits bzw. als Kopfumfangunterhalb der 3. Perzentile andererseits,wobei letzterer als extreme Mikrozephaliebezeichnet wird. Diese Definitionen inklu-dieren auch normale Individuen.
Die Ätiologie der Mikrozephalie ist viel-fältig. Wir unterscheiden eine primäre Mi-krozephalie, welche bereits im 7. Schwan-gerschaftsmonat sichtbar wird und durcheine Störung der Induktion und Migrationbedingt ist. Die primäre Mikrozephaliekann autosomal-rezessiv oder autosomal-dominant vererbt werden; das Wiederho-lungsrisiko ist relativ hoch, daher ist einepräzise Diagnostik entscheidend für die ge-netische Beratung der Eltern.
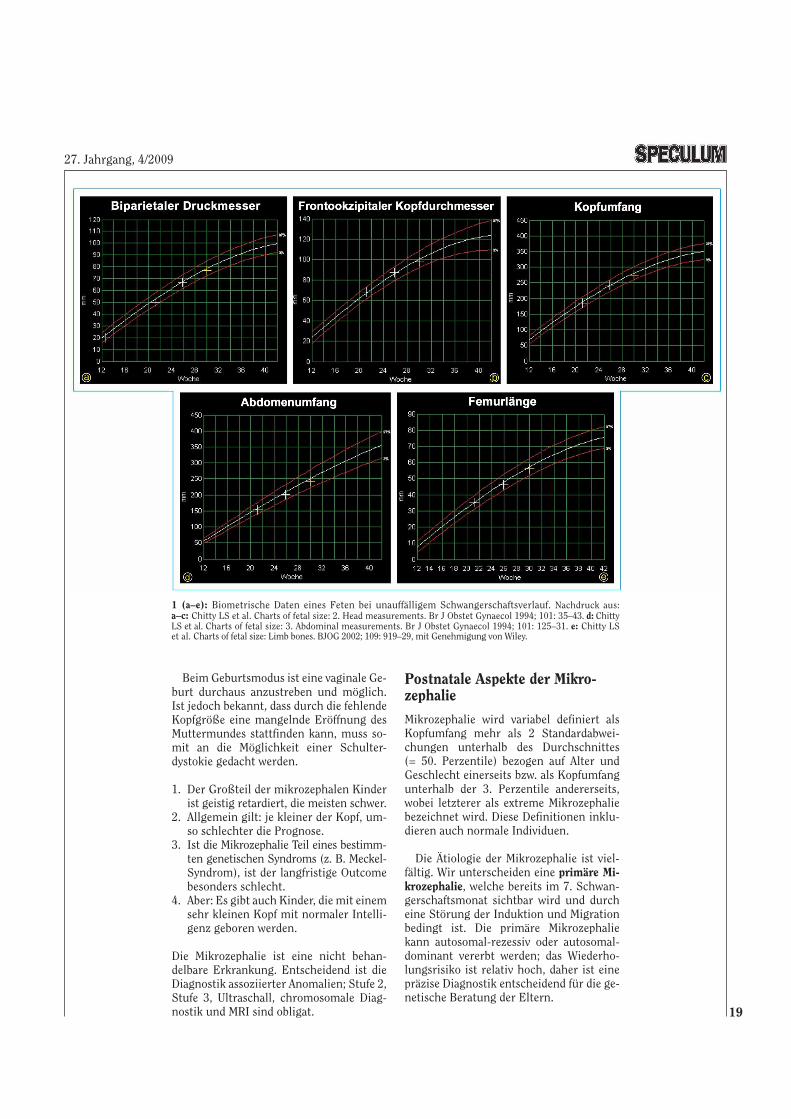
1 (a–e): Biometrische Daten eines Feten bei unauffälligem Schwangerschaftsverlauf. Nachdruck aus:a–c: Chitty LS et al. Charts of fetal size: 2. Head measurements. Br J Obstet Gynaecol 1994; 101: 35–43. d: ChittyLS et al. Charts of fetal size: 3. Abdominal measurements. Br J Obstet Gynaecol 1994; 101: 125–31. e: Chitty LSet al. Charts of fetal size: Limb bones. BJOG 2002; 109: 919–29, mit Genehmigung von Wiley.
20
27. Jahrgang, 4/2009
Die primäre Mikrozephalie inkludiert zahl-reiche Migrationsstörungen (Schizenzephalie,Lissenzephalie, Pachygyrie, Polymikrogy-rie, Agenesie des Corpus callosum), Hetero-topien der grauen Substanz, zahlenmäßigreduzierte bzw. abnorm konfigurierte Neu-ronen oder eine pathologische Schichtungdes Kortex.
Eine primäre Mikrozephalie kann ebensochromosomal bedingt sein (inklusive Tri-somien z. B. 13, 15, 18, 21, 22; Deletionen z. B.4p, 5p, 18p, 18q; Translokationen), oder imRahmen eines Dysmorphie-Syndroms mitRetardierung auftreten (mit normalemKaryotyp, vererbt oder nicht vererbt, z. B.Cornelia-de-Lange-Syndrom). Die zugrundeliegende Pathologie ist hierbei ungeklärt.
Eine Bestrahlung zwischen der 4. und20. Gestationswoche kann Ursache einerprimären Mikrozephalie sein. Der Zeitpunktder Bestrahlung korreliert mit der Größedes Gehirns und der neurologischen Schä-digung. Die diagnostische Bestrahlung
< 1 Gray bedeutet jedoch wenig Risikowährend der ersten 4 Schwangerschafts-monate.
Kongenitale Infektionen (Zytomegalie,Röteln, Toxoplasmose) sowie Drogen/Che-mikalien (Alkohol, Phenytoin, Aminopterin)können einen primären Mikrozephalus ver-ursachen.
Die sekundäre Mikrozephalie wird nachdem 7. Schwangerschaftsmonat, perinatalsowie in der frühen Kindheit manifest.Hierbei zeigt sich eine Schädigung des Ge-hirns (zystische Degeneration, Enzephalo-malazie, Porenzephalie, entzündlich glioti-sche Vernarbung und Schrumpfung), wel-che zu einem verminderten postnatalenWachstum sowie zu einem frühzeitigenVerschluss der Schädelnähte führt.
Die Ursachen der sekundären Mikroze-phalie sind folgende: hypoxisch-ischämi-sche Enzephalopathie, Meningitis/Enzepha-litis, mütterliche Erkrankungen (Diabetesmellitus, Hyperphenylalaninämie [PKU],
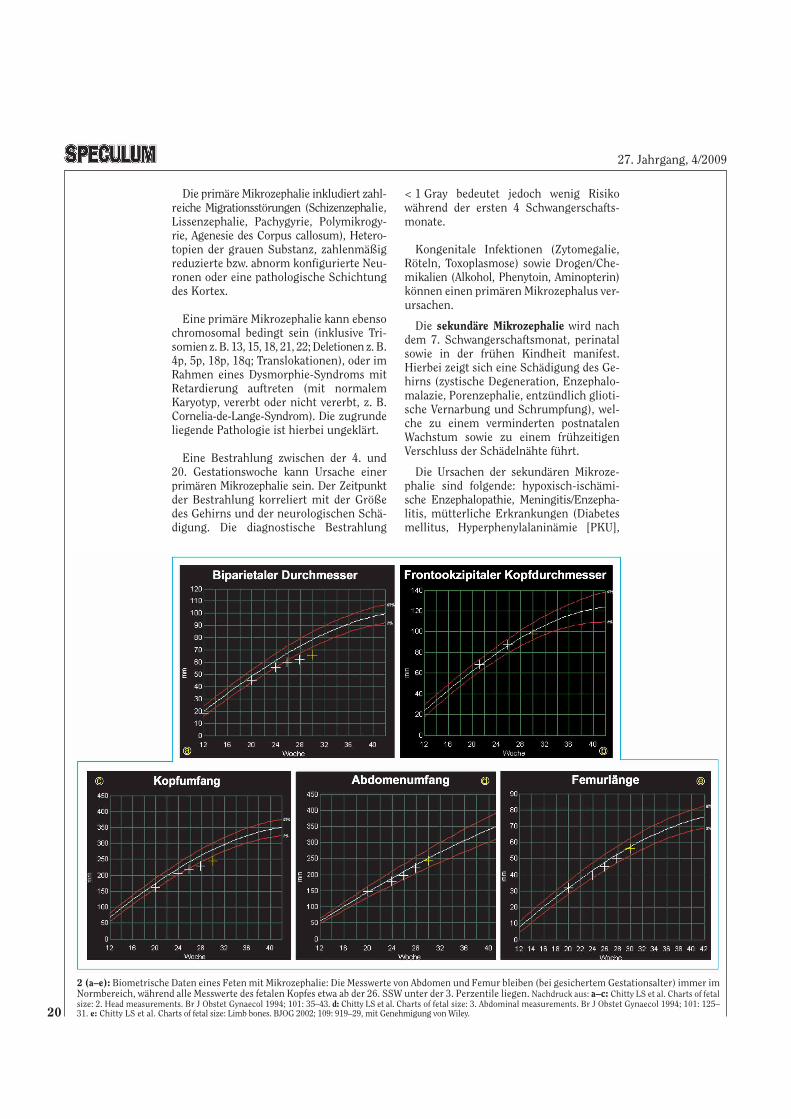
2 (a–e): Biometrische Daten eines Feten mit Mikrozephalie: Die Messwerte von Abdomen und Femur bleiben (bei gesichertem Gestationsalter) immer imNormbereich, während alle Messwerte des fetalen Kopfes etwa ab der 26. SSW unter der 3. Perzentile liegen. Nachdruck aus: a–c: Chitty LS et al. Charts of fetalsize: 2. Head measurements. Br J Obstet Gynaecol 1994; 101: 35–43. d: Chitty LS et al. Charts of fetal size: 3. Abdominal measurements. Br J Obstet Gynaecol 1994; 101: 125–31. e: Chitty LS et al. Charts of fetal size: Limb bones. BJOG 2002; 109: 919–29, mit Genehmigung von Wiley.

21
27. Jahrgang, 4/2009
Hyperthyreose) sowie Schädel-Hirn-Trau-ma und degenerative Erkrankungen des ZNS(z. B. Rett-Syndrom, Leukodystrophie).
Bei der Mikrozephalia vera handelt essich um eine genetische Form (autosomal-rezessiv vererbt), der Kopfumfang liegt mehrals 3–5 (–6) Standardabweichungen unterder 50. Perzentile. Die Kinder präsentierensich mit einer schmalen, fliehenden Stirnund einem prominenten Scheitel, jedochohne ausgeprägte klinisch-neurologischeSymptomatik. Neben Verhaltensauffälligkei-ten (hyperkinetisch) und einer verminder-ten feinmotorischen Koordination tretenbei etwa einem Drittel der Patientinnen ze-rebrale Anfälle auf. Die mentale Retardie-rung ist mild und die expressive Sprachevereinfacht. Dies ist insofern erstaunlich,da das Gehirn der Betroffenen ca. 500 gbeim Erwachsenen wiegt, dies entsprichtnormalerweise einem 3–5 Monate alten Fe-tus. Histologisch zeigt sich ein schwererVerlust von Neuronen.
Unter dem Begriff Mikrozephalie plusversteht man die Kombination aus Mikro-zephalie und einem Syndrom (z. B. geneti-sche Mikrozephalie + zerebrale Anfälle und/oder Spastizität) oder Abnormitäten der Re-tina, Kleinwuchs und assoziierte Anomalien(wie z. B. Corpus-callosum-Agenesie, kon-genitales nephrotisches Syndrom, schwereMigrationsstörung).
Die klinische Manifestation der Mikroze-phalie weist ein breites Spektrum auf hin-sichtlich motorischer Beeinträchtigung (ge-ringfügige Beeinträchtigung der Koordina-tion und der Feinmotorik vs. schwere mo-torische Beeinträchtigung), mentaler Re-tardierung (lernfähig milde Retardierungvs. schwere mentale Retardierung) sowiedes Verhaltens (milde Hyperkinesie vs.schwer autistisches Verhalten).
Die Diagnostik inkludiert neben einerausführlichen Familienanamnese die obli-
gate Messung des Kopfumfanges bei Ge-burt des Kindes (und regelmäßig im Ver-lauf beim Kinderfacharzt) sowie die Mes-sung des Kopfumfanges von Eltern und Ge-schwistern.
Die weiterführende Diagnostik wird be-stimmt durch die erhobene Anamnese unddie physikalische Untersuchung und kannneben einer Bestimmung von Phenylalaninim Serum der Kindesmutter (bei Verdachtauf Phenylketonurie = PKU), die Bestim-mung des Karyotyps (v. a. chromosomalesSyndrom), ein kraniales MRT (Vorliegenstruktureller Abnormitäten), ein CCT (Vorlie-gen intrazerebraler Verkalkungen) sowieAminosäuren/organische Säuren (Serum/Harn), Ammoniak im Serum, TORCH, HIV,Zytomegalie im Harn beim Kind inkludieren.
Die pränatale Diagnostik ist extrem schwie-rig, insbesondere, wenn assoziierte Fehl-bildungen fehlen. Zusätzlich besteht einegewisse Fehlerquelle durch die Messungselbst. Besonders zu achten ist auf die Dif-ferenzialdiagnose der sagittalen Kraniosyn-ostose: Hierbei kann der biparietale Durch-messer klein sein, daher sind wiederholteMessungen notwendig und es ist auf diecharakteristische Schädelform zu achten.
Eine kausale Therapie ist nur selten mög-lich (z. B. bei der Phenylketonurie oderchronischen Hypoglykämie der Kindesmut-ter). Bei bekannter Ursache ist eine geneti-sche Beratung der Eltern indiziert. Im All-gemeinen ist eine intensive Förderung derPatientinnen notwendig (kognitives Trai-ning, Integrationskindergarten/-schule), umeine optimale Entwicklung zu ermögli-chen.
Die Prognose ist variabel, hängt ab vonder zugrunde liegenden Ursache und kanneine milde bzw. schwere Entwicklungs-verzögerung (sowohl im mentalen als auchim motorischen Bereich) bedeuten.
3:Normale fetale
Schädelkonfiguration
4:DolichozephaleSchädelkonfigurationbei Beckenendlagedes Feten; der Kopf-umfang liegt jedochim Normbereich.Deswegen mussimmer auch derKopfumfang für dieDiagnose „Mikro-zephalie“ herange-zogen werden.

22
27. Jahrgang, 4/2009
LITERATUR
1. Twining P, Mc Hugo JM, Pilling DW (eds). Text-book of fetal abnormalities. Churchill Livingstone,London, 2000.
2. Brock DHJ, Rodeck CH, Ferguson-Smith MA(eds). Prenatal diagnosis and screening. ChurchillLivingstone, London, 1992.
3. Rodeck CH, Whittle MJ (eds). Fetal medicine.Basic science and clinical practice. Churchill Living-stone, London, 1999.
4. Nyberg DA, Mc Gahan JP, Pretorius DH (eds). Dia-gnostic imaging of fetal anomalies. LippincottWilliams & Wilkins, Philadelphia, 2003.
5. Romero R, Pilu G, Jeanty P (eds). Prenatal diag-nosis of congenital anomalies. Appleton & Lange,Norwalk, 1998.
6. Merz E (Hrsg). Sonographische Diagnostik inGynäkologie und Geburtshilfe: Lehrbuch und Atlas.Bd. 2: Geburtshilfe. Thieme, Stuttgart, 2002.
7. Aicardi J. Diseases of the nervous system inChildhood, 3rd ed. Mac Keith Press, London, 1998.
8. Menkes JH, Sarnat HB (eds). Child neurology, 6th
ed. Lippincott Williams & Wilkins, Philadelphia,2000.
9. Behrman RE, Kliegmann RM, Jenson HB (eds).Nelson Textbook of pediatrics. Saunders – Elsevier,Philadelphia, 2007.
Univ.-Prof. Dr. med. Josef DeutingerJahrgang 1954. 1975–1980 Medizinstudium und Promotion in Wien, praktische Ausbildung in diversenKrankenhäusern in Salzburg, 1982–1987 Fachausbildung an der II. Universitätsfrauenklinik Wien. 1987 Preisder deutschen Gesellschaft für Perinatologie, 1988 Hugo-Husslein-Preis, 1990 Habilitation (Vaginosono-graphische Dopplerströmungsmessungen). 1993–2003 stv. Leiter der Abteilung für Pränatale Diagnostik undTherapie, 2004–2006 Präsident der ÖGUM, seit 1991 stv. ärztlicher Leiter der Krankenanstalt Gyn Schall (Insti-tut für Pränatale Diagnostik, Ultraschall, Genetik), seit 1993 Vorstandsmitglied des Vereins der Freunde derpränatalen Diagnostik und Therapie.
Korrespondenzadresse:Univ.-Prof. Dr. med. Josef DeutingerUniversitätsfrauenklinik Wien, Abteilung für fetomaternale MedizinA-1090 Wien, Währinger Gürtel 18–20E-Mail: [email protected]

Mitteilungen aus der Redaktion
Haftungsausschluss
Die in unseren Webseiten publizierten Informationen richten sich ausschließlich an geprüfte und autorisierte medizinische Berufsgruppen und entbinden nicht von der ärztlichen Sorg-faltspflicht sowie von einer ausführlichen Patientenaufklärung über therapeutische Optionen und deren Wirkungen bzw. Nebenwirkungen. Die entsprechenden Angaben werden von den Autoren mit der größten Sorgfalt recherchiert und zusammengestellt. Die angegebenen Do-sierungen sind im Einzelfall anhand der Fachinformationen zu überprüfen. Weder die Autoren, noch die tragenden Gesellschaften noch der Verlag übernehmen irgendwelche Haftungsan-sprüche.
Bitte beachten Sie auch diese Seiten:
Impressum Disclaimers & Copyright Datenschutzerklärung
Abo-AktionWenn Sie Arzt sind, in Ausbildung zu einem ärztlichen Beruf, oder im Gesundheitsbereich tätig, haben Sie die Möglichkeit, die elektronische Ausgabe dieser Zeitschrift kostenlos zu beziehen.
Die Lieferung umfasst 4–6 Ausgaben pro Jahr zzgl. allfälliger Sonderhefte.
Das e-Journal steht als PDF-Datei (ca. 5–10 MB) zur Verfügung und ist auf den meisten der marktüblichen e-Book-Readern, Tablets sowie auf iPad funktionsfähig.
Bestellung kostenloses e-Journal-Abo
Besuchen Sie unserezeitschriftenübergreifende Datenbank
Bilddatenbank Artikeldatenbank Fallberichte