
Schnelle Orientierung in Genomdaten
22
Schnelle Orientierung in Genomdaten Dr. Fritz Schinkel Fujitsu München Dr. Matthias Schlesner DKFZ Heidelberg
-
Upload
fujitsu-central-europe -
Category
Health & Medicine
-
view
527 -
download
3
Transcript of Schnelle Orientierung in Genomdaten

0 Copyright 2016 FUJITSU
Schnelle Orientierung in Genomdaten
Dr. Fritz Schinkel Fujitsu München
Dr. Matthias Schlesner DKFZ Heidelberg
03.03.2016 Matthias Schlesner 1
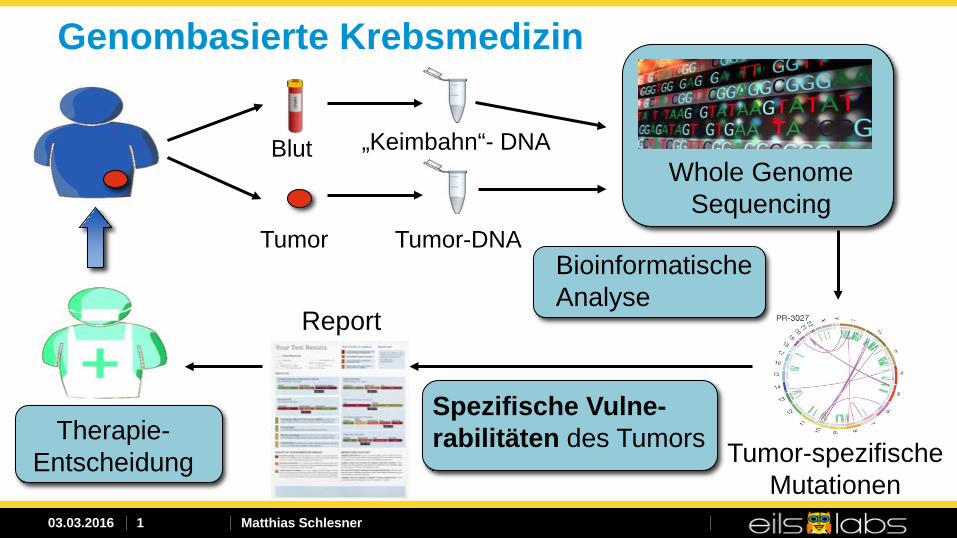
Blut
Tumor
„Keimbahn“- DNA
Tumor-DNA
Whole Genome Sequencing
Tumor-spezifische Mutationen
Spezifische Vulne-rabilitäten des Tumors
Report
Bioinformatische Analyse
Therapie- Entscheidung
Genombasierte Krebsmedizin

03.03.2016 Matthias Schlesner 2
Deutsches Krebsforschungszentrum betreibt Illumina X Ten
• Kapazität: 18.000 humane Genome (30x) pro Jahr
Personalisierte Onkologie
Illumina HiSeq X Ten
www.illumina.com
• Ziel: allen Tumorpatienten am Nationalen Centrum für Tumorerkrankungen in Heidelberg (~3.500 Patienten p.a.) die Sequenzierung (whole genome) des Tumorgenoms anbieten

03.03.2016 Matthias Schlesner 3
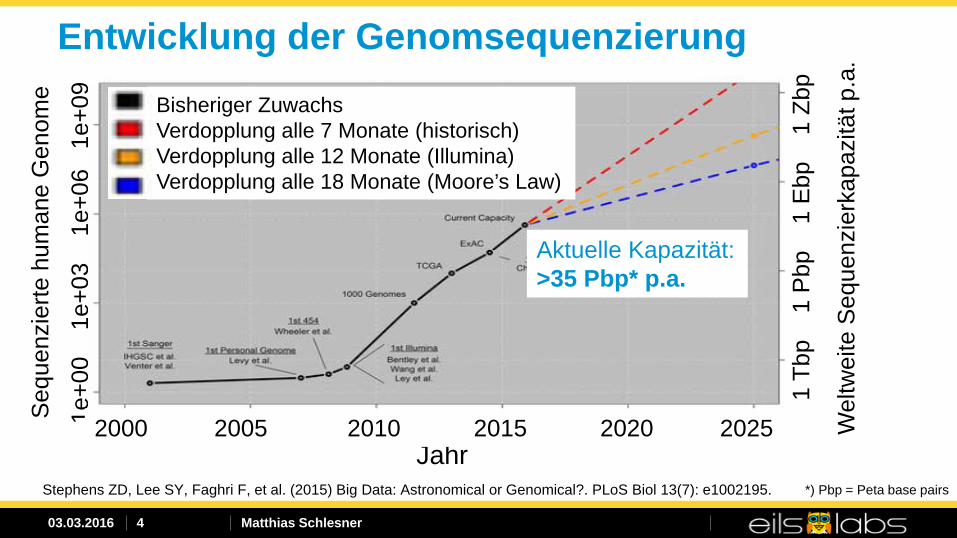
Big Data: Wachstumsraten
• 600 Terabytes pro Tag (Blog-Eintrag auf code.facebook.com von April 10th, 2014)
• 12 Terabytes pro Tag (Cloud Data Management; Liang Zhao et al., 2014)
• Sequencing@DKFZ ~10 Terabytes pro Tag
03.03.2016 Matthias Schlesner 4
Jahr
1e+0
0
1e+0
3
1e+0
6 1
e+09
1 Tb
p
1 Pb
p
1 Eb
p
1 Zb
p
Stephens ZD, Lee SY, Faghri F, et al. (2015) Big Data: Astronomical or Genomical?. PLoS Biol 13(7): e1002195.
Aktuelle Kapazität: >35 Pbp* p.a.
Wel
twei
te S
eque
nzie
rkap
azitä
t p.a
.
Sequ
enzi
erte
hum
ane
Gen
ome
2000 2005 2010 2015 2020 2025
Entwicklung der Genomsequenzierung Bisheriger Zuwachs Verdopplung alle 7 Monate (historisch) Verdopplung alle 12 Monate (Illumina) Verdopplung alle 18 Monate (Moore’s Law)
*) Pbp = Peta base pairs
03.03.2016 Matthias Schlesner 5
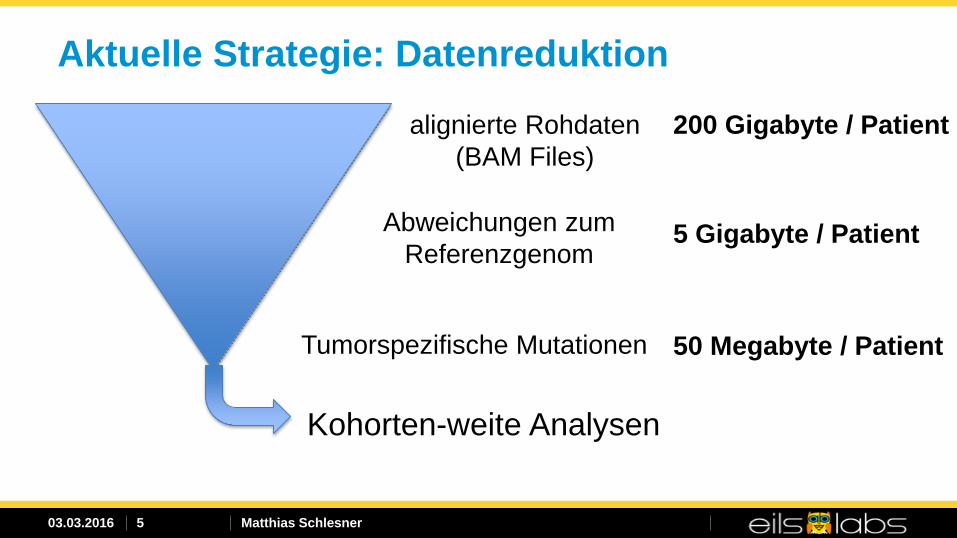
alignierte Rohdaten (BAM Files)
Kohorten-weite Analysen
Abweichungen zum Referenzgenom
Tumorspezifische Mutationen
200 Gigabyte / Patient
5 Gigabyte / Patient
50 Megabyte / Patient
Aktuelle Strategie: Datenreduktion
03.03.2016 Matthias Schlesner 6
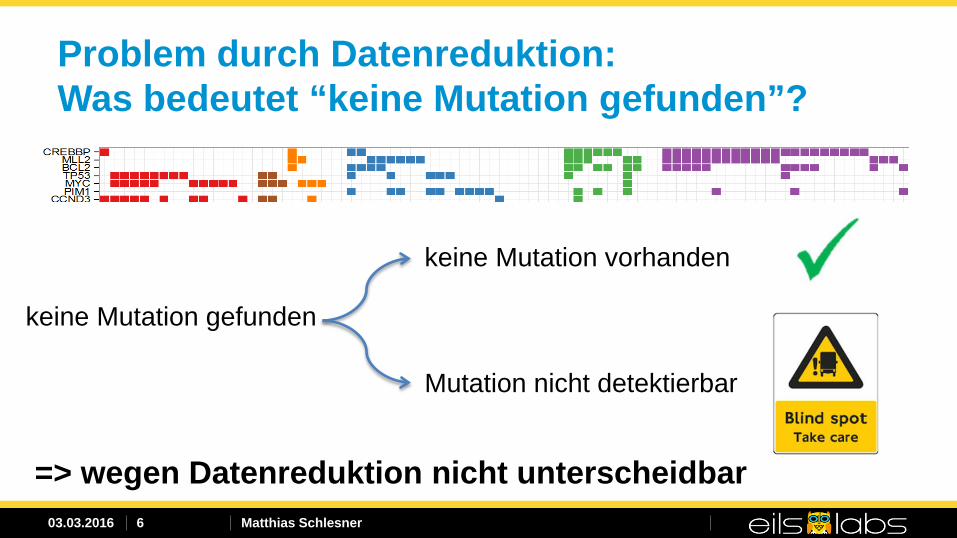
Problem durch Datenreduktion: Was bedeutet “keine Mutation gefunden”?
keine Mutation gefunden
keine Mutation vorhanden
Mutation nicht detektierbar
=> wegen Datenreduktion nicht unterscheidbar

03.03.2016 Matthias Schlesner 7
• Analyse einer Testkohorte (52 Patienten, je ein Tumor- und ein Kontrollgenom) ohne Datenreduktion
1. Performance-Vergleich bei der Analyse von ~900.000 Positionen in der gesamten Kohorte
2. Identifikation von Regionen in bekannten Krebsgenen ohne ausreichende Abdeckung zur verlässlichen Identifikation von Mutationen
03.03.2016
Zielsetzung
8 Copyright 2016 FUJITSU
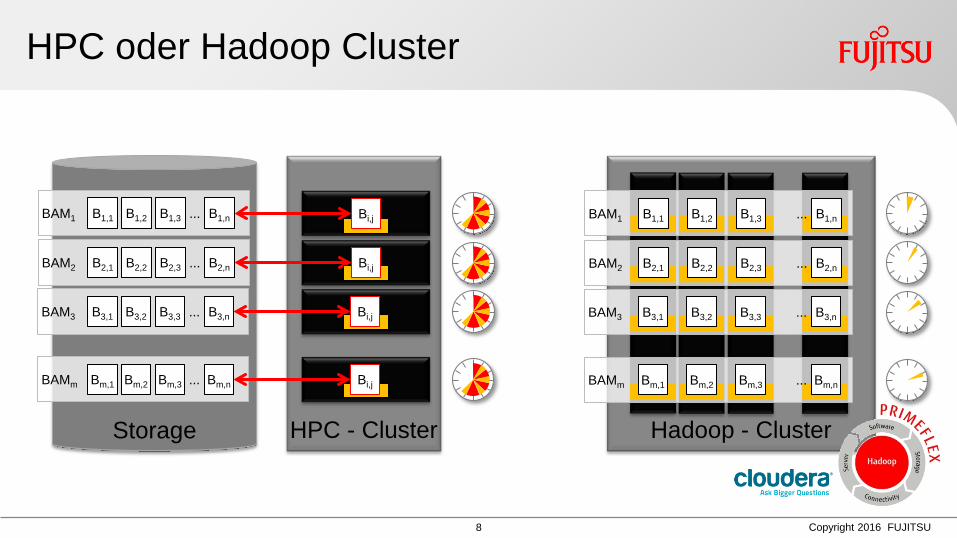
Storage
HPC oder Hadoop Cluster
BAM2 B2,1 B2,2 B2,3 B2,n ...
BAM1 B1,1 B1,2 B1,3 B1,n ...
BAM3 B3,1 B3,2 B3,3 B3,n ...
BAMm Bm,1 Bm,2 Bm,3 Bm,n ...
HPC - Cluster Hadoop - Cluster
BAM2
BAM1
BAM3
BAMm
Bi,j
Bi,j
Bi,j
Bi,j
...
...
...
B2,1
B3,1
Bm,1
B1,1
B2,2 B2,3 B2,n
B1,2 B1,3 B1,n
B3,2 B3,3 B3,n
Bm,2 Bm,3 Bm,n ...

9 Copyright 2016 FUJITSU
Bedienung: Daten statt Technik
Sammlung
Referenzgenom
Diagnose DNA Proben Analyse
10 Copyright 2016 FUJITSU
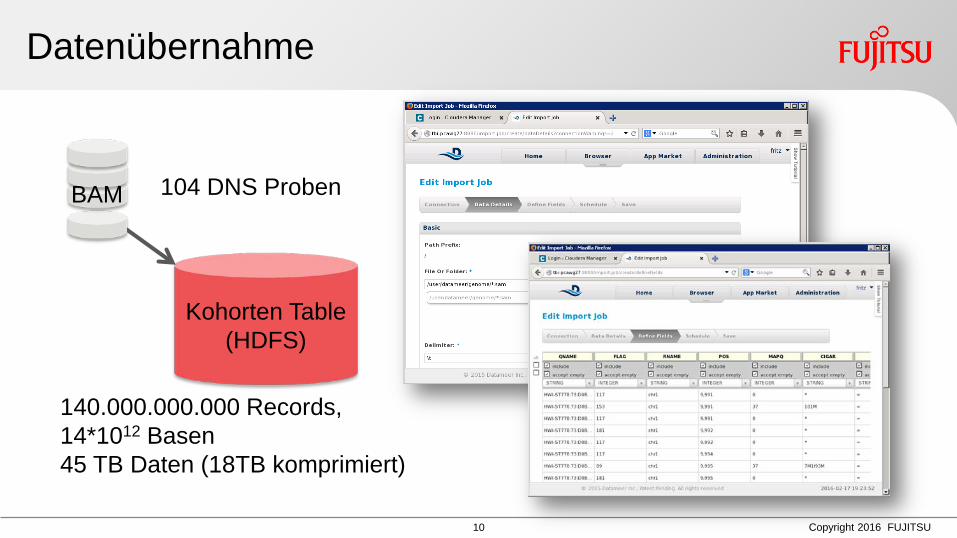
Datenübernahme
Kohorten Table (HDFS)
BAM 104 DNS Proben
140.000.000.000 Records, 14*1012 Basen 45 TB Daten (18TB komprimiert)
11 Copyright 2016 FUJITSU
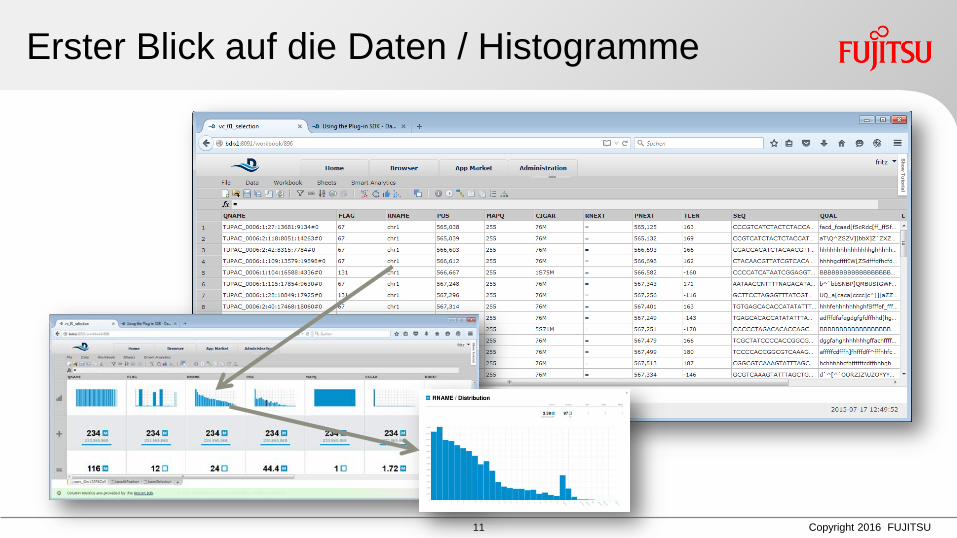
Erster Blick auf die Daten / Histogramme
12 Copyright 2016 FUJITSU
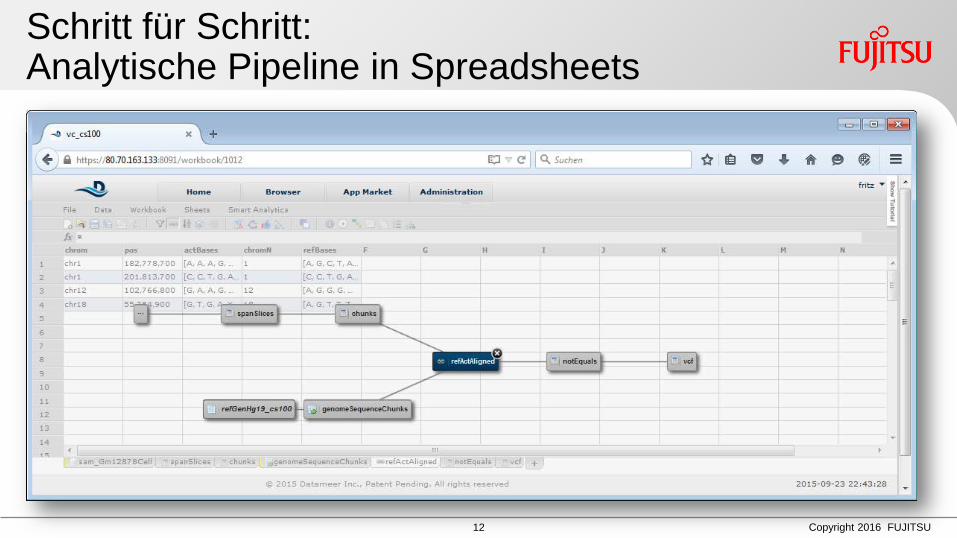
Schritt für Schritt: Analytische Pipeline in Spreadsheets
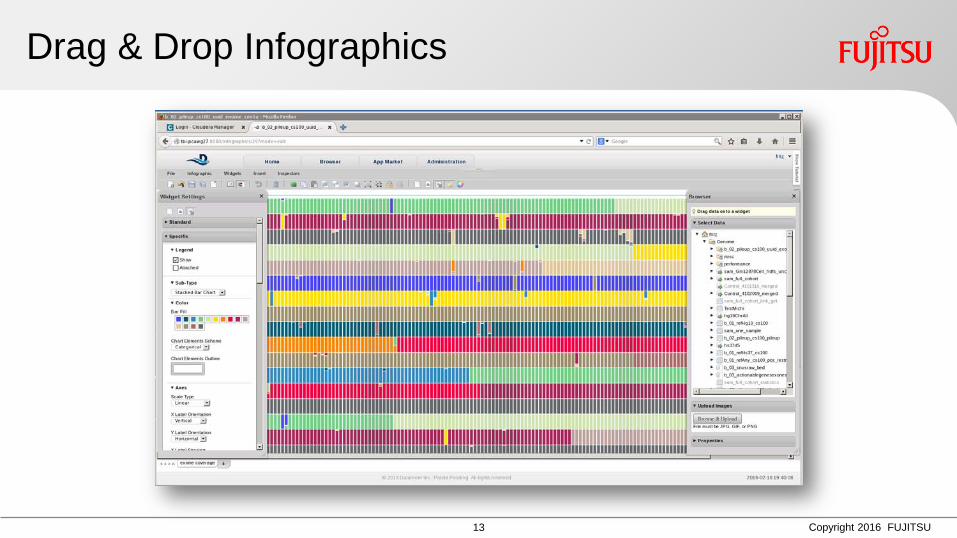
13 Copyright 2016 FUJITSU
Drag & Drop Infographics
14 Copyright 2016 FUJITSU
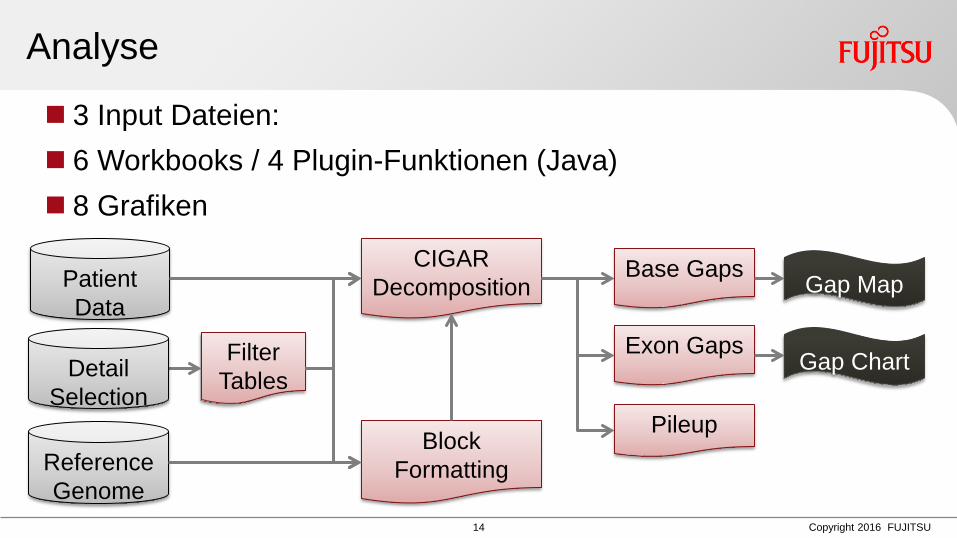
Analyse
3 Input Dateien: 6 Workbooks / 4 Plugin-Funktionen (Java) 8 Grafiken
Block Formatting Reference
Genome
Pileup
Base Gaps Gap Map
Filter Tables
Exon Gaps Gap Chart
CIGAR Decomposition
Detail Selection
Patient Data

03.03.2016 Matthias Schlesner 15
Performance Test: Selektiver Pileup
Basen / min * core: 1,6 Mrd Zeit pro BAM file: 8 Minuten (Parallelisierung auf Chromosomenebene)
Skalierung: problematisch (Bottlenecks: zentraler File-Server)
21 Knoten, (105 Cores) 104 BAM Files (14 Tbp) 898.677 Pileup Positionen
1 HPC Knoten (1 Core) 1 BAM File (0,14 Tbp) 898.677 Pileup Positionen
Basen / min * core: 1,9 Mrd Zeit pro BAM file: <2 Minuten (Parallelisierung auf Blockebene)
Skalierung: unproblematisch
HPC Hadoop
Laufzeit: 85 Minuten Laufzeit: 70 Minuten

03.03.2016 Matthias Schlesner 16
Große Patientenkohorten (Forschung) • Kostengünstige Server / Storage Konsolidierung • Ohne Datentransport kürzere Gesamtlaufzeit Beschleunigung von Analysen auf Rohdaten (~20%)
Analyse einzelner Patientendaten (Klinik) • Parallele Analyse einzelner Patientendaten Um Faktoren schnellere Voll- und Detailanalysen (~4 x)
Hadoop: Geeignete Plattform für Genomdaten
Kosten
Durchsatz
03.03.2016 Matthias Schlesner 17
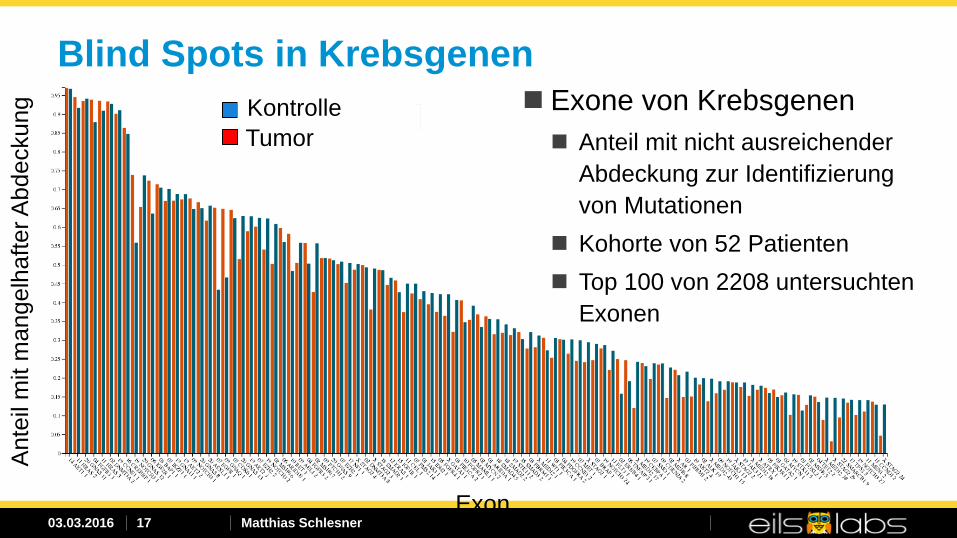
Blind Spots in Krebsgenen Exone von Krebsgenen Anteil mit nicht ausreichender
Abdeckung zur Identifizierung von Mutationen
Kohorte von 52 Patienten Top 100 von 2208 untersuchten
Exonen
Ante
il m
it m
ange
lhaf
ter A
bdec
kung
Kontrolle Tumor
Exon

03.03.2016 Matthias Schlesner 18
Blind Spots in Krebsgenen
03.03.2016 Matthias Schlesner 19
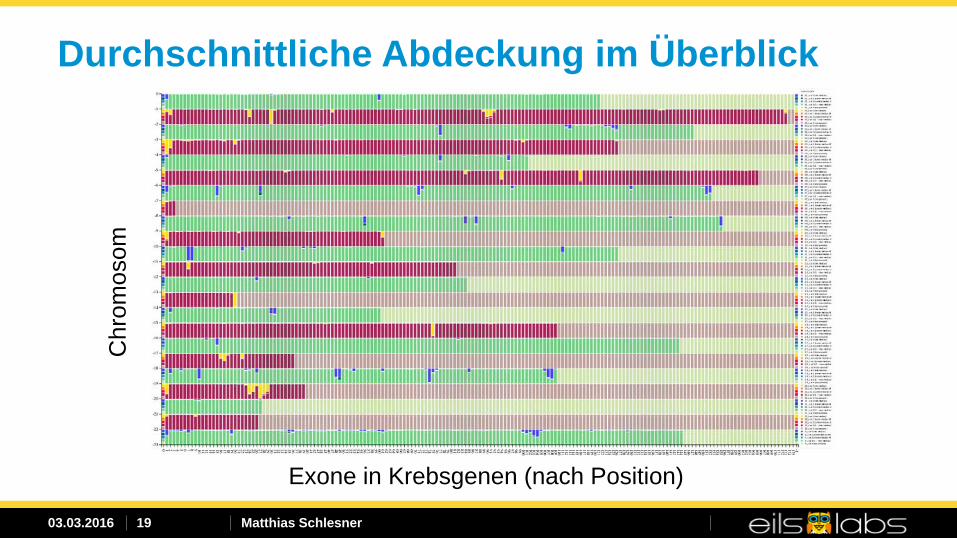
Durchschnittliche Abdeckung im Überblick C
hrom
osom
Exone in Krebsgenen (nach Position)

20 Copyright 2016 FUJITSU
Zusammenfassung und Ausblick
Genomweite Datensätze konnten im Hadoop-Cluster parallel und ohne Skalierungsprobleme analysiert werden
Spreadsheets erlauben Nutzern ohne Programmierkenntnisse die Analyse genomweiter Datensätze
Mehrere Regionen in Krebsgenen ohne ausreichende Sequenzierdaten zur Identifikation von Mutationen wurden identifiziert

21 Copyright 2016 FUJITSU


















