Selektive katalytische Reduktion von NO mit Methan an mit ... · IE Ion Exchange IR Infrarot LFC...
162
Selektive katalytische Reduktion von NO mit Methan an mit Indium und Cer modifizierten Zeolithkatalysatoren DISSERTATION zur Erlangung des Grades eines Doktors der Naturwissenschaften vorgelegt von Thomas Sowade aus Kobe Ruhr-Universit¨ at Bochum Lehrstuhl f¨ ur Technische Chemie 2002
Transcript of Selektive katalytische Reduktion von NO mit Methan an mit ... · IE Ion Exchange IR Infrarot LFC...

Selektive katalytische Reduktion von NO mit Methanan mit Indium und Cer modifizierten
Zeolithkatalysatoren
DISSERTATION
zur Erlangung des Grades
eines Doktors der Naturwissenschaften
vorgelegt von
Thomas Sowade
aus Kobe
Ruhr-Universitat Bochum
Lehrstuhl fur Technische Chemie
2002

Die vorliegende Arbeit wurde in der Zeit vom Januar 1999 bis Oktober 2002 am Lehrstuhl f¨ur
Technische Chemie der Ruhr-Universit¨at Bochum angefertigt.
Disputation am 12. Dezember 2002
Prufungskommission:
Vorsitzender: Prof. Dr. C. W¨oll
Referent: Prof. Dr. W. Gr¨unert
Korreferent: Prof. Dr. M. Muhler
Dritter Prufer: Prof. Dr. W. S. Sheldrick

Danksagung
Herrn Prof. Dr. Wolfgang Gr¨unert danke ich f¨ur die Moglichkeit zur Durchf¨uhrung dieser Ar-
beit, sowie fur die interessante Aufgabenstellung und seine st¨andige Diskussionsbereitschaft.
Herrn Prof. Dr. Martin Muhler m¨ochte ich danken f¨ur die freundlicheUbernahme des Korrefe-
rats.
An dieser Stelle sei auch Herrn Dipl.-Chem. Khalid Doukkali Attar f¨ur die im Rahmen seiner
Diplomarbeit durchgef¨uhrten kinetischen Messungen und Simulationen gedankt.
Gleichzeitig gilt mein Dank auch Herrn Dr. Heinz Berndt und Herrn Dr. Frank-Walter Sch¨utze
vom ACA fur dieUberlassung einer zweiten Katalyseapparatur zur Durchf¨uhrung kinetischer
Messungen.
Bedanken m¨ochte ich mich ferner bei Herrn Dr. Ulrich Bartmann f¨ur die Bereitstellung des
Optimierungsprogramms”OPTI“und bei Herrn Heinz Pfeiffer f¨ur seine EDV-technische Un-
terstutzung.
Weiterhin gilt mein Dank Frau Susanne Wiedemeyer f¨ur die Durchfuhrung von TPR-
und BET-Messungen, Frau Dr. Elke L¨offler und Frau Astrid Gomann f¨ur die Aufnahme der
IR-Spektren und XRD-Untersuchungen, Frau Dr. Olga Tkachenko f¨ur die Anfertigung von
XP-Spektren, sowie Frau Dr. Carmen Schmidt f¨ur die Realisation und Auswertung von
EXAFS- und XP-Spektren.
Allen Mitarbeitern des Lehrstuhls f¨ur Technische Chemie gilt mein Dank f¨ur die gute
Arbeitsatmosph¨are und die immerw¨ahrende Hilfsbereitschaft bei jeglicher Art von Problemen.
Frau Dr. Renate Aubel, Herrn Matthias Aubel und Herrn Dipl.-Chem. Markus Bergmann sei
fur die Durchsicht der Arbeit gedankt.
Teile dieser Arbeit wurden durch das Bundesministerium f¨ur Bildung und Forschung gef¨ordert
(Projekt-Nr.: FKZ 03C0271B6).

Zusammenfassung
Einleitung
Die selektive katalytische Reduktion von Stickoxiden mit Kohlenwasserstoffen stellt aus ¨oko-
nomischer und technologischer Sicht eine vielversprechende Alternative zur heute g¨angigen
Beseitigung von Stickoxiden mit stickstoffhaltigen Verbindungen wie Ammoniak und Harn-
stoff als Reduktionsmittel dar.
Als aktive Katalysatoren auf Zeolithbasis gelten unter anderen die mit Indium modifizierten
ZSM-5-Systeme, die sich durch einen hohen Methannutzungsgrad auszeichnen. Allerdings un-
terliegen sie einer starken Vergiftung durch Wasser. Nach Promotierung mit Edelmetallen wie
Platin, Iridium oder Rhodium lassen sich die Aktivit¨aten sowohl im trockenen als auch im
feuchten Gasstrom steigern, so dass diese im f¨ur eine technische Nutzung interessanten Be-
reich liegen. Vergleichbare Aktivit¨aten lassen sich mit CeO2 als Promotor erzielen.
Zielsetzung
Das Ziel dieser Arbeit bestand darin, in Zusammenarbeit mit dem Institut f¨ur angewandte Che-
mie in Berlin-Adlershof m¨oglichst aktive Katalysatoren zu pr¨aparieren. Gleichzeitig sollten die
aktiven Spezies durch eine Auffindung von Beziehungen zwischen Katalysatorstrukturen und
der Reaktivitat identifiziert werden. Besonders die Rollen der einzelnen Katalysatorkomponen-
ten (CeO2, Indium und die Protonen des Zeolithen) standen im Fokus des Interesses. Nach
der Aufstellung von Formalkinetiken f¨ur die einzelnen Systeme CeO2-ZSM-5, In-ZSM-5 und
CeO2-In-ZSM-5 war mit Hilfe der erhaltenen Parameter auf Unterschiede und Gemeinsamkei-
ten im Reaktionsablauf zu schließen.
Untersuchungsmethodik
In einem mikrokatalytischen Festbettreaktor wurde die selektive katalytische Reduktion von
NO mit Methan an unterschiedlich pr¨aparierten Katalysatoren untersucht (Standardreaktions-
bedingungen 1000 ppm Methan, 1000 ppm NO, 2 % O2, GHSV = 30000 h�1, 600 - 350ÆC).

Zusammenfassung III
Bei den zum Einsatz gekommenen Pr¨aparationsmethoden handelt es sich um den w¨assrigen Io-
nentausch, die F¨allung, die Sublimation, die Transportreaktion, den Festk¨orperionentausch und
die mechanische Mischung. F¨ur das In-ZSM-5-Grundsystem wurde die gesamte Bandbreite
der Praparationen ausgesch¨opft, wahrend sich die Zumischung von CeO2 auf eine mechanische
Beimengung oder F¨allung beschr¨ankte. Um die Probenanzahl sinnvoll einzugrenzen, wurde nur
ein Zeolith und ein Silikalit mit MFI-Struktur verwendet.
Ausgewahlte Katalysatoren wurden einer eingehenden Charakterisierung mittels unterschied-
licher Methoden (BET, XRD, DRIFTS, TPR) unterzogen. In einer parallelen Dissertation von
C. Schmidt erfolgte die Charakterisierung der Proben durch XPS und EXAFS.
Nach der Auswahl dreier f¨ur die Systeme CeO2-ZSM-5, In-ZSM-5 und CeO2-In-ZSM-5 re-
prasentativer Katalysatoren wurden an diesen kinetische Messungen durchgef¨uhrt. Die ein-
zelnen Bedingungen waren an die Aktivit¨at des jeweiligen Katalysatorsystems angepasst und
variierten deshalb. Es ergaben sich allerdingsUberschneidungen, die zum Vergleich der kine-
tischen Parameter herangezogen wurden.
Ergebnisse
Fur das Grundsystem In-ZSM-5 wurden Katalysatoren mit einer m¨aßigen Aktivitat prapariert.
Hierbei fuhren verschiedene Pr¨aparationsstrategien zu unterschiedlichen Indiumstrukturen, die
sich auch in ihrer katalytischen Aktivit¨at unterscheiden.
Als aktive Indiumkomponente wurden intrazeolithische Indiumoxospezies identifiziert.
Besitzen diese jedoch noch Chlor in ihrer Koordinationsph¨are, so zeigen sie eine niedrige Ak-
tivit at in der SCR-Reaktion, obwohl sie Methan aktivieren k¨onnen.
Bei mechanischen Mischungen gibt es Hinweise auf die Existenz einer bisher nicht beobach-
teten Indiumspezies, die vermutlich durch kovalente In-O-Si- und/oder In-O-Al-Bindungen
an der außeren Oberfl¨ache des Zeolithen verankert ist und eine hohe Aktivit¨at fur die
SCR-Reaktion aufweist.
Die Promotierung mit CeO2 laßt sich sehr einfach durch bloßes mechanisches Beimengen eines
CeO2 mit hoher Oberfl¨ache erreichen.
Da auch Katalysatoren mit sehr geringer SCR-Aktivit¨at wirkungsvoll promotiert werden
konnen, ist die Suche nach dem besten CeO2-In-ZSM-5-Katalysator nicht auf das Auffin-

Zusammenfassung IV
den des besten In-ZSM-5-Katalysators mit anschließender Promotierung zu vereinfachen. Die
Effektivitat der Promotierung ist abh¨angig von der Art der Indiumzentren; besonders ausge-
pragt lassen sich die an der ¨außeren Oberfl¨ache des Zeolithen lokalisierten Indiumzentren
promotieren.
Die in der Literatur bereits postulierte Rolle des CeO2 als NO-Oxidationskatalysator wird
bestatigt.
Gleichzeitig kann vorliegendes Ce3+ unter bestimmten Pr¨aparationsbedingungen als Redukti-
onsmittel fur einen Festk¨orperionentausch der Indiumkomponente fungieren.
Durch Versuche zur Vergiftung der Protonen des Zeolithen bzw. der Verwendung von Silikalit
gibt es Hinweise auf eine Beteiligung der Brønstedzentren des Zeolithen an der Reaktion, dies
kann jedoch nicht in letzter Konsequenz best¨atigt werden.
Die ermittelten kinetischen Parameter lassen sich in die Liste der in der Literatur zug¨anglichen
Werte einordnen. Sie best¨atigen die NO-Oxidation als anf¨anglichen Reaktionsschritt. Gleich-
zeitig zeigen sie, dass die Reaktanden einer Konkurrenzadsorption mit Wasser unterliegen.
Schlussfolgerung
Der aus den Grundsystemen CeO2-ZSM-5 und In-ZSM-5 kombinierte Katalysator
CeO2-In-ZSM-5 liefert eine hohe Aktivit¨at in der SCR-Reaktion mit Methan als Reduktions-
mittel.
In Langzeitversuchen bei 600ÆC unter 10 % Wasser im Eduktgas kommt es jedoch zu einer
erheblichen irreversiblen Deaktivierung bei Verwendung von Methan als Reduktionsmittel, so
dass eine technische Anwendung unter diesen Bedingungen nicht m¨oglich scheint. Diese Aus-
sage gilt jedoch nicht f¨ur mildere Reaktionsbedingungen oder andere Reduktionsmittel wie
beispielsweise Propan. Hier ist aus am ACA erhaltenen Ergebnissen ein großes Potential f¨ur
eine technische Anwendung ersichtlich.

Symbolverzeichnis
Akronyme und Abk urzungenACA Institut fur Angewandte Chemie Berlin-AdlershofDRIFTS Diffuse Reflectance Infrared Fourier Transform SpectroscopyEDX Energy Dispersive Analysis of X-raysESR Electron Spin ResonanceEXAFS Extended X-ray Absorption Fine StructureFCC Fluid Catalytic CrackingFQS absolute FehlerquadratsummeFX relative FehlerquadratsummeGHSV Gas Hourly Space Velocity [h�1]I Intensitat (Massenspektrometer)IE Ion ExchangeIR InfrarotLFC Liquid Flow ControllerMFC Mass Flow ControllerMTG Methanol to GasolineMW MegawattOCM Oxidative Coupling of MethaneP PrecipitationPSA Pressure-Swing-AdsorptionRSSIE Reductive Solid State Ion ExchangeRT Raumtemperatur [ÆC]S SublimationSCR Selective Catalytic ReductionSSIE Solid-State-Ion-ExchangeSTP Standard Temperature and PressureT Transport ReactionTA-Luft Technische Anleitung zur Reinhaltung der LuftTPR temperaturprogrammierte ReduktionXANES X-ray Absorption Near Edge StructureXAS X-ray Absorption SpectroscopyXPS X-ray Photoelectron SpectroscopyXRD X-ray Diffraction

Symbolverzeichnis VI
SymboleBo Bodensteinzahlc Konzentration [mol�m�3]d Durchmesser [m]D Diffusionskoeffizient [m2 � s�1]DaII Damkohler-Zahl 2.ArtF Flache (Gaschromatograph)Ea Aktivierungsenergie [kJ�mol�1]G Gibbs-Energie [kJ�mol�1]L Lange [m]_n Stoffstrom [mol� s�1]Pe Pecletzahlr Reaktionsgeschwindigkeit [mol�s�1 �m�3]Re Reynolds-ZahlSc Schmidt-ZahlSh Sherwood-ZahlX Umsatz [%]� Heizrampe [K� min�1]� Porositatsfaktor� Katalysatorwirkungsgrad, oder dynamische Viskosit¨at [kg�m�1 �s�1]� kinematische Viskosit¨at [m2 � s�1]� Dichte [kg� m�3]� Tortuositatsfaktor Kollisionsintegral Weisz-Modul
Indizes15 Masse 15 im Massenspektrometer30 Masse 30 im Massenspektrometereff effektivext externi,j Komponente i,jKat KatalysatorP PelletR ReaktorTag z.B. Tagesdruck oder -temperatur

Inhaltsverzeichnis
Zusammenfassung II
Symbolverzeichnis V
1 Einleitung und Zielsetzung 1
2 Stand des Wissens 4
2.1 Zeolithe . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2 Selektive katalytische Reduktion von NO mit Methan . . . . . . . . . . . . . . 6
2.2.1 Das System CeO2-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . 6
2.2.2 Das System In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.2.3 Das System CeO2-In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . 9
2.2.4 Formalkinetiken der SCR mit Methan . . . . . . . . . . . . . . . . . . 11
3 Untersuchungsmethodik 19
3.1 Experimentelles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
3.1.1 Versuchsapparatur . . .. . . . . . . . . . . . . . . . . . . . . . . . . 19
3.1.2 Durchfuhrung und Auswertung der katalytischen Messungen . . . . . . 23
3.1.3 Katalysatorpr¨aparation . . . . . . . . . . . . . . . . . . . . . . . . . . 25
3.1.3.1 Ionentausch . . . . . . . . . . . . . . . . . . . . . . . . . . 25
3.1.3.2 Fallung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
3.1.3.3 Ionentausch mit anschließender F¨allung . . . . . . . . . . . 26
3.1.3.4 Festk¨orperionentausch . . . . . . . . . . . . . . . . . . . . . 26
3.1.3.5 Sublimation . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3.1.3.6 Transportreaktion . . . . .. . . . . . . . . . . . . . . . . . 27
3.1.3.7 Mechanische Mischungen .. . . . . . . . . . . . . . . . . . 28
3.1.3.8 Schichtexperimente . . . . . . . . . . . . . . . . . . . . . . 28
3.1.4 Katalysatorcharakterisierung . . . . .. . . . . . . . . . . . . . . . . . 28
3.1.4.1 Spezifische Katalysatoroberfl¨ache . . . . . . . . . . . . . . . 28
3.1.4.2 Infrarot-Spektroskopie . . .. . . . . . . . . . . . . . . . . . 30
3.1.4.3 Rontgendiffraktometrie . .. . . . . . . . . . . . . . . . . . 32

Inhaltsverzeichnis VIII
3.1.4.4 Photoelektronenspektroskopie . . .. . . . . . . . . . . . . . 34
3.1.4.5 Rontgenabsorptionsspektroskopie .. . . . . . . . . . . . . . 36
3.1.4.6 Temperaturprogrammierte Reduktion . . . . . .. . . . . . . 38
3.2 Die Modellierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.2.1 Simulationsprogramm .. . . . . . . . . . . . . . . . . . . . . . . . . 40
3.2.2 Reaktormodell . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.2.3 Stofftransportlimitierung und axiale Dispersion . . . . . .. . . . . . . 42
4 Ergebnisse 48
4.1 Katalyse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
4.1.1 Das System CeO2-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . 48
4.1.2 Das System In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.1.2.1 Definition der Probenbezeichnungen . . . . . .. . . . . . . 51
4.1.2.2 Charakterisierungsergebnisse . . .. . . . . . . . . . . . . . 53
4.1.2.3 Katalytische Ergebnisse . . . . . . . . . . . . . . . . . . . . 60
4.1.2.4 Zusammenfassung . . . . .. . . . . . . . . . . . . . . . . . 75
4.1.3 Das System CeO2-In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . 76
4.1.3.1 Definition der Probenbezeichnungen . . . . . .. . . . . . . 76
4.1.3.2 Katalytische Ergebnisse . . . . . . . . . . . . . . . . . . . . 77
4.1.3.3 Zusammenfassung . . . . .. . . . . . . . . . . . . . . . . . 102
4.2 Modellierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
4.2.1 Das System CeO2-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . 104
4.2.2 Das System In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . . . . 114
4.2.3 Das System CeO2-In-ZSM-5 . . . . . . . . . . . . . . . . . . . . . . . 122
4.2.4 Zusammenfassung und Vergleich der Ergebnisse . . . . .. . . . . . . 138
5 Schlussfolgerungen und Ausblick 143
6 Literaturverzeichnis 145
A Anhang 151

1 Einleitung und Zielsetzung
Die Reduktion von Stickoxiden aus Abgasen stellt heutzutage eine der Hauptaufgaben des
Umweltschutzes dar.
Stickoxidemissionen entstehen haupts¨achlich bei Verbrennungsprozessen und enthalten auf-
grund des thermodynamischen Gleichgewichts zwischen NO und NO2 in Verbindung mit
den hohen Abgastemperaturen als Hauptkomponente Stickstoffmonoxid. Als Stickstoffquellen
kommen der Brennstoff selber, bei hohen Temperaturen aber auch der Stickstoff der Verbren-
nungsluft in Frage.
Stickstoffmonoxid ist als Radikal in der Lage, in der Atmosph¨are durch Reaktion von NO
mit Ozon die Ozonschicht der Erde abzubauen. Desweiteren wirkt es beim Einatmen wie eine
Saure, was zu Sch¨adigungen der Lunge f¨uhrt.
Auf Grund dieser Gefahren sind vom Gesetzgeber verschiedene Bestimmungen erlassen
worden, welche Grenzwerte f¨ur die Emissionen festlegen. F¨ur große Feuerungsanlagen lassen
sich diese Werte der TA-Luft [1] oder dem Bundesimmissionsschutzgesetz [2] entnehmen. So
gilt beispielsweise f¨ur neue Großfeuerungsanlagen mit einer Leistung< 300 MW ein Grenz-
wert fur gasformige Brennstoffe von 200 mg/m3 bzw. 100 ppm. F¨ur mobile Quellen wird der
zulassige Grenzwert in g/km angegeben, so z.B. f¨ur Ottomotoren 0,5 g/km NOx [3]. Allerdings
ist fur die weitere Zukunft eine drastische Absenkung der Grenzwerte geplant [4–6]. Besonders
rigide Grenzwertbestimmungen gelten in den Vereinigten Staaten von Amerika, insbesondere
in Kalifornien.
Zur Minderung der NOx-Emissionen kommen zwei M¨oglichkeiten in Frage: Prim¨armaß-
nahmen sollen die Entstehung von Stickoxiden vermindern. Dies l¨asst sich durch Einsatz stick-
stoffarmer Brennstoffe und/oder Absenkung der Verbrennungstemperaturen, aber auch durch
optimierte Verbrennungsvorg¨ange im Motoreninnenraum erzielen. Sekund¨armaßnahmen zielen
als sogenannte”end-of-pipe-technology“ auf die Umsetzung der schon gebildeten Stickoxide
und lassen sich grob in Nassverfahren und Trockenverfahren unterteilen. In Anbetracht der zu
erwartenden Grenzwertsenkungen k¨onnen in Zukunft nur durch ein gezieltes Zusammenspiel
von Primar- und Sekund¨armaßnahmen zufriedenstellende Ergebnisse erreicht werden, so dass
beide Moglichkeiten ihre Daseinsberechtigung haben und weiterentwickelt werden m¨ussen. In

1 Einleitung und Zielsetzung 2
der heterogenen Katalyse spielen nur die Trockenverfahren eine Rolle, eineUbersichtuber die
Nassverfahren ist in [7] gegeben.
Die eleganteste L¨osung zur Entfernung der Stickoxide w¨are die Zersetzung des
NO-Molekuls zu N2 und O2. Obwohl dies thermodynamisch m¨oglich ist [8], stehen bis heute
keine technisch geeigneten Katalysatoren zur Verf¨ugung, da die besten bisher entdeckten Ka-
talysatoren, z.B. Cu-ZSM-5, Ag-promotiertes Co3O4 und Pt/Al2O3, noch keine ausreichende
Aktivit at in Anwesenheit von Sauerstoff¨uberschuss aufweisen [9,10]. Gerade dies ist aber im
Hinblick auf die Entwicklung von Diesel- und Magermixmotoren essentiell, da in ihrem Ge-
misch einuberstochiometrisches Verh¨altnis Sauerstoff/Kohlenwasserstoffe enthalten ist. Auch
die bisherigen 3-Wege-Katalysatoren (Pt, Pd und Rh auf modifiziertem -Al 2O3) fur Ottomo-
toren versagen bei Sauerstoff¨uberschuss, so dass hier weiterer Forschungsbedarf besteht.
Bei stationaren Emissionsquellen wie Kraftwerken hat sich mittlerweile die Technik der se-
lektiven katalytischen Reduktion (SCR) durchgesetzt. Hier wird das NO selektiv mit NH3 an
V2O5/TiO2-Katalysatoren umgesetzt. Allerdings muss auf Probleme wie Ammoniakschlupf,
Ammoniaklogistik und Korrosion der Anlage geachtet werden. F¨ur einen mobilen Einsatz
erhalt das Problem der Ammoniaklogistik eine ganz neue Bedeutung, da sowohl im Fahrzeug
als auch an der Tankstelle neue Vorratsbeh¨alter fur Ammoniak oder alternativ Harnstoff zu
installieren waren. Abgesehen davon arbeiten die Katalysatoren nur in einem engen Tempera-
turfenster, das bei mobilen Quellen zeitweise sowohl unter- als auch ¨uberschritten wird.
Als im Jahr 1990 die beiden Arbeitsgruppen umHeld et al. [11] undIwamotoet al. [12]
unabhangig voneinander ¨uber den Einsatz von Kohlenwasserstoffen (außer Methan) als se-
lektives Reduktionsmittel f¨ur Stickoxide unter Sauerstoff¨uberschuss an einem Cu-ZSM-5 be-
richteten, setzte eine intensive Forschert¨atigkeit ein, da Kohlenwasserstoffe in ausreichender
Menge schon im Abgas vorhanden sind, aber problemlos auch zudosiert werden k¨onnen. Zwei
Jahre sp¨ater fandenLi et al. den ersten vielversprechenden Katalysator f¨ur die SCR von NO mit
Methan, einen Co-ZSM-5 [13]. Im darauffolgenden Jahrzehnt wurden als weitere Aktivkom-
ponenten Gallium, Indium, Palladium und Rhodium in Verbindung mit dem Zeolithen ZSM-5
identifiziert, aber auch analog modifizierte Ferrierite und Mordenite weisen Aktivit¨at auf.
Am Lehrstuhl fur Technische Chemie der Ruhr-Universit¨at Bochum wurde im Rahmen
der Untersuchung eines CeO2-ZSM-5 Komposit-Katalysators ein CeO2-In-ZSM-5-Katalysator
entwickelt [14], dessen Aktivit¨at im Bereich technischen Interesses liegt. Im Anschluss

1 Einleitung und Zielsetzung 3
daran formierte sich ein Forschungsverbund aus dem Institut f¨ur Angewandte Chemie in
Berlin-Adlershof (ACA), der Firma W.C.Heraeus in Hanau und der Ruhr-Universit¨at Bochum.
Aufgabe des Verbundes war eine angewandte Grundlagenforschung auf dem Gebiet neuer
Zeolith-Katalysatoren f¨ur die Aktivierung von Methan und Fl¨ussiggas-Kohlenwasserstoffen in
der selektiven katalytischen Reduktion von NOx. Durch die Integration eines industriellen Part-
ners flossen fr¨uhzeitig anwendungstechnische Aspekte in die Entwicklung des Katalysators ein.
In dieser Arbeit liegt der Schwerpunkt nicht so sehr auf einer technischen Entwick-
lung, sondern auf der Grundlagenforschung. Die Katalysatoren In-ZSM-5, CeO2-ZSM-5 und
CeO2-In-ZSM-5 sollen hinsichtlich der Funktion und der Struktur der einzelnen Komponen-
ten, besonders des Indiums, untersucht werden. Hierzu wurden verschiedene Pr¨aparations-
strategien mit dem Ziel verfolgt, Katalysatoren unterschiedlicher Strukturen zu generieren.
Nach der Charakterisierung der erhaltenen Strukturen haupts¨achlich durch die XAFS-Technik
(X-ray Absorption Fine Structure) in einer parallelen Arbeit [15] soll die Struktur der Ka-
talysatoren mit ihrer Reaktivit¨at korreliert werden. Im Anschluss an die Optimierung des
CeO2-In-ZSM-5 hinsichtlich Reaktivit¨at und Stabilitat erfolgen an allen drei Systemen kine-
tische Messungen und deren formalkinetische Simulation. Durch Vergleich der ermittelten ki-
netischen Parameter (Reaktionsordnungen, Aktivierungsenergien) soll auf Analogien oder Un-
terschiede in den zugrundeliegenden Reaktionsmechanismen geschlossen werden. Zus¨atzlich
kann die ermittelte Formalkinetik als Grundlage f¨ur eine sp¨atere Reaktorauslegung herangezo-
gen werden.

2 Stand des Wissens
2.1 Zeolithe
Zum besseren Verst¨andnis der nachfolgenden Kapitel ist es notwendig, die Struktur und Eigen-
schaften von Zeolithen, in diesem Fall speziell des ZSM-5, zu kennen.
Der Name Zeolith stammt aus dem Griechischen und bezeichnet die Eigenschaft, beim Er-
hitzen Wasser abgeben zu k¨onnen (zein = wallen, kochen,lithos = Stein). Die weitere No-
menklatur ist unsystematisch und orientiert sich an Mineralien oder aber auch an Projekten
zur Synthese neuer Tetraederger¨uste. So stammt der Name ZSM-5 vom f¨unften Produkt im
”Zeolite Socony-Mobil project“. Zeolithe bestehen aus regelm¨aßigen dreidimensionalen Git-
tern eckenverkn¨upfter Tetraeder TO4 (T = Si, Al, P, Be, Ga, Ge, Zn), wodurch sich Kan¨ale und
Hohlraume bilden, deren Struktur durch das bei der Synthese eingesetzte Templat bestimmt
werden kann.
Im ZSM-5 bildet sich durch das Templat Tetrapropylammoniumbromid (mittlerweile
existieren allerdings weitere Template und auch templatfreie Synthesen) eine Struktur geradli-
niger Kanale (5,1A x 5,6 A), die durch zickzackf¨ormige Kanale (5,4A x 5,6 A) gekreuzt wer-
den und denen als Grundbaustein ein f¨unfgliedriger Ring (Pentasil-Einheit) zugeordnet wird
(siehe Abbildung 2.1). Durch einen Einbau von Al3+ anstelle von tetraedrisch koordiniertem
Si4+ kommt es zu einer negativen Gesamtladung, die durch Kationen kompensiert wird. Das
Kation selber kann sich in dem Namen eines bestimmten Zeolith-Ger¨ustes wiederfinden; so be-
Abbildung 2.1: Gittergerust des Zeolithen ZSM-5 [16,17]

2 Stand des Wissens 5
schreibt z.B. NH4-ZSM-5 einen Zeolithen ZSM-5 mit einem Ammoniumion als ladungskom-
pensierendem Kation. F¨ur einen Zeolithen l¨asst sich demnach die folgende allgemeine Formel
aufstellen:
Mx=n[(AlO2)x(SiO2)y] � wH2O (2.1)
Handelt es sich bei dem Kation Mn+ um H+, so wird ein H-ZSM-5 mit Brønsted-Azidit¨at
erhalten. Gleichzeitig enthalten Zeolithe aber auch noch Lewis-azide Zentren z.B. durch
nicht vollstandig in das Gitter eingebaute Aluminiumatome oder durch Dehydratisierung der
Brønsted-Form. Genaueres ¨uber die einzelnen aziden Zentren wird sp¨ater im Zusammenhang
mit der DRIFT-Spektroskopie vorgestellt werden.
Durch diese aziden Eigenschaften sind Zeolithe nicht erst f¨ur die SCR-Reaktion katalytisch
interessant geworden, sondern es existieren seit langem eine große Anzahl technischer Anwen-
dungen [18].
Eine spezielle Eigenschaft ist ihre Formselektivit¨at fur Reaktanden und Produkte. Die Se-
lektivitat gegen¨uber Reaktanden ergibt sich aus der Eigenschaft, dass nur Molek¨ule unterhalb
einer bestimmten Gr¨oße die Kanal¨offnungen passieren k¨onnen, gr¨oßere Molek¨ule werden von
der Katalyse ausgeschlossen. Beim innerhalb der Kan¨ale gebildeten Produkt spielt bez¨uglich
der Formselektivit¨at ebenfalls die Gr¨oße des Molek¨uls die entscheidende Rolle. So liefert bei-
spielsweise die Isomerisierung von Dimethylbenzol als Hauptprodukt die 1,4-Form, die auf
Grund ihrer geringeren Gr¨oße schnell aus dem Zeolithen diffundieren kann, w¨ahrend die 1,2-
und 1,3-Form (ca. 0,6A großer) durch die h¨ohere Verweilzeit weiteren Isomerisierungsreak-
tionen unterworfen sind.
Diese Molekularsiebeffekte werden weiterhin ausgenutzt bei verschiedenen Adsorptions-
und Trennverfahren z.B. bei Gasreinigungsverfahren oder bei der n-/iso-Paraffintrennung.
Der Großteil der heutigen Zeolithproduktion endet als Waschmittelzusatz. Hier wird die hohe
Selektivitat des Zeoliths A gegen¨uber Calciumionen zur Wasserenth¨artung ausgenutzt.
Besonders in der Petrochemie werden die sauren Eigenschaften der Zeolithe genutzt.
Erwahnt sei hier das FCC Verfahren (FCC =”Fluid Catalytic Cracking“) zum Cracken h¨oher-
siedender Erd¨olfraktionen an Zeolith Y mit Zusatz von H-ZSM-5 zum Cracken von n-Alkanen,
das die thermischen Crackverfahren weitestgehend verdr¨angt hat. Interessant ist in diesem Zu-
sammenhang auch der an ZSM-5-Katalysatoren durchf¨uhrbare MTG-Prozeß (MTG =”Me-
thanol to Gasoline“), der noch nicht wirtschaftlich ist, und das Mobil-Badger-Verfahren zur
Erzeugung von Ethylbenzol aus Benzol und Ethylen an ZSM-5.

2 Stand des Wissens 6
2.2 Selektive katalytische Reduktion von NO mit Methan
In zahlreichenUbersichtsartikeln wird ¨uber die selektive katalytische Reduktion von NO mit
Kohlenwasserstoffen [19–23,8] berichtet, allerdings liegt hier der Hauptfokus auf den C2-, C3-
und C4-Kohlenwasserstoffen als Reduktionsmittel. Besonders intensiv befasst sichArmor mit
Methan als Reduktionsmittel [24], doch sind auch den vorhergenannten Artikeln zahlreiche
Informationen zur SCR mit Methan zu entnehmen. Bevor auf die einzelnen Katalysatorsysteme
eingegangen wird, soll hier erst eine allgemeineUbersichtuber die SCR mit Methan gegeben
werden.
Die allgemeine nichtst¨ochiometrische Reaktionsgleichung f¨ur die SCR-Reaktion lautet wie
folgt:
NO+HC +O2 ! N2 + (N2O) + CO2 + CO +H2O (2.2)
Daneben existiert als Hauptkonkurrenzreaktion die unselektive Kohlenwasserstofftotaloxidati-
on:
HC +O2 ! CO2 + CO +H2O (2.3)
Ein weiterer Gesichtspunkt bez¨uglich der Selektivit¨at liegt auf Seiten der stickstoffhaltigen Pro-
dukte. Die Reduktion soll hier ausschließlich zu N2 fuhren; N2O, das h¨aufig bei platinhaltigen
Katalysatoren gebildet wird, ist unerw¨unscht.
Nach der Entdeckung durchLi et al. 1992, dass Co-ZSM-5 mit Methan selektiv Stickoxi-
de reduzieren kann, wurden als weitere, das reaktionstr¨age Methan aktivierende Elemente Ga,
In, Ce, Mn, Ni, Pd, Pt, Ru und Rh gefunden [25–28]. Allerdings lassen sich die ermittelten
Aktivit aten nur qualitativ untereinander vergleichen, da i.A. keine einheitlichen Reaktionspa-
rameter angewendet wurden. F¨ur den interessierten Leser befindet sich in [14] eine Tabelle
aller bis dato bekannten Katalysatoren mit den jeweiligen Umsatzmaxima bei der zugeh¨ori-
gen Temperatur und Belastung. In den folgenden Abschnitten m¨ochte ich die in dieser Arbeit
behandelten Systeme vorstellen.
2.2.1 Das System CeO2-ZSM-5
Cer-haltige Zeolithe sind besonders intensiv von der Gruppe umMisonoet al. studiert worden.
Der Untersuchungsschwerpunkt lag hier auf Propen als Reduktionsmittel [29–31]. Der Zeolith

2 Stand des Wissens 7
wurde durch Ionentausch hergestellt, und als aktive Zentren wurden Cerionen auf Kationen-
platzen angenommen [31–33]. Es wurde herausgefunden, dass auf diesen die Oxidation des
NO zu NO2 stattfindet. Auf dem Tr¨ager ZSM-5 erfolgt dann die weitere Reaktion von Propen
mit NO2 zu Organonitroverbindungen, die im weiteren Verlauf mit NOx und O2 am Cer zu N2
und COx zerfallen.
Auf der Basis von quantenchemischen Simulationsrechnungen ist auch das Ceroxokation
[CeO+] als aktive Komponente vorgeschlagen worden [34].
Durch Beimengung von Mn2O3 oder CeO2 zur Forderung der Oxidation von NO zu NO2
konnte die Aktivitat gesteigert werden. Dies ist ein weiterer Hinweis darauf, dass die Oxidation
einen anfanglichen Reaktionschritt darstellt [35–37].
Uber die SCR mit Methan gibt es wenige Studien, die aber alle dem ionengetauschten
Ce-ZSM-5 eine geringe Aktivit¨at zuweisen [28,38]. Auch in den Arbeiten vonLieseundRu-
tenbeckan der Ruhr-Universit¨at Bochum wurde f¨ur einen ionengetauschten Ce-ZSM-5 eine
geringe Aktivitat mit Methan, aber eine h¨ohere mit Propen festgestellt. Allerdings kehrt sich
dieses Ph¨anomen um bei Verwendung eines ZSM-5-Katalysators, auf den das Cer als CeO2 auf
dieaußere Oberfl¨ache aufgef¨allt wurde [39,40]. Aus den Ergebnissen von Schichtexperimenten
an physikalischen Gemischen (CeO2 oberhalb, mittig, unterhalb und verteilt im H-ZSM-5) und
Versuche mit NO2 wurde der in Abbildung 2.2 dargestellte Reaktionsmechanismus postuliert:
NO wird am CeO2 zu NO2 oxidiert. Eine weitere am CeO2 gebildete Spezies (vermutlich
ein Methylradikal) reagiert noch am CeO2 mit NO2 zu Nitro- oder Nitrosomethan. Dieses dif-
fundiertuber die Gasphase in den Zeolithen und wird dort mit NO2 zu N2 und COx umgesetzt.
Gleichzeitig kann NO2 CH4 in einer homogenen Gasphasenreaktion oder am Zeolithen unse-
lektiv zu COx oxidieren.
NO + 1/2 O2CeO2
NO2
NO + CH2 4
CeO2
CH NO , (CH NO)3 2 3 ZeolithGasphase
CH NO , (CH NO) + NO3 2 3 2
ZeolithN + CO2 x
CH NO , (CH NO)3 2 3
Abbildung 2.2: Reaktionsmechanismus f¨ur CeO2-H-ZSM-5 [14]

2 Stand des Wissens 8
2.2.2 Das System In-ZSM-5
Von den Elementen der dritten Hauptgruppe haben sich Gallium und Indium als aktiv in der
SCR mit Methan erwiesen. Erste Untersuchungen vonKikuchiet al. befassten sich mit Gallium
als aktive Komponente [41], Indium wurde erst im Jahre 1994 entdeckt [27,42]. Beide Ele-
mente zeichnen sich durch eine hohe Selektivit¨at bezuglich des Kohlenwasserstoffverbrauchs
aus, zudem wurde kein N2O als Nebenprodukt entdeckt. Allerdings brechen beide Katalysa-
toren unter 10 % Wasser im Gasstrom bei einer Belastung von 16000 h�1 dramatisch in ihrer
NO-Reduktionsaktivit¨at ein (Ga-ZSM-5 von 65 % auf 0 % Umsatz, und In-ZSM-5 von 80 %
auf 10 % Umsatz, jeweils 1000 ppm NO, 2000 ppm Methan, 10 % O2). Verwendet man NO2
als zu reduzierende Spezies, so sinkt auch hier der NO-Umsatz am Ga-ZSM-5 von 90 % auf ca.
5 %, der In-ZSM-5 erreicht aber immer noch 50 % Umsatz von anf¨anglichen 90 %. Diese gerin-
gere Anfalligkeit des In-ZSM-5 gegen¨uber Wasser wird begr¨undet mit der geringeren Affinit¨at
des Wassers zu koordinativ unges¨attigten InO+-Kationen im Gegensatz zu GaO+-Kationen.
Bei letzteren kommt die Konkurrenzadsorption zwischen Wasser und den Reaktionspartnern
NO und HC starker zum Tragen.
Bestarkt wird diese Theorie durch Versuche vonTabataet al., die Adsorptionsversuche mit
Wasser und Methan an Ga- und In-ZSM-5 durchf¨uhrten. Bei vorheriger Adsorption von Wasser
nahm die dissoziative Adsorption von Methan an einem Ga-ZSM-5 signifikant ab, w¨ahrend sie
an einem In-ZSM-5 nur geringf¨ugig zuruckging [43,44].
Eine weitere Best¨atigung des großen Einflusses von Wasser auf die nukleophile Umgebung
der GaO+-Kationen lieferten quantenchemische Berechnungen [34].
Hinweise fur die Existenz dieser bisher angenommenen Oxo-Kationen liefern
IR-spektroskopische Untersuchungen vonKikuchi et al. [45–47]. Sie postulieren f¨ur ein
Gemisch aus In2O3 und H-ZSM-5 durch unterschiedliche thermische Behandlungen einen
SSIE (SSIE = solid-state-ion-exchange) nach folgender m¨oglicher Formel:
In2O3 + 2H+Zeolith� ! 2[InO]+Zeolith� +H2O (2.4)
Auch durch Zhang et al. wird ein SSIE an impr¨agnierten Katalysatoren vorgeschlagen;
sie berichten von hochdispersen In-Spezies, die eine Verbindung mit den Protonen der
Zeolithoberflache eingehen. Allerdings zeigten mechanische Mischungen von In2O3 mit
H-ZSM-5 nur geringe Aktivitat [48,49]. Die vonRichteret al. gefundene starke Lewis-Azidit¨at

2 Stand des Wissens 9
der eingetauschten In-Spezies best¨arkt die Annahme von Oxo-Kationen [50].
Unter Berucksichtigung all dieser Studien l¨asst sich auch hier ein Reaktionsschema f¨ur die
SCR mit Methan am In-ZSM-5 vorschlagen: NO wird an Lewis-aziden Zentren des Zeolithen
zu NO2 oxidiert und an [InO]+-Kationen adsorbiert. Hier erfolgt die weitere Umsetzung mit
CH4 und NO zu N2.
NO + 1/2 O2 NO2
Lewis-Zentren
des Zeolithen
NO2,ads
InO+
+ CH4 + NOxN2 + COx + H2O
Abbildung 2.3: Reaktionsmechanismus f¨ur In-ZSM-5 [51]
2.2.3 Das System CeO2-In-ZSM-5
Da anscheinend durch Wasser die Oxidation des NO unterdr¨uckt wird, habenKikuchi et al.
einen Ersatz f¨ur die vergiftungsanf¨alligen Lewis-aziden Zentren des Zeolithen gesucht, der
selbst unter Wasser die Oxidationsreaktion erm¨oglicht.
Getestet wurde die Impr¨agnierung des In-ZSM-5 mit einem Gewichtsprozent Pt, Rh, Ir, Cr,
Pd, Fe, Ce, Au, Co, Ag, Mn und Ni. Als besonders vielversprechend stellten sich Pt, Rh und Ir
heraus, Ce brachte kaum eine Verbesserung, und Co und Ni bewirkten sogar eine Verschlech-
terung der Aktivitat. Die maximalen NO-Ums¨atze betragen f¨ur den Pt-In-ZSM-5 bei 500ÆC
ca. 18 % (1000 ppm NO, 1000 ppm CH4, 10 % O2, 10 % Wasser, GHSV 41000 h�1) und
fur den Ir-In-ZSM-5 ca. 21 %. F¨ur die niedrigere Belastung von 16000 h�1 existiert fur den
Pt-In-ZSM-5 eine NO-Umsatzkurve bei 500ÆC in Abhangigkeit der Wasserkonzentration. So
betragt der NO-Umsatz mit 2 % Wasser 95 %, mit 5 % Wasser 75 % und mit 8 % Wasser ca.
50 %. Auffallig ist hier, dass sich f¨ur den NO-Umsatz bis zu diesen hohen Wasserkonzentratio-
nen von 8 % noch kein Minimum gefunden hat; daher muss von einer weiteren Aktivit¨atsein-
buße bei Erh¨ohung der Wasserkonzentration ausgegangen werden. Trotzdem handelt es sich
bei diesen Katalysatoren um die bis dato aktivsten Systeme auf Zeolith-Basis in Anwesenheit
von Wasser [51–55].
Im Jahr 1999 fandLiese bei der Verbesserung des CeO2-ZSM-5 den CeO2-In-ZSM-5.
Geplant war ein Ersatz der gegen¨uber Wasser vergiftungsanf¨alligen Protonen durch ande-

2 Stand des Wissens 10
re Kationen auf Indium- oder Silberbasis [14]. Die mit dem nun gefundenen Katalysator
CeO2-In-ZSM-5 bei 500ÆC erzielten Ums¨atze von 92 % im trockenen und 80 % im feuch-
ten (2 % Wasser) Medium bei einer Belastung von 10000 h�1 liegen im Bereich der guten Ir-
und Pt-In-ZSM-5-Katalysatoren. Eine m¨ogliche Erklarung fur die hohe katalytische Aktivit¨at
unter Verwendung von Cer ist die im Vergleich zu [51–55] sehr viel h¨ohere Cerbeladung von
8,2 Gewichtsprozent im Gegensatz zu 1,0 Gewichtsprozent. Allerdings zeigte der entwickel-
te CeO2-In-ZSM-5-Katalysator noch keine ausreichende Langzeitstabilit¨at in Anwesenheit von
Wasser. In einem Langzeittest (1000 ppm NO, 1000 ppm CH4, 2 % O2, 2 % Wasser, 13000 h�1,
460ÆC) stieg der NO-Umsatz innerhalb der ersten 12 Stunden von 50 % auf 65 % an, um dann
im weiteren Verlauf kontinuierlich zu sinken und nach 140 Stunden einen Wert von 22 % zu
erreichen. Der Methanumsatz sank im gleichen Zeitraum von 42 % auf 16 %.Uber ein Lang-
zeitverhalten des Pt- oder Ir-In-ZSM-5 wird nichts berichtet.
Fur die Katalysatorsysteme CeO2-In-ZSM-5 und Ir-In-ZSM-5 wird der in Abb 2.4 gezeigte
Mechanismus diskutiert. NachLiesewird NO an außerhalb des Zeolithen lokalisiertem CeO2
NO + 1/2 O2 NO2
Ir/Pt
NO2,ads
InO+
+ CH4 + NOxN2 + COx + H2O
NO + 1/2 O2CeO2
NO2
NO2 ZeolithGasphase
NO2 4+ CHZeolith
N + CO2 x
Abbildung 2.4: Reaktionsmechanismus f¨ur die promotierten In-ZSM-5-Systeme: links nach[14], rechts nach [51]
zu NO2 oxidiert. NO2 kann dann ¨uber eine weite Distanz zum Zeolithen transportiert und dort
mit CH4 umgesetzt werden.
Ein analoger, aber detaillierterer Reaktionsweg wird vonKikuchi et al. vorgeschlagen:
Die Ausgangsverbindungen NO, CH4 und O2 diffundieren in den Zeolithen. Hier erfolgt an
Ir-Spezies eine dissoziative Adsorption von Sauerstoff. NO wird zu NO2 oxidiert, gleichzei-
tig findet aber in einer Konkurrenzreaktion die Oxidation von Methan statt. NO2 wird an
[InO]+-Kationen chemisorbiert und reagiert mit CH4 zu einem Reaktionsintermediat.
Als Zwischenprodukte in der SCR mit Methan an diversen modifizierten Zeolithen werden
in der Literatur bisher immer ausgehend von der Bildung eines Methylradikals (nachgewie-

2 Stand des Wissens 11
sen durch einen kinetischen Isotopeneffekt und als geschwindigkeitsbestimmender Schritt pos-
tuliert) [56,57] und dessen weiteren Reaktionen organische Produkte wie R-NO2 [33,58,59],
R-CN [60,61], R-NCO [62,63] oder am Ende der Reaktionskette auch NH3 [64] vorgeschlagen,
wobei deren Existenz meistens durchin-situ IR-Spektroskopie oder Einsatz der Intermediate
(z.B. Nitromethan) als Edukte begr¨undet wird [65].Uber die genauen Reaktionswege besteht
noch Unklarheit, lediglich die NO-Oxidation als Startreaktion und die M¨oglichkeit, dass meh-
rere Folgereaktionen gleichzeitig aber in unterschiedlicher Gewichtung auftreten, ist akzeptiert.
Das bei der Reaktion von an [InO]+-Kationen chemisorbiertem NO mit Methan gebildete
Zwischenprodukt muss lautKikuchi nochmals mit NO reagieren, um N2 zu bilden. Allerdings
ist die Bildung von NO2 nicht die einzige Funktion der Ir-Spezies. Sie erh¨ohen gleichzei-
tig die Menge des an den [InO]+-Kationen chemisorbierten NO2. Da NO nach PSA-Studien
(PSA = Pressure-Swing-Adsorption) schneller in den Katalysator diffundieren kann als NO2,
ist eine Oxidation von NO in den Kan¨alen des Zeolithen g¨unstig.Kikuchi et al. nannten den
ablaufenden Katalysevorgang deshalb”intrapore catalysis“.
2.2.4 Formalkinetiken der SCR mit Methan
Bis jetzt sind die fur diese Arbeit wichtigen Katalysatorsysteme CeO2-ZSM-5, In-ZSM-5,
CeO2-In-ZSM-5 und Ir-In-ZSM-5 hinsichtlich ihrer geschichtlichen Entwicklung und den an
ihnen gewonnenen Erkentnissen ¨uber mogliche Reaktionsmechanismen vorgestellt worden. In
diesem Abschnitt sollen nun die f¨ur die SCR mit Methan in der Literatur zug¨anglichen Formal-
kinetiken aufgef¨uhrt und ihre Relevanz hinsichtlich der postulierten Reaktionsmechanismen
erlautert werden.
Mit Hilfe einer aufgestellten Formalkinetik l¨asst sich kein Reaktionsmechanismus beweisen,
sie kann lediglich unterst¨utzenden Charakter f¨ur einen schon durch andere Untersuchungen
postulierten Reaktionsmechanismus annehmen. In anderen Worten ausgedr¨uckt bedeutet dies,
dass eineUbereinstimmung zwischen Reaktionsmechanismus und Formalkinetik positiv, da-
hingegen aber eine Nicht¨ubereinstimmung nicht negativ zu werten ist. In den Tabellen 2.1,
2.2 und 2.3 sind nun alle bisher f¨ur die SCR mit Methan aufgestellten Formalkinetiken darge-
stellt.
Wie der Spalte”Kinetischer Ansatz“zu entnehmen ist, werden lediglich einfache Po-
tenzans¨atze verwendet. Dies liegt daran, dass in allen Literaturstellen außer bei [69] lediglich
2 Stand des Wissens 12
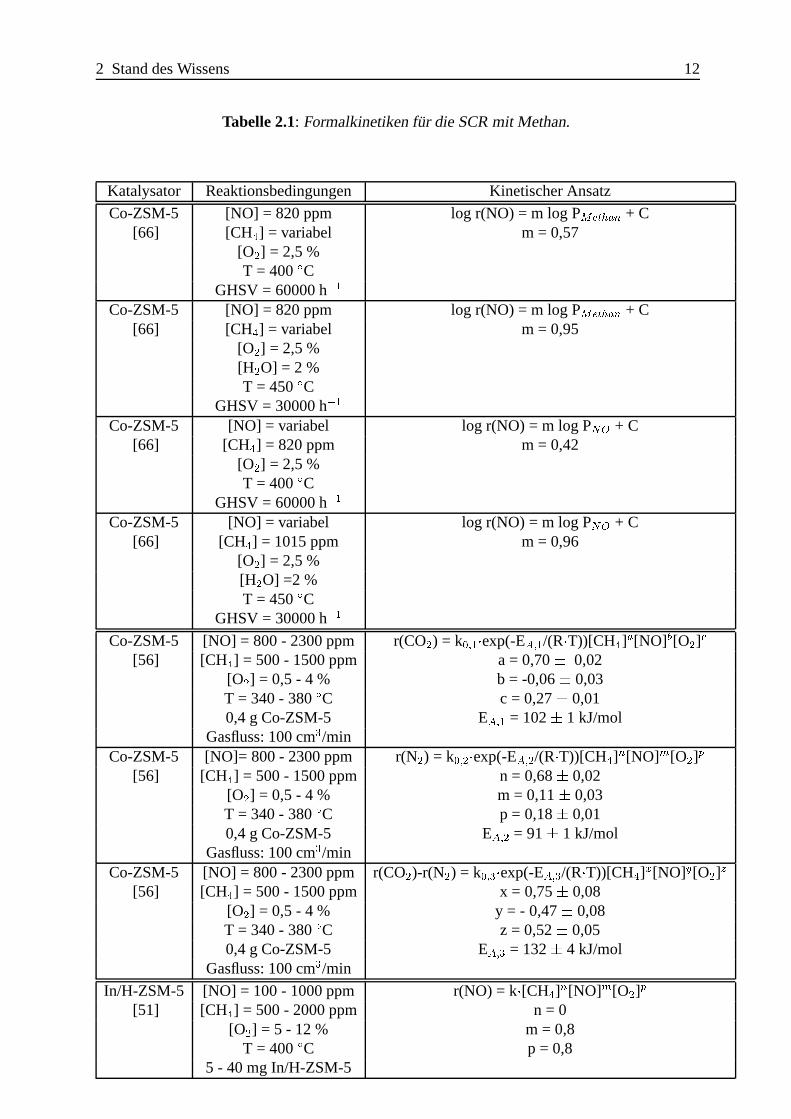
Tabelle 2.1: Formalkinetiken f¨ur die SCR mit Methan.
Katalysator Reaktionsbedingungen Kinetischer Ansatz
Co-ZSM-5 [NO] = 820 ppm log r(NO) = m log PMethan + C[66] [CH4] = variabel m = 0,57
[O2] = 2,5 %T = 400ÆC
GHSV = 60000 h�1
Co-ZSM-5 [NO] = 820 ppm log r(NO) = m log PMethan + C[66] [CH4] = variabel m = 0,95
[O2] = 2,5 %[H2O] = 2 %T = 450ÆC
GHSV = 30000 h�1
Co-ZSM-5 [NO] = variabel log r(NO) = m log PNO + C[66] [CH4] = 820 ppm m = 0,42
[O2] = 2,5 %T = 400ÆC
GHSV = 60000 h�1
Co-ZSM-5 [NO] = variabel log r(NO) = m log PNO + C[66] [CH4] = 1015 ppm m = 0,96
[O2] = 2,5 %[H2O] =2 %T = 450ÆC
GHSV = 30000 h�1
Co-ZSM-5 [NO] = 800 - 2300 ppm r(CO2) = k0;1�exp(-EA;1/(R�T))[CH4]a[NO]b[O2]c
[56] [CH4] = 500 - 1500 ppm a = 0,70� 0,02[O2] = 0,5 - 4 % b = -0,06� 0,03T = 340 - 380ÆC c = 0,27� 0,010,4 g Co-ZSM-5 EA;1 = 102� 1 kJ/mol
Gasfluss: 100 cm3/minCo-ZSM-5 [NO]= 800 - 2300 ppm r(N2) = k0;2�exp(-EA;2/(R�T))[CH4]n[NO]m[O2]p
[56] [CH4] = 500 - 1500 ppm n = 0,68� 0,02[O2] = 0,5 - 4 % m = 0,11� 0,03T = 340 - 380ÆC p = 0,18� 0,010,4 g Co-ZSM-5 EA;2 = 91� 1 kJ/mol
Gasfluss: 100 cm3/minCo-ZSM-5 [NO] = 800 - 2300 ppm r(CO2)-r(N2) = k0;3�exp(-EA;3/(R�T))[CH4]x[NO]y[O2]z
[56] [CH4] = 500 - 1500 ppm x = 0,75� 0,08[O2] = 0,5 - 4 % y = - 0,47� 0,08T = 340 - 380ÆC z = 0,52� 0,050,4 g Co-ZSM-5 EA;3 = 132� 4 kJ/mol
Gasfluss: 100 cm3/min
In/H-ZSM-5 [NO] = 100 - 1000 ppm r(NO) = k�[CH4]n[NO]m[O2]p
[51] [CH4] = 500 - 2000 ppm n = 0[O2] = 5 - 12 % m = 0,8
T = 400ÆC p = 0,85 - 40 mg In/H-ZSM-5
2 Stand des Wissens 13
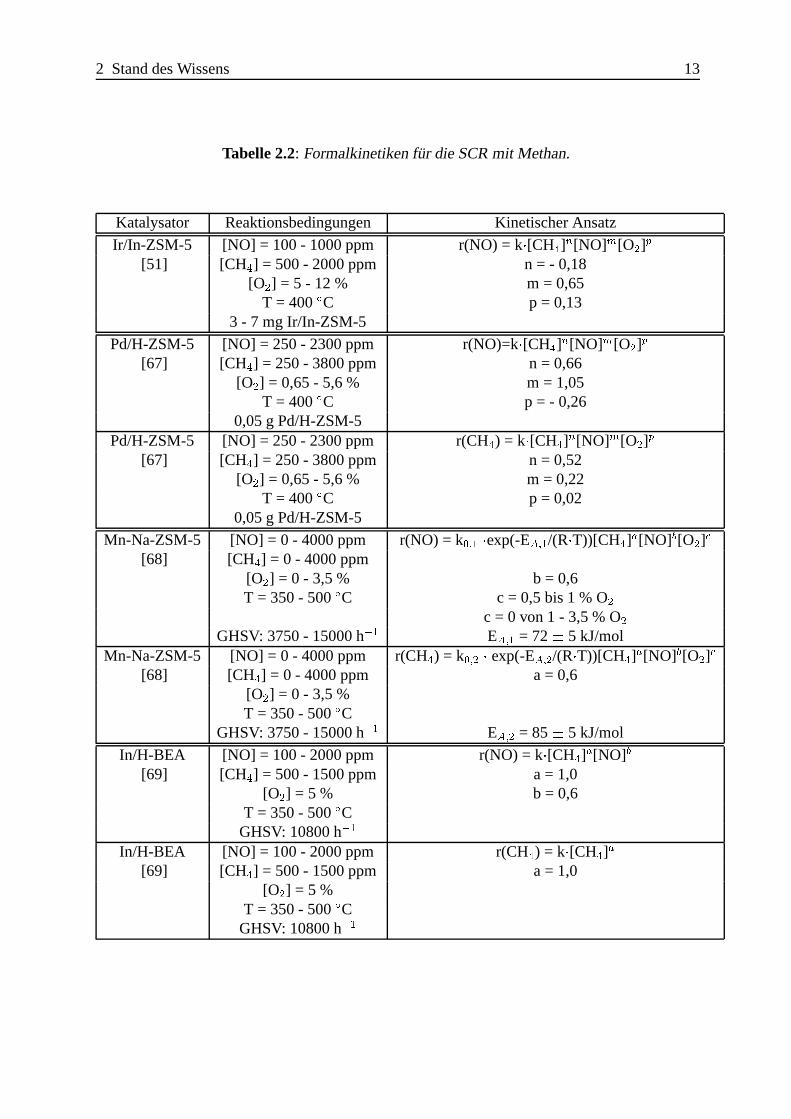
Tabelle 2.2: Formalkinetiken f¨ur die SCR mit Methan.
Katalysator Reaktionsbedingungen Kinetischer Ansatz
Ir/In-ZSM-5 [NO] = 100 - 1000 ppm r(NO) = k�[CH4]n[NO]m[O2]p
[51] [CH4] = 500 - 2000 ppm n = - 0,18[O2] = 5 - 12 % m = 0,65
T = 400ÆC p = 0,133 - 7 mg Ir/In-ZSM-5
Pd/H-ZSM-5 [NO] = 250 - 2300 ppm r(NO)=k�[CH4]n[NO]m[O2]p
[67] [CH4] = 250 - 3800 ppm n = 0,66[O2] = 0,65 - 5,6 % m = 1,05
T = 400ÆC p = - 0,260,05 g Pd/H-ZSM-5
Pd/H-ZSM-5 [NO] = 250 - 2300 ppm r(CH4) = k�[CH4]n[NO]m[O2]p
[67] [CH4] = 250 - 3800 ppm n = 0,52[O2] = 0,65 - 5,6 % m = 0,22
T = 400ÆC p = 0,020,05 g Pd/H-ZSM-5
Mn-Na-ZSM-5 [NO] = 0 - 4000 ppm r(NO) = k0;1 �exp(-EA;1/(R�T))[CH4]a[NO]b[O2]c
[68] [CH4] = 0 - 4000 ppm[O2] = 0 - 3,5 % b = 0,6T = 350 - 500ÆC c = 0,5 bis 1 % O2
c = 0 von 1 - 3,5 % O2GHSV: 3750 - 15000 h�1 EA;1 = 72� 5 kJ/mol
Mn-Na-ZSM-5 [NO] = 0 - 4000 ppm r(CH4) = k0;2 � exp(-EA;2/(R�T))[CH4]a[NO]b[O2]c
[68] [CH4] = 0 - 4000 ppm a = 0,6[O2] = 0 - 3,5 %T = 350 - 500ÆC
GHSV: 3750 - 15000 h�1 EA;2 = 85� 5 kJ/mol
In/H-BEA [NO] = 100 - 2000 ppm r(NO) = k�[CH4]a[NO]b
[69] [CH4] = 500 - 1500 ppm a = 1,0[O2] = 5 % b = 0,6
T = 350 - 500ÆCGHSV: 10800 h�1
In/H-BEA [NO] = 100 - 2000 ppm r(CH4) = k�[CH4]a
[69] [CH4] = 500 - 1500 ppm a = 1,0[O2] = 5 %
T = 350 - 500ÆCGHSV: 10800 h�1
2 Stand des Wissens 14
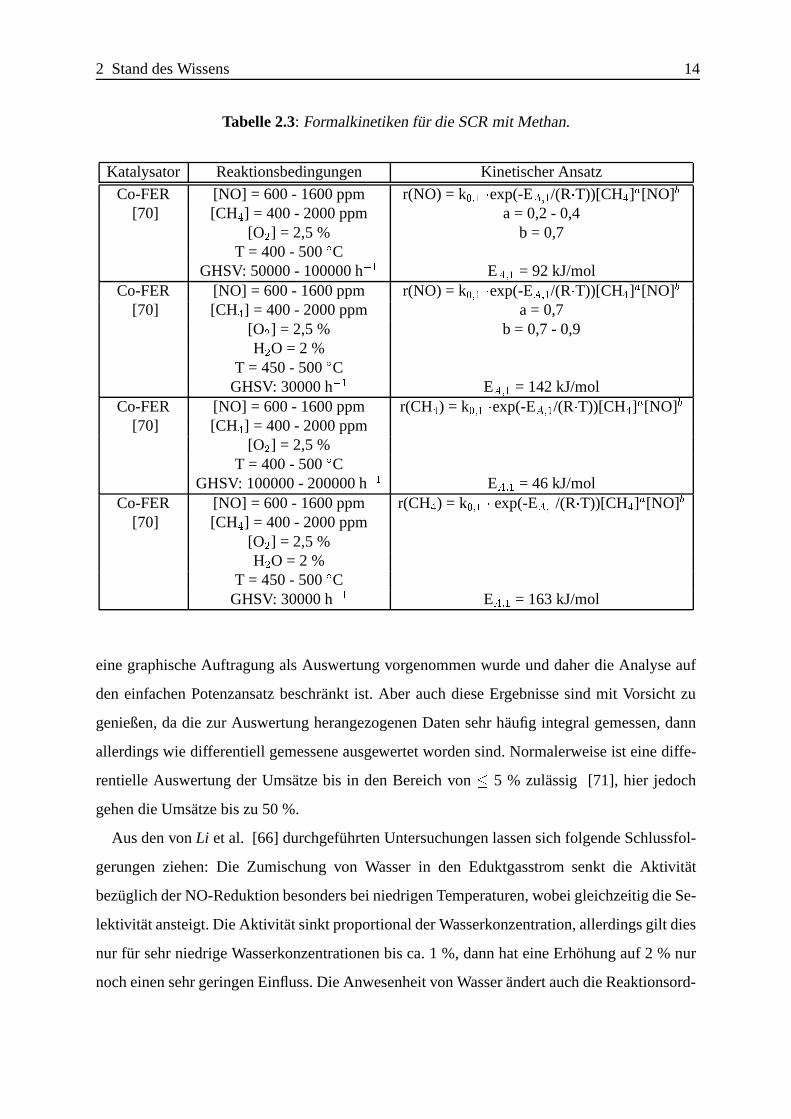
Tabelle 2.3: Formalkinetiken f¨ur die SCR mit Methan.
Katalysator Reaktionsbedingungen Kinetischer Ansatz
Co-FER [NO] = 600 - 1600 ppm r(NO) = k0;1 �exp(-EA;1/(R�T))[CH4]a[NO]b
[70] [CH4] = 400 - 2000 ppm a = 0,2 - 0,4[O2] = 2,5 % b = 0,7
T = 400 - 500ÆCGHSV: 50000 - 100000 h�1 EA;1 = 92 kJ/mol
Co-FER [NO] = 600 - 1600 ppm r(NO) = k0;1 �exp(-EA;1/(R�T))[CH4]a[NO]b
[70] [CH4] = 400 - 2000 ppm a = 0,7[O2] = 2,5 % b = 0,7 - 0,9H2O = 2 %
T = 450 - 500ÆCGHSV: 30000 h�1 EA;1 = 142 kJ/mol
Co-FER [NO] = 600 - 1600 ppm r(CH4) = k0;1 �exp(-EA;1/(R�T))[CH4]a[NO]b
[70] [CH4] = 400 - 2000 ppm[O2] = 2,5 %
T = 400 - 500ÆCGHSV: 100000 - 200000 h�1 EA;1 = 46 kJ/mol
Co-FER [NO] = 600 - 1600 ppm r(CH4) = k0;1 � exp(-EA;1/(R�T))[CH4]a[NO]b
[70] [CH4] = 400 - 2000 ppm[O2] = 2,5 %H2O = 2 %
T = 450 - 500ÆCGHSV: 30000 h�1 EA;1 = 163 kJ/mol
eine graphische Auftragung als Auswertung vorgenommen wurde und daher die Analyse auf
den einfachen Potenzansatz beschr¨ankt ist. Aber auch diese Ergebnisse sind mit Vorsicht zu
genießen, da die zur Auswertung herangezogenen Daten sehr h¨aufig integral gemessen, dann
allerdings wie differentiell gemessene ausgewertet worden sind. Normalerweise ist eine diffe-
rentielle Auswertung der Ums¨atze bis in den Bereich von� 5 % zulassig [71], hier jedoch
gehen die Ums¨atze bis zu 50 %.
Aus den vonLi et al. [66] durchgef¨uhrten Untersuchungen lassen sich folgende Schlussfol-
gerungen ziehen: Die Zumischung von Wasser in den Eduktgasstrom senkt die Aktivit¨at
bezuglich der NO-Reduktion besonders bei niedrigen Temperaturen, wobei gleichzeitig die Se-
lektivitat ansteigt. Die Aktivit¨at sinkt proportional der Wasserkonzentration, allerdings gilt dies
nur fur sehr niedrige Wasserkonzentrationen bis ca. 1 %, dann hat eine Erh¨ohung auf 2 % nur
noch einen sehr geringen Einfluss. Die Anwesenheit von Wasser ¨andert auch die Reaktionsord-

2 Stand des Wissens 15
nungen fur die NO-Reduktion. Die NO-Ordnung steigt von 0,42 auf 0,96 und die CH4-Ordnung
von 0,57 auf 0,95. Als Erkl¨arung dient die Annahme, dass Wasser mit NO um Adsorptions-
platze konkurriert. Unterst¨utzt wird die These durch TPD-Experimente an unterschiedlich stark
getrockneten Katalysatoren. Die trockenen Proben k¨onnen eine sehr viel gr¨oßere Menge NO
adsorbieren als die feuchten Proben.
Auch vonCowanet al. ist der vielversprechende Co-ZSM-5 weiter studiert worden [56].
Sie entdeckten bei Einsatz von CH4 im Vergleich mit CD4 einen kinetischen Isotopeneffekt
sowohl mit als auch ohne Wasser im Eduktgas und folgerten als geschwindigkeitsbestimmen-
den Schritt eine Wasserstoffabstraktion von CH4 bzw. den Bruch einer C-H-Bindung. Da die
kinetischen Ordnungen f¨ur Methan fur die selektive Reduktion (0,68) und die nichtselektive
Reaktion (0,75) nahezu gleich sind, kann ein gemeinsames Reaktionsintermediat angenommen
werden. Weil in Mischungen von CD4 und CH4 kein Isotopenaustausch stattfindet, schließen
die Autoren einen”dissociative preequilibrium step“aus. Auch hier werden die geringen Re-
aktionsordnungen f¨ur NO (-0,06 und 0,11) als Folge einer Adsorption von NO oder NO2
im ersten Reaktionsschritt interpretiert. Der Vorschlag f¨ur einen Reaktionsmechanismus ist
in Abb. 2.5 gegeben: NO2 wird an der Oberfl¨ache adsorbiert und an dieser Oberfl¨achenspe-
zies S erfolgt dann der geschwindigkeitsbestimmende Schritt der CH4-Aktivierung bzw. der
Bruch der Kohlenstoff-Wasserstoff-Bindung, wodurch ein Intermediat X, m¨oglicherweise ein
Methyl-Radikal, gebildet wird. Dieses Intermediat kann nun auf zwei konkurrierenden Reak-
tionspfaden weiter umgesetzt werden, einmal selektiv mit NO oder NO2 zu N2, Wasser und
Kohlendioxid, und zum anderen unselektiv mit Sauerstoff zu Kohlendioxid und Wasser (Total-
oxidation).
Kato et al. haben nun die Messungen vonCowanet al. am Co-ZSM-5 mit ihren eigenen
Slangsam
+CH4 ,-HX
+ NOoder
NO2
NO2-
Adsorption
+ O2
COx
N2+ COx
Abbildung 2.5: Reaktionsmechanismus f¨ur das Co-ZSM-5-System [56]

2 Stand des Wissens 16
Messungen an einem Pd/H-ZSM-5 verglichen. Auch f¨ur den Pd/H-ZSM-5 stellten sie einen
kinetischen Isotopeneffekt fest, allerdings in geringerem Ausmaß und mit sinkender Tempera-
tur von sinkender Auspr¨agung. Gleichzeitig wurde ein H-D Isotopenaustausch bei Verwendung
einer CD4+CH4-Mischung als Reduktionsmittel beobachtet, was zu der Annahme f¨uhrt, dass
die C-H-Dissoziation (i) zwar langsam, aber nicht der einzige geschwindigkeitsbestimmende
Schritt ist. Durch die Beobachtung weiterer Isotopeneffekte kommen sie zu dem Schluss, dass
die Oxidation von NO zu NO2 (ii) ein weiterer relativ langsamer Schritt ist, und bei niedri-
gen Temperaturen wird Schritt (iii), die Reaktion von NO2 mit CHx, langsamer als (i) und (ii).
Der zugeh¨orige Reaktionsmechanismus ist in Abb. 2.6 dargestellt. NO wird an Protonen des
NO NO2H+, Pd
CH4 CHxPd
N2+ COx
H+, (Pd)
(ii)
(i)
(iii)
Abbildung 2.6: Reaktionsmechanismus f¨ur das Pd/H-ZSM-5-System [67]
Zeolithen und/oder Pd zu NO2 oxidiert, gleichzeitig wird CH4 an Pd aktiviert zu CHx. Dann
reagieren NO2 und CHx weiter an Protonen, wobei zum weiteren Reaktionsablauf keine Vor-
schlage gemacht werden. Die hohe Reaktionsordnung f¨ur NO in der Reduktionsreaktion von
1,05 spricht auch f¨ur eine langsame NO-Oxidation und gegen eine Adsorption von NO2 wie
beim Co-ZSM-5. Beim Vergleich von NO-TPDs von Co-ZSM-5 und dem Pd/H-ZSM-5 zeigt
sich, dass an dem Co-ZSM-5 die 8-10fache Menge NO adsorbiert wird.
Zwei weitere Systeme sind vonKikuchi et al. untersucht und untereinander verglichen wor-
den: ein In/H-ZSM-5 und ein Ir/In-ZSM-5 [51]. Auff¨allig ist hier, dass der Zusatz von Ir zu
In/H-ZSM-5 ein Absinken der Reaktionsordnungen f¨ur die NO-CH4-O2-Reaktion fur alle drei
Komponenten NO (von 0,8 auf 0,65), CH4 (von 0,0 auf -0,18) und O2 (von 0,8 auf 0,13) be-
wirkt. Dadurch ist auch erkl¨arbar, warum gerade Ir/In-ZSM-5 NO noch bei sehr geringen Kon-
zentrationen reduzieren kann. Die negative Reaktionsordnung im Hinblick auf CH4 lasst eine
Konkurrenzoxidationsreaktion von NO und CH4 an Ir vermuten, wobei am Ir wahrscheinlich
der Sauerstoff dissoziativ adsorbiert wird, was die Absenkung der Reaktionsordnung f¨ur O2 bei
Zugabe von Ir zum In/H-ZSM-5 erkl¨art. Wie schon auf Seite 9 erw¨ahnt, steigert das Ir die Rate
der NO-Oxidation und die NO2-Adsorptionskapazit¨at von In/H-ZSM-5, wobei Ir selbst nahezu

2 Stand des Wissens 17
keine Adsorptionskapazit¨at aufweist. Die niedrigere NO-Reaktionsordnung von Ir-In-ZSM-5
im Vergleich zu In/H-ZSM-5 unterst¨utzt die These, dass die NO-Oxidation beschleunigt wird
und CH4 durch an Indiumoxokationen adsorbiertes NO2 aktiviert wird. Ein Vorschlag f¨ur die
Abfolge der einzelnen Reaktionen ist schon in Abb. 2.4 gezeigt worden.
Das System Mn-Na-ZSM-5 ist vonCampaet al. untersucht worden, allerdings legten sie den
Hauptgesichtspunkt auf die Variation des Mn-Gehaltes und aufin situIR-Messungen. Eine For-
malkinetik ergibt sich hier nur als”Nebenprodukt“und wird auch nicht weiter diskutiert [68].
Interessant ist die Verwendung von Na-ZSM-5 als Ausgangszeolith. Normalerweise wird die
H- oder die NH4-Form bevorzugt, da sp¨ater die Protonen des Zeolithen im Reaktionsmechanis-
mus eine Rolle spielen. Allerdings erfolgt die Beladung mit Mangan (2-75 % Austauschgrad)
durch Manganacetat bei einem pH� 5,5, so dass hier ein vorgelagerter Ionentausch von Na+
gegen H+ nicht ausgeschlossen werden kann, was aber von den Autoren nicht weiter disku-
tiert wird. Die ermittelten Aktivierungsenergien, Reaktionsordnungen und Selektivit¨aten sind
unabhangig vom Mangangehalt, lediglich die Aktivit¨at steigt mit dem Mangangehalt. Durch
ESR wird die aktive und auch einzig vorhandene Spezies als Mn2+ identifiziert, so dass die
Steigerung der Aktivit¨at auf eine h¨ohere Anzahl aktiver Zentren zur¨uckgefuhrt wird. An die-
sen aktiven Zentren wird eine Monodentatnitrat-Spezies gebildet, die eine hohe Reaktivit¨at
gegen¨uber Methan besitzt (durchin situFTIR beobachtet). Auch die geringe NO-Ordnung von
0,6 spricht fur eine adsorbierte stickstoffhaltige Spezies.
Die einzige Formalkinetik, die nicht auf Basis einer graphischen Auswertung gewonnen wur-
de, stammt vonHeinischet al. [69]. Bei dem vermessenen Katalysator handelt es sich um einen
In/H-BEA. Hier werden die formalkinetischen Ans¨atze durch eine Modellierung des Reaktors
gewonnen, wobei die berechneten Werte iterativ an integral gemessene Ums¨atze von NO und
CH4 angepasst werden. Als Simulationsgrundlage dient ein heterogenes Modell mit einer Ka-
talysatorphase und einer Gasphase, wobei der Massentransfer zwischen diesen beiden Phasen
mit Hilfe eines Massentransferkoeffizienten nach [71] berechnet wird. Im weiteren Verlauf
zeigt sich jedoch durch Test auf Filmdiffusionshemmung, dass auch ein homogenes Modell
zur Beschreibung der experimentellen Daten gereicht h¨atte, da keine Massentransportlimitie-
rung vorliegt. Die erhaltenen Reaktionsordnungen unterscheiden sich erheblich von denen in
[51] an einem In/H-ZSM-5 bestimmten. So erhalten die Autoren f¨ur die Reaktionsordnung von
NO statt 0,8 hier 0,6 in der NO-Reduktionsreaktion und f¨ur die CH4-Ordnung statt 0 den Wert

2 Stand des Wissens 18
1,0. Hier muss allerdings erw¨ahnt werden, dass es sich um Zeolithe unterschiedlicher Struktur
handelt, MFI und BEA.
AuchLi et al. fanden, dass sich f¨ur andere Zeolithe auf Grund der unterschiedlichen Topolo-
gien andere Reaktionsordnungen ergeben k¨onnen. So ergab eine Untersuchung von Co-ZSM-5
fur die NO-Reduktion eine NO-Ordnung von 0,42 ohne bzw. 0,96 mit 2 % Wasser und f¨ur
die CH4-Ordnung 0,57 ohne bzw. 0,95 mit Wasser [66]. Im Gegensatz hierzu ergibt sich bei
Verwendung eines Ferrieriten als Zeolith f¨ur die NO-Reduktionsreaktion f¨ur die NO-Ordnung
ein Wert von 0,2 - 0,4 ohne bzw. 0,7 mit Wasser und f¨ur die CH4-Ordnung Werte von 0,7 ohne
bzw. 0,7 - 0,9 mit Wasser [70]. Auch hier gibt es nach Ansicht der Autoren eine Konkurrenzad-
sorption zwischen Wasser, CH4 und NO an Co-Spezies, was die unter Wasser h¨oheren Re-
aktionsordnungen und Aktivierungsenergien plausibel erscheinen l¨asst. Die unterschiedlichen
Reaktionsordnungen bei Verwendung von ZSM-5 oder Ferrierit erkl¨aren sie mit unterschiedli-
chen Adsorptionsgleichgewichten an den einzelnen Zeolithen, die grundlegenden Prinzipien im
Reaktionsmechanismus sind jedoch gleich und schon in Abb. 2.5 dargestellt. Eine bisher noch
nicht vorgestellte Beobachtung ist die Tatsache, dass sich die einzelnen Reaktionsordnungen
uber die Temperaturen hinweg ver¨andern und so eine Verschiebung der Gewichtung einzelner
Reaktionsschritte vermuten lassen. So wird der Anstieg der NO-Ordnung von 0,19 bei 425ÆC
auf 0,38 bei 500ÆC mit einer thermodynamisch begr¨undeten geringeren NO2-Konzentration,
die zur Adsorption bereitsteht, erkl¨art. Im feuchten Reaktionsgas sinkt die CH4-Ordnung von
0,92 bei 450ÆC auf 0,75 bei 500ÆC, weil die Konkurrenzadsorption von Wasser bei h¨oheren
Temperaturen unterdr¨uckt wird.
Zusammenfassend l¨aßt sich sagen, dass mit der Ermittlung einer Formalkinetik dem
Beobachter ein weiteres Werkzeug zur Aufkl¨arung des Reaktionsmechanismus in die Hand
gelegt wird, dessen Nutzen nicht untersch¨atzt werden darf. Allerdings ist eine relativ große
Datenmenge n¨otig, um zu sinnvollen Ergebnissen gelangen zu k¨onnen. In den hier zitierten
Artikeln sind meistens nur Teilbereiche abgedeckt. So fehlen beispielsweise manchmal Akti-
vierungsenergien und/oder Reaktionsordnungen f¨ur die unselektive Totaloxidationsreaktion.
Deshalb ist es nun ein Ziel dieser Arbeit, f¨ur die beiden Grundysteme In-ZSM-5 und
CeO2-ZSM-5 und danach f¨ur das promotierte System CeO2-In-ZSM-5 eine moglichst breite
Datenbasis zu schaffen und diese dann mittels Computersimulationen auszuwerten, um auch
integrale Messungen ber¨ucksichtigen zu k¨onnen.

3 Untersuchungsmethodik
Bevor auf die Ergebnisse eingegangen wird, sollen in diesem Kapitel die zum weiteren
Verstandnis notwendigen Grundlagen erl¨autert werden. Es erfolgt eine Grobeinteilung in einen
experimentellen Teil und in einen die Modellierung katalytischer Ergebnisse beschreibenden
Teil. Im experimentellen Teil werden die Versuchsapparatur und die mit ihrer Hilfe durch-
gefuhrten Standardexperimente beschrieben. Einen weiteren Bestandteil stellen die Katalysa-
torpraparation und die m¨oglichen Katalysatorcharakterisierungsmethoden dar. Im Weiteren soll
dann die Vorgehensweise bei der Modellierung behandelt werden.
3.1 Experimentelles
3.1.1 Versuchsapparatur
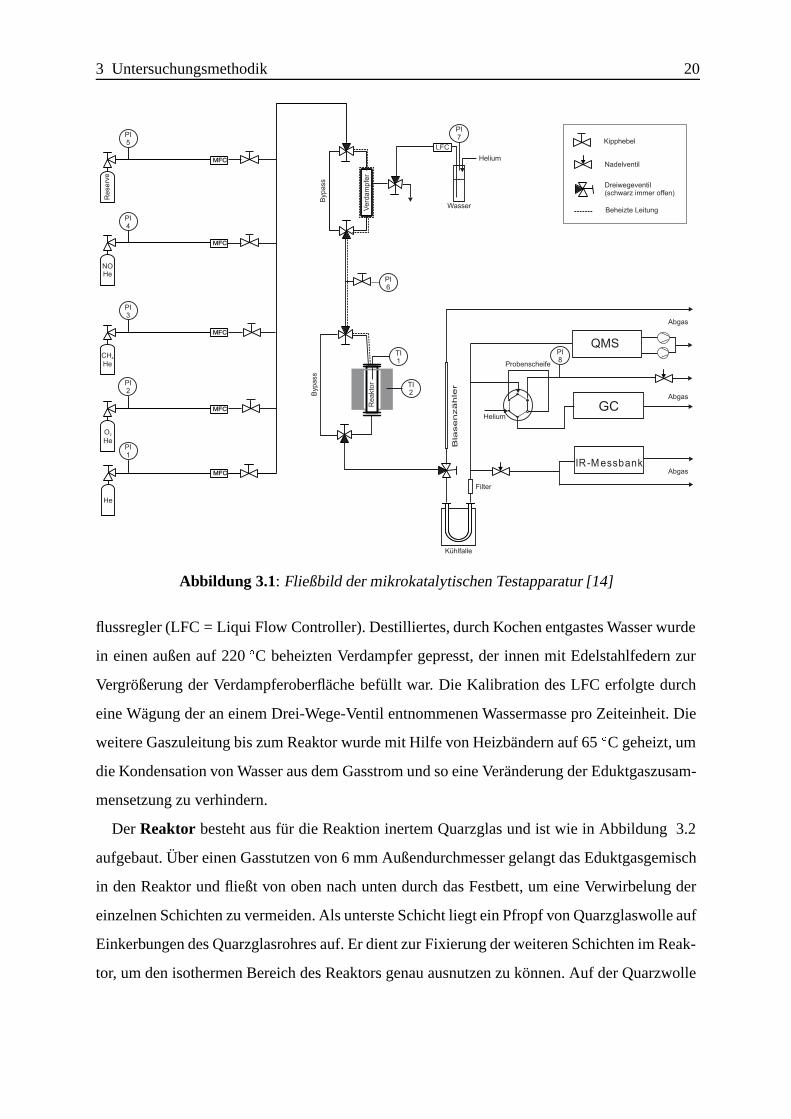
Die fur die station¨aren Experimente zur Verf¨ugung stehende Versuchsapparatur ist in Abbil-
dung 3.1 dargestellt. Es handelt sich um eine modifizierte Version der Anlage, die schon von
Liese[14] verwendet wurde; sie besteht aus Gasversorgung, Reaktor und Analytik.
Die Gasversorgungerfolgt durch Entnahme von kommerziell erh¨altlichen Gasmischungen
aus Druckgasflaschen ¨uber Druckminderer, das Helium zur Versorgung des Gaschromatogra-
phen (siehe Analytik) wird der zentralen Gasversorgung des Lehrstuhls entnommen. Die Gas-
mischungen werden ¨uber Massenflussregler (MFC = Mass Flow Controller) dosiert und mit
Helium verdunnt, um die fur die Messungen gew¨unschten Eduktgaszusammensetzungen zu
erhalten. Hierbei wird erst reines Helium zugegeben und danach Sauerstoff dosiert, darauf-
hin wird Methan als Reduktionsmittel zugegeben, und erst als letzter Schritt erfolgt die Bei-
mischung von NO. Dies soll gew¨ahrleisten, dass keine lokal hohen Sauerstoffkonzentrationen
mit zugegebenem NO vor dem Katalysator zu NO2 reagieren. EineUberprufung ergab, dass
ohne Katalysator selbst bei f¨ur die Oxidation g¨unstigen Bedingungen (z.B. hohe Sauerstoff-
konzentration, hohe Verweilzeit) keine Bildung von NO2 am Gasausgang beobachtet werden
konnte. Hinter den Massenflussreglern befinden sich Kipphebel zur vollst¨andigen Absperrung
der Zudosierung.
Neben der Gasdosierung erfolgte zus¨atzlich eine Fl¨ussigdosierung ¨uber einen Massen-
3 Untersuchungsmethodik 20
MFC
MFC
MFC
MFC
MFC
LFC
Ve
rda
mp
fer
Ve
rda
mp
fer
Ve
rda
mp
fer
Ve
rda
mp
fer
Bla
senzähle
r
Kühlfalle
IR-MessbankAbgas
GCAbgas
Abgas
QMS
Probenscheife
Helium
Reserv
e
Kipphebel
Nadelventil
Dreiwegeventil(schwarz immer offen)
Helium
Beheizte Leitung
Re
akto
r
Byp
ass
Byp
ass
Wasser
Filter
TI1
TI2
PI6
PI7
PI8
PI5
PI4
PI3
PI2
PI1
CH
He4
O
He2
He
NOHe
Abbildung 3.1: Fließbild der mikrokatalytischen Testapparatur [14]
flussregler (LFC = Liqui Flow Controller). Destilliertes, durch Kochen entgastes Wasser wurde
in einen außen auf 220ÆC beheizten Verdampfer gepresst, der innen mit Edelstahlfedern zur
Vergroßerung der Verdampferoberfl¨ache bef¨ullt war. Die Kalibration des LFC erfolgte durch
eine Wagung der an einem Drei-Wege-Ventil entnommenen Wassermasse pro Zeiteinheit. Die
weitere Gaszuleitung bis zum Reaktor wurde mit Hilfe von Heizb¨andern auf 65ÆC geheizt, um
die Kondensation von Wasser aus dem Gasstrom und so eine Ver¨anderung der Eduktgaszusam-
mensetzung zu verhindern.
Der Reaktor besteht aus f¨ur die Reaktion inertem Quarzglas und ist wie in Abbildung 3.2
aufgebaut.Uber einen Gasstutzen von 6 mm Außendurchmesser gelangt das Eduktgasgemisch
in den Reaktor und fließt von oben nach unten durch das Festbett, um eine Verwirbelung der
einzelnen Schichten zu vermeiden. Als unterste Schicht liegt ein Pfropf von Quarzglaswolle auf
Einkerbungen des Quarzglasrohres auf. Er dient zur Fixierung der weiteren Schichten im Reak-
tor, um den isothermen Bereich des Reaktors genau ausnutzen zu k¨onnen. Auf der Quarzwolle
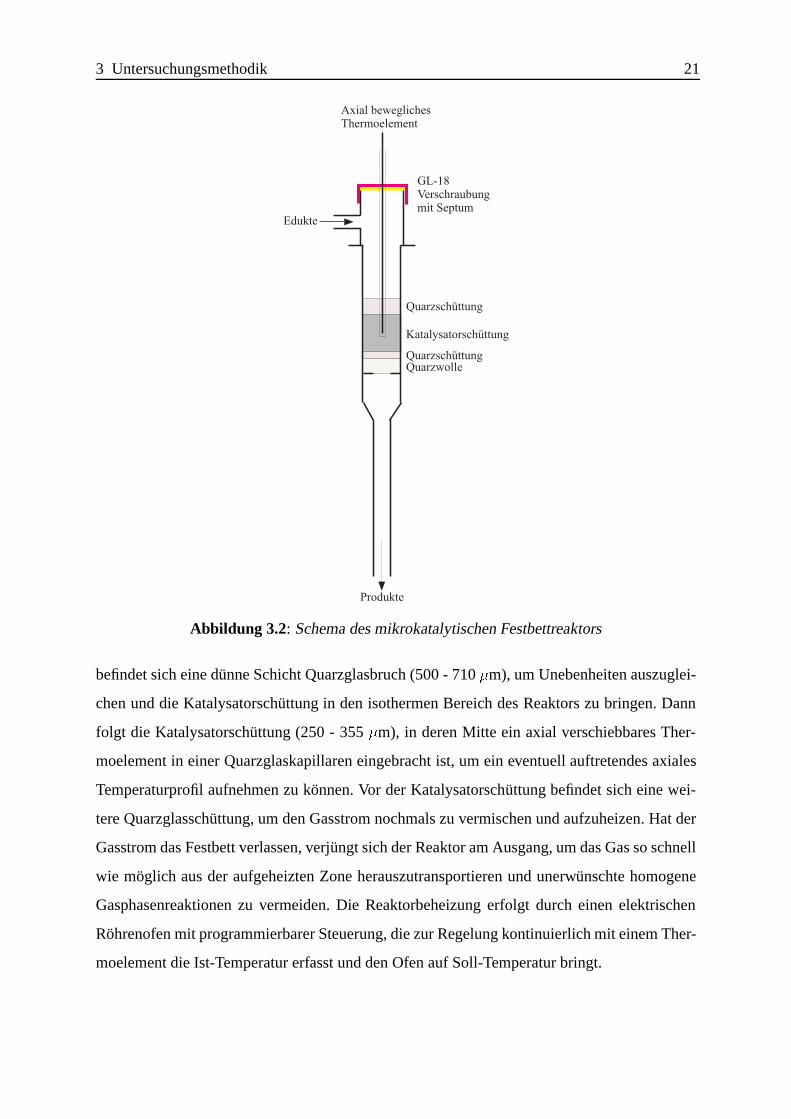
3 Untersuchungsmethodik 21
Edukte
Produkte
Axial beweglichesThermoelement
GL-18Verschraubungmit Septum
Quarzschüttung
Katalysatorschüttung
QuarzschüttungQuarzwolle
Abbildung 3.2: Schema des mikrokatalytischen Festbettreaktors
befindet sich eine d¨unne Schicht Quarzglasbruch (500 - 710�m), um Unebenheiten auszuglei-
chen und die Katalysatorsch¨uttung in den isothermen Bereich des Reaktors zu bringen. Dann
folgt die Katalysatorsch¨uttung (250 - 355�m), in deren Mitte ein axial verschiebbares Ther-
moelement in einer Quarzglaskapillaren eingebracht ist, um ein eventuell auftretendes axiales
Temperaturprofil aufnehmen zu k¨onnen. Vor der Katalysatorsch¨uttung befindet sich eine wei-
tere Quarzglassch¨uttung, um den Gasstrom nochmals zu vermischen und aufzuheizen. Hat der
Gasstrom das Festbett verlassen, verj¨ungt sich der Reaktor am Ausgang, um das Gas so schnell
wie moglich aus der aufgeheizten Zone herauszutransportieren und unerw¨unschte homogene
Gasphasenreaktionen zu vermeiden. Die Reaktorbeheizung erfolgt durch einen elektrischen
Rohrenofen mit programmierbarer Steuerung, die zur Regelung kontinuierlich mit einem Ther-
moelement die Ist-Temperatur erfasst und den Ofen auf Soll-Temperatur bringt.

3 Untersuchungsmethodik 22
Die Analytik schließt sich als aufwendigster Teil an den Reaktor an. Der Gasstrom wird
zur Auskondensation von Wasserdampf durch eine sich in einem Eisbad befindliche K¨uhl-
falle geleitet. Dies ist notwendig, da durch Wasser einmal die Trenns¨aule des Gaschromato-
graphen (Molsieb 5A) in ihrer Adsorptionskapazit¨at beeinflusst wird, und zum anderen die
IR-Messbank eine Querempfindlichkeit auf Wasser aufweist und die Messk¨uvette zudem noch
verschmutzt werden kann. Nach der K¨uhlfalle wird der Gasstrom aufgesplittet.
Ein Teil gelangt in die IR-Messbank der Firma Bionics, die sich den ben¨otigten Gasstrom
mittels integrierter Pumpe selbst ansaugt und deren Innenraum konstant temperiert ist. Detek-
tierbar ist hier N2O (querempfindlich auf CH4), CO und CO2. Das Gas gelangt in eine von
einer IR-Quelle durchstrahlte K¨uvette und je nach vorhandener Konzentration im Gas ¨andert
sich die Intensit¨at der durchkommenden IR-Strahlung. Hinter der K¨uvette befindet sich ein py-
roelektrischer Detektor. Dieser ist in drei Zonen aufgeteilt f¨ur die drei Wellenlangen der zu
bestimmenden Komponenten und wandelt die durch die einfallende IR-Strahlung entstehende
Warme in elektrische Spannung um. Die Spannung ist ein Maß f¨ur die Intensitat der einfallen-
den Strahlung und somit auch der Konzentration der jeweiligen Komponente im Gasstrom. Die
Spannungswerte werden ¨uber eine serielle Schnittstelle auf einen PC ¨ubertragen und durch das
Programm”IR29“von Liese[14] ausgewertet. Durch eine vorher erfolgte 4-Punkt-Kalibration
werden direkt die Konzentrationswerte in ppm angegeben.
Mit Hilfe eines Gaschromatographen wurden die Konzentrationen der Komponenten O2, N2
und CH4 bestimmt. Die Betriebsbedingungen und Retentionszeiten der Komponenten lassen
sich Tabelle A.1 im Anhang entnehmen. Die Integration der erhaltenen Signale des Gaschroma-
tographen erfolgte durch das Programm”Steuerung“, mit dem auch die Steuerung der MFC’s
durchgefuhrt wurde.
Fur die Bestimmung von NO und Methan dient ein Massenspektrometer
(Balzers Quadstar 420 QMS). Die Auswertung der Massen m/e: 30 (NO+) und
m/e: 15 (NO++ und CH+3 ) liefert die Konzentration an NO und Methan. Zur Bestim-
mung von NO2 stand leider kein Ger¨at zur Verfugung, aber anhand von N-Massenbilanzen
kann in den meisten F¨allen eine Bildung verneint werden.

3 Untersuchungsmethodik 23
3.1.2 Durchfuhrung und Auswertung der katalytischen Messungen
Als Standardreaktionsbedingungen wurden die Konzentrationen von 1000 ppm Stickstoff-
monoxid, 1000 ppm Methan, 2 % Sauerstoff und Rest Helium bei einer Katalysatorbelastung
von 30000 h�1 gewahlt (die Katalysatorbelastung errechnet sich aus dem Quotienten des Gas-
volumenstroms zum Katalysatorsch¨uttvolumen). Die genannte Belastung wird erreicht durch
ein Katalysatorsch¨uttvolumen von 0,44 ml und einen Gasvolumenstrom von 220 mlSTPmin�1,
wobei auf Grund unterschiedlicher Dichten der Katalysatoren die Einwaagen zwischen
300 - 350 mg variierten.
Vor Beginn jeder Messung wurden Tagesdruck und Tagestemperatur notiert und sp¨ater bei
der Auswertung ber¨ucksichtigt. Der eingebaute Katalysator wurde mit Hilfe eines Tempera-
turprogramms im Heliumstrom (220 mlSTPmin�1) kalziniert: Zuerst erfolgt eine moderate
Erwarmung von 2 K min�1 auf 120ÆC mit einem isothermen Abschnitt von 15 Minuten, um
das Wasser aus dem Zeolithen zu entfernen, ohne dabei das Zeolithger¨ust durch Dealuminie-
rung zu zerst¨oren. Mit 5 K min�1 wird danach auf 600ÆC aufgeheizt. Diese Temperatur von
600ÆC wird sodann 1 Stunde gehalten. W¨ahrend dieser Zeit wird das entmischungsfrei arbei-
tende Gaseinlassventil des Massenspektrometers ge¨offnet und ein Rezipientendruck von ca.
3; 1 � 10�7 mbar eingeregelt. Mit dem Gaschromatographen wird ein Testlauf durchgef¨uhrt.
Zeigt das Chromatogramm kein Sauerstoffsignal, so ist die Katalyseapparatur leckfrei und die
Messungen k¨onnen beginnen. Der Vordruck vor der Katalysatorsch¨uttung wird gemessen und
dann bei zeitlich konstanten Werten des Massenspektrometers eine Untergrundmessung ¨uber
100 Messsignale aufgenommen, da die Intensit¨aten niemals Null erreichen und dieser Grund-
wert bei der Auswertung von den sp¨ater gemessenen Werten abgezogen wird. Im Anschluss
wird der Reaktor auf”Bypass“geschaltet und das Eduktgasgemisch eingestellt. Nach Errei-
chen zeitlich konstanter Werte des Massenspektrometers werden wieder 100 Meßwerte auf-
gezeichnet und gleichzeitig ein Gaschromatogramm gestartet, indem ein Probenschleifenvolu-
men 3 Minuten lang eingesp¨ult wird. Mit der IR-Messbank erfolgt nach einem Nullabgleich
mit infrarotinaktivem N2 eine Aufzeichnung der CO, CO2 und N2O-Konzentrationen (hierbei
werden 20 Messwerte alle drei Sekunden aufgezeichnet und gemittelt). Ist auch im Eduktgas-
strom laut Gaschromatogramm kein Stickstoff enthalten, wird der Reaktor”online“geschaltet.
Haben sich die gemessenen Signalintensit¨aten des Massenspektrometers stabilisiert, so werden

3 Untersuchungsmethodik 24
wieder 100 Messzyklen aufgenommen, ein Gaschromatogramm gestartet und die CO, CO2 und
N2O-Konzentrationen der IR-Messbank aufgezeichnet. Sind alle Messungen beendet, wird per
Regler die Temperatur um 50ÆC gesenkt und nach oben beschriebener Prozedur wieder eine
Edukt- und eine Produktanalyse durchgef¨uhrt. So werden ¨uber einen gr¨oßeren Temperaturbe-
reich ausgew¨ahlte Temperaturen angefahren und jeweils die station¨are katalytische Aktivit¨at
des Katalysators vermessen.
Die zur Auswertung der gemessenen Daten verwendeten Gleichungen werden im Folgen-
den vorgestellt. Da sich w¨ahrend der Reaktion aufgrund der hohen Verd¨unnung der Kompo-
nenten nur eine minimale Volumen¨anderung ergab, wurde eine diesbez¨ugliche Korrektur nicht
berucksichtigt. Die Stickstoffmonoxid- und Methankonzentration wurden ¨uber die Intensit¨aten
der Signale m/e = 30 (NO+) und m/e = 15 (NO++ und CH3 +) gemessen, wobei ca. 5 % des
NO als doppelt positiv geladenes Kation auf der Masse 15 erscheint und daher die Methanin-
tensitat neben der Nullpunktskorrektur zus¨atzlich um diesen Betrag korrigiert werden muss.
cNO =I30;Produkt � I30;NullI30;Edukt � I30;Null
cNO;Edukt (3.1)
cCH4 =(I15;Produkt � 0; 05(I30;Produkt � I30;Null))� I15;Null(I15;Edukt � 0; 05(I30;Edukt � I30;Null))� I15;Null
cCH4;Edukt (3.2)
Die Konzentrationen von N2 und O2 werden mit Hilfe des Gaschromatographen bestimmt.
Die integrierten Fl¨achen werden mit Hilfe einer durch Kalibration erhaltenen Geradenglei-
chung in die Konzentrationen umgerechnet, wobei gleichzeitig eine Korrektur auf Tagesdruck
und Tagestemperatur (beeinflussen die Stoffmenge in der Probenschleife) durchgef¨uhrt wird.
cx = mFx760Torr TTag
pTag 273K+ C (3.3)
Hierbei ist cx die Konzentration der jeweiligen Komponente, m die Steigung der Geradenglei-
chung, Fx die integrierte Fl¨ache der jeweiligen Komponente, TTag die Tagestemperatur, pTag
der Tagesdruck und C der Achsenabschnitt der durch Kalibration erhaltenen Geradengleichung.
Als letztes soll die Auswertung der Daten der IR-Messbank vorgestellt werden. Mit Hilfe der
nichtdispersiven IR-Messungen wurden die Konzentrationen an CO, CO2 und N2O ermittelt,
wobei die charakteristische Wellenl¨ange des N2O Querempfindlichkeit auf Kohlenwasserstoffe
(hier speziell Methan) und Wasser aufweist und dahingehend eine Korrektur erfahren muss. Die
Konzentrationen werden mittels Kalibration durch ein Polynom 4. Grades aus den gemessenen

3 Untersuchungsmethodik 25
Daten direkt berechnet.
cN2O = cN2Ogemessen � (cN2OMethanEdukt(1� XCH4))� cN2OWasser
(3.4)
Der Umsatz von Methan wird aus den Werten des Massenspektrometers wie folgt errechnet:
XCH4 =cCH4;ein � cCH4;aus
cCH4;ein; (3.5)
wobei XCH4 der Methanumsatz, cCH4;ein die Methaneingangskonzentration und cCH4;aus die
Methanausgangskonzentration darstellt. Diese Art der Berechnung wird f¨ur alle Komponenten
verwendet und ist hier zul¨assig, da nur eine vernachl¨assigbar geringe Volumen¨anderung statt-
findet. Normalerweise m¨usste statt mit Konzentrationen mit Molenstr¨omen gerechnet werden.
Da fur die Komponente NO2 keine Analytik zur Verfugung stand, wurde eine eventuelle
Bildung durch die Aufstellung einer Stickstoffbilanz ¨uberpruft. Wird zu wenig Stickstoff ge-
funden, so ist vermutlich NO2 gebildet worden, allerdings wurde dies unter Ber¨ucksichtigung
einer Fehlergrenze von� 5 % bei keinem der Experimente festgestellt. Auch f¨ur die aufgestell-
ten Kohlenstoffbilanzen liegen die gefundenen Werte bei 95 - 105 %.
3.1.3 Katalysatorpraparation
In diesem Kapitel wird auf die verschiedenen m¨oglichen Praparationsrouten der Katalysato-
ren eingegangen, wobei auch die durch verschiedene Vorgehensweisen beabsichtigten Loka-
lisierungen (intra- und/oder extrazeolithisch) der Komponenten vorgestellt werden. Als Aus-
gangsmaterial dienten zwei industriell gefertigte Zeolithe. Hierbei handelt es sich um einen
NH4-ZSM-5 mit der Bezeichnung SM27 der Firma AlsiPenta mit einem Si/Al-Verh¨altnis
von 14, und einen H-ZSM-5 der Firma CK Bitterfeld mit der Bezeichnung T3 mit einem
Si/Al-Verhaltnis von 19.
3.1.3.1 Ionentausch
Der Ionentausch erfolgt nach einer Vorschrift zur Pr¨aparation eines Cu-ZSM-5 vonIwamo-
to [72]. 5 g Zeolith werden in 400 ml einer 13 mmolaren Cer- oder Indiumnitratl¨osung ca.
10 h bei RT ger¨uhrt. Anschließend wird der Zeolith abgenutscht und mit destilliertem Wasser
gewaschen. Der Ionentausch wird noch zweimal mit frischer Salzl¨osung wiederholt, um das
Gleichgewicht in der Austauschl¨osung zu verschieben und die Endbeladung des Zeolithen mit

3 Untersuchungsmethodik 26
Metallkationen zu erh¨ohen. Nach dem letzten Waschvorgang wird der Zeolith ¨uber Nacht bei
120ÆC im Trockenschrank getrocknet und am n¨achsten Tag 30 Sekunden bei einem Druck von
8 Tonnen gepresst, zerkleinert und eine Kornfraktion von 250 - 355�m ausgesiebt. Mit dieser
Praparationsmethode sollten Metallkationen auf intrazeolithischen Austauschpl¨atzen des Zeo-
lithen lokalisiert werden.
3.1.3.2 Fallung
2 g Zeolith werden unter R¨uhren in 100 ml einer 13 mmolaren Metallsalzl¨osung (0,56 g
Ce(NO3)3 �6 H2O; 0,5 g In(NO3)3 �5 H2O) suspendiert, und sofort werden 20 ml einer 25 %igen
NH3-Losung zugegeben. Die Suspension wird abgenutscht, mit 400 ml Wasser gewaschen und
bei 120ÆC im Trockenschrank ¨uber Nacht getrocknet. Durch die schnelle Zugabe des Ammo-
niaks sollte ein Ionentausch vermieden und die Cer- bzw. Indiumkomponente extrazeolithisch
auf deraußeren Oberfl¨ache lokalisiert werden. Bei vollst¨andiger Fallung ergibt sich ein theore-
tischer Indium- bzw. Cergehalt von 7 respektive 9 Massenprozent.
3.1.3.3 Ionentausch mit anschließender Fallung
Beim Ionentausch mit anschließender F¨allung handelt es sich um eine Kombination der beiden
oben beschriebenen Methoden mit leichten Modifikationen, wobei die Vorschrift vom Koope-
rationspartner am ACA ¨ubernommen wurde. Um eine Beladung mit 4 Massenprozent Indium
zu erreichen, werden 5 g Zeolith und 0,68 g In(NO3)3 �5 H2O in 250 ml Wasser suspendiert und
20 h bei 60ÆC geruhrt. Anschließend erfolgt durch Zugabe von 20 ml 0,1 molarer NH3-Losung
die Fallung des ¨ubersch¨ussigen In(NO3)3 �5 H2O, und nach weiteren 30 Minuten R¨uhren er-
folgt die Abnutschung und Trocknung des Katalysators bei 120ÆC im Trockenschrank. Hier
sollte gezielt eine Kombination extra- und intrazeolithischer Indiumspezies dargestellt werden,
um ein mogliches positives oder negatives Zusammenspiel der Komponenten in der Katalyse
beobachten zu k¨onnen.
3.1.3.4 Festkorperionentausch
Beim Festkorperionentausch (SSIE = Solid-State-Ion-Exchange) wird eine mechanische Mi-
schung von H-ZSM-5 mit InCl3 thermisch behandelt, wobei 5 g des vorher kalzinierten und
dabei von der NH4-Form in die H-Form ¨uberfuhrten Zeoliths mit der gew¨unschten Menge

3 Untersuchungsmethodik 27
InCl3 intensiv vermengt werden, um eine homogene Probe zu erhalten und Schichtenbildung
zu vermeiden. Dann folgt ein Temperaturprogramm von 2 K min�1 bis 120ÆC, nach einer
10-minutigen Haltezeit mit 5 K min�1 bis 550ÆC, danach 2 Stunden Haltezeit, wobei die Pro-
be unter dem Vakuum einer angeschlossenen Drehschieberpumpe steht. Im Idealfall sollten auf
diese Weise intrazeolithische Indiumspezies durch eine Festk¨orperreaktion des Halides mit den
Protonen des Zeolithen unter gleichzeitiger Entfernung des fl¨uchtigen Reaktionsproduktes HCl
prapariert werden.
3.1.3.5 Sublimation
Bei der Sublimationsmethode wird der H-ZSM-5 im Gegensatz zum SSIE nicht mit dem InCl3
vermengt, sondern die beiden Komponenten werden getrennt voneinander eingebracht. Ein
Stickstoffstrom streicht erst ¨uber das imUberschuss eingesetzte InCl3 und muss dann eine
Schuttung aus 5 g pelletisierten Zeolithk¨ornern (250 - 355�m) passieren. So ist gew¨ahrleistet,
dass nur fl¨uchtige Verbindungen bis zum Zeolithen vordringen k¨onnen und ein eventueller
Ruckstand nicht im Zeolithen zur¨uckbleibt, sondern im weiter vorne deponierten InCl3. Auch
hier wird wieder das Temperaturprogramm 2 K min�1 bis 120ÆC, 10-minutige Haltezeit, wei-
ter mit 5 K min�1 bis 550ÆC, und 2 Stunden Haltezeit, gefahren. Ziel dieser Pr¨aparationsweise
waren wiederum ausschließlich intrazeolitische Indiumspezies.
3.1.3.6 Transportreaktion
Die Transportreaktion wurde an zwei verschiedenen Matrizen durchgef¨uhrt [15], an einem
ZSM-5 und an einem Silikaliten mit MFI-Struktur. Der wichtigste Unterschied zwischen den
Transportproben und den SSIE-Proben besteht darin, dass bei den Transportproben m¨oglichst
unhydrolysiertes InCl3 verwendet wurde und die Reaktionstemperaturen sehr viel niedriger
lagen. An einer Vakuumlinie wurde die Matrix ausgeheizt, um Wasser aus den Poren zu ent-
fernen; die dann verschlossene Ampulle wurde in eine Glove-Box transferiert. Dort wurde eine
Schicht Glaswolle und danach trockenes InCl3 zugefugt, so dass die Indiumkomponente r¨aum-
lich getrennt von der Matrix vorlag. Nach Evakuierung an der Vakuumlinie wurde die Ampulle
abgeschmolzen und 3 Tage lang bei 400ÆC in einem Muffelofen belassen. Ziel dieser Pr¨apara-
tionsmethode war die Generierung ausschließlich intrazeolithischer Indium-Spezies unter Ver-
meidung eventuell st¨orender Einfl¨usse durch w¨ahrend der Pr¨aparation hydrolysiertes InCl3.

3 Untersuchungsmethodik 28
3.1.3.7 Mechanische Mischungen
Die mechanischen Mischungen verfolgten das Ziel, ausschließlich extrazeolithische Indium-
spezies zu generieren. Hierzu wurden 2 g des Zeolithen mit der entsprechenden Menge CeO2,
In(OH)3 oder In2O3 vermengt, um 7 (Indium) bzw. 9 (Cer) Massenprozent im Katalysator
zu erhalten. Hierbei konnten die einzelnen Komponenten vor der Zusammenmischung unter-
schiedlichen Kalzinationsbedingungen unterworfen werden.
3.1.3.8 Schichtexperimente
Die Schichtexperimente dienten wie beiLiese[14] zur Auffindung von Hinweisen ¨uber den Re-
aktionsmechanismus. Die Komponenten des Katalysators wurden r¨aumlich in Schichten aufge-
trennt. Aus der Wirkung dieser Anordnung auf das katalytische Verhalten wurden R¨uckschlusse
uber den Reaktionsmechanismus gezogen. Die konkrete Konzeption der jeweiligen Schichtex-
perimente wird im Ergebnisteil dargestellt.
3.1.4 Katalysatorcharakterisierung
In diesem Abschnitt werden die unterschiedlichen angewandten Charakterisierungsmethoden
fur Oberflachen- als auch f¨ur Volumeneigenschaften der Katalysatoren vorgestellt.
3.1.4.1 Spezifische Katalysatoroberflache
Bei vielen heterogenen Gas/Feststoffreaktionen spielt die spezifische Oberfl¨ache des Katalysa-
tors eine entscheidende Rolle, da hier Edukte, Intermediate und Produkte an aktiven Zentren
adsorbieren. Auch bei Feststoff/Feststoff-Reaktionen kann ein Einfluss der spezifischen Kata-
lysatoroberflache angenommen werden. Daher ist die Bestimmung der Katalysatoroberfl¨ache
ein wichtiges Werkzeug bei der Katalysatorcharakterisierung.
Die gebrauchlichste Methode zur Oberfl¨achenbestimmung ist die Physisorption von Gasen.
Verwendet werden unpolare Gase wie N2 oder Krypton (letzteres f¨ur Oberflachen< 2m2 g�1),
die eine unspezifische Wechselwirkung mit der Katalysatoroberfl¨ache eingehen. Nach Ermitt-
lung der fur eine Monolagenbedeckung notwendigen Adsorbatmenge kann aus der Anzahl
der Adsorbatmolek¨ule und dem Fl¨achenbedarf des Adsorbatmolek¨uls die Oberflache berechnet
werden.
SBET =n(N2)mon � NA � AN2
mKat(3.6)

3 Untersuchungsmethodik 29
Hierbei istSBET die spezifische Katalysatoroberfl¨ache, NA die Avogadrosche Konstante, AN2
der Flachenbedarf eines einzelnen adsorbierten N2-Molekuls (16; 2 � 10�20 m2) und mKat die
Katalysatormasse.
In der Praxis ist es schwierig, die Stoffmenge einer monomolekularen Schichtn(N2)mon
zu bestimmen, da diese durch Mehrschichtenadsorption ¨uberlagert wird. Zur Bestimmung
der Katalysatoroberfl¨ache wird meistens die von Brunauer, Emmet und Teller entwickelte
BET-Methode verwendet, die den Vorgang der Physisorption unter Ber¨ucksichtigung einer
Mehrschichtenadsorption beschreibt [73]. Hierbei erfolgt die Monolagenadsorption wie beim
Langmuir’schen Modell auf ¨aquivalenten Pl¨atzen [74]. Die Erweiterung der Theorie auf Mehr-
schichtenadsorption f¨uhrt zu folgenden weiteren Annahmen: Bei der Adsorption der Monolage
wird die Kondensationsenthalpie�Hads frei. Fur alle weiteren Lagen wird aufgrund fehlen-
der Adsorbat-Katalysator-Wechselwirkungen lediglich die geringere Kondensationsenthalpie
�Hkon pro Lage frei. Dieser Vorgang wird mathematisch f¨ur theoretisch unendlich viele Lagen
wie folgt beschrieben:
p=p0n(N2)ads � (1 � p=p0)
=1
n(N2)mon � C+
C � 1
n(N2)mon � C� pp0
(3.7)
Diese Gleichung ist im Bereich0; 05 < p=p0 < 0; 30 gultig (p0 = Sattigungsdampfdruck
des Adsorbates bei der entsprechenden Temperatur), in anderen Bereichen ist meistens keine
Linearitat mehr gegeben. Durch eine Auftragung von p=p0n(N2)ads � (1� p=p0)
gegen pp0
kann aus der
Steigung der Geraden und dem x-Achsenabschnitt die Monolagenstoffmengen(N2)mon und
die Konstante C abgelesen werden.
Die Bestimmung der Oberfl¨ache von Zeolithen ist schwierig, da in den Mikroporen von mo-
lekularer Dimension keine Mehrschichtenadsorption auftritt und die BET-Methode zu unge-
naueren Ergebnissen f¨uhrt. In dieser Arbeit sind deshalb lediglich Komponenten wie In(OH)3
auf ihre Oberflache hin untersucht worden und nicht die Zeolithe selber. Die Messungen er-
folgten mit N2 als Adsorbat bei einer Temperatur von 78 K in der statischen Sorptionsapparatur
Autosorb-1C der Firma Quantachrome. Die adsorbierte Stoffmenge wird aus der Druckabnah-
me des statisch dosierten Gases bei konstantem Volumen bestimmt.

3 Untersuchungsmethodik 30
3.1.4.2 Infrarot-Spektroskopie
Die Infrarotspektroskopie ist eine der ¨altesten Methoden zur Charakterisierung von Zeolithen
und Zeolith/Adsorbat-Systemen und dient zur Aufkl¨arung von Gittereigenschaften sowie zur
Untersuchung katalyserelevanter Stellen und adsorbierter Molek¨ule [75–77].
Die am haufigsten verwendete Strahlung liegt im mittleren Infrarotbereich (Wellenzahlen
von 4000 - 400 cm�1). Das Grundprinzip der Infrarotspektroskopie besteht darin, dass ein
IR-Strahl einer bestimmten Anfangsintensit¨at eine Probe bestrahlt bzw. durchstrahlt (Trans-
missionsspektroskopie) und dabei nach dem Lambert-Beerschen Gesetz an Intensit¨at verliert,
indem es Molek¨ule zu Schwingungen anregt.
I = I0 � e���c�d; (3.8)
wobei mit I0 und I die Eingangs- bzw. die Austrittsintensit¨at, mit � der Extinktionskoeffizient,
mit c die Konzentration und mit d die Schichtdicke der Probe bezeichnet wird.
Damit ein Molekul Strahlung absorbieren kann, muss sich sein Dipolmoment w¨ahrend der
Schwingung ¨andern (im Gegensatz hierzu muss sich bei der Ramanspektroskopie die Polari-
sierbarkeitandern, wodurch sich die beiden Messmethoden oft sinnvoll erg¨anzen). Da auch bei
der Schwingungsspektroskopie die Energieniveaus gequantelt sind, ergibt sich f¨ur die diskreten
Energiezust¨ande in der N¨aherung des harmonischen Oszillators die Gleichung
Ev = h�(v + 1=2): (3.9)
Dabei kennzeichnet v die Schwingungsquantenzahl (sie kann Null und alle ganzzahligen posi-
tiven Werte annehmen, allerdings sind nur dieUbergange�v =�1 erlaubt). Die Oszillatorfre-
quenz� wird gegeben durch
� = 1=2�pk=�: (3.10)
Das Symbol k beschreibt in dieser Gleichung die Kraftkonstante (bei Molek¨ulen ist dies die
Bindungsst¨arke) und� die reduzierte Masse mit
� =m1m2
m1 +m2: (3.11)
Allerdings muss beachtet werden, dass in der Realit¨at kein harmonischer Oszillator vorliegt und
es daher doch zu verbotenenUbergangen (� v > � 1) und somit Oberschwingungen kommen

3 Untersuchungsmethodik 31
kann. Theoretisch k¨onnen mit diesen Grundlagen dieUbergange in einem Spektrum zugeordnet
und Bindungsst¨arken berechnet werden.
In dieser Arbeit werden jedoch nur die Spektren der Zeolithmatrix aufgenommen und mit-
einander verglichen. Hierbei wird allerdings nicht die Transmissionsspektroskopie verwendet,
bei der ein selbsttragender Pressling durchstrahlt wird, sondern die DRIFT-Spektroskopie (Dif-
fuse Reflectance Infrared Fourier Transform Spectroscopy), die auch bei stark absorbieren-
den und streuenden Proben eingesetzt werden kann. In der DRIFT-Spektroskopie k¨onnen lose
Pulver vermessen werden, was auch f¨ur in situ Messungen g¨unstig ist, da eventuell an einem
Pressling st¨arkere Diffusionslimitierungen auftreten k¨onnen. Die Strahlung wird diffus an den
ersten Atomlagen gestreut und durch einen ellipsenf¨ormigen Spiegel geb¨undelt und auf den De-
tektor fokussiert. Das Absorptionsspektrum kann mit der Kubelka-Munk-Funktion beschrieben
werden:K
S=
(1� R1)2
2R1
(3.12)
mit dem Absorptionskoeffizienten K, dem Streukoeffizienten S und der Reflexion einer unend-
lich dicken Probe R1.
Ist der Streukoeffizient unabh¨angig von der Einstrahlfrequenz, so kann das gemesse-
ne Spektrum R1 (�) in das Absorptionsspektrum K(�) transformiert werden. Da die
Kubelka-Munk-Funktion nur f¨ur verdunnte Systeme angewendet werden darf, kommt hier zur
Analyse der Spektren unverd¨unnter Proben die vonOlinger undGriffithsentwickelte”Schein-
bare Absorption“-logR1 zum Einsatz [78]. Empirisch wurde von ihnen ein linearer Zusammen-
hang zwischen der Bandenintensit¨at und der Konzentration der untersuchten Probe festgestellt,
so dass sich die Spektren wie normale Absorptionsspektren auswerten lassen.
Im folgenden Abschnitt sollen nun die an einer zeolithischen Probe beobachtbaren und f¨ur
diese Arbeit wichtigen Absorptionsbanden vorgestellt werden.
Besonders interessant sind die Schwingungen der in Abbildung 3.3 beschriebenen Zen-
tren des Zeolithen, da diese Reaktionen mit anderen Ionen eingehen k¨onnen. Die soge-
nannten Brønstedzentren weisen die h¨ochste S¨aurestarke auf und kompensieren die nega-
tive Ladung der Si-O-Al-Einheit (3617 - 3600 cm�1), sie konnen mit benachbarten Sauer-
stoffatomen des Ger¨usts (3250 cm�1) in Wechselwirkung treten. Schw¨acher sauer sind die
an deraußeren Oberfl¨ache des Zeolithen liegenden Silanolgruppen (3745 - 3740 cm�1), dann
folgen die wechselwirkenden inneren Silanolgruppen (3735 - 3725 cm�1 sowie 3550 cm�1).

3 Untersuchungsmethodik 32
OAl
O
OOH
3250 cm
Si O H3745 - 3740 cm-1
SiO
Al
H
3617 - 3600 cm-1
SiO
H<b>
O
H<f>
Si3735 - 3725 cm <f>3550 cm <b>
-1
-1
Al O H3770-3800 cm-1
-1
Abbildung 3.3: OH-Schwingungen in Zeolithen [15]
Durch Dealuminierungsprozesse oder durch unvollst¨andigen Einbau w¨ahrend der Synthese
entstandene AlOH-Gruppen besitzen den geringsten S¨aurecharakter und haben eine Banden-
lage bei 3770 - 3800 cm�1. Weitere AlOH-Gruppen am Nichtger¨ustaluminium zeigen Banden
bei 3680 - 3665 cm�1. Im Wellenzahlenbereich von 2200 - 1700 cm�1 liegen die Oberschwin-
gungen des Zeolithgitters. Durch Normierung der Bandenintensit¨aten auf diese Gitterschwin-
gungen k¨onnen einzelne Spektren qualitativ miteinander verglichen werden; eine quantitative
Auswertung ist bei der DRIFTS-Methode auf Grund vieler schwer zu reproduzierender expe-
rimenteller Parameter nicht m¨oglich. Zudem liefern die Gitterschwingungen Anzeichen einer
eventuellen Zerst¨orung des Zeolithgitters.
Im Rahmen dieser Arbeit wurden unverd¨unnte Zeolithproben mit einem Nicolet Proteg´e 460
FTIR-Spektrometer mit einer DRIFT-Einrichtung vermessen. Die Proben wurden zur Entfer-
nung von adsorbiertem Wasser mit 5ÆC min�1 auf 550ÆC aufgeheizt und eine Stunde isotherm
gehalten. Danach wurde die Probe auf 150ÆC abgek¨uhlt und ein aus 1000 Messzyklen zusam-
mengesetztes Spektrum mit einer Aufl¨osung von 4 cm�1 aufgenommen.
3.1.4.3 Rontgendiffraktometrie
Die Rontgendiffraktometrie wird zur Bestimmung von Kristallstrukturen, zur Phasenanalyse
(Phasenumwandlungen durchin situ XRD bei z.B. Reduktions- oder Oxidationsreaktionen)
und zur Kristallitgroßenbestimmung eingesetzt. Es handelt sich hierbei um eine Volumenme-
thode [16,75].
Rontgenstrahlen wechselwirken mit den Elektronen der Materie und werden in alle Richtun-
gen gestreut. Da die Wellenl¨angen imAngstrømbereich und somit in der Gr¨oßenordnung der

3 Untersuchungsmethodik 33
Netzebenenabst¨ande der Kristallite liegen, kann es zu Interferenzerscheinungen und somit zur
Entstehung von Interferenzmaxima und -minima kommen. Hierf¨ur ist allerdings eine ausrei-
chende Fernordnung in den Kristalliten notwendig, so dass amorphe Phasen oder sehr kleine
Partikel aufgrund ihrer sehr breiten oder gar nicht vorhanden Signale nicht identifiziert werden
konnen.
Die Rontgenstrahlen selber werden erzeugt durch den Beschuss einer Anode mit energierei-
chen Elektronen. Durch das Abbremsen der Elektronen durch das Metall entsteht eine kontinu-
ierliche Bremsstrahlung als Untergrund im R¨ontgenstrahlspektrum. Gleichzeitig werden aber
Elektronen aus inneren Elektronenschalen des Metalls herausgeschlagen und durch Elektronen
eines hoherenergetischen Orbitals wieder aufgef¨ullt, wobei Strahlung emittiert wird. In einem
Kupferatom kann ein leerer Elektronenplatz des 1s-Orbitals von einem Elektron des 2p- oder
des 3p-Orbitals aufgef¨ullt werden. Die entstehende Strahlung wird als K�- bzw. K�-Strahlung
bezeichnet, wobei diese wegen der Spinmultiplizit¨at in Dubletts gespalten sind (z.B. K�1 und
K�2). Um nur eine einzige Wellenl¨ange zu verwenden, wird das R¨ontgenspektrum mit Hil-
fe eines Kristall-Monochromators oder eines Metallfolienfilters monochromatisiert. Wird der
Rontgenstrahl an den Netzebenen gebeugt, so entsteht eine konstruktive Interferenz, wenn die
Bragg-Gleichung
n� = 2dhkl sin� (3.13)
erfullt ist. Hierbei ist n ganzzahlig,� die Wellenlange der R¨ontgenstrahlung, dhkl der Netzebe-
nenabstand mit den Millerschen Indizes hkl und� der Glanzwinkel der einfallenden R¨ontgen-
strahlung. Durch die Beugungsmaxima und deren Position (bzgl.�) wird also eine Strukturin-
formation gewonnen. Eine weitere Auswertung ist mit Hilfe der Scherrer-Gleichung
d =K�
� cos�(3.14)
moglich. Mit Hilfe der Peakhalbwertsbreite� und der Konstanten K (je nach Kristallgestalt
nimmt sie Werte zwischen 0,89 und 1,39 an, n¨aherungsweise wird h¨aufig der Wert 1 einge-
setzt) ist es m¨oglich, die Große d der Kristallite zu bestimmen. An dieser Gleichung ist außer-
dem erkennbar, dass bei Verwendung einer kurzwelligeren Strahlung noch kleinere Kristallite
erfasst werden k¨onnen (z.B mit Mo K�-Strahlung mit� = 0,71A statt der Cu K�-Strahlung mit
� = 1,54A).

3 Untersuchungsmethodik 34
In dieser Arbeit wird mit Hilfe der R¨ontgendiffraktogramme ¨uberpruft, ob das Gitter der Zeo-
lithe wahrend der Modifizierung zerst¨ort wurde. Zudem lassen sich mit Hilfe der PDF-Kartei
(Powder Diffraction File) des JCPDS (Joint Committee on Powder Diffraction Standards) even-
tuell entstandene zus¨atzliche Phasen von Metallen oder Metalloxiden identifizieren. Gleichzei-
tig spricht eine eventuelleAnderung der Reflexintensit¨aten des Zeolithen f¨ur einen Einbau von
schwereren Ionen in den Zeolithen [18]. Dies wurde z.B. beim Einbau von Pt auf Kationen-
platze eines NH4-Y-Zeolithen durchHatjeet al. beobachtet [79].
Gemessen wurde an einem Siemens-Diffraktometer D500 mit einer Bragg-Brentano-
Geometrie. Die Proben wurden fein gem¨orsert und als Flachpr¨aparate eingesetzt.
3.1.4.4 Photoelektronenspektroskopie
Mit Hilfe der Rontgen-Photoelektronen-Spektroskopie (XPS = X-Ray Photoelectron Spectros-
copy) konnen Informationen ¨uber die elementare Zusammensetzung der Oberfl¨ache einer Pro-
be,uber den Oxidationszustand eines Elementes und unter g¨unstigen Bedingungen sogar ¨uber
die Dispersitat einer Phase ¨uber eine andere erhalten werden [75].
Die Grundlage der Photoelektronenspektroskopie stellt der photoelektrische Effekt dar, bei
dem ein Atom ein Photon mit der Energie h� absorbiert und dabei ein Rumpf- oder Valenz-
elektron der Bindungsenergie Eb mit der kinetischen Energie Ek herausgeschlagen wird.
Ek = h� � Eb � '; (3.15)
wobei' eine Korrektur fur die Austrittsarbeit des Spektrometers bezeichnet. Zur Anregung
werden normalerweise die R¨ontgenstrahlen Mg K� (1253,6 eV) und Al K� (1486,3 eV) be-
nutzt. Da die herausgeschlagenen Elektronen mit anderen Atomen kollidieren und dabei Ener-
gie verlieren, ergibt sich eine Ausdringtiefe aus der Probe von ca. 6 - 8 Monolagen. Allerdings
stammen ca. 20 - 30% des Messsignals aus der ¨außersten Atomlage, so dass diese Methode als
oberflachensensitiv betrachtet werden kann (Bei der Ionenstreuspektroskopie (ISS) hingegen
handelt es sich um eine rein oberfl¨achensensitive Methode, bei der monochromatische Edelgas-
ionen auf die Probe geschossen werden und dabei nach derUbertragung von Energie reflektiert
werden). Das XPS-Spektrum enh¨alt eine Auftragung der Photoelektronenintensit¨at als Funkti-
on der Bindungsenergie. Diese Bindungsenergie entspricht aber nicht exakt der Orbitalenergie,
aus der das Elektron entfernt wurde, da die anderen Elektronen des Atoms zur Erniedrigung

3 Untersuchungsmethodik 35
der absoluten Energie etwas relaxieren. Trotzdem gilt es nach dem Koopmans’schen Theorem
in erster Naherung.
Zusatzlich zu den Signalen der Photoelektronen enth¨alt das Spektrum noch Signale durch
Auger-Elektronen. Der Auger-Prozess ist ein Sekund¨areffekt der Photoemission. Das ionisier-
te Atom wird stabilisiert, indem ein Elektron aus einem h¨oheren Valenzband an die Stelle
des herausgeschlagenen Elektrons r¨uckt. Dabei wird Strahlung frei. Da diese freigewordene
Strahlung unabh¨angig von der verwendeten R¨ontgenstrahlung ist, k¨onnen bei Verwendung
zweier unterschiedlicher R¨ontgenstrahlenergien die Auger-Signale identifiziert werden. Die
XPS-Signale bleiben bei der gleichen Bindungsenergie, w¨ahrend sich die Auger-Signale ver-
schieben.
Mit Hilfe der XP-Spektroskopie lassen sich nun die vorhandenen Elemente in einer Probe be-
stimmen, weil jedes Element einen Satz charakteristischer Bindungsenergien aufweist. Zus¨atz-
lich konnen aber auch Informationen ¨uber verschiedene Oxidationszust¨ande einzelner Elemen-
te gewonnen werden. Im allgemeinen verschieben sich die Bindungsenergien bei h¨oheren Oxi-
dationszahlen zu h¨oheren Werten, bei gleichen Oxidationszahlen erh¨ohen sich die Bindungs-
energien mit zunehmender Elektronegativit¨at der Liganden. Allerdings gibt es Ausnahmen. Die
Verschiebungen liegen im Bereich von 0-3 eV. Da sich w¨ahrend der Messung isolierende Pro-
ben positiv aufladen und sich so die Bindungsenergien zu h¨oheren Werten verschieben, wird
die immer vorhandene Kohlenstoffkontaminierung (C 1s) als Kalibration verwendet (bei Kata-
lysatoren kann auch auf die Bindungsenergie des Tr¨agers kalibriert werden). Damit ist es dann
auch nicht notwendig, die Austrittsarbeit des Spektrometers zu kennen.
Da die XPS eine oberfl¨achensensitive Technik ist, kann mit ihr die Dispersit¨at von Partikeln
auf einem Trager untersucht werden. Bei gleicher Beladung zweier Katalysatoren und unter-
schiedlicher Dispersit¨at ergibt sich fur den Katalysator mit der h¨oheren Dispersit¨at ein hoheres
IPartikel/ITr�ager-Verhaltnis (I fur Intensitat des Signals), weil bei kleinen Partikeln fast alle Ato-
me an der Oberfl¨ache sind und eine große Fl¨ache des Tr¨agers bedeckt ist. Dementsprechend
weisen große Partikel ein niedriges IPartikel/ITr�ager-Verhaltnis auf. Die Intensit¨atverhaltnisse
werden bei XP-Spektren mit Hilfe von Scofield-Faktoren auf Konzentrationsverh¨altnisse um-
gerechnet [80], wobei zus¨atzlich der Abstand zwischen den Signalen im Spektrum ber¨ucksich-
tigt wird.
In einer parallelen Arbeit [15] wurden die hier pr¨aparierten Katalysatoren mit Hilfe der

3 Untersuchungsmethodik 36
XP-Spektroskopie auf ihre Dispersit¨at und ihre Oxidationszust¨ande untersucht. Da diese Er-
gebnisse zur Diskussion der Abh¨angigkeit zwischen katalytischer Aktivit¨at und Struktur n¨otig
sind, werden sie am Anfang der Kapitel ¨uber die entsprechenden Katalysatorsysteme in
tabellarischer Form vorgestellt. Interessant in diesem Zusammenhang ist die Beobachtung von
Kikuchi et al., die fur eine Mischung aus In2O3 mit H-ZSM-5 nach thermischer Behandlung
bei 500ÆC in Helium eine Verschiebung der In 3d5=2-Bande von 445 nach 446 eV beobach-
teten. Diese Erh¨ohung der Bindungsenergie begr¨undeten sie mit der Entstehung kationischer
Indiumspezies im Zeolithen [46].
3.1.4.5 Rontgenabsorptionsspektroskopie
Bei der Rontgenabsorptionsspektroskopie (XAS = X-Ray Absorption Spectroscopy) werden
wie in der Photoelektronenspektroskopie Elektronen mit Hilfe von R¨ontgenstrahlen angeregt.
Dabei wird der durch die Materie gehende R¨ontgenstrahl durch die Absorptionseigenschaf-
ten des Materials geschw¨acht, und es ergibt sich die dem Lambert-Beerschen Gesetz analoge
Beziehung
It = I0e��(E)x; (3.16)
mit dem linearen Absorptionskoeffizienten�(E), der Eingangsintensit¨at Io, der Austrittsinten-
sitat It und der Photonenenergie E. Bei einem XA-Spektrum wird der Absorptionskoeffizient
als Funktion der Photonenenergie aufgetragen. Die Absorption erfolgt durch die Anregung
von Rumpfatomen in unbesetzte h¨oherenergetische Zust¨ande oder in das Kontinuum. Erreicht
die Energie des Photons die Grenze zur Anregung, erfolgt ein scharfer Anstieg des Absorpti-
onskoeffizienten. Im Spektrum wird dies als Absorptionskante bezeichnet. Aus der H¨ohe des
Kantenhubs l¨asst sich die Atomkonzentration des jeweiligen Elementes bestimmen.
Bei der XAS werden zwei Regionen unterschieden. Die XANES-Region (XANES = X-Ray
Absorption Near Edge Structure) liegt rund um die Absorptionskante des Elementes und
enthalt Elektronen¨ubergange und Mehrfachstreuereignisse. Elektronen¨ubergange liegen zwi-
schen 20 eV vor bis 10 eV nach der Kante und Mehrfachstreuereignisse 10 - 50 eV nach
der Kante. Die Region>50 eV bis mehrere hundert eV nennt man EXAFS (EXAFS = Ex-
tended X-Ray Absorption Fine Structure), sie enth¨alt nur Einfachstreuungen. DieAnderun-
gen im Absorptionskoeffizienten ergeben sich aus konstruktiven oder destruktiven Interferen-
zen von an Nachbaratomen r¨uckgestreuten Photonenelektronenwellen mit ausgehenden Pho-

3 Untersuchungsmethodik 37
tonenelektronenwellen, wobei konstruktive Interferenz zu einer Erh¨ohung des Absorptionsko-
effizienten fuhrt und destruktive Interferenz zu einer Erniedrigung. Beide Regionen enthal-
ten also Informationen ¨uber Anzahl, Art und Abstand der streuenden Nachbaratome. Dies
ist bei XRD-Messungen auch der Fall, allerdings ist bei XAS keine Fernordnung n¨otig, so
dass auch amorphe oder teilkristalline Verbindungen, aber auch Gase und Fl¨ussigkeiten un-
tersucht werden k¨onnen. Zus¨atzlich erfordert die XAS keine Vakuumbedingungen, was die
Durchfuhrung vonin situMessungen erm¨oglicht. Auf Grund der komplexen Informationen im
XANES-Bereich (hier finden sich zus¨azlich Informationen ¨uber die Bandstruktur) wird dieser
meist nur zu qualitativen Analysen herangezogen: das Spektrum einer Verbindung bekannter
Struktur wird mit dem Spektrum der Probe verglichen. Aus Unterschieden oder Gemeinsam-
keiten der Spektren werden R¨uckschlusse auf die Struktur der Probe geschlossen. Anders sieht
es aus f¨ur die EXAFS-Region, in der es nur zur Einfachr¨uckstreuung kommt. DieUberlage-
rung der R¨uckstreuanteile der verschiedenen Nachbarn kann mathematisch ausgewertet wer-
den. Das Ergebnis kann als Auftragung der gemessenen Absorption gegen die Energie der
Strahlung im sogenannten”k-Raum“dargestellt werden, oder, nach einer Fouriertransformati-
on, als Auftragung der”Streukraft“gegen den Abstand r des absorbierenden Atoms im soge-
nannten”r-Raum“. Die genauere Vorgehensweise findet sich in [15]. Ein gravierender Nach-
teil der XA-Spektroskopie ist darin zu sehen, dass die ermittelten strukturellen Parameter den
Mittelwert fur verschiedene eventuell vorhandene Phasen bilden. Mit Hilfe der Phasenkorre-
lationsanalyse kann f¨ur den XANES-Bereich versucht werden, eindeutig identifizierte Phasen
aus dem Spektrum herauszurechnen bzw. ein Spektrum aus Kombination bekannter Phasen
aufzubauen.
Um ein hohes Signal/Rausch-Verh¨altnis zu erhalten, wird Synchroton-Strahlung verwendet,
deren Intensit¨at mehrere Gr¨oßenordnungen st¨arker ist als die Intensit¨at normaler R¨ontgenstrah-
lung. Ein weiterer Vorteil besteht darin, dass die Wellenl¨ange der Strahlung stufenlos eingestellt
werden kann.
In einer parallelen Arbeit [15] sind die hier pr¨aparierten Katalysatoren am DESY in Ham-
burg untersucht worden. Mittels der EXAFS-Auswertung und der Phasenkorrelationsanalyse
der XANES-Spektren wurde teilweise auf die Struktur der Indiumphase geschlossen. Aus dem
Kantenhub der Spektren ließ sich der Gehalt an Indium in der Probe bestimmen. In der vor-
liegenden Arbeit wurden die zur Struktur erhaltenen Informationen in Korrelation zur kata-
lytischen Aktivitat der Proben gesetzt.

3 Untersuchungsmethodik 38
3.1.4.6 Temperaturprogrammierte Reduktion
Mit Hilfe der temperaturprogrammierten Reduktion (TPR) lassen sich schnell metallische Ka-
talysatoren charakterisieren. Es werden Informationen ¨uber verschiedene nach der Pr¨aparation
vorhandene Phasen und ¨uber deren Oxidationszust¨ande erhalten. Bei bimetallischen Katalysa-
toren kann eventuell auf eine Mischung der Komponenten geschlossen werden. In g¨unstigen
Fallen ist es sogar m¨oglich, Aktivierungsenergien f¨ur die Reduktion zu bestimmen.
Bei der H2-TPR wird ein verd¨unnter H2-Strom (hier ca. 5 Volumenprozent) ¨uber den linear
aufgeheizten Katalysator geleitet und der Wasserstoffverbrauch gemessen. Die Lage und Form
der beobachteten Signale geben Aufschl¨usse ¨uber den Oxidationszustand des Metalls und des
Einflusses von Tr¨agern und Promotoren. Die auftretende Reduktionsreaktion wird durch fol-
gende Gleichung beschrieben:
MOn + nH2 ! M + nH2O: (3.17)
Ob die Reduktion thermodynamisch m¨oglich ist, bestimmt die Formel
�G = �G0 + n R T ln
�pH2OpH2
�; (3.18)
in der� G die Anderung der freien Gibbsenergie ,� G0 die Standard-Gibbsenergie , n den
stochiometrischen Koeffizienten , R die Gaskonstante , T die Temperatur und p den Partial-
druck angeben. Nur bei einer negativenAnderung der freien Gibbsenergie ist eine Reduktion
moglich. Bei vielen Metalloxiden ist� G0 negativ, bei anderen, wie z.B. Fe2O3 oder SnO2
hingegen nicht. Hier muss der zweite Term die positive freie Standard-Gibbsenergie kompen-
sieren. Das gelingt bei niedrigen Wasser- und hohen Wasserstoffpartialdr¨ucken. Die Gr¨oßen
in diesem Triebkraftterm sind beeinflussbar durch den Volumenstrom, durch die eingewoge-
nen Mengen des reduzierbaren Materials, durch die Wasserstoffkonzentration im Eduktgas und
die Geschwindigkeit der Heizrampe. Um geeignete Bedingungen einhalten zu k¨onnen, haben
Monti undBaikerein Kriterium entwickelt, mit dessen Hilfe vor dem Versuch die Bedingungen
abgesch¨atzt werden [81]:
K =S0_Vc0
: (3.19)
Hierbei ist S0 die Menge an reduzierbarer Substanz [mol],_V der Gesamtvolumenstrom [ml/s]
und c0 die Wasserstoffkonzentration im Eduktgas [mol�ml�1]. Das Kriterium K [s] sollte bei

3 Untersuchungsmethodik 39
Heizrampen zwischen 6 - 18 K min�1 zwischen 55 - 140 s liegen. Damit liegt der Wasserstoff-
verbrauch zwischen 10 - 66 %, um den Verbrauch detektieren zu k¨onnen und die Triebkraft
ausreichend groß zu halten. Durchgef¨uhrte Messungen lassen sich mit einem zweiten Kriteri-
um vonBosch, Kipp, van Ommen und Gellingsuberprufen:
mMOn < _n�T1=2 �MMOn
� �O=M : (3.20)
mMOn bezeichnet die Probenmenge [g],_n den Wasserstofffluss [mol�s�1], �T1=2 die Halb-
wertsbreite des Signals der Wasserstoffaufnahme [K],� die Heizrampe [K�s�1], MMOn die
Molmasse der Probe [g�mol�1] und O/M das Sauerstoff/Metallverh¨altnis.
In gunstigen F¨allen lassen sich auch Aktivierungsenergien f¨ur Reduktionsreaktionen nach
der Gleichung
ln
��
T2max
�= � Ered
RTmax+ ln
��R
Ered
�+ C (3.21)
bestimmen. Hierbei ist� der praexponentielle Faktor und Ered die Aktivierungsenergie der
Reduktion. Hierfur wird eine Serie von TPR-Messungen mit unterschiedlichen Heizrampen
� aufgenommen und der linke Gleichungsterm gegen 1/Tmax aufgetragen. Als Steigung der
Geraden ergibt sich der Wert -Ered/R. Durch geeignete Simulationen kann nun entschieden
werden, ob der Reduktionsmechanismus nach dem Modell der Nukleation oder dem Modell
”shrinking core“abl¨auft [82].
In dieser Arbeit wird die TPR verwendet, um durch unterschiedliche Reduktionstempera-
turen auf unterschiedliche Lokalisierungen der Aktivkomponenten (besonders des Indiums)
im/am Zeolithen (extra- oder intrazeolithisch) zu schließen. Die verwendete Versuchsapparatur
findet sich in der Diplomarbeit vonWapner[83].
150 mg Probe wurden in synthetischer Luft mit 10ÆC min�1 auf 600ÆC aufgeheizt und
1 Stunde kalziniert. Nach Abk¨uhlen auf Raumtemperatur und Aussp¨ulen der synthetischen
Luft mit Argon wird ein Gemisch aus 5,2 % H2 in Argon auf den Katalysator geleitet und eine
Temperaturrampe von 10ÆC min�1 auf 800ÆC gestartet. Mit Hilfe eines W¨armeleitfahigkeits-
detektors wurde der Wasserstoffverbrauch als Funktion der Temperatur aufgezeichnet.

3 Untersuchungsmethodik 40
3.2 Die Modellierung
Im folgenden Kapitel wird das zur Modellierung der kinetischen Daten eingesetzte Rechenpro-
gramm und die Vorgehensweise bei der Modellierung vorgestellt.
3.2.1 Simulationsprogramm
Verwendet wird das am Lehrstuhl f¨ur Technische Chemie auf Fortran-Basis entwickelte Pro-
gramm”OPTI“, das in mehreren Arbeiten seine gute Anwendbarkeit best¨atigen konnte [84–89].
Es wird ein pseudohomogener, eindimensionaler, isothermer Festbettreaktor mit Pfropfen-
stromung als Kaskade ideal durchmischter R¨uhrkessel simuliert, wobei die R¨uhrkesselanzahl
hier 500 betr¨agt (eine Erh¨ohung der Kesselzahl bewirkt nur eine Verl¨angerung der Rechenzeit,
das Ergebnis ist identisch). Um eventuell sehr hohe Anfangsreaktionsgeschwindigkeiten op-
timal berucksichtigen zu k¨onnen, wird der erste Kessel nochmals in 8 Unterkessel eingeteilt.
Fur jeden einzelnen Kessel werden die Bilanzgleichungen (Stoff- und W¨armebilanzen) iterativ
nach dem Wegstein-Algorithmus gel¨ost [90]. In diesem Algorithmus wird im Iterationsschritt k
ein neuer Sch¨atzwert einer Variablen x, die die Funktion F(x) erf¨ullen soll, nach der folgenden
Gleichung berechnet:
xk+1 = t � F(xk) + (1� t) � xk ; (3.22)
wobei
t = 1=(1� s)s =F(xk)� F(xk�1)
xk � xk�1: (3.23)
Im Falle t > 10 gilt t = 10, und im Fallet < � 10 gilt t = -10. Sind die Sch¨atzwerte
fur die Konzentrationen negativ, so werden sie auf Null gesetzt, da negative Konzentrationen
physikalisch sinnlos sind.
Um die gesuchten Modellparameter eines kinetischen Ansatzes bestimmen zu k¨onnen, muss
dem Programm zuallererst eine zu minimierende Zielfunktion eingegeben werden, in diesem
Fall die absolute Fehlerquadratsumme der Ums¨atze an NO und CH4:
FQS =nXi=1
(Xexperimentell � Xberechnet)2 (3.24)
Die absolute Fehlerquadratsumme bietet gegen¨uber der relativen Fehlerquadratsumme
FX =nXi=1
�Xexperimentell � Xberechnet
Xexperimentell
�2
(3.25)

3 Untersuchungsmethodik 41
den Vorteil, dass bei ihr die Abweichungen bei kleinen Ums¨atzen, die aus analytischen Gr¨unden
mit einem großeren Fehler behaftet sind, weniger stark ber¨ucksichtigt werden.
Dann berechnet das Programm mit Hilfe der eingegebenen Startparameter die Fehlerquadrat-
summe und variiert die Startparameter mit Hilfe eines Zufallszahlengenerators. Durch diesen
Random-Search-Algorithmus werden immer wieder zuf¨allig im Parameterraum verteilte ki-
netische Parameter erhalten. Wird die Fehlerquadratsumme besser, so beh¨alt der Algorithmus
seine Suchrichtung bei, bis im Rahmen eines vorgegebenen Grenzwertes keine Verbesserung
mehr erzielt wird und vollkommen willk¨urlich eine neue Richtung eingeschlagen wird. Um zu
verhindern, dass statt des globalen Minimums nur ein lokales Minimum gefunden wird, kann
die Schrittweite derAnderung der kinetischen Parameter vergr¨oßert oder das Programm an
verschiedenen Stellen des Parameterraums gestartet werden.
3.2.2 Reaktormodell
Wie im vorherigen Kapitel erw¨ahnt, wird hier ein pseudohomogener, eindimensionaler iso-
thermer Festbettreaktor mit Propfenstr¨omung als Kaskade idealer R¨uhrkessel simuliert. Das
Programm”OPTI“ erlaubt die Aufstellung homogener und heterogener Stoff- und W¨arme-
bilanzen. Auf Grund der geringen umgesetzten Stoffmengen konnte allerdings die W¨armebi-
lanz vernachl¨assigt und ein isothermer Reaktor gew¨ahlt werden, da auch experimentell keine
nennenswerte W¨armeentwicklung zu beobachten war. Als Stoffbilanz wurde ein homogenes
Modell gewahlt, so dass sich insgesamt als Reaktormodell ein pseudohomogener, isothermer
Festbettreaktor mit Pfropfenstr¨omung ergibt. NachRosahlund Gelbin [91] herrscht in einer
katalytischen Sch¨uttung Propfenstr¨omung bei G¨ultigkeit nachfolgender Gleichung:
dReaktordPellet
> 32 (3.26)
Als dPellet wird das arithmetische Mittel der Kornfraktion der Katalysatorsch¨uttung
(255< dP < 350�m) von 302,5�m verwendet. Bei einem Reaktorinnendurchmesser von
12 mm ergibt sich ein Wert von� 40, es herrscht also Pfropfenstr¨omung, und das eindimen-
sionale Modell des Reaktors ist anwendbar. F¨ur ein pseudohomogenes Modell gilt die in der
nachfolgenden Gleichung dargestellte homogene Stoffbilanz
0 = �@ _ni@z
+D2R�
4(1� �)�Kat
NRXj=1
�i;jrj : (3.27)

3 Untersuchungsmethodik 42
Hierbei steht der Index i f¨ur die jeweilige Komponente (NO, CH4, O2, CO, CO2, N2, H2O und
He) und der Index j f¨ur die jeweilige Reaktion.
Um das pseudohomogene Modell anwenden zu d¨urfen, mussen innere und ¨außere Stofftrans-
portlimitierungen ausgeschlossen werden. Dies kann mit Hilfe des Katalysatorwirkungsgrads
als Funktion des Weisz-Moduls f¨ur innere Stofftransportlimitierung (oder auch Porendiffusi-
onslimitierung genannt) und f¨ur außere Stofftransportlimitierung (auch Filmdiffusionslimitie-
rung genannt) ¨uber den externen Wirkungsgrad als Funktion vonDaII � �ext berechnet wer-
den [71]. Da es sich um einen eindimensionalen Reaktor handelt, sollte auch die axiale R¨uck-
vermischung der Gase gepr¨uft werden.Uber die Berechnung der Bodensteinzahl, die gr¨oßer als
100 sein sollte, l¨asst sich ¨uber eine axiale Dispersion und damit ¨uber eine Verweilzeitverteilung
entscheiden.
3.2.3 Stofftransportlimitierung und axiale Dispersion
Die axiale Dispersion tritt auf, wenn vergleichsweise langsame Gasgeschwindigkeiten im
katalytischen Bett auftreten und somit eine R¨uckvermischung der Gase m¨oglich ist. Um ei-
ne axiale Dispersion zu verhindern, sollte die Bodensteinzahl
Bo =Peax � LdP
> 100 (3.28)
großer als 100 sein. Die axiale P´ecletzahl lasst sich f¨ur eine Gasstr¨omung im Festbettreaktor
als Funktion vonRe � Sc graphisch ablesen [71] oder mit der folgenden Formel berechnen:
1
Peax=
0; 3
Re � Sc +0; 5
1 + 3; 8=(Re � Sc) : (3.29)
In beiden Fallen ergibt sich ein Wert von 0,1. Die Reynolds- und die Schmidt-Zahl werden
ebenfalls zurUberprufung auf Stofftransportlimitierung ben¨otigt; sie sind Tabelle 4.52 zu ent-
nehmen. Wird die kleinste eingesetzte Katalysatorbetth¨ohe von 4 mm als L¨ange L verwendet,
so ergibt sich eine Bodensteinzahl von 1,33, die bedeutend kleiner als 100 ist. Allerdings wird
in der Praxis davon ausgegangen, dass bei Bo� 7 eine gute Ann¨aherung der Propfenstr¨omung
tatsachlich gegeben ist [92]. Dieser Wert w¨urde in den hier durchgef¨uhrten Experimenten unter
Einbezug der vorgelagerten Quarzglasschicht zur L¨ange des Katalysatorbettes erf¨ullt werden.
Zudem ist auch die GleichungdReaktordPellet
> 32 (3.30)
erfullt, so dass eine axiale Dispersion vermutlich keine Rolle spielt.

3 Untersuchungsmethodik 43
Um eineaußere Stofftransportlimitierung ausschließen zu k¨onnen, muss der externe Wir-
kungsgrad des Katalysators ermittelt werden. Dieser ist zug¨anglich aus einer Auftragung von
�ext gegenDaII � �ext [71] (DaII = Damkohler-Zahl 2. Art), wobei letzteres Produkt rechne-
risch bestimmt werden kann:
DaII � �ext =re�
kg � a � cg: (3.31)
Hierbei bezeichnet reff die effektive Reaktionsgeschwindigkeit [mol�s�1 �m�3)], kg den
Stoffubergangskoeffizient [m�s�1], a die auf das Volumen bezogene Katalysatoroberfl¨ache
[m�1] und cg die Konzentration des Reaktanden in der Gasphase [mol� m�3]. Der Stoffuber-
gangskoeffizient l¨asst sich mit der Gleichung
kg =Sh �Di
dP(3.32)
berechnen, wobei Di den Diffusionskoeffizienten [m2 � s�1], dP den Pelletdurchmesser [m] und
Sh die Sherwoodzahl darstellen. Die Sherwoodzahl ist aus den empirischen Korrelationen
1; 15p�Re�0;5 = jm =
Sh
Re � Sc1=3 (3.33)
berechenbar [71]. Der Diffusionskoeffizient Di kann nach einer Formel vonHirschfelder [93]
ermittelt werden
D12 =0; 0018583 � T3=2((M1 +M2)=(M1 �M2))
p � �212i(3.34)
mit der Temperatur T [K], den Molmassen Mi [kg�mol�1], dem Gesamtdruck p [bar], dem Kol-
lisionsintegral und zusatzlich zur weiteren Berechnung den Lennard-Jones Kraftkonstanten�
und� und der Boltzmann-Konstante k [J�K�1]. Das Kollisionsintegral und die Lennard-Jones-
Kraftkonstanten sind tabelliert, wobei das Kollisionsintegral eine Funktion von�12 ist.
= f
�kT
�12
�(3.35)
mit
�12 =p�1 � �2 (3.36)
�12 = 0; 5 � (�1 + �2) : (3.37)

3 Untersuchungsmethodik 44
Die Reynolds- und die Schmidt-Zahl werden nach folgenden Formeln berechnet:
Re =u � dP�
(3.38)
Sc =�
Di(3.39)
mit der Leerrohrgeschwindigkeit u [m�s�1] und der kinematischen Viskosit¨at der Gasmischung
� [m2 �s�1]. Letztere kann aus der dynamischen Viskosit¨at nach
� =�
�(3.40)
ermittelt werden.
Die Uberprufung aufinnere Stofftransportlimitierung kann mit der Auftragung des Kata-
lysatorwirkungsgrads gegen das Weisz-Modul durchgefuhrt werden.
= L2cm+ 1
2
re�Dei cg
(3.41)
mit Lc als charakteristische L¨ange (fur eine Kugel rK/3), der Reaktionsordnung m, dem ef-
fektiven Diffusionskoeffizienten Dei und der Konzentration cg in der Gasphase. In den Me-
soporen zwischen den Zeolithkristallen kann von molekularer Diffusion ausgegangen wer-
den, daher muss der Diffusionskoeffizient noch um einen Porosit¨atsfaktor�P (die Flache der
Porenoffnungen macht nur einen bestimmten Anteil der gesamten Oberfl¨ache aus) und den
Tortuositatsfaktor� (die Poren haben eine unregelm¨aßige Gestalt) korrigiert werden:
Dei = Di �
�P�: (3.42)
In den Zeolithporen selber muss von konfigureller Diffusion ausgegangen werden, wobei im
H-ZSM-5 die Diffusionskoeffizienten von kurzkettigen Kohlenwasserstoffen in der Gr¨oßen-
ordnung von 10�10 m2 �s�1 liegen [94,95].
Die zur Berechnung notwendigen Parameter sind in Tabelle 4.52 aufgetragen. Besteht die
Auswahl zwischen verschiedenen Messbedingungen (z.B. sollte die h¨ochste effektive Reak-
tionsgeschwindigkeit gew¨ahlt werden), so wurden jeweils diejenigen gew¨ahlt, die eine Stoff-
transportlimitierung beg¨unstigen sollten.
Es zeigt sich, dass die Reaktion weder im Regime von ¨außerer noch von innerer Stofftrans-
portlimitierung steht. Das ist umso interessanter, da es in der Literatur Hinweise auf Diffusi-
onslimitierung fur die SCR mit Kohlenwasserstoffen gibt.
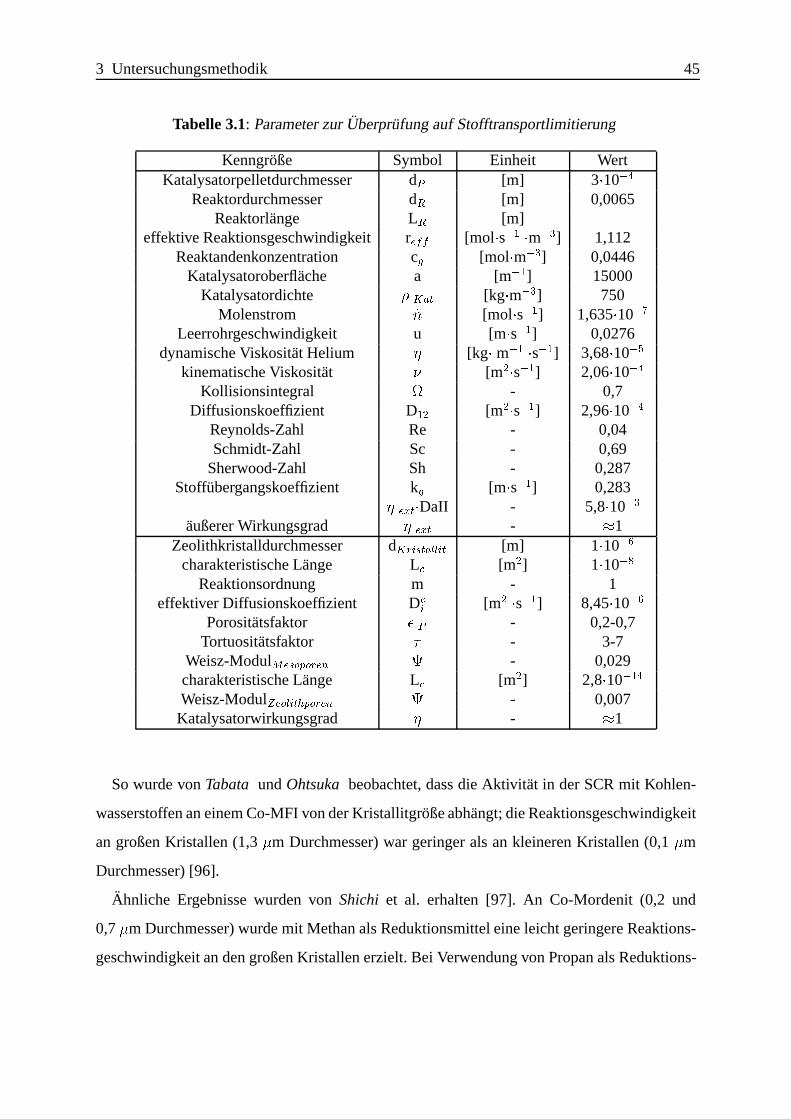
3 Untersuchungsmethodik 45
Tabelle 3.1: Parameter zurUberprufung auf Stofftransportlimitierung
Kenngroße Symbol Einheit WertKatalysatorpelletdurchmesser dP [m] 3�10�4
Reaktordurchmesser dR [m] 0,0065Reaktorlange LR [m]
effektive Reaktionsgeschwindigkeit reff [mol�s�1 �m�3] 1,112Reaktandenkonzentration cg [mol�m�3] 0,0446
Katalysatoroberfl¨ache a [m�1] 15000Katalysatordichte � Kat [kg�m�3] 750
Molenstrom _n [mol�s�1] 1,635�10�7
Leerrohrgeschwindigkeit u [m�s�1] 0,0276dynamische Viskosit¨at Helium � [kg� m�1 �s�1] 3,68�10�5
kinematische Viskosit¨at � [m2�s�1] 2,06�10�4
Kollisionsintegral - 0,7Diffusionskoeffizient D12 [m2�s�1] 2,96�10�4
Reynolds-Zahl Re - 0,04Schmidt-Zahl Sc - 0,69
Sherwood-Zahl Sh - 0,287Stoffubergangskoeffizient kg [m�s�1] 0,283
� ext�DaII - 5,8�10�3
außerer Wirkungsgrad � ext - �1Zeolithkristalldurchmesser dKristallit [m] 1�10�6
charakteristische L¨ange Lc [m2] 1�10�8
Reaktionsordnung m - 1effektiver Diffusionskoeffizient De
i [m2 �s�1] 8,45�10�6
Porositatsfaktor � P - 0,2-0,7Tortuositatsfaktor � - 3-7
Weisz-ModulMesoporen - 0,029charakteristische L¨ange Lc [m2] 2,8�10�14
Weisz-ModulZeolithporen - 0,007Katalysatorwirkungsgrad � - �1
So wurde vonTabata undOhtsuka beobachtet, dass die Aktivit¨at in der SCR mit Kohlen-
wasserstoffen an einem Co-MFI von der Kristallitgr¨oße abh¨angt; die Reaktionsgeschwindigkeit
an großen Kristallen (1,3�m Durchmesser) war geringer als an kleineren Kristallen (0,1�m
Durchmesser) [96].
Ahnliche Ergebnisse wurden vonShichi et al. erhalten [97]. An Co-Mordenit (0,2 und
0,7�m Durchmesser) wurde mit Methan als Reduktionsmittel eine leicht geringere Reaktions-
geschwindigkeit an den großen Kristallen erzielt. Bei Verwendung von Propan als Reduktions-

3 Untersuchungsmethodik 46
mittel war der Unterschied zwischen großen und kleinen Kristallen deutlicher. In einer weiteren
Untersuchung stellten sie fest, daß die intrakristalline Diffusion in Cu-MFI abh¨angig ist von der
Art des Reduktionsmittels. Der NO-Umsatz war bei Verwendung von Ethen abh¨angig von der
Kristallgroße, bei Propan aber nicht. Sie erkl¨aren dies mit einer st¨arkeren Wechselwirkung des
Ethens vermutlich aufgrund der Doppelbindung mit Kupferionen im Zeolithen [98].
Witzel et al. haben verschiedene Kohlenwasserstoffe (Methan, Propan, Isobutan, n-Pentan,
Neopentan, 3,3-Dimethylpentan, 2,2,4-Trimethylpentan und 3,3-Diethylpentan) an verschie-
denen Katalysatoren (H-, Co-, Ni-, Cu-, La-, Mn-, Ga-ZSM-5 und -Mordenit) getestet [99].
Eigentlich sollten Molek¨ule, die� Neopentan ausfallen, nicht in die Kan¨ale der Zeolithe pas-
sen. Allerdings zeigt erst 3,3-Diethylpentan keine NO-Ums¨atze mehr, dagegen findet eine
vollstandige Totaloxidation zu CO2 statt. Der von den Autoren vorgestellte Erkl¨arungsversuch
argumentiert mit einer Anfangsreaktion der großen Molek¨ule an der ¨außeren Oberfl¨ache oder
an den Poren¨offnungen. Sie werden zu kleineren Molek¨ulen aufgebrochen, die dann leicht in
den Zeolithen diffundieren k¨onnen.
Fur einen Pt/MCM-41 mit Propan als Reduktionsmittel habenShenet al. eineaußere Stoff-
transportlimitierung zur Erkl¨arung ansteigender Reaktionsordnungen f¨ur NO herangezogen, in-
nere Stofftransportlimitierung schlossen sie wegen der Mesoporen im MCM-41 von vornherein
aus [100].
Allerdings gibt es auch vonHeinischet al. einen Test auf ¨außere Stofftransportlimitierung f¨ur
einen In/H-BEA mit Methan als Reduktionsmittel, der eine Limitierung ausschließt. Er kommt
zu dem Schluss, dass die bei ihm mit einer heterogenen Massenbilanz mit einem Stofftransfer
von der Gasphase in die Katalysatorphase simulierte Reaktion auch mit Hilfe eines pseudoho-
mogenen Reaktormodells modelliert werden kann [69].
Auch in dieser Arbeit werden im Ergebnisteil einige Vorversuche zur Stoffdiffusionslimi-
tierung vorgestellt. Diese sind notwendig, um die Aussagen der theoretischen Berechnun-
gen auf Stofftransportlimitierung zu unterst¨utzen. Wahrend der Berechnung wurde h¨aufig am
Limit der gultigen Bereiche gearbeitet, da die verwendeten Gleichungen h¨aufig fur einen
technischen Maßstab ausgerichtet sind und hier mit einem mikrokatalytischen Festbettre-
aktor gearbeitet wurde. So ist z.B. eine axiale Dispersion anhand der Berechnungen nicht
vollig auszuschließen; Ein Kriterium spricht dagegen und ein zweites ist nicht eindeutig.
Andere kritische Gr¨oßen wie z.B. die Diffusionskoeffizienten von Molek¨ulen in den Zeo-

3 Untersuchungsmethodik 47
lithen sind nuraußerst schwierig zu bestimmen [94,95]. Gleichzeitig sind m¨oglicherweise
auch die Reaktionen der Zwischenprodukte innerhalb des Zeolithen diffusionskontrolliert,
allerdings gibt es keine M¨oglichkeit, dies mit einfachen Mitteln zu ¨uberprufen.

4 Ergebnisse
Dieses Kapitel ist in zwei Unterkapitel unterteilt. Zuerst werden Untersuchungen zur Funktion
der Komponenten der einzelnen Katalysatorsysteme unter Ber¨ucksichtigung unterschiedlicher
Praparationsmodi innerhalb des jeweiligen Systems vorgestellt, dann erfolgt im zweiten Unter-
kapitel die Modellierung der an den optimierten Katalysatoren erhaltenen Daten. Dabei wird
jeweils zuerst ¨uber die beiden Grundsysteme CeO2-ZSM-5 und In-ZSM-5 berichtet, ehe dann
spater das promotierte System CeO2-In-ZSM-5 folgt.
4.1 Katalyse
4.1.1 Das System CeO2-ZSM-5
Die Funktion des Systems CeO2-ZSM-5 ist aus der Arbeit vonLiesebekannt [14]. In dieser
Arbeit wurden daher nur Untersuchungen zur Reproduktion und zur Gewinnung von Details
uber die Katalysatorpr¨aparation durchgef¨uhrt.
Die Standardversuchsbedingungen beiLiese[14] waren 1000 ppm NO, 1000 ppm CH4, 2 %
O2 und eine Belastung von 10000 h�1. Die hochsten Ums¨atze konnten an dem durch F¨allung
praparierten System mit 64 % X(NO) und 90 % X(CH4) an dem Zeolithen H-ZSM-5 der Fir-
ma Sud Chemie (Si/Al = 14) erzielt werden. Der zweitbeste Katalysator mit dem Zeolithen
H-ZSM-5 T3 der Firma CK Bitterfeld (Si/Al = 19) erreicht Ums¨atze von 62 % X(NO) und
93 % X(CH4), wobei fur beide Katalysatoren das Umsatzmaximum im Temperaturmaximum
von 600ÆC liegt und der Umsatz danach kontinuierlich mit der Temperatur absinkt. Ein wei-
terer aktiver Katalysator kann mit dem Zeolithen NH4-ZSM-5 SM27 (Si/Al = 14) der Firma
Alsi Penta prapariert werden, allerdings liegt der NO-Umsatz mit 47 % X(NO) deutlich unter-
halb der NO-Ums¨atze der besten Katalysatoren. Zudem ist das Umsatzmaximum von 600ÆC
auf 550ÆC verschoben.
Ein erstes Ziel stellt hier die Reproduktion dieser Ergebnisse bzw. eine eventuelle Verbesse-
rung der Ums¨atze dar. F¨ur die beiden getesteten Zeolithe SM27 und T3 (siehe Abbildung 4.1)
ergibt sich eine guteUbereinstimmung mit den Ergebnissen vonLiese: Der T3 erreicht sein
Umsatzmaximum bei 600ÆC mit Werten von 62 % X(NO) und 94 % X(CH4), was in sehr guter
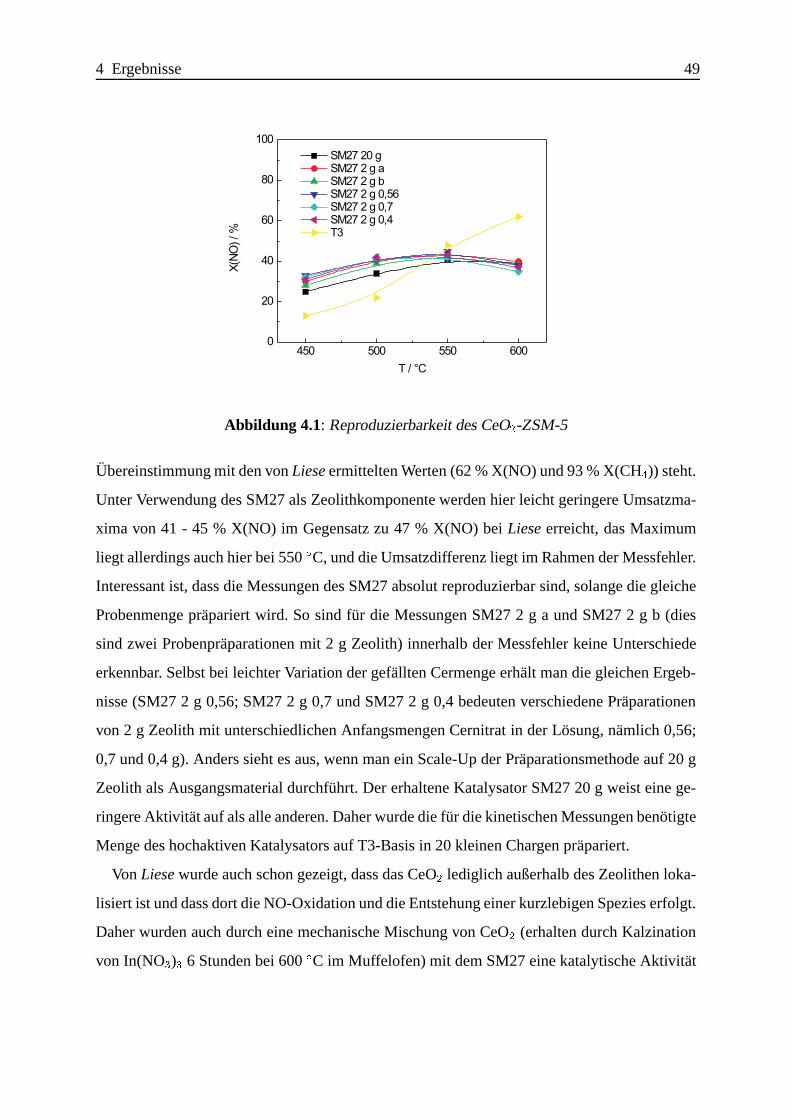
4 Ergebnisse 49
450 500 550 6000
20
40
60
80
100
SM27 20 gSM27 2 g aSM27 2 g bSM27 2 g 0,56SM27 2 g 0,7SM27 2 g 0,4T3
X(N
O)/%
T / °C
Abbildung 4.1: Reproduzierbarkeit des CeO2-ZSM-5
Ubereinstimmung mit den vonLieseermittelten Werten (62 % X(NO) und 93 % X(CH4)) steht.
Unter Verwendung des SM27 als Zeolithkomponente werden hier leicht geringere Umsatzma-
xima von 41 - 45 % X(NO) im Gegensatz zu 47 % X(NO) beiLieseerreicht, das Maximum
liegt allerdings auch hier bei 550ÆC, und die Umsatzdifferenz liegt im Rahmen der Messfehler.
Interessant ist, dass die Messungen des SM27 absolut reproduzierbar sind, solange die gleiche
Probenmenge pr¨apariert wird. So sind f¨ur die Messungen SM27 2 g a und SM27 2 g b (dies
sind zwei Probenpr¨aparationen mit 2 g Zeolith) innerhalb der Messfehler keine Unterschiede
erkennbar. Selbst bei leichter Variation der gef¨allten Cermenge erh¨alt man die gleichen Ergeb-
nisse (SM27 2 g 0,56; SM27 2 g 0,7 und SM27 2 g 0,4 bedeuten verschiedene Pr¨aparationen
von 2 g Zeolith mit unterschiedlichen Anfangsmengen Cernitrat in der L¨osung, n¨amlich 0,56;
0,7 und 0,4 g). Anders sieht es aus, wenn man ein Scale-Up der Pr¨aparationsmethode auf 20 g
Zeolith als Ausgangsmaterial durchf¨uhrt. Der erhaltene Katalysator SM27 20 g weist eine ge-
ringere Aktivitat auf als alle anderen. Daher wurde die f¨ur die kinetischen Messungen ben¨otigte
Menge des hochaktiven Katalysators auf T3-Basis in 20 kleinen Chargen pr¨apariert.
Von Liesewurde auch schon gezeigt, dass das CeO2 lediglich außerhalb des Zeolithen loka-
lisiert ist und dass dort die NO-Oxidation und die Entstehung einer kurzlebigen Spezies erfolgt.
Daher wurden auch durch eine mechanische Mischung von CeO2 (erhalten durch Kalzination
von In(NO3)3 6 Stunden bei 600ÆC im Muffelofen) mit dem SM27 eine katalytische Aktivit¨at
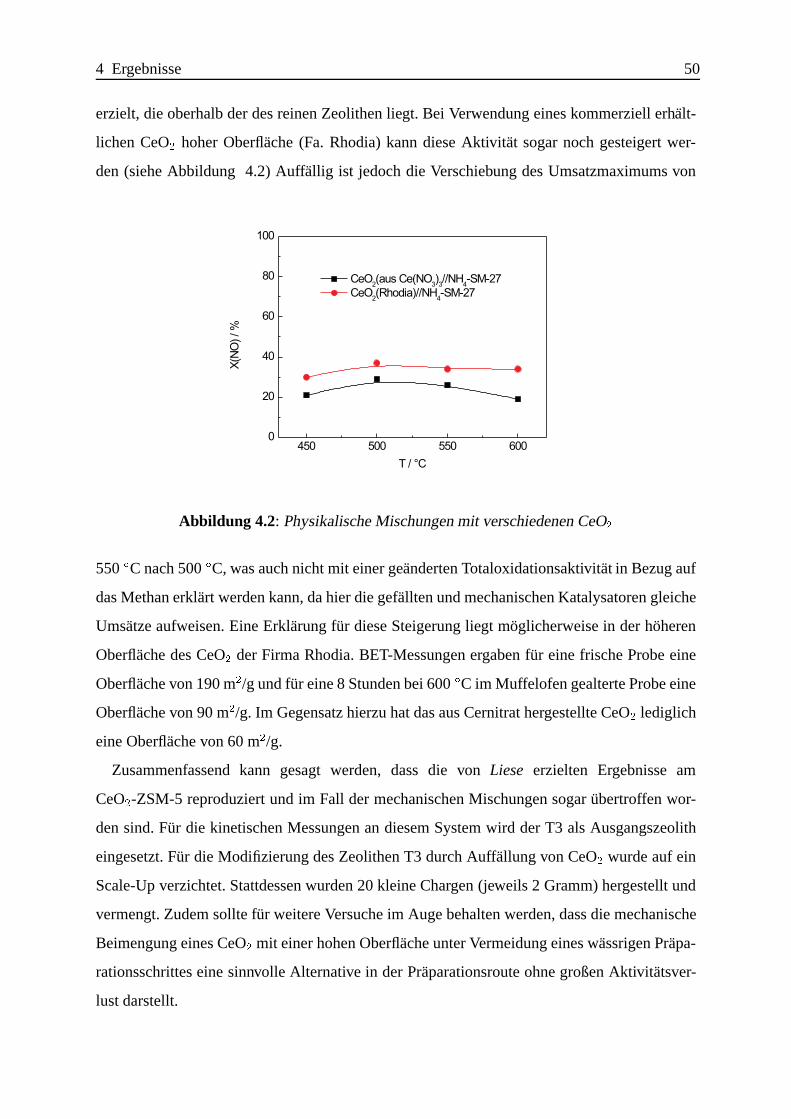
4 Ergebnisse 50
erzielt, die oberhalb der des reinen Zeolithen liegt. Bei Verwendung eines kommerziell erh¨alt-
lichen CeO2 hoher Oberfl¨ache (Fa. Rhodia) kann diese Aktivit¨at sogar noch gesteigert wer-
den (siehe Abbildung 4.2) Auff¨allig ist jedoch die Verschiebung des Umsatzmaximums von
450 500 550 6000
20
40
60
80
100
CeO2(aus Ce(NO
3)3//NH
4-SM-27
CeO2(Rhodia)//NH
4-SM-27
X(N
O)/%
T / °C
Abbildung 4.2: Physikalische Mischungen mit verschiedenen CeO2
550ÆC nach 500ÆC, was auch nicht mit einer ge¨anderten Totaloxidationsaktivit¨at in Bezug auf
das Methan erkl¨art werden kann, da hier die gef¨allten und mechanischen Katalysatoren gleiche
Umsatze aufweisen. Eine Erkl¨arung fur diese Steigerung liegt m¨oglicherweise in der h¨oheren
Oberflache des CeO2 der Firma Rhodia. BET-Messungen ergaben f¨ur eine frische Probe eine
Oberflache von 190 m2/g und fur eine 8 Stunden bei 600ÆC im Muffelofen gealterte Probe eine
Oberflache von 90 m2/g. Im Gegensatz hierzu hat das aus Cernitrat hergestellte CeO2 lediglich
eine Oberflache von 60 m2/g.
Zusammenfassend kann gesagt werden, dass die vonLiese erzielten Ergebnisse am
CeO2-ZSM-5 reproduziert und im Fall der mechanischen Mischungen sogar ¨ubertroffen wor-
den sind. F¨ur die kinetischen Messungen an diesem System wird der T3 als Ausgangszeolith
eingesetzt. F¨ur die Modifizierung des Zeolithen T3 durch Auff¨allung von CeO2 wurde auf ein
Scale-Up verzichtet. Stattdessen wurden 20 kleine Chargen (jeweils 2 Gramm) hergestellt und
vermengt. Zudem sollte f¨ur weitere Versuche im Auge behalten werden, dass die mechanische
Beimengung eines CeO2 mit einer hohen Oberfl¨ache unter Vermeidung eines w¨assrigen Pr¨apa-
rationsschrittes eine sinnvolle Alternative in der Pr¨aparationsroute ohne großen Aktivit¨atsver-
lust darstellt.

4 Ergebnisse 51
4.1.2 Das System In-ZSM-5
Das Grundsystem In-ZSM-5 ist ein vielversprechender Katalysator f¨ur die SCR mit Me-
than. Dies liegt allerdings haupts¨achlich an der Tatsache, dass es sich zusammen mit Edel-
metallen und anderen Promotoren gegen¨uber dem Katalysatorgift Wasser als weniger vergif-
tungsanfallig erweist als andere Systeme. Es existieren viele Untersuchungen f¨ur die Systeme
Cu-ZSM-5, Fe-ZSM-5 und Co-ZSM-5; ¨uber das In-ZSM-5-System selber existiert weniger Li-
teratur (vide supra). Dabei gehen die Autoren davon aus, dass die f¨ur die SCR aktive Spezies ein
Indiumoxokation ist, an dem NO2 reduziert wird, welches selber vorher durch NO-Oxidation
an Lewis-aziden Zentren des Zeolithen entsteht. Die NO-Oxidation kann aber auch von den
Edelmetallen oder anderen Promotoren (z.B. CeO2) ubernommen werden, wobei das CeO2 im
Kompositkatalysator CeO2-ZSM-5 selber Methan aktiviert [101].
Die nachfolgende Untersuchung des In-ZSM-5-Systems hat das Ziel, auf verschiedenen
Praparationswegen gezielt unterschiedliche In-Strukturen herzustellen und durch eine Zuord-
nung der Struktur zu den katalytischen Eigenschaften eine Aussage ¨uber die katalytisch aktive
Spezies zu treffen.
4.1.2.1 Definition der Probenbezeichnungen
Um denUberblick uber die Katalysatorproben zu erleichtern, werden zuerst kurz die Proben-
bezeichnungen erl¨autert. Die angewandten Pr¨aparationsmethoden untergliedern sich in nasse
und trockene Methoden, deren genaue Ausf¨uhrung dem Kapitel 3.1.3 zu entnehmen ist. Die
durch Ionentausch hergestellten Proben erhalten die Bezeichnung IE (IE = Ion Exchange), wo-
bei als weiteres K¨urzel der wahrend der Pr¨aparation durch HNO3 eingestellte pH-Wert im Na-
men enthalten ist. Demnach ergeben sich die Proben IE-1.5, IE-2.5, IE-6 und IEn (n = nat¨urli-
cher pH, der bei Verwendung von deionisiertem Wasser ca. 6,5 betr¨agt). Alle Proben wurden
mit dem Zeolithen NH4-ZSM-5 (SM27) der Firma Alsi Penta (Si/Al = 14) hergestellt. Bei
einer Verwendung der H-Form, die durch Kalzination der Ammoniumform in synthetischer
Luft erhalten wurde (2 K min�1 auf 120ÆC, 10 min halten, 5 K min�1 auf 600ÆC, 2 Stun-
den halten), wird dies durch ein (H) gekennzeichnet. Ein P (P = Precipitation) kennzeich-
net die durch F¨allung hergestellten Proben und IE+P stellt die Bezeichnung f¨ur Ionentausch
mit anschließender F¨allung dar. Bei den trockenen Pr¨aparationsmethoden wird zwischen SSIE
(SSIE = Solid State Ion Exchange), S (S = Sublimation) und T (T = Transport Reaction) un-
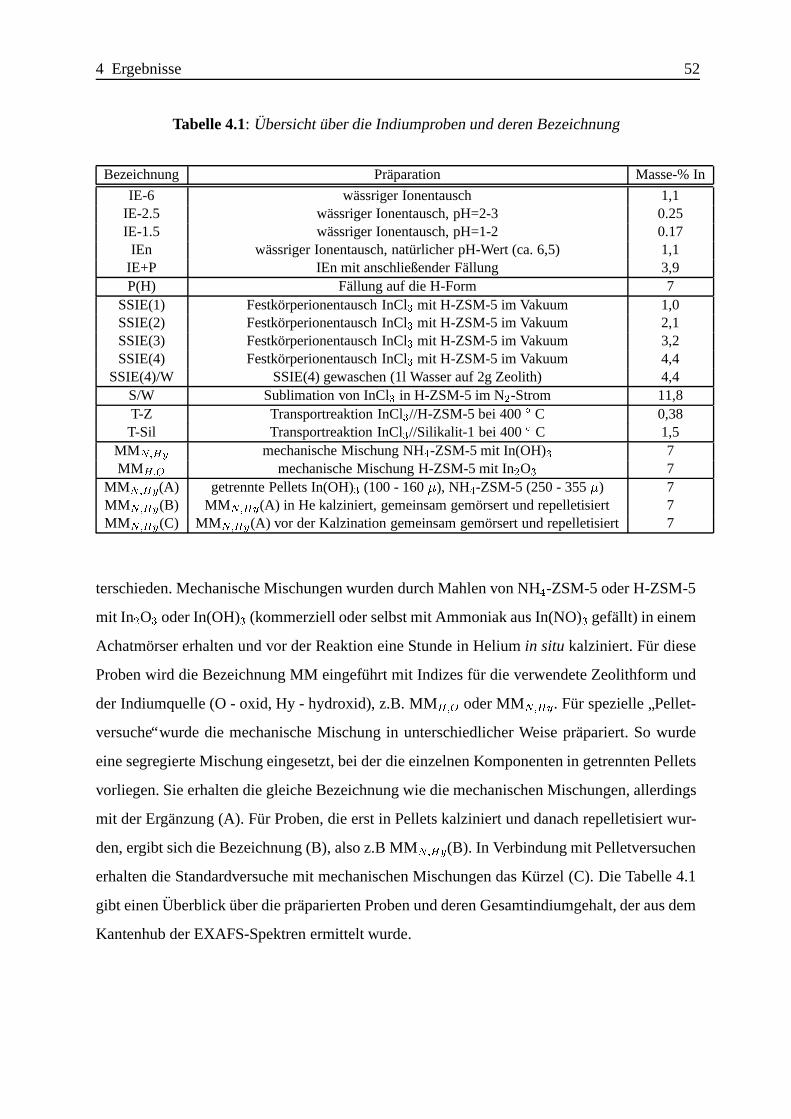
4 Ergebnisse 52
Tabelle 4.1: Ubersichtuber die Indiumproben und deren Bezeichnung
Bezeichnung Praparation Masse-% In
IE-6 wassriger Ionentausch 1,1IE-2.5 wassriger Ionentausch, pH=2-3 0.25IE-1.5 wassriger Ionentausch, pH=1-2 0.17IEn wassriger Ionentausch, nat¨urlicher pH-Wert (ca. 6,5) 1,1
IE+P IEn mit anschließender F¨allung 3,9P(H) Fallung auf die H-Form 7
SSIE(1) Festkorperionentausch InCl3 mit H-ZSM-5 im Vakuum 1,0SSIE(2) Festkorperionentausch InCl3 mit H-ZSM-5 im Vakuum 2,1SSIE(3) Festkorperionentausch InCl3 mit H-ZSM-5 im Vakuum 3,2SSIE(4) Festkorperionentausch InCl3 mit H-ZSM-5 im Vakuum 4,4
SSIE(4)/W SSIE(4) gewaschen (1l Wasser auf 2g Zeolith) 4,4S/W Sublimation von InCl3 in H-ZSM-5 im N2-Strom 11,8T-Z Transportreaktion InCl3//H-ZSM-5 bei 400Æ C 0,38T-Sil Transportreaktion InCl3//Silikalit-1 bei 400Æ C 1,5
MMN;Hy mechanische Mischung NH4-ZSM-5 mit In(OH)3 7MMH;O mechanische Mischung H-ZSM-5 mit In2O3 7
MMN;Hy(A) getrennte Pellets In(OH)3 (100 - 160�), NH4-ZSM-5 (250 - 355�) 7MMN;Hy(B) MMN;Hy(A) in He kalziniert, gemeinsam gem¨orsert und repelletisiert 7MMN;Hy(C) MMN;Hy(A) vor der Kalzination gemeinsam gem¨orsert und repelletisiert 7
terschieden. Mechanische Mischungen wurden durch Mahlen von NH4-ZSM-5 oder H-ZSM-5
mit In2O3 oder In(OH)3 (kommerziell oder selbst mit Ammoniak aus In(NO)3 gefallt) in einem
Achatmorser erhalten und vor der Reaktion eine Stunde in Heliumin situ kalziniert. Fur diese
Proben wird die Bezeichnung MM eingef¨uhrt mit Indizes fur die verwendete Zeolithform und
der Indiumquelle (O - oxid, Hy - hydroxid), z.B. MMH;O oder MMN;Hy. Fur spezielle”Pellet-
versuche“wurde die mechanische Mischung in unterschiedlicher Weise pr¨apariert. So wurde
eine segregierte Mischung eingesetzt, bei der die einzelnen Komponenten in getrennten Pellets
vorliegen. Sie erhalten die gleiche Bezeichnung wie die mechanischen Mischungen, allerdings
mit der Erganzung (A). F¨ur Proben, die erst in Pellets kalziniert und danach repelletisiert wur-
den, ergibt sich die Bezeichnung (B), also z.B MMN;Hy(B). In Verbindung mit Pelletversuchen
erhalten die Standardversuche mit mechanischen Mischungen das K¨urzel (C). Die Tabelle 4.1
gibt einenUberblickuber die praparierten Proben und deren Gesamtindiumgehalt, der aus dem
Kantenhub der EXAFS-Spektren ermittelt wurde.

4 Ergebnisse 53
Tabelle 4.2: EXAFS- und XPS-Ergebnisse f¨ur die Indiumproben
Bezeichnung Struktur der Indiumkomponente
IE-6 extra- und intrazeolithisches IndiumIE-2.5 ausschließlich intrazeolithisch, aber eine andere Koordination als in IE-6IE-1.5 intrazeolithisches Indium mit unbekannter KoordinationIEn extra- und intrazeolithisches Indium wie in IE6
IE+P extra- und intrazeolithisches Indium wie in IE6P(H) extra- und intrazeolithisches Indium
SSIE(1) ausschließlich intrazeolithisches Indium,�3,5 O bei 2,16A/�2,5Cl bei 2,36ASSIE(2) ausschließlich intrazeolithisches Indium,�3,5 O bei 2,16A/�2,5Cl bei 2,36ASSIE(3) ausschließlich intrazeolithisches Indium,�3,5 O bei 2,16A/�2,5Cl bei 2,36ASSIE(4) ausschließlich intrazeolithisches Indium,�3,5 O bei 2,16A/�2,5Cl bei 2,36A
SSIE(4)/W ausschließlich intrazeolithisches Indium,�5 O bei 2,16A/�1Cl bei 2,36AS/W ausschließlich intrazeolithisches Indium,�3,5 O bei 2,16A/�2,5Cl bei 2,36AT-Za ausschließlich intrazeolithisches Indium, kein ChlorT-Sila intra- und extrazeolithisches Indium, kein Chlor
anach der Katalyse
4.1.2.2 Charakterisierungsergebnisse
Die Charakterisierung erfolgte haupts¨achlich durch EXAFS- und XPS-Messungen in der Ar-
beit von C. Schmidt [15] (siehe auch [102]). Diese Ergebnisse werden hier in Tabellenform
zitiert (siehe Tabelle 4.2). Dar¨uberhinaus werden auch eigene mittels DRIFTS, TPR und XRD
erhaltene Charakterisierungsergebnisse vorgestellt.
Ziel deswassrigen Ionentauschsist der Eintausch von Kationen an die Austauschpositionen
des Zeolithen, um ausschließlich intrazeolitische Spezies zu generieren. Bei den beiden Pro-
ben IEn und IE-6 entsteht neben intrazeolithischem Indium allerdings auch extrazeolithisches
Indium. In der Literatur wird beschrieben, dass es beim Ionentausch mit vorzugsweise h¨oher-
wertigen Kationen zu Problemen kommen kann. Um ein Verstopfen der Zeolithporen zu ver-
meiden, sollte die Metallsalzkonzentration kleiner als 0,5-molar sein, was in unserem Fall mit
0,013-molar erf¨ullt ist [103]. Besonders schwierig erweist sich der Eintausch von trivalenten
Ionen [104–106], wobei der Eintausch sterisch durch die Gr¨oße des solvatisierten Kations als
auch durch elektrostatische Wechselwirkungen mit dem hydrophoben Gitter des Zeolithen be-
hindert wird. Bei geringen Metallsalzkonzentrationen k¨onnen noch Metallsalzhydroxidionen
wie [Me(OH)2]+ eingetauscht werden, bei h¨oheren Konzentrationen bilden sich allerdings po-

4 Ergebnisse 54
lynukleare Kationen, die zu groß f¨ur den Prozess des Ionentauschs sind und die zur Auff¨allung
der Hydroxide fuhren [107,108]. Um ein Ausf¨allen der Hydroxide zu verhindern, wurde der
pH-Wert der Metallsalzl¨osung durch Salpeters¨aure variiert, da der Solvatisierungsgrad multi-
valenter Kationen der Hauptgruppenelemente mit dem pH-Wert abnimmt und im stark sauren
Bereich nur noch isolierte Kationen vorliegen sollten. So liegen denn in den Proben IE-2.5 und
IE-1.5 wirklich nur intrazeolithische Indiumspezies vor. Das Absinken des Indiumgehalts mit
dem pH-Wert ist signifikant und l¨asst sich mit einer Verschiebung des Austauschgleichgewichts
in der Losung erklaren.
Bei derFallung soll die Indiumkomponente ausschließlich außerhalb des Zeolithen aufge-
bracht werden. In den EXAFS- und XPS-Untersuchungen konnte jedoch gezeigt werden, dass
sowohl intra- als auch extrazeolithisches Indium vorliegt. Best¨atigt wird dieses Ergebnis von am
ACA gemessenen TPR-Ergebnissen, die sowohl ein Tieftemperatursignal (� 250ÆC) als auch
ein Hochtemperatursignal (�480ÆC) aufweisen. Eine Zuordnung des Tieftemperatursignals
erfolgt zur Reduktion redoxaktiver InO+-Kationen auf zeolithischen Kationenpositionen; das
Hochtemperatursignal spiegelt die Reduktion von oxidischem In(III) an der ¨außeren Oberfl¨ache
des Zeolithen wider [109–111]. Interessant ist, dass sowohl f¨ur die Fallung auf die H-Form, als
auch fur die Fallung auf die Ammoniumform des Zeolithen die beiden Indiumformen erhalten
werden.
Bei der Ammoniumform kann als Erkl¨arung der sogenannte reduktive Festk¨orperionen-
tausch herangezogen werden. Hier wirkt das bei der Kalzination in dem Inertgas Helium freige-
setzte Ammoniak als Reduktionsmittel und reduziert In(III) zu In(I), welches in den Zeolithen
migrieren und dort Protonen austauschen kann. Bei Kontakt mit Luftsauerstoff erfolgt leicht
eine Oxidation des In+ zu InO+. Das gleiche Ph¨anomen ist als templatinduzierter reduktiver
Festkorperionentausch beobachtet worden, bei dem ein zur Synthese ben¨otigtes organisches
Templat zum reduktiven Festk¨orperionentausch f¨uhrt [112]. Eine weitere m¨ogliche Erklarung
liegt darin, dass der Zeolith durch das bei der F¨allung verwendete Ammoniak trotz z¨ugigen Ab-
nutschens teilweise in die Ammoniumform ¨uberfuhrt wurde oder aber auch nur Ammoniak an
dem gefallten Indiumhydroxid oder an den Protonen adsorbierte und unter Inertgasbedingun-
gen also doch ein reduktiver Festk¨orperionentausch erfolgen konnte. In diesem Zusammenhang
ist der vonKanazirevet al. eingef¨uhrte reduktive Festk¨orperionentausch von Interesse. Eine
mechanische Mischung von In2O3 und H-Zeolith wird im Wasserstoffstrom bis 500ÆC erhitzt,

4 Ergebnisse 55
wobei erst das Indium(III) zu In(I) reduziert wird und dieses in den Zeolithen einwandert und
mit den Protonen reagieren kann [113–115]. Hiermit l¨asst sich eine homogene Verteilung ¨uber
den gesamten Zeolithkritall erreichen, wie eine Schichtenanalyse eines 140 x 50 x 50�m Mo-
nokristalls zeigte, bei dem in einzelnen Schritten eine jeweils 15�m dicke Schicht wegpoliert
und mit Hilfe von EDX (Energy Dispersive Analysis of X-Rays) analysiert wurde. Bei diesem
Verfahren ist allerdings eine von außen zugef¨uhrte reduzierende Komponente entscheidend.
Diese Praparationsmethode wurde von unserem Kooperationspartner ACA in Berlin weiter ver-
folgt.
Allerdings ist auch in die H-Form des Zeolithen Indium eingetauscht worden, was schon
vonOguraet al. fur eine Mischung aus H-Zeolith und In2O3 beobachtet und als autoreduktiver
Festkorperionentausch bezeichnet worden ist [46].
Mit Hilfe desFestkorperionentauschs SSIEsollen ausschließlich intrazeolithische Indium-
spezies generiert werden. Dies geht zur¨uck aufRaboet al., die schon im Jahr 1973 ¨uber den
Festkorperionentausch zwischen mechanischen Mischungen aus Metalloxiden und -chloriden
mit Zeolithen sowohl der H-Form als auch der NH4-Form berichteten [116]. Hierbei erfolgt
ein Eintausch des ladungskompensierenden Kations im Zeolithen gegen das Kation der Reak-
tionskomponente unter Freisetzung einer fl¨uchtigen Reaktionskomponente wie HCl oder H2O.
Mittlerweile ist sogar eine Reaktion von metallischem Zink mit den brønstedsauren Zentren
von Faujasiten und Mordeniten bekannt, wobei der Eintauschgrad mit Hilfe der Wasserstoffent-
wicklung wahrend der Festk¨orperreaktion verfolgt werden kann [117]. F¨ur den Eintausch von
Indium wurde vonKikuchiet al. gefunden, dass oberhalb von 500ÆC ein Festk¨orperionentausch
zwischen In2O3 und sauren Gitterprotonen stattfindet, der ¨uber IR- und XPS-Spektroskopie
nachgewiesen werden kann [46]. Die Mobilit¨at der Indiumspezies wird aber nur dem einwerti-
gen Indium(I) zugeschrieben, das nach Reaktion mit den Protonen des Zeolithen leicht zu der
katalytisch aktiven und koordinativ unges¨attigten Spezies InO+ oxidiert werden kann.
In dieser Arbeit wird InCl3 als Indiumlieferant f¨ur den SSIE gew¨ahlt; Mechanische Mischun-
gen mit In2O3 oder In(OH)3 werden gesondert behandelt. Die theoretische Austauschmenge an
Indium liegt bei 4 Massenprozent (In/Al = 1/3). Daher werden in einer Konzentrationsreihe
Katalysatoren mit 25, 50, 75 und 100 % Austauschgrad respektive 1, 2, 3 und 4 Massenpro-
zent Indium prapariert, indem die entsprechende Menge InCl3 mit H-ZSM-5 im Vakuum auf
550ÆC erhitzt wird. Die durch SSIE erhaltenen Katalysatoren enthalten ausschließlich intra-

4 Ergebnisse 56
zeolithische Indiumspezies, allerdings enth¨alt das Indium in seiner ersten Koordinationssph¨are
außer Sauerstoff noch Chlor. Durch Waschen mit entionisiertem Wasser konnte ein Teil des
Chlors entfernt werden, ohne den Indiumgehalt zu verringern. Somit ist das Indium in den
ungewaschenen Proben von 3,5 O-Atomen und 2,5 Cl-Atomen koordiniert, w¨ahrend die gewa-
schene Probe Werte von 5 O-Atomen und 1 Cl-Atom aufweist. Die durch XP-Spektren ermit-
telten hohen In 3d5=2 Bindungsenergien von 446 eV m¨ussen aber nicht zwangsl¨aufig intrazeoli-
thischen Indiumspezies zugeordnet werden, da das InCl3 selber eine hohe Bindungsenergie auf-
weist. Erst die weitere Entfernung von Chlor aus der Probe durch Waschen mit entionisiertem
Wasser in Verbindung mit einer konstantbleibenden hohen Bindungsenergie liefert den Beweis,
dass diese intrazeolithischen Indiumspezies und nicht eventuell zur¨uckgebliebenem InCl3 zu-
zuordnen ist. Aber die EXAFS-Ergebnisse mit fehlenden In-In-Abst¨anden haben schon vorher
die Anwesenheit intrazeolithischer Indiumspezies bewiesen.
Die durchSublimation (S = Sublimation) dargestellte Probe ist in Bezug auf den Indiumge-
halt uberausgetauscht, wenn ein In/Al-Verh¨altnis von 1/3 angenommen wird. Ein 100 %iger
Austausch wird theoretisch mit 4 Massenprozent Indium erreicht. Da aber auch hier wie
bei den SSIE-Proben noch Chlor in der Koordinationssph¨are des Indiums gefunden wird, ist
es wahrscheinlich, dass InCl3�xx+-Spezies an den Brønsted-Zentren des Zeolithen oder so-
gar InCl3+x x�-Spezies an den Lewis-Zentren des Zeolithen eingebaut werden. Damit ist es
moglich, bei einem In/Al-Verh¨altnis von 1/1 bis zu 12 Massenprozent Indium einzubringen,
was mit dem gefundenen Wert von 11,8 beinahe erreicht wird. Trotzdem liegt eine rein intra-
zeolithische Indiumspezies vom Typ InClxOy vor.
Fur die durch Festk¨orperionentausch und Sublimation hergestellten Proben liegen auch
DRIFT-Untersuchungen vor, mit denen der Einfluss eines Indiumeintauschs auf die vom Zeo-
lithen bereitgestellten OH-Gruppen verfolgt werden kann. Abbildung 4.3 vergleicht die Region
der OH-Schwingungen des H-ZSM-5 mit den Proben SSIE(1), SSIE(4), S/W und S. In der
Abbildung 4.3 zeigt der H-ZSM-5 die f¨ur ihn typischen Bandenlagen f¨ur die Brønstedzentren
(3606 cm�1), die terminalen Al-OH-Gruppen an Extrager¨ustaluminium (3775 cm�1), die Si-
lanolgruppen in Wechselwirkung mit Extragitteraluminium oder Si-OH-Gruppen in Silanol-
nestern (3725 cm�1) und Si-OH-Gruppen an der ¨außeren Kristalloberfl¨ache (3740 cm�1).
Durch Einbringen von Indium mit Hilfe des Festk¨orperionentauschs oder der Sublimation
verliert die Bande der Brønstedzentren (3606 cm�1) proportional (nur qualitativ) zur einge-
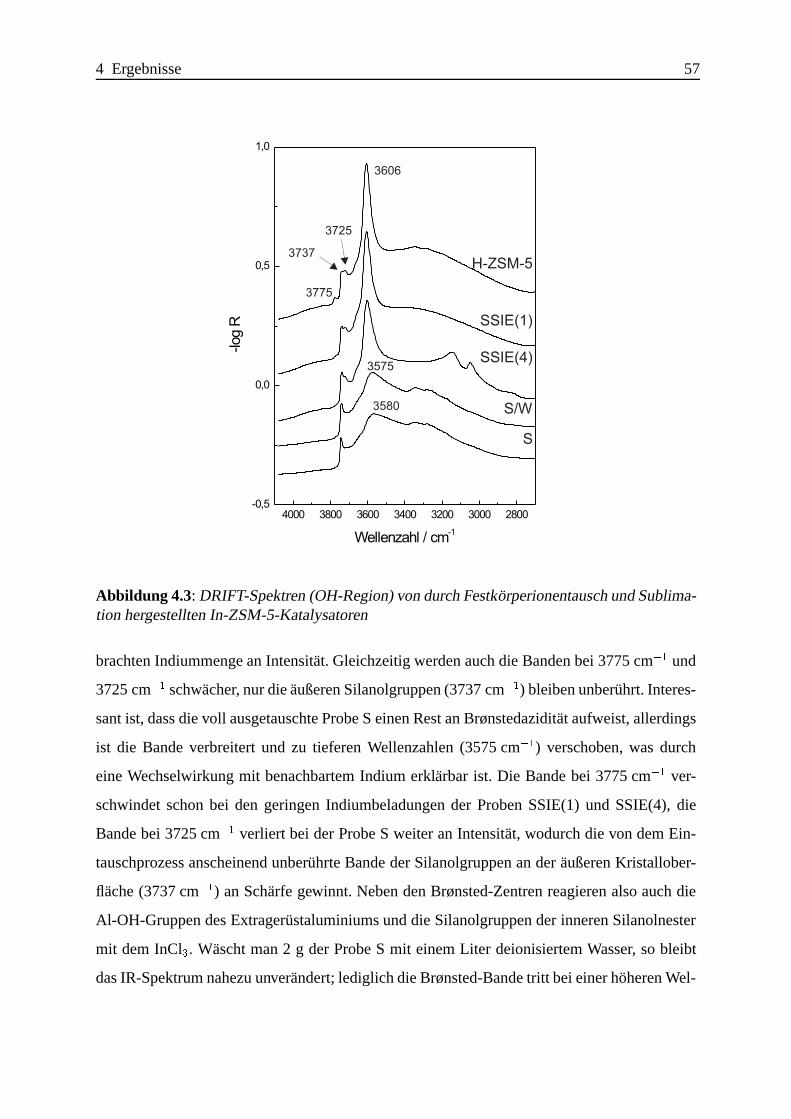
4 Ergebnisse 57
4000 3800 3600 3400 3200 3000 2800-0,5
0,0
0,5
1,0
-log
R
Wellenzahl / cm-1
H-ZSM-5
SSIE(1)
SSIE(4)
S/W
S
3606
3725
3737
3775
3575
3580
Abbildung 4.3: DRIFT-Spektren (OH-Region) von durch Festk¨orperionentausch und Sublima-tion hergestellten In-ZSM-5-Katalysatoren
brachten Indiummenge an Intensit¨at. Gleichzeitig werden auch die Banden bei 3775 cm�1 und
3725 cm�1 schwacher, nur die ¨außeren Silanolgruppen (3737 cm�1) bleiben unber¨uhrt. Interes-
sant ist, dass die voll ausgetauschte Probe S einen Rest an Brønstedazidit¨at aufweist, allerdings
ist die Bande verbreitert und zu tieferen Wellenzahlen (3575 cm�1) verschoben, was durch
eine Wechselwirkung mit benachbartem Indium erkl¨arbar ist. Die Bande bei 3775 cm�1 ver-
schwindet schon bei den geringen Indiumbeladungen der Proben SSIE(1) und SSIE(4), die
Bande bei 3725 cm�1 verliert bei der Probe S weiter an Intensit¨at, wodurch die von dem Ein-
tauschprozess anscheinend unber¨uhrte Bande der Silanolgruppen an der ¨außeren Kristallober-
flache (3737 cm�1) an Scharfe gewinnt. Neben den Brønsted-Zentren reagieren also auch die
Al-OH-Gruppen des Extrager¨ustaluminiums und die Silanolgruppen der inneren Silanolnester
mit dem InCl3. Wascht man 2 g der Probe S mit einem Liter deionisiertem Wasser, so bleibt
das IR-Spektrum nahezu unver¨andert; lediglich die Brønsted-Bande tritt bei einer h¨oheren Wel-
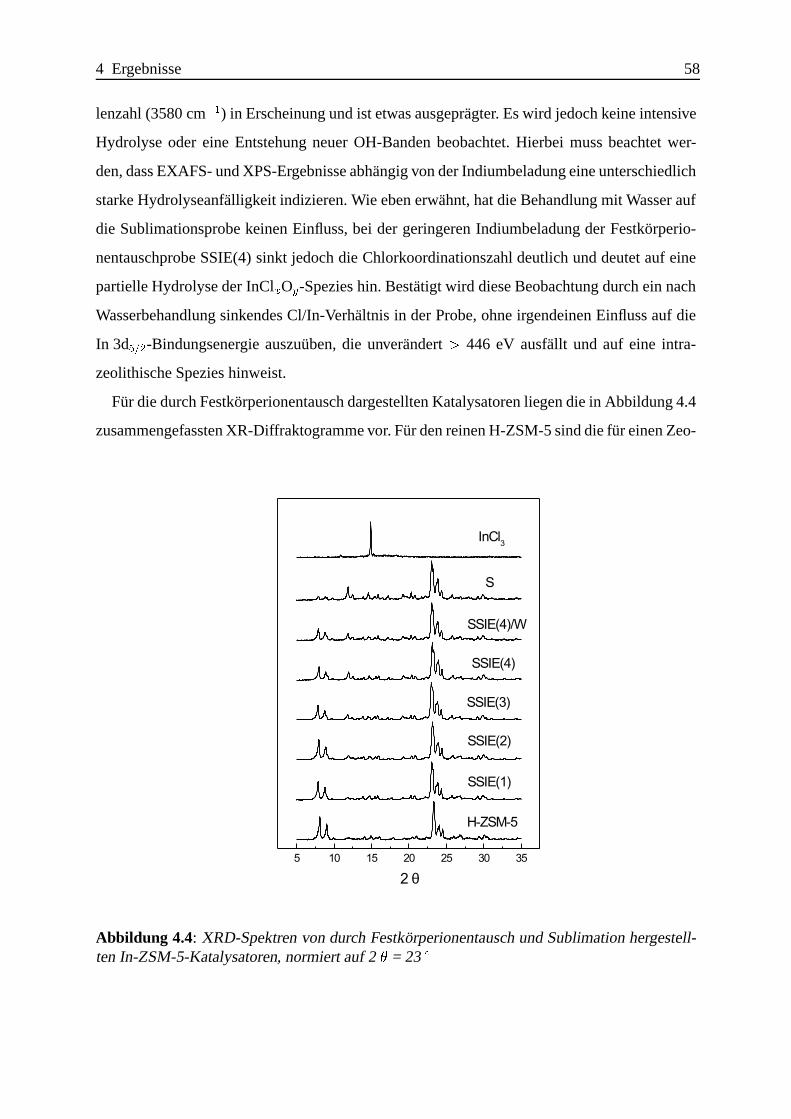
4 Ergebnisse 58
lenzahl (3580 cm�1) in Erscheinung und ist etwas ausgepr¨agter. Es wird jedoch keine intensive
Hydrolyse oder eine Entstehung neuer OH-Banden beobachtet. Hierbei muss beachtet wer-
den, dass EXAFS- und XPS-Ergebnisse abh¨angig von der Indiumbeladung eine unterschiedlich
starke Hydrolyseanf¨alligkeit indizieren. Wie eben erw¨ahnt, hat die Behandlung mit Wasser auf
die Sublimationsprobe keinen Einfluss, bei der geringeren Indiumbeladung der Festk¨orperio-
nentauschprobe SSIE(4) sinkt jedoch die Chlorkoordinationszahl deutlich und deutet auf eine
partielle Hydrolyse der InClxOy-Spezies hin. Best¨atigt wird diese Beobachtung durch ein nach
Wasserbehandlung sinkendes Cl/In-Verh¨altnis in der Probe, ohne irgendeinen Einfluss auf die
In 3d5=2-Bindungsenergie auszu¨uben, die unver¨andert> 446 eV ausf¨allt und auf eine intra-
zeolithische Spezies hinweist.
Fur die durch Festk¨orperionentausch dargestellten Katalysatoren liegen die in Abbildung 4.4
zusammengefassten XR-Diffraktogramme vor. F¨ur den reinen H-ZSM-5 sind die f¨ur einen Zeo-
5 10 15 20 25 30 35
S
InCl3
SSIE(4)/W
SSIE(4)
SSIE(3)
SSIE(2)
SSIE(1)
H-ZSM-5
2 θ
Abbildung 4.4: XRD-Spektren von durch Festk¨orperionentausch und Sublimation hergestell-ten In-ZSM-5-Katalysatoren, normiert auf 2� = 23 Æ

4 Ergebnisse 59
lithen des MFI-Typs charakteristischen Beugungsreflexe bei 2� von ungefahr 8Æund 9Æ, und
die bei 23Æ, 24 Æund 24,5Æzu erkennen; Beugungsreflexe anderer Zeolithtypen treten nicht
auf [118]. Bei Betrachtung der Diffraktogramme der Proben SSIE(1) bis SSIE(4) f¨allt auf, dass
alle Reflexe noch vorhanden sind, sich jedoch Verschiebungen in den Intensit¨aten ergeben. Be-
sonders auff¨allig ist dies fur zwei Reflexe bei 2� � 13Æ, die in dem reinen H-ZSM-5 nur erahnt
werden konnen, dann aber mit steigendem Indiumgehalt an Intensit¨at gewinnen. F¨ur die gewa-
schene Probe SSIE(4)/W geht die Intensit¨at etwas zur¨uck, obwohl der Indiumgehalt nicht sinkt.
Dies kann aber mit einer Ver¨anderung der Indiumspezies durch Chlorverlust erkl¨art werden,
der sich auch auf das Gitter des Zeolithen auswirken kann. Bei dem durch Sublimation herge-
stellten Katalysator (hier handelt es sich um eine ungewaschene und im Vakuum sublimierte
Probe) ist der Effekt der Intensit¨atsverschiebung so deutlich ausgepr¨agt, dass man die charak-
teristischen Reflexe bei 2� � 8 Æund 9Ænur noch schwer erkennt, die bei 2� � 13 Æhingegen
deutlich hervortreten. Durch Vergleich mit den JCPDS-Daten l¨asst sich kein Reflex außer denen
des ZSM-5 erkennen; es ist also kein Kristall einer Indiumchlorid- oder Indiumoxid- oder In-
diumoxochloridphase mit einer f¨ur die Identifikation durch XRD ausreichenden Kristallitgr¨oße
vorhanden. Gleichzeitig wird festgestellt, dass die Pr¨aparationsmethoden der Sublimation und
des Festk¨orperionentauschs, bei denen jeweils HCl als fl¨uchtige Komponente frei wird, das
Gerust des Zeolithen nicht zerst¨oren. Die Veranderung der relativen Reflexintensit¨aten ist ein
Zeichen fur den Eintausch eines schweren Elements (hier Indium) in die Zeolithstruktur [18];
allerdings sind Reflexintensit¨aten mit Vorsicht zu genießen, da sie auch durch die Probenpr¨apa-
ration vor der Messung beeinflusst werden. Da die Probenpr¨aparation immer von der gleichen
Person durchgef¨uhrt wurde und damit eventuelle Fehler sytematisch sind und weiterhin der ge-
fundene Trend mit der Indiumkonzentration eindeutig ist, k¨onnen die durch die Probenpr¨apa-
ration auftretenden Effekte als zu vernachl¨assigend betrachtet werden.
Die Transportproben T-Z (H-ZSM-5 als Ausgangsmaterial) und T-Sil (Silikalit-1 als Aus-
gangsmaterial) wurden unter Ausschluss von Luft in einer Glove-Box pr¨apariert, um eine Hy-
drolyse der zweiten Ausgangsverbindung InCl3 zu vermeiden. Dabei handelt es sich um eine
rein akademische Pr¨aparationsmethode; f¨ur eine technische Anwendung w¨are sie zu kosten-
intensiv. Die Reaktionstemperatur wird bei dieser Art der Pr¨aparation unter 400ÆC gehalten,
um einen isomorphen Einbau von Indiumspezies in das Gitter des Zeolithen bzw. Silikaliten
(Einbau in die tetraedrischen Defektpl¨atze, Silanolnester) zu verhindern, der vonYamagishiet

4 Ergebnisse 60
al. bei Temperaturen von 650ÆC fur die Elemente Aluminium, Gallium und Indium beobachtet
und von ihnen alsAtom Planting Methodbezeichnet wurde [119]. Durch die Transportreaktion
wird fur den H-ZSM-5 ein Einbau von Indium auf Kationenpl¨atzen erwartet. F¨ur den Silika-
liten kann wegen des Fehlens von Brønstedzentren eine Einlagerung des Indiumchloridmo-
lekuls als Ganzes in die Poren erwartet werden [120]. Außerdem sollte bei Einbau von Indium
in die Defektstellen des Silikaliten die Erzeugung weiterer Lewiszentren m¨oglich sein. Da in
dem Silikaliten keine Brønstedzentren vorhanden sind, geben m¨ogliche Unterschiede oder Ge-
meinsamkeiten im katalytischen Verhalten unter der Bedingung ¨ahnlich eingebauter Indium-
spezies Informationen ¨uber die Rolle der Brønstedzentren in der SCR von NO mit Methan. Die
durch EXAFS und XPS gewonnenen Daten f¨uhren zu dem Schluss, dass ausschließlich intra-
zeolithische Indiumspezies vorliegen. Die In 3d5=2- Bindungsenergien liegen nahe bei 446 eV
und weisen auf intrazeolithische Spezies hin. Die eingetauschte Indiummenge ist jedoch im
Vergleich zu den Proben aus Sublimation und Festk¨orperionentausch mit 0,34 Massenprozent
fur den T-Z bzw. mit 1,5 Massenprozent f¨ur den T-Sil sehr gering. Gr¨unde fur diesen geringen
Eintauschgrad sind m¨oglicherweise die niedrige Reaktionstemperatur und die Pr¨aparationsme-
thode in einem geschlossenen System, bei dem das entstehende HCl nicht abgef¨uhrt werden
kann. So kann es zu einem Austauschgleichgewicht ¨ahnlich dem des w¨assrigen Ionentauschs
kommen, wobei in letzterem Fall aber die w¨assrige Indiumnitratl¨osung dreimal erneuert wird.
Rontgendiffraktogramme der Proben zeigen die f¨ur einen Eintausch charakteristische Verschie-
bung der Reflexintensit¨atsverhaltnisse und DRIFT-Spektren best¨atigen den Einbau von Indium-
spezies an die Brønstedzentren des H-ZSM-5 bzw. in die Defektstellen des Silikaliten [15].
Fur die durchmechanische Mischunghergestellten Indiumproben liegen keine Ergebnisse
mittels EXAFS vor. An geeigneter Stelle werden die an diesen Proben erhaltenen katalytischen
Ergebnisse durch TPR-Messungen weiter analysiert.
4.1.2.3 Katalytische Ergebnisse
Soweit nicht anders angegeben, sind f¨ur die nachfolgenden Versuche die Standardreaktions-
bedingungen 1000 ppm NO, 1000 ppm CH4, 2 % O2 und eine Belastung von 30000 h�1
eingestellt worden. In Abbildung 4.5 werden die mit den verschiedenen pr¨aparierten Kata-
lysatoren erhaltenen Ums¨atze fur Methan und NO vorgestellt und mit den an einem reinen
H-ZSM-5 erzielten Ums¨atzen verglichen. Die NO-Ums¨atze sind f¨ur alle Katalysatoren nicht
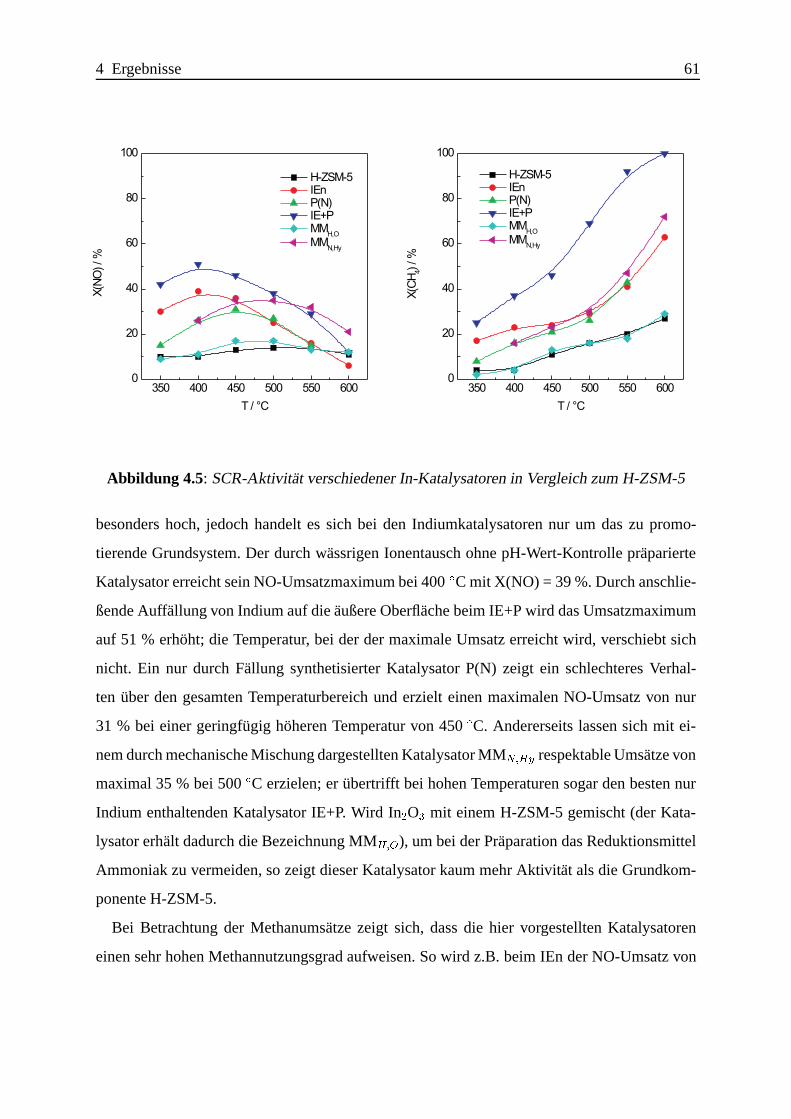
4 Ergebnisse 61
350 400 450 500 550 6000
20
40
60
80
100
H-ZSM-5IEnP(N)IE+PMM
H,O
MMN,Hy
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
H-ZSM-5IEnP(N)IE+PMM
H,O
MMN,Hy
X(C
H4)
/%
T / °C
Abbildung 4.5: SCR-Aktivitat verschiedener In-Katalysatoren in Vergleich zum H-ZSM-5
besonders hoch, jedoch handelt es sich bei den Indiumkatalysatoren nur um das zu promo-
tierende Grundsystem. Der durch w¨assrigen Ionentausch ohne pH-Wert-Kontrolle pr¨aparierte
Katalysator erreicht sein NO-Umsatzmaximum bei 400ÆC mit X(NO) = 39 %. Durch anschlie-
ßende Auffallung von Indium auf die ¨außere Oberfl¨ache beim IE+P wird das Umsatzmaximum
auf 51 % erh¨oht; die Temperatur, bei der der maximale Umsatz erreicht wird, verschiebt sich
nicht. Ein nur durch F¨allung synthetisierter Katalysator P(N) zeigt ein schlechteres Verhal-
ten uber den gesamten Temperaturbereich und erzielt einen maximalen NO-Umsatz von nur
31 % bei einer geringf¨ugig hoheren Temperatur von 450ÆC. Andererseits lassen sich mit ei-
nem durch mechanische Mischung dargestellten Katalysator MMN;Hy respektable Ums¨atze von
maximal 35 % bei 500ÆC erzielen; er ¨ubertrifft bei hohen Temperaturen sogar den besten nur
Indium enthaltenden Katalysator IE+P. Wird In2O3 mit einem H-ZSM-5 gemischt (der Kata-
lysator erhalt dadurch die Bezeichnung MMH;O), um bei der Pr¨aparation das Reduktionsmittel
Ammoniak zu vermeiden, so zeigt dieser Katalysator kaum mehr Aktivit¨at als die Grundkom-
ponente H-ZSM-5.
Bei Betrachtung der Methanums¨atze zeigt sich, dass die hier vorgestellten Katalysatoren
einen sehr hohen Methannutzungsgrad aufweisen. So wird z.B. beim IEn der NO-Umsatz von
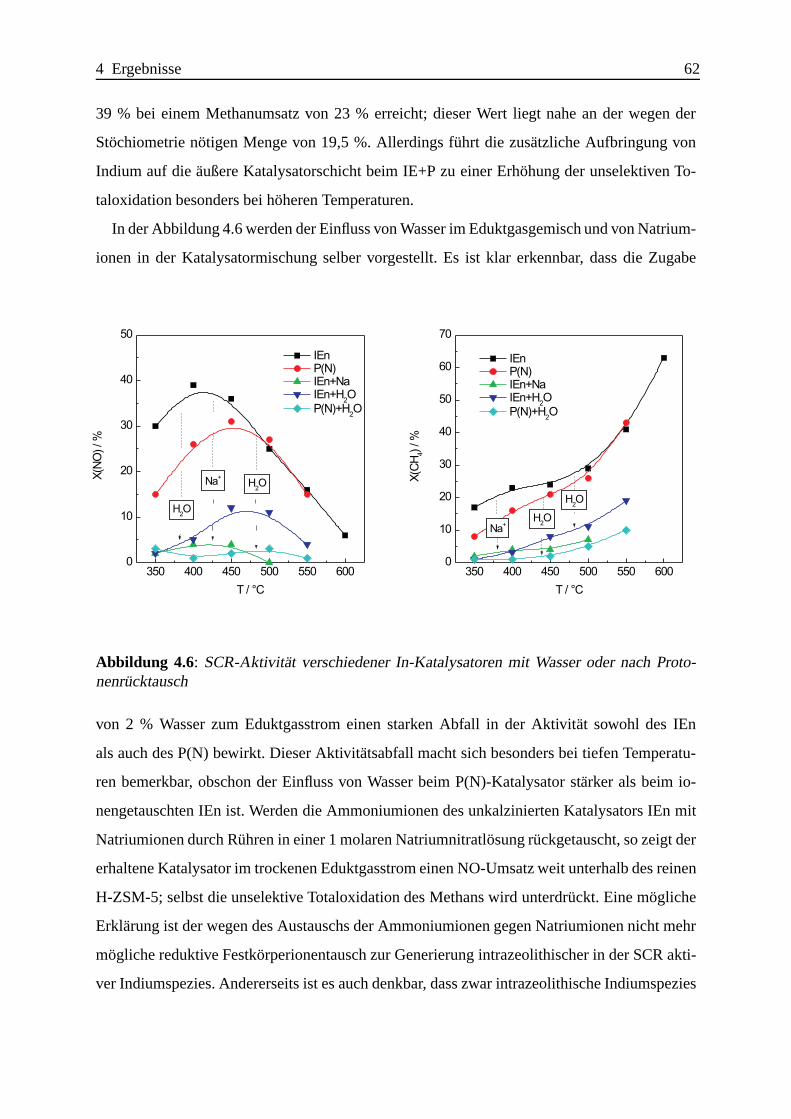
4 Ergebnisse 62
39 % bei einem Methanumsatz von 23 % erreicht; dieser Wert liegt nahe an der wegen der
Stochiometrie n¨otigen Menge von 19,5 %. Allerdings f¨uhrt die zus¨atzliche Aufbringung von
Indium auf dieaußere Katalysatorschicht beim IE+P zu einer Erh¨ohung der unselektiven To-
taloxidation besonders bei h¨oheren Temperaturen.
In der Abbildung 4.6 werden der Einfluss von Wasser im Eduktgasgemisch und von Natrium-
ionen in der Katalysatormischung selber vorgestellt. Es ist klar erkennbar, dass die Zugabe
350 400 450 500 550 6000
10
20
30
40
50
Na+
H2O
H2O
IEnP(N)IEn+NaIEn+H
2O
P(N)+H2O
X(N
O)/%
T / °C
350 400 450 500 550 6000
10
20
30
40
50
60
70
Na+
H2O
H2O
IEnP(N)IEn+NaIEn+H
2O
P(N)+H2O
X(C
H4)
/%
T / °C
Abbildung 4.6: SCR-Aktivitat verschiedener In-Katalysatoren mit Wasser oder nach Proto-nenrucktausch
von 2 % Wasser zum Eduktgasstrom einen starken Abfall in der Aktivit¨at sowohl des IEn
als auch des P(N) bewirkt. Dieser Aktivit¨atsabfall macht sich besonders bei tiefen Temperatu-
ren bemerkbar, obschon der Einfluss von Wasser beim P(N)-Katalysator st¨arker als beim io-
nengetauschten IEn ist. Werden die Ammoniumionen des unkalzinierten Katalysators IEn mit
Natriumionen durch R¨uhren in einer 1 molaren Natriumnitratl¨osung ruckgetauscht, so zeigt der
erhaltene Katalysator im trockenen Eduktgasstrom einen NO-Umsatz weit unterhalb des reinen
H-ZSM-5; selbst die unselektive Totaloxidation des Methans wird unterdr¨uckt. Eine mogliche
Erklarung ist der wegen des Austauschs der Ammoniumionen gegen Natriumionen nicht mehr
mogliche reduktive Festk¨orperionentausch zur Generierung intrazeolithischer in der SCR akti-
ver Indiumspezies. Andererseits ist es auch denkbar, dass zwar intrazeolithische Indiumspezies
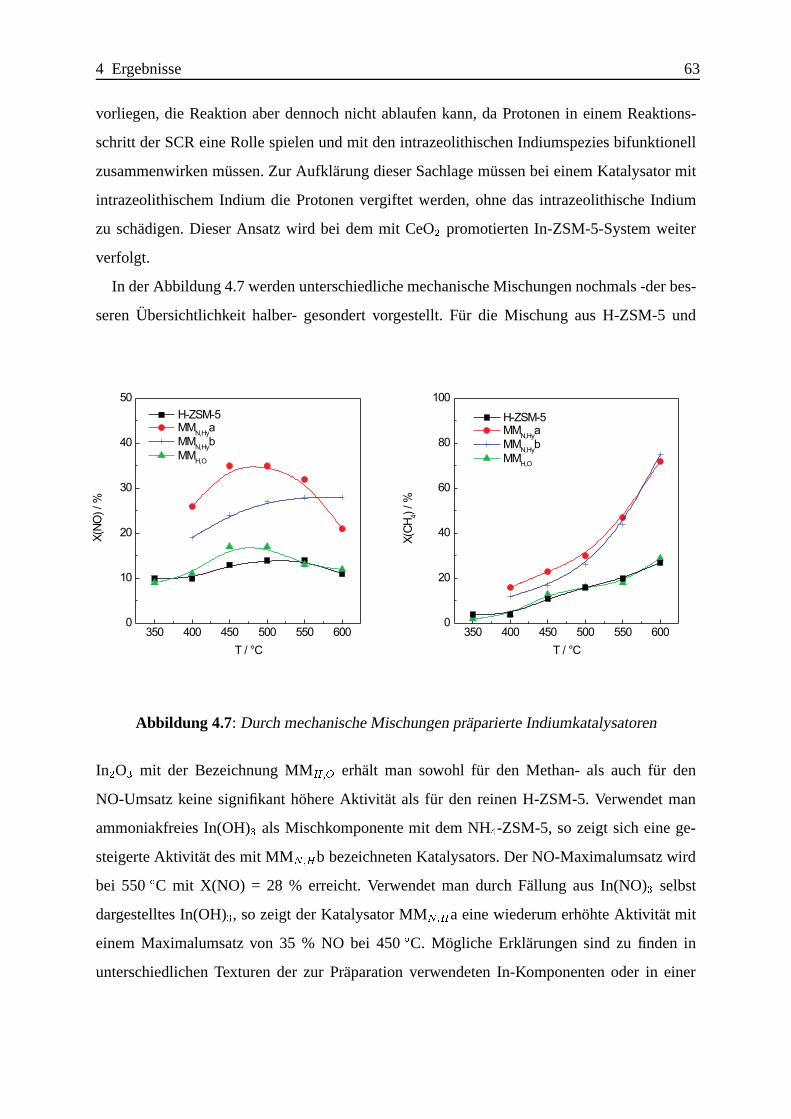
4 Ergebnisse 63
vorliegen, die Reaktion aber dennoch nicht ablaufen kann, da Protonen in einem Reaktions-
schritt der SCR eine Rolle spielen und mit den intrazeolithischen Indiumspezies bifunktionell
zusammenwirken m¨ussen. Zur Aufkl¨arung dieser Sachlage m¨ussen bei einem Katalysator mit
intrazeolithischem Indium die Protonen vergiftet werden, ohne das intrazeolithische Indium
zu schadigen. Dieser Ansatz wird bei dem mit CeO2 promotierten In-ZSM-5-System weiter
verfolgt.
In der Abbildung 4.7 werden unterschiedliche mechanische Mischungen nochmals -der bes-
serenUbersichtlichkeit halber- gesondert vorgestellt. F¨ur die Mischung aus H-ZSM-5 und
350 400 450 500 550 6000
10
20
30
40
50
H-ZSM-5MM
N,Hya
MMN,Hy
b
MMH,O
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
H-ZSM-5MM
N,Hya
MMN,Hy
b
MMH,O
X(C
H4)
/%
T / °C
Abbildung 4.7: Durch mechanische Mischungen pr¨aparierte Indiumkatalysatoren
In2O3 mit der Bezeichnung MMH;O erhalt man sowohl f¨ur den Methan- als auch f¨ur den
NO-Umsatz keine signifikant h¨ohere Aktivitat als fur den reinen H-ZSM-5. Verwendet man
ammoniakfreies In(OH)3 als Mischkomponente mit dem NH4-ZSM-5, so zeigt sich eine ge-
steigerte Aktivitat des mit MMN;Hb bezeichneten Katalysators. Der NO-Maximalumsatz wird
bei 550ÆC mit X(NO) = 28 % erreicht. Verwendet man durch F¨allung aus In(NO)3 selbst
dargestelltes In(OH)3, so zeigt der Katalysator MMN;Ha eine wiederum erh¨ohte Aktivitat mit
einem Maximalumsatz von 35 % NO bei 450ÆC. Mogliche Erklarungen sind zu finden in
unterschiedlichen Texturen der zur Pr¨aparation verwendeten In-Komponenten oder in einer
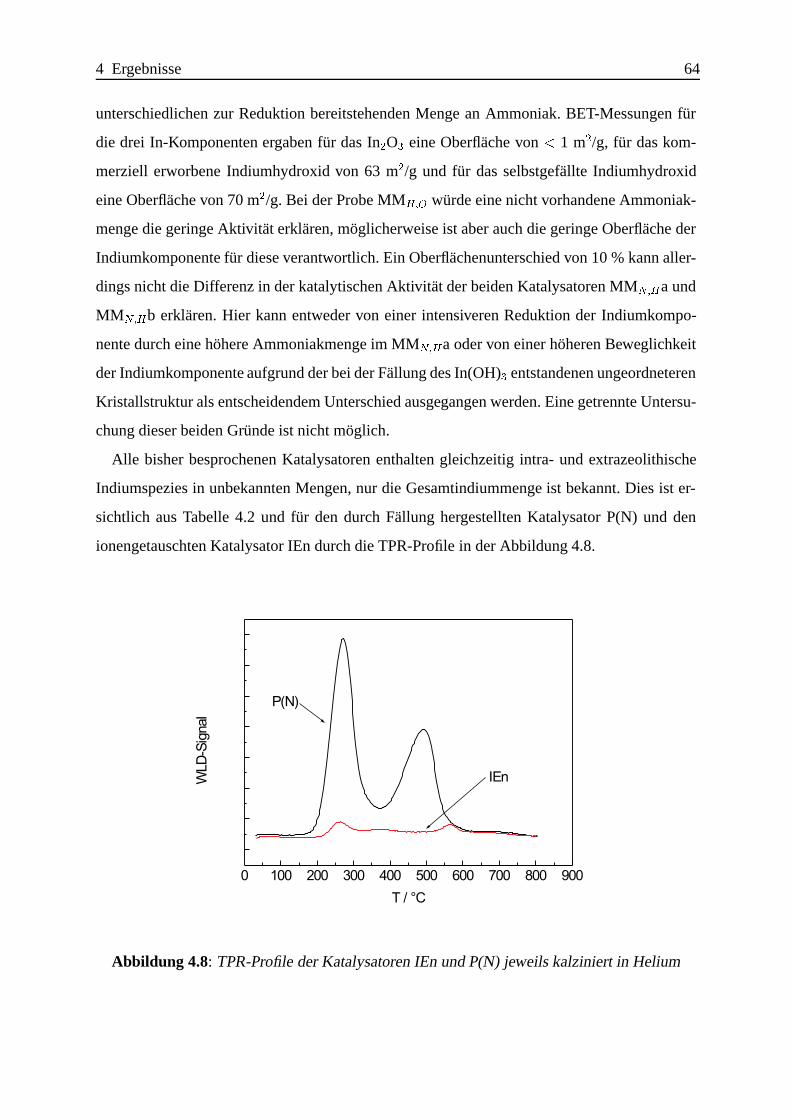
4 Ergebnisse 64
unterschiedlichen zur Reduktion bereitstehenden Menge an Ammoniak. BET-Messungen f¨ur
die drei In-Komponenten ergaben f¨ur das In2O3 eine Oberflache von< 1 m2/g, fur das kom-
merziell erworbene Indiumhydroxid von 63 m2/g und fur das selbstgef¨allte Indiumhydroxid
eine Oberflache von 70 m2/g. Bei der Probe MMH;O wurde eine nicht vorhandene Ammoniak-
menge die geringe Aktivit¨at erklaren, moglicherweise ist aber auch die geringe Oberfl¨ache der
Indiumkomponente f¨ur diese verantwortlich. Ein Oberfl¨achenunterschied von 10 % kann aller-
dings nicht die Differenz in der katalytischen Aktivit¨at der beiden Katalysatoren MMN;Ha und
MMN;Hb erklaren. Hier kann entweder von einer intensiveren Reduktion der Indiumkompo-
nente durch eine h¨ohere Ammoniakmenge im MMN;Ha oder von einer h¨oheren Beweglichkeit
der Indiumkomponente aufgrund der bei der F¨allung des In(OH)3 entstandenen ungeordneteren
Kristallstruktur als entscheidendem Unterschied ausgegangen werden. Eine getrennte Untersu-
chung dieser beiden Gr¨unde ist nicht m¨oglich.
Alle bisher besprochenen Katalysatoren enthalten gleichzeitig intra- und extrazeolithische
Indiumspezies in unbekannten Mengen, nur die Gesamtindiummenge ist bekannt. Dies ist er-
sichtlich aus Tabelle 4.2 und f¨ur den durch F¨allung hergestellten Katalysator P(N) und den
ionengetauschten Katalysator IEn durch die TPR-Profile in der Abbildung 4.8.
0 100 200 300 400 500 600 700 800 900
P(N)
IEnWLD
-Sig
nal
T / °C
Abbildung 4.8: TPR-Profile der Katalysatoren IEn und P(N) jeweils kalziniert in Helium
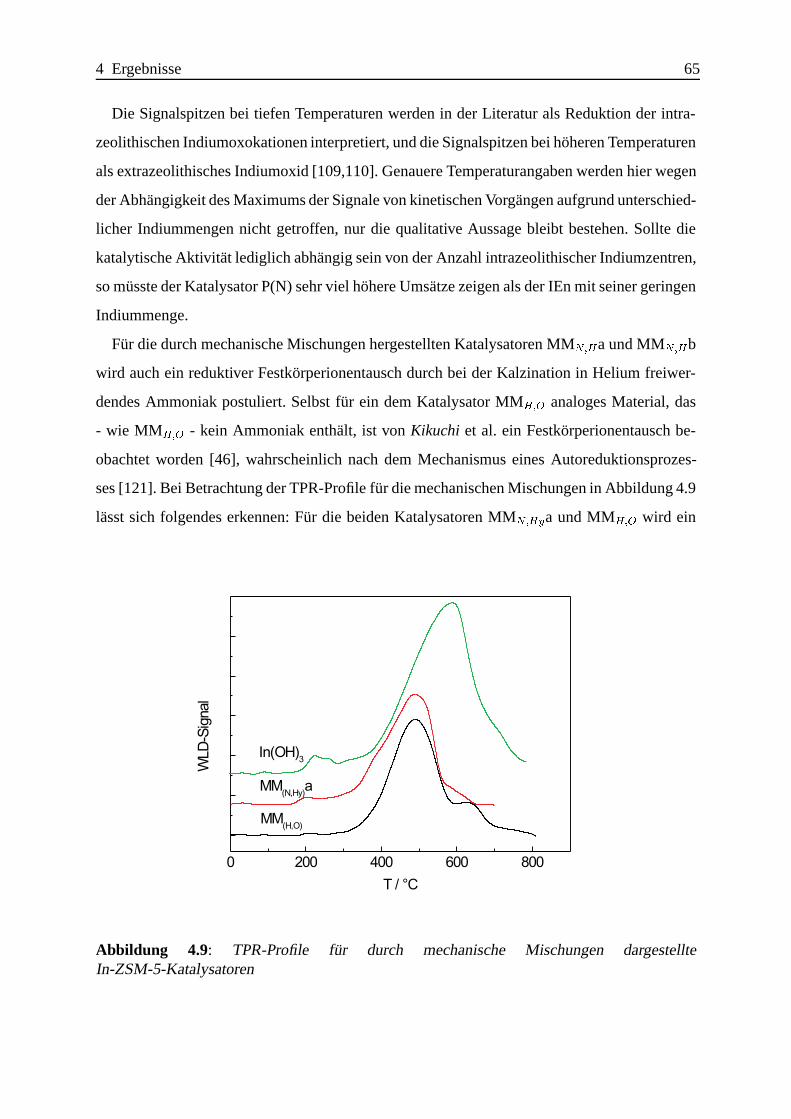
4 Ergebnisse 65
Die Signalspitzen bei tiefen Temperaturen werden in der Literatur als Reduktion der intra-
zeolithischen Indiumoxokationen interpretiert, und die Signalspitzen bei h¨oheren Temperaturen
als extrazeolithisches Indiumoxid [109,110]. Genauere Temperaturangaben werden hier wegen
der Abhangigkeit des Maximums der Signale von kinetischen Vorg¨angen aufgrund unterschied-
licher Indiummengen nicht getroffen, nur die qualitative Aussage bleibt bestehen. Sollte die
katalytische Aktivitat lediglich abh¨angig sein von der Anzahl intrazeolithischer Indiumzentren,
so musste der Katalysator P(N) sehr viel h¨ohere Ums¨atze zeigen als der IEn mit seiner geringen
Indiummenge.
Fur die durch mechanische Mischungen hergestellten Katalysatoren MMN;Ha und MMN;Hb
wird auch ein reduktiver Festk¨orperionentausch durch bei der Kalzination in Helium freiwer-
dendes Ammoniak postuliert. Selbst f¨ur ein dem Katalysator MMH;O analoges Material, das
- wie MMH;O - kein Ammoniak enth¨alt, ist vonKikuchi et al. ein Festk¨orperionentausch be-
obachtet worden [46], wahrscheinlich nach dem Mechanismus eines Autoreduktionsprozes-
ses [121]. Bei Betrachtung der TPR-Profile f¨ur die mechanischen Mischungen in Abbildung 4.9
lasst sich folgendes erkennen: F¨ur die beiden Katalysatoren MMN;Hya und MMH;O wird ein
0 200 400 600 800
MM(H,O)
MM(N,Hy)
a
In(OH)3
WLD
-Sig
nal
T / °C
Abbildung 4.9: TPR-Profile fur durch mechanische Mischungen dargestellteIn-ZSM-5-Katalysatoren

4 Ergebnisse 66
sehr niedriges Tieftemperatursignal erhalten, im Verh¨altnis zum Hochtemperatursignal ist es
fast nicht mehr zu erkennen. Allerdings stellt sich die Frage, ob diese kleine Andeutung eines
Tieftemperatursignals durch die Reduktion intrazeolithischen Indiums zustandekommt, da sich
auch im TPR-Profil des reinen In(OH)3 ein sogar etwas ausgepr¨agteres Signal finden l¨asst.
Um eine katalytische Aktivit¨at gezielt intra- oder extrazeolithischen Indiumspezies zuordnen
zu konnen, ist die Pr¨aparation von Katalysatoren mit ausschließlich einer Spezies n¨otig, solange
nicht fur eine der beiden Spezies eine katalytische Aktivit¨at ausgeschlossen werden kann. Die
Synthese von In-ZSM-5-Katalysatoren mit lediglich einer Spezies stellt sich hierbei als sehr
kompliziert heraus, da extrazeolithisches Indium leicht unter reduzierender Atmosph¨are in den
Zeolithen einwandern kann und Indium gleichzeitig bei der Pr¨aparation durch geringf¨ugige Be-
dingungsanderungen (pH-Wert, Abnutschzeit, ...) auf die ¨außere Zeolithoberfl¨ache aufgef¨allt
werden kann.
Wie weiter oben beschrieben (siehe Tabelle 4.2), ist es uns dennoch gelungen, Katalysatoren
herzustellen, welche definitiv nur intrazeolithisches Indium enthalten, wohingegen die Darstel-
lung rein extrazeolithischer In-ZSM-5-Katalysatoren nicht gl¨uckte.
Deshalb werden nun die katalytischen Eigenschaften der ausschließlich intrazeolithischen
Indiumkatalysatoren vorgestellt. Als Pr¨aparationsroute wurden der Festk¨orperionentausch
(SSIE), die Sublimation (S), die Transportreaktion (T-Z und T-Sil) und der w¨assrige Ionen-
tausch bei niedrigen pH-Werten (IE-1.5 und IE-2.5) eingesetzt. In Abbildung 4.10 werden die
an den durch Festk¨orperionentausch und Sublimation hergestellten Katalysatoren gemessenen
NO- und Methanums¨atze gezeigt.
Die NO-Umsatze liegen f¨ur alle Katalysatoren sehr niedrig. Dabei lassen sich die durch
SSIE praparierten Katalysatoren in zwei Gruppen aufteilen. In der ersten Gruppe befinden
sich die beiden Katalysatoren SSIE(1) und SSIE(2) mit Indiumgehalten von 1 % bzw. 2 %.
Bei niedrigen Temperaturen liegen die NO-Ums¨atze oberhalb des Umsatzes von einem reinen
H-ZSM-5, der hochste Wert wird bei 400ÆC mit 25 % erreicht. Bei hohen Temperaturen fallen
die NO-Umsatze unter die Referenzwerte f¨ur den reinen H-ZSM-5. Die zweite Gruppe von Ka-
talysatoren mit den Proben SSIE(3) und SSIE(4) weist eine h¨ohere Indiumbeladung von 3 %
bzw. 4 % auf und liegt mit ihren NO-Ums¨atzenuber den gesamten Temperaturbereich unter-
halb des H-ZSM-5. Die Methanums¨atze an diesen Katalysatoren zeigen einen gegenl¨aufigen
Trend, hier steigt der Methanumsatz mit steigendem Indiumgehalt an. Wird der Katalysator
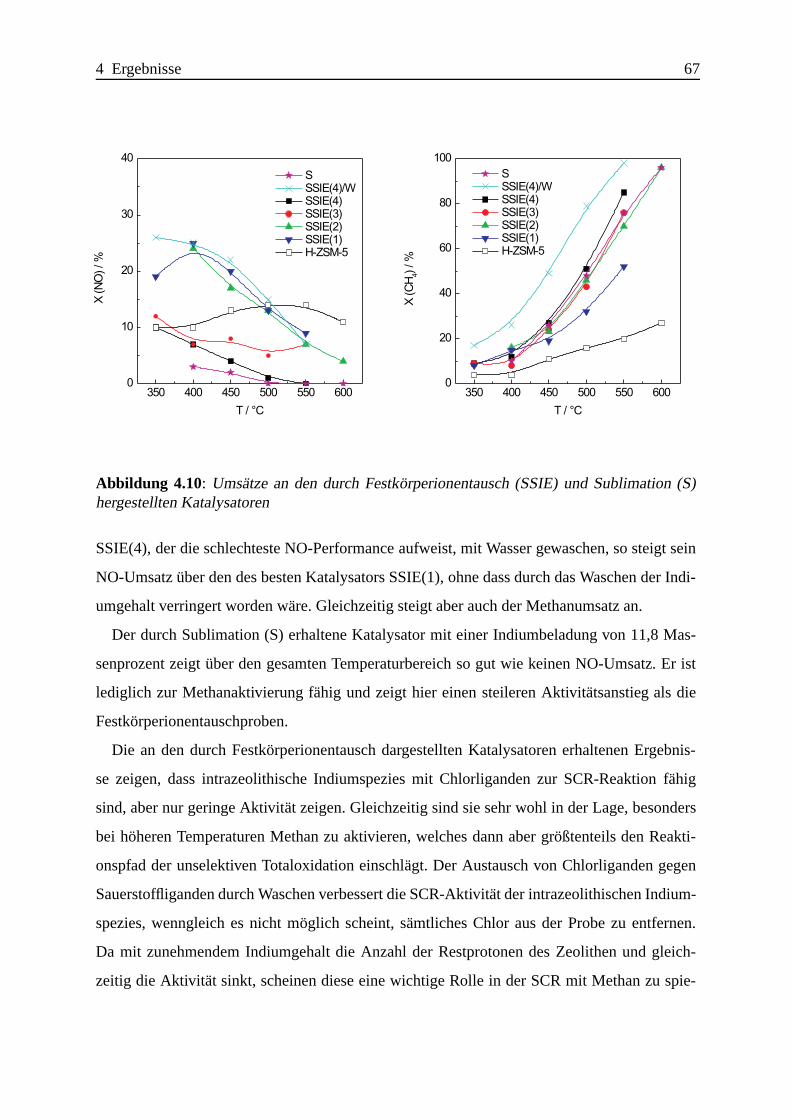
4 Ergebnisse 67
350 400 450 500 550 6000
10
20
30
40
SSSIE(4)/WSSIE(4)SSIE(3)SSIE(2)SSIE(1)H-ZSM-5
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
SSSIE(4)/WSSIE(4)SSIE(3)SSIE(2)SSIE(1)H-ZSM-5
X(C
H4)
/%
T / °C
Abbildung 4.10: Umsatze an den durch Festk¨orperionentausch (SSIE) und Sublimation (S)hergestellten Katalysatoren
SSIE(4), der die schlechteste NO-Performance aufweist, mit Wasser gewaschen, so steigt sein
NO-Umsatz ¨uber den des besten Katalysators SSIE(1), ohne dass durch das Waschen der Indi-
umgehalt verringert worden w¨are. Gleichzeitig steigt aber auch der Methanumsatz an.
Der durch Sublimation (S) erhaltene Katalysator mit einer Indiumbeladung von 11,8 Mas-
senprozent zeigt ¨uber den gesamten Temperaturbereich so gut wie keinen NO-Umsatz. Er ist
lediglich zur Methanaktivierung f¨ahig und zeigt hier einen steileren Aktivit¨atsanstieg als die
Festkorperionentauschproben.
Die an den durch Festk¨orperionentausch dargestellten Katalysatoren erhaltenen Ergebnis-
se zeigen, dass intrazeolithische Indiumspezies mit Chlorliganden zur SCR-Reaktion f¨ahig
sind, aber nur geringe Aktivit¨at zeigen. Gleichzeitig sind sie sehr wohl in der Lage, besonders
bei hoheren Temperaturen Methan zu aktivieren, welches dann aber gr¨oßtenteils den Reakti-
onspfad der unselektiven Totaloxidation einschl¨agt. Der Austausch von Chlorliganden gegen
Sauerstoffliganden durch Waschen verbessert die SCR-Aktivit¨at der intrazeolithischen Indium-
spezies, wenngleich es nicht m¨oglich scheint, s¨amtliches Chlor aus der Probe zu entfernen.
Da mit zunehmendem Indiumgehalt die Anzahl der Restprotonen des Zeolithen und gleich-
zeitig die Aktivitat sinkt, scheinen diese eine wichtige Rolle in der SCR mit Methan zu spie-
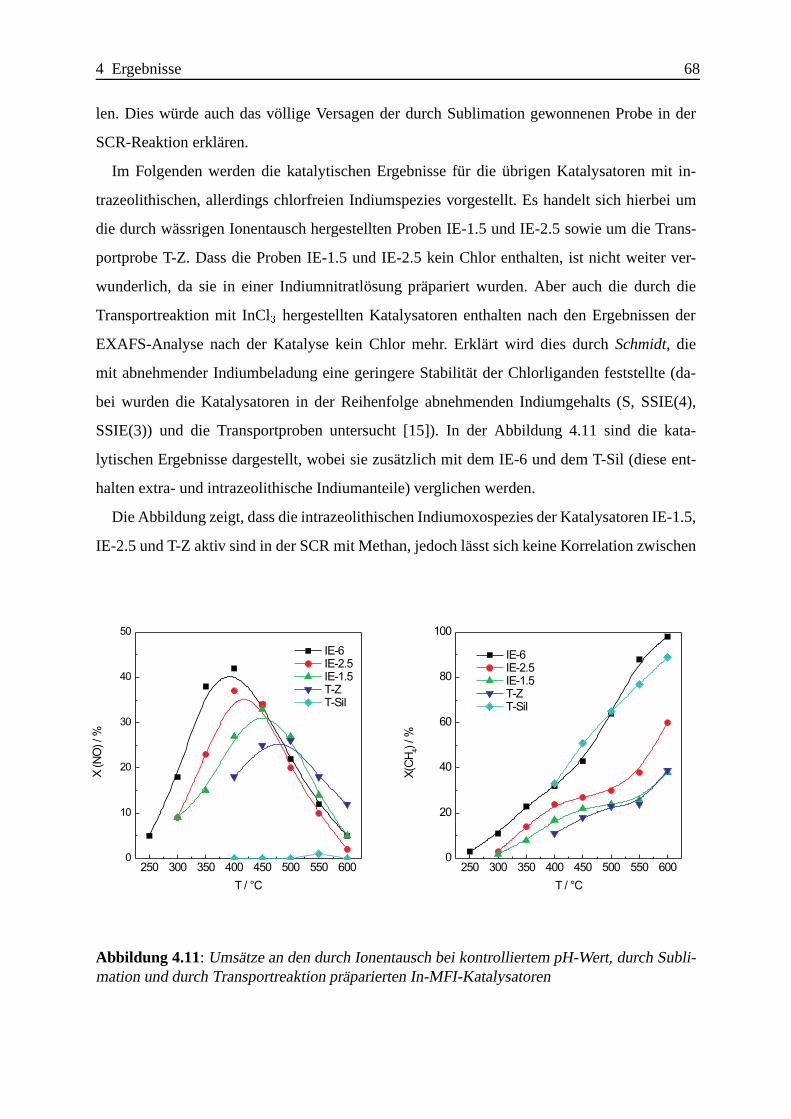
4 Ergebnisse 68
len. Dies wurde auch das v¨ollige Versagen der durch Sublimation gewonnenen Probe in der
SCR-Reaktion erkl¨aren.
Im Folgenden werden die katalytischen Ergebnisse f¨ur die ubrigen Katalysatoren mit in-
trazeolithischen, allerdings chlorfreien Indiumspezies vorgestellt. Es handelt sich hierbei um
die durch wassrigen Ionentausch hergestellten Proben IE-1.5 und IE-2.5 sowie um die Trans-
portprobe T-Z. Dass die Proben IE-1.5 und IE-2.5 kein Chlor enthalten, ist nicht weiter ver-
wunderlich, da sie in einer Indiumnitratl¨osung prapariert wurden. Aber auch die durch die
Transportreaktion mit InCl3 hergestellten Katalysatoren enthalten nach den Ergebnissen der
EXAFS-Analyse nach der Katalyse kein Chlor mehr. Erkl¨art wird dies durchSchmidt, die
mit abnehmender Indiumbeladung eine geringere Stabilit¨at der Chlorliganden feststellte (da-
bei wurden die Katalysatoren in der Reihenfolge abnehmenden Indiumgehalts (S, SSIE(4),
SSIE(3)) und die Transportproben untersucht [15]). In der Abbildung 4.11 sind die kata-
lytischen Ergebnisse dargestellt, wobei sie zus¨atzlich mit dem IE-6 und dem T-Sil (diese ent-
halten extra- und intrazeolithische Indiumanteile) verglichen werden.
Die Abbildung zeigt, dass die intrazeolithischen Indiumoxospezies der Katalysatoren IE-1.5,
IE-2.5 und T-Z aktiv sind in der SCR mit Methan, jedoch l¨asst sich keine Korrelation zwischen
250 300 350 400 450 500 550 6000
10
20
30
40
50
IE-6IE-2.5IE-1.5T-ZT-Sil
X(N
O)/%
T / °C
250 300 350 400 450 500 550 6000
20
40
60
80
100
IE-6IE-2.5IE-1.5T-ZT-Sil
X(C
H4)
/%
T / °C
Abbildung 4.11: Umsatze an den durch Ionentausch bei kontrolliertem pH-Wert, durch Subli-mation und durch Transportreaktion pr¨aparierten In-MFI-Katalysatoren

4 Ergebnisse 69
der Indiummenge und der katalytischen Aktivit¨at feststellen. W¨aren alle Indiumoxokationen
gleichartig, so m¨usste IE-2.5 ¨uber den gesamten Temperaturbereich h¨ohere NO-Ums¨atze auf-
weisen als IE-1.5, da sich die beiden Katalysatoren in ihrer Indiummenge unterscheiden. Nun
zeigt der Katalysator IE-1.5 f¨ur hohe Temperaturen sogar eine dem IE-2.5 ¨uberlegene Akti-
vitat bei geringerer Indiummenge. Im Umkehrschluss m¨usste der durch die Transportreaktion
praparierte Katalysator T-Z mit der h¨ochsten Indiummenge der drei Katalysatoren auch die
hochsten NO-Ums¨atze erzielen. Dies ist aber nur bei hohen Temperaturen der Fall; bei tiefe-
ren Temperaturen sinkt der NO-Umsatz und liegt unterhalb des NO-Umsatzes des Katalysators
IE-1.5.
Mogliche Erklarungen f¨ur diese Aktivitatsverschiebung hin zu h¨oheren Temperaturen unter
gleichzeitiger Abnahme der Aktivit¨at waren das Vorliegen verschiedener Indiumspezies oder
Schaden an der Zeolithstruktur (z.B. Verarmung an Protonen durch Dealuminierung). Mit Hilfe
von EXAFS ist die Koordinationssph¨are der Indiumspezies tats¨achlich als verschieden erkannt
worden. Gleichzeitig gibt es aus XPS-Untersuchungen auch Hinweise auf eine Dealuminie-
rung des Zeolithen w¨ahrend der Pr¨aparation bei niedrigen pH-Werten [15]. Da der Protonen-
eintausch bei einem pH-Wert von 1 - 2 eine Standardprozedur zurUberfuhrung von Zeolithen
der Na-Form in die H-Form darstellt, kann der Grund des Aktivit¨atsruckgangs nicht ein Mangel
an Protonen sein. Demnach m¨ussen die unterschiedlichen Indiumspezies f¨ur die Verschiebun-
gen in den katalytischen Ergebnissen verantwortlich sein.
Die beiden Katalysatoren, die extra- und intrazeolithische Indiumspezies enthalten, zeigen
bemerkenswerte Eigenschaften. W¨ahrend Katalysator IE-6 mit einem NO-Umsatz von 42 %
bei 400ÆC die hochste SCR-Aktivit¨at aller hier untersuchten Katalysatoren aufweist, ist der
NO-Umsatz des T-Sil mit maximal 2 % vernachl¨assigbar. Der Methanumsatz an beiden Ka-
talysatoren liegt jedoch in der gleichen Gr¨oßenordnung, so dass als Grund f¨ur die schlechte
Aktivit at des T-Sil das Fehlen von Brønstedzentren postuliert werden kann.
Beim Vergleich der Form der NO-Umsatzkurve des IE-6 mit denen der restlichen Kataly-
satoren fallt auf, dass der Anstieg des NO-Umsatzes und die Verschiebung des Maximums zu
tieferen Temperaturen ausgehend von den anderen Katalysatoren IE-2.5, IE-1.5 und T-Z ex-
trapoliert werden kann. Dieser eindeutige Trend deutet darauf hin, dass die katalytisch aktive
Spezies intrazeolithisch vorliegt und in einer strukturell intakteren Matrix als bei den ande-
ren Katalysatoren eingebettet ist. Demnach h¨atten die extrazeolithischen Indiumspezies keinen
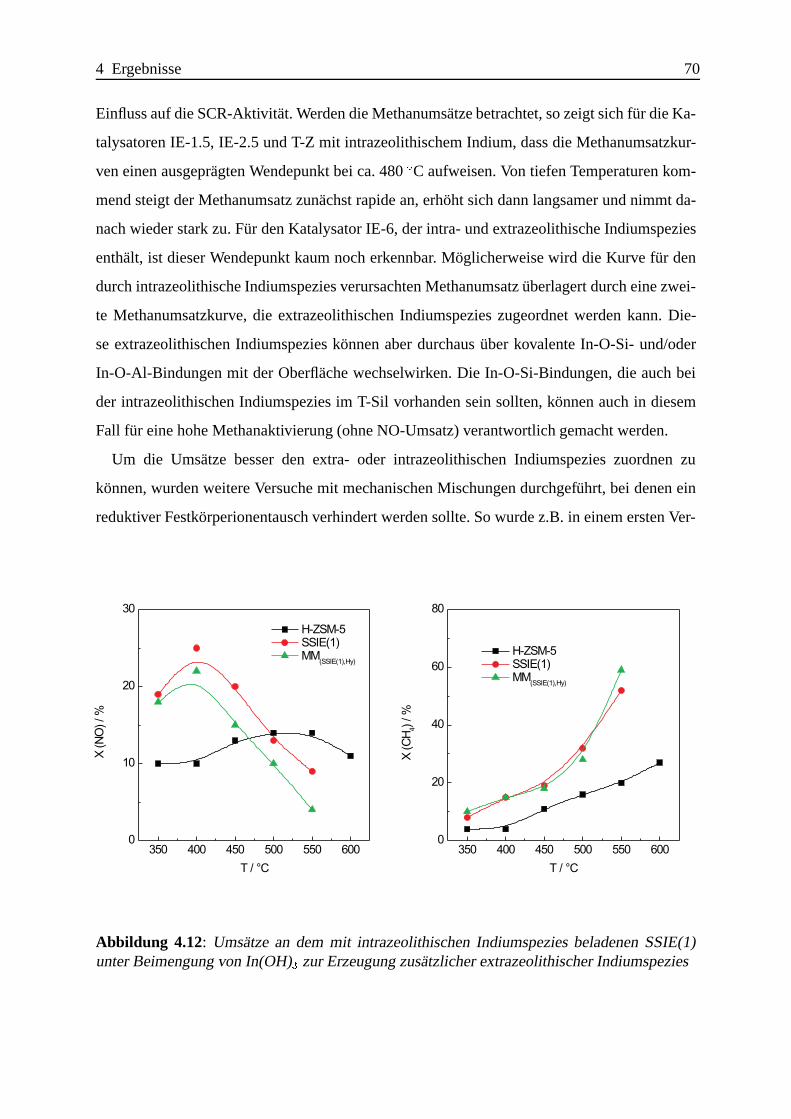
4 Ergebnisse 70
Einfluss auf die SCR-Aktivit¨at. Werden die Methanums¨atze betrachtet, so zeigt sich f¨ur die Ka-
talysatoren IE-1.5, IE-2.5 und T-Z mit intrazeolithischem Indium, dass die Methanumsatzkur-
ven einen ausgepr¨agten Wendepunkt bei ca. 480ÆC aufweisen. Von tiefen Temperaturen kom-
mend steigt der Methanumsatz zun¨achst rapide an, erh¨oht sich dann langsamer und nimmt da-
nach wieder stark zu. F¨ur den Katalysator IE-6, der intra- und extrazeolithische Indiumspezies
enthalt, ist dieser Wendepunkt kaum noch erkennbar. M¨oglicherweise wird die Kurve f¨ur den
durch intrazeolithische Indiumspezies verursachten Methanumsatz ¨uberlagert durch eine zwei-
te Methanumsatzkurve, die extrazeolithischen Indiumspezies zugeordnet werden kann. Die-
se extrazeolithischen Indiumspezies k¨onnen aber durchaus ¨uber kovalente In-O-Si- und/oder
In-O-Al-Bindungen mit der Oberfl¨ache wechselwirken. Die In-O-Si-Bindungen, die auch bei
der intrazeolithischen Indiumspezies im T-Sil vorhanden sein sollten, k¨onnen auch in diesem
Fall fur eine hohe Methanaktivierung (ohne NO-Umsatz) verantwortlich gemacht werden.
Um die Umsatze besser den extra- oder intrazeolithischen Indiumspezies zuordnen zu
konnen, wurden weitere Versuche mit mechanischen Mischungen durchgef¨uhrt, bei denen ein
reduktiver Festk¨orperionentausch verhindert werden sollte. So wurde z.B. in einem ersten Ver-
350 400 450 500 550 6000
20
40
60
80
H-ZSM-5SSIE(1)MM
(SSIE(1),Hy)
X(C
H4)
/%
T / °C
350 400 450 500 550 6000
10
20
30
H-ZSM-5SSIE(1)MM
(SSIE(1),Hy)
X(N
O)/%
T / °C
Abbildung 4.12: Umsatze an dem mit intrazeolithischen Indiumspezies beladenen SSIE(1)unter Beimengung von In(OH)3 zur Erzeugung zus¨atzlicher extrazeolithischer Indiumspezies

4 Ergebnisse 71
such dem SSIE(1)-Katalysator mit intrazeolithischem Indium noch In(OH)3 als mechanische
Mischung unter Verwendung eines Achatm¨orsers beigegeben, was zu der Katalysatorbezeich-
nung MMSSIE(1);Hy fuhrt. Die katalytischen Ergebnisse werden in Abbildung 4.12 vorgestellt,
zu Vergleichszwecken ist auch der reine H-ZSM-5 enthalten. Die ¨außeren Erscheinungsfor-
men der beiden NO-Umsatzkurven von SSIE(1) und MMSSIE(1);Hy sind gleich; jedoch ist die
NO-Umsatzkurve f¨ur den Katalysator MMSSIE(1);Hy parallel zu tieferen Ums¨atzen verscho-
ben. Die Methanums¨atze fur beide Katalysatoren zeigen f¨ur den gesamten Temperaturverlauf
im Rahmen der Fehlergrenzen eineUbereinstimmung. Die Zugabe von extrazeolithischem In-
dium bringt also keine Verbesserung in der SCR-Aktivit¨at, das Gegenteil ist der Fall. Allerdings
wird auch der Methanumsatz nicht erh¨oht, nur die Selektivit¨at zur SCR sinkt.
Bisher konnte nur indirekt gezeigt werden, dass extrazeolithische Indiumspezies nicht zur
SCR-Reaktion bef¨ahigt sind, da die Pr¨aparation ausschließlich extrazeolithischer Indium-
spezies nicht gelungen ist. Daher sind weitere Versuche mit Pellets der Komponenten durch-
gefuhrt worden, die in dieser Arbeit den mechanischen Mischungen zugerechnet werden. Das
Ziel der Versuche besteht darin, eine Reihe von Katalysatoren mit unterschiedlichem Gehalt an
intrazeolithischen Spezies zu generieren und dies mittels TPR zu dokumentieren, weil bei me-
chanischen Mischungen ein Festk¨orperionentausch nicht ausgeschlossen werden kann (siehe
oben). Da eine unmittelbare N¨ahe der Indiumkomponente zu dem Zeolithen die Voraussetzung
1. Mahlen
2. Verpressen
3. 600°C
inHe
1. 600 °C in He
2. Mahlen3. Verpressen
AB C
In(OH)3
NH4-ZSM-5
Abbildung 4.13: Graphische Darstellung der Pr¨aparation der Pelletexperimente

4 Ergebnisse 72
fur einen reduktiven Festk¨orperionentausch bildet, sollte mit Hilfe der Pelletversuche diese
Nahe variiert werden. Die unterschiedlichen Pr¨aparationsrouten werden in Abbildung 4.13 dar-
gestellt. Ausgehend von Pellets aus NH4-ZSM-5 der Siebfraktion 250 - 355�m und von Pellets
aus In(OH)3 der Siebfraktion 100 - 160�m (In(OH)3 mit Ammoniak gefallt aus In(NO)3) wer-
den die Mischungen MMN ;Hy (A), (B) und (C) prapariert. Katalysator MMN ;Hy (A) besteht
nur aus zusammengegebenen Pellets der beiden Komponenten nach Kalzination in Helium bei
600ÆC, wobei die Indiummenge wie f¨ur alle anderen Katalysatoren der Pelletversuche 7 Mas-
senprozent ausmacht. Durch diese Pr¨aparationsweise wird w¨ahrend der gesamten Pr¨aparations-
und Katalysezeit eine große Distanz zwischen der Zeolith- und der Indiumkomponente gew¨ahr-
leistet. Bei dem Katalysator MMN ;Hy (B), der aus Katalysator MMN ;Hy (A) durch Morsern
und Verpressen erhalten wird, sind die Indium- und die Zeolithkomponente nur w¨ahrend der
ersten Kalzinationsstufe, bei der das f¨ur den reduktiven Festk¨orperionentausch ben¨otigte Am-
moniak freigesetzt wird, getrennt. W¨ahrend der zweiten Kalzinationsstufe in Helium sind sie
wieder intensiv miteinander vermengt. Der Katalysator MMN ;Hy (C) ahnelt dem normalen Ka-
talysator MMN ;Hy, allerdings waren seine Ausgangskomponenten beide vorher pelletisiert. Er-
wartet wird nun fur diese Katalysatorreihe eine Variation der intrazeolithischen Indiummenge,
wobei diese in der Reihenfolge (A)< (B) < (C) ansteigen sollte. Abbildung 4.14 zeigt die
katalytische Aktivitat der eben beschriebenen Mischungen. Durch die Trennung der einzel-
nen Komponenten bei Katalysator MMN ;Hy (A) bricht sowohl der NO- als auch der Methan-
umsatz zusammen. Die beiden anderen Katalysatoren MMN ;Hy (B) und MMN ;Hy (C) zeigen
eine hohere Aktivitat mit dem Umsatzmaximum bei einer tieferen Temperatur, wobei zwi-
schen diesen Katalysatoren entgegen der Erwartungen innerhalb der Fehlergrenzen kein Un-
terschied festzustellen ist. DieUberprufung auf intrazeolithische Indiumspezies erfolgt durch
die Aufnahme von TPR-Profilen in Abbildung 4.15. Zum Vergleich ist hier der durch F¨allung
auf den NH4-ZSM-5 dargestellte Katalysator P(N) aufgezeichnet, der nach Kalzinierung bei
600ÆC in Helium die zwei charakteristischen Signale aufweist: das Tieftemperatursignal f¨ur
intrazeolithische Indiumspezies und das Hochtemperatursignal f¨ur extrazeolithische Indium-
spezies. F¨ur die mechanischen Mischungen zeigt sich nur ein Signal bei h¨oheren Temperaturen,
das allerdings verschoben ist und teilweise Schultern aufweist. Der Katalysator MMN ;Hy (A)
hat im Bereich der intrazeolithischen Indiumspezies ¨uberhaupt kein Signal, sondern erst das
Hochtemperatursignal bei ca. 570ÆC zeigt die Existenz von extrazeolithischem Indiumoxid
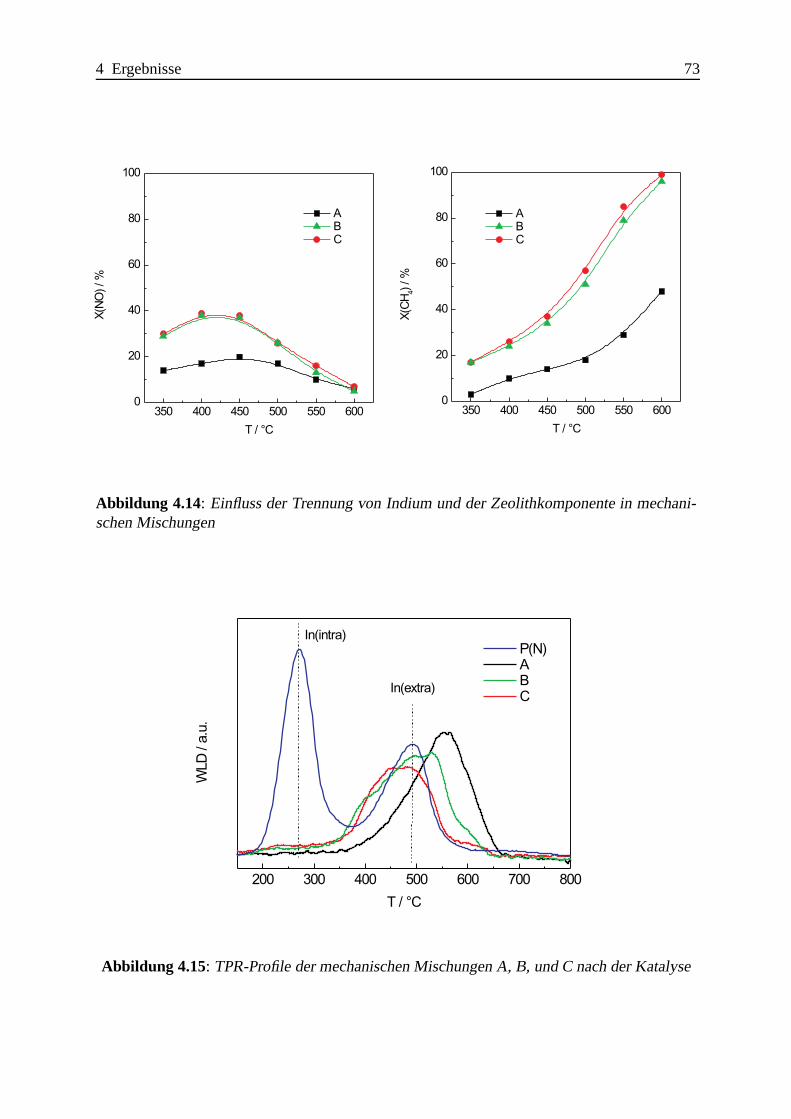
4 Ergebnisse 73
350 400 450 500 550 6000
20
40
60
80
100
ABC
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
ABC
X(C
H4)
/%
T / °C
Abbildung 4.14: Einfluss der Trennung von Indium und der Zeolithkomponente in mechani-schen Mischungen
200 300 400 500 600 700 800
In(extra)
In(intra)P(N)ABC
WLD
/a.u
.
T / °C
Abbildung 4.15: TPR-Profile der mechanischen Mischungen A, B, und C nach der Katalyse

4 Ergebnisse 74
an. Ein Testversuch mit In(OH)3 nach Kalzination in Sauerstoff ohne eine Vermengung mit
Zeolith zeigt ein Signal bei ca. 580ÆC und best¨atigt das Vorhandensein von Indiumoxid. Die
Verschiebung um 10ÆC zu niedrigeren Temperaturen kann durch eine leichte Wechselwirkung
mit dem Zeolithen erkl¨art werden. Beim Katalysator MMN ;Hy (B) liegt der Hochtemperatursi-
gnal bei ca. 510ÆC, ist aber gespalten in zwei Spitzen bei 490ÆC und 530ÆC und zeigt noch
eine Schulter bei ca. 410ÆC. Eine zweite Schulter im Tieftemperaturbereich bei ca. 230ÆC
macht nicht mehr als 10 % der Gesamtsignalfl¨ache aus, wodurch, bei einer Zurechnung dieser
Schulter zu intrazeolithischem Indium, f¨ur letzteres ein In/Si-Verh¨altnis von 0,002 berechnet
werden kann. Das liegt in der Gr¨oßenordnung der durch Ionentausch erhaltenen Proben IE-2.5
und IE-1-5. Die Aufspaltung des Hochtemperatursignals kann erkl¨art werden durch extrazeo-
lithische Indiumspezies mit engerem oder schlechterem Kontakt zum Zeolithen, entstanden
durch die mechanische Vermischung des Zeolithen. Die Schulter bei 410ÆC kann unter der
Annahme, dass eine Linearit¨at zwischen Reduktionstemperatur und der Intensit¨at der Indium-
wechselwirkung mit dem Zeolithen besteht, einer extrazeolithischen Indiumspezies mit Bin-
dung an die ¨außere Oberfl¨ache zugeordnet werden, z.B kovalent gebundenen In-Spezies ¨uber
In-O-Si- oder In-O-Al-Bindungen. In dem TPR-Profil des Katalysators MMN ;Hy (C), bei dem
die hochste Menge an intrazeolithischem Indium erwartet wurde, verschiebt sich das Zentrum
des Hochtemperatursignals zu einer noch tieferen Temperatur (ca. 460ÆC). Die Menge an in-
trazeolithischem Indium scheint zugenommen zu haben, da die Schulter im Tieftemperaturbe-
reich bei einer tieferen Temperatur startet und h¨oher lauft. Allerdings ist nicht der erwartete
große Unterschied zu sehen, was auch durch die gleichwertige katalytische Aktivit¨at in Abbil-
dung 4.14 best¨atigt wird.
Somit scheint es, dass intrazeolithische Indiumspezies der Katalysatoren MMN ;Hy (B) und
MMN ;Hy (C) fur die hohere katalytische Aktivit¨at im Gegensatz zu der getrennten Mischung
MMN ;Hy (A) verantwortlich ist. Da aber zwischen MMN ;Hy (B) und MMN ;Hy (C) trotz un-
terschiedlicher Mengen an intrazeolithischem Indium gleiche Aktivit¨aten erhalten werden,
ist nicht auszuschließen, dass eine extrazeolithische Indiumspezies mit kovalenten In-O-Si-
und/oder In-O-Al-Bindungen katalytisch aktiv ist. Diese Spezies ist allerdings schwer zu iden-
tifizieren, da sie nur als Schulter oder Teil des Hochtemperatursignals im TPR-Profil vorliegt.
Mit Sicherheit steht fest, dass eine enge Nachbarschaft zwischen der Zeolithkomponente und
der Indiumkomponente die Voraussetzung f¨ur eine hohe katalytische Aktivit¨at ist. Moglicher-

4 Ergebnisse 75
weise ist aber nicht ein reduktiver Eintausch der Indiumkomponente das entscheidende Kri-
terium, sondern nur eine m¨oglichst kurze Distanz zwischen den einzelnen Reaktionszentren
des extrazeolithischen Indiums und den Brønstedzentren des Zeolithen, um Diffusionslimitie-
rung zu vermeiden, wie es f¨ur das System CeO2-ZSM-5 vonLiesevorgeschlagen wurde [101].
Die mogliche Existenz eines solchen bifunktionalen Reaktionsmechanismus wird durch die
schlechten Aktivitaten von Katalysatoren, bei denen keine Protonen anwesend sind (z.B. der
mit Natrium vergiftete IEn-Na und der T-Sil ohne Brønstedazidit¨at), untermauert.
Auf die an deraußeren Katalysatorfl¨ache gebundenen Indiumspezies soll aber noch in ei-
nem spateren Abschnitt ¨uber das mit CeO2 promotierte In-ZSM-5-System n¨aher eingegangen
werden. Hierbei soll ihnen eine katalytische Aktivit¨at zugeordnet werden.
4.1.2.4 Zusammenfassung
Verschiedene Pr¨aparationsrouten f¨ur In-ZSM-5-Katalysatoren f¨uhren zu unterschiedlichen
Strukturen der Indiumkomponente und zu verschiedenen Aktivit¨aten in der SCR mit Methan.
Die durch wassrigen Ionentausch bei verschiedenen pH-Werten, durch F¨allung, Festk¨orperio-
nentausch, Sublimation, Transportreaktion und durch mechanisches Mischen pr¨aparierten Ka-
talysatoren sind in der Arbeit vonSchmidtdurch EXAFS und XPS charakterisiert worden,
um gezielt Systeme mit bekannter Indiumlokalisation (extra- oder/und intrazeolithisch) und
Koordinationssph¨are (ausschließlich Sauerstoffatome, Sauerstoff- und Chloratome) katalytisch
vermessen zu k¨onnen.
Nach den Ergebnissen zu urteilen, sind intrazeolithische Indiumoxospezies aktive Zentren
fur die SCR-Reaktion, wobei die katalytische Aktivit¨at stark von der Koordinationssph¨are
abhangt. Die intrazeolithischen InOxCly-Spezies zeigen eine niedrige SCR-Aktivit¨at, wobei
sie sehr wohl in der Lage sind, Methan zu aktivieren. Mit sinkender Chloratomzahl und stei-
gender Sauerstoffatomzahl in der Koordinationssph¨are steigt die SCR-Aktivit¨at. Durch die Zu-
gabe von extrazeolithischen Indiumspezies kann die SCR-Aktivit¨at nicht weiter gesteigert wer-
den, lediglich die Totaloxidationsaktivit¨at bezuglich des Methans wird erh¨oht. Die Versuche
mit mechanischen Mischungen zeigen, dass nur eine innige Vermischung der Indium- und
Zeolithkomponente zu Katalysatoren mit einer Aktivit¨at im Bereich der durch w¨assrigen Io-
nentausch pr¨aparierten f¨uhrt. Mit den bis zu diesem Punkt vorliegenden Ergebnissen gibt es
hierfur zwei Erklarungen: Zum einen kann die Aktivit¨at durch einen reduktiven oder auto-

4 Ergebnisse 76
reduktiven Festk¨orperionentausch des Indiums in den Zeolithen erzielt werden, zum anderen
besteht die M¨oglichkeit, dass keine Festk¨orperreaktion, sondern lediglich eine enge Nachbar-
schaft der Komponenten f¨ur einen bifunktionalen Reaktionsmechanismus, bei dem es zu Dif-
fusionslimitierungen kommen kann, ben¨otigt wird. Das Vorliegen eines bifunktionalen Reakti-
onsmechanismus wird durch Versuche mit Katalysatoren best¨atigt, die entweder von Anfang an
keine Brønstedazidit¨at besitzen (T-Sil) oder deren Brønstedzentren durch Natriumionen r¨uck-
getauscht wurden (IEn-Na).
Bei den mechanischen Mischungen wird auf der Grundlage von TPR-Versuchen eine in
der Literatur noch nicht beschriebene Indiumspezies vermutet, die durch kovalente In-O-Si-
und/oder In-O-Al-Bindungen an der ¨außeren Oberfl¨ache des Zeolithen verankert ist. Genauere
Untersuchungen hierzu werden aber beim CeO2-In-ZSM-5-System vorgestellt.
4.1.3 Das System CeO2-In-ZSM-5
4.1.3.1 Definition der Probenbezeichnungen
Die Probenbezeichnungen orientieren sich an denen, die wir oben f¨ur die
In-ZSM-5-Katalysatoren verwendet haben, weil diese als Grundlage f¨ur die Praparation
der CeO2-In-ZSM-5-Katalysatoren dienen. Da hier der Zeolith mit Ce und In modifiziert
wird, ist die Reihenfolge der Modifizierung den Katalysatorbezeichnungen entnehmbar.
Wie im folgenden Abschnitt berichtet wird, ist die einfachste Promotierung mit Cer durch
eine mechanische Mischung der In-ZSM-5-Katalysatoren mit CeO2 zu erreichen; demnach
werden die Katalysatoren mit K¨urzeln wie z.B. CeO2//IEn bezeichnet. Dieses K¨urzel steht
fur einen durch Ionentausch bei neutralem pH-Wert pr¨aparierten In-ZSM-5-Katalysator,
der mechanisch mit CeO2 vermengt wurde. Dabei bezeichnet die amAnfangdes Namens
dargestellte Komponente diezuletzt hinzugefugte Komponente. Die Namensgebung f¨ur
die In-ZSM-5-Katalysatoren wird aber nur f¨ur die Katalysatoren beibehalten, die aus dem
bekannten In-Grundsystem und aus dem mechanisch beigemengtem CeO2 entstehen. F¨ur die
restlichen Pr¨aparationsarten, besonders f¨ur verschiedene mechanische Mischungen, w¨urde
eine Weiterfuhrung dieser Namensgebung zu sehr komplizierten Abk¨urzungen f¨uhren. Hier
erfolgt eine Abweichung von der normalen Namensgebung. So ist z.B. der Katalysator
[(CeO2//NH4-ZSM-5 in He) + (In(OH)3 in NH3/He)] folgendermaßen definiert: Eine mecha-
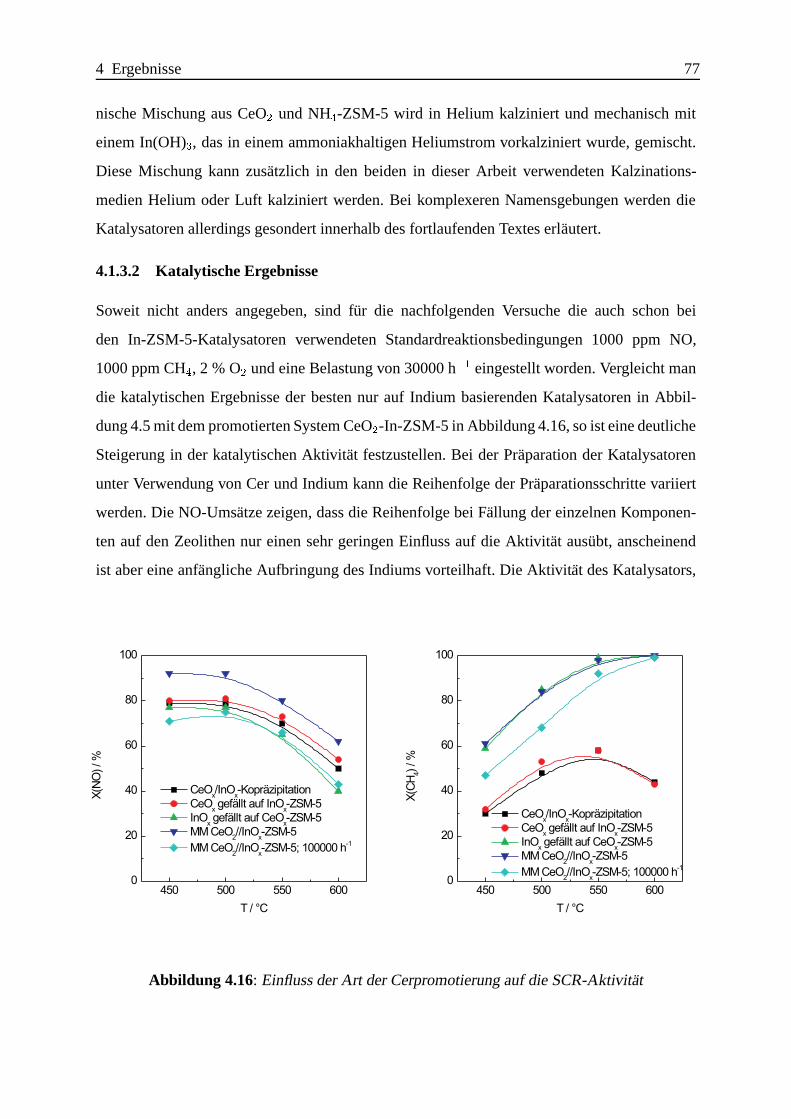
4 Ergebnisse 77
nische Mischung aus CeO2 und NH4-ZSM-5 wird in Helium kalziniert und mechanisch mit
einem In(OH)3, das in einem ammoniakhaltigen Heliumstrom vorkalziniert wurde, gemischt.
Diese Mischung kann zus¨atzlich in den beiden in dieser Arbeit verwendeten Kalzinations-
medien Helium oder Luft kalziniert werden. Bei komplexeren Namensgebungen werden die
Katalysatoren allerdings gesondert innerhalb des fortlaufenden Textes erl¨autert.
4.1.3.2 Katalytische Ergebnisse
Soweit nicht anders angegeben, sind f¨ur die nachfolgenden Versuche die auch schon bei
den In-ZSM-5-Katalysatoren verwendeten Standardreaktionsbedingungen 1000 ppm NO,
1000 ppm CH4, 2 % O2 und eine Belastung von 30000 h�1 eingestellt worden. Vergleicht man
die katalytischen Ergebnisse der besten nur auf Indium basierenden Katalysatoren in Abbil-
dung 4.5 mit dem promotierten System CeO2-In-ZSM-5 in Abbildung 4.16, so ist eine deutliche
Steigerung in der katalytischen Aktivit¨at festzustellen. Bei der Pr¨aparation der Katalysatoren
unter Verwendung von Cer und Indium kann die Reihenfolge der Pr¨aparationsschritte variiert
werden. Die NO-Ums¨atze zeigen, dass die Reihenfolge bei F¨allung der einzelnen Komponen-
ten auf den Zeolithen nur einen sehr geringen Einfluss auf die Aktivit¨at ausubt, anscheinend
ist aber eine anf¨angliche Aufbringung des Indiums vorteilhaft. Die Aktivit¨at des Katalysators,
450 500 550 6000
20
40
60
80
100
CeOx/InO
x-Kopräzipitation
CeOxgefällt auf InO
x-ZSM-5
InOxgefällt auf CeO
x-ZSM-5
MM CeO2//InO
x-ZSM-5
MM CeO2//InO
x-ZSM-5; 100000 h
-1
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
CeOx/InO
x-Kopräzipitation
CeOxgefällt auf InO
x-ZSM-5
InOxgefällt auf CeO
x-ZSM-5
MM CeO2//InO
x-ZSM-5
MM CeO2//InO
x-ZSM-5; 100000 h
-1
X(N
O)/%
T / °C
Abbildung 4.16: Einfluss der Art der Cerpromotierung auf die SCR-Aktivit¨at

4 Ergebnisse 78
bei dem erst Cer und dann Indium aufgef¨allt wird, ist geringer als bei dem durch Kopr¨azipita-
tion praparierten Katalysator; der durch F¨allung von Cer auf das vorher aufgebrachte Indium
dargestellte Katalysator ist wiederum aktiver. Erkl¨arbar ist dies mit Hilfe der Vermutung, dass
eine moglichst enge Nachbarschaft des Indiums zu dem Zeolithen f¨ur eine hohe SCR-Aktivit¨at
notig ist. Durch das vorher gef¨allte Cer wird ein Teil der Zeolithoberfl¨ache blockiert, und das
Indium wird von ihr abgeschirmt. Bei der Kopr¨azipitation stehen Cer und Indium in Konkur-
renz um die Zeolithoberfl¨ache; lediglich bei der Erstbeladung mit Indium steht die komplette
Zeolithoberflache zur Wechselwirkung mit dem Indium bereit, und dies ergibt den aktivsten
Katalysator.
Gleichzeitig zeigte der Katalysator, bei dem Indium zuletzt aufgebracht wurde, eine sehr
hohe Methanaktivierung. Letzteres ist ein Anzeichen daf¨ur, dass Indium auf der ¨außeren Ober-
flache des Zeolithen m¨oglicherweise zur unselektiven Methantotaloxidation beitragen kann.
Eine nachtr¨agliche mechanische Beimengung eines CeO2 mit hoher Oberfl¨ache
(ca. 200 m2/g) zeichnet sich als die Pr¨aparationsmethode der Wahl aus, da sie eine auch f¨ur die
Industrie einfache Promotierungsmethode darstellt und zu den aktivsten Katalysatoren f¨uhrt.
Im Temperaturbereich von 400 - 450ÆC werden NO-Ums¨atze oberhalb von 90 % erreicht, auch
bei einer erh¨ohten Belastung von 100000 h�1 werden noch NO-Ums¨atze von 70 % erzielt.
In der Abbildung 4.17 werden der Einfluss von Wasser im Eduktgasgemisch und von
Natriumionen in einer Katalysatormischung selber vorgestellt.
Der CeOx/InOx-ZSM-5-Katalysator zeigt eine erstaunlich hohe SCR-Aktivit¨at unter
2 % Wasser im Eduktgas. Bei 550ÆC betragt der NO-Umsatz immer noch 60 %. Nach R¨uck-
tausch der Protonen bzw. Ammoniumionen des Zeolithen mit Natriumnitrat bricht auch hier
wie beim In-ZSM-5-System die katalytische Aktivit¨at zusammen. Der Erkl¨arungsansatz ist
analog zu dem Deutungsversuch, den wir bei der Besprechung des In-ZSM-5-Systems vorge-
stellt haben. Entweder ist ein reduktiver Festk¨orperionentausch aufgrund fehlender Ammoni-
umionen nicht mehr m¨oglich, oder aber Protonen spielen in einem bifunktionellen Reaktions-
mechanismus eine Rolle (siehe Seite 62).
Um dieRolle des CeO2 zu untersuchen, wurden schon vonLieseVersuche mit unterschied-
lichen Verteilungen des CeO2 durchgefuhrt. Ziel ist, die Reichweite einer eventuellen bifunk-
tionellen Wechselwirkung zu testen. Ist die Rolle des CeO2 lediglich die NO-Oxidation, so
sollte bei Trennung der Komponenten der NO-Umsatz nicht vollst¨andig zusammenbrechen.
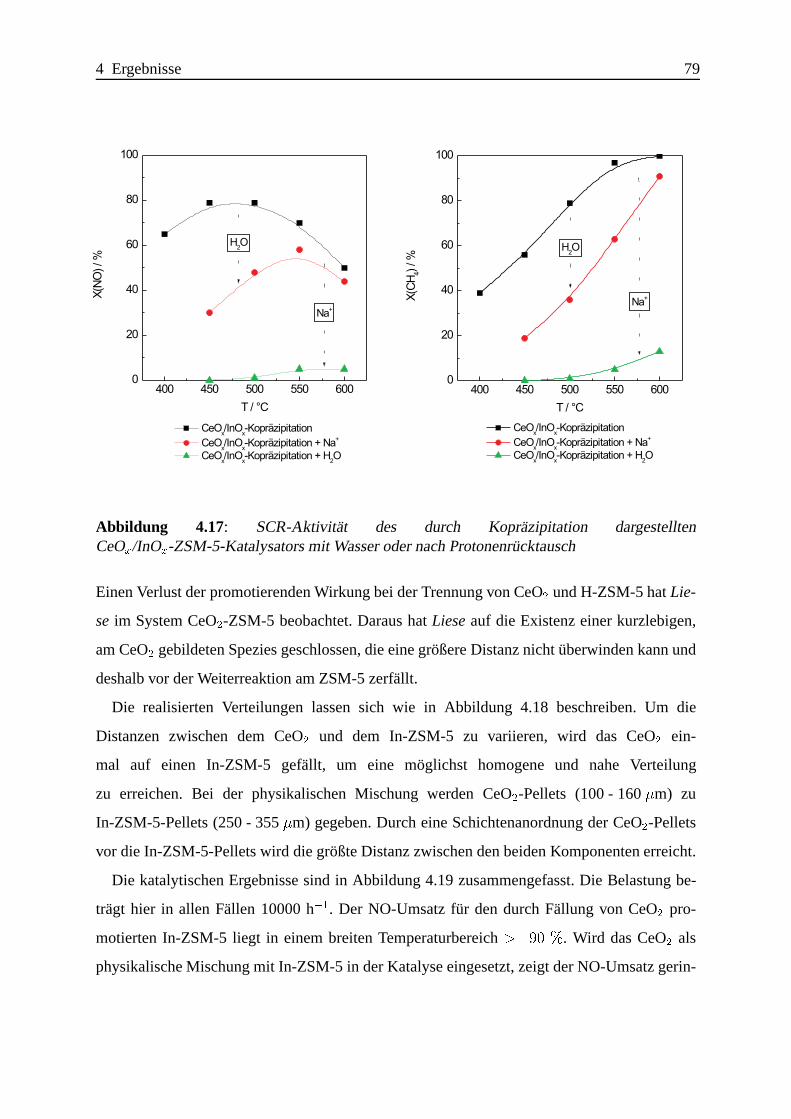
4 Ergebnisse 79
400 450 500 550 6000
20
40
60
80
100
Na+
H2O
CeOx/InO
x-Kopräzipitation
CeOx/InO
x-Kopräzipitation + Na
+
CeOx/InO
x-Kopräzipitation + H
2O
X(N
O)/%
T / °C
400 450 500 550 6000
20
40
60
80
100
Na+
H2O
CeOx/InO
x-Kopräzipitation
CeOx/InO
x-Kopräzipitation + Na
+
CeOx/InO
x-Kopräzipitation + H
2O
X(C
H4)
/%
T / °C
Abbildung 4.17: SCR-Aktivitat des durch Kopr¨azipitation dargestelltenCeOx/InOx-ZSM-5-Katalysators mit Wasser oder nach Protonenr¨ucktausch
Einen Verlust der promotierenden Wirkung bei der Trennung von CeO2 und H-ZSM-5 hatLie-
se im System CeO2-ZSM-5 beobachtet. Daraus hatLieseauf die Existenz einer kurzlebigen,
am CeO2 gebildeten Spezies geschlossen, die eine gr¨oßere Distanz nicht ¨uberwinden kann und
deshalb vor der Weiterreaktion am ZSM-5 zerf¨allt.
Die realisierten Verteilungen lassen sich wie in Abbildung 4.18 beschreiben. Um die
Distanzen zwischen dem CeO2 und dem In-ZSM-5 zu variieren, wird das CeO2 ein-
mal auf einen In-ZSM-5 gef¨allt, um eine m¨oglichst homogene und nahe Verteilung
zu erreichen. Bei der physikalischen Mischung werden CeO2-Pellets (100 - 160�m) zu
In-ZSM-5-Pellets (250 - 355�m) gegeben. Durch eine Schichtenanordnung der CeO2-Pellets
vor die In-ZSM-5-Pellets wird die gr¨oßte Distanz zwischen den beiden Komponenten erreicht.
Die katalytischen Ergebnisse sind in Abbildung 4.19 zusammengefasst. Die Belastung be-
tragt hier in allen F¨allen 10000 h�1. Der NO-Umsatz f¨ur den durch F¨allung von CeO2 pro-
motierten In-ZSM-5 liegt in einem breiten Temperaturbereich> 90 %. Wird das CeO2 als
physikalische Mischung mit In-ZSM-5 in der Katalyse eingesetzt, zeigt der NO-Umsatz gerin-
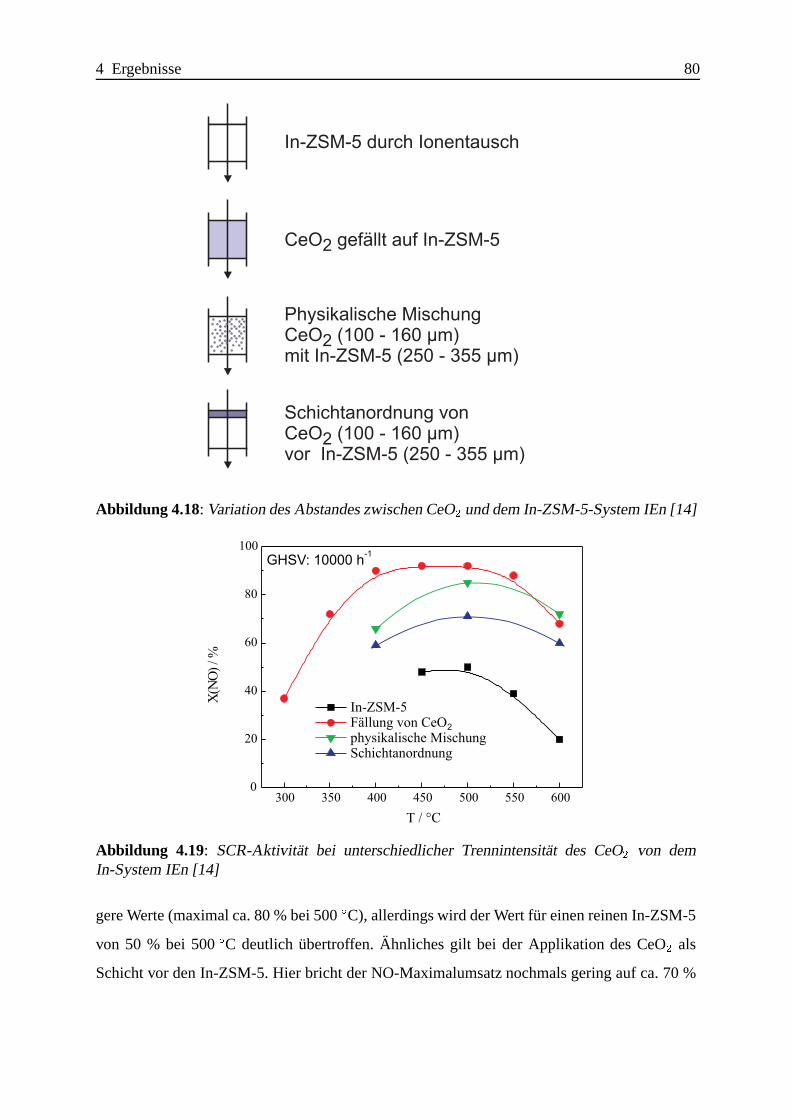
4 Ergebnisse 80
In-ZSM-5 durch Ionentausch
CeO gefällt auf In-ZSM-52
Physikalische MischungCeO (100 - 160 µm)mit In-ZSM-5 (250 - 355 µm)
2
Schichtanordnung vonCeO (100 - 160 µm)vor In-ZSM-5 (250 - 355 µm)
2
Abbildung 4.18: Variation des Abstandes zwischen CeO2 und dem In-ZSM-5-System IEn [14]
300 350 400 450 500 550 6000
20
40
60
80
100
2
-1GHSV: 10000 h
In-ZSM-5Fällung von CeOphysikalische MischungSchichtanordnung
X(N
O)
/%
T / °C
Abbildung 4.19: SCR-Aktivitat bei unterschiedlicher Trennintensit¨at des CeO2 von demIn-System IEn [14]
gere Werte (maximal ca. 80 % bei 500ÆC), allerdings wird der Wert f¨ur einen reinen In-ZSM-5
von 50 % bei 500ÆC deutlichubertroffen.Ahnliches gilt bei der Applikation des CeO2 als
Schicht vor den In-ZSM-5. Hier bricht der NO-Maximalumsatz nochmals gering auf ca. 70 %
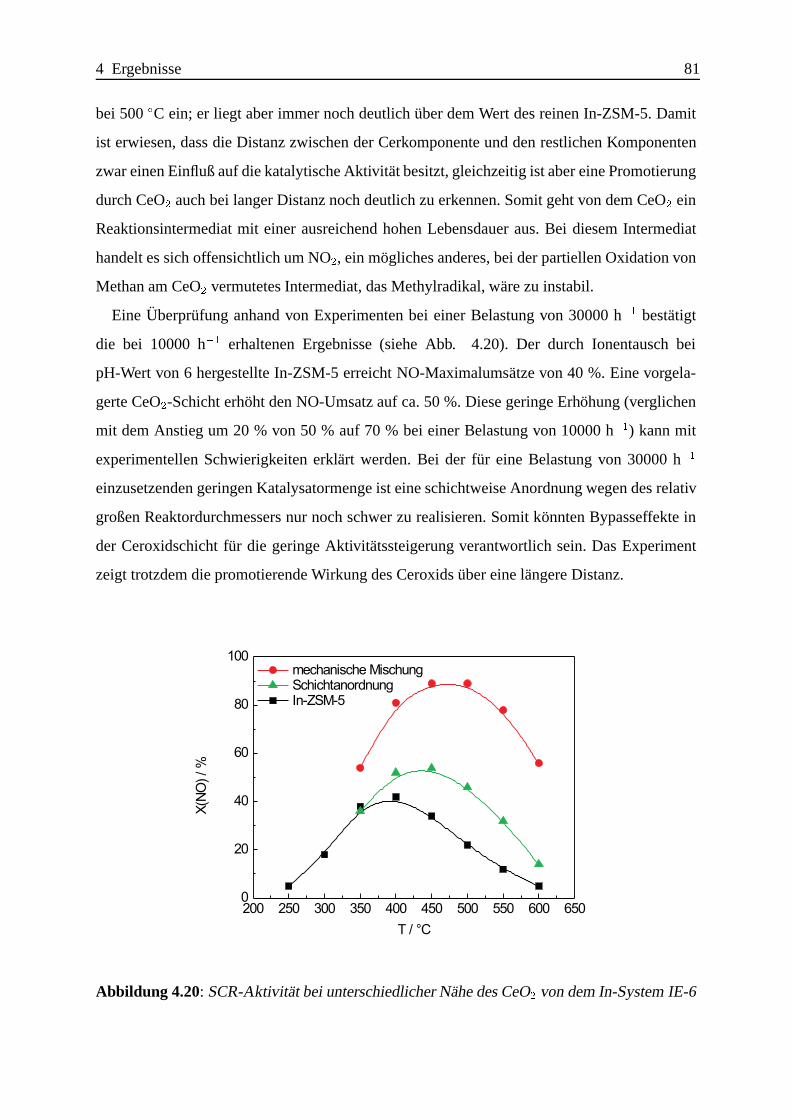
4 Ergebnisse 81
bei 500ÆC ein; er liegt aber immer noch deutlich ¨uber dem Wert des reinen In-ZSM-5. Damit
ist erwiesen, dass die Distanz zwischen der Cerkomponente und den restlichen Komponenten
zwar einen Einfluß auf die katalytische Aktivit¨at besitzt, gleichzeitig ist aber eine Promotierung
durch CeO2 auch bei langer Distanz noch deutlich zu erkennen. Somit geht von dem CeO2 ein
Reaktionsintermediat mit einer ausreichend hohen Lebensdauer aus. Bei diesem Intermediat
handelt es sich offensichtlich um NO2, ein mogliches anderes, bei der partiellen Oxidation von
Methan am CeO2 vermutetes Intermediat, das Methylradikal, w¨are zu instabil.
Eine Uberprufung anhand von Experimenten bei einer Belastung von 30000 h�1 bestatigt
die bei 10000 h�1 erhaltenen Ergebnisse (siehe Abb. 4.20). Der durch Ionentausch bei
pH-Wert von 6 hergestellte In-ZSM-5 erreicht NO-Maximalums¨atze von 40 %. Eine vorgela-
gerte CeO2-Schicht erh¨oht den NO-Umsatz auf ca. 50 %. Diese geringe Erh¨ohung (verglichen
mit dem Anstieg um 20 % von 50 % auf 70 % bei einer Belastung von 10000 h�1) kann mit
experimentellen Schwierigkeiten erkl¨art werden. Bei der f¨ur eine Belastung von 30000 h�1
einzusetzenden geringen Katalysatormenge ist eine schichtweise Anordnung wegen des relativ
großen Reaktordurchmessers nur noch schwer zu realisieren. Somit k¨onnten Bypasseffekte in
der Ceroxidschicht f¨ur die geringe Aktivitatssteigerung verantwortlich sein. Das Experiment
zeigt trotzdem die promotierende Wirkung des Ceroxids ¨uber eine langere Distanz.
200 250 300 350 400 450 500 550 600 6500
20
40
60
80
100mechanische MischungSchichtanordnungIn-ZSM-5
X(N
O)/%
T / °C
Abbildung 4.20: SCR-Aktivitat bei unterschiedlicher N¨ahe des CeO2 von dem In-System IE-6
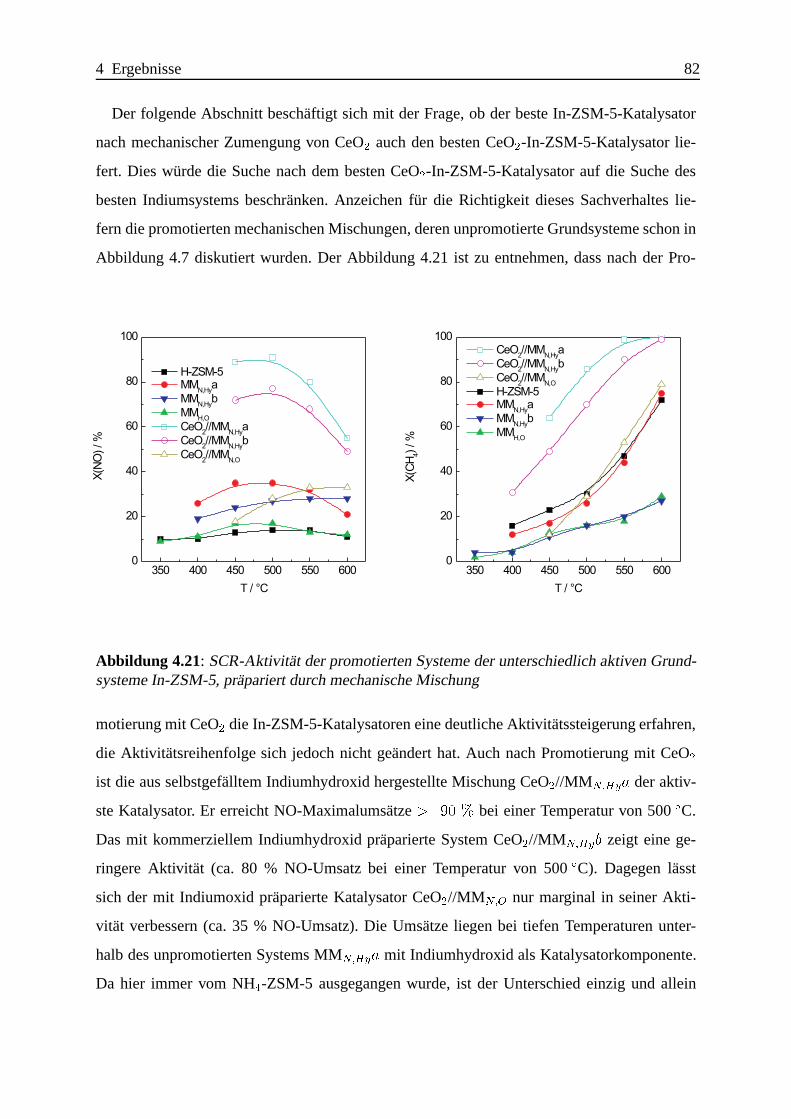
4 Ergebnisse 82
Der folgende Abschnitt besch¨aftigt sich mit der Frage, ob der beste In-ZSM-5-Katalysator
nach mechanischer Zumengung von CeO2 auch den besten CeO2-In-ZSM-5-Katalysator lie-
fert. Dies wurde die Suche nach dem besten CeO2-In-ZSM-5-Katalysator auf die Suche des
besten Indiumsystems beschr¨anken. Anzeichen f¨ur die Richtigkeit dieses Sachverhaltes lie-
fern die promotierten mechanischen Mischungen, deren unpromotierte Grundsysteme schon in
Abbildung 4.7 diskutiert wurden. Der Abbildung 4.21 ist zu entnehmen, dass nach der Pro-
350 400 450 500 550 6000
20
40
60
80
100
H-ZSM-5MM
N,Hya
MMN,Hy
b
MMH,O
CeO2//MM
N,Hya
CeO2//MM
N,Hyb
CeO2//MM
N,O
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100CeO
2//MM
N,Hya
CeO2//MM
N,Hyb
CeO2//MM
N,O
H-ZSM-5MM
N,Hya
MMN,Hy
b
MMH,O
X(C
H4)
/%
T / °C
Abbildung 4.21: SCR-Aktivitat der promotierten Systeme der unterschiedlich aktiven Grund-systeme In-ZSM-5, pr¨apariert durch mechanische Mischung
motierung mit CeO2 die In-ZSM-5-Katalysatoren eine deutliche Aktivit¨atssteigerung erfahren,
die Aktivitatsreihenfolge sich jedoch nicht ge¨andert hat. Auch nach Promotierung mit CeO2
ist die aus selbstgef¨alltem Indiumhydroxid hergestellte Mischung CeO2//MMN;Hya der aktiv-
ste Katalysator. Er erreicht NO-Maximalums¨atze> 90 % bei einer Temperatur von 500ÆC.
Das mit kommerziellem Indiumhydroxid pr¨aparierte System CeO2//MMN;Hyb zeigt eine ge-
ringere Aktivitat (ca. 80 % NO-Umsatz bei einer Temperatur von 500ÆC). Dagegen l¨asst
sich der mit Indiumoxid pr¨aparierte Katalysator CeO2//MMN;O nur marginal in seiner Akti-
vitat verbessern (ca. 35 % NO-Umsatz). Die Ums¨atze liegen bei tiefen Temperaturen unter-
halb des unpromotierten Systems MMN;Hya mit Indiumhydroxid als Katalysatorkomponente.
Da hier immer vom NH4-ZSM-5 ausgegangen wurde, ist der Unterschied einzig und allein
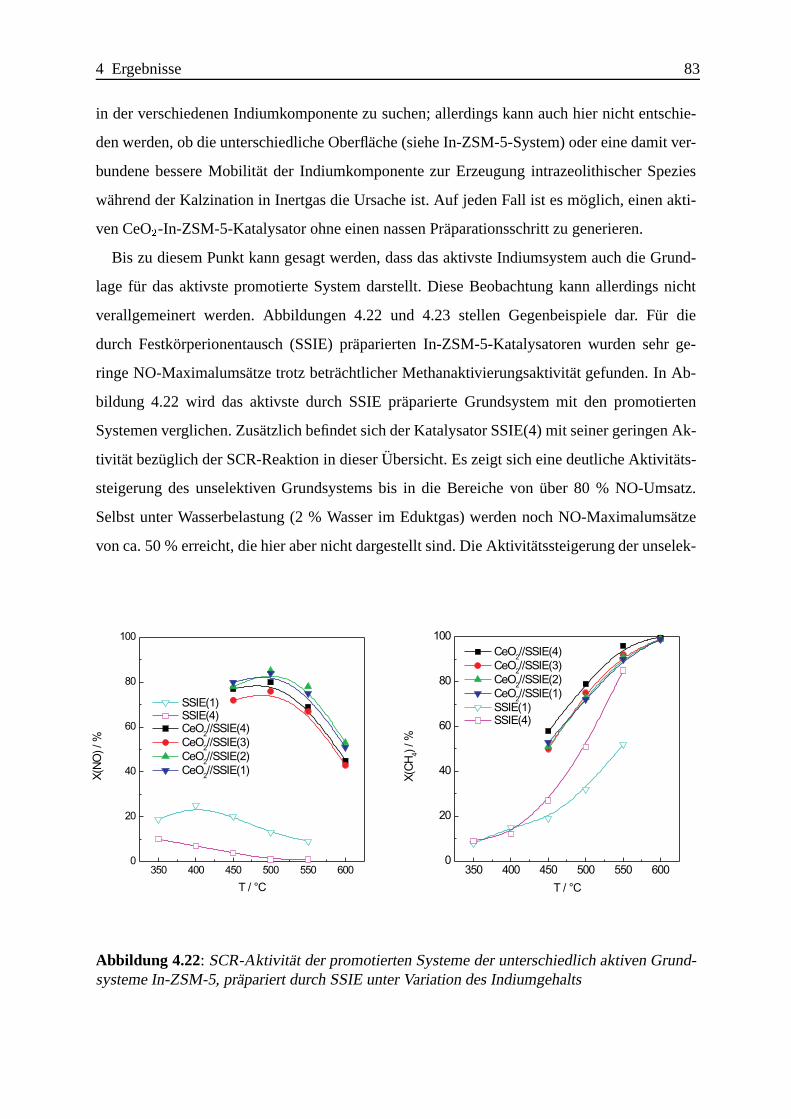
4 Ergebnisse 83
in der verschiedenen Indiumkomponente zu suchen; allerdings kann auch hier nicht entschie-
den werden, ob die unterschiedliche Oberfl¨ache (siehe In-ZSM-5-System) oder eine damit ver-
bundene bessere Mobilit¨at der Indiumkomponente zur Erzeugung intrazeolithischer Spezies
wahrend der Kalzination in Inertgas die Ursache ist. Auf jeden Fall ist es m¨oglich, einen akti-
ven CeO2-In-ZSM-5-Katalysator ohne einen nassen Pr¨aparationsschritt zu generieren.
Bis zu diesem Punkt kann gesagt werden, dass das aktivste Indiumsystem auch die Grund-
lage fur das aktivste promotierte System darstellt. Diese Beobachtung kann allerdings nicht
verallgemeinert werden. Abbildungen 4.22 und 4.23 stellen Gegenbeispiele dar. F¨ur die
durch Festk¨orperionentausch (SSIE) pr¨aparierten In-ZSM-5-Katalysatoren wurden sehr ge-
ringe NO-Maximalums¨atze trotz betr¨achtlicher Methanaktivierungsaktivit¨at gefunden. In Ab-
bildung 4.22 wird das aktivste durch SSIE pr¨aparierte Grundsystem mit den promotierten
Systemen verglichen. Zus¨atzlich befindet sich der Katalysator SSIE(4) mit seiner geringen Ak-
tivit at bezuglich der SCR-Reaktion in dieserUbersicht. Es zeigt sich eine deutliche Aktivit¨ats-
steigerung des unselektiven Grundsystems bis in die Bereiche von ¨uber 80 % NO-Umsatz.
Selbst unter Wasserbelastung (2 % Wasser im Eduktgas) werden noch NO-Maximalums¨atze
von ca. 50 % erreicht, die hier aber nicht dargestellt sind. Die Aktivit¨atssteigerung der unselek-
350 400 450 500 550 6000
20
40
60
80
100
CeO2//SSIE(4)
CeO2//SSIE(3)
CeO2//SSIE(2)
CeO2//SSIE(1)
SSIE(1)SSIE(4)
X(C
H4)
/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
SSIE(1)SSIE(4)CeO
2//SSIE(4)
CeO2//SSIE(3)
CeO2//SSIE(2)
CeO2//SSIE(1)
X(N
O)/%
T / °C
Abbildung 4.22: SCR-Aktivitat der promotierten Systeme der unterschiedlich aktiven Grund-systeme In-ZSM-5, pr¨apariert durch SSIE unter Variation des Indiumgehalts
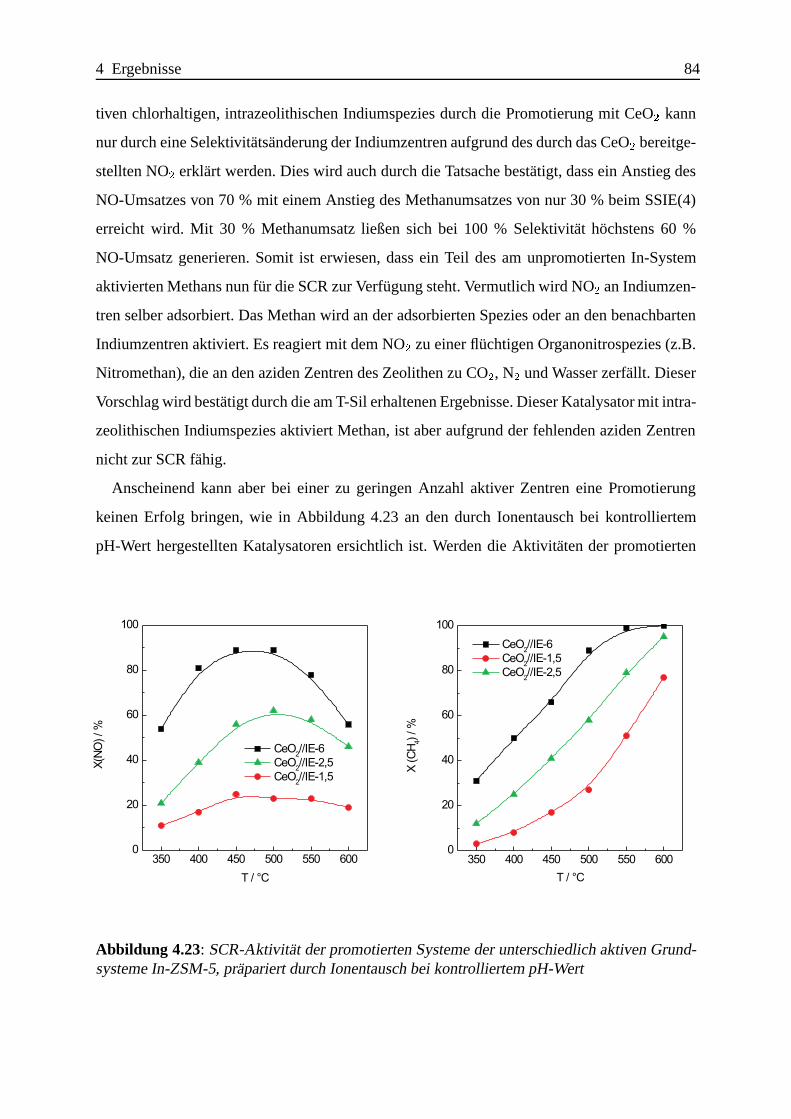
4 Ergebnisse 84
tiven chlorhaltigen, intrazeolithischen Indiumspezies durch die Promotierung mit CeO2 kann
nur durch eine Selektivit¨atsanderung der Indiumzentren aufgrund des durch das CeO2 bereitge-
stellten NO2 erklart werden. Dies wird auch durch die Tatsache best¨atigt, dass ein Anstieg des
NO-Umsatzes von 70 % mit einem Anstieg des Methanumsatzes von nur 30 % beim SSIE(4)
erreicht wird. Mit 30 % Methanumsatz ließen sich bei 100 % Selektivit¨at hochstens 60 %
NO-Umsatz generieren. Somit ist erwiesen, dass ein Teil des am unpromotierten In-System
aktivierten Methans nun f¨ur die SCR zur Verf¨ugung steht. Vermutlich wird NO2 an Indiumzen-
tren selber adsorbiert. Das Methan wird an der adsorbierten Spezies oder an den benachbarten
Indiumzentren aktiviert. Es reagiert mit dem NO2 zu einer fluchtigen Organonitrospezies (z.B.
Nitromethan), die an den aziden Zentren des Zeolithen zu CO2, N2 und Wasser zerf¨allt. Dieser
Vorschlag wird best¨atigt durch die am T-Sil erhaltenen Ergebnisse. Dieser Katalysator mit intra-
zeolithischen Indiumspezies aktiviert Methan, ist aber aufgrund der fehlenden aziden Zentren
nicht zur SCR fahig.
Anscheinend kann aber bei einer zu geringen Anzahl aktiver Zentren eine Promotierung
keinen Erfolg bringen, wie in Abbildung 4.23 an den durch Ionentausch bei kontrolliertem
pH-Wert hergestellten Katalysatoren ersichtlich ist. Werden die Aktivit¨aten der promotierten
350 400 450 500 550 6000
20
40
60
80
100
CeO2//IE-6
CeO2//IE-2,5
CeO2//IE-1,5
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
CeO2//IE-6
CeO2//IE-1,5
CeO2//IE-2,5
X(C
H4)
/%
T / °C
Abbildung 4.23: SCR-Aktivitat der promotierten Systeme der unterschiedlich aktiven Grund-systeme In-ZSM-5, pr¨apariert durch Ionentausch bei kontrolliertem pH-Wert

4 Ergebnisse 85
Systeme in Abbildung 4.23 mit dem unpromotierten System aus Abbildung 4.11 verglichen,
so lasst sich f¨ur IE-6 eine deutliche Aktivit¨atsteigerung von 43 % NO-Umsatz bei 400ÆC auf
90 % bei 500ÆC erkennen. F¨ur IE-2,5 ist diese Steigerung (von 38 % NO-Umsatz bei 400ÆC
auf 60 % bei 500ÆC) weniger stark ausgepr¨agt, und bei IE-1,5 liefert eine Promotierung mit
CeO2 einen NO-Maximalumsatz (23 % bei 500ÆC), der unterhalb des NO-Maximalumsatzes
des unpromotierten In-ZSM-5-Systems (33 %) liegt. Anscheinend reicht bei IE-1,5 die Anzahl
der aktiven Zentren nicht aus, um die Aktivit¨at zu steigern. M¨oglicherweise sind sie vollst¨andig
mit NO2 bedeckt und besitzen keine benachbarten Indiumzentren zur Methanaktivierung mehr;
allerdings spricht der immer noch hohe Methanumsatz gegen letzte Hypothese. Durch die Bil-
dung von NO2 konnte aber schon vonLieseeine gesteigerte unselektive Methanoxidation am
Zeolithen selber beobachtet werden, wobei NO2 als Oxidationsmittel wirkt und auf die Stufe
des NO reduziert wird. Daher ist es wahrscheinlich, dass das Methan in einer Konkurrenzreak-
tion unselektiv oxidiert wird, f¨ur die eigentliche SCR-Reaktion aber nicht gen¨ugend Indium-
zentren zur Verf¨ugung stehen. Neben diesem Erkl¨arungsansatz muss jedoch auch in Betracht
gezogen werden, dass die Katalysatoren vermutlich Indiumspezies mit unterschiedlicher Koor-
dinationssph¨are enthalten (vide supra).
Der folgende Abschnitt befasst sich mit derRolle der Zeolithprotonen. Erste Anzeichen
fur eine kritische Rolle finden sich in den Experimenten mit dem Katalysator T-Sil, der kei-
ne Protonen enth¨alt und trotz Methanumsatzes keine SCR-Aktivit¨at aufweist, sowie in dem
mit Natriumionen r¨uckgetauschten IEn, der weder NO- noch Methanums¨atze zeigt. Bei letzte-
rem Katalysator kann aber nicht ausgeschlossen werden, dass keine aktiven intrazeolithischen
Spezies mehr vorliegen, da diese beim R¨ucktausch verdr¨angt worden sein k¨onnten und der
Ruckweg per SSIE durch die Natriumionen behindert ist. Weitere Anzeichen finden sich in
den SSIE-Proben, bei denen mit sinkendem Protonengehalt auch eine sinkende SCR-Aktivit¨at
festgestellt werden konnte.
Mit Hilfe eines selektiven Vergiftungsexperiments sollte die postulierte kritische Rolle der
Protonen eingehender untersucht werden. Die Idee bestand darin, durch einen Festk¨orperionen-
tausch (SSIE) mit KBr die Protonen des Katalysators CeO2//IE-6 unter Abgabe von HBr gegen
Kaliumionen auszutauschen und somit zu defunktionalisieren.
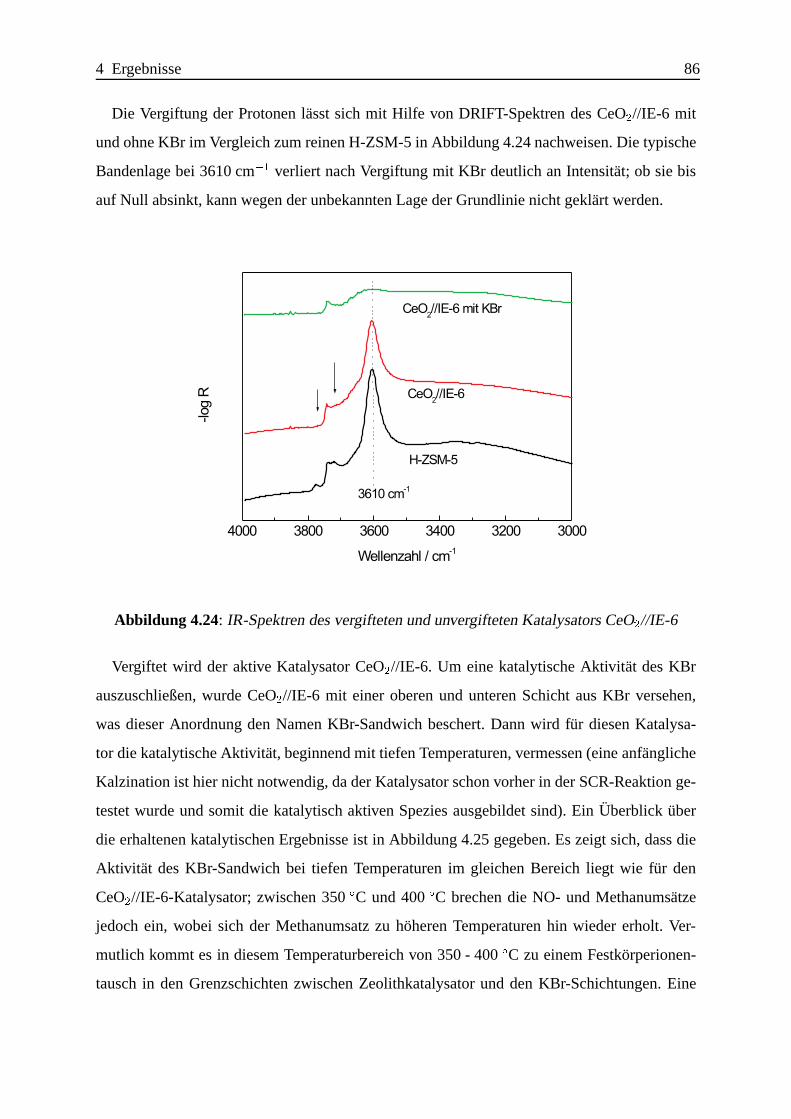
4 Ergebnisse 86
Die Vergiftung der Protonen l¨asst sich mit Hilfe von DRIFT-Spektren des CeO2//IE-6 mit
und ohne KBr im Vergleich zum reinen H-ZSM-5 in Abbildung 4.24 nachweisen. Die typische
Bandenlage bei 3610 cm�1 verliert nach Vergiftung mit KBr deutlich an Intensit¨at; ob sie bis
auf Null absinkt, kann wegen der unbekannten Lage der Grundlinie nicht gekl¨art werden.
4000 3800 3600 3400 3200 3000
3610 cm-1
H-ZSM-5
CeO2//IE-6
CeO2//IE-6 mit KBr
-log
R
Wellenzahl / cm-1
Abbildung 4.24: IR-Spektren des vergifteten und unvergifteten Katalysators CeO2//IE-6
Vergiftet wird der aktive Katalysator CeO2//IE-6. Um eine katalytische Aktivit¨at des KBr
auszuschließen, wurde CeO2//IE-6 mit einer oberen und unteren Schicht aus KBr versehen,
was dieser Anordnung den Namen KBr-Sandwich beschert. Dann wird f¨ur diesen Katalysa-
tor die katalytische Aktivit¨at, beginnend mit tiefen Temperaturen, vermessen (eine anf¨angliche
Kalzination ist hier nicht notwendig, da der Katalysator schon vorher in der SCR-Reaktion ge-
testet wurde und somit die katalytisch aktiven Spezies ausgebildet sind). EinUberblick uber
die erhaltenen katalytischen Ergebnisse ist in Abbildung 4.25 gegeben. Es zeigt sich, dass die
Aktivit at des KBr-Sandwich bei tiefen Temperaturen im gleichen Bereich liegt wie f¨ur den
CeO2//IE-6-Katalysator; zwischen 350ÆC und 400ÆC brechen die NO- und Methanums¨atze
jedoch ein, wobei sich der Methanumsatz zu h¨oheren Temperaturen hin wieder erholt. Ver-
mutlich kommt es in diesem Temperaturbereich von 350 - 400ÆC zu einem Festk¨orperionen-
tausch in den Grenzschichten zwischen Zeolithkatalysator und den KBr-Schichtungen. Eine
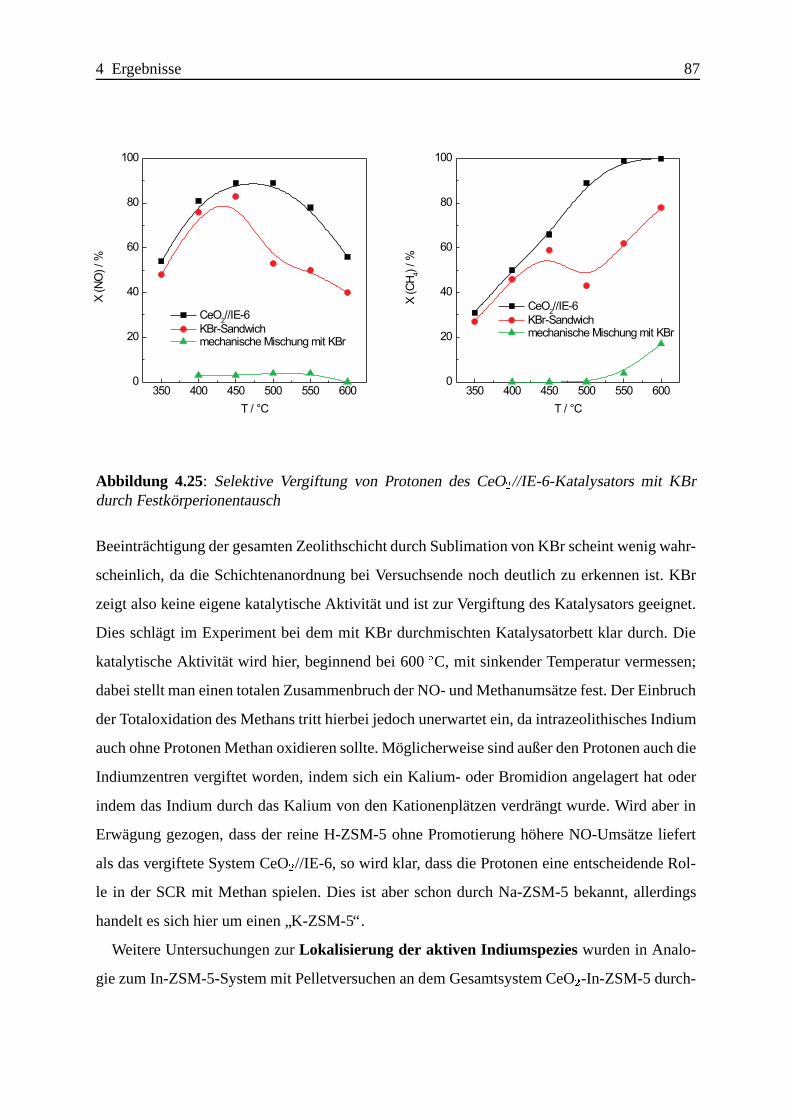
4 Ergebnisse 87
350 400 450 500 550 6000
20
40
60
80
100
CeO2//IE-6
KBr-Sandwichmechanische Mischung mit KBr
X(C
H4)
/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
CeO2//IE-6
KBr-Sandwichmechanische Mischung mit KBr
X(N
O)/%
T / °C
Abbildung 4.25: Selektive Vergiftung von Protonen des CeO2//IE-6-Katalysators mit KBrdurch Festk¨orperionentausch
Beeintrachtigung der gesamten Zeolithschicht durch Sublimation von KBr scheint wenig wahr-
scheinlich, da die Schichtenanordnung bei Versuchsende noch deutlich zu erkennen ist. KBr
zeigt also keine eigene katalytische Aktivit¨at und ist zur Vergiftung des Katalysators geeignet.
Dies schlagt im Experiment bei dem mit KBr durchmischten Katalysatorbett klar durch. Die
katalytische Aktivitat wird hier, beginnend bei 600ÆC, mit sinkender Temperatur vermessen;
dabei stellt man einen totalen Zusammenbruch der NO- und Methanums¨atze fest. Der Einbruch
der Totaloxidation des Methans tritt hierbei jedoch unerwartet ein, da intrazeolithisches Indium
auch ohne Protonen Methan oxidieren sollte. M¨oglicherweise sind außer den Protonen auch die
Indiumzentren vergiftet worden, indem sich ein Kalium- oder Bromidion angelagert hat oder
indem das Indium durch das Kalium von den Kationenpl¨atzen verdr¨angt wurde. Wird aber in
Erwagung gezogen, dass der reine H-ZSM-5 ohne Promotierung h¨ohere NO-Ums¨atze liefert
als das vergiftete System CeO2//IE-6, so wird klar, dass die Protonen eine entscheidende Rol-
le in der SCR mit Methan spielen. Dies ist aber schon durch Na-ZSM-5 bekannt, allerdings
handelt es sich hier um einen”K-ZSM-5“.
Weitere Untersuchungen zurLokalisierung der aktiven Indiumspezieswurden in Analo-
gie zum In-ZSM-5-System mit Pelletversuchen an dem Gesamtsystem CeO2-In-ZSM-5 durch-
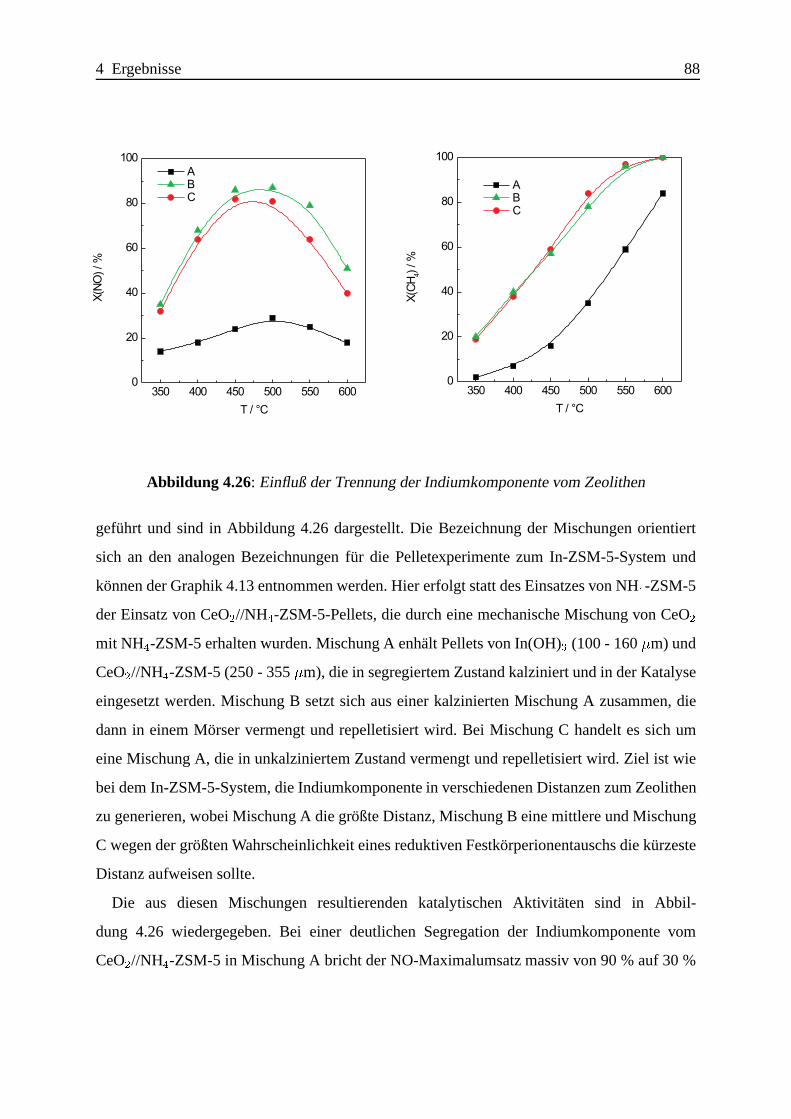
4 Ergebnisse 88
350 400 450 500 550 6000
20
40
60
80
100
ABC
X(C
H4)
/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
ABC
X(N
O)/%
T / °C
Abbildung 4.26: Einfluß der Trennung der Indiumkomponente vom Zeolithen
gefuhrt und sind in Abbildung 4.26 dargestellt. Die Bezeichnung der Mischungen orientiert
sich an den analogen Bezeichnungen f¨ur die Pelletexperimente zum In-ZSM-5-System und
konnen der Graphik 4.13 entnommen werden. Hier erfolgt statt des Einsatzes von NH4-ZSM-5
der Einsatz von CeO2//NH4-ZSM-5-Pellets, die durch eine mechanische Mischung von CeO2
mit NH4-ZSM-5 erhalten wurden. Mischung A enh¨alt Pellets von In(OH)3 (100 - 160�m) und
CeO2//NH4-ZSM-5 (250 - 355�m), die in segregiertem Zustand kalziniert und in der Katalyse
eingesetzt werden. Mischung B setzt sich aus einer kalzinierten Mischung A zusammen, die
dann in einem M¨orser vermengt und repelletisiert wird. Bei Mischung C handelt es sich um
eine Mischung A, die in unkalziniertem Zustand vermengt und repelletisiert wird. Ziel ist wie
bei dem In-ZSM-5-System, die Indiumkomponente in verschiedenen Distanzen zum Zeolithen
zu generieren, wobei Mischung A die gr¨oßte Distanz, Mischung B eine mittlere und Mischung
C wegen der gr¨oßten Wahrscheinlichkeit eines reduktiven Festk¨orperionentauschs die k¨urzeste
Distanz aufweisen sollte.
Die aus diesen Mischungen resultierenden katalytischen Aktivit¨aten sind in Abbil-
dung 4.26 wiedergegeben. Bei einer deutlichen Segregation der Indiumkomponente vom
CeO2//NH4-ZSM-5 in Mischung A bricht der NO-Maximalumsatz massiv von 90 % auf 30 %
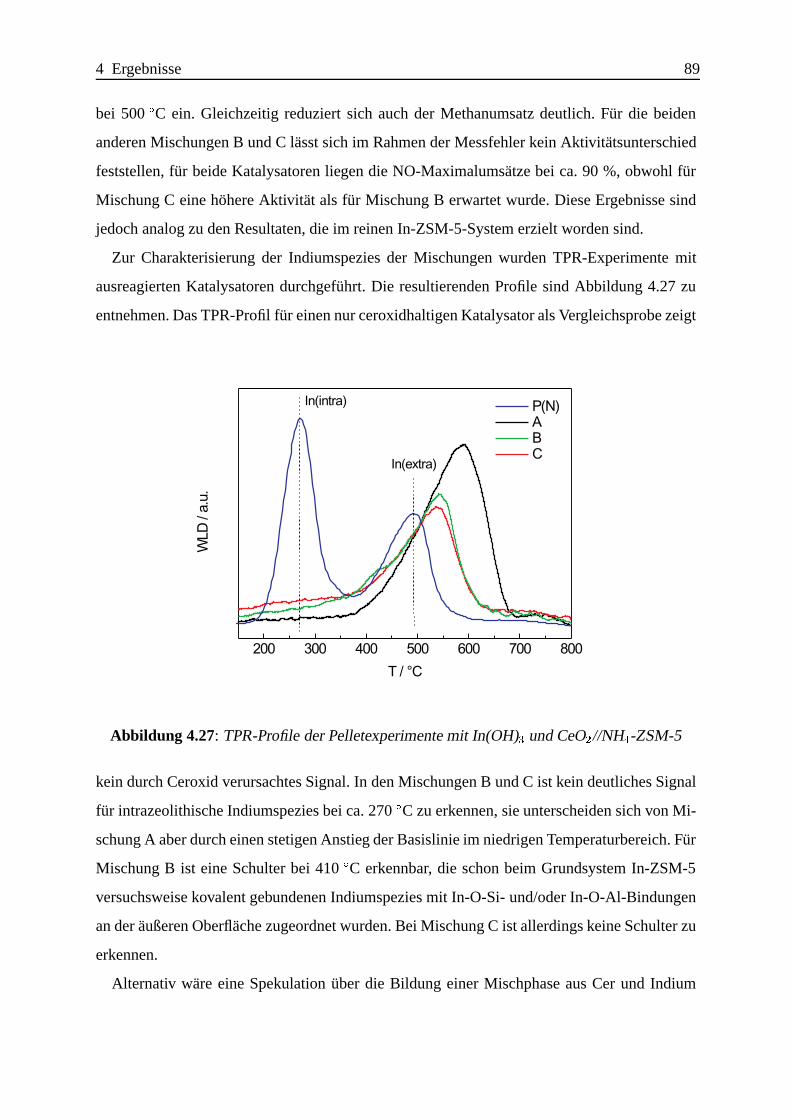
4 Ergebnisse 89
bei 500ÆC ein. Gleichzeitig reduziert sich auch der Methanumsatz deutlich. F¨ur die beiden
anderen Mischungen B und C l¨asst sich im Rahmen der Messfehler kein Aktivit¨atsunterschied
feststellen, f¨ur beide Katalysatoren liegen die NO-Maximalums¨atze bei ca. 90 %, obwohl f¨ur
Mischung C eine h¨ohere Aktivitat als fur Mischung B erwartet wurde. Diese Ergebnisse sind
jedoch analog zu den Resultaten, die im reinen In-ZSM-5-System erzielt worden sind.
Zur Charakterisierung der Indiumspezies der Mischungen wurden TPR-Experimente mit
ausreagierten Katalysatoren durchgef¨uhrt. Die resultierenden Profile sind Abbildung 4.27 zu
entnehmen. Das TPR-Profil f¨ur einen nur ceroxidhaltigen Katalysator als Vergleichsprobe zeigt
200 300 400 500 600 700 800
In(extra)
In(intra) P(N)ABC
WLD
/a.u
.
T / °C
Abbildung 4.27: TPR-Profile der Pelletexperimente mit In(OH)3 und CeO2//NH4-ZSM-5
kein durch Ceroxid verursachtes Signal. In den Mischungen B und C ist kein deutliches Signal
fur intrazeolithische Indiumspezies bei ca. 270ÆC zu erkennen, sie unterscheiden sich von Mi-
schung A aber durch einen stetigen Anstieg der Basislinie im niedrigen Temperaturbereich. F¨ur
Mischung B ist eine Schulter bei 410ÆC erkennbar, die schon beim Grundsystem In-ZSM-5
versuchsweise kovalent gebundenen Indiumspezies mit In-O-Si- und/oder In-O-Al-Bindungen
an deraußeren Oberfl¨ache zugeordnet wurden. Bei Mischung C ist allerdings keine Schulter zu
erkennen.
Alternativ ware eine Spekulation ¨uber die Bildung einer Mischphase aus Cer und Indium

4 Ergebnisse 90
moglich, wie sie z.B. vonSachtleret al. fur Palladium und Kobalt auf einem Mordeniten [122]
gefunden wurde. Nach der Charakterisierung mittels XRD, TPR, TEM und EXAFS postulie-
ren die Autoren die Existenz einer bimetallischen Pd-Co-Phase, die bei tiefen Temperaturen das
fur die Methanaktivierung in der SCR-Reaktion notwendige Pd0 stabilisiert. Bei h¨oheren Tem-
peraturen kommt es zur Bildung von PdO, welches lediglich die unselektive Totaloxidation
des Methans bewirkt. VonLombardoet al. wurde fur Kobalt und Platin auf einem Mordeni-
ten [123,124] durch TPR und XPS eine Wechselwirkung zwischen den beiden Komponenten
gefunden, die sich in einer geringeren Reduktionstemperatur des Kobalts zeigt. Auf diese Wei-
se erklaren die genannten Autoren die erh¨ohte katalytische Aktivit¨at eines Pt-Co-ZSM-5, die
bei einer mechanischen Mischung aus Co-ZSM-5 und Pt-ZSM-5 hingegen nicht zu beobachten
ist. Die Bildung einer Mischphase aus Cer und Indium scheint hier jedoch wenig wahrschein-
lich, da die erw¨ahnten Schultern im TPR-Diagramm auch im Grundsystem In-ZSM-5 ohne
Cerzusatz auftreten.
Die TPR-Profile zeigen jedoch, dass im Tieftemperaturbereich ein Unterschied zu der kata-
lytisch nicht aktiven Mischung A besteht; eine eindeutige Interpretation ist aber nicht m¨oglich,
da es sich nicht um ein klares Signal handelt, sondern, wie oben bereits erw¨ahnt, um einen
kontinuierlichen Anstieg der Basislinie.
Bei Untersuchungen zum Autoreduktionsverhalten mechanischer Mischungen unterver-
schiedenen Kalzinationsbedingungenwurden allerdings auch Tieftemperatursignale f¨ur ka-
talytisch wenig aktive Mischungen erhalten, wie den Abbildungen 4.28 und 4.29 zu entnehmen
ist.
Ab hier weichen die Katalysatorbezeichnungen von den anfangs definierten Bezeichnungen
ab. Es handelt sich ausschließlich um mechanische Mischungen aus drei Komponenten, die all-
gemein mit Cerkomponente//Indiumkomponente//Zeolithkomponente bezeichnet werden. Die
Mischung dieser Komponenten sowie die Komponenten selbst k¨onnen verschiedenen Vorbe-
handlungen unterzogen werden. Diese Vorbehandlung wird f¨ur jede Komponente angegeben;
keine Angabe bedeutet, dass die Komponente nicht vorbehandelt wurde.
Sowohl fur die in Helium als auch die in 10 % Sauerstoff kalzinierten mechanischen Mi-
schungen CeO2//In(OH)3//NH4-ZSM-5 zeigt sich im Tieftemperaturbereich eine deutliche
Schulter, trotzdem unterscheiden sie sich gravierend in ihrer katalytischen Aktivit¨at (siehe Ab-
bildung 4.29) . Kalzination in Helium f¨uhrt bei Verwendung der Ammoniumform des Kataly-
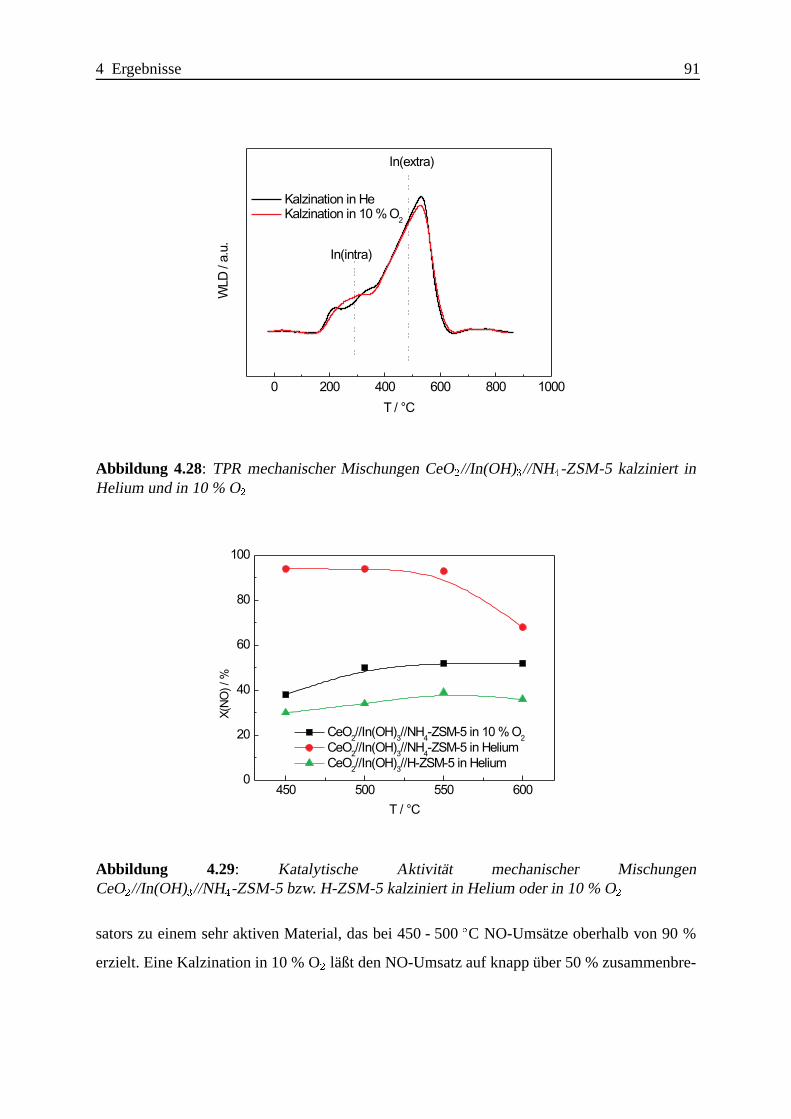
4 Ergebnisse 91
0 200 400 600 800 1000
In(extra)
In(intra)
Kalzination in HeKalzination in 10 % O
2W
LD
/a.u
.
T / °C
Abbildung 4.28: TPR mechanischer Mischungen CeO2//In(OH)3//NH4-ZSM-5 kalziniert inHelium und in 10 % O2
450 500 550 6000
20
40
60
80
100
X(N
O)/%
CeO2//In(OH)
3//NH
4-ZSM-5 in 10 % O
2
CeO2//In(OH)
3//NH
4-ZSM-5 in Helium
CeO2//In(OH)
3//H-ZSM-5 in Helium
T / °C
Abbildung 4.29: Katalytische Aktivitat mechanischer MischungenCeO2//In(OH)3//NH4-ZSM-5 bzw. H-ZSM-5 kalziniert in Helium oder in 10 % O2
sators zu einem sehr aktiven Material, das bei 450 - 500ÆC NO-Umsatze oberhalb von 90 %
erzielt. Eine Kalzination in 10 % O2 laßt den NO-Umsatz auf knapp ¨uber 50 % zusammenbre-
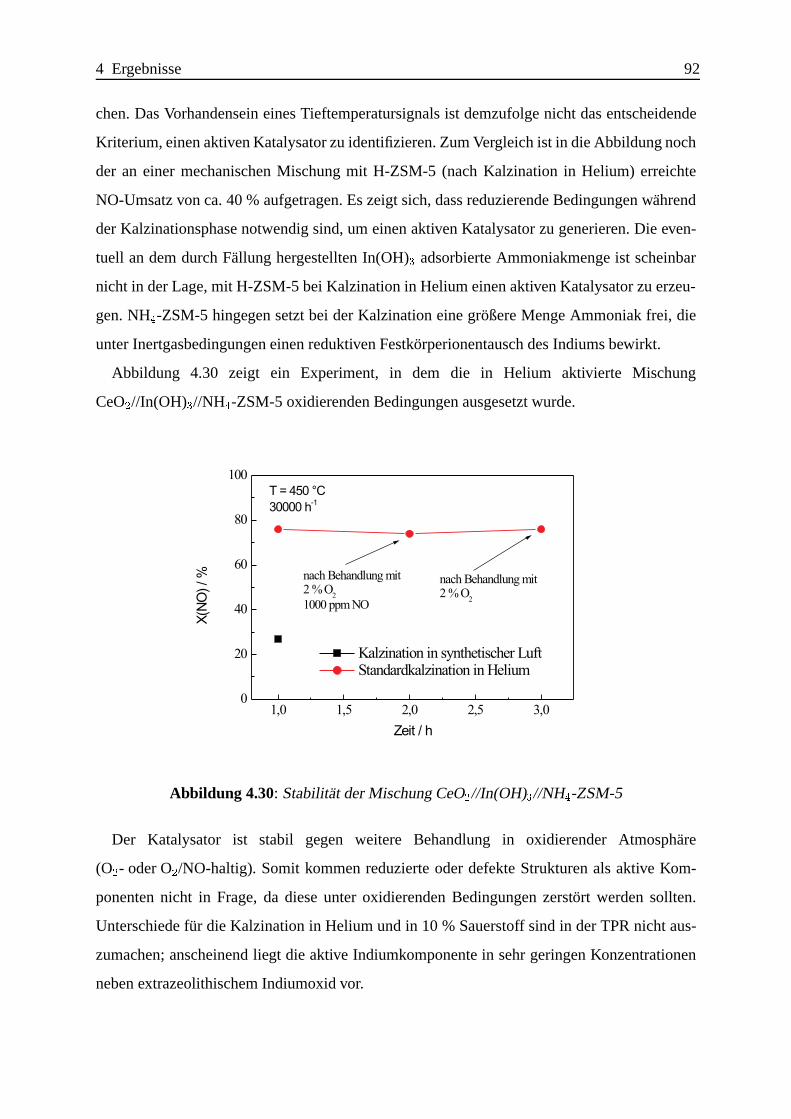
4 Ergebnisse 92
chen. Das Vorhandensein eines Tieftemperatursignals ist demzufolge nicht das entscheidende
Kriterium, einen aktiven Katalysator zu identifizieren. Zum Vergleich ist in die Abbildung noch
der an einer mechanischen Mischung mit H-ZSM-5 (nach Kalzination in Helium) erreichte
NO-Umsatz von ca. 40 % aufgetragen. Es zeigt sich, dass reduzierende Bedingungen w¨ahrend
der Kalzinationsphase notwendig sind, um einen aktiven Katalysator zu generieren. Die even-
tuell an dem durch F¨allung hergestellten In(OH)3 adsorbierte Ammoniakmenge ist scheinbar
nicht in der Lage, mit H-ZSM-5 bei Kalzination in Helium einen aktiven Katalysator zu erzeu-
gen. NH4-ZSM-5 hingegen setzt bei der Kalzination eine gr¨oßere Menge Ammoniak frei, die
unter Inertgasbedingungen einen reduktiven Festk¨orperionentausch des Indiums bewirkt.
Abbildung 4.30 zeigt ein Experiment, in dem die in Helium aktivierte Mischung
CeO2//In(OH)3//NH4-ZSM-5 oxidierenden Bedingungen ausgesetzt wurde.
1,0 1,5 2,0 2,5 3,00
20
40
60
80
100
T = 450 °C
30000 h-1
nach Behandlung mit2 % O
2
nach Behandlung mit2 % O
2
1000 ppm NO
Kalzination in synthetischer LuftStandardkalzination in Helium
X(N
O)/%
Zeit / h
Abbildung 4.30: Stabilitat der Mischung CeO2//In(OH)3//NH4-ZSM-5
Der Katalysator ist stabil gegen weitere Behandlung in oxidierender Atmosph¨are
(O2- oder O2/NO-haltig). Somit kommen reduzierte oder defekte Strukturen als aktive Kom-
ponenten nicht in Frage, da diese unter oxidierenden Bedingungen zerst¨ort werden sollten.
Unterschiede f¨ur die Kalzination in Helium und in 10 % Sauerstoff sind in der TPR nicht aus-
zumachen; anscheinend liegt die aktive Indiumkomponente in sehr geringen Konzentrationen
neben extrazeolithischem Indiumoxid vor.
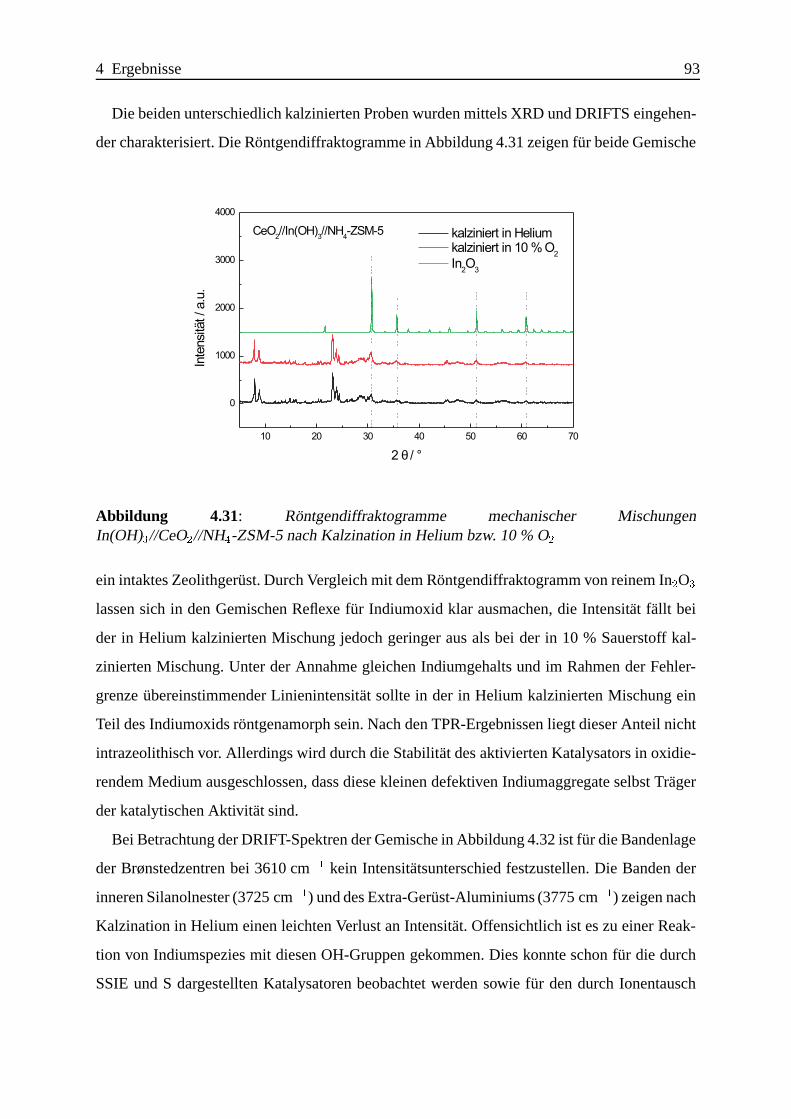
4 Ergebnisse 93
Die beiden unterschiedlich kalzinierten Proben wurden mittels XRD und DRIFTS eingehen-
der charakterisiert. Die R¨ontgendiffraktogramme in Abbildung 4.31 zeigen f¨ur beide Gemische
10 20 30 40 50 60 70
0
1000
2000
3000
4000
CeO2//In(OH)
3//NH
4-ZSM-5 kalziniert in Helium
kalziniert in 10 % O2
In2O
3
Inte
nsi
tät/a.u
.
2 θ / °
Abbildung 4.31: Rontgendiffraktogramme mechanischer MischungenIn(OH)3//CeO2//NH4-ZSM-5 nach Kalzination in Helium bzw. 10 % O2
ein intaktes Zeolithger¨ust. Durch Vergleich mit dem R¨ontgendiffraktogramm von reinem In2O3
lassen sich in den Gemischen Reflexe f¨ur Indiumoxid klar ausmachen, die Intensit¨at fallt bei
der in Helium kalzinierten Mischung jedoch geringer aus als bei der in 10 % Sauerstoff kal-
zinierten Mischung. Unter der Annahme gleichen Indiumgehalts und im Rahmen der Fehler-
grenzeubereinstimmender Linienintensit¨at sollte in der in Helium kalzinierten Mischung ein
Teil des Indiumoxids r¨ontgenamorph sein. Nach den TPR-Ergebnissen liegt dieser Anteil nicht
intrazeolithisch vor. Allerdings wird durch die Stabilit¨at des aktivierten Katalysators in oxidie-
rendem Medium ausgeschlossen, dass diese kleinen defektiven Indiumaggregate selbst Tr¨ager
der katalytischen Aktivit¨at sind.
Bei Betrachtung der DRIFT-Spektren der Gemische in Abbildung 4.32 ist f¨ur die Bandenlage
der Brønstedzentren bei 3610 cm�1 kein Intensitatsunterschied festzustellen. Die Banden der
inneren Silanolnester (3725 cm�1) und des Extra-Ger¨ust-Aluminiums (3775 cm�1) zeigen nach
Kalzination in Helium einen leichten Verlust an Intensit¨at. Offensichtlich ist es zu einer Reak-
tion von Indiumspezies mit diesen OH-Gruppen gekommen. Dies konnte schon f¨ur die durch
SSIE und S dargestellten Katalysatoren beobachtet werden sowie f¨ur den durch Ionentausch
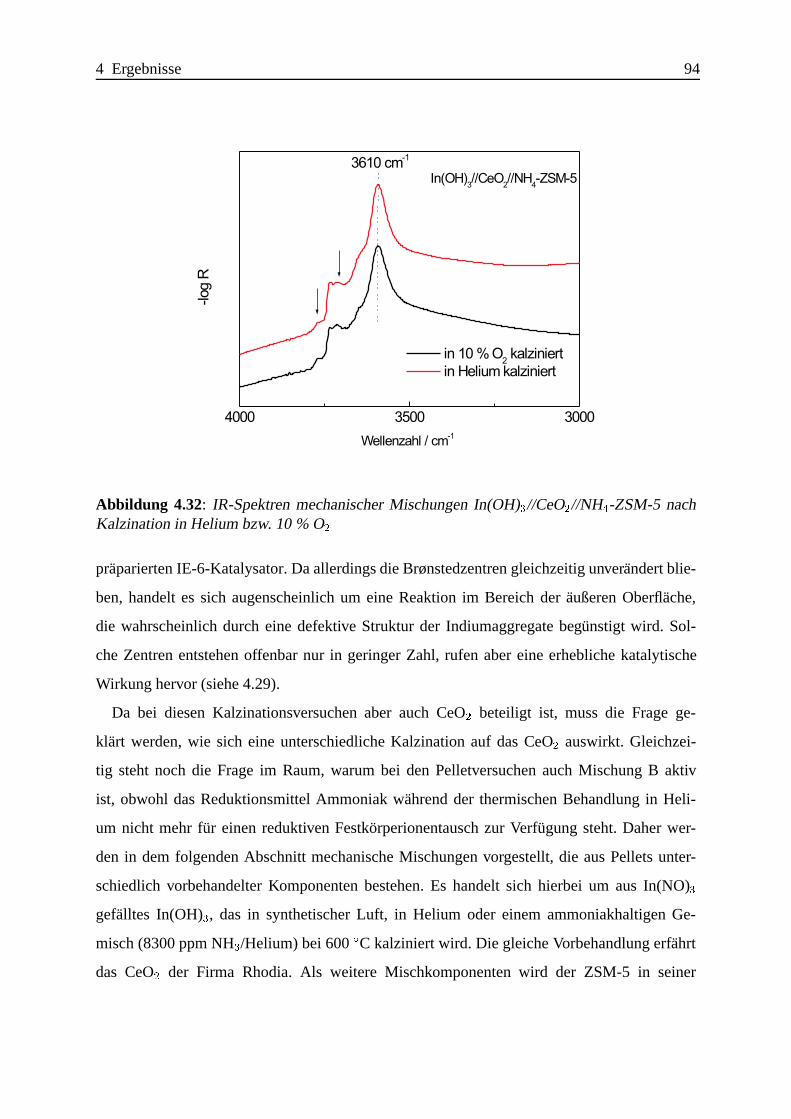
4 Ergebnisse 94
4000 3500 3000
Wellenzahl / cm-1
In(OH)3//CeO
2//NH
4-ZSM-5
3610 cm-1
in 10 % O2kalziniert
in Helium kalziniert
-log
R
Abbildung 4.32: IR-Spektren mechanischer Mischungen In(OH)3//CeO2//NH4-ZSM-5 nachKalzination in Helium bzw. 10 % O2
praparierten IE-6-Katalysator. Da allerdings die Brønstedzentren gleichzeitig unver¨andert blie-
ben, handelt es sich augenscheinlich um eine Reaktion im Bereich der ¨außeren Oberfl¨ache,
die wahrscheinlich durch eine defektive Struktur der Indiumaggregate beg¨unstigt wird. Sol-
che Zentren entstehen offenbar nur in geringer Zahl, rufen aber eine erhebliche katalytische
Wirkung hervor (siehe 4.29).
Da bei diesen Kalzinationsversuchen aber auch CeO2 beteiligt ist, muss die Frage ge-
klart werden, wie sich eine unterschiedliche Kalzination auf das CeO2 auswirkt. Gleichzei-
tig steht noch die Frage im Raum, warum bei den Pelletversuchen auch Mischung B aktiv
ist, obwohl das Reduktionsmittel Ammoniak w¨ahrend der thermischen Behandlung in Heli-
um nicht mehr f¨ur einen reduktiven Festk¨orperionentausch zur Verf¨ugung steht. Daher wer-
den in dem folgenden Abschnitt mechanische Mischungen vorgestellt, die aus Pellets unter-
schiedlich vorbehandelter Komponenten bestehen. Es handelt sich hierbei um aus In(NO)3
gefalltes In(OH)3, das in synthetischer Luft, in Helium oder einem ammoniakhaltigen Ge-
misch (8300 ppm NH3/Helium) bei 600ÆC kalziniert wird. Die gleiche Vorbehandlung erf¨ahrt
das CeO2 der Firma Rhodia. Als weitere Mischkomponenten wird der ZSM-5 in seiner
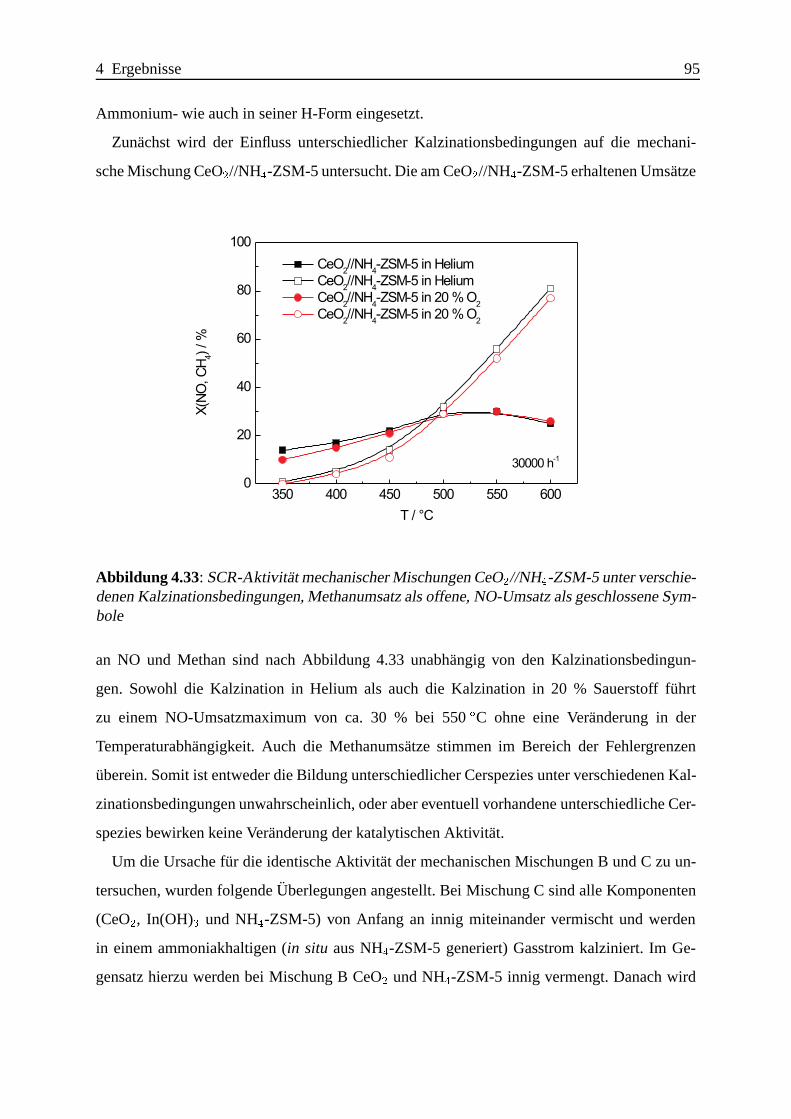
4 Ergebnisse 95
Ammonium- wie auch in seiner H-Form eingesetzt.
Zunachst wird der Einfluss unterschiedlicher Kalzinationsbedingungen auf die mechani-
sche Mischung CeO2//NH4-ZSM-5 untersucht. Die am CeO2//NH4-ZSM-5 erhaltenen Ums¨atze
350 400 450 500 550 6000
20
40
60
80
100
30000 h-1
CeO2//NH
4-ZSM-5 in Helium
CeO2//NH
4-ZSM-5 in Helium
CeO2//NH
4-ZSM-5 in 20 % O
2
CeO2//NH
4-ZSM-5 in 20 % O
2
X(N
O,C
H4)
/%
T / °C
Abbildung 4.33: SCR-Aktivitat mechanischer Mischungen CeO2//NH4-ZSM-5 unter verschie-denen Kalzinationsbedingungen, Methanumsatz als offene, NO-Umsatz als geschlossene Sym-bole
an NO und Methan sind nach Abbildung 4.33 unabh¨angig von den Kalzinationsbedingun-
gen. Sowohl die Kalzination in Helium als auch die Kalzination in 20 % Sauerstoff f¨uhrt
zu einem NO-Umsatzmaximum von ca. 30 % bei 550ÆC ohne eine Ver¨anderung in der
Temperaturabh¨angigkeit. Auch die Methanums¨atze stimmen im Bereich der Fehlergrenzen
uberein. Somit ist entweder die Bildung unterschiedlicher Cerspezies unter verschiedenen Kal-
zinationsbedingungen unwahrscheinlich, oder aber eventuell vorhandene unterschiedliche Cer-
spezies bewirken keine Ver¨anderung der katalytischen Aktivit¨at.
Um die Ursache f¨ur die identische Aktivit¨at der mechanischen Mischungen B und C zu un-
tersuchen, wurden folgendeUberlegungen angestellt. Bei Mischung C sind alle Komponenten
(CeO2, In(OH)3 und NH4-ZSM-5) von Anfang an innig miteinander vermischt und werden
in einem ammoniakhaltigen (in situ aus NH4-ZSM-5 generiert) Gasstrom kalziniert. Im Ge-
gensatz hierzu werden bei Mischung B CeO2 und NH4-ZSM-5 innig vermengt. Danach wird
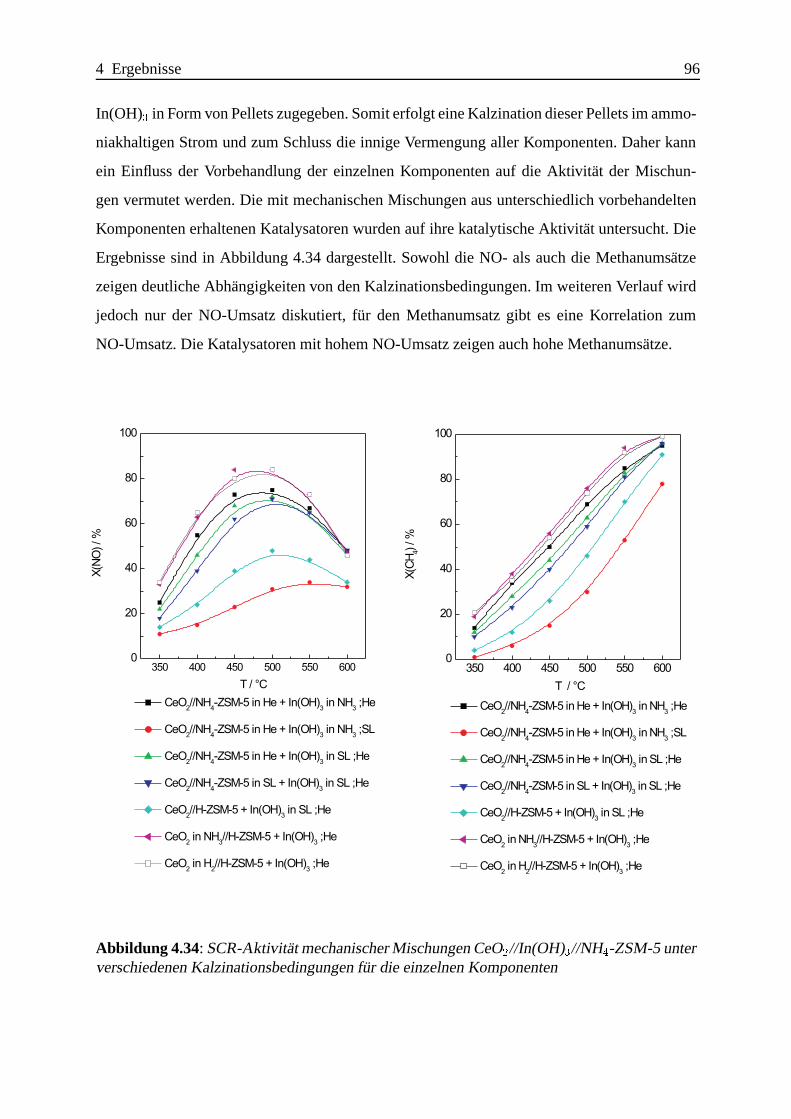
4 Ergebnisse 96
In(OH)3 in Form von Pellets zugegeben. Somit erfolgt eine Kalzination dieser Pellets im ammo-
niakhaltigen Strom und zum Schluss die innige Vermengung aller Komponenten. Daher kann
ein Einfluss der Vorbehandlung der einzelnen Komponenten auf die Aktivit¨at der Mischun-
gen vermutet werden. Die mit mechanischen Mischungen aus unterschiedlich vorbehandelten
Komponenten erhaltenen Katalysatoren wurden auf ihre katalytische Aktivit¨at untersucht. Die
Ergebnisse sind in Abbildung 4.34 dargestellt. Sowohl die NO- als auch die Methanums¨atze
zeigen deutliche Abh¨angigkeiten von den Kalzinationsbedingungen. Im weiteren Verlauf wird
jedoch nur der NO-Umsatz diskutiert, f¨ur den Methanumsatz gibt es eine Korrelation zum
NO-Umsatz. Die Katalysatoren mit hohem NO-Umsatz zeigen auch hohe Methanums¨atze.
350 400 450 500 550 6000
20
40
60
80
100
CeO2//NH
4-ZSM-5 in He + In(OH)
3in NH
3;He
CeO2//NH
4-ZSM-5 in He + In(OH)
3in NH
3;SL
CeO2//NH
4-ZSM-5 in He + In(OH)
3in SL ;He
CeO2//NH
4-ZSM-5 in SL + In(OH)
3in SL ;He
CeO2//H-ZSM-5 + In(OH)
3in SL ;He
CeO2in NH
3//H-ZSM-5 + In(OH)
3;He
CeO2in H
2//H-ZSM-5 + In(OH)
3;He
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
CeO2//NH
4-ZSM-5 in He + In(OH)
3in NH
3;He
CeO2//NH
4-ZSM-5 in He + In(OH)
3in NH
3;SL
CeO2//NH
4-ZSM-5 in He + In(OH)
3in SL ;He
CeO2//NH
4-ZSM-5 in SL + In(OH)
3in SL ;He
CeO2//H-ZSM-5 + In(OH)
3in SL ;He
CeO2in NH
3//H-ZSM-5 + In(OH)
3;He
CeO2in H
2//H-ZSM-5 + In(OH)
3;He
X(C
H4)
/%
T / °C
Abbildung 4.34: SCR-Aktivitat mechanischer Mischungen CeO2//In(OH)3//NH4-ZSM-5 unterverschiedenen Kalzinationsbedingungen f¨ur die einzelnen Komponenten

4 Ergebnisse 97
Als erstes wird der Katalysator [(CeO2//NH4-ZSM-5 in He) + (In(OH)3) in NH3)] bei Kal-
zination in Helium und in synthetischer Luft verglichen. Hierbei sind die Komponenten
CeO2//NH4-ZSM-5 in Helium sowie In(OH)3 im ammoniakhaltigen Gasstrom vorkalziniert
und dann mechanisch vermengt worden; diese Vorgehensweise entspricht also Katalysator B.
Kalzination des Gesamtgemisches in Helium liefert einen sehr aktiven Katalysator; es werden
NO-Umsatze von ca. 85 % bei 450 - 500ÆC erhalten. Wie erwartet, ist das in synthetischer
Luft kalzinierte Gesamtgemisch wenig aktiv, der maximale NO-Umsatz betr¨agt ca. 30 % bei
einer hoheren Temperatur von 550ÆC. Das durch Vorkalzination des CeO2 mit der Ammoni-
umform des Zeolithen in Helium erhaltene Material ist nach Mischen mit der Indiumkompo-
nente je nach Kalzinationsmedium Teil von Katalysatoren unterschiedlicher Aktivit¨at. Folglich
ist die Wirkung der unterschiedlichen Kalzinationsbedingungen in einem Einfluss auf die In-
diumkomponente und nicht auf den CeO2-Promotor zu suchen. Wie schon erw¨ahnt, scheint
die aktive Indiumkomponente durch eine Reaktion kleiner Indiumaggregate mit der ¨außeren
Oberflache des Zeolithen generiert zu werden. Diese Vermutung wird best¨arkt durch die an den
durch Ionentausch bei kontrolliertem pH-Wert pr¨aparierten Indiumkatalysatoren (siehe Abbil-
dung 4.23). Am wirkungsvollsten l¨asst sich der IE-6-Katalysator, der sowohl intra- als auch
extrazeolithische Indiumspezies enth¨alt, mit CeO2 promotieren. Die beiden anderen Katalysa-
toren IE-1,5 und IE-2,5 mit ausschließlich intrazeolithischem Indium zeigen keine bzw. nur
eine geringe promotierende Wirkung durch CeO2.
Da die Kalzination in synthetischer Luft offensichtlich zu Katalysatoren minderer Ak-
tivit at fuhrt und dies auf eine Einwirkung auf die Indiumkomponente zur¨uckzufuhren ist,
wurden mechanische Mischungen mit in synthetischer Luft vorkalziniertem Indiumhydroxid
durchgefuhrt. Die an den Katalysatoren [(CeO2//NH4-ZSM-5 in He) + (In(OH)3 in SL)] und
[(CeO2//NH4-ZSM-5 in SL) + (In(OH)3 in SL)] nach Kalzination der Gesamtmischung in He-
lium erreichten NO-Ums¨atze sind ebenfalls in Abbildung 4.34 dargestellt.Uberraschender-
weise zeigen beide Katalysatoren vergleichbar hohe NO-Ums¨atze von ca. 70 % bei 500ÆC;
allerdings liegen hier die NO-Ums¨atze unterhalb der NO-Ums¨atze der Katalysatormischung
[(CeO2//NH4-ZSM-5 in He) + (In(OH)3 in NH3)], die im ammoniakhaltigen Gasstrom vor-
kalziniertes Indiumhydroxid enth¨alt. Auch zwischen den beiden Katalysatoren mit in syn-
thetischer Luft vorkalziniertem Indiumhydroxid zeigt sich im Niedertemperaturbereich von
350 - 450ÆC ein Aktivitatsunterschied. Wird die Mischung CeO2//NH4-ZSM-5 in Helium

4 Ergebnisse 98
vorbehandelt, so werden h¨ohere NO-Ums¨atze generiert. F¨ur eine mechanische Mischung
[(CeO2//H-ZSM-5 + (In(OH)3 in SL)] erreicht der Katalysator in der SCR-Reaktion nur noch
NO-Umsatze von ca. 47 %. Dies ist ein erwartetes Ergebnis und best¨atigt die in Abbildung
4.29 erhaltenen Ergebnisse der mechanischen Mischung CeO2//In(OH)3//H-ZSM-5. Obwohl
durch die Fallung des In(OH)3 mit NH3 die Anwesenheit adsorbierten Ammoniaks nicht aus-
zuschließen ist, fehlt hier die f¨ur einen reduktiven Festk¨orperionentausch des Indiums auf Ka-
tionenplatze des Zeolithen notwendige Menge an Reduktionsmittel.
Wieso aber werden aktive Mischungen mit [(CeO2//NH4-ZSM-5 in He) + (In(OH)3 in SL)]
und [(CeO2//NH4-ZSM-5 in SL) + (In(OH)3 in SL)] erhalten, bei denen auch kein Ammoni-
ak mehr fur einen reduktiven Festk¨orperionentausch im letzten Schritt zur Verf¨ugung steht?
Der einzige Unterschied ist darin zu sehen, dass das CeO2 im Ammoniakstrom (in situ aus
NH4-ZSM-5) kalziniert und dabei m¨oglicherweise zum Ce3+ reduziert wurde. Tats¨achlich zei-
gen sich in den XP-Spektren aus Abbildung 4.35 der Mischungen CeO2//NH4-ZSM-5 sowohl
bei Kalzination in Helium als auch bei Kalzination in 20 % Sauerstoff Beitr¨age, die von Ce3+
stammen.
930 920 910 900 890 880 870 860 850 840
CeO2//NH
4-ZSM-5/
kalziniert in O2
CeO2
CeO2//NH
4-ZSM-5/
kalziniert in Helium
Beiträge von Ce3+
InCeZSM-5/vor SCR
InCeZSM-5/nach SCR
InCeZSM-5/Langzeit-Wasser
Bindungsenergie [eV]
Abbildung 4.35: XP-Spektren der unterschiedlich kalziniertenCeO2//NH4-ZSM-5-Mischungen im Vergleich zu Probenspektren vonLiese, die Ce3+
und Ce4+ enthalten, und im Vergleich zu reinem CeO2 mit ausschließlich Ce4+

4 Ergebnisse 99
Bei dem Vergleichskatalysator InCeZSM-5 vonLiesezeigt sich im Ausgangszustand (vor
der SCR) im Ce-3d-Spektrum die typische Satellitenstruktur des Ce4+. Auch fur reines CeO2
ist diese deutlich erkennbar. Nach der SCR-Reaktion, besonders nach einem Langzeittest mit
Wasser, sind Anzeichen von Ce3+ zu sehen.
Damit ware eine Erkl¨arung gegeben f¨ur die katalytische Aktivit¨at der Mischung
[(CeO2//NH4-ZSM-5 in SL) + (In(OH)3 in SL)].
Der Frage, ob Ce3+ fur die SCR n¨otig ist, wurde durch die Vorkalzinati-
on des CeO2 im ammoniakhaltigen Gasstrom nachgegangen. Der Katalysator
[(CeO2 in NH3//H-ZSM-5) + (In(OH)3)] nach Kalzination in Helium zeigt ¨uberraschen-
derweise eine sehr hohe Aktivit¨at und erreicht NO-Ums¨atze von ca. 85 %, obwohl sonst unter
Verwendung der H-Form des Zeolithen offenbar aufgrund des fehlenden Reduktionsmittels
keine aktiven Katalysatoren erhalten werden. Es liegt die Vermutung nahe, dass durch die
Behandlung in Ammoniak entstandenes Ce3+ als”Zwischenspeicher“dient und die Reduktion
des In(OH)3 auslost, die zu einer starken Wechselwirkung des Indiums mit der Oberfl¨ache
des Zeolithen f¨uhrt. Allerdings lasst sich adsorbiertes Ammoniak im IR-Spektrum eines im
ammoniakhaltigen Gasstrom kalzinierten CeO2 erkennen. Dieses adsorbierte Ammoniak
konnte fur die Reduktion w¨ahrend der weiteren Katalysatorpr¨aparation verantwortlich sein.
Ob die adsorbierte Ammoniakmenge ausreichend ist, kann nicht beurteilt werden, da das
IR-Spektrum nicht quantitativ auswertbar ist. Die Menge sollte jedoch gering sein, da die
Kalzination im Ammoniakstrom bei 600ÆC beendet wurde und danach eine Sp¨ulung in
Helium erfolgte, so dass die gr¨oßte Menge Ammoniak wieder desorbiert sein sollte.
Um ein Versuchsergebnis ohne anhaftendes Ammoniak zu gewinnen, wurde das CeO2 in
Wasserstoff vorkalziniert. Mit diesem Katalysator [(CeO2 in H2//H-ZSM-5) + (In(OH)3)] wer-
den wiederum NO-Ums¨atze bei ca. 85% erreicht. Unter der Voraussetzung, dass CeO2 keinen
Wasserstoff speichern kann, muss tats¨achlich auf eine Rolle des Ce3+ fur den aktiven Kataly-
sator geschlossen werden.
Es kann jedoch leider nicht entschieden werden, ob das Ce3+ zur Reduktion des Indiums
benotigt wird oder ob es eine eigene katalytische Wirkung entfaltet.
Zur Beantwortung dieser Frage wurde folgender Versuch durchgef¨uhrt: Eine
In(OH)3//NH4-ZSM-5-Mischung wird in Helium kalziniert und damit aktiviert. Dann er-
folgt die mechanische Zugabe von unbehandeltem CeO2 und die Mischung wird in 20 %
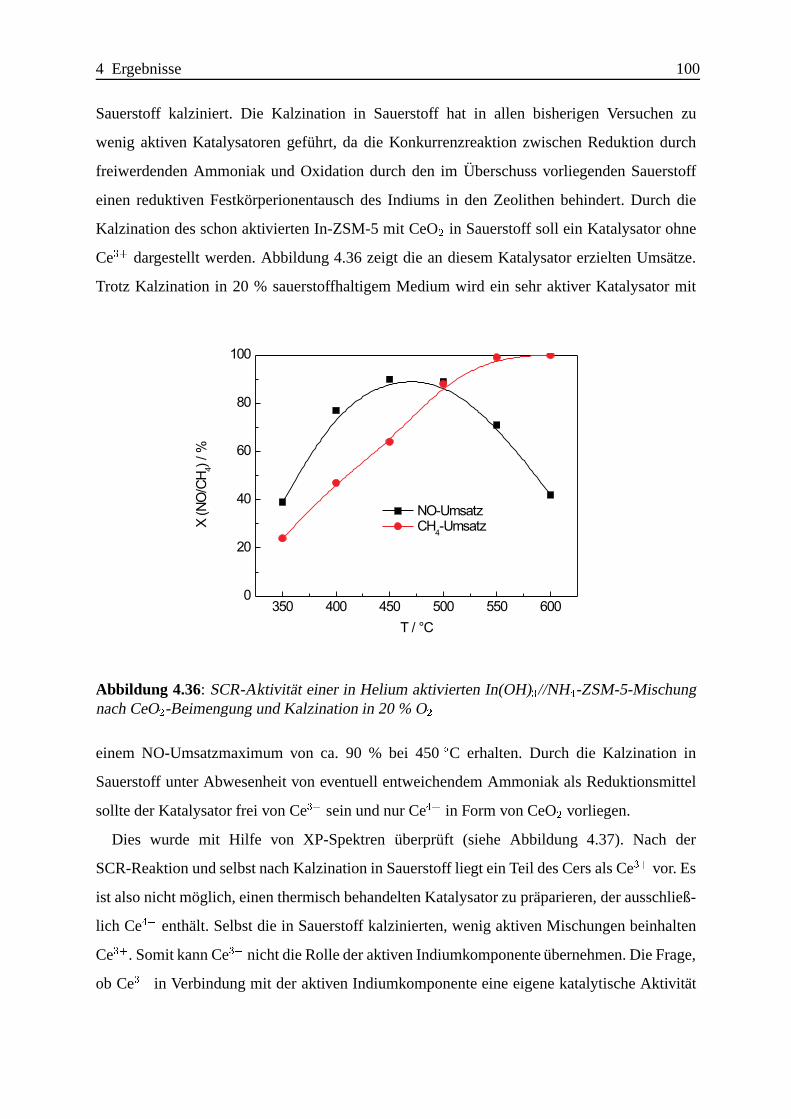
4 Ergebnisse 100
Sauerstoff kalziniert. Die Kalzination in Sauerstoff hat in allen bisherigen Versuchen zu
wenig aktiven Katalysatoren gef¨uhrt, da die Konkurrenzreaktion zwischen Reduktion durch
freiwerdenden Ammoniak und Oxidation durch den imUberschuss vorliegenden Sauerstoff
einen reduktiven Festk¨orperionentausch des Indiums in den Zeolithen behindert. Durch die
Kalzination des schon aktivierten In-ZSM-5 mit CeO2 in Sauerstoff soll ein Katalysator ohne
Ce3+ dargestellt werden. Abbildung 4.36 zeigt die an diesem Katalysator erzielten Ums¨atze.
Trotz Kalzination in 20 % sauerstoffhaltigem Medium wird ein sehr aktiver Katalysator mit
350 400 450 500 550 6000
20
40
60
80
100
NO-UmsatzCH
4-UmsatzX
(NO
/CH
4)/%
T / °C
Abbildung 4.36: SCR-Aktivitat einer in Helium aktivierten In(OH)3//NH4-ZSM-5-Mischungnach CeO2-Beimengung und Kalzination in 20 % O2
einem NO-Umsatzmaximum von ca. 90 % bei 450ÆC erhalten. Durch die Kalzination in
Sauerstoff unter Abwesenheit von eventuell entweichendem Ammoniak als Reduktionsmittel
sollte der Katalysator frei von Ce3+ sein und nur Ce4+ in Form von CeO2 vorliegen.
Dies wurde mit Hilfe von XP-Spektren ¨uberpruft (siehe Abbildung 4.37). Nach der
SCR-Reaktion und selbst nach Kalzination in Sauerstoff liegt ein Teil des Cers als Ce3+ vor. Es
ist also nicht m¨oglich, einen thermisch behandelten Katalysator zu pr¨aparieren, der ausschließ-
lich Ce4+ enthalt. Selbst die in Sauerstoff kalzinierten, wenig aktiven Mischungen beinhalten
Ce3+. Somit kann Ce3+ nicht die Rolle der aktiven Indiumkomponente ¨ubernehmen. Die Frage,
ob Ce3+ in Verbindung mit der aktiven Indiumkomponente eine eigene katalytische Aktivit¨at
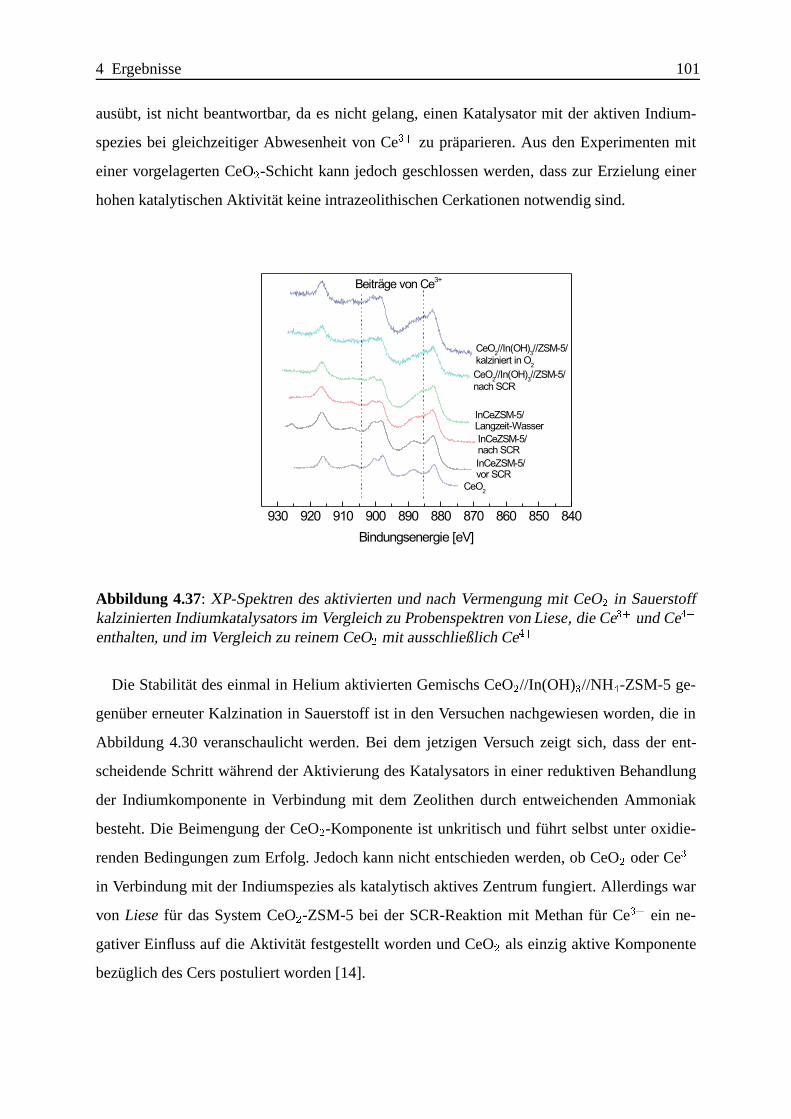
4 Ergebnisse 101
ausubt, ist nicht beantwortbar, da es nicht gelang, einen Katalysator mit der aktiven Indium-
spezies bei gleichzeitiger Abwesenheit von Ce3+ zu praparieren. Aus den Experimenten mit
einer vorgelagerten CeO2-Schicht kann jedoch geschlossen werden, dass zur Erzielung einer
hohen katalytischen Aktivit¨at keine intrazeolithischen Cerkationen notwendig sind.
930 920 910 900 890 880 870 860 850 840
CeO2
CeO2//In(OH)
3//ZSM-5/
kalziniert in O2
CeO2//In(OH)
3//ZSM-5/
nach SCR
Beiträge von Ce3+
InCeZSM-5/vor SCR
InCeZSM-5/nach SCR
InCeZSM-5/Langzeit-Wasser
Bindungsenergie [eV]
Abbildung 4.37: XP-Spektren des aktivierten und nach Vermengung mit CeO2 in Sauerstoffkalzinierten Indiumkatalysators im Vergleich zu Probenspektren vonLiese, die Ce3+ und Ce4+
enthalten, und im Vergleich zu reinem CeO2 mit ausschließlich Ce4+
Die Stabilitat des einmal in Helium aktivierten Gemischs CeO2//In(OH)3//NH4-ZSM-5 ge-
genuber erneuter Kalzination in Sauerstoff ist in den Versuchen nachgewiesen worden, die in
Abbildung 4.30 veranschaulicht werden. Bei dem jetzigen Versuch zeigt sich, dass der ent-
scheidende Schritt w¨ahrend der Aktivierung des Katalysators in einer reduktiven Behandlung
der Indiumkomponente in Verbindung mit dem Zeolithen durch entweichenden Ammoniak
besteht. Die Beimengung der CeO2-Komponente ist unkritisch und f¨uhrt selbst unter oxidie-
renden Bedingungen zum Erfolg. Jedoch kann nicht entschieden werden, ob CeO2 oder Ce3+
in Verbindung mit der Indiumspezies als katalytisch aktives Zentrum fungiert. Allerdings war
von Liesefur das System CeO2-ZSM-5 bei der SCR-Reaktion mit Methan f¨ur Ce3+ ein ne-
gativer Einfluss auf die Aktivit¨at festgestellt worden und CeO2 als einzig aktive Komponente
bezuglich des Cers postuliert worden [14].

4 Ergebnisse 102
4.1.3.3 Zusammenfassung
Durch die Promotierung von In-ZSM-5-Katalysatoren mit CeO2 lassen sich hochaktive
Systeme f¨ur die SCR-Reaktion von NO mit Methan erzeugen. Bei der Pr¨aparation der
CeO2-In-ZSM-5-Systeme ist die Pr¨aparation des Grundsystems In-ZSM-5 als kritischer Schritt
identifiziert worden. Die Beigabe des CeO2 selber kann auf nasschemischem Wege durch
Fallung vor der oder nach der Indiumkomponente oder als Kopr¨azipitat erfolgen; eine prim¨are
Zugabe der Indiumkomponente scheint jedoch auf Grund der dann gr¨oßeren N¨ahe der Indium-
komponente zur Zeolithmatrix zu marginal h¨oheren NO-Ums¨atzen zu f¨uhren. Durch mechani-
sche Beimengung eines CeO2 mit hoher Oberfl¨ache wurden deutlich h¨ohere Aktivitaten erzielt,
so dass letzteres als die Methode der Wahl zu betrachten ist.
Obwohl in begrenzten Versuchsreihen der Eindruck entstehen kann, dass die Promotie-
rung des aktivsten In-ZSM-5-Systems zum aktivsten CeO2-In-ZSM-5-System f¨uhrt, kann dies
nicht verallgemeinert werden. So werden aus durch Festk¨orperionentausch (SSIE) pr¨aparier-
ten In-ZSM-5-Katalysatoren mit sehr geringer SCR-Aktivit¨at, aber mit hohem Methanumsatz
durch Zusatz von CeO2 sehr aktive SCR-Katalysatoren pr¨apariert. Das aktivste Indiumsystem
liefert also nach Promotierung mit CeO2 nicht zwangsl¨aufig die aktivsten Katalysatoren.
Die Rolle des CeO2 wird durch Nachstellung der vonLiese[14] durchgefuhrten Schicht-
und Pelletexperimente (allerdings bei einer h¨oheren Belastung) als die eines Katalysators f¨ur
die NO-Oxidation zu NO2 bestatigt.
Gleichzeitig wird aber dem CeO2 bei der Praparation mechanischer Mischungen mit In(OH)3
und Zeolith eine zweite Aufgabe zugeschrieben: Unter bestimmten Pr¨aparationsbedingungen
fungiert es in Form von Ce3+ als Reduktionsmittel zur Aktivierung der Indiumkomponente f¨ur
die wahrend der Pr¨aparation ablaufende Festk¨orperreaktion. Ob Ce3+ bei gleichzeitiger Anwe-
senheit der aktiven Indiumkomponente eine eigene katalytische Aktivit¨at entfaltet, kann nicht
entschieden werden. Jedoch ist die Anwesenheit von intrazeolithischem Cer nicht erforderlich.
Jedenfalls lassen sich mit diesen mechanischen Mischungen auf einfache Art hohe
SCR-Aktivitaten erzielen, sofern w¨ahrend eines Aktivierungsschritts ein Reduktionsmittel zu-
gegen ist (z.B. NH+4 -Ionen im Zeolithen) und eine Aktivierung in Helium erfolgt. Die dabei
entstehende aktive Indiumspezies unterscheidet sich von den InO+-Ionen auf Kationenpl¨atzen.
Sie liegt in sehr geringer Anzahl im Bereich der ¨außeren Zeolithoberfl¨ache vor und ist vermut-

4 Ergebnisse 103
lich uber In-O-Si- oder In-O-Al-Bindungen am Interface zwischen kleinen InOx-Aggregaten
und der Oberfl¨ache des Zeolithen fixiert.
Versuche zur Vergiftung der Protonen durch Festk¨orperionentausch mit KBr oder nasschemi-
schem Ionentausch mit einer Natriumnitratl¨osung lassen vermuten, dass Protonen einen wichti-
gen Beitrag zur SCR-Reaktion leisten, da ohne sie die SCR-Aktivit¨at einbricht.Uber die genaue
Rolle der Protonen kann allerdings nur spekuliert werden, da die Protonenvergiftung sowohl
Einfluss auf den NO- als auch den Methanumsatz nimmt.
4.2 Modellierung
Im Folgenden wird f¨ur die drei Systeme CeO2-ZSM-5, In-ZSM-5 und CeO2-In-ZSM-5 eine
kinetische Modellierung durchgef¨uhrt. Durch Vergleich der an den Systemen erhaltenen kine-
tischen Parameter untereinander und zus¨atzlich mit Hilfe von Literaturergebnissen sind m¨ogli-
cherweise Unterschiede und/oder Analogien in den zugrundeliegenden Reaktionsmechanismen
zu erkennen.
Da in der Literatur bisher f¨ur die SCR mit Methan nur Parameter f¨ur einen einfachen Po-
tenzansatz vorliegen, wurde in dieser Arbeit auf eine Modelldiskriminierung verzichtet und die
Arbeit ebenfalls auf einen Potenzansatz fokussiert. Im Unterschied zur Literatur, in der gr¨oßten-
teils lediglich eine graphische Auswertung der experimentellen Daten unter Missachtung diffe-
rentieller Messbedingungen erfolgte, werden hier integrale Messungen mit Hilfe des Simulati-
onsprogramms”OPTI“ausgewertet. Als Reaktormodell fand ein homogenes, eindimensionales
und isothermes Modell Anwendung, dessen G¨ultigkeit schon in Abschnitt 3.2 nachgewiesen
wurde.
Die genaue Vorgehensweise bei der Modellierung wird eingehend im Unterabschnitt 4.2.1
fur das System CeO2-ZSM-5 vorgestellt; in den Unterabschnitten 4.2.2 und 4.2.3 erfolgt ledig-
lich die Darstellung der Ergebnisse f¨ur In-ZSM-5 bzw. CeO2-In-ZSM-5, da es sich um eine
analoge Vorgehensweise handelt, die lediglich zu f¨ur das jeweilige System spezifischen kineti-
schen Parametern f¨uhrt.
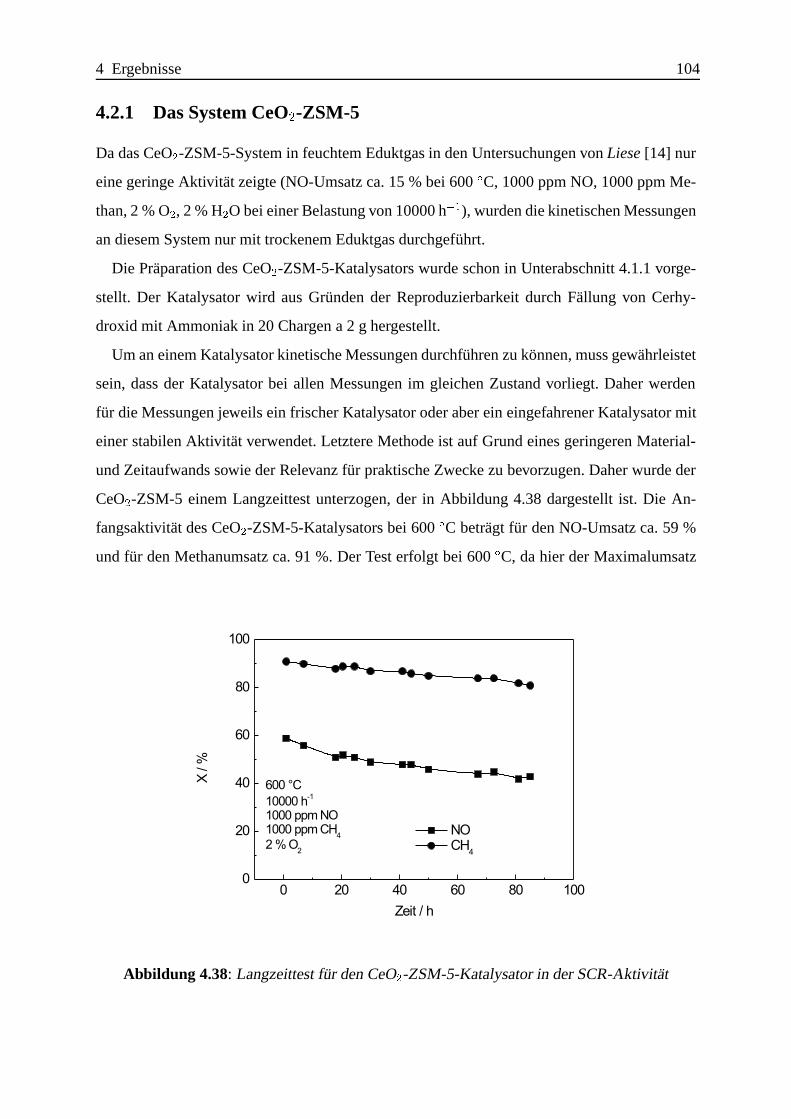
4 Ergebnisse 104
4.2.1 Das System CeO2-ZSM-5
Da das CeO2-ZSM-5-System in feuchtem Eduktgas in den Untersuchungen vonLiese[14] nur
eine geringe Aktivitat zeigte (NO-Umsatz ca. 15 % bei 600ÆC, 1000 ppm NO, 1000 ppm Me-
than, 2 % O2, 2 % H2O bei einer Belastung von 10000 h�1), wurden die kinetischen Messungen
an diesem System nur mit trockenem Eduktgas durchgef¨uhrt.
Die Praparation des CeO2-ZSM-5-Katalysators wurde schon in Unterabschnitt 4.1.1 vorge-
stellt. Der Katalysator wird aus Gr¨unden der Reproduzierbarkeit durch F¨allung von Cerhy-
droxid mit Ammoniak in 20 Chargen a 2 g hergestellt.
Um an einem Katalysator kinetische Messungen durchf¨uhren zu konnen, muss gew¨ahrleistet
sein, dass der Katalysator bei allen Messungen im gleichen Zustand vorliegt. Daher werden
fur die Messungen jeweils ein frischer Katalysator oder aber ein eingefahrener Katalysator mit
einer stabilen Aktivitat verwendet. Letztere Methode ist auf Grund eines geringeren Material-
und Zeitaufwands sowie der Relevanz f¨ur praktische Zwecke zu bevorzugen. Daher wurde der
CeO2-ZSM-5 einem Langzeittest unterzogen, der in Abbildung 4.38 dargestellt ist. Die An-
fangsaktivitat des CeO2-ZSM-5-Katalysators bei 600ÆC betragt fur den NO-Umsatz ca. 59 %
und fur den Methanumsatz ca. 91 %. Der Test erfolgt bei 600ÆC, da hier der Maximalumsatz
0 20 40 60 80 1000
20
40
60
80
100
600 °C
10000 h-1
1000 ppm NO1000 ppm CH
4
2 % O2
NOCH
4
X/%
Zeit / h
Abbildung 4.38: Langzeittest f¨ur den CeO2-ZSM-5-Katalysator in der SCR-Aktivit¨at

4 Ergebnisse 105
sowohl bez¨uglich des NO als auch des Methans liegt; außerdem sollten sich bei dieser hohen
Temperatur auftretende Deaktivierungsph¨anomene als am ausgepr¨agtesten erweisen. Es zeigt
sich, dass f¨ur beide Ums¨atze eine kontinuierliche Deaktivierung erfolgt, wobei diese f¨ur den
Methanumsatz ¨uber den gesamten Zeitraum ¨ahnlich stark verl¨auft. Von einem Anfangsumsatz
bei 91 % geht der Methanumsatz nach 20 Stunden auf 89 % zur¨uck, nach 40 Stunden werden
noch 86 % Umsatz erreicht und nach 80 Stunden 82 %. Beim NO-Umsatz sieht es hinge-
gen anders aus. Der Anfangsumsatz von 59 % geht relativ schnell nach 20 Stunden auf 52 %
zuruck, im weiteren Zeitverlauf erfolgt dann eine langsamere Deaktivierung, ohne dass sich
eine Bodenbildung erkennen l¨asst. Nach 40 Stunden betr¨agt der NO-Umsatz 48 % und nach
80 Stunden 44 %. Da diese Umsatzdifferenz zwischen den beiden letztgenannten Messwerten
schon im Bereich der Fehlergrenzen liegt, k¨onnen mit einem 40 Stunden deaktivierten Kata-
lysator kinetische Messungen ¨uber einen Zeitraum von mindestens 40 Stunden durchgef¨uhrt
werden.
Vorversuche auf Stofftransportlimitierung sind f¨ur dieses System nicht durchgef¨uhrt worden,
fur das aktivere System CeO2-In-ZSM-5 wurde dies experimentell getestet. Eine rechnerische
Uberprufung auf Stofftransportlimitierung in Abschnitt 3.2.3 konnte jedoch sowohl eine ¨außere
als auch innere Stofftransportlimitierung ausschließen, so dass ein pseudohomogenes Reaktor-
modell angewendet werden kann.
Da das genaue Reaktionsnetzwerk nicht bekannt ist, wurden lediglich die Geschwindigkeits-
ansatze fur die selektive Reduktion des Stickstoffmonoxids und die Totaloxidation des Methans
in Form der folgenden Potenzans¨atze verwendet:
rCH4 = k1 � caCH4 � cbO2� ccNO (4.1)
rNO = k2 � ceCH4 � cfO2� cgNO (4.2)
Im Falle der Totaloxidation wird auch NO im Geschwindigkeitsansatz ber¨ucksichtigt, daLiese
herausfand, dass NO diese Reaktion ¨uber aziden Zentren beg¨unstigt [14]. NO wird aber auch
allgemein zur Aktivierung von Methan in der OCM-Reaktion (OCM = Oxidative Coupling
of Methane) verwendet. EinUberblick uber die Aktivierung von Methan mit Sauerstoff oder
Stickoxiden findet sich beiTabataet al. [125].
Wie schon im Abschnitt 3.2 erw¨ahnt, verandert das Optimierungsprogramm die Parameter
k1, k2 und die Reaktionsordnungen mit Hilfe eines Zufallszahlengenerators, wobei die Such-

4 Ergebnisse 106
richtung bei einer verbesserten Anpassung beibehalten wird. Optimiert wird die Fehlerquadrat-
summe FQS aus gemessenen und simulierten NO- und Methanums¨atzen. Um eine ausreichend
große Anzahl an Messbedingungen zu erhalten, wurde der in Abbildung 4.39 dargestellte Para-
meterbereich untersucht. In diesem dreidimensionalen Parameterraum wurden die drei folgen-
250
500
750
1000
5000
10000
15000
20000
500
1000
1500
2000
Bela
stung
[h-1]
CH 4-K
onze
ntrati
on[pp
m]NO-Konzentration [ppm]
Abbildung 4.39: Messplan der kinetischen Messungen am CeO2-ZSM-5-Katalysator
den Parameter ge¨andert: die NO-Konzentration mit den Einstellungen 250, 500 und 1000 ppm,
die Methankonzentration mit 500, 1000 und 2000 ppm, und die Belastung mit 5000, 10000
und 20000 h�1, wobei die Sauerstoffkonzentration konstant bei 2 % lag. F¨ur die Eckpunk-
te des W¨urfels und den zentralen Punkt 1000 ppm NO, 1000 ppm CH4 bei einer Belastung
von 10000 h�1 wurden zus¨atzlich Sauerstoffkonzentrationen von 0,5, 4 und 10 % vermessen.
Außerdem wurde die Temperatur f¨ur jeden Messpunkt in 50ÆC-Schritten variiert, so dass f¨ur
das CeO2-ZSM-5-System bei den Temperaturen 450ÆC, 500ÆC, 550ÆC und 600ÆC gemessen
wurde. Die Messungen mit variierendem Sauerstoffgehalt erfolgten lediglich bei 500ÆC und
600ÆC.
Um fur die Simulation geeignete Startparameter zu finden, erfolgt zuerst eine konventio-
nelle graphische Auswertung der Experimente, wie sie beispielhaft in Abbildung 4.40 dar-
gestellt ist. Hierbei kann durch eine graphische Auftragung des nat¨urlichen Logarithmus der
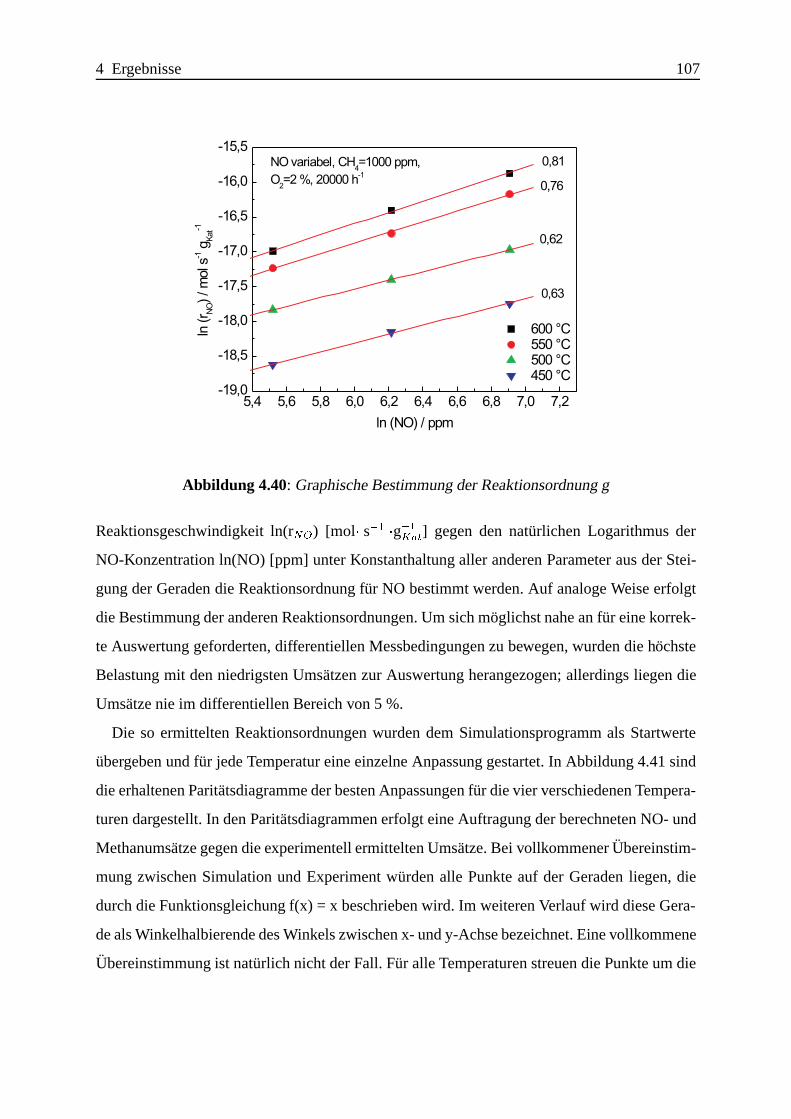
4 Ergebnisse 107
5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8 7,0 7,2-19,0
-18,5
-18,0
-17,5
-17,0
-16,5
-16,0
-15,5
600 °C550 °C500 °C450 °C
NO variabel, CH4=1000 ppm,
O2=2 %, 20000 h
-1
0,63
0,62
0,76
0,81
ln(r
NO)/m
ols
-1g
Kat-1
ln (NO) / ppm
Abbildung 4.40: Graphische Bestimmung der Reaktionsordnung g
Reaktionsgeschwindigkeit ln(rNO) [mol� s�1 �g�1Kat] gegen den nat¨urlichen Logarithmus der
NO-Konzentration ln(NO) [ppm] unter Konstanthaltung aller anderen Parameter aus der Stei-
gung der Geraden die Reaktionsordnung f¨ur NO bestimmt werden. Auf analoge Weise erfolgt
die Bestimmung der anderen Reaktionsordnungen. Um sich m¨oglichst nahe an f¨ur eine korrek-
te Auswertung geforderten, differentiellen Messbedingungen zu bewegen, wurden die h¨ochste
Belastung mit den niedrigsten Ums¨atzen zur Auswertung herangezogen; allerdings liegen die
Umsatze nie im differentiellen Bereich von 5 %.
Die so ermittelten Reaktionsordnungen wurden dem Simulationsprogramm als Startwerte
ubergeben und f¨ur jede Temperatur eine einzelne Anpassung gestartet. In Abbildung 4.41 sind
die erhaltenen Parit¨atsdiagramme der besten Anpassungen f¨ur die vier verschiedenen Tempera-
turen dargestellt. In den Parit¨atsdiagrammen erfolgt eine Auftragung der berechneten NO- und
Methanums¨atze gegen die experimentell ermittelten Ums¨atze. Bei vollkommenerUbereinstim-
mung zwischen Simulation und Experiment w¨urden alle Punkte auf der Geraden liegen, die
durch die Funktionsgleichung f(x) = x beschrieben wird. Im weiteren Verlauf wird diese Gera-
de als Winkelhalbierende des Winkels zwischen x- und y-Achse bezeichnet. Eine vollkommene
Ubereinstimmung ist nat¨urlich nicht der Fall. F¨ur alle Temperaturen streuen die Punkte um die
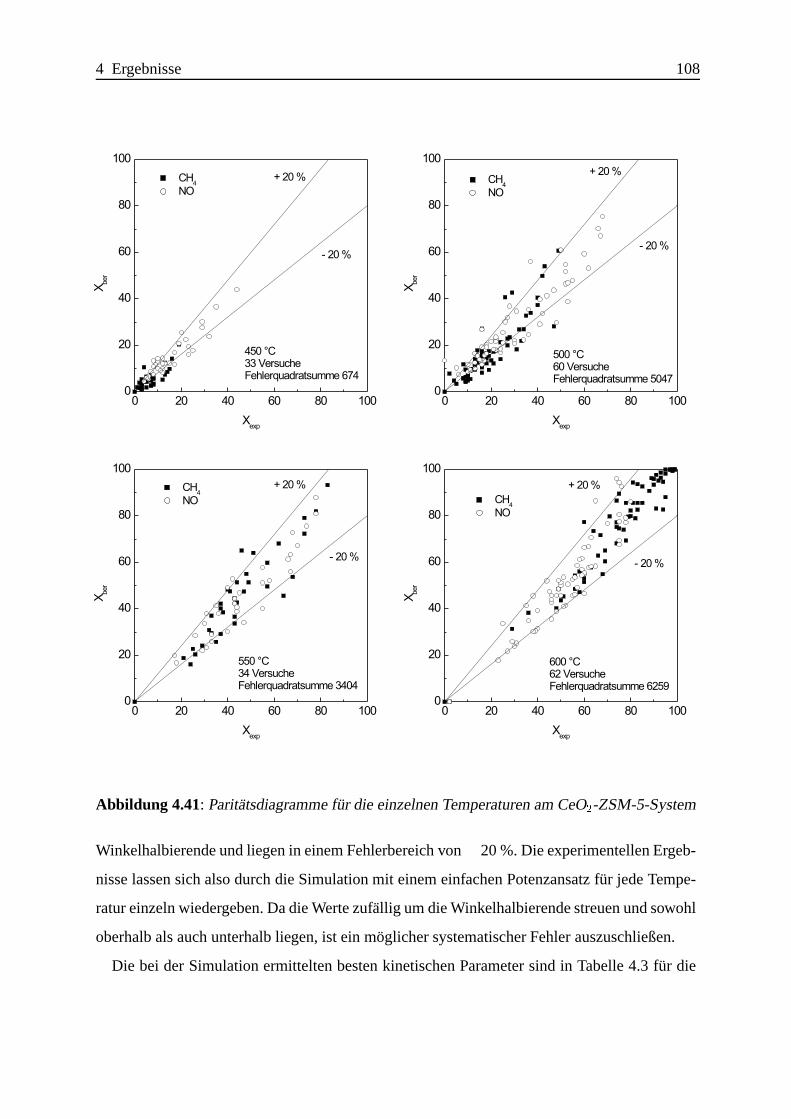
4 Ergebnisse 108
0 20 40 60 80 1000
20
40
60
80
100
600 °C62 VersucheFehlerquadratsumme 6259
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
550 °C34 VersucheFehlerquadratsumme 3404
- 20 %
+ 20 %CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
500 °C60 VersucheFehlerquadratsumme 5047
- 20 %
+ 20 %CH
4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
450 °C33 VersucheFehlerquadratsumme 674
- 20 %
+ 20 %CH4
NO
Xber
Xexp
Abbildung 4.41: Paritatsdiagramme f¨ur die einzelnen Temperaturen am CeO2-ZSM-5-System
Winkelhalbierende und liegen in einem Fehlerbereich von� 20 %. Die experimentellen Ergeb-
nisse lassen sich also durch die Simulation mit einem einfachen Potenzansatz f¨ur jede Tempe-
ratur einzeln wiedergeben. Da die Werte zuf¨allig um die Winkelhalbierende streuen und sowohl
oberhalb als auch unterhalb liegen, ist ein m¨oglicher systematischer Fehler auszuschließen.
Die bei der Simulation ermittelten besten kinetischen Parameter sind in Tabelle 4.3 f¨ur die
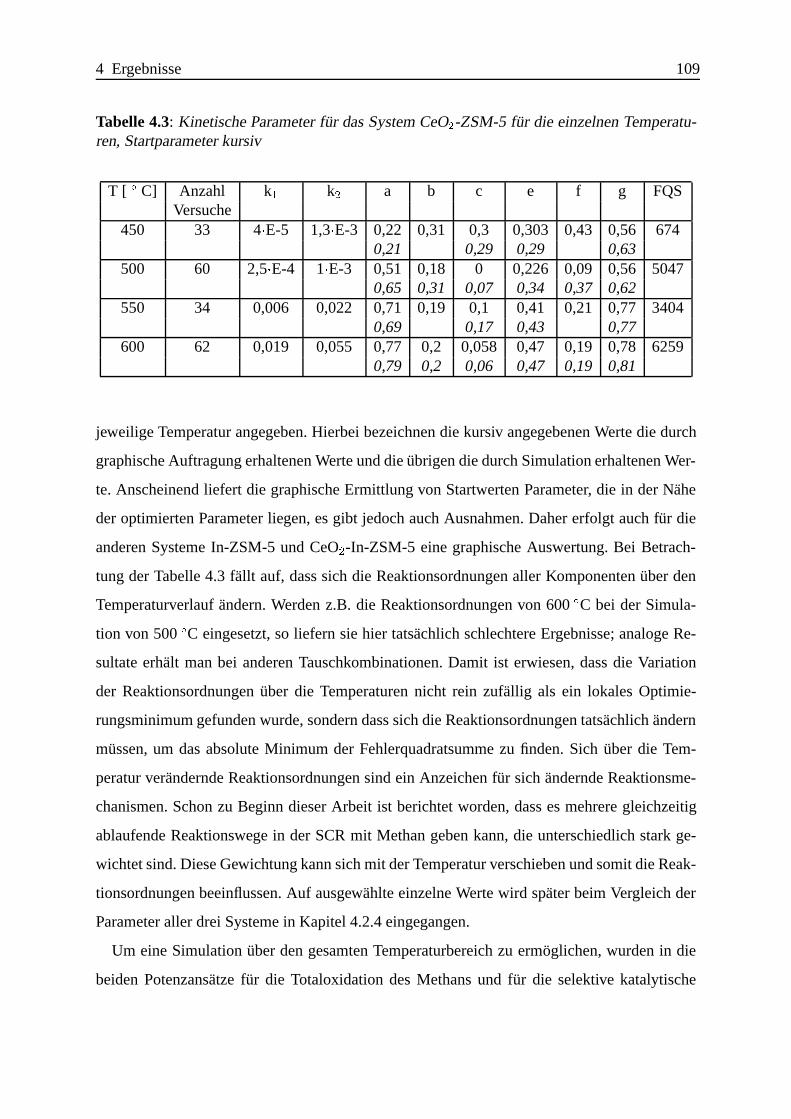
4 Ergebnisse 109
Tabelle 4.3: Kinetische Parameter f¨ur das System CeO2-ZSM-5 fur die einzelnen Temperatu-ren, Startparameter kursiv
T [ Æ C] Anzahl k1 k2 a b c e f g FQSVersuche
450 33 4�E-5 1,3�E-3 0,22 0,31 0,3 0,303 0,43 0,56 6740,21 0,29 0,29 0,63
500 60 2,5�E-4 1�E-3 0,51 0,18 0 0,226 0,09 0,56 50470,65 0,31 0,07 0,34 0,37 0,62
550 34 0,006 0,022 0,71 0,19 0,1 0,41 0,21 0,77 34040,69 0,17 0,43 0,77
600 62 0,019 0,055 0,77 0,2 0,058 0,47 0,19 0,78 62590,79 0,2 0,06 0,47 0,19 0,81
jeweilige Temperatur angegeben. Hierbei bezeichnen die kursiv angegebenen Werte die durch
graphische Auftragung erhaltenen Werte und die ¨ubrigen die durch Simulation erhaltenen Wer-
te. Anscheinend liefert die graphische Ermittlung von Startwerten Parameter, die in der N¨ahe
der optimierten Parameter liegen, es gibt jedoch auch Ausnahmen. Daher erfolgt auch f¨ur die
anderen Systeme In-ZSM-5 und CeO2-In-ZSM-5 eine graphische Auswertung. Bei Betrach-
tung der Tabelle 4.3 f¨allt auf, dass sich die Reaktionsordnungen aller Komponenten ¨uber den
Temperaturverlauf ¨andern. Werden z.B. die Reaktionsordnungen von 600ÆC bei der Simula-
tion von 500ÆC eingesetzt, so liefern sie hier tats¨achlich schlechtere Ergebnisse; analoge Re-
sultate erh¨alt man bei anderen Tauschkombinationen. Damit ist erwiesen, dass die Variation
der Reaktionsordnungen ¨uber die Temperaturen nicht rein zuf¨allig als ein lokales Optimie-
rungsminimum gefunden wurde, sondern dass sich die Reaktionsordnungen tats¨achlichandern
mussen, um das absolute Minimum der Fehlerquadratsumme zu finden. Sich ¨uber die Tem-
peratur ver¨andernde Reaktionsordnungen sind ein Anzeichen f¨ur sichandernde Reaktionsme-
chanismen. Schon zu Beginn dieser Arbeit ist berichtet worden, dass es mehrere gleichzeitig
ablaufende Reaktionswege in der SCR mit Methan geben kann, die unterschiedlich stark ge-
wichtet sind. Diese Gewichtung kann sich mit der Temperatur verschieben und somit die Reak-
tionsordnungen beeinflussen. Auf ausgew¨ahlte einzelne Werte wird sp¨ater beim Vergleich der
Parameter aller drei Systeme in Kapitel 4.2.4 eingegangen.
Um eine Simulation ¨uber den gesamten Temperaturbereich zu erm¨oglichen, wurden in die
beiden Potenzans¨atze fur die Totaloxidation des Methans und f¨ur die selektive katalytische

4 Ergebnisse 110
Reduktion des NO Aktivierungsenergien implementiert:
rCH4 = k0;1 � e�Ea;1R�T � caCH4 � cbO2
� ccNO (4.3)
rNO = k0;2 � e�Ea;2R�T � ceCH4 � cfO2
� cgNO (4.4)
Da die Aktivierungsenergien und die zugeh¨origen praexponentiellen Faktoren eine hohe Sensi-
tivit at in Bezug auf die resultierende Fehlerquadratsumme aufweisen, ist hier die Kenntnis guter
Startparameter noch wichtiger als bei den einzelnen Reaktionsordnungen. Eine graphische Auf-
tragung des nat¨urlichen Logarithmus der Reaktionsgeschwindigkeit ln(rCH4) gegen 1/T liefert
als Steigung der Geraden den Wert -Ea/R und somit nach Multiplikation mit der allgemeinen
Gaskonstante R die Aktivierungsenergie der Reaktion. Die f¨ur die Auftragung ben¨otigten An-
fangsreaktionsgeschwindigkeiten lassen sich durch Einsetzen der kinetischen Parameter aus
Tabelle 4.3 bestimmen. Eine direkte Auftragung der dort aufgelisteten Geschwindigkeitskon-
stanten k fuhrt nicht zu verwertbaren Ergebnissen, da k stark von den Reaktionsordnungen
abhangt. Letzteres ist unmittelbar ersichtlich aus der Einheit f¨ur die hier verwendeten konzen-
trationsabh¨angigen Geschwindigkeitskonstanten kc von [ m3�(x+y+z)
mol(x+y+z)�1�s�kgKat]. Selbst nach einer
Umrechnung der konzentrationsabh¨angigen kc-Werte in die molenbruchabh¨angigen ky-Werte
nach der Formel [126]
ky =kc
R�Tpt
(x+y+z)(4.5)
zeigen die ky-Werte mit der Einheit[ mols�kgKat
] in einer Testrechnung mit variierenden Reaktions-
ordnungen (x, y und z) eine Abh¨angigkeit von letzteren, so dass wirklich nur die Anfangsreak-
tionsgeschwindigkeiten zur Auswertung herangezogen werden k¨onnen.
Fur das System CeO2-ZSM-5 werden die Aktivierungsenergien f¨ur die Totaloxidations-
reaktion und die SCR-Reaktion mit Hilfe der Anfangsreaktionsgeschwindigkeiten in Abbil-
dung 4.42 ermittelt.
Nach Multiplikation der Steigung der Geraden mit der allgemeinen Gaskonstanten R er-
gibt sich fur die Totaloxidation eine Aktivierungsenergie von ca. 188 kJ/mol und f¨ur die
SCR-Reaktion eine Aktivierungsenergie von ca. 78 kJ/mol. Mit diesen Werten und dem Mit-
telwert der Reaktionsordnungen der einzelnen Komponenten ¨uber die Temperatur wird die Si-
mulation gestartet. Das optimierte Ergebnis ist in Abbildung 4.43 zu sehen. Sowohl die NO-
als auch die Methanums¨atze werden ¨uber den gesamten Bereich von 0 bis 100 %, rund um
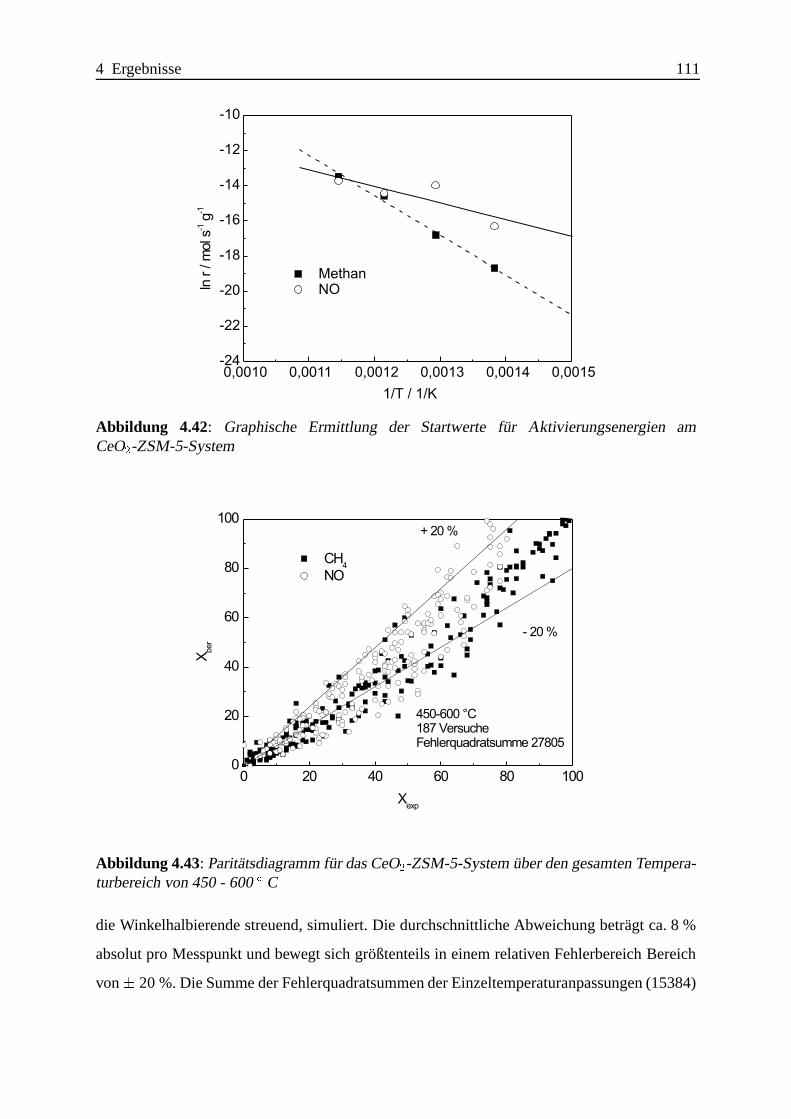
4 Ergebnisse 111
0,0010 0,0011 0,0012 0,0013 0,0014 0,0015-24
-22
-20
-18
-16
-14
-12
-10
MethanNOln
r/m
ols
-1g
-1
1/T / 1/K
Abbildung 4.42: Graphische Ermittlung der Startwerte f¨ur Aktivierungsenergien amCeO2-ZSM-5-System
0 20 40 60 80 1000
20
40
60
80
100
450-600 °C187 VersucheFehlerquadratsumme 27805
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
Abbildung 4.43: Paritatsdiagramm f¨ur das CeO2-ZSM-5-System ¨uber den gesamten Tempera-turbereich von 450 - 600Æ C
die Winkelhalbierende streuend, simuliert. Die durchschnittliche Abweichung betr¨agt ca. 8 %
absolut pro Messpunkt und bewegt sich gr¨oßtenteils in einem relativen Fehlerbereich Bereich
von� 20 %. Die Summe der Fehlerquadratsummen der Einzeltemperaturanpassungen (15384)

4 Ergebnisse 112
liegt allerdings um ca. 45 % niedriger als die Fehlerquadratsumme der Gesamttemperaturan-
passung, dies kann jedoch auf Grund mit der Temperatur stark variierender Reaktionsordnun-
gen erwartet werden.
Um einen Eindruck ¨uber die Qualit¨at derUbereinstimmung zwischen Experiment und Simu-
lation zu erhalten, sind in Abbildung 4.44 mehrere Vergleiche bei unterschiedlichen Methan-,
NO- und Sauerstoffkonzentrationen sowie bei verschiedenen Belastungen ¨uber den gesamten
Temperaturbereich dargestellt.
Trotz einiger Abweichungen z.B. bei Einsatz unterschiedlicher NO-Konzentrationen (Bild c)
ist die Gesamttemperaturanpassung als gelungen zu bezeichnen. Die gefundenen kinetischen
Parameter sind folgende:
rCH4 = k0;1 � e�Ea;1R�T � c0;7CH4 � c
0;2O2� c0;1NO (4.6)
rNO = k0;2 � e�Ea;2R�T � c0;315CH4
� c0;2O2� c0;8NO (4.7)
mit
k0;1 = 2 � 106 m3
mol0�s�kgKat
Ea;1 = 137 kJ=mol
k0;2 = 7500 m3;945
mol0;315 �s�kgKat
Ea;2 = 90 kJ=mol
Hierbei liegen die Aktivierungsenergien im physikalisch sinnvollen Bereich; sie unterschei-
den sich jedoch deutlich von den Startwerten. Eine genauere Diskussion erfolgt in Kapitel 4.2.4.
Mit Hilfe einer Sensitivitatsanalyseder erhaltenen kinetischen Parameter sollen diese wei-
ter bewertet werden. Dazu wird der Parameter um 5 % nach oben bzw. unten gesetzt und die
Auswirkung auf die Fehlerquadratsumme beobachtet. Normalerweise wird eine Sensitivit¨ats-
analyse bei sehr komplexen Reaktionsnetzwerken angewendet, um sensitive von insensitiven
Parametern zu unterscheiden. Hier sollten alle Parameter sensitiv sein; lediglich der Grad der
Anderung sollte sich unterscheiden. Die Ergebnisse sind in Tabelle 4.4 wiedergegeben. Die
Sensitivitatsanalyse zeigt, dass besonders die beiden Aktivierungsenergien und die Reaktions-
ordnung fur Methan in der Totaloxidationsreaktion und die f¨ur NO in der SCR-Reaktion bei
Veranderung einen Einfluss auf die Fehlerquadratsumme aus¨uben. Es kann also davon ausge-
gangen werden, dass f¨ur diese Parameter ein genauerer Wert bestimmt wurde als f¨ur die weniger
sensitiven Parameter. Allerdings konnte nicht f¨ur jedeAnderung die zugeh¨orige Abweichung
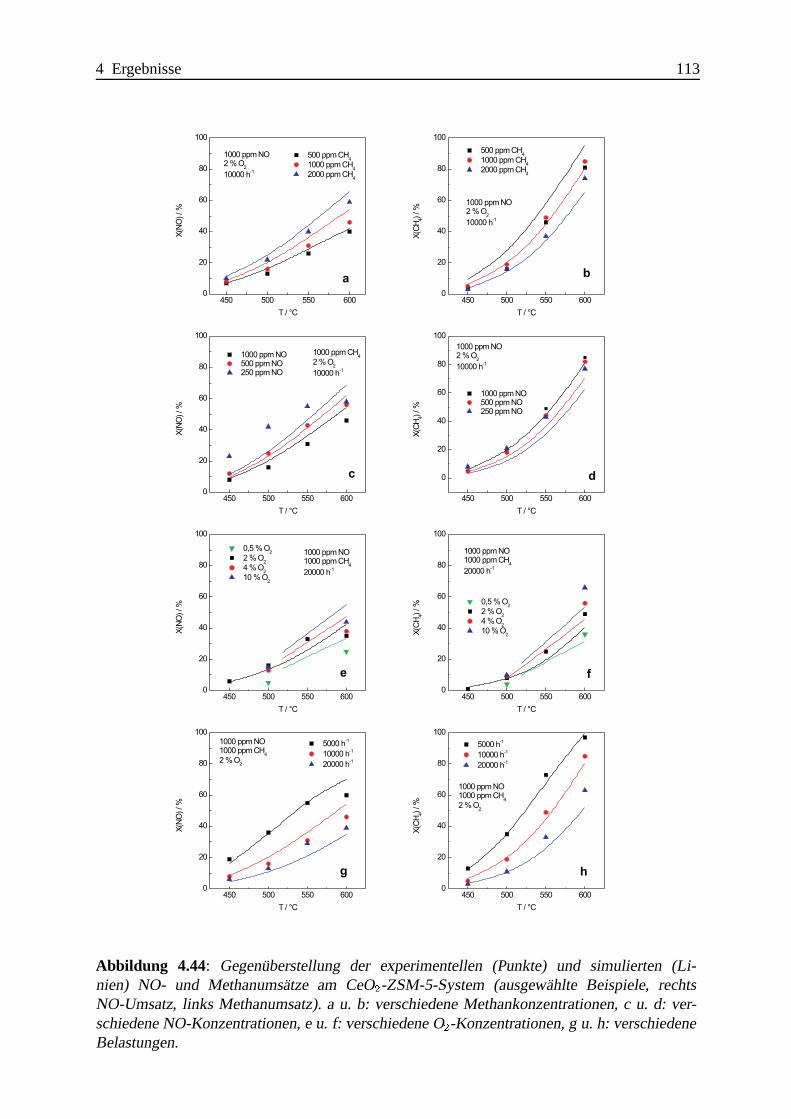
4 Ergebnisse 113
450 500 550 6000
20
40
60
80
100
a
1000 ppm NO2 % O
2
10000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(N
O)/%
T / °C
450 500 550 600
0
20
40
60
80
100
d
1000 ppm NO2 % O
2
10000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
e
1000 ppm NO1000 ppm CH
4
20000 h-1
0,5 % O2
2 % O2
4 % O2
10 % O2
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
f
1000 ppm NO1000 ppm CH
4
20000 h-1
0,5 % O2
2 % O2
4 % O2
10 % O2X
(CH
4)/%
T / °C
450 500 550 6000
20
40
60
80
100
h
1000 ppm NO1000 ppm CH
4
2 % O2
5000 h-1
10000 h-1
20000 h-1
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
b
1000 ppm NO2 % O
2
10000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
c
1000 ppm CH4
2 % O2
10000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
g
1000 ppm NO1000 ppm CH
4
2 % O2
5000 h-1
10000 h-1
20000 h-1
X(N
O)/%
T / °C
Abbildung 4.44: Gegen¨uberstellung der experimentellen (Punkte) und simulierten (Li-nien) NO- und Methanums¨atze am CeO2-ZSM-5-System (ausgew¨ahlte Beispiele, rechtsNO-Umsatz, links Methanumsatz). a u. b: verschiedene Methankonzentrationen, c u. d: ver-schiedene NO-Konzentrationen, e u. f: verschiedene O2-Konzentrationen, g u. h: verschiedeneBelastungen.

4 Ergebnisse 114
Tabelle 4.4: Sensitivitatsanalyse der kinetischen Parameter f¨ur das CeO2-ZSM-5-System
Parameter k0;1 k0;2 Ea;1 Ea;2 a b c e f g
+ 5 % 0,2 % 281 % 35 % 2,8 % 5 % 3,9 % 0,9 %- 5 % 8,5 % 31 % 1,9 % 0,5 % 21 %
bestimmt werden, da das Optimierungsprogramm in manchen F¨allen nicht konvergierte. An
dieser Stelle tritt ein leeres Feld in der Tabelle auf.
4.2.2 Das System In-ZSM-5
Die zur Durchfuhrung der Simulation ben¨otigten kinetischen Messungen sind im Rahmen ei-
ner Diplomarbeit vonK.D. Attar durchgefuhrt worden [127]. In dieser Arbeit erfolgten auch
erste Ans¨atze zur Modellierung der Einzeltemperturanpassungen mit zufriedenstellenden Er-
gebnissen; lediglich die Einbeziehung unterschiedlicher Sauerstoffkonzentrationen und eine
Gesamttemperaturanpassung fehlen. Wegen der geringen katalytischen Aktivit¨at mit Wasser
im Eduktgas wurden nur Messungen mit trockenem Eduktgas durchgef¨uhrt (siehe Seite 62).
Die Praparation des Katalysators erfolgte durch Kombination von Ionentausch mit
anschließender F¨allung auf die NH4-Form des Zeolithen ZSM-5 der Firma AlsiPenta. Diese
Praparationsmethode gefolgt von einer Kalzination in Helium liefert Katalysatoren, die ¨uber
ein breiteres Temperaturfenster von 350 - 450ÆC eine nahezu konstant hohe SCR-Aktivit¨at
zeigen, wahrend bei den ¨ubrigen Praparationen nach dem Umsatzmaximum ein steilerer Abfall
der NO-Umsatze zu beobachten ist. Um die katalytischen Messungen an einem stabilen Kataly-
sator durchzuf¨uhren, wurde zun¨achst ein Langzeittest durchgef¨uhrt, der in Abbildung 4.45 dar-
gestellt ist. Als Testpunkt wurden die Bedingungen 1000 ppm NO, 1000 ppm Methan, 2 % Sau-
erstoff und eine Belastung von 30000 h�1 bei einer Temperatur von 500ÆC gewahlt, die nur
eine Stufe unterhalb der Maximaltemperatur von 550ÆC wahrend der kinetischen Messungen
liegt. Uberraschenderweise ergibt sich f¨ur diesen Katalysator statt einer erwarteten Deaktivie-
rung eine Aktivierung bez¨uglich des NO-Umsatzes. Dieser steigt von 16 % nach einer Stunde
auf 25 % nach 20 Stunden. Im weiteren Verlauf verlangsamt sich die Steigerungsrate, so dass
nach 40 Stunden NO-Ums¨atze von ca. 27 % und nach 90 Stunden NO-Ums¨atze von ca. 29 %
erreicht werden. Diese Aktivit¨atssteigerung kann zur¨uckgefuhrt werden auf eine Deaktivie-
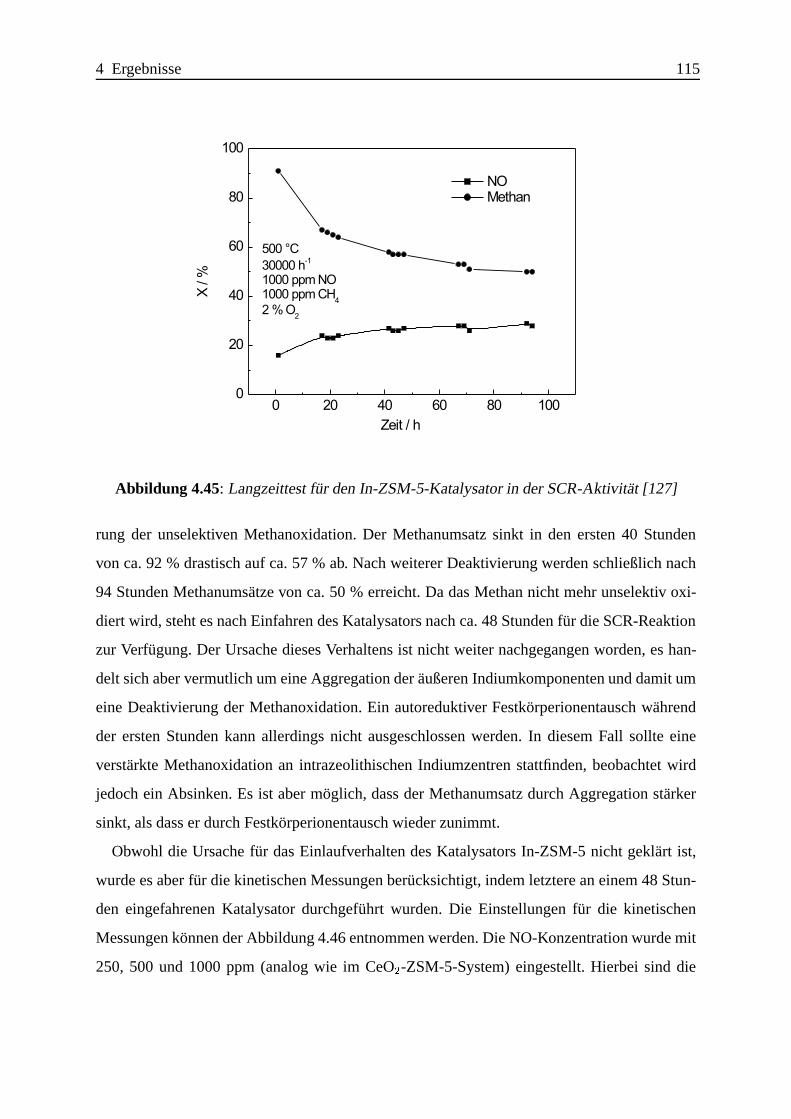
4 Ergebnisse 115
0 20 40 60 80 1000
20
40
60
80
100
500 °C
30000 h-1
1000 ppm NO1000 ppm CH
4
2 % O2
NOMethan
X/%
Zeit / h
Abbildung 4.45: Langzeittest f¨ur den In-ZSM-5-Katalysator in der SCR-Aktivit¨at [127]
rung der unselektiven Methanoxidation. Der Methanumsatz sinkt in den ersten 40 Stunden
von ca. 92 % drastisch auf ca. 57 % ab. Nach weiterer Deaktivierung werden schließlich nach
94 Stunden Methanums¨atze von ca. 50 % erreicht. Da das Methan nicht mehr unselektiv oxi-
diert wird, steht es nach Einfahren des Katalysators nach ca. 48 Stunden f¨ur die SCR-Reaktion
zur Verfugung. Der Ursache dieses Verhaltens ist nicht weiter nachgegangen worden, es han-
delt sich aber vermutlich um eine Aggregation der ¨außeren Indiumkomponenten und damit um
eine Deaktivierung der Methanoxidation. Ein autoreduktiver Festk¨orperionentausch w¨ahrend
der ersten Stunden kann allerdings nicht ausgeschlossen werden. In diesem Fall sollte eine
verstarkte Methanoxidation an intrazeolithischen Indiumzentren stattfinden, beobachtet wird
jedoch ein Absinken. Es ist aber m¨oglich, dass der Methanumsatz durch Aggregation st¨arker
sinkt, als dass er durch Festk¨orperionentausch wieder zunimmt.
Obwohl die Ursache f¨ur das Einlaufverhalten des Katalysators In-ZSM-5 nicht gekl¨art ist,
wurde es aber f¨ur die kinetischen Messungen ber¨ucksichtigt, indem letztere an einem 48 Stun-
den eingefahrenen Katalysator durchgef¨uhrt wurden. Die Einstellungen f¨ur die kinetischen
Messungen k¨onnen der Abbildung 4.46 entnommen werden. Die NO-Konzentration wurde mit
250, 500 und 1000 ppm (analog wie im CeO2-ZSM-5-System) eingestellt. Hierbei sind die

4 Ergebnisse 116
250
500
750
1000
10000
15000
20000
25000
30000
500
1000
1500
2000
Bela
stung
[h-1]
CH 4-K
onze
ntrati
on[pp
m]NO-Konzentration [ppm]
Abbildung 4.46: Messplan der kinetischen Messungen am In-ZSM-5-Katalysator
geringeren Konzentrationen interessanter, da eine NOx-Minderung im Normalfall bei gerin-
gen Konzentrationen anspruchsvoller ist. Die Methankonzentration wurde ebenfalls wie beim
CeO2-ZSM-5-System auf 500, 1000 oder 2000 ppm eingestellt. Lediglich f¨ur die Belastung
wurden unter Orientierung an der Aktivit¨at des Katalysators abweichende Werte von 10000,
15000 und 30000 h�1 gewahlt. Alle Messungen erfolgten mit 2 % Sauerstoff im Eduktgas in
50 ÆC-Temperaturschritten von 300 - 550ÆC. Fur die beiden zentralen Punkte 1000 ppm NO,
1000 ppm Methan bei Belastungen von 10000 h�1 und 30000 h�1 wurden zus¨atzlich Messun-
gen bei 0,5, 4 und 10 % Sauerstoff durchgef¨uhrt.
Nach graphischer Ermittlung der Reaktionsordnungen wird jeweils wieder eine Einzeltempe-
raturanpassung mit Hilfe des schon vorgestellten Potenzansatzes durchgef¨uhrt. Die erhaltenen
Paritatsdiagramme werden in der folgenden Abbildung 4.47 vorgestellt. DieUbereinstimmung
zwischen Simulation und Experiment bewegt sich in den relativen Fehlergrenzen von� 20 %
fur jede Temperatur, wobei bei niedrigen Temperaturen die Anpassung noch besser ist. Bei den
hohen Temperaturen von 500ÆC und 550ÆC vergroßert sich die Streuung der Messpunkte um
die Winkelhalbierende zwischen x- und y-Achse. Hier kommen anscheinend weitere Neben-
reaktionen ins Spiel, die durch die zwei einfachen Potenzans¨atze nur ungenau erfasst werden
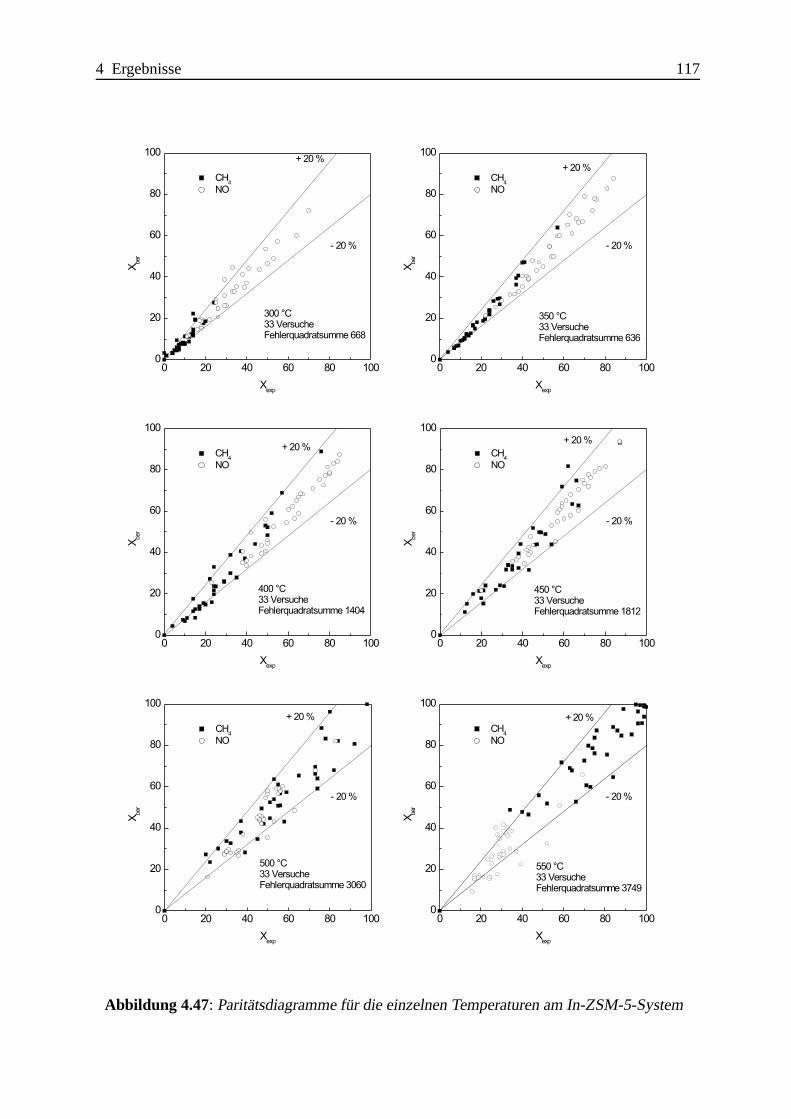
4 Ergebnisse 117
0 20 40 60 80 1000
20
40
60
80
100
550 °C33 VersucheFehlerquadratsumme 3749
- 20 %
+ 20 %CH
4
NO
Xbe
r
Xexp
0 20 40 60 80 1000
20
40
60
80
100
500 °C33 VersucheFehlerquadratsumme 3060
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
450 °C33 VersucheFehlerquadratsumme 1812
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
400 °C33 VersucheFehlerquadratsumme 1404
- 20 %
+ 20 %CH
4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
350 °C33 VersucheFehlerquadratsumme 636
- 20 %
+ 20 %CH
4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
300 °C33 VersucheFehlerquadratsumme 668
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
Abbildung 4.47: Paritatsdiagramme f¨ur die einzelnen Temperaturen am In-ZSM-5-System
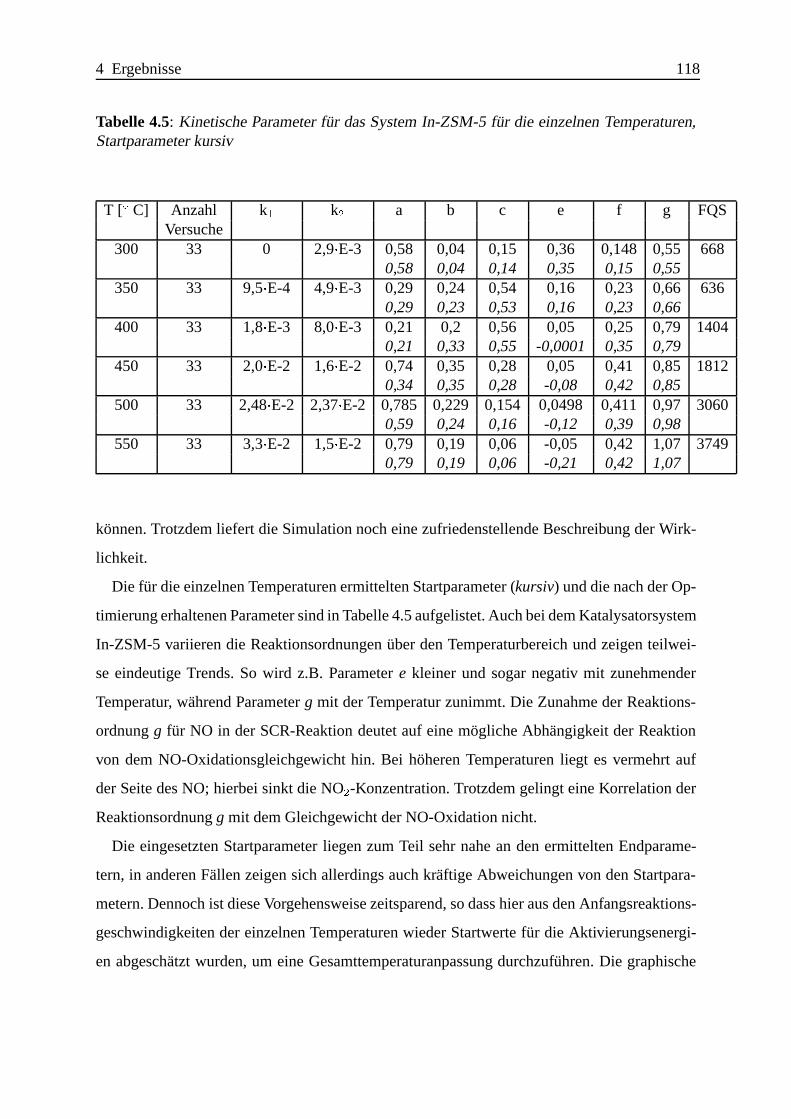
4 Ergebnisse 118
Tabelle 4.5: Kinetische Parameter f¨ur das System In-ZSM-5 f¨ur die einzelnen Temperaturen,Startparameter kursiv
T [Æ C] Anzahl k1 k2 a b c e f g FQSVersuche
300 33 0 2,9�E-3 0,58 0,04 0,15 0,36 0,148 0,55 6680,58 0,04 0,14 0,35 0,15 0,55
350 33 9,5�E-4 4,9�E-3 0,29 0,24 0,54 0,16 0,23 0,66 6360,29 0,23 0,53 0,16 0,23 0,66
400 33 1,8�E-3 8,0�E-3 0,21 0,2 0,56 0,05 0,25 0,79 14040,21 0,33 0,55 -0,0001 0,35 0,79
450 33 2,0�E-2 1,6�E-2 0,74 0,35 0,28 0,05 0,41 0,85 18120,34 0,35 0,28 -0,08 0,42 0,85
500 33 2,48�E-2 2,37�E-2 0,785 0,229 0,154 0,0498 0,411 0,97 30600,59 0,24 0,16 -0,12 0,39 0,98
550 33 3,3�E-2 1,5�E-2 0,79 0,19 0,06 -0,05 0,42 1,07 37490,79 0,19 0,06 -0,21 0,42 1,07
konnen. Trotzdem liefert die Simulation noch eine zufriedenstellende Beschreibung der Wirk-
lichkeit.
Die fur die einzelnen Temperaturen ermittelten Startparameter (kursiv) und die nach der Op-
timierung erhaltenen Parameter sind in Tabelle 4.5 aufgelistet. Auch bei dem Katalysatorsystem
In-ZSM-5 variieren die Reaktionsordnungen ¨uber den Temperaturbereich und zeigen teilwei-
se eindeutige Trends. So wird z.B. Parametere kleiner und sogar negativ mit zunehmender
Temperatur, w¨ahrend Parameterg mit der Temperatur zunimmt. Die Zunahme der Reaktions-
ordnungg fur NO in der SCR-Reaktion deutet auf eine m¨ogliche Abhangigkeit der Reaktion
von dem NO-Oxidationsgleichgewicht hin. Bei h¨oheren Temperaturen liegt es vermehrt auf
der Seite des NO; hierbei sinkt die NO2-Konzentration. Trotzdem gelingt eine Korrelation der
Reaktionsordnungg mit dem Gleichgewicht der NO-Oxidation nicht.
Die eingesetzten Startparameter liegen zum Teil sehr nahe an den ermittelten Endparame-
tern, in anderen F¨allen zeigen sich allerdings auch kr¨aftige Abweichungen von den Startpara-
metern. Dennoch ist diese Vorgehensweise zeitsparend, so dass hier aus den Anfangsreaktions-
geschwindigkeiten der einzelnen Temperaturen wieder Startwerte f¨ur die Aktivierungsenergi-
en abgesch¨atzt wurden, um eine Gesamttemperaturanpassung durchzuf¨uhren. Die graphische
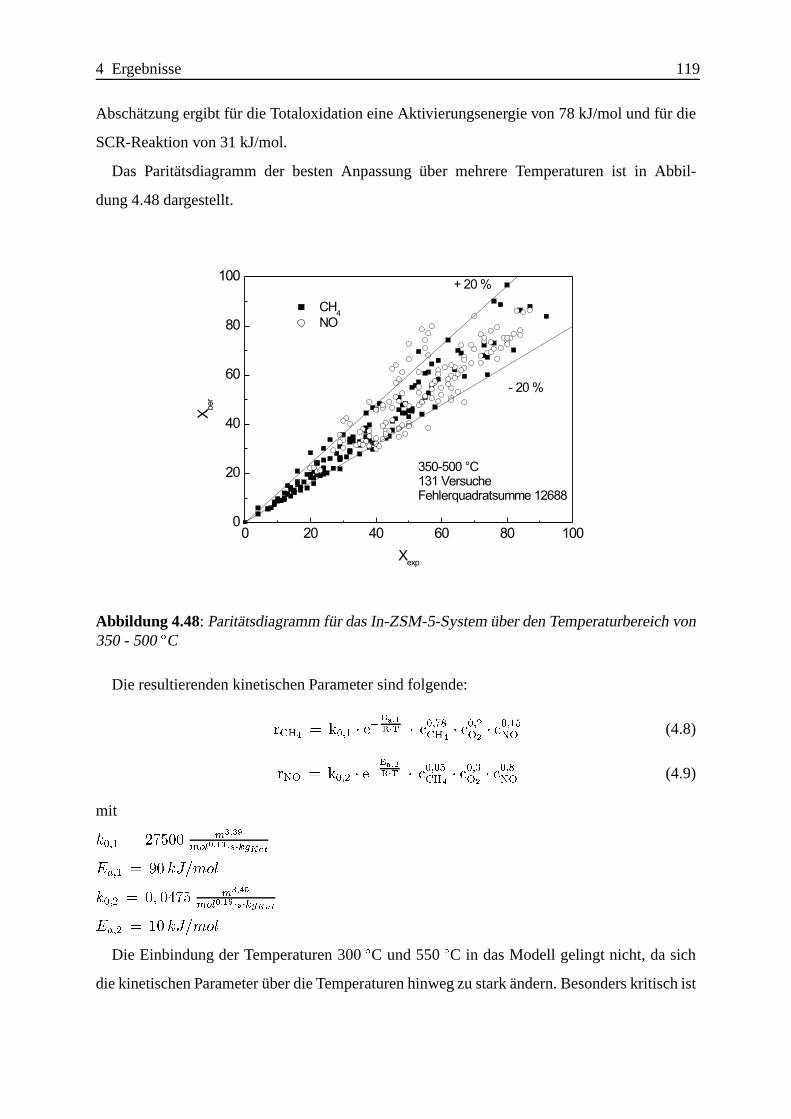
4 Ergebnisse 119
Abschatzung ergibt f¨ur die Totaloxidation eine Aktivierungsenergie von 78 kJ/mol und f¨ur die
SCR-Reaktion von 31 kJ/mol.
Das Paritatsdiagramm der besten Anpassung ¨uber mehrere Temperaturen ist in Abbil-
dung 4.48 dargestellt.
0 20 40 60 80 1000
20
40
60
80
100
350-500 °C131 VersucheFehlerquadratsumme 12688
- 20 %
+ 20 %
CH4
NO
Xber
Xexp
Abbildung 4.48: Paritatsdiagramm f¨ur das In-ZSM-5-System ¨uber den Temperaturbereich von350 - 500ÆC
Die resultierenden kinetischen Parameter sind folgende:
rCH4 = k0;1 � e�Ea;1R�T � c0;78CH4
� c0;2O2� c0;15NO (4.8)
rNO = k0;2 � e�Ea;2R�T � c0;05CH4
� c0;3O2� c0;8NO (4.9)
mit
k0;1 = 27500 m3;39
mol0;13�s�kgKat
Ea;1 = 90 kJ=mol
k0;2 = 0; 0475 m3;45
mol0;15�s�kgKat
Ea;2 = 10 kJ=mol
Die Einbindung der Temperaturen 300ÆC und 550ÆC in das Modell gelingt nicht, da sich
die kinetischen Parameter ¨uber die Temperaturen hinweg zu stark ¨andern. Besonders kritisch ist

4 Ergebnisse 120
die Einbindung der Versuche bei 550ÆC, da hier ein drastischer Abfall der selektiven Nutzung
des Methans f¨ur die SCR-Reaktion und ein starker Anstieg der Totaloxidation zu verzeichnen
ist.
Uber den immer noch weiten Temperaturbereich von 350 - 500ÆC werden die experimen-
tellen Ergebnisse gr¨oßtenteils innerhalb von� 20 % relativem Fehler simuliert. Ein ¨uber-
raschendes Ergebnis ist hierbei die sehr niedrige Aktivierungsenergie von 10 kJ/mol f¨ur die
SCR-Reaktion. Anhand des Startwertes von 31 kJ/mol wurde eine niedrige Aktivierungsener-
gie erwartet, aber nicht in diesem Maße. Bei einer einfachen Reaktion w¨urde ein Wert von
10 kJ/mol fur eine Stofftransportlimitierung sprechen, diese ist jedoch durch theoretische Be-
rechnungen ausgeschlossen worden. Da es sich jedoch um ein sehr komplexes Reaktionsnetz-
werk handelt, kommen auch andere Ursachen f¨ur diesen niedrigen Wert in Frage. Auch bei
vorgelagerten Gleichgewichten kann es zu niedrigen Aktivierungsenergien kommen. So weist
z.B. die NO-Oxidation eine scheinbare negative Aktivierungsenergie auf, da der Oxidations-
mechanismus ¨uber eine vorgelagerte Gleichgewichtseinstellung zwischen NO und N2O2, mit
anschließender Oxidation des N2O2, verlauft. Hierbei wirkt mit steigender Temperatur die
Verschiebung des Gleichgewichts zum NO st¨arker als die Aktivierungsenergie der Oxidati-
onsreaktion, was sich in einer negativen Aktivierungsenergie ausdr¨uckt [128]. Gerade in der
SCR-Reaktion k¨onnen Gleichgewichte dieser Art eine Rolle spielen, da als Anfangsreaktion
die NO-Oxidation postuliert wird. Bei den Stickoxiden k¨onnen sehr viele zus¨atzliche Gleich-
gewichte auftreten.
Bei Verwendung von Indium als Dotierkomponente auf einem sulfatierten Zirkonoxidtr¨ager
wurden auch vonPappet al. eine sehr niedrige Aktivierungsenergie von 15 kJ/mol im Ge-
gensatz zu 55 kJ/mol bei einem Ga-Katalysator gefunden, die Autoren treffen jedoch keine
Aussage ¨uber eine m¨ogliche Diffusionslimitierung [129].
Die Gute der Gesamttemperaturanpassung wird an ausgew¨ahlten Beispielen untersucht.
In Abbildung 4.49 sind mehrere Vergleiche bei unterschiedlichen Methan-, NO- und Sauer-
stoffkonzentrationen sowie bei verschiedenen Belastungen ¨uber den Temperaturbereich von
350 - 500ÆC dargestellt. Die Methanums¨atze werden f¨ur alle Abhangigkeiten recht gut si-
muliert. Bei den NO-Ums¨atzen wird der zu hohen Temperaturen hin auftretende Abfall des
Umsatzes von dem verwendeten Potenzmodell nur schlecht wiedergegeben. Auch durch eine
hohere Totaloxidation des Methans kann kein sinkender NO-Umsatz simuliert werden. Dies
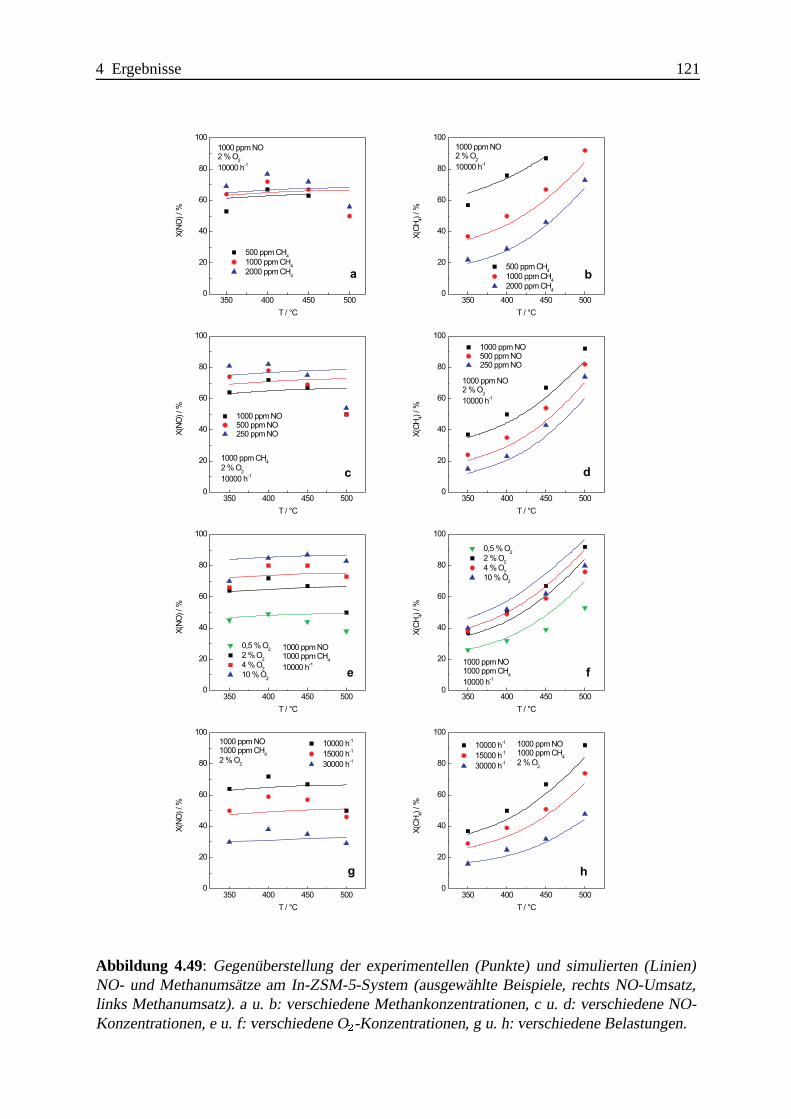
4 Ergebnisse 121
350 400 450 5000
20
40
60
80
100
a
1000 ppm NO2 % O
2
10000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(N
O)/%
T / °C
350 400 450 5000
20
40
60
80
100
b
1000 ppm NO2 % O
2
10000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
c
1000 ppm CH4
2 % O2
10000 h-1
1000 ppm NO500 ppm NO250 ppm NOX
(NO
)/%
T / °C
350 400 450 5000
20
40
60
80
100
d
1000 ppm NO2 % O
2
10000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
f1000 ppm NO1000 ppm CH
4
10000 h-1
0,5 % O2
2 % O2
4 % O2
10 % O2
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
e
1000 ppm NO1000 ppm CH
4
10000 h-1
0,5 % O2
2 % O2
4 % O2
10 % O2
X(N
O)/%
T / °C
350 400 450 5000
20
40
60
80
100
h
1000 ppm NO1000 ppm CH
4
2 % O2
10000 h-1
15000 h-1
30000 h-1
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
g
1000 ppm NO1000 ppm CH
4
2 % O2
10000 h-1
15000 h-1
30000 h-1
X(N
O)/%
T / °C
Abbildung 4.49: Gegen¨uberstellung der experimentellen (Punkte) und simulierten (Linien)NO- und Methanums¨atze am In-ZSM-5-System (ausgew¨ahlte Beispiele, rechts NO-Umsatz,links Methanumsatz). a u. b: verschiedene Methankonzentrationen, c u. d: verschiedene NO-Konzentrationen, e u. f: verschiedene O2-Konzentrationen, g u. h: verschiedene Belastungen.

4 Ergebnisse 122
Tabelle 4.6: Sensitivitatsanalyse der kinetischen Parameter f¨ur das In-ZSM-5-System
Parameter k0;1 k0;2 Ea;1 Ea;2 a b c e f g
+ 5 % 0 % 2,3 % 106 % 14,7 % 12,8 % 0,5 % 1,4 % 0,07 % 0,5 % 71 %- 5 % 1 % 4,9 % 7,1 % 0,3 % 0,2 % 0,01 % 1,1 % 81 %
spricht fur einen zusammengesetzten Reaktionsmechanismus, der mit Hilfe eines komplizier-
teren Ansatzes angepasst werden muss, was aber aus Zeitgr¨unden und Gr¨unden der Vergleich-
barkeit zwischen den einzelnen Systemen nicht durchgef¨uhrt wurde. Bei h¨oheren Belastungen
sind die Anpassungen jedoch besser, und f¨ur die Einzeltemperaturen wird mit Hilfe eines ein-
fachen Potenzansatzes eine guteUbereinstimmung erzielt.
Um die Aussagef¨ahigkeit der erhaltenen Parameter eingehender zu untersuchen, wurde auch
fur dieses System eine Sensitivit¨atsanalyse durchgef¨uhrt, deren Ergebnisse in Tabelle 4.6 auf-
gelistet sind. Besonders sensitiv auf eine Ver¨anderung reagieren die beiden Aktivierungsener-
gien, die Reaktionsordnung f¨ur Methan in der Totaloxidationsreaktion und die Reaktions-
ordnung fur NO in der SCR-Reaktion. Dies steht qualitativ inUbereinstimmung mit dem
CeO2-ZSM-5-System.
4.2.3 Das System CeO2-In-ZSM-5
Fur die kinetischen Messungen an dem CeO2-In-ZSM-5-System wurde ein durch reduktiven
Festkorperionentausch mit H2 als Reduktionsmittel dargestellter Katalysator verwendet. Dieser
Katalysator mit dem Namen 8Ce13In-RSSIE/H wurde von unseren Projektpartnern am Institut
fur angewandte Chemie in Berlin/Adlershof im Technikumsmaßstab in einer Menge von 3 kg
prapariert.
Bei der Praparation wurde zun¨achst Indium mittels Ionentausch in einer w¨assrigen Indi-
umnitratlosung bei 60ÆC als [In(OH)]2+ oder als [In(OH)2]+ in den Zeolithen NH4-ZSM-5
(Si/Al = 14, Zeolyst Int) eingebracht. Anschließend erfolgte die Auff¨allung des restlichen In-
diums als In(OH)3 mit 25 %iger Ammoniakl¨osung. Der modifizierte Zeolith wurde abgefiltert,
bei 140ÆC 6 Stunden getrocknet und f¨ur 2 Stunden bei 500ÆC in einem Muffelofen kalziniert.
Die Promotierung mit Cer wurde in einem weiteren Pr¨aparationsschritt durch rasche Auff¨allung
von Ce(OH)3 mit 25 %iger Ammoniakl¨osung aus einer Cernitratl¨osung realisiert. Nach Filte-

4 Ergebnisse 123
rung, Trocknung und Kalzination (vide supra) wurde der Katalysator drei reduktiv/oxidativen
Kalzinationszyklen unterworfen. Dabei wurde alle 30 Minuten bei 500ÆC von einer reduzie-
renden (5 Volumenprozent H2 in N2) auf eine oxidierende Gasmischung (5 Volumenprozent O2
in N2) umgeschaltet (vor jeder Umschaltung wurde die Apparatur mit Stickstoff gesp¨ult).
Durch die reduktive Behandlung werden, wie vonSchmidtgezeigt werden konnte [15], intra-
zeolithische Indiumspezies gebildet. Es handelt sich hier wegen der hohen Indiumbeladung um
polynukleare Kationen, die ¨uberwiegend in oberfl¨achennahen Regionen des Zeolithen lokali-
siert sind.
Der Katalysator 8Ce13In-RSSIE/H weist eine Beladung von 8-Massenprozent Cer und
13-Massenprozent Indium auf. Er hat sich bei hydrothermalen Alterungsversuchen am ACA
als besonders stabil erwiesen und ist damit f¨ur eine sp¨atere technische Anwendung von In-
teresse. Im weiteren Verlauf dieser Arbeit wird er hier als CeO2-In-ZSM-5-Katalysator oder
-System bezeichnet.
Die kinetischen Messungen wurden gr¨oßtenteils an der vom ACA in Berlin bereitgestellten
Messapparatur durchgef¨uhrt. Erganzende Versuche, besonders im Hinblick auf Messungen im
feuchten Eduktgas, erfolgten in Bochum.
Zunachst sollen hier die mit trockenem Eduktgas erhaltenen Ergebnisse vorgestellt werden,
um sie mit den Ergebnissen der anderen Systeme CeO2-ZSM-5 und In-ZSM-5 vergleichen zu
konnen. Das Einfahrverhalten des Katalysators wird in Abbildung 4.50 beschrieben. Da zu die-
sem Zeitpunkt der Messungen lediglich der Gaschromatograph f¨ur eine Analyse zur Verf¨ugung
stand, sind die Messwerte mit einer gr¨oßeren Fehlergrenze behaftet als bei Verwendung des
Massenspektrometers. Trotzdem kann dem Langzeitversuch mit trockenem Edukt entnommen
werden, dass der Katalysator sowohl bez¨uglich des Methanumsatzes, als auch bez¨uglich des
NO-Umsatzes einer stetigen Deaktivierung unterliegt. Der NO-Umsatz sinkt von anfangs ca.
85 % auf ca. 83 % nach 50 Stunden. Nach 100 Stunden betr¨agt er ca. 81 % und nach 150 Stun-
den ca. 79 %. Es erfolgt also alle 2 Tage ein Umsatzr¨uckgang um ca. 2 %. F¨ur den Methan-
umsatz wird eine etwas st¨arkere Deaktivierung beobachtet. Er sinkt von ca. 60 % auf ca. 55 %
nach 50 Stunden, erreicht nach 100 Stunden noch ca. 52 %, und nach 150 Stunden werden
immer noch ca. 49 % Methan umgesetzt. Aus diesem Grund wurde der f¨ur die kinetischen
Messungen eingesetzte Katalysator ¨uber Nacht vorbehandelt und danach 4 Tage lang zu Mes-
sungen eingesetzt. Hierbei betrug die Temperatur nat¨urlich nur in Extremfallen 500ÆC, so dass
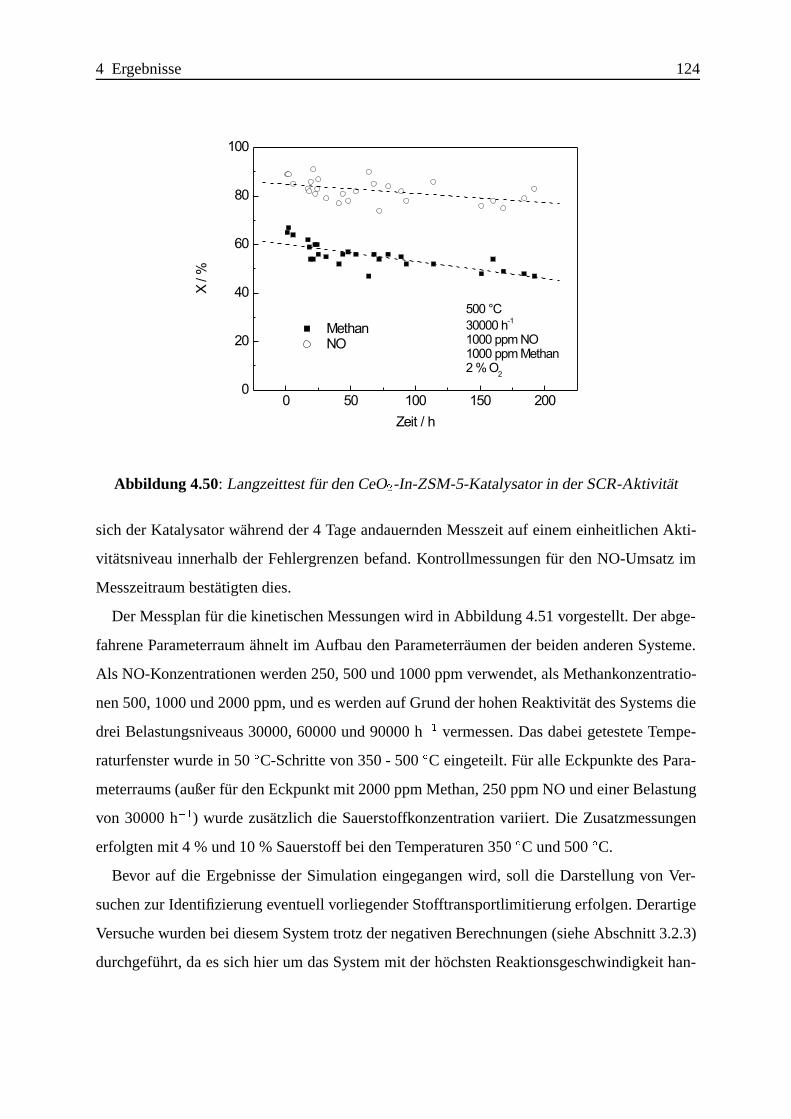
4 Ergebnisse 124
0 50 100 150 2000
20
40
60
80
100
500 °C
30000 h-1
1000 ppm NO1000 ppm Methan2 % O
2
MethanNO
X/%
Zeit / h
Abbildung 4.50: Langzeittest f¨ur den CeO2-In-ZSM-5-Katalysator in der SCR-Aktivit¨at
sich der Katalysator w¨ahrend der 4 Tage andauernden Messzeit auf einem einheitlichen Akti-
vitatsniveau innerhalb der Fehlergrenzen befand. Kontrollmessungen f¨ur den NO-Umsatz im
Messzeitraum best¨atigten dies.
Der Messplan f¨ur die kinetischen Messungen wird in Abbildung 4.51 vorgestellt. Der abge-
fahrene Parameterraum ¨ahnelt im Aufbau den Parameterr¨aumen der beiden anderen Systeme.
Als NO-Konzentrationen werden 250, 500 und 1000 ppm verwendet, als Methankonzentratio-
nen 500, 1000 und 2000 ppm, und es werden auf Grund der hohen Reaktivit¨at des Systems die
drei Belastungsniveaus 30000, 60000 und 90000 h�1 vermessen. Das dabei getestete Tempe-
raturfenster wurde in 50ÆC-Schritte von 350 - 500ÆC eingeteilt. F¨ur alle Eckpunkte des Para-
meterraums (außer f¨ur den Eckpunkt mit 2000 ppm Methan, 250 ppm NO und einer Belastung
von 30000 h�1) wurde zus¨atzlich die Sauerstoffkonzentration variiert. Die Zusatzmessungen
erfolgten mit 4 % und 10 % Sauerstoff bei den Temperaturen 350ÆC und 500ÆC.
Bevor auf die Ergebnisse der Simulation eingegangen wird, soll die Darstellung von Ver-
suchen zur Identifizierung eventuell vorliegender Stofftransportlimitierung erfolgen. Derartige
Versuche wurden bei diesem System trotz der negativen Berechnungen (siehe Abschnitt 3.2.3)
durchgefuhrt, da es sich hier um das System mit der h¨ochsten Reaktionsgeschwindigkeit han-

4 Ergebnisse 125
250
500
750
1000
30000
60000
90000
500
1000
1500
2000
Bela
stung
[h-1]
CH 4-K
onze
ntrati
on[pp
m]NO-Konzentration [ppm]
Abbildung 4.51: Messplan der kinetischen Messungen am CeO2-In-ZSM-5-Katalysator
delt und da somit die Wahrscheinlichkeit einer Stofftransportlimitierung am h¨ochsten ist. Die
Versuche sind in Abbildung 4.52 dargestellt. ZurUberprufung aufaußere Stofftransportlimitie-
rung wird die Gasgeschwindigkeit durch Ver¨anderung der Gasvolumenstr¨ome unter Beibehal-
tung der Belastung von 30000 h�1 variiert. In Abbildung 4.52 sind die NO- und die Methan-
umsatze fur die drei Volumenstr¨ome 55, 110 und 220 ml/min und damit f¨ur drei unterschied-
liche Gasstr¨omungsgeschwindigkeiten und Grenzschichtdicken aufgetragen. Die NO-Ums¨atze
zeigen fur alle drei Str¨omungsgeschwindigkeiten einen ¨ahnlichen Verlauf, und der Maximal-
umsatz wird bei 500ÆC erreicht; allerdings liegen die Werte des NO-Maximalumsatzes f¨ur
geringere Gasgeschwindigkeiten leicht unter dem Wert, der bei einem Volumenstrom von
220 ml/min beobachtet wird. Da aber mit 55 ml/min h¨ohere Ums¨atze erzielt werden als mit
110 ml/min, handelt es sich nicht um einen Diffusionseffekt. Die Ver¨anderung der Partikel-
große von der Standardkornfraktion 250 - 355�m auf eine Kornfraktion von 500 - 1000�m
bewirkt keineAnderung der Ums¨atze; Porendiffusionslimitierung in den Mesoporen des Zeo-
lithen kann somit ausgeschlossen werden. EineUberprufung auf Porendiffusionslimitierung in
den Zeolithporen selber kann nicht durchgef¨uhrt werden, da keine Zeolithkristalle mit unter-
schiedlicher Gr¨oße, aber mit ansonsten identischen Eigenschaften zur Verf¨ugung standen.
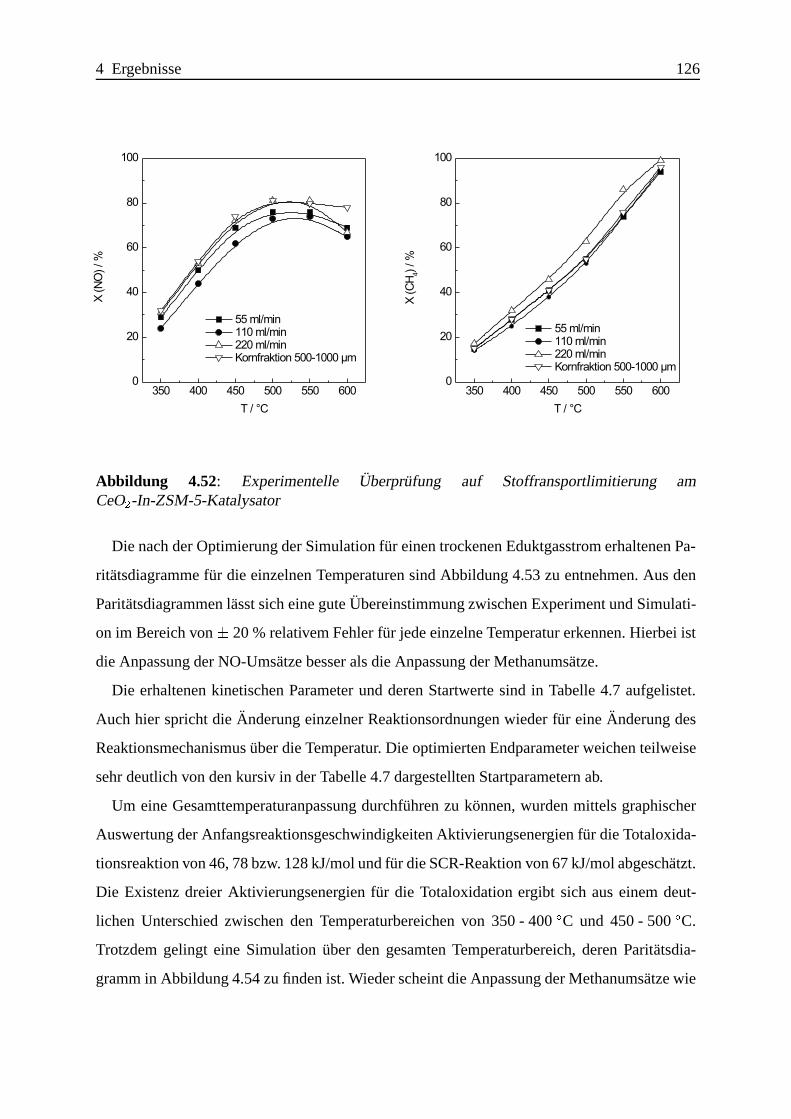
4 Ergebnisse 126
350 400 450 500 550 6000
20
40
60
80
100
55 ml/min110 ml/min220 ml/minKornfraktion 500-1000 µm
X(N
O)/%
T / °C
350 400 450 500 550 6000
20
40
60
80
100
55 ml/min110 ml/min220 ml/minKornfraktion 500-1000 µm
X(C
H4)
/%
T / °C
Abbildung 4.52: Experimentelle Uberprufung auf Stoffransportlimitierung amCeO2-In-ZSM-5-Katalysator
Die nach der Optimierung der Simulation f¨ur einen trockenen Eduktgasstrom erhaltenen Pa-
ritatsdiagramme f¨ur die einzelnen Temperaturen sind Abbildung 4.53 zu entnehmen. Aus den
Paritatsdiagrammen l¨asst sich eine guteUbereinstimmung zwischen Experiment und Simulati-
on im Bereich von� 20 % relativem Fehler f¨ur jede einzelne Temperatur erkennen. Hierbei ist
die Anpassung der NO-Ums¨atze besser als die Anpassung der Methanums¨atze.
Die erhaltenen kinetischen Parameter und deren Startwerte sind in Tabelle 4.7 aufgelistet.
Auch hier spricht dieAnderung einzelner Reaktionsordnungen wieder f¨ur eineAnderung des
Reaktionsmechanismus ¨uber die Temperatur. Die optimierten Endparameter weichen teilweise
sehr deutlich von den kursiv in der Tabelle 4.7 dargestellten Startparametern ab.
Um eine Gesamttemperaturanpassung durchf¨uhren zu konnen, wurden mittels graphischer
Auswertung der Anfangsreaktionsgeschwindigkeiten Aktivierungsenergien f¨ur die Totaloxida-
tionsreaktion von 46, 78 bzw. 128 kJ/mol und f¨ur die SCR-Reaktion von 67 kJ/mol abgesch¨atzt.
Die Existenz dreier Aktivierungsenergien f¨ur die Totaloxidation ergibt sich aus einem deut-
lichen Unterschied zwischen den Temperaturbereichen von 350 - 400ÆC und 450 - 500ÆC.
Trotzdem gelingt eine Simulation ¨uber den gesamten Temperaturbereich, deren Parit¨atsdia-
gramm in Abbildung 4.54 zu finden ist. Wieder scheint die Anpassung der Methanums¨atze wie
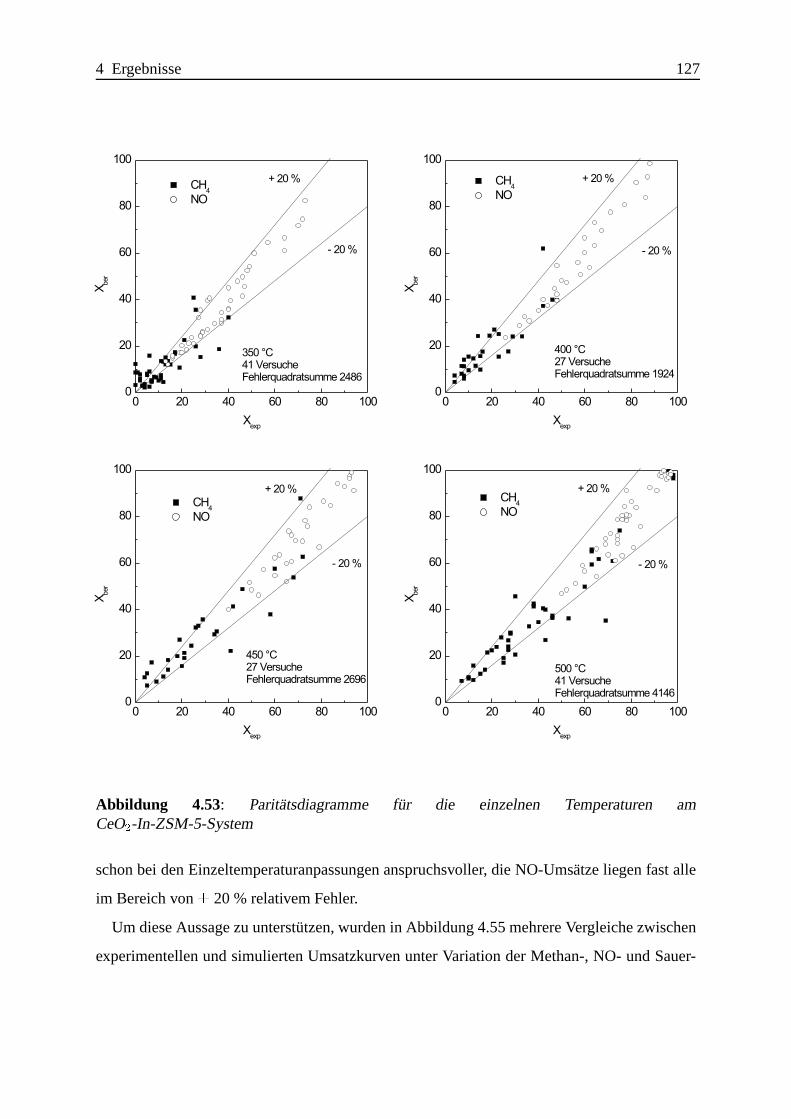
4 Ergebnisse 127
0 20 40 60 80 1000
20
40
60
80
100
- 20 %
+ 20 %
450 °C27 VersucheFehlerquadratsumme 2696
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
- 20 %
+ 20 %
400 °C27 VersucheFehlerquadratsumme 1924
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
- 20 %
+ 20 %
350 °C41 VersucheFehlerquadratsumme 2486
CH4
NO
Xber
Xexp
0 20 40 60 80 1000
20
40
60
80
100
- 20 %
+ 20 %
500 °C41 VersucheFehlerquadratsumme 4146
CH4
NO
Xber
Xexp
Abbildung 4.53: Paritatsdiagramme f¨ur die einzelnen Temperaturen amCeO2-In-ZSM-5-System
schon bei den Einzeltemperaturanpassungen anspruchsvoller, die NO-Ums¨atze liegen fast alle
im Bereich von� 20 % relativem Fehler.
Um diese Aussage zu unterst¨utzen, wurden in Abbildung 4.55 mehrere Vergleiche zwischen
experimentellen und simulierten Umsatzkurven unter Variation der Methan-, NO- und Sauer-
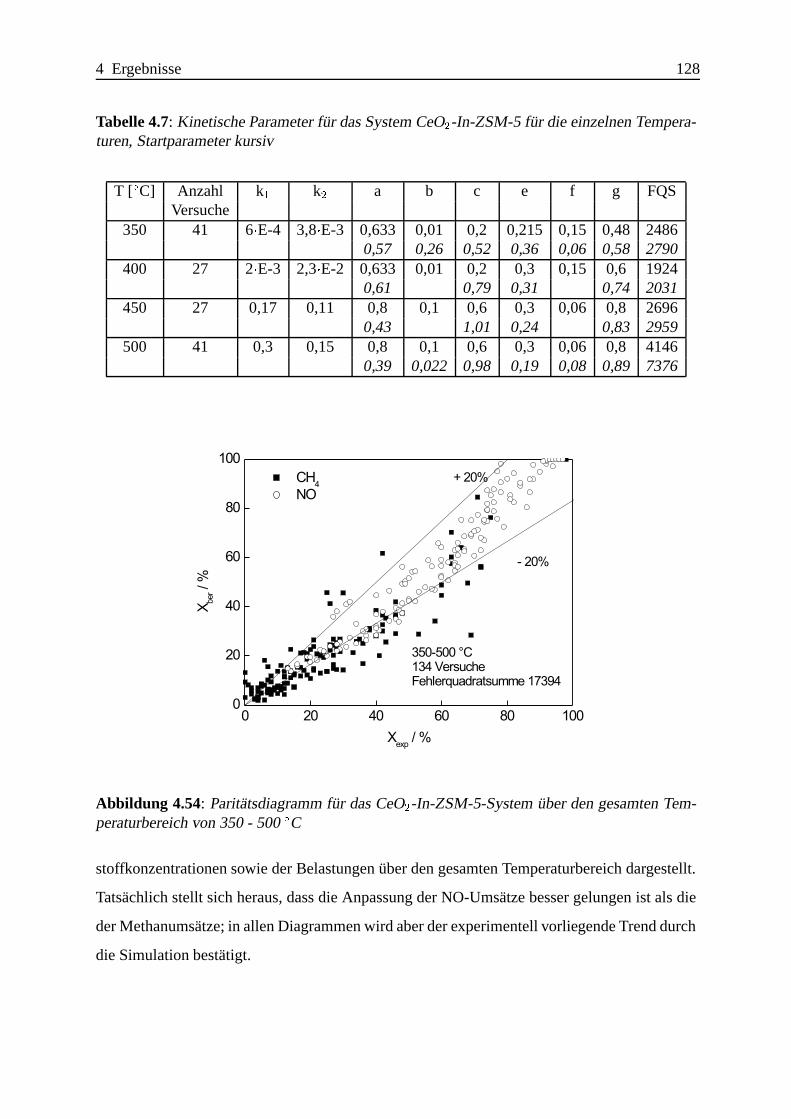
4 Ergebnisse 128
Tabelle 4.7: Kinetische Parameter f¨ur das System CeO2-In-ZSM-5 fur die einzelnen Tempera-turen, Startparameter kursiv
T [ÆC] Anzahl k1 k2 a b c e f g FQSVersuche
350 41 6�E-4 3,8�E-3 0,633 0,01 0,2 0,215 0,15 0,48 24860,57 0,26 0,52 0,36 0,06 0,58 2790
400 27 2�E-3 2,3�E-2 0,633 0,01 0,2 0,3 0,15 0,6 19240,61 0,79 0,31 0,74 2031
450 27 0,17 0,11 0,8 0,1 0,6 0,3 0,06 0,8 26960,43 1,01 0,24 0,83 2959
500 41 0,3 0,15 0,8 0,1 0,6 0,3 0,06 0,8 41460,39 0,022 0,98 0,19 0,08 0,89 7376
0
20
40
60
80
100
0 20 40 60 80 100
350-500 °C134 VersucheFehlerquadratsumme 17394
- 20%
+ 20%CH4
NO
Xexp
/ %
Xber/%
Abbildung 4.54: Paritatsdiagramm f¨ur das CeO2-In-ZSM-5-System ¨uber den gesamten Tem-peraturbereich von 350 - 500ÆC
stoffkonzentrationen sowie der Belastungen ¨uber den gesamten Temperaturbereich dargestellt.
Tatsachlich stellt sich heraus, dass die Anpassung der NO-Ums¨atze besser gelungen ist als die
der Methanums¨atze; in allen Diagrammen wird aber der experimentell vorliegende Trend durch
die Simulation best¨atigt.
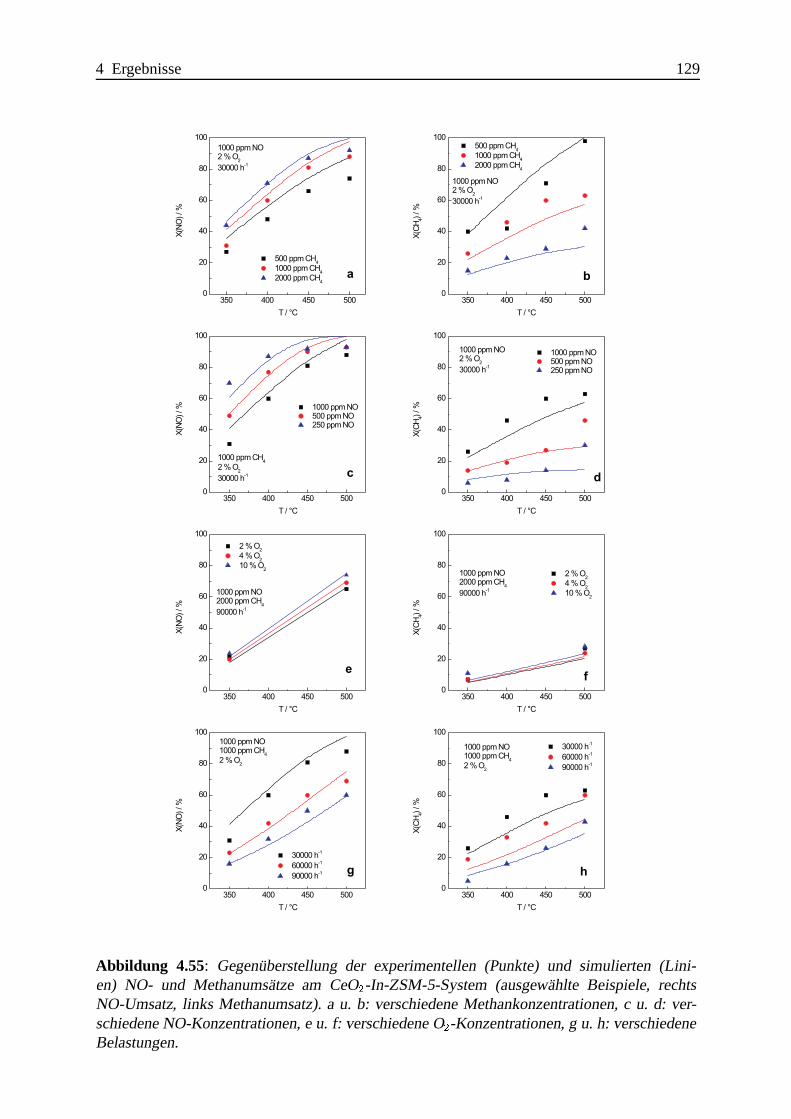
4 Ergebnisse 129
350 400 450 5000
20
40
60
80
100
a
1000 ppm NO2 % O
2
30000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(N
O)/%
T / °C
350 400 450 5000
20
40
60
80
100
b
1000 ppm NO2 % O
2
30000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
c
1000 ppm CH4
2 % O2
30000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(N
O)/%
T / °C
350 400 450 5000
20
40
60
80
100
d
1000 ppm NO2 % O
2
30000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
e
1000 ppm NO2000 ppm CH
4
90000 h-1
2 % O2
4 % O2
10 % O2
X(N
O)/%
T / °C
350 400 450 5000
20
40
60
80
100
f
1000 ppm NO2000 ppm CH
4
90000 h-1
2 % O2
4 % O2
10 % O2
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
h
1000 ppm NO1000 ppm CH
4
2 % O2
30000 h-1
60000 h-1
90000 h-1
X(C
H4)
/%
T / °C
350 400 450 5000
20
40
60
80
100
g
1000 ppm NO1000 ppm CH
4
2 % O2
30000 h-1
60000 h-1
90000 h-1
X(N
O)/%
T / °C
Abbildung 4.55: Gegen¨uberstellung der experimentellen (Punkte) und simulierten (Lini-en) NO- und Methanums¨atze am CeO2-In-ZSM-5-System (ausgew¨ahlte Beispiele, rechtsNO-Umsatz, links Methanumsatz). a u. b: verschiedene Methankonzentrationen, c u. d: ver-schiedene NO-Konzentrationen, e u. f: verschiedene O2-Konzentrationen, g u. h: verschiedeneBelastungen.

4 Ergebnisse 130
Tabelle 4.8: Sensitivitatsanalyse der kinetischen Parameter f¨ur das CeO2-In-ZSM-5-System
Parameter k0;1 k0;2 Ea;1 Ea;2 a b c e f g
+ 5 % 8,6 % 154 % 1,7 % 0,3 % 3,9 % 3,4 % 0,5 % 37 %- 5 % 0,8 % 3,9 %
Die ermittelten kinetischen Parameter sind folgende:
rCH4 = k0;1 � e�Ea;1R�T � c0;4CH4 � c
0;2O2� c0;8NO (4.10)
rNO = k0;2 � e�Ea;2R�T � c0;2CH4 � c
0;1O2� c0;65NO (4.11)
mit
k0;1 = 5100 m4;2
mol0;4�s�kgKat
Ea;1 = 72 kJ=mol
k0;2 = 70 m2;85
mol�0;05�s�kgKat
Ea;2 = 47 kJ=mol
Die Reaktionsordnungen werden im Abschnitt 4.2.4 eingehender diskutiert, die ermittelten Ak-
tivierungsenergien liegen im physikalisch sinnvollen Bereich.
Nach der in Tabelle 4.8 dargestellten Sensitivit¨atsanalyse zeigen sich wie bei den beiden
ubrigen Katalysatorsystemen auch die beiden Aktivierungsenergien und die Reaktionsordnung
g fur NO in der SCR-Reaktion besonders anf¨allig gegen¨uber einer Parameter¨anderung.
Bis hierher sind die Ergebnisse f¨ur das CeO2-In-ZSM-5-System mit trockenem Edukt-
gas vorgestellt worden, bei denen durch die vollst¨andige Umsetzung des Methans h¨ochstens
0,4 % Wasser entstehen. Interessanter unter dem Aspekt einer m¨oglichen technischen An-
wendung sind jedoch Aktivit¨atsmessungen, bei denen schon im Eduktgas h¨ohere Wasser-
konzentrationen enthalten sind. Daher wurden zus¨atzliche kinetische Messungen unter Zu-
gabe von 0,5 - 10 % Wasser durchgef¨uhrt. Die Uberprufung der Stabilit¨at des Katalysators
CeO2-In-ZSM-5 unter Wasserdampf ist in Abbildung 4.56 dargestellt. Zur Alterung des Ka-
talysators wurden dem Eduktgas 10 % Wasser zugegeben. Die Aktivit¨at wurde im Anschluss
ohne Wasser im Eduktgas vermessen. Hierbei dauert es ca. 1 Stunde, bis der urspr¨unglich mit
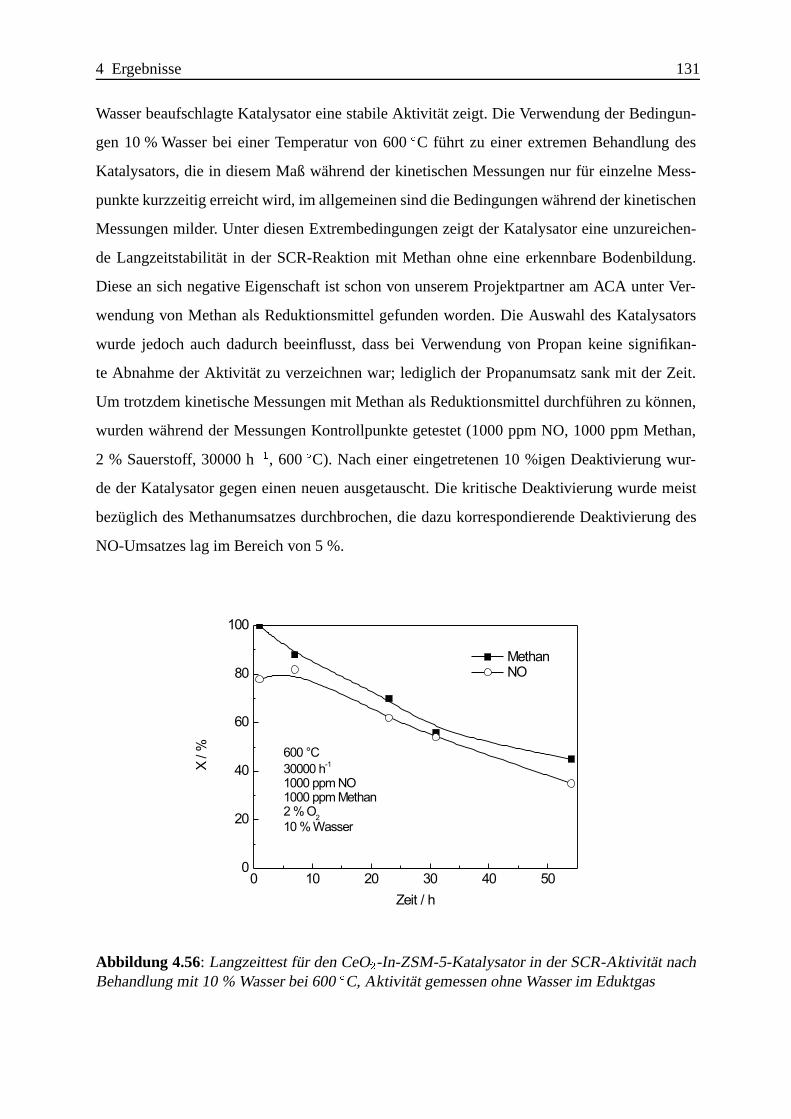
4 Ergebnisse 131
Wasser beaufschlagte Katalysator eine stabile Aktivit¨at zeigt. Die Verwendung der Bedingun-
gen 10 % Wasser bei einer Temperatur von 600ÆC fuhrt zu einer extremen Behandlung des
Katalysators, die in diesem Maß w¨ahrend der kinetischen Messungen nur f¨ur einzelne Mess-
punkte kurzzeitig erreicht wird, im allgemeinen sind die Bedingungen w¨ahrend der kinetischen
Messungen milder. Unter diesen Extrembedingungen zeigt der Katalysator eine unzureichen-
de Langzeitstabilit¨at in der SCR-Reaktion mit Methan ohne eine erkennbare Bodenbildung.
Diese an sich negative Eigenschaft ist schon von unserem Projektpartner am ACA unter Ver-
wendung von Methan als Reduktionsmittel gefunden worden. Die Auswahl des Katalysators
wurde jedoch auch dadurch beeinflusst, dass bei Verwendung von Propan keine signifikan-
te Abnahme der Aktivit¨at zu verzeichnen war; lediglich der Propanumsatz sank mit der Zeit.
Um trotzdem kinetische Messungen mit Methan als Reduktionsmittel durchf¨uhren zu konnen,
wurden wahrend der Messungen Kontrollpunkte getestet (1000 ppm NO, 1000 ppm Methan,
2 % Sauerstoff, 30000 h�1, 600ÆC). Nach einer eingetretenen 10 %igen Deaktivierung wur-
de der Katalysator gegen einen neuen ausgetauscht. Die kritische Deaktivierung wurde meist
bezuglich des Methanumsatzes durchbrochen, die dazu korrespondierende Deaktivierung des
NO-Umsatzes lag im Bereich von 5 %.
0 10 20 30 40 500
20
40
60
80
100
600 °C
30000 h-1
1000 ppm NO1000 ppm Methan2 % O
2
10 % Wasser
MethanNO
X/%
Zeit / h
Abbildung 4.56: Langzeittest f¨ur den CeO2-In-ZSM-5-Katalysator in der SCR-Aktivit¨at nachBehandlung mit 10 % Wasser bei 600ÆC, Aktivitat gemessen ohne Wasser im Eduktgas

4 Ergebnisse 132
Die zur Ermittlung der kinetischen Parameter durchgef¨uhrten Messungen sind in der Abbil-
dung 4.57 wiedergegeben. Am ACA in Berlin fanden Messungen bei Belastungen von 30000
und 90000 h�1 mit einem Wassergehalt von 1,5 % statt. Die Zusatzmessungen in Bochum
erfolgten unter Wasserkonzentrationen von bis zu 10 % und der geringeren Belastung von
30000 h�1, um im Hinblick auf die Messgenauigkeit akzeptable Ums¨atze zu erzielen. An al-
len Messpunkten wurden, soweit nicht anders angegeben, die Temperaturen 450, 500, 550 und
600ÆC getestet.
Um den Wassereinfluss auf die Totaloxidationsreaktion und die SCR-Reaktion modellieren
zu konnen, wurden die beiden Potenzans¨atze wie folgt modifiziert:
rCH4 = k1 � caCH4 � cbO2� ccNO � cdH2O (4.12)
rNO = k2 � ceCH4 � cfO2� cgNO � chH2O (4.13)
Von den bisherigen f¨ur die trockenen Systeme verwendeten Potenzans¨atzen unterscheiden sie
sich lediglich durch den Erg¨anzungsterm f¨ur die Wasserkonzentration. Somit ist es m¨oglich, die
Reaktionsordnung im trockenen Reaktionsmedium mit den Reaktionsordnungen im feuchten
Reaktionsmedium zu vergleichen.
Die nach einer anf¨anglichen Parametersch¨atzung und anschließender Optimierung erhal-
tenen Parit¨atsdiagramme sind in Abbildung 4.58 dargestellt. Trotz der Verwendung zweier
zusatzlicher Parameter streuen die Punkte in den Parit¨atsdiagrammen deutlicher um die Winkel-
halbierende, als es in den trockenen Eduktgasmessungen der Fall ist. Aufgrund der statistischen
Streuung ist ein systematischer Fehler auszuschließen, da sowohl zu hohe als auch zu niedrige
Methan- und NO-Ums¨atze simuliert werden und kein Trend zu erkennen ist.
Die ermittelten kinetischen Parameter finden sich in Tabelle 4.9. Auff¨allig ist, dass die op-
timierten Parameter teilweise erheblich von den gesch¨atzten Startparametern abweichen. Die
Reaktionsordnungen liegen hier insgesamt h¨oher als die Reaktionsordnungen, die f¨ur trockene
Eduktgasstr¨ome ermittelt wurden. Dieser Befund kann mit einer kompetitiven Adsorption von
Wasser und der entsprechenden Komponente erkl¨art werden. Eine genauere Diskussion mit
einem zus¨atzlichen Vergleich der Systeme untereinander und Literaturdaten erfolgt in Ab-
schnitt 4.2.4.
Mit den aus den optimierten Parametern berechenbaren Anfangsreaktionsgeschwindigkei-
ten konnen die Aktivierungsenergien f¨ur die Totaloxidationsreaktion zu 291 bzw. 78 kJ/mol
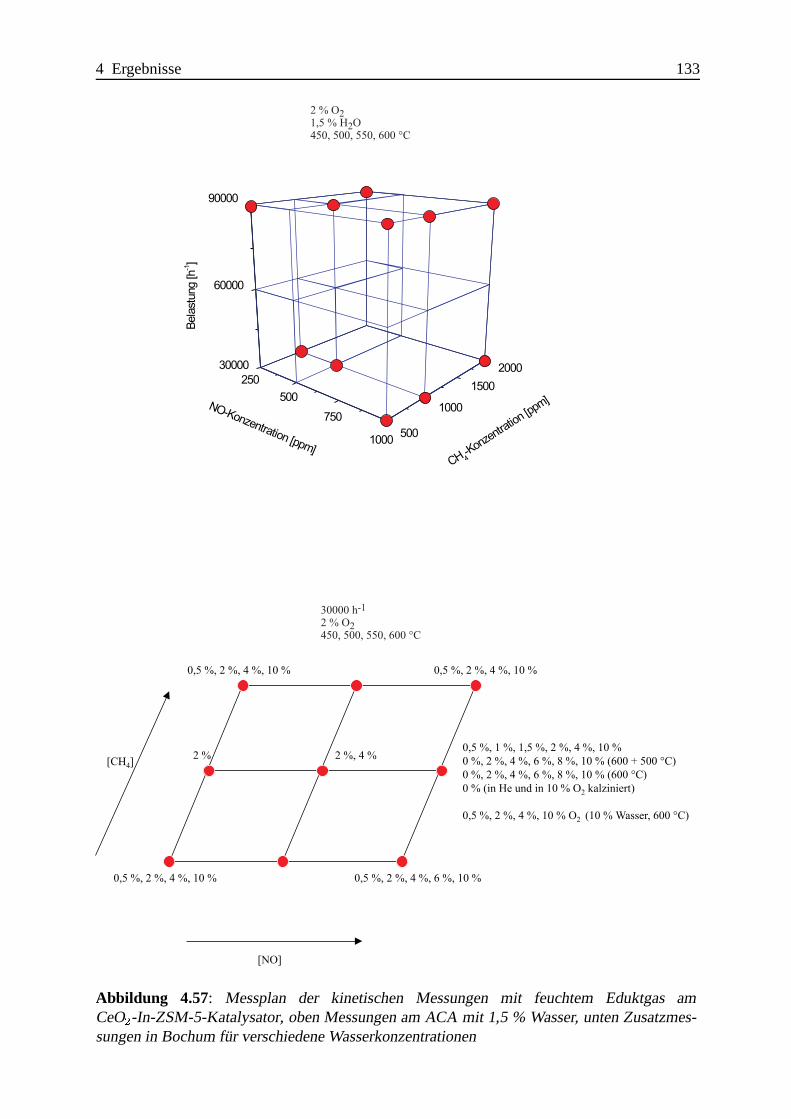
4 Ergebnisse 133
250
500
750
1000
30000
60000
90000
500
1000
1500
2000
Bela
stung
[h-1]
CH 4-K
onze
ntrati
on[pp
m]NO-Konzentration [ppm]
0,5 %, 2 %, 4 %, 10 %
2 % 2 %, 4 %
0,5 %, 2 %, 4 %, 10 %
0,5 %, 2 %, 4 %, 6 %, 10 %0,5 %, 2 %, 4 %, 10 %
0,5 %, 1 %, 1,5 %, 2 %, 4 %, 10 %
0 %, 2 %, 4 %, 6 %, 8 %, 10 % (600 + 500 °C)
0 %, 2 %, 4 %, 6 %, 8 %, 10 % (600 °C)
0 % (in He und in 10 % O2 kalziniert)
0,5 %, 2 %, 4 %, 10 % O2 (10 % Wasser, 600 °C)
[NO]
[CH4]
30000 h2 % O450, 500, 550, 600 °C
-1
2
2 % O1,5 % H O450, 500, 550, 600 °C
2
2
Abbildung 4.57: Messplan der kinetischen Messungen mit feuchtem Eduktgas amCeO2-In-ZSM-5-Katalysator, oben Messungen am ACA mit 1,5 % Wasser, unten Zusatzmes-sungen in Bochum f¨ur verschiedene Wasserkonzentrationen
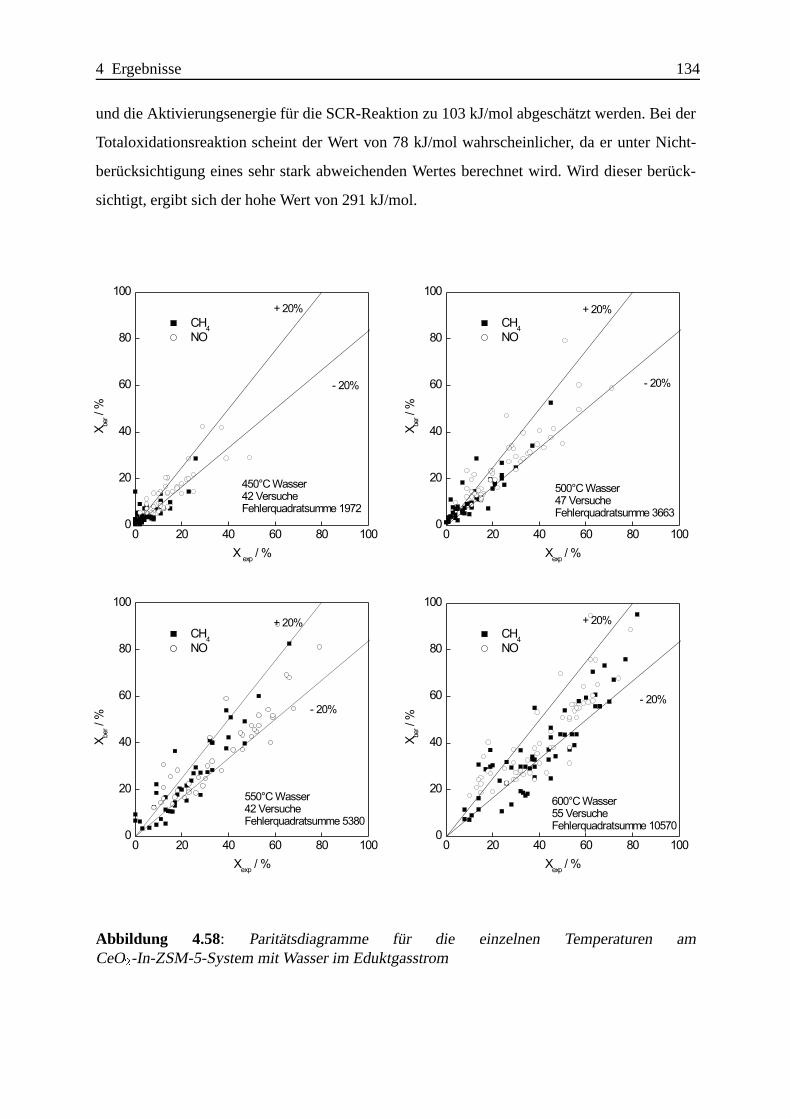
4 Ergebnisse 134
und die Aktivierungsenergie f¨ur die SCR-Reaktion zu 103 kJ/mol abgesch¨atzt werden. Bei der
Totaloxidationsreaktion scheint der Wert von 78 kJ/mol wahrscheinlicher, da er unter Nicht-
berucksichtigung eines sehr stark abweichenden Wertes berechnet wird. Wird dieser ber¨uck-
sichtigt, ergibt sich der hohe Wert von 291 kJ/mol.
0
20
40
60
80
100
0 20 40 60 80 100
600°C Wasser55 VersucheFehlerquadratsumme 10570
- 20%
+ 20%
CH4
NO
Xexp
/ %
Xbe
r/%
0
20
40
60
80
100
0 20 40 60 80 100
550°C Wasser42 VersucheFehlerquadratsumme 5380
- 20%
+ 20%CH
4
NO
Xexp
/ %
Xber/%
0
20
40
60
80
100
0 20 40 60 80 100
500°C Wasser47 VersucheFehlerquadratsumme 3663
- 20%
+ 20%
CH4
NO
Xexp
/ %
Xbe
r/%
0
20
40
60
80
100
0 20 40 60 80 100
450°C Wasser42 VersucheFehlerquadratsumme 1972
- 20%
+ 20%
CH4
NO
Xexp
/ %
Xbe
r/%
Abbildung 4.58: Paritatsdiagramme f¨ur die einzelnen Temperaturen amCeO2-In-ZSM-5-System mit Wasser im Eduktgasstrom
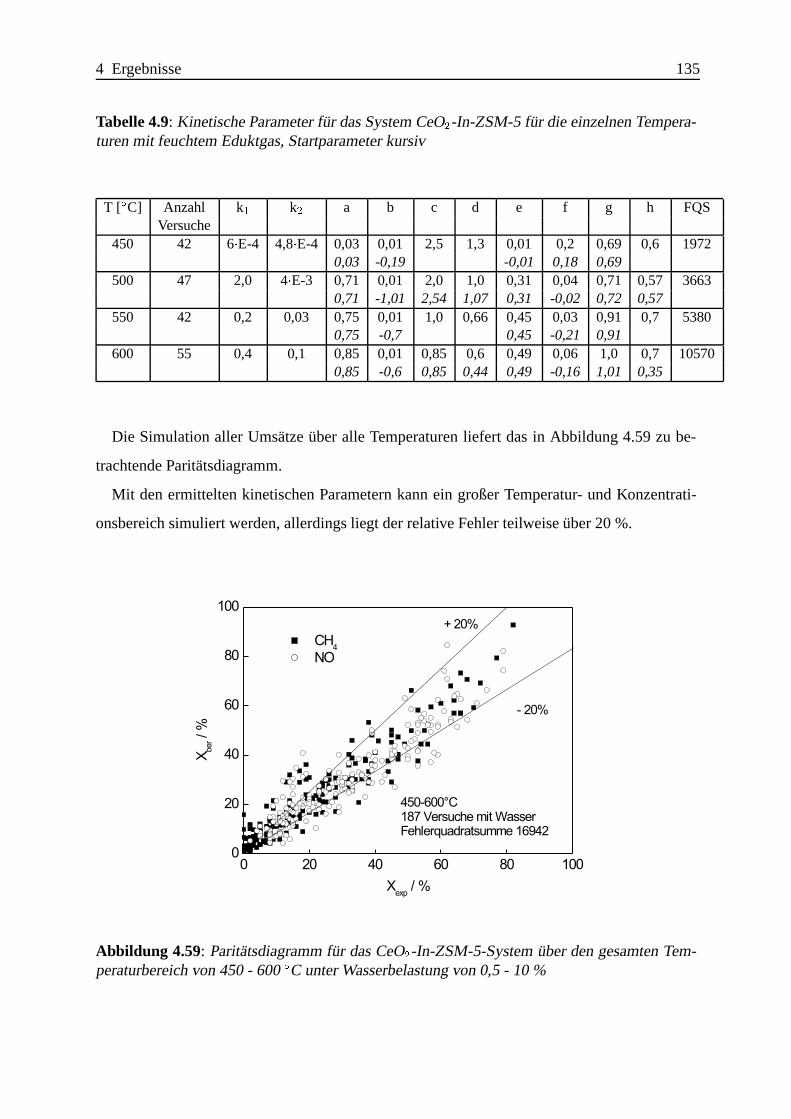
4 Ergebnisse 135
Tabelle 4.9: Kinetische Parameter f¨ur das System CeO2-In-ZSM-5 fur die einzelnen Tempera-turen mit feuchtem Eduktgas, Startparameter kursiv
T [ÆC] Anzahl k1 k2 a b c d e f g h FQSVersuche
450 42 6�E-4 4,8�E-4 0,03 0,01 2,5 1,3 0,01 0,2 0,69 0,6 19720,03 -0,19 -0,01 0,18 0,69
500 47 2,0 4�E-3 0,71 0,01 2,0 1,0 0,31 0,04 0,71 0,57 36630,71 -1,01 2,54 1,07 0,31 -0,02 0,72 0,57
550 42 0,2 0,03 0,75 0,01 1,0 0,66 0,45 0,03 0,91 0,7 53800,75 -0,7 0,45 -0,21 0,91
600 55 0,4 0,1 0,85 0,01 0,85 0,6 0,49 0,06 1,0 0,7 105700,85 -0,6 0,85 0,44 0,49 -0,16 1,01 0,35
Die Simulation aller Ums¨atzeuber alle Temperaturen liefert das in Abbildung 4.59 zu be-
trachtende Parit¨atsdiagramm.
Mit den ermittelten kinetischen Parametern kann ein großer Temperatur- und Konzentrati-
onsbereich simuliert werden, allerdings liegt der relative Fehler teilweise ¨uber 20 %.
0
20
40
60
80
100
0 20 40 60 80 100
450-600°C187 Versuche mit WasserFehlerquadratsumme 16942
- 20%
+ 20%
CH4
NO
Xexp
/ %
Xber/%
Abbildung 4.59: Paritatsdiagramm f¨ur das CeO2-In-ZSM-5-System ¨uber den gesamten Tem-peraturbereich von 450 - 600ÆC unter Wasserbelastung von 0,5 - 10 %
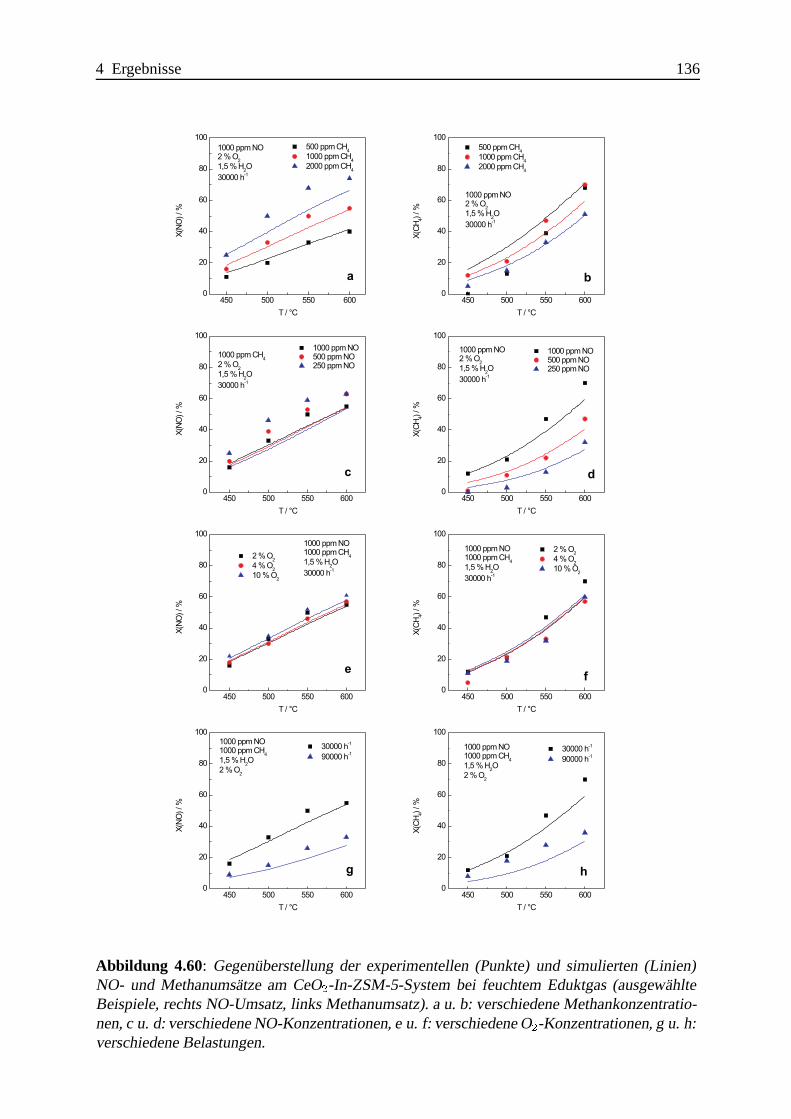
4 Ergebnisse 136
450 500 550 6000
20
40
60
80
100
a
1000 ppm NO2 % O
2
1,5 % H2O
30000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
b
1000 ppm NO2 % O
2
1,5 % H2O
30000 h-1
500 ppm CH4
1000 ppm CH4
2000 ppm CH4
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
c
1000 ppm CH4
2 % O2
1,5 % H2O
30000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
d
1000 ppm NO2 % O
2
1,5 % H2O
30000 h-1
1000 ppm NO500 ppm NO250 ppm NO
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
e
1000 ppm NO1000 ppm CH
4
1,5 % H2O
30000 h-1
2 % O2
4 % O2
10 % O2
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
f
1000 ppm NO1000 ppm CH
4
1,5 % H2O
30000 h-1
2 % O2
4 % O2
10 % O2
X(C
H4)
/%
T / °C
450 500 550 6000
20
40
60
80
100
g
1000 ppm NO1000 ppm CH
4
1,5 % H2O
2 % O2
30000 h-1
90000 h-1
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
h
1000 ppm NO1000 ppm CH
4
1,5 % H2O
2 % O2
30000 h-1
90000 h-1
X(C
H4)
/%
T / °C
Abbildung 4.60: Gegen¨uberstellung der experimentellen (Punkte) und simulierten (Linien)NO- und Methanums¨atze am CeO2-In-ZSM-5-System bei feuchtem Eduktgas (ausgew¨ahlteBeispiele, rechts NO-Umsatz, links Methanumsatz). a u. b: verschiedene Methankonzentratio-nen, c u. d: verschiedene NO-Konzentrationen, e u. f: verschiedene O2-Konzentrationen, g u. h:verschiedene Belastungen.
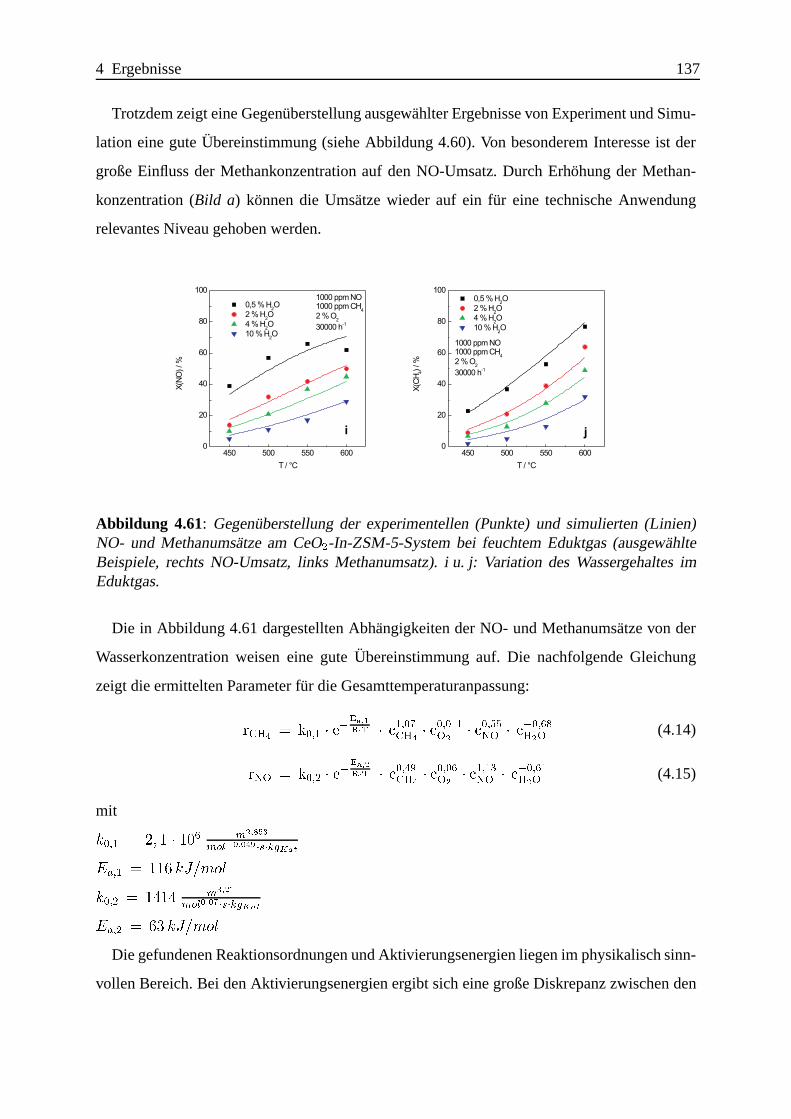
4 Ergebnisse 137
Trotzdem zeigt eine Gegen¨uberstellung ausgew¨ahlter Ergebnisse von Experiment und Simu-
lation eine guteUbereinstimmung (siehe Abbildung 4.60). Von besonderem Interesse ist der
große Einfluss der Methankonzentration auf den NO-Umsatz. Durch Erh¨ohung der Methan-
konzentration (Bild a) konnen die Ums¨atze wieder auf ein f¨ur eine technische Anwendung
relevantes Niveau gehoben werden.
450 500 550 6000
20
40
60
80
100
i
1000 ppm NO1000 ppm CH
4
2 % O2
30000 h-1
0,5 % H2O
2 % H2O
4 % H2O
10 % H2O
X(N
O)/%
T / °C
450 500 550 6000
20
40
60
80
100
j
1000 ppm NO1000 ppm CH
4
2 % O2
30000 h-1
0,5 % H2O
2 % H2O
4 % H2O
10 % H2O
X(C
H4)
/%
T / °C
Abbildung 4.61: Gegen¨uberstellung der experimentellen (Punkte) und simulierten (Linien)NO- und Methanums¨atze am CeO2-In-ZSM-5-System bei feuchtem Eduktgas (ausgew¨ahlteBeispiele, rechts NO-Umsatz, links Methanumsatz). i u. j: Variation des Wassergehaltes imEduktgas.
Die in Abbildung 4.61 dargestellten Abh¨angigkeiten der NO- und Methanums¨atze von der
Wasserkonzentration weisen eine guteUbereinstimmung auf. Die nachfolgende Gleichung
zeigt die ermittelten Parameter f¨ur die Gesamttemperaturanpassung:
rCH4 = k0;1 � e�Ea;1R�T � c1;07CH4
� c0;011O2� c0;55NO � c�0;68H2O
(4.14)
rNO = k0;2 � e�Ea;2R�T � c0;49CH4
� c0;06O2� c1;13NO � c�0;61H2O
(4.15)
mit
k0;1 = 2; 1 � 106 m2;853
mol�0;049�s�kgKat
Ea;1 = 116 kJ=mol
k0;2 = 1414 m3;21
mol0;07�s�kgKat
Ea;2 = 63 kJ=mol
Die gefundenen Reaktionsordnungen und Aktivierungsenergien liegen im physikalisch sinn-
vollen Bereich. Bei den Aktivierungsenergien ergibt sich eine große Diskrepanz zwischen den

4 Ergebnisse 138
Tabelle 4.10: Sensitivitatsanalyse der kinetischen Parameter f¨ur das CeO2-In-ZSM-5-Systemunter Wasserbelastung
Parameter k0;1 k0;2 Ea;1 Ea;2 a b c d e f g h
+ 5 % 1 % 0,6 % 51 % 114 % 6 % 0 % 1,7 % 2 % 10,7 % 2,7 % 57,3 % 0,7 %- 5 % 0,3 % 2 % 149 % 151 % 17 % 0 % 5,1 % 8,9 % 2,5 % 67,4 % 5,7 %
anfangs gesch¨atzten und letztlich optimierten Werten. Dabei zeigen gerade diese Parameter
in der Sensitivitatsanalyse in Tabelle 4.10 eine besonders große Anf¨alligkeit gegen¨uber einer
Parameter¨anderung. Allerdings ist eine Beurteilung der Sensitivit¨atsanalyse mit Vorsicht zu
genießen, da z.B. gerade die Aktivierungsenergien mit den pr¨aexponentiellen Faktoren kor-
relieren. Durch eine Ver¨anderung der pr¨aexponentiellen Faktoren kann eine Ver¨anderung der
Aktivierungsenergien kompensiert werden.
4.2.4 Zusammenfassung und Vergleich der Ergebnisse
Das katalytische Verhalten der drei Systeme CeO2-ZSM-5, In-ZSM-5 und CeO2-In-ZSM-5
lasst sich mit Hilfe eines einfachen Potenzansatzes ¨uber weite Messbereiche simulieren. Hier-
bei ist zur Auffindung guter Startparameter eine differentielle graphische Auswertung der in-
tegralen Messungen von großem Nutzen. Jedoch kann die graphische Auswertung eine Simu-
lation nicht ersetzen, da sich einige gesch¨atzte Parameter nach einer Optimierung deutlich von
den Startparametern unterscheiden. Daher ist der vielfach in der Literatur zur SCR-Reaktion
mit Methan beschrittene Weg der graphischen Auswertung mit Fehlern behaftet, solange diese
differentiell mit integralen Messungen erfolgt. Die Gr¨oße der Abweichung ist hierbei sicherlich
abhangig von der Diskrepanz zwischen den ausgewerteten integralen und den zur Auswertung
benotigten differentiellen Ums¨atzen.
Als besonders schwierig stellt sich die Absch¨atzung von Aktivierungsenergien heraus. In Ta-
belle 4.11 sind die gesch¨atzten und optimierten Werte gegen¨ubergestellt. Die Tendenz zwischen
den Aktivierungsenergien der einzelnen Systeme findet sich sowohl in den abgesch¨atzten als
auch in den optimierten Werten wieder, aber die absoluten Zahlen unterscheiden sich deutlich.
Einzelne Werte der Aktivierungsenergien werden im Folgenden zusammen mit ausgew¨ahlten
Reaktionsordnungen diskutiert.
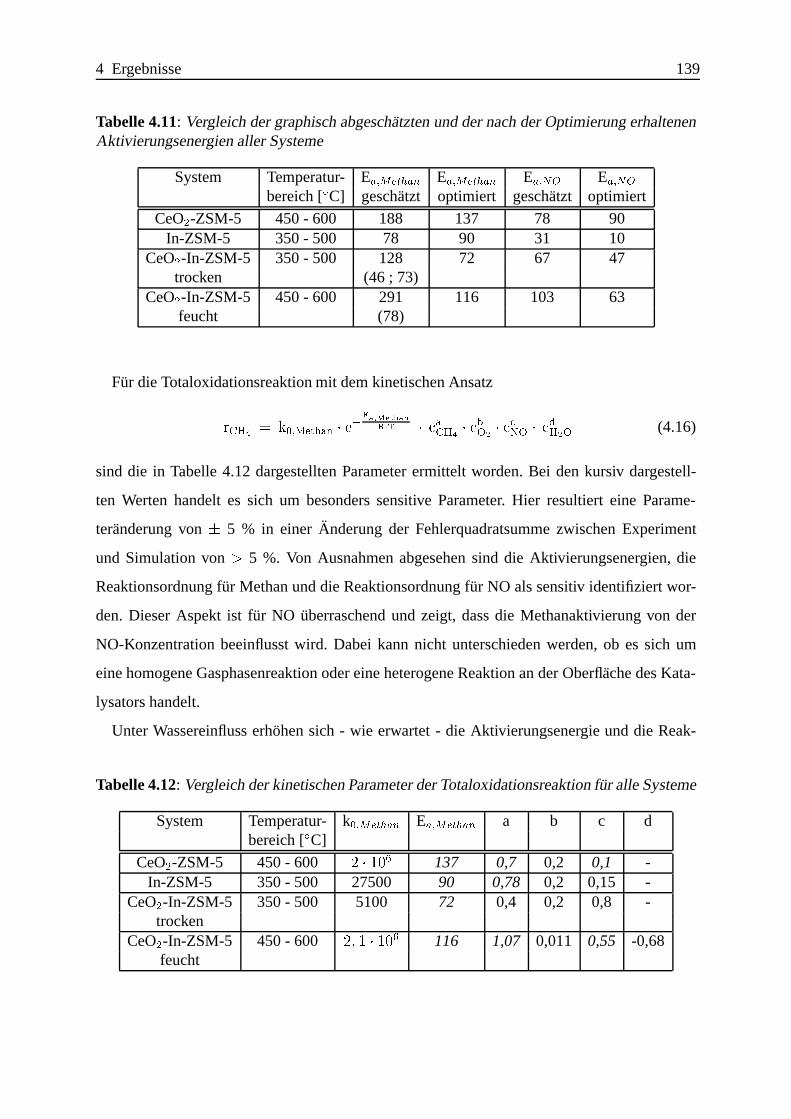
4 Ergebnisse 139
Tabelle 4.11: Vergleich der graphisch abgesch¨atzten und der nach der Optimierung erhaltenenAktivierungsenergien aller Systeme
System Temperatur- Ea;Methan Ea;Methan Ea;NO Ea;NO
bereich [ÆC] geschatzt optimiert geschatzt optimiert
CeO2-ZSM-5 450 - 600 188 137 78 90In-ZSM-5 350 - 500 78 90 31 10
CeO2-In-ZSM-5 350 - 500 128 72 67 47trocken (46 ; 73)
CeO2-In-ZSM-5 450 - 600 291 116 103 63feucht (78)
Fur die Totaloxidationsreaktion mit dem kinetischen Ansatz
rCH4 = k0;Methan � e�Ea;Methan
R�T � caCH4 � cbO2� ccNO � cdH2O (4.16)
sind die in Tabelle 4.12 dargestellten Parameter ermittelt worden. Bei den kursiv dargestell-
ten Werten handelt es sich um besonders sensitive Parameter. Hier resultiert eine Parame-
teranderung von� 5 % in einerAnderung der Fehlerquadratsumme zwischen Experiment
und Simulation von> 5 %. Von Ausnahmen abgesehen sind die Aktivierungsenergien, die
Reaktionsordnung f¨ur Methan und die Reaktionsordnung f¨ur NO als sensitiv identifiziert wor-
den. Dieser Aspekt ist f¨ur NO uberraschend und zeigt, dass die Methanaktivierung von der
NO-Konzentration beeinflusst wird. Dabei kann nicht unterschieden werden, ob es sich um
eine homogene Gasphasenreaktion oder eine heterogene Reaktion an der Oberfl¨ache des Kata-
lysators handelt.
Unter Wassereinfluss erh¨ohen sich - wie erwartet - die Aktivierungsenergie und die Reak-
Tabelle 4.12: Vergleich der kinetischen Parameter der Totaloxidationsreaktion f¨ur alle Systeme
System Temperatur- k0;Methan Ea;Methan a b c dbereich [ÆC]
CeO2-ZSM-5 450 - 600 2 � 106 137 0,7 0,2 0,1 -In-ZSM-5 350 - 500 27500 90 0,78 0,2 0,15 -
CeO2-In-ZSM-5 350 - 500 5100 72 0,4 0,2 0,8 -trocken
CeO2-In-ZSM-5 450 - 600 2; 1 � 106 116 1,07 0,011 0,55 -0,68feucht

4 Ergebnisse 140
tionsordnung f¨ur Methan. Demgegen¨uber sinkt die Reaktionsordnung f¨ur NO. Allerdings darf
diesem Befund f¨ur die beiden Reaktionsordnungen nicht allzuviel Gewichtung geschenkt wer-
den, da der Vergleich teilweise mit relativ insensitiven Parametern durchgef¨uhrt wird.
Die fur die SCR-Reaktion nach dem Potenzansatz
rNO = k0;NO � e�Ea;NOR�T � ceCH4 � cfO2
� cgNO � chH2O (4.17)
optimierten Parameter werden in Tabelle 4.13 vorgestellt. Bei den kursiv dargestellten Werten
handelt es sich analog zur Totaloxidationsreaktion um die Parameter, die durch eine 5 %ige Pa-
rameteranderung eineAnderung der Fehlerquadratsumme von> 5 % bewirken. Hier erweisen
sich die Aktivierungsenergien, die Reaktionsordnung von NO und sowie die Reaktionsordnung
von Wasser als besonders sensitiv. Es zeigt sich, dass die Zugabe von Wasser in den Edukt-
gasstrom bei dem Katalysatorsystem CeO2-In-ZSM-5 eine Erh¨ohung der Aktivierungsenergie
von 47 auf 63 kJ/mol sowie einen Anstieg der Reaktionsordnung f¨ur Methan von 0,2 auf 0,49
bzw. der Reaktionsordnung f¨ur NO von 0,65 auf 1,13 bewirkt. Dies deutet auf eine Konkur-
renzadsorption des Wassers sowohl bez¨uglich des NO als auch bez¨uglich des Methans hin.
Die NO-Oxidation wird von Wasser nicht beeinflusst [14] und kann daher keineAnderung der
Reaktionsordnung bewirken. Die durchgehend niedrige Reaktionsordnung f¨ur Sauerstoff zeigt,
dass bei allen Systemen eine Erh¨ohung der Sauerstoffkonzentration nur einen geringen Einfluss
auf die Aktivitat bewirkt.
Die Aktivierungsenergien aller Systeme liegen im physikalisch sinnvollen Bereich; lediglich
die sehr niedrige Aktivierungsenergie beim In-ZSM-5 f¨allt aus dem Rahmen. Sie kann dahinge-
hend interpretiert werden, dass im System In-ZSM-5 die NO-Oxidation (diese selber zeigt eine
Tabelle 4.13: Vergleich der kinetischen Parameter der SCR-Reaktion f¨ur alle Systeme
System Temperatur- k0;NO Ea;NO e f g hbereich [ÆC]
CeO2-ZSM-5 450 - 600 7500 90 0,315 0,2 0,8 -In-ZSM-5 350 - 500 0,0475 10 0,05 0,3 0,8 -
CeO2-In-ZSM-5 350 - 500 70 47 0,2 0,1 0,65 -trocken
CeO2-In-ZSM-5 450 - 600 1414 63 0,49 0,06 1,13 - 0,61feucht

4 Ergebnisse 141
scheinbare negative Aktivierungsenergie, siehe Seite 120) m¨oglicherweise den geschwindig-
keitsbestimmenden Schritt darstellt. Dies stimmt ¨uberein mit der Tatsache, dass dieses System
die hochste Abh¨angigkeit von der Sauerstoffkonzentration aufweist.
Im folgenden Abschnitt werden ausgew¨ahlte Ergebnisse mit den aus der Literatur bekann-
ten Werten verglichen. Eine erste Gegen¨uberstellung ist in Tabelle 4.14 gegeben. Hierbei be-
schrankt sich die Gegen¨uberstellung auf die durch den Potenzansatz
rNO = k0;NO � ceCH4 � cfO2� cgNO (4.18)
fur die SCR-Reaktion erhaltenen Werte, da die Modelle in der Literatur gr¨oßtenteils unvoll-
standig ausgewertet wurden und die Totaloxidationsreaktion vernachl¨assigt wurde. Zum Ver-
gleich der Literaturwerte mit den in dieser Arbeit ermittelten Parametern dienen die Einzeltem-
peraturanpassungen, wobei die Temperaturen verglichen werden, die m¨oglichst nahe bei den in
der Literatur vermessenen Temperaturen liegen. Wie der Tabelle entnommen werden kann, han-
delt es sich um die zentrale Temperatur von 400ÆC. Der Vergleich der in dieser Arbeit ermit-
telten NO-Ordnung f¨ur In-ZSM-5 liefert eine guteUbereinstimmung mit den Literaturwerten.
Die geringere NO-Ordnung von CeO2-ZSM-5 und CeO2-In-ZSM-5 zeigt eine Verschiebung
des geschwindigkeitsbestimmenden Schritts im Reaktionsmechanismus, die schon in ¨ahnlicher
Weise bei Ir/In-ZSM-5 beobachtet werden konnte. Bei der Promotierung von In-ZSM-5 mit
Iridium fandenKikuchi et al. die oben in der Tabelle angegebene Erniedrigung der Ordnung
fur NO. Sie geben zwei Erkl¨arungen: Zum einen wird NO an Iridium zu NO2 oxidiert, zum
anderen wird gleichzeitig die NO2-Adsorptionskapazit¨at des In-ZSM-5 drastisch erh¨oht [51].
Der Zusammenhang einer hohen NO-Adsorptionskapazit¨at mit einer niedrigen
Tabelle 4.14: Vergleich ausgew¨ahlter kinetischen Parameter der SCR-Reaktion f¨ur die in dieserArbeit untersuchten Systeme mit Systemen aus der Literatur (nur trockene Eduktgasstr¨ome)
System T [ÆC] e f g Literatur
Pd/H-ZSM-5 400 0,66 -0,26 1,05 [67]Co-ZSM-5 350 0,68 0,18 0,11 [56]
Ir/In-ZSM-5 400 -0,18 0,13 0,65 [51]In-ZSM-5 400 0 0,8 0,8 [51]In-ZSM-5 400 0,05 0,25 0,79 -
CeO2-ZSM-5 450 0,303 0,09 (500ÆC) 0,56 -CeO2-In-ZSM-5 400 0,30 0,06 (500ÆC) 0,6 -

4 Ergebnisse 142
NO-Reaktionsordnung wurde vorher f¨ur das System Co-ZSM-5 vonDumpelmannet al.
postuliert und nachgewiesen [56]. Sie schlossen daraus, dass der geschwindigkeitsbestimmen-
de Schritt der Reaktion die Aktivierung des Methans an einer durch Adsorption von NO2 und
weiteren Reaktionsschritten entstandenen Oberfl¨achenspezies ist [56].
In Ubereinstimmung hiermit fordernKato et al. fur das Pd/H-ZSM-5-System auf Grund der
sehr hohen NO-Reaktionsordnung einen anderen Reaktionsmechanismus, bei dem das Methan
nicht an adsorbiertem NO2 aktiviert wird. Hierbei legen sie sich nicht auf einen geschwindig-
keitsbestimmenden Schritt fest, sondern behaupten lediglich, dass dieser sich ¨uber die Tempe-
ratur hinwegandern kann [67].
Bezogen auf die in dieser Arbeit ermittelten Reaktionsordnungen kann folgendes postuliert
werden: Durch die Zugabe von CeO2 zum In-ZSM-5 wird der geschwindigkeitsbestimmen-
de Schritt von der NO-Oxidation verschoben. Das Ausmaß der Verschiebung, erkennbar an
der Anderung in der Reaktionsordnung, ist analog zu dem Ausmaß der Verschiebung beim
Ubergang vom In-ZSM-5 zum Ir/In-ZSM-5, aber weniger deutlich als beim Vergleich von
Co-ZSM-5 und Pd/H-ZSM-5. Gleichzeitig kann beiin situDRIFTS-Messungen keine Ver¨ande-
rung der Spektren beim Umschalten vom reinen Heliumstrom auf eine Gasmischung von
1000 ppm NO und 2 % Sauerstoff gefunden werden; daher muß hier eine Adsorption von NO
oder NO2, wie fur das Ir/In-ZSM-5-System vorgeschlagen, zumindest an der obersten Schicht
des Katalysatorbettes ausgeschlossen werden. Auch der Zusatz von Methan zu der letztgenann-
ten Gasmischung liefert keine ver¨anderten Spektren. Allerdings unterscheidet sich die verwen-
dete Apparatur f¨ur die in situ DRIFTS-Messungen, die genau in der Dissertation vonHein-
rich beschrieben ist [130], von der vonKikuchi et al. verwendeten Apparatur [55]. Im letzten
Fall wird der Messzelle ein katalytischer Reaktor vorgeschaltet, so daß in der Messzelle selber
nicht die oberste Katalysatorschicht, sondern eine gewissermaßen tiefere Katalysatorschicht
vermessen wird. Da in der von uns verwendeten Anordnung der Vorreaktor fehlt, kann nicht
ausgeschlossen werden, dass uns Produkte entgangen sind, die erst in tieferen Schichten des
Katalysatorbettes gebildet werden. Daher sind die durchgef¨uhrtenin situ DRIFTS-Messungen
kein Beweis daf¨ur, dass kein NO2 oder eine sonstige Spezies adsorbiert wird, da dies auch erst
an tieferen, f¨ur die Analyse unzug¨anglichen Katalysatorschichten erfolgen kann. Hier k¨onnten
Untersuchungen zur NO/NO2-Adsorptionskapazit¨at der einzelnen Katalysatoren weitere Ein-
blicke geben, die in dieser Arbeit aus Zeitgr¨unden nicht mehr durchgef¨uhrt wurden.

5 Schlussfolgerungen und Ausblick
In dieser Arbeit konnte gezeigt werden, dass unterschiedliche Pr¨aparationsmethoden des
In-ZSM-5-Systems in verschiedenen Strukturen der Indiumkomponente und gleichzeitig un-
terschiedlichen Aktivit¨aten in der SCR-Reaktion resultieren.
Intrazeolithische Indiumoxospezies wurden als aktive Zentren identifiziert. Die Aktivit¨at
zeigt eine starke Abh¨angigkeit von der Koordinationsph¨are. Ist in dieser Chlor enthalten, so
kommt es lediglich zu einer unselektiven Methanoxidation, die SCR-Reaktion ist unterdr¨uckt.
Bei Versuchen mit mechanischen Mischungen kann unter geeigneten Aktivierungsbedin-
gungen eine bisher unbekannte aktive Indiumspezies generiert werden. Sie ist an der ¨außeren
Oberflache des Zeolithen lokalisiert und liegt in sehr geringer Menge vor. Vermutlich erfolgt
eine Fixierung an der Oberfl¨acheuber kovalente In-O-Al- und/oder In-O-Si-Bindungen.
CeO2-promotierte In-ZSM-5-Katalysatoren sind hochaktiv in der SCR-Reaktion mit Me-
than. Selbst unter Wassereinfluss wird eine hohe Aktivit¨at erzielt, der Maximalumsatz an NO
verschiebt sich jedoch zu h¨oheren Temperaturen.
Die einfachste Art der Promotierung des In-ZSM-5-Grundsystems wird erreicht durch eine
mechanische Beimengung eines CeO2 mit einer hohen Oberfl¨ache. Aber auch durch F¨allung
von CeO2 wird die Aktivitat des Grundsystems erheblich gesteigert, wobei die Reihenfolge der
Cer- bzw. Indiumzugabe nur einen minimalen Einfluss auf die Aktivit¨at zeigt.
Auch Katalysatoren mit InOxCly-Spezies (durch Festk¨orperionentausch erhalten), die eine
geringe Aktivitat zeigen, lassen sich mit CeO2 wirkungsvoll promotieren. Somit liefert das
aktivste In-ZSM-5-System nicht den aktivsten CeO2-In-ZSM-5-Katalysator.
Am CeO2 wird NO zu NO2 oxidiert, das in weiteren Reaktionsschritten zu N2 reduziert wird.
Eine zweite Aufgabe des CeO2 wird bei der Praparation mechanischer Mischungen ver-
mutet. Ist wahrend der Pr¨aparation Ce3+ entstanden, so kann dieses als Reduktionsmittel f¨ur
einen reduktiven Festk¨orperionentausch der Indiumkomponente auf Kationenpl¨atze des Zeo-
lithen wirken. Als Reduktionsmittel fungiert ansonsten NH3, das wahrend der Kalzination in
Helium aus dem NH4-ZSM-5 entweicht.
Die Protonen des Zeolithen leisten vermutlich einen wichtigen Beitrag zur SCR-Reaktion.
So wird an einem Silikaliten, der keine Protonen enth¨alt, kein NO-Umsatz erzielt. Bei Ver-

5 Schlussfolgerungen und Ausblick 144
suchen zur Defunktionalisierung der Protonen im H-ZSM-5 durch Natriumionenr¨ucktausch
oder Festk¨orperionentausch mit KBr bricht der NO-Umsatz ebenfalls ein; allerdings kann ein
Einfluss der beiden Behandlungsmethoden auf die Indiumkomponente nicht ausgeschlossen
werden.
Bei den kinetischen Messungen zeigt sich eine irreversible Deaktivierung des
CeO2-In-ZSM-5-Katalysators unter extremen Bedingungen (600ÆC, 10 % Wasser).
Die ermittelten Reaktionsordnungen f¨ur die einzelnen Katalysatorsysteme sind konform zu
aus der Literatur bekannten Daten. Sie zeigen die NO-Oxidation als ersten Schritt des Reak-
tionsmechanismus. Durch Wasserzugabe kommt es zu einer kompetitiven Adsorption an den
aktiven Zentren.
Da unter extremen Bedingungen (600ÆC, 10 % Wasser) f¨ur eine technische Anwendung
mit Methan als Reduktionsmittel keine ausreichend hohe Stabilit¨at zu erzielen ist, m¨ussen an-
dere Bedingungen ¨uberpruft werden. Hier bieten sich die Wahl eines anderen Reduktionsmit-
tels und/oder tiefere Temperaturen unter unver¨anderter Wasserdampfbelastung an. Am ACA in
Berlin wurde fur mildere Bedingungen (500ÆC, 5 % Wasser) eine geringere Deaktivierungs-
tendenz bei Verwendung von Methan als Reduktionsmittel gefunden; mit Propan wurde eine
weitere Verbesserung erzielt, die f¨ur eine technische Anwendung Potential bietet.
Bei Verwendung von anderen Reduktionsmitteln sollte zus¨atzlich eine Variation des Tr¨agers
moglich sein.McCreadyet al. fanden f¨ur In2O3 auf -Al 2O3 eine hohe katalytische Akti-
vitat mit Propen (1000 ppm NO, 1000 ppm Propen, 9 % O2, 7 % H2O, 30000 h�1, 450ÆC,
60 % NO-Umsatz) [131]. Damit kann die hydrothermale Instabilit¨at des Zeolithen umgangen
werden.
Von Interesse ist zus¨atzlich der Einfluss von SO2 auf die Aktivitat des Katalysators, da diese
Komponente in den meisten Abgasen enthalten ist.
Bei der Modellierung der Ums¨atze konnen m¨oglicherweise verbesserte Anpassungen durch
Einsatz ver¨anderter kinetischer Ans¨atze erzielt werden.
Durch den Einsatz von Isotopen wie beispielsweise CD4 wahrend der kinetischen Messun-
gen zeigen sich eventuell kinetische Isotopeneffekte, die R¨uckschlusse auf geschwindigkeits-
bestimmende Schritte zulassen.
Weitere Moglichkeiten zur Best¨atigung von Reaktionsmechanismen an den untersuchten Ka-
talysatorsystemen liegen in TPD-Experimenten speziell mit NO bzw. NO2.

6 Literaturverzeichnis
[1] ”Technische Anleitung zur Reinhaltung der Luft,Heider Verlag, Bergisch-Gladbach”,
(1986).
[2] ”17. Verordnung zur Durchf¨uhrung des Bundes-Immissionsschutzgesetzes”, (1990).
[3] Grunert, W., Papp, H., Rottl¨ander, Chr., Baerns, M.,Chem. Technik47 (1995) 205.
[4] Konig, A., Herding, G., Hupfeld, B., Richter, Th., Weidmann, K.,Topics in Catalysis
16/17Nos.1-4 (2001) 23.
[5] Greening, P.,Topics in Catalysis16/17Nos.1-4 (2001) 5.
[6] Bertelsen, B.I.,Topics in Catalysis16/17Nos.1-4 (2001) 15.
[7] Kleifges, K.H.,Fortschrittsberichte VDIReihe 5, Nr. 153.
[8] Fritz, A., Pitchon, V.,Applied Catalysis B13 (1997) 1.
[9] Iwamoto, M., Hamada, H.,Catal. Today10 (1991) 57.
[10] Iwamoto, M., Yahiro, H.,Catal. Today22 (1994) 5.
[11] Held, W., Konig, A., Richter, T., Puppe, L.,SAE-Paper 900496(1990).
[12] Iwamoto, M., Yahiro, H., Shundo, S., Yu, Y., Mizuno, N.,Shokubai32 (1990) 430.
[13] Li, Y., Armor, J.N.,Appl. Catal. B1 (1992) 31.
[14] Liese, T., Dissertation, Bochum (1999).
[15] Schmidt, C., Dissertation, Bochum (2001).
[16] Weller, M.T.,Anorganische Materialien, VCH, Weinheim (1996).
[17] Shriver, D.F., Atkins, P.W., Langford, C.H.,Inorganic Chemistry, Oxford University
Press (1994).
[18] Puppe, L., in:Chemie in unserer Zeit20 Jahrg. Nr.4 (VCH Verlagsgesellschaft 1986)
117.
[19] Traa, Y., Burger, B., Weitkamp, J.,Microporous and Mesoporous Materials30 Nr.1
(1999) 3.
[20] Shelef, M.,Chem. Rev.95 (1995) 209.
[21] Amiridis, M.D., Zhang, T., Farrauto, R.J.,Applied Catalysis B10 (1996) 203.
[22] Iwamoto, M.,Studies in Surface Science and Catalysis130, ed. Corma, A., Melo, F.V.,
Mendioroz, S., Fierro, J.L.G., Elsevier, Amsterdam (2000) 23.

6 Literaturverzeichnis 146
[23] Kim, M.H., Nam, I.-S.,Korean J. Chem. Eng.18(5) (2001) 725.
[24] Armor, J.N.,Catalysis Today26 (1995) 147.
[25] Li, Y., Armor, J.N.,Appl. Catal. B2 (1993) 239.
[26] Li, Y., Armor, J.N.,J. Catal.145(1994) 1.
[27] Kikuchi, E., Yogo, K.,Catalysis Today22 (1994) 73.
[28] Nishizaka, Y., Misono, M.,Chem. Lett.(1993) 1295.
[29] Yokoyama, C., Misono, M.,Chem. Lett.(1992) 1669.
[30] Yokoyama, C., Misono, M.,Bull. Chem. Soc. Jpn.67 (1994) 557.
[31] Yokoyama, C., Misono, M.,Catalysis Today22 (1994) 59.
[32] Yokoyama, C., Misono, M.,J. Catal.160(1996) 95.
[33] Yokoyama, C., Misono, M.,J. Catal.150(1994) 9.
[34] Miyamoto, A., Himei, H., Oka, Y., Maruya, E., Katagiri, M., Vetrivel, R., Kubo, M.,
Catalysis Today22 (1994) 87.
[35] Yokoyama, C., Misono, M.,Catal. Lett.29 (1994) 1.
[36] Misono, M., Hirao, Y., Yokoyama, C.,Catalysis Today38 (1997)157.
[37] Misono, M., Niiro, H., Hirao, Y.,Res. Chem. Intermed.24 (1998)123.
[38] Li, Z., Flytzani-Stephanopoulos, M.,Appl. Catal. A165(1997) 15.
[39] Liese, T., Diplomarbeit, Bochum (1995).
[40] Rutenbeck, D., Diplomarbeit, Bochum (1996).
[41] Yogo, K., Tanaka, S., Ihara, M., Hishiki, T, Kikuchi, E.,Chem. Lett.(1992) 1025.
[42] Yogo, Kl, Kikuchi, E.,Stud. Surf. Sci. Catal.84 (1994) 1547.
[43] Tabata, T., Kokitsu, M., Okada, O.,Catal. Lett.25 (1994) 393.
[44] Tabata, T., Kokitsu, M., Okada, O.,Appl. Catal. B6 (1995) 225.
[45] Kikuchi, E., Ogura, M., Terasaki, I., Goto, Y.,J. Catal.161(1996) 465.
[46] Ogura, M., Aratani, N., Kikuchi, E., in:Progress in Zeolite and Microporous Materials
105, ed. Chon, H., Ihm, S.-K. and Uh, Y.S. (Studies in Surface Science and Catalysis,
Elsevier, Amsterdam 1997) 1593.
[47] Ogura, M., Ohsaki, T., Kikuchi, E.,Microporous Mater.21 (1998) 533.
[48] Zhou, X., Zhang, T., Xu, Z., Lin, L.,Catal. Lett.40 (1996) 35.
[49] Zhou, X., Xu, Z., Zhang, T., Lin, L.,J. Mol. Catal. A: Chemical122(1997) 125.
[50] Richter, M., Berndt, H., Fricke, R., L¨ucke, B.,DGMK-Ber.9601283.

6 Literaturverzeichnis 147
[51] Ogura, M., Kikuchi, E.,Studies in Surface Science and Catalysis101, ed. Hightower,
J.W., Delgass, W.N., Iglesia, E., Bell, A.T., Elsevier, Amsterdam (1996) 671.
[52] Kikuchi, E., Ogura, M., Aratani, N., Sugiura, Y., Hiramoto, S., Yogo, K.,Catalysis Today
27 (1996)35.
[53] Ogura, M., Hiromoto, S., Kikuchi, E.,Chem. Lett.(1995)1135.
[54] Ogura, M., Kikuchi, E.,Chem. Lett.(1996)1017.
[55] Ogura, M., Hayashi, M., Kikuchi, E.,Catal. Today42 (1998) 159.
[56] Cowan, A.D., Dumpelmann, R., Cant, N.W.,J. Catal.151(1995) 356.
[57] Li, Y., Slager, T.L., Armor, J.,J. Catal.150(1994) 388.
[58] Lombardo, E.A., Sill, G.A., D’Itri, J.L., Hall, W.K.,J. Catal.173(1998) 440.
[59] Aylor, A.W., Lobree, L.J., Reimer, J.A., Bell, A.T.,Studies in Surface Science and Cata-
lysis101, ed. Hightower, J.W., Delgass, W.N., Iglesia, E., Bell, A.T., Elsevier, Amster-
dam (1996) 661.
[60] Hayes, N.W., Gr¨unert, W., Hutchings, G.J., Joyner, R.W., Shpiro, E.S.,J. Chem. Soc.
Chem. Comm.(1994) 531.
[61] Beutel, T., Adelmann, B., Sachtler, W.H.M.,Catal. Lett.37 (1996) 125.
[62] Yahiro, H., Yuu, Y., Takeda, H., Mizuno, N., Iwamoto, M.,Shokubai35 (1993) 130.
[63] Ukisu, Y., Sato, S., Muramatsu, G., Yoshida, K.,Catal. Lett.11 (1991) 177.
[64] Shimizu, K., Okada, F., Nakamura, Y., Satsuma, A., Hattori, T.,J. Catal.195(2000) 151.
[65] Cant, N.W., Liu, I.,Catalysis Today63 (2000)133.
[66] Li, Y., Battavio, P.J., Armor, J.,J. Catal.142(1993) 561.
[67] Kato, H., Yokoyama, C., Misono, M.,Catalysis Today42 (1998)93.
[68] Campa, M., Pietrogiacomi, D., Tuti, S., Ferraris, G., Indovina, V.,Appl. Catal. B18
(1998) 151.
[69] Heinisch, R., Jahn, M., Langohe, J., Pohl, M.,Chem. Eng. Technol.24 (2001)381.
[70] Li, Y., Armor, J.,J. Catal.150(1994) 376.
[71] Baerns, M., Hofmann, H., Renken, A.,Chemische ReaktionstechnikLehrbuch der Tech-
nischen Chemie, Band 1, 2.Auflage, Georg Thieme Verlag, Stuttgart (1992).
[72] Iwamoto, M., Furukawa, H., Mine, Y., Uemura, F., Mikuriga, S., Kagawa, S.,J. Chem.
Soc., Chem. Commun.(1986) 1272.
[73] Brunauer, S., Emmett, P.H., Teller, E.,J. Am. Chem. Soc.60 (1938) 309.

6 Literaturverzeichnis 148
[74] Langmuir, I.,J. Am. Chem. Soc.40 (1918) 1361.
[75] Niemantsverdriet, J.W.,Spectroscopy in Catalysis(VCH, Weinheim 2000).
[76] Karge, H.G., Hunger, M., Beyer, H.K., in:Modification of zeolites, ed. Weitkamp, J.,
Lothar, P. (Springer, Berlin 1999) 198.
[77] Gunzler, H., Heise, H.M.,IR-Spektroskopie, VCH, Weinheim (1996).
[78] Olinger, J.M., Griffiths, P.R.,Anal. Chem.60 (1988) 2427.
[79] Hatje, U., Ressler, T., Petersen, S., F¨orster, H.,J. de Physique, IV (1996)141.
[80] Scofield, J.H.,J. Electr. Spectr. Rel. Phenom.8 (1976)129.
[81] Monti, D.A.M., Baiker, A.,J. Catal.83 (1994) 323.
[82] Hurst, N.W., Gentry, S.J., Jones, A., McNicol, B.D.,Catal. Rev.-Sci.Eng.25 (1983) 233.
[83] Wapner, K., Diplomarbeit, Bochum (2001).
[84] Stansch, Z., Mleczko, L., Baerns. M.,American Chemical Society(1997).
[85] Stansch, Z., Dissertation, Bochum (1995).
[86] Ostrowski, Th., Dissertation, Bochum (1998).
[87] Marschall, K.-J., Dissertation, Bochum (1999).
[88] Bartmann, U., Dissertation, Bochum (1999).
[89] Ratajczak, O., Dissertation, Bochum (2000).
[90] Myers, A.L., Seider, W.D.,Introduction to Chemical Engineering and Computer Calcu-
lations, Prentice-Hall, Englewood Cliffs, (1976).
[91] Rosahl, B., Gelbin, D.,Chem. Technik11 (1966) 647.
[92] Hagen, J.,Chemische Reaktionstechnik, VCH, Weinheim (1992).
[93] Hirschfelder, J.O., Curtiss, C.F., Bird, R.B.,Molecular Theorie of Gases, Jon Wiley &
Sons, New York (1954).
[94] Chen, N.Y., Degnan, T.F., Smith, C.M.,Molecular Transport and Reaction in Zeolites,
VCH, Weinheim (1994).
[95] Karger, J., Freude, D.,Chem. Eng. Technol.25 (8) (2002) 769.
[96] Tabata, T., Ohtsuka, H.,Catal. Lett.48 (1997) 203.
[97] Shichi, A., Satsuma, A., Iwase, M., Shimizu, K., Komai, S., Hattori, T.,Appl. Catal. B
17 (1998) 107.
[98] Shichi, A., Katagi, K.,Satsuma, A., Hattori, T.,Appl. Catal. B24 (2000) 97.
[99] Witzel, F., Sill, G.A., Hall., W.K.,J. Catal.149(1994) 229.

6 Literaturverzeichnis 149
[100] Shen, S.-C., Kawi, S.,Catalysis Today68 (2001)245.
[101] Liese, T., Loffler, E., Grunert, W.,J. Catal.197(2001) 123.
[102] Schmidt, C., Sowade, T., L¨offler, E., Birkner, A., Grunert, W.,J. Phys. Chem. B106
(2002) 4085.
[103] Barrer, R.M., Walker, A.J.,Trans Faraday Soc.60 (1964) 171.
[104] Kaliaguine, S., Lemay, G., Adnot, A., Burelle, S., Audet, R., Jean, G., Sawichi, J.A.,
Zeolites10 (1990) 559.
[105] Stakheev, A.Y., Khodakov, A.Y., Kustov, L.M., Kazansky, V.B., Minachev, Kh.M.,Zeo-
lites12 (1992) 866.
[106] Han, S., Schmitt, K.D., Chang, C.D.,Inorganica Chimica Acta304(2000) 297.
[107] Wade, K., Banister, J.A., in:Comprehensive Inorganic Chemistry, Chap.12 (Pergamon
Press, Elmsford, New York 1973).
[108] Berndt, H., Richter, M., Fricke, R., L¨ucke, B.,Proc. EuropaCat 3, Krakow1 (1997) 385.
[109] Mihalyi, R.M., Pal-Borbely, G., Beyer, H.K., Minchev, Ch., Neinska, Y., Karge, H.G.,
React. Kinet. Catal. Lett.60 No.1 (1997) 195.
[110] Beyer, H.K., Mihalyi, Minchev, Ch., Neinska, Y., Kanazirev, V.,Microporous Mater.7
(1996) 333.
[111] Mihalyi, R.M., Beyer, H.K., Mavrodinova, V., Minchev, Ch., Neinska, Y.,Microporous
Mater.24 (1998) 143.
[112] Neinska, Y., Mihalyi, R.M., Mavrodinova, V., Minchev, C., Beyer, H.K.,Phys. Chem.
Chem. Phys.1 (1999) 5761.
[113] Kanazirev, B., Neinska, Y., Tsoncheva, T., Kosova, L., in:Proc. 9th Int. Conf. Zeolite,
ed. Ballmooset al., (Butterworth-Heinemann, London 1993) 461.
[114] Kanazirev, B., Price, G.L.,Stud. Surf. Sci. Catal.84 (1994) 1935.
[115] Kanazirev, V., Valtchev, V., Tarassov, M.P.,J. Chem. Soc., Chem. Comm.(1994) 1043.
[116] Rabo, M.A., Pooutsma, M., Skeels, G.W., in:5th Int. Congr. Catalysis, ed. Hightower,
J.W., (North-Holland Publ., NewYork 1973) 1353.
[117] Beyer, H.K., Pal-Borbely, G., Keindl, M.,Microporous and Mesoporous Materials31
(1999) 333.
[118] van Koningsveld, H., van Bekkum, H., Janssen, J.C. ,Acta Cryst.43 (1987) 127.
[119] Yamagishi, K., Namba, S., Yashima, T.,Bull. Chem. Soc. Jpn.64 (1991) 949.

6 Literaturverzeichnis 150
[120] Pillep, B., (Dissertation, M¨unchen 1999).
[121] Mihalyi, M.R., Beyer, H.K.,Chem. Comm.(2001) 2242.
[122] Wen, B., Jia, J., Li, S., Liu, T., Chen, L.X., Sachtler, M.H.,Phys. Chem. Chem. Phys.4
(2002) 1983.
[123] Gutierrez, L., Boix, A., Lombardo, E.A., Fierro, J.L.G.,J. Catal.199(2001) 60.
[124] Gutierrez, L., Boix, A., Petunchi, J.,Catal. Today54 (1999) 451.
[125] Tabata, K., Teng, Y., Takamoto, T., Suzuki, E., Banares, M.A., Pena, M.A., Fierro,
J.L.G.,Cat. Reviews44 (2002) 1.
[126] Froment, G.F., Bischoff, K.B.,Chemical Reactor Analysis and Design, John Wiley &
Sons (1979).
[127] Attar, K.D., Diplomarbeit, Bochum (2001).
[128] Atkins, P.W.,Physikalische Chemie, VCH, Weinheim (1990).
[129] Suprun, W., Speer, V., Papp, H.,XXXV. Jahrestreffen deutscher Katalytiker(2002) 75.
[130] Heinrich, F., (Dissertation, Bochum 2001.
[131] Park, P.W., Ragle, C.S., Boyer, C.L., Balmer, M.L., Engelhard, M., McCready, D.,J.
Catal.210(2002) 97.
A Anhang
In der folgenden Tabelle A.1 sind die Betriebsparameter des Gaschromatographen wiedergege-ben:
Tabelle A.1: Betriebsparameter des GC Delsi IGC 11.
Trennsaule Molsieb 5A von Restek (3 m, 1/8 Zoll)Detektor Warmeleitfahigkeitsdetektor (Br¨uckenspannung 300 mA)Tragergas Helium (11 ml/min)
Analysenzeit 15 MinutenSaulentemperatur 30 CRetentionszeit O2 4,2 minRetentionszeit N2 8,5 min
Retentionszeit CH4 13,3 min
In der Tabelle A.2 finden sich die verwendeten Gase wieder, in der letzten Tabelle A.3 werdendie zu Praparationszwecken eingesetzten Chemikalien aufgelistet:
Tabelle A.2: Liste der verwendeten Reingase und Gasmischungen.
Gas Hersteller Reinheit
Helium MesserGriesheim 4.620 % O2 in He MesserGriesheim 4.5/4.6
O2 MesserGriesheim 4.52,5 % NO in He MesserGriesheim 2.5/4.62,5 % CH4 in He MesserGriesheim 2.5/4.6
Tabelle A.3: Liste der verwendeten Chemikalien.
Name Hersteller Reinheit
NaNO3 Riedel-de Ha¨en AG 99,5 %KBr Merck fur Spektroskopie
In(NO3)3 � 5H2O Alfa Aesar 99,99 %In2O3 Heraeus 99,999 %InCl3 ChemPur 99,99 %
In(OH)3 Alfa Aesar 99,99 %Ce(NO3)3 � 6H2O Alfa Aesar 99,99 %
CeO2 Rhodia -

Lebenslauf
Personliche Daten
Name: Frank ThomasSowade
Geburtsdatum und -ort: 12.01.1973 in Kobe (Japan)
Staatsangeh¨origkeit: Deutsch
Schulausbildung
08/1979 - 07/1983 Gemeinschaftsgrundschule Schildgen, Bergisch-Gladbach
08/1983 - 06/1992 Nicolaus-Cusanus-Gymnasium, Bergisch-Gladbach;Abschluss: Allgemeine Hochschulreife
Wehrdienst
07/1992 - 06/1993 Ableistung des Wehrdienstes in Hessen
Hochschulausbildung
10/1993 Beginn des Studiums der Chemie an der Universit¨at zu Koln
12/1995 Diplom-Vorpr¨ufung in Chemie
10/1998 Abschluss als Diplom-Chemiker
01/1998-10/1998 Diplomarbeit am Stiftungslehrstuhl f¨ur Technische Chemieunter Anleitung von Herrn Prof. Dr. K. Elgeti bei der Bayer AG Leverkusen
Untersuchung der Absorption von NOx-Gasen in wassrigen Losungen
mit Hilfe von H2O2
seit 01/1999 Promotionsstudium im Fach Technische Chemie an der Ruhr-
Universitat Bochum unter der Betreuung von Prof. Dr. W. Gr¨unert
Selektive katalytische Reduktion von NO mit Methan an mit Indium
und Cer modifizierten Zeolithkatalysatoren
Koln, Oktober 2002


















