
Spektroskopie - Medicinal Chemistry · Aufbau eines IR-Spektrometers Zwei Gerätetypen sind...
213
Spektroskopie • Allgemeines und Prinzip • Theoretische Grundlagen • IR-Spektroskopie – Raman-Spektroskopie • UV/Vis-Spektroskopie • Fluoreszenz-Spektroskopie • NMR-Spektroskopie • Massenspektrometrie • Atomabsorptions-Spektroskopie
Transcript of Spektroskopie - Medicinal Chemistry · Aufbau eines IR-Spektrometers Zwei Gerätetypen sind...

Spektroskopie
• Allgemeines und Prinzip • Theoretische Grundlagen • IR-Spektroskopie
– Raman-Spektroskopie • UV/Vis-Spektroskopie • Fluoreszenz-Spektroskopie • NMR-Spektroskopie • Massenspektrometrie • Atomabsorptions-Spektroskopie

Einsatzgebiete spektroskopischer Methoden
Identifikation und Nachweis von Stoffen Gehaltsbestimmungen von Verbindungen und
Elementen Charakterisierung von Substanzen Strukturaufklärung von Verbindungen

Einteilung der spektroskopischen Methoden
Atomspektroskopie Molekülspektroskopie Absorptionsspektroskopie Emissionsspektroskopie
Atomabsorptionsspektroskopische Methoden Atomabsorptionsspektroskopie (AAS)
Atomemissionsspektroskopische Methoden Atomemissionsspektroskopie
Molekülabsorptionsspektroskopische Methoden UV/Vis-Spektroskopie, IR-Spektroskopie (NMR-Spektroskopie)
Molekülemissionsspektroskopische Methoden Fluoreszenzspektroskopie (Raman-Spektroskopie)
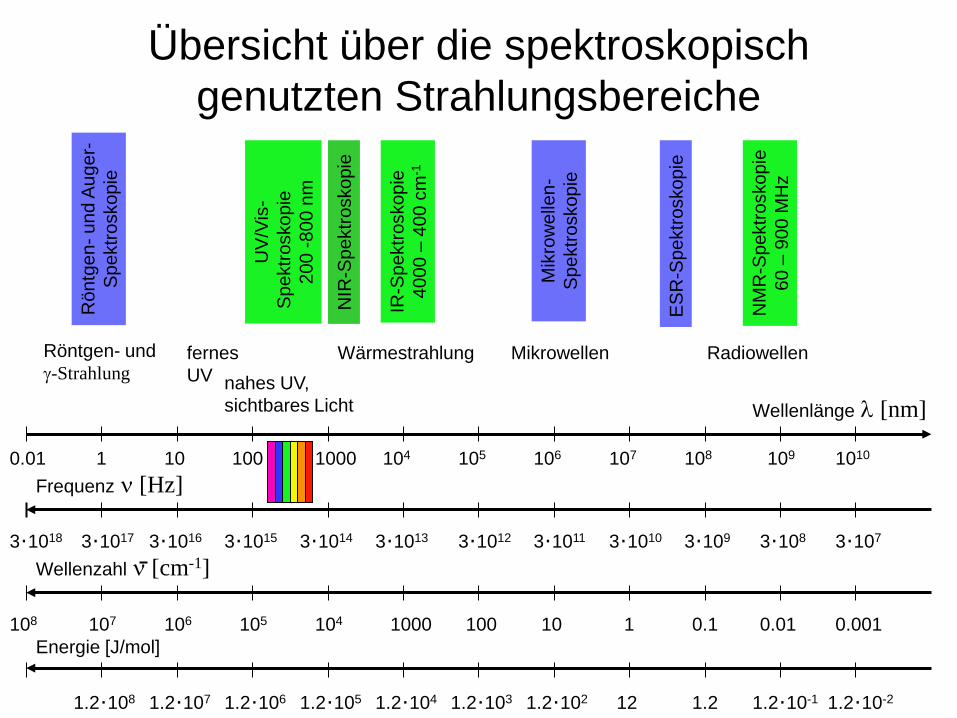
Übersicht über die spektroskopisch genutzten Strahlungsbereiche
Wellenlänge λ [nm]
Röntgen- und γ-Strahlung
fernes UV nahes UV,
sichtbares Licht
0.01 1 10 100 1000 104 105 106 107 108 109 1010
Wärmestrahlung Mikrowellen Radiowellen
Frequenz ν [Hz]
Rön
tgen
- und
Aug
er-
Spek
trosk
opie
UV/
Vis-
S
pekt
rosk
opie
20
0 -8
00 n
m
IR-S
pekt
rosk
opie
4000
– 4
00 c
m-1
NM
R-S
pekt
rosk
opie
60
– 9
00 M
Hz
ESR
-Spe
ktro
skop
ie
Mik
row
elle
n-
Spek
trosk
opie
3·107 3·109 3·108 3·1011 3·1010
Wellenzahl ν [cm-1]
Energie [J/mol] 0.001 0.01 0.1 1 10 1000 100 104 105 106
NIR
-Spe
ktro
skop
ie
3·1013 3·1012 3·1016 3·1015 3·1014 3·1018 3·1017
108 107
1.2·10-2 1.2·10-1 1.2 12 1.2·102 1.2·103 1.2·104 1.2·105 1.2·106 1.2·107 1.2·108

x
Amplitude (A)
Magnetisches Feld
Elektrisches Feld y = Asin(2πνt)
Elektromagnetische Welle

Absorption
Reflexion
Streuung
Brechung
Streuung
Emission
Wechselwirkungen zwischen Wellen und Materie
elastische Stöße
inelastische Stöße

Grundlagen der Spektroskopie
Es gelten die Beziehungen
E = h · ν = h · c / λ und ν = 1 / λ h: 6.6 · 10-34 Js, Planck-Konstante c: 3 · 108 m/s, Lichtgeschwindigkeit λ: Wellenlänge ν: Frequenz ν: Wellenzahl Die Energieaufnahme bzw. Abgabe gehorcht den
Regeln der Quantenmechanik

Grundlagen der Spektroskopie
Energieübergänge finden aus einem niedrigeren in ein höheres Energieniveau (Ab-sorption) oder von einem höheren in ein niedrigeres Energieniveau (Emission) statt.
Die Energie eines atomaren bzw. molekularen Systems ergibt sich aus der Lösung der Schrödinger-Gleichung.
Angeregter Zustand
Grundzustand
Absorption Emission

Schrödinger-Gleichung
Die Schrödinger-Gleichung ist eine Eigenwertgleichung, d. h.: Ĥψ = Eψ Die Anwendung des Hamilton-Operators auf die Wellenfunktion ψ ergibt den Energie- Eigenwert E. Der Hamilton-Operator setzt sich zusammen aus den Operatoren der kinetischen und der potentiellen Energie. H = T + V Sinnvolle Lösungen der Schrödinger-Gleichung sind nur für diskrete Werte von E er- laubt → Quantenzahlen!

Die Boltzmann-Gleichung
Die Atome bzw. Moleküle verteilen sich in Abhängigkeit von der Temperatur in die zu Verfügung stehenden Energieniveaus. Es gilt:
N N0
= k‘ · e - ΔE
k · T
N: Teilchen im angeregten Zustand N0: Teilchen im Grundzustand k: Boltzmann-Konstante k‘: systemabhängige Konstante ΔE: Energiedifferenz der beiden Energieniveaus T: absolute Temperatur [K]
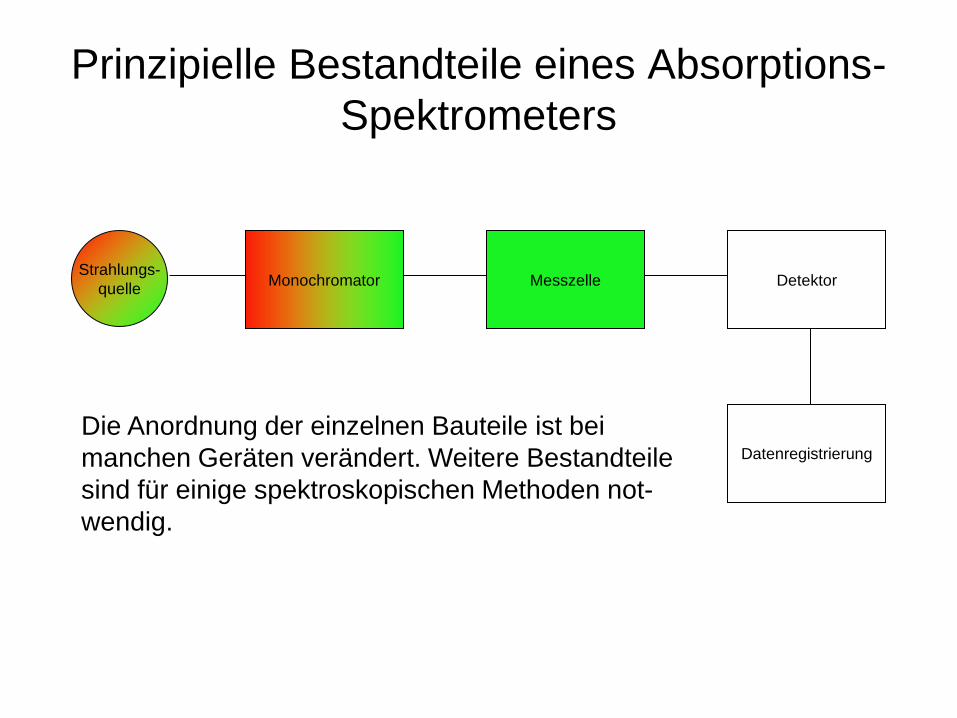
Prinzipielle Bestandteile eines Absorptions-Spektrometers
Monochromator Strahlungs- quelle Messzelle Detektor
Datenregistrierung Die Anordnung der einzelnen Bauteile ist bei manchen Geräten verändert. Weitere Bestandteile sind für einige spektroskopischen Methoden not- wendig.

Strahlenbang in einem Prisma
Strahlenbang an einem Gitter
α
G

Das Lambert-Beer‘sche Gesetz
Die Absorption (A) ist proportional zur Konzentration (c) des absorbierten Stoffs und zur durchstrahlten Strecke (d). Es gilt:
A = lg(I0/I) A = k · c A = k’ · d
A = ε · c · d ε: Absorptionskoeffizient

Infrarot-Spektroskopie
Anregung von Schwingungen und Rotationen
Anregungsenergie ca. 5۰104 – 1۰104 J/mol entsprechend 4000 – 600 cm-1
Schwingungen werden nur angeregt, wenn sich während der Schwingung das Dipolmoment ändert.

IR-Spektrum von Acetylsalicylsäure
KBr-Pressling

Molekül-Schwingungen
Valenzschwingungen υ
Deformationsschwingungen δ
symmetrische Valenzschwingung υsy
asymmetrische Valenzschwingung υas
Spreizschwingung δs Pendelschwingung ρ Torsionsschwingung τ
● ○
● Bewegung aus der Bildebene ○ Bewegung hinter die Bildebene Kippschwingung ω
● ●

Valenzschwingung eines zweiatomigen Moleküls
Die zur Anregung einer Schwingung notwendige Energie hängt von der Bindungsstärke und den an der Schwingung beteiligten Massen ab.

Das Modell des harmonischen Oszillators
Wird die Bindung als Feder, die die Massen m1 und m2 verbindet, betrachtet, so gilt:
√ k M
1 2π υ =
m1۰ m2 m1 + m2
mit M = der reduzierten Masse
Die Energie einer Schwingung und damit υ̃ wird größer:
Je stärker die Bindung Je kleiner die reduzierte Masse

Der harmonische Oszillator - quantenmechanisch
Ĥψ = Eψ
2M dx2 ћ2 d2
+ kx2
2 Ĥ = -
d2ψ dx2 +
ћ2 2ME - Mkx2
ћ2 = 0 ( ) ψ
√ k M
h 2π
E = (v + ½) v (Schwingungsquantenzahl) = 0, 1, 2, ...
ψ enthält die Hermiteschen Polynome.

Der harmonische Oszillator υ
r
Gleichgewichtabstand der schwingenden Atome (r0)
v = 0
E = h υ(v +1/2) v: Schwingungsquantenzahl Erlaubte Übergänge: v = ±1
√ k M
1 2π υ =

Der anharmonische Oszillator V
r
Die Abstände der Niveaus sind nicht mehr gleichmäßig. Bei großen Kernabständen nähert sich der entsprechende Parabelast der Dissoziationsenergie. Erlaubte Übergange: ±1, ±2, ±3,……
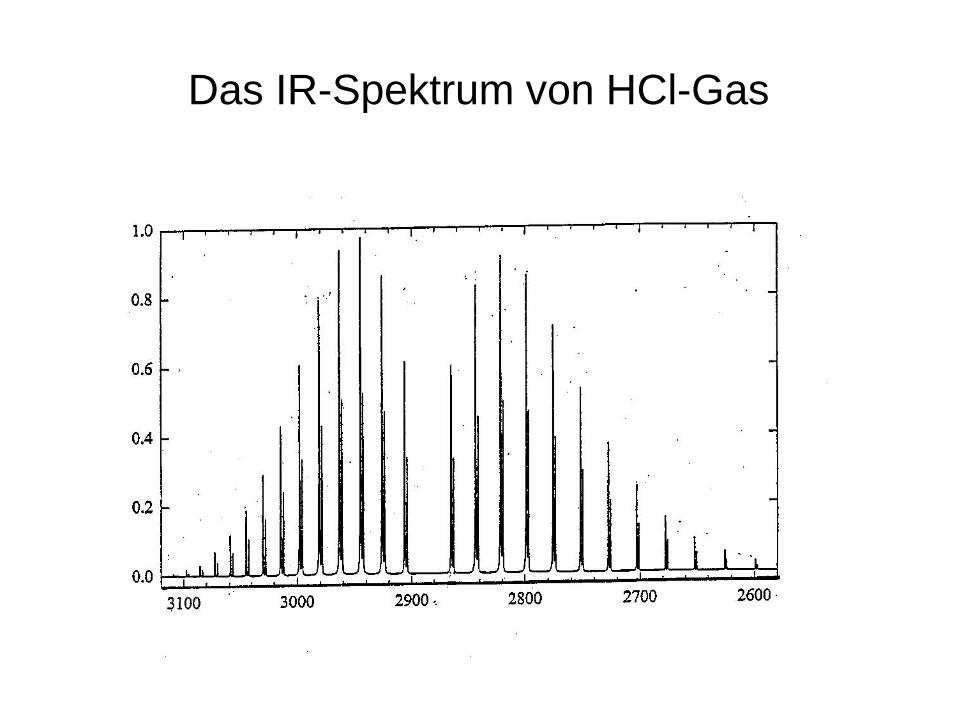
Das IR-Spektrum von HCl-Gas
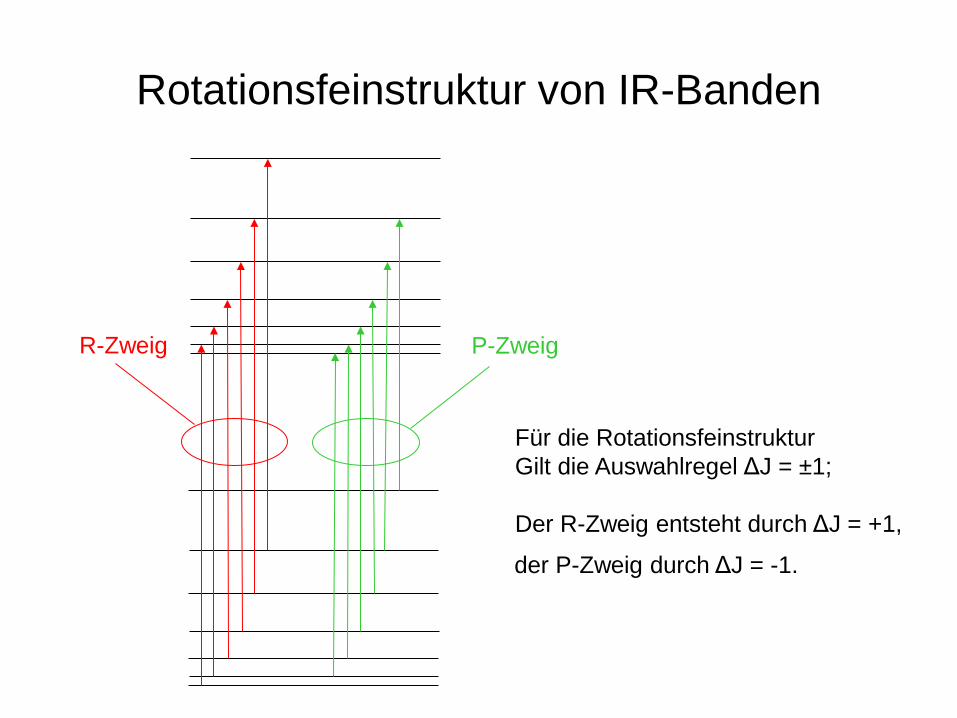
Rotationsfeinstruktur von IR-Banden
R-Zweig P-Zweig
Für die Rotationsfeinstruktur Gilt die Auswahlregel ΔJ = ±1; Der R-Zweig entsteht durch ΔJ = +1,
der P-Zweig durch ΔJ = -1.

Bereiche des IR-Spektrums
4000 3000 2000 1000
O-H N-H C-H
C≡C C≡N
X=Y=Z
1600
C=O C=N C=C N=O
„Fingerprint“-Bereich
Valenzschwingungen von Einfachbindungen (C−C, C−O etc. Deformations- schwingungen
ν̃ [cm-1]
δ
δ
δ
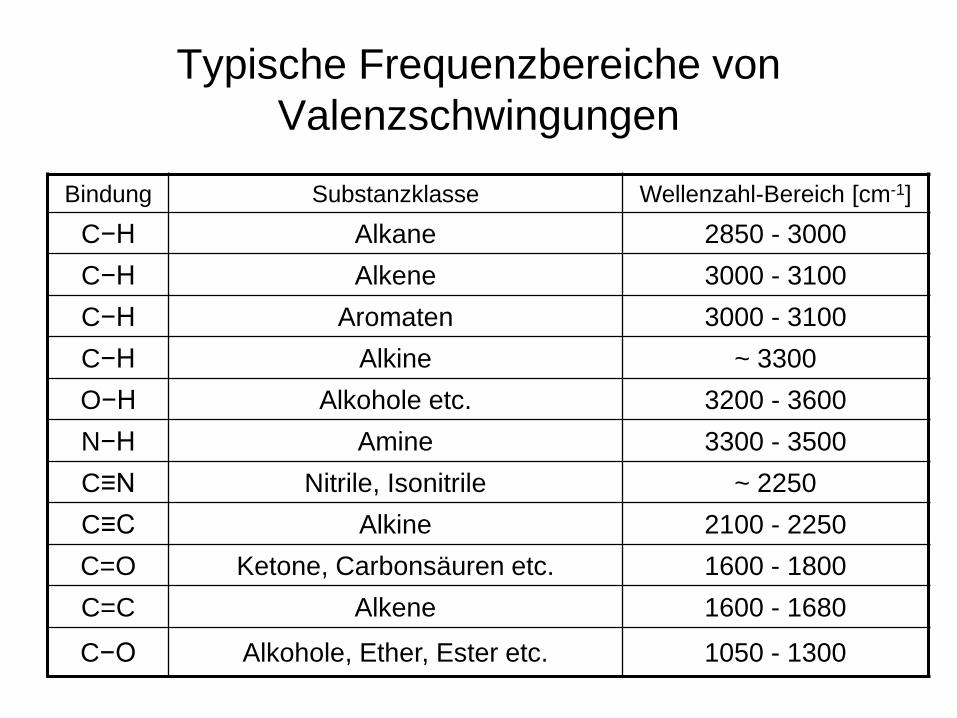
Typische Frequenzbereiche von Valenzschwingungen
Bindung Substanzklasse Wellenzahl-Bereich [cm-1]
C−H Alkane 2850 - 3000 C−H Alkene 3000 - 3100 C−H Aromaten 3000 - 3100 C−H Alkine ~ 3300 O−H Alkohole etc. 3200 - 3600 N−H Amine 3300 - 3500 C≡N Nitrile, Isonitrile ~ 2250 C≡C Alkine 2100 - 2250 C=O Ketone, Carbonsäuren etc. 1600 - 1800 C=C Alkene 1600 - 1680 C−O Alkohole, Ether, Ester etc. 1050 - 1300

Intensität von IR-Banden
Die Intensität einer IR-Bande hängt ab: Vom Besetzungsverhältnis der am Übergang beteiligten Energiezustände (Boltzmann). Vom Grad der Änderung des Dipolmoments Es wird beobachtet, dass: C=C-Banden schwächer sind als C=O-Banden, C–C-Banden schwächer sind als C–O bzw. C–N, C–H-Banden schwächer sind als O–H bzw.. N–H.
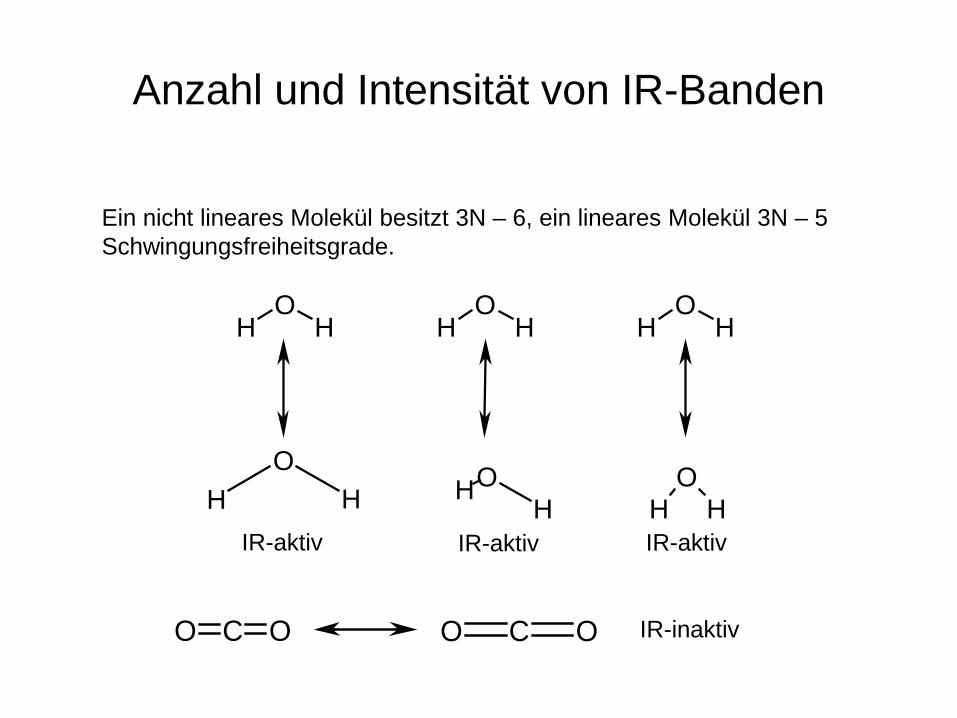
Anzahl und Intensität von IR-Banden
Ein nicht lineares Molekül besitzt 3N – 6, ein lineares Molekül 3N – 5 Schwingungsfreiheitsgrade.
HO
H HOH
HO
H HO
H HO
H
HO
HIR-aktiv IR-aktiv IR-aktiv
O C O O C O IR-inaktiv

Aufbau eines IR-Spektrometers
Zwei Gerätetypen sind gebräuchlich: Das Zweistrahl-IR-Spektrometer (Klassische Bauweise) Das Fourier-Transform (FT) IR-Spektrometer
Vorteile des FT-IR-Spektrometers: Sehr kurze Messzeit Verbessertes Signal/Rausch-Verhältnis Möglichkeit der Spektren-Addition bzw. Subtraktion (wichtig zur Hintergrundkompensation)

Das IR-Spektrometer
Nernst-Stift oder Globar
Strahlteiler
Messzelle
Vergleichszelle
Strahlungs- quelle
Prisma oder Gitter
Monochromator
Thermoelement Golay-Detektor Pyroelektrischer Detektor
Detektor

Michelson-Interferometer
Strahlungs- quelle
Strahlteiler
feststehender Spiegel
beweglicher Spiegel
Detektor
Durch die Bewegung des Spiegels kommt es am Strahlteiler zu zeit- Abhängigen Interferenzen. Während der Bewegungwerden alle möglichen Frequenzen erzeugt Das frequenzabhängige Spektrum wird durch Fourier-Transformation erhalten.

Interferenzen
konstruktive Interferenz
destruktive Interferenz

Die Bauteile des IR-Spektrometers
Alle im Strahlengang befindlichen Materialien dürfen IR-Strahlung nicht absorbieren. Sie sind aus NaCl, KBr, LiF, CsF etc. Strahlungsquelle: Beheizte Keramik (Zirkonoxid = Nernst-Stift) oder Siliziumcarbid (Globar). Detektor: Der Pyroelektrische Detektor besteht aus einem mit L-Alanin dotierten Glycinsulfat-Kristall. Die Wärmestrahlung verändert die Abstände der Gitterebenen des Kristalls und induziert dadurch einen Strom, der gemessen wird. Im Golay-Detektor wird die durch Wärme erzeugte Ausdehnung eines Gases gemessen. Im Thermoelement wird die Spannungsdifferenz gemessen, die durch Bestrahlung einer von zwei Verbindungsstellen zweier Drähte entsteht.

Die IR-Probe
Gase und Flüssigkeiten werden als Reinsubstanzen in geeigneten Küvetten mit entsprechenden Schichtdicken gemessen. Feststoffe werden als Lösung, Suspension, Pressling oder Film gemessen. Als Lösungsmittel ist alles geeignet was nicht mit der Probe bzw. den Küvetten- fenstern reagiert und im betrachteten Spektralbereich keine nicht kompensierbare Eigenabsorption besitzt. Suspensionsgrundlage sind zähflüssige Paraffine (Nujol). Presslinge werden aus KBr mit einem Substanzgehalt von 1 – 5% hergestellt. Zur Herstellung eines Substanzfilms wird eine Lösung der Substanz auf einem geeigneten Träger (z. B. NaCl-Platte) verdampft. Polymere können auch direkt zu Folien geeigneter Dicke geformt werden.

IR-Spektren von in verschiedenen Lösungsmitten

IR-Analyse im Praktikum
Aufnahme des IR-Spektrums (Zweistrahl IR-Gerät, KBr-Pressling) Interpretation aller gemessenen Banden im Bereich zwischen 4000 und 1600 cm-1 und Angabe der entsprechenden funktionellen Gruppen. Im Fingerprint-Bereich werden nur die intensiven bzw. besonders relevanten Banden interpretiert (z. B. C−O, aromatische C−H Deformations- Schwingungen).

Aufnahme von IR-Spektren durch abgeschwächte Totalreflektion (ATR)
Die Bandenintensität hängt von der Eindringtiefe der Strahlung in die Probe und damit von der Wellenlänge ab. Bei größeren Wellenlängen – kleineren Wellenzahlen – sind die Banden intensiver.
Detektor Kristall aus Material mit Großem Brechungsindex z. B. ZnSe
Probe
IR-Strahlung

Interpretation von IR-Spektren
Beurteilung von Bandenlage und Intensität Modell des harmonischen Oszillators Zusätzliche Informationen zu charakteristischen Banden; z. B. : zu υ N − H, O − H entsprechende δ N − H, O − H zu υ C=O eventuell υ C−O zu υ C−H oberhalb 3000 cm-1 υ C=C oder out of plane C−H

Interpretation von IR-Spektren Regeln zur Beurteilung von C=O Banden
υ C=O Säure > υ C=O Ester > υ C=O Keton, Aldehyd, Amid
O OO
O
O
O
O
O
OO
NH
O
NH
O
NHO
O
O
O
O
~1710 cm-1 ~1745 cm-1 ~1780 cm-1 ~1670 cm-1
~1720 cm-1 ~1760 cm-1 ~1840 cm-1 ~1775 cm-1 ~1750 cm-1
~1670 cm-1 ~1720 cm-1 ~1750 cm-1
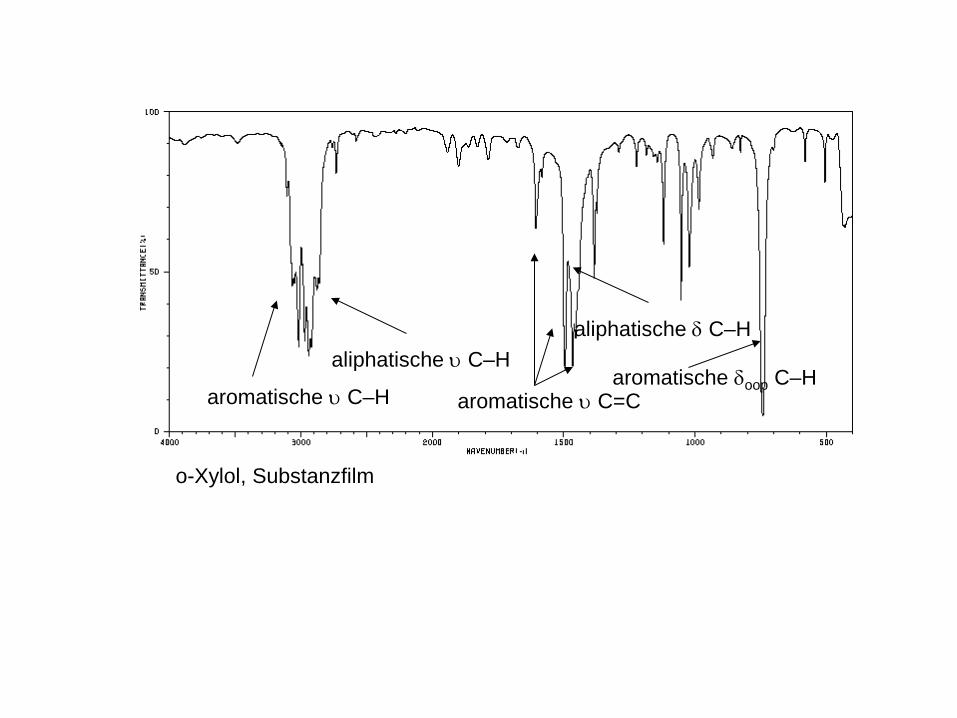
o-Xylol, Substanzfilm
aromatische υ C–H
aliphatische υ C–H
aromatische υ C=C
aliphatische δ C–H
aromatische δoop C–H

m-Xylol, Substanzfilm
Möglichkeit zur Unterscheidung von Substitutionsisomeren

p-Xylol, Substanzfilm

IR-Spektren von o-, m- und p-Xylol
o-Xylol
m-Xylol
p-Xylol p-Xylol

IR-Spektren von Kohlenwasserstoffen
Pentan
1-Penten
1-Pentin

IR-Spektren von Alkoholen
n-Butanol
2-Butanol
t-Butanol

IR-Spektren von Aminen
n-Butylamin
Diethylamin
Dimethylethylamin
υ N−H
υ N−H
δ N−H
ω N−H
δ N−H
δ C−H
ω N−H
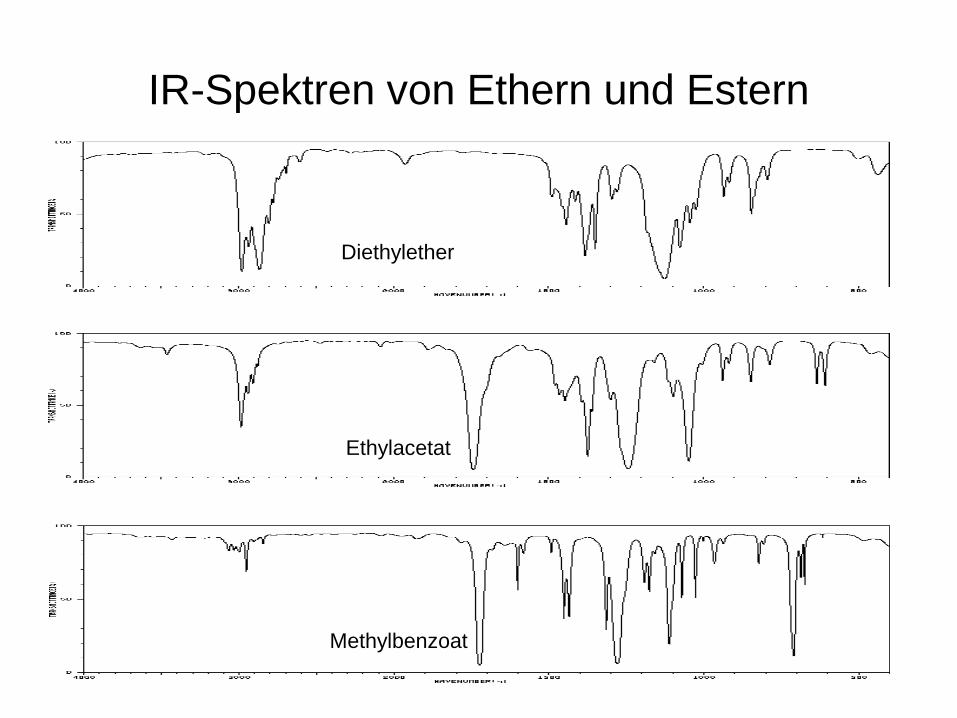
IR-Spektren von Ethern und Estern
Diethylether
Ethylacetat
Methylbenzoat
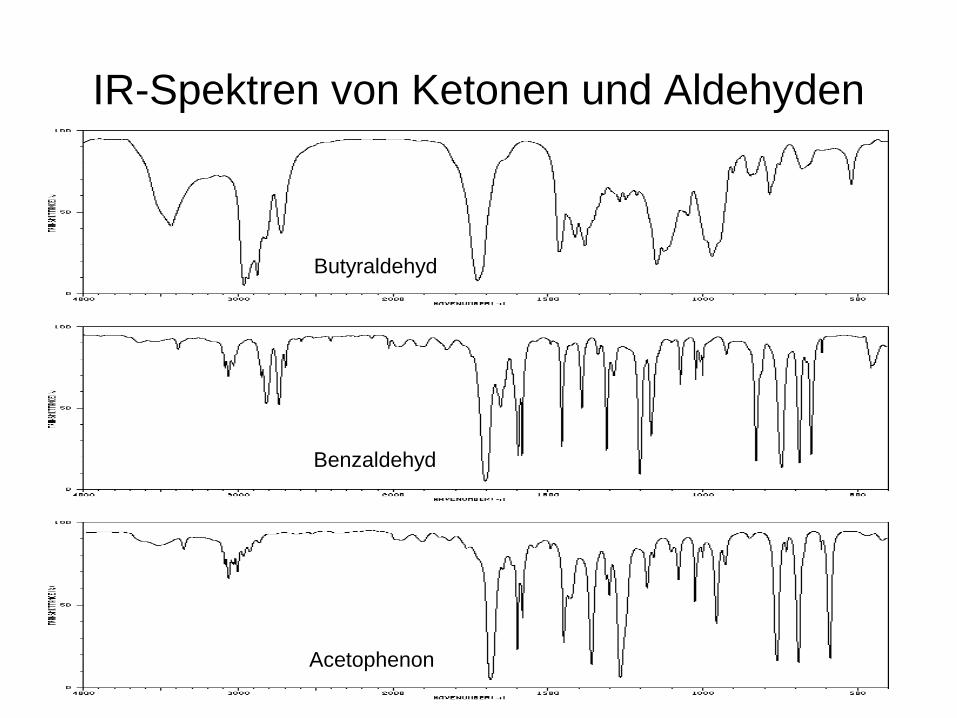
IR-Spektren von Ketonen und Aldehyden
Benzaldehyd
Butyraldehyd
Acetophenon

IR-Spektren von Carbon- und Aminosäuren
Essigsäure
L-Alanin
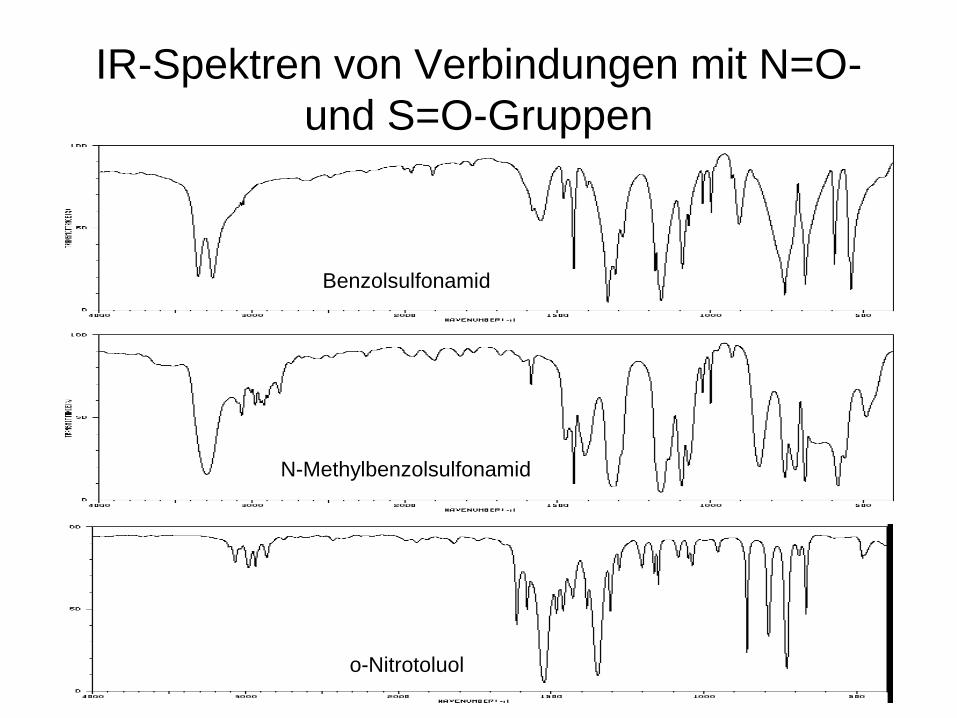
IR-Spektren von Verbindungen mit N=O- und S=O-Gruppen
N-Methylbenzolsulfonamid
Benzolsulfonamid
o-Nitrotoluol
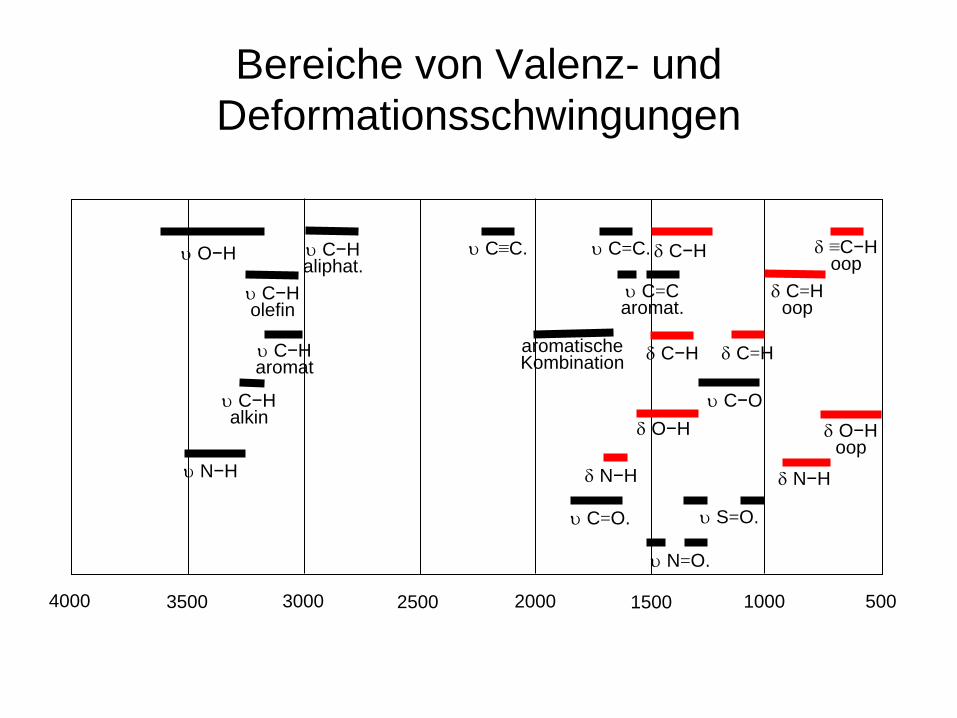
Bereiche von Valenz- und Deformationsschwingungen
4000 3500 3000 2500 2000 1500 1000 500
υ C−H aliphat.
υ C−H aromat
υ C−H olefin
υ C−H alkin
υ O−H υ C≡C. δ C−H υ C=C.
δ C=H oop
δ ≡C−H oop
υ C=C aromat.
aromatische Kombination δ C=H
υ C−O δ O−H
oop
δ C−H
δ O−H
δ N−H υ N−H δ N−H
υ C=O. υ S=O.
υ N=O.

Raman-Spektroskopie
Wie bei der IR-Spektroskopie werden Schwingungen und Rotationen angeregt. Im Gegensatz zur IR-Spektroskopie hängt die Bandenintensität jedoch nicht von der Änderung des Dipolmoments sondern von der Änderung der Polarisier- barkeit der Bindung ab. Dadurch treten im Raman-Spektrum häufig Banden auf, die im IR-Spektrum nicht beobachtet werden. Es gelten die quantenmechanischen Auswahlregeln der IR-Spektroskopie (Δυ = ±1). Etwa 1% des eingestrahlten Lichts wird unter Abgabe oder Aufnahme von Energie gestreut, das gestreute Licht besitzt also eine größere oder kleinere Wellenlänge als das eingestrahlte Licht (Raman Effekt). Die Differenz der Wellenlängen zwischen eingestrahltem und gestreutem Licht liegt im Bereich der Infrarot-Strahlung.

Prinzip der Raman-Spektroskopie
Strahlungs- quelle
gestreutes Licht
Raman-Streulicht
Detektor für Absorption
Detektor für Streulicht
Das Streulicht wird im 90° Winkel zum Eingangsstrahl gemessen. Die Lichtquelle muss hohe Intensität abstrahlen, es werden Laser eingesetzt. Es kann mit sichtbarem Licht gearbeitet werden; die Bauteile des Spektrometers werden aus Glas gefertigt.
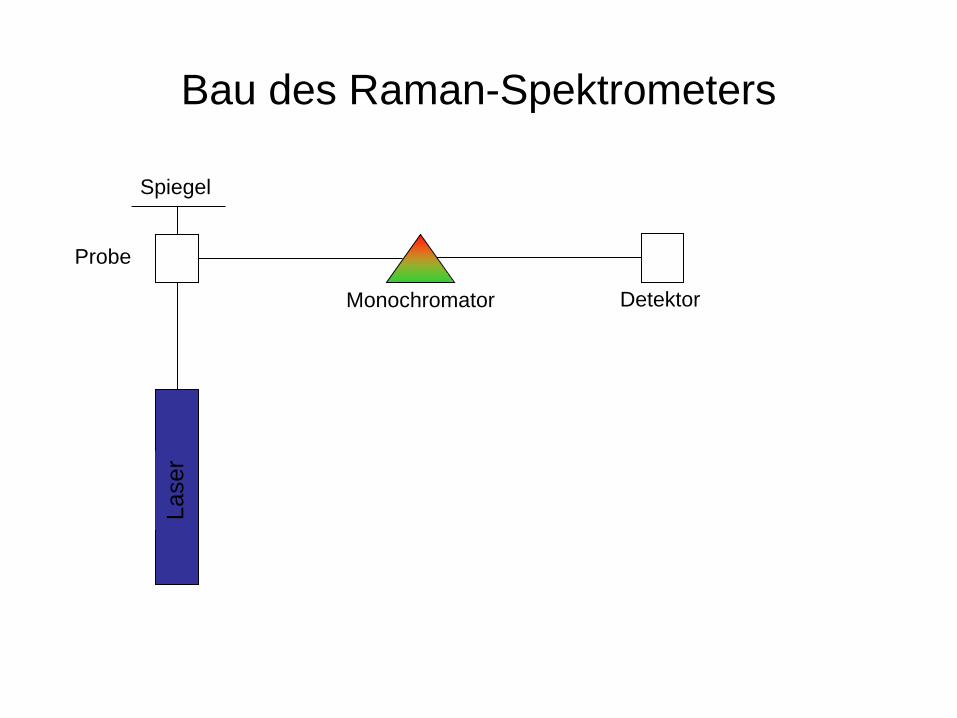
Bau des Raman-Spektrometers
Spiegel La
ser
Monochromator Detektor
Probe

Das Raman-Spektrum In
tens
ität
0 ν̃ [cm-1] -ν̃ [cm-1]
Rayleigh
anti-Stokes-Linie
Stokes-Linie
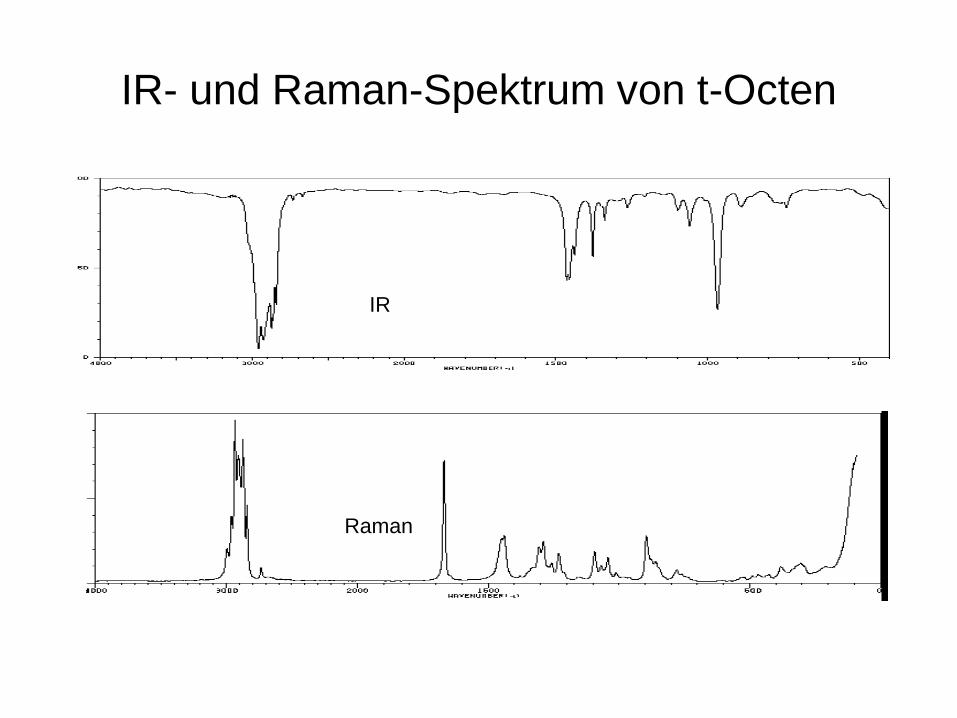
IR- und Raman-Spektrum von t-Octen
IR
Raman

UV/Vis-Spektroskopie
Die UV/Vis-Spektroskopie ist eine sehr empfindliche (Boltzmann), molekülabsorptionsspektroskopische Methode, die insbesondere zur quantitativen Bestimmung (Lambert-Beer) von Substanzen herangezogen wird.
Es werden Bindungselektronen angeregt.
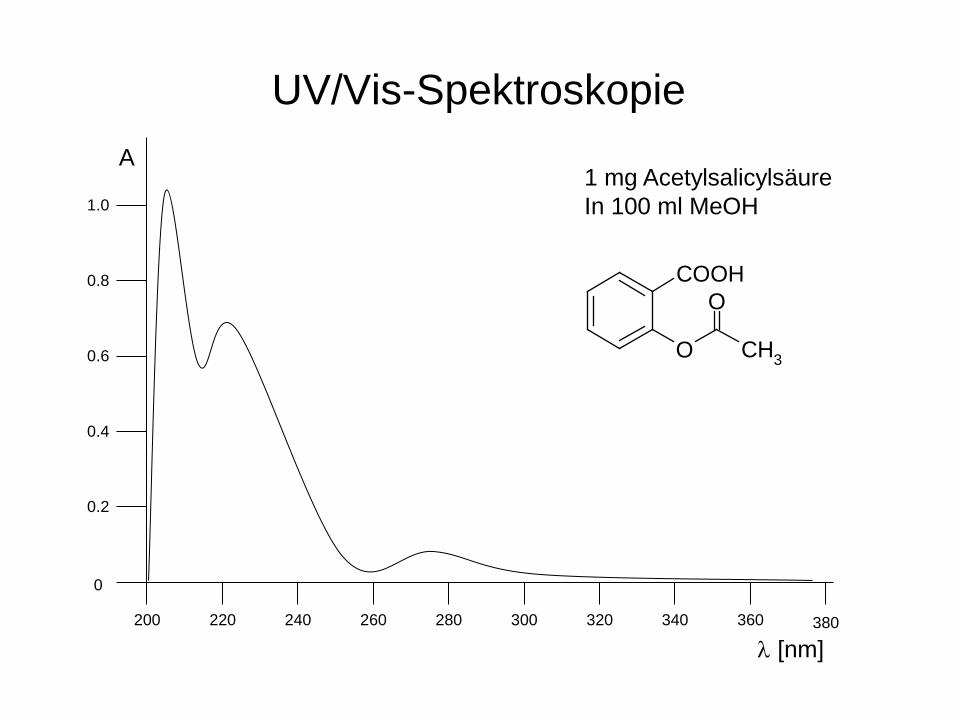
UV/Vis-Spektroskopie
0
380
0.4
A
200 240 220 260 300 280 340 360 320
0.2
0.6
1.0
0.8
λ [nm]
1 mg Acetylsalicylsäure In 100 ml MeOH
COOH
O
O
CH3

Das UV-Spektrometer UV/Vis-Spektren werden in Lösung gemessen. Lösungs-
mittel und im Strahlengang befindliche Materialien dürfen im interessierenden Bereich keine Eigenabsorption zeigen.
Monochromator Strahlungs- quelle
Messzelle
Detektor
Deuterium- oder Wolframlampe
Prisma oder Gitter
Küvette
Photozelle Photomultiplier
Vergleichszelle
Rotierender Spiegel
Eintritts- und Austrittsspalt

Strahlengang eines Zweistrahl UV/Vis-Spektrometers
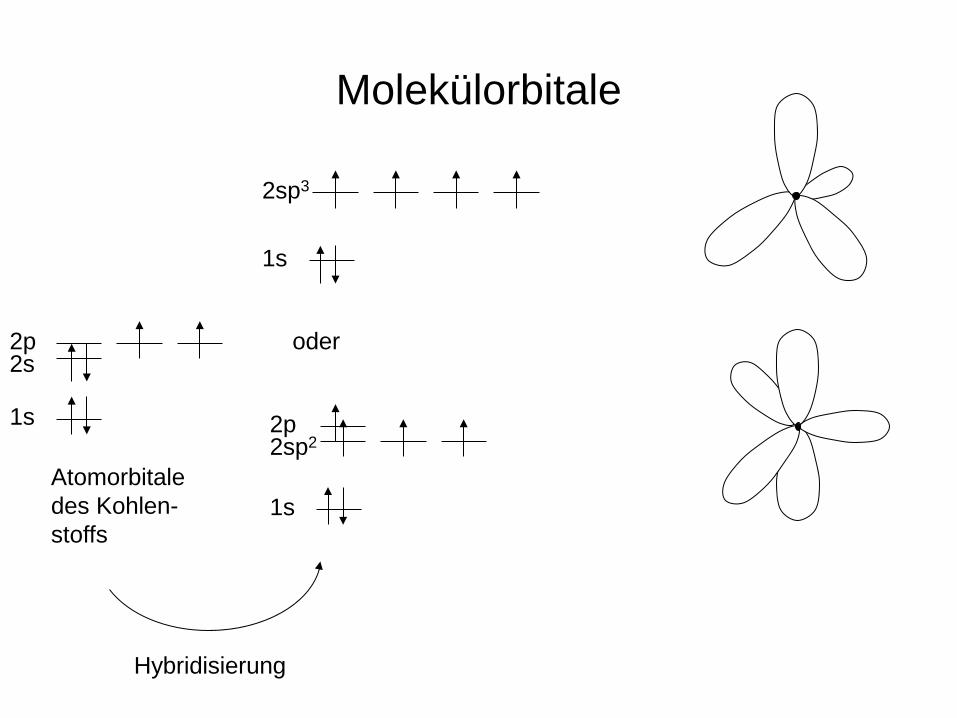
Molekülorbitale
1s
2p 2s
1s
2sp3
Atomorbitale des Kohlen- stoffs
1s
2sp2 2p
Hybridisierung
oder

Molekülorbitale
2sp2
1s
2p
1s
2sp2 2p
Molekülorbitale bindend
antibindend

Molekülorbitale und mögliche Übergänge
nicht bindend
σ
σ*
π
π*
σ
σ*
π
π*
n
Übergänge sind nur erlaubt, wenn sich Gesamtspin und Multiplizität nicht ändern, zwischen Orbitalen ungleicher Parität (gerade, ungerade), wenn sich die Orbitale ausreichend überlappen. Es kann immer nur ein Elektron angeregt werden.
HOMO
LUMO

Das Jablonski Termschema
Schwingungsniveaus
Rotationsniveaus
Auswahlregeln: Δn = ± 1 Ausreichende Überlappung
Gleiche Symmetrie

Einfache Chromophore H
H H
H
π - π∗ λmax = 162.5 nm in Heptan
ε = 16000
CH3
CH3
CH3
CH3
π - π∗ λmax = 196.5 nm in Heptan
ε = 12500
OCH3
CH3
n - π∗ λmax = 279 nm in Heptan
π − π∗
λmax = 188 nm in Heptan ε = 14, 950 CH3 CH3
σ - σ∗ λmax = 135 nm in Heptan
ε = groß
CH3 OH
n - σ∗ λmax = 177 nm in Heptan
ε = 200
CH3 Cln - σ∗
λmax = 173 nm in Hexan ε = 200
CH3 Brn - σ∗
λmax = 208 nm in Hexan ε = 260
CH3 I
n - σ∗ λmax = 259 nm in Hexan
ε = 380
Weitere Beispiele siehe Hesse, Meier, Zeeh

Konjugierte Chromophore
• Auxochrome • Polyene • Konjugierte Carbonylverbindungen • Aromaten

Definitionen
• Bathochromer Effekt (Rotverschiebung) Verschiebung eines Absorptionsmaximums zu größeren Wellenlängen
• Hypsochromer Effekt (Blauverschiebung) Verschiebung eines Absorptionsmaximums zu niedrigeren Wellenlängen
• Hyperchromer Effekt Erhöhung eines Absorptionsmaximums
• Hypochromer Effekt Erniedrigung eines Absorptionsmaximums

Auxochrome
• Funktionelle Gruppen, deren freie Elektronenpaare mit einem einfachen Chromophor konjugiert sind. Die n-Elektronenorbitale überlappen mit anderen Orbitalen.
OR NR2 Hal
OR+
-_OR

Der Einfluss von Auxochromen auf Chromophore
Absenkung der Energiedifferenz zwischen HOMO und LUMO
n π1
π2
π3
σ*
π
π*
σ
HOMO
LUMO
H
H H
H H
H
H
OMe
λmax: 162.5 nm ε: 16000
λmax: 191 nm ε: -
σ

Polyene
0.4
A
200 240 220 260 300 280 340 360 320
0.2
0.6
1.0
0.8
λ [nm]
0
380
2,4,6,8,10-Dodecapentaen
2,4,6,8-Octatetraen
H
H H
H
π - π∗ λmax = 162.5 nm in Heptan
ε = 16000
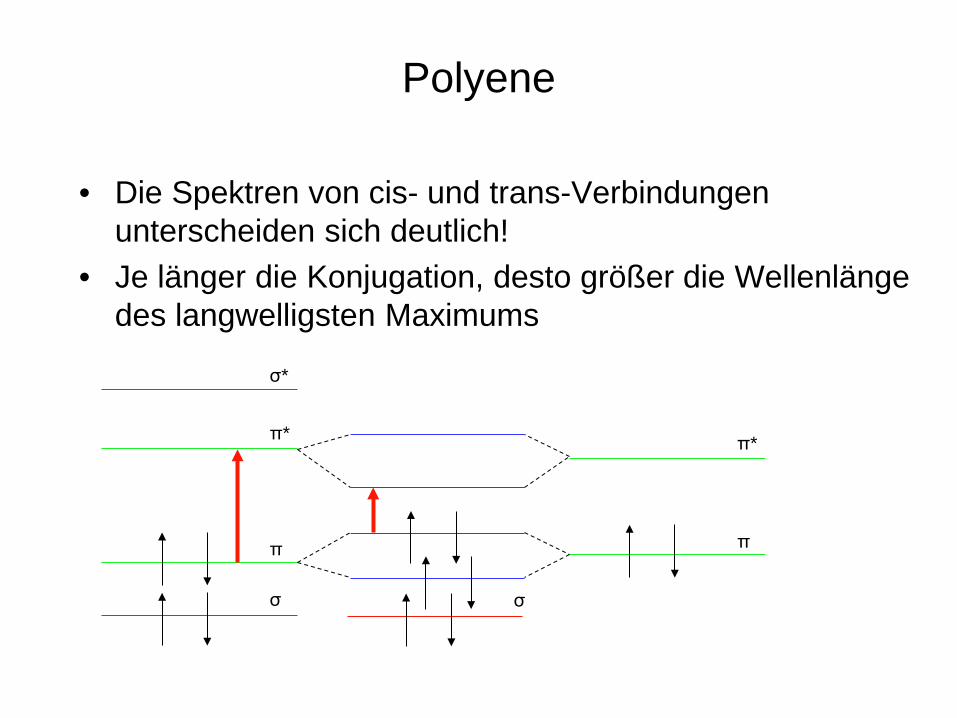
Polyene
• Die Spektren von cis- und trans-Verbindungen unterscheiden sich deutlich!
• Je länger die Konjugation, desto größer die Wellenlänge des langwelligsten Maximums
π
π*
σ
σ*
π
π*
σ
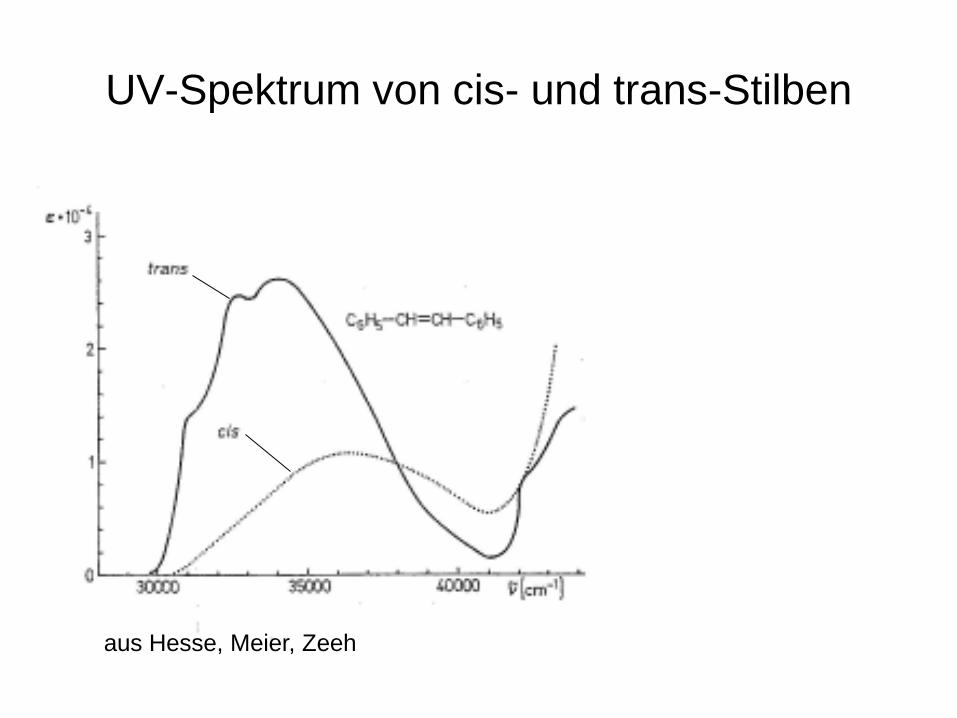
UV-Spektrum von cis- und trans-Stilben
aus Hesse, Meier, Zeeh

Regeln zur Abschätzung des langwelligsten Maximums konjugierter Olefine
Ausgangswert für ein heteroannulares bzw. acyclisches Dien: 214 nm Ausgangswert für ein homoannulares Dien: 253 nm Inkrement für einen Alkylsubstituenten: 5 nm Inkrement für eine weitere konjugierte Doppelbindung: 30 nm Inkrement für die exocyclische Lage einer Doppelbindung: 5 nm Inkremente für Auxochrome -O-Alkyl: 6 nm -O-Acyl: 0 nm -Cl, -Br: 5 nm -S-Alkyl: 30 nm -N-(Alkyl)2: 60 nm
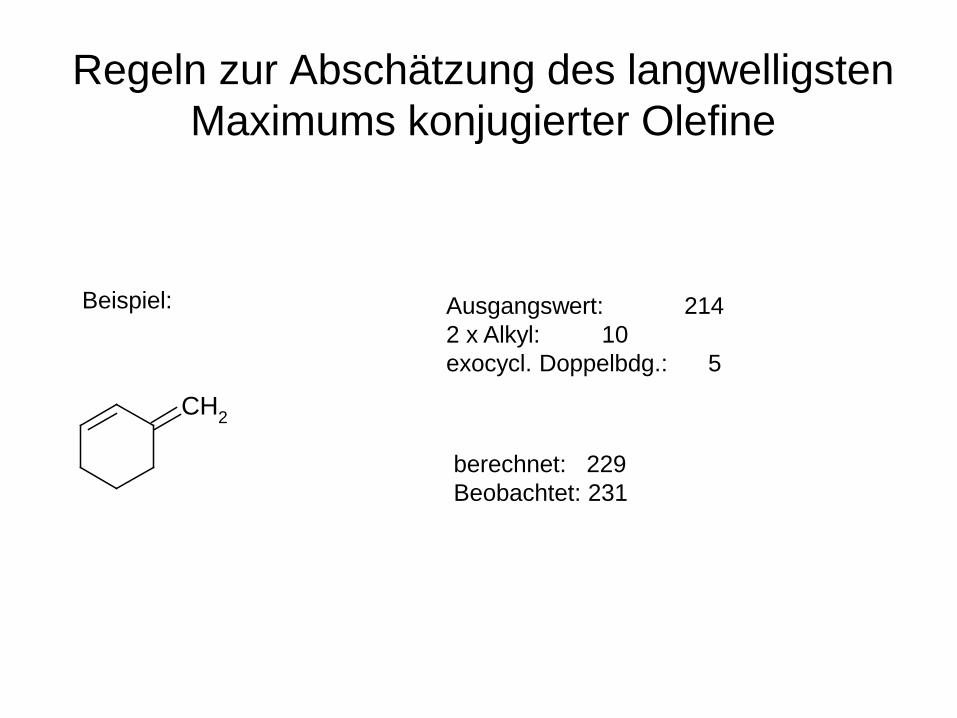
Regeln zur Abschätzung des langwelligsten Maximums konjugierter Olefine
Beispiel:
CH2
Ausgangswert: 214 2 x Alkyl: 10 exocycl. Doppelbdg.: 5
berechnet: 229 Beobachtet: 231

Konjugierte Carbonylverbindungen
Die Spektren von Carbonylverbindungen sind stark vom Lösungs-mittel abhängig (z. B. Aceton in Wasser: 265 nm, in Ethanol: 270 nm, in Dioxan: 277 nm, in Hexan: 280 nm).
σ
σ*
π
π*
π
π*
n
σ

Regeln zur Abschätzung des langwelligsten Maximums konjugierter Carbonylverbindungen
Ausgangswert für X = H: 207 nm Ausgangswert für X = alkyl oder 6-Ring: 215 nm Ausgangswert für 5-Ring: 202 nm Ausgangswert für X = OR: 193 nm Inkrement für eine weitere konjugierte Doppelbindung: 30 nm Inkrement für die exocyclische Lage einer Doppelbindung: 5 nm Inkrement für einen Alkylsubstituenten in α -Position: 10 nm Inkrement für einen Alkylsubstituenten in β -Position: 12 nm Inkrement für einen Alkylsubstituenten in γ- oder δ -Position: 18 nm Inkremente für Auxochrome -O-Alkyl in α, β, γ, δ -Position: 35, 30, 17, 31nm -O-Acyl in α, β, γ, δ -Position: 6 nm -Cl in α, β -Position: 15, 12 nm -OH in in α, β, δ -Position: 35, 30, 50 nm -N-(Alkyl)2 in β -Position: 95 nm
X
Oβ
α
δ
γ
nach Woodward bzw. Fieser; Weitere Angaben s. Hesse, Meier, Zeeh
Für Messungen in MeOH bzw EtOH
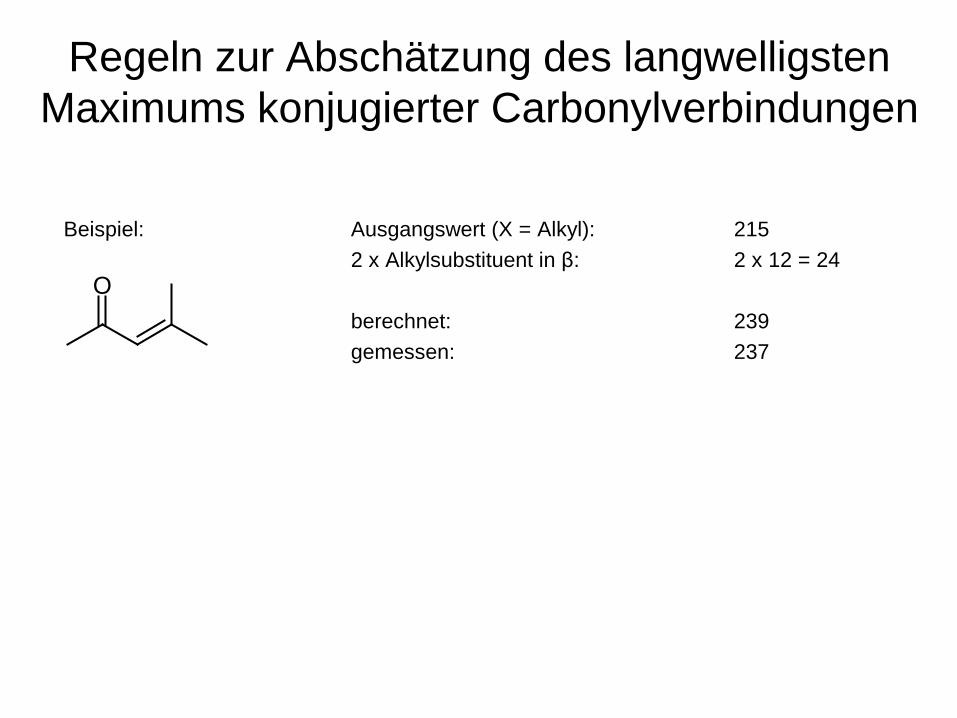
Regeln zur Abschätzung des langwelligsten Maximums konjugierter Carbonylverbindungen
Beispiel: Ausgangswert (X = Alkyl): 215 2 x Alkylsubstituent in β: 2 x 12 = 24 berechnet: 239 gemessen: 237
O
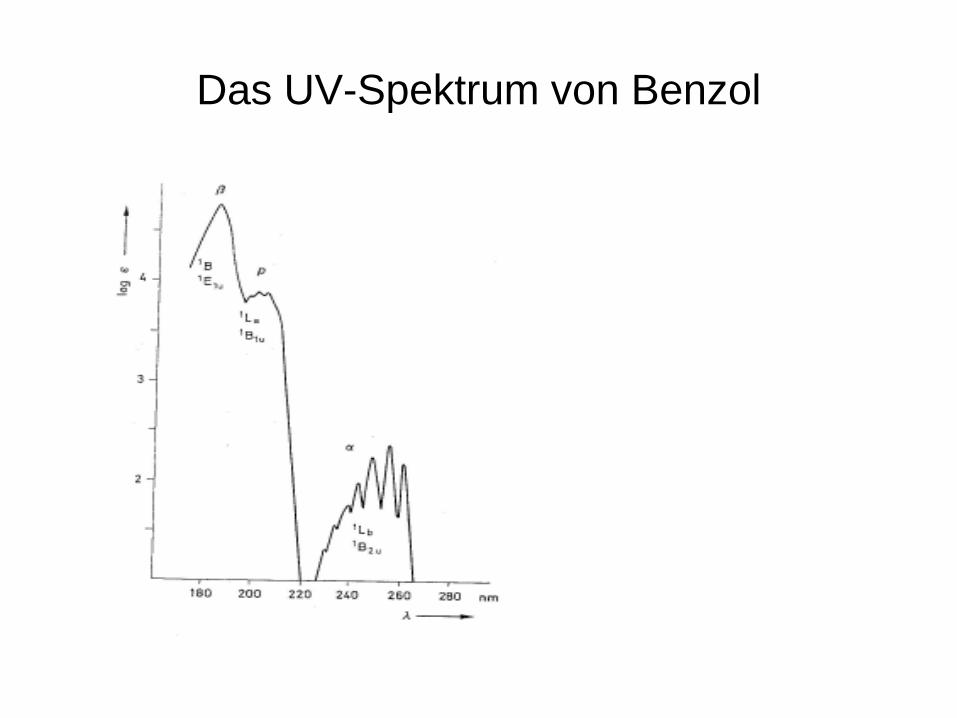
Das UV-Spektrum von Benzol

Energieniveaus des Benzols
π1
π2 π3
π5* π4
*
π6*
Nach Hückel LCAO
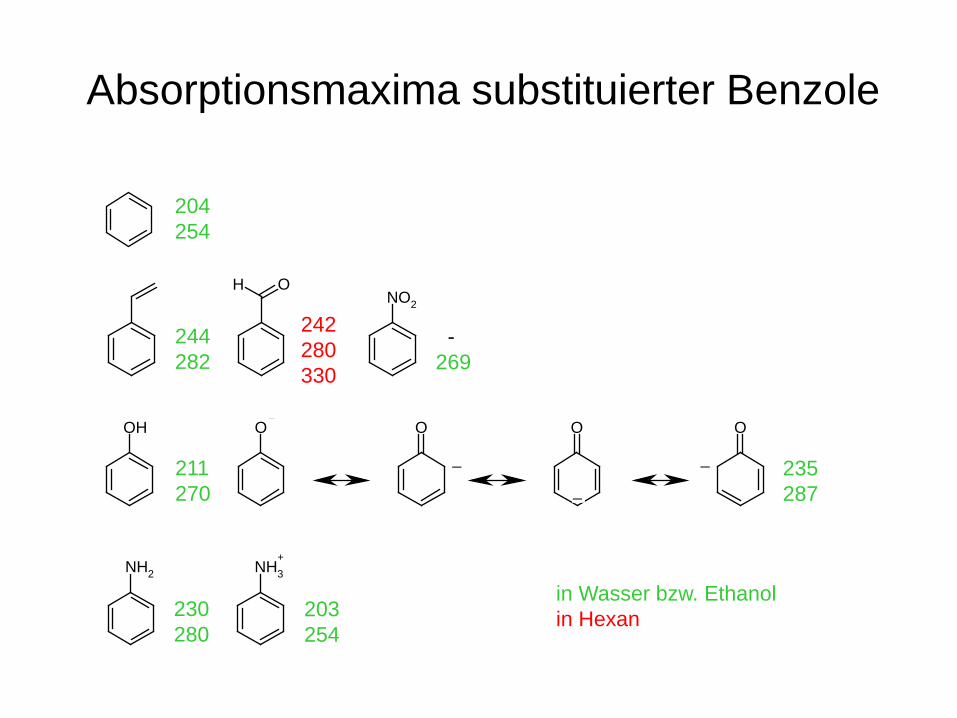
Absorptionsmaxima substituierter Benzole
204 254
OHNO2
244 282
242 280 330
- 269
in Wasser bzw. Ethanol in Hexan
OH O O O O
_
_
_211 270
235 287
NH2 NH3+
230 280
203 254

Konjugierte aromatische Verbindungen
OH
OH
OH
OH
Welche Verbindung hat ihr langwelligstes Maximum bei größter Wellenlänge?
COOH COOH204 254
230 273
230 273

Phenolphthalein OH OH
OO
O O
OO
O
COO
O
COO
OO
__
+ 2 OH
-2 H2O
-
farblos
violett

Das Lambert-Beer‘sche Gesetz
Die Absorption (A) ist proportional zur Konzentration (c) des absorbierten Stoffs und zur durchstrahlten Strecke (d). Es gilt:
A = lg(I0/I) A = k · c A = k’ · d
A = ε · c · d ε: Absorptionskoeffizient
Das Lambert-Beer‘sche Gesetz ist Grundlage der quantitativen UV/Vis-Analyse!
c = A ε · d

Quantitative UV/Vis-Spektroskopie
Erstellung einer Eichgeraden mit mindestens 2, besser mehreren Einwaagen
A
Konzentration
Gemessene Absorption
Bestimmte Konzentration
A = ε • c • d

Quantitative Bestimmungen von Mehrkompo-nenten-Gemischen durch UV-Spektroskopie
Substanz 1 Substanz 2
Gemisch aus 1 + 2
λ
1A = 1A1 + 1A2 2A = 2A1 + 2A2
Die UV-Spektren zweier Komponenten sind additiv

Für die Absorptionen bei Wellenlänge 1 (1A) bzw. 2 (2A) gilt: I: 1A = 1ε1c1d + 1ε2c2d und II: 2A = 2ε1c1d + 2ε2c2d
Löst man die Gleichungen I und II nach c1 bzw. c2 auf, so ergeben sich für die Konzentrationen c1 und c2 der beiden im Gemisch enthaltenen Verbindungen die Beziehungen:
1A - 1ε2c2d 1ε1d
III: c1 = 2A - 2ε2c2d
2ε1d IV: c1 =
1A - 1ε1c1d 1ε2d
V: c2 = 2A - 2ε1c1d
2ε2d VI: c2 =
Quantitative Bestimmungen von Mehrkomponenten-Gemischen durch UV-
Spektroskopie
1A: Absorption des Gemischs bei Wellenlänge 1 2A: Absorption des Gemischs bei Wellenlänge 2 nεm: Absorptionskoeffizient der Substanz m bei Wellenlänge n c1: Konzentration der Substanz 1 c2: Konzentration der Substanz 2 d: Schichtdicke der Probe

Aus III und IV bzw.V und VI lassen sich Gleichungen für c2 bzw. c1 herleiten, die nur noch von den aus den Messungen mit den entsprechenden Reinsubstanzen bestimmbaren Größen nεm und den Absorptionen der Analyse bei den Wellen- längen 1 und 2 abhängen.
1A2ε1 – 2A1ε1 (1ε2
2ε1 - 2ε21ε1)d c2 =
Quantitative Bestimmungen von Mehrkomponenten-Gemischen durch UV-
Spektroskopie
1A2ε2 – 2A1ε2 (1ε1
2ε2 - 2ε11ε2)d c1 =
Wird immer mit der selben Küvette gemessen, so dass d zu 1 gesetzt werden kann! ε muss mit Hilfe einer entsprechenden Standardlösung bestimmt werden.
1A: Absorption des Gemischs bei Wellenlänge 1 2A: Absorption des Gemischs bei Wellenlänge 2 nεm: Absorptionskoeffizient der Substanz m bei Wellenlänge n c1: Konzentration der Substanz 1 c2: Konzentration der Substanz 2 d: Schichtdicke der Probe [cm]

Fluoreszenz-Spektroskopie
Fluoreszenz: Die Emission von Licht auf Grund des Übergangs eines Elektrons aus einem angeregten Singulett-Zustand in den Grundzustand.
Die Fluoreszenz-Spektroskopie ist eine emissionsspektroskopische Methode. Es können Atome und Moleküle zur Fluoreszenz angeregt werden.
Phosphoreszenz: Die Emission von Licht auf Grund des Übergangs eines Elektrons aus einem angeregten Triplett-Zustand in den Grundzustand.
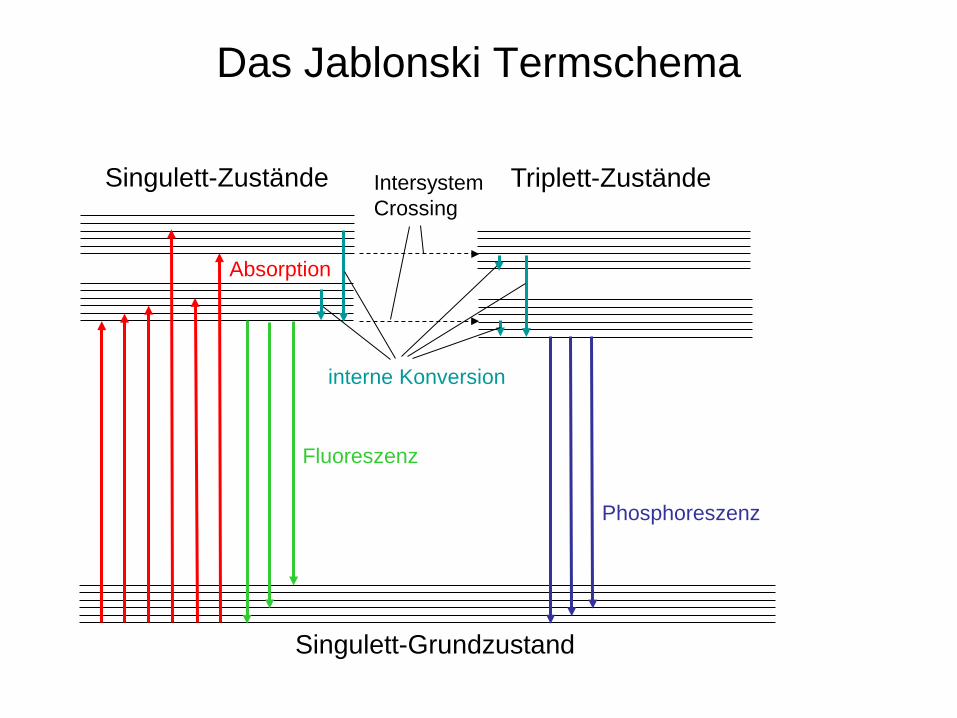
Das Jablonski Termschema
Singulett-Zustände Triplett-Zustände
Singulett-Grundzustand
Fluoreszenz
Phosphoreszenz
Absorption
interne Konversion
Intersystem Crossing

Fluoreszenz
Absorption

Fluoreszenz
interne Konversion

Fluoreszenz
Fluoreszenz

Phosphoreszenz
Absorption

Phosphoreszenz
interne Konversion
Phosphoreszenz
Intersystem Crossing
Phosphoreszenz
interne Konversion

Phosphoreszenz
Phosphores- zenz

Wechselwirkungen von Licht mit Lösungen
I0 I Absorption
Fluoreszenz
Streuung
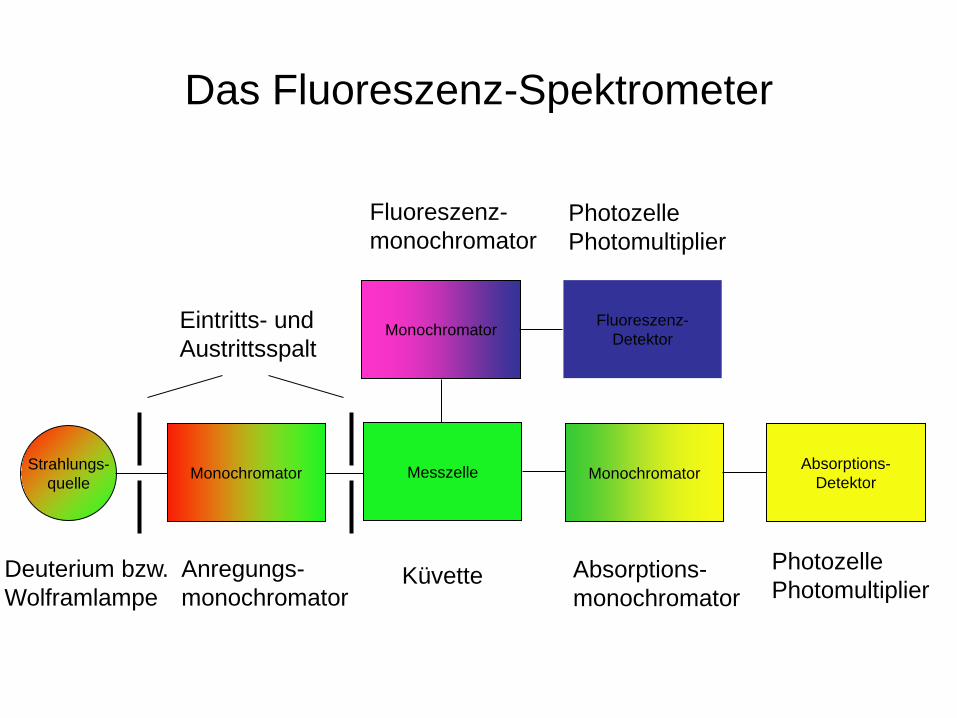
Das Fluoreszenz-Spektrometer
Absorptions- Detektor
Photozelle Photomultiplier
Deuterium bzw. Wolframlampe
Strahlungs- quelle Monochromator
Anregungs- monochromator
Eintritts- und Austrittsspalt
Küvette
Messzelle Monochromator
Absorptions- monochromator
Fluoreszenz- Detektor
Photozelle Photomultiplier
Monochromator
Fluoreszenz- monochromator

Absorptions- und Emissionsspektrum von Tryptophan
λ 240 280 320 360 400
I
200
Absorption
Emission

Fluoreszierende Verbindungen
Alle fluoreszierenden Verbindungen sind hoch konjugiert, weitgehend starr und planar gebaut.
O O
Cumarin
O
COOH
OH
Fluorescein
O
O
O
O O
OMe
Aflatoxin B1
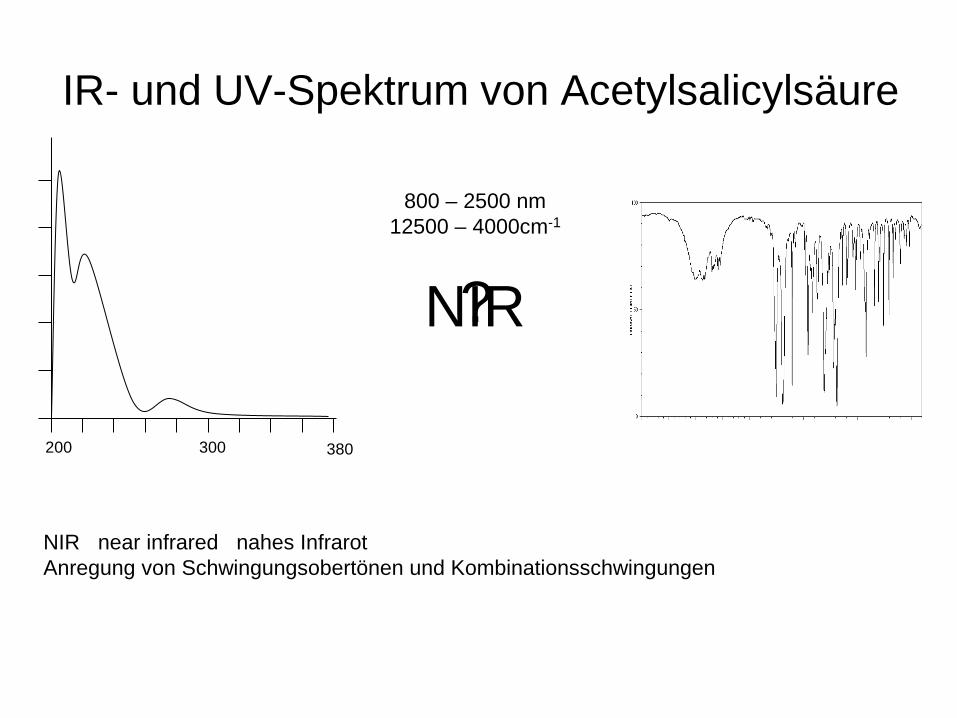
IR- und UV-Spektrum von Acetylsalicylsäure
380 200 300
? NIR
NIR near infrared nahes Infrarot Anregung von Schwingungsobertönen und Kombinationsschwingungen
800 – 2500 nm 12500 – 4000cm-1

Vor- und Nachteile der NIR-Spektroskopie
Vorteile: Es können Bauteile aus Quarz verwendet werden – leichte Handhabbarkeit. Verwendung von Lichtleitern ist möglich. Messung ohne besondere Proben- vorbereitung.
Nachteile: Die Banden haben geringe Intensität und überlappen stark – schwierige Kalibrierung.
Die meisten NIR-Spektren werden in Reflektionstechnik gemessen. Beispiel: ATR

Anwendungen der NIR-Spektroskopie
Eingangs- und Prozesskontrolle in der Pharma- und Lebensmittelindustrie. Z. B. Proteingehalt von Getreide Wassergehalt von Hilfsstoffen wie Talkum etc. Quantitative Bestimmung von Wirkstoffen in Anwesenheit der Hilfstoffe.

Die Skalierung
Fragmentionen
Basepeak
Das Massenspektrum Beispiel: Cumol
Molekülion
Isotopenpeak
Spektrum aus: http://riodb01.ibase.aist.jp/sdbs/

Was kann die Massenspektrometrie?
Bestimmung der Molmasse (Molekülion) Bestimmung der Elementarzusammensetzung (Isotopenmuster, hochauf- gelöste Massenspektrometrie) Strukturaufklärung, Hinweise auf funktionelle Gruppen und Partialstrukturen (Zerfallsmuster) Charakterisierung einer Substanz Nachweis und Identifikation einer Substanz (höchste Empfindlichkeit!)

Aufbau eines Massenspektrometers
Ionisator Analysator Detektor
Signal
Datenverarbeitung
Hochvakuum 10-5 – 10-8 torr
Probeneinlass

Probeneinlass
Vakuumschleuse (Schubstange)
GC-MS (Separatoren)
LC-MS (ESI. APCI, APPI)

Ionisationsmethoden
Beschuss der Substanz mit Teilchen oder Wellen Elektronenstoß (EI-MS) Atome (FAB-MS) Ionen (SI-MS) Laserstrahlung (MALDI) Einbringen der Substanz in ein ionisierendes Medium Chemische Ionisation Feldionisation, Felddesorption
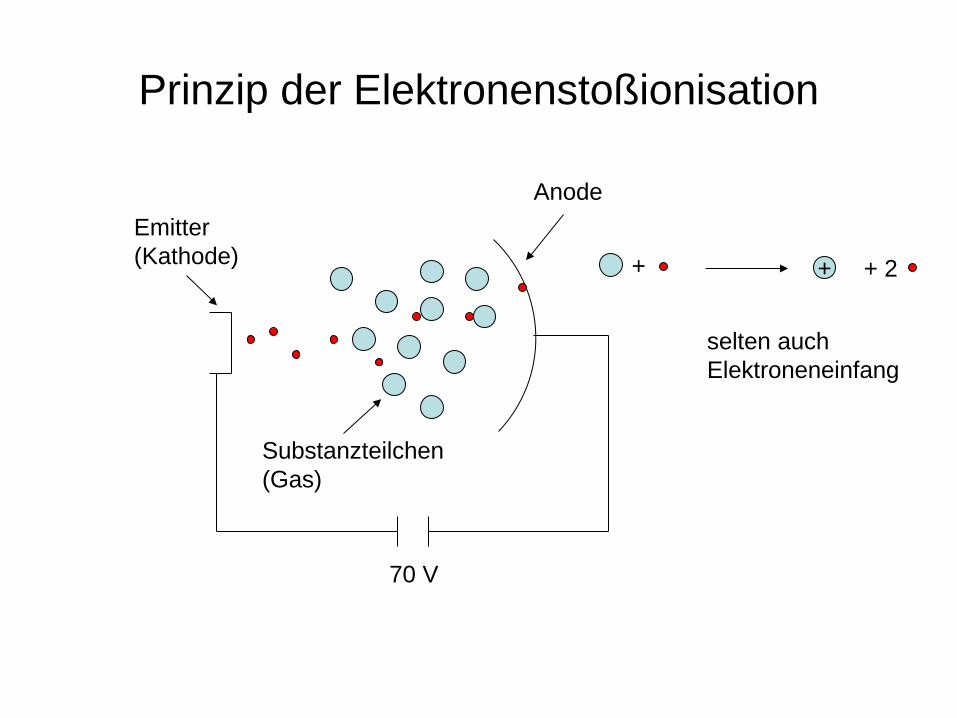
Prinzip der Elektronenstoßionisation
Emitter (Kathode)
70 V
Anode
+ + + 2
selten auch Elektroneneinfang
Substanzteilchen (Gas)

Chemische Ionisation
Prinzip:
1. Gas + e- Gas+ + 2e-
2b. Gas+• + M M+• + Gas (Redox-Typ)
2a. Gas+ + M MH+ + Gas - H (Säure-Base-Typ)
Als Reaktand-Gase kommen in Frage: Methan; iso-Butan, Ammoniak, Lachgas, …….

Prinzip der FAB-Ionisation
Ar(Xe)-Atome
Ionisierte Matrix- und Substanzmoleküle
Matrix für FAB und SIMS: Glycerin, DMSO,...... (ausreichende Flüchtigkeit der Matrix)

Prinzip der MALDI-Ionisation Matrix assisted laser desorption ionisation
Laser-Strahlung
Ionisierte Matrix- und Substanzmoleküle
Matrix für MALDI: Benzoesäure- und Zimtsäureester

Analysatoren
Sektorfeld-Analysator
Quadrupol-Analysator
Ionenfalle
Flugzeit-Analysator
Ionencyclotron

Der doppelfokussierende Sektorfeld-Analysator
Ionisator
Detektor
elektrisches Feld
Magnetfeld
Fokussierungs-spalte
Im elektrischen Feld gilt: z • U = mv2 1
2
Im Magnetfeld gilt: m • v
r = z • B
m z = r2 • B2
2U
z: Ionenladung; m: Ionenmasse; v: Ionengeschwindigkeit; r: Ablenkradius; U: Beschleunigungsspannung; B: Magnetfeldstärke

Der Quadrupol-Analysator
+
+ -
- -
- +
+
Ionenquelle Detektor
Trennung im mit einem Radiofrequenzfeld, das von einem Gleichstrom überlagert wird.

Die Ionenfalle Dreidimensionale Variante des Quadrupols
Ionenquelle Detektor
Ringelektrode
Endkappenelektroden

Der Flugzeit-Analysator
Ionenquelle
Reflektor
Detektor
2U m/z = v2
Es müssen alle Ionen die gleiche kinetische Energie besitzen.
U: Beschleunigungsspannung

Interpretation von Massenspektren Allgemeine Regeln
Bei der EI-Ionisation entsteht ein Radikal-Kation Ein Radikal-Kation zerfällt: in ein Kation und ein Radikal in ein Radikal-Kation und ein Neutral-Teilchen Ein einmal gebildetes Kation zerfällt in ein Kation und ein Neutral-Teilchen (nicht in ein Radikal-Kation und ein Radikal!) Zerfälle bei denen sich möglichst stabile Teilchen bilden sind bevorzugt. Massenspektrometrische Zerfälle sind monomolekulare Reaktionen des Typs:
RK + • K + + R •
RK‘ RK + • + N + •
+ K‘ + N K +

Interpretation von Massenspektren Allgemeine Regeln
Welche Zerfälle ablaufen richtet sich ausschließlich nach der freien Aktivierungs- enthalpie. Es wird davon ausgegangen, dass Elektronen aus dem höchsten besetzten Orbital (HOMO) entfernt werden. Die Zerfallsreaktionen sind radikal- oder ladungsinduziert.

Interpretation von Massenspektren Zerfallsmechanismen
Radikalinduzierte Zerfälle
+ •
+ •
R CR2 XR
R CR X
R2C XR
RC X
R +
R +
+
+
•
•
α-Zerfall: Ether, Amine, Ketone, Olefine (= Allyl-Spaltung)
+ • + R CR2 CR CR2 CR2 CR CR2R + •

Interpretation von Massenspektren Zerfallsmechanismen
Ladungsinduzierte Zerfälle
+
+
R X R X R
RX
RRC X
R X R X R
R +
R +
R +
•
•
+
+ •
+ •
+

Interpretation von Massenspektren Zerfallsmechanismen
Umlagerungen Es wird vorwiegend Wasserstoff umgelagert.
•
+ X
RH X
RH+ •

Interpretation von Massenspektren Beispiele
Benzylspaltung CH2R - R.
+
.+
O
R
O
- R.
.+
N RR
.+- R. N
R
OH
- R.H2O
.+.+
+
+
α-Spaltung
Abspaltung von Wasser oder ähnlichen Neutralteilchen

Interpretation von Massenspektren Mechanismus der Wasser-Abspaltung
OHR
R
HO
HR
R
H R
R
+ • + • •
+

Interpretation von Massenspektren Beispiele
McLafferty-Zerfall
X
ORH .+
X
OH .+
R
X
ORH .+
X
OH
R .+
.+ .+
.+ .+
+
+
+
+
Retro-Diels-Alder-Zerfall

•
+
+ •
Interpretation von Massenspektren Mechanismus der McLafferty-Umlagerung
+ •
+
•
OHR
R
OHR
R
R
RO
H
R
OH
R
+
+
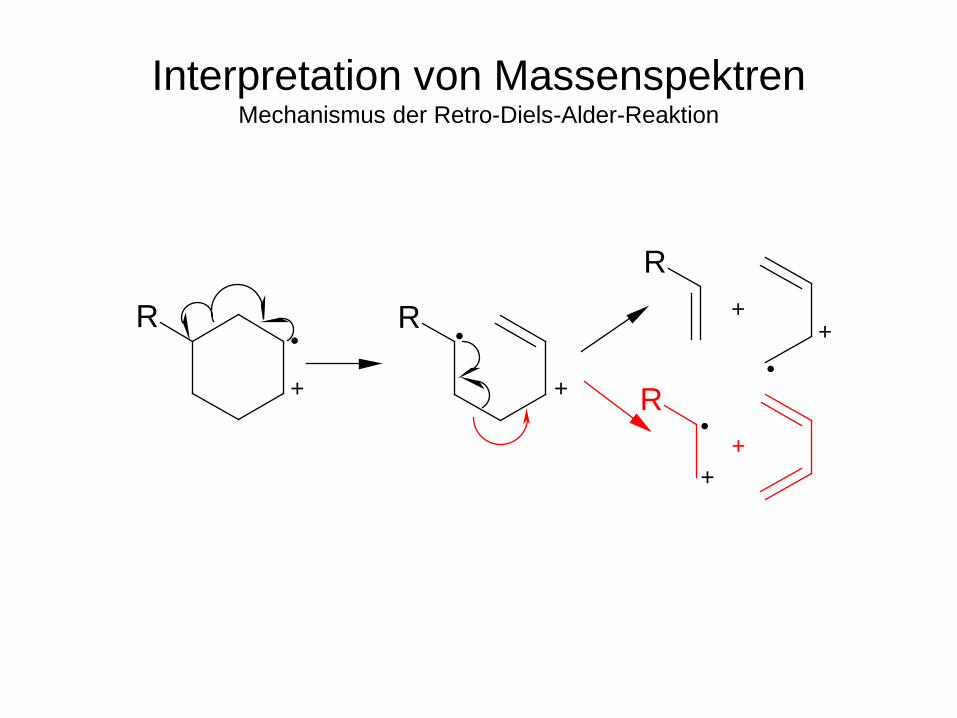
Interpretation von Massenspektren Mechanismus der Retro-Diels-Alder-Reaktion
+
+
• • R R
R
R+
+
+ •
+
•
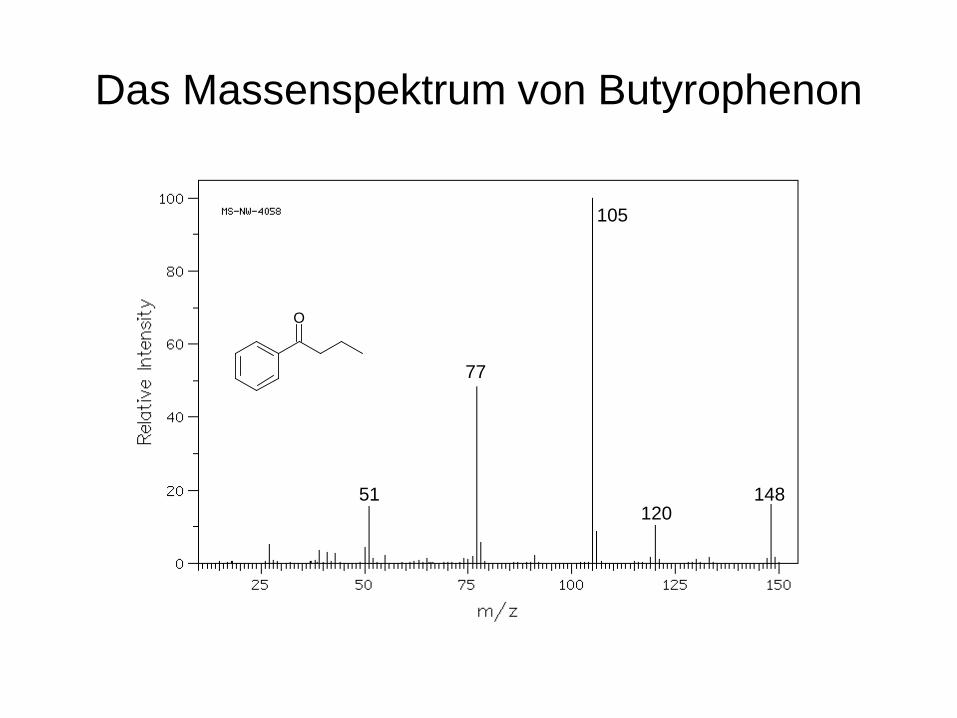
Das Massenspektrum von Butyrophenon
105
120 148
77
51
O

Der massenspektrometrische Zerfall von Butyrophenon
O
OO+.
+.
+
+ +
- C2H4 - C3H7
- C2H2
- CO
-COC3H7
m/z 148
m/z 120
m/z 77
m/z 105
m/z 51

GC-MS
Problem: Überdruck im GC – Hochvakuum im MS Problemlösung: Geeignete Separatoren (Jet-Separator, Watson-Bieman-Separator) Alle Ionisationstechniken die gasförmige Moleküle ionisieren (EI, CI) werden eingesetzt.

Der Jet-Separator
leichte Teilchen (Trägergas)
Zur Vakuumpumpe
vom GC zum MS
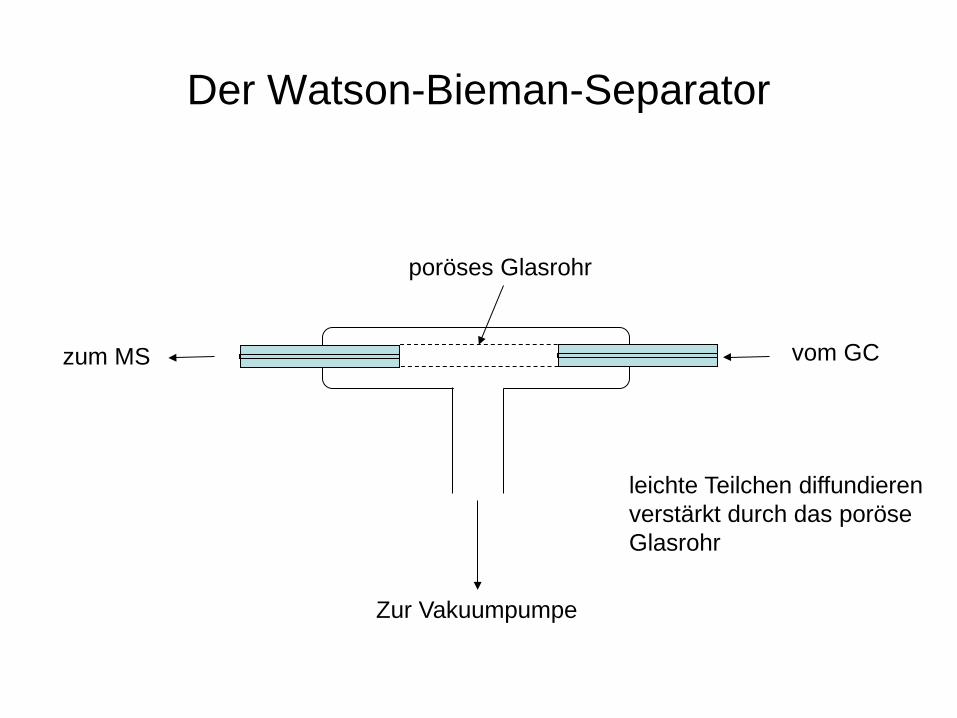
Der Watson-Bieman-Separator
vom GC zum MS
poröses Glasrohr
Zur Vakuumpumpe
leichte Teilchen diffundieren verstärkt durch das poröse Glasrohr

LC-MS
Problem: Überdruck in der LC-Anlage – Hochvakuum im MS Problemlösung: Ionenerzeugung bei Normaldruck außerhalb des MS (Atmospheric Pressure Ionization API; z. B.: ESI, APCI, APPI)
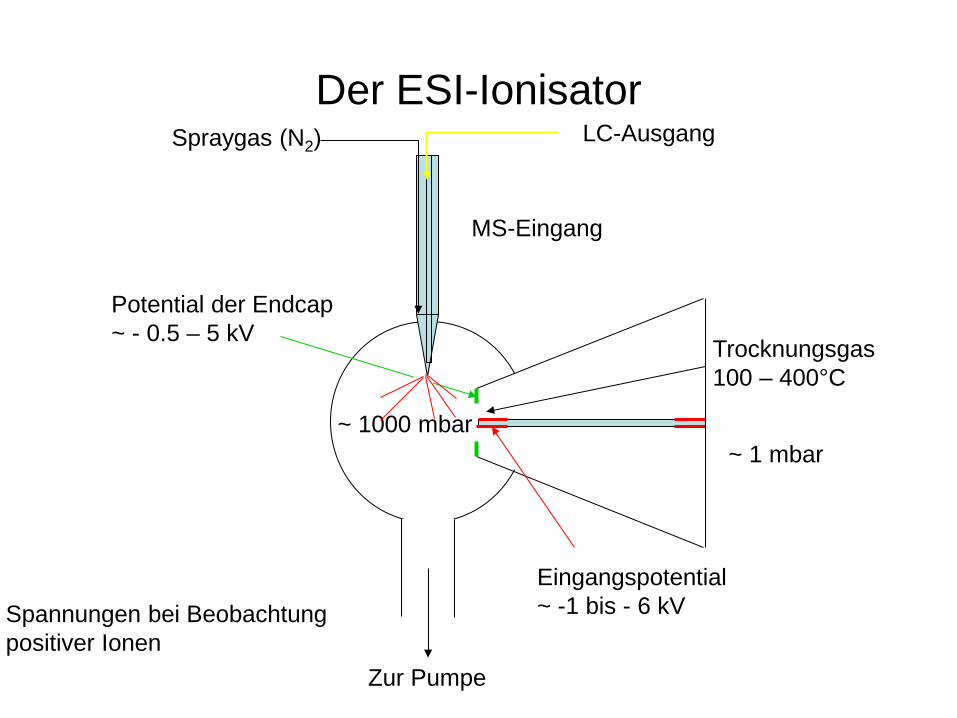
Der ESI-Ionisator
Zur Pumpe
MS-Eingang
Trocknungsgas 100 – 400°C
~ 1000 mbar ~ 1 mbar
Eingangspotential ~ -1 bis - 6 kV
Potential der Endcap ~ - 0.5 – 5 kV
Spannungen bei Beobachtung positiver Ionen
Spraygas (N2) LC-Ausgang

Ionenbildung im ESI-Ionisator
Coulomb-Explosion Eingangskapillare
Trocknungsgas
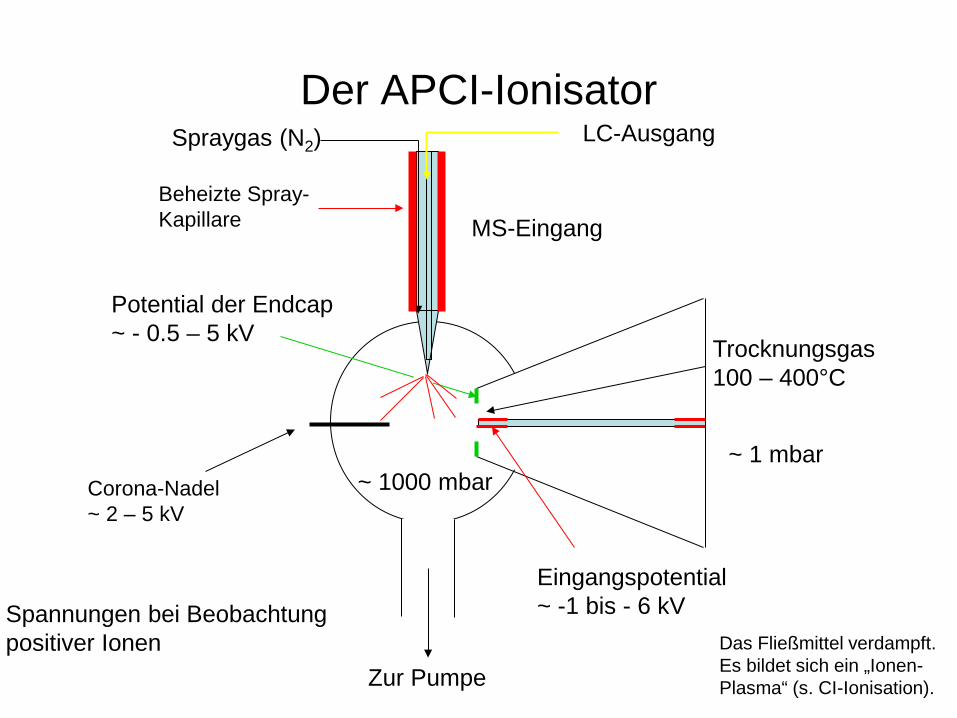
Der APCI-Ionisator
Zur Pumpe
MS-Eingang
Trocknungsgas 100 – 400°C
~ 1000 mbar ~ 1 mbar
Eingangspotential ~ -1 bis - 6 kV
Potential der Endcap ~ - 0.5 – 5 kV
Spannungen bei Beobachtung positiver Ionen
Spraygas (N2) LC-Ausgang
Corona-Nadel ~ 2 – 5 kV
Beheizte Spray- Kapillare
Das Fließmittel verdampft. Es bildet sich ein „Ionen- Plasma“ (s. CI-Ionisation).

Hochauflösende Massenspektrometrie
Die Massen der Ionen werden mit hoher Genauigkeit bestimmt. Nicht alle Analysatoren sind dazu in der Lage. Beispiel: Unterscheidung von Verbindungen gleicher Nominalmassen (m/z 28). 12C: 12.000000 1H: 1.007825 14N: 14.00307 16O: 15.99491 12C=16O: 27.99492, 14N2: 28.00615, 12C2
1H4: 28.03130 Beispiel: Bestimmung der Elementarzusammensetzung. Gemessen wurde eine exakte Masse von 374.2457 Bei einer möglichen Abweichung von ± 5 ppm wurden für Verbindungen, die nur aus C, H, N und O bestehen folgende Elementarzusammensetzungen berechnet: C26H32NO 374.2484 C24H30N4 374.2470 C23H34O4 374.2457 C21H32N3O3 374.2444 C19H30N6O2 374.2430

Isotopenmuster der MS-Peaks Die Verteilung der Isotope eines Elements ist im Massenspektrum erkennbar und wird zur Ermittlung der Elementarzusammensetzung herangezogen. Beispiel: Wie viele Chloratome befinden sich in einem Ion? Chlor besteht aus den Isotopen 35Cl (76%) und 37Cl (24%) entsprechend einem Verhältnis von 1 : 0.32.
35Cl
37Cl CH2Cl2 CHCl3
2 35Cl
1 35Cl + 1 37Cl
2 37Cl
Beispiel: Bestimmung der Elementarzusammensetzung mit Hilfe der Isotopenpeaks. Aus der relativen Häufigkeit der natürlich vorkommenden Isotope von C, H, N und O können die theoretischen Verhältnisse der Peaks des Molekülionen-Clusters berechnet werden. 12C : 13C = 100 : 1.112; 1H : 2H = 100 : 0.015; 14N : 15N = 100 : 0.367; 16O : 17O :18O = 100 : 0.038 : 0.200 Für Aceton (Nominalmasse 58) ergibt sich M+ : (M+1)+ = 100 : 3.37, für Butan (Nominalmasse 58) ergibt sich M+ : (M+1)+ = 100 : 4.47. Je höher der Anteil an C, desto größer (M+1)+; je höher der Anteil an O, desto größer (M+2)+.

Massenspektren verschiedener Dodekane
Molekülion M+ m/z 170
Molekülion M+ m/z 170
Molekülion M+ m/z 170
n-Dodekan
3-Methylundekan
2-Methylundekan
Spektren aus: http://riodb01.ibase.aist.jp/sdbs/
Die Spektren verschiedener Verbindungen unterscheiden sich, auch bei großer struktureller Ähnlichkeit, deutlich. Es wird ein Signal bei der Masse des Moleküls beobachtet. Es treten Molekül-charakteristische Signale auf.

Geschichte der NMR-Spektroskopie
• 1924 W. Pauli postuliert, dass Atomkerne einen Kernspin (I) und damit ein magnetisches Moment µ besitzen.
• 1946 Experimenteller Nachweis des NMR-Prinzips an kondensierter Materie (Wasser, Paraffin) durch F. Bloch und E. M. Purcell (Nobelpreis für Physik, 1952).
• 1951 Das erste hochauflösende 1H NMR-Spektrum wird gemessen (Arnold et al., “Entdeckung der chemischen Verschiebung”).
• 1953 Das erste kommerzielle NMR-Gerät wird von der Firma Varian präsentiert.
• 1957 Das erste 13C NMR-Spektrum wird von P. C. Lauterbur publiziert.
• 1966 R. R. Ernst entwickelt die FT NMR-Spektroskopie (Nobelpreis für Chemie 1991)

• 1973 Ein bilderzeugendes Verfahren auf der Basis der Kernspin- resonanz wird von P. C. Lauterbur (Nobelpreis 2003) präsentiert.
• 1976 R. R. Ernst etabliert die ersten zweidimensionalen NMR- Experimente (ursprünglich schon 1971 von J. Jeener vorgeschlagen).
• 1980 Die multidimensionale NMR-Spektroskopie wird möglich. • 1985 Inverse Detektion erhöht die Empfindlichkeit, gepulste
Feldgradienten verkürzen die Messzeit für die Aufnahme mehrdimensionaler NMR-Spektren.
• Stetige Weiterentwicklung der Magnet- und Aufnahmetechnik führen zu erheblichen Empfindlichkeitssteigerungen.

NMR-Spektroskopie Nuclear Magnetic Resonance Spectroscopy Kernmagnetische Resonanzspektroskopie
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
Zur Strukturaufklärung unbekannter Verbindungen bis zur (absoluten) Konfiguration.
Wichtigste spektroskopische Methode in der organischen Chemie
Zur eindeutigen Charakterisierung von Verbindungen.
Zur Bestimmung intra- und intermolekularer Wechselwirkungen. Zur Ermittlung von Reaktionsgeschwindigkeiten. Zur Beobachtung chemischer Austauschprozesse.
Zur Konformationsanalyse.
Zur Beobachtung chemischer Reaktionen.

Spektren von Haloperidol
IR
UV
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
19.1
3
51.7
6
3.25
19.3
9
6.48
1H NMR
200 180 160 140 120 100 80 60 40 20 0 ppm
-0.2
00.
000.
20
21.9
835
.58
37.7
138
.98
39.1
439
.31
39.4
839
.64
39.7
439
.81
39.9
039
.98
40.0
748
.80
57.2
069
.43
115.
4011
5.57
126.
6512
7.56
130.
6213
0.73
130.
8113
3.88
133.
90
149.
13
163.
7416
5.74
198.
18
13C NMR

Prinzip der NMR-Spektroskopie
Atomkerne besitzen einen Kernspin und damit verbun- den ein magnetisches Moment, das mit einem externen Magnetfeld in Wechselwirkung treten kann. Diese Wechselwirkungen folgen den Regeln der Quantenmechanik. Für die Kernspins sind nur bestimmte
Einstellungen definierter Energie möglich (für 1H und 13C z. B. nur 2, je einmal parallel bzw. antiparallel zur Feldachse).
Die Energien hängen von der Stärke des Magnetfelds und von
der Art des Kerns ab (E = -m γ h/2π B0). Durch Absorption einer geeigneten Energie können die
Kernspins aus dem Zustand niedrigerer Energie in den Zustand höherer Energie angeregt werden. Zur Anregung werden elektromagnetische Wellen mit Frequenzen zwischen 5 und 1000 MHz eingesetzt.

Theoretische Grundlagen der NMR-Spektroskopie
Der Hamilton-Operator für die Wechselwirkung eines Kernspins I mit einem Magnetfeld B0 lautet: Ĥ = -γ B0Îz
γ, die gyromagnetische Konstante (gyromagnetisches Verhältnis) ist eine für jeden Kern charakteristische Konstante.
Der Operator Îz repräsentiert die Komponente des Kernspins in Richtung des Magnetfelds B0 (z). Îz ist mit der Spinquantenzahl I verknüpft, die die Werte I = 0, 1/2, 1, 3/2,........7 annehmen kann.
Die Lösung der Schrödinger-Gleichung ergibt als Energie-Eigenwerte:
E = -mħγB0
m ist die magnetische Quantenzahl. Sie kann die Werte -I, -I+1 bis +I annehmen.

Theoretische Grundlagen der NMR-Spektroskopie
Für Kerne mit I = 1/2 sind also zwei (+1/2, -1/2), für Kerne mit I = 1 drei (+1, 0, -1) usw. Energiezustände möglich. Die Anregungsenergie (∆E) zwischen zwei Spinzuständen ist abhängig von der Stärke des äußeren Magnetfelds (B0) und der gyromagnetischen Konstante (γ). ∆E = ħγB0 Für 1H (γ = 2.67522 ∙ 108 rad s-1T-1) ergibt sich in einem Magnetfeld von 9.4 Tesla eine Energie von ca. 400 MHz (υ = ω/2π).

1H NMR-Spektren von Ethylacetat bei verschiedenen Feldstärken
PPM 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
90.130000 90.130360
9.4 Tesla
2.1 Tesla
14.1 Tesla
400.131600 400.130000
600.130000 600.132400
MHz

Magnetische Eigenschaften einiger Kerne
γ [107 rad s-1 T-1] I ∆E bei 11.7467 T Besetzungsverhältnis bei 300 K und 11.7467 T
1H 26.75222 1/2 500.130 MHz 1 : 1.00008
2D 4.10663 1 76.773 MHz
12C 0
13C 6.72829 1/2 125.758 MHz 1 : 1.00002
14N 1.93377 1 36.141 MHz
15N -2.71262 1/2 50.697 MHz 1 : 1.000008
16O 0
19F 25.16233 1/2 470.592 MHz
31P 10.83941 1/2 202.456 MHz

Klassische Beschreibung des NMR-Experiments
Ohne Magnetfeld Die Kernspins sind ungerichtet, ihre magnetischen Momente summieren sich zu null.
Mit Magnetfeld Die Kernspins orientieren sich parallel bzw. antiparallel zur Feldrichtung, um die sie mit der Larmor-Frequenz rotieren. Die energetisch günstigere, parallele Anordnung ist stärker besetzt.
z
y x
B0
Es bleibt eine Magnetisierung parallel zur Achse des äußeren Magnetfelds. (longitudinale Magnetisierung)
Vor der Anregung

Klassische Beschreibung des NMR-Experiments
z
y x
B0 Die Magnetisierung wird durch ein elektromagnetisches Feld (Puls) in die x-y-Ebene gedreht, wo sie mit der Larmor-Frequenz um B0 präzediert. Dabei wird in der Empfängerspule ein Strom (FID) induziert.
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
Fourier-Transformation ergibt daraus das NMR-Spektrum
FT
nach der Anregung

Wie ist ein NMR-Spektrometer aufgebaut?
Signal Spektrum
Sender Empfänger
Computer

Wie kommt das NMR-Spektrum zu Stande?
B B
Anregung
Relaxation
FID
Messung
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
Fourier- transformation
Ungeordnete Kernspins

1H NMR-Spektrum von Haloperidol
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm

Inhalt des 1H NMR-Spektrums
Integral Signalintensität
Chemische Verschiebung 345 ppm
Multiplizität
ppm

1H NMR-Spektrum von Haloperidol
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm

Das Integral im NMR-Spektrum
In der Regel werden nur 1H NMR-Spektren integriert
Die Stufenhöhe ist ein Maß für die Fläche unter dem jeweiligen Signal, diese ist proportional zur Menge der Wasserstoffe, die das Signal erzeugen.
-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm
2 6
1 4
1 6
7
Zur Bestimmung der Anzahl Wasserstoffe im Molekül
Zur Quantifizierung
Zur Abschätzung der Reinheit einer Probe

Energieniveaus zweier I = ½ Kerne (1H..)
-½ ħγB0
+½ ħγB0
E = hν = -m ħγB0
+½ ν0 [Hz]
-½ ν0 [Hz]
ν0: Larmorfrequenz ν0: = -γB0/2π
Beff = B0 – B0σ
Beff = B0 (1 – σ)
-½ ħγ Beff
+½ ħγ Beff

Die Skala des NMR-Spektrums
υSubst - υStand υStand
δ = 106
Um die in verschiedenen Magneten gemessenen Spektren vergleichen zu können, wird eine relative, frequenzunabhängige Skala definiert.
Der Nullpunkt der Skala wird durch einen internen (oder externen) Standard fest- gelegt.
CH3 SiCH3
CH3CH3Tetramethylsilan (TMS)

Anforderungen an einen internen Standard
chemische Inertheit hohe Signalintensität schmale Signalform geeignete Signallage leichte Entfernbarkeit

Die chemische Verschiebung
Es gilt: Beff = B0 – σ • B0 (σ: Abschirmungskonstante)
Verschiedene Faktoren verändern das Magnetfeld am Ort des Kerns
Für Wasserstoff hängt die chemische Verschiebung im Wesentlichen ab:
B0 Von der Elektronendichte
Von Anisotropie und Ringstromeffekten
Von Substitutionsgrad und Stereochemie
Von Lösungsmittel, Temperatur, pH-Wert und Konzentration

Die chemische Verschiebung Abhängigkeit von der Elektronendichte
0.23 4.27 3.06
HH
HH H
H
HF H
H
HCl H
H
HBr H
H
HI
HH
ClCl
HH
HNH2 H
Cl
ClCl
HH
HOH
2.69 2.16
3.39 5.33
7.26 2.87
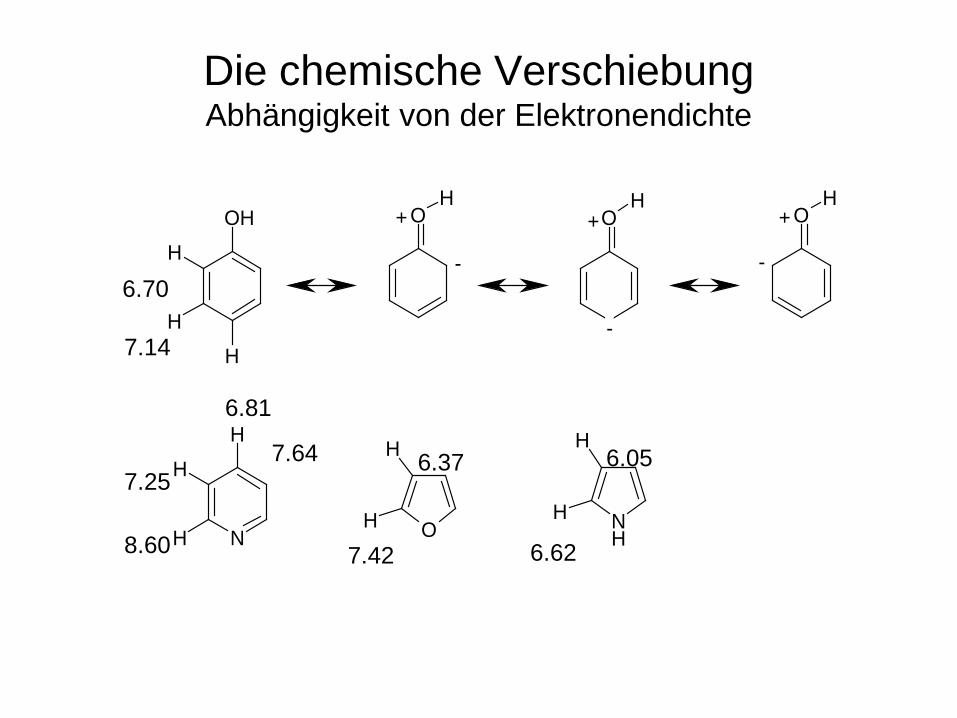
Die chemische Verschiebung Abhängigkeit von der Elektronendichte
NH
HH
OH
H
NH
H
H
OHH
HH
OH
+
-
OH
-
+OH
-
+
7.42
6.37
6.62
6.05
8.60
7.25 7.64
6.70
7.14
6.81

Die chemische Verschiebung Abhängigkeit von der Elektronendichte
aber!
Cl CH3 Br CH3 I CH3
3.47
1.33
3.37
1.66
3.16
1.88

Die chemische Verschiebung Abhängigkeit von Anisotropie- und Ringstromeffekten
O
H
H
H
HH H
1.80 5.25 7.26
HHHH
H
H
HH
H
H
HHH
H
H
H
HH
HO
OH
(CH2)n
0.22 2.58
9.28
-2.99
8.00
9.33 0.8
+ +
+
+

Die chemische Verschiebung Abhängigkeit von Substitutionsgrad und Stereochemie
HH
HH H
CH3
HH H
CH3
HCH3
HCH3
CH3
CH3
0.23 0.86 1.33 1.50
1.93
1.27
Häq: 1.57 Hax: 0.79
Häq: 1.40 Hax: 1.35
Häq: 1.78 Hax: 1.07
Häq: 1.58 Hax: 1.33
H
CH3
CH3
H
OH
H
H
OH
3.38
3.89

Die chemische Verschiebung von Wasserstoff
elektronenreiche Aromaten
elektronenarme Aromaten
Hochfeldverschiebung, Abschirmung, + Anisotropie
Tieffeldverschiebung, Entschirmung, - Anisotropie
-I, -M-Effekt +I, +M-Effekt
X = O, F, Cl
O TMS
5 10
aliphatische Verbindungen
X-CH COOH, OH, NH O=C-H
H-C≡C
olefinische Verbindungen
aromatische Verbindungen
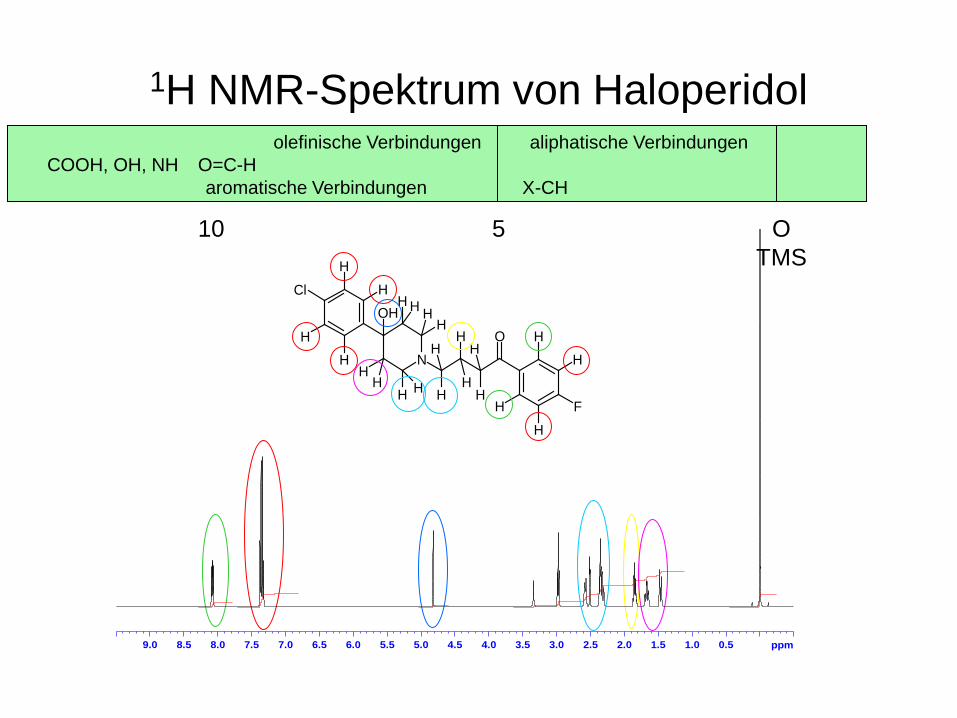
1H NMR-Spektrum von Haloperidol
N
OHCl
F
O
HH
HH
H
H
H
H
H
HH
HH
HH
H
H HH
H
H H
O TMS
5
aliphatische Verbindungen olefinische Verbindungen
aromatische Verbindungen
10
COOH, OH, NH X-CH
O=C-H
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm

1H NMR-Spektrum von Haloperidol in DMSO-d6 bzw. CDCl3
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 ppm
in CDCl3
in DMSO-d6
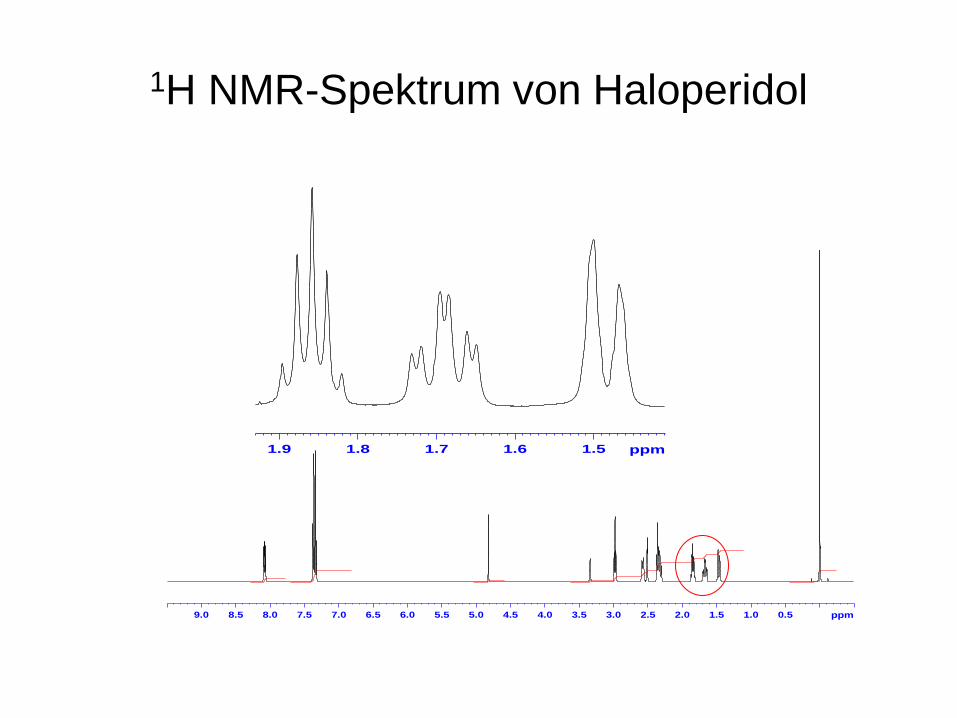
1H NMR-Spektrum von Haloperidol
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
1.51.61.71.81.9 ppm

Die Multiplizität (Aufspaltung) der Signale
Benachbarte Kerne können ein Signal aufspalten (Kopplung).
Für Wasserstoff gilt: Jeder Kopplungspartner erzeugt aus einer Linie zwei. Die Größe der Aufspaltung wird in Hz gemessen (Kopplungs- konstante J). Sie hängt von verschiedenen strukturellen Parametern aber nicht von der Stärke des äußeren Feldes ab.
Die Kopplung wird über die Bindungselektronen vermittelt- nicht durch den Raum

Die Bildung von Multipletts
Singulett
Duplett
Triplett
Quartett J
Singulett
Duplett J1
J2 doppeltes Duplett

Energieniveaus zweier I = ½ Kerne (1H..) (nicht gekoppelt)
-½ ħγB0 [J]
+½ ħγB0 [J]
E = hν = -m ħγB0
+½ ν0 [Hz]
-½ ν0 [Hz]
ν0: Larmorfrequenz ν0: = -γB0/2π
α
β
E0
α α
β β
αα
βα αβ
ββ
+½ ν0,1 + ½ ν0,2
-½ ν0,1 + ½ ν0,2 +½ ν0,1 - ½ ν0,2
-½ ν0,1 - ½ ν0,2

Energieniveaus zweier I = ½ Kerne (1H..) (gekoppelt)
αα
βα αβ
ββ
+½ ν0,1 + ½ ν0,2
+½ ν0,1 - ½ ν0,2 -½ ν0,1 + ½ ν0,2
-½ ν0,1 - ½ ν0,2
αα
βα αβ
ββ
+½ ν0,1 + ½ ν0,2 + 1/4 J
-½ ν0,1 + ½ ν0,2 - 1/4 J +½ ν0,1 - ½ ν0,2 - 1/4 J
-½ ν0,1 - ½ ν0,2 + 1/4 J
Zwei Linien für rot: ν0,1 + 1/2J ν0,1 – 1/2J
Zwei Linien für grün: ν0,2 + 1/2J ν0,2 – 1/2J

Wovon hängt die Größe der Kopplungskonstante (J) ab?
Da die Kopplungskonstante über die Bindungselektronen vermittelt wird, wird sie von der Elektronendichte und der Überlappung der Orbitale sowie der Entfernung der Kopplungspartner beeinflusst. Besonderen Einfluss haben:
Hybridisierung des Kohlenstoffs
Bindungs und Diederwinkel (Stereochemie!)
Elektronegativität von Substituenten
Sind alle Einflüsse identisch, so werden identische Kopplungskonstanten gemessen.

Faktoren, die die Kopplungskonstante beeinflussen
Entfernung der Kopplungspartner
~ -12 Hz ~ 7.5 Hz H
HH
HH
H < 0.5 Hz
H
H
H
H
H
H~ 8 Hz
~ 1.5 Hz < 0.5 Hz
Geminale Kopplung (2J)
Vicinale Kopplung (3J) Fernkopplung (>3J)

Faktoren, die die Kopplungskonstante beeinflussen
Substituenten
HH
HH
OH
~ 7.5 Hz ~ 6.5 Hz
H
H
R
O
OH
H
R~ -7.5 Hz ~ -10 Hz
HH
O
HH
OO
~ -14 Hz ~ -16 Hz

Faktoren, die die Kopplungskonstante beeinflussen
Hybridisierung des Kohlenstoffs
H
H
H
H~ -12 Hz ~ ±1 Hz

Faktoren, die die Kopplungskonstante beeinflussen
Bindungs- und Diederwinkel
H
H
H
HHH
H
H~ 10 Hz
~ 0 Hz ~ 7.5 Hz
~ 12 Hz
H
H
HH
~ 16 Hz ~ 10 Hz

Faktoren, die die Kopplungskonstante beeinflussen
Bindungs- und Diederwinkel – Die Karplus-Beziehung
J
10 Hz
0 Hz
0° 90° 180° Diederwinkel φ
3J = A + B cosφ + C cosφ
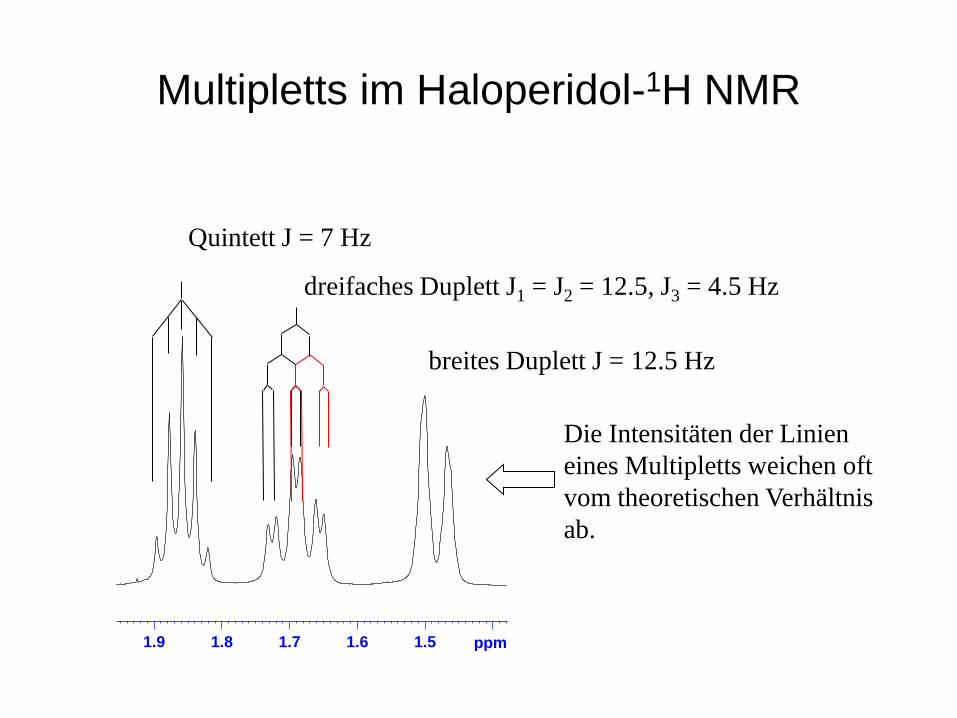
Multipletts im Haloperidol-1H NMR
Quintett J = 7 Hz
dreifaches Duplett J1 = J2 = 12.5, J3 = 4.5 Hz
1.51.61.71.81.9 ppm
breites Duplett J = 12.5 Hz
Die Intensitäten der Linien eines Multipletts weichen oft vom theoretischen Verhältnis ab.

Systeme höherer Ordnung
Systeme höherer Ordnung entstehen: Immer wenn die Differenz der chemischen Verschiebung zweier miteinander gekoppelter Kerne nicht wesentlich größer als die Kopplungskonstante ist. ∆δ << ∆J Wenn gekoppelte Kerne zwar chemisch aber nicht magnetisch äquivalent sind. Merkmale: Zusätzliche Linien, unterschiedliche Kopplungskonstanten für ein und dieselbe Kopplung im gleichen Multiplett, Intensitätsverzerrungen.

AX nach AB
AX-System AB-System
δ

Systeme höherer Ordnung AMX
8.02 8.00 7.98
62 7.60 7.58
2.02 2.00 1.98

2.02 2.00 1.98
8.04 8.02 8.00 7.98 7.96 7.94
Systeme höherer Ordnung ABX

Chemische Äquivalenz – magnetische Äquivalenz
Chemisch äquivalente Kerne haben identische Elektronenumgebungen also iden- tische chemische Verschiebungen. Auch bei unterschiedlicher elektronischer Umgebung kann es zu identischer chemischer Verschiebung kommen – Isochronie Beispiele: Protonen von Methylgruppen nicht diastereotope Protonen von Methylengruppen
R
R'
H
H
H
HR R
H H
H H
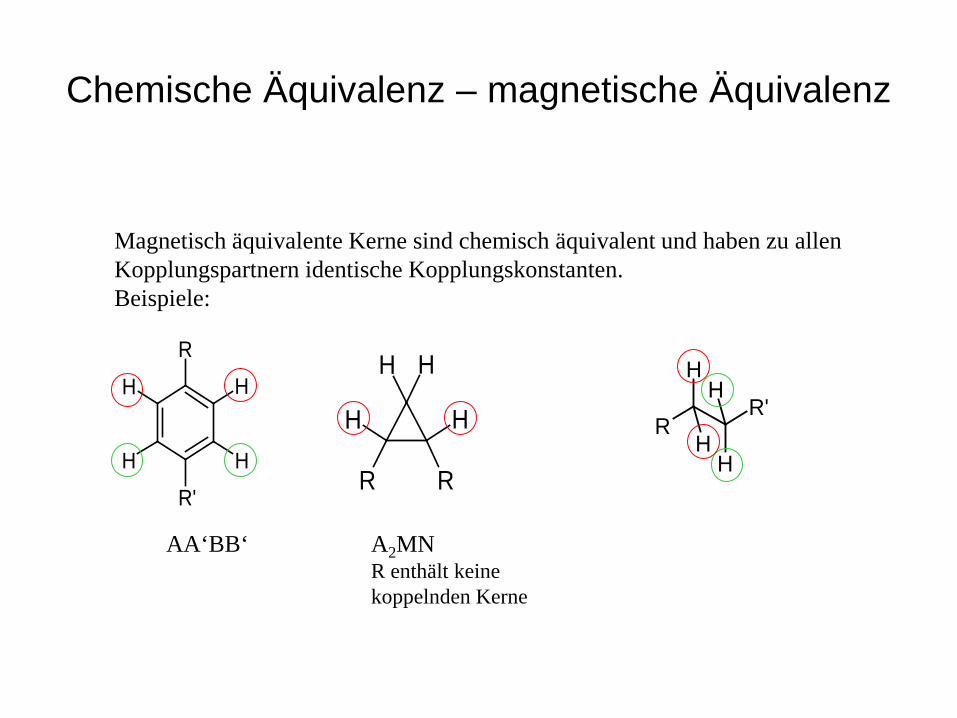
Chemische Äquivalenz – magnetische Äquivalenz
Magnetisch äquivalente Kerne sind chemisch äquivalent und haben zu allen Kopplungspartnern identische Kopplungskonstanten. Beispiele:
R
R'
H
H
H
HR R
H H
H H
AA‘BB‘ A2MN R enthält keine koppelnden Kerne
RR'
H
HH
H

3.183.193.203.21 ppm
Systeme höherer Ordnung
1.851.861.871.881.891.901.91 ppm

1.952.00 ppm
1.95 (ddddd, J = 11.6, 11.6, 4.4, 4.4, 3.2 Hz)
1.95 (m ?)
7.27.37.47.57.67.77.87.98.0 ppm
8.05, m, 2H
7.42, m, 2H 7.33, m, 2H
7.17, m, 2H

2.35
2.35 (ddd, J = 18.2, 4.0, 3.8 Hz, 1H, H-3)
Maximal 2 Stellen nach dem Komma
3.45 Immer wenn ein Signal eines Kerns beschrieben wird. 3.40 – 3.50 Wenn das überlagerte Signal mehrerer Kerne beschrieben wird.

1H NMR-Spektrum von Acetylsalicylsäure in DMSO-d6
14 13 12 11 10 9 8 7 6 5 4 3 2 1 ppm

7.207.257.307.357.407.457.507.557.607.657.707.757.807.857.907.95 ppm
1H NMR-Spektrum von Acetylsalicylsäure in DMSO-d6

8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
1H NMR-Spektrum von m-Hydroxyacetophenon in CDCl3

6.906.957.007.057.107.157.207.257.307.357.407.457.507.55 ppm
1H NMR-Spektrum von m-Hydroxyacetophenon in CDCl3
13 12 11 10 9 8 7 6 5 4 3 2 1 ppm
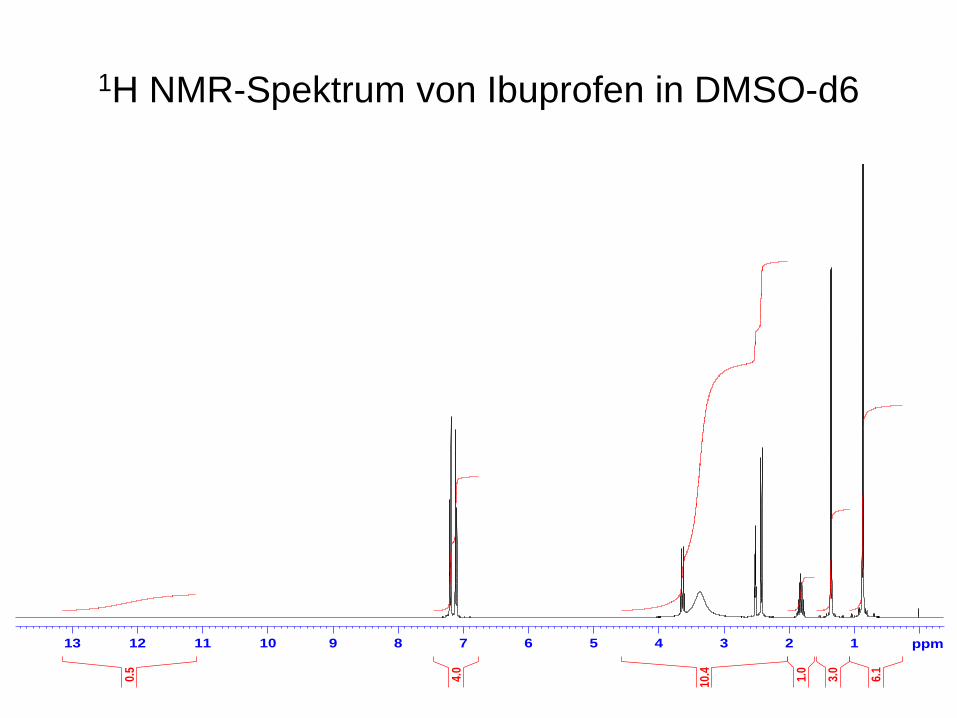
6.13.01.010.44.00.5
1H NMR-Spektrum von Ibuprofen in DMSO-d6
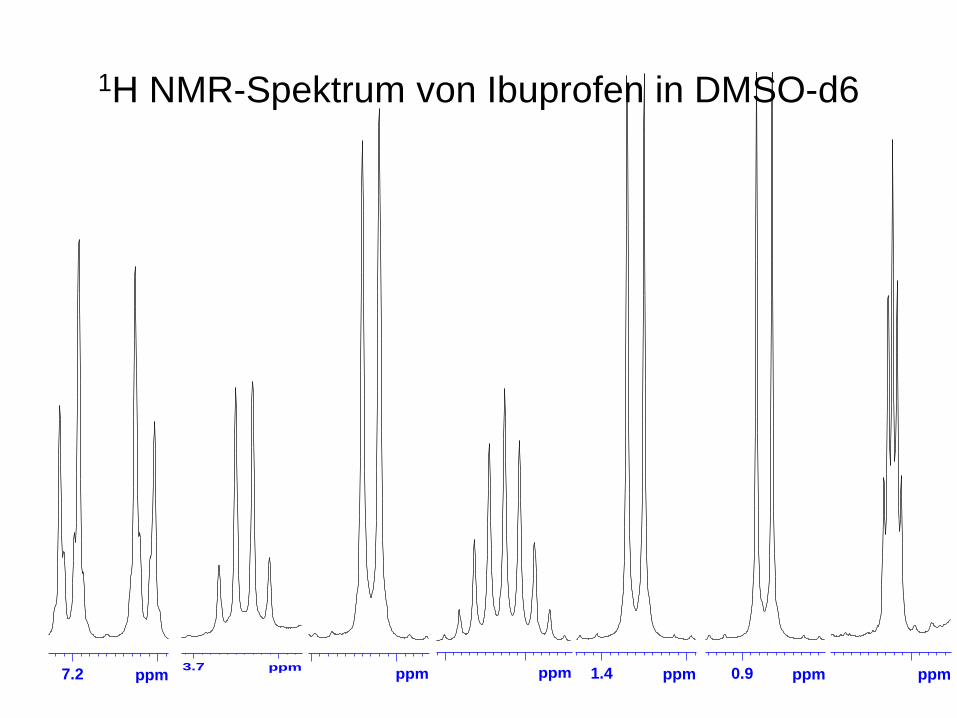
1H NMR-Spektrum von Ibuprofen in DMSO-d6
7.2 ppm 3.7 ppm ppm 1.4 ppm 0.9 ppm ppmppm

11 10 9 8 7 6 5 4 3 2 1 0 ppm
3.21.01.02.01.0
1H NMR-Spektrum von Vanillin in CDCl3

1H NMR-Spektrum von Vanillin in CDCl3
7.057.107.157.207.257.307.357.407.45 ppm

8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
3.2
4.1
2.0
2.0
1H NMR-Spektrum von Benzocain in CDCl3

6.76.86.97.07.17.27.37.47.57.67.77.87.9 ppm
1H NMR-Spektrum von Benzocain in CDCl3

4.14.24.3 ppm 1.31.41.5 ppm
1H NMR-Spektrum von Benzocain in CDCl3
9.5 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
6.24.21.02.0
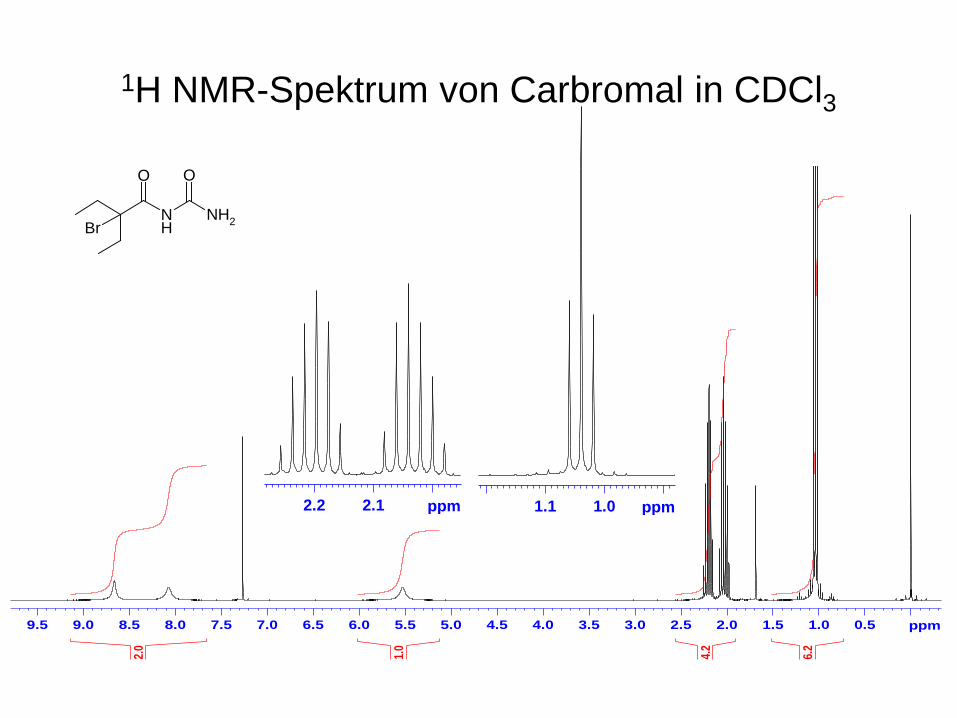
1H NMR-Spektrum von Carbromal in CDCl3
2.12.2 ppm 1.01.1 ppm
NH
NH2
O O
Br

13C NMR-Spektroskopie
Die Signale werden nicht integriert (Signalintensitäten variieren!).
Die Signale besitzen (im Normalfall) keine Multiplizität.
Die Skala umfasst einen mehr als 10-fach größeren Bereich als die der 1H NMR-Spektroskopie.
Unterschiede zur 1H NMR-Spektroskopie
Sehr viel unempfindlicher als 1H NMR nur 1% des natürlichen Kohlenstoffs nur ca.1/4 der gyromagnetischen Konstante des Wasserstoffs

1H NMR-Spektrum von Vanillin (in CDCl3) (Bereich der aromatischen Protonen)
6.906.957.007.057.107.157.207.257.307.357.407.457.507.557.60 pp

13C NMR-Spektren von Haloperidol
200 180 160 140 120 100 80 60 40 20 0 ppm
115116117 ppm
Breitbandentkopplung zur: Vereinfachung Intensitätssteigerung
Vorteile: Höhere Empfindlichkeit leichtere Interpretation Nachteile:Informationsverlust

Die Chemische Verschiebung von Kohlenstoff
ges. Ketone
Aldehyde
Säuren, Säurederivate
0 TMS
100 200
aliphatische Verbindungen aromatische und olefinische Verbindungen Carbonyl- verbindungen
Alkine
C≡N O-C-O C-O
Die Signallage hängt im Wesentlichen nur von der Hybridisierung und der Elektronendichte ab.
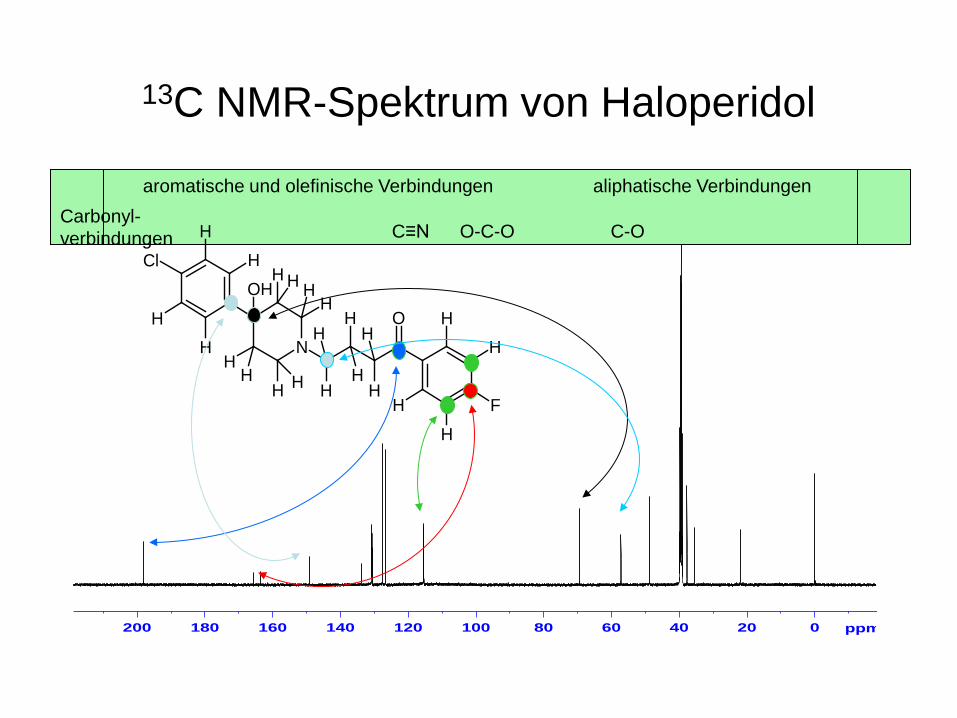
13C NMR-Spektrum von Haloperidol
200 180 160 140 120 100 80 60 40 20 0 ppm
-0.2
00.
000.
20
21.9
835
.58
37.7
138
.98
39.1
439
.31
39.4
839
.64
39.7
439
.81
39.9
039
.98
40.0
748
.80
57.2
069
.43
115.
4011
5.57
126.
6512
7.56
130.
6213
0.73
130.
8113
3.88
133.
90
149.
13
163.
7416
5.74
198.
18
aromatische und olefinische Verbindungen Carbonyl- verbindungen
aliphatische Verbindungen
C-O O-C-O C≡N
N
OHCl
F
O
HH
HH
H
H
H
H
H
HH
HH
HH
H
H HH
H
H H
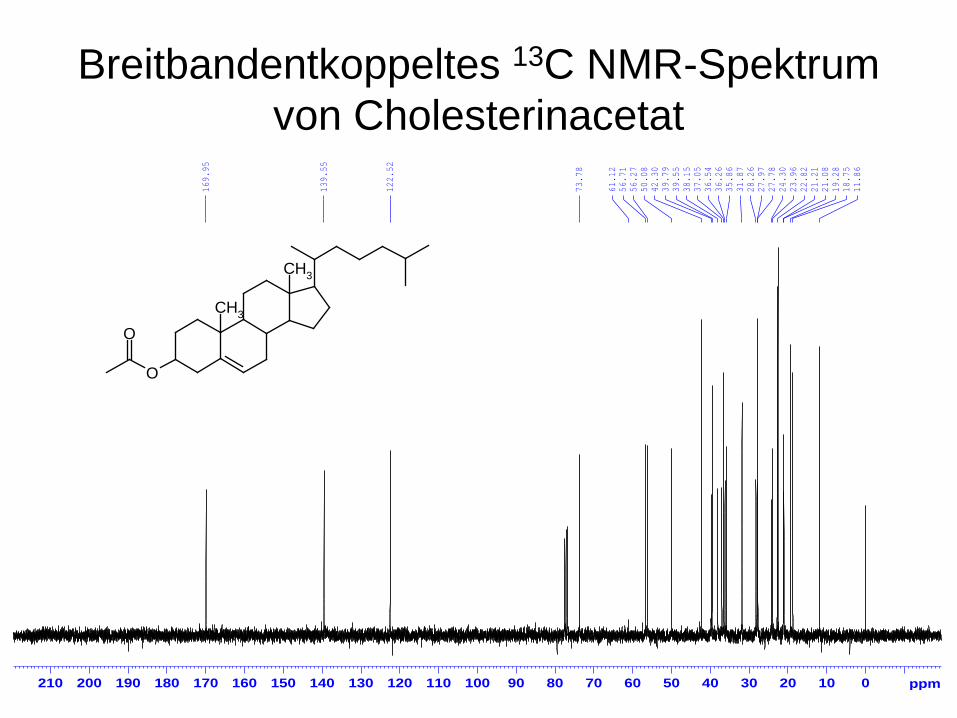
Breitbandentkoppeltes 13C NMR-Spektrum von Cholesterinacetat
210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
11.8
618
.75
19.2
821
.08
21.2
122
.82
23.9
624
.30
27.7
827
.97
28.2
631
.87
35.8
636
.26
36.5
437
.05
38.1
539
.55
39.7
942
.30
50.0
856
.27
56.7
161
.12
73.7
8
122.
52
139.
55
169.
95
CH3
CH3
O
O

210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
31.8
735
.86
36.2
636
.54
39.5
542
.31
50.0
8
56.2
756
.71
73.7
8
122.
52
DEPT90 13C NMR-Spektrum von Cholesterinacetat

210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm
11.8
519
.27
20.4
421
.07
21.2
122
.58
22.8
223
.96
24.2
927
.78
27.9
728
.26
31.8
731
.89
35.8
536
.26
37.0
538
.14
39.5
539
.78
42.3
050
.07
56.2
656
.71
73.7
8
122.
52
DEPT135 13C NMR-Spektrum von Cholesterinacetat
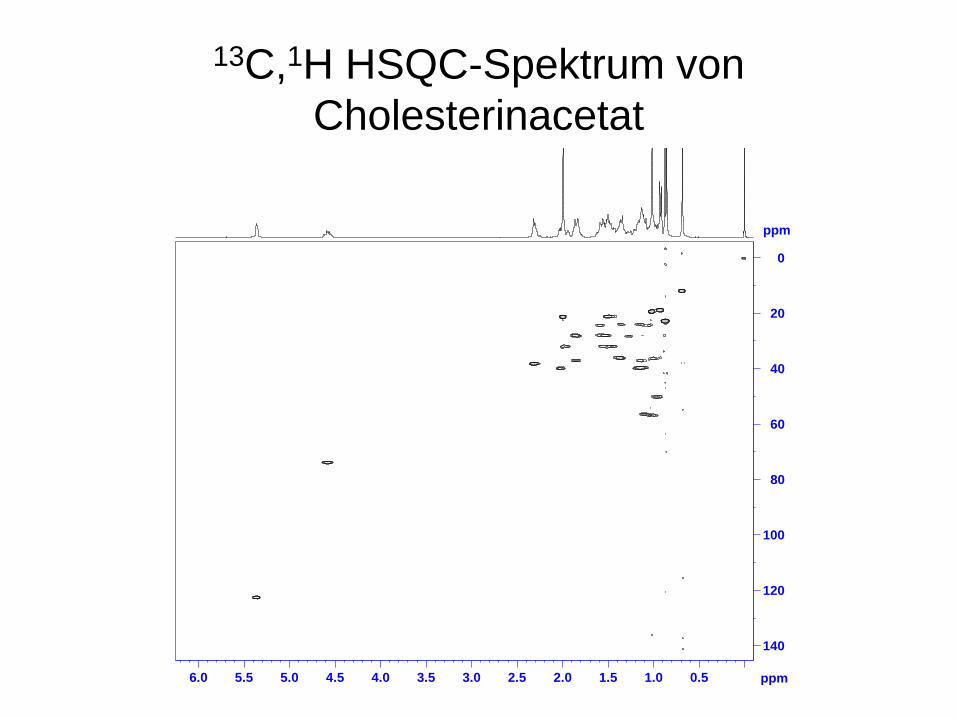
ppm
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm
140
120
100
80
60
40
20
0
13C,1H HSQC-Spektrum von Cholesterinacetat

ppm
5 4 3 2 1 0 ppm
200
180
160
140
120
100
80
60
40
20
0
13C,1H HSQC-Spektrum von Cholesterinacetat

Verwandte Methoden
Festkörper-NMR-Spektroskopie
NMR-Imaging
Kernspintomographie
Bedeutung für die Materialanalyse, Untersuchung von Polymeren
Medizinische Diagnose-Technik
Zerstörungsfreie Analyse verschiedener Materialien, in vivo Untersuchung von Labortieren…..

Kernspintomographie
Aufnahme eines Kniegelenks


















