
Teil 3 Additionsreaktionen - bcp.fu-berlin.de · 3.1.2 trans-Additionen über kationische...
28
Teil 3 Additionsreaktionen 69
Transcript of Teil 3 Additionsreaktionen - bcp.fu-berlin.de · 3.1.2 trans-Additionen über kationische...

Teil 3
Additionsreaktionen
69

3.1 Elektrophile Additionen (AE)
3.1.1 Einleitung und Grundbegriffe
Triebkraft von Additionsreaktionen. Die Grundlage der Reaktivität von Olefinen in Additionsreak-tionen ist die Tatsache, dass die C-C-π-Bindung eine recht schwache Bindung ist. Die beteiligten Kohlen-stoffatome sind sp2-hypridisiert. Eine laterale Überlappung der beiden 2pz-Orbitale ist weniger effizient alsdie frontale Überlappung zweier sp2- oder sp3-Orbitale einer C-C-σ-Bindung.
63 kcal/mol83 kcal/mol
H
HH
H
HH
H
H
H
H
H
H
H H
HH
73 kcal/mol
σ-C,C-Bindungin Ethan
π-C,C-Bindungin Ethylen
C,C-Bindungin Cyclopropan
Deswegen ist die Summe der Bindungsenergien der beiden neu gebildeten C-X-σ-Bindungen in aller Regeldeutlich größer als die der C-C-π-Bindung und der eventuell gebrochenen Bindungen zur Erzeugung derFragmente X und Y, und die Addition an Olefine oft stark exotherm. Dagegen wird die Addition anzyklische Verbindungen, bei der eine C-C-σ-Bindung gebrochen wird ungünstiger sein und im allgemeinennur an kleinen Ringen wie Drei- und Vierringen ablaufen, bei denen die Ringspannung (die Abweichungvom idealen Bindungswinkel) die Bindungsenergie der C-C-σ-Bindung erniedrigt.
cis- und trans-Additionen. Bei der Anlagerung zweier Fragmente X und Y an das planare Olefinekönnen zwei verschiedene Produkte entstehen, je nachdem ob beide Fragmente an dieselbe Seite der Dop-pelbindungsebene oder an verschiedene Seiten addiert werden. Man unterscheidet dementsprechend cis-und trans-Additionen. Die beiden möglichen Produkte sind (normalerweise) Diastereomere.
cis-Addition
syn-Eliminierung
trans-Addition
anti-Eliminerung
70

3.1.2 trans-Additionen über kationische Zwischenstufen
3.1.2.1 Mechanismen elektrophiler trans-Additionen
Bei der C-C-π-Bindung in Olefinen handelt es sich aufgrund ihrer Elektronendichte um eine Lewis-Base.Reaktionen unter Beteiligung von C-C-π-Bindungen werden daher zumeist durch den Angriff eines Elek-trophils (einer Lewis-Säure) eingeleitet. Die so entstandene elektrophile Zwischenstufe kann sich durch dieAddition eines Nucleophils stabilisieren.
R1
R2
H
R1
E
H
R2
H
R1
E
Nu
R2
HH
H E Nu
Merksatz: Die Reaktionsfolge bei elektrophilen Additionen ist also ein elektrophilerAngriff, gefolgt vom Abschlussangriff eines Nucleophils. Es handelt sich um einezweistufige Reaktion mit einer kationischen, reaktiven Zwischenstufe.
Mechanismen über offenkettige Zwischenstufen: Halogenwasserstoff-Addition. Die Additionvon Halogenwasserstoffen an Olefine wird durch den Angriff eines Protons als Elektrophil eingeleitet. Esentsteht ein offenkettiges Carbenium-Ion. Dieses reagiert anschließend mit einem Nucleophil zum Additi-onsprodukt weiter.
Rkt
E
‡1‡2
langsam schnell
∆G‡1
∆G‡2ZS
R1
R2
+ H
R1
H
H
R2Br H
R1
H
Br
R2
H
H
H
H Br
Der Reaktionsvrelauf zeigt folgende Charakteristika:
• Die Reaktion ist zweistufig mit einem Carbenium als echter, reaktiver Zwischenstufe.
• Der erste Schritt, die endothermen Bildung der reaktiven Zwischenstufe, ist geschwindigkeitsbestim-mend (Hammond-Postulat, Polanyi-Prinzip).
• Die Reaktion ist in diesem Schritt bimolekular.
71

• Die Reaktion folgt dementsprechend einer Kinetik 2. Ordnung:
vA = −d[Olefin]dt
= kA · [HX] · [Olefin]
• Die Reaktionsgeschwindigkeit ist sowohl von der Art und Konzentration des Olefins als auch von HXabhängig.
Anmerkung: Gelegentlich werden auch Kinetiken gefunden, die eher einen termolekularenVerlauf nahelegen, also eine konzertierte Addition von Elektrophil E und Nucleophil X aus zweiMolekülen EX.
vA = −d[Olefin]dt
= kA · [EX]2 · [Olefin]
Mechanismen über zyklische Zwischenstufen: Halogen-Addition. Die Reaktion von Olefinen mitHalogenen wird durch den elektrophilen Angriff des Halogens auf die Doppelbindung eingeleitet. Dabeibildet sich eine zyklische kationische Zwischenstufe, ein sogenanntes Halonium-Ion. Energieprofil und Re-aktionscharakteristika sind identisch mit dem obigen Mechanismus. Verschieden ist nur die Struktur derZwischenstufe, was schwerwiegende Auswirkungen auf den stereochemischen Verlauf der Reaktion hat.
Rkt
E
‡1‡2
langsam schnell
∆G‡1
∆G‡2ZS
Br
R2R1
R4R3R1
R2Br
R3
R4Br
R1 R3
R2 R4BrBr Br ++
Konkurrenz- und Nebenreaktionen. Der zweistufige Verlauf wird in beiden Fällen unter anderemdadurch belegt, dass bei Zusatz von Nucleophilen oder Verwendung nucleophiler Solventien diese um denAbschluss der Additionsreaktionen konkurrieren können. Zum Beispiel liefert die Addition von HCl anStyrol in Essigsäure ein Produktgemisch.
H
H
+H
H
H
H
Ph
Br
Br
H
Ph
H
H
H
Ph
HH Br
+ H OAc++ H OAc
+ H Br
+ H OAc
AcO
H
Ph
H
H
H
72

Anmerkung: Aufgrund der pkA-Werte sollte der Angriff durch HCl sehr viel schneller seinals durch Essigsäure. Wäre die Reaktion einstufig, dann hätte also die Addition von Essigsäurekeine Chance. Anscheinend jedoch können Chlorid- und Acetat-Anion erfolgreich im zweitenAdditionsschritt konkurrieren, da das Acetat sogar basischer und stärker nucleophil ist als dasChlorid. Die Reaktion kann wiederum durch Zusatz von anorganischen Chloriden auf die Seitedes chlorierten Produkts verschoben werden.
Auch andere, für das Vorliegen von Carbenium-Ionen typische Nebenreaktionen finden statt, so zum BeispielWagner-Meerweein-Umlagerungen und Pinakol-Umlagerungen, wie sie im Zusammenhang mit dem SN1-Mechanismus beschrieben wurden.
3.1.2.2 Regioselektivität elektrophiler trans-Additionen (Markovnikov-Regel)
Die Aktivierungsenergie des geschwindigkeitsbestimmenden Schritts, des Angriffs des Elektrophils, solltesich proportional zur Energie der Zwischenstufe, des Carbeniumions, verhalten. Deswegen begünstigt eineStabilisierung dieser Zwischenstufe die Reaktion mit entsprechender Orientierung.
H
H
H
H
H
H
Ph
Br
Br
H
Ph
H
H
H
Ph
H
H Br
H
H
Ph
H
H
Br
Br
H
H
H
Ph
H
Rkt
‡1b‡2b
langsam schnell
∆G‡1b
∆G‡2bZS
E
Rkt
‡1a‡2a
∆G‡1a
∆G‡2a ZS
sehr langsamschnell
Markovnikovbevorzugt
Dies wird in der Markovnikov-Regel zusammengefasst:
Merksatz: Der elektrophile Angriff erfolgt bevorzugt so, dass sich ein möglichststabilisiertes (d.h. höher substituiertes oder mesomeriestabilisertes) Carbeniumiondurchlaufen wird. Das sogenannte “Markovnikov-Produkt” ist also jenes, wo dasProton an das niedriger substituierte Kohlenstoffatom und das Halogenid an dashöher substituierte Kohlenstoffatom (und insbesondere in Benzyl- oder Allylposi-tion) addiert wird.
Die zeitgeäße Interpretation der Markovnikov-Regel lautet: “Wer viel hat, kriegtnoch mehr”.
73

Beispiele:
Br
H
H
Br
Br
H
H
Br
Br
H
H
Br
Br
H
H
BrHBr
HBr
HBr
HBr
+
+
+
+
Hauptprodukt Nebenprodukt
3.1.2.3 Stereoselektivität elektrophiler trans-Additionen
Da die Addition von Halogenwasserstoffen in zwei Schritten und über ein freies Carbeniumion verläuft,ist im Prinzip der abschließende Angriff des Nucleophils von beiden Seiten des Carbeniumions möglich,zumal es sich nicht um ein Nucleophil aus demselben Molekül handeln muss. Deswegen ist eigentlich keineStereoselektivität zu erwarten.
R1
R4
R3
R2
H
R2R1
Br
R3
R4
H
Y
H
R2R1
Br
R4R3
Br
R4
R3H
R2R1
Br
Br
cis-Produkt
trans-Produkt
In der Realität findet man aber in vielen Fällen eine bevorzugte Reaktion im Sinne einer trans-Addition.
H
CH3
CH3
HBrH
CH3
BrCH3
CH3
CH3
Br
+
CH3
H
DCl CH3
Cl
D Cl
CH3D
+
Offenkettige und zyklische Zwischenstufen. Der Grund dafür ist, dass die obigen Mechanismen überoffenkettige bzw. über zyklische kationische Zwischenstufen als Grenzfälle anzusehen sind, zwischen denenreale Reaktionen angesiedelt sind. Die Frage ist also, in welchem Ausmaß bei einer gegebenen Reaktion
74

eine Abschirmung der Seite des elektrophilen Angriffs durch das Elektrophil erfolgt. Die Ursache für dieAbschirmung ist, dass gewissermaßen ein Kampf um die π-Elektronen stattfindet zwischen dem angreifen-den Elektrophil und dem sich ausbildenden Carbenium-Ion, das selbst ein starkes Elektrophil (Lewis-Säure,Elektronenmangel-Verbindung) ist und nach Möglichkeiten sucht, seinen Elektronenbedarf zu sättigen. Ne-ben einer Stabilisierung durch σ- und π-Donoren ist eine weitere Möglichkeit der “Missbrauch” der sichbildenden Bindung zum Elektrophil als Lewis-Base.
Merksatz: Die in den beiden Mechanismen vorgestellten Zwischenstufen zyklischesOnium-Ion und offenkettiges Carbenium-Ion sind als Grenzstrukturen anzusehen,je nachdem wie weit die Bindungsbildung zum Elektrophil fortgeschritten ist.
Anmerkung: Allgemein unterscheidet man bei kationischen Spezies Onium-Ionen, die eine hö-here Bindigkeit aufweisen als das entsprechende Element normalerweise zeigt (Oxonium, Bro-monium etc.), und Enium-Ionen, die eine geringere Bindigkeit aufweisen, als das entsprechendeElement normalerweise zeigt (Carbenium etc.).
E
RR
RR
E
RH
RRR
H RH
E
HR
RR
E
RRR
H
E
RRR
H
E
RR
RH
E
zyklischesOnium-Ion
verzerrtesOnium-Ion
offenkettigesCarbenium-Ion
bei zunehmender Größe, Diffusität, Stabilisierung von E
bei zunehmender Polarisierbarkeit, Schwäche von C-E
bei zunehmender Stabilisierung von R
Der Vorgang lässt sich wie folgt zusammenfassen:
• Durch Überlappung der p-Orbitale mit dem Elektrophil entsteht eine Zwei-Elektronen-Dreizentren-Bindung (2e3c-Bindung), über die die positive Ladung delokalisiert ist. Es bildet sich also ein zykli-sches Onium-Ion. Diese Struktur ist am wahrscheinlichsten für schwache, weiche Elektrophile.
• Je stärker und härter das Elektrophil ist, d. h. je weniger gut stabilisiert und je kleiner es ist, undje stärker und weniger polarisierbar seine Bindung zum Kohlenstoff ist, desto weniger ist eine solcheÜberlappung günstig. Das Elektrophil beansprucht die Elektronen zunehmend für sich. Es entstehtein verzerrtes Onium-Ion miot einer kurzen und einer langen Bindung zwischen Kohlenstoff undElektrophil.
• In die gleiche Richtung wirkt auch eine Abschwächung der Elektrophilie des Carbokations durchzunehmende Stabilisierung durch σ- und π-Donoren. Man kann das verzerrte Onium-Ion nämlich auchals dative Bindung der (stärkeren) Kohlenstoff-Elektrophil-Bindung zum Carbenium-Ion betrachten.
75

• Im Extremfall erhält man schließlich ein offenkettiges Carbenium-Ion. Diese Struktur ist also amwahrscheinlichsten für starke, hart Elektrophile und gut stabilisierte Carbenium-Ionen.
• Die Richtung der Verzerrung des zyklischen Onium-Ions bis hin zum offenkettigen Carbenium-Ionerfolgt im Sinne der Markovnikov-Regel, also mit einer zunehmend längeren Bindung zum besserstabilisierten Kohlenstoff.
In diesem Sinne lässt sich folgende grobe Einteilung festhalten:
Cl
RR
RR
I
RH
RH
F
HR
RR
RR
Br
RH
Iodonium Bromonium
RR
XHg
RH
Mercurinium Chloronium
H
RRR
H
Hydronium
immer zyklischteilweise verzerrt (abhängig von Substituenten)
fallweise zyklisch (abhängig von Substituenten)verzerrt (abhängig von Substituenten)
nie zyklischimmer Carbenium
Folgen für die Stereoselektivität. Erstens ist also die Seite, von der aus das Elektrophil angegriffenhat, sterisch und elektronisch durch dieses abgeschirmt. Ferner ist die Rotation um die σ-C,C-Bindungdurch die Zyklisierung gehindert. Der abschließende Angriff des Nucleophils muss dann von der anderenSeite des Moleküls im Sinne eines Rückseitenangriffs erfolgen. Das Ergebnis ist eine Addition mit trans-Stereoselektivität.
Merksatz: Elektrophile Additionsreaktionen über kationische Zwischenstufen ver-laufen genau dann als trans-Additionen, wenn (verzerrte) zyklische Onium-Ionenals Zwischenstufe wahrscheinlich sind. Dann muss der abschließende nucleophileAngriff in Bezug auf den Angriff des Elektrophils von der Rückseite des Molekülserfolgen.
MeHHPh
Br
H
H
Ph
MeMe
H
HPh
Br
+MeH
Br
MeH
Br(E)
+BrBr Br
+Br
Br
Ph
PhH
H* *
* *
(RS) und (SR)aber nicht (RR) und (SS)
MePhHH
Br
H
Ph
H
MeMe
Ph
HH
Br
+MeH
Br
MeH
Br(Z)
+BrBr Br
+Br
Br
H
HPh
Ph* *
* *
(RR) und (SS)aber nicht (RS) und (SR)
Merksatz: Wenn eine Addition eindeutig als trans-Addition verläuft (und wenn beider Addition an beiden Kohlenstoffen zwei neue Stereozentren entstehen), dann istderen relative Konfiguration festgelegt, nicht aber ihre absolute Konfiguration. DieReaktion ist also diastereoselektiv.
76

Ein Sonderfall liegt vor, wenn die Konnektivität an beiden Stereozentren gleich ist, wenn also die Substituen-ten an beiden Seiten der Doppelbindung gleich sind und an beide Seiten das gleiche Fragment addiert wird.Dann entstehen aus (Z)-konfigurierten Edukten die beiden enantiomeren rac-Diastereomere im racemischenGemisch, aus (E)-konfigurierten Edukten dagegen nur eine Verbindung, das achirale meso-Diastereomere.
RHHR
Br
H
H
R
RR
H
HR
Br
+R
H
Br
RH
Br(E)
BrBr Br
+Br
Br
R
RH
H* *
* *
(RS) = (SR)meso
RRHH
Br
H
R
H
RR
R
HH
Br
+R
H
Br
RH
Br(Z)
+BrBr Br
+Br
Br
H
HR
R* *
* *
(RR) und (SS)rac
Ausnahmen von der trans-Selektivität treten dann auf, wenn das intermediäre Carbeniumion anderweitigstark stabilisiert ist, wie zum Beispiel durch Benzylsubstituenten, so dass seine Elektrophilie nicht mehrausreicht, um eine σ-C,H-Bindung zu polarisieren, also mindestens ein verzerrtes Onium-Ion zu erzeugen.In einem solchen Fall ist entweder keine Stereoselektivität vorhanden, oder es liegt sogar cis-Selektivitätvor, wenn der Kollaps des intermediären Ionenpaars schneller verläuft als die Rotation um die (nicht durcheine 2e3c-Bindung fixierten) σ-C,C-Bindung (bzw. falls diese anderweitig fixiert ist).
DBr
D Br
+
D Br
Ein interessantes Beispiel ist die Addition von HCl an 1,2-Dimethylcyclohexen (siehe oben), die bei ge-nauerer Betrachtung temperaturgesteuert mit cis- bzw. trans-Selektivität verläuft. Siehe Synthesis 1973,789.
HCH3
CH3
HClH
CH3
CH3
CH3
CH3-78°CCH2Cl2
H
HClH
CH3
ClCH3
CH3
CH3
Cl
0°C
Cl
Cl
+
+
88% syn
95% anti
77

3.1.2.4 Synthetisch relevante elektrophile trans-Additionen
Addition von HX. Durch Addition von Dihydropyran an Alkohole werden diese mit derTetrahydropyranyl-Schutzgruppe (THP) geschützt. Da ein Alkohol natürlich nicht sauer genug ist, wirdeine starke Säure als Katalysator benötigt.
O
O
H
O
H
OH
O
OEt+
[p-TsOH]CH2Cl2
O
H
O
H O
OEt
O
H
O
O
OEt
+ H - H
Durch Addition von Isobutylen an Alkohole oder Säuren werden diese als tert.-Butyl-Ether bzw. tert.-Butyl-Ester geschützt. Auch hier wird in beiden Fällen eine starke Säure als Katalysator benötigt.
NH3Cl
O
OH[p-TsOH]CH2Cl2
HO
NH2
O
OO
Völlig analog wird tert.-Butylmethylether (TBME) technisch hergestellt. TBME wird seit geraumer Zeitals Zusatz zu Treibstoffen zur Erhöhung der Klopffestigkeit verwendet, ist aber in den letzten Jahren alsUmweltgift, vor allem in Trinkwasser, ins Gerede gekommen.
[H2SO4], p
O+ MeOH
HO
H
Halogenierung von Olefinen. Die Reaktion von Olefinen mit Halogenen ist bereits behandelt worden.Die Reaktivität von Olefinen gegenüber Halogenen wird mit zunehmender Elektronendichte größer, alsodurch σ-Donoren und vor allem durch π-Donoren erhöht. Diese stabilisieren das intermediär gebildeteHalonium-Ion und setzen so die Aktivierungsenergie herab. Sterische Faktoren spielen dagegen kaum eineRolle.
R R COOH Cl> > >
R
PhR
R
R
R
R
ROR >>>>
Im Falle von Iod sind die einzelnen Reaktionsschritte vollkommen reversibel, und auch thermodynamischist die Rückreaktion wegen der schwachen C-I-Bindung nicht ungünstig. Zusammen mit einem Substitu-tionsschritt, und begünstigt durch die Zugabe anorganischer Iodide wie NaI im Überschuss kann das zurIsomerisierung von Olefinen bei Anwesenheit von Iod genutzt werden.
78

R R
I
HR I
HR
I I
R RH H
I
HR I
RH
R
R
A
E
A
E
S N 2
Halogenhydrinreaktion, Halogenlactonisierung, Halogenveretherung. Verwendet man Additi-onsreagenzien, bei denen die gebildeten Nucleophile extrem schwach sind, so kann man den nucleophilenAbschlussangriff gezielt mit einem zugesetzten Nucleophil (meistens fungiert das Lösungsmittel als Nucleo-phil) durchführen. Mögliche Reagenzien sind Iod, N-Iodsuccinimid (NIS), N-Bromsuccinimid (NBS) oderChloramin-T. Sie liefern die entsprechenden Halonium-Ionen, aber sehr schwach nucleophile Anionen.
I I N
BrCl
NH
S
CH3
O O
O O
N
I
O O
Iod N-IodsuccinimidNIS
N-BromsuccinimidNBS
N-Chlor-ToluolsulfonamidChloramin-T
So erhält man bei der Halogenhydrinreaktion in wasserhaltigen Medien (DMSO für NIS und NBS sowieAceton für Chloramin-T) sogenannte Halogenhydrine (β-Haloalkohole), die sehr nützliche Intermediate fürweitere Umsetzungen sind.
NHTsCl
H
Et
H
Cl
HO
H
HNHTs
Cl
+ + +Acetone, H2O
H H
EtEt Et
Et
H
Et
N
O
O
Br N
O
O
HN
O
O
+ + +BrDMSO, H2O Br
OH
NHTs
Anmerkung: Die Regioselektivität im zweiten Beispiel beruht auf der erläuterten Selektivi-tätsregel für SN2-Reaktionen: der Rückseitenangriff erfolgt niemals in Neopentenylstellung.
Auch Carboxylate und Alkohole sind geeignete Nucleophile, was man sich vor allem bei intramolekularenHalogenlactonisierungen und Halogenveretherungen zu Nutze macht.
79

CH3
O
HO
I IKHCO3, H2O
CH3
O
O
I
I O
HH IH
CH3
+ I+
OH NBS
OHBr
OHBr
O
Br
- H
Anmerkung: Im Prinzip gilt auch bei den beschriebenen Reaktionen die Markovnikov-Regel.Allerdings können auch andere Faktoren entscheidend sein. So werden in den obigen Beispielenzu Iodlactonisierung und Bromveretherung bei der Möglichkeit des Angriffs auf zwei sekun-däre Kohlenstoffatome bevorzugt die kleineren, fünfgliedrigen Ringsysteme gebildet (nicht diesechsgleidrigen). Bei möglicher Bildung von Sechs- oder Siebenring wird wiederum der kleinereSechsring bevorzugt gebildet
Anmerkung: Man beachte die Stereochemie im zweiten Beispiel, die dadurch zu erklären ist,dass nur eines der beiden Isomere einen günstigen Rückseitenangriff auf das Bromoniumionerlaubt.
Solvomercurierung. Als Elektrophile lassen sich auch andere Kationen, unter anderem auch Metallka-tionen einsetzen, bevorzugt weiche, starke Lewis-Säuren, die zur Aubildung von zyklischen Onium-Ionenbefähigt sind.
R1
R2
R3
R4
+ MLxn+
R2 R4R1 R3
MLxn+
R2 R4R1 R3
MLx(n-1)+
Nu LxM(n-1)+
R2R1
Nu
R3
R4
Besonders häufig werden Quecksilbersalze als weiche Lewis-Säuren verwendet, vor allem mit schwach nucleo-philen Gegenionen wie ClO−
4 , AcO− oder TfO−. Im allgemeinen ist das Lösungsmittel das angreifendeNucleophil (deswegen “Solvonmercurierung”). Am häufigsten führt man die Reaktion in Gegenwart vonWasser, Alkoholen oder Essigsäure durch. Da es sich in diesen Fällen um Sauerstoff-Nucleophile handelt,spricht man auch von Oxymercurierung (Hydroxymercurierung, Alkoxymercurierung).
+ Hg(OAc)2- AcO
Hg(OAc)ROH
R = H, Alkyl, Ac
Hg(OAc)
OR
Die Oxymercurierung verläuft über ein zyklisches Mercuriniumion, das entsprechend der Markovnikov-Regel verzerrt ist. Deswegen verlaufen Oxymercurierungen regioselektiv (Markovnikov) und diastereoselektiv(trans-Diastereoselektivität).
80

+ Hg(OAc)2- AcO
ROHHg(OAc)
Hg(OAc)
OR
+ +
ROHHg(OAc)
Hg(OAc)
OR
R = H, Alkyl, Ac
Die erhaltenen Organoquecksilberverbindungen sind stabil und isolierbar. Sie reagieren als Nucleophile,gehen Reaktionen ein ähnlich denen von Grignard-Verbindungen RMgX, sind aber viel weniger reaktiv.
R1 Hg(OAc)
OR2
R1 H
OR2
R1 Br
OR2
R1
OR2 O
R3
Br2
NaBH4
R3 Cl
O
- HgBr(OAc)
- HgCl(OAc)
(Reduktion)
Die häufigste Anwendung der Oxymercurierung ist die regioselektive Hydratisierung durch Additionin wässrigen Medien und anschließende Reduktion mit NaBH4.
HO OH
97% 3%
Markovnikov anti-Markovnikov
1. Hg(OAc)2, H2O2. NaBH4
Dabei erhält man als Produkt einen Alkohol, analog zur direkten Hydratisierung (Addition von Wasser)an die Doppelbindung. Im Unterschied zu dieser verläuft die Reaktionsfolge Oxymercurierung/Reduktionaber wie gesagt hochregioselektiv.
81

3.1.3 Elektrophile cis-Additionen mit konzertierten Mechanismen
Die Mechanismen der bekannten konzertierten cis-Additionen sind so verschieden, dass sie nicht gemeinsambehandelt werden können, mit Ausnahme der stereochemischen Konsequenzen.
3.1.3.1 Stereoselektivität bei elektrophilen cis-Additionen
Wenn, anders als bei den obigen Mechanismen, die Addition der beiden Fragmente X und Y an das Olefinals konzertierte Reaktion aus einem Substrat X-Y erfolgt (und nicht nacheinander), dann muss die Reaktionzwangsläufig stereochemisch im Sinne einer cis-Addition verlaufen.
Merksatz: Konzertierte Additionen aus einem Substrat X-Y verlaufen immer alscis-Additionen. Wenn bei der Addition an beiden Kohlenstoffen zwei neue Stereo-zentren entstehen, dann ist deren relative Konfiguration festgelegt, nicht aber ihreabsolute Konfiguration. Die Reaktion ist also diastereoselektiv.
R2 R4
R1 R3
X
R1
R4
R3
R2
R2 R4
R1 R3
X
+X Y
Y
Y
R2 R4
R1 R3
X
R2 R4
R1 R3
X
+
Y
Y
Enantiomere
Wenn die beiden übertragenen Fragmente identisch sind (siehe Epoxidierungen, Dihydroxylierungen, Hy-drierungen), dann erhält man bei gleicher Konnektivität an den beiden Zentren des Olefins weniger Diaste-reomere durch das Auftreten achiraler rac-Diastereomere.
H RR H
X
R
R
H
HH RR H
X
+X X
X
X
H RR H
X
H RR H
X
+
X
X
rac-Diastereomereschiral
Enantiomere
R RH H
X
H
R
H
RR RH H
X
+X X
X
X
R RH H
X
R RH H
X
X
X
meso-Diastereomeresachiral
(Z)
(E)
3.1.3.2 Hydroborierungen
Diboran B2H6 ist ein elektrophiles Reagenz, das sich an Olefine addieren kann. Genau genommen könnennur monomere Borane sich an Olefine addieren, so dass das eigentliche Reagenz das monomere BH3 ist, dasmit Diboran im Gleichgewicht steht. Da Diboran gasförmig, sehr giftig und pyrophor ist, verwendet mannormalerweise stabile BH3-Komplexe (Addukte mit neutralen Lewis-Basen wie THF, Dimethylsulfid oderTriphenylphosphin).
82

BHH
H
HB
HH
HB S
HHO
HB
HH
CH3
CH3
HB P
HH
Ph
Ph
Ph
Anmerkung: Herbert C. Brown erhielt den Nobel-Preis für Chemie 1979 für seine Arbeitenan Organoboranen als Reagenzien in der organischen Synthese. Literatur: Herbert C. Brown etal., Science 1980, 210, 485.
Sterisch anspruchsvolle Boran-Reagenzien. Das Produkt verfügt über zwei weitere Wasserstoffato-me. Da auch Organoborane an Olefine addiert werden können, ist also eine mehrfache Addition möglich,wobei der sterische Anspruch des Olefins entscheidend ist für den Fortschritt der Reaktion. Cyclische Dio-lefine wie Cyclooctadien (COD) können intramolekular an die zweite Doppelbindung addiert werden.
H3C
H3C
CH3
CH3
"BH3" H3C
H3C
CH3
CH3
BH2H
H3C
H3C
CH3
H
"BH3" H3C
H3C
CH3
H
BHCH3
CH3
CH3
H
H
Thexylboran
Disiamylboran
B
H
HB
H
HB
H
"BH3"
9-Borabicycl[3.3.1]nonan (9-BBN)
Anmerkung: Das Beispiel zeigt, dass der wachsende sterische Anspruch die Reaktivität derBorane herabsetzt. Die Verwendung von Organoboranen mit sehr unterschiedlichem sterischemAnspruch ist ein wichtiges Mittel, die Regio- und die Stereoselektivität der Hydroborierunggenau zu kontrollieren. Vor allem 9-BBN und Disiamylboran sind ihrerseits wichtige Hydrobo-rierungsreagenzien.
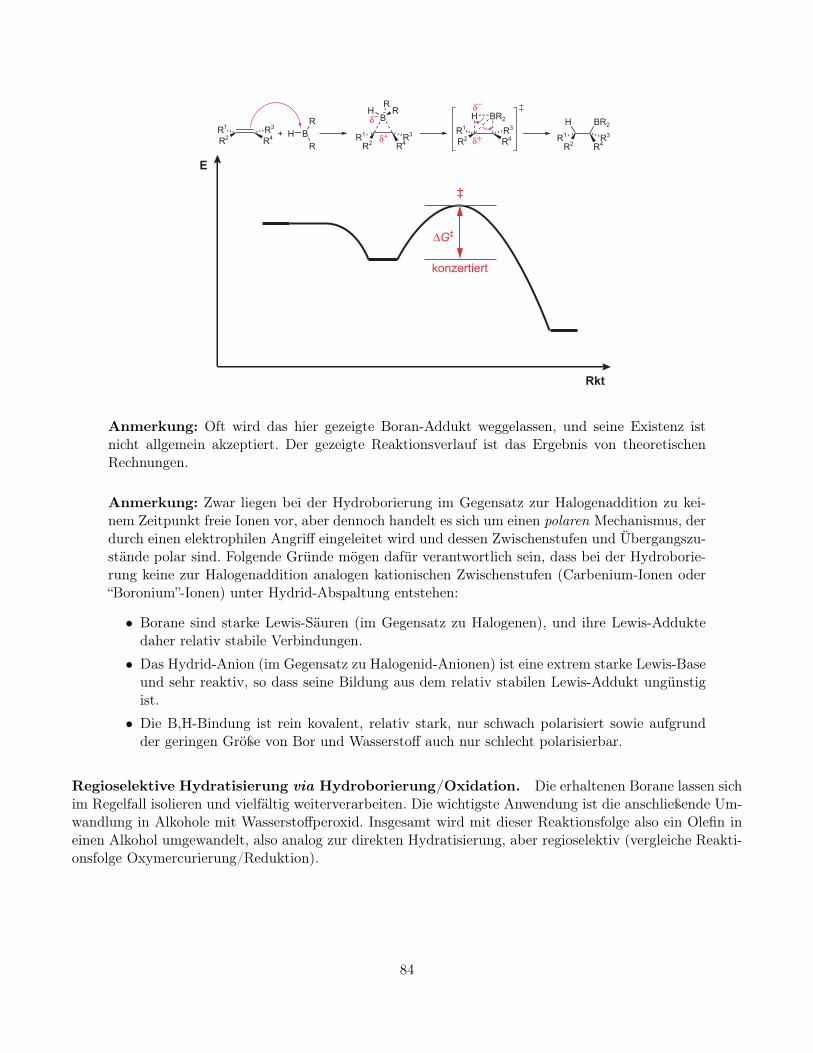
Mechanismus der Hydroborierung. Die Addition von Boranen wird durch den elektrophilen Angriffdes Bor-Zentrums (EN = 2, 0, eigentliches Elektrophil; Wasserstoff EN = 2, 2) auf das Olefin eingeleitet. DieReaktion verläuft wahrscheinlich über ein Boran-Addukt. Es handelt sich um eine einfache Lewis-Säure-Base-Reaktion, und die Bildung der Zwischenstufe ist im Gegensatz zur Bildung von Onium-Ionen exothermund mehr oder weniger ohne Aktivierungsbarriere, da weder Bindungen gebrochen werden, noch Arbeit zurLadungstrennung geleistet werden muss. Im folgenden Schritt wird der hydridische Wasserstoff des Boransauf das Olefin übertragen. Die beiden neuen Bindungen, die C,H-σ- und die C,B-σ-Bindung, werden dabeikonzertiert geknüpft.
83
δ+
δ−δ−
δ+ R4R3R1
R2
BR2HH
R2R1
R4R3R1
R2
BR2
R4R3
B
R1 R3
R2 R4H B+
H RR
R
R
Rkt
‡
konzertiert
∆G‡
E
Anmerkung: Oft wird das hier gezeigte Boran-Addukt weggelassen, und seine Existenz istnicht allgemein akzeptiert. Der gezeigte Reaktionsverlauf ist das Ergebnis von theoretischenRechnungen.
Anmerkung: Zwar liegen bei der Hydroborierung im Gegensatz zur Halogenaddition zu kei-nem Zeitpunkt freie Ionen vor, aber dennoch handelt es sich um einen polaren Mechanismus, derdurch einen elektrophilen Angriff eingeleitet wird und dessen Zwischenstufen und Übergangszu-stände polar sind. Folgende Gründe mögen dafür verantwortlich sein, dass bei der Hydroborie-rung keine zur Halogenaddition analogen kationischen Zwischenstufen (Carbenium-Ionen oder“Boronium”-Ionen) unter Hydrid-Abspaltung entstehen:
• Borane sind starke Lewis-Säuren (im Gegensatz zu Halogenen), und ihre Lewis-Adduktedaher relativ stabile Verbindungen.
• Das Hydrid-Anion (im Gegensatz zu Halogenid-Anionen) ist eine extrem starke Lewis-Baseund sehr reaktiv, so dass seine Bildung aus dem relativ stabilen Lewis-Addukt ungünstigist.
• Die B,H-Bindung ist rein kovalent, relativ stark, nur schwach polarisiert sowie aufgrundder geringen Größe von Bor und Wasserstoff auch nur schlecht polarisierbar.
Regioselektive Hydratisierung via Hydroborierung/Oxidation. Die erhaltenen Borane lassen sichim Regelfall isolieren und vielfältig weiterverarbeiten. Die wichtigste Anwendung ist die anschließende Um-wandlung in Alkohole mit Wasserstoffperoxid. Insgesamt wird mit dieser Reaktionsfolge also ein Olefin ineinen Alkohol umgewandelt, also analog zur direkten Hydratisierung, aber regioselektiv (vergleiche Reakti-onsfolge Oxymercurierung/Reduktion).
84

H2O2
BO
OH
BoranhydroperoxidR'
R'
BR'R
H
BR'
RR' R
R' R'
BOR'R- OH
OR'
BO OR'
R
Borsäuretriester
NaOH
ROH
+ 2 R'OH + NaB(OH)4
+
Bei der Regioselektivität der Reaktionsfolge Hydroborierung/Oxidation ist zu berücksichtigen, dass dasBor-Zentrum das eigentliche Elektrophil ist. Das Wasserstoffatom im Boran ist hydridisch, d.h. negativpolarisiert und somit eher nucleophil. Im Prinzip gilt daher das bisher Gesagte:
Merksatz: Auch bei der Hydroborierung gilt die Markovnikov-Regel unter Berück-sichtigung der Elektronegativität von Bor und Wasserstoff. Das Bor-Zentrum (daseigentliche Elektrophil) addiert sich an das Kohlenstoffatom, das mehr Wasserstof-fatome trägt. Das Hydrid wird an die höher substituierte Seite addiert.
BH+
B
HNaOH/H2O2
OH100% Markovnikov-Produkt
H
B
H
+
HNaOH/H2O2
OH94% Markovnikov-Produkt
H
H H
9-BBN
B3
δ+δ−
δ+δ−
Allerdings sitzt in jedem Fall (wegen der entgegengesetzten Polarität) der Wasserstoff gerade nicht dort, woer bei der HX-Addition (Hydratisierung, Halogenwasserstoffaddition, Oxymercurierung/Reduktion) sitzenwürde. Durch die nachfolgende Oxidation kommt man so also zu den “anti-Markovnikov”-Alkoholen, beidenen die OH-Funktion am niedriger substituierten Zentrum sitzt. Dies ist der entscheidende Wert derReaktionsfolge Hydroborierung/Oxidation. Man erhält jeweils hochregioselektiv durch die ReaktionsfolgenOxymercurierung/Reduktion bzw. Hydroborierung/Oxidation die komplementären Produkte.
B
H
9-BBN
NaBH4
NaOH/H2O2
Hg(OAc)
OHHg(OAc)2
H2O
B
H
OH
OH
H
H
97% Markovnikov-Alkohol
Markovnikov-Addukt
100% "anti-Markovnikov"-Alkohol (i. Vgl. z. oben)
Markovnikov-Addukt
Anmerkung: Häufig wird die Markovnikov-Regel von vorneherein so ausgelegt, dass sie sich nurauf die Position des übertragenen Wasserstoff-Atoms bezieht. In diesem Fall müsste man sagen,
85

dass die Hydroborierung anti-Markovnikov-Produkte liefert. Da aber die in der Markovnikov-Regel beschriebene Regioselektivität für alle elektrophilen Additionen gilt, auch solche, die garkeinen Wasserstoff übertragen, ist die hier gegebene Definition von allgemeinerer Gültigkeit unddeswegen zu bevorzugen.
Die durch elektronische Effekte verursachte Regioselektivität bei der Hydroborierung unsymmetrisch sub-stituierter Olefine wird verstärkt durch den Einfluss von sterischen Effekten. Sterisch gehinderte Boranezeigen bessere Regioselektivität. Es gilt für zunehmend sterisch gehinderte Hydroborierungs-Reagenzienfolgende, strengere Regel:
Merksatz: Bei gleicher Zahl von Wasserstoffen an beiden Seiten der Doppelbin-dung addiert sich das Bor-Zentrum an die Seite, die weniger sterisch gehindert ist.Diese durch sterische Effekte verursachte Regioselektivität nimmt drastisch zu mitwachsender sterischer Beladung des Borzentrums.
B
H
9-BBN
O
H
B
H
HH2O2
H2O2
OH
HO
erwartetesProdukt
H
H
bevorzugterAngriff des
Borzentrums
+
99,8 : 0,2
57 : 43
Stereoselektivität bei Hydroborierungen. Die Hydroborierung verläuft als konzertierte Reaktion unddamit notwendigerweise im Sinne einer cis-Addition. Dabei können an beiden Kohlenstoffen Stereozentrenentstehen. Da bei der Hydroborierung zwei unterschiedliche Gruppen an die beiden Kohlenstoffe übertragenwerden, können hier keine rac- und meso-Isomere gildet werden.
Merksatz: Hydroborierungen verlaufen immer als cis-Additionen. Wenn bei derAddition an beiden Kohlenstoffen zwei neue Stereozentren entstehen, dann ist de-ren relative Konfiguration festgelegt, nicht aber ihre absolute Konfiguration. Hy-droborierungen sind also diastereoselektiv.
86

Et MeMe Et
R2B
Me
Me
Et
EtEt MeMe Et
R2B
+
(E)
H BR2
Et EtMe Me
R2B
Me
Et
Me
EtEt EtMe Me
R2B
+
(Z)
H BR2
H
H
H
H
Et MeMe Et
R2B
Et MeMe Et
R2B
+
Et EtMe Me
R2B
Et EtMe Me
R2B
+
H
H
H
H
* *
* *
* *
* *
(SS) und (RR)aber nicht (RS) und (RS)
(RS) und (SR)aber nicht (RR) und (SS)
Et MeMe Et
HO
Et MeMe Et
HO
+
Et EtMe Me
HO
Et EtMe Me
HO
+
H
H
H
H
* *
* *
* *
* *
(SS) und (RR)aber nicht (RS) und (RS)
(RS) und (SR)aber nicht (RR) und (SS)
Anmerkung: Die scheinbare Umkehrung der Stereozentren im obigen Beispiel ist nur auf dieunterschiedliche Priorität von Bor bzw. Sauerstoff in der Cahn-Ingold-Prelog-Nomenklatur zu-rückzuführen.
Bei Verwendung sterisch gehinderter Borane wie 9-BBN lassen sich Hydroborierungen somit zugleich regio-und diastereoselektiv durchführen.
Me
B
H
9-BBN
B
HMe
B
MeH
OH
HMe
OH
MeH
87

3.1.3.3 Katalytische cis-Dihydroxylierung
Olefine lassen sich unter milden Bedingungen selektiv zu vicinalen Diolen oxidieren, wenn Metalloxidein hohen Oxidationsstufen wie KMnO4 oder OsO4 eingesetzt werden. Bei der anschließenden wässrigenAufarbeitung werden die zunächst erhaltenen Ester unter Erhalt der Stereochemie hydrolysiert.
OOs
O
O O+8
H3O
HydrolyseR+ R
OH
OH
OsmatEster
Diol
R
OOs
O
O O+6
Stereoselektivität bei Dihydroxylierungen. Dihydroxylierungen verlaufen konzertiert über eine zy-klische Zwischenstufe und somit notwendigerweise als cis-Addition. Dihydroxylierungen verlaufen deswegendiastereoselektiv ; die relative Konfiguration der beiden neuen Stereozentren ist also festgelegt, nicht aberihre absolute. Da zwei identische Fragmente übertragen werden spielt die Regiochemie keine Rolle.
R RH H
OOs
O
OOs
O
O O+8
O O+6
H2O
H
R
H
R R RH H
OOs
O
O O
+6
+ R RH H
OHHO
R RH H
OHHO(Z)
R HH R
OOs
O
OOs
O
O O+8
O O+6
H2O
H
H
R
R R HH R
OOs
O
O O
+6
+ R HH R
OHHO
R HH R
OHHO
+
(E)
- OsO2(OH)2
- OsO2(OH)2
meso
rac
Merksatz: Bei gleicher Konnektivität der Stereozentren erhält man aus (Z)-konfigurierten Olefinen das achirale meso-Diastereomere, aus (E)-konfiguriertenOlefinen ein racemisches Gemisch der beiden enantiomeren rac-Diastereomere.
Experimentelle Details. Die Reaktionen werden üblicherweise in Wasser durchgeführt, was die Artder einsetzbaren Olefine beschränkt. Allerdings können Kronenether oder quartäre Ammoniumsalze alsPhasentransferkatalysatoren eingesetzt werden, was die Durchführung in unpolaren Solventien wie Benzolerlaubt.
unpolar,wasserunlöslich
KMnO4R4N+Cl–
Benzol
Phasentransfer-Katalysator
OH
OH
88

Nebenreaktionen, vor allem bei drastischeren Bedingungen, sind die Weiteroxidation zu Ketonen bis hin zurSpaltung der C,C-Bindung. OsO4 ist besonders selektiv. Es erlaubt die Oxidation von Doppelbindungenneben einer Vielzahl anderer oxidationsempfindlicher funktioneller Gruppen wie Aldehyden oder Dreifach-bindungen, ist aber teuer und äußerst toxisch. Die Metallverbindung kann unter Umständen katalytischeingesetzt und mit Oxidationsmitteln wie Wasserstoffperoxid immer wieder regeneriert werden.
[OsO4] (kat.)
O NOCH3
HO OH
65%
Interessant ist auch die (von der Arbeitsgruppe Stark untersuchte) oxidative Cyclisierung von 1,5-Dienen zu Tetrahydrofuranen, die auch mit Polyenen als diastereoselektive Kaskadenreaktion zu Oli-go(tetrahydropyranen) durchgeführt werden kann:
ORu
O
O
O+VIII
[3+2] cycloaddition ORu
O
O O
[3+2] cycloaddition ORu
O
O O
H H
+VI
+IV
H
ruthenate ester
H3Ohydrolysis
OHO OHH H- RuO2
RuO2 (10 mol%),NaIO4 (8 eq),
30 min
50% O OO OOHO OHH H H HH H
squalene
3.1.3.4 Epoxidierung
Olefine können auch mit organischen Peroxiden oxidiert werden. Man erhält zunächst Epoxide (Oxirane),die im allgemeinen durch anschließende Hydrolyse im alkalischen Medium unter Ringöffnung in das dihy-droxylierte Produkt überführt werden können. Man erhält also zur Dihydroxylierung analoge Produkte.
R
O OH
HydrolyseSN2
RO
OH
O
R'+
Persäure
ROH
OH
Epoxid Diol
89

Stereoselektivität bei Epoxidierungen. Epoxidierungen verlaufen über einen zyklischen Übergangs-zustand, in dem die beiden neuen C,O-Bindungen des Epoxids konzertiert geknüpft werden. Deswegenerfolgen sie notwendigerweise als cis-Addition und sind diastereoselektiv. Die relative Konfiguration derbeiden neuen Stereozentren ist also festgelegt, nicht aber ihre absolute.
R RH H
H
R
H
R R RH H
O
+ R
RH
H
OH
HO
R
R
H
H
OH
HO(Z)
R HH R
O
H
H
R
R R HH R
O
+ R
HH
R
OH
HO
R
H
H
R
OH
HO
+
(E)
OO
H
O R
OO
H
O RO
OH
OH
meso
racSN2
Rückseiten-Angriffanti-Öffnung
Hydrolyse
Hydrolyse
Man gelangt zu den Diolen durch alkalische Hydrolyse des Epoxids unter Ringöffnung. Es handelt sich umeine SN2-Reaktion mit dem Hydroxid-Anion als Nucleophil, also unter Inversion der Konfiguration einesStereozentrums. Deswegen ist das stereochemische Ergebnis zu dem der Dihydroxylierung komplentär.
Merksatz: Bei gleicher Konnektivität der Stereozentren erhält man bei der Reak-tionsfolge Epoxidierung/Hydrolyse (in Umkehrung der Verhältnisse bei der Dihy-droxylierung) aus (E)-konfigurierten Olefinen das achirale meso-Diastereomere, aus(Z)-konfigurierten Olefinen ein racemisches Gemisch der beiden enantiomeren rac-Diastereomere.
Experimentelle Details. Typischerweise werden Persäuren als Reagenzien verwendet. Donorsubstiuen-ten am Olefin und Akzeptorsubstituenten an der Persäure begünstigen die Reaktion. Für akzeptorsubsti-tuierte Olefine muss Peroxytrifluoressigsäure eingesetzt werden.
O
OOH O
CH3
CH3
CH3
CH3
O
O
OOHCl
meta-Chlor-peroxybenzoesäure
(MCPBA)
Peroxybenzoesäure
O
O
F3C
O
OOH
Peroxytrifluor-essigsäure
O
O
O
90

3.1.3.5 Katalytische Hydrierungen
Olefine können mit Wasserstoff unter Verwendung von Edelmetallen (Ni, Pd, Pt, Rh, Ru) als heteroge-nen Katalysatoren hydriert werden. Meistens werden fein dispergierte Metall (Pd auf Aktivkohle, Raney-Nickel) verwendet. Andere funktionelle Gruppen können allerdings auch reduziert werden wie aromatischeHalogenide und Benzylether. Heterogen katalysierte Hydrierungen sind Oberflächenreaktionen und ihreMechanismen deswegen komplex, vielschrittig und in vielen Fällen nicht einmal genau geklärt.
CH3
CH3
CH3
CH3
H
H
H
CH3
CH3
H
Pt, H2
+
70-85% cis 15-30% trans
Die Hydrierung mit heterogenen Katalysatoren verläuft oft diastereoselektiv, und zwar bevorzugt im Sinneeiner cis-Addition. Auch induzierte Diastereoselektivität wird beobachtet, zum Beispiel bei Anwesenheitvon funktionellen Gruppen, die als Ankergruppen am Katalysator fungieren.
HO HO
HH
HOH H
Pt, H2
+
5% 95%
Homogene Katalysatoren (oft Rh- oder Ir-Komplexe), wie zum Beispiel der Wilkinson-Katalysator, erlaubensehr viel selektivere Reaktionen und sind zum Beispiel mit anderen reduzierbaren funktionellen Gruppenwie −C(O)OR, −NO2, −CN oder −C(O)R kompatibel. Der Mechanismus verläuft im allgemeinen über oxi-dative Addition und reduktive Eliminierung am Metall-Komplex, was in Vorlesungen über metallorganischeKomplexchemie behandelt wird.
R RH
H
(Ph3P)3RhCl
Wilkinson-Katalysator
H2
Interessant ist die Möglichkeit, die Hydrierung von Olefinen bei Verwendung chiraler Katalysatoren en-antioselektiv durchzuführen. Solche “asymmetrischen Hydrierungen” finden breite Anwendungen in derorganischen Synthese bis hin zu kommerziellen Anwendungen, wie zum Beispiel der Synthese von (l)-DOPA-Derivaten für die Behandlung von Parkinson.
O
OPh2P
PPh2
RhO
O
*
*
(+)-(diop)Rh(thf)2+
(reines Enantiomer)
COOH
RNHAc
THF, H2COOH
RNHAc
H97%
(L)-Dopa (-Derivat)
H
RNHAc
COOH3%
* *+
91

3.2 Radikalische Additionen (AR)
Mehrfachbindungen in Alkenen (und anderen ungesättigten Verbindungen) können auch durch Radikaleangegriffen werden, da diese auch im Normalfall wie schwache Elektrophile reagieren. Dies ist bereits alsNebenreaktion bei der allylischen Bromierung angesprochen worden. Analog zu den radikalischen Substi-tutionen erfordern radikalische Additionen die Erzeugung von Radikalen durch Erhitzen, Bestrahlung oderin aller Regel durch Verwendung typischer Radikalinititoren wie AIBN oder DBPO.
3.2.1 Mechanismus radikalischer Additionen
Beispiel: Bromwasserstoff-Addition. Ganz analog zu den radikalischen Substitutionen handelt es sichauch bei radikalischen Additionen um Radikalkettenreaktionen, bei denen sich Initiatorzerfall, Startreaktion,Kettenfortpflanzung und Abbruchreaktionen als Teilschritte unterscheiden lassen.
RIRI 2 RI
RI + HBr RI H + Br
Br +H
R
H
H
H
R
H
HBr
H
R
H
HBr + HBr
H
R
H
HBrH + Br
Initiatorzerfall
Initiierung
Kettenfortpflanzung
Kettenfortpflanzung
Abbruch z.B. durch Disproportionierung
H
R
H
HBr2
H
R
H
HBrH
H
R
Br
H
+
3.2.2 Regioselektivität radikalischer Additionen (anti-Markovnikov)
Wie bei den elektrophilen Addition diskutiert, kann auch der Angriff eines Radikals an beiden Kohlenstoffa-tomen einer Doppelbindung erfolgen. Von den beiden möglichen Reaktionswegen sollte wie immer derjenigebevorzugt sein, der über die stabilere radikalische Zwischenstufe erfolgt. Da aber im Unterschied zur elek-trophilen Addition von Halogenwasserstoffen, bei denen das Proton das angreifende Elektrophil ist, hierdas angreifende Radikal ein Brom-Radikal ist, erhält man als Ergebnis das sogenannte anti-Markovnikov-Produkt.
92

Merksatz: Der Angriff des Brom-Radikals erfolgt bevorzugt so, dass eine mög-lichst stabilisierte (d. h. höher substituierte oder durch Mesomerie stabilisierte)radikalische Zwischenstufe durchlaufen wird. Dadurch wird das sogenannte “anti-Markovnikov-Produkt” bevorzugt gebildet, bei dem sich Brom am niedriger substi-tuierten Kohlenstoffatom und Wasserstoff am höher substituierten Kohlenstoffatombefindet.
Rkt
‡1b‡2b
langsam schnell
∆G‡1b
∆G‡2bZS
E
Rkt
‡1a‡2a
∆G‡1a
∆G‡2a ZS
sehr langsamschnell
H
H
H
Br
H
H
PhH
H
Ph
Br
H
H
Ph
H BrH
Br
Ph
H
HH
H
H
Br
Ph
HBrH H
anti-Markovnikovbevorzugt
Die Regioselektivität der Addition ist umso größer, je größer der Unterschied in der Stabilisierung derbeiden möglichen radikalischen Zwischenstufen ist, und deswegen vor allem bei Mesomeriestabilisierung derZwischenstufe zu beobachten.
Me
Me
Me
HBrDBPO H
MeBr
H Me
MeH
55% anti-Markovnikov
OO
Me
Methylacrylat
HBrDBPO
>80% anti-Markovnikov
OO
MeBr
H
Regioselektivität und Reaktionsbedingungen. Man kann also mit dem gleichen Reagenz, nämlichHBr, zu unterschiedliche Regioisomeren gelangen je nachdem, ob man die Reaktion unter ionischen oderunter radikalischen Bedingungen durchführt. Deswegen ist die strikte Einhaltung der Arbeitsbedingungenentscheidend.
• Wenn man zum anti-Markovinkov-Produkt gelangen will, sollte man radikalische Bedingungen wählen,d. h. weniger polare Solventien verwenden, Radikalinitiatoren einsetzen und in der Hitze arbeiten.
• Wenn man zum Markovinkov-Produkt gelangen will, sollte man ionische Bedingungen wählen,d. h.möglichst polare Solventien verwenden, auf Sauberkeit achten, unter Luftausschluss, im Dun-keln und in der Kälte arbeiten.
93

3.2.3 Stereoselektivität radikalischer Additionen
Bei radikalischen Additionen findet man im Normalfall keine Stereoselektivität. Wenn überhaupt, dannverläuft die Addition im Sinne einer trans-Addition, was mit dem Vorliegen einer zyklischen radikalischenZwischenstufe erklärt werden kann. Da Radikale aber deutlich weniger elektrophil sind als Carbokationen, istihre Fähigkeit, eine benachbarte σ-Bindung zu polarisieren, deutlich geringer und damit auch die Bedeutungzyklischer radikalischer Zwischenstufen. Es gibt dennoch einige Beispiele, vor allem bei polyzyklischenVerbindungen mit sterischer Hinderung.
+ CCl3 CCl3DBPO
Cl
CCl3Cl
+
73% 4%
DBPO, 80°CCCl4
CCl3+ CCl4
- CCl3
CCl3
3.2.4 Beispiele für radikalische Additionen
Radikalische HX-Additionen. Verbindungen mit leicht homolytisch abspaltbaren Wasserstoffen lassensich an Olefine radikalisch addieren, wie zum Beispiel Aldehyde.
H
O
ROOC COOR+
DBPO
ROOC COOR
O
H
Radikalische Polymerisation. Die wichtigste radikalische Additionsreaktion ist die radikalische Poly-merisation von Olefinen. Im Unterschied zu den bisher besprochenen radikalischen Additionen (und Sub-stitutionen) ist hier ein leicht homolytisch spaltbares Reagenz wie HBr abwesend. Deswegen kann sich dieradikalische Zwischenstufe nur “stabilisieren”, indem sie ein weiteres Olefin-Molekül angreift. Dabei entstehtaber nur eine neue radaikalische Zwischenstufe. So wird aus der Kettenreaktion zugleich das Wachstum einerPolymerkette. Erst eine Abbruchreaktion beendet deren Wachstum.
O
OMeRi
O OMe O OMeOMeO
RiRi etc.
PMMA
Da die Lebensdauer der radikalischen Zwischenstufe groß genug sein muss, um viele Kettenwachstumsschrit-te pro Abbruch zu erlauben, wird die radikalische Polymerisation normalerweise mit mesomeriestabilisiertenOlefinen durchgeführt. Zu den wichtigsten Produkten gehören deswegen Poly(styrol) (PS, Pappbecher) undPoly(methylmethacrylat) (PMMA, Plexiglas).
94

3.3 Nucleophile Additionen
Im Normalfall sind C-C-π-Bindungen Lewis-Basen, die von Elektrophilen angegriffen werden. Dies gilt nichtmehr, wenn ihre Elektronendichte durch π-Akzeptor-Substituenten stark reduziert wird. Die wichtigsteGruppe von Substraten für die nucleophile Addition sind “Michael-Systeme” (d. h. Doppelbindungen inKonjugation mit Carbonylfunktionen).
O
R
O
OR
O
NR2
C N NO2 SO2R
3.3.1 Mechanismus nucleophiler Additionen
Elektronenarme Doppelbindungen können durch ein Nucleophil angegriffen werden. Die erzeugte negativeLadung wird durch Delokalisierung zum Substituenten stabilisiert. Der abschließende Angriff zum Additi-onsprodukt erfolgt durch ein Elektrophil, typischerweise ein Proton.
ONu
Nu O Nu O
+ NuH- Nu
OHNu Nu O
H
Keto/Enol
1,4-Addukt 3,4-Addukt
δ+ δ−
δ+ δ−
Im Gleichgewicht ist das Enolat bevorzugt. Das Proton addiert sich dann im Sinne einer 1,4-Addition. Dasentstandene Enol tautomerisiert jedoch quantitativ zum 3,4-Produkt. Bei Abwesenheit von protonenaktivenVerbindungen kann man die Zwischenstufe auch durch Substitutionsreaktionen abfangen und so selektiv zuhöher substituierten Derivaten gelangen.
O R O R O R O
R'
R' BrR2CuLi
3.3.2 Michael-Reaktionen
Wenn das Substrat ein Michael-System und das angreifende Nucleophil eine C,H-acide Verbindung ist,spricht man von einer Michael-Addition. Dies ist eine der wichtigsten Methoden zu C,C-Verknüpfung undwird detailliert im Teil über Reaktionen an Cabonylverbindungen behandelt werden.
95

CN
O
OEt
H
CNKOH CH2NH2
O
OEt+ CN+
CN
O OEt
CN
CN
O OEt
CN
HH2O
Im obigen Beispiel wird Cyclopentenon umgesetzt mit einem Esterenolat (die Trimethylsilylgruppe wirdaufgrund der starken Si,F-Bindung entfernt). Im folgenden Beispiel handelt es sich um eine sogenannte“Cyanethylierung” (Übertragung von “Ethylcyanid”).
O
H O
OMe
SiMe3 H O
OMe
H O
OMeKF
- F-SiMe3
- K
O
Me
O
OMe
H O
Me
O
OMe
HH2O
1
1, KF
H
96


















