
Trennverfahren Institut für Chemie, Karl-Franzens...
63
Vorlesungsskriptum Chromatographische Trennverfahren in der Organischen Chemie (SS 2004) Bernd Trathnigg HPLC Hardware und Grundlagen mit Schwerpunkt SEC Martin Mittelbach: Gaschromatographie Georg Uray Praktische Flüssigchromatographie und deren präparative Anwendung Wolfgang Stadlbauer Dünnschichtchromatographie Institut für Chemie, Karl-Franzens-Universität Graz
Transcript of Trennverfahren Institut für Chemie, Karl-Franzens...

Vorlesungsskriptum
Chromatographische
Trennverfahren
in der
Organischen Chemie(SS 2004)
Bernd Trathnigg
HPLC Hardware und Grundlagen
mit Schwerpunkt SEC
Martin Mittelbach:
Gaschromatographie
Georg Uray
Praktische
Flüssigchromatographie und
deren präparative Anwendung
Wolfgang Stadlbauer
Dünnschichtchromatographie
Inst
itut
für
Chem
ie,
Karl
-Fra
nzens-
Univ
ers
ität
Gra
z

Einleitung

Chromatographische Trennmethoden
Chromatographische Verfahren dienen zur Trennung von Stoffgemischen. Die getrennten Komponenten können identifiziert und ihre Menge quantitativ bestimmt werden. Die Trennung erfolgt in der Weise, daß eine mobile Phase an einer stationären phase vorbeiströmt, mit der die Probenkomponenten unterschiedlich stark wechselwirken. Damit kommt es zu einer Verteilung der Komponenten zwischen mobiler und stationärer Phase. Je nach dem Verteilungskoeffizienten erscheinen die einzelnen Komponenten früher oder später. Die mobile Phase kann gasförmig oder flüssig sein, die stationäre Phase flüssig oder fest. Esgibt allerdings auch Methoden, bei denen die stationäre Phase fehlt oder keine mobile Phase strömt. Damit ergeben sich folgende chromatographische Techniken:
Mobile Phase Stationäre Phase Methode
Gasförmig flüssig oder fest Gaschromatographie (GC) flüssig flüssig oder fest Flüssigkeitschromatographie (LC) überkritisch flüssig oder fest Supercritical Fluid Chromatography (SFC) flüssig keine Field Flow Fractionantion (FFF) keine flüssig oder fest Kapillarelektrophorese (CE)
Je nach der Natur der Probe eignen sich die verschiedenen Ausführungsvarianten unterschiedlich gut:
Probe Methode
verdampfbar Gaschromatographie (GC) Supercritical Fluid Chromatography (SFC)
nicht flüchtig, löslich Flüssigkeitschromatographie (LC)
geladen, löslich Kapillarelektrophorese (CE) löslich oder unlöslich Field Flow Fractionation (FFF)
Bei der Füssigkeitschromatographie gibt es je nach dem verwendeten Trennsystem verschiedene Ausführungsvarianten:Die stationäre Phase kann als dünne Schicht auf einem Träger (Dünnschichtchromatographie, DC oder Thin layer chromatography, TLC) oder an der inneren Oberfäche einer Kapillare vorliegen oder als gepackte Säule.
Bei der Säulenchromatographie bestimmt die Größe der Partikel die Dichte der Packung, und damit den Strömungswiderstand. Entsprechend steigt mit der Packungsdichte der erforderliche Druck, der benötigt wird, um die mobile Phase durch die Säule zu pumpen. Die gebräuchliche Abkürzung HPLC für High Performance Liquid Chromatography (=Hochleistungs-Flüssigkeitschromatographie) wird deshalb oft mißverstanden: der Druck (pressure) hat nichts mit der Trennleistung zu tun und ist nur eine unerwünschte Begleiterscheinung der dicht gepackten Säulen, die für hohe Trennleistung erforderlich sind !
Die wichtigsten Techniken in der HPLC unterscheiden sich in der chemischen Natur der stationären und mobilen Phase bzw. durch die Art der Wechselwirkung. Damit erfolgt die Trennung nach anderen Kriterien:

MobilePhase
Stationäre Phase
Trennkriterium Methode
unpolar polar polare Gruppen normal phase LC (NP-LC) polar unpolar unpolare Gruppen reversed phase LC (RP-LC) polar polar
unpolar unpolar
Gesamtgröße der Moleküle bzw. Molekulargewicht
Ausschlußchromatographie =Size exclusion chromatography (SEC), Gel-Permeations-Chromatographie (GPC)
Typische stationäre Phasen in der HPLC enthalten meist als Basis von Kieselgel, das an der Oberfläche mit polaren oder unpolaren Gruppen modifiziert sein kann:
Si OH
Si NH2
Si OOH
OH
SiCN
Si NO2
Si, Silica
NO2, Nitro
NH2, APS, AminoAminopropylsilyl
CN, CPS, PCN, Cyano,Cyanopropyl, Nitrile
OH, Diol, Glycerol
Si CH3
Si
Si
Si
Si
Si
Si
SiCN
Si
C1, TMS, Trimethyl
CN, CPS, PCN, Cyano, Cyanopropyl, Nitrile
C2, RP-2, Dimethyl
Phenyl, C6H5
C3, Propyl
C4, Butyl
C6, Hexyl
C8, MOS, RP-8, LC8, Octyl
C18, ODS, RP-18, LC18, Octadecyl
In der SEC werden meist vernetzte Polymere (z.B. Styrol-Divinylbenzol) verwendet.
Polymer-basierende Phasen können auch in der Adsorptionschromatographie vorteilhaft sein, wenn restliche Silanolgruppen die Trennung basischer Substanzen stören würden.
Je nach Chromatographieart (Adsorption, Verteilung, Ausschluss, ...) werden also ganz spezifische funktionelle Gruppen genützt.

Wolfgang Stadlbauer
Dünnschichtchromatographie

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 1 -
"Chromatographische Trennverfahren in der Organischen Chemie"
DÜNNSCHICHTCHROMATOGRAPHIEVorlesungs-Folien als PDF-Files: http://www-och.uni-graz.at/~sta/lehre/chrom/
1. Allgemeines
Geschichte: Vorläufer der DC sind Kapillarbilder (= Zirkularchromatogramme) von Farbstoffen (Runge, 1822).
Namensgebung für Chromatographie durch Tswett (russischer Botaniker, 1903, Auftrennung von Pflanzenfarb-
stoffen an Calciumcarbonatsäulen; chromatos = Farbe; graphein = schreiben).
Entwicklung der DC: Kuhn et al. (1931, Adsorptionschromatographie als Analysenmethode, Trennung von Caro-
tinoiden); Izmailov, Schraiber (Ringchromatogramme auf Al2O3 als Analysenmethode in der Pharmazie); Martin,
Synge (1944, Nobelpreis 1952, Papierchromatographie von Aminosäuren); Stahl (1958, Durchbruch der DC,
Namensgebung; engl. TLC); Halpaap, Ripphahn (1975, High Performance Thin Layer Chromatography, HPTLC).
Anwendung - WANN: Bei schwerflüchtigen Substanzen, bei polaren und unpolaren Substanzen. - Kostengün-
stige Bearbeitung grosser Probenanzahl in kurzer Zeit; - wenn Probenmaterial GC- und LC-Säulen beschädigen
würde; - wenn Detektion mit GC und LC nicht möglich oder sehr aufwendig sein würde; - wenn auch nach der
Chromatographie alle Probenbestandteile erkennbar sein müssen (z.B. an Start oder Front); - wenn getrennte
Bestandteile einzeln nachgewiesen oder detektiert werden müssen (z. B. Drogenscreening), - wenn keine elektri-
sche Energie vorhanden ist.
Anwendung - WO: In der Pharmazie und Drogenkunde, in der klinischen, forensischen und Biochemie (Wirk-
stoffe, Metaboliten), in der Kosmetologie (Farbstoffe, Konservierungs-, Parfüminhaltsstoffe, Tenside), in der Analy-
tik (Lebensmittel-, Umweltanalytik, Analytik anorganischer und organischer Substanzen), in der Prozess- und
Reinheitskontrolle im organischen Syntheselabor.
Methoden und Anwendungsformen: Analytische Methoden: qualitative bzw. quantitative Identifizierung mit
DC (veraltet: mit Papierchromatographie = PC)
Präparative Methode: Präparative Schicht-Chromatographie (PSC): Isolierung der Einzelkomponenten aus chro-
matographisch aufgetrennten Gemischen durch Extraktion aus der Schicht.
Vorteile der DC: Die DC ist eine rein physikalische Methode. Es tritt normalerweise kein Abbau oder Verlust und
keine Umwandlung ein, d.h., man kann prinzipiell nach der Durchführung der Chromatographie das untersuchte
Analysengut wieder quantitativ und unverändert zurückgewinnen.
Literatur: Dünnschichtchromatographie (E. Hahn-Deinstrop), Wiley-VCH 1998
Qualitative und Quantitative Dünnschichtchromatographie (H. P. Frey, K. Zieloff) Wiley- VCH 1992
Dünnschichtchromatographie (H. Jork, W. Funk, W. Fischer, H. Wimmer) Wiley-VCH 1993
Chromatographie (J. Becker), Vogel-Buchverlag, Würzburg, 1997 (auch für die anderen Teile der Chromatogra-
phievorlesung geeignet).
Chromatographische Trennmethoden (G. Schwedt) Thieme 1994
2. Trennprinzipien
Allgemeine Begriffe: Eine Lösung eines Substanzgemisches (mobile Phase) strömt über eine stationäre Phase
(oberflächenreicher Feststoff oder Flüssigkeit, die mit der mobilen Phase nicht mischbar ist). Die in der mobilen
Phase gelösten Substanzen haben unterschiedliche Affinität zur mobilen bzw. stationären Phase. Daraus resul-
tieren unterschiedliche Laufstrecken der einzelnen gelösten Substanzen und es kommt zu einer Auftrennung in
Einzelkomponenten.
Konzentration: Der Zusammenhang zwischen Stoffmengenkonzentration in der stationären und mobilen Phase
wird durch folgende Gleichung ausgedrückt. Diese Funktion wird als Sorptionsisotherme bezeich-
net:
Cs/Cm= K
Cs ist die Konzentration in der stationären Phase, Cm die Konzentration in der mobilen Phase.
Für geringe Stoffmengen gilt, dass das Verhältnis der beiden Konzentrationen konstant ist. Wenn
die Sättigungskonzentration der stationären Phase erreicht ist, knickt die Kurve ab. Die Konstante

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 2 -
K beschreibt im Geraden-Teil die Affinität der Substanz zur stationären Phase: Je grösser K ist, desto höher ist die
Affinität zur stationären Phase, d.h., desto langsamer ist die Laufgeschwindigkeit.
Trennprinzip Adsorption: Die Trennung erfolgt an der Oberfläche von feinverteilten stationären Phasen (polare
Materialien wie z.B. Kieselgel oder Aluminiumoxid). Die Adsorbate (in der mobilen Phase gelöste Stoffe) werden
umso stärker zurückgehalten, je polarer sie sind.
Eine Desaktivierung der stationären Phase erfolgt durch Wasser und polare Lösungsmittel.
Liste der Reihenfolge von polaren Gruppen nach steigender Polarität:
Kohlenwasserstoffe < Aromaten < Halogen < O-Alkyl < NO2 < NR2 < COOR < CR=O < NH2 < OH
Trennprinzip Verteilung: Die Trennung erfolgt zwischen zwei nichtmischbaren Flüssigkeiten. Eine davon ist auf
einem feinverteilten Träger fixiert (z. B. in den Kapillaren von Filterpapier, auf Cellulosepulver, Kieselgur, Dextran-
gel oder Polyamid). Die Trennung erfolgt nach dem Prinzip der multiplikativen Verteilung
Beschreibung des Laufverhaltens
Rf-Wert (retarding factor, relate to front) dient zur Beschreibung des Laufverhaltens
aufgetrennter Substanzen.
b b.... Laufstrecke der Substanz
Rf = ------
a a... Laufstrecke des Laufmittels
bis zum Mittelpunkt des Substanzflecks gemessen
bei unregelmässigen Flecken: Angabe von Rf-Bereichen
Der Rf-Wert ist nur ein Richtwert, er variiert durch unterschiedliche Trennbedingungen.
Der praktisch gefundene Wert ist daher oft sehr unterschiedlich. Bei einem Vergleich
zwischen zwei Substanzen muss daher immer mit einem Standard verglichen werden,
wobei beide auf derselben Platte aufgetragen werden müssen. Günstige Rf-Werte lie-
gen zwischen 0.2 und 0.8 (unter 0.1 = Sitzenbleiber, über 0.9 = Frontläufer, daher keine Aussage möglich).
Qualität der Trennung
Als TRENNLEISTUNG wird die Zonenverbreiterung des Substanzflecks entlang der Trennstrecke in Abhängigkeit
zur Bodenhöhe bezeichnet. Die Trennleistung ist eine Materialeigenschaft, sie wird durch die geometri-
sche Struktur beeinflusst, d.h. vom Partikeldurchmesser (Korngrösse) und der Korngrössenverteilung.
Die Trennstrecke hängt stark von der verwendeten Chromatographieart ab. So entspricht z.B. die Lauf-
strecke von 10 cm bei der HPTLC einer Laufstrecke von 2 m bei der Gaschromatographie.
Als SELEKTIVITÄT wird der Rf-Wert-Unterschied für die getrennten Substanzen bezeichnet, und sie
wird durch die chemische Struktur der stationären Phase und die Zusammensetzung des Laufmittels
beeinflusst.
Trennleistung und Selektivität ergeben zusammen die AUFLÖSUNG in einem Chromatogramm. Insgesamt ist
die Auflösung entscheidend für die praktische Anwendung, d.h., ob zwei ähnliche Substanzen getrennt oder nicht
getrennt werden (Substanzfleckverbreiterung oder zu kleine Rf-Wert-Unterschiede können eine Trennung verhin-
dern).
Quantitative Beziehung: Die Van-Deemter-Gleichung beschreibt diese Zusammenhänge mit drei Parametern:
Streudiffusion, Molekulardiffusion und Ad- bzw. Desorption bewirken breitere, unscharfe und verlängerte Flecken.
Empirische Einflussgrössen für die TRENNUNG
- Einflüsse auf die stationäre Phase: Aktivität des Adsorbens, Luftfeuchtigkeit, Korngrösse und Korngrössenvertei-
lung, Art des Bindemittels, Schichtdicke und Gleichmässigkeit der Schicht.
- Einflüsse auf die mobile Phase: Laufmittelgradient, Fliessgeschwindigkeit, Verunreinigungen im Laufmittel (Ein-
flüsse auf Aktivität durch polare Verunreinigungen, Änderung der Laufmitteleigenschaft durch saure oder basische
Verunreinigungen), Veränderung des Laufmittels durch Verdunsten und Auftrennung der Komponenten.
- Kammertyp und Konditionierung beeinflussen stark die Trennung, weiters beeinflusst die Kammersättigung die
Rf-Werte.

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 3 -
- Einflüsse durch unterschiedliche Fleckengrösse aufgrund der Trennleistung und der Auftragung, die Entfernung
Start - Laufmitteloberkante, durch die Länge der Trennstrecke und den pH-Wert des Laufmittels.
3. DIE STATIONÄRE PHASE
Die wichtigste Voraussetzung für die Dünnschichtchromatographie als Routinemethode sind kommerziell erhältli-
che Sorbentien von gleichbleibender Qualität. Die allgemeinen Eigenschaften werden durch Art und Aktivität der
Sorbentien bestimmt.
Chemische Zusammensetzung und Struktur: Ein allgemeine Einteilung unterscheidet zwischen polaren bzw.
hydrophilen Phasen (straight/normal phases) sowie unpolaren bzw. lipophilen Phasen (Umkehrphasen, reversed
phases, RP). Daneben gibt es noch mittelpolare Phasen durch modifizierte Sorbentien.
Praktische Bedeutung besitzen vor allem Kieselgel, modifizierte Kieselgelarten, Aluminiumoxid, Cellulose, modi-
fizierte Cellulose und in Spezialfällen Polyamid.
KIESELGEL wird für ca. 90% aller Trennungen verwendet. Kieselgel ist ein amorphes Pulver mit einigen Si-OH-
Gruppen pro 10 nm2 an der Oberfläche. Kieselgel bildet mit den Trennsubstanzen vor allem Wasserstoffbrücken
aus (Prinzip: Adsorptionschromatographie). Durch die Si-OH-Gruppen reagiert Kieselgel schwach sauer.
modifiziertes Kieselgel: Bedeutung haben hydrophob gemachte Kieselgele, hergestellt durch Reaktion mit Silanen,
die Amino-, Dihydroxy- oder Nitrilgruppen tragen (RP).
Durch geeignete Reaktion erhält man auch chirale Kieselgele, die optisch aktiv sind und zur Trennung optischer
Isomerer verwendet werden können.
Probleme bei der Verwendung von Kieselgel: Isomerisierungen durch den schwach sauren Einfluss und die feine
Verteilung auf einer grossen Oberfläche, Abbau (Ringöffnung) von -Lactamen, Kondensation von Aceton (das oft
als Laufmittel verwendet wird).
ALUMINIUMOXID (Prinzip: Adsorptionschromatographie) besitzt in der Dünnschichtchromatographie ähnliche
Eigenschaften wie in der Säulenchromatographie. Es gibt neutrales, basisches (AlO2-hältiges) und saures
(Al3+hältiges) Aluminiumoxid.
Weiters werden verschiedene Aktivitätsstufen (Stufe I-V) durch Desaktivierung mit Wasser hergestellt. Dabei wer-
den die aktivsten Stellen mit 0-18% Wasser belegt.
Probleme: Es kann zur Verseifung von Estern kommen, oder es tritt Autoxidation von Fettsäuren ein. Auch Wan-
derungen von C=C-Doppelbindungen, die bereits erwähnte Kondensation von Aceton sowie Isomerisierungen
können auftreten.
POLYAMID Prinzip: Verteilungschromatographie durch polare Wechselwirkungen (H-Brücken)
CELLULOSE (Prinzip: Verteilungschromatographie) verhält sich ähnlich wie die Papierchromatographie und ist
daher besonders für hydrophile Substanzen geeignet, während reines Kieselgel und Aluminiumoxid vorwiegend
für lipophile Substanzen geeignet ist (bei RP gelten die umgekehrten Verhältnisse). Typische Anwendungsbei-
spiele für Cellulose sind Aminosäuren und Kohlenhydrate. Acetylierte Cellulose (RP) wird für für polycyclische
Aromaten, Chinone, Carbonsäuren verwendet. Cellulose-Ionenaustauscher sind für Peptide, Enzyme, Nucleotide,
Nucleoside geeignet.
Vergleich mit Papierchromatographie: Vorteile der DC: die Trennung verläuft viel schneller, die Folien und Platten
sind auch mit aggressiven Lösungsmitteln besprühbar, es wird eine bessere Trennung (schärfere Flecken = bes-
sere Trennleistung) erzielt. Vorteile der PC: die Chromatogramme sind besser archivierbar, man hat eine grös-
sere Papierauswahl und die Materialien sind insgesamt billiger.
Korn- und Porencharakteristik: Die Trennleistung wird von der geometrischen Struktur beeinflusst, die Selekti-
vität hauptsächlich von der chemischen Struktur. Für eine gute Trennleistung
wird ein kleiner Partikeldurchmesser und eine enge Korngrössenverteilung
benötigt.
Partikelgrösse und Form: Kieselgel für DC-Zwecke hat einen Partikeldurch-
messer von 5-40 m. Die Oberfläche und das Porenvolumen beeinflussen die
in der Sorptionsisotherme dargestellten Adsorptionskraft. Die Oberfläche
beträgt durchschnittlich 400-600 m2 pro Gramm, das Porenvolumen ca. 6 nm.
Standardmaterial weist gebrochenes Material auf, sphärische Partikel
(LiChrospher) besitzen gesteigerte Trennleistung.

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 4 -
Schichteigenschaften: Die Schichtdicke beträgt ca. 0.1 - 0.5 mm in der Dünnschichtchromatographie und 0.5 - 2
mm in der präparativen Schichtchromatographie. Als Binder wird meist Gips verwendet, das Plattenmaterial ist je
nach Anforderungen Glas, Kunststoff oder Aluminium. Es gibt auch spezielle DC-Materialien, die eine Schicht mit
Konzentrierungszonen enthalten. Daraus resultieren kleinere Flecken und eine bessere Auflösung.
Schichtzusätze: Fluoreszenzindikatoren zur Detektion mittels Fluoreszenzlöschung (manganaktiviertes Zinksili-
kat...grün, säurestabile Wolframate...blau), Imprägnierungen zur schnellen Detektion (z. B. Coffein für polycycli-
sche Aromaten).
4. DIE MOBILE PHASE
Der Grund für das Strömen des Fliessmittels ist der Kapillardruck. Dieser hat seine Ursache darin, dass das
das Fliessmittel von der ebenen Oberfläche des Vorratsgefässes in die kapillaren Hohlräume mit engem Krüm-
mungsradius strömt, da dadurch die Oberflächenspannung verringert wird.
Die Wahl des Laufmittels hat einen entscheidenden Einfluss auf die Trennung. Je stärker das Laufmittel an der
stationären Phase adsorbiert wird, desto grösser ist seine Elutions- (Verdrängungs)-kraft, d.h., desto grösser wird
der Rf-Wert der zu trennenden Substanzen. Im Extremfall wird das Adsorbat gar nicht mehr adsorbiert, da das
Laufmittel bereits alle aktiven Stellen des Adsorbens besetzt hat.
Einfluss des Laufmittels: Das Laufmittel hat daher zwei wichtige Eigenschaften in der DC: es ist sowohl Konkur-
rent um aktive Adsorptionsstellen und bewirkt auf der anderen Seite die Solvatisierung von Substanzen, d.h., die
Lösung im Laufmittel.
Die Zusammensetzung des Laufmittelgemisches muss nicht mit der MOBILEN PHASE identisch sein, da auch
das Laufmittel chromatographiert wird (in der Verteilungschromatographie wird dem Laufmittel ausserdem noch z.
B. das Wasser für die Ausbildung der stationären Phase entzogen). Dazu kommen noch Verdunstungseffekte, die
das Auftreten von sog. -Fronten bewirken.
Die Eigenschaften verschiedener Laufmittel werden in der Eluotropen Reihe für die Adsorptionschromatogra-
phie zum Ausdruck gebracht. Die eluotrope Reihe ist eine empirische Ordnung der Laufmittel nach steigender
Elutionskraft (für Kieselgel und Aluminiumoxid) und entspricht im allgemeinen der Dielektrizitätskonstanten (in
Klammern angeführt), mit Ausnahme einiger "Ausreisser", die durch andere Eigenschaften (z.B. Ausbildung von
Wasserstoffbrückenbindungen) verursacht werden:
Cyclohexan (2) Tetrachlorkohlenstoff (2) Toluol (2) Chloroform (5) Dichlormethan (9) Acetonitril (37) 2-
Propanol (22) Essigester (6) Aceton (21) [Pyridin(12)] Ethanol (25) Dioxan (2) THF (7) Methanol (32)
Wasser (80)
Es gilt allgemein, dass ab Essigester eine Desaktivierung der stationären (Adsorbens-)Phase eintritt.
Umkehrung der eluotropen Reihe bei Polyamid als stationärer Phase (RP).
Das Laufmittelgemisch für Verteilungschromatographie (Papierchromatographie und Cellulose) enthält stets
einen unpolaren organischen Anteil und einen stark polaren Anteil (meist Wasser)
Die polare Phase wird am Träger angereichert und bildet dadurch die stationäre Phase, während die mobile
Phase lipophiler wird. Dadurch ist eine Auftrennung stark polarer Verbindungen möglich.
5. Kammersättigung: Ohne Kammersättigung verdampft ein Teil des
Lösungsmittels in Frontnähe. Dadurch ist zwar mehr Laufmittel nötig, was
besonders bei grossen Kammern zu berücksichtigen ist, auf der anderen
Seite steigen die Rf-Werte, was bei Trennproblemen mit kleinen Rf-Werten
von Vorteil sein kann. Gesättigte Kammern liefern lineare Rf-Werte, was bei
grossen Rf-Werten von Vorteil ist.
6. Einfluss verschiedener Laufmittel auf ein Trennproblem: In unpolaren oder stark polaren Laufmit-
teln ist oft keine Trennung zu beobachten, während mässig polare Laufmittel im mittleren Rf-Bereich die beste Auf-
trennung liefern.
Ermittlung geeigneter Trennbedingungen: Die Festlegung der Trennbedingungen resultiert aus der Wechsel-
wirkung der Faktoren MOBILE PHASE, STATIONÄRE PHASE und UNTERSUCHUNGSGUT. Man kann daraus

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 5 -
eine logische "Wahrheitstabelle" ableiten, die die Zusammenhänge grob widerspiegelt, obwohl natürlich jedes
Trennproblem am besten meist nur von einem Trennprinzip optimal gelöst wird.
Laufmittelwahl: bei komplizierten Problemen sind vor Beginn eigener Untersuchungen Literaturrecherchen von
Vorteil. Dabei sollten toxische Komponenten durch ähnliche aus der eluotropen Reihe ersetzt werden. Um einen
groben Überblick zu erhalten, welches Laufmittel für das Trennproblem geeignet ist, können durch einen Spot-
Test auf einer DC-Folie mehrere Laufmittel auf ihre Eignung untersucht werden. Dazu lässt man nach dem Auftra-
gen der Substanz das Lösungsmittel verdunsten. Dann setzt man eine Kapillare mit dem zu testenden Laufmittel
in die Mitte des Substanzflecks auf und lässt das Laufmittel auf diese Weise zufliessen. Dadurch erhält man ein
zirkulares Chromatogramm, das grob anzeigt, ob die Substanz in dem getesteten Laufmittel überhaupt läuft, und
ob eine Trennung zu beobachten ist.
Häufig verwendete Laufmittel: für die gängigen Trennprobleme haben sich die folgenden Laufmittelgemische
bewährt:
DC auf Kieselgel: Aceton-Toluol 1:10 (für eher unpolare Substanzen)
Chloroform-Aceton 7:3 (für eher polare Substanzen).
Bei sehr polaren Substanzen kann (wenig) Ethanol oder Methanol zugesetzt werden, Carbonsäuren laufen durch
Zusatz von einigen Tropfen Eisessig besser, bei basischen Verbindungen kann mit Pyridin oder Triethylamin eine
bessere Trennung erreicht werden. Wenn man vor der Entscheidung steht, welches Laufmittel(gemisch) man für
ein bestimmtes Trennproblem verwenden soll, so ist vor allem die Struktur der zu trennenden Substanzen zu
berücksichtigen. Hier kann mit der alten Chemikerweisheit "similia similibus solvuntur" eine grobe erste Entschei-
dung getroffen werden.
DC(=PC) auf Cellulose n-Butanol-Aceton-Eisessig-Wasser 3:3:7:2 (für saure Substanzen)
i-Propanol-Ammoniak-Wasser 6:3:1 (für basische Substanzen)
Auch hier gilt wieder, dass Säuren in stark sauren Laufmitteln getrennt werden, Basen in basischen Laufmitteln.
7. Praktische Hinweise zur Durchführung
Handhaben der Fertigschichten: Zuschneiden und Kanten versäubern, Platte möglichst nicht berühren. Die Akti-
vität entspricht der Luftfeuchigkeit des Labors, daher ist ein Voraktivieren normalerweise nicht notwendig.
Probenvorbereitung: kein besonderes Thema; bei der Adsorptionschromatographie keine wasser-feuchten Pro-
ben verwenden. Die Substanz muss vollständig gelöst sein. Wenn nur ein Teil gelöst ist, so würde das eine
vollkommen falsche Zusammensetzung der Probe vortäuschen. Daher ist es wichtig, ein gutes, aber leicht flüchti-
ges Lösungsmittel zu verwenden. Dieses sollte ausserdem ungiftig sein, nicht zu polar und nicht zu hoch siedend
sein. Polare Lösungsmittel erzeugen in der Startzone Ringchromatogramme, die im Extremfall 2 Substanzen vor-
täuschen können. In der Adsorptionschromatographie wird zum Lösen meistens ACETON verwendet. In der Ver-
teilungschromatographie auf Cellulose wird zum Lösen meistens WASSER oder ETHANOL verwendet
Probe und Vergleich müssen im gleichen Lösungsmittel gelöst sein, um keine nichtreproduzierbare Desakti-
vierung der Platte zu erhalten.
Auftragen der Probe: Da die Rf-Werte manchmal konzentrationsabhängig sind, müssen Vergleichssubstanzen
auf derselben Platte wie die Probe chromatographiert werden. Bei der Dünnschichtchromatogra-
phie wird die Substanz punktförmig aufgetragen, bei der Präparativen Schichtchromatographie
bandförmig. Die Substanz darf nicht zu konzentriert aufgetragen werden, da sonst Schwanzbil-
dung durch Übersättigung der Schicht auftritt. Erklärbar ist das durch gekrümmte Adsorptionsiso-
thermen.
Vor dem eigentlichen Chromatographievorgang muss das Lösungsmittel vollständig entfernt
werden, da sich sonst die Laufmittelzusammensetzung für die einzelnen Substanz-Probanden in nicht reproduzier-
barer Weise ändert (unterschiedliche Laufstrecken, -Fronten).
lipophil (+) Adsorption aktiv (+) unpolar (+)
lipophil Verteilung lipophil polar
hydrophil Adsorption inaktiv polar
hydrophil (+) Verteilung hydrophil (+) unpolar (+)
Untersuchungsgut Trennprinzip stationäre Phase mobile Phase

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 6 -
Analytische Chromatogramme: Zuerst wird die Startlinie markiert (bei der aufsteigenden DC ca. 5-10 mm vom
unteren Rand entfernt). Die Laufstrecke soll mindestens 7-10 cm betragen. Die Markierung wird mit einem wei-
chem Bleistift am äusseren Rand durchgeführt. Am oberen Rand können noch weitere Beschriftungen ange-
bracht werden (meist Probenbezeichnungen).
Präparative Trennungen werden meist auf 20x20cm-Platten mit 2 mm Schichtdicke durchgeführt. Im Abstand
von 2-3 cm vom unteren Rand wird eine Startlinie aufgetragen (mit einem Auftragegerät oder mit Lineal und Pipet-
te). Die Startlinie wird mit Auftragegeräten aufgebracht, man kann dabei bis ca. 200 mg Substanz auftrennen. Es
muss auf genügend Laufmittelvorrat geachtet werden.
Auftragemethoden: Man verwendet Kapillaren oder Spritzen, deren Volumen für qualitative Untersuchungen
nicht bekannt zu sein braucht. Diese Kapillaren kann man selbst aus Glasrohren oder Schmelzpunktsröhrchen
über dem Bunsenbrenner ziehen.
Auftragen für qualitative DC's: Kapillaren aufsetzen, Kapillaren sollten sich vollständig füllen und entleeren. Die
Konzentration soll möglichst so gewählt werden, dass ein einmaliges Auftragen ausreicht. Die Startzonen sind vor
dem nächsten Auftragen und dem Entwickeln vollständig zu trocknen.
Auftragemethoden für quantitative Untersuchungen: Man verwendet Mikrokapillaren mit definiertem Volumen.
Für DC verwendet man Volumina von 0.5-5 mL, für HPTLC Volumina von 0.1-0.5 mL. Zur reproduzierbaren Auf-
tragung gibt es spezielle Auftragegeräte.
Positionierung der Proben: bei quantitativen DC's wird doppelt aufgetragen (um eine Plattenhälfte versetzt), um
Ungleichmässigkeiten der Schicht auszugleichen. Bei Identitätsproblemen wird überlappend aufgetragen, weil
dadurch ein besserer Vergleich möglich ist als durch nebeneinanderliegende Flecken.
8. ENTWICKELN
Aufgabe des Laufmittels: Das Laufmittel soll nicht nur das Substanzgemisch lösen, sondern auch die
Trennsubstanzen über die Sorbensschicht transportieren und das anstehende Trennproblem durch geeignete
Trennstrecken (Rf-Werte) lösen.
Anforderung an das Laufmittel: Ausreichende Reinheit - ausreichende Stabilität - geringe Viskosität - lineare
Adsorptions/Verteilungsisotherme - mittlerer Dampfdruck - geringe Toxizität.
Reinheit des Laufmittels: Für qualitative Untersuchungen ist die Reinheit der üblichen Lösungsmittel ausrei-
chend (eventuelle Verunreinigen könnten auch aus der Entwicklungskammer stammen, wenn z.B. das Laufmittel
die Startflecken löst). Für quantitative Untersuchungen sind hochgereinigte Lösungsmittel für die Chromatographie
notwendig. ACHTUNG: manchen Lösungsmitteln sind Stabilisatoren zugesetzt (Chloroform kann z.B. Ethanol
enthalten).
Begriff der MOBILEN PHASE: Das Laufmittel wird bei der Wanderung in eine flüssige stationäre Phase (in die
Poren des Sobens) aufgetrennt. Bei der Verteilungschromatographie kommt es zur Verarmung an Wasser, bei der
Adsorptionschromatographie zur Verarmung an polaren Laufmittelbestandteilen. Der Rest = mobile Phase. Durch
unterschiedliche Wanderungsgeschwindigkeiten der Laufmittelbestandteile: Ausbildung von -Fronten.
Die Auswahl der Laufmittel erfolgt nach der eluotropen Reihe (gilt streng nur für Kieselgel); zuerst mit mittlerer
Elutionskraft und reinen Lösungsmitteln beginnen. Niedrige Viskosität ist vorteilhaft (Zeitfaktor). Die Toxizität ist zu
beachten, ebenso das Auftreten von -Fronten, denn diese täuschen falsche Rf-Werte vor und vermindern die
Selektivität.
Ansetzen und Aufbewahren von Laufmitteln: Die einzelnen Teilvolumina müssen einzeln abgemessen werden
und dann erst gemischt werden, sonst kommt es zu Fehlern durch Volumsänderungen. Die Zusammensetzung ist
in Volumsteilen angegeben. Das Mischen erfolgt bereits im Vorratsbehälter, erst dann wird das Laufmittel in die
Chromatographie-Kammer gegeben und gut verschlossen halten, um teilweises Verdampfen/Verdunsten zu ver-
hindern. Das Laufmittel in der Chromatographie-Kammer darf nicht zu oft wiederverwendet werden: maximal 2-3
mal, denn durch ungleiche Verdunstung, chemische Reaktion der Komponenten oder bevorzugte Adsorption einer
Komponente kann es zur Verarmung von Laufmittelanteilen kommen. Dadurch ändert sich die Laufmittelzusam-
mensetzung in unkontrollierter Weise. Bedenkliche Laufmittel (Benzen, Ether, CKW) und kritische Laufmittel (Säu-
ren, Alkohole, Ester) beachten.

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 7 -
Die gebräuchlichste Technik ist die Aufsteigende Entwicklung: Dabei wird die DC-Platte so in die Kammer
gestellt, dass die DC-Schicht unterhalb der Startpunkte benetzt wird (die Start-
punkte dürfen auf keinen Fall in das Laufmittel tauchen!). Durch die Kapillar-
kräfte steigt das Laufmittel max. ca. 20 cm hoch und transportiert dabei das
Trenngut. Die Startflecken vergrössern sich zur Front hin. Wenn die
gewünschte Laufhöhe erreicht ist, wird die Platte aus der Kammer genommen,
die Laufmittelfront am Rand durch Einritzen oder mit einem Bleistift markiert,
und die Platte getrocknet.
8.8 Mehrfachentwicklung: jeweils nach Zwischentrocknung und eventueller Rekonzentrierung erfolgt eine erneute
Entwicklung. Meist verwendet man verschiedene Laufmittel, um z.B. unpolare Verunreinigungen an die Front zu
befördern, bevor man das eigentliche Substanzgemisch auftrennt.
Durchlaufentwicklung: Dazu verwendet man eine DC-Kammer mit einem Schlitz im Deckel. Während der Entwick-
lung verdampft das Lösungsmittel kontinuierlich an der Front und man erreicht dadurch eine bessere Auflösung für
kleine Rf-Werte, während grosse Rf-Werte zusammengestaucht werden.
Horizontale Entwicklung: man verwendet eine eben liegende Platte, das Laufmittel wird z.B. über einen seitli-
chen Docht oder Kapillarspalten zugeführt.
9. DC-Trennkammern
Kammer-Arten: Hier gibt es zwei Arten: mit grossem Gasraum (Normal-Kammer) und kleinem Gasraum (Sand-
wich-Kammer). Der Grund dafür ist, dass bei grossen Kammern und präparativen Platten der Laufmittelverbrauch
oft so gross ist, dass das Laufmittel nicht immer bis zum Ende der Trennung ausreicht. Das wird durch die Sand-
wichtechnik vermieden.
Die gesättigte Normalkammer ist für 2 DC-Platten bis zum Format 20x20 cm geeignet. Die Wände werden mit
Filterpapier ausgekleidet, wobei das Filterpapier so zugeschnitten wird, dass seitlich ein Sichtfenster frei bleibt, um
den Trennvorgang beobachten zu können. Die Kammer wird 1 cm hoch mit Laufmittel gefüllt.
Bei der nichtgesättigter Kammer fliesst mehr Laufmittel durch Platte, da ein Teil des Laufmittels an der Front
verdampft. Daraus resultieren grössere Rf-Werte in der unteren Hälfte des Chromatogramms.
andere Kammerarten: Normalkammer für 2 Platten, Schräge Kammer (braucht weniger Laufmittel), runde Kam-
mern (z.B. für analytische DC's). Die Doppeltrogkammer hat eine Schwelle am Boden der Kammer. Man kommt
dadurch mit weniger Lösungsmittel aus, sie wird auch für Vorbeladung verwendet.
Kammern mit kleinem Gasraum: Die Sandwichkammer wird dann verwendet, wenn die Schicht nicht mit Lauf-
mitteldampf vorbeladen werden soll.
Die Linearkammer wird für die horizontale Entwicklung benötigt. Meist verwendet man diese Technik für die
HPTLC. Um den Laufmittelverbrauch klein zu halten, wird in einer Sandwichtechnik durch eine Gegenplatte der
Gasraum klein gehalten, der Laufmittelzufluss erfolgt seitlich über Kapillarspalten in einem Glasstreifen. Diese
Kammerart ist besonders für hohe Probenzahlen und schnelle Analysen geeignet (70 Parallelproben).
Durchführung der Entwicklung: Während der Entwicklung dürfen keine grossen Temperaturschwankungen auf-
treten, da es sonst zur Ausbildung unregelmässiger Fronten kommt. Die Platten sind auch stets vor Sonnenlicht zu
schützen, da sonst die Gefahr besteht, dass Photoreaktionen auf der Platte stattfinden.
Eindimensionale Entwicklung: häufigste Entwicklung, speziell bei quantitativen DC's.
Mehrdimensionale Entwicklung: Die erste Auftragung erfolgt in einer Ecke, zuerst wird aufsteigend nach oben
entwickelt und getrocknet. Dann wird die Platte um 90° gedreht und erneut in
einem anderem Laufmittel entwickeln. Dadurch kann auf einer einzigen Platte
durch die Vortrennung eine bessere Trennwirkung erzielt werden.
Für schwertrennbare Gemische, oder zum Nachweis von Reaktionen, die wäh-
rend der Entwicklung auf der Platte auftreten.
10. Trocknen: Das Laufmittel muss schnell von der Schicht entfernt werden, da sonst nachträgliche Diffusion
die Flecken vergössert und die Qualität des Chromatogramms verschlechtert. Auf der Platte befindliches Laufmit-
tel verschlechtert auch die nachfolgende Detektion. Das Trocknen erfolgt waagrecht, bei empfindlichen Substan-

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 8 -
zen mit kalter Luft, sonst mit warmer Luft (nicht zu heiss trocknen, da sonst durch Thermolyse und Abdampfen der
Substanzen die Qualität des Chromatogramms verschlechtert wird).
Entsorgen des Laufmittels in die entsprechenden Behälter; dann DC-Kammer gut reinigen, da sonst Ver-
schmutzungen Artefakte auf den nachfolgenden Chromatogrammen bewirken. Die Kammer muss gut getrocknet
werden, um eine unkontrollierte Veränderung des nächsten Laufmittels zu verhindern.
11. DETEKTION: Nach dem Entwickeln und Trocknen werden die getrennten Substanzen detektiert.
Visuelle Detektion: färbige Substanzen sind direkt auf dem Chromatogramm im sichtbaren Licht erkennbar
(Farbstoffe, färbige Verbindungen)
Detektion mit UV-Strahlung: Dazu verwendet man schwache UV-Lampen (ca. 15 W) mit Wellenlängen von 254
und 366 nm. Substanzen, die das UV-Licht von 254 nm absorbieren (Aromaten, längere konjugierte C=C-Syste-
me), verringern die Emission des auf der Platte aufgebrachten Fluoreszenzindikators F 254. Durch diese Flu-
oreszenzlöschung (quenchen) erscheinen die Substanzen als dunkle Flecken auf fluoreszierendem hellgrünem
oder hellblauen Untergrund. Da die Löschung bei UV-absorbierenden Substanzen proportional zur Substanz-
menge ist, kann man hier auch grobe quantitative Aussagen treffen.
Substanzen, die durch UV-Licht von 366 nm zur Eigenfluoreszenz angeregt werden, erscheinen als leuchtende
farbige Flecken am dunklen Chromatogramm. Diese Fluoreszenz ist stark strukturabhängig und kann keinesfalls
zu einer mengenmässigen Beurteilung herangezogen werden.
Derivatisierung ist immer dann notwendig, wenn die Substanzen nicht färbig sind und auch auf UV-Licht durch
quenching oder Fluoreszenz nicht (oder nicht genügend) ansprechen. Auch kann eine Steigerung der Selektivität
und eine Verbesserung der Nachweisempfindlichkeit erreicht werden.
Der häufigste Fall ist die postchromatographische Derivatisierung nach dem Entwickeln und Trocknen. Dies wird
durch Aufbringen von Nachweisreagenzien erreicht.
Buchhinweis: Dünnschichtchromatographie - Reagenzien und Nachweismethoden (H. Jork, W. Funk, W.
Fischer) Wiley - VCH 1993
Anfärbereagenzien für Dünnschicht- und Papierchromatographie (Reagenzien - Merck, Darmstadt, 1980).
Derivatisierungsreaktionen:
- Thermo- oder Photochemische Reaktion: Zersetzung, Reaktion mit modifizierten Kieselgelschichten
- Aufsprühen von Nachweisreagenzien (häufigste Technik)
- Tauchen von DC-Platten (umweltfreundlicher, billiger)
- Bedampfen von DC-Platten - Reaktion mit Iod, Chlor, ....
Derivatisierung durch Aufsprühen: Dazu ist ein Laborsprüher mit Druckluft oder Treibgas notwendig. Das
Besprühen muss immer im Abzug oder in einer Sprühbox durchgeführt werden, um
die giftigen, aggressiven Reagenznebel und Lösungsmitteldämpfe nicht einzuatmen
(Handschuhe und Schutzbrillen!). Das Chromatogramm wird schräg in die Sprühbox
gestellt, dann wird mit gleichmässigem Sprühnebel auf die Platte gesprüht. Es ist dar-
auf zu achten, dass sich keine Tröpfchen bilden. Es wird solange gesprüht, bis die
Schicht leicht glänzt. Oft muss nach dem Sprühen erwärmt werden, um die Reaktion
zwischen Substanzflecken und Reagens zu vervollständigen. Die entstandenen
Flecken müssen sofort mit Bleistift gekennzeichnet werden, denn oft verblassen diese nach kurzer Zeit.
Derivatisierung durch Tauchen: Statt Sprühen kann man auch Tauchvorrichtungen verwenden, die z.T. auto-
matisiert sind. Vorteile gegenüber Sprühen: gleichmässigere Belegung der Schicht - keine Abhängigkeit vom
Sprühgerät - exakte quantitative Auswertung - bessere Reproduzierbarkeit - keine Kontamination des Arbeitsplat-
zes - keine Sprüheinrichtung mit Abzug notwendig. Manuelles Tauchen ist nicht gut reproduzierbar, aber einfach.
Automatische Tauchgeräte: mit definierter Verweilzeit, Senken und Heben sind gut reproduzierbar. Tauchlösun-
gen sollten weniger konzentriert sein als Sprühlösungen, Wasser wird meist durch Alkohol ersetzt. Die Trennsubs-
tanzen dürfen nicht im Tauchmittel löslich sein. Grosse Substanzmengen und lange Tauchzeiten bilden "Kometen-
schweif". Nach dem Tauchen wird die Platte mit Luft abgeblasen und getrocknet. Oft muss nach dem Tauchen
noch erwärmt werden, um die Reaktion zu beschleunigen. Die entstandenen Flecken müssen auch hier sofort mit
Bleistift gekennzeichnet werden, denn oft verblassen diese nach kurzer Zeit.

Skriptum: Chromatographische Trennverfahren - Stadlbauer Seite - 9 -
Beispiele von Derivatisierungsreagenzien.
Ninhydrin für den Nachweis von Aminosäuren, Aminen, .....(0.2% Ninhydrin in Ethanol, Sprühen oder Tauchen)
Vanillin - Schwefelsäure für höhere Alkohole, Steroide, etherische Öle....(0.5-1% Vanillin in Ethanol/konz. Schwe-
felsäure [1:10], Sprühen oder Tauchen)
Ammonmolybdat-Cersulfat für höhere Alkohole, Steroide,....(4% Ammonmolybdat und 10% Cer(IV)sulfat in 10%
Schwefelsäure, Sprühen oder Tauchen)
Anisaldehyd-Schwefelsäure für Polyalkohole, Steroide, Terpene....(1% Anisaldehyd in konz. 2% Schwe-
felsäure/Eisessig, Sprühen oder Tauchen)
Kaliumpermanganat für Polyalkohole, Polycarbonsäuren, ungesättigte Verbindungen.....(1% KMnO4 in Wasser
oder Sodalösung, Sprühen oder Tauchen)
Bedampfen mit Ioddämpfen - unspezifischer Nachweis vieler Substanzen.....(Glasschale mit Iod in verschlos-
senem Glasgefäss)
Bedampfen mit Chlor und nachfolgende Reaktion mit KI/Stärke für den Nachweis von Säureamiden (Chlor aus
KMnO4/Salzsäure erzeugen, dann mit KI/Stärkelösung sprühen).
12. AUSWERTUNG
Visuelle Auswertung: qualitativ: Bei der Kontrolle von Syntheseansätzen zur Identifikation oder Reinheitsprü-
fung muss man immer mit Vergleichs-(Referenz)-substanzen am selben Chromatogramm arbeiten, denn die Rf-
Werte sind nicht genau reproduzierbar. Bei unklaren Ergebnissen müssen auch Konzentrationsreihen hergestellt
werden, denn die Rf-Werte ändern sich z.T. durch nichtlineare Sorptionsisothermen mit der Konzentration. Wenn
die Rf-Werte sehr ähnlich sind, so stellt man sich - wenn vorhanden - ein Gemisch aus den beiden zu untersu-
chenden Substanzen her und untersucht, ob sich beide trennen lassen. Eventuell muss noch mit geänderten Lauf-
mitteln experimentiert werden.
halbquantitativ: Man kann durch Konzentrationsreihen eine Untersuchung durchführen, ob Grenzwerte deutlich
unter- oder überschritten werden. Dazu wird die Probe und mehrere Konzentrationen der interessierenden Subs-
tanz am selben Chromatogramm aufgetragen. Die ähnlichsten Flecken werden nochmals nebeneinander aufgetra-
gen. Die Auswertung erfolgt durch visuellen Vergleich oder Messung der Fleckendurchmesser. Die Genauigkeit
beträgt dabei etwa 10%.
quantitative optische Auswertung: Zur direkten quantitativen Auswertung werden gleiche Volumina von Stan-
dard und Probe auf die Platte aufgetragen. Nach dem Entwickeln wird das Chromato-
gramm mit dem Spektralphotometer abgescannt. Dazu wird das Chromatogramm mit
UV/vis im Absorptionsmaximum in Reflexion vermesssen. Zusätzlich gibt es noch
Kopplungsmethoden zur Substanzidentifikation (z.B. FT-IR, RAMAN und MS)
Etwas ungenauer wird das (meist derivatisierte) Chromatogramm digitalisiert und
durch ein Computerprogramm ausgewertet.
13. DC-Auswertegeräte: UV-Vis DC-Scanner: analoges Spektralphotometer-System mit Photomultiplier,
sehr genau, aber empfindlich und störungsanfällig.
Auswertung mittels Digitalisierung: Digitaler Bildaufnahmeteil und Datenverarbeitung, aber derzeit noch keine
exakte quantitative Auswertung möglich; Vorteil: einfach, billig, robust.
14. DC-Chromatographiekurs (im Rahmen der organisch-chemischen Übungen)
- Photochemische Umlagerung von Azobenzen und DC-Auftrennung der (E)-
und (Z)-Isomeren
-Auftrennung eines Aminosäurengemisches und DC-Identi-
fizierung nach Derivatisierung mit Ninhydrin
- DC-Analyse eines unbekannten Substanzgemisches und Identifizierung durch UV-Detektion sowie Deriva-
tisierung durch Tauchen in Vanillin-Schwefelsäure
N N N N
trans (E) - cis (Z) -Azobenzen
R
NH2
O
OH+
O
O
O
O
N
OH
O
O

Martin Mittelbach:
Gaschromatographie

1
CHROMATOGRAFISCHE TRENNVERFAHREN IN DER ORGANISCHEN CHEMIE
GASCHROMATOGRAFIE
M.Mittelbach, 2003
EINLEITUNG
Die Gaschromatografie wurde im Jahre 1997 50 Jahre alt; im Juni 1947 wurde an der Universität
Innsbruck der erste Gaschromatograf von Erika Cremer und Fritz Prior vorgestellt. 1958 Patent wurde
Golay das Patent für Kapillartrennsäulen erteilt,
1958 beschrieb Kovac den Retentionsindex.
Die Gaschromatografie ist wie die HPLC eine Trennmethode zur Gewinnung reiner Stoffe sowie für
die qualitative und quantitative Analyse von Mischungen. Sie dient sowohl zum Nachweis bekannter
Verbindungen (target analysis) als auch zur Strukturaufklärung unbekannter Verbindungen (Hinweis
Kurs: Analyse eines Parfums!), sie ist die ideale Ergänzung zur HPLC, ist auch für Automatisation
geeignet.
Die mobile Phase ist immer gasförmig;
Die stationäre Phase kann sein:
fest: GAC oder GSC (Gasadsorptionschromatografie): Adsorption
flüssig: GLC (Gas-liquid-chromatography): Verteilung
1. Theoretische Grundlagen
1.1 Konzept der theoretischen Böden:
Martin und Synge, Nobelpreis 1952
Dynamisches System: Serie aufeinanderfolgender Verteilungsschritte, bei jeder Stufe
Gleichgewichtseinstellung zwischen mobiler und stationärer Phase.
Bei einer Vergrößerung der Trennstufenzahl befindet sich der gelöste Stoff in einem schmäleren
Bereich des Gesamtsystems.

2
Reale Chromatografie ist kontinuierlicher Prozeß mit einer Serie von Gleichgewichtseinstellungen.
Diese heißen "theoretische Böden" (Begriff aus der Destillation)
Teilt man die Länge der Säule durch die Anzahl der theoretischen Böden, so erhält man die Angabe
des
Höhenäquivalentes eines theoretischen Boden
Height Equivalent to a Theoretical Plate (HETP)
sogenannter H-Wert
Gute GC-Säule: H 0.1 mm, bis zu 1 Mill. theoret.Böden; eine übliche Säule hat 50.000 - 150.000
Böden; eine gepackte Säule hat ca. 2000 - 5000 theoretische Böden
Gute HPLC-Säule: H = 0.05 mm hat 20.000 theoretische Böden
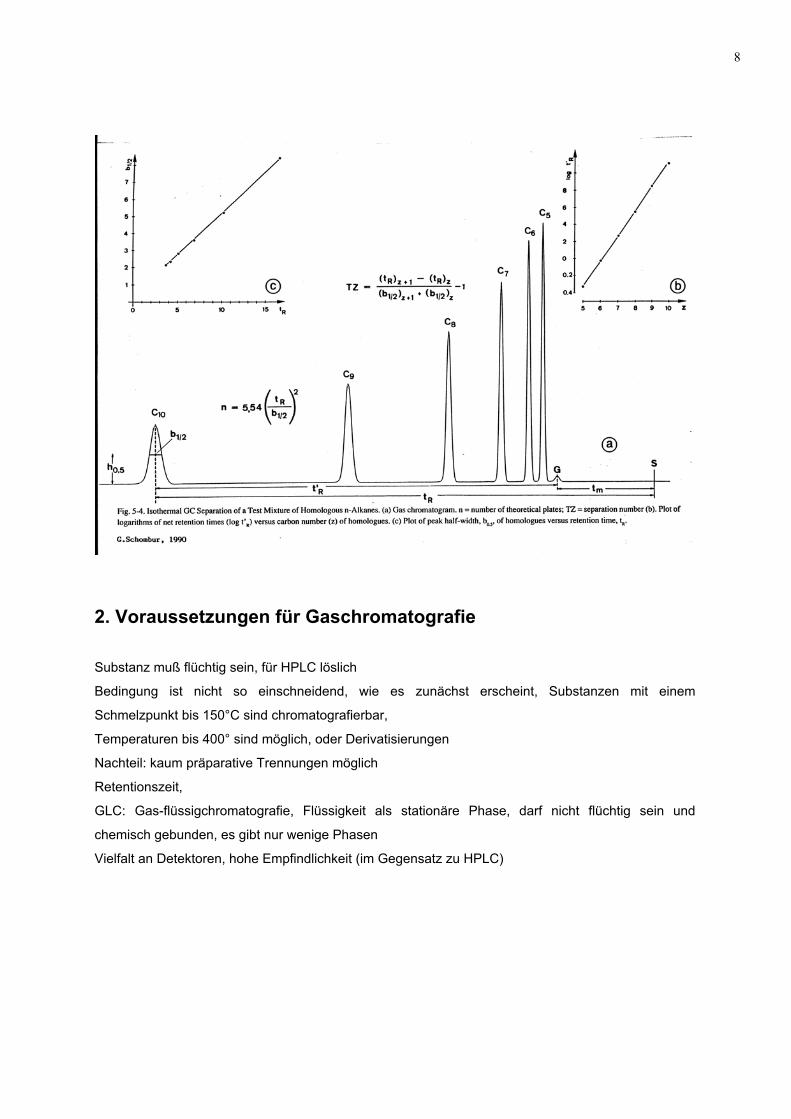
H = HETP = 0.18. L b 0.52 (m)
tr
L/H = N = 5.54 (tr / b0.5)2
L ... Länge der Trennsäule in m
b 0.5... Bandenbreite auf halber Höhe
tr ... Retentionszeit
N.... Bodenzahl
Verteilungskoeffizient: Ki = ci (l) / ci (g)
bezieht sich auf die Konzentration
Kapazitätsverhältnis: k´ = ni (l) / ni (g) ni = ci . v
bezieht sich auf die Menge
k´ = Ki / ß
ß = Ki / k´ = Ci(l). ni(g) /ci (g). ni(l) = V(g) / v(l)
Menge an mobiler Phase ist viel größer als Menge an stationärer Phase; deshalb muß man die
Verteilungskoeffizienten K korrigieren.
Dieser Korrekturfaktor ist das Verhältnis der Volumina der mobilen Phase und der stationären Phase
und wird als Phasenverhältnis ß bezeichnet:
ß = v (mobile Phase) / v (stationäre Phase)
ß für gepackte Säule zwischen 5 und 35
ß für Kapillarsäule zwischen 50 und 1000

3
Die Retentionskapazität ist die Säuleneigenschaft, die das generelle Verzögern der Komponente in
der Trennsäule bewirkt. Diese ist bei Kapillarsäulen bedeutend kleiner als bei gepackten Säulen,
deshalb müssen Kapillarsäulen länger sein.
1.2. Das Gaschromatogramm
Chromatografie ist ein kontinuierlicher Vorgang, sodaß Diffusion stattfindet, welche zu einer
Peakverbreiterung führt.
Je länger eine Substanz in der Säule ist, umso breiter wird die eluierte Substanzzone sein. Diese
Banden- oder Peakverbreiterung ist eine Funktion des chromatografischen Verteilungsprozesses und
kein Diffusionsphänomen. Zusätzlich kommt es wegen der Diffusion zu einer weiteren
Peakverbreiterung. Peakverbreiterung kann durch Temperaturprogrammierung in der GC oder durch
Gradientenelution in der HPLC in Grenzen gehalten werden.

4
1.3. Auflösung (Resolution):
Ein Maß für die Güte einer Trennung. Die Auflösung (R = resolution) entspricht dem Abstand zweier
peak-maxima dividiert durch das arithmetische Mittel der Basisbreiten der peaks. Ein R-Wert von
mindestens 1,5 beschreibt eine befriedigende Trennung.
R = 2 (t2 - t1) / (w1 + w2) w... Basisbreite
Die Auflösung wird mit der Wurzel der Säulenlänge erhöht, Säule mit 4-facher Länge liefert eine
doppelte Auflösung, führt jedoch auch zu einer 4-fachen Analysenzeit.
Auflösung wichtig für:
a) einwandfreie Ermittlung des Peakmaximums
b) störungsfreie Ermittlung der Peakfläche
c) präparative isolierung reiner Komponenten

5
Die Auflösung hängt von 2 Faktoren ab:
Selektivität:
Sie ist ein Maß für die Wechselwirkung der Probe mit der stationären Phase, deshalb gibt es für
bestimmte Substanzgruppen optimale stationäre Phasen. Bei falscher Wahl der stationären Phase
kann trotz Verwendung einer Säule mit hoher Bodenzahl oft keine Trennung erzielt werden.
Effizienz:
Die chromatografische Wirksamkeit einer Säule (Effizienz) ist in erster Näherung unabhängig von den
zu trennenden Stoffen, d.h. der H-Wert ist konstant.
1.4. Van Deemter-Gleichung
Die Diffusion wird umso kleiner, je höher die Geschwindigkeit ist, dabei wird jedoch die
Gleichgewichtseinstellung behindert. Da die beiden Forderungen gegenläufig sind, muß man einen
Kompromiß zwischen möglichst hoher Flußrate und bestmöglicher Gleichgewichtseinstellung suchen.
Die Verknüpfung zwischen Diffusion, Gleichgewichtseinstellung und Flußrate wird durch die Van
Deeter-Gleichung beschrieben:
H = A + B / u + C . u
u ... mittlere Geschwindigkeit der mobilen Phase
A ... Maß für die Bandenverbreiterung durch Umströmen der stationären Phase, abhängig von der
Güte der Packung und der Art der Säule, aber unabhängig von der Flußrate, Eddy-Diffusion, sie tritt
nur bei gepackten Säulen auf.
B... Bandenverbreiterung durch Longitudinal-Diffusion, mit Flußrate umgekehrt proportional
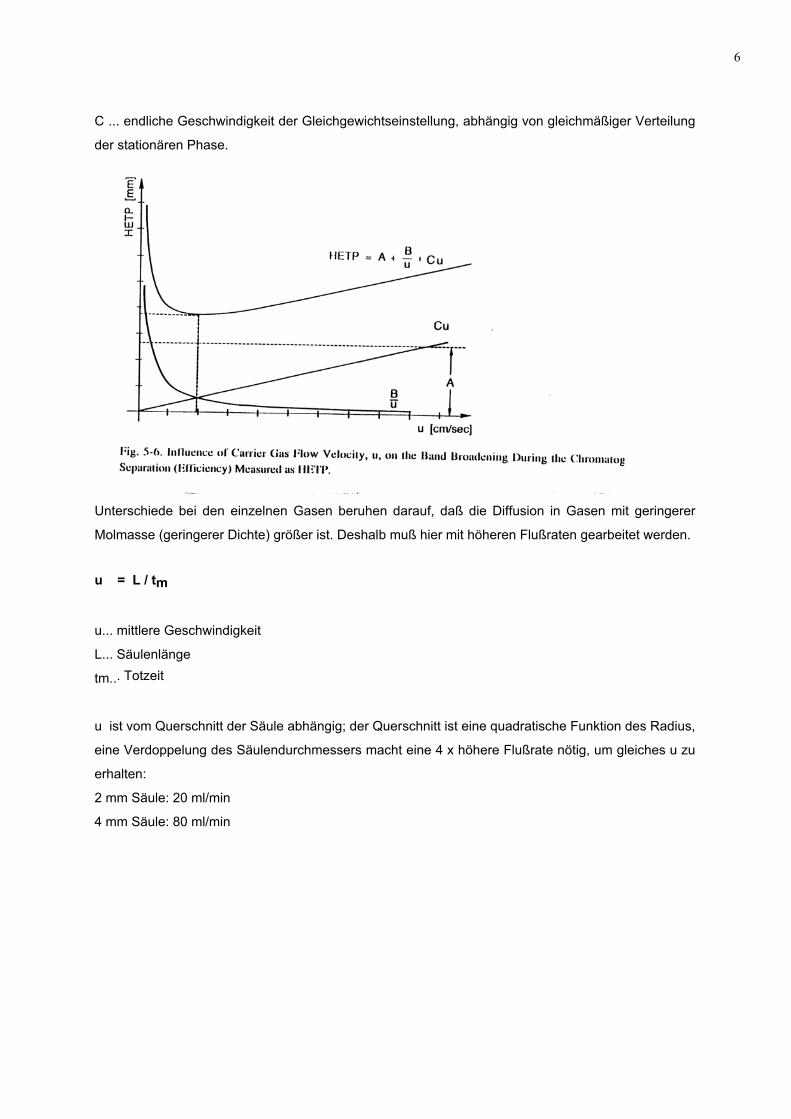
6
C ... endliche Geschwindigkeit der Gleichgewichtseinstellung, abhängig von gleichmäßiger Verteilung
der stationären Phase.
Unterschiede bei den einzelnen Gasen beruhen darauf, daß die Diffusion in Gasen mit geringerer
Molmasse (geringerer Dichte) größer ist. Deshalb muß hier mit höheren Flußraten gearbeitet werden.
u = L / tm
u... mittlere Geschwindigkeit
L... Säulenlänge
tm... Totzeit
u ist vom Querschnitt der Säule abhängig; der Querschnitt ist eine quadratische Funktion des Radius,
eine Verdoppelung des Säulendurchmessers macht eine 4 x höhere Flußrate nötig, um gleiches u zu
erhalten:
2 mm Säule: 20 ml/min
4 mm Säule: 80 ml/min
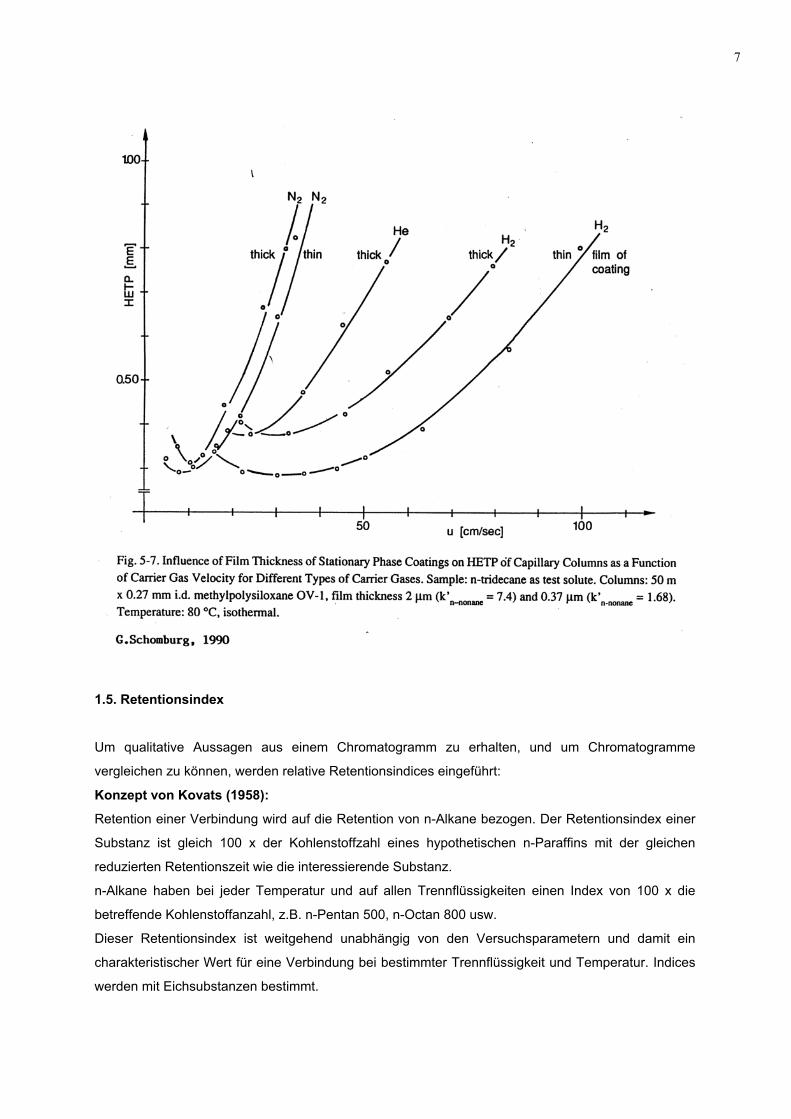
7
1.5. Retentionsindex
Um qualitative Aussagen aus einem Chromatogramm zu erhalten, und um Chromatogramme
vergleichen zu können, werden relative Retentionsindices eingeführt:
Konzept von Kovats (1958):
Retention einer Verbindung wird auf die Retention von n-Alkane bezogen. Der Retentionsindex einer
Substanz ist gleich 100 x der Kohlenstoffzahl eines hypothetischen n-Paraffins mit der gleichen
reduzierten Retentionszeit wie die interessierende Substanz.
n-Alkane haben bei jeder Temperatur und auf allen Trennflüssigkeiten einen Index von 100 x die
betreffende Kohlenstoffanzahl, z.B. n-Pentan 500, n-Octan 800 usw.
Dieser Retentionsindex ist weitgehend unabhängig von den Versuchsparametern und damit ein
charakteristischer Wert für eine Verbindung bei bestimmter Trennflüssigkeit und Temperatur. Indices
werden mit Eichsubstanzen bestimmt.
8
2. Voraussetzungen für Gaschromatografie
Substanz muß flüchtig sein, für HPLC löslich
Bedingung ist nicht so einschneidend, wie es zunächst erscheint, Substanzen mit einem
Schmelzpunkt bis 150°C sind chromatografierbar,
Temperaturen bis 400° sind möglich, oder Derivatisierungen
Nachteil: kaum präparative Trennungen möglich
Retentionszeit,
GLC: Gas-flüssigchromatografie, Flüssigkeit als stationäre Phase, darf nicht flüchtig sein und
chemisch gebunden, es gibt nur wenige Phasen
Vielfalt an Detektoren, hohe Empfindlichkeit (im Gegensatz zu HPLC)

9
In der GC gibt es 5 wichtige Variablen:
Trägergas
Probenaufgabe
Säule bzw. stationäre Phase
Temperaturbedingungen während der Trennung
Detektor
2.1. Trägergas
In HPLC mobile Phase am schwierigsten, Detektor am einfachsten
Natur des Gases ist eher sekundär,
wenig reaktiven kommen in Frage: Wasserstoff, Stickstoff, Helium, Argon
warum keine Luft oder Sauerstoff?
Bei höheren Temperaturen nimmt Gasfluß ab, deshalb viele Geräte mit elektronischer
Flußkontrolle.
Hilfsgase:
Make-up-Gas: Schönungsgas für Detektor bei Verwendung von Kapillarsäulen, wird vor dem Detektor
zugemischt, um optimalen Fluß für Detektor zu erreichen.
Detektorgase:
z.B. H und Luft bei FID

10
2.2. Probenaufgabe
Die Probe wird mittels Spritze in den Einspritzblock des Systems gebracht, dabei wird Gummidichtung
(Septum) durchstoßen. Einspritzblock muß Temperatur über der maximalen Säulentemperatur
besitzen. Probe verdampft und wird als Gas auf die Säule gespült.
Hauptprobleme: Zersetzung der Proben, Zurückbleiben von nichtflüchtigen Rückständen
(Geisterpeaks), zu viel Probe bei Kapillarsäulen, mit üblicher Spritze nicht so klein dosierbar, mehrere
Möglichkeiten:
Split-Injektion: bevor der Gasstrom mit der Probe auf die Säule kommt, wird er geteilt, das Verhältnis
der Gasströme wird durch ein Nadelventil kontrolliert. Ein Gasstrom geht ins Freie, der andere in die
Säule: Splitverhältnis
Probe wird in einen Glas-liner gespritzt.
Splitless: kein split, für ca. 30 sec wird split ausgeschaltet, Probe geht auf Säule, dann wieder split:
bewirkt Erhöhung der Empfindlichkeit. Säule soll niedrigere Temperatur als Siedepunkt des
Lösungsmittels haben, dann wird Probe in einer Zone aufkonzentriert.
On Column-Aufgabe: Auftragung erfolgt mit Spezialspritzen direkt in die Säule, Verdampfung findet
in der Säule direkt statt, alles gelangt in die Säule, keine Diskriminierung.
Headspace (Kopfraum)-Technik: es wird nur der Gasraum über einer nicht vollständig
verdampfbaren Probe zur GC-Trennung gebracht. Vor allem für flüchtige Komponenten. Probe wird in
eine geschlossene, thermostatisierte Ampulle gebracht, Probe wird vom Gasraum entnommen.
2.3 Säulen
Heute werden fast ausschließlich Kapillarsäulen eingesetzt, um hohe Trennleistung zu erzielen,
höhere Bodenzahl
Material ist dabei Quarz: Fused Silica-Säulen
Innendurchmesser: 0.2 - 0.5 mm (Widepore-Säulen, änlich wie gepackt), Länge 10-60 m. Um
Festigkeit und Biegsamkeit zu erreichen, werden sie mit einem Polyimidfilm beschichtet, macht
braune Farbe. Sie sind nur bis 350°C geeignet. Es gibt auch HT-Säulen, die bis 400° einsetzbar sind.
Früher wurde stationäre Phase auf Träger aufgebracht und in Säule (aus Glas) gepackt; es wurden
viele Phasen verwendet
heute hpts. Kapillarsäulen mit flüssigen, stationären Phasen, welche in LM gelöst werden, durch
Säule gespült und erhitzt, WCOT (wall coated open tubular)
Filmdicke ist entscheidend: 01 m - 5.0 m, bei dickerem Film kann mehr Probe getrennt werden.
Viele Säulen besitzen bereits chemisch gebundene stationäre Phasen
11
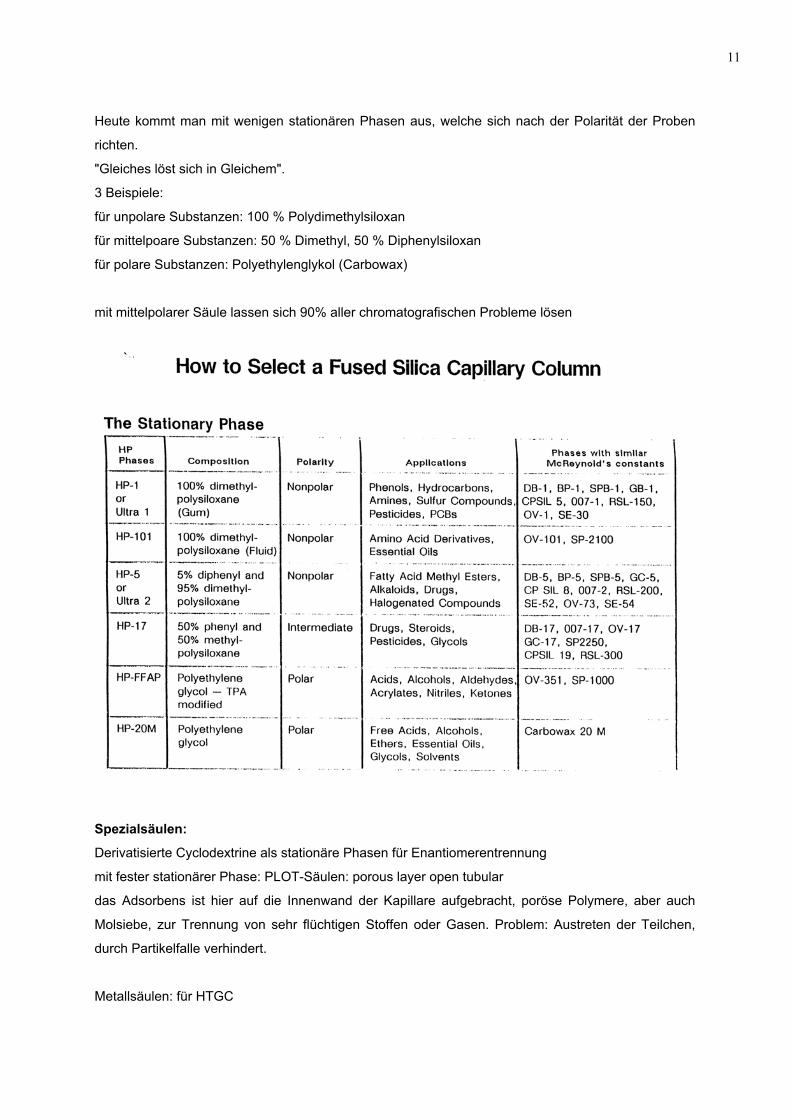
Heute kommt man mit wenigen stationären Phasen aus, welche sich nach der Polarität der Proben
richten.
"Gleiches löst sich in Gleichem".
3 Beispiele:
für unpolare Substanzen: 100 % Polydimethylsiloxan
für mittelpoare Substanzen: 50 % Dimethyl, 50 % Diphenylsiloxan
für polare Substanzen: Polyethylenglykol (Carbowax)
mit mittelpolarer Säule lassen sich 90% aller chromatografischen Probleme lösen
Spezialsäulen:
Derivatisierte Cyclodextrine als stationäre Phasen für Enantiomerentrennung
mit fester stationärer Phase: PLOT-Säulen: porous layer open tubular
das Adsorbens ist hier auf die Innenwand der Kapillare aufgebracht, poröse Polymere, aber auch
Molsiebe, zur Trennung von sehr flüchtigen Stoffen oder Gasen. Problem: Austreten der Teilchen,
durch Partikelfalle verhindert.
Metallsäulen: für HTGC

12
Behandlung von Kapillarsäulen:
Säulen müssen zunächst konditioniert werden, mehrmalige Erhitzung unter Trägergasstrom mit
Temperaturprogrammierung auf die maximale Arbeitstemperatur (Ausheizen der Säule, um
niedermolekulare Stoffe auszutreiben und stabile Basislinie zu erhalten)
Niemals Säule ohne Trägergasstrom erhitzen, da es sonst zur Zerstörung der stationären Phase
kommt, niemals überhitzen, kaputte Säule erkennt man an starkem Säulenbluten: hohes
Grundrauschen durch Zersetzungsprodukte der stationären Phase.
Aufbewahrung von Säulen: vor dem Ausbauen Ausheizen, an beiden Enden mit Septum
verschließen.
2.4 Temperaturprogrammierung
Die Trennung durch GC beruht auf 2 Eigenschaften der zu trennenden Substanzen: ihrer Löslichkeit
und ihrem Dampfdruck. Dampfdruck hängt von Temperatur ab, die verändert werden kann.
Säule befindet sich in Säulenofen, der beheizt ist; Einspritzblock- und Detektortemperatur müssen
über der maximalen Säulentemperatur liegen.
Isotherme Trennung: bei gleicher Temperatur, wenn sich Substanzen gut trennen, man erkennt
Homologe, welche nach regelmäßiger Retentionszeiterhöhung eluiert werden (Logarithmischer
Zusammenhang zwischen Retentionszeit und C-Zahl, siehe Abbildung).
Temperaturprogrammierung: analog der Gradienteneluierung bei HPLC; wenn Substanzgemische
stark unterschiedliche Siedetemperatur besitzen, um Analysenzeit zu verkürzen. Je länger die Probe
in der Säule ist, umso breiter wird der peak. Temperaturerhöhung um 30°C führt zu einer Halbierung
der Retentionszeiten.
Temperaturanstieg kann sein:
linear
stufenweise
zuerst isotherm und dann linear
Durch eine automatische Druckprogrammierung kann der Fluss konstant gehalten werden. Bei
Temperatursteigerung wird auch der Druck erhöht.
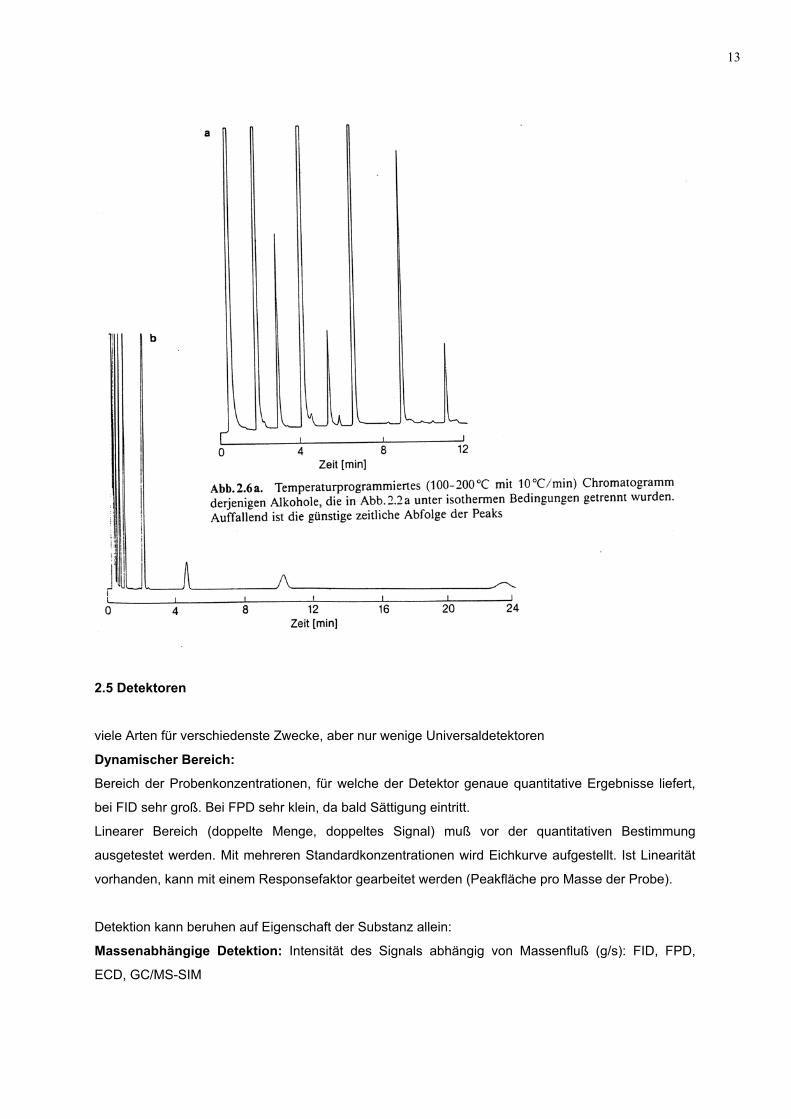
13
2.5 Detektoren
viele Arten für verschiedenste Zwecke, aber nur wenige Universaldetektoren
Dynamischer Bereich:
Bereich der Probenkonzentrationen, für welche der Detektor genaue quantitative Ergebnisse liefert,
bei FID sehr groß. Bei FPD sehr klein, da bald Sättigung eintritt.
Linearer Bereich (doppelte Menge, doppeltes Signal) muß vor der quantitativen Bestimmung
ausgetestet werden. Mit mehreren Standardkonzentrationen wird Eichkurve aufgestellt. Ist Linearität
vorhanden, kann mit einem Responsefaktor gearbeitet werden (Peakfläche pro Masse der Probe).
Detektion kann beruhen auf Eigenschaft der Substanz allein:
Massenabhängige Detektion: Intensität des Signals abhängig von Massenfluß (g/s): FID, FPD,
ECD, GC/MS-SIM

14
Detektion beruht auf Eigenschaft des Trägergas/Substanz-Gemisches:
Konzentrationsabhängige Detektion: Intensität des Signals abhängig von Konzentration (g/ml):
WLD, GC/FTIR
hier wird das Signal kleiner bei Erhöhung des Trägergasflusses
Universelle Detektoren:
sprechen auf alles an, was Säule verläßt,
schlechte chromatografische Trennung kann Detektion einer Komponente stören, sprechen auch auf
Säulenbluten an sowie auf Schwankungen wie Trägergasdruck und Temperatur
Selektive Detektoren:
können elementselektiv, strukturselektiv oder selektiv auf andere Eigenschaften sein. FID z.B. günstig
für wäßrige Proben. ECD für Umweltanalytik.

15

16
Wärmeleitfähigkeitsdetektor (WLD, TCD)
Prinzip:
gab es schon vor der GC,
für jede Verbindung, deren Wärmeleitfähigkeit sich von der des Trägergases unterscheidet. Gase mit
größter Wärmeleitfähigkeit haben niedrigste Massen. Helium besitzt besonders hohe
Wärmeleitfähigkeit, deshalb günstig.
Ein Heizdraht besitzt konstante Temperatur und konstanten Widerstand, kommt Probe durch, sinkt
Wärmeleitfähigkeit, der Draht erhitzt sich und erhöht den Widerstand, der gemessen und
aufgezeichnet wird.
Heizdraht aus W oder W-Legierungen, mit Oxidschichten passiviert.
Da WLD ein nicht destruktiver Detektor ist, kann man ihn mit anderen in Serie schalten und für
präparative GC einsetzen.
Heute gibt es Ein-Filament Geräte, auch nur eine Säule notwendig
Anwendung:
für Gasanalysen, Biogas, Mikro-Wasserbestimmung
Flammenionisationsdetektor:
in der GC der wichtigste und am meisten verwendete Detektor,
Prinzip:
In einer Wasserstoff/Luft-Flamme werden organische Verbindungen verbrannt und ionisiert. Ein
Kollektor (2 Hochspannungselektroden, 200 V) mit einer anliegenden Spannung wird in die Nähe der
Flamme gebracht, Elektronen werden von ihm angezogen und produzieren einen Strom, welcher
einen Spannungsabfall erzeugt und der proportional zur Probenmenge in der Flamme ist.
Die in der Flamme gebildeten Radikale reagieren mit Sauerstoffatomen unter Bildung von Ionen:
CH. + O CHO+ + e-
Voraussetzung sind C-H und C-C-Bindungen, org. Komponenten werden zunächst in der nicht
oxidierenden Zone zu Radikalen pyrolysiert und dann im oxidierenden Flammensaum zu
Molekülionen und Elektronen umgewandelt, welche am Kollektor (Anode) entladen werden. Das FID-
Signal wird einem Verstärker zugeführt.
Säuleneluat wird zuerst mit Wasserstoff gemischt und erreicht dann die Flamme. Flammenprofil ist
entscheidend für Empfindlichkeit, öfters putzen. Wichtig ist genaue Flußrate aller 3 verwendeten
Gase.
Anwendung:
registriert alle organischen Verbindungen, außer sie brennen oder ionisieren nicht, wie z.B.
hochhalogenierte Verbindungen.
nicht registriert wird Luft, Wasser, Kohlendi(mon)oxid

17
Elektroneneinfangdetektor (ECD):
wichtig in der Spurenanalytik, für Halogene, Pestizidanalysen
Prinzip:
Elektronegative Spezies können thermische Elektronen einfangen und negative Ionen bilden. Das
Trägergas wird mit ß-Teilchen aus einer radioaktiven Quelle ionisiert. Der entstehende Elektronenfluß
produziert einen kleinen Strom, der gemessen wird. Erreichen Probenmoleküle die Zelle, fangen sie
Elektronen ein, die sonst vom Kollektor gesammelt werden und einen Strom ergeben würden. Die
resultierende Stromabnahme kann gemessen werden. Man arbeitet heute meist mit Spannungspuls,
der Elektronen periodisch einfangt und registriert, die schwereren, negativen Ionen können in dieser
kurzen Zeit nicht gefangen werden und werden durch das Trägergas entfernt.
Radioaktive Quelle ist meist 63Ni, mit dem die Oberfläche der Zelle beschichtet ist.
Trägergas ist Stickstoff oder Argon mit 5 % Methan, welches durch energiereduzierende Kollisionen
mehr energiearme Elektronen erzeugt.
Anwendung:
Paraffine und einfache Kohlenwasserstoffe werden nicht detektiert.
Chemische Gruppe rel. Empfindlichkeit
Kohlenwasserstoffe 1
Ether, Ester 10
Alkohole, Amine 100
Br-, Cl- und F-Verbindungen 100 - 1.000.000
Detektor sehr empfindlich, deshalb auch schnell verunreinigt, muß vom Hersteller gereinigt werden.
Thermoionischer Detektor (NPD, N,P-FID)
Prinzip:
ähnich aufgebaut wie FID, nur weniger Luft und Wasserstoff, um Ionisation von normalen
Kohlenwasserstoffen zu minimieren. Eine Alkalisalzperle (Kalium- od. Rubidiumsalze) ist um Düse
positioniert, wodurch Alkalimetallionen in die Flamme geschleust werden. Dabei werden bevorzugt N-
und P-haltige organische Moleküle mit hoher Ausbeute ionisiert. Mechanismus nicht genau bekannt.
Alkalimetalle haben niedriges Ei, PO auch, Elektroneneinfangreaktionen sind beteiligt.
hohe Empfindlichkeit, Gefahr der Kontamination

18
Flammenphotometrischer Detektor (FPD)
Prinzip:
Schwefel- und phosphorhaltige Kohlenwasserstoffe werden in einer Flamme angeregt, und emittieren
Licht einer bestimmten Wellenlänge, das gefiltert und mit Photomultiplier verstärkt wird. Geringer
linearer Bereich
Anwendung:
vor allem in Biochemie, aber auch für P-haltige Pestizide
Photoionisationsdetektor (PID)
Prinzip:
Probenmoleküle werden durch Absorption von UV-Licht ionisiert, entsprechend ihrer
Ionisationspotentiale. Die geladenen Partikel werden zwischen 2 Elektroden gemessen. Die
verwendete Lampe bestimmt Energie der Photonen und damit auch die Verbindungen, die gemessen
werden.
Anwendung:
nicht destruktiv, vor allem aromatische Verbindungen
FT-Infrarot-Detektor (FTIRD):
Laufend wird im Gasstrom IR-Spektrum aufgenommen, für Spektrensuche geeignet, einziger Vorteil
gegenüber MS: Isomere können gefunden werden
ideale Kombination ist GC-MS-FTIR
Wasser in Probe muß vermieden werden, um Optik aus NaCl zu schonen.
Atomemissionsdetektor (AED)
Die Probe wird angeregt, z.B. durch mikrowelleninduziertes Plasma, beim Übergang in Grundniveau
wird Licht ausgesendet, die Wellenlängen werden mit einem Spektrometer gemessen. Es können
gleichzeitig mehrere Elemente gemessen werden, z.B. C, H, Cl, Br
Massenselektive Detektoren (MSD):
Prinzip:
schon lange bekannt, durch Einsatz von Computern bedienungsfreundlich, wurde zum Routinegerät.
Unterschiede zu IR: destruktiv, aber empfindlicher, eher für Homologe als Isomere.

19
1. Elektronenstoßionisation (EI = electron impact)
geschieht im Hochvakuum
2 Prinzipien:
Magnetfeldmassenspektrometer: Bruchstücke werden je nach Masse durch Magnetfeld in
verschiedene Flugbahnen gelenkt, nicht als Detektor geeignet.
Quadrupol-Massenspektrometer: zwischen 4 Stabmagneten wird ein elektrisches
Hochfrequenzwechselfeld erzeugt. Nur Ionen mit einem bestimmten Masse/Ladungs-Verhältnis
bewegen sich auf geraden Bahnen zum Detektor, die anderen werden auf den Magnetstäben
entladen. Durch Variation des elektrischen Feldes können stufenweise einzelne Massen gesucht und
detektiert werden.
SCAN-MODE:
für qualitative Aussagen; zunächst wird meist ein scan über den gesamten Massenbereich
durchgeführt, ca.1-3 scans/sec sind möglich. Dabei erhält man ein Massenspektrum für jeden scan,
welches man nachträglich mit Bibliotheken vergleichen kann, optimale Aussagen möglich
auch nicht getrennte Substanzen können so identifiziert werden
SIM-MODE:
für quantitative Messungen, hier wird nur auf wenige, charakteristische und intensive Ionen geschaut.
Dabei kann öfter gescant werden und damit die Empfindlichkeit erhöht werden. Quantifizierung ohne
Trennung ist möglich.
2.Chemische Ionisation (CI):
Bei EI sieht man vor allem bei labilen Verbindungen nicht die Molekülmasse, deshalb mildere
Ionisation notwendig.
Ein sog. Reaktandgas wird kontinuierlich in die Ionenquelle eingeführt , sodaß relativ hoher Druck von
0.3 mbar in der Ionenquelle entsteht, außerhalb ist Hochvakuum. Ionisation erfolgt bei ca. 200 eV,
weil höherer Druck herrscht
Aufgrund der hohen Reaktandgaskonzentration findet Ionisation der Gasmoleüle statt, wodurch
Reaktandionen gebildet werden. Probenmoleküle werden dabei nicht direkt ionisiert (Positive
chemische Ionisation PICI)
aus Methanmolekülen werden CH4+, CH3+, CH5+ usw.
CH4 + e- CH4+ + 2e-
CH4+ + CH4 CH5+ + .CH3
CH5+ + M MH+ + CH4
Durch Reaktionen mit Probenmolekülen entstehen "Quasimolekülionen":
M + RH MH+ + R
M + R+ MR+
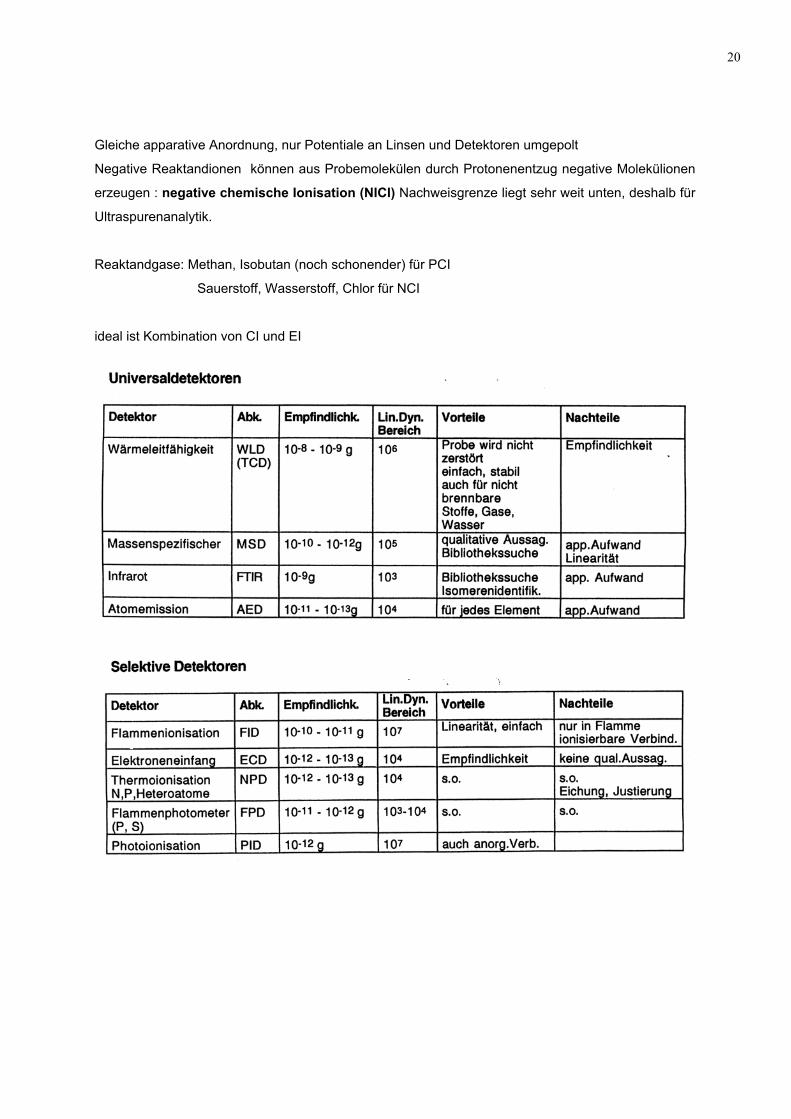
20
Gleiche apparative Anordnung, nur Potentiale an Linsen und Detektoren umgepolt
Negative Reaktandionen können aus Probemolekülen durch Protonenentzug negative Molekülionen
erzeugen : negative chemische Ionisation (NICI) Nachweisgrenze liegt sehr weit unten, deshalb für
Ultraspurenanalytik.
Reaktandgase: Methan, Isobutan (noch schonender) für PCI
Sauerstoff, Wasserstoff, Chlor für NCI
ideal ist Kombination von CI und EI

21
Derivatisierungen
Um schwer flüchtige Stoffe (meist polare) in den Gasraum zu bekommen und unzersetzt
chromatografieren zu können, wird derivatisiert.
meist für Verbindungen mit OH, NH und Carboxylgruppen
Silylierung:
Verbindungen mit aciden H-Atomen werden in entsprechende Trimethylsiliylderivate übergeführt.
N,O-bis(Trimethylsilyl)acetamid (BSA):
das gängigste, allerdings entsteht SiO2, welches sich im Detektor anlagert und den Detektor
verdreckt. Die silylierten Produkte geben auch schöne Fragmentierung im Massenspektrum.
N,O-bis(Trimethylsilyl)trifluoracetamid (BSTFA):
ist relativ flüchtig, geht mit LM weg, es entsteht dabei flüchtige Nebenprodukte, die sich nicht
ablagern.
Acetylierung:
für Hydroxyl- und Aminogruppen
Essigsäureanhydrid, Trifluoressigsäureanhydrid
Methylierung:
hpts. für Carbonsäuren, zur Überführung in Methylester, z.B. Fettsäuren
Diazomethan
BF3/MeOH
TMAH: Trimethylanilinhydroxid, nicht toxisch

Bernd Trathnigg
HPLC Hardware und Grundlagen
mit Schwerpunkt SEC
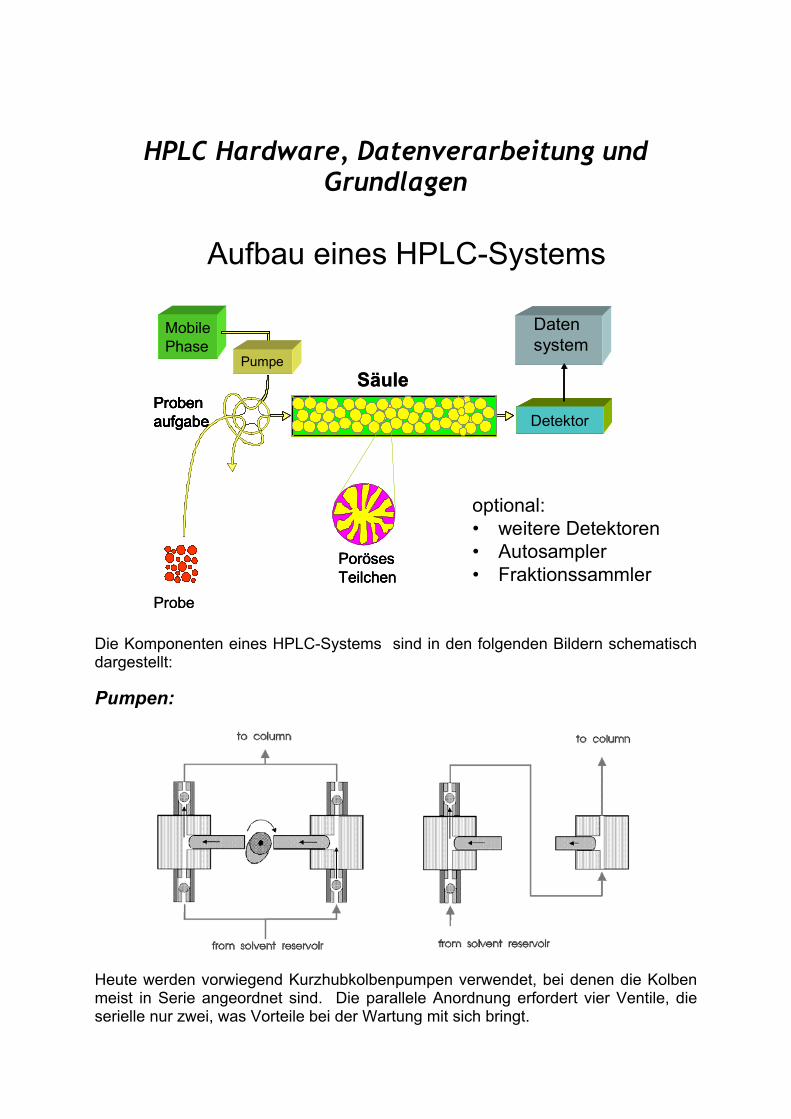
HPLC Hardware, Datenverarbeitung und Grundlagen
Aufbau eines HPLC-Systems
optional:• weitere Detektoren• Autosampler• Fraktionssammler
ProbeProbe
ProbenaufgabeProbenaufgabeProbenaufgabeProbenaufgabeProbenaufgabe
MobilePhase
PumpePumpe
DetektorDetektorDetektor
DatensystemDatensystem
SäuleSäule
PorösesTeilchenPorösesTeilchenPorösesTeilchen
Die Komponenten eines HPLC-Systems sind in den folgenden Bildern schematisch dargestellt:
Pumpen:
Heute werden vorwiegend Kurzhubkolbenpumpen verwendet, bei denen die Kolben meist in Serie angeordnet sind. Die parallele Anordnung erfordert vier Ventile, die serielle nur zwei, was Vorteile bei der Wartung mit sich bringt.
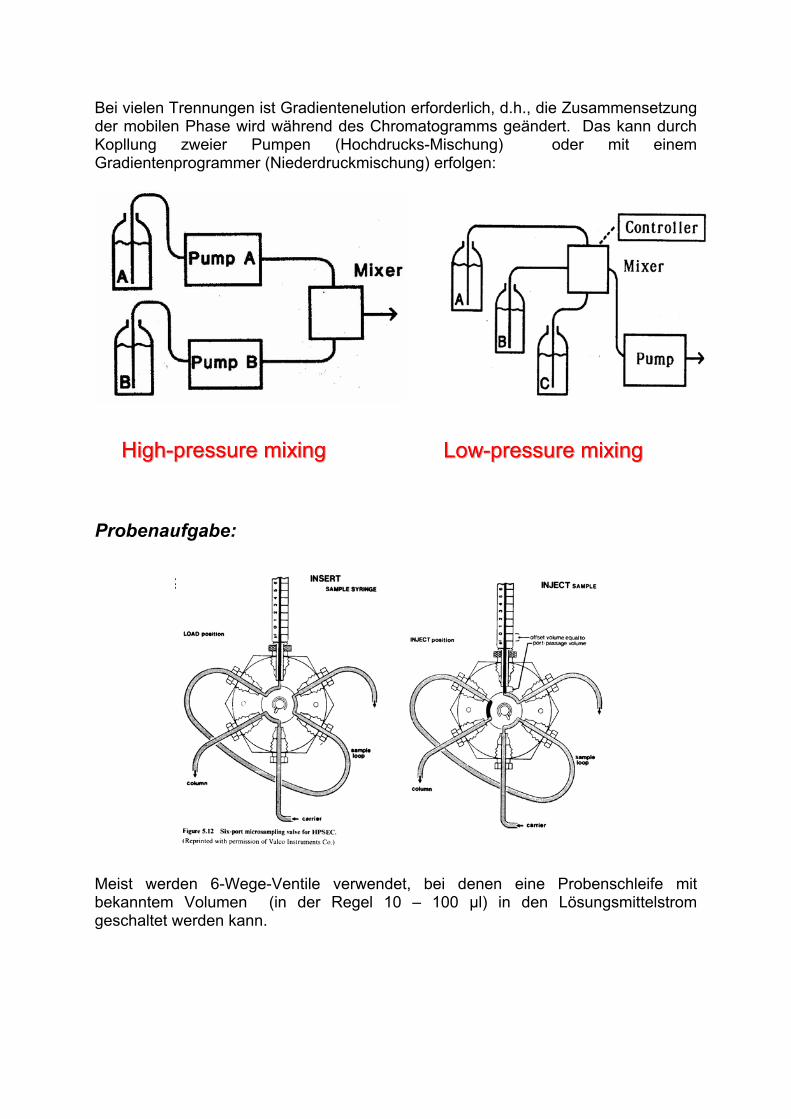
Bei vielen Trennungen ist Gradientenelution erforderlich, d.h., die Zusammensetzung der mobilen Phase wird während des Chromatogramms geändert. Das kann durch Kopllung zweier Pumpen (Hochdrucks-Mischung) oder mit einem Gradientenprogrammer (Niederdruckmischung) erfolgen:
LowLow--pressure mixingpressure mixingLowLow--pressure mixingpressure mixingHighHigh--pressure mixingpressure mixingHighHigh--pressure mixingpressure mixing
Probenaufgabe:
Meist werden 6-Wege-Ventile verwendet, bei denen eine Probenschleife mit bekanntem Volumen (in der Regel 10 – 100 µl) in den Lösungsmittelstrom geschaltet werden kann.

Konzentrationsdetektoren:
Der Detektor liefert ein Signal, das der Konzentration der getrennten Komponenten proportional sein soll.Dabei gibt es im Prinzip zwei Klassen von Konzentrations-Detektoren: Selektive Detektoren messen eine Eigenschaft der zu detektierenden Substanz, universelle (bulk property) Detektoren messen eine Eigenschaft des Eluats.
Selektive Detektoren “Bulk property” Detektoren
(Spektral) Photometer (UV-VIS) Differentialrefraktometer (RI-Detektor) Fluoreszenz-Detektor Leitfähigkeitsdetektor Elektrochemischer Detektor Evaporative Light Scattering Detector (ELSD)
Dichtedetektor
Kopplung von HPLC mit Massenspektrometern (HPLC-MS) und NMR- bzw. IR-Spektrometern ist ebenfalls möglich, aber sehr teuer und deshalb noch nicht als Routinegerät üblich.
Bei der Ausschlußchromatographie werden auch (zusätzlich zum Konzentrationsdetektor) Molmassen-Detektoren verwendet, deren Signal von Konzentration und Molmasse abhängig ist. Dazu gehören Lichtstreu-Detektoren verschiedener Bauart (Low-Angle bzw. Multi-Angle Laser Light Scattering Detector, LALLS bzw. MALLS) und Differentialviskometer.
RI Detektor:
Hier wird vor allem der “Deflection Type” RI-Detektor verwendet. Meßzelle und Referenzzelle sind thermisch gekoppelt, um Temperaturschwankungen auszugleichen. Optischer Abgleich dient zur Justierung des Lichtstrahls auf die Photozelle. Ändert sich der Brechungsindex in der Meßzelle zu stark, trifft der Lichtstrahl nicht mehr auf die Photodiode, ist keine detektion mehr möglich (der Detektor ist “out of range”).
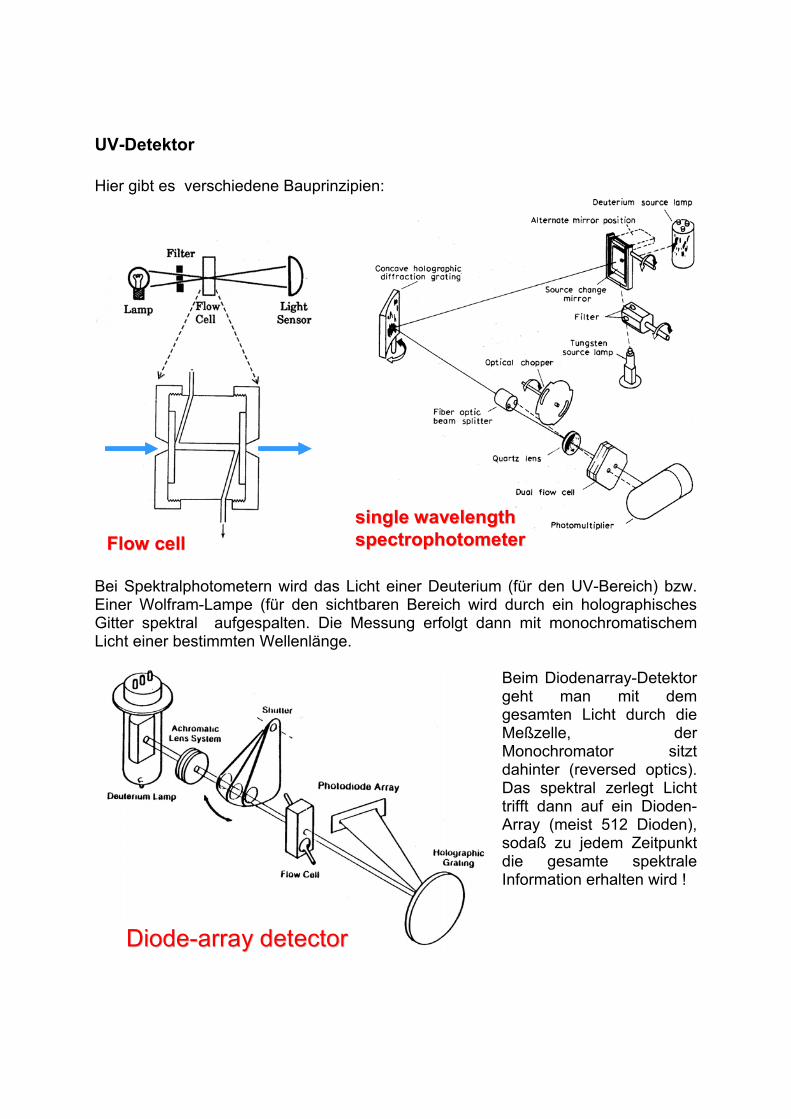
UV-Detektor
Hier gibt es verschiedene Bauprinzipien:
single wavelength single wavelength
spectrophotometerspectrophotometerFlow cellFlow cell
Bei Spektralphotometern wird das Licht einer Deuterium (für den UV-Bereich) bzw. Einer Wolfram-Lampe (für den sichtbaren Bereich wird durch ein holographisches Gitter spektral aufgespalten. Die Messung erfolgt dann mit monochromatischem Licht einer bestimmten Wellenlänge.
Beim Diodenarray-Detektor geht man mit dem gesamten Licht durch die Meßzelle, der Monochromator sitzt dahinter (reversed optics). Das spektral zerlegt Licht trifft dann auf ein Dioden-Array (meist 512 Dioden), sodaß zu jedem Zeitpunkt die gesamte spektrale Information erhalten wird !
DiodeDiode--array detectorarray detector
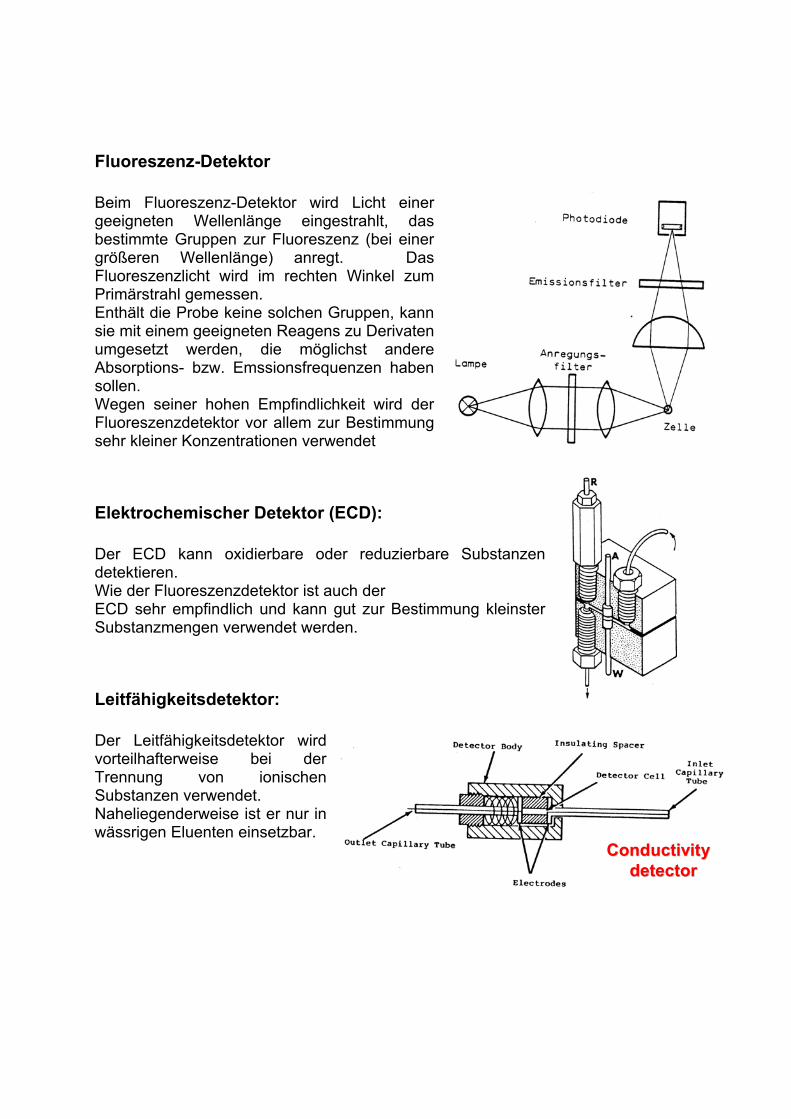
Fluoreszenz-Detektor
Beim Fluoreszenz-Detektor wird Licht einer geeigneten Wellenlänge eingestrahlt, das bestimmte Gruppen zur Fluoreszenz (bei einer größeren Wellenlänge) anregt. Das Fluoreszenzlicht wird im rechten Winkel zum Primärstrahl gemessen. Enthält die Probe keine solchen Gruppen, kann sie mit einem geeigneten Reagens zu Derivaten umgesetzt werden, die möglichst andere Absorptions- bzw. Emssionsfrequenzen haben sollen. Wegen seiner hohen Empfindlichkeit wird der Fluoreszenzdetektor vor allem zur Bestimmung sehr kleiner Konzentrationen verwendet
Elektrochemischer Detektor (ECD):
Der ECD kann oxidierbare oder reduzierbare Substanzen detektieren.Wie der Fluoreszenzdetektor ist auch derECD sehr empfindlich und kann gut zur Bestimmung kleinster Substanzmengen verwendet werden.
Leitfähigkeitsdetektor:
Der Leitfähigkeitsdetektor wird vorteilhafterweise bei der Trennung von ionischen Substanzen verwendet.Naheliegenderweise ist er nur in wässrigen Eluenten einsetzbar.
Conductivity Conductivity
detectordetector

Dichtedetektor:
Der Dichtedetektor ist eine Eigenentwicklung des Instituts und kann nicht als Standarddetektor betrachtet werden. Er ist vor allem bei der Chromatographie von Polymeren in Kombination mit anderen Detektoren sehr nützlich. Gemessen wird hier die Periode eine schwingenden Glasrohrs. Diese hängt mit der Dichte des Inhalts zusammen
Evaporative Light Scattering detector (ELSD)
Beim ELSD wird das Eluat mit Hilfe eines Trägergasstroms (meist Stickstoff) zu einem feinen Aerosol vernebelt, aus dem dann die mobile Phase verdampft wird. Am Ende des Verdampferrohrs passiert der Gasstrom einen Lichstrahl, der von den verbleibenden Partikeln gestreut wird. Die Intensität des gestreuten Lichts sollte der Menge an nichtflüchtigen Substanzen entsprechen. Der ELSD ist recht empfindlich und kann auch mit Gradienten verwendet werden. Wegen des komplexen Zusammenhangs zwischen Konzentration und Signal ist aber bei der Quantifizierung Vorsicht geboten.
Molmassendetektoren
Lichtstreudetektoren
Diese messen das Streulicht in Lösung. Die Messung kann bei einem (LALLS) oder bei mehreren Winkeln (MALLS) erfolgen. Während der LALLS nur die Molmasse liefert, kann beim MALLS auch Information über die Gestalt erhalten werden.

Viskositätsdetektoren
Hier gibt es verschiedene Bauarten. Bei allen wird der Druckabfall an einer Kapillare gemessen, der von der Flußrate und der Viskosität abhängt. Um die Pulsation der Pumpe zu kompensieren wird meist eine Referenzkapillare benützt, in der nur die mobile Phase strömt, während in der Meßkapillare der Peak erscheint. Das wird durch ein Holdup-Volumen (H) erreicht. DP sind Differenzdruckaufnehmer. Bei einem Gerät wird der Eluatstrom zunächst mit Hilfe zweier identischer Kapillaren im Verhältnis 1:1 geteilt. Dieser Aufbau entspricht dem Prinzip der Wheatstone´schen Brücke und hat den Vorteil, daß tatsächlich die durch das Polymer verursachte Druckänderung gemessen wird und nicht eine kleine Änderung eines großen Signals.
Datenerfassung und –verarbeitung:
Das Signal des Detektors ist in der Regel eine Spannung, d.h. analog. Somit muß es zuerst digitalisiert werden, was mit Hilfe eines AD-Wandlers geschieht. Die digitalen Daten werden dann zur Verarbeitung in den Computer übertragen.
Rauschen
Durch die Digitalisierung entsteht notwendigerweise eine Abstufung in ganzzahlige Werte d.h. ein Rauschen von 5.0 !Darüber hinaus können short-term und long term noise bzw. Drift auftreten.Naheliegenderweise wird die Nachweisgrenze durch das gesamte Rauschen begrenzt: ein Peak sollte ein Signal-Rausch-Verhältnis von 5:1 haben !
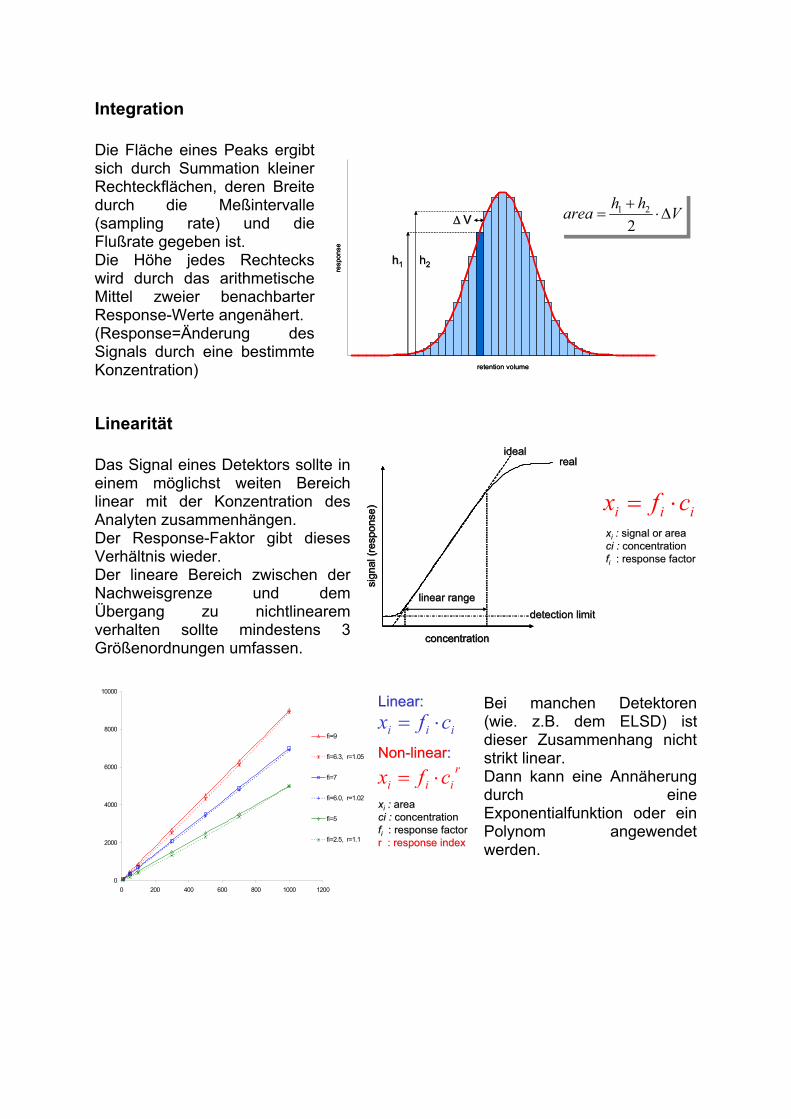
Integration
Die Fläche eines Peaks ergibt sich durch Summation kleiner Rechteckflächen, deren Breite durch die Meßintervalle (sampling rate) und die Flußrate gegeben ist.Die Höhe jedes Rechtecks wird durch das arithmetische Mittel zweier benachbarter Response-Werte angenähert. (Response=Änderung des Signals durch eine bestimmte Konzentration)
Linearität
Das Signal eines Detektors sollte in einem möglichst weiten Bereich linear mit der Konzentration des Analyten zusammenhängen. Der Response-Faktor gibt dieses Verhältnis wieder. Der lineare Bereich zwischen der Nachweisgrenze und dem Übergang zu nichtlinearem verhalten sollte mindestens 3 Größenordnungen umfassen.
Bei manchen Detektoren (wie. z.B. dem ELSD) ist dieser Zusammenhang nicht strikt linear.Dann kann eine Annäherung durch eine Exponentialfunktion oder ein Polynom angewendet werden.
retention volume
resp
on
se
Vhh
area2
21VV
hh11 hh22
retention volume
resp
on
se
Vhh
area2
21VV
hh11 hh22
sign
al (
resp
on
se)
sign
al (
resp
on
se)
concentrationconcentration
idealidealrealreal
linear rangelinear range
detection limitdetection limit
sign
al (
resp
on
se)
sign
al (
resp
on
se)
concentrationconcentration
sign
al (
resp
on
se)
sign
al (
resp
on
se)
concentrationconcentration
idealidealrealreal
linear rangelinear range
detection limitdetection limit
iii cfxxxii :: signal or areasignal or areaci : ci : concentrationconcentrationffii : response factor: response factor
iii cfx
r
iii cfx
xxii :: areaareaci : ci : concentrationconcentrationffii : response factor: response factorr : response indexr : response index
0
2000
4000
6000
8000
10000
0 200 400 600 800 1000 1200
fi=9
fi=6.3, r=1.05
fi=7
fi=6.0, r=1.02
fi=5
fi=2.5, r=1.1
Linear:Linear:
NonNon--linearlinear::

Trennmechanismen
Die Trennung von homologen Reihen kann durch verschiedene Mechanismen erfolgen, von denen je nach der Zusammensetzung der mobilen Phase der eine oder andere überwiegt.
Ein Molekül wandert durch die Säule, wenn es sich außerhalb der Poren der Stationären Phase befindet (wo die mobile Phase strömt), und es bewegt sich nicht, solange es in einer Pore ist. Dabei spielt es keine Rolle, ob es dort gelöst vorliegt oder an der Oberfläche der stationären Phase adsorbiert ist. Da die innere Oberfläche sehr viel größer ist als die äußere, kann diese vernachlässigt werden.
Es ist offensichtlich, daß zunächst das Zwischenkornvolumen Vi die Säule passieren muß, bevor der erste Peak erscheinen kann. Große Moleküle, die keinen Zugang zu den Poren haben, erscheinen somit bei Vi (Ausschlußgrenze).Kleinen Molekülen steht das gesamte Porenvolumen Vp zur Verfügung. Diese erscheinen deshalb beim sogenannten Totvolumen V0 = Vi + Vp, wenn sie nicht an der stationären Phase adsorbiert werden.In thermodynamisch guten Lösungsmitteln kann das als erfüllt gelten. Moleküle mittlerer Größe haben Zugang zu einem Teil des Porenvolumens. Sie eluieren deshalb zwischen Vi + Vp.In diesem Bereich arbeitet die Ausschlußchromatographie (Size Exclusion Chromatography, SEC). Synonyme, die nicht mehr verwendet werden sollten, sind Gel Permeation Chromatography (GPC) und (in der Biochemie) Gel Filtration Chromatography (GFC).
LACLACLACLAC
= Vo= Vo
LCCCLCCC
ViVi
FlowFlow
Mo
l.m
ass
Elution volume
Mo
l.m
ass
Elution volume+ Vp
SECSEC
+ Vp
SECSEC
+ Vp
SECSECSECSEC
ColumnColumn
mobilemobile
phasephase
mobilemobile
phasephase

Size Exclusion Chromatography, SEC
In der SEC ist die Trennung entropiekontrolliert. Die Polymerknäuel ändern ihre Form nicht. Je nach ihrer Größe eluieren sie zwischen Vi + Vp. In diesem Bereich besteht ein annähernd linearer Zusammenhang zwischen Elutionsvolumen und log M. Beim Totvolumen V0 ist die Trennung auf jeden Fall vorbei. Die Selektivität kommt nur aus dem Porenvolumen. Daraus ergibt sich, daß relativ lange Säulen benötigt werden.
Adsorptionschromatographie (LAC):
Kommt es zu (enthalpischen) Wechselwirkungen der Probenmoleküle mit der stationären Phase, eluieren diese nach dem Totvolumen, und zwar umso mehr, je stärker die Wechselwirkung und je größer die Zahl n der wechselwirkenden Gruppen ist. Die Wahrscheinlichkeit, adsorbiert zu werden, steigt exponentiell mit n.Die Moleküle verlieren ihre Knäuelform, die sie in Lösung annehmen, und werden an der Oberfläche der stationären Phase aufgefaltet.Für sehr großes n kann die Retention so groß werden, daß die Moleküle nicht mehr eluieren. Dann muß man im Verlauf des Chromatogramms die Lösungsmittelzusammensetzung so ändern, daß die Wechselwirkung schwächer wird (Gradientenelution).
Liquid Chromatography under Critical Conditions (LCCC):
Für eine Struktureinheit (repeat unit) können bei einer bestimmten Lösungsmittelzusammensetzung und Temperatur entropische und enthalpische Beiträge einander kompensieren, sodaß alle Ketten aus dieser Struktureinheit (unabhängig von ihrer Länge) zugleich eluieren. Sie werden quasi “chromatographisch unsichtbar”, sodaß eine Trennung nach anderen ´Gruppen erreicht werden kann.
Wichtige Kenngrößen
Martin´s rule
•• RetentionRetention factorfactor
(capacity factor)(capacity factor)
0
0)('
V
VVk e
nBAk 'ln•• Distribution coefficientDistribution coefficient
p
ie
V
VVK SEC: K<1 SEC: K<1 VVee << VV00
LCCC: K=1 LCCC: K=1 VVee = V= V00
LAC: K>1 LAC: K>1 VVee >> VV00pie VKVV
SEC: k´< 0SEC: k´< 0LCCC: k´= 0LCCC: k´= 0LAC: k´> 1LAC: k´> 1
Die Theorie der Chromatographie von polymerhomologen Reihen zeigt, daß der entscheidende Parameter der Verteilungskoeffizient K zwischen mobiler und stationärer Phase ist.K ist in der SEC kleiner als 1, in der LAC größer als 1, in der LCCC ist K=1.
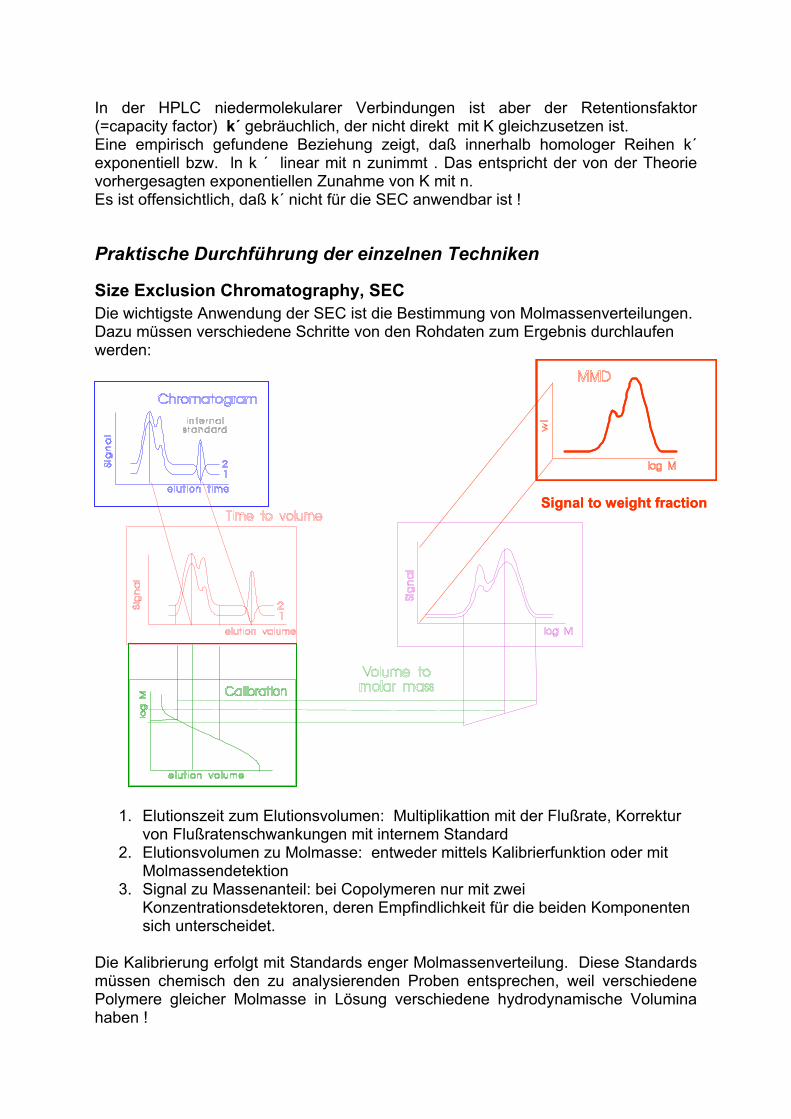
In der HPLC niedermolekularer Verbindungen ist aber der Retentionsfaktor(=capacity factor) k´ gebräuchlich, der nicht direkt mit K gleichzusetzen ist. Eine empirisch gefundene Beziehung zeigt, daß innerhalb homologer Reihen k´ exponentiell bzw. ln k ´ linear mit n zunimmt . Das entspricht der von der Theorie vorhergesagten exponentiellen Zunahme von K mit n. Es ist offensichtlich, daß k´ nicht für die SEC anwendbar ist !
Praktische Durchführung der einzelnen Techniken
Size Exclusion Chromatography, SEC
Die wichtigste Anwendung der SEC ist die Bestimmung von Molmassenverteilungen.Dazu müssen verschiedene Schritte von den Rohdaten zum Ergebnis durchlaufen werden:
Signal to weight fractionSignal to weight fraction
1. Elutionszeit zum Elutionsvolumen: Multiplikattion mit der Flußrate, Korrektur von Flußratenschwankungen mit internem Standard
2. Elutionsvolumen zu Molmasse: entweder mittels Kalibrierfunktion oder mit Molmassendetektion
3. Signal zu Massenanteil: bei Copolymeren nur mit zwei Konzentrationsdetektoren, deren Empfindlichkeit für die beiden Komponenten sich unterscheidet.
Die Kalibrierung erfolgt mit Standards enger Molmassenverteilung. Diese Standards müssen chemisch den zu analysierenden Proben entsprechen, weil verschiedene Polymere gleicher Molmasse in Lösung verschiedene hydrodynamische Volumina haben !
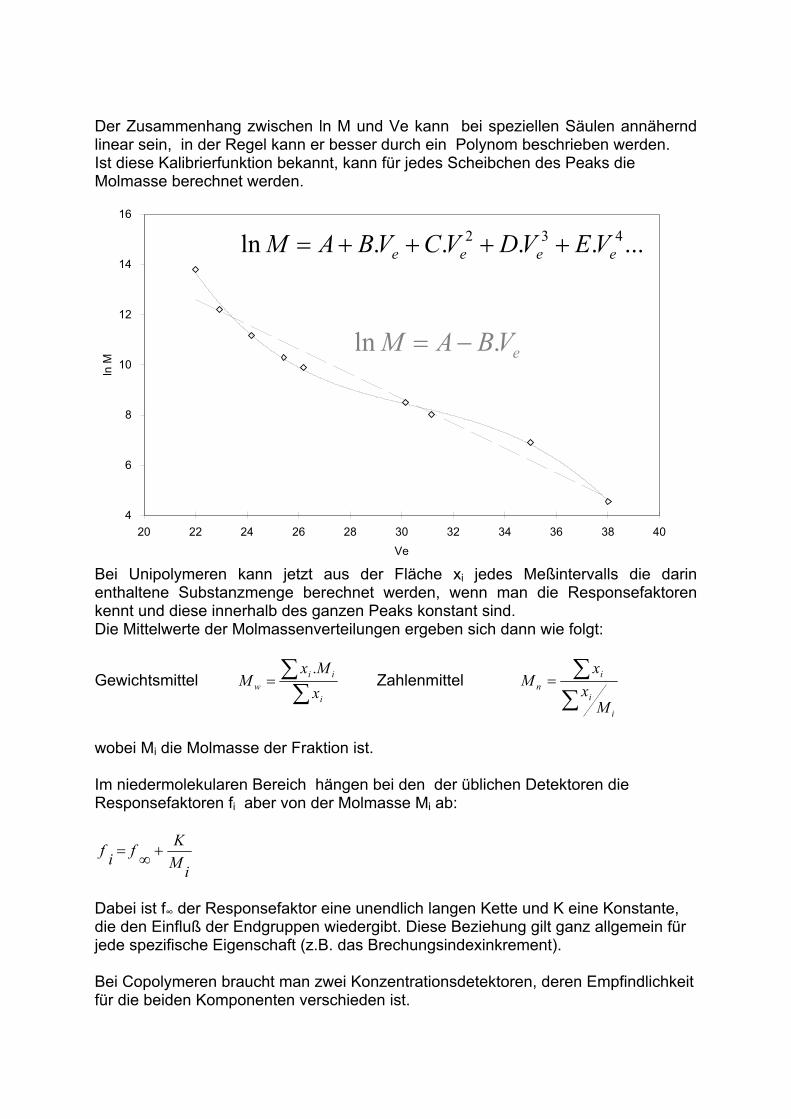
Der Zusammenhang zwischen ln M und Ve kann bei speziellen Säulen annähernd linear sein, in der Regel kann er besser durch ein Polynom beschrieben werden.Ist diese Kalibrierfunktion bekannt, kann für jedes Scheibchen des Peaks die Molmasse berechnet werden.
4
6
8
10
12
14
16
20 22 24 26 28 30 32 34 36 38 40
Ve
ln M
.......ln 432eeee VEVDVCVBAM
eVBAM .ln
Bei Unipolymeren kann jetzt aus der Fläche xi jedes Meßintervalls die darin enthaltene Substanzmenge berechnet werden, wenn man die Responsefaktoren kennt und diese innerhalb des ganzen Peaks konstant sind.Die Mittelwerte der Molmassenverteilungen ergeben sich dann wie folgt:
Gewichtsmitteli
ii
wx
MxM
. Zahlenmittel
i
i
i
n
Mx
xM
wobei Mi die Molmasse der Fraktion ist.
Im niedermolekularen Bereich hängen bei den der üblichen Detektoren die Responsefaktoren fi aber von der Molmasse Mi ab:
iM
Kf
if
Dabei ist f der Responsefaktor eine unendlich langen Kette und K eine Konstante, die den Einfluß der Endgruppen wiedergibt. Diese Beziehung gilt ganz allgemein für jede spezifische Eigenschaft (z.B. das Brechungsindexinkrement).
Bei Copolymeren braucht man zwei Konzentrationsdetektoren, deren Empfindlichkeit für die beiden Komponenten verschieden ist.
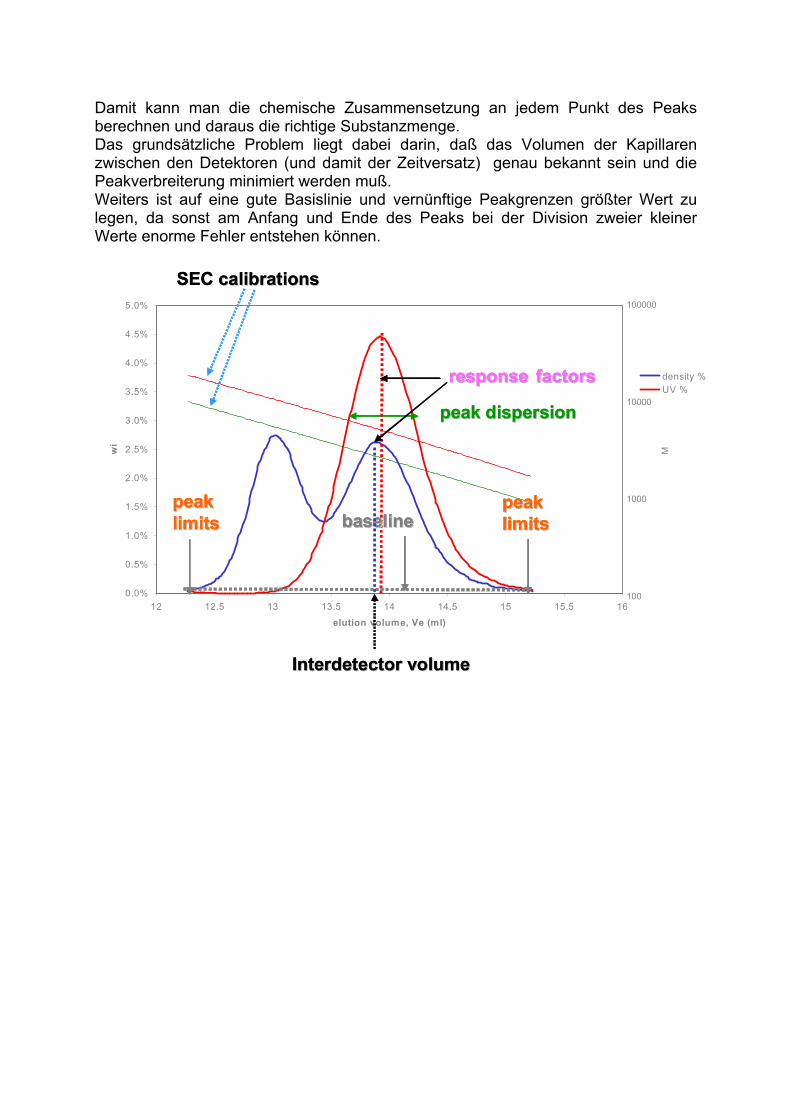
Damit kann man die chemische Zusammensetzung an jedem Punkt des Peaks berechnen und daraus die richtige Substanzmenge. Das grundsätzliche Problem liegt dabei darin, daß das Volumen der Kapillaren zwischen den Detektoren (und damit der Zeitversatz) genau bekannt sein und die Peakverbreiterung minimiert werden muß.Weiters ist auf eine gute Basislinie und vernünftige Peakgrenzen größter Wert zu legen, da sonst am Anfang und Ende des Peaks bei der Division zweier kleiner Werte enorme Fehler entstehen können.
100
1000
10000
100000
M
0.0%
0.5%
1.0%
1.5%
2.0%
2.5%
3.0%
3.5%
4.0%
4.5%
5.0%
12 12.5 13 13.5 14 14.5 15 15.5 16
elution volume, Ve (ml)
wi
density %
UV %
peak dispersionpeak dispersionpeak dispersionpeak dispersion
peak peak
limitslimitspeak peak
limitslimits
peak peak
limitslimits
peak peak
limitslimitspeak peak
limitslimitspeak peak
limitslimitsbaselinebaselinebaselinebaselinebaselinebaseline
responseresponse factorsfactorsresponseresponse factorsfactorsresponseresponse factorsfactors
Interdetector volumeInterdetector volumeInterdetector volumeInterdetector volume
SEC calibrationscalibrationsSEC calibrationscalibrations

Georg Uray
Praktische
Flüssigchromatographie und
deren präparative Anwendung

PRAKTISCHE FLÜSSIGCHROMATOGRAPHIE UND DEREN PRÄPARATIVE ANWENDUNG
Kommentar: die "Einleitung zur Chromatographie" wird je nach Reihenfolge wechselweise mit Prof.
Trathnigg vorgetragen!
Abb. sind über einen Link zu laden: http://www-ang.kfunigraz.ac.at/~uray/chrom_abb
empfehlenswertes Buch: Veronika Meyer "Praxis der Hochleistungsflüssigkeitschromatographie"
Zusätzliche Ergänzungen zur Einleitung in diesem Skriptum:
Der Begriff "Präparative Chromatographie" umfasst alle Methoden, bei denen (auch mikrogramm) Fraktionen isoliert werden. Definition der Bezeichnung Chromatographie: abgeleitet von chromatos (griech)- gefärbt. Man verstand ursprünglich darunter die Trennung von Substanzen und Identifizierung durch ihre Farbe. Heute gilt der Ausdruck für alle Arten von Trennungen, die etwas mit Wechselwirkungen einer aufgegebenen festen, flüssigen oder gasförmigen Probe mit festem oder flüssigem Material (stationäre Phase) zu tun haben. Die Identifizierung erfolgt dabei beliebig. Zum Transport dient die “mobile Phase”, die ebenso alle Aggregatzustände einnehmen kann. Zwischen Gas und Flüssigkeit liegt zB die Chromatographie mit überkritischen mobilen Phasen , davon wird meist Kohldioxid in Mischung mit Methanol benützt. Die Grenzbedingungen sind:CO2 : Tc 31 C; Pc=73 bar, d= 0.44. Diese Art der Chromatographie wird aber mit ähnlichen Trägermaterialien wie Flüssigkeitschromatographie durchgeführt. Sie hat den Vorteil, dass der Strömungswiderstand wesentlich geringer als bei einer Flüssigkeit ist und dass präparativ das Laufmittel sehr leicht durch Druckverringerung entfernt werden kann. (z.B. Entkoffeinierung v. Kaffee und Tee). Die Anwendungsbreite ist jedoch im Vergleich zu GC und LC ziemlich eingeschränkt, es wird dazu auch ein Spezialchromatograph benötigt.
ARTEN DER FLÜSSIGKEITSCHROMATOGRAPHIE NACH DEM TRENNPRINZIP
Die wichtigste Trennprinzip in der Organischen Chemie bei der Fest-Flüssig Chromatographie ist die
1a Adsorptionschromatographie
Dabei findet an einem festen Trägermaterial (feste stationäre Phase) ständig ein Adsorptions/ Desorptionsprozess statt, der durch stationäre Phase, mobile Phase und Art der Analyten gesteuert wird . Entscheidend sind dabei verschiedene im Vergleich zu ionischen Wechselwirkungen relativ schwächere Kräfte, die in Summe für das Trennergebnis entscheidend sind: Dazu gehören hauptsächlich Wasserstoffbrücken, -Säure- -Base Interaktion, Dipol-Dipol Wechselwirkung und sterische Einflüsse. Die Adsorptionschromatographie kann in Art der Normal- oder Umkehrphase betrieben werden (genaueres s. unten)

Ib Ionenpaarchromatographie (Abb.)
Ähnlich wie die Adsorptionschromatographie, dabei bleiben die Ionen des Analyten durch ein Gegenion (langkettiges Ammonium+ oder langkettige Sulfonate-) maskiert und es wird sozusagen das äußerlich neutrale Paar in solvatisierter Form auf einer Umkehrphase getrennt.
Ic Ionenaustauschchromatographie (Abb.) Die stationäre Phase (Ionenaustauscher-Gel) enthält ionische Gruppen (zB. -NR3+ oder SO3-), und entsprechende einfache aliphatische Gegenionen , welche mit gleichartig geladenen Gegenionen der Analyten im Gleichgewicht um die Ionenbindung konkurrieren können. Die I-Chromatographie erfolgt bei geringem Überdruck. Sie wird vor allem zur Trennung von Säuren, Aminen oder Aminosäuren verwendet und ist eine komplementäre Methode zur Adsorptionschromatographie, bei der gerade Amine nicht besonders gut getrennt werden können (Schlechte Peakform). Allerdings gibt es inzwischen moderne (metallfreie!) Kieselgele, die schon sehr gut für diese Problemanalyten geeignet sind)
Weitere Trennprinzipien, die in der Fest-Flüssig Chromatographie angewandt werden:
Id Affinitätschromatographie (Abb.) mit besonders hoher Spezifität:
Hier wird ein ganz spezielles Rezeptormolekül an die stationäre Phase gebunden (Biomolekül), das nur - meist höhermolekulare - Proben bindet, die ganz bestimmte räumliche und ladungsmäßige Voraussetzungen erfüllen. (Enzym-Substrat, Antigen-Antikörper). Sie dient zur Isolierung von Peptiden, Proteinen usw.
1e Elektrochromatographie
Diese und ähnliche Verfahren haben eines gemeinsam: sie nützen den elektrischen Strom für die Bewegung der Analyten. Der große Vorteil dabei ist das verbesserte Strömungsprofil gegenüber Druckbeförderung: Es sieht aus wie ein sich bewegender Stopfen, während es ansonsten immer an den Kapillarwänden Rückhalteeffekte gibt und der Fluss daher in der Mitte am größten ist. Man erzielt damit besonders große Bodenzahlen, typisch über 100000. An ca. 50cm und kürzere dünne (wegen Erhitzen!), 0.2 bis 0.02 mm Quarz-Kapillaren (offen, also nur an der Wand gecoated, oder mit 2-5um Silica Material gepackt) wird eine Potentialdifferenz von etwa 50 kilovolt/meter angelegt. Dabei wandern geladene Teilchen, die auch nur Teilladungen haben können, je nach Vorzeichen zur Kathode bzw Anode. Die Methode heißt “Kapillarelektrophorese” (CE) für geladenen Teilchen, “Micellare Elektrokinetische capillarchromatographie” MECC , wenn ungeladene Analyten teilweise in sich bewegende Micellen gehen. (Die treibende Kraft heißt “Elektro(end)osmotischer Flow”) Es können also auch neutrale Moleküle befördert werden, sofern sie an der Oberfläche Überschussladungen aufbauen oder sich mit einem “host”, der sich mit Strom bewegen lässt ist verbinden.
If Gelchromatographie (Ausschlusschromatographie) (Näheres im speziellen Vorlesungsteil von Prof. Trathnigg) Trennung nach der Größe der Teilchen - große schnell, kleine langsam, weil sie erst durch die passenden Hohlräume in der Phase müssen und daher zurückgehalten werden. Dies wird vor allem für Oligomere und Polymere angewandt. Es werden daher großporige Gele (300Å) verwendet.

ad Ia. DIE ADSORPTIONSCHROMATOGRAPHIE AN KIESELGEL UND ANDEREN FESTPHASEN.
Wir unterscheiden normale stationäre Phasen mit polarem Trägermaterial (“stationäre Phase”) die mit eher unpolarem Laufmittel (KW-Stoffe wie Hexan, Heptan mit etwas Essigester, Dioxan oder 2-Propanol etc) ) als mobile Phase betrieben werden und Umkehrphasen (“reversed phase material”) mit unpolarer, chemisch modifizierter Oberfläche, welche mit stark polarer mobiler Phase betrieben werden. (Man verwendet als Laufmittel meist Wasser , oft gepuffert, sowie Gemische von Wasser mit Methanol oder Acetonitril). Im ersteren Fall (normal phase) werden unpolare (lipophile) Stoffe leicht, polare Stoffe (Säuren, Amine) schwer oder gar nicht eluiert , im Umkehrfall kommen die letzteren Substanzen zuerst wieder von der Phase herunter. Hier gilt also,: "Ähnliches bindet sich besser an Ähnliches" (wie “similis similibus solvuntur” bei Löslichkeitsvergleichen.
Polare Materialien für normale Phasen sind: vor allem Kieselgel, aber auch Aluminiumoxid, Magnesiumsilikat; in speziellen Fällen (Beispiel: Enantiomerentrennung) werden auch natürliche organische Polymere wie Cellulose eingesetzt, die meist auch chemisch modifiziert und dann auf Kieselgel gecoated oder kovalent gebunden wird (s. unten).Kieselgel und diverse andere Materialien haben an der Oberfläche OH-Gruppen, die durch chemische Umsetzung mit unpolaren Gruppen (vorzugsweise C8 oder C18 Alkyl u.a.) lipophilisiert werden können und dann als stationäre Umkehrphasen verwendet werden können. Man verwendet aber auch künstliche Polymere und Copolymere sowie Graphit oder poröses Glas als zu modifizierendes Trägermaterial.
ABBILDUNGEN:
Modifikation der Materialien: Chemische Derivatisierung der Oberfläche Normalphasen und Umkehrphasen
SiOHBürstenoberfläche
Abb. von verschiedenen Gruppen, die an Kieselgel gebunden werden können. Wie bereits erwähnt, ist C8 und C18 die häufigste Modifikation. Sie wird auch in der präp. Chromatographie genützt.
Durch gezielte Umsetzung der OH-Funktionen und anderer reaktiver Funktionen mit weiteren, komplexeren polaren Gruppen können aber auch chemisch modifizierte Materialien hergestellt werden, die dann ganz spezielle Eigenschaften aufweisen.(Abb. chemische Modifikation und Phasentypen).
Als spezifisches Beispiel seien hier die Trennung von razemischem Phenylcyclohexanol an Kieselgel gezeigt, das durch kovalente Bindung von enantiomerenreinen “Selektoren” chiral modifiziert wurde. (Abb. Phenylcyclohexanoltrennung). Die meistverwendete natürliche Quelle von chiralen Selektoren ist modifizierte Zellulose (Chiracel®)

PRÄPARATIVE CHROMATOGRAPHIE:
Definition: Als präparative Chromatographie bezeichnet man jede chromatographische Methode, deren Ergebnis oder Ziel die Isolierung eines gereinigten Stoffes ist. Man bezeichnet weiters noch als “semipräparative” Verfahren solche die Substanzmengen von etwa 0.1 bis 10 mg bewältigen können. Präparative GC geht sogar in den ng-Bereich. (Beispiel Duftstoffe - Schnüffeldetektor!)
Am weitaus häufigsten in der (präparativen) Chromatographie wird normales Kieselgel als stationäre Phase verwendet. Es ist relativ billig, leicht zu standardisieren und auch zu modifizieren und wird jetzt näher besprochen.
(Abb. Kieselgel - Formen)
Unterschiede in der Form: Sphärisch oder gebrochen Erhöhter Gegendruck entsteht durch feine Anteile, die durch “floating” weggespült werden müssen. Uniformes Material gibt die beste Performance. Je kleiner die Teilchen, desto höher ist der Gegendruck.
Begriffe der Poren- und Teilchengröße:
Die typische Porendurchmesser in der Chromatographie einfacher organischer Stoffe ist 60 Å(ngström) = 6nm (6.10-3µ) ”Kieselgel 60” , größere Porenweiten werden bei Makromolekülen eingesetzt, z.B. 300A. Die Porenweite wird also meist in Angström angegeben, sie ist daher leicht mit der Molekülgröße in Verbindung zu bringen. Typisches HPLC Material hat 5µ Durchmesser Es ergibt gepackt in einem Stahlröhrchen von 4mm Innendurchmesser und 25cm Länge einen Gegendruck von etwa 50 bis 250 bar je nach Flussrate (1-2mL/min ) und Laufmittel. Polare LM entwickeln höheren Gegendruck, wobei große Unterschiede auftreten können zB zw. Isopropanol und Methanol (ersteres wesentlich höher!) Die Teilchengröße wird bei präparativen Anwendungen von 5 auf 10-40 Å. Wichtig ist auch noch die spezifische Oberfläche , die in m2/g angegeben wird (typisch: 300-500, bei Dünnschichtmaterial oft nur etwa 50 .
Abb.: Einfluss des Säulendurchmessers und der Größe der Phasenteilchen auf die Peakform.
Die Teilchengröße beeinflusst die Trennleistung, sie ist an sich unabhängig vom Säulendurchmesser, wenn man von Wechselwirkungen mit der glatten inneren Oberfläche absieht! Die Peaks werden aber mit engerem Durchmesser bei gleicher Konzentration höher, soferne nicht überladen wird. Die Verweilzeit im Detektor spielt ebenso eine Rolle. Eine dünnere Säule erlaubt daher eine kleinere Flussrate bei gleicher Detektorempfindlichkeit Daher verwendet man zunehmend auch Mikro- (1mm Durchmesser) und Kapillarsäulen (<0.07mm). Sogar Nanoliter - Chromatographie wird langsam so stabil, dass Routinegeräte erhältlich sind
Offene Säulen sind solche, die nur an den Innenwänden belegt sind, gepackte sind dagegen sehr dicht mit stationärer Phase gefüllt (mit etwa 400bar)
Probleme bei Mikrosäulen sind Detektion (geringste Volumina!) und Stabilität, Vorteile sind der geringere Lösungsmittelverbrauch, der in der Flüssigkeitschromatographie, auch in der analytischen HPLC, erheblich ist.

Methoden der präparativen Chromatographie, dazugehörige Geräte und Bedingungen:
Varianten der Säulenchromatographie: Flash, DryFlash und Mitteldruckchromatographie, SMB (simulated moving bed chromatography) .
Präparative Dünnschichtchromatographie s. Prof. Stadlbauer.
“alte” Säulen-Chromatographie: ca. 200 µ Kieselgel. Das Verfahren wurde wenig genützt wegen des großen Zeitaufwands: es arbeitet ohne Druck, nur mit der Schwerkraft: Abb.
Seit 1979 (Erfinder Clark Still) wird die Flash-Chromatographie als Routinemethode angewandt: ca. 30 - 60µ Material, ca. 1-2 bar Druck (Abb.).Dieses Verfahren wird im Chromatographiekurs geübt
Hauptunterschied zur gewöhnlichen Säulenchromatographie: Überdruck erfolgt aus Gasflasche mit Regelung von oben, das Flüssigkeitsreservoir ist die halbe Säule oder es wird zusätzlich in Form einer Kugel dazwischengeschaltet.
Trennbare Substanzmenge: von etwa 1/100 bis 1/25 der Phasenmenge je nach Rf-Wert , Ähnlichkeit und Menge in den Verunreinigungen. Diese Angabe gilt für die Flash- und Dryflash - Chromatographie, aber etwa auch für eine Mitteldrucksäule
Rf > 0.2 Rf<0.2 K-Gel S-Durchm Fraktionen Vol mg mg mL (g) cm (Gelhöhe) mL mL/min
100 40 6 (2.5) 1(8) 5 10900 360 140 (57) 3(20) 20 352500 1000 600 (235) 5(30) 50 100
Das Leervolumen ist etwa 80% des Raumbedarfs des Kieselgels. (Beispiel HPLC: Säule 4x250mm enthält ca. 2.3g Gel bei 2.5 mL Leervolumen) . Je schwieriger die Trennung, desto weniger Substanz kann pro gegebener Phase und Säule aufgetragen werden.
Packmethoden:
Eine ausführlicheAnleitung kann auch unter http://www.emdchemicals.com/ (suche dort nach flash
chromatographie gefunden werden. Es gibt dort auch noch andere recht gute Tipps. Trockenpackmethode: Verdichten der trocken gepackten Säule erfolgt durch vorsichtiges Aufstoßen auf Holz/Gummi Für die Flashchromatographie wird eher die Trockenpackmethode bevorzugt, aber dabei muss man die beträchtliche Hitzeentwicklung bei Benetzung des trockenen Materials beachten und daher rasch und zügig konditionieren (Selbstkühlung durch LM)!
Flash: Hahn offen, Gasauslaß komplett offen: dann Lösungmittel hinein, bis voll, Gas anlegen, mit dem

Zeigefinger Druck erzeugen. Nicht trockenlaufen lassen, sonst entstehen Risse.
Probenaufgabe mit langer Pipette, als LM das am wenigsten polare der Mischung, damit ein dünnes Startband entsteht. Eine Alternative vor allem bei schwerlöslichen Verbindungen ist das Mischen von einigen g Kieselgel mit einer Lösung der Probe und Einrotieren. Dann kann das Pulver aufgebracht werden.
Eine nachherige DC-Prüfung genügt. Fraktionssammler sind nicht nötig, weil der Vorgang nur etwa 30 min dauert. Es werden einfach nach bestimmten Volumina (zB 25 mL) die Vorlagen gewechselt. Dry-Flash Material ca. 15µm (etwa wie DC Kieselgel, hat aber eine wesentlich größere Oberfläche (3-500m2/g und ist ohne Gipszusatz (Kieselgel H statt G).
Abb. der Apparaturen
Hauptsächlicher Unterschied zur Säulen- und Flash: Das Bett darf trocken laufen, Gradientenelution ist so möglich. Die Methode wird im Kurs geübt. Probleme gibt es bezüglich Verdunstungskälte von niedrig siedenden Laufmitteln und die dadurch verursachte Kondensation von Wasser außen an der Glasapparatur.
Chromatogrammabbildung:
Präp. HPLC chiral modifizierten Säulen: Beispiel Halbmikrotrennung von R-und S-2-Phenylcyclohexanol....
SIMULATED MOVING BED CHROMATOGRAPHIE (SMB):
Das ist eine kontinuierlich präparative Chromatographiemethode, wie sie in der Feinchemikalienindustriezunehmend angewendet wird. Es sind dazu mindestens 6 Säulen notwendig, die über eine programmierbare Einlaß-Auslaßventilschaltung die laufende vollautomatische Zugabe/Abnahme von Probe/reinem Produkt und Laufmittel an jeder Verbindungsstelle ermöglicht. Es gibt Anlagen mitKapazitäten von 10g /tag bis zu über 1000 Jahrestonnen (siehe Bild mit Arbeiter)
Eine sehr anschauliche Darstellung findet sich auf
http://www.novasep.com/what/licosep.htm

Bedeutung der Desaktivierung von Kieselgel oder Al-oxid:
“NEUES” Trägermaterial besitzt zum Teil STARK aktive Stellen, die Substanzen zu stark festhalten könnten: daher werden diese meist durch Zusatz von Wasser zum LM deaktiviert. Die Trennung kann dadurch bedeutend verbessert werden UND ES VERSCHWINDET WENIGER SUBSTANZ auf nimmer Wiedersehen.
Für die Reproduzierbarkeit bei analytischen Methoden verwendet man besser zu 50% wassergesättigtes Laufmittel (natürlich bei solchen, die mit Wasser nicht mischbar sind), das durch Mischen von trockenem und mit Wasser gesättigten LM aus einer zweiten Flasche erzeugt werden kann. Bei Hexan verwendet man besser 0.05% Acetonitril wegen Mischbarkeitsproblemen. bei Al-oxid nur 25% Sättigung Aluminiumoxid gibt es in drei Arten: basisch, neutral und sauer, wobei man durch Zugabe von vorschriftsmäßigen Mengen Wasser Al2O3 von genau definierter Aktivitätsstufe (I-IV) erzielen kann. Selbstverständlich werden auf sauren Materialien saure Verbindungen (Carbonsäuren usw und auf basischen Materialien basische Verbindungen (Amine) getrennt - weil umgekehrt würde ja leicht Salzbildung eintreten und die Analyten würden nie mehr von der statt. Phase herunterkommen. Man hilft sich oft auch durch Zugabe von etwas Amin (zB Triethylamin) oder Säure (zB Eisessig, Ameisensäure, Trifluoressigsäure) zur mobilen Phase, damit derartige Analyten schärfer eluiert werden.
Nach Konditionieren der Säule mit einem Laufmittel sollen Lösungsmittel besser nur stufenweise ersetzt werden, (etwa Wasser - Alkohol -Essigester - Toluol - Hexan), weil sich die Gleichgewichte nur sehr langsam einstellen. Deswegen ist Gradientenelution bei Normalphasen HPLC schwierig.. Bei RP ist das viel besser (schneller) möglich. Hier ist also das ideale Umfeld für Gradientenelution gegeben. Dabei geht man immer vom schlechten LM zum besseren LM Vorteil: Peaks werden “zusammengeschoben”. Aufgetragen wird immer aus dem Laufmittel oder einem weniger guten, nie aus einem besseren LM, weil das zu Peakverbreiterungen führt.
RP-Phasen:Meist wird mit einem Gradienten eluiert (zB von 90% auf 10 % Wasseranteil) Proben werden umso stärker retardiert, je weniger polar sie sind, also kommen zB Alkane sehr spät oder gar nicht, dafür kommen z.B. Aminosäuren, welche mit einer Normalphase kaum zu bewegen sind , sehr früh, haben also kaum Wechselwirkungen mit der statt. Phase.
LM mit zunehmender Elutionskraft: Methanol<Acetonitril<Ethanol<Isopropanol<DMF<n-Propanol<Dioxan<THF
Gradientenelution ist im Gegensatz zu der Normalphase sehr gut möglich, den die Einstellungszeit des Gleichgewichts ist kurz. Oft werden wässrige Mischung mit Methanol und häufiger Acetonitril verwendet, oft gepuffert, zB mit Ammoniumacetat oder Ammoniumformiat, letzteres kann man durch Gefriertrocknen und Destillation entfernen.
Bei der LC spielt die Reinheit der LM eine sehr große Rolle, daher sind sie teuer. Selbst das Wasser muss besonders sorgfältig gereinigt werden.Es enthält neben Salzen immer organische Verunreinigungen, die auch beim Destillieren übergehen können: Bei präparativer Anwendung spielt das ebenso eine Rolle, denn die Volumina werden entsprechend größer!).

Gradienten sammeln Verunreinigungen am Säuleneingang und eluieren oft diese bei höherem org. LM-Anteil. Daher Vorsicht beim „Leerlauf“!
Das Chromatogramm und einige Aussagen dazu: (Abb. 14)
Anhand verschiedener Abb. ( normale Peaks und Überladungseffekte, Tailing, Fronting , soll die Aussagekraft von Kapazitätsfaktor k´, Trennfaktor alpha, Resolution R und Bodenzahl n besprochen werden.
Die Retentionszeit einer Komponente ist bei gleichen chromatographischen Bedingungen stets gleich groß.Die Retentionszeit ist die Zeit, die vom Einspritzen bis zum Maximum der Peakhöhe verstreicht
Chrom. Bedingungen sind: Handelsnamen, Säulenabmessungen, stationäre und mobile Phase, Fließgeschwindigkeit, ev. Probengröße und Temperatur.
Quantitative Analytik:
Fläche der Peaks ist der identen Stoffmenge proportional
Bei UV-Detektor: E= cd bei UV-Detektion, die Fläche ist daher auch Geräte und Substanz- bzw Wellenlängenabhängig.
Angaben anhand eines Chromatogramms über die Güte der Trennung und Säule:
Idealerweise sollte der Peak eine Gauß-Kurve (Glockenkurve) darstellen. Wenn überhaupt, sollte er eher Tailing als Fronting zeigen.
w: Basisbreite des peaks; w1/2 die Halbwertsbreite t0: Totzeit der Trennsäule, die mobile Phase benötigt, die Trennsäule zu durchwandern, ebenso eine nicht retardierte Substanz. An einem solchen peak wird die Leistung der Säule bezüglich Bodenzahl bestimmt
tR : Retentionszeit, vom Einspritzen bis zur Registrierung des Maximums
t´R Netto- Retentionszeit, t´R = tR - t0
Diese Zeitwerte sind von der Säulenlänge und der Fließgeschwindigkeit abhängig, daher gibt man die dimensionslose Größe k´ an, wodurch man auch aus jeder Kopie ohne Zeitskala die Trennparameter bestimmen kann.
k´: ist der Kapazitätsfaktor
(tR - t0)/t0 = t´R/t0
Das ist nichts anderes als das Molverhältnis des Stoffes X in der stationären Phase und mobilen Phase, k´x = nstat/nmob
was etwas sehr ähnliches ist wie der Verteilungskoeffizient

Kx = cstat/cmob
wobei statt c (conc) eigentlich die Aktivität stehen müsste Kapazitätsfaktoren zwischen 1 und 5 sind ideal, bei zu kleinen Werten erfolgt oft keine Trennung, weil zu wenig Wechselwirkung stattfindet, bei größeren Werten ist neben der Peakverbreiterung durch Diffusion die zu lange Analysendauer und der hohe Laufmittelverbrauch von Nachteil.
Zwei Komponenten werden nur dann getrennt, wenn sie sich in den k´- Werten unterscheiden.

Die relative Retention (oder der Trennfaktor) ist ein Maß für die Trennung, bzw die Eigenschaft des chromatographische Systems, zwei bestimmte Stoffe zu trennen.
= k´2/k´1
wobei k´2 der größere Wert ist (späterer peak) Ist = 1, ist keine Trennung gegeben
Damit ist die Trennung manchmal nur ungenau beschrieben, denn die Peakbreite ist hier nicht berücksichtigt. Daher verwendet man den Begriff Auflösung oder Resolution bei Peaks mit guter Symmetrie. Sind sie abweichend von der Gaußform, nennt man es Fronting oder Tailing(Abb. 15, 16). Bei zu starkem Fronting/Tailing ist die Flächenmessung leicht fehlerhaft. Tailing ist meistSubstanzabhängig (typisch: Amine , selbst häufig bei reversed phase!)
Die Auflösung (Resolution) berechnet sich unter Inkludierung der Basisbreite
tR2 - tR1
R = 2 ---------------- w1 + w2
oder wegen der bei teilw. Trennungen leichter zu messenden Halbwertsbreite:
tR2 - tR1
R = 1.18 ----------------- w1/2,(1) + w1/2,(2)
Was kann man sich unter Werten von R = 0.8; 1.0 oder 1.25 vorstellen?
ABB. 17
Trennstufen oder Bodenzahl:
tR2 2 N = 16 --------
w
t R2 2 N = 5.54 ----------
w1/2
Sie liegt bei 250 mm HPLC - Säulen im Bereich von etwa 6000 bis etwa 20000 , Bei Mitteldrucksäulen , die aber länger sind, bis etwa 7000 (15bar bei sehr geringer Beladung), sie ist ein Maß für die Güte der Packung einer Säule. Der Wert ist auch abhängig von der Testsubstanz, die auch nicht zu konzentriert sein

sollte (einige Mikrogramm/g Kieselgel)
Ein verwandter Begriff ist die Trennstufen- oder Bodenhöhe H, die strecke auf der das chromatographische Gleichgewicht einmal eingestellt ist =”Höhe eines Theoretischen Bodens” = englisch HETP
H=L/N L=Länge der Säule, N= Bodenzahl
Die Angabe zB: Die Säule hat 10000 Böden genügt nicht! man muss dabei auch die Länge und den Innendurchmesser wissen, die Fließgeschw. der mob. Phase, Probesubstanz und deren k´ - Wert, stationäre und mobile Phase
ÜBERLADUNG BEI PRÄPARATIVER CHROMATOGRAPHIE: (Abb. 18, 19)
Aus ökonomischen Gründen werden Säulen in der präparativen Variante prinzipiell überladen. Das bewirkt allerdings daß die Trennung immer schlechter wird und der k´-Wert kleiner wird. Dies wird bis jedoch bewusst toleriert, weil man wesentlich weniger häufig einspritzen muss, die Hauptfraktionen schneiden kann und weniger LM braucht. Der Mittelteil wird zurückgeführt. Richtwert: max 10% Verkürzung der analytischen Retentionszeit durch Überladung ist tolerierbar.
Allgemeine Hinweise:
Die Auflösung ist nur der Wurzel aus der Bodenzahl proportional Eine Verdoppelung der Böden um den Faktor 2 gibt nur eine 1.4 mal bessere Auflösung
Auch die Säulenlänge geht nur mit der Wurzel in die Resolution ein
Am meisten kann noch durch Veränderung der mobilen Phase erreicht werden, dadurch kann am ehesten verbessert werden. Durch Wechseln des LM ändern sich die Wechselwirkungseigenschaften mit der stationären Phase. k´ sollte aber im gleichen Bereich bleiben (1-5), daher muss das andere LM ähnlich polar sein wie das alte
Eine Derivatisierung der Analyten ist oft zur besseren Detektion (Fluoreszenz!) notwendig; sie kann pre-und postcolumn erfolgen.
Vorteile der Pre-Column Derivatisierung:
Freie Wahl der Reaktionsbedingungen Reaktion kann langsam sein Derivatisierung kann zugleich zur Reinigung und Aufarbeitung dienen Überschüssiges Reagens kann wieder entfernt werden Chromatographische Eigenschaften können verbessert werden (weniger tailing, bessere Trennparameter, zB. von Amin zu Amid

Nachteile: Nebenprodukte können entstehen,Derivatisierung ist oft nicht quantitativ und die chromatographische Selektivität kann auch sinken. Vorteile der post-column Methode (in einem sogenannten Reaktionsdetektor): Die Reaktion muss nicht quantitativ verlaufen, man kann zB. zwischen UV- und Flu-Detektor derivatisieren Nachteile: Die Reaktion muss in der verwendeten mobilen Phase reproduzierbar ablaufen Das Reagens, das in großem Überschuss zugegeben werden muss, darf nicht detektiert werden. Reaktionsdetektoren: offene Kapillaren 0.5 -1 min; knitted (= gebogene) tube - 5 min; Bettreaktoren (mit unporösem Material gefüllt) 0.5 bis 5 min Verweilzeit, die Packung kann auch aus einem Katalysator oder einem immobilierten Enzym bestehen In der Indutrieanalytik gibt es auch "segmentierte Systeme" für Reaktionen bis 30 min, Aufteilung in kleine Portionen durch Luftblasen oder nicht mischbares LM.








![Chromatographische, NMR-spektroskopische und ...€¦ · 2+ und cis-[PtCl 2(NH3)2] Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Chemie](https://static.fdokument.com/doc/165x107/5aff25557f8b9a444f8fc9c8/chromatographische-nmr-spektroskopische-und-2-und-cis-ptcl-2nh32-dissertation.jpg)









