
Über S-Oxalyl-thiole
11
1962 J. KOCH und L. JAENfCKE 129 UBER S-OXALYL-THIOLE von J~~RGEN KOCH und LOTHAR JAENICKE Aus dem Chemischen Laboratorium der Universitiit Miinchen, Institut f i r Biochemie Eingegangen am 6. November 1961 S-Oxalyl-thiole, die noch nicht untersuchten ersten Glieder der homologen Reihe von Thioestern der Dicarbonsauren, wurden durch Kondensation von Oxalylchlorid mit Thiolen dargestellt. Die zungchst erhaltenen Oxalyl-dithiole zeichnen sich durch eine auoerordentliche Labilitat im neutralen Medium aus. Sie hydrolysieren spontan zu den Mono-thioestern, die eine bemerkenswerte Stabilitat in saurer Lbsung und ein unerwartet langwelliges Absorptionsmaxi- mum besitzen. Die Ursachen dieser Eigenschaften werden in den Polarisations- verhiiltnissen der Thioester gesehen und diskutiert. Oxalyl-Coenzym A tritt im Stoffwechsel der Oxalsiiure bei Mikroorganismen a d . Es wird durch ein Codehydrase I1 benotigendes s p d s c h e s Enzymsystem glatt zu Glyoxylsaure und Coenzym A reduziert. Ein aus Faulschlamm isoliertes Bakterium der Klasse Pseudomonus wachst auf Oxalsaure als einziger Kohlenstoff- und Energiequelle. Extrakte dieses Bakteriums besitzen, wie unveroffentlichte Versuchevon Dr. P. C. CHAN ergeben haben, die Fahig- keit, Oxalsaure zu COZ und Formiat zu spalten. Als Cofaktoren sind Phosphat, Coenzym A (CoA) und Thiaminpyrophosphat erforderlich. Als Zwischenprodukt kOMte Glyoxylsaure in Verdiinnungsversuchen wahrscheinlich gemacht werden. Vor kurzem erschienene Publikationen von QUAYLEI-4) berichteten ebenfalls uber die Spaltung der Oxalsaure durch Pseudomonas oxuluticus und iiber eine in Extrakten dieses Mikroorganismus gefundene Dehydrogenase, die mittels TPN Glyoxylsaure zu Oxalsaure oxidiert. Auch diese Reaktion erfordert CoA. Aus beiden Beobachtun- gen ergibt sich die Moglichkeit, da13 Oxalsaure, an CoA aktiviert, im Stoffwechsel verwendet wird. Oxalyl-monothiole (Thiooxalsaure-S-ester, RS . CO . C02H) wurden bisher noch wenig beschrieben*). Im folgenden sol1 uber Darstellung und Eigen- schaften dieser interessanten Zwischenstufen des Stoffwechsels berichtet werden. Allgemein geht man zur Synthese von Acylthiolen vom Anhydrid oder Chlorid der entsprechenden Saure aus, die mit dem Thiol kondensiert wird. Auf diese Weise wurde *) R. STOLL~, Ber. dtsch. &em. Ges. 47, 1130 (1914). hat bei seinen Versuchen zur Dar- stellung des Methylthionaphthenchinons das Thiokresyloxalsaurechlorid und durch dessen Hydrolyse die Thiokresyloxalsiiure dargestellt. 1) J. R. QUAYLE und D. B. KEECH, Biochem. J. 75, 515 (1960). 2) J. R. QUAYLE, D. B. KEECH und G. A. TAYLOR, Biochem. J. 76.3 P (1960). 3) J. R. QUAYLE, D. B. KEECH und G. A. TAYLOR, Biochem. J. 78,225 (1961). 4) J. R. QUAYLE und G. A. TAYLOR, Biochem. J. 78,611 (1961). Licbign Ann. Chcm. Bd. 652 9
-
Upload
juergen-koch -
Category
Documents
-
view
212 -
download
0
Transcript of Über S-Oxalyl-thiole

1962 J. KOCH und L. JAENfCKE 129
UBER S-OXALYL-THIOLE
von J ~ ~ R G E N KOCH und LOTHAR JAENICKE
Aus dem Chemischen Laboratorium der Universitiit Miinchen, Institut f i r Biochemie
Eingegangen am 6. November 1961
S-Oxalyl-thiole, die noch nicht untersuchten ersten Glieder der homologen Reihe von Thioestern der Dicarbonsauren, wurden durch Kondensation von Oxalylchlorid mit Thiolen dargestellt. Die zungchst erhaltenen Oxalyl-dithiole zeichnen sich durch eine auoerordentliche Labilitat im neutralen Medium aus. Sie hydrolysieren spontan zu den Mono-thioestern, die eine bemerkenswerte Stabilitat in saurer Lbsung und ein unerwartet langwelliges Absorptionsmaxi- mum besitzen. Die Ursachen dieser Eigenschaften werden in den Polarisations- verhiiltnissen der Thioester gesehen und diskutiert. Oxalyl-Coenzym A tritt im Stoffwechsel der Oxalsiiure bei Mikroorganismen a d . Es wird durch ein Codehydrase I1 benotigendes s p d s c h e s Enzymsystem glatt zu Glyoxylsaure
und Coenzym A reduziert.
Ein aus Faulschlamm isoliertes Bakterium der Klasse Pseudomonus wachst auf Oxalsaure als einziger Kohlenstoff- und Energiequelle. Extrakte dieses Bakteriums besitzen, wie unveroffentlichte Versuche von Dr. P. C. CHAN ergeben haben, die Fahig- keit, Oxalsaure zu COZ und Formiat zu spalten. Als Cofaktoren sind Phosphat, Coenzym A (CoA) und Thiaminpyrophosphat erforderlich. Als Zwischenprodukt kOMte Glyoxylsaure in Verdiinnungsversuchen wahrscheinlich gemacht werden.
Vor kurzem erschienene Publikationen von QUAYLEI-4) berichteten ebenfalls uber die Spaltung der Oxalsaure durch Pseudomonas oxuluticus und iiber eine in Extrakten dieses Mikroorganismus gefundene Dehydrogenase, die mittels TPN Glyoxylsaure zu Oxalsaure oxidiert. Auch diese Reaktion erfordert CoA. Aus beiden Beobachtun- gen ergibt sich die Moglichkeit, da13 Oxalsaure, an CoA aktiviert, im Stoffwechsel verwendet wird. Oxalyl-monothiole (Thiooxalsaure-S-ester, RS . CO . C02H) wurden bisher noch wenig beschrieben*). Im folgenden sol1 uber Darstellung und Eigen- schaften dieser interessanten Zwischenstufen des Stoffwechsels berichtet werden.
Allgemein geht man zur Synthese von Acylthiolen vom Anhydrid oder Chlorid der entsprechenden Saure aus, die mit dem Thiol kondensiert wird. Auf diese Weise wurde
*) R. STOLL~, Ber. dtsch. &em. Ges. 47, 1130 (1914). hat bei seinen Versuchen zur Dar- stellung des Methylthionaphthenchinons das Thiokresyloxalsaurechlorid und durch dessen Hydrolyse die Thiokresyloxalsiiure dargestellt.
1) J. R. QUAYLE und D. B. KEECH, Biochem. J. 75, 515 (1960). 2 ) J. R. QUAYLE, D. B. KEECH und G. A. TAYLOR, Biochem. J. 76.3 P (1960). 3) J. R. QUAYLE, D. B. KEECH und G. A. TAYLOR, Biochem. J. 78,225 (1961). 4) J. R. QUAYLE und G. A. TAYLOR, Biochem. J. 78,611 (1961).
Licbign Ann. Chcm. Bd. 652 9

I30 J. KOCH und L. JAENICKE Bd. 652
in verschiedenen Arbeitskreisens-9) eine groDe Anzahl von einfachen und kompli- zierteren Acylthiolen dargestellt. Im Fall der Oxalsaure ist diese Methode aber nicht anwendbar. Das Halbchlorid der Oxalsaure ist instabil, ein Anhydrid existiert nicht 10).
Wir schlugen deshalb den Weg uber das Dichlorid ein, das sich in glatter Reaktion mit Thiolen zu den Dithioestern umsetztlo). Halbhydrolyse dieser Verbindungen fiihrt dann zu den gewiinschten Oxalyl-monothioestern. Fiir die Halbhydrolyse ver- wendeten wir anfangs die berechnete Menge Kaliumacetat oder Alkali, bis sich her- ausstellte, daB die Halbester bereits in wal3riger Liisung durch spontane Hydrolyse aus den Diestern entstehen.
Nach dem skizzierten Verfahren wurden S.S'-Oxalyl-bis-[N-acetyl-cysteamin] (I a) und S.S'-Oxalyl-bis-[N-capryloyl-cysteamin] (I b) sowie S-Oxalyl-N-acetyl-cysteamin (I1 a) und S-Oxalyl-N-capryloyl-cysteamin (I1 b) als Ausgangsprodukte fiir weitere Umacylierungen gewonnen.
R- S-C=O R- S - G O a: R = CH3.CO.NH.(CH2)2- I I
R-S-C=O HO-C=O b: R = CH3.(CHi)6*CO*NH.(CH2)2- I I1
Die physikalischen Eigenschaften der Verbindungen sind in Tabelle 1 zusammenge- stellt.
Tabelle I . Eigenschaften von S-Oxalyl-thiolen
Nr. Verbindung Eigenschaften Schmp.
I a
Ib
I1 a
I l b
S.9-Oxalyl-bis-[N-acetyl- cysteamin]
S.9-Oxalyl-bis-[N-capryloyl- cysteamin]
S-Oxalyl-N-acetyl- cysteamin
Natriumsalz
S-Oxalyl-N-caprylo yl- cysteamin
Natriumsalz
Cylohexylammoniumsalz S-Oxalyl-glutathion S-Oxalyl-Coenzym A
weiDe Nadeln aus Dioxan 176'
lange Nadeln aus Dioxan
durchsichtige Stabchen 159- 160' (Zers.) aus Dioxan
hygroskop. Kristalle aus Xthanol/Aceton
Bliittchen aus Essig- 60' esterlPetrol5ther
Kristalle aus khanol/Aceton
Nadeln aus Dioxan 108' (Zen.) hygroskopisches Pulver hygroskopisches Pulver
148 - 149"
5) W. MICHLER, Liebigs Ann. Chem. 176, 177 (1875). 6 ) E. J. SIMON und D. SHEMIN, J. h e r . chem. SOC. 75.2520 (1953). 7 ) TH. WIEUND und H. BONHARD, Liebigs Ann. Chem. 572, 190 (1951). 8) TH. WIEUND und E. BOKELMANN, Angew. Chem. 64,59 (1952). 9) TH. WIELAND und L. RUEFF, Angew. Chem. 65,186 (1953).
10) H. STAUDMGER, Ber. dtsch. chem. Ges. 41,3558 (1908).

1962 Uber S-Oxalyl-thiole 131
Zur Darstellung komplexer Acylthiole hat sich das Verfahren von ECK~ERER~~) bewiihrt. Es beruht auf der intermediaen Darstellung des Acylderivates des ather- loslichen N-Capryloyl-cysteamins, aus dem man durch Transacylierung im schwach alkalischen Medium z. B. das CoA-Derivat bereiten kann. Man setzt das Acceptor- Thiol mit einem UberschuD an S-Oxalyl-N-capryloyl-cysteamin in waBriger Losung bei pH 8 um. Kontinuierliche kherextraktion des freien N-Capryloyl-cysteamins verschiebt das Gleichgewicht zugunsten des hydrophilen CoA-Derivats. Durch Um- esterung nach dieser Methode wurden S-Oxalyl-glutathion und S-Oxalyl-CoA her- gestellt. Die Ausbeute an CoA- und Glutathion-Derivat ist quantitativ, wie durch Papierchromatographie und Papierelektrophorese gezeigt wurde.
S-Oxalyl-thiole geben, wie alle Thioester, mit Hydroxylamin die entsprechende Hydroxamsaure, mit Ammoniak das Amid. Die sich daraus ergebenden analytischen Verfahren sind S. 138/139 beschrieben.
Die Infrarotspektren der Oxalsaurethioester zeigen die starke C =0-Valenzschwin- gungsbande der Carbonylgruppe bei etwa 168Ocm-1 und eine sehr starke C-S- Schwingungsbande innerhalb der -CO-S-Gruppierung, deren Lage vom Rest des Molekiils abhangt und die zwischen 755 und 820 cm-1 liegt (la: 795 cm-1, Ib: 820 cm-1, IIb: 755 cm-1, Cyclohexylammoniumsalz vod IIb: 793 cm-1). Bei den Salzen der Halbester lindet man auhrdem noch die gekoppelte 0-C-0-Schwingung der Carboxylatgruppe bei 1370-1380 cm-1. Die entsprechenden Sauren zeigen die C-O- Schwingung der Carboxylgruppe bei 1250 cm-1.
Eine auffallende Eigenschaft der S-Oxalyl-thiole, durch die sie sich von den anderen Acylmercaptanen unterscheiden, ist ihre relativ langwellige Ultraviolett-Absorption. Die Absorptionskurve der Monothioester zeigt in 0,Ol m Suminatpuffer pH 5.75 ein Maximum bei 260 mp. Die molare Extinktion betragt fii S-Oxalyl-N-acetyl-cysteamin E = 3.4 - 103, fur S-Oxalyl-N-capryloyl-cysteamin E = 2.9.103. In 1 n HCl verschiebt sichdasMaximumnach270mp,dieExtinktionsteigtumetwa 10%(IIb: E = 3.1 elO3).
Das Absorptionsmaximum der Oxalsiiure-dithioester liegt sowohl in Wasser als auch in Dioxan bei 285 mp. Beim Stehen der waBrigen Losung verschwindet diese Bade jedoch in kurzer Zeit, und statt dessen tritt die 260-mp-Bande der Monothio- ester auf. Die Dithioester der Oxalsaure sind - wie erwahnt - aukrst empfindlich gegen Wasser und hydrolysieren rasch zu den Halbestern. Aus diesem Grunde -den die Extinktionskoeffizienten in trockenem Dioxan bestimmt. Sie sind etwa doppelt SO
hoch wie die der Halbester in neutraler Losung : S.S'-Oxalyl-bis-[N-acetyl-cysteaminl: E = 6.9- 103, S.S'-Oxalyl-bis-[N-cpryloyl-cysteamin]: E = 5.85.10, (vgl. Abb. 1).
Die Intensitaten der Banden entsprechen, bezogen auf einen Chromophor, denen anderer Carbonshrethioester. Fiir S-Formyl-N-acetyl-cysteaminlz) berechnet sich
11) H. EGGERER, unverbffentlicht. 12) J. KOCH, unvertbffentlicht.
9.
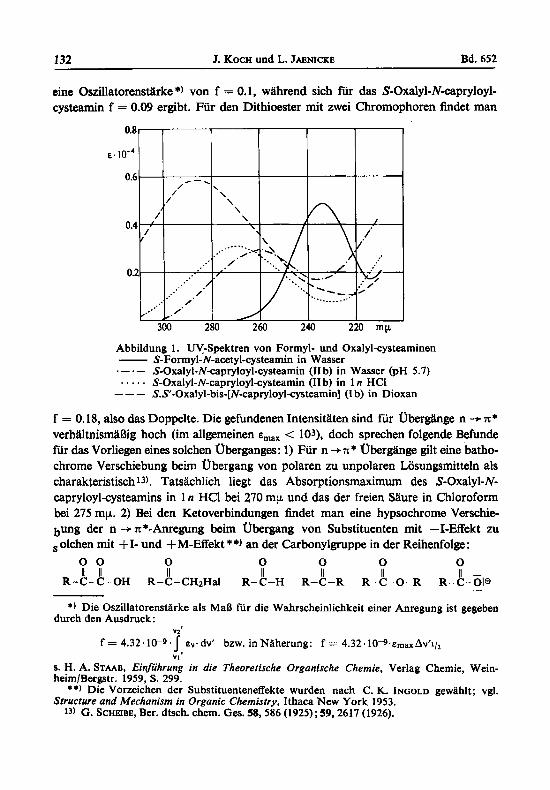
132 J. KOCH und L. JAEN~CKE Bd. 652
eine Oszillatorenstiirke *) von f = 0.1, wahrend sich fiir das S-Oxalyl-N-capryloyl- cysteamin f = 0.09 ergibt. Fur den Dithioester mit zwei Chromophoren findet man
Abbildung 1. UV4pektren von Formyl- und Oxalyl-cystearninen - S-Formyl-N-acetyl-cysteattin in Wasser .-.- S-Oxalyl-N-capryloyl-cysteamin (I1 b) in Wasser (PH 5.7) ..... S-Oxalyl-N-capryloyl-cysteamin (I1 b) in 1 n HCl --- S.S’-Oxalyl-bis-[N-capryloyl-cysteamin] (I b) in Dioxan
f = 0.18, also das Doppelte. Die gefundenen Intensitaten sind fur Ubergiinge n -+ x* verhiiltnismaBig hoch (im allgemeinen E,,, < lO3), doch sprechen folgende Befunde fur das Vorliegen eines solchen Uberganges: 1) Fur n +TC* Ubergange gilt eine batho- chrome Verschiebung beim ubergang von polaren zu unpolaren Losungsmitteln als charakteristisch 13). Tatsachlich liegt das Absorptionsmaximum des S-Oxalyl-N- capryloylcysteamins in 1 n HC1 bei 270 mp und das der freien Saure in Chloroform bei 275 mp. 2) Bei den Ketoverbindungen findet man eine hypsochrome Verschie- bU”g der n -+x*-Anregung beim Ubergang von Substituenten mit --I-Effekt zu olchen mit +I- und +M-Effekt **) an der Carbonylgruppe in der Reihenfolge:
R-C-C-OH R-C-CHZHal R-C-H R-C-R R-C-0-R R-k-Ole
0 0 0 0 0 0 0 II II II II I1 II
-
*) Die Oszillatorenstiirke als MaB fur die Wahrscheinlichkeit einer Anregung ist gegeben durch den Ausdruck :
v2’
v1’
S. H. A. S T U B , Einfiihrung in die Theoretische Organische Chemie, Verlag Chemie, Wein- heim/Bergstr. 1959, S. 299.
**) Die Vorzeichen der Substituenteneffekte wurden nach C. K. INGOLD gewahlt; vgl. Structure and Mechanism in Organic Chemistry, Ithaca New York 1953.
13) G. SCHEIBE, Ber. dtsch. chem. Ges. 58, 586 (1925); 59,2617 (1926).
f = 4.32810-9.5 ~y.dv’ bzw. inNaherung: f = 4.32.10-9.~maxA~’i/~

1962 Uber S-Oxalyl-thiole 133
0.8 E - 10-2
0.6
0.4 Abbildung 2 UV-Spektren von Estern und Aldehyden
--- Acetaldehyd in Athano1 1s) ...... Essigester in khanol14) 0.2 .---.
....
: . . . :. .?. - .’
14) E. A. FEHNEL und M. CARMACK. J. Amer. chem. SOC. 71,84 (1949). 1s) G. 0. BURR und E. S. MILLER, Chem. Reviews 29,419 (1941), und mar S. 421.

134 J. KOCH und L. JAENICKE Bd. 652
zende induktive Effekt der Alkylreste und die abschirmende negative Ladung der Carboxylatgruppe wegfallen. Mit dieser Tatsache stimmt auch die auflerordentliche Labilitat der Oxalsauremonothioester gegeniiber Alkalien iiberein. So wird in der ,,verzogerten" Nitroprussid-Reaktion16), die auf der Ammonolyse des Acylmercap- tans beruht, der Scheitelwert der Extinktion bei Raumtemperatur mit Oxalylthiolen bereits nach etwa 30 Sek. erreicht (Abb. 3). Einen Vergleich mit einigen anderen Thioestern gibt die Tabelle 2.
Tabelle 2. Ammonolysegeschwindigkeiten von S-Acyl-thiolen
Verbindung t [Sek.]*l
S-Oxalyl-N-capryloyl-cysteamin 30
S-Succinyl-N-acetyl-cysteamin 480
S-Propion yl-N-capryloyl-cy steamin 18018) 4) t = &it, in der die maximale Extinktion bei der ,,verzogerten" Nitroprussid-Reaktion erreicht wird.
S-Methylmalonyl-N-capryloyl-cy steamin 900 17)
S- Acetyl-N-acetyl-cysteamin 12018)
Die Hydrolysegeschwindigkeit bei verschiedenen pH-Werten 1aBt sich an der Ab- nahme der Extinktion bei 260mp als Funktion der Zeit verfolgen19). Bei dieser
Abbildung 3. ,,Verziigerte" Nitroprussid-Reaktion der 0xals;iuremono thioester
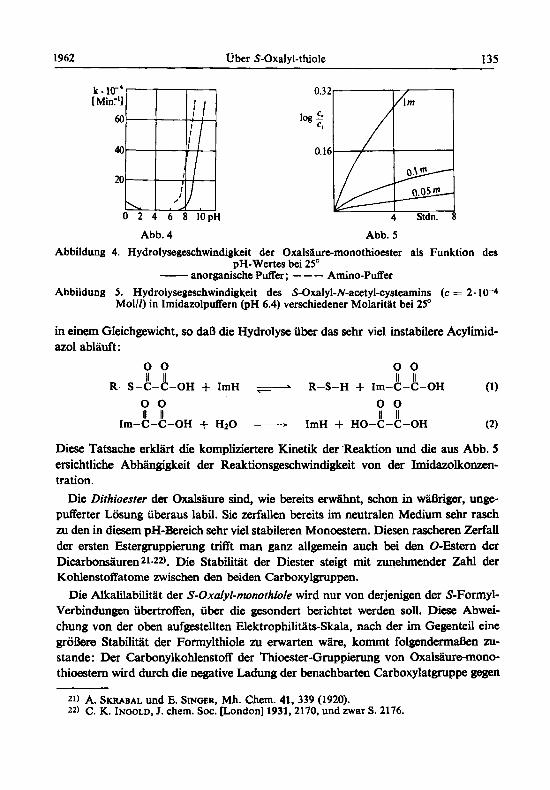
Wellenlange konnen die Absorptionen des bei der Hy- drolyse entstehenden Mercaptans und der Carbonsaure vernachlassigt werden. Abb. 4 zeigt, daB S-Oxalyl-thiole zwischen pH 3 und pH 6.5 in anorganischen Pufferge- mischen stabil sind. Bemerkenswert ist die Abhangigkeit nicht nur vom pH-Wert, sondern auch von der Art des Puffers. In Aminopuffern (Triathanolamin, Tris-hydro- xymethyl-amino-methan) findet man wesentlich hohere Zerfallskonstanten als in anderen Puffern gleicher Was- serstoffionenkonzentration. In Imidazolpuffern (ImH) zerfallen die Oxalylthiole selbst bei pH-Werten, bei denen sie in anderen Puffern vollig stabil sind. Dieser Zerfall ist
stark von der Pufferkonzentration abhangig und folgt nicht einer Funktion erster Ordnung (Abb. 5) , wie das fur die Hydrolyse in anderen Puffergemischen unter den gewiihlten, fur pseudomonomolekulare Reaktionen typischen Bedingungen der Fall ist. Es kann sich daher bei der Reaktion nicht mehr M eine einfache Hydrolyse des Oxalylthiols handeln. Nach STADTMAN 20) stehen die Thioester mit den Acylimidazolen
16) F. LYNEN, Liebigs Ann. Chem. 574,33 (1951). 17) P. OVERATH, Dissertation Univ. Mtinchen 1961. 18) H. EGGERER, Dissertation Univ. Miinchen 1957. 19) L. H. NODA, S. A. KUBY und H. A. LARDY, J. Amer. chem. SOC. 75,913 (1953). 20) E. R. STADTMAN, in W. D. MCELROY und B. GLASS, The Mechanism of Enzyme Action,
The Johns Hopkins Press, Baltimore 1954, S. 581.
1962 uber S-Oxalyl-thiole 135
k . lo-' 0.32 [ MinT'I
60 log:
40 0.16
20 0.05
0 2 4 6 8 10pH 4 Stdn. 8
Abb. 4 Abb. 5 Abbildung 4. Hydrolysegeschwindigkeit der Oxalsauremonothioester als Funktion des
pH-Wertes bei 25" - anorganische Puffer; - - - Amino-Puffer Abbildung 5. Hydrolysegeschwindigkeit des S-Oxalyl-N-acetyl-cystearnins (c = 2.10-4
MOW) in ImidazolpufFem (PH 6.4) verschiedener Molaritlt bei 25'
in einem Gleichgewicht, so daO die Hydrolyse iiber das sehr viel instabilere Acylimid- azol ablauft :
0 0 0 0 I1 I1 II II
R-S-C-C-OH + ImH R-S-H + Im-C-C-OH (1)
Irn-C-C-OH + HzO - -+ ImH + HO-C-C-OH (2)
Diese Tatsache erklart die kompliziertere Kinetik der .Reaktion und die aus Abb. 5 ersichtliche Abhiingigkeit der Reaktionsgeschwindigkeit von der Imidazolkonzen- tration.
Die Dirhioester der Oxalsaure sind, wie bereits erw&hnt, schon in waBnger, unge pufTerter Losung iiberaus labil. Sie zerfallen bereits im neutralen Medium sehr rasch N den in diesem pH-Bexeich sehr viel stabileren Monoestern. Diesen rascheren Zerfall der ersten Estergruppierung trSt man ganz allgemein auch bei den 0-Estern der Dicarbonsauren 21.22). Die Stabilitiit der Diester steigt mit mehmender Zahl der Kohlenstoffatome zwischen den beiden Carboxylgruppen.
Die Alkalilabilitat der S-Oxalyl-nwnothiole wird nur von derjenigen der S-Formyl- Verbindungen iibertroffen, iiber die gesondert berichtet werden SOU. Diese Abwei- chung von der oben aufgestellten Elektrophilitats-Skala, nach der im Gegenteil eine grokre Stabilitiit der Formylthiole zu erwarten ware, kommt folgendennakn zu- stande : Der Carbonylkohlenstoff der Thioester-Gruppierung von Oxalsiiuremono- thioestern wird durch die negative Ladung der benachbarten Carboxylatgruppe gegen
0 0 0 0 II I1 II II
21) A. SKRABAL und E. SINGER, Mh. Chem. 41, 339 (1920). 22) C. K. INGOLD, J. chem. SOC. [London] 1931,2170, und m a r S. 2176.

136 J. KOCH und L. JAENICKE Bd. 652
Hydroxylionen etwas abgeschirmt 23). Bei den Formylthiolen fehlt der Schutz durch den Feldeffekt eines Substituenten, und das OHe-Ion kann den stark positivierten Carbonylkohlenstoff leichter angreifen. Die Ursachen dieser Abweichung sind also kinetischer und nicht thermodynamischer Natur.
Der DEUTSCHEN FORSCHUNCSGEMEINSCHAFT, Bad Godesberg. sowie dem FONDS DER
CHEMISCHEN INDUSTRIE, Diisseldorf, und der Fa. C. H. BOEHRINGER SOHN, Ingelheim, die uns bei der Ausfilhrung der Arbeit durch Sach- und Personalmittel unterstiitzten, danken wir filr die stete groBziigige Hilfe.
B E S C H R E I B U N G D E R V E R S U C H E
N-Acetyl-cystearnin. - 100 ccrn Methanol werden auf -60' gekiihlt. Man leitet 30 Min. Schwefelwasserstof ein. Dann laDt man unter Riihren und bei kraftigem HzS-Strom 8.5 g Acetylariridin 24) zutropfen und das Gemisch auf Raumtemperatur kommen. AnscNieDend wird fraktioniert. Wasserhelle, schwer bewegliche Fliissigkeit, Sdp.o.4 1 12- 1 13". Ausbeute 11.3 g(95%).
S.S-Oxalyl-bis-[N-acetyl-cysteamin] (la). - Zu einer Lbsung von 24 g (0.2 Mol) N-Acetyl- cysteamin und 37 g (0.2 Mol) Tributylamin in 500 ccm trockenem cc14 IiiDt man unter Kiih- lung auf - 10" und kraftigem Riihren 12.7 g (0.1 Mol) Oxalylchlorid in'50 ccrn CCl4 zutropfen. Man laRt weitere 2 Stdn. bei Raumtemperatur stehen, saugt den ausgefallenen Dithioester ab und kristallisiert ihn aus trocknem Dioxan urn. WeiRe Nadeln, Schmp. 176'. Ausbeute
S-Oxalyl-N-acetyl-cysteamin (IIa). - Natriumsalz: 2.92 g (10 mMol) Diester l a werden in 500 ccm Aceton gelast und unter starkem Riihren 10 ccm 1 n NaOH zugegeben. Man lPDt 30 Min. bei Raumtemperatur stehen und zieht dann das Lasungsmittel i. Vak. (Umlaufver- dampfer) ab. Der Riickstand wird rnit lOccm kaltem absol. hhanol versetzt, die Lasung klar zentrifugiert und der uberstand in 200 ccrn kaltes Aceton gegossen. Der Niederschlag wird abzentrifugiert und im Zentrifugenglas im Exsikkator getrocknet. XuRerst hygroskopi- sches weiDes Kristallpulver. Ausbeute 1.6 g (75 %). Saure: Eine gekiihlte Lasung des Natriumsalzes in einer gerade ausreichenden Menge ge- sattigter (NH4)2S04-Lasung wird rnit 2n H2S04 angesiiuert und mehrmals rnit Essigester extrahiert. Nach dem Trocknen des Extrakts mit Natriumsulfat wird der Essigester abgezogen und der Ruckstand aus trockenem Dioxan umkristallisiert. Durchsichtige Stabchen, Schmp. 159- 160" (Zers.).
18 g (62%).
GH9N04S (191.8) Ber. C 37.69 H 4.74 N 7.29 Gef. C 37.96 H 4.80 N 7.23
S.S-Oxalyl-bis-[N-capry1oyl-cysteaminl (I b). - Analog I a aus N-Capryloyl-cysieamin 17)
und Oxalylchlorid. Aus trockenem Dioxan lange weiBe Nadeln, Schmp. 148- 149". Ausbeute 85% d. Th.
C ~ ~ H M N Z O ~ S Z (460.7) Ber. C 57.40 H 8.75 N 6.05 Gef. C 57.73 H 8.82 N 5.65 ~~
23) E. FISCHER, Ber. dtsch. chem. Ges. 31,3266 (1898). 24) H. BESTIAN, Liebigs Ann. Chem. 566,210 (1950), und zwar S. 218.

1962 Uber S-Oxatyl-thiole 137
S-Oxalyl-N-capryloyl-cysteamin (I1 b). - Natriumsalz: 2.3 g (5 mMol) Diester Ib werden in 750 ccm Aceton gelbst und bei Raumtemperatur unter starkem Riihren mit 5 ccm 1 n NaOH venetzt. Nach 30 Min. wird das Lbsungsmittel im Umlaufverdampfer abdestilliert. Man lost den Ruckstand in 10 ccm kaltem Wasser. filtriert vom ungelbsten N-Capryloyl-cystearnin ab und dunstet wieder ohne zu erwiirmen ein. Zum Ruckstand fugt man 20 ccm kaltes absol. Athanol, filtriert und gieDt in 500 ccm kaltes Aceton ein. Nach einstiindigem Stehenlassen im Kiihlschrank wird das ausgefallene Na-Salz abgesaugt. WeiDes Pulver, Ausbeute 0.73 g (50 %h
Saure: Eine Lbsung des Natriumsalzes in moglichst wenig Wasser wird in der Ktilte mit dem gleichen Volumen 2n HzSO4 versetzt und die ausgefallene SHure in Essigester aufgenommen. Man trocknet Uber NazS04, zieht das Lbsungmittel ab, nimmt nochmals in wenig Essigester auf und versetzt mit dem doppelten Volumen CCl4. Beim Stehen bei -20" fallen weiOe Kristalle aus. Aus Essigester/Petrolather erhalt man glanzende Blattchen, Schmp. 60".
Cyclohexylammoniumsalz: Die Essigesterlbsung der Saure wird mit einer Lbsung von Cyclo- hexylamin in Essigester versetzt, bis die Mischung gerade noch schwach sauer (auf pH-Papier getlipfelt) reagiert. Das ausgefallene Salz wird abgesaugt und aus trockenem Dioxan um- kristallisiert. WeiDe Nadeln, Schmp. 108".
C ~ ~ H ~ J N Z O ~ S (374.5) Ber. C 57.73 H 9.15 N 7.48 Gef. C 58.16 H 9.20 N 7.62
S-Oxalyl-glutathion. - 55.35 mg (50 pMol) Glutathion und 150 mg ( 5 0 0 pMol) Natrium- salz von IIb werden in 2 ccm 0.2m KHCO3 gelost und mit 1 ccm peroxidfreiem k h e r uber- schichtet. Man liiDt bei Raumtemperatur 1 Stde. stehen, perforiert sodann 1 Stde. mit Ather. k W t die wtiBrige Phase in Eis und gibt 2 ccm Amberlife ZR-I20 (Ha) zu. Unter Eisklihlung wird nochmals 3 Stdn. mit Ather perforiert. Der Austauscher wird abfiltriert, mit kaltem Wasser gewaschen und die saure L6sung nochmals ausgeathert. Die wdrige Phase wird gefriergetrocknet. Es hinterbleibt ein weiBes hygroskopisches Pulver. Die Verbindung wurde durch Bestimmung der aktiven Acylgruppe. durch Thioesterbestimmung und durch Amino- saurebestimmung nach ROSEN =) charakterisiert. Es ergab sich das molare VerhHltnis Acyl : SH : Glutathion = 1.04 : 1 .OO : 0.94. Die Verbindung ist elektrophoretisch einheitlich. Sie wandert schneller als Glutathion. Die Elektrophorese wurde in Pyridinacetat-Puffer pH 6.2 (9% Pyridin/l % Eisessig) bei einer Feldstarkevon etwa 40 V-cm-1 vorgenommen.
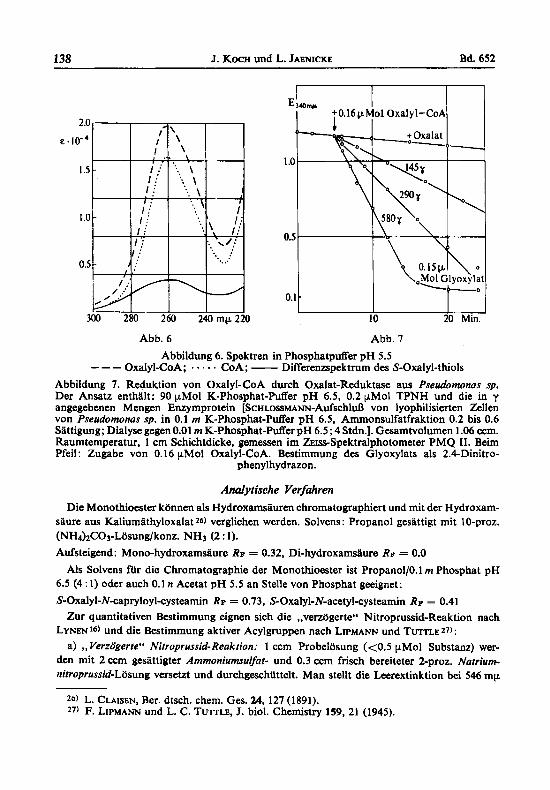
S-Oxalyl-CoA. - 10 mg CoA (C. F. BOEHRINCER & SOHNE) und 60 mg Natriumsalz von Ilb werden in der nir die Darstellung von S-Oxalyl-glutathion beschriebenen Weise umgesetzt. Das erhaltene Oxalyl-CoA verhielt sich bei der Chromatographie mit khanol/O.l m Acetat pH 5.5 (1 : 1) als Steigmittel einheitlich. Bei 2°C: CoA RF = 0.45, Oxalyl-CoA RF = 0.28. Aus den Differenzspektren vor und nach Hydrolyse der Thioesterverbindung ergab sich ein Verhiiltnis CoA : S-Acyl = 1 : 1.01 (Abb. 6).
Durch die in Pseudomonas sp. gefundene Oxalat-Reduktase wird Oxalyl-CoA mittels TPNH quantitativ zu Glyoxylat reduziert. Abb. 7 zeigt die Kinetik dieser Reaktion rnit einem Enzympraparat aus einem anfraktionierten Extrakt der Bakterien.
25) H. ROSEN, Arch. Biochem. Biophysics 67, 10 (1957).
J. KOCH und L. JAENICKE Bd. 652 138
10 20 Min.
Abb. 6 Abb. 7
Abbildung 6. Spektren in PhosphatpuEer pH 5.5 --- Oxalyl-CoA; . . . . . COA; - Differenzspektrum des S-Oxalyl-thiols Abbildung 7. Reduktion von Oxalyl-CoA durch Oxalat-Reduktase aus Pseudomonas sp. Der Ansatz enthalt: 90 pMol K-Phosphat-Puffer pH 6.5, 0.2 pMol TPNH und die in y angegebenen Mengen Enzymprotein [ScHLossmm-AufschluB von lyophilisierten Zellen von Pseudomonas sp. in 0.1 rn K-Phosphat-Puffer pH 6.5, Ammonsulfatfraktion 0.2 bis 0.6 Sattigung; Dialyse gegen 0.01 m K-Phosphat-puffer pH 6.5 ; 4 Stdn.]. Gesamtvolumen 1.06 ccm. Raumtemperatur, 1 cm Schichtdicke, gemessen im ZErss-Spektralphotometer PMQ 11. Beim Pfeil: Zugabe von 0.16 pMol Oxalyl-CoA. Bestimmung des Glyoxylats als 2.4-Dinitro-
phenylhydrazon . Analytische Verfahren
Die Monothioester kannen als Hydroxamsauren chromatographiert und mit der Hydroxam- saure aus Kaliumathyloxalat 26) verglichen werden. Solvens : Propanol gesattipt mit 10-proz. (NH4)2CO3-Lbsung/konz. NH3 (2 : 1).
Aufsteigend: Mono-hydroxamsaure RF = 0.32, Di-hydroxamsaure RF = 0.0
6.5 (4 : 1) oder auch 0.1 n Acetat pH 5.5 an Stelle von Phosphat geeignet: S-Oxalyl-N-capryloyl-cysteamin RP = 0.73, S-Oxalyl-N-acetyl-cysteamin RF = 0.41
Zur quantitativen Bestimmung eignen sich die ,,verzagerte" Nitroprussid-Reaktion nach L Y H E N ~ ~ ) und die Bestimmung aktiver Acylgruppen nach LIPMANN und TUTTLE 27):
a) ,, Verzogerte" Nitroprussid-Reaktion: 1 ccm Probelasung (<0.5 pMol Substanz) wer- den mit 2 ccm gesattigter Arnrnoniurnsulfat- und 0.3 ccrn frisch bereiteter 2-proz. Natrium- nitroprussid-Lasung versetzt und durchgeschiittelt. Man stellt die Leerextinktion bei 546 mv
Als Solvens fiir die Chromatographie der Monothioester ist Propanol/O. 1 rn Phosphat pH
26) L. CLAISEN, Ber. dtsch. chem. Ges. 24, 127 (1891). 27) F. LIPMANN und L. C. TUTTLE, J. biol. Chemistry 159, 21 (1945).

1962 G. OPITZ und W. MERZ 139
im Photometer EPPENDORP ein, iiberschichtet mit 0.3 ccrn konz. NH3, schuttelt durch und liest die maximale Extinktion ab. Diese wird nach etwa 30 Sek. erreicht. Bei einer Schicht- dicke von 1 cm entsprechen 0.1 pMol einem A L a X von 0.14.
b) Bestimmung der aktiven Acylgruppen: 1 ccrn Probelasung (<1 pMol) werden mit 0.6 ccrn einer Losung versetzt, die aus gleichen Teilen 4n Hydroxylamin-HCI und 3.511 NaOH be- steht. Nach 10 Min. werden 0.4 ccm FeCI3-Lasung zugegeben (10 g FeCl3.6 HzO in 90 g 5-proz. Trichloressigsaure-Losung und 20 ccrn 12n HCl). Falls es sich um das Oxalylderivat eines wasserunlaslichen Thiols handelt, wird dieses mit 1 ccrn peroxidfreiem Ather entfernt. In einer Ktivette mit 2 cm Lichtweg entspricht 1 pMol einem AEM mcI von 0.8.
Enamine, VIII * SALZE VON DIEN-AMINEN
von GUNTER OPITZ und WALTER MERZ
Aus dem Chemischen Institut der Universitat TUbingen Eingegangen am 4. November 1961
Die Salzbildung der aus sekundiiren Aminen und Crotonaldehyd, Z-Athyl- hexen-(Z)-al bzw. Isophoron gewonnenen Dienamine I mit Chlorwasserstoff wird IR- und UV-spektroskopisch sowie durch Umsetzung mit nucleophilen Partnern untersucht. Endprodukte sind die konjugierten En-immoniumsalze IIc. Sie entstehen, zumindest in einigen Fallen, nicht durch direkte C-CProtonierung, sondern durch Umlagerung der primiir gebildeten Dien-ammoniumsalze I1 a. Der Nachweis der 3 mOglichen Salzarten IIa, I l b und IIc gelingt, wenn man die Dienamin-hydrochloride zu verschiedenen Zeiten nach ihrer Darstellung
mit komplexen Hydriden oder metallorganischen Verbindungen umsetzt.
Enamine liefern mit starken Sauren bevorzugt Immoniumsalze. Beim Studiwn der Salzbildung in Abhangigkeit von der Zeit konnte in mehreren Fallen nachgewiesen werden, daB zunachst durch Proton-Aufnahme am Stickstoffatom a. P-ungesattigte Am- moniumsalze entstehen, die leicht in die isomeren Immoniumsalze iibergehen. In anderen Fallen lieD sich zwischen direkter und indirekter C-2-Protonierung nicht unterscheiden 1).
Bevor der Zweistufen-Mechanismus und die im allgemeinen rasche Umlagerung der En-ammoniumsalze in Immoniumsalze erkannt waren, untersuchten wir die Salzbildung von 1-Dialkylarnino-1.3-dienen (I). Aus derartigen Basen - hier der
*) VII. Mitteilung: G. OPITZ, Liebigs Ann. Chem. 650, 122 (1961). 1) G. OPITZ, A. GRIESINGER und H. W. SCKUBERT, in Vorbereitung.


















