
Überlebensfähigkeit von kryokonservierten Mäuseembryonen ... · Diese sind nach dem...
122
Aus dem Institut für Tierzucht und Haustiergenetik der Georg-August-Universität Göttingen Überlebensfähigkeit von kryokonservierten Mäuseembryonen nach Zwischenlagerung bei -18°C oder -79°C Masterarbeit im Fachbereich Fortpflanzungsbiologie und Biotechnik vorgelegt von Stephanie Buhtz Göttingen im Juli 2007
Transcript of Überlebensfähigkeit von kryokonservierten Mäuseembryonen ... · Diese sind nach dem...

Aus dem Institut für Tierzucht und Haustiergenetik
der Georg-August-Universität Göttingen
Überlebensfähigkeit von
kryokonservierten Mäuseembryonen
nach Zwischenlagerung bei
-18°C oder -79°C
Masterarbeit
im Fachbereich Fortpflanzungsbiologie und Biotechnik
vorgelegt von Stephanie Buhtz
Göttingen im Juli 2007

1. Prüfer: Prof. Dr. W. Holtz
2. Prüfer: Prof. Dr. M. Gauly
Tag des Kolloquiums: 26.07.2007

I
Inhaltsverzeichnis
Abbildungsverzeichnis .................................................................................. IV
Tabellenverzeichnis......................................................................................... V
Abkürzungen................................................................................................... VI
Verwendete Chemikalien ............................................................................. VIII
Verwendete Geräte und Verbrauchsmaterialen ............................................IX
1 Einleitung .......................................................................................................1
2 Literatur ..........................................................................................................3
2.1 Grundlagen der Kryobiologie ........................................................................ 3
2.2 Prozesse beim Gefrieren................................................................................. 4
2.3 Physikalische und chemische Abläufe beim Einfrieren von Zellen .... 6
2.4 Kryoprotektiva - Aufgabe und Wirkung ...................................................... 8
2.4.1 Penetrierende Kryoprotektiva ................................................................ 9
2.4.1.1 Glycerol ............................................................................................... 11
2.4.1.2 Dimethylsulfoxid (DMSO) ............................................................... 12
2.4.1.3 Ethylenglycol (EG)............................................................................ 14
2.4.2 Nicht penetrierende Kryoprotektiva.................................................... 16
2.5 Gefrierverfahren .............................................................................................. 19
2.5.1 Standardgefrierverfahren ...................................................................... 20
2.5.2 Langsames Kryokonservieren ............................................................. 22
2.5.3 Schnelles Kryokonservieren................................................................. 23
2.5.4 Ultraschnelles Kryokonservieren ........................................................ 24
2.5.5 Vitrifikation ................................................................................................ 25
2.6 Auftauen ........................................................................................................ 27
2.7 Ausverdünnungsmethoden.......................................................................... 29
2.7.1 Schrittweises Ausverdünnen und Ausverdünnen mit nicht
penetrierenden Kryoprotektiva .......................................................... 29
2.7.2 One-Step-Ausverdünnung..................................................................... 30
2.7.3 Direkte Ausverdünnung von Ethylenglycol...................................... 32
2.8 Kultivierung von Embryonen ....................................................................... 33

II
2.9 Beurteilungsmöglichkeiten für Embryonen ............................................. 34
2.9.1 Morphologische Beurteilung ................................................................ 34
2.9.2 Beurteilung nach Fluoreszenzanfärbung mittels FDA................... 35
2.9.3 Beurteilung nach Fluoreszenzanfärbung durch
Hoechst A 33342 .................................................................................... 36
3 Material und Methoden................................................................................37
3.1 Tiermaterial ....................................................................................................... 37
3.2 Haltungsbedingungen.................................................................................... 37
3.3 Medien ................................................................................................................ 38
3.3.1 Spülmedium .............................................................................................. 38
3.3.2 Kulturmedium ........................................................................................... 41
3.3.3 Kryoprotektivum ...................................................................................... 42
3.3.4 Auftaumedium .......................................................................................... 43
3.3.5 Überschichtungsöl für Kultivierung- und Waschdrops ................ 43
3.4 Behandlung der Embryonenspender......................................................... 44
3.5 Gewinnung der Embryonen.......................................................................... 46
3.6 Beurteilung der gewonnenen Embryonen................................................ 48
3.7 Einfrieren der Embryonen............................................................................. 49
3.7.1 Standartgefrierverfahren........................................................................ 50
3.7.1.1 Haake ................................................................................................... 50
3.7.1.2 Consartic............................................................................................. 51
3.8 Zwischenlagerung der eingefrorenen Embryonen ................................ 53
3.9 Auftauen der Embryonen .............................................................................. 54
3.10 In vitro Kultivierung der Embryonen ....................................................... 55
3.11 Beurteilung von Embryonen ...................................................................... 56
3.11.1 Morphologische Beurteilung .............................................................. 56
3.11.2 Kombinierte FDA-Hoechst-Färbung zur
Zytoplasmaaktivitätsbestimmung und Kernzählung ................... 60
3.12 Geräte und Gerätekontrolle........................................................................ 62
3.13 Photographien ............................................................................................... 64
3.14 Statistische Auswertung ............................................................................. 64

III
4 Ergebnisse ...................................................................................................65
4.1 Superovulation................................................................................................. 65
4.2 Embryonengewinnung, -stadium und -qualität....................................... 65
4.3 Zwischenlagerung........................................................................................... 66
4.4 In vitro Kultivierung ........................................................................................ 67
4.5 Vitalfärbung mit FDA und Hoechst 33341 ................................................ 69
4.6 Einfluss der Abstammung der Mutterherkunft auf die
Embryonenausbeute ............................................................................. 69
4.7 Einfluss der Abstammung der Mutter auf das Entwicklungsstadium
nach 48 Stunden in vitro Kultivierung.............................................. 70
5 Diskussion....................................................................................................71
5.1 Superovulation................................................................................................. 72
5.2 Überleben und Weiterentwicklung unter in vitro Bedingungen ......... 75
5.3 Zwischenlagerungseinfluss auf die Weiterentwicklung der
kryokonservierten Embryonen........................................................... 76
6 Zusammenfassung ......................................................................................79
7 Literaturverzeichnis.....................................................................................81
Danksagung ..................................................................................................109
Eidesstattliche Erklärung.............................................................................110

IV
Abbildungsverzeichnis
Abb. 1: Abhängigkeit der Überlebensrate von der Kühlrate..............................23
Abb. 2: Werktisch mit integriertem Wärmebereich (Varolab, Giessen) und
CO2 Kleininkubator C42 (Labotect) ......................................................43
Abb. 3: Ip-Injektion von eCG bzw. hCG ............................................................44
Abb. 4: Vaginal-Plaque: 1) positiv und 2) negativ am Morgen nach erfolgter
Bedeckung............................................................................................45
Abb. 5: Zervikale Dislokation ............................................................................47
Abb. 6: Eröffnete Bauchhöhle...........................................................................47
Abb. 7: Einfriersystem Firma HAAKE, Berlin ....................................................49
Abb. 8: Einfriersystem Firma CONSARTIC, Schöllkrippen ...............................49
Abb. 9: Befüllungsschema Kunststoffpaillette...................................................51
Abb. 10: Kunststoffpaillette mit Entwicklungsstadium, Spezies, Spültag und
Stückzahl............................................................................................51
Abb. 11: Zwischenlagerung bei -79°C (li.) und -18°C (re.)................................54
Abb. 12: Waschschale mit fünf Medium-Waschdrops unter Öl.........................55
Abb. 13: Kultivierungsschale mit 10 Kultivierungs-Mediumdrops unter Öl........56
Abb. 14: Inkubator C60, Labotect, Göttingen....................................................56
Abb. 15: Embryo a) "Sehr gut", b) "Gut", c) "Mäßig", d) degeneriert,
e) retadiert..........................................................................................59
Abb. 16: a) Schlüpfende Blastovyste, b) Geschlüpfte Blastocyste....................60
Abb. 17: Aktives (a) bzw. inaktives (b) Zytoplasma - Feststellung mittels FDA 61
Abb. 18: Lebende und tote Kerne - Sichtbarmachung mittels
Hoechst H 33342................................................................................61
Abb. 19: Arbeitsplatz mit Kleininkubator C42 (Easy Bench, Varolab, Giessen;
C42, Labotect, Göttingen) ..................................................................62
Abb. 20: Kleininkubator C42 (Labotect, Göttingen)...........................................62
Abb. 21: Messgerät In Control 1051 (Labotect, Göttingen) (Quelle: Labotect,
Göttingen) ..........................................................................................63
Abb. 22: Verwendeter Zeiss Axio Observer A1 (Mitte)
(Quelle: Zeiss, Göttingen) ..................................................................64

V
Tabellenverzeichnis
Tab. 1: Zusammensetzung Mäusealleinfutter...................................................38
Tab. 2: Zusammensetzung der verwendeten Stammlösungen.........................39
Tab. 3: Zusammensetzung des Spülmediums M2............................................40
Tab. 4: Zusammensetzung des Kulturmediums MediCult Universal
IVF Medium ..........................................................................................42
Tab. 5: Behandlungsschema zur Gewinnung von Mäuseembryonen...............46
Tab. 6: Schema Gefrierprogramm ....................................................................52
Tab. 7: Entwicklungsstadium und Verteilung gewonnener Eizellen und
Embryonen ...........................................................................................66
Tab. 8: Ergebnisse der in vitro Kultivierung von bei -18°C bzw. -79°C
zwischengelagerten kryokonservierten Blastocysten..........................68
Tab. 9: Ergebnisse der in vitro Kultivierung von bei -18°C bzw. -79°C
zwischengelagerten kryokonservierten Morulae ..................................68
Tab. 10: Reaktion des Zytoplasmas auf die FDA-Färbung ...............................69
Tab. 11: Embryonengewinnung aus MPI-Mäusen............................................70
Tab. 12: Embryonengewinnung aus institutseigenen Mäusen .......................70
Tab. 13: Mutterherkunft Institutseigen ............................................................70
Tab. 14: Mutterherkunft MPI .............................................................................70

VI
Abkürzungen
Bl. Blastocyste
BSA bovines Serumalbumin
bzw. beziehungsweise
ca. circa
Deg. Degeneriert
DMSO Dimethylsulfoxid
DNA Desoxyribonucleic acid
EBSS Earle´s Buffered Saltsolution
eCG equine Chorionic Gonadotropin
EG Ethylenglycol
ER Entwicklungsrate
et al. et altera
Exp. Expandiert
Exp. - Expandiert, mäßig
Exp. ! Expandiert, sehr gut
FDA Fluorescein-3`6`-diacetat
g Gramm
Geschl. Geschlüpft
hCG human Chorionic Gonadotropin
HSA humanes Serum Albumin
IE Internationale Einheiten
ip intraperitoneal
kg Kilogramm
LF Luftfeuchte
LN2 flüssiger Stickstoff (-196°C)
L ! sehr aktiv
L aktiv
L - schwach aktiv
M Mol
mg Milligramm
Mg Molekulargewicht
ml Milliliter

VII
mm Millimeter
Mo Morula
mOsmol Auf Osmolalität (Osmol/l) o. Osmolarität (Osmol/kg) bezogene Einheit
MPI Max Planck Institut
PBS Phosphate Buffered Saline
PG Propylenglycol
PROH Propandiol
PVA Polyvinylalkohol
PVP Polyvinylpyrrolidon
rel. relativ
RT Raumtemperatur
Schl. Schlüpfend
SR Schlupfrate
T tot
TR Trächtigkeitsraten
ÜR Überlebensrate
UV Ultra violett
VP Vaginal-Plaque
VP+ Vaginal-Plaque positiv
VP- Vaginal-Plaque negativ
z.B. zum Beispiel
µl Mikroliter

VIII
Verwendete Chemikalien
Aceton MERCK – Schuchardt, Hohenbrunn
Alkohol SIGMA-ALDRICH, Steinheim
eCG Intergonan; Intervet, Unterschleißheim
Ethylenglycol SIGMA-ALDRICH, Steinheim
FDA FDA-Färbung (3,6-Diacetoxyfluoran); SIGMA-ALDRICH,
Steinheim
Fermacidal® D IC Products SA, CH-Minusio; Vertrieb Labotect, Göttingen
hCG Ekluton; Intervet, Unterschleißheim
HEPES HEPES (4-(2-Hydroxyethyl)-1-piperazinthansulfonsäure
Hoechst A 33342 B2261-25MG; bisbenzimid; SIGMA-ALDRICH, Steinheim
Kulturmedium Universal IVF Medium mit Phenolrot, 60 ml; MediCult a/s,
DK-Jyllinge
M2 eigene Herstellung
Mineralöl Mineral oil; SIGMA-ALDRICH, Steinheim
Reinstwasser Ampuwa®, Fresenius, Bad Homburg
Saccharose Sucrose, S9378; SIGMA-ALDRICH, Steinheim
Stickstoff flüssiger Stickstoff (LN2)
Vaselin Weißes Vaselin Lichtenstein DAB; Lichtenstein Pharma-
zeutica GmbH & Co., Mülheim-Kärlich

IX
Verwendete Geräte und Verbrauchsmaterialen
Brutschrank C60 Labotect, Göttingen
Brutschrank CO2 Kleininkubator C42 Labotect, Göttingen
Center-Well Schale (60×15mm) Falcon, Heidelberg
Digital-pH-Meter pH 525 WTW, Weilheim i.OB
Einfriersystem Consartic BV 65/55 Consartic, Schöllkrippen
Einfriersystem Haake Kryosta, Typ F-3Q mit
Programmgeber Typ PG 20
Haake, Berlin
Einmalfilter Filtropur S (0,2 µm) Sarstedt, Nümbrecht
Eppendorfgefäß (1 ml) Eppendorf, Hamburg
Falcon-Röhrchen (50 ml) Sarstedt, Nümbrecht
Feinwaage 2007 MP6 Sartorius, Göttingen
Gefrierpailletten 0,25 ml Kunststoffpailletten Minitüb, Landshut
Kanüle (0,3×1,0 cm, 2R2) Unimed, CH-Lausanne
Kleenex KIMTECH Science, Kimberley-
Clark ® Professional
Luer-Einwegspritze (1 ml/2 ml) B-D Plastipak®, Becton Dickin-
son, Irland
Magnetrührer Heidolph, Schwabach
Mäusealleinfutter ssniff Spezialdiäten GmbH,
Soest
Maus-Polycarbonatkäfig (Typ II: 17×23×14
cm /Typ III: 23×39×15 cm)
Ebeco, Castrop-Rauxel
Messelektrode WTW pH-Elektrode SenTix
60
WTW, Weilheim i. OB
Messgerät In Control 1050 Labotect, Göttingen
Mikroskop Axio Observer A1 ZeissMicroImaging GmbH,
Göttingen
Mikroskop Axioskop (mit Filtersatz 02 u. 09) Zeiss, Göttingen
Mikroskop Stereomikroskop SZ 11 Olympus, Hamburg

X
Mikroskop Stereomikroskop M8 Wild, Heerebrugg, Wetzlar
Parafilm American National Can TM,
Greenwich
Petrischale (35×10mm) Greiner, Frickenhausen
Petrischale (85×15mm) Greiner, Frickenhausen
Software Octax EyeWare™ Imaging Soft-
ware
Octax, Herborn-MTG Altendorf
Sterilisator U 50 Memmert, Schwabach
Trockenschrank BE 40 Memmert, Schwabach
Versiegelungskitt Hämatokrit Brandt, Wertheim
Wärmeplatte HT 200 Minitüb, Landshut
Wärmeplatte MEDAX SP14 MEDAX Nagel GmbH, Kiel
Wärmeschrank Typ 85 Melag Brutschrank Incubat,
Berlin
Mikrobiologische Sicherheitswerkbank Typ
MRF
Steag Laminarflow-Prozeß-
technik, Pfullingen
Werktisch Easy Bench Varolab, Giessen

1
1 Einleitung
Die Kryokonservierung von Embryonen wird bei einigen Säugern bereits prakti-
ziert und hat schon seit langem in der praktischen Rinderzucht Einzug gehalten.
Eine Voraussetzung für eine konventionelle Anwendung der Kryokonservierung
ist, dass die Zellen den Einfrierprozess ohne Folgeschäden überstehen. Nur
dann kann die Tiefgefrierkonservierung innerhalb der Technik weitere Bedeu-
tung erlangen.
Erste erfolgreiche Kryokonservierungsversuche konnten 1972 von WHITTING-
HAM, LEIBO und MAZUR für die Maus beschrieben werden. Die Maus wird
häufig, wie auch im Falle der Kryotechnik, als Modell verwendet, da die Gewin-
nung von Embryonen größerer landwirtschaftlicher Nutztiere einen wesentlich
größeren Kosten- und Apparateaufwand verursacht.
Unter Berücksichtigung einiger spezies-spezifischer Modifikationen, können die
bei der Maus gewonnenen Erkenntnisse auf die größeren landwirtschaftlichen
Nutztiere übertragen werden.
Die Verwendung flüssigen Stickstoffs ist bisher eine übliche Methode, um tief-
gekühlte Zellen zu lagern oder auch zu transportieren. Da aber der Transport
von Behältern mit flüssigem Stickstoff (LN2) den Vorschriften und gesetzlichen
Gegebenheiten für Gefahrguttransporte unterliegt, wäre es speziell für diesen
Bereich eine Vereinfachung, könnten in LN2 konservierte Zellen auch bei weni-
ger niedrigen Temperaturen gelagert werden, ohne dass es dabei zu einer Ver-
ringerung der Überlebensraten käme. Der überregionale Transport von Game-
ten und Embryonen könnte somit sehr vereinfacht werden, ohne dass die Kos-
ten für diese Transporte Ausmaße annehmen, die die Ökonomie eines Embryo-
transfers in Frage stellen.
Ziel der vorliegenden Arbeit war es, den Einfluss einer 1-stündigen Zwischenla-
gerung bei unterschiedlichen Temperaturen deutlich über -196°C (flüssiger
Stickstoff), in Bezug auf die in vitro Überlebens- und Weiterentwicklungsrate zu
ermitteln.
Versuche zu diesem Thema wurden zwar bereits 1973 durch MAURER &
BANK und 1977 durch MAURER et al. durchgeführt. Laut Literaturrecherche
wurde dieses Thema aber erst 2002 wieder durch MATAR aufgegriffen. Im Un-

2
terschied zu der Arbeit von MATAR, wurde in dieser Arbeit sowohl auf ein
Refreezing der zwischengelagerten Embryonen als auch auf einen Transfer der
aufgetauten Embryonen auf Empfängertiere verzichtet, da zunächst geklärt
werden sollte, ob die Embryonen generell in der Lage sind eine Zwischenlage-
rung bei Temperaturen von -18°C oder -79°C zu überstehen.

3
2 Literatur
2.1 Grundlagen der Kryobiologie
Der Begriff Kryo stammt ursprünglich aus dem Griechischen. Dort bedeutet kry-
os soviel wie Kälte, Eis.
Heutzutage versteht man unter Kryokonservierung die Lagerung von Zellen und
Geweben in einem Temperaturbereich von unter -130°C mit dem Ziel, eine
möglichst vollständige Erhaltung der ursprünglichen Ausgangseigenschaften
des konservierten biologischen Materials zu erreichen. Ein besonderes Augen-
merk liegt hier auf der Erhaltung der Lebensfähigkeit des Materials (SCHEIWE
& RAU, 1981).
Die Herunterkühlung auf sehr tiefe Temperaturen geschieht in der Regel durch
die Verwendung von flüssigem Stickstoff (LN2). Der Einsatz flüssigen Stickstoffs
wurde durch die Entdeckung der Verflüssigung von Gasen ermöglicht.
Das Bemerkenswerte an dem Prozeß der Kryokonservierung ist, dass die Vitali-
tät der Zellen erhalten bleibt, obwohl das ganze biologische System der Zellen
in einen Festkörperzustand übergeht. Dadurch kommt der Zellstoffwechsel voll-
ständig zum Stillstand und die biochemischen Abläufe werden unterbrochen.
Bisher ist die Kryokonservierung die einzige Möglichkeit, tierische, pflanzliche
und menschliche Zellen und Zellverbände über eine beinahe unbegrenzte Zeit-
dauer in lebensfähiger Form einzulagern und dann zu gegebenem Zeitpunkt
wieder zu reanimieren. Von ersten erfolgreichen Versuchen zur Kryokonservie-
rung von Embryonen berichteten WHITTINGHAM et al. (1972) sowie WILMUT
(1972).
Die einzige bisher bekannte Gefahr für korrekt kryokonservierte Zellen sind
Strahlungsschäden an der DNS durch kosmische Strahlung. Da dieser Schädi-
gungsprozess aber erst nach vielen tausend Jahren eine Schädigung bewirken
würde, ist er als nicht unbedingt relevant anzusehen (ASHWOOD-SMITH &
FRIEDMANN, 1979; MAZUR, 1980; LYON et al., 1981; GLENISTER et al.,
1984).
Für eine längere Lagerungsdauer werden die zu lagernden Zellen in Stahlbe-
hältern aufbewahrt. Diese sind nach dem Dewar-Prinzip konstruiert, bei dem
zwei Behälter unter einem so gering wie möglich gehaltenem Abstand ineinan-
der montiert und durch einen Vakuumbereich voneinander isoliert werden. Sol-

4
len die Stahlbehälter zur Lagerung verwendet werden, muss der innere Behäl-
ter mindestens zu einem Viertel mit flüssigem Stickstoff gefüllt sein. Die Proben
werden dann in speziellen Metall- oder Plastikeinsätzen in der kalten Gasphase
gehalten bzw., wenn sie festverschlossen sind, in die flüssige Kühlphase einge-
taucht.
Die auf diese Weise konservierten Proben können Jahre, Jahrzehnte und unter
Umständen auch Jahrhunderte lang ohne Einschränkung ihrer Vitalität überste-
hen.
2.2 Prozesse beim Gefrieren
Das Vermögen eines Körpers zur Abkühlung wird durch physikalische Parame-
ter wie zum Beispiel die Fähigkeit zur Wärmeleitung, die Geometrie und auch
die Körpergröße beeinflusst. Dadurch ergibt sich eine zellspezifische Definition
der Abkühlrate.
Eine Kryokonservierung lebender Zellen ohne Kryoprotektiva und ohne, dass
die Zellen Schaden nehmen, ist bisher noch nicht möglich.
Durch die Fähigkeit der Zellen zur Wärmeleitung und die Diffusionsmöglichkeit
der Kryoprotektiva in die Zellen ergeben sich ganz spezielle Temperaturverläufe
während des Einfrierens.
Während des dynamischen Gefrierprozesses kommt es zwischen dem Intra-
und Extrazellulärraum der Zellen zu einer Reihe von biophysikalischen Reaktio-
nen. Diese Abläufe werden hauptsächlich von den Membraneigenschaften der
zu konservierenden Zellen und von den Veränderungen der Temperatur in Ab-
hängigkeit von der Zeit beeinflusst (SCHWARTZ & DILLER, 1983).
Von großer Bedeutung für die Zellen sind die während der Tiefgefrierung ablau-
fenden Konzentrationsveränderungen der umgebenden Medien und die me-
chanischen Änderungen in den Zellstrukturen, die durch sich bildende Eiskris-
talle entstehen (HOBBS, 1974). Eine bedeutende Rolle spielt dabei die Größe
der sich bildenden Eiskristalle. Diese wird vor allem durch die Abkühlrate beein-
flusst (MAZUR et al., 1974; MAZUR, 1977). Werden flüssige Lösungen mit ei-
ner schnellen Abkühlrate eingefroren, kommt es infolge einer unvollständigen
Wärmeabfuhr zu einer spontanen Bildung kleiner Eiskristalle. Bei einer langsa-

5
men Abkühlung hingegen kann die Wärme vollständig abgeführt werden und es
entstehen größere Kristalle (MAZUR et al., 1974; DILLER, 1975; FARRANT et
al., 1977).
Trotz einer abgeschlossenen Kristallisation ist es aber biophysikalisch dennoch
möglich, dass kleine Eiskristalle durch Zusammenlagerungen zu größeren Kris-
tallen verschmelzen, ihr Energiepotential erhöhen und durch sogenannte ther-
mische Stöße ihre Position in der Zelle ändern (MAZUR, 1966; LUYET & RA-
PATZ, 1970; MAZUR, 1977; RALL & POLGE, 1984; MEYBERG, 2007).
Ganz allgemein lässt sich der Prozess des Einfrierens in fünf verschiedene Ab-
schnitte unterteilen:
1. Equilibrierungsphase: Die Embryonen werden in der Gefrierschutzlösung
equilibriert.
2. Abkühlungsphase: Sie beginnt bei der Körpertemperatur der Zellen und
endet mit Erreichen der gewählten Lagerungstemperatur. Es müssen
dabei definierte Einfrierraten (°C pro Minute) des jeweiligen Gefriergutes
eingehalten werden.
3. Lagerungsphase: Hier werden die Zellen bei mindestens -130°C, in der
Regel aber bei -196°C aufbewahrt.
4. Auftau- und Wiedererwärmungsphase: In diese Phase werden die Zellen
von der Lagerungstemperatur wieder auf Körpertemperatur erwärmt. Die
dabei gewählte Auftaurate ist von der zuvor verwendeten Einfrierrate ab-
hängig.
5. Ausverdünnungsphase: In dieser letzten Phase müssen die verwendeten
Kryoprotektiva wieder möglichst vollständig und schnell von den Zellen
entfernt werden, da nur dann eine ungestörte Weiterentwicklung im An-
schluss an das Auftauen möglich ist (WILLADSEN et al., 1978; RALL et
al., 1984).

6
2.3 Physikalische und chemische Abläufe beim Einfrieren von
Zellen
Ganz ähnlich der fünf verschiedenen Prozessphasen beim Einfrieren, können
auch die intra- und extrazellulär ablaufenden Vorgänge während des Einfrierens
in sechs Schritte eingeteilt werden:
1. Im Bereich zwischen 37°C und 20°C befindet sich die Zelle in einem
physiologischen Bereich und die intra- und extrazellulären Prozesse lau-
fen aufeinander abgestimmt ab.
2. In der Temperaturzone von 20° bis -2°C ist der Stoffwechsel der Zelle
bereits stark eingeschränkt. Alle Vorgänge in den Zellen laufen deutlich
langsamer ab oder werden sogar gänzlich gestoppt. Aus dem Grunde ist
dieser Temperaturbereich von so großer Bedeutung in der Vorbereitung
der Zelle zum eigentlichen Einfrieren.
3. Die Temperaturen sinken von -2° auf -15°C ab. Die einsetzende Eisbil-
dung beginnt meistens an Kristallisationskeimen in der extrazellulären
Lösung. Die Temperatur, bei der die Kristallisation einsetzt, wird durch
die chemische Zusammensetzung der verwendeten Kryoprotektivumslö-
sung festgelegt (MAZUR, 1977; LEIBO & MAZUR, 1978). Als Folge des
osmotischen Effektes wird Wasser aus der Zelle herausgezogen. Da-
durch dehydriert die Zelle und beginnt zu schrumpfen. Dieses Schrump-
fen ist möglich, da ca. 90% des Wassers der Blastomeren in ungebun-
dener Form vorliegt (MAZUR, 1963; MERYMAN et al., 1984). Der Emb-
ryo ist in der Lage, bis auf etwa 40% seines ursprünglichen isotonischen
Ausgangsvolumens zu schrumpfen, ohne dass er dabei Schäden erleidet
(SCHNEIDER & MAZUR, 1984). Durch Auswahl möglichst optimaler Ab-
kühlraten kann den Prozess der intrazellulären Eiskristallbildung opti-
miert werden. Die Raten sollen langsam genug sein, um eine ausrei-
chenden Dehydrierung zu ermöglichen und schnell genug um die Zellen
nicht unnötig den Lösungseffekten der Kryoprotektiva auszusetzen.
4. Es erfolgt ein weiteres Absinken der Temperaturen im Bereich von -15°
auf -25°C. In diesem Temperaturbereich setzt auch die intrazelluläre
Eisbildung ein. Dabei kommt es zu einer Entmischung und Dislokalisati-
on der Bestandteile des Zytoplasmas. Durch Zugabe von Kryoprotektiva

7
kann die Wasserstruktur dahingehend beeinflusst werden, dass statt ei-
niger großer, viele kleine Eiskristalle gebildet werden, die etwas weniger
schädlich für die Zelle sind. Die zunehmende Eiskristallbildung führt zu
einer Konzentrationserhöhung gelöster Stoffe in der restlichen Flüssig-
keit. Das hat zur Folge, dass der Gefrierpunkt der verbleibenden Flüssig-
keit kontinuierlich sinkt (SCHIEWE et al., 1987).
5. Die Temperatur bewegt sich von -25° auf -130°C. Dieser Bereich ist
gekennzeichnet durch ein migratorisches Wachstum der großen
Eiskristalle auf Kosten der kleineren. Der Vorgang der Umkristallisation
läuft zwar mit den stetig geringer werdenden Temperaturen immer
langsamer ab, trotzdem hat der Prozess immense Auswirkungen auf die
Zelle. Die Beweglichkeit kleinerer Eiskristalle und die damit verbundene
Dynamik innerhalb der Zelle führt zu einer Erhöhung des
Energiepotentials der Kristalle (Thermische Stösse) und verursacht
demzufolge, dass Wassermoleküle immer noch in der Lage sind ihre
Positionen wechseln. Das hat wiederum zur Folge, dass bereits vorhan-
dene, größere Eisbezirke weiter wachsen und so zu einer Schädigung
der Zellen führen können. Dies ist einer der Gründe warum man
kryokonservierte Proben auch nicht über eine längere Zeit bei -80°C
aufbewahren und diesen Temperaturbereich möglichst schnell
überwinden sollte. 6. In diesem letzten Schritt kommen bei Temperaturwerten von unter -
130°C alle Vorgänge fast vollständig zum Stillstand. Das ist der Grund,
weshalb heute Langzeiteinlagerungen von lebenden Proben bei mindes-
tens -130°C unter Verwendung von LN2 als Kühlmittel üblich sind.
Während des Auftauvorganges laufen all diese Prozesse abermals ab, aller-
dings in umgekehrter Weise. Für die Zellen bedeutet dieser Umstand eine er-
neute enorme Belastung und zusätzlichen Stress.
Bis heute ist es, trotz der großen Fortschritte auf dem Sektor der Kryofor-
schung, noch immer nicht möglich, jede Art von Zellen mittels Kryokonservie-
rung haltbar zu machen.

8
2.4 Kryoprotektiva - Aufgabe und Wirkung
Um ein Überleben der eingefrorenen Zellen zu erreichen, ist das Vorhanden-
sein von Gefrierschutzmitteln (Kryoprotektiva) von entscheidender Bedeutung.
Die Gefrierschutzmittel werden in penetrierende, in die Zelle hineingelangende,
und nicht penetrierende, im extrazellulären Raum verbleibende (DOEBBLER,
1966; MERYMAN, 1971; McGANN, 1978; LEIBO, 1981) unterteilt.
Trotz intensiver Forschung besteht noch keine ganz gesicherte, genaue Infor-
mation über die Wirkungsweise der Kryoprotektiva (FAHY, 1986) Es sind aller-
dings einige Schutzmechanismen und -wirkungen in der Diskussion. Als relativ
wahrscheinlich sind die im Folgenden kurz genannten Wirkungen und Mecha-
nismen anzunehmen.
Durch den Einsatz von Kryoprotektiva wird eine Verringerung der Temperatur
erreicht, bei der die intrazelluläre Eisbildung einsetzt. Außerdem scheinen sie
die Zellmembranen zu stabilisieren, indem sie Schäden durch intrazelluläre
Kristallbildung sowie Lösungseffekte vermindern (NIEMANN, 1983).
SMITH et al. (1951) vermuteten, dass eine Zugabe von Gefrierschutzmitteln
dazu führt, dass vermehrt kleine und weniger große, schädigende Eiskristalle
gebildet werden. Durch die Wasserbindungsfähigkeit der Kryoprotektiva wird
analog der osmolytischen Gegebenheiten eine Verdünnung der Elektrolytkon-
zentration in der extrazellulären Flüssigkeit erreicht (MAZUR, 1970; FARRANT
& MORRIS, 1973), die eine verlangsamte Dehydrierung der Zellen zur Folge
hat. Das bedeutet, dass Zellschäden verursachende Vorgänge langsamer ab-
laufen und extra- und intrazelluläre Ionenkonzentrationen erst ab deutlich tiefe-
ren Temperaturen, und damit zu einem für die Zellen weniger kritischen Zeit-
punkt, auf schädigende Werte ansteigen (LOVELOCK, 1954; LEIBO, 1977;
MERYMAN et al., 1977; LEIBO & MAZUR, 1978; LEHN-JENSEN, 1981;
FRIEDLER et al., 1988). WHITTINGHAM konstatierte 1980, dass Kryoprotekti-
va zwar die Bildung intrazellulären Eises in den Zellen nicht vollständig verhin-
dern, wohl aber schädigende Effekte, verursacht durch zu hohe Ionenkon-
zentrationen, mildern können.
Im Vergleich zu Medien, die keine Kryoprotektiva enthalten, bewirken die penet-
rierenden und nicht penetrierenden Inhaltsstoffe der Kryoprotektiva während

9
der Konservierungsabläufe, dass es in Lösungen mit Gefrierschutzmittelzugabe
zu weniger Zellmembranveränderungen kommt (FARRANT & MORRIS, 1973).
Für die verwendeten Kryoprotektiva ist es elementar, dass sie weder während
der Abkühlungs- noch während der Auftauphase toxische Wirkungen auf die
Zellen ausüben (FARRANT, 1965). FRIEDLER et al. (1988) plädierten trotzdem
dafür, die Kryoprotektiva im Anschluss an das Auftauen schnellstmöglich aus
den Zellen zu entfernen, da sie bei zu langem Verbleib in den aufgetauten Zel-
len eine Schädigung bewirken können.
Als erstes geeignetes Gefrierschutzmittel für eine Tiefgefrierung von lebenden
Zellen und Geweben beschrieben POLGE et al. 1949 den Wirkstoff Glycerin.
LOVELOCK & BISHOP entdeckten 1959 Dimethylsulfoxid (DMSO) als Kryopro-
tektivum, welches aber heutzutage wegen seiner, im Vergleich mit anderen
Kryoprotektiva, erhöhten Zelltoxizität weniger Verwendung findet.
Seit dieser Zeit wurden diverse Substanzen auf ihre Eignung als Kryoprotekti-
vum untersucht. So zum Beispiel ein- und mehrfache Alkohole, Amide, Dextra-
ne, Mono- und Disaccharide, Polyvinylpyrrolidon (PVP), Polyvinylalkohol (PVA)
und Serumproteine. Von diesen Stoffen haben sich allerdings nur wenige als
praxistauglich erwiesen.
Die den Gefrierschutzmedien zugesetzten Serumproteine sorgen für einen zu-
sätzlichen Membranschutz der konservierten Zellen, scheinen aber weiter keine
direkten Schutzmechanismen zu erfüllen (FARRANT, 1981).
2.4.1 Penetrierende Kryoprotektiva
Das Kennzeichen penetrierender Gefrierschutzmittel ist das Vermögen, auf
Grund eines geringen Molekulargewichtes, durch die Zellmembranen in die Zel-
len hineinzugelangen (MERYMAN, 1971; LEIBO, 1988). Penetrierende Gefrier-
schutzmittel verhindern eine extreme intrazelluläre Eisbildung und werden des-
halb auch in sehr hohen Konzentrationen von ein bis vier Mol verwendet. Gera-
de bei diesen Konzentrationshöhen sollten sie möglichst keine toxischen Aus-
wirkungen auf die Zelle haben.

10
Die penetrierenden Kryoprotektiva müssen die Zellmembran durchdringen, um
eine osmotische Dehydrierung mit daraus resultierender hoher Elektrolytkon-
zentration im Inneren der Zelle zu verhindern (MERYMAN, 1971).
Nach Zugabe der Gefrierschutzsubstanzen besteht zunächst eine Osmolari-
tätsdifferenz zwischen der extra- und der intrazellulären Lösung. Durch das
Kryoprotektivum erhöht sich die Osmolarität in der extrazellulären Lösung. Um
dieses auszugleichen beginnt der Embryo zu schrumpfen. Dies geschieht so
lange, bis die Rate des Ausströmens von Wasser der Rate des Einströmens
von Kryoprotektivum in den intrazellulären Raum entspricht und so ein Konzent-
rationsausgleich erreicht wird. Das Ausmaß der anfänglichen Schrumpfung des
Embryos steht in Beziehung zur Permeabilität der Zellmembran. Je stärker eine
Penetration möglich ist, desto geringer ist die Schrumpfung, da mit dem Ge-
frierschutzmittel zugleich auch Wasser aufgenommen wird. Beim Schrump-
fungsprozess ist es den Zellen möglich, bis auf etwa ein Drittel ihrer Ausgangs-
größe zu schrumpfen ohne bleibende Schäden zu erleiden (SCHNEIDER &
MAZUR, 1984; LEIBO, 1989). Durch das Wasserbindungsvermögen der penet-
rierenden Kryoprotektiva wird eine zu starke Dehydrierung der Zellen verhin-
dert.
In welchem Umfang ein Gefrierschutzmittel in die Zellen gelangt und auch die
Zeitspanne bis zur abgeschlossenen Equilibrierung, ist von verschiedenen Fak-
toren abhängig. Zu diesen Faktoren gehören die Temperatur bei der die Equi-
librierung stattfindet, der Permeabilitätskoeffizient des Kryoprotektivums, der
Unterschied zwischen extra- und intrazellulärer Konzentration und die Beschaf-
fenheit der Zelloberfläche. Außerdem besitzt jede Spezies und jedes Embryo-
nalstadium ganz eigene, individuelle Charakteristika bezüglich der Permeabilität
(MAZUR, 1977; SCHNEIDER & MAZUR, 1984; FRIEDLER et al., 1988; PED-
RO et al., 2005). Die Temperatur hat einen entscheidenden Einfluss auf die
Geschwindigkeit der Equilibrierung, da diese bei höheren Temperaturen schnel-
ler abläuft als bei niedrigeren. In der Praxis hat sich eine Equilibrierung bei 20°C
bis 25°C Raumtemperatur etabliert.
Es wird auch vermutet, dass penetrierende Kryoprotektiva bewirken, dass die
intra- und extrazelluläre Ionenkonzentration der noch ungefrorenen Restflüssig-
keit sich verringert und somit der Gefrierpunkt der intrazellulären Flüssigkeiten
sinkt (LOVELOCK, 1954; LEIBO, 1977; MERYMAN et al., 1977; LEIBO & MA-

11
ZUR, 1978; MAZUR, 1980). Das hat zur Folge, dass unter Verwendung von
Kryoprotektiva zum Beispiel bei Mäuseblastomeren erst ab -35°C bis -45°C ein
Gefrieren der Intrazellularflüssigkeit eintritt (LEIBO et al., 1977).
Als penetrierende Kryoprotektiva finden Stoffe wie Glycerol, Dimethylsulfoxid
(DMSO), Ethylenglycol (EG), Propandiol (PROH), Ethanol und andere Alkohole
Verwendung. DMSO wird nach neueren Erkenntnissen weniger stark verwendet
als noch vor einigen Jahren, da es eine ausgesprochen hohe Toxizität besitzt.
1983 berichteten RALL et al. von sehr guten Ergebnissen bei der Kryokonser-
vierung von Mäuseembryonen unter der Verwendung des Alkohols Methanol.
Auch andere höherwertige Alkohole wie Ethylenglycol (EG) und Propylenglycol
(PG) wurden in der Folgezeit auf ihre Embryonenverträglichkeit getestet. Es
zeigte sich sowohl eine gute kryoprotektive Wirkung auf die tierischen Kanin-
chen- und Mausembryonen als auch auf humane Embryonen (KASAI et al.,
1981; MIYAMOTO & ISHIBASHI, 1981, 1983a; NGUYEN et al., 1983; RALL &
POLGE, 1984; RENARD et al., 1984; RENARD & BABINET, 1984; TESTART
et al., 1985). In der Rinderzucht wird heutzutage bei der Kryokonservierung
sehr häufig Ethylenglycol eingesetzt, da dessen Toxizität so gering ist, dass es
möglich ist, kryokonservierte Embryonen auch ohne vorherige Ausverdünnung
zu übertragen.
2.4.1.1 Glycerol
Glycerol als Kryoprotektivum wurde erstmals 1949 von POLGE et al. als Ge-
frierschutzmittel für eine Kryokonservierung von Geflügelsperma erwähnt. Seit
diesem Zeitpunkt ist die Eigenschaft, Toxizität und Dosierung von Glycerol fort-
laufend erforscht worden.
Bereits 1971 vermerkte MERYMAN, dass Glycerol im Gegensatz zu DMSO im
Zellinneren kaum toxische Wirkung hat. BILTON untersuchte 1980 den Einfluss
von Glycerol und DMSO auf Rinderembryonen. Es wurden dabei allerdings kei-
ne signifikanten Unterschiede festgestellt. In weiteren Versuchen zu der optima-
len Konzentrationshöhe stellte sich heraus, dass Glycerol in den Konzentratio-
nen von 1,3 M bis 1,4 M DMSO mit einer Konzentration von 1,3 M bis 1,5 M bei
konventionellen Kryokonservierungsverfahren gegenüber im Vorteil ist. Zu die-

12
sem Schlüssen kamen FALGE et al. (1982), RALL & POLGE (1984), TAKEDA
et al. (1987) nach Versuchen mit Mäuseembryonen sowie BOUYSSON &
CHUPIN (1982) und PRATHER et al. (1987) nach Versuchen mit Rinderembry-
onen. RALL & POLGE (1984) begründeten ihre Ergebnisse mit der Aussage,
dass DMSO-Lösungen eine geringere Viskosität aufwiesen als Glycerol-
Lösungen. Kommt Glycerol in konventionellen Gefrierverfahren zum Einsatz,
wird häufig eine Konzentration von ein bis zwei Mol gewählt. 1983 untersuchten
MERRY et al. die Auswirkungen von verschiedenen Glycerol-Konzentrationen
auf die in vitro-Entwicklungsraten (ER) nach einer Kryokonservierung und ei-
nem schrittweisen Auftauen. Die verwendeten Konzentrationen (1,0 M, 1,4 M,
1,8 M) führten bei den ermittelten in vitro-ER der Embryonen (49%, 44%, 52%)
allerdings nicht zu signifikanten Unterschieden.
Auch für anderen Tierrassen wurden Versuche zur Höhe der Gefrierschutzmit-
tel-Konzentration durchgeführt. So führten Versuche an Rinderembryonen mit
unterschiedlichen Glycerol-Konzentrationen durch KENNEDY et al. 1983 zu
keinen eindeutigen Unterschieden. Zu ähnliche Ergebnisse kam LEHN-
JENSEN (1986), als er Glycerol in den Konzentrationen 0,5 M, 1,0 M und 1,4 M
einsetzte. Bei 1,0 M und 1,4 Mol kam es zu keinen signifikanten Unterschieden.
Allerdings stellte er bei 0,5 M fest, dass diese Konzentration als nicht ausrei-
chend für einen sicheren Kryoschutz anzusehen ist.
An Schafembryonen testeten WARE & BOLAND 1987 die Auswirkungen ver-
schiedener Konzentrationen auf erfolgreiches Kryokonservieren und Auftauen
im Zusammenhang mit Saccharose. Dabei stellen sie fest, dass sich eine Kon-
zentration von 2,8 M Glycerol immer toxisch auswirkt; unabhängig davon in
welcher Geschwindigkeit das Ausverdünnen mit Saccharose erfolgte.
Bei den ultraschnellen Gefrierverfahren kommt Glycerol in multimolarer Kon-
zentration und bei der Vitrifikation in Kombination mit weiteren Kryoprotektiva
zum Einsatz.
2.4.1.2 Dimethylsulfoxid (DMSO)
LOVELOCK & BISHOP waren 1959 die ersten, die die kryoprotektiven Eigen-
schaften von DMSO bei der Gefrierkonservierung von Zellen beschrieben. 1972

13
gelang es WHITTINGHAM et al. erstmals eine erfolgreiche Kryokonservierung
von Mäuseembryonen durchzuführen. Dabei schien es, dass DMSO Glycerol in
seinen protektiven Eigenschaften überlegen sei.
In den folgenden Jahren gelang es, weitere Spezies unter Verwendung von
DMSO erfolgreich tiefzugefrieren. BANK & MAURER (1974) sowie MAURER &
HASEMAN (1976) berichteten von positiven Ergebnissen bei der Kryokonser-
vierung von Kaninchenembryonen. WILLADSEN et al. (1978) und BILTON
(1980) froren erfolgreich Rinderembryonen ein und TROUNSON & MOHR ge-
lang 1983 die Konservierung von Humanembryonen.
Glycerol und DMSO sind auf Grund ihres molekularen Aufbaus in der Lage, Zel-
len schneller zu penetrieren als Wasser. Im direkten Vergleich zwischen Glyce-
rol und DMSO ist DMSO jedoch noch deutlich schneller (TROUNSON &
MOHR, 1983; AGCA et al., 1997). 1971 stellte MERYMAN fest, dass DMSO die
Fähigkeit besitzt, vollständig in die Zelle einzudringen. Laut MIYAMOTO et al.
(1998) ist die vollständige Penetration elementare Voraussetzung für einen
wirksamen Schutz, da ein Vorhandensein von DMSO allein in der extrazellulä-
ren Lösung nicht ausreicht, um eine sichere Kryokonservierung zu gewährleis-
ten.
Verschiedene Versuche zur nötigen Konzentrationshöhe von DMSO führten
LEIBO & MAZUR 1978 zu der Feststellung, dass eine Verwendung von 1 M
DMSO keinen sicheren Schutz bietet. BANK & MAURER (1974), FORGRAVE
et al. (1977) und WILLADSEN (1980) kamen in ihren Versuchen an unter-
schiedlichen Spezies zu dem Schluss, dass DMSO in konventionellen Gefrier-
verfahren erst ab einer Konzentration von 1,5 M eine ausreichende Schutzwir-
kung hat.
LEHN-JENSEN stellte 1980 durch Messungen an pH-Wert und Osmolarität
fest, dass DMSO in Konzentrationen, wie sie beim Kryokonservieren zur Ver-
wendung kommen, deutlich größere pH-Wert Veränderungen und wesentlich
größeren osmotischen Stress für die Zellen hervorruft als es bei Glycerol der
Fall ist. Dadurch erklärt sich auch die höhere Toxizität von DMSO im Vergleich
zu Glycerol (FRIEDLER et al., 1988).
Allerdings besitzt DMSO eine gute Bereitschaft zu Glasbildung (FRIEDLER et
al., 1988; VALDEZ et al., 1992). und wird aus diesem Grunde häufig als Be-

14
standteil für Vitrifikationslösungen genutzt (ISHIMORI et al., 1992, 1993; VIN-
CENTE & GARCIA-XIMENEZ, 1994).
2.4.1.3 Ethylenglycol (EG)
Im Vergleich zu Glycerol (Mg 91,1), DMSO (Mg 78,1) und Propandiol (Mg 76,1)
besitzt Ethylenglycol mit 62,1 ein geringeres Molekulargewicht (Mg). Aus die-
sem Grunde ist es in der Lage, schneller in die Zelle hinein und wieder heraus
zu diffundieren, als es den anderen penetrierenden Kryoprotektiva möglich ist.
Besonders bei höheren Konzentrationen stellen das erhöhte Permeabilitäts-
vermögen und die sich so ergebende verringerte Toxizität, verglichen mit ande-
ren Kryoprotektiva, bedeutende Vorteile dar (MAHMOUDZADEH et al. 1993;
CSEH et al. 1997).
Dass EG eine kryoprotektive Wirkung besitzt, konnten MIYAMOTO & ISHI-
BASHI 1977 erstmalig anhand kontrollierter Kryokonservierungsversuche an
Ratten- und 8-Zell-Mäuseembryonen zeigen.
In diversen weiteren Versuchen wurden Unterschiede auf die Überlebensraten
(ÜR) von Rinder- und Schafembryonen bei Verwendung von EG oder Glycerol
als Gefrierschutzmittel bei konventioneller Kryokonservierung untersucht.
Dabei stellte man fest, dass EG im Vergleich zu Glycerol gleiche oder sogar
bessere Überlebensraten erzielte (COCERO et al. 1988; LANGE 1995; KLING
1997; BEAL et al. 1998).
Bei Versuchen zur Ermittlung verträglicher und wirkungsvoller EG-
Konzentrationen verwendeten VOELKEL & HU (1992a; 1992b) Rinderembryo-
nen in den Stadien Morula bis expandierte Blastocyste bei Ethylenglycolkon-
zentrationen zwischen 1,0 M bis 2,0 M. Dabei stellten sie fest, dass eine Ethy-
lenglycolkonzentration in Höhe von 1,5 M am besten geeignet war.
1999 equilibrierten SOMMERFELD & NIEMANN in vitro-produzierte Tag 7-
Rinderblastozysten 10 min in Ethylenglycollösungen bei Konzentrationen in Hö-
he von 1,8 bis 8,9 M bei Raumtemperatur. Die höchste Schlupfrate (SR) erziel-
ten nach 72 h in vitro-Kultivierung mit 98% diejenigen Embryonen, die bei einer
Konzentration von 3,6 M equilibriert worden waren. Auch beim one-step-
Equilibrieren und Ausverdünnen von zuvor kryokonservierten in vitro-

15
produzierten Rinderembryonen zeigte sich eine 3,6 M Ethylenglycolkonzentrati-
on als beste Konzentrationshöhe.
Versuche zu Auswirkungen von unterschiedlich vielen Equilibrierungstufen vor
und Ausverdünnungsstufen während des Auftauens nach Kryokonservierung
führten McGINNIS et al. 1989 durch. Sie stellten fest, dass es nach Kryokon-
servierung mit 1,5 M Ethylenglycol keine Unterschiede in den in vitro-ÜR gab.
In verschiedenen Untersuchungen wurde festgestellt, dass EG in der Lage ist
schneller durch Zellmembranen zu diffundieren als Glycerol. Zusätzlich hat E-
thylenglycol auch bei höheren Temperaturen offensichtlich wesentlich weniger
toxische Auswirkungen als Glycerol. Diese Eigenschaften führen dazu, dass auf
das zeitlich und arbeitstechnisch aufwendige Ausverdünnen des Gefrier-
schutzmittels beim Auftauen verzichtet werden kann.
VOELKER & HU (1992b) stellten in ihren Versuchen fest, dass ein direkter
Transfer von, mit Ethylenglycol tiefgefrorenen, Rinderembryonen möglich sei.
Diese verbesserte Handhabung (geringerer Material- und Zeitaufwand), beson-
ders für die praktische Umsetzung in der Routine bedeutsam, wurde in weiteren
Versuchen wiederholt und ebenfalls bestätigt (McINTOSH & HAZELAGER,
1994; BRACKE et al., 1995; DOCHI et al., 1995; JANOWITZ & HERMANNS,
1995; LANGE, 1995). Dieses sogenannte One-Step-Verfahren ist heute in der
Routine weit verbreitet.
Ethylenglycol wird aber nicht nur bei der konventionellen Kryokonservierung,
sondern auch bei der Vitrifikation und dem ultraschnellen Tieffrieren verwendet.
Bei Versuchen zur Vitrifikation zeigte es sich, dass EG weniger toxisch auf die
Embryonen wirkte als andere Kryoprotektiva (MAHMOUDZADEH et al., 1993).
Für Mäuse- und Rinderembryonen wurden auch beim ultraschnellen Tiefgefrie-
ren gute Schutzwirkungen durch Ethylenglycol nachgewiesen. RAYOS et al. er-
hielt 1992 sehr gute in vitro-ÜR, nachdem er Ein-Zell-Mauseembryonen für 10
Minuten in 3 M EG und 0,25 M Saccharose equilibriert hatte. NOWSHARI &
BREM (1998a) stellten deutlich höhere Schlupfraten (SR) bei ultraschnell tiefge-
frorenen, expandierten Mauseembryonen fest, wenn diese nach vorheriger De-
hydrierung mit Saccharose, unter Verwendung von 7 M EG anstatt 7 M PROH
kryokonserviert wurden.

16
2.4.2 Nicht penetrierende Kryoprotektiva
Im Gegensatz zu den penetrierenden Kryoprotektiva, dringen die nicht penetrie-
renden Gefrierschutzmittel nicht in die Zelle ein, sondern entfalten ihre schüt-
zenden Wirkung im extrazellulären Raum. Denn auch ohne Penetration scheint
von diesen Substanzen eine kryoprotektive Wirkung auszugehen. Mehrere Au-
toren vermuten, dass die Gefrierschutzmittel eine Stabilisierung der Zellmemb-
ranen bewirken (HEBER, 1968; MAZUR, 1970; LEIBO et al., 1974; MERYMAN,
1974; MAZUR, 1976; BARNETT, 1978; UTSUMI, 1978; RALL et al., 1983).
Die in der Kryokonservierung verwendeten nicht penetrierenden Kryoprotektiva
gehören zu den Gruppen der Monosaccharide (Galaktose, Fructose, Glucose),
den Disacchariden (Saccharose, Trehalose, Lactose), Proteinen, Lipoproteinen
sowie Polyvinylpyrrolidon (PVP). Da die Saccharose das in der Praxis am wei-
testen verbreitete Kryoprotektivum ist (LEIBO, 1988) und auch in der vorliegen-
den Arbeit verwendet wurde, wird schwerpunktmäßig auf dieses nicht penetrie-
rende Kryoprotektivum eingegangen.
Es ist ausreichend, die nicht penetrierenden Kryoprotektiva in einem niedrigen
molaren Konzentrationsbereich einzusetzen, wodurch die Toxizität dieser Sub-
stanzen verringert wird (NIEMANN & MEINEKE, 1993). Um die Phase der De-
hydration zu unterstützen, werden die nicht penetrierenden Gefrierschutzmittel
sowohl in der Equilibrierungsphase vor der Kryokonservierung als auch bei der
Ausverdünnung der Kryoprotektiva während des Auftauens eingesetzt, um ein
übermäßiges Anschwellen der Embryonen zu verhindern (FRIEDLER et al.,
1988).
MERYMAN stellt 1971 fest, dass durch den Einsatz von nicht penetrierenden
Kryoprotektiva die Zellen in der Lage sind, gelöste Stoffe unter osmotischem
Stress penetrieren zu lassen.
PVP und Acetamid, als für Embryonen hochtoxische Substanzen, werden heut-
zutage in der Regel nur noch bei der Vitrifikation eingesetzt (RALL & FAHY,
1985; RALL et al., 1985). In der assistierten Reproduktion kommt PVP zur Im-
mobilisierung der Spermien bei ICSI zur Verwendung - weniger als Schutzsub-
stanz bei der Kryokonservierung.

17
Dem Gefrierschutzmittel Saccharose selber wird so gut wie keine kryoprotektive
Wirkung zugeschrieben (WHITTINGHAM, 1971; WILMUT, 1972; MIYAMOTO &
ISHIBASHI, 1976; MERYMAN et al., 1977; KASAI et al., 1981). Sie wird aber
als Unterstützung für die Dilution der Kryoprotektiva und bei dem Verfahren der
Vitrifikation zur isothermen Dehydrierung verwendet (TAKEDA et al., 1984;
KRAG et al., 1985a, 1985b; PEURA et al., 1985; RALL & FAHY, 1985; RALL et
al., 1985; WILLIAMS & JOHNSON, 1985).
KASAI et al. (1981, 1983) entdeckten, dass es möglich ist, Embryonen über ei-
nen begrenzten Zeitraum hinweg in saccharosehaltigen Medien bei Temperatu-
ren oberhalb des Gefrierpunktes zu lagern.
SEIDEL (1989) nahm an, dass eine kryoprotektive Wirkung der Saccharose
schon vor dem eigentlichen Kühlprozess einsetzt. Er stellte fest, dass durch
Einsatz von 0,2 M bis 0,3 M Saccharose die Zellen soweit vordehydriert wer-
den, dass das Umsetzen in flüssigen Stickstoff (LN2) schon bei deutliche höhe-
ren Temperaturen durchgeführt werden kann, ohne das die Zellen beschädigt
werden. Diese These wurde auch von MASSIP et al. (1987) und MERMILLOD
et al. (1992) unterstützt, die ein Umsetzen bereits ab -25°C für möglich erachte-
ten.
Da Saccharose aber als alleiniges Protektivum keine ausreichende Schutzwir-
kung besitzt (LEIBO, 1989), wird sie in der Regel in Konzentrationen von 0,1 M
bis 0,3 M kombiniert mit penetrierenden Kryoprotektiva eingesetzt (LEHN-
JENSEN, 1984).
Um auch bei höheren Kühlraten einen ausreichenden Schutz zu gewährleisten,
ist offensichtlich zusätzlich zu den penetrierenden Gefrierschutzmitteln der Ein-
satz nicht penetrierender Kryoprotektiva sinnvoll, da nur so eine notwendige
und schnelle Dehydrierung der Zellen möglich ist (MERYMAN et al., 1977).
Beim Auftauen ist der Embryo im Vergleich zur Equilibrierungsphase in Vorfeld
der Kryokonservierung einer verringerten extrazellulären Kryoprotektiva-
Konzentration ausgesetzt, was zur Folge hat, dass Wasser relativ schnell in den
Embryo einströmt. Dabei kann unter Umständen Wasser so schnell und so
stark einströmen, dass der Embryo bis zu seinem Zerreißen anschwillt. Bleibt
die Volumenzunahme aber innerhalb der maximal tolerierbaren Grenzen, ent-
stehen an den Zellmembranen keine Schäden. Auf Grund der Molekülgröße ist
Saccharose nicht in der Lage, Zellwände zu penetrieren. Sie wird aber als os-

18
motischer Puffer bei der Entfernung penetrierender Gefrierschutzsubstanzen
verwendet, um ein zu starkes Anschwellen zu vermeiden (NIEMANN, 1982;
RENARD et al., 1982; LEIBO, 1984; MASSIP et al., 1987).
In Versuchen stellten SCHNEIDER & MAZUR (1984) fest, dass Mäuseembryo-
nen fähig sind, bei 23°C für 30 Minuten eine 2,7-fache Volumenzunahme zu
überstehen. Wird Saccharose als Puffer verwendet, können die Embryonen bis
auf 25% ihres ursprünglichen Volumens schrumpfen ohne das es zu Schäden
kommt. Nach abgeschlossener Equilibrierung und Umsetzung in zum Beispiel
ein Kulturmedium kehren die Embryonen zu ihrem normalen Volumen zurück
(SZELL & SHELTON, 1986; VOELKEL & HU, 1992a). Ein vollständig ablaufen-
der Schrumpfungs- und Reexpandierungszyklus ist neben der morphologischen
Beurteilung ein zusätzlicher Hinweis auf Unversehrtheit und Vitalität der Zellen
(BIELANSKY et al., 1986).
1989 untersuchten MAPLETOFT et al. die in vitro-Überlebensraten (ÜR) frisch
gespülter Mäuseembryonen in Abhängigkeit unterschiedlicher Saccharose-
Konzentrationen. Dazu wurden die Embryonen für 10 oder 30 Minuten bei 20°C
bzw. 35°C in den Saccharose-Lösungen equilibriert. Bei einer 0,5 M Saccharo-
se-Lösung schienen Zeit und Temperatur nur wenige Auswirkungen zu haben.
Bei 1,0 M Saccharose nahmen die in vitro-ÜR nach Verlängerung der Equi-
librierungsdauer und einer Temperaturerhöhung deutlich ab. Wurden die Mäu-
seembryonen einer 2,0 M Konzentration ausgesetzt, waren die ÜR sehr stark
verringert.
In Versuchen zur Verträglichkeit steigender Saccharose-Konzentrationen (0,0 M
bis 1,3 M) bei mit Glycerol kryokonservierten 8-Zell-Mauseembryonen, stellten
TAKEDA et al. 1987 im Anschluss an das Ausverdünnen bei steigenden Sac-
charose-Konzentrationen zunehmend Schäden an der Zona pellucida fest.
Durch die Entwicklung des sogenannten, 1983 von LEIBO beschriebenen One-
Step-Verfahrens, bei dem das Ausverdünnen bereits in der Gefrierpaillette statt-
findet, konnte der Arbeits- und Zeitaufwand bei Auftauen deutlich verringert
werden. Dazu werden außer dem Embryo und dem Kryoprotektivum auch zwei
Säulen mit Saccharose aufgezogen, die sich beim anschließenden Auftauen in
der Paillette vermischen und ein direktes Ausverdünnen ermöglichen. Für Rin-
derembryonen untersuchten HOOGENKAMP (1984) sowie SCHUBERT (1984)

19
die Glycerol/Saccharose-Verhältnisse und erzielten die besten ÜR bei einem
Verhältnis von 1:2. In 1994 von CSEH et al. veröffentlichten Untersuchungen
wurden die höchsten Trächtigkeitsraten nach Transfer (38%) bei einer Mi-
schung von 1:9 Glycerol/Saccharose erreicht.
Aber auch bei der ultraschnellen Kryokonservierung und der Vitrifikation wird
Saccharose erfolgreich als nicht penetrierendes Kryoprotektivum zur Unterstüt-
zung der Dehydrierung und als Zellschutz eingesetzt. Bei Untersuchungen zur
ultraschnellen Tiefgefrierung von Mäuseembryonen setzten RAYOS et al.
(1992a, b) eine Kombination von 0,5 M Saccharose und 3 M EG ein. THOMAS
et al. (1989) verwendeten ebenfalls 0,5 M Saccharose, aber statt EG 3,5 M
Glycerol. Die Ausverdünnung erfolgte unter Verwendung von Saccharose.
KASAI et al. (1990) und KASAI (1996) beschrieben eine zur Vitrifikation geeig-
nete Lösung, die als penetrierendes Protektivum Ethylenglycol, als nicht penet-
rierende Schutzsubstanz 0,5 M Saccharose und als Zusatzstoff das Makromo-
lekül Ficoll enthielt. Saccharose und Ficoll bewirken dabei, dass die EG-
Konzentration in den Zellen reduziert wird und die Embryonen schnell schrump-
fen. Das wiederum führt zu einer Verringerung der Toxizität der Lösung und da-
her zu einer besseren Überlebenswahrscheinlichkeit der Zellen.
2.5 Gefrierverfahren
Die heute verwendeten Kryokonservierungsverfahren werden in das konventio-
nelle, kontrollierte Tiefgefrierverfahren - auch als Standardgefrierverfahren be-
zeichnet - das ultraschnelle Kryokonservieren und die Vitrifikation eingeteilt.
Beim konventionellen Tiefgefrieren kommen die Embryonen vor ihrer Konser-
vierung in eine, das Kryoprotektivum enthaltende Lösung und verbleiben in die-
ser bis zur abgeschlossenen Equilibrierung. Im Gegensatz dazu wird den Emb-
ryonen beim ultraschnellen Tieffrieren und bei der Vitrifikation keine Möglichkeit
zur vollständigen Equilibrierung gegeben. Es handelt sich hierbei um eine so-
genannte Nonequilibrium-Kryokonservierung. Vor der Kryokonservierung erfolgt
eine Dehydrierung der Embryonen, indem sie für einen kurzen Moment in eine
Mischung aus nicht penetrierenden und hochkonzentrierten penetrierenden
Kryoprotektiva kommen (LEIBO, 1989).

20
Zur Aufbewahrung der kryokonservierten Embryonen wurden in den Anfangs-
zeiten der Kryokonservierung Glasampullen verwendet, die im Laufe der Zeit
dann allerdings durch die heute gebräuchlichen Kunststoffpailletten ersetzt
wurden. BIELANSKY et al. (1985) stellten in ihren Untersuchungen fest, dass
es keine Unterschiede zwischen den Embryoüberlebensraten von Glasampul-
len und Kunststoffpailletten gab. Die Kunststoffpailletten erwiesen sich aber in
der Handhabung von Kryokonservierung, Lagerung und spätere Transfers als
vorteilhafter und haben sich daher durchgesetzt (MASSIP et al., 1982).
2.5.1 Standardgefrierverfahren
Das Hauptmerkmal des Standardgefrierverfahrens ist, dass Embryonen und
Kryoprotektiva nach festgelegten und kontrollierten Schemata auf Lagerungs-
temperatur heruntergekühlt werden. Auf diese Weise wird versucht, irreparable
Schäden durch unkontrollierte intrazelluläre Eiskristallbildung und Lösungsef-
fekte zu verhindern, die zum Zelltod und damit zum Absterben des Embryos
führen können. Deshalb werden spezielle, optimierte Kühlraten verwendet, die
auf die unterschiedlichen Spezies und Entwicklungsstadien sowie verwendeten
Kryoprotektiva genau zugeschnitten sind. Zur Steuerung der Abkühlraten wer-
den programmierbare Temperaturregler verwendet, an denen die jeweils benö-
tigten Kühlraten eingestellt werden können.
Beim Standardgefrierverfahren wird bei einer Temperatur von -6°C oder -7°C
manuell oder automatisch die Kristallisation der extrazellulären Medien ausge-
löst. Durch diesen als „Seeding“ bezeichneten Vorgang wird die Bildung kleiner
intrazellulärer Eiskristalle unterstützt und es soll durch diesen Vorgang ebenso
verhindert werden, dass es zu einer für den Embryo schädlichen Unterkühlung,
dem Supercooling, kommt. Häufig wird die Temperatur im Anschluss an das
Seeding noch für einige Minuten auf Seeding-Temperatur gehalten, bevor sie
mit definierter langsamer Kühlrate bis auf etwas -32°C weiter abgesenkt und
erneut für einige Minuten dort gehalten wird. Im letzten Schritt werden die Zel-
len durch schnelles Einfrieren, welches in der Regel durch direktes Umsetzen in
LN2 erfolgt, auf die Lagerungstemperatur von -196°C gebracht.

21
Würde kein manuelles, oder das durch neuere Technik ermöglichte, automati-
sche Seeding erfolgen, wäre eine willkürliche, spontane Kristallbildung der Me-
dien die Folge. In Abhängigkeit vom verwendetem Kryoprotektivum tritt diese
spontane Eiskristallbildung ab etwa -10°C im extrazellulären und bei etwa -35°C
bis -45°C im intrazellulären Raum ein (MAZUR, 1970; BANK, 1973; LEIBO et
al., 1978; LEHN-JENSEN & RALL, 1983; RALL & POLGE, 1984; TONER et al.,
1991). Im Zuge dieser spontanen Kristallisation innerhalb der Zellen und des
Mediums kommt es zu einer Wärmefreisetzung und damit zu einem kurzfristi-
gen Temperaturanstieg. Die dabei entstehenden intrazellulären Eiskristalle
würde auf Grund ihrer Größe unweigerlich zum Tod der Embryonen führen
(WILLADSEN, 1980; PAVEL, 1985; SEIDEL, 1989; NIEMANN, 1991; STREI-
CHER, 1998; SEIBT, 2003).
SOMMERFELD (1997) war es möglich, eine Abhängigkeit von Seeding-
Temperatur und Kryoprotektivums-Konzentration festzustellen. Nach einer Er-
höhung der Ethylenglycol-Konzentration auf 3,6 M sank die Gefriertemperatur
auf -11°C ab. Wurde die Temperatur erhöht, konnte die Kristallisation nicht auf-
rechterhalten werden.
Der Umstieg von Glasampullen auf Kunststoffpailletten hatte auch für das er-
folgreiche Seeding Vorteile. Wegen des geringeren Paillettendurchmessers und
die dünnere Wand der Kunststoffpailletten ist das manuelle Seeding deutlich
schneller und präziser möglich, wodurch die Überlebenswahrscheinlichkeit der
Embryonen steigt (NIEMANN, 1983).
Um für den Embryo unnötigen Stress zu vermeiden, sollte der Kristallisations-
kern nicht direkt an der den Embryo enthaltenden Säule gesetzt werden, son-
dern es sollte dafür eine, durch eine Luftblase getrennte, zweite Mediumsäule
gewählt werden (DE PAZ et al., 1994).
Das Herunterkühlen auf Seeding-Temperatur kann mit unterschiedlichen Kühl-
raten erfolgen. Je nach Spezies werden die Kühlraten eingestellt. Bei humanen
Embryonen wird häufig eine Kühlrate von 2°C/min gewählt. Mäuseembryonen
hingegen können offensichtlich auch ohne nachteilige Effekte auf die spätere
Vitalität direkt in Seeding-Temperaturen eingesetzt werden. Arbeitstechnisch
bedeutet dieses eine deutliche Erleichterung, da die Dauer des Kryokonservie-
rungsprogramms verkürzt werden kann (WILLADSEN, 1980; SCHNEIDER &
MAZUR, 1984; PAVEL, 1985).

22
Die Unterscheidung zwischen dem langsamen und dem schnellen Verfahren
definiert sich in den Kühlraten im Anschluss an das Seeding.
2.5.2 Langsames Kryokonservieren
Das Kennzeichen der langsamen Konservierung sind die Kühlraten von
0,1°C/min bis 0,3°C/min, mit der die Temperatur auf -60 bis -120°C herabge-
kühlt wird (LEIBO et al., 1974; FALGE et al., 1982; NIEMANN, 1983; LEHN-
JENSEN, 1984).
WHITTINGHAM et al. (1972) sowie WILMUT (1972) gelang es erstmals auf
diese Weise ein Überleben von Mauseembryonen zu erreichen.
Diese niedrigen Kühlraten wurden gewählt, damit den Zellen die Möglichkeit zur
Dehydrierung und osmotischen Equilibrierung gegeben ist. Durch die beinahe
vollständige Dehydrierung der Zellen ist es möglich, die unerwünschte intrazel-
luläre Kristallisation zu umgehen (MAZUR, 1966; MAZUR & SCHNEIDER,
1986). Die Rate der Auftaugeschwindigkeit für dieses Verfahren liegt bei
2°C/min bis 10°C/min (NIEMANN, 1991).
Bei graphischer Darstellung von Kühlrate und Überlebensrate zeigt sich das
Bild einer Parabel (Abb. 1). Mit steigender Kühlrate steigt auch die Überlebens-
rate (ÜR). Bei einer mittleren Kühlrate erreicht die ÜR ihr Maximum, um an-
schließend wieder abzusinken. Als optimalste Kühlrate wird eine Rate angese-
hen, die langsam genug ist, der intrazellulären Eisbildung entgegenzuwirken,
aber wiederum schnell genug ist damit die Zellen nicht länger als unbedingt
notwendig den Lösungseffekten ausgesetzt sind (MAZUR, 1970, 1980)

23
Kühlrate (°C/min)
Übe
rleb
ensr
ate
(%)
Abb. 1: Abhängigkeit der Überlebensrate von der Kühlrate
2.5.3 Schnelles Kryokonservieren
Mit dem schnellen oder auch 2-stufigen Kryokonservierungsverfahren ist eine
Methode entwickelt worden, die deutlich schneller und so für die Routine we-
sentlich praktikabler ist.
Als erste konnten WOOD & FARRANT 1980 von erfolgreichen Versuchen mit
Mäuseembryonen berichten, die eine langsame Kühlung auf -20°C bzw. -25°C
und eine daran direkt anschließende Überführung in flüssigen Stickstoff (LN2)
überlebt hatten.
In Versuchen mit in DMSO kryokonservierten Rinderblastocysten stellen LEHN-
JENSEN & GREVE 1980 fest, dass es keinen Unterschied machte, ob die Emb-
ryonen nach dem langsamen oder dem schnellen Tiefgefrierverfahren eingefro-
ren wurden. Nach NIEMANN (1982) erklärt sich das dadurch, dass die Embryo-
nen noch weiterhin die Möglichkeit haben, ihr Zellwasser in das noch nicht
komplett gefrorene, sie umgebene extrazelluläre Medium abzugeben. Bei einer
Temperatur von -42°C sind erst ungefähr 74% der Lösung kristallisiert. Schließt
sich hier eine sehr schnelle Temperatursenkung auf bis -150°C an, führt das zu
einer Viskositätszunahme der Restflüssigkeit. Weitere Eiskristallbildungen sind
dann nicht mehr möglich und die Restflüssigkeit vitrifiziert (RALL, 1981; RALL
et al., 1984).

24
Diverse Untersuchungen beschäftigten sich damit, die optimale Temperatur
zum Umsetzen zu ermitteln. Dabei zeigte sich, dass der beste Umsetztempera-
turbereich in LN2 offensichtlich zwischen -30°C und -35°C liegt (BILTON, 1980;
LEHN-JENSEN & GREVE, 1982; FARRANT et al., 1985; SCHIEWE et al.,
1985).
MASSIP et al. (1987) sowie NIBART & HUMBLOT (1997) konnten von guten
Trächtigkeitsraten berichten, nachdem sie die Embryonen bei -25°C umgesetzt
hatten. Als Kryoprotektivum verwendeten sie Glycerol und Saccharose und e-
quilibrierten die Embryonen bei Raumtemperatur. Durch die Verwendung von
Saccharose dehydrieren die Embryonen soweit, dass es möglich ist, das Küh-
len auch schon bei höheren Temperaturen zu beenden.
MAZUR (1990) sah die anschließende Auftaugeschwindigkeit als Vorausset-
zung für ein Überleben von bei -20°C bis -40°C umgesetzten Embryonen an.
Nur wenn die Auftaurate schnell genug ist, um eine Rekristallisation zu verhin-
dern, haben die Embryonen eine Überlebenschance.
2.5.4 Ultraschnelles Kryokonservieren
Bei diesem Verfahren werden die Embryonen direkt in LN2 getaucht. Dabei bil-
den sich kleinere intra- und extrazelluläre Eiskristalle. Als verwendete Gefrier-
schutzsubstanzen kommen recht hoch dosierte penetrierende (2,0 M bis 3,5 M)
und nicht penetrierende (0,2 M bis 0,5 M) Kryoprotektiva zum Einsatz (NIE-
MANN, 1991b). Die nicht penetrierenden Kryoprotektiva, wie zum Beispiel Sac-
charose, bewirken, dass die Embryonen bereits vor dem Tiefgefrieren relativ
weit dehydrieren und dadurch das Risiko intrazellulärer Eisbildung sehr stark
reduziert wird (MAZUR, 1990).
Zu Beginn der Entwicklung des ultraschnellen Tiefgefrierens war es nur möglich
Mäuseembryonen tiefzugefrieren. Inzwischen ist es aber auch möglich weitere
Spezies nach diesem Verfahren zu konservieren.
Aber auch die Wahl des penetrierenden Kryoprotektivums hat bei diesem Ver-
fahren offensichtlich Auswirkungen auf die Überlebensraten der konservierten
Embryonen. So untersuchten GUTIERREZ et al. (1993) den Einfluss von Glyce-
rol, EG und Propandiol in der Konzentration von 3,0 M in Verbindung mit jeweils

25
0,25 M Saccharose auf die Überlebensrate von Mäusemorulae. Dabei stellte sie
signifikante Unterschiede zwischen Glycerol (76,1%) und EG (79,5%) im Ver-
gleich zu Propandiol mit 43,6% in vitro-Weiterentwicklungsrate fest.
NOWSHARI & BREM (1998a) verglichen in ihren Versuchen mit expandierten
Mäuseembryonen die Entwicklungsraten bei der ausschließlichen Verwendung
von EG bzw. Propandiol in unterschiedlich hohen Konzentrationen. Es zeigte
sich, dass eine Konzentration von 7,0 M den effektivsten Schutz bietet und dass
die Schlupfrate nach Verwendung von EG höher lag als nach Propandiolver-
wendung (84% zu 78%).
In wie weit die Equilibrierungsdauer Einfluss auf die Entwicklungsraten hat, un-
tersuchten RAYOS et al. (1992b). Sie ermittelten die optimalste Zeitdauer für in
3,0 M Ethylenglycol und 0,25 M Saccharose equilibrierte Mäuseembryonen. 10
Minuten führten zu den höchsten in vitro-Entwicklungsraten, während bei länge-
ren Zeitspannen von 20 bzw. 30 Minuten die Entwicklungsraten wieder absan-
ken.
LEIBO (1989) erklärte das damit, dass es durch die längere Equilibrierung zu
einer erhöhten intrazellulären EG-Konzentration kommt, die beim Ausverdün-
nen des Kryoprotektivums durch das schnelle Einströmen von Wasser zur os-
motischen Zellschädigung führt.
2.5.5 Vitrifikation
Ebenso wie bei dem ultraschnellen Tiefgefrieren erfolgt auch bei der Vitrifikation
ein direktes Umsetzung in LN2.
Bei der Vitrifikation wandelt sich die hochkonzentrierte Kryoprotektivums-
Lösung durch sehr schnelle Kühlraten von der flüssigen Phase in einen stabi-
len, glasartigen Zustand. Zunächst wird sie bei diesem Prozess zunehmend
visköser, bevor sie schließlich ganz erstarrt.
Ist die Vitrifikation korrekt verlaufen, kommt es zu keiner Eiskristallbildung im
Vitrifikationsmedium und auch im Anschluss an die Umsetzung in flüssigen
Stickstoff bleibt das Medium klar und durchsichtig. Da die Lösung beim Gefrie-
ren nicht durch die Bildung von Eiskristallen aufkonzentriert wird, können neben
den Schäden durch intrazelluläres Gefrieren auch Schäden durch Lösungsef-

26
fekte umgangen werden (FAHY et al., 1984; FRIEDLER et al., 1988; NIEMANN,
1991a; DOBRINSKY, 1996; STREICHER, 1998).
Im Gegensatz zu den anderen Kryokonservierungsverfahren, ist bei der Vitrifi-
kation keine programmierbare Gefriereinheit zwingend notwendig, da durch das
direkte Eintauchen in LN2 die sehr schnelle notwendige Kühlrate von bis zu
2000°C/min erreicht wird. Allerdings haben gesteuerte Geräte eine sehr viel ge-
nauere Abkühlrate und eine höherer Abkühlleistung von bis zu 130000°C/Min,
was wiederum den Einsatz der hochtoxischen Gefrierschutzmittel auf ein Mini-
mum reduzieren kann (Vitmaster™).
Denn ein großes Problem stellt bei dem Verfahren der Vitrifikation die Toxizität
der sehr hoch konzentrierten Kryoprotektiva dar, die sowohl während der Equi-
librierung als auch beim Ausverdünnen während des Auftauprozesses bei
Raumtemperatur starke Schäden an den Embryonen verursachen können
(FRIEDLER et al., 1988; DE LEEUW et al., 1991; ISHIMORI et al., 1992; RALL,
1992).
Von entscheidender Bedeutung für eine gelungene Vitrifikation ist es, diejenige
Konzentration an Protektiva zu finden, die maximalen Kryoschutz bei minimalen
toxischen Auswirkungen ermöglicht. Aus diesem Grunde hat es sich als positiv
erwiesen, Mischungen von verschiedenen Kryoprotektiva zu verwenden, da die
vitrifizierenden, aber unter Umständen sehr toxischen Eigenschaften des einen
Kryoprotektivums mit den weniger toxischen Eigenschaften eines anderen Kry-
oprotektivums ausgeglichen werden können (MASSIP et al., 1987).
RALL & FAHY waren 1985 die ersten, die von einer erfolgreichen Vitrifikation
an Mäuseembryonen berichteten. Als Vitrifikationslösung verwendeten sie eine
Mischung aus den Komponenten DMSO, Acetamid, Propandiol sowie Polyethy-
lenglycol und konnten nach einer 48-stündigen in vitro-Kultivierung eine Ent-
wicklungsrate von 87,8% vermerken.
Zum Gelingen einer erfolgreichen Vitrifikation tragen, neben der Kombination
der Kryoprotektiva, entscheidend die Equilibrierungsdauer der Embryonen in
der Vitrifikationslösung und die Temperatur bei der die Equilibrierung stattfindet
bei. In gewissem Masse lässt sich die Toxizität beeinflussen, indem man die
Equilibrierungstemperatur und auch die Verweilzeit der Embryonen in der Vitri-
fikationslösung auf ein Minimum reduziert.

27
Deshalb kommen die Embryonen in einem ersten Schritt zunächst in eine nied-
riger konzentrierte Lösung zum Equilibrieren und anschließend in einem zwei-
ten Schritt für einen sehr kurzen Moment (wenige Sekunden bis etwa eine hal-
be Minute) bei geringerer Temperatur (4°C) in die eigentliche, hochkonzentrier-
te Vitrifikationslösung (FRIEDLER et al., 1988; NIEMANN & MEINECKE, 1993).
Saccharose wurde erstmals durch KASAI et al. 1990 als Vitrifikationslösungs-
Bestandteil verwendet. Sie benutzten eine Mischung aus 40% Ethylenglycol,
30% Ficoll und 0,5 M Saccharose. In seiner Eigenschaft als schnell penetrie-
rendes Kryoprotektivum gelangt EG bei der Equilibrierung rasch in ausreichen-
der Konzentration in die Zellen und auch beim Ausverdünnen ist EG in der La-
ge, die Zellen schnell wieder zu verlassen. Das bedeutet, dass die Toxizität
deutlich gesenkt werden kann, was wiederum dazuführt, dass höhere Überle-
bensraten erreicht werden.
In den Versuchen von KASAI et al. (1990) erfolgte die Embryonenequilibrierung
bei Raumtemperatur in einem Schritt von zwei bzw. fünf Minuten. Die Entwick-
lungsraten zu expandierten Blastocysten erreichten dabei Werte von bis zu 97
bzw. 98%.
2.6 Auftauen
Während zum Einfrier- und Gefriervorgang die drei Phasen Vorbereitungspha-
se, Seeding und Gefrierphase sowie im weiteren Sinne noch die beiden Phasen
Schädigungsprozesse und Lagerungsphase gehören, ist der Auftauvorgang
selber und die Gefrierschutzmittelausverdünnung dem Prozess des Auftauvor-
ganges zuzuordnen.
Der Auftaurate kommt für die spätere Vitalität der Embryonen eine ebenso gro-
ße Bedeutung zu wie der Kühlrate.
WHITTINGHAM et al. (1972) haben Mäuseembryonen mit -0,3°C/min auf -78°C
bis -269°C nach dem langsamen Verfahren eingefroren und tauten sie an-
schließend mit Raten von 4, 25, 215, 450°C/min wieder auf. Die besten Überle-
bensraten erzielten diejenigen Embryonen, die mit den Auftauraten von 4°C/min
bzw. 25°C/min aufgetaut worden waren.

28
Vergleichbare Ergebnisse erhielten WILMUT (1972) aus Untersuchungen mit
Mausembryonen und WILLADSEN et al. (1976) nach Versuchen mit Schafemb-
ryonen. LEIBO & MAZUR (1978) stellten fest, dass langsam eingefrorene Emb-
ryonen auch sehr langsam wieder aufgetaut werden müssen, um eine möglichst
große Überlebensfähigkeit der Embryonen zu erreichen. Das erklärt auch, wa-
rum schnell kryokonservierte, ultraschnell tiefgefrorene und Embryonen nach
Vitrifikation Auftauraten von über 300°C/min benötigen. Bei zu langsamen Auf-
tauraten käme es andernfalls zur intrazellulären Kristallisation und damit zum
Zelltod.
RALL & POLGE führten 1984 Untersuchungen an 8-Zell-Mauseembryonen
durch. Sie equilibrierten die Embryonen in 1,5 M Glycerol und tauten sie nach
vorangegangenem Einfrieren mit unterschiedlichen Raten wieder auf. Dabei
überlebten die Embryonen, die langsam auf -20°C bis -40°C heruntergekühlt,
anschließend direkt in LN2 überführt und mit 2°C/min aufgetaut wurden eine
48-stündige in vitro-Kultivierung nur zu 4 bis 25%. Bei denjenigen Embryonen
aber, die ebenfalls langsam auf -25°C heruntergekühlt wurden, um mit
500°C/min schnell wieder aufgetaut zu werden, überlebten 74% der Embryonen
die in vitro-Kultivierung.
Eine Erklärung für die Abhängigkeit der Überlebensrate von Kühl- und Auftaura-
te liegt in der Fähigkeit der Eiskristalle zur Rekristallisation. Beim schnellen und
ultraschnellen Tiefgefrieren bilden sich viele kleine Eiskristalle, die eine geringe
Oberflächenenergie besitzen und aus diesem Grunde thermisch instabil sind.
Diese verschmelzen beim langsamen Auftauen in wenige aber größere und da-
durch schädigende Eiskristalle. Erfolgt das Auftauen aber ausreichend schnell,
schmelzen die kleinen Kristalle, bevor sie untereinander verschmelzen können
(MAZUR, 1965; BANK, 1973; RALL et al., 1980).
Zum schnellen Auftauen werden die eingefrorenen Pailletten in der Regel in ein
warmes Wasserbad getaucht. ELSDEN et al. (1982) tauten Rinderembryonen
im 25°C bzw. 37°C warmen Wasserbad auf und verglichen anschließend die
Trächtigkeitsraten im Anschluss an einen Transfer dieser Embryonen. Die
Trächtigkeitsraten der bei 37°C aufgetauten Embryonen lagen mit 36,5 % über
den Resultaten der bei 25°C aufgetauten Embryonen (23,5%). 1990 konservier-
ten SEIDEL et al. Rinderembryonen mit einem Kryoprotektivum, dem sie zu-
sätzlich ein Makromolekül zugesetzt hatten. Embryonen, die vor dem Auftauen

29
im Wasserbad sechs Sekunden an der Luft angetaut wurden, ergaben bessere
Trächtigkeitsraten als die Embryonen, die ausschließlich im Wasserbad aufge-
taut worden waren.
WHITTINGHAM berichtete 1981, dass nach Kryokonservierung von 8-Zell-
Mauseembryonen und einem schnellen Auftauen die besten Ergebnisse erzielt
werden, wenn beim Einfrieren das langsame Kühlen bei -40°C durch Umsetzen
in LN2 gestoppt wird.
2.7 Ausverdünnungsmethoden
Nahezu alle Kryoprotektiva wirken bei wärmeren Temperaturen toxisch auf den
Embryo und müssen aus diesem Grunde vor einer in vitro-Kultivierung oder ei-
nem Transfer möglichst vollständig wieder aus den Zellen entfernt werden. Je
nach verwendetem Kryoprotektivum und Equilibrierungsdauer enthalten die
aufgetauten embryonalen Zellen höhere Konzentrationen an Kryoprotektiva.
Kommen die Zellen aus dem Gefrierschutzmedium in physiologische Medien
oder Kulturmedien zurück, schwellen sie wegen des osmotischen Gradienten
nach Wassereinstrom an. Versuche zu Überlebensrate und maximal tolerierba-
re Volumenzunahme von Mausembryonen ergaben, dass Ausverdünnungsver-
fahren gewählt werden sollten, bei denen das Embryonenvolumen zwischen
50% und 200% des isotonischen Volumens liegt (MAZUR & SCHNEIDER,
1986).
2.7.1 Schrittweises Ausverdünnen und Ausverdünnen mit nicht
penetrierenden Kryoprotektiva
Um übermäßiges Anschwellen zu vermeiden kann auf zwei unterschiedliche
Verfahren ausverdünnt werden. Die erste Variante ist das schrittweise Ausver-
dünnen. Dabei reduziert man die den Embryo umgebende extrazelluläre Kon-
zentration des Kryoprotektivums immer weiter indem der Embryo fortlaufend in
Lösungen mit absteigender Konzentration des Kryoprotektivums umgesetzt

30
wird. So kann das Anschwellen des Embryonen in einem, für die Zellen tolerier-
baren Rahmen gehalten werden.
Ein anderes Verfahren verhindert das Überschreiten des isotonischen Volu-
mens, indem zum Ausverdünnen nicht penetrierende Kryoprotektiva verwendet
werden. Durch die nicht penetrierenden Stoffe wird die extrazelluläre Osmolari-
tät konstant gehalten. Das intrazelluläre Kryoprotektivum kann aus den Zellen
herausströmen, ohne dass es zu einem zerstörenden Anschwellen des Zellvo-
lumens kommt. Hat auf diese Weise genügend Kryoprotektivum die Zellen ver-
lassen, kann der Embryo, ohne weitere Schäden durch übermäßiges Anschwel-
len zu nehmen, in isotonisches physiologisches (Kultur-)Medium verbracht wer-
den (LEIBO & MAZUR, 1978; MAZUR, 1985).
Das Ausverdünnen mit nicht penetrierenden Kryoprotektiva wie zum Beispiel
Saccharose bietet zudem die Möglichkeit zu kontrollieren, ob der Embryo die
Kryokonservierung überlebt hat.
Ein vitaler Embryo schwillt in einer Saccharose-Lösung nicht übermäßig stark
an, schrumpf dafür aber fortlaufend, solange das Kryoprotektivum aus dem
Embryo herausdiffundiert. Dieses anfängliche Schrumpfen und anschließende
Anschwellen ist ein Zeichen dafür, dass die Funktionen der Zellmembranen
noch intakt sind (SCHNEIDER & MAZUR, 1984).
2.7.2 One-Step-Ausverdünnung
In diversen Untersuchungen konnte nachgewiesen werden, dass das penetrie-
rende Kryoprotektivum Glycerin mit Hilfe des nicht penetrierenden Kryoprotekti-
vum Saccharose auch in einem Schritt ausverdünnt werden kann. Die beste
Überlebensrate von Embryonen wurde bei Konzentrationen von 1 M Saccharo-
se erzielt. Mit Konzentrationen, die kleiner als 0,8 M Saccharose waren, konn-
ten hingegen keine zufriedenstellenden Überlebensraten mehr erreicht werden
(MERRY et al., 1983; HERNANDEZ-LEDEZMA et al., 1988b; DEL CAMPO et
al., 1990; SUZUKI et al., 1990).
BIELANSKY et al. (1985) untersuchten, inwieweit sich die Überlebensraten von
Rinderembryonen nach Ausverdünnen in einem Schritt mit Saccharose oder in

31
mehreren Schritten ohne Saccharose von einander unterschieden. Sie konsta-
tierten, dass die Überlebensrate bei der Ein-Schritt-Methode höher war.
MAPLETOFT et al. (1987) verglichen die Auswirkungen der Ein-Schritt-
Methode mit dem schrittweisen Ausverdünnen mit Saccharose bei Mäuseemb-
ryonen. LANDSVERK et al. (1992) und ARRESEIGOR et al. (1998) untersuch-
ten die Auswirkungen dieser beiden Methoden auf Rinderembryonen. Dabei
stellten sie fest, dass die zeitaufwendigere Methode der schrittweisen Ausver-
dünnung in mehreren Schritten keinerlei Vorteile gegenüber der One-Step-
Methode aufwies.
Eine erneute Vereinfachung des Ausverdünnens in einem Schritt ist die von
LEIBO (1983) und RENARD et al. (1983) entwickelte One-Step-Methode. Bei
diesem Verfahren wird der in Glycerol kryokonservierte Embryo vor dem Trans-
fer direkt in der Paillette ausverdünnt.
CHUPIN et al. (1984) und HOOGENKAMP (1984) zogen dazu in der Gefrier-
paillette vor dem Kryokonservieren drei unterschiedliche Säulen auf. Die mittle-
re Säule enthielt in einer Glycerolmenge den Embryo. Diese Säule wurde durch
jeweils eine Luftblase von den beiden äußeren Säulen getrennt. Diese äußeren
Säulen bestanden entweder alle beide aus einer Saccharose-Lösung oder die
eine Säule aus einer Saccharose-Lösung und die zweite Säule aus Medium.
CHUPIN et al. (1984) stellten zwischen den Trächtigkeitsraten nach direktem
Transfer bei den beiden verschiedenen Varianten keine signifikante Unter-
schiede fest. HOOGENKAMP (1984) jedoch erreichte mit denjenigen Embryo-
nen, deren Pailletten zwei Säulen Saccharose enthielten, höhere Trächtigkeits-
raten (46%) als mit Embryonen, deren Pailletten eine Säule Medium und eine
Säule Saccharose enthielten (21%).
In Versuchen zu eventuellen Einflüssen der Verweildauer, mit denen die Emb-
ryonen dem Gemisch beider Medien bei 37°C bis zum Transfer konfrontiert
wurden, warteten PAVEL (1985) und NOHNER (1986) nach dem Auftauen und
Schütteln der Pailletten, welches eine Durchmischung der Medien erreichen
sollte, unterschiedlich lange. Bei PAVEL (1985) betrug die Trächtigkeitsrate
nach einer weniger als zehnminütigen Wartezeit nur 18,5%. Nach einer Warte-
zeit von mehr als 10 Minuten stieg die Trächtigkeitsrate auf 100% an. NOHNER
(1986) dagegen stellte fest, dass die Trächtigkeitsrate mit zunehmender Warte-
zeit nach dem Ausverdünnen sank. Erzielte er bei 5 bis 15 min Wartezeit noch

32
eine Trächtigkeitsrate von 44,4%, waren es nach 30 bis 60 Minuten Wartezeit
lediglich noch 28,6%.
Trotz recht guter Trächtigkeitsergebnisse (von 25% bis über 45%) in Feldversu-
chen hat sich die One-Step-Methode in der Routine des Embryonentransfers
noch nicht flächendeckend durchsetzten können (VOELKEL & HU 1992a;
STREICHER 1998).
2.7.3 Direkte Ausverdünnung von Ethylenglycol
Ethylenglycol hat im Vergleich zu Glycerol den Vorteil, dass es auch bei höhe-
ren Temperaturen eine deutlich geringere toxische Wirkung auf Zellen besitzt.
Zusätzlich diffundiert Ethylenglycol wesentlich schneller als Glycerol durch die
Zellmembranen hindurch. VOELKEL & HU (1992a) untersuchten die in vitro-
Überlebensraten von Rinderembryonen nach konventionellem Tiefgefrieren in
Abhängigkeit von vier verschiedenen Kryoprotektiva bzw. direktem Ausverdün-
nen nach Auftauen in PBS. Dabei zeigte sich, dass die Überlebensraten von in
Ethylenglycol kryokonservierten Embryonen die Überlebensraten von in Glyce-
rol, DMSO oder Propandiol tiefgekühlten Embryonen überstiegen.
Diese Ergebnisse zeigten, dass das Zeit- und Materialaufwendige schrittweises
Ausverdünnen der Kryoprotektiva nach dem Auftauen nicht unbedingt notwen-
dig ist. JANOWITZ & GÖRLACH (1994) hielten fest, dass die Möglichkeit gege-
ben ist, die Embryonen im Anschluss an das Auftauen ohne Schäden direkt zu
übertragen.
Untersuchungen von ARRESEIGOR et al. (1998) kamen zu dem Ergebnis,
dass die Trächtigkeitsraten (TR) nach einem Direkttransfer von in Ethylenglycol
kryokonservierten Rinderembryonen mit denen in Glycerol tiefgefrorenen
Embryonen, die dann allerdings in drei Schritten ohne Saccharose oder einem
Schritt mit Saccharose ausverdünnt worden waren (47,4% TR gegenüber 45,9
bzw. 47,2% TR), zu vergleichen waren.
Durch die guten Trächtigkeitsergebnisse und die stark vereinfachte Handha-
bung ist dieses One-Step-Verfahren von der Routine in größerem Masse ange-
nommen worden.

33
2.8 Kultivierung von Embryonen
Das Weiterentwicklungsvermögen von Embryonen unter Kulturbedingungen ist
ein gutes Kriterium, um die Vitalität von Embryonen zu bewerten (SCHNEIDER
et al., 1974; TROUNSON et al., 1978b; NIEMANN, 1980).
POLGE & WILLADSEN (1978) wie auch LEHN-JENSEN et al. (1981) stellten
nach einer 24-stündigen in vitro Kultivierung von kryokonservierten und an-
schließend wieder aufgetauten Rinderembryonen bei 37°C keine signifikanten
Unterschiede zwischen in vitro und in vivo Entwicklungsraten. TROUNSON et
al. (1976a, 1978) und MASSIP et al. (1979) hingegen stellten fest, dass die
Trächtigkeitsraten deutlich verringert waren, wurden die Embryonen vor dem
Transfer einer in vitro Kultivierung ausgesetzt.
Eine erste erfolgreiche Kultivierung von Mäuseembryonen gelang HAMMOND
1949. Er kultivierte in seinen Versuchen Mäuseembryonen vom 8-Zellstadium
bis zur Blastocyste in einem biologischen Medium, das aus den anorganischen
Salzen NaCl, KCl, CaCl2, MgCl2 und NaH2PO4 bestand. Desweiteren setzte er
noch Glucose, 5 % Hühnereiweiß sowie Hühnereidotter zu.
WHITTEN (1956) war der erste, der über einen erfolgreichen Einsatz eines
chemischen Mediums berichtete. Bei dem Medium handelte es sich um eine
Krebs-Ringer-Lösung, in der das Hühnereiweiß durch bovines Serumalbumin
(BSA) ersetzt worden war. McLAREN & BIGGERS (1958) bewiesen, dass sich
in diesem Medium kultivierte Embryonen nach einem Transfer auf Empfänger-
tiere zu ganz normalen Föten weiterentwickelten.
BRINSTER (1963) stellte fest, dass es nach Zusatz von Laktat und Pyruvat zum
Kulturmedium sogar möglich ist Embryonen ab dem 2-Zellstadium zu kultivie-
ren.
Ebenfalls BRINSTER (1965) beschrieb, dass ein Wachstum von Mäuseembry-
onen vom 2-Zellstadium bis zum Stadium Blastocyste in einem Bereich von 200
bis 354 mOsmol und einem pH-Wert von 5,87 bis 7,78 möglich sei. Als opti-
malste Bedingungen ermittelte er eine Osmolalität von 276 mOsmol und einen
pH-Wert von 6,82.

34
Für eine in vitro Kultivierung von Embryonen wird am häufigsten die Mikrotrop-
fenmethode unter Öl angewandt. Dabei werden ein bis mehrere Mikrotropfen
Kultivierungsmedium (ca. 50 bis 100 µl) in einer Petrischale platziert und mit
Mineralöl überschichtet. Durch das Öl wird das Medium vor einer Verdunstung
geschützt; ein Gasaustausch mit der umgebenden Gasphase ist weiterhin mög-
lich (BRINSTER, 1963).
Während einer in vitro Kultivierung müssen laufend die folgenden Parameter
kontrolliert werden: Temperatur, Luftfeuchtegehalt und CO2-Gehalt in der Gas-
phase. Als optimale Werte für eine Mäuse- und Kanincheneizellen-Kultivierung
wurden von ALLISTON et al. (1965), ADAMS (1968) und BRINSTER (1969) ei-
ne Temperatur von 37°C, eine nahezu 100 %ige rel. Luftfeuchte sowie ein CO2-
Partialdruck von 5 % angesehen.
2.9 Beurteilungsmöglichkeiten für Embryonen
Es gibt die unterschiedlichsten Verfahren, um Embryonen in ihrem Erschei-
nungsbild zu beurteilen. Die einfachste und häufigste Methode ist die morpho-
logische Beurteilung, in der Regel unter zu Hilfenahme eines Mikroskopes. Wei-
terhin sind verschiedene Arten der Anfärbung von Embryonen und Zellen ent-
wickelt worden, durch die die Möglichkeit besteht auch morphologisch nicht
sichtbare, aber die Qualität beeinflussende Kriterien, sichtbar zu machen.
2.9.1 Morphologische Beurteilung
Die morphologische Beurteilung ist die wahrscheinlich am wenigsten aufwendi-
ge Methode Zellen und Zellkomplexe zu beurteilen. Eine Klassifizierung der
Embryonenqualität erfolgt mit mikroskopischer Hilfe durch eine subjektive Beur-
teilung in Anlehnung an die Verfahren von LINDNER & WRIGHT (1983) sowie
KAUFFHOLDT & THAMM (1985).
Die subjektive Beurteilung ist dabei der kritische Punkt in diesem Verfahren, da
eine genügend große Erfahrung im Erkennen von guten und schlechten Emb-
ryonen und deren verschiedener Stadien notwendig ist.

35
Entscheidende Kriterien bei der Bewertung sind Faktoren wie die Unversehrt-
heit der Zona pellucida, das Größenverhältnis der Blastomeren untereinander,
die Anzahl und Kompaktheit der Blastomeren. Auch sollte ein Entwicklungssta-
dium, das dem Zeitpunkt der Gewinnung entspricht, vorgefunden werden. Ist
dieses nicht der Fall, sollten die Embryonen als retardiert und damit als ein ge-
schädigter, nicht zu verwendender Embryo angesehen werden (MAURER, &
HASEMANN, 1976; TROUNSON et al., 1976a/b; ELSDEN et al., 1978; BO-
LAND et al., 1978).
2.9.2 Beurteilung nach Fluoreszenzanfärbung mittels FDA
Um eine Vitalitätsbestimmung von Embryonen mit Farbstoffen durchführen zu
können, ist es notwendig, dass der verwendete Farbstoff eine exakte Beurtei-
lung erlaubt und die Entwicklungsfähigkeit der Embryonen nicht verringert wird.
Ein fluoreszierender Farbstoff der dieses gewährleisten soll ist Fluorescein-3`6`-
diacetat, bekannt als FDA. Erste Berichte über die Verwendung von FDA zur
Überprüfung der Zellvitalität wurden 1966 durch ROTMAN & PAPERMASTER
veröffentlicht. Ende der siebziger Jahre beschrieben SCHILLING et al. die Mög-
lichkeiten einer Bestimmung der Vitalität durch FDA (SCHILLING & DÖPKE,
1978; SCHILLING et al., 1979; SCHILLING & SMIDT, 1979). Anhand des FDA-
Verfahrens kann einerseits die Integrität der Plasmamembranen, andererseits
auch eine positive zytoplasmatische Enzymaktivität nachgewiesen werden. Im
Anschluss an eine relativ kurze Inkubationsdauer im Fluoreszenzfarbstoff wei-
sen lebende Embryonen nach einer UV-Anregung eine leuchtende, hellgrüne
Farbe auf. Tote Embryonen dagegen lassen keine Reaktionen mehr erkennen.
Nach Meinung der zu diesem Thema veröffentlichten Berichte, besteht nach ei-
ner Färbung keine Verringerung der in vitro-Weiterentwicklungsrate, verursacht
durch Färbung und kurzfristige UV-Bestrahlung.

36
2.9.3 Beurteilung nach Fluoreszenzanfärbung durch Hoechst A
33342
Hinter dem Namen Hoechst A 33342 steht der Fluoreszenzfarbstoff Benzimid,
genauer 2`-[4Hydroxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5´bi-1H-bizimidazole.
Das Fluorochrome Hoechst A 33342 ist ein DNA-spezifischer Farbstoff, mit dem
es möglich ist, das Chromatin in Oozyten und Embryonen sichtbar zu machen.
Durch diese Färbemethode kann im Anschluss an eine morphologische Beurtei-
lung eine Aussage über die Anzahl aktiver Kerne und damit die Vitalität der
Embryonen gemacht werden (VIUFF et al., 1991). Bei UV-Bestrahlung werden
mitotisch aktive Kerne als leuchtend hellblaue und scharf umrandete Komplexe
erkennbar. Kerne ohne Aktivität sind nur undeutlich und ohne klare Konturen
schwach erkennbar.

37
3 Material und Methoden
3.1 Tiermaterial
Die vorliegenden Untersuchungen wurden unter Verwendung dreier verschie-
dener Mäuseherkünfte durchgeführt:
1. 24 weibliche Tiere entstammten der Linie C57/B6 (Black) aus der Zucht
des Max-Planck-Instituts für experimentelle Medizin.
2. 36 weibliche Tiere entstammten der Linie NMRI (Albino) aus der Zucht
des Max-Planck-Instituts für experimentelle Medizin.
3. 72 weibliche Tiere entstammten der Linie NMRI (Albino) aus institutseige-
ner Zucht.
3.2 Haltungsbedingungen
Die Tiere wurden in einem 10 m2 großen klimatisierten Raum gehalten, in dem
mittels Heizung und Klimaanlage die Temperatur bei 22°C bis 25°C gehalten
wurde. Durch einen Luftbefeuchter und offene Wasserwannen wurde eine rela-
tive Luftfeuchte von 50 - 60 % erreicht. Das Lichtprogramm von je 12 Stunden
Hell-/Dunkelphase wurde durch eine Zeitschaltuhr überwacht (Lichtphase von
7.00 bis 19.00 Uhr).
Die für Versuchszwecke benötigten weiblichen Tiere wurden in Gruppen von
mindestens zwei bis maximal zehn Tieren in Polycarbonatkäfigen von
17×23×14 cm Größe (Typ II) bzw. 23×39×15 cm Größe (Typ III) mit Stahldraht-
deckeln (Ebeco, Castrop-Rauxel) gehalten.
Die männlichen Tiere wurden einzeln in entsprechenden Käfigen (17×23×14
cm) gehalten.
Mindestens einmal wöchentlich wurden alle Tiere in frisch gereinigte Käfige mit
frischer Sägespäne-Einstreu umgesetzt.
Als ad-libitum-Fütterung war ein pelletiertes Mäusealleinfutter (Tab. 1, ssniff
Spezialdiäten GmbH, Soest) verfügbar. Trinkwasser erhielten die Tiere aus 25
ml Plastikflaschen mit Tränkdeckel, die mindestens einmal wöchentlich ausge-
tauscht wurden.

38
Tab. 1: Zusammensetzung Mäusealleinfutter
Inhaltsstoff % Zusatzstoffe Menge
Rohprotein 19,00 Vit.A 15.000 IE/kg
Rohfett 3,30 Vit.D3 1.000 IE/kg
Rohfaser 4,90 Vit.E 100 IE/kg
Rohasche 6,70 CU 12 mg/kg
3.3 Medien
Alle Chemikalien wurden, soweit in der Arbeit nicht anders ausgewiesen, von
der Firma Sigma-Aldrich, Steinheim, bezogen und waren zellkulturgetestet.
3.3.1 Spülmedium
Die zur Herstellung und anschließenden Aufbewahrung des Embryonenspül-
mediums M2 benötigten Glasgefäße wurden vor ihrer Verwendung zuerst mit
70 %igem Alkohol gereinigt und danach 5 x mit destilliertem Wasser aus- und
abgespült und im Trockenschrank (BE 40, Memmert, Schwabach) bei 55°C ge-
trocknet. Nach vollständiger Trocknung wurden sie mit Alufolie fest verschlos-
sen und unter trockener Hitze im Sterilisator (U 50, Memmert, Schwabach) für
mindestens 2h bei 200°C sterilisiert.
Zur Herstellung des Spülmediums M2 wurden verschiede Anteile der Stammlö-
sungen A, B, C, D, E (Tab. 2) und Reinstwasser (Ampuwa®, Fresenius, Bad
Homburg) verwendet.
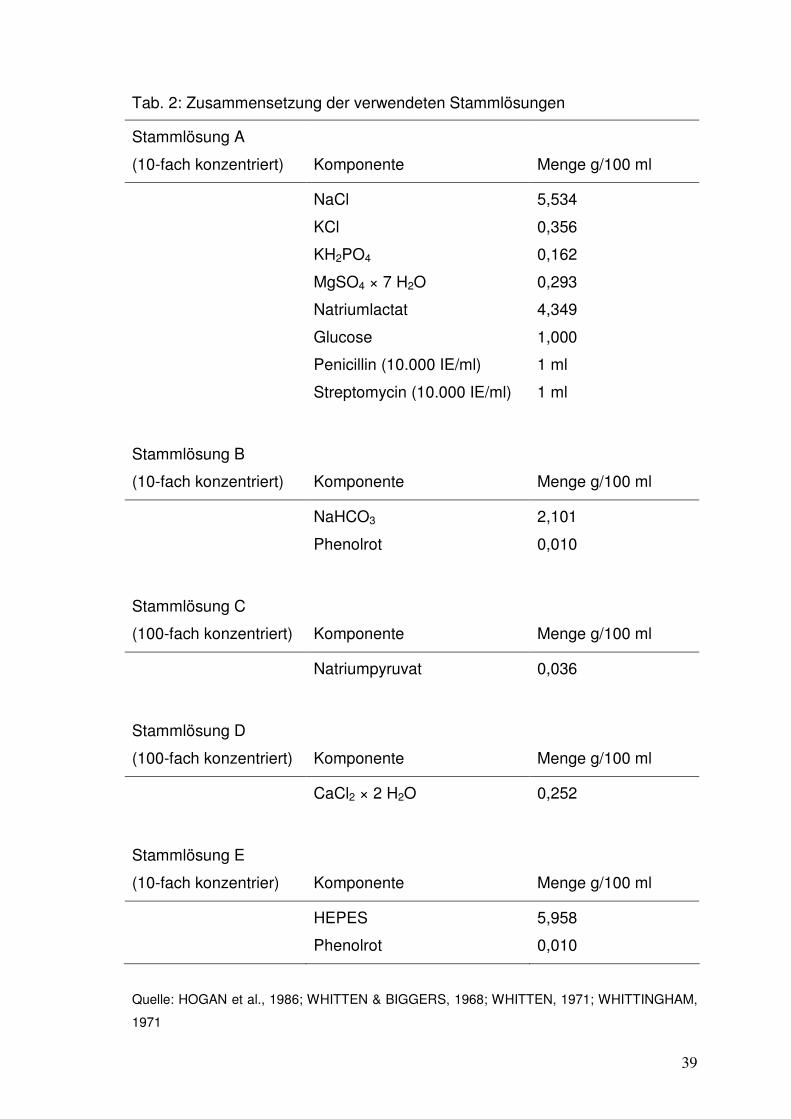
39
Tab. 2: Zusammensetzung der verwendeten Stammlösungen
Stammlösung A
(10-fach konzentriert) Komponente Menge g/100 ml
NaCl
KCl
KH2PO4
MgSO4 × 7 H2O
Natriumlactat
Glucose
Penicillin (10.000 IE/ml)
Streptomycin (10.000 IE/ml)
5,534
0,356
0,162
0,293
4,349
1,000
1 ml
1 ml
Stammlösung B
(10-fach konzentriert) Komponente Menge g/100 ml
NaHCO3
Phenolrot
2,101
0,010
Stammlösung C
(100-fach konzentriert) Komponente Menge g/100 ml
Natriumpyruvat 0,036
Stammlösung D
(100-fach konzentriert) Komponente Menge g/100 ml
CaCl2 × 2 H2O 0,252
Stammlösung E
(10-fach konzentrier) Komponente Menge g/100 ml
HEPES
Phenolrot
5,958
0,010
Quelle: HOGAN et al., 1986; WHITTEN & BIGGERS, 1968; WHITTEN, 1971; WHITTINGHAM,
1971

40
Fertige Stammlösungen wurden unter der sterilen Werkbank (Mikrobiologische
Sicherheitswerkbank Typ MRF, Steag Laminarflow-Prozeßtechnik, Pfullingen)
in 100 ml-Flaschen mit einem Einmalfilter steril filtriert (Filtropur S 0,2 µm, Sar-
stedt, Nümbrecht) und die ersten 5 ml der Filtrationen jeweils verworfen. An-
schließend wurden die Stammlösungen in sterile 1 ml-Eppendorfgefäße (Ep-
pendorf, Hamburg) pipettiert und bei -18°C im Gefrierschrank gelagert.
Die Stammlösungen A, D und E hielten sich aufgetaut bei +4°C über einen Zeit-
raum von drei Monaten, die Stammlösungen B und C jedoch nur eine Woche.
Daraus ergab sich eine maximale Haltbarkeit des gebrauchsfertig angesetzten
M2-Spülmediums von einer Woche bei +4°C.
Den benötigten Stammlösungsanteilen entsprechend wurde das M2-Medium
am Tag vor Gebrauch unter der sterilen Werkbank angesetzt und bis zur Ver-
wendung bei +4°C aufbewahrt.
Tab. 3: Zusammensetzung des Spülmediums M2
Komponenten Menge je 50 ml M2
Stammlösung A (5-fache Konzentration) 10,0 ml
Stammlösung B (10-fache Konzentration) 0,80 ml
Stammlösung C (100-fache Konzentration) 0,50 ml
Stammlösung D (100-fache Konzentration) 0,50 ml
Stammlösung E (10-fache Konzentration) 4,20 ml
H2O (Ampuwa®) 34,0 ml
BSA (Hitzeinaktiviert) 0,20 g
Nach Ansetzten des Mediums wurde die Osmolalität ermittelt. Dabei soll die
Osmolalität zwischen 270-302 mOsmol/kg-1 liegen (BOXBERGER, 2007).
(Dieser Wert gibt bei Flüssigkeiten die Teilchenzahl der osmotisch aktiven Sub-
stanzen pro Kilogramm Lösemittel an. Die Osmolalität, also das dynamische
Gleichgewicht von Wasser und Salzen ist von entscheidender Bedeutung für
die Aufrechterhaltung der Zellstruktur, die Anregung der Zellproliferation und
muß daher möglichst genau eingehalten werden (BOXBERGER, 2007). Sie er-
rechnet sich nach der Formel:
X = Q × (a-b) × a (KOPF, 1988)

41
wobei Q = Mediummenge (ml)
a = gemessene Osmolalität (mOsmol/kg-1)
b = gewünschte Osmolalität (mOsmol/kg-1)
X = Mediummenge (ml), die zur Korrektur entnommen und verworfen werden
muß und durch hochreines Wasser ersetzt wird.)
Fertiges M2-Medium (HEPES-gepuffert) wurde unter dem Laminarflow in sterile
Flaschen filtriert (Filtropur S 0,2 µm, Sarstedt, Nümbrecht), mit autoklavierten
Gummistopfen und Parafilm (American National Can TM, Greenwich) versiegelt
und im Kühlhaus bei +4°C bis zur Verwendung am nächsten Tag aufbewahrt.
(HEPES (4-(2-Hydroxyethyl)-1-piperazinthansulfonsäure) besitzt eine gute Puf-
ferkapazität, hat allerdings in höheren Konzentrationen zytotoxische Effekte und
ist stark von der Temperatur abhängig (BOXBERGER, 2007))
Vor der Verwendung wurde am Versuchstag mittels einer Glas-Elektrode (WTW
pH-Elektrode SenTix 60, WTW, Weilheim i. OB) der pH-Wert des M2-Mediums
gemessen (Digital-pH-Meter pH 525, WTW, Weilheim i.OB) und durch Zufü-
gung von HCl bzw. NaOH auf pH 7,3 bis 7,4 eingestellt. Anschließend wurde
das gebrauchsfertige Medium erneut in sterile 50ml-Glasflaschen filtriert, dann
erst die BSA Komponente hinzugefügt und mit autoklavierten Gummistopfen
und Parafilm verschlossen und im Wärmeschrank (Typ 85, Melag Brutschrank
Incubat, Berlin) ohne CO2 Begasung auf 37°C erwärmt.
3.3.2 Kulturmedium
Zur Kultivierung der aufgetauten Embryonen wurde gebrauchsfertig bezogenes
Universal IVF Medium mit Phenolrot (Medicult a/s, DK-Jyllinge, Fa. Stefan
Gück) verwendet.

42
Tab. 4: Zusammensetzung des Kulturmediums MediCult Universal IVF Medium
Komponenten
Earle’s Balanced Salts Solution (EBSS)
Synthetic Serum Replacement (SSR®) (USA: ART Sup-
plement)
Human serum albumin (HSA)
Glucose
Sodium pyruvate
Sodium bicarbonate
Penicillin 50.000 IE/l
Streptomycin 50 mg/l
Phenolrot
(Quelle: Fa. MediCult; Jyllinge; DK)
3.3.3 Kryoprotektivum
Um Gefrierschäden an den Embryonen so gering wie möglich zu halten, wurde
ein Gefrierschutzmedium mit den Substanzen Ethylenglycol (EG) (Sigma-
Aldrich, Steinheim) und Saccharose (Sigma-Aldrich, Steinheim) verwendet. E-
thylenglycol, als sehr leicht penetrierendes Protektivum, wurde in einer Kon-
zentration von 1,5 Mol, Saccharose, als nicht penetrierendes Protektivum, in
der Konzentration von 0,25 Mol zugefügt.
Die nötigen Mengen an EG und Saccharose wurden unter dem Laminarflow zu
M2-Medium zugegeben, mit einem Magnetrührer (Heidolph, Schwabach) ge-
löst, anschließend steril in Glasfläschen (20ml) filtriert (Filtropur S 0,2 µm, Sar-
stedt, Nümbrecht) und mit autoklavierten Gummistopfen und Parafilm ver-
schlossen.
Die Haltbarkeit des Kryoprotektivum beträgt bei +4°C maximal eine Woche, es
wurde aber vor jeder Verwendung neu angesetzt.
Bis zum Gebrauch wurde das Medium bei Raumtemperatur (RT) gehalten.

43
3.3.4 Auftaumedium
Als jeweils frisch angesetztes Auftaumedium diente Universal IVF Medium (Me-
diCult, DK-Jyllinge, Fa. Stefan Gück) versetzt mit 0,5 Mol Saccharose (Sigma-
Aldrich, Steinheim).
Die Menge an Saccharose wurde auf einer Feinwaage (2007 MP6, Sartorius,
Göttingen) abgewogen und unter Rühren mit einem Magnetrührer in Universal
IVF Medium gelöst. Das Auftaumedium wurde unter dem Laminarflow steril in
20ml Mediumflaschen filtriert. Gleich im Anschluss an den Filtrationsprozess
wurden 350µl Auftaumedium in die Mitte einer Center-Well Schale (60×15mm,
Falcon, Heidelberg) pipettiert; diese wurden auf dem 37°C warmen Werktisch
(Easy Bench, Varolab, Giessen) (Abb. 1) erwärmt.
Abb. 2: Werktisch mit integriertem Wärmebereich (Varolab, Giessen) und CO2
Kleininkubator C42 (Labotect)
3.3.5 Überschichtungsöl für Kultivierung- und Waschdrops
Als Überschichtungsöl für das verwendete Kulturmedium diente Mineral-Öl
(Sigma-Aldrich, Steinheim). Auf diese Weise wurde vermieden, dass Mikro-
Medium-Drops austrocknen. Weiterhin können Kontaminationen durch die Öl-
barriere reduziert werden. Es erfolgte 24 Stunden vor der Verwendung des Öles
eine Ölwaschung zur Equilibrierung des Öles mit dem Medium. Das hatte zur
Folge, dass eventuell vorhandene Schadstoffe im Öl in das Medium ausgewa-
schen wurden und gleichzeitig eine Anreicherung der fettlöslichen Vitamine aus
dem Medium in das Öl erfolgte, so dass während der Kultivierungsphase die Vi-

44
tamine und essentiellen Aminosäuren tatsächlich den Embryonen zur Verfü-
gung standen. Dazu wurde ein steriles 50ml Falcon-Röhrchen (Sarstedt, Nüm-
brecht) mit 20ml MediCult Universal IVF Medium und 30ml Mineral-Öl unter ste-
rilen Bedingungen befüllt. Anschließendes behutsames Schwenken des Röhr-
chens führte zu einer Vermischung beider Substanzen. Bis zum Gebrauch des
Öles wurde das Röhrchen mit lockerem Deckel zur Equilibrierung und Entmi-
schung der Phasen in den Brutschrank (C60, Labotect, Göttingen) bei 37°C, 6%
CO2 und gesättigter Luftfeuchte (LF) gestellt.
3.4 Behandlung der Embryonenspender
Zur Embryonengewinnung wurden die Spendertiere hormonell einer Superovu-
lation unterzogen. Dazu wurde den Weibchen im Alter von fünf bis 16 Wochen
47 Stunden vor dem Zusammensetzten der männlichen und weiblichen Tiere
zur Auslösung einer Superovulation 7,5 IE eCG (equine chorionic gonadotropin,
Intergonan, Intervet, Unterschleißheim) intraperitoneal (ip) an der Linea alba zur
Follikelstimulation injiziert (Abb. 2). 5,0 IE hCG (human chorionic gonadotropin,
Ekluton, Intervet, Unterschleißheim) wurden direkt vor dem Zusammensetzen
der Tiere ip injiziert.
Zu Beginn des Versuches wurde 24 Spendertieren um 17.00 Uhr eCG und 47
Stunden später um 16.00 Uhr hCG injiziert. Bei allen restlichen Tieren wurde
die eCG-Gabe auf 15.00 Uhr und die hCG-Gabe auf 14.00 Uhr vorgezogen.
Abb. 3: Ip-Injektion von eCG bzw. hCG

45
Da nach EDWARDS & GATES (1959) die Ovulation ungefähr 12 Stunden nach
Verabreichung des hCG erfolgt, wurden die Embryonenspender im direkten An-
schluss an die ip Injektion von 5,0 IE hCG einzeln zu einem Bock im Alter von
drei bis elf Monaten zur Verpaarung gesetzt. Bei allen weiblichen Tieren kamen
Böcke der NMRI-Linie aus MPI-Herkunft oder institutseigener Zucht zum Ein-
satz. Die Verpaarung erfolgte in der anschließenden Nacht.
Am folgenden Morgen konnte als Beweis für eine erfolgreiche Bedeckung bei
den gedeckten Weibchen ein Ejakulationspfropf (Vaginal-Plaque, VP) gefunden
werden. Für diese Kontrolle wurden die Spendertiere auf das Käfiggitter gesetzt
und am Schwanz angehoben. Der VP zeichnete sich als weißlich-gelblicher,
harter Pfropf im vorderen Abschnitt der Vagina ab (Abb. 3).
1) 2)
Abb. 4: Vaginal-Plaque: 1) positiv und 2) negativ am Morgen nach erfolgter Be-
deckung
Ein VP bildet sich etwa 20 bis 30 Minuten nach dem Deckakt, ist für ca. 12
Stunden sichtbar und besteht aus Seminalplasma und abgeschilferten Zellen
der Vaginalschleimhaut (HEINEKE & KLAUS, 1975). Da der Deckakt in der Re-
gel in der zweiten Hälfte der Dunkelphase stattfindet, ist der VP bis zum nächs-
ten Morgen zu erkennen. Beim vorliegenden Versuch fand die VP-Kontrolle und
Markierung der 'VP+'-Tiere im Zeitraum von 8.00 bis 9.00 Uhr an dem auf die
Verpaarung folgenden Morgen statt.
Als Tag 1 der Embryonalentwicklung wurde der Tag gewertet, am dem der VP
zu erkennen war (Tab. 5). Alle Tiere, sowohl VP+ also auch VP-, wurden bis zur
Embryonenspülung drei Tage später in ihren gewohnten Gruppen gehalten, um
für die Tiere keinen unnötigen Stress zu erzeugen.

46
Tab. 5: Behandlungsschema zur Gewinnung von Mäuseembryonen
Tag Behandlung Entwicklungsstadium der
Embryonen
Lokalisation der Embry-
onen
-2 eCG
-1 ―
0 hCG + ♂
+1 VP- Kontrolle 1- Zeller Eileiterampulle
+2 2- bis 4- Zeller Eileiter
+3 8- Zeller bis frühe Morula Eileiter und Uterushorn
+4 OP (Spülen) Morula bis Blastocyste Uterushorn
3.5 Gewinnung der Embryonen
In Abhängigkeit vom gewünschten Entwicklungszustand der Embryonen sind
unterschiedliche Spülzeiten und -orte zu beachten (s. Tab. 5). Um eine hohe
Überlebensrate der Embryonen nach dem Auftauen zu gewährleisten, sollten
die Stadien späte Morula bis späte Blastocyste verwendet werden (TAO et al.,
2001). Aus diesem Grund erfolgte die Embryonengewinnung am vierten Träch-
tigkeitstag zwischen 10.00 und 12.00 Uhr.
Die Tiere wurden durch zervikale Dislokation getötet (Abb. 4) und auf einer
37°C warmen Wärmeplatte (MEDAX SP14, MEDAX Nagel GmbH, Kiel) in Rü-
ckenlage positioniert. Das Operationsfeld wurde mit 70%igem Alkohol desinfi-
ziert, auch um zu verhindern, dass Fellbestandteile in die Operationsöffnung
gelangen (RAFFERTY, 1970).
Durch einen quer zur Linea alba in Höhe der Kniegelenke verlaufenen Haut-
schnitt wurde der Bauchraum geöffnet und durch manuelles Reißen großflächig
eröffnet (Abb. 5).

47
Abb. 5: Zervikale Dislokation
Abb. 6: Eröffnete Bauchhöhle
Darm und Bauchfett wurden seitlich verlagert, die beiden Uterushörner, als
weißlich bis rötliche Stränge erkennbar, freipräpariert und mit einer zweiten ste-
rilen Schere und sterilen Pinzette angehoben. Fettgewebe wurde vorsichtig ab-
getrennt und die Uterushörner mit der Schere zwischen Ovidukt und Ovar, so-
wie an der Bifurcatio uteri, entfernt. Anschließend wurden die Uterushörner in
einer Petrischale (35×10mm, Greiner, Frickenhausen) in 2 ml frischem, sterilfilt-
riertem Spülmedium M2 auf einer 37°C warmen Wärmeplatte (MEDAX Nagel
GmbH, Kiel sowie HT 200, Minitüb, Landshut) gelagert.
Direkt vor dem Spülprozeß wurden die Uterushörner zweimal in frischem M2-
Medium gespült, um eventuell noch anhaftende Verunreinigungen zu entfernen.
Der Eileiter wurde unter dem Stereomikroskop (M8, Wild, Heerebrugg, Wetzlar)

48
bei 100- bis 200-facher Vergrößerung mit einer sterilen Pinzette fixiert und mit
einer Skalpellklinge abgetrennt.
Das Ausspülen der Embryonen erfolgte dann in einer Petrischale (35×10mm,
Greiner, Frickenhausen) unter Kontrolle des Stereomikroskops auf der auf 37°C
eingestellten Heizplatte bei 300-facher Vergrößerung. Dazu wurde das zu spü-
lende Uterushorn mit einer Pinzette fixiert und nach Einführung der, auf eine 2-
ml Einwegspritze (B-D Plastipak®, Becton Dickinson, Irland) aufgesetzten Kanü-
le (0,3×1,0 cm, 2R2, Unimed, CH-Lausanne), von der Hornspitze in Richtung
Uteruskörper mit 1,0ml M2-Medium durchspült. Die Spülflüssigkeit wurde in ei-
ner Petrischale (35×10mm, Greiner, Frickenhausen) aufgefangen und nach ei-
ner kurzen Verweildauer unter dem Stereomikroskop bei 200- bis 500-facher
Vergrößerung auf vorhandene Embryonen durchsucht.
Alle gewonnenen Embryonen wurden nach morphologischen Kriterien (Kap.
2.9.1) beurteilt und die als gefriertauglich befundenen anschließend in einer mit
2 ml frischem Spülmedium (37°C) gefüllten Petrischale gesammelt und bis zum
Einfrieren im Wärmeschrank (Typ 85, Melag Brutschrank Incubat, Berlin) bei
37°C und gesättigter LF verwahrt.
Während des gesamten Versuches dienten zur Handhabung der Embryonen,
über einem Bunsenbrenner fein ausgezogene, sterile Pasteurpipetten, die an
einen etwa 50 cm langen Mundschlauch aus Silikon und zwischengeseztem
Einmalfilter adaptiert wurden.
3.6 Beurteilung der gewonnenen Embryonen
Da nach ATTRODT (1985) die Qualität der Embryonen ein wichtiges Kriterium
für die späteren Trächtigkeits- und Überlebensraten ist, sollten nach Möglichkeit
nur gute und sehr gute Embryonen kryokonserviert werden.
Im Anschluss an eine Kontrolle unter dem Stereomikroskop bei 500-facher Ver-
größerung erfolgte, je nach Erscheinungsbild, die Klassifizierung der gefunde-
nen Embryonen (Morulae und Blastozysten) in:
1. „gefriertauglich“: Embryonen, die im gewünschten Entwicklungsstadium
waren und intakte Morphologie, intakte Zona pellucida, gleichmäßige
Form und Struktur und gleichmäßiger Färbung aufwiesen.

49
oder
2. „nicht gefriertauglich“: Embryonen, entweder nicht im gewünschten Ent-
wicklungsstadium und/oder mit morphologisch erkennbaren Abweichun-
gen (z.B. Vakuolenbildung, defekte Zona pellucida, Unterschiede in den
Blastomeren).
3.7 Einfrieren der Embryonen
Für das Einfrieren der Embryonen fanden zwei programmgesteuerte Gefrierau-
tomaten Verwendung. Zu Beginn der Versuche wurde die Anlage der Firma
Haake (Kryosta, Typ F-3Q, Haake, Berlin) mit angeschlossenem Programmge-
ber (Typ PG 20, Haake, Berlin) (Abb. 6) verwendet. Als Kühlflüssigkeit diente
für dieses Gerät 70%iges Ethanol. Im weiteren Verlauf des Versuches wurde
auf Grund der vereinfachten Handhabung auf das Einfriersystem BV 65/55
(Consartic, Schöllkrippen), bestehend aus Temperaturregler CL 5500, Gefrier-
kammer FC5AT und Cryobad (alle Einheiten: Consartic, Schöllkrippen), ge-
wechselt (Abb. 7). Als Kühlflüssigkeit wurde in diesem Fall flüssiger Stickstoff
(LN2) verwendet.
Abb. 7: Einfriersystem Firma HAAKE, Berlin
Abb. 8: Einfriersystem Firma CONSARTIC, Schöllkrippen

50
3.7.1 Standartgefrierverfahren
Mittels des Standartgefrierverfahrens wurden die als gefriertauglich eingeordne-
ten Embryonen unter Verwendung des Kryoprotektivums (1,5 Mol EG und 0,25
Mol Saccharose in M2-Medium) eingefroren
Dazu wurde die Embryonen mit der Mundpipette unter dem Stereomikroskop
aus dem M2-Medium herausgenommen und in 10er-Gruppen in eine mit 350 µl
Gefrierschutzmedium gefüllte Center-well-Schale (60×15mm, Falcon, Heidel-
berg) umgesetzt.
3.7.1.1 Haake
Bei Verwendung des HAAKE-Einfrierautomaten verblieben die Embryonen für
20 Minuten bei Raumtemperatur (RT) auf der 37°C warmen Wärmeplatte (HT
200, Minitüb, Landshut) zum Equilibrieren im Kryoprotektivum. Kurz vor Ablauf
dieser Frist erfolgte das Aufziehen der equilibrierten Embryonen auf mit Ent-
wicklungsstadium, Spezies, Spültag und Stückzahl gekennzeichnete 0,25 ml
Kunststoffpailletten (Minitüb, Landshut) (Abb. 9). Dazu wurde zunächst eine
Mediumssäule von etwa 2cm, gefolgt von einer 1cm langen Luftsäule aufge-
nommen. Als nächstes kam das die Embryonen beinhaltende Kryoprotektivum,
wiederum gefolgt von einer 1cm langen Luftblase und einer abschließenden
Mediumssäule. Die letzte Kryoprotektivumssäule wurde solange aufgezogen,
bis am oberen Ende der Stopfen erreicht war. Die Luftblasen übernehmen da-
bei die Funktion eines Puffers und verhindern dadurch sowohl das Ansaugen
der Embryonen in den Stopfen, als auch ein versehentliches Kristallisations-
punktsetzen im Embryonen beinhaltenden Medium (Abb. 8).

51
Abb. 9: Befüllungsschema Kunststoffpaillette
Abb. 10: Kunststoffpaillette mit Entwicklungsstadium, Spezies, Spültag und
Stückzahl
Die Pailletten wurden abschließend am unteren Ende mit Hämatokrit-
Versiegelungskitt (Brandt, Wertheim) fest verschlossen und anschließend senk-
recht in das auf -6°C vorgekühlte Alkoholbad des Einfrierautomaten gegeben.
Nach fünf Minuten erfolgte manuell die Kristallisationsauslösung (Seeding).
Dieses geschah am oberen Ende der Paillette, am zwischen Stopfen und Luft-
blase eingeschlossenen Gefriermediumssegment, durch kurzes Berühren mit
einer in LN2 gekühlten Metallpinzette. Als Kontrolle für korrektes Seeding war
eine sich vom Berührungspunkt ausgehende Kristallisation zu beobachten. Im
Anschluß an das Seeding wurde die Temperatur noch für fünf weitere Minuten
bei -6°C gehalten. Nach dem Umstellen auf den externen Programmgeber und
starten desselben wurde das Alkoholbad mit einer konstanten Kühlrate von
0,3°C/min nun auf -32°C heruntergekühlt und nach Erreichen dieser Tempera-
turstufe für 10 Minuten dort gehalten. Danach erfolgte mit einer in LN2 gekühl-
ten Metallpinzette die direkte Umsetzung der Embryonen in LN2 zur weiteren
Lagerung.
3.7.1.2 Consartic
Bei Verwendung des CONSARTIC-Einfrierautomaten verblieben die Embryo-
nen für 7 Minuten bei Raumtemperatur (RT) auf der 37°C warmen Wärmeplatte
Versiegelungskitt
Luftblase Luftblase
Stopfen Medium mit Embryonen
. . . . . . .
Kristallisationskeim setzen

52
(HT 200, Minitüb, Landshut) zum Equilibrieren im Kryoprotektivum. Kurz vor Ab-
lauf dieser Frist erfolgt auch hier nach dem gleichen Prinzip das Aufziehen der
equilibrierten Embryonen auf mit Entwicklungsstadium, Spezies, Spültag und
Stückzahl gekennzeichnete 0,25 ml Kunststoffpailletten. Die Pailletten wurde
ebenfalls mit Hämatokrit-Versiegelungskitt verschlossen und in die, auf 20°C
gehaltene, Gefrierkammer gegeben. Durch drücken der Taste „Run“ am Pro-
grammgeber startete das Gefrierprogramm. Die Temperatur wurde mit konstant
2°C/min von +20°C auf -6°C abgesenkt. Nach Erreichen von -6°C und Halten
dieser Temperatur für fünf Minuten erfolgte das manuelle Seeding durch Berüh-
rung mit einer in LN2 gekühlten Metallpinzette und anschließendes Halten der
Temperatur auf -6°C für weitere fünf Minuten. Im Folgenden wurde bei einer
konstanten Kühlrate von 0,3°C/min die Temperatur im Inneren der Gefrierkam-
mer auf -32°C heruntergekühlt und nach Erreichen dieser Temperaturstufe für
zehn Minuten dort gehalten. Danach wurde die Temperatur im freien Fall auf
-163°C abgesenkt; von dieser Stufe aus fand die direkte Umsetzung der Emb-
ryonen in LN2 statt.
Bei allen folgenden Handgriffen wurden die Kunststoffpailletten ausschließlich
mit einer in LN2 gekühlten Metallpinzette am Verschlussstopfen angefasst, um
so eine Erwärmung zu vermeiden.
Tab. 6: Schema Gefrierprogramm
Programmschritt Haake Consarctic
Equilibrieren Embr. RT 20 Min. 7 Min.
Einsetzen in Kryostat ― ×
Kühlen auf -6°C × -2°C/min
Einsetzen in Kryostat × ―
Halten bei -6°C 5 Min. 5 Min.
Seeding × ×
Halten bei -6°C 5 Min. 5 Min.
Kühlen auf -32°C -0,3°C/min -0,3°C/min
Halten bei -32°C 10 Min. 10 Min.
Umsetzen in LN2 × ―
Freier Fall auf -163°C ― ×
Umsetzen in LN2 ― ×

53
3.8 Zwischenlagerung der eingefrorenen Embryonen
Es wurden insgesamt 573 Embryonen eingefroren. Diese Summe teilte sich in
413 Blastocysten und 160 Morulae auf. Die Unterteilung erfolgte in drei Grup-
pen (Kontrolle, -18°C, -79°C), die verschiedenen Versuchstemperaturen zu ge-
ordnet waren. So weit es Mengenmäßig möglich war, wurden den jeweiligen
Gruppen pro Versuchsdurchgang sowohl Blastocysten als auch Morulae eines
Embryonen-Spültages zugeordnet.
Gruppe Kontroll-Embryonen: Diese Embryonen wurden als Kontrollgruppe ver-
wendet. Sie verblieben bis zu dem Zeitpunkt, an dem auch die übrigen
Embryonen des Durchganges kultiviert wurden in LN2.
Gruppe -18°C-Embryonen: Embryonen aus dieser Gruppe lagerten für die Zeit-
spanne von 60 Minuten bei -18°C in der Gefrierzelle. Dazu wurden die
Pailletten in einem mit LN2 gefüllten Styroporbehälter in die Gefrierzelle
gebracht, mit einer in LN2 gekühlten Metallpinzette herausgenommen
und in einen temperierten offenen Styroporkasten gelegt. Hier verblie-
ben sie ab Ablegungszeitpunkt für 1 Stunde.
Gruppe -79°C-Embryonen: In dieser Gruppe überdauerten die Embryonen für
eine Stunde in Trockeneis bei -79°C. Dazu wurden mit einer heißen
Zange Rillen in die, in einem vorgekühlten Styroporkasten mit Deckel,
gelagerten Trockeneisplatten hineingebrannt. Um ein gleichmäßiges
Liegen der Pailletten zu gewährleisten, waren die Rillen geringfügig
länger als die Pailletten. Anschließend wurden die Pailletten mit der in
LN2 gekühlten Metallpinzette angefasst und in den fertigen Rillen abge-
legt. Eine zweite Trockeneisplatte übernahm die Funktion eines De-
ckels. So sollte erreicht werden, dass die Pailletten, ohne gequetscht
zu werden, von allen Seiten gleichmäßig bei -79°C lagerten. Ab dem
Moment des Auflegens der oberen Trockeneisplatte lief die Zwischen-
lagerungsdauer von 60 Minuten. Um ein schnelles Verdunsten des
Trockeneises zu verhindern wurde dieses ebenfalls in die auf -18°C
gekühlte Gefrierzelle gebracht.

54
Nach Ablauf der Zwischenlagerung von 60 Minuten wurden die Pailletten zum
Auftauen aus dem jeweiligen Lagerungsumfeld entfernt. Im Falle der Gruppe K
erfolgte die Herausnahme aus dem flüssigen Stickstoff.
Abb. 11: Zwischenlagerung bei -79°C (li.) und -18°C (re.)
3.9 Auftauen der Embryonen
Das Auftauen der Embryonen erfolgte nach einer Stunde Zwischenlagerung bei
-18°C bzw. -79°C oder im Falle der Kontrollgruppe ohne Zwischenlagerung di-
rekt aus -196°C kaltem flüssigen Stickstoff (LN2) heraus.
Dazu wurden die Pailletten zunächst für ca. fünf Sekunden bei Raumtemperatur
in der Luft geschwenkt und dann vorsichtig für 20 bis 30 Sekunden in ein
36,6°C bis 37,0°C warmes Wasserbad getaucht. Anschließend wurden die Pail-
letten mittels eines Kleenex (KIMTECH Science, Kimberley-Clark ® Professio-
nal) trockengewischt.
Über einer Petrischale (35×10mm, Greiner, Frickenhausen) wurde zunächst mit
einer Schere das untere Ende der aufgetauten Paillette abgeschnitten, an-
schließend unter leichter Schräghaltung der Paillette das obere Ende. Das Ge-
friermedium mit den Embryonen konnte dadurch herauslaufen. Der in der Pail-
lette verbleibende Rest wurde unter Zuhilfenahme einer 1ml-Luer-Einwegspritze
(B-D Plastipak®, Becton Dickinson, Irland) mit aufgesetzter Pipettenspitze vor-
sichtig herausgeblasen.
Die wiedergefundenen Embryonen wurden unter dem Stereomikroskop (SZ 11,
Olympus, Hamburg) bei 110-facher Vergrößerung mit einer ausgezogenen ste-

55
rilen Glaspipette aufgenommen und schnellstmöglich aus dem Gefriermedium
heraus in das Auftaumedium umgesetzt. In diesem verblieben sie bei Raum-
temperatur für fünf Minuten zum equilibrieren.
3.10 In vitro Kultivierung der Embryonen
Alle gewonnenen und kryokonservierten Embryonen wurden im Anschluss an
das Auftauen und Ausverdünnen des Kryoprotektivums für 48 Stunden bei
37°C, 6% CO2 und gesättigter LF kultiviert.
Nach Entnahme aus dem Auftaumediummedium erfolgte ein fünfmaliges Wa-
schen in Mineralöl überschichteten MediCult Universal IVF Medium-Drops (Abb.
11), die zuvor für 24 Stunden bei 37°C, 6% CO2 und gesättigter LF im Brut-
schrank (C60, Labotect, Göttingen) equilibriert worden waren. Im Anschluss
daran wurden die Embryonen einzeln oder maximal zu zweit in die Drops der
endgültigen Kultivierungspetrischalen (85×15mm, Greiner, Frickenhausen)
(Abb. 12) übergesetzt. Diese waren ebenso wie das Waschmedium vorher zur
24-stündigen Equilibrierung bei 37°C, 6% CO2 und gesättigter LF in den Brut-
schrank (C60, Labotect, Göttingen) (Abb. 13) gegeben worden. In diesem fand
auch die sich anschließende in vitro Kultivierung statt.
Abb. 12: Waschschale mit fünf Medium-Waschdrops unter Öl

56
Abb. 13: Kultivierungsschale mit 10 Kultivierungs-Mediumdrops unter Öl
Abb. 14: Inkubator C60, Labotect, Göttingen (Quelle: Labotect, Göttingen)
Die Embryonen wurden jeweils im Abstand von 12 Stunden auf die Weiterent-
wicklung kontrolliert. Dieses Schema ermöglichte eine gute Verfolgung der wei-
teren Entwicklung.
3.11 Beurteilung von Embryonen
3.11.1 Morphologische Beurteilung
In dieser Arbeit wurden Embryonen in der ersten Phase lediglich lichtmikrosko-
pisch beurteilt, am Ende zusätzliche einer fluoreszenzmikroskopischen Unter-
suchung mit FDA und Hoechst unterzogen. Die erste morphologische Beurtei-
lung erfolgte gleich bei der Gewinnung der Embryonen. Für die Versuche zuge-

57
lassen wurden die Stadien Morula und Blastozyste. Als nicht zulässig galten
jüngere Stadien, degenerierte Embryonen sowie unbefruchtete Eizellen.
Die nächsten morphologischen Klassifizierungen der Embryonen erfolgten be-
reits während der Rehydrierungsphase im Auftaumedium sowie während des
Waschens.
Die Weiterentwicklung der Embryonen in vitro bei 37°C, 6% CO2 und gesättig-
ter LF im Brutschrank C60 (Labotect, Göttingen) wurden nach 12/24/36/48
Stunden unter dem Stereomikroskop (SZ 11, Olympus, Hamburg sowie Axio
Observer. A1, ZeissMicroImaging GmbH, Göttingen) 50- bis 200-facher Ver-
größerung kontrolliert.
Die Morula- Embryonen wurden eingeteilt in:
Grade I
• „Sehr gut“: Embryonen mit guter Morphologie und Entwicklungsgeschwindig-
keit. Die Blastomeren sind gleichmäßig in Form und Farbe und haben eine
hervorragende Erscheinung mit intakter Zona pellucida.
Grade II
• „Gut“: Dem Entwicklungsstand entsprechende Embryonen mit geringfügigen
Abweichungen und Unregelmäßigkeiten in der morphologischen Beschaffen-
heit (z.B. einzelne ausgeschleuste Blastomeren, geringe Vesikelbildung, un-
regelmäßige Zellformen, Farbveränderungen).
Grade III
• „Mäßig“: gewisser Grad an Veränderungen (z.B. Schäden an der Zona pellu-
cida, mehrere ausgeschleuste Blastomere, geringe Vakuolenbildung in den
Blastomeren).
Grade IV
• „Degeneriert und retardiert“: Embryonen mit starken Abweichungen in der
morphologischen Beschaffenheit ((keine Rehydrierung, starke Zelllysis, zerfal-
lene Blastomere mit Vakuolen sowie dunklen Punkten, verlorene Zona pellu-
cida, kollabierte Embryonen).

58
Die Blastozysten – Embryonen wurde eingeteilt in:
Grade I
• „Sehr gut“: Gut expandierte Embryonen mit gut cavertiertem Blastocoel sowie
sehr guter Differenzierung zwischen ICM und Tropfektoderemzellen, die eine
sichelförmige Struktur haben. Die Zona pellucida ist ausgedünnt (Abb. 14a).
Grade II
• „Gut“: Embryonen mit geringerem Durchmesser und reduzierter Blastocoelbil-
dung und kaum verdünnter Zona pellucida (Abb. 14b).
Grade III
• „Mäßig“: Embryonen ohne Ausdünnung der Zona pellucida und einem insge-
samt dunklen Erscheinungsbild durch beginnende Degenerationen der Zellen
(Abb. 14c).
Grade IV
• „Degeneriert und retardiert“: Embryonen mit starken Abweichungen in der
morphologischen Beschaffenheit (keine Rehydrierung, starke Zelllysis, zerfal-
lene Blastomere mit Vakuolen sowie dunklen Punkten, verlorene Zona pellu-
cida, kollabierte Embryonen) (Abb.14d+e).

59
a) b)
c) d) e)
Abb. 15: Embryo a) "Sehr gut", b) "Gut", c) "Mäßig", d) degeneriert, e) retardiert
Zusätzlich erfolgte während der gesamten Kultivierungsdauer die Einteilung des
Entwicklungsverlaufes in die Gruppen
1. „kompaktierend“: Übergangsstadium von Morula zu Blastocyste
2. „expandierend, Beginn“: beginnende Expandierung der Blastocyste
3. „expandierend“: deutliche, aber noch nicht abgeschlossene Expandie-
rung der Blastocyste
4. „expandierend - “: deutliche, aber weniger stark ausgeprägte oder
gleichmäßige Expandierung der Blastocyste
5. „expandierend ! “: deutliche, nahezu abgeschlossene Expandierung der
Blastocyste
6. „schlüpfend“: sich öffnende Zona pellucida mit Freisetzung Embryo (Abb.
15a)
7. „geschlüpft“: Embryo der die Zona pellucida verlassen hat (Abb. 15b)
8. „kollabiert“: zusammengefallener Zellverband der Blastocyste
9. „degeneriert“: zerfallene Blastomere, sehr viele degenerierte Zellen, sehr
stark geschrumpfte innere Zellmasse oder Vakuolenbildung, Zelllysis

60
a) b)
Abb. 16:a) Schlüpfende Blastocyste, b) Geschlüpfte Blastocyste
3.11.2 Kombinierte FDA-Hoechst-Färbung zur Zytoplasmaaktivi-
tätsbestimmung und Kernzählung
Die FDA-Färbung (3,6-Diacetoxyfluoran, SIGMA-ALDRICH, Steinheim) ist eine
Möglichkeit, tote von lebenden Embryonen zu unterscheiden. Lebende Embry-
onen leuchten in einem sehr hellen Grün (Abb. 16a), während tote Embryonen
keine oder nur noch kaum sichtbare Signale aussenden (Abb. 16b) (SCHIL-
LING & DÖPKE, 1978; SCHILLING et al., 1979; SCHILLING & SMIDT, 1979).
Die zu färbenden Embryonen wurden zunächst dreimal in 37°C warmen Medi-
Cult Universal IVF Medium gewaschen und dann in FDA-haltiges MediCult Uni-
versal IVF Medium gegeben. Für die FDA-Lösung wurde zunächst eine FDA-
Stammlösung von 5mg FDA/ml Aceton (Merck, Darmstadt) angesetzt. Diese
wurde auf eine Verdünnung von 2µl/2ml MediCult Universal IVF Medium ge-
bracht (DIDION et al., 1990). Von der Verdünnung wurden anschließend 10µl
abgenommen und in einen Mediumdrops von 50µl eingeben. Die Embryonen
wurden darin für jeweils 1,5 Minuten auf der 37°C warmen Werkbank inkubiert.
Daran anschließend erfolgte eine erneute dreimalige Waschung in MediCult U-
niversal IVF Medium.
Im nächsten Schritt wurden die Embryonen zur Anfärbung der mitotisch aktiven
Kerne in eine Hoechst-Farbstoff H 33342 (Sigma-Aldrich, Steinheim) enthalten-
de Lösung umgesetzt. Aktive Kerne strahlen daraus folgend in leuchtendem
Hellblau. Inaktive tote Kerne zeigen keine Antwort mehr (Abb. 17) (VIUFF et al.,
1991).
Auch hier wurde zunächst eine Stammlösung in der Dosierung von 1mg
Hoechst H 33342/ml Aqua dest. (PURSEL et al., 1985) angesetzt. Diese wurde

61
dann auf 10µl/50µl MediCult Universal IVF Medium verdünnt. Zum Inkubieren
blieben die Embryonen im Hoechst-Farbstoff zwei Minuten auf der 37°C war-
men Werkbank. An diesen Schritt schloss sich wiederum ein dreimaliges Wa-
schen in MediCult Universal IVF Medium an.
Zur nachfolgenden mikroskopischen Beurteilung wurden die Embryonen auf ei-
nen Objektträger, auf den vorher zwei Vaseline-Streifen (Weißes Vaselin Lich-
tenstein DAB, Lichtenstein, Mülheim-Kärlich) aufgebracht waren, mittig positio-
niert und mittels eines Deckgläschens fixiert. Dabei war darauf zu achten, dass
die Embryonen nur leicht angequetsch wurden.
Die FDA-Beurteilung wurde bei 100-facher (für Photographien) bzw. 400-facher
Vergrößerung mit einem Axioskop (Axioskop, Zeiss, Göttingen), Filtersatz 09
(Zeiss, Göttingen), einer Anregung von 450-490nm (BP Erregerfilter) und einer
Emission von 520nm (Interferenzfilter FT 510, Sperrfilter LP 520) vorgenom-
men.
Die Klassifizierung durch Hoechst-Färbung erfolgte ebenfalls bei 100-facher
bzw. 400-facher Vergrößerung auf dem Axioskop (Axioskop, Zeiss, Göttingen)
unter Verwendung des Filtersatzes 02 (Zeiss, Göttingen), der Anregung von
365nm (Erregerfilter G 365) und einer Emission von 420nm (Interferenzfilter FT
395, Sperrfilter LP 420).
a) b)
Abb. 17: Aktives (a) bzw. inaktives (b) Zytoplasma - Feststellung mittels FDA
Abb. 18: Lebende und tote Kerne - Sichtbarmachung mittels Hoechst H 33342

62
3.12 Geräte und Gerätekontrolle
Der für die Kontrollen genutzte Arbeitsplatz zeichnet sich durch eine Edelstahl-
werkbank mit großem integriertem Wärmefeld aus (Easy Bench, Varolab, Gie-
sen).
Abb. 19: Arbeitsplatz mit Kleininkubator C42 (Easy Bench, Varolab, Giessen; C42, Labotect, Göttingen)
Ein kleiner Brutschrank (C 42, Labotect, Göttingen) mit 6% CO2, 37°C und ge-
sättigter LF sorgte auch in diesem Bereich für eine adäquate Zwischenkultur, so
dass die Embryonen kaum einem Kultivierungstress unterlagen.
Abb. 20: Kleininkubator C42 (Labotect, Göttingen)

63
Alle Werte der Inkubatoren und Wärmefelder wurde in dreitägigem Rhythmus
mit dem Messgerät In Control 1050 (In Control1050, Labotect, Göttingen) auf
Genauigkeit kontrolliert.
Abb. 21: Messgerät In Control 1051 (Labotect, Göttingen) (Quelle: Labotect, Göttin-gen)
Zur Temperaturmessung wurde der Temperaturfühler in den Innenraum des
Brutschrankes gehängt, die innere und äußere Tür geschlossen und das Mess-
programm gestartet. Nach gleicher Verfahrensweise erfolgte auch die Messung
des CO2-Gehalts. Zur Überprüfung der relativen Luftfeuchte wurde an dem für
externe Probenentnahmen vorgesehenen Anschluss (Sample port) ein Silikon-
schlauch mit zwischengeschalteter Kondensationsfalle angeschlossen und das
Messprogramm gestartet. Bei Abweichungen des Istwertes von Sollwert erfolg-
te die entsprechende Korrektur in der Programmierung des Brutschrankes.
Einmal wöchentlich erfolgte die Reinigung der Inkubatoren um möglichen
Keimbefall zu eliminieren. Dazu wurden die zuvor ausgeräumten Geräte innen
mit Fermacidal® D (Fermacidal® D, IC Products SA, CH-Minusio, Vertrieb Labo-
tect, Göttingen) ausgewischt. Der Inkubator C42 wurde mit neuem, zweifach
destilliertem Wasser aufgefüllt, in das vier Hübe Fermacidal® D gegeben wur-
den. Bei dem mit aktiver Sterilbefeuchtung ausgestatteten Brutschrank C60
wurde der externe Wasservorratsbehälter auf seinen Füllstand kontrolliert und
gegebenenfalls aufgefüllt.

64
3.13 Photographien
Die Bilder während der Entwicklungskontrollen wurden mit dem Zeissmikroskop
Axio Observer. A1 (ZeissMicroImaging GmbH, Göttingen) (Abb. 22) bei 200- bis
400-facher Vergrößerung und bei den Fluoreszenzaufnahmen mit dem Axi-
oskop (Zeiss, Göttingen) bei 100-facher Vergrößerung unter Verwendung von
Octax EyeWare™ Imaging Software (Octax, Herborn-MTG Altendorf) aufge-
nommen.
Abb. 22: Verwendeter Zeiss Axio Observer A1 (Mitte) (Quelle: Zeiss, Göttingen)
3.14 Statistische Auswertung
Die Ergebnisse und Unterschiede zwischen der Kontrollgruppe und den Ver-
suchsgruppen (-18°C und -79°C) hinsichtlich Überlebens- und Weiterentwick-
lungsrate wurden mit Hilfe des SAS-Programms 9.1 berechnet und auf Signifi-
kanzen mit dem Chi-Tests überprüft.

65
4 Ergebnisse
4.1 Superovulation
Es wurden 132 weibliche Mäuse im Alter von vier bis neun Wochen mittels der
Hormone eCG und hCG stimuliert.
83 Tiere (62,9 %) wiesen am Morgen nach der Zusammensetzung mit dem
Bock bei der Kontrolle auf eine erfolgte Bedeckung einen Vaginal-Plaque (VP)
auf.
Unabhängig davon ob die Tiere einen VP aufgewiesen hatten, wurden sie drei
Tage nach der Bedeckung getötet und die Embryonen gespült. Dieses Vorge-
hen wurde gewählt, da Erfahrungen der Arbeitsgruppe ergeben hatten, dass
auch Tiere ohne VP verwendbare Embryonen liefern können.
Von den 132 Spendermäusen hatten 80 ovuliert, das entspricht 60,6 % der
hormonell stimulierten und 96,4 % der positiv auf einen VP kontrollierten Tiere.
4.2 Embryonengewinnung, -stadium und -qualität
Die 132 gespülten Mäuse ergaben insgesamt 1023 Eizellen und Embryonen.
Das ergibt ein Mittel von 7,8 Eizellen und Embryonen je gespülter Maus (SD
12,2; Spanne: 0 bis 57).
Wurden nur die Mäuse zur Berechnung herangezogen, die ovuliert hatten, er-
gab sich ein Mittelwert von 12,8 Eizellen und Embryonen pro Maus (SD 13,49;
Spanne 0 bis 57).
Die zahlenmäßige Verteilung der insgesamt gewonnenen Eizellen und Embryo-
nen ist Tabelle 7 zu entnehmen.

66
Tab. 7: Entwicklungsstadium und Verteilung gewonnener Eizellen und Embryo-nen
Entwicklungsstadium Anzahl %
Blastocyste 531 51,9
Morula 279 27,3
Degenerierter Embryo 91 8,9
Unbefruchtete Eizelle 88 8,6
Sonstiges Stadium 34 3,3
Summe 1023 100
Von den insgesamt 531 gewonnenen Blastocysten wurden 410 (77,2 %) als ge-
friertauglich bewertet. Von den 279 Morulae wurden 167 (59,9 %) als gefrier-
tauglich beurteilt. Das bedeutet, dass pro behandelter Maus 3,1 gefriertaugliche
Blastocysten und 1,3 gefriertaugliche Morulae gewonnen wurden. Pro ovulierter
Maus ergab das im Mittel 5,1 gefriertaugliche Blastocysten sowie 2,1 gefrier-
taugliche Morulae. Insgesamt wurden 577 Embryonen kryokonserviert.
4.3 Zwischenlagerung
Die Kontrollgruppe umfasste 164 Embryonen, die sich aufteilten auf 109 Blasto-
cysten (Bl.) und 55 Morulae (Mo.).
Insgesamt 172 Embryonen (127 Bl., 45 Mo.) wurden bei -18°C zwischengela-
gert, weitere 170 (130 Bl., 40 Mo.) bei -79°C.
Überdies wurden 55 Embryonen (31 Bl., 24 Mo.) einer Zwischenlagerung von
72 Stunden bei -18°C, bzw. -79°C ausgesetzt oder als Kontrollgruppe verwen-
det.
Sechzehn Embryonen (13 Bl., 3 Mo.) waren bereits im Vorfeld aufgetaut wor-
den, um an ihnen die Technik des Auftauens zu üben.

67
4.4 In vitro Kultivierung
Für die Untersuchungen wurden insgesamt 430 Embryonen (309 Bl., 121 Mo.)
kultiviert. Diese teilten sich in 140 Embryonen (91 Bl., 49 Mo.) der Kontrollgrup-
pe, 142 (107 Bl., 35 Mo.) bei -18°C und 148 (111 Bl., 37 Mo.) bei -79°C gela-
gerte Embryonen auf.
Die Ergebnisse der in vitro Kultivierung von bei -18°C bzw. -79°C zwischenge-
lagerten Embryonen sowie für die Embryonen der Kontrollgruppe sind den Ta-
bellen 8 und 9 zu entnehmen.

68
Tab. 8: Ergebnisse der in vitro Kultivierung von bei -18°C bzw. -79°C zwischengelagerten kryokonservierten Blastocysten
Wiederfindungsrate Morphologischer Zustand nach in vitro Kultivierung (%)
Nach 12 h Nach 24 h Nach 48 h
Gruppe
Anzahl
Embryonen Anzahl %
Deg. Exp. Exp.! Schl. Geschl. Deg. Exp. Exp.! Schl. Geschl. Deg. Exp. Exp.! Schl. Geschl.
Kontrolle 109 91 83,5 60,4 18,7 20,9 - - 59,3 12,1 28,6 - - 73,6 4,5 15,4 3,3 2,2
-18°C 127 107 84,3 74,8 16,8 8,4 - - 79,4 5,6 15,0 - - 86,9 3,7 8,5 0,9 -
-79°C 130 111 85,4 74,8 13,5 11,7 - - 80,2 7,2 12,6 - - 85,6 4,5 8,1 0,9 0,9
Tab. 9: Ergebnisse der in vitro Kultivierung von bei -18°C bzw. -79°C zwischengelagerten kryokonservierten Morulae
Wiederfindungsrate Morphologischer Zustand nach in vitro Kultivierung (%)
Nach 12 h Nach 24 h Nach 48 h
Gruppe
Anzahl
Embryonen Anzahl %
Deg. Exp. Exp.! Schl. Geschl. Deg. Exp. Exp.! Schl. Geschl. Deg. Exp. Exp.! Schl. Geschl.
Kontrolle 55 49 89,1 65,3 22,5 10,2 2,0 - 69,4 24,5 4,1 2,0 - 85,7 4,1 6,1 - 4,1
-18°C 45 35 77,8 65,7 25,7 8,6 - - 71,4 11,8 17,1 - - 85,7 5,7 8,6 - -
-79°C 40 37 92,5 64,9 18,9 13,5 2,7 - 70,3 5,4 18,9 0,9 0,9 75,7 5,4 10,8 5,4 2,7

69
4.5 Vitalfärbung mit FDA und Hoechst 33341
Im Anschluss an die abschließende morphologische Beurteilung nach 48 Stun-
den in vitro Kultivierung wurden alle als expandiert sowie gut expandiert klassi-
fizierten Embryonen angefärbt. Das entsprach 131 der insgesamt 430 kultivier-
ten Embryonen.
Je nach Intensität der Reaktion des Zytoplasmas auf den FDA-Farbstoff wurde
dabei unterschieden in tot (T), schwach aktiv (L -), aktiv (L) und sehr aktiv (L !).
Die Ergebnisse sind Tabelle 10 zu entnehmen.
Als Reaktion auf Hoechst 33341 ließen sich im UV-Licht noch bei 120 Embryo-
nen Zellkerne erkennen. Im Mittel waren dies 13,1 sichtbare Zellkerne pro an-
gefärbtem Embryo (SD 13,0; Spanne 0 bis 65).
Tab. 10: Reaktion des Zytoplasmas auf die FDA-Färbung
T L - L L !
Anzahl % Anzahl % Anzahl % Anzahl %
80 61,1 25 19,1 10 7,6 16 12,2
4.6 Einfluss der Abstammung der Mutterherkunft auf die Emb-
ryonenausbeute
Da bei der Gewinnung der Embryonen aufgefallen war, dass die Mäuse, die
von einem MPI-Herkunft bezogen worden waren deutlich mehr und auch mor-
phologisch bessere Embryonen lieferten als die im Institutsbestand gezogenen,
wurden die Ergebnisse zusätzlich noch nach Herkunft der Mütter (MPI = M, In-
stitut = I) aufgeschlüsselt. Die Ergebnisse sind in Tabellen 11 und 12 angeben.

70
Tab. 11: Embryonengewinnung aus MPI-Mäusen
Stadium Mittelwert SD Min. Max.
Blastocyste 11,8 13,6 1 48
Morula 7,3 5,8 1 27
Degeneriert 2,7 1,3 1 5
Unbefruchtet 2,6 2,1 1 10
Tab. 12: Embryonengewinnung aus institutseigenen Mäusen
Stadium Mittelwert SD Min. Max.
Blastocyste 9,4 6,2 1 22
Morula 5,9 4,2 1 13
Degeneriert 3,1 2,9 1 11
Unbefruchtet 3,2 2,9 1 12
4.7 Einfluss der Abstammung der Mutter auf das Entwicklungs-
stadium nach 48 Stunden in vitro Kultivierung
Da sich die morphologische Entwicklung nach 48stündiger in vitro Kultivierung
unterschied, je nachdem welche Mutterherkunft die Embryonen hatten, wurden
die Ergebnisse danach differenziert (Tabelle 13 und 14).
Tab. 13: Mutterherkunft Institutseigen
Morphologischer Zustand nach 48 h in vitro Kultivierung in % Gruppe
Deg. Exp. Exp.! Schl. Geschl.
Kontrolle 83,2 10,5 6,3 - -
-18°C 86, 9,6 3,7 - -
-79°C 93,0 4,7 1,6 - 0,7
Tab. 14: Mutterherkunft MPI
Morphologischer Zustand nach 48 h in vitro Kultivierung in % Gruppe
Deg. Exp. Exp.! Schl. Geschl.
Kontrolle 59,9 8,6 26,4 2,7 2,4
-18°C 79,6 6,7 13,2 0,5 -
-79°C 75,6 6,9 15,1 1,5 0,9

71
Der Einfluss der Mutterherkunft hatte einen hoch signifikanten Einfluss
(p<0,001) bei dem Merkmal tot bzw. lebendig. Die Anzahl der toten Embryonen
bei Institutsherkunft war signifikant höher als bei Embryonen aus MPI-Zucht. Als
„tot“ wurden dabei alle degenerierten Embryonen und als „lebendig“ alle Emb-
ryonen der Stadien Exp., Exp.!, Exp.-, Schl. und Geschl. bezeichnet.
Bei der Überprüfung hinsichtlich der Signifikanzen zeigte sich, dass bei -18°C
die Herkunft der Mutter keinen signifikanten Einfluss (p>0,05) auf die Entwick-
lung ausübte. Bei den Gruppen Kontrolle sowie -79°C zeigte sich allerdings ein
hoch signifikanter Einfluss (p<0,001) der Herkunft auf das Entwicklungsstadium.
Ebenfalls von hoch signifikantem Einfluss war das Entwicklungsstadium zum
Zeitpunkt der Kryokonservierung (Blastocyste oder Morula). Die Überlebensrate
der Morulae war signifikant höher als bei den Blastocysten.
Wurde der Beobachtungszeitpunkt in Korrelation zu der Zwischenlagerungs-
temperatur gesetzt, ergab sich nur beim Beobachtungszeitpunkt 36 h nach Kul-
tivierungsbeginn ein niedrig signifikanter Einfluss auf die Entwicklung (p<0,01).
Zu den übrigen Zeitpunkten (12/24/48 Stunden) zeigten sich keine Unterschie-
de (p>0,05).

72
5 Diskussion
Das Ziel dieser Arbeit war es, festzustellen ob es möglich ist kryokonservierte
und in flüssigem Stickstoff gelagerte Mäuseembryonen über einen Zeitraum von
einer Stunde bei Temperaturen von -18°C oder -79°C z.B. für Transportzwecke
zwischenzulagern.
Dafür wurden Morulae und Blastocysten von superovulierten Spendern gewon-
nen, kryokonserviert und anschließend für eine Stunde bei -18°C bzw. -79°C
zwischengelagert
5.1 Superovulation
Wie schon von HOGAN et al. 1986 berichtet und von OZGUNEN et al. (2001)
bestätigt, hängt bei Mäusen eine erfolgreiche Superovulation maßgeblich von
dem Alter und Körpergewicht der Mäuse, der gespritzten Hormonmenge, der
Zuchtlinie sowie den Haltungsbedingungen ab.
HOOGENKAMP & LIWING (1982) gaben an, dass die optimale Hormonmenge
für jede Linie separat ermittelt werden muss, da das Optimum zwischen den un-
terschiedlichen Zuchtlinien teilweise stark schwankt.
Die anfangs in diesem Versuch angewandte Hormondosierung (7,5 IE eCG und
5,0 IE hCG) wurde auf Grund der positiven Ergebnisse der Arbeitsgruppe
Reischl & Al Yakoub (2006) gewählt. Wegen der teilweise sehr geringen Erfolge
der Superovulation wurde nach Erfahrungsaustausch mit anderen, ebenfalls
superovulierte Mäuse verwendenden Arbeitsgruppen (WOLF, 2006; NOWS-
HARI, 2006) auf eine leicht verringerte Hormondosierung (5,0 IE eCG und 5,0
IE hCG) gewechselt.
Der zum Kontrollzeitpunkt durch einen VP als gedeckt gekennzeichnete Anteil
Mäuse lag mit 62,9 Prozent bei einem eher unbefriedigenden Niveau. HOO-
GENKAMP & LEWING (1982) erreichten eine Deckrate von 85 %, SMORAG et
al. (1981) von 61 %, SCHMIDT et al. (1985) von 59 % und MATAR (2002) von
76 %, wobei allerdings andere Hormondosierungen und Mäuselinien verwendet
wurden.

73
Als nur bedingt zufriedenstellend anzusehen war der Anteil der gewonnenen
Eizellen und Embryonen mit 7,8 pro Spender auf die Gesamtheit der gespülten
Tiere bezogen. Die zur Weiterzucht verwendeten Vergleichstiere erreichten oh-
ne Hormonstimulation Wurfgrößen von bis zu 16 Jungen.
Zufriedenstellend war der Anteil der Mäuse die ovuliert hatten, bezogen auf die
als gedeckt gekennzeichneten Tiere mit 96,4 %.
Nur bedingt zufriedenstellend war die Höhe der gewonnenen Menge an Eizellen
und Embryonen mit im Mittel 12,8 je Tier. Gleiches gilt für die Höhe der Rate an
gewonnenen Morulae (27,3 %) und Blastocysten (51,9 %) aus den Spendertie-
ren die ovuliert hatten.
Die zuweilen nur bedingt akzeptablen Ergebnisse nach Superovulationsindukti-
on bei der Gewinnung der Embryonen könnten auf dem Alter und Gewicht, den
Haltungsbedingungen sowie dem Inzuchtgrad der verwendeten Mäuse beru-
hen.
GATES beobachtete 1971, dass das Alter, die physische Verfassung und auch
ein möglichst optimales Gewicht (13 bis 14 g) in großem Ausmaß die Anzahl
gewonnener Embryonen nach einer Superovulation beeinflusst. Auch MINTZ
(1967) gab an, dass es wichtig ist, präpuberale Mäuse zu verwenden, da Tiere
mit einem eigenen Zyklus nach einer Hormonbehandlung deutlich schlechtere
Ergebnisse liefern. HOOGEN (1968) gab als das geeignetste Alter für eine er-
folgreiche Superovulation den Zeitraum zwischen dritter und sechster Lebens-
woche an. Dieses wurde 1993 durch KOVACS et al. bestätigt, die ebenfalls
festgestellt hatten, dass postpuberale Tiere nach einer Hormonbehandlung we-
niger Eizellen ovulierten und auch eine geringere Embryonengewinnungsrate
aufwiesen. Das ist insofern interessant, als Mäuse in der Regel drei bis vier
Wochen nach der Geburt abgesetzt werden und mit etwa sechs bis acht Wo-
chen geschlechtsreif werden.
Die Tatsache, dass präpuberale Weibchen besser auf eine Hormongabe an-
sprechen als geschlechtsreife, konnte auch in der vorliegenden Untersuchung
beobachtet werden. Die bei Versuchsbeginn zur Verfügung stehenden bereits
geschlechtsreifen Weibchen wiesen nach einer hormonellen Stimulation deut-
lich weniger Embryonen bei der Spülung auf, als die zum Vergleich herangezo-

74
genen Wurfgrößen der ohne Hormonbehandlung zur Weiterzucht genutzten
Weibchen des selben Wurfes oder der gleichen Altersklasse verpaart mit den
identischen Zuchtböcken wie bei den gespülten Tieren.
Als optimales Haltungsklima wird für Mäuse eine Raumtemperatur (RT) zwi-
schen 22°C bis 25°C und eine rel. Luftfeuchte (LF) von 50 bis 60 % angenom-
men.
Ellendorf et al. (1970) berichteten, dass sowohl deutlich niedrigere oder höhere
Temperaturen als auch vom Optimalwert abweichende rel. LF sich deutlich ne-
gativ auf die Fruchtbarkeit der Mäuse auswirken. Auf Grund der baulichen und
technischen Gegebenheiten im Mäusehaus des Institutes waren die zu den
Versuchen herangezogenen Mäuse mitunter suboptimalen Haltungsbedingen
ausgesetzt. So waren die als optimal anzusehenden Werte nicht zu jeder Zeit
erfüllbar. Dieses könnte ein weiterer Grund für die schlechteren Ergebnisse bei
der Embryonengewinnung sein.
HOGAN et al. veröffentlichten 1986, dass die Linien CBA, C57 und BALB und
deren Kreuzungen sehr gute Ovulationsrate hervorbringen. Die im Versuch ein-
gesetzte NMRI-Linie wird häufig als Rezipient bei Transferversuchen einge-
setzt; sie ist aber eigentlich ebenfalls für eine gute Vitalität bekannt, die nach
einer Superovulation mit einer größeren Anzahl gewonnener Embryonen ein-
hergeht. Insofern ist die doch recht geringe Reaktion auf die Superovulations-
behandlung etwas verwunderlich.
Es ist allgemein bekannt, dass sich Inzucht auf bestimmte Merkmale wie z.B.
Wurfgröße und damit auf die Anzahl ovulierter Eizellen positiv auswirken kann.
Bei einer zu stark steigenden Homozygotie kann es jedoch zu einer Verände-
rung züchterisch relevanter Merkmale wie z.B. der Fruchtbarkeit, des Wachs-
tums oder der Vitalität im Allgemeinen kommen (KÖHLER, 1989).
Das kann dazu führen, dass die Tiere eher auf Krankheiten ansprechen und
damit unter Umständen eine geringere Lebenserwartung haben (WEIß et al.,
1996). SMIDT & MONZAVIFAR (1969) berichteten, dass es häufig in Folge von
zunehmender Inzucht zu einem Sinken der Fruchtbarkeit kommt.

75
Da der institutseigene Mäusebestand starke Inzucht aufwies, könnte dieser
Umstand auch eine Erklärung dafür sein, dass es nach Austausch der Mäuse-
population zu einem deutlichen Anstieg der gewonnenen und als tauglich beur-
teilten Embryonen kam.
5.2 Überleben und Weiterentwicklung unter in vitro Bedingun-
gen
Ein recht zuverlässiges und einfaches Beurteilungsverfahren für die Überprü-
fung der Unversehrtheit von Embryonen im Anschluss an das Auftauen ist die
Beobachtung der Weiterentwicklungsrate in vitro unter Verwendung eines ge-
eigneten Kulturmediums und geeigneter Kulturbedingungen (SCHNEIDER et
al., 1974; BILTON & MOORE, 1976a; TROUNSON et al., 1978; AL-HASANI et
al., 1986; LEIBO, 1990).
Das in den Versuchen zur Kultivierung verwendet verbrauchsfertige Medicult
Universal IVF-Medium ist ein im Bereich der humanen Reproduktion verwende-
tes Medium auf Basis Earle´s Gepufferter Salzlösung (EBSS). Bevor Medien im
humanen Bereich zum Einsatz kommen, müssen sie am Mausmodell erfolg-
reich getestet worden sein und dort gute Ergebnisse gebracht haben. Das in
den vorliegenden Versuchen verwendete Medium war zu Beginn der eigentli-
chen Untersuchung in Vorversuchen, die dem Erlernen der Techniken dienen
sollen, zur Kultivierung frischer wie auch kryokonservierter Mäuseembryonen
eingesetzt worden. Durch die dabei beobachteten guten bis sehr guten Ergeb-
nisse führten zu der Entscheidung dieses Medium auch für die eigentlichen
Versuche zu nutzen.
Bei dem Medium war, da es kein HEPES enthielt, eine CO2-Begasung in Höhe
von mindestens 5% notwendig. Die Einstellung des Brutschrankes wurde im
Abgleich mit dem dazugehörigen Messgerät InContol 1050 sehr genau justiert.
Dieser Brutschrank zeichnet sich besonders durch den sterile Wasserzufuhr
aus, die eine absolut kontaminationsfreie Atmosphäre durch ein neuartiges
Wassereinspritzsystem gewährleistet. Im Unterschied zu den sonst häufig in
den in vitro Kultivierungen muriner Embryonen eingesetzten Medien, enthielt

76
das verwendete Medium als Serumkomponente humanes Serum Albumin
(HSA). Das könnte ein Grund sein, warum weniger Blastocysten schlüpften als
es auf Grund ihrer Entwicklung anzunehmen war.
LADD et al. stellt 1982 in Versuchen fest, dass Mäuseembryonen in absteigen-
der Reihenfolge offensichtlich die besten Weiterentwicklungsraten bei Verwen-
dung von fetalem Kälberserum (FCS), Lämmerserum sowie bovinem Serumal-
bumin (BSA) aufweisen. ARIFF et al. (1988) dagegen erzielten bei Verwendung
von BSA als Serumkomponente eine Weiterentwicklungsrate von 90 % wäh-
rend einer in vitro Kultivierung von 2-Zell-Mäuseembryonen hin zum Blasto-
cystenstadium.
Beim Auftauen der Embryonen wurde sehr großer Wert auf eine vollständige
Entfernung des Kryoprotektivums gelegt, da wie SCHNEIDER & MAZUR (1984)
und CHUPIN (1986) feststellten, eine unzureichende Ausverdünnung von Kry-
oprotektiva zu einer verringerten Entwicklungsrate der aufgetauten Embryonen
führen kann.
5.3 Zwischenlagerungseinfluss auf die Weiterentwicklung der
kryokonservierten Embryonen
Anders als erwartet, entwickelten sich nach einer Stunde Zwischenlagerung
nicht nur Embryonen aus der Kontrollgruppe (ohne Zwischenlagerung) weiter,
sondern auch Embryonen aus den Zwischenlagerungstemperaturen -18°C und
-79°C. Auch fiel der erwartete Unterschied in Bezug auf überlebende und tote
Embryonen zwischen den Versuchsgruppen -18°C und -79°C erstaunlich gering
aus. Das lässt vermuten, dass es bei einer Temperaturerhöhung über den Zeit-
raum von einer Stunde zu weniger Schäden an den Embryonen kommt als
weithin angenommen wird. MAURER & Bank (1973) sowie MAURER et al
(1977) lagerten kryokonservierte 8-Zell-Mäuse-embryonen bei -44°C, -88°C und
-196°C ein. Dabei stellten sie nach anschließender in vitro Kultivierung fest,
dass je höher die Lagertemperatur ist, desto geringer die Überlebens- und Wei-
terentwicklungsrate der Embryonen ausfällt.
Die Ursachen für diese Beobachtungen können nicht definitiv benannt werden,
wahrscheinlich ist aber, dass viele einzelne Faktoren Einfluss haben. So könnte

77
einerseits die bei höheren Temperaturen stärker wirkende Toxizität der Kry-
oprotektiva sich negativ auswirken (KASAI et al., 1981), andererseits könnte
auch eine eventuell ablaufende Umkristallisation der beim schnellen Einfrieren
auf -196°C gebildeten kleinen intrazellulären Eiskristalle in größere schädliche
Eiskristalle Einfluss haben. Beim Prozess des schnellen Einfrierens bilden sich
viele kleine, für die Zellen unschädliche intrazelluläre Eiskristalle (LEIBO et al.,
1974; FARRANT et al., 1977; RALL, 1981; JONDET et al., 1984), die aber beim
Auftauvorgang ebenfalls eine schnelle Auftaurate (350°C bis 500°C/min) benö-
tigen, damit es nicht zu einer Umkristallisation in größere und damit schädigen-
de Kristalle kommt (WILLADSEN et al., 1978; WOOD et al., 1978; BILTON,
1980). Physikalischen Gesetzen nach, muss je nach verwendeter Lösung bis zu
einer Temperatur von etwa -80°C mit einer Positionsverschiebung der Eiskris-
talle gerechnet werden, die ein Zusammenschmelzen einzelner Kristalle in grö-
ßere Komplexe bewirkt (MEYBERG, 2007) und eine gewisse Dynamik in der
Zelle zu Schädigungen führt.
Ein zusätzlich weiterer, beeinflussender Faktor könnte die Qualität der kryokon-
servierten Embryonen sein. So stellte zum Beispiel MAHABIR (2000) fest, dass
je besser die Spendertiere auf eine Superovulation reagiert hatten, desto bes-
ser auch die Qualität der gewonnenen Embryonen ausfiel. Das würde sich mit
den Beobachtungen in diesem Versuch decken, die sich bei allen Behandlun-
gen (Kontrollgruppe, -18°C, -79°C) ein deutlich besseres Überleben derjenigen
Embryonen zeigen, die an Spültagen gewonnen worden waren, bei denen die
Spenderinnen eine sehr gute Reaktion auf die Superovulation gezeigt hatten.
Nicht ohne positiven Effekt könnte auch der Einsatz des moderneren Einfrier-
systems der Firma Consartic gewesen sein. Durch die Computerchip gesteuer-
te Programmierung des Ablaufes des Programmes ist eine extrem genaue Ein-
haltung der Einfrierraten gewährleistet, sowie auch die einheitliche Wiederhol-
barkeit der verschiedenen Kryozyklen.
Die Verwendung des Kleininkubators C42 wird sich ebenfalls positiv auf die Er-
haltung der Vitalität der Embryonen ausgeübt haben, denn alle Embryonen
wurden immer nur kurz zu Waschvorgängen aus der optimalen Atmosphäre he-
rausgenommen und waren somit nur ganz geringen Zellkulturstress unterwor-
fen. Die Gewinnung der Embryonen wurde sehr steril und temperaturstabil

78
durchgeführt, da die Werkbank aus gut zu säuberndem Edelstahl mit integrier-
tem Wärmefeld bestand.
Ähnliche Untersuchungen wie die der vorliegenden Untersuchungen waren
2002 durch MATAR durchgeführt worden, die allerdings eine 72-stündige Zwi-
schenlagerung bei -5°C, -18°C und -79°C mit sofort anschließendem Refree-
zing und im nächsten Schritt einen Transfer auf Empfängertiere bzw. in vitro
Kultivierung beinhalteten. Aus ihren Ergebnissen schlussfolgerte sie, dass eine
Versuchswiederholung mit der Temperaturstufe -79°C nur dann sinnvoll ist,
wenn ein optimiertes Kryoprotektivum, reduzierte Lagerungsdauer und anstatt
einem sofortigen Refreezing zuvor ein vollständiges Auftauen und danach er-
neutem Einfrieren eventuell sinnvoll wäre.
Die in dem eigenen Versuch gewonnenen Resultate lassen vermuten, dass die
kryokonservierten Embryonen in der Lage sind, eine kurzfristige Erhöhung der
Temperatur mit direkt anschließender in vitro Kultivierung zu tolerieren. Um die-
se Beobachtungen noch einmal zu überprüfen und gegebenenfalls zu festigen
wäre es sinnvoll, die Versuche mit Embryonen, ausschließlich von gut supero-
vulierten Spenderinnen gewonnen, zu wiederholen. In diesem Zusammenhang
wäre es auch interessant festzustellen, wie sich eine Verlängerung der Zwi-
schenlagerungsdauer auf das Überleben der Embryonen auswirken würde und
ob eine Übertragung der Technik auch auf andere Spezies möglich ist.

79
6 Zusammenfassung
Das Ziel der vorliegenden Arbeit war es, die Einflüsse einer 60-minütigen Zwi-
schenlagerung auf die Entwicklungsrate von bei -18°C und -79°C auf zuvor in
flüssigem Stickstoff (-196°C) kryokonservierte Mäuseembryonen zu untersu-
chen.
Um bei den Spendertieren ein Erhöhung der zu gewinnenden Embryonen pro
Tier zu erreichen wurden insgesamt 132 Spendertiere mit zu Beginn der Versu-
che 7,5 IE eCG und 5,0 IE hCG und im weiteren Verlauf der Versuche mit 5,0
IE eCG und 5,0 IE hCG hormonell behandelt. Von diesen Tieren wurden nach
Spülung der Eileiter und Uterushörner insgesamt 1023 Eizellen und Embryonen
gewonnen. 410 Blastocysten und 167 Morulae wurden als gefriertauglich ange-
sehen und mittels des Standartgefrierverfahrens folgendermaßen kryokonser-
viert:
Im Anschluss an eine 20-minütige Equilibrierung im Kryoprotektivum (1,5 M EG
und 0,25 M Saccharose) bei Raumtemperatur, bei der bereits ein Aufziehen der
Embryonen auf die Kryopailletten stattfand, wurden die Pailletten in ein auf -6°C
gekühltes Alkoholbad (Gerät Haake) bzw. in die auf -6°C gekühlte Gefrierkam-
mer (Gerät Consartic) gegeben und fünf Minuten bei dieser Temperatur gehal-
ten. Nach Ablauf der fünf Minuten erfolgte das Seeding durch eine in LN2 ge-
kühlte Metallpinzelle. Nach dem Seeding erfolgte ein weiteres Halten der Tem-
peratur für fünf Minuten bei -6°C. Anschließend wurde die Temperatur mit einer
Kühlrate von 0,3°C/min bis auf -32°C abgesenkt und dort für noch einmal zehn
Minuten gehalten, bevor ein direktes Umsetzen in LN2 (Gerät Haake) bzw. die
Absenkung der Temperatur in der Gefrierkammer in freiem Fall auf -196°C (Ge-
rät Consartic). Bis zum Beginn der Versuche verblieben die Embryonen zur La-
gerung in LN2.
Für die Versuche wurden die Embryonen zu gleichen Gruppen in Kontrollgrup-
pe, Lagerung bei -18°C und Lagerung bei -79°C aufgeteilt.
Nach Lagerung und Auftauen bzw. bei der Kontrollgruppe nur nach dem Auf-
tauen wurden die Embryonen unter in vitro Bedingungen über die Zeitspanne
von 48 Stunden auf ihre Entwicklungsraten beobachtet.

80
Die Kultivierung fand unter Verwendung des MediCult-Universal-IVF-Mediums
bei 37°C, 6% CO2 und gesättigter LF im Brutschrank (C60, Labotect, Göttingen)
statt.
Am Ende der 48-stündigen in vitro Kultivierung waren in der Kontrollgruppe
73,6 % der Blastocysten degeneriert und 26,4 % lebend. Bei dem ursprüngli-
chen Stadium Morula waren 85,7 % degeneriert und 14,3 % lebend.
Bei der Versuchsgruppe mit einer -18°C Zwischenlagerung waren aus der
Gruppe der Blastocysten 86,9 % degeneriert und 13,1 % lebend. Bei den Moru-
lae waren 85,7 % degeneriert und 14,3 % lebend.
In der Versuchsgruppe -79°C Zwischenlagerung waren nach 48 Stunden
85,6 % der Blastocysten degeneriert, 14,4 % lebend und bei den Morulae wa-
ren 75,7 % degeneriert und 24,3 % lebend.
Die Ergebnisse lassen vermuten, dass die Embryonen bei dem Prozess der
Zwischenlagerung zwar deutlich geschädigt werden, ein Überleben aber den-
noch möglich zu sein scheint. Eine Möglichkeit der Schädigung wäre eventuell
in der Veränderung der Eiskristallstruktur von kleinen zu größeren Kristallen
durch die Lagerung bei höheren Temperaturen zusehen und oder eine Toxizi-
tätszunahme der Kryoprotektiva, ebenfalls durch die Erhöhung der Temperatur
hervorgerufen. Es könnte als Überlegung ebenso noch hinzugezogen werden,
ob eventuell eine nicht optimale Embryonenqualität - verursacht durch eine
schlechte Reaktion der Spendertiere auf die Superovulationsbehandlung - wie
auch die mit Blastocyste und Morula schon relativ weit fortgeschrittene Embry-
onalentwicklung die insgesamt schlechten Ergebnisse zusätzlich gefördert ha-
ben.
Insofern wäre es interessant, die durchgeführten Ergebnisse noch einmal mit
Embryonen aus Spülvorgängen mit besseren Reaktionen auf eine Superovula-
tionsbehandlungen, früheren Embryonalstadien und eventuell auch anderen
Kryokonservierungsmethoden wie zum Beispiel der Vitrifikation durchzuführen.

81
7 Literaturverzeichnis
ADAMS, C.E. (1968): Probleme der Eiübertragung bei Haustieren. Z. Tierz.
Züchtungsbiol. (1968); 84: 319-329.
AL-HASANI, S., TOLKSDORF, A., DIEDRICH, K., VAN DE VEN, H., KREBS,
D. (1986): Successful in vitro fertilization of frozen-thawed rabbit emb-
ryos.
ALLISTON, C.W., HOWARTH jr., B., ULBERG, U.C. (1965): Embryonic morta-
lity following culture in vitro of one- and two-cell rabbit eggs at eleva-
ted temperatures. J. Reprod. Fertil. (1965); 9: 337-341.
ARIFF, B., NG, S.C., MOK, H., LIM, M.N., WONG, P.C., SHAN, R. (1988): E-
vaulation of Chang´s culture medium for mouse in vitro fertilization
and embryonic development. J. In vitro fert. Embr. transf. (1988); 2:
102-106.
ATTRODT, G (1985): Untersuchungen zur Tiefgefrierkonservierung und Flu-
orszenzmikroskopischen Beurteilung von Mäuseembryonen. Diss. U-
niversität Gießen.
AGCA, Y., LIU, J., PETER, A.T., CRITSER, E.S., CRITSER, J.K. (1997): Cry-
oprotectant and water permeability of immature and in vitro matured
bovine oocytes. Theriogenology (1997); 47: 340.
ARRESEIGOR, C.J., SISUL, A., ARRESEIGOR, A.E., STAHRINGER, R.C.
(1998): Effect of cryoprotectant, thawing method, embryo grade and
breed on pregnancy rates of cryopreserved bovine embryos. Therio-
genology (1998); 59: 160.
ASHWOOD-SMITH, M.J. u. FRIEDMANN, G.B. (1979): Lethal and chromoso-
mal effects of freezing , thawing, storage time and X-irradiation on

82
mammalian cells preserved at -196°C in Dimethyl Sulfoxide. Cryobio-
logy (1979); 16: 132-140.
BANK, H. (1973): Visualization of freezing damage. II. Strutural alterations du-
ring warming. Cryobiology (1973); 10: 157-170.
BANK, H., MAURER, R.R. (1974): Survival of frozen rabbit embryos. Experim.
Cell Research (1974); 89: 188-196.
BARNETT, R.E. (1978): The effect of dimethylsulfoxide and glycerol on Na+,
K+, ATP-ase and membrane structure. Cryobiology (1978); 15: 227-
229.
BEAL, W.E., HINSHAW, R.H., WHITMAN, S.S. (1998): Evaluating embryo
freezing method and the site of embryo deposition on pregnancy rate
in bovine embryo transfer. Theriogenology (1998); 49: 241.
BIELANSKY, A., SCHNEIDER, U., PAWLYSHYN, V.P., MAPLETOFT, R.J.
(1985): Effect of sample container and cryoprotectant removal method
on survival of deep frozen bovine embryos. Theriogenology (1985);
23: 179.
BIELANSKY, A., SCHNEIDER, U., PAWLYSHYN, V.P., MAPLETOFT, R.J.
(1986): Factors affecting survival of deep frozen bovine embryos in
vitro: the effect of freezing container and method of removing cryopro-
tectant. Theriogenology (1986); 25: 429-437.
BILTON, R.J. (1980): Preservation of embryos of the large domestic species.
9th Int. Congr. Anim. Reprod. and A.I., Madrid (1980); 2: 245-253.
BILTON, R.J., MOORE, N.W. (1976): In vitro culture, storage and transfer of
goat embryos. Austr. J. Bio. Sci.(1976); 29: 125-129.

83
BOLAND, M.P., YROSBY, T.E., GORDON, I. (1978): Morphological normality
of cattle embryos following superovulation. Theriogenology (1978); 10:
175-180.
BOUYSSON, B., CHUPIN, D. (1982): Two-step freezing of cattle blastocysts
with disulfoxyde (DMSO) or glycerol. Theriogenology (1982); 17: 159-
166.
BOXBERGER, H.J. (2007): Leitfaden für die Zell- und Gewebekultur. Wiley-
VCH Verlag, Weinheim; 86.
BRACKE, C., DÖPKE, H.H., NIEMANN, H. (1995): Untersuchungen zur Kryo-
konservierung von Rinderembryonen mit dem Ethylenglycol-One-
Step-Verfahren und durch Vitrifikation. AET-d Jahrestagung; 1./2. Juni
1995, Kleve.
BRINSTER, R.L. (1963): A method for in vitro cultivation of mouse ova from
two-cell to blastocyst. Exp. Cell. Res. (1963); 32: 205-208.
BRINSTER, R.L. (1965): Studies on the development of mouse embryos in
vitro. I. The effect of osmolarity and hydrogen ion concentration. J.
Exp. Zool. (1965); 158: 49-57.
BRINSTER, R.L. (1969): Incorporation of carbon from glucose and pyruvate in-
to the preimplantation mouse embryo. Exp. Cell. Res. (1969); 58: 153-
158.
CHUPIN, D. (1986): Quick freezing of bovine blastocysts. Theriogenology
(1986); 21: 230.
CHUPIN, D., FLORIN, B., PROCUREUR, R. (1984): Comparison of two me-
thods for one-step in straw thawing and direct transfer of cattle blasto-
cysts. Theriogenology (1984); 21: 455-459.

84
COCERO, M.J., PROCUREUR, R., DE LA FUENTE, J., CHUPIN, D. (1988):
Glycerol or ethylene glycol for cryopreservation of deep frozen ewe
embryos. Theriogenology (1988); 29: 238.
CSEH, S., SEREGI, J., SOLTI, L. (1994): Practical experiences with direct
transferred frozen embryos. Theriogenology (1994); 41: 185.
CSEH, S., CORSELLI, J., NEHLSEN-CANARELLA, S.L., BAILEY, L.L., SZA-
LAY,A.A. (1997): The effect of quick-freezing in ethylen glycol on
morphological survival and in vitro development of mouse embryos
frozen at different preimplantation stages. Theriogenology (1997); 48:
43-50.
DE LEEUW, A.M., RALL, W.F., DEN DAAS, J.H.G., KRUIP, TH.A.M. (1991):
Comparative studies of the efficiency of rapid cryopreservation me-
thods for bovine embryos. 7 e Reunion A.E.T.E., Cambridge; 77-86.
DE PAZ, P., SANCHEZ, A.J., FERNANDEZ, J.G., CARBAJO, M., DOMIN-
GUEZ, J.C., CHAMORRO, C.A., ANEL, L. (1994): Sheep embryo
cryopreservation by vitrification and conventional freezing. Therioge-
nology (1994); 42: 327-338.
DEL CAMPO, M.R., BECERRA, F., MAPLETOFT, R.J. (1990): Thawing and
glycerol removal from bovine embryos. Theriogenology (1990); 45:
178.
DIDION, B. A., POMP, D., MARTIN, M.J., HOMANICS, G.E., MARKERT, C.L.
(1990): Observation on the cooling and cryopreservation of pig oocy-
tes at the germinal vesicle stage. J. Anim. Sci. (1990); 68: 2803-2810.
DOCHI, O., IMAI, K., TAKAKURA, H. (1995): Birth of calves after direct trans-
fer of thawed bovine embryos stored frozen in ethylene glycol. Anim.
Reprod. Science (1995); 38: 179-185.

85
DOBRINSKY, J.R. (1996): Cellular approach to cryopreservation of embryos.
Theriogenology (1996); 45: 17-26.
DOEBBLER, G.F. (1966): Cryoprotective compounds: Review and discussion
of structure and function. Cryobiology (1966); 3: 2-11.
EDWARDS, R.G. u. GATES, A.H. (1959): Timing of the stages of the maturati-
on divisions, ovulation, fertilization and the first cleavage of eggs of
adult mice treated with gonadotrophin. J. Endocrinol. (1959); 18: 292-
304.
ELSDEN, R.P., NELSON, L.D., SEIDEL jr., G,E. (1978): Superovulation cows
with follicle stimulating hormon and pregnant mare´s serum gona-
dotropin. Theriogenology (1978); 9: 17-26.
ELSDEN, R.P., SEIDEL jr., G.E., TAKEDA, T., FARRAND, G.D. (1982): Field
experiments with frozen-thawed bovine embryos transferred nonsur-
gically. Theriogenology (1982); 17: 1-10.
EMILIANI, S., VAN DEN BERGH, M., VANNIN, A.-S., BIRAMANE, J., ENG-
LERT, Y. (2000): Comparison of ethylene glycol, 1,2-propanediol and
glycerol for cryopreservation of slow-cooled mouse zygotes, 4-cell
embryos and blastocysts. Hum. Reprod. (2000); 15 (4): 905-910.
FAHY, G.M. (1986): The relevance of cryoprotectans ´toxicity` to cryobiology.
Cryobiology (1986); 23: 1-13.
FAHY, G.M., MAC FARLANE, D.R., ANGELL, C.A., MERYMAN, H.T. (1984):
Vitrification as an approach to cryopreservation. Cryobiology (1984);
21: 407-426.
FALGE, R., ROMMEL, P., KAUFHOLD, P. (1982): Stand und Methoden der
Tiefgefrierungkonservierung bei Embryonen von Säugern. Arch. Tier-
zucht, Berlin (1982); 25: 289-294.

86
FARRAND, G.D., ELSDEN, R.P. SEIDEL jr., G.E. (1982): Effect of slow coo-
ling of bovine embryos prior to plunging in liquide nitrogen. Therioge-
nology (1982); 17: 88.
FARRAND, G.D., ELSDEN, R.P., SEIDEL jr., G.E. (1985): Effect of slow coo-
ling and point temperature on survival of frozen bovine embryos. J.
Anim. Sci. (1985); 61: 460-465.
FARRANT, G.M. (1965): Mechanism of cell damage during freezing and tha-
wing and its prevention. Nature (1965); 205: 1284-1287.
FARRANT, J., MORRIS, G.J. (1973): Thermal shock and dilution shock as the
cause of freezing injury. Cryobiology (1973); 10: 134-140.
FARRANT, J., LEE, H., WALTER, C.A., McGANN, L.E. (1977): Use of two-
step coolingprocedures to examine factor influencing cell survival fol-
lowing freezing and thawing. Cryobiology (1977); 14: 273-286.
FORGRAVE, L.F., RAJAMAHENDRAN, P., BAKER, R.D. (1977): Cryopreser-
vation of mouse embryos in Macdonald freeze-thaw appartus. Can. J.
Anim. Sci. (1977); 57: 389-394.
FRIEDLER, S., GIUDICE, L.C., LAMB, E.J. (1988): Cryopreservation of emb-
ryos and ova. Fertil. Steril. (1988); 49: 743-764.
GATES, A.H. (1971): Maximizing yield and development uniformity of eggs. In:
Methods in mammalian embryology. J.E. Daniel jr., Freeman and
Company, San Francisco.
GLENISTER, P.H., WHITTINGHAM, D.G., LYON, M.F. (1984): Further studies
on the effect of radiation during storage of frozen 8-cell mouse embry-
os at -196°C. J. Reprod. Fert. (1984); 70: 229-234.

87
GLENISTER, P. H., THORNTON, C.E. (2000): Cryoconservation - archiving for
the future. Mamm. Genome (2000); 11(7): 565-71.
GUTIÉRREZ, C.G., ARTIGA, J., GARDE, J., PINTADO, B. (1993): Effect of
equilibration period and cryoprotectant in quick freezing of mouse mo-
rulae. Theriogenology (1993); 39: 229.
HAMMOND, J. (1949): Recovery and culture of tubal mouse ova. Nature
(1949); 163: 28-29.
HEBER, U. (1968): Freezing injury in relation to loss of enzyme activities and
protection against freezing. Cryobiology (1968); 5: 188-201.
HEINEKE, H., KLAUS, S. (1975): Möglichkeiten zur Bestimmung der Trächtig-
keit bei der Maus. Nova Acta Leopoldiana, N. f. Leipzig (1975); 41.
HERNANDEZ-LEDEZMA, J.J., SELGRATH, J.P., WRIGHT jr., R.W. (1988):
Effect of one-step sucrose dilution of glycerol on in-vitro development
of frozen-thawed mouse embryos. Theriogenology (1988); 30: 529-
535.
HOBBS, P.V. (1974): Ice physics. Oxford Univ. Press (Clarendon), Lon-
don/New York (1974).
HOGAN, B., CONSTANTINI, F., LACY. E. (1986): Manipulation the mouse
embryo. Cold Spring Harbour Laboratory, New York (1986).
HOOGENKAMP, H. (1984): Transfer of frozen cow embryos without removing
glycerol. 10th Intern. Congr. Anim. Reprod. A.I., Illinois; 2: 229.
HOOGENKAMP, H., LEWING, P. (1982): Superovulation in mice in relation to
their age. Vet. Quart. (1982); 447-448.

88
ISHIMORI, H.K., SAEKI, K., INAI, M., NAGAO, Y., ITASAKA, J., MIKI, Y.,
SEIKE, N., KAINUMA, H. (1992): Vitrifikation of bovine embryos in a
mixture of ethylene glycol and dimethyl sulfoxid. Theriogenology
(1992); 40: 427-433.
ISHIMORI, H.K., TAKAHASHI (1992): Viability of vitrified mouse embryos using
various cryoprotectant mixtures. Theriogenology (1992); 37: 481-487.
JANOWITZ, U., GÖRLACH, A. (1994): Durchbruch beim Auftauen von Embry-
onen. Der Tierzüchter (1994); 8: 24-25.
JANOWITZ, U., HERMANNS, L. (1995): Erfahrungen und Ergebnisse mit dem
Direkttransferverfahren. AET-d Jahrestagung; 1./2. Juni 1995, Kleve.
JONDET, M., DOMINIQUE, S., SCHOLLER, R. (1984): Effects of freezing and
thawing on mammalian oocyte. Cryobiology (1984); 21: 192-199.
KASAI, M. (1996): Simple and efficient methods for vitrification of mammalian
embryos. Anim. Reprod. Science (1996); 2: 229.
KASAI, M., NIWA, K., IRITANI, A. (1981): Effect of various cryoprotective a-
gents on the survival of unfrozen and frozen mouse embryos. J. Re-
prod. Fertil. (1981); 63: 175-180.
KASAI, M., NIWA, K., IRITANI, A. (1983): Protective effect of sucrose on the
survival of mouse and rat embryos stored at 0°C. J. Reprod. Fertil.
(1983); 68: 377-380.
KASAI, M., KOMI, J.H., TAKAKAMO, A., TSUDERA, H., SAKURAI, T., MA-
CHIDA, R.W. (1990): A simple method for mouse embryo cryopreser-
vation in a low toxicity vitrification solution, without appreciable loss of
viability. J. Reprod. Fertil. (1990); 89: 91-97.

89
KASAI, M., ITO, K., EDASHIGE, K. (2002): Morphological appearance of the
cryopreserved mouse blastocyst as a tool to identify the type of cry-
oinjury. Hum. Reprod. (2002); 17 (7): 1863-1874.
KAUFFOLD, P., THAMM, I. (1985): Zustandsbeurteilung von Rinderembryo-
nen. Arbeitsanleitung für den Embryotransfer. Forschungszentrum für
Tierproduktion, Dummersdorf-Rostock.
KENNEDY, L.G., BOLAND, M.P., GORDON, I. (1983): The effect of embryo
quality at freezing on subsequent development of thawed cow embry-
os. Theriogenology (1983); 19: 823-832.
KLING, A. (1997): Vergleichende Analyse der Ergebnisse aus dem Transfer fri-
scher, in Glyzerin und Äthylenglykol kryokonservierter Rinderembryo-
nen. Diss. Universität Leipzig.
KOPF, G.S. (1988): Choice, preparation and use of culturemedium. In: Wolf,
D.P., Bavister, B.D., Gerrity, M., Kopf, G.S. (Hrsg.): In vitro fertilization
and embryotransfer. A manual of basic techniques. Plenum press,
New York; 47-56.
KOVACS, M.S., LOWE, L., KUEHN, M.R. (1993): Use of superovulated mice
as embryo donors for ES cell injection chimeras. Lab. Anim. Sci.
(1993); 1: 91-93.
KRAG, K.T., KÖHLER, J.-M., WRIGHT, R.W. (1985a): A method for freezing
early murine embryos by plunging directly into liquide nitrogen. Theri-
ogenology (1985); 23 (1): 199.
KRAG, K.T., KÖHLER, J.-M., WRIGHT, R.W. (1985b): Trehalose: A non per-
meable cryoprotectant for direct freezing of early stage murine embry-
os. Theriogenology (1985); 23 (1): 200.

90
LADD, P.C., PLAIR, A.C., BRADIILOW, B.K., SNYDER, D.A. (1982): Deve-
lopment of early mouse and sheep embryos in serum-supplemented,
phosphate-buffered saline. Theriogenology (1982); 17: 94.
LANDSVERK, K., REFSDAL, A.O., VATN, T. (1992): Pregnancy rates with
frozen bovine embryos using two different methods for dilution of gly-
cerol. Theriogenology (1992); 37: 241.
LANGE, H. (1995): Cryopreservation of bovine embryos and demi-embryos u-
sing ethylene glycol for direct transfer after thawing. Theriogenology
(1995); 43: 258.
LEE, R.K.-K., HO, H.-Y., YU, S.-L., LU, C.-H. (2003): Blastocyst development
after cryopreservation and subcutaneous transplantation of mouse
ovarian tissue. J. Ass. Reprod. Gene (2005); 22(2): 95-101.
LEHN-JENSEN, H. (1980): Bovine egg transplantation. Nord. Vet.-Med. (1980);
32: 523-532.
LEHN-JENSEN, H. (1981): Deep-freezing of cow embryos. Nord. Vet.-Med.
(1981); 33: 476-483.
LEHN-JENSEN, H. (1984): Deep freezing of cattle embryos. 10th Intern. Congr.
Anim. Reprod. and A.I., Illinois; 4: II-1 – II-12.
LEHN-JENSEN, H. (1986): Cryopreservation of bovine embryos. A/S Carl Fr.
Mortensen, Copenhagen.
LEHN-JENSEN, H., GREVE, T. (1980): Preservation of bovine blastocysts in li-
quid nitrogen using two different freezing/thawing rates and
DMSO/glycerol as cryoprotectans. Theriogenology (1980); 13: 100.

91
LEHN-JENSEN, H., GREVE, T. (1982): The survival of cow blastocysts frozen
in 1,4 M Glycerol after plunging between -15 and -60°C and rapid
thawing. Theriogenology (1982); 17: 95.
LEIBO, S.P. (1977): Fundamental cryobiology of mouse ova and embryos. In:
Elliot, K. & Whelan, J. (Hrsg.): The Freezing of Mammalian Embryos.
Elsevier, Amsterdam, Ciba Foundation Symposium (1977); 52: 69-92.
LEIBO, S.P. (1981a): Preservation of ova and embryos by freezing. In: Bra-
ckett, B.G., Seidel, G.E., Seidel, S.M. (Hrsg.): New Technologies in
Animal Breeding. Academic Press, New York (1981); 127-139.
LEIBO, S.P. (1981b): Introduction to embryo freezing. In: Zeilmaker, G.H.
(Hrsg.): Frozen storage of laboratory animals. G. Fischer Verlag,
Stuttgart (1981); 1-19.
LEIBO, S.P. (1983): Field trial of one-step frozen bovine embryos transferred
non-surgically. Theriogenology (1983); 19: 139.
LEIBO, S.P. (1984): A one-step method for direct nonsurgical transfer of fro-
zen-thawed bovine embryos. Theriogenology (1984); 21: 767-790.
LEIBO, S.P. (1988): Cryopreservation of embryos. 11th Int. Congr. Anim. Re-
prod. and A.I.. Dublin; 5: 370-377.
LEIBO, S.P. (1989): Equilibrium and nonequilibrium cryopreservation of embry-
os. Theriogenology (1989); 31: 85-93.
LEIBO, S.P., MAZUR, P., JACKOWSKI, S.C. (1974): Factors affecting survival
of mouse embryos during freezing and thawing. Experim. Cell Re-
search (1974); 89: 79-88.

92
LEIBO, S.P., RALL, W.F., MAZUR, P. (1977): Comparison of the observed and
calculated water loss from mouse ova at subzero temperatures. Cryo-
biology, Abstr. No. 65.
LEIBO, S.P., MAZUR, P. (1978): Methods for the preservation of mammalian
embryos by freezing. In: Daniels, J.C. (Hrsg.): Methods in Mammalian
Reproduction. Academic Press, New York (1978); 179-201.
LEIBO, S.P. (1990): The theory and practice of seeding in embryo cryopreser-
vation. Embryo Transfer Newsletter (1990); 8: 7-9.
LINDNER, G.M., WRIGHT jr., R.W. (1983): Bovine embryo morphology and
evaluation. Theriogenology (1983); 20: 407-416.
LOVELOCK, J.E. (1954): Physical instability and thermal shock in red cells.
Nature (1954); 173: 659-661.
LOVELOCK, J.E., BISHOP, M.W.H. (1959): Prevention of freezing damage to
living cells by dimethyl sulfoxide. Nature (1959); 183: 1394-1395.
LUDWIG, M., MUSCHALLA, H., AL-HASANI, S., DIEDRICH, K. (1998): The
effect of multiple cryopreservation procedures and blastomere biopsy
on the in-vitro development of mouse embryos. Hum. Reprod. (1998);
13 (2): 3165-3168.
LYON, M.F., GLENISTER, P.H., WHITTINGHAM, D.G. (1981): Long-term via-
bility of embryos stored under irradiation. In: Zeilmaker, G.H. (Hrsg.):
Frozen storage of laboratory animals. G. Fischer Verlag, Stuttgart,
New York (1981); 139-147.
MAHABIR, E. (2000): In vitro fertilization in the mouse and migration and in
vitro culture of primordial germ cells in the pig. Diss. Uni. Göttingen.

93
MAHMOUDZADEH, A.R., VAN SOOM, A., VAN VLAENDEREN, I., DE KRUIF,
A. (1993): A comparative study of the effect of one-step addition of
different vitrification solutions on in vitro survival of vitrified bovine
embryos. Theriogenology (1993); 39: 1291-1302.
MAPLETOFT, R.J., MOKER, J.S., HAGELE, W.C. (1987): Comparision of two
methods of removing glycerol from frozen-thawed mouse embryos
with sucrose. Theriogenology (1987); 27: 255.
MAPLETOFT, R.J., MOKER, J., PALASZ, A. (1989): The effect of sucrose
concentration, temperature and time on survival of fresh mouse emb-
ryos in culture. Theriogenology (1989); 31: 225.
MASSIP, A. (2000): Cryopreservation of embryos of farm animals. Reprod.
Dom. Anim. (2001); 36: 49-55.
MASSIP, A., VAN DER ZWALMEN, P., ECTORS, F., DE COSTER, R.,
D´IETEREN, G., HANZEN, C. (1979): Deep freezing of cattle embry-
os in glass ampules or fench straws. Theriogenology (1979); 12: 79-
84.
MASSIP, A., VAN DER ZWALMEN, P., HANZEN, C., ECTORS, F. (1982):
Fast freezing of cow embryos in french straws with an automatic pro-
gram. Theriogenology (1982); 18; 325-332.
MASSIP, A., VAN DER ZWALMEN, P. (1982): In virto survival of mouse emb-
ryos frozen in glycerol or glycerol-sucrose. Cryo-Letters (1982); 3:
326.
MASSIP, A., VAN DER ZWALMEN, P., ECTORS, F. (1987): Recent progress
in cryopreservation of cattle embryos. Theriogenology (1987); 27: 69-
79.

94
MATAR, S. (2002): Zwischenlagerung von tiefgefrorenen Mäuseembryonen bei
verschiedenen Temperaturen. Masterarbeit Uni. Göttingen.
MAURER, R.R., BANK, H. (1973): Freezing and storage of eight-cell mouse
embryos. Cryobiology (1973); 10 (6): 509.
MAURER, R.R., HASEMAN, J.K. (1976): Freezing morula stage rabbit embry-
os. Biol. of Reprod. (1976);14: 256-263.
MAURER, R.R., BANK, H., STAPLES, R.E. (1977): Pre- and postnatal deve-
lopment of mouse embryos after storage for different periods at cryo-
genic temperatures. Biol. Reprod. (1977); 16: 139-146.
MAZUR, P. (1965): Causes of injury in frozen and thawed cells. Fed. Proc. A-
mer. Soc. Exp. Biol. (1965); 24 (15): 175-182.
MAZUR, P. (1966): Physical and chemical basis of injury in single celled mic-
roorganisms subjected to freezing and thawing. In: Cryobiology H.T.
Meryman (ed.), London, Academic Press (1966); 6: 213-315.
MAZUR, P. (1970): Cryobiology: The freezing of biological systems. Science
(1970); 168: 939-949.
MAZUR, P. (1977): Slow-freezing injury in mammalian cells. In: Elliot, K. &
Whelan, J. (Hrsg.): The Freezing of Mammalian Embryos. Elsevier,
Amsterdam, Ciba Foundation Symposium (1977); 52: 19-42.
MAZUR, P. (1977): The role of intracellular freezing in the death of cells cooled
at supraoptimal rates. Cryobiology (1977); 14: 251-272.
MAZUR, P. (1980): Fundamental aspects of freezing of cells with emphasis on
mammalian ova and embryos. Proc. 9th Int. Congr. Anim. Reproducti-
on and A.I., Madrid (1980); 1: 99-114.

95
MAZUR, P. (1985): Basic concepts in freezing cells. First Int. Conf. On Deep
Freezing of Boar Semen, Uppsala; 91-111.
MAZUR, P. (1990): Equilibrium, quasi-equilibrium and non-equilibrium freezing
of mammalian embryos. Cell Biophysics (1990); 17: 53-92.
MAZUR, P., SCHNEIDER, U. (1986): Osmotic responses of preimplantation
mouse and bovine embryos and their cryobiological implications. Cell
Biophysics (1986); 8: 259-285.
McGANN, L.E. (1978): Differing actions of penetrating and non-penetrating
cryoprotective agents. Cryobiology (1978); 15: 382-390.
McGINNIS, L.K., DUPLANTIS jr., S.C., WALLER, S.L., YOUNGS, C.R.
(1989): The use of ethylene glycol for cryopreservation of sheep emb-
ryos. Theriogenology (1989); 31: 226.
McINTOSH, A., HAZELEGER, N.L. (1994): The use of ethylene glycol for cry-
opreservation of sheep embryos. Theriogenology (1994); 41: 253.
McLAREN, A., BIGGERS, J.D. (1958): Successful development and birth of
mice cultivated in vitro as early embryos. Nature (1958);182: 877-878.
MERMILLOD, P., WILS, C., MASSIP, A., DESSY, F. (1992): In vitro survival of
frozen-thawed in vitro produced cattle blastocysts. Se Reunion
A.E.T.E., Lyon, 11.-12. September 1992.
MERRY, D.A., ALLEN, R.L., KRAG, K., WRIGHT jr., R.W. (1983): Sucrose di-
lutionof frozen mouse embryos: interaction of glycerol and sucrose
concentrations. Theriogenology (1983); 20: 325-332.
MERYMAN, H.T. (1971): Cryoprotective agents. Cryobiology (1971); 8: 173-
183.

96
MERYMAN H.T. (1974): Freezing injury and ist prevention in living cells. Annu.
Rev. Biophys. Bioeng. (1974); 3: 341-363.
MERYMAN, H.T., WILLIAMS, R.J., DOUGLAS, M.ST.J. (1977): Freezing inju-
ry from „solution effects“ and its prevention by natural or artificial cry-
oprotection. Cryobiology (1977); 14: 287-302.
MEYBERG, M. (2007): Persönliche Gespräche, Russikon, Schweiz.
MINTZ, B. (1967): Gene control of mammalian pigment differentiantin. I. Clonal
origin of melanocytes. Proc. Natl. Acad. Sci. USA (1967); 58: 344-351.
MIYAMOTO, H., ISHIBASHI, T. (1976): Factors involved in the survival of mou-
se eight-cell and morula embryos frozen to -196°C. -Abstract in
english- Japanese Jounal of Zootechnical Science (1976); 47: 23-28.
MIYAMOTO, H., ISHIBASHI, T. (1977): Survival of froozen-thawed mouse and
rat embryos in the presence of ethylene glycol. J. Reprod. Fertil.
(1977); 50: 373-375.
MIYAMOTO, H., ISHIBASHI, T. (1981): Effects of the temperature of ice-
seeding on survival of frozen-and-thawed mouse morulae. Experimen-
tia (1981); 37: 187-188.
MIYAMOTO, H., ISHIBASHI, T. (1983a): Solid CO2 freezing of mouse embry-
os. J. Reprod. Fertil. (1983); 67: 107-111.
MIYAMOTO, H., ISHIBASHI, T. (1983b): Survival of mouse embryos frozen-
thawed slowly or rapidly in the presence of various cryoprotectans. J.
exp. Zoology (1983); 226: 123-127.
MIYAMOTO, H., SUGIMOTO, M., OKUDA, T., TAKAHAGI, Y., MANABE, N.
(1998): Assessment of the effect of dimethyl sulfoxide on the survival

97
of cryopreserved mouse morulae by single step exposure and rapid
freezing. Theriogenology (1998); 49: 171.
NIBART, M., HUMBLOT, P. (1997): Pregnancy rates following direct transfer of
glycerol sucrose or ethylene glycol cryopreserved bovine embryos.
Theriogenology (1997); 47: 371.
NIEMANN, H. (1980): Die fluoreszenzmikroskopische Beurteilung der Entwick-
lungsfähigkeit früher Embryonalstadien von Kaninchen und Rind mit
dem FDA- und DAPI-Test. Diss. Tierärztl. Hochschule, Hannover.
NIEMANN, H. (1982): Neuere Ergebnisse und Erkenntnisse aus Tiefgefrierver-
suchen mit Rinderembryonen. Der Tierzüchter (1982); 34: 412-413.
NIEMANN, H. (1983): Übersichtreferat: Theorie und Praxis des Tiefgefrierens
von Rinderembryonen. Dtsch. tierärztl. Wschr. (1983); 90: 81-120.
NIEMANN, H. (1991a): Cryopreservation of ova and embryos from livestock:
current status and research needs. Theriogenology (1991); 35: 109-
124.
NIEMANN, H. (1991b): Entwicklungsstand von Embryotransfer und assoziier-
ten Biotechniken bei landwirtschaftlichen Nutztieren. Züchtungskunde
(1991); 63: 183-190.
NIEMANN, H., MEINEKE, B. (1993): Embryotransfer und assoziierte Biotechni-
ken bei landwirtschaftlichen Nutztieren. Enke Verlag, Stuttgart.
NGUYEN, B.X., RENARD, J.-P., GARNIER, V. (1983): Rapid freezing and
thawing of 2-cell stage rabbit embryos after partial dehydration at
room temperature. Cryobiology (1983); 20: 742.

98
NOHNER, H.-P. (1986): Versuche zur Tiefgefrierkonservierung von Rinderemb-
ryonen unter besonderer Berücksichtigung verschiedener Ausverdün-
nungsverfahren. Diss. Tierärztl. Hochschule, Hannover.
NOWSHARI, M.A. (2006): Telefonische Gespräche, Bad Schwartau.
NOWSHARI, M.A. (2007): Telefonische Gespräche, Bad Schwartau.
NOWSHARI, M.A., BREM, G. (1998a): Cryopreservation of expended mouse
blastocysts by rapid-freezing procedure. Theriogenology (1998); 49:
172.
NOWSHARI, M.A., BREM, G. (1998b): Effect of cryoprotectans and their con-
centration on post-thaw survival and development of expended mouse
blastocysts frozen by a simple rapid-freezing procedure. Theriogeno-
logy (1998); 50:1001-1013.
NOWSHARI, M.A., BREM, G. (2001): Effect of freezing rate and exposure time
to cryoprotectant on the development of mouse pronuclear stage
embryos. Hum. Reprod. (2001); 16 (11): 2368-2373.
OKAMOTO, M., NAKAGATA, N., TOYODA Y. (2001): Cryopreservation and
transport of mouse spermatozoa at -79°C. Exp. Anim. (2001); 50 (1):
83-86.
OZGUNEN, K. T., ERDOGAN, S. (2001): Effect of gonadotrophin dose on oo-
cyte retrieval in superovulated BALB/c mice. Theriogenology (2001);
56 (3): 435-445.
PAVEL, G. (1985): Versuche zur Tiefgefrierkonservierung von Rinderembryo-
nen im One-Step-Verfahren unter Verwendung eines Kryostaten.
Diss. Tierärztl. Hochschule, Hannover.

99
PEDRO, P.B., YOKOYAMA, E., ZHU, S.E., YOSHIDA, N., VALDEZ, D.M., jr.,
TANAKA, M., EDASHIGE, K., KASAI, M. (2005): Permeabilität of
mouse oocytes and embryos at various developmental stages to five
cryoprotectans. J Reprod. Dev. (2005); 51(2): 235-246.
PEURA, A., TROUNSON, A.O., FREEMANN, L. (1985): Ultra rapid embryo
freezing. In: 4th World Congress of In Vitro Fertilization. Abstract
Handbook, Melbourne, 1985; 244.
POLGE, C., SMITH, A.U., PARKES, A.S. (1949): Revival of spermatozoa after
vitrification and dehydration at low temperatures. Nature (1949); 164:
666.
POLGE, C., WILLADSEN, S.M. (1978): Freezing eggs and embryos of farm a-
nimals. Cryobiology (1978); 15: 370-373.
PRATHER, R.S., SPIRE, M.F., SCHALLES, R.R. (1987): Evaluation of cry-
opreservation techniques for bovine embryos. Theriogenology (1987);
28: 195-204.
PURSEL, V. G., WALL, R.J., REXROAD, C.E., HAMMER, R.E., BRINSTER
R.L. (1985): A rapid whole-mount staining procedure for nuclei of
mammalian embryos. Theriogenology (1985); 24: 687-700.
RAFFERTY jr., K.A. (1970): Methods in experimental embryology of the mou-
se. The John Hopkins Press, Baltimore and London (1970).
RALL, W.F. (1981): The role of intracellular ice in the slow warming injury of
mouse embryos. In: Zeilmaker, G.H. (Hrsg.): Frozen storage of labo-
ratory animals. G. Fischer Verlag, Stuttgart, New York (1981); 33-44.
RALL, W.F. (1992): Cryopreservation of oocytes and embryos: methods and
applications. Anim. Reprod. Science (1992); 28: 237-245.

100
RALL, W.F., REID, D.S., FARRANT, J. (1980): Innocuous biological freezing
during warming. Nature (1980); 286: 511-514.
RALL, W.F., MAZUR, P., McGRATH, J.J. (1983): Depression of the ice-
nucleation temperature of rapid cooled mouse embryos by glycerol
and DMSO. Biophysical Journal (1983); 41: 1-12.
RALL, W.F., POLGE, C. (1984): Effect of warming rate on mouse embryos fro-
zen and thawed in glycerol. J. Reprod. Fertil. (1984); 70: 285-292.
RALL, W.F., FAHY, G.M. (1985): Ice-free cryopreservation of mouse embryos
at -196°C by vitrification. Nature (1985); 313: 573-575.
RALL, W.F., WOOD, M., WHITTINGHAM, D. (1985): Embryo preservation by
vitrification. In: Human Reproduction – Abstracts and Programme of
the 1st meeting of the European Society of Human Reproduction and
Embryology, Bonn, 1985; 34.
RAYOS, A.A., TAKAHASHI, Y., HISHINUMA, M., KANAGAWA, H (1992a):
Quick freezing of one-cell mouse embryos using ethylene glycol with
sucrose. Theriogenology (1992); 37: 595-603.
RAYOS, A.A., TAKAHASHI, Y., HISHINUMA, M., KANAGAWA, H. (1992b):
Quick freezing of mouse two-, four- and eight-cell embryos with ethy-
lene glycol plus sucrose or lactose: effects of development stage and
equilibration period on survival in vitro. Anim. Reprod. Science (1992);
27: 239-245.
REISCHL, J., AL YAKOUB, A. (2006): Arbeitsgruppe Fortpflanzung, Institut f.
Tierz. Haustiergenetik, Uni. Göttingen.
RENARD, J.-P., HEYMAN, Y., OZIL, J. (1982): Congélation del l’embryo bovin:
une nouvelle méthode de décongélation pour le transfert cervical

101
d’embryons conditionés une seule fois en pailettes. Ann. Méd. Vét.
(1982); 126: 23-32.
RENARD, J.-P., HEYMAN, Y., LEYMONIE, P., PLAT, J.-C. (1983): Sucrose di-
lution: A technique for field transfer of bovine embryos frozen in the
straw. Theriogenology (1983); 19: 145.
RENARD, J.-P., NGUYEN, B.-X., GARNIER, V. (1984): Two-step freezing of
two-cell rabbit embryos after partial dehydration at room temperature.
J. Reprod. Fertil. (1984); 71: 573-580.
RENARD, J.-P., BABINET, C. (1984): High survival of mouse embryos after
rapid freezing and thawing inside plastic straws with 1,2 propandiol as
cryoprotectant. J. exp. Zoology (1984); 230: 443-448.
REVEL, A., MOSHE, N., HELMAN, A., SAFRAN, A., SIMON, A., KOLER, M.
(2004): Mouse embryos generated from frozen-thawed oocytes can
successfully survive a second cryopreservation. Hum. Reprod. (2004);
19 (3): 666-669.
ROTMAN, B., PAPERMASTER, B.W. (1966): Membrane properties of living
mammalian cells as studied by enzymatic hydrolysis of fluorogenic es-
ters. Proc. nat. Acad. Sci. (1966); 55: 134-141.
SCHEIWE, M.W., RAU, G. (1981): Biokältetechnik: Verfahren der Gefrierkon-
servierung in der Medizin. Chem.-Ing.-Techn. (1981); 53 (10): 787-
797.
SCHIEWE, M.C., SCHMIDT, P.M., RALL, W.F., WILDT, D.E. (1985): Influence
of cryoprotectant and plunge temperature on post-thaw viability of bo-
vine embryos. Biol. Reprod. (1985); 32 (1): 98.

102
SCHIEWE, M.C., SCHMIDT, P.M., WILDT, D.E. (1987): A comparative study of
mouse embryo freezing-preservation including the examination of a
thermoelectric freezing device. Cryobiology (1978); 24: 238-246.
SCHILLING, E., DÖPKE, H.H. (1978): A rapid diagnostic test for the viability of
early cattle embryos using diacetylfluorescin. Naturwissenschaften
(1978); 65: 658-659.
SCHILLING, E., SMIDT, D. (1979): Beurteilung der Lebensfähigkeit früher
Embryonalstadien beim Rind. Tagung: „Physiologie und Pathologie
der Haustiere“, München. Zuchthygiene (1979); 14: 86.
SCHILLING, E., D. SMIDT, B. SACHER, S. EL KASCHAB (1979): Diagnosis
of the viability of early bovine embryos by fluorescence microscopy.
Ann. Biol. Anim. Bioch. Biophys. (1979);19 (5): 1625-1629.
SCHMIDT, P.M., HANSEN, C.T., WILDT, D.E. (1985): Viability of frozen-
thawed mouse embryos is affected by genotyp. Biol. Reprod. (1985);
32: 507-514.
SCHNEIDER, U., HAHN, J., SULZER, H. (1974): Erste Ergebnisse der Tiefge-
frierkonservierung von Mäuse- und Kanincheneizellen. Deut. Tierärztl.
Wochenschr. (1974); 81: 470-472.
SCHNEIDER, U., MAZUR, P. (1984): Osmotic consequences of cryoprotectant
permeability and its relation to the survival of frozen-thawed embryos.
Theriogenology (1984); 21: 68-79.
SCHUBERT, W. (1984): Tiefgefrierversuche mit Rinderembryonen zur metho-
dischen Verbesserung der Kryoprotektion, der Ausverdünnung und
des Auftauvorganges. Diss., Tierärztl. Hochschule, Hannover.
SCHWARTZ, G.J., DILLER, K.R. (1983): Osmotic response of individual cells
during freezing. Cryobiology (1983); 20: 61-77.

103
SEIBT, W. (2003): Physik für Mediziner. 5. Auflage; Georg Thieme Verlag,
Stuttgart.
SEIDEL, G.E. (1989): Methods and comparative aspects of embryo cryopreser-
vation in domestic animals. Equine Vet. Journal Suppl. (1989); 8: 77-
79.
SEIDEL, G.E., ELSDEN, R.P., BRINK, Z. (1990): Cryopreservation of bovine
embryos in media with chemically defined macromolecules. Therioge-
nology (1990); 33: 322.
SMIDT, D., MONZAVIFAR, H. (1969): Untersuchungen zur phänotypischen
Ausprägung unterschiedlicher Fruchtbarkeitsmerkmale bei Mäusen.
Zeitschr. f. Tierz. u. Züchtungsbiol. (1969); 86: 69-80.
SMITH, A.U., POLGE, C., SMILES, J. (1951): Microskopic observation of living
cells during freezing and thawing. J. Royal Microscopic Soc. (1951);
71: 186-195.
SMORAG, Z.L., KATSKE, M., WIERZBOWSK, I. (1981): Some facors affecting
the success of embryo-freezing: storage before freezing, superovula-
tion rate, PBS-concentration, cooling and thawing rates. In: Zeilmaker
(Hrsg.): Frozen storage of laboratory animals. 45-53.
SOMMERFELD, V. (1997): Untersuchungen zur Kryokonservierung in vitro
produzierter Rinderembryonen mit Ethylenglycol durch kontrollierte
Gefrierverfahren oder Vitrifikation. Diss. Tierärztl. Hochschule, Han-
nover.
SOMMERFELD, V., NIEMANN, H. (1999): Cryopreservation of bovine in vitro
produced embryos using ethylene glycol in controlled freezing or vitri-
fication. Cryobiology (1999); 38: 95-105.

104
STREICHER, M. (1998): Untersuchungen zur Feststellung der Befruchtungsfä-
higkeit von Sperma verschiedener Bullen nach Änderung des Swim-
up-Verfahrens, der Heparinzugabe und der Kapazitationsdauer bei
der In-Vitro-Befruchtung von Rinderoozyten sowie Untersuchungen
zur Kryokonservierung von in vitro produzierten Rinderembryonen mit
Hilfe von Ethylenglycol und Sucrose. Diss. Tierärztl. Hochschule,
Hannover.
SUZUKI,T., YAMAMOTO, M., OOE, M., SAKATA, A., MATSUOKA, M., NIS-
HIKATA, Y., OKAMOTO, K. (1990): Effect of sucrose concentration
used for one-step dilution upon in vitro and in vivo survival of bovine
embryos refrigerated in glycerol and 1,2-propandiol. Theriogenology
(1990); 34: 1051-1057.
SZELL, A., SHELTON, J.N. (1986): Sucrose dilution of glycerol from mouse
embryos frozen rapidly in liquid nitrogen vapour. J. Reprod. Fertil.
(1986); 76: 401-408.
TAKEDA, T., ELSDEN R.P., SEIDEL jr., G.E. (1984): Cryopreservation of
mouse embryos by direct plunging into liquide nitrogen. Theriogenolo-
gy (1984); 21 (1): 266.
TAKEDA, T., ELSDEN R.P., SEIDEL jr., G.E. (1987): Use of sucrose during
removal of cryoprotectans after thawing eight-cell mouse embryos.
Theriogenology (1987); 28: 101-108.
TAO, J., TAMIS, R., FINK, K. (2001): Cryopreservation of mouse embryos at
morula/compact stage. J. Assist. Reprod. Genet. (2001); 4: 235-243.
TESTART, J., LASSALLE, B., BERTHE, V., BELAISCH-ALLART, J., FRYD-
MAN, R. (1985): Cryopreservation of human embryos. In: 4th World
Congress of In Vitro Fertilization. Abstract Handbook, Melbourne,
1985; 248.

105
THOMAS, W.K., BIERY, K.A., KRAEMER, D.C. (1989): Effect of cryoprotec-
tant concentration on mouse embryo development following rapid
freezing. Theriogenology (1989); 31: 266.
TROUNSON. A.O., WILLADSEN, S.M., ROWSON, L.E.A., NEWCOMB, R.
(1976a): The storage of cow eggs at room temperature and low tem-
peratures. J. Reprod. Fertil. (1976); 46: 173-178.
TROUNSON, A.O., WILLADSEN, S.M., ROWSON, L.E.A. (1976b): The in-
fluence of in vitro culture and cooling on the survival and development
of cow embryos. J. Reprod. Fertil. (1976); 47: 367-370.
TROUNSON, A.O., BRAND, A., AARTS, M.H. (1978): Non-surgical transfer of
deep-froozen bovine embryos. Theriogenology (1978); 10: 111-115.
TROUNSON, A.O., MOHR, L. (1983): Human pregnancy following cryopreser-
vation, thawing and transfer of an eight-cell embryo. Nature (1983);
305: 707-709.
TROUNSON,A,O., BRAND, A., AARTS, M.H. (1978a): Non-surgical transfer of
deep-frozzen bovine embryos. Theriogenology (1978); 10: 111-115.
TROUNSON, A.O., SHEA, B.F., OLLIS, G.W., JACOBSON, M.E. (1978b):
Frozen storage and transfer of bovine embryos. J. Anim. Sci. (1978);
47: 667-681.
UTSUMI, K., YUHARA, M., YAMADA, M. (1978): Viability of frozen two-cell
eggs and blastocysts in rat. Cryobiology (1978); 15: 689.
VALDEZ, C.A., ABAS MANZI, O., TAKAHASHI, Y., FUJIKAWA, S., KANA-
GAWA, H. (1992): Sucsessful cryopreservation for mouse blastocysts
using a new vitrification solution. J. Repro. Fertil. (1994); 96: 793-802.

106
VINCENTE, J.S., GARCIA-XIMENEZ, F. (1994): Osmotic and cryoprotective
effects of a mixture of DMSO and ethylene glycol on rabbit morulae.
Theriogenology (1994); 42: 1205-1215.
VIUFF, D., MADISON,V., HYTTEL, P., AVERY, B., GREVE, T. (1991): Fluo-
rescent intravital staining of bovine oocytes and zygotes. Theriogeno-
logy (1991); 35: 291.
VOELKEL, S.A., HU, Y.X. (1992a): Direct transfer of frozen-thawed bovine
embryos. Theriogenology (1992); 37: 23-37.
VOELKEL, S.A., HU, Y.X. (1992b): The use of ethylene glycol as a cryoprotec-
tant for bovine embryos allowing direct transfer of frozen-thawed emb-
ryos to recipient females. Theriogeneology (1992); 37: 687-697.
WARE, C.B., BOLAND, M.P. (1987): Effect of varying glycerol ans sucrose
concentration combinations on embryo survival rate in a one-step cry-
oprotectant removal from frozen-thawed ovine embryos. Theriogeno-
logy (1987); 27: 721-728.
WEIGEL, M.M. (1989): Versuche zur „langsamen“ Kryokonservierung von Mäu-
seembryonen mit dem automatisierten „offenen System“ mit Selbst-
seeding. Diss. Uni Erlangen-Nürnberg, 1989.
WEIß, J., MAEß, J., NEBENDAHL, K., ROSSBACH, W. (1996): Haus- und
Versuchstierpflege. Gustav Fischer Verlag.
WHITTEN, W.K. (1956): Culture of tubal mouse ova. Nature (1956); 117: 96.
WHITTEN, W.K. (1971): Nutrient requirements for the culture of preimplantant
embryos in vitro. Adv. Biosci. (1971); 6: 129-141.

107
WHITTEN, W. K., BIGGERS, J. D. (1968): Complete development in vitro of
the pre-implantation stages of the mouse in a simple chemically de-
fined medium. J. Reprod. Fert. (1986); 17: 399-401.
WHITTINGHAM, D.G. (1971): Survival of mouse embryos after freezing and
thawing. Nature (1971); 233: 125-126.
WHITTINGHAM, D.G. (1980): Preservation of embryos of the laboratory ani-
mals. In: Proceedings of the 9th International Congress in Animal Re-
production and Artificial Insemination, Madrid (1980); 2: 237-243.
WHITTINGHAM, D.G. (1981): Sensitivity of mouse embryos to the rate of tha-
wing. In: Zeilmaker, G.H. (Hrsg.): Frozen storage of laboratory ani-
mals. G. Fischer Verlag, Stuttgart, New York (1981); 1-32.
WHITTINGHAM, D.G., LEIBO, S.P., MAZUR, P. (1972): Survival of mouse
embryos frozen to -196°C and -269°C. Science (1971); 178: 411-414.
WILLADSEN, S.M. (1980): Deep freezing of embryos in the large domestic
species. 9th Int. Congr. Anim. Reprod. and A.I., Madrid (1980); 2: 255-
261.
WILLADSEN, S.M., POLGE, C., ROWSON, L.E.A., MOOR, R.M. (1976): Deep
freezing of sheep embryos. J. Reprod. Fertil. (1976); 46: 151-154.
WILLADSEN, S.M., POLGE, C., ROWSON, L.E.A. (1978): The viability of
deep-froozen cow embryos. J. Reprod. Fertil. (1978); 52: 391-393.
WILLIAMS, T.J., JOHNSON, S.E. (1985): Quick-freezing of day four mouse
embryos. Theriogenology (1985); 23 (1): 235.
WILMUT, I. (1972): The effect of cooling rate, warming rate, cryoprotective a-
gent and stage of development on survival of mouse embryos during
freezing and thawing. Life Sci. (1972); 11 (2): 1071-79.

108
WOLF (2006): Mäusehaus Humangenetik - persönliche Gespräche.
WOOD, M.J., FARRANT, J., WHITTINGHAM, D.G., LEE, H., HALSLEY, A.
(1978): Survival of mouse embryos after rapid thawing. Cryobiology
(1978); 15: 680-681.
WOOD, M.J., FARRANT, J. (1980): Preservation of mouse embryos by two-
step freezing. Cryobiology (1980); 17: 178-180.

109
Danksagung
Herrn Professor Dr. W. Holtz möchte ich für die Überlassung des interessanten
Themas, der Möglichkeit zur Erlernung reproduktionsmedizinischer Techniken
und Methoden sowie für die vielfältigen Ideen und Anregungen während der An-
fertigungszeit dieser Arbeit danken.
Bei Herrn Professor Dr. M. Gauly möchte ich mich für das Interesse an meinem
Thema und der Bereitschaft zur Übernahme des Korreferates bedanken.
Unsagbar zum Gelingen dieser Arbeit hat die Unterstützung der Firmen Con-
sarctic, Gück Zellkulturbedarf, Labotect, MTG, Varolab und Zeiss MicroImaging
beigetragen. Durch Dauerleihgaben der Geräte und Bereitstellung von Medien
konnten die Versuche auf höchstem technischen Niveau erfolgen, was die Qua-
lität der Aussagekraft der Versuche sicher maßgeblich mit beeinflußt hat. In
diesem Zusammenhang sei auch die Firma Dragon IVF International genannt,
auf deren Initiative hin diese große Kooperation zwischen Universität und In-
dustrie erstmals stattfinden konnten.
Erste technische Einführungen zum Thema wurden von Frau Dr. Judith Reischl
gegeben, der ich auch zu großem Dank verpflichtet bin.
Herrn Azzam Al Yakub möchte ich für die freundschaftliche Zusammenarbeit
meinen besonderen Dank ausdrücken.
Für die schnelle und qualifizierte Hilfe bei der statistischen Auswertung möchte
ich Frau Bianca Lind danken.
Den Kollegen und Kolleginnen insbesondere Frau Elisabeth Stüwe, Herrn Peter
Ludewig und Frau Birgit Sohnrey möchte ich für die motivierende, freundschaft-
liche Arbeitsatmosphäre danken.
Ein ganz herzliches Dankeschön geht an Dr. Manzoor Nowshari, Frau Dr. Ma-
nuela Ropeter-Scharfenstein, Herrn Dr. Klaus Nebendahl, Herrn Wolf (Human-
genetik) sowie allen Freunden und Mitarbeitern am Institut für Tierzucht und
Haustiergenetik.

110
Eidesstattliche Erklärung
Mit meiner Unterschrift versichere ich, die vorliegende Arbeit selbständig ver-
fasst und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt zu
haben
Göttingen, den 23.07.2007 ...............................................
(Stephanie Buhtz)


















