
Warum diagnostizieren wir viele angeborene Immundefekte...
36
Warum diagnostizieren wir viele angeborene Immundefekte erst bei Erwachsenen? -wurde im Kindesalter nicht erkannt/verkannt -späte Manifestation PID Leitsymptom: Pathologische Infektanfälligkeit AWMF-Leitlinie Diagnostik von primären Immundefekten“
Transcript of Warum diagnostizieren wir viele angeborene Immundefekte...

Warum diagnostizieren wir viele angeborene Immundefekte erst bei Erwachsenen? -wurde im Kindesalter nicht erkannt/verkannt -späte Manifestation
PID Leitsymptom: Pathologische Infektanfälligkeit AWMF-Leitlinie Diagnostik von primären Immundefekten“

„ELVIS“ als Akronym pathologischer Infektanfälligkeit
E Erreger („opportunistische“)
L Lokalisation (polytop, atypisch)
V Verlauf (protrahiert, rekurrent)
I Intensität (sog. „Major-Infektionen“)
S Summe
AMWF-Leitlinien
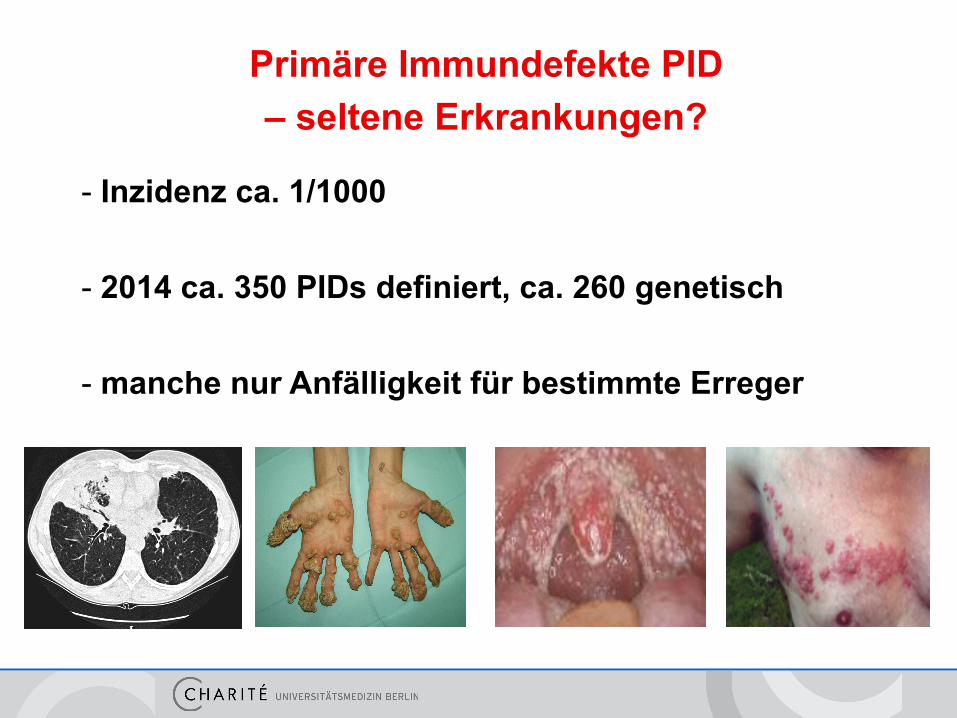
- Inzidenz ca. 1/1000
- 2014 ca. 350 PIDs definiert, ca. 260 genetisch
- manche nur Anfälligkeit für bestimmte Erreger
Primäre Immundefekte PID – seltene Erkrankungen?

PID – Klassifikation nach IUIS: 9 Hauptgruppen*
§ Combined immunodeficiencies (SCID, DOCK8 u.a.)
§ Well-defined syndromes with immunodeficiency (WAS, STAT3)
§ Predominantly antibody deficiencies (CVID u.a.)
§ Diseases of immune dysregulation (IPEX u.a.)
§ Congenital defects of phagocytes (CGD u.a)
§ Defects in innate immunity (MyD88 u.a.)
§ Autoinflammatory disorders (FMF u.a.)
§ Complement deficiencies (PNH u.a.)
§ Phenocopies of PID (somatische Neumutationen und Autoantikörper)
*Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies IUIS expert committee for primary immunodeficiency. Al-Herz W. et al. Front Immunol 2014
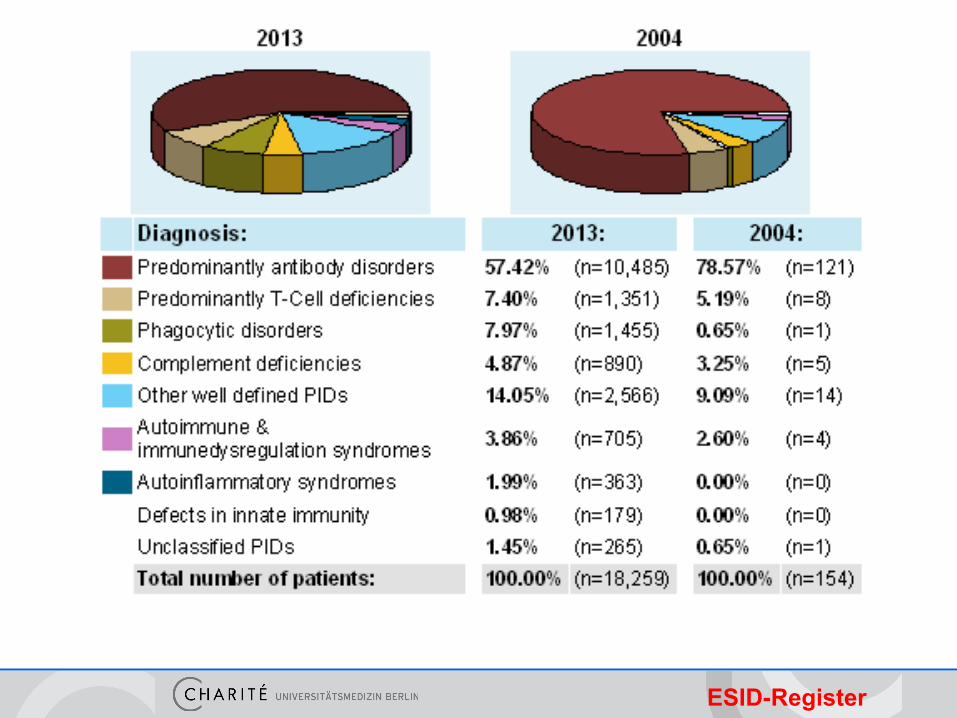
ESID-Register

PID – Klassifikation nach IUIS: 9 Hauptgruppen*
§ Combined immunodeficiencies (SCID, DOCK8 u.a.)
§ Well-defined syndromes with immunodeficiency (WAS, STAT3)
§ Predominantly antibody deficiencies (CVID u.a.)
§ Diseases of immune dysregulation (IPEX u.a.)
§ Congenital defects of phagocytes (CGD u.a)
§ Defects in innate immunity (MyD88 u.a.)
§ Autoinflammatory disorders (FMF u.a.)
§ Complement deficiencies (PNH u.a.)
§ Phenocopies of PID (somatische Neumutationen und Autoantikörper)
*Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies IUIS expert committee for primary immunodeficiency. Al-Herz W. et al. Front Immunol 2014

Pat. A.S., geb. 1987
Anamnese seit Kindheit Ekzeme am ganzen Körper als Kind häufige bronchopulmonale Infekte häufig kutane Abszesse
IgE 2580 U/l Eosinophilie 12%
Immundefekt Ambulanz für Erwachsene

Pat. A.C., geb. 1991
Anamnese 2010 ITP 2014 Ekzem am ganzen Körper Warzen
IgE 6900 U/l CD4 T-Zellen 0,07/nl
Immundefekt Ambulanz für Erwachsene

Hyper IgE Syndrome
Autosomal-dominantes Hyper-IgE-Syndrom (2007) STAT3 loss-of-function Mutation Ekzeme, kutane Infekte, Pneumonien, Gesichtsdysmorphie Autosomal-rezessives Hyper-IgE (2009) Dock8 loss-of-function Mutation Ekzeme, Warzen, Herpes schwerer T-Zell-Defekt
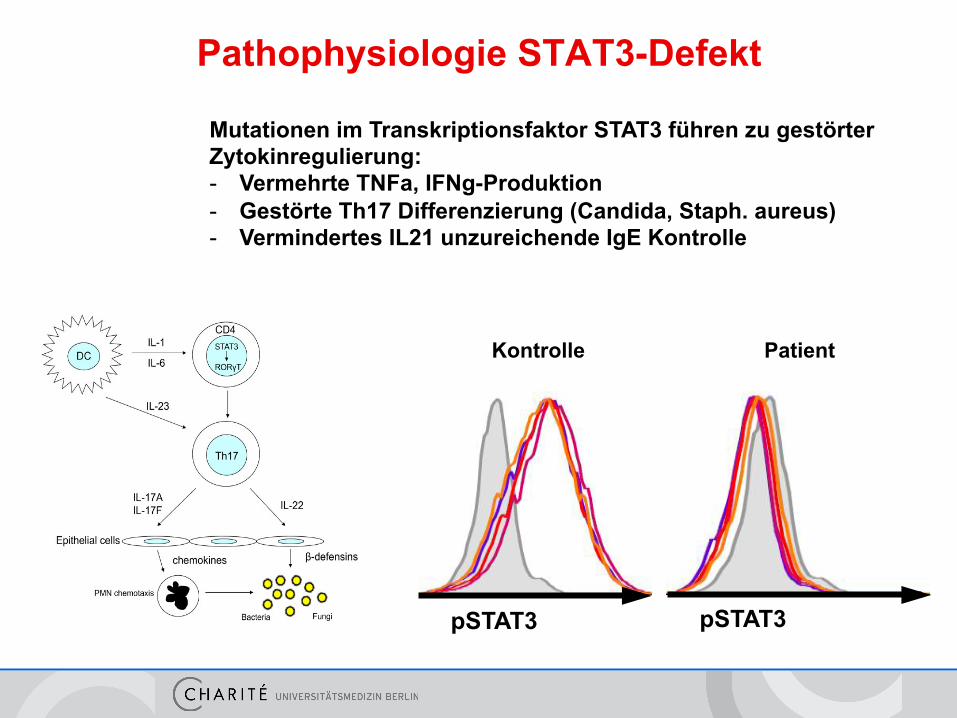
Pathophysiologie STAT3-Defekt
pSTAT3
Kontrolle Patient
pSTAT3
Mutationen im Transkriptionsfaktor STAT3 führen zu gestörter Zytokinregulierung: - Vermehrte TNFa, IFNg-Produktion - Gestörte Th17 Differenzierung (Candida, Staph. aureus) - Vermindertes IL21 unzureichende IgE Kontrolle

Hyper-IgE-Syndrome STAT3 DOCK8

Hyper IgE - Therapie
• Prophylaxe: TMPS 960 tgl., Diflucan,
Valaciclovir, HPV-Impfung
• IgG-Substitution bei bakteriellen Infekten
• ggfs HSCT bei DOCK8

PID – Klassifikation nach IUIS: 9 Hauptgruppen*
§ Combined immunodeficiencies (SCID, DOCK8 u.a.)
§ Well-defined syndromes with immunodeficiency (WAS, STAT3)
§ Predominantly antibody deficiencies (CVID u.a.)
§ Diseases of immune dysregulation (IPEX u.a.)
§ Congenital defects of phagocytes (CGD u.a)
§ Defects in innate immunity (MyD88 u.a.)
§ Autoinflammatory disorders (FMF u.a.)
§ Complement deficiencies (PNH u.a.)
§ Phenocopies of PID (somatische Neumutationen und Autoantikörper)
*Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies IUIS expert committee for primary immunodeficiency. Al-Herz W. et al. Front Immunol 2014
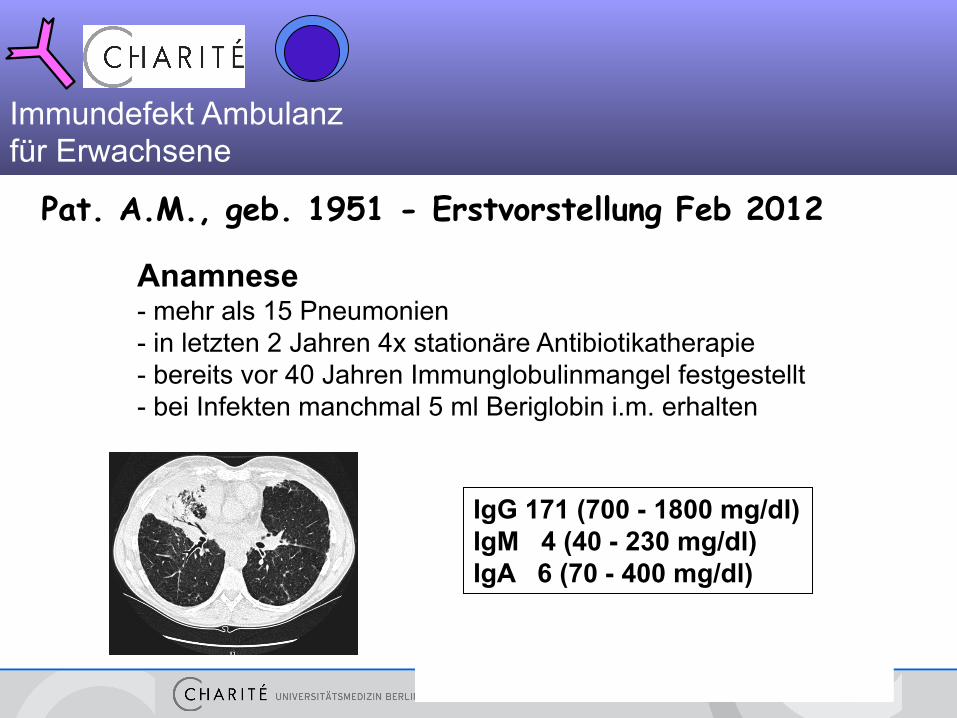
Pat. A.M., geb. 1951 - Erstvorstellung Feb 2012
Anamnese - mehr als 15 Pneumonien - in letzten 2 Jahren 4x stationäre Antibiotikatherapie - bereits vor 40 Jahren Immunglobulinmangel festgestellt - bei Infekten manchmal 5 ml Beriglobin i.m. erhalten
IgG 171 (700 - 1800 mg/dl) IgM 4 (40 - 230 mg/dl) IgA 6 (70 - 400 mg/dl)
Immundefekt Ambulanz für Erwachsene

Primäre Antikörpermangelerkrankungen
„Immunphänotyp“ Häufigkeit
Mangel aller 3 Ig-Hauptklassen und Fehlen von B-Zellen „Bruton Agammaglobulinämie“
1:100T
Mangel von IgG und IgA, zT. IgM und B-Zell-Reifungsstörung „CVID“
ca. 1:10T
IgG-Mangel geschätzt 1:20T IgA-Mangel 1:500 IgM-Mangel geschätzt 1:20T IgG-Subklassenmangel 1:100 Spezifischer Antikörpermangel (PcP) häufig Igl-Mangel bei syndromalen Erkrankungen (WAS) selten Igl-Mangel bei komplexen Erkrankungen (CFS) 0,1%

Common variable immune deficiency – CVID
• „peak“ Inzidenz 24. Lj • heterogene Erkrankung Diagnosekriterien • Mangel von IgG, IgA +/-IgM • verminderte Impfantwort oder verminderte Gedächtnis B-Zellen + Infekte • häufige Schleimhautinfekte • invasive v.a. bakterielle Infekte oder
• Autoimmunität • granulomatöse Entzündungen • Lymphoproliferation
erhöhtes Risko für Neoplasien
n= 52 CVID patients

„GARFIELD“ als Akronym gestörter Immunregulation
G Granulome („sarkoid-like lesions“)
A Autoimmunität (Autoimmun-Zytopenien, -Thyreoiditis, RA, u.a.)
R FI Rezidivierendes Fieber
E Ekzematöse Hautveränderungen
L Lymphoproliferation
D Darmentzündung
AMWF-Leitlinien
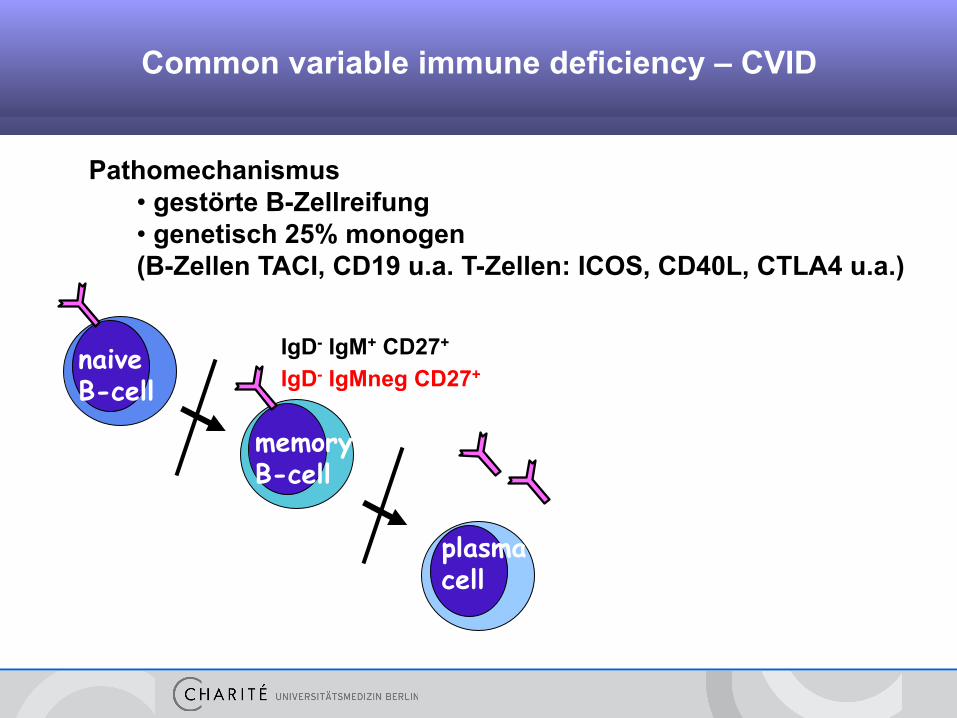
Pathomechanismus • gestörte B-Zellreifung • genetisch 25% monogen (B-Zellen TACI, CD19 u.a. T-Zellen: ICOS, CD40L, CTLA4 u.a.)
plasma cell
memory B-cell
naive B-cell
Background Common variable immune deficiency – CVID
IgD- IgM+ CD27+
IgD- IgMneg CD27+
Common variable immune deficiency – CVID
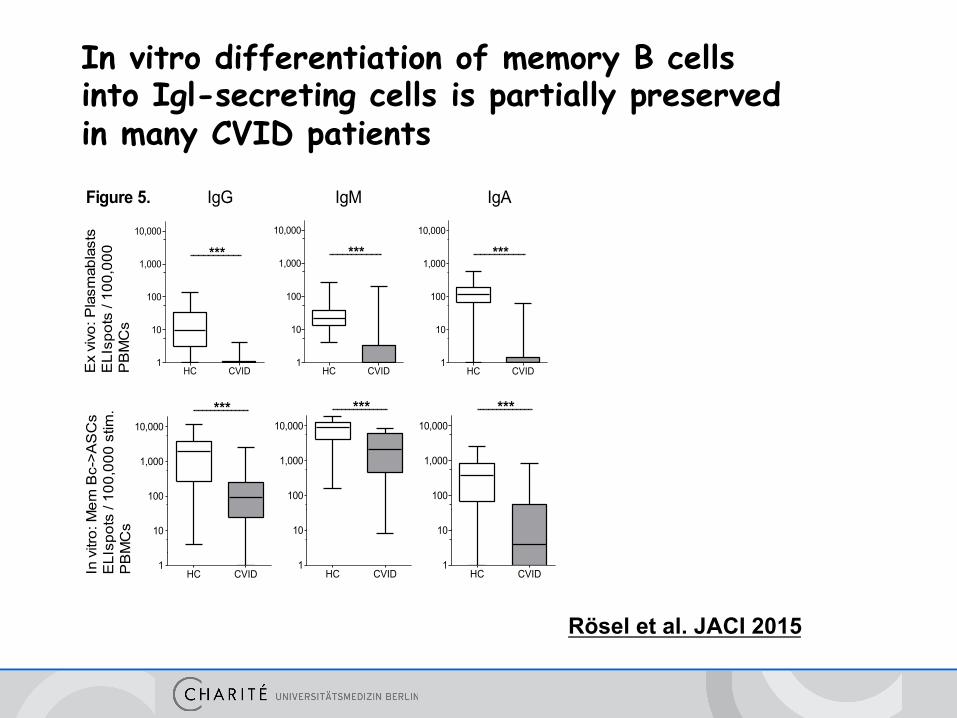
Rösel et al. JACI 2015
HC CVID1
10
100
1,000
10,000
***
HC CVID1
10
100
1,000
10,000
***
HC CVID1
10
100
1,000
10,000
***
HC CVID1
10
100
1,000
10,000***
HC CVID1
10
100
1,000
10,000***
HC CVID1
10
100
1,000
10,000***
Ex
vivo
: Pla
smab
last
sE
LIsp
ots
/ 100
,000
PB
MC
s
IgG IgM IgA
In v
itro:
Mem
Bc-
>AS
Cs
ELI
spot
s / 1
00,0
00 s
tim.
PB
MC
s
Figure 5.
In vitro differentiation of memory B cells into Igl-secreting cells is partially preserved in many CVID patients

Therapie des Antikörpermangel
Immunglobulinsubstitution (IgG) • absolute Indikation: IgG dauerhaft < 3-5g/l • relative Indikation: vermehrte Infekte und IgG 5-7g/l • spezifischer Ak-Mangel (spez. Pneumokokken-Polysaccharid-Ak) • IgG-Subklassenmangel
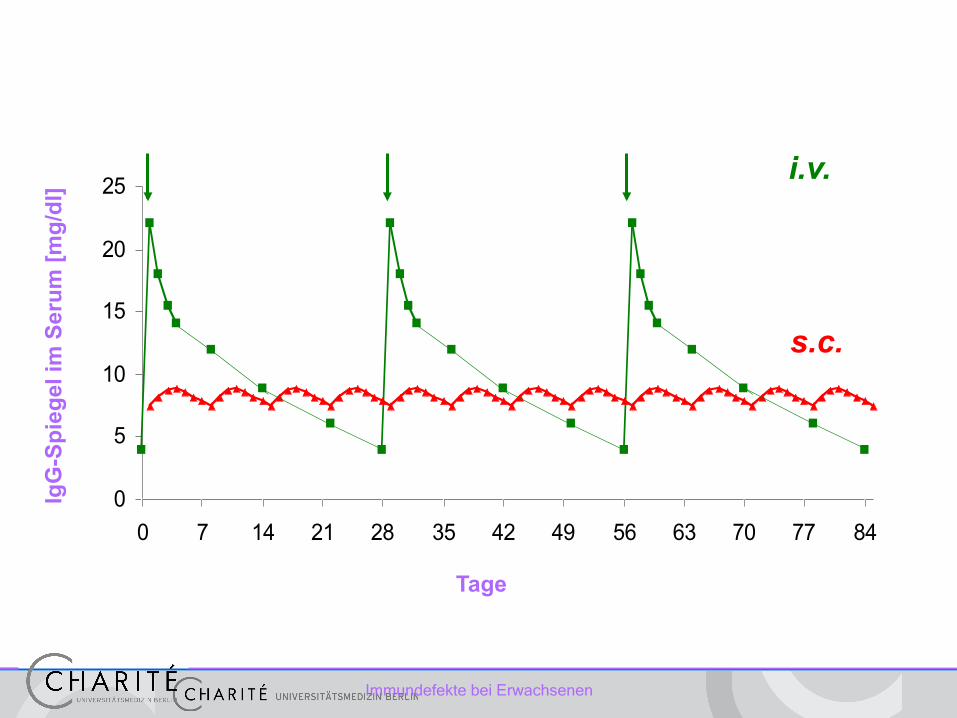
Immundefekte bei Erwachsenen
0
5
10
15
20
25
0 7 14 21 28 35 42 49 56 63 70 77 84
i.v.
s.c.
Tage
IgG
-Spi
egel
im S
erum
[mg/
dl]

PID – Klassifikation nach IUIS: 9 Hauptgruppen*
§ Combined immunodeficiencies (SCID, DOCK8 u.a.)
§ Well-defined syndromes with immunodeficiency (WAS, STAT3)
§ Predominantly antibody deficiencies (CVID u.a.)
§ Diseases of immune dysregulation (IPEX u.a.)
§ Congenital defects of phagocytes (CGD u.a)
§ Defects in innate immunity (MyD88 u.a.)
§ Autoinflammatory disorders (FMF u.a.)
§ Complement deficiencies (PNH u.a.)
§ Phenocopies of PID (somatische Neumutationen und Autoantikörper)
*Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies IUIS expert committee for primary immunodeficiency. Al-Herz W. et al. Front Immunol 2014
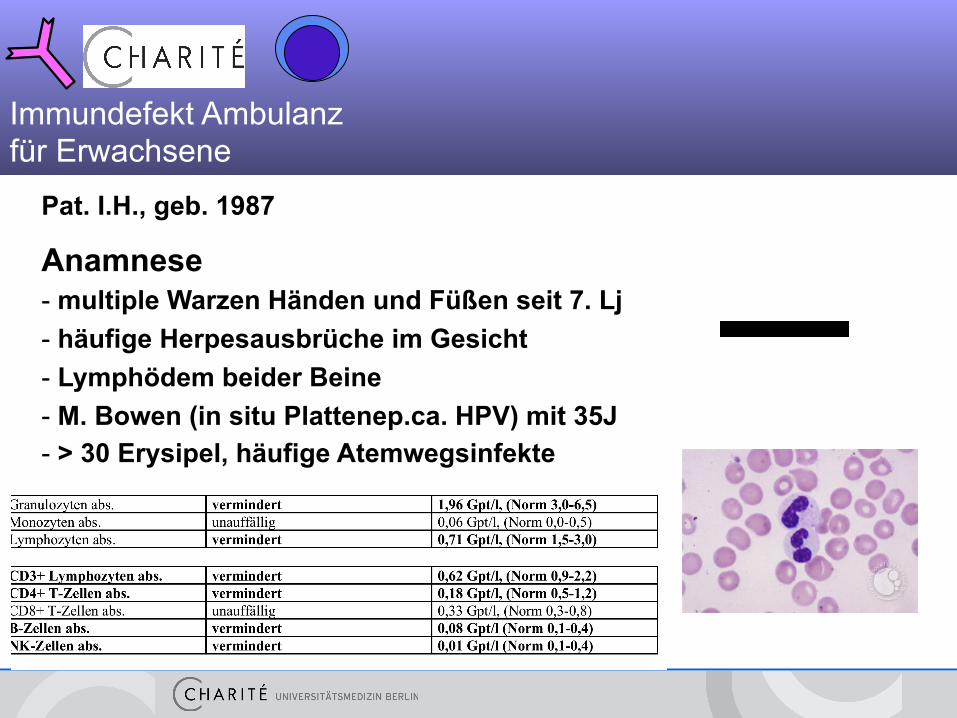
Pat. I.H., geb. 1987
Anamnese - multiple Warzen Händen und Füßen seit 7. Lj - häufige Herpesausbrüche im Gesicht - Lymphödem beider Beine - M. Bowen (in situ Plattenep.ca. HPV) mit 35J - > 30 Erysipel, häufige Atemwegsinfekte
Immundefekt Ambulanz für Erwachsene
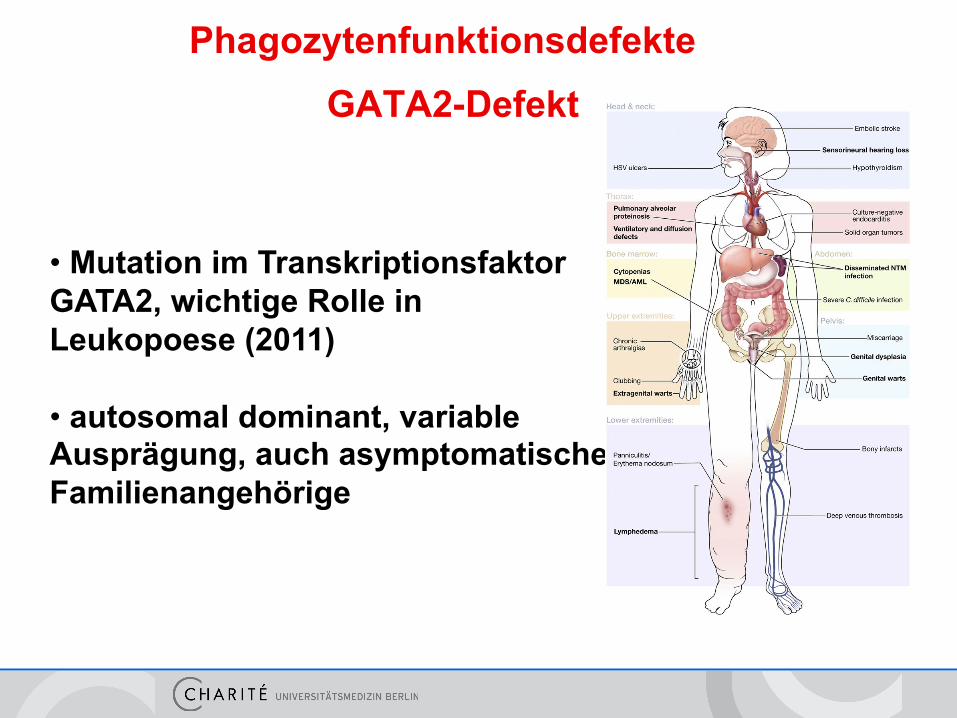
• Mutation im Transkriptionsfaktor GATA2, wichtige Rolle in Leukopoese (2011) • autosomal dominant, variable Ausprägung, auch asymptomatische Familienangehörige
GATA2-Defekt Phagozytenfunktionsdefekte

GATA2-Klinik
• Neutropenie, Myelodsyplasie (hypozelluläres KM), erhöhtes AML-Risiko
• Panlymphopenie, verminderte Monozyten und dendritische Zellen
• Infekte: Erhöhtes Risiko insbesondere für HPV (Warzen,
Tumore), Herpes und atypische Mykobakterien, auch typische bakterielle Infekte
• weitere Probleme:
Hörminderung, pulmonale Alveolarproteinose, Lymphödeme, Thrombosen, Fehlgeburten Mammakarzinom-Risiko, Endokarditis

GATA2- Therapie / Vorsorge
• HPV-Impfung, Interferon-a bei HPV, Valaciclovir bei HSV, antibiotische Prophylaxe
• Überwachung incl. KM und Zytogenetik, Lufu, Derma, Gyn-Vorsorge
• Frühe HSCT wird diskutiert • Screening auf GATA2 auch bei isolierter Neutropenie, CD4- Lymphopenie, MDS

Ursachen • GATA2 • DOCK8 • MagT1 • „hypomorphe SCID“ RAG1, ADA
Klinik opportunistische Infekte (Mykobakterien, Kryptokokken, PCP, Mykosen, EBV, HPV, auch bakterielle Pneumonien) Differentialdiagnose • Infekt (HIV) • Sjögren, SLE, Sarkoidose • Lymphom • Medikamententoxizität
Therapie PCP, Toxo, Pilz-Prophylaxe MagT1: Magnesium IgG Spez. Inhibitoren SCT
Differentialdiagnose CD4-Lymphopenie
CD4 < 0,3/nl > 3Monate
• PI3Kd
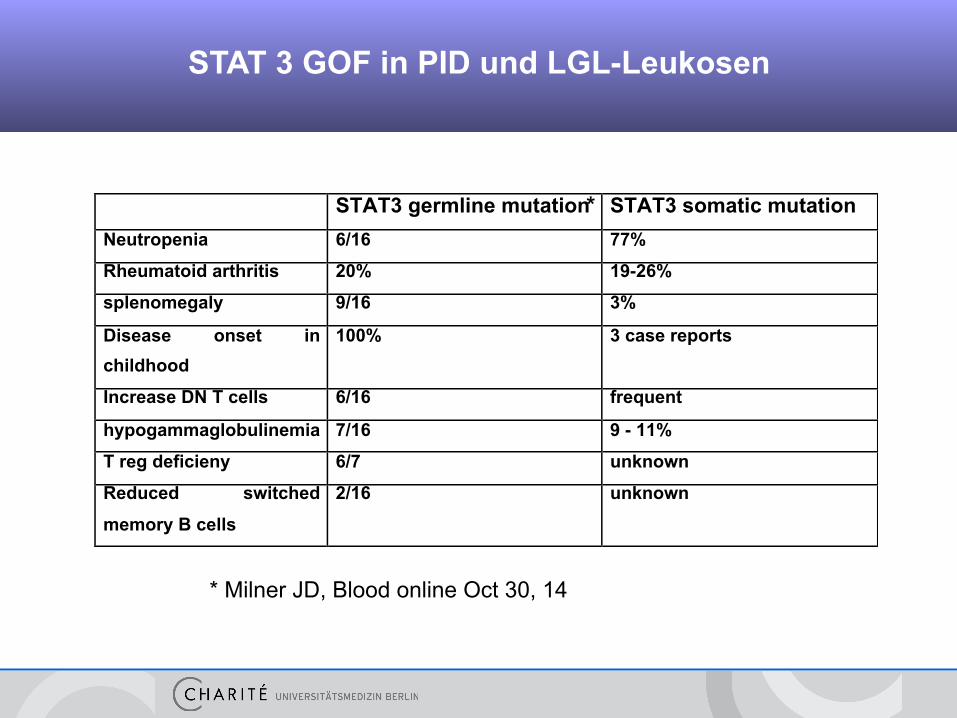
STAT 3 GOF in PID und LGL-Leukosen
STAT3 germline mutation STAT3 somatic mutation
Neutropenia 6/16 77%
Rheumatoid arthritis 20% 19-26%
splenomegaly 9/16 3%
Disease onset in
childhood
100% 3 case reports
Increase DN T cells 6/16 frequent
hypogammaglobulinemia 7/16 9 - 11%
T reg deficieny 6/7 unknown
Reduced switched
memory B cells
2/16 unknown
!
* Milner JD, Blood online Oct 30, 14
*

PID – Klassifikation nach IUIS: 9 Hauptgruppen*
§ Combined immunodeficiencies (SCID, DOCK8 u.a.)
§ Well-defined syndromes with immunodeficiency (WAS, STAT3)
§ Predominantly antibody deficiencies (CVID u.a.)
§ Diseases of immune dysregulation (IPEX u.a.)
§ Congenital defects of phagocytes (CGD u.a)
§ Defects in innate immunity (MyD88 u.a.)
§ Autoinflammatory disorders (FMF u.a.)
§ Complement deficiencies (PNH, MBL u.a.)
§ Phenocopies of PID (somatische Neumutationen und Autoantikörper)
*Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies IUIS expert committee for primary immunodeficiency. Al-Herz W. et al. Front Immunol 2014
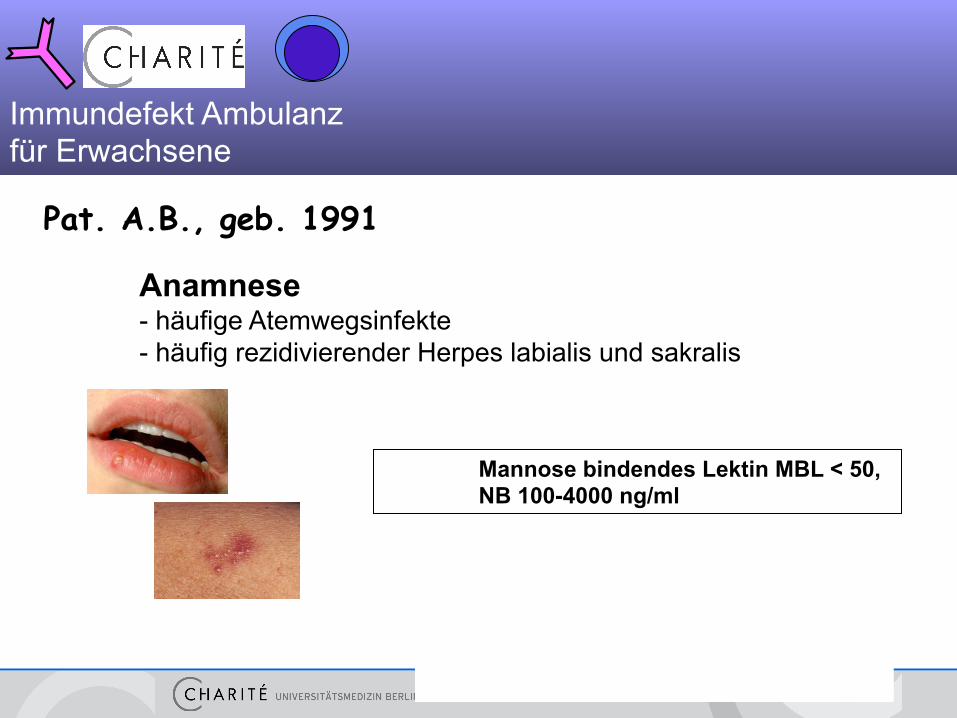
Pat. A.B., geb. 1991
Anamnese - häufige Atemwegsinfekte - häufig rezidivierender Herpes labialis und sakralis
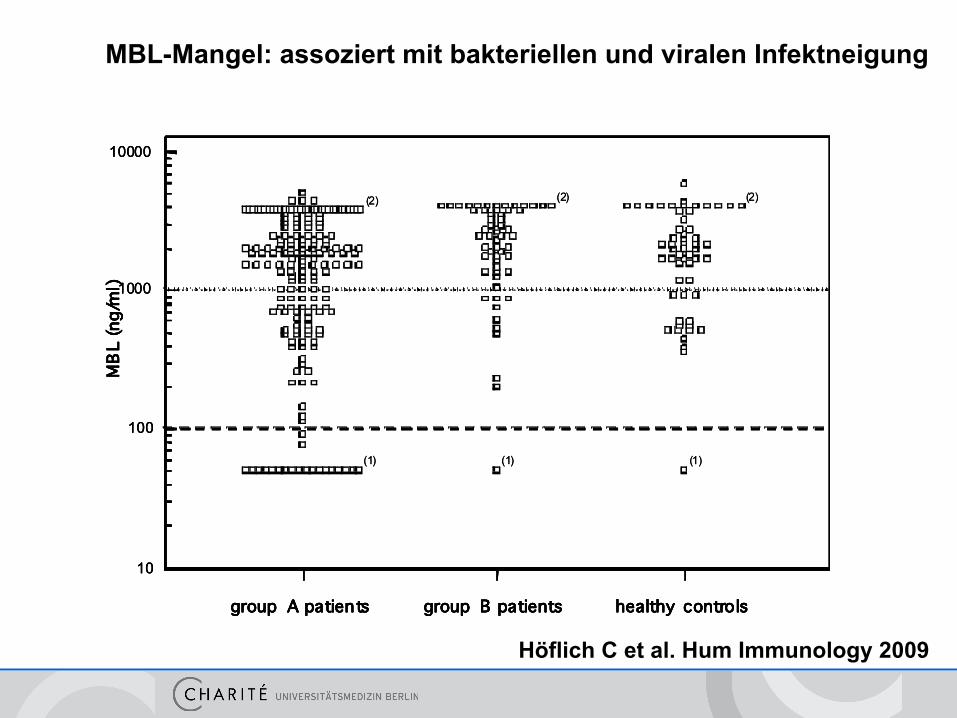
Mannose bindendes Lektin MBL < 50, NB 100-4000 ng/ml
Immundefekt Ambulanz für Erwachsene
B. Hoffmeister • Immundefekte bei Erwachsenen
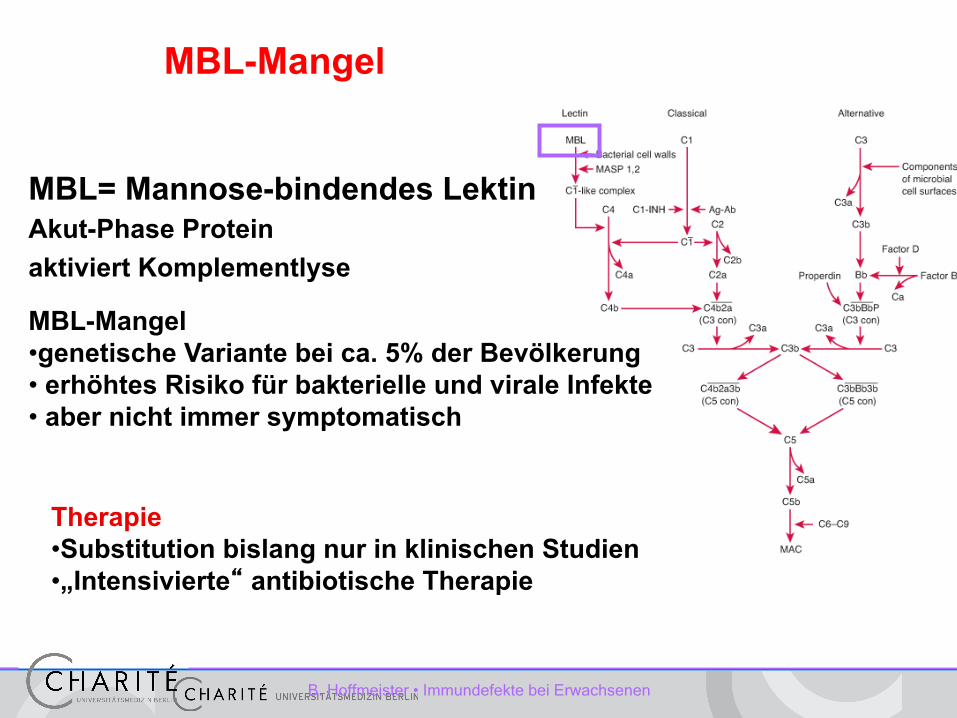
MBL= Mannose-bindendes Lektin Akut-Phase Protein aktiviert Komplementlyse
MBL-Mangel • genetische Variante bei ca. 5% der Bevölkerung • erhöhtes Risiko für bakterielle und virale Infekte • aber nicht immer symptomatisch
Therapie • Substitution bislang nur in klinischen Studien • „Intensivierte“ antibiotische Therapie
MBL-Mangel
10000
1000
100
10
group A patients group B patients healthy controls
MB
L (n
g/m
l)
(2)
(1) (1) (1)
(2) (2)
10000
1000
100
10
group A patients group B patients healthy controls
MB
L (n
g/m
l)
10000
1000
100
10
group A patients group B patients healthy controls
MB
L (n
g/m
l)
(2)
(1) (1) (1)
(2) (2)
MBL-Mangel: assoziert mit bakteriellen und viralen Infektneigung
Höflich C et al. Hum Immunology 2009

Immundefekte bei Erwachsenen
Sinnvolle Basisdiagnostik
– Differentialblutbild – Immunglobulinhauptklassen
• IgG, IgM, IgA
– Ferritin, Vitamin D, HbA1c, IgE
– Gezielte Erregerdiagnostik bei rezidivierenden Infektionen, unüblichen Lokalisationen

Immundiagnostik • IgG/A/M, Subklassen • spez. Antikörper (PcP, HSV u.w.) • MBL
• Lymphozytendifferenzierung • B-Zell-Differenzierung (CVID) • T-Zell-Funktion (IFNg/IL-2, LTT)
• Monozytenfunktion (TLR, Burst) • Spezialdiagnostik (NK, TLR, Adhäsion etc) • Genetik (Immun-Panel, NGS)

Zusammenfassung PID
• Infektanfälligkeit Leitsymptom „ELVIS“ Erreger, Lokalisation, Verlauf, Intensität
• Erstmanifestation nicht selten erst bei Erwachsenen • Viele seltene PID, aber:
Ø Antikörpermangelerkrankungen häufig Ø rekurrente AWI u.w. Ø „GARFIELD“ Granulome, Autoimmunität, rez Fieber, Lymphoprol. Diarrhoen
Ø IgG-Substitution und Antibiotika
Ø CD4-Mangel: opportunistische Infekte
Ø MBL-Mangel mild aber häufig 5%
• bei PID-Verdacht initiale Immundiagnostik: Diff-BB, IgG/A/M • Überweisung in PID-Zentrum • zukünftig Genetik „Immunopanel“ für > 200 monogene Defekte
FIND-ID: Immundefektzentren für Erwachsene
Hamburg UKE Infektiologie
Hannover MHH Immunologie/Rheuma Krefeld Helios Kinderklinik Siegen St. Marienkrankenhaus
Frankfurt Kinderklinik Ulm Kinderklinik Freiburg CCI
Berlin - Charité Immunologie
Leipzig
- Praxis Immundefizienz - IDCL Klinikum St. Georg
Dresden Kinderklinik
Erlangen Immunologie/ Rheuma München Klinikum Rheumatologie


















