
Zur Synthese und Analyse von Sterylglycosiden als ...
104
Zur Synthese und Analyse von Sterylglycosiden als nachwachsende Rohstoffe Diplomarbeit zur Erlangung des akademischen Grades eines Magisters der Naturwissenschaften vorgelegt von Christopher LUDWIG Am Institut für Chemie Arbeitsgruppe Nachwachsende Rohstoffe Begutachter: Ao.Univ.-Prof. Mag.rer.nat. Dr.phil.Martin Mittelbach Graz, 2015
Transcript of Zur Synthese und Analyse von Sterylglycosiden als ...

Zur Synthese und Analyse von Sterylglycosiden als nachwachsende Rohstoffe
Diplomarbeit
zur Erlangung des akademischen Grades eines Magisters der Naturwissenschaften
vorgelegt von
Christopher LUDWIG
Am Institut für Chemie Arbeitsgruppe Nachwachsende Rohstoffe
Begutachter: Ao.Univ.-Prof. Mag.rer.nat. Dr.phil.Martin Mittelbach
Graz, 2015

Danksagung
Ich möchte mich bei all jenen bedanken, die mich im Zuge meines Studiums als auch meiner
Diplomarbeit unterstützt haben.
Ganz besonders möchte ich mich bei Prof. Dr. Martin Mittelbach für die Bereitstellung des Themas
sowie der fachlichen Betreuung bedanken.
Weiters möchte ich mich bei der gesamten Arbeitsgruppe NAWARO für die fachliche Unterstützung,
vielmehr aber noch für einen neu erhaltenen Freundeskreis bedanken. Ganz besonders danke ich an
dieser Stelle DI Philipp Marco Neu, Stephanie Flitsch, MSc. und Aline Silva Muniz, MSc.
Mein Dank gilt außerdem Dr. Tibor Czabany, Institut für Biotechnologie und Bioprozesstechnik TU
Graz, für die fachliche Unterstützung.
Christina Rother, BSc. möchte ich ganz besonders für ihre Geduld, die fachliche als auch für die
mentale Unterstützung, beziehungsweise einfach für alles danken.
Außerdem möchte ich mich noch bei meinen Eltern bedanken, die mir das Studium erst ermöglicht
haben.
Abschließend danke ich noch meiner Familie sowie meinen Freunden: Danke fürs Dasein.

Kurzfassung
Sterylglycoside (SGe) sind natürliche, in Pflanzen vorkommende Sterolderivate, die sowohl in freier,
als auch in acylierter Form (ASGe) vorliegen können. In acylierter Form sind diese, aufgrund der
unpolaren Fettsäurekomponente, in Pflanzenölen wie Sojaöl, Palmöl oder Rapsöl, aus denen unter
anderem Biodiesel hergestellt wird, gut löslich. In der Biodieselproduktion werden Pflanzenöle,
genauer gesagt Triacylglyceride, mit Methanol in Fettsäuremethylester (FAME) umgeestert. Bei
dieser Reaktion entstehen, neben dem gewünschten Produkt, auch Nebenprodukte. Unter anderem
werden aus ASGen nicht acylierte SGe gebildet, welche bei der Lagerung von Biodiesel ausfallen.
Diese Präzipitate können zur Verstopfung von Treibstofffiltern führen. Durch den hohen
Schmelzpunkt dieser Verbindungen haben sie außerdem einen negativen Einfluss auf das
Fließverhalten von Biodiesel bei kalten Temperaturen und können damit zu Motorversagen führen.
In Zuge dieser Arbeit werden SGe auf drei verschiedene Arten synthetisiert, bereits bekannte
Methoden modifiziert beziehungsweise in der Literatur noch nicht beschriebene Methoden
angewendet. Des Weiteren werden die Vor- und Nachteile der Methoden anhand ihrer Ausbeute,
Reinheit und der effektiven Arbeitszeit verglichen, sowie ein grober Vergleich der entstehenden
Kosten durch Chemikalien getätigt. Die synthetisierten SGe sollen als Modellverbindungen für neue
Analysemethoden dienen. Neben der Synthese werden außerdem verschiedene Analysemethoden
wie NMR-Spektroskopie, MS-TOF sowie Gaschromatographie angewendet und beschrieben und
deren Vor- und Nachteile herausgehoben. Außerdem wurden verschiedene Versuche getätigt,
acylierte Sterylglycoside enzymatisch, mittels einer Lipase, aus Sterylglycosiden herzustellen. Weiters
wurde Rapsbiodiesel gaschromatographisch vermessen und dessen Gehalt an verschiedenen
Sterylglycosiden bestimmt.

Abstract
Sterylglycosides (SGs) are natural derivates of sterols that occur in various plants either as free SGs or
as acylated compounds (ASGs). The acylated form is, due to the non-polar fatty acid component,
soluble in edible oils like soybean oil, palm oil or rapeseed oil, which are inter alia raw materials for
biodiesel production. In biodiesel production, plant oils, or to be more precise, triacylglycerols are in
a transesterification reaction with methanol transformed into fatty acid methyl esters (FAME). In this
reaction, besides the favored product, various by products occur. One of these by-products are SGs,
that are made of acylated SGs, which precipitate during storage of biodiesel. These precipitations can
lead to problems like clogging of filters. Due to the high melting point of these compounds, they also
have a negative influence on the fluidity of biodiesel, especially at low temperatures, which can lead
to engine failures. In this paper, SGs were synthesized in three different ways. Already known
methods were modified respectively in literature not yet described methods were used. Next to the
description of synthesis, also the advantages and disadvantages of the different methods, concerning
the yield, the purity of the products as well as the working hours investigated are discussed.
Moreover, the methods are compared regarding the costs of the raw materials which were used. The
synthesized SG should be used as model compounds for new methods of analysis. Furthermore,
different methods of analysis, like NMR-spectroscopy, MS-TOF as well as gas chromatography are
described and utilized. Also the pros and cons of the analysis-methods are pointed out. Moreover,
different experiments for enzymatic production of ASGs from SGs were carried out. Besides,
rapeseed-fatty acid methyl ester was analyzed by gas chromatography and the content of SGs was
determined.

Inhalt
Abkürzungsverzeichnis ............................................................................................................................ 8
1 Einleitung ....................................................................................................................................... 10
2 Grundlagen und Theorie I: Sterylglycoside ................................................................................... 12
2.1 Sterole ................................................................................................................................... 12
2.1.1 Zoosterole ...................................................................................................................... 12
2.1.2 Phytosterole .................................................................................................................. 13
2.1.3 Mycosterole ................................................................................................................... 14
2.2 Sterylglycoside ....................................................................................................................... 15
2.2.1 Biosynthese von Sterylglycosiden ................................................................................. 15
2.2.2 Synthese von Sterylglycosiden ...................................................................................... 16
2.2.3 Vorkommen von Sterylglycosiden ................................................................................. 19
2.2.4 Die Biodieselproblematik .............................................................................................. 21
2.2.5 Entfernung von Sterylglycosiden aus Biodiesel ............................................................. 22
2.2.6 Analyse von Sterylglycosiden ........................................................................................ 24
3 Grundlagen und Theorie II: Analytische Methoden-Allgemein ..................................................... 28
3.1 Chromatographie .................................................................................................................. 28
3.1.1 Adsorptionschromatographie ....................................................................................... 30
3.1.2 Verteilungschromatographie ........................................................................................ 30
3.1.3 Größenausschlusschromatographie-GPC ...................................................................... 32
3.2 Massenspektrometrie ........................................................................................................... 33
3.2.1 Ionisierung ..................................................................................................................... 33
3.2.2 Analysatoren .................................................................................................................. 36
3.3 Kernspinresonanzspektroskopie-NMR .................................................................................. 38
4 Material und Methoden ................................................................................................................ 39
4.1 Lösungsmittel und Reagenzien .............................................................................................. 39
4.2 Geräte .................................................................................................................................... 41
4.2.1 Probenvorbereitung ...................................................................................................... 41
4.2.2 GC-MS ............................................................................................................................ 41
4.2.3 HPLC-TOF-MS................................................................................................................. 41
4.2.4 NMR-Spektroskopie ....................................................................................................... 41
4.2.5 GC-FID ............................................................................................................................ 41
4.3 Synthese von Cholesterylglycosiden ..................................................................................... 42

6
4.3.1 Synthese durch Koenigs-Knorr Methode ...................................................................... 42
4.3.2 Synthese durch Trichloracetimidat-Methode ............................................................... 44
4.3.3 Synthese von Cholesteryl-β-D-glucopyranosid mit Saccharose als Edukt..................... 47
4.4 Versuch der enzymatischen Herstellung von Cholesteryl(6´-O-palmitoyl)-β-D-
glucopyranosid .................................................................................................................................. 49
4.5 Probenvorbereitung für die qualitative und quantitative Analyse von Pflanzenölen und
Biodiesel auf Sterylglycoside ............................................................................................................. 50
4.6 Analytik .................................................................................................................................. 50
4.6.1 GC-MS ............................................................................................................................ 51
4.6.2 GC-FID ............................................................................................................................ 51
4.6.3 TOF-MS .......................................................................................................................... 51
4.6.4 NMR-Spektroskopie ....................................................................................................... 52
5 Ergebnisse und Diskussion ............................................................................................................ 52
5.1 Synthese durch Koenigs-Knorr Methode .............................................................................. 52
5.1.1 Synthese von β-D-Glucose-Pentaacetat [99] ................................................................ 53
5.1.2 Synthese von Acetobromoglucose ................................................................................ 53
5.1.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid durch Koenigs-
Knorr-Mechanismus ...................................................................................................................... 54
5.1.4 Synthese von Cholesteryl-β-D-glucopyranosid ............................................................. 55
5.2 Synthese durch Trichloracetimidat-Methode ....................................................................... 58
5.2.1 Synthese von 2,3,4,6-tetra-acetyl-D-glucose ................................................................ 58
5.2.2 Synthese von O-(2,3,4,6-tetra-O-acetyl-D-glucopyranosyl)-trichloracetimidat ............ 59
5.2.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid ......................... 61
5.2.4 Synthese von Cholesteryl-β-D-glucopyranosid ............................................................. 62
5.3 Synthese von Cholesteryl-β-D-glucopyranosid mit Saccharose als Edukt ............................ 65
5.3.1 Synthese von Octa-O-Benzylsaccharose ....................................................................... 66
5.3.2 Synthese von 2,3,4,6-Tetra-O-benzyl-D-glucopyranose ................................................ 67
5.3.3 Synthese von O-(2,3,4,6-Tetra-O-benzyl-D-glucopyranosyl)trichloracetimidat ........... 70
5.3.4 Synthese von Cholesteryl-(2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosid) ..................... 71
5.3.5 Synthese von Cholesteryl-β-D-glucopyranosid ............................................................. 72
5.4 Vergleich der verschiedenen SG-Synthesemethoden ........................................................... 75
5.5 Versuch der enzymatischen Herstellung von Cholesteryl(6´-O-palmitoyl)-β-D-
glucopyranosid .................................................................................................................................. 77
5.5.1 Versuch mit reinem Hexan als Lösungsmittel ............................................................... 78
5.5.2 Versuch mit Hexan nach vorherigem Lösen des Substrates in Chloroform .................. 81

7
5.5.3 Versuch mit THF/Pyridin 4:1 als Lösungsmittel ............................................................. 82
5.6 Qualitative und quantitative Analyse von Biodiesel auf den Gehalt von Sterylglycosiden .. 84
6 Zusammenfassung und Ausblick ................................................................................................... 86
8 Tabellenverzeichnis ....................................................................................................................... 98
9 Abbildungsverzeichnis ................................................................................................................... 98
10 Anhang..................................................................................................................................... 101

8
Abkürzungsverzeichnis
ABG Acetobromoglucose
ac Wechselspannung
APCI Chemische Ionisation bei Atmosphärendruck
ASG Acyliertes Sterylglycosid
CDP Cytidindiphosphat
CFPP Cold Filter Plugging Point
CP Cloud Point
DBU 1,8-Diazabicyclo[5.4.0]undec-7-en
DC Dünnschichtchromatographie, Dünnschichtchromatogramm
dc Gleichspannung
DCM Dichlormethan
GTATH Tetra-O-acetyl-β-D-glucopyranosyl-trichloracetimidat
Enz. Enzym
EI Elektronenstoßionisation
ELSD Evaporative Light Scattering Detector, Lichtstreudetektor
ESI Elektrosprayionisation
FAME Fettsäuremethylester
FG Frischgewicht
FID Flammenionisationsdetektor
GC Gaschromatographie, Gaschromatograph
GLC Gas-Liquid-Chromatography
GPA Glucosepentaacetat
GPC Gelpermeationschromatographie
GTA Glucosetetraacetat
HPLC High Performance Liquid Chromatography
HRMS High Resolution Mass Spectronomy
IS Interner Standard
LC Liquid-Chromatography
LLC Liquid-Liquid-Chromatography
m/z Masse zu Ladungs-Verhältnis
MALDI Matrix-assisted Laser Desorption/Ionization
MHz Megahertz

9
MS Massenspektrometer, Massenspektrum
MSTFA N-Methyl-N-(trimethylsilyl) trifluoroacetamid
NMR Kernspinresonanz
n.d. nicht detektierbar
NP Normalphase
PI Photoionisation
ppm parts per million
Rf Retentionsfaktor
RP Reversed Phase
rpm revolutions per minute
RT Raumtemperatur
SG Sterylglycosid
SN1 Nucleophile Substitution erster Ordnung
SN2 Nucleophile Substitution zweiter Ordnung
SEC Size Exclusion Chromatography
SPE Solid Phase Extraction
TBG Tetrabenzylglucose
TDP Thiamindiphosphat
TG Triacylglycerid
THF Tetrahydrofuran
TMS Trimethylsilan
TMSOTf Trimethylsilyl trifluoromethansulfonat
TOF Time of Flight
UDP Uridindiphosphat
UHPLC Ultra High Performance Liquid Chromatography
UV Ultraviolett
VIS Visible
WLD Wärmeleitfähigkeitsdetektor

10
1 Einleitung
Bei Biodiesel handelt es sich um einen häufig eingesetzten erneuerbaren Kraftstoff, der eine direkte
Alternative zu fossilen Energiequellen darstellt. Dieser wird aus in Pflanzenölen enthaltenen
Triacylglyceriden durch eine Umesterungsreaktion mit Methanol hergestellt, wodurch die Fettsäuren
vom Glyceringrundgerüst abgespalten werden und es in weiterer Folge zur Herstellung von
Fettsäuremethylestern und Glycerin kommt. In Pflanzen sind unter anderem auch freie
Sterylglycoside (SGe) und acylierte Sterylglycoside (ASGe), welche mitunter zur Membranstabilität
beitragen, enthalten. ASGe sind mit einer Fettsäure verestert und befinden sich, nach der Herstellung
des Pflanzenöls aus der Pflanze, in gelöster Form auch in diesem wieder [1]. Bei der
Umesterungreaktion, im Zuge der Biodieselherstellung, kommt es zur Abspaltung des
Fettsäurerestes, woraus Sterylglycoside in freier Form resultieren [1]. Bei ASGen handelt es sich
aufgrund der Fettsäurekomponente um sehr unpolare Verbindungen, die sowohl im Pflanzenöl als
auch im Biodiesel löslich sind. Nach der Abspaltung der Fettsäurekomponente stellt das daraus
resultierende Sterylglycosid (SG), aufgrund der Absenz der unpolaren Fettsäurekomponente, eine
polarere Verbindung dar. Da in der Chemie der Grundsatz „Similia similibus solvuntur“ also „Gleiches
löst Gleiches“ gilt, bedeutet das, dass SGe in unpolaren Lösungsmitteln, wie dies Biodiesel im
weitesten Sinne darstellt, nicht löslich sind. Diese benötigen jedoch einige Zeit zur Präzipitation,
wodurch eine Abtrennung im Zuge des Herstellungsprozesses nicht möglich ist. Durch die Lagerung
vergeht indes Zeit, in der sich diese Feststoffe bilden, und beispielsweise Filter und Düsen verstopfen
können. Zum einen tritt dieses Problem schon bei der Lagerung in den Tanks an, zum anderen kann
dies auch beim Endverbraucher noch zu Leistungseinschränkungen bis hin zu Motorschäden führen,
wobei dies vermehrt bei kalten Temperaturen zu beobachten ist. Die Konzentration, bei denen SGe
Probleme verursachen beträgt unter 20 ppm [2], jedoch existieren zur Entfernung von SGen aus
Biodiesel keine Methoden, welche eine ausreichende Wirkung hinsichtlich der negativen
Eigenschaften auf die Filtrierbarkeit aufweisen (vgl. Kapitel 2.2.5). Neben dem direkten negativen
Einfluss der SGe auf die Fluidität, stehen diese außerdem im Verdacht, für das Ausfallen anderer
Stoffe verantwortlich zu sein, also als Impfkristalle zu wirken, wodurch es zu einer weiteren
Verschlechterung des Fließverhaltens kommt [3]. Deshalb ist es wichtig, dass zur Untersuchung von
Biodieselproben Analysen zur Verfügung stehen, welche eine möglichst niedrige Nachweisgrenze
aufweisen. Zur Entwicklung selbiger bedarf es unter anderem an Modellverbindungen, um die
Quantifizierung von Sterylglycosiden durch den Vergleich mit internen Standards durchführen zu
können.

11
Zur Entwicklung solcher sensitiven Methoden war es Ziel der Diplomarbeit, eine neue, effizientere
Methode zur Herstellung von Sterylglycosiden zu entwickeln. Im weiteren Sinne, waren folgende
Aufgabenstellungen zu bewerkstelligen:
Literaturteil:
Das Vorkommen und die Unterteilung von Sterolen sowie deren biochemische Bedeutung
Was sind Sterylglycoside, wo und in welcher Form kommen sie vor? Was ist die biologische
beziehungsweise biochemische Bedeutung von Sterylglycosiden?
Die Problematik von Sterylglycosiden in Biodiesel. Warum kommt es zu Problemen? Welche
Möglichkeiten gibt es, um Sterylglycoside aus Biodiesel zu entfernen?
Welche Synthese- und Analysemethoden für Sterylglycoside sind bekannt beziehungsweise
werden angewendet?
Praktischer Teil:
Synthese von Cholesteryl-β-D-glucopyranosid mittels verschiedener Methoden.
Modifizierung der Methoden beziehungsweise Entwicklung einer neuen Methode zur
Ausbeutesteigerung und zur Ersparung zeitintensiver säulenchromatographischer
Trennungen.
Charakterisierung der hergestellten Verbindungen durch diverse instrumentell-analytische
Verfahren.
Versuch der Herstellung von acylierten Sterylglycosiden durch enzymatische Veresterung.
Bewertung
Bewertung der untersuchten Methoden hinsichtlich ihrer Effizienz bezüglich der Ausbeute,
sowie der investierten Zeit als auch den verwendeten Chemikalien.

12
2 Grundlagen und Theorie I: Sterylglycoside
2.1 Sterole
Sterole sind wichtige, in der Natur vorkommende Verbindungen, die der Gruppe der Steroide
angehören. Die Grundstruktur der Sterole ist das Steran, an dem sich eine 3β-Hydroxygruppe
befindet. Bei der Grundstruktur sind drei Cyclohexanringe, meist trans-verknüpft, mit einem
Cyclopentanring verbunden [4]. Die Einteilung wird, je nach Herkunft, in Zoosterole (in Tieren),
Phytosterole (in Pflanzen) und Mycosterole (in Pilzen), vorgenommen [4].
Abbildung 1: Grundgerüst der Sterole
2.1.1 Zoosterole
Zoosterole nehmen eine zentrale Rolle im Stoffwechsel von Tieren ein, viele von ihnen besitzen
physiologische Aktivität. Das am weitesten verbreitete Sterol ist das Cholesterol, welches,
zurückzuführen auf die Anzahl von acht Stereozentren, eine Anzahl von 256 Stereoisomeren besitzt,
wobei nur die Hälfte davon in der Natur vorkommt [5]. Cholesterol kommt bei Menschen und Tieren
in großen Mengen, vor allem im Gehirn und in Rückenmark, vor. So befinden sich im Körper eines
erwachsenen Menschen beispielsweise 200-300 g Cholesterol. Dieses ist Verursacher zahlreicher
Krankheiten, wie beispielsweise Arteriosklerose, Herzerkrankungen sowie ein nicht unwesentlicher
Bestandteil von Gallensteinen. Cholesterol fungiert jedoch auch als Ausgangsstoff für die Synthese
von Steroidhormonen im Körper, als auch für die Produktion von Gallensäure. Weiters werden aus
Cholesterol, über den Zwischenschritt der Pregnenolon-Synthese, die Geschlechtshormone Östradiol
und Progesteron gebildet [6]. Die zwei Hauptaufgaben im Organismus sind zu einem die Stabilisation
von Zellmembranen, zum anderen die Resorption von Fettsäuren im Darm [4].

13
Abbildung 2: Strukturformel von Cholesterol
2.1.2 Phytosterole
Phytosterole kommen in praktisch allen höheren Pflanzen vor und tragen einen wesentlichen Beitrag
zur Stabilität pflanzlicher Zellmembranen bei. Vom Cholesterol unterscheiden sich pflanzliche Sterole
dahingehend, dass sie eine zusätzliche Methyl- oder Ethylgruppe besitzen [5]. Aufgrund der
unterschiedlichen Lage von Doppelbindungen und variablen Seitenketten kommt in Pflanzen eine
Vielzahl unterschiedlicher Phytosterole vor. Die Stereoisomere mitinbegriffen, wurden bereits mehr
als 250 verschiedene Verbindungen nachgewiesen [7]. Diese Vielfalt lässt sich durch die oft nur
geringen Unterschiede erklären. So besitzen beispielsweise Sitosterol und Stigmasterol beide eine
Ethylgruppe am C-24 Atom und unterscheiden sich lediglich durch die Doppelbindung am C-22 Atom.
Bei den am häufigsten in Pflanzen vorkommenden Sterolen handelt es sich um β-Sitosterol,
Campesterol und Stigmasterol, welche, hinsichtlich der Aufnahme an Phytosterolen, 65%, 30%
beziehungsweise 3% der menschlichen Ernährung ausmachen [8]. Stigmasterol stellt beispielsweise
12-25% des unverseifbaren Anteils des Sojaöls dar [9]. Auch Cholesterol kommt in Pflanzen vor, spielt
aber aufgrund der geringen Konzentration in der Ernährung eine vernachlässigbar kleine Rolle [10].
Neben freien Phytosterolen kommen diese in der Natur unter anderem auch noch in veresterter
Form, wie beispielsweise als Sterylglycoside und acylierte Sterylglycoside vor.
Abbildung 3: Strukturformeln von a) Sitosterol, b) Stigmasterol, c) Campesterol
14
Das Vorkommen von Phytosterolen lässt sich vor allem in fettreichen Pflanzenteilen wie Samen oder
Nüssen beobachten, wohingegen in frischem Obst und Gemüse in der Regel nur wenige Sterole
enthalten sind. (vgl. Tabelle 1) [11]. Im Organismus haben Phytosterole, sowie auch einige Derivate,
eine cholesterolsenkende Wirkung, weshalb sie auch bei der Therapie und Vorbeugung von
Hypercholesterolämie eingesetzt werden [12]. Diese Eigenschaft beruht darauf, dass Phytosterole,
durch die cholesterolähnliche Struktur, an dessen Stelle absorbiert werden, wodurch es zu einer
vermehrten Ausscheidung und somit zu keinem Anstieg in der Gallensäureexkretion kommt [13].
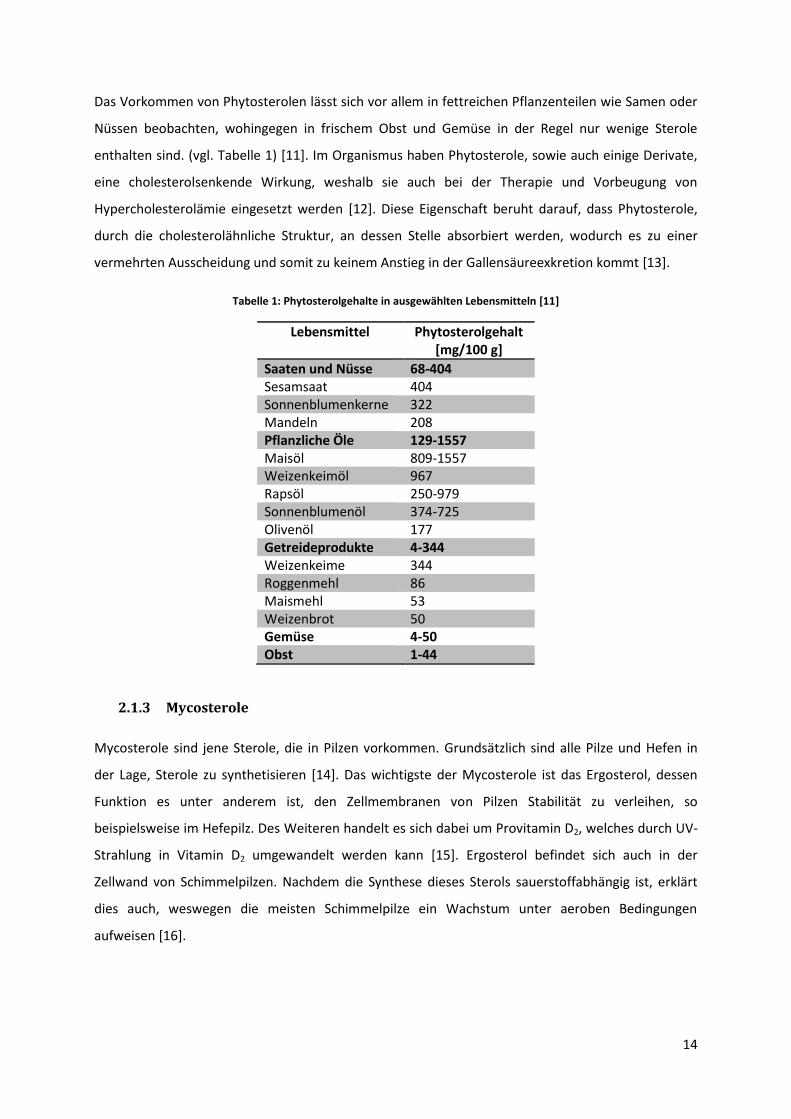
Tabelle 1: Phytosterolgehalte in ausgewählten Lebensmitteln [11]
Lebensmittel Phytosterolgehalt [mg/100 g]
Saaten und Nüsse 68-404 Sesamsaat 404 Sonnenblumenkerne 322 Mandeln 208 Pflanzliche Öle 129-1557 Maisöl 809-1557 Weizenkeimöl 967 Rapsöl 250-979 Sonnenblumenöl 374-725 Olivenöl 177 Getreideprodukte 4-344 Weizenkeime 344 Roggenmehl 86 Maismehl 53 Weizenbrot 50 Gemüse 4-50 Obst 1-44
2.1.3 Mycosterole
Mycosterole sind jene Sterole, die in Pilzen vorkommen. Grundsätzlich sind alle Pilze und Hefen in
der Lage, Sterole zu synthetisieren [14]. Das wichtigste der Mycosterole ist das Ergosterol, dessen
Funktion es unter anderem ist, den Zellmembranen von Pilzen Stabilität zu verleihen, so
beispielsweise im Hefepilz. Des Weiteren handelt es sich dabei um Provitamin D2, welches durch UV-
Strahlung in Vitamin D2 umgewandelt werden kann [15]. Ergosterol befindet sich auch in der
Zellwand von Schimmelpilzen. Nachdem die Synthese dieses Sterols sauerstoffabhängig ist, erklärt
dies auch, weswegen die meisten Schimmelpilze ein Wachstum unter aeroben Bedingungen
aufweisen [16].

15
Abbildung 4: Strukturformel von Ergosterol
2.2 Sterylglycoside
Sterylglycoside (SGe) sind chemisch gesehen, neben Saponinen, Estrogenglycosiden und
Herzglycosiden, eine Unterkategorie der Steroidglycoside [17]. Sterylglycoside kommen in der Natur
in einer Vielzahl von Pflanzen, aber auch in Pilzen, Bakterien, Algen und Tieren, beispielsweise in der
Epidermis von Schlangen vor [18]. Es handelt sich dabei um Sterole, die, meist β-D-glycosidisch, mit
einem Monosaccharid verbunden sind. Es kommen jedoch auch α-D-glycosidische Bindungen vor,
wie dies bei dem gram-negativen Bakterium Heliobacter pylori der Fall ist [19]. Als Zuckergrundgerüst
können verschiedene Einfachzucker wie Galactose, Mannose, Xylose oder Arabinose dienen, wobei
das Steryl-β-D-monoglucopyranosid das mit Abstand am häufigsten vorkommende Sterylglycosid
darstellt [20]. Sterylglycoside tragen, wie auch freie Sterole, zur Stabilisierung von Zellmembranen
bei, wobei eine Cholesterolkomponente eine bessere Wirkung aufweist, als dies bei Sitosterol oder
Stigmasterol der Fall ist [21]. Sitosteryl-Glycosid dient beispielsweise als Ausgangsstoff für die
Biosynthese von Cellulose, in dem es als Glucosedonator bei der Polymerisation von Glucanketten
fungiert [22]. Weiters ist bekannt, dass SGe eine positive Auswirkung auf die Membraneigenschaften,
ganz besonders auf jene der Proteininteraktionen, haben [23]. Außerdem wird vermutet, dass SGe
einen Einfluss auf die Umweltanpassung von Pflanzen hinsichtlich deren Hitze- und
Kälteempfindlichkeit haben [23]. Eine weitere Funktion, welche SGen im Organismus zukommt, ist
bei der Glycosylierung von Ceramidmolekülen, bei der sie als Glucosedonatoren wirken, indem sie als
Glucosecarrier zwischen UDP-Glucose und Ceramid fungieren [24]. Im Organismus haben SGe
außerdem einen positiven Einfluss auf das Immunsystem, da sie die Lymphozytenzahl erhöhen [25].
Außerdem besitzen SGe, durch die Inhibierung entzündungsfördernder Stoffe, eine
entzündungshemmende Wirkung [26].
2.2.1 Biosynthese von Sterylglycosiden
Die Biosynthese von Sterylglycosiden erfolgt durch eine enzymatische Reaktion, bei der die
Enzymklasse der Sterylglycosyltranferasen, die ihrerseits menbrangebundene Enzyme darstellen,

16
diese Reaktion katalysiert. Sterylglycosyltranferasen übertragen hierbei ein aktiviertes
Zuckernucleotid, meist in Form der UDP-Glucose auf eine Sterolkomponente. Genauer kommt es bei
dieser Reaktion zu einer Glycosylierung des Sterols an der 3β-Hydroxygruppe, woraus das 3β-D-
Glycosid resultiert [27].
Abbildung 5: Biosynthese von Stigmasteryl-β-D-glucopyranosid
Diese Sterylglycosyltransferasen besitzen hinsichtlich des Glycosyldonors eine hohe
Substratspezifität. So findet beispielsweise bei der Anwesenheit von UDP-Galaktose als auch bei
UDP-Mannose keine Umsetzung statt. Die Substratspezifität der Sterolkomponente betreffend ist
jedoch weniger ausgeprägt. Die in Abbildung 5 ersichtliche Reaktion kann unter anderem auch mit
Sitosterol, Cholesterol und Ergosterol erfolgen [28]. Glycosyltransferasen lassen sich, anhand der
stereochemischen Ausrichtung ihrer Produkte, in inverting- und in retaining-Glycosyltransferasen
einteilen [29]. Da der UDP-Teil stets α-ständig an die Glucopyranose gebunden ist, spricht man bei
der Biosynthese eines α-SGs von retaining-Glycosyltransferasen, also dem Aufrechterhalten der
anomeren Konfiguration (z.B.: UDP-α-Glucose α-SG). Häufiger kommen hingegen die inverting-
Glycosyltransferasen vor, bei denen es zu einer Umkehrung der axialen Ausrichtung des Zuckers
kommt (z.B.: UDP-α-Glucoseβ-SG). Hierbei kommt es in Folge einer nucleophilen Substitution
zweiter Ordnung, bei der der Akzeptor durch eine Brønsted-Base deprotoniert wird, zu einem
nucleophilen Angriff am anomeren C-Atom, wodurch es zu einer Umkehr der anomeren Ausrichtung
kommt [29] [30].
In tierischen Zellen wurde das Vorhandensein der β-Glycosyltransferasen noch nicht beobachtet,
jedoch wurde herausgefunden, dass hierbei nicht UDP-Glucose, sondern Glycosylceramid, bei einer
durch β-Glucosidase katalysierten Reaktion, als Glycosyldonor fungiert [31].
2.2.2 Synthese von Sterylglycosiden
Zur Synthese komplizierter Glycoside, wie beispielsweise jener von SGen, haben sich zwei Methoden
bewährt. Zum einen die Koenigs-Knorr-Methode, zum anderen die Trichloracetimidat-Methode. Auf
diese beiden Varianten wird hier nur kurz eingegangen, da diese im weiteren Verlauf der Arbeit noch

17
genauer beleuchtet, beziehungsweise die Synthesen mittels dieser Methoden durchgeführt wird.
Diese sind in Kapitel 4.3 beziehungsweise Kapitel 5 beschrieben.
Bei der Koenigs-Knorr-Methode wird über das peracetylierte Glucopyranosid das entsprechende, am
C-1 halogenierte, acetylierte Glucopyranosid durch Umsetzung mit Bromwasserstoffsäure
hergestellt. Dieses wird in weiterer Folge in Gegenwart von Silbercarbonat und dem entsprechenden
Sterol zum peracetylierten SG umgesetzt. Eine Abspaltung der Acetylgruppen erfolgt mittels
Zemplén-Verseifung.
Abbildung 6: Mechanismus der Zemplén-Verseifung
Die Synthese durch die Trichloracetimidat-Methode erfolgt ebenfalls über das peracetylierte
Glucopyranosid. Dieses wird in Gegenwart einer Säure in das Alkoholanion umgewandelt. Genauer
gesagt wird die anomere Schutzgruppe durch ein Nitril ersetzt. In weiterer Folge wird in Gegenwart
einer Lewis-Säure und dem entsprechenden Sterol das SG gebildet [5]. Die Abspaltung erfolgt
wiederum durch Zemplén-Verseifung. Hierbei kommt es, durch den basischen pH-Wert, zuerst zu
einer Deprotonierung des Methanols, so dass Methanolat vorliegt. In weiterer Folge greift dieses an
den Carbonyl-Kohlenstoffatomen der Acetyl-Schutzgruppen an wodurch es zur Bildung von
Essigsäuremethylestern als auch des entschutzten Cholesterylglycosids kommt.
Andere Methoden beschreiben die Verwendung diverser Modifikationen der Koenigs-Knorr- als auch
der Trichloracetimidat-Methode, bei denen für die Synthese von SGen Benzoylschutzgruppen [32],
oder für das α-Anomer auch Benzylschutzgruppen verwendet werden [33]. Als Edukt fungiert jeweils
Glucose [34]. Den Vorteil, den Benzoylschutzgruppen gegenüber Benzylschutzgruppen aufweisen, ist
die leichtere Abspaltung durch Zemplén-Verseifung im Vergleich mit zeit- und kostenintensiveren
Hydrierungen.
Eine weitere Methode, welche in der Literatur von Murui et al. (1993) [35] beschrieben wird, ist die
Umsetzung von Cholesterol mit Glucosepentaacetat in Gegenwart von Zinnchlorid. Bei diesem
Verfahren handelt es sich um eine Modifikation der Helferich-Synthese [5]. Dieses Verfahren basiert
auf einer durch Zinnchlorid, als Lewis-Säure fungierend, katalysierten SN1 Reaktion bei der vom
anomeren C-Atom eine Acetoxygruppe abgespalten, und anschließend das Sterol angelagert wird.
Dichlormethan wird als Lösungsmittel verwendet. Nach vollständiger Umsetzung, nach
sechsstündigem Rühren, und anschließender Zugabe von gesättigter Natriumchlorid-Lösung wird die
Entstehung eines Niederschlages beschrieben, der in weiterer Folge abgesaugt wird. Die organische
Phase wird daraufhin mit Wasser nachgewaschen und anschließend am Rotavapor eingeengt.
Anschließend wird zur Schutzgruppenabspaltung Natriummethoxid zugegeben, also wiederum durch

18
Zemplén-Verseifung, durchmischt, das Produkt in Ethanol gelöst und sechs Stunden bei 70°C
gelagert. Nach dem Abziehen des Lösungsmittels und der Zugabe von Wasser wird ein Waschschritt
mit Aceton, n-Hexan sowie heißem Aceton durchgeführt, um das Cholesterylglycosid zu erhalten. Die
Aufreinigung erfolgt mittels Säulenchromatographie. Neben dem gewünschten Produkt entstehen
jedoch außerdem noch Glucopyranosylchlorid sowie das α-Anomer. Ausbeuten werden zu dieser
Methode von Murui et al. (1993) [35] jedoch nicht beschrieben. Aufgrund der entstehenden
Nebenprodukte als auch der nicht gegebenen anomeren Kontrolle ist jedoch mit einer nicht
zufriedenstellenden Ausbeute zu rechnen. Weitere Synthesen von SGen nach diesem Mechanismus
finden sich in der Literatur nicht.
Abbildung 7: Mechanismus der Helferich-Synthese
Eine Methodik, auf die im Zuge dieser Arbeit praktisch nicht genauer eingegangen wird, ist die
Herstellung von SGen durch enzymatische Prozesse. Zu einem besteht die Möglichkeit, SGe mittels
Sterol 3-β-Glycosyltransferasen herzustellen. Dieses Verfahren wird analog zur Biosynthese von SGen
praktiziert, in dem ein aktivierter Nucleosid-Diphosphat-Zucker als Glycosyldonor dient. In der Regel
wird dafür UDP-Glucose verwendet, da diese höhere Ausbeuten als beispielsweise CDP- oder TDP-
Glucose liefert [36]. Das SG wird in weiterer Folge durch die Umsetzung mit einem freien Sterol in
Gegenwart des Enzyms dargestellt. Potocka et al. (2008) [36] beschrieben hier ein Verfahren, bei
dem sie dies mit dem entsprechendem Enzym, welches zuvor aus Melanzani isoliert wurde,
experimentell durchführten. Zur Herstellung von größeren Mengen an SGen eignet sich diese
Methode, aufgrund der hohen Kosten für UDP-Glucose jedoch nicht. So erfolgt die Darstellung auf
diese Weise, wie beispielsweise bei Potocka et al. [36], im Nanomol-Bereich. Neben der Anwendung
von Transferasen, wird in der Literatur auch der Einsatz von Glycosylasen beschrieben. Da
Glycosylasen, eine Untergruppe der Hydrolasen, in Verbindung mit Wasser für gewöhnlich
glycosidische Bindungen hydrolysieren, handelt es sich hierbei eher um eine ungewöhnliche
Methode [37]. Wird die Reaktion jedoch in organischen Lösungsmitteln ausgeführt, können diese

19
Enzyme auch eine Kondensationsreaktion, in diesem Fall auch als umgekehrte Hydrolyse (reversed
hydrolysis) bezeichnet, in der die Zucker- und die Sterolkomponente verbunden werden, katalysieren
[38]. Durch die Wahl von α- beziehungsweise β-Glycosylasen besteht hier der Vorteil der anomeren
Kontrolle. Als Substrate für die Reaktion dienen Glucose sowie das entsprechende Sterol, somit muss
also kein aktivierter Zucker vorliegen [39]. Dadurch, dass diese Methode noch wenig erforscht ist,
besteht jedoch noch die Problematik, ein geeignetes Lösungsmittel zu finden, in dem die Wasserhülle
des Enzyms im organischen Lösungsmittel erhalten bleibt, ohne dabei zusätzlich Wasser im
Lösungsmittel vorliegen zu haben, und die Reaktion somit optimal von statten gehen kann [39].
Neben der Synthese von SGen, ist die Darstellung auch durch die Isolierung aus höheren Pflanzen
möglich [40]. In der Literatur finden sich hier beispielsweise Sojabohnen [41], Erbsen, Avocado,
Blumenkohl oder Spinat als verwendete Pflanzen [42]. Weiters kann die Isolierung aus Pflanzenölen
erfolgen, wobei sich zur Trennung und Aufreinigung verschiedene Techniken existieren. Für die
Extraktion können beispielsweise Waschschritte mit organischen Lösungsmittelgemischen wie
Chloroform/Methanol 2:1 [43] oder Soxhletverfahren mit Aceton als Lösungsmittel verwendet
werden [44], wobei hier jeweils eine vorherige Homogenisierung der Pflanzen notwendig ist. Zur
Deaktivierung der enthaltenen Glycosidasen wird des Weiteren heißes Ethanol zugegeben [43]. Zur
weiteren Aufarbeitung können verschiedene chromatographische Methoden angewendet werden.
Bei den wichtigsten handelt es sich hierbei um gewöhnliche säulenchromatographische Methoden
wie Normalphasen- (NP), Umkehrphasen (RP)- sowie Silberionenchromatograpie. Weiters finden
auch präparative dünnschichtchromatographische Methoden wie NP-DC und RP-DC Verwendung. Bei
den am häufigsten eingesetzten chromatographischen Methoden handelt es sich um diverse HPLC-
Verfahren, welche gegenüber der gewöhnlichen Säulenchromatographie die Vorteile einer
geringeren Dauer, sowie eines verminderten Verbrauchs an Laufmittel aufweisen.
2.2.3 Vorkommen von Sterylglycosiden
Da SGe in einer Vielzahl von Pflanzen vorkommen, liegt es auf der Hand, dass diese auch über die
Ernährung zugeführt werden. Besonders zu erwähnen sind an dieser Stelle Sojabohnen, deren
Lecithin SGe in einer Konzentration von 3-4% beinhaltet [45]. Bezogen auf das Trockengewicht von
Pflanzen sind Blumenkohl, Zucchini, Brokkoli, Melanzani und Gurken die Lebensmittel, die mit einem
SG-Gehalt von 490, 462, 241, 234 beziehungsweise 217 µg/g Trockenmasse die höchsten
Konzentrationen enthalten [45]. Neben freien SGen kommen diese auch in Form von verschiedenen
Derivaten vor. Diese können polyhydroxyliert, sulfatiert sowie acyliert vorliegen [43]. Die Acylierung,
gewöhnlicherweise am 6´O-Atom des Zuckers, stellt die häufigste Derivatisierung der SGe dar. Meist
handelt es sich dabei um eine Veresterung mit Palmitinsäure. In geringeren Mengen kommen jedoch
auch Veresterungen mit Ölsäure, Stearinsäure, Linolsäure und Linolensäure vor [46]. In Abbildung 9
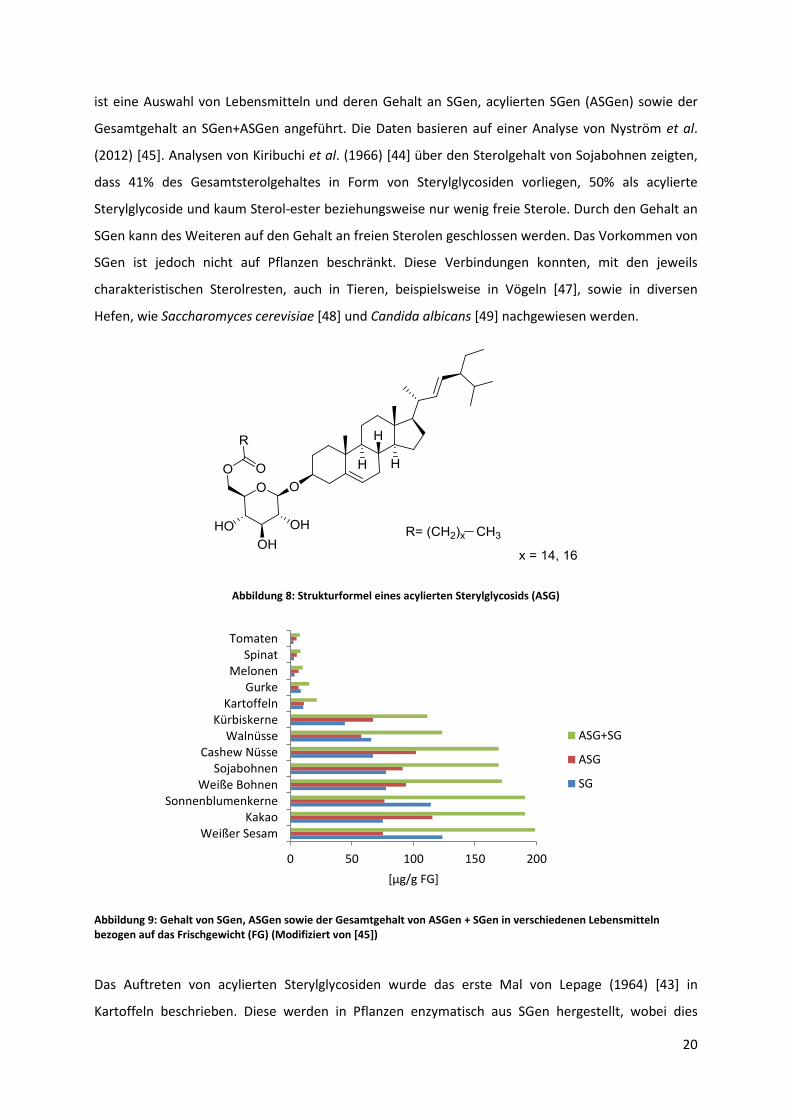
20
ist eine Auswahl von Lebensmitteln und deren Gehalt an SGen, acylierten SGen (ASGen) sowie der
Gesamtgehalt an SGen+ASGen angeführt. Die Daten basieren auf einer Analyse von Nyström et al.
(2012) [45]. Analysen von Kiribuchi et al. (1966) [44] über den Sterolgehalt von Sojabohnen zeigten,
dass 41% des Gesamtsterolgehaltes in Form von Sterylglycosiden vorliegen, 50% als acylierte
Sterylglycoside und kaum Sterol-ester beziehungsweise nur wenig freie Sterole. Durch den Gehalt an
SGen kann des Weiteren auf den Gehalt an freien Sterolen geschlossen werden. Das Vorkommen von
SGen ist jedoch nicht auf Pflanzen beschränkt. Diese Verbindungen konnten, mit den jeweils
charakteristischen Sterolresten, auch in Tieren, beispielsweise in Vögeln [47], sowie in diversen
Hefen, wie Saccharomyces cerevisiae [48] und Candida albicans [49] nachgewiesen werden.
Abbildung 8: Strukturformel eines acylierten Sterylglycosids (ASG)
Abbildung 9: Gehalt von SGen, ASGen sowie der Gesamtgehalt von ASGen + SGen in verschiedenen Lebensmitteln bezogen auf das Frischgewicht (FG) (Modifiziert von [45])
Das Auftreten von acylierten Sterylglycosiden wurde das erste Mal von Lepage (1964) [43] in
Kartoffeln beschrieben. Diese werden in Pflanzen enzymatisch aus SGen hergestellt, wobei dies
0 50 100 150 200
Weißer SesamKakao
SonnenblumenkerneWeiße Bohnen
SojabohnenCashew Nüsse
WalnüsseKürbiskerne
KartoffelnGurke
MelonenSpinat
Tomaten
[µg/g FG]
ASG+SG
ASG
SG

21
durch die Enzymklasse der Sterylglycosid-Acyltransferasen katalysiert wird [17]. Durch diese Enzyme
wird der Zucker am 6´O-Atom mit einer Fettsäure verestert, wobei Phosphoglyceride, Mono- und
Diacylglyceride sowie Monogalactosyldiacylglyceride häufige Donatoren der Fettsäurekomponente
darstellen [50]. Ein weiterer Biosyntheseweg für ASGe wurde von Frasch et al. (1976) [51] nach der
Untersuchung von Sterolen mittels radioaktiver Markierung des Cholesterols in den Setzlingen von
Nicotiana Tabaccum L. vorgeschlagen. Hierbei soll das Sterol mit einem bereits acylierten
Glycosylrest über ein unbekanntes Zwischenprodukt zum ASG reagieren.
2.2.4 Die Biodieselproblematik
Der Gehalt von SGen und ASGen in Pflanzenölen beträgt, abhängig von der Art der Pflanze, bis zu
einigen hundert Milligramm pro Kilogramm Öl, wobei der ASG-Gehalt in der Regel jenen der freien
SGe übersteigt (vgl. Tabelle 2) [2].
Tabelle 2: Analyse von SG und ASG in Pflanzenölen und Biodieselproben nach Lacoste et al. [2].
Probe SG Gehalt [mg/kg] ASG Gehalt [mg/kg]
Soja-FAME 10 n.d. Raps-FAME 12 n.d. Jatropha-FAME 18 n.d. Rapsöl n.d. n.d. Jatrophaöl 24 95 Palmöl 11 219 Sojaöl 13 21
n.d.: nicht detektierbar
Biodiesel (Fettsäuremethylester) wird im Zuge einer Umesterungsreaktion von Triacylglyceriden mit
Methanol hergestellt. Bei dieser Reaktion kommt es, als Nebenreaktion, zur Abspaltung des
Fettsäurerestes vom 6´O-Atom des acylierten Sterylglycosides, wodurch es zur Bildung freier SGe
kommt (siehe Abbildung 10).
Abbildung 10: Bildung von SGen aus ASGen durch Umesterung bei der Biodieselproduktion

22
ASGe sind in Pflanzenölen aufgrund des unpolaren Fettsäurerestes löslich, SGe hingegen besitzen nur
eine polare Zuckerkomponente ohne den angehängten Fettsäurerest und sind deshalb in der
unpolaren Biodieselmatrix nicht löslich, weshalb sie langsam ausfallen und in weiterer Folge als
Feststoff vorliegen. Dieses Verhalten der SGe wird durch kalte Temperaturen begünstigt [1]. Die
Temperatur, bei der Inhaltsstoffe im Biodiesel sichtbare Kristalle bilden, wird als cloud point (CP)
bezeichnet. Die Sichtbarkeit der Kristalle ist mit einem Durchmesser von > 0,5 µm definiert [52].
Diese Unlöslichkeit führt nun weiterführend dazu, dass diese Präzipitationen sowohl bei der Lagerung
des Biodiesels in Tanks, als auch bei der Verwendung in Motoren zu Verstopfung von Filtern und
Düsen führen können, was zur Folge unter anderem das Versagen von Motoren haben kann.
Außerdem liegen Vermutungen vor, dass die ausgefallenen SGe die Kristallisation und
Komplexbildung weiterer Verbindungen, wie beispielsweise jener von gesättigten Monoglyceriden,
begünstigen [3]. Labortechnisch wird die Auswirkung der SGe hinsichtlich der Verstopfung von Filtern
durch die Messung der Temperaturgrenze der Filtrierbarkeit1, auch als cold filter plugging point
(CFPP) bezeichnet, ermittelt. Der CFPP beschreibt die niedrigste Temperatur, bei der 20 mL Treibstoff
durch einen Filter mit einer Maschenweite von 45 µm bei einem Unterdruck von 0,019 Atmosphären
in 60 Sekunden noch problemlos durchgepumpt werden können [53].
2.2.5 Entfernung von Sterylglycosiden aus Biodiesel
2.2.5.1 Filtration und mechanische Methoden
Um den Problemen, welche SGe in Biodiesel hervorrufen, entgegenzuwirken, wurden in den
vergangenen Jahren zahlreiche Methoden entwickelt, um diese aus dem Treibstoff zu entfernen.
Viele dieser Methoden beschäftigen sich damit, die ausgefallenen Bestandteile abzufiltrieren. Lee et
al. (2006) [54] haben beispielsweise eine Methode entwickelt, bei welcher der Biotreibstoff über
Aktivkohle filtriert wird, um störende Bestandteile zu entfernen. Ein modifiziertes Verfahren wurde
von Danzer et al. (2007) [55] vorgestellt, bei dem direkt nach der Umesterungsreaktion zur
Herstellung von FAME auf 38°C gekühlt wurde, um unlösliche Stoffe wie SGe auszufällen. Durch
Zugabe von Aktivkohle und anschließender Filtration konnten ausgefallene SGe entfernt werden.
Sohling et al. (2011) [56] haben als Adsorbens ein Gemisch aus Smektit-Kieselgel (SiO2 > 60 Gew.%)
verwendet, dessen spezifische Oberfläche 120 m²/g beträgt. Dieses Verfahren hat den Vorteil, dass
das Adsorbens natürlich vorkommt und somit relativ kostengünstig bereitgestellt werden kann. Der
Nachteil ist jedoch, dass sich diese Methode lediglich für die Nachreinigung von Biodiesel eignet.
Durch dieses Verfahren konnte der Anteil freier SGe unter 20 ppm reduziert werden. Die
1 Bezeichnung laut DIN EN 116:1998-01

23
beschriebenen Methoden stellen jedoch das Problem dar, dass sie nur schwer in größerem Maßstab
durchführbar sind. Außerdem handelt es sich hierbei um kostenintensive Verfahren, bei denen ein
zusätzlicher Aufreinigungsschritt zur Entfernung des Adsorbens notwendig ist. Nicht zu
vernachlässigen ist des Weiteren, dass bei Filtrationsmethoden die Präzipitation von SGen
abgewartet werden muss, wodurch der Biodiesel vor der schlussendlichen Aufreinigung einem
Lagerungsprozess unterzogen werden muss, was sich sowohl als zeit- sowie auch als kostenintensiv
erweist. Weiters geht bei den Filtrationsprozessen jeweils ein Teil des Produktes verloren. Tang et al.
(2010) [57] haben verschiedene Strategien, nämlich Kaltfiltration, die Filtration bei Raumtemperatur,
die Zugabe von Adsorbens, Zentrifugieren sowie die Vakuumdestillation zur Entfernung von freien
SGen aus Biodiesel verglichen. Die Vakuumdestillation zeigte sich als die effektivste Methode, bei der
neben den freien SGen auch lösliche SGe bis auf einen Gehalt von 20 ppm abgetrennt werden
konnten. Die anderen untersuchten Methoden hatten keinen Einfluss auf gelöste SGe, es wurde
lediglich die Menge an bereits ausgefallenen SGen reduziert. Neben dem hohen technischen
Aufwand, den die Vakuumdestillation mit sich bringt, sowie den hohen Kosten, findet des Weiteren
eine drastische Qualitätsminderung des Biodiesels durch Reduktion der Oxidationsstabilität statt,
weshalb auch diese Methode, abgesehen von den hohen Kosten, keine optimale Lösung darstellt.
Diese Abnahme beruht auf der Entfernung der im Biodiesel natürlich enthaltenen Antioxidantien wie
Tocopherolen und Carotinoiden. Kass et al. (2011) [58] patentierten ein Verfahren, bei welchem
Biodiesel, bei dem bereits Präzipitate vorlagen, Ultraschall mit einer Energie zwischen 2000 und 7000
Joule ausgesetzt wurde. Außerdem wird beschrieben, dass einer Bildung von SG-Agglomeraten,
beispielsweise mit Monoglyceriden, die durch eine unvollständige Umesterungsreaktion im Biodiesel
enthalten sind, durch die vorherige Behandlung mit Ultraschall vorgebeugt werden kann. Um eine
übermäßige Erhitzung bei diesem Verfahren zu verhindern, geschieht die Ultraschallbehandlung in
Intervallen mit einer Dauer von je etwa 40 Sekunden. Durch dieses Verfahren kommt es zu einer
irreversiblen Dissoziation der Präzipitationen, so dass sich auch nach dem Abkühlen keine
Niederschläge mehr bilden können. Durch diese Behandlung kann die anschließende
Filtrationsdauer, verglichen mit jener ohne Ultraschallbehandlung, um 19% verkürzt werden.
2.2.5.2 Enzymatische Entfernung von SG
Neben der Möglichkeit der Filtration finden sich in der Literatur auch Methoden, in denen die
unlöslichen SGe in lösliche ASGe umgewandelt werden. Brask et al. (2012) [59]. beschreiben eine
Methode, in der die SGe in einer Umesterungsreaktion durch Lipasen, beispielsweise Novozyme 435,
in ASGe umgewandelt werden. Die Effizienz dieser Methode ist jedoch dahingehend begrenzt, dass
eine bestmögliche Umsetzung nur dann funktioniert, wenn sich das Substrat in Lösung befindet. Da
diese Enzyme nur eine begrenzte Thermostabilität aufweisen und eine optimale

24
Reaktionstemperatur von 40-60°C haben, ist eine vollständige Lösung des Produktes in unpolaren,
organischen Lösungsmitteln, wie Biodiesel eines darstellt, jedoch nicht möglich. Eine Erhöhung der
Reaktionstemperatur würde zu einer Minimierung der Aktivität beziehungsweise zu einer
Denaturierung des Proteins führen. Da sich in Biodiesel bei einer Temperatur von 50°C SGe jedoch
nur in einer Menge von 50 ppm lösen, und in Rohbiodiesel in der Regel 10-480 ppm [60] vorliegen, ist
deren Entfernung jedoch nur begrenzt möglich. Eine weitere Möglichkeit, die von Menzella et al.
(2013) [61] beschrieben wurde, ist der Einsatz von neu designten thermostabilen Sterylglycosidasen,
welche SGe durch Hydrolyse der glycosidischen Bindung in Glucose sowie in das Sterol spalten. Durch
die Thermostabilität der Enzyme kann die Reaktionstemperatur bei dieser Methode erhöht werden,
wodurch sich ein größerer Anteil der enthaltenen SGe in Lösung befindet und so enzymatisch
umgewandelt werden kann. Zu beachten ist jedoch, dass es sich bei enzymatischen Prozessen in der
Regel um relativ teure Methoden handelt, wodurch es hier noch an Forschung für die optimalen
Reaktionsbedingungen, um den Einsatz der Enzymmenge auf ein Minimum zu begrenzen, bedarf. Es
werden jedoch Experimente hinsichtlich des Recyclings von Enzymen durchgeführt, in dem diese
nach der Reaktion abfiltriert und anschließend durch Aufreinigungsschritte und Trocknungsprozesse
wieder aktiviert werden [62] [63].
2.2.5.3 Präprozedurale Prozesse
Bei den bisherig beschriebenen Methoden handelt es sich jeweils um Prozesse, die nach der
eigentlichen Biodieselherstellung durchgeführt werden. Weiters besteht jedoch die Möglichkeit, SGe
als auch ASGe, aus denen bei der Umesterung zu Biodiesel SGe hergestellt werden können (vgl.
Abbildung 10), bereits aus dem Rohöl zu entfernen. Murui et al. (1997) [64] haben beispielsweise ein
Verfahren entwickelt, mit denen 90% der freien SGe sowie 50% der ASGe durch die Zugabe von
Bleicherde zu Palmöl und anschließender Filtration entfernt werden konnten.
2.2.6 Analyse von Sterylglycosiden
2.2.6.1 Gaschromatographische Methoden
Aufgrund der geringen Flüchtigkeit von SGen und ASGen eignen sich gaschromatographische
Methoden nicht für deren Analyse, ohne die Verbindungen einer vorherigen Derivatisierung zu
unterziehen. Diese geschieht in der Regel durch eine Silylierung mittels MSTFA, welche zur
Stabilisation der thermolabilen Moleküle beiträgt, sowie die Auflösung verbessert [45]. Bei den GC-
Methoden wird grundsätzlich zwischen zwei Arten unterschieden. Zum einen gibt es die Möglichkeit
einer „indirekten GC“ andererseits jene der „direkten GC“. Bei der indirekten Methode, wird der
Sterolester durch alkalische Hydrolyse in den Sterolteil, in den Zuckerteil sowie die eventuell
angehängte Fettsäure gespalten. Die Fettsäure kann nun als das korrespondierende Salz, welches

25
durch die Verseifung entsteht, isoliert werden, anschließend beispielsweise durch Methylierung
derivatisiert, und so gaschromatographisch mittels FID-Detektion vermessen werden [65]. Bei der
Analyse der Sterole handelt es sich um die offizielle Methode für Sterolanalysen (ISO 12228). Die SGe
werden nach der alkalischen Hydrolyse durch Festphasenextraktion (SPE) oder
Dünnschichtchromatographie aufgereinigt, silyliert und anschließend am Gaschromatographen
analysiert [66]. Als Referenzwert dient in der Regel Cholesterol, welches eine ähnliche Retentionszeit
wie andere Sterole aufweist, wodurch somit deren relative Retentionszeit im Vergleich zum
Cholesterol ermittelt werden kann. Weiters birgt Cholesterol als interner Standard den Vorteil, dass
es in Pflanzen kaum vorkommt und somit der Peak ausschließlich auf das hinzugegebene Cholesterol
zurückgeführt werden kann [67]. Außerdem ist es kommerziell in großen Mengen verfügbar und
stellt das kostengünstigste Sterol dar. Für die direkte Methode hingegen ist keine vorhergehende
Hydrolyse notwendig. Hierbei wird das SG silyliert und anschließend direkt vermessen, wodurch
dieses Verfahren sowohl eine Zeit- als auch eine Kostenersparnis, verglichen mit der indirekten
Methode, birgt [68].
Durch die Ionisierung bei der Messung mittels GC-MS kommt es zum einen zur Spaltung der
glycosidischen Bindung, wodurch zum einen der Sterolteil als Ladungsträger analysiert werden kann.
Die Unterscheidung zwischen den verschiedenen Sterolen ist somit, durch die Kopplung mit einem
Massenspektrometer, leicht möglich. Zum anderen kommt es zur Abspaltung der Silylgruppen der
Zuckerkomponente. Diese bilden charakteristische, ladungstragende Trimethylsilylester und -ether
welche im Massenspektrum ersichtlich sind, und auf die Anwesenheit des Zuckers schließen lassen
[69]. Verglichen mit anderen verwendeten instrumentellen Methoden, wie TOF-
Massenspektroskopie oder NMR-Spektroskopie, ergibt sich bei gaschromatographischen Messungen
die längste Analysendauer. Außerdem ist, bedingt durch die Derivatisierung, eine Vorlaufdauer von
etwa einer Stunde bei der Messung einzuberechnen, wodurch die Dauer der Gesamtanalyse etwa
zwei Stunden beträgt. Den Vorteil, den diese Methode jedoch birgt, ist die exakte Berechnung der
Reinheit durch Integration der Peakflächen bei Verwendung eines Flammenionisationsdetektors
(FID). Weiters ist eine Unterscheidung von Anomeren aufgrund der unterschiedlichen
Retentionszeiten möglich. Ein Vorteil von gaschromatographischen Messungen ist die
Quantifizierung sowie die Differenzierung zwischen den einzelnen, strukturell sehr ähnlichen,
Sterolkomponenten. Positiv ist außerdem, dass aufgrund der hohen Sensitivität der Methode, nur
wenig Probe verwendet werden muss. Ist das Ziel der GC-Analyse lediglich die Quantifizierung von
SGen, so hat sich zur Detektion die Verwendung eines FID-Detektors bewährt, welcher den Vorteil
einer hohen Robustheit und Empfindlichkeit mit sich bringt. Bei Verwendung derselben Säule ist
außerdem eine GC-Messung mit MS-Kopplung zur Identifikation der Struktur und in weiterer Folge
eine Quantifizierung durch FID-Kopplung möglich. Lacoste et al. (2009) [2] haben mit einer GC-FID-

26
Methode, nach der vorhergegangenen Aufreinigung der Probe mittels Säulenchromatographie, eine
Nachweisgrenze von 4 ppm erreicht, die Quantifizierung gelang bis zu einer Konzentration von 10
ppm [2].
2.2.6.2 NMR-Spektroskopie
Die NMR-Spektroskopie stellt eine zuverlässige Methode zur qualitativen Analyse von SGen dar. Zu
einem handelt es sich, bei der Messung an sich, um eine schnelle Methode. So dauert die Erstellung
eines 1H-NMR-Spektrums etwa zwei Minuten, jene eines 13C-NMR-Spektrums zirka 30 Minuten. Zum
anderen sind durch molekülspezifische Shifts sichere Analyseaussagen möglich. Weiters ist, im
Gegensatz zur Gaschromatographie, keine Derivatisierung nötig. Negativ anzumerken ist jedoch,
dass, verglichen mit TOF-MS und GC-MS, mit einer Menge von ungefähr 20 mg viel Probe für die
Analyse nötig ist. Dadurch, dass für die Analyse deuterierte Lösungsmittel verwendet werden
müssen, ergibt sich außerdem noch ein nicht unwesentlicher Kostenfaktor. Eine Unterscheidung von
Anomeren ist bei der SG-Synthese, aufgrund der ähnlichen Signale, nur schwer möglich. Außerdem
ist die Auswertung der Ergebnisse, ohne vorliegende NMR-Referenzspektren, sehr zeitaufwändig.
Hinsichtlich der Reinheit ist anzumerken, dass sich diese lediglich durch das Überprüfen von
eventuell auftretenden Nebensignalen abschätzen lässt. Eine Berechnung der Reinheit ist somit nicht
möglich.
2.2.6.3 TOF-MS
Die Analyse mittels Flugzeitmassenspektrometer stellt mit einer Messdauer von etwa einer Minute
die schnellste der beschriebenen Methoden dar. Die Probe muss sich lediglich in einem geeigneten
Lösungsmittel, welches eine Mischbarkeit mit Acetonitril aufweist, lösen, was einen großen Vorteil
im Vergleich zur Gaschromatographie darstellt. Weiters ist keine Derivatisierung notwendig. Für die
Analyse ist, aufgrund der geringen Nachweisgrenze im Femto- bis Attogrammbereich nur eine
äußerst geringe Menge an Probe notwendig [70]. Durch den hochauflösenden Charakter des TOF-
Massenspektrometers lässt sich außerdem die Summenformel der Verbindung generieren, wodurch
eine sichere Identifizierung gegeben ist. Außerdem lässt sich eine Berechnung der Abweichung der
erhaltenen Masse von der theoretischen Masse in ppm vornehmen, wobei Abweichungen unter 5
ppm akzeptabel sind. Durch die milde Ionisierung bei Atmosphärendruck (APCI (Atmospheric
Pressure Chemical Ionization)) oder auch durch Elektrospray-Ionisation (ESI) findet des Weiteren
keine Fragmentierung in den Zucker- und den Sterolteil statt, wodurch das gesamte Molekül
analysiert werden kann. Der Nachteil, den die Methode darstellt ist jedoch, dass keine
Unterscheidung zwischen Anomeren vorgenommen werden kann. Weiters lässt sich, aufgrund von

27
eventueller selektiver Ionisationsfähigkeit verschiedener Moleküle, keine Berechnung der Reinheit
vornehmen.
2.2.6.4 HPLC
Der Einsatz von HPLC-Methoden kann sowohl für die Trennung von Sterylglycosiden, als auch für die
Quantifizierung erfolgen. Grundsätzlich finden sowohl Normalphasen- (NP) als auch Umkehrphasen-
(Reversed Phase (RP))-HPLC Verwendung. Mittels RP-HPLC ist jedoch, aufgrund der strukturellen
Ähnlichkeit, eine Unterscheidung verschiedener SGe nur schwer möglich, es kann somit nur der
Gesamt-SG-Gehalt bestimmt werden [71]. Eine gute Auftrennung zwischen Sterolen und SGen ist
jedoch möglich [72]. Mittels NP-HPLC hingegen können auch ähnliche SGe voneinander getrennt und
analysiert werden [67]. Ein Vorteil ist, dass für HPLC-Methoden in der Regel keine Derivatisierung
notwendig ist, was die Dauer der Probenvorbereitung minimiert. Die Messdauer ist kürzer als jene
der Gaschromatographie und beträgt etwa 20 Minuten und kann bei Verwendung von UHPLC-
Methoden auf wenige Minuten verkürzt werden. Weiters handelt es sich um eine nicht destruktive
Methode, weshalb Sterylglycoside nach der Messung im aufgetrennten Zustand isoliert werden
können.
In der Praxis werden verschiedene Detektortypen für die SG-Analyse verwendet. Eine HPLC-Methode
zur Trennung und Quantifizierung von SGen und ASGen wurde von Sugawara et al. (1999) [71]
beschrieben, wobei zur Detektion ein Lichtstreudetektor (ELSD, Evaporative Light Scattering
Detector) angewendet wurde. Mit dieser Methode wurde für SGe eine Nachweisgrenze von 0,2 µg
erreicht, für ASGe 0,5 µg.
Murui et al. (1993) [35] beschreiben zur Messung von SGen und ASGen eine Methode mit einer
Nachweisgrenze von 0,5-50 ng die auf der Derivatisierung mittels 1-Anthroylnitril beruht. Bei dieser
Methode werden Sterylglycoside zu Beginn mittels Säulenchromatographie aus der Probe isoliert,
durch dünnschichtchromatographische Trennung in SGe und ASGe separiert und anschließend bei
100°C für 20 Minuten, in Gegenwart von Quinuclidin (1-Azabicyclo[2.2.2]octan) als Katalysator, mit 1-
Anthroylnitril derivatisiert. Diese UV-sensitiven Derivate können in weiterer Folge durch HPLC mittels
UV-Detektor analysiert werden. Die UV-Absorption von Sterolen ist hierbei bei 200-210 nm zu
beobachten beziehungsweise bei 282 nm bei konjugierten Dienen wie Stigmasterol oder Ergosterol
[73]. Moreau et al. (2008) [74] beschreiben eine Methode, in der sich durch HPLC mit APCI-MS
Sterole in Biodiesel nachweisen und mit anschließender RP-HPLC mit Elektronenspray-MS eine
Unterscheidung zwischen Sitosterylglycosid und Campesterylglycosid vornehmen konnten. Mit NP-
HPLC und isokratischer Elution und anschließender Detektion mittels ELSD beziehungsweise UV-
Detektor wurde anschließend die Quantifizierung vorgenommen.

28
2.2.6.5 Dünnschichtchromatographie
SGe können durch dünnschichtchromatographische (DC-) Methoden durch Auftragung der Probe
gegen einen Standard analysiert werden. Die Detektion kann durch destruktive Weise, wie durch die
Verkohlung mittels Schwefelsäure, als auch durch nicht destruktive Weise, zum Beispiel durch die
Detektion mittels Ioddampf oder UV-Licht erfolgen. Verwendung finden auch Sprühreagenzien wie
Fluorescein und Rhodamin 6G [75]. Somit kann die Methode der nicht destruktiven Detektion neben
der qualitativen Analyse auch für die Quantifizierung herangezogen werden. Diese erfolgt durch das
Abschaben der SG-Bande von der DC-Platte und anschließender Trennung des Kieselgels durch
Waschschritte in organischen Lösungsmittelgemischen (z.B. Chloroform/Methanol 4:1) mit
anschließender Filtration sowie Zentrifugation [67]. Die Vorteile, die DC-Methoden bieten, sind zum
einen eine schnelle qualitative Analyse sowie die simultane Auftrennung mehrerer Proben. Weiters
handelt es sich um eine kostengünstige Methode, da die verwendeten Gerätschaften in der Regel
billiger sind als GC-, HPLC-Systeme etc. sind. Durch die anschließende Aufreinigung bei der
Quantifizierung sind DC-Methoden jedoch zeit- und arbeitsaufwändig. Weiters kommt es hierbei zu
Produktverlusten wodurch die Genauigkeit der Analyse abnimmt. Durch die geringere Anzahl an
theoretischen Böden, verglichen mit HPLC-Systeme, ist mitunter keine absolute Trennung recht
ähnlicher Substanzen, wie dies bei den verschiedenen Sterylglycosiden der Fall ist, gegeben.
Außerdem können nur Mengen im Milligramm-Bereich quantifiziert werden, wodurch diese
Methode eher für qualitative Bestimmungen brauchbar ist.
3 Grundlagen und Theorie II: Analytische Methoden-Allgemein
In Kapitel drei werden die analytischen Methoden, welche im Zuge der Diplomarbeit verwendet
wurden, vorgestellt und deren theoretischer Hintergrund genauer beschrieben. Auf nicht
verwendete Methoden wird nicht genauer eingegangen.
3.1 Chromatographie
Das Prinzip der Chromatographie beruht auf der Trennung von Komponenten mit verschiedenen
chemischen oder physikalischen Eigenschaften zwischen zwei nicht mischbaren Phasen.
Grundsätzlich wird zwischen einer stationären und einer mobilen Phase unterschieden. Die
stationäre Phase ist in der Regel an eine Säule gebunden, die mobile Phase läuft durch sie in eine
definierte Richtung hindurch. Die Trennung beruht auf der Wechselwirkung der Analyten mit der
stationären Phase, wodurch diese zu unterschiedlichen Zeiten eluieren. Zur Analyse wird die zu

29
analysierende Probe in der flüssigen, gasförmigen oder superkritischen mobilen Phase gelöst und auf
die Säule aufgetragen.
In Abbildung 11 ist eine schematische Darstellung der chromatographischen Auftrennung ersichtlich.
Das Schema zeigt eine höhere Wechselwirkung des Moleküls A mit der stationären Phase, wodurch A
sich mit einer langsameren Geschwindigkeit als B bewegt und somit länger in der Säule verbleibt. Bei
automatisierten Systemen kann sich am Probenauslass noch ein Detektor befinden.
Abbildung 11: Schematische Darstellung der Säulenchromatographie [76]
Eine Unterscheidung der unterschiedlichen Chromatographiearten kann durch die verschiedenen
Arten der Interaktion zwischen der stationären Phase und dem Analyten vorgenommen werden. In
der Regel unterteilt man in Adsorptions-, Verteilungs-, Ionenaustausch-, Größenausschluss- und
Affinitätschromatographie. Außerdem kann eine Unterteilung aufgrund des Aggregatzustandes der
stationären Phase in Gaschromatographie (GC) und Flüssigkeitschromatographie (LC, Liquid
Chromatography) getätigt werden. Bei der Flüssigkeitschromatographie hat sich mittlerweile die
Methodik der High Performance Liquid Chromatography (HPLC) als schnelle und automatisierte
Methode durchgesetzt. Am Ende der chromatographischen Auftrennung finden in der Regel
verschiedene Detektorarten Verwendung. Die Detektion kann beispielsweise anhand chemischer
Derivatisierung erfolgen. Bei instrumentellen Methoden finden unter anderem Flammenionisations-,
Lichtstreu-, UV-VIS, Brechungsindex und Wärmeleitfähigkeitsdetektoren Verwendung. Auf die in
dieser Arbeit angewendeten Methoden wird im Zuge dieses Kapitels noch genauer eingegangen.

30
3.1.1 Adsorptionschromatographie
Bei der Adsorptionschromatographie wird als stationäre Phase ein Adsorbens eingesetzt. Die
Auftrennung beruht auf der wiederholten Adsorption und Desorption des Analyten an der
stationären Phase. Als stationäre Phase kann beispielsweise Silikagel dienen. Van-der-Waals-Kräfte
sowie Wasserstoffbrückenbindungen sind für unterschiedliche Retentionszeiten verantwortlich. So
verbleiben, bei Silikagel als stationäre Phase, polare Substanzen länger in der Säule, unpolare
Substanzen eluieren früher. Somit können durch diese Methode polare von unpolaren Substanzen
getrennt werden. Je nach Größe der Säule beziehungsweise nach der Methode, können
Probenmengen im Bereich weniger Milligramm, beispielsweise bei Festphasenextraktion (SPE, Solid-
Phase-Extraction) bis zu einigen Gramm aufgetrennt werden. Durch die verschiedenen
Retentionszeiten der Analyten können somit die Proben, beispielsweise durch den Vergleich mit der
Retentionszeit interner Standards, charakterisiert werden. Des Weiteren kann noch zwischen
Normalphasen (NP)- und Umkehrphasenchromatographie (RP, Reversed-Phase) unterschieden
werden. Bei NP-Chromatographie werden polare stationäre Phasen, wie Silikagel, und unpolare
Laufmittel eingesetzt, bei RP-Chromatographie fungieren beispielsweise langkettige
Kohlenwasserstoffe als unpolare, stationäre Phasen [77].
3.1.2 Verteilungschromatographie
Die Auftrennung bei der Verteilungschromatographie ist auf die unterschiedliche Löslichkeit der
Analyten in einer der beiden Phasen zurückzuführen. Bei besserer Wechselwirkung mit der
stationären Phase bleibt der Analyt also länger in der Säule. Ist hingegen eine schlechte
Wechselwirkung mit der stationären Phase und dafür eine bessere mit der mobilen Phase gegeben,
so verlässt der Analyt schneller die Säule und wird somit von anderen Stoffen getrennt. Bei der
stationären Phase handelt es sich um eine Flüssigkeit, die an ein festes Trägermaterial gebunden ist.
Ist die mobile Phase flüssig, so handelt es sich um LLC (Liquid Liquid Chromatography), liegt sie
hingegen gasförmig vor, so spricht man von GLC (Gas Liquid Chromatography) oder einfach
Gaschromatographie (GC).
Auf GC-Methoden wird in diesem Kapitel noch genauer eingegangen, LLC-Methoden hingegen
werden nicht weiter behandelt.
3.1.2.1 Gaschromatographie
Die Gaschromatographie stellt einen Sonderfall dar, da es sich hierbei sowohl um eine Verteilungs-
als auch um eine Adsorptionschromatographie handelt. Die Auftrennung erfolgt hierbei anhand der
unterschiedlichen Siedepunkte der Analyten. Diese werden verdampft und anschließend nach der

31
Wechselwirkung mit der stationären Phase getrennt. Als mobile Phasen dienen Inertgase wie
Stickstoff oder Helium, aber auch Wasserstoffgas findet Verwendung. Die Säulen, in der analytischen
Chemie in der Regel Kapillarsäulen, sind als stationäre Phase meist mit Polysiloxanharzen
ausgekleidet. In der präparativen Chemie werden hingegen meist gepackte Säulen, welche unter
anderem einen größeren Durchmesser und geringere Längen als Kapillarsäulen aufweisen,
verwendet. Der Innendurchmesser von Kapillarsäulen beträgt zwischen 0,1 und 0,75 mm, Längen
zwischen 10 und 100 m finden Verwendung. Die Säule befindet sich im Säulenofen in dem der Analyt
auf Temperaturen bis 400°C aufgeheizt wird [78]. Für die Probenauftragung können verschiedene
Injektorsysteme verwendet werden, wobei die beiden am häufigsten verwendeten Injektoren
Split/Splitless für thermostabile, beziehungsweise für thermolabile Verbindungen Cool on Column-
Injektoren darstellen [79].
Abbildung 12: Schema eines Gaschromatographen. (Übernommen von [80])
Detektoren, die in der Gaschromatographie verwendet werden, können in zwei Unterkategorien
eingeteilt werden. Zum einen handelt es sich um konzentrationsabhängige Detektoren, die die
Anzahl der Teilchen, welches sich in einem definierten Volumen des Trägergases befindet, detektiert.
So zum Beispiel Wärmeleitfähigkeitsdetektoren (WLD). Andererseits existieren
massenstromabhängige Detektoren, welche die Anzahl der Teilchen, die in einer gewissen Zeit zum
Detektor gelangen, messen [81]. Ein Beispiel dafür stellt der Flammenionisationsdetektor (FID) dar.
Durch die Anwendung in der vorliegenden Diplomarbeit, wird auf diesen im Zuge dieses Kapitels
noch genauer eingegangen. Weiters können GC-Systeme mit Massenspektrometern gekoppelt
werden (vgl. Kapitel 4.2 bzw. 4.7).
Die Detektion mittels FID basiert auf der Ionisierung des Analyten in einer Flamme, wodurch
messbarer Strom entsteht. Der Einsatzbereich ist vor allem die Quantifizierung von oxidierbaren
Kohlenstoffen, also Verbindungen die C-C und C-H Bindungen enthalten. Bei diesen Substanzen
zeichnet sich der FID durch seine hohe Sensitivität aus, der lineare Bereich beträgt 107 [82] . Nicht
verbrennbare Substanzen können nicht detektiert werden. Auch für die Detektion von funktionellen

32
Gruppen ist diese Methode wenig geeignet. Weitere Nachteile sind, dass es sich hierbei um eine
destruktive sowie nicht selektive Methode handelt. Die Probe wird durch ein Trägergas in eine, durch
die Verbrennung von Wasserstoffgas erzeugte Flamme, die sich in einem elektrischen Feld befindet,
eingebracht. Durch die Verbrennung der kohlenstoffhältigen Verbindungen kommt es zur Pyrolyse
und somit zur Bildung von Radikalen. Diesen wird in weiterer Folge Luft zugeführt, wodurch es durch
die Reaktion mit Sauerstoff zur Oxidation und somit zur Bildung von positiv geladenen Ionen sowie
zur Bildung von Elektronen kommt.
Abbildung 13: Pyrolyse und Ionisation im FID
Über der Flamme befindet sich eine positiv geladene Sammelelektrode, welche gleichzeitig die
Anode des Systems darstellt. Die Flammenspitze sowie die Düse, aus der Wasserstoffgas strömt,
fungieren als Kathode. Die erzeugten Elektronen werden an der Anode gesammelt und der daraus
resultierende Stromfluss gemessen.
Abbildung 14: Schematische Darstellung eines Flammenionisationsdetektors (Modifiziert von [83])
3.1.3 Größenausschlusschromatographie-GPC
Bei der Größenausschlusschromatographie (SEC, engl.: Size Exclusion Chromatography) oder auch
Gelpermeationschromatographie (GPC) handelt es sich um eine Unterkategorie der
Flüssigchromatographie, in der Moleküle anhand ihrer Größe, genauer durch das hydrodynamische
Volumen aufgetrennt werden. In der Säule befindet sich hierbei als stationäre Phase ein poröses Gel.
Kleine Moleküle verbleiben länger in der Säule, da sie in die porösen Partikel eindringen können und
sich darin nur mehr durch Diffusion bewegen können, große Moleküle hingegen strömen an den
Partikeln vorbei und haben demzufolge eine kürzere Retentionszeit. Die Bestimmung der
Molekülmasse wird anschließend durch den Vergleich mit Kalibriersubstanzen vorgenommen. Die
Detektion kann beispielsweise über die Ermittlung des Brechungsindexes erfolgen.

33
3.2 Massenspektrometrie
Die Methodik der Massenspektrometrie (MS) beruht auf der Analyse von Atomen und Molekülen
aufgrund ihrer Masse. Die Proben werden verdampft und ionisiert, woraufhin geladene Moleküle
entstehen. Die entstandenen Ionen werden anschließend hinsichtlich deren Masse zu
Ladungsverhältnis (m/z) analysiert.
3.2.1 Ionisierung
Die Ionisierung kann auf verschiedene Weisen erfolgen und hängt von der chemischen und
physikalischen Beschaffenheit der Proben ab. Häufig kommen unter anderem Elektronenstoß-
Ionisation (EI), Photo-Ionisation (PI), Elektrospray-Ionisation (ESI), Chemische-Ionisation (CI) sowie
Matrix-unterstützte Laser-Desorption/Ionisation (MALDI) vor. Hierbei kann noch zwischen harten (EI)
und weichen (ESI, MALDI) Ionisierungen unterteilt werden, wobei bei der weichen Ionisierung nur
wenige Fragment-Ionen entstehen [84]. Als Möglichkeit der weichen Ionisierung hat sich des
Weiteren die chemische Ionisation bei Atmosphärendruck (APCI) etabliert, welche bei weniger
polaren Substanzen eine bessere Effektivität als zum Beispiel ESI aufweist [85]. Eine
Zusammenfassung der Ionisationsmöglichkeiten für die Massenspektrometrie sowie eine
Unterteilung anhand der Eigenschaften der zu ionisierenden Proben sind in Abbildung 15 dargestellt
[86].
Abbildung 15: Ionisationsmöglichkeiten für die Massenspektroskopie [86]

34
3.2.1.1 Elektronenstoßionisation
Bei dieser Methode handelt es sich um das Standardverfahren bei gaschromatographischen
Massenanalysen, wobei es bei dieser Methode zu einer weitgehenden Fragmentbildung kommt.
Durch das Erhitzen eines kathodischen Filaments, meist aus Wolfram oder Rhenium, werden durch
thermoionische Emission Elektronen erzeugt. Diese Elektronen werden zu einem Strahl gebündelt
und durch das Zuführen von Energie, etwa 70 eV, durch eine Anode beschleunigt. Die Probe wird
senkrecht zum Elektronenstrahl aufgetragen, wodurch Elektronen und Probenmoleküle miteinander
in Wechselwirkung treten. Auf diese Weise kann es zur Ionisation der Moleküle kommen, wobei
durch elektrostatische Abstoßung Elektronen aus der Elektronenhülle der Moleküle geschlagen
werden und es so zur Bildung von radikalischen Molekülkationen kommt. Diese werden daraufhin
durch ein schwaches elektrisches Feld zum Analysator transportiert [84].
Abbildung 16: Bildung von radikalischen Molekülkationen
Durch überschüssige Energie kann es weiters zum Zerfall der Molekülionen kommen. Die Fragmente
sind für jedes Molekül charakteristisch und sind, sofern sie eine Ladung tragen, im Massenspektrum
ersichtlich.
3.2.1.2 Elektrospray-Ionisation
Bei der Elektrospray-Ionisation handelt es sich um eine Ionisation bei Atmosphärendruck, welche bei
LC-MS Verfahren Anwendung findet. Durch diese milde Ionisierung zählt die ESI zu den weichen
Ionisationsmethoden und wird deshalb, aufgrund der geringen Fragmentierung, oft für die Analyse
von Biomolekülen eingesetzt. Zu Beginn dieser Ionisationstechnik wird die Probe bei
Atmosphärendruck durch eine Metallkapillare gepumpt, an deren Spitze eine Spannung angelegt ist.
Durch den Spannungsunterschied zur Gegenelektrode, etwa 3-6 kV, kommt es zur Ausbildung eines
starken elektrischen Feldes [84]. Aufgrund der elektrischen Spannung bewegen sich die Ionen in der
Analytlösung in Richtung der Gegenelektrode, wodurch sich ein Überschuss an gleichartig geladenen
Ionen bildet, welche sich gegenseitig abstoßen. Beim Austritt aus der feinen Spitze der Kapillare
bilden diese einen sogenannten Taylor-Cone. Das heißt, dass sich diese unter der Bildung feiner
Tröpfchen explosionsartig in Richtung der Gegenelektrode bewegen [87]. Der zweite Schritt der
Ionisation, die Vernebelung (engl. Nebulisation) wird durch ein inertes Trägergas, in der Regel
erhitzter Stickstoff, unterstützt. Der Durchmesser der Tröpfchen nimmt immer weiter ab, bis das
„Rayleigh limit“ erreicht ist und es kommt zur Abstoßung der Ionen aufgrund der Ladungsgleichheit
(„Coulomb-Explosion“), wodurch wiederum kleinere Tröpfchen entstehen. Aus den Tröpfchen
werden nun die Ionen in die Gasphase emittiert. Durch die Spannungsdifferenz zwischen der

35
Kapillare und der Gegenelektrode werde die Ionen nun, durch eine Öffnung in der Gegenelektrode in
das MS transportiert [88].
Abbildung 17: Elektrospray-Ionisation (Modifiziert von [87])
3.2.1.3 Chemische Ionisation bei Atmosphärendruck, APCI
Bei der chemischen Ionisation bei Atmosphärendruck handelt es sich um eine besonders
analytenschonende Modifizierung der chemischen Ionisation. Sie wird, wie auch ESI unter
Atmosphärendruck durchgeführt, jedoch handelt es sich bei APCI um eine etwas härtere Methode,
wodurch es zu vermehrter Fragmentbildung kommt [89]. Diese Methodik findet beispielsweise bei
thermisch stabilen und verdampfbaren Analyten, welche durch ESI schlecht ionisierbar sind, da sie
zum Beispiel wenige funktionelle Gruppen besitzen, Anwendung. Der gelöste Analyt wird auf eine
Kapillare aufgetragen und durch einen Stickstoffstrom zerstäubt, wodurch ein Spray entsteht der in
weiterer Folge zum Verdampfen des im Überschuss vorliegenden Lösungsmittels in einen erhitzten
Quarzkanal (Vaporizer) gelangt. Durch Anlegen einer Hochspannung an die Corona-Nadel entsteht
ein Elektronenstrahl, durch welchen Primärionen aus der gasförmigen Lösungsmittelphase erzeugt
werden. Durch Kollision mit den im Dampf befindlichen Analytmolekülen beziehungsweise durch den
Energieunterschied zu den Lösungsmittelionen werden die Analytenmoleküle in weiterer Folge
ionisiert. [84] Beim Lösungsmittel handelt es sich oft um einen Ammoniumformiat-Puffer. Die
Ionisierung kann im Positiv- oder im Negativ-Modus erfolgen, wodurch es zur Bildung von positiven
beziehungsweise negativen Ionen kommt. Im Positiv-Modus kommt die Ionisation entweder durch
Protonentransfer zwischen dem Pufferion, bei Ammoniumformiat demzufolge Ammonium, und dem
Analyten oder durch Adduktbildung zustande. Im Negativ-Modus kommt es entweder zur Abspaltung
eines Protons oder wiederum zur Adduktbildung, bei Ammoniumformiat also der Anlagerung des
Formiat-Ions [84]. Bei den gebildeten Ionen handelt es sich jeweils um Primärionen. Die Ionen
werden daraufhin durch einen dünnen Einlass fokussiert und zum MS transportiert.

36
Abbildung 18: Schematische Darstellung der Chemischen Ionisation bei Atmosphärendruck. (Modifiziert von [90])
3.2.2 Analysatoren
Die Analyse von Molekülen beruht auf der Trennung von Ionen anhand verschiedener physikalischer
Prinzipien, wobei das Masse zu Ladungsverhältnisses (m/z) entscheidend ist. Analysatoren können
grundsätzlich in zwei Arten eingeteilt werden. Zum einen in simultan messende Analysatoren, bei
denen alle Ionen gleichzeitig gemessen werden (zum Beispiel Flugzeitanalysatoren (TOF)) sowie
gepulste Analysatoren, bei denen Ionen nacheinander gemessen werden (zum Beispiel
Quadrupolanalysatoren). Weiters kann eine Unterscheidung aufgrund der physikalischen Prinzipien
erfolgen. Hierbei wird anhand der Analyse der unterschiedlichen Flugzeiten der im feldfreien Raum
(z.B. TOF), der Selektion anhand der Ionenmassen in elektrischen Wechselfeldern (z.B. Quadrupol)
und der Ablenkung der Ionen in elektrischen oder magnetischen Feldern (Sektorfeldanalysatoren)
unterschieden [84] [91].
3.2.2.1 Flugzeitanalysatoren-TOF
Das Prinzip von Flugzeitanalysatoren beruht auf der Trennung der Ionen aufgrund ihrer Masse, wobei
alle Ionen simultan analysiert werden können. Ionen werden, von der Ionenquelle kommend, in
einem elektrischen Feld gepulst beschleunigt, bevor die Ionen mit äquivalenter kinetischer Energie in
einen feldfreien Bereich mit der definierten Länge d eintreten. Durch die Korrelation der Masse mit
der kinetischen Energie durch die Formel Ekin=0,5*(m*v2) lässt sich somit berechnen, dass Ionen mit
höheren Massen eine längere Verweildauer in der feldfreien Zone aufweisen. In der Regel
unterscheidet man zwischen linearen TOFs, bei denen die Ionen eine lineare Flugbahn durch das
Flugrohr haben, und Reflektron-TOFs. Reflektrons, auch Ionenspiegel, kehren am Ende des
Flugrohres die Richtung der Ionen um, wodurch der Einfluss der kinetischen Energieverteilung auf die

37
Flugzeit minimiert wird, was in einer Erhöhung der Auflösung, im Vergleich zu linearen TOFs
resultiert [84].
Abbildung 19: Schematische Darstellung eines Reflektron-TOFs (Modifiziert von [92])
3.2.2.2 Quadrupol
Quadrupolanalysatoren werden häufig in Verbindung mit GC-EI-MS eingesetzt, wobei der Quadrupol
das Herzstück des Massenspektrometers darstellt [93]. Der Quadrupol besteht aus vier parallel
angeordneten Stabelektroden, welche durch Keramikklemmen zusammengehalten werden. Es liegen
sich jeweils zwei gleich geladene Stabelektroden gegenüber (vgl. Abbildung 20). Zur Trennung wird
ein elektrisches Feld, durch Anlegen einer Gleichspannung (dc) und einer oszillierenden
hochfrequenten Wechselspannung (ac), erzeugt [93]. Die von der Ionisationsquelle erzeugten Ionen
werden vor der Analyse beschleunigt, und können dann in den Quadrupol eintreten. Negativ
geladene Ionen treten danach mit den positiv geladenen Stabelektroden in Wechselwirkung und
positive Ionen mit den negativ geladenen. Diese Wechselwirkung variiert je nach Stärke des
angelegten elektrischen Feldes. Durch Veränderung des elektrischen Feldes werden Ionen anhand
der verschiedenen Massen nacheinander auf stabile, sinusförmige Flugbahnen gebracht und können
so den Quadrupol passieren [84].
Abbildung 20: Schema eines Quadrupol-Analysators (Modifiziert von [94])

38
3.3 Kernspinresonanzspektroskopie-NMR
Die NMR-Spektroskopie ist eine Untergruppe der Spektroskopie. Spektroskopische Methoden
basieren im Allgemeinen darauf, dass verschiedene Strahlungsarten nach definierten Eigenschaften,
wie zum Beispiel Wellenlänge, Energie oder Masse zerlegt werden. Bei der NMR-Spektroskopie
werden die Atomkerne anhand ihres Eigendrehimpulses (Kernspin) untersucht. Durch diesen
Eigendrehimpuls erzeugen die Atomkerne ein Magnetfeld, welches willkürlich ausgerichtet ist. Durch
das Anlegen eines Magnetfeldes, durch die Einbringung von resonanten elektromagnetischen
Wellen, kommt es zu einer Ausrichtung der Spins, entweder mit dem angelegten Magnetfeld, oder in
die entgegengesetzte Richtung [95]. Es kommt also zum Übergang von einem relaxierten
Energieniveau in ein angeregtes (Resonanz). Atome gleicher Art haben in der Regel die gleiche
Resonanz. Sind diese an verschiedene andere Atome gebunden, ändert sich diese geringfügig, da die
Elektronendichte um den Kern verschoben ist, wodurch unterschiedliche Bindungspartner stärkere
oder schwächere Abschirmungen des angelegten Magnetfeldes vom Atomkern hervorrufen. Durch
diesen Rückschluss auf die Elektronendichte eignet sich die NMR-Spektroskopie zur
Strukturaufklärung von Molekülen.
Abbildung 21: Schematische Darstellung eines Kernspinspektrometers (Modifiziert von [96])
Die Probe wird hierbei, um eine Beeinflussung des Spektrums durch das Lösungsmittel, durch darin
enthaltene Wasserstoffatome, zu vermeiden, in deuteriertem Lösungsmittel gelöst und auf das
veränderbare Magnetfeld aufgebracht. Dieses ist von einer Induktionsspule umgeben, welches
senkrecht zum bestehenden Magnetfeld ein elektromagnetisches Wechselfeld erzeugt [97]. Das
veränderbare Magnetfeld wird nun so lange verstärkt, bis es zu einer Resonanz kommt, wodurch die
Atomkerne Energie aus dem Wechselfeld aufnehmen und es somit zu einer messbaren Veränderung
der Stromstärke kommt. Die Auswertung erfolgt anhand der Berechnung der Verschiebung zu einem
Standard (Shift), bei 1H- sowie 13C-Messungen wird hierzu häufig Tetramethylsilan (TMS) eingesetzt
[95].

39
4 Material und Methoden
4.1 Lösungsmittel und Reagenzien
1,8-Diazabicyclo[5.4.0]undec-7-en (DBU) (98%), Sigma-Aldrich
Aceton (chem. Rein), Brenntag
Acetonitril (HPLC- grade ≥ 99,9%), Fisher-Scientific
Benzylbromid (98%), Fluka
Biodiesel, ADM-Hamburg
Bromwasserstoff (33% in AcOH), Merck
Calciumchlorid (wasserfrei geschmolzen granuliert, reinst), Fisher
Chloroform (99%), Merck
Cholesterol (from sheep wool, ≥ 92.5%), Sigma-Aldrich
Cyclohexen (99%), Riedel-de-Haen
DC Kieselgel 60 F254, Merck
Dichlormethan (HPLC-grade), Acros
Diethylether (rektifiziert), Prolab
Dimethylformamid (99%), Fluka
Dioxan (99,5%), Acros
Essigsäureanhydrid (98%), Riedel-de-Haen
Ethanol (96%), VWR
Ethanol (abs.), Merk
Kaliumhydroxid (p.A.), Fisher
Kieselgel60 (0,015-0,040 mm), Merck
Lipase acrylic resin from Candida antarctica (Novozyme 435), Sigma Aldrich
Magnesiumsulfat (97% wasserfrei pur.), Acros
Methanol (techn. > 98%), VWR
Methanol (HPLC-grade ≥ 99,5%), VWR
Molekularsieb 3Å, Merck
Molekularsieb 4Å, Merck
MSTFA (N-Methyl-N-(trimethylsilyl) trifluoroacetamid) (97%), Acros
Natrium (In Paraffinöl), Merck
Natriumacetat (wasserfrei, 99%), Riedel-de-Haen
Natriumchlorid (techn.), Brenntag
Natriumhydrid (dry, 95%), Sigma-Aldrich

40
Natriumhydrogencarbonat(≥ 99%), Riedel-de-Haen
Natriumhydroxid (97%), Acros
Natriumsulfat (≥ 99%, wasserfrei), Carl Roth
n-Hexan (HPLC- grade ≥ 97%), VWR
n-Hexan (techn. 95%), Carl Roth
Palladiumhydroxid/C (Pearlman´s Catalyst), Sigma Aldrich
Palmitinsäure(puriss. p.a), Riedel-de-Haen
Petrolether (40-65°C bp.), Acros
Phosphorpentoxid (≥ 98,5%), Riedel-de-Haen
Piperidin (99%), Sigma-Aldrich
Pyridin (≥ 99%), Acros
Rapsöl, Spar
Saccharose (Staubzucker), Dr. Oetker
Salzsäure (37%), IfC
Silbercarbonat (99%), Sigma-Aldrich
Sojaöl, Kunela Feinkost
Tetrahydrofuran (HPLC-grade ≥ 99,5%), VWR
Trichloracetonitril (98%), Sigma-Aldrich
Triethylamin (99%), Riedel-de-Haen
Trimethylsilyl trifluoromethanesulfonate (TMSOTf) (99%), Sigma-Aldrich
α-D-Glucose (wasserfrei), Sigma-Aldrich
β-D-Glucosepentaacetat (98%), Sigma-Aldrich

41
4.2 Geräte
4.2.1 Probenvorbereitung
Rotavapor Heidolph VV2000 mit Wasserbad Heidolph WB 2000 und Membranvakumpumpe ABM MZ
2C, Schwabach, Deutschland
Autovortex SA6, Stuart Scientific, Staffordshire, United Kingdom
Laborschüttler IKA KS 130 basic, Staufen, Deutschland
4.2.2 GC-MS
Agilent Technologies 7693 Autosampler
Agilent Technologies 7890A GC-System
Agilent Technologies 5975C inert XL MSD with Triple-Axis-Detector
Software: MSD-Chemstation E.02.00.493
Säule: DB5-HT (400°C max, 30 m, 0,25 mm, 0,10 µm)
4.2.3 HPLC-TOF-MS
TOF-MS: Agilent 6230 TOF LC/MS G6230B
Agilent 1260 Infinity Series
HiP Degasser G4225A
Bin Pump G1312B
ALS Autosampler G1329B
TCC Columnthermostat G1316A
DAD Detector G4212B
Software: MassHunter Workstation Rev.B.05.01 SP2
4.2.4 NMR-Spektroskopie
Spektrometer: Bruker Avance III
Probenkopf: Broadband Observe
4.2.5 GC-FID
HP 7683 Series Injector
HP 6890 Series GC System
Software: GC ChemStation Rev.B. 03.01.
Säule: DB5-HT (400°C max, 15 m, 0,32 mm, 0,10 µm)

42
4.3 Synthese von Cholesterylglycosiden
In diesem Kapitel wird die Synthese von Cholesterylglycosiden durch drei unterschiedliche Methoden
beschrieben. Bei diesen handelt es sich um die Koenigs-Knorr-Methode, die Trichloracetimidat-
Methode und eine weitere Trichloracetimidatmethode, bei der Saccharose als Edukt dient.
4.3.1 Synthese durch Koenigs-Knorr Methode
4.3.1.1 Synthese von β-D-Glucose-Pentaacetat [98]
Bei der Synthese von Glucosepentaacetat wurde Glucose (25,00 g; 138,8 mmol) und Natriumacetat
(20,00 g; 243,8 mmol) in einen 250 mL Rundkolben eingewogen, in Essigsäureanhydrid (125 mL; 1,13
mol) gelöst und der Rundkolben anschließend mit einem Rückflusskühler inklusive eines CaCl2-
Trockenrohrs verbunden. Die Lösung wurde 90 min lang unter Rückfluss und unter Rühren erhitzt
und anschließend wurde die Reaktion auf Raumtemperatur abgekühlt. Das entstandene
Produktgemisch wurde anschließend langsam auf 1250 mL Eiswasser gegossen, woraufhin sich
farblose Kristalle bildeten, die mit der Vakuumpumpe abgesaugt wurden. Anschließend wurde aus
Methanol umkristallisiert. Dazu wurden die Kristalle in wenig heißem Methanol gelöst und heiß
filtriert. Nach wenigen Stunden bildeten sich bei Raumtemperatur farblose Kristalle, die wieder
abgesaugt wurden. Das Produkt der Reaktion wurde bei 45°C über Nacht im Trockenschrank
getrocknet. Die Analyse erfolgte mittels Dünnschichtchromatographie und
Schmelzpunktbestimmung.
Rf-Wert: (Laufmittel Toluol/Aceton 3:1) 0,57
Ausbeute: 32,75 g (83,90 mmol), (60% d. Th., 97% d. Lit.)
4.3.1.2 Synthese von Acetobromoglucose
Glucosepentaacetat (15,00 g; 38,4 mmol) und 33%-ige Bromwasserstofflösung in Eisessig (34 mL;
192,7 mmol HBr) aus dem Kühlschrank wurden in einen 250 mL Rundkolben durch Rühren und durch
kräftiges Umschütteln unter Eiswasserkühlung in eine homogene Mischung überführt. Der
Rundkolben wurde mit einem CaCl2-Trockenrohr versetzt und die Reaktion anschließend zwei
Stunden bei Raumtemperatur gerührt, bis sich eine klare Flüssigkeit ergab. Das Gemisch wurde
daraufhin unter Rühren mit einem Glasstab in ein Becherglas mit 700 mL Eiswasser gegossen. Es
bildete sich ein farbloser Niederschlag, der mit dem Glasstab gründlich unter Eiswasser zerrieben
wurde. Dieser wurde mit der Vakuumpumpe abgesaugt und mit Wasser nachgewaschen. Die
farblosen Kristalle wurden in Diethylether gelöst und das ausgeschiedene Wasser mit einem
Schütteltrichter entfernt. Die Lösung wurde in einen 500 mL Rundkolben mit CaCl2-Trockenrohr
überführt und über Nacht über Natriumsulfat getrocknet. Die Lösung wurde anschließend filtriert

43
und der Ether unter Vakuum am Rotavapor abgezogen, woraufhin sich eine viskose Flüssigkeit
bildete. Zu dieser wurde Diethylether zugegeben, bis sich die Flüssigkeit gerade wieder löste.
Daraufhin wurde solange Petrolether zugetropft, woraufhin sich Schlieren bildeten, die beim
Umschütteln wieder verschwanden. Dieser Vorgang wurde so lange wiederholt, bis sich die Schlieren
beim Umschütteln nicht mehr lösten. Die so erhaltene Lösung wurde zum Auskristallisieren über
Nacht in den Kühlschrank gestellt, woraufhin sich farblose Kristalle bildeten die mit Hilfe der
Vakuumpumpe abgesaugt wurden. Das Produkt wurde mit einem eiskalten Gemisch aus
Petrolether/Diethylether im Verhältnis 1:1 nachgewaschen. Die Trocknung erfolgte im evakuierten
Exsikkator über Natriumsulfat im Kühlschrank. Die Analyse erfolgte mittels
Dünnschichtchromatographie und Schmelzpunktbestimmung.
Rf-Werte: (Laufmittel Petrolether/Ethylacetat 4:5): ABG: 0,70; GPA: 0,53
Ausbeute: 12,95 g (31,49 mmol), (82% d. Th., 90% d. Lit.)
4.3.1.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid durch
Koenigs-Knorr-Mechanismus
Acetobromoglucose (4,00 g; 9,7 mmol), Cholesterol (4,71 g; 12,18 mmol) und Silbercarbonat (3,02 g;
11,0 mmol) wurden in einen 250 mL Rundkolben eingewogen und etwa 17 g Molekularsieb 3Å
zugegeben. Dem Gemisch wurden 72 mL Diethylether zugegeben und es wurde über Nacht unter
Luft- und Feuchtigkeitsausschluss gerührt. Hierzu wurde dem Rundkolben ein mit Stickstoff gefüllter
Ballon aufgesetzt. Die Reaktion wurde dünnschichtchromatographisch verfolgt, und nachdem das
gesamte Edukt verbraucht war, wurde das entstandene Silberbromid sowie das Molekularsieb
abfiltriert und mit Diethylether nachgewaschen. Die organische Phase wurde mit gesättigter
Natriumhydrogencarbonat-Lösung und mit Wasser gewaschen, in einen 500 mL Rundkolben mit
CaCl2-Trockenrohr überführt und über Natriumsulfat über Nacht getrocknet. Anschließend wurde das
Natriumsulfat abfiltriert und das Lösungsmittel am Rotavapor unter Vakuum abgezogen. Es verblieb
ein Produktgemisch (7,23 g) aus farblosen Kristallen, welches aus Cholesterol, Cholesterylacetat
sowie dem acetylierten Cholesterylglycosid und dem Orthoester bestand.
2 g des Produktgemisches wurden auf eine Chromatographiesäule (Ø 2 cm), welche 40 cm hoch mit
Kieselgel 60 (40-63 µm) befüllt wurde, aufgetragen und getrennt. Als Laufmittel diente
Petrolether/Aceton 4:1. Die letzten erhaltenen Fraktionen entsprachen jenen, die das acetylierten
Produkt enthielten. Diese wurden anschließend vereint und das Laufmittel am Rotavapor unter
Vakuum abgezogen. Es verblieb ein farbloser Feststoff, der über Nacht im evakuierten Exsikkator
über Natriumsulfat getrocknet wurde. Die Analyse erfolgte mittels Dünnschichtchromatographie,

44
durch Derivatisierung mit Schwefelsäure und anschließendem Erwärmen, sowie durch
Schmelzpunktbestimmung.
Rf-Werte: Glycosid: 0,14, Orthoester: 0,24; Cholesterol: 0,33; Cholesterylacetat: 0,75
Ausbeute: 0,70 g (0,97 mmol), (36% d. Th., 88% d. Lit.)
Schmelzpunkt:156-157°C (Literatur: 156-158°C).
4.3.1.4 Synthese von Cholesteryl-β-D-glucopyranosid
Das acetylierte Cholesterylglycosid (0,70 g; 0,97 mmol) wurde in einen 100 mL Rundkolben, der mit
einem CaCl2-Trockenrohr versetzt wurde, eingewogen und in 36 mL THF/MeOH 1:1 gelöst. Es wurde
1 mL MeO--Lösung (10 mg Natrium in 1 mL Methanol gelöst) zugegeben und der Reaktionsansatz
gerührt, bis die DC-Kontrolle (Laufmittel Petrolether/Aceton 1:1) das Verschwinden des acetylierte
Edukts zeigte. Außerdem wurde die vollständige Deacetylierung durch die Analyse mittels MS-TOF
überprüft. Die Reaktion wurde anschließend durch Zutropfen von HCl bis zu einem pH-Wert von 7
beendet und auf 50 mL Eiswasser gegossen woraufhin sich ein farbloser Niederschlag bildete. Dieser
wurde mit Hilfe der Vakuumpumpe abgesaugt und mit Wasser gründlich nachgewaschen. Die
Analyse erfolgte mittels Dünnschichtchromatographie, Gaschromatographie, MS-TOF und
Schmelzpunktbestimmung.
Rf-Wert: (Laufmittel Petrolether/Aceton 1:1): 0,14
Ausbeute: 0,43 g (0,78 mmol), (80% d. Th., 87% d. Lit.)
Schmelzpunkt: 276-278°C (Literatur: 276-278°C)
4.3.2 Synthese durch Trichloracetimidat-Methode
4.3.2.1 Synthese von 2,3,4,6-tetra-acetyl-D-glucose
Glucosepentaacetat (10,00 g; 25,6 mmol) wurde in einen 250 mL Rundkolben, welcher mit einem
CaCl2-Trockenrohr versetzt wurde, eingewogen und in 130 mL wasserfreiem THF gelöst. Unter
Feuchtigkeitsausschluss wurden 5 mL Piperidin zugegeben und die Lösung bei Raumtemperatur
gerührt. Die Reaktion wurde dünnschichtchromatographisch verfolgt (Laufmittel Hexan/Ethylacetat
1:1). Nach vier Stunden zeigte sich bereits ein signifikanter Umsatz von Glucosepentaacetat zu
Glucosetetraacetat. Es wurden weitere 2 mL Piperidin zugegeben und die Lösung über Nacht in den
Kühlschrank gegeben. Nach dem vollständigen Verschwinden des Edukts und der Präsenz von zwei
tieferliegenden Spots am Chromatogramm, welche das α- und das β- Anomer des
Glucosetetraacetats darstellen, wurde die Lösung mit Ethylacetat verdünnt, 10%-ige HCl zugegeben.
Die wässrige Phase wurde mit dem Schütteltrichter abgetrennt und noch zweimal ausgeschüttelt. Die
organischen Phasen wurden vereint und mit gesättigter NaHCO3-Lösung und Wasser nachgewaschen.

45
Anschließend wurde die Lösung in einen 250 mL Rundkolben überführt und über Natriumsulfat
getrocknet. Nach dem das Lösungsmittel am Rotavapor unter Vakuum abgezogen wurde, verblieb
eine farblose Flüssigkeit. Die Analyse erfolgte mittels Dünnschichtchromatographie.
Rf-Werte (Laufmittel Hexan: Ethylacetat 1/1): GPA: 0,48; Anomere GTA: 0,30 und 0,24
Ausbeute: 7,84g (2,53mmol) (88% d.Th. bzw. 103% d. Lit.)
4.3.2.2 Synthese von O-(2,3,4,6-tetra-O-acetyl-D-glucopyranosyl)-trichloracetimidat
Die beiden Anomere von 2,3,4,6-Tetra-acetyl-D-glucose (6,27 g; 18,01 mmol) wurden in 67 mL
Dichlormethan gelöst und die Lösung im Eiswasserbad auf 0°C gekühlt. Dem Gemisch wurde
anschließend eine katalytische Menge DBU (140 µL; 0,94 mmol) sowie Trichloracetonitril (20 mL; 199
mmol) hinzu pipettiert, das Eiswasserbad entfernt und die Reaktion bei Raumtemperatur gerührt.
Die dünnschichtchromatographische Verfolgung der Reaktion zeigte nach einiger Zeit, neben den
Edukten, zwei höherliegende Spots, welche das α- und das β-Anomer des Trichloracetimidat-
Produktes darstellten (Laufmittel Hexan/Ethylacetat 1:1). Nach vier Stunden war kein weiterer
Umsatz von Edukt zu Produkt mehr zu beobachten, jedoch zeigte sich, dass die Intensität des α-Spots
immer mehr zunahm, während der β-Spot beinahe zur Gänze verschwand. Die Reaktion wurde
daraufhin abgebrochen. Zur Entfernung des verbliebenen Eduktes wurde aus Hexan/Ethylacetat 1:1
umkristallisiert, woraufhin nur noch die beiden Anomere des Produktes, nachweisbar waren. Das
Lösungsmittel wurde am Rotavapor unter Vakuum abgezogen. Es verblieben farblose Kristalle,
welche über Nacht im Exsikkator über Natriumsulfat getrocknet wurden. Die Analyse erfolgte mittels
Dünnschichtchromatographie sowie durch NMR-Spektroskopie.
Rf-Werte: (Laufmittel Hexan/Ethylacetat 1:1): α- und β-Anomer: 0,50 und 0,62.
Ausbeute: 7,97 g (16,23 mmol) (90% d.Th. bzw. 102 % d.Lit.)
4.3.2.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid
Trichloracetimidat (8,41 g; 17,13 mmol) und Cholesterol (4,40 g; 11,38 mmol) wurden in
wasserfreiem Dichlormethan gelöst. Weiters wurde Molekularsieb 4Å zugegeben und das Gemisch
bei Raumtemperatur 30 Minuten lang unter Feuchtigkeitsausschluss gerührt. Anschließend wurde
eine 0,02 M Lösung von TMSOTf in DCM (215 µL TMSOTf in 59 mL DCM) tropfenweise zugegeben
und die Reaktion dünnschichtchromatographisch verfolgt. Nach zehn Minuten war das gesamte
Edukt verbraucht. Am Chromatogramm waren, neben diversen Verunreinigungen, das Produkt sowie
das Cholesterylacetat ersichtlich. Die Reaktion wurde durch Zutropfen von Triethylamin bis zum
Eintreten einer Gelbfärbung beendet. Anschließend wurde das Lösungsmittel am Rotavapor

46
abgezogen, woraufhin ein zähes, braunes Gemisch erhalten wurde, welches aus dem acetylierten
Cholesterylglycosid, dem Cholesterylacetat sowie diversen Verunreinigungen bestand. Die Analyse
erfolgte mittels Dünnschichtchromatographie und MS-TOF. Eine Berechnung der Ausbeute wurde für
diesen Schritt nicht als sinnvoll erachtet, da sich zahlreiche Verunreinigungen im Produkt befanden.
Außerdem wurden keine Aufreinigungsschritte getätigt, sondern mit dem erhaltenen Gemisch
unverändert weitergearbeitet.
Rf-Werte(Laufmittel Hexan/Ethylacetat 70:30): Produkt: 0,50; Cholesterylacetat: 0,81
Ausbeute: 12,48 g
4.3.2.4 Synthese von Cholesteryl-β-D-glucopyranosid
Das acetylierte Cholesterylglucopyranosid enthaltende Produktgemisch (5 g) wurde in einen 500 mL
Rundkolben eingewogen und in einem Gemisch aus THF/MeOH 1:1 durch Rühren mit dem
Rührknochen unter Luftausschluss gelöst. Nachdem sich das Produkt vollständig gelöst hatte, wurden
7 mL MeO--Lösung, welche durch Lösen von Natrium in Methanol (68 mg Na in 7 mL MeOH)
hergestellt wurde, zugegeben und das Gemisch über Nacht gerührt. Die
dünnschichtchromatographische Kontrolle zeigte die Absenz des acetylierten Ausgangsmaterials und
einen tieferliegenden Spot, welcher das deacetylierte Cholesteryl-β-D-glucopyranosid darstellte
(Laufmittel Hexan/Ethylacetat 70:30). Die Beendigung der Reaktion erfolgte durch Zutropfen von
konzentrierter Essigsäure bis zu einem pH-Wert von 7. Anschließend wurde das Lösungsmittel am
Rotavapor unter Vakuum abgezogen und der braune, zähe Rückstand zur Aufreinigung mit
Hexan/Methanol 1:1 bis zur Lösung versetzt. Das Gemisch wurde im Eiswasserbad auf 0°C gekühlt
und mehrere Stunden am Magnetrührer gerührt, bis sich farblose Kristalle abschieden. Diese wurden
mit der Vakuumpumpe abgesaugt und mit Hexan mehrere Male gründlich nachgewaschen, bis auf
der DC-Platte nur noch das gewünschte Produkt erkennbar war. Es verblieb ein farbloser Feststoff,
welcher über Nacht im Exsikkator über Natriumsulfat getrocknet wurde. Die Analyse erfolgte mittels
Dünnschichtchromatographie, MS-TOF, Gaschromatographie, NMR-Spektroskopie sowie durch
Schmelzpunktbestimmung.
Rf: (Laufmittel: Petrolether/Aceton 1:1): 0,14
Ausbeute: 1,20 g (2,19 mmol) (38% d.Th. bzw. 138 % d.Lit.)
Schmelzpunkt: 268-270°C (Literatur: 276-278°C)

47
4.3.3 Synthese von Cholesteryl-β-D-glucopyranosid mit Saccharose als Edukt
4.3.3.1 Synthese von Octa-O-Benzylsaccharose
Saccharose, in Form von Staubzucker (10,00 g; 29,22 mmol) und Natriumhydrid (10,00 g; 41,67
mmol) wurden unter Feuchtigkeitsausschluss in einen 500 mL Rundkolben eingewogen, gut
durchmischt, in 300 mL Dimethylformamid gelöst und im Eisbad auf 0°C gekühlt. Anschließend wurde
Benzylbromid (34 mL; 286,25 mmol) hinzugegeben, der Rundkolben mit einem CaCl2-Trockenrohr
versetzt und der Reaktionsansatz drei Stunden lang bei Raumtemperatur gerührt. Zur Beendigung
der Reaktion wurden 30 mL Methanol zugegeben und das Lösungsmittel am Rotavapor unter
Vakuum bei 90°C Badtemperatur abgezogen. Das verbliebene Rohprodukt wurde in Dichlormethan
und Wasser gelöst und die organische Phase noch zweimal im Schütteltrichter mit Wasser
nachgewaschen. Die Lösung wurde über Nacht über Magnesiumsulfat getrocknet, anschließend
filtriert und der entstandene Dibenzylether und andere flüchtige Verunreinigungen am Rotavapor bei
90°C unter Vakuum abgezogen. Das Produkt war ein zähes, hellbraunes Öl.
Ausbeute: 23,50 g (22,12 mmol) (76% d. Th. bzw. 80% d. Lit.)
4.3.3.2 Synthese von 2,3,4,6-Tetra-O-benzyl-D-glucopyranose
Octa-O-Benzylsaccharose (23,5 g; 22,12 mmol) wurden in einen 500 mL Rundkolben eingewogen, in
240 mL Aceton gelöst und mit 12 mL konzentrierter HCl versetzt. Die Lösung wurde 30 Minuten bei
55°C unter Rückfluss erhitzt. Der erhaltene Ansatz wurde am Rotavapor unter Vakuum einrotiert, in
Dichlormethan wieder gelöst und im Schütteltrichter dreimal mit Wasser gewaschen. Zur Entfernung
des verbliebenen Wassers wurde über Nacht über Magnesiumsulfat getrocknet, filtriert und das
Lösungsmittel am Rotavapor abgezogen. Der verbliebene Rückstand wurde in wenig heißem
Ethylacetat gelöst und zur Fällung des benzylierten Glucosederivates mit Hexan versetzt. Die Lösung
wurde anschließend über Nacht bei -25°C gelagert, die entstandenen farblosen Kristalle mit der
Vakuumpumpe abgesaugt, in wenig heißem Methanol gelöst, filtriert und anschließend wieder
bei -25°C gelagert. Die ausgefallenen Kristalle wurden wiederum abgesaugt und im Exsikkator über
Natriumsulfat getrocknet. Die Analyse erfolgte durch Dünnschichtchromatographie,
Schmelzpunktbestimmung sowie mittels NMR-Spektroskopie.
Rf-Wert (Laufmittelgemisch Hexan/Ethylacetat 20:80): 0,85
Ausbeute: 6,12 g (11,33 mmol) (51% d.Th. bzw. 73% d. Lit.)

48
4.3.3.3 Synthese von O-(2,3,4,6-Tetra-O-benzyl-D-glucopyranosyl)trichloracetimidat
Tetrabenzylglucose (1,20 g; 2,22 mmol) wurde in 10 mL absolutem Dichlormethan gelöst und unter
Feuchtigkeitsausschluss und intensivem Rühren bei Raumtemperatur 1 mL Trichloracetonitril und
5 mg Natriumhydrid zugegeben. Die Reaktion wurde mit aufgesetztem CaCl2 –Trockenrohr 15
Minuten gerührt, bis die dünnschichtchromatographische Kontrolle das Verschwinden des Eduktes
und das Entstehen zweier neuer Spots, welche das α- und das β-Anomer darstellten, zeigte. Zur
Beendigung der Anomerisierung wurden noch 70 mg Natriumhydrid zugegeben und das
Reaktionsgemisch nach 2,5 Stunden zur Entfernung des überschüssigen Natriumhydrids über eine
mit Kieselgel 60 bedeckte Glasfritte abfiltriert. Das Filtrat wurde am Rotavapor einrotiert, in
Petrolether/Diethylether 3:2 gelöst und wieder über Kieselgel filtriert. Der Filterkuchen wurde
gründlich nachgewaschen. Nach dem Abrotieren des Lösungsmittelgemisches verblieb eine ölige
Flüssigkeit, welche zum Auskristallisieren im Kühlschrank gelagert wurden. Das Ergebnis waren
farblose Kristalle. Die Analyse erfolgte mittels Dünnschichtchromatographie sowie MS-TOF.
Rf-Werte: (Laufmittel Petrolether/Diethylether 1:1): α-Anomer: 0,71, β-Anomer: 0,50
Ausbeute: 1,29 g (1,88 mmol) (85% d. Th. bzw. 89% d. Lit.)
4.3.3.4 Synthese von Cholesteryl-(2,3,4,6-tetra-O-benzyl-β-D-glucopyranosid)
Tetrabenzyl-β-D-glucopyranosyl-trichloracetimidat (1,29 g; 1,89 mmol) und Cholesterol (0,61 g; 1,58
mmol) wurden in abs. Dichlormethan gelöst, 7,5 g Molekularsieb 4Å zugegeben und das Gemisch 15
Minuten lang unter Feuchtigkeitsausschluss gerührt. Anschließend wurde eine 0,02 M TMSOTf-
Lösung (12 µL TMSOTf in 2 mL Dichlormethan) zugegeben. Die Reaktion wurde
dünnschichtchromatographisch verfolgt und nach dem Verschwinden des Eduktes wurde die
Reaktion durch Zugabe von 0,75 mL Triethylamin beendet. Nach dem Abfiltrieren des Molekularsiebs
und dem Abziehen des Lösungsmittels am Rotavapor verblieben 1,90 g eines hellbraunen Öles.
Dieses wurde mit einem Gemisch aus Methanol/Dioxan 10:1 versetzt und auf der Magnetrührplatte
gerührt. Nach einiger Zeit fiel ein farbloser Feststoff aus. Dieser wurde mit der Vakuumpumpe
abgesaugt und anschließend über Natriumsulfat im Exsikkator getrocknet. Die Analyse erfolgte
mittels Dünnschichtchromatographie sowie MS-TOF.
Rf-Wert: (Laufmittel: Hexan/Ethylacetat 70:30): 0,79
Die Ausbeute betrug 0,89 g (0,98 mmol), (52% d. Th.)

49
4.3.3.5 Synthese von Cholesteryl-β-D-glucopyranosid
Die benzylierte Cholesterylglucopyranose (0,30 g; 0,33 mmol) wurde in einen Zweihalskolben
eingewogen und mit einem Gemisch aus 15 mL Ethanol/Cyclohexen 2:1 versetzt. Der Rundkolben
wurde mit einem Rückflusskühler mit Trockenrohr, sowie einem Verbindungsstück für Thermometer
verbunden. Durch das Verbindungsstück wurde mit Hilfe einer Glaspipette etwa 20 Minuten lang
Stickstoff durch die Lösung geleitet, bevor diese mit einer katalytischen Menge Pd(OH)2 (20% auf
Kohlenstoff) versetzt wurde. Das Reaktionsgemisch wurde unter Stickstoffeinleitung unter ständigem
Rühren mit einem Rührknochen unter Rückfluss erhitzt und von Zeit zu Zeit wurde wieder ein
Gemisch aus Ethanol/Cyclohexen zugegeben, um den Lösungsmittelverlust durch Verdampfung
auszugleichen. Die Reaktion wurde solange fortgesetzt, bis die dünnschichtchromatographische
Kontrolle die Abspaltung der Benzylgruppen zeigte. Das Produkt war ein farbloser Feststoff. Die
Analyse erfolgte mittels Dünnschichtchromatographie, TOF-MS sowie Gaschromatographie.
Rf-Wert: (Laufmittel: Petrolether/Aceton 1:1): 0,14
Ausbeute betrug 0,18 g (0,33 mmol) (99% d. Th.).
4.4 Versuch der enzymatischen Herstellung von Cholesteryl(6´-O-
palmitoyl)-β-D-glucopyranosid
Cholesteryl-β-D-glucopyranosid (100,0 mg; 182 µmol) und Palmitinsäure (1,00 g; 3,899 mmol)
wurden in 69 mL Hexan gelöst und Molekularsieb (3Å, 5,15 g) hinzugegeben. Anschließend wurde die
Lipase Novozyme 435 (0,86 g) hinzugegeben und die Mischung bei 45°C im Trockenschrank am
Laborschüttler bei 320 rpm umgesetzt. Nach 48 Stunden wurde die Reaktion abgebrochen, die
Mischung in einen 500 mL Rundkolben überführt und das Lösungsmittel am Rotavapor abgezogen.
Anschließend wurde das Lösungsmittel abdekantiert, das Gemisch aus Enzym, Molekularsieb und
den verbliebenen Kristallen mit Dichlormethan/Methanol versetzt, und gaschromatographisch als
auch mittels TOF-MS analysiert. Die abdekantierte Lösung wurde mit 1%-iger NaOH solange mit Hilfe
eines Schütteltrichters ausgeschüttelt, bis sich kein Natriumpalmitat mehr bildete und ebenfalls
analysiert.
Ein weiterer Versuch mit n-Hexan als Lösungsmittel wurde durchgeführt, wobei dieses zuvor mit
Molekularsieb getrocknet wurde. Cholesteryl-β-D-glucopyranosid (50 µmol, 27,4 mg) wurde in einer
kleinen Menge an Chloroform gelöst. Palmitinsäure (1 mmol, 260 mg) wurde in n-Hexan gelöst, das
gelöste Cholesterylglycosid sowie Molekularsieb (3Å, 3 g) zugegeben und auf 45°C temperiert.
Anschließend wurde die Lipase Novozyme 435 (260 mg) zugegeben und sofort durch Umschwenken
in Bewegung gebracht. Die Reaktion wurde 24 Stunden bei 45°C und 240 rpm am Laborschüttler

50
durchgeführt. Anschließend wurden das Enzym sowie das Molekularsieb über Glaswolle abfiltriert,
mit Chloroform/Methanol 2:1 nachgewaschen und dann das Lösungsmittel abrotiert. Zum Rückstand
wurden 100 mL Chloroform gegeben und dieser wurde so lange mit 1%-iger NaOH ausgeschüttelt, bis
sich kein Natriumpalmitat mehr bildete. Die organische Phase wurde einrotiert und
gaschromatographisch und mit TOF-MS vermessen.
Weiters wurde die Synthese in einem Lösungsmittelgemisch von THF/Pyridin 4:1 ausgeführt. THF
wurde zuvor destilliert und über Natriumdraht gelagert. Das fertige Lösungsmittelgemisch wurde
noch über Molekularsieb getrocknet. Cholesteryl-β-D-glucopyranosid (50 µmol, 27,4 mg) und
Palmitinsäure (300 µmol, 78 mg) wurden in THF/Pyridin gelöst und Molekularsieb (3Å, 1 g)
zugegeben. Anschließend wurde auf 45°C temperiert, die Lipase Novozyme 435 (25 mg) zugegeben
und sofort durch Umschwenken in Bewegung gebracht. Die Reaktion wurde 48 Stunden bei 45°C und
240 rpm am Laborschüttler durchgeführt. Anschließend wurden das Enzym sowie das Molekularsieb
über Glaswolle abfiltriert, mit Chloroform/Methanol 2:1 nachgewaschen und dann das Lösungsmittel
abrotiert. Das erhaltene Gemisch wurde im Anschluss gaschromatographisch und mittels TOF-MS
analysiert.
4.5 Probenvorbereitung für die qualitative und quantitative Analyse von
Pflanzenölen und Biodiesel auf Sterylglycoside
Die Analyse der Biodieselprobe erfolgte in Anlehnung auf die Vorschrift von Pieber et al. (2010) [99].
Raps- und Sojaöl sowie Rapsbiodiesel wurden anhand der in Tabelle 3 angeführten Parameter für die
Messung am GC-MS vorbereitet. Zur eingewogenen Probe wurden 200 µL MSTFA zugegeben. Zur
Biodieselprobe wurde außerdem noch ein Cholesterolstandard (IS)mit einer Konzentration von
10,22 mg/kg hinzugefügt. Anschließend wurden die Proben 60 Minuten bei 60°C gelagert, mit Pyridin
aufgefüllt und anschließend gaschromatographisch analysiert.
Die Biodieselprobe wurde vor der Messung außerdem noch 1:50 verdünnt.
Tabelle 3: Probenvorbereitung für die Analyse am GC-MS
Probenmenge [mg] MSTFA [µL] Pyridin [g] IS [µl]
Rapsöl 46,4 200 1,20 - Sojaöl 38,9 200 1,14 -
Rapsbiodiesel 80,1 200 1,00 50
4.6 Analytik
In diesem Kapitel werden die im Zuge der Arbeit verwendeten Analysemethoden hinsichtlich der
Probenvorbereitung en als auch der Messparameter beschrieben.

51
4.6.1 GC-MS
Die zu messenden Proben wurden vor der Analyse am GC-MS mit MSTFA silyliert. Hierzu wurde ein
etwa 1 mg der Probe in ein GC-Vial gegeben, in 150 µL Pyridin gelöst, 200 µL MSTFA zugegeben und
verschlossen. Daraufhin wurde die Lösung am Vortex durchmischt und anschließend bei 60°C im
Trockenschrank gelagert. Nach 60 Minuten wurde das Vial mit Pyridin aufgefüllt und vermessen. In
Tabelle 4 sind die Parameter für die Messungen angeführt. Bei den Messungen mittels SIM-Methode
wurden die Fragmente 147 m/z, 204 m/z und 217 m/z zur Charakterisierung der Zuckerkomponente
verwendet.
Tabelle 4: GC-MS Parameter für die Messung von Cholesterylglycosid
Säule DB-5HT
Starttemperatur 100°C Hold time 2 min Gradient 10°C/min
Endtemperatur 370°C Hold time 15 min Trägergas He Flussrate 1 mL/min
4.6.2 GC-FID
Die Probenanalyse am GC-FID erfolgte analog zur Analyse nach Lacoste et al. (2009) [2].
Die Probenvorbereitung für die Messung erfolgte analog zur Probenvorbereitung für die GC-
Massenspektroskopie. In Tabelle 5 sind die Parameter für die Messung angeführt.
Tabelle 5: GC-FID Parameter für die Messung von Cholesterylglycosid
Säule DB-5HT
Starttemperatur 100°C Gradient 10°C/min
Endtemperatur 370°C Hold time 18 min Trägergas H2
Flussrate 1,2 mL/min
4.6.3 TOF-MS
Ein kleiner Teil der zu messenden Probe wurde in ein GC-Vial gegeben und in Acetonitril gelöst. In
Acetonitril nicht lösliche Proben wurden zuvor in einem geeigneten Lösungsmittel gelöst, ein Tropfen
davon entnommen und in Acetonitril gelöst. Anschließend wurde die Probe mittels Vortex
durchmischt und am TOF-MS vermessen.

52
Tabelle 6: TOF-MS Parameter für die Messung von Cholesterylglycosid
APCI Positiv/ Negative* mode
Gas Temp. (N2) 325°C
Vaporizer 350°C
Drying Gas 8 L/min
Nebulizer 40 psig
Fragmentor 175 V
Skimmer 65 V
OCT 1 RF Vpp 750 V
Vcap 3500 V
Corona* 4 µA/25* µA
Capallary 0.078 µA
Corona 176 V
Chamber 5.33 µA
Injection 0.5-2 µL
Solvent 30% H2O, 70%( 90% ACN 10%H2O (0,1% Amminoumformiat))isocratic
Flow 0,3 mL/min
Column Temp. 30°C
4.6.4 NMR-Spektroskopie
Die zu messenden Proben (20 mg) wurden in ein NMR-Röhrchen eingewogen und mit deuteriertem
Lösungsmittel, Chloroform beziehungsweise Pyridin, aufgefüllt und bis zur vollständigen Lösung des
Produktes stehengelassen. Anschließend wurde die Probe am NMR-Spektrometer vermessen.
Tabelle 7: NMR-Parameter für durchgeführte Messungen
1H-NMR 13C-NMR DEPT
Prozessierung % Auswertung Topspin® 2.1
Lösungsmittel CDCl3 bzw. C5D5
5 Ergebnisse und Diskussion
5.1 Synthese durch Koenigs-Knorr Methode
Die Darstellung mittels Koenigs-Knorr Methode wurde in Anlehnung an die von Pieber et al. (2010)
[99] beschriebene Synthesevorschrift durchgeführt, wobei bei dieser als Edukt Glucosepentaacetat
verwendet wurde. In der hier durchgeführten Synthese wurde dieses hingegen zuvor aus Glucose
hergestellt. Des Weiteren wurde zur Herstellung von Acetobromoglucose eine alternative Methode
gewählt.

53
5.1.1 Synthese von β-D-Glucose-Pentaacetat [98]
Die Synthese von Glucosepentaacetat beruht auf der Veresterung von alkoholischen Gruppen durch
ein Carbonsäureanhydrid. Die alkoholischen Gruppen sind in diesem Fall von der Glucose, das
Carbonsäureanhydrid wird durch Essigsäureanhydrid dargestellt. Durch die Wahl von Natriumacetat
als Katalysator beziehungsweise durch die kinetische Kontrolle der Reaktion wird ausschließlich das
β-Anomer gebildet.
Abbildung 22: Darstellung von β-D-Glucose-Pentaacetat mit Natriumacetat als Katalysator
Die erhaltene Ausbeute betrug 60% der Theorie beziehungsweise 97% der Literatur. Das Produkt
stellt einen farblosen Feststoff dar. Der Schmelzbereich der Substanz betrug 130-132°C, welcher sich
exakt mit den Literaturangaben deckt [98]. Bei dünnschichtchromatographischer Analyse zeigt das
Produkt, bei einem Laufmittelgemisch aus Toluol/Aceton 3:1, einen Rf-Wert von 0,57.
Rf-Wert: (Laufmittel Toluol/Aceton 3:1) 0,57
Schmelzpunkt: 130-132°C
Ausbeute: 32,75 g (83,90 mmol), (60% d. Th., 97% d. Lit.)
5.1.2 Synthese von Acetobromoglucose
Die Synthese von Acetobromoglucose nach Ravindranathan-Kartha et al. (1990) [100] beruht auf
dem Mechanismus einer nucleophilen Substitution am gesättigten Kohlenstoffatom. Die anomere
Acetylgruppe wird durch den anorganischen Säurerest der Bromwasserstoffsäure ersetzt.
Abbildung 23: Umsetzung von Glucosepentaacetat mit Bromwasserstoffsäure zur Herstellung von Acetobromoglucose

54
Die erhaltene Ausbeute betrug 82% der Theorie. Das Produkt war ein farbloser Feststoff mit einem
Schmelzpunkt von 88°C, was sich mit den gefundenen Literaturschmelzpunkten deckt. Bei
dünnschichtchromatographischer Analyse zeigt das Produkt, bei einem Laufmittelgemisch aus
Petrolether/Ethylacetat 4:5, einen Rf-Wert von 0,53.
Rf-Werte: (Laufmittel Petrolether/Ethylacetat 4:5): ABG: 0,70; GPA: 0,53
Ausbeute: 12,95 g (31,49 mmol), (82% d. Th., 90% d. Lit.)
5.1.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid durch
Koenigs-Knorr-Mechanismus
Bei dieser Reaktion handelt es sich um eine Substitutionsreaktion eines Glycosylhalogenids durch
einen Alkohol. In diesem Fall werden diese beiden Komponenten durch Acetobromoglucose und
Cholesterol dargestellt.
Abbildung 24: Darstellung des tetraacetylierten Glucopyranosids mittels Koenigs-Knorr Methode
Die Synthese wurde nach der Methodik von Kunz et al. [101] durchgeführt. Die Problematik dieser
Methode liegt unter anderem in der Orthoesterbildung, welche eine Aufreinigung durch
anschließende Säulenchromatographie unerlässlich macht. Neben dem zeitlichen Aufwand, den die
Säulung beansprucht, ist auch ein Produktverlust durch den Verbleib des Produktes in der Säule zu
verzeichnen. Außerdem führt die Bildung von Cholesterylacetat zu weiteren Produktverlusten und

55
erschwert, durch die Präsenz mehrerer Nebenprodukte, die säulenchromatographische Aufreinigung.
Neben dem im experimentellen Teil beschriebenen Laufmittelgemisch Petrolether/Aceton 4:1 wurde
auch ein Versuch mit einem 12:1 Verhältnis, wie dies in der Vorschrift von Pieber et al. (2010) [99]
beschrieben wurde, durchgeführt. Dieser war jedoch, aufgrund der geringen Löslichkeit des
Gemisches, nicht erfolgreich. Diese Faktoren führen in Summe zu geringen Ausbeuten bei hohem
zeitlichen Aufwand. Ein weiterer Faktor der bei dieser Methode zu berücksichtigen ist, ist die Menge
an Silbercarbonat, welche in etwa in äquimolarem Verhältnis zu Acetobromoglucose und Cholesterol
eingesetzt werden muss, und deshalb einen nicht zu vernachlässigenden Kostenfaktor darstellt. Die
geringe Löslichkeit des Produktes erschwert außerdem die säulenchromatographische Trennung. Für
die Herstellung größerer Mengen ist diese Methode somit nicht geeignet.
Vor der säulenchromatographischen Trennung wurde ein Produktgemisch aus dem acetylierten
Cholesterylglycosid, dem Orthoester, Cholesterolacetat sowie freiem Cholesterol in Form eines
farblosen Feststoffes erhalten. Nach der Trennung mittels Säule verblieb ein farbloser Feststoff. Die
erhaltene Ausbeute betrug 0,70 g. Dies entspricht 36% der Theorie beziehungsweise 88% der
Literatur. Der Schmelzpunkt betrug 156-157°C (Literatur: 156-158°C). Bei
dünnschichtchromatographischer Analyse zeigt das Produkt, bei einem Laufmittelgemisch aus
Laufmittel Petrolether/Aceton 4:1, einen Rf-Wert von 0,14.
Rf-Werte: (Laufmittel Petrolether/Aceton 4:1): Acetyliertes Cholesterylglycosid: 0,14; Orthoester:
0,24; Cholesterol: 0,33; Cholesterylacetat: 0,75
Ausbeute: 0,70 g (0,97 mmol), (36% d. Th., 88% d. Lit.)
5.1.4 Synthese von Cholesteryl-β-D-glucopyranosid
Die Darstellung des Cholesterylglucopyranosids basiert auf der Abspaltung der Acetylgruppen nach
Zemplén [102]. Konkret handelt es sich um eine basenkatalysierte Verseifung, bei der die
Acetylgruppen des Glucopyranosids durch Hydroxylgruppen substituiert werden.
Abbildung 25: Darstellung von Cholesteryl-β-D-glucopyranosid durch Zemplén-Verseifung
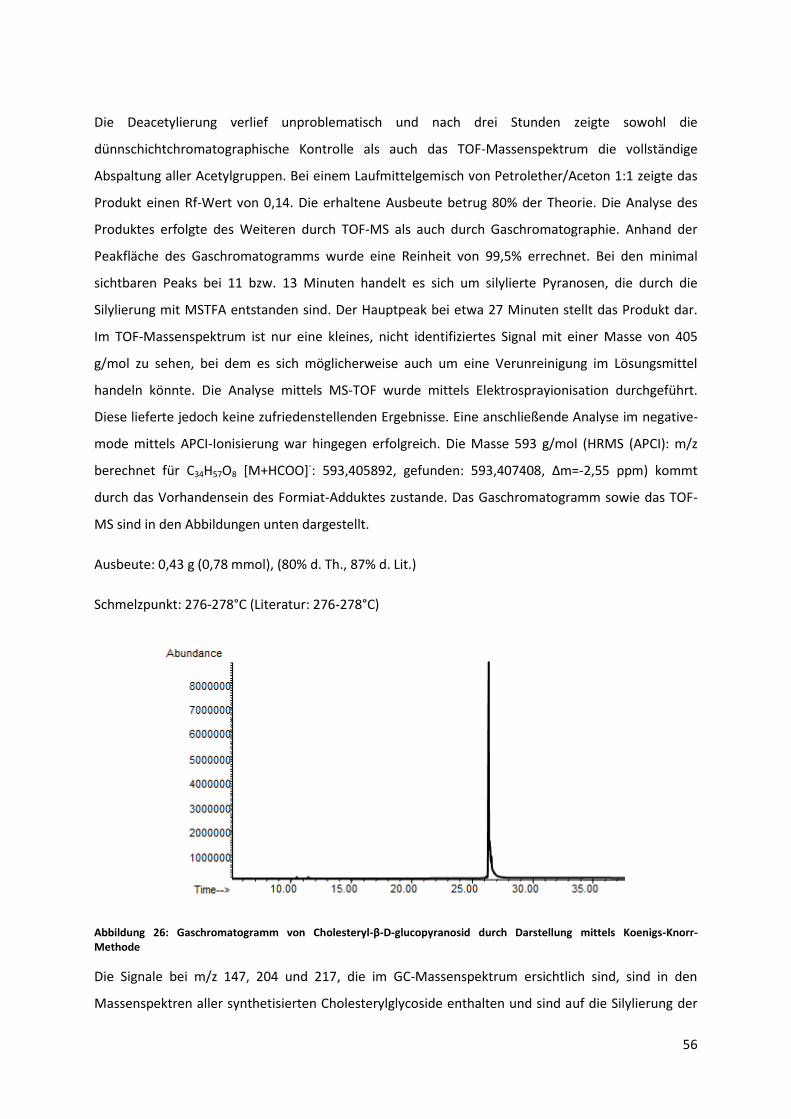
56
Die Deacetylierung verlief unproblematisch und nach drei Stunden zeigte sowohl die
dünnschichtchromatographische Kontrolle als auch das TOF-Massenspektrum die vollständige
Abspaltung aller Acetylgruppen. Bei einem Laufmittelgemisch von Petrolether/Aceton 1:1 zeigte das
Produkt einen Rf-Wert von 0,14. Die erhaltene Ausbeute betrug 80% der Theorie. Die Analyse des
Produktes erfolgte des Weiteren durch TOF-MS als auch durch Gaschromatographie. Anhand der
Peakfläche des Gaschromatogramms wurde eine Reinheit von 99,5% errechnet. Bei den minimal
sichtbaren Peaks bei 11 bzw. 13 Minuten handelt es sich um silylierte Pyranosen, die durch die
Silylierung mit MSTFA entstanden sind. Der Hauptpeak bei etwa 27 Minuten stellt das Produkt dar.
Im TOF-Massenspektrum ist nur eine kleines, nicht identifiziertes Signal mit einer Masse von 405
g/mol zu sehen, bei dem es sich möglicherweise auch um eine Verunreinigung im Lösungsmittel
handeln könnte. Die Analyse mittels MS-TOF wurde mittels Elektrosprayionisation durchgeführt.
Diese lieferte jedoch keine zufriedenstellenden Ergebnisse. Eine anschließende Analyse im negative-
mode mittels APCI-Ionisierung war hingegen erfolgreich. Die Masse 593 g/mol (HRMS (APCI): m/z
berechnet für C34H57O8 [M+HCOO]-: 593,405892, gefunden: 593,407408, Δm=-2,55 ppm) kommt
durch das Vorhandensein des Formiat-Adduktes zustande. Das Gaschromatogramm sowie das TOF-
MS sind in den Abbildungen unten dargestellt.
Ausbeute: 0,43 g (0,78 mmol), (80% d. Th., 87% d. Lit.)
Schmelzpunkt: 276-278°C (Literatur: 276-278°C)
Abbildung 26: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mittels Koenigs-Knorr-Methode
Die Signale bei m/z 147, 204 und 217, die im GC-Massenspektrum ersichtlich sind, sind in den
Massenspektren aller synthetisierten Cholesterylglycoside enthalten und sind auf die Silylierung der
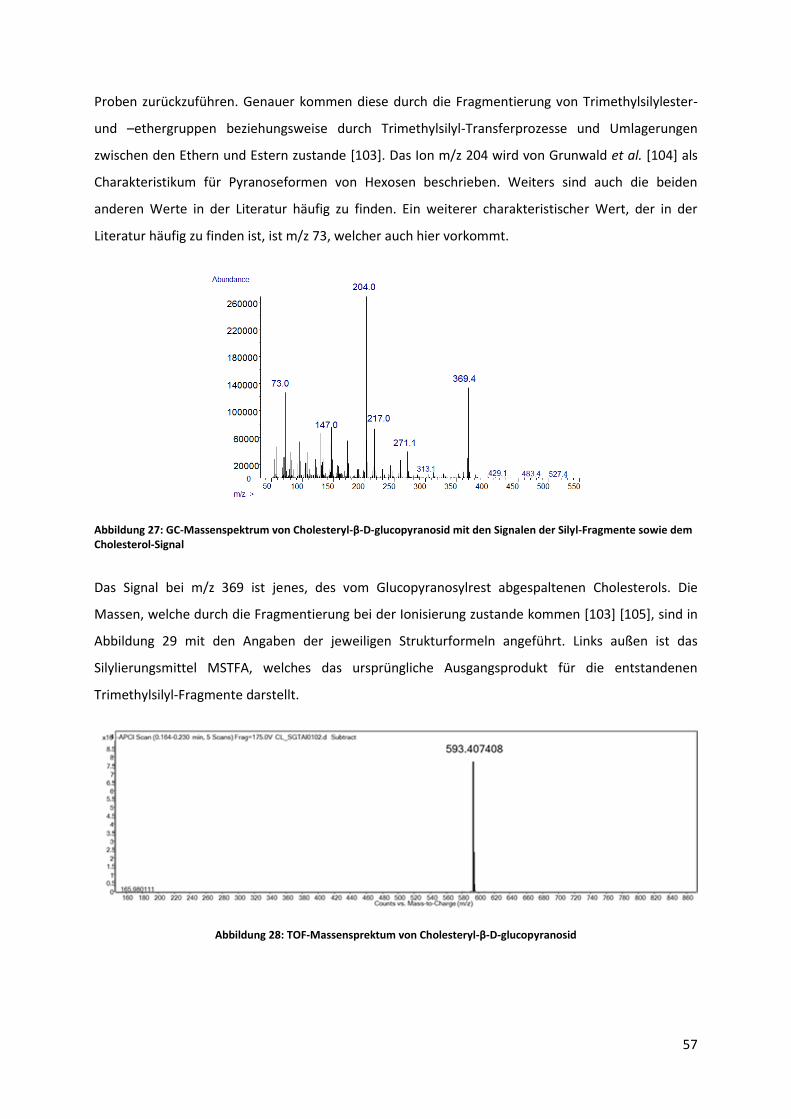
57
Proben zurückzuführen. Genauer kommen diese durch die Fragmentierung von Trimethylsilylester-
und –ethergruppen beziehungsweise durch Trimethylsilyl-Transferprozesse und Umlagerungen
zwischen den Ethern und Estern zustande [103]. Das Ion m/z 204 wird von Grunwald et al. [104] als
Charakteristikum für Pyranoseformen von Hexosen beschrieben. Weiters sind auch die beiden
anderen Werte in der Literatur häufig zu finden. Ein weiterer charakteristischer Wert, der in der
Literatur häufig zu finden ist, ist m/z 73, welcher auch hier vorkommt.
Abbildung 27: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid mit den Signalen der Silyl-Fragmente sowie dem Cholesterol-Signal
Das Signal bei m/z 369 ist jenes, des vom Glucopyranosylrest abgespaltenen Cholesterols. Die
Massen, welche durch die Fragmentierung bei der Ionisierung zustande kommen [103] [105], sind in
Abbildung 29 mit den Angaben der jeweiligen Strukturformeln angeführt. Links außen ist das
Silylierungsmittel MSTFA, welches das ursprüngliche Ausgangsprodukt für die entstandenen
Trimethylsilyl-Fragmente darstellt.
Abbildung 28: TOF-Massensprektum von Cholesteryl-β-D-glucopyranosid

58
Abbildung 29: Entstehende Fragmente bei der Ionisierung im GC-MS
5.2 Synthese durch Trichloracetimidat-Methode
5.2.1 Synthese von 2,3,4,6-tetra-acetyl-D-glucose
Bei dieser Reaktion handelt es sich um einen basenkatalysierten Additions-
Eliminierungsmechanismus. Formal handelt es sich um eine Substitutionsreaktion, bei der die
Acetylgruppen abgespalten werden.
Abbildung 30: Basenkatalysierte Darstellung von Tetraacetylglucose aus Glucosepentaacetat
Bei dieser Reaktion war darauf zu achten, dass sie unter absolut wasserfreien Bedingungen ablief,
weswegen das Lösungsmittel zuvor über KOH unter Rückfluss gekocht, anschließend destilliert und
über Natriumdraht gelagert wurde. Die Ausbeute der Reaktion lieferte bei der Darstellung analog zur
internen Vorschrift [106] keine zufriedenstellenden Ergebnisse. Erst nach signifikanter Erhöhung der
Piperidinmenge konnte ein vollständiger Umsatz der Reaktion beobachtet werden. Das Produkt war
nach dem Eindampfen eine farblose, ölige Flüssigkeit. Die erhaltene Ausbeute betrug 88% der

59
Theorie. Die dünnschichtchromatographische Analyse zeigte die beiden Anomere bei einem
Laufmittelgemisch Hexan/Ethylacetat 1:1 mit Rf-Werten von 0,30 bzw. 0,24.
Rf-Werte (Laufmittel Hexan: Ethylacetat 1:1): GPA: 0,48; Anomere GTA: 0,30 und 0,24
Ausbeute: 7,84g (2,53mmol) (88% d.Th. bzw. 103% d. Lit.)
5.2.2 Synthese von O-(2,3,4,6-tetra-O-acetyl-D-glucopyranosyl)-trichloracetimidat
Die Darstellung Glucopyranosyl-Trichloracetimidates beruht auf dem Prinzip der nucleophilen
Addition eines Alkoholatanions an ein Nitril.
Abbildung 31: Basenkatalysierte Darstellung von O-(2,3,4,6-tetra-O-acetyl-D-glucopyranosyl)-trichloracetimidat aus Tetraacetylglucose
Bei dieser Reaktion war wieder darauf zu achten, dass sie unter möglichst wasserfreien Bedingungen
ablief, weswegen Dichlormethan zuvor über P2O5 unter Rückfluss gekocht, und über Molekularsieb
gelagert wurde. Nach vier Stunden konnte durch die dünnschichtchromatographische Kontrolle kein
weiterer Umsatz von Edukt zu Produkt mehr festgestellt werden, weshalb die Reaktion abgebrochen
wurde. Im Gegensatz zu der, in der internen Vorschrift vorgeschlagenen säulenchromatographischen
Auftrennung, wurde eine Umkristallisation mit Hexan/Ethylacetat vorgezogen, wodurch das Edukt
beziehungsweise Verunreinigungen zur Gänze entfernt werden konnten. Das Anomerengemisch,
welches hauptsächlich aus dem Anomer mit dem Rf-Wert 0,62 bestand, war nach dem Abrotieren
ein farbloser Feststoff mit teilweise etwas bräunlicher Färbung. Dies lässt sich dadurch erklären, dass
hauptsächlich das α-Anomer, welches in Reinform einen farblosen Feststoff [106] darstellt, im
erhaltenen Produkt vorhanden war. Das β-Anomer hingegen ist in Reinform ein braunes Öl [106]. Der
Überschuss an α-Trichloracetimidat beruht darauf, dass durch die schnelle Addition zuerst das β-
Anomer gebildet wird, dieses jedoch in der langsameren basenkatalysierten Reaktion beinahe
Vollständig zum α-Anomer anomerisiert [107]. Die erhaltene Ausbeute betrug 90% der Theorie. Die
dünnschichtchromatographische Analyse zeigte die beiden Anomere bei einem Laufmittelgemisch
Hexan/Ethylacetat 1:1 bei 0,50 und 0,62. Weiters bestätigte die NMR-spektroskopische Analyse den
Erfolg der Methode, wodurch die Säulung vermieden werden kann.
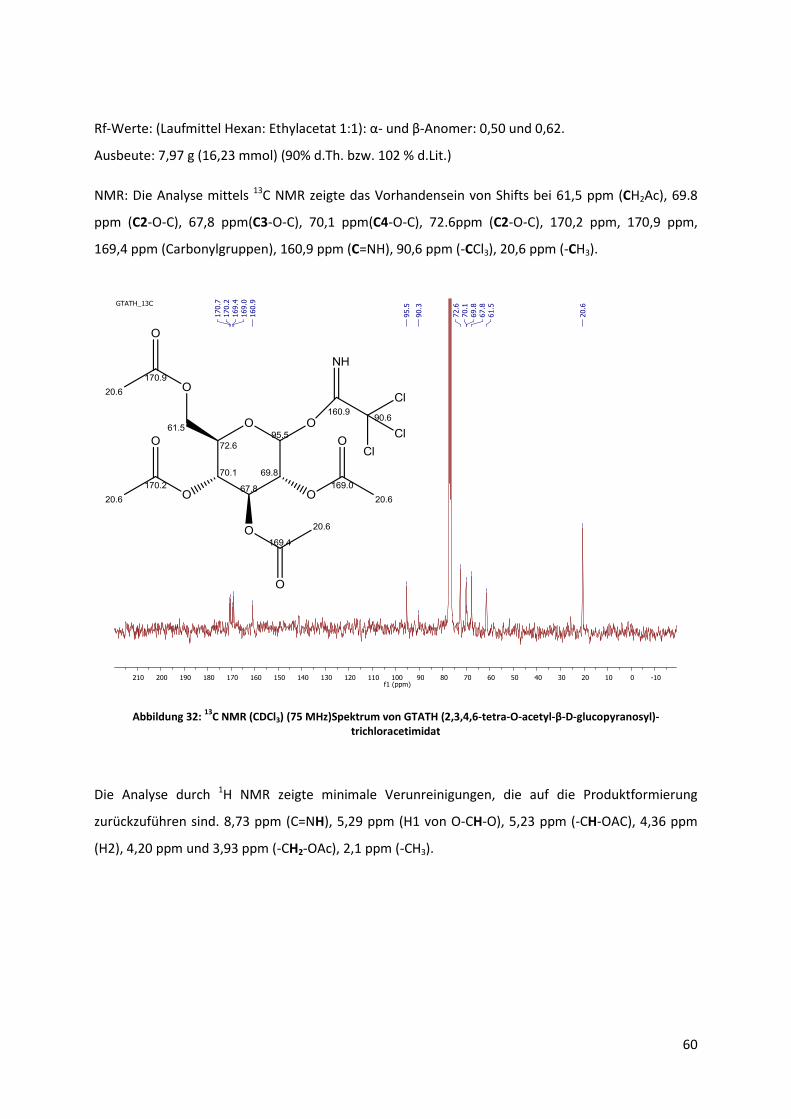
60
Rf-Werte: (Laufmittel Hexan: Ethylacetat 1:1): α- und β-Anomer: 0,50 und 0,62.
Ausbeute: 7,97 g (16,23 mmol) (90% d.Th. bzw. 102 % d.Lit.)
NMR: Die Analyse mittels 13C NMR zeigte das Vorhandensein von Shifts bei 61,5 ppm (CH2Ac), 69.8
ppm (C2-O-C), 67,8 ppm(C3-O-C), 70,1 ppm(C4-O-C), 72.6ppm (C2-O-C), 170,2 ppm, 170,9 ppm,
169,4 ppm (Carbonylgruppen), 160,9 ppm (C=NH), 90,6 ppm (-CCl3), 20,6 ppm (-CH3).
Abbildung 32: 13
C NMR (CDCl3) (75 MHz)Spektrum von GTATH (2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-trichloracetimidat
Die Analyse durch 1H NMR zeigte minimale Verunreinigungen, die auf die Produktformierung
zurückzuführen sind. 8,73 ppm (C=NH), 5,29 ppm (H1 von O-CH-O), 5,23 ppm (-CH-OAC), 4,36 ppm
(H2), 4,20 ppm und 3,93 ppm (-CH2-OAc), 2,1 ppm (-CH3).
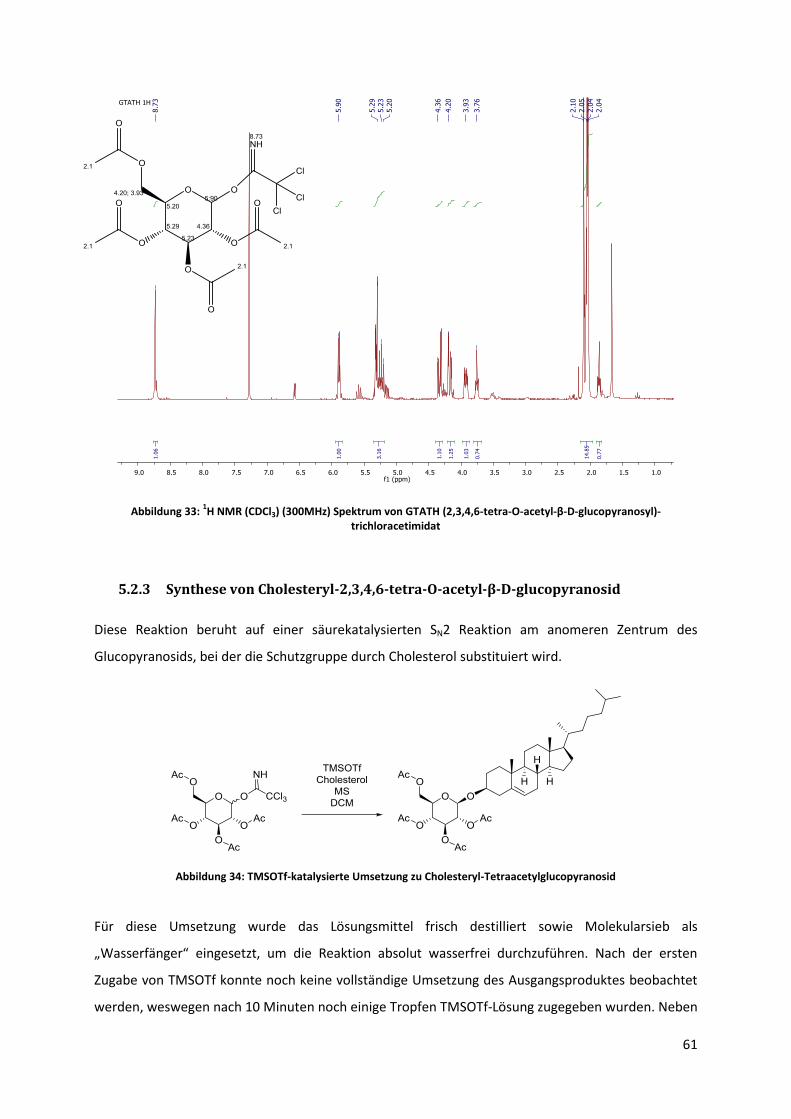
61
Abbildung 33: 1H NMR (CDCl3) (300MHz) Spektrum von GTATH (2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-
trichloracetimidat
5.2.3 Synthese von Cholesteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranosid
Diese Reaktion beruht auf einer säurekatalysierten SN2 Reaktion am anomeren Zentrum des
Glucopyranosids, bei der die Schutzgruppe durch Cholesterol substituiert wird.
Abbildung 34: TMSOTf-katalysierte Umsetzung zu Cholesteryl-Tetraacetylglucopyranosid
Für diese Umsetzung wurde das Lösungsmittel frisch destilliert sowie Molekularsieb als
„Wasserfänger“ eingesetzt, um die Reaktion absolut wasserfrei durchzuführen. Nach der ersten
Zugabe von TMSOTf konnte noch keine vollständige Umsetzung des Ausgangsproduktes beobachtet
werden, weswegen nach 10 Minuten noch einige Tropfen TMSOTf-Lösung zugegeben wurden. Neben

62
dem gewünschten Produkt bildeten sich noch Cholesterylacetat sowie einige nicht identifizierte
Nebenprodukte. Nach Beendigung der Reaktion wurde ein zähes braunes Gemisch erhalten, die
Ausbeute betrug 12,58 g. Die dünnschichtchromatographische Analyse zeigte bei einem
Laufmittelgemisch von Hexan/Ethylacetat 70:30 das Produkt mit einem Rf-Wert von 0,5, das
Cholesterylacetat bei 0,81 sowie diverse Verunreinigungen. Weiters war nach der Messung am TOF-
MS das Produkt im Massenspektrum zu sehen. Eine Berechnung der Ausbeute wurde für diesen
Schritt nicht als sinnvoll erachtet, da sich zahlreiche Verunreinigungen im Produkt befanden.
Außerdem wurden keine Aufreinigungsschritte getätigt, sondern mit dem erhaltenen Gemisch
unverändert weitergearbeitet.
Rf-Werte( Laufmittel Hexan/Ethylacetat 70:30): Produkt: 0,5; Cholesterylacetat: 0,81
5.2.4 Synthese von Cholesteryl-β-D-glucopyranosid
Bei dieser Darstellung wurde, wie auch schon bei der Synthese desselben Produktes im Zuge der
Koenigs-Knorr Methode, wieder die Zemplén-Reaktion angewendet. Die Acetylgruppen wurden
basenkatalysiert durch Hydroxylgruppen ersetzt.
Abbildung 35: Darstellung von Cholesterylglucopyranosid durch Zemplén-Verseifung
Die Reaktion zeigte bei dünnschichtchromatographischer Kontrolle nach etwa drei Stunden bereits
das gewünschte Produkt. Die Messung mittels TOF-MS zeigte jedoch, dass nach drei Stunden, wie
dies in der internen Vorschrift [106] beschrieben wird, in dem Reaktionsgemisch noch ein nicht
unwesentlicher Teil mit einer verbleibenden Acetylgruppe befand. Da dieses Zwischenprodukt
offenbar einen recht ähnlichen Rf-Wert wie das Produkt aufweist, war eine
dünnschichtchromatographische Unterscheidung nicht möglich. Zur Entfernung der letzten
Acetylgruppe wurde noch einige Stunden gerührt, wobei sich jedoch keine Veränderung mehr zeigte.
Erst nach Zugabe von weiterer MeO--Lösung und dem Rühren über Nacht konnte die Abspaltung der
verbleibenden Acetylgruppe beobachtet werden. Da die Methode bei der Darstellung mittels
Koenigs-Knorr Methode gut funktioniert hat, (vgl. Kapitel 4.3.1) ist die längere Reaktionsdauer auf

63
das unreine Ausgangsprodukt zurückzuführen. Außerdem wurde weniger Lösungsmittel als auch
weniger Natriummethanolat eingesetzt, als dies für dieselbe Menge an reinem Edukt der Fall
gewesen wäre, da ansonsten im Liter Maßstab gearbeitet hätte werden müssen. Durch die weitere
Zugabe von MeO--Lösung und durch das längere Rühren konnte jedoch schlussendlich die Abspaltung
aller Acetylgruppen erreicht werden. Bei der Aufreinigung wurde, nicht wie in der internen Vorschrift
empfohlen, eine säulenchromatographische Trennung durchgeführt, sondern das Produkt mit Hexan
und Methanol versetzt, auf Eis gestellt und mehrere Stunden gerührt, woraufhin sich das Produkt an
der Phasengrenze als farbloser Feststoff absetzte und so abgesaugt werden konnte. Eine
Schwierigkeit trat bei der Isolierung des gesamten Produktes auf, da sich in der Methanolphase das
Produkt teilweise löste und auch durch mehrmaliges Wiederholen des Rührens auf Eis nicht zur
Gänze isoliert werden konnte, was auch den Verlust an Ausbeute zum Teil erklärt. Durch
Nachwaschen mit Hexan, sowie durch Erhitzen in Hexan, gleichzeitigem Rühren und anschließendem
Absaugen konnten die durch Dünnschichtchromatographie nachweisbaren Verunreinigungen
komplett entfernt werden. Die Charakterisierung des Produktes erfolgte mittels TOF-MS, NMR-
Spektroskopie, GC, DC sowie durch Ermittlung des Schmelzpunktes. Am Dünnschichtchromatogramm
zeigte das Produkt bei einem Laufmittelgemisch Petrolether/Aceton 1:1 einen Rf-Wert von 0,14. Die
Ausbeute betrug 1,20 g, was bezogen auf die eingewogene Trichloracetimidat-Menge einer Ausbeute
von 38 % der Theorie bzw. 138% d. Literatur entspricht. Der Schmelzbereich betrug 276-278°C und
deckt sich mit dem Literaturwert.
Im Gaschromatogramm sind neben den Hauptpeak bei 27 Minuten wieder zwei kleinere Peaks bei
Minute 10 bzw. 11 zu erkennen. Bei diesen handelt es sich, wie schon bei der Synthese mittels
Koenigs-Knorr-Methode, um silylierte Glucopyranosen, die durch Silylierung mittels MSTFA
entstanden sind. Der Hauptpeak bei etwa 27 Minuten stellt das Produkt dar. Des Weiteren ist vor
den Hauptpeak ein kleinerer Peak erkennbar, der jedoch beinahe dieselbe Retentionszeit sowie exakt
dasselbe GC-Massenspektrum wie Cholesteryl-β-D-glucopyranosid aufweist, weshalb davon
auszugehen ist, dass hier zum Teil das α-Anomer entstanden ist. Dies ist auf eine nicht vollständige
Anomerisierung des β-Anomers zum α-Anomer, welches für die Synthese des β-
Cholesterylglucopyranosids von Nöten ist, bei der Herstellung des Trichloracetimidates
zurückzuführen. Dies stellt auch den Nachteil der Trichloracetimidatmethode dar, da eine 100%-ige
Stereoselektivität nicht erreicht werden kann. Bei der Berechnung der Peakfläche ergibt sich ein
Cholesterylglucopyranosidanteil von 98%, wobei zu 85% das β-Anomer, und zu 13% das α-Anomer
vorliegt. Die Verunreinigung durch silylierten Glucopyranosen beträgt etwa 2%. Das GC-
Massenspektrum zeigt, wie auch jenes der Darstellung durch Koenigs-Knorr Methode Signale bei m/z
73, 147, 204, 217 sowie 369. Die Erklärungen der Signale sind in Kapitel 5.1.4 nachzulesen, das
Massenspektrum befindet sich im Anhang. Die Analyse mittels TOF-MS wurde im negative-Mode
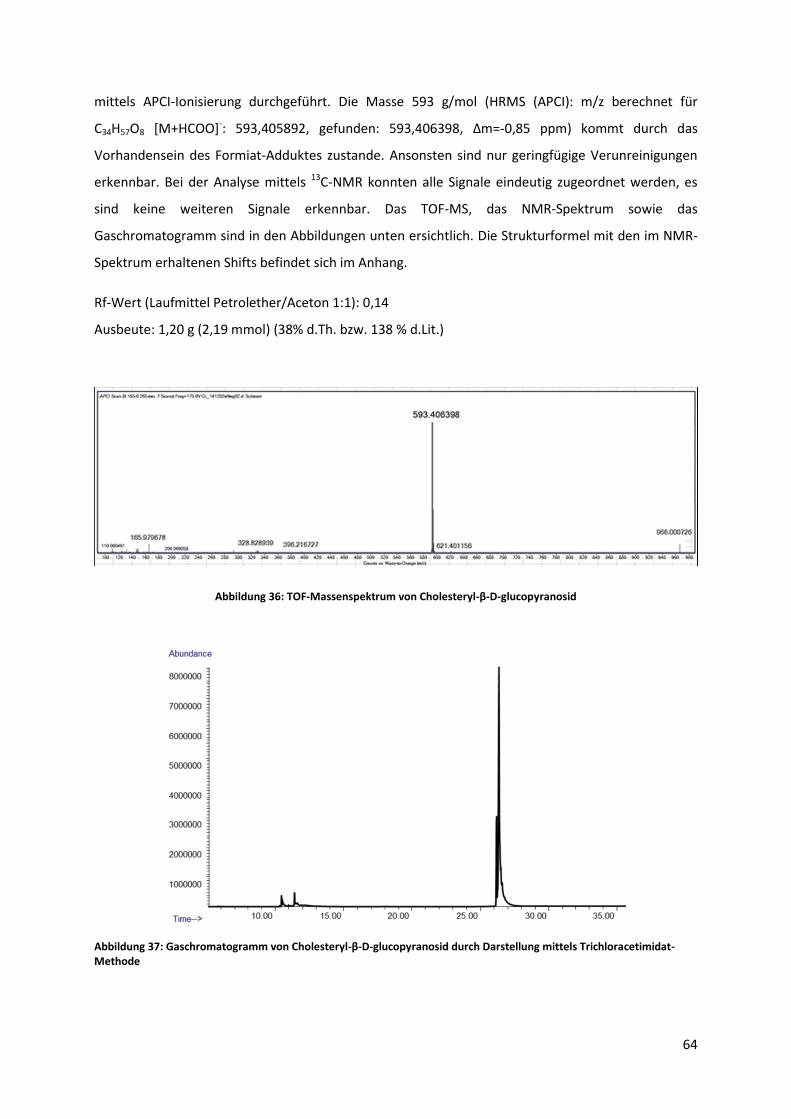
64
mittels APCI-Ionisierung durchgeführt. Die Masse 593 g/mol (HRMS (APCI): m/z berechnet für
C34H57O8 [M+HCOO]-: 593,405892, gefunden: 593,406398, Δm=-0,85 ppm) kommt durch das
Vorhandensein des Formiat-Adduktes zustande. Ansonsten sind nur geringfügige Verunreinigungen
erkennbar. Bei der Analyse mittels 13C-NMR konnten alle Signale eindeutig zugeordnet werden, es
sind keine weiteren Signale erkennbar. Das TOF-MS, das NMR-Spektrum sowie das
Gaschromatogramm sind in den Abbildungen unten ersichtlich. Die Strukturformel mit den im NMR-
Spektrum erhaltenen Shifts befindet sich im Anhang.
Rf-Wert (Laufmittel Petrolether/Aceton 1:1): 0,14
Ausbeute: 1,20 g (2,19 mmol) (38% d.Th. bzw. 138 % d.Lit.)
Abbildung 36: TOF-Massenspektrum von Cholesteryl-β-D-glucopyranosid
Abbildung 37: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mittels Trichloracetimidat-Methode
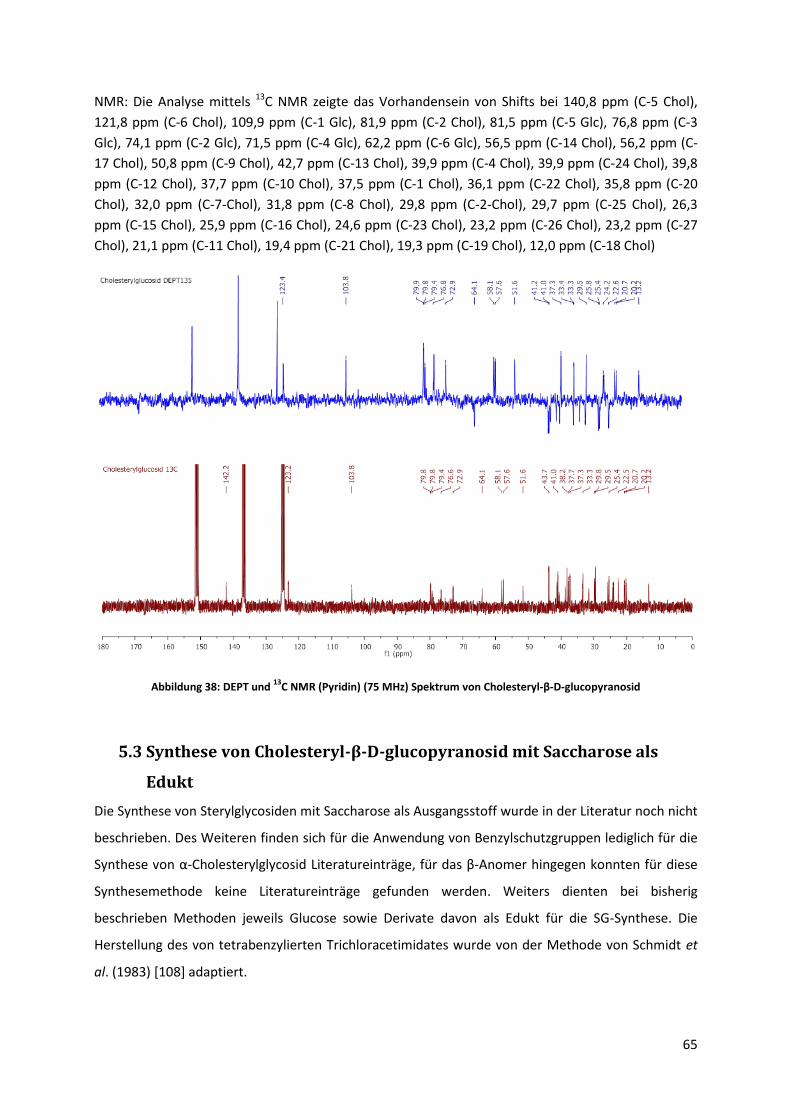
65
NMR: Die Analyse mittels 13C NMR zeigte das Vorhandensein von Shifts bei 140,8 ppm (C-5 Chol),
121,8 ppm (C-6 Chol), 109,9 ppm (C-1 Glc), 81,9 ppm (C-2 Chol), 81,5 ppm (C-5 Glc), 76,8 ppm (C-3
Glc), 74,1 ppm (C-2 Glc), 71,5 ppm (C-4 Glc), 62,2 ppm (C-6 Glc), 56,5 ppm (C-14 Chol), 56,2 ppm (C-
17 Chol), 50,8 ppm (C-9 Chol), 42,7 ppm (C-13 Chol), 39,9 ppm (C-4 Chol), 39,9 ppm (C-24 Chol), 39,8
ppm (C-12 Chol), 37,7 ppm (C-10 Chol), 37,5 ppm (C-1 Chol), 36,1 ppm (C-22 Chol), 35,8 ppm (C-20
Chol), 32,0 ppm (C-7-Chol), 31,8 ppm (C-8 Chol), 29,8 ppm (C-2-Chol), 29,7 ppm (C-25 Chol), 26,3
ppm (C-15 Chol), 25,9 ppm (C-16 Chol), 24,6 ppm (C-23 Chol), 23,2 ppm (C-26 Chol), 23,2 ppm (C-27
Chol), 21,1 ppm (C-11 Chol), 19,4 ppm (C-21 Chol), 19,3 ppm (C-19 Chol), 12,0 ppm (C-18 Chol)
Abbildung 38: DEPT und 13
C NMR (Pyridin) (75 MHz) Spektrum von Cholesteryl-β-D-glucopyranosid
5.3 Synthese von Cholesteryl-β-D-glucopyranosid mit Saccharose als
Edukt
Die Synthese von Sterylglycosiden mit Saccharose als Ausgangsstoff wurde in der Literatur noch nicht
beschrieben. Des Weiteren finden sich für die Anwendung von Benzylschutzgruppen lediglich für die
Synthese von α-Cholesterylglycosid Literatureinträge, für das β-Anomer hingegen konnten für diese
Synthesemethode keine Literatureinträge gefunden werden. Weiters dienten bei bisherig
beschrieben Methoden jeweils Glucose sowie Derivate davon als Edukt für die SG-Synthese. Die
Herstellung des von tetrabenzylierten Trichloracetimidates wurde von der Methode von Schmidt et
al. (1983) [108] adaptiert.

66
5.3.1 Synthese von Octa-O-Benzylsaccharose
Bei der Einführung der Schutzgruppen an das Saccharose-Molekül wurde die Methodik der
Williamson-Ethersynthese [109] gewählt. Bei dieser Methode handelt es sich um einen zweistufigen
Prozess, bei dem durch Natriumhydrid zuerst die alkoholischen Gruppen der Saccharose in
Alkoholate umgewandelt wurden. Anschließend wurden diese mit Benzylbromid umgesetzt und so
im Zuge einer SN2 Reaktion mit Benzylgruppen verethert.
Abbildung 39: Darstellung von Octa-O-Benzylsaccharose durch Williamson-Ethersynthese
Der Vorteil dieses Verfahrens nach Kessler et al. [110] mit Saccharose als Edukt liegt unter anderem
darin, dass Saccharose, in diesem Fall in Form von Staubzucker, einen recht kostengünstigen und
nachwachwachsenden Ausgangsstoff darstellt. Weitaus bedeutender als der Kostenfaktor ist jedoch,
dass die anomere Hydroxylgruppe nicht zusätzlich geschützt werden muss, da diese Aufgabe durch
den angehängten Fructofuranosylrest erfüllt wird. Die Benzylierung wurde zuerst unter Luft- und
Feuchtigkeitsausschluss mit einem aufgesetzten Stickstoff-Ballon durchgeführt, wobei die
gewünschte Reaktion jedoch nicht ablief und sich das gewünschte Produkt nicht bildete. Bei einem
zweiten Versuch hingegen wurde die Umsetzung nur mit einem aufgesetzten CaCl2-Trockenrohr
durchgeführt, bei der die Reaktion mit der unten angeführten Ausbeute ablief. Ein weiteres Problem
wurde bei der Reaktivität des Natriumhydrids festgestellt. Ein Syntheseversuch erfolgte mit NaH,
welches schon länger gelagert wurde, wobei jedoch keine Umsetzung beobachtet werden konnte.
Die Reaktivität des Natriumhydrids wurde daraufhin dadurch getestet, dass eine minimale Menge an
NaH mit Wasser in Kontakt gebracht wurde, wobei es im Normalfall zu einer stark exothermen
Reaktion kommt. Bei diesem Versuch war dies nicht der Fall, weshalb das Misslingen eindeutig auf
das NaH zurückzuführen war. Das Abziehen des Lösungsmittels unter Vakuum bei 90°C
Badtemperatur beruht auf der Tatsache, dass bei der Benzylierungsreaktion Dibenzylether als
Nebenprodukt entsteht, welcher laut Literatur [111] einen Siedepunkt von 158-160°C aufweist.
Dieser konnte durch das entstandene Vakuum (90 mbar) auf 80°C erniedrigt, und so zur Gänze
abdestilliert werden. Nach der Aufarbeitung verblieben 23,50 g einer hellbraunen, öligen Flüssigkeit,
was einer Ausbeute von 76 % entspricht. Die Abweichung von der Literaturausbeute lässt sich durch

67
Verluste bei der Aufreinigung erklären, wobei der größte Teil vermutlich bei der Filtration über
Aktivkohle verloren ging
Die dünnschichtchromatographische Kontrolle zeigte keinerlei Verunreinigungen und lieferte bei
einem Laufmittelgemisch Benzol/Diethylether 6:1 einen Rf-Wert von 0,83.
Ausbeute: 23,50 g (22,12 mmol) (76% d. Th. bzw. 80% d. Lit.)
5.3.2 Synthese von 2,3,4,6-Tetra-O-benzyl-D-glucopyranose
Die Darstellung der tetrabenzylierten Glucose beruht auf der säurekatalysierten hydrolytischen
Spaltung der Octa-O-benzylsaccharose.
Abbildung 40: Hydrolytische Spaltung von Octabenzylsaccharose in das Glucose- und das Fructosederivat
Die Entschützung der anomeren OH-Gruppe des Glucosederivat wurde nach dem Verfahren von
Kessler et al. [110] durchgeführt und verlief recht unproblematisch. Bei der Fällung des Produkts war
darauf zu achten, dass Hexan in großem Überschuss zuzugeben wurde. Bei der Zugabe von zu wenig
Hexan konnte nur die Fällung eines kleinen Teils der benzylierte Glucose beobachtet werden. Der
Vorgang der Fällung und des Auskristallisierens wurde deshalb mehrere Male durchgeführt, um eine
Maximierung der Ausbeute zu erhalten. Das Fructosederivat verblieb, aufgrund des weniger polaren
Charakters, in Lösung, wodurch das gewünschte Produkt ohne das störende Nebenprodukt
abgesaugt werden konnte. Dünnschichtchromatographisch konnte das Fructosederivat in der
Mutterlauge nachgewiesen werden. Die Charakterisierung des Produktes erfolgte mittels NMR, MS-
TOF, DC sowie durch Ermittlung des Schmelzpunktes. Am Dünnschichtchromatogramm zeigte das
Produkt bei einem Laufmittelgemisch Hexan/Ethylacetat 20:80 einen Rf-Wert von 0,85. Die Ausbeute
betrug 6,12g, was einer Ausbeute von 51% entspricht. Die, verglichen mit dem Literaturwert
geringere Ausbeute lässt sich unter anderem darauf zurückführen, dass nicht das gesamte Produkt
zur Auskristallisation gebracht werden konnte, und so zum Teil in der Mutterlauge verblieb. Weiters
traten Probleme beim Absaugen der Kristalle auf. Das heißt, dass bei jeder Filtration ein Teil der recht
feinen Kristalle durch das Filterpapier gezogen wurde, und so nicht das gesamte Produkt isoliert
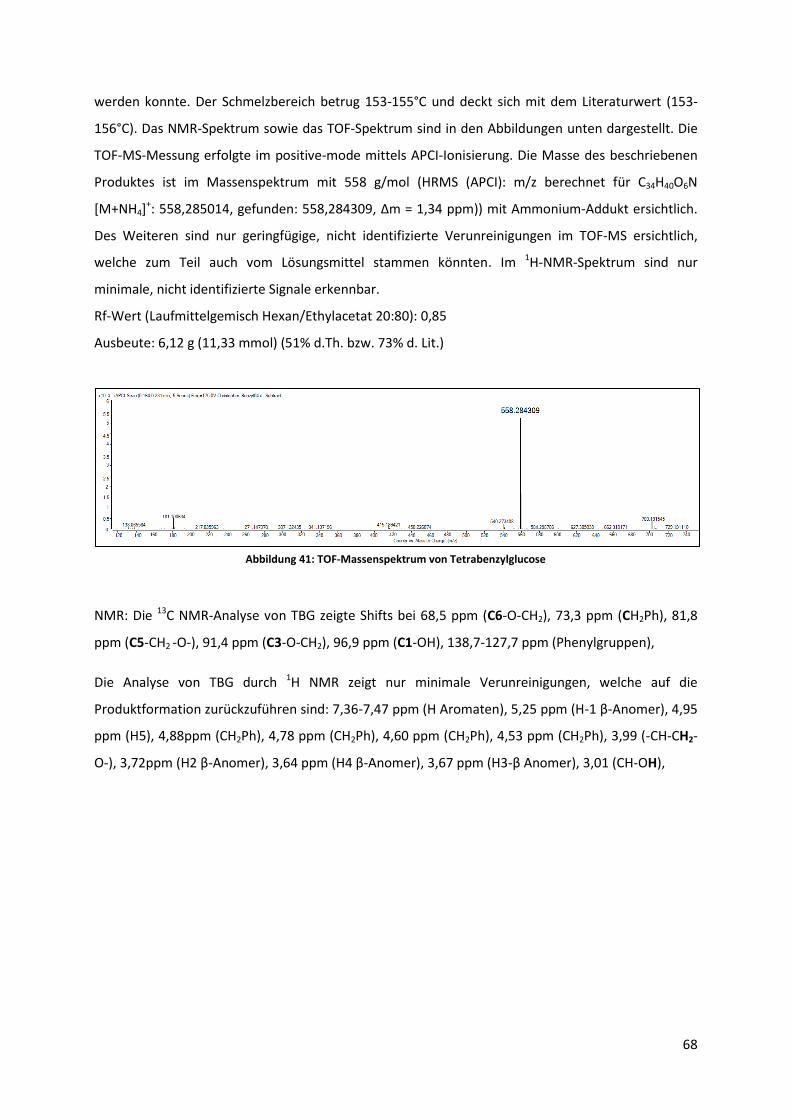
68
werden konnte. Der Schmelzbereich betrug 153-155°C und deckt sich mit dem Literaturwert (153-
156°C). Das NMR-Spektrum sowie das TOF-Spektrum sind in den Abbildungen unten dargestellt. Die
TOF-MS-Messung erfolgte im positive-mode mittels APCI-Ionisierung. Die Masse des beschriebenen
Produktes ist im Massenspektrum mit 558 g/mol (HRMS (APCI): m/z berechnet für C34H40O6N
[M+NH4]+: 558,285014, gefunden: 558,284309, Δm = 1,34 ppm)) mit Ammonium-Addukt ersichtlich.
Des Weiteren sind nur geringfügige, nicht identifizierte Verunreinigungen im TOF-MS ersichtlich,
welche zum Teil auch vom Lösungsmittel stammen könnten. Im 1H-NMR-Spektrum sind nur
minimale, nicht identifizierte Signale erkennbar.
Rf-Wert (Laufmittelgemisch Hexan/Ethylacetat 20:80): 0,85
Ausbeute: 6,12 g (11,33 mmol) (51% d.Th. bzw. 73% d. Lit.)
Abbildung 41: TOF-Massenspektrum von Tetrabenzylglucose
NMR: Die 13C NMR-Analyse von TBG zeigte Shifts bei 68,5 ppm (C6-O-CH2), 73,3 ppm (CH2Ph), 81,8
ppm (C5-CH2 -O-), 91,4 ppm (C3-O-CH2), 96,9 ppm (C1-OH), 138,7-127,7 ppm (Phenylgruppen),
Die Analyse von TBG durch 1H NMR zeigt nur minimale Verunreinigungen, welche auf die
Produktformation zurückzuführen sind: 7,36-7,47 ppm (H Aromaten), 5,25 ppm (H-1 β-Anomer), 4,95
ppm (H5), 4,88ppm (CH2Ph), 4,78 ppm (CH2Ph), 4,60 ppm (CH2Ph), 4,53 ppm (CH2Ph), 3,99 (-CH-CH2-
O-), 3,72ppm (H2 β-Anomer), 3,64 ppm (H4 β-Anomer), 3,67 ppm (H3-β Anomer), 3,01 (CH-OH),
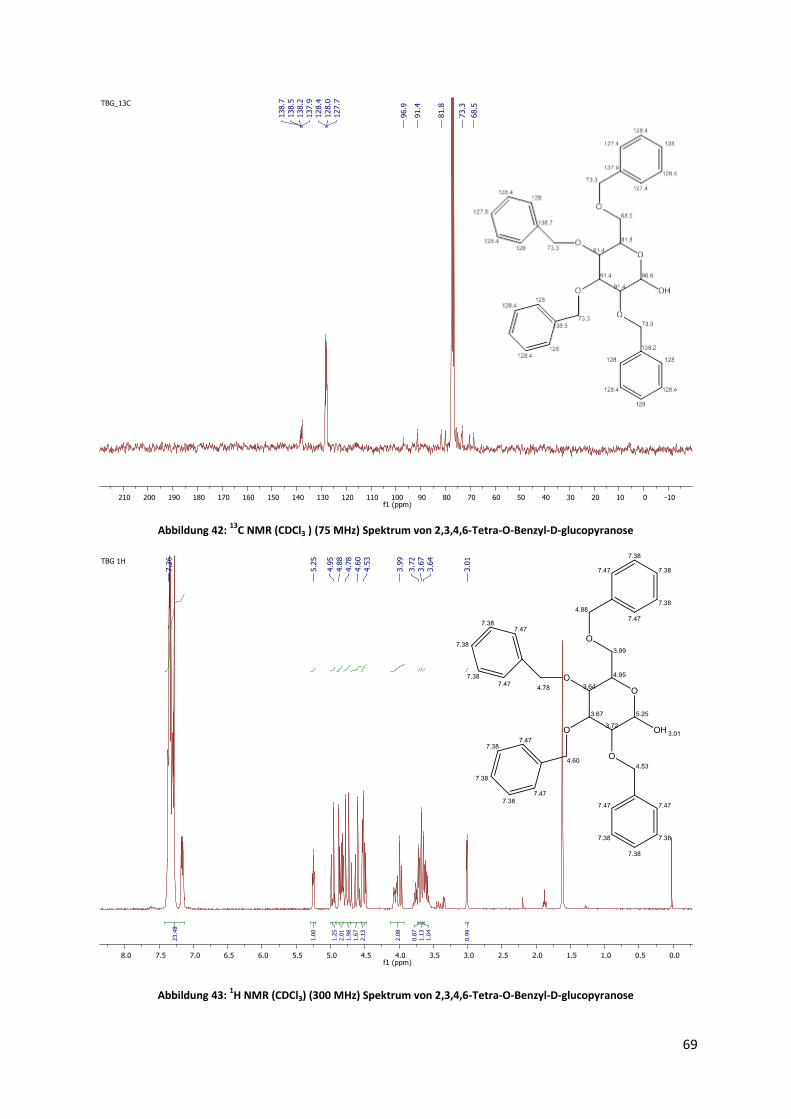
69
Abbildung 42: 13
C NMR (CDCl3 ) (75 MHz) Spektrum von 2,3,4,6-Tetra-O-Benzyl-D-glucopyranose
Abbildung 43: 1H NMR (CDCl3) (300 MHz) Spektrum von 2,3,4,6-Tetra-O-Benzyl-D-glucopyranose

70
5.3.3 Synthese von O-(2,3,4,6-Tetra-O-benzyl-D-glucopyranosyl)trichloracetimidat
Die Synthese des benzylierten Glucopyranosyl-Trichloracetimidates basiert auf der nucleophilen
Addition eines Alkoholanions an ein elektronenarmes Nitril.
Abbildung 44: Basenkatalysierte Darstellung von Tetrabenzyl-Glucopyranosyltrichloracetimidat aus Tetrabenzylglucose
Die Darstellung der Trichloracetimidates erfolgte nach einem Verfahren, welches von Schmidt et al.
(1983) [108] beschrieben wurde. Zur Synthese des nachfolgenden β-Glycosids musste zuerst das α-
Anomer des Trichloracetimidates hergestellt werden, um einen nucleophilen Angriff des Cholesterols
am anomeren Zentrum der geschützten Glucopyranose zu ermöglichen. Dies wurde durch die
Trichloracetimidatmethode bewerkstelligt. Die dünnschichtchromatographische Kontrolle zeigte kurz
nach dem Start der Umsetzung des Eduktes mit NaH und Trichloracetonitril bereits zwei neue Spots,
welche das α- und das β-Anomer darstellten, wobei der Spot für das β-Anomer bei weitem stärker
ausgeprägt vorlag. Durch Zugabe von weiterem NaH wurde die basenkatalysierte Anomerisierung
von β-Imidat zu α-Imidat erreicht. Nach längerem Stehenlassen konnte beobachtet werden, dass die
Intensität des β-Anomeren-Spots immer mehr abnahm und der des α-Anomers, durch die
Stabilisation des anomeren Effekts, zunahm. (vgl. Kapitel 5.2.2). Bei der Filtration über Kieselgel zur
Entfernung von überschüssigem NaH musste darauf geachtet werden, nicht zu viel Kieselgel zu
verwenden, um einen übermäßigen Produktverlust vorzubeugen. Deswegen wurde hier auch die
Methode gewählt, die Filtration mittels Saugflasche und unter Vakuum in einem modifizierten Dry-
Flash-Verfahren durchzuführen. Außerdem wurde gründlich mit Lösungsmittel nachgespült. Nach der
weiteren Aufreinigung wurde ein farbloses Öl erhalten welches laut Vorschrift nach wenigen Stunden
durchkristallisieren sollte. Die Kristallisation wurde jedoch erst nach dem Stehenlassen für mehrere
Tage beobachtet. Das Produkt zeigte sich daraufhin als farblose, kristalline Substanz. Die Ausbeute
betrug 1,29 g bzw. 85% der Theorie. Produktverluste sind durch Filtrationsprozesse zu erklären, bei
dem sich ein Teil des Produkts nicht mehr aus der Kieselgel-Matrix löste. Die
dünnschichtchromatographische Analyse zeigte bei einem Laufmittelgemisch von Petrolether/Ether
1:1 das α -Anomer mit einem Rf-Wert von 0,71, das β-Anomer mit einem minimalen Spot mit einem
Rf-Wert von 0,5. Weiters wurde ein Schmelzbereich von 75-76°C ermittelt, welcher nur gering vom
Literaturwert (77°C) abweicht.

71
Ausbeute: 1,29 g (1,88 mmol) (85% d. Th. bzw. 89% d. Lit.)
5.3.4 Synthese von Cholesteryl-(2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosid)
Bei dieser Reaktion wird die Schutzgruppe am anomeren Zentrum des Glycopyranosids in einer
säurekatalysierten SN2 Reaktion durch Cholesterol substituiert.
Abbildung 45: Darstellung des benzylierten Cholesterylglycosids durch nucleophile Substitution
Bei dieser Umsetzung war auf absolute Wasserfreiheit zu achten, weshalb Dichlormethan zuvor
destilliert wurde, außerdem wurde Molekularsieb eingesetzt. Bereits 15 Minuten nach der TMSOTf
Zugabe war der Umsatz der Ausgangssubstanz zum gewünschten Produkt vollendet, welches nach
Beendigung der Reaktion und dem Abrotieren als braunes Öl vorlag. Die Auskristallisierung im
Kühlschrank brachte auch nach mehreren Tagen nicht den gewünschten Erfolg. Erst nach der Zugabe
eines Gemisches aus Methanol/Dioxan 10:1 und anschließendem Rühren konnte das Ausfallen eines
farblosen Feststoffes beobachtet werden. Nach den weiteren Aufreinigungsschritten verblieben 0,89
g eines farblosen Feststoffes, was 52% der Theorie entspricht. Das Produkt wurde mittels
Dünnschichtchromatographie als auch TOF-MS analysiert. Bei einem Laufmittelgemisch von
Hexan/Ethylacetat 70:30 ergab sich Rf-Wert von 0,79. Weiters war ein minimaler Spot bei 0,70 zu
erkennen, welcher mittels TOF-MS nicht detektiert werden konnte und vermutlich das α-Anomer
darstellte. Das TOF-Massenspektrum ist in Abbildung 46 ersichtlich. Da bei der
dünnschichtchromatographischen Kontrolle kein Cholesterol detektiert werden konnte, ist der
markierte Cholesterol-Peak, der im Spektrum dargestellt ist, auf die Fragmentierung bei der
Ionisation zurückzuführen. Die TOF-MS-Messung erfolgte im positive-Mode mittels APCI-Ionisierung.
Die Masse des beschriebenen Produktes ist im Massenspektrum mit 926 g/mol (HRMS (APCI): m/z
berechnet für C61H84O6N [M+NH4]+: 926,629316, gefunden: 926,629023, Δm = -0,32 ppm)) mit
Ammonium-Addukt ersichtlich. Da die Reaktion mit dieser Methodik für die Herstellung des
tetrabenzylierten β-Cholesterylglycosids in der Literatur noch nicht beschrieben wurde, liegen auch
keine Referenzwerte hinsichtlich der Ausbeute vor.
72
Die Ausbeute betrug 0,89 g (0,98 mmol), (52% d. Th.)
Abbildung 46: TOF-Massenspektrum des tetrabenzylierten Cholesterylglycosids
5.3.5 Synthese von Cholesteryl-β-D-glucopyranosid
Die Darstellung des Cholesterylglucopyranosids beruht auf der hydrogenolytischen Abspaltung der
Benzylgruppen, wobei Cyclohexen als Wasserstoffdonor, und Palladiumhydroxid auf Aktivkohle als
Katalysator dienen [112].
Abbildung 47: Benzylgruppenabspaltung durch katalytische Hydrierung
Bei der Abspaltung der Benzylgruppen ergab sich das Problem, dass bei einer Hydrierung mittels
Pd/C und Wasserstoff nicht nur die Benzylgruppen, sondern auch die Doppelbindung am C5-Atom
hydriert worden wäre. Somit wäre als Produkt Cholestanol-β-D-glucopyranosid entstanden. Deshalb
wurde die relativ milde Hydrierungsmethode mit Palladiumhydroxid als Katalysator und Cyclohexen
als Wasserstoffdonor angewendet. Die Abspaltung wurde von Duivenvoorden (2011) [33] lediglich
für das α-Anomer des Cholesterylglycosids beschrieben, für das β-Anomer wurde in der Literatur
keine Beschreibung gefunden. Nachdem die Vorschrift zur Benzylgruppenabspaltung von jener der
für das α-Anomer des benzylierten Cholesterylglycosids adaptiert wurde, kam es zu der Problematik,
dass sich das Edukt, im Gegensatz zum α-Anomer, welches ein farbloses Öl darstellt, im
Lösungsmittel nicht beziehungsweise nur schlecht löste. Die dünnschichtchromatographische
Verfolgung der Reaktion zeigte nach fünfstündigem Kochen unter Rückfluss bei etwa 80°C

73
Badtemperatur keine Veränderungen, woraufhin das Volumen des Lösungsmittels verdoppelt, die
Reaktionstemperatur auf 150°C gesteigert sowie die Menge an Katalysator erhöht wurde. Die
Verfolgung der Reaktion mittels TOF-MS ließ sich die Reaktion besser verfolgen als durch
Dünnschichtchromatographie. Am Dünnschichtchromatogramm waren nach vier Stunden insgesamt
acht Spots erkennbar, wobei immer zwei davon, ein stärkerer und ein schwächerer, ein Duplett
bildeten (vgl. Abbildung 48).Bei den Dupletts handelte es sich jeweils um das Cholesterylglycosid mit
einer verschiedenen Anzahl an Benzylgruppen, wobei die beiden oberen Spots mit Rf-Werten von
0,72 bzw. 0,70 das Edukt darstellten, und die darunterliegenden Spots jeweils nach Abspaltung einer
gewissen Anzahl an Benzylgruppen. Durch die Analyse mittels TOF-MS, welche für die vorhandenen
Spots jeweils nur eine Masse generierte, wurden die schwächeren, unteren Spots als die jeweiligen
α-Anomere der teilbenzylierten Cholesterylglucopyranosen identifiziert. Die erste Messung mittels
TOF-MS erfolgte 12 Stunden nach Reaktionsbeginn. Im Massenspektrum war die teilweise
Abspaltung der Benzylgruppen erkennbar. Nach weiteren 48 Stunden war das
vollständig benzylierte Edukt beinahe verschwunden, woraufhin noch eine
Spatelspitze des Katalysators zugegeben wurde. Nach vier Tagen war die Reaktion
beendet. Die anschließende dünnschichtchromatographische Kontrolle als auch
das TOF-Massenspektrum zeigten keinerlei Nebenprodukte. Am DC zeigte das
Produkt bei einem Laufmittelgemisch Petrolether/Aceton 1:1 einen Rf-Wert von
0,14, die Verfolgung mittels TOF-MS ist in Abbildung 50 ersichtlich. Die Analyse
mittels MS-TOF wurde im negative-Mode mittels APCI-Ionisierung durchgeführt.
Die Masse 593 g/mol (HRMS (APCI): m/z berechnet für C34H57O8 [M+HCOO]-:
593,405892, gefunden: 593,405705, Δm = 0,32 ppm) kommt durch das
Vorhandensein des Formiat-Adduktes zustande. Weiters ist, nach der Abspaltung
der Benzylgruppen, fast ausschließlich das Signal des Cholesterylglycosids sichtbar. Die minimalen
Signale sind möglicherweise auf Verunreinigungen des Lösungsmittels zurückzuführen. Weiters
wurde eine Analyse mittels Gaschromatographie durchgeführt, das Chromatogramm ist in Abbildung
49 ersichtlich. Der Hauptpeak, bei dem es sich um das Produkt handelt, befindet sich bei etwa 27
Minuten. Wie im Chromatogramm ersichtlich, ist dem Hauptpeak noch ein zweiter Peak vorgelagert,
bei dem es sich, aufgrund des äquivalenten Massenspektrums, vermutlich um das α-Anomer handelt.
Das GC-Massenspektrum zeigt, wie auch jenes der Darstellung durch Koenigs-Knorr Methode Signale
bei m/z 73, 147, 204, 217 sowie 369. Die Erklärungen der Signale sind in Kapitel 5.1.4 nachzulesen,
das Massenspektrum befindet sich im Anhang. Die Entstehung des α-Anomers ist auf die nicht
vollständige Anomerisierung von β- zu α-Anomer im Zuge der Darstellung des Trichloracetimidates
zurückzuführen, da eine 100%-ige stereochemische Kontrolle bei dessen Bildung nicht gegeben ist.
Die Reinheit wurde durch Integration des Peakflächen berechnet und ergab, dass zu ≥ 99,5
Abbildung 48: DC-Benzylgrupppen-
abspaltung
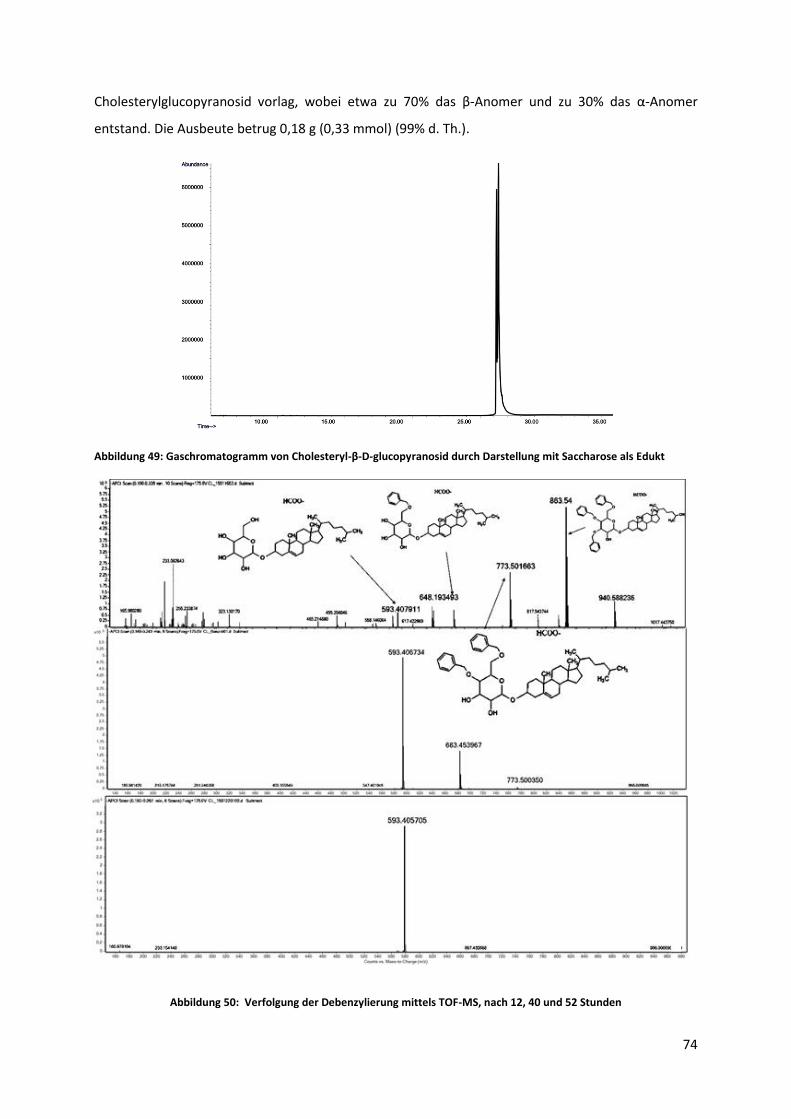
74
Cholesterylglucopyranosid vorlag, wobei etwa zu 70% das β-Anomer und zu 30% das α-Anomer
entstand. Die Ausbeute betrug 0,18 g (0,33 mmol) (99% d. Th.).
Abbildung 49: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mit Saccharose als Edukt
Abbildung 50: Verfolgung der Debenzylierung mittels TOF-MS, nach 12, 40 und 52 Stunden
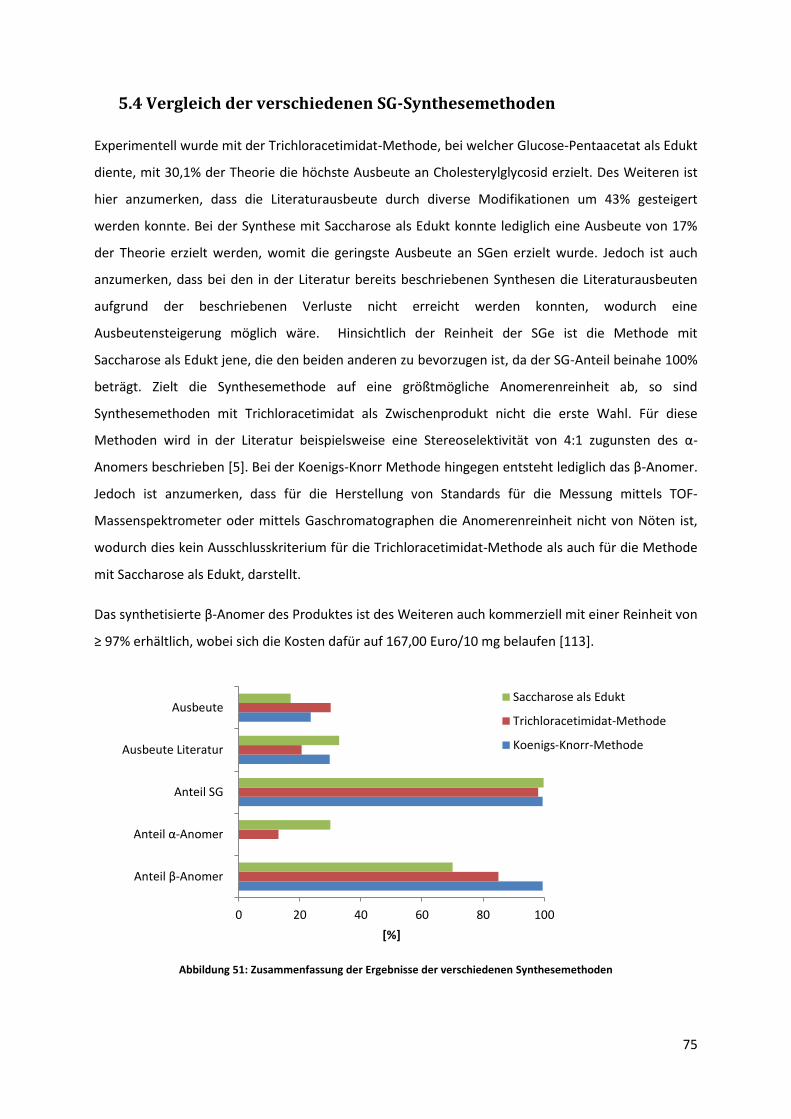
75
5.4 Vergleich der verschiedenen SG-Synthesemethoden
Experimentell wurde mit der Trichloracetimidat-Methode, bei welcher Glucose-Pentaacetat als Edukt
diente, mit 30,1% der Theorie die höchste Ausbeute an Cholesterylglycosid erzielt. Des Weiteren ist
hier anzumerken, dass die Literaturausbeute durch diverse Modifikationen um 43% gesteigert
werden konnte. Bei der Synthese mit Saccharose als Edukt konnte lediglich eine Ausbeute von 17%
der Theorie erzielt werden, womit die geringste Ausbeute an SGen erzielt wurde. Jedoch ist auch
anzumerken, dass bei den in der Literatur bereits beschriebenen Synthesen die Literaturausbeuten
aufgrund der beschriebenen Verluste nicht erreicht werden konnten, wodurch eine
Ausbeutensteigerung möglich wäre. Hinsichtlich der Reinheit der SGe ist die Methode mit
Saccharose als Edukt jene, die den beiden anderen zu bevorzugen ist, da der SG-Anteil beinahe 100%
beträgt. Zielt die Synthesemethode auf eine größtmögliche Anomerenreinheit ab, so sind
Synthesemethoden mit Trichloracetimidat als Zwischenprodukt nicht die erste Wahl. Für diese
Methoden wird in der Literatur beispielsweise eine Stereoselektivität von 4:1 zugunsten des α-
Anomers beschrieben [5]. Bei der Koenigs-Knorr Methode hingegen entsteht lediglich das β-Anomer.
Jedoch ist anzumerken, dass für die Herstellung von Standards für die Messung mittels TOF-
Massenspektrometer oder mittels Gaschromatographen die Anomerenreinheit nicht von Nöten ist,
wodurch dies kein Ausschlusskriterium für die Trichloracetimidat-Methode als auch für die Methode
mit Saccharose als Edukt, darstellt.
Das synthetisierte β-Anomer des Produktes ist des Weiteren auch kommerziell mit einer Reinheit von
≥ 97% erhältlich, wobei sich die Kosten dafür auf 167,00 Euro/10 mg belaufen [113].
Abbildung 51: Zusammenfassung der Ergebnisse der verschiedenen Synthesemethoden
0 20 40 60 80 100
Anteil β-Anomer
Anteil α-Anomer
Anteil SG
Ausbeute Literatur
Ausbeute
[%]
Saccharose als Edukt
Trichloracetimidat-Methode
Koenigs-Knorr-Methode
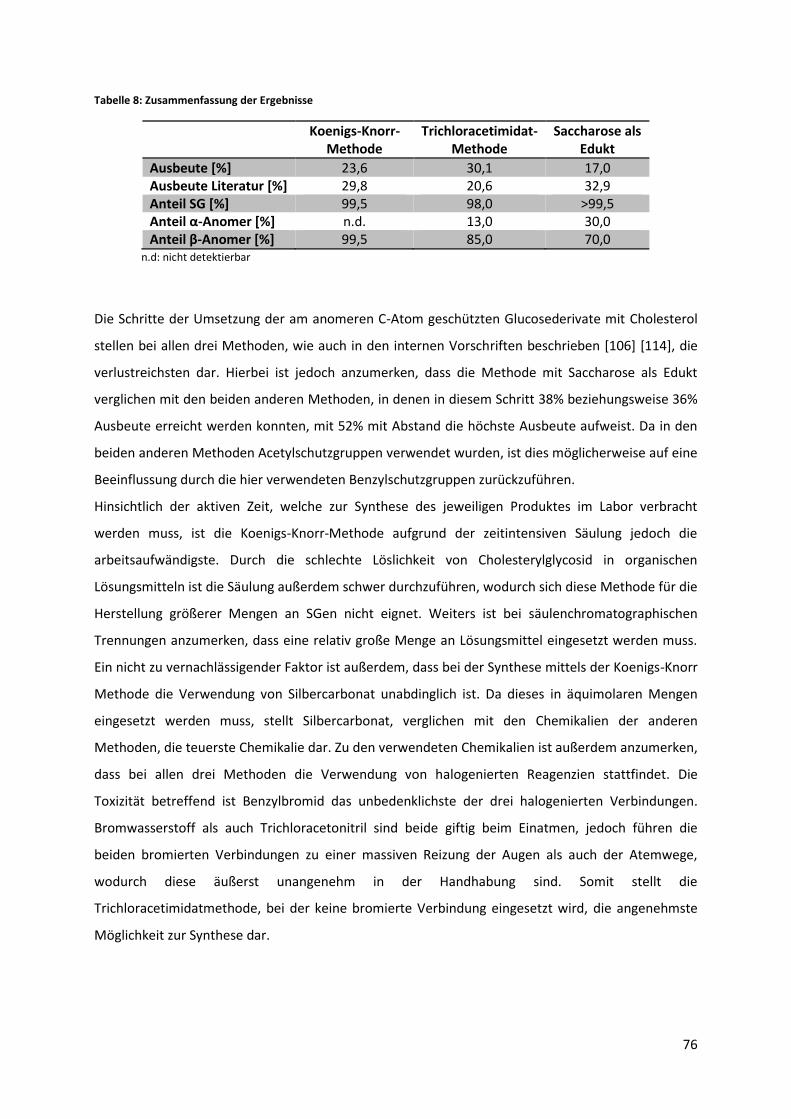
76
Tabelle 8: Zusammenfassung der Ergebnisse
Koenigs-Knorr-Methode
Trichloracetimidat-Methode
Saccharose als Edukt
Ausbeute [%] 23,6 30,1 17,0 Ausbeute Literatur [%] 29,8 20,6 32,9 Anteil SG [%] 99,5 98,0 >99,5 Anteil α-Anomer [%] n.d. 13,0 30,0 Anteil β-Anomer [%] 99,5 85,0 70,0
n.d: nicht detektierbar
Die Schritte der Umsetzung der am anomeren C-Atom geschützten Glucosederivate mit Cholesterol
stellen bei allen drei Methoden, wie auch in den internen Vorschriften beschrieben [106] [114], die
verlustreichsten dar. Hierbei ist jedoch anzumerken, dass die Methode mit Saccharose als Edukt
verglichen mit den beiden anderen Methoden, in denen in diesem Schritt 38% beziehungsweise 36%
Ausbeute erreicht werden konnten, mit 52% mit Abstand die höchste Ausbeute aufweist. Da in den
beiden anderen Methoden Acetylschutzgruppen verwendet wurden, ist dies möglicherweise auf eine
Beeinflussung durch die hier verwendeten Benzylschutzgruppen zurückzuführen.
Hinsichtlich der aktiven Zeit, welche zur Synthese des jeweiligen Produktes im Labor verbracht
werden muss, ist die Koenigs-Knorr-Methode aufgrund der zeitintensiven Säulung jedoch die
arbeitsaufwändigste. Durch die schlechte Löslichkeit von Cholesterylglycosid in organischen
Lösungsmitteln ist die Säulung außerdem schwer durchzuführen, wodurch sich diese Methode für die
Herstellung größerer Mengen an SGen nicht eignet. Weiters ist bei säulenchromatographischen
Trennungen anzumerken, dass eine relativ große Menge an Lösungsmittel eingesetzt werden muss.
Ein nicht zu vernachlässigender Faktor ist außerdem, dass bei der Synthese mittels der Koenigs-Knorr
Methode die Verwendung von Silbercarbonat unabdinglich ist. Da dieses in äquimolaren Mengen
eingesetzt werden muss, stellt Silbercarbonat, verglichen mit den Chemikalien der anderen
Methoden, die teuerste Chemikalie dar. Zu den verwendeten Chemikalien ist außerdem anzumerken,
dass bei allen drei Methoden die Verwendung von halogenierten Reagenzien stattfindet. Die
Toxizität betreffend ist Benzylbromid das unbedenklichste der drei halogenierten Verbindungen.
Bromwasserstoff als auch Trichloracetonitril sind beide giftig beim Einatmen, jedoch führen die
beiden bromierten Verbindungen zu einer massiven Reizung der Augen als auch der Atemwege,
wodurch diese äußerst unangenehm in der Handhabung sind. Somit stellt die
Trichloracetimidatmethode, bei der keine bromierte Verbindung eingesetzt wird, die angenehmste
Möglichkeit zur Synthese dar.

77
5.5 Versuch der enzymatischen Herstellung von Cholesteryl(6´-O-
palmitoyl)-β-D-glucopyranosid
Diese Reaktion basiert auf der enzymatischen Veresterung einer primären Alkoholgruppe mit der
Lipase Novozyme 435, wobei das Enzym hierbei regioselektiv am 6´-O-Atom des Glucoserestes
angreift.
Abbildung 52: Darstellung von durch Veresterung von Cholesterylglycosid mittels Novozyme 435
Zunächst wird bei dieser Reaktion, nach Michaelis-Menten, zwischen Fettsäure und Enzym ein
Enzym-Substrat-Komplex gebildet, wobei Wasser frei wird, welches durch die Zugabe von
Molekularsieb aus der Reaktion entfernt wird, um Hydrolysereaktionen zu vermeiden. Durch den
nucleophilen Angriff des Alkohols, in diesem Fall also der 6-OH-Gruppe des Cholesterylglycosids, auf
den Enzym-Substratkomplex, sollte es zur Bildung des acylierten SGs sowie der Bildung des Enzyms in
seiner nativen Form kommen.
Abbildung 53: Enzymkinetik nach Michaelis-Menten. E=Enzym, S= Substrat, P=Produkt.

78
Abbildung 54: Reaktionsmechanismus der Veresterung von Sterylglycosiden mit Palmitinsäure. Enz = Enzym Novozyme 435
Den Vorteil, den diese Methode mit sich bringt, ist, dass es durch die Regioselektivität, zu keiner
Veresterung der anderen freien Hydroxylgruppen kommt, wodurch keine Einführung von
Schutzgruppen notwendig ist. Die Problematik hingegen ist, dass sich das das Substrat nicht,
beziehungsweise nur begrenzt (vgl. Löslichkeit in Biodiesel (50 ppm bei 50°C)) in unpolaren,
organischen Lösungsmitteln, die für das Enzym empfohlen werden, löst [60]. Lipasen sind zwar
gleichermaßen in wässrigen Medien aktiv, jedoch kommt es in diesem Fall bevorzugt zu
Hydrolysereaktionen, was in diesem Fall einer Spaltung eines acylierten Sterylglycosids in das freie
Sterylglycosid sowie der Fettsäure entsprechen würde. Das Enzym benötigt zwar eine kleine Menge
Wasser, welches jedoch durch die Immobilisierung auf Acrylharz mit einem Wassergehalt von 1-2%
ohnehin gegeben ist. Jedoch ist bei der Reaktion an sich darauf zu achten, dass das Enzym nicht zu
lange, in direktem Kontakt mit dem Molekularsieb, ohne dass das Reaktionssystem in Bewegung ist,
kommt, da ansonsten das benötigte Wasser aus dem Enzym entzogen werden kann, was in einer
Minderung des Umsatzes beziehungsweise in einer Zerstörung des Enzyms resultieren würde [62]. Im
Gegensatz zum Substrat löst sich das acylierte SG im organischen Lösungsmittel. Durch den Umsatz
kann zwar somit erreicht werden, dass sich immer wieder ein Teil des Substrates im Lösungsmittel
löst, jedoch in so geringem Maße, dass nach wie vor der Großteil als ungelöster Feststoff vorliegt. So
gelangt jeweils nur ein kleiner Teil des Cholesterylglycosids in das aktive Zentrum des Enzyms
wodurch sich auch der geringe Umsatz erklären lässt.
5.5.1 Versuch mit reinem Hexan als Lösungsmittel
In der Literatur wurde dieser Versuch von Paczkowski et al. (2007) [63] im Maßstab für 1 µmol
beschrieben, jedoch handelt es sich dabei um das einzige Literaturzitat, welches die enzymatische
Veresterung durch eine freie Fettsäure beschreibt. Eine Umsetzung mit Fettsäureestern, zum Beispiel
mit Palmitinsäurevinylester [115] ist hingegen des Öfteren zu finden. Das Experiment wurde in
Anlehnung an die interne Vorschrift durchgeführt [114].
Bei der Umsetzung in Hexan ist es zu einem Versagen des Laborschüttlers während der Nacht
gekommen. Es wurde daraufhin die Schütteldauer noch verlängert, jedoch kann es möglicherweise
dennoch zu einer Anlagerung des Enzyms an das Edukt beziehungsweise zu einer Dehydratisierung
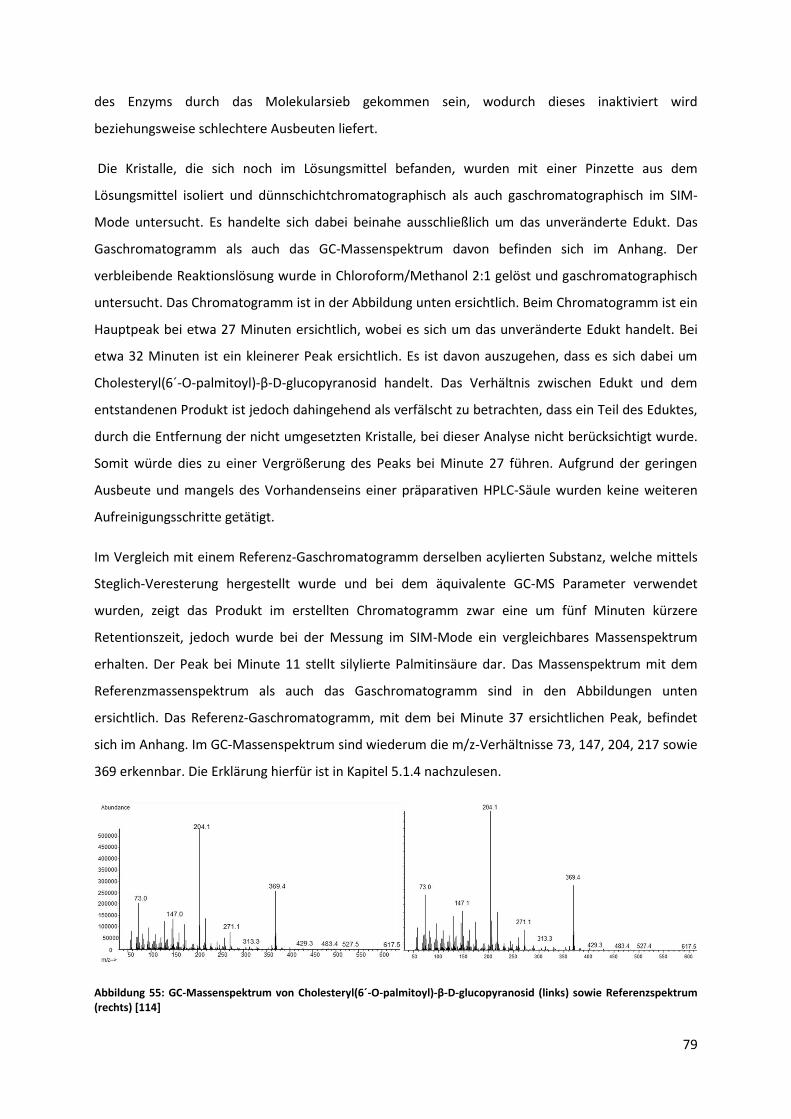
79
des Enzyms durch das Molekularsieb gekommen sein, wodurch dieses inaktiviert wird
beziehungsweise schlechtere Ausbeuten liefert.
Die Kristalle, die sich noch im Lösungsmittel befanden, wurden mit einer Pinzette aus dem
Lösungsmittel isoliert und dünnschichtchromatographisch als auch gaschromatographisch im SIM-
Mode untersucht. Es handelte sich dabei beinahe ausschließlich um das unveränderte Edukt. Das
Gaschromatogramm als auch das GC-Massenspektrum davon befinden sich im Anhang. Der
verbleibende Reaktionslösung wurde in Chloroform/Methanol 2:1 gelöst und gaschromatographisch
untersucht. Das Chromatogramm ist in der Abbildung unten ersichtlich. Beim Chromatogramm ist ein
Hauptpeak bei etwa 27 Minuten ersichtlich, wobei es sich um das unveränderte Edukt handelt. Bei
etwa 32 Minuten ist ein kleinerer Peak ersichtlich. Es ist davon auszugehen, dass es sich dabei um
Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid handelt. Das Verhältnis zwischen Edukt und dem
entstandenen Produkt ist jedoch dahingehend als verfälscht zu betrachten, dass ein Teil des Eduktes,
durch die Entfernung der nicht umgesetzten Kristalle, bei dieser Analyse nicht berücksichtigt wurde.
Somit würde dies zu einer Vergrößerung des Peaks bei Minute 27 führen. Aufgrund der geringen
Ausbeute und mangels des Vorhandenseins einer präparativen HPLC-Säule wurden keine weiteren
Aufreinigungsschritte getätigt.
Im Vergleich mit einem Referenz-Gaschromatogramm derselben acylierten Substanz, welche mittels
Steglich-Veresterung hergestellt wurde und bei dem äquivalente GC-MS Parameter verwendet
wurden, zeigt das Produkt im erstellten Chromatogramm zwar eine um fünf Minuten kürzere
Retentionszeit, jedoch wurde bei der Messung im SIM-Mode ein vergleichbares Massenspektrum
erhalten. Der Peak bei Minute 11 stellt silylierte Palmitinsäure dar. Das Massenspektrum mit dem
Referenzmassenspektrum als auch das Gaschromatogramm sind in den Abbildungen unten
ersichtlich. Das Referenz-Gaschromatogramm, mit dem bei Minute 37 ersichtlichen Peak, befindet
sich im Anhang. Im GC-Massenspektrum sind wiederum die m/z-Verhältnisse 73, 147, 204, 217 sowie
369 erkennbar. Die Erklärung hierfür ist in Kapitel 5.1.4 nachzulesen.
Abbildung 55: GC-Massenspektrum von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid (links) sowie Referenzspektrum (rechts) [114]
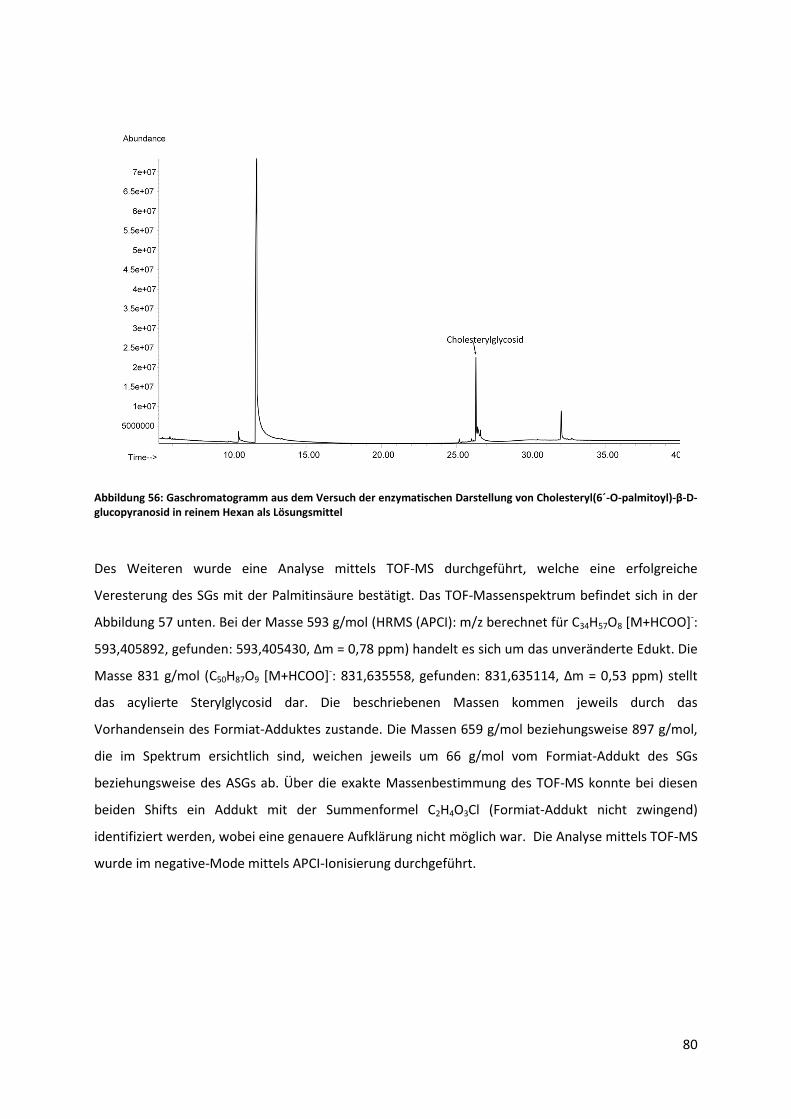
80
Abbildung 56: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel
Des Weiteren wurde eine Analyse mittels TOF-MS durchgeführt, welche eine erfolgreiche
Veresterung des SGs mit der Palmitinsäure bestätigt. Das TOF-Massenspektrum befindet sich in der
Abbildung 57 unten. Bei der Masse 593 g/mol (HRMS (APCI): m/z berechnet für C34H57O8 [M+HCOO]-:
593,405892, gefunden: 593,405430, Δm = 0,78 ppm) handelt es sich um das unveränderte Edukt. Die
Masse 831 g/mol (C50H87O9 [M+HCOO]-: 831,635558, gefunden: 831,635114, Δm = 0,53 ppm) stellt
das acylierte Sterylglycosid dar. Die beschriebenen Massen kommen jeweils durch das
Vorhandensein des Formiat-Adduktes zustande. Die Massen 659 g/mol beziehungsweise 897 g/mol,
die im Spektrum ersichtlich sind, weichen jeweils um 66 g/mol vom Formiat-Addukt des SGs
beziehungsweise des ASGs ab. Über die exakte Massenbestimmung des TOF-MS konnte bei diesen
beiden Shifts ein Addukt mit der Summenformel C2H4O3Cl (Formiat-Addukt nicht zwingend)
identifiziert werden, wobei eine genauere Aufklärung nicht möglich war. Die Analyse mittels TOF-MS
wurde im negative-Mode mittels APCI-Ionisierung durchgeführt.

81
Abbildung 57: TOF-Massenspektrum aus dem Versuch der enzymatischen Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel
Nachdem die enzymatische Veresterung in der internen Vorschrift nicht erfolgreich durchgeführt
werden konnte, ist die Entstehung des gewünschten Produktes möglicherweise auf die Reduzierung
der Menge an Palmitinsäure, welches in dieser im 70-fachen Überschuss verwendet wurde,
zurückzuführen. Weiters wurde auf absolute Wasserfreiheit geachtet. Weitere Faktoren, welche
möglicherweise eine positive Auswirkung auf die Reaktion hatten, waren die Verwendung von n-
Hexan an der Stelle von n-Oktan, sowie die Erhöhung der Reaktionstemperatur von 40°C auf 45°C.
5.5.2 Versuch mit Hexan nach vorherigem Lösen des Substrates in Chloroform
Um das Substrat in Lösung zu bringen, um es in weiterer Folge für das aktive Zentrum des Enzyms
zugänglich zu machen, wurde dieses im Vorhinein in Chloroform gelöst [116]. Weiters wurde die
Menge an Enzym im Vergleich zur internen Vorschrift reduziert, da das Enzym eine extrem hohe
Aktivität von >5000 U/g aufweist. Nichtsdestoweniger wurde das Enzym dennoch in großem
Überschuss eingesetzt. Nach der Aufreinigung wurde eine gaschromatographische Analyse
durchgeführt, welche zeigte, dass nur mehr eine kleine Menge des Cholesterylglycosids vorlag. Dies
lässt darauf schließen, dass durch die Zugabe von Chloroform und der damit verbundenen Löslichkeit
des Eduktes, ein höherer Umsatz im Vergleich zu reinem Hexan erreicht werden konnte. Weiters
entstand eine Reihe von Nebenprodukten, die weder durch die Analyse am GC-MS noch durch
Analyse am TOF-MS identifiziert werden konnten. Der Peak bei Minute 10 stellt, Methylpalmitat dar,
welches möglicherweise bei der Filtration und dem anschließenden Nachwaschen mit
Chloroform/Methanol 2:1 sowie dem darauffolgenden Abrotieren entstanden ist. Das gewünschte
Produkt wurde jedoch nicht gebildet. Auffällig hingegen ist beim Hauptpeak des entstandenen
Produktgemisches ein ausgeprägtes Signal mit m/z 399, welches jedoch nicht identifiziert werden
konnte.
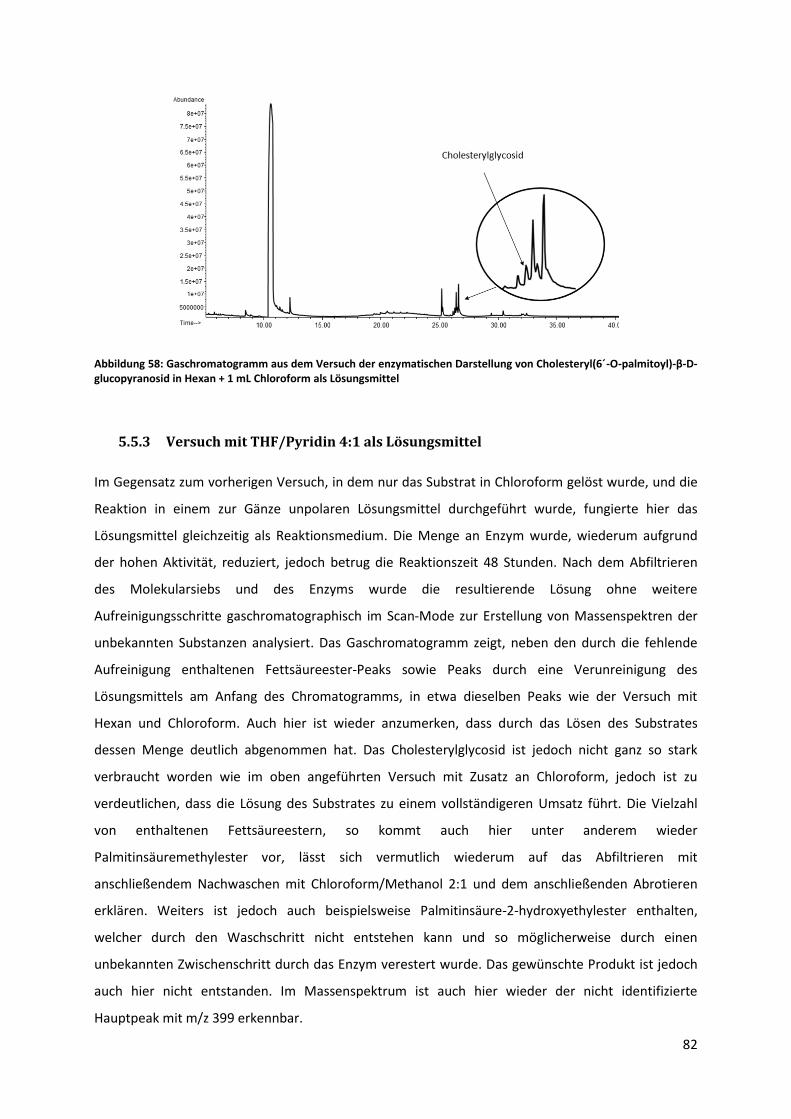
82
Abbildung 58: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in Hexan + 1 mL Chloroform als Lösungsmittel
5.5.3 Versuch mit THF/Pyridin 4:1 als Lösungsmittel
Im Gegensatz zum vorherigen Versuch, in dem nur das Substrat in Chloroform gelöst wurde, und die
Reaktion in einem zur Gänze unpolaren Lösungsmittel durchgeführt wurde, fungierte hier das
Lösungsmittel gleichzeitig als Reaktionsmedium. Die Menge an Enzym wurde, wiederum aufgrund
der hohen Aktivität, reduziert, jedoch betrug die Reaktionszeit 48 Stunden. Nach dem Abfiltrieren
des Molekularsiebs und des Enzyms wurde die resultierende Lösung ohne weitere
Aufreinigungsschritte gaschromatographisch im Scan-Mode zur Erstellung von Massenspektren der
unbekannten Substanzen analysiert. Das Gaschromatogramm zeigt, neben den durch die fehlende
Aufreinigung enthaltenen Fettsäureester-Peaks sowie Peaks durch eine Verunreinigung des
Lösungsmittels am Anfang des Chromatogramms, in etwa dieselben Peaks wie der Versuch mit
Hexan und Chloroform. Auch hier ist wieder anzumerken, dass durch das Lösen des Substrates
dessen Menge deutlich abgenommen hat. Das Cholesterylglycosid ist jedoch nicht ganz so stark
verbraucht worden wie im oben angeführten Versuch mit Zusatz an Chloroform, jedoch ist zu
verdeutlichen, dass die Lösung des Substrates zu einem vollständigeren Umsatz führt. Die Vielzahl
von enthaltenen Fettsäureestern, so kommt auch hier unter anderem wieder
Palmitinsäuremethylester vor, lässt sich vermutlich wiederum auf das Abfiltrieren mit
anschließendem Nachwaschen mit Chloroform/Methanol 2:1 und dem anschließenden Abrotieren
erklären. Weiters ist jedoch auch beispielsweise Palmitinsäure-2-hydroxyethylester enthalten,
welcher durch den Waschschritt nicht entstehen kann und so möglicherweise durch einen
unbekannten Zwischenschritt durch das Enzym verestert wurde. Das gewünschte Produkt ist jedoch
auch hier nicht entstanden. Im Massenspektrum ist auch hier wieder der nicht identifizierte
Hauptpeak mit m/z 399 erkennbar.
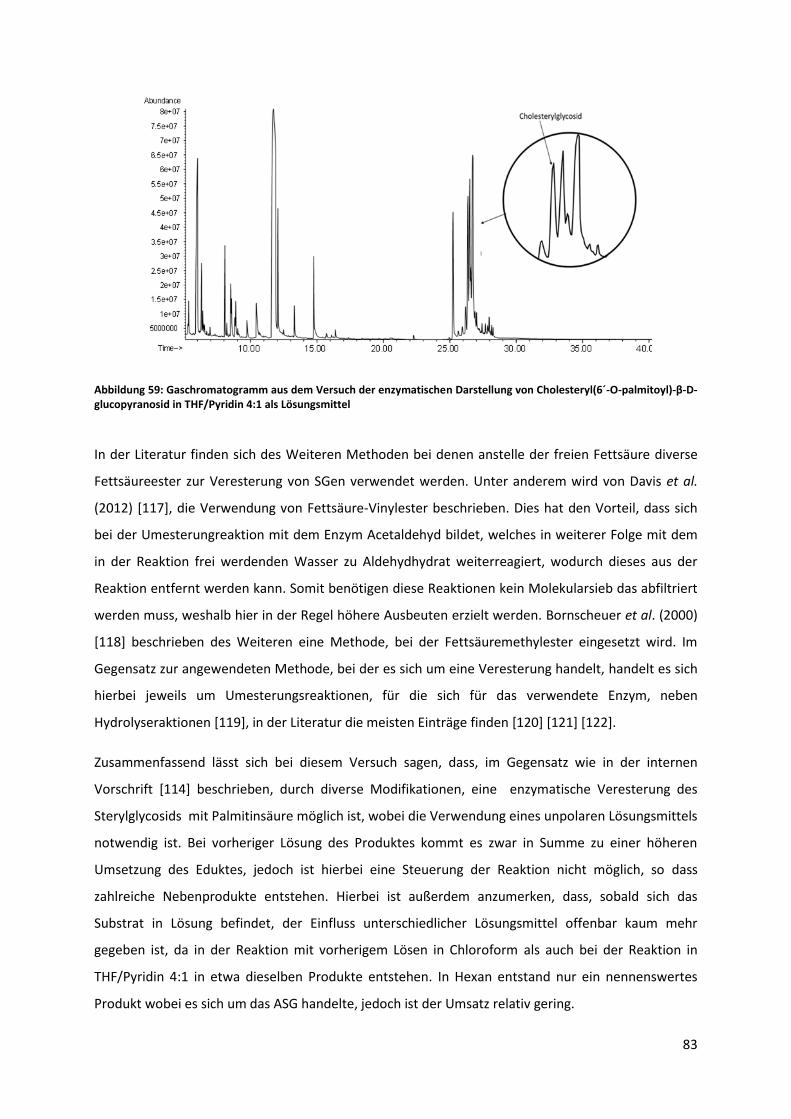
83
Abbildung 59: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in THF/Pyridin 4:1 als Lösungsmittel
In der Literatur finden sich des Weiteren Methoden bei denen anstelle der freien Fettsäure diverse
Fettsäureester zur Veresterung von SGen verwendet werden. Unter anderem wird von Davis et al.
(2012) [117], die Verwendung von Fettsäure-Vinylester beschrieben. Dies hat den Vorteil, dass sich
bei der Umesterungreaktion mit dem Enzym Acetaldehyd bildet, welches in weiterer Folge mit dem
in der Reaktion frei werdenden Wasser zu Aldehydhydrat weiterreagiert, wodurch dieses aus der
Reaktion entfernt werden kann. Somit benötigen diese Reaktionen kein Molekularsieb das abfiltriert
werden muss, weshalb hier in der Regel höhere Ausbeuten erzielt werden. Bornscheuer et al. (2000)
[118] beschrieben des Weiteren eine Methode, bei der Fettsäuremethylester eingesetzt wird. Im
Gegensatz zur angewendeten Methode, bei der es sich um eine Veresterung handelt, handelt es sich
hierbei jeweils um Umesterungsreaktionen, für die sich für das verwendete Enzym, neben
Hydrolyseraktionen [119], in der Literatur die meisten Einträge finden [120] [121] [122].
Zusammenfassend lässt sich bei diesem Versuch sagen, dass, im Gegensatz wie in der internen
Vorschrift [114] beschrieben, durch diverse Modifikationen, eine enzymatische Veresterung des
Sterylglycosids mit Palmitinsäure möglich ist, wobei die Verwendung eines unpolaren Lösungsmittels
notwendig ist. Bei vorheriger Lösung des Produktes kommt es zwar in Summe zu einer höheren
Umsetzung des Eduktes, jedoch ist hierbei eine Steuerung der Reaktion nicht möglich, so dass
zahlreiche Nebenprodukte entstehen. Hierbei ist außerdem anzumerken, dass, sobald sich das
Substrat in Lösung befindet, der Einfluss unterschiedlicher Lösungsmittel offenbar kaum mehr
gegeben ist, da in der Reaktion mit vorherigem Lösen in Chloroform als auch bei der Reaktion in
THF/Pyridin 4:1 in etwa dieselben Produkte entstehen. In Hexan entstand nur ein nennenswertes
Produkt wobei es sich um das ASG handelte, jedoch ist der Umsatz relativ gering.
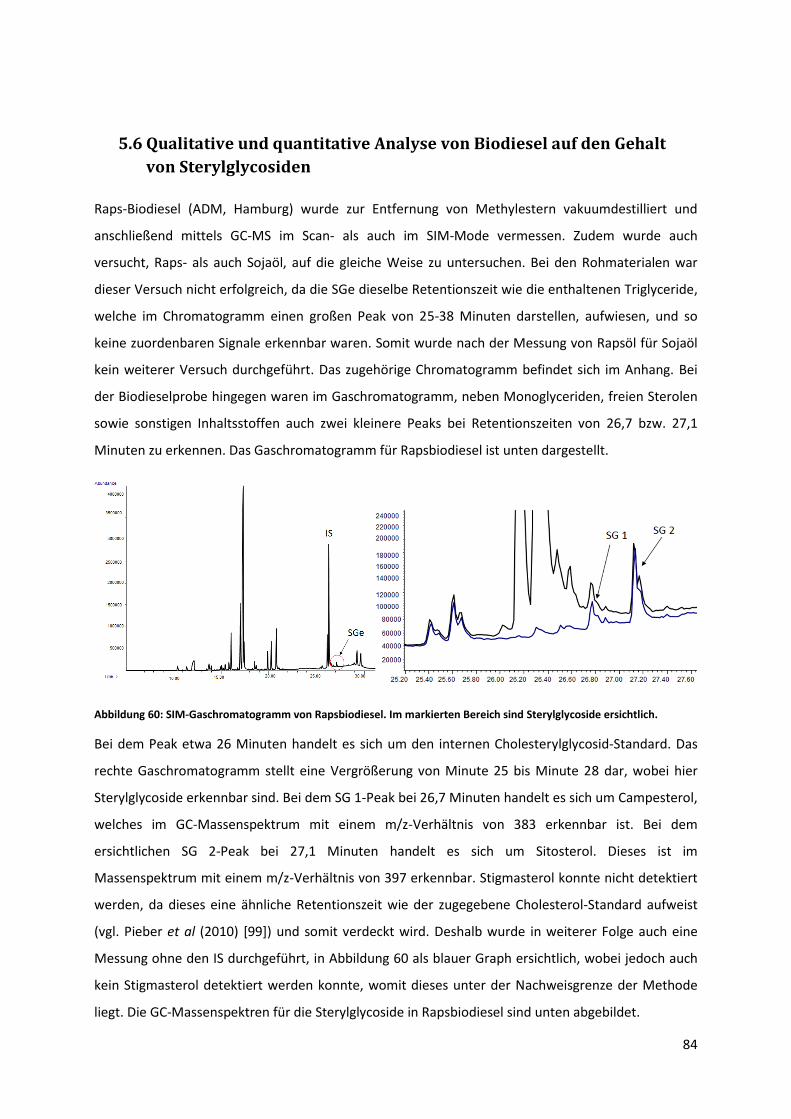
84
5.6 Qualitative und quantitative Analyse von Biodiesel auf den Gehalt
von Sterylglycosiden
Raps-Biodiesel (ADM, Hamburg) wurde zur Entfernung von Methylestern vakuumdestilliert und
anschließend mittels GC-MS im Scan- als auch im SIM-Mode vermessen. Zudem wurde auch
versucht, Raps- als auch Sojaöl, auf die gleiche Weise zu untersuchen. Bei den Rohmaterialen war
dieser Versuch nicht erfolgreich, da die SGe dieselbe Retentionszeit wie die enthaltenen Triglyceride,
welche im Chromatogramm einen großen Peak von 25-38 Minuten darstellen, aufwiesen, und so
keine zuordenbaren Signale erkennbar waren. Somit wurde nach der Messung von Rapsöl für Sojaöl
kein weiterer Versuch durchgeführt. Das zugehörige Chromatogramm befindet sich im Anhang. Bei
der Biodieselprobe hingegen waren im Gaschromatogramm, neben Monoglyceriden, freien Sterolen
sowie sonstigen Inhaltsstoffen auch zwei kleinere Peaks bei Retentionszeiten von 26,7 bzw. 27,1
Minuten zu erkennen. Das Gaschromatogramm für Rapsbiodiesel ist unten dargestellt.
Abbildung 60: SIM-Gaschromatogramm von Rapsbiodiesel. Im markierten Bereich sind Sterylglycoside ersichtlich.
Bei dem Peak etwa 26 Minuten handelt es sich um den internen Cholesterylglycosid-Standard. Das
rechte Gaschromatogramm stellt eine Vergrößerung von Minute 25 bis Minute 28 dar, wobei hier
Sterylglycoside erkennbar sind. Bei dem SG 1-Peak bei 26,7 Minuten handelt es sich um Campesterol,
welches im GC-Massenspektrum mit einem m/z-Verhältnis von 383 erkennbar ist. Bei dem
ersichtlichen SG 2-Peak bei 27,1 Minuten handelt es sich um Sitosterol. Dieses ist im
Massenspektrum mit einem m/z-Verhältnis von 397 erkennbar. Stigmasterol konnte nicht detektiert
werden, da dieses eine ähnliche Retentionszeit wie der zugegebene Cholesterol-Standard aufweist
(vgl. Pieber et al (2010) [99]) und somit verdeckt wird. Deshalb wurde in weiterer Folge auch eine
Messung ohne den IS durchgeführt, in Abbildung 60 als blauer Graph ersichtlich, wobei jedoch auch
kein Stigmasterol detektiert werden konnte, womit dieses unter der Nachweisgrenze der Methode
liegt. Die GC-Massenspektren für die Sterylglycoside in Rapsbiodiesel sind unten abgebildet.
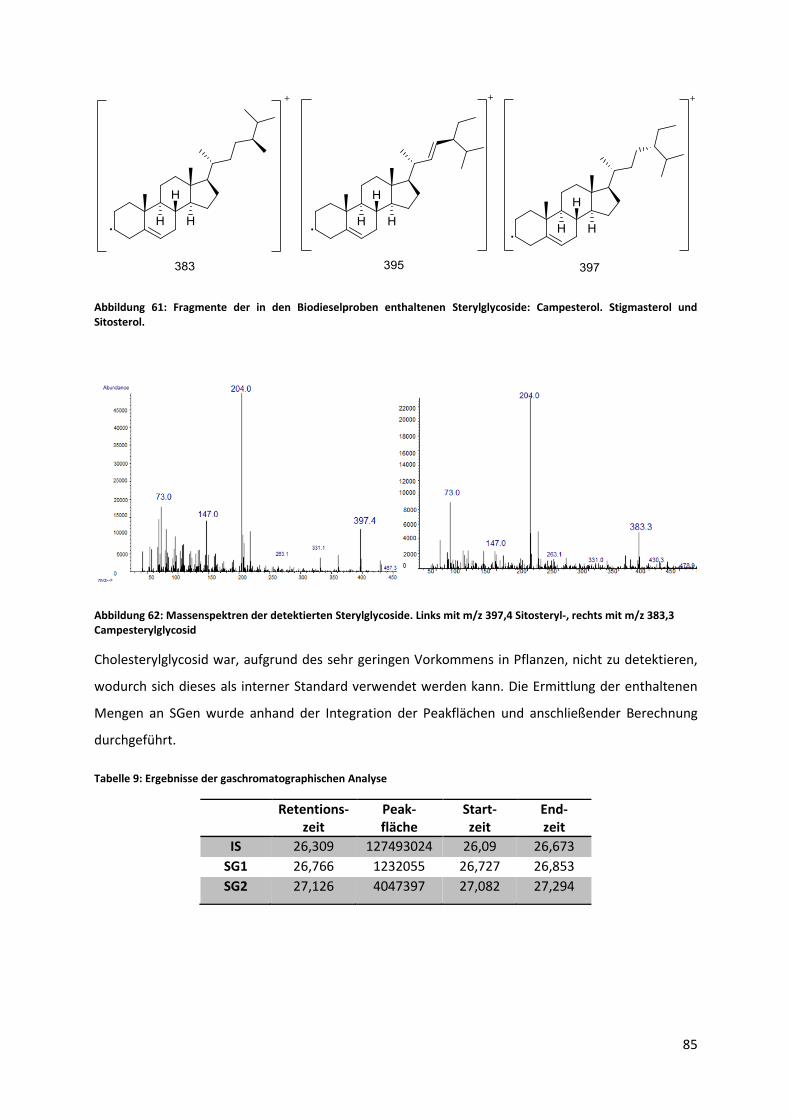
85
Abbildung 61: Fragmente der in den Biodieselproben enthaltenen Sterylglycoside: Campesterol. Stigmasterol und Sitosterol.
Abbildung 62: Massenspektren der detektierten Sterylglycoside. Links mit m/z 397,4 Sitosteryl-, rechts mit m/z 383,3 Campesterylglycosid
Cholesterylglycosid war, aufgrund des sehr geringen Vorkommens in Pflanzen, nicht zu detektieren,
wodurch sich dieses als interner Standard verwendet werden kann. Die Ermittlung der enthaltenen
Mengen an SGen wurde anhand der Integration der Peakflächen und anschließender Berechnung
durchgeführt.
Tabelle 9: Ergebnisse der gaschromatographischen Analyse
Retentions- zeit
Peak- fläche
Start- zeit
End- zeit
IS 26,309 127493024 26,09 26,673
SG1 26,766 1232055 26,727 26,853
SG2 27,126 4047397 27,082 27,294

86
Formel 1: Formel zur Berechnung des Sterylglycosid-Gehalts
Tabelle 10: SG-Gehalt in Raps-Biodiesel
Campesterylglycosid
[ppb]
Sitosterylglycosid-
Glycosid [ppb]
Gesamtgehalt
[ppb]
Rapsöl-Biodiesel 0,062 0,202 0,268
Laut Literatur ist im Rapsöl mit 800 mg/kg in der Regel ein höherer Gehalt an freien SGen enthalten,
der Gehalt an ASGen liegt in Rapsöl bei etwa 200 mg/kg [123]. Für den Gesamtgehalt an
Sterylglycosiden in Rapsbiodiesel .finden sich in der Literatur Werte um 10 mg/kg, der gemessene
Wert bei Lacoste et al. [2] beträgt beispielsweise 12 mg/kg. Die große Abweichung von diesen
Werten kann jedoch nicht erklärt werden.
6 Zusammenfassung und Ausblick
Wie in der Einleitung beschrieben, war das Ziel der Arbeit, die Synthese von Sterylglycosiden unter
dem Aspekt der Steigerung der Ausbeute durchzuführen. Zu diesem Zweck wurden zwei bekannte
Methoden, die Synthese mittels Koenigs-Knorr-Mechanismus sowie die Trichloracetimidat-Methode
modifiziert, und eine neue Methode, nämlich die Herstellung aus Saccharose, entwickelt.
Bei der Synthese nach Koenigs-Knorr konnte keine Steigerung der Ausbeute erzielt werden, wobei es
bei dieser Methode vielmehr das Ziel war, eine Vergleichssubstanz für die beiden anderen Methoden
herzustellen. Von der Anomerenreinheit, die jedoch für die Erstellung interner Standards nicht das
primäre Auswahlkriterium darstellt, liefert diese Methode das beste Ergebnis. Die Reinheit des
Produktes belief sich auf 99,5%.
Bei der Trichloracetimidatmethode konnte, aufgrund einiger Modifizierungen der internen
Synthesevorschrift [106] sowie der Umgehung von säulenchromatographischen Auftrennungen, eine
Steigerung auf 146% des Literaturwertes verzeichnet werden. Weiters wurde mit dieser Methode, im
Vergleich zu den beiden anderen, die höchste Ausbeute erzielt. Hinsichtlich der Anomerenreinheit
CIS Konzentration des IS in mg/kg
ASG Peakfläche der SGe
AIS Peakfläche IS
SG Konzentration der SGe in mg/kg
mFAME Gewicht von Biodiesel in mg
mIS Gewicht des IS in mg

87
entstanden bei der Synthese 13% des α-Anomers und 85% des β-Anomers. Die Gesamtreinheit des
erhaltenen Produktes betrug 98%.
Die Synthese von Sterylglycosiden mit Saccharose als Ausgangssubstanz wurde in der Literatur noch
nicht beschrieben, somit handelt es sich hierbei um eine neue Methode. Im Vergleich mit den beiden
anderen Methoden wurde hier die niedrigste Ausbeute erhalten, was jedoch teilweise auf
vermeidbare Produktverluste zurückzuführen ist. Theoretische Berechnungen der Ausbeute durch
den Vergleich mit diversen, in der Literatur beschriebenen, Zwischenschritten, zeigen jedoch, dass
mit dieser Methode die höchste Ausbeute erzielt werden könnte. Weiters wurde bei dem Schritt der
Substitution der Schutzgruppe durch Cholesterol, der beiden anderen Methoden den
verlustreichsten Zwischenschritt darstellte, hier mit einer vergleichsweise hohen Ausbeute vollzogen.
Bei der Synthese entstand zu 30% das α-Anomer und zu 70% das β-Anomer. Bei der Gesamtreinheit
wurde mit dieser Methode mit > 99,5% der beste Wert erzielt.
Somit ist zu sagen, dass dieses Ziel der Arbeit erfolgreich abgeschlossen werden konnte, da durch die
Modifikation eine Ausbeutensteigerung vorliegt. Weiters wurde durch die Methode mit Saccharose
als Edukt eine vielversprechende Grundlage für eine weitere Ausbeutensteigerung gelegt.
Die enzymatische Veresterung von Sterylglycosiden konnte, durch diverse Modifikationen der
internen Vorschrift [114], im Gegensatz zu dieser, zumindest dahingehend erfolgreich durchgeführt
werden, dass das gewünschte Produkt analytisch nachgewiesen werden konnte. Weiters wurde
gezeigt, dass durch vorherige Lösung des SGs andere Produkte entstehen, als dies beim ungelösten
Edukt der Fall ist, wobei das eingesetzte Lösungsmittel, nach der erfolgten Lösung, keinen Einfluss
auf die Reaktion hat. Außerdem ist aus den Versuchen ersichtlich, dass eine Umsetzung auch bei
einer drastischen Reduzierung der eingesetzten Enzymmenge als auch der Menge an Palmitinsäure
stattfindet, was zu einer Kostenreduktion für eventuelle weitere Versuche führen könnte. Eine
Reduktion von SGen konnte in allen drei Versuchen beobachtet werden, auch wenn in zwei davon
nicht das gewünschte Produkt entstand, wodurch solche Methoden möglicherweise auch zur
Reduktion von SGen in Biodiesel verwendet werden könnten.
Für zukünftige Experimente wäre es möglicherweise interessant, jeweils die Schritte der Umsetzung
der Glucopyranosyl-Trichloracetimidate beziehungsweise der Acetobromoglucose mit Cholesterol zu
modifizieren, da hierbei bei allen drei Methoden der höchste Ausbeutenverlust verzeichnet wird.
Außerdem wäre eine Modifizierung der Methode mit Saccharose als Edukt dahingehend einen
Versuch wert, die Benzylschutzgruppen durch Benzoylschutzgruppen zu ersetzen, da diese, in
weiterer Folge, leicht durch Zemplén-Verseifung abgespalten werden könnten. Bei den
Anomerisierungen bei Trichloracetimidat-Methoden könnte außerdem noch hinsichtlich der Dauer

88
der Anomerisierung experimentiert werden. Empfehlenswert wäre hierbei vielleicht diese über
Nacht durchzuführen, um eine möglichst vollständige Anomerisierung zum α-Trichloracetimidat zu
erreichen. Zur enzymatischen Synthese von acylierten Sterylglycosiden könnten noch Versuche mit
Fettsäureestern durchgeführt werden, bei denen diese anstelle der freien Fettsäuren eingesetzt
werden. Eine weitere Möglichkeit, die versucht werden könnte, ist die vorherige Veresterung am O-6
Atom [124] und eine anschließende Umsetzung mit Cholesterol. Summa summarum könnten weitere
Untersuchungen des Enzyms hinsichtlich der Entfernung freier Sterylglycoside zu einer Minderung
der Sterylglycosidproblematik im Biodiesel beitragen.

89
7 Literaturverzeichnis
[1] Arbeitsgemeinschaft Qualitätsmanagement Biodiesel e.V., „Sterylglycoside und acylierte
Sterylglycoside in Pflanzenölen und Fettsäuremethylestern und ihre Auswirkungen auf die
Filtrierbarkeit von Biodiesel. Studie zum SG- und ASG-Gehalt von Pflanzenölen und FAME
Untersuchungen zur Korrelation der Filtrierbarkeit,“ 2011.
[2] Lacoste F., Dejean F., Griffon H., „Quantification of Free and Esterfied Steryl Glucosides in
Vegetetable Oils and Biodiesel,“ Eur. J. Lipid Sci. Technol., Bd. 111, pp. 822-828, 2009.
[3] Lee I., Pfalzgraf L.M., Poppe G.B., Powers E., Haines T., „The Role of Sterol Glucosides on Filter
Plugging,“ Biodiesel Magazine, Bd. 4, pp. 105-112, 2007.
[4] Vollhardt K. P. C., Schore N. E., Organische Chemie, Weinheim: Wiley-VCH-Verlag, 2000.
[5] Habermehl G., Hammann P.E., Krebs H.C., Ternes W., Naturstoffchemie. Eine Einführung,
Heidelberg: Springer-Verlag, 2008.
[6] Voet D.J., Voet J.G., Pratt C.W., Lehrbuch der Biochemie, Heidelberg: Wiley-VCH-Verlag, 2002.
[7] Heupel, R.C., „Isolation and Primary Characterization of Sterols,“ in Analysis of Sterols and
Other Biologically Significant Steroids, San Diego, Academic Press Inc., 1989, pp. 1-27.
[8] Weihrauch J.L., Gardner J.M., „Sterol Content of Foods of Plant Origin,“ Journal of the
American Dietetic Association, Bd. 73, pp. 39-47, 1978.
[9] Steglich W., Fugmann B., Lang-Fugmanns S., Römpp Lexikon Naturstoffe, Stuttgart: Thieme,
1997.
[10] Bergenstrahle A., Borga P., Jonsson L., „Sterol Composition and Synthesis in Potato Tuber Discs
in Relation to Glycoalkaloid Synthesis,“ Phytochemistry, Bd. 41, pp. 155-160, 1996.
[11] Barnsteiner A., Analytik von Fettsäureestern pflanzlicher Sterole/Stanole in angereicherten
Lebensmitteln, Technische Universität München, 2012.
[12] Thompson G. R., Grundy S. M., „History and Development of Plant Sterol and Stanol Esters for
Cholesterol-Lowering Purposes,“ Am. J. Cardiol., Bd. 96, Nr. 1A, pp. 3D-9D, 2005.
[13] Miettinen T., Gylling H., „Plant stanol and sterol esters in prevention of cardiovascular
diseases.,“ Ann. Med., Bd. 36, Nr. 2, pp. 126-134, 2004.
[14] Kusmakow O.V., Versuche zur intrazellulären Lokalisierung der Sterol-Glucosyltransferase und
der Glucosylceramid-Synthase in Zellen von Allium fistulosum L., Universität Hamburg, 2005.
[15] Weete J.D., Abril M., Blackwell M., „Phylogenetic Distribution of Fungal Sterols,“ PLoS ONE,
Online Quelle:

90
http://www.plosone.org/article/fetchObject.action?uri=info:doi/10.1371/journal.pone.001089
9&representation=PDF, Bd. 5, Nr. 5, pp. 1-6, 2010.
[16] Weidenbörner M., Lexikon der Lebensmittelmykologie, Berlin, Heidelberg: Springer-Verlag,
2000.
[17] Grunwald C., „Steryl Glycoside Biosynthesis,“ Lipids, Bd. 13, pp. 697-703, 1978.
[18] Abraham W., Wert P.W., Burken R.R., Downing, D.T., „Glucosylsterol and acylglucosylsterol of
snake epidermis. Structure determination.,“ J Lipid Res, Bd. 28, Nr. 4, pp. 446-449, 1987.
[19] Tannaes T., Bukholm G., „Cholesteryl-6-O-acyl-α-D-glucopyranoside of Helicobacter Pylori
Relate to Relative Lysophospholipid Content,“ FEMS Microbiology Letters, Bd. 244, Nr. 1, pp.
117-120, 2005.
[20] Wojciechowski Z.A., „Biochemistry of Phytosterol Conjugates,“ in Physiology and Biochemistry
of Sterols, Champaign, American Oil Chemists´ Society, 1992, pp. 361-395.
[21] Grunwald C., „Effect of Sterols on the Permeability of Alcohol-Treated Red Beet Tissue,“ Plant
Physiol., Bd. 43, pp. 484-488, 1968.
[22] Peng L., Kawagoe Y., Hogan P., Delmer D., „Sitosterol-β-glucoside as Primer for Cellulose
Synthesis in Plants,“ Science, Bd. 295, pp. 147-150, 2002.
[23] Grille S., Glycolipid-Engineering zur Aufklärung der biologischen Funktion von Sterylglycosiden
in der Pexophagie von Pichia pastoris, Universität Hamburg, 2010.
[24] Read .SM., Bacic T., „Prime Time for Cellulose,“ Science, Bd. 295, pp. 59-60, 2002.
[25] Donald P.R., Lamprecht J.H., Freestone M., Albrecht C.F., Bouic P.J., Kotze D., van Jaarsveld
P.P., „A Randomised Placebo-Controlled Trial of the Efficacy of Beta-Sitosterol and Its
Glucoside as Adjuvants in the Treatment of Pulmonary Tuberculosis,“ International Journal of
Tuberculosis and Lung Disease, Bd. 1, Nr. 6, pp. 518-522, 1997.
[26] Kim J.A., Son J.H., Song S.B., Yang S.Y., Kim Y.H., „Sterols Isolated from Seeds of Panax Ginseng
and their Antiinflammatory Activities,“ Pharmacognosy Magazine, Bd. 9, pp. 182-185, 2013.
[27] Hartmann-Bouillon M.A., Benveniste P., „Sterol Biosynthetic Capacity of Purified Membrane
Fractions from Maize Coleoptiles,“ Phytochemistry, Bd. 17, p. 1037–1042, 1978.
[28] Warnecke D.C., Heinz, E., „Purification of a Membrane-Bound UDPGlucose: Sterol β-D-
Glucosyltransferase Based on Its Solubility in Diethyl Ether,“ Plant Physiol., Bd. 105, pp. 1067-
1073, 1994.
[29] Sinnott M.L., „Catalytic Mechanism of Enzymic Glycosyl Transfer,“ Chem. Rev., Bd. 90, pp.
1171-1202, 1990.

91
[30] Lairson L.L., Henrissat B., Davies G.J., Withers S.G., „Glycosyltransferases: Structures, Functions
and Mechanisms,“ Annual Review of Biochemistry, Bd. 77, pp. 521-555, 2008.
[31] Akiyama H., Kobayashi S., Hirabayashi Y., Murakami-Murofushi K., „Cholesterol Glucosylation
Is Catalyzed by Transglucosylation Reaction of Beta-Glucosidase,“ Biochemical and Biophysical
Research Communications, Bd. 1, Nr. 441, pp. 838-843, 2013.
[32] Deng S., Yu B., Jianming X., Yongzheng Hui, „Highly Efficient Glycosylation of Sapogenins,“ J.
Org. Chem., Nr. 64, pp. 7265-7266, 1999.
[33] Duivenvoorden B.A., Synthesis & Biological Applications of Glycosylated Iminosugars, Leiden
University, 2011.
[34] Mbadugha B.N.A., Menger F.M., „Sugar/Steroid/Sugar Conjugates: Sensitivity of Lipid Binding
to Sugar Structure,“ Organic Letters, Bd. 5, Nr. 22, pp. 4041-4044, 2003.
[35] Murui T., Wanaka K., „Measurement of Sterylglycosides by High Performance Liquid
Chromatography with 1-Anthroylnitrile Derivatives,“ Bioscience, Biotechnology, and
Biochemistry, Bd. 57, Nr. 4, pp. 614-617, 1993.
[36] Potocka A., Zirnowski J., „Metabolism of Conjugated Sterols in Eggplant. Part 1. UDP-Glucose:
Sterol Glucosyltransferase,“ Acta Biochimica Polonica, Bd. 55, pp. 127-134, 2008.
[37] Nomenclature Committee of the International Union of Biochemistry and Molecular Biology
(NC-IUBMB), „Enzyme Nomenclature. Recommendations. EC.3.2: Glycosylases,“
www.chem.qmul.ac.uk/iubmb/enzyme/EC3/2/, Aufgerufen am 19.02.2015.
[38] Cameleyre X., Bouchou A., Guibert A., Combes D., „Reverse Hydrolysis Reaction in Aqueous
Medium without any Cosolvent,“ Applied Biochemistry and Biotechnology, Bd. 62, Nr. 1, pp.
61-69, 1997.
[39] Bornscheuer U.T., Kazlauskas R.J., Hydrolases in Organic Synthesis, Weinheim: Wiley-VCH-
Verlag, 2006.
[40] Bush P.B., Grunwald C., „Steryl Glycoside Formation in Seedlings of Nicotiana tabacum L.,“
Plant Physiol., Bd. 53, Nr. 1, pp. 131-135, 1974.
[41] Galliard T., „Aspects of Lipid Metabolism in Higher Plants,“ Phytochemistry, Bd. 7, pp. 1907-
1914, 1968.
[42] Mudd D.J.B., „The Biosynthesis of Steryl Glucosides in Plants,“ Plant Physiol., Bd. 45, pp. 255-
262, 1970.
[43] Lepage, M., „Isolation and characterization of an esterified form of steryl glucoside,“ Journal of
lipid research, Bd. 5, pp. 587-592, 1964.
[44] Kiribuchi T., Mizunaga T., Funahashi S., „Separation of Soybeansterols by Florisil

92
Chromatography and Characterization of Acylated Steryl Glucoside,“ Agr. Biol. Chem., Bd. 30,
pp. 770-778, 1966.
[45] Nyström L., Schär A., Lampi A.M., „Steryl Glycosides and Acylated Steryl Glycosides in Plant
Foods Reflect Unique Sterol Patterns,“ European Journal of Lipid Science and Technology, Bd.
114, pp. 656-669, 2012.
[46] Kovganko N.V., Kashkan Z.N., „Sterol Glycosides and Acylglycosides,“ Chem. Nat. Compd., Bd.
35, pp. 479-497, 1999.
[47] Wertz P.W., Stover P.M., Abraham W., Downing D.T., „Lipids of Chicken Epidermis,“ J Lipid Res,
Bd. 27, p. 427–435, 1986.
[48] Parodi A.J., „Synthesis of Steryl Glucoside in Bakers Yeast,“ Acta Physiol. Lat. Am., Bd. 26, pp.
430-433, 1976.
[49] Ghannoum M.A., Janin G., Khamis, L., Radwan S.S., „Dimorphism-associated Variations in the
Lipid Composition of Candida albicans,“ J. Gen. Microbiol., Bd. 132, pp. 2367-2375, 1986.
[50] Misiak M., Kalinowska M., Wojciechowskie W., „Characterization of Acyllipid: Sterol Glucoside
Acyltransferase from Oat (Avena Sativa L.) Seedlings,“ Acta Biochim. Pol., Bd. 38, pp. 43-48,
1991.
[51] Frasch W., Grunwald C., „Acylated Steryl Glycoside Synthesis in Seedlings of Nicotiana
tabacum L.,“ Plant Physiol, Bd. 58, Nr. 6, pp. 744-748, 1976.
[52] Chandler J. E., Hornbeck F.G., Brown, G.I., „The Effect of Cold Flow Additives on Low
Temperature Operability of Diesel Fuels,“ SAE Technical Paper Series, Paper No. 922186, San
Francisco, 1992.
[53] Westbrook S.R., „Fuels for Land and Marine Diesel Engines and for Non-Aviation Gasturbines,“
Significance of Tests for Petroleum Products, pp. 63-81, Jan. 2003.
[54] Lee I., Mayfield, J.L., Pfalzgraf L. M., Solheim L., Bloomer S., „Processing and Producing
Biodiesel and Biodiesel Produced therefrom,“ US patent application 20070151146, 2006.
[55] Danzer M.F., Ely T.L., Kingery S.A., „Biodiesel Cold Filtration Process,“ US patent application
20070175091, 2007.
[56] Sholing U., Ruf F., Ortiz Niembro J.A., Vargas R.C., Bello J., „Process for Removing Steryl
Glucosides from Biodiesel,“ US Patent Appl. 20110154723, 2011.
[57] Tang H., De Guzman R.C., Salley O.S., Ng S.K.Y., „Comparing Process Efficiency in Reducing
Steryl Glucosides in Biodiesel,“ J Am Oil Chem Soc, Bd. 87, pp. 337-345, 2010.
[58] Kass M.D., Lewis S.A., Connaster R.M., „Method for Removing Precipitates in a Biofuel,“ US
Patent Appl.20110308933, 2011.

93
[59] Brask J., Nielsen P.M., „Enzymatic Removal of Steryl Glycosides in Fatty Acid Alkyl Esters,“ US
Patent Appl. 20120009659, 2012.
[60] Ringwald S.C., „Biodiesel Characterization in the QC Environment,“ in The 98th AOCS Annual
Meeting Abstracts. AOCS Press, Urbana 15, 2007.
[61] Menzelle H., Peiru S., Vetcher L., „Enzymatic Removal of Steryl Glycosides,“ US Patent Appl.
2013031769, 2013.
[62] Yahya A.R.M., Andersona W.A., Moo-Younga M., „Ester synthesis in lipase-catalyzed
reactions,“ Enzyme Microb. Technol., Bd. 23, Nr. 7-8, pp. 438-450, 1998.
[63] Paczkowski C., Arkadiusz M., Wlodkowski L., „Lipase Catalyzed Regioselective Synthesis of
Steryl (6´o-acyl)-glucosides,“ Biotechnol Lett, Bd. 29, pp. 1403-1408, 2007.
[64] Murui T., Siew Y.H., „Effect of Refining Process on the Content of Steryl Glycosides and
Alcohols in Palm Oil,“ Journal of JOCS, Bd. 46, pp. 683-686, 1997.
[65] Chui P.L., Patterson G.W., Fenner G.P., „Sterols of Bryophytes,“ Phytochemistry, Bd. 24, pp.
263-266, 1985.
[66] European Committee for Standardization 1999. ISO 12228 Bestimmung der individuellen und
der Gesamtsterine., Brüssel: European Committee for Standardization, 1999.
[67] Lorenz R.T., Haeckler K., Fenner G., Parks L.W., „Analysis of Steryl Esters,“ in Analysis of Sterols
and Other Biologically Significant Steroids, San Diego, Academic Press Inc., 1989, pp. 33-44.
[68] Phillips K.M., Ruggio D.M., Ashraf-Khorassani, M., „Analysis of Steryl Glucosides in Foods and
Dietary Supplements by Solid-Phase Extraction and Gas Chromatography,“ Journal of Food
Lipids, Bd. 12, pp. 124-140, 2005.
[69] Rontani, J.-F., Aubert C., „Trimethylsilyl Transfer during Electron Ionization Mass Spectral
Fragmentation of some ω-Hydroxycarboxylic and ω-Dicarboxylic Acid Trimethylsilyl Derivatives
and the Effect of Chain Length,“ Rapid Commun. Mass Spectrom, Bd. 18, pp. 1889-1895, 2004.
[70] Agilent Technologies, „Einsatzgebiete der LC/MS(MS). Was bietet die Massenspektrometrie
für den HPLC Anwender?,“
www.chem.agilent.com/Library/slidepresentation/Public/MSDetektion.pdf, Aufgerufen am
19.02.2015.
[71] Sugawara T., Miyazawa T., „Separation and Determination of Glycolipids from Edible Plant
Sources by High-Performance Liquid Chromatography and Evaporative Light-Scattering
Detection,“ Lipids, Bd. 34, pp. 1231-1237, 1999.
[72] Nes W.D., Yoniv Z., Heftmann E., „Translocation of Cholesterol from Leaves to Ripening Fruits
of Solanum Khasianum.,“ Phytochemistry, Nr. 21, pp. 581-583, 1982.

94
[73] Patterson G.W., „Chemical and Physical Methods in the Analysis of Plant Sterols,“ in
Isopentenoids in Plants: Biochemistry and Function, New York, Dekker, 1984, pp. 293-323.
[74] Moreau R.A., Scott K.M., Haas M.J., „The Identification and Quantification of Steryl Glucosides
in Precipitates from Commercial Biodiesel,“ J Am Oil Chem Soc, Bd. 85, pp. 761-770, 2008.
[75] Vioque E., „Spray Reagents for Thin-Layer Chromatography (TLC) and Paper Chromatography
(PC),“ in CRC Handbook of Chromatography. Vol. II. Lipids, M. H., Hrsg., Boca Raton, CRC Press,
1984, pp. 309-317.
[76] Freie Universität Berlin, „E-Learning Chemie-Chemie in Fragen und Antworten,“ www.e-
learning.chemie.fu-berlin.de/fragen/media/ggw_saeule.jpg?1339778913, Aufgerufen am
18.02.2014.
[77] Schwetlick K., Organikum. Organisch-Chemisches Grundpraktikum, 21 Hrsg., Weinheim: Wiley-
VCH-Verlag, 2001.
[78] Otto M., Analytische Chemie, 2 Hrsg., Weinheim: Wiley-VCH-Verlag, 2000.
[79] Hübschmann H.J., Handbook of GC/MS. Fundamentals and Applications., Weinheim: Wiley-
VCH-Verlag, 2009.
[80] „Gaschromatographie,“ Technik Lexikon.
www.techniklexikon.net/d/gaschromatographie/gaschromatographie.htm, Aufgerufen am
19.02.2015.
[81] Staufer D., „Das Chromatogramm,“ in Chromatogramme richtig integrieren und bewerten. Ein
Praxishandbuch für die HPLC und GC, Weinheim, Wiley-VCH-Verlag, 2008, pp. 3-53.
[82] Venn R.F., Principles and Practice of Bioanalysis, 2 Hrsg., Boca Raton: CRC Press, 2008.
[83] Sheffield Hallam University, „Gas Chromatography,“
teaching.shu.ac.uk/hwb/chemistry/tutorials/chrom/gaschrm.htm, Aufgerufen am 15.02.2015.
[84] De Hoffmann E., Stroobant V., Mass Spectrometry. Principles and Applications, Chichester:
John Wiley & Sons Ltd, 2002.
[85] ETH-Zürich, MS Service, „www.locms.ethz.ch/ionsource.html,“ Aufgerufen am 13.02.2015.
[86] Gross J.H., Mass Spectronomy. A Textbook, 2 Hrsg., Heidelberg: Springer, 2011.
[87] Banerjee S., Mazumdar S., „Electrospray Ionization Mass Spectrometry: A Technique to Access
the Information beyond the Molecular Weight of the Analyte,“ Int. J. Anal. Chem., pp. 1-40,
2012.
[88] Budzikiewicz H., Schäfer M., Massenspektrometrie. Eine Einführung, 6 Hrsg., Weinheim: Wiley-
VCH-Verlag, 2012.

95
[89] Zaikin V.G., Halket J.M., „Derivatization in Mass Spectrometry-8. Soft Ionization Mass
Spectrometry of Small Molecules,“ Europ. J. Mass Spectrom., Bd. 12, Nr. 2, pp. 79-115, 2006.
[90] Kos G., Krska R., „Separation and Detection Techniques for the Determination of Mycotoxins.
In: http://services.leatherheadfood.com/eman/FactSheet.aspx?ID=62,“ Aufgerufen am
14.02.2015.
[91] Brunnee C., „The Ideal Mass Analyzer: Fact or Fiction?,“ Int. J. Mass Spectrom., Bd. 76, Nr. 2,
pp. 125-237, 1987.
[92] Lohninger H., Fröhlich J., Mizaikoff B., Rosenberg E., „Flugzeit-Massenspektrometer,“ Teach/
Me. Instrumentelle Analytik. www.vias.org/tmanalytik_germ/hl_ms_tof.html, Aufgerufen am
14.02.2015.
[93] McMaster M.C., GC/MS. A Practical User´s Guide, Hoboken: John Wiley & Sons Inc., 2008.
[94] Universität Münster, Org. Chem. Institut., „Ionenanalyse im Quadrupol,“ www.uni-
muenster.de/Chemie.oc/service/mass/Methoden/Quadrupol/quadrupol.html, Aufgerufen am
14.02.2015.
[95] Friebolin H., Ein- und Zweidimensionale NMR-Spektroskopie. Eine Einführung, 5 Hrsg.,
Weinheim: Wiley-VCH-Verlag, 2013.
[96] Carey F., „Spectroscopy,“
www.mhhe.com/physsci/chemistry/carey/student/olc/ch13nmr.html, Aufgerufen am
15.02.2015.
[97] Badertscher M., Pretsch E., Seibl J., Simon W., Zenobi R., Amrein W., Oggenfuss P., Analytische
Chemie für Biologen und Pharmazeuten. Einführung in spektroskopische Methoden der
Strukturaufklärung organischer Verbindungen. Vorlesungsskript., Zürich: ETH-Zürich, 2011.
[98] Ault A., Techniques and Experiments for Organic Chemistry, 6 Hrsg., Sausalito: University
Science Books, 1998.
[99] Pieber B., Schober S., Goebl C., Mittelbach M., „Novel Sensitive Determination of Steryl
Glycosides in Biodiesel by Gas Chromatography-Mass Spectroscopy,“ J Chromatogr A, Bd.
1217, Nr. 42, pp. 6555-6561, 2010.
[100] Ravindranathan-Kartha K.P., Jennings H.J., „A Simplified, One-Pot Preparation of Aceto Bromo
Sugars from Reducing Sugars,“ J. Carboydr. Chem., Bd. 9, Nr. 5, pp. 777-781, 1990.
[101] Kunz H., Harreus A., „Glycosidsynthese mit 2,3,4,6-Tetra-O-pivaloyl-a-D-
glucopyranosylbromid,“ Liebigs Ann. d. Chem., Nr. 1, pp. 41-48, 1982.
[102] Zemplén G., Pacsu E., „Über die Verseifung acetylierter Zucker und verwandter Substanzen,“
Ber. Dtsch. Chem. Ges, Bd. 62, Nr. 6, pp. 1613-1618, 1929.

96
[103] Rotani J.-F., Aubert C., „Trimethylsilyl Transfer during Electron Ionization Mass Spectral
Fragmentation of some ω-Hydroxycarboxylic and ω-Dicarboxylic Acid Trimethylsilyl Derivatives
and the Effect of Chain Length,“ Rapid Commun. Mass Spectrom., Nr. 18, pp. 1889-1895, 2004.
[104] Grunwald C., Huang L.-S., „Analysis of Steryl Glycosids,“ in Analysis of Sterols and Other
Biologically Significant Steroids, San Diego, Academic Press Inc., 1989, pp. 49-58.
[105] Diekman, J., Thomson, J.B., Djerassi, C., „Mass Spectrometry in Structural and Stereochemical
Problems. CLV. Electron Impact Induced Fragmentations and Rearrangements of Some
Trimethylsilyl Ethers of Aliphatic Glycols and Related Compunds,“ Journal of Organic
Chemistry, Bd. 33, Nr. 6, pp. 2271-2284, 1968.
[106] Jäger S., Interne Kommunikation, University of Rosaria, 2013.
[107] Wibisino G., Darstellung von Terpenacyl-β-D-glucopyranosiden–Spaltung von
Benzylschutzgruppen mittels katalytischer Transferhydrogenolyse in Gegenwart
multifunktioneller Aglyca, Rheinischen Friedrich-Wilhelm-Universität Bonn, 2008.
[108] Schmidt R.R., Stumpp M., „Glycosylimidate, 8. Synthese von 1-Thioglycosiden,“ Liebigs
Annalen der Chemie, Bd. 7, pp. 1249-1256, 1983.
[109] Becker H.G.O., Domschke G., Organikum, Berlin: VEB Deutscher Verlag der Wissenschaften
Berlin, 1986.
[110] Kessler H., „Neues Verfahren zur Herstellung von 2,3,4,6-Tetra-O-benzyl-D-glucopyranose und
2,3,4,6-Tetra-O-allyl-D-glucopyranose,“ DE Patent: 19534366 A1, 2000.
[111] Sigma Aldrich, Dibenzyl ether, In:
www.sigmaaldrich.com/catalog/product/fluka/33630?lang=de®ion=AT, Aufgerufen am
21.02.2015.
[112] Forsyth J.C., Bellus D., Jacobsen E.N., Ley S.V., Noyori R., Regitz M., Reider P.J., Schaumann E.,
Shinkai I., Thomas E.J., Trost B.M., Science of Synthesis: Houben-Weyl Methods of Molecular
Transformations. Ethers, Stuttgart, New York: Georg Thieme Verlag, 2008.
[113] Sigma Aldrich, „Cholesterol β-D-glucoside,“
www.sigmaaldrich.com/catalog/product/sigma/28609?lang=de®ion=AT, Aufgerufen am
26.02.2015.
[114] Pieber B., Mittelbach M., Synthese und Analyse von Sterolglycosiden, IfC Graz, 2009.
[115] Davis R.A., Fettinger J.C., Gervay-Hague J., „Tandem Glycosyl Iodide Glycosylation and
Regioselective Enzymatic Acylation Affords 6-O-Tetradecanoyl-α-D-cholesterylglycosides,“ J
Org Chem, Bd. 79, Nr. 17, pp. 8447-8452, 2014.
[116] Czabany T., „Persönliches Gespräch,“ Institut für Biotechnologie und Bioprozesstechnik Graz,
06.02.2015.

97
[117] Davis R.A., Lin C.H., Gervay-Hague J., „Chemoenzymatic Synthesis of Cholesteryl-6-O-
tetradecanoyl-a-D-glucopyranoside: A Product of Host Cholesterol Efflux Promoted by
Helicobacter Pylori,“ Chem Commun, Bd. 48, Nr. 72, pp. 9083-9085, 2012.
[118] Bornscheuer U., Otto R., Schmid R.D., Syldatk C., Cao L., Verfahren zur selektiven Veresterung
von Polyolen, Patentschrift WO2000068408 A1, 2000.
[119] Nordblad M., Adlercreutz P., „Immobilisation Procedure and Reaction Conditions for Optimal
Performance of Candida Antarctica Lipase B in Transesterification and Hydrolysis,“ Biocatal.
and Biotransform., Bd. 31, Nr. 5, pp. 237-245, 2013.
[120] Kramer M., Cruz J.C., Pfromm P.H., Rezac M.E., Czermak P., „Enantioselective
Transesterification by Candida antarctica Lipase B Immobilized on Fumed Silica,“ Journal of
Biotechnology, Bd. 150, pp. 80-86, 2010.
[121] Uppenberg J., Hansen M.T., Patkar S., Jones T.A., „The Sequence, Crystal Structure
Determination and Refinement of Two Crystal Forms of Lipase B from Candida Antarctica,“
Structure, Bd. 2, Nr. 4, pp. 293-308, 1994.
[122] Yu Y., Lutz S., „Improved Triglyceride Transesterification by Circular Permuted Candida
Antarctica Lipase B.,“ Biotechnol Bioeng., Bd. 105, Nr. 1, pp. 44-50, 2010.
[123] Haupt J., Brankatschk G., Wilharm T., „Sterol Glucoside Content in Vegetable Oils as a Risk for
the Production of Biodiesel – Study of the Technological Chain Impact,“ OVID, Berlin, 2009.
[124] Pöhnlein M., Ulrich J., Kirschhöfer F., Nusser M., Muhle-Goll C., Kannengiesser B., Brenner-
Weiß G., Luy B., Liese A., Syldatk C., Hausmann R., „Lipase-Catalyzed Synthesis of Glucose-6-O-
hexanoate in Deep Eutectic Solvents,“ Eur. J. Lipid Sci. Technol., Bd. 117, pp. 161-166, 2015.
[125] Yahya A.R.M., William A.A., Moo-Young M., „Ester Synthesis in Lipase-Catalyzed Reactions,“
Enzyme and Microbial Technology, Bd. 23, Nr. 7-8, pp. 438-450, 1998.

98
8 Tabellenverzeichnis
Tabelle 1: Phytosterolgehalte in ausgewählten Lebensmitteln [10] ..................................................... 14
Tabelle 2: Analyse von SG und ASG in Pflanzenölen und Biodieselproben nach Lacoste et al. [1]. ..... 21
Tabelle 3: Probenvorbereitung für die Analyse am GC-MS .................................................................. 50
Tabelle 4: GC-MS Parameter für die Messung von Cholesterylglycosid ............................................... 51
Tabelle 5: GC-FID Parameter für die Messung von Cholesterylglycosid ............................................... 51
Tabelle 6: TOF-MS Parameter für die Messung von Cholesterylglycosid ............................................. 52
Tabelle 7: NMR-Parameter für durchgeführte Messungen .................................................................. 52
Tabelle 8: Zusammenfassung der Ergebnisse ....................................................................................... 76
Tabelle 9: Ergebnisse der gaschromatographischen Analyse ............................................................... 85
Tabelle 10: SG-Gehalt in Raps-Biodiesel ............................................................................................... 86
9 Abbildungsverzeichnis
Abbildung 1: Grundgerüst der Sterole .................................................................................................. 12
Abbildung 2: Strukturformel von Cholesterol ....................................................................................... 13
Abbildung 3: Strukturformeln von a) Sitosterol, b) Stigmasterol, c) Campesterol ............................... 13
Abbildung 4: Strukturformel von Ergosterol ......................................................................................... 15
Abbildung 5: Biosynthese von Stigmasterol-β-D-glucopyranosid ......................................................... 16
Abbildung 6: Mechanismus der Zemplén-Verseifung ........................................................................... 17
Abbildung 7: Mechanismus der Helferich-Synthese ............................................................................. 18
Abbildung 8: Strukturformel eines acylierten Sterylglycosids (ASG) .................................................... 20
Abbildung 9: Gehalt von SGen, ASGen sowie der Gesamtgehalt von ASGen + SGen in verschiedenen
Lebensmitteln bezogen auf das Frischgewicht (FG) (Modifiziert von [44]) .......................................... 20
Abbildung 10: Bildung von SGen aus ASGen durch Umesterung bei der Biodieselproduktion ............ 21
Abbildung 11: Schematische Darstellung der Säulenchromatographie [76] ........................................ 29
Abbildung 12: Schema eines Gaschromatographen. (Übernommen von [80]) .................................... 31
Abbildung 13: Pyrolyse und Ionisation im FID ....................................................................................... 32
Abbildung 14: Schematische Darstellung eines Flammenionisationsdetektors (Modifiziert von [83]) 32
Abbildung 15: Ionisationsmöglichkeiten für die Massenspektroskopie [86] ........................................ 33
Abbildung 16: Bildung von radikalischen Molekülkationen .................................................................. 34
Abbildung 17: Elektrospray-Ionisation (Modifiziert von [87]) ............................................................... 35
Abbildung 18: Schematische Darstellung der Chemischen Ionisation bei Atmosphärendruck.
(Modifiziert von [90]) ............................................................................................................................ 36

99
Abbildung 19: Schematische Darstellung eines Reflektron-TOFs (Modifiziert von [92]) ...................... 37
Abbildung 20: Schema eines Quadrupol-Analysators (Modifiziert von [94])........................................ 37
Abbildung 21: Schematische Darstellung eines Kernspinspektrometers (Modifiziert von [96]) .......... 38
Abbildung 22: Darstellung von β-D-Glucose-Pentaacetat mit Natriumacetat als Katalysator ............. 53
Abbildung 23: Umsetung von Glucosepentaacetat mit Bromwasserstoffsäure zur Herstellung von
Acetobromoglucose .............................................................................................................................. 53
Abbildung 24: Darstellung des tetraacetylierten Glucopyranosids mittels Koenigs-Knorr Methode ... 54
Abbildung 25: Darstellung von Cholesteryl-β-D-glucopyranosid durch Zemplén-Verseifung .............. 55
Abbildung 26: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mittels
Koenigs-Knorr-Methode ........................................................................................................................ 56
Abbildung 27: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid mit den Signalen der Silyl-
Fragmente sowie dem Cholesterol-Signal ............................................................................................. 57
Abbildung 28: TOF-Massensprektum von Cholesteryl-β-D-glucopyranosid ......................................... 57
Abbildung 29: Entstehende Fragmente bei der Ionisierung im GC-MS ................................................ 58
Abbildung 30: Basenkatalysierte Darstellung von Tetraacetylglucose aus Glucosepentaacetat ......... 58
Abbildung 31: Basenkatalysierte Darstellung von O-(2,3,4,6-tetra-O-acetyl-D-glucopyranosyl)-
trichloracetimidat aus Tetraacetylglucose ............................................................................................ 59
Abbildung 32: 13C NMR (CDCl3) (75 MHz)Spektrum von GTATH (2,3,4,6-tetra-O-acetyl-β-D-
glucopyranosyl)-trichloracetimidat ....................................................................................................... 60
Abbildung 33: 1H NMR (CDCl3) (300MHz) Spektrum von GTATH (2,3,4,6-tetra-O-acetyl-β-D-
glucopyranosyl)-trichloracetimidat ....................................................................................................... 61
Abbildung 34: TMSOTf-katalysierte Umsetzung zu Cholesteryl-Tetraacetylglucopyranosid................ 61
Abbildung 35: Darstellung von Cholesterylglucopyranosid durch Zemplén-Verseifung ...................... 62
Abbildung 36: TOF-Massenspektrum von Cholesteryl-β-D-glucopyranosid ......................................... 64
Abbildung 37: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mittels
Trichloracetimidat-Methode ................................................................................................................. 64
Abbildung 38: DEPT und 13C NMR (Pyridin) (75 MHz) Spektrum von Cholesteryl-β-D-glucopyranosid 65
Abbildung 39: Darstellung von Octa-O-Benzylsaccharose durch Williamson-Ethersynthese .............. 66
Abbildung 40: Hydrolytische Spaltung von Octabenzylsaccharose in das Glucose- und das
Fructosederivat ..................................................................................................................................... 67
Abbildung 41: TOF-Massenspektrum von Tetrabenzylglucose ............................................................. 68
Abbildung 42: 13C NMR (CDCl3 ) (75 MHz) Spektrum von 2,3,4,6-Tetra-O-Benzyl-D-glucopyranose ... 69
Abbildung 43: 1H NMR (CDCl3) (300 MHz) Spektrum von 2,3,4,6-Tetra-O-Benzyl-D-glucopyranose ... 69
Abbildung 44: Basenkatalysierte Darstellung von Tetrabenzyl-Glucopyranosyltrichloracetimidat aus
Tetrabenzylglucose ................................................................................................................................ 70

100
Abbildung 45: Darstellung des benzylierten Cholesterylglycosids durch nucleophile Substitution ..... 71
Abbildung 46: TOF-Massenspektrum des tetrabenzylierten Cholesterylglycosids ............................... 72
Abbildung 47: Benzylgruppenabspaltung durch katalytische Hydrierung ............................................ 72
Abbildung 48: DC-Benzylgrupppen-abspaltung .................................................................................... 73
Abbildung 49: Gaschromatogramm von Cholesteryl-β-D-glucopyranosid durch Darstellung mit
Saccharose als Edukt ............................................................................................................................. 74
Abbildung 50: Verfolgung der Debenzylierung mittels TOF-MS, nach 12, 40 und 52 Stunden ........... 74
Abbildung 51: Zusammenfassung der Ergebnisse der verschiedenen Synthesemethoden ................. 75
Abbildung 52: Darstellung von durch Veresterung von Cholesterylglycosid mittels Novozyme 435 ... 77
Abbildung 53: Enzymkinetik nach Michaelis-Menten. E=Enzym, S= Substrat, P=Produkt. .................. 77
Abbildung 54: Reaktionsmechanismus der Veresterung von Sterylglycosiden mit Palmitinsäure. Enz =
Enzym Novozyme 435 ........................................................................................................................... 78
Abbildung 55: GC-Massenspektrum von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid (links) sowie
Referenzspektrum (rechts) [114] .......................................................................................................... 79
Abbildung 56: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von
Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel .......................... 80
Abbildung 57: TOF-Massenspektrum aus dem Versuch der enzymatischen Darstellung von
Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel .......................... 81
Abbildung 58: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von
Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in Hexan + 1 mL Chloroform als Lösungsmittel ....... 82
Abbildung 59: Gaschromatogramm aus dem Versuch der enzymatischen Darstellung von
Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in THF/Pyridin 4:1 als Lösungsmittel ....................... 83
Abbildung 60: SIM-Gaschromatogramm von Rapsbiodiesel. Im markierten Bereich sind
Sterylglycoside ersichtlich. .................................................................................................................... 84
Abbildung 61: Fragmente der in den Biodieselproben enthaltenen Sterylglycoside: Campesterol.
Stigmasterol und Sitosterol. .................................................................................................................. 85
Abbildung 62: Massenspektren der detektierten Sterylglycoside. Links mit m/z 397,4 Sitosteryl-,
rechts mit m/z 383,3 Campesterylglycosid ........................................................................................... 85
Abbildung 63: Gaschromatogramm der Kristalle aus der Darstellung von Cholesteryl(6´-O-palmitoyl)-
β-D-glucopyranosid in reinem Hexan als Lösungsmittel ..................................................................... 101
Abbildung 64: Massenspektrum der Kristalle aus der Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-
glucopyranosid in reinem Hexan als Lösungsmittel ............................................................................ 101
Abbildung 65: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid Trichloracetimidatmethode
............................................................................................................................................................. 102
Abbildung 66: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid Saccharose als Edukt ...... 102
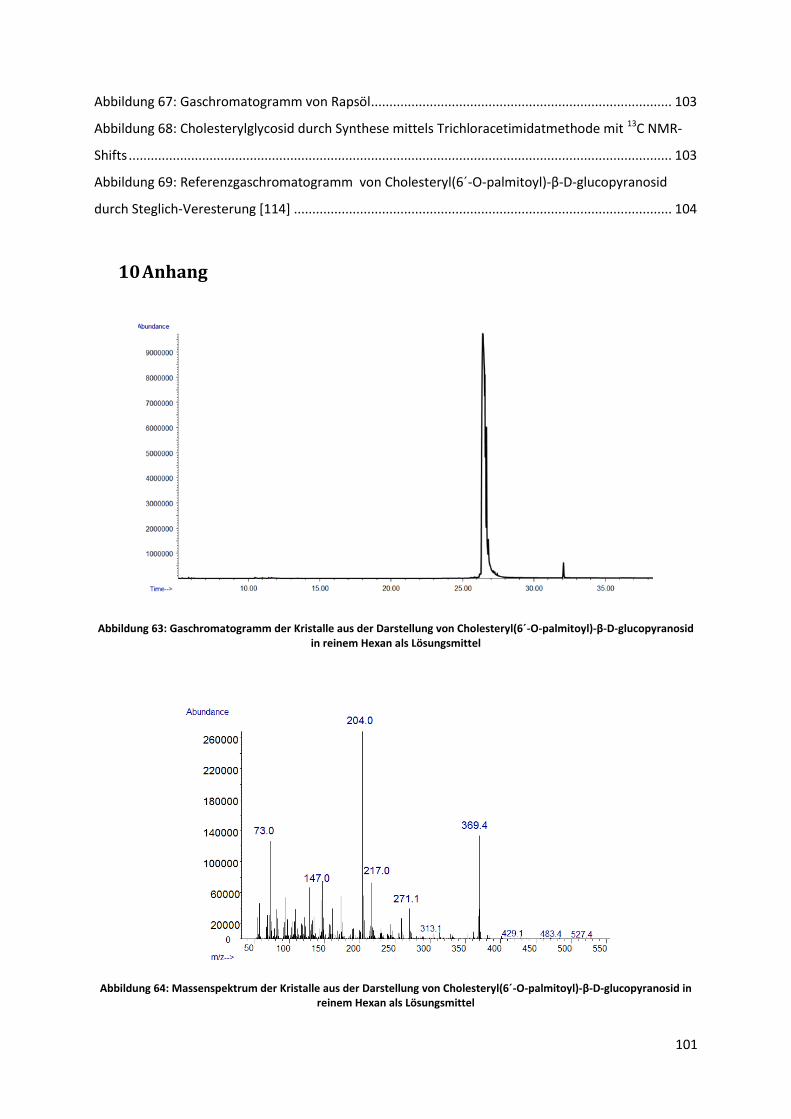
101
Abbildung 67: Gaschromatogramm von Rapsöl .................................................................................. 103
Abbildung 68: Cholesterylglycosid durch Synthese mittels Trichloracetimidatmethode mit 13C NMR-
Shifts .................................................................................................................................................... 103
Abbildung 69: Referenzgaschromatogramm von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid
durch Steglich-Veresterung [114] ....................................................................................................... 104
10 Anhang
Abbildung 63: Gaschromatogramm der Kristalle aus der Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel
Abbildung 64: Massenspektrum der Kristalle aus der Darstellung von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid in reinem Hexan als Lösungsmittel
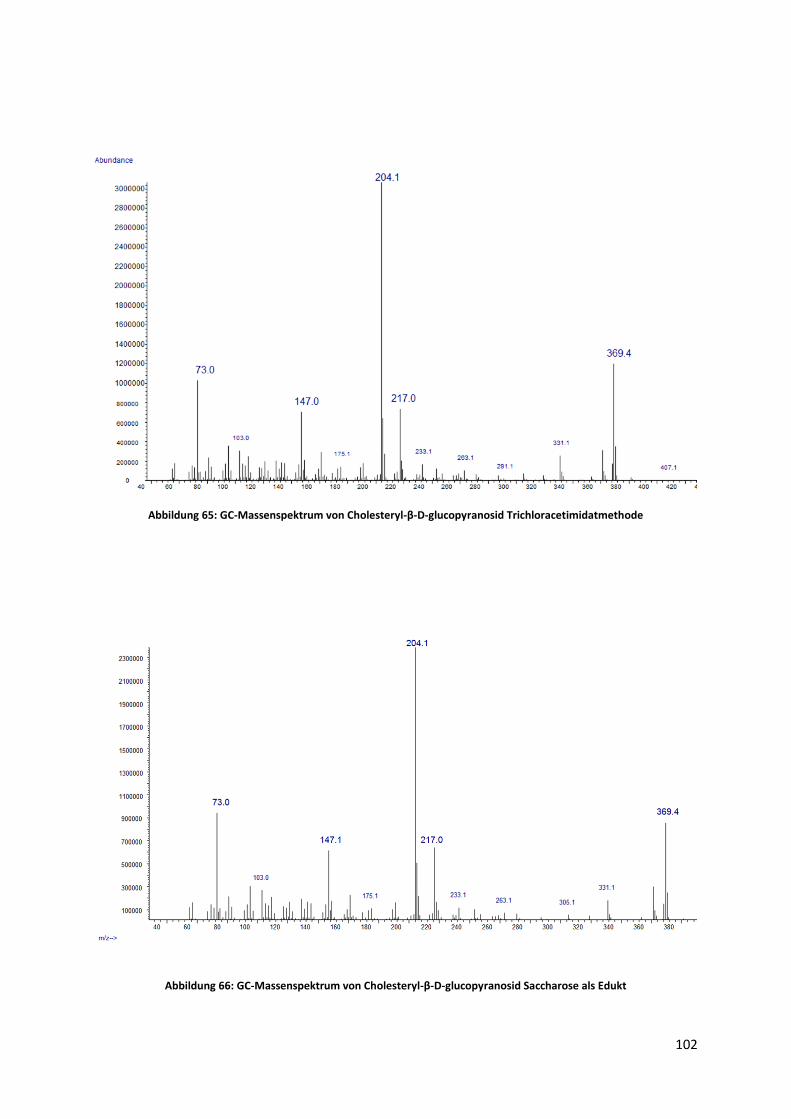
102
Abbildung 65: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid Trichloracetimidatmethode
Abbildung 66: GC-Massenspektrum von Cholesteryl-β-D-glucopyranosid Saccharose als Edukt
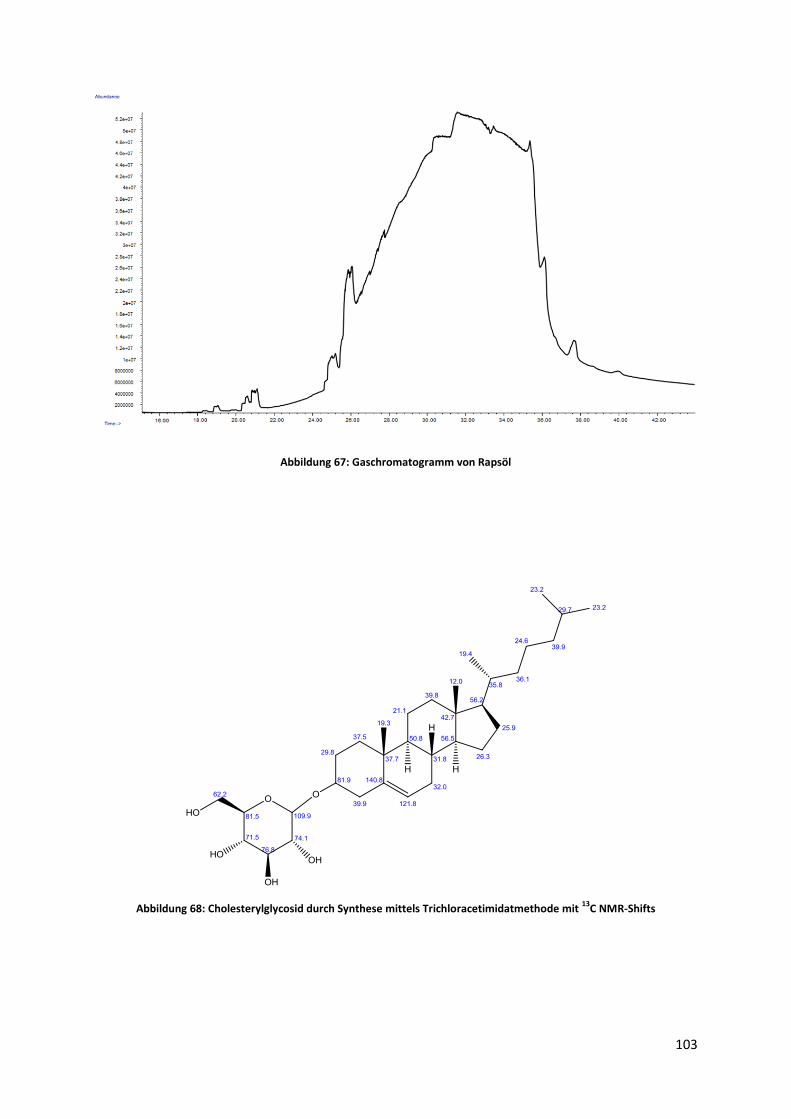
103
Abbildung 67: Gaschromatogramm von Rapsöl
Abbildung 68: Cholesterylglycosid durch Synthese mittels Trichloracetimidatmethode mit 13
C NMR-Shifts
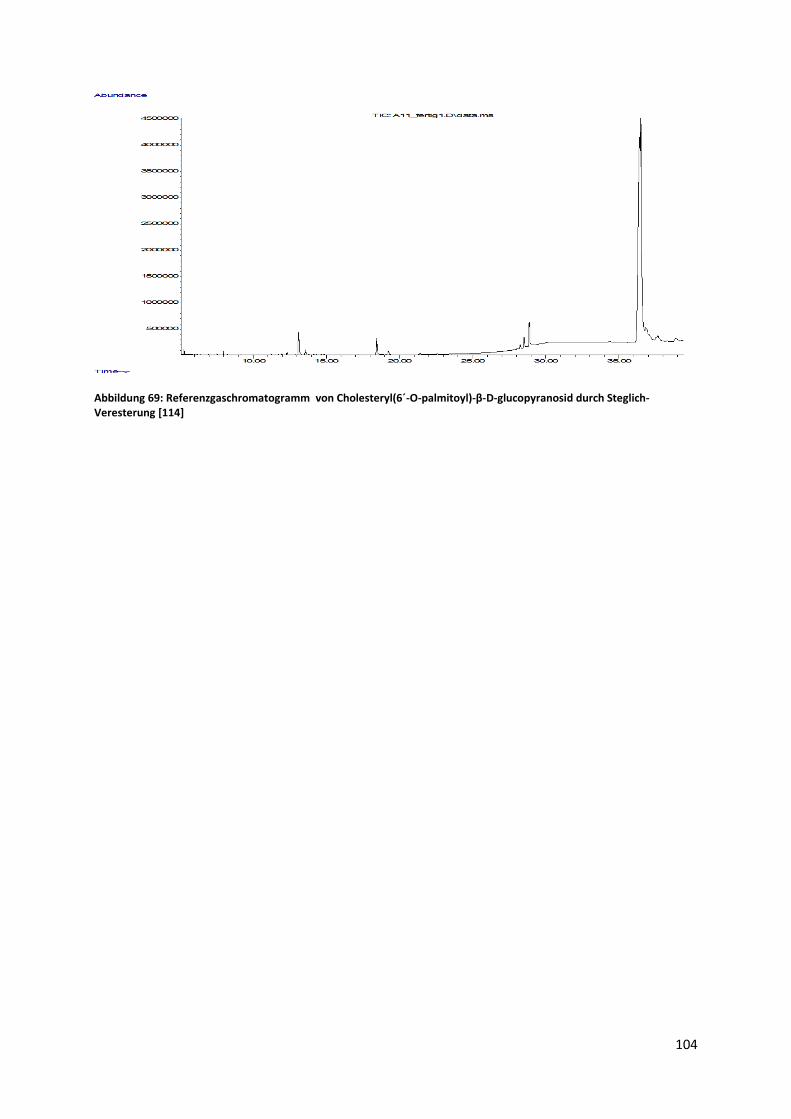
104
Abbildung 69: Referenzgaschromatogramm von Cholesteryl(6´-O-palmitoyl)-β-D-glucopyranosid durch Steglich-Veresterung [114]
















![Synthese neuer Benzo[c]phenanthridin-Derivate und deren ... · Synthese neuer Benzo[c]phenanthridin-Derivate . und deren Stickstoff-Analoga als potentielle Zytostatika . Dissertation](https://static.fdokument.com/doc/165x107/5e20f36f5b31f87be65362b3/synthese-neuer-benzocphenanthridin-derivate-und-deren-synthese-neuer-benzocphenanthridin-derivate.jpg)

