







Sprachen
Seiten
Rechtliche


Studien zur Reaktivität der Vinylcarben-Komplexe
in der Metathese Reaktion.
Darstellung des Wolfram-Allenylidenkomplexes
Inaugural-Dissertation zur Erlangung des Doktorgrades der Naturwissenschaftlichen Fachbereiche
(Fachbereich Biologie und Chemie) der Justus-Liebig-Universität Giessen
vorgelegt von
Parham Rooshenas aus Teheran
Giessen 2005

Dekan: Prof. Dr. Jürgen Mayer
1. Berichterstatter: Prof. Dr. Junes Ipaktschi
2. Berichterstatter: Prof. Dr. Rainer Askani
Tag der mündlichen Prüfung: 10.Feb.2006

Die vorliegende Arbeit entstand auf Anregung und unter Leitung von Herrn Prof. Dr. Ipaktschi
am Institut für Organische Chemie der Justus-Liebig-Universität Giessen in der Zeit von April
2001 bis Oktober 2005.
Mein herzlicher Dank gilt meinem geschätzten Doktorvater, Herrn Prof. Dr. Ipaktschi für die
interessante und herausfordernde Themenstellung, für seinen wissenschaftlichen Rat, sein
stetes Interesse am Fortgang der Arbeit und Vertiefung meiner chemischen Kenntnisse, die stets
vorhandene Diskussionsbereitschaft und die ausgezeichneten Arbeitsbedingungen.
Den analytischen Abteilungen des Institutes für Organische Chemie der Justus-Liebig-Universität
danke ich für die prompte Erledigung der Analysen. Besonders Frau Dr. H. Hausmann und Herr
Dr. H. P. Reisenhauer standen mir hier bei diversen NMR und IR-spektroskopischen
Strukturaufklärungen sehr hilfreich zur Seite.
Insbesondere bin ich Herrn Dipl. Chem. A. Dülmer, Dr. M. Serafin und G. Koch, Institut für
Anorganische und Analytische Chemie der Justus-Liebig-Universität, für die Anfertigung der
Röntgenstrukturanalysen.
Außerdem danke ich allen Mitarbeitern der zentralen Einrichtung des Instituts für Organische
Chemie herzlich für die Hilfe bei der Durchführung dieser Arbeit.
Allen Mitgliedern unseres Arbeitskreises danke ich für ein angenehmes Arbeitsklima.
Ein ganz besonderer Dank gilt meiner Frau Shaghayegh für ihre immerwährende Unterstützung
und ihre Hilfe im Labor, sowie bei der Korrektur dieses Textes.

Teile dieser Arbeit wurden bereits veröffentlicht:
• Ipaktschi, Junes; Rooshenas, Parham; Duelmer, Ansgar „Synthesis of η2-Allene
Complexes by the Reaction of η1-Vinylidene Tungsten Complexes with Diazoalkanes“
Eur. J. Inorg. Chem. 2006, xxxx.
• Ipaktschi, Junes; Rooshenas, Parham; Duelmer, Ansgar „Reaction of Tungsten
Vinylcarbene Complexes with Enamines” Organometallics, 2005, 24, 6239.
• Ipaktschi, Junes; Rooshenas, Parham; Klotzbach, Thomas; Duelmer, Ansgar;
Hueseynova, Elmira „Synthesis of Bridged Oxo-Tungsten Complexes“ Organometallics
2005, 24, 1351.

Meinen Eltern und
Shaghayegh

Inhaltsverzeichnis
A Einleitung
1 Organometallchemie 1
1.2 Carbenkomplexe 3
1.3 Vinylidenkomplexe 5
2 Problemstellung 10
B Allgemeiner Teil
B1 Die Reaktion von den Vinylcarbenkomplexen des Wolframs mit Enaminen
1.1 Vormerkungen 12
1.2 Darstellung der Vinylcarben Komplexe 16a-b 14
1.3 Die Umsetzung der Wolfram-Carben-Komplexe 16a-b mit Enaminen 14a-b.
Darstellung der Wolfram-η1-Acyl-Komplexe 18, 19 und 20 15
1.4 Darstellung von dem η2-Alken-Komplex 21. Die Diskussion spektroskopischer
Daten der Verbindung 21 21
1.5 Schlussfolgerung 25
B2 Darstellung des η1-Allenylidenkomplexes des Wolframs
2.1 Vormerkungen 26
2.1 Versuche zur Darstellung eines η1-Allenylidenkomplexes des Wolframs 27
2.2 Die nucleophile Addition an dem Allenylidenkomplex 26 32
2.3 Schlussfolgerung 36
B3 Die Reaktion von Diazoalkanen mit dem η1-Vinylidenkomplex des
Wolframs: Darstellung der η2-Allenkomplexe des Wolframs
3.1 Vormerkungen 39
3.2 Umsetzung vom Vinylidenkomplex 10 mit Diazomethan. Darstellung
und Diskussion der spektroskopischen Daten des η2 -Allenkomplexes 31 39
3.3 Darstellung der η2-Allenkomplexe 32 und 33. 46
3.4 Schlussfolgerung 49

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs
4.1 Vormerkungen 50
4.2 Darstellung von Phosphorkomplexen des Wolframs 11a-c 50
4.3 Die Hydrolysereaktionen der Phosphorkomplexe des Wolframs 11a-c 51
4.4 Schlussfolgerung 56
C Zusammenfassung 57
D Experimenteller Teil 62
E Daten der Röntgen-Kristallstrukturanalyse 101
F Spektren 133
G Literaturverzeichnis 162

Abkürzungsverzeichnis
Å Angström (10-10m) arom. aromatisch Ar Aryl ber. berechnet Bu Butyl n-BuLi n-Butyllithium bzw. beziehungsweise Cp Cyclopentadienyl °C Grad Celsius d Dublett DC Dünnschicht-Chromatographie dd Dublett von Dublett
δ chemische Verschiebung in ppm
E Elektrophil
ηX (X = 1 bis 8) Haptizität
Fa. Firma FID Free Induction Decay FW Formular Weight gef. gefunden Hz Hertz IPDS Image Plate Diffraction System IR Infrarot konz. konzentriert L Ligand Lsg. Lösung m Multiplett mm Millimeter mmol Millimol M Metall Me Methyl MS Massenspektrum
μ Absorbtionskoeffizient

μX (X = 2, 3) Art der Verbrückung
NMR kernmagnetische Resonanz (Nuclear Magnetic Resonance)
Nuc Nucleophil ν~ Wellenzahl
Ph Phenylgruppe ppm parts per million R Rest RT Raumtemperatur
ρ Dichte
s Singulett Smp. Schmelzpunkt TMS Tetramethylsilan THF Tetrahydrofuran THF-d8 Deuteriertes THF z. B. zum Beispiel Zers. Zersetzung

A Einleitung 1
A Einleitung
1 Metallorganische Chemie
Metallorganische Verbindungen (Organometallverbindungen) zeichnen sich durch
eine mehr oder weniger polare Bindung Mδ+-Cδ- zwischen Metall und Kohlenstoff aus. Zu den
Metallen werden in diesem Fall auch die Halbmetalle und Metalle B, Si, Ge, (P), As, Se, Te)
hinzu gezählt, daher spricht man auch von Elementorganischer Chemie.
Die erste metallorganische Verbindung wurde im Jahre 1827 von dem dänischen
Pharmazeuten Zeise entdeckt.[1] Nach 40 Jahren wurde der erste Metall-Carbonylkomplex, ein
Platinium-Chlorid-Komplex dargestellt.[2] Seit dem hat sich die Metallorganik zu einem
selbständigen Zweig in der Chemie entwickelt. Dieser rasche Fortschritt der Metallorganik ist
vor allem auf den detaillierten Strukturinformationen zurückzuführen, die durch verbesserte
und entwickelte spektroskopischen Methoden, wie Röntgenstrukturanalyse und IR- sowie
NMR-Spektroskopie, erzielt wurden. Die Entdeckung des Ferrocens durch Realy und Pauson
sowie Miller kann als ein Wendepunkt in dieser Entwicklung bezeichnet werden.[3] Die
Forschungen von Wilkinson, Rosenblum und Woodward und E.O.Fischer führten zur
Aufklärung der Sandwichstruktur des Ferrocens und Erläuterung der Benzolähnlichen
elektrophilen Substitutionsreaktionen dieser Verbindung.[4] 1964 berichteten E.O.Fischer und
Massböl die Darstellung des ersten Metall-Carbenkomplexes.[5] Die Leistungen von
E.O.Fischer und Wilkinson brachte ihnen im Jahre 1973 den Nobelpreis. Die
metallorganischen Verbindungen haben auch von Anfang an eine unentbehrliche Stellung in
der industriellen Chemie genommen. Unter anderem kann man folgende Prozesse benennen:
Das Fischer-Tropsch-Verfahren zur Herstellung von flüssigen Alkanen, Olefinen, Methanol
und höheren Alkoholen aus Synthesegas (CO und H2);[6] die Oxosynthese für die Darstellung
von Aldehyden durch Hydroformylierung von Olefinen mit Synthesegas;[7] das Ziegler-Natta-
Verfahren für Polymerisation von Alkenen;[8] das Wacker-Verfahren zur Darstellung von
Acetaldehyd und Vinylacetat;[9] das Monsanto-Verfahren zur Synthese von Essigsäure durch
Carbonylierung von Methanol;[10] der Halcon-Prozeß zur Epoxidierung von Olefinen;[11] das
Mond Verfahren zur Herstellung von hochreinem Nickel[12] und die Synthese von L-Dopa,
ein Medikament gegen Parkinson Krankheit, in der Firma Monsanto.[13]
Bindungsmodelle
Die Übergangsmetalle in ihren Komplexen weisen partiell besetzte d-Orbitale und
hingegen leere s- und p-Orbitale auf. Im Gegensatz dazu besitzen die meisten Liganden
besetzte spn-Hybridorbitale und manchmal unbesetzte antibindende π*-Orbitale. Die d-
Orbitale der Metalle haben oftmals die gleiche Symmetrie und eine vergleichbare Energie wie

A Einleitung 2
die π*-Orbitale der Liganden. Übergangsmetalle können dementsprechend zwei Arten der
Bindungen eingehen: σ-Donor-Bindungen, und π-Acceptor-Bindungen.
σ-Donor-Bindungen entstehen durch die Überlappung von besetzten spn-
Hybridorbitalen oder bindenden π-Orbitalen mit dem unbesetzten dsp-Hybridorbital des
Metallatoms. Liganden wie der Carbonyl-Ligand erhöhen die Elektrondichte am
Metallzentrum. Allerdings der Carbonyl-Ligand ist in der Lage durch eine π-Rückbindung die
Elektrondichte vom Metallzentrum zu entziehen (Abbildung 1). Dieser Konzept des σ-
Donor/π-Acceptor-Synergismus stellt einen wichtigen Ansatz zur Diskussion der Struktur-
und Bindungsverhältnisse in den metallorganischen Verbindungen.
bbildung 1. Die MO Darstellung der Bindungen in M=C=O.
π-Acceptorliganden in zwei Gruppen
unterte
A
Bezüglich der π-Rückbindungen können die
ilt werden: (a) die longitudinale (endgebundene) Acceptoren wie Kohlenmonoxid oder
Vinyliden-Ligand und (b) perpendikulare (seitengebunden) Acceptoren wie Alkene und
Alkine (Abbildung 2).

A Einleitung 3
M+
+
_
_ M+
+
_
_+ C O+
+
+_
_
C
C+
+
+
_
+__
_
+
endgebunden seitengebunden
unbesetztesdspn-Orbital
Das freie Elektronenpaaram Carbonyl-Kohlenstoffdient als σ-Donor
besetztesMetall-d-Orbital
leeres COπ*-Orbital unbesetztes
dsp-Orbital
besetztesMetall-d-Orbital
leeres Olefinπ*-Orbital
besetztes Olefin π-Orbitaldient als σ-Donor
Abbildung 2. Die zwei Arten der π-Rückbindung bei π-Acceptor-Liganden.
Aufgrund der Vielzahl der metallorganischen Verbindungsarten werden im Folgenden
nur die für diese Arbeit wichtige Komplexklassen, kurz besprochen.
1.2 Carbenkomplexe
Carbenkomplexe werden mit einer formalen Metall-Kohlenstoff-Doppelbindung
gekennzeichnet. Sie werden grob in zwei Gruppen unterteilt, die unterschiedliche
Polarisierungen der M=C Doppelbindung aufweisen. So differenziert man zwischen den
elektrophilen Heteroatom-stabilisierten Fischer-Carbenen und den nucleophilen
Alkylidenkomplexen, die auch Schrock-Carbene genannt werden.
Der erste Heteroatom-stabilisierte Metall-Carben-Komplex 1 wurde durch die
Umsetzung von Phenyllithium mit dem Wolframhexacarbonyl und nachfolgende Behandlung
des entstandenen Acylanions mit Diazomethan dargestellt (Abbildung 3).[5] Es folgten danach
Veröffentlichungen über Hunderte von Verbindungen, die einen Heteroatom-stabilisierten
(gewöhnlich ein N oder O) Carben-Liganden trugen.[14]
W(CO)6 PhLi (CO)5WO
Ph Li
+ CH2N2(CO)5W
Ph
OMe+
1 Abbildung 3.Die erste Darstellung eines Metall-Carbenkomplexes.

A Einleitung 4
Die Metallatome in den Fischer-Carbenen sind normalerweise elektronreich und in
niederen Oxidationsformen (mitte bis zu späten Übergangsmetalle), und enthalten π-
Accerptor-Liganden. Es wird angenommen, dass das Heteroatom in solchen Komplexen eine
positive Ladung am Carben-Kohlenstoffatom stabilisiert, weshalb sie auch als „elektrophile
Carbene“ charakterisiert wurden. Die M=C Bindung ist darin mit einer negativen Teilladung
(δ¯) am Metallzentrum und einer positive Teilladung (δ+) am Carben-Kohlenstoffatom
polarisiert. Es ist angemessen, ein Carben-Ligand, der eins oder zwei an das Carben-
Kohlenstoffatom gebundene Heteroatome enthält, als neutral mit einer Ordnung der Metall-
Kohlenstoff-Bindung zwischen eins und zwei anzusehen. Das MO-Bild des Carben-Liganden
verdeutlicht diese Überlegung; ein freies Elektronpaar wird vom Singulett-Carben zum leeren
d-Orbital des Metallatoms gegeben und gleichzeitig wird die Ladungsdichte von einem
gefüllten d-Orbitals des Metalls zu dem leeren pz-Orbital des Carben-Liganden
zurückgeflossen, wobei entsprechend der Resonanzstruktur 2c, das freie Elektronpaar auf dem
Heteroatom auch dieses leere pz-Orbital beansprucht (Abbildung 4).[15] Die Bedeutung dieser
Resonanzstruktur (2c) in der Beschreibung von den Bindungsverhältnissen in dem Carben-
Liganden wird von den Daten der Röntgenstrukturanalyse bestätigt. Die M=C Bindungslänge
in diesen Komplexen ist langer als eine normale Metall-Kohlenstoff-Doppelbindung und
kürzer als einfache Metall-Kohlenstoff-Bindung.[16]
M C R
O R'
[M] C
OR'
R[M] C
OR'
R[M] C
OR'
R
+
+_ _
2a 2b 2c Abbildung 4. MO Bild und Resonanzstrukturen eines Fischer-Carbenkomplexes.
Die Carbenkomplexe werden hauptsächlich mit der vom E.O.Fischer entwickelten
Methode dargestellt.[5] Die Umsetzung der entsprechenden Carbonylkomplexe mit
lithiumorganylen oder Gringard-Reagenzien führt zu dem Acylkomplex, der dann
anschließend an dem Sauerstoff alkyliert wird (Abbildung 5).[17]

A Einleitung 5
Fe(CO)5PhLi (CO)4Fe
O
Ph Li
+(CO)4Fe
Ph
OEtEtOSO2F+
3 Abbildung 5. Darstellung des Eisen-Carbenkomplexes 3.
Eine alternative Darstellung für die Carbenkomplexe ergibt sich aus der Addition
eines Alkohols an den kationischen Vinylidenkomplexen (Abbildung 6).[18]
Fe
Ph3PCO
Ph Fe+
Ph3PCO H
PhFe
+
Ph3PCO OMe
PhMeOHH+
4 Abbildung 6. Darstellung des Eisen-Carbenkomplexes 4 aus einem Alkinylkomplex.
Über die Anwendungen der Fischer-Carbenen in der Synthese, sind mehrere
Übersichtsartikeln erschienen.[19] Eine der ältesten, synthetisch genützten Reaktionen dieser
Komplexen ist die Cyclopropanierung elektrophiler Olefine.[20] Eine weitere thermische
Reaktion der Fischer-Carbenen ist die Dötz-Reaktion, bei der α,β-ungesättigte Alkoxycarben-
Komplexe mit Alkinen zu Hydrochinonderivaten umgesetzt werden.[21]
Die nucleophilen Carbenkomplexe wurden zum ersten Mal von R.R.Schrock
dargestellt.[22] Das Carben-Kohlenstoffatom besitzt einen oder zwei Alkylsubstituenten. Sie
werden als Carbenkomplexe des Schrock-Typs oder auch als Alkylidenkomplexe bezeichnet.
Die hohe Oxidationsstufe des Metallatoms in diesen Komplexen hat die Nucleophilie des
Carben-Kohlenstoffatoms zur Folge. Diese Komplexe spielen eine große Rolle bei den
katalytischen Metathese Reaktionen (Siehe Abschnitt B1).
1.3 Vinylidenkomplexe
Die Vinylidenkomplexe mit der allgemeinen Formel LnM=C=CRR', sind mit den
Carbenkomplexen eng verwandt. Die höhere thermodynamische Stabilität dieser Komplexe
ist auf ihren ausgezeichneten π-Acceptor-Eigenschaften zurückzuführen, denn außer π*-
Orbital der C=C-Bindung kann sich auch das freie p-Orbital am Cα-Atom an der Rückbindung
zu dem Metallatom beteiligen.[23] Ähnlich wie die Carbenkomplexe, weist die M=C-Bindung
eine Bindungsordnung zwischen eins und zwei auf (Abbildung 7). Die C=C-Bindungslänge
liegt zwischen 1.25 und 1.41 Å und entspricht einer formalen Bindungsordnung zwischen

A Einleitung 6
zwei und drei Die Ebene des Vinyliden-Fragmentes ist perpendikular zur Ebene des
Metallkomplexes.
[M] C CR
R[M] C
+C
R
R Abbildung 7. Polarisierung der M=C-Bindung in dem Vinylidenkomplex.
Die Berechnungen ergeben eine Rotationsbarriere für die Komplexe des Typs [(η5-
C5H5)(M=C=CR2)(L2)] in der Größenordnung von 15 Kcal/mol-1.[24] Unterscheiden sich die
beiden Substituten des Vinyliden Liganden, entstehen durch gehinderte Rotation um die M–
C–C-Achse, zwei Rotationsisomere; beispielsweise beträgt diese Rotationsbarriere für den
Komplex [(η5-Cp)(CO)(NO)Mo=C=C(Me)(Ph)] 20.5 (±0.3) kcal/mol.[25]
Aus fast allen Übergangsmetallen sind bis jetzt Vinylidenkomplexe dargestellt
worden.[26] King und Saran berichteten die Darstellung des ersten Vinylidenkomplexes 6,
durch die Umlagerung des Chloratoms von einem α-Chloroalkenyl-Liganden in der
Verbindung 5 auf das Metallzentrum (Abbildung 8).[27]
Mo
COOC
COCl
CN
CNPPh3
Mo
CN
CNPh3P
Ph3PClRückfluß in Benzol
5 6 Abbildung 8. Darstellung des ersten Vinylidenkomplexes 6.
Es sind mehrere Methoden für die Synthese der Vinylidenkomplexe in der Literatur
vorgestellt worden. Zu ihnen gehören: (a) die Deoxygenierung von der Metall-Acylkomplexe
durch das Einwirken von (CF3SO2)2O,[28] (b) Deprotonierung von Metall-Carbinkomplexen
(LnM≡C−CHR'R),[29] (c) Addition von verschiedenen Elektrophilen an dem β-
Kohlenstoffatom eines Metall-Alkinylkomplexes wie Alkylgruppen,[30] und Halogenen.[31]
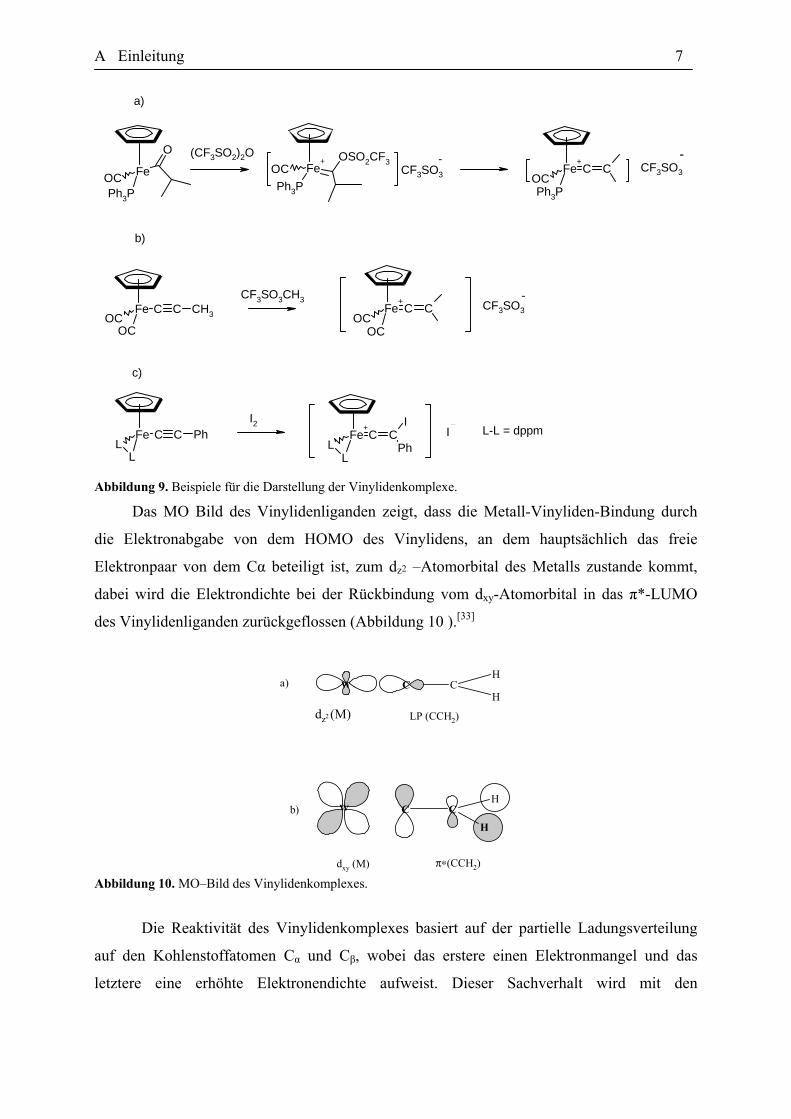
Mancher dieser Reaktionen sind in der Abbildung 9 präsentiert worden. Allerdings stellt die
Isomerisierung eines η2-Alkinkomplexes zum Vinylidenkomplexes eine wichtige Route für
die Synthese dieser Verbindungen dar.[32]
A Einleitung 7
Fe+ OSO2CF3
Ph3POC
Fe
+C C
Ph3POC
Fe C C
OCOC
CH3 Fe+C C
OCOC
CF3SO3CH3
Fe C C
LL
Ph Fe+C C
I
LL Ph
I2I
Fe
Ph3POC
O (CF3SO2)2OCF3SO3
-CF3SO3
-
a)
b)
CF3SO3
c)
L-L = dppm
-
Abbildung 9. Beispiele für die Darstellung der Vinylidenkomplexe.
Das MO Bild des Vinylidenliganden zeigt, dass die Metall-Vinyliden-Bindung durch
die Elektronabgabe von dem HOMO des Vinylidens, an dem hauptsächlich das freie
Elektronpaar von dem Cα beteiligt ist, zum dz2 –Atomorbital des Metalls zustande kommt,
dabei wird die Elektrondichte bei der Rückbindung vom dxy-Atomorbital in das π*-LUMO
des Vinylidenliganden zurückgeflossen (Abbildung 10 ).[33]
C CH
HW
W C CH
H
dz2 (M) LP (CCH2)
dxy (M) π∗(CCH2)
a)
b)
Abbildung 10. MO–Bild des Vinylidenkomplexes.
Die Reaktivität des Vinylidenkomplexes basiert auf der partielle Ladungsverteilung
auf den Kohlenstoffatomen Cα und Cβ, wobei das erstere einen Elektronmangel und das
letztere eine erhöhte Elektronendichte aufweist. Dieser Sachverhalt wird mit den

A Einleitung 8
entsprechenden Resonanzstrukturen veranschaulicht und auch mit theoretischen
Berechnungen belegt (Abbildung 11).[34]
M=C=CR'
RM-C=C
R'
R+ _
Abbildung 11. Mesomere Strukturen für einen Metall-Vinyliden-Komplex. Dieses Muster widerspiegelt sich in der Reaktion des Vinylidenkomplexes mit den
Nucleophilen wie Amine oder Phosphinchloride (Abbildung 12).[35, 36]
W C CH
NC
O
O
NH
W CC
H
NC
O
OH
N
W C CH
C6H5N
CO
OPCl
C6H5
C6H5
W CC H
NCl
O P C6H5
C6H5
C6H5
+
+
7
8
Abbildung 12. Reaktion der Nucleophilen mit den Vinylidenkomplexen 7, 8.
In dieser Arbeit wurden die Vinylidenkomplexe durch die Umsetzung der
entsprechenden Lithiumacetylide mit dem [(Cp)W(CO)3(NO)] (9) in THF bei –30 ºC zu dem.
grünen Anion und anschließende Protonierung dargestellt. Die Darstellung des
Vinylidenkomplexes 10 ist in der Abbildung 13 präsentiert.
W
NCO
O
CO
C C(CH3)3Si Li W C
NC
OO
C Si(CH3)3 H
W CNC
O O
CHNaHCO3/H2O
Li+
+
9 10 Abbildung 13. Darstellung des Vinylidenkomplexes 10.
Im Bereich von ν = 2000 cm-1 zeigen die IR-Spektren der Vinylidenkomplexe 7, 8
und 10 die charakteristischen Valenzschwingung für CO- und im Bereich von 1640 cm-1 die
für NO-Liganden. Bei 1620 cm-1 erscheint eine Bande für die Vinyliden-C=C-Schwingung,
sofern sie nicht durch die im nahen Bereich liegende NO-Schwingungsbande verdeckt ist. Die

A Einleitung 9
1H- und 13C-NMR-Spektren weisen doppelte Signalsätze für die Vinylidenkomplexe 7 und 8
auf, das auf dem Vorhandensein von zwei Rotameren zurückzuführen ist. Das
Produktverhältnis dieser Isomere lässt sich aus den Integralen im 1H-NMR-Spektrum
bestimmen. In den 13C-NMR-Spektren der Verbindungen 7, 8 und 10 sind die extreme
Tieffeldverschiebung des α-Kohlenstoffatoms charakteristisch. Sie liegen im Bereich von
δ 340 ppm. Für die β-Kohlenstoffatome findet man die Signale um 130 ppm.

A Einleitung 10
2 Problemstellung:
Folgende Aspekte der Chemie der Vinylidenkomplexe wurden im Rahmen der
vorliegenden Dissertation untersucht.
a) Auf der Suche nach neuen Katalysatoren für die Metathese Reaktion, sollte durch
die Umsetzung von Vinylidenkomplexe mit Enaminen die entsprechende Vinylcarben-
Komplexe hergestellt und auf potentielle Aktivität bei der Metathesereaktion studiert werden.
Die Metathese hat sich zu einer der sehr wirkungsvollen synthetischen Methoden in
der organischen und metallorganischen Chemie entwickelt, so dass die Suche nach neuen
Katalysatoren für diese Reaktion ein großes Interesse bei den Chemikern geweckt hat. Ein
erheblicher Anteil an diesen Katalysatoren gehört den Carbenkomplexen. In der
Arbeitsgruppe Ipaktschi wurde eine Methode zur Darstellung von Vinylcarben-Komplexen
aus den Vinylidenkomplex 10 und Iminiumsalzen beziehungsweise Enaminen entwickelt.[37]
Die Frage nach der eventuellen Reaktivität dieser Verbindungen in der Metathese Reaktion ist
im ersten Abschnitt dieser Arbeit bei der Umsetzung des Vinylcarben-Komplexes mit
elektronreichen Alkenen untersucht worden (Abbildung 14).
R1
R1
W C
NC
O
O
H
HN
NW
NC
OO
C CC
H
HH
R1
R1 C
C C
H R1
R1
HW N
CO
O
N
+
Abbildung 14. Die Metathesereaktion mit dem Vinylcarben-Komplex.
b) Weiterhin sollte ein Zugang zu dem bislang unbekannten und vermutlich reaktiven
Allenylidenkomplexe des Wolframs mit Cylopentadienylliganden erschlossen werden. Da die
Reaktion von η1-Acetylidkomplexen mit den Iminiumsalzen zu den Vinylidenkomplexen der
Konstitution [M=C=C(H)-C(NRR')R''2] führt, die durch die Eliminierung eines Amins einen
Allenylidenkomplex bilden können, wurde die Reaktion von η1-Acetylidkomplexen mit den
Iminiumsalzen zur Darstellung dieser Verbindungen in dem zweiten Abschnitt studiert
(Abbildung 15).

A Einleitung 11
[W] C C H C NR
R
R'
R'[W] C C
H
CN
R R
R'
R'[W] C C C
R
RNH
R'R'+
++
Abbildung 15. Eine Synthesestrategie für die Darstellung eines Allenylidenkomplexes.
c) Weiterhin sollte die Reaktion von Vinylidenkomplexe mit Diazoalkanen studiert
werden. Hier war es beabsichtigt einen Zugang zu den sonst schwer zugänglichen
substituierten Vinylidenkomplexen zu eröffnen.
d) Schließlich sollte die Hydrolyse der zuvor im Arbeitskreis dargestellten
Chlorhaltigen Komplexe 11a-c studiert werden. Hier wurde erhofft Hydroxy-
Wolframkomplexe des Typs 12a-c zu erhalten (Abbildung 16).
W P
NClO
HH
R
RW P
NOHO
HH
R
RNaOH
(CH3)3C-
11a-c 12a-c
Ph
(CH3)2C
R11, 12
a
b
c
Abbildung 16. Darstellung der Hydroxy-substituierten Komplexe 12a-c.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 12
1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen
1.1 Vormerkungen
Seit der Entdeckung der stabilen Carbenkomplexe der Übergangsmetalle durch E.O.
Fischer,[5] haben diese Verbindungen eine bereite Anwendung in der organischen Synthese
gefunden, unter anderem bei der Synthese der kleinen organischen Moleküle, Olefin-
Isomerisierung und Metathese Reaktion.[38] Die Metathese Reaktion hat sich mittlerweile zu
einem der wertvollsten Methoden in der Organischen Synthese entwickelt.[39] Eine Unzahl
von kleinen, mittleren und großen Carbo- und Heterocyclen sowie eine breite Auswahl an
acyclischen Verbindungen sind durch diesen wichtigen Reaktionstyp leicht zugänglich.[40]
Der Begriff Metathesis kommt aus dem griechischen Methatithemi und bedeutet
„Dazwischenbringen“ und im Prinzip beinhaltet eine Umalkylidenierung der linearen Alkene
(Abbildung 17).
R'R
MR'
R'R'
MR
RR'R R
MR'
R'R
MR
RR'
R
R´
R'
R'
R'
R
R
R
13a 13a'13b
13b'
+ +2
Metallalkyliden-Katalysator
propagierender Metallalkyliden
4
Abbildung 17. Der Metathese-Zyklus.
Der Schlüsselschritt der Olefinmetathese ist eine [2+2] Addition eines Olefins (z.B.
RCH=CHR', Abbildung 17) an einer Metall-Kohlenstoff-Doppelbindung (z.B. in 13a) zu
einem Metallacyclobutankomplex (13b). Das entstandene Metallacyclobutan kann in einer
Retro-[2+2] wieder zu 13a und RCH=CHR' zerfallen oder ein neues Metallalkyliden 13a' und
R'CH=CHR' ergeben. Die weitere Reaktion von 13a' mit RCH=CHR' liefert dann 13b', das
zu 13a und RCH=CHR zerfallen kann. Die R- und R'-Substituenten in den gebildeten
Metallacyclen können sowohl eine cis- als auch trans- Stellung zueinander haben. Deshalb

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 13
wird trans-RCH=CHR', wie in der Abbildung 17 dargestellt, katalytisch in einer
Gleichgewichtsmischung von ungefähr zwei Teilen RCH=CHR' (cis und trans) und jeweils
einem Teil RCH=CHR (cis und trans) und R'CH=CHR' (cis und trans) umgewandelt. Diese
Reaktion wird mit vielen Carbenkomplexen katalysiert. Die Pentacarbonylwolfram-Carben-
Komplexe waren einer der ersten dieser Gruppe der Komplexe, die vom Katz bei der
Metathese Reaktionen eingesetzt wurden.[38h] Zu den bekanntesten dieser Komplexen gehört
der Schrock-Katalysator,[41] Grubbs’sche Katalysator[42] und der von Hoveyda[43]]
vorgestellter Katalysator (Abbildung 18).
RuR
PCy3
PCy3
Cl
O Ru
Cl
P(Cy)3
Cl
CF3
CF3
OMoONCF3
MeCF3
MeMe
Me
iPr Cl
iPr
Grubbs'sche Katalysator
Hoveyda'scheKatalysator
Schrok'scheKatalysator
Abbildung 18. Die wichtigen Metathese-Katalysatoren.
In der Arbeitsgruppe Ipaktschi wurde neulich über die Umsetzung des Wolfram-
Vinylidenkomplexes 10 mit einer Reihe der von Pyrrolidin hergeleiteten Enaminen zu
Vinylcarben-Komplexen 16 berichtet.[37] Diese Reaktion beginnt mit einem Säure/Base-
Prozess an (Reaktionsschritt a), welcher zu einem Aggregat aus dem Wolfram-η1-
Acetylidkomplex und Iminiumion führt. Die darauf folgende Mannich-Typ Reaktion führt zu
der Aminoalkylierung des β-Kohlenstoffatoms des anionischen Wolfram-η1-
Acetylidkomplexes (Schritt b), welche die Bildung des intermediären reaktiven
Vinylidenkomplexes 15 zur Folge hat. Der letzte Schritt der Reaktion ist eine Retro-En-
Reaktion, die mit dem Transfer des Wasserstoffatoms von dem α-Kohlenstoffatom des
Pyrrolidinringes zu dem α-Kohlenstoffatom η1-Vinylidenkomplexes anfängt und schließlich
zur Fragmentierung des Komplexes zu dem Vinylcarben-Komplex 16 und 1-Pyrrolin führt
(Abbildung 19).

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 14
W C
CNO
C
O
H
NH R
R
W C
CNO
C
O
H
HW C
CN
O
C
O
H
W CC
NO
C
O
H
H
R
R
N
RR
N+
R R
N
+
-
+
10 14
15 16
(a) (b)
(c)1
2
1
2
1 2
2
1
Abbildung 19. Die Bildung des Vinylcarben-Komplexes durch die Retro-En-Reaktion.
Um die potentielle Reaktivität dieser Vinylcarben-Komplexe in einer Metathese
Reaktion zu studieren, wurden die Vinylcarben-Komplexe 16a und 16b dargestellt und mit
den Enaminen 14a-b als elektronreiche Alkene umgesetzt (Abbildung 20).
W C
CNO
C
O
H
H
W C
CNO
C
O
H
HN N
16a 16b 14a 14b Abbildung 20.
1.2 Darstellung der Vinylcarben-Komplexe 16a-b
Der grüne η1-Acetylidkomplex 10a wurde durch die Umsetzung des Komplexes 10
mit der n-Butyllithium Lösung in Hexan bei –78 °C dargestellt. Nach dem Aufwärmen der
Reaktionsmischung auf –30 °C wurde das entsprechende Iminiumsalz 17a (bzw. 17b) in
einem leichten Überschuss addiert. Nach kürzen Zeit trat eine rasche Farbänderung zum rot
auf. Danach wurde die Reaktionsmischung langsam auf die Raumtemperatur erwärmt und
nach der Aufarbeitung der Vinylcarben-Komplex 16a (bzw. 16b) in 70% (bzw. 65%)
Ausbeute erhalten (Abbildung 21).

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 15
NN
ClO4ClO4
W C CN
CO
OH
Li
+
--
-
10a17a 17b
+ +
Abbildung 21.
1.3 Die Umsetzung der Vinylcarben-Komplexe 16a-b mit Enaminen 14a-b,
Darstellung der Wolfram-η1-Acyl-Komplexe 18, 19 und 20
Eine Lösung des Vinylcarben-Komplexes 16a wurde bei –20 °C mit einem
Überschuss an dem Enamin 14b umgesetzt. Nach vier Stunden schlug die Farbe der Lösung
von rot nach gelb um und die Carbonyl Absorption der Verbindung 16a, bei 1980 cm-1, im
IR-Spektrum der Reaktionsmischung verschwand. Nach der Aufarbeitung und
Umkristallisierung wurde das Produkt, der Wolfram-η1-Acyl-Komplex 18, mit einer Ausbeute
von 70% isoliert (Abbildung 22).
W C
CNO
C
O
H
HN
WOON
HH
HN
16a 14b
+
18
THF, -20 oC 5
67
8
Abbildung 22. Darstellung des Wolfram-η1-Acyl-Komplexes 18.
Die Umsetzung der Wolfram-Carben-Komplexe 16a-b mit dem Enamin 14a, unter
den gleichen Bedingungen führte entsprechend zu den Wolfram-η1-Acyl-Komplexen 19 und
20 mit einer Ausbeute von 68% bzw. 62% (Abbildung 23).
WOON
HH
HN
WOON
H
HH
N
19 20
5 5
6
67 78 8
Abbildung 23. Die Wolfram-η1-Acyl-Komplexe 19 und 20.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 16
Diskussion charakteristischer und spektroskopischer Daten der Wolfram-η1-Acyl-
Komplexe 18-20: Die vorgeschlagene Struktur für die Verbindung 18 wurde sowohl mit den
spektroskopischen Analysemethoden als auch durch die Einkristallstrukturanalyse belegt. Das
IR-Spektrum des Komplexes 18 zeigt eine breite Nitrosyl Bande bei 1577 cm-1 und eine
Carbonyl Absorption bei 1648 cm-1, die die Absorption eines η1-Acyl-Komplexes
entspricht.[44] Das 1H-NMR Spektrum (400 MHz, THF-d8, –20 °C) der Verbindung 18 stimmt
also durch das charakteristische Muster des η3-Allyl Systems mit der angegebenen Struktur
überein.[45] Der Komplex 18 zeigt eine Dublett bei δ 5.50 ppm für das am zentralen
Kohlenstoffatom des η3-Allyl-Systems (C6) gebundene Proton, und eine Dublett von Dublett
bei δ 4.20 ppm für das Proton am C7. Die anti-Orientierung der beiden erwähnten Protonen
zu einander verursacht eine große Kupplungskonstante 3JH6-H7 von 12.8 Hz (Abbildung 24).[46]
Das 13C-NMR Spektrum (100 MHz, THF-d8, –20 °C) zeigt unter anderem die
folgenden Signale bei δ 91.7, 111.3 und 116.6 ppm, die dem η3-Allyl-Ligand zugeordnet
werden, nämlich C7, C6 und C5. Die Zuordnung wird durch C,H-COSY-NMR und DEPT-
NMR Experimente belegt. Sie stimmen mit den berichteten charakteristischen Tieffeld-
0.51.01.52.02.53.03.54.04.55.05.5
4.184.204.224.244.26
5.445.465.485.505.52
J=12.850
J=12.850
Abbildung 24. Des 1H-NMR Spektrum der Verbindung 18 (400 MHz, THF-d8, –20 °C).
signalen für die an dem Übergangsmetall gebundenen sp2-hybridisierten Kohlenstoffatome
überein.[47] Erwartungsgemäß absorbieren die endständigen Kohlenstoffatome in dem Bereich
der δ 80 - 90 ppm und das Zentral Kohlenstoffatom zwischen δ 110 - 130 ppm. Der
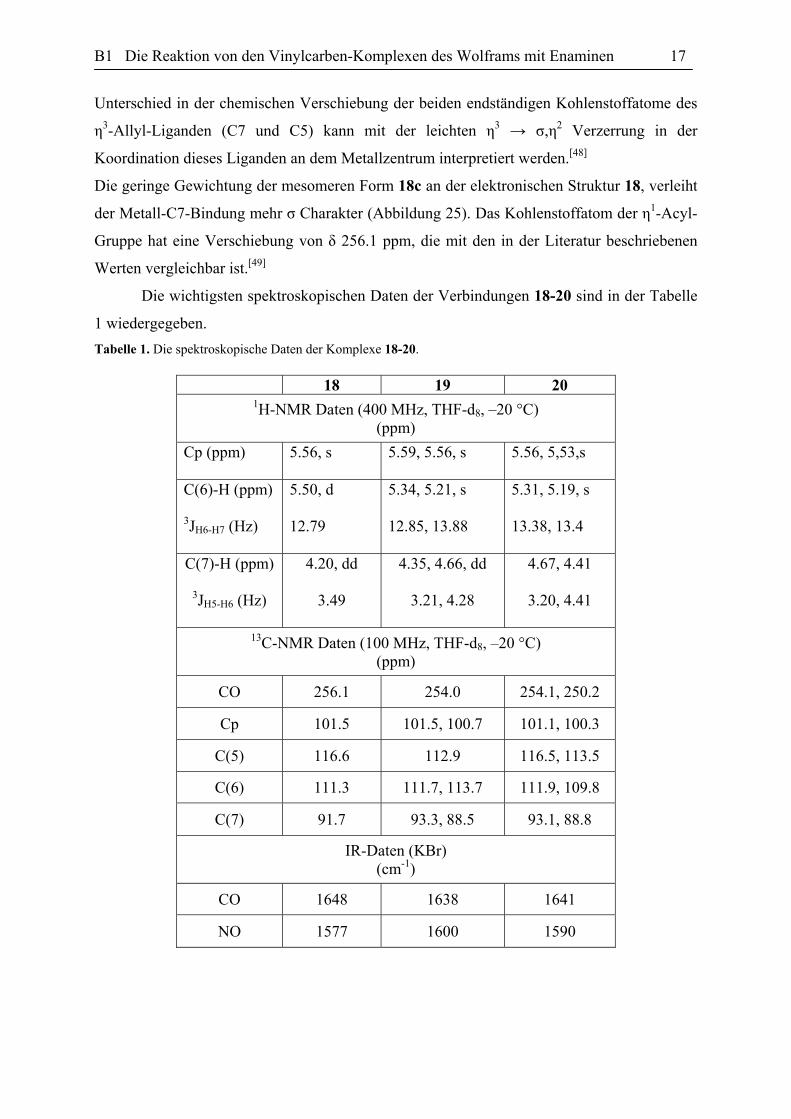
B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 17
Unterschied in der chemischen Verschiebung der beiden endständigen Kohlenstoffatome des
η3-Allyl-Liganden (C7 und C5) kann mit der leichten η3 → σ,η2 Verzerrung in der
Koordination dieses Liganden an dem Metallzentrum interpretiert werden.[48]
Die geringe Gewichtung der mesomeren Form 18c an der elektronischen Struktur 18, verleiht
der Metall-C7-Bindung mehr σ Charakter (Abbildung 25). Das Kohlenstoffatom der η1-Acyl-
Gruppe hat eine Verschiebung von δ 256.1 ppm, die mit den in der Literatur beschriebenen
Werten vergleichbar ist.[49]
Die wichtigsten spektroskopischen Daten der Verbindungen 18-20 sind in der Tabelle
1 wiedergegeben. Tabelle 1. Die spektroskopische Daten der Komplexe 18-20.
18 19 20 1H-NMR Daten (400 MHz, THF-d8, –20 °C)
(ppm) Cp (ppm)
5.56, s 5.59, 5.56, s
5.56, 5,53,s
C(6)-H (ppm) 3JH6-H7 (Hz)
5.50, d 12.79
5.34, 5.21, s 12.85, 13.88
5.31, 5.19, s 13.38, 13.4
C(7)-H (ppm)
3JH5-H6 (Hz)
4.20, dd
3.49
4.35, 4.66, dd
3.21, 4.28
4.67, 4.41
3.20, 4.41
13C-NMR Daten (100 MHz, THF-d8, –20 °C) (ppm)
CO 256.1 254.0 254.1, 250.2
Cp 101.5 101.5, 100.7 101.1, 100.3
C(5) 116.6 112.9 116.5, 113.5
C(6) 111.3 111.7, 113.7 111.9, 109.8
C(7) 91.7 93.3, 88.5 93.1, 88.8
IR-Daten (KBr) (cm-1)
CO 1648 1638 1641
NO 1577 1600 1590

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 18
R
W
HR
H
W
H
W
R
18a 18b 18c Abbildung 25. Die mesomere Strukturen für einen η3-Allyl-Liganden.
Röntgenstrukturanalyse des Wolfram-η1-Acyl-Komplexes 18: Geeignete
Einkristalle wurden aus einer Pentanlösung der Verbindung 18 bei 4 °C gezüchtet. Die
kristallografischen Daten sind in der Tabelle E.1.1 (Abschnitt E dieser Arbeit)
zusammengefasst. Der ORTEP Plot der Verbindung 18 ist in der Abbildung 27 zusehen.
Zu den wichtigsten strukturellen Eigenschaften des Komplexes 18 gehört die
Anwesenheit des η3-Allyl-Liganden. Die η3-Allyl-Komplexe sind in den letzten Jahren viel
studiert worden.[50] Ein η3-Allyl-Ligand kann als endo- oder exo-Isomere, angesichts der
Orientierung des zentralen Kohlenstoffatoms des η3-Allyl-Liganden zu einem vordefinierten
Referenzliganden an dem Metallzentrum, vorliegen. Bei dem exo-Isomer schaut der C2 des
Allylsystems zu dem Referenzligand (hier Cp Ligand) hin, bei dem endo-Liganden zeigt
dieser in der Gegenrichtung an (Abbildung 26).[51]
M M
endo Isomer exo Isomer
Abbildung 26. Definition der Endo- und Exo-Isomerie beim η3-Allyl-Liganden.
Bei dem η1-Acyl-Komplex 18 nimmt der η3-Allyl-Ligand eine endo-Stellung zum
Cyclopentadienyl-Liganden an. Der η3-Allyl-Ligand setzt sich aus den Kohlenstoffatomen
C7, C6 und C5 (Abbildung 23) zusammen. Die Bindungslängen C5-C6 und C6-C7 sind fast
gleich (1.421 bzw. 1.427 Ǻ). Die C-C-C Bindungswinkeln des η3-Allyl Fragmentes deuten
auf der sp2-Hybridisierung dieser Kohlenstoffatomen an: C8-C7-C6, 120.39(19)˚; C5-C6-C7,
119.2˚; C4-C5-C6, 118.7(2)˚. Die anti-Stellung der Protonen, sowie die endo-Position des η3-
Allyl-Liganden, lässt sich ebenfalls aus den entsprechenden Torsionswinkeln entnehmen.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 19
Abbildung 27. ORTEP Plot des Komplexes 18 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt. Die Protonen wurden für Übersichtlichkeit nicht nummeriert. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen): W(1)-N(1) 1.758(6), W(1)-C(13) 2.215(4), W(1)-C(6) 2.292(4), W(1)-C(5) 2.293(4), W(1)-C(7) 2.337(4), N(2)-C(12) 1.478(9), O(1)-C(13) 1.200(7), C(7)-C(6) 1.421, C(7)-C(8) 1.484(5), C(6)-C(5) 1.427, C(13)-C(12) 1.562(8), N(1)-W(1)-C(13)-85.7(2), C(6)-W(1)-C(5) 36.27(6), C(6)-C(7)-C(8) 120.39(19), C(7)-C(6)-C(5) 119.2, C(6)-C(5)-C(4) 120.6(2), C(6)-C(5)-C(1) 116.8(2), O(1)-C(13)-C(12) 118.2(5), O(1)-C(13)-W(1) 119.2(4).
Die W-C5 und W-C6 Bindungen sind gleich lang, nämlich 2.297(2) Ǻ, die mit den in
der Literatur berichteten Werten für η3-Allyl Komplexe übereinstimmen.[52] Dabei ist die
Bindung des anderen η3-Allyl Terminus zum Wolframatom leicht länger [2.337(4) Ǻ] und
verdeutlicht nochmals die leichte η3 → σ,η2 Verzerrung des η3-Allyl-Fragmentes. Die relativ
lange Bindungslänge des W-C(O) von 2.215(4) Ǻ deutet auf einer W-C-Einfachbindung hin
und der Winkel C12-C13-W (122.5˚) beweist zusätzlich die Existenz des η1-Acyl-Liganden.
Ein Vorschlag zur Entstehung des Wolfram-η1-Acyl-Komplexes 18: Der
nucleophile Angriff des β-Kohlenstoffatom des Enamins 14b an dem α-Kohlenstoffatom des
Vinylcarben-Komplexes 16a führt zur Bildung des intermediären Zwitterions 18a. An der
Stelle der erwarteten Ringbildung zu einem Metallacyclobutan-Derivativen und seine
anschließende Cycloreversion zum Metatheseprodukt, dem Aminocarben-Komplex 18c,[53]
führt der nucleophile Angriff des Kohlenstoffatoms der Carbonyl Gruppe an dem
Kohlenstoffatom der Iminum-Funktionalität zum beobachteten η1-Acyl-Komplex 18
(Abbildung 28).

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 20
W C
NC
O
O
H
HN WO
ON
N
HH
HW
NCO O
C C CN
+
HHH
NW
NC
OO
C CC
H
HHC
C C
H
HW N
C O
ON
+
16a 14b 18a
18b18c
18
Abbildung 28. Der postulierte Reaktionsweg für die Bildung des Komplexes 18. Da der Vinylcarben-Komplex 16a selbst aus der Reaktion von dem
Vinylidenkomplexes 10 mit dem Enamin 14a dargestellt wird, erschien es sinnvoll die
Möglichkeit einer direkten Umsetzung des Vinylidenkomplexes 10 mit einem Überschuss
vom Enamin 14a zu prüfen, um somit den Aufwand der Isolierung und Reinigung des
Vinylcarben-Komplexes 16a zu vermeiden.
Die durchgeführten Experimente zeigten, dass tatsächlich die Ein-Topf-Reaktion des
Vinylidenkomplexes 10 mit Enamin 14a in THF bei Raumtemperatur eine saubere und
einfachere Methode für die Synthese des Wolfram-η1-Acyl-Komplexen 19 (83%) darstellt
(Abbildung 29).
W C CH
HNC
O
O
N WOON
N
HH
H
1910 14a
+ 2
Abbildung 29. Die Ein-Topf-Reaktion zur Darstellung der Verbindung 19.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 21
1.4 Umsetzung des Vinylidenkomplexes 10 mit dem Enamin 14b
Analog zur Darstellung der Verbindung 19, wurde für eine Ein-Topf-Synthese der
Verbindung 18, das Enamin 14b mit dem Vinyliden Komplex 10 in THF umgesetzt. Nach 40
Minuten trat die vollständige Farbänderung zum hellrot auf. Überraschenderweise zeigte die
chemische Analyse des Rohproduktes, dass der erwartete Wolfram-η1-Acyl-Komplex 18 nur
als ein Nebenprodukt entstanden war. Das Hauptprodukt der Reaktion wurde auf der
Grundlage der analytischen und spektroskopischen Standardmethoden als der η2-
Alkenkomplex 21 identifiziert. Die Zuordnung der postulierten Struktur wurde auch mittels
der Röntgenstrukturanalyse bestätigt. Das Verhältnis der Verbindungen 18 und 21 in dem
Rohprodukt war 1:7 (Abbildung 30).
W C CH
HNC
O
O
N WO
N
N
H H
H
O
W NC
O
OCC
H H
HN
1810 14b
+ +
21
1:7
Raumtemp.
THF
1
2
3
7
Abbildung 30. Die Reaktion von der Verbindung 10 mit dem Enamin 14b in THF.
Das IR-Spektrum der Verbindung 21 zeigt eine Carbonyl Bande bei 1952 cm-1 und
eine bereite Nitrosyl Bande bei 1571 cm-1. Die 1H-NMR und 13C-NMR Daten der Verbindung
21 stimmen mit den Daten der in der Literatur beschriebnen η2-Olefinkomplexe überein.[54]
Bei dem 1H-NMR Spektrum (400 MHz, CDCl3, Raumtemperatur) der Verbindung 21
erscheint das Cp-Signal bei δ 5.66 ppm und das Methinproton (C2) zeigt eine Dublett von
Dublett bei δ 4.36 ppm. Dabei wird dieses Proton durch die Methylenprotonen des C1 mit
Kupplungskonstanten 3JH-Hcis = 9.1 und 3JH-Htrans = 12.3 Hz gespalten. Die Methylenprotonen
der Diethylamino-Gruppe sind chemisch verschieden und erscheinen als zwei Multipletts bei
δ 2.85 und 2.87 ppm. Im 13C-NMR Spektrum erscheinen die Signale für die Carbonylgruppe
bei δ 223.5 ppm, die Signale für C7 und C3 bei δ 143.1 und 139.3 ppm und das Cp Signal bei
95.6 ppm. Die Signale für C1 und C2 liegen bei δ 30.1 und 39.9 ppm, wobei die Zuordnung
des Letzteren mit der Hilfe des deuterierten Derivaten der Verbindung 21 geschieht, wie es
später beschrieben wird.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 22
Eine mechanistische Betrachtung der Entstehung des Komplexes 21: Im
Gegensatz zu den Enaminen mit einem Pyrrolidin Ring, zeigt das Enamin 14b in der Reaktion
mit dem Vinylidenkomplex 10 ein anderes Reaktivitätsmuster (Abbildung 31). Das Enamin
14b reagiert einmal als ein C-Nucleophil, was zu der Bildung der Verbindung 21 führt
(Reaktionsweg a, Abbildung 31). Der nucleophile Angriff des Enamins 14b an dem α-
Kohlenstoffatom des Vinylidenkomplexes 10 bildet das Zwitterion 21a. Die Migration des
Protons an dem C3-Kohlenstoffatom zum elektronreichen und basischen Metallzentrum führt
zur Bildung des Metallhydrids 21b. Durch die anschließende reduktive Eliminierung entsteht
der η2-Alkenkomplex 21. Der zweite Reaktionsweg basiert auf der basischen Eigenschaft des
Enamins 14b, welcher den Anlass zur Bildung des Nebenprodukts 18 gibt (Reaktionsweg b,
Abbildung 31). Die Säure-Base Reaktion führt über das Ionnenpaar-Aggregat 18d, zur
Bildung des Vinylcarben-Komplexes 16a. Dieser reagiert dann mit dem noch vorhandenen
Enamin 14b zum Wolfram-η1-Acyl-Komplex 18.
N+
W C CN
CO
OH
W C CH
HNC
O
O
W C
CNO
C
O
H
H
N
WOON
HH
HN
CC
H H
W NC
O
O
N+
H
CC
HH
NW N
CO
O
HW N
CO
OC
CH
H
HN
THF
10
16a
14b
+
18
21 21a21b
18d
(a)
(b)
14b+-
Abbildung 31. Der vorgeschlagene Mechanismus für die Reaktion des Enamins 14b mit dem Komplex 10 in THF.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 23
Um die Bildung der Verbindung 18 zu unterdrücken, kann die Polarität der
Reaktionslösung herabgesetzt werden, denn dadurch wird die Aggregatbildung 18d
ungünstig. Das Durchführen der Reaktion in Diethylether ergibt quantativ die Verbindung 21.
Als weiterer Hinweis zur Strukturbestätigung wurde der deuterierte Vinylidenkomplex
10b dargestellt und mit dem Enamin 14b in Diethylether zum Wolframkomplex 22 umgesetzt
(Abbildung 32).
W NC
O
OC
CD
D
HN
W C CD
DNC
O
O
2210b
1
2
45
6
Abbildung 32.
In dem 1H-NMR Spektrum der Verbindung 22 wird das, dem Methinproton am
Kohlenstoffatom C2 zugeordnete Dublett von Dublett, zu einem Singulett reduziert. In dem
DEPT Spektrum der Verbindung 21 werden die vier Methylenkohlenstoffatome- C1, C4, C5
und C6- bei δ 30.1, 29.8, 26.7 und 21.2 ppm beobachtet. Das 13C-NMR Spektrum des
deuterierten Komplexes 22 weist in diesem Bereich jedoch nur drei Signale bei δ 29.8, 26.7
und 21.2 ppm auf. Daher kann die Absorption bei δ 30.1 ppm dem C1 zugeordnet werden,
denn diese wird durch die beiden Deuteriumatome zu einem Quintett aufgespaltet und wird
wegen geringer Intensität nicht mehr detektiert (Abbildung 33).
Röntgenstrukturanalyse der Verbindung 21: Geeignete Einkristalle für
Röntgenstrukturanalyse wurden aus Ether bei -20 °C gezüchtet. Die kristallografische Daten
sind in der Tabelle E 2.2 zusammengefasst. Der ORTEP Plot der Verbindung 21 ist in der
Abbildung 34 wiedergegeben.

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 24
27282930313233343536373839404142434445
27282930313233343536373839404142434445
W NC
O
OC
CD
D
HN
22
W NC
O
OC
CH
H
HN
21
C1
Abbildung 33. Hochfeld-Ausschnitt des DEPT-Spektrums der Verbindung 21 (oben) und des 13C-NMR Spektrums der Verbindung 22 (unten).
Abbildung 34. ORTEP Plot von dem Komplex 21 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen): W-N 1.786(7), W-C 1.972(6), W-C(1) 2.270(7), W-C(2) 2.286(6), W-C(14) 2.298(8), C(16)-C(12) 1.327(18), C(1)-C(2 )1.393(11), C(2)-C(3) 1.467(10), C(3)-C(7) 1.30b9(10), C(7)-N(2) 1.449(11), C(7)-C(6) 1.515(12), C-W-C(1) 74.1(3), C(1)-W-C(2) 35.6(3), C(2)-C(1)-W 72.8(4), C(1)-C(2)-C(3) 124.5(7), C(1)-C(2)-W 71.6(4), C(3)-C(2)-W 117.1(4), C(7)-C(3)-C(2) 126.2(7), C(7)-C(3)-C(4) 111.1(7), C(2)-C(3)-C(4) 122.5(7), C(3)-C(7)-N(2) 124.3(8).

B1 Die Reaktion von den Vinylcarben-Komplexen des Wolframs mit Enaminen 25
1.5 Schlussfolgerung
In diesem Abschnitt wurde die Reaktion der Vinylcarben Komplexe 16a-b mit dem
Enamin 14a untersucht. Es wurde anstelle der erwarteten Metathese Reaktion, eine Addition
des Enamins an dem Vinylcarben-Komplex zu den Wolfram-η1-Acyl-Komplexen 18-20
beobachtet (Abbildung 23 und 28). Eine Ein-Topf-Reaktion des Vinylidenkomplexes 10 mit
einem Überschuss an dem Enamin 14a in THF führt ebenso zur Bildung der Verbindung 19.
Anderes verhält sich das Enamin 14b bei der Reaktion mit der Verbindung 10. Dieses Enamin
zeigt zwei verschiedene Reaktivitätsmuster (Abbildung 31):
(a) Es reagiert als eine Base und bildet das Ionenpaar 18d, das nach einer Mannich-Typ-
Allkylierung und anschießenden Retro-En-Reaktion zur Bildung des Vinylcarben-Komplexes
16a, mit dem Überschuss vom Enamin zum Nebenprodukt dieser Reaktion, nämlich der
Verbindung 18 führt,
(b) Hauptsächlich reagiert das Enamins 14b als ein Nucleophil mit dem
Vinylidenkomplex 10 zur Alkylierung der Verbindung 10 an dem α-Kohlenstoffatom. Die
nachfolgende Protonumlagerung und reduktive Eliminierung führt zu dem η2-Alken-Komplex
21.

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 26
2 Darstellung des η1-Allenylidenkomplexes des Wolframs
2.1 Vormerkungen
Die Allenylidenkomplexe [LnM]=C=C=CR1R2 gehören als nicht gesättigte Carben-
Derivaten, zu der Gruppe der Kummulenkomplexe {[MLn]=C(=C)n=CR1R2, n>0}. Das
Interesse an diesen Verbindungen ist in vergangnen Jahren gewachsen.[55] Die Forschungen
beschränken sich in zwei Bereichen der Materialwissenschaften und der organischen
Synthese.
Im Bereich der neuen Materialen sind die Allenylidenkomplexe von Interesse, weil sie
eine mit einem Metall verbundenen sp-hybridisierten Kohlenstoffkette enthalten. Diese
strukturelle Eigenschaft weist sie als interessante Stoffe unter anderem für die molekulare
Drähte der allgemeinen Konstitution M=C(=C)n=C=M auf.[56]
Die Anwendung der Allenylidenkomplexe in der Synthese beruht auf der
alternierenden Anordnung der elektrophilen und nucleophilen Zentren in diesen Systemen. In
den Allenylidenkomplexen sind Cα und Cγ elektrophil und Cβ nucleophil. Diese
Reaktivitätsmuster ermöglichte in den letzten Jahren die Einsetzung der Allenylidenkomplexe
als Wertvolle "Building-Blocks" für verschiedene Synthesen. Beispielsweise die Umsetzung
von in situ hergestelltem Ruthenium-Allenylidenkomplex mit Phenolen führt zu 1H-
Benzopyrane.[57] Andere Variationen dieser Reaktion sind für die Synthese von anderen
Heterocyclen benutzt worden.[58] Manche Allenylidenkomplexe sind ebenfalls als Vorstufe
für Ringschlussmetathese-Katalysatoren eingesetzt worden.[59]
Die Darstellung der ersten Allenylidenkomplexe wurden von E.O.Fischer[60] und
Berke[61] berichtet. Die allgemeine Methode zur Darstellung dieser Verbindungen, beruht
inzwischen auf den Arbeiten von J.P.Selegue,[62] der durch Aktivierung von
Propargylalkoholen erstmals einen direkten und einfachen Zugang zu diesen Verbindungen
vorstellte. Dabei wird durch die Koordination eines geeigneten Propargylalkohols zunächst
ein η2-Alkinkomplex gebildet, der dann zu einem Vinylidenkomplex sich umlagert. Dieser
Komplex wird anschließend – spontan oder im Gegenwart von Lewis-Säuren – unter Bildung
von einem Allenylidenkomplex dehydratisiert (Abbildung 35).[63]
CCH CR2(OH) CC[M]C
OH
RR
HCC[M] C
R
R[M] [M]
C
C
H
CR2(OH)
+
Abbildung 35. Allgemeine Methode zur Darstellung eines Allenylidenkomplexes.

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 27
Als eine andere Variante wird ein η2-Alkinkomplex, der eine Estergruppe enthält, in
einem η1-Acetylidkomplex überführt. Addition von Lithiumorganylen und anschließende
Protonierung und Dehydrastisierung führt zum gewünschten Produkt (Abbildung 36).[64]
[M]C
C
H
CO2MeLiNPr2
CC[M] CO2MeLiR
CC[M] CO
RR
CC[M] CR
R
HCl dann COCl2
Abbildung 36.
2.2 Versuche zur Darstellung eines η1-Allenylidenkomplexes des Wolframs
Wie es im ersten Abschnitt beschrieben wurde, kommt es im Laufe der Reaktion eines
Iminiumsalz mit dem η1-Acetylidkomplex 10a zur Bildung eines intermediären
Vinylidenkomplexes 15, der eine Retro-En-Reaktion eingeht. Gelingt es die Retro-En-
Reaktion zu unterbinden, dürfte das Abspalten eines Amins aus diesem Intermediat zur
Bildung eines Allenylidenkomplexes führen (Abbildung 37, Reaktionsweg b).
W C
CNO
C
O
H W C
CNO
C
O
H
NR
R
H R
R
W C
CNO
C
O
H
H
R
R
N+
RR
R R
W C
CNO
C
O
CR
R
Li+
X
NH
R R
+
-
10b
+
(b)
(a)
15
1 2
3 4
1
23 4
12
3
4
1
2
Abbildung 37. Mögliche Synthese-Strategie für die Darstellung eines Allenylidenkomplexes des Wolframs.
Im Rahmen dieser Untersuchungen zur Darstellung eines Allenylidenkomplexes
wurden folgende Versuche durchgeführt.
Reaktion des η1-Acetylidkomplexes 10a mit dem Iminiumsalz 23a: Die Umsetzung
des Iminiumsalzes 23a mit einer Lösung von dem Acetylidkomplex 10a in THF bei -30 °C
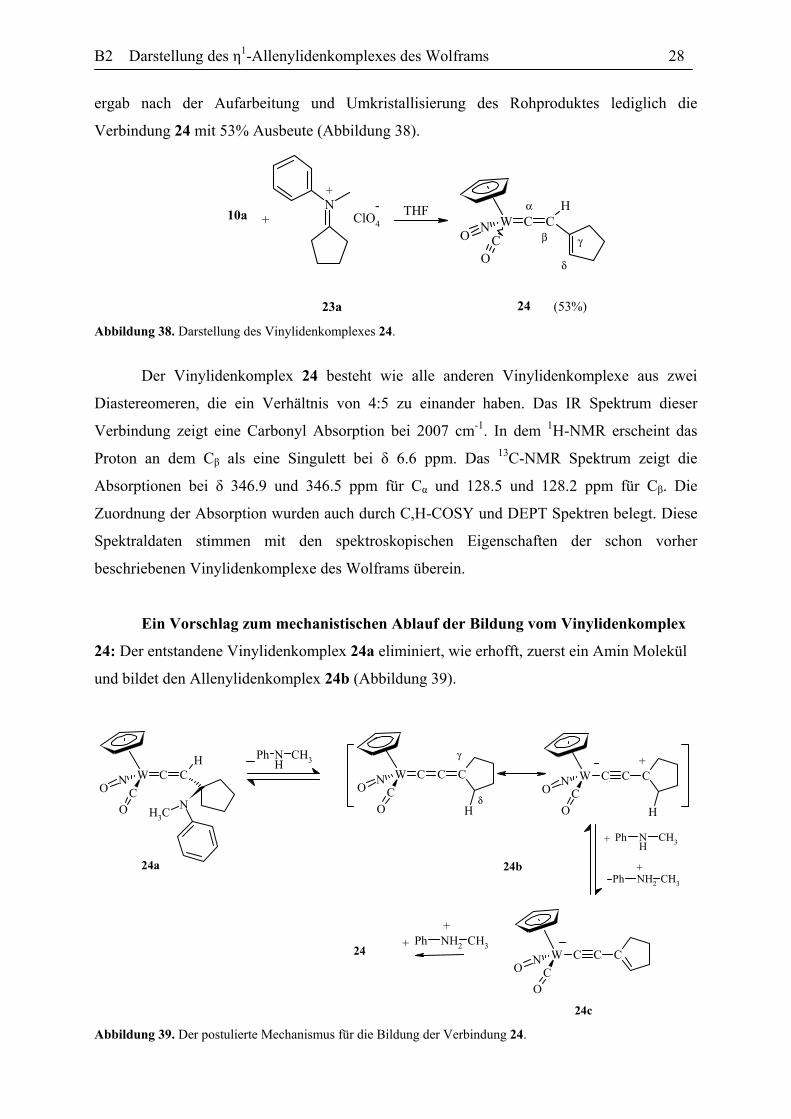
B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 28
ergab nach der Aufarbeitung und Umkristallisierung des Rohproduktes lediglich die
Verbindung 24 mit 53% Ausbeute (Abbildung 38).
W CNC
O
OC
HNClO4
10a +THF
2423a (53%)
α
β γ
δ
-+
Abbildung 38. Darstellung des Vinylidenkomplexes 24.
Der Vinylidenkomplex 24 besteht wie alle anderen Vinylidenkomplexe aus zwei
Diastereomeren, die ein Verhältnis von 4:5 zu einander haben. Das IR Spektrum dieser
Verbindung zeigt eine Carbonyl Absorption bei 2007 cm-1. In dem 1H-NMR erscheint das
Proton an dem Cβ als eine Singulett bei δ 6.6 ppm. Das 13C-NMR Spektrum zeigt die
Absorptionen bei δ 346.9 und 346.5 ppm für Cα und 128.5 und 128.2 ppm für Cβ. Die
Zuordnung der Absorption wurden auch durch C,H-COSY und DEPT Spektren belegt. Diese
Spektraldaten stimmen mit den spektroskopischen Eigenschaften der schon vorher
beschriebenen Vinylidenkomplexe des Wolframs überein.
Ein Vorschlag zum mechanistischen Ablauf der Bildung vom Vinylidenkomplex
24: Der entstandene Vinylidenkomplex 24a eliminiert, wie erhofft, zuerst ein Amin Molekül
und bildet den Allenylidenkomplex 24b (Abbildung 39).
W CNC
O
OC
H
NCH3
W CNC
O
OC C
H
W CNC
O
OC C
H
Ph NH
CH3
Ph NH2 CH3
W CNC
O
OC C
Ph NH2 CH3
Ph NH
CH3
+
++24
24a 24b
24c
γ
δ
+
+
Abbildung 39. Der postulierte Mechanismus für die Bildung der Verbindung 24.

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 29
Das Proton an dem Cδ ist aufgrund der abgebildeten mesomeren Struktur acide.
Deswegen kann die anwesende Base das Proton abspalten und den η1-Acetylidkomplex 24c
bilden, der noch mal an Cβ unter Bildung der Verbindung 24 protoniert wird.
Der saure Charakter der Cδ-H ist schon für andere Allenyidenkomplexe, wie z.B. der
unten stehenden Eisenderivat, in der Literatur beschrieben worden (Abbildung 40).[65]
Fe C C CEt
EtBr
H+
H+
Fe C C CEt
BrC CH3
H+
= Diethylphosphinoethan
+
Abbildung 40. Isomerisierung von dem Allenylidenkomplex zum Vinylvinyliden-Komplex.
Um die Isomerisierung des Allenylidenkomplexes in einem Vinylidenkomplex zu
unterbinden, wurde das Iminiumsalz 23b mit einem Adamantanrest dargestellt.
Die Reaktion des η1-Acetylidkomplexes 10a mit dem Iminiumsalz 23b: Zu der
grünen Lösung des η1-Acetylidkomplexes 10a wurde das Iminiumsalz 23b bei -30 °C addiert.
Nach dem die grüne Farbe verschwunden war, wurde die Reaktionsmischung aufgearbeitet.
Die Umkristallisierung des Rohprodukts ergab die orangen Kristalle des Aminocarben-
Komplexes 25 mit einer Ausbeute von 62% (Abbildung 41).
W CNC
O
O N
HN
ClO4
10a +THF
2523b (62%)
α
β γ
+
Abbildung 41. Darstellung des Aminocarben-Komplexes 25.
Das IR-Spektrum der Verbindung 25 zeigt eine NO-Absorption bei 1565 cm-1 und
eine Carbonyl-Absorption bei 1903 cm-1, die für die Aminocarben-Komplexe charakteristisch
ist.[35] Das 1H-NMR Spektrum der Verbindung 25 zeigt unter anderem das Proton an dem Cβ
bei δ 6.0 ppm. Die charakteristischen Signale im 13C-NMR sind die Absorptionen für Cα (δ
263.8 ppm), CO (δ 231.9 ppm).
Eine mechanistische Betrachtung für die Bildung des Aminocarben-Komplexes
25: Die Reaktion von 10a mit dem Iminiumsalz 23b führt zur Bildung des intermediären

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 30
Vinylidenkomplexes 25a. Für die Bildung der Verbindung 25 aus dem Komplex 25a sind
zwei Reaktionswege möglich. Entweder eliminiert 25a ein N-Methylanilin Molekül und
bildet den Allenylidenkomplex 26. Die nachfolgende nucleophile Addition des Amins an dem
Allenylidenkomplex 26 führt zur Bildung des Aminocarben-Komplexes 25 (Abbildung 42,
Reaktionsweg a).
W CNC
O
OC
H
N
CH3NPh
H
W CN
CO
OC C
10a + 23b
+
25a
26
(a)
(b)25
25
Abbildung 42. Möglicher mechanistischer Verlauf für die Bildung der Verbindung 25.
Die andere Möglichkeit ist eine [1,3] Verschiebung des Stickstoffes in dem
Vinylidenkomplex 25a, die dann direkt zum Produkt führt (Abbildung 42, Reaktionsweg b).
Die weiteren Untersuchungen, wie es später beschrieben wird, belegen den zweiten
Reaktionsweg.
Reaktion des η1-Acetylidkomplexes 10a mit dem Iminiumsalz 23c: Zu einer
Lösung des Vinylidenkomplexes 10 wurde n-Butyllithium Lösung in Hexan in einem leichten
Überschuss addiert. Nach dem sich der grüne η1-Acetylidkomplex 10a gebildet hatte, wurde
bei -30 ˚C das Iminiumsalz 23c addiert. Die Farbe des Reaktionsgemisches änderte sich sofort
zum rot. Danach wurde die Rektionsmischung auf 0 °C aufgewärmt, die Reaktionsfarbe
änderte sich langsam wieder zum grün. Nach drei Stunden war die Reaktion vollständig
(Abbildung 43). Nach der Aufarbeitung wurde das Rohprodukt chromatographiert. Die
Ausbeute der grünen Kristalle des Allenylidenkomplexes 26 betrug 70%.
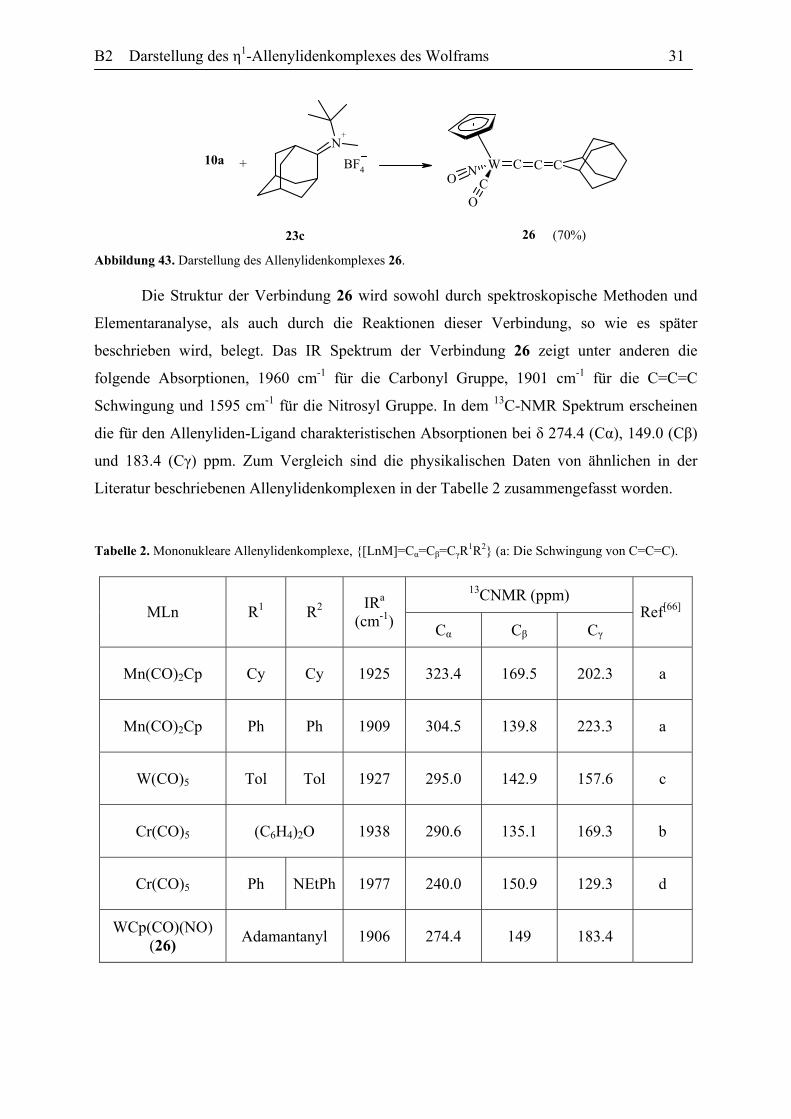
B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 31
N+
W CNC
O
OC CBF4+10a
26 (70%)23c Abbildung 43. Darstellung des Allenylidenkomplexes 26.
Die Struktur der Verbindung 26 wird sowohl durch spektroskopische Methoden und
Elementaranalyse, als auch durch die Reaktionen dieser Verbindung, so wie es später
beschrieben wird, belegt. Das IR Spektrum der Verbindung 26 zeigt unter anderen die
folgende Absorptionen, 1960 cm-1 für die Carbonyl Gruppe, 1901 cm-1 für die C=C=C
Schwingung und 1595 cm-1 für die Nitrosyl Gruppe. In dem 13C-NMR Spektrum erscheinen
die für den Allenyliden-Ligand charakteristischen Absorptionen bei δ 274.4 (Cα), 149.0 (Cβ)
und 183.4 (Cγ) ppm. Zum Vergleich sind die physikalischen Daten von ähnlichen in der
Literatur beschriebenen Allenylidenkomplexen in der Tabelle 2 zusammengefasst worden.
Tabelle 2. Mononukleare Allenylidenkomplexe, {[LnM]=Cα=Cβ=CγR1R2} (a: Die Schwingung von C=C=C).
13CNMR (ppm) MLn R1 R2 IRa
(cm-1) Cα Cβ Cγ
Ref[66]
Mn(CO)2Cp Cy Cy 1925 323.4 169.5 202.3 a
Mn(CO)2Cp Ph Ph 1909 304.5 139.8 223.3 a
W(CO)5 Tol Tol 1927 295.0 142.9 157.6 c
Cr(CO)5 (C6H4)2O 1938 290.6 135.1 169.3 b
Cr(CO)5 Ph NEtPh 1977 240.0 150.9 129.3 d
WCp(CO)(NO) (26) Adamantanyl 1906 274.4 149 183.4
B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 32
2.3 Die nucleophile Addition an dem Allenylidenkomplex 26.
Die mesomeren Strukturen der Verbindung 26 ordnen eine positive Ladungsdichte den
Kohlenstoffatomen Cα und Cγ zu.[67] Dementsprechend führt die Addition eines Nucleophils
an dieser Verbindung entweder zur Bildung eines Carbenkomplexes (26c), oder eines
Vinylidenkomplexes (26d) (Abbildung 44). Im weiteren Verlauf wurde die Reaktion der
Verbindung 26 mit verschiedenen Nucleophilen untersucht.
[W] C C C [W] C C C[W] C C C
Nu-H Nu-H
C[W] C
C
Nu
H
[W] C CH
CNu
+
26b 26 26a
[W] = W(Cp)(CO)(NO)
26Addition an Cα Addition an Cγ
26c 26d
+
Abbildung 44. Die nucleophile Addition an dem Allenylidenkomplex.
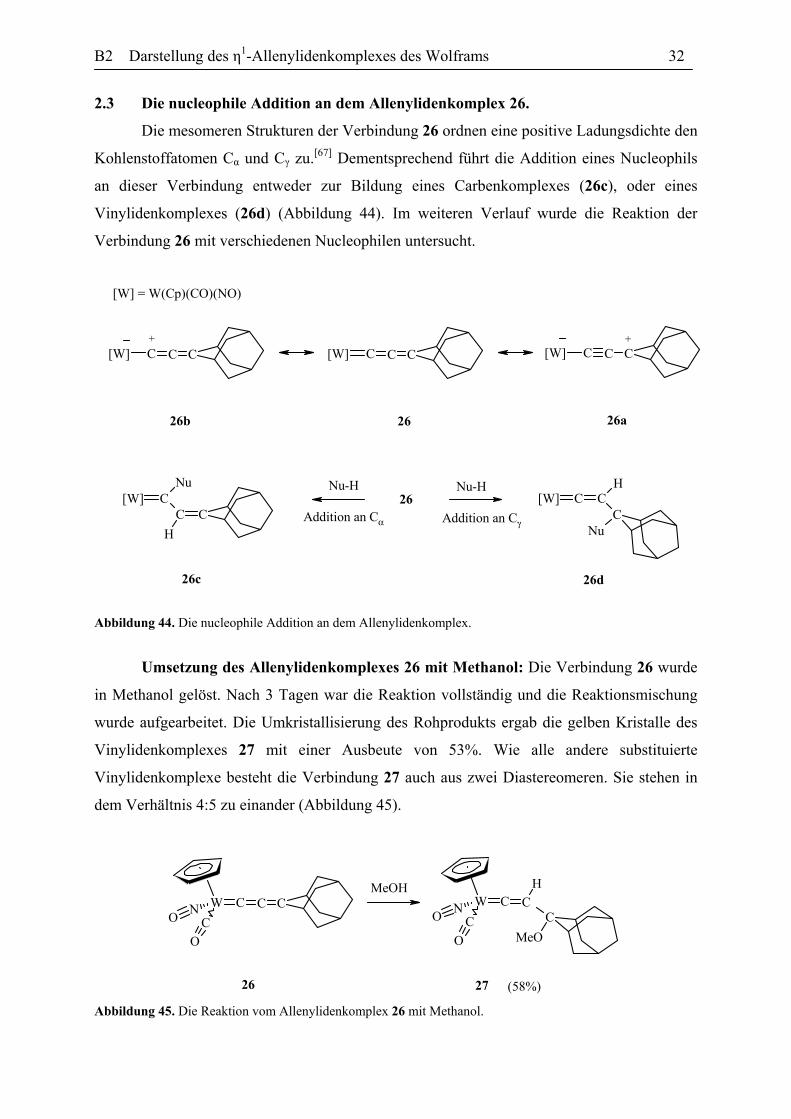
Umsetzung des Allenylidenkomplexes 26 mit Methanol: Die Verbindung 26 wurde
in Methanol gelöst. Nach 3 Tagen war die Reaktion vollständig und die Reaktionsmischung
wurde aufgearbeitet. Die Umkristallisierung des Rohprodukts ergab die gelben Kristalle des
Vinylidenkomplexes 27 mit einer Ausbeute von 53%. Wie alle andere substituierte
Vinylidenkomplexe besteht die Verbindung 27 auch aus zwei Diastereomeren. Sie stehen in
dem Verhältnis 4:5 zu einander (Abbildung 45).
W CNC
O
OC C W CN
CO
OC
H
CMeO
26 27 (58%)
MeOH
Abbildung 45. Die Reaktion vom Allenylidenkomplex 26 mit Methanol.

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 33
Die vorgeschlagene Vinylidenstruktur für die Verbindung 27 wird durch die Spektral-
Daten und Elementaranalyse bestätigt. Das IR Spektrum zeigt die Absorptionen für den
Carbonyl-Liganden bei 2007 und 1996 cm-1 und für Nitrosyl-Liganden bei 1641 und 1600
cm-1. In dem 13C-NMR Spektrum erscheinen die Signale für Cα bei 340.9 und 333.5 ppm und
die Signale für Cβ bei 131.1 und 131.2 ppm. Diese Daten stimmen mit den spektroskopischen
Daten anderer Vinylidenkomplexe überein (s. Tabelle 3).
Tabelle 3. Vergleich der Spektroskopischen Daten der η1-Vinyliden-Komplexe, 7, 24 und 27.
7 24 27
1H-NMR, CDCl3 (ppm)
CβH s, 5.73, 5.69 s, 6.66, 6.60 s, 5.37, 5.34
13CNMR, CDCl3 (ppm)
Cα 338.7, 338.4 346.9, 346.5 340.9, 333.5
Cβ 149.0, 139.5 128.5, 128.2 131.2, 131.1
IR( KBr, cm-1)
CO 1994 2007 2007, 1996
Es wurde auch versucht die Struktur der Verbindung 27 durch eine
Kristallstrukturanalyse zu belegen. Leider ergaben die Kristallisationsversuche nur Zwillinge
anstelle der Einkristalle. Daher ergab die Bewertung der Daten hohe R-Werte und des
Weiteren könnte die Raumgruppe nicht vermittelt werden. Der ORTEP Plot der Verbindung
27 ist in der Abbildung 46 zusehen.
Die Reaktion der Verbindung 27 mit Methanol zeigte, dass die Reaktion mit dem
Nucleophilen an dem Cγ Kohlenstoffatom des Allenylidenkomplexes stattfindet.

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 34
Abbildung 46. ORTEP Plot der Verbindung 27.
Umsetzung des Allenylidenkomplexes 26 mit Aminen: Eine Lösung vom Pyrrolidin
in THF wurde einer Lösung der Verbindung 26 in THF bei -10 °C zugetropft. Nach einer
kurzen Zeit änderte sich die Farbe der Reaktionsmischung zum Rot. Nach der Aufarbeitung
ergab die Chromatographie des Rohproduktes den Carbenkomplex 28 mit einer Ausbeute von
35%. Die anderen potentiellen Nebenprodukte konnten leider nicht identifiziert werden. Die
Verbindung 28 besteht aus zwei Diastereomeren mit dem Verhältnis 8:1. Die
spektroskopischen Methoden, besonderes NMR Daten, bestätigen die eindeutige Zuordnung
der vorgeschlagenen Struktur der Verbindung 28 und stimmen mit den ähnlichen in der
Literatur beschriebenen Strukturen.[68] Das 1H-NMR Spektrum des Carbenkomplexes 28 zeigt
zwei Dubletten bei δ 14.20 und 8.03 ppm, mit einer Kopplungskonstante 3JH-H = 13.29 Hz.
Die erste Absorption gehört dem Carbenproton an dem Cα und die zweite wird dem Cβ-Proton
zugeordnet. Das 13C-NMR Spektrum der Verbindung 28 zeigt auch die charakteristische
Signale für Carbenkomplexe unter anderem die Absorptionen bei δ 274 ppm für Cα, 138.0
und 138.6 ppm für Cβ und 153.5 ppm für Cγ.
Aus mechanistischer Sicht führt die nucleophile Addition des Pyrrolidins an dem Cγ
der Verbindung 26 zur Bildung des intermediären Vinyliden Komplexes 28b. Dieser
Komplex bildet in Folge einer Retro-En-Reaktion die Verbindung 28 (Abbildung 47).

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 35
W CN
CO
O H
C CH
W CNC
O
OC C N
H
W CN
CO
OC
C
H
N
α
β γ
28 (35%)
THF
28b
26
+
Abbildung 47. Die Reaktion des Allenylidenkomplexes mit Pyrrolidin.
Um diesen Mechanismus zu prüfen, wurde das Iminiumsalz 23d dargestellt und mit
der grünen Lösung des Acetylidkomplex 10a umgesetzt, was eine rasche Farbänderung zum
Rot zur Folge hatte. Nach der Aufarbeitung und Umkristallisierung erwies sich das Produkt
als Carbenkomplex 28 (61%) (Abbildung 48).
W CN CO
OH
C CH
W C CN C
OO
H
Li+
N+
ClO4
-
10a
+α
β
γ
23d 28 (61%)
THF
Abbildung 48. Darstellung des Carbenkomplexes 28.
Als nächstes wurde die Reaktion der Allenylidenkomplex 26 mit N-Methylanilin
untersucht. Diese Reaktion war besonderes interessant. Denn es konnte den Mechanismus der
Bildung des Aminocarben-Komplexes 25 aus der Reaktion vom Acetylidkomplex 10a und
das Iminiumsalz 23c erleuchten (Abbildung 42). Da wurde postuliert, dass es entweder die
Verbindung 25 durch die Reaktion des Amins mit dem gebildeten Allenylidenkomplex
zustande kommt, oder durch eine direkte [1,3] Verschiebung des Amins aus dem
intermediären Vinylidenkomplex 25a.
Die Reaktion von N-Methylanilin mit einer Lösung des Allenylidenkomplexes 26 in
THF zeigte keinen Umsatz. Nach zwei Tagen wurde der unreagierte Allenylidenkomplex

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 36
quantitativ isoliert. Dieses Ergebnis zeigte, dass die Nucleophilie des N-Methylanilins für die
Reaktion mit der Verbindung 26 nicht ausreicht. Daher kann die [1,3] Verschiebung des
Stickstoffes in dem intermediären Vinyliden Komplex 25a der einzige plausible
Mechanismus für die Bildung der Verbindung 25 sein (Abbildung 42 Reaktionsweg b).
Die Umsetzung einer Lösung der Verbindung 26 in THF mit Diethylamin führte zu
einer schnellen Änderung der Farbe der Reaktionslösung zum rot. Die Aufarbeitung des
Rohproduktes und die anschließende Umkristallisierung ergab die rote Kristalle des
Aminocarben-Komplexes 29 mit einer Ausbeute von 65% (Abbildung 49).
W CN CO
ON
C CH
W CN C
OO
C C N
H
W CN
CO
OC
C
H
N
α
β γ
THF+
26 29 (65%)
29b Abbildung 49. Die Reaktion der Verbindung 26 mit Diethylamin.
Die Struktur der Verbindung 29 ist eindeutig durch die spektroskopischen Daten
belegt worden. Das IR-Spektrum der Verbindung 29 zeigt das charakteristische Signal der
Carbonyl Gruppe bei 1895 cm-1. In dem 13C-NMR erscheinen die Signale für Cα bei 255.4, für
Cβ bei 122.8 und für Cγ bei 141.6 ppm. Auch hier, analog zu den vorhin beschriebenen
nucleophilen Additionen an dem Allenylidenkomplex 26, dürfte das Amin erst an dem Cγ
angreifen, um den intermediären η1-Vinylidenkomplex 29b zu bilden. Dann die [1,3]
Verschiebung des Stickstoffs führt zum Produkt.
2.4 Schlussfolgerung
Die Reaktion des η1-Acetylidkomplexes 10a mit dem Iminiumsalz bildet den
intermediären Vinylidenkomplex 15 (Abbildung 37). Ziel dieser Untersuchungen war es
durch die Wahl geeignet substituierter Iminiumsalze, die Retro-En-Reaktion zu unterdrücken,
damit eine Amineliminierung zum entsprechenden Allenylidenkomplex führt. Dabei hat es

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 37
sich herausgestellt, dass die Retro-En-Reaktion gegenüber Änderung des Aminfragmentes
sehr empfindlich beeinflusst wird.
W CN CO
O
CH
NPh
ClO4
W CN
CO
O N
H
Ph
N
Ph
ClO4-
N+
W CN CO
OH
C CH
N+
W CNC
O
OC C
BF4
ClO4-
W C CN C
OO
H
Li+
23a
25
23b
24
-
+
+
23d
28
23c
26
-
10a
Abbildung 50. Die Zusammenfassung der in dieser Arbeit berichteten Reaktionen von 10a und Iminiumsalzen.
Somit wurden bei der Umsetzung der Verbindung 10a mit den Iminiumsalzen 23a,
23b oder 23c keine Carbenkomplexe gebildet. Allerdings geht die Reaktion von 23b mit 10a
eine [1,3] Amin-Verschiebung zum Aminocarben-Komplex 25 ein. Die Umsetzung des
Iminiumsalzes 23a mit 10a führt zu einem Allenylidenkomplex, der auf Grund des aciden Cδ
Protons zu dem Vinylidenkomplex 24 umlagert. Nur das Iminiumsalz 23c reagiert mit der
Verbindung 10a zur Bildung eines stabilen Allenylidenkomplexes 26. Diese Ergebnisse sind
in der Abbildung 50 zusammengefasst.
Des Weiteren wurde die Addition verschiedener Nucleophilen an dem
Allenylidenkomplex 26 untersucht (Abbildung 51). Das Nucleophil greift die Verbindung 26
an dem Cγ an und es entsteht ein substituierter Vinylidenkomplex, dessen weitere
Entwicklung von den chemischen Eigenschaften des agierenden Nucleophils abhängt. Wenn

B2 Darstellung des η1-Allenylidenkomplexes des Wolframs 38
Methanol als Nucleophil eingesetzt wird, ist der entstandene Vinylidenkomplex stabil und
lässt sich isolieren (die Verbindung 27). Die Umsetzungen der Verbindung 26 mit dem
Diethylamin beziehungsweise Pyrrolidin, widerspiegelt das bekannte Reaktionsmuster des
intermediär entstandenen η1-Vinylidenkomplexes:
- Das mit Pyrrolidin substituierte Zwischenprodukt geht eine Retro-En-Reaktion zum
Carbenkomplex 28.
- Für das mit Diethylamin substituierte Intermediat ist eine Retro-En-Reaktion nicht günstig
(Abschnitt 2.4), daher lagert es mit einer [1,3] Verschiebung des Amins zum Aminocarben-
Komplex 29 um.
W CNCO
C C
O
W CNC
O
OC
H
CMeO
W CN
CO
O H
C CH
W CN
CO
O N
C CH
CH3OH
NH
NH
26
2728
29
Abbildung 51. Die nucleophile Addition des Amins an dem Allenylidenkomplex.

B3 Darstellung der η2-Allenkomplexe des Wolframs 39
3 Die Reaktion von Diazoalkanen mit dem η1-Vinylidenkomplex des Wolframs;
Darstellung der η2-Allenkomplexe des Wolframs
3.1 Vormerkungen
Die Vinylidenkomplexe [(η5-C5H5)(CO)(NO)W=C=CHR] können durch die
Umsetzung des entsprechenden Lithiumacetylid mit dem [(η5-C5H5)(CO)2(NO)W] (9)
hergestellt werden.[69] Um die Probleme mit der Handhabung des Propins für die Herstellung
des [(η5-C5H5)(CO)(NO)W=C=CH(CH3)] (30) zu vermeiden, wurde das Lithiumpropinylid
durch die Dehydrobrominierung des 2-Brompropen und anschließende Lithierung des
entstandene Propins in situ hergestellt und mit dem Wolframkomplex [(η5-
C5H5)W(CO)2(NO)] (9) umgesetzt und den entstandenen η1-Acetylidkomplex protoniert.[36]
Allerdings ist die Ausbeute dieser Reaktion ist gering.
Der acide Charakter des Protons in den Vinylidenkomplexen der Konstitution
M=C=C(H)R ist belegt und ergiebig studiert.[70] Der nach der Deprotonierung entstandene η1-
Acetylidkomplex kann dann mit den Elektrophilen reagieren.[70c,d] Als eine einfachere
Methode für die Darstellung der Vinylidenkomplexe [(η5-C5H5)(CO)(NO)W=C=CHCH3]
wurde die Reaktion des Vinylidenkomplexes 10 mit Diazomethan näher studiert.
W C CH
HNC
OO
CH2N2 W C CH
CH3N
CO
O
10
+
30
?
Abbildung 52. Die Reaktion von der Verbindung 10 mit Diazomethan.
3.2 Umsetzung von Vinylidenkomplex 10 mit Diazomethan: Darstellung und die
Diskussion der spektroskopischen Daten des η2 -Allenkomplexes 31
Die Umsetzung des Vinylidenkomplexes 10 mit einer etherischen Lösung von
Diazomethan erfolgte bei –30 °C. Der Verlauf der Reaktion wurde mit der IR-Spektroskopie
verfolgt. Nach der Aufarbeitung und Umkristallisierung wurden die hellgelben Kristalle des
η2-Allenkomplexes 31 mit einer Ausbeute von 61% isoliert. Diese Verbindung ist in den
meisten organischen Lösungsmitteln gut löslich und wurde eindeutig mit den gängigen
Spektroskopischen Methoden als auch Elementaranalyse charakterisiert. Die
spektroskopischen Eigenschaften der Verbindung 31 stimmen mit den Spektraldaten der

B3 Darstellung der η2-Allenkomplexe des Wolframs 40
anderen in der Literatur beschriebenen η2-Allen-Metall-Komplexe überein.[71] Die Einkristall-
Röntgensstruktur-Analyse bestätigt des Weiteren die vorgeschlagene Struktur der Verbindung
31. Das IR Spektrum dieser Verbindung zeigt unter anderem eine Carbonylabsorption bei
1988 cm-1 und eine breite NO-Absorption bei 1599 cm-1. Aufgrund der 1H-NMR Daten
besteht die Verbindung 31 aus zwei Diastereomeren in einem Verhältnis von 2:1. Das 1H-
NMR Spektrum (400MHz, CDCl3) zeigt für Hauptdiastereomer unter anderen drei
Signalsätze bei δ 1.86, 6.09 und 6.91 ppm. Entsprechende Signale wurden auch in den
Spektraldaten anderer η2-Allenkomplexe, wie z.B. [(η5-C5Me5)Re(CO)2 (CH2=C=CH2)][71c]
und [(η5-C5H5)Fe(CO)(PPh3)(CH2=C=CH2)]+BF4-.[71d] beobachtet. Verglichen mit der
Absorption des freien Allens, H2C=C=CH2 (δ 4.45, CCl4) erscheint das erste Triplet im
Hochfeld,[72] deswegen wird der an dem Metal koordinierten Doppelbindung zugeordnet. Die
beiden ins Tieffeld verschobenen Tripletts bei δ 6.09 und 6.91 ppm werden den beiden
geminalen Protonen an der freien Doppelbindung des η2-Allenkomplexes zugeordnet. Die
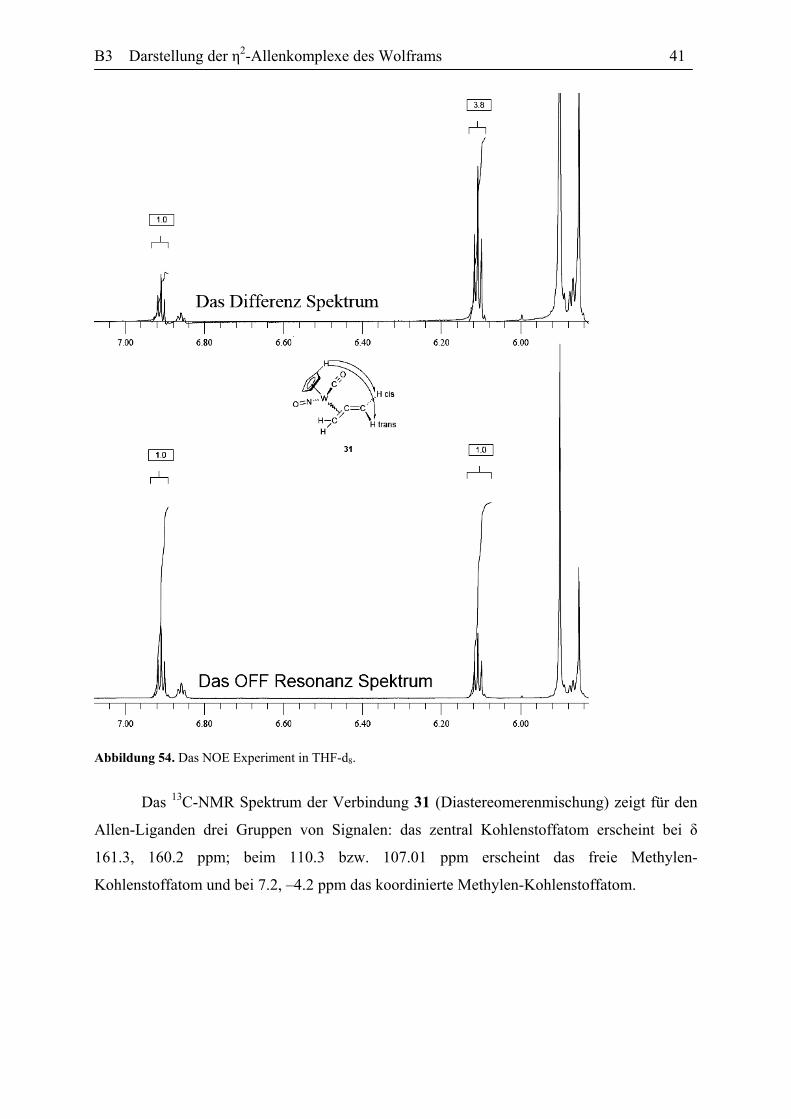
Differenz-Kern-Overhauser-NMR Versuche zeigen, dass die Absorption bei δ 6.09 ppm dem
Proton in der cis Stellung zum Metal Zentrum und die Absorption bei δ 6.91 ppm zu dem
trans positionierten Proton angehören. Durch die Sättigung der Cyclopentadienyl-Protonen ist
die Intensitätszunahme in der Absorption für das dem Cyclopentadienyl-Ligand näher
gelegene cis Proton größer. Das OFF-Resonanz und Differenz-Spektrum sind in der
Abbildung 54 wiedergegeben.
Die Protonen an der freien C=C-Doppelbindung zeigen eine long-range Kupplung mit
einer Kupplungskonstante von 4JH-H = 3.5 Hz. Der zweite Diastereomer zeigt vier Gruppen
von Signalen für die Protonen des Allen-Liganden. Dabei weisen die Protonen an der
koordinierten Doppelbindung des Allen-Liganden eine geminale Kupplung von 2JH-H = 11 Hz.
Eine ähnliche geminale Kupplung für das Hochfeld-Signal des Hauptdiastereomers wird nicht
beobachtet.
B3 Darstellung der η2-Allenkomplexe des Wolframs 41
Abbildung 54. Das NOE Experiment in THF-d8.
Das 13C-NMR Spektrum der Verbindung 31 (Diastereomerenmischung) zeigt für den
Allen-Liganden drei Gruppen von Signalen: das zentral Kohlenstoffatom erscheint bei δ
161.3, 160.2 ppm; beim 110.3 bzw. 107.01 ppm erscheint das freie Methylen-
Kohlenstoffatom und bei 7.2, –4.2 ppm das koordinierte Methylen-Kohlenstoffatom.

B3 Darstellung der η2-Allenkomplexe des Wolframs 42
Die Bildung des η2-Allenkomplexes 31 wird durch einen nucleophilen Angriff des
Diazomethans auf dem Cα des Vinylidenkomplexes 10 ausgelöst der dann über die Betaine
31a bzw. 31b zum Produkt führt (Abbildung 55).
W CC
H
H
NC
O
O C N2HH
W CC
H
H
NC
O
O CHH
W CC
H
H
NC
O
O CHH
WC
N
O
OC C
H
HCHH
WC
N
O
OC C
H
HCH
H
++
+
10
31a
-N2CH2N2
+
31b
+
31 Abbildung 55. Der postulierte Mechanismus für die Bildung der Verbindung 31.
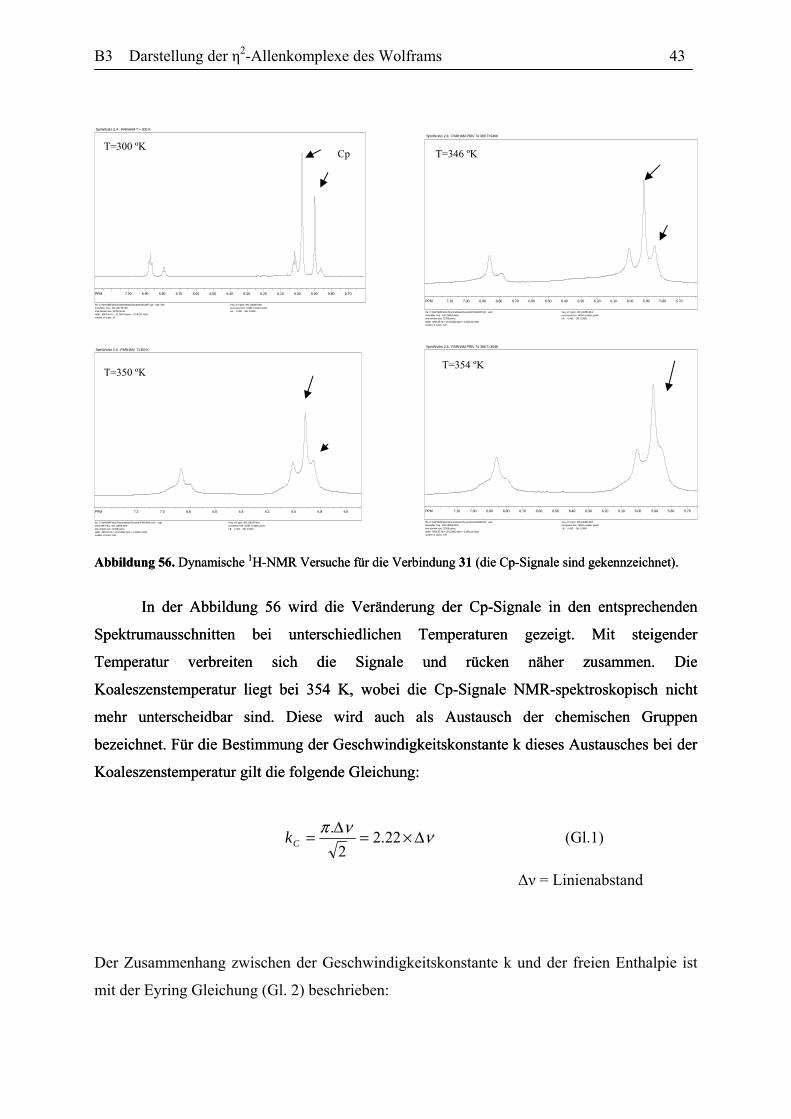
Dynamisches Verhalten des η2-Allenkomplexes 31: Wie bereits erwähnt wurde,
besteht die Verbindung 31 aus zwei Diastereomerenpaare, die sich in der relativen Anordnung
des Allen-Liganden zum Carbonyl- und Nitrosyl-liganden am Wolframatom unterscheiden
(Abbildung 55). Da die Trennungsversuche der beiden Isomerenpaare, sowohl durch die
Säulenchromatographie als auch mit der HPLC erfolglos blieben, erschien es sinnesvoll
anzunehmen, dass die beiden Isomerenpaare in einem Gleichgewicht zu einander stehen, und
die Energiebarriere für diese Reaktion soweit niedrig ist, dass es die Trennung bei
Raumtemperatur nicht gestattet.
Für die Bestimmung der Freien Enthalpie dieses Prozesses wurden 1H-NMR von der
Verbindung 31 in DMSO-d6 bei verschiedenen Temperaturen aufgenommen.
B3 Darstellung der η2-Allenkomplexe des Wolframs 43
SpinWorks 2.4: PARHAM T = 300 K
PPM 7.00 6.90 6.80 6.70 6.60 6.50 6.40 6.30 6.20 6.10 6.00 5.90 5.80 5.70
file: C:\Nmr\NMR labor\Diazomethane\Dynamik\OK200F.118 expt: X00transmitter freq.: 400.138778 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154414 ppm = 0.246111 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.136286 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
SpinWorks 2.4: PARHAM PRIV 74 368 T=346K
PPM 7.10 7.00 6.90 6.80 6.70 6.60 6.50 6.40 6.30 6.20 6.10 6.00 5.90 5.80 5.70
file: C:\Nmr\NMR labor\Diazomethane\Dynamik\PARHAM7.001 expt: transmitter freq.: 400.138035 MHztime domain size: 32768 pointswidth: 8064.55 Hz = 20.154423 ppm = 0.246111 Hz/ptnumber of scans: 128
freq. of 0 ppm: 400.136295 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
SpinWorks 2.4: PARHAM T=350 K
Cp
Abbildung 56. Dynamische 1H-NMR Versuche für die Verbindung 31 (die Cp-Signale sind gekennzeichnet). Abbildung 56. Dynamische
In der Abbildung 56 wird die Veränderung der Cp-Signale in den entsprechenden
Spektrumausschnitten bei unterschiedlichen Temperaturen gezeigt. Mit steigender
Temperatur verbreiten sich die Signale und rücken näher zusammen. Die
Koaleszenstemperatur liegt bei 354 K, wobei die Cp-Signale NMR-spektroskopisch nicht
mehr unterscheidbar sind. Diese wird auch als Austausch der chemischen Gruppen
bezeichnet. Für die Bestimmung der Geschwindigkeitskonstante k dieses Austausches bei der
Koaleszenstemperatur gilt die folgende Gleichung:
In der Abbildung 56 wird die Veränderung der Cp-Signale in den entsprechenden
Spektrumausschnitten bei unterschiedlichen Temperaturen gezeigt. Mit steigender
Temperatur verbreiten sich die Signale und rücken näher zusammen. Die
Koaleszenstemperatur liegt bei 354 K, wobei die Cp-Signale NMR-spektroskopisch nicht
mehr unterscheidbar sind. Diese wird auch als Austausch der chemischen Gruppen
bezeichnet. Für die Bestimmung der Geschwindigkeitskonstante k dieses Austausches bei der
Koaleszenstemperatur gilt die folgende Gleichung:
1H-NMR Versuche für die Verbindung 31 (die Cp-Signale sind gekennzeichnet).
ννπ Δ×=Δ= 22.22
.Ck (Gl.1)
Δν = Linienabstand
Der Zusammenhang zwischen der Geschwindigkeitskonstante k und der freien Enthalpie ist
mit der Eyring Gleichung (Gl. 2) beschrieben:
PPM 7.2 7.0 6.8 6.6 6.4 6.2 6.0 5.8 5.6
SpinWorks 2.4: PARHAM PRIV 74 368 T=354K
PPM
file: tr
C:\Nmr\NMR labor\Diazomethane\Dynamik\PARHAM11.001 expt: ansmitter freq.: 400.138035 MHz
e domain size: 32768 pointsh: 8064.55 Hz = 20.154423 ppm = 0.246111 Hz/ptber of scans: 128
freq. of 0 ppm: 400.136295 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000tim
widtnum
7.10 7.00 6.90 6.80 6.70 6.60 6.50 6.40 6.30 6.20 6.10 6.00 5.90 5.80 5.70
T=300 ºK T=346 ºK
T=354 ºK T=350 ºK
file: C:\Nmr\NMR labor\Diazomethane\Dynamik\PARHAM9.001 expt: transmitter freq.: 400.138035 MHztime domain size: 32768 pointswidth: 8064.55 Hz = 20.154423 ppm = 0.246111 Hz/ptnumber of scans: 128
freq. of 0 ppm: 400.136295 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000

B3 Darstellung der η2-Allenkomplexe des Wolframs 44
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−
= CTRG
CB ehTk
xk .
#
..
. (Gl. 2)
kB = Boltzmann-Faktor,
x = Transmissionkoeffizient
(in der Regel x=1),
Tc = Koalesznestemperatur,
h = Planksches Wirkungsquantum.
Daraus resultiert die folgende Gleichung:
1# .log32.1014.19 −⎟⎟⎠
⎞⎜⎜⎝
⎛+=Δ molJ
kT
TGC
CCC (Gl. 3)
Bei der Koaleszenstemperatur von Tc = 354 K und kC = 44.66 s-1, ist die frei Enthalpie 77
KJ.mol-1.
Die Kombination der Gleichung 3 mit der klassischen Gleichung der freien Enthalpie
(Gl. 4), setzt das Wertepaar (TC, kC) mit der Aktivierungsenthalpie ΔH# und ΔS# in einer
Beziehung (Gl.5): ### STHG Δ−Δ=Δ (Gl. 4),
4.1914.1932.10log
## ST
HTk Δ+Δ−= (Gl. 5).
Durch das Auftragen von log (k/T) gegen (1/T) können die ΔH# und ΔS# grafisch
ermittelt werden. Mit einer Linearregression ergeben sich die entsprechenden Werte:
ΔH# -272.51 J.mol-1
ΔS# -212.8 J.mol-1
ΔG# 77 KJ.mol-1
Die Größenordnung des berechneten Werts für die freie Enthalpie verdeutlicht es, dass
eine Trennung der Isomere bei Raumtemperatur nicht möglich ist. Die beiden Diastereomere
können mit einem ionischen oder einem radikalischen Prozess in einander überführt werden
(Abbildung 61).

B3 Darstellung der η2-Allenkomplexe des Wolframs 45
WC
N
O
OC C
H
HCH
H
WC
N
O
O
C C
H
HCH
H
W C
N
O
O C CH
HC
HH
WC
N
O
O CC H
HC
HH
W C
N
O
O CC
H
HC
HH
W
C
N
O
O C CH
HC
HH
WC
N
O
O C C
H
HC
HH
WC
N
O
O CC
+H
H
CHH+
+
Abbildung 57. Die mögliche Reaktionswege, die die beiden Diastereomerenpaare der Verbindung 31 ineinander überführen.
Röntgenstrukturanalyse der Verbindung 31
Die geeignete Einkristalle wurde von einer Pentanlösung der Verbindung 31 bei 4 °C
gezüchtet. Die kristallografische Daten sind in der Tabelle E 4.1 zusammengefasst. Der
ORTEP Plot der Verbindung 31 ist in der Abbildung 58 zusehen. Die molekulare Struktur des
η2-Allenkomplexes 31 ist ähnlich wie andere η2-Allenkomplexe.[71] Der Allen-Ligand ist
gebogen, der C1-C2-C3 Winkel beträgt 144.9(15)˚. Dieser Wert für C=C=C Winkel fällt in
der berichtete Grenze für Allenkomplexe von 134.5˚ bis 160˚.[73] Die Bindungslänge der
koordinierten Doppelbindung des Allens (C1=C2) ist mit 1.46(2) Ǻ länger als die freie
Doppelbindung (C2=C3) mit 1.330(7) Ǻ. Der Allen-Ligand ist fast symmetrisch an das
Wolframzentrum koordiniert, der Unterschied in der Bindungslänge von W-C1 und W-C2
beträgt nur 0.03 Ǻ.

B3 Darstellung der η2-Allenkomplexe des Wolframs 46
Abbildung 58. ORTEP Plot von dem Komplex 31 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen): W1 N1 1.767(10), W1 C 2.027(14), W1 C2 2.185(14), W1 C1 2.188(14), W1 C11 2.310(13), C11 C12 1.36(2), C11 C15 1.44(3), C1 C2 1.46(2), C2 C3 1.31(2), O1 N1 1.234(13), N1 W1 C 91.3(5), N1 W1 C2 103.8(5), C W1 C2 70.6(6), N1 W1 C1 93.1(5), C W1 C1 108.1(6), C2 W1 C1 38.9(6).
3.3 Darstellung der η2-Allenkomplexe 32 und 33.
Die Umsetzung von Diazoessigsäureethylester mit einer Lösung des
Vinylidenkomplexes 10 erfolgte in THF bei 45 °C. Die Aufarbeitung der Reaktionsmischung
ergab eine Mischung von zwei Diastereomeren η2-Allenkomplexe 32 und 33 in einem
Verhältnis von 5:1 mit einer Ausbeute von 87% (Abbildung 59).
N2CH2COOCH2CH3W
C
N
O
O C C
H
COOC2H5CH
H
WC
N
O
OC C
H
COOC2H5
CH
H
10 +THF, 450 C
+
32 33
1
2
3 4
Abbildung 59. Darstellung der Verbindungen 32 und 33.
Die Verbindungen 32 und 33 wurden mit Hilfe der HPLC getrennt. Die Zuordnung der
angegebenen Strukturen 32 und 33 sind durch die spektroskopischen Daten als auch mit Hilfe
der Einkristall-Röntgenstrukturanalysen belegt. Der wichtigste strukturelle Unterschied der
beiden Verbindungen liegt in der relativen Anordnung der Estergruppe zum Wolframatom.
B3 Darstellung der η2-Allenkomplexe des Wolframs 47
Dementsprechend ist die Verbindung 32 E und die Verbindung 33 Z konfiguriert. Die
Verbindung 32 selbst besteht aus zwei Diastereomeren mit einem Verhältnis 3:5. Die
wichtigsten spektroskopischen Daten der Verbindungen 31-33 sind in der Tabelle 4
aufgelistet worden:
Tabelle 4. Die spektroskopischen Daten der Verbindungen 31-33.
31[74] 32[76] 33 1H-NMR Daten (400 MHz, CDCl3)
(ppm)
Cp 5.66, s 5.72, s 5.80, s
C(3)-H 6.91, 6.86, t 6.67, 6.47, t 7.43, t
C(1)-H 1.77, t 2.58, 2.24, dd 1.77, 1.33, dd 13C-NMR Daten (100 MHz, CDCl3)
(ppm)
CO 213.7, 215 213.0, 210.8 212.9
Cp 95.9, 95.5 96.0, 95.6 96.7
C(1) 110.3, 107.1 10.4, -0.2 3.9
C(2) 161.3, 160.2 165.9, 164.9 167.8
C(3) 7.2, -4.2 118.9, 115.7 117.0
C(4) - 179.1, 176.4 180.0
IR-Daten (KBr-Prssling) (cm-1)
CO 1988 2006 2030b
NO 1599 1681, 1598 1671, 1625
CO (Ester) - 1717 1703
Kristallstrukturanalyse der Verbindungen 32 und 33: Die passenden Einkristalle
der Verbindungen 32 und 33 sind aus ihrer Pentanlösung bei 4 °C gezüchtet worden. Die
kristallografische Daten sind in der Tabelle E 5.1 sowie E 6.1 zusammengefasst. Der ORTEP
Plot der Verbindung 32 ist in der Abbildung 60 und der von Verbindung 33 in der Abbildung
61 zusehen.

B3 Darstellung der η2-Allenkomplexe des Wolframs 48
Abbildung 60. ORTEP Plot von dem Komplex 32 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen: W1-N1 1.795(5), W1-C 2.009(5), W1-C2 2.112(5), W1-C1 2.250(6), N1-O1 1.188(6), C-O2 1.121(6), C1-C2 1.402(7), C2-C3 1.330b(7), C3-C4 1.454(7), C4-O3 1.203(6), N1-W1-C 90.7(2), N1-W1-C2 90.0(2), C-W1-C2 108.9(2), N1-W1-C1 102.2(2), C-W1-C1 73.6(2), C2-W1-C1 37.3(2), O1-N1-W1 176.7(5), O2-C-W1 179.4(5), C2-C1-W1 66.0(3), C3-C2-C1 139.7(5), C3-C2-W1 143.0(4), C1-C2-W1 76.7(3), C2-C3-C4 124.2(5).
Abbildung 61. ORTEP Plot von dem Komplex 33 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen: W1-N1-1.783(5), W1-C 1.964(7), W1-C2 2.175(5), W1-C1 2.220(6), C1-C2 1.393(8), C2-C3 1.314(8), C3-C4 1.478(9), N1-O1 1.212(6), N1-W1-C 92.6(3), N1-W1-C2 99.5(2), C-W1-C2 74.9(3), N1-W1-C1 92.0(2), C-W1-C1 111.4(3), C2-W1-C1 37.0(2), C2-C1-W1 69.8(3), C3-C2-C1 142.1(6), C3-C2-W1 144.5(5), C1-C2-W1 73.3(3), C2-C3-C4 122.4(7), O1-N1-W1 173.8(4), O2-C-W1 178.1(7).

B3 Darstellung der η2-Allenkomplexe des Wolframs 49
3.4 Schlussfolgerung
Der Vinylidenkomplex 10 reagiert mit Diazomethan und Diazoessigsäureethylester, zu
den entsprechenden η2-Allenkomplexen. Wobei Diazomethan bei –30 °C schon die
Umsetzung zur Verbindung 31 eingeht, reagiert der weniger reaktive
Diazoessigsäureethylester mit dem Vinylidenkomplex 10 bei 45 °C zu 32 und 33. Die
Molekularstrukturen der Verbindungen 31-33 sind durch Röntgenspektroskopie gesichert.
Ferner wurde die Energiebarriere für Umwandlung der beiden Diastereomer der Verbindung
31 in einander mit der Hilfe der Dynamischen NMR-Methoden bestimmt.

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 50
4 Die Hydrolyse von Phosphorkomplexen des Wolframs
4.1 Vormerkungen
Innerhalb der Arbeitsgruppe Ipaktschi wurde über die Darstellung von den
chlorhaltigen η2-Phosphinovinylidenkomplexe des Wolframs 11a-c berichtet.[36] Mit der
Absicht einen Zugang zu den Metallkomplexen mit einem Hydroxy-Liganden, LnM-OH, zu
eröffnen, wurden die Hydrolyse der Verbindung 11a-c studiert (Abbildung 62).
W PN ClO
HH
R
RW P
N OO
HH
R
ROH-
?11a-c
Ph
(CH3)3C-11b
11c (CH3)2CH-
R
11a
Abbildung 62.
4.2 Darstellung von Phosphorkomplexen des Wolframs 11a-c
Die Phosphorkomplexe des Wolframs 11a-c wurden durch die Umsetzung des
jeweiligen Chlorophosphins 34a-c mit dem Vinylidenkomplex 10 dargestellt (Abbildung 63).
W PN ClO
HH
R
RW C
CNO
C
O
H
HP
RR
Cl+
10
Ph
(CH3)3C-b
c (CH3)2CH-
R
34a-c
α
β
11a-c
a-CO
Abbildung 63. Darstellung der η2-Phosphinovinyllidenkomplexe des Wolframs 11a-c.
Die Spektraldaten der Verbindungen 11a-c sind in der Tabelle 5 zusammengefasst.
Die wichtigsten charakteristischen Spektraldaten der Verbindung 11c sind hier besprochen.
Das IR-Spektrum (KBr) der Verbindung 11c zeigt unter anderen zwei NO Schwingungen bei
ν 1611 und 1591 cm-1. Das 31P-NMR Spektrum hat eine Absorption bei δ -57.6 ppm, die eine
W-P Kupplung von 131.0 Hz aufweist. Bei dem 1H-NMR Spektrum sind die Signale der
olefinischen Protonen charakteristisch für die magnetisch nicht äquivalenten Protonen. Die
Zuordnung der Signale erfolgte durch den Vergleich des Jeweiligen 3JP-H
Kopplungskonstanten. Somit gehört das Signal bei δ 7.42 ppm (3JP-H = 12,6 Hz) dem Proton

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 51
an, welches dem Phosphoratom cis steht, und das dem P-Atom trans-ständige Proton zeigt
eine Verschiebung bei δ 6.43 ppm (3JP-H = 31.6 Hz).
Tabelle 5. Wichtige spektroskopische Daten der Verbindungen 11a-c.
11a 11b 11c 1H-NMR Daten (400 MHz, CDCl3)
(ppm)
Cp (ppm) 5.85 5.82 5.83
C=CH2
7.65 (d, 1H,
cis-3JH–P = 15 Hz) 6.55 (d, 1H,
trans-3JH–P = 35 Hz)
7.47 (d, 1H,
cis-3JH−P = 13 Hz) 6.49 (d, 1H,
trans-3JH–P = 32 Hz)
7.42 (d, 1H, cis-3JP–H = 12.6 Hz)
6.43 (d, 1H, trans3JP–H = 32 Hz).
13C-NMR Daten (100 MHz, CDCl3) (ppm)
W-Cα163.1
(d, 1JC–P = 51 Hz) 165.5
(d, 1JC–P = 58 Hz) 161.9
(d, 1JP-C = 51.88 Hz)
W-C=Cβ133.5
(d, 2JC–P = 9 Hz) 132.8
(d, 2JC–P = 9 Hz) 132.7
(d, 2JP-C = 9.15 Hz)
Cp 102.7 (d, 2JC–P = 1.5 Hz)
103.4 (d, 2JC–P = 1.5 Hz)
102.5
(d, 2JC–P = 1.5 Hz ) 31P-NMR Daten (162 MHz, CDCl3, H3PO4 als externer Standard, 1H-Entkoppelt)
(ppm)
–85.5 (1JP–W = 131 Hz)
–46.0 (1JP–W = 118 Hz)
-57.6 (1JP-W = 130 Hz)
IR-Daten (KBr-Prssling) (cm-1)
NO 1610 1620 und 1528 1611 und 1591.
4.3 Die Hydrolysereaktionen der Phosphorkomplexe des Wolframs 11a-c
Die Verbindung 11a wurde in THF gelöst und mit einer Suspension von NaOH in
einem Wasser/THF Gemisch umgesetzt. Nach dem Verschwinden des Edukts wurde die
Reaktionsmischung aufgearbeitet. Das Produkt bestand aus einer Mischung der
Diastereomeren 35 und 36 (18:82). Der markante strukturelle Unterschied zwischen den
Komplexen 35 und 36 ist die relative Anordnung Cp-Liganden. Bei der Verbindung 35 sind
die beiden Cp Ringe auf einer Seite des mit Sauerstoff verbrückten Bicyclus (Abbildung 64),

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 52
somit Z konfiguriert; und in der Verbindung 36 haben sie entsprechend eine E Konfiguration
zu einander.
WN
P
ClO
HH
W
W
O
NO
NOP
PH
H
HH
HH
ON
PW
OW
NO
P
HH
11a
αβ
Der Z-Komplex 35 (13.5%)
Der E-Komplex 36 (61.5%)
NaOH Suspensionin THF/H2O
Abbildung 64. Hydrolyse der Verbindung 11a.
Die beiden Cp-Liganden in 35 sind durch eine C2 Symmetrie Operation in einander
überführbar, was ein einziges 1H-NMR Signal bei δ 5.29 ppm zur Folge hat. Im Gegensatz
dazu, die Verbindung 36 erweist zwei differenzierbaren Signale für die beiden nicht
äquivalenten Cp-Liganden, nämlich bei δ 5.70 und 5.24 ppm. Das Verhältnis der Integrale der
beiden Signal(-gruppen) wurde zur quantitativen Bestimmung der Verbindungen 35 und 36
benutzt.
Umsetzung der Verbindung 11c mit NaOH. Analog zu der oben beschriebenen
Vorgehensweise wurde der η2-Phosphinovinylliden-Komplex 11c hydrolysiert. Aufarbeitung
und anschließende Umkristallisierung des Rohprodukts lieferte die Verbindung 37 mit einer
Ausbeute von 60% (Abbildung 65).
W PN ClO
HH
W
W
O
NO
NOP
PH
H
HH
NaOH Suspension inTHF/H2O
11c 37
αβ
Abbildung65. Darstellung der Verbindung 37.

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 53
Alle Versuche zur Hydrolyse der Verbindung 11b sind gescheitert und die
Aufarbeitung der Reaktionsmischung führte zur qualitativen Wiedergewinnung des
eingesetzten Edukts 11b.
Diskussion charakteristischer und spektroskopischer Daten des Komplexes 37.
Das IR-Spektrum der Verbindung 37 zeigt eine Nitrosyl- Schwingung bei 1585 cm-1. Das 1H-
NMR zeigt zwei Gruppen von Methylen Protonen. Sie haben sowohl Kopplung zum Wolfram
als auch zum Phosphor. Allerdings aufgrund der natürlichen Häufigkeit von 183W von 14.4%,
macht sich die Kopplung zum Wolfram nur in form der Satelliten-Signale bemerkbar. Die
erste Gruppe der Methylenprotonen, trans-ständig zum Phosphor und cis-ständig zum
Wolfram, erscheint als ein Dublett bei δ 6.15 ppm, mit einer Kopplungskonstanten von 3JP-H =
40.8 Hz und 3JW-H = 7.4 Hz. Die zweite Gruppe ist auch ein Dublett mit einer chemischen
Verschiebung von δ 6.53 ppm, mit 3JP-H = 18.4 Hz und 3JW-H = 12.3 Hz (Abbildung 66).
6.106.206.306.406.506.60
J=18.704
J=12.305
J=40.854
J=7.383
P
W P
W
H
H
H
Ha
a
b
b
Abbildung 66. Das 1H-NMR Spektrum der Verbindung 12 (Ausschnitt).
In dem 13C-NMR Spektrum erscheint das Cα als ein Dublett von Dublett bei δ 173.1
ppm. Auffallend sind hierbei die Kopplungen zum Wolfram, 1JW–C = 18.31 Hz, und zum
Phosphor, 1JP-C = 59.9 Hz. Cβ bei δ 134.2 ppm zeigt keine Kopplung zum Phosphor bzw.
Wolfram. 31 P-NMR Spektrum der Verbindung 37 zeigt nur ein Signal bei δ 31.3 ppm, welche
von mehreren Satelliten begleitet ist. Die relevanten Wechselwirkungen sind zwischen
Wolfram und Phosphoratomen und führen zu einem AA’X Spinsystem. Die äußeren
Satelliten sind Dublett von Dublett. Sie kommen durch die Kopplung der Phosphoratome mit
benachbarten Wolframatom (1JW–p = 214.9 Hz) und die Kopplung jedes Phosphoratoms mit
dem anderen magnetisch inäquivalenten Phosphoratom (3JP–P = 5.08 Hz) zustande (Abbildung
67).

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 54
SpinWorks 2.4: PrV 55 31PNMR
PPM 32.0 31.8 31.6 31.4 31.2 31.0 30.8 30.6 30.4 30.2
32.
0227
31.
9877
31.
3965
31.
3399
31.
2821
30.
6918
30.
6570
file: C:\Nmr\NMR labor\Phosphor\PRI 55 expt: transmitter freq.: 161.985000 MHztime domain size: 65536 pointswidth: 41668.77 Hz = 257.238452 ppm = 0.635815 Hz/ptnumber of scans: 8026
freq. of 0 ppm: 161.977364 MHzprocessed size: 65536 complex pointsLB: 1.272 GB: 0.0000
Abbildung 67: Das 31P-NMR Spektrum der Verbindung 37.
Am Fuß des Hauptsignals werden auch Satelliten beobachtet. Diese Linien gehören zu
einem AA'X Spinsystem und kommen durch zwei Kopplungen 3JP-P und 2JP-W zustande. Die
Simulation mit SPINWORKS® ergibt die Größe der zweiten Kopplungskonstanten wieder: 2JW–P = 14.7 Hz. Die Simulation mit dem AA'X Spinsystem mit der dazugehörigen
Spinanalyse ist in der Abbildung 68 präsentiert.
SpinWorks 2.4: Simulation PrV55 PNMr
Hz 5180.0 5160.0 5140.0 5120.0 5100.0 5080.0 5060.0 5040.0 5020.0 5000.0 4980.0 4960.0 4940.0 4920.0
file: C:\Nmr\NMR labor\Phosphor\PRI 55 expt: transmitter freq.: 161.985000 MHztime domain size: 65536 pointswidth: 41668.77 Hz = 257.238452 ppm = 0.635815 Hz/pt
1 J P - W = 2 1 4 . 9 H z
3 J P - P = 5 . 0 8 H z
2 J P - W = 1 4 . 7 6 H z
freq. of 0 ppm: 161.977364 MHzprocessed size: 65536 complex pointsLB: 1.272 GB: 0.0000
number of scans: 8026
Abbildung 68. 31P-NMR Simulation der Verbindung 37.

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 55
Röntgenstrukturanalyse der Verbindung 37. Die geeigneten Einkristalle wurden
durch das Überschichten einer konzentrierten Lösung des Komplexes 37 in CH2Cl2 mit
Pentan bei 4 °C gezüchtet. Die Kristallografische Daten sind in der Tabelle E 7.1
zusammengefasst. Ein ORTEP Plot von der Verbindung 37 ist in der Abbildung 69
präsentiert.
Abbildung 69: ORTEP Plot von dem Komplex 37 einschließlich Benennung aller Nicht-Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide mit einer Aufenthaltswahrscheinlichkeit von 30b% dargestellt. Die Protonen an iso-Propyl- und Cp-Liganden wurden für Übersichtlichkeit nicht angegeben. Die ausgewählten Bindungslängen [Ǻ] und Bindungswinkeln [Gerade] (mit den Standardabweischungen): W(1)-W(2) 3.542, W(1)-N(1) 1.772(4), W(1)-O(12) 1.989(2), W(1)-C(11) 2.209(4), W(1)-P(1) 2.528(12), W(2)-N(2) 1.755(4), W(2)-O(12) 1.995(2), W(2)-C(13) 2.217(5), W(2)-P(2) 2.5256(11), N(1)-W(1)-O(12) 117.71(13), N(1)-W(1)-C(11) 83.76(16), O(12)-W(1)-C(11) 76.52(13), N(1)-W(1)-P(1) 89.80(12), W(2)-O(12)-W(1) 125.48(13), C(13)-P(1)-W(1) 104.27(15).
Die Verbindung 37 hat ein ähnliches Gerüst wie Norbornan. An den Brückenköpfen
liegen die beiden Wolframatomen, deren Abstand (W1-W2) 3.542 Ǻ beträgt. Der μ2-Oxo-
Ligand bildet die Brücke zwischen den beiden Metallzentren mit einem Winkel von
125.48(13)˚ (W1-O12-W2). Bei der Verbindung 37 liegen die Cp-Liganden auf einer Seite
und sind „Z“ zu einander orientiert. Diese Stellung verleiht dem Molekül eine C2-
Symmetrieachse. Die Beide Cp Liganden liegen auf derselben Seite wie die Sauerstoffbrücke.
Der sechsgliedrige Ring nimmt eine Wannenstruktur an, bei der die vier Heteroatome auf
einer Ebene liegen, und die zwei C=C Einheiten sind Endo gerichtet (bezüglich des
Norbornan Gerüstes). Die W1-C11 Bindungslänge beträgt 2.209 Ǻ und die W1-P1 Bindung

B4 Die Hydrolyse von Phosphorkomplexen des Wolframs 56
ist 2.529Ǻ. Verglichen mit den Werten für die Verbindung 11a (W1-C11: 2.171 und W1-P1:
2.437 Ǻ, Abbildung 63),[36] ist die Wolfram –Phosphorbindung in der Verbindung 37
verkürzt.
Das kann mit dem geringeren Ausmaß an Phosphan π-Rückbindung interpretiert
werden. Die Phosphan-Liganden sind σ-Donoren und π-Acceptoren. Der π-Acceptor-
Charakter wird im Wesentlichen von dem antibindenden P-C σ* Orbitale übernommen, an
deren Bildung nur die p-Orbitale am Phosphor beteiligt sind. Dabei kommt auf den d-Orbitale
des Phosphors eine untergeordnete Rolle zu (Abbildung 70).[75]
Abbildung 70. MO-Wechselwirkung zwischen dem Metall und einem Phosphan Liganden.
4.4 Schlussfolgerung
Es wurde gezeigt, dass die Substitution des am Metal σ-gebundene Chloratoms durch
einen Hydroxid-Liganden in der Verbindungen 11a und 11c möglich ist, jedoch anstelle der
erwarteten Hydroxy-substituierten Komplexe, resultieren durch Wasserabspaltung die µ-Oxo-
Verbrücken Komplexe 35-37.

C Zusammenfassung 57
C Zusammenfassung
Als Modellreaktion für die homogen katalysierte Olefin-Metathese wurde im Rahmen
der vorliegenden Dissertation die Umsetzung der Vinylcarben-Komplexe 16a und 16b mit
Enaminen 14a und 14b, als Beispiele für elektronreiche Alkene, untersucht. Als Produkt der
Reaktion wurden die η1-Acyl-Komplexe 18-20 in hoher Ausbeute isoliert und mit Hilfe der
spektroskopischen Daten identifiziert. Die Struktur der Verbindung 18 wurde zusätzlich mit
Hilfe der Einkristallstrukturanalyse belegt. Mechanistisch betrachtet, lässt sich die Bildung
der η1-Acyl-Komplexe 18-20 durch einen nukleophiler Angriff des Enamins an dem α-
Kohlenstoffatom des Vinylcarben-Komplexes beschreiben. Dies führt, wie am Beispiel der
Komplex 18 in Abbildung 71 formuliert, zur Bildung des Iminiumions 18a. Dieses
Zwischenprodukt lagert sich dann unter Einschiebung der Carbonylgruppe und gleichzeitiger
σ → η3-Umlagerung zur Verbindung 18 um.
W C
CNO
C
O
H
HN
WOON
HH
HN
W
NCO
O
C C CHH
H
N+
WOON
HH
HN
WOON
HH
HN
W C
CNO
C
O
H
HN
NW C
CNO
C
O
H
H
+
16a 14b
19
18
2016b
18a
14a
+
14a
+
16a
Abbildung 71. Darstellung der η1-Acyl-Komplexe 18-20.
Als nächstes wurde die Reaktion der Enamine mit dem Vinylidenkomplex 10
untersucht. Interessanterweise wird das Ergebnis dieser Reaktion sehr stark durch die Struktur
des Enamines beeinflusst. Während das Enamin 14a, ein Pyrrolidinderivat, mit dem
Vinylidenkomplex 10 zum η1-Acyl-Komplex 19 reagiert, wird bei der Umsetzung des
Enamins 14b ein vollständig anderes strukturiertes Produkt erhalten. Hier wird quantitativ der
Enamin-substituierte η2-Alkenkomplex 21 gebildet. Dieser divergierende Unterschied

C Zusammenfassung 58
zwischen den Enaminen 14a und 14b gegenüber Vinylidenkomplex 10, lässt sich vermutlich
auf die unterschiedliche Basizität bzw. Nuclephilie der Enamine zurückführen.
Das Enamin 14a fungiert bei der Reaktion mit dem Vinylidenkomplex 10 als eine
Base. Der nucleophile Angriff des Acetylidkomplexes auf das sich gebildete Iminium-Kation
liefert den intermediären Vinylidenkomplex 15. Dieser ergibt über eine Retro-En-Reaktion
den Vinylcarben-Komplexe16a, der dann mit dem Überschuss vom Enamin zum η1-Acyl-
Komplex 19 weiterreagiert.
Im Gegensatz zudem, wirkt Enamin 14b im ersten Schritt der Reaktion als ein
Nucleophil und führt über den Zwischenprodukt 21a nach einer Protonverschiebung und
anschließenden reduktiven Eliminierung zum η2-Alkenkomplex 21 (Abbildung 72). Die
Struktur von der Verbindung 21 wurde sowohl mit den spektroskopischen Methoden als auch
Röntgenstrukturanalyse belegt.
W C CH
HNC
O
O
NC
CH H
H
W NC
O
O
N+
H
CC
HH
NW N
CO
O
H
W NC
O
OC
CH
H
HN
Ether
W C CH
HNC
O
O
NW C C H
NC
O
O
W C CH
NC
OO
NH
+
+
2110
16a 19
14b 21a
14a
10
+14
15
Abbildung 72. Die Umsetzung der Enaminen 14a-b mit dem Vinylidenkomplex 10.
In dem zweiten Abschnitt der Dissertation, wurde die Synthese des
Allenylidenkomplexes 26 beschrieben. Nach vielen vergeblichen Versuchen zur Synthese der
Allenylidenkomplexe gelang es schließlich die Verbindung 26 durch die Umsetzung vom
Acetylidkomplex 10a mit dem Iminiumsalz 23c als grüne Kristalle zu erhalten und mit Hilfe
der Spektroskopischen Methoden zu charakterisieren. Bei dieser Reaktion führt die
Aminoalkyllierung der Verbindung 10a zum intermediären Bildung der Vinylidenkomplex
26a, der nach der Eliminierung des tert-Butylmethylamins den Allenylidenkomplex 26 liefert.
Dabei war es sehr wichtig, dass ein geringer Überschuss an n-Butyllithium als Base eingesetzt
wurde (Abbildung 73).

C Zusammenfassung 59
W C CN C
OO
H
N+
W CN C
OO
C CBF4
Li+
W CN C
OO
CH
N
NH
26
-
10a 23c 26
+
Abbildung 73: Darstellung des Allenylidenkomplexes 26.
Weniger erfolgsreich für die Synthese des Allenylidenkomplexes war die Reaktion
von 10a mit dem Iminiumsalz 23a. Hier wurde zwar zuerst den erwarteten
Allenylidenkomplex 24b gebildet, dieser jedoch isomerisierte sich rasch unter der
Reaktionsbedingungen durch Protonenwanderung zu dem vinylsubstituierten
Vinylidenkomplex 24 um (Abbildung 74).
W CN C
OO
CHN
ClO4 W CN C
OO
CH
N
W CN C
OO
C C 10a
2423 24
+
24b
+-PhNH(CH3)
Abbildung 74. Darstellung des Vinylidenkomplexes 24.
Ganz anderes verlief die Umsetzung des Iminuimsalzes 23b mit dem Acetylidkomplex
10a. Hier erleidet der als Zwischenprodukt gebildete Vinylidenkomplex 25a anstelle einer
Eliminierung zu dem erhofften Allenylidenkomplex einen [1,3]-Verschiebung des N-
Methylanilins zu dem Aminocarben-Komplex 25. Ebenso lieferte die Umsetzung von
Iminiumsalz 23d mit 10a anstelle eines Allenylidenkomplexes, über den intermediär
gebildeten Vinylidenkomplex 28a durch eine Retro-En-Reaktion zum Vinylcarben-Komplex
28 (Abbildung 75).
Des Weiteren wurde die Reaktivität des Allenylidenkomplexes 26 gegenüber
Nucleophilen untersucht. Es konnte gezeigt werden, dass der Angriff des Nucleophils
ausschließlich an dem Cγ des Allenylidenkomplexes stattfindet. Ein Beispiel hierfür ist die
Umsetzung mit Methanol, die bei der Raumtemperatur ausschließlich zu dem stabilen
Vinylidenkomplex 27 führt.

C Zusammenfassung 60
N+
ClO4
W CN C
OOH
C CH
W CN C
OON
HN
+
ClO4W C
N COO
CH
N
W CN C
OO
CC
H
N
23d 28
2523b 25a
28a
10a +
10a +
Abbildung 75. Die Reaktionen von der Verbindung 10a mit den Iminiumsalzen 23b und 23d.
Bei der Umsetzung des Pyrrolidins mit der Verbindung 26 reagiert der primär
entstandene Vinylidenkomplex 28a wie erwartet in einer Retro-En-Reaktion zu dem
Vinylcarben-Komplex 28. Schließlich liefert die Umsetzung von Diethylamin mit der
Verbindung 26 über eine [1,3] Aminverschiebung zur Bildung des Aminocarben-Komplexes
29. In diesem Fall wurde eine Retro-En-Reaktion nicht beobachtet. Diese Ergebnisse sind in
der Abbildung 76 zusammengefasst worden.
CC
MeO
W CNC
O
O
H
W CN
CO
O H
C CH
W CN
CO
O N
C CH
CH3OHNH
NH
W CNC
O
OC
H
C
N
W CN
CO
O
CH
C
N
26 2728b
29
28
Abbildung 76. Die Reaktionen des Allenylidenkomplexes 26 mit Nucleophilen.
In dem dritten Abschnitt dieser Arbeit wurde über die Darstellung der η2-
Allenkomplexe 31, 32 und 33 durch die Umsetzung der Verbindung 10 und entsprechenden
Diazoalkanen berichtet. Diese Komplexe wurden kristallographisch studiert. Die
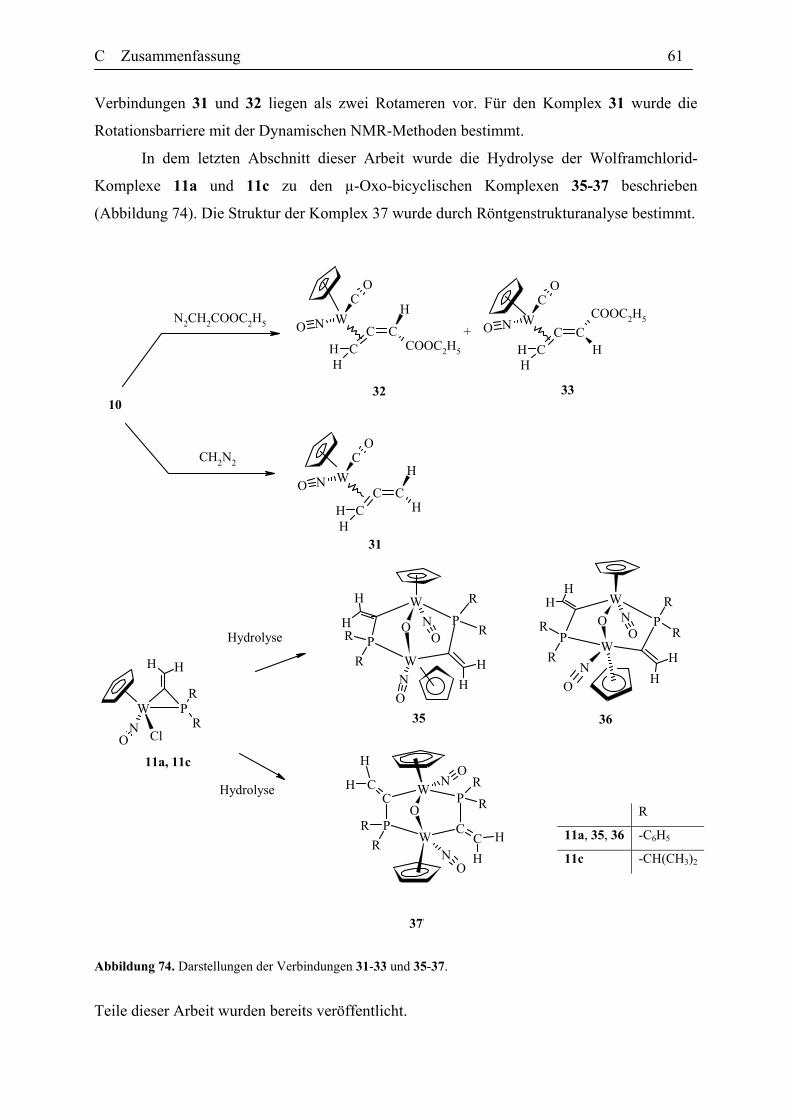
C Zusammenfassung 61
Verbindungen 31 und 32 liegen als zwei Rotameren vor. Für den Komplex 31 wurde die
Rotationsbarriere mit der Dynamischen NMR-Methoden bestimmt.
In dem letzten Abschnitt dieser Arbeit wurde die Hydrolyse der Wolframchlorid-
Komplexe 11a und 11c zu den µ-Oxo-bicyclischen Komplexen 35-37 beschrieben
(Abbildung 74). Die Struktur der Komplex 37 wurde durch Röntgenstrukturanalyse bestimmt.
Abbildung 74. Darstellungen der Verbindungen 31-33 und 35-37.
eile dieser Arbeit wurden bereits veröffentlicht.
O
T
R
11a, 35, 36 -C6H5
11c -CH(CH3)2
WC
NO
O
WC
C C
H
COOC2H5CH
H
NO C CCOOC2H5N2CH2COOC2H5
HCHH
CH2N2
WC
N
O
O C C
H
+
333210
HCHH
31
W PN ClO
HH
R
R
WC
PW C
PO
C
C
H
H
HH
N
N RR
RR
O
O
W
W
O
NO
OPPH
H
HH
R
R
N
R
R
HH
ON
PW
OW
NO
P
HH
R
R
R
RHydrolyse
35 36
11a, 11c
Hydrolyse
3737

D Experimenteller Teil 62
D Experimenteller Teil
1 Allgemeine Hinweise
Spektrometer
IR BRUKER FT-IR IFS 85
1H-NMR BRUKER AM 400, BRUKER AC 200
13C-NMR BRUKER AM 400, BRUKER AC 200
Die chemischen Verschiebungen (δ-Werte) der NMR-
Messungen sind in ppm, bezogen auf Tetramethylsilan
(TMS) als interner Standard, angegeben.
31P-NMR BRUKER AM 400
Die chemischen Verschiebungen (δ-Werte) der NMR-
Messungen sind in ppm, bezogen auf 85%ige
Phosphorsäure als externer Standard, angegeben.
MS VARIAN MAT 311-A, ITD Finnigan MAT
Röntgenkristallstrukturanalyse
IPDS (Image Plate
Diffraction System)
STOE & CIE GmbH, D-64295 Darmstadt
Analysen, Schmelzpunkte
Elementaranalysen CARLO-ERBA-Elementaranalyse MODELL 1104
Schmelzpunkte Apparatur nach Dr. TOTTOLI der Fa. BUECHI;
MODELL SMP-20

D Experimenteller Teil 63
Chromatographiematerialien
Säulen-Chromatographie Kieselgel S 0.063−0.2 mm der
Fa. RIEDEL-DE HÄEN, Seelze
Neutrales Aluminiumoxid, aktiviert, 50−200 Micron
F 0
Fa. ACROS ORGANICS; New Jersey, USA; Ceel, Belgium
Basisches Aluminiumoxid, aktiviert, 50−200 Micron
F SA; Ceel, Belgium
Dünnschicht-Chromato-
graphie P
F
Schicht: 0.2 mm Al O3 mit Fluoreszenzindikator
Fa. MACHEREY-NAGEL, Macherey-Nagel GmbH &
Co.KG
POLYGRAM Sil G / UV254
Fertigfolien für die DC (40
Schicht: 0.25 mm Kieselgel mit Fluoreszenzindikator
ACHEREY-NAGEL, Macherey-Nagel GmbH &
Co.KG
icht-
atographie
Glasplatten (20 × 20 cm),
Schicht: 2 mm, Kieselgel 60 PF254 für die präparative
Schicht-Chromatographie (mittlere Korngröße: 0.040–0.063
mm), Fa. MERCK EUROLAB GmbH
W = 101.96 ρ = 3.970
a. ACROS ORGANICS; New Jersey, U
OLYGRAM® ALOX N / UV254
ertigfolien für die DC (40 × 80 mm)
2
®
× 80 mm)
Fa. M
Präparative Sch
Chrom

D Experimenteller Teil 64
Schutzgas, Lösungsmittel, Reagenzien
Als Schutzgas diente Schweiß-Argon (99.99%) der Fa. MESSER-GRIESHEIM. Alle
R A t. Die verwendeten Apparaturen
wurden vor jeder Umsetzung lt.
Die für die Experim ungsmittel wurden wie folgt gereinigt und
getrocknet:
Tetrahydrofuran
-Pentan Destillation über KOH
iethylether
Dichloromethan
R
2-Adamantanon
, 98%
-1-propen (cis+trans) FLUKA FEINCHEMIKALIEN GmbH, Neu-Ulm,
purum
n-Butyllithium R ew Jersey, USA; Ceel,
P-Chlordiphenyl-
Phosphin
-Chlor-di-iso-
P-Chlor-di-tert-
butylphosphin
m,
FLUKA FEINCHEMIKALIEN GmbH, Neu-Ulm,
17a)
17b)
Darstellung nach der Literatur[76].
Darstellung nach der Literatur[76].
eaktionen wurden unter rgon-Atmosphäre durchgeführ
ausgeheizt und mit Argon gespü
ente verwendeten Lös
Destillation über KOH und Natrium
n
D Destillation über KOH und Natriumdraht
Destillation über CaH2
eagenzien:
ACROS ORGANICS; New Jersey, USA; Ceel,
Belgium
1-Brom
ACROS O GANICS; N
Belgium, 96%
P
propylphosphin
FLUKA FEINCHEMIKALIEN GmbH, Neu-Ul
~97%ig (GC)
~97%ig (GC)
ACROS ORGANICS; New Jersey, USA; Ceel,
Belgium, 96%
1-Cyclopenten-pyrrolidinium
Perchlorat (
1-Cyclopenten-pyrrolidinium
Perchlorat (

D Experimenteller Teil 65
Diazald® ACROS ORGANICS; New Jersey, USA; Ceel,
iazo
lbutin LANCASTER, Mühlheim am Main, 98%
ino-1-cyclopenten Darstellung nach der Literatur[78]
lester New Jersey, USA; Ceel,
; New Jersey, USA; Ceel,
Belgium, 98%
Darstellung nach der Literatur[78]
ethylsilylacetylen Darstellung nach der Literatur[79]
onyl
Belgium, 96%
Diazomethan
Darstellung nach der Literatur[77a]
D essigsäureethylester Darstellung nach der Literatur[77b]
3,3-Dimethy
Diethylam
(14b)
Glycinethy
hydrochlorid
ACROS ORGANICS;
Belgium, 98%
Phenylacetylen ACROS ORGANICS
Pyrrolidino-1-cyclopenten
(14a)
Trim
Wolframhexacarb ACROS ORGANICS; New Jersey, USA; Ceel,
Belgium, 99%

D Experimenteller Teil 66
2 hreibung der Vers
2.1 Darstellung von (η5-Cyclopentadienyl)-dicarbonyl-nitrosyl-wolfram (9)
n v in 300 ml THF wurde eine
L (186 mmo ml THF zugetropft.
W ng nahe dem Gefrierpunkt
gehalten. Anschließend wurde acht Stunden bei der Raumtemperatur gerührt bis sich das
N tändig gelöst bonyl
zur Reaktionsmischung gegeben. Die Reaktionsmischung wurde 10 Tage unter Rückfluss
erhitzt. Nach dem die Reaktio urde
mittels eines Tropftrichters ei 33.0 g (144 mmol) N-Methyl-N-nitroso-p-
t in 8 unden wurde die Lösung im
Vakuum mit einem Rotationsverdampfer zur Hälfte eingengt und der Rückstand in 500 ml
E ch der tel im Vakuum entfernt. Das
U ckstan n der
eingeengten Mutterlösung über ilicagel Säule mit Pentan/Ether (10:1) lieferte
usätzlich noch 2.2 g (6.6 mmol) von dem Komplex 9. Insgesamt wurde 42 g (Ausbeute 88%)
on der Verbindung 9 erhalten. 5-Cyclopentadienyl)-dicarbonyl-nitrosyl-wolfram (9)
Ausbeute: 88%
9
1H-NMR (400 MHz,
CDCl3):
δ: 5.68 (s, 5H, Cp).
13C-NMR (100 MHz,
CDCl3):
δ: 217.2 (CO); 92.0 (Cp).
IR (KBr)
Besc uche zur Darstellung der organometallischen Edukten
Zu einer Suspensio on 4.23 g (184 mmol) Natrium
ösung von 12.33 g l) frisch destilliertem Cyclopentadien in 40
ährend der Zugabe wurde die Temperatur mittels Eiskühlu
atrium fast volls hatte .Dann wurde 50.0 g (142 mmol) Wolframhexacar
nsmischung sich auf Raumtemperatur abgekühlt hatte, w
ne Lösung von
oluolsulfonamid (Diazald) 0 ml THF zuaddiert. Nach 10 St
ther aufgenommen. Na Filtration wurde das Lösungsmit
mkristallisieren des Rü des aus Ether ergab 39.8 g (118.9 mmol) und die Elutio
einer kurzen S
z
v
(η
WC
CNO O
O
ν = 2001 und 1901 cm–1 (CO); 1636 cm-1 (NO).

D Experimenteller Teil 67
2.1 Darstellung vom tert-Butyl-Vinylidenkomplex 7
Zu einer auf –78 °C abgekühlten Lösung von 2.5 ml (1.64 g, 20 mmol) tert-
Butylacetylen in 100 ml THF wurde 11.3 ml (18 mmol) einer n-Buthyllithium Lösung(1.59 M
en
Lösung
Hz,
3)
δ: 5.83 und 5.81 (s, 5H, Cp); 5.73 und 5.69 (s, 1H, CβH,
5:4); 1.10 und 1.08 [s, 9H, C(CH3)3].
C-NMR (100 MHz, δ 338.7 und 338.4 (C ); 211.8 und 210.8 (CO); 140.0
β
[C(CH ) ];31.2 und 31.0 [C(CH3)3].
IR (KBr)
in Hexan) zugetropft. Nach einer Stunde wurde diese Lösung mit einer abgekühlten orang
des Komplexes 9 (4 g, 12 mmol) in 50 ml THF bei –40 °C versetzt. Nachdem die
tiefgrüne Reaktionslösung drei Stunden bei –30 °C gerührt hatte, wurde sie mit 20 ml einer
HCl Lösung (1M) neutralisiert. Dabei schlug die Farbe auf hellrot um. Nach einer Stunde
Rühren bei Raumtemperatur wurde das Volumen der Reaktionsmischung unter vermindertem
Druck auf 1/5 reduziert und zweimal mit Ether extrahiert. Die Etherphasen wurden mit einer
gesättigten NaHCO3 Lösung und anschließend mit einer gesättigten NaCl Lösung gewaschen
und über MgSO4 getrocknet. Einengen der Lösung und anschließende Umkristallisierung des
Rückstandes aus Ether/Pentan (1:3) führte zu 4.2 g (90 %) der orangen Kristalle des
VinylidenKomplexes 7.
tert-Butyl-Vinylidenkomplex 7
Ausbeute: 4.2 g (91%)
W C
CNO
C
7
1H-NMR (400 M
CDCl
13
CDCl3) α
und 139.5 (C ); 96.2 und 96.0 (Cp); 34.6 und 33.0
3 3
ν = 1994 cm–1 (CO), 1606 cm–1 (NO).
O
H

D Experimenteller Teil 68
2.2 Darstellung vom Phenyl-Vinylidenkomplex 8
Zu einer auf –78 °C abgekühlten Lösung von 2.0 ml (1.83 g, 18 mmol)
Phenylacetylen in 50 ml THF wurde 10.1 ml (16 mmol) einer n-Butyllithium Lösung (1.59 M
in Hexan) zugetropft. Nach einer Stunde wurde diese Lösung mit einer abgekühlten orangen
Lösung von 4 g (12 mmol) [( 5-C5H5)(CO)2(NO)W] (9) in THF bei –50 °C versetzt und vier
Stunden gerührt. Dann wurde die tiefgrüne Reaktionslösung mit 17 ml einer HCl Lösung (1
M) versetzt. Dabei schlug die Farbe auf tiefrot um. Nach einer Stunde Rühren bei
Raumtemperatur wurde das Volumen der Reaktionsmischung unter vermindertem Druck auf
1/5 reduziert und zweimal mit Ether extrahiert. Die Etherphasen wurden mit einer gesättigten
NaHCO3 Lösung und anschließend mit einer gesättigten NaCl Lösung gewaschen und über
MgSO4 getrocknet. Nach dem Einengen wurde der Rückstand bei –10 °C mit 5 ml Pentane
gewaschen, und schnell filtriert. Umkristallisierung des Niederschlags aus Ether/Pentan
Mischung führte zu 3.9 g (80 %) der orangen Kristalle des Vinylidenkomplexes 8.
olframs 8
8
1 (400 MHz,
CDCl3)
δ: (s, 1H, CβH); 5.84,
5.81 (s, 5H, Cp). 13 (100 MHz,
CDCl3)
δ:
13
und 95.9 (Cp).
Phenyl-Vinylidenkomplex des W
Ausbeute: 80%
W C
CN
O
C
O
H
H-NMR 7.22-7.03 (m, 5H, Ph); 6.59 und 6.54
C-NMR 344.6 und 344.3 (Cα); 210.7 und 209.2 (CO); 131.3 und
0.9 (Cβ); 133.3, 128.7, 126.7, 126.1 und 125.9 (Ph); 96.2
IR (KBr) ν = 2019 cm (CO), 1653 cm (NO).
–1 -1

D Experimenteller Teil 69
2.3 Darstellung vom Vinylidenkomplex 10
Zu einer auf –78 °C abgekühlten Lösung von 2.9 ml (1.96 g, 20 mmol)
Trimethylsilylacetylen in 40 ml THF wurde 11.3 ml (18 mmol) einer n-Butyllithium Lösung
(1.59 M in Hexan) zugetropft. Nach 45 min wurde diese Lösung mit einer abgekühlten
orangen Lösung der Verbindung 9 (4 g, 12 mmol) in THF bei –40 °C versetzt. Nachdem die
tiefgrüne Reaktionslösung 3h bei –30 °C gerührt hatte, wurde sie mit 40 ml einer gesättigten,
wässrigen NaHCO3-Lösung desilyliert und neutralisiert. Nach zwei Stunden Rühren bei
Raumtemperatur war dieser Vorgang abgeschlossen (DC kontrolliert). Das Volumen der
Reaktionsmischung wurde unter vermindertem Druck auf 1/5 reduziert und zweimal mit
Ether extrahiert. Die Etherphasen wurden mit einer gesättigten NaHCO3 Lösung und
anschließend dreimal mit einer gesättigten NaCl Lösung gewaschen und über MgSO4.
Einengen der Lösung und anschließende Umkristallisierung des Rückstandes aus Ether ergab
3.5 g (87 %) von gelb-orangen Kristallen des Vinylidenkomplexes 10.
Vinylidenkomplex 10
Ausbeute: 87%
W C
CNO
C
O
H
H
10
IR (KBr)
1H-NMR (400 MHz,
CDCl3)
δ: 5.87 (s, 5H, Cp); 5.28 (3JW–H = 6 Hz) und 5.21 (zwei d, 2H, CβH2).
13C-NMR (100 MHz,
CDCl ) 3
δ: 341.2 (Cα); 209.9 (CO); 112.2 (Cβ); 96.3 (Cp).
ν = 1998 cm–1 (CO), 1635 cm–1 (NO), 1591 cm–1(C=C).

D Experimenteller Teil 70
2.4 Darstellung des deuterierten Vinylidenkomplexes 10c
D
C-NMR (100 MHz,
3)
δ: 341.2 (Cα); 209.9 (CO); 112.2 (Cβ); 96.3 (Cp).
ie Synthese erfolgte wie 2.3, allerdings wurde die Reaktionsmischung mit einer
gesättigten NaHCO3 Lösung in D2O neutralisiert und desilyliert. Die Aufarbeitung ergab 3.7 g
von der Verbindung 10c in einer Ausbeute von 91%
Deuterierter Vinylidenkomplex 10c
Ausbeute: 91%
10c
1H-NMR (400 MHz, δ: 5.87 (s, 5H, Cp).
W C CD
DNC
O
O
CDCl3)
13
CDCl

D Experimenteller Teil 71
3 Die Reaktionen der Vinylcarben-Komplexe des Wolfr
W C
CNO
C
O
H
H
ams mit Enaminen.
a
g 10 (1.0 g, 3 mmol) in 10 ml THF wurde 1.9 ml (3
thium (1.6 M, in Hexan) bei –78 °C zugetropft. Nach einer Stunde Rühren
urde die Reaktionsmischung auf –30 °C erwärmt und mit 1-Cyclopenten-
yrrolidiniumPerchlorat (22a, 831 mg, 3.5 mmol) versetzt. Die Reaktionsmischung wurde auf
aumtemperatur erwärmt und noch eine Stunde gerührt. Dann wurde die Reaktionslösung
nter heftiges Rühren zu 450 ml Pentan tropfenweise addiert. Nach zwei Stunden wurde die
ingeengt
nd bei –20 °C mehrfach auskristallisiert, um 835 mg der roten Kristalle der Verbindung 16a
m 70% z
Vinylcarben-Komplex 16a
Ausbeute: 70%
16a
1H-NMR (400 MHz,
CDCl3)
δ: 14.68 und 13.87 (zwei d, CαH); 8.18 und 7.35 (zwei d,
1H, CβH); 5.93 und 5.92 (s, 5H, Cp); 2.12-2.03 (m, 4H);
1.76-1.64 (m, 4H). 13C-NMR (100 MHz,
CDCl3)
δ: 276.3 (Cα); 214.3 (CO); 150.0 (Cβ); 141.7 und 141.0
(Cγ); 97.3 und 97.1 (Cp); 34.7, 34.5, 31.1, 30.8, 25.9,
25.8 und 25.7 (-CH2-).
IR (KBr)
3.1 Darstellung der Vinylcarben-Komplexe 16a-b
3.1.1 Darstellung des Vinylcarben-Komplexes 16
Zu einer Lösung der Verbindun
mmol) n-Butylli
w
p
R
u
entstandene Suspension filtriert. Das Filtrat wurde um ein Viertel des Volumens e
u
it einer Ausbeute von u ergeben.
ν = 1965 cm–1 (CO), 1571 und 1561 cm–1 (NO).

D Experimenteller Teil 72
3.1 Darstellung des Vinylcarben-Komplexes 16b .2
Wie unter 3.1.1 beschrieben, erfolgte die Umsetzung von 1-Cyclohexen-pyrrolidinium
gestellten Lösung von 10a (3 mmol,
ng ergab 810 mg des Vinylcarben-
Kompl
δ: 15.03 und 14.20 (zwei d, 1H, CαH); 8.04 und 7.21
(zwei d, 1H, CβH); 5.91 und 5.90 (zwei s, 8:1, 5H, Cp);
2.28-1.94 (m, 4H,); 1.74-1.44 (m, 6H).
C-NMR (100 MHz,
3)
δ: 274.4 (Cα); 214.2 (CO); 144.5 (Cβ); 142.7 und 142.0
(Cγ); 97.2 und 96.3 (Cp); 38.3, 37.8, 30.4, 30.2, 28.5,
I
Perchlorat (22b, 3.5 mmol, 881 mg) mit einer in situ dar
1g) in THF. Die Aufarbeitung der Reaktionsmischu
exes 16b mit einer Ausbeute von 65%.
Vinylcarben-Komplex 16b
Ausbeute: 65%
16b
W C
CNO
C
O
H
H
1H-NMR (400 MHz,
CDCl3)
13
CDCl
28.3, 27.7 und 26.8 (-CH2-).
R (KBr)
ν = 1994 cm–1 (CO), 1606 cm–1 (NO).
3. Vinyl
3 ie Darstellung des W
Zu einer hellroten Lös mmol) des Vinylcarben-Komplexes 16a
in 5 ml THF wurde eine Lösung von 139 mg (1.0 mmol) von Cyclopent-1-enyl-diethylamin
( ml THF bei –20 °C a pektroskopie
überwacht. Nach vier Stunden war das Edukt verbraucht. Das Lösungsmittel wurde im
akuum entfernt und der Rückstand aus einem Pentan/Ether Gemisch (3:1) bei –20 °C
umkristallisiert, um 166 mg der gelben Kristalle der Verbindung 18 mit einer Ausbeute von
70% zu erhalten.
2 Umsetzung der
.2.1 D
carben-Komplexe 16a-b mit den Enaminen 14a-b
olfram-η1-Acyl-Komplexes 18
ung von 200 mg (0.44
14b) in 1 ddiert. Der Reaktionsverlauf wurde mit der IR-S
V

D Experimenteller Teil 73
Wolfra
18
H-NMR (400MHz, δ: 5.58 (s, 5H, Cp); 5.50 (d, 3JH-H = 12.79 Hz, 1H, C6); 4.20
.67 (m, 1H,
C8); 2.55-2.62 (m, 2H); 2.38-2.55 (m, 2H); 2.30-2.41 (m,
,
3C-NMR (100 MHz, : 256.1 (CO); 116.6 (C5); 111.3 (C6); 101.5 (Cp); 91.7
7,
EPT (100 MHZ,
0 °C)
: 111.3 (C6), 101.5 (Cp), 91.7 (C7), 71.3 (C12), 44.2 (C8),
1
δ: 45.1 [N(CH2)CH3]; 39.7, 37.7, 32.0, 29.2, 26.9 und 24.3
(-CH2-).
m-η1-Acyl-Komplex 18
Ausbeute: 70%
1
THF-d8, -20 °C) (dd, 3JH-H = 12.8 Hz, 3JH-H = 3.9 Hz, 1H, C7); 2
3H); 2.0-2.06 (m, 1H); 1.76-1.96 (m, 4H); 1.52-1.69 (m
7H); 1.32-1.24 (m, 1H); 0.92 (t, CH3).
1
THF-d8, -20 °C)
δ
(C7); 71.3 (C12); 44.2 (C8); 45.1 [N(CH2)CH3]; 39.7, 37.
32.0, 29.2, 26.9 und 24.3 (-CH2-), 15.7 [N(CH2)CH3].
D
THF-d8,-2
δ
5.7 [N(CH2)CH3].
IR (KBr)
ν = 1648 cm-1(C=O), 1577 cm-1(N=O)
Elementaranalyse Berechnet für C22H32N2O2W: C 48.90, H 5.96, N 5.18.
Gefunden: C 49.04, H 5.94, N 5.21.
Kristallstrukturanalyse Siehe Abschnitt E
WOON
N
HH
H
5
12
7 68

D Experimenteller Teil 74
3.2.2 Die Darstellung des Wolfram-η1-Acyl-Komplexes 19
g (0.44 mmol) Vinylcarben-Komplex 16a in 5 ml THF mit
ol) Cyclopent-1-enyl-1-pyrrolidin (14a) in 1 ml
eschrieben. Die Aufarbeitung ergab 146 mg (62% Ausbeute) von den gelben Kristallen des
olfram-η1-Acyl-Komplexes des Wolframs 19. Die Verbindung 19 besteht aus zwei
iastereomeren in einem Verhältnis von 10:1.
olfram-η1-Acyl-Komplex 19.
usbeute: 62%
H-NMR (400MHz, δ 5.59 und 5.56 (s, 5H, Cp); 5.34 und 5.21 (d, 3JH-H = 12.85
H 3 3
H
d 2.05-
2.11 (m, 1H); 1.76-1.90 (m, 2H); 1.45-1.67 (m, 13H), 1.19-
1
13C-NMR (100 MHz,
THF-d8, -20 °C)
δ
100.7(Cp); 93.3 (C7); 71.4 (C12); 42.24 (C8); 48.6, 48.0,
39.5, 38.0, 33.2, 29.3, 26.7, 24.7, 24.4, 24.3 und 22.0 (-CH2-).
EPT (100 MHZ,
HF-d8,-20 °C)
δ: 111.7 und 113.7 (C6); 101.5 und 100.7(Cp); 93.3 (C7);
42.24 (C8).
δ
2
Die Umsetzung von 200 m
137 mg (1.0 mm THF erfolgt wie unter 3.2.1
b
W
D
W
A
19
1
THF-d8, -20 °C)
:
z, 1H, C6); 4.66 und 4.35 (dd, JH-H = 12.9 Hz, JH-H = 3.2
z Hauptdiastereomer und dd, 3JH-H = 13.9 Hz, 3JH-H = 4.2 Hz
er andere Diastereomer, 1H, C7); 2.33-2.60 (m, 9H),
.12 (m, 1H).
: 254 (CO); 112.9 (C5); 111.7 und 113.7 (C6); 101.5 und
D
T
: 48.6, 48, 39.5, 38.0, 33.2, 29.3, 26.7, 24.7, 24.4, 24.3 und
2.0 (-CH2-).
IR (KBr)
ν = 1638 cm-1(C=O), 1600 cm-1(N=O).
WOON
HH
HN
5
6
78

D Experimenteller Teil 75
Elem ntaranalyse Ber. Ce 1, N 5.20.
) Vinylcarben-Komplex 16b in 5 ml THF mit
enyl-1-pyrrolidin (14a) in 1 ml THF erfolgt wie unter 3.2.1
Aufarbeitung ergab 167 mg (68% Ausbeute) von den gelben Kristallen des
olfram-η1-Acyl-Komplexes 20. Die Verbindung 20 besteht aus zwei Diastereomeren in
inem Verhältnis von 10:3.
olfram-η1-Acyl-Komplex des Wolframs 20
usbeute: 68%
1H-NMR (400MHz,
THF-d8, -20 °C) 4.67 und 4.41 (dd, 3JH-H = 13.4 Hz, 3JH-H = 3.2
Hz Hauptkomponente und dd, 3JH-H = 13.4 Hz, 3JH-H = 4.4
3C-NMR (100 MHz, δ: 254.1 und 250.2 (CO); 116.5 und 113.5 (C5); 111.9 und
12); 45.8 und 42.8 (C8); 48.6, 47.9, 41.4, 41.5,
.3, 22.8 und 21.8 (-CH2-).
0 MHZ,
HF-d8,-20 °C)
00.3 (Cp); 93.1 und
88.8 (C7); 45.8 und 42.8 (C8).
δ: 48.6, 47.9, 41.4, 41.5, 39.2, 39.0, 35.4, 35.1, 33.6, 33.1,
22H30N2O2W: C 49.08, H 5.6
Gef.: C 48.94, H 5.29, N 5.32.
3.2.3 Die Darstellung der Wolfram-η1-Acyl-Komplexes 20.
Die Umsetzung von 200 mg (0.43 mmol
137 mg (1.0 mmol) Cyclopent-1-
beschrieben. Die
W
e
W
A
20
δ: 5.56 und 5.53 (s, 5H, Cp); 5.31 und 5.19 (d, 3JH-H = 13.4
Hz , 1H, C6);
Hz MinorKomponente, 1H, C7); 2.69-2.11 (m, 6H), 1.53-
2.08 (m, 17H); 1.15-1.4 (m, 4H).
1
THF-d8, -20 °C) 109.8 (C6); 101.1 und 100.3 (Cp); 93.1 und 88.8 (C7); 74.0
und 73.5 (C
39.2, 39.0, 35.4, 35.1, 33.6, 33.1, 27.9, 27.8, 27.2, 24.6,
24.5, 24.2, 23
DEPT (10
T
δ: 111.9 und 109.8 (C6); 101.1 und 1
WOON
HH
HN
5
678

D Experimenteller Teil 76
27.9, 27.8, 27.2, 24.6, 24.5, 24.2, 23.3, 22.8 und 21.8
(-CH2-).
IR (KBr) ν
= 1641 cm-1(C=O), 1590 cm-1(N=O)
Eleme
s 10 mit dem Enamin 14a, eine
zur Darstellung des Wolfram-η1-Acyl-Komplexes 19.
Eine Lösung von 345 mg (2.5 mmol) des Cyclopent-1-enyl-1-pyrrolidins (14a) in 2 ml
HF wurde tropfenweise mit einer Lösung des Vinylidenkomplexes 10 (330 mg, 1 mmol) in
ml THF bei der Raumtemperatur versetzt. Die Farbe der Reaktionsmischung änderte sich
ngsam von rot zu gelb. Der Reaktionsablauf wurde mit der IR-Spektroskopie überwacht.
ach vier Stunden war die CO Absorption des intermediär entstandenen Vinylcarben-
ft. Der
ückstand wurde mit 2 ml Pentan bei –30 °C für 15 min. gewaschen und sofort danach kalt
a odukt w 5
mg von der Verbindung 19 m
3.2.4 Die Umsetzung des V namin 14b: Darstellung
des Wolfram-η2-Alken-Komplexes 21.
306 m er
wurde tropfenweise mit einer Lösung vom l
Ether bei –30 °C versetz ur
Raumtemperatur erwärmt. D r
Reaktionsverlauf wurde mit DC übe
das Lösungsmittel eingeengt und mit Pentan überschichtet. Nach einer Nacht wurden 840 mg
v 21 in eine
ntaranalyse Ber.für C23H32N2O2W: C 50.01, H 5.83, N 5.07.
Gef.: C 50.25, H 5.90, N 4.98.
3.2.3 Die Umsetzung des Vinylidenkomplexe
Eintopfreaktion
T
5
la
N
Komplexes 16a verschwunden. Das Lösungsmittel wurde im Vakuum abgedamp
R
bgefiltert. Das Rohpr urde übernacht aus Pentan bei –20 °C auskristallisiert, um 37
it einer Ausbeute von 70% zu ergeben.
inylidenkomplexes 10 mit dem E
Ein Lösung von g (2.2 mmol) Cyclopent-1-enyldiethylamin (14b) in 1 ml Eth
Vinylidenkomplex 10 (666 mg,2 mmol) in 40 m
t. Dann wurde langsam die Reaktionsmischung bis z
ie Farbe schlug langsam von orange ins rot um. De
rwacht. Nach dem das Edukt verschwunden war, wurde
on der Verbindung r Ausbeute von 90% isoliert.

D Experimenteller Teil 77
Wolfram-η2-Alken-Komplex
Ausbeute: 90%
21
H-NMR (400MHz, δ: 5.72 (s, 5H, Cp); 4.36 (dd, 3JH-H = 12.3 Hz und 3JH-H = 9.1
.52 (m,
1.09 (t, 6H).
21
1
THF-d8, -20 °C) Hz, 1H, C2); 2.8 (2m, 4H, [N(CH2)CH3], 2.30 – 2
4H); 1.75-1.90 (m, 3H); 1.63 – 1.69 (m, 1H);
13C-NMR (100 MHz,
THF-d8, -20 °C)
δ: 223.7 (CO); 143.1 und 139.3 (C7 und C3); 95.6 (Cp);
48.5 [N(CH2)CH3]; 39.9 (C2); 30.1 (C1); 29.8, 26.7 und
21.2 (CH2); 14.2 [N(CH2)CH3].
DEPT (100 MHZ,
THF-d8,-20 °C)
δ: 95.6 (Cp); 39.9 (C2); 14.2 [N(CH2)CH3].
δ: 48.5 [N(CH2)CH3]; 30.1 (C1); 29.8, 26.7 und 21.2 (CH2).
IR (KBr)
ν = 1952 cm (C=O), 1571 cm (N=O).
Elementaranalyse Ber. für C H N O W: C 43.23, H 5.93, N 4.96.
Krista
25 ml Ether erfolgte, wie unter
3.2.4 beschrieben und ergab nach der Aufarbeitung und Umkristallisierung 260 mg der
Verbindung 22 (73% Ausbeute).
W N
-1 -1
17 24 2 2
Gef.: C 43.54, H 5.92, N 5.12.
llstruktur Daten Siehe Abschnitt E
3.2.5 Die Umsetzung des deuterierten Vinylidenkomplexes 10b mit dem Enamin 14b.
Darstellung des Wolfram-η2-Alken (d2)-Komplexes 22.
Die Umsetzung einer Lösung von 140 mg (1 mmol) Enamin 14b in 1 ml Ether mit
einer Lösung der Verbindung 10c (250 mg, 0.75 mmol) in
CO
O
NH
CC
HH
27
3
1

D Experimenteller Teil 78
Wolfram-η2-Alken (d2)-Komplex 22
22
H-NMR (400MHz, δ: 5.70 (s, 5H, Cp); 4.35 (s, 1H, C2); 2.8 (2m, 4H,
C-NMR (100 MHz, : 223.8 (CO); 143.3 und 139.6 (C7 und C3); 95.9 (Cp);
3 des Vin n THF.
ng des Ena er
Raumtemperatur zu einer Lösung der Verbindung 10 (660 mg, 2 mmol) in 5 ml THF
tropfenweise addiert. Die Reak 45 min war das Edukt
verbraucht. Das Lösungsmittel wurde im Vakuum entfernt. Der Rückstand wurde mit Pentan
b hen und eben. Dieses
Rohprodukt wurde NMR-sp bestand ausschließlich aus den
Verbindungen 18 und 21. Der Vergleich der Cp-Signale bei δ 5.67 und 5.56 ppm, die der
K ugeor das Verhältnis 7:1 für 21:18. Die Ausbeute
ieser Reaktion anhand des umgesetzten Edukts 10 betrug 83%.
W NC
O
OC
CD
D
HN
Ausbeute:73%
1
THF-d8, -20 °C) [N(CH2)CH3]; 2.30-2.52 (m, 2H); 1.63-1.90 (m, 4H); 1.09
(t, 6H).
13
THF-d8, -20 °C)
δ
48.7 [N(CH2)CH3)]; 39.9 (C2); 29.9, 26.8 und 21.3 (CH2);
14.4 [N(CH2)CH3].
.2.6 Die Reaktion ylidenkomplexes 10 mit dem Enamin 14b i
Eine Lösu mins 14b (417 mg, 3 mmol) in 2 ml THF wurde bei d
tion wurde mit DC kontrolliert. Nach
ei –30 °C gewasc filtriert, um 740 mg eines roten Pulvers zu erg
ektroskopisch untersucht und
omplexen 18 und 21 z dnet werden, zeigte
d

D Experimenteller Teil 79
4 Darstellung des η1-Allenyliden
N+
ClO
komplexes des Wolfram
ng der Iminiumsalze 23a-d
.1.1 Darstellung des Iminiumsalzes 23a, 23b und 23d
Bei der Darstellung der Iminiumsalz 23a, 23b und
ntsprechende Aminsalz durch das Umsetzen des Amins mit Perchlorsäure dargestellt.[76]
20.6 g von N-Methylanilium Perchlorat (100 mmol) wurde mit einer Mischung von
0 und C e
1 rt. Danach w n
und aus Isopropanol umkrista 9 g (34%) des Iminiumsalzes 23a zu erhalten.
Smp: 170 °C.
I
4.1 Darstellu
4
23d, wurde zuerst das
e
Cyclopentyliden-N-phenyl-methanamonium Perchlorat (23a)
.1 ml N-Methylanilin yclopentanon (250 mmol, 21g) versetzt. Die Suspension wurd
urde die Reaktionsmischung eingeengt, mit Pentan gewasche
llisiert, um
0 Stunden gerüh
R (KBr-Pressling): ν =
4
_
N+
CH3
ClO4
1671
Smp: 190 °C
23b
cm-1 (C=N).
23a
Adamantanyliden-N-phenylmethanaminum Perchlorat (23b)
N-Methylanilium-Perchlorat (18 mmol, 3.87 g) und eine katalytische Menge an N-
Methylanilin wurden zu einer Lösung von Adamantanon (22 mmol, 3.3 g) in 100 ml Toluol
addiert und unter Rückfluss erhitzt. Das entstandene Wasser wurde mit einem
Wasserabscheider entfernt. Nach 12 Stunden wurde das Volumen im Vakuum zur Hälfte
reduziert, und der Rückstand filtriert und aus Isopropanol umkristallisiert, um 2 g vom
Iminiumsalz 23b mit einer Ausbeute 32% zu erhalten.

D Experimenteller Teil 80
1H-NMR (200 MHz, δ:7.4-7.6 (5H ,Phenylrest); 3.8 (3H,
CD N) (2H); 2.0-2.3. 3C
NCH3); 3.5 und 2.6
13C-N
(Adamantanylidenrest)
MR (100 MHz,
CD3CN)
δ: 204 (C=N); 131.7, 131.6 und 124.3 (Phenylring); 46.8
(NCH3); 40.4, 40.2, 39.9, 38.5 und 35.5
IR (KBr) ν = 1641 cm-1 (C=N).
Adamantanyliden-pyrrolidinium Perchlorat (23d)
3.1 g) und eine katalytische Menge an
ilin wurden zu einer Lösung von Adamantanon (22 mmol, 3.3 g) in 100 ml
Die Aufarbeitung erfolgte, wie für das
iniumsalz 23b beschrieben, und ergab 2.5 g vom Iminiumsalz 23d mit einer Ausbeute
p: 205 °C
(100 MHz,
CD3CN)
δ: 195 (C=N); 54.3 (N-CH2); 39.8, 39.1, 35.6, 27.3 und
27.4.
IR (KBr)
N-Pyrrolidiniumperchlorat (18 mmol,
N-Methylan
Benzol addiert und unter Rückfluss erhitzt.
Im
46%.
Sm
N+
ClO4
23d
1H-NMR (200 MHz,
CD3CN)
δ: 3.9 (N-CH2); 2.2-1.9 (Adamantanylidenprotonen und
N-CH2-CH2). 13C-NMR
ν = 1680 cm-1 (C=N).

D Experimenteller Teil 81
4 n Adam
einer Lösung von ol) Adamantanon und 12 g (165 mmol) tert-
Butylamin in 125 ml Toluol wurde bei -20 °C langsam 3.64 ml (33 mmol) von TiCl4
z tunden hung
f unden unter Rück er
Rückstand im Pentan aufgen s Filtrat wurde dann eingeengt um ein
gelbes zehflüssiges Öl zu erhalten. Dieses Öl wurde dann durch Kugelrohr-Destillation
gereinigt um 5 g von der Verbindung 38 zu ergeben (77%).
Adamantanyliden-tert-butylam
) Butylgruppe).
13C-NMR (100 MHz,
CDCl3)
δ: 178 (C=N); 54.3 [C(CH3)3]; 39.2, 38.1 und 36.5
(-CH2-); 44.9 37.1 und 27.5 (-CH-); 30.8 [C(CH3)3].
4.1 dama
In 100 ml Dichloromethan wurde 0.735 g (5 mmol) Trimethyloxonium
Tetraflouroborat solange gerührt, bis es sich auf ol)
Adam -butylamin ( ösung versetzt. Nach einem Tag wurde die
Reaktion abgebrochen. Das Lösungsmittel wurde auf 10 ml eingeengt und filtriert. Der
Rückstand wurde dann aus Isopropanol umkristallisiert, um 0.95 g von 23c zu ergeben
(Ausbeute 62%).
.1.2 Darstellung vo
Zu
antanyliden-tert-butylamin 38[80]
4.95 g (33 mm
ugetropft. Nach drei S
ür 12 St
wurde dann das Kältebad entfernt und die Reaktionsmisc
fluss erhitzt. Das Lösungsmittel wurde eingeengt und d
ommen und filtriert. Da
in
N
38
1H-NMR (400 MHz,
CDCl
δ: 3.16 (1H); 2.4 (1H); 1.75-1.97 (12H); 1.23 (9H, tert-
3
.3 Darstellung von A
Tetraflouroborat (23c)
ntanyliden-N-tert-butyl-methanamonium
gelöst hatte. Dann wurde 1.12 g (5.5 mm
38) zu dieser Lantanyliden-tert

D Experimenteller Teil 82
Adamantanyliden-N-tert-butyl-methanamonium Tetraflouroborat (23c)
p.: 1
CD3CN)
3.8(s, 1H);3.4 (s, 1H); 3.6 (s, 3H, NCH3); 2.1-2.4
(12H); 1.73 (9H, tert-Butylgruppe). 13C-NMR (100 MHz,
CD3CN)
δ 205 (C=N); 69.9, 41.2, 41.1, 40, 39.6, 35.9, 29.6, 26.7.
IR (KBr)
Sm 90 °C
23c
N+
BF4
1H-NMR (400 MHz, δ:
ν = 1613 cm (C=N). -1
4.2 Darstellung des (1-Cyclopentenyl)-vinylidenkomplexes des Wolframs 24
s Viny e
0.5 m n-Butyllithium
einer Stunde wurde dieser grünen Lösung Cyclopentyliden-N-phenyl-methanamonium-
erchlorate (23a) (240 mg, 0.88 mmol) bei –30 °C zugesetzt. Nachdem die Farbe der Lösung
s rot umgeschlagen hatte, wurde die Reaktion abgebrochen. Das Lösungsmittel wurde im
gereinigt. Die
Umkristallisierung des Rohproduktes aus Pentan bei –20 °C ergab 172 mg der orangen
Zu einer Lösung de
l (0.8 mmol)
lidenkomplexes 10 (260 mg, 0.8 mmol) in 10 ml THF wurd
(1.6 M in Hexan) bei –78 °C tropfenweise addiert. Nach
P
in
Vakuum entfernt. Das Rohprodukt wurde über Silica mit Pentan/Ether
Kristalle der Verbindung 24 in einer Ausbeute von 53%. Die Verbindung 24 besteht aus zwei
Diastereomeren im Verhältnis von 4:5.

D Experimenteller Teil 83
(1-Cyclopentenyl)-vinylidenkomplex des Wolframs
Ausbeute: 53%
1H-NMR (400 MHz,
CD
δ: 6.66, 6.60 (s, 1H, W=C=CH); 5.89 und 5.88 (Cp); 5.52
(m
13C-NMR (100 MHz,
CD
δ: 346.9 und 346.5 (Cα); δ 210.5 und 209.6 (CO); 136.1 und
135. 2 (Cβ); 127.2 und 126.9 (Cδ); 96.5
und 96.4 (Cp); 33.2, 33.1, 32.6, 32.5 und 23.5 (-CH2-).
(100 M
5 und
24
Cl3) , 1H, C=CH-C); 2.36-2.40 und 1.90-1.94 (m, 6H, -CH2-).
Cl3) 1 (Cγ); 128.5 und 128.
DEPT δ: 136.1 und 135.1 (C
HZ, CDCl3) 96.4 (Cp).
δ: 33.2, 33.1, 32.6, 32.5 und 23.5 (-CH
γ); 128.5 und 128.2 (Cβ); 96.
2-).
IR (KBr)
ν = 2007 cm-1(CO), 1583 cm-1(NO), 1603 und 1618 cm-1
(C=C).
Elementaranalyse Ber. für C13H13NO2W: C 39.12, H 3.28, N 3.52.
Gef.: C 39.05, H 3.11, N 3.53.
W CNC
O
OC
Hβγ
α
δ

D Experimenteller Teil 84
Darstellung des Adamantyliden-Aminocarben-Komplexes 25
Lösung von 500 mg (1.5 mmol) des Vinylidenkomplexes 10 in 15 ml THF
urde 0.94 ml (1.5 mmol) n-Buthyllithium (1.6 M in Hexan) bei –78 °C tropfenweise addiert.
ach einer Stunde wurde bei –50 °C dieser grünen Lös ol)
damantanyliden-N-phenylmethanaminum Perchlorat (23b) zugesetzt. Die
eaktionsmischung wurde auf –20 °C erwärmt und ihre Farbe schlug langsam in rot um. Die
eaktion wurde mit DC kontrolliert. Nach zwei Stunden war das Edukt nicht mehr zu
rkennen. Die Reaktionsmischung wurde zu 450 ml gut gerührten Pentan getropft und nach
llisierung
es Rohproduktes aus Ether/Pentan (1:9) bei –20 °C ergab 532 mg der hellroten Kristalle der
V Ausbe
A tyliden-Aminocarbe
Ausbeute 62%
25
0 °C)
: 7.04 – 7.40 (m, 5H, aromatische Protonen); 6.0 (s, 1H,
W=C
z,
THF-d8, -20 °C)
29.6, 127.5
Cβ); 95.5 (Cp); 51.6 (NCH3);
40.6, 33.8, 29.6 und 29.2 (CH, Adamantanyliden); 40.9,
40.1, 38.9, 38.7 und 38.1 (CH2).
DEPT (100 MHZ,
THF-d8,-20 °C)
δ: 139.6, 129.6, 127.5 und 127.3 (phenyl.); 127.8 (Cβ); 95.5
(Cp); 51.6 (NCH3); 40.6, 33.8, 29.6 und 29.2 (CH,
Adamantanyliden).
δ: 40.9, 40.1, 38.9, 38.7 und 38.1 (CH2, Adamantanyliden).
Zu einer
w
N ung 546 mg (1.6 mm
A
R
R
e
einer Stunde filtriert. Das Lösungsmittel wurde im Vakuum entfernt. Die Umkrista
d
erbindung 25 in einer
daman
ute von 62%.
n-Komplex 25
1H-NMR (400MHz,
THF-d8, -2
δ
H); 5.79 (s, 5H, cp), 3.90 (s, 3H, N-CH3), 2.27 - 0.8
(Adamantanyliden).
13C-NMR (100 MH δ: 263.8 (Cα); 231.9 (CO); 150.8 (Cδ); 139.6, 1
und 127.3 (Phenylrest); 127.8 (
W CNC
O
O N
H

D Experimenteller Teil 85
IR (KBr) ν = 1903 cm-1 (CO), 1565 cm-1(NO).
at (23c) addiert. Die grüne Farbe der Lösung
onsmischung auf 0 °C erwärmt und für weitere
rührt. Dabei änderte sich die Farbe wieder zu grün um. Das Lösungsmittel
urde im Vakuum entfernt. Das Rohprodukt wurde über Silicagel chromatographiert. Nach
er Pre-Elution mit Pentan, wurde die Verbindung 26 mit 5% Ether/Pentan eluiert. Das
ösungsmittel wurde entfernt, um 195 mg der tiefgrünen Kristalle des η1-
llenylidenkomplexes 26 zu ergeben (70% Ausbeute). 1-Allenylidenkomplex des Wolframs
1H-NMR
(400 MHz, CDCl3)
l3) 2
Elementaranalyse Ber. für C18H21NO2W: C 52.46, H 4.93, N 4.89.
Gef.: C 52.02, H 4.69, N 4.32.
4.6 Darstellung des η1-Allenylidenkomplex des Wolframs 26
0.45 ml (0.625 mmol) von einer n-Butyllithium Lösung (1.6 M in Hexan) wurde einer
Lösung von 200 mg (0.6 mmol) des Vinyliden Komplexes 10 in 5 ml THF bei –78 °C
zugetropft. Nach einer Stunde wurde bei –30 °C 200 mg (0.65 mmol) von Adamantanyliden-
N-tert-butyl-methanamonium Tetraflourobor
änderte sich in rot um, dann wurde die Reakti
drei Stunden ge
w
d
L
A
η
Ausbeute: 70%
26
δ: 5.70 (s, Cp); 2.84, 2.64 und 190 - 1.99
(Adamantanylidenrest).
13C-NMR
(100 MHz, CDC
δ: 274.4 (Cα); 214.0 (CO); 183.4 (Cγ); 149.0 (Cβ); 96.8 (Cp);
36.9, 39.5 und 39.7 (-CH -); 43.2, 42.8 und 28.0 (CH).
W CNC
O
OC Cβ
γα

D Experimenteller Teil 86
DEPT
(100 MHZ, CDCl3)
δ: 96.8 (Cp); 43.2, 42.8 und 28.0 (CH).
: 36.9, 39.5 und 39.7 (-CH2-).
ss-
Spektroskopie
δ
Hochaufgelöste Ma Ber. fürC18H19NO2W (M+) m/e: 463.09026.
Gef.: 463.09015.
IR (KBr) ν
= 1971 cm-1(CO), 1906 cm-1(C=C=C), 1609 cm-1(NO)
UV-V
mg (1.1 mmol) Adamantanyliden-
. Die Reaktionsmischung wurde auf –20 °C erwärmt.
lug langsam in rot um. Die Reaktion wurde mit der IR-Spektroskopie
berwacht; nach zwei Stunden war das Edukt vollständig verbraucht. Die Reaktionsmischung
urde zu 250 ml gut gerührten Pentan getropft und nach einer Stunde filtriert. Das
ösungsmittel wurde im Vakuum entfernt. Das Umkristallisieren des Rohproduktes aus
ther/Pentan bei –20 °C ergab 285 mg der roten Kristalle der Verbindung 28 in einer
usbeute von 61%. Die Verbindung besteht aus zwei Diastereomeren im Verhältnis von 10:1.
dama tanyl en-C benk mplex 28
usbeute 61%
28
is (CH2Cl2)
λmax (log ε): 340 nm (4.04) und 474 nm (4.29).
Elementaranalyse Ber. für C18H19NO2W: C 46.72, H 4.20, N 3.03.
Gef.: C 46.80, H 4.14, N 3.05.
4.5 Umsetzung des Vinylidenkomplexes 10 mit dem Iminiumsalz 23d
Zu einer Lösung von 333 mg (1 mmol) der Verbindung 10 in 10 ml THF wurde 0.62
ml (1 mmol) n-Buthyllithium (1.6 M in Hexan) bei –78 °C tropfenweise addiert. Nach einer
Stunde wurde bei –50 °C dieser grünen Lösung 345
pyrrolidinium Perchlorat (23d) zugesetzt
Die Farbe sch
ü
w
L
E
A
A n id ar o
A
W CN
CO
O H
C CH β
γα

D Experimenteller Teil 87
z, Hauptdiastereomer, 1H,
3JH-H = 14.77
= 13.29 Hz, Hauptdiastereomer, 1H,
CH=C) und 7.21 (der andere Diastereomer 3JH-H = 14.77
H s,
m, m, Adamantanyliden, 14 H).
C-NMR δ: 274 (Cα); 214.6 (CO); 153.5 (Cγ); 138.0 und 138.6 (Cβ);
, 38.6, 28.2
en); 40.2, 39.7, 39.5, 39.2,
39.0, 38.7, 37.0 und 36.2 (CH2).
(100 M
4 (Cp); 46.7,
1H-NMR
(400 MHz, CDCl3)
δ: 14.20 (d, 3JH-H = 13.29 H
W=CH) und 15.05 (der andere Diastereomer,
Hz); 8.03 (d, 3JH-H
z); 5.90 (s, 5H, Cp); 3.0, 2.46, 186 - 1.95, 1.67 - 1.74 (s,
13
(100 MHz, CDCl3) 97.1 und 97.4 (Cp); 46.7, 41.8, 41.2, 34.1, 33.9
und 27.4 (CH, Adamantanylid
DEPT δ: 274 (Cα); 138.0 und 138.6 (Cβ); 97.1 und 97.
HZ, CDCl3) 41.8, 41.2, 34.1, 33.9, 38.6, 28.2 und 27.4 (CH,
Adamantanyliden).
δ: 40.2, 39.7, 39.5, 39.2, 39.0, 38.7, 37.0 und 36.2 (CH2).
IR (KBr)
ν = 1975 cm-1 (CO), 1592 und 1565 cm-1(NO)
Elementaranalyse Ber. für C18H21NO2W: C 46.27, H 4.53, N 3.00.
Gef.: C 46.28, H 4.30, N 2.84.
UV-Vis (CH2Cl2) λmax (log ε): 379.1 nm (3.16).
4.7 Umsetzung des η1-Allenylidenkomplexes 26 mit Methanol: Darstellung des
Vinylidenkomplexes 27
00 mg (0.21 mmol) vom Allenylidenkomplex 26 wurde in 8 ml entgasten und mit Argon
esättigten Methanol in einem Schleck Rohr für 3 Tagen gerührt. Die Farbe änderte sich von
rün ins gelb um. Das Lösungsmittel wurde entfernt und das Rohprodukt über eine kurze
entan bei –20 °C
ergab 59 mg von den gelben Kristallen der Verbindung 27 (Ausbeute 58%). Der
Vinylidenkomplex 27 besteht aus zwei Diastereomeren im Verhältnis 4:5.
1
g
g
Silica Säule mit Pentan filtriert. Die anschließende Umkristallisierung aus P

D Experimenteller Teil 88
Vinylidenkomplex 27
Ausbeute: 58%
27
3C-NMR
z, CDCl3)
: 340.9 und 333.5 (Cα); 214.3 und 213.2 (CO); 131.2 und
39.2, 39.1, 36.3, 36.2, 36.1, 33.8, 33.7,
00 MHZ, CDCl3)
δ: .3 und 49.0
(OCH3); 37.0 und 36.7, 29.4, 29.3, 28.7 und 28.6 (CH,
, 33.8, 33.7, 33.6 und 33.5
H2, Adamantanyliden).
(KBr)
CC
MeO
W CNC
O
O
H
αγ
β
1H-NMR
(400 MHz, CDCl3)
δ: 5.88 und 5.87 (s, 5H, cp); 5.37 und 5.34 (s, 1H, Cβ); 3.34
und 3.29 (s, 3H, OCH3); 2.22 - 1.48 (m, 14H,
Adamantanyliden).
1
(100 MH
δ
131.1 (Cβ); 97.4 und 97.3 (Cp); 83.8 und 83.2 (Cδ), 49.3 und
49.0 (OCH3); 37.0 ,36.7, 29.4, 29.3, 28.7 und 28.6 (CH,
Adamantanyliden);
33.6 und 33.5 (CH2, Adamantanyliden).
DEPT
(1
131.2 und 131.1 (Cβ); 97.4 und 97.3 (Cp); 49
Adamantanyliden).
δ: 39.2, 39.1, 36.3, 36.2, 36.1
(C
IR ν = 2007 und 1996 cm-1 (CO), 1641 und 1600 cm-1(NO).
f.: C 46.04, H 4.53, N 2.98.
Elementaranalyse Ber. für C19H23NO3W: C 45.89, H 4.66, 2.82.
Ge

D Experimenteller Teil 89
4.8 Umsetzung des η1-Allenylidenkomplexes 26 mit N-Methylanilin.
Lösung von 100 mg (0.21 mmol) des Allenylidenkomplexes 26 in 1 ml THF
urde 1 ml (0.21 mmol) einer Lösung von N-Methylanilin in THF (0.28 M) bei
aumtemperatur zugetropft. Nach 2 Tagen wurde der Allenylidenkomplex 26 quantitativ
oliert.
lung des
mantanyliden-Am
ng von 3
wurde 2.27 ml (0.75 mmol) einer Lösung von Diethylamin in THF (0.28 M) bei 0 ºC
zugetropft. Die Farbe der Reaktionsmischung schlug von grün in hellrot um. Das
L el wurde entfernt 1)
b llisiert, er
Ausbeute von 65% zu ergeben
Adamantanyliden-Aminocar
Ausbeute: 65%
29
00 MHz, CDCl3)
δ: 5.65 (s, 1H, C
3.40 (m, 4H, N-CH2-CH3); 2.47, 2.34 und 1.61 - 1.95 (m,
tanyliden; 1.37 und 1.14 (t, 6H, N-CH2-CH3).
C-NMR
(100 MHz, CDCl3)
δ: 255.4 (Cα); 234.7 (CO); 141.6 (Cγ); 122.8 (Cβ); 94.5 (Cp);
52.8 und 47.1 (N-CH2-CH3); 39.7, 32.5, 28.1 und 28.0 (CH,
Adamantanyliden); 39.7, 37.9, 37.8 und 36.9 (CH2,
Adamantanyliden); 14.1 und 12.8 (N-CH2-CH3).
Zu einer
w
R
is
4.8 Umsetzung des Allenylidenkomplexes 26 mit Diethylamin, Darstel
Ada inocarben-Komplexes 29
00 mg (0.64 mmol) des Allenylidenkomplexes 26 in 1 ml THFZu einer Lösu
ösungsmitt . Das Rohprodukt wurde aus einer Pentan/Ether Mischung (4:
um 350 mg der roten Kristalle der Verbindung 29 in ein
.
ben-Komplex 29
ei –20 ºC umkrista
1H-NMR
(4
β); 5.60 (s, 5H, Cp); 4.10, 3.94, 3.79 und
14H, Adaman
13
W CN
CO
O N
C CH β γ
α

D Experimenteller Teil 90
DEPT δ: 122.8 (C
(100 M
nd 28.0 (CH,
β); 94.5 (Cp); 39.7, 32.5, 28.1 u
HZ, CDCl3) Adamantanyliden); 14.1 und 12.8 (N-CH2-CH3).
δ: 52.8 und 47.1 (N-CH2-CH3); 39.7, 37.9, 37.8 und 36.9
(CH2, Adamantanyliden).
IR (KBr) ν = 1895 cm-1(CO), 1575 cm-1(NO).
e
6.
schlug von grün in hellrot um. Das Lösungsmittel wurde
it einer Pentan/Ether Mischung (9:1)
rt, um 85 mg von dem Adamantanyliden-Carben-Komplex 28 zu ergeben
4%).
Elem ntaranalyse Ber für C20H30N2O2W: C 49.08, H 5.62, N 5.20.
Gef.: C 48.68, H 5.43, N 4.6
4.9 Umsetzung des Allenylidenkomplexes 26 mit Pyrrolidin
Zu einer Lösung von 233 mg (0.5 mmol) des Allenylidenkomplexes 26 in 3 ml THF
wurde eine Lösung von 35 mg (0.5 mmol) Pyrrolidin in 2 ml THF bei –10 °C zugesetzt. Die
Farbe der Reaktionsmischung
entfernt. Der Rückstand wurde über eine Silica Säule m
chromatographie
(3

D Experimenteller Teil 91
4 Umsetzung des Vinyl
4.1 Umsetzung von dem
W(η5-C5H5)(CO)(NO)(η2-1,
Zu einer Lösung von 990 mg des Vinylidenkomplexes 10 (3.3 mmol) in 10 ml THF
wurde 230 ml (33 mmol) von ei bei –30 °C zugetropft.
Der Reaktionsverlauf wurde mittels IR-Spektroskopie kontrolliert. Nach dem die etherische
D g addiert zwei Stunden
gerührt, bis das Edukt vollstän ittel wurde im Vakuum entfernt.
as Rohprodukt wurde in Pentan gelöst und über eine kleine Silica Säule filtriert. Die eluierte
elbe Lösung wurde im Vakuum abgedampft und ergab 884 mg von dem Komplex 31, was
zwei Diastereomerpaaren.
Das NM
31
1H-NMR
(400 MHz, CDCl3)
δ: 6.91 und 6.86 (t, 4JH-H = 3.5 Hz für beide Diastereomers,
1H, C3); 6.09 und 5.81 (t, 4JH-H = 3.5 Hz für beide
Diastereomere, 1H, C3); 5.71 und 5.66 (Cp); 1.86 und 2.19
und 1.77 (2H, C1, der Hauptdiastereomer: t, 4JH-H = 3.5 Hz;
der andere Diastereomer, zwei dt, 2JH-H = 11 Hz, 4JH-H = 3.5,
Hz).
13C-NMR
(100 MHz, CDCl3)
δ: 213.7 und 215.0 (CO); 161.3 und 160.2 (C2); 110.3 und
107.1(C1); 95.9 und 95.5 (Cp); 7.2 und –4.1 (C3).
WC
idenkomplexes des Wolframs mit Diazoalkanen.
Vinylidenkomplex 10 mit Diazomethan, Darstellung von
2-H2C=C=CH2) (31)
ner CH2N2 Lösung in Ether (0.14 M)
iazomethan Lösun worden war, wurde die Reaktionsmischung noch
dig abreagierte. Das Lösungsm
D
g
eine Ausbeute von 85% entsprach. Die Verbindung 31 bestand aus
R spektroskopisch vermitteltes Verhältnis der beiden Diastereomeren betrug bei der
Raumtemperatur 2:1.
Carbonyl-(η5-cyclopentadienyl)-nitrosyl-(η2-1,2-H2C=C=CH2) Wolfram (31)
Ausbeute: 85%
O
NOH
C CHC
HH

D Experimenteller Teil 92
DEPT δ: 95.9 und 95.5 (Cp), 7.2 und -4.1 (C3).
(100 MHZ, CDCl3)
IR (KBr) ν = 1988 cm-1 (C≡O), 1599 cm-1 (N≡O).
on Diazoessigsäureethylester (464 mg, 4 mmol) in 10 ml THF wurde
er Rate von
nde versetzt. Der Reaktionsablauf wurde mit IR-Spektroskopie überwacht.
ach 12 Stunden war die Reaktion vollendet. Das Lösungsmittel wurd
as Rohprodukt wurde über eine kürze Silca Säule chromatographiert. Die Elution der gelben
ande mit Dichloromethan/Pentan (1:2) ergab 182 mg von einer Mischung der zwei
iastereomeren 32 und 33 mit dem Verhältnis von 5:1 (87% Ausbeute). Die Mischung von
iastereomeren wurde mit HPLC über Diol Phase mit TBME/Hexan getrennt.
ef.: C 34.34; H 3.15; N 3.31.
ristalle g
isoliert. Diese Verbindung bes
(E)-{(η 5-C5H5)(CO)(NO)W[
32
WC
Elementaranalyse Ber. für C9H9O2NW: C 31.15; H 2.61; N 4.04.
Gef.: C 31.52; H 2.28; N 3.89.
Kristallstrukturanalyse Siehe Abschnitt E
4.2 Umsetzung von dem Vinylidenkomplex 4 mit Diazoessigsäureethylester,
Darstellung vom η2-Allen-Komplex des Wolframs 32 und 33
Eine Lösung v
einer Lösung der Verbindung 10 (1 mmol, 333 mg) in 10 ml THF bei 45 °C in ein
1 ml in der Stu
N e im Vakuum entfernt.
D
B
D
D
Ber. für C12H13O4NW: C 34.39; H 3.13; N 3.34.
G
Die gelben K der Verbindung 32 wurden aus einer Pentan/Ether Lösun
teht aus zwei Diastereomeren im Verhältnis 3:5.
η2(2,3)-H2C=C=CHC(O)OCH2CH3] (32)
O
NO C C
H
COOC H2 5CH
H

D Experimenteller Teil 93
00 MHz, CDCl3)
: 6.67 und 6.47 (t, 4JH-H = 3.0 Hz, 4JH-H = 3.6 Hz, C3, X-
eil des ABX Spinsystems, 1H); 5.72 und 5.70 (s, 5H, Cp),
4 Teil, 2JH-H = 12.8
z, 4JH-H = 3.0 Hz, 1H, C1); 2.24 und 2.17 (dd, 2JH-H = 12.8
6.9 Hz, 3H,
3)
(CO); 179.1 und 176.4 (C4); 165.9 und
164.9 (C2); 118.9 und 115.7 (C3); 96.0 und 95.6 (Cp); 60.2
und 60.6 (C5); 14.4 und 14.3 (C6); 10.4 und -0.2 (C1).
100 M
95.6 (Cp); 14.4 und 14.3
1H-NMR
(4
δ
T
.24 (m, 2H, C4); 2.58 und 2.29 (dd, AB
H
Hz, 4JH-H = 3.0 Hz, 1H, C1); 1.31 (t, 3JH-H =
C6).
13C-NMR δ: 213.0 und 210.8
(100 MHz, CDCl
DEPT δ: 118.9 und 115.7 (C3); 96.0 und
( HZ, CDCl3) (C6).
δ: 60.2 und 60.6 (C5); 10.4 und -0.2 (C1).
IR (KBr): ν = 2006 cm-1 (W-C O), 1717 cm-1 (C=O ester), 1681 cm-1
(C=C) und 1598 (N=O) cm-1.
Kristallstrukturanalyse Siehe Abschnitt E
Die Kristalle der Verbindung 33 wurden aus einer Pentan Lösung gezüchtet.
)-{(η 5-C5H5)(CO)(NO)W[η2(2,3)-H2C=C=CHC(O)OCH2CH3]} (33)
H-NMR
δ: 7.43 (t, X Teil vom ABX Spinsystem, 1H, C3); 5.80 (s,
5H, Cp); 4.24 (m, 2H, C5); 2.07 (dd, 2JH-H = 12.1 Hz, 4JH-H
1); 1.77 (dd,
W
(Z
O
C
33
1
(400 MHz, CDCl3):= 2.7 Hz, B Teil vom ABX Spinsystem, 1H, C
NO C CCOOC2H5
HCHH

D Experimenteller Teil 94
2JH-H = 12.1 Hz, 4JH-H = 3.2 Hz, A Teil vom ABX
13C-NMR
(100 MHz, CDCl3)
DEPT
(100 MHZ, CDCl3)
.0(C3); 96.7 (Cp); 14.5 (C6).
: 59.6 (C5); 3.9 (C1).
Spinsystem, 1H, C1); 1.33 (t, 3H, C6).
δ: 212.9 (CO); 180.0 (C4); 167.8 (C2); 117.0(C3); 96.7
(Cp); 59.6 (C5); 14.5 (C6); 3.9 (C1).
δ: 117
δ
IR (KBr) ν = 2030 cm-1 (W-C O), 1703 cm-1 (C=O ester), 1671 cm-1
(C=C) und 1625 cm-1 (N=O).
Kristallstrukturanalyse
Siehe Abschnitt E

D Experimenteller Teil 95
5 Hydrolyse der Phosp
5.1 Darstellung der Phosphorkomplexe des Wolframs
5 tellung von dem
urde ein ordiphenylphosphin in 20
ml THF zu einer orangen Lösung vom Vinylidenkomplex 10 (1.5 g, 4.5 mmol) in THF (40
m tropft. Der Fortschr ktroskopie überwacht. Die
M Raumte ährend dieser
Zeit änderte sich die Farbe de von Orange nach weinrot. Das Lösungsmittel
wurde unter Vakuumbedingungen entfernt und di K 2
und Pentan ergab eine Ausbeute von 1.56 g (66%) an orm orange Kristalle.
Diphenylphosphor-Komplex des Wolframs 11a
Ausbeute 66%
11a
1H-NMR
(400 MHz, CDCl3)
δ: 7.30–7.80 (m, 10 H, Ph); 7.65 (d, 1H, cis-3JH–P = 15
Hz, C=CH2); 6.55 (d, 1H, trans-3JH–P = 35 Hz, C=CH2);
5.85 (s, 5H, Cp).
13C-NMR
(100 MHz, CDCl3)
δ: 163.1 (d, 1JC–P = 51 Hz, 1JC–W = 51 Hz, W–Cα); 133.5
(d, 2JC–P = 9 Hz, Cβ); 133.8 und 133.6 (zwei d, 2JC–P = 12
Hz, Phenyl); 131.2 und 130.8 (zwei d, 4JC–P = 3 Hz,
Phenyl); 129.0 und 128.9 (zwei d, 3JC–P = 12 Hz,
Phenyl); 128.5 und 127.0 (zwei d, 1JC–P = 50 Hz,
Phenyl), 102.7 (d, 2JC–P = 1.5 Hz, Cp).
IR (KBr)
horkomplexe des Wolframs
.1.1 Dars Diphenylphosphor-Komplex des Wolframs 11a
e Lösung von 0.84 ml (4.5 mmol) ChlBei –30 °C w
l) zuge itt der Reaktion wurde mit IR-Spe
ischung wurde auf mperatur erwärmt und für eine Stunde gerührt. W
r Reaktionslösung
e ristallisation des Rückstands aus CH2Cl
11a in F
W
N
P
ClO
HH
ν = 1610 cm–1 (NO).
3.1.2 Darstellung von Di-tert-butylphosphor-Komplex des Wolframs 11b

D Experimenteller Teil 96
Wie unter 2.3.1 beschrieben erfolgte die Umsetzung von 812 mg ( 4.5 mmol) Chlor-
i-tert-butylphosphin mit einer äquimolaren Menge an dem Vinylidenkomplex 10 (1.5 g). Die
tographie an Silicagel mit 3:1
mkrista
11b
H-NMR
3)
δ: 7.47 (d, 1H, C=CH2), 6.49 (d, 1H, C=CH2), 5.82 (s, 5H,
Cp), 1.48 und 1.36 [zwei d, 18H, P(CMe3)2].
C-NMR
(100 MHz, CDCl3)
δ: 165.5 (d, W-Cα), 132.8 (d, Cβ), 103.4 (d, Cp), 40.1 und
33.7 [zwei d, C(CH3)3], 31.0 (zwei d, CH3).
IR (KBr)
d
anschließende Reinigung des Rückstandes mit der Chroma
Pentan / Ether ergab 1.91 g (87%) des Komplexes 11b, die aus CH2Cl2 und Pentan
u llisiert wurde.
Di-tert-butylphosphor-Komplex des Wolframs 11b
Ausbeute: 87%
W PN ClO
HH
1
(400 MHz, CDCl
13
ν = 1528 cm–1 (NO).
3.1.3 Darstellung von Di-iso-propylphosphor-Komplex des Wolframs 11c
Eine Lösung von 0.8 ml (5 mm
L .82 mmo . Das
Lösungsmittel wurde abgedam ntan
/ Ether chromatographiert, Die
Umkristallisierung aus Ether b
ol) Diisopropylchlorophosphane, in 10 ml. THF wurde einer
l) Vinylidenkomplex 10 in 60 ml THF bei 0˚C zugetropft
pft und das Rohprodukt schließlich an Silicagel mit 3:1 Pe
um 1.441 g (65%) des Komplexes 11c zu erhalten.
ei –20 °C führte zu leuchtend orangen Kristallen.
ösung von 1.6 g (4

D Experimenteller Teil 97
D ropylphosphor-Komplex des Wolframs 11c
Ausbeute: 65%
i-iso-p
H NMR
3)
δ: 7.42 (d, 1H, 3JP–Hcis = 12.6 Hz, 3JH–Wtrans = 8.95 Hz, CβH);
6.43 (d, 1H, 3JP–Htrans = 31.64 Hz, 3J W–Hcis = 5.12 Hz CβH);
5.83 (s, 5H, Cp); 2.8 und 2.6 [m, 4H, PCH )2];1.1-1.5
[m, 12H, PCH(CH3)2].
100 MHz, CDCl3) P-C = 9.15 Hz,
β); 102.5 (d, Cp); 26.9 ,26.7, 22.3 und 22.1 [PCH(CH3)2];
1P-NMR
, CDCl3,
H3PO4 als externer
Standard, 1H-Entkoppelt)
: -57.63 (s, 1JP-W = 130.97 Hz).
W PN ClO
HH
11c
1
(400 MHz, CDCl
(CH3
δ:161.9 (d, 13C-NMR
(
1JP-C = 51.88 Hz, Cα); 132.7 (d, 2J
C
20.9, 20.8, 20.7, 20.7, 19.6 [PCH(CH3)2]
3
(162 MHz
δ
IR (KBr) ν = 1611 und 1591 cm-1(N=O).
Hochaufgel. MS Ber. m/z =455.05315
Gef. m/z = 455.04924
Elementaranalyse Ber. Für C13H21NOPClW: C 34.24; H 4.62; N 3.06.
Gef.: C 34.66; H 4.65; N 2.99.

D Experimenteller Teil 98
3.2 Hydrolyse der Phosphor komplexe des Wolframs
.2.1 Umsetzung von der Verbindung 11a mit NaOH
Zu einer Suspension von 1 g NaOH in 30 ml THF und 2 ml destilliertes Wasser wurde
ine Lösung von 527 mg (1 mmol) Komplex 11a in 30 ml THF bei 0 °C addiert. Die
eaktionsmischung wurde dann auf Raumtemperatur erwärmt. Der Verlauf der Reaktion
urde mittels DC kontrolliert. Nach sechs Stunden war das Edukt auf der DC-Karte
erschw nden. Das Lösungsmittel wurde dann zu ein viertel des Gesamtvolumen abgedampft
nd mit CH2Cl2 extrahiert. Dann wurde über MgSO4 getrocknet. Das einengen des
L 90 m 2W
m von 95 ukt setzte sich au zwei und 36
zusammen. Anhand des 1H- 35 :
36 von 18 : 82 bestimmt. Die beiden Verbindungen wurden mit der präparativen Schicht-
Chromatographie an Silicagel mit Ether getrennt.
D
35
1
3)
δ: 8.02 (m, 2H, Phenyl); 7.75 (m, 2H, Phenyl), 7.25-
und 6.39 (zwei d, 4H,
C H), 5.70 und 5.24 (d, 10H, Cp).
13C-NMR δ: 174.5 und 171.4 (zwei dd, 2C, 1JC–P = 43 Hz, 2JC–P
z 133.3
130.6, 129.2, 128.9, 128.2,
127.7 (Phenylgruppen); 103.2 und 101.9 (zwei s, Cp).
3
e
R
w
v u
u
ösungsmittels ergab 4 g von einem Feststoff der Zusammensetzung C38H34N2O3P
%. Das Prod s Isomeren 35
NMR Spektrums wurde das Verhältnis der beiden Isomeren
it einer Ausbeute
ie Verbindung 35
H-NMR
(400 MHz, CDCl 7.47 (m, 18H, Phenyl), 6.60
β
(100 MHz, CDCl3,)
= 21 H , Cα); 139.8 und 136.3 Cβ; 141.1, 137.7,
134.9, 133.9, 133.0, 132.4,
IR (KBr)
ν = 1553 cm–1 (zwei NO).
W
WO
NO
NOP
PH
HH
Hβ
α

D Experimenteller Teil 99
Die Verbindung 36
WC
PW
C
PO
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
H
H
C
C
H
H
H
H
H
H
NO
NO
3.4 132.9, 129.4, 129.15, 128.2,
127.5 (Phenylgruppen); 99.8 (s, zwei Cp).
36
1H-NMR
(400 MHz, CDCl3)
δ: 7.30 – 7.62 (m, 20H, Ph); 6.70 und 6.24 [d, 4H,
CβHb)]; 5.29 (s, 10H, Cp).
13C-NMR
(100 MHz, CDCl3)
δ: 176.1 (dd, 2C, 1JC–P = 30.5 Hz, 2JC–P = 30.5 Hz, Cα);
136.6, 134.2 (Cβ); 13
H
ON
P
W
O NO P
HH
HW
IR (KBr): ν = 1613 cm–1 (NO).
Umsetzung von der Verbindung 11c mit
des Wolframs 37
Die Umsetzung erfolgte wie unter 3.2.1 beschrieben, allerdings an der Stelle des
mischung ergab die Verbindung 37, die nach der
U ation aus mit Pentan der
r der Verbind
µ-Oxo-Dimer des Wolframs
Ausbeute: 60%
37
3.2.2 NaOH: Darstellung der µ-Oxo-Dimer
Komplexes 11a wurde 2.02 g (4.82 mmol) der Verbindung 11c und 1.5 g NaOH eingesetzt.
Die Aufarbeitung der Reaktions
mkristallis überschichteten Chloroform Lösung zu 498 mg (60%)
ung 37 führte. ot-orangen Kristalle

D Experimenteller Teil 100
1H-NMR
(400 MHz, CDCl3)
δ: 6.53 (d, 2H, 3J 3
5.91 Hz, Cβ
3JW-H cis =3.94 Hz, C
und 2.11 [m, 8H,
24H PCH(CH3)2].
C-NMR
CDCl3)
δ: 173.1 (dd, 1JP-C = 59.9 Hz, 1JW–C = 18.31 Hz,
25.5
5 [PCH(CH3)2].
3, H3PO4 als
xterner Standard, 1H-
)
IR (KBr)
P-H cis = 12.74 Hz, JW-H trans =
H); 6.15 (d, 2H, 3JP-H trans = 40.36 Hz,
βH); 5.73 (s, 10H, Cp); 2.62
PCH(CH3)2]; 0.93 - 1.25 [m,
13
(100 MHz, Cα), 134.2 (s, Cβ); 99.8 (s, Cp); 25.26 -
[PCH(CH3)2]; 18 - 19.4
δ: 31.34 (s, 31P-NMR
(162 MHz, CDCl
e
Entkoppelt
1JP-W = 209.81 Hz).
ν = 1585 cm (zwei NO).
+ 182
–1
Hocha
; N 3.04.
Gef: C 34.20; H 4.48; N 3.26.
llstruktur Siehe Abschnitt E
ufgel. MS (M , W) Ber. m/z = 856.1635.
Gef. m/z = 856.16339.
Elementaranalyse Ber. für (C26H42N2O3P2W)2 CHCl3:
C 34.60; H 4.65
Krista
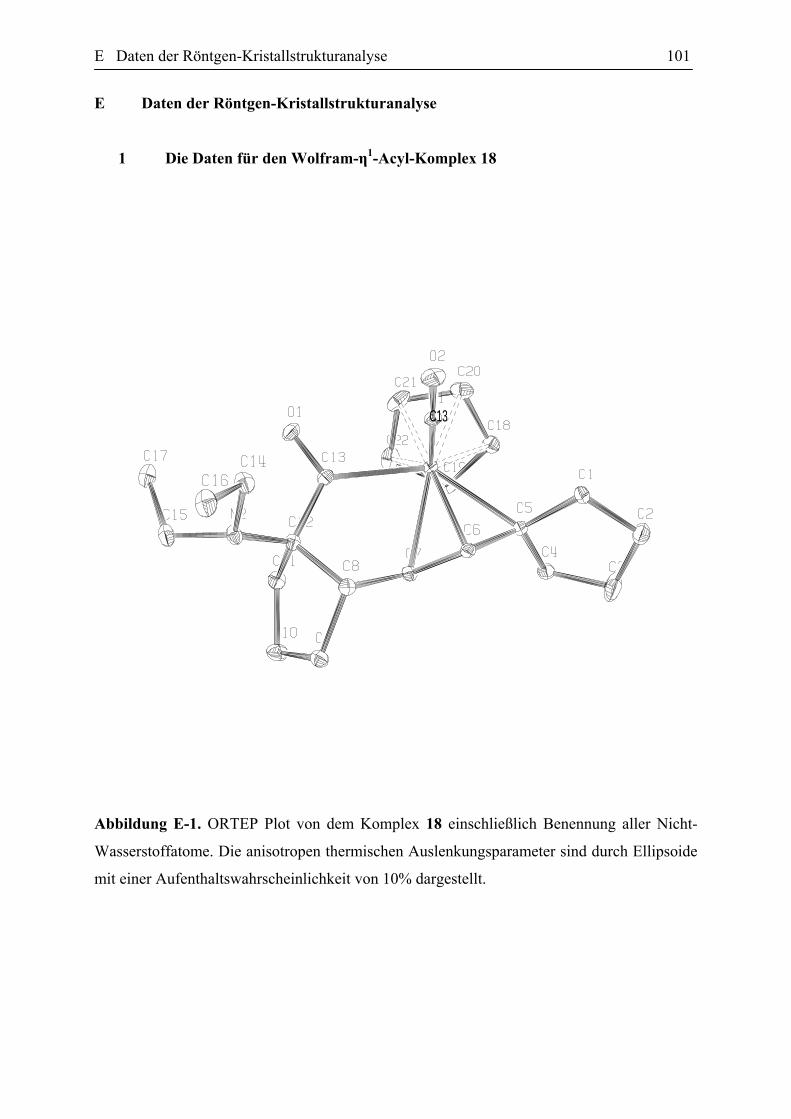
E Daten der Röntgen-Kristallstrukturanalyse 101
E n der Röntgen-Kris
1 Die Daten für den Wolfram-
Abbildung E-1. ORTEP Plot von dem Komplex 18 einschließlich Benennung aller Nicht-
W e. Die anisotrop lenkungsparameter sind durch Ellipsoide
it einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.
Date tallstrukturanalyse
η1-Acyl-Komplex 18
asserstoffatom en thermischen Aus
C13
m

E Daten der Röntgen-Kristallstrukturanalyse 102
Tabelle E-1.1. Kristallografische Daten der Verbindung 18.
Summenformel C H N O W
gruppe P21/c (No.14)
Zellparameter
a = 11.330b(2) Å,
b = 13.6604(10) Å,
c =13.711(2)Å,
β = 92.02(10)°.
Volumen 2120.8(5) Å3
Z Z = 1
Dichte (berechnet) 1.5913(4)g/cm3
Diffraktometer Image Plate Diffractometer System (STOE)
Strahlung MoKα (λ = 0.71069 Å)
Monochromator
Temperatur
Grafit
293(2) K
Messbereich
2.73 ≤ θ ≤ 28.05°
-14 ≤ h ≤ 14, -17 ≤ k ≤ 17,
-16 ≤I ≤ 18
Gemessene Reflexe
Unabhängige Reflexe
int
18720
4961
0.0526
arameter
R2
1
1 [F0 > 4σ(F0)]
190
0.0652
0.1347
0.0652
trukturberechnung
nd Verfeinerung
itionierung von
Wasserstoffatomen mit der Petterson Synthese
(Programm SHELEXS-86);[81] Wasserstoff
Positionen mit dem Ritter Model.
22 32 2 2
Formelgewicht 540.358
Kristallsystem Monoklin
Raum
R
P
w
R
R
Max und min in Δσ 5.007 und -2.505 e.Å-3
Korrekturen Lorentz und Polarisationkooeffizienten
Parameter zur Pos
S
u

E Daten der Röntgen-Kristallstrukturanalyse 103
Tabelle E-1.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 18.
____________ _ ____________________________________ z U(eq)
_____________ _________________________________________ 433(1) 9088(1) 2053(1) 37(1) 245(5) 1998(4) 43(1)
N(2) -3530(5) 9280(5) 2370(5) 49(2) O(1) -1829(5) 866(4) 68(2)
129(7) 1879(5) 71(2) C(1) -563(3) 8618(3) 3440(3) 40(1) C(2) 313(3) 9324(3) 3701(3) 41(1)
1516(3) 9129(3) 3496(3) 43(2) ) 2413(4) 9975(3) 3686(3) 57(2)
3384(4) 9530(3) 4356(3) 74(3) 2991(4)2117(4) 8178(3) 3860(3) 54(2)
-1483(4) 9005(3) 1682(3) 46(2) -2413(6) 8721(6) 2446(5) 42(1) -1836(2) 8866(2) 3483(2) 45(2)
C(11) -2553(2) 8162(2) 4128(2) 60(2) -2979(2) 7304(2) 3438(2) 69(2)
C(13) -2592(2) 7607(2) 2408(2) 54(2) C(14) -3352(2) 10327(2) 2198(2) 63(2)
-4560(2) 8863(2) 1838(2) 66(2) -4263(2) 2723(2) 78(3)
7) -4511(2) 778(2) 708(2) 85(3) C(18) 2156(2) 8316(2) 1501(2) 58(2)
1323(2) 7563(2) 1595(2) 56(2) ) 1738(2) 962(2) 780(2) 76(3)
1) 632(2) 606(2) 439(2) 80(4) 369(2) 767(2) 938(2) 73(3)
____________ __________________________
______________ x
_____ _______y
_____________ ________W(1) N(1) 10364(5)
9195(5) O(2) 11240(5)
C(3) C(4C(5) C(6) 8556(3) 4609(3) 133(7) C(7) C(8) C(9) C(10)
C(12)
C(15) C(16) 10949(2) C(1 8
C(19) C(20 8C(2 8C(22) 7____________________ ______________________

E Daten der Röntgen-Kristallstrukturanalyse 104
Tabelle E-1.3. Bindungslängen (Å) für 18.
W(1)-N(1) 1.758(6) C(3)-C(7) 1.541(6) W(1)-C(8) 2.215(4) C(3)-C(4) 1.555(6) W(1)-C(3) 2.293(4) C(4)-C(5) 15.341 W(1)-C(2) 2.292(4) C(5)-C(6) 14.498 W(1)-C(21) 2.326(3) C(6)-C(7) 14.932 W(1)-C(20) 2.332(3) C(8)-C(9) 1.562(8) W(1)-C(1) 2.337(4) C(9)-C(13) 1.536(8) W(1)-C(22) 2.365(3) C(9)-C(10) 1.556(7) W(1)-C(18) 2.367(3) C(10)-C(11) 15.551 W(1)-C(19) 2.407(3) C(11)-C(12) 15.708 N(1)-O(2) 1.214(10) C(12)-C(13) 15.495 N(2)-C(14) 1.465(7) C(14)-C(16) 15.360 N(2)-C(15) 1.470(7) C(15)-C(17) 15.562 N(2)-C(9) 1.478(9) C(18)-C(20) 13.960 O(1)-C(8) 1.200(7) C(18)-C(19) C(1)-C(2) 14.210 C(19)-C(22) 14.112
14.049
indungswin
119.9(5)
70.41(11)
71.89(14) 120.6(2)
(1)-C(20) 35.2 C(6)-C(5)-C(4) 106.7 (1)-C(1) 104.2(2) C(5)-C(6)-C(7) 110.8 (1)-C(1) 71.05(16) C(6)-C(7)-C(3) 101.9(2) (1)-C(1) 64.07(10) O(1)-C(8)-C(9) 118.2(5) (1)-C(1) 35.74(6) O(1)-C(8)-W(1) 119.2(4)
(1)-C(1) 139.68(13) C(9)-C(8)-W(1) 122.5(3) (1)-C(1) 157.82(13) N(2)-C(9)-C(13) 113.4(5)
(1)-C(22) 136.79(19) N(2)-C(9)-C(10) 109.3(5) (1)-C(22) 78.76(15) C(13)-C(9)-C(10) 102.0(5)
C(1)-C(10) 1.484(5) C(20)-C(21) 14.081 C(2)-C(3) 14.270 C(21)-C(22) 13.732 W(1)-N(1) 1.758(6) C(3)-C(7) 1.541(6)
Tabelle E-1.4. B keln (˚) für 18.
N(1)-W(1)-C(8) 85.7(2) C(15)-N(2)-C(9) N(1)-W(1)-C(3) 94.2(2) C(2)-C(1)-C(10) 120.39(19) C(8)-W(1)-C(3) 133.62(17) C(2)-C(1)-W(1) N(1)-W(1)-C(2) 83.7(2) C(10)-C(1)-W(1) 117.8(2) C(8)-W(1)-C(2) 98.14(18) C(1)-C(2)-C(3) 119.2 C(3)-W(1)-C(2) 36.27(6) C(1)-C(2)-W(1) 73.85(12) N(1)-W(1)-C(21) 104.8(2) C(3)-C(2)-W(1) C(8)-W(1)-C(21) 83.93(16) C(2)-C(3)-C(7) C(3)-W(1)-C(21) 139.56(12) C(2)-C(3)-C(4) 116.8(2) C(2)-W(1)-C(21) 171.36(13) C(7)-C(3)-C(4) 107.1(3) N(1)-W(1)-C(20) 96.9(2) C(2)-C(3)-W(1) 71.84(12) C(8)-W(1)-C(20) 117.80(14) C(7)-C(3)-W(1) 118.4(3) C(3)-W(1)-C(20) 108.28(12) C(4)-C(3)-W(1) 119.3(3) C(2)-W(1)-C(20) 144.03(12) C(5)-C(4)-C(3) 104.8(2) C(21)-WN(1)-WC(8)-WC(3)-WC(2)-WC(21)-WC(20)-WN(1)-WC(8)-W
E Daten der Röntgen-Kristallstrukturanalyse 105
C(3)-W(1)-C(22) 125.46(13) C(2)-W(1)-C(22) 138.03(13)
N(2)-C(9)-C(8) 114.9(6) C(13)-C(9)-C(8) 108.4(5)
-C(22) 108.1(4) -C(22) 108.2(3) C(22) ) 114.1(2) C(18) 103.3(3)
(18) ) )-C(12) 105.9 (18) )-C(11) 105.0 (18) ) 106.0(3) C(18) 111.5(3) C(18) 117.8(3) (18) ) (19) 108.4 C(18) (1) 71.38(8) (19) (1) 74.47(7) (19) ) (22) 107.4 (19) (1) 71.31(7)
-C(19) ) (1) 71.16(6) )-C(19) 107.0 -C(19) (1) 74.06(7) (19) ) (1) 72.17(8)
)-C(19) 109.2 -C(19) )-W(1) 74.52(8)
(1) 174.6(5) C(20)-C(21)-W(1) 72.64(10) C(21)-C(22)-C(19) 107.9
C(14)-N(2)-C(9) 113.2(5) C(21)-C(22)-W(1) 71.44(7) C(19)-C(22)-W(1) 74.45(7)
C(21)-W(1) 34.0 C(10)-C(9)-C(8) C(20)-W(1)C(1)-W(1)-
57.73(6) 108.18(13
C(1)-C(10)-C(9) C(1)-C(10)-C(11)
N(1)-W(1)- 121.9(2) C(9)-C(10)-C(11) C(8)-W(1)-C 135.80(14 C(10)-C(11C(3)-W(1)-C
C82.19(13) C(13)-C(12
C(2)-W(1)-C(21)-W(1)
117.15(1457.42(9)
C(9)-C(13)-C(12) N(2)-C(14)-C(16) -
C(20)-W(1)- 34.6 N(2)-C(15)-C(17) C(1)-W(1)-C 124.42(14 C(20)-C(18)-CC(22)-W(1)- 57.33(7) C(20)-C(18)-WN(1)-W(1)-CC(8)-W(1)-C
154.0(2) 108.20(13
C(19)-C(18)-WC(18)-C(19)-C
C(3)-W(1)-C 91.69(13) C(18)-C(19)-WC(2)-W(1) 114.76(13 C(22)-C(19)-WC(21)-W(1 56.78(7) C(18)-C(20)-C(21) C(20)-W(1)C(1)-W(1)-C
57.25(6) 101.13(13
C(18)-C(20)-WC(21)-C(20)-W
C(22)-W(1 34.4 C(22)-C(21)-C(20) C(18)-W(1) 34.2 C(22)-C(21O(2)-N(1)-WC(14)-N(2)-C(15) 114.2(5)

E Daten der Röntgen-Kristallstrukturanalyse 106
2 Die Daten für d -η2-Alken
Abbildung E-2. ORTEP Plot von dem Komplex 21 einschließlich Benennung aller Nicht-
Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide
mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.
en Wolfram -Komplex 21

E Daten der Röntgen-Kristallstrukturanalyse 107
Tab
Summenformel C17H24N2O2W Formelgewicht 472.24 Kristallsystem Monoklin Raumgruppe P21/c (No.14)
Zellparam
VolumZ
Diffraktom
Strahlung
Temp
Gemessene Reflexe Unabhängige Reflexe Rint
15178 4007 0.0286
Max und min in Δσ 1,259 und -0.522 e.Å-3
Korrekturen Lorentz und Polarisationkooeffizienten
Strukturberechnung und Verfeinerung
Parameter zur Positionierung von Wasserstoffatomen mit der Petterson Synthese (Programm SHELEXS-86); Wasserstoff Positionen mit dem Ritter Model.[81]
elle E-2.1. Kristallografische Daten der Verbindung 21.
eter
a = 16.0910(18) Å, b = 6.7543(5) Å, c =16.8063(18) Å, β = 104.422(13)°.
en 1769.03Å3
Z = 1 Dichte (berechnet) 1.769 g/cm3
eter Image Plate Diffractometer System (STOE)
MoKα (λ = 0.71069 Å)
Monochromator eratur
Grafit 293(2) K
Messbereich 3.40 ≤ θ ≤ 28.04° -21 ≤ h ≤ 21, -8 ≤ k ≤ 8, -22 ≤I ≤ 22
parameter wR2R1
R1 [F0 > 4σ(F0)]
173 0.0831 0.0477 0.0312

E Daten der Röntgen-Kristallstrukturanalyse 108
Tabelle E-2.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 21.
__________________ _______________________________
x z U(eq) ____________________________________________________________________ W 1443(1) 3703(1) 4524(1) 49(1) N 1608(3) 4937(3) 62(1) O(1) 1628(5) 5153(5) 102(2) C 1409(5) 5605(4) 64(2) O 1356(5) 6210(4) 107(2) C(11) 1004(8) 3118(5) 118(5) C(12) 925(8) (20) 3478(7) 120(4) C(13) 354(7) 3910(7) 110(4) C(14) 21(5) 3867(6) 113(5) C(15) 371(9) 3371(8) 136(5) C 2734(5) C 2804(4) 8(5) 63(2) C(21) 3321(4) 178(11) 4418(4) 59(2) C(22) 3531(4) (13) 3809(5) 70(2) C(23) 4106(6) 4102(7) 96(3) C 4299(5) (7) 97(3) C(25) 3735(5) 6) 5222(5) 88(2) N(2) 3235(6) 5517(13) 2954(5) 102(3) C 3046(13) 7050(30) 2388(13) 94(5) C 2298(11) 8060(30) 2434(11) 84(4) C(41) 3507 3631 2706 195(15) C(42) 4257 3685 2543 136(9) _ ____________________ ___________________________________________ T ngen (Å) für 21
1.786(7) 1.786(7) 1.972(6) 972(6)
W-C(1) 2.270(7) W-C(1) 2.270(7) W-C(2) 2.286(6)
2.300(8) W-C(11) 2.329(8) 2.329(8) W-C(13) 2.355(9) W-C(13) 2.355(9)
-C(12) 2.355(9) W-C(12) 2.355(9) N-O(1) 1.207(8) N-O(1) 1.207(8)
1.126(8) C-O(2) 1.126(8) 1.327(18) C(11)-C(12) 1.327(18)
___________________ ____________
y
6147(10)7855(11) 2624(12)
(2) 1988(12) 3080(30) 13601450(20) 3270(30) 4400(20)
(1) 2224(14) 5027(6) 79(2) (2) 3371(11) 435
560907856(17)
(24) 7755(17) 49956131(1
(31) (32)
___________ _____
abelle E-2.3. Bindungslä .
W-N W-N W-C W-C 1.
W-C(2) 2.286(6) W-C(14) 2.298(8) W-C(15) 2.300(8) W-C(11)
W-C(14) 2.298(8) W-C(15)
W
C-O(2) C(11)-C(12)
E Daten der Röntgen-Kristallstrukturanalyse 109
C(11)-C(15) 1.49(2) C(11)-C(15) 1.49(2) C(12)-C(13) 1.305(16) C(12)-C(13) 1.305(16) Tabelle E-2.4. Bindungswinkeln (˚) für 21.
N-W-C 91.1(3) O(1)-N-W 172.2(6) N-W-C(1) 103.2(3) O(2)-C-W 177.2(7) C-W-C(1) 74.1(3) C(12)-C(11)-C(15) 104.1(10) N-W-C(2) 94.8(2) C(12)-C(11)-W 74.6(5) C-W-C(2) 108.9(3) C(15)-C(11)-W 70.1(5) C(1)-W-C(2) 35.6(3) C(13)-C(12)-C(11) 112.0(14) N-W-C(14) 109.8(5) C(13)-C(12)-W 73.9(6) C-W-C(14) 98.2(4) C(11)-C(12)-W 72.4(6) C(1)-W-C(14) 146.3(6) C(12)-C(13)-C(14) 109.9(13) C(2)-W-C(14) 142.9(3) C(12)-C(13)-W 73.9(6) N-W-C(15) 98.3(4) C(14)-C(13)-W 71.0(6) C-W-C(15) 131.7(5) C(13)-C(14)-C(15) 109.4(12) C(1)-W-C(15) 146.0(6) C(13)-C(14)-W 75.7(5) C(2)-W-C(15) 117.2(5) C(15)-C(14)-W 73.0(5) C(14)-W-C(15) 34.3(5) C(14)-C(15)-C(11) 104.6(13) N-W-C(11) 122.7(5) C(14)-C(15)-W 72.8(5) C-W-C(11) 142.9(4) C(11)-C(15)-W 72.2(5) C(1)-W-C(11) 108.5(5) C(2)-C(1)-W 72.8(4) C(2)-W-C(11) 85.2(4) C(1)-C(2)-C(21) 124.5(7) C(14)-W-C(11) 58.4(4) C(1)-C(2)-W 71.6(4) C(15)-W-C 37.7(5) C(21)-C(2)-W 117.1(4) (11)
-C(13) 56.2(5) N(2)-C(22)-C(23) 123.8(7) -C(13) 55.5(4) C(24)-C(23)-C(22) 104.8(7)
C(23)-C(24)-C(25) 107.6(8) C(21)-C(25)-C(24) 104.0(7)
-C(12) 96.0(4) (2)-C(41) (12) )-C(22)
12) 55.3(5) )-C(22) (12) 57.2(5) -C(32) (12) 32.9(4) )-N(2) (12) 32.2(4)
N-W-C(13) 142.2(4) C(22)-C(21)-C(2) 126.2(7) C-W-C(13) 88.7(4) C(22)-C(21)-C(25) 111.1(7) C(1)-W-C(13) 113.0(5) C(2)-C(21)-C(25) 122.5(7) C(2)-W-C(13) 120.9(4) C(21)-C(22)-N(2) 124.3(8) C(14)-W-C(13) 33.3(5) C(21)-C(22)-C(23) 111.8(7) C(15)-WC(11)-WN-W-C(12) 154.3(4) C-W-C(12) 110.7(5) C(1)-W C(31)-N 119.3(10) C(2)-W-C 90.9(4) C(31)-N(2 116.3(12) C(14)-W-C( C(41)-N(2 118.3(7) C(15)-W-C N(2)-C(31) 111.6(16) C(11)-W-C C(42)-C(41 113.6(4) C(13)-W-C

E Daten der Röntgen-Kristallstrukturanalyse 110
3 Die Daten für d ylidenkomplex des Wolframs 27
ORTEP Plot von dem Komp h B ller Nicht-
e. Die anisotropen thermischen eter sind durch Ellipsoide
altswa ichkeit von 20%
en η1-Vin
Abbildung E-3. lex 27 einschließlic enennung a
Wasserstoffatom Auslenkungsparam
mit einer Aufenth hrscheinl dargestellt.

E Daten der Röntgen-Kristallstrukturanalyse 111
Tab
ummenformel C19H23NO3W
ormelgewicht 497.25
eter
a = 24.535(4) Å, b = 6.7289(11) Å, c = 24.603(7) Å, β = 119.966(15)°.
Z
R
arameter R2
1
1 [F0 > 4σ(F0)]
191 0.1818 0.3773 0.1623
trukturberechnung und g
Parameter zur Positionierung von Wasserstoffatomen mit der Petterson Synthese (Programm SHELEXS-86); Wasserstoff Positionen mit dem Ritter Model.[81]
elle E-3.1. Kristallografische Daten der Verbindung 27.
S
F
Zellparam
Volumen 3518(13)Å3
Z = 2
Dichte (berechnet) 1.877 g/cm3
Diffraktometer Image Plate Diffractometer System (STOE)
Strahlung MoKα (λ = 0.71069 Å)
Monochromator Temperatur
Grafit 293(2) K
Messbereich 2.53 ≤ θ ≤ 24.05° -28 ≤ h ≤ 27, -7 ≤ k ≤ 7, -28 ≤I ≤ 28
Gemessene Reflexe Unabhängige Reflexe
int
20639 5453 0.1483
pwRR
Max und min in Δσ 21.348 und -3.713 e.Å-3
Korrekturen Lorentz und Polarisationkooeffizienten
SVerfeinerun
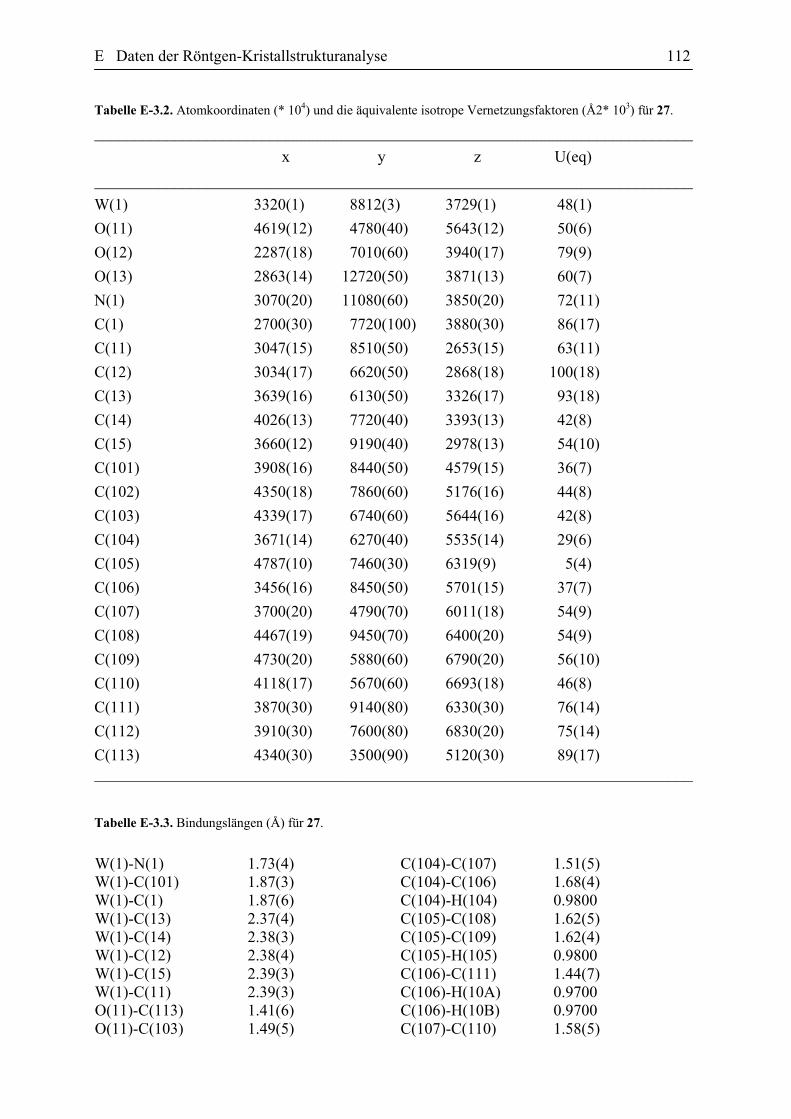
E Daten der Röntgen-Kristallstrukturanalyse 112
Tabelle E-3.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 27.
__________________________________________________________________________ x z U(eq)
_______________ ________________________________________ W(1) 3320(1) 8812(3) 3729(1) 48(1) O(11) 4619(12) 4780(40) 5643(12) 50(6)
2287(18) 7010(60) 3940(17) 79(9) O(13) 2863(14) 3871(13) 60(7) N(1) 3070(20) 11080(60) 3850(20) 72(11)
2700(30) 0) 3880(30) 86(17) 1) 3047(15) 8510(50) 2653(15) 63(11)
3034(17) 6620(50) 2868(18) 100(18) 3639(16) 6130(50) 3326(17) 93(18) 4026(13) 3660(12) 9190(40) 2978(13) 54(10) 3908(16) 8440(50) 4579(15) 36(7) 4350(18) 7860(60) 5176(16) 44(8)
C(103) 4339(17) 6740(60) 5644(16) 42(8) C(104) 3671(14) 6270(40) 5535(14) 29(6)
4787(10) 7460(30) 6319(9) 5(4) C(106) 3456(16) 8450(50) 5701(15) 37(7) C(107) 3700(20) 4790(70) 6011(18) 54(9)
4467(19) 9450(70) 6400(20) 54(9) 4730(20) 5880(60) 6790(20) 56(10)
0) 4118(17) 5670(60) 6693(18) 46(8) C(111) 3870(30) 9140(80) 6330(30) 76(14)
3910(30) 7600(80) 6830(20) 75(14) 4340(30) 3500(90) 5120(30) 89(17)
________________________ ________________________________________
Tabelle E-3.3. Bindungslängen (Å) für 27.
1.73(4) (5) 1.87(3) (4)
W(1)-C(1) 1.87(6) C(104)-H(104) 0.9800 W(1)-C(13) 2.37(4)
2.38(4) W(1)-C(15) 2.39(3) W(1)-C(11) 2.39(3) C(106)-H(10A) 0.9700
(11)-C(113) 1.41(6) C(106)-H(10B) 0.9700 (11)-C(103) 1.49(5) C(107)-C(110) 1.58(5)
_ y _____________ _______
O(12) 12720(50)
C(1) 7720(10C(1C(12) C(13) C(14) 7720(40) 3393(13) 42(8) C(15) C(101) C(102)
C(105)
C(108) C(109) C(11
C(112) 3) C(11
____ _______
W(1)-N(1) W(1)-C(101)
C(104)-C(107) 1.51C(104)-C(106) 1.68
C(105)-C(108) 1.62(5) C(105)-C(109) 1.62(4) C(105)-H(105) 0.9800 C(106)-C(111) 1.44(7)
W(1)-C(14) 2.38(3) W(1)-C(12)
OO
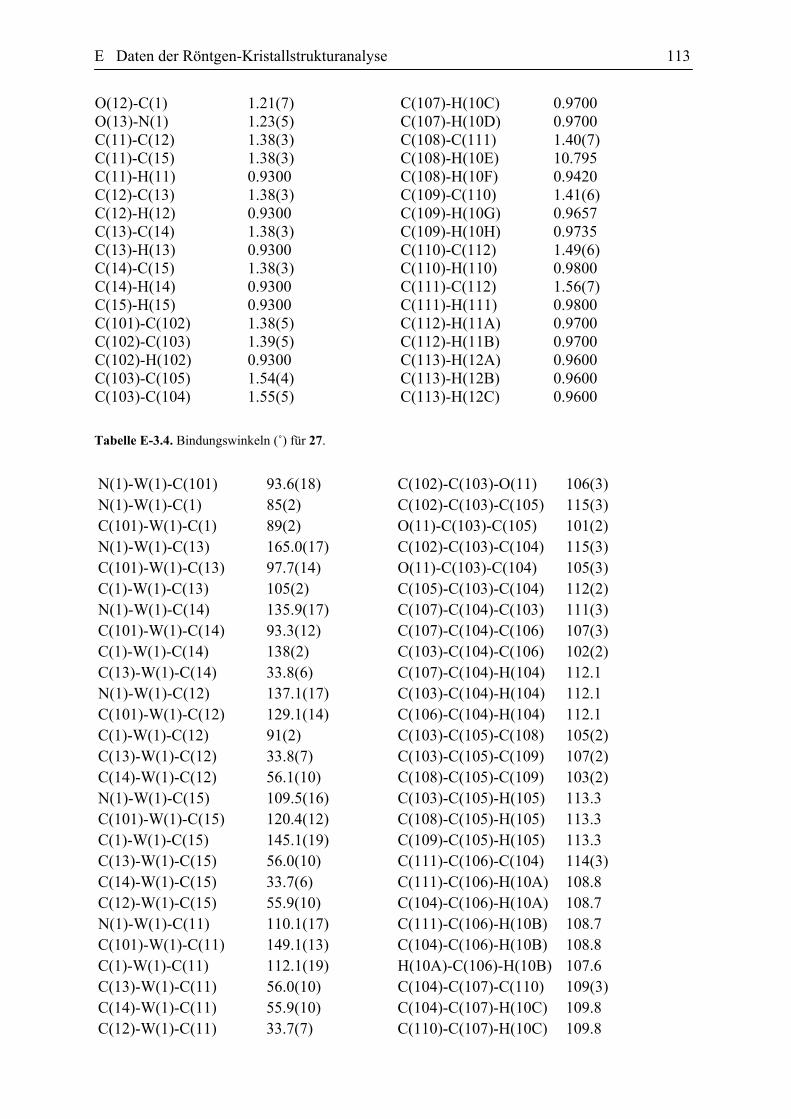
E Daten der Röntgen-Kristallstrukturanalyse 113
O(12)-C(1) 1.21(7) C(107)-H(10C) 0.9700 O(13)-N(1) 1.23(5) C(107)-H(10D) 0.9700 C(11)-C(12) 1.38(3) C(108)-C(111) 1.40(7) C(11)-C(15) 1.38(3) C(108)-H(10E) 10.795 C(11)-H(11) 0.9300 C(108)-H(10F) 0.9420 C(12)-C(13) .38(3) C(109)-C(110) 1.41(6) C(12)-H(12) .9300 C(109)-H(10G) 0.9657
1 0
1 0
3) 10.9300
5) 14) 1
Bindungswink
(1)-C(15) 109.5(16) C(103)-C(105)-H(105) 113.3 C(108)-C(105)-H(105) 113.3
(1)-C(15) 145.1(19) C(109)-C(105)-H(105) 113.3 C(15) 0) (104)
(15) ) (10A) -C(15) 0) (10A)
(11) (17) (10B) -C(11) (13) (10B) (11) (19)
(11) 0) 110) 11) 0) (10C) 11) ) (10C)
C(13)-C(14) .38(3) C(109)-H(10H) 0.9735 C(13)-H(13) .9300 C(110)-C(112) 1.49(6) C(14)-C(15) 1.38(3) C(110)-H(110) 0.9800 C(14)-H(14) 0.9300 C(111)-C(112) 1.56(7) C(15)-H(15) 0.9300 C(111)-H(111) 0.9800 C(101)-C(102) 1.38(5) C(112)-H(11A) 0.9700 C(102)-C(10 .39(5) C(112)-H(11B) 0.9700 C(102)-H(102) C(113)-H(12A) 0.9600 C(103)-C(10 .54(4) C(113)-H(12B) 0.9600 C(103)-C(10 .55(5) C(113)-H(12C) 0.9600 Tabelle E-3.4. eln (˚) für 27.
N(1)-W(1)-C(101) 93.6(18) C(102)-C(103)-O(11) 106(3) N(1)-W(1)-C(1) 85(2) C(102)-C(103)-C(105) 115(3) C(101)-W(1)-C(1) 89(2) O(11)-C(103)-C(105) 101(2) N(1)-W(1)-C(13) 165.0(17) C(102)-C(103)-C(104) 115(3) C(101)-W(1)-C(13) 97.7(14) O(11)-C(103)-C(104) 105(3) C(1)-W(1)-C(13) 105(2) C(105)-C(103)-C(104) 112(2) N(1)-W(1)-C(14) 135.9(17) C(107)-C(104)-C(103) 111(3) C(101)-W(1)-C(14) 93.3(12) C(107)-C(104)-C(106) 107(3) C(1)-W(1)-C(14) 138(2) C(103)-C(104)-C(106) 102(2) C(13)-W(1)-C(14) 33.8(6) C(107)-C(104)-H(104) 112.1 N(1)-W(1)-C(12) 137.1(17) C(103)-C(104)-H(104) 112.1 C(101)-W(1)-C(12) 129.1(14) C(106)-C(104)-H(104) 112.1 C(1)-W(1)-C(12) 91(2) C(103)-C(105)-C(108) 105(2) C(13)-W(1)-C(12) 33.8(7) C(103)-C(105)-C(109) 107(2) C(14)-W(1)-C(12) 56.1(10) C(108)-C(105)-C(109) 103(2) N(1)-WC(101)-W(1)-C(15) 120.4(12)C(1)-WC(13)-W(1)- 56.0(1 C(111)-C(106)-C 114(3)C(14)-W(1)-C 33.7(6 C(111)-C(106)-H 108.8 C(12)-W(1) 55.9(1 C(104)-C(106)-H 108.7 N(1)-W(1)-C 110.1 C(111)-C(106)-H 108.7 C(101)-W(1) 149.1 C(104)-C(106)-H 108.8 C(1)-W(1)-C 112.1 H(10A)-C(106)-H(10B) 107.6 C(13)-W(1)-C 56.0(1 C(104)-C(107)-C( 109(3)C(14)-W(1)-C( 55.9(1 C(104)-C(107)-H 109.8 C(12)-W(1)-C( 33.7(7 C(110)-C(107)-H 109.8
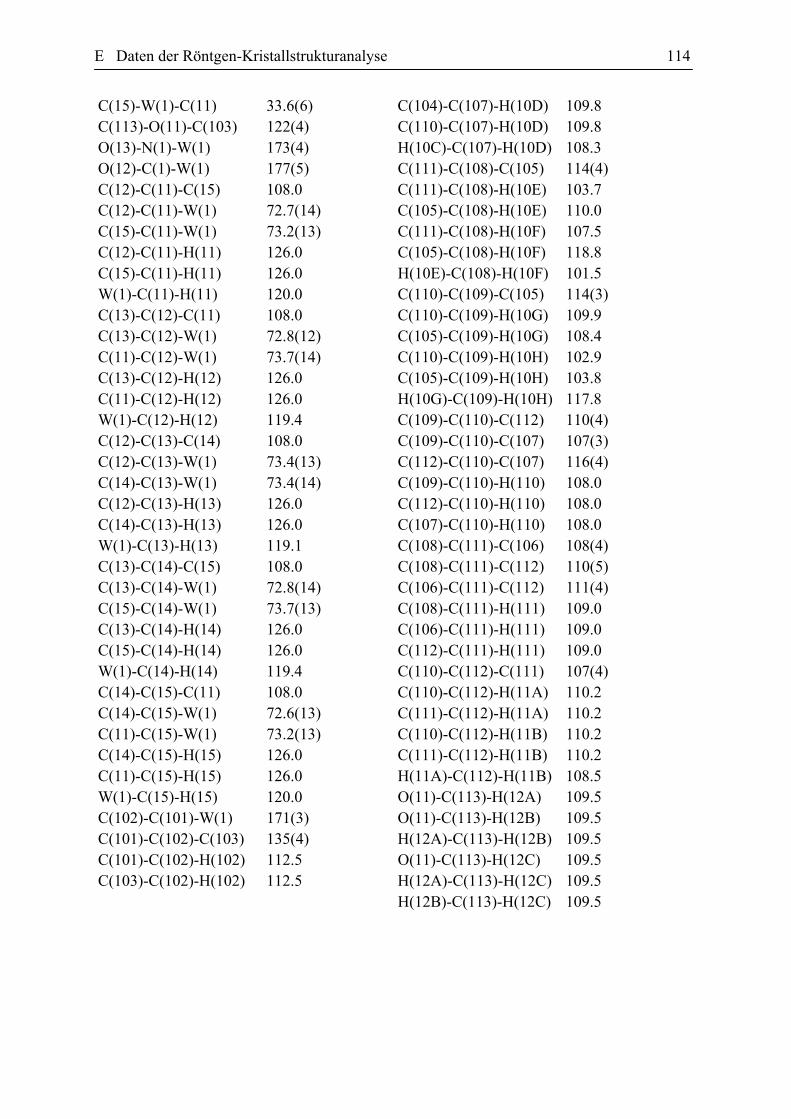
E Daten der Röntgen-Kristallstrukturanalyse 114
C(15)-W(1)-C(11) ) (10D) -C(103) ) C(110)-C(107)-H(10D) (1) 173(4) (1) 177(5) (105) (15) (10E) (1) 72.7(14) 10E) (1) 73.2(13) 10F) (11) (10F) (11) (10F) (11) (105)
1) 10G) (10G) 10H)
2) (10H) 2) (10H)
(1)-C(12)-H(12) 119.4 C(109)-C(110)-C(112) 110(4) C(109)-C(110)-C(107) 107(3)
(1) 73.4(13) C(112)-C(110)-C(107) 116(4)
3)
)
)
3)
03) 02) 02)
33.6(6 C(104)-C(107)-H 109.8 C(113)-O(11) 122(4 109.8 O(13)-N(1)-W H(10C)-C(107)-H(10D) 108.3 O(12)-C(1)-W C(111)-C(108)-C 114(4)C(12)-C(11)-C 108.0 C(111)-C(108)-H 103.7 C(12)-C(11)-W C(105)-C(108)-H( 110.0 C(15)-C(11)-W C(111)-C(108)-H( 107.5 C(12)-C(11)-H 126.0 C(105)-C(108)-H 118.8 C(15)-C(11)-H 126.0 H(10E)-C(108)-H 101.5 W(1)-C(11)-H 120.0 C(110)-C(109)-C 114(3)C(13)-C(12)-C(1 108.0 C(110)-C(109)-H( 109.9 C(13)-C(12)-W(1) 72.8(12) C(105)-C(109)-H 108.4 C(11)-C(12)-W(1) 73.7(14) C(110)-C(109)-H( 102.9 C(13)-C(12)-H(1 126.0 C(105)-C(109)-H 103.8 C(11)-C(12)-H(1 126.0 H(10G)-C(109)-H 117.8 WC(12)-C(13)-C(14) 108.0 C(12)-C(13)-WC(14)-C(13)-W(1) 73.4(14) C(109)-C(110)-H(110) 108.0 C(12)-C(13)-H(1 126.0 C(112)-C(110)-H(110) 108.0 C(14)-C(13)-H(13) 126.0 C(107)-C(110)-H(110) 108.0 W(1)-C(13)-H(13) 119.1 C(108)-C(111)-C(106) 108(4) C(13)-C(14)-C(15) 108.0 C(108)-C(111)-C(112) 110(5) C(13)-C(14)-W(1) 72.8(14 C(106)-C(111)-C(112) 111(4) C(15)-C(14)-W(1) 73.7(13) C(108)-C(111)-H(111) 109.0 C(13)-C(14)-H(14) 126.0 C(106)-C(111)-H(111) 109.0 C(15)-C(14)-H(14 126.0 C(112)-C(111)-H(111) 109.0 W(1)-C(14)-H(14) 119.4 C(110)-C(112)-C(111) 107(4)C(14)-C(15)-C(11) 108.0 C(110)-C(112)-H(11A) 110.2 C(14)-C(15)-W(1) 72.6(13) C(111)-C(112)-H(11A) 110.2 C(11)-C(15)-W(1) 73.2(1 C(110)-C(112)-H(11B) 110.2 C(14)-C(15)-H(15) 126.0 C(111)-C(112)-H(11B) 110.2 C(11)-C(15)-H(15) 126.0 H(11A)-C(112)-H(11B) 108.5 W(1)-C(15)-H(15) 120.0 O(11)-C(113)-H(12A) 109.5 C(102)-C(101)-W(1) 171(3) O(11)-C(113)-H(12B) 109.5 C(101)-C(102)-C(1 135(4) H(12A)-C(113)-H(12B) 109.5 C(101)-C(102)-H(1 112.5 O(11)-C(113)-H(12C) 109.5 C(103)-C(102)-H(1 112.5 H(12A)-C(113)-H(12C) 109.5 H(12B)-C(113)-H(12C) 109.5

E Daten der Röntgen-Kristallstrukturanalyse 115
4 Die Daten für den -η2-Allen-
P Plot des Komplex en aller Nicht-
ni n thermische s ch Ellipsoide
it einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.
Wolfram Komplex 31
Abbildung E-4. ORTE es 31 einschließlich B ennung
Wasserstoffatome. Die a sotrope n Auslenkungsparameter ind dur
m

E Daten der Röntgen-Kristallstrukturanalyse 116
abelle E-4.1. Kristallografische Daten der Verbindung 31.
ummenformel C18H9NO2W
Formelgewicht 347.02 Kristallsystem Monoklin Raumgruppe P21/c (No.14)
Zellparameter
a = 7.3511(7) Å, b = 6.1294(6) Å, c = 21.249(3)Å, β = 97.683(13)°.
Volumen 948.85(17) Å3
Z Z = 1 Dichte (berechnet) 2.,429 g/cm3
Diffraktometer Image Plate Diffractometer System (STOE) Strahlung MoKα (λ = 0.71069 Å)
Monochromator Tempertatur
Grafit 293(2) K
Messbereich 4.29 ≤ θ ≤ 25.90° -8 ≤ h ≤ 8, -7 ≤ k ≤ 7, -25 ≤I ≤ 26
Gemessene Reflexe Unabhängige Reflexe Rint
6223 1712 0.0210
arameter 2
1
1 [F0 > 4σ(F0)]
119 0.1234 0.0385 0.0343
ax und min in Δσ 1.483 und -0.925 e.Å-3
LEXS-86); Wasserstoff Positionen mit dem Ritter Model.[81]
T S
pwRRR
MKorekturen Lorentz und Polarisationkooeffizienten
Srukturberechnung und Verfeinerung
Parameter zur Positionierung vonw mit derPetterson Synthese (Program SHE

E Daten der Röntgen-Kristallstrukturanalyse 117
Tabelle E-4.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 31.
_______________________________
x z U(eq)
__________________ ______________________________________ 9144(1) ) 1181(1) 37(1)
6800(20) 1068(7) 67(4)
C(12) 7560(20) -880(30) 517(8) 65(4)
C(13) 7390(20) 211(7) 76(6)
6420(40) 538(13) 110(10)
C(15) 5880(20) 1087(9) 77(5)
1130(20) 1456(7) 56(3)
0640(20) 2023(7) 63(4)
0870(20) 2645(7) 64(4)
11924(16) ) 661(5) 71(3)
10800(14)
9050(20) (7) 56(3)
8960(20) (20) 2344(6) 102(5) _____________________ __________________________________
Tabelle E-4.3. Bindungslängen (Å) für 31
1.767(10) C(11)-C(12) 1.36(2) 2.027(14) C(11)-C(15) 1.44(3) 2.185(14) C(12)-C(13) 1.30(3)
W(1)-C(1) 2.188(14) C(13)-C(14) 1.36(4) ) 2.30(2) C(14)-C(15) 1.44(3)
)-C(11) 2.310(13) C(1)-C(2) 1.46(2) (1)-C(13) 2.332(14) C(2)-C(3) 1.31(2)
2.345(14) O(1)-N(1) 1.234(13) 2.400(16) 1.096(18)
_________________________________________________ y
________________ ________
W(1) 1749(1
C(11) -790(30)
960(40)
C(14) 2340(40)
1270(40)
C(1) 1 -850(20)
210(20) C(2) 1
C(3) 1
O(1)
190(30)
4571(18
N(1) 3441(15) 897(5) 39(2)
C 3710(20) 1945
O(2) 4820_____________ ____________
.
W(1)-N(1) W(1)-C W(1)-C(2)
W(1)-C(14W(1WW(1)-C(12) W(1)-C(15)
C-O(2)
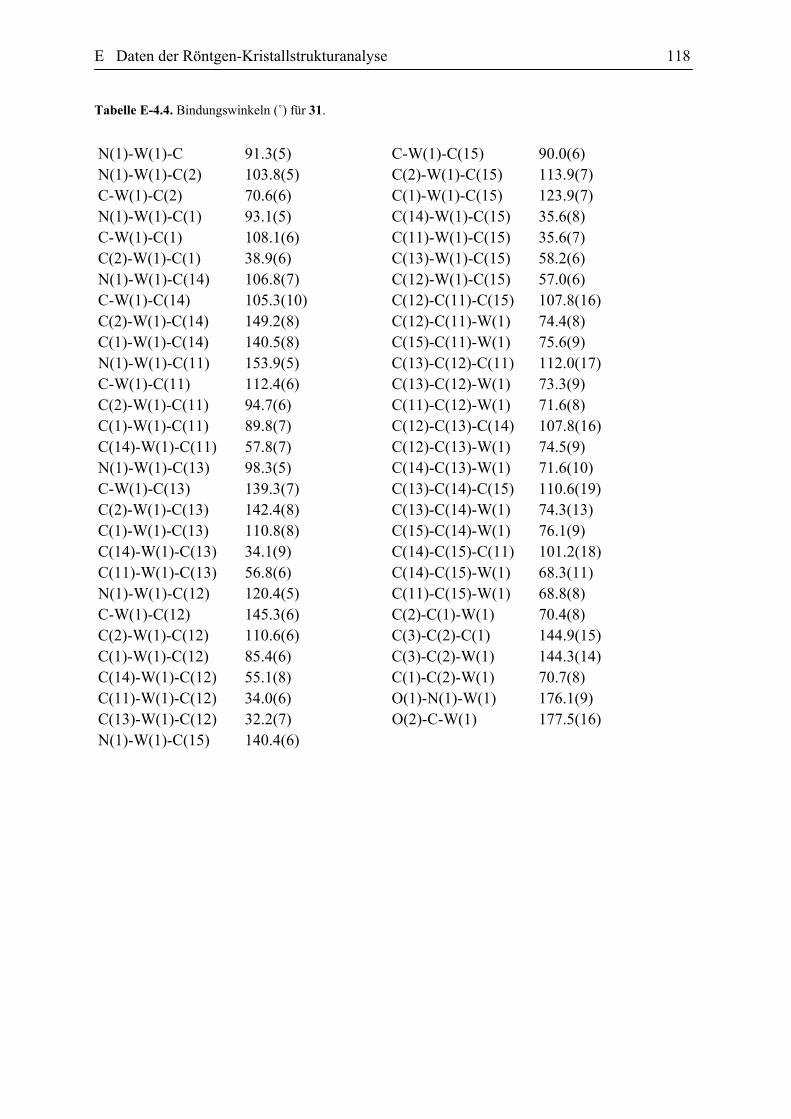
E Daten der Röntgen-Kristallstrukturanalyse 118
Tabelle E-4.4. Bindungswinkeln (˚) für 31.
N(1)-W(1)-C 91.3(5) C-W(1)-C(15) 90.0(6)
5
71.6(10)
(1)-C(13) 110.8(8) C(15)-C(14)-W(1) 76.1(9) (1)-C(13) 34.1(9) C(14)-C(15)-C(11) 101.2(18) (1)-C(13) 56.8(6) C(14)-C(15)-W(1) 68.3(11)
(1)-C(12) 120.4(5) C(11)-C(15)-W(1) 68.8(8) C(2)-C(1)-W(1) 70.4(8)
(1)-C(12) 110.6(6) C(3)-C(2)-C(1) 144.9(15) (12) ) )
) C(12) C(12) )
15)
N(1)-W(1)-C(2) 103.8(5) C(2)-W(1)-C(15) 113.9(7) C-W(1)-C(2) 70.6(6) C(1)-W(1)-C(15) 123.9(7) N(1)-W(1)-C(1) 93.1(5) C(14)-W(1)-C(15) 35.6(8) C-W(1)-C(1) 108.1(6) C(11)-W(1)-C(15) 35.6(7) C(2)-W(1)-C(1) 38.9(6) C(13)-W(1)-C(15) 58.2(6) N(1)-W(1)-C(14) 106.8(7) C(12)-W(1)-C(15) 57.0(6) C-W(1)-C(14) 105.3(10) C(12)-C(11)-C(15) 107.8(16) C(2)-W(1)-C(14) 149.2(8) C(12)-C(11)-W(1) 74.4(8) C(1)-W(1)-C(14) 140.5(8) C(15)-C(11)-W(1) 7 .6(9) N(1)-W(1)-C(11) 153.9(5) C(13)-C(12)-C(11) 112.0(17) C-W(1)-C(11) 112.4(6) C(13)-C(12)-W(1) 73.3(9) C(2)-W(1)-C(11) 94.7(6) C(11)-C(12)-W(1) 71.6(8) C(1)-W(1)-C(11) 89.8(7) C(12)-C(13)-C(14) 107.8(16) C(14)-W(1)-C(11) 57.8(7) C(12)-C(13)-W(1) 74.5(9) N(1)-W(1)-C(13) 98.3(5) C(14)-C(13)-W(1) C-W(1)-C(13) 139.3(7) C(13)-C(14)-C(15) 110.6(19) C(2)-W(1)-C(13) 142.4(8) C(13)-C(14)-W(1) 74.3(13) C(1)-WC(14)-WC(11)-WN(1)-WC-W(1)-C(12) 145.3(6) C(2)-WC(1)-W(1)-C 85.4(6) C(3)-C(2)-W(1 144.3(14C(14)-W(1)-C(12) 55.1(8) C(1)-C(2)-W(1 70.7(8) C(11)-W(1)- 34.0(6) O(1)-N(1)-W(1) 176.1(9)C(13)-W(1)- 32.2(7) O(2)-C-W(1) 177.5(16N(1)-W(1)-C( 140.4(6)

E Daten der Röntgen-Kristallstrukturanalyse 119
5 Die Daten für den η -Allen-K2 omplex des Wolframs 32
bbildung E-5. ORTEP Plot des Komplexes 32 einschließlich Benennung aller Nicht-
asserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide
it einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.
A
W
m
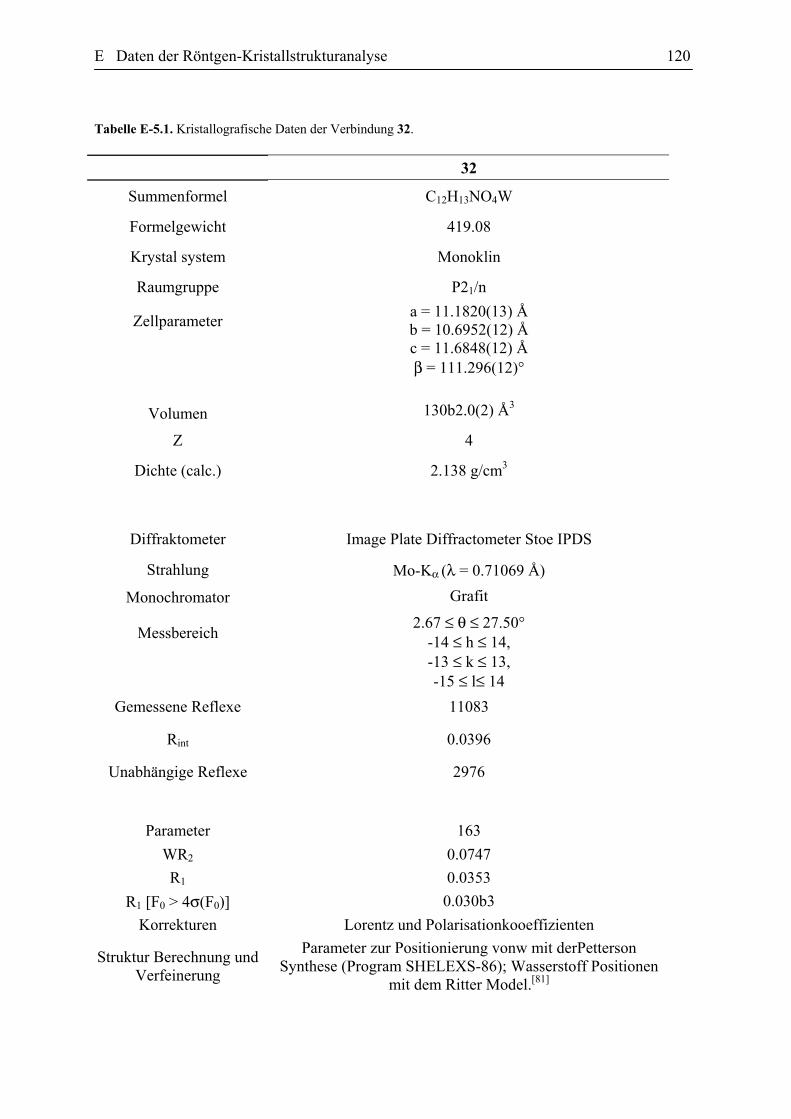
E Daten der Röntgen-Kristallstrukturanalyse 120
abelle E-5.1. Kristallografische Daten der Verbindung 32.
32
T
Summenformel C12H13NO4W
Formelgewicht 419.08
Krystal system Monoklin
Raumgruppe P21/n
Zellparameter
a = 11.1820(13) Å b = 10.6952(12) Å c = 11.6848(12) Å β = 111.296(12)°
Volumen 130b2.0(2) Å3
Z 4
Dichte (calc.) 2.138 g/cm3
Diffraktometer Image Plate Diffractometer Stoe IPDS
Strahlung Mo-Kα (λ = 0.71069 Å)
Monochromator Grafit
Messbereich
2.67 ≤ θ ≤ 27.50° -14 ≤ h ≤ 14, -13 ≤ k ≤ 13, -15 ≤ l≤ 14
Gemessene Reflexe 11083
Rint 0.0396
WR2 0.0747 R1 0.0353
R1 [F0 > 4σ(F0)] 0.030b3 Korrekturen Lorentz und Polarisationkooeffizienten
Struktur Berechnung und Verfeinerung
Parameter zur Positionierung vonw mit derPetterson Synthese (Program SHELEXS-86); Wasserstoff Positionen
mit dem Ritter Model.[81]
Unabhängige Reflexe 2976
Parameter 163
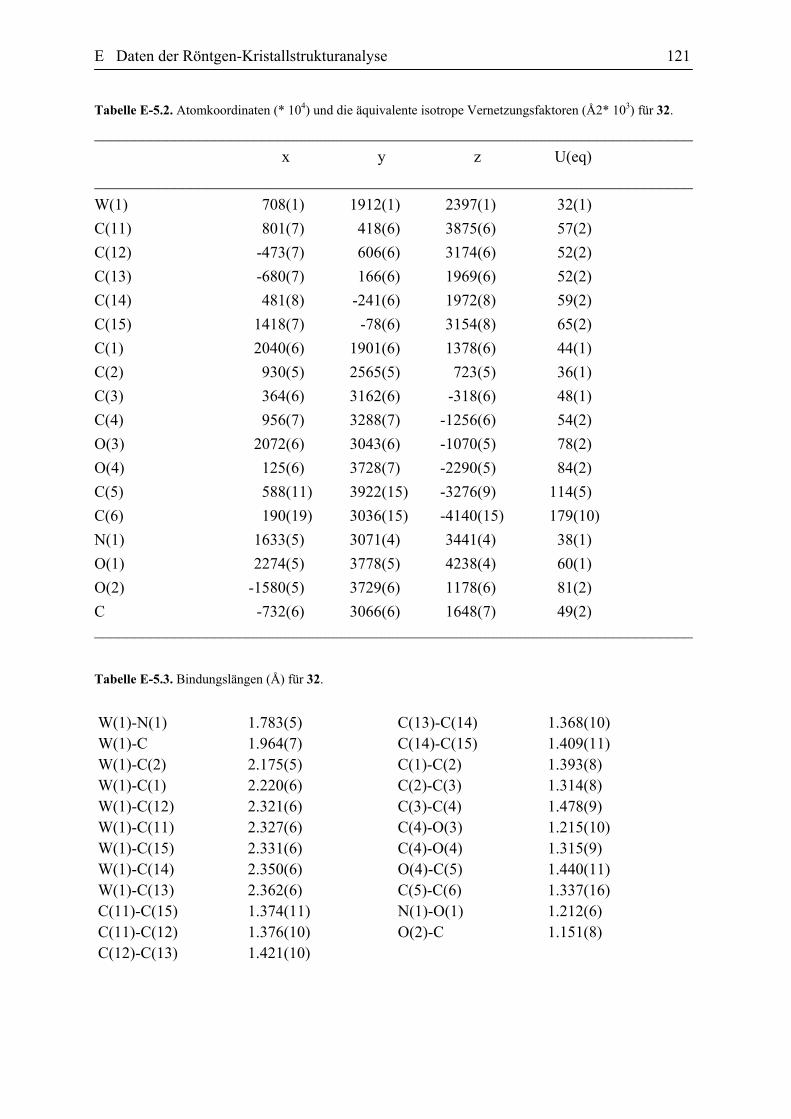
E Daten der Röntgen-Kristallstrukturanalyse 121
Tabelle E-5.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 32.
__________________________________ x y U(eq)
___________________________________________________________________________ W(1) 708(1) 1912(1) 2397(1) 32(1) C(11) 801(7) 418(6) 57(2) C(12) -473(7) 606(6) 52(2) C(13) -680(7) 166(6) 1969(6) 52(2) C(14) 481(8) -241(6) 59(2) C(15) 1418(7) -78(6) 65(2) C(1) 2040(6) 1901(6) 1378(6) 44(1) C(2) 930(5) 2565(5) 36(1) C(3) 364(6) 3162(6) 48(1) C(4) 956(7) 3288(7) 54(2) O(3) 2072(6) 3043(6) -1070(5) 78(2) O(4) 125(6) 3728(7) 84(2) C(5) 588(11) 3922(15) -3276(9) 114(5) C(6) 190(19) 3036(15) -4140(15) 179(10) N(1) 1633(5) O(1) 2274(5) 3778(5 60(1) O(2) -1580(5) 3729(6) 1178(6) 81(2) C -732(6) 3066(6) 1648(7) 49(2) ___________________________________________________________________________ Tabelle E-5.3. indungslängen (Å) für 32.
W(1)-N(1) 1.783(5) C(13)-C(14) 1.368(10) W(1)-C 1.964(7) C(14)-C 1.409(11) W .175(5) C(1)-C( 1.393(8) W C(2)-C(3) 1.314(8) W(1)-C(12) 2.321(6) C(3)-C(4) 1.478(9) W(1)-C 2.327(6) C(4)-O(3 1.215(10) W(1)-C(1 2.331(6) C(4)-O 1.315(9) W(1)-C(14 2.350(6) O(4)-C 1.440(11) W(1)-C(13) 2.362(6) C(5)-C 1.337(16) C(1 1.374(11) C(11) 1.376(10)
21
_________________________________________ z
3875(6) 3174(6)
1972(8) 3154(8)
723(5) -318(6)
-1256(6)
-2290(5)
3071(4) 3441(4) 38(1) ) 4238(4)
B
(15) (1)-C(2) 2(1)-C(1) 2.220(6)
2)
(11) )
5) (4) ) (5)
(6) (1) 1)-C(15) N(1)-O 1.212(6)
O(2)-C 1.151(8)-C(12) C(12)-C(13) 1.4 (10)

E Daten der Röntgen-Kristallstrukturanalyse 122
Tabelle E-5.4. Bindungswinkeln (˚) für 32. N(1)-W(1)-C 92.6(3) C(12)-W(1)-C(13) 35.3(2) N(1)-W(1)-C(2) 99.5(2) C(11)-W(1)-C(13) 57.6(2) C-W(1)-C(2) 74.9(3) C(15)-W(1)-C(13) 57.7(2) N(1)-W(1)-C(1) 92.0(2) C(14)-W(1)-C(13) 33.8(3) C-W(1)-C(1) 111.4(3) C(15)-C(11)-C(12) 109.4(6) C(2)-W(1)-C(1) 37.0(2) C(15)-C(11)-W(1) 73.0(4) N(1)-W(1)-C(12) 115.0(2) C(12)-C(11)-W(1) 72.5(4) C-W(1)-C(12) 93.6(3) C(11)-C(12)-C(13) 107.6(7) C(2)-W(1)-C(12) 144.3(2) C(11)-C(12)-W(1) 73.0(4) C(1)-W(1)-C(12) 142.6(2) C(13)-C(12)-W(1) 73.9(4) N(1)-W(1)-C(11) 96.4(2) C(14)-C(13)-C(12) 106.8(6) C-W(1)-C(11) 124.8(3) C(14)-C(13)-W(1) 72.6(4) C(2)-W(1)-C(11) 154.1(3) C(12)-C(13)-W(1) 70.8(3) C(1)-W(1)-C(11) 122.5(3) C(13)-C(14)-C(15) 109.3(7) C(12)-W(1)-C(11) 34.4(2) C(13)-C(14)-W(1) 73.6(4) N(1)-W(1)-C(15) 110.0(2) C(15)-C(14)-W(1) 71.8(4) C-W(1)-C(15) 148.7(3) C(11)-C(15)-C(14) 106.8(6) C(2)-W(1)-C(15) 120.0(3) C(11)-C(15)-W(1) 72.7(4) C(1)-W(1)-C(15) 89.8(3) C(14)-C(15)-W(1) 73.2(4) C(12)-W(1)-C(15) 57.7(3) C(2)-C(1)-W(1) 69.8(3) C(11)-W(1)-C(15) 34.3(3) C(3)-C(2)-C(1) 142.1(6) N(1)-W(1)-C(14) 144.8(3) C(3)-C(2)-W(1) 144.5(5) C-W(1)-C(14) 120.9(3) C(1)-C(2)-W(1) 73.3(3) C(2)-W(1)-C(14) 99.4(2) C(2)-C(3)-C(4) 122.4(7) C(1)-W(1)-C(14) 85.6(2) O(3)-C(4)-O(4) 125.3(7)
O(3)-C(4)-C(3) 123.8(7) (1)-C(14) 57.1(3) O(4)-C(4)-C(3) 110.9(7)
C(14) 3 C(4)-O(4)-C(5) 11(13) 1 C(6)-C(5)-O(4) 112.6(10)
3) 9 O 173.8(4) (13) 1 O 178.1(7) 13) 1
C(12)-W(1)-C(14) 57.3(3) C(11)-WC(15)-W(1)-
)-C5.0(3) 6.6(8)
N(1)-W(1 50.2(2) C-W(1)-C(1 1.9(3) (1)-N(1)-W(1) C(2)-W(1)-C 10.1(2) (2)-C-W(1) C(1)-W(1)-C(
13.5(3)
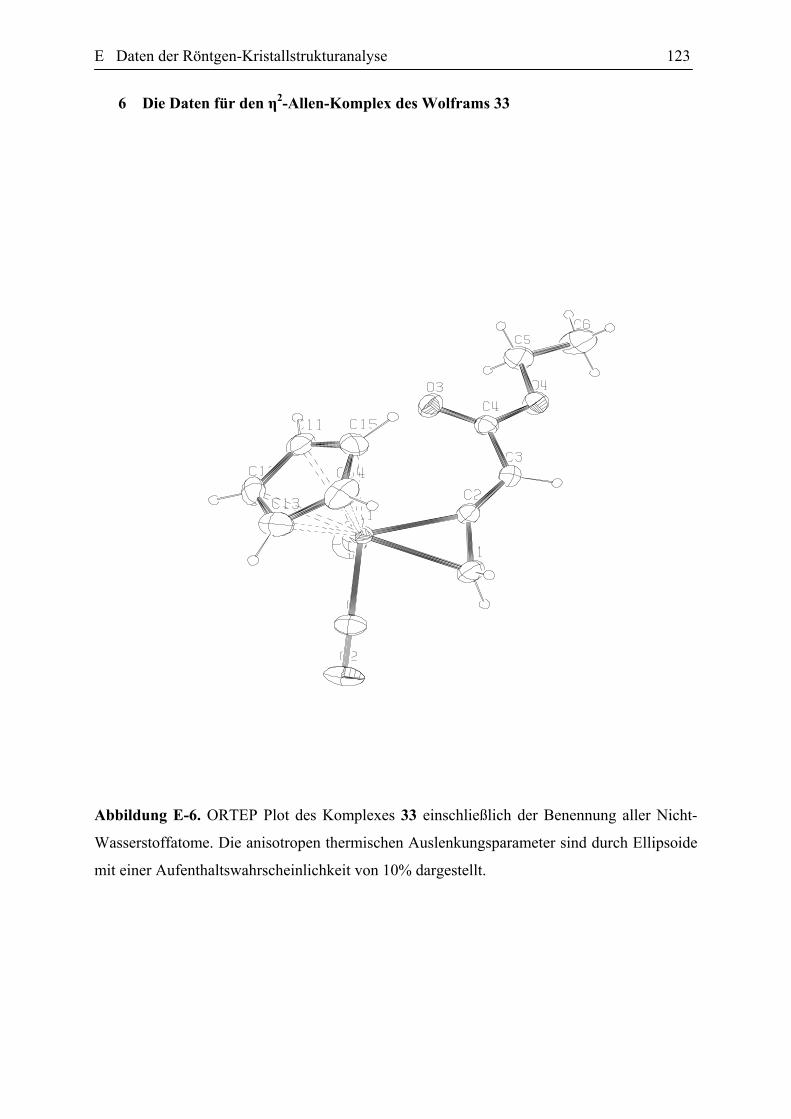
E Daten der Röntgen-Kristallstrukturanalyse 123
6 Die Daten für den η -Allen-K2 omplex des Wolframs 33
bbildung E-6. ORTEP Plot des Komplexes 33 einschließlich der Benennung aller Nicht-
asserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide
mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.
A
W

E Daten der Röntgen-Kristallstrukturanalyse 124
abelle E-6.1. Kristallografische Daten der Verbindung 33.
33
T
Summenformel C22H32N2O2W
Formelgewicht 540.358
Krystal system Monoklin
Raumgruppe P21/c Zellparameter
a = 6.7864(5) Å, b = 21.0383(18) Å, c = .9.2890(8) Å, β = 92.02(10)°.
Volumen 1314.36(19) Å3
Z 4
Dichte (calc.) 2.118 g/cm3
Diffraktometer Image Plate Diffractometer Stoe IPDS
Strahlung Mo-Kα (λ = 0.71069 Å) Monochromator Grafit
Messbereich
2.21 ≤ θ ≤ 25.97° -8 ≤ h ≤ 8,
-25 ≤ k ≤ 25, -11 ≤I ≤ 11
Gemessene Reflexe 9598
Rint 0.0526
Unabhängige Reflexe 4961
Parameter 163
nkooeffizienten
Struktur Berechnung und Verfeinerung
Parameter zur Positionierung vonw mit derPetterson Synthese (Program SHELEXS-86); Wasserstoff Positionen
mit dem Ritter Model.[81]
WR2 0.0652 R1 0.030b0
R1 [F0 > 4σ(F0)] 0.0235 Korrekturen Lorentz und Polarisatio

E Daten der Röntgen-Kristallstrukturanalyse 125
Tabelle E-6.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 33. __________________________________
x y U(eq) ___________________________________________________________________________ W(1) 1181(1) 4227(1) 2134(1) 46(1) C(11) 242(11) 3250(3) 75(2) C(12) 881(12) 3622(3) 29(6) 79(2) C(13) 2907(12) 3742(3) 83(2) C(14) 3471(11) 3429(3) 82(2) C(15) 1824(13) 3131(3) 78(2) N(1) -1151(7) 4636(2) 2062(5) 60(1) O(1) -2735(8) 4884(2) 1967(6) 96(2) C 2543(9) 5039(2) 1697(7) 64(1) O(2) 3314(7) 5489(2) 1447(6) 90(1) C(1) 2828(9) 4569(3) 4256(6) 66(1) C(2) 1306(8) 4133(2) 4409(6) 53(1) C(3) 357(8) 3886(2) 5441(5) 58(1) C(4) -1404(8) O(3) -2119(7) 3267(2 76(1) O(4) -2210(6) 3395(2) 6375(4) 71(1) C(5) -4048(11) 3035(4) 86(2) C(6) -4588(15) 2982(4) 7722(10) 119(3) ___________________________________________________________________________ Tab en (Å) für 33.
W 795(5) C(13)-C 1.402(10) W C(14)-C(15) 1.369(10) W(1)-C(2) 2.112(5) N(1)-O(1) 1.188(6) W(1)-C 2.250(6) C-O(2) 1.121(6) W(1)-C(12 2.319(5) C(1)-C 1.402(7) W(1)-C(11) 2.326(5) C(2)-C 1.330(7) W(1 2.335(5) C(3)-C 1.454(7) W(1)-C 2.347(5) W(1)-C(14) 2.349(5C( .36C(1 1.373(9)
1.397(10)
_________________________________________ z
1088(7)
418(8)
1741(8) 2124(7)
3488(2) 5161(6) 55(1) ) 4014(5)
6250(8)
elle E-6.3. Bindungsläng
(1)-N(1) 1.(1)-C 2.009(5)
(14)
(1) ) (2) (3)
)-C(13) (4) (15) C(4)-O(3) 1.203(6)
) C(4)-O(4) 1.332(6) 5(10) O(4)-C(5) 1.450(7)
C(5)-C(6) 1.467(11) 11)-C(15) 11)-C(12)
C(12)-C(13)
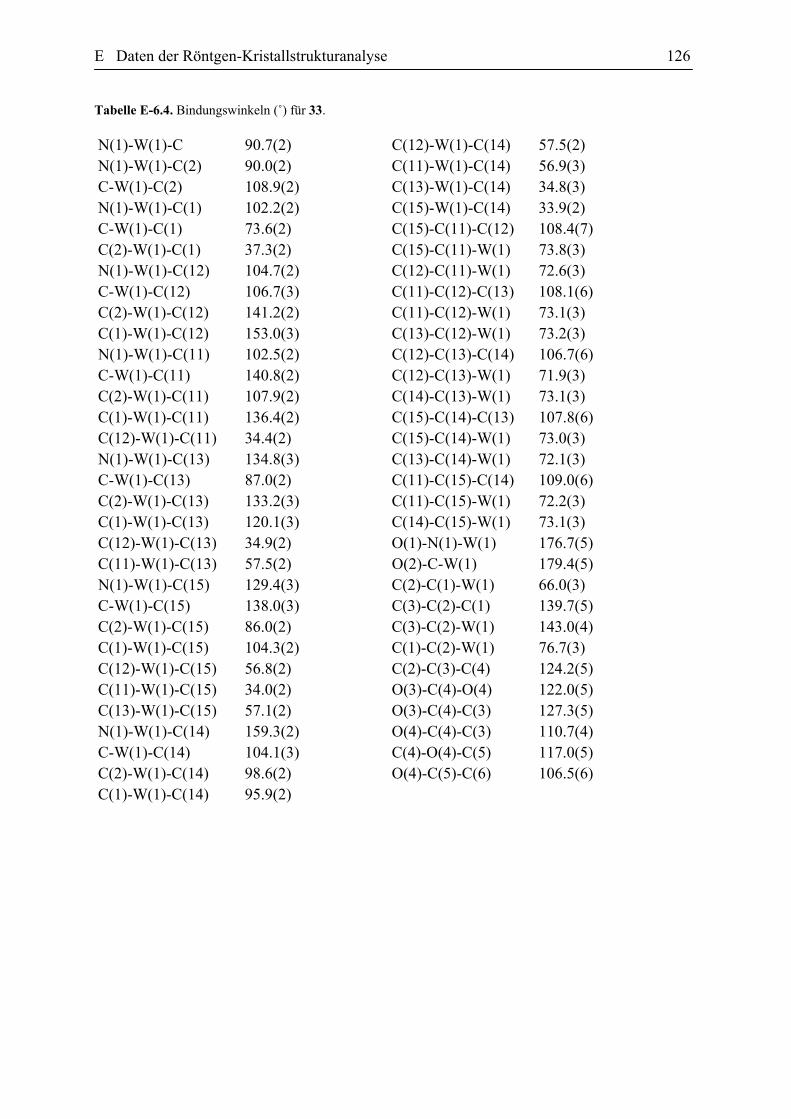
E Daten der Röntgen-Kristallstrukturanalyse 126
Tabelle E-6.4. Bindungswinkeln (˚) für 33. N(1)-W(1)-C 90.7(2) C(12)-W(1)-C(14) 57.5(2) N(1)-W(1)-C(2) 90.0(2) C(11)-W(1)-C(14) 56.9(3) C-W(1)-C(2) 108.9(2) C(13)-W(1)-C(14) 34.8(3) N(1)-W(1)-C(1) 102.2(2) C(15)-W(1)-C(14) 3 .9(2) 3
3
3032
2
77
(1)-C(15) 104.3(2) C(1)-C(2)-W(1) 76.7(3) C(2)-C(3)-C(4) 124.2(5)
(1)-C(15) 34.0(2) O(3)-C(4)-O(4) 122.0(5) C(15) O(3)-C(4)-C(3) 127.3(5)
)-C(14) O(4)-C(4)-C(3) 110.7(4) 4) C(4)-O(4)-C(5) 117.0(5) (14) O(4)-C(5)-C(6) 106.5(6) 14)
C-W(1)-C(1) 73.6(2) C(15)-C(11)-C(12) 108.4(7) C(2)-W(1)-C(1) 37.3(2) C(15)-C(11)-W(1) 73.8(3) N(1)-W(1)-C(12) 104.7(2) C(12)-C(11)-W(1) 72.6(3) C-W(1)-C(12) 106.7(3) C(11)-C(12)-C(13) 108.1(6) C(2)-W(1)-C(12) 141.2(2) C(11)-C(12)-W(1) 73.1(3) C(1)-W(1)-C(12) 153.0(3) C(13)-C(12)-W(1) 7 .2(3) N(1)-W(1)-C(11) 102.5(2) C(12)-C(13)-C(14) 106.7(6) C-W(1)-C(11) 140.8(2) C(12)-C(13)-W(1) 71.9(3) C(2)-W(1)-C(11) 107.9(2) C(14)-C(13)-W(1) 7 .1(3) C(1)-W(1)-C(11) 136.4(2) C(15)-C(14)-C(13) 1 7.8(6) C(12)-W(1)-C(11) 34.4(2) C(15)-C(14)-W(1) 7 .0(3) N(1)-W(1)-C(13) 134.8(3) C(13)-C(14)-W(1) 7 .1(3) C-W(1)-C(13) 87.0(2) C(11)-C(15)-C(14) 109.0(6) C(2)-W(1)-C(13) 133.2(3) C(11)-C(15)-W(1) 7 .2(3) C(1)-W(1)-C(13) 120.1(3) C(14)-C(15)-W(1) 73.1(3) C(12)-W(1)-C(13) 34.9(2) O(1)-N(1)-W(1) 1 6.7(5) C(11)-W(1)-C(13) 57.5(2) O(2)-C-W(1) 1 9.4(5) N(1)-W(1)-C(15) 129.4(3) C(2)-C(1)-W(1) 66.0(3) C-W(1)-C(15) 138.0(3) C(3)-C(2)-C(1) 139.7(5) C(2)-W(1)-C(15) 86.0(2) C(3)-C(2)-W(1) 143.0(4) C(1)-WC(12)-W(1)-C(15) 56.8(2) C(11)-WC(13)-W(1)- 57.1(2) N(1)-W(1 159.3(2) C-W(1)-C(1 104.1(3) C(2)-W(1)-C 98.6(2) C(1)-W(1)-C( 95.9(2)
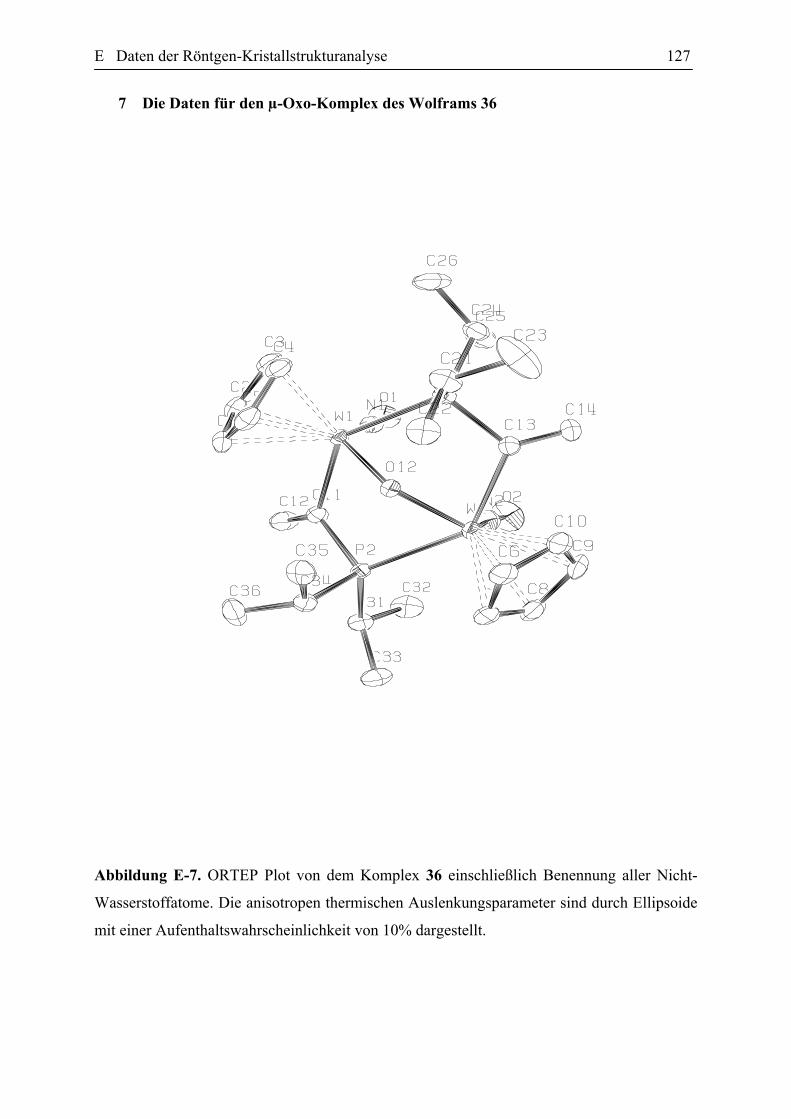
E Daten der Röntgen-Kristallstrukturanalyse 127
7 Die Daten für den µ-Oxo-Komplex des Wolframs 36
Abbildung E-7. ORTEP Plot von dem Komplex 36 einschließlich Benennung aller Nicht-
Wasserstoffatome. Die anisotropen thermischen Auslenkungsparameter sind durch Ellipsoide
mit einer Aufenthaltswahrscheinlichkeit von 10% dargestellt.

E Daten der Röntgen-Kristallstrukturanalyse 128
abelle E-7.1. Kristallografische Daten der Verbindung 36.
ummenformel C26 H42 N2 O3 P2 W2
ormelgewicht 860.27 g/mol
Kristallsystem Monoklin
Raumgruppe P21/n (No.14)
Zellparameter a = 10.1365(12) Å; b = 14.4120(16) Å c = 21.374(2) Å, β = 109.257(14)°.
Volumen 2947.8(6) Å3
Z 1
Dichte (berechnet) 1.938 Mg/m3
Diffraktometer Image Plate Diffraktometer System (STOE)
Strahlung Mo Kα
Monochromator Temperatur
Graphit 293(2) K
Messbereich 4.80˚ ≤ 2θ ≤ 51.67˚ -12<=h<=12, -17<=k<=16, -26<=l<=26
Gemessene Reflexe Unabhängige Reflexe Unabhängige Reflexe mit F0 > 4 σ (F0) Rint
20908 5687 5077 0.0501
arameter ,wR2, R1
1 [F0 > 4σ(F0)] 316, 0.0596, 0.0274 0.0226
ax und min in Δσ 1.271 und -0.735 e.Å-3
orrekturen Lorentz und Polarisationkooeffizienten
trukturberechnung und Verfeinerung Parameter zur Positionierung von
Positionen mit dem Ritter Model.[81]
T
S
F
PR
M
K
SWasserstoffatomen mit der Petterson Synthese (Program SHELEXS-86); Wasserstoff
E Daten der Röntgen-Kristallstrukturanalyse 129
Tabelle E-7.2. Atomkoordinaten (* 104) und die äquivalente isotrope Vernetzungsfaktoren (Å2* 103) für 36.
_______________________________
x y U(eq) ________________________________________________________________________________
-102(1) 2723(1) 4375(1) 27(1) 2(4) 3860(3) 4081(2) 36(1)
-96(4) 4660(2) 3887(2) 56(1) -1984(5) 2150(4)
C(2) -1852(5) 3120(4)C(3) -573(6) 3275(4) 5304(2) 49(1)
65(6) 2412(4) 5499(2) 49(1) ) -827(6) 1714(4) 5128(2) 49(1)
2537(1) 2641(1) 4819(1) 34(1) 3042(5) 2505(3) 4085(2) 4195(6) 2900(5) 4063(3) 3310(6) 1621(4) 5363(3) 52(1) 2728(6) 697(4) 86(3) 57(1) 4897(7) 1612(6) 5556(5) 115(3) 3553(5) 3630(4) 5296(2) 48(1) 3113(6) 4532(4)
C(26) 3413(8) 3717(6) 5986(3) 692(3) 1692(2) 3991(1) 28(1)
1452(1) 1810(1) 3245(1) 28(1) 915(3) 2881(2) 44(1)
) 1289(5) 3658(3) 2605(2) 76(1) C(6) 2327(5) 227(3) 3297(3) 47(1)
1323(5) 357(3) 46(1) 1800(5) 1061(4) 2349(2) 52(1) 3101(5) 1368(4) 2771(3) 57(1) 3406(5) 848(4)
-1176(1) 1658(1)2519(3)
C(12) -2802(5) 3033(4)C(31) -2235(5) 1895(3)C(32) -1682(7) 2762(4)C(33) -2276(6) 1085(4) 1534(2) 55(1) C(34) -1899(5) 522(3) 3012(2) 38(1)
(35) -1104(6) 40(4) 3658(2) 51(1) (36) -3433(5) 580(4) 2938(3) 64(2)
________________________________________________________________________________
_________________________________________________
z
W(1) N(1) O(1) C(1) 4694(3) 47(1)
4800(2) 47(1)
C(4) C(5P(1) C(13) C(14)
38(1) 70(2)
C(21) C(22) 50C(23) C(24) C(25) 4922(3) 67(2)
85(2) O(12) W(2) N(2) 1270(4) 2 O(2
C(7) 2673(2) C(8) C(9) C(10) 3363(3) 52(1) P(2) 2874(1) 28(1) C(11) -1671(4) 3384(2) 34(1)
3128(3) 54(1) 1996(2) 39(1) 1757(3) 59(2)
CC

E Daten der Röntgen-Kristallstrukturanalyse 130
Tabelle E-7.3. Bindungslängen (Å) für 36. W(1)-N(1) 1.772(4) C(24)-C(26) 1.531(7) W(1)-O(12) 1.989(2) O(12)-W(2) 1.995(2) W(1)-C(11) .209(4) W(2)-N(2) .755(4) 2 1
2.31 22. 22. 2
22
2 21 11.3 11 11 11.400(7) 1.401(8) 1 11 11 1
2 1 1 1 1 1 1
indungswinke
1
7
99
1
122.79(19) 113.26(16) 79.51(17) 35.24(19) 57.73(19)
W(1)-C(2) 5(4) W(2)-C(9) .310(4) W(1)-C(3) 328(4) W(2)-C(8) .323(4) W(1)-C(1) 377(4) W(2)-C(10) .361(4) W(1)-C(4) 2.396(5) W(2)-C(7) .406(4) W(1)-C(5) 2.456(4) W(2)-C(6) .437(5) W(1)-P(1) .5289(12) W(2)-P(2) .5256(11) N(1)-O(1) .219(5) N(2)-O(2) .227(5) C(1)-C(5) 83(8) C(6)-C(10) .384(7) C(1)-C(2) .416(7) C(6)-C(7) .398(7) C(2)-C(3) .404(7) C(7)-C(8) .402(7) C(3)-C(4) C(8)-C(9) C(4)-C(5) .409(8) C(9)-C(10) .415(8) P(1)-C(13) .813(4) P(2)-C(11) .825(4) P(1)-C(24) .855(5) P(2)-C(34) .857(4) P(1)-C(21) 1.878(5) P(2)-C(31) 1.861(4) C(13)-C(14) 1.315(7) C(11)-C(12) 1.323(6) C(13)-W(2) .217(5) C(31)-C(33) 1.521(6) C(21)-C(22) .498(8) C(31)-C(32) .526(7) C(21)-C(23) .523(8) C(34)-C(36) .513(6) C(24)-C(25) .515(8) C(34)-C(35) .518(7) Tabelle E-7.4. B ln (˚) für 36. N(1)-W(1)-O(12) 117.71(13) N(2)-W(2)-O(12) 14.92(15) N(1)-W(1)-C(11) 83.76(16) N(2)-W(2)-C(13) 83.76(19) O(12)-W(1)-C(11) 76.52(13) O(12)-W(2)-C(13) 6.17(13) N(1)-W(1)-C(2) 93.11(17) N(2)-W(2)-C(9) 92.3(2) O(12)-W(1)-C(2) 144.28(15) O(12)-W(2)-C(9) 148.50(17) C(11)-W(1)-C(2) 90.39(17) C(13)-W(2)-C(9) 2.33(18) N(1)-W(1)-C(3) 92.29(17) N(2)-W(2)-C(8) 4.5(2) O(12)-W(1)-C(3) 146.07(15) O(12)-W(2)-C(8) 145.36(16) C(11)-W(1)-C(3) 125.31(17) C(13)-W(2)-C(8) 27.53(17) C(2)-W(1)-C(3) 35.19(18) C(9)-W(2)-C(8) 35.21(19) N(1)-W(1)-C(1) 124.86(17) N(2)-W(2)-C(10) O(12)-W(1)-C(1) 109.30(15) O(12)-W(2)-C(10) C(11)-W(1)-C(1) 80.62(17) C(13)-W(2)-C(10) C(2)-W(1)-C(1) 35.09(18) C(9)-W(2)-C(10) C(3)-W(1)-C(1) 57.62(18) C(8)-W(2)-C(10) N(1)-W(1)-C(4) 122.47(17) N(2)-W(2)-C(7) 125.73(18) O(12)-W(1)-C(4) 111.64(15) O(12)-W(2)-C(7) 111.14(14) C(11)-W(1)-C(4) 136.67(17) C(13)-W(2)-C(7) 135.02(17) C(2)-W(1)-C(4) 57.48(19) C(9)-W(2)-C(7) 57.60(19)
E Daten der Röntgen-Kristallstrukturanalyse 131
C(3)-W(1)-C(4) 34.43(18) C(8)-W(2)-C(7) 34.43(18) C(10)-W(2)-C(7) 56.41(18)
(1)-C(5) 147.88(15) N(2)-W(2)-C(6) 148.55(17) C(5) (6)
(5) ) (6) ) 5) (6)
(5) (6) (5) (6) (5) (6) (1) (2)
-P(1) -P(2) P(1) ) -P(2) ) (1) ) (2) ) (1) ) (2) (1) ) -P(2) ) (1) (2) (1) ) (2) ) (1) 171.6(3) (2) 171.9(4) 2) (7)
) (2) )
) ) ) )
(1) 74.8(2) C(8)-C(7)-W(2) 69.5(3) C(9)-C(8)-C(7) 108.4(5)
(1) 75.4(3) C(9)-C(8)-W(2) 71.9(3)
)
C(1)-W(1)-C(4) 56.25(18) N(1)-WO(12)-W(1)- 94.40(13) O(12)-W(2)-C 96.48(14)C(11)-W(1)-C 105.87(18 C(13)-W(2)-C 103.23(17C(2)-W(1)-C( 56.99(18) C(9)-W(2)-C 57.18(19)C(3)-W(1)-C 56.87(17) C(8)-W(2)-C 56.76(18)C(1)-W(1)-C 33.21(18) C(10)-W(2)-C 33.48(17)C(4)-W(1)-C 33.75(18) C(7)-W(2)-C 33.56(17)N(1)-W(1)-P 89.80(12) N(2)-W(2)-P 89.71(13)O(12)-W(1) 66.30(8) O(12)-W(2) 66.28(8) C(11)-W(1)- 133.72(10 C(13)-W(2) 134.71(11C(2)-W(1)-P 135.80(14 C(9)-W(2)-P 132.78(15C(3)-W(1)-P 100.65(14 C(8)-W(2)-P 97.60(13)C(1)-W(1)-P 136.56(14 C(10)-W(2) 137.96(14C(4)-W(1)-P 84.21(13) C(7)-W(2)-P 83.67(12)C(5)-W(1)-P 103.58(14 C(6)-W(2)-P 105.23(12O(1)-N(1)-W O(2)-N(2)-WC(5)-C(1)-C( 109.1(5) C(10)-C(6)-C 108.1(5) C(5)-C(1)-W(1 76.5(3) C(10)-C(6)-W 70.3(3) C(2)-C(1)-W(1) 70.1(2) C(7)-C(6)-W(2 72.0(3) C(3)-C(2)-C(1 107.1(5) C(6)-C(7)-C(8 107.9(5) C(3)-C(2)-W(1 72.9(2) C(6)-C(7)-W(2 74.5(3) C(1)-C(2)-WC(4)-C(3)-C(2) 107.9(5) C(4)-C(3)-WC(2)-C(3)-W(1) 71.9(2) C(7)-C(8)-W(2) 76.0(2) C(3)-C(4)-C(5) 108.6(5) C(8)-C(9)-C(10) 106.8(5) C(3)-C(4)-W(1) 70.1(3) C(8)-C(9)-W(2) 72.9(2) C(5)-C(4)-W(1) 75.5(3) C(10)-C(9)-W(2) 74.4(2) C(1)-C(5)-C(4) 107.4(5) C(6)-C(10)-C(9) 108.7(5) C(1)-C(5)-W(1) 70.3(2) C(6)-C(10)-W(2) 76.3(3) C(4)-C(5)-W(1) 70.8(3) C(9)-C(10)-W(2) 70.4(3) C(13)-P(1)-C(24) 107.1(2) C(11)-P(2)-C(34) 107.67(19) C(13)-P(1)-C(21) 105.9(2) C(11)-P(2)-C(31) 107.2(2) C(24)-P(1)-C(21) 102.6(3) C(34)-P(2)-C(31) 101.3(2) C(13)-P(1)-W(1) 104.27(15) C(11)-P(2)-W(2) 102.85(14) C(24)-P(1)-W(1) 119.82(17) C(34)-P(2)-W(2) 117.27(15) C(21)-P(1)-W(1) 116.26(18) C(31)-P(2)-W(2) 119.86(15)C(14)-C(13)-P(1) 119.6(4) C(12)-C(11)-P(2) 120.3(4) C(14)-C(13)-W(2 125.7(4) C(12)-C(11)-W(1) 125.0(3) P(1)-C(13)-W(2) 114.2(2) P(2)-C(11)-W(1) 114.2(2) C(22)-C(21)-C(23) 110.0(5) C(33)-C(31)-C(32) 110.2(4) C(22)-C(21)-P(1) 114.9(4) C(33)-C(31)-P(2) 113.4(3) C(23)-C(21)-P(1) 111.2(4) C(32)-C(31)-P(2) 109.6(3)

E Daten der Röntgen-Kristallstrukturanalyse 132
C(25)-C(24)-C(26) 5)
109.5(5) C(36)-C(34)-C(3 109.8(4) C(25)-C(24)-P(1) 110.8(4) C(36)-C(34)-P(2) 112.4(4) C(26)-C(24)-P(1) 112.4(4) C(35)-C(34)-P(2) 115.4(3) W(1)-O(12)-W(2) 125.48(13)
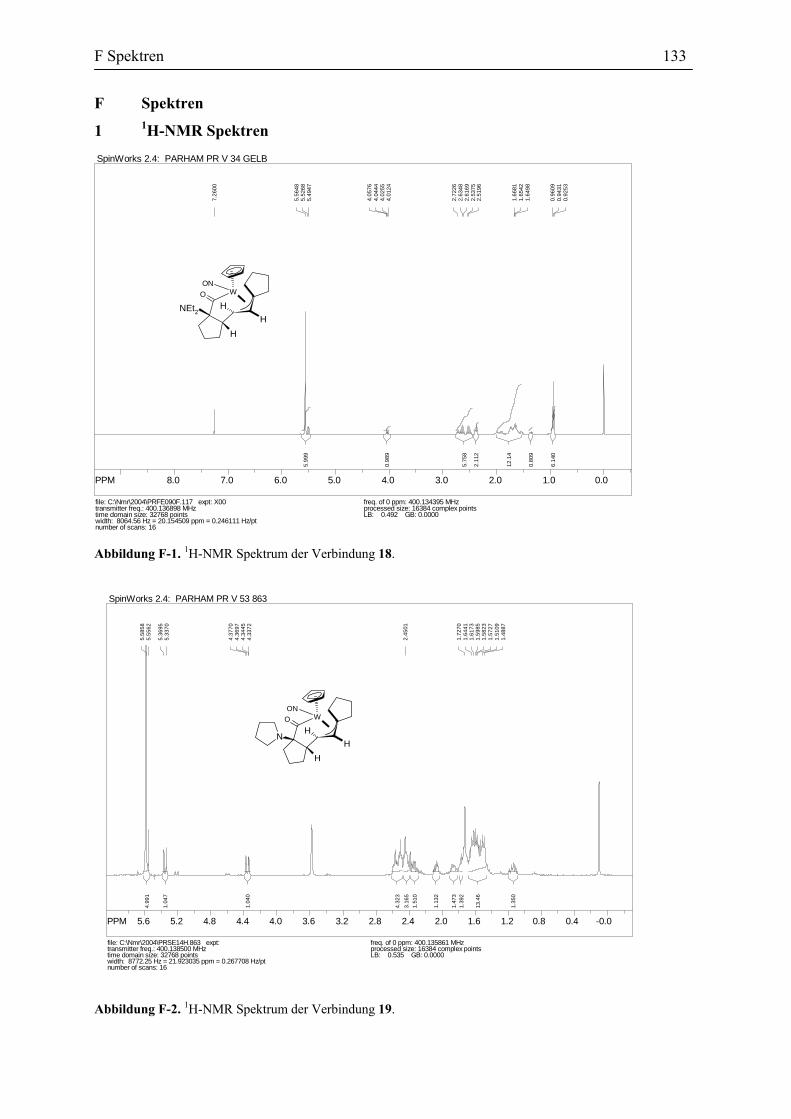
F Spektren 133
F Spektren
1 1H-NMR Spekt
Abbildung F-1. 1H-NMR Spektrum der Verbindung 18.
Abbildung F-2. 1H-NMR Spektrum der Verbindung 19.
ren SpinWorks 2.4: PARHAM PR V 34 GELB
7.
2600
5.
5648
5.
5268
5.
4947
4.
0576
4.
0444
4.
0255
4.
0124
2.
7226
2.
6348
2.
6169
2.
5375
2.
5196
1.
6681
1.
6542
1.
6498
0.
9609
0.
9431
PPM 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0
5.
999
0.
989
5.
758
2.
112
12
.14
6.
140
0.
809
0.
9253
e: C:\Nmr\2004\PRFE090F.117 expt: X00mitter freq.: 400.136898 MHz
e domain size: 32768 pointsdth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
number of scans: 16
freq. of 0 ppm: 400.134395 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
WOON
NEt2
HH
H
filtranstimwi
SpinWorks 2.4: PARHAM PR V 53 863
PPM 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
1.
047
4.
991
1.
040
4.
323
3.16
5
1.
510
1.
132
1.
473
1.39
2
13
.46
1.
350
5.
5858
5.
5562
5.
3695
5.
3370
4.
3770
4.
3697
4.
3445
4.
3372
2.
4501
1.
7270
1.
6441
1.
6173
1.
5985
1.
5823
1.
5727
1.
5109
1.
4887
file: C:\Nmr\2004\PRSE14H.863 expt: transmitter freq.: 400.138500 MHztime domain size: 32768 pointswidth: 8772.25 Hz = 21.923035 ppm = 0.267708 Hz/pt
freq. of 0 ppm: 400.135861 MHzprocessed size: 16384 complex pointsLB: 0.535 GB: 0.0000
number of scans: 16
WOON
HH
HN

F Spektren 134
Sp 2.4: PARHAM PR V 154 894inWorks
PPM
5.
5652
5.
5375
5.
3308
5.
2978
4.
4384
4.
4300
4.
4054
4.
3971
3.
5759
2.
5571
1.
7919
1.
6896
1.
5964
1.
5914
1.
5793
1.
5750
1.
5600
1.
5378
5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4
5.
000
0.
861
0.
150
0.
131
0.
902
1.
130
0.
456
1.
138
0.
921
2.
079
3.
900
2.
905
2.
059
3.
017
1.25
0
7.
161
2.
019
1.52
0
1.
2854
file: C:\Nmr\2004\PRSE20H.894 expt: transmitter freq.: 400.138500 MHztime domain size: 32768 pointswidth: 8771.98 Hz = 21.922372 ppm = 0.267700 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.136607 MHzprocessed size: 16384 complex pointsLB: 0.535 GB: 0.0000
WO
ON
HH
HN
Abbildung F-3. 1H-NMR Spektrum der Verbindung 20. SpinWorks 2.4: PARHAM PR V 62 279
PPM
5.
7252
4.
3813
4.
3565
4.
3516
4.
3269
2.
7901
2.
7722
1.
7273
1.
1044
1.
0866
8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0
5.
002
0.
833
3.
914
1.
895
1.21
3
3.
153
1.24
8
5.
961
1.
0686
file: C:\Nmr\2004\PRAP22H5.279 expt: transmitter freq.: 400.137300 MHztime domain size: 32768 pointswidth: 6024.12 Hz = 15.055126 ppm = 0.183841 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.135861 MHzprocessed size: 16384 complex pointsLB: 0.368 GB: 0.0000
W NC
O
OC
CH
H
HN
bbildung F-4. 1H-NMR Spektrum der Verbindung 21. A
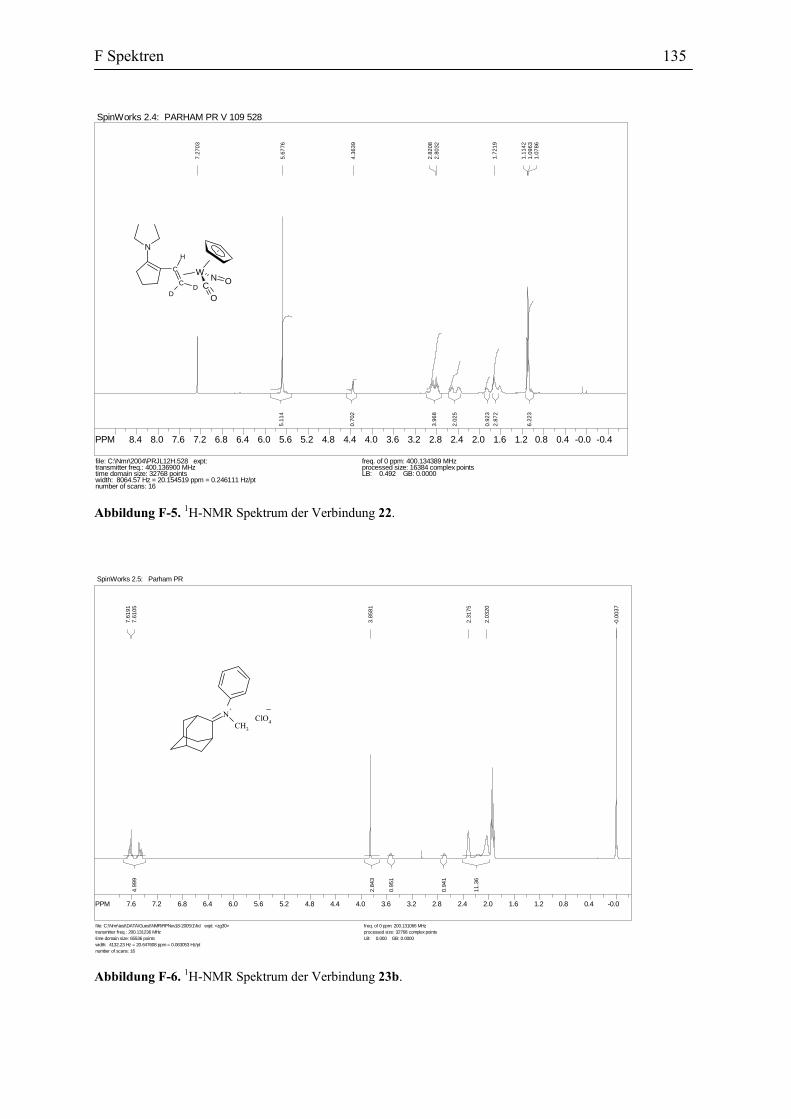
F Spektren 135
SpinWorks 2.4: PARHAM PR V 109 528
PPM 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0 -0.4
5.
114
0.
702
3.
968
2.
025
0.
923
2.87
2
6.
223
7.
2703
5.
6776
4.
3639
2.
8208
2.
8032
1.
7219
1.
1142
1.
0963
1.
0786
file: C:\Nmr\2004\PRJL12H.528 expt: transmitter freq.: 400.136900 MHztime domain size: 32768 pointswidth: 8064.57 Hz = 20.154519 ppm = 0.246111 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.134389 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
W NC
O
OC
CD
D
HN
Abbildung F-5. 1H-NMR Spektrum der Verbindung 22. SpinWorks 2.5: Parham PR
PPM 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
4.
999
2.
843
0.
951
0.
941
11
.36
7.
6191
7.
6105
3.
8581
2.
3175
2.
0320
-0.
0037
file: C:\Nmr\test\DATA\Guest\NMR\RPNov18-2005\1\fid expt: <zg30>transmitter freq.: 200.131236 MHztime domain size: 65536 pointswidth: 4132.23 Hz = 20.647608 ppm = 0.063053 Hz/ptnumber of scans: 16
freq. of 0 ppm: 200.131066 MHzprocessed size: 32768 complex pointsLB: 0.000 GB: 0.0000
N+
Abbildung F-6. 1H-NMR Spektrum der Verbindung 23b.
CH3
ClO4

F Spektren 136
SpinWorks 2.5: PARHAM PR V 197 997
PPM 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4
1.
002
3.
402
1.
042
14
.29
10
.99
3.
7606
3.
7540
3.
4691
3.
3094
3.
3024
2.
1684
1.
6214
file: C:\Nmr\2005\Feb\PRFE21H.997 expt: transmitter freq.: 400.138800 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154411 ppm = 0.246111 Hz/pt
N+
CH3
BF4
freq. of 0 ppm: 400.136528 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
number of scans: 16
Abbildung F-7. 1H-NMR Spektrum der Verbindung 23c. SpinWorks 2.5: Parham PrIII
PPM 8.8 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 0.0
5.
997
2.
999
16
.53
3.
9209
3.
9032
3.
8854
3.
1438
2.
1567
2.
1288
2.
1219
2.
1118
2.
1036
2.
0930
2.
0854
2.
0526
2.
0035
file: C:\Nmr\test\DATA\Guest\NMR\1\fid expt: <zg30>transmitter freq.: 400.132471 MHztime domain size: 65536 pointswidth: 8223.68 Hz = 20.552404 ppm = 0.125483 Hz/pt
N+
ClO4
freq. of 0 ppm: 400.130059 MHzprocessed size: 32768 complex pointsLB: 0.300 GB: 0.0000
number of scans: 16
Abbildung F-8. 1H-NMR Spektrum der Verbindung 23d.

F Spektren 137
SpinWorks 2.4: PARHAM PR V 190 442
PPM 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0
4.
997
1.
047
0.
923
4.
653
2.
030
6.
6661
6.
6005
5.
8893
5.
8858
5.
5328
5.
5275
5.
5225
2.
4039
2.
3957
2.
3854
2.
3683
1.
9421
1.
9235
1.
9047
1.
5382
file: C:\Nmr\2005\Mai\PRMY02H.442 expt: transmitter freq.: 400.136900 MHztime domain size: 32768 pointswidth: 8064.57 Hz = 20.154517 ppm = 0.246111 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.134395 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
W CNC
O
OC
H
Abbildung F-9. 1H-NMR Spektrum der Verbindung 24. SpinWorks 2.4: PRV157
PPM 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4
1.
001
4.
961
5.
174
2.
964
3.
698
0.
930
1.
083
1.
172
8.
743
5.
826
file: C:\Nmr\2004\PROK1910.950 expt: transmitter freq.: 400.138500 MHztime domain size: 32768 pointswidth: 8772.05 Hz = 21.922543 ppm = 0.267702 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.135836 MHzprocessed size: 16384 complex pointsLB: 0.535 GB: 0.0000
W CNC
O
O N
H
Abbildung F-10. 1H-NMR Spektrum der Verbindung 25.
F Spektren 138
SpinWorks 2.4: PARHAM PR V 209
PPM 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
4.
985
1.
174
1.
236
19
.39
5.
8898
2.
8350
2.
6413
1.
9785
file: C:\Nmr\2005\März\PRMZ090F.117 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
W CNC
O
OC C
freq. of 0 ppm: 400.134400 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
number of scans: 16
Abbildung F-11. 1H-NMR Spektrum der Verbindung 26. SpinWorks 2.4: PARHAM PR V 207
PPM 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0 -1.0
0.
886
2.
513
4.
406
2.
586
3.
008
4.
991
6.
070
5.
8768
5.
8687
5.
3716
5.
3446
3.
3446
3.
2910
2.
1004
1.
8727
1.
6886
1.
5275
file: C:\Nmr\2005\Feb\PRFE28H.066 expt: transmitter freq.: 400.136900 MHztime domain size: 32768 pointswidth: 8064.78 Hz = 20.155049 ppm = 0.246118 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.134395 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
W CNC
O
OC
H
C
MeO
Abbildung F-12. 1H-NMR Spektrum der Verbindung 27.

F Spektren 139
SpinWorks 2.4: PARHAM PR V 291 017
PPM 14.0 13.0 12.0 11.0 10.0 9.0 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0
4.
994
0.
812
0.
813
0.
953
0.
853
2.
403
10.3
0
14.
2325
14.
1995
8.
0481
8.
0151
5.
8986
3.
0032
2.
4624
1.
9514
1.
9293
1.
9243
1.
9022
1.
8934
1.
8863
1.
8794
1.
8588
1.
7471
1.
7114
1.
7039
file: C:\Nmr\2005\august\PRAG220F.102 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.134395 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
W CN
C
O
O H
C C
H
Abbildung F-13. 1H-NMR Spektrum der Verbindung 28. SpinWorks 2.4: PARHAM PR V 295 033
PPM 8.8 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 0.0
0.
975
0.92
5
1.
016
0.
925
1.
121
1.04
5
6.
231
5.
804
3.
501
3.
038
4.
948
0.82
5
5.
6480
5.
6006
4.
1058
3.
9431
3.
7904
3.
7889
3.
7745
3.
4052
2.
4742
2.
3375
1.
3684
1.
1387
file: C:\Nmr\2005\august\PRAG220F.114 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
W CN
C
O
O N
C C
H
mber of scans: 16
freq. of 0 ppm: 400.134395 MHzprocessed size: 16384 complex pointsLB: 0.000 GB: 0.0000
nu
Abbildung F-14. 1H-NMR Spektrum der Verbindung 29.
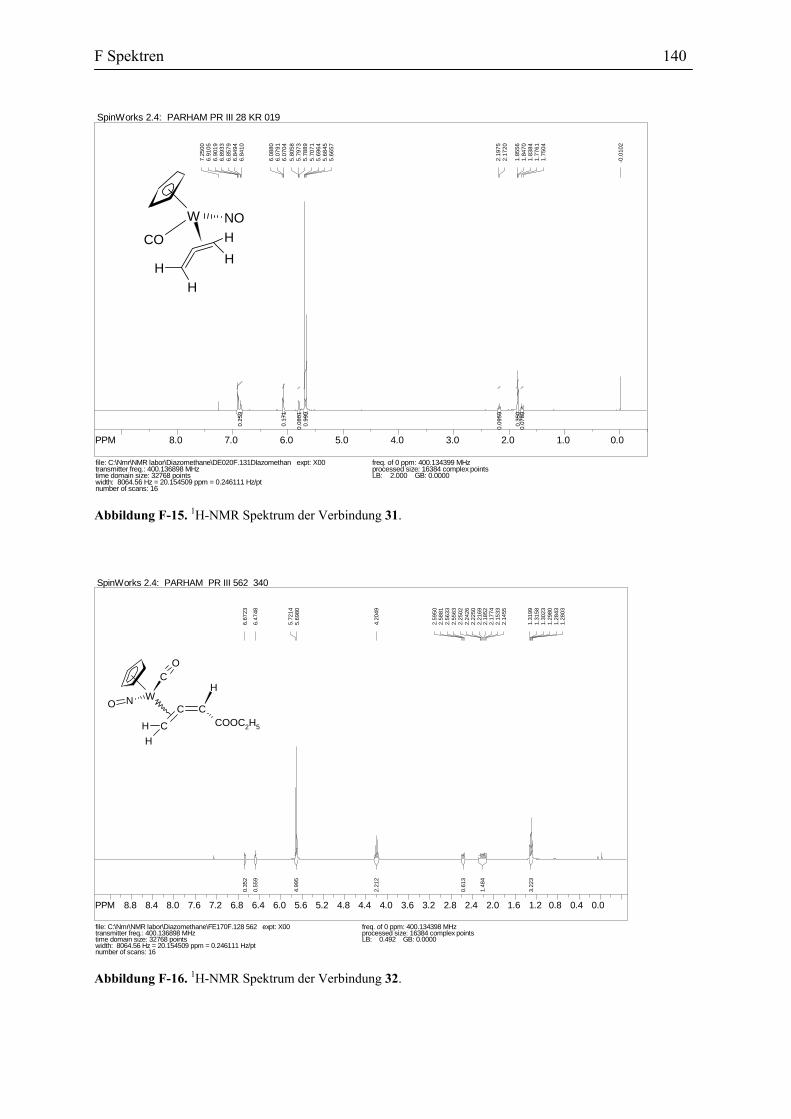
F Spektren 140
SpinWorks 2.4: PARHAM PR III 28 KR 019
PPM 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0
0.
999
0
.080
1
0.
171
0.
259
0.
350
0
.078
9
0.0
959
7.
2500
6.
9105
6.
9019
6.
8933
6.
8579
6.
8494
6.
8410
6.
0880
6.
0791
6.
0704
5.
8058
5.
7973
5.
7889
5.
7071
5.
6964
5.
6845
5.
6657
2.
1975
2.
1720
1.
8556
1.
8470
1.
8384
1.
7761
1.
7504
-0.
0102
file: C:\Nmr\NMR labor\Diazomethane\DE020F.131DIazomethan expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
W
CONO
HH
HH
number of scans: 16
freq. of 0 ppm: 400.134399 MHzprocessed size: 16384 complex pointsLB: 2.000 GB: 0.0000
Abbildung F-15. 1H-NMR Spektrum der Verbindung 31. SpinWorks 2.4: PARHAM PR III 562 340
PPM 8.8 8.4 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 0.0
4.
995
0.
559
0.
352
2.
212
0.
613
1.
484
3.
223
6.
6723
6.
4748
5.
7214
5.
6980
4.
2049
2.
5950
2.
5881
2.
5633
2.
5563
2.
2502
2.
2426
2.
2250
2.
2169
2.
1852
2.
1774
2.
1533
2.
1455
1.
3199
1.
3158
1.
3023
1.
2980
1.
2843
1.
2803
file: C:\Nmr\NMR labor\Diazomethane\FE170F.128 562 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
O
W
C
number of scans: 16
freq. of 0 ppm: 400.134398 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
Abbildung F-16. 1H-NMR Spektrum der Verbindung 32.
NOH
C CCOOC2H5CH
H

F Spektren 141
SpinWorks 2.4: PARHAM PR III 561 339
PPM 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0
0.
930
4.
986
2.
222
1.
100
1.
058
3.
416
7.
4380
7.
2600
5.
7987
4.
2515
4.
2470
4.
2336
4.
2292
2.
0611
2.
0544
1.
7878
1.
7802
1.
3525
1.
3346
1.
3168
-0.
0030
file: C:\Nmr\NMR labor\Diazomethane\FE170F.127 561 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/ptnumber of scans: 16
freq. of 0 ppm: 400.134394 MHzprocessed size: 32768 complex pointsLB: 0.492 GB: 0.0000
W
C
N
O
O C C
H
COOC2H5
CH
H
Abbildung F-17. 1H-NMR Spektrum der Verbindung 33. SpinWorks 2.4: PARHAM PR 39 730
PPM 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
5.
000
0.
986
0.
970
1.
858
12
.82
7.
4333
7.
3699
6.
4812
6.
3228
5.
8107
2.
8172
2.
7698
2.
5724
2.
5270
1.
4755
1.
3781
1.
3022
1.
2663
1.
2304
1.
2130
1.
2058
1.
1772
1.
1685
1.
1318
1.
1204
file: C:\Dokumente und Einstellungen\All Users\Dokumente\Parham Data\Phosphor\NMR\NO14H.730 expt: ERNAtransmitter freq.: 200.133500 MHztime domain size: 32768 pointswidth: 2994.03 Hz = 14.960161 ppm = 0.091371 Hz/ptnumber of scans: 16
freq. of 0 ppm: 200.132344 MHzprocessed size: 16384 complex pointsLB: 0.763 GB: 0.0000
W PN ClO
HH
Abbildung F-18. 1H-NMR Spektrum der Verbindung 11c.

F Spektren 142
SpinWorks 2.4: PARHAM PR 55 403
PPM 8.0 7.6 7.2 6.8 6.4 6.0 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0
1.
000
0.
993
5.
183
1.
005
0.
994
3.
023
9.
938
6.
5288
6.
4821
6.
1767
6.
0738
5.
7515
5.
7488
2.
6027
2.
0921
1.
2291
1.
2132
1.
1968
1.
1783
1.
1601
1.
1166
0.
9625
0.
9324
file: C:\Dokumente und Einstellungen\All Users\Dokumente\Parham Data\Phosphor\NMR\OK010F.132 expt: X00transmitter freq.: 400.136898 MHztime domain size: 32768 pointswidth: 8064.56 Hz = 20.154509 ppm = 0.246111 Hz/pt
WC
PW
C
PO
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
H
H
C
C
H
H
H
H
H
H
NO
NO
freq. of 0 ppm: 400.134405 MHzprocessed size: 16384 complex pointsLB: 0.492 GB: 0.0000
number of scans: 16
Abbildung F-19. 1H-NMR Spektrum der Verbindung 37.

F Spektren 143
2 13C-NMR Spektren
SpinWorks 2.4: PrV149 841 CNMR
PPM 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
111
.270
6
101
.525
5
91.
7172
71.
2985
45.
0818
44.
2463
39.
7268
37.
7920
32.
0847
29.
2489
26.
9216
24.
3562
256
.101
2
15.
8031
e: C:\Nmr\2004\PRSE13C.841 expt: freq. of 0 ppm: 100.614124 MHze: 32768 complex points
B: 0.0000
filtransmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 45458.89 Hz = 451.724940 ppm = 0.693648 Hz/ptumber of scans: 3001
processed sizLB: 11.500 G
n
bbildung F-20. 13C-NMR Spektrum der Verbindung 18. A SpinWorks 2.4: PRV54 SE14C.863
PPM 230.0 210.0 190.0 170.0 150.0 130.0 110.0 90.0 70.0 50.0 30.0 10.0
254
.064
2
112
.408
4 1
11.6
725
101
.506
7 1
00.6
910
93.
2972
71.
4389
48.
4958
47.
9048
42.
1942
39.
6607
37.
9461
33.
0928
29.
3005
26.
6987
26.
0507
24.
6420
24.
3667
24.
1726
22.
0049
file: C:\Nmr\2004\PRSE14C.863 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 45458.89 Hz = 451.724940 ppm = 0.693648 Hz/ptnumber of scans: 3990
freq. of 0 ppm: 100.614123 MHzprocessed size: 32768 complex pointsLB: 1.500 GB: 0.0000
Abbildung F-21. 13C-NMR Spektrum der Verbindung 19.
WOON
HNEt2H
H
WOON
HH
HN

F Spektren 144
SpinWorks 2.4: PV154
PPM
253
.826
9 2
49.8
194
116
.219
0 1
13.2
239
111
.657
5 1
09.5
354
100
.900
0 1
00.0
588
98.
8077
92.
8231
88.
5745
73.
7204
73.
2021
48.
3279
47.
5751
45.
5800
42.
5003
41.
2231
41.
1136
38.
8867
38.
7583
35.
1085
34.
8457
33.
3538
32.
8032
27.
6075
27.
4877
26.
9400
24.
3119
24.
2271
23.
9681
23.
0299
W NC
O
OC
CH
H
HN
21
1
2
3
7
260.0 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
22.
5567
file: C:\Nmr\2004\PRSE20C.894 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 45457.20 Hz = 451.708213 ppm = 0.693622 Hz/ptnumber of scans: 6388
freq. of 0 ppm: 100.614149 MHzprocessed size: 32768 complex pointsLB: 2.000 GB: 0.0000
WO
ON
HH
HN
Abbildung F-22. 13C-NMR Spektrum der Verbindung 20. SpinWorks 2.4: PrV62 13CNMR
PPM
223
.773
0
143
.325
8 1
39.4
994
95.
8234
48.
6475
40.
1116
30.
2679
29.
9525
21.
3450
230.0 210.0 190.0 170.0 150.0 130.0 110.0 90.0 70.0 50.0 30.0 10.0
14.
4029
file: C:\Nmr\2004\PRSE06C.813 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41669.11 Hz = 414.065957 ppm = 0.635820 Hz/ptnumber of scans: 14656
freq. of 0 ppm: 100.614127 MHzprocessed size: 32768 complex pointsLB: 1.272 GB: 0.0000
Abbildung F-23. 13C-NMR Spektrum der Verbindung 21.
F Spektren 145
W NC
O
OC
CD
D
HN
22
SpinWorks 2.4: PRV 109 13 CNMR
PPM 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
223
.849
9
143
.281
4 1
39.5
967
95.
8645
48.
6810
40.
0286
29.
9518
26.
7810
21.
3548
14.
4101
file: C:\Nmr\2004\PRSE06C4.826 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41669.11 Hz = 414.065957 ppm = 0.635820 Hz/ptnumber of scans: 29109
freq. of 0 ppm: 100.614125 MHzprocessed size: 32768 complex pointsLB: 1.272 GB: 0.0000
Abbildung F-24. 13C-NMR Spektrum der Verbindung 22. SpinWorks 2.5: Parham Pr
PPM 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
203
.625
6
131
.069
0 1
30.9
349
123
.587
1
117
.559
1
46.
1747
39.
7938
39.
4908
39.
2807
37.
7851
34.
8690
26.
5561
file: C:\Nmr\test\DATA\Guest\NMR\PRNov18-2005a\2\fid expt: <zgpg30>transmitter freq.: 50.327761 MHztime domain size: 65536 pointswidth: 12019.23 Hz = 238.819105 ppm = 0.183399 Hz/pt
N+
number of scans: 512
freq. of 0 ppm: 50.322984 MHzprocessed size: 32768 complex pointsLB: 0.000 GB: 0.0000
Abbildung F-25. 13C-NMR Spektrum der Verbindung 23b.
CH3
ClO4
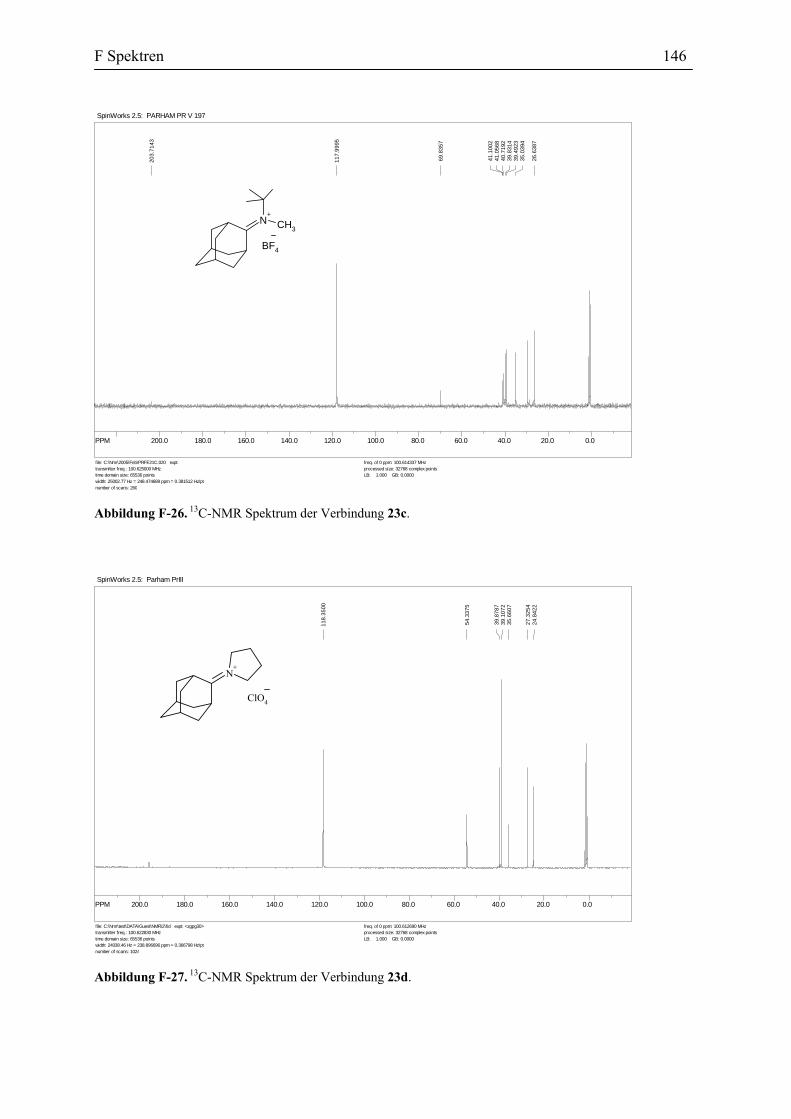
F Spektren 146
SpinWorks 2.5: PARHAM PR V 197
PPM 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
203
.714
3
117
.999
5
69.
8357
41.
1002
41.
0568
40.
7192
39.
8314
39.
4923
35.
0394
26.
6387
file: C:\Nmr\2005\Feb\PRFE21C.020 expt: transmitter freq.: 100.625000 MHztime domain size: 65536 pointswidth: 25002.77 Hz = 248.474689 ppm = 0.381512 Hz/pt
N+
number of scans: 260
freq. of 0 ppm: 100.614337 MHzprocessed size: 32768 complex pointsLB: 1.000 GB: 0.0000
Abbildung F-26. 13C-NMR Spektrum der Verbindung 23c. SpinWorks 2.5: Parham PrIII
PPM 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
118
.350
0
54.
3375
39.
8787
39.
1072
35.
6607
27.
3254
24.
8422
file: C:\Nmr\test\DATA\Guest\NMR\2\fid expt: <zgpg30>transmitter freq.: 100.622830 MHztime domain size: 65536 pointswidth: 24038.46 Hz = 238.896696 ppm = 0.366798 Hz/ptnumber of scans: 1024
freq. of 0 ppm: 100.612690 MHzprocessed size: 32768 complex pointsLB: 1.000 GB: 0.0000
Abbildung F-27. 13C-NMR Spektrum der Verbindung 23d.
CH3
BF4
N+
ClO4
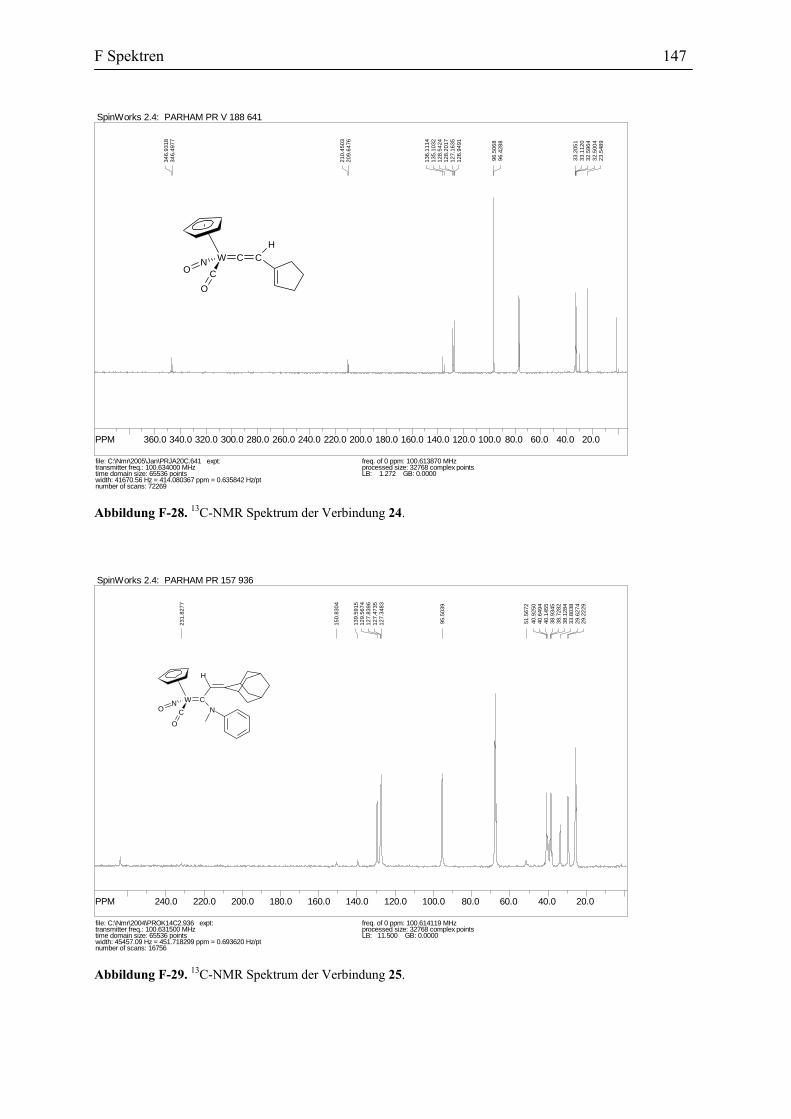
F Spektren 147
SpinWorks 2.4: PARHAM PR V 188 641
PPM 360.0 340.0 320.0 300.0 280.0 260.0 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
346
.931
8 3
46.4
977
210
.450
3 2
09.6
476
136
.111
4 1
35.1
032
128
.542
4 1
28.2
017
127
.163
5 1
26.9
491
96.
5068
96.
4288
33.
2051
33.
1120
32.
5964
32.
5004
23.
5489
file: C:\Nmr\2005\Jan\PRJA20C.641 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41670.56 Hz = 414.080367 ppm = 0.635842 Hz/ptnumber of scans: 72269
freq. of 0 ppm: 100.613870 MHzprocessed size: 32768 complex pointsLB: 1.272 GB: 0.0000
W CNC
O
OC
H
Abbildung F-28. 13C-NMR Spektrum der Verbindung 24. SpinWorks 2.4: PARHAM PR 157 936
PPM 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
231
.827
7
150
.830
4
139
.591
5 1
29.5
674
127
.839
6 1
27.4
735
127
.348
3
95.
5039
51.
5672
40.
9250
40.
6494
40.
1455
38.
9345
38.
7282
38.
1284
33.
8038
29.
6274
29.
2229
file: C:\Nmr\2004\PROK14C2.936 expt: transmitter freq.: 100.631500 MHztime domain size: 65536 pointswidth: 45457.09 Hz = 451.718299 ppm = 0.693620 Hz/ptnumber of scans: 16756
freq. of 0 ppm: 100.614119 MHzprocessed size: 32768 complex pointsLB: 11.500 GB: 0.0000
W CNC
O
O N
H
Abbildung F-29. 13C-NMR Spektrum der Verbindung 25.
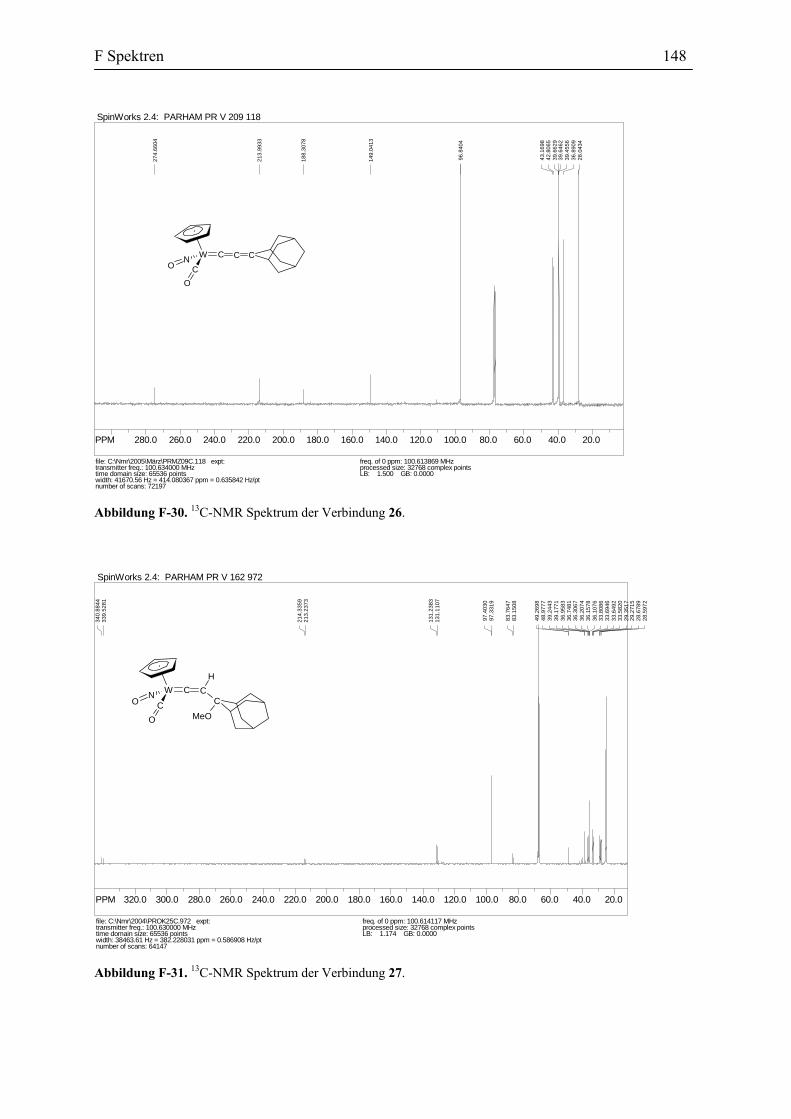
F Spektren 148
SpinWorks 2.4: PARHAM PR V 209 118
PPM 280.0 260.0 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
274
.660
4
213
.993
3
188
.307
8
149
.041
3
96.
8404
43.
1698
42.
8065
39.
6629
39.
6462
39.
4556
36.
8909
28.
0434
file: C:\Nmr\2005\März\PRMZ09C.118 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41670.56 Hz = 414.080367 ppm = 0.635842 Hz/ptnumber of scans: 72197
freq. of 0 ppm: 100.613869 MHzprocessed size: 32768 complex pointsLB: 1.500 GB: 0.0000
W CNC
O
OC C
Abbildung F-30. 13C-NMR Spektrum der Verbindung 26. SpinWorks 2.4: PARHAM PR V 162 972
PPM 320.0 300.0 280.0 260.0 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
340
.884
4 3
39.5
281
214
.335
9 2
13.2
373
131
.238
3 1
31.1
107
97.
4030
97.
3319
83.
7647
83.
1508
49.
2698
48.
9777
39.
2443
39.
1771
36.
9583
36.
7481
36.
3067
36.
2074
36.
1578
36.
1076
33.
8086
33.
6946
33.
6492
33.
5820
29.
3517
29.
2715
28.
6789
28.
5972
file: C:\Nmr\2004\PROK25C.972 expt: transmitter freq.: 100.630000 MHztime domain size: 65536 pointswidth: 38463.61 Hz = 382.228031 ppm = 0.586908 Hz/ptnumber of scans: 64147
freq. of 0 ppm: 100.614117 MHzprocessed size: 32768 complex pointsLB: 1.174 GB: 0.0000
W CNC
O
OC
H
C
MeO
Abbildung F-31. 13C-NMR Spektrum der Verbindung 27.
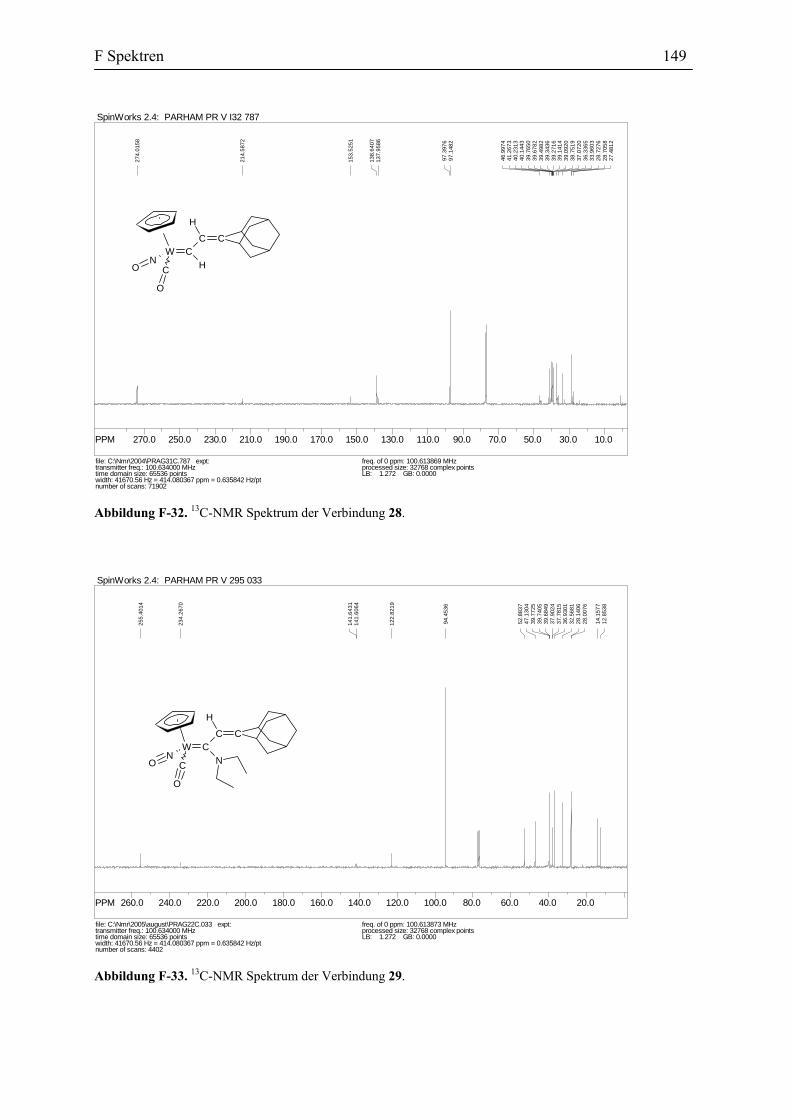
F Spektren 149
SpinWorks 2.4: PARHAM PR V I32 787
PPM 270.0 250.0 230.0 210.0 190.0 170.0 150.0 130.0 110.0 90.0 70.0 50.0 30.0 10.0
274
.015
8
214
.587
2
153
.525
1
138
.640
7 1
37.9
586
97.
3976
97.
1482
46.
9974
41.
2673
40.
2313
40.
1443
39.
7650
39.
6782
39.
4982
39.
3436
39.
2716
39.
1414
39.
0920
38.
7519
37.
0720
36.
3365
33.
9603
28.
7276
28.
7058
27.
4812
file: C:\Nmr\2004\PRAG31C.787 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41670.56 Hz = 414.080367 ppm = 0.635842 Hz/ptnumber of scans: 71902
freq. of 0 ppm: 100.613869 MHzprocessed size: 32768 complex pointsLB: 1.272 GB: 0.0000
W CN
C
O
O H
C C
H
Abbildung F-32. 13C-NMR Spektrum der Verbindung 28. SpinWorks 2.4: PARHAM PR V 295 033
PPM 260.0 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0
255
.401
4
234
.267
0
141
.643
1 1
41.6
064
122
.821
9
94.
4536
52.
8837
47.
1304
39.
7725
39.
7405
39.
6849
37.
9024
37.
7815
36.
9301
32.
5681
28.
1406
28.
0076
14.
1577
12.
8538
file: C:\Nmr\2005\august\PRAG22C.033 expt: transmitter freq.: 100.634000 MHztime domain size: 65536 pointswidth: 41670.56 Hz = 414.080367 ppm = 0.635842 Hz/pt
W CN
C
O
O N
C C
H
number of scans: 4402
freq. of 0 ppm: 100.613873 MHzprocessed size: 32768 complex pointsLB: 1.272 GB: 0.0000
Abbildung F-33. 13C-NMR Spektrum der Verbindung 29.
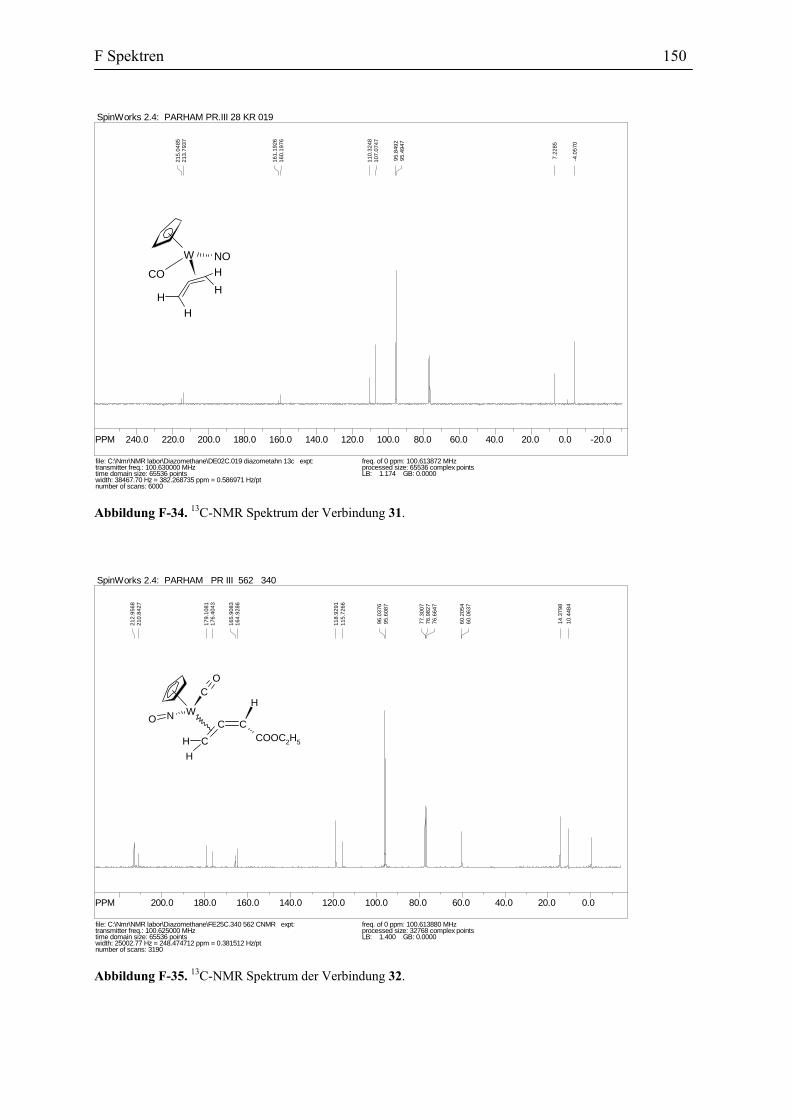
F Spektren 150
SpinWorks 2.4: PARHAM PR.III 28 KR 019
PPM 240.0 220.0 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0 -20.0
215
.048
5 2
13.7
937
161
.192
6 1
60.1
976
110
.324
8 1
07.0
747
95.
8492
95.
4947
7.
2285
-4.
0570
file: C:\Nmr\NMR labor\Diazomethane\DE02C.019 diazometahn 13c expt: transmitter freq.: 100.630000 MHztime domain size: 65536 pointswidth: 38467.70 Hz = 382.268735 ppm = 0.586971 Hz/ptnumber of scans: 6000
freq. of 0 ppm: 100.613872 MHzprocessed size: 65536 complex pointsLB: 1.174 GB: 0.0000
W
CONO
HH
HH
Abbildung F-34. 13C-NMR Spektrum der Verbindung 31. SpinWorks 2.4: PARHAM PR III 562 340
PPM 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
212
.956
8 2
10.8
427
179
.108
1 1
76.4
043
165
.908
3 1
64.9
286
118
.929
1 1
15.7
266
96.
0376
95.
6087
77.
3007
76.
9827
76.
6647
60.
2054
60.
0637
14.
3798
10.
4484
file: C:\Nmr\NMR labor\Diazomethane\FE25C.340 562 CNMR expt: transmitter freq.: 100.625000 MHztime domain size: 65536 pointswidth: 25002.77 Hz = 248.474712 ppm = 0.381512 Hz/ptnumber of scans: 3190
freq. of 0 ppm: 100.613880 MHzprocessed size: 32768 complex pointsLB: 1.400 GB: 0.0000
O
W
C
NO C C
H
COOC2H5CH
H
Abbildung F-35. 13C-NMR Spektrum der Verbindung 32.
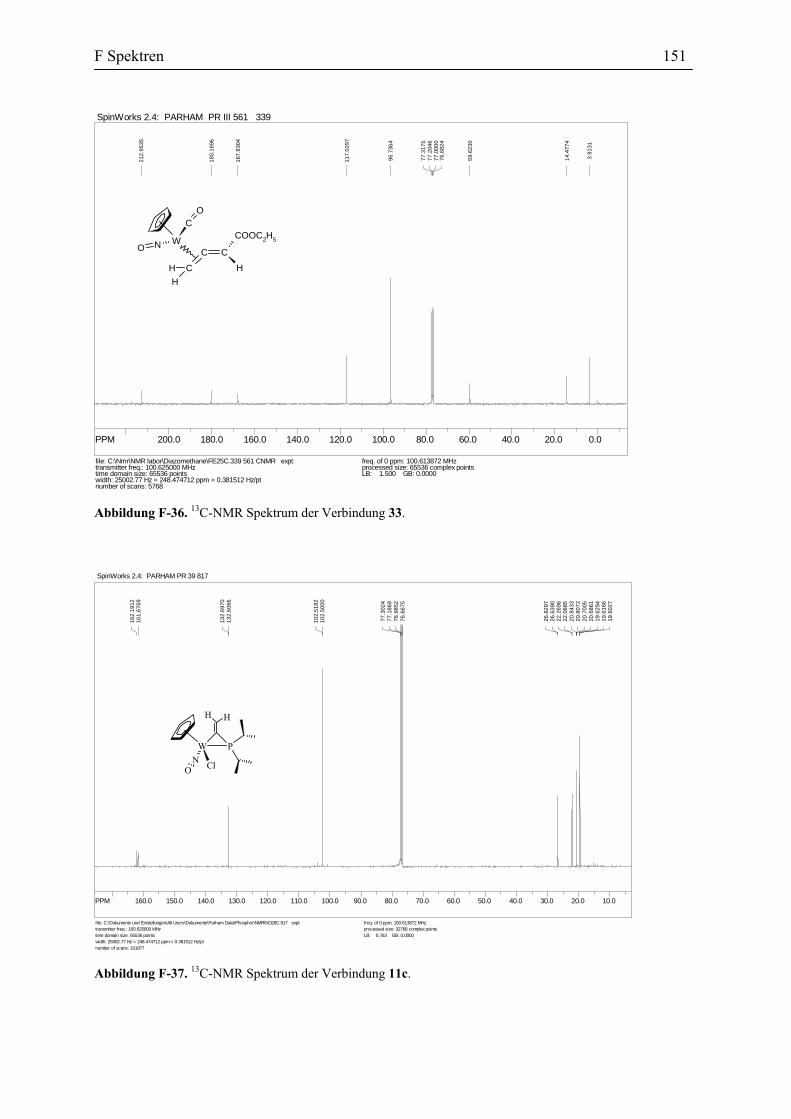
F Spektren 151
SpinWorks 2.4: PARHAM PR III 561 339
PPM 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
212
.953
5
180
.169
6
167
.830
4
117
.029
7
96.
7364
77.
3175
77.
2046
77.
0000
76.
6824
59.
6230
14.
4774
3.
9131
file: C:\Nmr\NMR labor\Diazomethane\FE25C.339 561 CNMR expt: transmitter freq.: 100.625000 MHztime domain size: 65536 pointswidth: 25002.77 Hz = 248.474712 ppm = 0.381512 Hz/ptnumber of scans: 5768
freq. of 0 ppm: 100.613872 MHzprocessed size: 65536 complex pointsLB: 1.500 GB: 0.0000
W
C
N
O
O C C
H
COOC2H5
CH
H
Abbildung F-36. 13C-NMR Spektrum der Verbindung 33. SpinWorks 2.4: PARHAM PR 39 817
PPM 160.0 150.0 140.0 130.0 120.0 110.0 100.0 90.0 80.0 70.0 60.0 50.0 40.0 30.0 20.0 10.0
162
.191
2 1
61.6
769
132
.697
0 1
32.6
066
102
.518
2 1
02.5
000
77.
3024
77.
1869
76.
9852
76.
6675
26.
8297
26.
6390
22.
2696
22.
0885
20.
8433
20.
8072
20.
7005
20.
6861
19.
6294
19.
6166
19.
6027
file: C:\Dokumente und Einstellungen\All Users\Dokumente\Parham Data\Phosphor\NMR\NO26C.817 expt: transmitter freq.: 100.625000 MHztime domain size: 65536 pointswidth: 25002.77 Hz = 248.474712 ppm = 0.381512 Hz/ptnumber of scans: 101877
freq. of 0 ppm: 100.613872 MHzprocessed size: 32768 complex pointsLB: 0.763 GB: 0.0000
W PN ClO
HH
Abbildung F-37. 13C-NMR Spektrum der Verbindung 11c.

F Spektren 152
SpinWorks 2.4: PARHAM PR 55 918
PPM 200.0 180.0 160.0 140.0 120.0 100.0 80.0 60.0 40.0 20.0 0.0
172
.398
1 1
72.2
161
171
.987
6 1
71.8
044
134
.226
9
99.
8665
77.
3062
76.
9887
76.
6712
25.
4785
25.
2519
19.
4423
19.
4209
19.
3974
18.
6654
18.
6471
18.
6301
18.
3117
18.
0150
17.
9933
17.
9685
file: C:\Dokumente und Einstellungen\All Users\Dokumente\Parham Data\Phosphor\NMR\DE06C.918 expt: transmitter freq.: 100.625000 MHztime domain size: 65536 pointswidth: 25002.77 Hz = 248.474712 ppm = 0.381512 Hz/pt
WC
PW
C
PO
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
H
H
C
C
H
H
H
H
H
H
NO
NO
freq. of 0 ppm: 100.613872 MHzprocessed size: 32768 complex pointsLB: 0.763 GB: 0.0000
8number of scans: 4422
Abbildung F-38. 13C-NMR Spektrum der Verbindung 37.
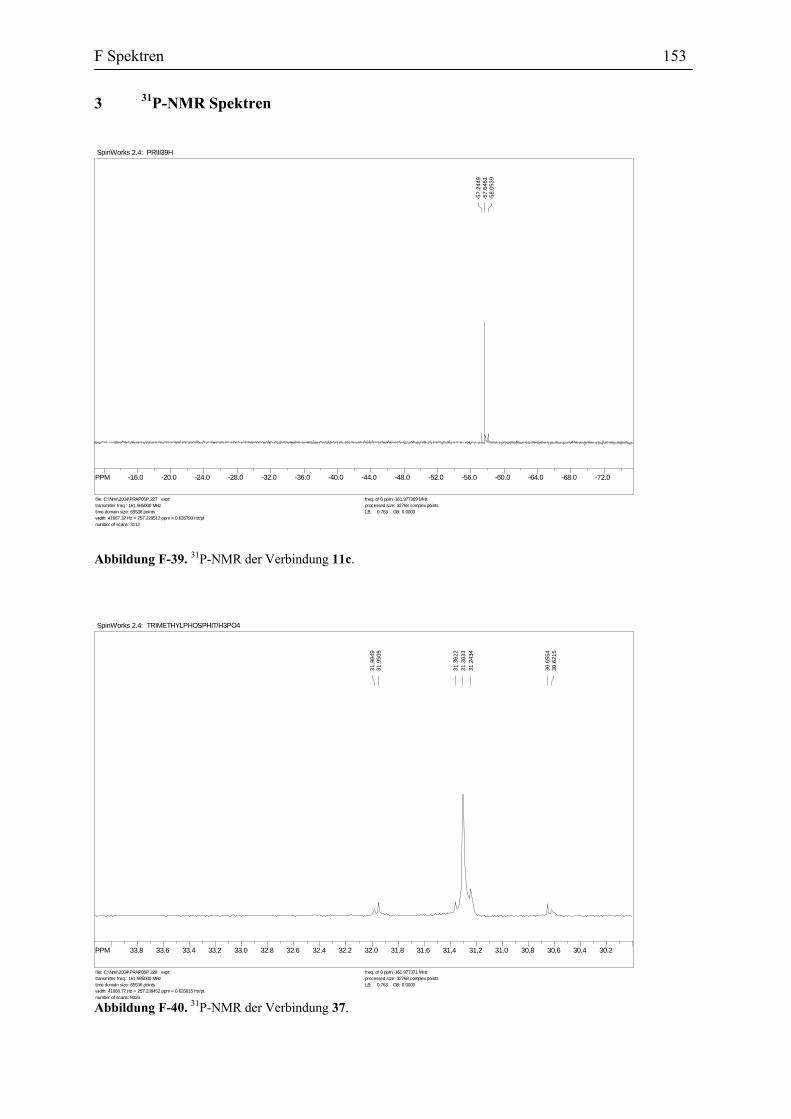
F Spektren 153
3 31P-NMR Spektren
SpinWorks 2.4: PRIII39H
PPM -16.0 -20.0 -24.0 -28.0 -32.0 -36.0 -40.0 -44.0 -48.0 -52.0 -56.0 -60.0 -64.0 -68.0 -72.0 -5
7.24
49 -5
7.64
61 -5
8.05
39file: C:\Nmr\2004\PRAP05P.227 expt: freq. of 0 ppm: 161.977369 MHz
omplex pointstransmitter freq.: 161.985000 MHztime domain size: 65536 pointswidth: 41667.32 Hz = 257.229512 ppm = 0.635793 Hz/pt
mber of scans: 3112
processed size: 32768 cLB: 0.763 GB: 0.0000
9. 31P-NMR der Verbindung 11c.
nu
Abbildung F-3 SpinWorks 2.4: TRIMETHYLPHOSPHIT/H3PO4
M
31.
9849
31.
9508
31.
3622
31.
3033
31.
2434
30.
6554
PP 33.8 33.6 33.4 33.2 33.0 32.8 32.6 32.4 32.2 32.0 31.8 31.6 31.4 31.2 31.0 30.8 30.6 30.4 30.2
30.
6215
file: C:\Nmr\2004\PRAP05P.228 expt: transmitter freq.: 161.985000 MHztime domain size: 65536 pointswidth: 41668.77 Hz = 257.238452 ppm = 0.635815 Hz/ptnumber of scans: 802
freq. of 0 ppm: 161.977371 MHzprocessed size: 32768 complex pointsLB: 0.763 GB: 0.0000
6 Abbildung F-40. 31P-NMR der Verbindung 37.

F Spektren 154
4 IR-Spektren
Abbildung F-41. IR-Spektrum der Verbindung 18.
Abbildung F-42. IR-Spektrum der Verbindung 19.
P arham / P r V 110 /P arh374 K B r-P ress ling
3448
2956
1649
1578
1448
1373
1261
1020
959
820
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
WOON
HH
HN
5
6
78
18
1
P arham / P r V 111 /P a rh375 K B r-P ress ling
3443
3114
2943
2864
2805
1638
1600
1445
1007
817
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
00
Tran
smitt
ance
[%]
WOON
HH
HN
5
6
78
19
F Spektren 155
A
bbildung F-43. IR-Spektrum der Verbindung 20.
bildung F-43. IR-Spektrum der Verbindung 20.
Abbildung F-44. IR-Spektrum der Verbindung 21. Abbildung F-44. IR-Spektrum der Verbindung 21.
P rV 29a A TR-Technik
2841
1952
1571
1440
1371
1332
1293
1227
1183
1110
1087
1059
1001
896
811
610
590
574
559
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
10 0
Tran
smitt
ance
[%]
W NC
O
OC
CH
H
HN
21
1
2
3
7
P arham / P r V 146 /P arh371 K B r-P ressling
3449
3101
2927
2847
1641
1590
1466
1447
1269
1127
1016
977
866
820
594
521
456
500100015002000250030003500W avenumber cm-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
WO
ON
H
HH
N
20
5
678
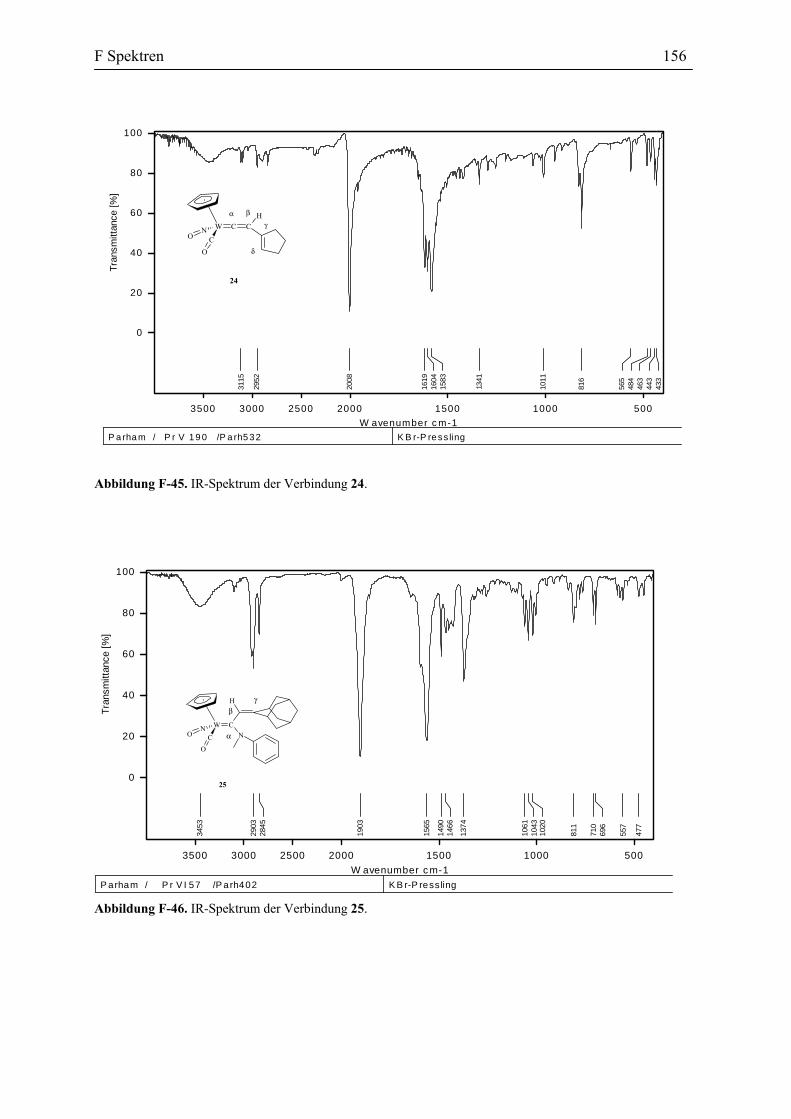
F Spektren 156
Abbildung F-45. IR-Spektrum der Verbindung 24.
Abbildung F-46. IR-Spektrum der Verbindung 25.
P a rha m / P r V 1 9 0 /P a rh5 32 K B r-P re ss ling
3115
2952
2008
1619
1604
1583
1341
1011
816
565
484
463
443
433
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
W CNC
O
OC
H
24
α β
γ
δ
80
60
40
P arham / P r V I 57 /P arh402 K B r-P ressling
3453
2903
2845
1903
1565
1490
1466
1374
1061
1043
1020
811
710
696
557
477
500100015002000250030003500W avenumber cm-1
0
20
100
Tran
smitt
ance
[%]
W CNC
O
O N
H γβ
α
25

F Spektren 157
Abbildung F-47. IR-Spektrum der Verbindung 26.
Abbildung F-48. IR-Spektrum der Verbindung 27.
P arham / P r V 207 /P arh486 K B r-P ressling
3422
2925
2853
2007
1641
1600
1469
1112
1078
1061
1048
1004
916
815
762
617
574
483
450
500100015002000250030003500W avenumber cm-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
P arham / P r V 209 /P arh488 K B r-P ressling
3423
2924
2848
1971
1906
1609
1446
1331
1084
948
869
815
570
533
447
500100015002000250030003500W avenumber cm-1
20
40
60
80
100
Tran
smitt
ance
[%]
W CNC
O
OC C
26
W CNC
O
OC
H
C
MeO
27

F Spektren 158
W CN
C
O
O N
C C
H
α
β γ
29
Abbildung F-49. IR-Spektrum der Verbindung 28.
bbildung F-50. IR-Spektrum der Verbindung 29.
A
Parham / Pr V132 /Parh373 KBr-Pressling
3441
2910
2851
1975
1592
1450
1343
1261
1097
815
500100015002000250030003500W avenumber cm-1
30
40
50
60
70
80
90
100
Tran
smitt
ance
[%]
P arham / PR V 295 /Parh555 Film, in C H2C l2 gel”st, wieder verdampft
2906
2849
1895
1575
1489
1431
1379
1351
1268
1202
1149
1098
1046
1023
948
802
736
703
573
472
500100015002000250030003500W avenumber cm-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
W CN
C
O
O H
C C
H β
γα
28

F Spektren 159
Abbildung F-52. IR-Spektrum der Verbindung 32.
Abbildung F-51. IR-Spektrum der Verbindung 31.
P arham / P r I V 612 /P a rh413 K B r-P ress ling
3455
3104
3083
2962
2006
1717
1681
1598
1463
1420
1369
1339
1248
1174
1099
1006
841
827
565
548
513
436
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
100
Tran
smitt
ance
[%]
P a rham / P r III 28 K r /P arh105 K B r-P ressling
3438
3107
1989
1599
1420
1073
1005
978
877
816
570
548
449
500100015002000250030003500W avenumber c m-1
0
20
40
60
80
100Tr
ansm
ittan
ce [%
]
W
C
N
O
O C C
H
HCH
H
31
1
2
3
W
C
N
O
O C C
H
COOC2H5CH
H
32

F Spektren 160
Abbildung F-53. IR-Spektrum der Verbindung 33.
Abbildung F-54. IR-Spektrum der Verbindung 11c.
P arham/ P r39/P arh39 K B r-P ellet
500100015002000250030003500W avenumber cm-1
0
10
20
30
40
50
60
70
80
Tran
smitt
ance
[%]
P a rha m / P r III 5 6 1 /P a rh1 7 4 In C H 2 C l2
3087
2991
2030
1704
1672
1625
1423
1361
1314
1157
1082
1008
964
903
857
824
734
578
563
504
449
412
5 0 010 001 50 02 00 02 50 03 0003 500W a ven um b er c m -1
0
2 0
4 0
6 0
8 0
1 00
Tran
smitt
ance
[%]
W
C
N
O
O C C
H
COOC2H5
CH
H
33
W P
NClO
HH
11c

F Spektren 161
bbildung F-55. IR-Spektrum der Verbindung 37.
A
Parham/ Pr39/Parh39 KBr-Pellet
500100015002000250030003500Wavenumber cm-1
0
10
20
30
40
50
60
70
80Tr
ansm
ittan
ce [%
]
WC
PW
C
PO
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
H
H
C
C
H
H
H
H
H
H
NO
NO
37
α
β

G Literaturverzeichnis 162
G Literaturverzeichnis
[1] Zeise, W. C. Ann. Phys. 1827, 9, 932.
[2] Schutzenberger, M. P. Annalen 1868, 15, 100.
[3] (a) Miller, S. A.; Tebboth, J. A.; Tremaine, J. F. J. Chem. Soc. 1952, 632. (b) Kealy, T.
J.; Pauson, P. J. Nature (London) 1951, 168, 1039.
[4] (a) Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. J. Am. Chem.
Soc. 1952, 74, 2125. (b) Fischer, E. O.; Pfab, W.Z. Naturforsch. 1952, 7B, 377. (c)
Wilkinson, G J. Am. Chem. Soc. 1952, 74, 6148.
[5] Fischer, E.O.; Maasböl, A. Angew. Chem. 1964, 14, 645.
[6] (a) Fischer, F.; Tropsch, DRP 411416, 1922. (b) Maitlis, P. M. J. Organomet. Chem.
2004, 689, 4366.
[7] (a) Roelen, O. Angew. Chem. 1948, 60, 62. (b) Jones, W.D.; Bergman, R. G. J. Am.
Chem. Soc. 1979, 101, 5447.
[8] (a) Ziegler, K. Adv. Organomet. Chem. 1968, 6, 1. (b) Goodall, B. L. J. Chem. Educ.
1986, 63, 191.(c) Natta, G. Scientific American 1961, 205, 33. (d) Ziegler, K.
Angew. Chem. 1955, 67, 541.
] Smid, J.; Hafner, W.; Jira, R.; Sedlmeier, J.; Sieber, R.; Rüttinger, R.; Kojer, H.
Angew. Chem. 1959, 71, 176.
[10] Forster, D. J. Am. Chem. Soc. 1975, 97, 951.
[11] Parshall, G. W. Homogenous Catalysis; J. Wiley & Sons, New York, 1980.
[12] Mond, L.; Langer, C.; Quincke, F. J. Chem. Soc. Dalton Trans. 1890, 57, 749.
[13] Knowles, W. S. Acc. Chem. Res. 1983, 16, 106.
[14] (a) Fischer, E. O. Pure Appl. Chem. 1970, 42, 407. (b) Fischer, E. O. Pure Appl.
Chem. 1972, 44, 353; (c) Fischer, E. O. Adv. Organomet. Chem. 1976, 14, 1. (d)
Cardin, D. J.; Cetinkaya, B.; Lappert; M. F. Chem. Rev. 1972; 72; 545.
[15] Taylor, T. E.; Hall, M. B. J. Am. Soc. Chem. 1984, 106, 1576.
[16] Mills, O. S.; Redhouse, A. D. J. Chem. Soc. A 1986, 642.
[17] Semmelhack, M. F.; Tamura, R. J. Am. Soc. Chem. 1983, 105, 4099.
[18] Chisholm, M. H.; Clark, H. C. Acc. Chem. Res. 1973, 6, 202.
[19] (a) Barluenga, J.; Fernandez-Rodriguez, M. A.; Aguilar, E. J. Organomet. Chem.
2005, 690, 539. (b) Barluenga, J.; Santamaria, J.; Tomas, M. Chem. Rev. 2004, 104,
2259. (c) Barluenga, J.; Fananas, F. J. Tetrahedron 2000, 56, 4597. (d) Herndon, J. W.
Tetrahedron 2000, 56, 1257.
[20] Wienand, A.; Reissig, H. U. Organometallics 1990, 9, 3133.
;
Holzkamp, E.; Breil, H.; Martin, H.
[9

G Literaturverzeichnis 163
[21 (a) Dötz, K. H. Angew.] Chem., Int. Ed. Engl. 1982, 21, 929. (b) Brookhart, M.;
, 411. (c) Aumann, R. Angew. Chem., Int. Ed.
[23] 0, 435. (b) Schilling, B. E. R;
[24]
94, 13, 1044.
,
I. Chem. Rev. 1991, 91,197. (c) Ouzzine, K.; Le Bozec, H.;
ometallics 1997, 16, 3902.
[28]
Organometallics
[29] een, M.; Welch, A. J. J. Am. Chem. Soc. 1980, 102,
94,
rga,
, N.; Toupet,
ios, I.; Jimenez
1997, 119, 6529. (f)
bezyuk, N. S.
[34]
[35]
[36] ulmer, A. Organometallics 2000, 19, 5281.
Studebaker, W. B. Chem. Rev. 1987, 87
Engl. 1988, 27, 1456.
[22] Schrock, R. R. Acc. Chem. Res. 1979, 12, 98.
(a) Werner, H. Nachr. Chem. Technol. Lab. 1992, 4
Hoffmann. R.; Lichtenberger, D. L. J. Am. Chem. Soc. 1979, 101, 585.
Schillin, B. E. R.; Hoffmann, R.; Lichtenberger, D. L. J. Am. Chem. Soc. 1979,
101,585.
[25] Ipaktschi, J.; Müller, B. G.; Glaum, R. Organometallics 19
[26] (a) Venkatesan, K.; Fox, T.; Schmalle, H. W.; Berke, H. Eur. J. Inorg. Chem. 2005, 5
901. (b) Bruce, M.
Dixneuf, P. H. J. Organomet. Chem. 1986, 317, C25. (d) Beckhaus, R.; Oster, J.;
Ganter, B.; Englert, U. Organ
[27] King, R. B.; Saran, M. S. J. Chem.Soc., Chem. Commun. 1972, 1053.
(a) Boland-Lussier, B. E.; Churchill, M. R.; Hugues, R. P.; Rheingold, A. L.
Organometallics 1982, 1, 628. (b) Bly, R. S.; Raya, M.; Bly, R. K.
1992, 11, 1220.
Baker, P. K.; Barber, G. K.; Gr
7811.
[30] Davidson, A.; Selegue, J. P. J. Am. Chem. Soc. 1978, 100, 7763.
[31] Gamasa, M. P.; Gimeno, J.; Lastra, E.; Martin, B. M.; Anillo, A.; Tiripicchio,.
Organometallics 1992, 11, 1373.
[32] (a) Wakatsuki, Y.; Koga, N.; Yamazaki, H.; Morokuma, K. J. Am. Chem. Soc. 19
116, 8105. (c) Jimenez Tenorio, M. A.; Jimenez Tenorio, M.; Puerta, M. C.; Vale
P. Organometallics 1997, 16, 5528. (d) Touchard, D.; Haquette, P.; Pirio
L.; Dixneuf, P. H. Organometallics 1993, 12, 3132. (e) de los R
Tenorio, M.; Puerta, M. C.; Valerga, P. J. Am. Chem. Soc.
Antonova, A. B.; Kolobova, N. E.; Petrovsky, P. V.; Lokshin, B. V.; O
J. Organomet. Chem. 1977, 137, 55.
[33] Stegmann, R.; Neuhaus, A.; Frenking, G. J. Am. Chem. Soc. 1993, 115, 11930.
Kostic, N. M.; Fenske, R. F. Organometallics 1982, I , 974.
Ipaktschi, J.; Uhlig, S.; Dulmer, A.; Organometallics 2001, 20, 4840.
Ipaktschi, J.; Klotzbach, T.; D

G Literaturverzeichnis 164
[37] Ipaktschi, J.; Mohsseni-Ala, J; Dülmer, A.; Loschen, C.; Frenking, G.,
Organometallics 2005, 24, 977.
(a) Kremzow, D.; Seid[38] el, G.; Lehmann, C. W.; Fürstner, A. Chem. Eur. J. 2005, 11,
znar, F. Org. Lett. 2005, 7, 1235. (c)
.
;
. Organometallics, 2002, 21, 2736. (g) de Meijere, A.; Schirmer, H.;
2005, 117,
[39] . C. G.
ge,
em. Commun. (Cambridge,
6.(f) Diver S.
[40] .
, 2124. (c) Grubbs, R. H.; Chang, S. Tetrahedron 1998, 54, 4413. (d)
g.
1999, 3, 211.
, 4592. (b)
c) Jernelius, J. A.; Schrock, R. R.; Hoveyda, A. H.
[42]
[43] . J.
oc. 2005, 127, 6877. (c) Gillingham, D. G.;
. (d)
, A. H.; Schrock, R. R. J.
1833. (b) Barluenga, J.; Faňanás-Mastral, M.; A
Diver, S. T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317. (d) Barluenga, J.;
Santamarýa, T. Chem. Rev. 2004, 104, 2259. (e) Zhang, J.; Gunnoe, T. B
Organometallics, 2003, 22, 2291. (f) Aumann, R.; Fu, X.; Vogt, D.; Fröhlich, R.
Kataeva, O
Duetsch, M. Angew. Chem. 2000, 112, 4124.(h) Katz,T.J. Angew. Chem.
3070.
(a) Connon, J. S.; Blechert, S. Angew. Chem. 2003, 115, 1944. (b) Soderberg, B
Coord. Chem. Rev. 2004, 248, 1085.(c) Schrock, R. R. Chem. Commun. (Cambrid
U.K.) 2005, 22, 2773. (d) Fuerstner, A.; Davies, P. W. Ch
U.K.) 2005, 18, 2307. (e) Wallace, D. J. Angew. Chem. 2005, 117, 194
T.; Giessert, A. J. Chem. Rev. 2004, 104, 1317.
(a) Fürstner, A. Angew. Chem. 2000, 112, 3140. (b) Schuster, M.; Blechert, S. Angew
Chem. 1997, 109
Phillips, J. A.; Abell, D. Aldrichimica Acta 1999, 32, 75. (e) Wright, D. L. Curr. Or
Chem.
[41] (a) Schrock, R. R.; Hoveyda, A. H. Angew. Chem. Int. Ed. 2003, 42
Schrock, R. R. J. Molec. Catal A 2004, 213, 21. (f) Sinha, A.; Schrock, R. R.
Organometallics 2004, 23, 1643. (
Tetrahedron 2004, 60, 7345.
(a) Despagnet-Ayoub, E.; Grubbs, R. H. Organometallics 2005, 24, 338.(b) Grubbs,
R. H. Tetrahedron 2004, 60, 7117. (c) Rutenberg, M.; Scherman, O. A.; Grubbs, R. H;
Jiang, W.; Garfunkel, E.; Bao, Z. J. Am. Chem. Soc. 2004, 126, 4062.
(a) Sattely, E. S.; Cortez, G. A.; Moebius, D. C.; Schrock, R. R.; Hoveyda, A. H
Am. Chem. Soc. 2005, 127, 8526. (b) Van Veldhuizen, J. J.; Campbell, J. E.; Guidici,
R. E.; Hoveyda, A. H. J. Am. Chem. S
Kataoka, O.; Garber, S. B.; Hoveyda, A. H. J. Am. Chem. Soc. 2004, 126, 12288
Dolman, S. J.; Hultzsch, K. C.; Pezet, F.; Teng, X.; Hoveyda
Am. Chem. Soc., 2004, 126, 10945.
[44] Hermes, A. R.; Girolami, G. S. Organometallics 1988, 7, 394.

G Literaturverzeichnis 165
[45] (a) Ipaktschi, J.; Mirzaei, F.; Demuth-Eberle, G. J.; Beck, J.; Serafin, M
Organometallics 1997, 16, 3965.
.
(b) Greenhough, J. T.; Legzdins; P.; Martin, D. T.;
shi, S.
ang, Y.
ler-
des,
[46] , P.; Bi, S.; Sung, H. H. Y.; Williams, I. D.; Lin, Z.; Jia, G. Organometallics
[47]
y. University Science Books; Mill
[48]
[49] ins, P.; Lundmark, P. J.; Riesen, A.; Einstein, F. W. B.
[50] a,
s 2004, 23, 3763. (c) Yih, K. H.; Lee, G. H.; Huang, S.
S.; Penner-
[51]
[52] 37.
[53] . Soc. 1980, 102, 422. (b) Sanford, M. S.; Ulman,
Trotter, J. Inorg.Chem. 1979, 18, 3268. (c) Kondo, H.; Yamaguchi, Y.; Nagashima,
H.; Chem. Commun. 2000, 1075. (d) Liu, G.; Beetstra, D. J.; Meetsma, A.; Hessen, B.
Organometallics 2004, 23, 3914. (e) Matsushima, Y.; Onitsuka, K.; Takaha
Organometallics 2004, 23, 3763. (f) Yih, K. H.; Lee, G. H.; Huang, S. L.; W
Organometallics 2002, 21, 5767. (g) van Staveren, D. R.; Weyhermüller, T.; Metz
Nolte, N. Organometallics 2000, 19, 3730. (h) Ascenso, J. R.; Dias, R. A.; Fernan
J. A.; Martins A. M.; Rodrigues S. S. Inorg. Chim. Acta 2003, 356, 279.
(a) Xue
2004, 23, 4735. (b) Sasabe, H.; Nakanishi, S.; Takata,T.; Inorg.Chem. Commun. 2002,
5, 177. (c) Cadierno, V.; Crochet, P.; Diez, J.; Garcia-Garrido, S. E.; Gimeno, J.
Organometallics 2003, 22, 5226.
(a) Collman, J. P., Hegedus, L. S.; Norton, J. R.; Finke, G., R. Principles and
Applications of Organotransition Metal Chemistr
Valley, CA, 1987. (b) Mann, B. E., Adv. Organomet. Chem. 1974, 12, 135.
Ward, D.Y.; Villanueva., L. A.; Allred, G. D.; Payne, S. C.; Semones, M. A.;
Liebeskind, L. S. Organometallics 1995, 14, 4132.
Dryden, N. H.; Legzd
Organometallics 1993, 12, 2085.
Als Beispiel für aktuellere Arbeiten siehe: (a) Liu, G.; Beetstra, D. J.; Meetsm
A.;Hessen, B. Organometallics 2004, 23, 3914. (b) Matsushima, Y.; Onitsuka, K.;
Takahashi, S. Organometallic
L.; Wang, Y. Organometallics 2002, 21, 5767. (d) Cedeno, L. D.; Weitz, E.
Organometallics 2003, 22, 2652. (e) Suzuki, T.; Okada,G.; Hioki, Y.; Fujimoto, H.
Organometallics 2003, 22, 3649. (f) Clot, E.; Eisenstein, O.; Weng, T.
Hahn, J.; Caulton, K. G. J.Am. Chem. Soc. 2004, 126, 9079.
Ariafard, A.; Lin, Z.; Organometllics 2005; 24, 2241.
(a) Frohnapfel, D. S.; White, P. S.; Templeton, J. L. Organometallics 1997, 16, 37
(b) Morales, D.; Navarro Clemente, M. E.; Pérez, J.; Riera, L.; Riera, V.
Organometallics 2003, 22, 4124.
(a) Katz, T. J.; Lee, S.J. J. Am. Chem
M.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 749.

G Literaturverzeichnis 166
[54] (a) Mayr, A.; Dorries, A. M.; Rheingold, A. L.; Geib, S. J., Organometall
964. (b) Alvarez, C.; Pacreau, A.; Parlier, A.; Rudler, H.; Daran, J. C. Organometallic
1987, 6, 1057.
(a) Gervasio, G.; Marabello, D.; Sappa, E.; Secco, A J. Organomet. Chem. 2005, 690,
3755. (b) Drexler, M.; Haas, T.; Yu, S.; Beckmann, H.; Weibert, B.; Fischer, H. J.
Organomet. Chem. 2005, 690, 3700. (c) Venancio, A.; Guedes da Silva, M.; Guedes;
M.; Frausto da Silva, J. F.; Pombeiro, A. J. L. Organometallics 2005, 24, 4654. (d)
Nishibayashi, Y.; Imajima, H.; Onodera, G.; Uemura, S. Organometallics 2005, 24,
4106. (e) Ammal, S. C.; Yoshikai, N.; Inada, Y.; Nishibayashi, Y.; Nakam
ics 1990, 9,
s
[55]
ura, E. J.
rt,
2005, 358, 1645. (i) Diez, J.; Gamasa, M. P.;
ni, N.;
683. (b)
ngania, K.; Fabrizi de Biani, F.;
ling, J.
hr, W.; Martin-Alvarez, J. M.; Hampel, F.; Gladysz,
. (f)
S.;
04.
2004, 23, 5893. (j) Zheng,
23, 5896.
[58] M.; Inada, Y.; Hidai, M.; Uemura, S. J. Org. Chem.
Am. Chem. Soc. 2005, 127, 9428. (f) Pavlik, S.; Mereiter, K.; Puchberger, M.;
Kirchner, K. Organometallics 2005, 24, 3561. (g) Cadierno, V.; Diez, J.; Garcia-
Garrido, S. E.; Gimeno, J. Organometallics 2005, 24, 3111. (h) Szesni, N.; Weibe
B.; Fischer, H. Inorg. Chim. Acta
Gimeno, J.; Lastra, E.; Villar, A. Organometallics 2005, 24, 1410. (j) Mantova
Brugnati, M.; Gonsalvi, L.; Grigiotti, E.; Laschi, F.; Marvelli, L.; Peruzzini, M.;
Reginato, G.; Rossi, R.; Zanello, P. Organometallics 2005, 24, 405.
[56] (a) Ceccon, A.; Santi, S.; Orian, L.; Bisello, A. Coord. Chem. Rev. 2004, 248,
Skibar, W.; Kopacka, H.; Wurst, K.; Salzmann, C.; O
Zanello, P.; Bildstein, B. Organometallics 2004, 23, 1024. (c) Stahl, J.; Boh
C.; Bauer, E. B.; Peters, T. B.; Mo
J. A. Angew. Chem. 2002, 114, 1951. (d) Le Stang, S.; Paul, F.; Lapinte, C.
Organometallics 2000, 19, 1035. (e) Bruce, M. I. Coord. Chem. Rev. 1997, 166, 91
Paul, F.; Lapinte, C. Coord. Chem. Rev. 1998, 178-180, 427. (g) Swager, T. M. Acc.
Chem. Res. 1998, 31, 201. (d) Creager, S.; Yu, C. J.; Bamdad, C.; O’Connor,
MacLean, T.; Lam, E.;Chong, Y.; Olsen, G. T.; Luo, J.; Gozin, M.; Kayyem, J. F. J.
Am. Chem.Soc. 1999, 121, 1059. (h) Henning, T.; Salama, F. Science 1998, 282, 22
(i) Owen, G. R.; Hampel, F.; Gladysz, J. A. Organometallics
Q.; Hampel, F.; Gladysz, J. A. Organometallics 2004,
[57] Nishibayashi, Y.; Inada, Y.; Hidai, M.; Uemura, S. J. Am. Chem. Soc. 2002, 124,
7900.
(a) Nishibayashi, Y.; Yoshikawa,
2004, 69, 3408. (b) Trost, B. M.; Frederiksen, M. U.; Rudd, M.T. Angew. Chem. 2005,
117, 6788.

G Literaturverzeichnis 167
[59] (a) Bassetti, M.; Centola, F.; Semeril, D.; Bruneau, C.; Dixneuf, P. H.
Organometallics, 2003, 22, 4459. (b) Fürstner, A.; Liebl, M.; Lehmann, C. W.;
Picquet, M.; Kunz, R.; Bruneau, C.; Touchard, D.; Dixneuf, P. H. Chem. Eur. J. 2000,
[60]
[61]
[62]
[63] dierno,
.
Carreno, E.
[64]
[65]
[66]
[67]
(b)
S.; Geoffroy, G. L.; White, C. A.; Rheingold, A. L. J. Am. Chem. Soc. 1993,
[69]
llics 1995, 14, 3335.
6, 1847. (c) Harlow, K. J.; Hill, A. F.; Wilton-Ely, J. D. E. T. J.Chem. Soc., Dalton
Trans. 1999, 285. (d) Jafarpour, L.; Huang, J.; Stevens,E. D.; Nolan, S. P.
Organometallics 1999, 18, 3760.
Fischer, E. O.; Kalder A.; Köhler, F. H.; Huttner, G. Angew. Chem. 1976, 88,683.
Berke, H. Angew. Chem. 1976, 88, 684.
Selegue, J. P. Organometallics 1982, 1, 217.
(a) Matsuzaka, H.; Takagi, Y.; Hidai, M. Organometallics 1994, 13, 13. (b) Ca
V.; Gamasa, M. P.; Gimeno, J.; Lastra, E.; Borge, J.;Garcıa-Granda, S.
Organometallics 1994, 13, 745. (c) Selegue, J. P. J. Am. Chem. Soc. 1983, 105, 5921
(d) Young, B. A.; Logan, S. L. Organometallics 1991, 10, 1972. (e) Pilette, D.;
Ouzzine, K.; Le Bozec, H.; Dixneuf, P. H.; Rickard, C. E. F.; Roper, W. R.
Organometallics 1992, 11, 809. (f) Cadierno, V.; Gamasa, M. P.; Gimeno, J.;
Gonzalez-Cueva, M.; Lastra, E.; Borge, J.; Garcıa-Granda, S.; Perez-
Organometallics 1996, 15, 2137. (g) Touchard, D.; Guesmi, S.; Bouchaib, M.;
Haquette, P.; Daridor, A.; Dixneuf, P. H. Organometallics 1996, 15, 2579.
Berke, H. Z. Naturforsch. 1980, 35b, 86.
Venancio, A. I. F.; Guedes da Silva, M. F. C.; Martins, L. M. D. R. S.; Frausto da
Silva, J. J. R.; Pombeiro, A. J. L.; Organometallics 2005; 24; 4654.
(a) Berke, H. Chem. Ber. 1980, 113, 1370.(b) Fischer, H.; Roth, G.; Reindl, D.; Troll,
C. J. Organomet. Chem.1993, 454, 133. (c) Fischer, H.; Reindl, D.; Roth, G. Z.
Naturforsch. 1994, 49b, 1207. (d) Aumann, R.; Jasper, B.; Fröhlich, R.
Organometallics 1995, 14, 3173.
(a) Für theoretische Berechnungen siehe: Berke, H.; Huttner, H.G.; von Seyerl, J. Z.
Naturforsch. B 1981, 36, 1277. (b) Marrone, A.; Re, N. Organometallics 2002, 21,
3562.
[68] a) Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1993, 115, 8130.
Yi, C.
115, 3806.
Ipaktschi, J.; Demuth-Eberle, G. J.; Mirzaei, F. Mueller, B.G.; Beck, J.; Serafin, M.
Organometa

G Literaturverzeichnis 168
[70] (a) Cadierno, V.; Gamasa, M. P.; Gimeno, J.; Gonzalez-Bernardo, C.; P
E.; Garcia-Granda, S. Organometallics 2001, 20, 5177. (b) Bianchini, C.; Innoce
P.; Meli, A.; Peruzzini, M.; Zanobini, F.; Zanello, P. Organometallics 1990, 9, 2514.
(c) Ipaktschi, J.; Mirzaei, F.; Mueller, B. G.; Beck, J.; Serafin, M. J.Organomet. Che
1996, 526(2), 363. (d) Ipaktschi, J.; Mirzaei, F.; Demuth-Eberle, G. J.; Bec
Serafin, M. Organometallics 1997
erez-Carreno,
nti,
m.
k, J.;
, 16, 3965.
.;
ics 1992, 11, 3232.(c) Lundquist, E. G.;
nstein, O.; Caulton, K. G. J. Am. Chem.
etallics 1991,
[72]
[73]
[74]
[75]
[76]
nth., Coll. Vol.4, 1963, 250. (b) Searle, N.
[78]
[80]
[81] tures,
oms from F-synthesis (program SHELXL-93:
ure for all non-hydrogen atoms. Atomic scattering factors from: Int. Tables for
[71] (a) Zhuang, J. M.; Sutton, D. Organometallics 1991, 10, 1516. (b) Pu, J.; Peng, T. S
Arif, A. M.; Gladysz, J. A. Organometall
Folting, K.; Streib, W.E.; Huffman, J. C.; Eise
Soc. 1990, 112, 855. (d) Reger, D. L.; Coleman, C. J.; McElligott, P. J. J.
Orgonomet.Chem. 1979, 9, 1191. (e) Zhuang, J. M.; Sutton, D. Organom
10, 1516. (f) Chacon, S. T.; Chisholm, M. H.; Folting, K.; Huffman, J. C.; Hampden
Smith, M. J. Organometallics 1991, 10, 3722.
Fantazier, R. M.; Poutama, M. L. J. Am. Chem. Soc. 1968, 90, 5490.
Bowden, F. L.; Giles, R., Coord. Chem. Rev. 1976, 20, (1), 81. 1H-NMR Angaben für Hauptdiastereomer.
Pacchioni, G;. Bagus, P. S. Inorg. Chem. 1992, 31, 4391.
Leonard, N. J.; Paukstelis; J. V. J. Org. Chem., 1963, 28, 3021.
[77] (a) De Boer, Th. J.; Backer, H. J., Org. Sy
E., Org. Synth., Coll. Vol.4, 1963, 424.
White, W. A.; Weingarten, H. J. Org. Chem, 1967, 32, 213.
[79] Holmes, B.; Sporikou, C. N. Org. Synth. 1983, Coll. Vol. VIII, 606.
Bjorgo, J.; Boyd, D. R. J. Chem. Soc, Perkin Transactions 2, 1973, 1575.
G.M. Sheldrick, SHELXS-86; Program for the Solution of Crystal Struc
Universität Göttingen, 1986); further at
G.M. Sheldrick, SHELXL-93; Program for Crystal Structure Refinement, Universität
Göttingen, 1993); structure refinement by the anisotropic full-matrix least-squares
proced
Crystallography, Volume C 1992, Ed. A.J.C. Wilson, Kluwer Academic Publishers,
Dordrecht.

169
Hierm t versichere ich an Eides statt, dass ich die vorliegende Arbeit selbstständig verfasst
verwendeten Hilfsmitteln angegeben habe.
, den ……………… ………………………….
Unterschrift
i
und die
Giessen

Formelverzeichnis
(CO)5WPh
OMe
1
[M] C
OR'
R[M] C
OR'
R[M] C
OR'
R
+
+_ _
2a 2b 2c
Mo
COOC
COCl
CN
CN
5
Fe+
Ph3PCO OMe
Ph
4
(CO)4FePh
OMe
3
W C CH
C6H5N
CO
O
8
Mo
CN
CNPh3P
Ph3PCl
6
W C CH
NC
O
O
7
W
NC
O
O
CO
9
H
W C
NCO
O
C
H
10
W C CNC
O
OH
Li
+-
10a
W C CD
DNC
O
O
10c
W P
NClO
HH
R
R
11a-c
(CH3)3C-
Ph
(CH3)2CH-
R
11a
11b
11c
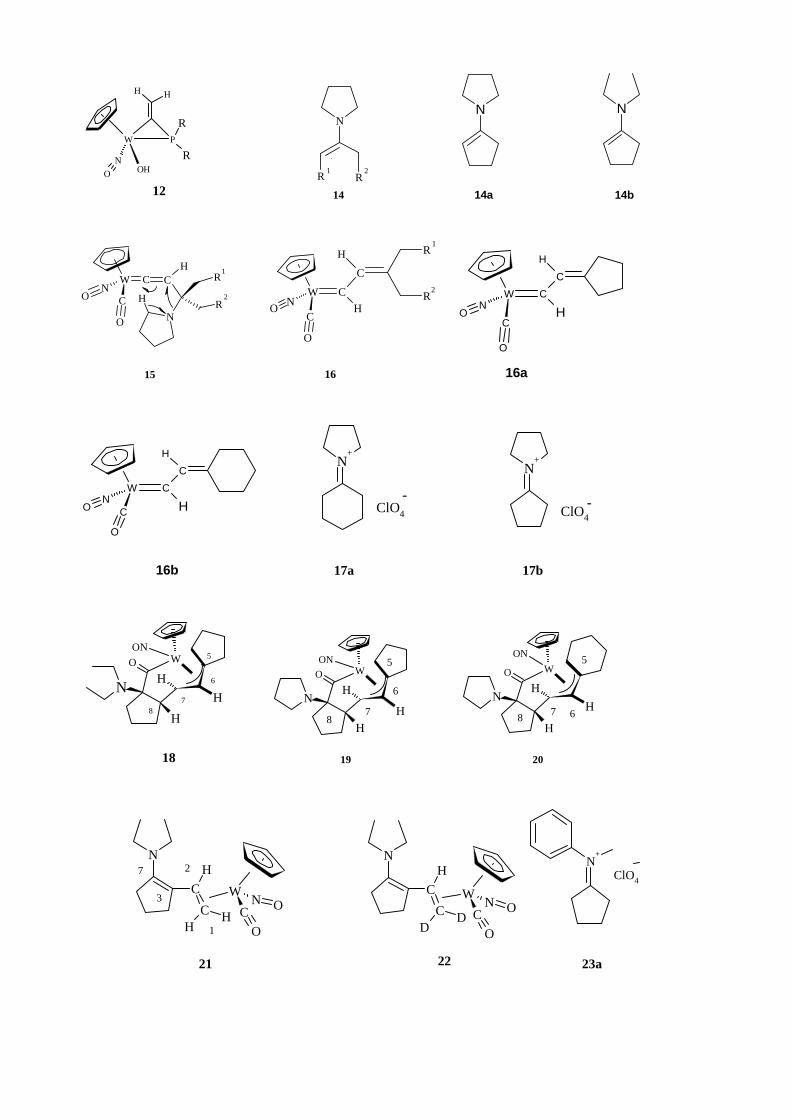
W P
NOHO
HH
R
R
12
N
RR
14
21
N
14a
N
14b
W C
CN
OC
O
H
N
H R
R
15
1
2
W C
CNO
C
O
H
H
R
R
16
1
2
W C
C
NO
C
O
H
H
16a
W C
CN
O
C
O
H
H
16b
N+
ClO4
-
17a
N+
ClO4-
17b
WOON
HH
HN
18
5
6
78
WOON
HH
HN
19
5
6
78
WO
ON
H
HH
N
20
5
678
W NC
O
OC
CH
H
HN
21
1
2
3
7
W NC
O
OC
CD
D
HN
22
N+
ClO4
23a

N+
CH3
ClO4
23b
N+
CH3
BF4
23c
N+
ClO4
23d
W CNC
O
OC C
26
W CNC
O
OC
H
24
α β
γ
δ
W CNC
O
O N
H
25
α
βγ
W CNC
O
OC
H
C
MeO
27
W CN
C
O
O H
C C
H
α
β
γ
28
W CN
C
O
O N
C C
H
α
β γ
29
W C CH
CH3N
CO
O
30
W
C
N
O
O C C
H
HCH
H
31
1
2
3
W
C
N
O
O C C
H
COOC2H5CH
H
32
W
C
N
O
O C C
H
COOC2H5
CH
H
33
PRR
ClPh
(CH3)3C-b
c (CH3)2CH-
R
34a-c
a
W
WO
NO
NOP
PHH
HH
α
β
35
HH
ON
P
W
O
WNO P
HH
36
WC
PW
C
PO
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
H
H
C
C
H
H
H
H
H
H
NO
NO
37
α
β
N
38
Top Related