
7. Emissionen 7.1 Kohlendioxid - uni-magdeburg.de · erforderlich, um die Selektivität,...
25
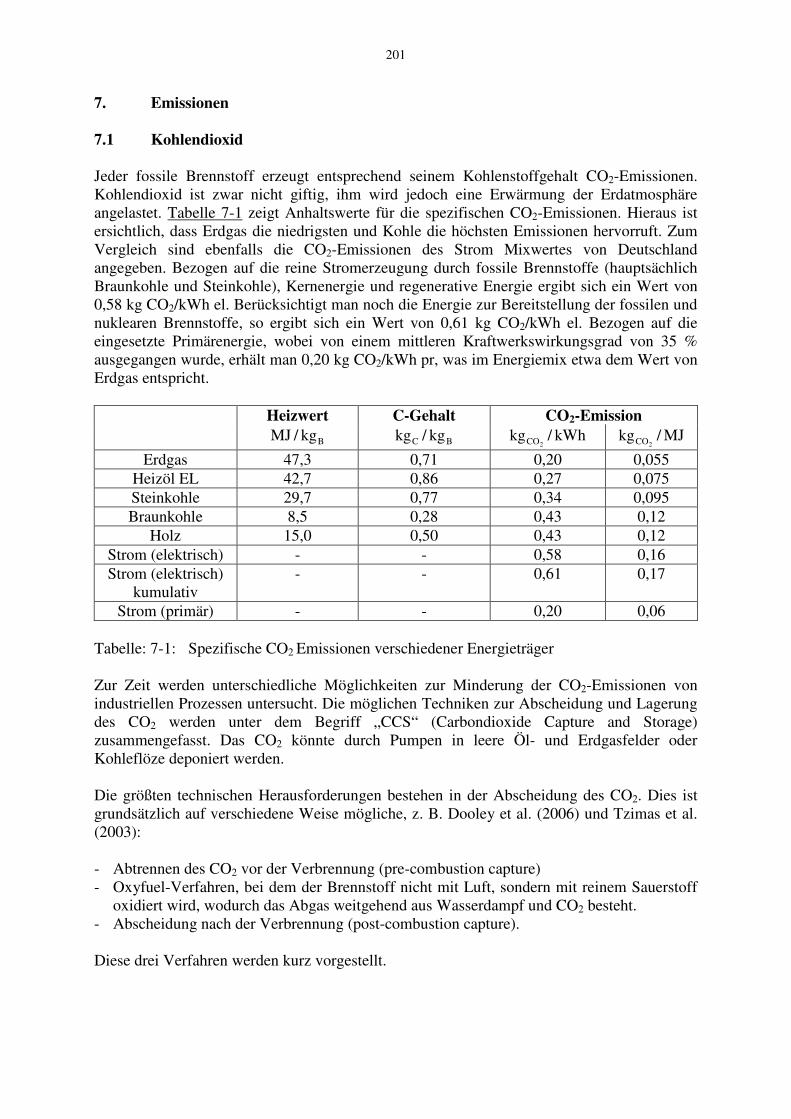
201 7. Emissionen 7.1 Kohlendioxid Jeder fossile Brennstoff erzeugt entsprechend seinem Kohlenstoffgehalt CO 2 -Emissionen. Kohlendioxid ist zwar nicht giftig, ihm wird jedoch eine Erwärmung der Erdatmosphäre angelastet. Tabelle 7-1 zeigt Anhaltswerte für die spezifischen CO 2 -Emissionen. Hieraus ist ersichtlich, dass Erdgas die niedrigsten und Kohle die höchsten Emissionen hervorruft. Zum Vergleich sind ebenfalls die CO 2 -Emissionen des Strom Mixwertes von Deutschland angegeben. Bezogen auf die reine Stromerzeugung durch fossile Brennstoffe (hauptsächlich Braunkohle und Steinkohle), Kernenergie und regenerative Energie ergibt sich ein Wert von 0,58 kg CO 2 /kWh el. Berücksichtigt man noch die Energie zur Bereitstellung der fossilen und nuklearen Brennstoffe, so ergibt sich ein Wert von 0,61 kg CO 2 /kWh el. Bezogen auf die eingesetzte Primärenergie, wobei von einem mittleren Kraftwerkswirkungsgrad von 35 % ausgegangen wurde, erhält man 0,20 kg CO 2 /kWh pr, was im Energiemix etwa dem Wert von Erdgas entspricht. Heizwert C-Gehalt CO 2 -Emission B kg / MJ B C kg / kg kWh / kg 2 CO MJ / kg 2 CO Erdgas 47,3 0,71 0,20 0,055 Heizöl EL 42,7 0,86 0,27 0,075 Steinkohle 29,7 0,77 0,34 0,095 Braunkohle 8,5 0,28 0,43 0,12 Holz 15,0 0,50 0,43 0,12 Strom (elektrisch) - - 0,58 0,16 Strom (elektrisch) kumulativ - - 0,61 0,17 Strom (primär) - - 0,20 0,06 Tabelle: 7-1: Spezifische CO 2 Emissionen verschiedener Energieträger Zur Zeit werden unterschiedliche Möglichkeiten zur Minderung der CO 2 -Emissionen von industriellen Prozessen untersucht. Die möglichen Techniken zur Abscheidung und Lagerung des CO 2 werden unter dem Begriff „CCS“ (Carbondioxide Capture and Storage) zusammengefasst. Das CO 2 könnte durch Pumpen in leere Öl- und Erdgasfelder oder Kohleflöze deponiert werden. Die größten technischen Herausforderungen bestehen in der Abscheidung des CO 2 . Dies ist grundsätzlich auf verschiedene Weise mögliche, z. B. Dooley et al. (2006) und Tzimas et al. (2003): - Abtrennen des CO 2 vor der Verbrennung (pre-combustion capture) - Oxyfuel-Verfahren, bei dem der Brennstoff nicht mit Luft, sondern mit reinem Sauerstoff oxidiert wird, wodurch das Abgas weitgehend aus Wasserdampf und CO 2 besteht. - Abscheidung nach der Verbrennung (post-combustion capture). Diese drei Verfahren werden kurz vorgestellt.
Transcript of 7. Emissionen 7.1 Kohlendioxid - uni-magdeburg.de · erforderlich, um die Selektivität,...
201
7. Emissionen
7.1 Kohlendioxid
Jeder fossile Brennstoff erzeugt entsprechend seinem Kohlenstoffgehalt CO2-Emissionen. Kohlendioxid ist zwar nicht giftig, ihm wird jedoch eine Erwärmung der Erdatmosphäre angelastet. Tabelle 7-1 zeigt Anhaltswerte für die spezifischen CO2-Emissionen. Hieraus ist ersichtlich, dass Erdgas die niedrigsten und Kohle die höchsten Emissionen hervorruft. Zum Vergleich sind ebenfalls die CO2-Emissionen des Strom Mixwertes von Deutschland angegeben. Bezogen auf die reine Stromerzeugung durch fossile Brennstoffe (hauptsächlich Braunkohle und Steinkohle), Kernenergie und regenerative Energie ergibt sich ein Wert von 0,58 kg CO2/kWh el. Berücksichtigt man noch die Energie zur Bereitstellung der fossilen und nuklearen Brennstoffe, so ergibt sich ein Wert von 0,61 kg CO2/kWh el. Bezogen auf die eingesetzte Primärenergie, wobei von einem mittleren Kraftwerkswirkungsgrad von 35 % ausgegangen wurde, erhält man 0,20 kg CO2/kWh pr, was im Energiemix etwa dem Wert von Erdgas entspricht.
Heizwert C-Gehalt CO2-Emission
Bkg/MJ BC kg/kg kWh/kg
2CO MJ/kg2CO
Erdgas 47,3 0,71 0,20 0,055 Heizöl EL 42,7 0,86 0,27 0,075 Steinkohle 29,7 0,77 0,34 0,095 Braunkohle 8,5 0,28 0,43 0,12
Holz 15,0 0,50 0,43 0,12 Strom (elektrisch) - - 0,58 0,16 Strom (elektrisch)
kumulativ - - 0,61 0,17
Strom (primär) - - 0,20 0,06 Tabelle: 7-1: Spezifische CO2 Emissionen verschiedener Energieträger Zur Zeit werden unterschiedliche Möglichkeiten zur Minderung der CO2-Emissionen von industriellen Prozessen untersucht. Die möglichen Techniken zur Abscheidung und Lagerung des CO2 werden unter dem Begriff „CCS“ (Carbondioxide Capture and Storage) zusammengefasst. Das CO2 könnte durch Pumpen in leere Öl- und Erdgasfelder oder Kohleflöze deponiert werden. Die größten technischen Herausforderungen bestehen in der Abscheidung des CO2. Dies ist grundsätzlich auf verschiedene Weise mögliche, z. B. Dooley et al. (2006) und Tzimas et al. (2003): - Abtrennen des CO2 vor der Verbrennung (pre-combustion capture) - Oxyfuel-Verfahren, bei dem der Brennstoff nicht mit Luft, sondern mit reinem Sauerstoff
oxidiert wird, wodurch das Abgas weitgehend aus Wasserdampf und CO2 besteht. - Abscheidung nach der Verbrennung (post-combustion capture). Diese drei Verfahren werden kurz vorgestellt.

202
Pre-combustion Verfahren Bei dem pre-combustion Verfahren wird der Brennstoff in einem ersten Schritt vergast, so dass ein Synthesegas aus Kohlenmonoxid und Wasserstoff entsteht. Die Energie für die endotherme Vergasung wird durch eine Teilverbrennung des fossilen Brennstoffs erzeugt. In einem anschließenden katalytischen Reaktor („shift converter“) wird das Kohlenmonoxid mit Wasserdampf entsprechend CO + H2O → CO2 + H2 (homogene Wassergasreaktion) zu CO2 oxidiert, wobei weiterer Wasserstoff gebildet wird. Das Synthesegas bestehend nun aus CO2 und H2 wird getrennt. Das CO2 wird deponiert und das H2 als Brennstoff verwendet. Durch den Einsatz von Wasserstoff anstatt wie bisher von fossilen Brennstoffen müssen die Industrieprozesse umgestellt oder angepasst werden, da sich andere Flammenverläufe, Strömungsverhältnisse und Temperaturfelder ergeben. Oxyfuel-Verfahren Beim Oxyfuel-Verfahren wird der Brennstoff mit reinem Sauerstoff verbrannt. Das Verbrennungsgas besteht dadurch aus CO2 und H2O. Das Abgas wird gekühlt, so dass der Wasserdampf auskondensiert. Die Separierung des CO2 aus dem Abgas ist dadurch zwar relativ einfach, jedoch muss die Luft zuvor zerlegt werden. Da der Stickstoffanteil fehlt, würden sich bei der Verbrennung sehr hohe Temperaturen und kleine Volumenströme ergeben. Daher muss ein erheblicher Teil des Abgases in die Verbrennungszone zurückgeführt werden, so dass ein hoher Kreislaufstrom aufrecht erhalten werden muss. Zur Beschreibung solcher Verfahren sei z. B. auf Anheden et al. (2005) und Anderson et al. (2003) verwiesen. Post-Combustion Verfahren Bei den Post-Combustion Verfahren wird das CO2 nach dem Verbrennungsprozess mittels einer End-of-Pipe Maßnahme abgetrennt. Der Vorteil dieses Verfahrens liegt darin, dass ein Einsatz hinter verschiedensten Industriepozessen möglich ist, ohne diese stofflich zu beeinflussen. Allerdings ist die CO2 Abscheidung relativ aufwändig. Man unterscheidet drei verschiedene Arten der CO2-Abscheidung - Absorption - Adsorption - Membrantrennung. Absorptionsverfahren Absorption in flüssigen Lösungsmitteln ist eine industriell erprobte CO2-Abtrenntechnik, mit der hohe Reinheiten und Abtrenngrade erreicht werden, wie beispielsweise bei Spasova et al. (2004), Draxler et al. (2004) und Rolker et al. (2006) beschrieben ist. Aktuelle Forschungsprojekte haben u. a. das Ziel, dieses Verfahren für verschiedene Industriebereiche weiterzuentwickeln und insbesondere die Kosten zu senken. Bisher sind nicht nur sehr hohe Investitionen erforderlich, sondern es entstehen auch hohe Betriebskosten durch Verbrauch an Absorptionsmittel (Degradation) und für Energie (Dampf) zur Regeneration des Lösemittels. Bei den Verfahren mit physikalischer Absorption wird das CO2 in Lösemitteln wie „Selexol“ (Dimethylether des Polyethylenglykols) oder „Rectisol“ (kaltes Methanol) aufgenommen. Dabei ist ein hoher Partialdruck des CO2 erforderlich.

203
Bei mittleren und niedrigen CO2-Partialdrücken werden chemische Absorptionsverfahren eingesetzt. Dabei werden basische Lösungsmittel eingesetzt, meist Amine, wie Ethanolamin und Monoethanolamin (MEA), eingesetzt. Die verschiedenen Waschmittel auf Aminbasis sind in der Regel toxisch, karzinogen, explosiv und somit umweltgefährdend. Als Waschmittel kommen auch Lösungen mit Alkalikarbonaten wie Kaliumkarbonat (Pottasche) und Natriumkarbonat in Frage. Das CO2 wird entsprechend CO2 + K2 CO3 + H2O → 2 KHCO3 CO2 + Na2CO3 + H2O → 2 NaHCO3 unter Bildung von Hydrogenkarbonaten abgeschieden. Da das CO2 hierbei in einem Feststoff gebunden ist, ergibt sich eine einfache Deponierung. Adsorptionsverfahren Bei den Adsorptionsverfahren wird CO2 selektiv an porösen Feststoffen mit großer Oberfläche gebunden, z. B. an Zeolithen, Aluminium- oder Silicagelen oder aktiviertem Kohlenstoff [Gauer et al. (2004) und Siriwardane et. Al. (2001)]. Der Adsorber wird über Druck- und Temperaturänderungen im Reaktionsbehälter regeneriert. Der große Vorteil der physikalischen Adsorption im Vergleich zur chemischen Absorption sind der einfache und energieeffiziente Betrieb und sowie die Regeneration. Nachteilig ist die begrenzte Verfügbarkeit von CO2-selektiven Feststoffen. Membran Verfahren Bei den Membranverfahren passiert der Abgasstrom eine Membran, die das CO2 vom restlichen Gas abtrennt. Dabei können organische und anorganische Materialien eingesetzt werden, z. B. organische Polymere, Palladium, Zeolithe. Bisher sind die Selektivität der Membranen und die Beständigkeit gegen aggressive Substanzen sowie Schwefelverbindungen und Spurenelemente jedoch noch nicht ausreichend, z. B. Siriwardane et al. (2001). Bis zu einer Anwendung im industriellen Maßstab ist weitere Forschung und Entwicklung erforderlich, um die Selektivität, Permeabilität und Dauerhaftigkeit zur verbessern. Mit Membranverfahren lassen sich keine hohen Abtrennraten erzielen, so dass Mehrfach-trennungen erforderlich sind.

204
7.2 Stickoxide
Die Stickoxidbildung lässt sich nach der Stickstoffquelle in drei Mechanismen unterteilen: - Thermisches NO - Prompt NO - Brennstoff NO. 7.2.1 Thermisches NO
Das Thermische NO wird nach Zeldovich, der diesen Mechanismus 1946 erstmals postulierte, aus dem molekularen Stickstoff der Verbrennungsluft nach drei Reaktionen gebildet. Zuerst reagiert Stickstoff mit atomarem Sauerstoff gemäß NNOON2 +→+ . (7-1)
Der atomare Stickstoff reagiert weiter mit O2 und OH gemäß ONOON 2 +→+ (7-2)
HNOOHN +→+ . (7-3) Für die NO-Bildung gilt entsprechend den drei Reaktionsgleichungen
OHNIIIONIINOINO x~x~kx~x~kx~x~k
dt
x~d22
⋅⋅+⋅⋅+⋅⋅= (7-4)
und für die Änderung des atomaren Stickstoffs
OHNIIIONIINOIN x~x~kx~x~kx~x~k
dt
x~d22
⋅⋅−⋅⋅−⋅⋅= . (7-5)
Die Reaktionskoeffizienten betragen nach Warnatz et al. 1996:
( ) ( )
⋅
⋅
⋅−⋅⋅=
−
skmol
m
TR
molkJ318exp108.1k
3111
I (7-6)
( ) ( )
⋅
⋅
⋅−⋅⋅=
−
skmol
m
TR
molkJ27exp100.9k
316
II (7-7)
( )
⋅⋅=
skmol
m108.2k
310
III . (7-8)
Der Reaktionskoeffizient kI der ersten Reaktion hat wegen der hohen thermischen Stabilität der N2-Dreifachbindung und demzufolge hohen Aktivierungsenergie den kleinsten Wert. Auf Grund der daher relativ schnellen Weiterreaktion der Stickstoffatome gemäß den Gleichungen (7-2) und (7-3) kann deren Konzentration als quasistationär angesehen werden. Mit

205
0dt/x~d N = folgt aus den beiden Gleichungen (7-4) und (7-5) für die NO-Bildung der
einfachere Zusammenhang
2NOI
NO x~x~k2dt
x~d⋅⋅⋅= . (7-9)
Zur Berechnung der NO-Bildung werden nach der obigen Gleichung die N2- und die O-Konzentration benötigt. Die N2-Konzentration ist aus der Verbrennungsgaszusammensetzung bekannt. Der atomare Sauerstoff wird im wesentlichen durch die drei Gleichungen OOHOH 2 +→+ (7-10)
OHOHH 2 +→+ (7-11)
HOHHOH 22 +→+ (7-12)
gebildet. Die Reaktionsgeschwindigkeiten betragen (Warnatz et al.1996)
⋅⋅=
skmol
m
mol T R
kJ3.70exp100.2k
311
O-H 2 (7-13)
⋅
⋅⋅−⋅⋅⋅=− skmol
m
molTR
kJ3.26expT101.5k
367.21
OHH (7-14)
und
⋅
⋅⋅−⋅⋅⋅=− skmol
m
molTR
kJ8.13expT100.1k
36.15
HOH 2. (7-15)
Bei Temperaturen oberhalb 1300 °C kann man in guter Näherung ein Gleichgewicht zwischen den obigen drei Gleichungen annehmen. Die Gleichgewichtsbeziehung lautet dann entsprechend Abschnitt 2.3.2 nach Gl. (2-36)
2O
O5 x~
x~)T(K 2= . (7-16)
Die Konzentration des atomaren Sauerstoffs berechnet sich dann nach
2/1
5
OO K
x~x~ 2
= (7-17)
mit der Gleichgewichtszahl K5 nach Tabelle 2-6. Für die NO-Bildung folgt aus Gl. (7-9)
.x~x~Kk2dt
x~d22 N
2/1
O2/1
51NO ⋅⋅⋅⋅= − (7-18)
Die Gleichgewichtszahl hat entsprechend Tabelle 2-6 eine sehr hohe Reaktionsenthalpie. In Verbindung mit der hohen Aktivierungsenergie des Reaktionskoeffizienten nach Gl. (7-6) ergibt sich somit eine überaus starke Temperaturabhängigkeit der NO-Bildung. Daher läuft
206
diese Reaktion erst bei ausreichend hohen Temperaturen schnell ab, weshalb dieser Mechanismus Thermische NO-Bildung genannt wird.
1,E-03
1,E-04
1,E-05
1,E-06
1,E-07
1,E-080,7 0,8 0,9 1 1,1 1,2 1,3 1,4
Luftzahl λ
Ko
nze
ntr
atio
n O
1800 C°
1600 C°1500 C°1400 C°1300 C°1200 C°
ϑG
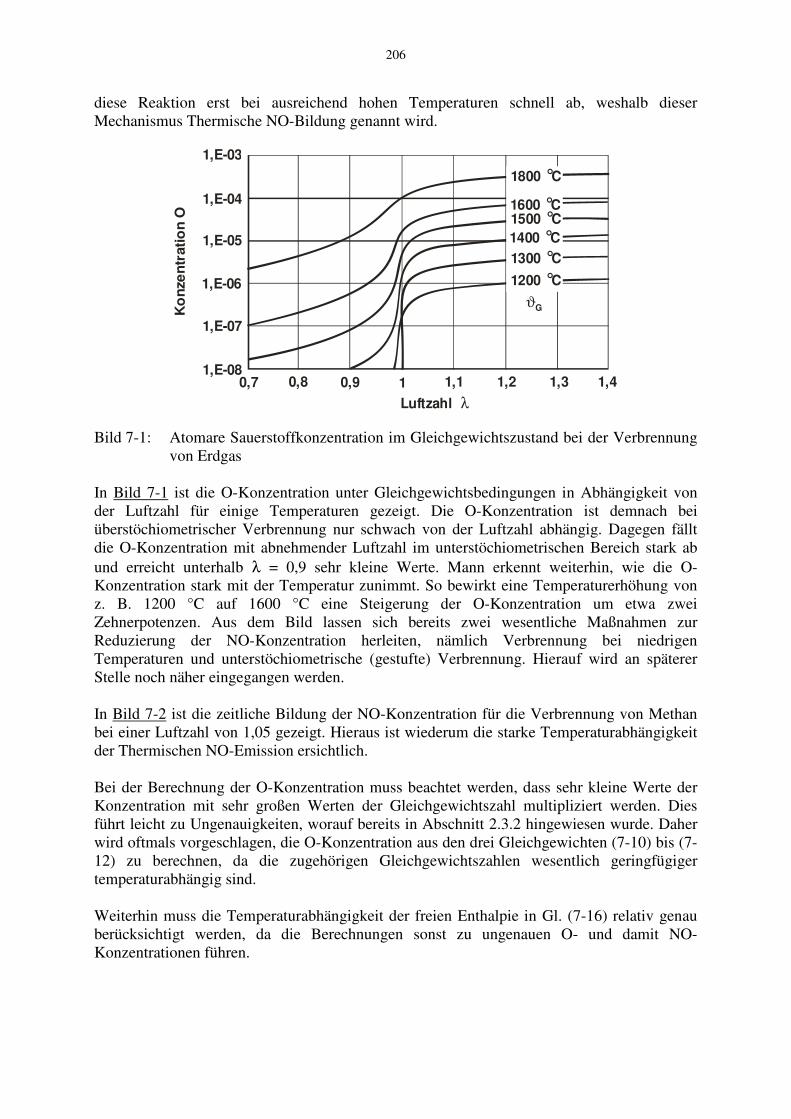
Bild 7-1: Atomare Sauerstoffkonzentration im Gleichgewichtszustand bei der Verbrennung
von Erdgas In Bild 7-1 ist die O-Konzentration unter Gleichgewichtsbedingungen in Abhängigkeit von der Luftzahl für einige Temperaturen gezeigt. Die O-Konzentration ist demnach bei überstöchiometrischer Verbrennung nur schwach von der Luftzahl abhängig. Dagegen fällt die O-Konzentration mit abnehmender Luftzahl im unterstöchiometrischen Bereich stark ab und erreicht unterhalb λ = 0,9 sehr kleine Werte. Mann erkennt weiterhin, wie die O-Konzentration stark mit der Temperatur zunimmt. So bewirkt eine Temperaturerhöhung von z. B. 1200 °C auf 1600 °C eine Steigerung der O-Konzentration um etwa zwei Zehnerpotenzen. Aus dem Bild lassen sich bereits zwei wesentliche Maßnahmen zur Reduzierung der NO-Konzentration herleiten, nämlich Verbrennung bei niedrigen Temperaturen und unterstöchiometrische (gestufte) Verbrennung. Hierauf wird an späterer Stelle noch näher eingegangen werden. In Bild 7-2 ist die zeitliche Bildung der NO-Konzentration für die Verbrennung von Methan bei einer Luftzahl von 1,05 gezeigt. Hieraus ist wiederum die starke Temperaturabhängigkeit der Thermischen NO-Emission ersichtlich. Bei der Berechnung der O-Konzentration muss beachtet werden, dass sehr kleine Werte der Konzentration mit sehr großen Werten der Gleichgewichtszahl multipliziert werden. Dies führt leicht zu Ungenauigkeiten, worauf bereits in Abschnitt 2.3.2 hingewiesen wurde. Daher wird oftmals vorgeschlagen, die O-Konzentration aus den drei Gleichgewichten (7-10) bis (7-12) zu berechnen, da die zugehörigen Gleichgewichtszahlen wesentlich geringfügiger temperaturabhängig sind. Weiterhin muss die Temperaturabhängigkeit der freien Enthalpie in Gl. (7-16) relativ genau berücksichtigt werden, da die Berechnungen sonst zu ungenauen O- und damit NO-Konzentrationen führen.
207
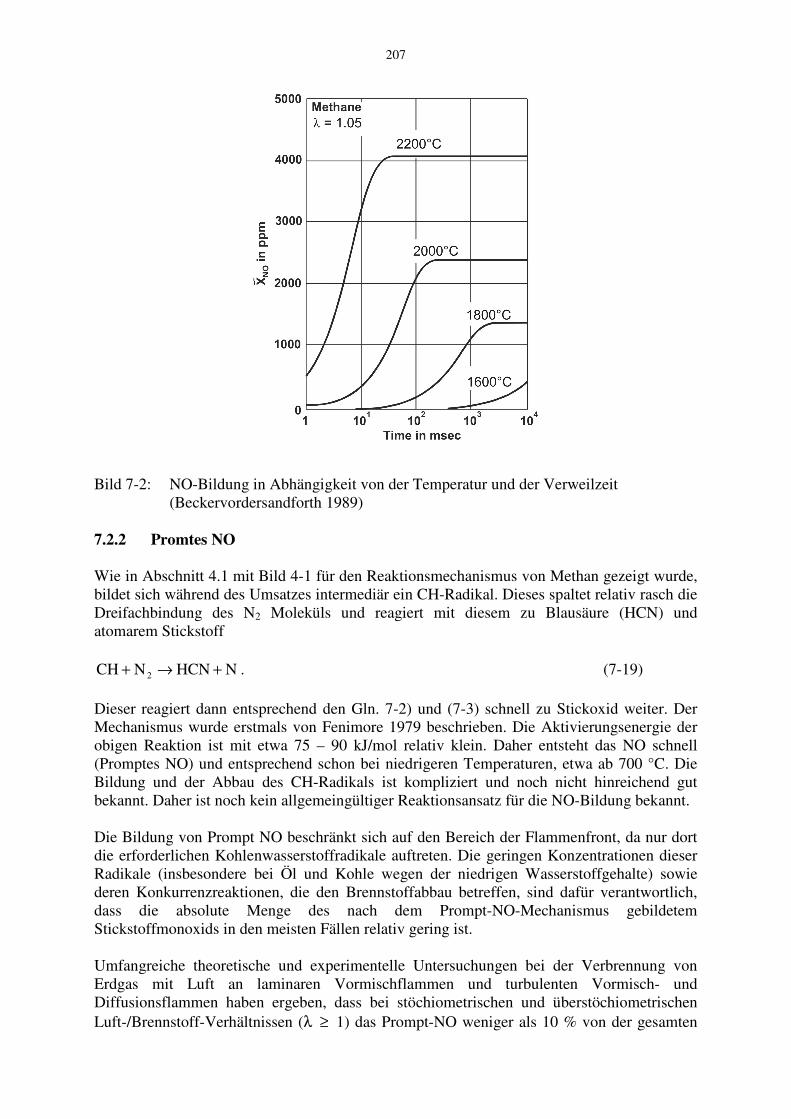
Bild 7-2: NO-Bildung in Abhängigkeit von der Temperatur und der Verweilzeit (Beckervordersandforth 1989) 7.2.2 Promtes NO
Wie in Abschnitt 4.1 mit Bild 4-1 für den Reaktionsmechanismus von Methan gezeigt wurde, bildet sich während des Umsatzes intermediär ein CH-Radikal. Dieses spaltet relativ rasch die Dreifachbindung des N2 Moleküls und reagiert mit diesem zu Blausäure (HCN) und atomarem Stickstoff
NHCNNCH 2 +→+ . (7-19)
Dieser reagiert dann entsprechend den Gln. 7-2) und (7-3) schnell zu Stickoxid weiter. Der Mechanismus wurde erstmals von Fenimore 1979 beschrieben. Die Aktivierungsenergie der obigen Reaktion ist mit etwa 75 – 90 kJ/mol relativ klein. Daher entsteht das NO schnell (Promptes NO) und entsprechend schon bei niedrigeren Temperaturen, etwa ab 700 °C. Die Bildung und der Abbau des CH-Radikals ist kompliziert und noch nicht hinreichend gut bekannt. Daher ist noch kein allgemeingültiger Reaktionsansatz für die NO-Bildung bekannt. Die Bildung von Prompt NO beschränkt sich auf den Bereich der Flammenfront, da nur dort die erforderlichen Kohlenwasserstoffradikale auftreten. Die geringen Konzentrationen dieser Radikale (insbesondere bei Öl und Kohle wegen der niedrigen Wasserstoffgehalte) sowie deren Konkurrenzreaktionen, die den Brennstoffabbau betreffen, sind dafür verantwortlich, dass die absolute Menge des nach dem Prompt-NO-Mechanismus gebildetem Stickstoffmonoxids in den meisten Fällen relativ gering ist. Umfangreiche theoretische und experimentelle Untersuchungen bei der Verbrennung von Erdgas mit Luft an laminaren Vormischflammen und turbulenten Vormisch- und Diffusionsflammen haben ergeben, dass bei stöchiometrischen und überstöchiometrischen Luft-/Brennstoff-Verhältnissen (λ ≥ 1) das Prompt-NO weniger als 10 % von der gesamten

208
NOx-Emission ausmacht (Stapf, Leuckel 1996). Folglich kann diesem NO-Bildungsmechanismus bei technischen Verbrennungsprozessen, die unter Luftüberschuss betrieben werden, nur eine geringe Bedeutung beigemessen werden. Unter brennstoffreichen Bedingungen, wie sie z. B. bei einer zweistufigen Verbrennung in der Primärstufe vorliegen, kann die Prompt-NO-Bildung demgegenüber einen wesentlichen Einfluss auf die Höhe der gesamten NOx-Emission haben (z. B. Tomeczek und Gradon 1997 sowie Glassman 1996). Dieser Befund wird auch durch Messergebnisse an vorgemischten Methan-Luft-Flammen bestätigt, wobei das Maximum der Prompt-NO-Bildung mit etwa 50 ppm im Luftzahlbereich 0,7 8,0≤λ≤ liegt.
In Heizkesselfeuerungen, bei denen die Flammentemperatur relativ niedrig gehalten werden kann, ist die Thermische NO-Bildung gering. Bei der Ermittlung der gesamten NO-Emission muss hier das Prompt-NO berücksichtigt werden. In Industriefeuerungen dagegen ist das Prompt NO gegenüber dem Thermischen NO und insbesondere bei flüssigen und festen Brennstoffen auch gegenüber dem Brennstoff-NO vernachlässigbar. 7.2.3 Brennstoff-NO
Enthält der Brennstoff chemisch gebundenen Stickstoff (Brennstoff-Stickstoff) in organischen (z. B. Amine, Amide, Nitride, Pyritin) oder anorganische Stickstoffverbindungen (z. B. Ammoniak, HCN), wird während des Verbrennungsprozesses nach einem weiteren Mechanismus Stickoxid gebildet. Sowohl bei der Verbrennung fossiler Brennstoffe (wie Kohle, Öle) als auch bei der thermischen Entsorgung von gasförmigen, flüssigen und festen stickstoffhaltigen Prozessrückständen (Abfällen) kommt der NO-Bildung nach diesem Brennstoff-NO-Mechanismus eine besondere Bedeutung zu, da der Brennstoff-Stickstoff in diesen Fällen die Hauptquelle der NOx-Emissionen darstellen kann. Während bei Kohlen und Rückstandsölen die Brennstoff-N-Gehalte bis zu 2 Ma% betragen, können Rest- und Abfallstoffe aus der chemischen Industrie auch mehr als 50 Ma% an chemisch gebundenen Stickstoff z. B. in Form von NH3 enthalten. Ist der Brennstoff-Stickstoff in organischen Verbindungen enthalten, so bildet sich hieraus zunächst in mehreren sehr schnellen Zerfallsreaktionen unter Abspaltung von Wasserstoffatomen die Zwischenverbindung HCN. Diese wird mit Radikalen zu CO und NHi umgewandelt. Die wesentlichen Reaktionen hierfür sind HCN + O → NCO + H (7-20) NCO + H → NH + CO (7-21) HCN + O → NH + CO (7-22) HCN + OH → NH2 + CO . (7-23) Anorganische Stickstoffverbindungen werden demgegenüber direkt in NHi-Radikale überführt. Hierbei ist die Reaktion NH3 + OH → NH2 + H2O (7-24) maßgebend. Die NHi-Bildung aus den anorganischen Verbindungen ist dabei schneller als aus den organischen. Die NH2-Radikale werden anschließend mit H und OH in schnellen Reaktionen abgebaut gemäß

209
NH2 + H → NH + H2 (7-25) NH + H → N + H2 (7-26) NH2 + OH → NH + H2O (7-27) NH + OH → N + H2O . (7-28) Die weitere Reaktionsfolge hängt davon ab, ob oxidierende oder reduzierende Bedingungen vorliegen. Unter oxidierenden Bedingungen wird Stickoxid entsprechend den Reaktionsgleichungen N + O2 → NO + O (7-29) NH + O2 → NO + OH (7-30) N + OH → NO + H (7-31) NH + OH → NO + H2 (7-32) NH + O → NO + H (7-33) NH2 + O → NO + H2 . (7-34) gebildet. Unter sauerstoffarmen Bedingungen wird die NO-Bildung auf Grund der geringen Konzentration an oxidierenden Radikalen unterdrückt. Die NHi-Radikale werden dann bevorzugt mit Stickoxid zu molekularem Stickstoff umgewandelt NH2 + NO → N2 + H2O (7-35) NH + NO → N2 + OH . (7-36) Welche der NHi-Radikale dabei hauptsächlich an den Reaktionen zur Bildung zu NO und N2 beteiligt sind, hängt im wesentlichen von den thermischen und stöchiometrischen Verbrennungsbedingungen ab.
Brennstoff-Stickstoff
No-Recycle
+CHi
NH3
NH2
NH
+O
+NO
+O,OH
N
N2
NO
HCN+O,OH,H
+CHi
+CHi
Prompt-NO-Mechanismus
Brennstoff -NO-Mechanismus
Zeldovich-Mechanismus
molekularerStickstoff
Bild 7-3: Prinzip der Brennstoff-NO-Bildung Der Gesamtmechanismus der Brennstoff-NO-Bildung ist in Bild 7-3 dargestellt. Für eine detailliertere Beschreibung des Mechanismus sei z. B. auf Jahnson et al. 1988 und 1989, Jahnson 1991, Kolb 1990 und Sybon 1994 verweisen. Darüber hinaus können noch

210
katalytische und nichtkatalytische, heterogene Gas-Feststoffreaktionen mit Asche, Kohle-, Koks- oder anderen Feststoffpartikeln bei der Verbrennung von Kohle und flüssigen Brenn-stoffen von Bedeutung für die NOx-Emissionen sein (Kremer und Schulz 1984 sowie Kremer, Schulz, et al. 1985). Die Brennstoff-NO-Bildung ist über die Radikale mit der Brennstoff-oxidation gekoppelt. Auf Grund des komplizierten gesamten Reaktionsmechanismus ist für technische Verbrennungssysteme eine exakte Berechnung der Stickstoffoxidemission noch nicht möglich. Unter bestimmten Bedingungen ist jedoch eine vereinfachte, quantitative Beschreibung der NOx-Bildung bei der Verbrennung von stickstoffhaltigen Brennstoffen möglich, wie Untersuchungen von Fenimore und De Soete gezeigt haben. Beide nutzten für ihre Experimente laminare Vormischflammen, da bei diesem Flammentyp die Kinetik der Brennstoff-NO-Bildung und –Reduktion weitgehend unabhängig vom Mischungsprozess zwischen Brennstoff und Oxidationsmittel beurteilt werden kann. Hierauf wird im Folgenden eingegangen. Globaler NO-Bildungs-Mechanismus von Fenimore Fenimore (1972, 1976, 1979, 1980) formulierte aus seinen Verbrennungsversuchen mit verschiedenen Stickstoffverbindungen einen globalen Reaktionsmechanismus, nach dem der Brennstoff-Stickstoff unabhängig von seiner Bindungsart vollständig über die Zwischenverbindung HCN in eine Spezies der NHi-Radikale umgewandelt wird. Die NHi-Radikale reagieren dann entweder mit OH-Radikalen entsprechend den Reaktionen (7-31) und (7-32) zu Stickstoffmonoxid oder werden gemäß den Reaktionen (7-35) und (7-36) mit NO zu molekularem Stickstoff umgesetzt. Hieraus entwickelte Fenimore 1972 die Beziehung
+−−=
NOgl
NOanfNO
NOgl
NO
x~x~x~
2
1exp1
x~x~
(7-37)
zur Berechnung der Konzentration NOx~ von Brennstoff-Stickoxid. In dieser Gleichung
bedeuten NOanfx~ die Anfangskonzentration an Brennstoff-Stickstoff, die der theoretischen
NO-Konzentration bei vollständiger Umwandlung des im Brennstoff enthaltenen N zu NO entspricht, und NOglx~ die NO-Gleichgewichtskonzentration, für die die Näherung
)T
EAexp(x~NOgl −= (7-38)
gilt. Die Größen A und E sind von der Luftzahl abhängig und müssen aus Experimenten bestimmt werden. Die Brennstoff-NO-Bildung nach Fenimore wird also mit diesem Globalmechanismus unabhängig von der Reaktionskinetik und damit von der Verweilzeit in Verbrennungsprozessen beschrieben. Mit diesem Fenimore-Mechanismus konnte beispielsweise von Scheuer 1987 und Gardeik 1985 die Bildung und der Abbau von NO in Zementofenanlagen und von Klöppner et al. 1993 und 1995 die NO-Konzentrationen von Rückstandsölen in Drallbrennkammersystemen beschrieben werden.

211
Globaler NO-Bildungs-Mechanismus nach De Soete De Soete 1974 und 1981 unterscheidet im Gegensatz zu Fenimore verschiedene, sogenannte sekundäre Stickstoffverbindungen (NH3, HCN und (CN)2), die aus den mit dem Brennstoff eingebrachten primären Stickstoffverbindungen durch Pyrolysereaktionen gebildet werden. Auf der Grundlage von Ergebnissen aus Verbrennungsversuchen hat er für die o. g. sekundären Stickstoffverbindungen die Reaktionsgeschwindigkeiten entweder zu Stickstoffmonoxid oder zu molekularem Stickstoff nach dem folgenden globalen Reaktionsmechanismus bestimmt: NX + O2 → NO + ... (7-39) NX + NO → N2 + ... . (7-40) Für die Bildungsgeschwindigkeit von NO gibt er die Gleichung
)x~kx~k(x~dt
x~dNONO
nOONX
NO
22⋅−⋅⋅= (7-41)
an und für die Abbaugeschwindigkeit der Brennstoff-Stickstoff-Verbindungen
)x~kx~k(x~dt
x~dNONO
nOONX
NX
22⋅+⋅⋅−= . (7-42)
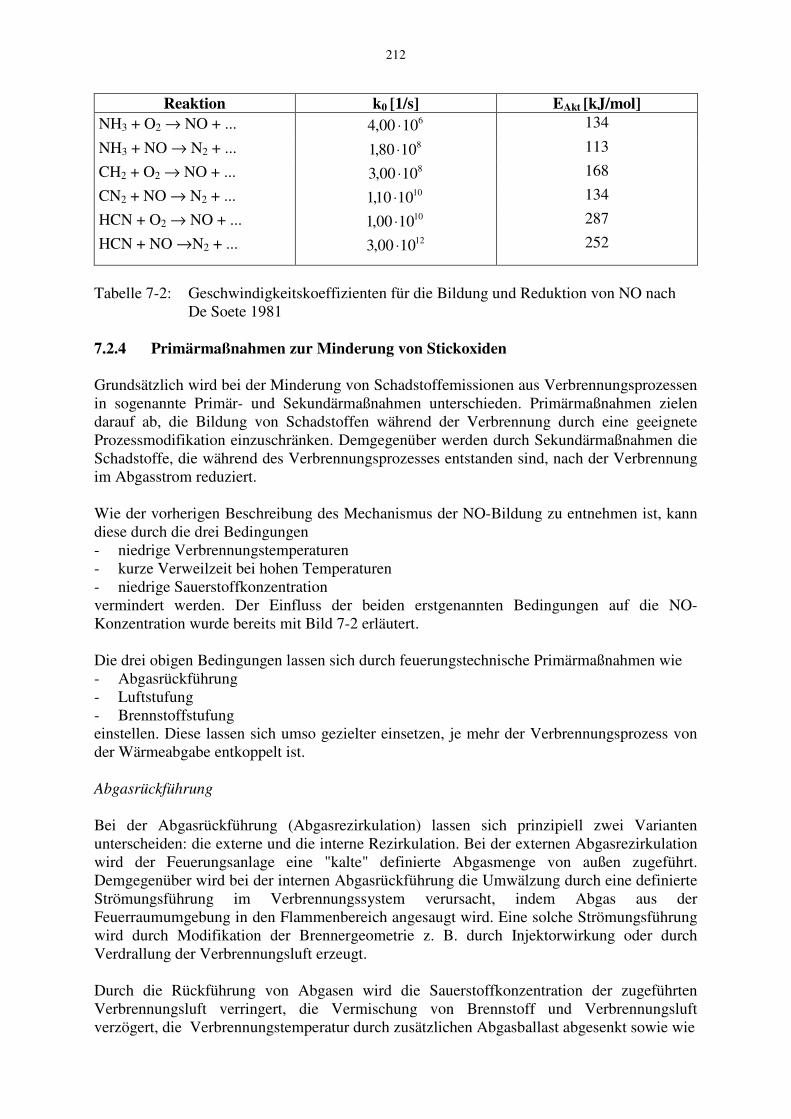
Die von De Soete für unterschiedliche Stickstoffverbindungen ermittelten Reaktionskoeffizienten
2Ok und KNO sind in Tabelle 7-2 angegeben. Für den Exponenten n gilt
die Näherung
−−=
−b1
O
a
x~lnexp1n 2 . (7-43)
Bei niedrigen O2-Konzentrationen ist demnach der Exponent gleich eins und bei hohen O2-Konzentrationen gleich null. Die Konstanten a und b sind für jeden Anwendungsfall experimentell zu ermitteln. Mit dem De Soete-Mechanismus konnte beispielsweise die NO-Bildung ebenfalls in einer Zementofenanlage und in einem Drallbrennkammersystem von Jeschar, Jennes et al. 1996 und 1999 bzw. Malek, Scholz et al. 1993 beschrieben werden. Letztendlich benötigen sowohl der Globalmechanismus nach De Soete als auch der nach Fenimore für jeden Anwendungsfall experimentell zu ermittelnde Anpassungsparameter.
212
Reaktion k0 [1/s] EAkt [kJ/mol]
NH3 + O2 → NO + ... 61000,4 ⋅ 134
NH3 + NO → N2 + ... 81080,1 ⋅ 113
CH2 + O2 → NO + ... 81000,3 ⋅ 168
CN2 + NO → N2 + ... 101010,1 ⋅ 134
HCN + O2 → NO + ... 101000,1 ⋅ 287
HCN + NO →N2 + ... 121000,3 ⋅ 252
Tabelle 7-2: Geschwindigkeitskoeffizienten für die Bildung und Reduktion von NO nach De Soete 1981 7.2.4 Primärmaßnahmen zur Minderung von Stickoxiden
Grundsätzlich wird bei der Minderung von Schadstoffemissionen aus Verbrennungsprozessen in sogenannte Primär- und Sekundärmaßnahmen unterschieden. Primärmaßnahmen zielen darauf ab, die Bildung von Schadstoffen während der Verbrennung durch eine geeignete Prozessmodifikation einzuschränken. Demgegenüber werden durch Sekundärmaßnahmen die Schadstoffe, die während des Verbrennungsprozesses entstanden sind, nach der Verbrennung im Abgasstrom reduziert. Wie der vorherigen Beschreibung des Mechanismus der NO-Bildung zu entnehmen ist, kann diese durch die drei Bedingungen - niedrige Verbrennungstemperaturen - kurze Verweilzeit bei hohen Temperaturen - niedrige Sauerstoffkonzentration vermindert werden. Der Einfluss der beiden erstgenannten Bedingungen auf die NO-Konzentration wurde bereits mit Bild 7-2 erläutert. Die drei obigen Bedingungen lassen sich durch feuerungstechnische Primärmaßnahmen wie - Abgasrückführung - Luftstufung - Brennstoffstufung einstellen. Diese lassen sich umso gezielter einsetzen, je mehr der Verbrennungsprozess von der Wärmeabgabe entkoppelt ist. Abgasrückführung
Bei der Abgasrückführung (Abgasrezirkulation) lassen sich prinzipiell zwei Varianten unterscheiden: die externe und die interne Rezirkulation. Bei der externen Abgasrezirkulation wird der Feuerungsanlage eine "kalte" definierte Abgasmenge von außen zugeführt. Demgegenüber wird bei der internen Abgasrückführung die Umwälzung durch eine definierte Strömungsführung im Verbrennungssystem verursacht, indem Abgas aus der Feuerraumumgebung in den Flammenbereich angesaugt wird. Eine solche Strömungsführung wird durch Modifikation der Brennergeometrie z. B. durch Injektorwirkung oder durch Verdrallung der Verbrennungsluft erzeugt. Durch die Rückführung von Abgasen wird die Sauerstoffkonzentration der zugeführten Verbrennungsluft verringert, die Vermischung von Brennstoff und Verbrennungsluft verzögert, die Verbrennungstemperatur durch zusätzlichen Abgasballast abgesenkt sowie wie
213
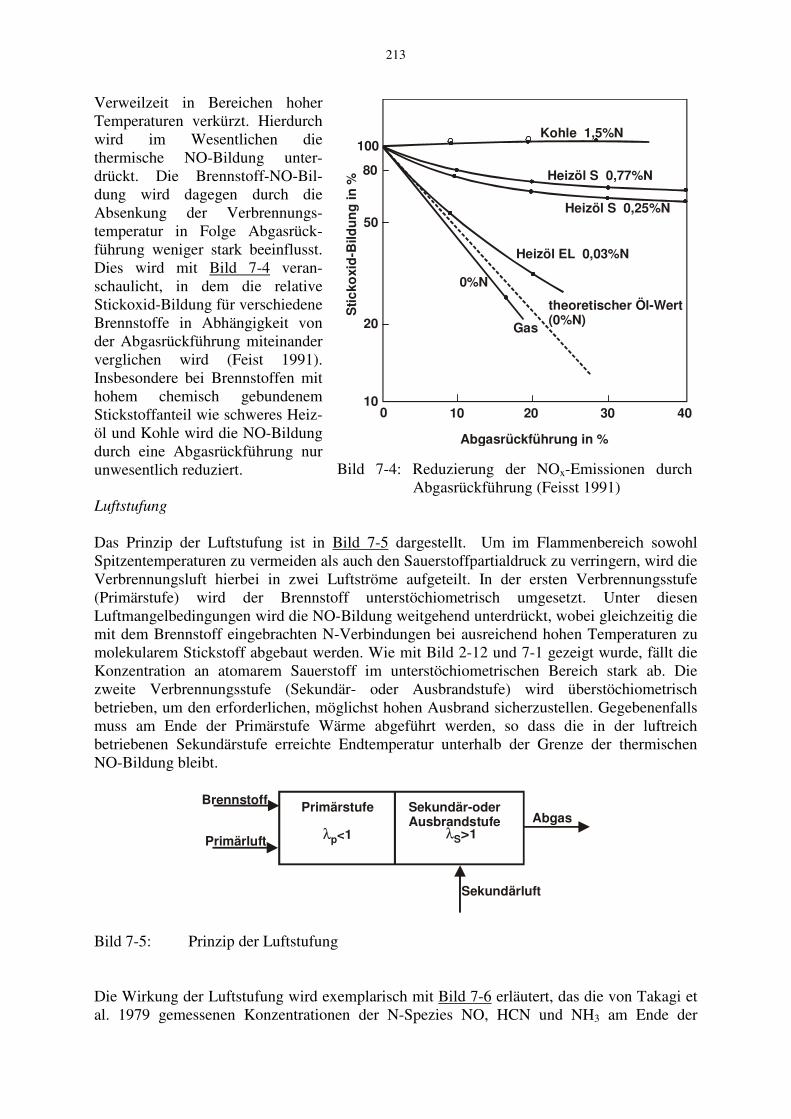
Verweilzeit in Bereichen hoher Temperaturen verkürzt. Hierdurch wird im Wesentlichen die thermische NO-Bildung unter-drückt. Die Brennstoff-NO-Bil-dung wird dagegen durch die Absenkung der Verbrennungs-temperatur in Folge Abgasrück-führung weniger stark beeinflusst. Dies wird mit Bild 7-4 veran-schaulicht, in dem die relative Stickoxid-Bildung für verschiedene Brennstoffe in Abhängigkeit von der Abgasrückführung miteinander verglichen wird (Feist 1991). Insbesondere bei Brennstoffen mit hohem chemisch gebundenem Stickstoffanteil wie schweres Heiz-öl und Kohle wird die NO-Bildung durch eine Abgasrückführung nur unwesentlich reduziert.
100
80
50
20
100 10 20 30 40
Kohle 1,5%N
Heizöl S 0,77%N
Heizöl S 0,25%N
Heizöl EL 0,03%N
theoretischer Öl-Wert(0%N)
Gas
0%N
Sti
cko
xid
-Bild
un
g in
%
Abgasrückführung in %
Bild 7-4: Reduzierung der NOx-Emissionen durch Abgasrückführung (Feisst 1991)
Luftstufung
Das Prinzip der Luftstufung ist in Bild 7-5 dargestellt. Um im Flammenbereich sowohl Spitzentemperaturen zu vermeiden als auch den Sauerstoffpartialdruck zu verringern, wird die Verbrennungsluft hierbei in zwei Luftströme aufgeteilt. In der ersten Verbrennungsstufe (Primärstufe) wird der Brennstoff unterstöchiometrisch umgesetzt. Unter diesen Luftmangelbedingungen wird die NO-Bildung weitgehend unterdrückt, wobei gleichzeitig die mit dem Brennstoff eingebrachten N-Verbindungen bei ausreichend hohen Temperaturen zu molekularem Stickstoff abgebaut werden. Wie mit Bild 2-12 und 7-1 gezeigt wurde, fällt die Konzentration an atomarem Sauerstoff im unterstöchiometrischen Bereich stark ab. Die zweite Verbrennungsstufe (Sekundär- oder Ausbrandstufe) wird überstöchiometrisch betrieben, um den erforderlichen, möglichst hohen Ausbrand sicherzustellen. Gegebenenfalls muss am Ende der Primärstufe Wärme abgeführt werden, so dass die in der luftreich betriebenen Sekundärstufe erreichte Endtemperatur unterhalb der Grenze der thermischen NO-Bildung bleibt.
Brennstoff
Primärluft
Primärstufe
λp<1
Sekundär-oderAusbrandstufe
λS>1
Sekundärluft
Abgas
Bild 7-5: Prinzip der Luftstufung
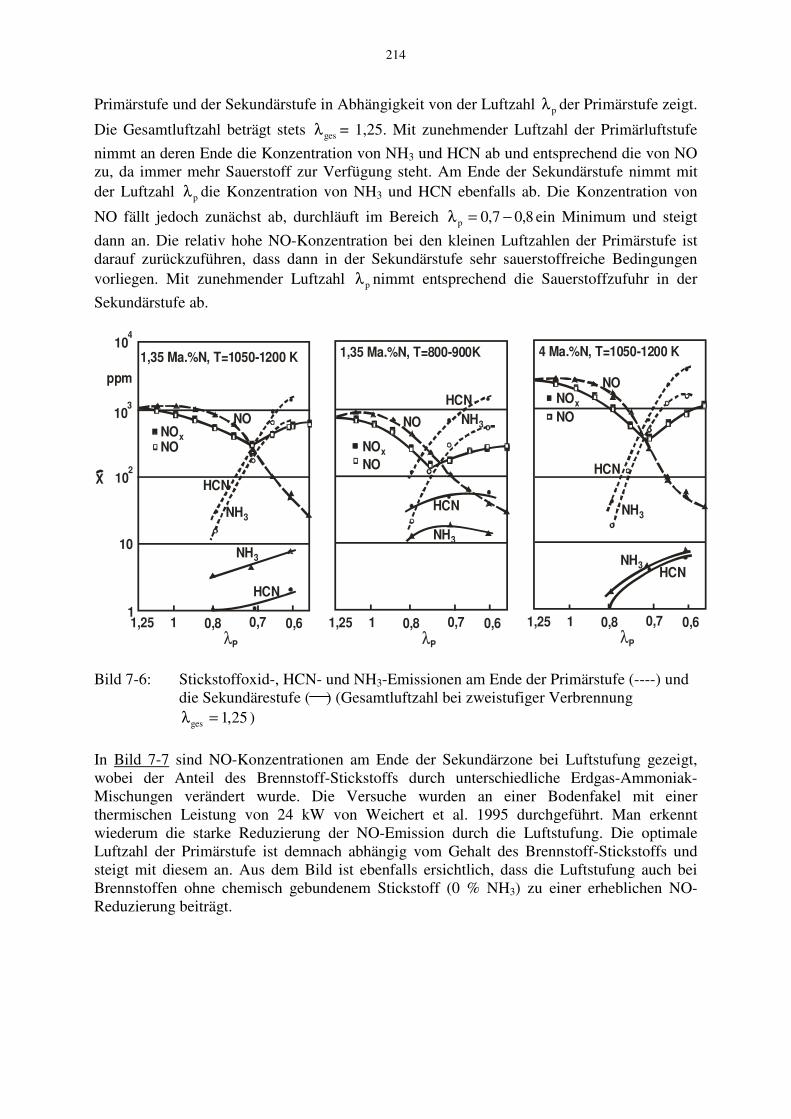
Die Wirkung der Luftstufung wird exemplarisch mit Bild 7-6 erläutert, das die von Takagi et al. 1979 gemessenen Konzentrationen der N-Spezies NO, HCN und NH3 am Ende der
214
Primärstufe und der Sekundärstufe in Abhängigkeit von der Luftzahl pλ der Primärstufe zeigt.
Die Gesamtluftzahl beträgt stets gesλ = 1,25. Mit zunehmender Luftzahl der Primärluftstufe
nimmt an deren Ende die Konzentration von NH3 und HCN ab und entsprechend die von NO zu, da immer mehr Sauerstoff zur Verfügung steht. Am Ende der Sekundärstufe nimmt mit der Luftzahl pλ die Konzentration von NH3 und HCN ebenfalls ab. Die Konzentration von
NO fällt jedoch zunächst ab, durchläuft im Bereich 8,07,0p −=λ ein Minimum und steigt
dann an. Die relativ hohe NO-Konzentration bei den kleinen Luftzahlen der Primärstufe ist darauf zurückzuführen, dass dann in der Sekundärstufe sehr sauerstoffreiche Bedingungen vorliegen. Mit zunehmender Luftzahl pλ nimmt entsprechend die Sauerstoffzufuhr in der
Sekundärstufe ab.
1,35 Ma.%N, T=1050-1200 K10
4
ppm
103
102
10
1
NONOxNO
HCN
NH3
NH3
HCN
1,25 1 0,8 0,7 0,6λP
1,35 Ma.%N, T=800-900K
NO
NOx
NO
HCNNH3
NH3
4 Ma.%N, T=1050-1200 K
NONOx
NO
HCNNH3
HCN
NH3HCN
1,25 1 0,8 0,7 0,6λP
1,25 1 0,8 0,7 0,6λP
X
-
Bild 7-6: Stickstoffoxid-, HCN- und NH3-Emissionen am Ende der Primärstufe (----) und die Sekundärestufe ( ) (Gesamtluftzahl bei zweistufiger Verbrennung 25,1ges =λ )
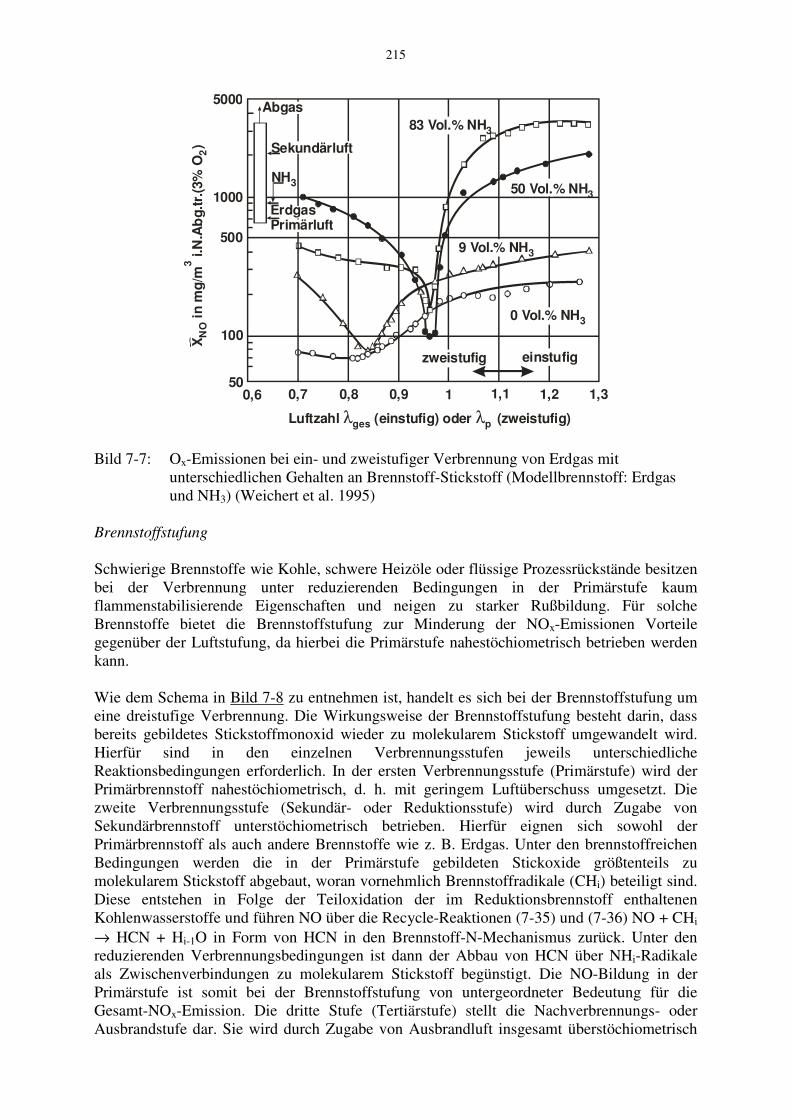
In Bild 7-7 sind NO-Konzentrationen am Ende der Sekundärzone bei Luftstufung gezeigt, wobei der Anteil des Brennstoff-Stickstoffs durch unterschiedliche Erdgas-Ammoniak-Mischungen verändert wurde. Die Versuche wurden an einer Bodenfakel mit einer thermischen Leistung von 24 kW von Weichert et al. 1995 durchgeführt. Man erkennt wiederum die starke Reduzierung der NO-Emission durch die Luftstufung. Die optimale Luftzahl der Primärstufe ist demnach abhängig vom Gehalt des Brennstoff-Stickstoffs und steigt mit diesem an. Aus dem Bild ist ebenfalls ersichtlich, dass die Luftstufung auch bei Brennstoffen ohne chemisch gebundenem Stickstoff (0 % NH3) zu einer erheblichen NO-Reduzierung beiträgt.
215
5000
1000
500
100
500,6 0,7 0,8 0,9 1 1,1 1,2 1,3
Luftzahl (einstufig) oder (zweistufig)λ λges p
50 Vol.% NH3
9 Vol.% NH3
0 Vol.% NH3
zweistufig einstufig
83 Vol.% NH3
X i
n m
g/m
i.N
.Ab
g.t
r.(3
% O
)N
O2
3NH3
Sekundärluft
Abgas
ErdgasPrimärluft
Bild 7-7: Ox-Emissionen bei ein- und zweistufiger Verbrennung von Erdgas mit unterschiedlichen Gehalten an Brennstoff-Stickstoff (Modellbrennstoff: Erdgas und NH3) (Weichert et al. 1995) Brennstoffstufung
Schwierige Brennstoffe wie Kohle, schwere Heizöle oder flüssige Prozessrückstände besitzen bei der Verbrennung unter reduzierenden Bedingungen in der Primärstufe kaum flammenstabilisierende Eigenschaften und neigen zu starker Rußbildung. Für solche Brennstoffe bietet die Brennstoffstufung zur Minderung der NOx-Emissionen Vorteile gegenüber der Luftstufung, da hierbei die Primärstufe nahestöchiometrisch betrieben werden kann. Wie dem Schema in Bild 7-8 zu entnehmen ist, handelt es sich bei der Brennstoffstufung um eine dreistufige Verbrennung. Die Wirkungsweise der Brennstoffstufung besteht darin, dass bereits gebildetes Stickstoffmonoxid wieder zu molekularem Stickstoff umgewandelt wird. Hierfür sind in den einzelnen Verbrennungsstufen jeweils unterschiedliche Reaktionsbedingungen erforderlich. In der ersten Verbrennungsstufe (Primärstufe) wird der Primärbrennstoff nahestöchiometrisch, d. h. mit geringem Luftüberschuss umgesetzt. Die zweite Verbrennungsstufe (Sekundär- oder Reduktionsstufe) wird durch Zugabe von Sekundärbrennstoff unterstöchiometrisch betrieben. Hierfür eignen sich sowohl der Primärbrennstoff als auch andere Brennstoffe wie z. B. Erdgas. Unter den brennstoffreichen Bedingungen werden die in der Primärstufe gebildeten Stickoxide größtenteils zu molekularem Stickstoff abgebaut, woran vornehmlich Brennstoffradikale (CHi) beteiligt sind. Diese entstehen in Folge der Teiloxidation der im Reduktionsbrennstoff enthaltenen Kohlenwasserstoffe und führen NO über die Recycle-Reaktionen (7-35) und (7-36) NO + CHi → HCN + Hi-1O in Form von HCN in den Brennstoff-N-Mechanismus zurück. Unter den reduzierenden Verbrennungsbedingungen ist dann der Abbau von HCN über NHi-Radikale als Zwischenverbindungen zu molekularem Stickstoff begünstigt. Die NO-Bildung in der Primärstufe ist somit bei der Brennstoffstufung von untergeordneter Bedeutung für die Gesamt-NOx-Emission. Die dritte Stufe (Tertiärstufe) stellt die Nachverbrennungs- oder Ausbrandstufe dar. Sie wird durch Zugabe von Ausbrandluft insgesamt überstöchiometrisch

216
betrieben, um einen möglichst hohen Ausbrand sicherzustellen. Da in dieser Stufe, bedingt durch die Luftzugabe und in Folge des Wärmeverlustes in den beiden vorangegangenen Verbrennungsstufen, weitaus geringere Temperaturen vorliegen, wird die erneute thermische NO-Bildung weitgehend unterdrückt.
Brennstoff
Primärluft
Primärstufe
λp <1
Sekundärstufe
λS >1
Sekundär-Brennstoff
Abgas
Tertiärluft
Tertiärstufe
λT >1
Bild 7-8: Prinzip der Brennstoffstufung Die Brennstoffstufung wird wegen des hohen Aufwandes in der Praxis noch sehr selten realisiert. Für weitergehende Informationen sei auf das Schrifttum wie beispielsweise Chen et al. 1986, Mechenbier 1989, Kolb 1990 und Sybon 1994 verwiesen. 7.2.5 Sekundärmaßnahmen zur NO-Minderung
Als sekundäre Maßnahmen zur Senkung der NO-Emission stehen selektive homogene Reduktion (als SHR oder thermisches DeNOx bezeichnet) und die selektive katalytische Reduktion (als SCR bezeichnet) zur Verfügung. SHR
Bei der selektiven homogenen Reduktion wird den Verbrennungsgasen Ammoniak (NH3) zugemischt, das durch OH zu NH2 abgebaut wird OHNHOHNH 223 +→+ . (7-44)
Dieses NH2 reagiert mit dem NO gemäß OHNNONH 222 +→+ (7-45)
OHHNNONH 22 +→+ . (7-46)
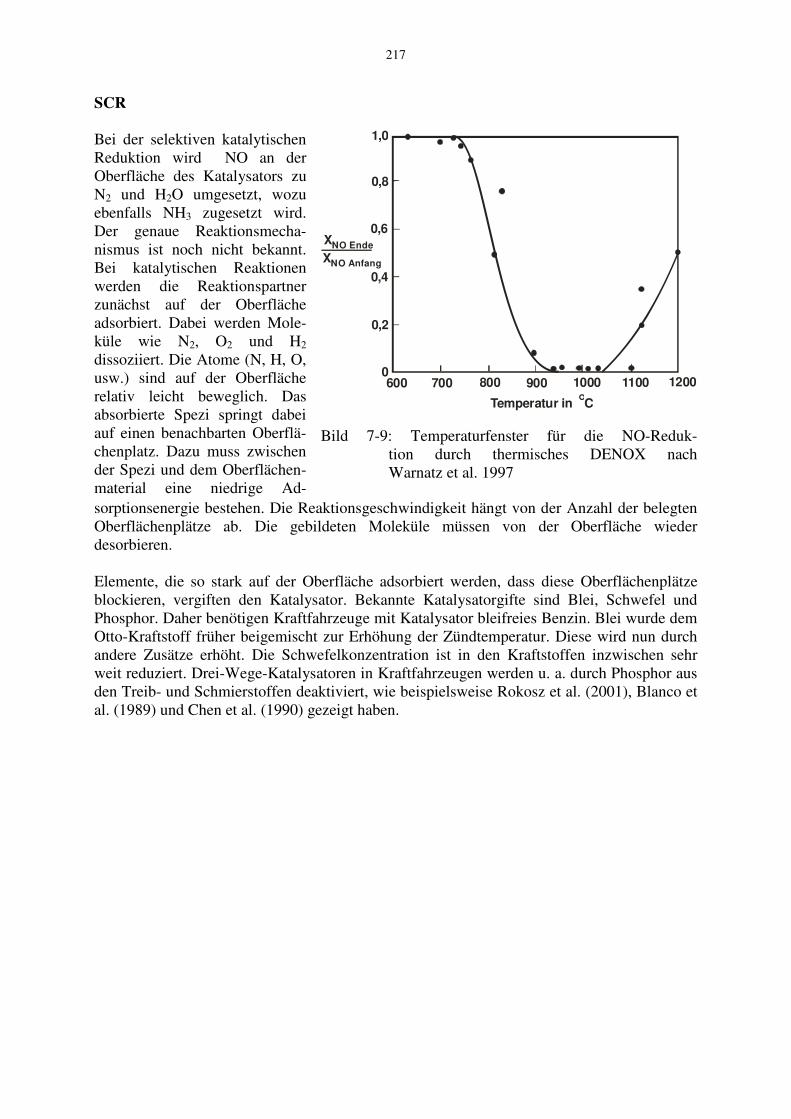
Diese drei Reaktionen sind die wichtigsten. Darüber hinaus laufen eine Vielzahl weiterer Elementarreaktionen ab, bei denen schließlich auch das N2H zu N2 umgesetzt wird. Ist die Temperatur nicht ausreichend hoch, so reagiert NH3 nicht mit OH nach Gl. (7-44). Bei zu hohen Temperaturen wird das NH3 oxidiert. Daher ist die homogene Reduktion nur in einem relativ engen Temperaturfenster möglich. In Bild 7-9 ist für ein Beispiel die NO-Reduktion in Abhängigkeit von der Temperatur gezeigt. Hieraus ist ersichtlich, dass das Temperaturfenster etwa im Bereich von 900 °C bis 1000 °C liegt. Außerdem darf der Überschuss des Ammoniaks gegenüber dem NO nicht zu hoch sein, da sonst dieser Überschuss in der Atmosphäre zur NO-Bildung führt. Als Anhaltswert gilt x~ NH3
/ x~ NO < 1,5.
217
SCR
Bei der selektiven katalytischen Reduktion wird NO an der Oberfläche des Katalysators zu N2 und H2O umgesetzt, wozu ebenfalls NH3 zugesetzt wird. Der genaue Reaktionsmecha-nismus ist noch nicht bekannt. Bei katalytischen Reaktionen werden die Reaktionspartner zunächst auf der Oberfläche adsorbiert. Dabei werden Mole-küle wie N2, O2 und H2 dissoziiert. Die Atome (N, H, O, usw.) sind auf der Oberfläche relativ leicht beweglich. Das absorbierte Spezi springt dabei auf einen benachbarten Oberflä-chenplatz. Dazu muss zwischen der Spezi und dem Oberflächen-material eine niedrige Ad-
1,0
0,8
0,6
0,4
0,2
0600 700 800 900 1000 1100 1200
Temperatur in °C
XNO Anfang
XNO Ende
Bild 7-9: Temperaturfenster für die NO-Reduk- tion durch thermisches DENOX nach Warnatz et al. 1997
sorptionsenergie bestehen. Die Reaktionsgeschwindigkeit hängt von der Anzahl der belegten Oberflächenplätze ab. Die gebildeten Moleküle müssen von der Oberfläche wieder desorbieren. Elemente, die so stark auf der Oberfläche adsorbiert werden, dass diese Oberflächenplätze blockieren, vergiften den Katalysator. Bekannte Katalysatorgifte sind Blei, Schwefel und Phosphor. Daher benötigen Kraftfahrzeuge mit Katalysator bleifreies Benzin. Blei wurde dem Otto-Kraftstoff früher beigemischt zur Erhöhung der Zündtemperatur. Diese wird nun durch andere Zusätze erhöht. Die Schwefelkonzentration ist in den Kraftstoffen inzwischen sehr weit reduziert. Drei-Wege-Katalysatoren in Kraftfahrzeugen werden u. a. durch Phosphor aus den Treib- und Schmierstoffen deaktiviert, wie beispielsweise Rokosz et al. (2001), Blanco et al. (1989) und Chen et al. (1990) gezeigt haben.
218
7.3 Schwefeldioxid
7.3.1 Schwefelsäurebildung
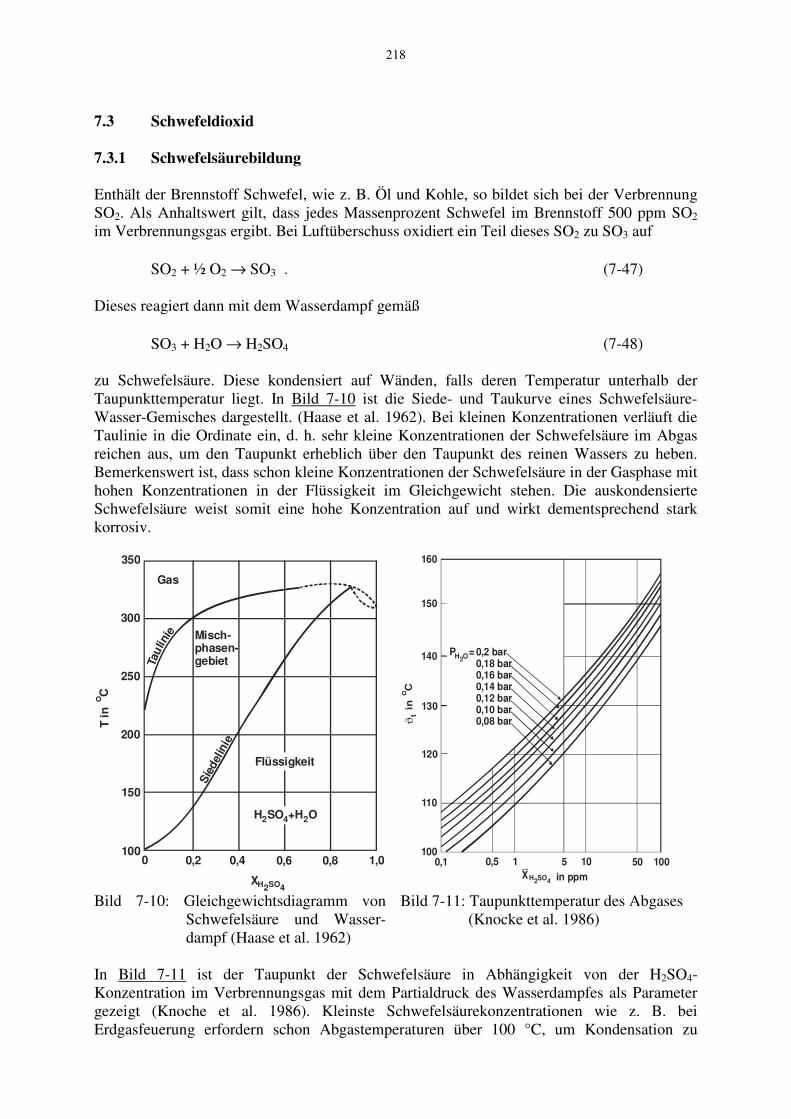
Enthält der Brennstoff Schwefel, wie z. B. Öl und Kohle, so bildet sich bei der Verbrennung SO2. Als Anhaltswert gilt, dass jedes Massenprozent Schwefel im Brennstoff 500 ppm SO2 im Verbrennungsgas ergibt. Bei Luftüberschuss oxidiert ein Teil dieses SO2 zu SO3 auf SO2 + ½ O2 → SO3 . (7-47) Dieses reagiert dann mit dem Wasserdampf gemäß SO3 + H2O → H2SO4 (7-48) zu Schwefelsäure. Diese kondensiert auf Wänden, falls deren Temperatur unterhalb der Taupunkttemperatur liegt. In Bild 7-10 ist die Siede- und Taukurve eines Schwefelsäure-Wasser-Gemisches dargestellt. (Haase et al. 1962). Bei kleinen Konzentrationen verläuft die Taulinie in die Ordinate ein, d. h. sehr kleine Konzentrationen der Schwefelsäure im Abgas reichen aus, um den Taupunkt erheblich über den Taupunkt des reinen Wassers zu heben. Bemerkenswert ist, dass schon kleine Konzentrationen der Schwefelsäure in der Gasphase mit hohen Konzentrationen in der Flüssigkeit im Gleichgewicht stehen. Die auskondensierte Schwefelsäure weist somit eine hohe Konzentration auf und wirkt dementsprechend stark korrosiv.
350
300
250
200
150
1000,2 0,4 0,6 0,8 1,00
T i
n
C°
XH SO2 4
Gas
Tau
linie Misch-
phasen-gebiet
Sie
del n
iei
Flüssigkeit
H SO +H O2 4 2
160
150
140
130
120
110
1000,1 0,5 1 5 10 50 100
ϑt
°in
C
in ppmXH SO2 4
0,2 bar0,18 bar0,16 bar0,14 bar0,12 bar0,10 bar0,08 bar
P =H O2
Bild 7-10: Gleichgewichtsdiagramm von Schwefelsäure und Wasser- dampf (Haase et al. 1962)
Bild 7-11: Taupunkttemperatur des Abgases (Knocke et al. 1986)
In Bild 7-11 ist der Taupunkt der Schwefelsäure in Abhängigkeit von der H2SO4-Konzentration im Verbrennungsgas mit dem Partialdruck des Wasserdampfes als Parameter gezeigt (Knoche et al. 1986). Kleinste Schwefelsäurekonzentrationen wie z. B. bei Erdgasfeuerung erfordern schon Abgastemperaturen über 100 °C, um Kondensation zu

219
vermeiden. Bei der Verbrennung von Kohle mit höheren Schwefelgehalten müssen Abgastemperaturen über 160 °C eingehalten werden. Als Maßnahmen zur Entschwefelung werden unterschieden - Heißentschwefelung, bei der Kalksteinmehl im Temperaturbereich um 1000 °C
eingeblasen wird - Trockenverfahren, bei dem Kalkhydratmehl im Niedertemperaturbereich eingeblasen wird - Halbtrockenverfahren, bei dem eine Suspension aus Kalkhydratmehl und Wasser im
Niedertemperaturbereich eingeblasen wird und - Nassverfahren, bei dem das Verbrennungsgas durch eine Kalkmilch geleitet wird. 7.3.2 Heißentschwefelung
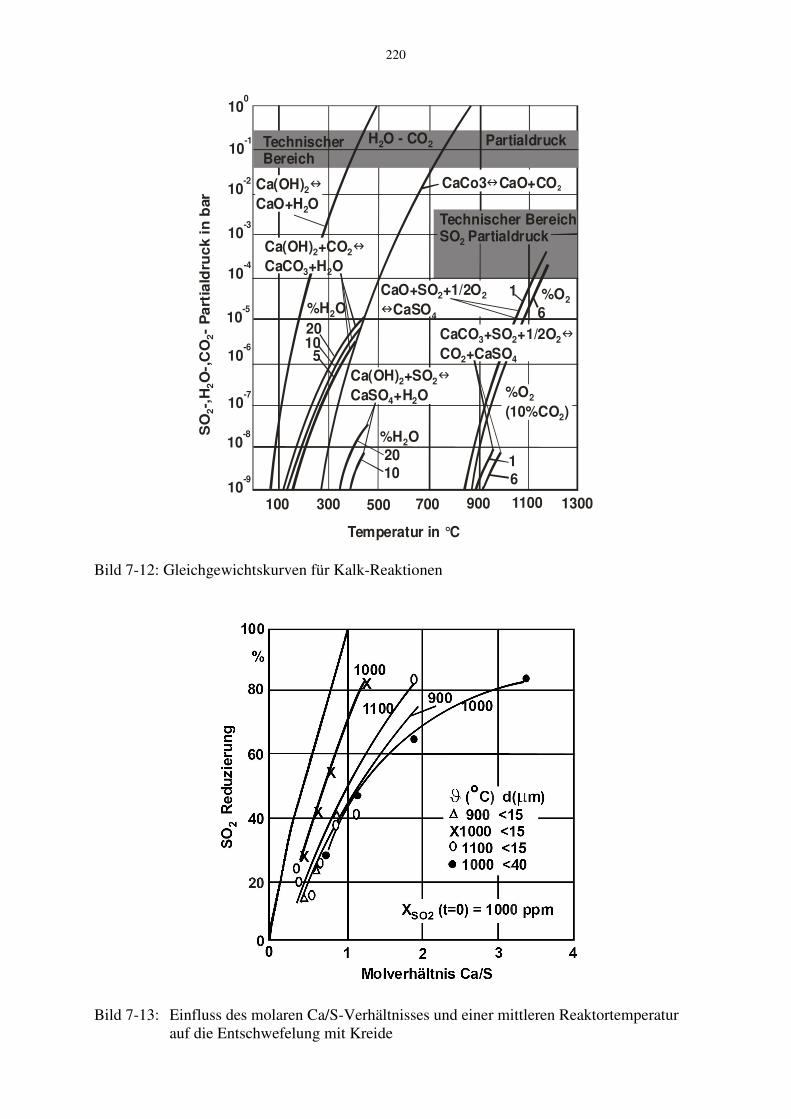
Bei der Heißentschwefelung wird Kalksteinmehl in den Hochtemperaturbereich des Brennraums eingeblasen. Dieses dekarbonatisiert entsprechend CaCO3 → CaO + CO2 , (7-49) so dass poröse CaO-Partikel entstehen. Diese nehmen dann das SO2 gemäß CaO + SO2 + ½O2 → CaSO4 (7-50) auf. Es bildet sich dabei eine dichte Sulfathülle um das Partikel, die mit fortschreitender Umsatzzeit die Diffusion des SO2 zum verbleibenden CaO-Kern behindert. Je poröser und je kleiner ein Partikel ist, umso mehr SO2 kann folglich eingebunden werden. Die beiden Reaktionen laufen nur in einem bestimmten Temperaturbereich ab. Dies wird mit Bild 7-12 erläutert, in dem die Gleichgewichtskurve der beiden Reaktionen dargestellt ist. Bei üblichen CO2-Konzentrationen im Verbrennungsgas um 10 % muss demnach die Temperatur höher als 750 °C sein, damit das CO2 abgespalten werden kann. Damit die Entsäuerungsreaktion ausreichend schnell abläuft, müssen nach Kainer et al. 1986 die Temperaturen oberhalb 900 °C liegen. Bei Temperaturen oberhalb etwa 1150 °C sintert das CaO-Partikel, wodurch die innere Oberfläche und damit die Reaktivität abnimmt. Das Partikel wird dann "totgebrannt". Die Sulfatreaktion des CaO mit SO2 läuft nur bei Temperaturen unterhalb von etwa 1170 °C (6 % O2, 1000 ppm SO2) in der gewünschten Richtung ab. Bei höheren Temperaturen würde sich bereits gebildetes Kalziumsulfat wieder zersetzen. Aus diesen Gleichgewichten ergibt sich, dass man für die Heißentschwefelung mit CaCO3 einen Temperaturbereich von etwa 900 °C bis 1150 °C einhalten muss. Darüber hinaus muss vermieden werden, dass das Additiv auch nur kurzzeitig Temperaturen oberhalb 1200 °C ausgesetzt wird, um dessen Inaktivierung zu vermeiden.
220
100
10-1
10-2
10-3
10-4
10-5
10-6
10-7
10-8
10-9
TechnischerBereich
SO
-,H
O-,
CO
- P
art
iald
ruc
k i
n b
ar2
22
Temperatur in °C
100 300 500 700 900 1100 1300
H O - CO2 2 Partialdruck
Technischer BereichSO Partialdruck2
2010
5
2010
%H O2
1
6%O2
16
Ca(OH)CaO+H O
2
2
�
Ca(OH) +COCaCO +H O
2 2
3 2
�
CaO+SO +1/2OCaSO
2 2
4�
Ca(OH) +SOCaSO +H O
2 2
4 2
�
CaCO +SO +1/2OCO +CaSO
3 2 2
2 4
�
%O(10%CO )
2
2
CaCo3 CaO+CO� 2
%H O2
Bild 7-12: Gleichgewichtskurven für Kalk-Reaktionen
Bild 7-13: Einfluss des molaren Ca/S-Verhältnisses und einer mittleren Reaktortemperatur
auf die Entschwefelung mit Kreide

221
In Bild 7-13 sind als Beispiel von Schopf et al. 1985 erzielte Entschwefelungsraten gezeigt. Die Versuche wurden an einer Drallbrennkammer mit SO2-dotierten Erdgasflammen durchgeführt. Man erkennt, dass die SO2-Einbindung mit dem molaren Ca/S-Verhältnis zunimmt. Bei Werten von 1 bis 2 ergeben sich demnach Entschwefelungsraten von etwa 80 %. Die optimale Brennraumtemperatur beträgt 1000 °C. Bei einer Abweichung von ± 100 K ist die Entschwefelung schon deutlich geringer und man benötigt für den gleichen Entschwefelungsgrad schon etwa die doppelte Menge an Kalkstein. In Bild 7-13 ist der Entschwefelungsgrad für die beiden Körnungen < 15 µm und < 40 µm jeweils für die optimale Brennraumtemperatur von 1000 °C verglichen. Hieraus ist ersichtlich, dass insbesondere für höhere Entschwefelungsraten sehr feine Körnungen, möglichst kleiner als 15 µm, benötigt werden. Zusätzlich zur Körnung übt auch die Art des Kalksteins einen Einfluss auf die Entschwefelung aus. Hierzu sei beispielsweise auf die Untersuchungen von Mehlmann et al. 1987 verwiesen. 7.3.3 Niedertemperaturentschwefelung
Zur Entschwefelung im Niedertemperaturbereich wird beim sogenannten Trockenverfahren ein Kalkhydratmehl Ca(OH)2 und beim sogenannten Halbtrockenverfahren eine Suspension aus Kalkhydratmehl und Wasser eingeblasen. Das Gleichgewicht der Dehydratisierung OHCaO)OH(Ca 22 +→ (7-51)
ist im Bild 7-12 dargestellt. Bei üblichen Wasserdampfgehalten im Abgas zwischen 5 und 20 % liegt demnach die Zersetzungstemperatur des Kalkhydrats bei 375 °C bzw. 425 °C. Da die Niedertemperaturentschwefelung bei Temperaturen unterhalb von 300 °C durchgeführt wird, findet also keine direkte Dehydratation des Mehls statt. Möglich ist jedoch eine Rekarbonatisierung entsprechend OHCaCOCO)OH(Ca 2322 +→+ . (7-52)
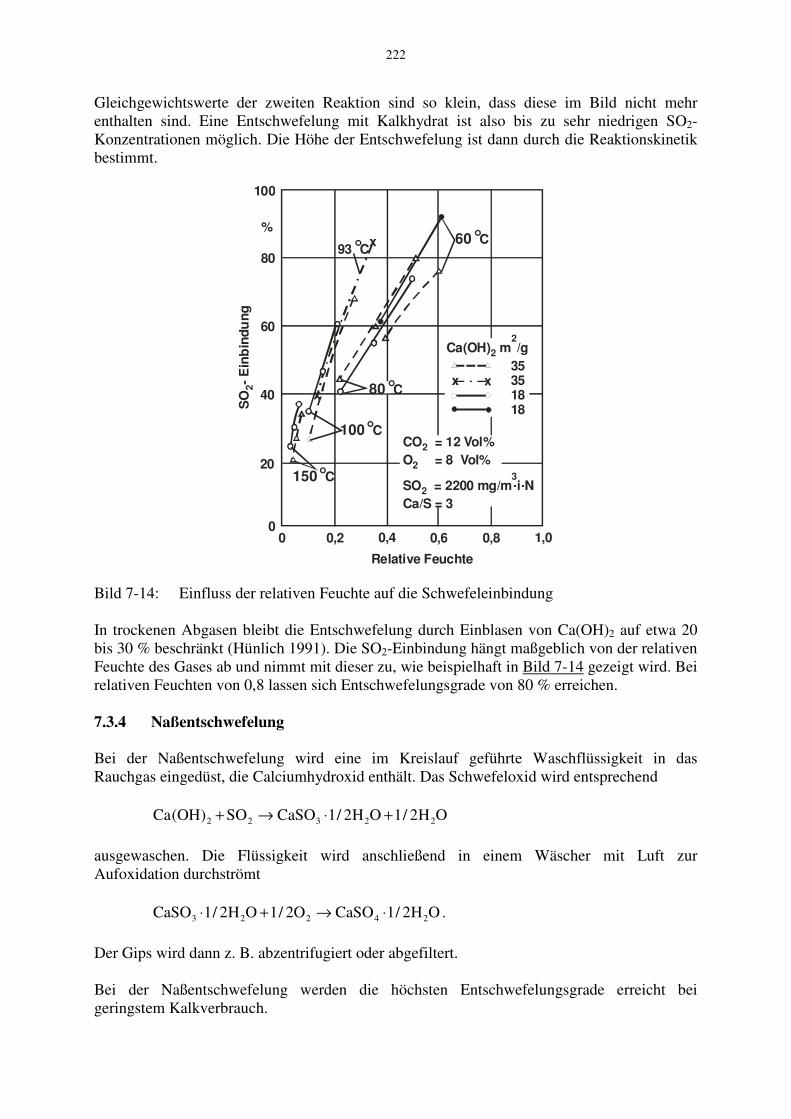
Die Gleichgewichtslinie ist exemplarisch für die drei Wasserdampfpartialdrücke von 5,10 und 20 % wiedergegeben. Bei CO2-Partialdrücken oberhalb den Gleichgewichtswerten läuft die Reaktion in der oben angegebenen Richtung ab. Die Kurvenschar ist durch die Gleichgewichtslinie der Kalksteinzersetzung begrenzt. Die Rekarbonatisierung entsprechend der obigen Reaktion ist von der Härtung des aus Ca(OH)2 bestehenden Mörtels bekannt. Im Vordergrund für die Entschwefelung stehen die Reaktionen OHCaSOSO)OH(Ca 2322 +→+ (7-53)
und
OHCaSOO2
1SO)OH(Ca 24222 +→++ . (7-54)
In Bild 7-12 ist die Gleichgewichtskurve der oberen Reaktion für die beiden Wasserdampfkonzentrationen 10 und 20 % dargestellt. Bei SO2-Partialdrücken oberhalb den Gleichgewichtswerten laufen die Reaktionen wiederum in der angegebenen Richtung ab. Die
222
Gleichgewichtswerte der zweiten Reaktion sind so klein, dass diese im Bild nicht mehr enthalten sind. Eine Entschwefelung mit Kalkhydrat ist also bis zu sehr niedrigen SO2-Konzentrationen möglich. Die Höhe der Entschwefelung ist dann durch die Reaktionskinetik bestimmt.
x
x
100
80
60
40
20
00 0,2 0,4 0,6 0,8 1,0
Relative Feuchte
SO
- E
inb
ind
un
g2
93 C° 60°C
100°C
150°C
35351818
Ca(OH) m /g2
2
x x80°C
%
CO = 12 Vol%O = 8 Vol%
SO = 2200 mg/m i NCa/S = 3
2
2
2
3. .
Bild 7-14: Einfluss der relativen Feuchte auf die Schwefeleinbindung In trockenen Abgasen bleibt die Entschwefelung durch Einblasen von Ca(OH)2 auf etwa 20 bis 30 % beschränkt (Hünlich 1991). Die SO2-Einbindung hängt maßgeblich von der relativen Feuchte des Gases ab und nimmt mit dieser zu, wie beispielhaft in Bild 7-14 gezeigt wird. Bei relativen Feuchten von 0,8 lassen sich Entschwefelungsgrade von 80 % erreichen. 7.3.4 Naßentschwefelung Bei der Naßentschwefelung wird eine im Kreislauf geführte Waschflüssigkeit in das Rauchgas eingedüst, die Calciumhydroxid enthält. Das Schwefeloxid wird entsprechend OH2/1OH2/1CaSOSO)OH(Ca 22322 +⋅→+
ausgewaschen. Die Flüssigkeit wird anschließend in einem Wäscher mit Luft zur Aufoxidation durchströmt OH2/1CaSOO2/1OH2/1CaSO 24223 ⋅→+⋅ .
Der Gips wird dann z. B. abzentrifugiert oder abgefiltert. Bei der Naßentschwefelung werden die höchsten Entschwefelungsgrade erreicht bei geringstem Kalkverbrauch.

223
7.4 Kohlenwasserstoffe und Ruß
Bei den Kohlenwasserstoffen unterscheidet man unverbrannte Kohlenwasserstoffe, polyzyklisch aromatisierte Kohlenwasserstoffe und Ruß. Die Bildung dieser Schadstoffe ist zwar experimentell vielfach untersucht worden, ein ausreichendes theoretisches Verständnis liegt jedoch noch nicht vor. Unverbrannte Kohlenwasserstoffe Unverbrannte Kohlenwasserstoffe entstehen durch örtliche Flammenlöschung, entweder an kalten Wänden oder durch Streckung. Wird an kalten Wänden der Löschabstand, der die Größenordnung der Flammenfrontdicke besitzt, unterschritten, so ist die Wärmeabfuhr so hoch, dass zum einem die Reaktion einfriert und zum anderen die Radikale durch Oberflächenreaktionen an der Wand zerstört werden. In Industrieöfen sind die Wandtemperaturen in der Regel so hoch, dass dieser Effekt nicht auftritt. Werden Flammenfronten stark gesteckt, hervorgerufen beispielsweise durch eine starke Turbulenz, so treten örtliche Flammenlöschungen auf. Findet keine erneute Zündung statt, so verlässt der Brennstoff die Reaktionszone mit unvollständigem Ausbrand. Polyzyklische aromatische Kohlenwasserstoffe Polyzyklische aromatische Kohlenwasserstoffe (PAK) werden aus kleinen Kohlenwasserstoff Bausteinen aufgebaut. Der wichtigste Vorläufer ist das Ethin (C2H2), das insbesondere in brennstoffreichen Flammen oder Bereichen in höheren Konzentrationen gebildet wird. Die aromatischen Ringstrukturen entstehen dann durch Reaktion von C2H2 mit CH oder CH2 unter Bildung von C3H3, das dann durch Umlagerungen den ersten Ring bilden kann. Die nachfolgenden Ringe entstehen dann durch weitere Anlagerungen von C2H2. Ruß Ruß wird durch ein weiteres Wachstum der PAK gebildet. Die Rußpartikel können mit Molekülen aus der Gasphase, wie z. B. Hydroxylradikale, reagieren, so dass die Partikel wachsen. Mehrere Rußpartikel können auch agglomerieren (Joos 2006). Die Struktur des Rußes ist nur schwer zu charakterisieren. Das molare C/H-Verhältnis liegt ungefähr bei eins. Nur die sehr feinen und unsichtbaren Rußpartikel sind lungengängig und karzinogen. Ruß wird im brennstoffreichen Teil des Gemisches gebildet, da dann die unvollständig verbrannten Komponenten CO und H2 relativ hoch sind. Bei örtlichen Luftzahlen kleiner als 1/3 wird die Konzentration von CO2 und H2O null. Dann reicht das O2-Angebot nur zur teilweisen Oxidation zu CO aus, so dass sich unter dieser Bedingung zwangsläufig Ruß bilden muss, u. a. durch 2 CO � C + CO2. Der atomare Kohlenstoff kann als Ruß sublimieren. Bereiche mit starkem örtlichem Luftmangel treten in nichtvorgemischten Flammen auf, wie z. B. bei der Kerzenflamme, der Verbrennung von Flüssigkeitstropfen und in Dieselmotoren. Der Ruß gibt Festkörperstrahlung ab. Daher sind die Flammen von Kerzen und Ölbrennern gelblich leuchtend sichtbar. Gelangt der Ruß in heiße, sauerstoffreiche Bereiche, so verbrennt er vollständig. Lediglich im Bereich kalter Wände, wie z. B. bei der motorischen Verbrennung, und durch Streckung auf Grund hoher Turbulenz kann der Ruß nicht mehr vollständig reagieren, was folglich zu unerwünschten Emissionen führt.

224
7.5 Angabe von Emissionen
Die Höhe der Konzentration von Emissionen im Abgas hängt von der Luftzahl ab. Je höher diese ist, umso niedriger ist die Konzentration. Zur Vergleichbarkeit der Emission von verschiedenen Anlagen müssen diese daher auf definierte Luftzahlen bezogen werden. Da die Luftzahl in der Regel aus der gemessenen O2-Konzentrationen ermittelt wird, werden die Emissionen zweckmäßigerweise auf eine definierte O2-Konzentration bezogen. Ist iAx~ die
Konzentration einer emittierten Komponente (NO, SO2, CO usw.) im Abgas mit der O2-Konzentration Ao2
x~ , so gilt für die Bezugskonzentration iBx~ bei der Bezugs-O2-Konzentration
BO2x~
iA
Ao
Bo
iB x~x~21,0
x~21,0x~
2
2 ⋅−
−= . (7-55)
Als Bezugs-O2-Konzentration werden meistens 3 % oder 6 % genommen. Dies entspricht Luftzahlen entsprechend Gl. (2-4) von λ = 1,15 bzw. 1,4 .
Grenzwerte werden vielfach als Partialdichte ∗ρi (Masse i pro Volumen Gas), z. B. mgi/m3
G,
angegeben. Mit der Volumenkonzentration ix~ besteht der Zusammenhang
iii x~ ρ⋅=ρ∗ , (7-56)
wobei iρ die Dichte der reinen Komponente i ist.
Unter den Stickoxidemissionen (NOx) wird das Gemisch aus Stickstoffmonoxid (NO) und Stickstoffdioxid (NO2) verstanden. Vereinbarungsgemäß wird die NOx-Konzentration als Partialdichte von NO2 angegeben. In vielen Messgeräten wird dazu das NO vor der Messung in NO2 konvertiert. In Tabelle 7-3 sind die Umrechnungsfaktoren von Volumenkonzentration in Partialdichte für die wichtigsten Emissionen angegeben.
ix~ ∗ρi
1 ppm CO
1 ppm NOx
1 ppm SO2
1,25 mg CO/m3 i. N.
2,05 mg NO2/m3 i. N.
2,93 mg SO2/m3 i. N.
Tabelle 7-3: Umrechnungsfaktoren von Vol. Konzentration in Partialdichte Bei vergleichbaren Anlagen, wie Wärmeerzeugungsanlagen (Heizkessel), werden die Emissionen auch auf die Nutzwärme (in kWh) bezogen. Dadurch werden die Anlagen unabhängig vom Bezugssauerstoffgehalt. In Tabelle 7-4 sind beispielhaft einige Grenzwerte für Emissionen angegeben.
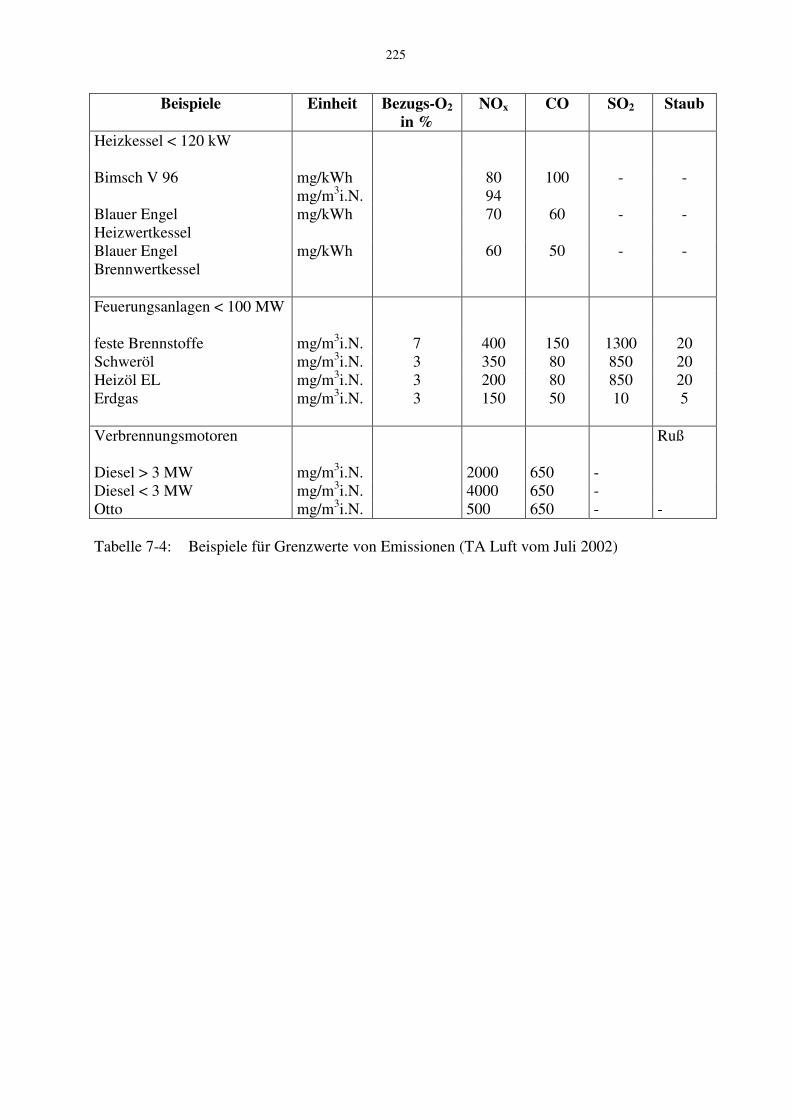
225
Beispiele Einheit Bezugs-O2
in %
NOx CO SO2 Staub
Heizkessel < 120 kW
Bimsch V 96 mg/kWh mg/m3i.N.
80 94
100 - -
Blauer Engel mg/kWh 70 60 - - Heizwertkessel Blauer Engel mg/kWh 60 50 - - Brennwertkessel
Feuerungsanlagen < 100 MW
feste Brennstoffe mg/m3i.N. 7 400 150 1300 20 Schweröl mg/m3i.N. 3 350 80 850 20 Heizöl EL mg/m3i.N. 3 200 80 850 20 Erdgas
mg/m3i.N. 3 150 50 10 5
Verbrennungsmotoren
Ruß
Diesel > 3 MW mg/m3i.N. 2000 650 - Diesel < 3 MW mg/m3i.N. 4000 650 - Otto mg/m3i.N. 500 650 - - Tabelle 7-4: Beispiele für Grenzwerte von Emissionen (TA Luft vom Juli 2002)


















