
AG Cypionka/Paläomikrobiologie Stand 10.11 · Quantitative PCR 60 Literatur 66. Methodenskript AG...
68
Methodenskript AG Cypionka/Paläomikrobiologie Stand 10.11.2003
Transcript of AG Cypionka/Paläomikrobiologie Stand 10.11 · Quantitative PCR 60 Literatur 66. Methodenskript AG...

Methodenskript
AG Cypionka/Paläomikrobiologie
Stand 10.11.2003

Methodenskript AG Cypionka - 1 -
I N H A L T S V E R Z E I C H N I S
Seite
Herstellung von Wachstumsmedien 3 Mineralisches Grundmedium für Anaerobier 4 Mineralisches Grundmedium für marine Aerobier 5 Spurenelementlösungen 5 Selenit-Wolframat-Lösung 6 Vitaminlösungen 6 Colorimetrische Bestimmung von Ammonium 7 Colorimetrische Bestimmung von Nitrit 7 Colorimetrische von Nitrat 7 Sulfidbestimmung nach Cline 10 Schnellnachweis von Schwefelwasserstoff 10 Turbidometrische Bestimmung von Sulfat 11 Colorimetrische Bestimmung von Thionaten 14 Colorimetrische Bestimmung von Sulfit 13 Colorimetrische Bestimmung von Schwefel 13 Colorimetrische Bestimmung von Orthophosphat 14 Proteinbestimmung nach Lowry 15 Proteinbestimmung nach Schmidt et al. 15 Proteinbestimmung nach Bradford 16 Bestimmung der Trockenmasse von Bakterienkulturen 17 Bestimmung der Carotinoide phototropher Bakterien 17 Bestimmung der Bacteriochlorophylle a, c, d und e 18 Berechnung der Zellzahlen aus MPN-Reihen 19 Berechnung der Zellzahlen aus Verdünnungsreihen 20 Herstellung von Agar-beschichteten Objektträgern 20 Bestimmung des Gram-Typs 21 Isolierung von Reinkulturen anaerober Bakterien 22 Geißelfärbung 23 Katalase-Test 23 Oxidase-Test 23 Substratspektren aerober Bakterienisolate 24 Aufnahme von Wachstumskurven 25 ß-Glucosidase-Aktivität 26 ß-Glucosaminidase-Aktivität 27 Leucin-Aminopeptidase 27 ß-Glucosidase-Aktivität in Mikrotiterplatten 28 Bestimmung der Gesamtzellzahl 28 Gesamtzellzahl in einer Wasserprobe 29 ATP-Bestimmung 30 Isolierung von DNA 32 DNA/RNA-Extraktion aus Sedimentproben 33 Schnelltest zur Quantifizierung von DNA 36 DNA-Extraktion mittels eines Kit von BioGene 36 Quantifizierung von DNA mit Picogreen 37 DNA-Quantifizierung am Microtiterplattenreader 38 PCR 40

Methodenskript AG Cypionka - 2 -
Agarose-Gelektrophorese 42 Sequenzieren von DNA 45 Denaturierende Gradienten-Gelelektrophorese 51 Fluoreszenz-in situ-Hybridisierung (FISH) 53 Quantitative PCR 60 Literatur 66

Methodenskript AG Cypionka - 3 -
Herstellung von Wachstumsmedien
Die Wachstumsmedien werden in speziell dafür angefertigten Glasgefäßen nach WIDDEL (1980,Abb. 1) zubereitet. Die Chemikalien wurden in angegebener Reihenfolge (siehe Tabellen) einge-wogen und unter Rühren gelöst. Die Abfüllglocke wird in Filztuch und Alufolie eingewickelt, derGasansatzstutzen mit dem Wattefilter mit einem Gummistopfen verschlossen. Die seitlichenSchraubverschlüsse werden ebenfalls verschlossen, allerdings wird während des Autoklavierensein seitlicher Schraubverschluß um etwa eine halbe Umdrehung geöffnet, um ein Zerplatzen desGefäßes durch Überdruck zu verhindern. Das Medium wird 50 min bei 121°C autoklaviert.
Abb. 1: Glaskolben zur Herstellung Medium für Anaerobier (nach WIDDEL 1980).
Nach dem Autoklavieren werden Supplementlösungen im Gasgegenstrom durch einen seitlichenStutzen zugegeben.
Oxische Medien:Nach dem Autoklavieren werden die seitlichen Schraubverschlüsse dicht verschlossen und derGummistopfen aus dem Gasansatzstutzen entfernt. Luft kann jetzt nur durch den Wattefilter inden Kolben eindringen. Um dem abgekühlten Medium die Supplementlösungen zugeben zu kön-nen wird der Kolben an N2 angeschlossen 5 kPa. Der Kolben mit dem fertigen Medium bleibt amStickstoff angeschlossen, da der Gasüberdruck zum Abfüllen benötigt wird. Das fertige Mediumwird unter der Abfüllglocke in sterile Röhrchen oder Flaschen abgefüllt.
Anoxische Medien:Nach dem Autoklavieren wird der Gasraum über dem Medium kurz mit N2/CO2 (80/20, v/v) mitN2 gespült, die Schraubverschlüsse dicht verschlossen und das Medium unter Rühren bei 5 kPaunter N2/CO2 (80/20, v/v) abgekühlt. Dem abgekühltem Medium wurden aseptisch im Gasgegen-strom die Supplementlösungen zugegeben. Der pH-Wert der Medien wurde (falls notwendig) mitsteriler 1 N Na2CO3 oder 1 N HCl auf 7.2 - 7.4 eingestellt. Das fertige Medium wird unter derAbfüllglocke in sterile Röhrchen oder Flaschen abgefüllt.

Methodenskript AG Cypionka - 4 -
Mineralisches Grundmedium für viele anaerobe Bakterien (nach Cypionka &Pfennig 1986)
(S => süß, B => Brackwasser, M => marin)S B M S B M
Substanz MW mM g/l
KH2PO4 136.09 1.5 1.5 1.5 0.20 0.20 0.20NH4Cl 53.49 5.0 5.0 5.0 0.25 0.25 0.25KCl 74.55 4.0 4.0 4.0 0.30 0.30 0.30CaCl2 * 2 H2O 147.02 1.0 1.0 1.0 0.15 0.15 0.15MgCl2 * 6 H2O 203.30 2.5 10 15 0.50 2.00 3.00NaCl 58.44 --- 222 342 --- 13 20Resazurin (0.5 mg/ml) ml 0.50 0.50 0.50
Autoklavieren und unter N2 abkühlen lassen danach aus sterilen Stammlösungen zusetzen:
Spurenelementlösung SL10 1.0 mlVitaminlösung (7 Vit.) 1.0 mlSe+W-Lösung1) (0.1 mM) 2*10-8 M 0.2 mlNaHCO3
2) (1 M => 30 mM) 30 mlDithionit (kristallin) wenig, bis zur Entfärbung < 17 mgpH3) einstellen mit steriler 1 M HCl oder Na2CO3
1) Nicht für alle Stämme benötigt 2) Vorsicht! Schutzgefäß, keine heißen Fläschchen berühren! 3) 6.8 - 7.0 für Medium S, 7.0 - 7.3 für Medium B und Medium M
Anmerkung: Die Medien B und M unterscheiden sich von S nur durch die Konzentrationen vonNaCl und MgCl2. Durch Zugabe eines Salzkonzentrats (NaCl, 5 M, + MgCl2, 0.2 M, ≈30 %Salz) lassen sich die Medien B und M aus weniger salzhaltigem Medium herstellen:
45 ml/l S-Medium für S => B 68 ml/l S-Medium für S => M 23 ml/l B-Medium für B => M
Häufig zu verwendetende Elektronendonatoren und -akzeptoren (mM)
H2 (80 %) + CO2 (20 %) + Acetat (2 mM), Lactat (20), Na2SO4 (10)Na2S2O3 (10), Na2S2O5 (5), NaNO3 (10)
Methodenskript AG Cypionka - 5 -
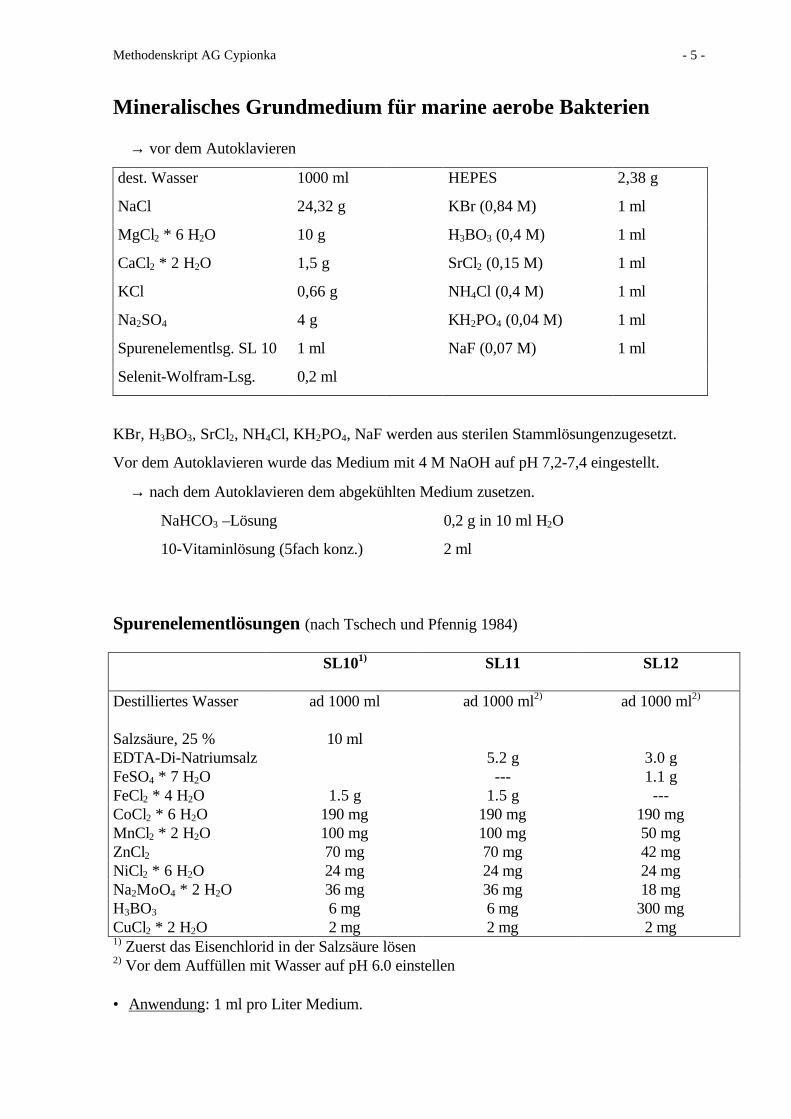
Mineralisches Grundmedium für marine aerobe Bakterien
→ vor dem Autoklavieren
dest. Wasser 1000 ml HEPES 2,38 g
NaCl 24,32 g KBr (0,84 M) 1 ml
MgCl2 * 6 H2O 10 g H3BO3 (0,4 M) 1 ml
CaCl2 * 2 H2O 1,5 g SrCl2 (0,15 M) 1 ml
KCl 0,66 g NH4Cl (0,4 M) 1 ml
Na2SO4 4 g KH2PO4 (0,04 M) 1 ml
Spurenelementlsg. SL 10 1 ml NaF (0,07 M) 1 ml
Selenit-Wolfram-Lsg. 0,2 ml
KBr, H3BO3, SrCl2, NH4Cl, KH2PO4, NaF werden aus sterilen Stammlösungenzugesetzt.
Vor dem Autoklavieren wurde das Medium mit 4 M NaOH auf pH 7,2-7,4 eingestellt.
→ nach dem Autoklavieren dem abgekühlten Medium zusetzen.
NaHCO3 –Lösung 0,2 g in 10 ml H2O
10-Vitaminlösung (5fach konz.) 2 ml
Spurenelementlösungen (nach Tschech und Pfennig 1984)
SL101) SL11 SL12
Destilliertes Wasser ad 1000 ml ad 1000 ml2) ad 1000 ml2)
Salzsäure, 25 % 10 mlEDTA-Di-Natriumsalz 5.2 g 3.0 gFeSO4 * 7 H2O --- 1.1 gFeCl2 * 4 H2O 1.5 g 1.5 g ---CoCl2 * 6 H2O 190 mg 190 mg 190 mgMnCl2 * 2 H2O 100 mg 100 mg 50 mgZnCl2 70 mg 70 mg 42 mgNiCl2 * 6 H2O 24 mg 24 mg 24 mgNa2MoO4 * 2 H2O 36 mg 36 mg 18 mgH3BO3 6 mg 6 mg 300 mgCuCl2 * 2 H2O 2 mg 2 mg 2 mg1) Zuerst das Eisenchlorid in der Salzsäure lösen2) Vor dem Auffüllen mit Wasser auf pH 6.0 einstellen
• Anwendung: 1 ml pro Liter Medium.
Methodenskript AG Cypionka - 6 -
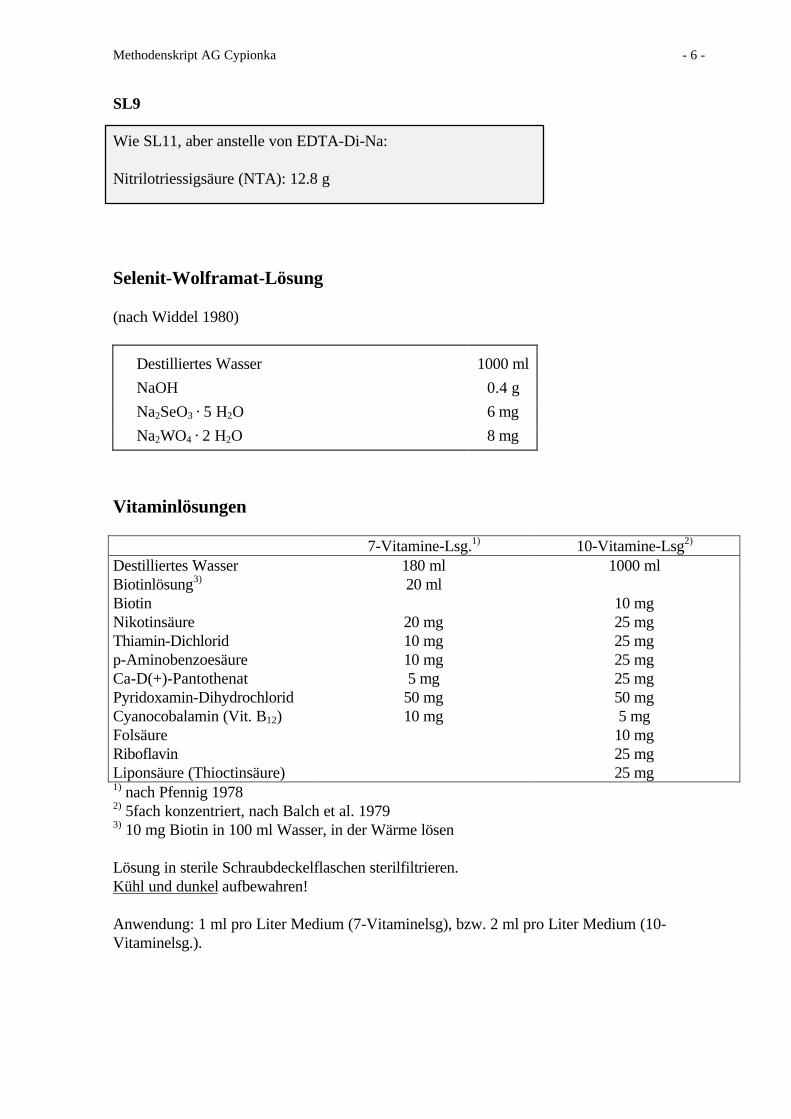
SL9
Wie SL11, aber anstelle von EDTA-Di-Na:
Nitrilotriessigsäure (NTA): 12.8 g
Selenit-Wolframat-Lösung
(nach Widdel 1980)
Destilliertes Wasser 1000 ml
NaOH 0.4 g
Na2SeO3 · 5 H2O 6 mg
Na2WO4 · 2 H2O 8 mg
Vitaminlösungen
7-Vitamine-Lsg.1) 10-Vitamine-Lsg2)
Destilliertes Wasser 180 ml 1000 mlBiotinlösung3) 20 mlBiotin 10 mgNikotinsäure 20 mg 25 mgThiamin-Dichlorid 10 mg 25 mgp-Aminobenzoesäure 10 mg 25 mgCa-D(+)-Pantothenat 5 mg 25 mgPyridoxamin-Dihydrochlorid 50 mg 50 mgCyanocobalamin (Vit. B12) 10 mg 5 mgFolsäure 10 mgRiboflavin 25 mgLiponsäure (Thioctinsäure) 25 mg1) nach Pfennig 19782) 5fach konzentriert, nach Balch et al. 19793) 10 mg Biotin in 100 ml Wasser, in der Wärme lösen
Lösung in sterile Schraubdeckelflaschen sterilfiltrieren.Kühl und dunkel aufbewahren!
Anwendung: 1 ml pro Liter Medium (7-Vitaminelsg), bzw. 2 ml pro Liter Medium (10-Vitaminelsg.).

Methodenskript AG Cypionka - 7 -
Bestimmung von Ammonium-Stickstoff (nach Chaney und Marbach)
Lösungen:A) 3 g Phenol + 3 mg Na-nitroprussid (Na-pentacyanonitrosylferrat (III) in 100 ml H2O dest. Ge-kühlt ca. 2 Wochen haltbar.
B) 2 g NaOH in 80 ml H2O dest., abkühlen lassen, 0.5 ml NaClO-Lösung mit 13% wirksamemChlor zusetzen und auf 100 ml auffüllen. (Vorsicht! Stark ätzend!).
Vorgehen:10 ml Probe werden in ein Reagenzglas pipettiert, 1 ml Lösung A zugesetzt, gemischt, 1 ml Lö-sung B zugesetzt und erneut gemischt. Eine Stunde bei Zimmertemperatur abgedunkelt stehenlassen. Bei zu starker Farbreaktion muß die Bestimmung mit verdünnter Probe durchgeführt wer-den. Anschließend Messung der Absorption bei 635 nm Wellenlänge gegen Ammonium-freienLeerwert. Niederschlag vorher abzentrifugieren! Der Nachweis ist sehr empfindlich. Die Glasge-räte müssen sauber sein; evtl. vorher nochmals spülen. Eichkurve: mit (NH4)2SO4 im Bereich 0 -100 µM aufnehmen (Achtung: 2 x Ammonium!).
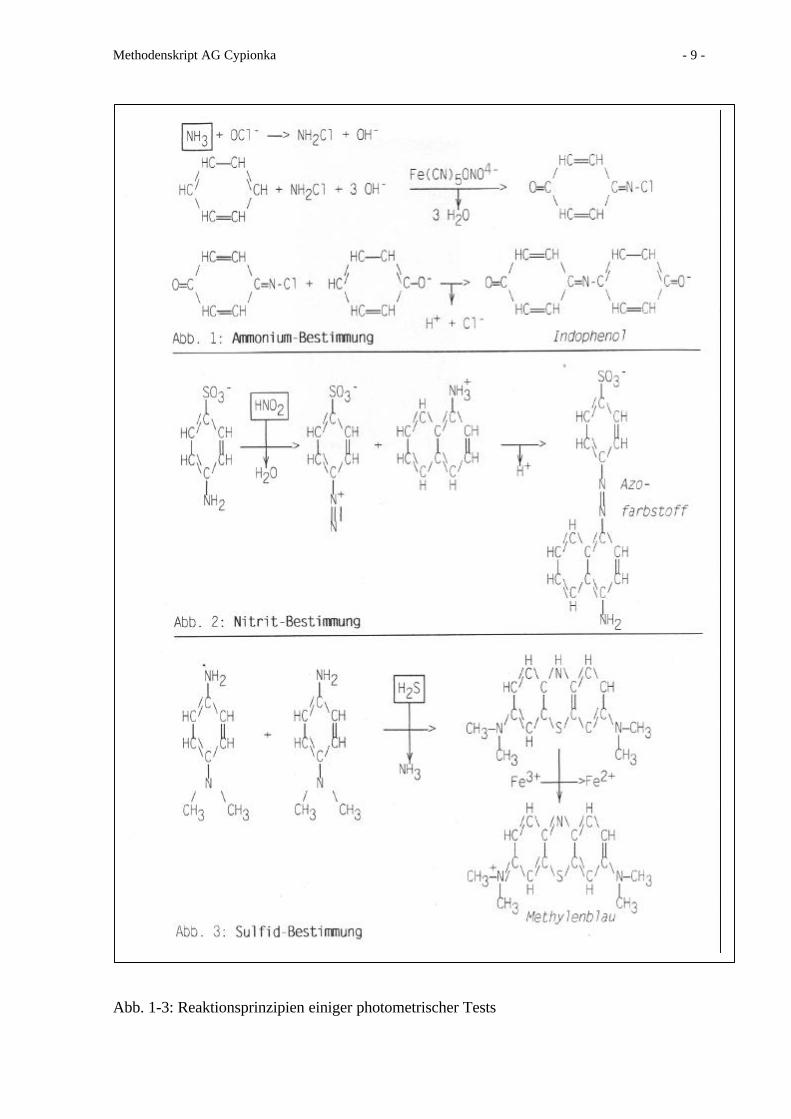
Prinzip der Reaktion siehe Abb.1
Bestimmung von Nitrit-Stickstoff (nach Göttinger Kursskript)
Lösungen:A) 1.65 g Sulfanilsäure wird in 375 ml heißem Wasser gelöst und mit 125 ml Eisessig versetzt.B) 0.5 g a-Naphthylamin wird in 100 ml Wasser suspendiert, 125 ml Eisessig zugesetzt, bis zurLösung gerührt und mit H2O ad 500 ml aufgefüllt. Vorsicht, stark carcinogen! Nicht mit denHänden berühren und nichts verschütten. Pipettierhilfe verwenden und benutzte Pipetten und Ge-fäße sogleich gut spülen.
Vorgehen:0.5 ml Probe in Reagenzglas, 0.5 ml Lsg. A zusetzen, mischen, 2.5 ml Lsg. B zusetzen. Messungnach 10 min. bei 530 nm.
Eichkurve: 0 - 100 µM mit KNO2 (Vorsicht, carcinogen!)
Prinzip der Reaktion siehe Abb.2
Bestimmung von Nitrat-Stickstoff (nach Goltermann)
Lösungen:A) 1 N HCl
B) 1 N NaOH

Methodenskript AG Cypionka - 8 -
C) Reduktionsmischung: C1) 0.039 g CuSO4 * 5 H2O in 100 ml H2O. C2) 0.12 g Hydrazinsulfat N2H4 * H2SO4 in 25 ml H2O.
5 ml C1 werden mit 25 ml C2 gemischt und mit H2O ad 50 ml aufgefüllt. Diese Reduktionsmi-schung und die Lösung C2 sind nicht haltbar und müssen täglich frisch hergestellt werden.
Vorgehen:10 ml zentrifugierte, partikel- und sulfidfreie Probe (wenn nötig, Sulfid mit CO2 austreiben) wirdmit 0.25 ml Lsg. B versetzt, 0.25 ml Lsg. C zugefügt, gemischt und 30 min bei 28-30 °C stehen-gelassen. Anschließend wird 0.25 ml Aceton und nach 5 Min. 0.25 ml HCl zugefügt. Das durchReduktion gebildete Nitrit wird anschließend nach der obigen Methode bestimmt. Vorsichtsmaß-nahmen wie bei der Nitritbestimmung!
Eichkurve: 0, 5 10, 20, 50, 100 und 200 µM KNO3.
Prinzip der Reaktion:Nitrat wird zu Nitrit reduziert. Die Reaktion verläuft allerdings nicht ausschließlich bis zum Nitrit.Daher Nitrit-Eichkurve nicht einfach übernehmen, sondern Eichkurve von Nitrat ausgehend er-stellen!
Methodenskript AG Cypionka - 9 -
Abb. 1-3: Reaktionsprinzipien einiger photometrischer Tests

Methodenskript AG Cypionka - 10 -
Colorimetrische Sulfidbestimmung (nach Cline)
Reagenz:1 g N,N-Dimethyl-p-Phenylendiammonium-Dichlorid (DMPD) und 1.5 g FeCl3 * 6 H2O in25%iger HCl lösen, mit 25%iger HCl auf 50 ml auffüllen. Vorsicht! Die Lösung ist stark sauerund carcinogen! Reagenz nicht mit Händen berühren, stets Pipettierhilfe verwenden, auf anhaften-de Reste achten! Benutzte Pipetten und Gefäße nicht stehen lassen, sondern alsbald spülen.
Vorgehen:In verschraubbaren 15 ml-Reagenzgläsern 0.4 ml Reagenz (mit Eppendorfpipette) vorlegen. Dazu5 ml Probe pipettieren. Reagenzglas verschließen, sofort mischen und nach frühestens 20 min dieExtinktion bei 670 nm messen. Sollte die Extinktion größer als 1 werden, muß weniger Probeeingesetzt werden (und dann nach Entwicklung der blauen Farbe das Volumen mit dest. Wasserauf 5 ml ergänzt werden).
Eichkurve: Als Standard dient eine Na2S-Lösung: 500 ml H2O wird in einem Meßkolben miteinem NaOH-Plätzchen versetzt und für mindestens 20 min durch eine lange Kanüle mit N2
durchspült. Danach wird ein in dest. H2O gewaschener und getrockneter, genau gewogener Na2S* 9 H2O, von etwa 0.6 g, hinzugegeben und der Kolben (N2-begast) mit einem Gummiseptumverschlossen. Diese Stammlösung enthält etwa 5 mM Sulfid (= 5 nmol/µl bei 0.6 g Na2S * 9 H2O)und ist nur unter Stickstoff maximal 1 Tag haltbar. Für die Eichkurve werden in 12 Reagenzröhr-chen 5 ml H2O vorgelegt, mit einem Gummiseptum verschlossen und 20 Minuten mit Stickstoffausgeblasen. Mit einer Hamiltonspritze (Vorsicht beim Durchstechen des Gummiseptums!) wer-den 0, 2, 5, 10, 20 und 50 µl (0 bis 250 nmol Sulfid) der Sulfidstammlösung zugesetzt, gemischtund sofort 0.4 ml Reagenz mit einer 1 ml-Spritze hinzugefügt (Doppelansätze). Nach 20 Minutenwird die Extinktion bei 670 nm gegen einen Ansatz ohne Sulfidlösung gemesssen. Zur Berech-nung der molaren Konzentration der Na2S-Lösung wird das Formel-Gewicht für Na2S * 9 H2Oangenommen, was sich bisher bewährt hat. Für sehr genaue Messungen müßte ein Aliquot derNa2S-Lösung in eine frische, genau eingestellte saure J-KJ-Lösung gegeben und das überschüssigeJod mit einer Na2S2O3-Lösung zurücktitriert werden.
Prinzip der Reaktion siehe Abb.3
Schnellnachweis von Schwefelwasserstoff (nach Widdel)
Reagenz: HCl 50 mM mit CuSO4 5 mM
für den Leerwert: HCl 50 mM
Vorgehen:Mit bis zu 0.5 ml Probe eine 3 ml-Küvette mit Reagenz (unter heftiger Rührung mit einem Mi-krorührfisch) auf 2.0 ml auffüllen. Innerhalb einer Minute Extinktion bei 436 nm gegen Wassermessen. Als Leerwert wird dieselbe Menge der Probe mit 50 mM HCl ohne CuSO4 versetzt undphotometriert.

Methodenskript AG Cypionka - 11 -
Eichkurve aufnehmen im Bereich bis 2 µmol/Ansatz
Dieser Test kann auch qualitativ zur Überprüfung der Sulfidbildung in Bakterienkulturen verwen-det werden. Auch ohne Rührung und Photometer zeigt das kolloidal ausfallende Kupfersulfid einekräftig braune Färbung.
Turbidometrische Bestimmung von anorganischem Sulfat (nach Cypionka undPfennig, Tabatabai)
Reagentien:A) 10 g Citronensäure * H2O, H2O ad 80 ml, lösen und mit 120 ml Glycerin (99.5%) mischen.B) 0.5 g BaCl2 * 2 H2O, 5 g Citronensäure * H2O, H2O ad 50 ml.
Vorgehen:2 ml Probe (wenn nötig, klarzentrifugiert) mit 2 ml Lösung A schlierenfrei mischen. 0.5 ml Lö-sung B zusetzen und sofort schlierenfrei mischen. Nach 30 - 45 Minuten erneut mischen und dieTrübung bei 436 nm gegen sulfatfreie Kontrolle messen. Immer Doppelbestimmungen mit ver-schieden verdünnten Proben durchführen! Stets Eichstandards über den erwarteten Konzentrati-onsbereich der Proben mitmessen!
Eichkurve im Bereich von 0.1 - 5.0 µmol Sulfat pro Testansatz anlegen. Für die Erstellung derTiefenprofile werden die von Sulfid und Partikeln befreiten Wasserproben (1 l) verwendet.
Prinzip der Reaktion: Ba2+ + SO42- → BaSO4 (unlöslich)
Citronensäure säuert den Ansatz an und komplexiert die Ba2+-Ionen. Aus den Komplexen entste-hen bei der Reaktion mit Sulfat sehr kleine Kristalle mit hoher optischer Dichte. Glycerin verlang-samt die Sedimentation der Kristalle.
Sulfat-Schnelltest mit 0.2 M HCl und 0.2 M BaCl2
1 ml Kultur mit 2 Tropfen HCl ansäuern, umschütteln, 2 Tropfen BaCl2 zusetzen. SofortigeTrübung zeigt Sulfat, langsam auftretende evtl. Thiosulfat (=> Schwefel) an.
Photometrische Analyse von Thionaten (nach Kelly et al. 1969; Fitz und Cypionka 1990)
Der photometrische Nachweis von Thiosulfat, Trithionat und Tetrathionat beruht auf der alkali-schen Cyanolyse der Thionate, die zu Thiocyanatäquivalenten umgesetzt werden. Zur Unterschei-dung der Thionate wird die Cyanolyse-Reaktion bei verschiedenen Temperaturen und zum Teilmit CuSO4 als Katalysator durchgeführt. Die dabei entstehenden Thiocyanate können dann als rot-brauner Komplex mit Eisen (III) photometrisch quantifiziert werden.

Methodenskript AG Cypionka - 12 -
(0°C)I S4O6
2- + 3 CN- + H2O → S2O32- + SO4
2- + 2 HCN + SCN-
(0°C, CuSO4)II S2O3
2- + CN- → SO32- + SCN-
(100°C, CuSO4)III S3O6
2- + 3 CN- + H2O → SO32- + SO4
2- + 2 HCN + SCN-
Daraus folgt, daß bei der Messung von Ansatz I lediglich die Konzentration von Tetrathionat er-mittelt wird. Bei der Messung des Ansatzes II werden die Konzentrationen von Tetrathionat, desbei der Cyanolyse entstehenden Thiosulfates und des bereits im Ansatz befindlichen Thiosulfateserfaßt. Bei der Messung des Ansatzes III wird die Gesamtkonzentration aller im Ansatz vorhan-denen Thionate (Thiosulfat, Tri- und Tetrathionat) ermittelt.
Lösungen:
1.) NaH2PO4 - NaOH - Puffer 1 M, pH 7.42.) KCN 1.25 M3.) CuSO4 * 5 H2O 0.375 M4.) Fe(NO3)3 * H2O 1.5 M gelöst in 4 M HClO4, Volumenzunahme beim Lösen! => 100 ml HClO4 + 92 g Fe(NO3)3 => 150 ml Gesamtvol.
Als Standards werden 1 mM Lösungen von Thiosulfat, Tetrathionat und Trithionat täglich frischangesetzt.
Ansätze:
In jedes Reagenzglas werden 0.06 ml der Lösung 1.) vorgelegt. Dann wird die Probe hinzupipet-tiert (max. 2.25 ml) und mit Aqua bidest. auf 2.31 ml Gesamtvolumen aufgefüllt. Der Standardwird als Mix mit allen drei Thionaten eingesetzt. Es wird für jeden Ansatz eine Nullprobe ge-macht, mit welcher der Nullabgleich bei der photometrischen Messung durchgeführt wird. 3 ver-schiedene Ansätze (I,II u. III)
Ansatz I zur Bestimmung von Tetrathionat:Ansatz I wird 10 min auf 0°C abgekühlt, bevor 0.06 ml der Lösung 2.) und 0.06 ml Aqua bidest.hinzugefügt werden (Mischen!). Der Ansatz bleibt dann ca. 20 min im Eisbad.
Ansatz II zur Bestimmung von Thiosulfat:Ansatz II wird 10 min auf 0°C abgekühlt. Dann wird 0.06 ml der Lösung 2.) hinzupipettiert (Mi-schen!). Nach einer 10-minütigen Inkubationszeit werden 0.06 ml der Lösung 3.) hinzupipettiert(Mischen!), bevor der Ansatz weitere 10 min im Eisbad verbleibt.

Methodenskript AG Cypionka - 13 -
Ansatz III zur Bestimmung von Trithionat:Nachdem dem Ansatz III 0.06 ml der Lösung 2.) hinzugefügt werden, wird er 45 min im Wasser-bad gekocht. Die Reagenzgläser sind dabei durch Glasmurmeln verschlossen. Danach wird derAnsatz auf 0°C abgekühlt (ca. 10 min) bevor 0.06 ml der Lösung 3.) hinzupipettiert werden (Mi-schen!). Danach bleibt der Ansatz weitere 10 - 15 min im Eisbad.Abschließend werden zu allen Ansätzen 1 ml der Lösung 4.) hinzugefügt (Mischen!). Wenn dieAnsätze dann auf Raumtemperatur wieder erwärmt sind, kann die Extinktion 460 nm gemessenwerden. Entsprechend den oben angegebenen Reaktionsgleichungen sind die Konzentrationen derThionate wie folgt zu ermitteln:
Konzentration von Tetrathionat: Ansatz I
Konzentration von Thiosulfat: Ansatz II - 2 x Ansatz I
Konzentration von Trithionat: Ansatz III - Ansatz II
Colorimetrischer Sulfitnachweis (nach Pachmayr)
Reagenz A: Säureentfärbte Fuchsinlösung
400 mg Fuchsin werden mit Aqua bidest. und 125 ml Schwefelsäure (konz.) versetzt und mitAqua bidest. auf einen Liter aufgefüllt.
Reagenz B: Formaldehyd 32 %ige Lösung
Ansatz:Die Probe wird mit Aqua bidest. auf 8.9 ml aufgefüllt. Dann erfolgt die Zugabe von 1 ml ReagenzA und 0.1 ml Reagenz B (mischen!).
10 min nach der letzten Zugabe erfolgt die photometrische Messung bei 570 nm gegen einen An-satz ohne Sulfit.
Colorimetrischer Schwefel-Nachweis (nach Chan und Suzuki, 1993)
Lösungen:
(1) 10 ml A. dest + 190 ml Aceton(2) 0.2 g NaCN + 125 ml Lösung (1)(3) 0.4 g FeCl3 * 6 H2O + 5 ml A. dest(4) Aceton(5) Petrolether(6) 6.4 mg S° in 10 ml DMSO (Endkonzentration 20 mM)(7) 3.2 mg S° in 10 ml Petrolether

Methodenskript AG Cypionka - 14 -
Vorgehen:
- Herstellen einer S°-Eichreihe mit Lösung (6) (weißer Niederschlag) und Puffer von 5-1000 µM 0 µM = Leerwert- Extraktion: 0.5 ml Bakteriensuspension (oder Eichlösung) + 1.0 ml Lösung (5) in Eppendorf-Caps- mischen (30 sec)- Zentrifugation: 14 000 Upm, 10 min in der Eppendorfzentrifuge der Überstand wird klar- Ansatz: 0.5 ml Überstand + 1.0 ml Lösung (2) in E.-Caps- mischen u. 2 min reagieren lassen- Meßansatz: 0.95 ml Lösung (4) + 0.05 ml Lösung (3) + 0.50 ml "Ansatz" in E.-Caps
- mischen, es bildet sich ein bräunlicher Niederschlag- Zentrifugation: 14 000 Upm, 1 min in der Eppendorfzentrifuge- Extinktion des Überstands messen bei 464 nm
Photometrische Bestimmung von Orthophosphat
Reagentien:A) Molybdatschwefelsäurereagenz:14.4 ml konz. H2SO4 (d = 1.84) werden in 30 ml H2O dest. gelöst und nach dem Abkühlenfolgende Lösungen zugesetzt:
1 g Amidosulfonsäure in 10 ml H2O;1.25 g (NH4)6 Mo7O24 * 4 H2O in 20 ml H2O;34.4 mg Kalium- antimontartrat in 10 ml H2O.Es wird mit H2O dest. auf 100 ml aufgefüllt.
B) 1.0 g Ascorbinsäure in 10 ml H2O dest. Täglich frisch herstellen.
Vorgehen:10 ml Wasserprobe (filtriert) werden im Reagenzglas mit 0.4 ml Reagenz A und 0.25 ml ReagenzB versetzt. Nach mindestens 10 min wird die Absorption bei 865 nm Wellenlänge in 1 cm-Küvetten gegen einen Reagenzienleerwert mit H2O gemessen.
Eichkurve:Mit KH2PO4 im Bereich von 0.2 - 40 µmol/l. Vergleichsweise auch Eichwerte mit 400 und 4000µmol/l einsetzen.
Prinzip der Reaktion: Das Molybdän in der entstandenen Molybdato-phosphorsäure wird mitAscorbinsäure zu Mo(+IV) reduziert, das mit dem übrigen Mo(+VI) eine blaue Verbindung ausgemischten Wertigkeitsstufen bildet.

Methodenskript AG Cypionka - 15 -
Proteinbestimmungen
1. Proteinbestimmung nach Lowry
Reagenzien:• Kupferreagenz: 0.1 g CuSO4 * 5 H2O in 20 ml 1%-iger K-Na-Tartratlösung lösen.1 ml dieser Lösung mit 50 ml Na2CO3-Lösung (2%) vermischen. Die Lösung täglich frisch anset-zen.
• Folin-Reagenz: 1 Teil Folin-C.-Reagenz (Merck) mit 2 Teilen dest. Wasser mischen.
• NaOH: 0.3 M
Vorgehen:10 ml Zellsuspension in der Kühlzentrifuge abzentrifugieren (6000g, 10 min) und einmal mit 0.6%-iger Kochsalzlösung waschen. Das Sediment wird sorgfältig in Kochsalzlösung resuspendiertund auf 10 ml aufgefüllt. In drei Proben von je 1 ml wird das Gesamtprotein nach LOWRY et al.bestimmt.
Um die Zellen aufzuschließen, 1 ml Probe mit 0.50 ml 0.3 M NaOH versetzen und im Wasserbadbei 60 °C in verschlossenem Reagenzglas während 90 min erhitzen. Nach dem Abkühlen 5 mlKupferreagenz unter ständigem Schütteln zufügen und 10 min im Dunkeln stehen lassen. Dannwird 0.5 ml Folin-Reagenz zugesetzt, sofort geschüttelt und 30 min im Dunkeln gehalten. Dannwird zentrifugiert (6000g, 10 min) und anschließend bei 623 nm gegen einen Blindwert photome-triert (d = 1cm). Mit Serumalbumin wird eine Eichkurve aufgestellt 10 - 200 µg pro Ansatz.
2. Proteinbestimmung nach Schmidt et.al. (1963) (abgewandelte Biuretmethode nach La Riviére 1958)
Reagenzien:(A) NaOH 4 M (= 160 g/l)(B) K-Na-Tartrat 5 g NaOH 4 g CuSO4 * 5 H2O 1 g KJ 2.5 g in H2O 400 ml
Vorgehen:- 10 ml Zellsuspension mit 0.9 % NaCl waschen und abzentrifugieren- Röhrchen mit 0.9 % NaCl auf 5 ml auffüllen- 0.5 ml Reagenz A zugeben, schütteln- genau 10 Minuten in kochendes Wasserbad (Glaskugeln auf die Reagenzgläser)- sofort in kaltem Wasser abkühlen- 2 ml Reagenz B zugeben, schütteln- 30 Minuten in 37 °C Wasserbad- Bei Trübung Partikel abzentrifugieren- Extinktion bei 546 nm messen

Methodenskript AG Cypionka - 16 -
- Eichkurve mit BSA (Stammlösung: 50 mg/ml) im Bereich von 0 bis 10 mg/Ansatz
3. Proteinbestimmung nach Bradford (1976)
Diese Proteinbestimmungsmethode beruht auf der Bindung von Coomassie Brilliant Blue G-250an Protein. Die Bindung des Farbstoffs verursacht eine Verschiebung des Absorptionsmaximumdes Farbstoffes von 465 zu 595 nm. Es wird der Anstieg der Absorption bei 595 nm gemessen.Dieser Test ist leicht reproduzierbar und schnell, weil schon nach 2 Minuten eine stabile Farbeauftritt, die ihrerseits für eine Stunde stabil ist. Es besteht nur eine geringe oder keine Beeinträch-tigung durch Natrium- oder Kalium-Ionen, ebensowenig durch Kohlenhydrate, wie z.B. Saccha-rose. Eine geringe Färbung entsteht bereits in der Gegenwart von alkalischer Puffer, jedoch kann derTest genau durchgeführt werden, wenn entsprechende Pufferkontrollen gemacht werden. Die ein-zigen Verbindungen, die eine starke Störung der Färbung im Test hervorrufen, sind relativ großeMengen an Detergenzien, wie Natriumdodecylsulfat, Triton X-100 und handelsübliche Glaswa-rendetergentien. Durch geeignete Kontrollen kann jedoch die Störung von geringen Mengen anDetergentien ausgeglichen werden. Coomassie Brilliant Blue G-250 kommt in zwei verschiedenen Farben vor, rot und blau. Die roteFarbe wird nach der Bindung des Farbstoffs an das Protein in die blaue Farbe umgewandelt.
Reagenz: 100 mg Coomassie Brilliant Blue G-250 werden in 50 ml 95% Ethanol gelöst. Zu die-ser Lösung werden 100 ml 85% (w/v) Phosphorsäure gegeben. Diese Lösung wird dann auf 1Liter aufgefüllt. Die Endkonzentration im Reagenz sind 0.01% (w/v) Coomassie Brilliant Blue G-250, 4.7% (w/v) Ethanol, und 8.5% (w/v) Phosphorsäure.
Standardmethode: Proteinlösungen, die 10 bis 100 µg Protein in 0.1 ml enthalten, werden in12*100 mm-Reagenzgläser pipettiert. Das Volumen in den RG wird mit entsprechendem Pufferauf 0.1 ml aufgefüllt. 5 ml Reagenz hinzugeben und mischen. Nach 2 Minuten und vor Ablaufeiner Stunde die Absorption bei 595 nm gegen den Leerwert messen.
Mikro-Test: Proteinlösungen, die 1 bis 10 µg Protein in einem Volumen von 0.1 ml enthalten,werden sofort in Halbmikro- Küvetten pipettiert. Das Volumen wird entsprechend mit geeignetemPuffer auf 0.1 ml aufgefüllt. 1 ml Reagenz wird zugegeben und gemischt. Die Absorption wirdebenfalls bei 595 nm gegen die Referenz gemessen.
Eichkurven mit Rinderserumalbumin (BSA) in 0.15 M NaCl herstellen.

Methodenskript AG Cypionka - 17 -
Bestimmung der Trockenmasse von Bakterienkulturen
(nach Widdel, modifiziert nach Cypionka und Pfennig)
1. Wägegefäße (abgeschnittene Reagenzgläser mit etwa 3 ml Volumen) gründlich reinigen (amSchluß nicht mehr mit den Fingern, sondern nur mit Pinzette berühren), in saubere kleine Alu-Zylinder stellen und bei 80 °C trocknen. Es wird keine Eichkurve erstellt, jedoch sollte immer einleeres Gefäß als Reserve und zur Kontrolle der Wägungen parallel eingesetzt werden.
2. Sulfidhaltige Bakterienkulturen mit CO2 durchperlen, um H2S auszutreiben, auf jeden FallKultur gründlich homogenisieren, optische Dichte (bei 436 nm oder einer anderen Wellenlänge,bei der keine spezifische Absorption gegeben ist) messen und notieren.3. Ein genau bestimmtes (und notiertes!) Volumen (500 - 1000 ml) in 250 ml-Zentrifugenbechernfür mindestens 20 min zentrifugieren (12000 upm), austarieren mit dest. Wasser.
4. 300 ml Ammonium-Acetat-Puffer, etwa 50 mM, pH 6.5 (mit Essigsäure einstellen), herstellen.Dieser Puffer verdampft bei Erhitzen vollständig als NH3 und Essigsäure!
5. Sofort nach Stillstand der Zentrifuge Überstand vorsichtig absaugen, Zellen aus mehreren Be-chern in einem einzigen sammeln, in Ammonium-Acetat-Puffer waschen, erneut abzentrifugieren.
6. Die trockenen Wägegefäße auf der oberschaligen Waage vorwiegen, die auf Null geeichteAnalysenwaage entsprechend voreinstellen und Gefäße genau wiegen, Leergewicht notieren.
7. Sofort nach Stillstand der Zentrifuge Überstand absaugen, Zellen (behutsam!) in 0.5 ml dest.Wasser mit Hilfe einer Pasteurpipette in das Wägegefäß überführen; mindestens zweimal mit 0.5ml dest. Wasser nachspülen.
8. Bei 80 °C bis zur Gewichtskonstanz trocknen (ein bis zwei Tage).
9. Die abschließende Wägung muß unmittelbar nach Entnahme der Proben aus dem heißenTrockenschrank erfolgen, da die Zellen Wasser absorbieren. Dazu Analysenwaage entsprechendvoreinstellen.
10. Wägegefäße gründlich reinigen! Trockenmasse pro OD-Einheit und Volumen ausrechnen.
Chromatographische Analyse der Carotinoide phototropher Bakte-rien
(nach Eichler und Pfennig)
a) Ernte der BakterienmasseGut gewachsene Kulturen werden abzentrifugiert (9000 rpm, 20 min), das überstehende Kul-turmedium vorsichtig dekantiert und verworfen.

Methodenskript AG Cypionka - 18 -
b) Extraktion der Carotinoide
Carotinoide sind in Gegenwart von Luft und Licht unbeständig!
Extraktionsmittel: Ethanol abs.: Aceton = 1:1.
Das Pellet von a) wird in dem restlichen Überstand (evtl. 0,2 ml dest. Wasser zusetzen) resuspen-diert und vollständig in ein 10 ml-Zentrifugenglas überführt. Dann werden 8 ml Extraktionsmittelzugegeben, gut durchgemischt und das Röhrchen nach Begasen mit N2 mit einem Butylgummi-stopfen verschlossen. Röhrchen etwa 1 Stunde bei Zimmertemperatur extrahieren lassen, noch-mals durchmischen und Zellreste abzentrifugieren; Gummistopfen vorher abnehmen, Tischzentri-fuge, 15 min. Farbigen Überstand bei 25 °C im Rotationsverdampfer unter schwarzem Tuch ein-engen. Farbigen Satz mit 0,75 ml Extraktionsmittel aufnehmen, in ein Röhrchen überführen, mitN2 begasen und mit Butylgummistopfen verschließen; im Dunkeln aufbewahren.
c) Chromatographie auf Dünnschicht-Platten
Laufmittel: Petroleumbenzin : Aceton = 9:1
Chromatographiekammer mit Fließpapier auslegen, mit N2 begasen und schließen. Dann 100_mlLaufmittel einfüllen und 1 h sättigen lassen. Den Extrakt von b) mit Hamilton-Spritze vorsichtigauf die Startlinie einer Kieselgel-Dünnschicht-Platte unter einem N2-Strom zum Trocknen auftra-gen (etwa 100 µl auf 3 cm Strecke auftragen, daneben etwa 50 µl auf 3 cm). Vergleichsextrakteebenso behandeln. Platte in der abgedunkelten Kammer entwickeln. Wenn Laufmittel etwa 16 cmhoch gestiegen ist (mit Bleistift markieren), Platte kurz trocknen mit N2 und nochmals entwickeln.Dann Platte mit N2 trocknen. Auf der trocknen Platte Banden mit Spatel vorsichtig markieren(Laufhöhe!), Platte mit Glasplatte abdecken und möglichst rasch auf durchsichtige Folie und Pa-pier kopieren. Farben der Banden genau aufschreiben und anhand der Banden der Vergleichsor-ganismen identifizieren.
Bestimmung der Bacteriochlorophylle a, c, d und e(nach Oelze, bzw. Steenbergen und Korthals)
Bakterienkultur (2 bis 5 ml) wird über ein Glasfaserfilter (f 25 mm) bzw. (bei Stämmen mit be-sonders kleinen Zellen) über Membranfilter (Porenweite 0,2 µm , f 2,5 cm) abfiltriert. NachÜberführen in 3 ml Aceton wird im Dunkeln bei 4°C über Nacht extrahiert. Die Messung erfolgtgegen reines Extraktionsmittel für Bchl a bei 771 nm Bchl c bei 663 nm Bchl d bei 652 nm Bchl ebei 647 nm. Die Pigmentkonzentration errechnet sich nach dem Lambert-Beer'schen Gesetz
E = c * d * εmit E = Extinktion am Absorptionsmaximum c = Pigmentkonzentration d = Schichtdicke der Küvette (1 cm) ε = Extinktionskoeffizient am Absorptionsmaximum für
Bchl a = 92.3 ml * mg-1 * cm-1
Bchl c = 92.6 ml * mg-1 * cm-1
Bchl d = 98.0 ml * mg-1 * cm-1 (dito anzunehmen für Bchl e)
Methodenskript AG Cypionka - 19 -
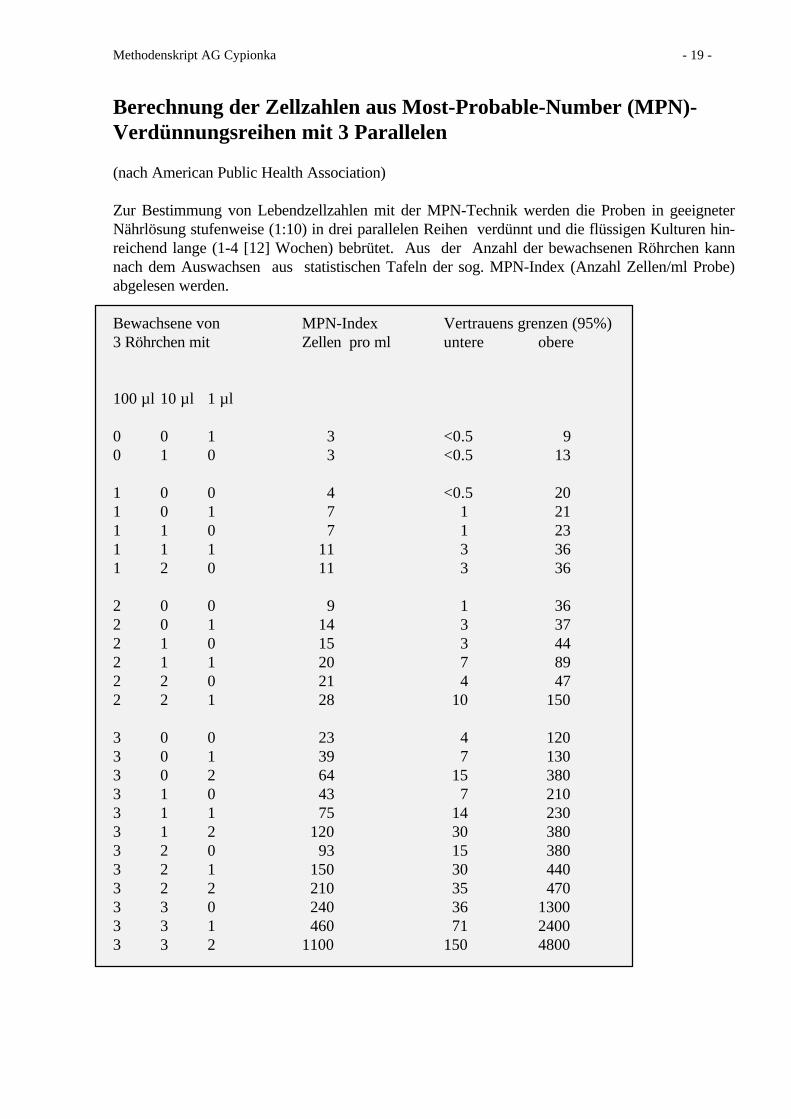
Berechnung der Zellzahlen aus Most-Probable-Number (MPN)-Verdünnungsreihen mit 3 Parallelen
(nach American Public Health Association)
Zur Bestimmung von Lebendzellzahlen mit der MPN-Technik werden die Proben in geeigneterNährlösung stufenweise (1:10) in drei parallelen Reihen verdünnt und die flüssigen Kulturen hin-reichend lange (1-4 [12] Wochen) bebrütet. Aus der Anzahl der bewachsenen Röhrchen kannnach dem Auswachsen aus statistischen Tafeln der sog. MPN-Index (Anzahl Zellen/ml Probe)abgelesen werden.
Bewachsene von MPN-Index Vertrauens grenzen (95%)3 Röhrchen mit Zellen pro ml untere obere
100 µl 10 µl 1 µl
0 0 1 3 <0.5 90 1 0 3 <0.5 13
1 0 0 4 <0.5 201 0 1 7 1 211 1 0 7 1 231 1 1 11 3 361 2 0 11 3 36
2 0 0 9 1 362 0 1 14 3 372 1 0 15 3 442 1 1 20 7 892 2 0 21 4 472 2 1 28 10 150
3 0 0 23 4 1203 0 1 39 7 1303 0 2 64 15 3803 1 0 43 7 2103 1 1 75 14 2303 1 2 120 30 3803 2 0 93 15 3803 2 1 150 30 4403 2 2 210 35 4703 3 0 240 36 13003 3 1 460 71 24003 3 2 1100 150 4800

Methodenskript AG Cypionka - 20 -
Berechnung der Zellzahlen aus Kolonien in oder auf Agar mit verschiedenenVerdünnungsstufen
(nach Cavalli-Sforza)
ΣiCi
x = ——— Σi(ni*zi)
x = mittlere Zellzahl im BeimpfungsvolumenC1, C2, ... , Ci = Anzahl der auf den einzelnen Platten gezählten Kolonienn1, n2, ... , ni = Platten/Verdünnungsstufez1, z2, ... , zi = Verdünnungen
Beispiel:
Verdünnung: 10-6: 303, 290, 285 Kolonien " 10-7: 32, 21 "
Mittlere Zellzahl x = 931/(3*10-6 + 2*10-7) = 2.91*108
Herstellung von Agar-beschichteten Objektträgern für die Mikrophotographie
(nach Pfennig und Wagener)
- Agar waschen2 g Difco Agar werden in ca. 500 ml H2O dest. 5 mal gewaschen, um den Agar von Staub undkleinen Partikeln zu reinigen. Zum Waschen wird die Agarlösung 10 min. auf dem Magnetrührergerührt und dann ca. 20 min. stehen gelassen bis der Agar sich vollständig abgesetzt hat. DerÜberstand wird vorsichtig dekantiert und der Agar erneut mit destilliertem Wasser gewaschen.Nach dem letzten Waschen wird die Suspension auf 100 ml mit H2O aufgefüllt (Endkonzentrationvon 2 %). Diese Agarlösung wird 20 min. bei 120 °C autoklaviert.
- Beschichten der ObjektträgerSorgfältig gereinigte Objektträger werden mit Agar beschichtet. Dazu werden die Objektträgerauf eine horizontale Platte gelegt (diese ggf. mit Wasserwaage tarieren). Mit Hilfe zweier Infra-rotlampen werden die Objektträger erwärmt, um eine gleichmäßige Beschichtung zu erhalten. Dieautoklavierte Agarlösung wird in einem Wasserbad bei 45 °C gehalten. 2 ml Agarlösung werdenauf einem Objektträger vorsichtig verteilt. Dazu wird mit der Pipettenspitze eine Zickzack-Linievon einem Ende des Objektträgers zum anderen gezogen. Dabei sorgfältig ein Herabfließen desAgars vom Objektträger zu vermeiden. Die Agarobjektträger müssen einige Tage trocknen. An-schließend sollten sie in einem geschlossenen Kasten staubfrei aufbewahrt werden.

Methodenskript AG Cypionka - 21 -
- Photographieren von BakterienZur photographischen Darstellung sollten die Bakterienkulturen eine hohe Zelldichte aufweisen;ist diese zu gering, wird die Zellsuspension durch Zentrifugation konzentriert. Drei Tropfen von20, 22 und 25 µl werden mit einer 0.1 ml- oder 0.2 ml-Pipette auf den Agar-beschichteten Ob-jektträger pipettiert. Jeder Tropfen wird sofort mit einem Deckglas (18x18 mm) abgedeckt. Umein Verdunsten der Flüssigkeit zu verhindern, werden alle vier Seiten des Deckglases mit Paraf-finlösung abgedichtet. Dazu wird ein Metallspatel erhitzt, mit der flachen Seite in das Paraffineingetaucht und anschließend am Deckglasrand vorsichtig angesetzt.
Bestimmung des Gram-Typs
a) Färbung nach GRAM (Modifiziert nach Bartholomew)
1) Ausstrich der Bakterien aus frischer Kultur (schnellwüchsige Bakterien maximal 24 h alt) aufsauberem Objektträger (oder Deckglas) herstellen und an der Luft trocknen lassen. Ausstrich fi-xieren durch dreimaliges Ziehen durch die Flamme (nicht Pyrolysieren!). Auf demselben Objekt-träger Vergleichsausstriche mit Referenzbakterien herstellen (mit einem Gram-negativen und ei-nem Gram-positiven Stamm).2) Fixierte Bakterien 1 min mit Huckers Ammoniumoxalat-Kristallvioett-Lösung färben.3) Farblösung ablaufen lassen, Ausstrich 5 sec mit fließendem Wasser waschen.4) Kaliumjodid-Jodlösung 1 min einwirken lassen.5) 5 sec mit fließendem Wasser abwaschen.6) Differenzieren: 3 x je 1/2 min in 3 Töpfen n-Propanol entfärben.7) 5 sec waschen8) Ausstrich eventuell mit Wasser und Deckglas mikroskopieren, um Qualität zu prüfen9) Eventuell Gegenfärbung 1 min mit Safranin. Farblösung ablaufen lassen und Ausstrich kurzmit fließendem Wasser abwaschen.10) Ausstrich trocknen, angefärbte Bakterien mit 10- oder 40- facher Vergrößerung suchenund dann direkt mit Immersionsöl ohne Deckglas im Hellfeld im Objektiv Ölimmersion, 100fache Vergr., bei offener Blende mikroskopieren.11) Resultat mit Gram-negativen und Gram-positiven Referenzbakterien vergleichen.
b) Kalilauge-Test (nach Gregersen)
1 Tropfen einer 3%igen KOH-Lsg. wird auf einen Objektträger gebracht. Mit einer Impföse wirdZellmaterial (Kolonie oder abzentrifugierte Zellen) in diesen Tropfen etwa 5 - 10 sec eingerührt.Zieht man bei vorichtigem Anheben der Platinöse einen schleimigen Faden nach, handelt es sichum Gram-negative Zellen. Beim Einrühren Gram-positiver Zellen in die KOH wird kein schleimi-ges Material gebildet.

Methodenskript AG Cypionka - 22 -
Isolierung von Reinkulturen anaerober Bakterien aus Tiefagar-Verdünnungsreihen
Herstellung sog. "Shake-Röhrchen":In einen 300 ml-Erlenmeyerkolben 100 ml dest. Wasser geben und Wasserstand markieren. Ge-mahlenen Agar (3.3 g) zusetzen, auffüllen und Suspension 10 min rühren. Danach Agar absetzenlassen, Wasser vorsichtig dekantieren. Dieser Waschvorgang dient dazu, wachstumshemmendeBestandteile und Hydrolyseprodukte zu entfernen, und wird fünfmal wiederholt. Anschließend auf100 ml auffüllen und autoklavieren (5 min bei 120 °C).
AGAR-PIPETTEN SOFORT MIT HEIßEM WASSER SPÜLEN:
Nach dem Autoklavieren die 3%ige Agarlösung heiß halten (60-70 °C) und mit einer 10 ml-Pipette in 3 ml-Portionen auf Reagenzgläser verteilen, die anschließend mit Wattestopfen ver-schlossen und erneut autoklaviert werden (15 min). Mit Verdunstungsschutz können die Röhr-chen im Kühlraum einige Zeit gelagert werden.
Agarverdünnungsreihe
Benötigt werden:• Pro Reihe 7 durchnumerierte und beschriftete Reagenzröhrchen mit je 3 ml 3%igem sterilemAgar (vor dem Sterilisieren 5fach mit dest. Wasser gewaschen), mit Wattestopfen verschlossen.• Sterile Gummistopfen, passend zu den Röhrchen• 1 Wasserbad mit 42 °C• 1 Wasserbad mit kaltem Wasser• 1 Bunsenbrenner mit Dreifuß und Konservendose mit 4 cm Wasserstand• 1 50 ml-Fläschchen mit komplettem Medium• sterile Pipetten
Vorgehen:• Das Fläschchen mit Medium im Wasserbad erwärmen (lange genug...)• Den Agar in den Röhrchen durch Kochen verflüssigen.• Die Röhrchen in das Wasserbad (42 °C) stellen, und je 6 ml warmes Medium zusetzen• Die Wattestopfen gegen Gummistopfen austauschen.• 1 Tropfen der Kultur in das 1. Röhrchen geben, einmal umschwenken• 1 Tropfen in das zweite Röhrchen gießen, das erste Röhrchen in das kalte Wasserbad stellenusw. (Das Röhrchen, aus dem gegossen wird, vorher außen abtrocknen, damit nicht Wassertrop-fen ausverdünnt werden.)• Möglichst zügig die Röhrchen mit N2/CO2 (80/20) begasen und bei 28 °C inkubieren.
Im Agar gewachsene Einzelkolonien können mit ausgezogenen Pasteurpipetten ausgestochen,mikroskopiert und zur Gewinnung von Reinkulturen erneut in Agar verdünnt werden.

Methodenskript AG Cypionka - 23 -
Geißelfärbung (nach Ryu)
Zunächst wird ein mikroskopisches Präparat einer Bakteriensuspension hergestellt. Sobald der
Großteil der Zellen angeheftet ist, wird die Färbelösung zu dem Präparat gegeben. Nach einer
Einwirkzeit von 5-15 min sollten die Geißeln sichtbar sein.
Lösung I: 5%-ige Phenollösung 10 ml
Tanninsäure 2 g
AlK(SO4)2 x 12 H2O, ges. Lösung 10 ml
Lösung II: ges. Lösung Kristallviolett (12 g/100 ml MeOH)
Färbelösung: Lsg.I : Lsg.II = 10 : 1
Katalase-Test
Reagenz: 5%ige H2O2-Lösung, aus 30%iger durch Verdünnen mit dest. H2O herstellen. Kühlund dunkel lagern. Nicht mit den Händen berühren. Verunreinigungen (Staub, Metalle etc.) be-wirken Zersetzung; daher sauber handhaben.
Vorgehen:Mit Impföse Bakterienmasse, möglichst aus dem Zentrum einer frischen Kolonie entnehmen undauf einen sauberen Objektträger geben. Einen Tropfen der verdünnten H2O2-Lösung mit saubererPasteur-Pipette darübergeben. Gasentwicklung (O2) zeigt Katalase an. H2O2 nicht direkt aufdieAgarplatte geben, da sonst die Bakterien abgetötet und nicht mehr für weitere Überimpfungenverwendet werden können!
Oxidase-Test
Reagenz:H2O 100 mlAscorbinsäure 0.1 gTetramethyl-p-phenylendiamin-HCl 1.0 g
Vorsicht, carcinogen! Reagenz (wird gestellt, täglich frisch ansetzen!) nicht mit Händen berührenund nichts verschütten! Benutzte Gefäße und Pipetten nicht liegen lassen, sondern gleich gut spü-len!
Vorgehen:Schmale Filterpapierstreifen (Cellulose) mit einigen Tropfen Reagenz tränken (nicht zu nass!).Bakterienmasse aus einer Kolonie mit einer Impföse auf den Papierstreifen geben und (evtl. mitabgerundetem Glasstab) verreiben. Blaufärbung zeigt Oxidaseaktivität. Vergleichspräparat mit E.coli.

Methodenskript AG Cypionka - 24 -
Analyse des Substratspektrums aerober Bakterienisolate
Der Substrattest stellt den Mittelpunkt der physiologischen Charakterisierung von Bakterieniso-laten dar. Getestet wird das bakterielle Wachstum auf 59 verschiedenen Kohlenstoffverbindungen(Angabe in Klammern = Endkonzentration in mM):
Komplexsubstrate: Pepton (0.05 %), Casamino Acids (0.05 %), Hefeextrakt (0.005 %)Polymersubstrate: Cellulose (0.05%), Stärke (0.1 %), Chitin (0.05 %), Xylan (0.05 %), La-
minarin (0.05 %)Disaccharide: Saccharose (5), Cellobiose (5), Maltose (5), Trehalose (5)Monosaccharide: Arabinose (5), Rhamnose (5), Xylose (5), Fructose (5), Glucose (5),
Mannose (5),Zuckerabkömmlinge: Mannitol (5), Gluconat (5), Glucosamin (5)Carbonsäuren: Formiat (5), Acetat (5), Propionat (1), Butyrat (2.5), Valerat (0.5), Ca-
pronat (0.5), Caprylat (0.5), Crotonat (0.2)Dicarbonsäuren: Malonat (5), Succinat (10), Fumarat (5), Malat (5), Tartrat (2)Andere org. Säuren: Glycolat (5), Pyruvat (5), Lactat (10), 2-Ketoglutarat (5), Citrat (2)Alkohole: Methanol (2), Ethanol (5), Propanol (5), Butanol (5), Glycol (5), Glycerin
(5), Tween 80 (0.001 %)Aminosäuren: Alanin (2), Arginin (2), Asparagin (2), Cystein (2), Glutamin (2), Isoleu-
cin (2), Phenylalanin (2), Tryptophan (2), Prolin (2)Amine: Betain (2)Aromaten: Benzoat (2), Salicylat (2)Heterozyklen: Nicotinat (2)
Der Test wird in 200 µl-Mikrotiterplatten (siehe Schema) unter aseptischen Bedingungen ange-setzt. Dazu werden 140 µl Medium (s.o.), 40 µl Substratlösung (aus einer Masterplatte) und 20 µlder Bakterienflüssigkultur in jedes „Well“ pipettiert und bei 20°C inkubiert. Es müssen eine zell-freie und eine subtratfreie Kontrolle angesetzt werden!WICHTIG: Zellen vor dem Überimpfen waschen, um den Eintrag von „fremden“ Substraten zuverhindern! Dafür 1.5 ml der Flüssigkultur in einer Tischzentrifuge abzentrifugieren, den Über-stand mit einer Pipette abnehmen und mit frischem Medium wieder auf 1.5 ml auffüllen. Alle Ar-beitsschritte werden in einer Sterilbank durchgeführt. Die Auswertung erfolgt über die durch bakterielles Wachstum hervorgerufene Trübung bzw.entstehende Bakterienpellets in den einzelnen Wells. Allerdings sollten Stichproben mikroskopischüberprüft werden. WICHTIG: Es muss beachtet werden, dass einige der eingesetzten Substanzenselbst eine Trübung hervorrufen!
Abb. Inkubationsschema für Sub-
strattest auf Mikrotiterplatten

Methodenskript AG Cypionka - 25 -
Aufnahme von Wachstumskurven
Wichtigster Parameter der Wachstumsversuche sind die Erträge pro umgesetzten Substrat. Diesewerden als Trockenmasse und Zellzahl dargetellt. Die Wachstumsrate einer Bakterienkultur wirdaus der Messung der optischen Dichte bestimmt. Diese wird mit dem Ratio/XR Turbidimeter vonHACH (Labor 2) gemessen. Um die OD auf Zelzahlen umrechnen zu können, wir an verdünntenProben die Zellzahl pro OD ermittelt. Gleichung zur Bestimmung der Wachstumsrate:
µ = Wachstumsrate (h-1)
x = OD (aktueller Zeitpunkt)
x0 = OD (zu Beginn des exponentiellen Wachstums)
t – t0 = betrachteter Zeitraum (h)
Lithotrophes Wachstum soll mit den folgenden Substraten getestet werden.
Serumflaschen: H2/O2 (70/30, v/v), S2O32- (5 mM)/ Luft, H2 (100 %)/NO3
- (10 mM)
Gradientenröhrchen: HS-/O2 (Stichkultur)
Schraubdeckelröhrchen: HS- (1 mM)/NO3- (10 mM), S2O3
2- (5 mM)/NO3- (10 mM)
Bestimmung der ββ-Glucosidase-Aktivität
Die β-Glucosidase (= Cellulase) hydrolysiert die β(1,4)-glucosidische Bindung zwischen den Glucoseein-heiten der Cellulose.
TestprinzipDie in der Probe enthaltenen β-Glucosidasen hydrolysieren das nicht fluoreszierende Substratanalogon 4-Methylumbelliferyl-β-D-Glucosid. Das abgespaltene 4-Methylumbelliferon wird fluorimetrisch gemessen.
)()ln()ln(
0
0
ttxx
µ−−
=
O
HOH2C
O
HO
HO
OH
O O
CH3
4-Methylumbelliferyl-β-D-Glucosid
β-Glucosidase
HO O O
CH3
4-Methylumbelliferon
+
O
HOH2C
HO
HO OH
OH
β-D-Glucose

Methodenskript AG Cypionka - 26 -
Geräte Reagenzien
Spektrofluorophotometer 4-Methylumbelliferyl-β-D-GlucosidReagenzgläser und Ständer 4-MethylumbelliferonVortexer GlycinMagnetrührer NatriumchloridpH-Messgerät NatriumhydroxidAutomatikpipetten Ethylenglykol-Monomethylether25°C WasserbadZentrifuge
Substratanalogon-Stammlösung3 mg 4-Methylumbelliferyl-β-D-Glucosid in 0.5 ml Ethylenglykol-Monomethylether vollständiglösen und 0.5 ml steriles bidest. H2O (in ausgeglühter Flasche) zusetzen. Die Substratanalogon-Lösung muss vor jedem Messtag frisch angesetzt werden.
Fluorophor-Stammlösung10 mg 4-Methylumbelliferon in 5 ml Ethylenglykol-Monomethylether lösen und 5 ml steriles bi-dest. H2O (in ausgeglühter Flasche) zusetzen. 100 µl dieser Lösung werden mit sterilem bidest.H2O (in ausgeglühter Flasche) auf 100 ml aufgefüllt (Endkonzetration 1 µg/ml). Die Lösung ist imDunkeln bei 4°C lagerbar.
Glycinpuffer0.75 g Glycin und 0.58 g Natriumchlorid in 100 ml sterilem bidest. H2O (in ausgeglühter Flasche)lösen. Der pH-Wert wird mit 1 M Natronlauge auf pH 10 eingestellt.
VersuchsdurchführungPro Probe werden 3 Parallelen angesetzt. 1 ml Probe plus 100 µl Substratanalogon-Lösung.Blindwert: 1 ml steriles bidest. H2O (in ausgeglühter Flasche) plus 100 µl Substratanalogon-Lösung.
Inkubation abgedeckt im Wasserbad bei 25°C für 2 h. Zum Abstoppen der Reaktion werdenjedem Ansatz 750 µl 0.1 M Glycinpuffer (pH 10) zugesetzt, gut durchmischt und sofort bei einerAnregungswellenlänge von 360 nm und einer Emissionswellenlänge von 440 nm in Spektrofluo-rophotometer gegen den Blindwert gemessen.
Bei hoher Eigenfluoreszenz der Probe wird ein Probenblindwert aus 1 ml Probe und 100 µlsterilem bidest. H2O (in ausgeglühter Flasche) angesetzt, genauso behandelt wie die Proben undgegen einen fluorimetrischen Blindwert (3.3 ml steriles bidest. H2O) gemessen. Die Fluoreszenzdieses Probenblindwertes wird von der Fluoreszenz der Probe subtrahiert.
EichreiheFür die den Test begleitende Eichreihe werden zwischen 10 und 70 µl MUF-Standardlösung zu3.3 ml sterilem bidest. H2O (in ausgeglühter Flasche) zugefügt. Dem Ansatz werden 750 µlGlycinpuffer zugegeben, gut durchmischt und gegen einen Blindwert (3.3 ml steriles bidest. H2Oplus 750 µl Glycinpuffer) gemessen. Die Eichreihe muss bei jeder Messerie mitgemessen werden. Die Aktivitätstests der β-Glucosaminidase, der Leucin-Aminopeptidase sowie derβ-Glucosidase im Mikrotiterplattentest werden wie oben beschrieben, mit einigen Änderungen,durchgeführt.

Methodenskript AG Cypionka - 27 -
Bestimmung der ββ-Glucosaminidase-Aktivität
Die β-Glucosaminidase (= Chitinase) hydrolysiert die β(1,4)-glucosidische Bindung zwischen denN-Acetyl-Glucosamineinheiten des Chitins.
Substratanalogon: 4-Methylumbelliferyl-N-Acetyl-β-D-Glucosaminid
Bestimmung der Leucin-Aminopeptidase-Aktivität
Die Leucin-Aminopeptidase spaltet Pepton, welches ein Gemisch von Peptiden und Aminosäurenaus tierischen oder pflanzlichen Proteinen ist.
Substratanalogon-Stammlösung- Herstellung 100 ml einer 2 mM L-Leucin-7-Amido-4-Methylcoumarin-Lösung in sterilem bidest
H2O (in ausgeglühter Flasche; ausglühen: 12 h bei 180°C)- Lagerung bei –20°C im Dunkeln möglich- nach dem Auftauen Herstellung der benötigten Konzentrationen durch Verdünnung
Fluorophor-Stammlösung- Herstellung 1 l einer 0.1 mM 7-Amino-4-Methylcoumarin-Lösung in 10 ml Ethylenglykol-
Monomethylether und 990 ml sterilem bidest. H2O (in ausgeglühter Flasche)- Lagerung als Teilmengen bei –20°C im Dunkeln- nach dem Auftauen Herstellung der benötigten Konzentrationen durch Verdünnung
Ammonium-Glycin-Puffer- Herstellung 1 l einer 0.2 M Ammoniumhydroxid / 0.05 M Glycin-Lösung in sterilem bidest. H2O
(in ausgeglühter Flasche); pH 10.5 mit 5 M Natronlauge einstellen
O
HOH2C
O
HO
HO
NH
O O
CH3
C
O
CH3
4-Methylumbelliferyl-N-Acetyl−β-D-Glucosaminid
β-Glucosaminidase
HO O O
CH3
4-Methylumbelliferon
+
O
HOH2C
HO
HO NH
OH
C
O
CH3
N-Acetyl-β-D-Glucosamin
O O
CH3
HNC
O
C
H2N H
H3CHCH2C
H3C
L-Leucin-7-Amido-4-Methylcoumarin
Leucin-Aminopeptidase
O
CH3
OH2N
7-Amino-4-Methylcoumarin
+COOHC
H2N H
H3CHCH2C
H3C
L-Leucin

Methodenskript AG Cypionka - 28 -
Versuchsdurchführung1 ml Probe plus 1 ml Substratanalogon-Lösung (zur Eichung 1 ml Probe plus 1ml FluorophorLösung). Blindwert: 1 ml steriles bidest. H2O (in ausgeglühter Flasche) plus 1 ml Substratanalo-gon-Lösung (zur Eichung 1 ml Probe plus 1 ml steriles bidest. H2O).
Zum Abstoppen der Reaktion werden die Proben 3 min gekocht und auf 25°C abgekühlt. Da-nach Zugabe von 1ml Ammonium-Glycin-Puffer, Sedimentation der groben Partikel, Überführungdes Überstandes und Zentrifugation 15 min 13000 rpm.
Bestimmung der ββ-Glucosidase-Aktivität in Mikrotiterplatten
Notwendige Geräte: Mikrotiterplatten mit 96 KavitätenAutomatik-MehrkanalpipetteMikrotiterplatten-Fluoreszenzreader
Herstellung der Substratanalogon-Stammlösung in 10facher Menge.Herstellung 1 l einer 0.14 M Natriumchloridlösung.
VersuchsdurchführungJeweils 200 µl Probe werden in drei Kavitäten der Mikrotiterplatte pipettiert und mit 20 µl Sub-stratanalogon-Lösung versetzt. Für den Probenblindwert werden zu 20 µl Natriumchloridlösung200 µl Probe pipettiert. Für den photometrischen Blindwert werden zu 200 µl Natriumchloridlö-sung 20 µl Substratanalogon-Lösung pipettiert. Die Mikrotiterplatten werden abgedeckt und bei30 °C im Brutschrank 24 h inkubiert. Nach der Inkubation wird der pH-Wert durch Zugabe von50 µl Glycinpuffer auf pH 10 in den einzelnen Kavitäten eingestellt. Die Platten werden sofort ineinem Mikrotiterplatten-Fluoreszenzreader bei einer Anregungswellenlänge von 355 nm und einerEmissionswellenlänge von 460 nm gemessen.
EichreiheFür die den Test begleitende Eichreihe werden zwischen 10 und 70 µl der MUF-Standardlösung(mindestens drei Konzentrationsstufen) mit sterilem bidest. H2O (in ausgeglühter Flasche) auf 200µl aufgefüllt und mit 20 µl Substratanalogon-Lösung versetzt. Die Eichreihe wird parallel zu jederMessreihe angesetzt und wie die Testansätze inkubiert. Auch hier wird nach der Inkubation derpH-Wert durch Zugabe von 50 µl Glycinpuffer auf pH 10 in den einzelnen Kavitäten eingestellt.Die Platten werden in einem Mikrotiterplatten-Fluoreszenzreader bei einer Anregungswellenlängevon 355 nm und einer Emissionswellenlänge von 460 nm gemessen.
Bestimmung der Gesamtzellzahl
Als Standard-Methode zur direkten Zählung von Mikroorganismen wird heute eine Kombinationaus Membranfiltertechnik und Epifluoreszenzmikroskopie verwendet. Dabei werden die Mikroor-ganismen vor oder nach der Filtration mit Fluoreszenzfarbstoffen wie Acridinorange oder DAPI(= 4´,6-Diamidino-2-phenylindol) gefärbt. Acridinorange bindet an die Phosphatgruppen vonNucleinsäuren. Gefärbte Bakterien fluoreszieren grün, z.T. orange, Fremdpartikel rot, orangeoder gelb. DAPI reagiert mit doppelsträngiger DNA. Gefärbte Bakterien fluoreszieren blassblau,Fremdpartikel gelb. Zur Filtration werden Membranfilter z.B. aus Polycarbonat oder anorganischeaus Aluminiumoxid (Anodisc) verwendet. Damit die Zellen nicht ins Innere der Filter eindringen,sollte deren Porenweite maximal 0.2 µm betragen.

Methodenskript AG Cypionka - 29 -
Materialien- Filtrieraufsatz (Millipore 20 mm Glasfilterhalter)- 0.2 µm Anodisc-Filter (Whatman)
Chemikalien- 0.5% TWEEN 80 (sterilfiltriert)- 0.22 µm sterilfiltriertes ddH2O- 0.22 µm sterilfiltrierter PBS-Puffer (130 mM NaCl, 5 mM NaH2PO4; pH 7.2)- Fixativ (4% Paraformaldehyd + 0.1% Triton X100in 1 x PBS; pH 7.25)- DAPI-Lösung (10 µg/ml; sterilfiltriert)- Einbettungsmittel DABCO (25 mg Diazabicyclo-octan + 1 ml PBS + 9 ml Glycerin)
- Färbelösung:1ml Fixativ
+ 930 µl ddH2O (sterilfiltriert)+ 70 µl DAPI-Lösung
MethodeZu den mit Glutardialdehyd (6 ml, 3%) fixierten Sedimentproben 100 µl TWEEN 80 zugeben unddie Proben mit Ultraschall behandeln (5x5 sec). In ausgeglühten Reagenzgläsern 10 ml PBS-Puffer vorlegen. Vorbehandelte Proben nochmals vortexen und 10 µl davon in den PBS-Pufferüberführen. Proben erneut vortexen und über mit Glasfaser-Filter (GF) unterlegte Anodisc-Membranfilter filtrieren. Reagenzglas und Filtrieraufsatz mit sterilfiltriertem Wasser nachspülen.Membran trockenfiltrieren und während des Vakuums abnehmen. Filtrieraufsatz vor jedem neuenFiltrationsvorgang mit Spülmittel, ddH2O und steril filtriertem ddH2O säubern. In einem kleinen Wägeschälchen DAPI-Lösung ansetzen. Filter mit der Probenseite nach obenvorsichtig auf die Färbelösung auflegen und 5 min im Dunkeln färben. Filter(rückseite) auf Kü-chenpapier im Dunkeln trocknen und danach auf einem Objektträger in DABCO einbetten, Deck-glas auflegen. Filter unterm Epifluoreszenzmikroskop auszählen.
Gesamtzellzahl in einer Wasserprobe (nach Hobbie et al.)
2-5 ml einer Wasserprobe mit Bakterien werden in einem Reagenzglas mit ca. 200 µl einer parti-kelfreien 0.1% Acridin-Orange Lösung 2 min lang gefärbt und dann durch einen mit Irgalan-schwarzlösung (0.1%) vorgefärbten 0.2 µm Nucleporefilter abfiltriert. Als Unterlage kommt aufden Filteruntersatz ein Cellulosemembranfilter. Der Nucleporefilter wird dann auf einem Objekt-träger getrocknet, mit einem fluoreszenzfreien Immersionsöl (z.B. Siliconöl) versehen und miteinem Deckglas bedeckt. Die Probe wird bei fluoreszenzfreier Ölimmersion unter dem Epifluores-zenzmikroskop bei Blaulichtanregung betrachtet. Es werden pro Filter 10 Zählquadrate ausge-zählt. Die Probenmenge sollte so gewählt sein, dass pro Zählquadrat ca. 30-50 Bakterien sichtbarsind Berechnung der Bakterienzahl/ml:
Zahl/ml = (y • A)/(a • V)
y: mittlere Zellzahl pro ZählquadratA: effektive Filtrationsfläche (µm2)a: Zählquadratfläche (µm2)V: Filtrationsvolumen (ml)

Methodenskript AG Cypionka - 30 -
ATP-Bestimmung (nach Bergmeyer)
Das Prinzip der ATP-Bestimmung mittels Luciferase-Assay beruht auf der Reaktion von ATPmit Luciferin und Sauerstoff in Gegenwart des Enzyms Luciferase gemäß:
LuciferaseATP + Luciferin + O2 → Oxyluciferin + AMP + PPi + CO2 + Licht
Wenn die übrigen Reagenzien im Überschuß vorliegen, ist die Lichtemission proportional zurATP-Menge. In einem geeichten Meßsystem kann also von der gemessenen Lichtintensität aufden ATP-Gehalt einer Probe geschlossen werden. Die Lichtemission wird in einem Luminometer(LKB Wallac) bestimmt, das mit einem internen 14C-Strahler auf die gewünschte Empfindlichkeiteingestellt wird. Am Ausgang des Luminometers ist ein Schreiber angeschlossen, der die Meß-werte in mV registriert.
Um den ATP-Gehalt von Zellen bestimmen zu können, müssen diese erst aufgeschlossen werden.
Extraktion des ATP (nach Blaut und Gottschalk 1984): 100 µl auf Eis gelagerte 3 M Perchlor-säure wird in einem Eppendorf-Reaktionsgefäß vorgelegt, 100 µl Probe zugegeben, einmal kurzund dann jede halbe Stunde erneut geschüttelt. Nach 1½ Stunden Extraktionszeit auf Eis werden50 µl 1 M Tes-Puffer pH 7.4 und 112 µl 3 M KOH zugegeben, sodaß der pH-Wert zwischen7.0 und 8.0 liegt. Die Kaliumkonzentration im neutralisierten Extrakt reicht aus, um das Perchlo-rat auszufällen, das die ATP-Test-Reaktion stören würde. Die Ausfällung wird in einer Tischzen-trifuge kurz abzentrifugiert.
Luciferase-Assay:Für den Luciferase-Assay werden folgende Chemikalien eingesetzt:
Tris-Puffer (EDTA-Tris-Acetat, pH 7.75):Es werden 12.12 g Tris-(hydroxy-methyl)aminomethan und 0.74 g Na2-EDTA eingewogen, mitdestilliertem Wasser auf 900 ml aufgefüllt, der pH-Wert mit 2 M Essigsäure auf 7.75 eingestelltund schließlich destilliertes Wasser bis auf 1000 ml Endvolumen dazugegeben.
Test-Reagenz:Das Test-Teagenz (1243-102 Monitoring Kit) der Firma LKB enthält:
I.) Firefly-LuciferaseII.) D-LuciferinIII.) 50 mg RinderserumalbuminIV). 0.5 mmol MagnesiumacetatV) 0.1 µmol anorganisches Pyrophosphat
Die gefriergetrocknete Testreagenz-Fertigmischung wird unter vorsichtigem Schütteln in 10 mlUltrapure-Wasser (durch Umkehrosmose hochgradig gereinigtes Wasser mit einer Leitfähigkeitvon 0.05-0.07 µS) gelöst, in Portionen zu je 1 ml in Cryo-Tubes gefüllt und in flüssigem Stick-stoff bis zu weiteren Verwendung aufbewahrt.

Methodenskript AG Cypionka - 31 -
ATP-Standard (0.5 µM):30.26 mg ATP (ATP Na2 H2 * 3 H2O, Boehringer, Mannheim) werden ad 100 ml in Tris-Puffer(siehe oben) gelöst und zu je 1 ml in Cryo-Tubes portioniert und in flüssigem Stickstoff aufbe-wahrt. An jedem Versuchstag wird diese ATP-Stammlösung aufgetaut und in 3 Schritten insge-samt 1:1000 in Tris-Puffer verdünnt. Diese ATP-Standard-Lösung hat eine Konzentration von0.5 µM i.e. 0.5 pmol/µl.
Durchführung der Messung:160 µl Tris-puffer werden mit 40 µl Test-Reagenz in einer Polystyren Küvette durch langsamesDrehen gemischt und der Leerwert der Strahlung im Luminometer gemessen. Die Küvette wirdnun wieder herausgenommen, 10 µl Zellextrakt werden zugesetzt (resultierendes Volumen 210µl), gemischt und wieder die Lichtemission ermittelt. Darauf wird die Messung zweimal durchZugabe von je 5 µl ATP-Standard (2.5 pmol) in dieselbe Küvette geeicht (resultierende Volumi-na: 215 µl und 220 µl). Durch diese interne Standardisierung werden Fehler, die durch die unter-schiedlichen Komponenten der einzelnen Proben verursacht werden können, ausgeschlossen.

Methodenskript AG Cypionka - 32 -
Isolierung von DNA
EinleitungNucleinsäuren können mit verschiedenen Methoden isoliert werden. Die gewählte Methode istabhängig von der Art der zu isolierenden Nucleinsäuren und ihrem späteren Verwendungszweck.Einige einfache Verfahren beruhen auf einem enzymatischen (mit Lysozym) oder mechanischen(mittels freeze and thaw, Beadbeater, Ultraschall) Aufschluß. Da man mit diesen Methoden i.d.R.stark kontaminierte Nucleinsäuren erhält, können Reinigungsverfahren, wie z.B. Extraktionen mitorganischen Lösungsmitteln, angeschlossen werden. Beim Zellaufschluß spielt die Herkunft der Proben eine große Rolle. Bakterienzellen aus Um-weltproben sind in einer Sedimentmatrix z.T. vor mechanischen Kräften und Enzymeinwirkungengeschützt. In Kultur gewachsene Bakterien würden in einem Beadbeater zu großen Scherkräftenausgesetzt. Neben diesen würde die Wärmeentwicklung zu einer Destabilisierung/Zerstörung derNucleinsäuren führen. In diesem Fall ist ein Aufschluß mittels freeze and thaw vorzuziehen, wobeidie Zellen durch plötzliches Einfrieren und Aufkochen physikalisch auseinandergebrochen werden. Weiterhin ist zu beachten, daß dem mechanischen Aufschluß grampositiver Bakterien eineEnyzmbehandlung mit z.B. Lysozym voranzustellen ist. Die Zellwand grampositiver Bakterien istmit 25 – 40 untereinander verknüpften Peptidoglycanlagen bis zu 10-mal dicker als die gramnega-tiver. Ihre Zellwand ist mechanisch wesentlich stabiler, die Stabilität nimmt mit dem Verknüp-fungsgrad des Peptidoglycans zu. Mittels Lysozym kann die β-1,4-glykosidische Bindung zwi-schen N-Acetylglucosamin und N-Acetylmuraminsäure, die abwechselnd miteinander verknüpftdas Rückgrat der Mureinstruktur bilden, gespalten werden.
Materialien- Biozentrifuge- Gefrierschrank (-70°C)- Heizblock
Chemikalien- Lysozym-Lösung (0,8 mg⋅ml-1)- 10 mM Tris-HCl, pH 8- 10% SDS-Lösung (9,6 ml 20% SDS + 2,4 ml 0,5 M Natrium-Acetat (pH 7,5) + 66,4 ml
ddH2O; autoklavieren)- 3 M Natrium-Acetatlösung
MethodeDie in Eppendorfcups bei –20°C eingefrorenen Proben werden aufgetaut und bei 19000 rpm30 min bei 4°C zentrifugiert. Die Überstände werden verworfen. Den Proben werden 100 µl Ly-sozym-Lösung sowie 100 µl Tris-HCl zugesetzt. Die Proben werden zehnmal invertiert und an-schließend 10 min auf Eis inkubiert. Danach werden 40 µl SDS-Mix und 60 µl Natrium-Acetatlösung zugegeben und eine Stunde auf Eis inkubiert.Für freeze and thaw werden die Proben in 5 Cyclen abwechselnd je 3 min bei –70°C eingefrorenund anschließend aufgekocht. Danach werden die Proben einer Phenol/Chloroform-Extraktionunterzogen.

Methodenskript AG Cypionka - 33 -
DNA/RNA Extraktion aus Sedimentproben
Um in der PCR Nukleinsäuren als Template einsetzen zu können, ist es erforderlich sich diese ersteinmal zu beschaffen. Dabei spielt nicht nur der Aufschluss der Zellen eine entscheidende Rolle,sondern auch die Aufreinigung und der Schutz der Nukleinsäuren vor DNasen und RNasen. BeiSedimentproben ist dabei ein Aufschluss der Bakterien durch „Freeze and Thaw“ (s. „Freeze andThaw“) meistens nicht möglich. Hierfür müssen stärkere Aufschlussmethoden verwendet werden,da das Sediment den Bakterien einen Schutz bietet. Das Problem bei alternativen Aufschlussme-chanismen ist aber nicht der Aufschluss der Zellen, sondern der Schutz der Nukleinsäuren. Sokönnen durch eine zu starke mechanische Beanspruchung die Nukleinsäuren dermaßen geschertwerden, dass eine weitere Untersuchung nicht mehr möglich ist. Zudem müssen die Nukleinsäurenbeim Aufschluss vor DNasen und RNasen geschützt werden, die beim Aufschluss der Bakterienebenfalls freigesetzt werden. Eine Aufreinigung der Nukleinsäuren ist ebenfalls notwendig, da die extrahierten Proben jenach Sediment sehr viele Huminsäuren oder andere für die PCR inhibierende Substanzen enthaltenkönnen. Eine Aufreinigung kann mittels der Phenol/Chloroform Reinigung erfolgen. Ebenso ist esmöglich Nukleinsäuren über Kits aufzureinigen, was in der Regel Kostenaufwendiger und nichtunbedingt effektiver ist.Die Fällung der Nukleinsäuren dient abschließend dazu, die Nukleinsäuren in ein bestimmtes Vo-lumen und geeigneten Puffer aufzunehmen. Nach Abschluss der Extraktion und gegebenenfallseiner Quantifizierung der Nukleinsäuren, steht einer Amplifikation mittels PCR/RT-PCR nichtsmehr im Wege.
Material
- Beadbeater- Eppendorf-Reaktionsgefäß Ständer- Sterile Kryroröhrchen + Deckel- Kühlfalle- Pumpe für die Speed Vac- Speed Vac- Sterile Eppendorf-Reaktionsgefäße (1,5 ml und 2,0 ml)- Sterile Zirkoniumperlen (∅ 0,1 mm)- Tischzentrifuge (Biofuge 13 R)- Wasserbad- Zentrifuge (Biofuge 15 R)
Chemikalien und Lösungen
- Chloroform- DEPC behandeltes ddH2O (0,1% Diethylpyrocarbonat (DEPC) in ddH2O lösen und gut mi-
schen. Über Nacht bei 37 °C inkubieren und anschließend autoklavieren. DEPC löst sich voll-ständig in CO2 und Ethanol auf).
- DNase (1 U/µl)- DNase-Puffer (pH 7,5) (40 mM Tris (Base), 6 mM MgCl2 in ddH2O ansetzen, pH-Wert mit
HCl einstellen und autoklavieren)- Ethanol (70 %)- Fällungsmix (125 ml Ethanol (abs.), 5 ml Na-Acetat (3M))

Methodenskript AG Cypionka - 34 -
- Phenol -Wasser gesättigt- (pH 4,0 und 7,5)- Phenol/Chloroform -Wasser gesättigt- (pH 4,0 und 7,5)- SDS-Extraktions-Mix (9,6 ml 20 % Sodium Dodecyl Sulfat (SDS), 2,4 ml 0,5 M Na-Acetat
(pH 7,5) und 66,4 ml ddH2O in eine 100 ml Pfennigflasche pipettieren und autoklavieren)- TE-Puffer (pH 8,0) (10 mM Tris (Base), 1 mM EDTA in ddH2O ansetzen, pH-Wert mit HCl
einstellen und autoklavieren)- Sämtliche Lösungen sind mit DEPC behandeltes ddH2O angesetzt, um DNasen und RNasen
zu eliminieren- Autoklaviert wird 20 min bei 121 °C
Zellaufschluss mittels Beadbeater
Je 1 g Sediment, 1 g Zirkoniumperlen und 1 ml SDS-Extraktions-Mix werden für jede Probe inein Kryroröhrchen überführt. In einem Beadbeater werden die Proben für 1 min geschüttelt (beica. 5.000 rpm), 30 sec gewartet und eine weitere Minute geschüttelt. Das Sediment und die Zir-koniumperlen werden in der Biofuge 15 R abzentrifugiert (5 min, 15.000 rpm). Der wässrigeÜberstand wird in ein 1,5 ml Eppendorf-Reaktionsgefäß überführt und auf Eis gestellt (dabeimöglichst kein Sediment mit überführen). In das Kryroröhrchen werden 500 µl Phenol (pH 7,5)hinzupipettiert und ein weiteres Mal dem Beadbeater und der Zentrifuge unterzogen. Der wässri-ge Überstand wird mit dem Vorherigem vereinigt. Nun werden in das Kryroröhrchen 250 µl SDS-Extraktions-Mix und 250 µl Phenol (pH 7,5) hinzugeführt und die Prozedur ein weiteres Malwiederholt.Für eine kombinierte DNA/RNA Extraktion wird der Überstand, bevor er einer Phe-nol/Chloroform Reinigung unterzogen wird, aufgeteilt.
Phenol/Chloroform Reinigung
Zu der aufzureinigenden Nukleinsäurelösung wird das einfache Volumen an Phenol (pH 4,0 fürRNA und pH 7,5 für DNA) hinzupipettiert. Nach einer Inversion (10 x) und einer Zentrifugation(13.000 rpm, 2 min), wird die wässrige Phase (in der Regel der Überstand) in ein neues 1,5 mlEppendorf-Reaktionsgefäß überführt. Die organische Phase und die Interphase beinhalten dieProteine und Huminsäuren die durch diese Reinigung entfernt werden sollen.Zur wässrigen Phase wird nun das einfache Volumen an Phenol/Chloroform (pH 4,0 für RNA undpH 7,5 für DNA) hinzupipettiert. Es folgt wieder eine Inversion und Zentrifugation (s.o.). Derwässrige Überstand wird in ein neues Reaktionsgefäß überführt (s.o.). Der Probe wird als letztesdas einfache Volumen an Chloroform zugegeben und die Probe erneut wie oben beschrieben be-handelt.Die nun erhaltene wässrige Lösung, mit den gereinigten Nukleinsäuren, wird in ein 2 ml Eppen-dorf-Reaktionsgefäß überführt. In der anschließenden Ethanol-Fällung werden die, für die PCR,störenden Salze entfernt und die Nukleinsäuren in einen geeigneten Puffer aufgenommen.
Ethanol-Fällung
Der Probe wird das 2,6-fache Volumen des Fällungsmixes (immer frisch ansetzen, da Na-Acetatbei längerer Lagerung ausfällt) beigefügt und die Nukleinsäuren über Nacht bei - 20 °Cgefällt (alternativ 4 h bei 4 °C). Am nächsten Tag werden die gefällten Nukleinsäuren abzentrifu-giert (15.000 rpm, 30 min). Hierbei ist auf eine Ausrichtung der Eppendorf-Reaktionsgefäße zu

Methodenskript AG Cypionka - 35 -
achten, damit diese beim nächsten Zentrifugationsschritt ebenfalls in der gleichen Position zentri-fugiert werden. Nach der Zentrifugation wird der Überstand verworfen und 500 µl 70 %iges Et-hanol auf die Proben zum Waschen gegeben. Es folgt eine erneute Zentrifugation (15.000 rpm, 10min). Der Überstand wird verworfen und die Proben werden für 5 - 10 min in der Speed Vac(Unterdruck wird durch eine Pumpe erreicht), bei einer mittleren Temperatureinstellung, zentrifu-giert. Hierdurch wird das restliche Ethanol entfernt. Mindestens 10 min vor Inbetriebnahme derSpeed Vac sollte die Kühlfalle angestellt werden, um die Pumpe vor Flüssigkeiten zu schützen.Die Nukleinsäuren können nach Trocknung in 50 µl TE-Puffer aufgenommen werden. Nach einerInkubation von 30 min bei Zimmertemperatur können die Proben dann bei 4 °C für den Gebrauchgelagert werden. Proben die längere Zeit nicht benötigt werden und dementsprechend gelagertwerden sollen, können wieder im Fällungsmix (2,6 fache Volumen) aufgenommen werden und bei– 70 °C eingefroren werden.
RNA-Extraktion und DNase-Verdau
Um reine RNA zu erhalten ist es notwendig Kontaminationen durch DNA vorzubeugen und/oderzu entfernen. Dies ist Notwendig, da in einer PCR zusätzlich vorhandene DNA mitamplifiziertwird. Bei der Extraktion von RNA wird die Phenol/Chloroform Reinigung bei einem pH-Wertvon 4,0 durchgeführt. Bei diesem pH-Wert fällt ein Teil der DNA in der Interphase aus. Die RNAlöst sich dagegen weiterhin vollständig in der wässrigen Phase. Da nach der Extraktion, Reinigungund der Fällung immer noch DNA in der Probe vorhanden ist, muss die Probe einem DNase-Verdau unterzogen werden, um somit die letzten Spuren von DNA zu entfernen.Dafür werden die RNA Proben nach der Ethanol-Fällung nicht in TE-Puffer aufgenommen, son-dern in 500 µl DNase-Puffer. Weiter werden 5 µl DNase hinzupipettiert und die Proben 60 minbei 37 °C in einem Wasserbad inkubiert. Anschließend werden die Schritte ab der Phe-nol/Chloroform Reinigung wiederholt (dient der Entfernung der DNase).Um später nachzuweisen ob sich noch DNA in den RNA Proben befindet, werden die Proben ineiner PCR eingesetzt. Sollte sich in dieser PCR Nukleinsäuren amplifizieren lassen (RNA wirdnicht amplifiziert), ist die Probe noch mit DNA kontaminiert. Sollten keine Produkte zu erkennensein, kann man davon ausgehen, daß die Probe DNA frei ist.
Kurzdarstellung der kombinierten DNA/RNA Extraktion
- Aufschluss der Zellen im Sediment mittels Beadbeater- Aufteilung der Proben in DNA und RNA Proben- Phenol/Chloroform Reinigung (pH Werte beachten)- Ethanol-Fällung- Aufnahme der DNA in TE-Puffer- DNase-Verdau mit den RNA Proben- Phenol/Chloroform Reinigung (pH 4,0)- Ethanol-Fällung- Aufnahme der RNA in TE-Puffer

Methodenskript AG Cypionka - 36 -
Schnelltest zur Quantifizierung von DNA (Ethidiumbromid (EtBr)-Platten Test)
Je 1 µl Probe wird auf eine EtBr-Platte pipettiert. (1,5 %iges Agarosegel in 1 x TAE Puffer an-setzen und in der Mikrowelle aufkochen. Nach Abkühlung (auf ca. 60 °C) werden 15 µl Ethidi-umbromid (10 mg/ml) pro 100 ml Agarose hinzupipettiert und die Agarose in Petrischalen gegos-sen). Zusätzlich werden je 1 µl Heringssperma in verschiedenen Konzentrationen (z.B. 10, 30, 50,100, 150 ng/µl) aufgetragen. Unter einem Transiluminator kann der DNA-Gehalt an Hand desStandards abgeschätzt werden.
Kurzdarstellung einer DNA-Extraktion mittels eines Kit von Bio-Gene
Um Nukleinsäuren schnell zu extrahieren lohnt sich häufig der Einsatz eines Kits, da eine Extrak-tion mit anschließender Phenol/Chloroformreinigung mindestens 2 Tage und länger dauern kann.Zudem arbeiten Kits bei der Aufreinigung der Nukleinsäuren häufig mit Filtern, die somit den Ein-satz von giftigen Substanzen wie Phenol und Chloroform minimieren. Der Nachteil der Kits liegtmeistens bei den hohen Kosten.Für die DNA-Extraktion wird ein Fast DNA-Extraktionskit for soil von BioGene verwendet.Zusätzlich zu dem im Kit enthaltenen Säulen, Tubes und Chemikalien, werden pro Probe zweisterile 1,5 ml und ein 2 ml Reaktionsgefäß benötigt, sowie Ethanol und Na-Acetat. Wenn nichtanders angegeben wird immer bei 13.000 rpm in einer Tischzentrifuge zentrifugiert.
- 0,5 g Sediment, 978 µl Sodium Phosphat Puffer und 122 µl MT-Puffer werden in ein LysingMatrix E Tube überführt
- Der Aufschluss der Zellen erfolgt durch einen Beadbeater (1 min bei 5000 rpm schütteln undnach 30 sec warten erneut 1 min schütteln)
- Zentrifugieren (Kühlzentrifuge: 15.000 rpm, 15 min, 4 °C)- Überstand in ein steriles 1,5 ml Reaktionsgefäß überführen und 250 µl PPS hinzupipettieren- Die Probe 10 mal invertieren und 5 min zentrifugieren- Überstand in ein steriles 2 ml Reaktionsgefäß überführen und mit 1 ml Binding Matrix Suspen-
sion versetzen. Die Binding Matrix Suspension muss vorher gut gemischt werden.- Probe 2 min von Hand schütteln und anschließend 3 min stehen lassen- 500 µl vom Überstand verwerfen und 750 µl der resuspendierten Lösung auf einen Spin Filter
geben, 1 min zentrifugieren und Filtrat verwerfen- Restliches Volumen der Probe auf den Spin Filter geben und erneut 1 min zentrifugieren- Filtrat verwerfen und die DNA mit 500 µl SEWS-M von der Binding Matrix auf den Filter
waschen (Zentrifugation: 1min)- Filtrat erneut verwerfen und 2 min zentrifugieren- Spin Filter in ein neues Catch Tube stellen und 5 min unter der Cleanbench im geöffneten Zu-
stand trocknen lassen- 50 µl DES auf den getrockneten Filter geben und 1 min zentrifugieren
Die DNA ist nun amplifikationsfertig. Da man in der Regel nicht die gesamten 50 µl benötigt,können 30 µl in ein weiteres steriles 1,5 ml Eppendorfreaktionsgefäß überführt werden. Mit Zu-gabe von 90 µl Ethanol/Na-Acetat (siehe Ethanol-Fällung) wird die DNA gefällt und bei - 70°C eingefroren. Im denaturierten Zustand und eingefroren ist die DNA vor Einflüssen die die

Methodenskript AG Cypionka - 37 -
DNA abbauen könnte, gut geschützt. Für eine erneute Verwendung der DNA siehe das Protokollder Ethanol-Fällung.Die restlichen 20 µl DNA dienen als Template in der PCR oder können für weitere molekularbio-logische Untersuchungen verwendet werden.
Protokoll zur DNA-Extraktion aus Wasserproben und Flüssigkulturen
Die Wasserproben wurden über einem 0,2 µm-Polycarbonat-Filter abfiltriert. Im Eppendorfgefäßwurden dem Filter 0,5 g Zirkonium Perlen, 20 µl SDS 25%, 600 µl Phosphatpuffer und 600 µlPhenol-Chloroform-Isoamylalkohol zugesetzt. Um die Zellen aufzuschließen, und um Fremdstoffewie Verunreinigungen durch das Probenwasser, sowie Zellbestandteile von der vorhandenenBakterien-DNA zu entfernen wurde das Eppendorfgefäß drei Minuten gevortext, danach 10 minbei 60°C in einem Wasserbad inkubiert und wieder drei Minuten lang gevortext. Die organischePhenolphase der Probe wurde durch 10 minütige Zentrifugation bei Raumtemperartur und10.000 rpm von der wässrigen Phasen getrennt. Die DNA befand sich nun in der oberen, wässri-gen Phase, die in ein neues Eppendorfgefäß überführt wurde. Zur ursprünglichen Probe wurdenweitere 300 µl Phosphatpuffer zugegeben, erneut eine Minute gevortext und wie vorher 10 minzentrifugiert. Der wässrige Überstand wurde zum ersten dazugegeben. Den Überständen wurdenun wieder 1 ml Phenol-Chloroform-Isoamylalkohol zugesetzt, um die restlichen Verunreinigun-gen von der DNA zu entfernen. Nach wiederholtem Vortexen und Zentrifugation konnte wiederder Überstand abgenommen und in ein neues Gefäß überführt werden. Sofern noch ein durchProteine verursachtes, weißes Präzipitat an der Interphase zu sehen war, wurde der Vorgang miterneuter Zugabe von 1 ml Phenol-Chloroform-Isoamylalkohol wiederholt.Anschließend wurde die wässrige Lösung, in der sich die DNA befand, nach Hinzufügen von30 µl Natriumacetat und 2,25 ml Isopropanol über Nacht gefällt. Am nächsten Tag konnte dieDNA der Lösung durch Zentrifugation bei 4°C und 13000 rpm für 30 min pelletiert werden. DerÜberstand wurde abpipettiert und das Pellet wurde mit 1,5 ml Ethanol (70%) gewaschen. DieLösung wurde wiederum unter gleichen Bedingungen 10 min zentrifugiert, der Überstand abge-nommen und das DNA-Pellet für drei Minuten in der Speed Vac getrocknet. Die präzipitierte undgetrocknete DNA wurde in 50 µl PCR-Wasser aufgenommen und stand für weitere molekular-biologische Untersuchungen zur Verfügung.
Quantifizierung von DNA mit Pico Green
Je 1 µl Probe werden in 899 µl TE-Puffer aufgenommen. Zur Quantifizierung werden zu jederProbe 100 µl Pico Green Reagenz (Pico Green 1:40 in TE-Puffer angesetzt) hinzupipettiert. Mit-tels eines Blindwertes und eines Standards (Blindwert: 900 µl TE-Puffer + 100 µl Pico-GreenReagenz; Standard: 899 µl TE-Puffer + 1 µl Heringssperma (100 ng/µl) + 100 µl Pico GreenReagenz) kann der DNA Gehalt in der Probe bestimmt werden. Dabei werden die Proben in ei-nem Spektrofluorophotometer (Shimadzu) bei einer Extinktion von 460 nm und einer Emissionvon 540 nm gemessen. Das Gerät liefert dabei Werte in ng/µl. Hierbei ist darauf zu achten, daßder Standard nicht von 100 ng/µl abweichen sollte (im Ergebnis). Vor der Messung müssen dieeinzelnen Proben nach Zugabe des Pico Green Reagenz gevortext und 5 Minuten dunkel inku-biert werden. Während der gesamten Messung ist mit Handschuhen und Kittel zu arbeiten. Die Abfälle wer-den im Färbeabfall entsorgt.

Methodenskript AG Cypionka - 38 -
DNA-Quantifizierung am Microtiterplattenreader
Da für die Sequenzierung, Real-Time PCR und DGGE die genaue Menge an DNA bekannt seinmuss, ist es notwendig den DNA-Gehalt zu quantifizieren. Die Quantifizierung erfolgt durch einenDNA-Fluoreszenzfarbstoff PicoGreen (hiermit vorsichtig arbeiten, da der Farbstoff in jeglicheDNA interkalieren kann) und einem Fluoreszenzreader. Die Berechnung erfolgt durch eine Eich-gerade die mittels einer bestimmten DNA Konzentration erstellt wird.
1) Pipettierung DNA-Standards
DNA-Konzentration TE-PufferpH 7,5
DNA-Stammlösungen PicoGreen 1:200 verdünnt mitTE-Puffer pH 7,5
100 ng/µl 99 µl 1 µl 100 µl50 ng/µl 99 µl 1 µl 100 µl25 ng/µl 99 µl 1 µl 100 µl0 ng/µl 100 µl 0 µl 100 µl
2) Pipettierung DNA-Probe
TE-PufferpH 7,5
DNA-Probe PicoGreen 1:200 verdünnt mitTE-Puffer pH 7,5
99 µl 1 µl 100 µl
è PicoGreen erst kurz vor der Messung dazu pipettierenè Inkubation 5 min im Reader
1) Microtiterplattenreader anschalten2) Computer anschalten3) Programm FLUOstar Optima starten4) Einloggen in das Program als „user“ und mit „Run“ bestättigen5) in Menüleiste Setup -> Reader Configuration -> Fluorescence einstellen6) Warnung wegklicken7) am Reader die silbernen Kabel nach oben stellen8) Button Test Protocols drückenè Test name: Reinhard Versuch durch Doppelklick startenè Einstellungen:
a) Basic ParametersTest name: Reinhard VersuchMicroplate: Nunc F96 Microwell 96Positioning delay: 0,1No. of kinetic windows: 1No. of cycles: 3Measurement start time: 0,0No. of flashes per cycle: 20Cycle time: =Variable (abhängig von Probenanzahl)x Fluorescence intensityNo. Of multichromatics: 1Excitation filter: 485 nmEmission filter: 520 nmGain: =Variable (abhängig von höchster DNA-Konzentration)Start: 1

Methodenskript AG Cypionka - 39 -
Stop: 1Pause before cycle: 0
b) LayoutEintragung der Proben- und Standard-Anordnung auf der Plattedanach Button Check timing drücken (durchführen unter Menü Basic Parameters)-> Festlegung der Cycle time
c) Concentrations/Volumes/ShakingNicht wichtig!
d) Injection/TimingNicht wichtig!
f) Timing Overview (One cycle)
9) Button Ampel anklicken -> Test name: Reinhard Versuchnklickenè Einstellungen:
a) Plate IDsID1, ID2, ID3 Beschreibung des Laufs eintragenz.B. ID1 DNA-Bestimmung ID2 DGGE AB1 ID3 PCR AB30No. Of executed runs since program start: (Zahl steigend)
b) Gain AdjustmentRequired value: 90%Excitation Emission Gain485 520 =Variablenach 4,5 min Inkubation mit PicoGreen S1 (=höchster DNA-Standard) markierenund Button Gain Adjustment (well) klicken -> Gain-Berechnung
c) Sample IDsProbenbezeichnungen eingeben
10) Starten der Messung durch anklicken des Buttons Start test run (nach 5 min)11) Auswertung
Button Excel drücken -> Testname wählen -> Anzeige der Raw dataz.B. X1 0 0 0 für 485/520 nmDatei öffnen C:\Reinhard\DNA-Bestimmung Berechnungstabelle.xls (bitte einen eigenenPfad anlegen und diese Datei nur kopieren)Raw data und Evaluation 96 Angaben reinkopieren

Methodenskript AG Cypionka - 40 -
PCR (Polymerase Chain Reaction)
Einleitung
Mit Hilfe der Polymerase-Kettenreaktion (polymerase chain reaction, PCR) lassen sich spezifischeDNA-Sequenzen gezielt vervielfältigen (amplifizieren). Dazu wird die doppelsträngige DNA imersten Schritt bei 94 - 96 °C zu DNA-Einzelsträngen aufgeschmolzen (Denaturierung). DasSchmelzen der DNA ist Voraussetzung dafür, daß sich im zweiten Schritt spezifische Primer andie DNA binden können (Annealing). Man benutzt dabei zwei unterschiedliche Primer, die dem 5'-sowie dem 3'-Ende des zu amplifizierenden DNA-Segments entsprechen. Im dritten Schritt kanneine thermostabile DNA-Polymerase (aus Thermus aquaticus, Taq-Pol.) von den Primern ausge-hend die DNA bei 72 °C replizieren (Elongation). Führt man diese drei Schritte viele Male hinter-einander aus, so verläuft die Amplifikation annähernd exponentiell (Faktor 1,6 – 1,8), da nicht nurdie Original-DNA, sondern auch deren Kopien vervielfältigt werden. Welche Bereiche der Pro-ben-DNA (Template) amplifiziert werden, läßt sich dabei durch die Wahl der Primer bestimmen.So kann z.B. das bakterielle Gen für die 16S rRNA (d.h. die 16S rDNA) gezielt vermehrt werden. Die RT-PCR wird verwendet um RNA amplifizieren zu können. So ist die Taq-Pol. nur in derLage, DNA zu replizieren. Um RNA (hier die 16S rRNA der Ribosomen) zu amplifizieren ist einzusätzlicher Schritt notwendig. Durch die Reverse Transcriptase (RT) wird die RNA in komple-mentäre DNA (cDNA) umgeschrieben und kann anschließend durch die PCR amplifiziert werden.Das Umschreiben der RNA in cDNA und die PCR werden bei einer RT-PCR hintereinander ineinem Arbeitsdurchgang durchgeführt. Alternativ kann man die gesamte extrahierte RNA durchZugabe einer Reversen Transcriptase in cDNA umschreiben (DNA ist stabiler als RNA) und diesefür weitere Untersuchungen genauso wie andere DNA-Proben behandeln.
Allgemeine Arbeitsanleitung für die PCR und RT-PCR
Die zur PCR/RT-PCR Amplifikation verwendeten Lösungen, Gefäße und Pipettenspitzen müssensteril und DNA/RNA-frei sein (und bleiben!), da bei einer Kontamination durch Fremd-DNA/RNA auch diese exponentiell vermehrt werden. Die Möglichkeit einer solchen Kontaminati-on muß immer durch eine DNA/RNA-freie Negativkontrolle, d.h. durch einen Ansatz ohneDNA/RNA-Template, ausgeschlossen werden. Bei Verdacht auf Kontamination sofort den Be-treuenden benachrichtigen, um weiteren Schaden zu vermeiden. Zusätzlich ist es erforderlich im-mer eine Positivkontrolle in einer PCR/RT-PCR einzusetzen (bekannte DNA/RNA) um die Funk-tionsweise der PCR/RT-PCR zu kontrollieren. Zudem ist es erforderlich, dass sämtliche Lösungenund Gefäße DNasen- und RNasen-frei sind, um ein Verdau der Nukleinsäuren im Reaktionsansatzzu verhindern.

Methodenskript AG Cypionka - 41 -
Beispiel für einen PCR-Ansatz: Lösung Menge [µl] H2O (Ampuwa, sauberes PCR-Wasser) von Fresenius 35 10fach konz. PCR Puffer für RedTaq von Sigma 5 dNTP-Lösung (enthaltend je 2,5 Mm der vier Desoxynucleosidtriphosphate)
4
Primer 1 (10 pmol/µl Stammlösung) 1 Primer 2 (10 pmol/µl Stammlösung) 1 BSA (10 mg/ml) 1 MgCl2 (20 mM) 1 RedTaq-Polymerase von Sigma (1U/µl) 1 DNA (ca. 50 ng/µl) 1Gesamtvolumen: 50
- Hat die DNA-Probe eine zu hohe Ausgangskonzentration, oder zu viele inhibierende Faktoren,so muß sie zuvor mit PCR-Wasser entsprechend verdünnt werden.- Der 10fach konzentrierte PCR-Puffer enthält 100 mM Tris-HCl (pH 8,3), 500 mM KCl, 11 mMMgCl2, 0,1 % (w/v) Gelatine.
- Die dNTP-Lösung wird aus 10 mM Stammlösungen der einzelnen Desoxynucleosidtriphosphate(dATP, dCTP, dTTP, dGTP) im Verhältnis (1:1:1:1) hergestellt.
- Die Primer: (Als Beispiel ist hier nur ein Primerpaar angegeben)16S rDNA-Amplifikation:
a) Primer GC357f (Universeller Eubakterien Primer) (Sequenz: 5'-CGC CCG CCG CGC CCCGCG CCC GGC CCG CCG CCC CCG CCC CCC TAC GGG AGG CAG CAG-3')b) Primer 907r (Universeller Primer) (Sequenz 5'-CCG TCA ATT CCT TTG AGT TT-3')Zielsequenz für diese Primer ist ein 626 bp langes Fragment (auf E. coli bezogen) im 16S rRNA-Gen des bakteriellen Genoms. Die GC-Klammer am 5'-Ende der Amplifikate wird benötigt, umdie DNA später in einem denaturierenden Gel (DGGE) auftrennen zu können.- RedTaq DNA Polymerase: 1 Unit/µl in einem Puffer laut Beipackzettel.
Beispiel für einen RT-PCR-Ansatz:
Lösung Menge [µl] H2O (RNase freies Wasser) von Qiagen 32 5fach konz. RT-PCR Puffer von Qiagen 10 RT-PCR dNTP-Lösung (enthaltend je 2.5 mM der vier Desoxynucleosidtriphosphate) von Qiagen
2
Primer 1 (10 pmol/µl Stammlösung) 1 Primer 2 (10 pmol/µl Stammlösung) 1 BSA (10mg/ml) 1 RT-PCR Enzymmix (1U/µl) von Qiagen 2 RNA (ca. 50 ng/µl) 1Gesamtvolumen: 50
- Hat die RNA-Probe eine zu hohe Ausgangskonzentration, oder zu viele inhibierende Faktoren, so muß sie zuvor mit PCR-Wasser entsprechend verdünnt werden.- Der 5fach konzentrierte RT-PCR-Puffer enthält Tris-HCl (pH 8,7), KCl, 12,5 mM MgCl2, 0,01 % (w/v) Gelatine, (NH4)2SO4, DTT.- Die Primer: Es werden die gleichen Primer wie für die PCR verwendet.- RT-PCR Enzymmix: Omniscript und Sensiscript Reverse Transcriptase und HotStarTaq DNA- Polymerase in einem Puffer laut Beipackzettel.
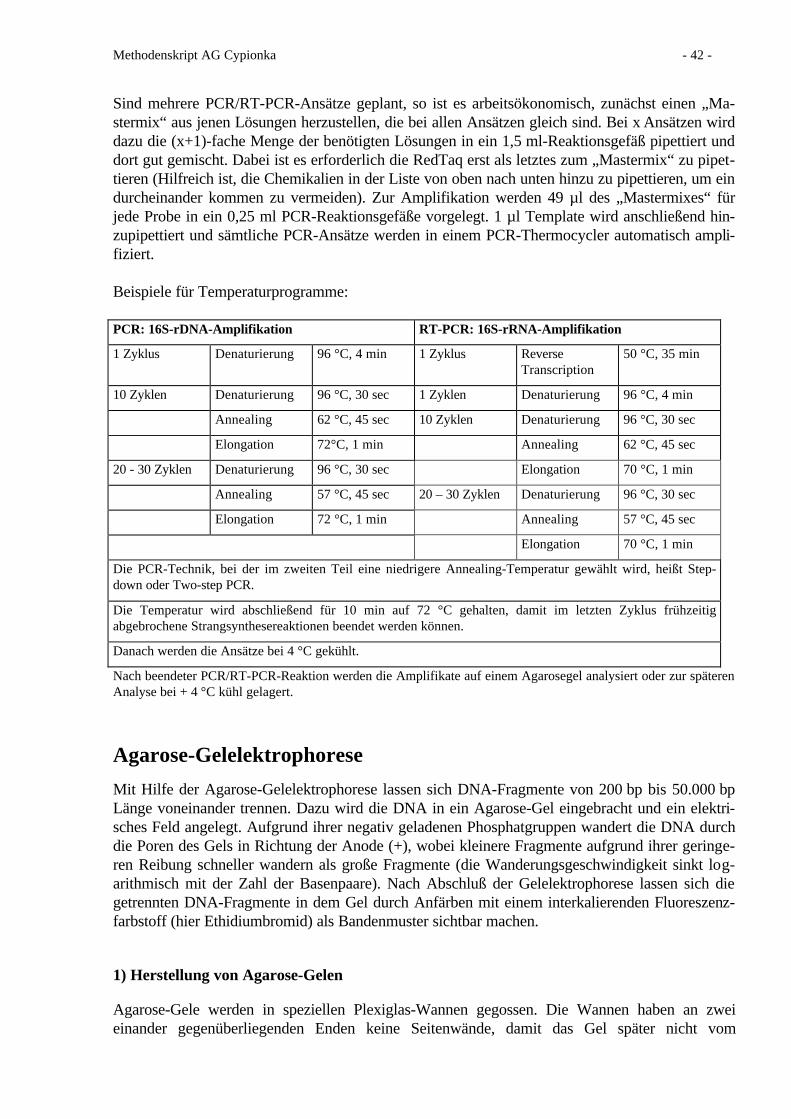
Methodenskript AG Cypionka - 42 -
Sind mehrere PCR/RT-PCR-Ansätze geplant, so ist es arbeitsökonomisch, zunächst einen „Ma-stermix“ aus jenen Lösungen herzustellen, die bei allen Ansätzen gleich sind. Bei x Ansätzen wirddazu die (x+1)-fache Menge der benötigten Lösungen in ein 1,5 ml-Reaktionsgefäß pipettiert unddort gut gemischt. Dabei ist es erforderlich die RedTaq erst als letztes zum „Mastermix“ zu pipet-tieren (Hilfreich ist, die Chemikalien in der Liste von oben nach unten hinzu zu pipettieren, um eindurcheinander kommen zu vermeiden). Zur Amplifikation werden 49 µl des „Mastermixes“ fürjede Probe in ein 0,25 ml PCR-Reaktionsgefäße vorgelegt. 1 µl Template wird anschließend hin-zupipettiert und sämtliche PCR-Ansätze werden in einem PCR-Thermocycler automatisch ampli-fiziert.
Beispiele für Temperaturprogramme:
PCR: 16S-rDNA-Amplifikation RT-PCR: 16S-rRNA-Amplifikation
1 Zyklus Denaturierung 96 °C, 4 min 1 Zyklus ReverseTranscription
50 °C, 35 min
10 Zyklen Denaturierung 96 °C, 30 sec 1 Zyklen Denaturierung 96 °C, 4 min
Annealing 62 °C, 45 sec 10 Zyklen Denaturierung 96 °C, 30 sec
Elongation 72°C, 1 min Annealing 62 °C, 45 sec
20 - 30 Zyklen Denaturierung 96 °C, 30 sec Elongation 70 °C, 1 min
Annealing 57 °C, 45 sec 20 – 30 Zyklen Denaturierung 96 °C, 30 sec
Elongation 72 °C, 1 min Annealing 57 °C, 45 sec
Elongation 70 °C, 1 min
Die PCR-Technik, bei der im zweiten Teil eine niedrigere Annealing-Temperatur gewählt wird, heißt Step-down oder Two-step PCR.
Die Temperatur wird abschließend für 10 min auf 72 °C gehalten, damit im letzten Zyklus frühzeitigabgebrochene Strangsynthesereaktionen beendet werden können.
Danach werden die Ansätze bei 4 °C gekühlt.
Nach beendeter PCR/RT-PCR-Reaktion werden die Amplifikate auf einem Agarosegel analysiert oder zur späterenAnalyse bei + 4 °C kühl gelagert.
Agarose-Gelelektrophorese
Mit Hilfe der Agarose-Gelelektrophorese lassen sich DNA-Fragmente von 200 bp bis 50.000 bpLänge voneinander trennen. Dazu wird die DNA in ein Agarose-Gel eingebracht und ein elektri-sches Feld angelegt. Aufgrund ihrer negativ geladenen Phosphatgruppen wandert die DNA durchdie Poren des Gels in Richtung der Anode (+), wobei kleinere Fragmente aufgrund ihrer geringe-ren Reibung schneller wandern als große Fragmente (die Wanderungsgeschwindigkeit sinkt log-arithmisch mit der Zahl der Basenpaare). Nach Abschluß der Gelelektrophorese lassen sich diegetrennten DNA-Fragmente in dem Gel durch Anfärben mit einem interkalierenden Fluoreszenz-farbstoff (hier Ethidiumbromid) als Bandenmuster sichtbar machen.
1) Herstellung von Agarose-Gelen
Agarose-Gele werden in speziellen Plexiglas-Wannen gegossen. Die Wannen haben an zweieinander gegenüberliegenden Enden keine Seitenwände, damit das Gel später nicht vom

Methodenskript AG Cypionka - 43 -
elektrischen Feld isoliert wird. Um das Gel gießen zu können, müssen diese Seiten daher zunächstmit einem Klebestreifen zugeklebt werden.Es werden 1,5 % (w/v) Agarose in TAE-Puffer 1 facher Stärke gegeben (1 x TAE-Puffer: 40 mMTris(hydroxymethyl)aminomethan; 1 mM EDTA, pH 7,4 eingestellt mit Acetat). Für einStandard-Gel benötigt man 0,6 g Agarose in 40 ml Puffer. Die Agarose wird in einer Mikrowelleso lange erhitzt, bis eine klare, schlierenfreie Lösung entstanden ist. Diese wird anschließend heißin die vorbereitete Wanne gegossen. Anschließend wird über ein Ende und in der Mitte der Wanneein Kunststoffkamm gehängt, und zwar so, daß dessen Zähne einen Abstand von ca. 1 mm(Schichtdicke eines Objekträgers) zum Wannenboden haben. Auf diese Weise bekommt dasfertige Gel an einem Ende und in der Mitte kleine Taschen die der Aufnahme der PCR-Amplifikate dienen. Nach etwa 20 min ist das Gel erstarrt und die Kämme können vorsichtig (!)herausgezogen werden. Zum Schluß werden die Klebebandstreifen entfernt. Wird das Gel nichtsofort benutzt, so muß es in einem mit 1 x TAE-Puffer gefüllten, geschlossenem Behälter vor demAustrocknen geschützt werden.
2.) Elektrophorese
Die Wanne mit dem Gel wird in eine Elektrophorese-Kammer gestellt (die offenen Enden inFeldrichtung). Anschließend wird die Elektrophorese-Kammer so hoch mit 1 x TAE-Puffergefüllt, daß das Gel vollständig eintaucht.Für PCR-Produkte die nicht mit der RedTaq erstellt wurden wird auf einem Stück Parafilm™1 µl-Tropfen Loading-Puffer 6 facher Stärke vorgelegt (6 x Loading-Puffer: 0,25% (w/v)Bromphenolblau; 40 % (v/v) Glyzerin in 1 x TAE). Je 5 µl DNA-Probe werden mit je einemTropfen Loading-Puffer gemischt, indem mit einer Pipette mehrfach hoch und runter gesogenwird. Anschließend wird jede Probe vorsichtig, präzise und luftblasenfrei in eine leere Tasche desGels pipettiert. Von PCR-Produkten die mittels der RedTaq amplifiziert wurden, werden 5 µlohne weitere Zugabe eines Loading-Puffers in die Kammern des Gels pipettiert. Die RedTaqenthält einen Loading-Puffer der das sofortige Auftragen aufs Gel ermöglicht. Als Standard wirdeine 100 bp Ladder verwendet (Konz. 125 ng/µl), die schon mit einem Loading-Puffer versehenist. Vom Standard werden jeweils 5 µl auf das Gel aufgetragen.

Methodenskript AG Cypionka - 44 -
Abschließend wird eine Spannungsquelle an die Kontakte der Elektrophorese-Kammerangeschlossen (korrekte Polung beachten!). Die sich anschließende Elektrophorese erfolgt bei100 V für 40 min (wahlweise 90 V für 45 min).Nach dem Ende der Elektrophorese wird das Gel in einen mit Ethidiumbromid in 1 x TAE-Puffer(0,8 µg/ml Endkonzentration des Ethidiumbromids) gefüllten, vor Licht geschützten,geschlossenen Behälter überführt und auf einem Schüttler für 20 min gefärbt.
–––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––Achtung: Ethidiumbromid ist ein in die DNA interkalierender Fluoreszenzfarbstoff. Es unterscheidet dabei nichtzwischen der Proben-DNA und der DNA unachtsamer Experimentatoren, wo es karzinogen wirkt. Beim Umgangmit Ethidiumbromid ist daher Bedacht angesagt und Handschuhe und Kittel selbstverständlich!
–––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––Nach dem Färben wird das Gel mit Hilfe einer Plastikzange in ein geschlossenen Behälter mitddH2O überführt. Anschließend wird das Gel auf einen Transiluminator im UV-Licht betrachtetund zur Dokumentation mit einer Digitalkamera fotographiert und digital gespeichert.
Aufreinigung von PCR-Produkten
Die PCR-Produkte werden mit dem Purification Kit der Firma Qiagen und einer Tischzentrifugeaufgereinigt: Das PCR-Produkt wird mit 5 Volumina PB-Puffer versetzt und auf eine QIAquick-Säule gegeben. Dann wird bei Raumtemperatur für 1 min zentrifugiert (13000 rpm). Der Durch-fluß wird verworfen und die Säule mit 750 µl PE-Puffer gewaschen; anschließend für 1 min ab-zentrifugieren (13000 rpm). Dieser Durchfluß wird ebenfalls verworfen und die Säule eine weitereMinute bei 13000 rpm zentrifugiert. Die QIAquick-Säule wird danach in ein steriles 1,5 ml-Capgestellt. Die aufgereinigte DNA wird durch Zugabe von 30 - 50 µl EB-Puffer (direkt auf dasZentrum der Membran geben und 1 min inkubieren lassen) von der Membran der Säule gelöst(Mit dem MinElute Kit von Qiagen kann das PCR-Produkt auf 10 – 30 µl eingeengt werden). DieAufreinigung bei der Sequenzierung wird mit PCR-Wasser anstatt EB-Puffer durchgeführt. Durcheine weitere Zentrifugation (13000 rpm, 1 min) wird die DNA eluiert.

Methodenskript AG Cypionka - 45 -
DNA-Sequenzierung nach Sanger (Kettenabbruch-Methode) amLiCor DNA Sequencing System 4200 von MWG Biotech
EinleitungEine Sequenzierreaktion nach Sanger ist im Prinzip eine PCR mit nur einem Primer. Da hierebenfalls mit verschiedenen Temperaturzyklen gearbeitet wird spricht man auch von (engl.) „Cy-cle Sequencing“. Die Entschlüsselung einer einzelnen Sequenz erfordert die Verwendung von vierverschiedenen Ansätzen, bei denen jeder ein anderes Didesoxynukleotid (ddNTP) als „Ab-bruchnukleotid“ enthält. Die Didesoxynukleotide sind zu 3% der „normalen“ dNTPs im Reakti-onsansatz enthalten. Es gibt also je einen Ansatz mit einem Anteil an ddATP, ddGTP, ddCTP,oder ddTTP. Die ddNTPs werden von der DNA-Polymerase wie die normalen dNTPs eingebaut,führen jedoch, da ihnen die OH-Gruppe am 3´-Ende fehlt, zu einem Kettenabbruch. Nach einerstatischen Verteilung des Einbaus der ddNTPs, erhält man somit in jeder Reaktion ein Gemischverschieden langer DNA-Fragmente, die alle auf ein bestimmtes Nukleotid enden (im Ansatz mitddATP also auf A). Die vier Ansätze werden nebeneinander auf ein Gel aufgetragen und das Gemisch der DNA-Fragmente elektrophoretisch ihrer Größe nach getrennt (kurze Fragmente laufen schneller als lan-ge Fragmente). Die Verwendung eines fluoreszierenden Primers ermöglicht die computergesteu-erte Detektion der DNA-Banden über einen Laser, der ähnlich wie ein Scanner über das Gel läuft.Die an ihm vorbeilaufenden Banden werden zur Fluoreszenz angeregt und können somit detektiertwerden. Das Ergebnis wird an einen Computer weitergeleitet und in ein Bild des Gels übersetzt.Ein Vergleich der Lauflängen der Banden aus den vier Ansätzen führt zur Aufdeckung der unbe-kannten Sequenz. Der schematische Ablauf einer Sequenzierungsreaktion ist in der nachfolgendenAbbildung dargestellt. Die Sequenz kann über das Internet an eine Datenbank (Bsp. EMBL, RDP) übermittelt wer-den, wo ein Vergleich mit dort abgelegten Sequenzen erfolgt. Als Ergebnis erhält man die pro-zentuale Ähnlichkeit zu bekannten Sequenzen.
Material
Geräte:ThermocyclerLiCor DNA Sequencing System 4200 (incl. Zubehör)
Chemikalien und Reagenzien:DYEnamic Direct Cycle Sequencing Kit (Amersham)Sequenzierprimer (IRD-markiert)
Stammlösungen:TBE-Puffer (10 ×)Tris Base (890 mM) 108 gBoric Acid (890 mM) 55 gNa2EDTA × 2H20 (20mM) 7,44 gH20 (dest.) ad. 1 l

Methodenskript AG Cypionka - 46 -
Arbeitslösungen:Gellösung:dest Wasser 14,5 mlUrea NF 12,6 g10x TBE-Buffer 3,6 mlsorgfältig rühren bis der Harnstoff gelöst ist"Long Ranger 50% gel solution" 3,6 ml
Abb. Schematische Darstellung der Sequenzierung nach Sanger.
Sequenzier-Reaktion in 4 Ansätzen
(Puffer, Polymerase, MgCl2, dNTPs)
ddATP
+ je 3%
ddGTP ddCTPddTTP
TTGAACAGCCTGACGC
Sequenziergel
6 bp
15 bp16 bp
5´
5´
3´
3´
unbekannte Sequenz
Sequenzier-Primer
bekannte Sequenz
fluoreszenz -markiert
Sequenzierung nach Sanger
T T G A A C A G C C T G A C G C
T G C G
Primer
Primeranlagerung
T T G A A C A G C C T G A C G C
A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 6 bp
A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 15 bp
A A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 16 bp
Sequenzier-Ansatz mit ddATPSequenzier-Reaktion in 4 Ansätzen
(Puffer, Polymerase, MgCl2, dNTPs)
ddATP
+ je 3%
ddGTP ddCTPddTTP
TTGAACAGCCTGACGC
Sequenziergel
6 bp
15 bp16 bp
Sequenzier-Reaktion in 4 Ansätzen
(Puffer, Polymerase, MgCl2, dNTPs)
ddATP
+ je 3%
ddGTP ddCTPddTTP
TTGAACAGCCTGACGC
Sequenziergel
6 bp
15 bp16 bp
5´
5´
3´
3´
unbekannte Sequenz
Sequenzier-Primer
bekannte Sequenz
fluoreszenz -markiert
Sequenzierung nach Sanger
5´
5´
3´
3´
unbekannte Sequenz
Sequenzier-Primer
bekannte Sequenz
fluoreszenz -markiert
Sequenzierung nach Sanger
T T G A A C A G C C T G A C G C
T G C G
Primer
Primeranlagerung
T T G A A C A G C C T G A C G C
A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 6 bp
A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 15 bp
A A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 16 bp
Sequenzier-Ansatz mit ddATP
T T G A A C A G C C T G A C G C
T G C G
Primer
Primeranlagerung
T T G A A C A G C C T G A C G C
A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 6 bp
A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 15 bp
A A C T T G T C G G A C T G C G
Einbau eines ddATP
Kettenabbruch, Fragmentlänge = 16 bp
Sequenzier-Ansatz mit ddATP

Methodenskript AG Cypionka - 47 -
Durchführung
SequenzierreaktionDie Sequenzierreaktion erfolgt mit dem DYEnamic Direct Cycle Sequencing Kit der Firma Amer-sham. Für die zu sequenzierende Probe werden vier 0,2 ml-Caps für die vier Nukleotide G, A, T,C verwendet (G = gelb, A = grün, T = rot, C = blau). Die zu sequenzierenden DNA-Probe (je 2µl) wird in jedes der vier Caps pipettiert. Anschliessend wird ein Mastermix vorbereitet. Pro Pro-be werden ddNTPs (1µl) und die IRD-Primer-Lösung (2 µl; 1pmol/µl) zusammenpipettiert (>10Proben n+2; <10 Proben n+1). Hierbei ist zu beachten, dass die verwendeten Primer fluoreszenz-markiert und daher vor Licht zu schützen sind. Ein Aliquod des Mastermix (3 µl) wird zu jedemAnsatz gegeben, anschließend kurz zentrifugiert, in den Thermocycler gestellt und das Gerät ge-startet (das zum Primer passende Programm wählen!).
Sequenzier-Programm:Temp.[C°] Zeit [min] Zyklen [n]
95 5 195 0,5* 0,5 3072 14 ∞
*Die Annealing-Temperatur ist abhängig von der Schmelztemperatur (Tm) des Primers.
Nach Beendigung des Programms wird Formamid Ladepuffer (5 µl) zu jedem Ansatz geben, ge-mischt und kurz abzentrifugiert. Bis zum Auftrag auf das Sequenziergel können die Proben bei –20°C gelagert werden. Vor dem Auftragen werden die Ansätze im Thermocycler denaturiert(70°C, 5 min), direkt auf Eis gestellt und bis zum Beladen des Gels im Dunkeln gelagert.
Elektrophorese mit dem LiCor DNA Sequencing System 4200:
Vorbereitung der Glasplatten:Die Elektrophorese wird mit einem "Gel-Sandwich" durchgeführt (Länge 33 cm, Gel-Dicke 0,25mm). Bei allen Arbeiten werden Handschuhe und Kittel getragen (Acrylamid ist kanzerogen). Beiden Glasplatten muß auf Markierungen geachtet werden, da Innen- und Aussenseiten besondersgekennzeichnet sind. Die Platten müssen gründlich mit SDS (25%) und Wasser (dest.) gereinigtsowie mit Ethanol (98%) nachgewaschen. Anschließend werden sie in die Halterung des Sequen-ziergeräts eingebaut. Ethanol muss vollständig entfernt werden, da Ethanol die Polymerisationvon Acrylamid behindert.
Gießen des Gels:Die Gel-Lösung wird immer frisch angesetzt und leicht (unter dem Abzug!) erwärmt, bis sich derHarnstoff gelöst hat. Dazu werden die Chemikalien in der oben angegebenen Reihenfolge in einemspeziellen Becherglas mit einem Rührfisch auf einem Magnetrührer gemischt. Um die Polymerisie-rung des Gels zu starten, werden TEMED (20 µl) und APS (200 µl) dazupipettiert und die Gellö-sung mit einer 50 ml-Spritze über einen 0,2 µm-Filter zwischen die waagerecht liegenden Plattengespritzt. Die Bildung von Luftblasen kann durch leichtes Klopfen auf die Glasplatten verhindertwerden. Sobald die Gellösung den unteren Rand erreicht hat, wird der Vorkamm eingesetzt. Dievollständige Polymerisierung dauert etwa 1,5 Stunden.

Methodenskript AG Cypionka - 48 -
Einbau des Gel-Sandwiches:Nach der Polymerisierung wird der Vorkamm vorsichtig herausgezogen und der obere Rand desGels von Acrylamidresten gereinigt. Die Platten werden im Bereich des Detektors nocheinmalgründlich mit Ethanol gesäubert. Am fertigen Gel-Sandwich wird die obere Pufferkammer ange-bracht und das Ganze in das Sequenziergerät mit der unteren Pufferkammer eingesetzt. In beideKammern wird TBE-Puffer (1 ×) bis zur "Max."-Markierung eingefüllt (insges. etwa 1 Liter) unddie Geltasche mit Hilfe einer Spritze mit Puffer gespült. Schließlich werden Deckel und Elektro-kabel angebracht, wobei darauf geachtet werden muss, daß die Platin-Drähte nicht berührt undzerrissen werden! Weitere Informationen stehen im Handbuch DNA Sequencing and GeneticAnalysis Manual, Kapitel Gel Preparation and Electrophoresis.
Steuerung der Elektrophorese:Die Steuerung des Sequenziergeräts erfolgt über den angeschlossenen Computer. Die Einstellun-gen sind dem DNA Analysis Systems Training Manual entnommen. Unterstrichene Begriffe be-schreiben anzuwählende Programme, Optionen oder Keybordtasten: Der Vorgang beginnt mit dem benennen des files über: Base Image IRV, Data Collection, File,New (z. B. FP02, etc.). Über Scanner Control werden die Betriebsdaten überprüft. Soll-Datensind gelb unterlegt, links daneben stehen die Ist-Werte im Gel. Wenn alle Vorgaben geprüft sind,scan status on und high voltage status on. Dann unter Parameters send anklicken. So wird dieVerbindung von Rechner zu Sequencer hergestellt. Jetzt muß die rote Leuchtschrift am Sockeldes Sequenzierers aufleuchten. Zur Equilibrierung des Gels erfolgt zunächst ein Vorlauf ohneProben (etwa eine Stunde).
Einstellungen des Sequenzierers für die Elektrophorese:Spannung 1500 VStromstärke 35 mALeistung 45 WattTemperatur 50°CMotor speed 2Signal Channel 2Rahmen/Frames 25
Wenn der Vorlauf beendet ist können über Options mit Auto Gain die Backround-Noise-Datenüberprüft werden. Danach werden die Autofunktion ausgewählt. Über Options und Focus dieLasereinstellung optimieren und Autofunktion wählen. Danach wieder über Options Auto Gaindurchführen und erneut Autofunktion wählen. Über Options mit Cross Sections muß sich dannfolgendes Bild ergeben:
DunkelgrauMittelgrauHellgrauMittelgrauDunkelgrau
Nach erneutem Autofunktion wählen muß das Gel in 25 Leserahmen (frames) eingeteilt sein.
Beladen des Gels:Nach der Beendigung des Vorlaufs wird die Elektrophorese gestoppt und über view und deleteimage die Daten der Vorlaufzeit gelöscht und über image Notpad (eigenes Icon) ausgefüllt. DerDeckel des oberen Puffertanks wird abgenommen und die Geltasche erneut gespült. Danach wirdder Haifischzahn-Kamm (64-sharkstooth-comb) eingesetzt, so dass sich die abgeschnittenen Zäh-

Methodenskript AG Cypionka - 49 -
ne an beiden Seiten des Kamms auf Höhe der vorderen Glasplatte befinden (die Spitzen der ande-ren Zähne berühren dann gerade die obere Seite des Gels). Vor dem Auftragen werden schließlichalle Proben im Thermocycler denaturiert (70°C, 3-5 min), sofort auf Eis gestellt und erst dann miteiner speziellen Spritze (8-channel-Hamilton syringe) aufgetragen (nicht mehr als 0,8 µl Probepro Tasche). Nun den Deckel des Puffertanks wieder aufsetzen und die Elektrophorese fortsetzen.Der Lauf erfolgt über Nacht und dauert je nach Länge der zu sequenzierenden Fragmente etwa 5 -10 Stunden. Die erzeugte Datei wird automatisch gespeichert. Das Programm Data Collectionkann somit einfach beendet werden.
Auswertung des Gels:Die Auswertung der Proben erfolgt am selben Computer (Software LI-COR 4200), nach denVorgaben des DNA Analysis Systems Training Manual über das Programm: Image Analysis. Fallsdie Sequenzier-Elektrophorese noch läuft, kann man per Alt und Tab-Taste zum Ausgangsmenüwechseln, ohne die Sequnezierung abzubrechen und dort das Program Image Analysis anklicken.Über File und Open Image kann die entsprechende Datei ausgewählt werden. Danach Image No-tepad (eigenes Icon) anklicken, um am Ende des Notpads die Auflistung der Probennummern undihre zugehörigen Geltaschen zu ermitteln. Unbedingt notieren: Gegenüberstellung file-name zuPrimer-Probencodierung. Jedes Gelspurquartett erhält einen eigenen Filenamen durch Vergabevon Buchstaben. Buchstabe m für Spur 1-4, auf der eine bestimmte DNA mit einem bestimmtenPrimer läuft; Buchstabe n für Spur 5-8, auf der eine andere DNA mit einem anderen Primer läuftetc. (Buchstabe j auslassen wegen Verwechselungsgefahr!). Unter Enhancement können Kontrast(s.u.) und Helligkeit verändert werden. Unter Sequence über Lane Definition werden jeder Gel-spur die aufgetragene Base so zugeordnet, wie sie aufgetragen wurden, also ACGT. Die Zuord-nung erfolgt wie im folgenden Beispiel: Doppelklick z. B. auf A mit der linken Maustaste; halten und die entstehende Linie an den lin-ken Rand der A-Spur schieben und loslassen (Definition des linken Randes). Dann links dieserLinie wieder linke Maustaste betätigen, halten, verschieben bis zum rechten Rand der A-Spur,loslassen. (Definition des rechen Randes). Damit ist Spur A fertig definiert und kann per OK be-stätigt werden. Achtung: Bei reverse Primern ist die A-Probe mit T zu benennen usw.! Weiter istnoch folgendes zu beachten: Unter Sequence über Band Spacing kontrollieren, ob die vorgegebenen Linienmitten durch dieeinzelnen Banden gehen; ansonsten entsprechend verändern. Unter Enhancement über Back-ground Substraction oder Bands Kontrast optimieren. Unter Sequence kann nun semi-automatisches Sequenzieren gewählt werden. Die Bestätigungder vom Rechner vorgeschlagenen Banden erfolgt per Doppelklick mit der linken Maustaste. Fallszweifelhaft mit rechter Taste klicken, eine Korrektur erfolgt mit Backspace. Für Basen die derComputer nicht eindeutig identifizieren kann, werden bestimmte Platzhalter gesetzt, die für be-stimmte Kombinationen von Basen (sog. Ambiguities, Mehrdeutigkeiten) stehen. Die Nomenkla-tur nach IUPAC ist in der unteren Tabelle angegeben. Die Speicherung der Sequencen erfolgtunter File mit Save as...Path ist z.B. DNA4000/DATA/FPRAKT. Der File lautet z.B. FP02i. Fi-lenamen eingeben! Er wird dann auf der Festplatte als smp-file (sample-file) gespeichert. Um denfile auf Diskette zu speichern, unter Report File New anklicken, dann den File als rpt- file (report-file) unter a: abspeichern. Wenn die alte Sequenz wieder aufgerufen werden soll: Im ProgrammImage Analysis, in dem wir uns befinden, über File und Open Sample die gewünschte Datei öff-nen. Dabei auf Dateinnamen der Sample-files achten, da nur ein Dateiname für das Bild/Gel exi-stiert (z.B. FP02), aber mehrere Dateinamen für mehrere Sequencen auf dem Bild/Gel exisitieren(z.B. FP02A, FP02B etc.). Soll eine neue Sequenz auf selbem Gel erstellt werden: File und NewSample anwählen, soll eine neue Sequenz auf anderem Gel erstellt werden: File und Open Imageanwählen. Insbesondere die äußeren Sequenzen zeigen oft keine gradlinige Bandenspur. Per Smi-le-Correction unter Enhancement kann per Anklicken von Define und dem manuellen Verschieben
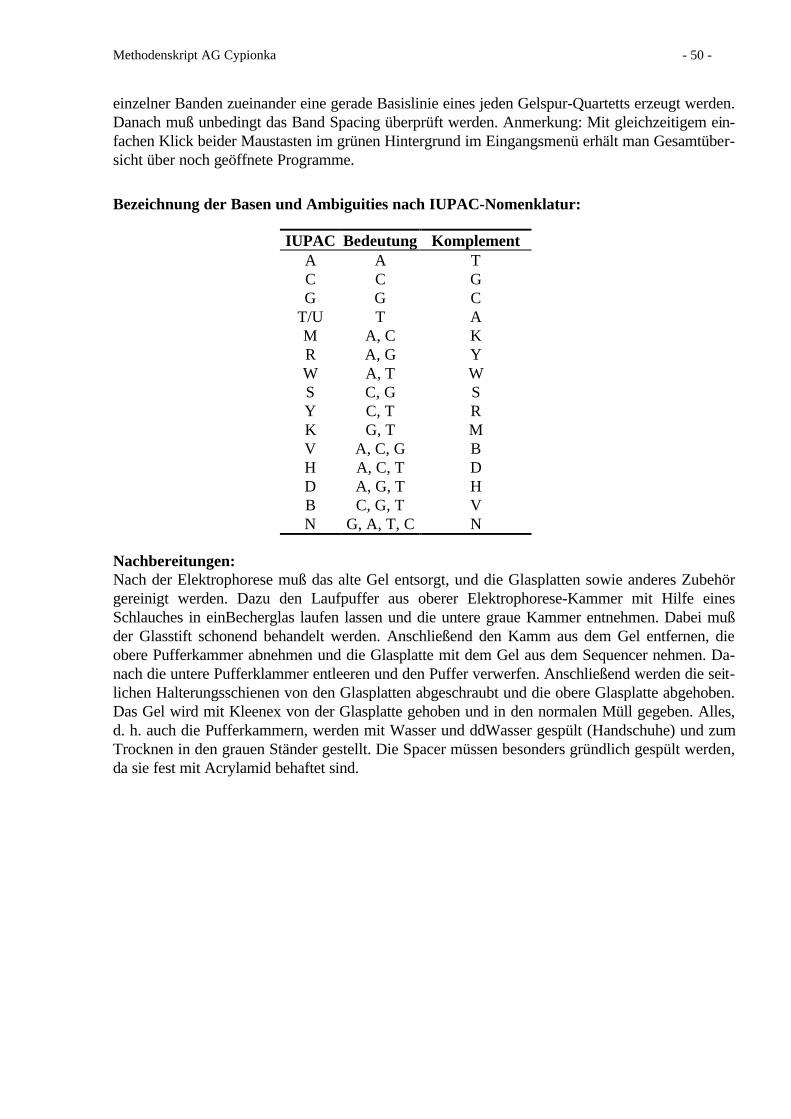
Methodenskript AG Cypionka - 50 -
einzelner Banden zueinander eine gerade Basislinie eines jeden Gelspur-Quartetts erzeugt werden.Danach muß unbedingt das Band Spacing überprüft werden. Anmerkung: Mit gleichzeitigem ein-fachen Klick beider Maustasten im grünen Hintergrund im Eingangsmenü erhält man Gesamtüber-sicht über noch geöffnete Programme.
Bezeichnung der Basen und Ambiguities nach IUPAC-Nomenklatur:
IUPAC Bedeutung KomplementA A TC C GG G C
T/U T AM A, C KR A, G YW A, T WS C, G SY C, T RK G, T MV A, C, G BH A, C, T DD A, G, T HB C, G, T VN G, A, T, C N
Nachbereitungen:Nach der Elektrophorese muß das alte Gel entsorgt, und die Glasplatten sowie anderes Zubehörgereinigt werden. Dazu den Laufpuffer aus oberer Elektrophorese-Kammer mit Hilfe einesSchlauches in einBecherglas laufen lassen und die untere graue Kammer entnehmen. Dabei mußder Glasstift schonend behandelt werden. Anschließend den Kamm aus dem Gel entfernen, dieobere Pufferkammer abnehmen und die Glasplatte mit dem Gel aus dem Sequencer nehmen. Da-nach die untere Pufferklammer entleeren und den Puffer verwerfen. Anschließend werden die seit-lichen Halterungsschienen von den Glasplatten abgeschraubt und die obere Glasplatte abgehoben.Das Gel wird mit Kleenex von der Glasplatte gehoben und in den normalen Müll gegeben. Alles,d. h. auch die Pufferkammern, werden mit Wasser und ddWasser gespült (Handschuhe) und zumTrocknen in den grauen Ständer gestellt. Die Spacer müssen besonders gründlich gespült werden,da sie fest mit Acrylamid behaftet sind.

Methodenskript AG Cypionka - 51 -
DGGE (Denaturing Gradient Gel Electrophoresis)
Die DGGE ist eine molekularbiologische Methode, die verschiedene DNA Sequenzen gleicherLänge voneinander trennt. Die Trennung erfolgt in einem Polyacrylamidgel, in das ein Gradientdenaturierender Agenzien (Harnstoff und Formamid) eingegossen wird. In diesem denaturieren-den Gel wandern verschiedene DNA Sequenzen auf unterschiedliche Weise (abhängig vom GC-Gehalt und sich bildenen Schmelzdomänen) und werden dadurch voneinander getrennt. Weiteretheoretische Grundlagen s. Muyzer et al. 1993.
I. Materialien
Der Arbeitsgruppe stehen zwei DGGE-Apparaturen zur Verfügung. Dabei handelt es sich um eineältere Apparatur der Firma BioRad und um ein neues Model der Firma Ingeny. Da die wesentli-chen Schritte des Protokolls für beide Apparaturen identisch sind, sind nur Abweichungen für dieeinzelnen Geräte mit (alt) für BioRad und (neu) für Ingeny gekennzeichnet.
DGGE-Apparratur von BioRad (alt): DGGE-Apparatur von Ingeny (neu):
a) Gel
- Harnstoff (Urea)- Formamid- 50 x TAE Puffer pH 7,4 (242 g Trisbase, 57,1 ml konz. Essigsäure, 100 ml 0,5 M EDTA pH
7,4, mit doppelt destilliertem Wasser (ddH20) auf fast 1000 ml auffüllen, titrieren mit konz. Es-sigsäure auf pH 7,4, Volumen mit ddH20 auf 1 l auffüllen.
- Acrylamid/Bisacrylamid 40 %ig (37,5:1)- ddH20- 10 % Ammoniumpersulfat (APS), frisch/eingefroren- Tetramethylethylendiamin (TEMED )- Laufpuffer (alt): 6,6 l 1 x TAE pH 7,4, hergestellt aus 50 x TAE pH 7,4: 132 ml 50 x TAE +
6.468 ml ddH20- Laufpuffer (neu): 17 l 1 x TAE pH 7,4, hergestellt aus 50 x TAE pH 7,4: 340 ml 50 x TAE +
16.660 ml ddH20
b) Sonstiges
- Ethanol konz.- 3 x 50 ml Bechergläser- Kanüle und Schläuche

Methodenskript AG Cypionka - 52 -
- 1 große und 1 kleine Glasplatte- 2 Spacer (alt); 1 Spacer (neu)- 4 Halterungen mit Schrauben (alt)- Schraubenschutz (neu)- Gießstand- Kamm mit 20 - 48 Zähnen- Gradientenmischer- DGGE-Apparatur- Magnetrührer- Rührfisch, Gegenvolumen- Dreiwegehahn
c) Färbung
- 2 schwarze Wannen- 500 ml 1 x Sybr Gold Färbelösung (50 µl Sybr Gold (10.000 fach Konzentriert) werden in
500 ml 1 x TAE-Puffer pH 7,4 gelöst)- 500 ml ddH2O- 50 ml Spritze- Schüttler
II. Vorbereitung
Sauberes Arbeiten ist erforderlich:- Eine große und eine kleine Glasplatte, zwei Spacer (alt), ein Spacer (neu) und der Kamm mit
ddH2O säubern. Bei den Glasplatten mit Ethanol das ddH2O entfernen.- Glasplatten mit zwei Spacern und zwei Halterungen zusammenschrauben. Nach dem Zusam-
menbauen die Bündigkeit der Spacer mit dem unteren Plattenrand überprüfen (alt).- Glasplatten mit Spacer und Schraubenschutz zusammenlegen (neu)- Die Glasplatten in den dafür vorgesehenen Ständer einspannen- Kamm ebenfalls zwischen den Glasplatten stecken- Schläuche des Gradientenmischers zusammenstecken. Den Rührfisch in die vordere und das
Gegenvolumen in die hintere Kammer geben.- Magnetrührer anstellen- 3 Bechergläser bereitstellen. Das APS (Gefrierfach), TEMED und die Stammlösungen (Kühl-
schrank) bereit stellen.-
III. Gießen des Gels
- Lösung A, B und C (s. u.) unter dem Abzug herstellen und in den bereitgestellten Becherglä-sern mischen
- Erst kurz vor dem Gießen wird APS und dann TEMED dazugeben (erst nur die Lösungen Bund C mit den Chemikalien versetzen)
- Lösung C in die vordere Kammer des Gradientenmischers geben (höhere Konzentration!).Dreiwegehahn am Gradientenmischer muß geöffnet sein, aber die Kanüle darf noch nicht aufdem Schlauch stecken (zu hoher Wiederstand).
- Hahn des Gradientenmischers kurz öffnen, damit die Luftblase entweichen kann

Methodenskript AG Cypionka - 53 -
- Lösung B in die hintere Kammer des Gradientenmischers geben (niedrigere Konzentration!),dabei den Hahn des Gradientenmischers geschlossen halten
- Gradientenmischer auf den Magnetrührer stellen, dabei den Schlauch so hoch halten wie mög-lich (mindestens über den Gradientenmischer, Dreiwegehahn des Schlauches ist offen!)
- Die Kanüle auf den Schlauch stecken und die beiden Kammern durch Umlegen des Hahnsverbinden
- Die Kanüle zwischen die Glasplatten stecken und das Gel zwischen die Glasplatten laufenlassen. Dabei sollte eine Vermischung der Lösungen in der vorderen Kammer des Gradien-tenmischers zu beobachten sein
- Die komplette Flüssigkeit, auch aus dem Schlauch auslaufen lassen. Dabei ist darauf zu ach-ten, daß die Kanüle während des Gießens nicht ins Gel eintaucht. Zudem darf die Lösung desGradienten den Kamm nicht erreichen. Vorher die Kanüle entfernen und restliche Lösung ineinen Becher laufen lassen.
- Lösung A mit APS und TEMED versetzen- Mit einer 10 ml Spritze Lösung A aufnehmen und die Kanüle auf die Spritze stecken- Lösung A vorsichtig an einer Seite aufs Gel geben. Der Gradient darf dabei den Kamm nicht
berühren, deshalb die Seiten häufiger wechseln.- Das Gel bis an den Rand gießen, so dass es fast überläuft- Mindestens 2 - 4 h warten, bis das Gel auspolymerisiert ist- Sämtliche Becher, Schläuche und Geräte gut mit ddH2O ab- und durchspülen
Vorschrift zur Herstellung der Stammlösungen 0 % und 80 %:
Substanz Stammlösung 0 % denaturierend Stammlösung 80 % denaturierendHarnstoff (Urea) - 50,4 gFormamid - 48 ml50 x TAE, pH 7,4 3,0 ml 3,0 mlAcrylamid/Bisacrylamid 22,5 ml 22,5 mlAuffüllen mit ddH20 auf 150 ml 150 ml
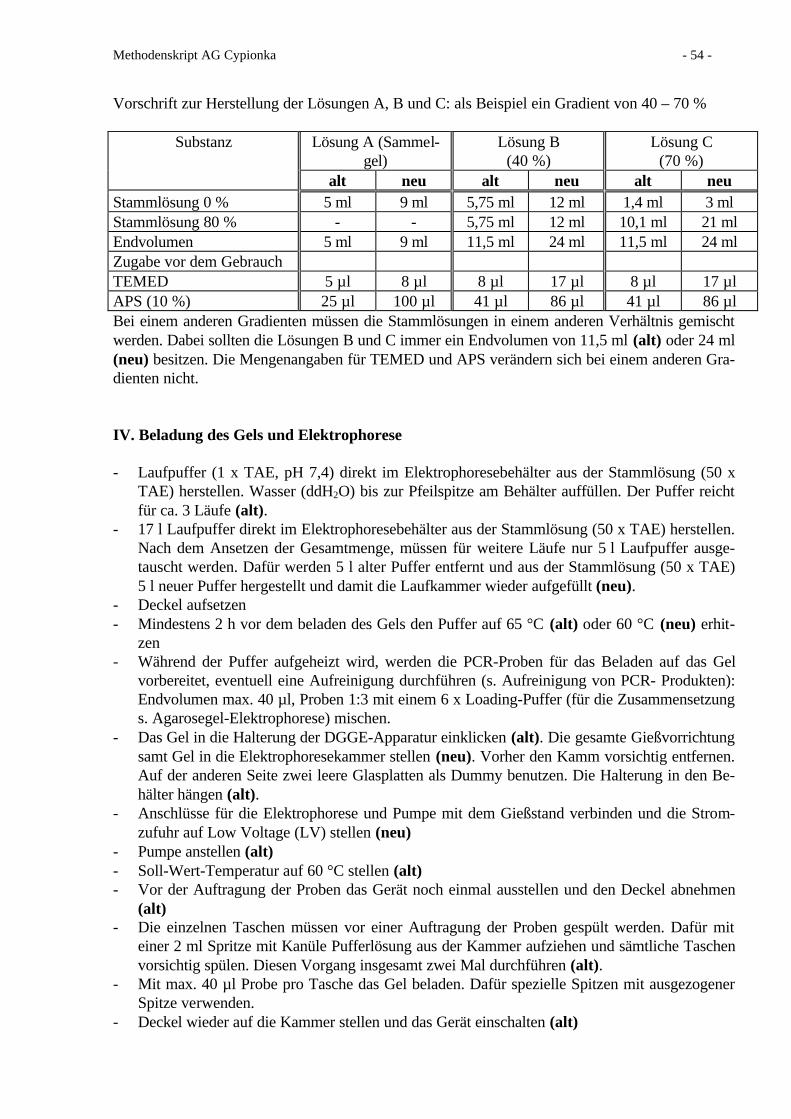
Methodenskript AG Cypionka - 54 -
Vorschrift zur Herstellung der Lösungen A, B und C: als Beispiel ein Gradient von 40 – 70 %
Substanz Lösung A (Sammel-gel)
Lösung B(40 %)
Lösung C(70 %)
alt neu alt neu alt neuStammlösung 0 % 5 ml 9 ml 5,75 ml 12 ml 1,4 ml 3 mlStammlösung 80 % - - 5,75 ml 12 ml 10,1 ml 21 mlEndvolumen 5 ml 9 ml 11,5 ml 24 ml 11,5 ml 24 mlZugabe vor dem GebrauchTEMED 5 µl 8 µl 8 µl 17 µl 8 µl 17 µlAPS (10 %) 25 µl 100 µl 41 µl 86 µl 41 µl 86 µlBei einem anderen Gradienten müssen die Stammlösungen in einem anderen Verhältnis gemischtwerden. Dabei sollten die Lösungen B und C immer ein Endvolumen von 11,5 ml (alt) oder 24 ml(neu) besitzen. Die Mengenangaben für TEMED und APS verändern sich bei einem anderen Gra-dienten nicht.
IV. Beladung des Gels und Elektrophorese
- Laufpuffer (1 x TAE, pH 7,4) direkt im Elektrophoresebehälter aus der Stammlösung (50 xTAE) herstellen. Wasser (ddH2O) bis zur Pfeilspitze am Behälter auffüllen. Der Puffer reichtfür ca. 3 Läufe (alt).
- 17 l Laufpuffer direkt im Elektrophoresebehälter aus der Stammlösung (50 x TAE) herstellen.Nach dem Ansetzen der Gesamtmenge, müssen für weitere Läufe nur 5 l Laufpuffer ausge-tauscht werden. Dafür werden 5 l alter Puffer entfernt und aus der Stammlösung (50 x TAE)5 l neuer Puffer hergestellt und damit die Laufkammer wieder aufgefüllt (neu).
- Deckel aufsetzen- Mindestens 2 h vor dem beladen des Gels den Puffer auf 65 °C (alt) oder 60 °C (neu) erhit-
zen- Während der Puffer aufgeheizt wird, werden die PCR-Proben für das Beladen auf das Gel
vorbereitet, eventuell eine Aufreinigung durchführen (s. Aufreinigung von PCR- Produkten):Endvolumen max. 40 µl, Proben 1:3 mit einem 6 x Loading-Puffer (für die Zusammensetzungs. Agarosegel-Elektrophorese) mischen.
- Das Gel in die Halterung der DGGE-Apparatur einklicken (alt). Die gesamte Gießvorrichtungsamt Gel in die Elektrophoresekammer stellen (neu). Vorher den Kamm vorsichtig entfernen.Auf der anderen Seite zwei leere Glasplatten als Dummy benutzen. Die Halterung in den Be-hälter hängen (alt).
- Anschlüsse für die Elektrophorese und Pumpe mit dem Gießstand verbinden und die Strom-zufuhr auf Low Voltage (LV) stellen (neu)
- Pumpe anstellen (alt)- Soll-Wert-Temperatur auf 60 °C stellen (alt)- Vor der Auftragung der Proben das Gerät noch einmal ausstellen und den Deckel abnehmen
(alt)- Die einzelnen Taschen müssen vor einer Auftragung der Proben gespült werden. Dafür mit
einer 2 ml Spritze mit Kanüle Pufferlösung aus der Kammer aufziehen und sämtliche Taschenvorsichtig spülen. Diesen Vorgang insgesamt zwei Mal durchführen (alt).
- Mit max. 40 µl Probe pro Tasche das Gel beladen. Dafür spezielle Spitzen mit ausgezogenerSpitze verwenden.
- Deckel wieder auf die Kammer stellen und das Gerät einschalten (alt)

Methodenskript AG Cypionka - 55 -
- Am Power-Pack eine Spannung von 50 V einstellen. Nach 30 min die Spannung auf 100 Verhöhen (alt). Stromzufuhr auf High Voltage (HV) umstellen und die Spannung auf 100 Veinstellen (neu). Die Laufzeit des Gels beträgt zwischen 18 h und 22 h.
V. Auswertung der Elektrophorese
- DGGE-Apparatur und Spannungsquelle ausstellen- Gel aus der Halterung entnehmen und Schrauben lösen- Obere Glasplatte vorsichtig abheben- Stege der Geltaschen mit einem Spacer vorsichtig abtrennen und bei einer symetrischen Pro-
benauftragung eine Ecke des Gels entfernen, um die Auftragungsrichtung später rekonstruie-ren zu können
- Gel mit der unteren Glasplatte über der Färbelösung in die Glasschale (in einer schwarzenWanne) halten und vorsichtig das Gel von der Platten lösen lassen, so dass das Gel sich in derFärbelösung befindet
- Zur Färbung der DNA im Gel 1 x Sybr Gold als Färbelösung verwenden- Glasschale mit der zweiten schwarzen Wanne abdecken (Färbelösungen sind Lichtempfind-
lich)- 1 - 2 h leicht schütteln- Färbelösung mit einer 50 ml Spritze vollständig entfernen (die Färbelösung kann wiederver-
wendet werden)- Das Gel mit 500 ml ddH2O mindestens 20 min wässern- Das Gel mit einer Bildanalyse dokumentieren und speichern
Während der Färbung unbedingt Handschuhe und Kittel tragen!
VI. Reinigung und Entsorgung
- Kamm, Spacer und der Gießstand werden sorgfältig mit VE-Wasser und ddH2O gereinigt- Die Glasplatten werden mit min. 10 %igen SDS (Sodium Dodecyl Sulfat) gründlich von
sämtlichen Acrylamidresten gereinigt (Acrylamid ist nicht Alkohol- und Wasserlöslich). An-schließend wird das SDS auf den Glasplatten mit VE-Wasser gründlich entfernt (SDS würdeeine erneute Polymerisation des Acrylamids verhindern). Um Wasserflecken und Salzreste zuentfernen, werden die Glasplatten anschießend mit Ethanol abgespühlt und getrocknet.
- Das Gel sowie verwendete Handschuhe und Tücher im Sonderabfall entsorgen- Die Färbelösung und verwendetes ddH2O, welches mit dem Farbstoff in Berührung kam, im
Färbeabfall entsorgen

Methodenskript AG Cypionka - 56 -
Fluoreszenz-in situ-Hybridisierung (FISH) mit rRNA-Oligonukleo-tid-Sonden zum spezifischen Nachweis verschiedener phylogeneti-scher Gruppen
EinleitungDie Technik der Fluoreszenz-in situ-Hybridisierung (FISH) mit rRNA-Oligonukleotid-Sondenwurde Ende der 80er-Jahre zum ersten Mal beschrieben (Giovannoni et al., 1988; DeLong et al.,1998; Amann et al., 1990) und wird seitdem als ein „Durchbruch“ in der mikrobiellen Ökologiegefeiert. Sie bietet die Möglichkeit einer Charakterisierung von Prokaryoten (Bakterien und Ar-chaeen) auf verschiedenen phylogenetischen Ebenen ohne vorherige Kultivierung. Es handelt sichbei dieser Methode um eine Färbetechnik an fixierten Zellen mit anschließender mikroskopischerAuswertung. Die Selektivität der Färbung beruht auf der besonderen Organisation der ribosoma-len RNA. Durch eine vergleichende Sequenzanalyse können meist auf der kleinen Untereinheit derribosomalen RNA (16S bzw. 18S rRNA) Bereiche ermittelt werden, die auf unterschiedlichenphylogenetischen Ebenen (Gattung bis Domäne) einheitlich sind. Diese, etwa 18 Basenpaare lan-gen Genabschnitte dienen als Zielsequenz für Oligonukleotide, die mit einem Fluoreszenz-Farbstoff (z.B. Cy3) markiert sind. Im Verlauf verschiedener Arbeitsschritte werden zunächst dieZellen von Reinkulturen oder bakteriellen Gemeinschaften aus Umweltproben auf einem Objek-träger fixiert. Danach werden sie mit der Oligonukleotid-Sonde in Kontakt gebracht, die in dieZellen gelangt und an der Zielsequenz auf der ribosomalen RNA bindet (hybridisiert). Die spezifi-schen Bedingungen hierfür müssen jede Sonde ausgetestet werden. Nach der eigentlichen Hybri-disierung werden unspezifisch gebundene Sonden abgewaschen und die Zellen mit einem her-kömmlichen Farbstoff (z.B. DAPI) gegengefärbt. Die fertigen Präparate werden mittels Epifluo-reszenz-Mikroskopie analysiert. Die Wahl verschiedener Filter erlaubt die Unterscheidung vonunspezifisch- (DAPI) und spezifisch (FISH) gefärbten Zellen. Probleme liegen insbesondere imDesign und Testen neuer Sonden, sowie in der Analyse von natürlichen Gemeinschaften. Zellenaus natürlichen Standorten liegen oft in einem physiologischen Zustand vor, in dem sie eher einegeringe Zahl an Ribosomen aufweisen und zeigen daher eine geringere Fluoreszenz. Ein weiteresProblem können störende Komponenten des Standortes darstellen (z.B. Sediment oder Ausfällun-gen), die ebenfalls eine Fluoreszenz besitzen oder die Hybridisierung behindern. Weitere Aspektezur Anwendung der Methode in der mikrobiellen Ökologie können im Review von Ammann &Ludwig, 2000 nachgelesen werden.
Material:
Geräte:Objekträger, Epoxy-beschichtet, mit acht Kammern, ∅ 6mmHybridisierungskammern (Greiner Röhrchen oder Falcon Tubes)HybridisierungsofenWasserbadBlockthermostat3 FärbekammernFluoreszenz-Mikroskop mit Filtersatz für DAPI und Cy3Greiner Röhrchen oder Falcon Tubes zum Waschen
Chemikalien und Reagenzien:Oligonukleotid-Probes, Cy3 markiertFixierte Zellen von Bakterien-Reinkulturen zum Testen der Spezifität der Probes
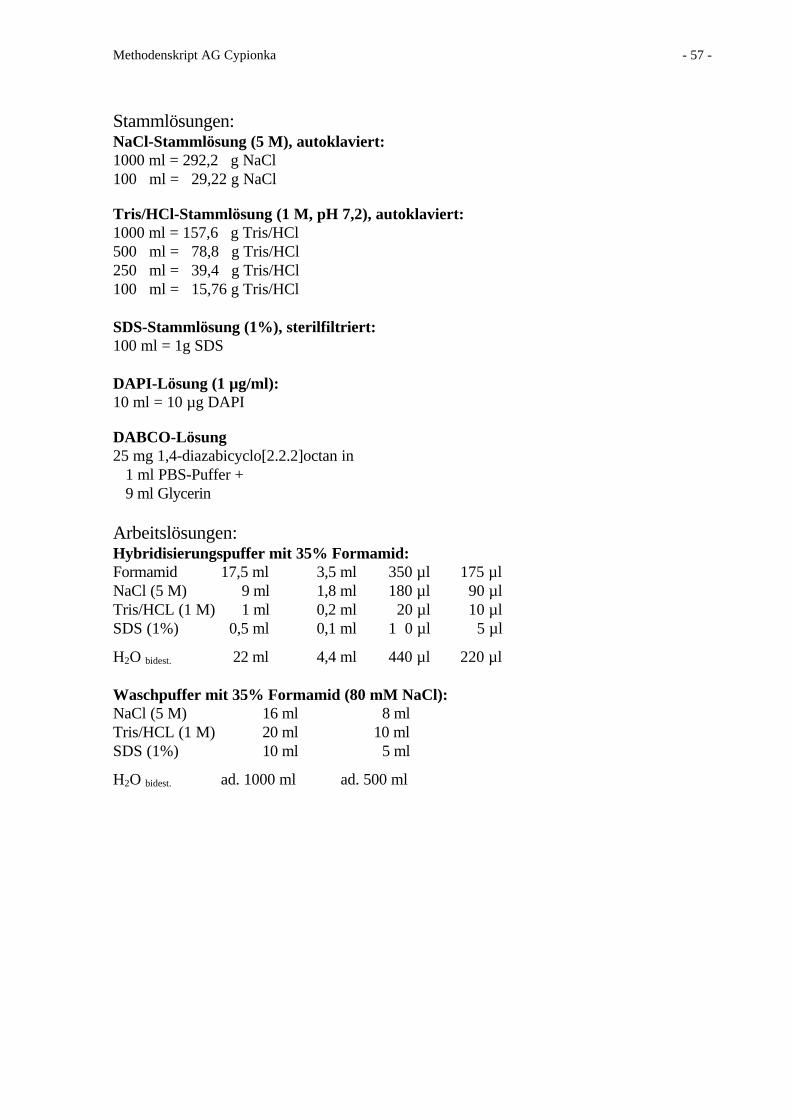
Methodenskript AG Cypionka - 57 -
Stammlösungen:NaCl-Stammlösung (5 M), autoklaviert:1000 ml = 292,2 g NaCl100 ml = 29,22 g NaCl
Tris/HCl-Stammlösung (1 M, pH 7,2), autoklaviert:1000 ml = 157,6 g Tris/HCl500 ml = 78,8 g Tris/HCl250 ml = 39,4 g Tris/HCl100 ml = 15,76 g Tris/HCl
SDS-Stammlösung (1%), sterilfiltriert:100 ml = 1g SDS
DAPI-Lösung (1 µg/ml):10 ml = 10 µg DAPI
DABCO-Lösung25 mg 1,4-diazabicyclo[2.2.2]octan in 1 ml PBS-Puffer + 9 ml Glycerin
Arbeitslösungen:Hybridisierungspuffer mit 35% Formamid:Formamid 17,5 ml 3,5 ml 350 µl 175 µlNaCl (5 M) 9 ml 1,8 ml 180 µl 90 µlTris/HCL (1 M) 1 ml 0,2 ml 20 µl 10 µlSDS (1%) 0,5 ml 0,1 ml 1 0 µl 5 µl
H2O bidest. 22 ml 4,4 ml 440 µl 220 µl
Waschpuffer mit 35% Formamid (80 mM NaCl):NaCl (5 M) 16 ml 8 mlTris/HCL (1 M) 20 ml 10 mlSDS (1%) 10 ml 5 ml
H2O bidest. ad. 1000 ml ad. 500 ml

Methodenskript AG Cypionka - 58 -
Konzentration von NaCl im Waschpuffer bei verschiedenen Konzentrationen von Forma-mid im Hybridisierungspuffer:
Formamid im NaCl im
Hybridisierungspuffer [%] Waschpuffer [mM]
0 9005 636
10 45015 31820 22525 15930 11235 8040 5645 4050 2855 2060 1465 1070 775 580 3,5
Durchführung:
Objekträger vorbereiten:Die Vertiefungen der Objekträger (acht Kammern, ∅ 6mm) mit Beschichtungslösung benetzenund die so behandelten Objekträger an der Luft trocknen lassen (längere Lagerung möglich).
Auftragen der Proben:Zell- oder Sedimentsuspension (3-4 µl) in die Vertiefungen der beschichteten Objekträger gebenund an der Luft trocknen lassen (15 min). Bei Standortproben bis zu 10 µl möglich, evtl. ein-trocknen lassen (auf Thermoblock, 50-60°C) und erneut mit Probe versehen.Lysozymbehandlung (nur bei Gram-positiven Bakterien):
Nach dem Antrocknen der Bakteriensuspension werden 10 µl einer Lysozymlösung (10 mg proml PBS-Puffer = 1% Lysozym) getropft und bei Raumtemperatur mit einer Schale als Staubschutzabgedeckt inkubieren (15 min). Danach Lysozymlösung mit ddH20 (Spritzflasche) abspülen.
Hybridisierungskammer (Falcon Tube) vorbereiten:Zellstoff falten, in das Röhrchen legen und mit Hybridisierungspuffer (1,5 ml) beträufeln. An-schließend das Röhrchen zur Einstellung eines konstanten Dampfdruckes in den Hybridisierungs-ofen legen (30 min) und inkubieren (46°C).
Ethanolbehandlung in Färbekammer:Die Objekträger in das erste Ethanolbad legen (50%, 3 min), danachdie Objekträger in das zweite Ethanolbad legen (80%, 3 min), anschließend die Objekträger indas dritte Ethanolbad legen (96-98%, 3 min) und die Präparate an der Luft trocknen lassen. DieEthanolbäder können mehrfach verwendet werden.
Hybridisierung der Proben:

Methodenskript AG Cypionka - 59 -
Je Ansatz werden 7 µl Hybridisierungspuffer (frisch ansetzen!) und 1 µl Sonde (50 ng/µl) benö-tigt. Mastermix für die entsprechende Probenmenge herstellen und vorheizen (46°C). Danach dieHybridisierungslösung (je 8 µl) in die Vertiefungen pipettieren. Die Objekträger vorsichtig in dievorbereiteten Hybridisierungskammern legen und inkubieren (46°C, 2 h).
Stoppen der Reaktion:Je zwei Objekträger Rücken an Rücken in ein Falcon Tube geben und in kaltem ddH20 schwen-ken. Anschließend das Wasser verwerfen.
Entfernen unspezifisch gebundener Sonden:Je zwei Objekträger Rücken an Rücken in ein Falcon Tube geben und in vorgewärmten Wasch-puffer schwenken (48°C, 20 min). Anschließend den Waschpuffer verwerfen, die Objekträgererneut mit ddH20 abspülen (Spritzflasche benutzen) und die Präparate an der Luft trocknen lassen.
Einbetten und Gegenfärbung:In die Vertiefungen wird je ein Tropfen (1 µl) eines 1:1-Gemisches bestehend aus DAPI- undDABCO-Lösung (= Einbettungsmedium) pipettiert. Der Objekträger wird mit einem Deckgläs-chen abgedeckt und nach Einwirken der Färbelösung (ca. 5 min) mikroskopiert oder dunkel gela-gert.

Methodenskript AG Cypionka - 60 -
Quantitative PCR
EinleitungDie quantitative PCR, auch Real-Time-PCR genannt, ist eine Methode, die es ermöglicht dieAmplifizierung eines spezifischen Sequenzabschnittes (target) während des Experiments überFluoreszenzmessungen zu verfolgen. Zu welchem Zeitpunkt während der cyclischen Amplifizie-rung die Konzentration der PCR-Produkte messbar wird, hängt ab von der Zahl an Zielsequenzen(targets) in der eingesetzten DNA (template). Dies bildet die Grundlage für die Quantifizierungvon spezifischen Zielsequenzen und somit der Zahl an Organismen in der Ausgangsprobe (z.B.Umweltprobe). Grundsätzlich gibt es zwei verschiedene Möglichkeiten der Durchführung:
• Einsatz von spezifischen Primern in Kombination mit interkalierenden Substanzen (z.B.SYBRGreen I), die nach Einlagerung in doppelsträngige DNA fluoreszieren.
• Einsatz von fluoreszenzgelabelten spezifischen Sonden (FRET- und TaqMan-Sonden, Molecu-lar Beacons etc.).
In diesem Skript soll die erste Methode beschrieben werden.
DurchführungGrundsätzlich gelten für die Real-Time-PCR die selben allgemeinen Arbeitsanleitungen wie für diePCR. Es gibt jedoch einige Abwandlungen. So werden in der Real-Time-PCR nur „halbe“ Ansät-ze (25µl) gefahren. Es werden 10µl stärker verdünnter DNA-Lösungen eingesetzt. Dies verringertden Pipettierfehler und verbessert die Quantifizierung. Bei der Real-Time-PCR wird eine farb-stofffreie DNA-Polymerase eingesetzt (New England Biolab Taq DNA-Polymerase, 5U/µl). Da-durch kann eine störungsfreie Fluoreszenzmessung garantiert werden. Der 10fach konzentriertePCR-Puffer für die NEB Taq Polymerase enthält: 10mM KCl, 10mM (NH4)2SO4, 20mM Tris-HCl, 2mM MgSO4, 0,1% Triton X-100 (pH=8,8). Als Fluoreszenzmittel soll SYBRGreen ITM
genutzt werden. Pro Ansatz wird 1µl einer 400fachen Verdünnung der SYBRGreen I-Stammlösung eingesetzt. Da SYBRGreen I, wie alle interkalierenden Substanzen mutagen ist,müssen bei der Herstellung der Verdünnung grüne Gifthandschuhe getragen werden.
Lösung Volumen [µl]10 x Puffer(2mM MgCl2) 2,5dNTPs ( 2,5mM each) 2BSA(20mg/ml) 0,25MgCl2 (10mM) 0,75PCR H2O dest. 7,4357f (10pmol/µl) 0,5907R (10pmol/µl) 0,5SYBRGreen I (1:400) 1NEB Taq-Polymerase (5U/µl) 0,1DNA-Template 10Endvolumen 25
Beispiel für einen PCR-Ansatz und das dazugehörige Temperaturprogramm
Temp. [°C] Time [min] Cycles96 4 194 155 1 4072 2 Fl.-Messung72 10 125 1 1
Die Fluoreszenz wird in jedemZyklus nach der Elongationgemessen

Methodenskript AG Cypionka - 61 -
Der ThermocyclerDurchgeführt wird die Real-Time-PCR am Thermocycler „ROTOR-GENE 2000/3000“ der Firma„Corbett-Research“. Der Cycler wird durch einen angeschlossenen Computer über die ROTOR-GENE Software gesteuert. Es existiert ein Handbuch in dem alle Details ausführlich dargestelltsind. Hier sollen nur die wichtigsten Punkte aufgeführt werden. Der Cycler verfügt über ein36/72er Dual-Kanal-Rotorsystem, wobei für unsere Zwecke der 36er Rotor (s.Abb), der mit 36Flachdeckel PCR-Tubes (0,2ml) beladen werden kann, ausreicht. Der Rotor muß immer voll be-stückt sein, leere Positionen werden mit leeren Tubes aufgefüllt. Der ROTOR-GENE-Cycler istmit 2 Detektions-Einheiten ausgestattet. Kanal (1) detektiert bei 510nm und Kanal (2) detektiertbei 555nm. Dies ermöglicht Multiplex-Detektionen mit 2 Farbstoffen gleichzeitig. Sobald das Probenröhrchen die Detektions-Einheit passiert, wird es durch hochenergetischeBlitze der entsprechenden Wellenlänge angeregt, und das entstehende Emissionslicht durch einenPhotomultiplier gemessen. Diese Daten werden an den PC gesandt und dort graphisch dargestellt(Fluoreszenzeinheiten / Cyklusnummer, s.Abb.).
Die ROTOR-GENE SoftwareVor dem Start der ROTOR-GENE Software muß der Cycler eingeschaltet werden, da die Soft-ware sonst nur im virtuellen Modus läuft.
Nach dem Start des ROTOR-GENE Programms erscheint zuerst das Experiment-Menüfenster
Die Option NEW öffnet das Auswahlfenster, um ein neues Experiment vorzubereiten und zu starten.
Vor dem Start der PCR: Die Toolbar-Arbeitsoberfläche
EXP. INFO (SETTINGS)Hier wird der Name des Operators und des Experiments eingegeben, außerdem werden das Re-aktionsvolumen, der Detektionskanal für die Fluoreszenzmessung und der Rotor des Cyclers aus-gewählt.

Methodenskript AG Cypionka - 62 -
PROFILEHier wird das Temperaturprogramm für die PCR geschrieben, dabei können die Optionen HOLD,CYCLING und MELT ausgewählt werden.
HOLD: Auswahl einer Temperatur, die über einen bestimmten Zeitraum gehalten wird(DENATURE: Extraoption für den ersten Deaturierungsvorgang)
CYCLING: Auswahl von max. 5 nacheinander ablaufenden Temperaturen und Zeiträumen (Dea-turierung, Annealing, Elongation, siehe PCR-Methodenteil). Unter dieser Option wird auch derZeitpunkt der Fluoreszenzmessung bestimmt. Da durch interkalierende Substanzen nur doppel-strängige DNA detektiert werden kann, erfolgt die Fluoreszenzmessung direkt nach der Elongati-on (bei 72°C). An dieser Stelle muß beachtet werden, dass durch den Einsatz interkalierenderSubstanzen auch unspezifische Produkte (z.B. Primerdimere) detektiert werden. Durch Auswahleiner Temperatur bei der z.B. Primerdimere wieder aufgeschmolzen sind (über 80°C) kann dieDetektion von unspezifischen Produkten von vorneherein ausgeschlossen werden. Außerdemkönnen hier auch die Darstellungen für die Rohdaten (Normal/High Sensitive/Low Sensitive) aus-gewählt werden (man lässt sich meistens alle 3 anzeigen).
MELT: Im Anschluss an das PCR-Programm oder separat kann eine Schmelzkurve der PCR Pro-dukte aufgenommen werden. Dabei wird die Temperatur langsam von einer niedrigen Ausgang-stemperatur (meist 50°C) über 1°C-Schritte auf 99°C erhöht. Dabei wird kontinuierlich die Fluo-reszenz für alle Proben gemessen. Vor dem Lauf muß die Anfangstemperatur der Schmelzkurveüber einen Zeitraum von mind. 1 min konstant gehalten werden.
MELT CYCLING
HOLD bzw.
DENATURE

Methodenskript AG Cypionka - 63 -
SAMPLESHier werden die Namen des Proben eingetragen. Man kann zwischen SAMPLE, NTC (non tem-plate controle), STANDARD und NONE wählen. Für die Standards können auch Konzentratio-nen bzw. Kopienzahlen angegeben werden.
STARTBeim Starten des PCR-Laufs erscheint nochmals das PCR-Profil. Außerdem wird das Experimentgespeichert. Die Rohdaten der Fluoreszenzmessungen und der Temperaturverlaufkönnen während des gesamten PCR-Laufs auf dem Bildschirm verfolgt werden.
Nach Beendigung der PCR: Das AnalysemenüDurch Klicken auf den ANALYSIS-Button im Toolbarmenü wird das Analysenmenü geöffnet.
QUANTITATION Bei Doppelklick auf diese Analysenfunktion werden drei Fenster geöffnet, das Hauptfenster indem die normalisierten Kurven der Rohdaten zu sehen sind, das Standardkurvenfenster (wennStandards bei der PCR definiert wurden) und das Ergebnisfenster, in dem sämtliche Daten dereinzelnen Proben aufgelistet sind.
HauptfensterDie normalisierten Rohdaten werden hier linear bzw. logarithmisch dargestellt (Button: LINEARSCALE/LOG. SCALE)
CT-CALCULATIONDer Ct-Wert (cycle time-Wert) ist der Punkt, an dem der Schwellenwert (Threshold) die Amplifi-kationskurven schneidet. Er gibt den Startzeitpunkt der exponentiellen Amplifikation an. Er kann
Das „Analysis“-Fenster ermöglicht es neue Analysen durchzufüh-ren bzw. zeigt bereits durchgeführte an. Als Analysen stehen:COMP. QUANTITATION, ALLELIC DISC., MELT undQUANTITATION zur Verfügung, wobei nur die beiden letzterenfür uns interessant sind. Bei Auswahl einer der Analysen erscheintdie Liste der möglichen Kanäle in den die Auswertungen durch-geführt werden können. Um die Analyse sichtbar zu machen, ge-nügt ein einfacher Doppelklick auf dem Kanal-Namen.

Methodenskript AG Cypionka - 64 -
direkt in Bezug zur Ausgangskopienzahl gesetzt werden. Der Threshold kann automatisch überden AUTO-FIND THRESHOLD-Button (nach Eingabe von Minimum und Maximum) oder ma-nuell über den Button rechts neben dem THRESHOLD eingestellt werden. Über den Buttonrechts neben ELIMINATE CYCLES BEFORE können die ersten PCR-Zyklen, die meist höhereSchwankungen in der Hintergrundfluoreszenz aufweisen, aus den Berechnungen eliminiert wer-den.
StandardkurveIn der Standardkurve sind die Ct-Werte gegen die für die Standards ausgewählten Einheiten(Konzentration, Kopienzahl etc.) aufgetragen. Die Standardkurve kann durch hinzufügen bzw.entfernen von Standards verändert werden. Die blauen Punkte in der Kurve zeigen die als Stan-dard definierten Proben an, die roten die Werte der unbekannten Proben.
CONC=.... : gibt das Verhältnis zwischen Ct-Wert und target-Konzentration im Template an.
TYPE FLOATING: Bei Auswahl dieser Option wird die Standardkurve bei Veränderung desThresholds neu berechnet.
TYPE FIXED: Bei Auswahl dieser Option bleibt die Standardkurve auch bei Veränderung desThreshold unverändert. Dies ist sinnvoll, wenn die Standardkurve auch für andere Experimentegenutzt werden soll.
IMPORT CURVE: hier kann eine Standardkurve aus einem anderen Experiment importiert undzur Berechnung herangezogen werden.
Ergebnisfenster
R-value: Korrelations-Koeffizient, Zahl zwischen 1und –1, die angibt wie gut eine Kurve die Bezie-hung zwischen 2 Variablen (in diesem Fall: y =mx+b) ausdrückt. Ein guter Wert liegt bei 0,99bzw. –0,99.
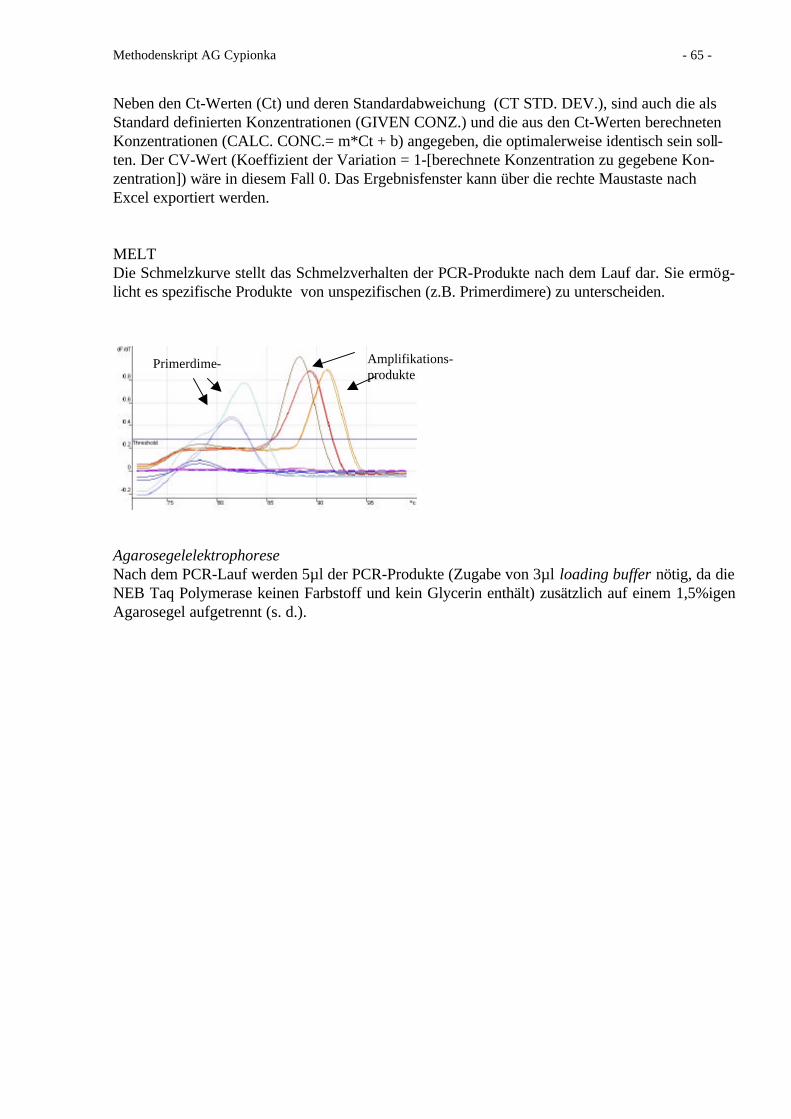
Methodenskript AG Cypionka - 65 -
Neben den Ct-Werten (Ct) und deren Standardabweichung (CT STD. DEV.), sind auch die alsStandard definierten Konzentrationen (GIVEN CONZ.) und die aus den Ct-Werten berechnetenKonzentrationen (CALC. CONC.= m*Ct + b) angegeben, die optimalerweise identisch sein soll-ten. Der CV-Wert (Koeffizient der Variation = 1-[berechnete Konzentration zu gegebene Kon-zentration]) wäre in diesem Fall 0. Das Ergebnisfenster kann über die rechte Maustaste nachExcel exportiert werden.
MELTDie Schmelzkurve stellt das Schmelzverhalten der PCR-Produkte nach dem Lauf dar. Sie ermög-licht es spezifische Produkte von unspezifischen (z.B. Primerdimere) zu unterscheiden.
AgarosegelelektrophoreseNach dem PCR-Lauf werden 5µl der PCR-Produkte (Zugabe von 3µl loading buffer nötig, da dieNEB Taq Polymerase keinen Farbstoff und kein Glycerin enthält) zusätzlich auf einem 1,5%igenAgarosegel aufgetrennt (s. d.).
Amplifikations-produkte
Primerdime-ree

Literatur
Amann, R., L. Krumholz, et al. (1990). Fluorescent-oligonucleotide probing of whole cells fordeterminative, phylogenetic, and environmental studies in microbiology. J. Bacteriol 172: 762-770.
Amann, R. and W. Ludwig (2000). Ribosomal RNA-targeted nucleic acid probes for studies inmicrobial ecology. FEMS Microbiol Rev 24: 555-565.
American Public Health Association (1969) Standard Methods for the examination of water andwaste water including bottom sediments and sludge. pp. 604-609
Balch W E, Fox G E, Magrum L J, Woese C R, Wolfe R S (1979) Methanogens: reevaluation ofa unique biological group. Microbiol Rev 43: 260-296
Bergmeyer H U (1983) Methods of enzymatic analysis. Vol. 3: 357-364Blaut M, Gottschalk G (1984) Coupling of ATP synthesis and methane formation from methanol
and molecular hydrogen in Methanosarcina barkeri. Eur. J. Biochem. 141: 217-222Bradford M M (1976) A rapid and sensitive method for the quantitation of microgram quantities
of protein utilizing the principle of protein dye-binding. Anal Biochem 72:248-254Cavalli-Sforza, L. (1972) Biometrie, G. Fischer Verlag StuttgartChan C. W., Suzuki I., (1993) Quantitative extraction and determination of elemental sulfur an
stoichiometric oxidation of sulfide to elemental sulfur by Thiobacillus thiooxidans. Can J Mi-crobiol 39:1166-1168
Chaney und Marbach (1962) Chemical Analyses Vol. 8, 2nd ed. Colorimetric Determination ofNonmetals (eds.: D.F. Boltz, J.A. Howell) John Wiley & Sons, New York.
Cline E (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. LimnolOceanogr 14:454-458
Cypionka H, Pfennig N (1986) Growth yields of Desulfotomaculum orientis with hydrogen inchemostat culture. Arch Microbiol 143:396-399
DeLong, E. F., G. S. Wickham, et al. (1989). Phylogenetic stains: ribosomal RNA-based probesfor the identification of single cells. Science 243: 1360-1363.
Eichler B Pfennig N (1986) Characterization of a new platelet-forming purple sulfur bacterium,Amoebobacter pedioformis sp. nov. Arch Microbiol 146:295-300
Fitz R M Cypionka H (1990) Formation of thiosulfate and trithionate during sulfite reduction bywashed cells of Desulfovibrio desulfuricans. Arch Microbiol 154:400-406
Giovannoni, S. J., E. F. Delong, et al. (1988). Phylogenetic, groupe-specific oligonucleotideprobes for identification of single microbial cells. J. Bacteriol. 170: 720-726.
Goltermann HL. Methods for chemical analysis of fresh waters, International biological pro-gramme handbook, Blackwell, Oxford
Gregersen, T. Rapid method for distinction of Gram-negative from Gram-positive bacteria. Eur. J.Appl. Microbiol. Biotechnol. 5:123-127
Hobbie, JE, Daley, RJ, Jasper, S. 1977. Use of Nuclepore filters for counting bacteria by epifluo-rescence microscopy. Appl. Environ. Microbiol. 33: 1225-1229
Knippers, Molekulare Genetik, 5. AuflageKelly D P Chambers L A Trudinger P A (1969) Cyanolysis and spectrophotometric estimation of
trithionate in mixture with thiosulfate and tetrathionate. Anal Chem 41:889-901La Riviére J W M (1958) On the microbial metabolism of the tartaric acid isomers. Dissertation,
Universität Delft, NiederlandeLowry O H, Rosebrough N J, Farr A L, Randall R J (1951) Protein measurement with the folin
phenol reagent. J Biol Chem 193:265-275

Projekt G - 67 -
Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of Complex Microbial Popu-lations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700.
Oelze J (1985) in: Methods in Microbiology (Ed. Gottschalk G), Academic Press, Vol. 18, pp257-284
Pachmayr F (1960) Vorkommen und Bestimmung von Schwefelverbindungen in Mineralwasser.Dissertation, Universität München
Pfennig N (1978) Rhodocyclus purpureus gen. nov. and sp. nov., a ring-shaped vitamin B12-requiring member of the family Rhodospirillaceae. Int J Syst Bacteriol 28:283-288
Pfennig N, Wagener S (1986) An improved method of preparing wet mounts for photomicro-graphs of microorganisms. J Microbiol Meth 4:303-306
Schmidt K, Liaaen-Jensen S, Schlegel H G (1963) Die Carotinoide der Thiorhodaceae. I. Okenonals Hauptcarotinoid von Chromatium okenii Perty. Arch Mikrobiol 46:117-126
Steenbergen C L, Korthals HJ (1982) Limnol Oceanogr 27:883-895Tabatabai M A (1974) Determination of sulfate in water samples. Sulphur Inst J 10:11-13Tschech A Pfennig N (1984) Growth yield increase linked to caffeate reduction in Acetobacte-
rium woodii. Arch Microbiol 137:163-167Widdel F (1980) Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten
Sulfat-reduzierender Bakterien. Dissertation, Universität Göttingen


















