
Antikörperanbindung auf plasmapolymerisierte ...€¦ · Shankaran, D.R. and N. Miura, Trends in...
87
Antikörperanbindung auf plasmapolymerisierte Maleinsäureanhydrid-Filme Diplomarbeit zur Erlangung des Grades eines Diplomchemikers dem Fachbereich Chemie der Johannes Gutenberg-Universität Mainz vorgelegt von Véronique Schwartz, geboren in Bernkastel-Kues Mainz 2008
Transcript of Antikörperanbindung auf plasmapolymerisierte ...€¦ · Shankaran, D.R. and N. Miura, Trends in...

Antikörperanbindung auf
plasmapolymerisierte
Maleinsäureanhydrid-Filme
Diplomarbeit
zur Erlangung des Grades
eines Diplomchemikers
dem Fachbereich Chemie der
Johannes Gutenberg-Universität Mainz
vorgelegt von
Véronique Schwartz,
geboren in Bernkastel-Kues
Mainz 2008

ii
Die vorliegende Arbeit wurde in der Zeit von Februar 2008 bis September 2008 am Max-
Planck-Institut für Polymerforschung unter der wissenschaftlichen Betreuung von Prof. Dr.
Wolfgang Knoll und Dr. Renate Förch angefertigt.
Beginn der Diplomarbeit: 01. Februar 2008
Diplomarbeit beim Prüfungsausschuss eingereicht: 30. September 2008
Ich versichere, dass ich die Diplomarbeit selbstständig verfasst habe und keine anderen als die
angegebenen Quellen und Hilfsmittel verwendet habe.
Mainz, den 30. September 2008
Véronique Schwartz

iii
Die Neugier steht immer an erster Stelle eines Problems, das gelöst werden will.
Galileo Galilei

iv
Inhaltsverzeichnis
1 Einleitung……………………………………………………………………..1
1.1 Motivation………………………………………………………………………….2
1.2 Zielsetzung…………………………………………………………………………3
Literaturverzeichnis…………………………………………………………………….4
2 Theoretische Grundlagen……………………………………………………5
2.1 Plasmapolymerisation……………………………………………………………...5
2.2.1 Plasmen…………………………………………………………………..5
2.2.2 Erzeugung von Niederdruckplasmen………………………………….…6
2.2.3 Plasmapolymerisation……………………………………………………6
2.2 Oberflächenplasmonenspektroskoptie……………………………………………10
2.2.1 Theoretischer Hintergrund……………………………………………....11
2.2.2 Messprinzip…………………………………………………………......13
2.3 Optische Wellenleitermodenspektroskopie……………………………………….15
2.4 Oberflächenplasmonenfluoreszenzspektroskopie…………………………..…….17
Literaturverzeichnis…………………………………………………………………...20
3 Materialien und Methoden…………………………………………………21
3.1 Plasmapolymerisation…………………………………………………………….21
3.1.1 Aufbau der Plasmapolymerisationsanlage…………………………..….21
3.1.2 Probenherstellung…………………………………………………….…22
3.1.3 Flussratenbestimmung des Monomers……………………………...…..22
3.2 Probenvorbereitung……………………………………………………………….24
3.2.1 Substrate………………………………………………………………...24
3.2.2 Substratreinigung………………………………………………………..24
3.2.3 Substratbedampfung………………………………………………...…..24
3.2.4 Assemblierungslösungen…………………………………………….….25
3.3 Chemikalien und experimentelle Methoden……………………………………...25
3.3.1 Chemikalien…………………………………………………………..…25
3.3.2 Antikörper……………………………………………………………….26

v
3.3.3 Experimentelle Methoden……………………………………………....26
3.4 Charakterisierungsmethoden……………………………………………………...27
3.4.1 SPR/OWS/SPFS………………………………………………………...27
3.4.2 Bestimmung der Filmdicke mit dem Oberflächenprofilometer……...…30
3.4.3 Kontaktwinkelmessungen…………………………………….…………31
3.4.4 Fourier-Transformation-Infrarot-Reflektions-Absorptions-Spektroskopie
(FT-IRRAS)…………………………………………………………...………32
3.4.5 UV-VIS-Spektroskopie………………………………………………....32
3.4.6 Rasterkraftmikroskopie…………………………………………………33
Literaturverzeichnis…………………………………………………………………...34
4 Ergebnisse und Diskussion………………………………………………....35
4.1 Allgemeine Eigenschaften plasmapolymerisierterMaleinsäureanhydridfilme in
Luft und wässriger Lösung…………………………………………………………....35
4.1.1 Charakterisierung plasmapolymerisierter Maleinsäureanhydrid-Filme mit
Hilfe von Schichtdicken-, AFM-, Kontaktwinkelmessungen und FT-IR-
Spektroskopie in Luft…………………………………………………………36
4.1.2 Charakterisierung plasmapolymerisierter Maleinsäureanhydrid-Filme mit
UV-VIS-Spektroskopie, SPR- und OWS-Messungen in Luft und wässriger
Lösung ………………………………………………………………………..44
4.2 Immobilisierung von IgG-Antikörpern auf plasmapolymerisierten
Maleinsäureanhydridfilmen…………………………………………………………..60
4.2.1 Kopplungsstrategien…………………………………………………….61
4.2.2 SPR-Messoptimierung…………………………………………………..63
4.3.3 Ergebnisse……………………………………………………………….65
Literaturverzeichnis…………………………………………………………………...77
5 Fazit………………………………………………………………………….80
6 Danksagung………………………………………………………………….81
7 Abkürzungsverzeichnis……………………………………………………..82

Kapitel 1 - Einleitung
1
1 Einleitung
1.1 Motivation
Plasmabeschichtungsverfahren haben sich in den letzten Jahrzehnten in vielfältigen
industriellen Anwendungsprozessen etabliert [1], u. A. in der Fahrzeugtechnik als
Vorbehandlung zu Lackierungsprozessen, in der Textilindustrie bei der Erzeugung von
wasserabweisenden Oberflächen, bei der Abscheidung von isolierenden Filmen auf
mikroelektronischen Bauteilen sowie in der Medizintechnik zur Sterilisierung von
medizinischen Geräten. Bei der Plasmapolymerisation handelt es sich um einen Spezialfall
der Plasmabeschichtung, bei dem überwiegend organische Precursormoleküle bei niedrigen
Temperaturen plasmaaktiviert und als dünne Filme auf einer Substratoberfläche abgeschieden
werden. Der aktuelle Stand der Technik erlaubt eine weitgehend zerstörungsfreie
Fragmentierung des Monomergases unter Erhaltung struktureller Eigenschaften. Durch das
breite Spektrum an Precursorn wird eine chemisch strukturelle Vielseitigkeit in einem
sauberen, lösemittelfreien Prozess gewährleistet. Die resultierenden Filme zeichnen sich
durch eine, durch den hohen Vernetzungsgrad bedingte, gute mechanische und chemische
Stabilität, gute Adhesionseigenschaften zum Substrat sowie eine weitgehend flache, lochfreie,
hoch spezifische funktionelle Oberfläche aus. Aufgrund dieser Eigenschaften bieten
Plasmapolymere eine hervorragende Grundlage zum Design von Biosensorgrenzflächen [1-3].
Die Entwicklung von SPR-Biosensoren zur Detektion chemischer und biologischer Spezies in
der medizinischen Diagnostik, in der Lebensmittelindustrie sowie der Umweltanalytik hat in
den letzten Jahrzehnten rapide zugenommen. Der grundlegende Aufbau eines Biosensors
umfasst ein Signalumwandlungsbauteil, auf dem ein biologisches Erkennungselement
assembliert ist, welches mit einem spezifischen Analyten interagieren kann. Ein SPR-
Biosensor nutzt das evaneszente Feld des an einem dünnen Metallfilm generierten
Oberflächenplasmons, um die jeweilige Zunahme des Brechungsindexes zu detektieren,
welche die Anlagerung eines spezifischen Analyts auf dem an der Metalloberfläche
immobilisierten biologischen Erkennungselement bewirkt. SPR-Immunosensoren gehören zu
den am häufigsten verwendeten Sensortypen, da sie eine hohe Affinität und Spezifität zum
Zielanalyten aufweisen.[4, 5].

Kapitel 1 - Einleitung
2
Das menschliche C-reaktive Protein (CRP) spielt eine bedeutende Rolle in der medizinischen
Diagnostik als biologischer Marker für infektiöse und nichtinfektiöse Erkrankungen. Im Falle
einer akuten Entzündung ist eine rapide Zunahme der CRP-Konzentration im Blutplasma zu
verzeichnen. Klinische Studien zeigten, dass das Risiko zur Ausbildung von u. A.
Herzgefäßerkrankungen bei Patienten mit nachweislich erhöhten CRP-Plasma-Werten steigt.
CRP wird standardmäßig über ELISA-Tests (Enzyme-linked Immunosorbent Assay)
nachgewiesen. Diese Methode zeichnet sich zwar durch eine hohe Sensitivität aus, ist aber
relativ teuer und zeitaufwendig [6-8]. Schnellere, ebenso sensitive Nachweistechniken, z. B.
auf der Basis von SPR-Biosensoren, sind daher gefragt. Bisherige Arbeiten berichten über
erste Erfolge modifizierter Dextran-, SAM- und Gold-basierter Sensoroberflächen [7-9].
Über SPR-Biosensoren, insbesondere Immunosensoren, auf der Basis von Plasmapolymeren
ist in der Literatur relativ wenig bekannt [10]. Bisherige Arbeiten berichten über die
Verwendung plasmapolymerisierter Hexamethyldisiloxan- sowie Ethylendiamin-basierter
Immunosensoren zur Detektion von BSA und HSA [11-13].
Plasmapolymerisierte Maleinsäureanhydridfilme (pp-MA) haben sich in der Vergangenheit
als geeignete Plattform für die Anbindung von Biomaterialien bewährt. Die
Polymeroberfläche konnte dabei erfolgreich als Unterlage für Lipid-Doppelschichten [14]
sowie für die Anbindung und Freisetzung von in Phospholipid-Doppelschicht-Vesikeln
eingekapselten Fluoreszenzfarbstoffen genutzt werden [15]. Über die erfolgreiche
Verwendung von pp-MA-Filmen als Grundlage für einen Immunosensor zum Nachweis von
CRP ist in der Literatur nichts bekannt.
1.2 Zielsetzung
Im Rahmen dieser Arbeit sollen pp-MA-Filme auf ihre Kapazität als Plattform für potentielle
SPR-CRP-Sensoren untersucht werden. Hierfür sollen pp-MA-Filme mit einer hoch
funktionalisierten Oberfläche synthetisiert werden, die sich durch eine gute Stabilität in
wässriger Lösung auszeichnen (Kapitel 4.1). Im Anschluss sollen Anti-CRP-IgG-Proteine auf
der Polymeroberfläche immobilisiert und auf ihre Stabilität hin getestet werden (Kapitel 4.2).
Das entwickelte pp-MA-anti-CRP-System soll als Modellsystem eine Grundlage für
nachfolgende Arbeiten zur Entwicklung eines SPR-CRP-Biosensors schaffen.

Kapitel 1 - Einleitung
3
Ein Erfolgsschlüssel für die Entwicklung eines SPR-Biosensors ist die Kopplungseffizienz
des biologischen Erkennungselements auf der Metalloberfläche [10]. Die Kopplungseffizienz
ist stark von der Benetzbarkeit und Ladungsdichte der Oberfläche abhängig [2]. Das
Plasmapolymer stellt hierbei das Bindeglied zwischen Metalloberfläche und Antikörper dar.
Der Polymerfilm muss daher durch gezielte Einstellung der Prozessbedingungen im Plasma
entsprechend funktionalisiert werden und gleichzeitig einen ausreichenden Vernetzungsgrad
beibehalten, um die Stabilität des Grundgerüsts in wässriger Lösung zu gewährleisten.
Proteine tendieren dazu unspezifisch, infolge von attraktiven elektrostatischen, van-der-
Waals– oder über Wasserstoffbrücken induzierte Wechselwirkungen, auf einer festen
Oberfläche zu adsorbieren [5]. Physikalisch adsorbierte IgG-Moleküle bieten eine schlechte
Sensorgrundlage, da die Antikörper weder einheitlich orientiert noch stabil auf der pp-MA-
Oberfläche aufliegen. Im Rahmen dieser Arbeit wird daher eine kovalente Verknüpfung der
Antikörper mit den funktionellen Gruppen der pp-MA-Oberfläche angestrebt, auf deren
Grundlage Orientierung bzw. Aktivität und Belegungsdichte des Antikörpers weiter optimiert
werden können.

Kapitel 1 - Einleitung
4
Literaturverzeichnis
1. R. Förch, Z.Z., W. Knoll, Soft Plasma Treated Surfaces: Tailoring of Structure and
Properties for Biomaterial Applications. Plasma Processes and Polymers, 2005. 2(5): p. 351-372.
2. R. Förch, A.N.C., A. Bousquet, H. L. Khor, M. Jungblut, L. Q. Chu, Z. Zhang, I. Osey-Mensah, E.-K. Sinner, W. Knoll, Recent and Expected Roles of Plasma-
Polymerized Films for Biomedical Applications. Chemical Vapor Deposition, 2007. 13(6-7): p. 280-294.
3. Muguruma, H. and I. Karube, Plasma-polymerized films for biosensors. Trac-Trends in Analytical Chemistry, 1999. 18(1): p. 62-68.
4. Homola, J., Surface Plasmon Resonance Sensors for Detection of Chemical and
Biological Species. Chem. Rev., 2008. 108(2): p. 462-493.
5. Shankaran, D.R. and N. Miura, Trends in interfacial design for surface plasmon
resonance based immunoassays. Journal of Physics D-Applied Physics, 2007. 40(23): p. 7187-7200.
6. Shrive, A.K., et al., Three dimensional structure of human C-reactive protein. Nat Struct Mol Biol, 1996. 3(4): p. 346-354.
7. Albrecht, C., N. Kaeppel, and G. Gauglitz, Two immunoassay formats for fully
automated CRP detection in human serum. Analytical and Bioanalytical Chemistry, 2008. 391(5): p. 1845-1852.
8. Hu, W.P., et al., Immunodetection of pentamer and modified C-reactive protein using
surface plasmon resonance biosensing. Biosensors and Bioelectronics, 2006. 21(8): p. 1631-1637.
9. Juk J. S., Y.S.-J., Jung S.-H., Han J.-A., Kim Y.-M., Ha K.-S., Ex Situ Analysis of
Protein Arrays by SPR Spectroscopy for the Application of Immunorsensors. J. Kor. Phys. Soc., 2004. 44(4): p. 967-972.
10. Muguruma, H., Plasma-polymerized films for biosensors II. TrAC Trends in Analytical Chemistry, 2007. 26(5): p. 433-443.
11. Akimoto, T., K. Ikebukuro, and I. Karube, A surface plasmon resonance probe with a
novel integrated reference sensor surface. Biosensors & Bioelectronics, 2003. 18(12): p. 1447-1453.
12. Nakamura, R., et al., A Plasma-Polymerized Film for Surface Plasmon Resonance
Immunosensing. Anal. Chem., 1997. 69(22): p. 4649-4652.
13. Muguruma, H., et al., Sensor Chip Using a Plasma-polymerized Film for Surface
Plasmon Resonance Biosensors: Reliable Analysis of Binding Kinetics. Analytical Sciences, 2000. 16(4): p. 347-348.
14. Bender K., F.S., Förch R., Jenkins A. T. A., Köper I., Naumann R., Schiller S. M. S. , Knoll W., ed. Plasma Polymer Supported Lipid Bilayers. Plasma Polymers & Related Materials. 2005, Hacettepe University Press 2008. 32-34.
15. Chifen, A.N., et al., Attachment and Phospholipase A2-Induced Lysis of Phospholipid
Bilayer Vesicles to Plasma-Polymerized Maleic Anhydride/SiO2 Multilayers. Langmuir, 2007. 23(11): p. 6294-6298.

Kapitel 2 – Theoretische Grundlagen
5
2 Theoretische Grundlagen
Im folgenden Kapitel werden die theoretischen Grundlagen der im Rahmen dieser Arbeit
verwendeten Prozesstechniken vorgestellt.
2.1 Plasmapolymerisation
2.1.1 Plasmen
Als Plasma bezeichnet man ein quasineutrales Gasgemisch wechselwirkender Spezies
aus Elektronen, Ionen und Molekülen oder Molekülfragmenten. Die physikalischen
Eigenschaften dieses sogenannten „vierten Aggregatzustandes“ werden durch die Anzahl
an freien Ladungsträgern bestimmt, welche für die simultan ablaufenden Prozesse wie die
Ionisation von Gasmolekülen und daraus resultierenden Fragmentierungs- und
Rekombinationsprozessen verantwortlich sind. Die charakteristische Farbe eines
bestimmten Plasmas lässt sich auf die Emission von Strahlung relaxierender angeregter
Zustände zurückführen.
Man unterscheidet zwischen thermischen Hochdruckplasmen, bei denen Elektronen und
Ionen sich im thermischen Gleichgewicht befinden, und nicht-thermischen
Niederdruckplasmen, bei denen die Ionentemperatur im Vergleich zur
Elektronentemperatur um einiges geringer ist. Hochdruckplasmen finden insbesondere
Anwendung bei thermischen Spritz- und Schweißtechniken, Niederdruckplasmen werden
in einer Vielfalt von Beschichtungsprozessen eingesetzt, wie z. B. in Sputterprozessen,
PECVD, Plasmaätzprozessen und insbesondere der in dieser Arbeit verwendeten
Plasmapolymerisation [1].

Kapitel 2 – Theoretische Grundlagen
6
2.1.2 Erzeugung von Niederdruckplasmen
Niederdruckplasmen werden technisch durch elektronenstoßinduzierte, photo- oder
thermisch induzierte Ionisation von Gasmolekülen bei niedrigen Drücken (p<1mbar)
erzeugt. Dabei werden die freien Ladungsträger entweder durch eine
Gleichstromentladung oder eine Hochfrequenzanregung generiert.
Bei der Gleichstromentladung wird eine Gleichspannung zwischen zwei Elektroden
angelegt. Bei genügend hoher Spannung sind die aus der Glühkathode
herausgeschlagenen Elektronen auf ihrem Weg zur Anode in der Lage, Gasmoleküle zu
ionisieren bzw. Sekundärelektronen herauszureißen.
Die Hochfrequenzentladung kann zum einen kapazitiv mit Hilfe von zwei Elektroden
oder induktiv über ein von einer Spule erzeugtes Wirbelfeld eingeleitet werden. Da bei
der HF-Wechselstromentladung nur Verschiebungsströme fließen, so dass der
Nettostromfluss nach außen hin annähernd Null beträgt, ist es im Gegensatz zur
Gleichstromentladung möglich, die Elektroden bzw. Spulen durch dielektrische
Reaktorwände vom Plasma zu trennen, beispielsweise über eine außen an einem
Glasreaktor anliegende Induktionsspule. Die HF-Methode wird vorwiegend für PECVD-
Prozesse oder auch Plasmapolymerisationsprozesse eingesetzt, da das Plasma nur
minimal durch im Zuge der Reaktion auf den Elektroden abgeschiedenes Material
beeinträchtigt wird. Die verwendete Arbeitsfrequenz von 13,56 MHz liegt oberhalb der
Ionenplasmafrequenz, so dass nur die Elektronen in der Lage sind dem zeitlichen Verlauf
der Spannung zu folgen [1].
2.1.3 Plasmapolymerisation
Die Plasmapolymerisation ist ein Verfahren zur Abscheidung organischer und
anorganischer Filme auf unterschiedlichsten Oberflächen. Dabei wird ein
organisches/anorganisches Precursorgas in einer speziell konstruierten Reaktionskammer
plasmaaktiviert, so dass die hierbei erzeugten reaktiven Spezies ein sich ebenfalls im
Reaktor befindendes Substrat bombardieren. An der Substratoberfläche konkurrieren

Kapitel 2 – Theoretische Grundlagen
7
simultan ablaufende Prozesse, die sowohl Oberflächenabtragungsreaktionen, durch
Bindungsdissoziation an der Oberfläche bzw. Oberflächenätzprozesse, als auch eine
Filmabscheidung, durch chemische Reaktionen zwischen aktivierten Oberflächenspezies
und plasmaaktivierten Molekülfragmenten an Radikalen, Ionen, metastabilen Spezies und
Elektronen, ermöglichen. Welcher der beiden Prozesse bevorzugt stattfindet, hängt von
den gewählten Prozessbedingungen, d. h. der Monomerflussrate, dem Monomerdruck,
der Eingangsleistung und der Reaktorgeometrie ab [2].
Die Auswahl der zu behandelnden Substrate wird nur durch die Reaktortauglichkeit des
Materials eingegrenzt. Typische Substrate sind Glas-Objektträger, Si-Wafer,
Kunststoffteile oder auch Textilstreifen.
Im Gegensatz zu konventionellen Polymerisationsverfahren, in denen die
Monomerstruktur den entsprechenden Polymerisationsmechanismus diktiert, ist die
Plasmapolymerisation ein mechanistisch unspezifisches Verfahren, welches keine
besonderen Anforderungen an die Monomerstruktur stellt. Prinzipiell ist jedes
organisches Molekül plasmachemisch polymerisierbar, sofern es sich beim
entsprechenden Arbeitsdruck in die Gasphase überführen lässt. Gasmischungen des
Monomers mit einem Aktivatorgas (O2, Ar) sind ebenfalls möglich. Das Aktivatorgas
kann, aber muss dabei nicht zwangläufig an der Polymerisation beteiligt sein. Schwer
sublimierbare Substanzen können mit Hilfe eines inerten Carriergases in den Reaktor
überführt werden. Im Gegensatz zu den klassischen Polymeren genau definierter
Kettenlängen und Wiederholungseinheiten ähnelt die Filmmorphologie der resultierenden
Plasmapolymere eher einem bunt zusammengewürfelten Netzwerk aus unterschiedlichen
Fragmenten, welches jedoch gleichzeitig einen hohen Anteil an funktionalen
Wiederholungseinheiten enthält.
Der Polymerisationsmechanismus lässt sich am besten mit Hilfe eines schnellen
bizyklischen Stufenwachstumsprozesses beschreiben, welcher simultan über
monofunktional aktivierte Spezies •iM (Zyklus 1, Abb.2.1) und bifunktional aktivierte
Spezies •• iM (Zyklus 2, Abb.2.1) abläuft, deren Interaktion in einem 3D-Netzwerk
willkürlicher Knotenpunkte resultiert [3]. Die Bindungsbildung erfolgt gleichzeitig zu
Ionisierungsprozessen, welche für einen Nachschub an reaktiven Spezies sorgen. Mit

Kapitel 2 – Theoretische Grundlagen
8
steigender eingespeister Leistung erhöht sich auch das Ausmaß an
Oberflächenmodifizierung. Nach dem Abbruch der Plasmareaktion enthält das
resultierende Netzwerk u. A. noch freie Radikalstellen sowie eingelagerte, nichtkovalent
gebundene Fragmente, welche in Kontakt mit Luft oder Flüssigkeit weitere Reaktionen
und/oder Konformationsänderungen eingehen können.
Abb. 2.1 Schematische Darstellung des bicyclischen Stufenwachstumsmechanismus der
Plasmapolymerisation
Mx: neutrales Monomermolekül oder Fragment; •M : monofunktional aktivierte Spezies
(Radikal, Kation, Anion ) ; •• M : bifunktional aktivierte Spezies (Radikalkation, -anion)
i,j,k: unterschiedliche Kettenlängen
Die Filmmorphologie wird maßgeblich von der Reaktorgeometrie sowie den
Prozessbedingungen beeinflusst. Die Reaktorgeometrie definiert sich hauptsächlich über
den Durchmesser, Art und Abstand der Elektroden, dem Monomereinlass und dem
Plasmafluss. Letztere beide Faktoren üben einen nicht zu vernachlässigenden Einfluss auf
die Filmhomogenität aus. Für die Synthese homogener Schichten ist es wichtig, dass alle
Monomermoleküle nach Einlass in den Reaktor die gleiche Wegstrecke bis zur
Abscheidung auf dem Substrat zurücklegen.
Neben der Reaktorgeometrie wirkt sich die Position des Substrats im Reaktor ebenfalls
auf die Filmmorphologie aus, da zwischen den Elektroden Zonen unterschiedlicher

Kapitel 2 – Theoretische Grundlagen
9
Plasmaaktivität existieren. Daher sollte die Probe nach Möglichkeit immer an der
gleichen Stelle in der Reaktionskammer platziert werden. Je nachdem, ob das Substrat
parallel oder senkrecht zum Plasmastrom angeordnet ist, sind laterale Dickegradienten
auf der Probe nicht zu vermeiden.
Eine gewisses Maß an Kontrolle über strukturelle und chemische Zusammensetzung der
Filme lässt sich durch gezielte Einstellung der Prozessbedingung realisieren,
insbesondere dem Monomerbasisdruck sowie der eingespeisten Leistung. Das Plasma
kann kontinuierlich oder nach Einführung einer Frequenzmodulation der HF-Leistung im
gepulsten Zustand betrieben werden.
Grundsächlich sind in Abhängigkeit von der Eingangsleistung bei kontinuierlichem
Plasmabetrieb einige Trends zu beobachten: Mit steigender Eingangsleistung erhöht sich
die Fragmentierung des Monomers sowie der Vernetzungsgrad der Polymerschicht. Die
resultierenden Filme weisen daher wenig Ähnlichkeit zur ursprünglichen
Monomerstruktur auf. Umgekehrt verhält es sich für niedrige Eingangsleistungen: hier
bleibt die Monomerstruktur im Hinblick auf Funktionalität teilweise erhalten, allerdings
auf Kosten des Vernetzungsgrades bzw. der Formstabilität [2].
Bei der gepulsten Plasmapolymerisation wird die Eingangsleistung mit Hilfe eines
Pulsgenerators in „Plasma-An-Phasen“ (ton) und „Plasma-Aus-Phasen“(toff) im Bereich
von Millisekunden aufgeteilt. Quantitativ lässt sich dies durch den Betriebszyklus („duty
cycle“, DC) beschreiben, welcher folgendermaßen definiert ist:
offon
on
tt
tDC
+= (1)
Das Substrat ist somit einer equivalenten Leistung ausgesetzt, die sich aus der
Eingangsleistung, multipliziert mit dem Betriebszyklus, errechnet:
Peakeq PDCP ⋅= (2)
Peq: equivalente Leistung; PPeak: Eingangsleistung
Im Vergleich zum kontinuierlichen Plasmabetrieb nimmt der Ablauf der gepulsten
Plasmapolymerisation etwas geordnetere Formen an: Während der ton-Phase werden die
Reaktionsvorgänge an der Substratoberfläche durch das volle Maß an generierten aktiven

Kapitel 2 – Theoretische Grundlagen
10
Spezies dominiert. In der toff-Phase dagegen sind nur noch Spezies relativ langer
Lebensdauer aktiv. Dabei handelt es sich vorwiegend um Radikalteilchen, welche die
Polymerisation an der Oberfläche fortsetzen. Aufgrund der relativ niedrigen
Konzentration an reaktiven Teilchen ist der zur Filmbildung simultan ablaufende
Abtragungsprozess an der Substratoberfläche vernachlässigbar klein. Zusätzlich ist die
Fragmentierung des Monomers im Vergleich zum kontinuierlichen Plasmabetrieb nicht
so ausgeprägt, da bevorzugt labile Stellen des Monomers, insbesondere
Doppelbindungen, angegriffen werden, so dass Monomerfunktionalitäten bis zu einem
gewissen Grad erhalten bleiben. Hier sind Parallelen zur klassischen radikalischen
Polymerisation erkennbar.
Aufgrund der im vorigen Abschnitt beschriebenen vielseitigen Abhängigkeiten von
äußeren Parametern sind bei der Erforschung plasmapolymerisierter Filme streng
genommen nur Ergebnisse vergleichbar, die unter den gleichen Bedingungen sowie unter
Verwendung von Reaktoren gleicher Bauart zustande gekommen sind.
2.2 Oberflächenplasmonenspektroskopie (SPR)
Bei der Oberflächenplasmonenspektroskopie handelt es sich um ein analytisches
Verfahren zur Untersuchung von Prozessen an Metall/Dielektrikumsgrenzflächen.
Die Anregung des Oberflächenplasmons erfolgt durch das evaneszente Feld eines
totalreflektierten parallel zur Einfallsebene polarisierten Lasers, welcher auf eine an ein
Prisma angrenzende Metallschicht trifft. Die Intensität des reflektierten Lichts wird
hierbei in Abhängigkeit vom Einfallswinkel detektiert. Mit einer Sensitivität von bis zu
150 nm über der Metalloberfläche lassen sich so Brechungsindex- und Dickeänderungen
einer an die Metallschicht angrenzenden Analytschicht erfassen sowie dynamische
Prozesse an der Oberfläche zeitaufgelöst aufnehmen.

Kapitel 2 – Theoretische Grundlagen
11
2.2.1 Theoretischer Hintergrund
Trifft ein Lichtstrahl auf die Grenzfläche zweier Medien mit unterschiedlichem
Brechungsindex wird dieser teilweise gebrochen bzw. reflektiert. Oberhalb eines
charakteristischen Winkels, dem sogenannten Grenzwinkel der Totalreflektion θc , erfolgt
beim Übergang von einem optisch dichteren zu einem optisch dünneren Medium nur
noch eine Reflektion des Lichtstrahls.
Die totalreflektierte Lichtwelle dringt dabei zu einem gewissen Anteil in das optisch
dünnere Medium ein und propagiert in Form einer Oberflächenwelle entlang der
Grenzfläche. Das elektromagnetische Feld dieser Oberflächenwelle, auch Evaneszentfeld
genannt, nimmt dabei sowohl in Ausbreitungsrichtung (x-Richtung) als auch in
Abhängigkeit von der Eindringtiefe (z-Richtung) exponentiell ab.
Unter bestimmten Vorraussetzungen lässt sich das Evaneszentfeld an einer
Metall/Dielektrikumsgrenzfläche zur Anregung von kollektiven longitudinalen
Schwingungen definierter Ausbreitungslänge und exponentiell abfallender
Feldamplitude, sogenannten Oberflächenplasmonen, im angrenzenden
Leitungselektronengas nutzen. Aus Mangel an direkten Nachbarn bzw. Atomrümpfen an
der Grenzfläche breiten sich diese nur entlang der Oberfläche aus. Daher kann die
Anregung auch ausschließlich mit parallel zur Einfallsrichtung polarisiertem (p-
polarisiertem) Licht erfolgen. Hierbei gelten Energie- und Impulserhaltung, d. h. die x-
Komponente des einfallenden Lichtimpulses wird am Resonanzwinkel direkt auf das
Plasmon übertragen. Wie man Abb. 2.2a entnehmen kann reicht der Impuls eines freien
Photons phk in Luft betragsmäßig nicht aus, um ein entsprechendes Plasmon spk anregen
zu können. In Abb. 2.2b sind die Dispersionskurven eines Photons in Luft (1) sowie eines
Oberflächenplasmons (2) an der Metall/Luft-Grenzfläche mit den zugehörigen
Ausbreitungsgeschwindigkeiten im jeweiligen Medium eingetragen. Für die resonante
Anregung eines Oberflächenplasmons müssen sich beiden Kurven schneiden. Durch
Einfügen eines hochbrechenden Mediums, z. B. eines Prismas, erfolgt eine
Impulsangleichung des Photons an das Oberflächenplasmon (Abb. 2.2c), so dass eine
Überlappung der Dispersionskurven realisiert werden kann (Abb. 2.2b (3)).
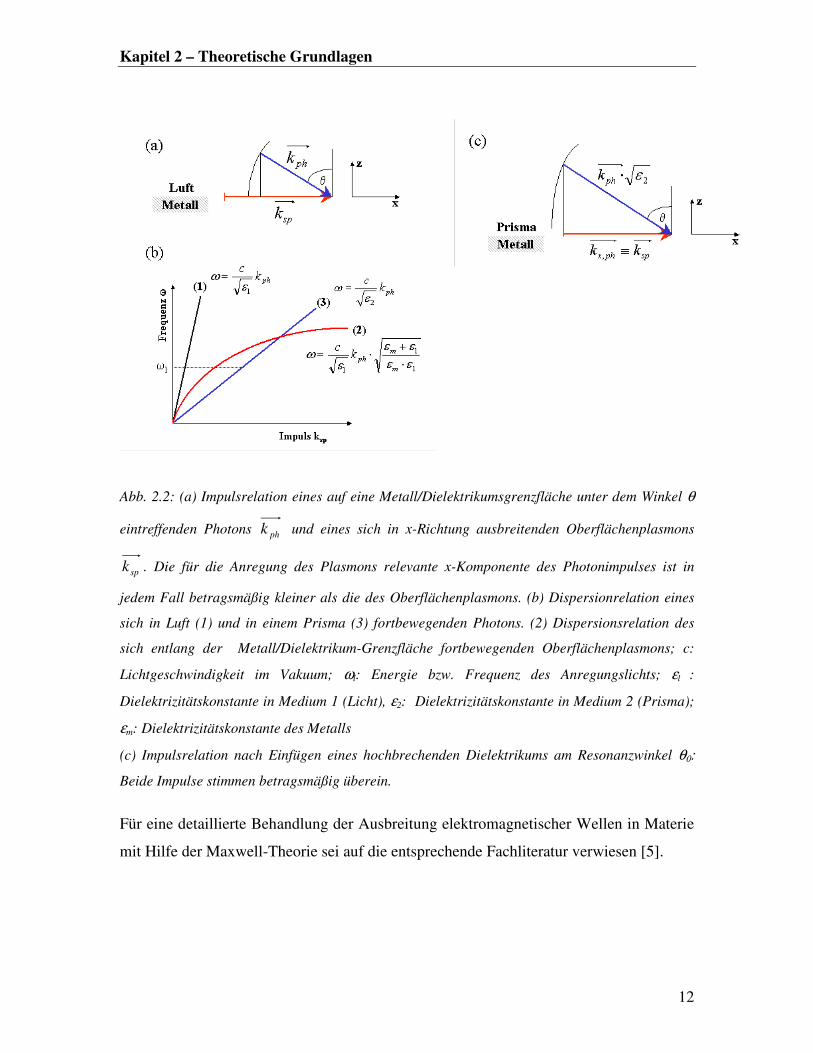
Kapitel 2 – Theoretische Grundlagen
12
Abb. 2.2: (a) Impulsrelation eines auf eine Metall/Dielektrikumsgrenzfläche unter dem Winkel θ
eintreffenden Photons phk und eines sich in x-Richtung ausbreitenden Oberflächenplasmons
spk . Die für die Anregung des Plasmons relevante x-Komponente des Photonimpulses ist in
jedem Fall betragsmäßig kleiner als die des Oberflächenplasmons. (b) Dispersionrelation eines
sich in Luft (1) und in einem Prisma (3) fortbewegenden Photons. (2) Dispersionsrelation des
sich entlang der Metall/Dielektrikum-Grenzfläche fortbewegenden Oberflächenplasmons; c:
Lichtgeschwindigkeit im Vakuum; ωl: Energie bzw. Frequenz des Anregungslichts; εl :
Dielektrizitätskonstante in Medium 1 (Licht), ε2: Dielektrizitätskonstante in Medium 2 (Prisma);
εm: Dielektrizitätskonstante des Metalls
(c) Impulsrelation nach Einfügen eines hochbrechenden Dielektrikums am Resonanzwinkel θ0:
Beide Impulse stimmen betragsmäßig überein.
Für eine detaillierte Behandlung der Ausbreitung elektromagnetischer Wellen in Materie
mit Hilfe der Maxwell-Theorie sei auf die entsprechende Fachliteratur verwiesen [5].

Kapitel 2 – Theoretische Grundlagen
13
2.2.2 Messprinzip
Die resonante Kopplung des einfallenden Photons mit einem Oberflächenplasmons an der
Metall/Dielektrikumsgrenzfläche kann nur unter Einbau bestimmter experimenteller
Konfigurationen zur Impulsangleichung realisiert werden. Die Impulsangleichung kann
sowohl über Gitter- als auch Prismenkopplung erfolgen.
Bei der Prismenkopplung unterscheidet man zwischen der weniger verbreiteten Otto-
Konfiguration und der von Kretschmann und Raether eingeführten am weitesten
verbreiteten Methode [6]. Da im Rahmen dieser Arbeit ausschließlich die Kretschmann-
Konfiguration verwendet wurde, sei hier für eine detaillierte Beschreibung der anderen
Methoden auf weiterführende Literatur verwiesen [5].
Abb. 2.3 zeigt einen schematischen Aufbau der Kretschmann-Konfiguration. Der
Anregungslaser trifft auf ein Glasprisma, an dessen Unterseite ein dünner Metallfilm von
ca. 50 nm aufgebracht ist, so dass dessen Evaneszentfeld noch in der Lage ist, ein
Oberflächenplasmon auf der dem Prisma abgewandten Metallseite zu induzieren. ( In der
Praxis wird der Metallfilm meist nicht direkt auf das Prisma, sondern auf ein dem
Brechungsindex des Prismas angepasstes Glassubstrat aufgebracht.)
Abb. 2.3: Schematischer Aufbau der Kretschmann-Konfiguration
Der Detektor zeichnet die Lichtintensität des reflektierten Strahls in Abhängigkeit vom
Einfallswinkel θ auf. In Abb. 2.4 sind zwei typische Reflektivitätskurven dargestellt.
Kurve a in Abb. 2.4 zeigt eine Reflektivitätsaufnahme in Luft eines mit 2 nm Chrom und
50 nm Gold bedampften LaSFN9-Glases. Nähert man sich dem Grenzwinkel der
Totalreflektion θc, steigt die reflektierte Intensität an, erreicht dann ihr Maximum bei θc
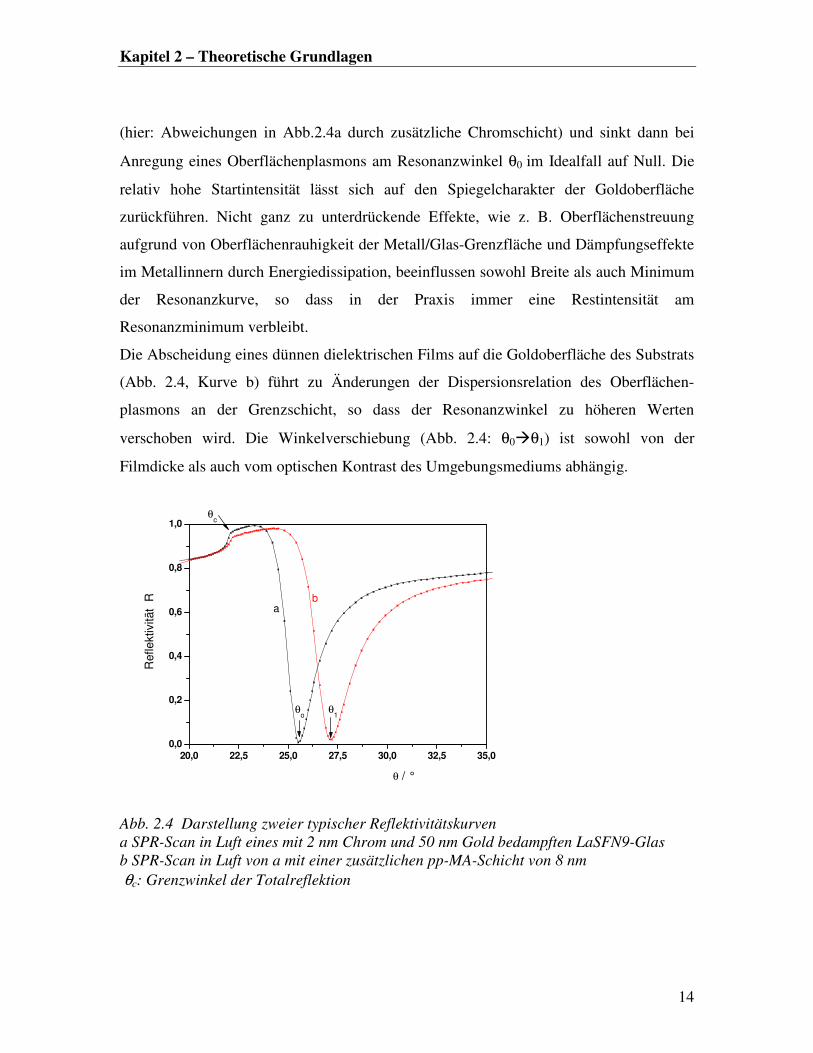
Kapitel 2 – Theoretische Grundlagen
14
(hier: Abweichungen in Abb.2.4a durch zusätzliche Chromschicht) und sinkt dann bei
Anregung eines Oberflächenplasmons am Resonanzwinkel θ0 im Idealfall auf Null. Die
relativ hohe Startintensität lässt sich auf den Spiegelcharakter der Goldoberfläche
zurückführen. Nicht ganz zu unterdrückende Effekte, wie z. B. Oberflächenstreuung
aufgrund von Oberflächenrauhigkeit der Metall/Glas-Grenzfläche und Dämpfungseffekte
im Metallinnern durch Energiedissipation, beeinflussen sowohl Breite als auch Minimum
der Resonanzkurve, so dass in der Praxis immer eine Restintensität am
Resonanzminimum verbleibt.
Die Abscheidung eines dünnen dielektrischen Films auf die Goldoberfläche des Substrats
(Abb. 2.4, Kurve b) führt zu Änderungen der Dispersionsrelation des Oberflächen-
plasmons an der Grenzschicht, so dass der Resonanzwinkel zu höheren Werten
verschoben wird. Die Winkelverschiebung (Abb. 2.4: θ0�θ1) ist sowohl von der
Filmdicke als auch vom optischen Kontrast des Umgebungsmediums abhängig.
20,0 22,5 25,0 27,5 30,0 32,5 35,0
0,0
0,2
0,4
0,6
0,8
1,0
Refle
ktivität
R
θ / °
ab
θo
θ1
θc
Abb. 2.4 Darstellung zweier typischer Reflektivitätskurven
a SPR-Scan in Luft eines mit 2 nm Chrom und 50 nm Gold bedampften LaSFN9-Glas
b SPR-Scan in Luft von a mit einer zusätzlichen pp-MA-Schicht von 8 nm
θc: Grenzwinkel der Totalreflektion

Kapitel 2 – Theoretische Grundlagen
15
Mit Hilfe der Fresnel-Theorie, welche die Kurve als Komplex homogener Multischichten
behandelt, lassen sich Brechungsindex und Dicke des auf das Metall aufgebrachten
Analytfilms in Abhängigkeit voneinander berechnen. Ist z. B. die Dicke des Analyts aus
unabhängigen Messungen bekannt, lässt sich somit der Brechungsindex bestimmen und
umgekehrt. Alternativ können beide Parameter mit Hilfe eines Variationsexperiments
unabhängig voneinander bestimmt werden, indem man eine Messung in zwei Medien
unterschiedlicher optischer Eigenschaften, z. B. Luft und Wasser, durchführt. Dies ist
allerdings nur für Filme möglich, die ihre strukturellen Eigenschaften in beiden Medien
nicht ändern [7].
Für Details zur Fresnel-Theorie sei auf die entsprechenden Fachliteratur verwiesen [8, 9].
2.3 Optische Wellenleitermodenspektroskopie (OWS)
Die optische Wellenleitermodenspektroskopie stellt einen Spezialfall der im vorigen
Abschnitt beschriebenen Oberflächenplasmonenspektroskopie dar, die sich insbesondere
zur Charakterisierung anisotroper Filme eignet.
Für den experimentellen Aufbau gelten die in Abschnitt 2.2.2 beschriebenen Prinzipien
mit dem Unterschied, dass nun auf der Goldoberfläche dickere Analytfilme (d > 200 nm)
abgeschieden werden, die als Wellenleiter im Substrat-Metall/Film/Luft-System
(Abb. 2.4a ) fungieren. Je nach Dicke und dielektrischen Eigenschaften des
abgeschiedenen Films lassen sich Wellenleitermoden verschiedener Ordnung (m) in
Abhängigkeit vom Einfallswinkel θ detektieren (Abb.2.4b ).
Das Prinzip der Wellenleitung beruht auf der Fortpflanzung von Licht innerhalb eines
optisch abgegrenzten, transparenten Mediums durch Totalreflektion an dessen
Grenzflächen. Für die Realisierung einer derartigen Konstruktion gibt es mehrere
Möglichkeiten, auf deren detaillierte Beschreibung mit Verweis auf die entsprechende
umfangreiche Fachliteratur verzichtet wird [10, 11].

Kapitel 2 – Theoretische Grundlagen
16
Im Folgenden beschränken wir uns auf die im Rahmen dieser Arbeit relevante Struktur
des asymmetrischen planaren Wellenleiters. Abb. 2.5a zeigt die Geometrie eines solchen
Wellenleiters: Der Wellenleiterfilm ist auf beiden Seiten durch optisch dünnere Medien
eingegrenzt, so dass für den Brechungsindex des Wellenleiters n2 an jeder Stelle folgende
Randbedingungen gelten: 312 , nnn > . Nach Einkopplung des Lichts oberhalb von θc
propagiert dieses nun entlang der Grenzflächen.
Abb. 2.4
(a) Schematische Darstellung der
experimentellen Konfiguration für
OWS. Zusätzlich zum Oberflächen-
plasmon sind Wellenleitermoden
der 1.-3. Ordnung dargestellt.
(b) Zugehöriger Reflektivitätscan zu
dem in Abb.2.4a gezeigten
Wellenleiter. Die verschiedenen
Wellenleitermoden erscheinen als
scharfe Minima im Spektrum, wobei
die Mode höchster Ordnung den
niedrigsten Resonanzwinkel θm
besitzt.
θc: Grenzwinkel der Totalreflektion
Abb. 2.5
(a)Schematischestrahlenoptische
Darstellungeines asymmetrischen
planaren Wellenleiters folgender
Geometrie:
Substrat(Glas/Au)/Film/Luft.
Es gilt: 312 , nnn > ; 21 nn ≠ ;
(b) Optische Feldverteilung des in
Abb.2.5adargestelltenWellen-
leiters für die s-polarisierten
Moden 1.-3. Ordnung

Kapitel 2 – Theoretische Grundlagen
17
Eine genauere Analyse des geschilderten 3-Lagen-Systems unter Einbeziehung der
Maxwell-Gleichungen zeigt, dass konstruktive Interferenz der elektromagnetischen
Wellen unter den gegebenen Randbedingungen und bei fixierter Wellenlänge nur für
bestimmte Kombinationen an optischen Eigenschaften, mathematisch mit Hilfe der
Eigenwertgleichung für Wellenleitermoden m’ter Ordnung beschrieben, existieren kann
[5]. Die sich so ergebenen Eigenmoden entsprechen der in Abb. 2.5b dargestellten
optischen Feldverteilung senkrecht zur Wellenleiterachse (z-Achse). Genauer betrachtet
zerlegt man eine Mode in zwei Komponenten unterschiedlicher Polarisation, sogenannte
TE- (transversal elektrisch, p-) und TM-Moden (transversal magnetisch, s-), da diese zur
Erfüllung der Phasenregel für konstruktive Interferenz unterschiedliche
Ausbreitungsgeschwindigkeiten im Wellenleiter besitzen. Dies lässt sich insbesondere für
die Charakterisierung anisotroper Filme nutzen:
Zur Illustration dieses Sachverhaltes kehren wir zu Abb. 2.4a zurück. Die ‚nullte’ Mode
entspricht dem Oberflächenplasmon, dessen Resonanzwinkel aufgrund des
exponentiellen Abfalls in z-Richtung nur von nz, dem mittleren Brechungsindex des
Filmdurchmessers, abhängig ist. Die Anregung von Moden höherer Ordnung erfolgt mit
s- oder p-polarisierten Licht. Daher hängen diese sowohl von den Brechungsindices aller
drei Raumrichtungen nx, ny, und nz, als auch der Filmdicke d ab. Die Mode höchster
Ordnung hat aufgrund ihrer erhöhten Feldverteilung an den Grenzflächen zu den optisch
dünneren Medien (vgl. Abb. 2.4a, m = 3) die höchste Sensitivität in Bezug auf die
Filmdicke. Die Auswertung unter Bestimmung der Parameter n und d ermöglicht die
Erstellung eines Profils optischer Eigenschaften für anisotrope Filme.
2.4 Oberflächenplasmonenfluoreszenzspektroskopie (SPFS)
In bestimmten Fällen reicht die Sensitivität der Oberflächenplasmonenspektroskopie
allein nicht aus, um Prozesse an Grenzflächen zu untersuchen. Dies trifft vor allem für
die Untersuchung von Grenzflächenanbindungsprozessen niedrig konzentrierter, relativ
kleiner Analytmoleküle geringen Molekulargewichts zu. Hier ist es notwendig, das
Signal mit Hilfe von Fluoreszenzmarkierung zu erhöhen [12].
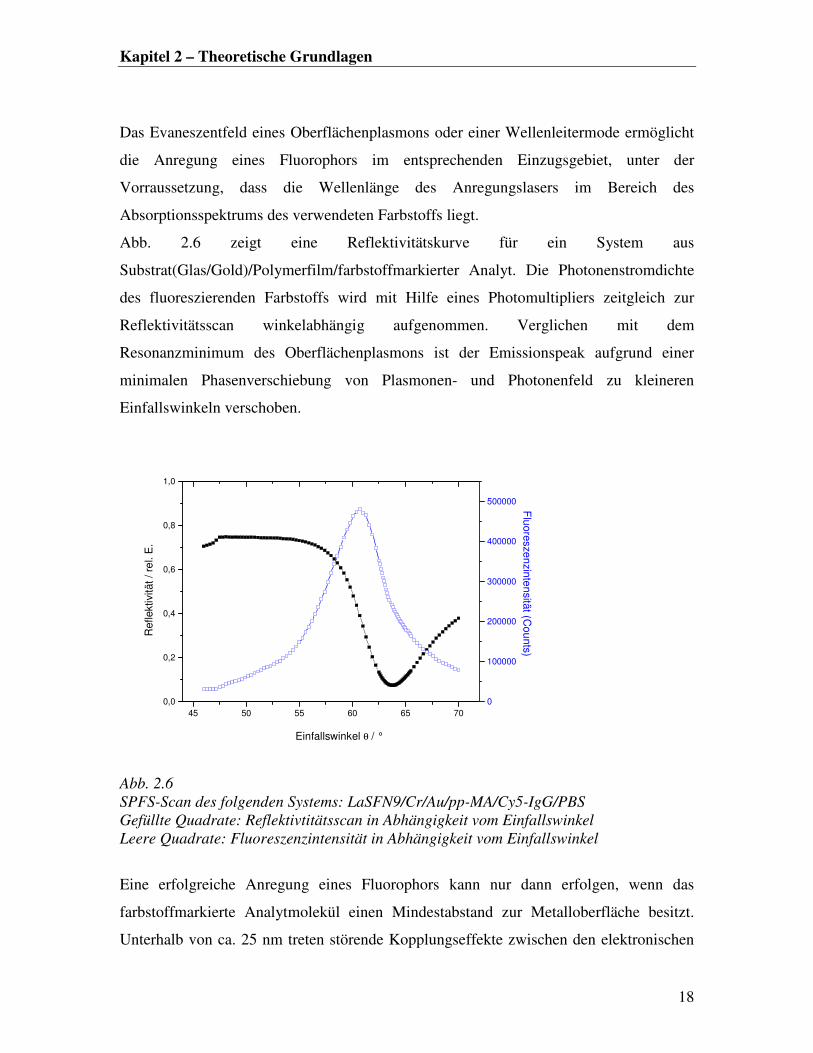
Kapitel 2 – Theoretische Grundlagen
18
Das Evaneszentfeld eines Oberflächenplasmons oder einer Wellenleitermode ermöglicht
die Anregung eines Fluorophors im entsprechenden Einzugsgebiet, unter der
Vorraussetzung, dass die Wellenlänge des Anregungslasers im Bereich des
Absorptionsspektrums des verwendeten Farbstoffs liegt.
Abb. 2.6 zeigt eine Reflektivitätskurve für ein System aus
Substrat(Glas/Gold)/Polymerfilm/farbstoffmarkierter Analyt. Die Photonenstromdichte
des fluoreszierenden Farbstoffs wird mit Hilfe eines Photomultipliers zeitgleich zur
Reflektivitätsscan winkelabhängig aufgenommen. Verglichen mit dem
Resonanzminimum des Oberflächenplasmons ist der Emissionspeak aufgrund einer
minimalen Phasenverschiebung von Plasmonen- und Photonenfeld zu kleineren
Einfallswinkeln verschoben.
45 50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Einfallswinkel θ / °
Reflektivität / re
l. E
.
0
100000
200000
300000
400000
500000
Flu
ore
sze
nzin
ten
sitä
t (Co
unts
)
Abb. 2.6
SPFS-Scan des folgenden Systems: LaSFN9/Cr/Au/pp-MA/Cy5-IgG/PBS
Gefüllte Quadrate: Reflektivtitätsscan in Abhängigkeit vom Einfallswinkel
Leere Quadrate: Fluoreszenzintensität in Abhängigkeit vom Einfallswinkel
Eine erfolgreiche Anregung eines Fluorophors kann nur dann erfolgen, wenn das
farbstoffmarkierte Analytmolekül einen Mindestabstand zur Metalloberfläche besitzt.
Unterhalb von ca. 25 nm treten störende Kopplungseffekte zwischen den elektronischen

Kapitel 2 – Theoretische Grundlagen
19
Metallzuständen und den Molekülorbitalen des Chromophors auf, die in Quenching der
Fluoreszenz bzw. Energiedissipation im Metall resultieren.
Für die kinetische Untersuchung von Bindungsprozessen an der Filmoberfläche ist zu
beachten, dass die aufgenommene Fluoreszenzintensität nicht linear mit einer
korrespondierenden Dickenzunahme bei Anbindung des farbstoffmarkierten Analyten
korreliert.

Kapitel 2 – Theoretische Grundlagen
20
Literaturverzeichnis
1. Haeter, R.A., ed. Oberflächen- und Dünnschichttechnologie. 1987, Springer-Verlag: Berlin. 56 ff.
2. Renate Förch, Z.Z.W.K., Soft Plasma Treated Surfaces: Tailoring of Structure
and Properties for Biomaterial Applications. Plasma Processes and Polymers, 2005. 2(5): p. 351-372.
3. Yasuda, H., ed. Plasma polymeristation. 1985, Academic Press, Inc: Orlando.
4. Hirotsugu Yasuda, T.Y., The competitive ablation and polymerization (CAP)
principle and the plasma sensitivity of elements in plasma polymerization and
treatment. Journal of Polymer Science Part A: Polymer Chemistry, 2000. 38(6): p. 943-953.
5. Knoll, W., Interfaces and thin films as seen by bound electromagnetic waves. Ann. Rev. Phys. Chem., 1998. 49: p. 569-638.
6. Kretschmann E., R.H., Z. Naturforsch. Teil A, 1968. 23: p. 2135-2136.
7. Bunjes, N., et al., Thiopeptide-Supported Lipid Layers on Solid Substrates. Langmuir, 1997. 13(23): p. 6188-6194.
8. E., H., ed. Optik. 3. Auflage ed. 2001, Oldenbourg Wissenschaftsverlag GmbH: Wien.
9. H., R., ed. Surface Plasmons on Smooth and Rough Surfaces and on Gratings. Vol. 111. 1988, Springer-Verlag Berlin Heidelberg.
10. Boisdé G., H.A., ed. Chemical and biochemical sensing with optical fibers and
waveguides. 1996, Artech House, Inc.: Norwood.
11. H.-G., U., ed. Optische Wellenleiter. Vol. Teil 1. 1984, Hüthig: Heidelberg.
12. Liebermann, T. and W. Knoll, Surface-plasmon field-enhanced fluorescence
spectroscopy. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2000. 171(1-3): p. 115-130.

Kapitel 3 – Materialien und Methoden
21
3 Materialien und Methoden
3.1 Plasmapolymerisation
3.1.1 Aufbau der Plasmapolymerisationsanlage
Die in dieser Arbeit verwendeten Plasmapolymere wurden in einem hausgebauten
zylinderförmigen, ca. 30 cm langen Pyrex-Glaskolben mit einem Durchmesser von 10 cm,
hergestellt (siehe Abb. 3.1). Die Einspeisung der 13,56 MHz-HF-Spannung erfolgt induktiv
über eine um die Reaktionskammer gewickelte Spule mit Hilfe eines Verstärkers RFG-150
Coaxial Power System Ltd mit einer maximalen Leistung von 150 W. Der Puls wird durch
einen hausgebauten Pulsgenerator eingestellt. Eine Leybold BCS 16 Vakuumpumpe evakuiert
die Reaktionskammer, deren Basisdruck von ca. 3106 −⋅ mbar durch ein MKS-Baratron,
welches in der Nähe des Monomergaseinlasses angebracht ist, gemessen wird. Eine mit
flüssigem Stickstoff gekühlte Kühlfalle zwischen Reaktor und Pumpe verhindert das eventuell
verbleibende Monomerrückstände in die Vakuumpumpe gelangen können.
Abb. 3.1 : Schematischer Aufbau der verwendeten Plasmapolymerisationsanlage

Kapitel 3 – Materialien und Methoden
22
Fein gemörsertes Maleinsäureanhydrid-Pulver wurde in einen Kolben eingefüllt und dieses
direkt an das System angeschlossen. Die verwendeten Substrate wurden auf eine Glasplatte
ungefähr in der Mitte der Plasmazone platziert. Vor bzw. nach jedem Polymerisationszyklus
wurde der Reaktor bei kontinuierlichem Plasmabetrieb mit einer O2/Ar-Gasmischung (1:9)
zwischen 20-90 Minuten, in Abhängigkeit vom Verschmutzungs-grad, gereinigt. Die
Gasflussbestimmung erfolgte an einem ähnlich aufgebauten Reaktor mit Hilfe eines dort
angeschlossenen MKS 647 Gasflusskontrollsystems.
3.1.2 Probenherstellung
Maleinsäureanhydrid wurde je nach Kolbenfüllstand bei Drücken zwischen 0,5-0,8 mbar
direkt in das System eingespeist. Für den Plasmabetrieb wurden Spitzenleistungen von 50 W
bzw. 100 W bei einem Betriebszyklus von DC=1/41 gewählt. Dies entspricht einer
Equivalentleistung von 1,22 W bzw. 2,44 W.
Für die Herstellung dünner pp-MA-Filme von ca. 25-30 nm genügten Polymerisationszeiten
von 5-10 Minuten in Abhängigkeit vom Monomerausgangsdruck. Dickere pp-MA-Filme im
Bereich von 0,5-1 µm erforderten Polymerisationszeiten von 2-3 Stunden.
Die mit pp-HMDSO-beschichteten Proben wurden zunächst 8 s lang mit Sauerstoff bei einem
Druck von ca. 0,3 mbar aktiviert, anschließend ca. 18 s bei einer Eingangsleistung von 120-
130 W einem kontinuierlichen HMDSO/O2-Plasma im Verhältnis 1:10 ausgesetzt. Dies ergab
Filmdicken von 50-60 nm. Der HMDSO-Fluss wurde mit Hilfe eines Nadelventils der Firma
Edwards kontrolliert und auf 0,1 mbar eingestellt. Nach erneuter Aktivierung mit Sauerstoff
wurden mikrometerdicke pp-MA-Filme auf die pp-HMDSO-Unterlage abgeschieden. Die
frisch hergestellten Proben wurden in PS-Gelboxen gelagert und innerhalb von 2 Stunden für
weitere Reaktionen verwendet.
3.1.3 Flussratenbestimmung des Monomers
Die Monomerflussrate lässt sich mit Hilfe einer Leckgasflussmessung bestimmen, deren
Grundlage das ideale Gasgesetz darstellt. Da in der Plasmapolymerisation bei niedrigen
Drücken gearbeitet wird, ist die Behandlung aller Reaktorgase als ideale Gase zulässig. Bei
vorgegebenem Reaktorvolumen VReaktor und Raumtemperatur T sind demnach der

Kapitel 3 – Materialien und Methoden
23
Systemdruck p sowie dessen zeitliche Änderung dp/dt direkt proportional zur Anzahl der
Moleküle n bzw. zur zeitlichen Änderung der Molekülanzahl, dem Fluss dn/dt :
RTV
dt
dp
dt
dnaktor
1Re ⋅⋅
=
R: universelle Gaskonstante 3.1
Die praktische Bestimmung der Monomerflussrate funktioniert folgendermaßen: Nach Öffnen
des Monomerventils strömt das Monomer in den Reaktor ein, bis sich ein konstanter
Basisdruck einstellt. Anschließend trennt man das System von der Vakuumpumpe und
registriert den resultierenden Druckanstieg im Reaktor in Abhängigkeit von der Zeit, welcher
zunächst linear ansteigt, bis sich ein Sättigungsgleichgewicht einstellt. Setzt man die Steigung
des linearen Bereichs (dp/dt) in Gleichung 3.1 ein und multipliziert 3.1 zusätzlich mit einem
Kalibrierungsfaktor ΘRT zur Einhaltung der Standardbedingungen, so erhält man den
Monomerfluss dn/dt in [ ]slmbar /⋅ :
T
TV
dt
dp
dt
dnaktor
Θ
⋅⋅
= Re 3.2
ΘT : Temperatur bei Standardbedingungen (273K); T : Raumtemperatur (293K)
Dabei wird die Flussrate eines Gases üblicherweise in cm3STP/min bzw. sccm („standard cubic
centimeters per minute) bei Standardbedingungen angegeben. Es gilt:
s
lmbarcmatmsccm
⋅⋅=
⋅= −2
3
1069,1min1
111 bzw. sccm
s
lmbar2,591 =
⋅
Bei einem Basisdruck von 0,06 mbar erhält man für Maleinsäureanhydrid eine mittlere
Flussrate von 2107,3 −⋅ sccm. Für Hexamethyldisiloxan beträgt der mittlere Fluss 2,5 sccm
bei einem Basisdruck von 0,1mbar.

Kapitel 3 – Materialien und Methoden
24
3.2 Probenvorbereitung
3.2.1 Substrate
In Tabelle 3.1 sind die verwendeten Substrate für die jeweilige Charakterisierungsmethode
aufgelistet.
Substrat Größe Anwendung
Einfach polierter Si-Wafer 10 x 20 mm2
Kontaktwinkel- und Dicke-
messung, AFM
BK7-Glas (Firma Berliner Glas, B270)
mit 2 nm Cr und 80 nm Au
20 x 35 mm2 FT-IR-Messung
Quarzglas 20 x 35 mm2 UV-VIS-Messung
LaSFN9-Glas (Firma Hellma, n = 1,845)
mit 2 nm Cr und 50 nm Au
20 x 25 mm2
SPR/OWS/SPFS- Messung
Tabelle 3.1: Verwendete Substrate
3.2.2 Substratreinigung
Die Glas- und Siliziumsubstrate wurden für jede Anwendung sorgfältig nach folgendem
Protokoll gereinigt:
1. Wiederholtes Spülen (10x) mit Milli-Q-Wasser in der Färberbox,
2. 15 Minuten Ultraschallbehandlung in einer alkalischen Tensidlösung (Firma Hellma,
Hellmanex II),
3. Spülen mit Milli-Q-Wasser, bis keine Schaumbildung mehr zu beobachten ist,
4. Abschließendes Spülen mit absolutem Ethanol (Chromasolv-Qualität)
Die gereinigten Gläser wurden anschließend im Stickstoffstrom getrocknet und über Nacht im
Trockenschrank bei 50°C aufbewahrt. Die LaSFN9-Gläser wurden nach jeder Anwendung
durch mehrstündige Einlagerung in einer Gold- (wässrige KI/I2-Lösung) sowie einer
Chromentfernungslösung (wässrige Ammoniumcer(IV)-nitrat-Lösung) regeneriert.
Anschließend wurden sie nach obigem Protokoll gereinigt und neu bedampft.
3.2.3 Substratbedampfung
Die trockenen BK7- und LaSFN9-Gläser wurden in einer Aufdampfanlage (Firma Edwards,
Model FL 400) bei einem Druck von ca. 8 x 10-6 mbar mit einer Aufdampfrate von 0,2 nm/s
zunächst mit 2 nm Chrom und anschließend, je nach Anwendung, mit 50 bzw. 80 nm Gold
bedampft. Die so präparierten Substrate wurden, wenn möglich, sofort verwendet bzw. bis zur

Kapitel 3 – Materialien und Methoden
25
Anwendung bei maximaler Lagerungszeit von einer Woche unter Inertgasatmosphäre (Ar, N2)
aufbewahrt.
3.2.4 Assemblierungslösungen
Zur Verbesserung der Filmadhesion wurden die mit Gold bedampften Substrate für
Messungen in wässrigen Medien mit einer SAM („self-assembled-monolayer“)-Schicht
belegt. Die Substrate wurden hierfür 24 h in eine 5 mM Allylmercaptan/Ethanollösung oder
alternativ in eine 5 mM 1-Dodecanethiol/Ethanollösung eingetaucht. Anschließend wurden
sie mit Ethanol gespült, im Stickstoffstrom trocken geblasen und direkt in den Plasmareaktor
überführt.
3.3 Chemikalien und experimentelle Methoden
3.3.1 Chemikalien
Monomere
Maleinsäureanhydrid-Plättchen wurden von der Firma Fluka bezogen; die
Maleinsäureanhydrid-Presslinge wurden von Sigma-Aldrich verwendet. Hexamethyldisiloxan
wurde von der Firma Alfa Aesar bezogen. Die Monomere wurden ohne weitere Aufreinigung
verwendet.
Thiole
1-Dodecanthiol und 1-Octanthiol wurden von Sigma-Aldrich, Allylmercaptan wurde von Alfa
Aesar bezogen.
Crosslinker
TSTU (O-(N-Succinimidyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborat ) und Sulfo-NHS
(N-hydroxysulfosuccinimide) wurden von der Firma Fluka bezogen - EDC (N-3-
Dimethylaminopropyl)-N-ethylcarbodiimid Hydrochlorid) wurde Sigma Aldrich geliefert.
Detergenzlösung

Kapitel 3 – Materialien und Methoden
26
Puffer
- PBS (Tablets, Zymed Laboratories):10 mM Phosphat, 150 mM NaCl; pH = 7,2-7,3
- MES : 0,05 M MES (2-(N-Morpholino)ethansulfonsäure, Sigma-Aldrich), 0,5 M
NaCl; pH = 5-6
3.3.2 Antikörper
Anti-Human C-Reactive Protein
Entwickelt im Hase; IgG Fraktion aus Antiserum; lyophiliertes Pulver; Firma: Sigma-Aldrich
Rabbit anti-Mouse IgG
Cy5 Konjugat , polyklonaler sekundärer Antikörper; Fluorophor/Protein-Verhältnis: ca. 2,2;
Absorptionsmaximum = 650 nm, Emissionsmaximum = 680 nm; lyophiliertes Pulver; Firma:
Chemicon International
3.3.3 Experimentelle Methoden
Kopplungsprotokolle mit TSTU :
(a) Nach 12 h Hydrolyse in Milli-Q-Wasser wurde die pp-MA-Probe in einer Petrischale unter
zehnminütiger Einwirkzeit einer Lösung aus 60 mg (0,2 mmol) TSTU, 85 µl (0,5 mmol)
DIPEA in 5 ml einer 2:2:1-DMF/Dioxan/Milli-Q-Wasser-Mischung aktiviert. Die Probe
wurde kurz mit Milli-Q-Wasser abgespült und unter Stickstoffstrom getrocknet. Nach Einbau
in das SPR-Setup wurden 5 ml IgG-Lösung ( 0,001 mg/ml; 0,2 mg/ml) in Milli-Q-Wasser bei
einer Pumprate von 10 µl/min unter Rücklauf 1 h lang durch die Flusszelle gepumpt. Die
Probenoberfläche wurde nachfolgend 1 h mit Milli-Q-Wasser gespült.
(Protokoll angepasst nach Bannwarth [1])
(b) Die in das SPR-Setup eingebaute pp-MA-Probe wurde 12 h lang mit Milli-Q-Wasser bei
einer Pumprate von 90 µl/min hydrolysiert. Im Anschluss wurden 5 ml einer Lösung aus 5 mg
(16,6 µmol) TSTU, 1,39 µl (10 µmol) Triethylamin in einer 4:1 Dioxan/Milli-Q-Wasser-
Mischung unter Rücklauf bei einer Pumprate von 10 µl/min 40 min lang zugegeben. Nach 10
min Spülen mit Milli-Q-Wasser wurden 5 ml einer IgG-Lösung ( 1 mg/ml) in Milli-Q-Wasser
bei 10 µl/min unter Rücklauf 1 h lang durch die Flusszelle gepumpt. Die Probenoberfläche

Kapitel 3 – Materialien und Methoden
27
wurde nachfolgend 15-60 min mit Milli-Q-Wasser gespült. (Protokoll angepasst nach
Andersson [2])
Kopplungsprotokolle mit EDC/Sulfo-NHS
Die in das SPR-Setup eingebaute pp-MA-Probe wurde 4-12 h lang mit PBS bei einer
Pumprate von 30 µl/min hydrolysiert. Im Anschluss wurden 5 ml einer Lösung aus 96 mg
(0,2 mol) EDC, 27 mg (0,05 mol) Sulfo-NHS in MES (pH = 5-6) oder PBS (pH= 7,2-7,3)
unter Rücklauf bei einer Pumprate von 30 µl/min 30-50 min lang zugegeben. Nach 20 min
Spülzeit (10 min mit MES, 10 min mit PBS) wurden 5 ml einer IgG-Lösung ( 0,1 mg/ml) in
PBS bei 30 µl/min unter Rücklauf 1 h lang durch die Flusszelle gepumpt. Die
Probenoberfläche wurde nachfolgend 1-12 h mit PBS bzw. nachfolgend mit 0,1%iger
Tween20-Lösung (Polyoxyethylen-(20)-sorbitanmonolaurat) gespült.
(Protokoll angepasst nach [3, 4])
3.4 Charakterisierungsmethoden
3.4.1 SPR/OWS/SPFS
Aufbau der Messapparatur
Ein schematischer Aufbau des im Rahmen dieser Arbeit hauptsächlich genutzten
kombinierten SPR/OWS/SPFS-Setups ist in Abb. 3.2 dargestellt.
Ein Helium-Neon-Laser (λ=633 nm) wird durch einen Chopper, zwei Polarisatoren und eine
Blende auf die Probe fokussiert. Der Chopper dient dazu den Anregungsstrahl in zeitgleiche
Licht- und Dunkelphasen aufzuteilen, damit der Lock-In-Verstärker das Detektorsignal im
gleichen Takt auslesen kann. Mit Hilfe der beiden Polarisatoren wird p-polarisiertes Licht für
SPR- bzw. p- und s-polarisiertes Licht für OWS-Messungen eingestellt sowie die
Lichtintensität des Anregungsstrahls reguliert. Die Probe ist in Kretschmann-Konfiguration
(vgl. Kapitel 2, Abb. 2.3) in einer Halterung auf einem schrittmotorgesteuerten Drehtisch
(Goniometer) aufgebracht. Der an der Prisma/Gold-Grenzfläche reflektierte Laserstrahl wird
von einer Linse auf die Detektorfläche fokussiert. Vor Beginn jeder Messung ist der Laser
innerhalb des Drehwinkelbereichs des Goniometers (20-90°) auf den Detektoreinlass zu
justieren.
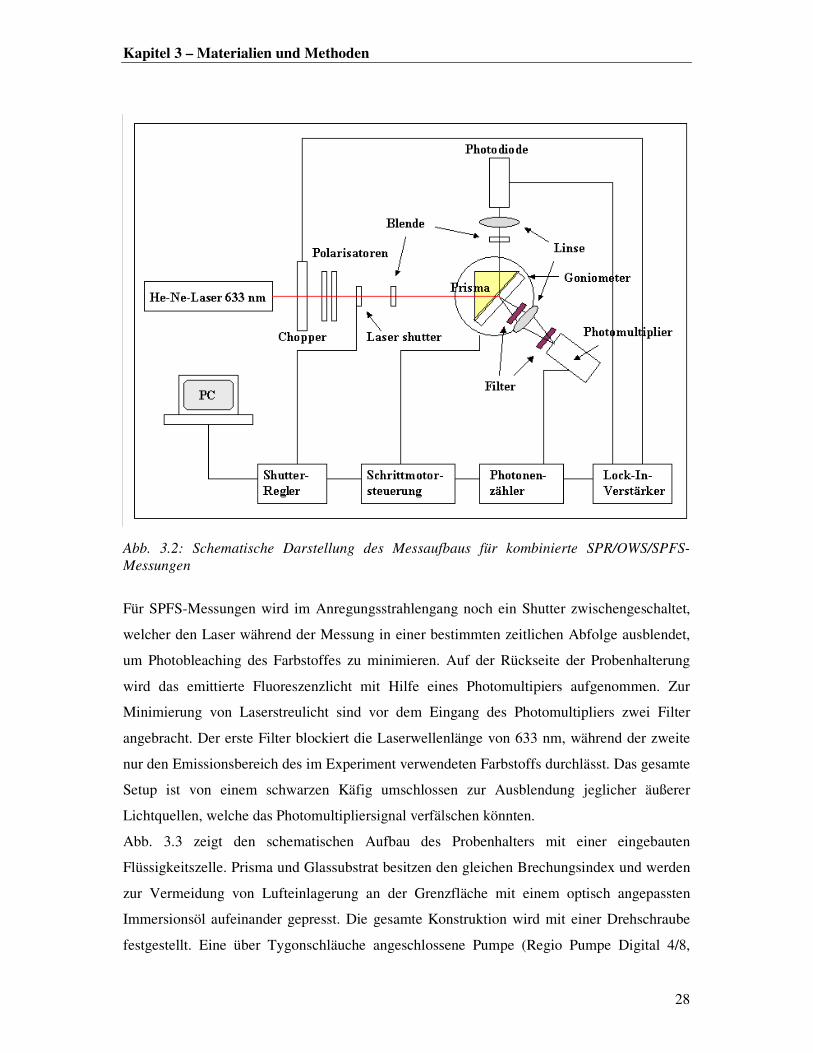
Kapitel 3 – Materialien und Methoden
28
Abb. 3.2: Schematische Darstellung des Messaufbaus für kombinierte SPR/OWS/SPFS-
Messungen
Für SPFS-Messungen wird im Anregungsstrahlengang noch ein Shutter zwischengeschaltet,
welcher den Laser während der Messung in einer bestimmten zeitlichen Abfolge ausblendet,
um Photobleaching des Farbstoffes zu minimieren. Auf der Rückseite der Probenhalterung
wird das emittierte Fluoreszenzlicht mit Hilfe eines Photomultipiers aufgenommen. Zur
Minimierung von Laserstreulicht sind vor dem Eingang des Photomultipliers zwei Filter
angebracht. Der erste Filter blockiert die Laserwellenlänge von 633 nm, während der zweite
nur den Emissionsbereich des im Experiment verwendeten Farbstoffs durchlässt. Das gesamte
Setup ist von einem schwarzen Käfig umschlossen zur Ausblendung jeglicher äußerer
Lichtquellen, welche das Photomultipliersignal verfälschen könnten.
Abb. 3.3 zeigt den schematischen Aufbau des Probenhalters mit einer eingebauten
Flüssigkeitszelle. Prisma und Glassubstrat besitzen den gleichen Brechungsindex und werden
zur Vermeidung von Lufteinlagerung an der Grenzfläche mit einem optisch angepassten
Immersionsöl aufeinander gepresst. Die gesamte Konstruktion wird mit einer Drehschraube
festgestellt. Eine über Tygonschläuche angeschlossene Pumpe (Regio Pumpe Digital 4/8,

Kapitel 3 – Materialien und Methoden
29
Ismatec) befördert die verwendete Flüssigkeit durch die Flusszelle. Für nähere Informationen
zum Design der verwendeten Flusszellen vergleiche Kapitel 4.2.2 unter Messoptimierung.
Abb. 3.3: Schematischer Aufbau der Probenhalterung mit eingebauter Flüssigkeitszelle
Durchführung einer Messung
Bei der Messung eines Reflektivitätsscans werden Proben- und Detektormotor des
Goniometers über den zu untersuchenden Winkelbereich gerastert und das reflektierte Licht
wird im Detektor registriert. Die Motorsteuerung erfolgt über eine hauseigene Software
(Wasplas). Bei den Messungen in Luft wird die Probe direkt mit dem Prisma in der
Probenhalterung festgeschraubt (vgl. Abb. 3.3), so dass die Filmoberfläche an der Luft liegt.
Bei den Messungen in Flüssigkeit wird zusätzlich, wie in Abb. 3.3 dargestellt, eine
Flüssigkeitszelle in die Probenhalterung eingebaut, welche mit wässriger Lösung gesättigt
wird.
Für kinetische Messungen gibt es zwei Möglichkeiten:
a) Bei fixierter Winkelposition wird die Änderung der reflektierten Intensität
zeitabhängig registriert. Hierfür wird ein Winkel gewählt, welcher sich im linearen
Kurvenbereich gerade unterhalb des Resonanzwinkels befindet. Für kleine
zeitabhängige Verschiebungen des Resonanzminimums ist die Reflektivitätsänderung
direkt proportional zu Änderung der optischen Dicke (∆nd).
b) Man verfolgt die zeitliche Änderung des Resonanzminimums.

Kapitel 3 – Materialien und Methoden
30
Methode (a) eignet sich besser zur Untersuchung von breiten Resonanzkurven, eines
Oberflächenplasmons, während Methode (b) leichter die zeitabhängige Verfolgung schmaler
Wellenleitermoden gestattet.
Auswertung der SPR-Reflektivitätskurven
Zur Auswertung der Messergebnisse wird eine hauseigene Software namens Winspall
verwendet. Die gemessenen Reflektivitätskurven lassen sich manuell oder iterativ mit einem
auf der Fresnel-Theorie basierenden Multischichtsystem anfitten. Eine typische Multischicht
beinhaltet das Prismenmaterial, die Metallbeschichtung, eine Thiolmonolage, das
Plasmapolymer und das umgebende Medium (Luft oder Flüssigkeit). Gibt man für jede
einzelne Lage Schichtdicke, Real- und Imaginärteil der Dielektrizitätskonstanten an, so
erstellt die Software unter Anwendung der Maxwellgleichungen einen simulierten
Intensitätsverlauf der Reflexion. Nach Anpassung an den experimentellen Intensitätsverlauf,
erhält man die optische Dicke (∆nd) des interessierenden Plasmapolymers. Es ist sinnvoll eine
Referenzmessung des Substrats vor Abscheidung des Polymers durchzuführen, so dass die
Substratparameter für die nachfolgende Simulation mit Polymerschicht schon bekannt sind.
3.4.2 Bestimmung der Filmdicke mit dem
Oberflächenprofilometer
Die Schichtdicke der pp-Filme wurden mit Hilfe eines Nadelprofilometers der Firma Tencor
(Modell P 10 Surface Profilometer α-Stepper) bestimmt. Hierbei wird der auf einem Si-Wafer
abgeschiedene Film mit einer Kanüle bis zur Si-Oberfläche angeritzt. Alternativ wird der Si-
Wafer vor der Filmabscheidung teilweise mit Kaptonklebeband abgedeckt, so dass man nach
Entfernen des Klebestreifens ein Höhenprofil zwischen behandeltem und abgedecktem,
unbehandeltem Substrat erhält. Anschließend tastet die Nadel des Step-Profilers, welche mit
einem kapazitiven Messsensor versehen ist, die Substratoberfläche ab und liefert die
entsprechende Höheninformation an einen angeschlossenen Computer. Der Fehler bei der
Schichtdickenbestimmung beträgt mindestens 2 nm und steigt mit zunehmender
Oberflächenrauhigkeit.

Kapitel 3 – Materialien und Methoden
31
3.4.3 Kontaktwinkelmessungen
Der Kontaktwinkel zwischen einer Flüssigkeit und einem Festkörper ist ein Maß für die
energetische Wechselwirkung zwischen dem Festkörper und der Flüssigkeit. Mit dieser
Methode lassen sich demnach Rückschlüsse auf die Benetzbarkeit der Substratoberfläche
ziehen, so dass Oberflächenmorphologie und –polarität beurteilt werden können. Der
Kontaktwinkel ist als Winkel zwischen der Festkörperoberfläche und der and den
Dreiphasenpunkt von Flüssigkeit, Luft und Festkörperoberfläche angelegten Tangente
definiert (Abb. 3.4).
�
Die in dieser Arbeit experimentelle Bestimmung des Wasserkontaktwinkels erfolgte nach der
Methode des liegenden Tropfens. Die Messungen wurden von einem Gerät der Firma Krüss,
dem Krüss Drop Shape Analysis System DSA 10-Mk2, aufgenommen. Mit Hilfe einer Nadel
wurde ein Wassertropfen (4 µl Volumen) mit einer Rate von ca. 20 µl pro Minute auf einen
plasmapolymer-beschichteten Si-Wafer aufgetragen. Nach Aufnahme der Tropfenform mit
einer CCD-Kamera bestimmte eine entsprechende Software (Drop Shape Analysis) den
Kontaktwinkel durch Mittelung der beiden Werte, die man durch Anlegen einer Tangente auf
beiden Seiten der Tropfenkontur erhält. Diese Prozedur wurde an mindestens vier Stellen auf
dem Substrat wiederholt und die Ergebnisse für den Kontaktwinkel gemittelt.
Alle Messungen wurden bei Raumtemperatur bei ca. 30-40% Luftfeuchtigkeit durchgeführt.
Die Proben, insbesondere die pp-MA-Filme, wurden bis zum Messzeitpunkt in möglichst
trockener Umgebung gelagert (Reaktor bzw. Gelbox unter N2-Atmosphäre), um eine
Verfälschung der Messergebnisse durch Aufnahme von Luftfeuchtigkeit zu minimieren.
Abb. 3.4: Schematische Darstellung der
Flüssigkeit/Festkörpergrenzflächezur-
Bestimmung des Kontaktwinkel θ nach
Young �

Kapitel 3 – Materialien und Methoden
32
3.4.4 Fourier-Transformation-Infrarot-Reflektions-Absorptions-
Spektroskopie (FT-IRRAS)
Eine qualitative Analyse der Oberflächenfunktionalität der hergestellten Plasmapolymere
erfolgte mittels FT-IR-Spektroskopie. Hierfür wurde ein Fourier-Transform-Spektrometer
(Magna IR850 Spectrometer Series II) der Firma Nicolet verwendet. Dessen Kernstück, ein
Michelson-Interferometer, erzeugt ein Interferenzmuster im Detektor, welches durch
Fouriertransformation in ein frequenzabhängiges Spektrum umgewandelt wird. Die Anregung
der zu untersuchenden Proben liegt im mittleren Infrarotbereich bei einem
Wellenzahlenbereich von 400-4000 cm-1. Durch ein spezielles Belüftungssystem werden
Temperatur und relative Feuchtigkeit in der Messkammer konstant gehalten.
Die Aufnahme der Spektren erfolgt nach der Reflektions-Absorptions-Methode (Abb. 3.5).
Vorraussetzung dafür ist ein reflektierendes Substrat, auf dem die zu untersuchende Probe
(Plasmapolymer) aufgebracht wird. Als reflektierendes Medium diente ein mit 80 nm Gold
bedampftes Glassubstrat.
3.4.5 UV-VIS-Spektroskopie
Die in dieser Arbeit aufgenommenen UV-VIS-Spektren wurden mit einem UV/VIS/NIR-
Spektrometer (Lambda 900, Boston USA) auf Quarzglas aufgenommen. Dabei wurde die
Transmission T = I/I0 der filmbeschichteten Gläser gemessen. Mit einem Monochromator
wurde der relevante Bereich von 200-860 nm abgefahren. Das Spektrometer wurde über eine
Software angesteuert und ausgelesen. Bei der Auftragung der Spektren wurde die Extinktion
verwendet, die sich aus der Transmission gemäß A = -log(T) = log (I0/I) berechnet.
Abb. 3.5: Schematische Darstellung
des Strahlengangs bei der IRRAS

Kapitel 3 – Materialien und Methoden
33
3.4.6 Rasterkraftmikroskopie
Die auf die Rauhigkeit untersuchten Proben wurden von Sascha Pihan mit einem AFM-Gerät
(Dimension 3100 CL) vermessen. Die 3x3 und 5x5 µm-Scans eines dicken und dünnen pp-
MA-Films auf Si-Wafern wurden im Tapping Mode aufgenommen.

Kapitel 3 – Materialien und Methoden
34
Literaturverzeichnis
1. Bannwarth, W. and R. Knorr, Formation of carboxamides with N,N,N',N'-tetramethyl
(succinimido) uronium tetrafluoroborate in aqueous / organic solvent systems. Tetrahedron Letters, 1991. 32(9): p. 1157-1160.
2. Andersson, M., S. Oscarson, and L. Öhberg, Synthesis of oligosaccharides with
oligoethylene glycolspacers and their conversion into glycoconjugates using
N,N,N′,N′-tetramethyl(succinimido) tetrafluoroborate as coupling reagent. Glycoconjugate Journal, 1993. 10(6): p. 461-465.
3. Hermanson, G.T., ed. Bioconjugate Techniques. 2nd ed. 1996, Elsevier Science, USA.
4. Su X., W.Y.-J., Robelek R., Knoll W., Surface Plasmon Resonance Spectroscopy and
Quartz Crystal Microbalance Study of Streptavidin Film Structure Effects on
Biotinylated DNA Assembly and Target DNA Hybridization. Langmuir, 2005. 21: p. 348-353.

Kapitel 4 – Ergebnisse und Diskussion
35
4 Ergebnisse und Diskussion
4.1 Allgemeine Eigenschaften plasmapolymerisierter-
Maleinsäureanhydridfilme in Luft und wässriger Lösung
Die Immobilisierungseffizienz biologischer Moleküle auf einer Analytoberfläche ist stark von
deren Oberflächeneigenschaften abhängig. Benetzbarkeit und Ladungsdichte der Oberfläche
werden durch die Natur und Anzahl der funktionellen Gruppen determiniert. Für die Synthese
der pp-MA-Filme bedeutet dies konkret, die Prozessbedingungen so zu steuern, dass eine
hohe Funktionalität erhalten bleibt. Schiller et al. stellten fest, dass die strukturellen
Eigenschaften von pp-MA-Filmen stark von den Prozessbedingungen im Plasma abhängig
sind [1]. Mit sinkender Eingangsspannung und Einführung eines niedrigen Betriebszyklus
wurde eine Erhöhung der Funktionalität, allerdings auf Kosten des Vernetzungsgrades,
beobachtet. Die Einstellung der Prozessbedingung ist daher keinesfalls trivial, da ein
ausreichender Vernetzungsgrad zur Stabilisierung der Gesamtstruktur beibehalten werden
muss. In Anlehnung an frühere Arbeiten wurde für die Synthese der pp-MA-Filme eine
Eingangsleistung von 50 W und ein Betriebszyklus von 1/41 ausgewählt [1-3].
Der nachfolgende Abschnitt dient dazu, einen Überblick über die strukturellen Eigenschaften
der hergestellten Plasmapolymere in Luft und in wässriger Lösung zu verschaffen. Hierfür
wurden die pp-MA-Filme mittels Dicken- und Kontaktwinkelmessungen, FT-IR-, SPR-/OW-
und UV-VIS-Spektroskopie sowie AFM-Messungen charakterisiert. Im Hinblick auf die
nachfolgend angestrebte IgG-Anbindung wurde eine detaillierte Analyse des Lösungs-
verhaltens nm- und µm-dicker pp-MA-Filme durchgeführt.
Kapitel 4 – Ergebnisse und Diskussion
36
4.1.1. Charakterisierung plasmapolymerisierter Maleinsäure-
anhydrid-Filme mit Schichtdicken-, AFM-, Kontakt-
winkelmessungen und FT-IR-Spektroskopie in Luft
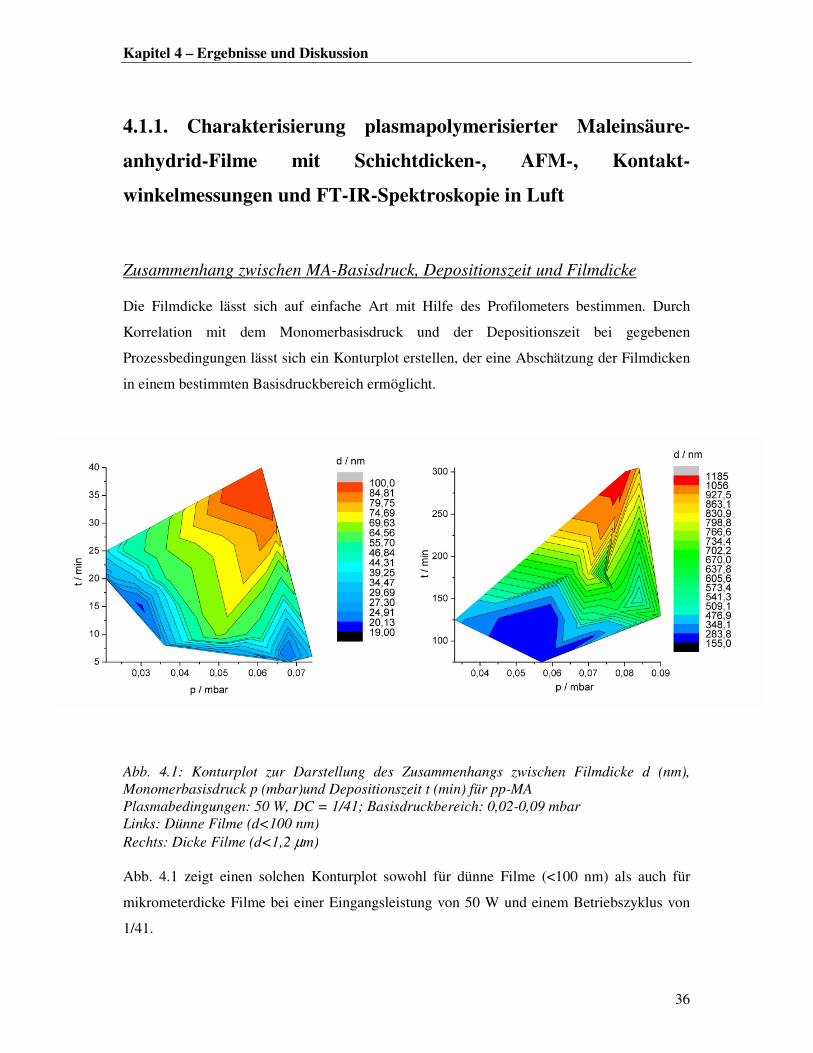
Zusammenhang zwischen MA-Basisdruck, Depositionszeit und Filmdicke
Die Filmdicke lässt sich auf einfache Art mit Hilfe des Profilometers bestimmen. Durch
Korrelation mit dem Monomerbasisdruck und der Depositionszeit bei gegebenen
Prozessbedingungen lässt sich ein Konturplot erstellen, der eine Abschätzung der Filmdicken
in einem bestimmten Basisdruckbereich ermöglicht.
Abb. 4.1: Konturplot zur Darstellung des Zusammenhangs zwischen Filmdicke d (nm), Monomerbasisdruck p (mbar)und Depositionszeit t (min) für pp-MA Plasmabedingungen: 50 W, DC = 1/41; Basisdruckbereich: 0,02-0,09 mbar Links: Dünne Filme (d<100 nm) Rechts: Dicke Filme (d<1,2 µm) Abb. 4.1 zeigt einen solchen Konturplot sowohl für dünne Filme (<100 nm) als auch für
mikrometerdicke Filme bei einer Eingangsleistung von 50 W und einem Betriebszyklus von
1/41.

Kapitel 4 – Ergebnisse und Diskussion
37
Betrachtet man zunächst nur die dünnen Filme (Abb. 4.1 links), so lässt sich ein innerhalb der
Fehlergrenzen stetiger Schichtdickenzuwachs mit steigendem Druck und steigender
Polymerisationszeit erkennen, was auf eine konstante Depositionsrate hindeutet. Dies steht im
Einklang mit früheren Ergebnissen, bei denen die Filmformation im Plasma in situ mittels
WaMS für Filmdicken von maximal 4 nm untersucht wurde. Jacobsen et al. [4] nahmen an,
dass die anfängliche Filmformation durch Ausbildung und Zusammenwachsen kleiner
Fragmentinseln auf der Substratoberfläche erfolgt. Daraus folgt, dass sich zur Ausbildung
eines homogenen Films ein gewisses dynamisches Gleichgewicht zwischen
Oberflächenbeschuss und Vernetzung bzw. Zusammenwachsen der Inseln einstellen muss.
Betrachtet man das Filmwachstum im Bereich von 0,5-1 µm lässt sich der Trend hinsichtlich
einer konstanten Depositionsrate nur noch begrenzt bestätigen, da hier größere Abweichungen
von der Stetigkeit zu verzeichnen sind (Abb. 4.1 rechts).
In den in Abb. 4.1 dargestellten Konturplots sind aus Übersichtsgründen keine Fehler
eingetragen. Für eine quantitative Auswertung der Dickenmessung mit dem Profilometer ist
ein Fehler von mindestens 2 nm zu berücksichtigen, welcher tendenziell mit zunehmender
Rauhigkeit steigt. Bei längerer Plasmaexposition wurde eine erhöhte Rauhigkeit der
Filmoberfläche registriert (siehe nächster Abschnitt). Daher muss bei der Korrelation der
Filmdicken im Mikrometerbereich eine mittlere Standardabweichung von 32 nm einkalkuliert
werden. Außerdem sollte man bedenken, dass bei der in dieser Arbeit verwendeten
Reaktorgeometrie nur eine waagerechte Platzierung des Substrats parallel zum
Monomerstrom möglich ist. Des Weiteren ist die zurückgelegte Wegstrecke der Teilchen im
Plasma aufgrund des seitlichen Monomereinlasses in die Reaktionskammer unterschiedlich.
Daher ist die Ausbildung eines lateralen Beschichtungsgradienten auf dem Substrat nicht zu
vermeiden. Die Filmdicke variiert somit in Abhängigkeit von der Messposition der
Profilometernadel, was insbesondere bei der Auswertung der dicken Filme ins Gewicht fällt.
Für statistische Betrachtungen sei angemerkt, dass der Korrelationsgraph in Abb. 4.1 links aus
27 Probendaten und in Abb. 4.2 rechts aus 19 Probendaten erstellt wurde.

Kapitel 4 – Ergebnisse und Diskussion
38
Charakterisierung der Oberflächenmorphologie
und Oberflächenrauhigkeit mittels AFM
Die Oberflächenrauhigkeit eines Plasmapolymers wird hauptsächlich von drei Faktoren
beeinflusst:
i) den Prozessbedingungen,
ii) dem verwendeten Monomer,
iii) der Plasmaexpositionsdauer.
I) und iii) sind darauf zurückzuführen, dass sich mit steigender Eingangsleistung und
Plasmadauer zum einen der Fragmentierungsgrad des Monomers und zum anderen die Anzahl
und Vielfalt aktiver Spezies im Plasma erhöht, so dass die Oberfläche insgesamt einem
stärkeren Materialbeschuss an unterschiedlichsten Formen ausgesetzt ist.
Bei der Interpretation der nachfolgenden Messergebnisse richtet sich das Hauptaugenmerk auf
die Expositionsdauer, da i) und ii) konstant gehalten wurden. Daher können Rückschlüsse auf
das Wachstumsverhalten des Films in Abhängigkeit von der Zeit gezogen werden.
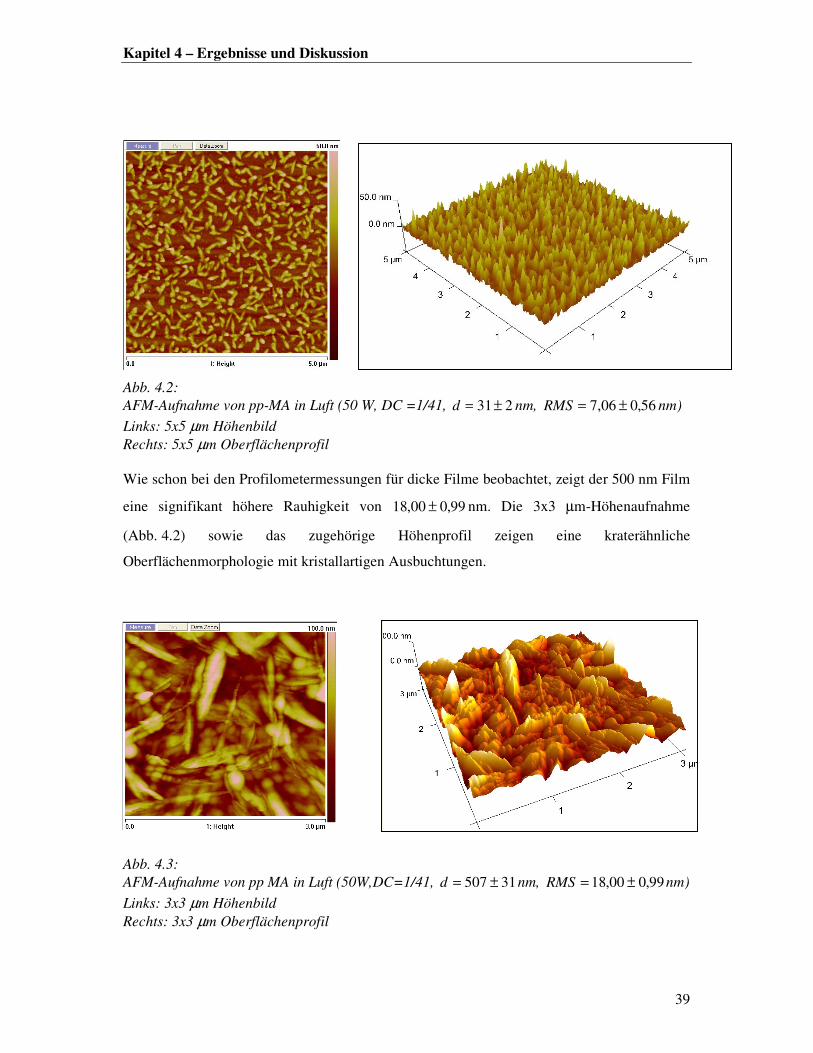
Erste Eindrücke zur Oberflächenbeschaffenheit lassen sich aus den Profilometeraufnahmen
der Proben gewinnen. Hierbei wurde eine Zunahme der Oberflächenrauhigkeit mit steigender
Filmdicke registriert. Eine genauere AFM-Analyse der Oberflächenrauhigkeit eines dünnen
und eines dicken Films bestätigt diesen Trend.
In Abb. 4.2 ist eine 5x5 µm-Höhenaufnahme eines 30 nm dicken Films mit dem zugehörigen
Höhenprofil dargestellt. Die Abbildung zeigt speerspitzenförmige Strukturen, die ca. 50 nm
aus der Oberfläche herausragen, bei einer RMS-Rauhigkeit von 56,004,7 ± nm. In früheren
Arbeiten wurden die pp-MA-Filme als blumenkohlartige Strukturen mit einer mittleren
Rauhigkeit von 0,641 nm in Luft beschrieben [5]. Diese Angaben sind mit den aktuellen
Ergebnissen allerdings nur begrenzt vergleichbar, da die Aufnahme auf einem kleineren
Ausschnitt (1x1 µm) erstellt wurde und die Literaturproben bei anderen Prozessbedingungen
(90 W, DC=5/100) und anderen Plasmaexpositionszeiten synthetisiert wurden.
Kapitel 4 – Ergebnisse und Diskussion
39
Abb. 4.2: AFM-Aufnahme von pp-MA in Luft (50 W, DC =1/41, 231±=d nm, 56,006,7 ±=RMS nm)
Links: 5x5 µm Höhenbild Rechts: 5x5 µm Oberflächenprofil Wie schon bei den Profilometermessungen für dicke Filme beobachtet, zeigt der 500 nm Film
eine signifikant höhere Rauhigkeit von 99,000,18 ± nm. Die 3x3 µm-Höhenaufnahme
(Abb. 4.2) sowie das zugehörige Höhenprofil zeigen eine kraterähnliche
Oberflächenmorphologie mit kristallartigen Ausbuchtungen.
Abb. 4.3: AFM-Aufnahme von pp MA in Luft (50W,DC=1/41, 31507 ±=d nm, 99,000,18 ±=RMS nm)
Links: 3x3 µm Höhenbild Rechts: 3x3 µm Oberflächenprofil

Kapitel 4 – Ergebnisse und Diskussion
40
Korreliert man die AFM-Aufnahmen mit den Konturplots für dünne und dicke Filme, so
lassen sich gewisse Parallelen erkennen. Die teilweise sehr unregelmäßig verlaufende
Schichtdickenzunahme der dicken Filme in Abb. 4.1. links zeugt von einer hohen Rauhigkeit,
die durch die AFM-Aufnahme in Abb. 4.3 bestätigt werden kann. Dagegen deuten die im
Vergleich dazu gleichmäßig verteilten Spitzen auf der Oberfläche des dünnen Films (vgl.
Abb. 4.2) auf ein kontrolliertes Filmwachstum hin, welches durch die stetig verlaufenden
Schichtdickenzunahme in Abb. 4.1 links bestätigt werden kann.
Damit lässt sich für den Filmwachstumsprozess im Plasma folgende Theorie aufstellen: Nach
anfänglicher Filmformation durch Inselbildung und Vernetzung [4] verläuft die Anlagerung
von Molekülfragmenten an die Oberfläche durch den fortlaufenden Beschuss an reaktiven
Spezies schneller als der Vernetzungsprozess. Dies hat zur Folge, dass die so aufgestapelten
Molekülfragmente stalagmitenähnlich aus der Oberfläche herauswachsen. Mit steigender
Plasmaexpositionszeit ist die Oberfläche einer zunehmenden Belastung durch im Plasma
formierte große Molekülbrocken ausgesetzt. Diese schlagen auf der Oberfläche auf und
werden erst nachträglich vernetzt, so dass die Oberfläche einer wie in Abb. 4.3 dargestellten
Kraterlandschaft ähnelt.
Chemische Zusammensetzung der pp-MA-Filme - Untersuchung der pp-MA-
Filmeigenschaften mit Kontaktwinkelmessungen und FT-IR-Spektroskopie
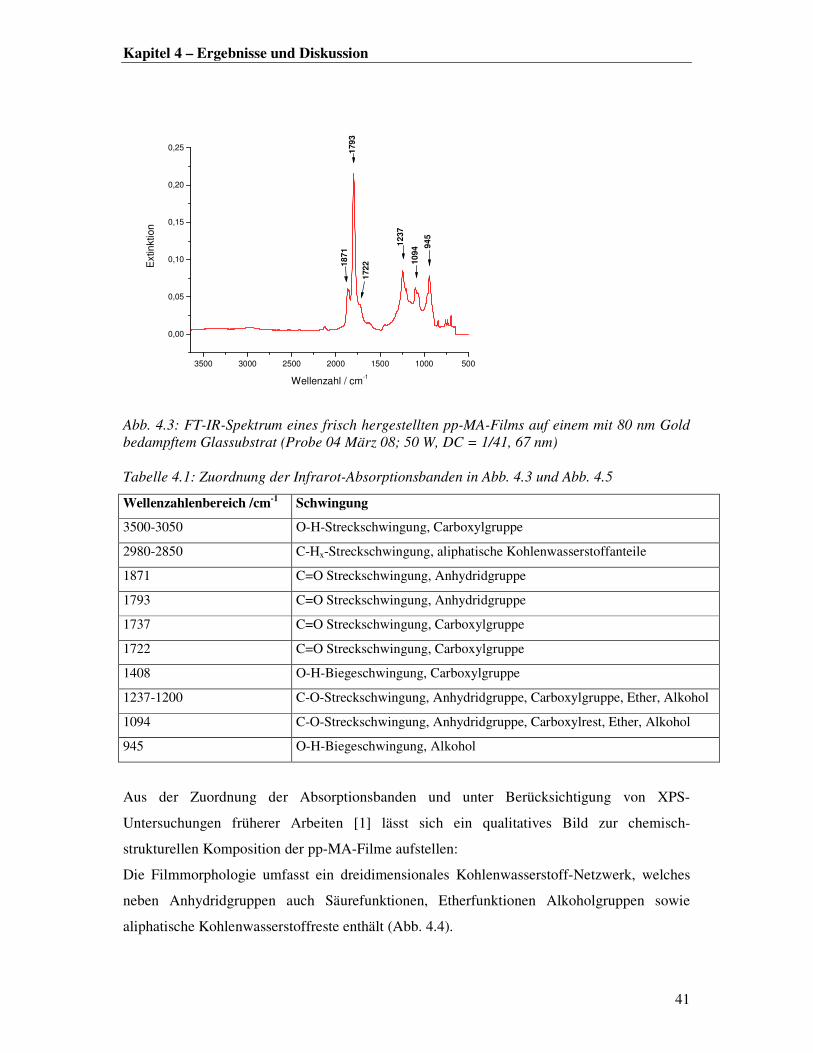
In Abb. 4.4 ist ein typisches FT-IR-Spektrum eines frisch hergestellten pp-MA-Films
dargestellt. In Übereinstimmung mit der Literatur sind die beiden für Anhydride
charakteristischen Absorptionsbanden bei 1871 und 1793 cm-1 im Spektrum mit einer hohen
Signalintensität vertreten [1]. Weiterhin sind Säureanteile (1722 cm-1) sowie in geringem
Maße CHx- Absorptionsbanden gesättiger Kohlenwasserstoffreste (<3000 cm-1) erkennbar.
Eine Zuordnung der auftretenden Schwingungsbanden ist in Tabelle 4.1 zusammengestellt.
Kapitel 4 – Ergebnisse und Diskussion
41
3500 3000 2500 2000 1500 1000 500
0,00
0,05
0,10
0,15
0,20
0,25E
xtinktion
Wellenzahl / cm-1
18
71
179
3
172
2
12
37
10
94 9
45
Abb. 4.3: FT-IR-Spektrum eines frisch hergestellten pp-MA-Films auf einem mit 80 nm Gold bedampftem Glassubstrat (Probe 04 März 08; 50 W, DC = 1/41, 67 nm) Tabelle 4.1: Zuordnung der Infrarot-Absorptionsbanden in Abb. 4.3 und Abb. 4.5
Wellenzahlenbereich /cm-1
Schwingung
3500-3050 O-H-Streckschwingung, Carboxylgruppe
2980-2850 C-Hx-Streckschwingung, aliphatische Kohlenwasserstoffanteile
1871 C=O Streckschwingung, Anhydridgruppe
1793 C=O Streckschwingung, Anhydridgruppe
1737 C=O Streckschwingung, Carboxylgruppe
1722 C=O Streckschwingung, Carboxylgruppe
1408 O-H-Biegeschwingung, Carboxylgruppe
1237-1200 C-O-Streckschwingung, Anhydridgruppe, Carboxylgruppe, Ether, Alkohol
1094 C-O-Streckschwingung, Anhydridgruppe, Carboxylrest, Ether, Alkohol
945 O-H-Biegeschwingung, Alkohol
Aus der Zuordnung der Absorptionsbanden und unter Berücksichtigung von XPS-
Untersuchungen früherer Arbeiten [1] lässt sich ein qualitatives Bild zur chemisch-
strukturellen Komposition der pp-MA-Filme aufstellen:
Die Filmmorphologie umfasst ein dreidimensionales Kohlenwasserstoff-Netzwerk, welches
neben Anhydridgruppen auch Säurefunktionen, Etherfunktionen Alkoholgruppen sowie
aliphatische Kohlenwasserstoffreste enthält (Abb. 4.4).

Kapitel 4 – Ergebnisse und Diskussion
42
Abb. 4.4: Schematische Darstellung eines auf einem mit Chrom und Gold bedampften Glassubstrat abgeschiedenen pp-MA-Films
Das im FT-IR-Spektrum auffallende Fehlen der für Doppelbindungen charakteristischen
Absorptionsbanden bei 3095-3075 cm-1 (=C-H-Streckschwingung) und 1660-1580 cm-1
(C=C-Streckschwingung) impliziert, dass die im Monomer enthaltene Doppelbindung im
Plasma vorwiegend fragmentiert wird. Dies zeugt von einer gewissen Selektivität der im
Plasma generierten reaktiven Spezies. Die Wahrscheinlichkeit für einen selektiven Angriff
steigt, wenn die Konzentration an aktiven Spezies relativ gering gehalten wird. Dies wird
durch den Pulsbetrieb des Plasmas bzw. die Einführung einer toff-Phase (vgl. Kapitel 2)
gewährleistet, so dass hauptsächlich Radikale an der Modifizierung der Substratoberfläche
beteiligt sind. Die in Kapitel 2 diskutierten mechanistischen Parallelen zwischen der gepulsten
Plasmapolymerisation und der klassischen radikalischen Polymerisation werden hier deutlich.
Grundsätzlich zeigen frisch synthetisierte Plasmapolymere Alterungserscheinungen, sobald
sie äußeren Einflüssen wie Luftfeuchtigkeit, Temperatur, UV-Licht, Druck, etc. ausgesetzt
werden. Die Plasmapolymerfilme sind als eine Art dynamisches Netzwerk zu verstehen, in
dem freie Kettenenden Konformationsänderungen und eingelagerte Radikale oder andere
aktive Spezies Reaktionen mit der äußeren Umgebung eingehen können [5].
Sobald die pp-MA-Filme aus dem Vakuum in die Atmosphärenluft gelangen, absorbieren sie
Luftfeuchtigkeit. Dieser Vorgang vollzieht sich innerhalb von wenigen Stunden, wie sich mit
Hilfe von Kontaktwinkelmessungen beweisen lässt. So beträgt der mittlere Kontaktwinkel
über neun verschiedene pp-MA-Proben, die bis wenige Minuten vor der Messung in trockener
Umgebung gelagert wurden, °± 1,66,52 . Zum Vergleich wurde der Kontaktwinkel einer
Probe, die bereits sechs Stunden der Luftfeuchtigkeit ausgesetzt worden war, mit °± 4,00,37
gemessen. Die Benetzbarkeit steigt demnach mit zunehmender Expositionsdauer in Luft.
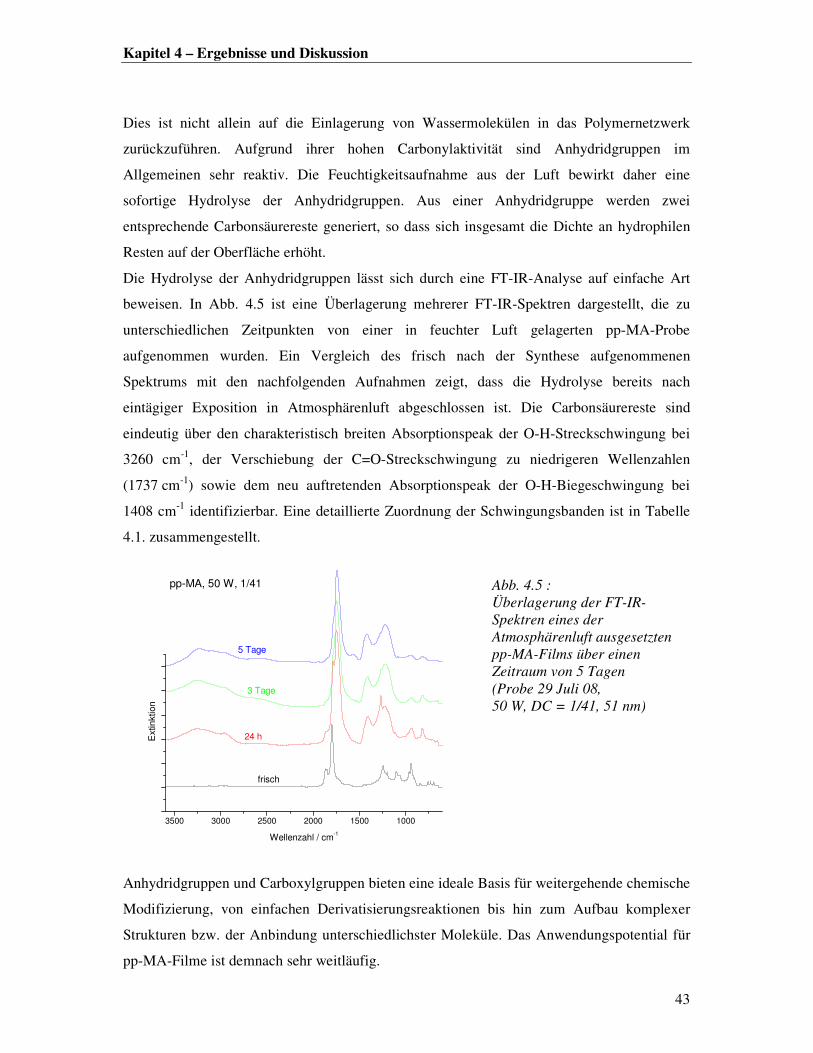
Kapitel 4 – Ergebnisse und Diskussion
43
Dies ist nicht allein auf die Einlagerung von Wassermolekülen in das Polymernetzwerk
zurückzuführen. Aufgrund ihrer hohen Carbonylaktivität sind Anhydridgruppen im
Allgemeinen sehr reaktiv. Die Feuchtigkeitsaufnahme aus der Luft bewirkt daher eine
sofortige Hydrolyse der Anhydridgruppen. Aus einer Anhydridgruppe werden zwei
entsprechende Carbonsäurereste generiert, so dass sich insgesamt die Dichte an hydrophilen
Resten auf der Oberfläche erhöht.
Die Hydrolyse der Anhydridgruppen lässt sich durch eine FT-IR-Analyse auf einfache Art
beweisen. In Abb. 4.5 ist eine Überlagerung mehrerer FT-IR-Spektren dargestellt, die zu
unterschiedlichen Zeitpunkten von einer in feuchter Luft gelagerten pp-MA-Probe
aufgenommen wurden. Ein Vergleich des frisch nach der Synthese aufgenommenen
Spektrums mit den nachfolgenden Aufnahmen zeigt, dass die Hydrolyse bereits nach
eintägiger Exposition in Atmosphärenluft abgeschlossen ist. Die Carbonsäurereste sind
eindeutig über den charakteristisch breiten Absorptionspeak der O-H-Streckschwingung bei
3260 cm-1, der Verschiebung der C=O-Streckschwingung zu niedrigeren Wellenzahlen
(1737 cm-1) sowie dem neu auftretenden Absorptionspeak der O-H-Biegeschwingung bei
1408 cm-1 identifizierbar. Eine detaillierte Zuordnung der Schwingungsbanden ist in Tabelle
4.1. zusammengestellt.
3500 3000 2500 2000 1500 1000
24 h
3 Tage
Extin
ktio
n
Wellenzahl / cm-1
5 Tage
frisch
pp-MA, 50 W, 1/41
Anhydridgruppen und Carboxylgruppen bieten eine ideale Basis für weitergehende chemische
Modifizierung, von einfachen Derivatisierungsreaktionen bis hin zum Aufbau komplexer
Strukturen bzw. der Anbindung unterschiedlichster Moleküle. Das Anwendungspotential für
pp-MA-Filme ist demnach sehr weitläufig.
Abb. 4.5 : Überlagerung der FT-IR-Spektren eines der Atmosphärenluft ausgesetzten pp-MA-Films über einen Zeitraum von 5 Tagen (Probe 29 Juli 08, 50 W, DC = 1/41, 51 nm)

Kapitel 4 – Ergebnisse und Diskussion
44
4.1.2. Charakterisierung plasmapolymerisierter Maleinsäure-
anhydrid-Filme mit UV-VIS-Spektroskopie, SPR- und OWS-
Messungen in Luft und wässriger Lösung
Wie bereits im vorangegangenen Abschnitt festgestellt wurde, hydrolysieren die
Anhydridgruppen der pp-MA-Filme beim Kontakt mit Luftfeuchtigkeit innerhalb von
mehreren Stunden. Bei der Immersion der pp-MA-Filme in wässrige Lösung erfolgt die
Hydrolyse der Anhydridgruppen bereits innerhalb von wenigen Minuten [6]. Gleichzeitig
werden kleine Fragmente an nichtkovalent gebundenem, im Polymer eingelagertem Material,
aus den Zwischenräumen herausgespült. Durch Einlagerung von Lösemittelmolekülen in das
Polymernetzwerk lässt sich ein über die Dicke messbarer Schwellungsprozess registrieren. Je
nach pH-Wert und Salzkonzentration der verwendeten Lösung können ladungskontrollierte
Reorganisationsprozesse zusätzliche Konformationsänderungen innerhalb der Ketten einleiten
[5]. Das Verhalten der pp-MA-Filme weist hierbei markante Ähnlichkeiten zu einem
Polyelektrolyten auf.
Je nach Art und Effizienz des verwendeten Adhesionsvermittlers sind Delaminationsprozesse
zwischen Substrat und Polymer möglich. Dies ist ein nicht zu unterschätzendes Problem, da
für die Anbindung von Biomolekülen ein stabiles Grundgerüst in wässriger Lösung
unerlässlich ist. In der Vergangenheit hat sich die Verwendung einer Thiolmonolage bewährt
[3]. Wie in Abb. 4.6 schematisch dargestellt, nutzt man auf der einen Seite die Affinität des
Schwefels zur Goldoberfläche des Substrats aus, während auf der anderen Seite eine
plasmachemische Vernetzung des Alkylrests mit dem Polymer angestrebt wird.
Abb. 4.6: Schematische Darstellung eines pp-MA-Films, der über ein Thiol mit der Goldoberfläche des Substrats verlinkt ist.

Kapitel 4 – Ergebnisse und Diskussion
45
SPRS und OWS haben sich in der Vergangenheit bei der Untersuchung der pp-MA-
Filmeigenschaften in wässriger Lösung sowie bei der Erforschung von Anbindungsprozessen
biologischer Moleküle vielfach bewährt [5-8]. Hierbei wurden die pp-MA-Eigenschaften in
wässriger Lösung bereits umfangreich diskutiert. Nichtsdestotrotz ist es sinnvoll, das
Lösungsverhalten der eigens synthetisierten Filme zu untersuchen. Zum einen ist es wichtig,
die Konformität der eigenen Ergebnisse mit der Literatur zu überprüfen. Zusätzlich dient die
Erfassung des eigenen Datensatzes als Basis für die Auswertung weiterführender Experimente
sowie zur Demonstration einer stabilen Unterlage bei der Anbindung von Biomolekülen (vgl.
Abschnitt 4.2). Daher wurde das Verhalten dünner und dicker pp-MA-Filme in Luft und
wässriger Lösung mittels SPR- und OWS-Messungen charakterisiert.
Verglichen mit SPRS-Messungen liefert eine OWS-Analyse der korrespondierenden µm-
dicken Proben genauere Erkenntnisse über die Filmmorphologie. Durch die unterschiedlichen
Anregungsmöglichkeiten mittels p- und s-polarisiertem Lichts lassen sich Rückschlüsse auf
die optische Homogenität und Isotropie der untersuchten Schichten ziehen. Zusätzlich können
bei einer Anregung von mindestens zwei Wellenleitermoden Schichtdicke und
Brechungsindex unabhängig voneinander bestimmt werden. Bei der Auswertung von
Oberflächenplasmonen-resonanzkurven dünner Filme kann der zuvor ermittelte
Brechungsindex zur Kalkulation der Schichtdicke eingesetzt werden. Die mit Hilfe des
Surface Profilers gemessene Dicke dient nur als Richtwert und ist nicht direkt mit den SPR-
Simulationswerten für die Schichtdicke vergleichbar, da für beide Messtechniken Substrate
unterschiedlicher Oberflächenrauhigkeit (Si, Au) verwendet werden. Beide Substrate wurden
zwar in einem Plasmazyklus behandelt, wurden allerdings an unterschiedlichen Stellen im
Reaktor positioniert. Zusätzlich ist noch der Beschichtungsgradient im Reaktor zu
berücksichtigen. Unter diesen Gesichtspunkten, können beide Substrate durchaus
abweichende Schichtdicken aufweisen.
Charakterisierung dünner und dicker pp-MA-Filme
mittels UV-VIS-Spektroskopie
Bei der Anregung eines Oberflächenplasmons bzw. einer Wellenleitermode können
Dämpfungseffekte aufgrund von Absorption und Streuung die Resonanz und Breite der
Reflektivitätskurve beeinträchtigen [9]. Für pp-MA-Filme sind Absorptionseffekte im
Wellenlängenbereich des für SPR und OWS-Messungen verwendeten Anregungslasers
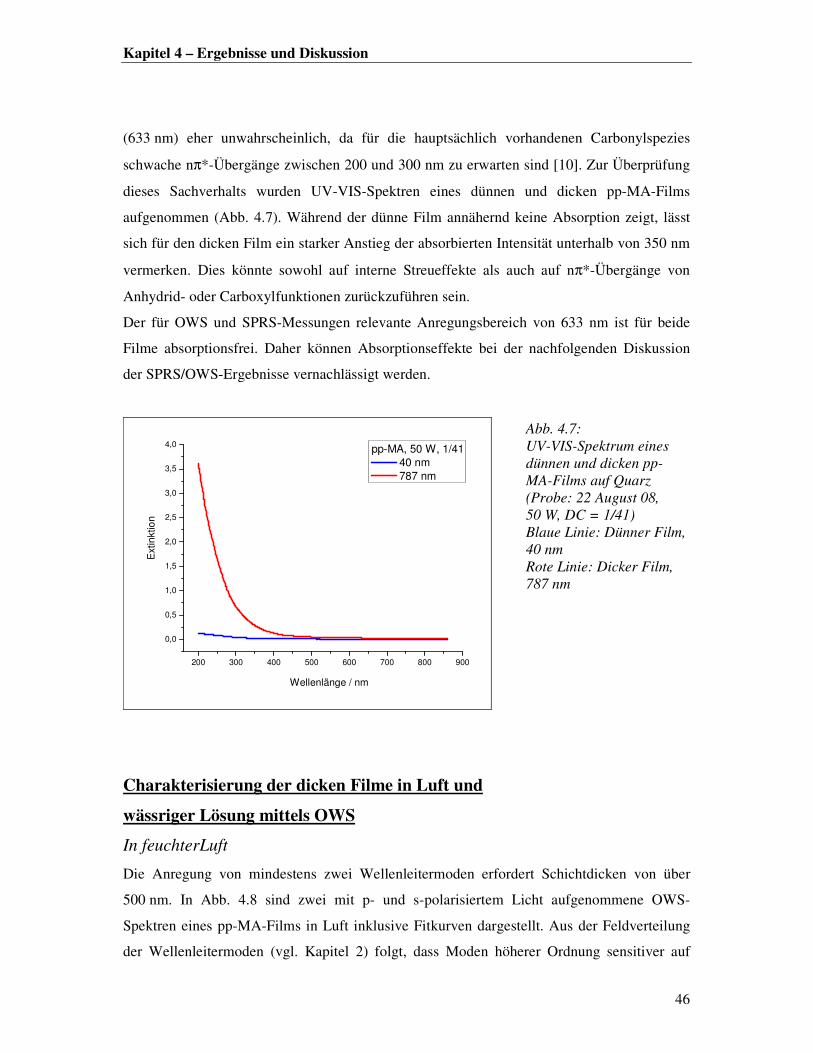
Kapitel 4 – Ergebnisse und Diskussion
46
(633 nm) eher unwahrscheinlich, da für die hauptsächlich vorhandenen Carbonylspezies
schwache nπ*-Übergänge zwischen 200 und 300 nm zu erwarten sind [10]. Zur Überprüfung
dieses Sachverhalts wurden UV-VIS-Spektren eines dünnen und dicken pp-MA-Films
aufgenommen (Abb. 4.7). Während der dünne Film annähernd keine Absorption zeigt, lässt
sich für den dicken Film ein starker Anstieg der absorbierten Intensität unterhalb von 350 nm
vermerken. Dies könnte sowohl auf interne Streueffekte als auch auf nπ*-Übergänge von
Anhydrid- oder Carboxylfunktionen zurückzuführen sein.
Der für OWS und SPRS-Messungen relevante Anregungsbereich von 633 nm ist für beide
Filme absorptionsfrei. Daher können Absorptionseffekte bei der nachfolgenden Diskussion
der SPRS/OWS-Ergebnisse vernachlässigt werden.
200 300 400 500 600 700 800 900
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
Extinktio
n
Wellenlänge / nm
pp-MA, 50 W, 1/41
40 nm
787 nm
Charakterisierung der dicken Filme in Luft und
wässriger Lösung mittels OWS
In feuchterLuft
Die Anregung von mindestens zwei Wellenleitermoden erfordert Schichtdicken von über
500 nm. In Abb. 4.8 sind zwei mit p- und s-polarisiertem Licht aufgenommene OWS-
Spektren eines pp-MA-Films in Luft inklusive Fitkurven dargestellt. Aus der Feldverteilung
der Wellenleitermoden (vgl. Kapitel 2) folgt, dass Moden höherer Ordnung sensitiver auf
Abb. 4.7: UV-VIS-Spektrum eines dünnen und dicken pp-MA-Films auf Quarz (Probe: 22 August 08, 50 W, DC = 1/41) Blaue Linie: Dünner Film, 40 nm Rote Linie: Dicker Film, 787 nm
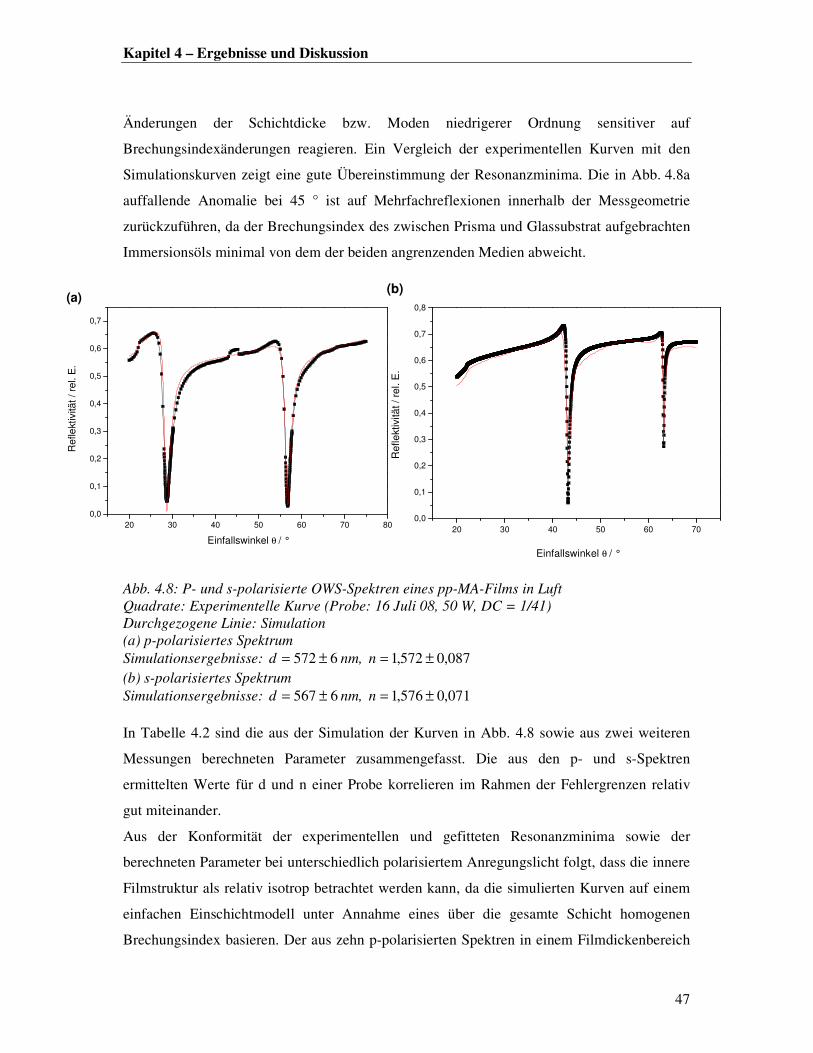
Kapitel 4 – Ergebnisse und Diskussion
47
Änderungen der Schichtdicke bzw. Moden niedrigerer Ordnung sensitiver auf
Brechungsindexänderungen reagieren. Ein Vergleich der experimentellen Kurven mit den
Simulationskurven zeigt eine gute Übereinstimmung der Resonanzminima. Die in Abb. 4.8a
auffallende Anomalie bei 45 ° ist auf Mehrfachreflexionen innerhalb der Messgeometrie
zurückzuführen, da der Brechungsindex des zwischen Prisma und Glassubstrat aufgebrachten
Immersionsöls minimal von dem der beiden angrenzenden Medien abweicht.
Abb. 4.8: P- und s-polarisierte OWS-Spektren eines pp-MA-Films in Luft Quadrate: Experimentelle Kurve (Probe: 16 Juli 08, 50 W, DC = 1/41) Durchgezogene Linie: Simulation (a) p-polarisiertes Spektrum Simulationsergebnisse: 6572 ±=d nm, 087,0572,1 ±=n
(b) s-polarisiertes Spektrum Simulationsergebnisse: 6567 ±=d nm, 071,0576,1 ±=n
In Tabelle 4.2 sind die aus der Simulation der Kurven in Abb. 4.8 sowie aus zwei weiteren
Messungen berechneten Parameter zusammengefasst. Die aus den p- und s-Spektren
ermittelten Werte für d und n einer Probe korrelieren im Rahmen der Fehlergrenzen relativ
gut miteinander.
Aus der Konformität der experimentellen und gefitteten Resonanzminima sowie der
berechneten Parameter bei unterschiedlich polarisiertem Anregungslicht folgt, dass die innere
Filmstruktur als relativ isotrop betrachtet werden kann, da die simulierten Kurven auf einem
einfachen Einschichtmodell unter Annahme eines über die gesamte Schicht homogenen
Brechungsindex basieren. Der aus zehn p-polarisierten Spektren in einem Filmdickenbereich
20 30 40 50 60 70 80
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Refle
ktivität / re
l. E
.
Einfallswinkel θ / °
(a)
20 30 40 50 60 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
R
efle
ktivitä
t / re
l. E
.
Einfallswinkel θ / °
(b)

Kapitel 4 – Ergebnisse und Diskussion
48
von 530-1200 nm berechnete mittlere Brechungsindex beträgt 012,0581,1 ± . Dies steht im
Einklang zu früheren Studien, in denen der Brechungsindexbereich zwischen 1,575 und 1,600
für bei 15 W und DC = 10/90 [4] und bei 37 W und DC =1/41 [8] hergestellte pp-MA-Filme
festgelegt wurde.
Tabelle 4.2: d- und n-Parameter aus der Simulation der Reflektivitätskurven in Abb. 4.8 und zwei weiteren Messungen Schichtdicke d / nm Brechungsindex n
Probe 16/07/08 p-Spektrum s-Spektrum
6572 ± 6567 ±
087,0572,1 ±
071,0576,1 ±
Probe 21/07/08 p-Spektrum s-Spektrum
7668 ± 7658 ±
084,0572,1 ±
071,0585,1 ±
Probe 31/07/08 p-Spektrum s-Spektrum
6587 ± 6577 ±
100,0573,1 ±
077,0582,1 ±
Eine detaillierte Analyse der Filmstruktur ist durch Anregung zusätzlicher Wellenleitermoden
möglich. In Abb. 4.9 links ist ein p-polarisiertes OWS-Spektrum eines µm-dicken pp-MA-
Films unter Anregung von vier Wellenleitermoden dargestellt. Ein Vergleich der
experimentellen Daten mit der Simulation zeigt, dass die Positionen der Resonanzminima
zwar gut korrelieren, Breite und Minimumsreflektivität jedoch stark voneinander abweichen.
Mit steigender Ordnung lässt sich eine signifikante Verbreiterung der Moden erkennen.
Derartige Beobachtungen lassen durch Dämpfungseffekte innerhalb des Evaneszentfeldes
erklären. Eine Dämpfung der resonanten Anregung kann grundsätzlich durch folgende
Faktoren hervorgerufen werden: i) Absorption, ii) Oberflächenstreuung, iii)
Volumenstreuung. Bei der Untersuchung der Filme mittels UV-VIS-Spektroskopie konnte
eine Absorption im Anregungsbereich ausgeschlossen werden. Es sind daher nur
Streuungseffekte zu berücksichtigen.
In der Maxwell-Betrachtung der Filmgeometrie werden Dämpfungseffekte im Imaginärteil
der Dielektrizitätskonstante ε’’ berücksichtigt. Durch Variation von ε’’ lässt sich, wie in
Abb. 4.9 links für die Mode niedrigster Ordnung dargestellt, jeweils nur eine Mode
zufriedenstellend simulieren. Beim Versuch alle vier Moden einzeln anzufitten, ergibt sich für
jede Mode ein unterschiedlicher Wert für ε’’ (Abb. 4.9, rechts). Das Dämpfungsprofil ist
demnach relativ inhomogen. Aus der Auftragung geht hervor, dass die Moden höherer
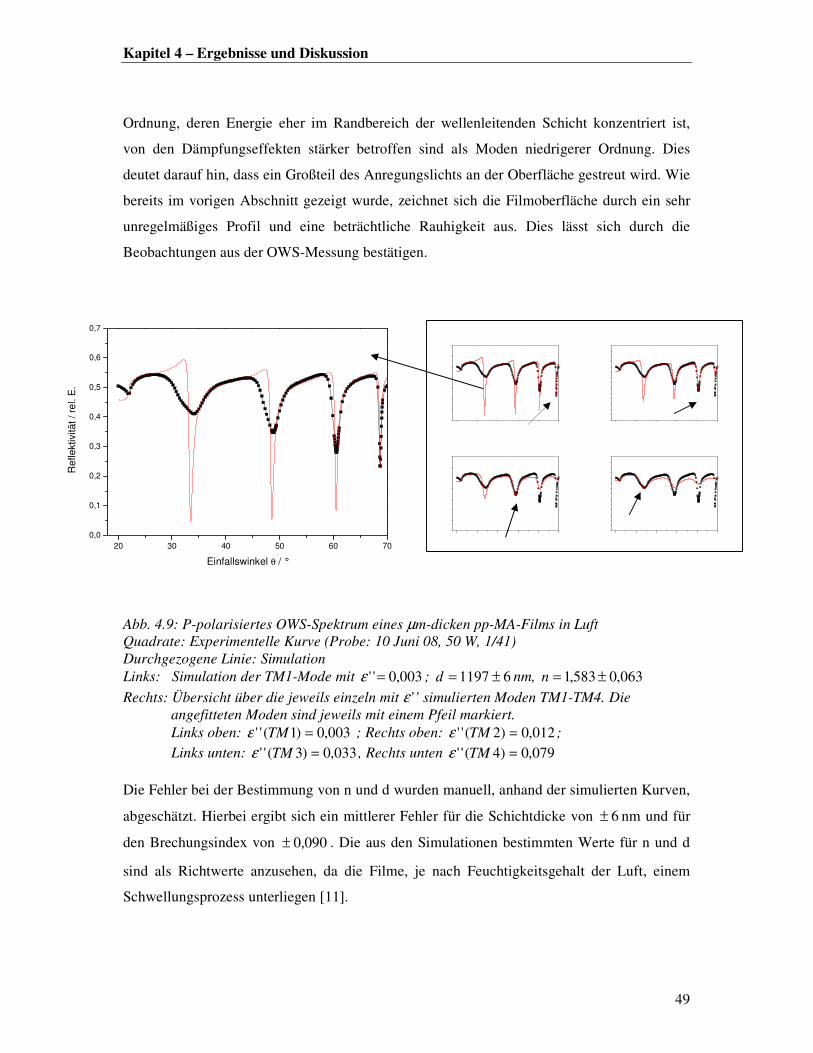
Kapitel 4 – Ergebnisse und Diskussion
49
Ordnung, deren Energie eher im Randbereich der wellenleitenden Schicht konzentriert ist,
von den Dämpfungseffekten stärker betroffen sind als Moden niedrigerer Ordnung. Dies
deutet darauf hin, dass ein Großteil des Anregungslichts an der Oberfläche gestreut wird. Wie
bereits im vorigen Abschnitt gezeigt wurde, zeichnet sich die Filmoberfläche durch ein sehr
unregelmäßiges Profil und eine beträchtliche Rauhigkeit aus. Dies lässt sich durch die
Beobachtungen aus der OWS-Messung bestätigen.
Abb. 4.9: P-polarisiertes OWS-Spektrum eines µm-dicken pp-MA-Films in Luft Quadrate: Experimentelle Kurve (Probe: 10 Juni 08, 50 W, 1/41) Durchgezogene Linie: Simulation Links: Simulation der TM1-Mode mit 003,0'' =ε ; 61197 ±=d nm, 063,0583,1 ±=n
Rechts: Übersicht über die jeweils einzeln mit ε’’ simulierten Moden TM1-TM4. Die angefitteten Moden sind jeweils mit einem Pfeil markiert. Links oben: 003,0)1('' =TMε ; Rechts oben: 012,0)2('' =TMε ;
Links unten: 033,0)3('' =TMε , Rechts unten 079,0)4('' =TMε
Die Fehler bei der Bestimmung von n und d wurden manuell, anhand der simulierten Kurven,
abgeschätzt. Hierbei ergibt sich ein mittlerer Fehler für die Schichtdicke von 6± nm und für
den Brechungsindex von 090,0± . Die aus den Simulationen bestimmten Werte für n und d
sind als Richtwerte anzusehen, da die Filme, je nach Feuchtigkeitsgehalt der Luft, einem
Schwellungsprozess unterliegen [11].
20 30 40 50 60 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Reflektivität / re
l. E
.
Einfallswinkel θ / °

Kapitel 4 – Ergebnisse und Diskussion
50
In wässriger Lösung
Zhang et al. registrierten in einer früheren SPR-Studie zum Schwellungsverhalten von niedrig
gepulsten pp-MA-Filmen in PBS-Lösung einen Anstieg der optischen Dicke in Abhängigkeit
von der Immersionszeit. Da eine detaillierte Interpretation auf der Grundlage der SPR-
Messergebnisse nicht möglich war, gingen die Autoren von einem relativ komplexen
Schwellungsmechanismus aus [6].
Abb. 4.10 zeigt zwei Reflektivitätsaufnahmen der in Abb. 4.9 dargestellten Probe in Milli-Q-
Wasser bei Hydrolysestart (Abb. 4.10a) und nach mehrstündiger Immersion in Lösung
(Abb. 4.10b). Die zugehörigen Simulationswerte, inklusive prozentualen Abweichungen, sind
in Tabelle 4.3 zusammengefasst. Unter Annahme eines einfachen Schwellungsprozesses ist
eine Zunahme der Schichtdicke unter gleichzeitiger Abnahme des Brechungsindexes zu
erwarten, da die Kettenenden im expandierenden Netzwerk weniger dicht angeordnet sind
bzw. die zuvor von Polymerfragmenten eingenommenen Zwischenräume mit optisch
dünneren Wassermolekülen besetzt werden. Dies lässt sich durch die experimentellen
Ergebnisse bestätigen: Im Vergleich zu den Scanaufnahmen in Luft ist zu Beginn der
Hydrolyse bereits ein Dickenzuwachs von 3,2 %, deutlich außerhalb der Fehlergrenzen sowie
ein minimaler Abfall im Brechungsindex zu verzeichnen. Die Wasseraufnahme in das
Polymernetzwerk erfolgt demnach innerhalb von wenigen Minuten. Eine kinetische
Untersuchung des Lösemittelverhaltens schlug fehl, da innerhalb der ersten Minuten bereits
eine beträchtliche Verschiebung der Resonanzwinkelposition auftrat, welcher die einstellbare
Motorschrittweite des Messprogramms überstieg. Die in früheren Studien [8] beobachtete
Ablösung von nichtkovalent gebundenem Material lässt sich hier nicht bestätigen, da über die
Reflektivitätskurven nur der Schwellungsprozess registriert werden konnte. Die
Resonanzkurven wurden jedoch in einem Zeitintervall von mehreren Stunden aufgenommen,
so dass eventuelle Ablösungsprozesse von nichtkovalent gebundenen Fragmenten nicht
erfasst werden konnten und daher nicht ausgeschlossen werden können.
Bei längerer Exposition in Wasser über mehrere Stunden ist ein weiteres Anschwellen mit
einem Gesamtdickenzuwachs von über 15 % sowie einem Brechungsindexabfall von 3 %
(Abb. 4.10 rechts) zu beobachten. Betrachtet man die Scanaufnahme in Abb. 4.10 rechts, so
ist trotz übereinstimmender Minimumspositionen von experimenteller und simulierter
Auftragung eine deutliche Diskrepanz in Bezug auf Breite und Tiefe der Resonanzkurven zu
erkennen. Die Wellenleitermoden erscheinen asymmetrisch verbreitert, was auf beträchtliche
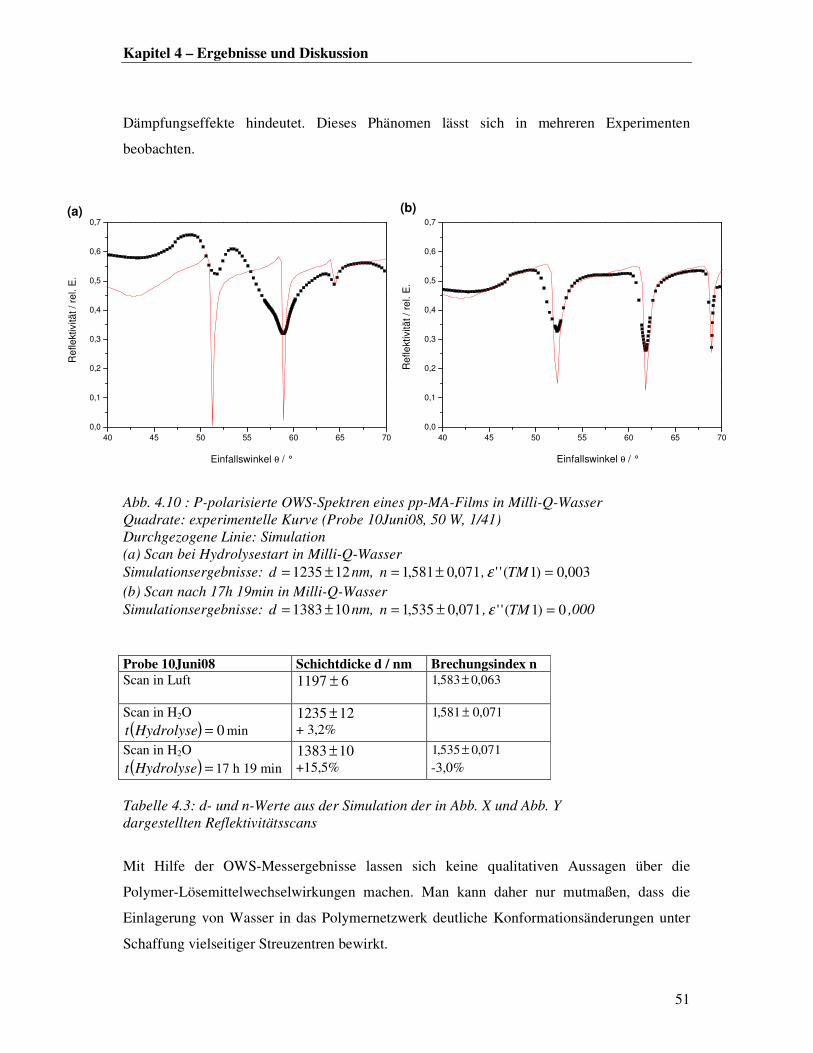
Kapitel 4 – Ergebnisse und Diskussion
51
Dämpfungseffekte hindeutet. Dieses Phänomen lässt sich in mehreren Experimenten
beobachten.
Abb. 4.10 : P-polarisierte OWS-Spektren eines pp-MA-Films in Milli-Q-Wasser Quadrate: experimentelle Kurve (Probe 10Juni08, 50 W, 1/41) Durchgezogene Linie: Simulation (a) Scan bei Hydrolysestart in Milli-Q-Wasser Simulationsergebnisse: 121235 ±=d nm, 071,0581,1 ±=n , 003,0)1('' =TMε
(b) Scan nach 17h 19min in Milli-Q-Wasser Simulationsergebnisse: 101383 ±=d nm, 071,0535,1 ±=n , 0)1('' =TMε ,000
Probe 10Juni08 Schichtdicke d / nm Brechungsindex n
Scan in Luft
61197 ± 063,0583,1 ±
Scan in H2O
( ) 0=Hydrolyset min 121235 ±
+ 3,2% 071,0581,1 ±
Scan in H2O
( ) =Hydrolyset 17 h 19 min 101383 ±
+15,5% 071,0535,1 ±
-3,0%
Tabelle 4.3: d- und n-Werte aus der Simulation der in Abb. X und Abb. Y dargestellten Reflektivitätsscans
Mit Hilfe der OWS-Messergebnisse lassen sich keine qualitativen Aussagen über die
Polymer-Lösemittelwechselwirkungen machen. Man kann daher nur mutmaßen, dass die
Einlagerung von Wasser in das Polymernetzwerk deutliche Konformationsänderungen unter
Schaffung vielseitiger Streuzentren bewirkt.
40 45 50 55 60 65 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Re
fle
ktivitä
t / re
l. E
.
Einfallswinkel θ / °
(a)
40 45 50 55 60 65 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
R
efle
ktivitä
t /
rel. E
.
Einfallswinkel θ / °
(b)
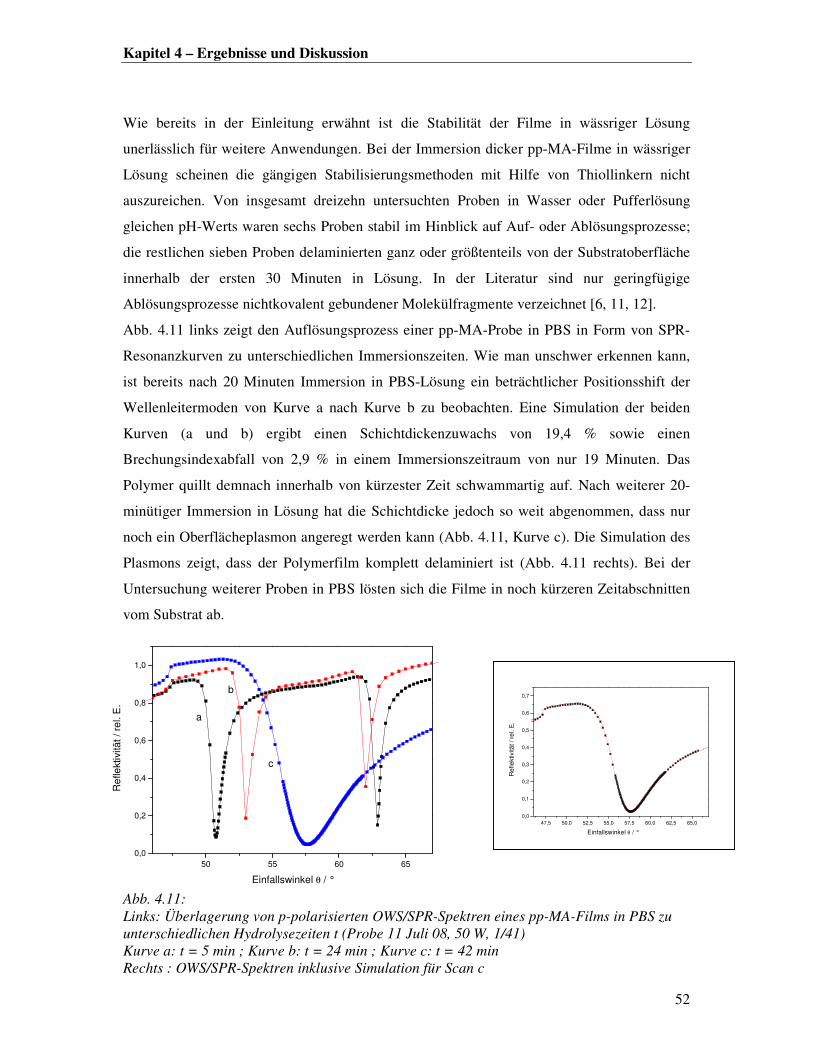
Kapitel 4 – Ergebnisse und Diskussion
52
Wie bereits in der Einleitung erwähnt ist die Stabilität der Filme in wässriger Lösung
unerlässlich für weitere Anwendungen. Bei der Immersion dicker pp-MA-Filme in wässriger
Lösung scheinen die gängigen Stabilisierungsmethoden mit Hilfe von Thiollinkern nicht
auszureichen. Von insgesamt dreizehn untersuchten Proben in Wasser oder Pufferlösung
gleichen pH-Werts waren sechs Proben stabil im Hinblick auf Auf- oder Ablösungsprozesse;
die restlichen sieben Proben delaminierten ganz oder größtenteils von der Substratoberfläche
innerhalb der ersten 30 Minuten in Lösung. In der Literatur sind nur geringfügige
Ablösungsprozesse nichtkovalent gebundener Molekülfragmente verzeichnet [6, 11, 12].
Abb. 4.11 links zeigt den Auflösungsprozess einer pp-MA-Probe in PBS in Form von SPR-
Resonanzkurven zu unterschiedlichen Immersionszeiten. Wie man unschwer erkennen kann,
ist bereits nach 20 Minuten Immersion in PBS-Lösung ein beträchtlicher Positionsshift der
Wellenleitermoden von Kurve a nach Kurve b zu beobachten. Eine Simulation der beiden
Kurven (a und b) ergibt einen Schichtdickenzuwachs von 19,4 % sowie einen
Brechungsindexabfall von 2,9 % in einem Immersionszeitraum von nur 19 Minuten. Das
Polymer quillt demnach innerhalb von kürzester Zeit schwammartig auf. Nach weiterer 20-
minütiger Immersion in Lösung hat die Schichtdicke jedoch so weit abgenommen, dass nur
noch ein Oberflächeplasmon angeregt werden kann (Abb. 4.11, Kurve c). Die Simulation des
Plasmons zeigt, dass der Polymerfilm komplett delaminiert ist (Abb. 4.11 rechts). Bei der
Untersuchung weiterer Proben in PBS lösten sich die Filme in noch kürzeren Zeitabschnitten
vom Substrat ab.
Abb. 4.11: Links: Überlagerung von p-polarisierten OWS/SPR-Spektren eines pp-MA-Films in PBS zu unterschiedlichen Hydrolysezeiten t (Probe 11 Juli 08, 50 W, 1/41) Kurve a: t = 5 min ; Kurve b: t = 24 min ; Kurve c: t = 42 min Rechts : OWS/SPR-Spektren inklusive Simulation für Scan c
50 55 60 65
0,0
0,2
0,4
0,6
0,8
1,0
Re
fle
ktivität / re
l. E
.
Einfallswinkel θ / °
a
b
c
47,5 50,0 52,5 55,0 57,5 60,0 62,5 65,0
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
R
efle
ktivitä
t /
rel. E
.
Einfallswinkel θ / °
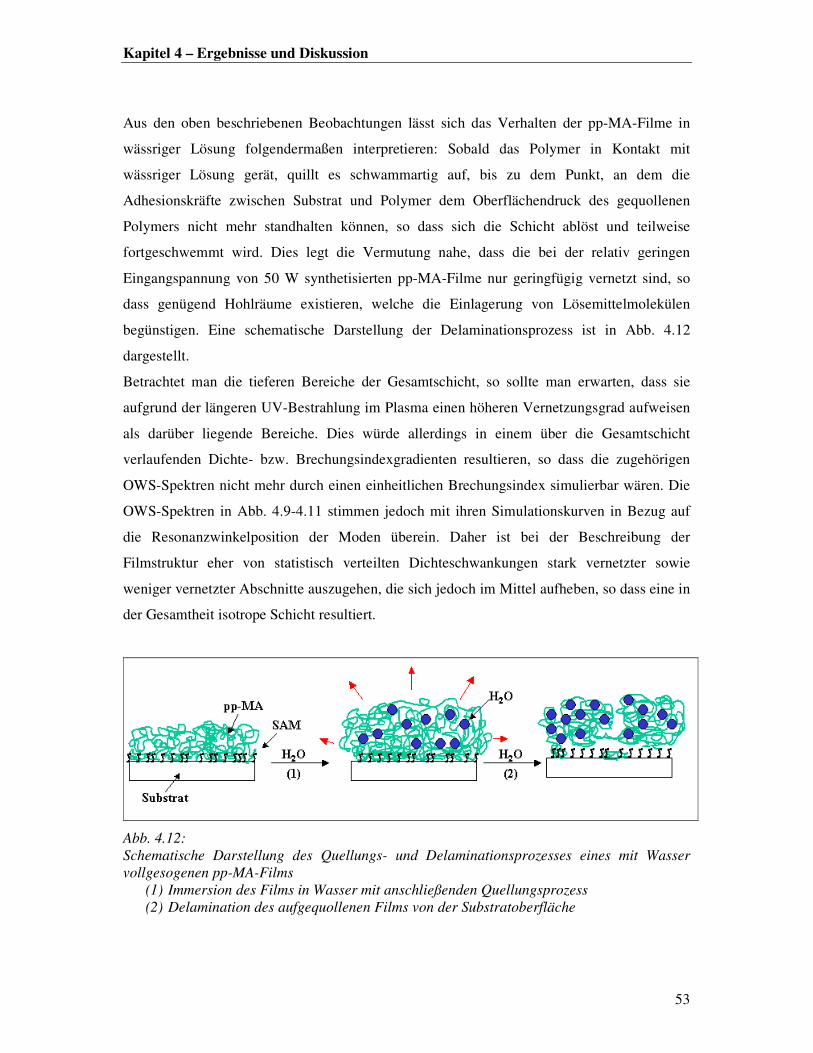
Kapitel 4 – Ergebnisse und Diskussion
53
Aus den oben beschriebenen Beobachtungen lässt sich das Verhalten der pp-MA-Filme in
wässriger Lösung folgendermaßen interpretieren: Sobald das Polymer in Kontakt mit
wässriger Lösung gerät, quillt es schwammartig auf, bis zu dem Punkt, an dem die
Adhesionskräfte zwischen Substrat und Polymer dem Oberflächendruck des gequollenen
Polymers nicht mehr standhalten können, so dass sich die Schicht ablöst und teilweise
fortgeschwemmt wird. Dies legt die Vermutung nahe, dass die bei der relativ geringen
Eingangspannung von 50 W synthetisierten pp-MA-Filme nur geringfügig vernetzt sind, so
dass genügend Hohlräume existieren, welche die Einlagerung von Lösemittelmolekülen
begünstigen. Eine schematische Darstellung der Delaminationsprozess ist in Abb. 4.12
dargestellt.
Betrachtet man die tieferen Bereiche der Gesamtschicht, so sollte man erwarten, dass sie
aufgrund der längeren UV-Bestrahlung im Plasma einen höheren Vernetzungsgrad aufweisen
als darüber liegende Bereiche. Dies würde allerdings in einem über die Gesamtschicht
verlaufenden Dichte- bzw. Brechungsindexgradienten resultieren, so dass die zugehörigen
OWS-Spektren nicht mehr durch einen einheitlichen Brechungsindex simulierbar wären. Die
OWS-Spektren in Abb. 4.9-4.11 stimmen jedoch mit ihren Simulationskurven in Bezug auf
die Resonanzwinkelposition der Moden überein. Daher ist bei der Beschreibung der
Filmstruktur eher von statistisch verteilten Dichteschwankungen stark vernetzter sowie
weniger vernetzter Abschnitte auszugehen, die sich jedoch im Mittel aufheben, so dass eine in
der Gesamtheit isotrope Schicht resultiert.
Abb. 4.12: Schematische Darstellung des Quellungs- und Delaminationsprozesses eines mit Wasser vollgesogenen pp-MA-Films
(1) Immersion des Films in Wasser mit anschließenden Quellungsprozess (2) Delamination des aufgequollenen Films von der Substratoberfläche

Kapitel 4 – Ergebnisse und Diskussion
54
Diese Theorie lässt sich bestätigen, wenn man das Substrat im Anschluss an die Messung
bzw. nach Entfernen der Flusszelle betrachtet. Auf der Substratoberfläche sind kleine
Filmbruchstücke mit dem bloßen Auge zu erkennen. Bläst man diese im Stickstoffstrom
herunter, so zeichnet sich in dem zuvor von der Flusszelle eingenommenen Bereich eine
makellose Goldoberfläche von der restlichen dunkleren Polymerfläche auf dem gesamten
Substrat ab.
Chifen et al. stellten eine verbesserte Stabilität von pp-MA-Filmen in wässriger Lösung fest,
wenn anstelle des Thiolinkers ein mit Sauerstoff aktivierter, bei hoher Eingangsleistung
abgeschiedener, pp-HMDSO/O2-Film als Adhesionsvermittler zwischen pp-MA und der
Goldoberfläche eingesetzt wird. Infolge der doppelten Sauerstoffaktivierung der Gold- und
der pp-HMDSO-Oberfläche werden reaktive Radikalstellen generiert, welche im folgenden
Plasmapolymerisationsprozess kovalent mit dem pp-MA-Film vernetzt werden können [12].
Abb. 4.13a zeigt die Reflektivitätskurve eines mit pp-HMDSO unterlegten pp-MA-Films in
Luft. Die Bestimmung der optischen Dicke des pp-HMDSO/O2-Films erfolgte mittels SPR-
Scanaufnahmen eines aus dem gleichen Plasmazyklus wie die eigentliche Probe beschichteten
Zweitsubstrats. Für die Simulation der Resonanzkurve wurde der Brechungsindex von
Quarzglas eingesetzt, da den pp-HMDSO/O2-Filme nachweislich eine SiOx-artige Struktur
zugeschrieben wird [12]. Die berechneten Werte dienten als Basis für den Fitprozess der
eigentlichen pp-MA-Resonanzkurven. Wie man Abb. 4.13a entnehmen kann, weist das
Resonanzminimum der mittleren Mode eine geringfügige Abweichung von der simulierten
Minimumsposition auf, welche jedoch im Rahmen der Fehlergrenzen tolerierbar ist.
In Abb. 4.13b ist eine Überlagerung der Reflektivitätsaufnahmen mit unterschiedlich
polarisiertem Licht in PBS-Lösung bei Immersionsstart dargestellt. Die aus den
Luftaufnahmen abgeleiteten zugehörigen Simulationskurven für wässrige Lösung sind
ebenfalls eingetragen, um die Abweichung zwischen erwarteten und experimentellen Befund
zu verdeutlichen. Anstelle der aufgrund der Schichtdicke in Luft zu erwarteten
Wellenleitermoden, wird nur noch ein Oberflächenplasmon angeregt. Die Ablösung des
Polymerfilms erfolgt demnach sofort nach Immersion in Lösung. Zum Beweis, dass das hier
dargestellte Reflektivitätsminimum wirklich auf der Anregung eines Oberflächenplasmons
basiert bzw. nicht einer durch Streueffekte verbreiterten Wellenleitermode entspricht, wurde
ein Spektrum mit s-polarisiertem Licht aufgenommen. Wie erwartet zeigt das s-Spektrum
keine Veränderung in der reflektierten Intensität, da Oberflächenplasmonen nur mit p-
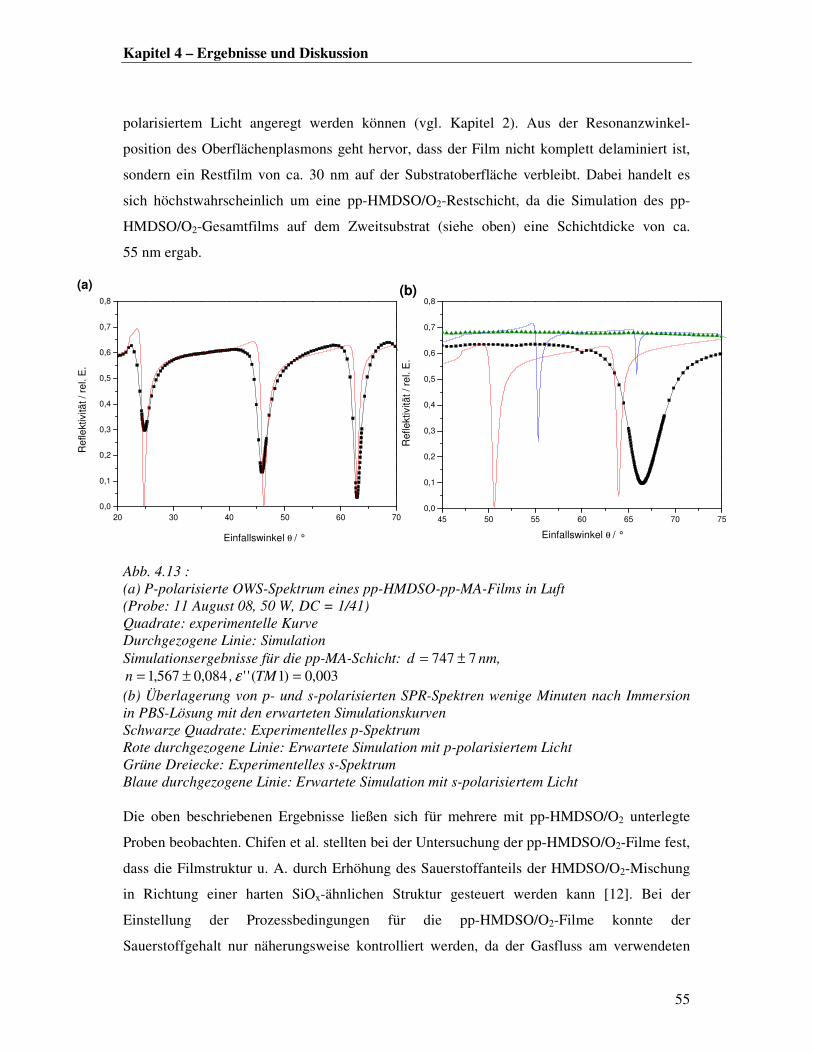
Kapitel 4 – Ergebnisse und Diskussion
55
polarisiertem Licht angeregt werden können (vgl. Kapitel 2). Aus der Resonanzwinkel-
position des Oberflächenplasmons geht hervor, dass der Film nicht komplett delaminiert ist,
sondern ein Restfilm von ca. 30 nm auf der Substratoberfläche verbleibt. Dabei handelt es
sich höchstwahrscheinlich um eine pp-HMDSO/O2-Restschicht, da die Simulation des pp-
HMDSO/O2-Gesamtfilms auf dem Zweitsubstrat (siehe oben) eine Schichtdicke von ca.
55 nm ergab.
Abb. 4.13 : (a) P-polarisierte OWS-Spektrum eines pp-HMDSO-pp-MA-Films in Luft (Probe: 11 August 08, 50 W, DC = 1/41) Quadrate: experimentelle Kurve Durchgezogene Linie: Simulation Simulationsergebnisse für die pp-MA-Schicht: 7747 ±=d nm,
084,0567,1 ±=n , 003,0)1('' =TMε
(b) Überlagerung von p- und s-polarisierten SPR-Spektren wenige Minuten nach Immersion in PBS-Lösung mit den erwarteten Simulationskurven Schwarze Quadrate: Experimentelles p-Spektrum Rote durchgezogene Linie: Erwartete Simulation mit p-polarisiertem Licht Grüne Dreiecke: Experimentelles s-Spektrum Blaue durchgezogene Linie: Erwartete Simulation mit s-polarisiertem Licht Die oben beschriebenen Ergebnisse ließen sich für mehrere mit pp-HMDSO/O2 unterlegte
Proben beobachten. Chifen et al. stellten bei der Untersuchung der pp-HMDSO/O2-Filme fest,
dass die Filmstruktur u. A. durch Erhöhung des Sauerstoffanteils der HMDSO/O2-Mischung
in Richtung einer harten SiOx-ähnlichen Struktur gesteuert werden kann [12]. Bei der
Einstellung der Prozessbedingungen für die pp-HMDSO/O2-Filme konnte der
Sauerstoffgehalt nur näherungsweise kontrolliert werden, da der Gasfluss am verwendeten
20 30 40 50 60 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Re
fle
ktivitä
t /
rel. E
.
Einfallswinkel θ / °
(a)
45 50 55 60 65 70 75
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Reflektivität / re
l. E
.
Einfallswinkel θ / °
(b)

Kapitel 4 – Ergebnisse und Diskussion
56
Reaktor nur manuell eingestellt werden konnte. FT-IR-Spektren der so synthetisierten pp-
HMDSO/O2-Filme ließen einen beträchtlichen Kohlenwasserstoffanteil erkennen, der als
Indiz für einen zu geringen Sauerstoffanteil gewertet werden kann [12]. Die in dieser Arbeit
synthetisierten pp-HMDSO/O2-Filme waren demnach nicht hart genug, um eine nachhaltige
Stabilisierung der dicken pp-MA-Filme zu gewährleisten. Versuche zur Verbesserung der
Adhesionseigenschaften für dicke pp-MA-Filme über pp-HMDSO/O2-Filme sind daher noch
ausbaufähig. Alternativ wäre auch die Synthese von pp-MA-Gradientenfilmen denkbar.
Hierbei wird ein Plasma bei einer hohen Eingangsleistung initiiert, welche dann im Laufe der
Plasmaexposition kontinuierlich verringert wird, so dass die resultierenden Filme in den
tieferen Schichtbereichen härtere Eigenschaften aufweisen als in der oberen Randschichten
[5].
Charakterisierung der dünnen Filme in Luft
und wässriger Lösung mittels SPR
Da sich bei SPR-Messungen immer nur Änderungen der kombinierten Eigenschaften von
Filmdicke und Brechungsindex erfassen lassen, ist eine verlässliche Interpretation der
Ergebnisse nur bei Kenntnis von mindestens einem der Parameter möglich. Die Auswertung
der pp-MA-OWS-Analyse liefert einen Brechungsindexbereich von 012,0581,1 ± , welcher im
Folgenden verwendet wird, um die Schichtdicke der dünnen Filme zu bestimmen. Unter
Einbeziehung der Standardabweichung für den Brechungsindex lässt sich die Schichtdicke
mit einer Genauigkeit von 0,1 nm berechnen.
Abb. 4.14 zeigt eine Überlagerung der Resonanzkurven eines unbehandelten Substrats (a)
sowie eines mit pp-MA-beschichteten Substrats (b). Bei der Simulation der Kurven lässt sich
ein gewisses Trendverhalten beobachten, welches sich über mehrere Proben erstreckt: Die
berechneten Schichtdicken sind im Schnitt etwa 6-7 nm geringer als die mit dem Surface
Profiler gemessenen Werte. Wie bereits erwähnt, lässt sich dieses Phänomen zum Teil auf die
Verwendung unterschiedlicher Substrate für beide Messprozesse zurückzuführen. Auffällig
ist, dass die Schichtdickenabweichung zwischen beiden Messtechniken in etwa im Bereich
der im vorigen Abschnitt gemessenen Oberflächenrauhigkeit liegt. Die Schichtdicken aus den
Profilometermessungen werden durch den direkten Kontakt von Nadel und Oberfläche stärker
von der Rauhigkeit beeinflusst. Daher liegt die Abweichung der aus beiden Messtechniken
ermittelten Schichtdicken durchaus im Bereich der Bestimmungsgrenzen.
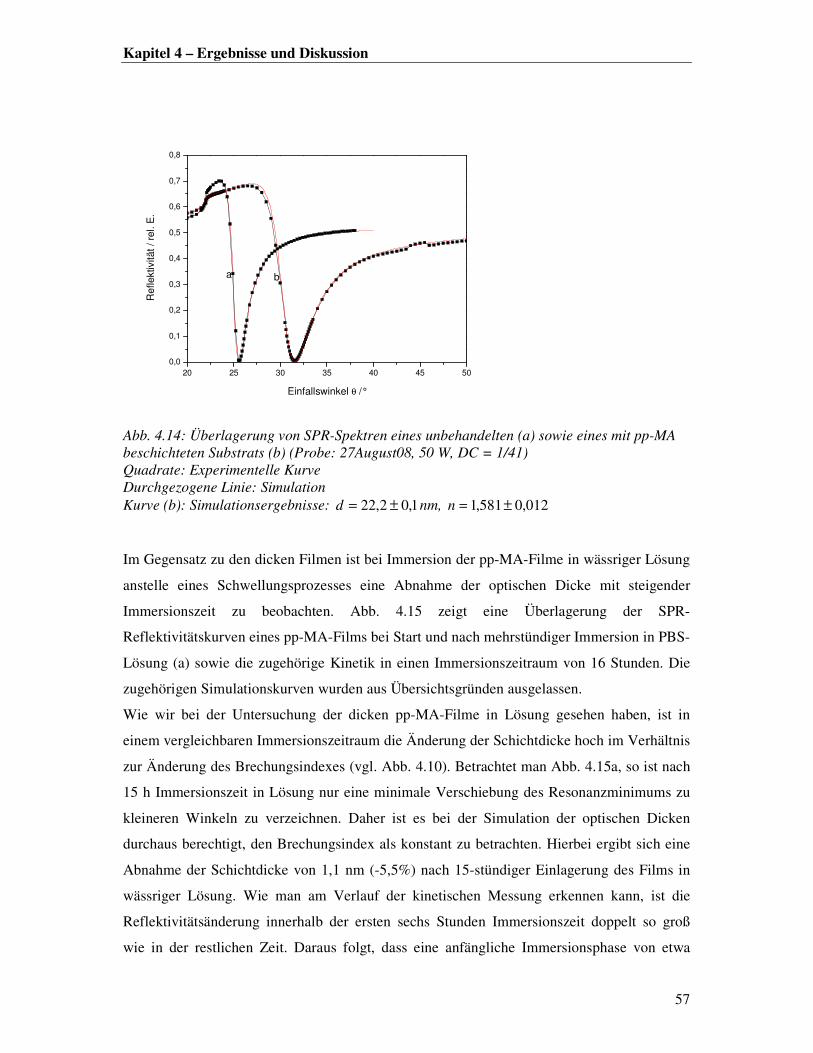
Kapitel 4 – Ergebnisse und Diskussion
57
20 25 30 35 40 45 50
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
R
eflektivität / re
l. E
.
Einfallswinkel θ /°
a b
Abb. 4.14: Überlagerung von SPR-Spektren eines unbehandelten (a) sowie eines mit pp-MA beschichteten Substrats (b) (Probe: 27August08, 50 W, DC = 1/41) Quadrate: Experimentelle Kurve Durchgezogene Linie: Simulation Kurve (b): Simulationsergebnisse: 1,02,22 ±=d nm, 012,0581,1 ±=n
Im Gegensatz zu den dicken Filmen ist bei Immersion der pp-MA-Filme in wässriger Lösung
anstelle eines Schwellungsprozesses eine Abnahme der optischen Dicke mit steigender
Immersionszeit zu beobachten. Abb. 4.15 zeigt eine Überlagerung der SPR-
Reflektivitätskurven eines pp-MA-Films bei Start und nach mehrstündiger Immersion in PBS-
Lösung (a) sowie die zugehörige Kinetik in einen Immersionszeitraum von 16 Stunden. Die
zugehörigen Simulationskurven wurden aus Übersichtsgründen ausgelassen.
Wie wir bei der Untersuchung der dicken pp-MA-Filme in Lösung gesehen haben, ist in
einem vergleichbaren Immersionszeitraum die Änderung der Schichtdicke hoch im Verhältnis
zur Änderung des Brechungsindexes (vgl. Abb. 4.10). Betrachtet man Abb. 4.15a, so ist nach
15 h Immersionszeit in Lösung nur eine minimale Verschiebung des Resonanzminimums zu
kleineren Winkeln zu verzeichnen. Daher ist es bei der Simulation der optischen Dicken
durchaus berechtigt, den Brechungsindex als konstant zu betrachten. Hierbei ergibt sich eine
Abnahme der Schichtdicke von 1,1 nm (-5,5%) nach 15-stündiger Einlagerung des Films in
wässriger Lösung. Wie man am Verlauf der kinetischen Messung erkennen kann, ist die
Reflektivitätsänderung innerhalb der ersten sechs Stunden Immersionszeit doppelt so groß
wie in der restlichen Zeit. Daraus folgt, dass eine anfängliche Immersionsphase von etwa
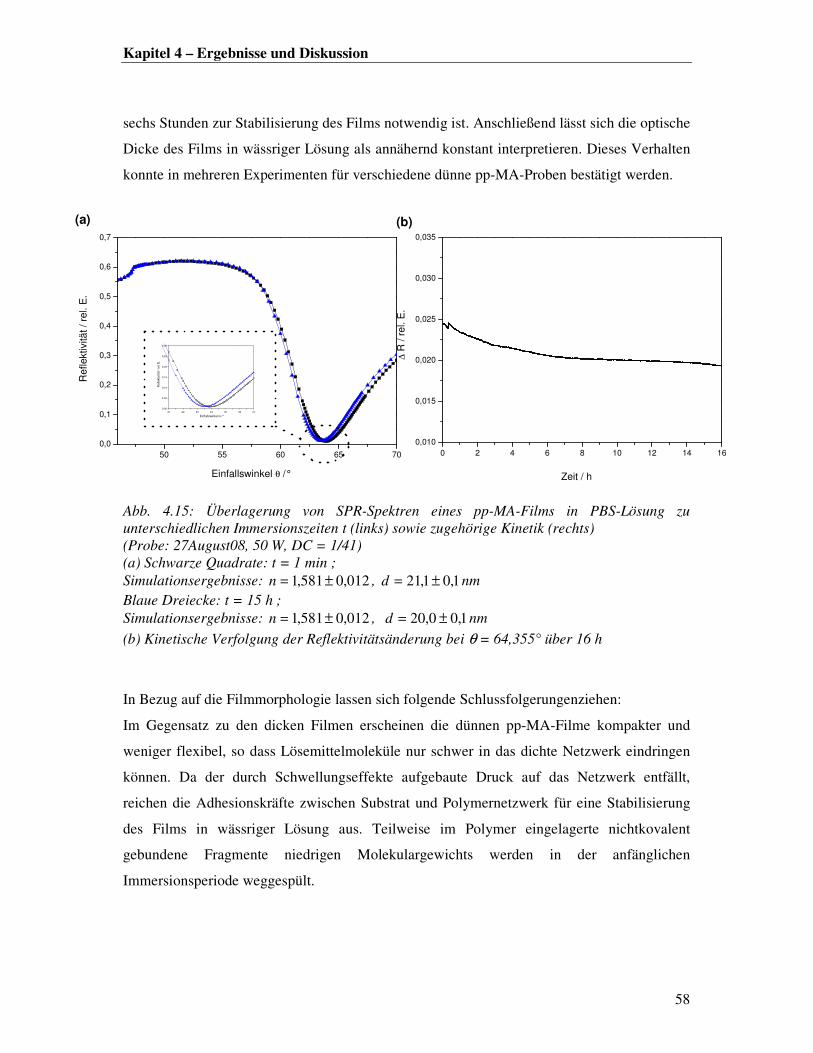
Kapitel 4 – Ergebnisse und Diskussion
58
sechs Stunden zur Stabilisierung des Films notwendig ist. Anschließend lässt sich die optische
Dicke des Films in wässriger Lösung als annähernd konstant interpretieren. Dieses Verhalten
konnte in mehreren Experimenten für verschiedene dünne pp-MA-Proben bestätigt werden.
Abb. 4.15: Überlagerung von SPR-Spektren eines pp-MA-Films in PBS-Lösung zu unterschiedlichen Immersionszeiten t (links) sowie zugehörige Kinetik (rechts) (Probe: 27August08, 50 W, DC = 1/41) (a) Schwarze Quadrate: t = 1 min ; Simulationsergebnisse: 012,0581,1 ±=n , 1,01,21 ±=d nm
Blaue Dreiecke: t = 15 h ; Simulationsergebnisse: 012,0581,1 ±=n , 1,00,20 ±=d nm
(b) Kinetische Verfolgung der Reflektivitätsänderung bei θ = 64,355° über 16 h
In Bezug auf die Filmmorphologie lassen sich folgende Schlussfolgerungenziehen:
Im Gegensatz zu den dicken Filmen erscheinen die dünnen pp-MA-Filme kompakter und
weniger flexibel, so dass Lösemittelmoleküle nur schwer in das dichte Netzwerk eindringen
können. Da der durch Schwellungseffekte aufgebaute Druck auf das Netzwerk entfällt,
reichen die Adhesionskräfte zwischen Substrat und Polymernetzwerk für eine Stabilisierung
des Films in wässriger Lösung aus. Teilweise im Polymer eingelagerte nichtkovalent
gebundene Fragmente niedrigen Molekulargewichts werden in der anfänglichen
Immersionsperiode weggespült.
0 2 4 6 8 10 12 14 16
0,010
0,015
0,020
0,025
0,030
0,035
∆ R
/ r
el. E
.
Zeit / h
(b)
50 55 60 65 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
61 62 63 64 65 66 67
0,00
0,05
0,10
0,15
0,20
0,25
0,30
Reflektiv
ität
/ re
l. E
.
Einfallswinkel θ /°
Reflektivitä
t / re
l. E
.
Einfallswinkel θ /°
(a)

Kapitel 4 – Ergebnisse und Diskussion
59
Zusammenfassung der strukturellen Eigenschaften der pp-MA-Filme
Die chemische Komposition der pp-MA-Filme wurde mittels FT-IR-Spektroskopie als ein
dreidimensionales Kohlenwasserstoffnetzwerk mit hoher Dichte an Anhydridgruppen neben
Carboxy-, Alkohol- und Etherfunktionen charakterisiert. Die Filme weisen stark hydrophile
Eigenschaften auf: Bei Kontakt mit Luftfeuchtigkeit oder wässriger Lösung hydrolysieren die
Anhydridgruppen in die entsprechende Carbonsäure.
Die Oberflächenbeschaffenheit zeigt eine relativ hohe Rauhigkeit, welche mit dem
Wachstumsmechanismus im Plasma korreliert werden kann.
Bei der Charakterisierung der strukturellen Eigenschaften dünner (d<100 nm) und dicker pp-
MA-Filme (0,5-1 µm) mittels SPR und OWS wurde ein teilweise abweichendes Verhalten in
Lösung festgestellt. Die Makrostruktur der im Rahmen dieser Arbeit synthetisierten dicken
Filme scheint einem kovalent verbundenen porösen Netzwerk zu ähneln. Inhomogenitäten im
Vernetzungsgrad sind dabei statistisch verteilt, so dass der Film nach außen hin isotrop
erscheint. Durch Diffusion von Lösemittelmolekülen in die Hohlräume und damit verbundene
Konformationsänderungen quillt das Polymer schwammartig auf und löst sich von der
Substratoberfläche, da die Adhesionskräfte dem steigenden Druck des vollgesogenen
Netzwerks nicht mehr standhalten können. Im Gegensatz dazu erscheinen die im Rahmen
dieser Arbeit synthetisierten dünnen Filme komprimiert und gleichmäßig vernetzt zu sein. Sie
sind stabil in wässriger Lösung und zeigen keine Quelleigenschaften. Vielmehr werden kurze
nichtkovalent gebundene Fragmente in Lösung herausgespült.
Die Abweichungen im Aufbau dünner und dicker Filme lassen sich auf die unterschiedlich
lange Plasmaexpositionszeit zurückführen. Mit steigender Plasmabehandlungszeit erhöht sich
die Vielfalt an aktiven Spezies im Plasma und damit die Unordnung des Systems. Das
bedeutet konkret, dass ständig neu im Plasma generierte Fragmente durch Kollision mit
anderen Spezies dichter und weniger dicht gepackte unregelmäßige Strukturen formen, die
sich auf der Substratoberfläche niederschlagen. Die Substratoberfläche ist damit einer
größeren Belastung ausgesetzt.

Kapitel 4 – Ergebnisse und Diskussion
60
4.2 Immobilisierung von IgG-Antikörpern auf
plasmapolymerisierten Maleinsäureanhydridfilmen
Der Immobilisierungsprozess von Antikörpern auf Substratoberflächen unter Erhaltung der
biologischen Aktivität ist nicht trivial. Wie bereits erwähnt, tendieren biologische Moleküle
dazu, unspezifisch auf einer festen Oberfläche zu adsorbieren. Im Hinblick auf potentielle
Sensorfunktionen ist aus Stabilitätsgründen eine kovalente Verknüpfung des Antikörpers mit
der Polymermatrix zu bevorzugen. In beiden Fällen kann durch Konformationsänderungen,
chemische Modifizierung oder eine ungünstige Orientierung auf oder in der Polymermatrix
das Epitop des Antikörpers deaktiviert werden. Daher ist eine sorgfältige Planung der
Immobilisierungsstrategie erforderlich [13].
Die Struktur eines IgG-Antikörpers umfasst zwei
armähnliche Antigen-Bindungsfragmente (Fab-
Region), welche über die sogenannte Hinge-
Region mit dem Fc-Stamm der insgesamt Y-
förmigen Struktur verbunden sind (vgl.
Abb. 4.16). Die Fab-Fragmente setzen sich aus
zwei identischen schweren Ketten und zwei
ebenfalls identischen leichten Ketten zusammen,
welche über Disulfidbrücken miteinander
verknüpft sind. Die Höhe der Y-förmigen
Struktur beträgt in etwa 12 nm bei einem
mittleren Molekulargewicht von 156 kDa [14].
Die Effizienz einer Antigendetektion ist von der
Orientierung bzw. der Zugänglichkeit der antigenbindenden Fab-Regionen der, auf den pp-
MA-Filmen immobilisierten, Antikörper abhängig.
Wie wir im vorangegangenen Abschnitt gesehen haben, zeigen dünne pp-MA-Filme eine gute
Stabilität in wässriger Lösung. Zusätzlich sind sie im hydrolysierten Zustand mit einer hohen
Dichte an Carboxylfunktionen innerhalb und an der Oberfläche der Polymermatrix
ausgestattet. Die Carboxylgruppen bieten eine exzellente Basis für biochemische
Kopplungsreaktionen mit N-terminalen Aminosäuren bzw. Aminosäureresten des
Antikörpers. Bei der im Folgenden beschriebenen Immobilisierungsstrategie von IgG-
Abb. 4.16: Schematische Darstellung der IgG-Struktur

Kapitel 4 – Ergebnisse und Diskussion
61
Antikörpern auf pp-MA-Filmen wird unser Modellsystem in Bezug auf Stabilität bzw.
Bindungsart und Orientierung hin untersucht.
4.2.1 Kopplungsstrategien
Standardverfahren zur kovalenten Kopplung von Carboxyl- an Aminogruppen bedienen sich
der Aktivesterchemie, da die Carbonylaktivität der Carboxylgruppe für die
Kondensationsreaktion zum entsprechenden Amid nicht ausreicht. Die am häufigsten
verwendeten Kopplungsreagenzien zur Aktivierung von Carboxylgruppen ( engl. Crosslinker)
basieren auf Pentahalogenphenyl-, und Hydroxysuccinimidoestern [15]. Diese sogenannten
„Zero-Length-Crosslinker“ leiten einen direkte kovalente Verknüpfung zwischen den
beteiligten Partnern ein, ohne dass zusätzliche Brückenatome eingebaut werden müssen[16].
Die Auswahl des Crosslinkers richtet sich nach der chemischen Spezifität der zu koppelnden
Reagenzien und der Kompatibilität der Reaktion in Bezug auf die Anwendung. Bei der
Immobilisierung von Antikörpern ist es im Hinblick auf spätere Antigensensoraktivität
notwendig, die native Struktur des Proteins weitgehend zu erhalten. Die Kopplungsreaktion
erfordert daher das Arbeiten bei physiologischen pH-Werten in gepufferten Systemen. Ein
IgG-Molekül enthält typischerweise mindestens 60 Aminosäurereste [17]. Die Konzentration
des Crosslinkers spielt daher eine entscheidende Rolle für die Retention der biologischen
Aktivität des Antikörpers, da eine hohe Anzahl an erfolgreichen Kopplungsprozessen
innerhalb des Proteins Denaturierungsvorgänge induzieren kann. Die Menge an verwendetem
Crosslinker sollte jedoch nicht zu gering sein, da eine hohe Proteinbelegungsdichte erwünscht
ist, zur Prävention unspezifischer Analytadsorption bei nachfolgender Antigenzugabe. Bei der
Konzentrationsabstimmung des Crosslinkers wurde sich an Literaturbespielen orientiert [18,
19].
Im Rahmen dieser Arbeit wurden zwei verschiedene Crosslink-Routen getestet, die im
Folgenden beschrieben werden. Für die Aktivierung der Carboxylgruppen auf der pp-MA-
Oberfläche ist es nicht unbedingt erforderlich in wässrigem Medium zu arbeiten, sofern der
resultierende Aktivester noch ausreichend hydrophil ist. Eventuell verbleibende organische
Lösemittelrückstände lassen sich leicht von der Substratoberfläche waschen. Bannwarth und
Knorr demonstrierten die Effizienz von TSTU-Estern (O-(N-Succinimidyl)-N,N,N’,N’-
tetramethyluronium tetrafluoroborat) in einem DMF/Dioxan/Wasser-Gemisch für die schnelle

Kapitel 4 – Ergebnisse und Diskussion
62
Kopplung von u. A. Biotin mit Aminoverbindungen innerhalb von nur zehn Minuten
Reaktionszeit [20]. Auf dieser Vorlage sowie einer Variation nach Andersson et al.
[21]basierend wurden erste Kopplungsversuche mit IgG-Antikörpern unternommen. Abb.
4.17 zeigt eine auf die pp-MA-Filmgeometrie angepasste schematische Darstellung der
Kopplungsreaktion mit Hilfe von TSTU.
O
O
O
R
O
OH
R
O
OHR
O
NH-IgG
R
O
NH-IgGRO
O
N O
O
R
O
ON
O
O
H2O TSTU/DIPEA
DMF/Dioxan/H2O
2:2:1
H2N-IgG
H2O
N
OC
+
N
O O
CH3CH3
N
CH3
CH3
BF4-
TSTU
Die Aktivierungs- und Kopplungsergebnisse wurden mittels FT-IR-Spektroskopie untersucht;
allerdings konnte eine IgG-Anbindung nicht eindeutig festgestellt werden (vgl. 4.2.3). Im
Folgenden wurde die Kopplungsstrategie auf das standardisierte, in wässriger Lösung
bewährte System aus EDC (N-3-Dimethylaminopropyl)-N-ethylcarbodiimid Hydrochlorid)
und Sulfo-NHS (N-hydroxysulfosuccinimid) umgestellt. Beide Komponenten sind
wasserlöslich. Durch die Zugabe von Sulfo-NHS lässt sich die Stabilität und
Wasserlöslichkeit des EDC-Aktivesters auf mehrere Stunden erhöhen, da auf EDC-basierende
Aktivester andernfalls innerhalb kürzester Zeit hydrolysieren. Die Amidierungsreaktion
funktioniert in einem pH-Bereich von 4,5-7,5. Detaillierte Studien zeigten, dass die Hydrolyse
des Esters für pH-Werte unterhalb von 6 langsamer verläuft. Der optimale pH-Bereich für die
Aktivierung liegt demnach im schwach sauren Bereich, während für die Kopplungsreaktion
mit Proteinen ein pH-Optimum von 7-7,5 festgestellt wurde [15, 22]. Der pH-Wert des
Reaktionsmediums für den Aktivierungsprozess wurde daher im Laufe der Arbeit von pH = 7
(Milli-Q-Wasser, PBS-Lösung) auf pH = 5 (MES-Pufferlösung) umgestellt. Abb. 4.18 zeigt
eine schematische Darstellung für die Aktivierung und Kopplung von pp-MA-Filmen mit
IgG-Antikörpern.
Abb. 4.17: Schematische Darstellung der Aktivierungs- und Kopplungsreaktion hydrolysierter pp-MA-Filme mit IgG-Antikörpern unter Verwendung von TSTU TSTU : O-(N-Succinimidyl)-N,N,N’,N-tetramethyluronium tetrafluoroborat DIPEA: Diisopropylethylamin

Kapitel 4 – Ergebnisse und Diskussion
63
O
O
O
PBS
RO
-Na
+
O
R
O
O-Na
+EDC/Sulfo - NHS
MES
CH3
N N
Cl-HN+
CH3
CH3
NO O
SO3-Na
+
OH
R
O
O
NHEt
N
NHMe2
+Cl
-
R
O
O NHEt
N
NHMe2+Cl
-
Sulfo-NHS
EDC Sulfo-NHS
RO
O
N O
O
SO3-Na
+
R
O
ON
O
O
SO3-Na
+
R
O
NH-IgG
RNH-IgG
O
H2N-IgG
PBS
Abb. 4.18: Schematische Darstellung der Aktivierung- und Kopplungsreaktion von hydrolysierten pp-MA-Filmen mit IgG-Antikörpern unter Vewendung von einer EDC/Sulfo-NHS-Mischung EDC: (N-3-Dimethylaminopropyl)-N-ethylcarbodiimid Hydrochlorid) Sulfo-NHS: (N-hydroxysulfosuccinimid)
4.2.2 SPR-Messoptimierung
Die Auswahl einer Flüssigkeitzelle für SPR-Messungen in Lösung erfolgt in Abstimmung auf
das jeweilige System, da die Sensitivität der Messung durch den Einsatz einer geeigneten
Zellgeometrie entschieden verbessert werden kann. Für die in situ-Verfolgung von Protein-
Immobilisierungsprozessen haben sich insbesondere kleine Zellvolumina bewährt, da diese
eine gute Durchmischung von beschichtetem Substrat und Analyt an der Grenzfläche
erlauben, so dass auch mit sehr geringen Proteinkonzentrationen gearbeitet werden kann.
Außerdem können eventuell auftretende Luftblasen leichter aus dem System gepumpt werden.
Im Rahmen dieser Arbeit wurden für die Messungen in wässriger Lösung zwei verschiedene
Flüssigkeitszellen verwendet. Abb. 4.19 zeigt eine schematische Zeichnung beider Modelle.
Modell A besteht aus einer in der Mitte kreisförmig ausgeschnittenen Teflonplatte mit
angebohrten Kanüleneinlässen an zwei Seiten. Die Zelle wird von beiden Seiten mit einem
Gummi-Ring abgedichtet und auf der Rückseite mit einem Deckglas verschlossen. Das
Substrat wird mit der polymerbeschichteten Seite auf dem O-Ring aufgelegt und die gesamte
Konstruktion wird festgeschraubt (vgl. Kapitel 3, Abb. 3.3). Dieses zunächst eingesetzte
Modell besitzt ein relativ großes Volumen, in dem sich leicht Luftblasen ausbilden können.

Kapitel 4 – Ergebnisse und Diskussion
64
Daher wurde es im Laufe der Arbeit durch Modell B ersetzt. Modell B setzt sich aus einer
dünnen quadratischen Quarzplatte zusammen, die mit zwei Kanüleneinlässe durchbohrt ist.
Auf der Vorderseite der Quarzplatte liegt eine dünne PDMS-Membran auf, welche
gleichzeitig als Zellwand und als Dichtungselement fungiert.
Abb. 4.19: Schematische Zeichnung der verwendeten Flüssigkeitszellen
Der Einfluss der Flusszellengeometrie auf die Sensitivität der Messung wird beim Vergleich
von kinetischen Studien zweier ähnlicher Experimente deutlich. Abb. 4.20 zeigt zwei
Messdurchläufe einer SPR-in-situ-Verfolgung des Immobilisierungsprozesses von IgG-
Antikörpern auf zwei pp-MA-Filmen (Probe A und B). Bis auf die Tatsache, dass Probe B ca.
3 Stunden länger in PBS-Lösung eingelagert wurde als Probe A, ist die Behandlung beider
Proben identisch. Die Messung von Probe A wurde in der oben beschriebenen
Flüssigkeitszelle des Modelltyps A durchgeführt und Probe B wurde in Modell B vermessen.
Ein rein optischer Vergleich beider Kurvenverläufe, ohne auf qualitative Details einzugehen,
zeigt, dass in Versuch B sehr viel schärfere Übergänge zwischen den einzelnen
Behandlungsschritten zu erkennen sind. Demnach trägt die Verwendung einer
Flüssigkeitszelle des Modelltyps B zur Verbesserung der Messsensitivität bei. Für eine
qualitative Erklärung der Versuchsergebnisse sei auf den nächsten Abschnitt verwiesen.
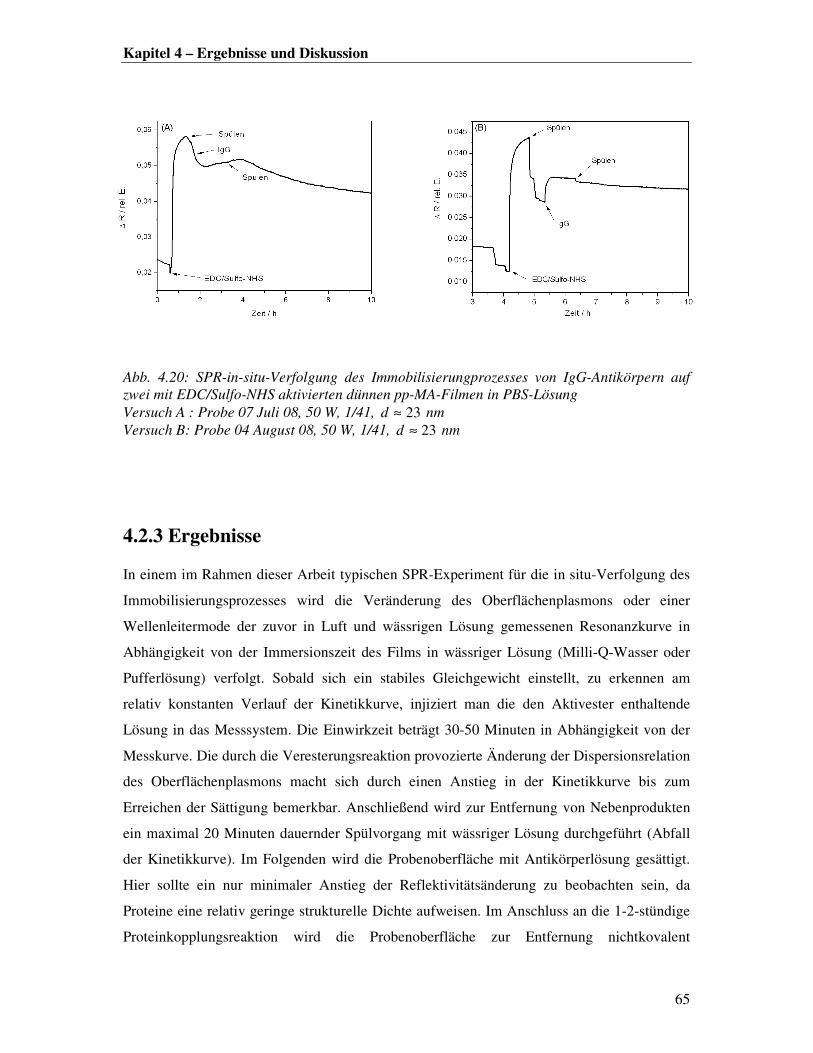
Kapitel 4 – Ergebnisse und Diskussion
65
Abb. 4.20: SPR-in-situ-Verfolgung des Immobilisierungprozesses von IgG-Antikörpern auf zwei mit EDC/Sulfo-NHS aktivierten dünnen pp-MA-Filmen in PBS-Lösung Versuch A : Probe 07 Juli 08, 50 W, 1/41, 23≈d nm Versuch B: Probe 04 August 08, 50 W, 1/41, 23≈d nm
4.2.3 Ergebnisse
In einem im Rahmen dieser Arbeit typischen SPR-Experiment für die in situ-Verfolgung des
Immobilisierungsprozesses wird die Veränderung des Oberflächenplasmons oder einer
Wellenleitermode der zuvor in Luft und wässrigen Lösung gemessenen Resonanzkurve in
Abhängigkeit von der Immersionszeit des Films in wässriger Lösung (Milli-Q-Wasser oder
Pufferlösung) verfolgt. Sobald sich ein stabiles Gleichgewicht einstellt, zu erkennen am
relativ konstanten Verlauf der Kinetikkurve, injiziert man die den Aktivester enthaltende
Lösung in das Messsystem. Die Einwirkzeit beträgt 30-50 Minuten in Abhängigkeit von der
Messkurve. Die durch die Veresterungsreaktion provozierte Änderung der Dispersionsrelation
des Oberflächenplasmons macht sich durch einen Anstieg in der Kinetikkurve bis zum
Erreichen der Sättigung bemerkbar. Anschließend wird zur Entfernung von Nebenprodukten
ein maximal 20 Minuten dauernder Spülvorgang mit wässriger Lösung durchgeführt (Abfall
der Kinetikkurve). Im Folgenden wird die Probenoberfläche mit Antikörperlösung gesättigt.
Hier sollte ein nur minimaler Anstieg der Reflektivitätsänderung zu beobachten sein, da
Proteine eine relativ geringe strukturelle Dichte aufweisen. Im Anschluss an die 1-2-stündige
Proteinkopplungsreaktion wird die Probenoberfläche zur Entfernung nichtkovalent
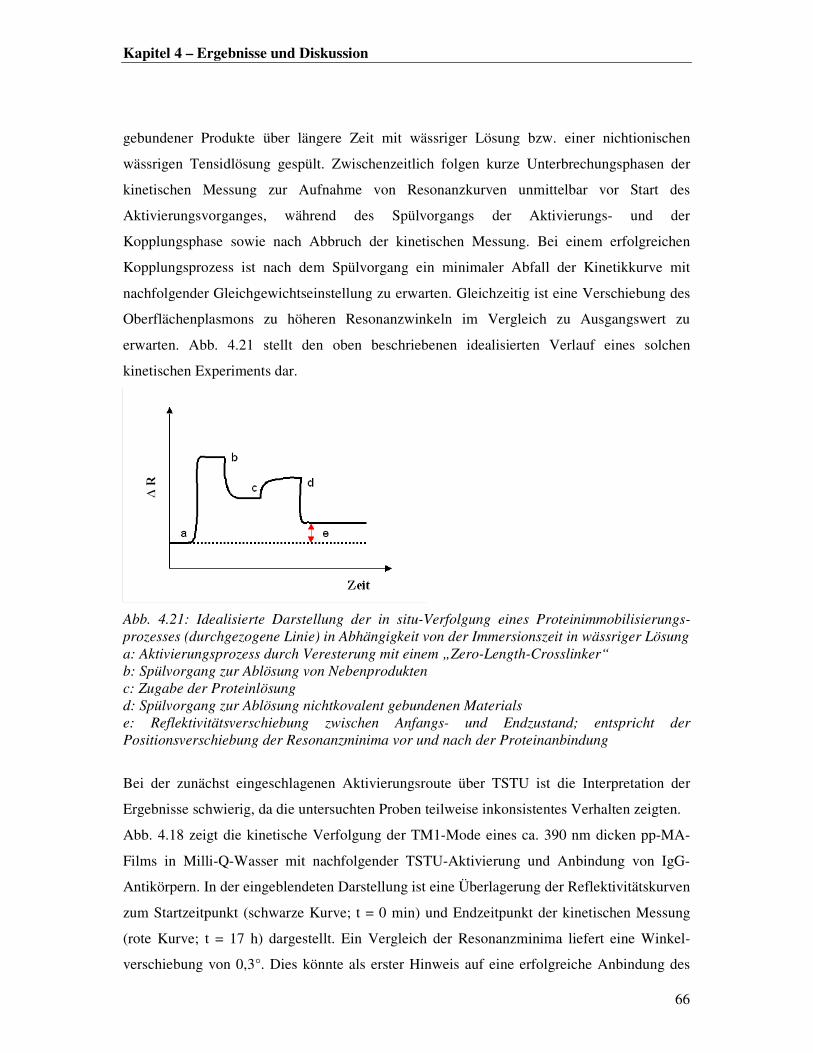
Kapitel 4 – Ergebnisse und Diskussion
66
gebundener Produkte über längere Zeit mit wässriger Lösung bzw. einer nichtionischen
wässrigen Tensidlösung gespült. Zwischenzeitlich folgen kurze Unterbrechungsphasen der
kinetischen Messung zur Aufnahme von Resonanzkurven unmittelbar vor Start des
Aktivierungsvorganges, während des Spülvorgangs der Aktivierungs- und der
Kopplungsphase sowie nach Abbruch der kinetischen Messung. Bei einem erfolgreichen
Kopplungsprozess ist nach dem Spülvorgang ein minimaler Abfall der Kinetikkurve mit
nachfolgender Gleichgewichtseinstellung zu erwarten. Gleichzeitig ist eine Verschiebung des
Oberflächenplasmons zu höheren Resonanzwinkeln im Vergleich zu Ausgangswert zu
erwarten. Abb. 4.21 stellt den oben beschriebenen idealisierten Verlauf eines solchen
kinetischen Experiments dar.
Abb. 4.21: Idealisierte Darstellung der in situ-Verfolgung eines Proteinimmobilisierungs-prozesses (durchgezogene Linie) in Abhängigkeit von der Immersionszeit in wässriger Lösung a: Aktivierungsprozess durch Veresterung mit einem „Zero-Length-Crosslinker“ b: Spülvorgang zur Ablösung von Nebenprodukten c: Zugabe der Proteinlösung d: Spülvorgang zur Ablösung nichtkovalent gebundenen Materials e: Reflektivitätsverschiebung zwischen Anfangs- und Endzustand; entspricht der Positionsverschiebung der Resonanzminima vor und nach der Proteinanbindung
Bei der zunächst eingeschlagenen Aktivierungsroute über TSTU ist die Interpretation der
Ergebnisse schwierig, da die untersuchten Proben teilweise inkonsistentes Verhalten zeigten.
Abb. 4.18 zeigt die kinetische Verfolgung der TM1-Mode eines ca. 390 nm dicken pp-MA-
Films in Milli-Q-Wasser mit nachfolgender TSTU-Aktivierung und Anbindung von IgG-
Antikörpern. In der eingeblendeten Darstellung ist eine Überlagerung der Reflektivitätskurven
zum Startzeitpunkt (schwarze Kurve; t = 0 min) und Endzeitpunkt der kinetischen Messung
(rote Kurve; t = 17 h) dargestellt. Ein Vergleich der Resonanzminima liefert eine Winkel-
verschiebung von 0,3°. Dies könnte als erster Hinweis auf eine erfolgreiche Anbindung des
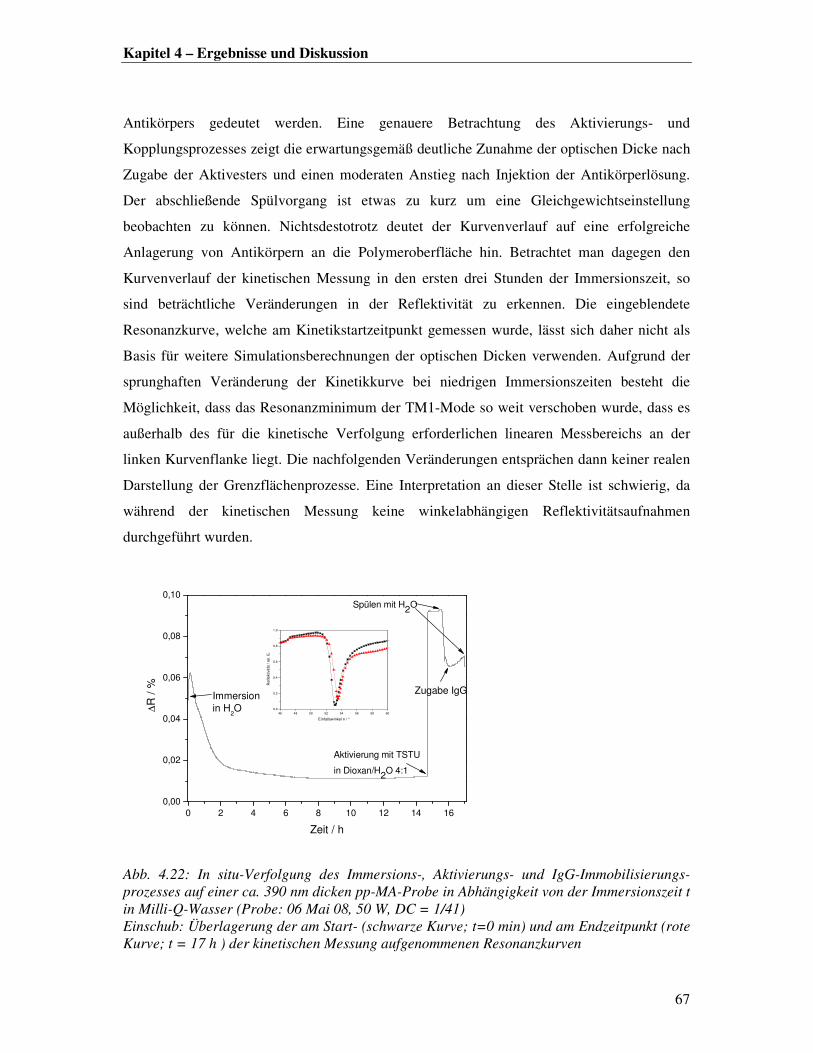
Kapitel 4 – Ergebnisse und Diskussion
67
Antikörpers gedeutet werden. Eine genauere Betrachtung des Aktivierungs- und
Kopplungsprozesses zeigt die erwartungsgemäß deutliche Zunahme der optischen Dicke nach
Zugabe der Aktivesters und einen moderaten Anstieg nach Injektion der Antikörperlösung.
Der abschließende Spülvorgang ist etwas zu kurz um eine Gleichgewichtseinstellung
beobachten zu können. Nichtsdestotrotz deutet der Kurvenverlauf auf eine erfolgreiche
Anlagerung von Antikörpern an die Polymeroberfläche hin. Betrachtet man dagegen den
Kurvenverlauf der kinetischen Messung in den ersten drei Stunden der Immersionszeit, so
sind beträchtliche Veränderungen in der Reflektivität zu erkennen. Die eingeblendete
Resonanzkurve, welche am Kinetikstartzeitpunkt gemessen wurde, lässt sich daher nicht als
Basis für weitere Simulationsberechnungen der optischen Dicken verwenden. Aufgrund der
sprunghaften Veränderung der Kinetikkurve bei niedrigen Immersionszeiten besteht die
Möglichkeit, dass das Resonanzminimum der TM1-Mode so weit verschoben wurde, dass es
außerhalb des für die kinetische Verfolgung erforderlichen linearen Messbereichs an der
linken Kurvenflanke liegt. Die nachfolgenden Veränderungen entsprächen dann keiner realen
Darstellung der Grenzflächenprozesse. Eine Interpretation an dieser Stelle ist schwierig, da
während der kinetischen Messung keine winkelabhängigen Reflektivitätsaufnahmen
durchgeführt wurden.
0 2 4 6 8 10 12 14 16
0,00
0,02
0,04
0,06
0,08
0,10
46 48 50 52 54 56 58 60
0,0
0,2
0,4
0,6
0,8
1,0
Refle
ktivität
/ re
l. E
.
Einfallswinkel θ / °
∆R
/ %
Zeit / h
Spülen mit H2
O
Immersion
in H2O
Aktivierung mit TSTU
in Dioxan/H2O 4:1
Zugabe IgG
Abb. 4.22: In situ-Verfolgung des Immersions-, Aktivierungs- und IgG-Immobilisierungs-prozesses auf einer ca. 390 nm dicken pp-MA-Probe in Abhängigkeit von der Immersionszeit t in Milli-Q-Wasser (Probe: 06 Mai 08, 50 W, DC = 1/41) Einschub: Überlagerung der am Start- (schwarze Kurve; t=0 min) und am Endzeitpunkt (rote Kurve; t = 17 h ) der kinetischen Messung aufgenommenen Resonanzkurven
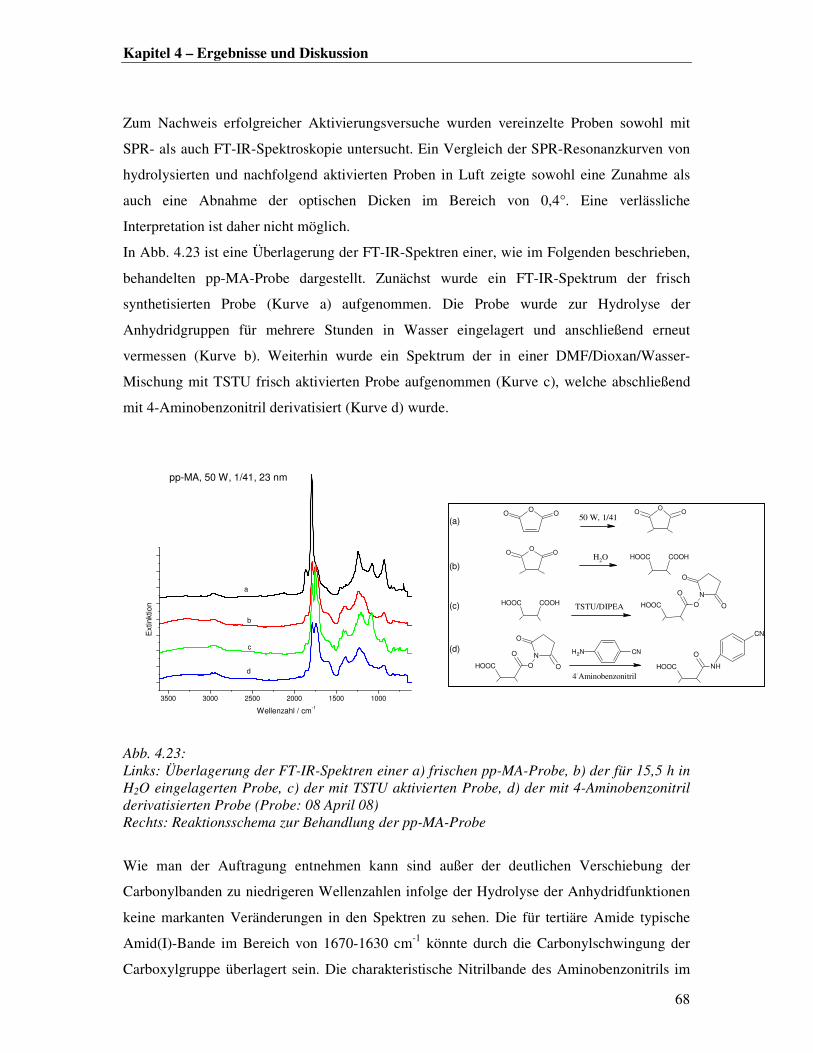
Kapitel 4 – Ergebnisse und Diskussion
68
Zum Nachweis erfolgreicher Aktivierungsversuche wurden vereinzelte Proben sowohl mit
SPR- als auch FT-IR-Spektroskopie untersucht. Ein Vergleich der SPR-Resonanzkurven von
hydrolysierten und nachfolgend aktivierten Proben in Luft zeigte sowohl eine Zunahme als
auch eine Abnahme der optischen Dicken im Bereich von 0,4°. Eine verlässliche
Interpretation ist daher nicht möglich.
In Abb. 4.23 ist eine Überlagerung der FT-IR-Spektren einer, wie im Folgenden beschrieben,
behandelten pp-MA-Probe dargestellt. Zunächst wurde ein FT-IR-Spektrum der frisch
synthetisierten Probe (Kurve a) aufgenommen. Die Probe wurde zur Hydrolyse der
Anhydridgruppen für mehrere Stunden in Wasser eingelagert und anschließend erneut
vermessen (Kurve b). Weiterhin wurde ein Spektrum der in einer DMF/Dioxan/Wasser-
Mischung mit TSTU frisch aktivierten Probe aufgenommen (Kurve c), welche abschließend
mit 4-Aminobenzonitril derivatisiert (Kurve d) wurde.
3500 3000 2500 2000 1500 1000
Extin
ktio
n
Wellenzahl / cm-1
pp-MA, 50 W, 1/41, 23 nm
a
b
c
d
Abb. 4.23: Links: Überlagerung der FT-IR-Spektren einer a) frischen pp-MA-Probe, b) der für 15,5 h in H2O eingelagerten Probe, c) der mit TSTU aktivierten Probe, d) der mit 4-Aminobenzonitril derivatisierten Probe (Probe: 08 April 08) Rechts: Reaktionsschema zur Behandlung der pp-MA-Probe
Wie man der Auftragung entnehmen kann sind außer der deutlichen Verschiebung der
Carbonylbanden zu niedrigeren Wellenzahlen infolge der Hydrolyse der Anhydridfunktionen
keine markanten Veränderungen in den Spektren zu sehen. Die für tertiäre Amide typische
Amid(I)-Bande im Bereich von 1670-1630 cm-1 könnte durch die Carbonylschwingung der
Carboxylgruppe überlagert sein. Die charakteristische Nitrilbande des Aminobenzonitrils im
H2O HOOC COOH
HOOC COOH TSTU/DIPEA HOOC
O
O
N
O
O
HOOC
O
O
N
O
O
4-Aminobenzonitril
CNNH2
HOOC
O
NH
CN
(b)
(c)
(d)
OO O
50 W, 1/41O
O O
OO O
(a)

Kapitel 4 – Ergebnisse und Diskussion
69
Bereich von 2260-2210 cm-1 ist nicht zu erkennen. Die Aktivierung scheint demnach nicht
funktioniert zu haben; allerdings ist eine eindeutige Aussage schwer zu treffen, da aufgrund
von sterischer Hinderung möglicherweise nur an der Oberfläche liegende Carboxylfunktionen
derivatisiert wurden. Diese stellen nur einen kleinen Anteil an der Gesamtdichte der
funktionellen Gruppen dar, so dass sie außerhalb der Sensitivitätsgrenzen der FT-IR-Messung
liegen.
Aufgrund der oben beschriebenen Unklarheiten in Bezug auf eine erfolgreiche Aktivierung
und Kupplung wurde der TSTU-basierte Ester durch die wasserlösliche Kombination aus
EDC und Sulfo-NHS ersetzt. Die Effizienz beider Reagenzien wurde in einem Testdurchlauf
zur Anbindung von Ethanolamin an hydrolysierte pp-MA-Filme bewiesen. Abb. 4.24 zeigt
die Kinetik des Testdurchlaufs in PBS-Lösung. In der eingeblendeten Darstellung ist eine
Überlagerung der Reflektivitätsaufnahmen vor der Aktivierung mit EDC und Sulfo-NHS
(schwarze Quadrate) und nach der Anbindung von Ethanolamin (rote Dreiecke) mit einer
deutlichen Verschiebung zu höheren Resonanzwinkeln zu sehen. Unter Annahme des
allgemein für organische Moleküle eingesetzten Brechungsindexes von n = 1,581, lässt sich
die Positionsverschiebung des Minimums mit einem Dickenzuwachs von ca. 5 nm
quantifizieren. Der Kurvenverlauf entspricht ziemlich genau dem in Abb. 4.21 beschriebenen
erwarteten Verlauf. Die Anbindung von Ethanolamin auf der pp-MA-Oberfläche hat demnach
einwandfrei funktioniert. Der markante Abfall der Kinetikkurve beim Pufferwechsel von PBS
zu MES ist auf die abrupte pH-Erniedrigung (7,4 auf 5) zurückzuführen. Im schwach sauren
Bereich ist ein geringerer Anteil an Carboxylgruppen deprotoniert, so dass die elektrostatische
Abstoßung zwischen den funktionalisierten Resten abnimmt. Infolgedessen ist eine dichtere
Anordnung der Carboxylreste in der Polymermatrix favorisiert. Die Triebkraft dieser
Konformationsänderungen liegt in der Reduktion der freien Oberflächenenergie [7, 8].
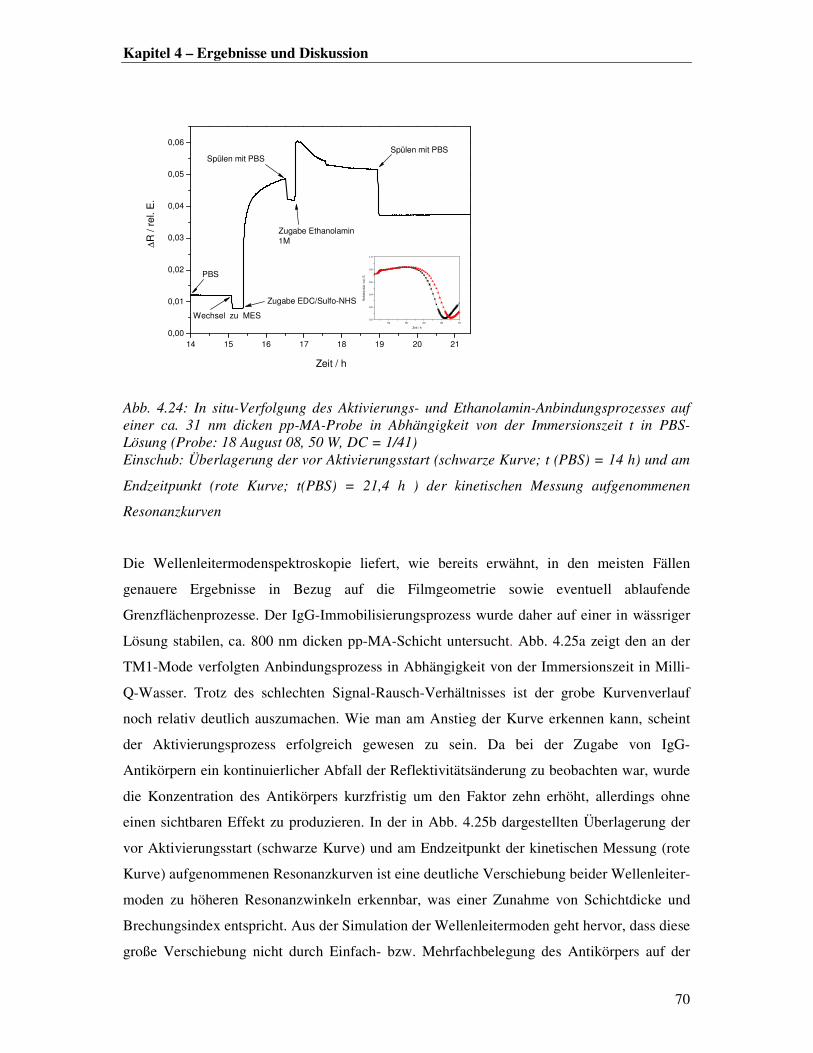
Kapitel 4 – Ergebnisse und Diskussion
70
14 15 16 17 18 19 20 21
0,00
0,01
0,02
0,03
0,04
0,05
0,06
50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Re
flektivi
tät
/ re
l. E
.
Zeit / h
Spülen mit PBS
∆R
/ r
el. E
.
Zeit / h
PBS
Wechsel zu MES
Zugabe EDC/Sulfo-NHS
Spülen mit PBS
Zugabe Ethanolamin
1M
Abb. 4.24: In situ-Verfolgung des Aktivierungs- und Ethanolamin-Anbindungsprozesses auf einer ca. 31 nm dicken pp-MA-Probe in Abhängigkeit von der Immersionszeit t in PBS-Lösung (Probe: 18 August 08, 50 W, DC = 1/41) Einschub: Überlagerung der vor Aktivierungsstart (schwarze Kurve; t (PBS) = 14 h) und am
Endzeitpunkt (rote Kurve; t(PBS) = 21,4 h ) der kinetischen Messung aufgenommenen
Resonanzkurven
Die Wellenleitermodenspektroskopie liefert, wie bereits erwähnt, in den meisten Fällen
genauere Ergebnisse in Bezug auf die Filmgeometrie sowie eventuell ablaufende
Grenzflächenprozesse. Der IgG-Immobilisierungsprozess wurde daher auf einer in wässriger
Lösung stabilen, ca. 800 nm dicken pp-MA-Schicht untersucht. Abb. 4.25a zeigt den an der
TM1-Mode verfolgten Anbindungsprozess in Abhängigkeit von der Immersionszeit in Milli-
Q-Wasser. Trotz des schlechten Signal-Rausch-Verhältnisses ist der grobe Kurvenverlauf
noch relativ deutlich auszumachen. Wie man am Anstieg der Kurve erkennen kann, scheint
der Aktivierungsprozess erfolgreich gewesen zu sein. Da bei der Zugabe von IgG-
Antikörpern ein kontinuierlicher Abfall der Reflektivitätsänderung zu beobachten war, wurde
die Konzentration des Antikörpers kurzfristig um den Faktor zehn erhöht, allerdings ohne
einen sichtbaren Effekt zu produzieren. In der in Abb. 4.25b dargestellten Überlagerung der
vor Aktivierungsstart (schwarze Kurve) und am Endzeitpunkt der kinetischen Messung (rote
Kurve) aufgenommenen Resonanzkurven ist eine deutliche Verschiebung beider Wellenleiter-
moden zu höheren Resonanzwinkeln erkennbar, was einer Zunahme von Schichtdicke und
Brechungsindex entspricht. Aus der Simulation der Wellenleitermoden geht hervor, dass diese
große Verschiebung nicht durch Einfach- bzw. Mehrfachbelegung des Antikörpers auf der
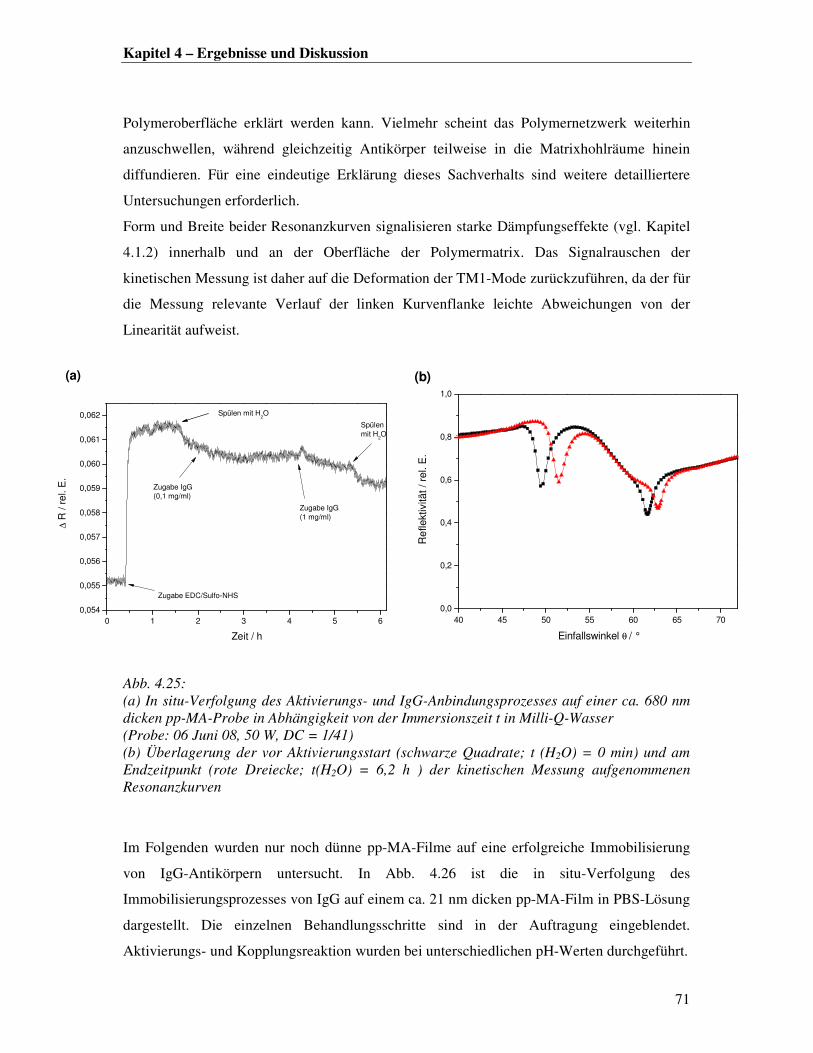
Kapitel 4 – Ergebnisse und Diskussion
71
Polymeroberfläche erklärt werden kann. Vielmehr scheint das Polymernetzwerk weiterhin
anzuschwellen, während gleichzeitig Antikörper teilweise in die Matrixhohlräume hinein
diffundieren. Für eine eindeutige Erklärung dieses Sachverhalts sind weitere detailliertere
Untersuchungen erforderlich.
Form und Breite beider Resonanzkurven signalisieren starke Dämpfungseffekte (vgl. Kapitel
4.1.2) innerhalb und an der Oberfläche der Polymermatrix. Das Signalrauschen der
kinetischen Messung ist daher auf die Deformation der TM1-Mode zurückzuführen, da der für
die Messung relevante Verlauf der linken Kurvenflanke leichte Abweichungen von der
Linearität aufweist.
Abb. 4.25: (a) In situ-Verfolgung des Aktivierungs- und IgG-Anbindungsprozesses auf einer ca. 680 nm dicken pp-MA-Probe in Abhängigkeit von der Immersionszeit t in Milli-Q-Wasser (Probe: 06 Juni 08, 50 W, DC = 1/41) (b) Überlagerung der vor Aktivierungsstart (schwarze Quadrate; t (H2O) = 0 min) und am Endzeitpunkt (rote Dreiecke; t(H2O) = 6,2 h ) der kinetischen Messung aufgenommenen Resonanzkurven
Im Folgenden wurden nur noch dünne pp-MA-Filme auf eine erfolgreiche Immobilisierung
von IgG-Antikörpern untersucht. In Abb. 4.26 ist die in situ-Verfolgung des
Immobilisierungsprozesses von IgG auf einem ca. 21 nm dicken pp-MA-Film in PBS-Lösung
dargestellt. Die einzelnen Behandlungsschritte sind in der Auftragung eingeblendet.
Aktivierungs- und Kopplungsreaktion wurden bei unterschiedlichen pH-Werten durchgeführt.
40 45 50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Re
flektivitä
t / re
l. E
.
Einfallswinkel θ / °
(b)
0 1 2 3 4 5 6
0,054
0,055
0,056
0,057
0,058
0,059
0,060
0,061
0,062
Spülen mit H2O
Zugabe IgG
(0,1 mg/ml)
Spülen
mit H2O
Zugabe IgG
(1 mg/ml)
Zugabe EDC/Sulfo-NHS
∆ R
/ r
el. E
.
Zeit / h
(a)
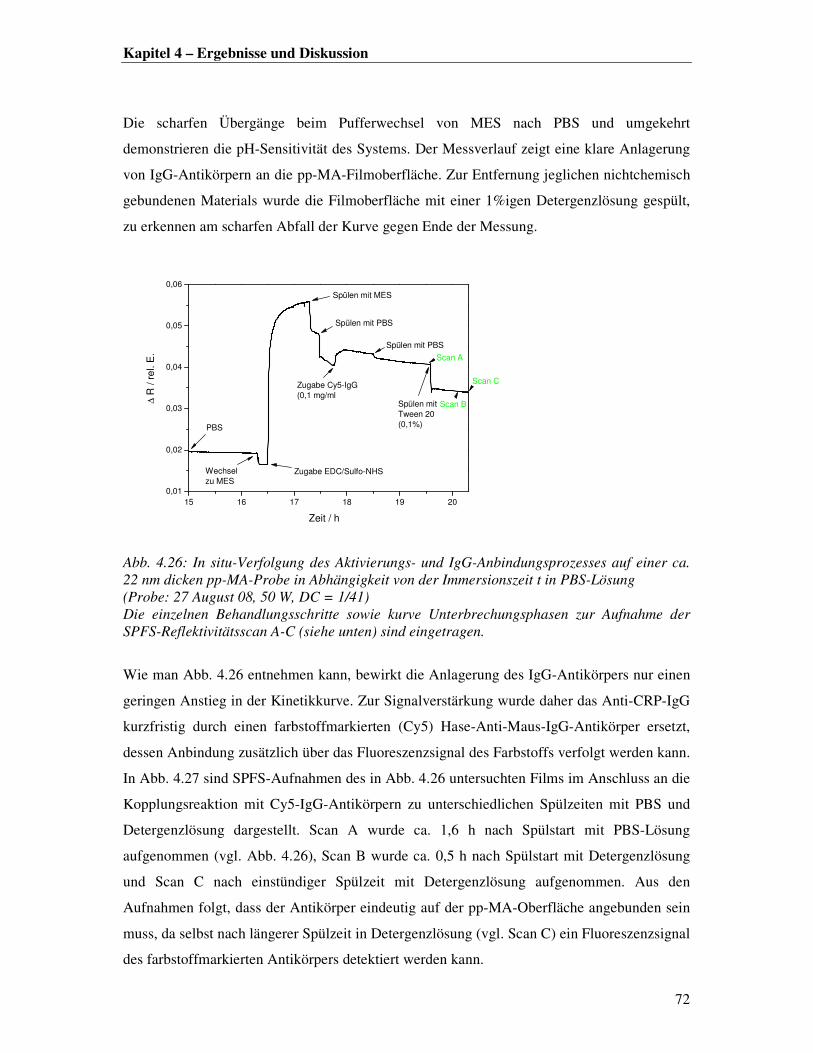
Kapitel 4 – Ergebnisse und Diskussion
72
Die scharfen Übergänge beim Pufferwechsel von MES nach PBS und umgekehrt
demonstrieren die pH-Sensitivität des Systems. Der Messverlauf zeigt eine klare Anlagerung
von IgG-Antikörpern an die pp-MA-Filmoberfläche. Zur Entfernung jeglichen nichtchemisch
gebundenen Materials wurde die Filmoberfläche mit einer 1%igen Detergenzlösung gespült,
zu erkennen am scharfen Abfall der Kurve gegen Ende der Messung.
15 16 17 18 19 20
0,01
0,02
0,03
0,04
0,05
0,06
∆ R
/ r
el. E
.
Zeit / h
PBS
Wechsel
zu MESZugabe EDC/Sulfo-NHS
Spülen mit PBS
Spülen mit MES
Zugabe Cy5-IgG
(0,1 mg/ml
Spülen mit PBS
Spülen mit
Tween 20
(0,1%)
Scan A
Scan B
Scan C
Abb. 4.26: In situ-Verfolgung des Aktivierungs- und IgG-Anbindungsprozesses auf einer ca. 22 nm dicken pp-MA-Probe in Abhängigkeit von der Immersionszeit t in PBS-Lösung (Probe: 27 August 08, 50 W, DC = 1/41) Die einzelnen Behandlungsschritte sowie kurve Unterbrechungsphasen zur Aufnahme der SPFS-Reflektivitätsscan A-C (siehe unten) sind eingetragen.
Wie man Abb. 4.26 entnehmen kann, bewirkt die Anlagerung des IgG-Antikörpers nur einen
geringen Anstieg in der Kinetikkurve. Zur Signalverstärkung wurde daher das Anti-CRP-IgG
kurzfristig durch einen farbstoffmarkierten (Cy5) Hase-Anti-Maus-IgG-Antikörper ersetzt,
dessen Anbindung zusätzlich über das Fluoreszenzsignal des Farbstoffs verfolgt werden kann.
In Abb. 4.27 sind SPFS-Aufnahmen des in Abb. 4.26 untersuchten Films im Anschluss an die
Kopplungsreaktion mit Cy5-IgG-Antikörpern zu unterschiedlichen Spülzeiten mit PBS und
Detergenzlösung dargestellt. Scan A wurde ca. 1,6 h nach Spülstart mit PBS-Lösung
aufgenommen (vgl. Abb. 4.26), Scan B wurde ca. 0,5 h nach Spülstart mit Detergenzlösung
und Scan C nach einstündiger Spülzeit mit Detergenzlösung aufgenommen. Aus den
Aufnahmen folgt, dass der Antikörper eindeutig auf der pp-MA-Oberfläche angebunden sein
muss, da selbst nach längerer Spülzeit in Detergenzlösung (vgl. Scan C) ein Fluoreszenzsignal
des farbstoffmarkierten Antikörpers detektiert werden kann.
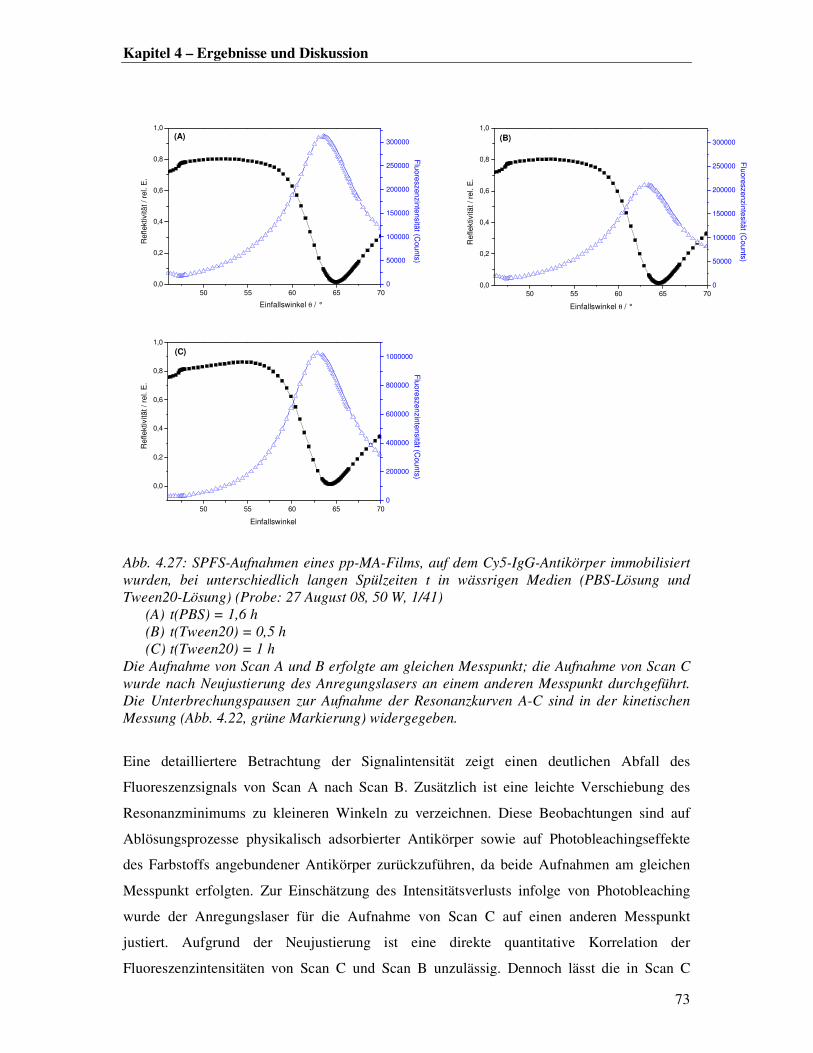
Kapitel 4 – Ergebnisse und Diskussion
73
Abb. 4.27: SPFS-Aufnahmen eines pp-MA-Films, auf dem Cy5-IgG-Antikörper immobilisiert wurden, bei unterschiedlich langen Spülzeiten t in wässrigen Medien (PBS-Lösung und Tween20-Lösung) (Probe: 27 August 08, 50 W, 1/41)
(A) t(PBS) = 1,6 h (B) t(Tween20) = 0,5 h (C) t(Tween20) = 1 h
Die Aufnahme von Scan A und B erfolgte am gleichen Messpunkt; die Aufnahme von Scan C wurde nach Neujustierung des Anregungslasers an einem anderen Messpunkt durchgeführt. Die Unterbrechungspausen zur Aufnahme der Resonanzkurven A-C sind in der kinetischen Messung (Abb. 4.22, grüne Markierung) widergegeben.
Eine detailliertere Betrachtung der Signalintensität zeigt einen deutlichen Abfall des
Fluoreszenzsignals von Scan A nach Scan B. Zusätzlich ist eine leichte Verschiebung des
Resonanzminimums zu kleineren Winkeln zu verzeichnen. Diese Beobachtungen sind auf
Ablösungsprozesse physikalisch adsorbierter Antikörper sowie auf Photobleachingseffekte
des Farbstoffs angebundener Antikörper zurückzuführen, da beide Aufnahmen am gleichen
Messpunkt erfolgten. Zur Einschätzung des Intensitätsverlusts infolge von Photobleaching
wurde der Anregungslaser für die Aufnahme von Scan C auf einen anderen Messpunkt
justiert. Aufgrund der Neujustierung ist eine direkte quantitative Korrelation der
Fluoreszenzintensitäten von Scan C und Scan B unzulässig. Dennoch lässt die in Scan C
50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Re
fle
ktivitä
t /
rel. E
.
Einfallswinkel
0
200000
400000
600000
800000
1000000
Flu
ore
sze
nzin
ten
sitä
t (Co
un
ts)
(C)
50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Einfallswinkel θ / °
Re
flektivitä
t / re
l. E
.(A)
0
50000
100000
150000
200000
250000
300000
Flu
ore
szen
zin
ten
sitä
t (Cou
nts
)
50 55 60 65 70
0,0
0,2
0,4
0,6
0,8
1,0
Einfallswinkel θ / °
Re
fle
ktivitä
t / re
l. E
.
(B)
0
50000
100000
150000
200000
250000
300000
Flu
ore
szen
zin
tesitä
t (Co
un
ts)
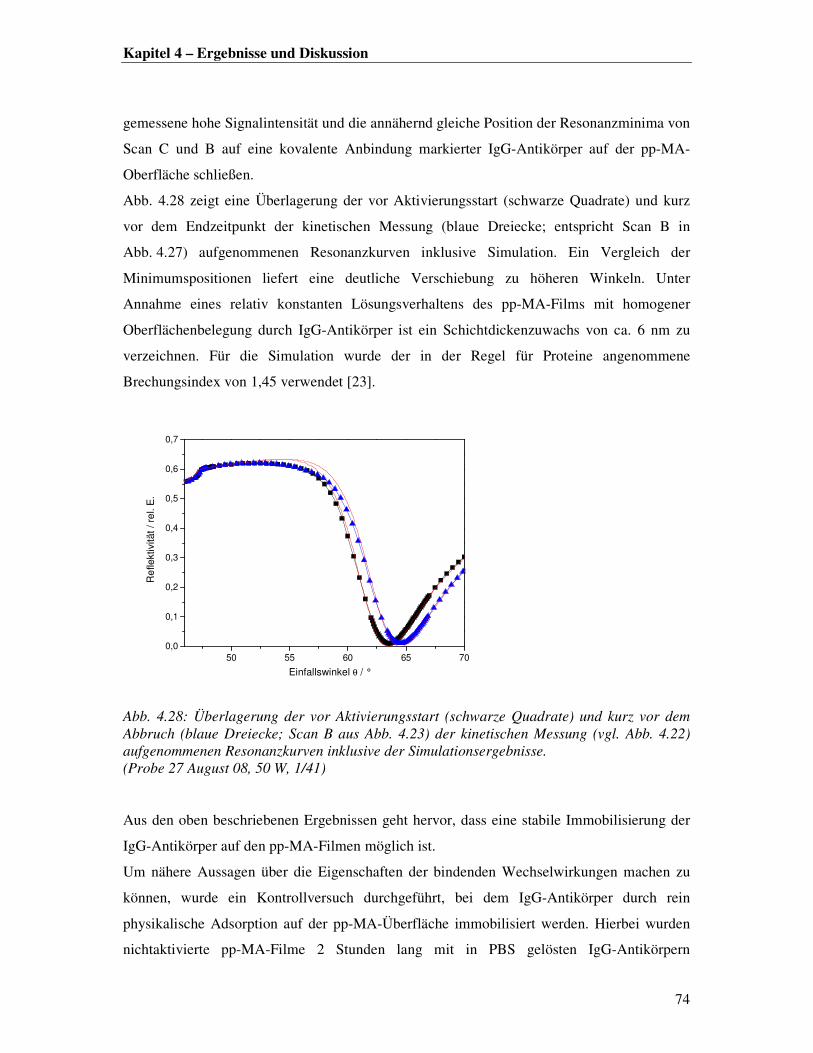
Kapitel 4 – Ergebnisse und Diskussion
74
gemessene hohe Signalintensität und die annähernd gleiche Position der Resonanzminima von
Scan C und B auf eine kovalente Anbindung markierter IgG-Antikörper auf der pp-MA-
Oberfläche schließen.
Abb. 4.28 zeigt eine Überlagerung der vor Aktivierungsstart (schwarze Quadrate) und kurz
vor dem Endzeitpunkt der kinetischen Messung (blaue Dreiecke; entspricht Scan B in
Abb. 4.27) aufgenommenen Resonanzkurven inklusive Simulation. Ein Vergleich der
Minimumspositionen liefert eine deutliche Verschiebung zu höheren Winkeln. Unter
Annahme eines relativ konstanten Lösungsverhaltens des pp-MA-Films mit homogener
Oberflächenbelegung durch IgG-Antikörper ist ein Schichtdickenzuwachs von ca. 6 nm zu
verzeichnen. Für die Simulation wurde der in der Regel für Proteine angenommene
Brechungsindex von 1,45 verwendet [23].
50 55 60 65 70
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Re
flektivität / re
l. E
.
Einfallswinkel θ / °
Abb. 4.28: Überlagerung der vor Aktivierungsstart (schwarze Quadrate) und kurz vor dem Abbruch (blaue Dreiecke; Scan B aus Abb. 4.23) der kinetischen Messung (vgl. Abb. 4.22) aufgenommenen Resonanzkurven inklusive der Simulationsergebnisse. (Probe 27 August 08, 50 W, 1/41) Aus den oben beschriebenen Ergebnissen geht hervor, dass eine stabile Immobilisierung der
IgG-Antikörper auf den pp-MA-Filmen möglich ist.
Um nähere Aussagen über die Eigenschaften der bindenden Wechselwirkungen machen zu
können, wurde ein Kontrollversuch durchgeführt, bei dem IgG-Antikörper durch rein
physikalische Adsorption auf der pp-MA-Überfläche immobilisiert werden. Hierbei wurden
nichtaktivierte pp-MA-Filme 2 Stunden lang mit in PBS gelösten IgG-Antikörpern

Kapitel 4 – Ergebnisse und Diskussion
75
gesättigt und anschließend 15 Stunden lang mit PBS-Lösung gespült. Aus der Verschiebung
der Resonanzminima der vor und nach Antikörperzugabe und subsequenten Spülvorgang
aufgenommen Reflektivitätskurven ist ein Schichtdickenzuwachs von ebenfalls 6 nm zu
verzeichnen. Dies entspricht den Ergebnissen der zuvor untersuchten kovalenten
Immobilisierungsversuche. Eine Unterscheidung zwischen physikalisch adsorbierten und
kovalent gebundenen Antikörpern scheint demnach zunächst nicht möglich zu sein. Die pp-
MA-Oberfläche wurde allerdings nicht mit Detergenzlösung gespült. Wie man der kinetischen
Messung des chemisch eingeleiteten Kopplungsversuchs in Abb. 4.24 entnehmen kann, ist ein
abschließendes Spülen mit PBS-Lösung nicht ausreichend, um physikalisch adsorbierte
Spezies von der pp-MA-Oberfläche abzulösen, da bei der nachfolgenden Zugabe von
Detergenzlösung nochmals ein deutlicher Abfall in der Kurve zu verzeichnen ist, der nur auf
eine Abtrennung nichtchemisch gebundener Spezies zurückzuführen ist. Die verbleibenden,
auf der pp-MA-Oberfläche immobilisierten, Antikörper sind demnach eindeutig kovalent mit
der Polymermatrix verknüpft.
Wie bereits in der Einleitung erwähnt, beträgt die Längsausdehnung der Antikörpermoleküle
in etwa 12 nm. Die oben beschriebenen Kopplungsversuche wurden bei einer relativ hohen
IgG-Konzentration von 0,1 mg/ml durchgeführt, so dass auch von einem relativ hohen
Belegungsgrad der Filmoberfläche ausgegangen werden kann. Ein mittlerer
Schichtdickenzuwachs von 6 nm deutet daher auf eine statistische Orientierung der
Antikörper auf der Filmoberfläche hin. Die reale Orientierung der Antikörper auf der pp-MA-
Oberfläche lässt sich aus den experimentellen Daten nicht determinieren, da diese von vielen
Faktoren abhängig ist, welche u. A. genauere Informationen zur Aminosäuresequenz bzw. zur
strukturellen Anordnung und Zugänglichkeit der Lysinreste erfordern.
4.2.4 Zusammenfassung und Ausblick
Aus den SPR-Untersuchungen geht hervor, dass eine kovalente Anbindung von IgG-
Antikörpern nach vorangegangener Aktivierung über EDC/Sulfo-NHS-basierte Aktivester auf
dünnen pp-MA-Filmen möglich ist. Plasmapolymerisierte Maleinsäureanhydridfilme sind
demnach als Bausteine für potentielle Biosensoren geeignet. Auf der Grundlage des
entwickelten pp-MA/IgG-Systems lassen sich nun im nächsten Schritt die
Sensoreigenschaften des Systems unter Zugabe von Antigenen untersuchen. Hierbei ist noch
viel Arbeit in die Optimierung des Kopplungsprozesses in Bezug auf eine Erhöhung der

Kapitel 4 – Ergebnisse und Diskussion
76
Proteinbelegungsdichte, ein optimales Crosslinker/Proteinverhältnis sowie eine kontrollierte
Orientierung der Antikörper zu investieren.
Die Proteinbelegungsdichte lässt sich z. B. durch elektrostatische Voradsorption des Proteins
unterhalb seines isoelektrischen Punkts realisieren. Die kovalente Verknüpfung der
Sensorbasis unter Orientierung der Antikörper ist nicht trivial. Mögliche Ansätze werden im
Folgenden beschrieben: Eine bereits an anderen Systemen erfolgreich getestete Route
beinhaltet die Immobilisierung von Protein A oder G auf der Sensoroberfläche mit
subsequenter IgG-Anbindung, da diese aus Bakterien gewonnenen Proteine spezifische
bindende Wechselwirkungen mit der Fc-Region des Antikörpers eingehen, so dass beide
antigenbindende Fab-Fraktionen nach außen zeigen [13]. Für die Anwendung auf einer pp-
MA-Unterlage ist zu bedenken, dass der Erfolg dieser Methode von der Kontrolle über die
Orientierung und Aktivität des immobilisierten Proteins A oder G abhängt. Eine weitere
Möglichkeit besteht darin, sich die biochemische Affinität des Avidin-Biotin-Systems zunutze
zu machen. Hierbei wird Avidin auf der Sensoroberfläche immobilisiert, welches dann die
Bindungsstellen für einen biotinylierten Antikörper zur Verfügung stellt. Die Effizienz des
Systems ist wiederum von der Orientierung der Avidin-Moleküle und der
Biotinylierungsmethode des Antikörpers abhängig [24]. Eine alternative, vielversprechende
Route stellt die gezielte Verknüpfung aminderivatisierter Plasmapolymerreste mit dem, an der
Fc-Region anhängenden, modifizierten Kohlenhydratrestes dar, da dieser zu weit von den
antigenbindenden Fab-Regionen angeordnet ist, um deren Aktivität beeinflussen zu können
[17].

Kapitel 4 – Ergebnisse und Diskussion
77
Literaturverzeichnis
1. S. Schiller, J.H., A. T. A. Jenkins, R. B. Timmons, F. S. Sanchez-Estrada, and R.F. W. Knoll, Chemical Structure and Properties of Plasma-Polymerized Maleic Anhydride Films. Chem. Mater., 2002. 14: p. 235-242.
2. Ryan, M.E., A.M. Hynes, and J.P.S. Badyal, Pulsed Plasma Polymerization of Maleic Anhydride. Chem. Mater., 1996. 8(1): p. 37-42.
3. Jenkins, A.T.A., et al., Pulsed Plasma Deposited Maleic Anhydride Thin Films as Supports for Lipid Bilayers. Langmuir, 2000. 16(16): p. 6381-6384.
4. Jacobsen, V., et al., In-situ thin film diagnostics using waveguide mode spectroscopy. Thin Solid Films, 2002. 409(2): p. 185-193.
5. R. Förch, Z.Z., W. Knoll, Soft Plasma Treated Surfaces: Tailoring of Structure and Properties for Biomaterial Applications. Plasma Processes and Polymers, 2005. 2(5): p. 351-372.
6. Zhang, Z., et al., Effect of aqueous solution on functional plasma polymerized films. Surface and Coatings Technology, 2003. 174-175: p. 588-590.
7. R. Förch, A.N.C., A. Bousquet, H. L. Khor, M. Jungblut, L. Q. Chu, Z. Zhang, I. Osey-Mensah, E.-K. Sinner, W. Knoll, Recent and Expected Roles of Plasma-Polymerized Films for Biomedical Applications. Chemical Vapor Deposition, 2007. 13(6-7): p. 280-294.
8. Chu, L.Q., R. Forch, and W. Knoll, Pulsed plasma polymerized maleic anhydride films in humid air and in aqueous solutions studied with optical waveguide spectroscopy. Langmuir, 2006. 22(6): p. 2822-2826.
9. Knoll, W., Interfaces and thin films as seen by bound electromagnetic waves. Ann. Rev. Phys. Chem., 1998. 49: p. 569-638.
10. Organikum. 2004, WILEY-VCH Verlag GmbH, Weinheim.
11. Chu, L.Q., H.Q. Mao, and W. Knoll, In situ characterization of moisture sorption/desorption in thin polymer films using optical waveguide spectroscopy. Polymer, 2006. 47(21): p. 7406-7413.
12. Chifen A. N. , J.A.T.A., Knoll W., Förch R., Adhesion Improvement of Plasma-Polymerized Maleic Anhydride Films on Gold Using HMDSO/O2 Adhesion Layers. Plasma Processes and Polymers, 2007. 4(9): p. 815-822.
13. Shankaran, D.R. and N. Miura, Trends in interfacial design for surface plasmon resonance based immunoassays. Journal of Physics D-Applied Physics, 2007. 40(23): p. 7187-7200.
14. Green, R.J., et al., Surface plasmon resonance for real time in situ analysis of protein adsorption to polymer surfaces. Biomaterials, 1997. 18(5): p. 405-413.
15. Hermanson, G.T., ed. Bioconjugate Techniques. 2nd ed. 1996, Elsevier Science, USA.
16. Kunkel, G., M. Mehrabian, and H. Martinson, Contact-site cross-linking agents. Molecular and Cellular Biochemistry, 1981. 34(1): p. 3-13.
17. Vogel, C.-W., Preparation of Immunoconjugates Using Antibody Oligosaccharide Moieties. 2004. p. 87-108.

Kapitel 4 – Ergebnisse und Diskussion
78
18. Johnsson, B., S. Löfås, and G. Lindquist, Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Analytical Biochemistry, 1991. 198(2): p. 268-277.
19. Su X., W.Y.-J., Robelek R., Knoll W., Surface Plasmon Resonance Spectroscopy and Quartz Crystal Microbalance Study of Streptavidin Film Structure Effects on Biotinylated DNA Assembly and Target DNA Hybridization. Langmuir, 2005. 21: p. 348-353.
20. Bannwarth, W. and R. Knorr, Formation of carboxamides with N,N,N',N'-tetramethyl (succinimido) uronium tetrafluoroborate in aqueous / organic solvent systems. Tetrahedron Letters, 1991. 32(9): p. 1157-1160.
21. Andersson, M., S. Oscarson, and L. Öhberg, Synthesis of oligosaccharides with oligoethylene glycolspacers and their conversion into glycoconjugates usingN,N,N′,N′-tetramethyl(succinimido)u roniu mtetrafluoroborate as coupling reagent. Glycoconjugate Journal, 1993. 10(6): p. 461-465.
22. Sinz, A., Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. Journal of Mass Spectrometry, 2003. 38(12): p. 1225-1237.
23. Liebermann, T. and W. Knoll, Surface-plasmon field-enhanced fluorescence spectroscopy. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2000. 171(1-3): p. 115-130.
24. Homola, J., Surface Plasmon Resonance Sensors for Detection of Chemical and Biological Species. Chem. Rev., 2008. 108(2): p. 462-493.

Kapitel 5 - Fazit
79
5 Fazit
In der vorliegenden Arbeit wurden die strukturellen Eigenschaften niedrig gepulster
Maleinsäureanhydrid-Plasmapolymere auf ihre Eignung als Biosensorunterlage untersucht.
Im Anschluss daran wurde eine Biosensorplattform durch kovalente Immobiliserung von
Anti-CRP-Antikörpern auf der pp-MA-Oberfläche zur nachfolgenden Detektion von CRP-
Molekülen entwickelt.
Die bei milden Prozessbedingungen synthetisierten pp-MA-Filme zeigten eine relativ raue,
hydrophile Oberfläche mit einer hohen Dichte an funktionellen Gruppen, die eine
nachfolgende chemische Modifizierung zur Anbindung unterschiedlichster Biomoleküle
ermöglichen. Die Stabilität der dünnen pp-MA-Filme in wässriger Lösung wurde in
kinetischen SPR-Aufnahmen untersucht. Hierbei konnte gezeigt werden, dass die dünnen pp-
MA-Filme trotz anfänglichem Materialverlust an unvernetzten Polymerfragmenten eine
stabile, kompakte Struktur aufweisen. Eine Vergleichsanalyse µm-dicker pp-MA-Filme in
wässriger Lösung zeigte, dass die Stabilisierung der Filmstruktur auf einen
Schichtdickenbereich von unter 100 nm begrenzt ist. Im Gegensatz zu den dünnen Filmen
zeigten die µm-dicken Filme poröse Strukturen, die in wässriger Lösung gelartig aufquellen.
Die Abweichungen im strukturellen Aufbau dünner und dicker pp-MA-Filme konnten mit der
unterschiedlich langen Plasmaexpositionszeit erklärt werden.
In nachfolgenden SPR-Experimenten konnte die chemische Immobilisierung von Anti-CRP-
IgG-Molekülen und farbstoffmarkierter Anti-Maus-IgG-Moleküle auf der Oberfläche dünner
pp-MA-Filme in situ verfolgt werden. Bei der kovalenten Verknüpfung der Antikörper mit
den Carbonsäureresten der hydrolysierten pp-MA-Filme wurde sich der Aktivesterchemie
bedient. Hierbei wurden verschiedene Crosslinkerreagenzien getestet, unter denen ein auf
EDC und Sulfo-NHS-basiertes System in wässriger Lösung die besten Ergebnisse erzielte.
Die Stabilität des pp-MA/IgG-Systems wurde durch mehrstündiges Spülen mit
Detergenzlösung demonstriert. Aus der Simulation der Resonanzkurven geht hervor, dass die
angebundenen Antikörper eine statistisch orientierte Monolage ausbilden.
Die pp-MA/Anti-CRP-Biosensorplattfom kann in nachfolgenden Testversuchen auf ihre
Effizienz bei der Detektion von CRP untersucht werden. Bei der entwickelten Biosensorbasis
handelt es sich um einen Prototypen, welcher weiter ausbaufähig ist.
Optimierungsmöglichkeiten in Bezug auf Belegungsdichte und Orientierung der Antikörper
auf der pp-MA- Oberfläche wurden vorgestellt.

Kapitel 5 - Fazit
80
Die Weiterentwicklung der pp-MA/Anti-CRP-Biosensorplattfom bis hin zum
funktionierenden CRP-Biosensor erfordert noch viel Arbeit, die jedoch im Hinblick auf das
hohe Anwendungspotential des Sensors im medizinischen Bereich lohnenswert ist.

81
6 Danksagung
In erster Linie gilt mein herzlichster Dank meinem Professor, Wolfgang Knoll, für die
freundliche Ermöglichung dieser Arbeit in seinem Arbeitskreis. Im Folgenden möchte ich
mich bei meiner Projektleiterin, Renate Förch, bedanken, für ihre Unterstützung und
Motivation und ihr jederzeit offenes Ohr für meine vielen Fragen und das schnelle
Korrekturlesen dieser Arbeit.
Weiterhin möchte ich mich bei Natalie Horn und Bernhard Menges für ihre Geduld und
Hilfestellung beim SPR-Messen sowie bei der Interpretation und Auswertung meiner
Ergebnisse bedanken.
Sascha Pihan möchte ich insbesondere für die fachmännische Einführung in die
Plasmatechnik, die Messung meiner AFM-Proben und das Korrekturlesen meiner Arbeit
danken. Weiterhin möchte ich an dieser Stelle noch Matthias Tamm und Sabir Okba nennen,
die mir bei diversen „Justageproblemen“ geholfen haben. Andreas Unger möchte ich
insbesondere für die Beantwortung all meiner physikalischen Fragen, für das Korrekturlesen
und die Hilfestellung beim UV-VIS-Spektrometer danken.
Bei Alex Lotz möchte ich mich für seine Hilfe und Geduld beim Formatieren dieser Arbeit,
Andreas Scheller und Marcus Schmelzeisen für ihre freundliche Unterstützung bei der
Behebung diverser Netzwerkprobleme und Walter Scholdei für seine Hilfe beim FT-IR-
Messen bedanken.
Mein herzlichster Dank gilt der Knoll-Gruppe für die freundliche Aufnahme in ihren Kreis,
besonders den Mitgliedern der „Plasmon Kitties“ für den unvergesslichen Spaß unserer
Trainingseinheiten und natürlich der Plasma Gruppe für die nette Arbeitsatmosphäre und
einen unvergesslichen Aufenthalt in Hirschegg.
Nicht zuletzt bedanke ich mich bei meinen Eltern, Stefan, Anne und Edith für ihre
unermüdliche Geduld und Motivation und ihren Glauben an mich und mein Projekt.

82
7 Abkürzungsverzeichnis
Abb. : Abbildung
AFM : Atomic Force Microscope
CRP: C-reactive Protein
DC: Duty cycle
FT-IR-Spektroskopie: Fourier-Transformations-Infrarot-Spektroskopie
HF-: Hochfrequenz-
IgG: Immunglobulin G
OWS: Optical Waveguide Spectroscopy
PECVD: Plasma enhanced Chemical Vapour Deposition
pp-HMDSO: plasmapolymerisiertes Hexamethyldisiloxan
pp-MA: plasmapolymerisiertes Maleinsäureanhydrid
SAM: Self-assembled-Monolayer
SPFS: Surface-Plasmon Field-enhanced Fluorescence Spectrocopy
SPRS: Surface Plasmon Resonance Spectroscopy
WaMS : Waveguide Mode Spectroscopy














