Argon-Matrix Isolation of Bis(carbomethoxy)thiirene:...
5
This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution 4.0 International License. Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung 4.0 Lizenz. Argon-Matrix Isolation of Bis(carbomethoxy)thiirene: Formation of Acyl and Carbalkoxythioketenes M. Torres, A. Clement, and 0. P. Strausz* Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2 This article is dedicated to Professor G. 0. Schenck on the occasion of his 70th birthday Z. Naturforsch. 88b, 1208-1212 (1983); received June 6, 1983 Thiirene, Thioketene, 1,2,3-Thiadiazole, Thio-Wolff Rearrangement Photolysis of argon-matrix isolated 4,5-bis(carbomethoxy)-l,2,3-thiadiazole with A = 254 or 265 nm resulted in the almost quantitative formation of bis(carbomethoxy)- thiirene (3g). Photolysis of 3g could only be carried out by long irradiation times at short wavelength A = 210nm and resulted mainly in extensive fragmentation instead of the formation of bis(carbomethoxy)thioketene, confirming earlier predictions that substituents, especially electron-withdrawing ones, should stabilize the thiirene ring. Methylcarboethoxy and methylacetylthioketene were obtained, however, in the argon-matrix photolysis or flow thermolysis of 4-methyl-5-carboethoxy- or 4-carboethoxy-5-methyl-l,2,3-thiadiazole and 4-acetyl-5-methyl-l,2,3-thiadiazole, respectively. Introduction The simplest examples of unsaturated hetero- cycles, oxirene, thiirene and 2-azirene, are highly unstable molecules whose syntheses have only been achieved recently in low temperature argon-matrices [1-3], although kinetic evidence for their transient existence had been available since the mid sixties [4, 5]. Their instability, apart from the inherent ring strain, has been generally attributed to their antiaromaticity whereby cyclic electron derealiza- tion raises the energy. Parallel to the kinetic and synthetic studies on these 4 n electron systems their stabilities have also been studied in recent years within the framework of molecular orbital theory. Large basis set ab initio type SCF-MO calculations, some including CI, have been carried out on the oxirene [6-9] and thiirene [10, 11] molecules along with their isomeric struc- tures. The computational results on the former lead to the conclusion that oxirene is indeed antiaro- matic and it is thermodynamically less stable than its ring opened diradical form. Thiirene on the other hand is somewhat more stable than its ring opened structure, but its C-S bond energy is markedly reduced compared to its saturated analog, the thiirane molecule. This phenomenon has been termed pseudoantiaromaticity and thiirene is there- fore a pseudoantiaromatic molecule [11]. Calcu- lations on 2-azirine, although indicative of pseudo- * Reprint requests to Prof. O. P. Strausz. 0340-5087/83/1000-1208/$ 01.00/0 antiaromaticity, have not yet been done with a similar degree of sophistication [4]. Using the photolysis of the two excellent source compounds 1,2,3-thiadiazoles and vinylene trithio- carbonates several thiirene molecule have been isolated in low temperature inert matrices in the past few years [1, 2; 12, 13]. Formation of thiirenes takes place simultaneously and in competition with the formation of thioketenes. Secondary photolysis of the parent thiirene and its trifluoromethyl deri- vative results in their rearrangement to ethynyl- thiol and trifluoromethylethynylthiol, respectively [1, 2], a rearrangement reminiscent of the cyclo- propene -> methylacetylene rearrangement [14]. However, except for bis(trifluoromethyl)thiirene which undergoes loss of sulfur with the generation of hexafluorobutyne-2 [12], photolysis of all the other thiirenes prepared thus far proceeds exclu- sively via Wolff-type rearrangements to thiokete- nes. Therefore the stability of thiirenes, as well as that of oxirenes and 2-azirines, should increase with substituents that have a low migrational aptitude, especially those that are also electron-withdrawing since these will lower the Tr-electron density on the carbon-carbon double bond and consequently rele- ase some of the "antiaromatic" character of the molecule. Good examples of these stabilizing effects are the cases of carboethoxymethylthiirene, tri- fluoromethylthiirene, and bis(trifluoromethyl)thi- irene [2] as well as the recent isolation of an inter- mediate [3] to which the bis (trifluoromethyl)-oxire- ne structure has been assigned.
Transcript of Argon-Matrix Isolation of Bis(carbomethoxy)thiirene:...
-
This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Argon-Matrix Isolation of Bis(carbomethoxy)thiirene: Formation of Acyl and Carbalkoxythioketenes
M. Torres, A. Clement, and 0 . P. Strausz* Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2
This article is dedicated to Professor G. 0. Schenck on the occasion of his 70th birthday
Z. Naturforsch. 88b, 1208-1212 (1983); received June 6, 1983
Thiirene, Thioketene, 1,2,3-Thiadiazole, Thio-Wolff Rearrangement Photolysis of argon-matrix isolated 4,5-bis(carbomethoxy)-l,2,3-thiadiazole with
A = 254 or 265 nm resulted in the almost quantitative formation of bis(carbomethoxy)-thiirene (3g). Photolysis of 3g could only be carried out by long irradiation times at short wavelength A = 210nm and resulted mainly in extensive fragmentation instead of the formation of bis(carbomethoxy)thioketene, confirming earlier predictions that substituents, especially electron-withdrawing ones, should stabilize the thiirene ring. Methylcarboethoxy and methylacetylthioketene were obtained, however, in the argon-matrix photolysis or flow thermolysis of 4-methyl-5-carboethoxy- or 4-carboethoxy-5-methyl-l,2,3-thiadiazole and 4-acetyl-5-methyl-l,2,3-thiadiazole, respectively.
Introduction
The simplest examples of unsaturated hetero-cycles, oxirene, thiirene and 2-azirene, are highly unstable molecules whose syntheses have only been achieved recently in low temperature argon-matrices [1-3], although kinetic evidence for their transient existence had been available since the mid sixties [4, 5]. Their instability, apart from the inherent ring strain, has been generally attributed to their antiaromaticity whereby cyclic electron derealiza-tion raises the energy.
Parallel to the kinetic and synthetic studies on these 4 n electron systems their stabilities have also been studied in recent years within the framework of molecular orbital theory. Large basis set ab initio type SCF-MO calculations, some including CI, have been carried out on the oxirene [6-9] and thiirene [10, 11] molecules along with their isomeric struc-tures. The computational results on the former lead to the conclusion that oxirene is indeed antiaro-matic and it is thermodynamically less stable than its ring opened diradical form. Thiirene on the other hand is somewhat more stable than its ring opened structure, but its C-S bond energy is markedly reduced compared to its saturated analog, the thiirane molecule. This phenomenon has been termed pseudoantiaromaticity and thiirene is there-fore a pseudoantiaromatic molecule [11]. Calcu-lations on 2-azirine, although indicative of pseudo-
* Reprint requests to Prof. O. P. Strausz. 0340-5087/83/1000-1208/$ 01.00/0
antiaromaticity, have not yet been done with a similar degree of sophistication [4].
Using the photolysis of the two excellent source compounds 1,2,3-thiadiazoles and vinylene trithio-carbonates several thiirene molecule have been isolated in low temperature inert matrices in the past few years [1, 2; 12, 13]. Formation of thiirenes takes place simultaneously and in competition with the formation of thioketenes. Secondary photolysis of the parent thiirene and its trifluoromethyl deri-vative results in their rearrangement to ethynyl-thiol and trifluoromethylethynylthiol, respectively [1, 2], a rearrangement reminiscent of the cyclo-propene -> methylacetylene rearrangement [14]. However, except for bis(trifluoromethyl)thiirene which undergoes loss of sulfur with the generation of hexafluorobutyne-2 [12], photolysis of all the other thiirenes prepared thus far proceeds exclu-sively via Wolff-type rearrangements to thiokete-nes. Therefore the stability of thiirenes, as well as that of oxirenes and 2-azirines, should increase with substituents that have a low migrational aptitude, especially those that are also electron-withdrawing since these will lower the Tr-electron density on the carbon-carbon double bond and consequently rele-ase some of the "antiaromatic" character of the molecule. Good examples of these stabilizing effects are the cases of carboethoxymethylthiirene, tri-fluoromethylthiirene, and bis(trifluoromethyl)thi-irene [2] as well as the recent isolation of an inter-mediate [3] to which the bis (trifluoromethyl)-oxire-ne structure has been assigned.
-
M. Torres et al. • Bis(carbomethoxy)thiirene: Acyl and Carbalkoxythioketenes 1209
Thioketenes have been prepared in increasing number since the first reported synthesis of thio-ketene by Howard in 1962 [15]. However, essen-tially all the current methods of synthesis employed [16-20] have only been successful for the prepara-tion of alkyl or aryl substituted thioketenes, scheme 1 (R = alkyl or aryl) and most of them
R ^ S y ^ R R - C = C = S ( 1 )
R — r — n p . S ; c=c = o 10 - ^C = C = S R
^C = C = S
; c=c=s
(2)
(3)
(4)
(5)
(6)
involve flash thermolysis of the source compound, eq. (3) to (5). Attempts to prepare acyl or carbal-koxythioketenes by these methods have failed and instead COS and alkynes were obtained, most likely through the mechanism [19]:
RCO R' /
Rx
> ^ c " C 0 S RC=CR' (7)
Experimental
An Air Products Model CS-202 Displex, closed-cycle helium refrigeration system was employed. The shroud was equipped with two cesium iodide windows and two spectrosil quartz windows and the optical cell was connected to a standard high vacuum apparatus. The matrix deposit was made on a cesium iodide plate for the IR studies, and on a spectrosil plate for UV studies. The light source was a Hanovia 30,620 medium pressure mercury arc aquipped with either a 7910 Vycor filter, a 265 or a 310 nm interference filter. Alternatively, photolyses at 254 nm were carried out with a Hanovia low-pressure mercury resonance lamp equipped with a 7910 Vycor filter.
Flow thermolyses were carried out in a 6 mm ID X 100 mm quartz tube wrapped with a heating filament, maintained at the thermolysis temperature and placed before the cryostat. The temperature was monitored by an iron versus constantan thermo-couple. The residence time at the deposition flow rate was measured to be ~0.7 sec.
A Varian Aerograph (90 P) GC was employed for preparative purposes and a Hewlett Packard 5750 instrument for analysis. IR spectra of matrix isolated species were obtained in a Nicolet 7199 FT infrared spectrophotometer at 1 cm - 1 resolution and the UV spectra on a Unicam SP 800 B ultraviolet spectrophotometer.
NMR spectra were obtained in either a Bruker WP-60, Bruker 100 or Bruker 200 MHz instruments equipped with proton (XH) or carbon (13C) probes. TMS was used as internal standard and chemical shift values are given in the d scale for LH NMR. CDCI3 was used as solvent and a internal standard for 13C NMR and chemical shifts are given in ppm.
Matheson UHP argon and commercial research grade chemicals were used without further purifica-tion. 1,2,3-thiadiazoles l a - f were prepared accord-ing to known methods [19, 21-23] and their spectral data are given in Tables I, II and III. 4-f-Butyl-l,2,3-thiadiazole-5-d (60%) was prepared from 3,3-dimethyl-2-butanone-l-d3 (60%) obtained from 3,3-dimethyl-2-butanone by exchange with D20/Na0H.
Table I. *H NMR of 1,2,3-thiadiazoles (1).
Flow thermolysis with trapping of the products in a low temperature IR cryostat, although involv-ing a longer residence time, has the advantage of allowing better monitoring of the thermolysis tem-perature. We would like to report here the prepara-tion and rearrangements of some thioketenes genera-ted by flow thermolysis and also by argon-matrix photolysis of 1,2,3-thiadiazoles and, in the search for more stable thiirenes, the synthesis of bis(carbo-methoxy)thiirene.
R R' Chemical shift (6)
l a H C(CH3)3 1.50(s) 9H; 8.12(s) 1H l b C(CH3)3 H 1.48(s) 9H; 8.37(s) 1H l c H H AB system centered at 8.8 l d COOEt CH3 1.38(t) 3H; 2.95(s) 3H;
4.38g (2H) l e CH3 COOEt 1.47(t) 3H; 2.90(s) 3H;
4.48g (2H) 1! CH3 CH3CO 2.83(s) 3H; 2.88(s) 3H H CH3OCO CH3OCO 4.0(s) 3H; 4.1 (s) 3H
-
1210 M. Torres et al. • Bis(carbomethoxy)thiirene: Acyl and Carbalkoxythioketenes 1210
Table II. 13C NMR of 1,2,3-thiadiazoles (1).
R R' Chemical shift (ppm) C(4) C(5) alkyl groups CO
la H C(CH3)3 173.4 128.9 33.8, 30.7 _ lb C(CH3)3 H 170.0 144.8 33.4, 32.7 lc H H 147.9 136.2 - — Id COOEt CH3 159.8 139.6 62.5, 14.2, 13.9 162.2 l e CH3 COOEt 159.9 150.3 61.9, 14.3, 11.0 160.9 lf CH3 COCH3 159.4 156.4 29.9, 11.14 193.7 lg CH3OCO CH3OCO 159.1 154.0 54.1, 53.5 160.1
Table III. Mass spectra of 1,2,3-thiadiazoles (1).
R R' m/e (% M+)*
la H C(CH3)3 114(8), 113(57), 99(100), 81(12), 79(13), 65(19), 57(9) D (60%) C(CH3)3 115(11), 114(63), 113(51), 100(100), 99(91), 82(12), 81(12), 80(11), 79(14), 66(16),
65(20), 58(9), 57(15) lb C(CH3)3 H 127(22), 114(5), 113(15), 99(47), 57(80), 41(100), 39(71) lc H H 86(23), 58(100), 57(25), 45(7), 32(10) Id COOEt CH3 172(6), 144(35), 127(17), 72(55), 71(55), 45(41), 29(100) l e CH3 COOEt 172(29), 144(23), 127(16), 72(32), 71(66), 45(34), 29(100) l f CH3 COCHg 142(3), 114(27), 72(36), 71(27), 43(100) 1 g COOMe COOMe 202(8), 174(48), 143(22), 99(18), 85(44), 84(55), 71(21), 59(100), 45(30)
* At 70 eV ionization potential.
Procedure For samples having sufficient vapour pressure at
room temperature (i.e. > 2 torr), the sample and the host (argon) were mixed together in a reservoir and stirred constantly. The mixture was then alloAved to enter the cell and deposited on the plate through a nozzle positioned in front of the plate and kept at 10 K throughout the deposition. The rates of deposition were measured by means of a rota-meter, typical deposition rates being 27 /zmolmin -1 and the ratios of guest to host were between 1:300 to 1:700. Similarly, thermolyses were carried out by passing the argon-sample mixture through the heated thermolysis furnace and then condensed on the cryostat plate previously cooled at 10 K. Samples having low vapour pressures were placed in a finger trap close to the cell and warmed to 313-328 K ; they were then swept into the cell by a flow of argon and monitored as before. Irradiation of the matrix isolated samples was followed by I R or UV, and was carried out until the spectrum of the course compound had disappeared.
Results and Discussion
The various substituted thiadiazoles, thioketenes and thiirenes examined are summarized below.
Photolysis (A 265 nm) of argon-matrix isolated 4 or 5-£-butyl-l,2,3-thiadiazole, l a or l b respectively, at 10 K, resulted in the formation of £-butylthio-ketene 2 a as identified by IR spectroscopy
:c=c=s V a b c d e f g
R H C(CH3)3H COOEt CH3 CH3 COOCH3 R 'C(CH3)3H H CH3 COOEt COCH3 COOCHS
(2974(ms), 2955(m), 1777(vs), 1764(vs), 1478(m), 1368(s), 1364(s), 1242(B), 1163(m), 954(m), 667(m), 385(w) cm - 1) . No f-butylthiirene could be detected and at the present time this is the first 1,2,3-thia-diazole that has failed to yield thiirene in a 10 K argon-matrix photolysis. Seybold and Heilb [19] reported the synthesis of 2 a by flash thermolysis of l a at 773-873 °K. In our flow thermolysis experiments however, thermolysis of either l a or l b at 813 °K resulted instead in the formation of isobutylene and parent thioketene 2 c, identified by comparison with the IR spectrum of an authen-tic sample of isobutylene under the same conditions and the I R spectrum of 2 c obtained by flow thermo-lysis of 1,2,3-thiadiazole ( lc ) at 703 °K. Flow thermolysis of l a and l b at lower temperatures, 623 and 723 °K respectively, did result in the for-mation of £-butylthioketene 2 a which has an IR spectrum identical to the one obtained in the photo-
-
M. Torres et al. • Bis(carbomethoxy)thiirene: Acyl and Carbalkoxythioketenes 1211
lysis of l a or l b . In a different approach, flow thermolysis of l a using two consecutive furnaces at 623 and 813 °K respectively resulted in the same decomposition to isobutylene and thioketene indi-cating that the decomposition takes place in the thioketene 2a rather than in l a and most likely via an ethynylthiol intermediate:
C H - H CH, V 2 t > 8 1 3 ° K r -1 C H C , > = C = S , HC=CSH
H -(CHJ)2C=CH2 L J 2A H 2 C=C=S
2C The mass spectra of 1 a, 1 b or 2 a all show a peak
at m/e 113 (M-N2 -H; M-N 2 -H, M-H, respectively). The mass spectrum of 4-£-butyl-l,2,3-thiadiazole-5-d, Table III, revealed that the loss of hydrogen originates at the t-butyl group, indicating that this hydrogen is labile, and thus lends support to the proposed mechanism for the decomposition.
We have already reported [2 a] that photolysis of argon-matrix isolated 4-methyl-5-carbethoxy-1,2,3-thiadiazole ld or 4-carbethoxy-5-methyl-1,2,3-thiadiazole 1 e at A = 265 nm results in the formation of methylcarbethoxythiirene, 3d, char-acterized by a C = C st at 1875 cm - 1 . Subsequent photolysis at X > 210 nm converted 3d into methyl -carbethoxythioketene 2d, characterized by its 1785 cm - 1 C = C st absorption, which represents the first example of the isolation of a carbalkoxy sub-stituted thioketene. Flow thermolysis of ld at 683 °K resulted in the formation of 2d, identified by comparison with the previously obtained I R spectrum of 2d, along with some decomposition products, among them COS (2060 cm - 1) . No clear evidence for the formation of CH3C = COEt could be obtained. At 643 °K, however, flow thermolysis of ld resulted mainly in the formation of thioketene 2d and only traces of decomposition products were observed, together with small quantities of ld.
Similarly, we have already described [13] the formation of methylacetyl-thiirene (3f) and its subsequent photolysis to methylacetylthioketene (2f) in the photolysis of argon-matrix isolated 4-acetyl-5-methyl-l,2,3-thiadiazole ( l f ) . We have also been able to obtain 2 f in the 633 °K flow ther-molysis of l f (IR 2980(broad), 2940(w), 1760(s), 1680(vs), 1250(m), 1240(s), 1140(m), 960(m), 660(w), 575(w) cm"1). At 863 CK, however, in addition to
2f, COS (2060 cm-1) and 2-butyne (identified by comparison with the IR spectrum of an authentic sample under the same conditions) were detected in agreement with the proposed mechanism, eq. (7).
The known resistance of the C02Me group to migrate in the Wolff rearrangement and its excep-tional electron-withdrawing properties made bis-(carbomethoxy)thiirene a good candidate in our search for more stable thiirenes.
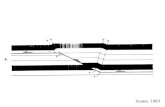
Photolysis of 4,5-bis(carbomethoxy)-l,2,3-thia-diazole ( lg ) , Fig. l a , isolated in an argon-matrix at 10 K with X = 265 nm or 254 nm radiation resulted in the formation, almost exclusively, of bis(carbomethoxy)thiirene (3g), Fig. l b , having significant absorptions at 1850 ( C = C st) and 1740 ( C = 0 st) cm - 1 . Conversion was essentially quan-titative and only traces of bis(carbomethoxy)thio-ketene (2g) could be observed. Subsequent photo-lysis of the photolyzate at X = 310 nm resulted in no change in the IR spectrum indicating that bis-(carbomethoxy)thiirene 3g is stable at this wave-length. 3g could be photolyzed very slowly at X > 210 nm and only after prolonged photolysis (16 h) had most of the thiirene disappeared. Some thioketene 2g ( C = C s t 1750 cm-1) was detected
2OO5 isbo i6oo [Too i2bo looo e5ö iEo "Too WAVENUMBERS
Fig. 1. 2000-400 cm-1 FTIR spectrum of argon-matrix isolated 4,5-bis(carbomethoxy)-l,2,3-thiadiazole (lg) at 10 K ; a) before irradiation; b) after 45 min irradiation, X — 254 nm.
-
1212 M. Torres et al. • Bis(carbomethoxy)thiirene: Acyl and Carbalkoxythioketenes 1212
but extensive fragmentation had also taken place, as indicated by strong absorptions at 2340(C(>2), 2240, 2140(00) and a weaker one at 2060(COS) cm-*. An absorption at 2200 cm - 1 was observed by Sey-bold and Heilb in their attempts to obtain thio-ketene 2 g by flash thermolysis of 1 g, and this was tentatively assigned to CH3OC = CCOOCH3 [19].
In our case, however, fragmentation does not appear to be due to the secondary photolysis and rearrangement of thioketene 2 g but rather to the photostability of thiirene 3g with regard to the thiirene-thioketene rearrangement. Thus, prolonged photolysis of acyl or carbalkoxythioketenes 2 d and 2f in an argon-matrix did not lead to loss of COS.
No thioketene (2g) could be generated by flow-thermolysis of l g in the range 565-651 °K and only decomposition products were obtained.
When 1 g was argon-matrix isolated and deposited on a spectrosil plate, photolysis under the same conditions as described above allowed the obser-vation of a UV spectrum assigned to thiirene 3g, showing absorptions at Amax 240, 290, and an ab-sorption at 460 nm, probably due to traces of thio-ketene 2g, was also observed.
The high photostability of 3g resembles that of bis(trifluoromethyl)thiirene and must be a conse-quence of the low migrational aptitude of the carbalkoxy groups. This molecule is the most stable thiirene prepared to date and attempts to prepare it at higher temperatures are currently underway.
We thank the Natural Sciences and Engineering Research Council for continuing financial support, Dr. E. M. Lown for helpful discussions and R. Swindlehust and J. Hoyle for assistance in taking the FTIR spectra.
[1] J. Laureni, A. Krantz, and R. H. Hajdu, J. Am. Chem. Soc. 98, 7872 (1976); A. Krantz and J. Laureni, J. Org. Chem. 44, 2730 (1979); J. Am. Chem. Soc. 103, 486 (1981).
[2] a) M. Torres, A. Clement, J. E. Bertie, H. E. Gunning, and O. P. Strausz, J. Org. Chem. 43, 2490 (1978); M. Torres, E. M. Lown, and O. P. Strausz, Heterocycles 11, 697 (1978); b) M. Torres, I. Safarik, A. Clement, J. E. Bertie, and O. P. Strausz, Nouv. J. Chim. 3, 365 (1979); I. Safarik, M. Torres, and O. P. Strausz, Chem. Phys. Lett. 72, 388 (1980).
[3] M. Torres, J. L. Bourdelande, A. Clement, and O. P. Strausz, J. Am. Chem. Soc. 105, 1698 (1983).
[4] For a recent review see: M. Torres, E. M. Lown, H. E. Gunning, and O. P. Strausz, Pure Appl. Chem. 52, 1623 (1980).
[5] O. P. Strausz, J. Font, E. L. Dedio, P. Kebarle, and H. E. Gunning, J. Am. Chem. Soc. 89, 4805 (1967); O. P. Strausz, Pure Appl. Chem. 4, 165 (1971); J. Font, M. Torres, H. E. Gunning, and O. P. Strausz, J. Org. Chem. 43, 2487 (1978).
[6] I. G. Csizmadia, H. E. Gunning, R. K. Gosavi, and O. P. Strausz, J. Am. Chem. Soc. 95, 133 (1973); O. P. Strausz, R. K. Gosavi, A. S. Denes, and I. G. Csizmadia, J. Am. Chem. Soc. 98, 4784 (1976); O. P. Strausz, R, K. Gosavi, and H. E. Gunning, Chem. Phys. Lett. 54, 510 (1978).
[7] C. E. Dykstra, J. Chem. Phys. 68, 4244 (1978). [8] K. Tanaka and M. Yoshimine, J. Am. Chem. Soc.
102, 7655 (1980). [9] W. J. Bouma, R. H. Nobes, L. Radom, and C.
Woodward, J. Org. Chem. 47, 1869 (1982).
[10] M. Yoshimine and J. Pacansky, J. Chem. Phys. 78, 1384 (1983).
[11] R. K. Gosavi and O. P. Strausz, Can. J. Chem., in press.
[12] M. Torres, A. Clement, H. E. Gunning, and O. P. Strausz, Nouv. J. Chim. 3, 149 (1979).
[13] M. Torres and O. P. Strausz, Nouv. J. Chim. 4, 703 (1980).
[14] K. B. Wiberg and W. J. Bartley, J. Am. Chem. Soc. 82, 6375 (1960).
[15] E. G. Howard, U. S. Pat. 3035030 (C. A. 57, 13617f (1962)).
[16] M. S. Raasch, Chem. Commun. 1966, 577; J. Org. Chem. 35, 3470 (1970); ibid. 37, 1347 (1972).
[17] A. Krantz and J. Laureni, J. Am. Chem. Soc. 96, 6768 (1974).
[18] E. U. Elam, F. H. Rash, J. T. Dougherty, V. W. Goodlett, and K. C. Brannock, J. Org. Chem. 33, 2738 (1968).
[19] G. Seybold and C. Heilb, Angew. Chem. Int. Ed. Engl. 14, 248 (1975); idem., Chem. Ber. 110, 1225 (1977).
[20] E. Schaumann and F. Grabley, Liebigs Ann. Chem. 1979, 1702; idem. 1979, 1715; E. Schauman, J. Ehlers, and H. Mrotzek, ibid. 1979, 1734.
[21] C. D. Hurd and R, I. Mori, J. Am. Chem. Soc. 77, 5359 (1955).
[22] U. Schmidt, E. Heymann, and K. Kabitzke, Chem. Ber. 96, 1478 (1963).
[23] S. J. Rämsby, S. O. 0 . Green, S. B. Ross, and W. E. Stjernstrom, Acta Pharm. Suecica 10, 285 (1973).
[24] H. Wieland and S. Bloch, Chem. Ber. 39, 1488 (1906).