
Charakterisierung der Verteilungen in präparativen...
135
Charakterisierung der Verteilungen in präparativen chromatographischen Säulen vorgelegt von Dipl.-Ing. Tobias Laiblin aus Berlin Von der Fakultät III - Prozesswissenschaften - der Technischen Universität Berlin zur Erlangung des akademischen Grades Doktor der Ingenieurwissenschaften - Dr.-Ing. - genehmigte Dissertation Promotionsausschuss: Vorsitzender: Prof. Dr.-Ing. M. Kraume Gutachter: Prof. Dr.-Ing. W. Arlt Gutachter: Prof. Dr.-Ing. G. Wozny Tag der wissenschaftlichen Aussprache: 03. Mai 2002 Berlin 2002 D83
Transcript of Charakterisierung der Verteilungen in präparativen...

Charakterisierung der Verteilungen in präparativenchromatographischen Säulen
vorgelegt von
Dipl.-Ing. Tobias Laiblin
aus Berlin
Von der Fakultät III - Prozesswissenschaften -
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Ingenieurwissenschaften
- Dr.-Ing. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr.-Ing. M. Kraume
Gutachter: Prof. Dr.-Ing. W. Arlt
Gutachter: Prof. Dr.-Ing. G. Wozny
Tag der wissenschaftlichen Aussprache: 03. Mai 2002
Berlin 2002
D83

Diese Seite ist leer

Danksagung
Danksagung
Diese Arbeit ist während meiner Tätigkeit als wissenschaftlicher Mitarbeiter am Fach-gebiet Thermodynamik und thermische Verfahrenstechnik der Technischen UniversitätBerlin entstanden.
Bei meinem Doktorvater Prof. Dr.-Ing. W. Arlt möchte ich mich für das anspruchsvollePromotionsthema, die Freiheiten in der Gestaltung der Arbeit, die vielen anregendenDiskussionen und die Förderung während der Promotion bedanken. Auch für dieMöglichkeit, die englischen Thermodynamikvorlesungen zu betreuen, bin ich ihm sehrdankbar.
Danken möchte ich meiner Frau Andrea, die mir die nötigen Freiräume für diese Arbeitgelassen hat, mir jederzeit Motivation gespendet hat und immer zu Diskussionen überdie Arbeit bereit war.
Für die ausgezeichnete Zusammenarbeit auf allen Gebieten dieses Themas möchte ichmich bei Henning Boysen bedanken. Die Zusammenarbeit mit ihm hat bis zum Endemeiner Zeit an der TU Berlin sehr viel Spaß gemacht und war sowohl in fachlicher wiein breitensportlicher Hinsicht sehr erfolgreich.
Für die ausgezeichnete Zusammenarbeit in der Vorbereitung der Arbeit (DFG-Antrag,Forschungs- und Untersuchungsschwerpunkte) und beim Aufbau der ersten Mess-apparaturen möchte ich mich bei Mark Lisso bedanken. Für die erfolgreicheVeröffentlichung und ausgezeichnete Zusammenarbeit in der Bestimmung derTemperaturverteilungen danke ich Yanxia Wu.
Carsten Jork möchte ich für seine Arbeiten für mich im Rahmen seiner Diplomarbeitdanken, da diese in Umfang und Qualität neue Maßstäbe gesetzt hat und damitentscheidenden Anteil am Gelingen der Experimente zum Referenzsystem hatte.
Sehr herzlich möchte ich mich bei meinen Mitarbeitern Thinh Nguyen-Xuan, SonjaGiesen und Johannes Gerhard bedanken, deren motiviertes Arbeiten und große Eigen-initiative mir immer eine große Hilfe gewesen sind. Dieser Dank gilt auch meinenStudien- und Diplomarbeitern Peter Schaarschuh, Mina Kavarnou, Cordin Arpagausund Andreas Hoppe. Ohne sie wäre die Arbeit nicht in diesem Umfang möglichgewesen.
Herrn Dr-med. R. Kaiser und Rainer Kirsch möchte ich für die angenehme undmotivierte Zusammenarbeit bei der Messung der Geschwindigkeitsprofile danken, diesich eigentlich immer ausserhalb der normalen Arbeitszeit abgespielt hat. Ohne siewären diese Experimente nicht durchführbar gewesen.
York Beste und Konstantin Lenz möchte ich für die erfolgreiche Arbeit innerhalb derChromatographiegruppe am Institut danken, die mir immer Anregungen und Verbes-serungsmöglichkeiten gebracht hat.
Für die Lösung kleinerer und größerer Probleme, für die andauernde Unterstützung unddas ausgezeichnete Arbeitsklima möchte ich mich bei meinen Kollegen Andreas

Danksagung
Böhme, Oliver Spuhl, Feelly Tumakaka, Marko Tischmeyer, Thomas Schneider, GötzFischer, Matthias Seiler, Steffi Hiller und Irina Smirnova bedanken. Weiter gilt meinDank Herrn Stübing, Lothar, Klaus, Shorty, Dietmar und Uwe in der Werkstatt,Christina, Susanne, Sylva und Herrn Schmidt im Labor und Birgit und Manuela imSekretariat. Die Feste und Feiern am Institut sind unerreicht und die Promotionsfeiernimmer wieder ein Genuß.
Prof. Dr.-Ing. G. Wozny möchte ich für die durch gute Zusammenarbeit der beidenLehrstühle entstandenen Anregungen und Diskussionen und für die Tätigkeit als zweiterGutachter danken.
Herrn Prof. Dr.-Ing. M. Kraume danke ich für die Übernahme des Vorsitzes desPromotionsausschusses.
Bei meinen Eltern möchte ich mich für die Unterstützung und Förderung währendmeines gesamten Lebens bedanken, ohne die ich es nicht soweit geschafft hätte.

Abstract
Abstract
Diese Arbeit beschäftigt sich mit der Charakterisierung der Verteilungen inchromatographischen Festbetten durch Bestimmung des anlagenspezifischen Anteils deraxialen Dispersion als integrales Maß der Verteilung einerseits und durch Messung derVerteilungen von Temperatur und Geschwindigkeit andererseits.
Je nach Zielsetzung in der Modellierung von chromatographischen Säulen werdenunterschiedliche Ansätze gewählt: Für die grobe Auslegung eines Trennprozesses fürbestehende Systeme ist normalerweise eine Lösung der axialen Komponentenbilanz derSäule ausreichend. Eine zwei- oder höherdimensionale Modellierung wird für den Falleiner zusätzlich zu betrachtenden radialen Verteilung, wie z.B. beim Design dergesamten Anlage oder ihrer einzelnen Komponenten (z.B. neuartiger Verteilersysteme),eingesetzt. Für beide Ansätze sind experimentelle Daten zur Auslegung bzw.Überprüfung der Modelle notwendig.
Zur Verbesserung der eindimensionalen, axialen Modellierung wird eine Unterteilungder axialen Dispersion in einen Anteil, der durch die stationäre Phase bestimmt wird,und einen anlagenspezifischen Anteil vorgenommen. Diese Unterteilung ermöglicht es,durch Versuche mit einem Referenzsystem in den verschiedenen chromatographischenAnlagen den Anteil der Anlagenkomponenten, wie z.B. der Ventile, Messfühler undFritten, an der axialen Dispersion zu bestimmen. Mit Hilfe dieses Ansatzes werden indieser Arbeit Versuche mit verschiedenen stationären Phasen (Glaskugeln, Uetikon,Kromasil) in zwei unterschiedlichen Anlagen durchgeführt. Modellierungen ingPROMS liefern die unterschiedlichen Anteile der axialen Dispersion für beideAnlagen. Es zeigt sich, dass ein anlagenspezifischer Anteil der axialen Dispersionexistiert, der innerhalb jeder einzelnen Anlage für ähnliche stationäre Phasen gleich ist.Mit Hilfe dieses Anteils wird die Übertragung von Trennprozessen z.B. aus kleinenLaboranlagen auf präparative Anlagen verbessert, da zusätzlich zum Einfluss derstationären Phase nun auch der Einfluss der Anlagenkomponenten berücksichtigtwerden kann.
Für den Fall der mehrdimensionalen Modellierung in FLUENT sind in dieser Arbeit alsValidierung Methoden zur Messung der radialen Temperatur- und Geschwindigkeits-verteilung in chromatographischen Säulen entwickelt worden. Es wird ein System zurMessung der Temperaturen an verschiedenen radialen Positionen bei gleicher axialerPosition aufgebaut. Mit diesem System werden die Temperaturprofile in drei stationärenPhasen (Glaskugeln, Uetikon, Kromasil) mit Methanol als Eluent für verschiedeneUnterkühlungen des Eluenten gemessen und der Einfluss der Unterkühlung auf dasPeakverhalten untersucht. Mit Hilfe dieser Ergebnisse konnte das in FLUENTimplementierte Modell erfolgreich validiert werden. Zur Messung der Geschwindig-keiten in der Säule als weitere Validierung wird die Einsatzmöglichkeit einesmedizinischen Magnet-Resonanz-Tomographen untersucht. In einer portablen, metall-frei gestalteten HPLC werden die Geschwindigkeiten an verschiedenen radialenPositionen in einer Schicht quer zur Säule bestimmt. Die Ergebnisse stimmen mit den inFLUENT berechneten überein.

Abstract

Inhaltsverzeichnis I
Inhaltsverzeichnis
1 EINLEITUNG 1
2 GRUNDLAGEN 3
2.1 Grundlagen des Energie-, Impuls- und Stofftranspor ts in Festbetten 3
2.1.1 Grundlegende Transportgleichungen 3
2.1.2 Massenerhaltung / Kontinuitätsgleichung 4
2.1.3 Impulsbilanz 5
2.1.4 Energiebilanz 7
2.1.5 Komponentenbilanz 10
2.1.6 Gleichungen für die HPLC 10
2.1.6.1 Druckverlust 12
2.1.6.2 Eindimensionale Komponentenbilanz 12
2.2 Grundlagen der Chromatographie 13
2.2.1 Chromatographische Kenngrößen 14
2.2.2 Porosität 17
2.2.3 Adsorptionsisothermen 18
2.2.4 Höhe einer Übertragungseinheit (Van Deemter Kurve) 21
2.2.5 Axiale Dispersion 22
2.2.6 Stofftransportwiderstand 24
2.2.7 Verfahrenstechnische Konzepte und Anlagen 25
2.2.7.1 Diskontinuierliche Verfahren 26
2.2.7.2 Kontinuierliche Verfahren 26
2.3 Scale-Up M ethoden für die Batch-Chromatographie 27
2.3.1 Methode nach Lode et al. 27
2.3.2 Methode nach Heuer und Seidel-Morgenstern 28
2.3.3 Methode nach Dwyer 30
2.3.4 Bewertung der Methoden 31
2.4 Magnet-Resonanz-Tomographie (M RT) 31
2.5 M essung von radialen Ver teilungen 35
2.5.1 Temperaturprofile 35
2.5.2 Konzentrationsprofile 36
2.5.2.1 Modellierung in FLUENT 40
2.5.3 Geschwindigkeitsprofile 41
3 VERWENDETE SYSTEME / APPARATUREN 43
3.1 Stationäre Phasen 43

II Inhaltsverzeichnis
3.1.1 Glaskugeln 43
3.1.2 Uetikon 44
3.1.3 Kromasil 45
3.1.4 Bestimmung der Porositäten 46
3.1.4.1 Messung der Gesamtporosität 46
3.1.4.2 Messung der internen Porosität 47
3.1.4.3 Messung der externen Porosität 48
3.1.4.4 Übersicht der Porositäten 48
3.2 Semipräparative HPLC 48
3.2.1 Allgemeine Beschreibung 48
3.2.2 Messtechnik 49
3.2.3 Packen der Säulen 50
3.2.4 Standardsystem 52
3.2.5 System zur Messung der Geschwindigkeitsprofile 52
3.2.6 Kernspintomograph 53
3.3 Präparative HPLC 53
3.3.1 Allgemeine Beschreibung 53
3.3.2 Messtechnik 55
3.3.3 Packen des NW100 56
3.3.4 Standardsystem 57
3.3.5 System zur Messung der Temperatur-Profile 57
4 MODELLIERUNG DER HPLC-SÄULEN 61
4.1 Eindimensionales axiales M odell (gPROM S) 61
4.2 Rigoroses Modell (FLUENT) 63
4.2.1 Grundsätzliches Vorgehen 64
4.2.2 Beschreibung des in FLUENT verwendeten Modells 65
5 BESTIMMUNG DER ANTEILE DER AXIALEN DISPERSION 69
5.1 Exper imente 70
5.1.1 System Glaskugeln - Methanol - Diethylphthalat 72
5.1.2 System Uetikon - Methanol - Diethylphthalat 73
5.1.3 System Kromasil - Methanol - Toluol 74
5.1.4 System Kromasil - n-Heptan - Toluol 75
5.1.5 Vergleich der experimentellen Ergebnisse 76
5.2 Modellierung in gPROM S 77
5.2.1 Vergleich der Peaks in Experiment und Modellierung 78
5.2.2 Ergebnisse für die Systemparameter 79
5.2.3 Ergebnis für die axiale Dispersion 80

Inhaltsverzeichnis III
5.2.4 Auslegung von HPLC-Systemen 82
6 MESSUNG DER RADIALEN VERTEILUNGEN 85
6.1 Temperaturprofile 85
6.1.1 Einfluss der Pt100 85
6.1.2 Säule Prepbar2: System Uetikon, Methanol, Toluol 88
6.1.3 NW100: Glaskugeln - Uetikon - Kromasil, Methanol, Diethylphthalat 93
6.1.4 Vergleich der Prepbar2 und des NW100 96
6.1.5 FLUENT 98
6.2 Geschwindigkeitsprofile 100
6.2.1 Experimentelle Ergebnisse 101
6.2.2 Bewertung der MRT-Ergebnisse 105
6.2.3 Modellierung in FLUENT 105
6.3 Ergebnis der M essungen 106
7 ZUSAMMENFASSUNG UND AUSBLICK 107
7.1 Zusammenfassung 107
7.2 Ausblick 108
8 FORMELZEICHEN, ABKÜRZUNGEN, KENNZAHLEN 111
8.1 Formelzeichen 111
8.2 Gr iechische Formelzeichen 114
8.3 Indizes 115
8.4 Abkürzungen 115
8.5 Kennzahlen 116
8.6 Tensorschreibweise 116
9 LITERATURVERZEICHNIS 117
10 ANHANG 123
10.1 GPROM S® Code zur Lösung der axialen Komponentenbilanz 123
10.2 GPROM S® Code zum Parameter fitting 125

IV Inhaltsverzeichnis

1 Einleitung 1
1 Einleitung
Die Flüssigchromatographie ist ein thermisches Trennverfahren, in dem zweiHilfsphasen (eine feste und eine flüssige Phase) dazu genutzt werden, besondersähnliche Substanzen zu trennen. Die Trennung der Substanzen kann bei niedrigenTemperaturen erfolgen und ermöglicht so eine schonende Trennung. Durch Auswahlder Hilfsphasen bzw. im Fall der festen Phasen durch deren spezielles Design kann dieChromatographie an jede Trennaufgabe angepasst werden.
Dieses Verfahren findet in Industrie und Wissenschaft immer dann Anwendung, wenndie anderen, "einfacheren" thermischen Trennverfahren wegen eines zu geringenTrennfaktors keine Trennung mehr erreichen, z.B. bei der Isomerentrennung, oder wenndie Produkte sehr temperaturempfindlich sind.
Die Chromatographie ist jedoch komplexer und aufwendiger als die anderen Trenn-verfahren, da die Produkte als Lösung im Eluent gewonnen werden und anschließendaufbereitet werden müssen. Diese höheren Kosten werden durch die häufig sehr hohenWertschöpfungen der Produkte gerechtfertigt. Aus diesem Grund hat sich dieChromatographie speziell in der pharmazeutischen Industrie etabliert.
Die zur Beschreibung der chromatographischen Trennung notwendige Thermodynamikwird in einer axialen Komponentenbilanz über die chromatographische Säule berück-sichtigt. Durch Lösung dieser Bilanz können die Peakverläufe und damit auch dieStofftrennung sowohl im analytischen als auch im präparativen Maßstab modelliertwerden. Diese Modellierung ist z.B. bei Seidel-Morgenstern (1995) beschrieben.Innerhalb dieser Modellierungen werden jedoch Einflüsse der chromatographischenAnlagen und ihrer unterschiedlichen Komponenten auf den Peakverlauf nichtberücksichtigt. So führt der unterschiedliche Aufbau einer Laboranlage (Leitungen,Ventile und Einbauten) im Vergleich zu einer präparativen Anlage zu unterschiedlichenEinflüssen auf die axiale Dispersion. Diese Einflüsse werden in Kapitel 0 an einersemipräparativen Laboranlage und einer präparativen Produktionsanlage untersucht, diesich in ihrem Aufbau unterscheiden. Die Laboranlage ist beispielsweise auf geringeTotvolumina optimiert, während die präparative Anlage größere Totvolumina zulässt,um den Druckverlust über die Anlage gering zu halten.
Sollen jedoch innerhalb einer Modellierung auch radiale Effekte innerhalb derchromatographischen Säulen berücksichtigt und untersucht werden, reicht eine reinaxiale, also eindimensionale Betrachtung der Anlage nicht mehr aus. Die Modellierungmuss auf zwei Dimensionen unter Ausnutzung der Achsensymmetrie derchromatographischen Säule oder drei Dimensionen erweitert werden. Zu diesem Zweckwurde in einem Kooperationsprojekt beim Projektpartner ein Modell innerhalb derCFD-Software FLUENT etabliert (Lisso, 2001; Boysen et al., 2001a, 2001b). ZurValidierung der mit Hilfe dieses Modells berechneten Verteilungen von Temperatur,Geschwindigkeit und Konzentration innerhalb der chromatographischen Säule sindexperimentelle Daten notwendig. In Kapitel 6 werden zwei Methoden zur Messung derVerteilungen der Temperatur und der Geschwindigkeit vorgestellt sowie deren Profile

2 Einleitung 1
gemessen. Diese Ergebnisse werden zur Überprüfung des in FLUENT entwickeltenModells verwendet. Zusätzlich wird in Kapitel 2.5 eine Übersicht über Methoden zurMessung von radialen Verteilungen in chromatographischen Säulen gegeben.
Um das Verständnis der innerhalb des chromatographischen Betts auftretendenStrömungen und ihrer mathematischen Modellierung zu erhöhen, werden in denGrundlagen (Kapitel 2.1) auch die notwendigen Bilanzierungen kurz hergeleitet. Die füreinen Anlagenvergleich notwendige Übersicht über Scale-up Verfahren schliesst dieGrundlagen ab. Ein innerhalb dieser Arbeit entwickeltes Verfahren zur Bestimmung derPorositäten der stationären Phasen ist in Kapitel 3.1.4 beschrieben.

2 Grundlagen 3
2 Grundlagen
2.1 Grundlagen des Energie-, Impuls- und Stofftransports inFestbetten
Die zur Modellierung der Transportvorgänge in chromatographischen Festbetten zulösenden Gleichungen sind die Kontinuitätsgleichung, die Impuls-, die Energie- und dieKomponentenbilanz. Eine einfache Methode zur Simulation chromatographischerTrennungen ist die eindimensionale Lösung der Komponentenbilanz nachOrtskoordinate und Zeit. Diese Lösung kann z.B., wie in dieser Arbeit geschehen, unterVerwendung eines Gleichungslösers wie gPROMS berechnet werden. Um jedochradiale Verteilungen der Temperatur oder der Geschwindigkeit innerhalb einerchromatographischen Säule zu modellieren, ist die Lösung der oben erwähntenGleichungen in allen drei Raumrichtungen notwendig (z.B. mit der CFD-SoftwareFLUENT). Zum besseren Verständnis der innerhalb dieser Arbeit verwendetenGleichungen und Vereinfachungen werden im Folgenden aus den grundsätzlichenTransportgleichungen die Gleichungen für den Transport in Festbetten hergeleitet. EineÜbersicht der Gleichungen der Transportvorgänge in Festbetten ist in Tabelle 2.1gegeben.
2.1.1 Grundlegende Transportgleichungen
Eine detaillierte Übersicht über die Phänomene und Gleichungen von Transport-prozessen ist unter anderem bei Baehr und Stephan (1998) und bei Deen (1998) zufinden.
Um Transportvorgänge in chromatographischen Festbetten zu beschreiben müssen alserstes die Ströme, die aufgrund von Gradienten innerhalb des Systems entstehen,berücksichtigt werden. Der Zusammenhang zwischen diesen Strömen und den lokalenStoffeigenschaften wird durch Grundgleichungen hergestellt. Diese Ströme bestehengrundsätzlich aus zwei Komponenten, einem konvektiven und einem diffusiven Anteil.Für die Chromatographie werden normalerweise das Fouriersche Gesetz (für denWärmetransport) und das Ficksche Gesetz (für den diffusiven Stofftransport) alsGrundgleichungen herangezogen:
T∇−= �q (2.1)
(Fouriersches Gesetz für isotrope Stoffe, wie z.B. Fluide)
iiji xD� ∇−=j mit ��x ii = (2.2)
(Ficksches Gesetz für die Diffusion in einem binären Gemisch)
Als Zweites müssen die Erhaltungsgleichungen berücksichtigt werden: Massen-,Energie- und Stoffbilanz. Allgemein lassen sich die folgenden Erhaltungsgleichungenformulieren:

4 Grundlagen 2
Für ein festes Kontrollvolumen (konstantes Volumen V und konstante Oberfläche O ,keine Oberflächengeschwindigkeit) ergibt sich für eine beliebige Erhaltungsgröße ( )tb ,reiner Größe mit ihrem gesamten Fluss ( )t,rF in das Kontrollvolumen und ihrer
Bildungsrate ( )tBV ,r die Erhaltungsgleichung für ein beliebiges makroskopisches
Volumen:
��� +⋅−=V
vSV
dVBdOdVbdtd
nF (2.3)
Für ein bewegliches Kontrollvolumen wird ein Term hinzugefügt, der die Bewegungder Oberfläche berücksichtigt (Oberflächengeschwindigkeit 0≠Ow ):
( ) ( ) ( ) ( )���� ⋅++⋅−=
tOO
tVv
tOtV
dObdVBdOdVbdtd
nwnF (2.4)
Die Konzentration ( )tb ,r muss nur innerhalb des Volumens V mit der Oberfläche
O kontinuierlich sein. Wenn man ein beliebig kleines Volumen betrachtet ( 0→V ),erhält man die Erhaltungsgleichung für einen einzelnen Punkt innerhalb einesKontinuums:
VBtb +⋅−∇=
∂∂
F (2.5)
Mit dem Fluss ausgedrückt durch seinen konvektiven und diffusiven Anteil
fwF +≡ b (2.6)ergibt sich
( ) VBbtb +⋅−∇=⋅∇+
∂∂
fw (2.7)
als allgemeine Form der Erhaltungsgleichung an einem Punkt innerhalb einesKontinuums. Unter Verwendung dieser allgemeinen Gleichungen können jetzt dieGleichungen für die einzelnen Erhaltungsgrößen Masse, Energie, Impuls und dieKomponenten hergeleitet werden.
2.1.2 Massenerhaltung / Kontinuitätsgleichung
Laut Definition gibt es keinen Massenstrom relativ zur über die gesamte Massegemittelten Geschwindigkeit w , deshalb gibt es keinen diffusiven Massenstrom für diegesamte Masse. Unter der Voraussetzung, dass keine Quellen oder Senken existieren( 0=VB ), ergibt sich aus Gl. (2.7) mit �b = als Dichte die lokale Massenerhaltung
oder Kontinuitätsgleichung als
( ) 0=⋅∇+∂∂
w�t
�(2.8)
Für inkompressible Fluide reduziert sich die Gleichung zu

2 Grundlagen 5
0=∇w (2.9)Da die Flüssigkeiten in der HPLC inkompressibel sind, ist die Dichte nur eine Funktionder Temperatur, die Abhängigkeit vom Druck kann vernachlässigt werden.
An dieser Stelle ist es sinnvoll, einen weiteren differentiellen Operator, die materielleoder auch substantielle Ableitung, einzuführen:
∇⋅+∂∂≡ wtDt
D(2.10)
Er repräsentiert die Geschwindigkeit der Änderung einer Grösse, wie sie ein mit demSystem mitbewegter Betrachter beobachten würde. Unter Verwendung dieses Operatorslässt sich aus Gl. (2.7) eine weitere Form der Erhaltungsgleichung für Fluide mitkonstanter Dichte herleiten:
VBDtDb +⋅−∇= f (2.11)
2.1.3 Impulsbilanz
Wenn man einen Körper mit der konstanten Masse m betrachtet, auf den eine Kraft Fwirkt, lässt sich das zweite Newtonsche Gesetz als
Fw =
dtd
m (2.12)
schreiben. Für ein materielles Volumen erhält man dann (gezeigt z.B. in Deen (1998)):
( )( )� � ==
tV tVM M
dVDtD�dV�
dtd
Fw
w (2.13)
Auf Fluide wirkende Kräfte können generell in Kräfte eingeteilt werden, die direkt aufeine Masse oder ein Volumen wirken, wie z.B. die Gravitationskraft, und in Kräfte, dieals Spannungen ( )ns ausgedrückt werden, wie z.B. Oberflächenkräfte. Unter Berück-
sichtigung dieser Kräfte erhält man die makroskopische (integrale) Formulierung derImpulsbilanz für beliebige Kontrollvolumina:
( ) ( )( )
( )��� +=
tOtVtV
dOdV�dVDtD� nsg
w(2.14)
Unter Verwendung des Spannungstensors � zum Ausdruck der auf die Oberflächewirkenden Spannungen erhält man:
( ) �� ⋅=⋅=i j
jiij�
�neenns (2.15)
( ) ( ) ( )��� ⋅+=
tOtVtV
dOdV�dVDtD�
�ngw
(2.16)

6 Grundlagen 2
Einsetzen des Divergenz-Theorems und Umstellen der Gleichung liefert:
( )0=�
�
���
� ⋅∇−−� dV�DtD�
tV
�gw
(2.17)
Für ein beliebiges Fluid, das als Kontinuum betrachtet werden kann, folgt unterBerücksichtigung der Gravitation als einziger global wirkenden Kraft:
�gw ⋅∇+= �
DtD�
(2.18)
An dieser Stelle ist eine Unterscheidung der Anteile des Druckes an den Spannungenvon den anderen Anteilen von Vorteil, da der Druck die einzige Spannung auf einruhendes Fluid darstellt und isotrop ist, normal zur Oberfläche wirkt und ein positivesVorzeichen besitzt, wenn er komprimierend wirkt. Daher lässt sich für denSpannungstensor und seine Divergenz mit Hilfe der Identität � und des Zähigkeits-spannungstensors � schreiben:
��� +−= P (2.19)
( ) ���� ⋅∇+−∇=⋅∇+−⋅∇=⋅∇ PP (2.20)Hieraus lässt sich die allgemeine Formulierung der Impulsbilanz (Cauchy-Moment)ableiten:
�gw ⋅∇+∇−= P�
DtD�
(2.21)
Unter Verwendung der Scher- bzw. dynamischen Viskosität � , der Volumenviskosität� ′ und des Zähigkeitstensors � erhält man einen Ausdruck für den Zähigkeits-
spannungstensor:
( ) �w�� ⋅∇��
���
� −′+= 32
2 (2.22)
Für ein inkompressibles Newtonsches Fluid reduziert sich dies zu:
( )[ ]t ww�� ∇+∇== 2 (2.23)
Dies führt unter Berücksichtigung konstanter Dichte und Viskosität zu:
[ ] ( )( )[ ] ( )[ ] wwwww��222 ∇=∇∇+∇=∇+∇⋅∇=⋅∇=⋅∇ t (2.24)
Eingesetzt in Gl. (2.21) erhält man für die Impulsbilanz die Navier Stokes Gleichung:
wgw 2∇+∇−= P�
DtD�
(2.25)
Die Navier-Stokes-Gleichung (Gl. (2.25)) und die Kontinuitätsgleichung (Gl. (2.8)) sindder Ausgangspunkt für alle rigorosen Modellierungen von HPLC-Säulen.

2 Grundlagen 7
In diesem Zusammenhang ist es angebracht, auf eine interssante Tatsache bei CFD-Modellierungen hinzuweisen: Die Bedeutung des Druckes. Diese Bedeutung weichtetwas von der reinen thermodynamischen Betrachtungsweise ab. Innerhalb der CFD-Simulationen wird der Druck nicht aus einer Zustandsgleichung berechnet, sondern alsmechanische Variable, die herangezogen wird, um die Kontinuitätsgleichung und dieImpulsbilanz zu erfüllen:
Der Druck hat innerhalb der numerischen Lösung dieselbe grundsätzliche Bedeutungwie in der Thermodynamik (als Variable lokal bestimmt aus einer Zustandsgleichungder Form ),( T�PP = ). Betrachtet man die isotherme Strömung eines Fluids bestehendnur aus einer Komponente, so sind die Unbekannten w , P und � , die bestimmenden
Gleichungen die Kontinuitätsgleichung, die Impulsbilanz (inkl. einer konstituierendenGleichung) und die Zustandsgleichung. Für inkompressible Fluide (und daherkonstante, bekannte Dichte � ) ist die Anzahl der Variablen um eine reduziert. Daher
wird die Zustandsgleichung zur Bestimmung des Druckes überflüssig, da der Druckjetzt eine direkte Funktion der Dichte geworden ist. Wenn der Druck nicht an einerSystemgrenze angegeben wird, bleibt sein tatsächlicher Wert unbestimmt. Dies stelltkein Problem dar, da inkompressible Strömungen nur vom Gradienten des Druckesbeeinflusst werden.
2.1.4 Energiebilanz
Wenn nur thermische Einflüsse berücksichtigt werden, kann Gl. (2.11) mit T� Cb V= ,
qf = und VV HB = in eine geeignete Formulierung der Energiebilanz umgeschrieben
werden:
VV HDtDT� C +⋅−∇= q (2.26)
Einsetzen des Fourierschen Gesetzes (Gl. (2.1)) mit der Annahme einer von Druck undTemperatur unabhängigen Wärmeleitfähigkeit und konstanter Dichte und Wärme-kapazität führt zu:
VP HT�
DtDT� C +∇= 2 (2.27)
Dies ist die Formulierung der Energiebilanz für reine, inkompressible Fluide unterausschliesslicher Berücksichtigung von thermischen Effekten.
Für eine allgemeine Formulierung müssen mechanische Effekte berücksichtigt werden,
wie der Wärmeeintrag Q� aus der Umgebung in das Fluid und die Arbeit W , die dasFluid an der Umgebung verrichtet. Als weitere Änderung wird die Enthalpie durch diespezifische innere Energie u ersetzt, und die gesamte Energie soll sich nur aus innererEnergie und kinetischer Energie 22w zusammensetzen. Der Anteil der Gravitation
wird im Arbeitsterm berücksichtigt. Es ergibt sich die Energiebilanz für ein definiertesKontrollvolumen für die Summe aus innerer Energie und kinetischer Energie:

8 Grundlagen 2
WQdOu dVu dtd
OV
�� −+⋅���
����
�+−=��
�
����
�+ �� nw
ww22
22
(2.28)
Unter Verwendung des Wärmestromes (und Fourierschen Gesetzes) als
� ⋅−=O
dOQ nq�(2.29)
und dem Arbeitseintrag unterteilt in den Anteil, den das System durch Veränderung dergeodätischen Höhe erhält,
( )� ⋅−=V
G dV�W gw�(2.30)
und dem Anteil der Oberflächenarbeit des Systems an der Umgebung
( ) ( )� � � ⋅⋅−⋅=⋅⋅−=O O O
S dOdOPdOW �nwnw�nw�(2.31)
mit der Unterteilung des Spannungstensors in Anteil des Druckes und viskosen Anteil(Gl. (2.19)). Wenn zusätzlich Quellterme existieren, können diese durch Einfügen eines
VH -Terms berücksichtigt werden. Durch Einsetzen dieser Gleichungen für die Arbeits-
und Wärmeströme erhält man unter Verwendung mathematischer Umformungen einenallgemeinen Ausdruck der Energiebilanz an einem beliebigen Punkt innerhalb einesreinen Fluids:
( ) ( ) ( )gww�wqw ⋅+⋅⋅∇+⋅∇−⋅−∇=��
�
����
�+ �Pu
DtD�
2
2
(2.32)
Es ist jedoch sinnvoller, die Energiebilanz als Funktion der Temperatur auszudrücken.Deshalb wird der Term ( ) Dt� D 22w ausgewertet und der sich ergebende Ausdruck
von Gl. (2.32) abgezogen. Die Auswertung des Terms ergibt die mechanische Energie-erhaltung (nach Bird et al. (1960)):
( ) ( )gw�www ⋅+⋅∇⋅+∇⋅−=��
�
����
� �PDtD�
2
2
(2.33)
Subtraktion von Gl. (2.33) von Gl. (2.32) führt zu folgender Gleichung, in der derOperator : das doppelt skalare Produkt darstellt:
( ) ( )w�wq ∇+⋅∇−⋅−∇= :PDtDu�
(2.34)
Die spezifische innere Energie und die spezifische Enthalpie hängen über Druck undspezifisches Volumen �1 wie folgt zusammen:
�Phu −= (2.35)
Mit )P(f� ≠ folgt unter Verwendung von

2 Grundlagen 9
DtD��PDt
DPDtDh��PDt
D�DtDh�
DtDu� +−=��
�
����
�−= (2.36)
und durch Umstellung der Kontinuitätsgleichung (Gl. (2.8)) nach der substantiellenAbleitung der Dichte
( )w⋅∇−= �DtD�
(2.37)
die allgemeine Form der Energiebilanz als Ausdruck der spezifischen Enthalpie:
( )w�q ∇++⋅−∇= :DtDP
DtDh�
(2.38)
In diesem Fall kann die Temperaturabhängigkeit der spezifischen Enthalpiefolgendermaßen ausgedrückt werden:
( )dP
T
�T�dTCdh
PP �
�
���
���
�
�
∂∂−+= 11
(2.39)
Dies erlaubt es, die linke Seite von Gl. (2.38) umzuschreiben:
( )DtDP
T
�� TDtDT� C
DtDh�
PP �
�
���
���
�
�
∂∂−+= 1
1 (2.40)
Aus diesen Umformungen erhält man dann die allgemeine Form der Energiebilanz fürein beliebiges reines Fluid mit einem symmetrischen Spannungstensor und )P(f� ≠ :
( )w�q ∇+��
���
�
∂∂−⋅−∇= :
DtDP
TP�TDt
DT� CP
P (2.41)
Für ein Newtonsches Fluid ist die Dissipationsleistung proportional zur Viskosität:
( ) ���=∇w� : (2.42)� ist die Dissipationsleistung und abhängig von verschiedenen partiellen Ableitungen
der Geschwindigkeit (nachzulesen z.B. bei Deen (1998) oder Schade und Kunz (1980)).Die viskose Wärmeentwicklung ist immer eine Wärmequelle, da Reibungseffekteimmer irreversible Verluste an mechanischer Energie darstellen. Für ein NewtonschesFluid lässt sich die Energiebilanz schreiben als:
���DtDP
TP�TDt
DT� CP
P +��
���
�
∂∂−⋅−∇= q (2.43)
Der Vollständigkeit halber sei hier noch eine andere Formulierung erwähnt, dieallgemeine Form der Energiebilanz in einem Vielkomponentensystem, dargestellt alsmolare Enthalpiebilanz (Deen, 1998):
���===
⋅+∇++���
����
� +⋅−∇=���
����
�
∂∂ n
iii
n
iii
n
iii Dt
DPhchc
t 111
: gjw�qw (2.44)

10 Grundlagen 2
2.1.5 Komponentenbilanz
Der molare Stoffstrom einer Komponente i relativ zu festgelegten Koordinaten sei iN ,
ic die molare Konzentration von i und ViR sei die Bildungsrate von i. Dann ist die
einfachste Form der Komponentenbilanz:
Viii R
tc +−∇=∂
∂N (2.45)
Ersetzt man für den Stoffstrom seinen konvektiven Anteil wic und seinen diffusiven
Anteil iJ :
iii Jc += wN (2.46)Für ein binäres Gemisch mit konstaner Dichte kann iJ durch den Diffusions-
koeffizienten ausgedrückt werden:
iiji cDJ ∇−= (2.47)
Hieraus folgt die Komponentenbilanz für eine Komponente eines binären Gemisches:
Viiiji RcD
DtDc +∇= 2 (2.48)
Die Auflösung der substantiellen Ableitung führt dann zur Komponentenbilanz, wie sieals Ansatz in der Chromatographie verwendet wird:
Viiiiji RccD
tc +∇⋅−∇=∂∂
w2 (2.49)
2.1.6 Gleichungen für die HPLC
Die für eine CFD-Simulation notwendigen Gleichungen / Bilanzen sind inTensorschreibweise in Tabelle 2.1 zusammengefasst.
Tabelle 2.1: Modellgleichungen für die HPLC in Tensorschreibweise
GleichungKontinuitätsgleichung
Gl. (2.8) ( ) 0=⋅∇+∂∂
w�t
�
Navier Stokes GleichungGl. (2.25) wg
w 2∇+∇−= P�DtD�
EnergiebilanzGl. (2.43) ���
DtDP
TP�TDt
DT� CP
P +��
���
�
∂∂−⋅−∇= q
KomponentenbilanzGl. (2.49) Viiiij
i RccDt
c +∇⋅−∇=∂∂
w2
Diese Gleichungen sind die den Modellierungen dieser Arbeit in FLUENT zu Grundeliegenden Gleichungen. Allerdings macht man sich bei der numerischen Implemen-

2 Grundlagen 11
tierung die Achsensymmetrie der chromatographischen Säulen zu Nutze, und modelliertdie Gleichungen in Zylinderkoordinaten, d.h. das System wird zweidimensionalberechnet. Die explizite Formulierung der Gleichungen in Zylinderkoordinaten ist inTabelle 2.2 aufgeführt.
Tabelle 2.2: Modellgleichungen für die HPLC in Zylinderkoordinaten
Kontinuitätsgleichung:
0=∂∂+
∂∂+
∂∂+
∂∂
z
�w�
�r
wr
�w
t
�z
�r
Navier Stokes Gleichung ( z,�
,r Koordinaten):
r - Koordinate: rP� g
zw
wr
w�wr
wr
ww
tw�
rr
zr
�r
rr �
∂∂−=
���
�
���
�
∂∂+−
∂∂+
∂∂+
∂∂ 2
( ) ��
���
�
∂∂+
∂∂−
∂∂+�
�
�
�
∂∂
∂∂+
2
2
22
2
2
211zw�w
r�w
rrw
rrr� r
�r
r
�- Koordinate: �P
r� g
zw
vrww�w
rw
rw
wt
w� ��z
�r
���r
�∂∂−=�
�
���
�
∂∂++
∂∂+
∂∂+
∂∂ 1
( ) ��
���
�
∂∂+
∂∂−
∂∂+�
�
�
�
∂∂
∂∂+
2
2
22
2
2
211zw�w
r�w
rrw
rrr� �
r��
z - Koordinate: zP� g
zw
w�wr
wr
ww
tw�
zz
zz
�z
rz
∂∂−=�
�
���
�
∂∂+
∂∂+
∂∂+
∂∂
��
���
�
∂∂+
∂∂+�
�
�
�
∂∂
∂∂+
2
2
2
2
2
11zw�w
rrw
rrr
� zzz
Energiebilanz (nur thermische Effekte berücksichtigt):
��
���
�
∂∂+
∂∂+�
�
�
�
∂∂
∂∂=
∂∂+
∂∂+
∂∂+
∂∂
2
2
2
2
2
11zT� T
rrT
rrr� C
�zT
w�Tr
wrT
wtT
Pz
�r
Pz
�r
PP� C��
zP
v�Pr
vrP
vtP
TP
C� T +��
���
�
∂∂+
∂∂+
∂∂+
∂∂⋅�
�
�
�
∂∂− 2 , mit der Dissipationsfunktion
( )2222222
32111
2 v⋅∇−��
���
�
∂∂+
∂∂+�
�
���
�
∂∂+
∂∂+�
�
���
�
∂∂+�
�
�
�
∂∂+
���
�
���
���
�
�
∂∂+�
�
�
� +∂∂+�
�
�
�
∂∂=
rv
zv
zv�v
r�v
rrv
rr
zv
rv�v
rrv�
zr�
zr�
zr�
r
Komponentenbilanz ( ( )tz�
rcc ii ,,,= :
Viiii
iji
zi
�i
ri R
zc� c
rrc
rrr
Dzc
w�cr
wrc
wtc +�
�
���
�
∂∂+
∂∂+�
�
�
�
∂∂
∂∂=
∂∂+
∂∂+
∂∂+
∂∂
2
2
2
2
2
11

12 Grundlagen 2
Die Implementierung der Gleichungen in die Simulationsumgebungen FLUENT undgPROMS werden in Kapitel 4 erläutert.
2.1.6.1 Druckver lust
Um den Fluss durch die poröse stationäre Phase innerhalb der chromatographischenSäule zu modellieren, wird Darcy’s Gesetz angewendet. Es korrelliert dieLeerrohrgeschwindigkeit und den Druckverlust über der Säule mit Hilfe derPermeabilität � der festen Phase:
P �w ∇−=0 (2.50)
Die Permeabilität ist eine Funktion der externen Porosität ext! und des Partikel-
durchmessers Pd der festen Phase, bestimmt durch die Blake-Kozeny Gleichung (Bird
et al. 1960):
( )2
32
1150 ext
extP ""
d#−
= (2.51)
Die Anwendbarkeit des Gesetzes von Darcy ist durch den Strömungszustand in derchromatographischen Säule gerechtfertigt. Für die Reynoldszahl in der Säule
�dw P0Re = (2.52)
ergibt sich für den Grenzfall (hohe Zwischenkorngeschwindigkeit, große Partikeldurch-messer, hohe Dichte und niedrige dynamische Viskosität) ein Wert von 0,63. Da fürRohr- und Zylinderströmungen der laminare Bereich der Reynoldszahl zwischen 0 und40 liegt, ist die Annahme einer Kriechströmung durch die stationäre Phase gerecht-fertigt.
2.1.6.2 Eindimensionale Komponentenbilanz
Ein weit verbreiteter Ansatz zur Modellierung der Chromatographie ist die numerischeBerechnung nur der örtlichen und zeitlichen Konzentrationsverläufe in einerchromatographischen Säule. Insbesondere der Konzentrationsverlauf am Ende der Säule(der Peakverlauf) ist für die Berechnung und den Vergleich mit den gemessenenexperimentellen Chromatogrammen wichtig. Hierfür muss lediglich die Komponenten-bilanz (Gl. (2.49)) in geeigneter Weise gelöst werden. Es existieren verschiedeneModelle zur Beschreibung des auftretenden Stofftransports, der linearen und nicht-linearen Adsorption und zur Beschreibung der Peakverbreiterung mit Hilfe des axialenDispersionskoeffizienten (Kapitel 2.2). Eine dataillierte Übersicht ist in der Literatur,z.B. bei Seidel-Morgenstern (1995), zu finden.
Da in dieser Arbeit das Dispersionsmodell zur eindimensionalen Simulation mitgPROMS verwendet wird, soll es hier beispielhaft abgeleitet werden. Gl. (2.47) wirdhierbei durch das 1. Ficksche Gesetz ersetzt, das die durch Dispersion hervorgerufene

2 Grundlagen 13
Stoffstromdichte über den axialen Dispersionskoeffizienten mit dem Konzentrations-verlauf korreliert:
zc
DJ iaxi ∂
∂= (2.53)
Der Quellterm lässt sich durch die Adsorption als Änderung der Beladung der Partikelder festen Phase ausdrücken, mit dem Phasenverhältnis F ergibt sich:
tq
FR iVi ∂
∂−= (2.54)
ext
ext""
F−= 1
(2.55)
Es ergibt sich die folgende Komponentenbilanz des Dispersionsmodells für eindifferentielles Volumenelement:
tq!
!zc
wzc
Dtc i
ext
extiz
iax
i
∂∂−−
∂∂−
∂∂=
∂∂ 1
2
2
(2.56)
Die Bestimmung der mittleren Beladung der Partikel erhält man über die differentielleKomponentenbilanz um die stationäre Phase, wobei eine Diffusion innerhalb derstationären Phase über die Grenzflächen des differentiellen Volumenelements hinausvernachlässigt wird (Festfilmmodell):
( )( )ii*iieff
i qcqkt
q −=∂
∂, (2.57)
Wenn zusätzlich noch die Gleichgewichtsbeladung der stationären Phase durch einegeeignete Adsorptionsisotherme (Kapitel 2.2.3) berücksichtigt wird, reicht diese Bilanzzur Simulation von Peakverläufen aus.
2.2 Grundlagen der Chromatographie
Die Chromatographie ist ein thermisches Trennverfahren, bei dem ein Stoffgemischzwischen zwei Hilfsphasen verteilt wird. Bei der Säulenchromatographie ruht die festeHilfsphase (stationäre Phase) in der Trennsäule, während die andere, fluide Hilfsphase(mobile Phase) durch diese hindurchströmt. Wird als mobile Phase eine Flüssigkeiteingesetzt, so spricht man von Flüssigchromatographie. Die Stofftrennung erfolgtaufgrund des Stoffaustausches zwischen stationärer und mobiler Phase. Mit der mobilenPhase werden die zu trennenden Komponenten durch die Säule transportiert. Hierbei istdie Verweildauer in der stationären Phase für unterschiedliche Moleküle verschiedengroß, so dass sie zeitlich versetzt die Säule verlassen.
Eingeteilt werden die flüssigchromatographischen Trennverfahren nach demMechanismus der Wechselwirkungen, der zwischen den zu trennenden Komponentenund der stationären Phase wirkt (Meyer, 1992), z.B.:

14 Grundlagen 2
- Affinitätschromatographie ist das Verfahren mit der größten Selektivität bei demdie Wechselwirkungen biochemischer Natur sind (z.B. Schlüssel-Schloss-Wechselwirkung). Die Bindungen sind am stärksten und können meist nur durchLösungsmittelwechsel oder pH-Wert Änderung gelöst werden.
- Adsorptionschromatographie mit normalen Phasen (normal phase) oderUmkehrphasen (reversed phase): Die Komponenten werden an der stationären Phaseunterschiedlich stark adsorbiert. Die Verfahren unterscheiden sich durchunterschiedliche Polaritäten der stationären Phase.
- Ionenaustauschchromatographie: Die stationäre Phase enthält ionische Gruppen,welche mit den zu trennenden Komponenten in Wechselwirkung treten.
- Ausschlußchromatographie: Die Komponenten werden nach der Molekülgrößegetrennt, da die großen Moleküle nicht oder nur in einen Teil der stationären Phaseeindringen können und dadurch schneller eluieren.
Die Chromatographie wird sehr häufig in der Analytik eingesetzt. Die aufgegebenenMengen sind hierbei sehr gering. Im präparativen Maßstab steht dagegen dieGewinnung reiner Stoffe im Vordergrund. Um dabei größere Durchsätze zu erzielen,wird in der Regel auf eine Säule größeren Durchmessers ein größeres Probevolumen(Volumenüberladung) mit höherer Konzentration (Konzentrationsüberladung)aufgegeben.
Die Grundlagen der Chromatographie, die zum Verständnis der eingesetzten Analytik,der verwendeten Modellierung, der durchgeführten Messungen und der Bestimmungder Modellparameter erforderlich sind, werden im Folgenden erläutert. Dieverschiedenen Verfahren der Adsorptionschromatographie werden abschließend kurzgegenübergestellt. Eine ausführliche Beschreibung des Energie-, Impuls und Stoff-transports in chromatographischen Festbetten ist bereits in Kapitel 2.1 erfolgt.
2.2.1 Chromatographische Kenngrößen
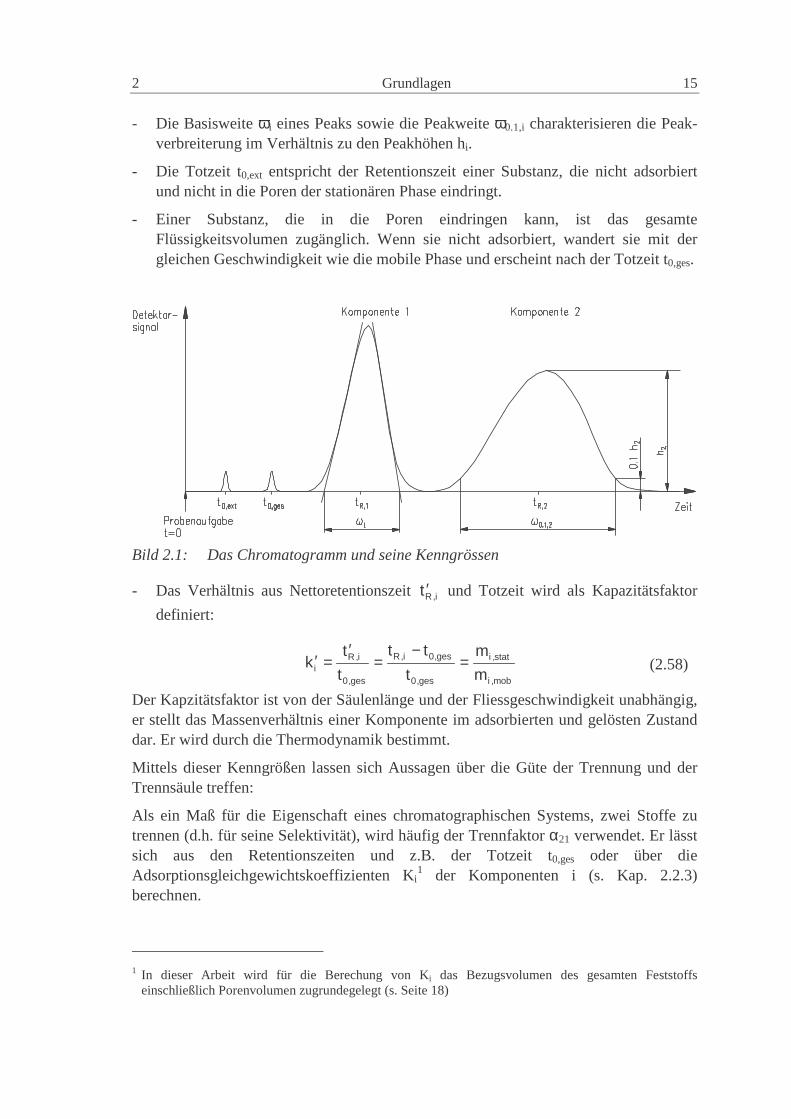
Zur Beurteilung der Trennung werden die Komponenten nach dem Verlassen derTrennsäule durch die mobile Phase in einen Detektor transportiert, der sie aufgrundbestimmter physikalischer oder chemischer Eigenschaften als Peaks erfasst. DieGesamtheit der Peaks wird Chromatogramm genannt (Bild 2.1). Die Peaks liefern in derAnalytik qualitative und quantitative Informationen über das getrennte Stoffgemisch:
- Die Fläche unter einem Peak und die Peakhöhe hi sind proportional zureingespritzten Menge der Komponente. Für jeden Detektor muss zurQuantifizierung der Menge eine Kalibrierung durchgeführt werden.
- Die Retentionszeit tR,i einer Substanz i ist für einen symmetrischen Peak die Zeit,die von der Peakaufgabe zum Zeitpunkt t = 0 bis zur Detektion des Peakmaximumsvergeht. Sie gibt eine qualitative Information über die Komponente und kann zuihrer Identifikation herangezogen werden.
2 Grundlagen 15
- Die Basisweite ωi eines Peaks sowie die Peakweite ω0.1,i charakterisieren die Peak-verbreiterung im Verhältnis zu den Peakhöhen hi.
- Die Totzeit t0,ext entspricht der Retentionszeit einer Substanz, die nicht adsorbiertund nicht in die Poren der stationären Phase eindringt.
- Einer Substanz, die in die Poren eindringen kann, ist das gesamteFlüssigkeitsvolumen zugänglich. Wenn sie nicht adsorbiert, wandert sie mit dergleichen Geschwindigkeit wie die mobile Phase und erscheint nach der Totzeit t0,ges.
Bild 2.1: Das Chromatogramm und seine Kenngrössen
- Das Verhältnis aus Nettoretentionszeit i,Rt ′ und Totzeit wird als Kapazitätsfaktor
definiert:
mob,i
stat,i
ges,
ges,i,R
ges,
i,Ri m
m
t
tt
t
tk =
−=
′=′
0
0
0
(2.58)
Der Kapzitätsfaktor ist von der Säulenlänge und der Fliessgeschwindigkeit unabhängig,er stellt das Massenverhältnis einer Komponente im adsorbierten und gelösten Zustanddar. Er wird durch die Thermodynamik bestimmt.
Mittels dieser Kenngrößen lassen sich Aussagen über die Güte der Trennung und derTrennsäule treffen:
Als ein Maß für die Eigenschaft eines chromatographischen Systems, zwei Stoffe zutrennen (d.h. für seine Selektivität), wird häufig der Trennfaktor α21 verwendet. Er lässtsich aus den Retentionszeiten und z.B. der Totzeit t0,ges oder über dieAdsorptionsgleichgewichtskoeffizienten K i
1 der Komponenten i (s. Kap. 2.2.3)berechnen.
1 In dieser Arbeit wird für die Berechung von K i das Bezugsvolumen des gesamten Feststoffs
einschließlich Porenvolumen zugrundegelegt (s. Seite 18)

16 Grundlagen 2
1
2
1
2
0,extR,1
0,extR,2
KK
kk
tt
tt=��
�
����
�
′′
=−−
=21α (2.59)
Die Güte der Trennung wird durch die Auflösung RS (von engl. resolution) beschrieben.Sie ist definiert durch den Quotienten aus dem zeitlichen Abstand zweier benachbarterPeakmaxima und dem arithmetischen Mittel aus den Basisweiten ω1 und ω2.
)$($21
ttR
21
R,1R,2S
+
−=
(2.60)
Zur Charakterisierung der Packungsgüte und der stationären Phase, d.h. der Effektivitätder Säule, kann die Trennstufenzahl NTUi (engl.: number of transfer units) herange-zogen werden. Die Bezeichnung NTUi ist ein Analogieschluß aus der Rektifikation,wenn man sich eine Trennsäule wie eine Rektifikationskolonne mit Trennstufenvorstellt. Aus dem Chromatogramm lässt sich für Peaks, die die Form einer Gaußkurvebesitzen, die Trennstufenzahl einer Säule wie folgt bestimmen (Aced und Möckel,1991).
2
i
R,i%t ���
����
�= 16NTUi (2.61)
In der Realität weisen chromatographische Peaks oft eine nicht zu vernachlässigendeUnsymmetrie auf (Tailing, Fronting). In diesem Fall ist die zuverlässigste Methode zurBestimmung der Trennstufenzahl und der Retentionszeit die Momentenmethode: DieRetentionszeit tR,i entspricht dann dem ersten bezogenen Anfangsmoment µ'1,i. Um dieMethode anwenden zu können, muss das Chromatogramm in digitaler Form vorliegen.Die Integrale können dann durch Summen über die n Datenpunkte desChromatogramms ersetzt werden.
�
�
�
�
=
=∞
∞
∆
∆≈=≡
n
jjji
n
jjjji
i
i
iRi
tc
ttc
dtc
dtct
t&
1,
1,
0
0,
',1 (2.62)
Die Trennstufenzahl NTUi ist über das erste zentrale Anfangsmoment und das zweitezentrale Moment µ2,i (die Varianz σ) definiert.
2
,2
',1NTU �
�
�
�
��
�
�=
i
ii &
&(2.63)
Hierbei berechnet sich das zweite zentrale Moment wie folgt:

2 Grundlagen 17
j
n
1ji,j
n
1jjR,iji,j
0i
0
2R,ii
2i2,i '
tc
't)t(tc
dtc
dt)t(tc(&�
�
�
�
=
=∞
∞
⋅−≈
−=≡ (2.64)
Je geringer die Trennstufenzahl NTUi einer Säule ist, desto höher muss die Selektivitätα21 des chromatographischen Systems sein, um das gleiche Trennergebnis (gleicheAuflösung RS) zu erreichen. Alle Auswertungen von Peakverläufen dieser Arbeiterfolgen nach der Momentenmethode.
Neben Peaks werden auch Konzentrationssprünge oder -stufen auf Säulen gegeben, umdie Säuleneigenschaften zu untersuchen. Aus den dabei entstehendenDurchbruchskurven können die Retentionszeit und die Varianz wie folgt bestimmtwerden.
( )( )I
iIIi
n
1jji,j
IIi
0
i
0
i
R,i,front cc
'tcc
dttc
dtttc
t−
⋅−≈
⋅∂∂
⋅∂∂
=�
�
�=
∞
∞
(2.65)
( )2
R,iIi
IIi
n
1jjji,j
IIi
0
i
0R,i
i
2i,front t
cc
)ttcc
dttc
dt)t(ttc
* −−
⋅−≈
⋅∂
∂
⋅−⋅∂
∂
=�
�
�=
∞
∞
(2.66)
2.2.2 Porosität
Zur Beschreibung des Phasenverhältnisses fest - flüssig wird die Porosität ε herange-zogen. Ausgehend vom Gesamtvolumen VS einer Säule können drei Teilvoluminadefiniert werden (Seidel-Morgenstern, 1995).
• externes Volumen (Volumen der mobilen Phase außerhalb der Partikel)
Sextext V+V = (2.67)
• Partikelvolumen (Volumen der Partikel inklusive Porensystem)
SextPar V)+(1V −= (2.68)
• internes Volumen (Volumen der mobilen Phase in den Poren der Partikel)
Parintint V+V = (2.69)
Über die Totzeiten t0,ext und t0,ges sowie den Volumenstrom der mobilen Phase �Vkönnen die externe Porosität εext und die gesamte Porosität εges bestimmt werden.
S
0,ext
S
extext V
Vt
VV, �
== (2.70)

18 Grundlagen 2
S
0,ges
S
intextges V
Vt
VVV+ �
=+= (2.71)
Über die nachfolgende Beziehung lässt sich die interne Porosität εint berechnen.
intextextges,),(1,, −+= (2.72)
Die Gesamtporosität weist nach Meyer (1992) häufig Werte von εges = 0.7...0.85 auf.Für die externe Porosität monodisperser Kugelschüttungen ergeben sich Wertezwischen εext = 0.36...0.41 (Tsotsas, 1987). Methoden zur experimentellen Bestimmungder Porositäten werden in Kapitel 3.1.4 beschrieben.
Das Phasenverhältnis Φ wird in dieser Arbeit als Verhältnis des Partikelvolumens zumexternen Flüssigkeitsvolumen definiert.
ext
ext
ext
Par ++1
VV- −== (2.73)
2.2.3 Adsorptionsisothermen
Bei der Adsorptions- und der Ionenaustauschchromatographie beruht der Trenneffektauf unterschiedlich starker, aber reversibler Adsorption an der stationären Phase. DasPhasengleichgewicht wird in der Chromatographie in den meisten Fällen durchAdsorptionsisothermen beschrieben. Sie geben für eine feste Temperatur denZusammenhang zwischen der Beladung des Feststoffs qi
* mit einer Komponente i undden Konzentrationen der in der mobilen Phase gelösten k Komponenten c1...ck an.
Die lineare Einstoffisotherme („Henry“-Gesetz) ist die einfachste Form der
Adsorptionsisotherme, bei der die Beladung q i* des Feststoffs mit der Komponente i
linear ausschließlich von der Konzentration ci dieser Komponente abhängt. LineareEinstoffisothermen treten häufig bei geringen Konzentrationen, z.B. in der analytischenChromatographie, auf.
ii*i cKq = (2.74)
An dieser Stelle sei darauf hingewiesen, dass entsprechend der gewählten Bezugsgröße(wie Feststoffmasse oder -volumen) der Beladung qi
* der Adsorptionsgleichgewichts-koeffizient K i unterschiedliche Werte und auch Dimensionen besitzt. In dieser Arbeitwird die adsorbierte Masse der Komponente i immer auf das Volumen des Feststoffsinklusive dem Porensystem bezogen. Damit wird auch die Masse der Komponente i alsadsorbiert betrachtet, die sich in der Flüssigkeit in den Poren des Feststoffs befindet.Diese Bezugsgröße vereinfacht die mathematische Modellierung (vergleiche Kap. 2.1.5)dahingehend, dass für die stoffliche Bilanzierung neben der Beladung nicht zusätzlichdie mittlere Flüssigphasenkonzentration betrachtet werden muss.
Bei hohen Konzentrationen, wie sie beispielsweise in der präparativen Chromatographieeingesetzt werden, ist der Adsorptionsgleichgewichtskoeffizient K i häufig eine Funktion
2 Grundlagen 19
der Konzentration ci, so dass eine nichtlineare Adsorptionsisotherme qi* = K i(ci)⋅ci
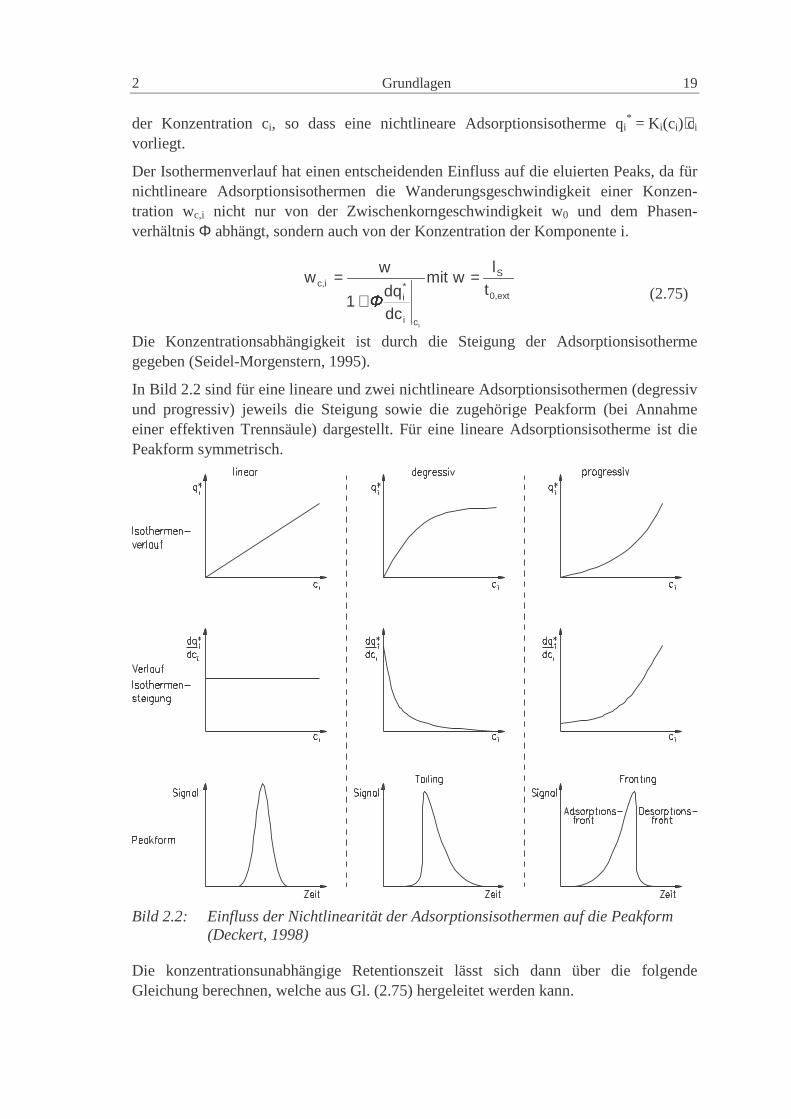
vorliegt.
Der Isothermenverlauf hat einen entscheidenden Einfluss auf die eluierten Peaks, da fürnichtlineare Adsorptionsisothermen die Wanderungsgeschwindigkeit einer Konzen-tration wc,i nicht nur von der Zwischenkorngeschwindigkeit w0 und dem Phasen-verhältnis Φ abhängt, sondern auch von der Konzentration der Komponente i.
0,ext
S
ci
*i
c,i tl
w
dcdq.
1
ww
i
=+
= mit(2.75)
Die Konzentrationsabhängigkeit ist durch die Steigung der Adsorptionsisothermegegeben (Seidel-Morgenstern, 1995).
In Bild 2.2 sind für eine lineare und zwei nichtlineare Adsorptionsisothermen (degressivund progressiv) jeweils die Steigung sowie die zugehörige Peakform (bei Annahmeeiner effektiven Trennsäule) dargestellt. Für eine lineare Adsorptionsisotherme ist diePeakform symmetrisch.
Bild 2.2: Einfluss der Nichtlinearität der Adsorptionsisothermen auf die Peakform(Deckert, 1998)
Die konzentrationsunabhängige Retentionszeit lässt sich dann über die folgendeGleichung berechnen, welche aus Gl. (2.75) hergeleitet werden kann.

20 Grundlagen 2
)K/
(1tt i0,extR,i += (2.76)
Liegt eine degressive Adsorptionsisotherme (Langmuir förmig, häufigster Fall) vor, ver-schiebt sich das Peakmaximum in Richtung der Totzeit. Gleichzeitig wird die vordereFront (Adsorptionsfront) steiler und die Desorptionsfront flacher, d.h. disperser. DiesesPhänomen wird Tailing genannt. Für eine progressive Adsorptionsisotherme (anti-Langmuir förmig, tritt selten auf) weist der Peak sogenanntes Fronting auf. DasMaximum wird zu späteren Elutionszeiten hin verschoben, gleichzeitig wird dieAdsorptionsfront disperser und die Desorptionsfront steiler (Guiochon et al., 1994).
Eine Überladung der Trennsäule, wie sie bei der präparativen Chromatographiebetrieben wird, führt meist zu einer Verschlechterung der Auflösung RS. DieElutionszeiten der Peaks werden kürzer, gleichen sich einander an und die Peakswerden dabei breiter. Es können mittels der Überladung jedoch größere Mengen desProdukts gewonnen werden (Unger, 1994).
Ist die Beladung darüber hinaus auch von den Konzentrationen der weiterenKomponenten abhängig, so spricht man von gekoppelten Adsorptionsisothermen qi
* =K i(c1...cr) ci. Aus der Vielzahl der Modelle für nichtlineare, gekoppelte Adsorptions-isothermen seien hier nur einige exemplarisch erläutert (Seidel-Morgenstern, 1995),(Kast, 1988):
Mit der Multi-Langmuir Gleichung (2.77) kann der Verlauf von realen Adsorptionsiso-thermen gut wiedergegeben werden, da diese meistens einen degressiven Verlaufzeigen.
��==
+=
+=
r
1jjj
iir
1jjj
iiS,i
*i
cb1
cn
cb1
cbqq
(2.77)
Das Produkt der Parameter Sättigungsbeladung qS,i der Komponente i undKrümmungsparameter bi entspricht dem Anfangsanstieg ni der Isotherme (ni = qS,i⋅bi),denn für kleine Konzentrationen geht die Langmuir-Isotherme in eine lineareAdsorptionsisotherme über. Um einen konzentrationsabhängigen Trennfaktor αij
beschreiben zu können, wird Gl. (2.77) um einen linear von der Konzentrationabhängigen Term additiv ergänzt und man erhält die Modifizierte-Langmuir-Isotherme.
�=
++=
r
1jjj
iii
*i
cb1
cnkq
(2.78)
Schließlich führt die Kombination von zwei Multi-Langmuir Termen zur gekoppeltenBi-Langmuir-Isotherme (Gl.(2.79)). Ihr liegt die Annahme zugrunde, dass zweiunterschiedliche Zentren vorhanden sind, an denen jeweils eine konkurrierendeAdsorption stattfindet. In der Praxis führt die Zunahme der anpassbaren Parameter zueiner größeren Flexibilität der Gleichung. Dadurch kann sie die unterschiedlichenGekrümmtheiten realer Isothermenverläufe sehr gut beschreiben.
2 Grundlagen 21
��==
++
+=
r
1jj
IIj
iIIi
r
1jj
Ij
iIi*
i
cb1
cn
cb1
cnq
(2.79)
Eine empirische Gleichung, mit der sowohl progressive als auch degressive gekoppelteZweistoffisothermen beschrieben werden können, wird von Ching et al. (1993) vor-geschlagen:
injij
miii
*i c)cBcA(Kq iji ++= (2.80)
Hierbei sind K i, A i und mi Reinstoffparameter (die feste Phase nicht mitgezählt). Diebinären Parameter Bij und nij müssen aus Gemischdaten ermittelt werden. Mit dieserGleichung kann ebenfalls ein konzentrationsabhängiger Trennfaktor αij beschriebenwerden.
2.2.4 Höhe einer Übertragungseinheit (Van Deemter Kurve)
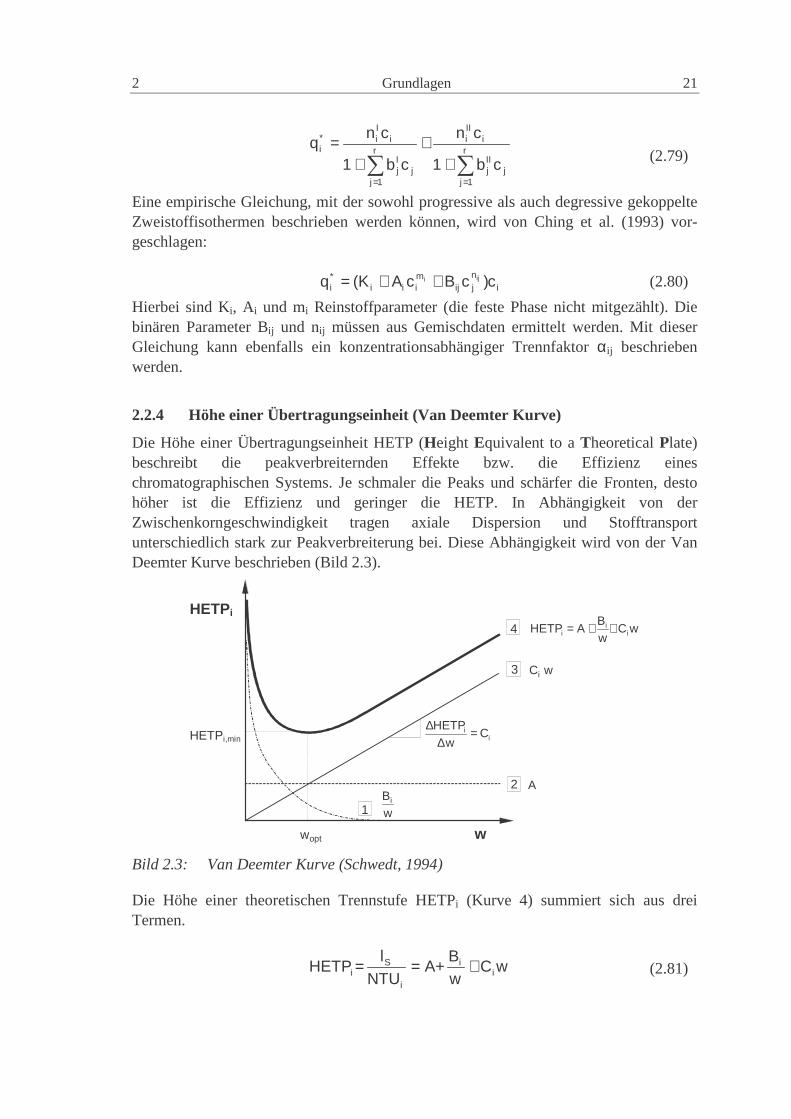
Die Höhe einer Übertragungseinheit HETP (Height Equivalent to a Theoretical Plate)beschreibt die peakverbreiternden Effekte bzw. die Effizienz eineschromatographischen Systems. Je schmaler die Peaks und schärfer die Fronten, destohöher ist die Effizienz und geringer die HETP. In Abhängigkeit von derZwischenkorngeschwindigkeit tragen axiale Dispersion und Stofftransportunterschiedlich stark zur Peakverbreiterung bei. Diese Abhängigkeit wird von der VanDeemter Kurve beschrieben (Bild 2.3).
2
3
1
4
w
HETPi
HETPi,min
wopt
Bw
i
wCwB
AHETP ii
i ++=
ii C
wHETP =∆
∆
C wi
A
Bild 2.3: Van Deemter Kurve (Schwedt, 1994)
Die Höhe einer theoretischen Trennstufe HETPi (Kurve 4) summiert sich aus dreiTermen.
wCwB
A+l
= ii
i
Si +=
NTUHETP (2.81)

22 Grundlagen 2
Die Parameter der Van Deemter Gleichung (1956) A, Bi und Ci sind für ein gegebenesSystem (Trennsäule, mobile Phase, zu trennende Komponente) und eine Temperaturkonstant.
• Kurve 1 erfasst den Anteil der molekularen Diffusion in axialer Richtung, derenEinfluss mit zunehmender Zwischenkorngeschwindigkeit abnimmt. In der Flüssig-keitschromatographie ist der Anteil der molekularen Diffusion so gering, dass dieserTerm meist vernachlässigt werden kann.
• Kurve 2 beschreibt den Anteil der Eddy-Diffusion, der Zu- und Abflusseffekte undder Strömungsungleichverteilung. Sie sind unabhängig von der Zwischenkorn-geschwindigkeit und der Molekülsorte. Der Wert der Konstante A wird durch diePartikelgröße und die Packungsgüte der stationären Phase bestimmt.
• Kurve 3 beschreibt den Anteil der Stoffaustauschphänomene, die mit steigenderZwischenkorngeschwindigkeit linear zunehmen (Meyer, 1992).
Der Zusammenhang zwischen der Van Deemter Kurve und den Parametern axialeDispersion Dax, Stoffübergangskoeffizient keff,i und Adsorptionsgleichgewichts-koeffizient K i wird durch die Gleichung (2.82) wiedergegeben. Sie wurde durchAnwendung der Momentenmethode (vergl. Gl. (2.62) - (2.64)) auf das Stufenmodellhergeleitet (Ruthven, 1984) und gilt daher nur im linearen Bereich der Adsorptions-isotherme.
( ) iext
ext
ext
ext
ieff,fest,i
ax
2
'1,i
2,iSi K�1
�1
�1�
Kkw2
wD2
�
�l
−
���
����
�
−+��
�
����
�
−+=�
�
�
�
��
�
�=HETP (2.82)
Wird die axiale Dispersion über Gl. (2.84) beschrieben, lässt sich ein Zusammenhang zuden Parametern der Van Deemter Gleichung herstellen.
( )
w C wB
A
K01
0101
0Kk
w2w
D21d21
ii
i
iext
ext
ext
ext
ieff,fest,i
m,i1P2i
++=
���
����
�
−+��
�
����
�
−++=
−
HETP
HETP������� �������� ��
�����
���
(2.83)
Die Messung der Van Deemter Kurve für einen Stoff, der nicht adsorbiert (der Term iC
fällt weg), ermöglicht die Bestimmung der Konstanten A und Bi. Die axiale Dispersion,die auch zur Beschreibung der Packungsgüte dient, ist dadurch unabhängig von denStoffaustauschphänomenen messbar.
2.2.5 Axiale Dispersion
Ein Peak erfährt beim Durchlaufen der Anlage eine zunehmende Verbreiterung, die miteiner gleichzeitigen Reduzierung der Peakhöhe einhergeht. Die Peakverbreiterung führtzu einer Verminderung der Trennleistung, da die Trennstufenzahl NTUi verringert und

2 Grundlagen 23
die Auflösung RS verschlechtert wird. Dieser als axiale Dispersion bezeichnete Effektfasst Stofftransportvorgänge zusammen, die in der mobilen Phase zusätzlich zumkonvektiven Transport auftreten. Dazu gehören:
- Strömungsungleichverteilungen: Durch Wandhaftung treten in der Nähe der Säulen-wand und auch zwischen den Partikeln Geschwindigkeitsunterschiede auf, so dassMoleküle, die sich in Wandnähe befinden, eine niedrigere Geschwindigkeit gegenüberdenjenigen im Kern der Strömung aufweisen. Eine zusätzliche Ursache einerUngleichverteilung ist das Verteilungssystem der Säule.
- Zu- und Abflusseffekte: An den Übergängen von kleinen (Zu- und Abflussleitungen)zu großen Strömungsquerschnitten (Trennsäule) wird das Geschwindigkeitsprofilverändert. Mit zunehmendem Säulenquerschnitt führt die radiale Verteilung zu einerVerzögerung der Moleküle, die an den Säulenrand transportiert werden.Untersuchungen haben ergeben, dass diese Effekte hauptsächlich zur Peakver-breiterung beitragen (Brandt, 1997), (Lisso et al., 2000).
- Eddy-Diffusion: Die Moleküle nehmen unterschiedliche Wege durch die Säule, da dieFlüssigkeit vielmals aufgeteilt und zusammengeführt wird. Daraus resultierenunterschiedliche Weglängen.
- Molekulare Diffusion: Aufgrund der hervorgerufenen Konzentrationsunterschiedetritt molekulare Diffusion sowohl in axialer als auch in radialer Richtung auf.
- Rückvermischungseffekte: Hervorgerufen z.B. durch Ventile oder plötzlicheLeitungsquerschnittsänderungen vor der Säule.
Die axiale Dispersion wird durch den axialen Dispersionskoeffizienten Dax charakteri-siert. Er ist von der Zwischenkorngeschwindigkeit w0, der Konzentration ci und von derTemperatur T abhängig. In der Literatur sind zahlreiche empirische Bestimmungsglei-chungen für die axiale Dispersion angegeben. Ruthven (1984) gibt eine Gleichung an,die ursprünglich für Gase entwickelt wurde. Sie berücksichtigt den Einfluss dermolekularen Diffusion Dm,i und einen vom Partikeldurchmesser dP und derZwischenkorngeschwindigkeit w abhängigen Term.
wd2D2D P2m,i1ax += (2.84)
Die Konstanten γ1 und γ2 haben für poröse Partikel normalerweise Werte von 50 und 0.5(Ruthven, 1984).
Eine gute Möglichkeit zur Vorausberechnung der axialen Dispersion geht von einerVereinfachung der van Deemter Gleichung (2.83) für nicht adsorbierende Substanzenaus (Altenhöhner et al., 1997), für die C Null wird und
w
D3d3
wB
A imP
i ,12i
22HETP +=+= (2.85)
Mit der von Ruthven (1984) vorgeschlagenen Gleichung (2.84) ergibt sich dann einZusammenhang für die Berechnung der axialen Dispersion aus den HETP derexperimentellen Peaks als

24 Grundlagen 2
2w
HETP ⋅=axD (2.86)
Die so berechneten Werte können als gute Startwerte für die axiale Dispersionverwendet werden. Die genauen Werte müssen aber durch den Vergleich mit denexperimentellen Peaks numerisch bestimmt werden (Altenhöhner et al., 1997), da soauch Anlageneffekte, wie z.B. Rückvermischung, berücksichtigt werden.
Für Flüssigkeiten kann die molekulare Diffusion imD , im allgemeinen vernachlässigt
werden, so dass die axiale Dispersion für alle Komponenten gleich groß ist. In dieserArbeit wird deshalb der axiale Dispersionskoeffizient nur als Funktion derZwischenkorngeschwindigkeit dargestellt:
wCD Daxax ⋅= (2.87)
Die Größe der Konstante des axialen Dispersionskoeffizienten ist hierbei von derstationären Phase und von den Anlagenkomponenten (Verteiler, Kapillarleitungen,Ventile, etc.) abhängig. Eine Unterteilung in einen phasenspezifischen Anteil und einenanlagenspezifische Anteil ist sinnvoll. Dieses Vorgehen wird in Kap. 0 dargestellt.
2.2.6 Stofftransportwiderstand
Der Stoffaustausch zwischen mobiler und stationärer Phase wird durch denStofftransportwiderstand behindert, der zu einer zeitlichen Verzögerung derGleichgewichtseinstellung führt. Durch die Adsorption wird ein Molekül einige Zeit ander Feststoffoberfläche festgehalten. Die mit der Strömung transportierten Molekülewandern den in den Poren befindlichen voraus, so dass die Stoffaustauschphänomene(Adsorption und Stofftransportwiderstand) mit zunehmender Fluidgeschwindigkeitverstärkt zu einer Peakverbreiterung beitragen.
Bild 2.4: Konzentrationsverlauf nach der Zweischichthypothese
Für den Stofftransport in das Innere der Partikel existieren in der frei strömendenFlüssigkeit (Bulkphase) und in den Partikeln Stofftransportwiderstände. In der Zwei-schichthypothese (Brauer, 1971) werden vereinfachend die Konzentrationsdifferenzen

2 Grundlagen 25
(an Stelle der realen chemischen Potentiale) als treibende Kraft für den Stofftransportangenommen. Eine Konzentrationsänderung tritt nur in den sogenanntenGrenzschichten (in der festen und der flüssigen Phase) in der Nähe derPhasengrenzfläche auf (Bild 2.4). An der hypothetischen Phasengrenzfläche herrscht einGleichgewicht, welches durch die Adsorptionsisotherme vorgegeben ist. In der Praxiswird der Stofftransportwiderstand häufig über einen linearisierten effektiven Stoffüber-gangskoeffizienten keff,i erfasst. Es werden dann die in den beiden Phasen auftretendenWiderstände zu einem effektiven Widerstand zusammengefasst, der entweder der festen(Festfilmmodell) oder der flüssigen Phase (Fluidfilmmodell) zugeordnet wird.
Die zeitliche Änderung der über die Partikel gemittelten Beladung, d.h. der Stoffstromin die Partikel bezogen auf das Partikelvolumen, wird wie folgt berechnet:
Festfilmmodell: )q(qkt
qi
*ieff,fest,i
i −=∂
∂(2.88)
Fluidfilmmodell: )c(cVO
kt
q *ii
Par
Pareff,fl,i
i −=∂
∂(2.89)
Der Stoffstrom �mi durch beide Grenzschichten muss betragsmäßig in beiden gleich
groß sein, so dass die effektiven Stoffübergangskoeffizienten der beiden Modelle fürlineare Adsorptionsisothermen ineinander überführt werden können.
)c(cOk)c(cKVkm *iiPareff,fl,ii
*iiPareff,fest,ii −−=−=� (2.90)
Pari
Pareff,fl,ieff,fest,i VK
Okk = (2.91)
Empirische Korrelationsgleichungen zur Berechnung der effektiven Stoffübergangs-koeffizienten sind in Ruthven (1984) und Deckert (1998) aufgelistet. Peev undTzibranska (1997) verwenden z.B. die Abhängigkeit der Sherwoodzahl von Reynolds-und Schmidtzahl für die Strömung in Flüssigkeiten bei kleinen Reynoldszahlen zurBerechnung von effk .
Der effektive Stofftransportwiderstand ist im einfachsten Fall von der Konzentration,der Geschwindigkeit und der Temperatur abhängig. Der Widerstand im Fluid ist von derZwischenkorngeschwindigkeit abhängig, da sie die Dicke der Grenzschicht beeinflusst.Der Widerstand in den Makroporen dagegen ist geschwindigkeitsunabhängig. Dereffektive Widerstand kann deshalb als geschwindigkeitsunabhängig betrachtet werden,wenn der Widerstand in den Poren der Partikel dominiert. Da diese Annahme für diebetrachteten Systeme vorausgesetzt wird, wird im weiteren Verlauf der Arbeit effk
geschrieben, obwohl eigentlich ifesteffk ,, gemeint ist.
2.2.7 Verfahrenstechnische Konzepte und Anlagen
Eine umfassende Darstellung der technischen Verfahren zur Umsetzung der adsorptivenund chromatographischen Trennprozesse ist z.B. bei Ganetsos und Barker (1993) zufinden. Die Verfahren lassen sich grundsätzlich nach diskontinuierlichen und

26 Grundlagen 2
kontinuierlichen Verfahren unterscheiden (dargestellt in Bild 2.5). Eine gute Übersichtist z.B. bei Seidel-Morgenstern (1995) zu finden.
Bild 2.5: Prinzip der a) diskontinuierlichen, b) kontinuierlichen Gegenstrom- und c)simulierten Gegenstromchromatographie (Broughton, 1984)
2.2.7.1 Diskontinuier liche Verfahren
Die diskontinuierliche chromatographische Trennung ist ein Chargenprozess, in demeine definierte Probenmenge aufgegeben und getrennt wird (Batch-Chromatographie).Je nach Probenmenge unterscheidet man nach Elutions- und Frontalchromatographie.Bei der Frontalchromatographie wird die Säule durch eine größere Probenmengevollständig gesättigt, es kommt zur Ausbildung einer Plateaukonzentration und einemDurchbruch der Substanz am Säulenende. In der Elutionschromatographie ist dieProbenmenge so gewählt, dass die Konzentration der Probe am Ausgang der Säule nichtdie Eingangskonzentration erreicht. Die Experimente dieser Arbeit wurden als Elutions-chromatographie durchgeführt.
Es existieren Weiterentwicklungen dieser Umsetzung in Hinblick auf dieProbenaufgabe (z.B. die Verdrängungschromatographie), die Pumprichtung (Flip-Flop-Chromatographie) oder auf die Verschaltung der chromatographischen Anlage(Rezyklierungstechniken, wie z.B. das Closed-Loop-Verfahren oder das Alternate-Pumping-Recycling).
2.2.7.2 Kontinuier liche Verfahren
Um die produzierte Menge des Produktes zu erhöhen, werden kontinuierliche Verfahreneingesetzt. Hierzu ist es notwendig, eine relative Bewegung der fluiden und der festenPhase zueinander zu erreichen.
Eine mögliche Realisierung ist die annulare Chromatographie, bei der Eluent undstationäre Phase im Kreuzstrom zueinander geführt werden. Das Verfahren und seine

2 Grundlagen 27
Anwendungen ist in den Arbeiten von Bart et al. (1996), Brozio und Barth (2000) undHerbsthofer et al. (2001) ausführlich beschrieben.
Eine andere Art der Realisierung ist der Gegenstrom. Es wird zwischen einemtatsächlichen Gegenstrom der festen und flüssigen Phasen und einem simuliertenGegenstrom durch geschickte Verschaltung von Chromatographiesäulen und Ventilenunterschieden (Bild 2.5). Ein umfassender Vergleich der tatsächlichen und dersimulierten Gegenstromchromatographie ist z.B. bei Deckert (1998) und Beste (2001a)zu finden.
2.3 Scale-Up Methoden für die Batch-Chromatographie
Die Maßstabsübertragung (Scale-up) dient zur Auslegung präparativer Anlagen aufBasis von Experimenten in möglichst kleinem Maßstab. Da das Verhalten vonProduktionsanlagen nur in wenigen Fällen ausreichend genau auf Basis vonLaborexperimenten berechnet werden kann, finden vor der endgültigen Auslegunghäufig Versuche im Technikumsmaßstab (semi-präparativ) Anwendung.
Im Folgenden werden Methoden zur Durchführung der Maßstabsübertragung vomanalytischen Maßstab zum Produktionsmaßstab vorgestellt.
2.3.1 Methode nach Lode et al.
Nach Lode et al. (1998) werden bei großen Strömungsgeschwindigkeiten und schnellerAdsorption chromatographische Säulen durch zwei Zeitkonstanten ausreichendbeschrieben. Die erste Zeitkonstante ist die Totzeit, bezogen auf eine nichtadsorbierende und nicht porengängige Komponente:
wl
wdld
V
Vt
ext
S
extS
SSext
⋅=
⋅⋅⋅⋅⋅⋅==
εεππ
2
2
0
44�
(2.92)
Die zweite betrachtete Zeitkonstante ist die Zeitkonstante der Diffusion:
i,m
pDiff D
dt
⋅=
6
2
(2.93)
Die Berechnung der intrapartikulären molekularen Diffusion kann nach Yamamoto(1995) vorgenommen werden. Im Fall der langsamen Adsorption (z.B. in Folge vonÜberladung der Säule), die dann den geschwindigkeitsbestimmenden Schritt darstellt,ist der Diffusionskoeffizient in der Partikel durch eine geeignete Adsorptionszeit-konstante zu ersetzen. Für ein Scale-up bei gleichbleibender Anzahl an Trennstufenmüssen dann die folgenden Größen konstant gehalten werden:
• Der Quotient aus den nach Gl. (2.92) und Gl. (2.93) bestimmten Zeitkonstanten,
• die dimensionslose Durchströmgeschwindigkeit im
P
Dwd
,
⋅ und
• die Länge der Säule.

28 Grundlagen 2
Zur Erfassung der Einflüsse der Systemperipherie (Ventile, Kapillarrohre, Verteiler undSammler) schlagen Lode et al. (1998) Versuche mit einer einzelnen Säule und mehrerenhintereinander geschalteten Säulen gleicher Trennstufenzahlen in der gleichenApparatur vor. Aus den erhaltenen Chromatogrammen werden die additiven zweitenzentralen Momente ermittelt und voneinander subtrahiert. Das resultierende Momentdient zum Anpassen einer Kurve, die nur den Säuleneinfluss wiedergibt. Zur Bestim-mung der mikroskopischen, irreversiblen Einflüsse innerhalb des Festbettes (wie z.B.Rückvermischungseffekte) schlagen sie das folgende Experiment vor: Nach Aufgabedes Peaks auf die Säule wird nach der halben Retentionszeit der Durchfluss gestoppt,die Säule umgedreht und weiter betrieben. Auf diese Weise werden die reversiblenEinflüsse aus dem Ergebnis eliminiert. Diese Ergebnisse sind in den obigen Regelnberücksichtigt worden.
2.3.2 Methode nach Heuer und Seidel-Morgenstern
Heuer und Seidel-Morgenstern (1996) schlagen für das Scale-up die folgende Vor-gehensweise vor:
Nach Festlegung der Methode im analytischen Maßstab wird für das analytische Systemdie Gesamtporosität (εges) durch die Auswertung disperser Fronten (ECP - Elution by
Characteristic Point) vermessen. Bedingung hierfür ist das Vorliegen einer großen Zahlan Trennstufen (> 1000). Die Adsorptionsisothermen werden experimentell vermessenund durch eine geeignete Isothermengleichung (z.B. bi-Langmuir) approximiert. AlsRegeln für das Scale-Up ergeben sich:
• Die Volumenströme werden ausgehend von der analytischen Methode um dasVerhältnis der Säulenquerschnittsflächen AS1 und AS2 vergrößert:
21
22
11
212
S
S
S
S
dd
VAA
VV ��� == (2.94)
• Die Masse des injizierten Stoffgemisches ist im gleichen Verhältnis wie dasSäulenvolumen zu vergrößern:
12
1
22
21
1
212
SS
SSinjinjinj lD
lDm
VV
mm⋅⋅== (2.95)
Zur Beschreibung des Verhaltens der präparativen Säule wird ausgehend von denexperimentell ermittelten Daten eine Simulationsrechnung basierend auf demDispersionsmodell (Seidel-Morgenstern, 1995) durchgeführt:
Komponentenbilanz:
2
2
zc
Dzc
wt
qF
tc i
axiii
∂∂=
∂∂+
∂∂+
∂∂
(2.96)
Axiale Dispersion:

2 Grundlagen 29
NTUlu
D Sax ⋅
⋅=2
(2.97)
Der axiale Dispersionskoeffizient dient zur Quantifizierung aller Störeinflüsse, die zurBandenverbreiterung führen (2.2.5). Störungen in radialer Richtung, vor allemverursacht durch Verteilungsprobleme der Aufgabesysteme, wirken sich letztendlichauch als bandenverbreiternd aus. Dies führt zu einer Ausdehnung des Peaks in axialerRichtung in der Rohrleitung zwischen Sammler der Säule und Detektor. Im axialenDispersionskoeffizienten sind somit integral die axialen Störeinflüsse und die axialenProjektionen der radialen Störungen zusammengefasst.
Die Berechnungen nach Gl. (2.97) beruhen auf dem Vergleich zweier mathematischerModelle der Chromatographie (dem Zellenmodell und dem Dispersionsmodell) für einegroße Anzahl an Trennstufen, schnelle Adsorption, lineare Adsorptionsisothermen undeine Peakaufgabe in Form eines Dirac-Stoßes. Gemischisothermen werden durchAnwendung der "Ideal Adsorbed Solution theory" (IAS) unter Verwendung der imanalytischen System bestimmten Einzelisothermen berechnet:
( )iii cccqq ,,, 21 �= (2.98)Lineare Geschwindigkeit:
Sges
S
AV
tl
u⋅
==ε
�
0
(2.99)
Phasenverhältnis:
Vt
VtVV
VF S
gestFlüssigkei
Feststoff
ges
gesex �
�
⋅⋅−==
−=
0
0
,
1
εε
(2.100)
Da die Bestimmung dieses experimentellen Phasenverhältnisses durch Messung derRetentionszeit einer nicht adsorbierenden aber porengängigen Substanz bei großenSäulen zur zeitlichen Verschiebung der simulierten Chromatogramme gegenüber dengemessenen Peakverläufen des Trennsystems führt, wird eine auf der Vermessung derinteressierenden Komponente beruhende Vorgehensweise verwendet:
Hierfür wird auf die präparative Säule eine analytische Probenmenge der reineninteressierenden Komponente aufgegeben. Aus dem resultierenden Peakverlauf erhältman die Trennstufenzahl für diese Komponente und hieraus den axialen Dispersions-koeffizienten (nach Gl. (2.97)). Desweiteren kann mit der folgenden Gleichung ein fürdie Simulation geeigneteres Phasenverhältnis bestimmt werden:
SrefthR
RSth VKVt
VtVF
⋅−⋅⋅−=
,�
�
(2.101)
Kth,ref stellt in dieser Gleichung die Henry-Konstante bzw. Anfangssteigung der
Adsorptionsisothermen dar, welche aus Referenzversuchen mit der analytischen Säulezur Verfügung steht.
30 Grundlagen 2
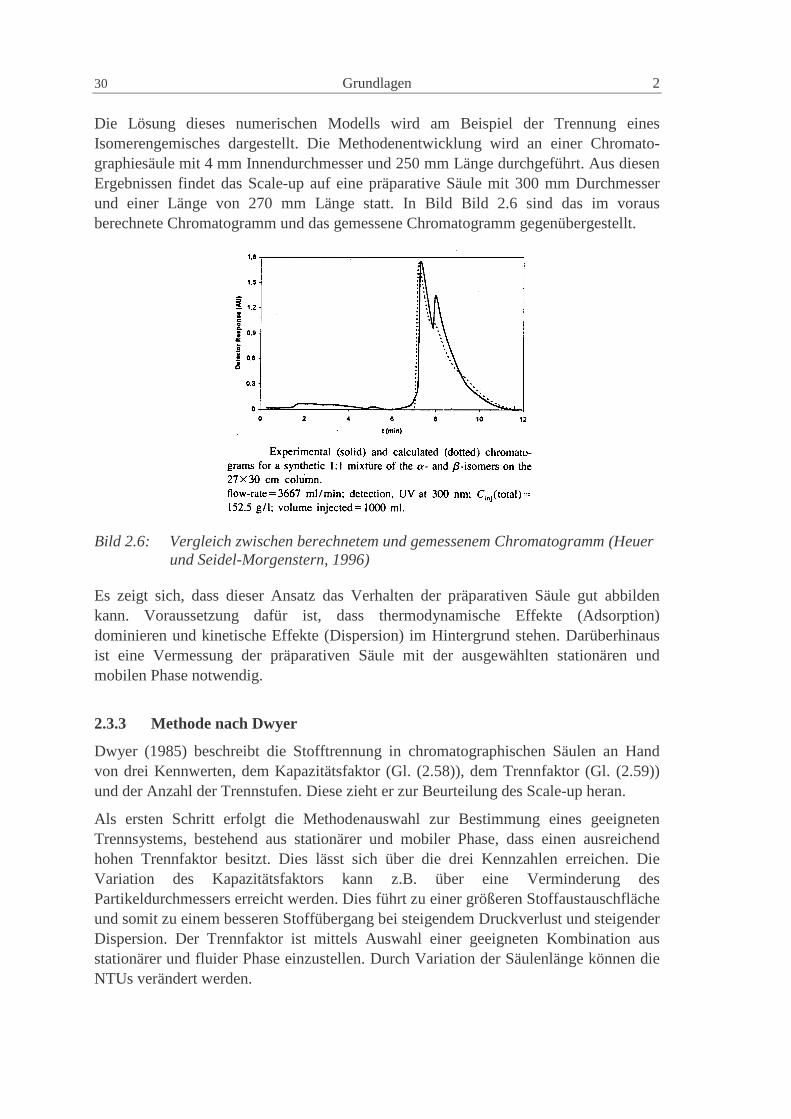
Die Lösung dieses numerischen Modells wird am Beispiel der Trennung einesIsomerengemisches dargestellt. Die Methodenentwicklung wird an einer Chromato-graphiesäule mit 4 mm Innendurchmesser und 250 mm Länge durchgeführt. Aus diesenErgebnissen findet das Scale-up auf eine präparative Säule mit 300 mm Durchmesserund einer Länge von 270 mm Länge statt. In Bild Bild 2.6 sind das im vorausberechnete Chromatogramm und das gemessene Chromatogramm gegenübergestellt.
Bild 2.6: Vergleich zwischen berechnetem und gemessenem Chromatogramm (Heuerund Seidel-Morgenstern, 1996)
Es zeigt sich, dass dieser Ansatz das Verhalten der präparativen Säule gut abbildenkann. Voraussetzung dafür ist, dass thermodynamische Effekte (Adsorption)dominieren und kinetische Effekte (Dispersion) im Hintergrund stehen. Darüberhinausist eine Vermessung der präparativen Säule mit der ausgewählten stationären undmobilen Phase notwendig.
2.3.3 Methode nach Dwyer
Dwyer (1985) beschreibt die Stofftrennung in chromatographischen Säulen an Handvon drei Kennwerten, dem Kapazitätsfaktor (Gl. (2.58)), dem Trennfaktor (Gl. (2.59))und der Anzahl der Trennstufen. Diese zieht er zur Beurteilung des Scale-up heran.
Als ersten Schritt erfolgt die Methodenauswahl zur Bestimmung eines geeignetenTrennsystems, bestehend aus stationärer und mobiler Phase, dass einen ausreichendhohen Trennfaktor besitzt. Dies lässt sich über die drei Kennzahlen erreichen. DieVariation des Kapazitätsfaktors kann z.B. über eine Verminderung desPartikeldurchmessers erreicht werden. Dies führt zu einer größeren Stoffaustauschflächeund somit zu einem besseren Stoffübergang bei steigendem Druckverlust und steigenderDispersion. Der Trennfaktor ist mittels Auswahl einer geeigneten Kombination ausstationärer und fluider Phase einzustellen. Durch Variation der Säulenlänge können dieNTUs verändert werden.

2 Grundlagen 31
Nun wird die Methode an die vorhandene Anlage im Labormaßstab angepasst. Hierfürgibt Dwyer Erfahrungswerte für den Eluentenstrom (Ersatz des Säulenleervolumensinnerhalb von 5 bis 10 Minuten), die Beladung (erhöhen, bis die Peaks aufgrund derauftretenden Überschneidungen gerade noch als Einzelpeaks zu erkennen sind; in derRegel 10 bis 100 mg Stoffgemisch auf 1 g stationäre Phase) und die Produktentnahme-zeiten (durch Probennahme zu verschiedenen Zeitpunkten).
Für das Scale-up ist der Volumenstrom der mobilen Phase so anzuheben, dass die Leer-rohrgeschwindigkeit konstant bleibt (vergl. Gl. (2.94)). Die Beladung ist direktproportional zur Masse der stationären Phase zu erhöhen. Die Chromatogramme derpräparativen Anlage sollen denen der analytischen Anlage stark ähneln. Die Schnitte inder präparativen Säule müssen noch durch Probennahme zu geeigneten Zeitpunktenüberprüft werden.
2.3.4 Bewertung der Methoden
Der Ansatz von Dwyer ist ein reiner Erfahrungsansatz und ohne Modellierung derSäulen zu lösen, allerdings mit hohem experimentellen Aufwand verbunden. Für den fürtechnische Trennprozesse am häufigsten auftretenden Fall, dass die thermodynamischenEffekte gegenüber den kinetischen Aspekten dominieren, bietet sich die Methode nachSeidel-Morgenstern an. Ausgehend von Laborversuchen zur Auslegung des Trenn-prozesses ist mit den Gleichungen (2.94) und (2.95) ein einfaches und effektives Scale-up auf den präparativen Maßstab möglich. Unter Berücksichtigung anlagenspezifischerEinflüsse (wie z.B. in Kapitel 0 beschrieben) lässt sich das Verhalten der grösseren,präparativen Anlage sehr gut mit Hilfe der axialen Komponentenbilanz beschreiben.Alternativ kann jedoch auch ein Ausschluss der anlagenspezifischen Effekte nach Lodeund Lightfoot durchgeführt werden, welcher aber für präparative Systeme einen nichtunwesentlichen Umbauaufwand bedeutet. Innerhalb dieser Arbeit wird der Ansatz vonSeidel-Morgenstern verwendet.
2.4 Magnet-Resonanz-Tomographie (MRT)
Die Magnet-Resonanz-Tomographie (engl.: Nuclear Magnetic Resonance tomography,NMR) beruht auf der Magnetisierung der Wasserstoffatome durch ihren Spin. Dies istdie Drehung des Atoms um seine Achse, was im Fall des Wasserstoffs bedeutet, dasssich ein Proton und ein Elektron umeinander drehen. Diese Drehung erzeugt einmagnetisches Feld. Durch ein starkes äußeres Dauermagnetfeld ordnen sich die Spinsparallel oder antiparallel zum Dauermagnetfeld an, wobei die parallele Ausrichtungwegen ihres geringeren energetischen Zustands bevorzugt ist. Zusätzlich führen dieSpins eine Kreiselbewegung mit der Achse parallel zum Magnetfeld aus. Die Frequenzdieser Kreiselbewegung wird von der Larmor-Gleichung beschrieben:
00 B4 ⋅=ϖ (2.102)
( 5 : magnetische Präzessionsrate, B: Feldstärke, ω: Resonanzfrequenz)

32 Grundlagen 2
Die Summe der Präzessionen ihrer Spins zeigt parallel in z-Richtung (longitudinaleMagnetisierung). Die Protonen werden durch einen magnetischen Impuls im MHz-Bereich mit ihrer Resonanzfrequenz angeregt.
Bild 2.7: Anregung der Spins der Wasserstoffatome
Durch diesen Impuls werden einige der Spins der Wasserstoffatome in die antiparallelePräzession gehoben (höheres Energieniveau). Die Summe aller Präzessionen zeigt jetztnicht nur betragsmäßig weniger in z-Richtung (kleinere longitudinale Magnetisierung),sondern es kommt noch ein Anteil in y-Richtung hinzu (transversale Magnetisierung,siehe Bild 2.7). Nach dieser Anregung kehren die Protonen vom hohen Energieniveauwieder auf das niedrigere Niveau zurück. Den Verlauf der Magnetisierung über derZeit, bis alle Protonen wieder auf dem ursprünglichen Energieniveau gelandet sind,nennt man Relaxationszeit. Die longitudinale Magnetisierung erreicht wieder ihren altenZustand (longitudinale Relaxationszeit, T1), die transversale Magnetisierungverschwindet wieder (transversale Relaxationszeit T2 oder FID: Free Induction Decay).Diese Methode wird auch als Pulsed Field Gradient Magnet-Resonanz-Tomographie(PFG-MRT) bezeichnet. Sie liefert je nach Messeinstellungen entweder statische Spin-verschiebungen (wie z.B. die Konzentration an Protonen) oder dynamischeSpinverschiebungen (wie die relativen Geschwindigkeiten der Protonen).
Eine Weiterentwicklung ist die sogenannte Pulsed Field Gradient Spin-Echo Magnet-Resonanz-Tomographie (PFGSE-MRT). Sie ist in der Lage, gleichzeitig statische unddynamische Informationen über die Spinverschiebungen zu liefern. Hierbei werden zurFlussquantifizierung getaktete 90° und 180° Pulse aufgegeben, die ein Spinechoerzeugen, das zur Auswertung herangezogen wird:

2 Grundlagen 33
Bild 2.8: Spinechoerzeugung in der MRT
Ein Spinensemble, das mit hoher Geschwindigkeit senkrecht zur Messschicht fliesst,gibt ein schwaches Signal ab, ein mit geringer Geschwindigkeit fließendes Ensembleein starkes Signal. Die Signale werden vom Magnetom auf Grauwerte (z.B. für dasMagnetom Harmony 4096) abgebildet ( limmin ww −= ; limmax ww = ). Die Signale sind
hauptsächlich abhängig von der Schichtstärke d, der Echozeit TE und derGeschwindigkeit des Ensembles. Die Flussgeschwindigkeit limw , bei der eine maximale
Einstromverstärkung (beste Auflösung) auftritt, hängt von der Schichtdicke d und derWiederholzeit TR ab:
TRd
w =lim (2.103)
Die Ergebnisse können im Amplitudenbild oder im Phasenbild dargestellt werden.Beide unterscheiden sich in ihrem Informationsgehalt: Das Amplitudenbild liefert dieAmplituden und Richtungen der Bildgradienten (stationäre Verschiebung - staticdisplacement distribution, k-space), während das Phasenbild die dynamischenVerschiebungen der Bildgradienten innerhalb der Messzeit (dynamic diplacementdistribution, q-space) enthält. Durch eine sehr spezialisierte Auswertungssoftwarekönnen diese Signale der Spinrelaxation und der Spinbewegung in Geschwindigkeitensenkrecht zu einer gegebenen Schicht konvertiert werden.
Für das grundsätzliche Verständnis der Datenauswertung zur Bestimmung der makros-kopischen Geschwindigkeiten soll hier noch ein kurzer Überblick der Gleichungengegeben werden (nach Chang und Watson, 1999):
Die räumliche Information erhält man durch die Einführung eines zeitabhängigen,räumlichen Gradienten (t)g des Magnetfeldes in der Art, dass die Oszillationsfrequenz
der transversalen Magnetisierung über die Probenschicht variiert:
( ) ( ) rg ⋅=′ t6rϖ (2.104)Dessen Signal besitzt die folgende Form (Callaghan, 1991):
( ) ( )� ⋅= rrkr di7kS exp)( (2.105)
mit

34 Grundlagen 2
( )� ′′=t
tdt60
gk (2.106)
und der Spindichte ( )r8 an der Koordinate r mit der magnetischen Präzessionsrate 9 .
Der Zusammenhang zwischen Messsignal und Spindichte als Fouriertransformationstellt die Grundlage jeder Auswertung dar. Innerhalb des Signals ist die räumlicheInformation phasenkodiert in der x-Richtung und frequenzkodiert in der z-Richtungenthalten. In der Zeit : zwischen den geschwindigkeitskodierten Gradientenimpulsenwerden die Bahnen der Flüssigkeitsmoleküle verfolgt. Für zwei geschwindigkeits-kodierte Gradientenimpulse der Amplitude G und der Breite ; erhält man die Phasen-verschiebung Φ durch einen Spin zum Zeitpunkt t , der innerhalb eines Gradienten
( )tg ′ dem Weg ( )t ′r folgt, als
( ) ( ) ( )� ′′⋅′=t
tdtt6t0
rgΦ (2.107)
Unter der Annahme, dass die Dauer ; eines Pulses klein genug ist, um die molekulareVerschiebung in diesem Zeitraum gegenüber der Verschiebung in der Beobachtungs-zeit : vernachlässigen zu können, erzeugt der erste Impuls eine Phasenverschiebung
0rG ⋅6=< gegenüber eines Spins an der Position 0r . Diese Phasenverschiebung wird
durch die folgenden beiden 90° Impulse der Echosequenz invertiert. Nimmt manweiterhin an, dass der Spin nach dem zweiten Impuls die Position 0rR + erreicht hat,
ergibt sich die Nettoverschiebung für den Spin als RG ⋅> ; . Wie in Gl. (2.106) wird einweiterer Wellenvektor q eingeführt, um die durch molekulare Bewegung hervor-
gerufene Phasenmodulation zu beschreiben:
Gq > ;= (2.108)Während k mit der Position r des Spins verbunden ist (k-space), stellt q die
Verbindung zur Verschiebung R der Spins her (q-space). Mit der Kombination ausgeschwindigkeitskodierten Gradienten und statischer Bilderfassung ist es möglich, eineVerteilungsfunktion der Spinbewegungen in Volumenelementen (Voxel) der Probe zuerhalten. Die Spindichte ( )r8 kann nach Moran (1982) zur kombinierten Spindichte-
funktion vereinfacht werden:
( ) ( ) ( )rRrRr ,P8,8 ?? ′= (2.109)wobei ( )rR,P? ′ die normierte Verteilungsfunktion der Spinverschiebungen der Zeit :in einem Voxel an der Position r ist. In der Definition von ( )Rr ,8 ? sind die
Relaxationen der Spins enthalten. Diese Funktion ist der Anzahl an Spins im räumlichenVolumenelement proportional. Das gemessene Signal wird durch die beiden Wellen-vektoren k und q moduliert und kann ausgedrückt werden als
( ) ( ) ( ) ( ) RrRqrkRrqk ddiexpiexp,@,S A ⋅⋅= �� (2.110)
Für die Geschwindigkeitsbestimmung definiert man sich den zeitlichen Mittelwert derGeschwindigkeit jedes Spins, z.B. in z-Richtung, als

2 Grundlagen 35
( ) ( ):
z:z:Z
w0−== (2.111)
wobei der Nenner die Verschiebung in z-Richtung wiedergibt. Das MRT-Signal für einVoxel an der Position r ist dann die Fouriertransformation von Gl. (2.110) nach k :
( ) ( ) ( ) ( )�∞
∞−
′=′ ZdZiexp,ZP,S B qrrqr ρ
( ) ( ) ( ) ( )�∞
∞−
=′ wd:wiwPS B qrrqr exp,, ρ(2.112)
Die Geschwindigkeitsverteilungsfunktion ( )r,wPB ′ ist die Wahrscheinlichkeitsdichte an
Spins, die sich mit einer Geschwindigkeit von w bis wdw + bewegen. Die mittlereGeschwindigkeit für ein Voxel ist dann:
( )�∞
∞−
= wdwPww Br
r, (2.113)
Zur Bestimmung der mittleren Geschwindigkeit eignet sich einerseits die Phasen-verschiebung des gemessenen Signals (Gl. (2.112)) unter Verwendung einer Referenzmit der Geschwindigkeit 0, oder andererseits eine Auswertung der Geschwindigkeits-verteilungsfunktion unter Verwendung von Gl. (2.113).
Für detailliertere Beschreibungen der PFGMRT und der PFGSEMRT sei auf dieArbeiten von Tallarek et al. (1995, 1998a) und Chang und Watson (1999) verwiesen.
2.5 Messung von radialen Verteilungen
2.5.1 Temperaturprofile
Messungen der Temperaturprofile innerhalb von chromatographischen Säulen sindnicht nur zur Validierung der numerischen Modellierung sinnvoll sondern gewähreneinen guten Einblick in die Vorgänge, die bei der Unterkühlung des Eluenten gegenüberder Säulenwand auftreten und zu einer Verbesserung der Trennleistung führen.Ausserdem lassen sie einen Rückschluss auf die Verteilungsgüten der eingesetztenSäule zu.
Eine Temperaturdifferenz innerhalb einer präparativen Säule kann auch durchexotherme oder endotherme Mischung der Lösungsmittel, Veränderung der Lagerungs-temperatur in den Vorlagen oder der Umgebungstemperatur (bei nicht temperiertenSäulen) und eine Erwärmung der Lösungsmittel durch von der Pumpe erzeugte Wärmeerzeugt werden. Da die Wärmeleitfähigkeit des Bettes gering ist und innerhalb der Säuleeine laminare Plugflow Strömung mit wenig radialer Vermischung herrscht, ist derWärmetransport in der Säule schlecht und es stellen sich radiale Temperaturgradienten

36 Grundlagen 2
ein. Diese führen zu einer Verbreiterung der Peaks, da sie sowohl die lokalenGeschwindigkeiten als auch die Adsorptions- und Desorptionseigenschaften verändern.
In der Literatur wurden deshalb die meisten Methoden zur Messung der Temperatur-profile zur Untersuchung des Einflusses der Unterkühlung des Eluenten auf die Trenn-leistung entwickelt. Dieser Einfluss wird von O. Dapremont et al. (1998) beschrieben.Anhand eines mathematischen Modells unter Berücksichtigung linearer Adsorptions-isothermen beschreibt er die Auswirkungen eines nicht-isothermen Betriebs auf dasPeakverhalten. Das mathematische Modell beruht auf der Lösung der Energiebilanz inZylinderkoordinaten (siehe Tabelle 2.2). Der Einfluss der Grösse der Temperatur-differenz, des Volumenstroms und der Säulengeometrie (Länge und Durchmesser)führen ihn zum Ergebnis, dass eine gute Regelung der Temperaturen des Eluenten undder Säulenwand während der Trennung für das Erreichen einer guten Trennungnotwendig sind. Kleine Differenzen zwischen Eluent und Wand können zu einerKompensation der Ungleichverteilung bei der Aufgabe auf das Festbett und damit zueiner Optimierung der Trennleistung genutzt werden.
Temperaturfühler wurden von A. Brandt (1997) in eine präparative Säule (ProchromPackstand, Säulendurchmesser 60 mm, Bettlängen 35 bis 300 mm) eingebaut. ZumEinbau hat er den Säulendeckel so umgebaut, dass die 6 Fühler in Schutzhülsen überden Radius verteilt 30 mm in das gepackte Bett ragen. Der Einfluss der Messfühler aufdie HETP der Säule wird von A. Brandt mit 55 % bei optimaler linearer Geschwindig-keit angegeben. Mit Hilfe dieses Systems hat er den Einfluss der Temperatur aufThermodynamik und Stoffaustausch, die kalorische Erwärmung im chromatogra-phischen Bett und die Auswirkungen von Temperaturgradienten im Säulenbettuntersucht. Die entstehenden Temperaturfelder innerhalb der Säule hat er durch Lösungeiner vereinfachten Energiebilanz (Gl. (2.43), Vereinfachungen siehe Brandt, 1997),aufgestellt von Poppe et al. (1981), berechnet:
zP
wzT
C8wrT
rrTC
zTC
tT8 C PmradaxP ∂
∂+∂∂−��
�
����
�
∂∂+
∂∂+
∂∂=
∂∂ 1
2
2
2
2
(2.114)
Den Term zP
∂∂
hat er nicht mit dem Gesetz von Darcy (Gl. (2.50)) beschrieben, sondern
die gemessenen Werte des Druckabfalls über das chromatographische Bett verwendet.
2.5.2 Konzentrationsprofile
Die in der Literatur angewandten Methoden lassen sich grob in Messungen an derlaufenden Anlage (zeitliche Verläufe) und in Messungen nach Abschalten der Anlage(nur ein Zeitpunkt) unterteilen.
Der von Jones et al. (1996) gewählte Aufbau zur Messung der Konzentrationen (Bild2.9) über den Radius am Säulenaustritt ist analog der Messung der Temperaturprofileausgelegt, nur das die Messfühler nicht in das Säulenbett ragen.

2 Grundlagen 37
Bild 2.9: Experimenteller Aufbau des Systems von Jones et al. (1996)
Die Experimente wurden in einem vertikalen Bett (L=81 bzw. 165 mm) aus PyrexGlaskugeln einheitlicher Form und Grösse (3,0 mm, 1,5 mm, 0,275 mm) in einemspeziell für die Messungen gebauten Zylinder (ID=175 mm) durchgeführt. DieMessfühler mit einem Gesamtdurchmesser von 0,23 mm bestehen aus jeweils zweioptischen Fasern, von denen die eine durch einen Argon-Ionen-Laser (λ=488 nm)gespeist wird und am Säulenbett endet. Die zweite führt das durch das Laser-Lichtangeregte Fluoreszenssignal des als Probe verwendeten Farbstoffs Rhodamine 6G aneinen Empfänger, der nach Durchlaufen eines Bandfilters (λ=590 nm) die Intensität desSignals an einer Photodiode detektiert. Durch die Konstruktion der Messfühler wird nurein kleines Volumen direkt am Ende der Fühler angeregt. Über den Säulenradiuskommen 61 Messfühler zum Einsatz, die durch Bohrungen in der Auslassfritte dasBettende erreichen.
Experimente wurden mit deionisiertem Wasser und 5 ml (1000 ppm) Rhodamine 6G alsTracer bei Volumenströmen von 12,5 bis 53 ml/min und einer Temperatur von 25 °Cdurchgeführt. Sie lieferten den zeitlichen Verlauf der Tracerkonzentrationen an jedemMesspunkt vor der Austrittsfritte, wobei defekte Sensoren durch Vergleich mitReferenzmessungen ohne stationäre Phase festgestellt und ausgeschlossen wurden.
Die Ergebnisse wurden von Jones zur Charakterisierung der Strömung innerhalb derstationären Phase und zur Validierung des Modells der idealen Dispersion (segregatedflow) verwendet. Das wichtigste Ergebnis ist, dass Jones zu einer Abschätzung kommt,nach der 17-33 % der auftretenden Dispersion durch Veränderung der Retentionszeitdurch querlaufende Effekte, die auf einem Maßstab grösser als der Partikelradiuswirken, entsteht.
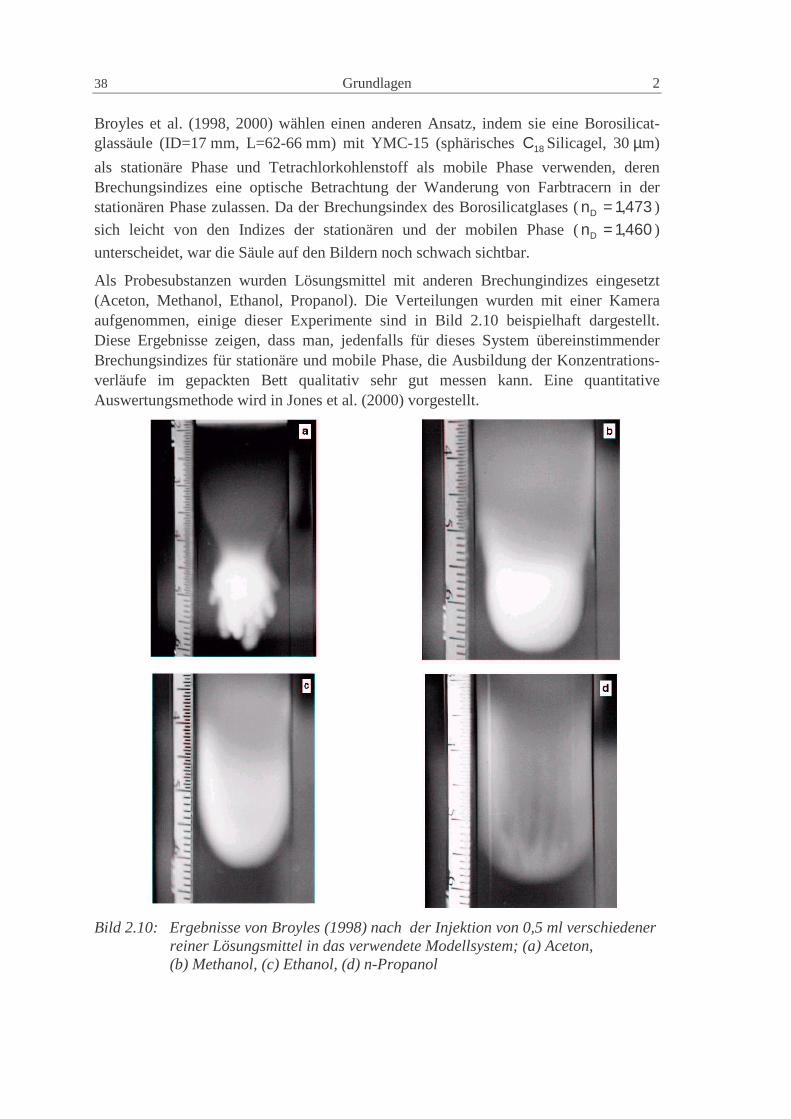
38 Grundlagen 2
Broyles et al. (1998, 2000) wählen einen anderen Ansatz, indem sie eine Borosilicat-glassäule (ID=17 mm, L=62-66 mm) mit YMC-15 (sphärisches 18C Silicagel, 30 µm)
als stationäre Phase und Tetrachlorkohlenstoff als mobile Phase verwenden, derenBrechungsindizes eine optische Betrachtung der Wanderung von Farbtracern in derstationären Phase zulassen. Da der Brechungsindex des Borosilicatglases ( 4731,nD = )
sich leicht von den Indizes der stationären und der mobilen Phase ( 4601,nD = )
unterscheidet, war die Säule auf den Bildern noch schwach sichtbar.
Als Probesubstanzen wurden Lösungsmittel mit anderen Brechungindizes eingesetzt(Aceton, Methanol, Ethanol, Propanol). Die Verteilungen wurden mit einer Kameraaufgenommen, einige dieser Experimente sind in Bild 2.10 beispielhaft dargestellt.Diese Ergebnisse zeigen, dass man, jedenfalls für dieses System übereinstimmenderBrechungsindizes für stationäre und mobile Phase, die Ausbildung der Konzentrations-verläufe im gepackten Bett qualitativ sehr gut messen kann. Eine quantitativeAuswertungsmethode wird in Jones et al. (2000) vorgestellt.
Bild 2.10: Ergebnisse von Broyles (1998) nach der Injektion von 0,5 ml verschiedenerreiner Lösungsmittel in das verwendete Modellsystem; (a) Aceton,(b) Methanol, (c) Ethanol, (d) n-Propanol

2 Grundlagen 39
Hauptziel der Untersuchungen von Broyles war die Untersuchung der Ausbildung vonviskosen Ungleichverteilungen (viscous fingering). Viskose Fingerbildung trittbesonders bei grossen Unterschieden (> 10 %) in der Viskosität der Lösungsmittel auf(vergl. Bild 2.10, A). Je geringer die Viskositätsunterschiede, desto mehr bildet sich einungestörtes Plug-Flow Profil innerhalb des Bettes aus. Dieses Phänomen trittunabhängig von der Güte der Packung in der Säule auf. Der Einfluss der Bettqualität,der Lösungsmittel, des Probenvolumens und des Volumenstroms wird von Broylesausführlich dargestellt. Experimente, in denen nach Ausbildung der viskosen Ungleich-verteilungen die Strömungsrichtung in der Säule umgekehrt wurde, zeigen, dass dieseUngelichverteilungen reversibel sind und nur der Einfluss der axialen Diffusion und derEddy-Dispersion irreversibel sind. Eine viskose Ungleichverteilung ist gut an einemstark erhöhten Pumpenvordruck der Säule zu erkennen. Der Einfluss auf die Trennungist zusätzlich abhängig von der Adsorptionsisothermen und kann z.B. für eineLangmuir-Isotherme vernachlässigt werden. Probleme können durch Reduzieren desVolumenstroms oder Verringerung der Viskosität der Probe beseitigt werden.
Diese Methode bietet ein großes Potenzial zur Messung von Konzentrationsprofilen inchromatographischen Säulen, ist allerdings gleichzeitig durch die Bedingung derVerwendung von stationärer und mobiler Phase gleicher Brechungsindizes auf wenigeKombinationen eingeschränkt. Zusätzlich reduziert nach Broyles die geringeDruckbelastbarkeit der Glasssäulen die möglichen Kombinationen aus Volumenströmenund viskosen Lösungsmitteln (Probe und Eluent). Die quantitative Auswertung (Broyleset al., 2000) zeigt, dass eine Erweiterung der Methode ein Ansatz zur indirektenMessung der Geschwindigkeiten der Bandprofile ist.
Bild 2.11: Photo der Farbverteilungen in der Säule nach Brandt (1997)

40 Grundlagen 2
Brandt (1997) bestimmt die Konzentrationsprofile innerhalb des gepackten Betts, indem er ein Farbgemisch viermal in kurzen zeitlichen Abständen bei jeweiligerUnterbrechung der Pumpe injiziert. Bevor das zuerst injizierte Gemisch die Säuleverlässt, hat er die Pumpe gestoppt und das gepackte Bett aus der Säule gepresst. DurchAufschneiden des Kerns in der Mitte sind die Konzentrationsverteilungen durchverschiedene Farbzonen zu erkennen (Bild 2.11). Dargestellt sind die sich ergebendenVerläufe für gleiche Mantel- und Eluenttemperatur und für Eluentunterkühlung.
Zur Untersuchung der Konzentrationsverläufe innerhalb des chromatographischenBettes wurde auch die Magnet-Resonanz-Tomographie eingesetzt, z.B. von Tallarek etal. (1995) und Bayer et al. (1995). In seiner Arbeit verwendet er einen Magnetom 63(Fa. Siemens, Erlangen) zur Messung der Gadolinium-Ionen Verteilungen innerhalbeiner Merck Superformance Säule (ID=26mm, L=135mm), gepackt mit Lichrospher15 µm (Fa. Merck, Darmstadt). Ziel der Untersuchungen ist der Einfluss vonfehlerhaften Injektionen, verschmutzten Fritten und eines fehlerhaften Säuleeinlassesauf die Konzentrationsverteilung in der Säule. Hierbei kann die Wanderung derGadoliniumionen durch die Säule beobachtet werden, da diese Ionen die Relaxationszeitder Protonen in ihrer Nähe deutlich reduzieren. Allerdings ist eine Kalibration diesesEinflusses schwierig. Für die Messungen hat er eine Solenoid-Spule verwendet. DieMesseinstellungen waren 1 mm für die Schichtdicke der Messebene entlang derSäulenachse, ein Field of View (FOV) von 166*166mm, eines Säulenquerschnitts von26*135 mm mit einer Auflösung von 256*256 Graustufen (ergibt eine Pixelgrösse von0,65*0,61 mm), bei einer Messzeit von 7 s. Die Auswertung der Messdaten gibt er alsschwierig an, kommt aber dennoch zu folgenden Ergebnissen: Für die fehlerfreie Säuleerhält er eine gleichmässige Verteilung (Plug-Flow Profil) der Gadoliniumfrontinnerhalb des Bettes. Die Einflüsse der eingestellten Fehler sind gut zu erkennen undführen zu einer deutlichen Ungleichverteilung der Front und damit zu einer Erhöhungder HETP der Trennung. Allgemein stellt er folgendes klar:
Die MRT eignet sich gut, um die lokalen Konzentrationen innerhalb der Säule sichtbarzu machen. Für diese statische Messung ist der Einfluss der Geschwindigkeit auf dasMessergebnis durch geeignete Einstellung der Relaxationszeit gering. Für einedynamische Auswertung, z.B. zur Bestimmung der relativen Geschwindigkeiten, isteine andere Relaxationszeit erforderlich, allerdings reicht die Auflösung seinerExperimente nicht für eine Auswertung aus.
2.5.2.1 Modellierung in FLUENT
Die Ergebnisse der Konzentrationsmessungen von A. Brandt (1997) wurden in denArbeiten von Lisso et al. (2000), Wu (2000) und Lisso (2001) zur Validierung derModellierung in FLUENT V4 und von Boysen et al. (2001a und 2001b) in FLUENT V5verwendet und werden hier nicht weiter betrachtet. Als Ausblick an dieser Stelle bietetsich eine zusätzliche Auswertung der Arbeiten von Broyles et al. (1998, 2000) für dieModellierung in FLUENT V5 an.

2 Grundlagen 41
2.5.3 Geschwindigkeitsprofile
Die PFG-MRT ist von Tallarek et al. (1995) und Bayer et al. (1995) (Kapitel 2.5.2) auchzur Bestimmung der relativen Geschwindigkeiten in gepackten Säulen eingesetztworden. Allerdings ließ die Auswertung der Ergebnisse keine ausreichende Auflösungzur Bestimmung der absoluten Geschwindigkeiten in der Säule zu. Die Ergebnisse vonTallarek et al. ergeben ein Plug-flow Profil für die Konzentration an Gadolinium,welches auf eine gleichmässige Verteilung der Geschwindigkeit innerhalb desuntersuchten Systems hindeutet.
Ein Ansatz, die PFG-MRT zur direkten Messung von Geschwindigkeiten in gepacktenFestbetten zu verwenden, stammt von Roper (1994; Lode et al., 1998). Er verwendetedie MRT zur Messung von Geschwindigkeiten an verschiedenen radialen Positionen ineiner kleinen, mit geschichteten Membranen gefüllten Säule. Die Übertragung seinerErgebnisse auf chromatographische Festbetten ist schwierig, da sich die Strömungenwesentlich unterscheiden. Im Fall der Membranen existieren nahezu paralleleStrömungslinien, im Fall des gepackten Festbettes sind es jedoch sehr deterministischeKurvenzüge.
Für eine Säule ohne Durchfluß haben Lode et al. (1998, Lode 1997) die MRTerfolgreich zur Messung der Packungsgüte innerhalb einer Säule angewandt. AlsErgebnis erhielten sie Informationen über Inhomogenitäten und Porositätsverteilungenim Festbett.
Wahrscheinichkeiten für die axiale Ungleichverteilung innerhalb einer PEEK-Säule( mm15044 ×, ), die mit porösen, sphärischen Silica-Gel Partikeln ( �m50=Pd )
gepackt war, wurden von Tallarek et al. (1998b, 1998c) mit Hilfe der PFG-MRT(pulsed field gradient magnetic resonance tomography) vermessen. Die Messungenwurden volumetrisch über den Säulenquerschnitt und 40 mm Säulenlänge gemittelt.Aus den Messungen konnten Stofftransportkoeffizienten für den Fall derDiffusionshemmung bestimmt werden. Tallarek et al. konnten mit den Ergebnissendieser Untersuchungen (direkt: relative Anteile an ruhender und fliessender Flüssigkeit,Kinetiken des Stofftransports. Abgeleitet: Porositäten, mittlere Geschwindigkeiten) diebreiten Einsatzmöglichkeiten der PFG-MRT zur Untersuchung der Hydrodynamik inHPLC-Säulen demonstrieren.
Tallarek et al. (1998a) konnten diese Ergebnisse noch erweitern, indem sie die PFGSEmit der Multiecho (ME) und der Turbospin (TSE) Bilderfassung kombinierten. Durchdiese Kombination erreichen sie die notwendige räumliche Auflösung, um dieGeschwindigkeiten und die Verteilung der scheinbaren Dispersion innerhalb einer2 mm starken Schicht in der Säule zu bestimmen (Bild 2.12).
Hierbei erlaubt die PFGTSE die Bestimmung der dynamischen Verschiebungen derSpins (dynamic displacement distribution, q-space imaging) an jedem Punkt desstationären Bildraumes, während die PFGME die gleichzeitige Bestimmung vontransversaler Relaxation, Flüssiganteil und Verschiebung der Spins liefert unterMinimierung der SNR (signal to noise ratio). Für diese Untersuchungen haben Tallareket al. einen eigenen Tomographen um eine axial komprimierbare Säule aufgebaut.
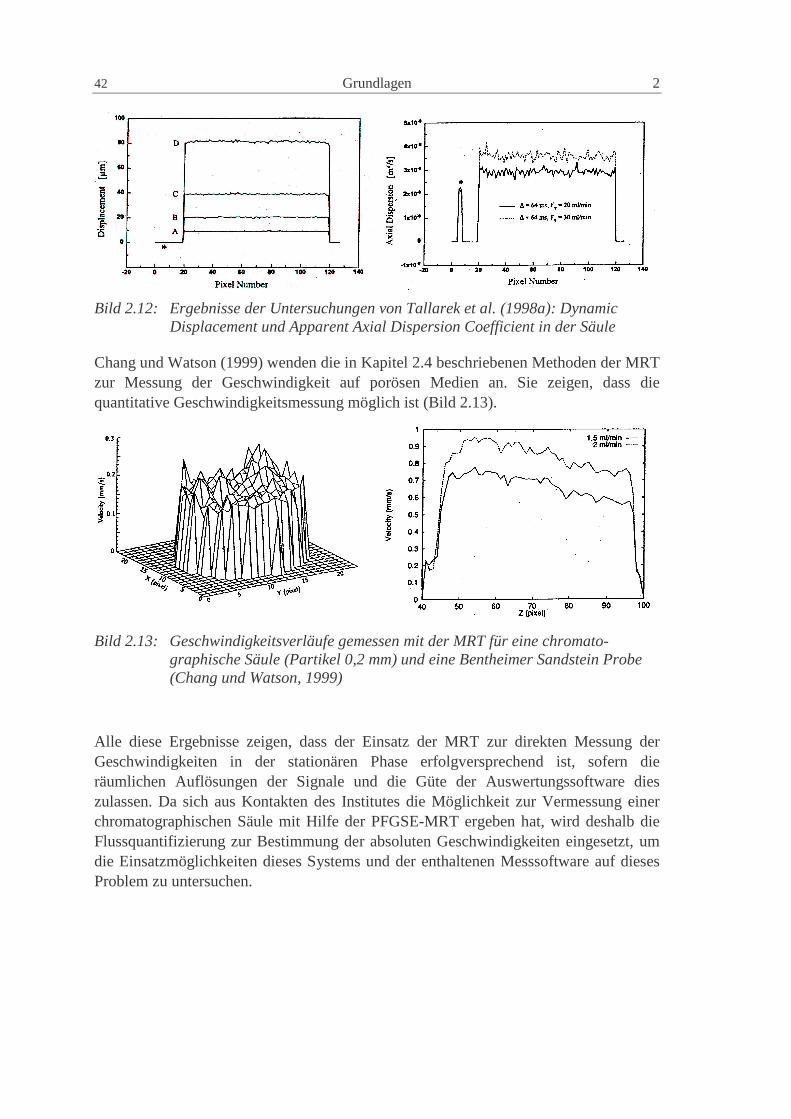
42 Grundlagen 2
Bild 2.12: Ergebnisse der Untersuchungen von Tallarek et al. (1998a): DynamicDisplacement und Apparent Axial Dispersion Coefficient in der Säule
Chang und Watson (1999) wenden die in Kapitel 2.4 beschriebenen Methoden der MRTzur Messung der Geschwindigkeit auf porösen Medien an. Sie zeigen, dass diequantitative Geschwindigkeitsmessung möglich ist (Bild 2.13).
Bild 2.13: Geschwindigkeitsverläufe gemessen mit der MRT für eine chromato-graphische Säule (Partikel 0,2 mm) und eine Bentheimer Sandstein Probe(Chang und Watson, 1999)
Alle diese Ergebnisse zeigen, dass der Einsatz der MRT zur direkten Messung derGeschwindigkeiten in der stationären Phase erfolgversprechend ist, sofern dieräumlichen Auflösungen der Signale und die Güte der Auswertungssoftware dieszulassen. Da sich aus Kontakten des Institutes die Möglichkeit zur Vermessung einerchromatographischen Säule mit Hilfe der PFGSE-MRT ergeben hat, wird deshalb dieFlussquantifizierung zur Bestimmung der absoluten Geschwindigkeiten eingesetzt, umdie Einsatzmöglichkeiten dieses Systems und der enthaltenen Messsoftware auf diesesProblem zu untersuchen.

3 Verwendete Systeme / Apparaturen 43
3 Verwendete Systeme / Apparaturen
Innerhalb dieser Arbeit kamen Anlagen der diskontinuierlichen HPLC zum Einsatz,eine semipräparative und eine präparative HPLC. Für die Experimente wurden für beideAnlagen entsprechende Säulen mit den stationären Phasen gepackt, charakterisiert undmit den Anlagen verwendet. Im Rahmen dieses Kapitels werden nur die stationärenPhasen und die verwendeten Anlagen beschrieben. Die eingesetzten Eluentien undProbensubstanzen werden in den Kapiteln 0 und 6 behandelt, in denen die experimen-tellen Messungen und Ergebnisse dargestellt sind.
3.1 Stationäre Phasen
Für die Experimente kamen verschiedene, am Institut vorhandene stationäre Phasenzum Einsatz. Sie unterscheiden sich im Wesentlichen durch ihre Partikelgröße, ihrePorositäten und ihre Adsorptionseigenschaften. Da die Porositäten der Phasen einewesentliche Rolle für Experimente und Modellierungen spielen, soll das folgendeKapitel genauer auf die für diese Arbeit verwendeten Verfahren zur Bestimmung derPorositäten eingehen.
3.1.1 Glaskugeln
Als Referenzphase, die keine Poren und keine Adsorption aufweisen soll, wurdenMikroglaskugeln der Firma Worf Glaskugeln (Mainz) ausgewählt. Innerhalb derArbeiten wurden Experimente mit unterschiedlichen Korngrößenverteilungen durch-geführt: 70-110 µm, 100-300 µm und 250-420 µm. Alle Korngrößenverteilungenzeigten das gleiche, nicht adsorbierende Verhalten gegenüber allen eingesetztenProbensubstanzen. Um eine möglichst grosse Ähnlichkeit zu den eingesetzten Normal-Phasen (NP) zu erzielen, wurde die kleinste Partikelgrössenverteilung (70-110 µm) alsReferenzphase ausgewählt.
Bild 3.1: REM-Aufnahmen der Glaskugeln (70-110 µm)

44 Verwendete Systeme / Apparaturen 3
Rasterelektronenmiskropische (REM) Aufnahmen zeigen, dass mehr als 98 % derPartikel eine absolut runde Form besitzen (Bild 3.1). In diesem Bild kann manerkennen, dass sich nur sehr wenige Glaskugeln in der Phase befinden, derenDurchmesser sich deutlich oberhalb der Korngrössenverteilung befindet. Allerdingsenthält die Phase herstellungs- und trennungsbedingt eine grosse Anzahl an Kugeln mitDurchmessern von 1-8 µm. Um einen Eindruck davon zu erhalten, ob die Glaskugelnwirklich unporös sind, wurden weitere REM-Aufnahmen bei den höchsten, für dieGeräte innerhalb der TU Berlin möglichen, Auflösungen der Oberfläche der Glaskugelnaufgenommen. Die Aufnahmen sind in Bild 3.2 dargestellt. Die Auswertung ist jedochproblematisch, da die Glaskugeln für die EM-Aufnahmen mit Gold bedampft werdenmüssen. Diese Goldschicht kann zu einer Veränderung der Porenstruktur bzw. zu einerÜberdeckung kleiner Poren (< 1 nm) führen. Die Aufnahmen stellen somit dieGoldschicht an der Oberfläche dar, nicht die eigentliche Glasoberfläche. OhneGoldbedampfung ist die Messung jedoch nicht möglich, da der einfallende Elektronen-strahl den unbedampften und damit unpolaren Glaskugeln eine starke Wanderungs-bewegung aufzwingt, die eine Aufnahme unmöglich macht. Dies gilt auch für die REM-Aufnahmen der anderen Phasen.
Bild 3.2: REM-Aufnahmen der Oberfläche einer Glaskugel
Insbesondere im Vergleich mit den NP-Phasen, die einen hohen Porendurchmesseraufweisen, ist jedoch zu erkennen, dass die Oberfläche der Glaskugeln sehr glatterscheint. Ein Vorhandensein von Poren kann daher mit hoher Wahrscheinlichkeitausgeschlossen werden. Innerhalb der Arbeit sind mit der Bezeichnung Glaskugelnohne Grössenangabe immer die Glaskugeln mit der Partikelgrösse 70-110 µm gemeint.
3.1.2 Uetikon
Bei diesem Material handelt es sich um ein Silicagel C560 (Kieselgel) der Firma CUChemie Uetikon AG (Schweiz) mit einer Partikelgrössenverteilung von 40-63 µm. DiePartikel besitzen eine signifikante Schwankungsbreite um die mittlere Partikelgrösse(Bild 3.3). Insbesondere variieren Partikel in ihrer geometrischen Form (vomlanggezogenen Quader bis zur quasi-sphärischen Form). Die Form der Partikel ist

3 Verwendete Systeme / Apparaturen 45
unsymmetrisch (gebrochenes Material). Sie besitzen eine ausgeprägte Porenstruktur miteinem mittleren Porendurchmesser von 6 nm (Bild 3.4).
Bild 3.3: REM-Aufnahmen der Uetikon-Partikel
Die Oberfläche beträgt 518,6 m2/g und ist mit polaren Gruppen besetzt (NP-Phase), diedurch Ausbildung von Van-der-Waals-Kräften mit dem Adsorptiv wechselwirken.Diese Art der stationären Phase findet in der Flüssigchromatographie und derGrössenausschlusschromatographie Verwendung (Meyer, 1992).
Bild 3.4: REM-Aufnahmen der Oberfläche der Uetikon-Partikel
3.1.3 Kromasil
Kromasil ist ein Silicagel mit einer sehr engen Partikelgrössenverteilung von 6-12 µm(Herstellerangabe 10 µm). Es besitzt eine poröse Oberfläche mit einem mittlerenPorendurchmesser von 10 nm. Die Partikel sind rund in ihrer Form, jedoch mit eineretwas unebeneren Oberfläche als die der Glaskugeln (Bild 3.5). Von dieser stationärenPhase konnten aufgrund der hohen, vom Elektronenstrahl aufgeprägten Wanderungs-geschwindigkeit keine Aufnahmen der Oberfläche (ohne Goldbedampfung) erstelltwerden. Die Partikel besitzen wegen ihrer Grösse eine zu geringe Trägheit, andereRasterelektronenmikroskope stehen an der TU Berlin nicht zur Verfügung.

46 Verwendete Systeme / Apparaturen 3
Bild 3.5: REM-Aufnahmen der Kromasil-Partikel
Kromasil ist eine in der Flüssigchromatographie verbreitete Hochleistungsphase undstand im Institut zur Verfügung.
3.1.4 Bestimmung der Porositäten
Eine Unterscheidung der Porosität in interne, externe und Gesamtporosität ist bereits inKapitel 2.2.2 erfolgt. Hier wird ausschliesslich die zur Charakterisierung der festenPhasen notwendige experimentelle Bestimmung ihrer Porositäten beschrieben.
3.1.4.1 Messung der Gesamtporosität
Die Messung der Gesamtporosität kann mit der gravimetrische Bestimmung durchLösungsmittelwechsel erfolgen. Als Ansatz dient die Tatsache, dass sich die Gesamt-masse der chromatographischen Säule aus den Massen der leeren Säule (inkl.Verschraubungen), des Festbetts und des Lösungsmittels berechnet:
PhasemobPhasestatleerSgesamtS mmmm ..,, ++= (3.1)
Die Masse der mobilen Phase lässt sich als Produkt der Lösungsmitteldichte und desLösungsmittelvolumens darstellen, sodass sich für die Gesamtmasse als lineareFunktion der Lösungsmitteldichte mit dem Lösungsmittelvolumen als Steigung und derMasse an stationärer Phase und leerer Säule als Achsenabschnitt ergibt:
PhasemobPhasemobPhasestatleerSgesamtS DVmmm ...,, ++= (3.2)
Durch Befüllen der gepackten Säule mit Lösungsmitteln unterschiedlicher Dichte undAuftragung der Gesamtmassen über den Lösungsmitteldichten erhält man die Gesamt-porosität als Quotienten aus Lösungsmittel- und des Säulenvolumen.
Diese Methode ist zur Bestimmung der Porosität von nicht-quellenden stationärenPhasen geeignet. Problematisch ist sie für quellende Phasen, wie z.B. Harze, bei denendas Quellverhalten bekannt sein muss. Desweiteren muss gewährleistet sein, dass diestationäre Phase keine Restbeladung eines vorherigen Lösungsmittels mehr besitzt.Auch sollte ein Lösungsmittel mit starken adsorptiven Wechselwirkungen zur festen

3 Verwendete Systeme / Apparaturen 47
Phase möglichst nicht verwendet werden, da eine anschliessende Desorption kaum bzw.überhaupt nicht möglich ist und die Phase dadurch für einen Einsatz als Trennsystemungeeignet wird. Da viele feste Phasen hygroskopisch sind, soll eine Trocknung derfesten Phase im Rahmen ihrer Materialeigenschaften vor dem Packen erfolgen. DieGenauigkeit dieser Methode hängt insbesondere von der Wägegenauigkeit dereingesetzten Waagen ab. Sie betrug für die innerhalb der Arbeit eingesetztemechanische Balkenwaage der Firma Mettler ± 0,005 g. Dies entspricht für dieeingesetzten Chromatographiesäulen mit 10 mm Innendurchmesser (ID) einerAbweichung von 0,004 % des Messwerts.
Eine weitere Methode besteht in der Verwendung eines wenig adsorbierenden Tracersmit einem stark adsorbierenden Eluens. Unter der Voraussetzung, das beideKomponenten (Eluens und Tracer) komplett porengängig sind, lässt sich aus demPeakverlauf des nicht adsorbierenden Tracers die Gesamtporosität bestimmen.Nachteilig an dieser Methode ist, dass durch die Belegung der Oberfläche der Partikeldurch das Eluens das dem Tracer zugängliche Porenvolumen reduziert wird unddadurch die Gesamtporosität zu klein berechnet wird (Siehe auch Lenz (2000)).
gesSTracerREluentges EVtVV == ,� (3.3)
3.1.4.2 Messung der internen Porosität
Zur experimentellen Bestimmung der internen Porosität wird eine von Mottlau undFisher (1962) beschriebene Methode für stationäre Phasen der Chromatographievariiert:
Ein festgelegtes Volumen der stationären Phase wird in ein verschliessbares Becherglasgefüllt und im Vakuumofen getrocknet. Sobald keine Restbeladung mehr vorliegt, wirddas Becherglas auf einem Magnetrührer plaziert und ein Rührfisch hineingegeben. Eswird nun solange ein porengängiges Lösungsmittel in das Becherglas titriert, bis sichdas Erscheinungsbild der festen Phase ändert: Solange das Lösungsmittel in die Porenhineindiffundieren kann, bildet sich an der Oberfläche der Partikel kein Flüssigkeitsfilmaus und die Partikel bewegen sich frei. Wenn jedoch die Poren keine weitere Flüssigkeitmehr aufnehmen können, bildet sich ein Lösungsmittelfilm auf der Oberfläche und eskommt zur Agglomeration der Partikel. Das Erscheinungsbild im Becherglas schlägtum, da jetzt hauptsächlich Agglomerate bewegt werden. Bei Bildung der Agglomeratemuss die Titration gestoppt werden. Wenn sich die Agglomerate nicht schnell wiederauflösen (Transport des Lösungsmittels zu noch unbenetzten Partikeln), ist derUmschlagpunkt erreicht und das hineintitrierte Lösungsmittelvolumen liefert mit demzuvor gemessenen Volumen der stationären Phase die interne Porosität.
Die so gemessenen internen Porositäten entsprechen den aus den Werten der externenund der Gesamtporosität bestimmten Werten.

48 Verwendete Systeme / Apparaturen 3
3.1.4.3 Messung der externen Porosität
Zur Messung der externen Porosität verwendet man einen nicht porengängigen, nichtadsorbierenden Tracer, dessen Peakverläufe bestimmt werden. Für diese Substanz istnur das externe Volumen zugänglich, aus ihrer Retentionszeit lässt sich analog zuGl. (3.3) die externe Porosität berechnen:
extSTracerREluentext EVtVV == ,� (3.4)
Man kann geeignete Tracer experimentell durch Vergleich der Retentionszeiten inFrage kommender Substanzen ermitteln. Je geringer die Retentionszeit der Substanz ist,desto geeigneter ist sie zur Bestimung der externen Porosität. Damit eine Substanz nichtporengängig ist, muss sie eine beträchtliche Molekülgrösse aufweisen. Dextran(C6H10O5)n ist z.B. mit einem Molekulargewicht von 2*105 g/mol eine geeignete
Substanz für ein Kunststoffharz wie Lewatit. Um zu überprüfen, ob ein nichtporengängiger Stoff adsorbiert wird, werden Chromatogramme bei unterschiedlichenSystemtemperaturen aufgenommen. Im Falle vorhandener Adsorption verändert sich dieRetentionszeit mit der Temperatur, da die Adsorptionsgleichgewichte temperaturab-hängig sind.
3.1.4.4 Übersicht der Porositäten
In Tabelle 3.1 sind die gemessenen Porositäten der stationären Phasen gegenüber-gestellt.
Tabelle 3.1: Übersicht der Porositäten der stationären Phasen
Porosität: Gesamtporosität Interne Porosität Externe PorositätStat. Phase:
gemgesF
, int,gemF
gem,extF
Glaskugeln 0,41 0 0,59Ueticon 0,82 0,32 0,50Kromasil 0,80 0,48 0,32
3.2 Semipräparative HPLC
3.2.1 Allgemeine Beschreibung
In Bild 3.6 ist das Schema der semipräparativen HPLC dargestellt. Diese Anlage lässtden Einsatz von analytischen Säulen (ID 10 mm, Hibar oder Superformance) und semi-präparativen Säulen (ID 26 und 50 mm, Hibar oder SF) zu. Sie setzt sich im Einzelnenaus den folgenden Komponenten zusammen:
Aus der Lösungsmittelvorlage wird der Eluent mit einer Hochdruckpumpe (Fa. KnauerWellchrom K-1800) gefördert. Die Lösungsmittelvorlage ist in einem Ultraschallbad(Fa. Bandelin, Sonorex TK52) gelagert, welches eine Entgasung des Eluenten

3 Verwendete Systeme / Apparaturen 49
ermöglicht. Am Probeaufgabeventil (Fa. Knauer, Sechs-Wege Ventil) kann dem Eluentein durch eine Probenschleife definiertes Volumen an Substanzgemisch beigemengtwerden. Hinter dem Probenaufgabeventil werden über Kapillarverschraubungen dieHPLC-Säulen angeschlossen. Die Hibar-Säulen der Firma Merck können mit einemUmluftofen und die Superformance-Säulen über ihren Mantelwärmetauscher mit einemThermostatbad temperiert werden.
Bild 3.6: Anlagenschema der semipräparativen HPLC
Nachfolgend werden die Substanzen in einem UV-Detektor (Fa. Knauer, UV-Detektor64) detektiert. Dieser Detektor kann durch ein Refraktometer (Fa. Knauer,Refraktometer 64) ersetzt oder ergänzt werden, wenn es die Versuchsbedingungenerfordern. Der Detektor ist über ein Digitalmultimeter (Fa. Conrad, Voltcraft ME-22T)mit einem PC verbunden, an den alle anfallenden Messdaten weitergeleitet und dortausgewertet werden. Als Verbindungs- und Auswertungssoftware kommt FlexPro-Control 5.0 (Weisang GmbH & Co.) zum Einsatz. Schliesslich wird der Stoffstrom inseine Fraktionen zerlegt bzw. recycliert.
3.2.2 Messtechnik
Wie in der Anlagenbeschreibung bereits erwähnt, werden die Messdaten mit demDigitalmultimeter ME-22T erfasst. Das Gerät kann einen Kanal mit einer Schaltzeit von0,1 s bis 10 Minuten aufzeichnen. Zur Aufzeichnung der Chromatogramme wird einSpannungssignal von 0 bis 2 V mit einer Abtastrate von 2 Hz registriert. Die Genauig-keit und die Auflösung des Spannungssignals betragen in dieser Einstellung 0,001 V,was einer Genauigkeit von 1 mAU (milli Adsorption Unit) entspricht. DieMesswertaufzeichnung mit FlexPro-Control erzielt über das eingesetzte DasyLab-

50 Verwendete Systeme / Apparaturen 3
Komprimierungsverfahren sehr kleine Datendateien. Die weitere Bearbeitung der Datenwurde mit der Tabellenkalkulation Microsoft Excel durchgeführt.
3.2.3 Packen der Säulen
Das Packen der analytischen und der semipräparativen Säulen erfolgte analog. Zuerstwird die zu packende Säule wie in Bild 3.7 dargestellt aufgebaut und in ein Stativeingespannt. Auf der oberen Seite werden das Stirnstück (1) und die Adapterverstellung(2) noch nicht montiert. In die Säule wird von oben ein Teflon-Trichter eingesteckt, deran den Innenwänden der Glassäule dicht abschliesst.
Bild 3.7: Skizze zum Packen der analytischen und semipräparativen Säulen
Die untere Auslasskapillare wird über mehrere Kühlfallen mit einer Vakuumpumpeverbunden und die Säule in ein Ultraschallbad RK255S der Firma Sonorex eingetaucht.Die Säule wird mit dem Lösungsmittel befüllt, mit dem auch die Slurry angesetztwurde. Als Lösungsmittel sind Acetonitril und Isopropanol für NP-Phasen undMethanol für RP-Phasen und Glaskugeln gut geeignet. Von Merck werden Aceton fürRP-Phasen und Methanol für unmodifizierte Kieselgelträger empfohlen. Die Slurrysollte für die eingesetzten Phasen zumindest aus gleichen Anteilen an Feststoff undLösungsmittel bestehen, um eine hohe Packungsqualität zu erzielen. In der Literaturwerden abhängig von der eingesetzten stationären Phase noch weit geringere Feststoff-anteile empfohlen.
Nun wird das Vakuum angelegt und die notwendige Menge an Slurry in den Trichtergefüllt. Hierbei ist das Vakuum so zu regulieren, dass das Bett zu keiner Zeit amunteren (dem Vakuum zugewandten) Ende trockenfällt. Die Säule wird anschliessendverschlossen und das Bett für 24 bis 48 Stunden ruhen gelassen. Das Stirnstück wirdentsprechend nachgeführt und das Bett durch einen möglichst hohen Volumenstrom für
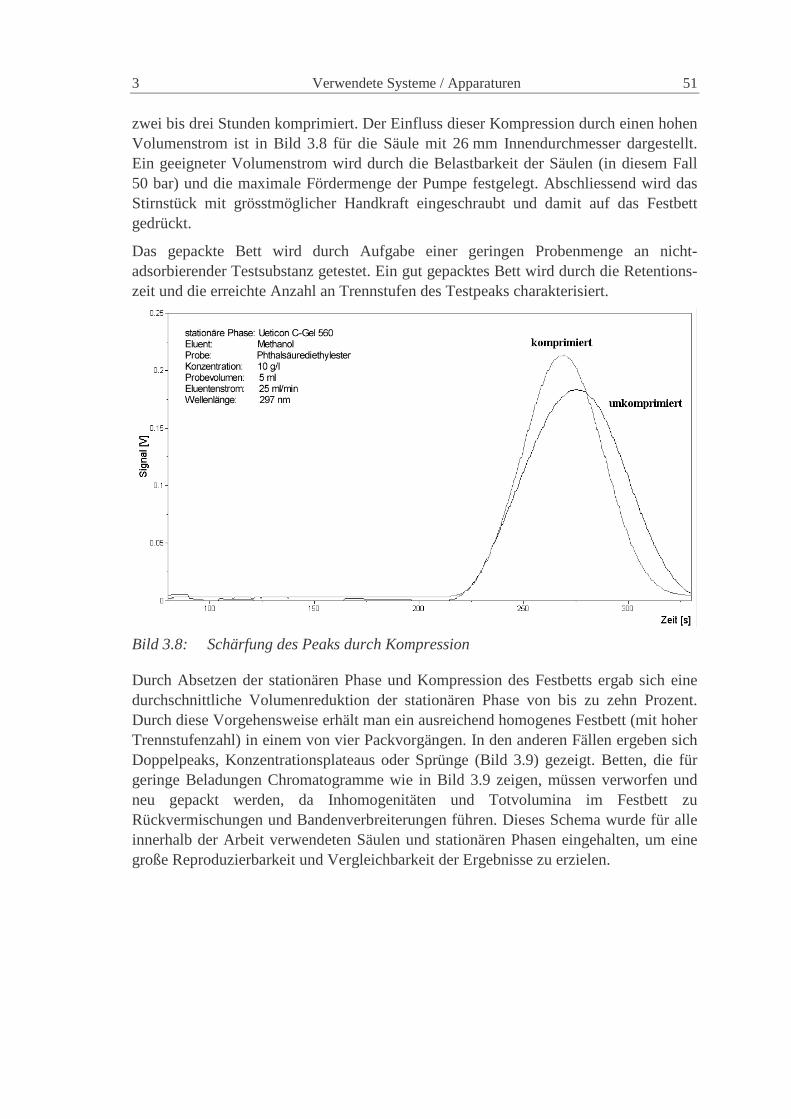
3 Verwendete Systeme / Apparaturen 51
zwei bis drei Stunden komprimiert. Der Einfluss dieser Kompression durch einen hohenVolumenstrom ist in Bild 3.8 für die Säule mit 26 mm Innendurchmesser dargestellt.Ein geeigneter Volumenstrom wird durch die Belastbarkeit der Säulen (in diesem Fall50 bar) und die maximale Fördermenge der Pumpe festgelegt. Abschliessend wird dasStirnstück mit grösstmöglicher Handkraft eingeschraubt und damit auf das Festbettgedrückt.
Das gepackte Bett wird durch Aufgabe einer geringen Probenmenge an nicht-adsorbierender Testsubstanz getestet. Ein gut gepacktes Bett wird durch die Retentions-zeit und die erreichte Anzahl an Trennstufen des Testpeaks charakterisiert.
Bild 3.8: Schärfung des Peaks durch Kompression
Durch Absetzen der stationären Phase und Kompression des Festbetts ergab sich einedurchschnittliche Volumenreduktion der stationären Phase von bis zu zehn Prozent.Durch diese Vorgehensweise erhält man ein ausreichend homogenes Festbett (mit hoherTrennstufenzahl) in einem von vier Packvorgängen. In den anderen Fällen ergeben sichDoppelpeaks, Konzentrationsplateaus oder Sprünge (Bild 3.9) gezeigt. Betten, die fürgeringe Beladungen Chromatogramme wie in Bild 3.9 zeigen, müssen verworfen undneu gepackt werden, da Inhomogenitäten und Totvolumina im Festbett zuRückvermischungen und Bandenverbreiterungen führen. Dieses Schema wurde für alleinnerhalb der Arbeit verwendeten Säulen und stationären Phasen eingehalten, um einegroße Reproduzierbarkeit und Vergleichbarkeit der Ergebnisse zu erzielen.
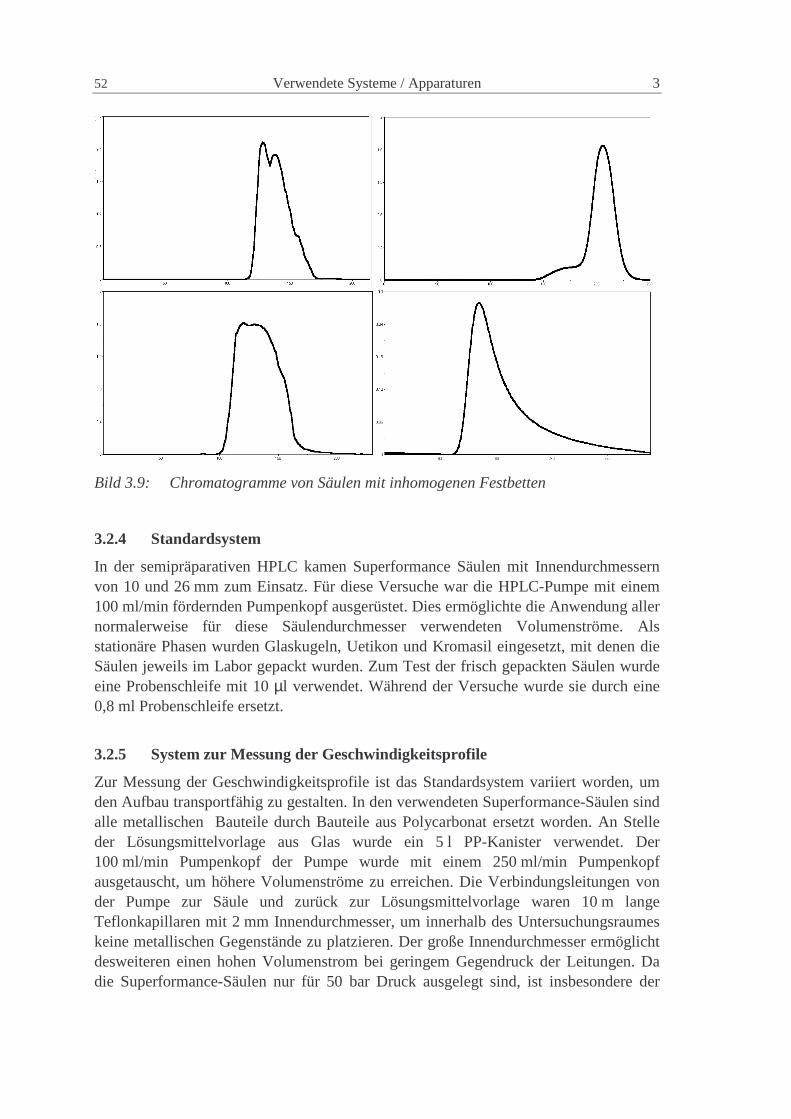
52 Verwendete Systeme / Apparaturen 3
Bild 3.9: Chromatogramme von Säulen mit inhomogenen Festbetten
3.2.4 Standardsystem
In der semipräparativen HPLC kamen Superformance Säulen mit Innendurchmessernvon 10 und 26 mm zum Einsatz. Für diese Versuche war die HPLC-Pumpe mit einem100 ml/min fördernden Pumpenkopf ausgerüstet. Dies ermöglichte die Anwendung allernormalerweise für diese Säulendurchmesser verwendeten Volumenströme. Alsstationäre Phasen wurden Glaskugeln, Uetikon und Kromasil eingesetzt, mit denen dieSäulen jeweils im Labor gepackt wurden. Zum Test der frisch gepackten Säulen wurdeeine Probenschleife mit 10 µl verwendet. Während der Versuche wurde sie durch eine0,8 ml Probenschleife ersetzt.
3.2.5 System zur Messung der Geschwindigkeitsprofile
Zur Messung der Geschwindigkeitsprofile ist das Standardsystem variiert worden, umden Aufbau transportfähig zu gestalten. In den verwendeten Superformance-Säulen sindalle metallischen Bauteile durch Bauteile aus Polycarbonat ersetzt worden. An Stelleder Lösungsmittelvorlage aus Glas wurde ein 5 l PP-Kanister verwendet. Der100 ml/min Pumpenkopf der Pumpe wurde mit einem 250 ml/min Pumpenkopfausgetauscht, um höhere Volumenströme zu erreichen. Die Verbindungsleitungen vonder Pumpe zur Säule und zurück zur Lösungsmittelvorlage waren 10 m langeTeflonkapillaren mit 2 mm Innendurchmesser, um innerhalb des Untersuchungsraumeskeine metallischen Gegenstände zu platzieren. Der große Innendurchmesser ermöglichtdesweiteren einen hohen Volumenstrom bei geringem Gegendruck der Leitungen. Dadie Superformance-Säulen nur für 50 bar Druck ausgelegt sind, ist insbesondere der

3 Verwendete Systeme / Apparaturen 53
Gegendruck der Rücklaufleitung gering zu halten. Auf den Einsatz eines Detektorswurde verzichtet, da die Messung des Geschwindigkeitsprofils eines Tracers keinewesentliche Zusatzinformation liefern würde. Als Eluent kam aufgrund derDetektierbarkeit Wasser zum Einsatz, als stationäre Phasen Uetikon (ein Ionenaus-tauscherharz) und Glaskugeln.
3.2.6 Kernspintomograph
Die Messungen der Geschwindigkeitsprofile erfolgten in der Praxis von Dr. R. Kaiser.Es wurde das in Kapitel 3.2.5 beschriebene chromatographische System mit Hilfe einesMagnetom Harmony (Fa. Siemens Medical, Erlangen, Bild 3.10) untersucht.
Bild 3.10: Kernspintomograph Siemens Harmony 1T
Als Messspulen kamen eine CP Flexspule und eine CP Kopf-Arrayspule (Fa. Siemens)zum Einsatz. Zur Auswertung wurde das von der Fa. Siemens zur Berechnung derFließgeschwindigkeiten des menschlichen Blutes in grossen Gefäßen, wie z.B. derAorta und des Liquorflusses, entwickelte Applikationspaket zur Flussquantifizierungverwendet. Es besteht hauptsächlich aus einer segmentierten 2D-FLASH-Sequenz. DieMesseinstellungen und Ergebnisse der Messungen sind in Kapitel 6.2.1 beschrieben.
3.3 Präparative HPLC
3.3.1 Allgemeine Beschreibung
Die präparative HPLC beruht grundsätzlich auf einem Merck-Prepbar2-System, das ineinigen Positionen (verwendete Säule, Probenaufgabe und Messwertaufnahme) ange-passt worden ist. Es unterscheidet sich wenig von der semipräparativen HPLC und ist inBild 3.11 schematisch dargestellt:
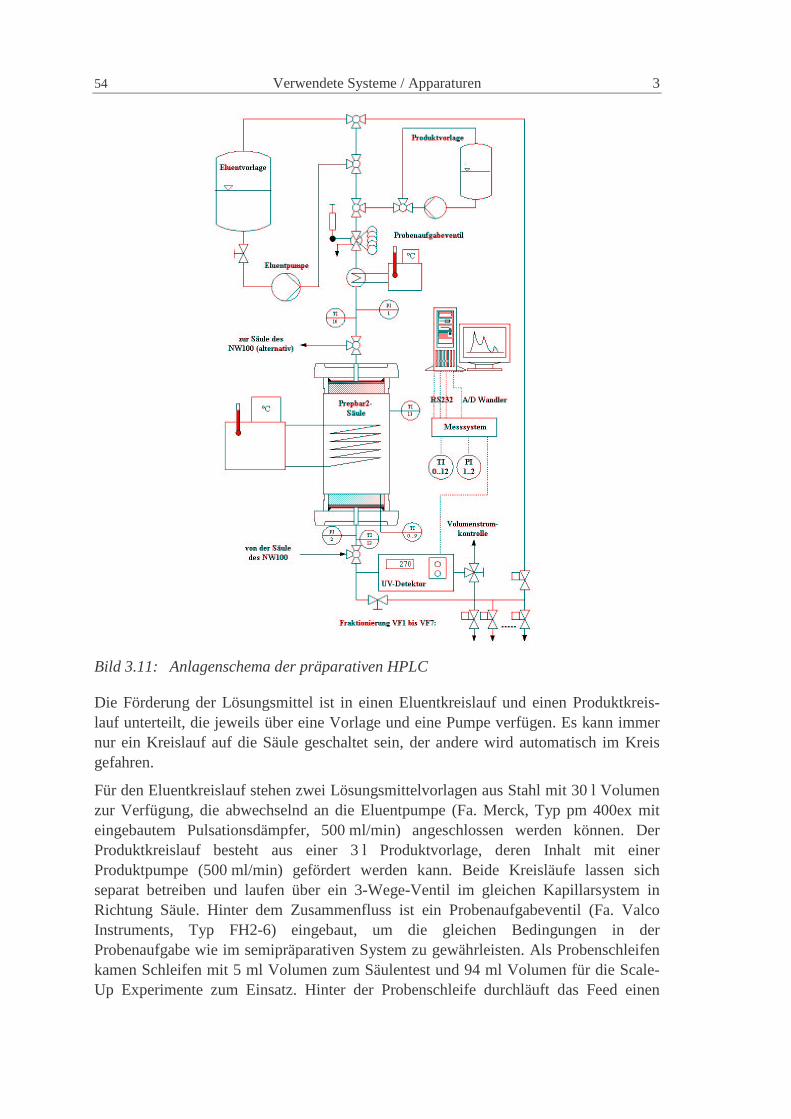
54 Verwendete Systeme / Apparaturen 3
Bild 3.11: Anlagenschema der präparativen HPLC
Die Förderung der Lösungsmittel ist in einen Eluentkreislauf und einen Produktkreis-lauf unterteilt, die jeweils über eine Vorlage und eine Pumpe verfügen. Es kann immernur ein Kreislauf auf die Säule geschaltet sein, der andere wird automatisch im Kreisgefahren.
Für den Eluentkreislauf stehen zwei Lösungsmittelvorlagen aus Stahl mit 30 l Volumenzur Verfügung, die abwechselnd an die Eluentpumpe (Fa. Merck, Typ pm 400ex miteingebautem Pulsationsdämpfer, 500 ml/min) angeschlossen werden können. DerProduktkreislauf besteht aus einer 3 l Produktvorlage, deren Inhalt mit einerProduktpumpe (500 ml/min) gefördert werden kann. Beide Kreisläufe lassen sichseparat betreiben und laufen über ein 3-Wege-Ventil im gleichen Kapillarsystem inRichtung Säule. Hinter dem Zusammenfluss ist ein Probenaufgabeventil (Fa. ValcoInstruments, Typ FH2-6) eingebaut, um die gleichen Bedingungen in derProbenaufgabe wie im semipräparativen System zu gewährleisten. Als Probenschleifenkamen Schleifen mit 5 ml Volumen zum Säulentest und 94 ml Volumen für die Scale-Up Experimente zum Einsatz. Hinter der Probenschleife durchläuft das Feed einen

3 Verwendete Systeme / Apparaturen 55
Rohrschlangenwämetauscher (Eigenbau), einen Druckfühler (Fa. Jumo, Typ 4AD-30)und einen Temperaturfühler (Fa. Conatex, Typ Pt100, 2 mm Aussendurchmesser, diesgilt für alle Temperaturfühler der Anlage), bevor es in die angeschlossene Säule geführtwird. Der Wärmetauscher wird mit einem Thermostatbad (Fa. Haake, Typ D8/GH)betrieben.
Das System wurde mit verschiedenen Säulen betrieben: Während des ersten Teils derArbeit eine Merck Prepbar2 100 mm Säule und nach der Weiterentwicklung desSystems ein Merck Füllstand NW100. Beide Säulensysteme sind axial komprimierbar,haben 100 mm Innendurchmesser und besitzen einen Mantelwärmetauscher mitTemperaturfühler. Die Mantelwärmetauscher werden durch ein Thermostatbad (Fa.Haake, Typ F3/K) gespeist. Die Prepbar2 Säule wurde von der Fa. Muder und WocheleChromatographietechnik mit Uetikon 40-63 µm Material gepackt. Der Füllstand wurdemit allen stationären Phasen selbst gepackt. Hinter den Säulen schliessen sich wiederein Temperatur- und ein Druckfühler an, die Detektion erfolgt mit Hilfe eines UV-Detektors (Knauer UV-Detektor 64). Abschliessend erfolgt die Aufteilung des Produkt-stromes mit Hilfe der Fraktionierventile in Fraktionen und den Kreislaufstrom, der indie Eluentvorlage zurückgeführt wird. Alle Messstellen inklusive des Detektorsignalswerden mit einem speziell für die Anlage erstellten Messsystem erfasst und am PCausgewertet.
3.3.2 Messtechnik
Zur Erfassung und Auswertung der anfallenden Messwerte der Druck- und Temperatur-fühler und des Detektionssignals wurde ein eigenes Messsystem aufgebaut, dass diegleichzeitige Aufnahme aller Messwerte erlaubt. Es besteht aus einer unabhängigenKonstantspannungsversorgung und verschiedenen Modulen zur Messung dereingehenden elektrischen Grössen. Diese Module wandeln die eingehenden Signale inSpannungen von 0 bis 5 V und geben sie an den angeschlossenen PC weiter. ZurErfassung der Daten der Pt100 Temperaturfühler kommen 6 Eingangsmodule (Fa.NuDAM, Typ NuDAM 6013) zum Einsatz, an die je 3 Pt100 Widerstandsthermometerangeschlossen werden können. Je zwei dieser Module sind mit einem Übertragungs-modul (Fa. NuDAM, Schnittstellenkonverter RS485-RS232 Typ NuDAM 6520)verbunden, das über eine RS232-Verbindung die Daten an den PC weitergibt. Eineingesetzter A/D Wandler (Fa. Adlink, Typ ACL8112DG) ermöglicht überverschiedene aufsteckbare Module sowohl die Erfassung und Wandlung der Strom-signale der Druckfühler (4-20 mA) sowie die Durchführung des Spannungsignal desDetektors an eine PC-Steckkarte. Alle Module sind in einem 19"-Rack untergebrachtund die Anschlüsse der Fühler nach aussen geführt.
Das Ansprechen der zum PC gelangenden Signale erfolgt wie an der semipräparativenHPLC mit FlexPro-Control 5.0. Die Auswertung erfolgt mit der TabellenkalkulationMicrosoft Excel. Durch diesen Aufbau wird sowohl eine hohe Messgenauigkeit als aucheine häufige, gleichzeitige Aufnahme aller Messstellen sichergestellt. Es ergeben sichsie folgenden Auflösungen und Genauigkeiten bei einer Messfrequenz von 1 Hz:

56 Verwendete Systeme / Apparaturen 3
Tabelle 3.2: Genauigkeit und Auflösung des Messaufbaus
Messauflösung MessgenauigkeitTemperatur 0,03 K 0,15 KDruck 0,05 bar 0,10 barDetektorsignal 0,001 V 0,001 V
3.3.3 Packen des NW100
Das Packen des Füllstandes NW100 erfolgte weitestgehend nach dem in der Betriebs-anleitung (Merck, 1996) empfohlenen Vorgehen.
Zuerst wird den physikalischen Parametern des Packungsmaterials entsprechend vielMaterial in einem 5 l Kanister abgefüllt und mit Lösungsmittel auf 4,5 l aufgefüllt.Nach Absetzen der stationären Phase in der Slurry kann das zum Erreichen einerbestimmten Bettlänge benötigte Volumen durch weiteres Zufüllen oder durch Entnahmean Slurry eingestellt werden. Nach Einstellung des benötigten Volumens wird die Slurryerneut suspendiert (Schütteln und anschliessendes Homogenisieren im Ultraschallbad).
Auf den geöffneten Füllstand wird das Füllrohr aufgesetzt und mit der Spannketteverschraubt. Der PTFE-Schlauch am Kompressionkolben wird über zwei Kühlfallenzum Auffangen des Lösungsmittels mit einer Vakuumquelle verbunden. Dassuspendierte Packungsmaterial wird mit Hilfe eines Trichters in das mit der Säuleverbundene Füllrohr eingefüllt. Anschliessend wird das Vakuum angelegt. Die Saugzeitund ihre Einstellung (Intervalle) sind von der Art und der Korngrösse der stationärenPhase abhängig. Ein zu lange angelegtes Vakuum kann zum Trockenlegen des unterenBettbereichs und damit zur Bildung von Brücken führen, die eine optimale Bettstrukturverhindern. Nach Einsaugen der Suspension wird das Füllrohr entfernt und die Säulemit dem Säulendeckel verschlossen.
Zur endgültigen Herstellung des Bettes wird das Bett nun durch Anlegen einesmässigen Stempeldruckes (ca. 200 bar) mit der Hydraulik stabilisiert. Die optimalePackungsstabilität und Peaksymmetrie wird dann durch Equilibrieren des Säulenbettesunter hohem Durchfluss (höchster mit der Anlage möglicher Volumenstrom,500 ml/min) über eine Zeit von 30 bis 60 Minuten erreicht. Abschliessend wird das Bettnachkomprimiert (wieder ca. 200 bar) und der Kolben durch die Anschlagschraubeverriegelt. Vor Inbetriebnahme wird die Säule 3-4 mal für ca. eine Minute mit höchstemVolumenstrom betrieben und das Bett jedes Mal durch Nachführen des Hydraulik-zylinders und des Kolbenanschlags erneut stabilisiert. Nun wird die Packung durchAufgabe eines analytischen Peaks getestet und charakterisiert.
Entleeren und erneutes Wiederbefüllen wurde ausschliesslich wie im Handbuch desFüllstandes (Merck, 1996) erläutert durchgeführt.

3 Verwendete Systeme / Apparaturen 57
3.3.4 Standardsystem
Als Standardsystem kamen sowohl die Prepbar2-Säule (im Bild noch ohne Mantel-wärmetauscher) als auch die unmodifizierte Säule des Füllstand NW100 zum Einsatz:
Bild 3.12: Fotos Prepbar2-Säule und Füllstand NW100 unmodifiziert
Zur Messung der Temperaturprofile innerhalb der Säulen wurden in beiden Systemendie Sammler und Auslaufflansche (jeweils der obere Teil in Bild 3.12) angepasst,nachdem die Systeme gepackt worden sind und alle Experimente ohne Fühlerabgeschlossen waren.
3.3.5 System zur Messung der Temperatur -Profile
Die Temperaturfühler wurden in die hinteren Flansche der Säulen eingebaut, uminnerhalb der Säulen das voll ausgebildete Temperaturprofil zu messen. Um diegrundsätzliche Anwendbarkeit zu demonstrieren, wurden zuerst Temperaturfühler in diePrepbar2-Säule eingebaut. Eine detaillierte Beschreibung des Einbaus in die Prepbar2-Säule ist in der Arbeit von P. Schaarschuh (2000) zu finden. Einem axialen Einbau derTemperaturfühler wurde wegen der geringeren Beeinflussung der Strömung und einereinfacheren mechanischen Umsetzung innerhalb der bestehenden Systeme der Vorzugvor einem radialen Einbau gegeben.
Mit dem Prepbar2 System ist allerdings kein Packen der stationären Phase um dieTemperaturfühler herum möglich. Dieses Problem wurde durch den Einbau derTemperaturfühler in die Säule des Füllstandes NW100 behoben. Der Einfluss der Fühlerauf die Strömung konnte durch diese Massnahme weiter reduziert werden (dieErgebnisse sind ausführlich in Kapitel 6.1 beschrieben).

58 Verwendete Systeme / Apparaturen 3
Für beide Systeme erfolgte der Einbau der Pt100 Temperaturfühler nach dem gleichenPrinzip: Der Endflansch der Säule wurde in der institutseigenen Werkstatt modifiziertbzw. modifiziert nachgebaut. Der grundsätzliche Aufbau ist in Bild 3.13 dargestellt.
Bild 3.13: Einbauschema der Pt100-Temperaturfühler
Die Eindringtiefe der Pt100 in die stationäre Phase beträgt für die Prepbar2-Säule70 mm und für die NW100-Säule 50 mm. Sie ist für die NW100-Säule von 10 mm bis100 mm variabel einzustellen. Die Anordnung der Temperaturfühler auf den Flanschenist in Bild 3.14 dargestellt, ihr jeweiliger Abstand vom Mittelpunkt der Säule ist Tabelle3.3 zu entnehmen.
Vor dem Einbau wurden alle Pt100 im Temperaturbereich von 10 bis 80 °C gegen einRosemount-Standard Pt25 kalibriert. Laut Hersteller liefern die Pt100 eine Genauigkeitvon ± 0,15 K bei einer Auflösung von 0,03 K.
Die Abstände der Temperaturfühler vom Mittelpunkt wurden für die NW100-Säulegegenüber dem Prepbar2 System weiter an den Säulenrand verlagert, da die Ergebnissedes Prepbar2 Systems ein sehr gleichmässiges Temperaturprofil in der Säulenmitteergeben haben (siehe Kapitel 6.1). Dies ist auch vorteilhaft, da das Verteilungssystemdes NW100 gegenüber der Prepbar2 durch eine weitere gelaserte Lochscheibe zwischenFlansch und Fritte verbessert wurde und ein Durchbohren dieser Scheibe zu deutlichverschlechterten Verteilungen geführt hat.

3 Verwendete Systeme / Apparaturen 59
Bild 3.14: Anordnung der Verschraubungen der Pt100 (Prepbar2-, NW100-Säule)
Tabelle 3.3: Abstände der Pt100 vom Mittelpunkt der Säule
Abstand vom MittelpunktSensornummer Prepbar2 NW100
0 15,0 mm 28,0 mm1 18,5 mm 30,0 mm2 22,0 mm 32,0 mm3 25,5 mm 34,0 mm4 29,0 mm 36,0 mm5 32,5 mm 38,0 mm6 36,0 mm 40,0 mm7 39,5 mm 42,0 mm8 43,0 mm 44,0 mm9 46,5 mm 46,0 mm
Für die Pt100 wurden Flansch und Fritte zusammengesetzt mit 2 mm Bohrungenversehen, wobei die Passung so gewählt wurde, dass keine stationäre Phase an denFühlern entlang die Fritte passieren kann. Die Dichtung der Pt100-Schäfte wird mitRohrverschraubungen 1/8" aus Edelstahl (Fa. Swagelok, Typ 1/8" 2MO) undKlemmringen aus PTFE (Fa. Swagelok) sichergestellt. Sowohl für das Prepbar2-Systemals auch für den Füllstand NW100 existierten keine Probleme mit der Dichtigkeit derPt100-Verschraubungen. Die in den Flansch und die Fritte des NW100 eingebautenPt100 sind in Bild 3.15 dargestellt. Auf eine Darstellung der Pt100 in der Prepbar2-Säule wurde verzichtet, da der Einbau bis auf die Positionierung der Pt100 analogerfolgt ist.

60 Verwendete Systeme / Apparaturen 3
Bild 3.15: Montierte Pt100 im NW100 Kopfstück
Der Einfluss der Pt100 auf die Qualität des chromatographischen Betts wurde durchVergleich der Peak-Verläufe vor und nach Einbau der Pt100 bestimmt. Für eineDarstellung der Einflüsse sei auf Kapitel 6.1.1 verwiesen.

4 Modellierung der HPLC-Säulen 61
4 Modellierung der HPLC-Säulen
Die Modellierung der chromatographischen Systeme erfolgte je nach notwendigemFreiheitsgrad:
• Für den Fall der Messungen zum Referenzsystem (Kap. 0) ist nur eine Modellierungder Peakverläufe notwendig. Hierfür ist die Lösung der Komponentenbilanzen inder Flüssigkeit (Gleichung (2.56)) und um die Partikel (Gleichung (2.57)) in einerRaumrichtung (axial) ausreichend. Als Gleichungslöser kam das kommerziellePaket gPROMS zum Einsatz. Eine detaillierte Übersicht zum Einsatz von gPROMSzur Simulation von chromatographischen Systemen, speziell der simuliertenGegenstromchromatographie, ist bei Scheer (2001) zu finden.
• Zur Modellierung der Messungen der Verteilungen (Temperatur- und Geschwindig-keitsprofile, Kap. 0) innerhalb der HPLC-Säulen wurden die Gleichungen ausTabelle 2.2 mit Hilfe der CFD-Simulationsumgebung FLUENT gelöst und dieVerteilungen berechnet.
• Eine Untersuchung der Einsatzmöglichkeit von FEMLAB, einem Simulations-aufsatz auf MATLAB, zur Modellierung chromatographischer Säulen ist währenddieser Arbeit durchgeführt worden. FEMLAB bietet insbesondere eine komfortableDarstellungsmöglichkeit von Strömungslinien in chromatographischen Festbetten.Eine Implementierung der Adsorption in FEMLAB war mit den derzeit in FEMLABverfügbaren Modellen nicht möglich. Für die Ergebnisse dieser Arbeiten sei auf dieArbeit von C. Arpagaus (2000) verwiesen.
Innerhalb dieses Kapitels soll sich die Darstellung auf die wichtigsten Informationenbeschränken, die notwendig sind, um die verwendeten Programme (gPROMS undFLUENT) und die Implementierung der Systeme zu verstehen. Die Methoden werdenals gegeben hingenommen, für eine detaillierte Beschreibung der Probleme(Implementierung der Adsorption, numerische Lösungsprobleme, Optimierung derGitter, etc.) sei auf die folgenden Stellen verwiesen: Lisso et al. (1999, 2000), Lisso(2001), Boysen et al. (2001a, 2001b) und Guillaume (2001).
4.1 Eindimensionales axiales Modell (gPROMS)
Zur Lösung der Gleichungen (2.56) und (2.57) unter Berücksichtigung einer geeignetenAdsorptionsisotherme (Kapitel 2.2.3) findet in gPROMS eine Orts- und Zeitdiskre-tisierung innerhalb der Chromatographiesäule statt, die mit der sog. "Method of Lines"gelöst wird. Hierdurch werden die partiellen Differentialgleichungen in ein Systemgewöhnlicher Differentialgleichungen überführt. Sowohl die zeitlichen Ableitungen wieauch die örtlichen Ableitungen werden linearisiert, d.h. der Differentialquotient wirddurch den Differenzenquotient ersetzt:

62 Modellierung der HPLC-Säulen 4
• vorderer Differenzenquotient (1. Ableitung):
( ) ( )z
zczczc ini
z
i
�
1 −≈∂∂ + (4.1)
• hinterer Differenzenquotient (1. Ableitung):
( ) ( )z
zczczc nii
z
i
�
1−−≈∂∂
(4.2)
• zentraler Differenzenquotient (1. Ableitung):
( ) ( )z
zczczc nini
z
i
�211 −+ −≈
∂∂
(4.3)
• zentraler Differenzenquotient (2. Ableitung):
( ) ( ) ( )2
112
2
�
2z
zczczczc niini
z
i −+ +−≈∂∂
(4.4)
Es werden für einen Anfangszeitpunkt Startwerte für die Konzentrationen in derFlüssigphase und die mittlere Beladung der stationären Phase an jeder Stützstellegegeben, wobei von einer nicht vorbeladenen Säule ausgegangen wird:
( ) 00 == z,tc i (4.5)Gleichungen (2.56) und (2.57) dienen nun zur Bestimmung der zeitlichen Ableitungender Konzentration � t� ic . Für die Berechnung dieses Differenzenquotienten sind
sowohl explizite wie implizite Verfahren verfügbar (z.B. das Euler-Verfahren oder dasRunge-Kutta-Verfahren). Die impliziten Lösungsverfahren benötigen für jeden Zeit-schritt zur Berechnung des Gradienten die Lösung eines Iterationsvorganges. Es istnicht bekannt, welche Lösungsverfahren innerhalb des Gleichungslösers gPROMS zumEinsatz kommen.
Zur Simulation einer Peakaufgabe wird als Randbedingung für jeden weiteren Zeitpunkteine Eintrittskonzentration vorgegeben:
( ) ( )tcz,tc Ei == 0 (4.6)
Wobei für die Eintrittskonzentration gilt:
( ) injinji
Ei ttctc ≤= für (4.7)
( ) injEi tttc >= für0 (4.8)
gPROMS ignoriert die Vorgabe einer zweiten Randbedingung. Auch auf dieZeitschrittweite kann kein Einfluss genommen werden. Für die Anzahl an Stützstellenwird das Kriterium angelegt, dass sich das Chromatogramm mit steigender Anzahl anStützstellen nicht mehr verändern soll. In diesem Fall ist eine ausreichende Anzahl anStützstellen gewählt worden. Dieses Kriterium ist in Bild 4.1 am Beispiel derModellierung der Experimente mit Glaskugeln veranschaulicht.
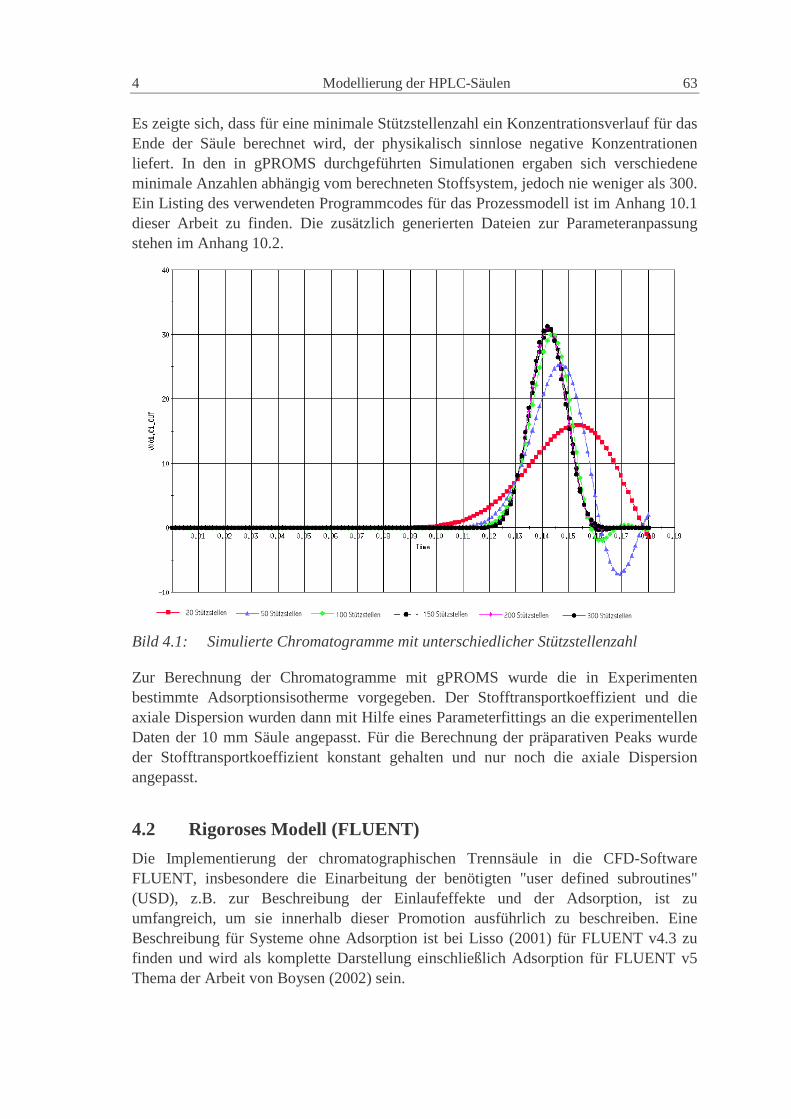
4 Modellierung der HPLC-Säulen 63
Es zeigte sich, dass für eine minimale Stützstellenzahl ein Konzentrationsverlauf für dasEnde der Säule berechnet wird, der physikalisch sinnlose negative Konzentrationenliefert. In den in gPROMS durchgeführten Simulationen ergaben sich verschiedeneminimale Anzahlen abhängig vom berechneten Stoffsystem, jedoch nie weniger als 300.Ein Listing des verwendeten Programmcodes für das Prozessmodell ist im Anhang 10.1dieser Arbeit zu finden. Die zusätzlich generierten Dateien zur Parameteranpassungstehen im Anhang 10.2.
Bild 4.1: Simulierte Chromatogramme mit unterschiedlicher Stützstellenzahl
Zur Berechnung der Chromatogramme mit gPROMS wurde die in Experimentenbestimmte Adsorptionsisotherme vorgegeben. Der Stofftransportkoeffizient und dieaxiale Dispersion wurden dann mit Hilfe eines Parameterfittings an die experimentellenDaten der 10 mm Säule angepasst. Für die Berechnung der präparativen Peaks wurdeder Stofftransportkoeffizient konstant gehalten und nur noch die axiale Dispersionangepasst.
4.2 Rigoroses Modell (FLUENT)
Die Implementierung der chromatographischen Trennsäule in die CFD-SoftwareFLUENT, insbesondere die Einarbeitung der benötigten "user defined subroutines"(USD), z.B. zur Beschreibung der Einlaufeffekte und der Adsorption, ist zuumfangreich, um sie innerhalb dieser Promotion ausführlich zu beschreiben. EineBeschreibung für Systeme ohne Adsorption ist bei Lisso (2001) für FLUENT v4.3 zufinden und wird als komplette Darstellung einschließlich Adsorption für FLUENT v5Thema der Arbeit von Boysen (2002) sein.

64 Modellierung der HPLC-Säulen 4
4.2.1 Grundsätzliches Vorgehen
Das generelle Vorgehen zum Aufbau eines Modells in FLUENT ist in Bild 4.2dargestellt. Ausgehend von der physikalischen Problemstellung und der enthaltenenAbläufe wird ein mathematisches Beschreibungsmodell erstellt. Es umfasst alle zurAbbildung des Systems notwendigen Bilanzen, repräsentiert durch ihre mathematischenGleichungen (Kap. 2.1), sowie alle notwendigen Parameter (Stoffdaten der betrachtetenKomponenten etc.).
Bild 4.2: Schema der Implementierung eines Modells in FLUENT
Die Geometrie des zu modellierenden Systems wird geeignet in die Simulations-umgebung übertragen: Alle Aussenkanten werden entweder als undurchdringlicheWände oder als Grenzen definiert, die die Eckpunkte der Geometrie verbinden. Eswerden auch alle internen Grenzen (wie z.B. Phasengrenzen) festgelegt, sofern sieeindeutig geometrisch bestimmbar sind. Anschliessend wird diese Geometrie durch einGitter diskretisiert, um auftretende große Gradienten der betrachteten Variablen(Konzentration, Geschwindigkeit) numerisch ausreichend gut abbilden zu können. Alsallgemeine Regel wird das Gitter so lange verfeinert, bis sich die berechnetenErgebnisse nicht mehr mit einer weiteren Verfeinerung des Gitters ändern. DieÜbertragung der Geometrie und die Erstellung des Gitters erfolgt für FLUENT mitHilfe des Programms gambit, das im FLUENT-Paket enthalten ist. Für dieBeschreibung der Grenzen kommen folgende Randbedingungen zum Einsatz:

4 Modellierung der HPLC-Säulen 65
• Dirichlet-Randbedingung: Am Einstromrand wird die Geschwindigkeit derStrömung vorgegeben, womit gleichzeitig auch der Druck festgelegt ist. An realenWänden wird die Wandhaftung miteinbezogen.
• Neumann-Randbedingung: Am Ausstromrand werden die Normalableitungen allerGeschwindigkeitskomponenten Null gesetzt, um eine Überbestimmtheit desProblems zu vermeiden. An den Symmetrielinien wird verlangt, dass dieStrömungsgrössen normal zur Symmetrielinie verschwinden.
Zur Berechnung des Systems müssen noch die Rand- und Anfangsbedingungenvorgegeben werden. Eine geeignete Diskretisierung der Gleichungen wird vonFLUENT nach der Finiten-Volumen-Methode (FVM) übernommen. Anschliessendwerden die numerischen Berechnungen zur Lösung der Gleichungssysteme und ihrerParameter durchgeführt, auftretende Konvergenzprobleme werden durch geeigneteVerfeinerung des Gitters an kritischen Punkten gelöst.
Nach erfolgreicher Berechnung des Systems, d.h. der Existenz einer konvergiertenLösung des mathematischen Modells für das Gitter mit den festgelegten Bedingungenund Parametern, werden die Ergebnisse ausgegeben und visualisiert. Für eine weiter-gehende Auswertung werden die Ergebisse in Microsoft Excel übertragen und mitexperimentellen Ergebnissen verglichen. Zum Erreichen einer angemessenen Überein-stimmung von numerischen und experimentellen Ergebnissen wurden diemathematischen Modelle in FLUENT angepasst und erweitert (über User DefinedFunctions, UDF), physikalische Parameter bestimmt und numerische Parameterangepasst. Diese Anpassung ist zusammen mit der experimentellen Validierung deraufwendigste und umfangreichste Schritt der Entwicklung einer Modellumgebung inFLUENT. Sie ist Inhalt der Arbeiten von Boysen (2001a, 2001b und 2002).
4.2.2 Beschreibung des in FLUENT verwendeten Modells
Zum besseren Verständnis ist in Bild 4.3 die in FLUENT über gambit erstellteGeometrie der modellierten HPLC-Säule dargestellt:
Bild 4.3: Geometrie und Gitter der HPLC-Säule in FLUENT
Die Linien sind die Ränder (boundaries) der Geometrie, d.h. sie repräsentieren sowohlundurchlässige Grenzen, wie z.B. die Wände der Säule, als auch durchlässige
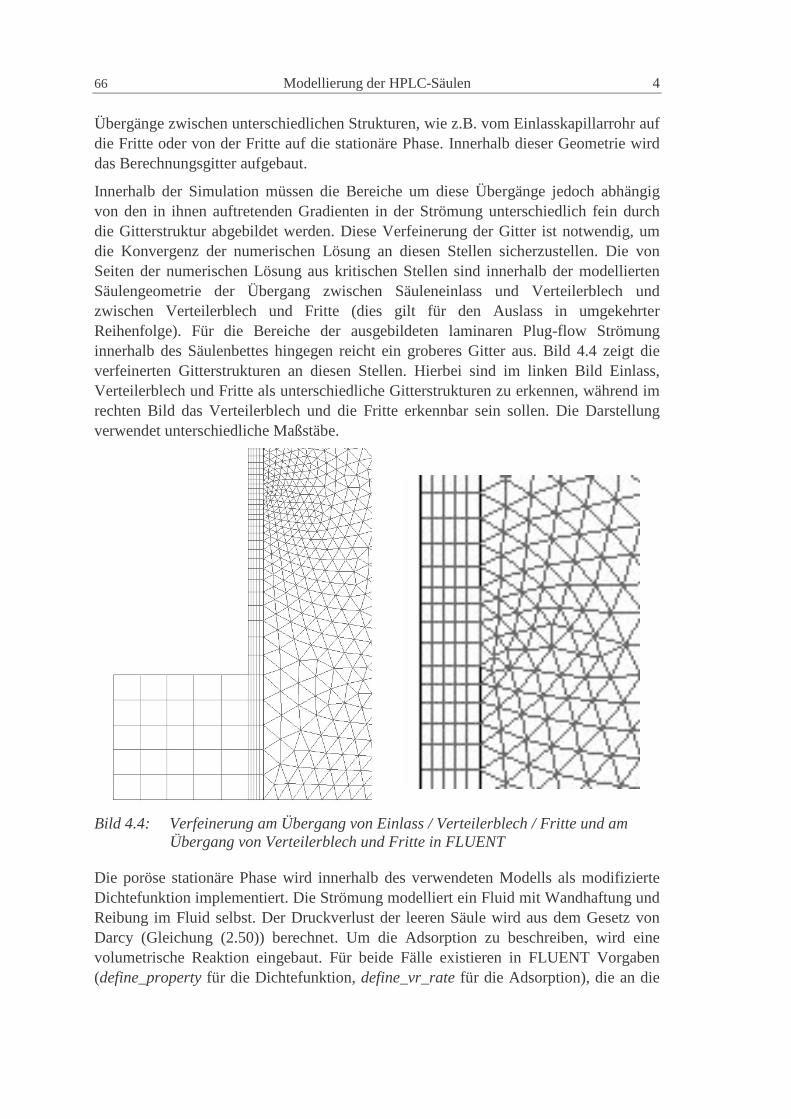
66 Modellierung der HPLC-Säulen 4
Übergänge zwischen unterschiedlichen Strukturen, wie z.B. vom Einlasskapillarrohr aufdie Fritte oder von der Fritte auf die stationäre Phase. Innerhalb dieser Geometrie wirddas Berechnungsgitter aufgebaut.
Innerhalb der Simulation müssen die Bereiche um diese Übergänge jedoch abhängigvon den in ihnen auftretenden Gradienten in der Strömung unterschiedlich fein durchdie Gitterstruktur abgebildet werden. Diese Verfeinerung der Gitter ist notwendig, umdie Konvergenz der numerischen Lösung an diesen Stellen sicherzustellen. Die vonSeiten der numerischen Lösung aus kritischen Stellen sind innerhalb der modelliertenSäulengeometrie der Übergang zwischen Säuleneinlass und Verteilerblech undzwischen Verteilerblech und Fritte (dies gilt für den Auslass in umgekehrterReihenfolge). Für die Bereiche der ausgebildeten laminaren Plug-flow Strömunginnerhalb des Säulenbettes hingegen reicht ein groberes Gitter aus. Bild 4.4 zeigt dieverfeinerten Gitterstrukturen an diesen Stellen. Hierbei sind im linken Bild Einlass,Verteilerblech und Fritte als unterschiedliche Gitterstrukturen zu erkennen, während imrechten Bild das Verteilerblech und die Fritte erkennbar sein sollen. Die Darstellungverwendet unterschiedliche Maßstäbe.
Bild 4.4: Verfeinerung am Übergang von Einlass / Verteilerblech / Fritte und amÜbergang von Verteilerblech und Fritte in FLUENT
Die poröse stationäre Phase wird innerhalb des verwendeten Modells als modifizierteDichtefunktion implementiert. Die Strömung modelliert ein Fluid mit Wandhaftung undReibung im Fluid selbst. Der Druckverlust der leeren Säule wird aus dem Gesetz vonDarcy (Gleichung (2.50)) berechnet. Um die Adsorption zu beschreiben, wird einevolumetrische Reaktion eingebaut. Für beide Fälle existieren in FLUENT Vorgaben(define_property für die Dichtefunktion, define_vr_rate für die Adsorption), die an die

4 Modellierung der HPLC-Säulen 67
physikalischen Modelle angepasst werden. Die in Kapitel 2.1.6 beschriebenenGleichungen werden dann unter Berücksichtigung der veränderten Dichte und einesQuellterms aus Kapitel 2.1.6.2 gelöst.

68 Modellierung der HPLC-Säulen 4

5 Bestimmung der Anteile der axialen Dispersion 69
5 Bestimmung der Anteile der axialen Dispersion
Für die rein thermodynamische Modellierung der Peakverläufe wird nur dieKomponentenbilanz in axialer Richtung (Gl. (2.56)) durch die Säule und eine geeigneteAdsorptionsisotherme (Gl. (2.57)) betrachtet. In diese Gleichungen gehen sowohl allestofflichen Einflüsse als auch alle Anlageneinflüsse ein. Die stofflichen Einflüssewerden im Wesentlichen durch die Adsorptionsisotherme *
iq , den Stofftransportkoef-
fizienten effk und die Porosität wiedergegeben. Alle Einflüsse des Anlagenaufbaus und
des Verteilungssystems der Säule hingegen beeinflussen nur den Wert der axialenDispersion axD . Allerdings hat auch die Struktur und die Form der stationären Phase
einen Einfluss auf die axiale Dispersion des Systems (Kapitel 2.2.5). Aus diesem Grundist eine Unterteilung der axialen Dispersion in einen anlagenspezifischen und einenphasenspezifischen Anteil sinnvoll.
Dies wird bei der folgenden Betrachtung deutlich: In der üblichen Reihenfolge derMethodenentwicklung einer chromatographischen Trennung stehen Laborexperimentean erster Stelle. In ihnen wird in ersten Experimenten das Trennsystem ausgewählt undauf seine Eignung zur Trennung der gewünschten Produkte hin untersucht. Ist diestationäre Phase festgelegt, werden die thermodynamischen Eigenschaften desTrennsystems bestimmt (Adsorptionsisothermen, Stofftransportkoeffizienten undPorositäten). Die Methode wird auf einer analytischen Anlage durch Probenaufgabenund deren Auswertung etabliert. Aus diesen Experimenten erhält man auch die axialeDispersion dieses Systems (in dem ein Anlageneinfluss normalerweise klein ist, dadiese Anlagen auf kleine Totvolumina etc. optimiert sind), z.B. über Gleichung (2.86)oder durch Anpassung der axialen Dispersion in der Modellierung. Mit dengewünschten Produktionsparametern (Produktivität, Reinheit) wird, falls dieEntscheidung für eine Batch-Trennung gefallen ist, ein präparatives System zurtechnischen Trennung ausgewählt. Diese präparativen Systeme besitzen jedoch eineandere axiale Dispersion, da sie sich im Anlagenaufbau wesentlich von der analytischenAnlage unterscheiden:
• Grösseres Anlagenholdup, mehr Messstellen (Druck und Temperatur vor, an undnach der Säule), meist eigenständiger Feedkreislauf an Stelle eines Probenaufgabe-ventils. Dies führt zu größeren Rückvermischungseffekten.
• Andere Verteilerbauart, da die Flüssigkeit auf einen wesentlich grösseren Säulen-querschnitt verteilt werden muss.
• Zusätzlich wird die stationäre Phase axial komprimiert, um eine hohe Bettqualität zugarantieren.
Um die Ergebnisse der analytischen Methodenentwicklung auf die präparativeTrennung zu übertragen, ist es sinnvoll, den Einfluss der unterschiedlichen Anlagen aufdie axiale Dispersion quantifizieren zu können. Durch Versuche mit einemReferenzsystem in allen Anlagen (analytisch und präparativ) und durch Vergleich derErgebnisse in Bezug auf die auftretende axiale Dispersion kann der Unterschied der
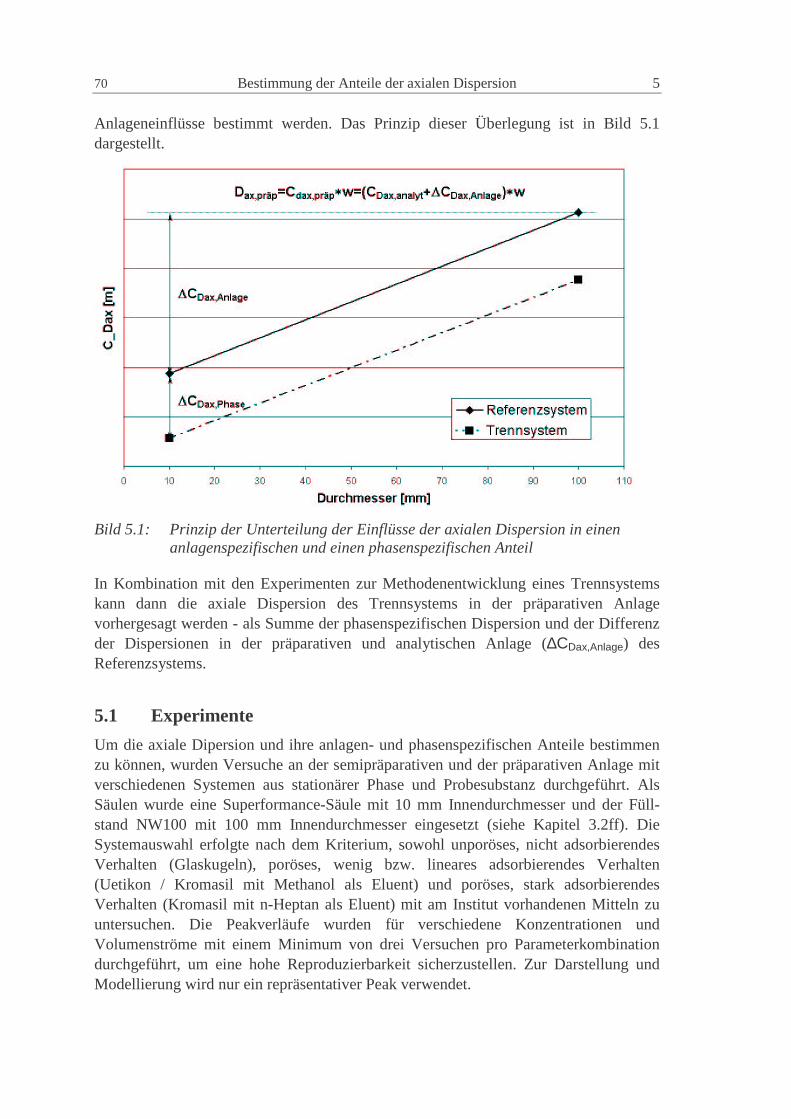
70 Bestimmung der Anteile der axialen Dispersion 5
Anlageneinflüsse bestimmt werden. Das Prinzip dieser Überlegung ist in Bild 5.1dargestellt.
Bild 5.1: Prinzip der Unterteilung der Einflüsse der axialen Dispersion in einenanlagenspezifischen und einen phasenspezifischen Anteil
In Kombination mit den Experimenten zur Methodenentwicklung eines Trennsystemskann dann die axiale Dispersion des Trennsystems in der präparativen Anlagevorhergesagt werden - als Summe der phasenspezifischen Dispersion und der Differenzder Dispersionen in der präparativen und analytischen Anlage (∆CDax,Anlage) desReferenzsystems.
5.1 Exper imente
Um die axiale Dipersion und ihre anlagen- und phasenspezifischen Anteile bestimmenzu können, wurden Versuche an der semipräparativen und der präparativen Anlage mitverschiedenen Systemen aus stationärer Phase und Probesubstanz durchgeführt. AlsSäulen wurde eine Superformance-Säule mit 10 mm Innendurchmesser und der Füll-stand NW100 mit 100 mm Innendurchmesser eingesetzt (siehe Kapitel 3.2ff). DieSystemauswahl erfolgte nach dem Kriterium, sowohl unporöses, nicht adsorbierendesVerhalten (Glaskugeln), poröses, wenig bzw. lineares adsorbierendes Verhalten(Uetikon / Kromasil mit Methanol als Eluent) und poröses, stark adsorbierendesVerhalten (Kromasil mit n-Heptan als Eluent) mit am Institut vorhandenen Mitteln zuuntersuchen. Die Peakverläufe wurden für verschiedene Konzentrationen undVolumenströme mit einem Minimum von drei Versuchen pro Parameterkombinationdurchgeführt, um eine hohe Reproduzierbarkeit sicherzustellen. Zur Darstellung undModellierung wird nur ein repräsentativer Peak verwendet.
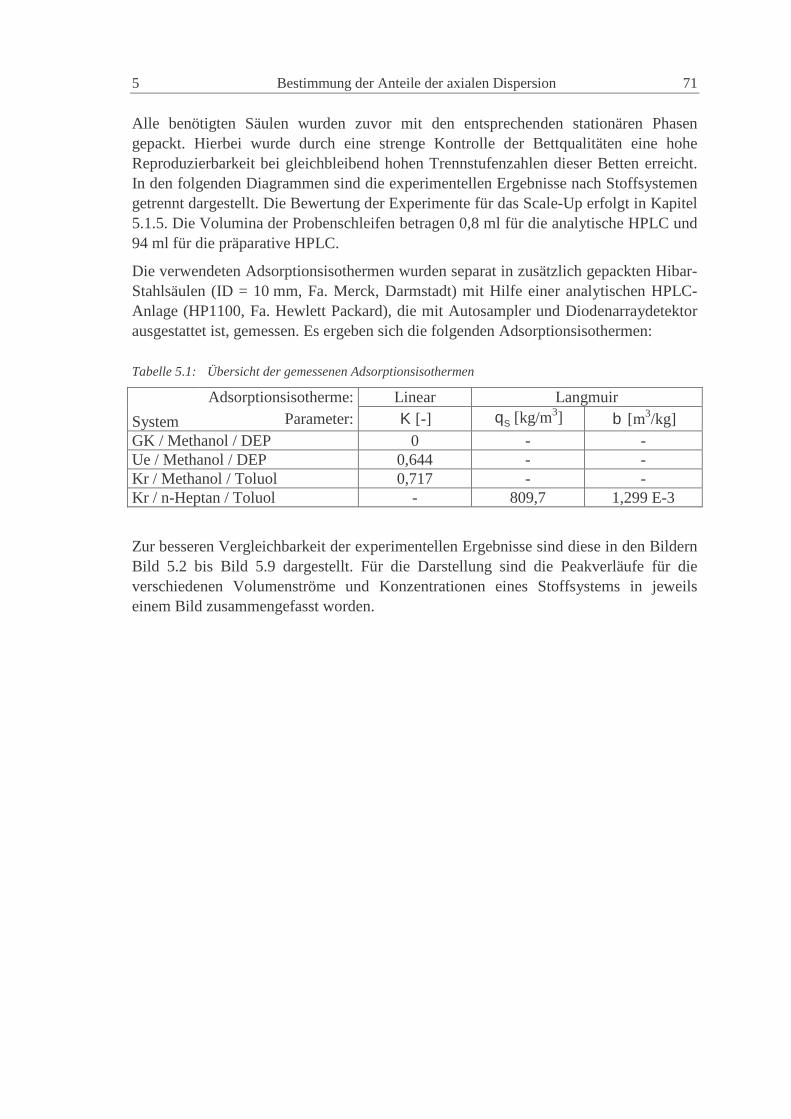
5 Bestimmung der Anteile der axialen Dispersion 71
Alle benötigten Säulen wurden zuvor mit den entsprechenden stationären Phasengepackt. Hierbei wurde durch eine strenge Kontrolle der Bettqualitäten eine hoheReproduzierbarkeit bei gleichbleibend hohen Trennstufenzahlen dieser Betten erreicht.In den folgenden Diagrammen sind die experimentellen Ergebnisse nach Stoffsystemengetrennt dargestellt. Die Bewertung der Experimente für das Scale-Up erfolgt in Kapitel5.1.5. Die Volumina der Probenschleifen betragen 0,8 ml für die analytische HPLC und94 ml für die präparative HPLC.
Die verwendeten Adsorptionsisothermen wurden separat in zusätzlich gepackten Hibar-Stahlsäulen (ID = 10 mm, Fa. Merck, Darmstadt) mit Hilfe einer analytischen HPLC-Anlage (HP1100, Fa. Hewlett Packard), die mit Autosampler und Diodenarraydetektorausgestattet ist, gemessen. Es ergeben sich die folgenden Adsorptionsisothermen:
Tabelle 5.1: Übersicht der gemessenen Adsorptionsisothermen
Adsorptionsisotherme: Linear Langmuir
System Parameter: K [-] Sq [kg/m3] b [m3/kg]GK / Methanol / DEP 0 - -Ue / Methanol / DEP 0,644 - -Kr / Methanol / Toluol 0,717 - -Kr / n-Heptan / Toluol - 809,7 1,299 E-3
Zur besseren Vergleichbarkeit der experimentellen Ergebnisse sind diese in den BildernBild 5.2 bis Bild 5.9 dargestellt. Für die Darstellung sind die Peakverläufe für dieverschiedenen Volumenströme und Konzentrationen eines Stoffsystems in jeweilseinem Bild zusammengefasst worden.
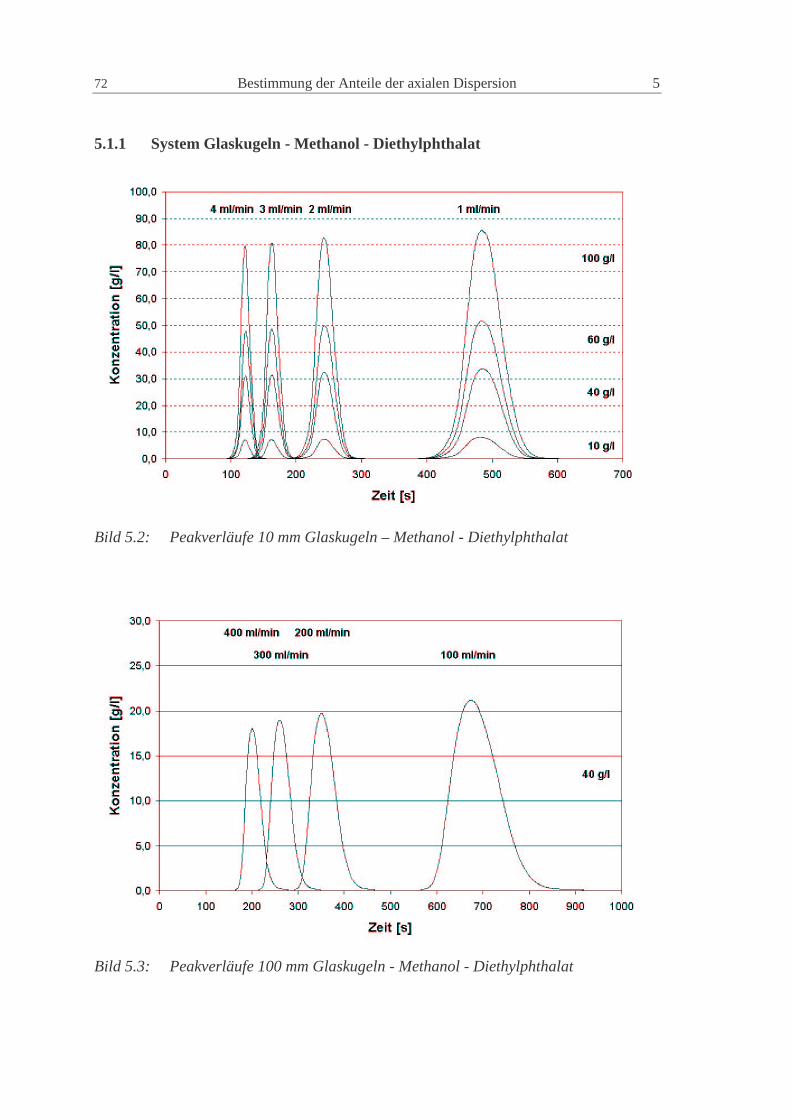
72 Bestimmung der Anteile der axialen Dispersion 5
5.1.1 System Glaskugeln - Methanol - Diethylphthalat
Bild 5.2: Peakverläufe 10 mm Glaskugeln – Methanol - Diethylphthalat
Bild 5.3: Peakverläufe 100 mm Glaskugeln - Methanol - Diethylphthalat
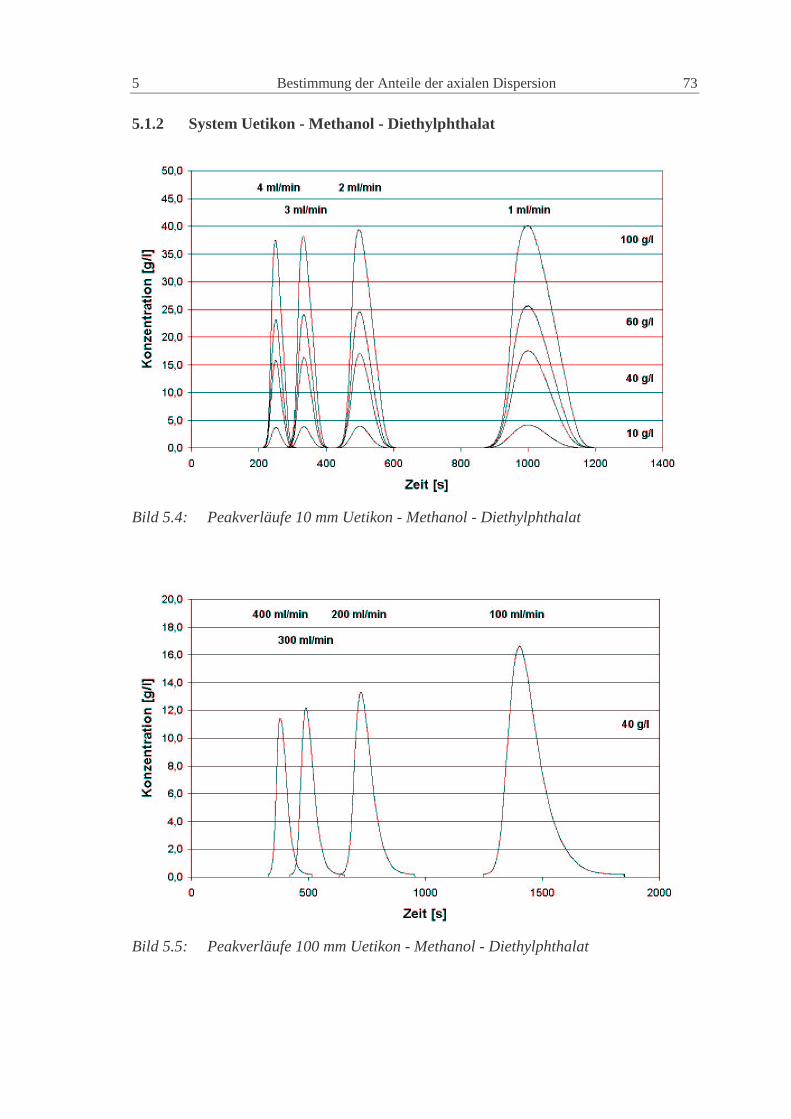
5 Bestimmung der Anteile der axialen Dispersion 73
5.1.2 System Uetikon - Methanol - Diethylphthalat
Bild 5.4: Peakverläufe 10 mm Uetikon - Methanol - Diethylphthalat
Bild 5.5: Peakverläufe 100 mm Uetikon - Methanol - Diethylphthalat
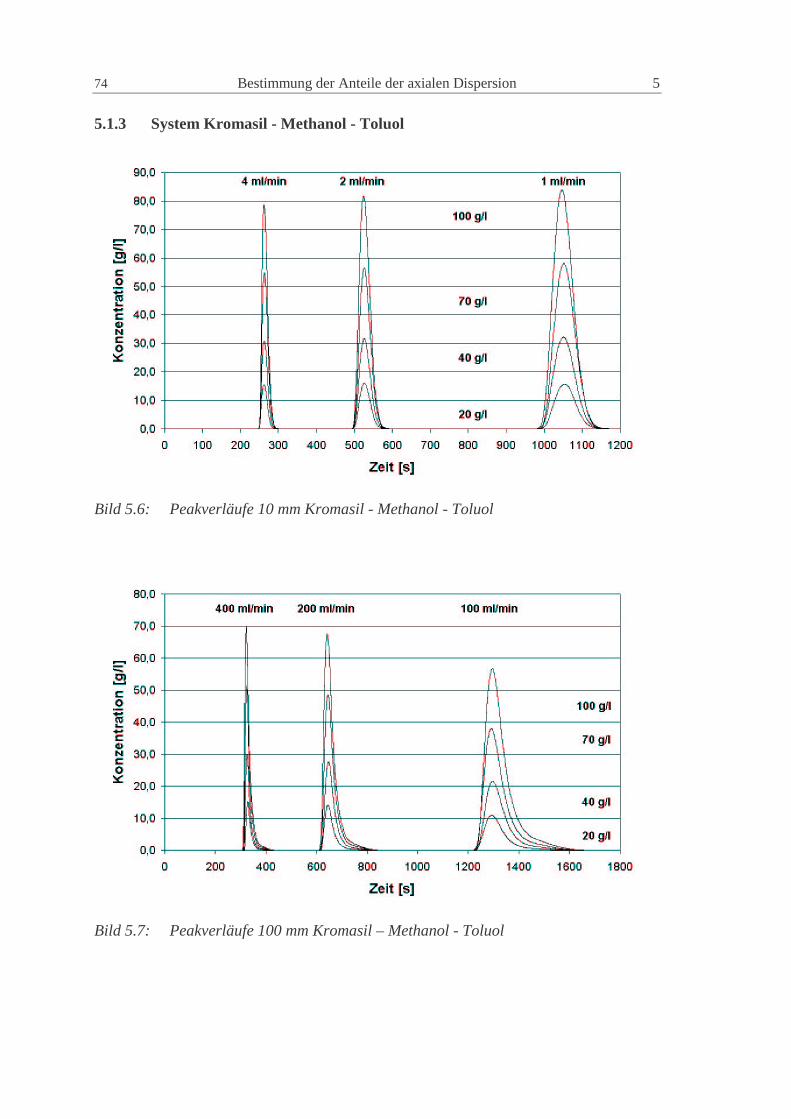
74 Bestimmung der Anteile der axialen Dispersion 5
5.1.3 System Kromasil - Methanol - Toluol
Bild 5.6: Peakverläufe 10 mm Kromasil - Methanol - Toluol
Bild 5.7: Peakverläufe 100 mm Kromasil – Methanol - Toluol
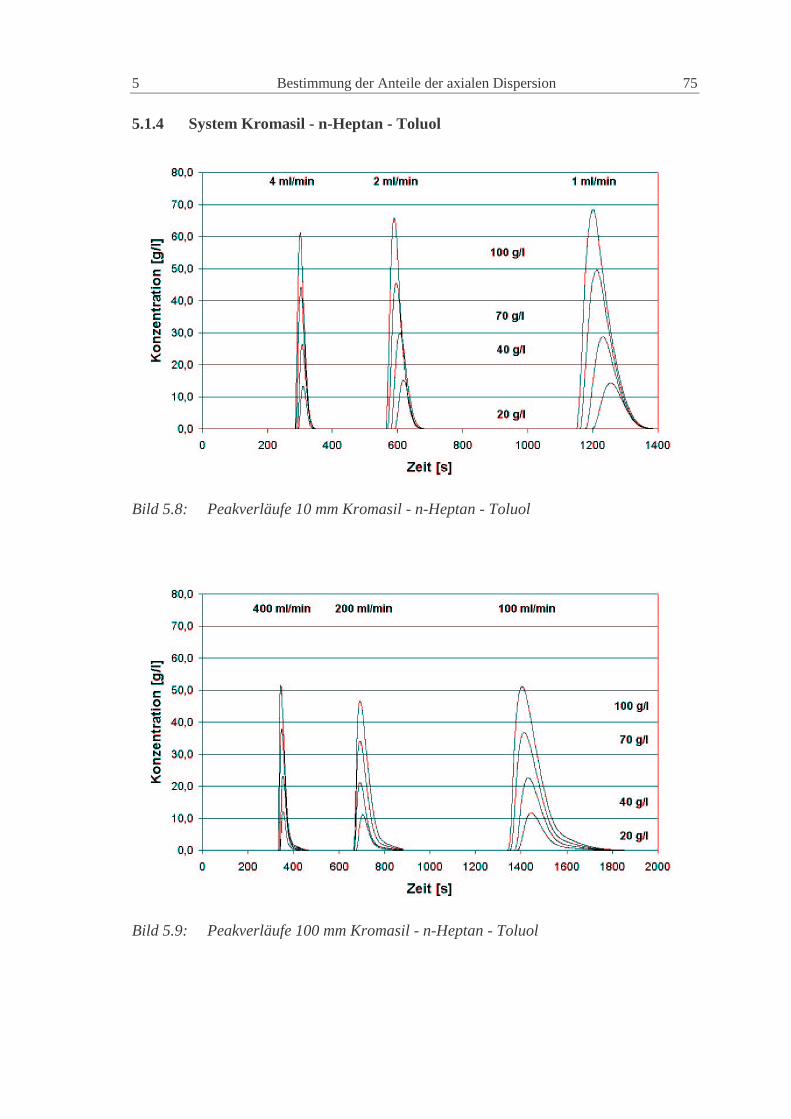
5 Bestimmung der Anteile der axialen Dispersion 75
5.1.4 System Kromasil - n-Heptan - Toluol
Bild 5.8: Peakverläufe 10 mm Kromasil - n-Heptan - Toluol
Bild 5.9: Peakverläufe 100 mm Kromasil - n-Heptan - Toluol
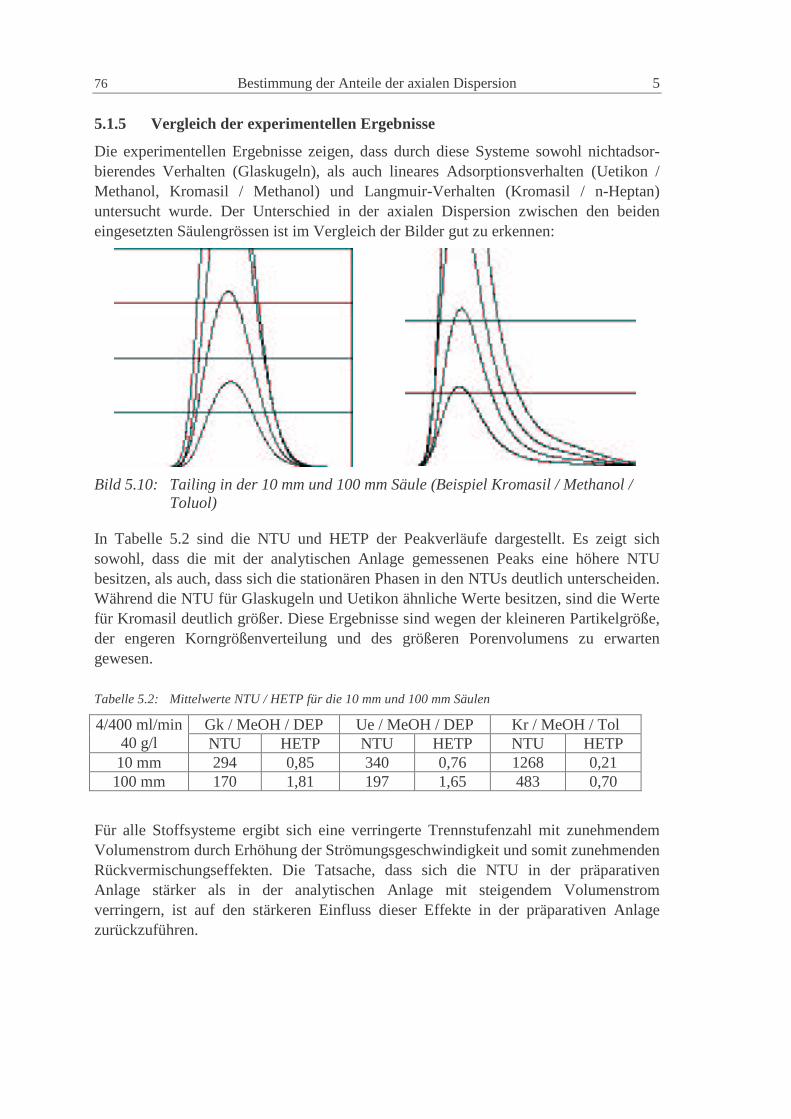
76 Bestimmung der Anteile der axialen Dispersion 5
5.1.5 Vergleich der exper imentellen Ergebnisse
Die experimentellen Ergebnisse zeigen, dass durch diese Systeme sowohl nichtadsor-bierendes Verhalten (Glaskugeln), als auch lineares Adsorptionsverhalten (Uetikon /Methanol, Kromasil / Methanol) und Langmuir-Verhalten (Kromasil / n-Heptan)untersucht wurde. Der Unterschied in der axialen Dispersion zwischen den beideneingesetzten Säulengrössen ist im Vergleich der Bilder gut zu erkennen:
Bild 5.10: Tailing in der 10 mm und 100 mm Säule (Beispiel Kromasil / Methanol /Toluol)
In Tabelle 5.2 sind die NTU und HETP der Peakverläufe dargestellt. Es zeigt sichsowohl, dass die mit der analytischen Anlage gemessenen Peaks eine höhere NTUbesitzen, als auch, dass sich die stationären Phasen in den NTUs deutlich unterscheiden.Während die NTU für Glaskugeln und Uetikon ähnliche Werte besitzen, sind die Wertefür Kromasil deutlich größer. Diese Ergebnisse sind wegen der kleineren Partikelgröße,der engeren Korngrößenverteilung und des größeren Porenvolumens zu erwartengewesen.
Tabelle 5.2: Mittelwerte NTU / HETP für die 10 mm und 100 mm Säulen
Gk / MeOH / DEP Ue / MeOH / DEP Kr / MeOH / Tol4/400 ml/min40 g/l NTU HETP NTU HETP NTU HETP
10 mm 294 0,85 340 0,76 1268 0,21100 mm 170 1,81 197 1,65 483 0,70
Für alle Stoffsysteme ergibt sich eine verringerte Trennstufenzahl mit zunehmendemVolumenstrom durch Erhöhung der Strömungsgeschwindigkeit und somit zunehmendenRückvermischungseffekten. Die Tatsache, dass sich die NTU in der präparativenAnlage stärker als in der analytischen Anlage mit steigendem Volumenstromverringern, ist auf den stärkeren Einfluss dieser Effekte in der präparativen Anlagezurückzuführen.

5 Bestimmung der Anteile der axialen Dispersion 77
5.2 Modellierung in gPROMS
Die Modellierung in gPROMS erfolgte nach der in Kapitel 4.1 beschriebenen Methodemit der externen Porosität als Maß der Porosität.
Die Startwerte für die Konstante der axialen Dispersion DaxC der analytischen Anlage
wurden aus den Werten der axialen Dispersion, die sich nach Auswertung derPeakverläufe nach der Momentenmethode aus Gleichung (2.86) ergaben, berechnet. Fürjedes Stoffsystem werden die entsprechenden, mit der HP1100 gemessenen,Adsorptionsisothermen eingesetzt und anschliessend der Wert für die Konstante desaxialen Dispersionskoeffizienten an die Experimente der analytischen Anlageangepasst. Für die Experimente mit Glaskugeln als Referenzsystem wurde dann analogder Wert der Konstante der axialen Dispersion bestimmt. Die Differenz dieser Wertecharakterisiert den unterschiedlichen Einfluss der Anlagen.
Für die Modellierung der weiteren Systeme wurden ausgehend vom aus den NTUberechneten Startwert der axialen Dispersion die Werte des Stofftransportkoeffizienten
effk und DaxC für die analytische Anlage angepasst. Zur Modellierung der präparativen
Peakverläufe wurde dann DaxC nach dem in Bild 5.1 dargestellten Prinzip um die
Differenz der Konstanten der axialen Dispersion der Anlagen vergrössert.Anschliessend wurden DaxC und effk über eine Optimierungsroutine an die
experimentellen Peakverläufe angepasst. Der Vergleich der berechneten undgemessenen Peakverläufe erfolgte im Hinblick auf möglichst genaue Abbildung derRetentionszeiten und Trennstufenzahlen (Abweichungen < 2 %).
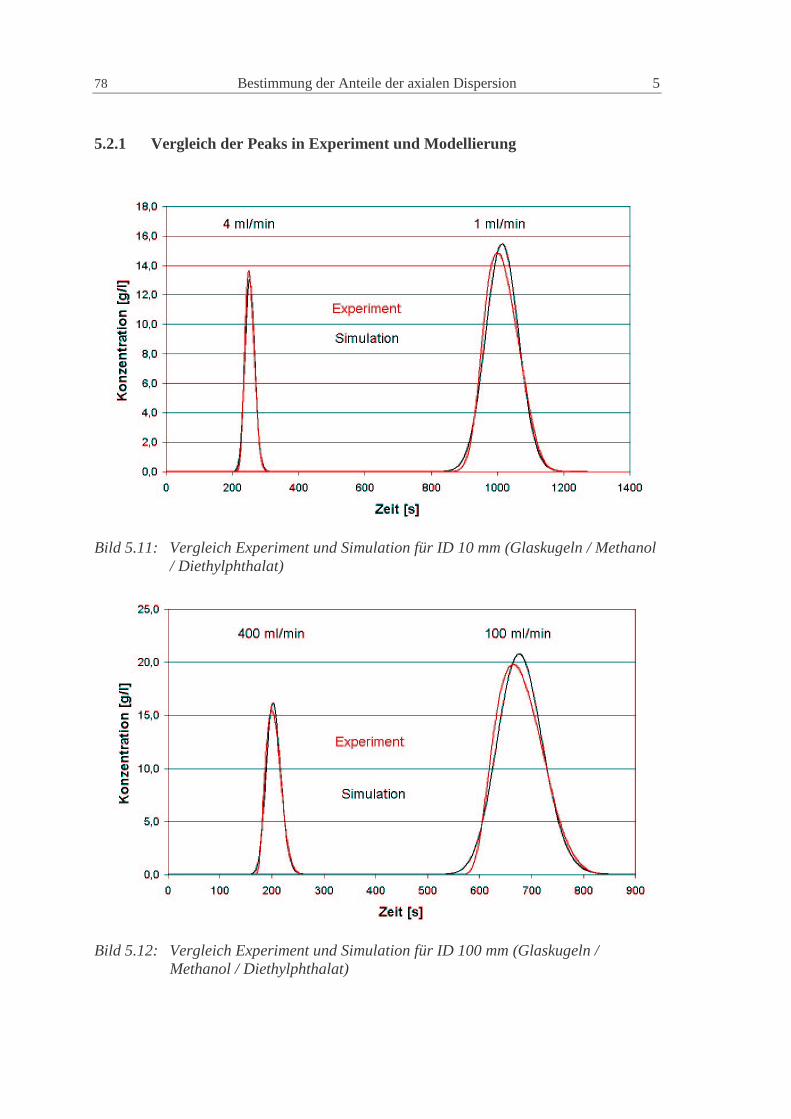
78 Bestimmung der Anteile der axialen Dispersion 5
5.2.1 Vergleich der Peaks in Exper iment und Modellierung
Bild 5.11: Vergleich Experiment und Simulation für ID 10 mm (Glaskugeln / Methanol/ Diethylphthalat)
Bild 5.12: Vergleich Experiment und Simulation für ID 100 mm (Glaskugeln /Methanol / Diethylphthalat)
5 Bestimmung der Anteile der axialen Dispersion 79
Ein Vergleich der berechneten und gemessenen Peakverläufe ist beispielhaft für dieExperimente mit Glaskugeln in der analytischen HPLC mit Methanol als Eluent undDiethylphthalat als Probe in Bild 5.11 dargestellt. Bild 5.12 enthält den Vergleich fürdas gleiche Stoffsystem in der präparativen Anlage.
Diese Bilder zeigen die gute qualitative Übereinstimmung der Ergebnisse derModellierung mit den Experimenten. Die quantitative Übereinstimmung in Retentions-zeit und NTU ist die Bedingung der Anpassung der Parameter der Simulation gewesen.
5.2.2 Ergebnisse für die Systemparameter
Aus den Simulationen ergeben sich die in Tabelle 5.3 dargestellten Parameter allereingesetzten Systeme.
Tabelle 5.3: Übersicht der Simulationsparameter
SystemVolumenstrom
[ml/min]eext uo [mm/s] ds [mm] keff [1/s] Cdax [m]
MittelwertCdax [m]
1 0,54 -- 2,800E-042 1,08 -- 3,580E-043 1,61 -- 4,510E-044 2,15 -- 4,100E-04
100 0,54 -- 7,570E-04200 1,08 -- 8,423E-04300 1,61 -- 1,150E-03400 2,15 -- 1,354E-03
1 0,44 1 1,880E-042 0,88 1 1,140E-043 1,33 1 3,540E-054 1,77 1 --
100 0,44 1 7,768E-04200 0,88 1 7,848E-04300 1,33 1 6,192E-04400 1,77 1 8,340E-04
1 0,66 -- 8,727E-052 1,32 83,96 9,623E-054 2,65 100,00 1,162E-04
100 0,66 -- 3,769E-04200 1,32 6,68 3,974E-04400 2,65 27,05 4,832E-04
1 0,66 -- 1,138E-042 1,32 98,14 1,228E-044 2,65 633,60 1,615E-04
100 0,66 -- 4,176E-04200 1,32 66,83 5,283E-04400 2,65 270,50 5,909E-04
GK-MeOH
GK-MeOH
Uet-MeOH
Uet-MeOH
Krom-MeOH
Krom-MeOH
Krom-nHept
Krom-nHept
0,3942
0,3942
0,4800
0,4800
0,3204
0,3204
0,3204
0,3204
10
100
10
100
10
100
10
100
3,748E-04
1,026E-03
1,125E-04
7,537E-04
9,990E-05
4,191E-04
1,327E-04
5,123E-04
An dieser Stelle wird aus den aus den Modellierungen für die verschiedenen Volumen-ströme erhaltenen Werten der axialen Dispersion ein Mittelwert für jedes Stoffsystemgebildet. Diese Mittelwertbildung erlaubt es, alle Versuchsergebnisse als großeDatenbasis in den Ansatz zu integrieren. Allerdings ergeben sich aus denModellierungen für die verschiedenen Volumenströme eines Stoffsystemsunterschiedliche Werte der Konstanten der axialen Dispersion. So beträgt z.B. die
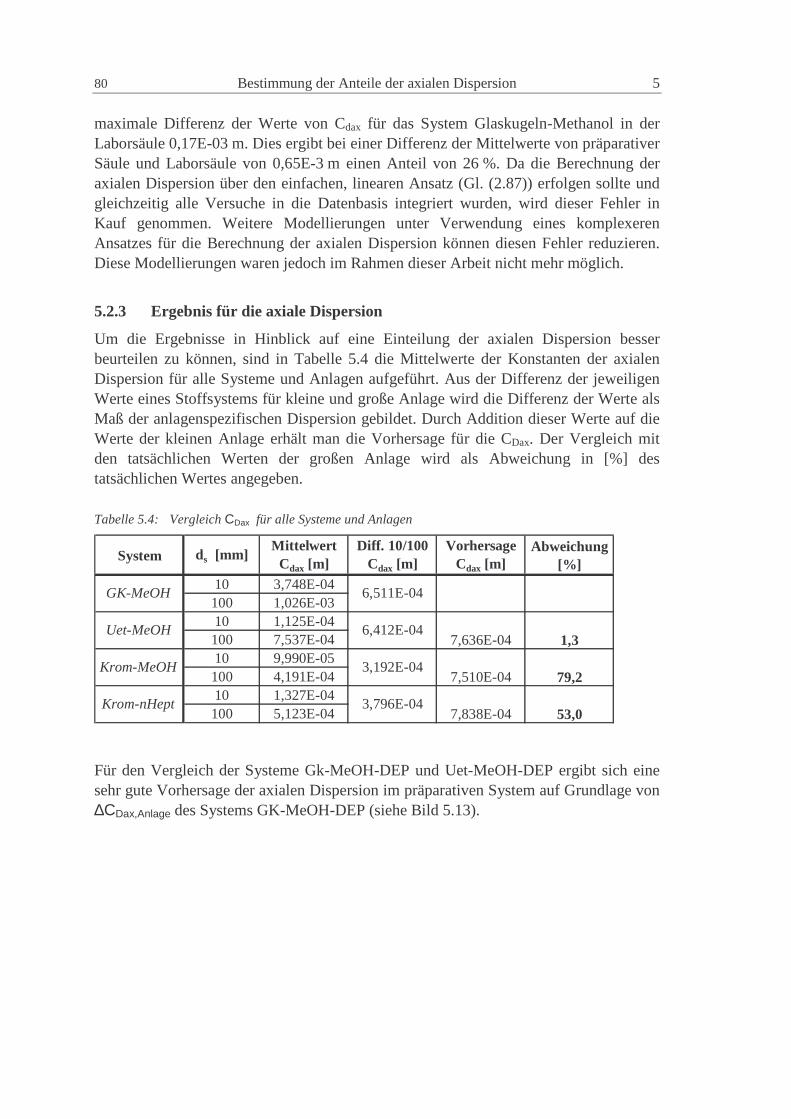
80 Bestimmung der Anteile der axialen Dispersion 5
maximale Differenz der Werte von Cdax für das System Glaskugeln-Methanol in derLaborsäule 0,17E-03 m. Dies ergibt bei einer Differenz der Mittelwerte von präparativerSäule und Laborsäule von 0,65E-3 m einen Anteil von 26 %. Da die Berechnung deraxialen Dispersion über den einfachen, linearen Ansatz (Gl. (2.87)) erfolgen sollte undgleichzeitig alle Versuche in die Datenbasis integriert wurden, wird dieser Fehler inKauf genommen. Weitere Modellierungen unter Verwendung eines komplexerenAnsatzes für die Berechnung der axialen Dispersion können diesen Fehler reduzieren.Diese Modellierungen waren jedoch im Rahmen dieser Arbeit nicht mehr möglich.
5.2.3 Ergebnis für die axiale Dispersion
Um die Ergebnisse in Hinblick auf eine Einteilung der axialen Dispersion besserbeurteilen zu können, sind in Tabelle 5.4 die Mittelwerte der Konstanten der axialenDispersion für alle Systeme und Anlagen aufgeführt. Aus der Differenz der jeweiligenWerte eines Stoffsystems für kleine und große Anlage wird die Differenz der Werte alsMaß der anlagenspezifischen Dispersion gebildet. Durch Addition dieser Werte auf dieWerte der kleinen Anlage erhält man die Vorhersage für die CDax. Der Vergleich mitden tatsächlichen Werten der großen Anlage wird als Abweichung in [%] destatsächlichen Wertes angegeben.
Tabelle 5.4: Vergleich CDax für alle Systeme und Anlagen
System ds [mm]M ittelwert
Cdax [m]Diff. 10/100
Cdax [m]Vorhersage
Cdax [m]Abweichung
[%]10 3,748E-04100 1,026E-0310 1,125E-04100 7,537E-0410 9,990E-05100 4,191E-0410 1,327E-04100 5,123E-04
GK-MeOH
Uet-MeOH
Krom-MeOH
Krom-nHept
6,511E-04
6,412E-04
3,192E-04
3,796E-04
7,636E-04
7,510E-04
7,838E-04
1,3
79,2
53,0
Für den Vergleich der Systeme Gk-MeOH-DEP und Uet-MeOH-DEP ergibt sich einesehr gute Vorhersage der axialen Dispersion im präparativen System auf Grundlage von∆CDax,Anlage des Systems GK-MeOH-DEP (siehe Bild 5.13).
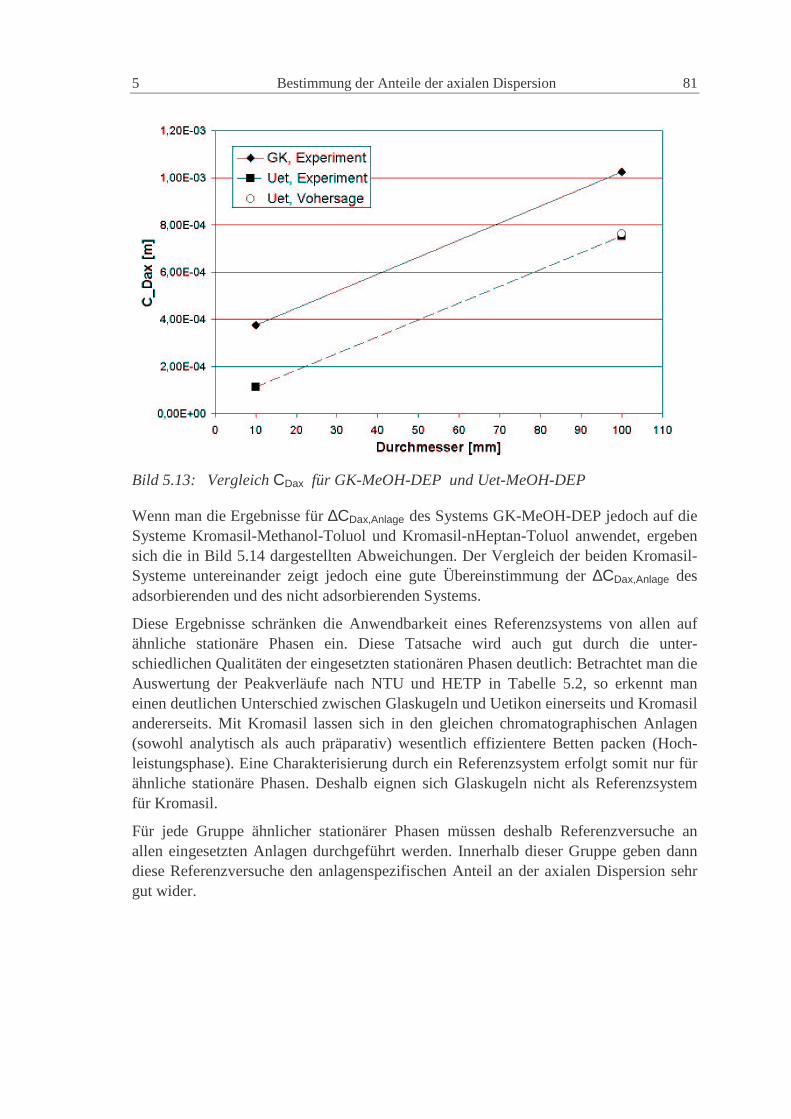
5 Bestimmung der Anteile der axialen Dispersion 81
Bild 5.13: Vergleich CDax für GK-MeOH-DEP und Uet-MeOH-DEP
Wenn man die Ergebnisse für ∆CDax,Anlage des Systems GK-MeOH-DEP jedoch auf dieSysteme Kromasil-Methanol-Toluol und Kromasil-nHeptan-Toluol anwendet, ergebensich die in Bild 5.14 dargestellten Abweichungen. Der Vergleich der beiden Kromasil-Systeme untereinander zeigt jedoch eine gute Übereinstimmung der ∆CDax,Anlage desadsorbierenden und des nicht adsorbierenden Systems.
Diese Ergebnisse schränken die Anwendbarkeit eines Referenzsystems von allen aufähnliche stationäre Phasen ein. Diese Tatsache wird auch gut durch die unter-schiedlichen Qualitäten der eingesetzten stationären Phasen deutlich: Betrachtet man dieAuswertung der Peakverläufe nach NTU und HETP in Tabelle 5.2, so erkennt maneinen deutlichen Unterschied zwischen Glaskugeln und Uetikon einerseits und Kromasilandererseits. Mit Kromasil lassen sich in den gleichen chromatographischen Anlagen(sowohl analytisch als auch präparativ) wesentlich effizientere Betten packen (Hoch-leistungsphase). Eine Charakterisierung durch ein Referenzsystem erfolgt somit nur fürähnliche stationäre Phasen. Deshalb eignen sich Glaskugeln nicht als Referenzsystemfür Kromasil.
Für jede Gruppe ähnlicher stationärer Phasen müssen deshalb Referenzversuche anallen eingesetzten Anlagen durchgeführt werden. Innerhalb dieser Gruppe geben danndiese Referenzversuche den anlagenspezifischen Anteil an der axialen Dispersion sehrgut wider.
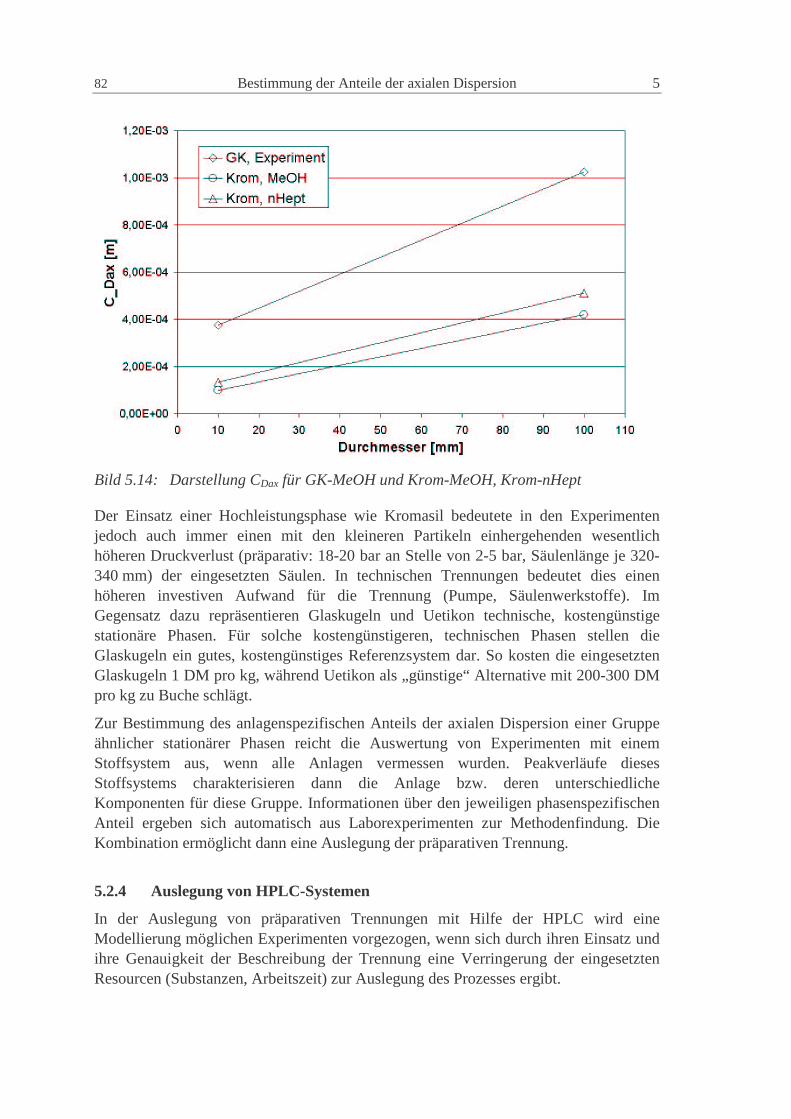
82 Bestimmung der Anteile der axialen Dispersion 5
Bild 5.14: Darstellung CDax für GK-MeOH und Krom-MeOH, Krom-nHept
Der Einsatz einer Hochleistungsphase wie Kromasil bedeutete in den Experimentenjedoch auch immer einen mit den kleineren Partikeln einhergehenden wesentlichhöheren Druckverlust (präparativ: 18-20 bar an Stelle von 2-5 bar, Säulenlänge je 320-340 mm) der eingesetzten Säulen. In technischen Trennungen bedeutet dies einenhöheren investiven Aufwand für die Trennung (Pumpe, Säulenwerkstoffe). ImGegensatz dazu repräsentieren Glaskugeln und Uetikon technische, kostengünstigestationäre Phasen. Für solche kostengünstigeren, technischen Phasen stellen dieGlaskugeln ein gutes, kostengünstiges Referenzsystem dar. So kosten die eingesetztenGlaskugeln 1 DM pro kg, während Uetikon als „günstige“ Alternative mit 200-300 DMpro kg zu Buche schlägt.
Zur Bestimmung des anlagenspezifischen Anteils der axialen Dispersion einer Gruppeähnlicher stationärer Phasen reicht die Auswertung von Experimenten mit einemStoffsystem aus, wenn alle Anlagen vermessen wurden. Peakverläufe diesesStoffsystems charakterisieren dann die Anlage bzw. deren unterschiedlicheKomponenten für diese Gruppe. Informationen über den jeweiligen phasenspezifischenAnteil ergeben sich automatisch aus Laborexperimenten zur Methodenfindung. DieKombination ermöglicht dann eine Auslegung der präparativen Trennung.
5.2.4 Auslegung von HPLC-Systemen
In der Auslegung von präparativen Trennungen mit Hilfe der HPLC wird eineModellierung möglichen Experimenten vorgezogen, wenn sich durch ihren Einsatz undihre Genauigkeit der Beschreibung der Trennung eine Verringerung der eingesetztenResourcen (Substanzen, Arbeitszeit) zur Auslegung des Prozesses ergibt.

5 Bestimmung der Anteile der axialen Dispersion 83
Aus den Ergebnissen der Charakterisierung ergibt sich die folgende Strategie zurAuslegung präparativer flüssigchromatographischer Trennungen:
1. In Laborexperimenten wird das Trennsystem bestimmt (Methodenentwicklung). Diefür eine Modellierung notwendigen Parameter für Porosität und Adsorptions-isothermen ergeben sich aus diesen Experimenten.
2. Durch Auswertung der Experimente (z.B. nach der Momentenmethode unterVerwendung von Gleichung (2.86)) ergeben sich Startwerte für die axialeDispersion. Durch Modellierung der axialen Komponentenbilanz erhält man dannreale Werte für die axiale Dispersion und den Stofftransport.
3. Unter Verwendung der Ergebnisse von Messungen mit einem Referenzsystem ergibtsich die Differenz der anlagenspezifischen Anteile an der axialen Dispersionzwischen analytischer und präparativer Anlage. Deren Berücksichtigung ergibt dieaxiale Dispersion des Trennsystems in der präparativen Anlage.
4. Abschliessend erfolgt eine Anpassung des Stofftransportkoeffizienten an erstepräparative Ergebnisse.
5. Die präparative Trennung kann mit Hilfe der Modellierung komplett ausgelegt undoptimiert werden.
Dieses Vorgehen war bereits Inhalt der Vorträge von Laiblin (2000), Boysen (2001b)und von Beste et al. (2001b).

84 Bestimmung der Anteile der axialen Dispersion 5

6 Messung der radialen Verteilungen 85
6 Messung der radialen Ver teilungen
Um die Ergebnisse der Simulationen und damit die Anwendbarkeit bzw. die Voll-ständigkeit der mathematischen Modelle und numerischen Lösungsmethoden beurteilenzu können, sind Experimente notwendig. Für die präparative HPLC sind besondersMessungen zur Bestimmung der Verteilungsgüte innerhalb der chromatographischenSäule wichtig. Im einfachsten Fall können diese Experimente als Pulsexperimente(Peakaufgabe und Messung des Konzentrationsverlaufes am Ende der Säule) zurValidierung der eindimensionalen axialen Lösung und zur Bestimmung einer anlagen-spezifischen Dispersion durchgeführt werden. Dieser Weg wird in Kapitel 0beschrieben.
Komplizierter ist die direkte Messung der radialen Verteilungen der Temperatur,Geschwindigkeiten oder Konzentrationen innerhalb des gepackten Festbettes. SolcheErgebnisse eignen sich allerdings besonders gut zur Validierung der Ergebnisse dernumerischen Modellierung in FLUENT, da hier auch innerhalb der Anlage auftretendeEffekte (Ungleichverteilungen, Temperaturgradienten zwischen Eluent und Wand, etc.)berücksichtigt werden. Im Folgenden werden die innerhalb dieser Arbeit eingesetztenMethoden zur Messung der Temperatur- und Geschwindigkeitsverteilungen innerhalbder chromatographischen Säulen beschrieben und ihre Ergebnisse dargestellt. In derLiteratur angeführte Methoden zur Messung der Konzentrationen in der stationärenPhase sind in Kapitel 2.5.2 aufgeführt, wurden aber nicht angewendet.
6.1 Temperaturprofile
6.1.1 Einfluss der Pt100
Der Einfluss der Temperaturfühler auf die Bettqualität wurde sowohl für die Prepbar2-Säule als auch für die NW100-Säule durch Vergleich der HETP der Säulen vor undnach dem Einbau bestimmt. Zur Charakterisierung wurden die Peaks einer 5 ml ToluolProbe in Methanol als Eluent auf die mit Uetikon gepackte Säule aufgegeben undausgewertet. Beide Lösungsmittel entsprechen der Qualität Lichrosolv (Fa. Merck). DerEinfluss der Pt100 auf die Peakverläufe ist für verschiedene Geschwindigkeiten in Bild6.1 dargestellt, die entsprechenden Änderungen der NTU sind in Tabelle 6.1 aufgeführt.Da die Bettlänge sich durch den Einbau nicht verändert hat (die Säule ist nicht axialkomprimiert) reicht eine Darstellung der NTU zur Bewertung der Bettqualität aus.
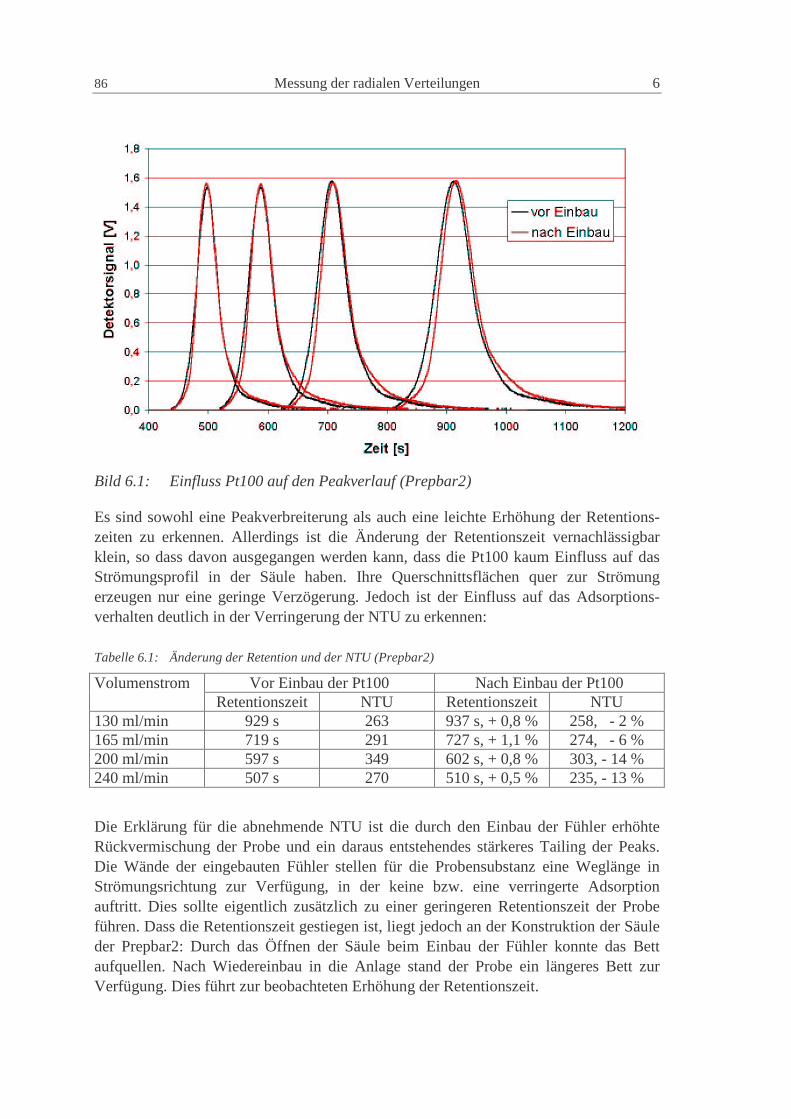
86 Messung der radialen Verteilungen 6
Bild 6.1: Einfluss Pt100 auf den Peakverlauf (Prepbar2)
Es sind sowohl eine Peakverbreiterung als auch eine leichte Erhöhung der Retentions-zeiten zu erkennen. Allerdings ist die Änderung der Retentionszeit vernachlässigbarklein, so dass davon ausgegangen werden kann, dass die Pt100 kaum Einfluss auf dasStrömungsprofil in der Säule haben. Ihre Querschnittsflächen quer zur Strömungerzeugen nur eine geringe Verzögerung. Jedoch ist der Einfluss auf das Adsorptions-verhalten deutlich in der Verringerung der NTU zu erkennen:
Tabelle 6.1: Änderung der Retention und der NTU (Prepbar2)
Vor Einbau der Pt100 Nach Einbau der Pt100VolumenstromRetentionszeit NTU Retentionszeit NTU
130 ml/min 929 s 263 937 s, + 0,8 % 258, - 2 %165 ml/min 719 s 291 727 s, + 1,1 % 274, - 6 %200 ml/min 597 s 349 602 s, + 0,8 % 303, - 14 %240 ml/min 507 s 270 510 s, + 0,5 % 235, - 13 %
Die Erklärung für die abnehmende NTU ist die durch den Einbau der Fühler erhöhteRückvermischung der Probe und ein daraus entstehendes stärkeres Tailing der Peaks.Die Wände der eingebauten Fühler stellen für die Probensubstanz eine Weglänge inStrömungsrichtung zur Verfügung, in der keine bzw. eine verringerte Adsorptionauftritt. Dies sollte eigentlich zusätzlich zu einer geringeren Retentionszeit der Probeführen. Dass die Retentionszeit gestiegen ist, liegt jedoch an der Konstruktion der Säuleder Prepbar2: Durch das Öffnen der Säule beim Einbau der Fühler konnte das Bettaufquellen. Nach Wiedereinbau in die Anlage stand der Probe ein längeres Bett zurVerfügung. Dies führt zur beobachteten Erhöhung der Retentionszeit.
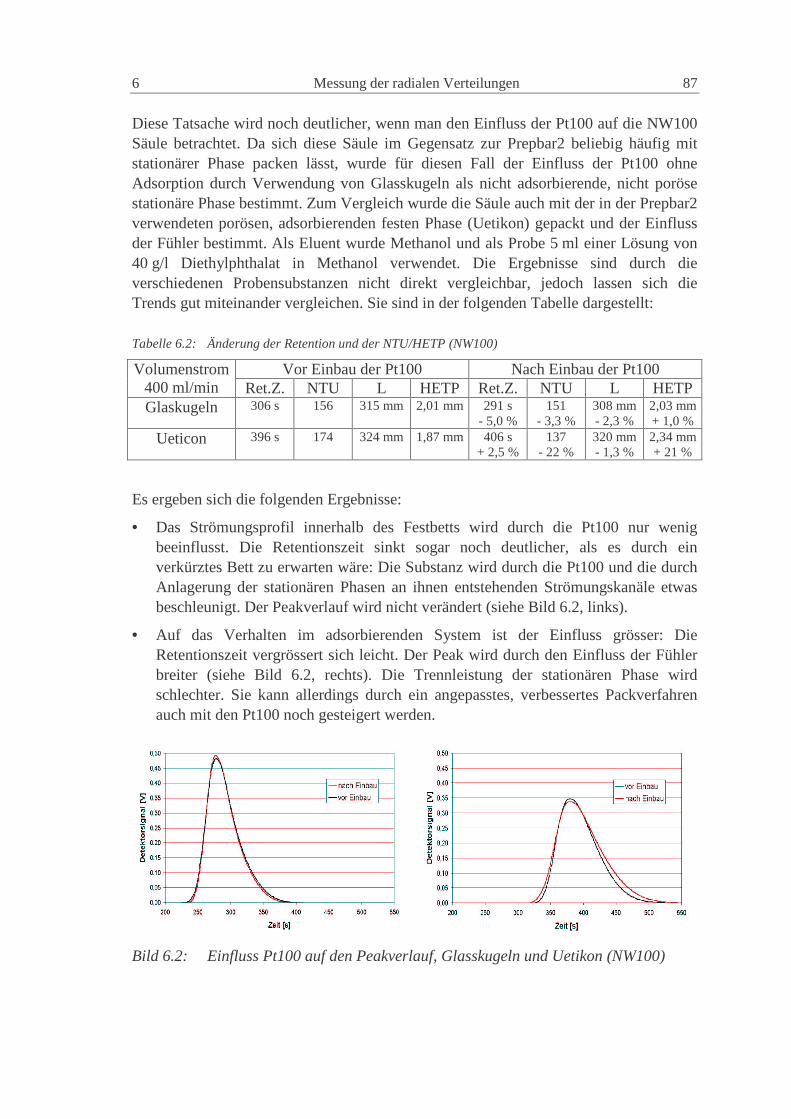
6 Messung der radialen Verteilungen 87
Diese Tatsache wird noch deutlicher, wenn man den Einfluss der Pt100 auf die NW100Säule betrachtet. Da sich diese Säule im Gegensatz zur Prepbar2 beliebig häufig mitstationärer Phase packen lässt, wurde für diesen Fall der Einfluss der Pt100 ohneAdsorption durch Verwendung von Glasskugeln als nicht adsorbierende, nicht porösestationäre Phase bestimmt. Zum Vergleich wurde die Säule auch mit der in der Prepbar2verwendeten porösen, adsorbierenden festen Phase (Uetikon) gepackt und der Einflussder Fühler bestimmt. Als Eluent wurde Methanol und als Probe 5 ml einer Lösung von40 g/l Diethylphthalat in Methanol verwendet. Die Ergebnisse sind durch dieverschiedenen Probensubstanzen nicht direkt vergleichbar, jedoch lassen sich dieTrends gut miteinander vergleichen. Sie sind in der folgenden Tabelle dargestellt:
Tabelle 6.2: Änderung der Retention und der NTU/HETP (NW100)
Vor Einbau der Pt100 Nach Einbau der Pt100Volumenstrom400 ml/min Ret.Z. NTU L HETP Ret.Z. NTU L HETPGlaskugeln 306 s 156 315 mm 2,01 mm 291 s
- 5,0 %151
- 3,3 %308 mm- 2,3 %
2,03 mm+ 1,0 %
Ueticon 396 s 174 324 mm 1,87 mm 406 s+ 2,5 %
137- 22 %
320 mm- 1,3 %
2,34 mm+ 21 %
Es ergeben sich die folgenden Ergebnisse:
• Das Strömungsprofil innerhalb des Festbetts wird durch die Pt100 nur wenigbeeinflusst. Die Retentionszeit sinkt sogar noch deutlicher, als es durch einverkürztes Bett zu erwarten wäre: Die Substanz wird durch die Pt100 und die durchAnlagerung der stationären Phasen an ihnen entstehenden Strömungskanäle etwasbeschleunigt. Der Peakverlauf wird nicht verändert (siehe Bild 6.2, links).
• Auf das Verhalten im adsorbierenden System ist der Einfluss grösser: DieRetentionszeit vergrössert sich leicht. Der Peak wird durch den Einfluss der Fühlerbreiter (siehe Bild 6.2, rechts). Die Trennleistung der stationären Phase wirdschlechter. Sie kann allerdings durch ein angepasstes, verbessertes Packverfahrenauch mit den Pt100 noch gesteigert werden.
Bild 6.2: Einfluss Pt100 auf den Peakverlauf, Glasskugeln und Uetikon (NW100)
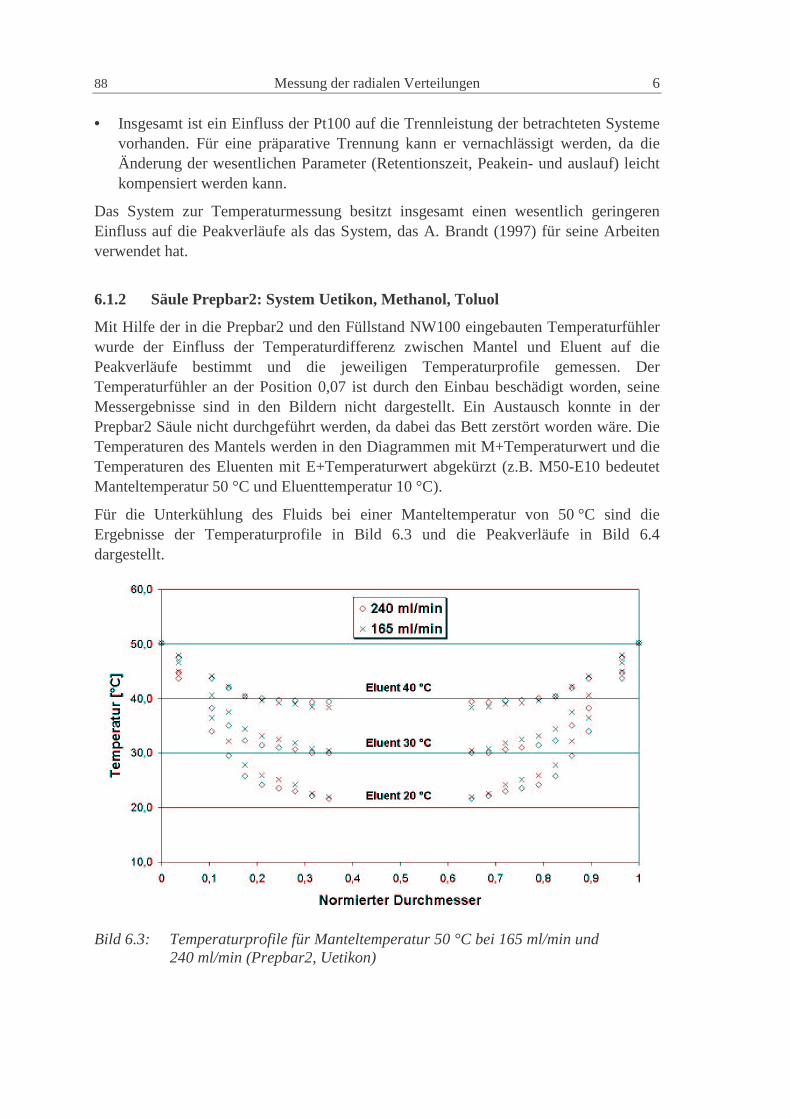
88 Messung der radialen Verteilungen 6
• Insgesamt ist ein Einfluss der Pt100 auf die Trennleistung der betrachteten Systemevorhanden. Für eine präparative Trennung kann er vernachlässigt werden, da dieÄnderung der wesentlichen Parameter (Retentionszeit, Peakein- und auslauf) leichtkompensiert werden kann.
Das System zur Temperaturmessung besitzt insgesamt einen wesentlich geringerenEinfluss auf die Peakverläufe als das System, das A. Brandt (1997) für seine Arbeitenverwendet hat.
6.1.2 Säule Prepbar2: System Uetikon, Methanol, Toluol
Mit Hilfe der in die Prepbar2 und den Füllstand NW100 eingebauten Temperaturfühlerwurde der Einfluss der Temperaturdifferenz zwischen Mantel und Eluent auf diePeakverläufe bestimmt und die jeweiligen Temperaturprofile gemessen. DerTemperaturfühler an der Position 0,07 ist durch den Einbau beschädigt worden, seineMessergebnisse sind in den Bildern nicht dargestellt. Ein Austausch konnte in derPrepbar2 Säule nicht durchgeführt werden, da dabei das Bett zerstört worden wäre. DieTemperaturen des Mantels werden in den Diagrammen mit M+Temperaturwert und dieTemperaturen des Eluenten mit E+Temperaturwert abgekürzt (z.B. M50-E10 bedeutetManteltemperatur 50 °C und Eluenttemperatur 10 °C).
Für die Unterkühlung des Fluids bei einer Manteltemperatur von 50 °C sind dieErgebnisse der Temperaturprofile in Bild 6.3 und die Peakverläufe in Bild 6.4dargestellt.
Bild 6.3: Temperaturprofile für Manteltemperatur 50 °C bei 165 ml/min und240 ml/min (Prepbar2, Uetikon)
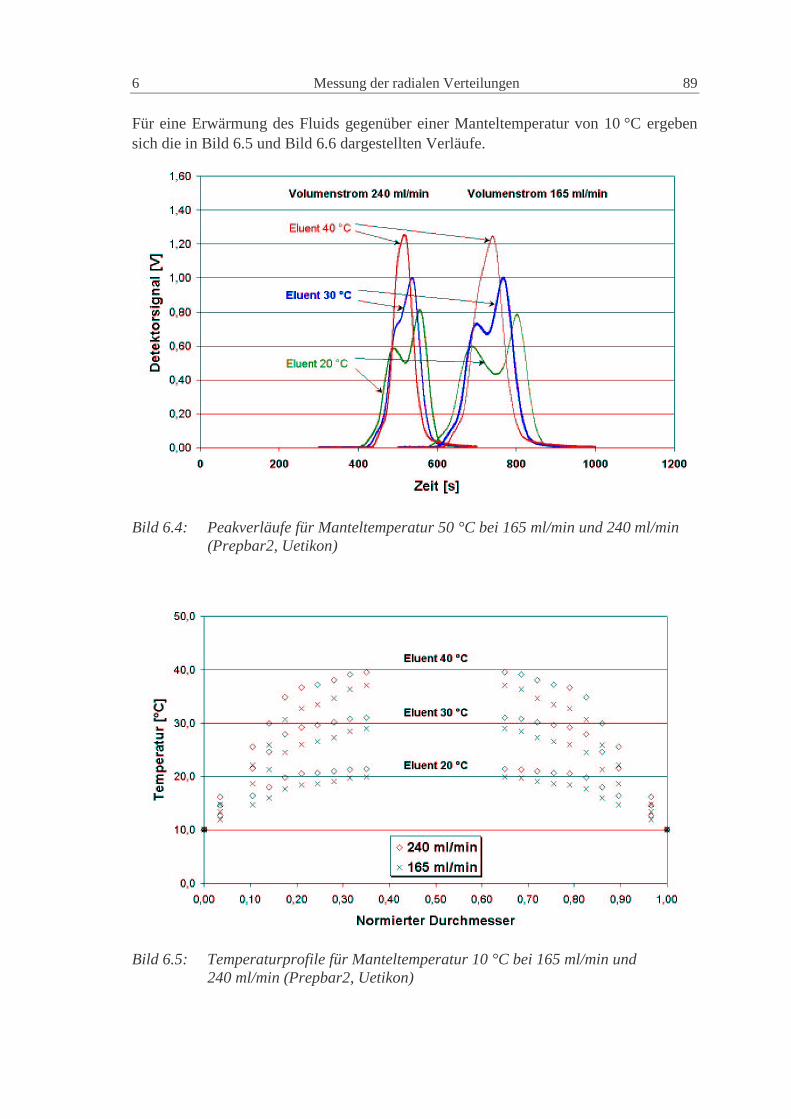
6 Messung der radialen Verteilungen 89
Für eine Erwärmung des Fluids gegenüber einer Manteltemperatur von 10 °C ergebensich die in Bild 6.5 und Bild 6.6 dargestellten Verläufe.
Bild 6.4: Peakverläufe für Manteltemperatur 50 °C bei 165 ml/min und 240 ml/min(Prepbar2, Uetikon)
Bild 6.5: Temperaturprofile für Manteltemperatur 10 °C bei 165 ml/min und240 ml/min (Prepbar2, Uetikon)
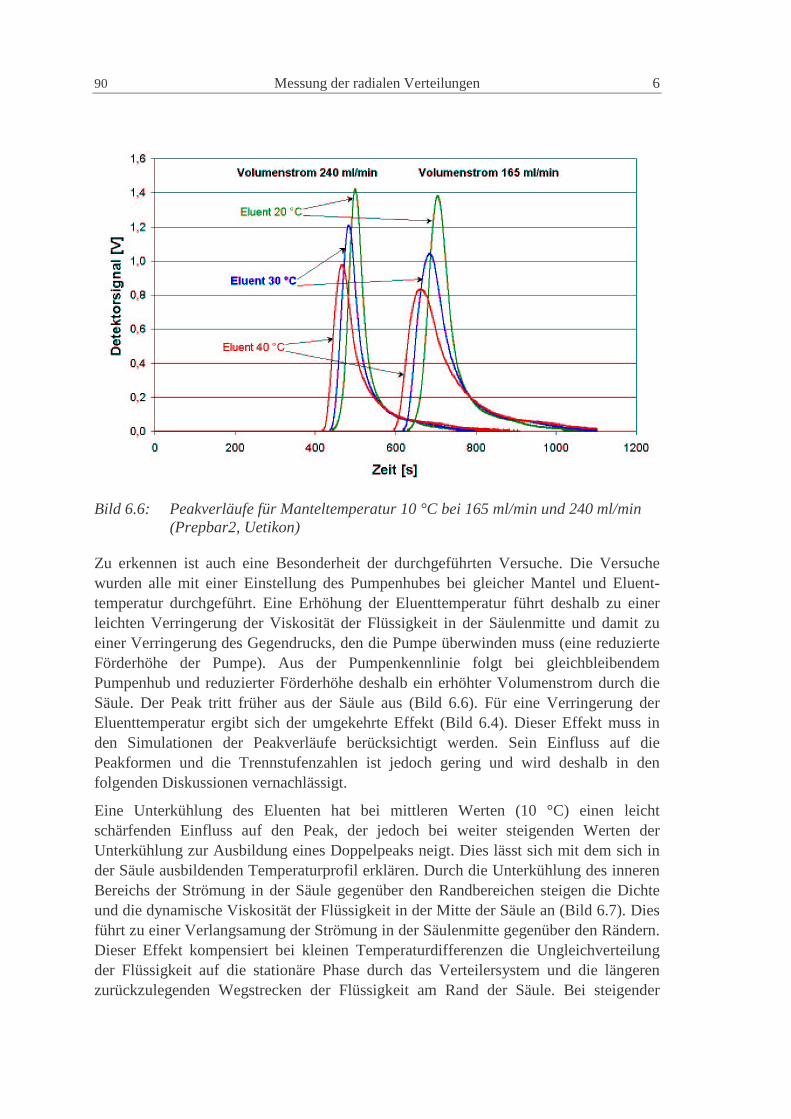
90 Messung der radialen Verteilungen 6
Bild 6.6: Peakverläufe für Manteltemperatur 10 °C bei 165 ml/min und 240 ml/min(Prepbar2, Uetikon)
Zu erkennen ist auch eine Besonderheit der durchgeführten Versuche. Die Versuchewurden alle mit einer Einstellung des Pumpenhubes bei gleicher Mantel und Eluent-temperatur durchgeführt. Eine Erhöhung der Eluenttemperatur führt deshalb zu einerleichten Verringerung der Viskosität der Flüssigkeit in der Säulenmitte und damit zueiner Verringerung des Gegendrucks, den die Pumpe überwinden muss (eine reduzierteFörderhöhe der Pumpe). Aus der Pumpenkennlinie folgt bei gleichbleibendemPumpenhub und reduzierter Förderhöhe deshalb ein erhöhter Volumenstrom durch dieSäule. Der Peak tritt früher aus der Säule aus (Bild 6.6). Für eine Verringerung derEluenttemperatur ergibt sich der umgekehrte Effekt (Bild 6.4). Dieser Effekt muss inden Simulationen der Peakverläufe berücksichtigt werden. Sein Einfluss auf diePeakformen und die Trennstufenzahlen ist jedoch gering und wird deshalb in denfolgenden Diskussionen vernachlässigt.
Eine Unterkühlung des Eluenten hat bei mittleren Werten (10 °C) einen leichtschärfenden Einfluss auf den Peak, der jedoch bei weiter steigenden Werten derUnterkühlung zur Ausbildung eines Doppelpeaks neigt. Dies lässt sich mit dem sich inder Säule ausbildenden Temperaturprofil erklären. Durch die Unterkühlung des innerenBereichs der Strömung in der Säule gegenüber den Randbereichen steigen die Dichteund die dynamische Viskosität der Flüssigkeit in der Mitte der Säule an (Bild 6.7). Diesführt zu einer Verlangsamung der Strömung in der Säulenmitte gegenüber den Rändern.Dieser Effekt kompensiert bei kleinen Temperaturdifferenzen die Ungleichverteilungder Flüssigkeit auf die stationäre Phase durch das Verteilersystem und die längerenzurückzulegenden Wegstrecken der Flüssigkeit am Rand der Säule. Bei steigender
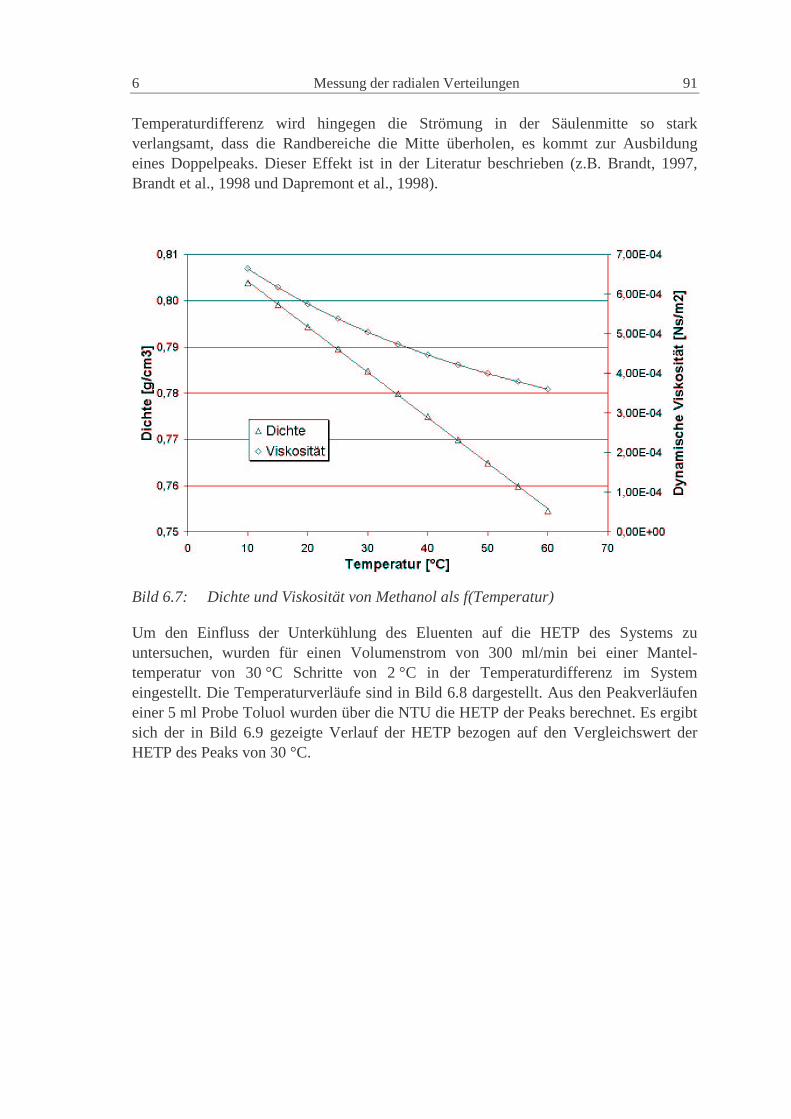
6 Messung der radialen Verteilungen 91
Temperaturdifferenz wird hingegen die Strömung in der Säulenmitte so starkverlangsamt, dass die Randbereiche die Mitte überholen, es kommt zur Ausbildungeines Doppelpeaks. Dieser Effekt ist in der Literatur beschrieben (z.B. Brandt, 1997,Brandt et al., 1998 und Dapremont et al., 1998).
Bild 6.7: Dichte und Viskosität von Methanol als f(Temperatur)
Um den Einfluss der Unterkühlung des Eluenten auf die HETP des Systems zuuntersuchen, wurden für einen Volumenstrom von 300 ml/min bei einer Mantel-temperatur von 30 °C Schritte von 2 °C in der Temperaturdifferenz im Systemeingestellt. Die Temperaturverläufe sind in Bild 6.8 dargestellt. Aus den Peakverläufeneiner 5 ml Probe Toluol wurden über die NTU die HETP der Peaks berechnet. Es ergibtsich der in Bild 6.9 gezeigte Verlauf der HETP bezogen auf den Vergleichswert derHETP des Peaks von 30 °C.
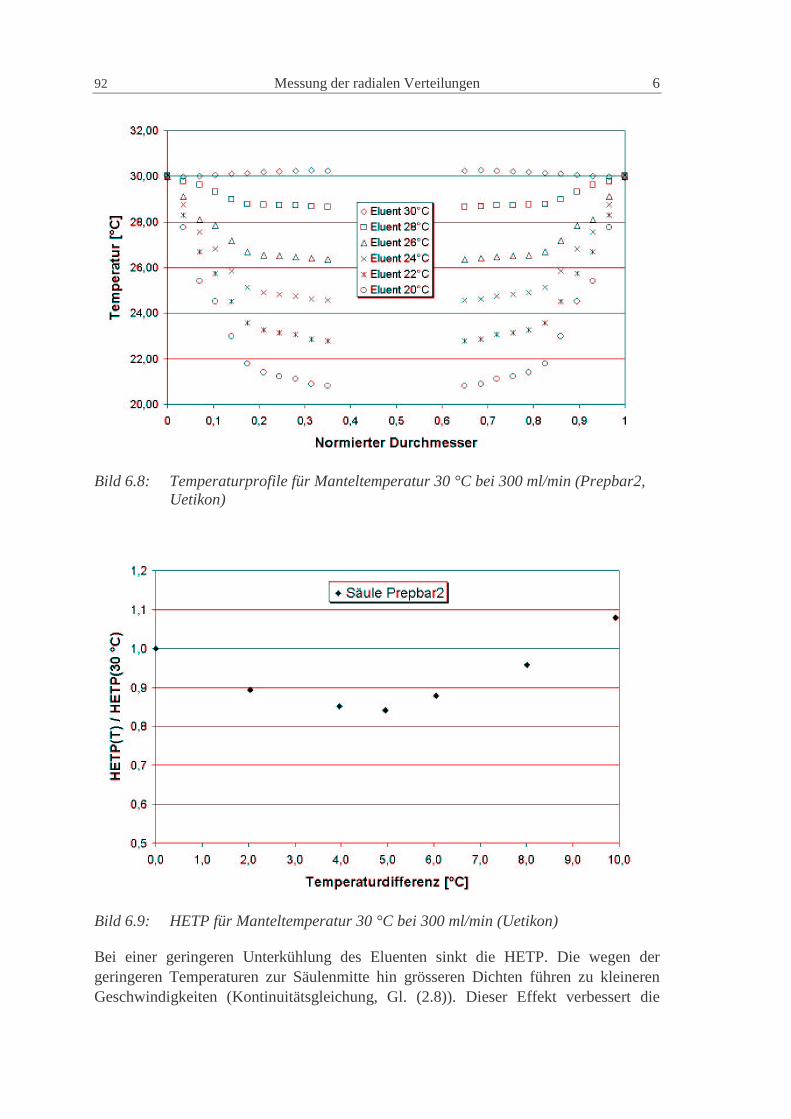
92 Messung der radialen Verteilungen 6
Bild 6.8: Temperaturprofile für Manteltemperatur 30 °C bei 300 ml/min (Prepbar2,Uetikon)
Bild 6.9: HETP für Manteltemperatur 30 °C bei 300 ml/min (Uetikon)
Bei einer geringeren Unterkühlung des Eluenten sinkt die HETP. Die wegen dergeringeren Temperaturen zur Säulenmitte hin grösseren Dichten führen zu kleinerenGeschwindigkeiten (Kontinuitätsgleichung, Gl. (2.8)). Dieser Effekt verbessert die
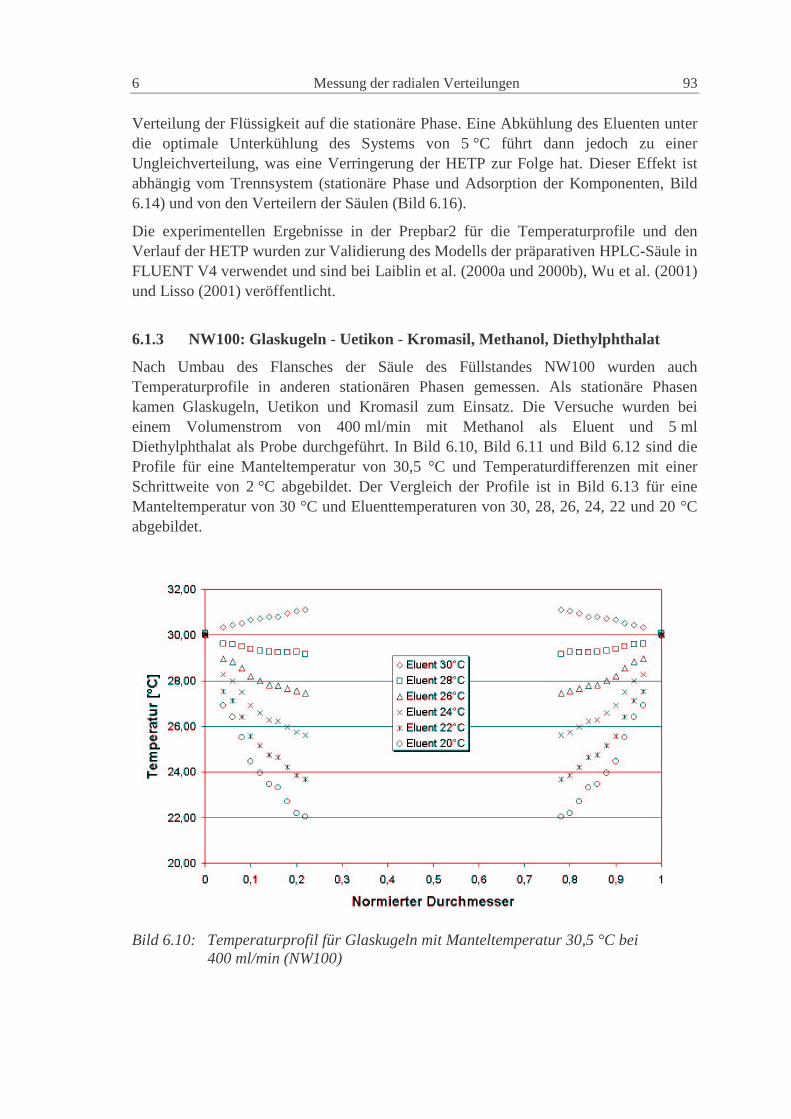
6 Messung der radialen Verteilungen 93
Verteilung der Flüssigkeit auf die stationäre Phase. Eine Abkühlung des Eluenten unterdie optimale Unterkühlung des Systems von 5 °C führt dann jedoch zu einerUngleichverteilung, was eine Verringerung der HETP zur Folge hat. Dieser Effekt istabhängig vom Trennsystem (stationäre Phase und Adsorption der Komponenten, Bild6.14) und von den Verteilern der Säulen (Bild 6.16).
Die experimentellen Ergebnisse in der Prepbar2 für die Temperaturprofile und denVerlauf der HETP wurden zur Validierung des Modells der präparativen HPLC-Säule inFLUENT V4 verwendet und sind bei Laiblin et al. (2000a und 2000b), Wu et al. (2001)und Lisso (2001) veröffentlicht.
6.1.3 NW100: Glaskugeln - Uetikon - Kromasil, Methanol, Diethylphthalat
Nach Umbau des Flansches der Säule des Füllstandes NW100 wurden auchTemperaturprofile in anderen stationären Phasen gemessen. Als stationäre Phasenkamen Glaskugeln, Uetikon und Kromasil zum Einsatz. Die Versuche wurden beieinem Volumenstrom von 400 ml/min mit Methanol als Eluent und 5 mlDiethylphthalat als Probe durchgeführt. In Bild 6.10, Bild 6.11 und Bild 6.12 sind dieProfile für eine Manteltemperatur von 30,5 °C und Temperaturdifferenzen mit einerSchrittweite von 2 °C abgebildet. Der Vergleich der Profile ist in Bild 6.13 für eineManteltemperatur von 30 °C und Eluenttemperaturen von 30, 28, 26, 24, 22 und 20 °Cabgebildet.
Bild 6.10: Temperaturprofil für Glaskugeln mit Manteltemperatur 30,5 °C bei400 ml/min (NW100)
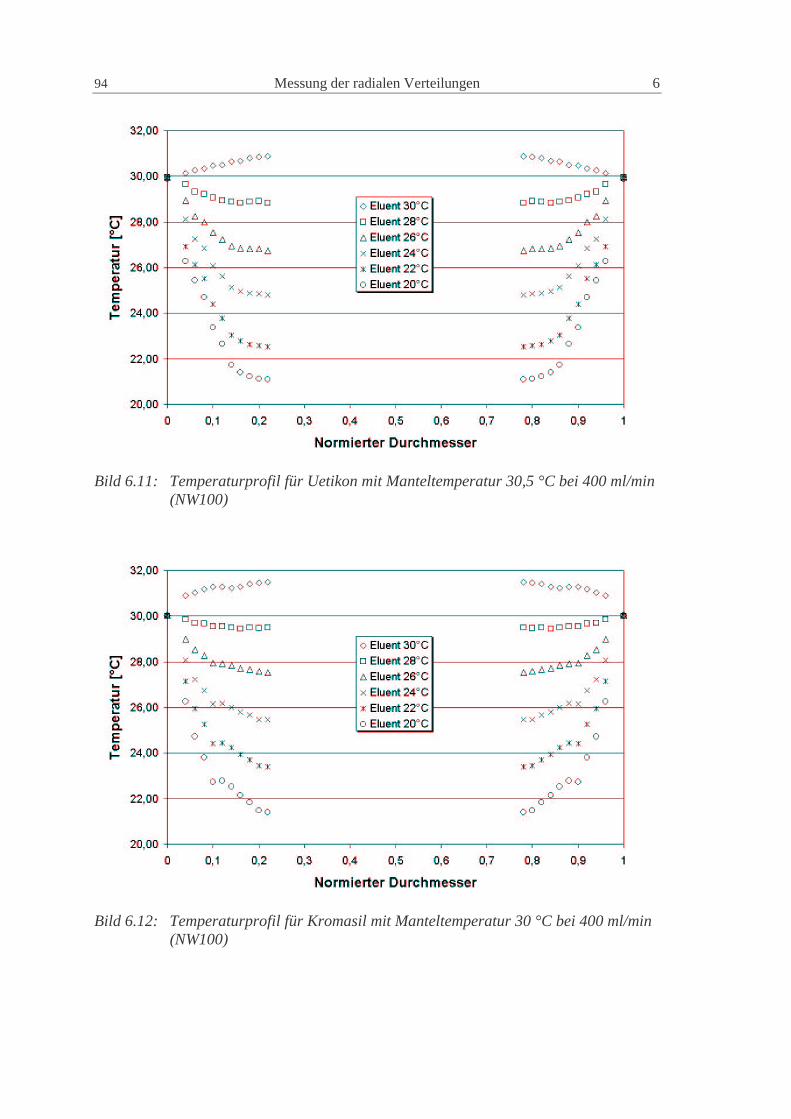
94 Messung der radialen Verteilungen 6
Bild 6.11: Temperaturprofil für Uetikon mit Manteltemperatur 30,5 °C bei 400 ml/min(NW100)
Bild 6.12: Temperaturprofil für Kromasil mit Manteltemperatur 30 °C bei 400 ml/min(NW100)
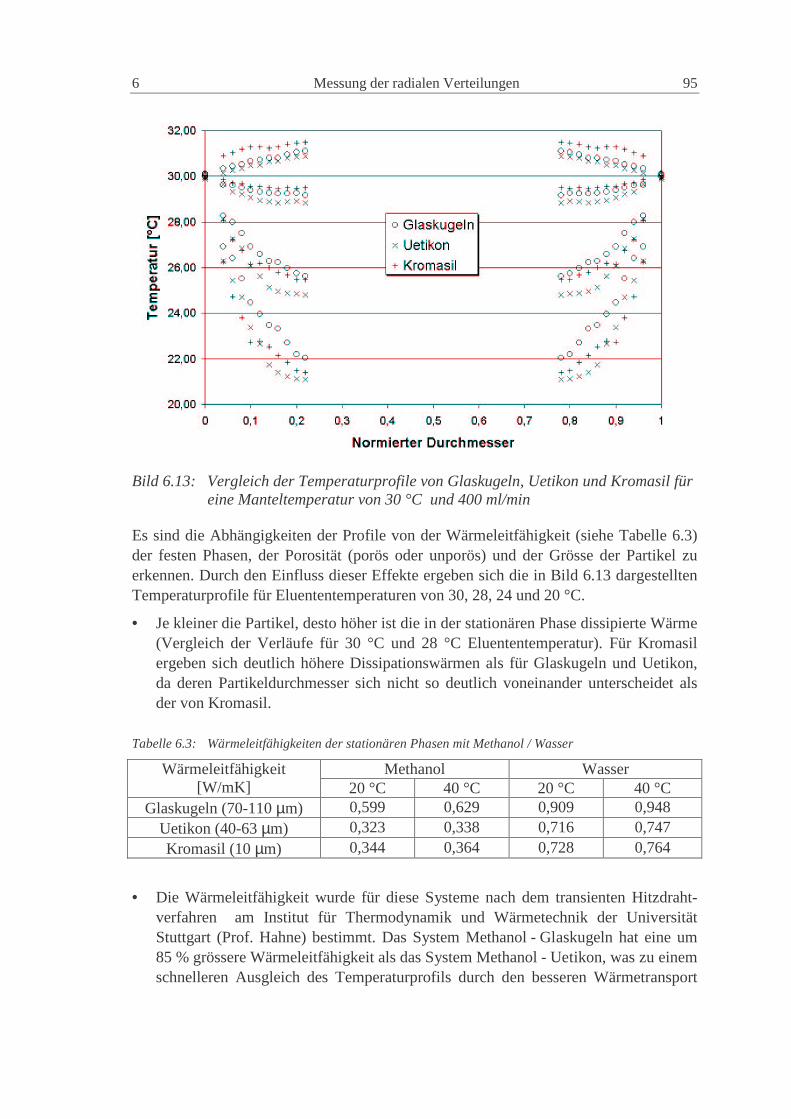
6 Messung der radialen Verteilungen 95
Bild 6.13: Vergleich der Temperaturprofile von Glaskugeln, Uetikon und Kromasil füreine Manteltemperatur von 30 °C und 400 ml/min
Es sind die Abhängigkeiten der Profile von der Wärmeleitfähigkeit (siehe Tabelle 6.3)der festen Phasen, der Porosität (porös oder unporös) und der Grösse der Partikel zuerkennen. Durch den Einfluss dieser Effekte ergeben sich die in Bild 6.13 dargestelltenTemperaturprofile für Eluententemperaturen von 30, 28, 24 und 20 °C.
• Je kleiner die Partikel, desto höher ist die in der stationären Phase dissipierte Wärme(Vergleich der Verläufe für 30 °C und 28 °C Eluententemperatur). Für Kromasilergeben sich deutlich höhere Dissipationswärmen als für Glaskugeln und Uetikon,da deren Partikeldurchmesser sich nicht so deutlich voneinander unterscheidet alsder von Kromasil.
Tabelle 6.3: Wärmeleitfähigkeiten der stationären Phasen mit Methanol / Wasser
Methanol WasserWärmeleitfähigkeit[W/mK] 20 °C 40 °C 20 °C 40 °C
Glaskugeln (70-110 µm) 0,599 0,629 0,909 0,948Uetikon (40-63 µm) 0,323 0,338 0,716 0,747Kromasil (10 µm) 0,344 0,364 0,728 0,764
• Die Wärmeleitfähigkeit wurde für diese Systeme nach dem transienten Hitzdraht-verfahren am Institut für Thermodynamik und Wärmetechnik der UniversitätStuttgart (Prof. Hahne) bestimmt. Das System Methanol - Glaskugeln hat eine um85 % grössere Wärmeleitfähigkeit als das System Methanol - Uetikon, was zu einemschnelleren Ausgleich des Temperaturprofils durch den besseren Wärmetransport
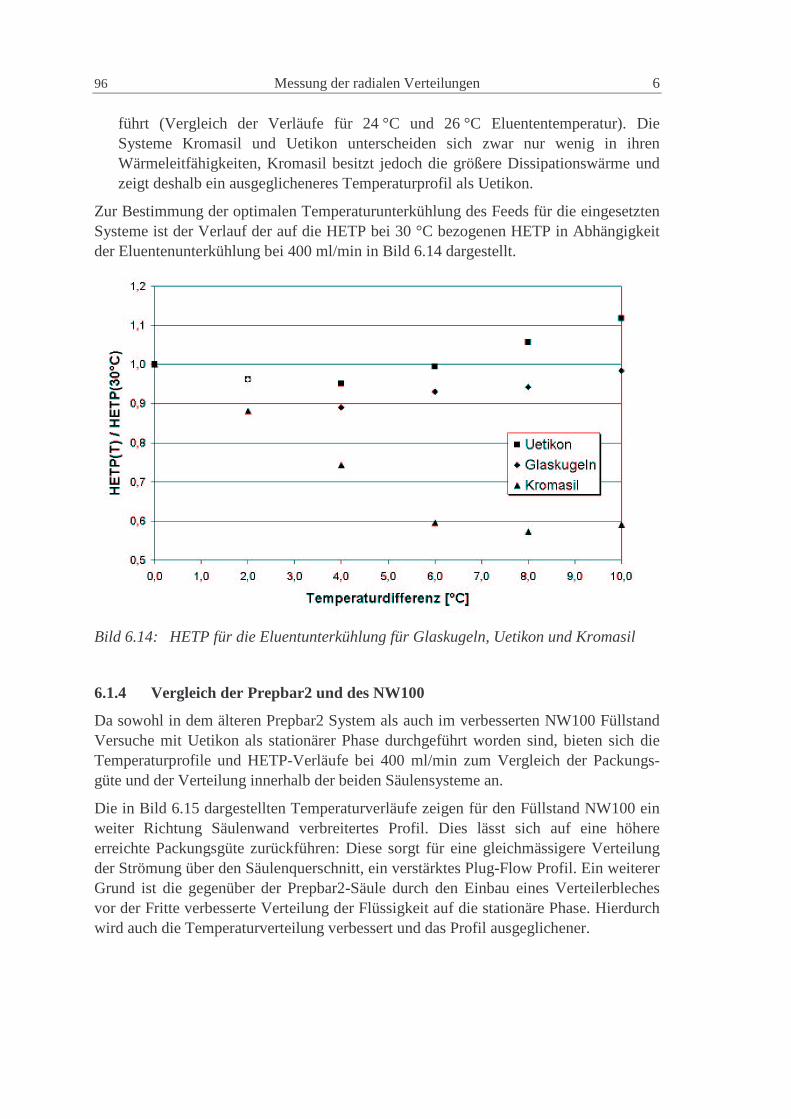
96 Messung der radialen Verteilungen 6
führt (Vergleich der Verläufe für 24 °C und 26 °C Eluententemperatur). DieSysteme Kromasil und Uetikon unterscheiden sich zwar nur wenig in ihrenWärmeleitfähigkeiten, Kromasil besitzt jedoch die größere Dissipationswärme undzeigt deshalb ein ausgeglicheneres Temperaturprofil als Uetikon.
Zur Bestimmung der optimalen Temperaturunterkühlung des Feeds für die eingesetztenSysteme ist der Verlauf der auf die HETP bei 30 °C bezogenen HETP in Abhängigkeitder Eluentenunterkühlung bei 400 ml/min in Bild 6.14 dargestellt.
Bild 6.14: HETP für die Eluentunterkühlung für Glaskugeln, Uetikon und Kromasil
6.1.4 Vergleich der Prepbar2 und des NW100
Da sowohl in dem älteren Prepbar2 System als auch im verbesserten NW100 FüllstandVersuche mit Uetikon als stationärer Phase durchgeführt worden sind, bieten sich dieTemperaturprofile und HETP-Verläufe bei 400 ml/min zum Vergleich der Packungs-güte und der Verteilung innerhalb der beiden Säulensysteme an.
Die in Bild 6.15 dargestellten Temperaturverläufe zeigen für den Füllstand NW100 einweiter Richtung Säulenwand verbreitertes Profil. Dies lässt sich auf eine höhereerreichte Packungsgüte zurückführen: Diese sorgt für eine gleichmässigere Verteilungder Strömung über den Säulenquerschnitt, ein verstärktes Plug-Flow Profil. Ein weitererGrund ist die gegenüber der Prepbar2-Säule durch den Einbau eines Verteilerblechesvor der Fritte verbesserte Verteilung der Flüssigkeit auf die stationäre Phase. Hierdurchwird auch die Temperaturverteilung verbessert und das Profil ausgeglichener.
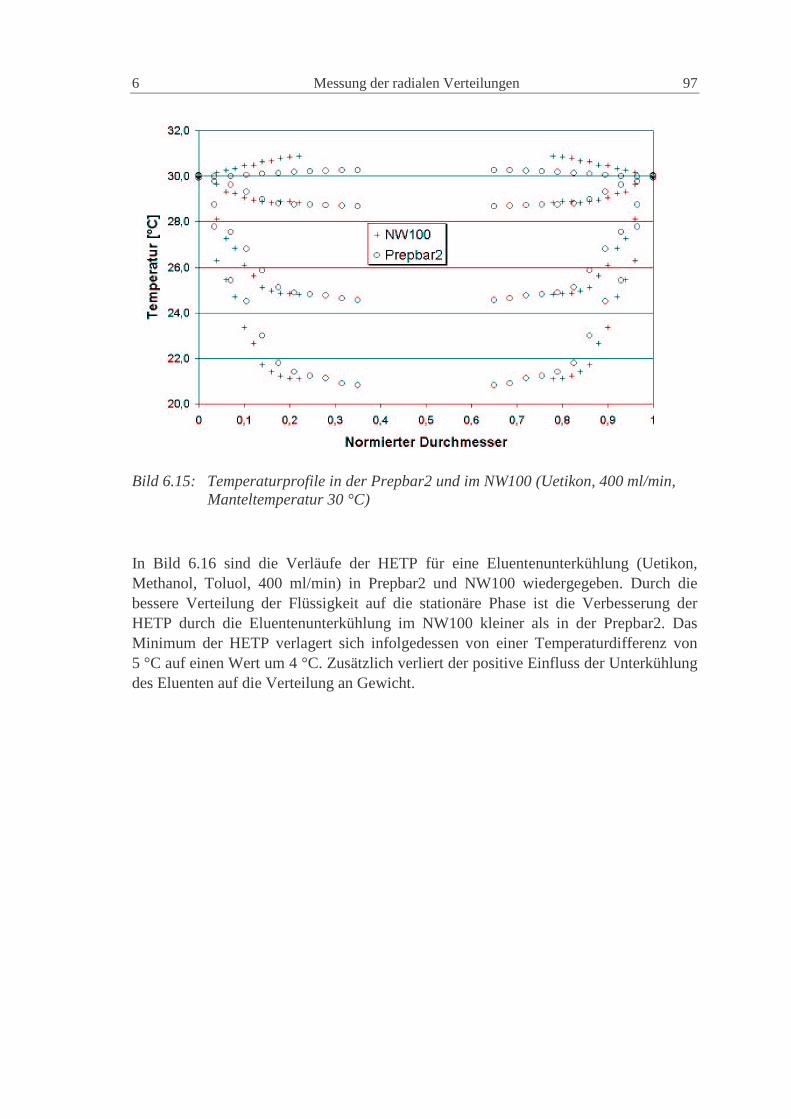
6 Messung der radialen Verteilungen 97
Bild 6.15: Temperaturprofile in der Prepbar2 und im NW100 (Uetikon, 400 ml/min,Manteltemperatur 30 °C)
In Bild 6.16 sind die Verläufe der HETP für eine Eluentenunterkühlung (Uetikon,Methanol, Toluol, 400 ml/min) in Prepbar2 und NW100 wiedergegeben. Durch diebessere Verteilung der Flüssigkeit auf die stationäre Phase ist die Verbesserung derHETP durch die Eluentenunterkühlung im NW100 kleiner als in der Prepbar2. DasMinimum der HETP verlagert sich infolgedessen von einer Temperaturdifferenz von5 °C auf einen Wert um 4 °C. Zusätzlich verliert der positive Einfluss der Unterkühlungdes Eluenten auf die Verteilung an Gewicht.
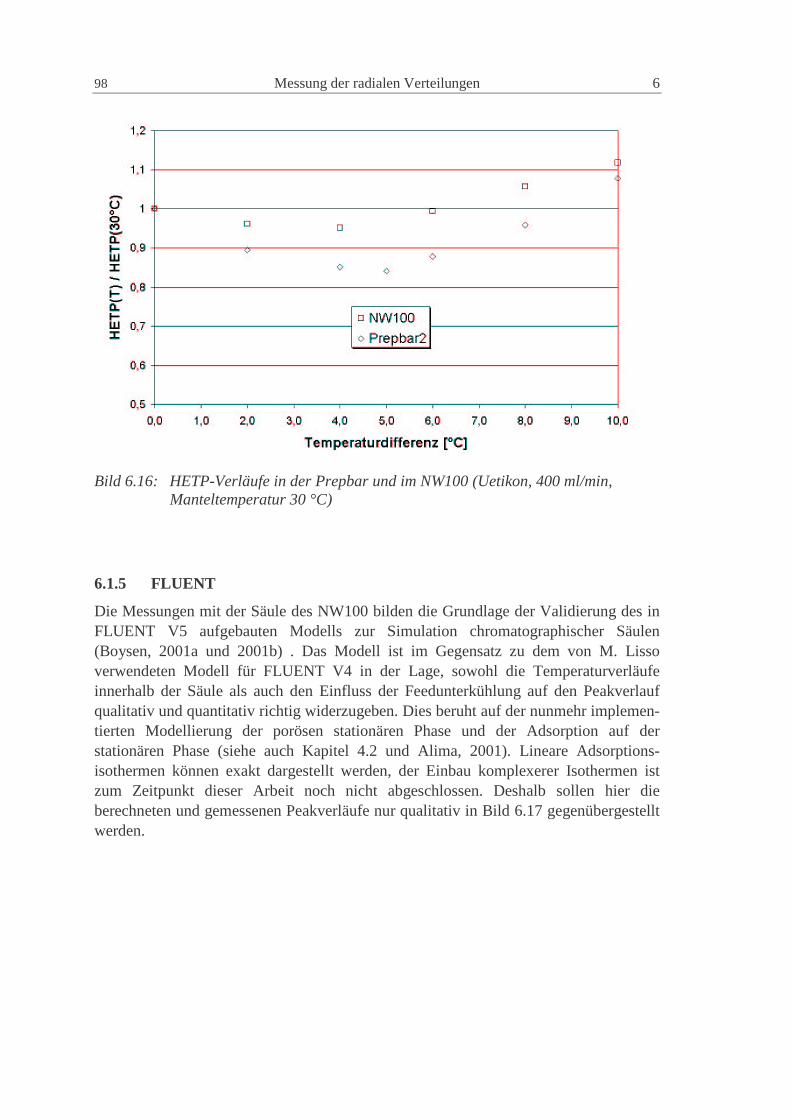
98 Messung der radialen Verteilungen 6
Bild 6.16: HETP-Verläufe in der Prepbar und im NW100 (Uetikon, 400 ml/min,Manteltemperatur 30 °C)
6.1.5 FLUENT
Die Messungen mit der Säule des NW100 bilden die Grundlage der Validierung des inFLUENT V5 aufgebauten Modells zur Simulation chromatographischer Säulen(Boysen, 2001a und 2001b) . Das Modell ist im Gegensatz zu dem von M. Lissoverwendeten Modell für FLUENT V4 in der Lage, sowohl die Temperaturverläufeinnerhalb der Säule als auch den Einfluss der Feedunterkühlung auf den Peakverlaufqualitativ und quantitativ richtig widerzugeben. Dies beruht auf der nunmehr implemen-tierten Modellierung der porösen stationären Phase und der Adsorption auf derstationären Phase (siehe auch Kapitel 4.2 und Alima, 2001). Lineare Adsorptions-isothermen können exakt dargestellt werden, der Einbau komplexerer Isothermen istzum Zeitpunkt dieser Arbeit noch nicht abgeschlossen. Deshalb sollen hier dieberechneten und gemessenen Peakverläufe nur qualitativ in Bild 6.17 gegenübergestelltwerden.
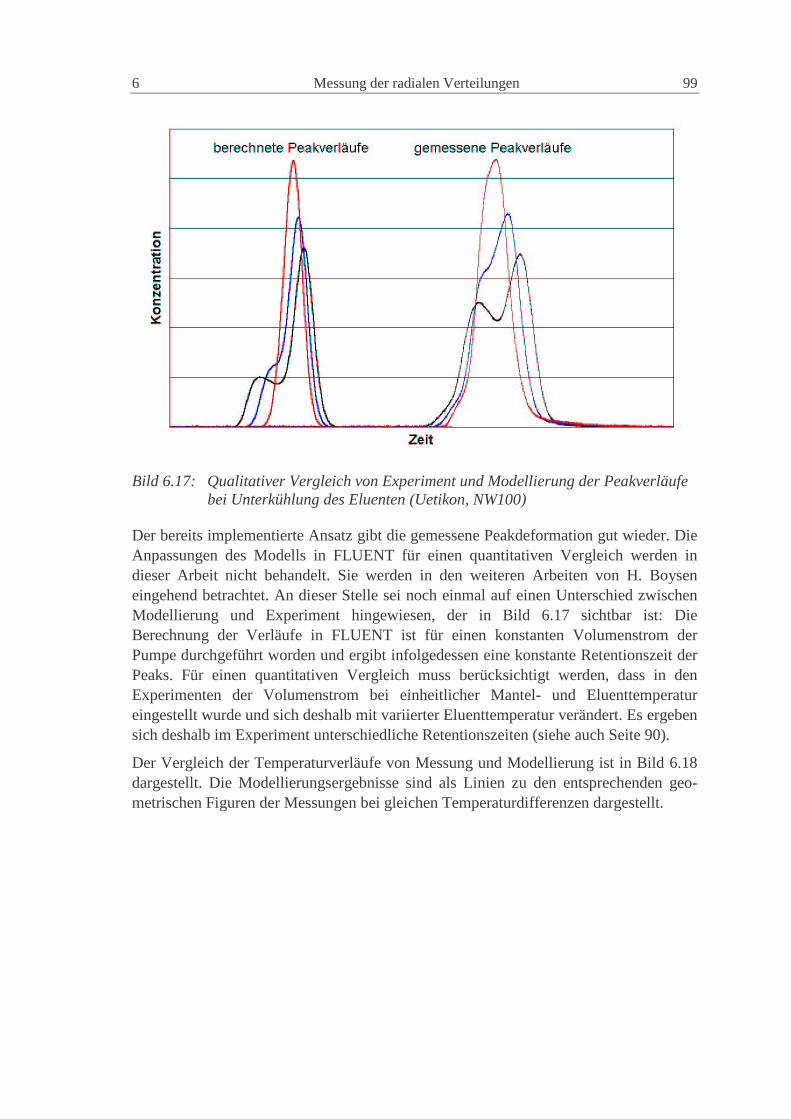
6 Messung der radialen Verteilungen 99
Bild 6.17: Qualitativer Vergleich von Experiment und Modellierung der Peakverläufebei Unterkühlung des Eluenten (Uetikon, NW100)
Der bereits implementierte Ansatz gibt die gemessene Peakdeformation gut wieder. DieAnpassungen des Modells in FLUENT für einen quantitativen Vergleich werden indieser Arbeit nicht behandelt. Sie werden in den weiteren Arbeiten von H. Boyseneingehend betrachtet. An dieser Stelle sei noch einmal auf einen Unterschied zwischenModellierung und Experiment hingewiesen, der in Bild 6.17 sichtbar ist: DieBerechnung der Verläufe in FLUENT ist für einen konstanten Volumenstrom derPumpe durchgeführt worden und ergibt infolgedessen eine konstante Retentionszeit derPeaks. Für einen quantitativen Vergleich muss berücksichtigt werden, dass in denExperimenten der Volumenstrom bei einheitlicher Mantel- und Eluenttemperatureingestellt wurde und sich deshalb mit variierter Eluenttemperatur verändert. Es ergebensich deshalb im Experiment unterschiedliche Retentionszeiten (siehe auch Seite 90).
Der Vergleich der Temperaturverläufe von Messung und Modellierung ist in Bild 6.18dargestellt. Die Modellierungsergebnisse sind als Linien zu den entsprechenden geo-metrischen Figuren der Messungen bei gleichen Temperaturdifferenzen dargestellt.
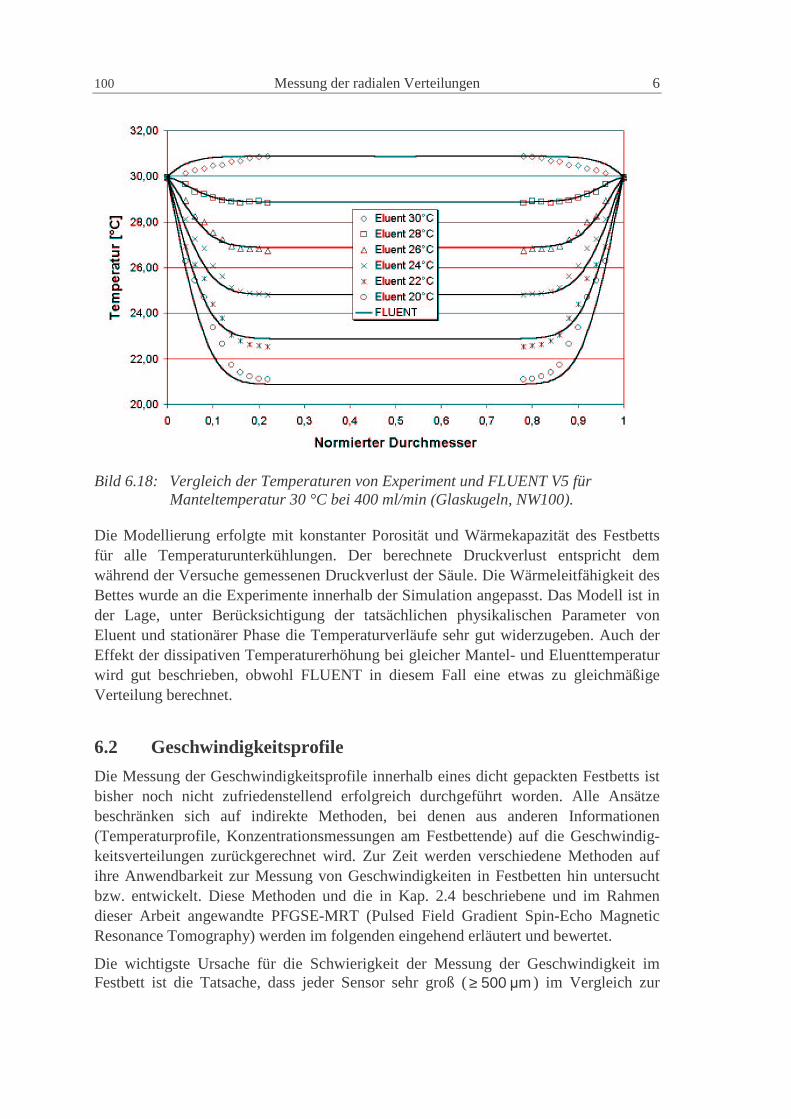
100 Messung der radialen Verteilungen 6
Bild 6.18: Vergleich der Temperaturen von Experiment und FLUENT V5 fürManteltemperatur 30 °C bei 400 ml/min (Glaskugeln, NW100).
Die Modellierung erfolgte mit konstanter Porosität und Wärmekapazität des Festbettsfür alle Temperaturunterkühlungen. Der berechnete Druckverlust entspricht demwährend der Versuche gemessenen Druckverlust der Säule. Die Wärmeleitfähigkeit desBettes wurde an die Experimente innerhalb der Simulation angepasst. Das Modell ist inder Lage, unter Berücksichtigung der tatsächlichen physikalischen Parameter vonEluent und stationärer Phase die Temperaturverläufe sehr gut widerzugeben. Auch derEffekt der dissipativen Temperaturerhöhung bei gleicher Mantel- und Eluenttemperaturwird gut beschrieben, obwohl FLUENT in diesem Fall eine etwas zu gleichmäßigeVerteilung berechnet.
6.2 Geschwindigkeitsprofile
Die Messung der Geschwindigkeitsprofile innerhalb eines dicht gepackten Festbetts istbisher noch nicht zufriedenstellend erfolgreich durchgeführt worden. Alle Ansätzebeschränken sich auf indirekte Methoden, bei denen aus anderen Informationen(Temperaturprofile, Konzentrationsmessungen am Festbettende) auf die Geschwindig-keitsverteilungen zurückgerechnet wird. Zur Zeit werden verschiedene Methoden aufihre Anwendbarkeit zur Messung von Geschwindigkeiten in Festbetten hin untersuchtbzw. entwickelt. Diese Methoden und die in Kap. 2.4 beschriebene und im Rahmendieser Arbeit angewandte PFGSE-MRT (Pulsed Field Gradient Spin-Echo MagneticResonance Tomography) werden im folgenden eingehend erläutert und bewertet.
Die wichtigste Ursache für die Schwierigkeit der Messung der Geschwindigkeit imFestbett ist die Tatsache, dass jeder Sensor sehr groß ( �m500≥ ) im Vergleich zur

6 Messung der radialen Verteilungen 101
Teilchengröße ( �m10010 ↔ ) ist und damit ein starke Störung der Geschwindigkeit
darstellt. Durch die dichte Packung der Partikel verändert der Sensor das eigentlich zumessende Profil so stark, dass die Messwerte letztendlich nichts über das Profilaussagen können. Erfolgversprechend sind deshalb zur Zeit nur nicht invasiveMethoden, wie z.B. die Magnet-Resonanz-Tomographie oder die von Broyles et al.(1998) angewandte Methode (siehe Kapitel 2.5.2) zur Messung der Konzentrations-profile.
Durch den Einsatz der MRT ergeben sich allerdings weitere Probleme, die bis zurEtablierung eines funktionsfähigen Meßaufbaus gelöst werden müssen:
• Die Meßergebnisse müssen komplex ausgewertet werden. Das Meßgerät (derMagnetom) ist teuer und die Auswertungssoftware muss von der Vermessungmenschlicher Organe an die Vermessung von durchströmten Festbetten angepasstwerden.
• Hieraus ergibt sich direkt das nächste Problem, da die Auflösung der Messung hochgenug sein muss, um die sich ständig ändernden Vektoren der lokalen Geschwindig-keiten zu erfassen. Dies ist insbesondere wichtig, da die lokale Geschwindigkeit undnicht nur die "mittlere" Geschwindigkeit über mehrere Partikelradien von Interesseist.
• Alle eingesetzten Materialien müssen metallfrei sein, damit sie nicht mit demstarken Dauermagnetfeld der MRT (üblicherweise 1 T) wechselwirken. Dasschränkt die Einsatzgebiete auf Glas- oder Polymerwerkstoffe für Säulen, Leitungenund Verbinder ein, was zu einer reduzierten Druckbelastbarkeit insbesondere derSäulen führt.
• Die eingesetzten chromatographischen Systeme müssen Wasser oder eine wässrigeLösung als Eluent verwenden, da nur die Wasserstoffatome stimuliert werden.Verbesserungen können durch Zugabe von z.B. Gadolinium-Ionen erzielt werden(Tallarek et al. 1995).
6.2.1 Exper imentelle Ergebnisse
Für die Experimente wurden die in Kapitel 3.2.5 beschriebenen Säulen in einer CP-Flexspule auf dem Behandlungstisch des Tomographen von Dr. R. Kaiser zentriert. Alsstationäre Phase erwies sich nur das Uetikon Material als geeignet, da sich für dieunporösen Glaskugeln das Gesamtsignal an Wasser in der Säule als zu gering erwiesenhat.
Ein Bild der Strömung in der Säulenebene ist sowohl für die 10 mm als auch für die26 mm Säule in Bild 6.19 dargestellt. Für die ersten Messungen zur Anwendbarkeit derMethode wurde die Strömung in der 26 mm Säule vermessen. Bei einem Volumenstromvon 200 ml/min wurde mit der vaskularen Messung ein Bild der Schnittebene der Säule,durch die das Wasser strömt, aufgenommen (Bild 6.20). Die zwei zur Erhöhung desMesssignals eingesetzten Wasserphantome (1 l Container) sind als helle Flächen obenim Bild erkennbar. Zur Auswertung wird die Flussquantifizierung verwendet. Es
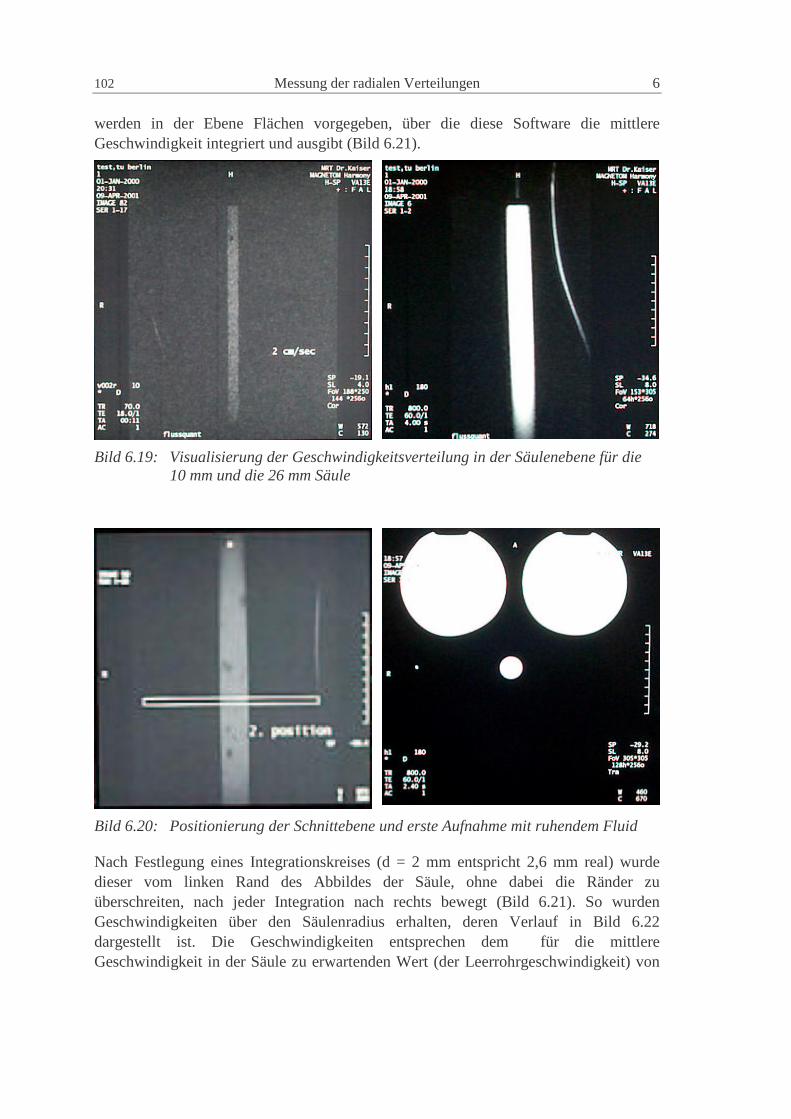
102 Messung der radialen Verteilungen 6
werden in der Ebene Flächen vorgegeben, über die diese Software die mittlereGeschwindigkeit integriert und ausgibt (Bild 6.21).
Bild 6.19: Visualisierung der Geschwindigkeitsverteilung in der Säulenebene für die10 mm und die 26 mm Säule
Bild 6.20: Positionierung der Schnittebene und erste Aufnahme mit ruhendem Fluid
Nach Festlegung eines Integrationskreises (d = 2 mm entspricht 2,6 mm real) wurdedieser vom linken Rand des Abbildes der Säule, ohne dabei die Ränder zuüberschreiten, nach jeder Integration nach rechts bewegt (Bild 6.21). So wurdenGeschwindigkeiten über den Säulenradius erhalten, deren Verlauf in Bild 6.22dargestellt ist. Die Geschwindigkeiten entsprechen dem für die mittlereGeschwindigkeit in der Säule zu erwartenden Wert (der Leerrohrgeschwindigkeit) von
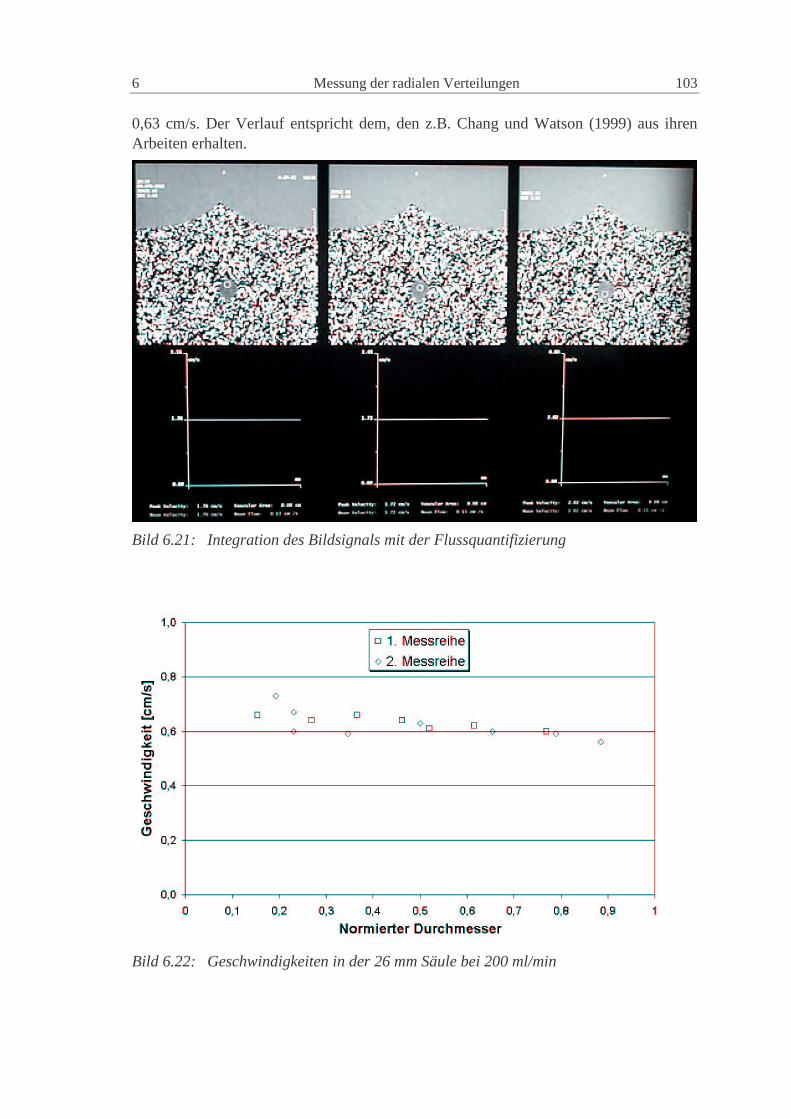
6 Messung der radialen Verteilungen 103
0,63 cm/s. Der Verlauf entspricht dem, den z.B. Chang und Watson (1999) aus ihrenArbeiten erhalten.
Bild 6.21: Integration des Bildsignals mit der Flussquantifizierung
Bild 6.22: Geschwindigkeiten in der 26 mm Säule bei 200 ml/min

104 Messung der radialen Verteilungen 6
Aufbauend auf diesen ersten, guten Ergebnissen wurden weitere Messungen durch-geführt. In diesen sollten unterschiedliche Volumenströme, Säulendurchmesser, dieReproduzierbarkeit der Messungen und die Genauigkeit und Auflösung derAuswertungssoftware bestimmt werden.
Bild 6.23: Messung in der 26 mm Säule mit Referenz
Für diese Messungen wurde eine mit Wasser gefüllte Injektionsspritze als Referenz derruhenden Flüssigkeit zusammen mit der Säule in die Messspule eingesetzt: In Bild 6.23ist die Säule als hellgrauer Kreis, die Referenz als dunkler Kreis zu erkennen. Überdiese Referenz kann die Auswertungssoftware das Hintergrundrauschen der Messungbei der Auswertung der Geschwindigkeiten reduzieren, da sie das Messsignal derruhenden Flüssigkeit in der Referenz zur Normierung verwenden kann.
Für diesen Aufbau wurden dann die Geschwindigkeiten in der Säulen fürVolumenströme von 100, 150, 200 und 250 ml/min gemessen. Das Ergebnis ist in Bild6.24 dargestellt. Zum Vergleich sind in Tabelle 6.4 die zu erwartenden mittlerenGeschwindigkeiten in der Säule angegeben.
Tabelle 6.4: Volumenströme und jeweilige mittlere Geschwindigkeiten in der 26 mm Säule
Volumenstrom [ml/min] 100 150 200 250mittl. Geschwindigkeit [cm/s] 0,31 0,47 0,63 0,78
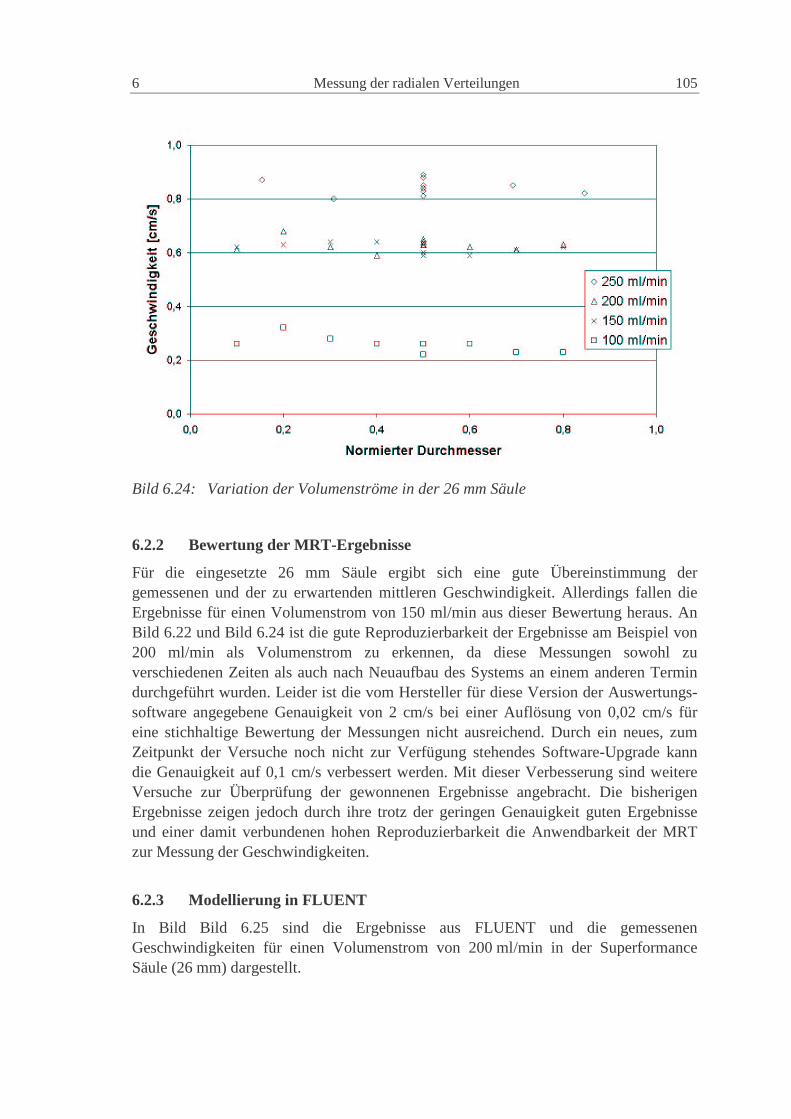
6 Messung der radialen Verteilungen 105
Bild 6.24: Variation der Volumenströme in der 26 mm Säule
6.2.2 Bewertung der MRT-Ergebnisse
Für die eingesetzte 26 mm Säule ergibt sich eine gute Übereinstimmung dergemessenen und der zu erwartenden mittleren Geschwindigkeit. Allerdings fallen dieErgebnisse für einen Volumenstrom von 150 ml/min aus dieser Bewertung heraus. AnBild 6.22 und Bild 6.24 ist die gute Reproduzierbarkeit der Ergebnisse am Beispiel von200 ml/min als Volumenstrom zu erkennen, da diese Messungen sowohl zuverschiedenen Zeiten als auch nach Neuaufbau des Systems an einem anderen Termindurchgeführt wurden. Leider ist die vom Hersteller für diese Version der Auswertungs-software angegebene Genauigkeit von 2 cm/s bei einer Auflösung von 0,02 cm/s füreine stichhaltige Bewertung der Messungen nicht ausreichend. Durch ein neues, zumZeitpunkt der Versuche noch nicht zur Verfügung stehendes Software-Upgrade kanndie Genauigkeit auf 0,1 cm/s verbessert werden. Mit dieser Verbesserung sind weitereVersuche zur Überprüfung der gewonnenen Ergebnisse angebracht. Die bisherigenErgebnisse zeigen jedoch durch ihre trotz der geringen Genauigkeit guten Ergebnisseund einer damit verbundenen hohen Reproduzierbarkeit die Anwendbarkeit der MRTzur Messung der Geschwindigkeiten.
6.2.3 Modellierung in FLUENT
In Bild Bild 6.25 sind die Ergebnisse aus FLUENT und die gemessenenGeschwindigkeiten für einen Volumenstrom von 200 ml/min in der SuperformanceSäule (26 mm) dargestellt.
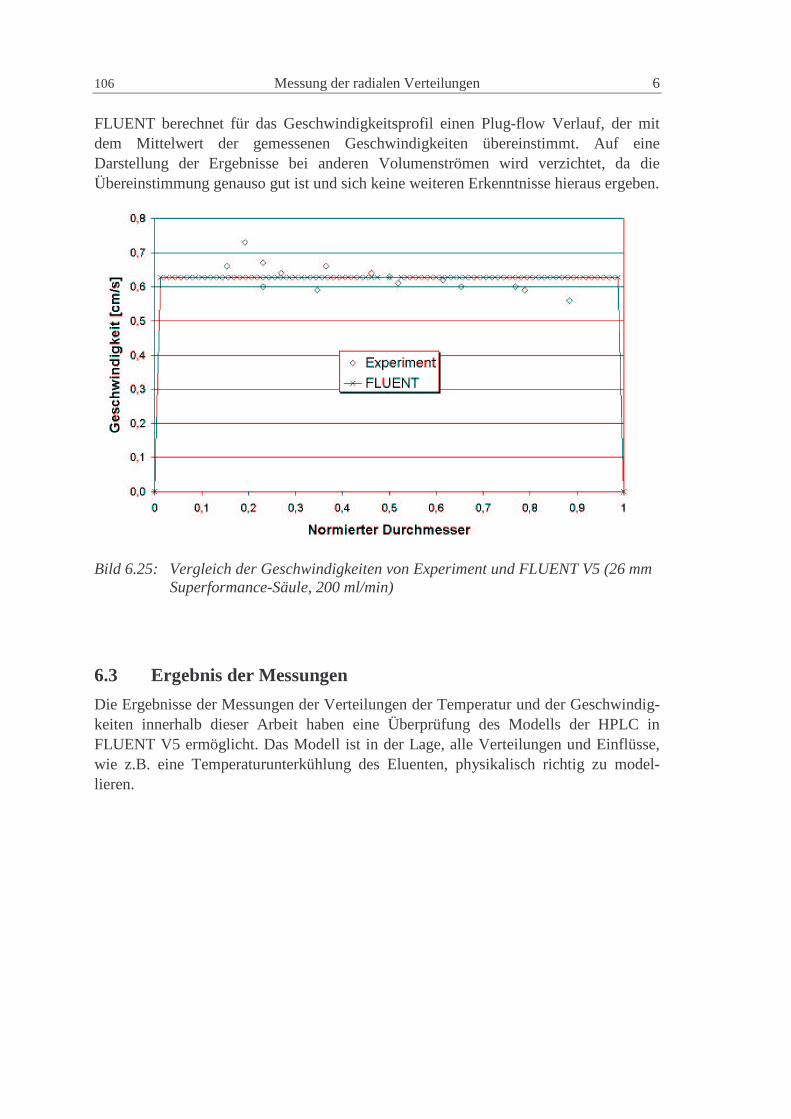
106 Messung der radialen Verteilungen 6
FLUENT berechnet für das Geschwindigkeitsprofil einen Plug-flow Verlauf, der mitdem Mittelwert der gemessenen Geschwindigkeiten übereinstimmt. Auf eineDarstellung der Ergebnisse bei anderen Volumenströmen wird verzichtet, da dieÜbereinstimmung genauso gut ist und sich keine weiteren Erkenntnisse hieraus ergeben.
Bild 6.25: Vergleich der Geschwindigkeiten von Experiment und FLUENT V5 (26 mmSuperformance-Säule, 200 ml/min)
6.3 Ergebnis der Messungen
Die Ergebnisse der Messungen der Verteilungen der Temperatur und der Geschwindig-keiten innerhalb dieser Arbeit haben eine Überprüfung des Modells der HPLC inFLUENT V5 ermöglicht. Das Modell ist in der Lage, alle Verteilungen und Einflüsse,wie z.B. eine Temperaturunterkühlung des Eluenten, physikalisch richtig zu model-lieren.

7 Zusammenfassung und Ausblick 107
7 Zusammenfassung und Ausblick
7.1 Zusammenfassung
Diese Arbeit beschäftigt sich mit der Charakterisierung der Verteilungen vonTemperatur, Geschwindigkeit und Substanzen in chromatographischen Systemen.Experimentelle Charakterisierungen der Verteilungen sind insbesondere zur Validierungvon Modellrechnungen notwendig. Die Charakterisierung erfolgt in dieser Arbeit durchzwei unterschiedliche Ansätze, einen direkten und einen indirekten Ansatz:
Im indirekten Ansatz wird die Verteilung der Probesubstanzen über die Bestimmung deraxialen Dispersion aus Pulsexperimenten als Maß der Verteilungsgüte charakterisiert.Die Ergebnisse verbessern die eindimensionale Modellierung der HPLC-Säulen, z.B.zur Verfahrensauslegung.
Die Vorteile einer Unterteilung der axialen Dispersion in anlagenspezifische undphasenspezifische Anteile wurden an Hand der Trennsysteme Glaskugeln/Methanol/Diethylphthalat und Uetikon/Methanol/Diethylphthalat demonstriert. Aus dieser Unter-teilung ergeben sich deutliche Vorteile für die Auslegung präparativer HPLC-Systeme.Auswertungen einer Referenz ermöglichen eine verbesserte Vorausbestimmung deraxialen Dispersion im präparativen Maßstab für ein neues Trennsystem, da so auch dieAnlageneinflüsse berücksichtigt werden können. Aus den Experimenten undSimulationen mit der Hochleistungsphase Kromasil ergab sich allerdings eineBegrenzung der Einsetzbarkeit der Ergebnisse der Referenz auf qualitativ gleichwertigestationäre Phasen (Gruppen ähnlicher Phasen). Jedoch ergab sich auch für dieExperimente mit Kromasil mit unterschiedlichen Eluentien ein gut bestimmbarerEinfluss der Anlagen.
Im direkten Ansatz werden die Verteilungen innerhalb der gepackten Säule gemessen.Es wurden Messsysteme zur Messung der Temperatur und der Geschwindigkeit mitdem Ziel der Validierung von Simulationsergebnissen aus FLUENT entwickelt undaufgebaut. Ergebnisse dieser Messungen sind z.B. zur Überprüfung von zwei- oderdreidimensionalen Modellierungen von HPLC-Säulen (z.B. zur Entwicklung neuerVerteilersysteme) von großem Interesse.
Zur Messung der Temperaturprofile wurde ausgehend von den Erfahrungen von A.Brandt (1997) ein System entwickelt, das den Einfluss der Fühler auf die Strömung unddie Trennung innerhalb des chromatographischen Bettes reduziert. Durch seinen Einbauin einen axial komprimierbaren Füllstand ist es flexibel genug, um beliebigeTrennsysteme zu untersuchen und gleichzeitig den jeweiligen Einfluss der Fühler aufdie Trennung zu quantifizieren. Drei am Fachgebiet von Prof. Arlt vorhandenestationäre Phasen (Glaskugeln, Uetikon, Kromasil) wurden auf die Ausbildung vonTemperaturprofilen bei Unterkühlung und Erwärmung des Eluenten gegenüber der

108 Zusammenfassung und Ausblick 7
Säulenwand hin untersucht und der Einfluss der Fühler bestimmt. Für einfacheProbenaufgaben wurde die Verbesserung der NTU durch Eluentenunterkühlunguntersucht und die Ergebnisse dargestellt. An Hand dieser Ergebnisse wurden inFLUENT aufgebaute Modelle zur Beschreibung der HPLC untersucht und erfolgreichvalidiert.
Um eine Überprüfung der Geschwindigkeitsprofile innerhalb der chromatographischenBetten zu ermöglichen, wurde der Einsatz eines Magnet-Resonanz-Tomographenuntersucht. Der Tomograph wurde von Dr.-med. R. Kaiser für diese Messungen zurVerfügung gestellt. Mit Hilfe der für medizinische Zwecke installierten Software zurFlussquantifizierung wurde eine semipräparative HPLC-Säule untersucht, in der zuvoralle metallischen Komponenten durch nichtmetallische ersetzt worden sind. MitUetikon als stationärer Phase wurden die mittleren Geschwindigkeiten durch eindefiniertes Kontrollvolumen bestimmt. Trotz Einschränkungen der vom Herstellergarantierten Genauigkeiten ergaben sich gute Ergebnisse sowohl in Reproduzierbarkeitals auch Größe der gemessenen Geschwindigkeiten.
Mit Hilfe dieser direkten Messungen ist es möglich gewesen, das in FLUENTimplementierte Modell zu validieren und zu erweitern.
7.2 Ausblick
Für den Einsatz des Referenzsystems sind weitere Versuche mit weiteren stationärenPhasen unterschiedlicher Leistungsfähigkeit sinnvoll, um eine mögliche Einteilung inAnwendungsklassen und die Anwendbarkeit der vorgeschlagenen Bestimmung derAnlageneinflüsse zu überprüfen. Die Ansätze sollten mit Erfahrungen derHauptanwender der HPLC (der Industrie) verglichen werden, da hiermit eine großeDatenmenge zur Auswertung zur Verfügung stehen würde. Dies ist insbesondere vonVorteil, da Experimente kosten- und zeitintensiv sind. Experimentelle Daten bereitsdurchgeführter Trennungen unterschiedlichster Systeme können jedoch zur Auswertungherangezogen werden.
Ein wesentlicher Punkt zukünftiger Arbeit ist eine Untersuchung, ob sich einzelneKomponenten der Anlagen, wie z.B. eine bestimmte Fritte, untereinander in ihrenEinflüssen signifikant unterscheiden. Das bedeutet, ob ein Einbau einer neuen Fritte,durch ihre gegenüber der ersetzten Fritte unterschiedlichen Verteilungseigenschaften,die anlagenspezifische Dispersion verändert.
Um die Genauigkeit der Beschreibung der axialen Dispersion zu erhöhen, können dieUntersuchungen zu den Referenzsystemen mit komplexeren Ansätzen zur Berechnungder axialen Dispersion an Stelle des linearen Ansatzes untersucht werden.
Die Einsatzmöglichkeit der Messung der Temperaturprofile zur online Kontrolle vonProzessbedingungen sollte untersucht werden. So kann die Vermessung in größereAnlagen Aufschluss auf Verteilungsprobleme geben, z.B. bei Aufgabe an verschiedenen

7 Zusammenfassung und Ausblick 109
Positionen mit unterschiedlicher Temperatur. Das registrierte Temperaturprofil gibtdann Auskunft über Veränderungen in der Verteilung.
Zur Messung von Geschwindigkeitsverteilungen könnte die Methode von Broyles et al.(1998) (durchsichtige Säule und Bett) durch Aufnahme der Wanderung der Probendurch die Säule auf Videomaterial erweitert werden. Der Einsatz der NMR muss durchUpgrade der Software bzw. Anpassung an das Messsystem Chromatographie verbessertwerden, um die Ergebnisse bestätigen zu können. Die Auflösung und dieEmpfindlichkeit können eventuell durch den Einsatz geeigneter Kontrastmittel nochverbessert werden.
Bei der Modellierung ist eine Erweiterung der bereits erstellten Modelle in FLUENTwünschenswert, so dass Adsorption uneingeschränkt modellierbar wird. Mit Hilfe deskompletten Modells sollte dann auch eine Entwicklung neuer, verbesserter Verteiler-systeme möglich sein.

110 Zusammenfassung und Ausblick 7

8 Formelzeichen, Abkürzungen, Kennzahlen 111
8 Formelzeichen, Abkürzungen, Kennzahlen
8.1 Formelzeichen
Buchstabe Einheit Bedeutung
______________________________________________________________________
A m Parameter der Van Deemter Kurve
A m2 Fläche
Ai - Parameter der Adsorptionsisotherme nach Ching
Ab % Ausbeute
B0 T magnetische Feldstärke (NMR)
Bi m²/s Parameter der Van Deemter Kurve
Bij - Parameter der Adsorptionsisotherme nach Ching
Bv * /s Bildungsrate, allgemein
b * /m3 Konzentration, allgemein
bi m³/kg Krümmungsparameter der Langmuir-Gleichung
Ci s Parameter der Van Deemter Kurve
CDax m Konstante des axialen Dispersionskoeffizienten
Cp J/kgK Wärmekapazität
ci kg/m3 Flüssigphasenkonzentration der Komponente i
Dax m2/s axialer Dispersionskoeffizient
Dij m2/s binärer Diffusionskoeffizient
Dm,i m2/s molekularer Diffusionskoeffizient
d mm Durchmesser, Schichtdicke (NMR)
F N Kraft
F * /s Fluss
f * /s diffusiver Anteil des Flusses
G m Amplitude der Gradientenimpulse (NMR)
g m/s2 Erdbeschleunigung, spez. Gravitationskraft
g i m/s2 spezifische Volumenkraft auf Komponente i durch äußereFelder (elektrische Felder, Gravitationsfeld, ...)

112 Formelzeichen, Abkürzungen, Kennzahlen 8
g(t) Gradient des Magnetfeldes (NMR)
Hv J Energiebildungsrate
HETP m Höhe einer theoretischen Trennstufe (NTU)
h J/kg spezifische Enthalpie
h J/mol partiell molare Enthalpie
hi m Peakhöhe
J kg/s Anteil des diffusiven Stoffstroms
j i kg/s Stoffstrom relativ zur mittleren Geschwindigkeit
Ki - Adsorptionsgleichgewichtskoeffizient
keff,i 1/s effektiver Stoffübergangskoeffizient (Festfilmmodell)
ik ′ - Kapazitätsfaktor
Lb - Lösungsmittelbedarf
l m Länge
m kg Masse
mi - Parameter der Adsorptionsisotherme nach Ching
Ni mol/s molarer Stoffstrom
NTUi - Anzahl der theoretischen Trennstufen
n - Normalenvektor
ni,j - Parameter der Adsorptionsisotherme nach Ching
O m2 Oberfläche
P bar Druck
∆P bar Druckverlusst
Pd kg/(hm3) Produktivität
Q J Wärme
q J/s Wärmestrom relativ zur mittleren Geschwindigkeit
q m Wellenvektor (NMR)
qi kg/m3 Beladung der stationären Phase mit i, Bezugsvolumen:VPar
qi* kg/m3 Beladung der stationären Phase im Gleichgewicht mit ci
qS,i kg/m³ Sättigungsbeladung, Parameter der Langmuir-Gleichung
Rh % Reinheit
RS - Auflösung

8 Formelzeichen, Abkürzungen, Kennzahlen 113
Rv 1/s Standardbildungsrate
r m Ortsvektor
S(t) - Signal (NMR)
s(n) Pa Spannung
T °C Temperatur
TF,i - Tailingfaktor
TE s Echozeit (NMR)
TR s Wiederholzeit (NMR)
t s Zeit
t0, t0,ext s Retentionszeit einer nicht retardierten Substanz, die nichtin die Poren eindringt, Totzeit
t0,ges s Retentionszeit einer nicht retardierten Substanz, die in diePoren eindringt, Nettoretentionszeit
tdiff s Zeitkonstante der Diffusion
tR,i s Retentionszeit
tverz s Verzögerungsszeit
u J/kg spezifische innere Energie
V m³ Volumen
Vext m³ Zwischenkornvolumen
Vint m³ intrapartikuläres Flüssigkeitsvolumen
Vverz m³ Verzögerungsvolumen
V� m3/s Volumenstrom der flüssigen Phase
W J Arbeit
w m/s Geschwindigkeit, Zwischenkorngeschwindigkeit
w m/s Leerrohrgeschwindigkeit
wo m/s Oberflächengeschwindigkeit
wc,i m/s Konzentrationswanderungsgeschwindigkeit
wfl m/s Geschwindigkeit der flüssigen Phase
wrel m/s Relativgeschwindigkeit
z m Koordinate in axialer Richtung
∆z m Ortsschrittweite (Länge einer Stützstelle)

114 Formelzeichen, Abkürzungen, Kennzahlen 8
8.2 Griechische Formelzeichen
Buchstabe Einheit Bedeutung
______________________________________________________________________
αij - Trennfaktor
β - Stabilitätsfaktor
δδδδ - Identität / Einheitsvektor
δ s Breite / Läge des Gradientenimpulses (NMR)
εext - externe Porosität
εint - interne Porosität
εges - Gesamtporosität
κ m2 Permeabilität
λλλλ W/mK Wärmeleitfähigkeit
µ ? Scherviskosität
γ 1/s gyromagnetische Präzessionsrate (NMR)
γ1, γ2 - Konstanten zur Berechnung von Dax
η Pa⋅s dynamische Viskosität, Scherviskosität
η′ Pa⋅s Volumenviskosität
ΦΦΦΦ W Dissipationsleistung, viscous dissipation rate
Φ - Phasenverhältnis, Phasenverschiebung (NMR)
µ1,'
i s erstes bezogenes Anfangsmoment
µ2,i s² zweites zentrales Moment
ω 1/s Resonanzfrequenz (NMR)
ρ kg/m3 Dichte
ρ(r) - Spindichte (NMR)
ρi kg/m3 Massenanteil
ν m²/s kinematische Viskosität
σσσσ Pa Spannungstensor
σ i2 s² Varianz
� Pa Zähigkeitsspannungstensor
ωi min Basisweite eines Peaks

8 Formelzeichen, Abkürzungen, Kennzahlen 115
ω0.1,i min Peakweite bei 1/10 der Peakhöhe hi
ΓΓΓΓ Pa Zähigkeits- / Viskositätstensor
8.3 Indizes
Buchstabe Bedeutung
______________________________________________________________________
exp experimentell
ext extern (ohne Partikel)
E Wert am Eintritt in die Säule
fest feste Phase
fl flüssige Phase
ges gesamt, extern + intern
i,j Komponente i,j
inj Wert der Injektion / Probenaufgabe
int intern (innerhalb der Partikel)
max maximal
min minimal
mob mobile Phase
P, Par Partikel
S Säule
s Sättigung
stat stationäre Phase
verz Verzögerung
8.4 Abkürzungen
Abkürzung Bezeichnung
______________________________________________________________________
DEP Di-Ethylphthalat
GK Glaskugeln
HPLC Hochdruckflüssigchromatographie

116 Formelzeichen, Abkürzungen, Kennzahlen 8
Krom Kromasil
MeOH Methanol
nHept n-Heptan
Uet Uetikon
8.5 Kennzahlen
Symbol Definition Bezeichnung
______________________________________________________________________
Pew l
Dfl S
ax
Peclet-Zahl
Red wP extε
νReynolds-Zahl
Sciν
D m,i
Schmidt-Zahl
8.6 Tensorschreibweise
w Skalar, z.B. Geschwindigkeit in eine Raumrichtung
�w , Vektor (Geschwindigkeit), Tensor (Spannungstensor)
e Einheitsvektor
gF ⋅ Skalarprodukt
we Dyadisches Produkt
∇ Nabla-Operator oder Gradient
w⋅∇ Divergenz
∇⋅∇=∇2 Laplace-Operator
∇⋅+∂∂= wtDt
DSubstantielle Ableitung
( )w∇:G Doppelt skalares Produkt, Double-Dot Produkt

9 Literaturverzeichnis 117
9 Literaturverzeichnis
Aced G., Möckel H. J.,
Liquidchromatographie,
VCH Verlagsgesellschaft, Weinheim, 1991
Alima, G. F.,
Diplomarbeit Technische Universität Berlin,
Institut für Prozess- und Anlagentechnik, 2001
Altenhöhner U., Meurer M., Strube J., Schmidt-Traub H.,
J. Chromat. A, 769, 59-69, 1997
Arpagaus C.
Semesterarbeit, Technische Universität Berlin,
Institut für Verfahrenstechnik, 2000
Baehr H.D., Stephan K.,
Wärme- und Stoffübertragung,
Springer Verlag, Berlin 1998
Bart H.J., Messenböck R.C., Byers C.H., Prior A., Wolfgang J.
Chem. Eng. Process. 35, 459-471, 1996
Bayer E., Baumeister E., Tallarek U., Albert K., Guiochon G.
J. Chromat. A 704, 37-44, 1995
Beste Y.
Dissertation, Technische Universität Berlin, 2001a
Beste Y., Laiblin T., Lenz K., Arlt W., Boysen H., Wozny G.,
ECCE (European Conference on Chemical Engineering) 2001
Nürnberg, 26.-28.06. 2001b
R.B. Bird, W.E. Steward, E.N. Lightfoot
Transport Phenomena,
Wiley, New York 1960
Boysen H., Wozny G., Laiblin T., Arlt W.
FORTWIHR (Forschungsbereich Technisch-WissenschaftlichesHochleistungsrechnen) 2001 conference,
Erlangen, 12. - 14.03. 2001a

118 Literaturverzeichnis 9
Boysen H., Wozny G., Laiblin T., Arlt W.
GVC/DECHEMA Fachausschusssitzung Adsorption,
Bamberg, 05. - 06.04. 2001b
Boysen H.
Dissertation, Technische Universität Berlin, 2002
Brandt A.,
Dissertation, Technische Universität Berlin, 1997
Brandt A., Mann G., Arlt W.,
J Chromat. A 796, 223-228, 1998
Brauer H.,
Grundlagen der Einphasen- und Mehrphasenströmungen,
Verlag H. R. Sauerländer & Co., Frankfurt/Main, 1971
Broughton D.B.,
Sep. Sci. Technol., 19(11&12), 723-736, 1984
Broyles B.S., Shalliker R.A., Cherrak D.E., Guiochon G.,
J. Chromat. A, 822, 173-187, 1998
Broyles B.S., Shalliker R.A., Guiochon G.,
J. Chromat. A 867, 71-92, 2000
Brozio J., Bart H.J.
Chem. Ing. Tech. 72, 708-713, 2000
Callaghan P.T.
Priciples of Nuclear Magnetic Resonance Microscopy,
Clarendon, Oxford, 1991
Chang C.T., Watson A.T.
AIChE Journal Vol. 45, No. 9, 437-444, 1999
Ching C. B., Chu K. H., Hidajat K.,
Chem. Eng. Sci., 48(7), 1343, 1993
Dapremont O., Cox G.B., Martin M., Hilaireau P., Colin H.
J. Chromat. A 796, 81-99, 1998
Deckert P.,
Dissertation, Technische Universität Berlin, 1998
Deen W.M.,
Analysis of Transport Phenomena,
Oxford University Press, Oxford 1998

9 Literaturverzeichnis 119
Dwyer J.L.,
Kemia-Kemi, No.3, 1985
Ganetsos G., Barker P.E. (Editors)
Preparative and production scale chromatography,
Marcel Dekker, New York, 1993
Guiochon G., Golshan Shirazi S., Katti A. M.,
Fundamentals of Preparative and Nonlinear Chromatography,
Academic Press, Boston, 1994
Herbsthofer R., Bart H.J., Wolfgang J., Prior A.
Chem. Ing. Tech. 73, 56-59, 2001
Heuer C., Seidel-Morgenstern A.,
J. Chromat. A, 752, 19, 1996
Jones M.C., Nassimbene R.D., Wolfe J.D.
Chem. Eng. Sci. 51(7), 1009-1021, 1996
Kast W.,
Adsorption aus der Gasphase,
Verlag Chemie, Weinheim, 1988
Laiblin T., Arlt W., Lisso M., Boysen H., Wozny G.
GVC-Jahrestagung 2000,
Karlsruhe, 20. - 22.09. 2000a
Laiblin T., Arlt W., Lisso M., Boysen H., Wozny G.
International Symposium on Preparative and Industrial Chromatography andAllied Techniques (SPICA) 2000,
Zürich, 09. - 11.10. 2000b
Lisso M., G. Wozny, Y. Beste, T. Laiblin, W. Arlt,
AIChE Annual Meeting 1999,
Dallas/TX, USA, 1999
Lisso M., Wozny G., Arlt W., Beste Y. A.
Chem. Ing. Tech., 72, 494, 2000
Lisso M.
Dissertation, Technische Universität Berlin, 2001
Moran P.R.,
Magn. Reson. Imaging, 1, 197, 1982

120 Literaturverzeichnis 9
Lode F.G., Rosenfeld A., Yuan Q.S., Root T.W., Lightfoot E.N.,
J. Chromat. A, 796, 3, 1998
Lode F.G.,
M.Sc. Thesis, University of Winsconsin, 1997
Merck KGaA
Betriebsanleitung zum Selbstfüllstand NW100,
Version vom 25.04.1996, LPRO CHROM 2, Darmstadt 1996
Meyer V.,
Praxis der Hochleistungsflüssigchromatographie,
Otto Salle Verlag, Frankfurt a.M., 7. Auflage, 1992
Mottlau A.Y., Fisher N.E.
Anal. Chem. 34(6), 714-715, 1962
Peev G., Tzibranska I.
Chem. Eng. Proc. 36, 317-325, 1997
Poppe H., Kraak J.C., Huber J.F.K., van den Berg J.H.M.
Chromatographia, 14(9), 515, 1981
Roper D.K.,
Ph.D. Thesis, University of Wisconsin, 1994
Ruthven D.M.,
Principles of Adsorption and Adsorption Processes,
Wiley, New York, 1984
Schaarschuh P.
Studienarbeit, Technische Universität Berlin,
Institut für Verfahrenstechnik, 2000
Schade H., Kunz E.,
Strömungslehre
Walter de Gruyter, Berlin 1980
Scheer J.
Studienarbeit, Technische Universität Berlin,
Institut für Verfahrenstechnik, 2001
Schwedt G.
Chromatographische Trennmethoden
Georg Thieme Verlag Stuttgart, 1994

9 Literaturverzeichnis 121
Seidel-Morgenstern A.,
Mathematische Modellierung der präparativen Flüssigchromatographie,
Deutscher Universitäts-Verlag GmbH, Wiesbaden 1995
Tallarek U., Baumeister E., Albert K., Bayer E., Guiochon G.
J. Chromat. A 696, 1-18, 1995
Tallarek U., Bayer E., van Dusschoten D., Scheenen T., van As H., Guiochon G., Neue
AIChE J., 44(9), 1962-1975 1998a
Tallarek U., van Dusschoten D., van As H., Bayer E., Guiochon G.
J. Phys. Chem. B 102, 3486-3497, 1998b
Tallarek U., van Dusschoten D., van As H., Guiochon G., Bayer E.
Magn. Res. Imag. 16, Nos 5/6, 699-702, 1998c
Tsotsas E.,
Über die Wärme- und Stoffübertragung in durchströmten Festbetten
VDI Fortschrittsberichte, Reihe 3, Nr. 223, 1987
Unger K. K.,
Handbuch der HPLC, Teil 2: Präparative Säulenflüs-sigchromatographie
GIT Verlag, Darmstadt, 1994
van Deemter J.J., Zuiderweg F.J., Klinkenberg A.,
Chem. Eng. Sci., 5, 271, 1956
Wu Y.W., Ching C.B., Lisso M., Wozny G., Laiblin T., Arlt W.
J Chrom. A., eingereicht, noch nicht veröffentlicht, 2001
Yamamoto S.,
Biotech. Bioeng. 48, 444-451, 1995

122 Literaturverzeichnis 9

10 Anhang 123
10 Anhang
10.1 GPROMS® Code zur Lösung der axialen Komponentenbilanz
#---------------------------------------------------------------------------# Declarations#---------------------------------------------------------------------------
DECLARE TYPE # name =initial: lower : upper limit unit # --------------------------------------------------------------------------
Concentration =0 : -1.0 : 1.0e10 unit="kg/m3" Velocity =0 : -1.0e10 : 1.0e10 unit="m/s" Axiale_dispersion =0 : 0.0 : 1.0e10 unit="m2/s" NoType =0 : -1e9 : 1e9END
#---------------------------------------------------------------------------# Model description#---------------------------------------------------------------------------
MODEL Saeule
PARAMETER ReaktorLenght AS REAL DEFAULT 0.271 # Saeulenlaenge [m] A_s AS REAL DEFAULT 7.854e-5 # Querschnittsflaeche der Saeule [m2] # eps_ges AS REAL DEFAULT 0.7962 # Gesamtporositaet [-] eps_ext AS REAL DEFAULT 0.3204 # Externe Porositaet [-] V_Probe AS REAL DEFAULT 0.8e-6 # Probevolumen [m3] # C_dax AS REAL DEFAULT 6.160e-5 # Dispersionkonstante [m] D_add AS REAL DEFAULT 0 # Dispersionskoeffizient [m2/s] q_s AS REAL DEFAULT 1621.510 # Saettigungskapazitaet Methanol [kg/m3] b AS REAL DEFAULT 4.577e-4 # Langmuir-Paramter Methanol bezogen aufeps_ext [m3/kg] L_in AS REAL DEFAULT 6.67e-8 # Eluentenstrom 4 ml/min [m3/s] # k_ggw AS REAL DEFAULT 5.076e1 # Stoffübergangskoeffizient [1/s]
DISTRIBUTION_DOMAIN Axial AS [ 0 : ReaktorLenght ]
VARIABLE u_s AS Velocity # Zwischenkorngeschwindigkeit [m/s] D_ax AS Axiale_dispersion # axiale Dispersion [m2/s] c1_in AS Concentration # Eintrittskonzentration [kg/m3] c1_out AS Concentration # Austrittskonzentration [kg/m3] F AS NoType # Phasenverhaeltnis [-] c1 AS DISTRIBUTION(Axial) OF Concentration # Konzentration [kg/m3] q1_ggw AS DISTRIBUTION(Axial) OF Concentration # Gleichgewichtsbeladung derstationaeren Phase [kg/m3] q1 AS DISTRIBUTION(Axial) OF Concentration # Beladung der stationaeren Phase[kg/m3]

124 Anhang 10
C_dax AS NoType # Dispersionkonstante [m] k_ggw AS NoType # Stoffübergangskoeffizient [1/s]
SET Axial := [CFDM,2,8000]; # Loesungsmethode, 2.Ordnung, 8000 Stützstellen
BOUNDARY c1(0) = c1_in ; # Randbedingungen c1_out = c1(ReaktorLenght);
EQUATION u_s=L_in/(eps_ext*A_s); # Geschwindigkeit bestimmen D_ax=u_s*C_dax+D_add; # axiale Dispersion bestimmen F=(1-eps_ext)/eps_ext; # Phasenverhaeltnis bestimmen
FOR z := 0|+ TO ReaktorLenght DO # Konzenration an der Stelle z berechnen $c1(z)=D_ax*PARTIAL(c1(z),Axial,Axial)-u_s*PARTIAL(c1(z),Axial)-F*k_ggw*(q1_ggw(z)-q1(z)); END
FOR z := 0 TO ReaktorLenght DO $q1(z)=k_ggw*(q1_ggw(z)-q1(z)); # Beladung berechnen END
FOR z := 0 TO ReaktorLenght DO q1_ggw(z)=q_s*(b*c1(z))/(1+b*c1(z)); # Gleichgewichtsbeladung berechnen ENDEND
#---------------------------------------------------------------------------# Process description#---------------------------------------------------------------------------
PROCESS Batch_40g4ml_est
UNIT T001 AS Saeule
ASSIGN WITHIN T001 DO c1_in := 0; C_dax := 6.160e-5; k_ggw := 5.076e1; END
INITIAL FOR z := 0|+ to T001.ReaktorLenght DO T001.c1(z) = 0.0; END
FOR z := 0 to T001.ReaktorLenght DO T001.q1(z) = 0.0; END
SOLUTIONPARAMETERS AbsoluteAccuracy := 1.0e-9 # absolute Genauigkeit einstellen RelativeAccuracy := 1.0e-9 # relative Genauigkeit einstellen EffectiveZero := 1.0e-5 # Nullwert einstellen

10 Anhang 125
InitAccuracy := 1.0e-5 # Initialisierungsgenauigkeit einstellen ReportingInterval := 2 # Schrittweite 2 s # Monitor := OFF # Ausgabe auf Monitor ausschalten
SCHEDULE SEQUENCE # MONITOR OFF # Ausgabe unterdrücken RESET T001.c1_in := 40; # Probenkonzentration 40 g/l bzw. [kg/m3](Sprung beginnt) END CONTINUE FOR T001.V_Probe/T001.L_in RESET T001.c1_in := 0; # Probenmenge injetziert (Sprung beendet) END CONTINUE FOR 540 # Rechenzeit 9 min # CONTINUE FOR 180 # Rechenzeit 3 min # MONITOR ON # Ausgabe einschalten # CONTINUE FOR 360 # Rechenzeit 6 min ENDEND
10.2 GPROMS® Code zum Parameter fitting
ESTIMATET001.C_dax6.160e-5 1e-6 1e-4
ESTIMATET001.k_ggw5.076e1 1e-3 1e3
RUNSMESSUNG_40G4ML 1


















