
Claudia Schelenz - km-potsdam.de · Die akuten myeloischen Leukämien sind klonale...
37
Akute myeloische Leukämie Claudia Schelenz 1
Transcript of Claudia Schelenz - km-potsdam.de · Die akuten myeloischen Leukämien sind klonale...

Akute myeloische Leukämie
Claudia Schelenz
1

Die akuten myeloischen Leukämien sind klonale Stammzellerkrankungen, die durch einen Reifungsarrest mit Expansion meist unreifer myeloischer Zellen charakterisiert sind. Zum Zwecke der morphologischen Einteilung können die myeloischen Zellen in vier Gruppen unterteilt werden.

Morphologische Einteilung myeloischer Zellen
Granulopoetische Zellreihe
Monopoetische Zellreihe
Erythropoetische Zellreihe
Megakaryopoetische Zellreihe

Die klinische Manifestation der akuten myeloischen Leukämie beruht auf der hämatologischen Insuffizienz, die durch eine Blasteninfiltration des Knochenmarkes entsteht.

Die reife Zelle kann eine Funktion ausüben, Phagozytose findet statt und Antikörper werden gebildet.
Die unreife Zelle kann keine Funktion ausüben. Der Patient verstirbt ohne Therapie an nicht beherrschbaren Infekten oder Blutungen, an Zellabfallprodukten oder durch zu viele Zellen an gestörter Viskösität = hämatologischer Herzinfarkt

Befallsmuster
Generell ist das Knochenmark betroffen. Seltener und je nach Subtyp die Milz Lymphknoten, Haut Gingiva, Meningen (AMoL, AM4eoL).
Gehäuft Koagulopathien (plasmatisch) mit Blutungsneigung finden wir bei Promyelozyten-Leukämie (APL).

BefallsmusterAkute Promyelozytenleukämie M3
Häufig kommt es durch Freisetzung von prokoagulatorisch wirkenden zytoplasmatischen Granula zu einer Koagulopathie mit Verbrauchskoagulopathie.
Es kommt zur Freisetzung einer Proteolyse, die den vWillebrand-Faktor abbaut. Durch den Mangel an vWF kommt es zur Blutungsneigung, denn dieser ist für die Adhäsion der Thrombozyten am Subendothel erforderlich.

Differentialdiagnose
Akute lymphatische Leukämie
Agranulozytose
Aplastische Anämie
Myelodysplatisches Syndrom
Chronisch myeloische Leukämie im Blastenschub

Diagnostik
9
Die Diagnostik der AML beruht auf den folgenden Methoden•Zytomorphologie mit Zytochemie •Immunphänotypisierung•Klassische Chromosomenanalyse•FISH-Analyse•Molekulargenetik

Diagnostik
10
•Zytomorphologie mit Zytochemie (Peroxidase und Esterase)Sie dient zur Sicherung der Diagnose, zurKlassifikation nach FAB und WHO und ist immer noch Standarduntersuchung zur Remissionsbeurteilung unter laufender Therapie.

FAB-Klassifikation
11
Um die Diagnose AML stellen zu können ist die zytomorphologische und zytochemische Untersuchung von Ausstrichen des Knochenmarkes und des peripheren Blutes erforderlich. Die FAB-Klassifikation (French-American-British) teilt die AML in 11 morphologisch unterschiedliche Formen ein.
Um die Diagnose AML zu stellen muss der Anteil der Blasten an allen kernhaltigen Zellen mindestens 30% betragen. Mindestens 3% der Blasten müssen Peroxidase positiv sein.

Diagnostik
12
• Immunphänotypisierung Sie dient zur Abgrenzung der AML von der ALL.Sie sichert die Diagnose der AML M7, diemorphologisch häufig schwer zu erkennen istund bestimmt zum Zeitpunkt der Diagnose densogenannten „Leukämie-assoziiertenImmunphänotyp (LAIP), der unter Therapie zumNachweis der minimalen Restkrankheit dient

Diagnostik
13
• Klassische ChromosomenanalyseSie bestimmt den Karyotyp der leukämischen Blasten und gehört bei jedem Patienten mit AML zur Standard-Diagnostik dazu. Sie ist erforderlich zur Klassifikation nach WHO. Der Karyotyp ist zur Zeit der wichtigste unabhängige prognostische Parameter bei der AML. Aus ihm leiten sich therapeutische Konsequenzen ab.

Diagnostik
14
50-75 % der Erwachsenen und 75-85% der Kinder weisen klonale Chromosomenveränderungen auf.In der WHO-Klassfikation wurden die spezifischen Chromosomenveränderungen als entscheidendes Klassifikatonskriterium aufgenommen

Diagnostik
15
Kenntnisse über die Zusammenhänge von genetischen Veränderungen und Therapieansprechen haben dazu geführt, dass bei der AML die Therapieentscheidung vom Karyotyp getroffen wird.Die prognostische Bedeutung des Karyotypes ist altersunabhängig und findet sich auch bei der therapieassoziierten AML.

Zytogenetik
t(8;21) - AML-M2
t(15;17) - AML-M3 und M3 Variante
Inversion 16- AML-M4Eo
t(8;16) - AML-M5 mit Erythrophagozytose
t(9;22) - 1/3 der ALL-Fälle, M1
inv(3) - AML-M1
t(6;9) - M2 und M4 mit Basophilie
Myeloische Marker CD13, CD33, CD117, MPO
Monozytäre Marker CD14, CD 64

Diagnostik
17
• FISH-AnalyseSie erfolgt in Ergänzung zur klassischen Chromosomenanalyse um nachgewiesene Aberationen zu bestätigen. Sie dient im weiteren Verlauf zum Nachweis eventuell vorhandener Resterkrankung unter Therapie Da die Veränderungen des Karyotypes sehr vielfältig sind lässt sich mittels FISH nur ein kleiner Teil der möglichen Aberationen erfassen.FISH ersetzt nicht die klassische Chromosomenanalyse.

Diagnostik
18
• MolekulargenetikSie dient dem Nachweis prognostisch relevanter Fusionstranskripten und von molekularen Mutationen in AML-relevanten Genen. Sie stellt die sensitivste Methode zum Nachweis einer minimalen Resterkrankung dar.

Diagnostik
19
In den letzten Jahren wurden Mutationen und kleine Genrearrangements beschrieben, die zytogenetisch nicht erkennbar sind. Zu diesen Mutationen gehört z.B. die partielle Tandemduplikation im MLL-Gen (MLL-PTD) und die Längenmutation im FLT-Gen.Diese Mutationen werden bei den AML mit zytogenetisch normalem Karyotyp gefunden, haben aber eine ungünstige Prognose.
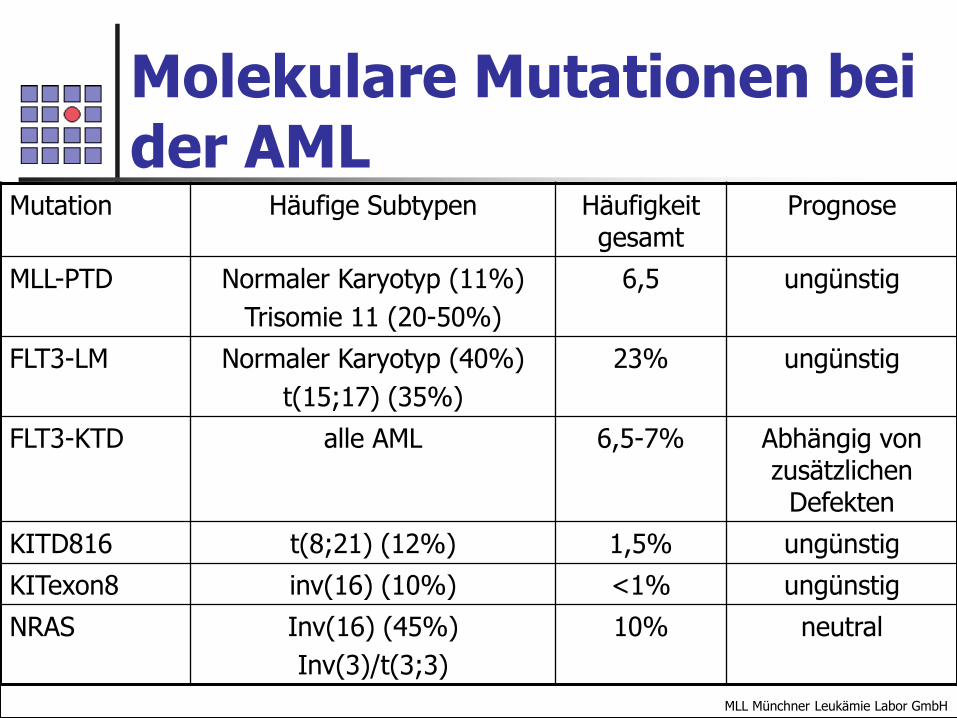
Molekulare Mutationen bei der AML
Mutation Häufige Subtypen Häufigkeit gesamt
Prognose
MLL-PTD Normaler Karyotyp (11%)
Trisomie 11 (20-50%)
6,5 ungünstig
FLT3-LM Normaler Karyotyp (40%)
t(15;17) (35%)
23% ungünstig
FLT3-KTD alle AML 6,5-7% Abhängig von zusätzlichen
Defekten
KITD816 t(8;21) (12%) 1,5% ungünstig
KITexon8 inv(16) (10%) <1% ungünstig
NRAS Inv(16) (45%)
Inv(3)/t(3;3)
10% neutral
MLL Münchner Leukämie Labor GmbH
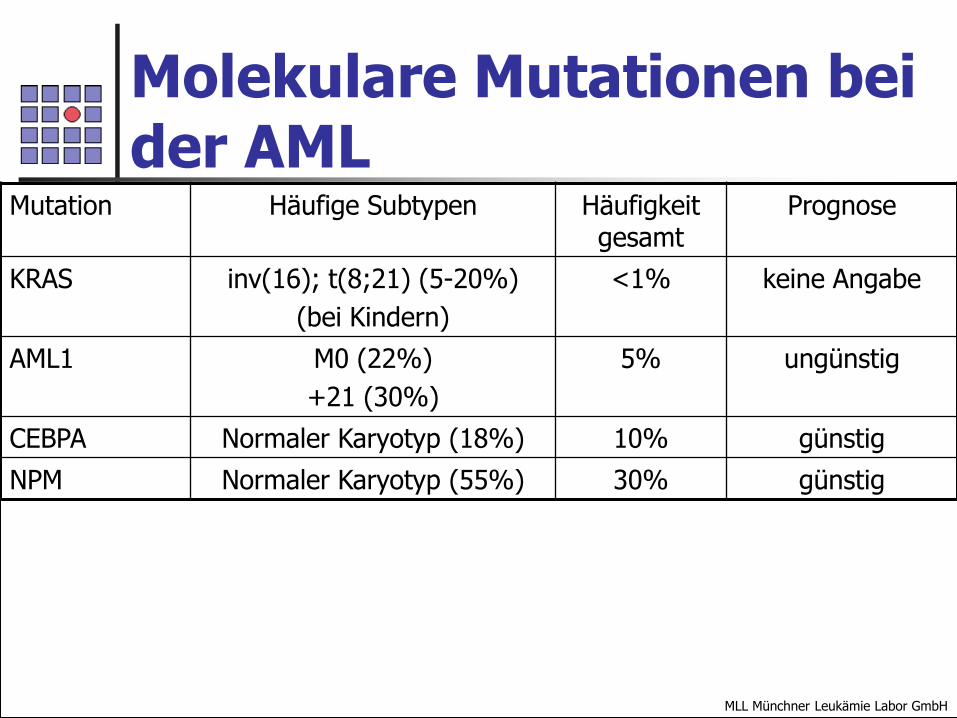
Molekulare Mutationen bei der AML
Mutation Häufige Subtypen Häufigkeit gesamt
Prognose
KRAS inv(16); t(8;21) (5-20%)
(bei Kindern)
<1% keine Angabe
AML1 M0 (22%)
+21 (30%)
5% ungünstig
CEBPA Normaler Karyotyp (18%) 10% günstig
NPM Normaler Karyotyp (55%) 30% günstig
MLL Münchner Leukämie Labor GmbH

WHO-KlassifikationAML mit wiederkehrenden zytogenetischen Abnormalitäten
Akute myeloische Leukämie mit t(8;21)(q22;q22), (AML 1/ETO)
Akute Promyelozytenleukämie, t(15;17)(q22;q21) (M3) (PML/RAR) und Varianten
Akute myeloische Leukämie, inv(16)(p13;q22) oder t(16;16)(p13;22), (CBFb/MYH11)
Akute myeloische Leukämie (v;11q13)

WHO-KlassifikationAML ohne weitere Kategorie
Akute Myeloblasten-Leukämie, minimal differenziert (M0)
Akute Myeloblasten-Leukämie, ohne Ausreifung (M1)
Akute Myeloblasten-Leukämie, mit Ausreifung (M2)
Akute Promyelozytenleukämie (M3)
Akute myelomonoblastäre Leukämie (M4)
Akute monoblastäre Leukämie (M5)
Akute Monoblastenleukämie (M5a)
Akute Monozyten-Leukämie, differenziert (M5b)

WHO-KlassifikationAML ohne weitere Kategorie
Akute erythroblastäre Leukämie (M6)
Akute Erythroleukämie (M6a)
Akute erythroblastäre Leukämie (M6b)
Akute Megakaryoblasten-Leukämie (M7)
Akute Basophilenleukämie
Akute Panmyelose mit Markfibrose

WHO-Klassifikationmit myelodysplasie-assoziierten Merkmalen
Akute myeloische Leukämie mit vorausgegangenem MDS
Akute myeloische Leukämie mit multilineärer Dysplasie ohne anamnestisch vorausgegangenem MDS

WHO-KlassifikationAML, therapieassoziiert
Assoziation mit Alkylantientherapie
Assoziation mit Tropoisomerase Typ II

Therapieansätze
27
1. Die Einteilung der Patienten erfolgt nach den Risikofaktoren in 3 Risikogruppen (Niedrig-, Intermediär-, Hochrisiko) mit entsprechend angepasster Therapie
2. Die intensive Chemotherapie besteht aus Induktion/ Doppelinduktion, Konsilidierung/ intensiver Konsilidierung, oder Erhaltungstherapie im Rahmen eines Studienprotokolls
3. Stammzelltransplantation mit peripheren Blutstammzellen oder Knochenmark
4. Rezidivtherapie nach Studienprotokollen. Bei schlechtem Allgemeinzustand NW-arme blastenreduzierende Schemata und/oder Supportivtherapie.

TherapieansätzeAm Tag 15 der Induktionstherapie DA-Schema (Daunorubicin/Cytarabin) erfolgt eine Knochenmarkuntersuchung.
Blastenreduktion (<10% Blasten im KM) -> 2. Identischer Induktionskurs grundsätzlich am Tag 22
Blastenreduktion (>10 % Blasten Im KM) -> bei fehlender medizinischer Kontrainduktion sofortiger Beginn der 2. Induktionstherapie und Einstufung des Patienten in die Hochrisikogruppe

Postremissionstherapie
Niedrigrisiko
3 Kurse HD-Cytarabin, der 2. und 3. Postremissionszyklus startet eine Woche nach erreichen der CR-Kriterien, frühestens am Tag 28 des vorherigen Zyklus

PostremissionstherapieStandartrisiko
Mit Familien-Stammzellspender -> allogene SZT
Ohne Familienspender -> 1x Cytarabin-Schema + SZ-Separation, autologe SZT
ohne Spender oder autologe SZ -> 2 Zyklen Cytarabin oder MAMC (Cytarabin/Amsacarin), MAC (Cytarabin/Mitoxantron)

Postremissionstherapie
Hochrisiko
allogene Familien- oder Fremdspender SZT nach 2x DA
ohne Spender -> Cytarabin oder MAC-Schema SZ-Separation, autologe SZT
ohne Spender oder autologe SZ -> 2 Zyklen Cytarabin oder MAMC, MAC

Akute Promyelozytenleukämie (M3)
Die Promyelozytenleukämie wird durch eine Proliferation atypischer Promyelozyten definiert und zeigt eine typische t(15;17) Translokation. Der Gehalt an typischen Myeloblasten liegt im Durchschnitt nur bei 5-8%.

Akute Promyelozytenleukämie (M3)
Die Bruchstelle auf dem Chromosom 17 ist Ort des Retinolsäurerezeptors-Alpha (RAR-Alpha).
Hier ist auch der Angriffpunkt für die Therapie mit ATRA.
ATRA bewirkt eine Induktion zur Differenzierung der Blasten zu reifen Granulozyten.

Therapie
Sofortige Einleitung der Therapie, da mit letalen Komplikationen zu rechnen ist und die kurativen Chance sehr hoch ist.
Hämatologischer Notfall! Sofortiger Beginn mit der supportiven Therapie.

Prognose
35
Im Gesamtkollektiv aller an einer Akuten myeloischen Leukämie erkrankten Erwachsenen werden im Rahmen der Primärtherapie anhaltende Remissionsdaten von 70 % über 5 Jahre erreicht. Damit ist eine Heilung bei etwa 30% der Patienten möglich.
Nach allogener SZT (Familienspender) in der ersten Remission beträgt das krankheitsfreie Überleben 50%.

Nachsorge
Alle 3 Monate durch einen Hämatologen !

Vielen Dank für Ihre Aufmerksamkeit










